
三酸化硫黄分解用触媒、及び水素生成方法
【課題】三酸化硫黄分解触媒、特にI−Sサイクル法で水素を生成する際に必要とされる温度を低下させることができる三酸化硫黄分解触媒を提供する。
【解決手段】本発明の三酸化硫黄分解触媒は、銅とバナジウムとの複合酸化物がシリカ担体に担持されてなり、かつラマン分光分析において、バナジウム−酸素(V−O)結合に起因する920cm−1付近のピークの高さが、(a)920cm−1付近のピークの高さが、バナジウム−酸素(V−O)結合に起因する他のピークの最大高さの3.0倍以下、及び(b)920cm−1付近のピークの半値幅が、30cm−1以上の少なくとも一方の条件を満たす。
【解決手段】本発明の三酸化硫黄分解触媒は、銅とバナジウムとの複合酸化物がシリカ担体に担持されてなり、かつラマン分光分析において、バナジウム−酸素(V−O)結合に起因する920cm−1付近のピークの高さが、(a)920cm−1付近のピークの高さが、バナジウム−酸素(V−O)結合に起因する他のピークの最大高さの3.0倍以下、及び(b)920cm−1付近のピークの半値幅が、30cm−1以上の少なくとも一方の条件を満たす。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、三酸化硫黄(SO3)分解用触媒に関する。また、本発明は、三酸化硫黄分解用触媒を用いて三酸化硫黄を分解する工程を含む水素生成方法に関する。
【背景技術】
【0002】
近年、地球温暖化等の問題から、燃焼時に二酸化炭素を生成しないクリーンエネルギーとしての水素が注目されている。
【0003】
水素の生成のためには一般に、下記式(A1)及び(A2)で示される炭化水素燃料の水蒸気改質が用いられている:
(A1)CnHm + nH2O → nCO + (n+m/2)H2
(A2)CO + H2O → CO2 + H2
全反応:CnHm + 2nH2O → nCO2 + (2n+m/2)H2
【0004】
したがって、水素の燃焼自体は二酸化炭素を生成させないものの、水素の生成においては二酸化炭素を発生させていることが一般的であった。
【0005】
これに関して、炭化水素燃料を用いずに水素を生成させるための方法として、太陽熱エネルギー又は原子力熱エネルギーを用いることが提案されている(特許文献1、非特許文献1)。
【0006】
熱エネルギーを利用して水から水素を生成させる方法としては、下記式(B1)〜(B3)で示されるI−S(ヨウ素−イオウ)サイクル法と呼ばれる方法が提案されている:
(B1)H2SO4(液体)
→ H2O(気体) + SO2(気体) + 1/2O2(気体)
(反応温度=約950℃、ΔH=188.8kJ/mol−H2)
(B2)I2(液体) + SO2(気体) + 2H2O(液体)
→ 2HI(液体) + H2SO4(液体)
(反応温度=約130℃、ΔH=−31.8kJ/mol−H2)
(B3)2HI(液体) → H2(気体) + I2(気体)
(反応温度=約400℃、ΔH=146.3kJ/mol−H2)
【0007】
上記式(B1)〜(B3)で示されるI−S(ヨウ素−イオウ)サイクル法の全反応は下記のとおりである:
H2O → H2 + 1/2O2
(ΔH=286.5kJ/mol−H2(高位発熱量基準)
(ΔH=241.5kJ/mol−H2(低位発熱量基準)
【0008】
ここで、上記式(B1)の反応は、下記式(B1−1)及び(B1−2)の2つの素反応に分けることができる:
(B1−1)H2SO4(液体) → H2O(気体) + SO3(気体)
(反応温度=約300℃、ΔH=90.9kJ/mol−H2)
(B1−2)SO3(気体) → SO2(気体) + 1/2O2(気体)
(反応温度=約950℃、ΔH=97.9kJ/mol−H2)
【0009】
すなわち、I−Sサイクル法で水素を生成する場合、式(B1−2)の三酸化硫黄(SO3)分解反応において最も高い温度を必要とし、この反応で必要とされる高温を得ることが容易でなかった。
【0010】
このような問題に関して、非特許文献1では、熱源として太陽熱エネルギーを用いつつ、必要に応じて天然ガスを燃焼させて、追加の熱エネルギーを得るとしている。
【0011】
また、式(B1−2)の三酸化硫黄分解反応において必要とされる温度を低下させるために、白金触媒を用いることが提案されている。しかしながら、この反応において白金触媒を用いる場合、触媒の使用開始時には高い特性を有するものの、反応によって生成する酸素によって白金が酸化され、白金粒子が粗大化することにより触媒活性が低下することが知られている。また、白金触媒は高価であることから、産業的な規模においては用いることが難しい。
【0012】
これに関して、非特許文献2では、三酸化硫黄分解反応において必要とされる温度を低下させるために、白金(Pt)、クロム(Cr)、鉄(Fe)、及びそれらの酸化物からなる群より選択される触媒をアルミナ担体に担持させて用いることを提案している。
【0013】
また、I−Sサイクル法に関して、特許文献2では、上記式(B2)で表される反応、すなわちヨウ素、二酸化硫黄及び水から、ヨウ化水素及び硫酸を得る反応において、二酸化硫黄と水との反応をカチオン交換膜の正極側で行わせ、かつヨウ素の反応をカチオン交換膜の負極側で行わせることによって、その後の分離操作を省略することを提案している。
【0014】
なお、I−Sサイクル法以外にも、熱エネルギーを利用して水素を生成する方法として、ウエスティングハウス・サイクル、Ispra−Mark 13サイクル法、ロスアラモス・サイエンスラボラトリ・サイクル法等が知られているが、これらの方法においても、式(B1−2)でのようにして、三酸化硫黄を二酸化硫黄と水素とに分解することが必要とされている。
【先行技術文献】
【特許文献】
【0015】
【特許文献1】特開2007−218604号公報
【特許文献2】特開2005−041764号公報
【非特許文献】
【0016】
【非特許文献1】A.Giaconia, et al., International Journal of Hydrogen Energy, 32, 469−481(2007)
【非特許文献2】H.Tagawa, et al.,International Journal of Hydrogen Energy, 14, 11−17(1989)
【発明の概要】
【発明が解決しようとする課題】
【0017】
本発明では、三酸化硫黄分解触媒、特に水から水素を生成する際に必要とされる温度を低下させることができる三酸化硫黄分解触媒を提供する。
【課題を解決するための手段】
【0018】
本件発明者は、鋭意検討の結果、下記の本発明に想到した。
【0019】
〈1〉銅とバナジウムとの複合酸化物がシリカ担体に担持されてなり、かつラマン分光分析において、バナジウム−酸素(V−O)結合に起因する920cm−1付近のピークの高さが、下記の(a)及び(b)の少なくとも一方の条件を満たす、三酸化硫黄分解触媒:
(a)前記920cm−1付近のピークの高さが、バナジウム−酸素(V−O)結合に起因する他のピークの最大高さの3.0倍以下、及び
(b)前記920cm−1付近のピークの半値幅が、30cm−1以上。
〈2〉前記複合酸化物において、銅とバナジウムとの原子比が、1:9〜9:1である、上記〈1〉項に記載の触媒。
〈3〉条件(a)を少なくとも満たす、上記〈1〉又は〈2〉項に記載の触媒。
〈4〉条件(b)を少なくとも満たす、上記〈1〉又は〈2〉項に記載の触媒。
〈5〉条件(a)において、前記920cm−1付近のピークの高さが、バナジウム−酸素(V−O)結合に起因する他のピークの最大高さの1.5倍以下である、上記〈1〉〜〈4〉項のいずれか一項に記載の触媒。
〈6〉条件(b)において、前記920cm−1付近のピークの半値幅が、40cm−1以上である、上記〈1〉〜〈5〉項のいずれか一項に記載の触媒。
〈7〉前記シリカ担体が、細孔構造を有する多孔質シリカ担体である、上記〈1〉〜〈6〉項のいずれか一項に記載の触媒。
〈8〉前記複合酸化物が、前記多孔質シリカ担体の細孔構造内に担持されており、且つ
前記多孔質シリカ担体の細孔分布において、シリカの一次粒子間の間隙に起因するピークが、細孔径5〜50nmの範囲にあり、且つシリカ粒子内の細孔構造に起因するピークが、細孔径1〜5nmの範囲にある、
上記〈7〉項に記載の触媒。
〈9〉(a)銅塩の水溶液及びバナジウム塩の水溶液の一方の水溶液を、シリカ担体に吸水させ、乾燥及び仮焼成すること、
(b)工程(a)の後で、銅塩の水溶液及びバナジウム塩の水溶液の他方の水溶液を、前記シリカ担体に吸水させ、乾燥及び仮焼成すること、そして
(c)工程(b)の後で、得られた前記シリカ担体を700℃以上の温度で焼成すること
を含み、かつ
工程(c)における前記熱処理の前に、前記シリカ担体が700℃以上の熱処理を受けていない、
三酸化硫黄分解触媒の製造方法。
〈10〉(a)銅塩及びバナジウム塩を含有する水溶液を、シリカ担体に吸水させ、乾燥及び仮焼成すること、
(b)工程(a)の後で、得られた前記シリカ担体を700℃以上の温度で焼成すること
を含み、かつ
工程(b)における前記熱処理の前に、前記シリカ担体が700℃以上の熱処理を受けていない、
三酸化硫黄分解触媒の製造方法。
〈11〉上記〈1〉〜〈8〉項のいずれか一項に記載の三酸化硫黄分解触媒を用いて、三酸化硫黄を二酸化硫黄と酸素とに分解することを含む、二酸化硫黄の生成方法。
〈12〉前記分解を800℃以下の温度で行う、上記〈11〉項に記載の方法。
〈13〉水を、水素及び酸素に分解することを含む、水素生成方法であって、下記式(X1)で示される反応で、硫酸を、水、二酸化硫黄、及び酸素に分解することを含み、且つ下記式(X1)で示される反応の素反応である式(X1−1)及び(X1−2)の素反応のうち、式(X1−2)の素反応を、上記〈11〉又は〈12〉項に記載の方法によって行う、水素生成方法:
(X1)H2SO4 → H2O + SO2 + 1/2O2
(X1−1)H2SO4 → H2O + SO3
(X1−2)SO3 → SO2 + 1/2O2
〈14〉I−Sサイクル法、ウエスティングハウス・サイクル法、Ispra−Mark 13サイクル法、又はロスアラモス・サイエンスラボラトリ・サイクル法である、上記〈13〉項に記載の水素生成方法。
【発明の効果】
【0020】
本発明の三酸化硫黄分解触媒によれば、三酸化硫黄分解反応に必要とされる温度を低下させることができる。また、本発明の二酸化硫黄生成方法によれば、比較的低い温度において、三酸化硫黄を分解して二酸化硫黄を得ることができる。さらに、本発明の水素生成方法によれば、比較的低い温度において、水を分解して水素を得ることができる。
【図面の簡単な説明】
【0021】
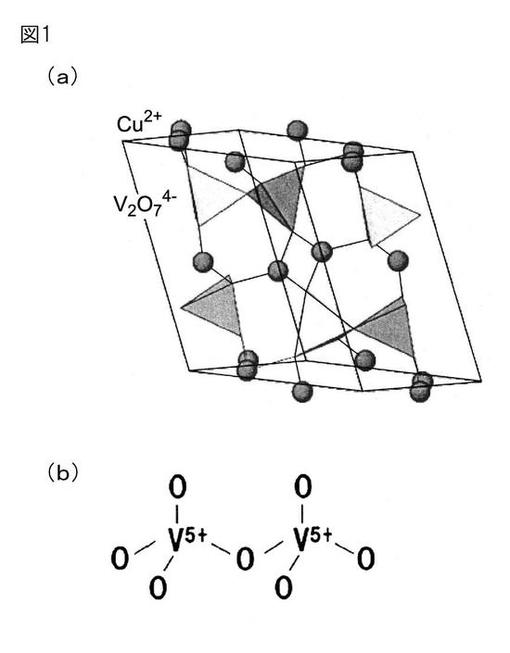
【図1】図1は、ピロバナジン酸銅の(a)結晶構造及び(b)バナジン酸結合を示す図である。
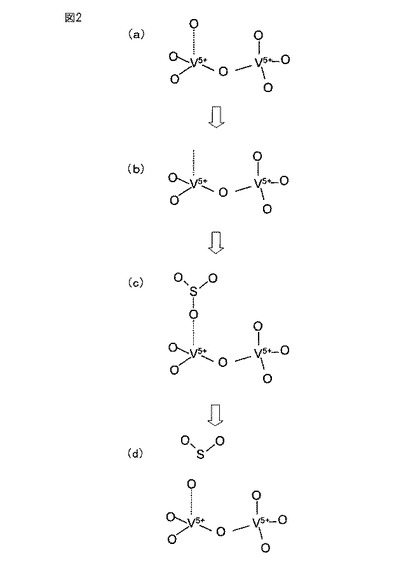
【図2】図2は、本発明の三酸化硫黄分解触媒による触媒反応過程を示す概念図である。
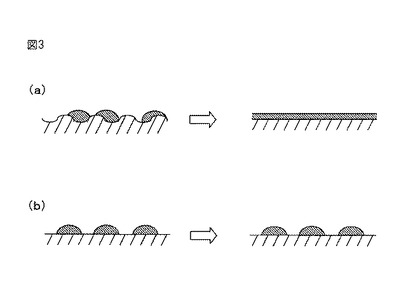
【図3】図3は、(a)本発明の三酸化硫黄分解触媒の製造過程、及び(b)従来の三酸化硫黄分解触媒の製造過程を説明するための図である。
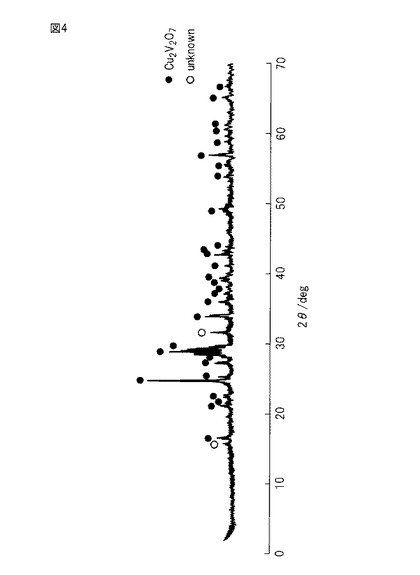
【図4】図4は、参考例1で単身触媒として用いた複合金属酸化物についてのX線回折分析の結果を示す図である。
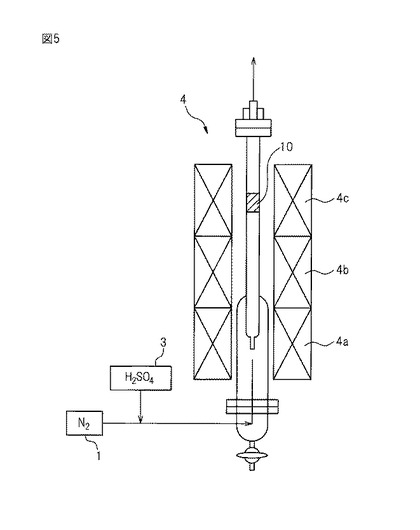
【図5】図5は、実施例、比較例及び参考例の三酸化硫黄分解触媒の評価のために用いた装置を示す図である。
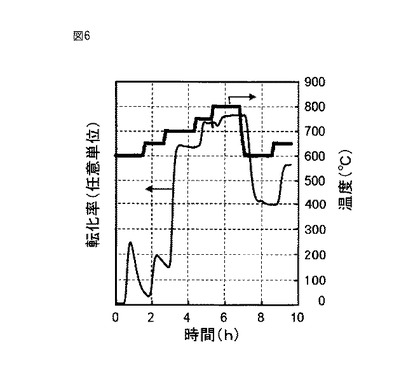
【図6】図6は、実施例1の三酸化硫黄分解触媒の評価における温度変化と転化率変化との関係を示す図である。
