
中枢神経系疾患および障害の治療に有効な新規化合物
抗痙攣作用を有する一連の新規化合物が記載される。このような医薬活性化合物はまた、不安、抑鬱、不眠、片頭痛、統合失調症、パーキンソン病、痙性、アルツハイマー病や双極性障害など、その他の中枢神経系(CNS)疾患や障害の治療にも有用である可能性がある。さらに、このような化合物は鎮痛剤(例えば、慢性あるいは神経因性頭痛のための)として、脳卒中、慢性神経変性性疾患(アルツハイマー病およびハンチントン病など)の、そしてまたは脳や脊髄の損傷の治療において有用な神経保護薬として有用である可能性がある。さらに、このような化合物はまた癲癇重積状態の治療、そしてまたは化学的対応策としても有用である可能性がある。

【発明の詳細な説明】
【技術分野】
【0001】
関連出願
本出願は2008年3月19日に出願の米国特許仮出願番号 61/037,987 の「中枢神経系疾患および障害の治療に有効な新規化合物」の利益を主張し、当該出願の内容を引用することによりその全てがここに組み込まれることとする。
【0002】
発明の背景
1. 発明の技術分野
本発明は、実験動物の中枢神経系(CNS)に作用を示す新規化合物に関連する。さらに具体的には、本発明は、現存する中枢神経系(CNS)治療薬に比べて、同等あるいはより優れた生物学的作用(すなわち有効性)を発揮しながら、毒性学的安全性の増大/改善(すなわち毒性の低減)、代謝安定性の増大/改善、半減期の延長および/または優れた副作用プロファイルと抗痙攣作用を示す新規化合物に関連する。
【背景技術】
【0003】
2. 関連技術
数多くの病理学的症状(例えば、癲癇、脳卒中、双極性感情障害、片頭痛、不安、抑鬱、不眠、統合失調症、慢性または神経因性疼痛、痙性、脊椎損傷および慢性神経変性障害など)および疾患(例えば、パーキンソン病、ハンチントン病およびアルツハイマー病など)は、中枢神経系(CNS)の正常な機能の異常として特徴づけられる。これらの症状や疾患は、一般的にはCNSの正常な機能を調節する化合物や物質による薬理学的治療に対して反応する。そのような活性を有する化合物には、癲癇などのCNSの異常を治療するために本明細書に開示される本発明の化合物が含まれる。現在利用できる治療薬はしばしば良好なCNS活性を有するが、慢性毒性、重度の、および/または不快な副作用、短い薬理学的半減期のような不十分な薬物動態特性などその他の好ましくない特性を示すものが多い。例えば、CNS治療薬の短い半減期は、有害事象を発現させずに治療濃度を維持するためには、頻回の入院を必要とし、頻繁な投与スケジュールが必要となり治療コストが増加する可能性がある。さらに、必要とされる投与回数が増加すると患者の服薬コンプライアンスが低下する傾向がある。従って、現在利用できる治療薬に比べて、例えば半減期の延長、活性の増大(すなわち有効性の改善)および/または代謝安定性の向上(例えば、毒性代謝物の低減など)などの優れた特性を有し、CNS活性を調節できる新たな化合物の提供が望まれている。さらに、より幅広く多くの患者人口に本発明の有効な化合物の提供を可能にする、改善された、より簡単な/簡略化された、合成および化学的製造工程を開発することも可能である。
【0004】
関連技術は、米国特許番号第 5,463,125号 (Sandovalら、「抗痙攣作用を有するフェニルアルコールアミド類」(Phenyl alcohol amides having anticonvulsant activity))、WO9941229 (Carvajal Sandoval ら、「抗痙攣作用を有するハロゲン化フェニルアルコールアミド類(GABAB受容体のリガンド)」(Halogenated phenyl alcohol amides (ligands of GABAB receptor) having an anticonvulsant activity))、WO03091201(Carvajal Sandovalら、「抗痙攣作用を有するDL-ヒドロキシーアルキルーフェニルアミド類」(DL−Hydroxy−alkyl−phenylamides having anticonvulsive activity))、WO2005085182 (Meza Toledo、「抗痙攣作用を有するDL-ヒドロキシベンズアミド類」(DL−Hydroxybenzamides having anticonvulsive activity))および米国特許出願番号第20060287397号 (Meza Toledo、「抗痙攣作用を有するDL-ヒドロキシーアルキルーフェニルアミド類」(Dl−Hydroxy−alkyl−phenylamides having anticonvulsive activity))に記載されている。上述の先行技術および本発明の技術の重要な違いは、先行技術はγヒドロキシ酪酸(γヒドロキシ酪酸塩またはGHB)と構造上関連する化合物を含む、または関連するのに対し、本発明の化合物は、3-メチル酪酸アミドに構造上関連することである。
【発明の概要】
【課題を解決するための手段】
【0005】
発明の簡単な概要
本明細書では、抗痙攣作用を有する一連の新規アミド類を開示するが、その多くは、短い、多様に分岐/置換された脂肪族リンカーを介してフェニル基がアミド部分へ結合している。本発明のその他の化合物(以下に示す)は、アラニン (Z = CH3 以下)、バリン [Z = CH(CH3)2]、ロイシン [Z = CH2CH(CH3)2]、イソロイシン [Z = CH(CH3)CH2CH3]またはフェニルアラニン (Z = CH2C6H5)などの光学的に活性なアミノ酸(例えば、 DまたはL)の、あるいはグリシン(Z=H)またはタウリン [R2 = (CH2)2SO3H、 以下に示す] などの光学的に不活性なアミノ酸の誘導体であるアミド類である。そのような化合物は以下の式で例示される:
式Iを有するCNS活性化合物:
【0006】
【化1】
【0007】
式Iにおいて、Arは、任意に置換されたフェニル、任意に置換されたナフチル、任意に置換されたテトラヒドロナフチル、任意に置換されたインダンであって良く、あるいは任意に置換されたヘテロサイクリックアリルでも良く、ここで5つまでの置換基を含んでも良い。Ar上の各置換基は、アルキル、シクロアルキル、ハロゲン、アルコキシ、チオアルキル、スルホキシアルキル、 スルホニルアルキル、アルキレンジオキシ、ハロアルキル、ハロアルコキシ、OH、CH2OH、CONH2、CN、アセトキシ、N(アルキル)2、ベンジル、ベンジロキシ、α,α-ジメチルベンジル、 NO2、CHO、CH3CH(OH)、アセチル、OCH2COOHから成る群から独立して選ばれてもよく、またはそれらの組み合わせであっても良い。また、Ar には、Arの1個または2個の原子に結合する任意に置換された芳香環系が含まれても良く、そのような芳香環系は、フェニル、フェノキシ、ヘテロサイクリックアリルから選択されるか、またはその組み合わせである。上記のArおよび/またはその置換基は、5個までの置換基を含んで良く、各置換基は、アルキル、シクロアルキル、ハロゲン、アルコキシ、チオアルキル、スルホキシアルキル、 スルホニルアルキル、アルキレンジオキシ、ハロアルキル、ハロアルコキシ、OH、CH2OH、CONH2、CN、アセトキシ、N(アルキル)2、NO2、CHO、CH3CH(OH)、アセチル、およびOCH2COOHからなる群から独立に選択される基であって良い。さらに、Arおよび/または芳香環基は、Arおよび/または芳香環基と芳香環を構成する二官能置換基を含んで良く、該二官能置換基は、アルキル、シクロアルキル、メチレンジオキシ、エチレンジオキシまたは他のアルキレンジオキシの任意の置換基またはそれらの組み合わせでも良い。
【0008】
式Iにおいて、R1 および R2は各々独立して、H、長鎖または短鎖の置換された、あるいは置換されていないアルキル、置換された、あるいは置換されていないシクロアルキル、CW2フェニル、のうちの少なくとも1つであって良い。各Wは独立してH、メチルおよびエチルからなる群から選択される基である(Wが共にエチルにはならない限り)。5個までの置換基がフェニル基に含まれて良く、各置換基は、ハロゲン、アルコキシ、チオアルキル、スルホキシアルキル、スルホニルアルキル、ハロアルキル、ハロアルコキシ、CONH2、CN、アセトキシ、N(アルキル)2、NO2 およびアセチルからなる群から独立して選択される。
別の実施態様では、R1は H であり、R2 は(CH2)2SO3HまたはCHZCOOHであり、Zは、H、 CH3、CH(CH3)2、CH2C6H5、CH2CH(CH3)2および CH(CH3)CH2CH3からなる群から独立して選択される。任意に、R1 および R2が共にシクロアルキルである。
式Iにおいて、R3は、ヒドロキシ、置換されたあるいは置換されていないアルキル、置換されたあるいは置換されていないシクロアルキルのいずれかであるか、あるいはR4も共にシクロアルキルである。任意に、R3 が OHの場合には R4 はエチルではない。
【0009】
式Iにおいて、R4は、置換されたあるいは置換されていないアルキル、置換されたあるいは置換されていないシクロアルキルのうちの一つであるか、またはR3も共にシクロアルキルである。
【0010】
式Iにおいて、Xは無置換、置換されたあるいは置換されていないアルキレン、メチレン、ケトン、CHOH、酸素、NR1、硫黄、スルホンまたはスルホキシドのうちの1つである。
式IIを有するCNS活性化合物:
【0011】
【化2】
【0012】
式IIにおいて、R1 および R2は各々独立して、H、長鎖または短鎖の置換されたあるいは置換されていないアルキル、置換されたあるいは置換されていないシクロアルキル、CW2フェニルのうちの少なくとも一つ、またはそれらの組み合わせである。Wは独立してH、メチルおよびエチルから選択されるが、Wが共にエチルであることはできない。フェニル基またはシクロアルキル基には5個までの置換基が含まれて良く、各置換基は、ハロゲン、アルコキシ、チオアルキル、スルホキシアルキル、スルホニルアルキル、ハロアルキル、ハロアルコキシ、CONH2、CN、アセトキシ、N(アルキル)2、NO2 およびアセチルからなる群から独立して選択される。任意に、R1は H およびR2 は(CH2)2SO3HまたはCHZCOOHを含み、ZはH、CH3、CH(CH3)2、CH2C6H5、CH2CH(CH3)2および CH(CH3)CH2CH3からなる群のうちの1つである。また別の選択として、R1 およびR2は共にシクロアルキルである。
【0013】
R4および R5は、各々独立して任意に置換されたフェニルまたは任意に置換されたヘテロサイクリックアリルのいずれかであり、ここで5個までの置換基があっても良く、各置換基はハロゲン、アルコキシ、チオアルキル、スルホキシアルキル、 スルホニルアルキル、ハロアルキル、ハロアルコキシ、CH2OH、CONH2、CN、アセトキシ、N(アルキル)2、NO2、アセチルおよび OCH2COOHからなる群から独立して選択される。
【0014】
Yは、無置換、置換された、あるいは置換されていないメチレン、あるいは他の置換されたあるいは置換されていないアルキレンのいずれかである。
実施態様の一つにおいては、本発明は以下の式のうちの一つを有するCNS活性化合物を含む:
【0015】
【化3】
【0016】
実施態様の一つにおいては、R1 および R2は各々独立してH、長鎖または短鎖の置換されたあるいは置換されていないアルキル、置換されたあるいは置換されていないシクロアルキル、CW2フェニルのうちのの少なくとも一つであるか、またはそれらの組み合わせである。WはH、メチルおよびエチルからなる群から独立して選択されるが、Wが共にエチルであることはできない。フェニル基またはシクロアルキル基には5個までの置換基が含まれて良く、各置換基はハロゲン、アルコキシ、チオアルキル、スルホキシアルキル、スルホニルアルキル、ハロアルキル、ハロアルコキシ、CONH2、CN、アセトキシ、N(アルキル)2、NO2 およびアセチルからなる群からから独立して選択される。任意にR1が H でありR2 が(CH2)2SO3HまたはCHZCOOHであって、ここで、Zは、H、CH3、CH(CH3)2、CH2C6H5、CH2CH(CH3)2および CH(CH3)CH2CH3 からなる群のうちの一つである。また別の選択としては、R1 およびR2が共にシクロアルキルである。
【0017】
実施態様の一つにおいては、R1 は H であり、R2 は(CH2)2SO3Hまたは CHZCOOHであり、ここでZはH、CH3、CH(CH3)2、CH2C6H5、CH2CH(CH3)2および CH(CH3)CH2CH3 からなる群のうちの一つである。任意に、R1 および R2は共にシクロアルキルである。
【0018】
実施態様の一つにおいては、R3は、ヒドロキシ、置換されたあるいは置換されていないアルキル、置換されたあるいは置換されていないシクロアルキルのうちのいずれかであるか、R4 も共にシクロアルキルである。任意に、R3が OHの場合には R4 はエチルではない。
【0019】
実施態様の一つにおいては、R4は、置換されたあるいは置換されていないアルキル、置換されたあるいは置換されていないシクロアルキルのうちの一つであるか、またはR3 も共にシクロアルキルである。
【0020】
実施態様の一つにおいては、R5 は H、Cl、F、CF3、CN、C1-C5 アルキル、 C1-C5 アルコキシ、OCF3、CONR1R2、ハロゲン置換アルキル、ハロゲン置換アルコキシのうちの一つである。
【0021】
実施態様の一つにおいては、Xは無置換、置換されたあるいは置換されていないアルキレン、メチレン、ケトン、CHOH、酸素、NR1、硫黄、スルホンまたはスルホキシドのうちの一つである。
【0022】
実施態様の一つにおいては、nは0〜5、より好ましくは1〜3であり最も好ましくは1〜2であって良い。
実施態様の一つにおいては、式1〜8のいずれも以下の特長を有しても良い:R1 は H、CH3、(CH2)2SO3Hまたは CHZCOOHのうちの一つであり、ここでZは、H、CH3、CH(CH3)2、CH2C6H5、CH2CH(CH3)2および CH(CH3)CH2CH3、これらの組み合わせまたはこれらの誘導体からなる群のうちの一つであり;R2、R3およびR4 は、独立してH、CH3、置換または未置換のアルキル、OH、OCH3、置換または未置換のアルコキシ、それらの組み合わせまたはそれらの誘導体のうちの一つであり;R5は、H、Cl、F、CF3、CN、C1-C5 置換または未置換のアルキル、C1-C5 置換または未置換のアルコキシ、OCF3CONR1R2、またはこれらの組み合わせあるいはこれらの誘導体のうちの一つであり;nは1〜5、好ましくは1〜3であり;および X は、O、NR1、無置換、C=O、S、SO2、これらの組み合わせまたはこれらの誘導体のうちの一つである。
【0023】
本発明のこれらおよびその他の実施態様や特長は、以下の説明および付加された特許請求の範囲からさらに明らかとなるかあるいは、以下に説明する本発明の実施例によって理解され得る。
【図面の簡単な説明】
【0024】
【図1】図1は本発明の(例えば)哺乳類などの中枢神経系(CNS)に薬理学的活性な新規化合物の化学構造を示し、本発明の実施態様を例示するものである。
【図2】図2は本発明に記載される化合物の相対的な生物学的な活性を示すもので、好ましい(第2類、ED50 < 300 mg/kg)および最も好ましい(第1類、ED50 < 100 mg/kg)化合物について具体的に記載する。
【図3A】図3Aは、さらに本発明の最も好ましい化合物類のその他の化合物の化学構造を示す。
【図3B】図3Bは、さらに本発明の最も好ましい化合物類のその他の化合物の化学構造を示す。
【図4A】多様な化合物および主要な中間体の実施例を示す。
【図4B】多様な化合物および主要な中間体の実施例を示す。
【図4C】多様な化合物および主要な中間体の実施例を示す。
【図4D】多様な化合物および主要な中間体の実施例を示す。
【図4E】多様な化合物および主要な中間体の実施例を示す。
【図4F】多様な化合物および主要な中間体の実施例を示す。
【図4G】多様な化合物および主要な中間体の実施例を示す。
【図4H】多様な化合物および主要な中間体の実施例を示す。
【図4I】多様な化合物および主要な中間体の実施例を示す。
【図4J】多様な化合物および主要な中間体の実施例を示す。
【図4K】多様な化合物および主要な中間体の実施例を示す。
【図4L】多様な化合物および主要な中間体の実施例を示す。
【図4M】多様な化合物および主要な中間体の実施例を示す。