【図7】図7は、実施例1の三酸化硫黄分解触媒の走査透過型電子顕微鏡(STEM)写真を示す図である。なお、図7(a)〜(c)はそれぞれ、600℃、750℃及び800℃の温度での三酸化硫黄分解反応に用いた後の触媒についての写真である。
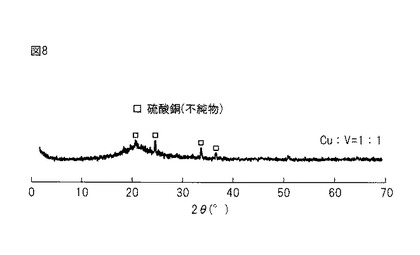
【図8】図8は、実施例1のXRD(X線回折)分析結果を示す図である。なお、図8は、800℃の温度での三酸化硫黄分解反応に用いた後の触媒についての評価結果である。
【図9】図9は、ケイ素(Si)、バナジウム(V)、及び銅(Cu)についての実施例1の三酸化硫黄分解触媒のEDS(エネルギー分散形X線分光)分析結果を、STEM−HAADF(走査透過電子顕微鏡(高角度散乱暗視野法))による分析結果と併せて示す図である。なお、図8は、800℃の温度での三酸化硫黄分解反応に用いた後の触媒についての評価結果である。
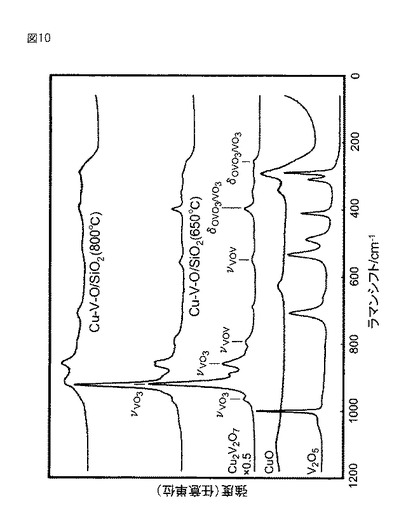
【図10】図10は、実施例1の三酸化硫黄分解触媒のラマン散乱分析結果を、比較のためのバナジン酸銅(Cu2V2O7)、酸化銅(CuO)及び五酸化バナジウム(V2O5)についての分析結果と併せて示す図である。なお、図10は、650℃及び800℃の温度での三酸化硫黄分解反応に用いた後の触媒(Cu−V−O/SiO2(650℃)及びCu−V−O/SiO2(800℃))についての評価結果である。
【発明を実施するための形態】
【0022】
(三酸化硫黄分解触媒)
本発明の三酸化硫黄分解触媒は、銅とバナジウムとの複合酸化物がシリカ担体に担持されてなる。ここで、この本発明の三酸化硫黄分解触媒では、複合酸化物の結晶の対称性が低下している。すなわち、本発明の三酸化硫黄分解触媒では、銅とバナジウムとの複合酸化物の結晶が歪んでいる。このような結晶の歪みは例えば、銅とバナジウムとの複合酸化物が、充分に薄い層としてシリカ担体に担持されており、かつシリカ担体が、この複合酸化物に化学的に影響を与えることによって達成されると考えられる。
【0023】
理論に限定されるものではないが、本発明の三酸化硫黄分解触媒でのように、銅とバナジウムとの複合酸化物の結晶が歪んでいる場合、バナジウム−酸素(V−O)結合の一部が、他のバナジウム−酸素(V−O)結合よりも切れやすくなっていると考えられる。このようにバナジウム−酸素(V−O)結合が切れやすくなっている場所では、比較的容易に酸素が脱離して、酸素欠陥が形成され、この酸素欠陥において三酸化硫黄の吸着及び分解が進行すると考えられる。
【0024】
これに関して、本発明の三酸化硫黄分解触媒では、銅とバナジウムとの複合酸化物が図1に示すようなピロバナジン酸銅(Cu2V2O7)構造を有すると考えられる。なお、図1では、ピロバナジン酸銅の(a)結晶構造及び(b)バナジン酸結合を示している。
【0025】
したがって、本発明の三酸化硫黄分解触媒では、図2に示すようにして三酸化硫黄の分解反応が促進されると考えられる。具体的には、銅とバナジウムとの複合酸化物の結晶が歪んでいる場合、この複合酸化物において、バナジウム−酸素(V−O)結合の一部(点線)が、他のバナジウム−酸素(V−O)結合(実線)よりも長く、それによって切れやすくなっており(図2(a))、この結合が切れて酸素が脱離すると、その位置が酸素欠陥(図2(b))となり、三酸化硫黄がこの酸素欠陥に吸着し(図2(c))、そして分解されて、二酸化硫黄が生成する(図2(d))と考えられる。
【0026】
このように銅とバナジウムとの複合酸化物の結晶が歪んでいることは、ラマン分光分析において、対称性の低下、すなわちピークの高さの低下及び/又はピークのブロード化として確認できる。したがって例えば、本発明の三酸化硫黄分解触媒は、ラマン分光分析において、バナジウム−酸素(V−O)結合に起因する920cm−1付近のピークが、下記の(a)及び(b)の少なくとも一方の条件を満たしている:
(a)上記920cm−1付近のピークの高さが、バナジウム−酸素(V−O)結合に起因する他のピークの最大高さの3.0倍以下、2.5倍以下、2.0倍以下、1.5倍以下、又は1.0倍以下、及び
(b)上記920cm−1付近のピークの半値幅が、30cm−1以上、40cm−1以上、50cm−1以上。
【0027】
本発明の三酸化硫黄分解触媒によれば、式(B1−2)の三酸化硫黄分解反応に必要とされる温度を低下させ、例えば700℃以下程度の温度において、実質的な速度で三酸化硫黄分解反応を進行させることができる。
【0028】
上記記載のように、三酸化硫黄を分解する従来の方法では、1000℃近い温度を用いることが一般的であった。しかしながら、このような高温に耐えられる材料は非常に限定されており、またかなり高価なものであった。
【0029】
また、1000℃近い高温は、太陽エネルギーからは安価に得ることが困難であった。すなわち例えば、太陽熱エネルギーを得る集光装置としては、パラボリックディッシュ型集光装置、ソーラータワー型集光装置、及びパラボリックトラフ型集光装置が知られているが、これらのうちで構造が簡単で、コストが安く、且つ大規模なプラントに適しているパラボリックトラフ型集光装置では、太陽エネルギーの収集と放射によるエネルギーの散逸との釣り合いから、1000℃近い高温での太陽エネルギーの収集は非現実的である。
【0030】
したがって、本発明の三酸化硫黄分解触媒によって、三酸化硫黄分解反応に必要とされる温度を低下させ、例えば700℃程度の温度において、実質的な速度で三酸化硫黄分解反応を進行するようにすることは、産業的な価値が非常に大きい。
【0031】
(三酸化硫黄分解触媒−複合酸化物)
本発明の三酸化硫黄分解触媒の複合酸化物において、銅とバナジウムとの原子比(銅:バナジウム)は例えば、1:9〜9:1、2:8〜8:2、3:7〜7:3、又は4:6〜6:4にすることができる。
【0032】
本発明の三酸化硫黄分解触媒では例えば、銅及びバナジウムの合計量が担体100gに対して、0.01mol以上、0.05mol以上、0.10mol以上となる割合で、複合金属酸化物をシリカ担体に担持することができる。また、この担持量は例えば、担体100gに対して、2.00mol以下、1.00mol以下、又は0.50mol以下の割合にすることができる。
【0033】
(三酸化硫黄分解触媒−製造方法)
本発明の三酸化硫黄分解触媒は、任意の方法で得ることができる。
【0034】
例えば、本発明の三酸化硫黄分解触媒は、銅塩の水溶液を、担体に吸水させ、乾燥及び仮焼成し、バナジウム塩の水溶液を、担体に吸水させ、乾燥及び仮焼成し、そしてその後で、得られた担体を焼成することによって得ることができる。また、これとは反対に、本発明の三酸化硫黄分解触媒は、バナジウム塩の水溶液を先に担体に吸水させ、乾燥及び仮焼成し、銅塩の水溶液を、担体に吸水させ、乾燥及び仮焼成し、そしてその後で、得られた担体を焼成することによって得ることができる。また、銅塩及びバナジウム塩を、これらの共沈が可能なように選択する場合、銅塩及びバナジウム塩の両方を含有する水溶液を、担体に吸水させ、乾燥及び仮焼成し、そしてその後で、得られた担体を焼成することによって、本発明の三酸化硫黄分解触媒を得ることもできる。
【0035】
なお、本発明の三酸化硫黄分解触媒の製造においては、銅とバナジウムとの複合酸化物が、充分に薄い層としてシリカ担体に担持されており、かつシリカ担体に少なくとも部分的に影響され、それによってこの複合酸化物の結晶の対称性が低下するようにすることが必要である。
【0036】
これに関して、本発明の三酸化硫黄分解触媒を製造する上記の方法においては、銅塩及びバナジウム塩の水溶液を先に担体に吸水させ、乾燥及び仮焼成して得られたシリカ担体を、比較的高温で焼成すること、並びにこのようにして焼成されるシリカ担体が、この焼成の前に比較的高温での熱処理を受けていないことが好ましい。
【0037】
このように、高温での熱処理を受けていないシリカ担体では、水酸基、タングリングボンド、微細な凹凸形状等の不安定部分が比較的多く表面に残されている。したがって、図3(a)に示すように、このような表面上に複合酸化物前駆体を担持し、これらを共に焼成する場合、これらの不安定部分が、焼成の間に、その上に担持された複合酸化物前駆体に作用し、複合酸化物を充分に薄い層状にし、かつ/又は得られる複合酸化物の結晶の対称性を低下させると考えられる。これに関して、微細な凹凸形状が焼成の間に平坦化される場合、シリカ担体の表面と、その上に担持された複合酸化物前駆体及び複合酸化物との混和が促進されると考えられる。
【0038】
これに対して、高温での熱処理を受けているシリカ担体では、水酸基、タングリングボンド、微細な凹凸形状等の不安定部分が、高温での熱処理によって減少している。したがって、図3(b)に示すように、このような表面上に複合酸化物前駆体を担持し、これらを共に焼成する場合であっても、シリカ担体の表面がその上に担持された複合酸化物前駆体に対して与える影響が小さいと考えられる。
【0039】
本発明の方法において多孔質シリカ担体を焼成する温度は、600℃以上、650℃以上、700℃以上、750℃以上、800℃以上であってよい。
【0040】
また、本発明の方法において多孔質シリカ担体を焼成する雰囲気は、任意の雰囲気であってよく、例えば空気のような酸素含有雰囲気、窒素又はアルゴンのような不活性雰囲気、又は硫酸雰囲気若しくは三酸化硫黄雰囲気のような酸化雰囲気であってよい。
【0041】
なお、本発明の方法における焼成は、本発明の三酸化硫黄分解触媒の使用の間に行ってもよい。すなわち、最終的な焼成を行わずに本発明の三酸化硫黄分解触媒を装置に装填し、そして三酸化硫黄分解反応を少なくとも一時的に、上記の焼成温度よりも高い温度において行うことによって、焼成を達成することもできる。
【0042】
(三酸化硫黄分解触媒−担体)
本発明の三酸化硫黄分解触媒において用いられるシリカ担体は、銅とバナジウムとの複合酸化物に少なくとも部分的に影響を与え、それによってこの複合酸化物の結晶の対称性を低下させる範囲で、任意のシリカ担体を用いることができる。
【0043】
したがって、三酸化硫黄分解触媒を製造する本発明の方法において用いられるシリカ担体は、水酸基、タングリングボンド、微細な凹凸形状等の不安定部分を表面に有するシリカ担体、例えば700℃以上、650℃以上、600℃以上、又は550℃以上の熱処理を受けていないシリカ担体であってよい。
【0044】
このようなシリカ担体としては特に、細孔構造を有する多孔質シリカ担体を用いることができる。この場合、好ましくは、複合酸化物が多孔質シリカ担体の細孔構造内に担持されている。
【0045】
このような多孔質シリカ担体は、メソポーラスシリカ担体、例えばKIT−6のような立方晶メソポーラスシリカであってよい。また、このような多孔質シリカ担体は例えば、多孔質シリカ担体の細孔分布において、シリカの一次粒子間の間隙に起因するピークが、細孔径5〜50nm、特に細孔径5〜30nmの範囲にあり、且つシリカ粒子内の細孔構造に起因するピークが、細孔径1〜5nm、特に細孔径2〜4nmの範囲にある多孔質シリカ担体であってよい。
【0046】
このように、細孔構造を有する多孔質シリカ担体を用いる場合、複合酸化物が、多孔質シリカ担体の細孔構造表面近傍に担持され、それによって複合酸化物粒子のシンタリングが抑制される。理論に限定されるわけではないが、このように非常に微細な状態で維持されている複合酸化物粒子では、触媒の微粒子化によって、触媒の表面積が100倍程度に増大されるだけでなく、触媒の表面の性質が変化して、複合酸化物の触媒性能が改良される場合もあると考えられる。
【0047】
また、細孔構造を有する多孔質シリカ担体の細孔分布において、二元の細孔分布となることにより細孔径が数nmの表面積の広い活性部位に、十〜数十nmの細孔から拡散速度の速い気相ガスが高速に供給されることによって、複合酸化物粒子と三酸化硫黄との接触の機会が多く、それによって触媒性能を改良すると考えられる。
【0048】
なお、細孔構造を有する多孔質シリカ担体は例えば、特開2008−12382に記載の方法によって得ることができる。
【0049】
(二酸化硫黄の生成方法)
二酸化硫黄を生成する本発明の方法は、本発明の三酸化硫黄分解触媒を用いて、三酸化硫黄を二酸化硫黄と酸素とに分解することを含む。ここで、この方法は、本発明の三酸化硫黄分解触媒を用いることによって、三酸化硫黄を分解する従来の方法よりも低い温度、例えば800℃以下、750℃以下、700℃以下、650℃以下の温度で実施することができる。
【0050】
(水素生成方法)
水素を生成する本発明の方法は、水を、水素及び酸素に分解すること、例えばI−Sサイクル法、ウエスティングハウス・サイクル法、Ispra−Mark 13サイクル法、又はロスアラモス・サイエンスラボラトリ・サイクル法によって、水を水素及び酸素に分解することを含む。ここで、この本発明の方法は、下記式(X1)で示される反応で、硫酸を、水、二酸化硫黄、及び酸素に分解することを含み、且つ下記式(X1)で示される反応の素反応である式(X1−1)及び(X1−2)の素反応のうち、式(X1−2)の素反応を、二酸化硫黄を生成する本発明の方法によって行う:
(X1)H2SO4 → H2O + SO2 + 1/2O2
(X1−1)H2SO4 → H2O + SO3
(X1−2)SO3 → SO2 + 1/2O2
【0051】
すなわち、例えば、水素を生成する本発明の方法は、下記式(X1)〜(X3)で示されるI−S(ヨウ素−イオウ)サイクル法において、式(X1)の反応の素反応である式(X1−1)及び(X1−2)の素反応のうち、式(X1−2)の素反応を、二酸化硫黄を生成する本発明の方法によって行うことを含む:
(X1)H2SO4 → H2O + SO2 + 1/2O2
(X1−1)H2SO4 → H2O + SO3
(X1−2)SO3 → SO2 + 1/2O2
(X2)I2 + SO2 + 2H2O → 2HI + H2SO4
(X3)2HI → H2 + I2
全反応:H2O → H2 + 1/2O2
【0052】
また、例えば、水素を生成する本発明の方法は、下記式(X1)、(X4)及び(X5)で示されるウエスティングハウス・サイクル法において、式(X1)の反応の素反応である式(X1−1)及び(X1−2)の素反応のうち、式(X1−2)の素反応を、二酸化硫黄を生成する本発明の方法によって行うことを含む:
(X1)H2SO4 → H2O + SO2 + 1/2O2
(X1−1)H2SO4 → H2O + SO3
(X1−2)SO3 → SO2 + 1/2O2
(X4)SO2 + 2H2O → H2SO3
(X5)H2SO3 + H2O+ → H2 + H2SO4(電気分解)
全反応:H2O → H2 + 1/2O2
【0053】
さらに、例えば、水素を生成する本発明の方法は、下記式(X1)、(X6)及び(X7)で示されるIspra−Mark 13サイクル法において、式(X1)の反応の素反応である式(X1−1)及び(X1−2)の素反応のうち、式(X1−2)の素反応を、二酸化硫黄を生成する本発明の方法によって行うことを含む:
(X1)H2SO4 → H2O + SO2 + 1/2O2
(X1−1)H2SO4 → H2O + SO3
(X1−2)SO3 → SO2 + 1/2O2
(X6)2HBr → Br2 + H2
(X7)Br2 + SO2 + 2H2O+ → 2HBr + H2SO4
全反応:H2O → H2 + 1/2O2
【0054】
さらに、例えば、水素を生成する本発明の方法は、下記式(X1)、及び(X8)〜(X10)で示されるロスアラモス・サイエンスラボラトリ・サイクル法において、式(X1)の反応の素反応である式(X1−1)及び(X1−2)の素反応のうち、式(X1−2)の素反応を、二酸化硫黄を生成する本発明の方法によって行うことを含む:
(X1)H2SO4 → H2O + SO2 + 1/2O2
(X1−1)H2SO4 → H2O + SO3
(X1−2)SO3 → SO2 + 1/2O2
(X8)Br2 + SO2 + 2H2O+ → 2HBr + H2SO4
(X9)2CrBr3 → 2CrBr2 + Br2
(X10)2HBr + 2CrBr2 → 2CrBr3 + H2
全反応:H2O → H2 + 1/2O2
【実施例】
【0055】
《参考例1及び比較例1〜3》
以下の参考例及び比較例では、銅(Cu)とバナジウム(V)との複合金属酸化物が、三酸化硫黄分解触媒として優れた性質を有することを示す。
【0056】
《参考例1》
参考例1では、銅(Cu)とバナジウム(V)との複合金属酸化物(Cu−V−O)を単身触媒として用いた。
【0057】
(単身触媒の製造)
参考例1の単身触媒は、それぞれの金属の原子比が1:1である酸化銅及び酸化バナジウムを、乳鉢で粉砕し、良く混合し、アルミナ性るつぼに入れ、そして750℃で12時間にわたって焼成して得た。得られた単身触媒についてのX線回折分析(XRD)結果を図4に示す。
【0058】
《比較例1》
比較例1では、銅(Cu)の酸化物(Cu−O)を単身触媒として用いた。ここでは、参考例1で原料として用いた酸化銅をそのまま単身触媒として用いた。
【0059】
《比較例2》
比較例2では、バナジウム(V)の酸化物(V−O)を触媒として用いた。ここでは、参考例1で原料として用いた酸化バナジウムをそのまま単身触媒として用いた。
【0060】
《比較例3》
比較例3では触媒を用いなかった。
【0061】
(評価(転化率))
図5に示す固定床流通反応装置を用いて、参考例1及び比較例1〜3の単身触媒について、下記式(X1−2)の三酸化硫黄分解反応の転化率を評価した:
(X1−2)SO3 → SO2 + 1/2O2
【0062】
具体的には、三酸化硫黄分解反応の転化率は、図5に関して下記で説明するようにして評価した。
【0063】
14〜20メッシュに調整した0.5gの単身触媒又は担持触媒を、触媒床10として、石英製反応管4(内径10mm)に充填した。窒素(N2)(100mL/分)及び47重量%硫酸(H2SO4)水溶液(50μL/分)を、それぞれ窒素供給部1及び硫酸供給部3から、石英製反応管4の下段に供給した。
【0064】
石英製反応管4の下段に供給された硫酸(H2SO4)は、石英製反応管4の下段及び中段において加熱されて、三酸化硫黄(SO3)及び酸素(O2)に分解し、そして触媒床10に流入した(SO3:4.5mol%、H2O:31mol%、N2:残部、0℃換算ガス流量:148.5cm3/分、重量流量比(W/F比):5.61×10−5g・h/cm3、気体時空間速度(GHSV:Gas Hourly Space Velocity):約15,000h−1)。
【0065】
ここで、石英製反応管4は、下段がヒーター4aによって約400℃に加熱されており、かつ中段がヒーター4bによって約600℃に加熱されていた。また、石英製反応管4の上段は、ヒーター4cによって初めに約600℃に加熱されており、定常状態になった後で、650℃に加熱した。
【0066】
石英製反応管4の上段をヒーター4cによって650℃に加熱した後で、石英製反応管4からの流出ガスを、空冷し、その後で、0.05Mのヨウ素(I2)溶液にバブリングして、ヨウ素溶液に二酸化硫黄(SO2)を吸収させた。0.025Mのチオ硫酸ナトリウム(Na2S2O3)溶液を用いて、二酸化硫黄を吸収したヨウ素溶液にヨードメトリー滴定を行って、吸収された二酸化硫黄の量を求めた。
【0067】
また、ヨウ素溶液にバブリングした後の流出ガスは、ドライアイス・エタノール混合物で冷却し、残留している二酸化硫黄及び三酸化硫黄をミストアブソーバー及びシリカゲルで完全に除去し、その後で、磁気圧力酸素計(堀場製作所のMPA3000)及びガスクロマトグラフ(島津製作所のGC8A、モレキュラーシーブ5A、TCD検出器)を用いて、酸素(O2)の量を求めた。
【0068】
三酸化硫黄(SO3)から二酸化硫黄(SO2)への平衡転化率に対する到達率は、上記のようにして求めた二酸化硫黄及び酸素の量から計算した。
【0069】
参考例及び比較例についての評価結果を、下記の表1に示す。
【0070】
【表1】

【0071】
表1からは、参考例1の触媒が、比較例1〜3の触媒と比較して、650℃という比較的低い温度において、有意に好ましい三酸化硫黄分解特性を有していることが理解される。
【0072】
なお、上記の比較例2で用いられている酸化バナジウム、特に五酸化バナジウム(V2O5)は、下記式(C−1)〜(C−3)で示される反応で硫酸を製造する接触法と呼ばれる方法において、二酸化硫黄を酸化させて三酸化硫黄を得る式(C−2)の反応を促進するために用いられている:
(C−1)S(固体) + O2(気体) → SO2(気体)
(C−2)2SO2(気体) + O2(気体) → 2SO3(気体)
(C−3)SO3(気体) + H2O(液体) → H2SO4(液体)
【0073】
しかしながら、酸化バナジウムを用いている比較例2は、参考例1と比較して有意に劣った転化率を示していた。
【0074】
《実施例1、並びに参考例2及び3》
実施例1及び参考例2では、550℃において焼成して得たシリカ担体を原料として用いる場合(実施例1)と、800℃において焼成して得たシリカ担体を原料として用いる場合(比較例1)の違いについて評価した。また、参考例3では、触媒金属として白金を用いて評価を行った。
【0075】
《実施例1》
実施例1では、銅(Cu)とバナジウム(V)との複合金属酸化物(Cu−V−O)が多孔質シリカ担体に担持されてなる触媒を用いた。ここで、用いられている多孔質シリカ担体は、空気中において550℃で焼成して得たものである。
【0076】
具体的には、実施例1の触媒は下記のようにして製造した。
【0077】
(多孔質シリカ担体の製造)
多孔質シリカ担体は、立方晶メソポーラスシリカ(KIT−6)であり、下記のようにして製造した。
【0078】
(1)蒸留水144mLに、7.9gの35質量%塩酸(HCl)及び4.0gの非イオン性界面活性剤(Pluronic(商標)P−123)を添加し、得られた水溶液を35℃の温度において撹拌して、成分を溶解。
(2)得られた混合物に、4.0gの1−ブタノールを添加し、混合物が透明になるまで、35℃の温度において撹拌し、それによって、非イオン性界面活性剤を自己配列。
(3)得られた混合物に、シリカ源としての8.6gのテトラエトキシシラン(TEOS)を添加し、35℃の温度において24時間にわたって強撹拌し、自己整列している非イオン性界面活性剤をテンプレートとして、テトラエトキシシラン(TEOS)を加水分解。
(4)100℃の温度において24時間にわたって静置し、その後、洗浄せずにそのまま110℃の温度において24時間にわたって乾燥。
(6)8mlの35質量%塩酸と120mLのエタノールとの混合物中において、1.5時間にわたって撹拌して洗浄。
(7)110℃の温度において24時間にわたって乾燥し、そして3℃/分の昇温速度で550℃まで加熱して、この温度で5時間にわたって焼成して、立方晶メソポーラスシリカ(KIT−6)を取得。
【0079】
(複合金属酸化物の担持)
複合酸化物は、吸水担持法によって、多孔質シリカ担体に担持した。具体的には、初めに、銅の硝酸塩を水に溶解した水溶液を作り、この水溶液を担体に吸水させ、150℃で乾燥し、350℃で1時間にわたって仮焼成した。次に、メタバナジン酸アンモニウムを水に溶解し、この水溶液を担体に吸水させ、150℃で乾燥し、350℃で1時間にわたって仮焼成した。最後に、得られた担体を600℃で2時間にわたって焼成して、複合酸化物を担持している多孔質シリカ担体を得た。
【0080】
なお、担持量は、銅が0.12mol/100g−担体、かつバナジウムが0.12mol/100g−担体とした。
【0081】
《参考例2》
参考例2では、銅(Cu)とバナジウム(V)との複合金属酸化物(Cu−V−O)が多孔質シリカ担体に担持されてなる触媒を用いた。ここで、用いられている多孔質シリカ担体は、空気中において800℃で焼成して得たものである。