【図4N】多様な化合物および主要な中間体の実施例を示す。
【図4O】多様な化合物および主要な中間体の実施例を示す。
【発明を実施するための形態】
【0025】
図示による実施態様の説明
1. 概観
本発明者は、本発明の化合物および薬理学活性のある化合物類似体および同類のあるものは、in vivoの投与によってCNS活性の調節に有効であることを発見した。すなわち、これらの薬剤は神経系の活性の全てを完全に抑制することなく、中枢神経系の神経伝達の抑制の増強または興奮の抑制によってCNS活性を調節する。従って、本発明に準じて、この薬剤を投与される患者は、例えば、癲癇発作の緩和(無麻酔で)、筋緊張の緩和(麻痺を起こさないで)、静穏化作用(鎮静作用なしで)または痙性のような歩行系症状の改善(虚弱化、あるいは弛緩状態にすることなく)のような状況下において、過度な鎮静、麻酔、あるいは麻痺をもたらされない。
【0026】
痙攣(癲癇発作)、痙性、双極性感情障害のような気分障害、頭痛(慢性、群発性、片性)、静止不能症候群、神経因性疼痛、運動障害などで示される多くの病変は、CNS活性の調節によって緩和できる少なくとも一つの症状を有する。従って、このような病変に悩む個人は、本発明に準じて、主要な活性成分として、本発明の化合物、あるいはその構造関連化合物類または同類を含む医薬製剤処方または組成の投与を受ける治療の候補者である。
【0027】
2. 中枢神経系(CNS)活性の調節によって改善される病変の好例
痙攣:癲癇は、多くの原因による一般的な疾患であり、臨床上の管理が非常に困難な場合があり、発作を管理するために何年もの治療を必要とすることが多い。研究者達は、現時点では、患者の多くにとって有効な癲癇の治療法は存在しないと述べている。臨床試験において、患者が同様な種類の癲癇を有し、薬剤が同様な作用機序を有していても、他の薬剤よりもある薬剤に良好に反応する患者があることが示された。副作用の頻度及び重症度にも大きな差が存在する。このように癲癇の治療には、癲癇の治癒が可能となるか、あるいは、広範な活性を持った強力で安全な新薬が発見および開発されるまで、異なる作用機序と異なる副作用を持った複数の薬剤による治療が必要であろう。Dichter ら、 Drug Therapy 334:1583 (1996)。
【0028】
かなり予測的で実験的に使用可能な癲癇の動物モデルによって、数多くの臨床上有効な抗痙攣薬が調製され開発されてきている。例えば、Cereghino ら、「抗癲癇薬」(ANTIEPILEPTIC DRUGS), 第4版、1-11 ページ (Rave Press 1995)の「序章」を参照、「多くの患者においては癲癇は現存の抗癲癇薬で管理が可能であるが、患者の25〜30パーセントは最適な治療を実施しても癲癇を抑えることができず、また患者の多くは容認できない副作用を経験している。」Dichter ら(1996) 同上。
【0029】
このように多くの抗痙攣薬の臨床使用は、やっかいな日中の鎮静、筋肉の虚弱化、耐性、歯肉増殖症、致命的な可能性のある悪液質および肝毒性などの重大な副作用が悩みとなっている。これらの副作用の多くは、特に小児癲癇の臨床管理(治療)において問題となっている。
【0030】
本発明は、癲癇のような痙攣性疾患を治療するために使用できる。すなわち、本発明の組成物、医薬製剤処方および組成は「抗痙攣作用」を示すが、それは動物癲癇モデルにおける痙攣の重症度、発作回数あるいは期間の軽減によって実証されている。痙攣の緩和には、患者における痙攣発作の重症度、回数や期間の軽減が含まれる。従って新規組成物および医薬製剤処方および組成は、これらに限定されるものではないが、全身性強直・間代性発作、欠神発作、ミオクローヌス発作、単純部分発作、複雑部分発作、二次性全般性部分発作、癲癇重積状態、および頭部の外傷や外科手術による外傷性癲癇発作などの症状の治療に有効である。
痙性:痙性は、伸張反射の過剰興奮による過剰な腱反射を伴う緊張性反射(筋緊張)の増強を特長とする障害である。Lance、シンポジウム抄録、「痙性-運動制御障害」(SPASTICITY--DISORDERED MOTOR CONTROL)、Feldman ら(Eds.)(1980). 痙性に付随する主要な疾患及び症状には、多発性硬化症、脳性麻痺、脳卒中、脊髄の外傷または損傷および頭部外傷がある。痙性に起こる症状には疼痛を伴う屈筋および伸筋痙攣、亢進したあるいは過剰の深部腱反射、クローヌス、筋肉の虚弱化、疲労、器用さの損失、全般的な運動機能の様々な程度の損失、麻痺及び睡眠障害などが含まれる。
【0031】
痙性にみられる病理学的症状は、一般的にみられる局部的な外部からの傷害によって特定の筋肉に起こる、よくある筋肉の急性の痛み、緊張および捻挫、すなわち、CNS外部または末梢における症状、とは生理学的レベルで基本的に異なる。これらの病理学的症状はまた血管痙縮、膀胱痙縮および気管支痙縮のような比較的一般的に起こる不随意の平滑筋の痙縮とも異なる。このような非痙縮性(non-spastic, CNS性でない)、末梢性のまたは局所性の症状は通常、いわゆる「鎮痙薬」または「痙縮緩解薬」によって治療されるが、これらの薬剤は一般的に痙性の治療には有効ではない。GOODMAN AND GILMAN'S「治療薬の薬理学的基礎」(THE PHARMACOLOGICAL BASIS OF THERAPEUTICS)、第8版、「パーキンソン病、痙性および急性痙攣の治療薬(Drugs for Parkinson's Disease, Spasticity and Acute Muscle Spasms)」Cedarbaum および Schleifer。(以降GOODMAN AND GILMAN'Sと掲載)、ページ 463-484 ページ(Pergamon Press 1990年)。
【0032】
本発明によって用いられる組成物及び医薬製剤処方および組成は、筋緊張の中枢を介した軽減に有効であり、痙性による症状または副作用の一つ以上についての急性または慢性の緩和に有効である。これに関連して「痙性」は、これらに限定されるものではないが、疼痛を伴う屈筋または伸筋の痙攣、亢進されたあるいは過剰の深部腱反射、過反射、器用さの損失、筋肉の虚弱化、過剰な腱反射およびクローヌスなどの症状に表れる骨格筋の増強した緊張を意味する。本明細書における「抗痙性剤」とは、以下の痙性による症状または副作用の少なくとも一つを緩和することで示される痙性の症候の治療に有効な組成を意味する:それらの症状とは、疼痛を伴う屈筋または伸筋の痙攣、亢進したあるいは過剰の深部腱反射、反射亢進、器用さの喪失、筋肉の虚弱化、過剰な腱反射およびクローヌス、であり、あるいは治療はこれらの症状の発現や副作用の頻度の低減を含む。
【0033】
従って、本明細書の痙性の「緩和」とは、これらに限定されないが、疼痛を伴う屈筋または伸筋の痙攣、亢進されたあるいは過剰の深部腱反射、反射亢進、器用さの損失、筋肉の虚弱化、過剰な腱反射およびクローヌスなどの痙性の症状の一つ以上の軽減あるいはこれらの症状または副作用の回数の減少を意味する。
情緒障害:情緒障害には、例えば、躁病、総合失調感情障害、脳外傷性攻撃性、心的外傷後ストレス障害、双極性感情障害、パニック症状および行動調整不良症候群のような抑鬱症から不機嫌躁病まで含まれる。「J.感情障害」(J. Affective Disorders) 8:243-250 (1985)、 Emrich ら、 および 「感情性障害における抗痙攣剤(ANTICONVULSANTS IN AFFECTIVE DISORDERS)」 14-32 ページ、Bernasconi ら (Excerpta Medica 1984)。本発明による新規化合物および医薬製剤処方および組成は、これらの疾患、障害及び症状の治療に有効であり、またこの治療薬分類における現存の治療薬に比較して改善された副作用プロフィールを示す。
【0034】
神経障害性疼痛症候群:この「神経障害性疼痛」に分類される症状は、脳卒中、外傷、多発性硬化症および糖尿病のような脳または脊髄の障害で苦しむ多くの患者に影響を与えている。「痛みと中枢神経系疾患(PAIN AND CENTRAL NERVOUS SYSTEM DISEASE)」、 (Raven 1991)、 Casey。多様な疼痛症状の治療のための抗痙攣薬の使用は、広く文献で報告されている。J. Clin. Neuropharmacol. 7、Swendlow:51-82 (1984). したがって、本発明の新規化合物および医薬製剤処方および組成は、神経障害性疼痛の緩和に同じように適用することが可能である。
頭痛:片頭痛 [Hering と Kuritzky, Cephalagia 12: 81-84 (1992)]、群発性頭痛 [Hering とKuritzky, loc. cit. 9:195-198 (1989)]および慢性頭痛といった頭痛 [Mathew とSabiha, Headache 31: 71-74 (1991)] は抗痙攣剤によって治療されてきた。従って本発明の組成物および医薬製剤処方は、これらのタイプの頭痛の症状の緩和に、現存の治療にみられる有害な副作用なしで使用することが可能である。
【0035】
静止不能症候群:身体的な(非精神性の)動揺だけでなく不随意の四肢の動きを特長とする「静止不能症候群」とは、体性の(非精神性の)静止不能症状が特徴であり、感情からは独立したもので、従って、精神的不穏症とは本質的に区別される。[Sachev ら、 Austral. New Zealand J. Psychiatry30:38-53 (1996)を参照]。
【0036】
静止不能症候群は多くの症状を含め、多くの器質性および非器質性精神疾患との関連性が見られる。例えば、薬剤起因性錐体外路症状のような薬剤に起因する静止不良症状(遅発性、慢性および離脱性アカシジア)は、神経遮断薬治療の最も一般的な副作用の一つである。静止不能症候群類には、頭部や脊髄外傷、および脊髄の病巣に関連する可能性のある病変である、いわゆる「下肢静止不能症候群」および「睡眠時周期性下肢運動障害」が含まれる。特発性下肢静止不能症候群は、多様な臨床症状発現を伴う常染色体の優性遺伝から起こる。O'Keefe, Arch. Intern. Med. 156: 243-248 (1996); Danek ら、 「神経学的障害:経過と治療(NEUROLOGICAL DISORDERS: COURSE AND TREATMENT)」、819-823 ページ (Academic Press 1996); Mellick や Mellick、Neurology 45(補足): 285-286 (1995)を参照。本発明は、最低限の副作用の静止不能症候群の有効な治療を提供する。
【0037】
運動障害:パーキンソン病、ハンチントン舞踏病、アルツハイマー病、遅発性ジスキネディア、ステッフマン症候群などの運動障害を軽減する多様な薬剤が知られている。Lloyd および Morselli、「精神薬理学:三代目の進歩(PSYCHOPHARMACOLOGY: THE THIRD GENERATION OF PROGRESS )」(Raven Press 1987)。本発明の治療は、運動障害の一つ以上の症状を緩和する。
【0038】
本発明の化合物は、不安軽減薬(抗不安薬)としても有効である可能性がある。
「神経障害または疾患」とは、これらに限定されないが、癲癇、不安症、多発性硬化症、脳卒中、頭部外傷、脊髄傷害およびパーキンソン病およびハンチントン病、アルツハイマー病および筋萎縮性側索硬化症のような慢性神経変性疾患を含む神経系の障害および疾患を意味する。さらに「神経障害または疾患とは、抗痙性剤または抗痙攣剤による治療が適用され、有効性を示し、推奨されるおよび/または処方される疾患または症状を意味する。
【0039】
「神経変性疾患」とは、これらに限定されないが、ハンチントン病、パーキンソン病、アルツハイマー病および筋萎縮性側索硬化症(ALS)のような疾患を意味する。
「抗痙攣剤」とは全身性強直・間代発作、欠神発作、単純部分発作、複雑部分発作、二次性全般性部分発作、癲癇重積状態および頭部外傷や外科手術後の外傷起因性痙攣などの症状に発症し、観察され、見られる痙攣の重症度、回数や期間を軽減することのできる化合物を意味する。
【0040】
「抗痙攣作用」とは全身性強直・間代発作、欠神発作、単純部分発作、複雑部分発作、二次性全般性部分発作、痙攣重積状態および頭部外傷や外科手術後の外傷起因性痙攣のような症状に起こる、観察される、みられる痙攣発作の重症度および回数や期間を軽減する効果を意味する。
【0041】
「治療用量」とは、患者において、疾患や症状の一つ以上をある程度緩和する化合物の量を意味する。さらに、「治療用量」は、疾患または症状に付随する生理学的または生物化学的パラメタを部分的にまたは完全に正常値に戻す量を意味する。一般的に、患者の年齢、体格および疾患によって0.1-15-20-30 mg/kg体重の量である。投薬回数は1日1回から4回であっても良い。
【0042】
「医薬組成分」とは、本発明の化合物を溶解させるまたは化合物の投与を促進するために添加する医薬品として許容できる担体中に含まれる治療有効量の本発明の化合物を意味する。医薬品として許容できる担体の例には、水、生理食塩水、生理学的緩衝食塩水が含まれる。これらの医薬品組成物は、適切な投与量で提供される。これらの組成物は一般に特定の疾患の治療への使用をFDAまたは米国外の同等の機関によって承認されたものである。
【0043】
本発明の上記の化合物のあるものは一つ以上の螺旋対象性の立体中心を有することが知られている。そのような化合物は、ラセミ(あるいはジアステレオ異性の)混合体、R およびSエナンチオマー混合体(あるいはジアステレオマー)または純粋なエナンチオマー(RまたはS)(あるいはジアステレオマー)として、好ましい生物学的活性を示す場合がある。 ある純粋なエナンチオマーが好ましい生物学的活性を示す場合、この好ましいエナンチオマーはユートマーと呼ばれ、より好ましくない、生物学的活性がより低いエナンチオマーはジストマーと呼ばれる。
【0044】
医薬製剤処方および組成分の調製方法と投与方法:本明細書に示されるように、本発明の有用な医薬製剤処方および組成は神経系障害または疾患の治療に使用することができる。これらの製剤は、一般に人の患者の治療に使用されるが、その他の霊長類、飼育動物、豚、牛および家禽などの家畜動物や馬、犬や猫などのなどスポーツ動物やペットなどの他の脊椎動物の同様な、あるいは同じ疾患の治療に使用することも可能である。
【0045】
本発明は、また上記の二つ以上の活性化合物の組み合わせを含む医薬製剤処方や組成分にも向けられる。本発明の化合物は、既知の方法に従って医薬製剤として有効な組成を調製(処方)することが可能であり、それによって活性成分は医薬品として許容できる一つのあるいは複数の担体と組み合わされる。例えば、Gennaro (Ed.)、「レミントンの薬理科学(REMINGTON'S PHARMACEUTICAL SCIENCES)」、第18版 (Mack Publishing Co., Easton, PA, 1990) および「グッドマンとギルマンの治療薬の薬理学的基礎(GOODMAN AND GILMAN'S THE PHARMACOLOGICAL BASIS OF THERAPEUTICS)」を参照のこと。化合物および/または組成は、投与される患者が認容できる場合には「医薬品として許容できる担体」中に含まれるべきであるとされる。滅菌リン酸緩衝生理食塩水は医薬品として許容できる担体の1例である。その他の適切な担体(例えば、生理食塩水およびリンガー液)は、当業者には周知である(以下を参照)。
【0046】
医薬品として許容できる担体には、投与して患者が認容できる適切な賦形剤および/または補助剤が含まれる。先行技術にて既知の医薬品として許容な担体には、カルボン酸カルシウム、リン酸カルシウム、硫酸カルシウム、ショ糖、デキシトロース、ラクトース、フルクトース、キシリトール、ソルビトール、デンプン、デンプンペースト、セルロース誘導体、ゼラチン、ポリビニルピロリドン、塩化ナトリウム、デキストリン、ステアリン酸、ステアリン酸マグネシウム、ステアリン酸カルシウム、植物オイル、ポリエチレングリコール、滅菌リン酸緩衝生理食塩水、生理食塩水、リンガー液およびこれらの混合物が含まれるがこれらに限定されるものではない。例えば、「レミントンの薬理学的科学(REMINGTON'S PHARMACEUTICAL SCIENCES)」、同上、を参照。
【0047】
米国食品医薬品局(FDA)によって市販が承認された医薬品として許容できる有機酸(アミノ酸など)の塩には、ナトリウム、カリウム、リチウム、亜鉛、アルミニウム、カルシウムおよびマグネシウムの塩が含まれる。「レミントンの薬理学的科学(REMINGTON'S PHARMACEUTICAL SCIENCES)」、 第18版、1445ページ (Mack Publishing Co. 1990) を参照。
【0048】
本発明の化合物およびその医薬品製剤組成は先行技術で調剤できる。例えば、本発明の化合物は、医薬品として許容できる担体と組み合わせて望ましい剤形処理をして良い。本発明の医薬製剤組成は、本発明の医薬品製剤組成および医薬品として許容できる担体を共に含む、従来の混合、溶解、顆粒化、糖衣錠作成、磨砕、乳化、カプセル封入、封入または凍結の工程のような自明の方法で生成または製造してもよい。
【0049】
一般的には、本明細書に記載される上記の化合物の投与量、処方および組成は、患者の年齢、体重、身長、性別、健康状況および既往歴などの因子によって異なる。治療を目的として、本発明の化合物および医薬品として許容できる担体は、治療の必要な患者に治療有効量が投与される。活性成分および担体の組み合わせ(処方または組成)は、投与量が生理学的に有意な量であれば、「治療有効量」を投与するとされる、。医薬製剤組成は、投与された患者に、結果として生理学的に検出可能な変化がある場合には、生理学的に有意である。例えば、このような文脈からすると、抗痙攣活性組成を投与することによって癲癇発作および/または痙攣のような癲癇の症状の一つ以上が緩和される場合は、本組成は生理学的に有意である。さらに、用量およびおそらく投与回数も各患者の年齢、体重および反応に応じて異なってくる。上記に考察されたような同等の計画は獣医学的薬剤においても用いることが可能である。
【0050】
本発明の化合物は、例えば、腸溶性錠剤、カプレット、ジェルカップ、散剤、あるいはカプセル剤などの固形剤形またはシロップやエリキシル剤などの液状剤形によって経口的に投与され得る。個体経口投与単位剤形は、錠剤またはカプセル中に1日最大2回の投与により、1回に1〜2個の服用ですむような活性成分量が含まれることが好ましい。液状製剤もまた、1回の投与用量あたり小さじ1〜2杯となる量の活性成分が含まれるようにすることが可能である。さらに、相当する低用量の小児用の咀嚼可能および液体経口投与剤形を調製し投与することも可能である。これらの化合物は、経口投与用に、滴剤(「濃縮」調剤の滴下用から)を食品や飲料へ添加することも可能である。さらに、本発明の上記化合物を経口送達と吸収を促進させるためにチューイングガムに入れた剤形で調剤することも可能である。本発明の処方や組成に使用される各化合物の適切な用量は、当業者には前述の説明で明らかとなり得る。
【0051】
あるいは、本発明の上記の化合物は、注射または経皮投与または、例えば、経鼻、舌下、経頬、経膣投与などの経粘膜投与、あるいは座薬などの経腸投与などのその他の全身投与経路によって投与されることも可能である。その他の投与経路(例えば、獣医学的適用に有用な)には、腸内や非経口投与があり、筋肉内、皮下および/または脊髄内投与ばかりでなくクモ膜下腔内、脳室内、静脈内、腹腔内、鼻腔内または眼内注射などが含まれる。しかしながら、経口投与が非常に便利であるために好ましい。
【0052】
本発明はこのように、経口、非経口、経皮、経粘膜、鼻腔内、舌下、経頬または経直腸投与に適する多様な化合物を意図している。本発明の化合物は他の薬理学的な活性成分と組み合わせて、さらに新規な医薬製剤組成を調製し得ることも理解されている。
【0053】
治療に関連の、治療的に価値のある活性の実証:上述のように、症状の緩和のためのある医薬製剤処方や組成の適合性および治療有効性は、例えば米国特許番号第6,589,994、6,617,358および7,265,155号に記載され、本明細書にその全体が組み込まれるが、動物モデルの使用や、試験およびスクリーニング法を使用して実証することが可能である。
【0054】
上記の本発明の化合物の治療有効性は、一般的な毒性の無さと併せて、本発明の化合物を上述の症状の治療のための理想的な薬剤とする。このような背景に基づいて、本発明は以下の実施例を参照することで当業者にはさらに容易に理解されると思われるが、それらの図示された実施例は図解の目的で提供されるもので、本発明の範囲を限定する意図はない。
【0055】
実施例
実施例1 化合物A[3−(4−クロロフェニル)−3−メチルブチルアミド]の調製。
【0056】
【化4】
【0057】
3−(4−クロロフェニル)−3−メチル酪酸(6.1g、41.9mmol) 溶液をCH2Cl2(100mL)およびDMF(0.2mL)溶液中で、塩化オキサリル(5.2mL、7.45mmol)を使って0℃窒素静圧下で処理した。反応液を窒素下で1晩室温で撹拌した。減圧下で過剰なジクロロメタンを除去した。入手した残渣をトルエン(50mL)で共沸蒸留した。
【0058】
アンモニア(ガス)を泡化させて無水THF(100mL)中の前記酸塩化物[3−(4−クロロフェニル)−3メチルブチリルクロリド]溶液に5℃で15分間通した。反応混合物を静圧窒素下で1晩室温で撹拌した。
【0059】
白色沈殿物(塩化アンモニウム)を濾過しTHF(100mL)で洗浄した。濾過液および洗浄液を合わせて減圧下で蒸発させた。その結果入手した白色固体をエチルアセテートに再溶解させた(300mL)。エチルアセテート層をH2O、1.0MのHCL、炭酸水素ナトリウム飽和溶液および塩水で洗浄した。次にエチルアセテート溶液を硫酸マグネシウム上で乾燥させ、濾過し、減圧下で蒸発させた。その結果得られた白色固体を冷却したジエチルエーテルおよびヘキサン(50:50)溶液を加えて粉末にした。この結果4.22gの白色の剥片[3−(4−クロロフェニル)−3−メチルブチルアミド]が得られた(収率69%)。GC/MS分析によりこの物質は100%純粋であった。1H NMR 分光法によって生成物の構造と一致するシグナルが得られ、98%以上の純度が示された。
【0060】
実施例2 化合物B[3−(4−クロロフェニル)−3,N−ジメチルブチルアミド]の調製。
【0061】
【化5】
【0062】
3−(4−クロロフェニル)−3−メチル酪酸(5.95g、28mmol) をCH2Cl2(100mL)およびDMF(0.2mL)溶液中で塩化オキサリル(5.2mL、7.45mmol)によって0℃窒素静圧下で処理した。反応液を窒素下で1晩室温で撹拌した。減圧下で過剰なジクロロメタンを除去した。入手した残渣をトルエン(50mL)を使って共沸蒸留した。
【0063】
残渣を150mLの乾燥THFに溶解させ、メチルアミン溶液(THFの2.0M溶液、45mL、84mmol)で5℃で処理した。反応混合物を静圧窒素下で1晩室温で撹拌した。
白色沈殿物を濾過しTHF(100mL)で洗浄した。濾過物および洗浄液を合わせて減圧下で蒸発させた。その結果入手した白色固体をジエチルエーテル中で再溶解させた(300mL)。エーテル層をH2O、1.0M HCL、炭酸水素ナトリウム飽和溶液および塩水で洗浄した。次にエーテル溶液を硫酸マグネシウム上で乾燥させ、濾過し、減圧下で蒸発させた。その結果得られた白色固体を冷却したジエチルエーテルおよびヘキサン(50:50)溶液を加えて粉末とした。この結果5.46gの白色の剥片[3−(4−クロロフェニル)−3,N−ジメチルブチルアミド]が得られた(収率86%)。 GC/MS分析によりこの物質は100%純粋であった。 1H NMR 分光法によって生成物の構造と一致するシグナルが得られ、98%以上の純度が示された。
【0064】
実施例3 化合物C [(R)-3-フェニルブチルアミド]の調製。
【0065】
【化6】
【0066】
(R)-3−フェニル酪酸(4g、24.36mmol)の CH2Cl2(75mL)およびDMF(0.1mL)溶液を塩化オキサリル(3.0mL、34.0mmol)で0℃、窒素静圧下で処理した。反応液を窒素下で室温にて1晩撹拌した。減圧下で過剰なジクロロメタンを除去した。入手した残渣をトルエン(50mL)を使って共沸蒸留した。
【0067】
アンモニア(ガス)を泡化させて酸塩化物[(R)-3−フェニルブチリルクロライド] の無水THF(100mL)溶液に5℃で15分間通した。反応混合物を静圧窒素下で1晩室温で撹拌した。
【0068】
白色沈殿物(塩化アンモニウム)を濾過しTHF (100mL)で洗浄した。濾過物および洗浄液を合わせて減圧下で蒸発させた。その結果入手した白色固体をエチルアセテートに再溶解させた(300mL)。エチルアセテート層をH2O、1.0M HCL、炭酸水素ナトリウム飽和溶液および塩水で洗浄した。次にエチルアセテート溶液を硫酸マグネシウム上で乾燥させ、濾過し、減圧下で蒸発させた。粗物質をBiostage SP4システム(カラムSi 40+M 0344−1,95:5, CH2Cl2: MeOH)を使用して精製した。 その結果得られた灰色がかった白色の固体を冷却したジエチルエーテルおよびヘキサン(50:50)溶液で共沸蒸留した。2.9gの白色固体[(R)-3-フェニルブチルアミド]を入手した(収率73%)。GC/MS分析によりこの物質は100%純粋であった。 1H NMR 分光法によって生成物の構造と一致するシグナルが得られ、98%以上の純度が示された。
【0069】
実施例4 化合物D[3−(3−フルオロフェニル)ブチルアミド]の調製。
【0070】
【化7】
【0071】
500mL容量の三つ口丸底フラスコを使って、水素化ナトリウム(オイル中60%、1.1eq.80mmol、3.20g)をN,N-ジメチルホルムアミド(DMF、100mL)に懸濁させ、、窒素下でトリエチルホスホノアセテートのDMF(50 mL)溶液(1.2eq.87mmol、19.50g)を滴下して処理した。添加後に反応混合液を水素化ナトリウムが目視で確認できなくなるまで水浴(100℃)を使って加熱した(30分間)。混合液を室温まで冷却し、次に3’−フルオロアセトフェノン(1.0eq.72.4mmol、10g)のDMF溶液(50mL)で処理した。反応混合液を室温で2時間撹拌し、1mLの分画を取り出し水中(〜2mL)で冷却した。ここへジエチルエーテル(〜2mL)を添加し、混合液を平衡化させた。GC/MS分析によって有機層を分析したところ、最初のベンゾフェノンは完全に消費されていた。その結果として、反応混合液に水を添加して冷却した。混合液を大きな丸底フラスコへ移し回転蒸発装置で溶媒のほとんどを除去した。混合液を冷却し[a]ジエチルエーテル(500mL)および水(250mL)を使用して分液ロートへ移した。混合液を平衡化させ水層を除去した。有機層をさらに3回水で洗浄した(3x250mL)。この溶液のGC/MS分析により単一の生成物が得られたことが示された(ホスホノアセテートの残留は無かった)。有機溶液を無水MgSO4上で乾燥させ、濾過および濃縮により18.09gの粗物質(水素化ナトリウムからのオイルを含む)を入手した。
【0072】
【化8】
【0073】
粗[3−(3−フルオロフェニル)−ブタ−2−エン酸エチルエステル]溶液(9g、0.015mmol)をPd/C(10%、450mg)でメタノール(75mL)中で処理した。反応混合液を45psiで1時間水素化した。反応混合物を炭素上のパラジウムを除去するためCeliteプラグを通過させた。減圧下で濾過液を濃縮した。この結果無色のオイル[3−(3−フルオロフェニル)酪酸エチルエステル]10.1gを入手した。GC/MS分析によってこの物質が94%純粋であることを確認した。本生成物を精製(鉱物油をの除去のための)しないで使用した。
【0074】
【化9】
【0075】
[3−(3−フルオロフェニル)−酪酸エチルエステル](10.1g、48mmol)のエタノール(50mL)粗溶液を10M NaOH溶液(50mL、857mmol)で処理した。反応混合液を一晩還流した。エチルアルコールを除去するために減圧下で反応混合液を乾燥させた。結果として入手した残渣を水150mLで再溶解させた。混合液を冷却し、ジエチルエーテル(200mL)および水(50mL)を使用して分液ロートへ移した。混合液を平衡化させ、エーテル層を除去した。HCL溶液(pH〜2)を使って、水層を酸性化しジエチルエーテル(300mL)で抽出した。次に有機層を硫酸マグネシウム上で乾燥させ、濾過し、減圧下で濃縮した。この結果、橙色の粘性オイルである3−(3−フルオロフェニル)酪酸8.1gを入手した(収率92.6%)。GC/MS分析により、この物質は100%純粋であった。
【0076】
【化10】
【0077】
3−(3−フルオロフェニル)酪酸(8.1g、44.46mmol)のCH2Cl2(100mL)とDMF(0.7mL)との溶液を塩化オキサリル(5.43mL、7.9mmol)で0℃窒素静圧下で処理した。反応液を窒素下で1晩室温で撹拌した。減圧下で過剰なジクロロメタンを除去した。入手した残渣をトルエン(70mL)を使って共沸蒸留した。
【0078】
アンモニア(ガス)を泡化によって酸塩化物[3−(3−フルオロフェニル)ブチリルクロライド]の無水THF(100mL)溶液に5℃で15分間通した。反応混合物を静圧窒素下で1晩室温で撹拌した。
【0079】
白色沈殿物(塩化アンモニウム)を濾過しTHF(200mL)で洗浄した。濾過液および洗浄液を合わせて減圧下で蒸発させた。その結果入手した白色固体をエチルアセテートに再溶解させた(350mL)。エチルアセテート層をH2O、1.0M HCL、炭酸水素ナトリウム飽和溶液および塩水で洗浄した。次にエチルアセテート溶液を硫酸マグネシウム上で乾燥させ、濾過し、減圧下で蒸発させた。その結果得られた白色固体を冷却したジエチルエーテルおよびヘキサン(50:50)溶液を用いて粉末にした。この結果、乳白色の粉末3−(3−フルオロフェニル)ブチルアミド 6.8gを入手した(収率84%)。GC/MS分析によりこの物質は100%純粋であった(RおよびSの鏡像異性体の混合物)。 1H−NMR 分光法によって生成物の構造と一致するシグナルが得られ98%以上の純度が示された。
【0080】
実施例5−14 化合物E−Nの調製
対応するアセトフェノン(以下の表1)から上述の実施例4の化合物Dの調製方法を使って化合物E−Nを調製した。さらに、化合物M(実施例13)および化合物N(実施例14)を対応するアミン、すなわち各々メチルアミンおよびジメチルアミン、から調製した。すべての最終生成物はGC/MS分析により100%純粋であった。1H−NMR 分光法によって各々の最終生成物の構造と一致するシグナルが得られ98%以上の純度が示された。
【0081】
【表1】
【0082】
実施例15 化合物O[3−(4−シアノフェノキシ)ブチルアミド]の調製。
【0083】
【化11】
【0084】
水酸化ナトリウム4g(0.1mol)を水(100mL)に溶かした溶液、および4−シアノフェノール11.9g(0.1mol)を還流させ15分間加熱した。βブチロラクトン(8.6g 0.1mol)を還流液へ15時間以上にわたって添加した。次に反応液を室温に冷却した。反応液をジエチルエーテル(200mL)および水(200mL)を使って分液ロートへ移した。混合液を平衡化し、エーテル層を除去した。HCL溶液(pH−2)を使って、水層を酸性化しエチルアセテート(300mL)で抽出した。エチルアセテート層を硫酸マグネシウム上で乾燥させ、濾過し、減圧下で濃縮し粗生成物10.78gを入手した。
【0085】
粗生成物をBiostage SP4システム(カラムSi 65i、9:1 CH2Cl2: MeOH)を使用して精製し、淡黄色の粘性オイル[3−(4−シアノフェノキシ)酪酸]9.87gを入手し、室温で放置し固化した。GC/MS分析によりこの物質は96%純粋であった。本化合物はさらに精製しないで使用した。
【0086】
【化12】
【0087】
3−(4−シアノフェノキシ)酪酸の粗溶液(10.8g、52.6 mmol)をCH2Cl2 (100mL)およびDMF(0.2mL)溶液中で塩化オキサリル(6mL、68.4mmol)によって0℃窒素静圧下で処理した。反応液を窒素下で1晩室温で撹拌した。減圧下で過剰なジクロロメタンを除去した。
【0088】
アンモニア(ガス)を泡化させて無水CH2Cl2(150mL)中の酸塩化物[3−(4−シアノフェノキシ)ブチリルクロライド]溶液に5℃で15分間通した。反応混合物を静圧窒素下で1晩室温で撹拌した。
【0089】
白色沈殿物(塩化アンモニウム)を濾過し、CH2Cl2(100mL)で洗浄した。濾過液および洗浄液を合わせて減圧下で蒸発させた。その結果入手した白色固体をエーテルに再溶解させた(250mL)。エーテル層をH2O、HCL 1.0M、炭酸水素ナトリウム飽和溶液および塩水で洗浄した。次にエーテル溶液を硫酸マグネシウム上で乾燥させ、濾過し、減圧下で蒸発させた。粗物質をBiostage SP4システム(カラムSi 40+M0344−1,95:5, CH2Cl2: MeOH) を使用して精製した。 この結果、灰色がかった白色の固体3−(4−シアノフェノキシ)ブチルアミド 2.987gを入手した(収率29%)。 GC/MS分析によりこの物質は97%純粋であった。 1H NMR 分光法によって製品の構造と一致するシグナルが得られ98%以上の純度が示された。