【0082】
具体的には、参考例2の触媒は下記のようにして製造した。
【0083】
(多孔質シリカ担体の製造)
多孔質シリカ担体は、特開2008−12382に記載の方法と類似の方法によって製造した。すなわち、多孔質シリカ担体は、下記のようにして製造した。
【0084】
蒸留水6L(リットル)に、セチルトリメチルアンモニウムクロライド1kgを溶解した。得られた水溶液を2時間にわたって撹拌して、セチルトリメチルアンモニウムクロライドを自己配列させた。次に、セチルトリメチルアンモニウムクロライドを自己配列させた溶液に、テトラエトキシシランとアンモニア水を添加して、溶液のpHを9.5にした。
【0085】
この溶液中において、テトラエトキシシランを30時間にわたって加水分解して、配列したヘキサデシルアミンの周りにシリカを析出させて、ナノサイズの細孔を有する一次粒子からなる二次粒子を形成し、多孔質シリカ担体前駆体を得た。
【0086】
その後、得られた多孔質シリカ担体前駆体を、エタノール水で洗浄し、ろ過し、乾燥して、800℃の空気中で2時間にわたって焼成して、多孔質シリカ担体を得た。
【0087】
ここで得られた多孔質シリカ担体は、シリカの細孔構造に起因する2.7nm付近の細孔、及びシリカの一次粒子間の間隙に起因する10nm強の細孔を有していた。
【0088】
(複合金属酸化物の担持)
実施例1と同様にして、銅とバナジウムの複合酸化物を多孔質シリカ担体に担持した。
【0089】
《参考例3》
参考例3では、γ−アルミナ担体に白金を担持して、担持触媒を製造した。ここでは、担持量を0.5g−Pt/100g−担体とした。
【0090】
(評価(転化率))
上記参考例1等でのようにして評価を行った。ただし、実施例1についての評価では、石英製反応管4の上段が約600℃で定常状態になった後で、図6で示すようにして加熱温度を変化させて評価を行った。加熱温度の変化と併せて、三酸化硫黄(SO3)から二酸化硫黄(SO2)への転化率の変化を図6に示している。
【0091】
図6で示されているように、加熱温度600℃及び650℃においては、一時的に転化率が大きくなるものの、この転化率はすぐに小さくなった。
【0092】
その後、加熱温度を700℃、750℃及び800℃にすると、転化率が有意に大きくなった。これらの温度での転化率は、対応する温度における平衡転化率に対して100%近かった。
【0093】
その後、再び加熱温度を600℃及び650℃まで加熱温度を低下させると、予想外に、転化率は最初に加熱温度を600℃及び650℃にしたときの転化率よりも有意に大きくなった。600℃の温度での転化率は、この温度での平衡転化率に対して79.8%であった。また、評価開始から10時間後の加熱温度650℃の状態での転化率は、対応する温度における平衡転化率に対して100%近かった。なお、650℃の温度での転化率をその後27時間にわたって評価したところ、転化率がほぼ維持されていることが確認された。
【0094】
参考例2についての評価では、図6で示すようにして加熱温度を変化させたところ、評価開始から10時間後の加熱温度650℃の状態において、この温度における平衡転化率に対する到達率は88.5%であった。実施例1では、対応する到達率は100%近かったので、参考例2の触媒と比較して、実施例1の三酸化硫黄分解触媒が有意に優れていることが理解される。
【0095】
参考例3についての評価では、図6で示すようにして加熱温度を変化させたところ、評価開始から10時間後の加熱温度650℃の状態において、対応する温度における平衡転化率に対して62.7%であった。実施例1では、対応する到達率は100%近かったので、参考例3の触媒と比較して、実施例1の三酸化硫黄分解触媒が有意に優れていることが理解される。
【0096】
(評価(STEM分析))
実施例1で用いた三酸化硫黄分解触媒について、600℃、750℃及び800℃の温度での三酸化硫黄分解反応に用いた後で、走査透過型電子顕微鏡(STEM)よる分析を行った。得られた結果をそれぞれ図7(a)〜(c)に示す。
【0097】
600℃の温度での三酸化硫黄分解反応に用いた後の触媒(図7(a))では、シリカ担体の細孔に由来する孔(白く抜けた部分)、及び担体に担持されている銅とバナジウムとの複合酸化物(Cu−V−O)が確認できた。
【0098】
また、750℃の温度での三酸化硫黄分解反応に用いた後の触媒(図7(b))では、シリカ担体の細孔に由来する孔が潰れ、全体として収縮していた。
【0099】
さらに、800℃の温度での三酸化硫黄分解反応に用いた後の触媒(図7(c))では、シリカ担体のシンタリングが大きく進行し、それによって表面積が小さく、かつシリカゲルのような形態となっていた。
【0100】
これら図7(a)〜(c)から明らかなように、シリカ担体は加熱によって大きく劣化していた。しかしながら、三酸化硫黄分解触媒としての性質は、800℃の温度での使用後に改良されていたことから、担体に担持された銅とバナジウムとの複合酸化物の物性が改良されていたことが理解される。
【0101】
(評価(XRD分析))
800℃の温度での三酸化硫黄分解反応に用いた後の触媒を、XRD(X線回折)分析で評価した。評価結果を図8に示す。
【0102】
図8のXRD分析結果からは、硫酸銅(CuSO4)に帰属される弱いピークが観測され、これは、極微量の硫酸銅の存在を示唆している。また、XRD分析結果では、それ以外の明確なピークが示されていないことから、大部分の成分が非常に均一に分散していると考えられる。なお、650℃の温度での三酸化硫黄分解反応に用いた後の触媒についてん6XRD分析でも、800℃の場合と同様な結果が得られた。
【0103】
(評価(EDS分析))
800℃の温度での三酸化硫黄分解反応に用いた後の触媒を、ケイ素(Si)、バナジウム(V)、及び銅(Cu)についてのEDS(エネルギー分散形X線分光)分析で評価した。評価結果を、STEM−HAADF(走査透過電子顕微鏡(高角度散乱暗視野法))による分析結果と併せて、図9に示す。
【0104】
図9のESD分析結果からは、担体に担持されている触媒成分であるCu及びVの分布が、担体の構成元素であるSiの分布と同様であることが分かる。これは、触媒成分である銅とバナジウムとの複合酸化物が、シリカ担体上に薄膜状で堆積していることを示している。なお、担体上に担持されているCuは部分的に凝縮している箇所があるが、これは、硫酸銅として析出した部分であると考えられる。
【0105】
(評価(ラマン散乱分析))
650℃及び800℃の温度での三酸化硫黄分解反応に用いた後の触媒(Cu−V−O/SiO2(650℃)及びCu−V−O/SiO2(800℃))を、ラマン散乱分析で評価した。評価結果を、比較のためのバナジン酸銅(Cu2V2O7)、酸化銅(CuO)及び五酸化バナジウム(V2O5)についての分析結果と併せて、図10に示す。なお、バナジン酸銅(Cu2V2O7)についての分析結果は、高さを0.5倍(×0.5)して表している。
【0106】
図10のラマン散乱分析結果によれば、650℃の温度での三酸化硫黄分解反応に用いた後の触媒(Cu−V−O/SiO2(650℃))では、バナジン酸銅(Cu2V2O7)と同様なラマン散乱プロファイルが得られた。
【0107】
このプロファイルでは、920cm−1付近に存在するピーク、すなわちピロバナジン酸(V2O7)構造の両端のVO3の対称収縮振動(対称性最大)に由来するピークが、最大のピークであった。具体的には、このプロファイルでは、920cm−1付近のピークの高さが、バナジウム−酸素(V−O)結合に起因する他のピークの最大高さの約3.9倍になっていた。また、この触媒(Cu−V−O/SiO2(650℃))では、920cm−1付近のピークの半値幅が、約25cm−1になっていた。
【0108】
これに対して、800℃の温度での三酸化硫黄分解反応に用いた後の触媒(Cu−V−O/SiO2(800℃))では、920cm−1付近のこのピークが低く、かつブロードになっていた。具体的には、このプロファイルでは、920cm−1付近のピークの高さが、バナジウム−酸素(V−O)結合に起因する他のピークの最大高さの約0.9倍になっていた。また、この触媒(Cu−V−O/SiO2(800℃))では、920cm−1付近のピークの半値幅が、約59cm−1になっていた。
【0109】
これは、ピロバナジン酸(V2O7)構造が変化して結晶が歪み、それによってピロバナジン酸(V2O7)構造の両端のVO3の対称性が低下したことを示している。この対称性の低下は、バナジウムと酸素との間の結合の一部が、バナジウムと酸素との間の他の結合よりも長く、それによって切れやすくなっていることを意味すると考えられる。
【符号の説明】
【0110】
1 窒素供給部
3 硫酸供給部
4 石英製反応管
4a、4b、4c ヒーター
10 触媒床
【技術分野】
【0001】
本発明は、三酸化硫黄(SO3)分解用触媒に関する。また、本発明は、三酸化硫黄分解用触媒を用いて三酸化硫黄を分解する工程を含む水素生成方法に関する。
【背景技術】
【0002】
近年、地球温暖化等の問題から、燃焼時に二酸化炭素を生成しないクリーンエネルギーとしての水素が注目されている。
【0003】
水素の生成のためには一般に、下記式(A1)及び(A2)で示される炭化水素燃料の水蒸気改質が用いられている:
(A1)CnHm + nH2O → nCO + (n+m/2)H2
(A2)CO + H2O → CO2 + H2
全反応:CnHm + 2nH2O → nCO2 + (2n+m/2)H2
【0004】
したがって、水素の燃焼自体は二酸化炭素を生成させないものの、水素の生成においては二酸化炭素を発生させていることが一般的であった。
【0005】
これに関して、炭化水素燃料を用いずに水素を生成させるための方法として、太陽熱エネルギー又は原子力熱エネルギーを用いることが提案されている(特許文献1、非特許文献1)。
【0006】
熱エネルギーを利用して水から水素を生成させる方法としては、下記式(B1)〜(B3)で示されるI−S(ヨウ素−イオウ)サイクル法と呼ばれる方法が提案されている:
(B1)H2SO4(液体)
→ H2O(気体) + SO2(気体) + 1/2O2(気体)
(反応温度=約950℃、ΔH=188.8kJ/mol−H2)
(B2)I2(液体) + SO2(気体) + 2H2O(液体)
→ 2HI(液体) + H2SO4(液体)
(反応温度=約130℃、ΔH=−31.8kJ/mol−H2)
(B3)2HI(液体) → H2(気体) + I2(気体)
(反応温度=約400℃、ΔH=146.3kJ/mol−H2)
【0007】
上記式(B1)〜(B3)で示されるI−S(ヨウ素−イオウ)サイクル法の全反応は下記のとおりである:
H2O → H2 + 1/2O2
(ΔH=286.5kJ/mol−H2(高位発熱量基準)
(ΔH=241.5kJ/mol−H2(低位発熱量基準)
【0008】
ここで、上記式(B1)の反応は、下記式(B1−1)及び(B1−2)の2つの素反応に分けることができる:
(B1−1)H2SO4(液体) → H2O(気体) + SO3(気体)
(反応温度=約300℃、ΔH=90.9kJ/mol−H2)
(B1−2)SO3(気体) → SO2(気体) + 1/2O2(気体)
(反応温度=約950℃、ΔH=97.9kJ/mol−H2)
【0009】
すなわち、I−Sサイクル法で水素を生成する場合、式(B1−2)の三酸化硫黄(SO3)分解反応において最も高い温度を必要とし、この反応で必要とされる高温を得ることが容易でなかった。
【0010】
このような問題に関して、非特許文献1では、熱源として太陽熱エネルギーを用いつつ、必要に応じて天然ガスを燃焼させて、追加の熱エネルギーを得るとしている。
【0011】
また、式(B1−2)の三酸化硫黄分解反応において必要とされる温度を低下させるために、白金触媒を用いることが提案されている。しかしながら、この反応において白金触媒を用いる場合、触媒の使用開始時には高い特性を有するものの、反応によって生成する酸素によって白金が酸化され、白金粒子が粗大化することにより触媒活性が低下することが知られている。また、白金触媒は高価であることから、産業的な規模においては用いることが難しい。