【0090】
実施例16 化合物P[1R,2R]-トランス−2−フェニルシクロプロパン−1−カルボキシアミド]の調製
【0091】
【化13】
【0092】
(1R,2R)−トランス-2−フェニルシクロプロパン−1−カルボン酸(2.1g、12.8 mmol) をCH2Cl2 (50mL)およびDMF(0.2mL)の溶液中で塩化オキサリル(1.5mL、16.7 mmol)で0℃窒素静圧下で処理した。反応液を窒素下で1晩室温で撹拌した。減圧下で過剰なジクロロメタンを除去した。
【0093】
アンモニア(ガス)を泡化させながら酸塩化物[(1R,2R)−トランス-2−フェニルシクロプロパン−1−カルボン酸クロライド]の無水CH2Cl2溶液に5℃で15分間通した。反応混合液を室温で一夜、窒素下に攪拌した。
【0094】
反応混合物を減圧下で蒸発させ、その結果入手した残渣をエチルアセテート/水の混合液中に再溶解させた。混合液をエチルアセテート(100mL)および水(60mL)を使用して分液ロートへ移した。混合液を平衡化し水層を除去した。有機層を1.0 M のHCl (10 mL)、H2O (70 mL)および塩水で連続して洗浄した。 有機層を無水硫酸マグネシウム上で乾燥させ、濾過し、減圧下で余剰な溶媒を蒸発させた。その結果入手した薄茶色の固体をBiostage SP4システム(カラムSi 40+S 90:10 CH2Cl2: MeOH) を使用して精製し、白色粉末[(1R,2R)-トランス−2−フェニルシクロプロパン−1−カルボキシアミド]1.127gを入手した(54%の収率)。GC/MS分析によりこの物質は100%純粋であった。1H−NMR 分光法によって製品の構造と一致するシグナルが得られ98%以上の純度が示された。
【0095】
実施例17 化合物Q[(1R,2R)-2−フェニルシクロプロパン−カルボン酸―((S)−1―カルバモイル−3−メチルブチル)アミド]の調製
【0096】
【化14】
【0097】
(1R,2R)−トランス-2−フェニルシクロプロパン−1−カルボン酸(0.905g、5.6 mmol) を CH2Cl2 (30mL)およびDMF(0.05mL、16.7mmol)の溶液中で塩化オキサリル(0.65mL、7.23 mmol)で0℃窒素静圧下で処理した。反応液を窒素下で1晩室温で撹拌した。減圧下で過剰なジクロロメタンを除去した。
【0098】
酸塩化物[(1R,2R)−トランスー2−フェニルシクロプロパンー1−カルボン酸クロライド]のCH2Cl2 (50mL)溶液 を H−Leu−NH2[L−ロイシンアミド、(S)―2−アミノー4―メチル−n―バレルアミド](0.761g、5.8mmol)およびトリエチレンアミン(1.13g、11.1mmol)のCH2Cl2(60mL)溶液に0℃で滴下して加えた。反応混合物を窒素下で1晩室温で撹拌した。
【0099】
反応混合物を減圧下で蒸発させ、その結果入手した残渣をエチルアセテート/水の混合液に再溶解させた。混合液をエチルアセテート(80mL)および水(50mL)を使用して分液ロートへ移した。混合液を平衡化し水層を除去した。有機層を1.0 M HCl(20 mL)、H2O(90 mL)および塩水(120mL)で連続して洗浄した。次に有機層を無水硫酸マグネシウム上で乾燥させ、濾過し、減圧下で余剰な溶媒を蒸発させた。その結果入手した橙茶色の固体をBiostage SP4システム(カラムSi 40+M 90:10 CH2Cl2: MeOH) を使用して精製し、白色粉末[(1R,2R)−2−フェニルシクロプロパンカルボン酸((S)−1−カルバモイル−3−メチルブチル)−アミド]0.365gを入手した(24%の収率)。GC/MS分析によりこの物質は100%純粋であった。1H−NMR 分光法によって製品の構造と一致するシグナルが得られ98%以上の純度が示された。
【0100】
実施例18 化合物U[2−[1−(4−メトキシフェニル)シクロプロピル]−アセタミド]の調製。
【0101】
【化15】
【0102】
水素化リチウムアルミニウム(0.211mol)を無水エーテル(200mL)中に撹拌した懸濁液を1−(4−メトキシフェニル)−1−シクロプロパンカルボン酸(0.1406mol)で0℃でエーテル100mL中で処理する。次に反応混合物を窒素下で1晩室温で撹拌する。反応混合液を脱イオン水100mLを滴下し冷却する。混合液を濾過し、ケーキ状の固体をジエチルエーテル(1L)で洗浄する。濾過した混合液(エーテルおよび水)を分液ロートへ移す。有機層を水層から分離し塩水で洗浄する。次にエーテル層を硫酸マグネシウム上で乾燥させ、濾過し、室温減圧下で濃縮させる。その結果、[1−(4−メトキシフェニル)シクロプロピル]‐メタノールを入手する。
【0103】
[1−(4−メトキシ−フェニル)シクロプロピル]メタノール(0.074mol)の未希釈液を三臭化リン(0.081mol)を静圧窒素下に0℃で滴下して処理する。反応液を130℃まで加熱し、その温度を6時間維持する。反応混合液を室温まで冷却し橙色の沈殿物を濾過する。橙色の沈殿物をジエチルエーテル200mLで洗浄する。水150mLおよびジエチルエーテル200mLを使用して濾液を分液ロートへ移す。混合液を平衡化し水層をジエチルエーテル200mLでもう1回抽出する。エーテル抽出物およびエーテル洗浄液を合わせて、炭酸水素ナトリウム飽和溶液および食塩水で洗浄する。次にエーテル抽出物を硫酸マグネシウム上で乾燥させ、余剰のジエチルエーテルを30℃減圧下で除去する。この結果、1−(1−ブロモメチル−シクロプロピル)−4−メトキシベンゼンを入手する。この粗物質をさらに精製しないで対応するニトリルへ変換させる。
【0104】
【化16】
【0105】
1−(1−ブロモメチル−シクロプロピル)−4−メトキシベンゼン(60.3mmol)のジメチルスルホキシド(60mL)粗溶液をシアン化ナトリウム(180.7mmol)で処理する。反応混合液を、窒素下で1晩95℃で加熱する。反応液を塩水(150mL)およびクロロホルム(300mL)を使用して分液ロートへ移す。反応混合液を平衡化し水層を除去する。さらに2回クロロホルム(2x300mL)を使って水層を抽出する。有機抽出液を合わせて無水硫酸マグネシウム上で乾燥させ、濾過し、減圧下で濃縮させ[1−(4−メトキシフェニル)シクロプロピル]アセトニトリルを入手する。この粗物質をさらに精製しないで次の段階で(ニトリルの対応するアミドへの加水分解)使用する。[または、本物質から、酸加水分解(例えば、 硫酸を使用)によって対応するカルボン酸を入手できる。]
[1−(4−メトキシ−フェニル)シクロプロピル)アセトニトリル(60.4mmol)のDMSO(75mL)溶液をH2O2(50%w/w)(434mmol)および炭酸カリウム(121mmol)を使って0°Cで処理する。反応混合液を室温で週末中、撹拌する。反応混合液を水(100mL)およびCH2Cl2(200mL)を使用して分液ロートへ移す。反応混合液を平衡化しCH2Cl2層を除去する。水層をさらに2回CH2Cl2(2x300mL)で抽出する。CH2Cl2抽出液を合わせて、水で5回連続して洗浄した後(5x200mL)、塩水(500mL)で洗浄し、無水硫酸マグネシウム上で乾燥させ、濾過し、減圧下で濃縮して、2−[1−(4−メトキシフェニル)−シクロプロピル]―アセトアミドを入手する。
【0106】
実施例19 化合物V[3−(4−クロロフェノキシ)−3−メチルブチル−アミド]の調製。
【0107】
【化17】
【0108】
水酸化リチウムアルミニウム(0.211mol)を無水エーテル(200mL)中で撹拌した懸濁液を2−(4−クロロフェノキシ)−2−メチルプロパン酸(0.1406mol)を使って0℃エーテル100mL中で処理する。反応混合物を窒素下で1晩室温で撹拌する。反応混合液を脱イオン水100mLを滴下し冷却する。混合液を濾過し、ケーキ状固体をジエチルエーテル(1L)で洗浄する。濾過した混合液(エーテルおよび水)を分液ロートへ移す。有機層を水層から分離し塩水で洗浄する。次にエーテル層を硫酸マグネシウム上で乾燥させ、濾過し、室温減圧下で濃縮させる。この結果、2−(4−クロロフェノキシ)−2−メチルプロパン−1−オールを入手する。
【0109】
2−(4−クロロフェノキシ)−2−メチルプロパン−1−オール(0.074mol)の未希釈液を三臭化リン(0.081mol)で静圧窒素下0℃で滴下によって処理する。反応液を130℃まで加熱しその温度を6時間維持する。反応混合液を室温へ冷却し橙色の沈殿物を濾過する。橙色の沈殿物をジエチルエーテル200mLで洗浄する。水150mLおよびジエチルエーテル200mLを使用して濾液を分液ロートへ移す。混合液を平衡化し、水層をジエチルエーテル200mLでもう1回抽出する。エーテル抽出液およびエーテル洗浄液を合わせて、炭酸水素ナトリウム飽和溶液および塩水で洗浄する。次にエーテル抽出物を硫酸マグネシウム上で乾燥させ、余剰のジエチルエーテルを30℃、減圧下で除去する。この結果、1−(2−ブロモ−1,1−ジメチルエトキシ)−4−クロロベンゼンを入手する。1−(2−ブロモ−1,1−ジメチルエトキシ)−4−クロロベンゼンの粗生成物をさらに精製しないで対応するニトリルへ変換する。
【0110】
【化18】
【0111】
1−(2−ブロモ−1,1−ジメチルエトキシ)−4−クロロベンゼン(60.3mol)のジメチルスルホキシド(60mL)粗溶液をシアン化ナトリウム(180.7mmol)で処理する。反応混合液を、窒素下で1晩95℃に加熱する。反応混合液を塩水(150mL)およびクロロホルム(300mL)を使用して分液ロートへ移す。反応混合液を平衡化し水層を除去する。水層をさらに2回クロロホルム(2x300mL)を使って抽出する。有機抽出液を合わせて無水硫酸マグネシウム上で乾燥させ、濾過し、減圧下で濃縮させ3−(4−クロロフェノキシ)−3−メチルブチロニトリルを入手する。この粗物質をさらに精製しないで次の段階で(ニトリルの対応するアミドへの加水分解)使用する。[または、本物質から、酸加水分解(例えば、 硫酸を使用)によって対応するカルボン酸を入手できる。]
(3−(4−クロロフェノキシ)−3−メチルブチロニトリル(60.4mmol)のDMSO(75mL)溶液をH2O2(50%w/w)(434mmol)および炭酸カリウム(121mmol)を使って0°Cで処理する。反応混合液を室温で週末の間、撹拌する。反応混合液を水(100mL)およびCH2Cl2(200mL)を使用して分液ロートへ移す。反応混合液を平衡化しCH2Cl2層を除去する。さらに水層を2回CH2Cl2(2x300mL)を使って抽出する。CH2Cl2抽出液を合わせて水で5回連続して洗浄した後(5x200mL)、塩水(500mL)で洗浄し、無水硫酸マグネシウム上で乾燥させ、濾過し、減圧下で濃縮し、3−(4−クロロフェノキシ)−3−メチルブチルアミドを入手する。
【0112】
実施例20 化合物AG[(R)-3−(4−トリフルオロメチルフェニル)―ブチルアミド]の調製。
【0113】
【化19】
【0114】
アセチルアセトナートビス(エチレン)ロジウム(I)(0.3mmol)、(s)−(−)−2,2’―ビス(ジフェニルホスフィノ)−1,1’−ビナフタレン(0.045mmol)、4−(トリフルオロメチル)フェニルボロン酸(2mmol)、K2CO3 (0.5mmol)および ブタ-2-エン酸アミド(1 mmol)を、磁気撹拌棒、セプタム注入口および還流冷却器のある25mL丸底フラスコへ加える。フラスコをアルゴンでフラッシュし、1,4−ジオキサン(3mL)および脱イオン水(0.5mL)で充填する。得られた反応混合液を100℃で16時間攪拌する。(R)−3−(4−トリフルオロメチルフェニル)ブチルアミドをエチルアセテートで抽出し、塩水で洗浄し無水硫酸マグネシウム上で乾燥させる。シリカゲル上のクロマトグラフィーによって所望の生成物が得られる。
【0115】
実施例21 化合物AA[3−(4−トリフルオロメチルフェニル)―ペンタンアミド]の調製。
【0116】
【化20】
【0117】
ビス(トリメチルシリル)アミドリチウム(1.0M、50mL)の冷却溶液に温度を10℃以下に維持しながらトリメチルホスホノアセテート溶液を滴下した。次に、本溶液を室温まで暖め、さらに5分間撹拌し、4’−(トリフルオロメチル)プロピオフェノンのTHF(25mL)溶液を一度に加える。本溶液をゆっくりと50℃に温め、8時間加熱した。本溶液を室温まで冷却し、10%NH4Cl溶液(100mL)で希釈し、エチルアセテート(2x100mL)で抽出した。有機層を合わせて、硫酸マグネシウム上で乾燥させ、濾過し、濾液をヘキサン/エチルアセテートから白色固体が再結晶するまで濃縮し、5.42gの3−(4−トリフルオロメチルフェニル)ペント−2−エン酸メチルエステル中間体を得た(85%の収率)。
【0118】
3−(4−トリフルオロメチルフェニル)ペント−2−エン酸メチルエステルのTHF/MeOH溶液に水酸化ナトリウム水溶液(15mL)を添加した。その結果得られる水溶液を室温で12時間撹拌し、酢酸(3g)を添加した。次に、溶液を濃縮しオイルを入手した。オイルを酢酸エチル(100mL)へ溶解させ水(3x100mL)で洗浄した。酢酸エチル抽出液を合わせて、硫酸マグネシウム上で乾燥させ、濾過し、濃縮させて半固体を入手し、MeOH/THF(2:1,50mL)へ溶解させ10%Pd/Cと水素圧下50psiで24時間振盪した。TLCにより反応が不完全であったことが示された。10%Pd/Cを追加(500mg)し懸濁液をさらに24時間振盪した。懸濁液を濾過し、濾液を濃縮し半固体を入手した(4.97g)。固体をCH2Cl2(30mL)に溶解し、その結果得られた溶液を0℃まで冷却した。この溶液へ塩化オキサリルを加え、9インチ使い捨てピペットを使ってDMFを1滴添加した。この溶液を8時間撹拌し、濃縮して固体とし、追加のCH2Cl2(30mL)に溶解させた。この溶液を再度濃縮して半固体を得、追加のCH2Cl2(50mL)に溶解させ、得られた溶液を、機械的に撹拌させた冷却(5℃)NH4OH (10 mL) 溶液へ約5分間かけて滴下した。次に懸濁液を濃縮し、粘性のある混合液を入手しエチルアセテート(2x100mL)で抽出した。エチルアセテート抽出液を合わせて、硫酸マグネシウム上で乾燥させ、濾過し、濾液を濃縮し、琥珀色の粗固体を入手しCH2Cl2/THFを使用してシリカゲル(50g)へ吸収させた。この固体をシリカゲル(EtOAc/ヘキサン)のクロマトグラフィーで処理し、2.25gの灰色がかった白色の固体を入手した(37%の収率)。この物質はLC/MSにより100%純粋であった。H-NMR により、物質の構造と一致するシグナルが得られ98%以上の純度が示された。
【0119】
実施例22 化合物AW[3−(4−イソプロピルフェニル)ブチルアミド]の調製。
【0120】
【化21】
【0121】
ビス(トリメチルシリル)アミドリチウム(1.0M、56mL)の冷却溶液(0°C)に温度を10℃以下に維持しながらトリメチルホスホノアセテート溶液を滴下した。次に、本溶液を室温まで暖め、さらに5分間撹拌した後、p−イソブチルアセトフェノンのTHF(25mL)溶液をを一度に加えた。この溶液をゆっくりと65℃まで加熱し30時間還流させた。本溶液を室温まで冷却し、10%NH4Cl溶液(100mL)で希釈し、エチルアセテート(2x100mL)で抽出した。有機層を合わせて、硫酸マグネシウム上で乾燥させ、濾過し、濾液を濃縮して白色固体(4.98g)を得、さらにそれをヘキサン/エチルアセテートから再結晶させて、5.93gの3−(4−イソブチルフェニル)ブタ−2−エン酸メチルエステル中間体を得た(85%の収率)。
【0122】
3−(4−イソブチルフェニル)ブタ−2−エン酸メチルエステルのTHF/MeOH溶液に水酸化ナトリウム水溶液(15mL)を加えた。その結果得られる水溶液を室温で12時間撹拌し、酢酸(3g)を添加した。この溶液を濃縮しオイルを入手した。このオイルを酢酸エチル(150mL)へ溶解させ水(3x100mL)で洗浄した。酢酸エチル抽出液を合わせて、硫酸マグネシウム上で乾燥させ、濾過し、濃縮させて固体(4.98g)を入手し、MeOH/THF(2:1,50mL)に溶解させ10%Pd/Cと水素圧下50psiで24時間振盪した。TLCにより反応が不完全であったことが示された。10%Pd/Cを追加(500mg)し懸濁液をさらに24時間振盪した。この懸濁液を濾過し、濾液を濃縮し固体を入手した(4.75g)。この固体をCH2Cl2(30mL)に溶解し、その結果得られた溶液を0℃に冷却した。この溶液に塩化オキサリルを加えた後、9インチ使い捨てピペットを使ってDMFを1滴添加した。この溶液を6時間撹拌し、濃縮して固体を得、追加のCH2Cl2(30mL)に溶解させた。この溶液を再度濃縮して半固体を得、追加のCH2Cl2(50mL)に溶解させ、得られた溶液を、機械的に撹拌しながら冷却した(5℃)NH4OH (10 mL) 溶液へ約5分間かけて滴下した。次にこの懸濁液を濃縮し、固体/水の混合物を入手しエチルアセテート(2x100mL)で抽出した。エチルアセテート抽出液を合わせて、硫酸マグネシウム上で乾燥させ、濾過し、濾液を濃縮し、粗固体を入手しCH2Cl2/THFを使用してシリカゲル(50g)へ吸収させた。この固体をシリカゲル(EtOAc/ヘキサン)を使ったクロマトグラフィーで処理し、2.5gの灰色がかった白色の固体を入手した(40%の収率)。この物質はLC/MSにより100%純粋であった。. H-NMR により、物質の構造と一致するシグナルが得られ98%以上の純度が示された。
【0123】
実施例23 化合物AE[3−(6−メトキシナフタレン−2−シル)―ブチルアミド]の調製。
【0124】
【化22】
【0125】
ビス(トリメチルシリル)アミドリチウム(1.0M、50mL)の冷却溶液にトリメチルホスホノアセテート溶液を温度10℃以下に維持しながら滴下した。次に、この溶液を室温まで温め、さらに10分間撹拌し、2−アセチル−6−メトキシナフタレンのTHF(20mL)溶液を一度に加えた。この溶液をゆっくりと50℃まで温め、14時間加熱した。この溶液を室温に冷却し、10%NH4Cl溶液(100mL)で希釈し、エチルアセテート(2x100mL)で抽出した。有機層を合わせて、硫酸マグネシウム上で乾燥させ、濾過し、濾液を濃縮し白色固体を得、ヘキサン/エチルアセテートから再結晶して、5.88gの3−(6−メトキシナフタレン−2−イル)ブタ−2−エン酸メチルエステル中間体を得た(92%の収率)。
【0126】
3−(6−メトキシナフタレン−2−イル)ブタ−2−エン酸メチルエステルのTHF/MeOH溶液に水酸化ナトリウム水溶液(15mL)を加えた。その結果得られた水溶液を室温で12時間撹拌し、酢酸(3g)を添加した。この溶液を濃縮し固体残渣を入手した。この固体を酢酸エチル(150mL)に溶解させ水(3x100mL)で洗浄した。酢酸エチル抽出液を合わせて、硫酸マグネシウム上で乾燥させ、濾過し、濃縮させて固体を入手し、MeOH/THF(2:1,50mL)に溶解させ10%Pd/Cと水素圧下50psiで24時間振盪した。TLCにより反応が不完全であったことが示された。10%Pd/Cを追加(500mg)し懸濁液をさらに24時間振盪した。この懸濁液を濾過し、濾液を濃縮し半固体を入手した(4.97g)。この固体をCH2Cl2(30mL)に溶解し、その結果得られた溶液を0℃まで冷却した。この溶液へ塩化オキサリルを加えた後、9インチ使い捨てピペットを使ってDMFを一度に加えた。この溶液を6時間撹拌し、濃縮して固体を得、追加のCH2Cl2(30mL)に溶解させた。この溶液を再度濃縮して半固体を得、追加のCH2Cl2(50mL)に溶解させ、得られた溶液を、機械的に撹拌した冷却(5℃)NH4OH (10 mL) 溶液へ約15分間かけて滴下した。次にこの懸濁液を濃縮し、粘性物質と水の混合液を入手しエチルアセテート(2x100mL)で抽出した。エチルアセテート抽出液を合わせて、硫酸マグネシウム上で乾燥させ、濾過し、濾液を濃縮し、粗固体を入手しCH2Cl2/THFを使用してシリカゲル(50g)に吸収させた。この固体をシリカゲル(EtOAc/ヘキサン)を使ったクロマトグラフィーにより処理し、1.87gの灰色がかった白色の固体を入手した(32%の収率)。LC/MSによりこの物質は100%純粋であった。H-NMR により、物質の構造と一致するシグナルが得られ98%以上の純度が示された。
【0127】
実施例24 化合物AX[3−(2−フルオロ−ビフェニル−4−イル)ブチルアミド]の調製。
【0128】
【化23】
【0129】
冷却した(0℃)2−(2−フルオロ−ビフェニル−4−イル)プロピオン酸のTHF溶液にイソブチルクロロホルメートを加え、TEAを滴下した。その結果得られた白色のスラリーを1時間撹拌しTHF(50mL)で希釈して濾過した。濾過したケーキ状固体を追加のTHF(50mL)で洗浄し、濾液を回転式蒸発器で約50mLまで濃縮した。濃縮された濾液を−20℃で撹拌し、 NaBH4 水溶液(20mL)を15分間かけて滴下した。その結果得られた懸濁液を0℃で2時間撹拌し、水(200mL)で希釈し、エチルアセテート(2x100mL)で抽出した。エチルアセテート層を合わせて、1.0N HCl溶液(100mL)で洗浄し5%炭酸水素塩溶液で洗浄した(100mL)。このエチルアセテート溶液を濃縮しオイル状の残渣である2−(2−フルオロービフェニル−4−イル)プロパノールを入手した(4.47g、95%の収率)。
【0130】
冷却した(0℃)2−(2−フルオロ−ビフェニル−4−イル)プロパノールのCH2Cl2溶液にメタンスルホニルクロライドを加え、TEAを滴下により添加した。その結果得られた白色のスラリーを1時間撹拌し水(200mL)で希釈した。この懸濁液をCH2Cl2 (2 x 100 mL)で抽出した。CH2Cl2層を合わせて、水(2x100mL)および5%NH4OH (100mL)を加えた。CH2Cl2 層を追加の水(200mL)で洗浄し、硫酸マグネシウム上で乾燥させた。CH2Cl2層を濃縮しオイル状の残渣を入手し無水DMF(50mL)に溶解させた。この溶液をNaCNで処理し60℃で14時間撹拌した。TLCにより1個の主要な、より極性の低い溶出生成物(メシレートに比較して)と複数の微量の極性が主要生成物およびメシレートに比較して、より低い溶出生成物が示された。反応を室温まで冷却し水(100mL)で希釈した。この溶液をエチルアセテート(2x100mL)で抽出し、硫酸マグネシウム上で乾燥させ濃縮しオイル状残渣を入手し、シリカゲルを使ったクロマトグラフィー(90% Hex、10%EtOAc)により3−(2−フルオロ−ビフェニル−4−イル)ブチロニトリルをオイルとして入手したが、これはゆっくりと室温で固化した(2.05g、49%の収率)。
【0131】
3−(2−フルオロ−ビフェニル−4−イル)ブチロニトリルのtert-ブチルアルコール溶液に水酸化カリウムの微粉末1.87gを添加した。その結果得られた懸濁液を撹拌し、70℃まで温め、2.5時間加熱し室温まで冷却した。反応懸濁液を1.0N HCl溶液(100mL)で希釈しエチルアセテート(2x100mL)で抽出した。エチルアセテート抽出液を合わせて5%炭酸水素塩溶液(100mL)で、続いて水(100mL)で洗浄した。次に有機物を硫酸マグネシウム上で乾燥させ濃縮して白色固体を得、EtOAc/Hexで数回再結晶させ白色の剥片状のプリズム結晶を入手した(1.62g、75%収率)。
【0132】
実施例25 化合物AY[3−(4−モルホリン−4−イルーフェニル)―ブチルアミド]の調製。
【0133】
【化24】
【0134】
冷却(0℃)したカリウム tert-ブトキシド(1.0M、37.1mL)溶液に温度を25℃以下に維持しながらトリメチルホスホノアセテート溶液を滴下した。次に、この溶液を室温まで温め、さらに5分間撹拌した後、4―モルホリノアセトフェノンのTHF(20mL)溶液を一度に加えた。この溶液をゆっくりと60℃まで温め、36時間加熱した。この溶液を室温まで冷却し、1.0N HCl溶液(100mL)で希釈し、エチルアセテート(2x100mL)で抽出した。有機層を合わせて、硫酸マグネシウム上で乾燥させ、濾過し、濾液を濃縮して白色固体を得、ヘキサン/エチルアセテートから再結晶させて、3.33gの3−(4−モルホリノフェニル)ブタ−2−エン酸メチルエステル中間体を得た(69.9%の収率)。
【0135】
3−(4−モルホリノフェニル)ブタ−2−エン酸メチルエステルのTHF/MeOH溶液(1:1)に水酸化ナトリウム水溶液(15mL)を加えたた。その結果得られた溶液を室温で15時間撹拌し、酢酸(3g)を加えた。この溶液のpHは、6.5であった。この溶液を濃縮しオイルを入手した。オイルを酢酸エチル(150mL)に溶解させ水(3x100mL)で洗浄した。酢酸エチル抽出液を合わせて、硫酸マグネシウム上で乾燥させ、濾過し、濃縮して不定形の固体を入手し、MeOH(50mL)に溶解させ10%Pd/Cを加えて水素圧下50psiで8時間振盪した。TLCにより反応が完了したことが示された。この懸濁液を濾過し、濾液を濃縮し半固体を入手した(2.77g)。この半固体をCH2Cl2(30mL)に溶解させ、その結果得られた溶液を0℃に冷却した。この溶液に塩化オキサリルを加えた後、9インチ使い捨てピペットを使ってDMFを1滴添加した。この溶液を4時間撹拌し、濃縮して固体を得、追加のCH2Cl2(30mL)に溶解させた。この溶液を再度濃縮して半固体を得、、追加のCH2Cl2(50mL)に溶解させ、得られた溶液を、機械的に撹拌させた冷却(5℃)NH4OH (15mL) 溶液へ約5分間かけて滴下した。次にこの溶液を濃縮し、固体/水の混合物を入手しエチルアセテート(2x100mL)で抽出した。エチルアセテート抽出液を合わせて、硫酸マグネシウム上で乾燥させ、濾過し、濾液を濃縮し、粗固体を入手しCH2Cl2/THFを使用してシリカゲル(50g)に吸収させた。この固体をシリカゲル(EtOAc/ヘキサン)のクロマトグラフィーで処理し、2.1gのベージュ色の平板状固体を入手した(46%の収率)。LC/MSにより、この物質は100%純粋であった。. H-NMR で測定し、物質の構造と一致するシグナルが得られ98%以上の純度が示された。
【0136】
実施例26 化合物Q−1[1R,2R]-2−フェニルシクロプロパンーカルボン酸―((S)−1―カルバモイル−プロピル)アミド]の調製。
【0137】
【化25】
【0138】
トランスー2−フェニルシクロプロパンカルボニルクロライドのCH2Cl2 (20mL)溶液をL−2−アミノブタンアミドヒドロクロライド(1.61g、11.6mmol)およびトリエチレンアミン(3.36g、33.2mmol)のCH2Cl2 (60mL)溶液中に、0℃で滴下した。反応混合物を窒素下で1晩室温で撹拌した。
【0139】
この反応混合物を減圧下で蒸発させ、その結果入手した残渣をエチルアセテート/水混合液に再溶解させた。この混合液をエチルアセテート(80mL)および水(50mL)を使用して分液ロートへ移した。混合液を平衡化し水層を除去した。有機層をHCl 1.0 M(20 mL)、 H2O (90 mL)および塩水(120mL)で連続して洗浄した。この有機層を無水硫酸マグネシウム上で乾燥、濾過し、減圧下で余剰な溶媒を蒸発させた。その結果入手した橙茶色の固体をBiostage SP4システム(カラムSi 40+M 90:10 CH2Cl2/ MeOH)を使用して精製し、白色粉末0.365gを入手した(24%の収率)。GC/MSによりこの物質は100%純粋であった。. 1H-NMRで測定し、物質の構造と一致するシグナルが得られ98%以上の純度が示された。
【0140】
図4A-4Oは、合成有機化学文献から引用した様々な化合物および主要中間体(図解 1−15)の合成の追加の実施例を示し、それらから当業者は本発明のさまざまな追加化合物の調製について想像することが可能である。
【0141】
実施例27 齧歯類の癲癇モデルによる抗痙攣性の生物学的活性の実証。
本発明の多様な化合物の抗痙攣作用は、多種の齧歯類(マウスおよびラット)癲癇モデルにおいてin vivoで証明された。動物実験をWhite ら、「抗癲癇薬の発見と非臨床試験による開発」(Discovery and preclinical development of antiepileptic drugs), in Antiepileptic Drugs, 5th ed., Levy et al.(Eds.), Lippincott Williams and Wilkins, Philadelphia, PA, 2002 (968 pp.), pp. 36-48 の記載に従って実施し、参照により その全体が本明細書に組み込まれる。化合物A、G、H、IおよびFの結果を以下の表2および3に要約して示す。
【0142】
【表2】
【0143】
【表3】
【0144】
実施例28 ラットの癲癇重積状態モデルによる抗痙攣性生物学的活性の実証
本発明のさまざまな化合物の抗痙攣活性はまた、2つのラットの癲癇重積状態モデルにおいてin vivoで証明された。米国国立衛生研究所(NIH)の米国国立神経疾患脳卒中研究所(NINDS)の抗痙攣剤スクリーニングプログラム(ASP)で開発されたプロトコルに従って動物実験を実施した。表4に結果の要約を示す。
【0145】
【表4】
【0146】
実施例29 In Vitroアッセイ(LDHおよび細胞増殖)における無毒性の実証。
化合物A、I、HおよびFについてStem Cell Innovations, Inc. (Houston, TX) のヒト肝細胞に基づくアッセイACTIVTox(R)(C3A肝細胞を使用)で試験を実施した。 具体的には、試験化合物を多種類の濃度で、乳酸脱水素酵素LDHの放出を測るLDH放出アッセイ(細胞死の指標)で試験した。
【0147】
試験化合物の100μMという濃度は、生理学的状況下で予測される肝細胞への暴露量よりも非常に高い。従って100μM量の標準試験濃度(比較の目的で)を使用して、陰性対照群の吸光度に対する試験化合物存在下での吸光度(LDH放出レベルの測定)の比は、「平均倍数」(平均倍数=平均吸光度/陰性対照の平均吸光度)値として知られている。1.75以下のAVFは、LDH放出アッセイでは試験化合物は、細胞毒性または肝毒性作用が無いことを意味する。
【0148】
ACTIVToxデータ(以下の表5を参照)は、化合物A、I、HおよびFには、生理学的濃度( < 100 μM)では細胞毒性または肝毒性がないことを示す。
【0149】
【表5】
【0150】
化合物AおよびIはまたACTIVTox 細胞増殖アッセイを使って試験し、各々平均倍数値が1.15および1.25であった100μM濃度では増殖中の細胞に対する毒性がないことが示された。
【0151】
本発明は、その真意や本質的な特徴から逸脱することなくその他の具体的な態様で実施されうる。本明細書に記載される実施態様は、あらゆる面で例証的なものでありこれらに限定されるものではない。従って本発明の範囲は、前述の説明によってよりもむしろ、付記された特許請求の範囲によって示される。本特許請求に意味するものと、それに同等なものの範囲内の全ての変更は、本特許請求に組み込まれる。本明細書に引用される参照は、その全体を本明細書に具体的な参照として組み込まれる。
【技術分野】
【0001】
関連出願
本出願は2008年3月19日に出願の米国特許仮出願番号 61/037,987 の「中枢神経系疾患および障害の治療に有効な新規化合物」の利益を主張し、当該出願の内容を引用することによりその全てがここに組み込まれることとする。
【0002】
発明の背景
1. 発明の技術分野
本発明は、実験動物の中枢神経系(CNS)に作用を示す新規化合物に関連する。さらに具体的には、本発明は、現存する中枢神経系(CNS)治療薬に比べて、同等あるいはより優れた生物学的作用(すなわち有効性)を発揮しながら、毒性学的安全性の増大/改善(すなわち毒性の低減)、代謝安定性の増大/改善、半減期の延長および/または優れた副作用プロファイルと抗痙攣作用を示す新規化合物に関連する。
【背景技術】
【0003】
2. 関連技術