【0012】
これに関して、非特許文献2では、三酸化硫黄分解反応において必要とされる温度を低下させるために、白金(Pt)、クロム(Cr)、鉄(Fe)、及びそれらの酸化物からなる群より選択される触媒をアルミナ担体に担持させて用いることを提案している。
【0013】
また、I−Sサイクル法に関して、特許文献2では、上記式(B2)で表される反応、すなわちヨウ素、二酸化硫黄及び水から、ヨウ化水素及び硫酸を得る反応において、二酸化硫黄と水との反応をカチオン交換膜の正極側で行わせ、かつヨウ素の反応をカチオン交換膜の負極側で行わせることによって、その後の分離操作を省略することを提案している。
【0014】
なお、I−Sサイクル法以外にも、熱エネルギーを利用して水素を生成する方法として、ウエスティングハウス・サイクル、Ispra−Mark 13サイクル法、ロスアラモス・サイエンスラボラトリ・サイクル法等が知られているが、これらの方法においても、式(B1−2)でのようにして、三酸化硫黄を二酸化硫黄と水素とに分解することが必要とされている。
【先行技術文献】
【特許文献】
【0015】
【特許文献1】特開2007−218604号公報
【特許文献2】特開2005−041764号公報
【非特許文献】
【0016】
【非特許文献1】A.Giaconia, et al., International Journal of Hydrogen Energy, 32, 469−481(2007)
【非特許文献2】H.Tagawa, et al.,International Journal of Hydrogen Energy, 14, 11−17(1989)
【発明の概要】
【発明が解決しようとする課題】
【0017】
本発明では、三酸化硫黄分解触媒、特に水から水素を生成する際に必要とされる温度を低下させることができる三酸化硫黄分解触媒を提供する。
【課題を解決するための手段】
【0018】
本件発明者は、鋭意検討の結果、下記の本発明に想到した。
【0019】
〈1〉銅とバナジウムとの複合酸化物がシリカ担体に担持されてなり、かつラマン分光分析において、バナジウム−酸素(V−O)結合に起因する920cm−1付近のピークの高さが、下記の(a)及び(b)の少なくとも一方の条件を満たす、三酸化硫黄分解触媒:
(a)前記920cm−1付近のピークの高さが、バナジウム−酸素(V−O)結合に起因する他のピークの最大高さの3.0倍以下、及び
(b)前記920cm−1付近のピークの半値幅が、30cm−1以上。
〈2〉前記複合酸化物において、銅とバナジウムとの原子比が、1:9〜9:1である、上記〈1〉項に記載の触媒。
〈3〉条件(a)を少なくとも満たす、上記〈1〉又は〈2〉項に記載の触媒。
〈4〉条件(b)を少なくとも満たす、上記〈1〉又は〈2〉項に記載の触媒。
〈5〉条件(a)において、前記920cm−1付近のピークの高さが、バナジウム−酸素(V−O)結合に起因する他のピークの最大高さの1.5倍以下である、上記〈1〉〜〈4〉項のいずれか一項に記載の触媒。
〈6〉条件(b)において、前記920cm−1付近のピークの半値幅が、40cm−1以上である、上記〈1〉〜〈5〉項のいずれか一項に記載の触媒。
〈7〉前記シリカ担体が、細孔構造を有する多孔質シリカ担体である、上記〈1〉〜〈6〉項のいずれか一項に記載の触媒。
〈8〉前記複合酸化物が、前記多孔質シリカ担体の細孔構造内に担持されており、且つ
前記多孔質シリカ担体の細孔分布において、シリカの一次粒子間の間隙に起因するピークが、細孔径5〜50nmの範囲にあり、且つシリカ粒子内の細孔構造に起因するピークが、細孔径1〜5nmの範囲にある、
上記〈7〉項に記載の触媒。
〈9〉(a)銅塩の水溶液及びバナジウム塩の水溶液の一方の水溶液を、シリカ担体に吸水させ、乾燥及び仮焼成すること、
(b)工程(a)の後で、銅塩の水溶液及びバナジウム塩の水溶液の他方の水溶液を、前記シリカ担体に吸水させ、乾燥及び仮焼成すること、そして
(c)工程(b)の後で、得られた前記シリカ担体を700℃以上の温度で焼成すること
を含み、かつ
工程(c)における前記熱処理の前に、前記シリカ担体が700℃以上の熱処理を受けていない、
三酸化硫黄分解触媒の製造方法。
〈10〉(a)銅塩及びバナジウム塩を含有する水溶液を、シリカ担体に吸水させ、乾燥及び仮焼成すること、
(b)工程(a)の後で、得られた前記シリカ担体を700℃以上の温度で焼成すること
を含み、かつ
工程(b)における前記熱処理の前に、前記シリカ担体が700℃以上の熱処理を受けていない、
三酸化硫黄分解触媒の製造方法。
〈11〉上記〈1〉〜〈8〉項のいずれか一項に記載の三酸化硫黄分解触媒を用いて、三酸化硫黄を二酸化硫黄と酸素とに分解することを含む、二酸化硫黄の生成方法。
〈12〉前記分解を800℃以下の温度で行う、上記〈11〉項に記載の方法。
〈13〉水を、水素及び酸素に分解することを含む、水素生成方法であって、下記式(X1)で示される反応で、硫酸を、水、二酸化硫黄、及び酸素に分解することを含み、且つ下記式(X1)で示される反応の素反応である式(X1−1)及び(X1−2)の素反応のうち、式(X1−2)の素反応を、上記〈11〉又は〈12〉項に記載の方法によって行う、水素生成方法:
(X1)H2SO4 → H2O + SO2 + 1/2O2
(X1−1)H2SO4 → H2O + SO3
(X1−2)SO3 → SO2 + 1/2O2
〈14〉I−Sサイクル法、ウエスティングハウス・サイクル法、Ispra−Mark 13サイクル法、又はロスアラモス・サイエンスラボラトリ・サイクル法である、上記〈13〉項に記載の水素生成方法。
【発明の効果】
【0020】
本発明の三酸化硫黄分解触媒によれば、三酸化硫黄分解反応に必要とされる温度を低下させることができる。また、本発明の二酸化硫黄生成方法によれば、比較的低い温度において、三酸化硫黄を分解して二酸化硫黄を得ることができる。さらに、本発明の水素生成方法によれば、比較的低い温度において、水を分解して水素を得ることができる。
【図面の簡単な説明】
【0021】
【図1】図1は、ピロバナジン酸銅の(a)結晶構造及び(b)バナジン酸結合を示す図である。
【図2】図2は、本発明の三酸化硫黄分解触媒による触媒反応過程を示す概念図である。
【図3】図3は、(a)本発明の三酸化硫黄分解触媒の製造過程、及び(b)従来の三酸化硫黄分解触媒の製造過程を説明するための図である。
【図4】図4は、参考例1で単身触媒として用いた複合金属酸化物についてのX線回折分析の結果を示す図である。
【図5】図5は、実施例、比較例及び参考例の三酸化硫黄分解触媒の評価のために用いた装置を示す図である。
【図6】図6は、実施例1の三酸化硫黄分解触媒の評価における温度変化と転化率変化との関係を示す図である。
【図7】図7は、実施例1の三酸化硫黄分解触媒の走査透過型電子顕微鏡(STEM)写真を示す図である。なお、図7(a)〜(c)はそれぞれ、600℃、750℃及び800℃の温度での三酸化硫黄分解反応に用いた後の触媒についての写真である。
【図8】図8は、実施例1のXRD(X線回折)分析結果を示す図である。なお、図8は、800℃の温度での三酸化硫黄分解反応に用いた後の触媒についての評価結果である。
【図9】図9は、ケイ素(Si)、バナジウム(V)、及び銅(Cu)についての実施例1の三酸化硫黄分解触媒のEDS(エネルギー分散形X線分光)分析結果を、STEM−HAADF(走査透過電子顕微鏡(高角度散乱暗視野法))による分析結果と併せて示す図である。なお、図8は、800℃の温度での三酸化硫黄分解反応に用いた後の触媒についての評価結果である。
【図10】図10は、実施例1の三酸化硫黄分解触媒のラマン散乱分析結果を、比較のためのバナジン酸銅(Cu2V2O7)、酸化銅(CuO)及び五酸化バナジウム(V2O5)についての分析結果と併せて示す図である。なお、図10は、650℃及び800℃の温度での三酸化硫黄分解反応に用いた後の触媒(Cu−V−O/SiO2(650℃)及びCu−V−O/SiO2(800℃))についての評価結果である。
【発明を実施するための形態】
【0022】
(三酸化硫黄分解触媒)
本発明の三酸化硫黄分解触媒は、銅とバナジウムとの複合酸化物がシリカ担体に担持されてなる。ここで、この本発明の三酸化硫黄分解触媒では、複合酸化物の結晶の対称性が低下している。すなわち、本発明の三酸化硫黄分解触媒では、銅とバナジウムとの複合酸化物の結晶が歪んでいる。このような結晶の歪みは例えば、銅とバナジウムとの複合酸化物が、充分に薄い層としてシリカ担体に担持されており、かつシリカ担体が、この複合酸化物に化学的に影響を与えることによって達成されると考えられる。
【0023】
理論に限定されるものではないが、本発明の三酸化硫黄分解触媒でのように、銅とバナジウムとの複合酸化物の結晶が歪んでいる場合、バナジウム−酸素(V−O)結合の一部が、他のバナジウム−酸素(V−O)結合よりも切れやすくなっていると考えられる。このようにバナジウム−酸素(V−O)結合が切れやすくなっている場所では、比較的容易に酸素が脱離して、酸素欠陥が形成され、この酸素欠陥において三酸化硫黄の吸着及び分解が進行すると考えられる。
【0024】
これに関して、本発明の三酸化硫黄分解触媒では、銅とバナジウムとの複合酸化物が図1に示すようなピロバナジン酸銅(Cu2V2O7)構造を有すると考えられる。なお、図1では、ピロバナジン酸銅の(a)結晶構造及び(b)バナジン酸結合を示している。
【0025】
したがって、本発明の三酸化硫黄分解触媒では、図2に示すようにして三酸化硫黄の分解反応が促進されると考えられる。具体的には、銅とバナジウムとの複合酸化物の結晶が歪んでいる場合、この複合酸化物において、バナジウム−酸素(V−O)結合の一部(点線)が、他のバナジウム−酸素(V−O)結合(実線)よりも長く、それによって切れやすくなっており(図2(a))、この結合が切れて酸素が脱離すると、その位置が酸素欠陥(図2(b))となり、三酸化硫黄がこの酸素欠陥に吸着し(図2(c))、そして分解されて、二酸化硫黄が生成する(図2(d))と考えられる。
【0026】
このように銅とバナジウムとの複合酸化物の結晶が歪んでいることは、ラマン分光分析において、対称性の低下、すなわちピークの高さの低下及び/又はピークのブロード化として確認できる。したがって例えば、本発明の三酸化硫黄分解触媒は、ラマン分光分析において、バナジウム−酸素(V−O)結合に起因する920cm−1付近のピークが、下記の(a)及び(b)の少なくとも一方の条件を満たしている:
(a)上記920cm−1付近のピークの高さが、バナジウム−酸素(V−O)結合に起因する他のピークの最大高さの3.0倍以下、2.5倍以下、2.0倍以下、1.5倍以下、又は1.0倍以下、及び
(b)上記920cm−1付近のピークの半値幅が、30cm−1以上、40cm−1以上、50cm−1以上。
【0027】
本発明の三酸化硫黄分解触媒によれば、式(B1−2)の三酸化硫黄分解反応に必要とされる温度を低下させ、例えば700℃以下程度の温度において、実質的な速度で三酸化硫黄分解反応を進行させることができる。
【0028】
上記記載のように、三酸化硫黄を分解する従来の方法では、1000℃近い温度を用いることが一般的であった。しかしながら、このような高温に耐えられる材料は非常に限定されており、またかなり高価なものであった。
【0029】
また、1000℃近い高温は、太陽エネルギーからは安価に得ることが困難であった。すなわち例えば、太陽熱エネルギーを得る集光装置としては、パラボリックディッシュ型集光装置、ソーラータワー型集光装置、及びパラボリックトラフ型集光装置が知られているが、これらのうちで構造が簡単で、コストが安く、且つ大規模なプラントに適しているパラボリックトラフ型集光装置では、太陽エネルギーの収集と放射によるエネルギーの散逸との釣り合いから、1000℃近い高温での太陽エネルギーの収集は非現実的である。