数多くの病理学的症状(例えば、癲癇、脳卒中、双極性感情障害、片頭痛、不安、抑鬱、不眠、統合失調症、慢性または神経因性疼痛、痙性、脊椎損傷および慢性神経変性障害など)および疾患(例えば、パーキンソン病、ハンチントン病およびアルツハイマー病など)は、中枢神経系(CNS)の正常な機能の異常として特徴づけられる。これらの症状や疾患は、一般的にはCNSの正常な機能を調節する化合物や物質による薬理学的治療に対して反応する。そのような活性を有する化合物には、癲癇などのCNSの異常を治療するために本明細書に開示される本発明の化合物が含まれる。現在利用できる治療薬はしばしば良好なCNS活性を有するが、慢性毒性、重度の、および/または不快な副作用、短い薬理学的半減期のような不十分な薬物動態特性などその他の好ましくない特性を示すものが多い。例えば、CNS治療薬の短い半減期は、有害事象を発現させずに治療濃度を維持するためには、頻回の入院を必要とし、頻繁な投与スケジュールが必要となり治療コストが増加する可能性がある。さらに、必要とされる投与回数が増加すると患者の服薬コンプライアンスが低下する傾向がある。従って、現在利用できる治療薬に比べて、例えば半減期の延長、活性の増大(すなわち有効性の改善)および/または代謝安定性の向上(例えば、毒性代謝物の低減など)などの優れた特性を有し、CNS活性を調節できる新たな化合物の提供が望まれている。さらに、より幅広く多くの患者人口に本発明の有効な化合物の提供を可能にする、改善された、より簡単な/簡略化された、合成および化学的製造工程を開発することも可能である。
【0004】
関連技術は、米国特許番号第 5,463,125号 (Sandovalら、「抗痙攣作用を有するフェニルアルコールアミド類」(Phenyl alcohol amides having anticonvulsant activity))、WO9941229 (Carvajal Sandoval ら、「抗痙攣作用を有するハロゲン化フェニルアルコールアミド類(GABAB受容体のリガンド)」(Halogenated phenyl alcohol amides (ligands of GABAB receptor) having an anticonvulsant activity))、WO03091201(Carvajal Sandovalら、「抗痙攣作用を有するDL-ヒドロキシーアルキルーフェニルアミド類」(DL−Hydroxy−alkyl−phenylamides having anticonvulsive activity))、WO2005085182 (Meza Toledo、「抗痙攣作用を有するDL-ヒドロキシベンズアミド類」(DL−Hydroxybenzamides having anticonvulsive activity))および米国特許出願番号第20060287397号 (Meza Toledo、「抗痙攣作用を有するDL-ヒドロキシーアルキルーフェニルアミド類」(Dl−Hydroxy−alkyl−phenylamides having anticonvulsive activity))に記載されている。上述の先行技術および本発明の技術の重要な違いは、先行技術はγヒドロキシ酪酸(γヒドロキシ酪酸塩またはGHB)と構造上関連する化合物を含む、または関連するのに対し、本発明の化合物は、3-メチル酪酸アミドに構造上関連することである。
【発明の概要】
【課題を解決するための手段】
【0005】
発明の簡単な概要
本明細書では、抗痙攣作用を有する一連の新規アミド類を開示するが、その多くは、短い、多様に分岐/置換された脂肪族リンカーを介してフェニル基がアミド部分へ結合している。本発明のその他の化合物(以下に示す)は、アラニン (Z = CH3 以下)、バリン [Z = CH(CH3)2]、ロイシン [Z = CH2CH(CH3)2]、イソロイシン [Z = CH(CH3)CH2CH3]またはフェニルアラニン (Z = CH2C6H5)などの光学的に活性なアミノ酸(例えば、 DまたはL)の、あるいはグリシン(Z=H)またはタウリン [R2 = (CH2)2SO3H、 以下に示す] などの光学的に不活性なアミノ酸の誘導体であるアミド類である。そのような化合物は以下の式で例示される:
式Iを有するCNS活性化合物:
【0006】
【化1】
【0007】
式Iにおいて、Arは、任意に置換されたフェニル、任意に置換されたナフチル、任意に置換されたテトラヒドロナフチル、任意に置換されたインダンであって良く、あるいは任意に置換されたヘテロサイクリックアリルでも良く、ここで5つまでの置換基を含んでも良い。Ar上の各置換基は、アルキル、シクロアルキル、ハロゲン、アルコキシ、チオアルキル、スルホキシアルキル、 スルホニルアルキル、アルキレンジオキシ、ハロアルキル、ハロアルコキシ、OH、CH2OH、CONH2、CN、アセトキシ、N(アルキル)2、ベンジル、ベンジロキシ、α,α-ジメチルベンジル、 NO2、CHO、CH3CH(OH)、アセチル、OCH2COOHから成る群から独立して選ばれてもよく、またはそれらの組み合わせであっても良い。また、Ar には、Arの1個または2個の原子に結合する任意に置換された芳香環系が含まれても良く、そのような芳香環系は、フェニル、フェノキシ、ヘテロサイクリックアリルから選択されるか、またはその組み合わせである。上記のArおよび/またはその置換基は、5個までの置換基を含んで良く、各置換基は、アルキル、シクロアルキル、ハロゲン、アルコキシ、チオアルキル、スルホキシアルキル、 スルホニルアルキル、アルキレンジオキシ、ハロアルキル、ハロアルコキシ、OH、CH2OH、CONH2、CN、アセトキシ、N(アルキル)2、NO2、CHO、CH3CH(OH)、アセチル、およびOCH2COOHからなる群から独立に選択される基であって良い。さらに、Arおよび/または芳香環基は、Arおよび/または芳香環基と芳香環を構成する二官能置換基を含んで良く、該二官能置換基は、アルキル、シクロアルキル、メチレンジオキシ、エチレンジオキシまたは他のアルキレンジオキシの任意の置換基またはそれらの組み合わせでも良い。
【0008】
式Iにおいて、R1 および R2は各々独立して、H、長鎖または短鎖の置換された、あるいは置換されていないアルキル、置換された、あるいは置換されていないシクロアルキル、CW2フェニル、のうちの少なくとも1つであって良い。各Wは独立してH、メチルおよびエチルからなる群から選択される基である(Wが共にエチルにはならない限り)。5個までの置換基がフェニル基に含まれて良く、各置換基は、ハロゲン、アルコキシ、チオアルキル、スルホキシアルキル、スルホニルアルキル、ハロアルキル、ハロアルコキシ、CONH2、CN、アセトキシ、N(アルキル)2、NO2 およびアセチルからなる群から独立して選択される。
別の実施態様では、R1は H であり、R2 は(CH2)2SO3HまたはCHZCOOHであり、Zは、H、 CH3、CH(CH3)2、CH2C6H5、CH2CH(CH3)2および CH(CH3)CH2CH3からなる群から独立して選択される。任意に、R1 および R2が共にシクロアルキルである。
式Iにおいて、R3は、ヒドロキシ、置換されたあるいは置換されていないアルキル、置換されたあるいは置換されていないシクロアルキルのいずれかであるか、あるいはR4も共にシクロアルキルである。任意に、R3 が OHの場合には R4 はエチルではない。
【0009】
式Iにおいて、R4は、置換されたあるいは置換されていないアルキル、置換されたあるいは置換されていないシクロアルキルのうちの一つであるか、またはR3も共にシクロアルキルである。
【0010】
式Iにおいて、Xは無置換、置換されたあるいは置換されていないアルキレン、メチレン、ケトン、CHOH、酸素、NR1、硫黄、スルホンまたはスルホキシドのうちの1つである。
式IIを有するCNS活性化合物:
【0011】
【化2】
【0012】
式IIにおいて、R1 および R2は各々独立して、H、長鎖または短鎖の置換されたあるいは置換されていないアルキル、置換されたあるいは置換されていないシクロアルキル、CW2フェニルのうちの少なくとも一つ、またはそれらの組み合わせである。Wは独立してH、メチルおよびエチルから選択されるが、Wが共にエチルであることはできない。フェニル基またはシクロアルキル基には5個までの置換基が含まれて良く、各置換基は、ハロゲン、アルコキシ、チオアルキル、スルホキシアルキル、スルホニルアルキル、ハロアルキル、ハロアルコキシ、CONH2、CN、アセトキシ、N(アルキル)2、NO2 およびアセチルからなる群から独立して選択される。任意に、R1は H およびR2 は(CH2)2SO3HまたはCHZCOOHを含み、ZはH、CH3、CH(CH3)2、CH2C6H5、CH2CH(CH3)2および CH(CH3)CH2CH3からなる群のうちの1つである。また別の選択として、R1 およびR2は共にシクロアルキルである。
【0013】
R4および R5は、各々独立して任意に置換されたフェニルまたは任意に置換されたヘテロサイクリックアリルのいずれかであり、ここで5個までの置換基があっても良く、各置換基はハロゲン、アルコキシ、チオアルキル、スルホキシアルキル、 スルホニルアルキル、ハロアルキル、ハロアルコキシ、CH2OH、CONH2、CN、アセトキシ、N(アルキル)2、NO2、アセチルおよび OCH2COOHからなる群から独立して選択される。
【0014】
Yは、無置換、置換された、あるいは置換されていないメチレン、あるいは他の置換されたあるいは置換されていないアルキレンのいずれかである。
実施態様の一つにおいては、本発明は以下の式のうちの一つを有するCNS活性化合物を含む:
【0015】
【化3】
【0016】
実施態様の一つにおいては、R1 および R2は各々独立してH、長鎖または短鎖の置換されたあるいは置換されていないアルキル、置換されたあるいは置換されていないシクロアルキル、CW2フェニルのうちのの少なくとも一つであるか、またはそれらの組み合わせである。WはH、メチルおよびエチルからなる群から独立して選択されるが、Wが共にエチルであることはできない。フェニル基またはシクロアルキル基には5個までの置換基が含まれて良く、各置換基はハロゲン、アルコキシ、チオアルキル、スルホキシアルキル、スルホニルアルキル、ハロアルキル、ハロアルコキシ、CONH2、CN、アセトキシ、N(アルキル)2、NO2 およびアセチルからなる群からから独立して選択される。任意にR1が H でありR2 が(CH2)2SO3HまたはCHZCOOHであって、ここで、Zは、H、CH3、CH(CH3)2、CH2C6H5、CH2CH(CH3)2および CH(CH3)CH2CH3 からなる群のうちの一つである。また別の選択としては、R1 およびR2が共にシクロアルキルである。
【0017】
実施態様の一つにおいては、R1 は H であり、R2 は(CH2)2SO3Hまたは CHZCOOHであり、ここでZはH、CH3、CH(CH3)2、CH2C6H5、CH2CH(CH3)2および CH(CH3)CH2CH3 からなる群のうちの一つである。任意に、R1 および R2は共にシクロアルキルである。
【0018】
実施態様の一つにおいては、R3は、ヒドロキシ、置換されたあるいは置換されていないアルキル、置換されたあるいは置換されていないシクロアルキルのうちのいずれかであるか、R4 も共にシクロアルキルである。任意に、R3が OHの場合には R4 はエチルではない。
【0019】
実施態様の一つにおいては、R4は、置換されたあるいは置換されていないアルキル、置換されたあるいは置換されていないシクロアルキルのうちの一つであるか、またはR3 も共にシクロアルキルである。
【0020】
実施態様の一つにおいては、R5 は H、Cl、F、CF3、CN、C1-C5 アルキル、 C1-C5 アルコキシ、OCF3、CONR1R2、ハロゲン置換アルキル、ハロゲン置換アルコキシのうちの一つである。
【0021】
実施態様の一つにおいては、Xは無置換、置換されたあるいは置換されていないアルキレン、メチレン、ケトン、CHOH、酸素、NR1、硫黄、スルホンまたはスルホキシドのうちの一つである。
【0022】
実施態様の一つにおいては、nは0〜5、より好ましくは1〜3であり最も好ましくは1〜2であって良い。
実施態様の一つにおいては、式1〜8のいずれも以下の特長を有しても良い:R1 は H、CH3、(CH2)2SO3Hまたは CHZCOOHのうちの一つであり、ここでZは、H、CH3、CH(CH3)2、CH2C6H5、CH2CH(CH3)2および CH(CH3)CH2CH3、これらの組み合わせまたはこれらの誘導体からなる群のうちの一つであり;R2、R3およびR4 は、独立してH、CH3、置換または未置換のアルキル、OH、OCH3、置換または未置換のアルコキシ、それらの組み合わせまたはそれらの誘導体のうちの一つであり;R5は、H、Cl、F、CF3、CN、C1-C5 置換または未置換のアルキル、C1-C5 置換または未置換のアルコキシ、OCF3CONR1R2、またはこれらの組み合わせあるいはこれらの誘導体のうちの一つであり;nは1〜5、好ましくは1〜3であり;および X は、O、NR1、無置換、C=O、S、SO2、これらの組み合わせまたはこれらの誘導体のうちの一つである。
【0023】
本発明のこれらおよびその他の実施態様や特長は、以下の説明および付加された特許請求の範囲からさらに明らかとなるかあるいは、以下に説明する本発明の実施例によって理解され得る。
【図面の簡単な説明】
【0024】
【図1】図1は本発明の(例えば)哺乳類などの中枢神経系(CNS)に薬理学的活性な新規化合物の化学構造を示し、本発明の実施態様を例示するものである。
【図2】図2は本発明に記載される化合物の相対的な生物学的な活性を示すもので、好ましい(第2類、ED50 < 300 mg/kg)および最も好ましい(第1類、ED50 < 100 mg/kg)化合物について具体的に記載する。
【図3A】図3Aは、さらに本発明の最も好ましい化合物類のその他の化合物の化学構造を示す。
【図3B】図3Bは、さらに本発明の最も好ましい化合物類のその他の化合物の化学構造を示す。
【図4A】多様な化合物および主要な中間体の実施例を示す。
【図4B】多様な化合物および主要な中間体の実施例を示す。
【図4C】多様な化合物および主要な中間体の実施例を示す。
【図4D】多様な化合物および主要な中間体の実施例を示す。
【図4E】多様な化合物および主要な中間体の実施例を示す。
【図4F】多様な化合物および主要な中間体の実施例を示す。
【図4G】多様な化合物および主要な中間体の実施例を示す。
【図4H】多様な化合物および主要な中間体の実施例を示す。
【図4I】多様な化合物および主要な中間体の実施例を示す。
【図4J】多様な化合物および主要な中間体の実施例を示す。
【図4K】多様な化合物および主要な中間体の実施例を示す。
【図4L】多様な化合物および主要な中間体の実施例を示す。
【図4M】多様な化合物および主要な中間体の実施例を示す。
【図4N】多様な化合物および主要な中間体の実施例を示す。
【図4O】多様な化合物および主要な中間体の実施例を示す。
【発明を実施するための形態】
【0025】
図示による実施態様の説明
1. 概観
本発明者は、本発明の化合物および薬理学活性のある化合物類似体および同類のあるものは、in vivoの投与によってCNS活性の調節に有効であることを発見した。すなわち、これらの薬剤は神経系の活性の全てを完全に抑制することなく、中枢神経系の神経伝達の抑制の増強または興奮の抑制によってCNS活性を調節する。従って、本発明に準じて、この薬剤を投与される患者は、例えば、癲癇発作の緩和(無麻酔で)、筋緊張の緩和(麻痺を起こさないで)、静穏化作用(鎮静作用なしで)または痙性のような歩行系症状の改善(虚弱化、あるいは弛緩状態にすることなく)のような状況下において、過度な鎮静、麻酔、あるいは麻痺をもたらされない。
【0026】
痙攣(癲癇発作)、痙性、双極性感情障害のような気分障害、頭痛(慢性、群発性、片性)、静止不能症候群、神経因性疼痛、運動障害などで示される多くの病変は、CNS活性の調節によって緩和できる少なくとも一つの症状を有する。従って、このような病変に悩む個人は、本発明に準じて、主要な活性成分として、本発明の化合物、あるいはその構造関連化合物類または同類を含む医薬製剤処方または組成の投与を受ける治療の候補者である。
【0027】
2. 中枢神経系(CNS)活性の調節によって改善される病変の好例
痙攣:癲癇は、多くの原因による一般的な疾患であり、臨床上の管理が非常に困難な場合があり、発作を管理するために何年もの治療を必要とすることが多い。研究者達は、現時点では、患者の多くにとって有効な癲癇の治療法は存在しないと述べている。臨床試験において、患者が同様な種類の癲癇を有し、薬剤が同様な作用機序を有していても、他の薬剤よりもある薬剤に良好に反応する患者があることが示された。副作用の頻度及び重症度にも大きな差が存在する。このように癲癇の治療には、癲癇の治癒が可能となるか、あるいは、広範な活性を持った強力で安全な新薬が発見および開発されるまで、異なる作用機序と異なる副作用を持った複数の薬剤による治療が必要であろう。Dichter ら、 Drug Therapy 334:1583 (1996)。
【0028】