【0030】
したがって、本発明の三酸化硫黄分解触媒によって、三酸化硫黄分解反応に必要とされる温度を低下させ、例えば700℃程度の温度において、実質的な速度で三酸化硫黄分解反応を進行するようにすることは、産業的な価値が非常に大きい。
【0031】
(三酸化硫黄分解触媒−複合酸化物)
本発明の三酸化硫黄分解触媒の複合酸化物において、銅とバナジウムとの原子比(銅:バナジウム)は例えば、1:9〜9:1、2:8〜8:2、3:7〜7:3、又は4:6〜6:4にすることができる。
【0032】
本発明の三酸化硫黄分解触媒では例えば、銅及びバナジウムの合計量が担体100gに対して、0.01mol以上、0.05mol以上、0.10mol以上となる割合で、複合金属酸化物をシリカ担体に担持することができる。また、この担持量は例えば、担体100gに対して、2.00mol以下、1.00mol以下、又は0.50mol以下の割合にすることができる。
【0033】
(三酸化硫黄分解触媒−製造方法)
本発明の三酸化硫黄分解触媒は、任意の方法で得ることができる。
【0034】
例えば、本発明の三酸化硫黄分解触媒は、銅塩の水溶液を、担体に吸水させ、乾燥及び仮焼成し、バナジウム塩の水溶液を、担体に吸水させ、乾燥及び仮焼成し、そしてその後で、得られた担体を焼成することによって得ることができる。また、これとは反対に、本発明の三酸化硫黄分解触媒は、バナジウム塩の水溶液を先に担体に吸水させ、乾燥及び仮焼成し、銅塩の水溶液を、担体に吸水させ、乾燥及び仮焼成し、そしてその後で、得られた担体を焼成することによって得ることができる。また、銅塩及びバナジウム塩を、これらの共沈が可能なように選択する場合、銅塩及びバナジウム塩の両方を含有する水溶液を、担体に吸水させ、乾燥及び仮焼成し、そしてその後で、得られた担体を焼成することによって、本発明の三酸化硫黄分解触媒を得ることもできる。
【0035】
なお、本発明の三酸化硫黄分解触媒の製造においては、銅とバナジウムとの複合酸化物が、充分に薄い層としてシリカ担体に担持されており、かつシリカ担体に少なくとも部分的に影響され、それによってこの複合酸化物の結晶の対称性が低下するようにすることが必要である。
【0036】
これに関して、本発明の三酸化硫黄分解触媒を製造する上記の方法においては、銅塩及びバナジウム塩の水溶液を先に担体に吸水させ、乾燥及び仮焼成して得られたシリカ担体を、比較的高温で焼成すること、並びにこのようにして焼成されるシリカ担体が、この焼成の前に比較的高温での熱処理を受けていないことが好ましい。
【0037】
このように、高温での熱処理を受けていないシリカ担体では、水酸基、タングリングボンド、微細な凹凸形状等の不安定部分が比較的多く表面に残されている。したがって、図3(a)に示すように、このような表面上に複合酸化物前駆体を担持し、これらを共に焼成する場合、これらの不安定部分が、焼成の間に、その上に担持された複合酸化物前駆体に作用し、複合酸化物を充分に薄い層状にし、かつ/又は得られる複合酸化物の結晶の対称性を低下させると考えられる。これに関して、微細な凹凸形状が焼成の間に平坦化される場合、シリカ担体の表面と、その上に担持された複合酸化物前駆体及び複合酸化物との混和が促進されると考えられる。
【0038】
これに対して、高温での熱処理を受けているシリカ担体では、水酸基、タングリングボンド、微細な凹凸形状等の不安定部分が、高温での熱処理によって減少している。したがって、図3(b)に示すように、このような表面上に複合酸化物前駆体を担持し、これらを共に焼成する場合であっても、シリカ担体の表面がその上に担持された複合酸化物前駆体に対して与える影響が小さいと考えられる。
【0039】
本発明の方法において多孔質シリカ担体を焼成する温度は、600℃以上、650℃以上、700℃以上、750℃以上、800℃以上であってよい。
【0040】
また、本発明の方法において多孔質シリカ担体を焼成する雰囲気は、任意の雰囲気であってよく、例えば空気のような酸素含有雰囲気、窒素又はアルゴンのような不活性雰囲気、又は硫酸雰囲気若しくは三酸化硫黄雰囲気のような酸化雰囲気であってよい。
【0041】
なお、本発明の方法における焼成は、本発明の三酸化硫黄分解触媒の使用の間に行ってもよい。すなわち、最終的な焼成を行わずに本発明の三酸化硫黄分解触媒を装置に装填し、そして三酸化硫黄分解反応を少なくとも一時的に、上記の焼成温度よりも高い温度において行うことによって、焼成を達成することもできる。
【0042】
(三酸化硫黄分解触媒−担体)
本発明の三酸化硫黄分解触媒において用いられるシリカ担体は、銅とバナジウムとの複合酸化物に少なくとも部分的に影響を与え、それによってこの複合酸化物の結晶の対称性を低下させる範囲で、任意のシリカ担体を用いることができる。
【0043】
したがって、三酸化硫黄分解触媒を製造する本発明の方法において用いられるシリカ担体は、水酸基、タングリングボンド、微細な凹凸形状等の不安定部分を表面に有するシリカ担体、例えば700℃以上、650℃以上、600℃以上、又は550℃以上の熱処理を受けていないシリカ担体であってよい。
【0044】
このようなシリカ担体としては特に、細孔構造を有する多孔質シリカ担体を用いることができる。この場合、好ましくは、複合酸化物が多孔質シリカ担体の細孔構造内に担持されている。
【0045】
このような多孔質シリカ担体は、メソポーラスシリカ担体、例えばKIT−6のような立方晶メソポーラスシリカであってよい。また、このような多孔質シリカ担体は例えば、多孔質シリカ担体の細孔分布において、シリカの一次粒子間の間隙に起因するピークが、細孔径5〜50nm、特に細孔径5〜30nmの範囲にあり、且つシリカ粒子内の細孔構造に起因するピークが、細孔径1〜5nm、特に細孔径2〜4nmの範囲にある多孔質シリカ担体であってよい。
【0046】
このように、細孔構造を有する多孔質シリカ担体を用いる場合、複合酸化物が、多孔質シリカ担体の細孔構造表面近傍に担持され、それによって複合酸化物粒子のシンタリングが抑制される。理論に限定されるわけではないが、このように非常に微細な状態で維持されている複合酸化物粒子では、触媒の微粒子化によって、触媒の表面積が100倍程度に増大されるだけでなく、触媒の表面の性質が変化して、複合酸化物の触媒性能が改良される場合もあると考えられる。
【0047】
また、細孔構造を有する多孔質シリカ担体の細孔分布において、二元の細孔分布となることにより細孔径が数nmの表面積の広い活性部位に、十〜数十nmの細孔から拡散速度の速い気相ガスが高速に供給されることによって、複合酸化物粒子と三酸化硫黄との接触の機会が多く、それによって触媒性能を改良すると考えられる。
【0048】
なお、細孔構造を有する多孔質シリカ担体は例えば、特開2008−12382に記載の方法によって得ることができる。
【0049】
(二酸化硫黄の生成方法)
二酸化硫黄を生成する本発明の方法は、本発明の三酸化硫黄分解触媒を用いて、三酸化硫黄を二酸化硫黄と酸素とに分解することを含む。ここで、この方法は、本発明の三酸化硫黄分解触媒を用いることによって、三酸化硫黄を分解する従来の方法よりも低い温度、例えば800℃以下、750℃以下、700℃以下、650℃以下の温度で実施することができる。
【0050】
(水素生成方法)
水素を生成する本発明の方法は、水を、水素及び酸素に分解すること、例えばI−Sサイクル法、ウエスティングハウス・サイクル法、Ispra−Mark 13サイクル法、又はロスアラモス・サイエンスラボラトリ・サイクル法によって、水を水素及び酸素に分解することを含む。ここで、この本発明の方法は、下記式(X1)で示される反応で、硫酸を、水、二酸化硫黄、及び酸素に分解することを含み、且つ下記式(X1)で示される反応の素反応である式(X1−1)及び(X1−2)の素反応のうち、式(X1−2)の素反応を、二酸化硫黄を生成する本発明の方法によって行う:
(X1)H2SO4 → H2O + SO2 + 1/2O2
(X1−1)H2SO4 → H2O + SO3
(X1−2)SO3 → SO2 + 1/2O2
【0051】
すなわち、例えば、水素を生成する本発明の方法は、下記式(X1)〜(X3)で示されるI−S(ヨウ素−イオウ)サイクル法において、式(X1)の反応の素反応である式(X1−1)及び(X1−2)の素反応のうち、式(X1−2)の素反応を、二酸化硫黄を生成する本発明の方法によって行うことを含む:
(X1)H2SO4 → H2O + SO2 + 1/2O2
(X1−1)H2SO4 → H2O + SO3
(X1−2)SO3 → SO2 + 1/2O2
(X2)I2 + SO2 + 2H2O → 2HI + H2SO4
(X3)2HI → H2 + I2
全反応:H2O → H2 + 1/2O2
【0052】
また、例えば、水素を生成する本発明の方法は、下記式(X1)、(X4)及び(X5)で示されるウエスティングハウス・サイクル法において、式(X1)の反応の素反応である式(X1−1)及び(X1−2)の素反応のうち、式(X1−2)の素反応を、二酸化硫黄を生成する本発明の方法によって行うことを含む:
(X1)H2SO4 → H2O + SO2 + 1/2O2
(X1−1)H2SO4 → H2O + SO3
(X1−2)SO3 → SO2 + 1/2O2
(X4)SO2 + 2H2O → H2SO3
(X5)H2SO3 + H2O+ → H2 + H2SO4(電気分解)
全反応:H2O → H2 + 1/2O2
【0053】
さらに、例えば、水素を生成する本発明の方法は、下記式(X1)、(X6)及び(X7)で示されるIspra−Mark 13サイクル法において、式(X1)の反応の素反応である式(X1−1)及び(X1−2)の素反応のうち、式(X1−2)の素反応を、二酸化硫黄を生成する本発明の方法によって行うことを含む:
(X1)H2SO4 → H2O + SO2 + 1/2O2
(X1−1)H2SO4 → H2O + SO3
(X1−2)SO3 → SO2 + 1/2O2
(X6)2HBr → Br2 + H2
(X7)Br2 + SO2 + 2H2O+ → 2HBr + H2SO4
全反応:H2O → H2 + 1/2O2
【0054】
さらに、例えば、水素を生成する本発明の方法は、下記式(X1)、及び(X8)〜(X10)で示されるロスアラモス・サイエンスラボラトリ・サイクル法において、式(X1)の反応の素反応である式(X1−1)及び(X1−2)の素反応のうち、式(X1−2)の素反応を、二酸化硫黄を生成する本発明の方法によって行うことを含む:
(X1)H2SO4 → H2O + SO2 + 1/2O2
(X1−1)H2SO4 → H2O + SO3
(X1−2)SO3 → SO2 + 1/2O2
(X8)Br2 + SO2 + 2H2O+ → 2HBr + H2SO4
(X9)2CrBr3 → 2CrBr2 + Br2
(X10)2HBr + 2CrBr2 → 2CrBr3 + H2
全反応:H2O → H2 + 1/2O2
【実施例】
【0055】
《参考例1及び比較例1〜3》
以下の参考例及び比較例では、銅(Cu)とバナジウム(V)との複合金属酸化物が、三酸化硫黄分解触媒として優れた性質を有することを示す。
【0056】
《参考例1》
参考例1では、銅(Cu)とバナジウム(V)との複合金属酸化物(Cu−V−O)を単身触媒として用いた。
【0057】
(単身触媒の製造)
参考例1の単身触媒は、それぞれの金属の原子比が1:1である酸化銅及び酸化バナジウムを、乳鉢で粉砕し、良く混合し、アルミナ性るつぼに入れ、そして750℃で12時間にわたって焼成して得た。得られた単身触媒についてのX線回折分析(XRD)結果を図4に示す。
【0058】
《比較例1》
比較例1では、銅(Cu)の酸化物(Cu−O)を単身触媒として用いた。ここでは、参考例1で原料として用いた酸化銅をそのまま単身触媒として用いた。
【0059】
《比較例2》