かなり予測的で実験的に使用可能な癲癇の動物モデルによって、数多くの臨床上有効な抗痙攣薬が調製され開発されてきている。例えば、Cereghino ら、「抗癲癇薬」(ANTIEPILEPTIC DRUGS), 第4版、1-11 ページ (Rave Press 1995)の「序章」を参照、「多くの患者においては癲癇は現存の抗癲癇薬で管理が可能であるが、患者の25〜30パーセントは最適な治療を実施しても癲癇を抑えることができず、また患者の多くは容認できない副作用を経験している。」Dichter ら(1996) 同上。
【0029】
このように多くの抗痙攣薬の臨床使用は、やっかいな日中の鎮静、筋肉の虚弱化、耐性、歯肉増殖症、致命的な可能性のある悪液質および肝毒性などの重大な副作用が悩みとなっている。これらの副作用の多くは、特に小児癲癇の臨床管理(治療)において問題となっている。
【0030】
本発明は、癲癇のような痙攣性疾患を治療するために使用できる。すなわち、本発明の組成物、医薬製剤処方および組成は「抗痙攣作用」を示すが、それは動物癲癇モデルにおける痙攣の重症度、発作回数あるいは期間の軽減によって実証されている。痙攣の緩和には、患者における痙攣発作の重症度、回数や期間の軽減が含まれる。従って新規組成物および医薬製剤処方および組成は、これらに限定されるものではないが、全身性強直・間代性発作、欠神発作、ミオクローヌス発作、単純部分発作、複雑部分発作、二次性全般性部分発作、癲癇重積状態、および頭部の外傷や外科手術による外傷性癲癇発作などの症状の治療に有効である。
痙性:痙性は、伸張反射の過剰興奮による過剰な腱反射を伴う緊張性反射(筋緊張)の増強を特長とする障害である。Lance、シンポジウム抄録、「痙性-運動制御障害」(SPASTICITY--DISORDERED MOTOR CONTROL)、Feldman ら(Eds.)(1980). 痙性に付随する主要な疾患及び症状には、多発性硬化症、脳性麻痺、脳卒中、脊髄の外傷または損傷および頭部外傷がある。痙性に起こる症状には疼痛を伴う屈筋および伸筋痙攣、亢進したあるいは過剰の深部腱反射、クローヌス、筋肉の虚弱化、疲労、器用さの損失、全般的な運動機能の様々な程度の損失、麻痺及び睡眠障害などが含まれる。
【0031】
痙性にみられる病理学的症状は、一般的にみられる局部的な外部からの傷害によって特定の筋肉に起こる、よくある筋肉の急性の痛み、緊張および捻挫、すなわち、CNS外部または末梢における症状、とは生理学的レベルで基本的に異なる。これらの病理学的症状はまた血管痙縮、膀胱痙縮および気管支痙縮のような比較的一般的に起こる不随意の平滑筋の痙縮とも異なる。このような非痙縮性(non-spastic, CNS性でない)、末梢性のまたは局所性の症状は通常、いわゆる「鎮痙薬」または「痙縮緩解薬」によって治療されるが、これらの薬剤は一般的に痙性の治療には有効ではない。GOODMAN AND GILMAN'S「治療薬の薬理学的基礎」(THE PHARMACOLOGICAL BASIS OF THERAPEUTICS)、第8版、「パーキンソン病、痙性および急性痙攣の治療薬(Drugs for Parkinson's Disease, Spasticity and Acute Muscle Spasms)」Cedarbaum および Schleifer。(以降GOODMAN AND GILMAN'Sと掲載)、ページ 463-484 ページ(Pergamon Press 1990年)。
【0032】
本発明によって用いられる組成物及び医薬製剤処方および組成は、筋緊張の中枢を介した軽減に有効であり、痙性による症状または副作用の一つ以上についての急性または慢性の緩和に有効である。これに関連して「痙性」は、これらに限定されるものではないが、疼痛を伴う屈筋または伸筋の痙攣、亢進されたあるいは過剰の深部腱反射、過反射、器用さの損失、筋肉の虚弱化、過剰な腱反射およびクローヌスなどの症状に表れる骨格筋の増強した緊張を意味する。本明細書における「抗痙性剤」とは、以下の痙性による症状または副作用の少なくとも一つを緩和することで示される痙性の症候の治療に有効な組成を意味する:それらの症状とは、疼痛を伴う屈筋または伸筋の痙攣、亢進したあるいは過剰の深部腱反射、反射亢進、器用さの喪失、筋肉の虚弱化、過剰な腱反射およびクローヌス、であり、あるいは治療はこれらの症状の発現や副作用の頻度の低減を含む。
【0033】
従って、本明細書の痙性の「緩和」とは、これらに限定されないが、疼痛を伴う屈筋または伸筋の痙攣、亢進されたあるいは過剰の深部腱反射、反射亢進、器用さの損失、筋肉の虚弱化、過剰な腱反射およびクローヌスなどの痙性の症状の一つ以上の軽減あるいはこれらの症状または副作用の回数の減少を意味する。
情緒障害:情緒障害には、例えば、躁病、総合失調感情障害、脳外傷性攻撃性、心的外傷後ストレス障害、双極性感情障害、パニック症状および行動調整不良症候群のような抑鬱症から不機嫌躁病まで含まれる。「J.感情障害」(J. Affective Disorders) 8:243-250 (1985)、 Emrich ら、 および 「感情性障害における抗痙攣剤(ANTICONVULSANTS IN AFFECTIVE DISORDERS)」 14-32 ページ、Bernasconi ら (Excerpta Medica 1984)。本発明による新規化合物および医薬製剤処方および組成は、これらの疾患、障害及び症状の治療に有効であり、またこの治療薬分類における現存の治療薬に比較して改善された副作用プロフィールを示す。
【0034】
神経障害性疼痛症候群:この「神経障害性疼痛」に分類される症状は、脳卒中、外傷、多発性硬化症および糖尿病のような脳または脊髄の障害で苦しむ多くの患者に影響を与えている。「痛みと中枢神経系疾患(PAIN AND CENTRAL NERVOUS SYSTEM DISEASE)」、 (Raven 1991)、 Casey。多様な疼痛症状の治療のための抗痙攣薬の使用は、広く文献で報告されている。J. Clin. Neuropharmacol. 7、Swendlow:51-82 (1984). したがって、本発明の新規化合物および医薬製剤処方および組成は、神経障害性疼痛の緩和に同じように適用することが可能である。
頭痛:片頭痛 [Hering と Kuritzky, Cephalagia 12: 81-84 (1992)]、群発性頭痛 [Hering とKuritzky, loc. cit. 9:195-198 (1989)]および慢性頭痛といった頭痛 [Mathew とSabiha, Headache 31: 71-74 (1991)] は抗痙攣剤によって治療されてきた。従って本発明の組成物および医薬製剤処方は、これらのタイプの頭痛の症状の緩和に、現存の治療にみられる有害な副作用なしで使用することが可能である。
【0035】
静止不能症候群:身体的な(非精神性の)動揺だけでなく不随意の四肢の動きを特長とする「静止不能症候群」とは、体性の(非精神性の)静止不能症状が特徴であり、感情からは独立したもので、従って、精神的不穏症とは本質的に区別される。[Sachev ら、 Austral. New Zealand J. Psychiatry30:38-53 (1996)を参照]。
【0036】
静止不能症候群は多くの症状を含め、多くの器質性および非器質性精神疾患との関連性が見られる。例えば、薬剤起因性錐体外路症状のような薬剤に起因する静止不良症状(遅発性、慢性および離脱性アカシジア)は、神経遮断薬治療の最も一般的な副作用の一つである。静止不能症候群類には、頭部や脊髄外傷、および脊髄の病巣に関連する可能性のある病変である、いわゆる「下肢静止不能症候群」および「睡眠時周期性下肢運動障害」が含まれる。特発性下肢静止不能症候群は、多様な臨床症状発現を伴う常染色体の優性遺伝から起こる。O'Keefe, Arch. Intern. Med. 156: 243-248 (1996); Danek ら、 「神経学的障害:経過と治療(NEUROLOGICAL DISORDERS: COURSE AND TREATMENT)」、819-823 ページ (Academic Press 1996); Mellick や Mellick、Neurology 45(補足): 285-286 (1995)を参照。本発明は、最低限の副作用の静止不能症候群の有効な治療を提供する。
【0037】
運動障害:パーキンソン病、ハンチントン舞踏病、アルツハイマー病、遅発性ジスキネディア、ステッフマン症候群などの運動障害を軽減する多様な薬剤が知られている。Lloyd および Morselli、「精神薬理学:三代目の進歩(PSYCHOPHARMACOLOGY: THE THIRD GENERATION OF PROGRESS )」(Raven Press 1987)。本発明の治療は、運動障害の一つ以上の症状を緩和する。
【0038】
本発明の化合物は、不安軽減薬(抗不安薬)としても有効である可能性がある。
「神経障害または疾患」とは、これらに限定されないが、癲癇、不安症、多発性硬化症、脳卒中、頭部外傷、脊髄傷害およびパーキンソン病およびハンチントン病、アルツハイマー病および筋萎縮性側索硬化症のような慢性神経変性疾患を含む神経系の障害および疾患を意味する。さらに「神経障害または疾患とは、抗痙性剤または抗痙攣剤による治療が適用され、有効性を示し、推奨されるおよび/または処方される疾患または症状を意味する。
【0039】
「神経変性疾患」とは、これらに限定されないが、ハンチントン病、パーキンソン病、アルツハイマー病および筋萎縮性側索硬化症(ALS)のような疾患を意味する。
「抗痙攣剤」とは全身性強直・間代発作、欠神発作、単純部分発作、複雑部分発作、二次性全般性部分発作、癲癇重積状態および頭部外傷や外科手術後の外傷起因性痙攣などの症状に発症し、観察され、見られる痙攣の重症度、回数や期間を軽減することのできる化合物を意味する。
【0040】
「抗痙攣作用」とは全身性強直・間代発作、欠神発作、単純部分発作、複雑部分発作、二次性全般性部分発作、痙攣重積状態および頭部外傷や外科手術後の外傷起因性痙攣のような症状に起こる、観察される、みられる痙攣発作の重症度および回数や期間を軽減する効果を意味する。
【0041】
「治療用量」とは、患者において、疾患や症状の一つ以上をある程度緩和する化合物の量を意味する。さらに、「治療用量」は、疾患または症状に付随する生理学的または生物化学的パラメタを部分的にまたは完全に正常値に戻す量を意味する。一般的に、患者の年齢、体格および疾患によって0.1-15-20-30 mg/kg体重の量である。投薬回数は1日1回から4回であっても良い。
【0042】
「医薬組成分」とは、本発明の化合物を溶解させるまたは化合物の投与を促進するために添加する医薬品として許容できる担体中に含まれる治療有効量の本発明の化合物を意味する。医薬品として許容できる担体の例には、水、生理食塩水、生理学的緩衝食塩水が含まれる。これらの医薬品組成物は、適切な投与量で提供される。これらの組成物は一般に特定の疾患の治療への使用をFDAまたは米国外の同等の機関によって承認されたものである。
【0043】
本発明の上記の化合物のあるものは一つ以上の螺旋対象性の立体中心を有することが知られている。そのような化合物は、ラセミ(あるいはジアステレオ異性の)混合体、R およびSエナンチオマー混合体(あるいはジアステレオマー)または純粋なエナンチオマー(RまたはS)(あるいはジアステレオマー)として、好ましい生物学的活性を示す場合がある。 ある純粋なエナンチオマーが好ましい生物学的活性を示す場合、この好ましいエナンチオマーはユートマーと呼ばれ、より好ましくない、生物学的活性がより低いエナンチオマーはジストマーと呼ばれる。
【0044】
医薬製剤処方および組成分の調製方法と投与方法:本明細書に示されるように、本発明の有用な医薬製剤処方および組成は神経系障害または疾患の治療に使用することができる。これらの製剤は、一般に人の患者の治療に使用されるが、その他の霊長類、飼育動物、豚、牛および家禽などの家畜動物や馬、犬や猫などのなどスポーツ動物やペットなどの他の脊椎動物の同様な、あるいは同じ疾患の治療に使用することも可能である。
【0045】
本発明は、また上記の二つ以上の活性化合物の組み合わせを含む医薬製剤処方や組成分にも向けられる。本発明の化合物は、既知の方法に従って医薬製剤として有効な組成を調製(処方)することが可能であり、それによって活性成分は医薬品として許容できる一つのあるいは複数の担体と組み合わされる。例えば、Gennaro (Ed.)、「レミントンの薬理科学(REMINGTON'S PHARMACEUTICAL SCIENCES)」、第18版 (Mack Publishing Co., Easton, PA, 1990) および「グッドマンとギルマンの治療薬の薬理学的基礎(GOODMAN AND GILMAN'S THE PHARMACOLOGICAL BASIS OF THERAPEUTICS)」を参照のこと。化合物および/または組成は、投与される患者が認容できる場合には「医薬品として許容できる担体」中に含まれるべきであるとされる。滅菌リン酸緩衝生理食塩水は医薬品として許容できる担体の1例である。その他の適切な担体(例えば、生理食塩水およびリンガー液)は、当業者には周知である(以下を参照)。
【0046】
医薬品として許容できる担体には、投与して患者が認容できる適切な賦形剤および/または補助剤が含まれる。先行技術にて既知の医薬品として許容な担体には、カルボン酸カルシウム、リン酸カルシウム、硫酸カルシウム、ショ糖、デキシトロース、ラクトース、フルクトース、キシリトール、ソルビトール、デンプン、デンプンペースト、セルロース誘導体、ゼラチン、ポリビニルピロリドン、塩化ナトリウム、デキストリン、ステアリン酸、ステアリン酸マグネシウム、ステアリン酸カルシウム、植物オイル、ポリエチレングリコール、滅菌リン酸緩衝生理食塩水、生理食塩水、リンガー液およびこれらの混合物が含まれるがこれらに限定されるものではない。例えば、「レミントンの薬理学的科学(REMINGTON'S PHARMACEUTICAL SCIENCES)」、同上、を参照。
【0047】
米国食品医薬品局(FDA)によって市販が承認された医薬品として許容できる有機酸(アミノ酸など)の塩には、ナトリウム、カリウム、リチウム、亜鉛、アルミニウム、カルシウムおよびマグネシウムの塩が含まれる。「レミントンの薬理学的科学(REMINGTON'S PHARMACEUTICAL SCIENCES)」、 第18版、1445ページ (Mack Publishing Co. 1990) を参照。
【0048】
本発明の化合物およびその医薬品製剤組成は先行技術で調剤できる。例えば、本発明の化合物は、医薬品として許容できる担体と組み合わせて望ましい剤形処理をして良い。本発明の医薬製剤組成は、本発明の医薬品製剤組成および医薬品として許容できる担体を共に含む、従来の混合、溶解、顆粒化、糖衣錠作成、磨砕、乳化、カプセル封入、封入または凍結の工程のような自明の方法で生成または製造してもよい。
【0049】
一般的には、本明細書に記載される上記の化合物の投与量、処方および組成は、患者の年齢、体重、身長、性別、健康状況および既往歴などの因子によって異なる。治療を目的として、本発明の化合物および医薬品として許容できる担体は、治療の必要な患者に治療有効量が投与される。活性成分および担体の組み合わせ(処方または組成)は、投与量が生理学的に有意な量であれば、「治療有効量」を投与するとされる、。医薬製剤組成は、投与された患者に、結果として生理学的に検出可能な変化がある場合には、生理学的に有意である。例えば、このような文脈からすると、抗痙攣活性組成を投与することによって癲癇発作および/または痙攣のような癲癇の症状の一つ以上が緩和される場合は、本組成は生理学的に有意である。さらに、用量およびおそらく投与回数も各患者の年齢、体重および反応に応じて異なってくる。上記に考察されたような同等の計画は獣医学的薬剤においても用いることが可能である。
【0050】
本発明の化合物は、例えば、腸溶性錠剤、カプレット、ジェルカップ、散剤、あるいはカプセル剤などの固形剤形またはシロップやエリキシル剤などの液状剤形によって経口的に投与され得る。個体経口投与単位剤形は、錠剤またはカプセル中に1日最大2回の投与により、1回に1〜2個の服用ですむような活性成分量が含まれることが好ましい。液状製剤もまた、1回の投与用量あたり小さじ1〜2杯となる量の活性成分が含まれるようにすることが可能である。さらに、相当する低用量の小児用の咀嚼可能および液体経口投与剤形を調製し投与することも可能である。これらの化合物は、経口投与用に、滴剤(「濃縮」調剤の滴下用から)を食品や飲料へ添加することも可能である。さらに、本発明の上記化合物を経口送達と吸収を促進させるためにチューイングガムに入れた剤形で調剤することも可能である。本発明の処方や組成に使用される各化合物の適切な用量は、当業者には前述の説明で明らかとなり得る。