比較例2では、バナジウム(V)の酸化物(V−O)を触媒として用いた。ここでは、参考例1で原料として用いた酸化バナジウムをそのまま単身触媒として用いた。
【0060】
《比較例3》
比較例3では触媒を用いなかった。
【0061】
(評価(転化率))
図5に示す固定床流通反応装置を用いて、参考例1及び比較例1〜3の単身触媒について、下記式(X1−2)の三酸化硫黄分解反応の転化率を評価した:
(X1−2)SO3 → SO2 + 1/2O2
【0062】
具体的には、三酸化硫黄分解反応の転化率は、図5に関して下記で説明するようにして評価した。
【0063】
14〜20メッシュに調整した0.5gの単身触媒又は担持触媒を、触媒床10として、石英製反応管4(内径10mm)に充填した。窒素(N2)(100mL/分)及び47重量%硫酸(H2SO4)水溶液(50μL/分)を、それぞれ窒素供給部1及び硫酸供給部3から、石英製反応管4の下段に供給した。
【0064】
石英製反応管4の下段に供給された硫酸(H2SO4)は、石英製反応管4の下段及び中段において加熱されて、三酸化硫黄(SO3)及び酸素(O2)に分解し、そして触媒床10に流入した(SO3:4.5mol%、H2O:31mol%、N2:残部、0℃換算ガス流量:148.5cm3/分、重量流量比(W/F比):5.61×10−5g・h/cm3、気体時空間速度(GHSV:Gas Hourly Space Velocity):約15,000h−1)。
【0065】
ここで、石英製反応管4は、下段がヒーター4aによって約400℃に加熱されており、かつ中段がヒーター4bによって約600℃に加熱されていた。また、石英製反応管4の上段は、ヒーター4cによって初めに約600℃に加熱されており、定常状態になった後で、650℃に加熱した。
【0066】
石英製反応管4の上段をヒーター4cによって650℃に加熱した後で、石英製反応管4からの流出ガスを、空冷し、その後で、0.05Mのヨウ素(I2)溶液にバブリングして、ヨウ素溶液に二酸化硫黄(SO2)を吸収させた。0.025Mのチオ硫酸ナトリウム(Na2S2O3)溶液を用いて、二酸化硫黄を吸収したヨウ素溶液にヨードメトリー滴定を行って、吸収された二酸化硫黄の量を求めた。
【0067】
また、ヨウ素溶液にバブリングした後の流出ガスは、ドライアイス・エタノール混合物で冷却し、残留している二酸化硫黄及び三酸化硫黄をミストアブソーバー及びシリカゲルで完全に除去し、その後で、磁気圧力酸素計(堀場製作所のMPA3000)及びガスクロマトグラフ(島津製作所のGC8A、モレキュラーシーブ5A、TCD検出器)を用いて、酸素(O2)の量を求めた。
【0068】
三酸化硫黄(SO3)から二酸化硫黄(SO2)への平衡転化率に対する到達率は、上記のようにして求めた二酸化硫黄及び酸素の量から計算した。
【0069】
参考例及び比較例についての評価結果を、下記の表1に示す。
【0070】
【表1】
【0071】
表1からは、参考例1の触媒が、比較例1〜3の触媒と比較して、650℃という比較的低い温度において、有意に好ましい三酸化硫黄分解特性を有していることが理解される。
【0072】
なお、上記の比較例2で用いられている酸化バナジウム、特に五酸化バナジウム(V2O5)は、下記式(C−1)〜(C−3)で示される反応で硫酸を製造する接触法と呼ばれる方法において、二酸化硫黄を酸化させて三酸化硫黄を得る式(C−2)の反応を促進するために用いられている:
(C−1)S(固体) + O2(気体) → SO2(気体)
(C−2)2SO2(気体) + O2(気体) → 2SO3(気体)
(C−3)SO3(気体) + H2O(液体) → H2SO4(液体)
【0073】
しかしながら、酸化バナジウムを用いている比較例2は、参考例1と比較して有意に劣った転化率を示していた。
【0074】
《実施例1、並びに参考例2及び3》
実施例1及び参考例2では、550℃において焼成して得たシリカ担体を原料として用いる場合(実施例1)と、800℃において焼成して得たシリカ担体を原料として用いる場合(比較例1)の違いについて評価した。また、参考例3では、触媒金属として白金を用いて評価を行った。
【0075】
《実施例1》
実施例1では、銅(Cu)とバナジウム(V)との複合金属酸化物(Cu−V−O)が多孔質シリカ担体に担持されてなる触媒を用いた。ここで、用いられている多孔質シリカ担体は、空気中において550℃で焼成して得たものである。
【0076】
具体的には、実施例1の触媒は下記のようにして製造した。
【0077】
(多孔質シリカ担体の製造)
多孔質シリカ担体は、立方晶メソポーラスシリカ(KIT−6)であり、下記のようにして製造した。
【0078】
(1)蒸留水144mLに、7.9gの35質量%塩酸(HCl)及び4.0gの非イオン性界面活性剤(Pluronic(商標)P−123)を添加し、得られた水溶液を35℃の温度において撹拌して、成分を溶解。
(2)得られた混合物に、4.0gの1−ブタノールを添加し、混合物が透明になるまで、35℃の温度において撹拌し、それによって、非イオン性界面活性剤を自己配列。
(3)得られた混合物に、シリカ源としての8.6gのテトラエトキシシラン(TEOS)を添加し、35℃の温度において24時間にわたって強撹拌し、自己整列している非イオン性界面活性剤をテンプレートとして、テトラエトキシシラン(TEOS)を加水分解。
(4)100℃の温度において24時間にわたって静置し、その後、洗浄せずにそのまま110℃の温度において24時間にわたって乾燥。
(6)8mlの35質量%塩酸と120mLのエタノールとの混合物中において、1.5時間にわたって撹拌して洗浄。
(7)110℃の温度において24時間にわたって乾燥し、そして3℃/分の昇温速度で550℃まで加熱して、この温度で5時間にわたって焼成して、立方晶メソポーラスシリカ(KIT−6)を取得。
【0079】
(複合金属酸化物の担持)
複合酸化物は、吸水担持法によって、多孔質シリカ担体に担持した。具体的には、初めに、銅の硝酸塩を水に溶解した水溶液を作り、この水溶液を担体に吸水させ、150℃で乾燥し、350℃で1時間にわたって仮焼成した。次に、メタバナジン酸アンモニウムを水に溶解し、この水溶液を担体に吸水させ、150℃で乾燥し、350℃で1時間にわたって仮焼成した。最後に、得られた担体を600℃で2時間にわたって焼成して、複合酸化物を担持している多孔質シリカ担体を得た。
【0080】
なお、担持量は、銅が0.12mol/100g−担体、かつバナジウムが0.12mol/100g−担体とした。
【0081】
《参考例2》
参考例2では、銅(Cu)とバナジウム(V)との複合金属酸化物(Cu−V−O)が多孔質シリカ担体に担持されてなる触媒を用いた。ここで、用いられている多孔質シリカ担体は、空気中において800℃で焼成して得たものである。
【0082】
具体的には、参考例2の触媒は下記のようにして製造した。
【0083】
(多孔質シリカ担体の製造)
多孔質シリカ担体は、特開2008−12382に記載の方法と類似の方法によって製造した。すなわち、多孔質シリカ担体は、下記のようにして製造した。
【0084】
蒸留水6L(リットル)に、セチルトリメチルアンモニウムクロライド1kgを溶解した。得られた水溶液を2時間にわたって撹拌して、セチルトリメチルアンモニウムクロライドを自己配列させた。次に、セチルトリメチルアンモニウムクロライドを自己配列させた溶液に、テトラエトキシシランとアンモニア水を添加して、溶液のpHを9.5にした。
【0085】
この溶液中において、テトラエトキシシランを30時間にわたって加水分解して、配列したヘキサデシルアミンの周りにシリカを析出させて、ナノサイズの細孔を有する一次粒子からなる二次粒子を形成し、多孔質シリカ担体前駆体を得た。
【0086】
その後、得られた多孔質シリカ担体前駆体を、エタノール水で洗浄し、ろ過し、乾燥して、800℃の空気中で2時間にわたって焼成して、多孔質シリカ担体を得た。
【0087】
ここで得られた多孔質シリカ担体は、シリカの細孔構造に起因する2.7nm付近の細孔、及びシリカの一次粒子間の間隙に起因する10nm強の細孔を有していた。
【0088】
(複合金属酸化物の担持)
実施例1と同様にして、銅とバナジウムの複合酸化物を多孔質シリカ担体に担持した。
【0089】
《参考例3》
参考例3では、γ−アルミナ担体に白金を担持して、担持触媒を製造した。ここでは、担持量を0.5g−Pt/100g−担体とした。
【0090】
(評価(転化率))
上記参考例1等でのようにして評価を行った。ただし、実施例1についての評価では、石英製反応管4の上段が約600℃で定常状態になった後で、図6で示すようにして加熱温度を変化させて評価を行った。加熱温度の変化と併せて、三酸化硫黄(SO3)から二酸化硫黄(SO2)への転化率の変化を図6に示している。
【0091】
図6で示されているように、加熱温度600℃及び650℃においては、一時的に転化率が大きくなるものの、この転化率はすぐに小さくなった。
【0092】
その後、加熱温度を700℃、750℃及び800℃にすると、転化率が有意に大きくなった。これらの温度での転化率は、対応する温度における平衡転化率に対して100%近かった。
【0093】
その後、再び加熱温度を600℃及び650℃まで加熱温度を低下させると、予想外に、転化率は最初に加熱温度を600℃及び650℃にしたときの転化率よりも有意に大きくなった。600℃の温度での転化率は、この温度での平衡転化率に対して79.8%であった。また、評価開始から10時間後の加熱温度650℃の状態での転化率は、対応する温度における平衡転化率に対して100%近かった。なお、650℃の温度での転化率をその後27時間にわたって評価したところ、転化率がほぼ維持されていることが確認された。
【0094】
参考例2についての評価では、図6で示すようにして加熱温度を変化させたところ、評価開始から10時間後の加熱温度650℃の状態において、この温度における平衡転化率に対する到達率は88.5%であった。実施例1では、対応する到達率は100%近かったので、参考例2の触媒と比較して、実施例1の三酸化硫黄分解触媒が有意に優れていることが理解される。
【0095】
参考例3についての評価では、図6で示すようにして加熱温度を変化させたところ、評価開始から10時間後の加熱温度650℃の状態において、対応する温度における平衡転化率に対して62.7%であった。実施例1では、対応する到達率は100%近かったので、参考例3の触媒と比較して、実施例1の三酸化硫黄分解触媒が有意に優れていることが理解される。
【0096】
(評価(STEM分析))
実施例1で用いた三酸化硫黄分解触媒について、600℃、750℃及び800℃の温度での三酸化硫黄分解反応に用いた後で、走査透過型電子顕微鏡(STEM)よる分析を行った。得られた結果をそれぞれ図7(a)〜(c)に示す。
【0097】
600℃の温度での三酸化硫黄分解反応に用いた後の触媒(図7(a))では、シリカ担体の細孔に由来する孔(白く抜けた部分)、及び担体に担持されている銅とバナジウムとの複合酸化物(Cu−V−O)が確認できた。
【0098】
また、750℃の温度での三酸化硫黄分解反応に用いた後の触媒(図7(b))では、シリカ担体の細孔に由来する孔が潰れ、全体として収縮していた。
【0099】
さらに、800℃の温度での三酸化硫黄分解反応に用いた後の触媒(図7(c))では、シリカ担体のシンタリングが大きく進行し、それによって表面積が小さく、かつシリカゲルのような形態となっていた。
【0100】
これら図7(a)〜(c)から明らかなように、シリカ担体は加熱によって大きく劣化していた。しかしながら、三酸化硫黄分解触媒としての性質は、800℃の温度での使用後に改良されていたことから、担体に担持された銅とバナジウムとの複合酸化物の物性が改良されていたことが理解される。
【0101】
(評価(XRD分析))
800℃の温度での三酸化硫黄分解反応に用いた後の触媒を、XRD(X線回折)分析で評価した。評価結果を図8に示す。
【0102】
図8のXRD分析結果からは、硫酸銅(CuSO4)に帰属される弱いピークが観測され、これは、極微量の硫酸銅の存在を示唆している。また、XRD分析結果では、それ以外の明確なピークが示されていないことから、大部分の成分が非常に均一に分散していると考えられる。なお、650℃の温度での三酸化硫黄分解反応に用いた後の触媒についてん6XRD分析でも、800℃の場合と同様な結果が得られた。
【0103】
(評価(EDS分析))
800℃の温度での三酸化硫黄分解反応に用いた後の触媒を、ケイ素(Si)、バナジウム(V)、及び銅(Cu)についてのEDS(エネルギー分散形X線分光)分析で評価した。評価結果を、STEM−HAADF(走査透過電子顕微鏡(高角度散乱暗視野法))による分析結果と併せて、図9に示す。
【0104】
図9のESD分析結果からは、担体に担持されている触媒成分であるCu及びVの分布が、担体の構成元素であるSiの分布と同様であることが分かる。これは、触媒成分である銅とバナジウムとの複合酸化物が、シリカ担体上に薄膜状で堆積していることを示している。なお、担体上に担持されているCuは部分的に凝縮している箇所があるが、これは、硫酸銅として析出した部分であると考えられる。
【0105】
(評価(ラマン散乱分析))
650℃及び800℃の温度での三酸化硫黄分解反応に用いた後の触媒(Cu−V−O/SiO2(650℃)及びCu−V−O/SiO2(800℃))を、ラマン散乱分析で評価した。評価結果を、比較のためのバナジン酸銅(Cu2V2O7)、酸化銅(CuO)及び五酸化バナジウム(V2O5)についての分析結果と併せて、図10に示す。なお、バナジン酸銅(Cu2V2O7)についての分析結果は、高さを0.5倍(×0.5)して表している。
【0106】
図10のラマン散乱分析結果によれば、650℃の温度での三酸化硫黄分解反応に用いた後の触媒(Cu−V−O/SiO2(650℃))では、バナジン酸銅(Cu2V2O7)と同様なラマン散乱プロファイルが得られた。
【0107】
このプロファイルでは、920cm−1付近に存在するピーク、すなわちピロバナジン酸(V2O7)構造の両端のVO3の対称収縮振動(対称性最大)に由来するピークが、最大のピークであった。具体的には、このプロファイルでは、920cm−1付近のピークの高さが、バナジウム−酸素(V−O)結合に起因する他のピークの最大高さの約3.9倍になっていた。また、この触媒(Cu−V−O/SiO2(650℃))では、920cm−1付近のピークの半値幅が、約25cm−1になっていた。
【0108】
これに対して、800℃の温度での三酸化硫黄分解反応に用いた後の触媒(Cu−V−O/SiO2(800℃))では、920cm−1付近のこのピークが低く、かつブロードになっていた。具体的には、このプロファイルでは、920cm−1付近のピークの高さが、バナジウム−酸素(V−O)結合に起因する他のピークの最大高さの約0.9倍になっていた。また、この触媒(Cu−V−O/SiO2(800℃))では、920cm−1付近のピークの半値幅が、約59cm−1になっていた。
【0109】
これは、ピロバナジン酸(V2O7)構造が変化して結晶が歪み、それによってピロバナジン酸(V2O7)構造の両端のVO3の対称性が低下したことを示している。この対称性の低下は、バナジウムと酸素との間の結合の一部が、バナジウムと酸素との間の他の結合よりも長く、それによって切れやすくなっていることを意味すると考えられる。
【符号の説明】
【0110】
1 窒素供給部
3 硫酸供給部
4 石英製反応管
4a、4b、4c ヒーター
10 触媒床
【特許請求の範囲】
【請求項1】
銅とバナジウムとの複合酸化物がシリカ担体に担持されてなり、かつラマン分光分析において、バナジウム−酸素(V−O)結合に起因する920cm−1付近のピークの高さが、下記の(a)及び(b)の少なくとも一方の条件を満たす、三酸化硫黄分解触媒:
(a)前記920cm−1付近のピークの高さが、バナジウム−酸素(V−O)結合に起因する他のピークの最大高さの3.0倍以下、及び
(b)前記920cm−1付近のピークの半値幅が、30cm−1以上。
【請求項2】
前記複合酸化物において、銅とバナジウムとの原子比が、1:9〜9:1である、請求項1に記載の触媒。
【請求項3】
条件(a)を少なくとも満たす、請求項1又は2に記載の触媒。
【請求項4】
条件(b)を少なくとも満たす、請求項1又は2に記載の触媒。
【請求項5】
条件(a)において、前記920cm−1付近のピークの高さが、バナジウム−酸素(V−O)結合に起因する他のピークの最大高さの1.5倍以下である、請求項1〜4のいずれか一項に記載の触媒。
【請求項6】
条件(b)において、前記920cm−1付近のピークの半値幅が、40cm−1以上である、請求項1〜5のいずれか一項に記載の触媒。
【請求項7】
前記シリカ担体が、細孔構造を有する多孔質シリカ担体である、請求項1〜6のいずれか一項に記載の触媒。
【請求項8】
前記複合酸化物が、前記多孔質シリカ担体の細孔構造内に担持されており、且つ
前記多孔質シリカ担体の細孔分布において、シリカの一次粒子間の間隙に起因するピークが、細孔径5〜50nmの範囲にあり、且つシリカ粒子内の細孔構造に起因するピークが、細孔径1〜5nmの範囲にある、
請求項7に記載の触媒。
【請求項9】
(a)銅塩の水溶液及びバナジウム塩の水溶液の一方の水溶液を、シリカ担体に吸水させ、乾燥及び仮焼成すること、
(b)工程(a)の後で、銅塩の水溶液及びバナジウム塩の水溶液の他方の水溶液を、前記シリカ担体に吸水させ、乾燥及び仮焼成すること、そして
(c)工程(b)の後で、得られた前記シリカ担体を700℃以上の温度で焼成すること
を含み、かつ
工程(c)における前記熱処理の前に、前記シリカ担体が700℃以上の熱処理を受けていない、
三酸化硫黄分解触媒の製造方法。
【請求項10】
(a)銅塩及びバナジウム塩を含有する水溶液を、シリカ担体に吸水させ、乾燥及び仮焼成すること、
(b)工程(a)の後で、得られた前記シリカ担体を700℃以上の温度で焼成すること
を含み、かつ
工程(b)における前記熱処理の前に、前記シリカ担体が700℃以上の熱処理を受けていない、
三酸化硫黄分解触媒の製造方法。
【請求項11】
請求項1〜8のいずれか一項に記載の三酸化硫黄分解触媒を用いて、三酸化硫黄を二酸化硫黄と酸素とに分解することを含む、二酸化硫黄の生成方法。
【請求項12】
前記分解を800℃以下の温度で行う、請求項11に記載の方法。
【請求項13】
水を、水素及び酸素に分解することを含む、水素生成方法であって、下記式(X1)で示される反応で、硫酸を、水、二酸化硫黄、及び酸素に分解することを含み、且つ下記式(X1)で示される反応の素反応である式(X1−1)及び(X1−2)の素反応のうち、式(X1−2)の素反応を、請求項11又は12に記載の方法によって行う、水素生成方法:
(X1)H2SO4 → H2O + SO2 + 1/2O2
(X1−1)H2SO4 → H2O + SO3
(X1−2)SO3 → SO2 + 1/2O2
【請求項14】
I−Sサイクル法、ウエスティングハウス・サイクル法、Ispra−Mark 13サイクル法、又はロスアラモス・サイエンスラボラトリ・サイクル法である、請求項13に記載の水素生成方法。
【請求項1】
銅とバナジウムとの複合酸化物がシリカ担体に担持されてなり、かつラマン分光分析において、バナジウム−酸素(V−O)結合に起因する920cm−1付近のピークの高さが、下記の(a)及び(b)の少なくとも一方の条件を満たす、三酸化硫黄分解触媒:
(a)前記920cm−1付近のピークの高さが、バナジウム−酸素(V−O)結合に起因する他のピークの最大高さの3.0倍以下、及び
(b)前記920cm−1付近のピークの半値幅が、30cm−1以上。
【請求項2】
前記複合酸化物において、銅とバナジウムとの原子比が、1:9〜9:1である、請求項1に記載の触媒。
【請求項3】
条件(a)を少なくとも満たす、請求項1又は2に記載の触媒。
【請求項4】
条件(b)を少なくとも満たす、請求項1又は2に記載の触媒。
【請求項5】
条件(a)において、前記920cm−1付近のピークの高さが、バナジウム−酸素(V−O)結合に起因する他のピークの最大高さの1.5倍以下である、請求項1〜4のいずれか一項に記載の触媒。
【請求項6】
条件(b)において、前記920cm−1付近のピークの半値幅が、40cm−1以上である、請求項1〜5のいずれか一項に記載の触媒。
【請求項7】
前記シリカ担体が、細孔構造を有する多孔質シリカ担体である、請求項1〜6のいずれか一項に記載の触媒。
【請求項8】
前記複合酸化物が、前記多孔質シリカ担体の細孔構造内に担持されており、且つ
前記多孔質シリカ担体の細孔分布において、シリカの一次粒子間の間隙に起因するピークが、細孔径5〜50nmの範囲にあり、且つシリカ粒子内の細孔構造に起因するピークが、細孔径1〜5nmの範囲にある、
請求項7に記載の触媒。
【請求項9】
(a)銅塩の水溶液及びバナジウム塩の水溶液の一方の水溶液を、シリカ担体に吸水させ、乾燥及び仮焼成すること、
(b)工程(a)の後で、銅塩の水溶液及びバナジウム塩の水溶液の他方の水溶液を、前記シリカ担体に吸水させ、乾燥及び仮焼成すること、そして
(c)工程(b)の後で、得られた前記シリカ担体を700℃以上の温度で焼成すること
を含み、かつ
工程(c)における前記熱処理の前に、前記シリカ担体が700℃以上の熱処理を受けていない、
三酸化硫黄分解触媒の製造方法。
【請求項10】
(a)銅塩及びバナジウム塩を含有する水溶液を、シリカ担体に吸水させ、乾燥及び仮焼成すること、
(b)工程(a)の後で、得られた前記シリカ担体を700℃以上の温度で焼成すること
を含み、かつ
工程(b)における前記熱処理の前に、前記シリカ担体が700℃以上の熱処理を受けていない、
三酸化硫黄分解触媒の製造方法。
【請求項11】
請求項1〜8のいずれか一項に記載の三酸化硫黄分解触媒を用いて、三酸化硫黄を二酸化硫黄と酸素とに分解することを含む、二酸化硫黄の生成方法。
【請求項12】
前記分解を800℃以下の温度で行う、請求項11に記載の方法。
【請求項13】
水を、水素及び酸素に分解することを含む、水素生成方法であって、下記式(X1)で示される反応で、硫酸を、水、二酸化硫黄、及び酸素に分解することを含み、且つ下記式(X1)で示される反応の素反応である式(X1−1)及び(X1−2)の素反応のうち、式(X1−2)の素反応を、請求項11又は12に記載の方法によって行う、水素生成方法:
(X1)H2SO4 → H2O + SO2 + 1/2O2
(X1−1)H2SO4 → H2O + SO3
(X1−2)SO3 → SO2 + 1/2O2
【請求項14】
I−Sサイクル法、ウエスティングハウス・サイクル法、Ispra−Mark 13サイクル法、又はロスアラモス・サイエンスラボラトリ・サイクル法である、請求項13に記載の水素生成方法。
【図2】

【図3】
【図4】
【図5】
【図6】
【図8】
【図10】
【図1】
【図7】

【図9】

【図3】
【図4】
【図5】
【図6】
【図8】
【図10】
【図1】
【図7】
【図9】
【公開番号】特開2013−111542(P2013−111542A)
【公開日】平成25年6月10日(2013.6.10)
【国際特許分類】
【出願番号】特願2011−260754(P2011−260754)
【出願日】平成23年11月29日(2011.11.29)
【出願人】(000003207)トヨタ自動車株式会社 (59,920)
【出願人】(504159235)国立大学法人 熊本大学 (314)
【Fターム(参考)】
【公開日】平成25年6月10日(2013.6.10)
【国際特許分類】
【出願日】平成23年11月29日(2011.11.29)
【出願人】(000003207)トヨタ自動車株式会社 (59,920)
【出願人】(504159235)国立大学法人 熊本大学 (314)
【Fターム(参考)】
[ Back to top ]