【0051】
あるいは、本発明の上記の化合物は、注射または経皮投与または、例えば、経鼻、舌下、経頬、経膣投与などの経粘膜投与、あるいは座薬などの経腸投与などのその他の全身投与経路によって投与されることも可能である。その他の投与経路(例えば、獣医学的適用に有用な)には、腸内や非経口投与があり、筋肉内、皮下および/または脊髄内投与ばかりでなくクモ膜下腔内、脳室内、静脈内、腹腔内、鼻腔内または眼内注射などが含まれる。しかしながら、経口投与が非常に便利であるために好ましい。
【0052】
本発明はこのように、経口、非経口、経皮、経粘膜、鼻腔内、舌下、経頬または経直腸投与に適する多様な化合物を意図している。本発明の化合物は他の薬理学的な活性成分と組み合わせて、さらに新規な医薬製剤組成を調製し得ることも理解されている。
【0053】
治療に関連の、治療的に価値のある活性の実証:上述のように、症状の緩和のためのある医薬製剤処方や組成の適合性および治療有効性は、例えば米国特許番号第6,589,994、6,617,358および7,265,155号に記載され、本明細書にその全体が組み込まれるが、動物モデルの使用や、試験およびスクリーニング法を使用して実証することが可能である。
【0054】
上記の本発明の化合物の治療有効性は、一般的な毒性の無さと併せて、本発明の化合物を上述の症状の治療のための理想的な薬剤とする。このような背景に基づいて、本発明は以下の実施例を参照することで当業者にはさらに容易に理解されると思われるが、それらの図示された実施例は図解の目的で提供されるもので、本発明の範囲を限定する意図はない。
【0055】
実施例
実施例1 化合物A[3−(4−クロロフェニル)−3−メチルブチルアミド]の調製。
【0056】
【化4】
【0057】
3−(4−クロロフェニル)−3−メチル酪酸(6.1g、41.9mmol) 溶液をCH2Cl2(100mL)およびDMF(0.2mL)溶液中で、塩化オキサリル(5.2mL、7.45mmol)を使って0℃窒素静圧下で処理した。反応液を窒素下で1晩室温で撹拌した。減圧下で過剰なジクロロメタンを除去した。入手した残渣をトルエン(50mL)で共沸蒸留した。
【0058】
アンモニア(ガス)を泡化させて無水THF(100mL)中の前記酸塩化物[3−(4−クロロフェニル)−3メチルブチリルクロリド]溶液に5℃で15分間通した。反応混合物を静圧窒素下で1晩室温で撹拌した。
【0059】
白色沈殿物(塩化アンモニウム)を濾過しTHF(100mL)で洗浄した。濾過液および洗浄液を合わせて減圧下で蒸発させた。その結果入手した白色固体をエチルアセテートに再溶解させた(300mL)。エチルアセテート層をH2O、1.0MのHCL、炭酸水素ナトリウム飽和溶液および塩水で洗浄した。次にエチルアセテート溶液を硫酸マグネシウム上で乾燥させ、濾過し、減圧下で蒸発させた。その結果得られた白色固体を冷却したジエチルエーテルおよびヘキサン(50:50)溶液を加えて粉末にした。この結果4.22gの白色の剥片[3−(4−クロロフェニル)−3−メチルブチルアミド]が得られた(収率69%)。GC/MS分析によりこの物質は100%純粋であった。1H NMR 分光法によって生成物の構造と一致するシグナルが得られ、98%以上の純度が示された。
【0060】
実施例2 化合物B[3−(4−クロロフェニル)−3,N−ジメチルブチルアミド]の調製。
【0061】
【化5】
【0062】
3−(4−クロロフェニル)−3−メチル酪酸(5.95g、28mmol) をCH2Cl2(100mL)およびDMF(0.2mL)溶液中で塩化オキサリル(5.2mL、7.45mmol)によって0℃窒素静圧下で処理した。反応液を窒素下で1晩室温で撹拌した。減圧下で過剰なジクロロメタンを除去した。入手した残渣をトルエン(50mL)を使って共沸蒸留した。
【0063】
残渣を150mLの乾燥THFに溶解させ、メチルアミン溶液(THFの2.0M溶液、45mL、84mmol)で5℃で処理した。反応混合物を静圧窒素下で1晩室温で撹拌した。
白色沈殿物を濾過しTHF(100mL)で洗浄した。濾過物および洗浄液を合わせて減圧下で蒸発させた。その結果入手した白色固体をジエチルエーテル中で再溶解させた(300mL)。エーテル層をH2O、1.0M HCL、炭酸水素ナトリウム飽和溶液および塩水で洗浄した。次にエーテル溶液を硫酸マグネシウム上で乾燥させ、濾過し、減圧下で蒸発させた。その結果得られた白色固体を冷却したジエチルエーテルおよびヘキサン(50:50)溶液を加えて粉末とした。この結果5.46gの白色の剥片[3−(4−クロロフェニル)−3,N−ジメチルブチルアミド]が得られた(収率86%)。 GC/MS分析によりこの物質は100%純粋であった。 1H NMR 分光法によって生成物の構造と一致するシグナルが得られ、98%以上の純度が示された。
【0064】
実施例3 化合物C [(R)-3-フェニルブチルアミド]の調製。
【0065】
【化6】
【0066】
(R)-3−フェニル酪酸(4g、24.36mmol)の CH2Cl2(75mL)およびDMF(0.1mL)溶液を塩化オキサリル(3.0mL、34.0mmol)で0℃、窒素静圧下で処理した。反応液を窒素下で室温にて1晩撹拌した。減圧下で過剰なジクロロメタンを除去した。入手した残渣をトルエン(50mL)を使って共沸蒸留した。
【0067】
アンモニア(ガス)を泡化させて酸塩化物[(R)-3−フェニルブチリルクロライド] の無水THF(100mL)溶液に5℃で15分間通した。反応混合物を静圧窒素下で1晩室温で撹拌した。
【0068】
白色沈殿物(塩化アンモニウム)を濾過しTHF (100mL)で洗浄した。濾過物および洗浄液を合わせて減圧下で蒸発させた。その結果入手した白色固体をエチルアセテートに再溶解させた(300mL)。エチルアセテート層をH2O、1.0M HCL、炭酸水素ナトリウム飽和溶液および塩水で洗浄した。次にエチルアセテート溶液を硫酸マグネシウム上で乾燥させ、濾過し、減圧下で蒸発させた。粗物質をBiostage SP4システム(カラムSi 40+M 0344−1,95:5, CH2Cl2: MeOH)を使用して精製した。 その結果得られた灰色がかった白色の固体を冷却したジエチルエーテルおよびヘキサン(50:50)溶液で共沸蒸留した。2.9gの白色固体[(R)-3-フェニルブチルアミド]を入手した(収率73%)。GC/MS分析によりこの物質は100%純粋であった。 1H NMR 分光法によって生成物の構造と一致するシグナルが得られ、98%以上の純度が示された。
【0069】
実施例4 化合物D[3−(3−フルオロフェニル)ブチルアミド]の調製。
【0070】
【化7】
【0071】
500mL容量の三つ口丸底フラスコを使って、水素化ナトリウム(オイル中60%、1.1eq.80mmol、3.20g)をN,N-ジメチルホルムアミド(DMF、100mL)に懸濁させ、、窒素下でトリエチルホスホノアセテートのDMF(50 mL)溶液(1.2eq.87mmol、19.50g)を滴下して処理した。添加後に反応混合液を水素化ナトリウムが目視で確認できなくなるまで水浴(100℃)を使って加熱した(30分間)。混合液を室温まで冷却し、次に3’−フルオロアセトフェノン(1.0eq.72.4mmol、10g)のDMF溶液(50mL)で処理した。反応混合液を室温で2時間撹拌し、1mLの分画を取り出し水中(〜2mL)で冷却した。ここへジエチルエーテル(〜2mL)を添加し、混合液を平衡化させた。GC/MS分析によって有機層を分析したところ、最初のベンゾフェノンは完全に消費されていた。その結果として、反応混合液に水を添加して冷却した。混合液を大きな丸底フラスコへ移し回転蒸発装置で溶媒のほとんどを除去した。混合液を冷却し[a]ジエチルエーテル(500mL)および水(250mL)を使用して分液ロートへ移した。混合液を平衡化させ水層を除去した。有機層をさらに3回水で洗浄した(3x250mL)。この溶液のGC/MS分析により単一の生成物が得られたことが示された(ホスホノアセテートの残留は無かった)。有機溶液を無水MgSO4上で乾燥させ、濾過および濃縮により18.09gの粗物質(水素化ナトリウムからのオイルを含む)を入手した。
【0072】
【化8】
【0073】
粗[3−(3−フルオロフェニル)−ブタ−2−エン酸エチルエステル]溶液(9g、0.015mmol)をPd/C(10%、450mg)でメタノール(75mL)中で処理した。反応混合液を45psiで1時間水素化した。反応混合物を炭素上のパラジウムを除去するためCeliteプラグを通過させた。減圧下で濾過液を濃縮した。この結果無色のオイル[3−(3−フルオロフェニル)酪酸エチルエステル]10.1gを入手した。GC/MS分析によってこの物質が94%純粋であることを確認した。本生成物を精製(鉱物油をの除去のための)しないで使用した。
【0074】
【化9】
【0075】
[3−(3−フルオロフェニル)−酪酸エチルエステル](10.1g、48mmol)のエタノール(50mL)粗溶液を10M NaOH溶液(50mL、857mmol)で処理した。反応混合液を一晩還流した。エチルアルコールを除去するために減圧下で反応混合液を乾燥させた。結果として入手した残渣を水150mLで再溶解させた。混合液を冷却し、ジエチルエーテル(200mL)および水(50mL)を使用して分液ロートへ移した。混合液を平衡化させ、エーテル層を除去した。HCL溶液(pH〜2)を使って、水層を酸性化しジエチルエーテル(300mL)で抽出した。次に有機層を硫酸マグネシウム上で乾燥させ、濾過し、減圧下で濃縮した。この結果、橙色の粘性オイルである3−(3−フルオロフェニル)酪酸8.1gを入手した(収率92.6%)。GC/MS分析により、この物質は100%純粋であった。
【0076】
【化10】
【0077】
3−(3−フルオロフェニル)酪酸(8.1g、44.46mmol)のCH2Cl2(100mL)とDMF(0.7mL)との溶液を塩化オキサリル(5.43mL、7.9mmol)で0℃窒素静圧下で処理した。反応液を窒素下で1晩室温で撹拌した。減圧下で過剰なジクロロメタンを除去した。入手した残渣をトルエン(70mL)を使って共沸蒸留した。
【0078】
アンモニア(ガス)を泡化によって酸塩化物[3−(3−フルオロフェニル)ブチリルクロライド]の無水THF(100mL)溶液に5℃で15分間通した。反応混合物を静圧窒素下で1晩室温で撹拌した。
【0079】
白色沈殿物(塩化アンモニウム)を濾過しTHF(200mL)で洗浄した。濾過液および洗浄液を合わせて減圧下で蒸発させた。その結果入手した白色固体をエチルアセテートに再溶解させた(350mL)。エチルアセテート層をH2O、1.0M HCL、炭酸水素ナトリウム飽和溶液および塩水で洗浄した。次にエチルアセテート溶液を硫酸マグネシウム上で乾燥させ、濾過し、減圧下で蒸発させた。その結果得られた白色固体を冷却したジエチルエーテルおよびヘキサン(50:50)溶液を用いて粉末にした。この結果、乳白色の粉末3−(3−フルオロフェニル)ブチルアミド 6.8gを入手した(収率84%)。GC/MS分析によりこの物質は100%純粋であった(RおよびSの鏡像異性体の混合物)。 1H−NMR 分光法によって生成物の構造と一致するシグナルが得られ98%以上の純度が示された。
【0080】
実施例5−14 化合物E−Nの調製
対応するアセトフェノン(以下の表1)から上述の実施例4の化合物Dの調製方法を使って化合物E−Nを調製した。さらに、化合物M(実施例13)および化合物N(実施例14)を対応するアミン、すなわち各々メチルアミンおよびジメチルアミン、から調製した。すべての最終生成物はGC/MS分析により100%純粋であった。1H−NMR 分光法によって各々の最終生成物の構造と一致するシグナルが得られ98%以上の純度が示された。
【0081】
【表1】
【0082】
実施例15 化合物O[3−(4−シアノフェノキシ)ブチルアミド]の調製。
【0083】
【化11】
【0084】
水酸化ナトリウム4g(0.1mol)を水(100mL)に溶かした溶液、および4−シアノフェノール11.9g(0.1mol)を還流させ15分間加熱した。βブチロラクトン(8.6g 0.1mol)を還流液へ15時間以上にわたって添加した。次に反応液を室温に冷却した。反応液をジエチルエーテル(200mL)および水(200mL)を使って分液ロートへ移した。混合液を平衡化し、エーテル層を除去した。HCL溶液(pH−2)を使って、水層を酸性化しエチルアセテート(300mL)で抽出した。エチルアセテート層を硫酸マグネシウム上で乾燥させ、濾過し、減圧下で濃縮し粗生成物10.78gを入手した。
【0085】
粗生成物をBiostage SP4システム(カラムSi 65i、9:1 CH2Cl2: MeOH)を使用して精製し、淡黄色の粘性オイル[3−(4−シアノフェノキシ)酪酸]9.87gを入手し、室温で放置し固化した。GC/MS分析によりこの物質は96%純粋であった。本化合物はさらに精製しないで使用した。
【0086】
【化12】
【0087】
3−(4−シアノフェノキシ)酪酸の粗溶液(10.8g、52.6 mmol)をCH2Cl2 (100mL)およびDMF(0.2mL)溶液中で塩化オキサリル(6mL、68.4mmol)によって0℃窒素静圧下で処理した。反応液を窒素下で1晩室温で撹拌した。減圧下で過剰なジクロロメタンを除去した。
【0088】
アンモニア(ガス)を泡化させて無水CH2Cl2(150mL)中の酸塩化物[3−(4−シアノフェノキシ)ブチリルクロライド]溶液に5℃で15分間通した。反応混合物を静圧窒素下で1晩室温で撹拌した。
【0089】
白色沈殿物(塩化アンモニウム)を濾過し、CH2Cl2(100mL)で洗浄した。濾過液および洗浄液を合わせて減圧下で蒸発させた。その結果入手した白色固体をエーテルに再溶解させた(250mL)。エーテル層をH2O、HCL 1.0M、炭酸水素ナトリウム飽和溶液および塩水で洗浄した。次にエーテル溶液を硫酸マグネシウム上で乾燥させ、濾過し、減圧下で蒸発させた。粗物質をBiostage SP4システム(カラムSi 40+M0344−1,95:5, CH2Cl2: MeOH) を使用して精製した。 この結果、灰色がかった白色の固体3−(4−シアノフェノキシ)ブチルアミド 2.987gを入手した(収率29%)。 GC/MS分析によりこの物質は97%純粋であった。 1H NMR 分光法によって製品の構造と一致するシグナルが得られ98%以上の純度が示された。
【0090】
実施例16 化合物P[1R,2R]-トランス−2−フェニルシクロプロパン−1−カルボキシアミド]の調製
【0091】
【化13】
【0092】
(1R,2R)−トランス-2−フェニルシクロプロパン−1−カルボン酸(2.1g、12.8 mmol) をCH2Cl2 (50mL)およびDMF(0.2mL)の溶液中で塩化オキサリル(1.5mL、16.7 mmol)で0℃窒素静圧下で処理した。反応液を窒素下で1晩室温で撹拌した。減圧下で過剰なジクロロメタンを除去した。
【0093】
アンモニア(ガス)を泡化させながら酸塩化物[(1R,2R)−トランス-2−フェニルシクロプロパン−1−カルボン酸クロライド]の無水CH2Cl2溶液に5℃で15分間通した。反応混合液を室温で一夜、窒素下に攪拌した。
【0094】
反応混合物を減圧下で蒸発させ、その結果入手した残渣をエチルアセテート/水の混合液中に再溶解させた。混合液をエチルアセテート(100mL)および水(60mL)を使用して分液ロートへ移した。混合液を平衡化し水層を除去した。有機層を1.0 M のHCl (10 mL)、H2O (70 mL)および塩水で連続して洗浄した。 有機層を無水硫酸マグネシウム上で乾燥させ、濾過し、減圧下で余剰な溶媒を蒸発させた。その結果入手した薄茶色の固体をBiostage SP4システム(カラムSi 40+S 90:10 CH2Cl2: MeOH) を使用して精製し、白色粉末[(1R,2R)-トランス−2−フェニルシクロプロパン−1−カルボキシアミド]1.127gを入手した(54%の収率)。GC/MS分析によりこの物質は100%純粋であった。1H−NMR 分光法によって製品の構造と一致するシグナルが得られ98%以上の純度が示された。
【0095】
実施例17 化合物Q[(1R,2R)-2−フェニルシクロプロパン−カルボン酸―((S)−1―カルバモイル−3−メチルブチル)アミド]の調製
【0096】
【化14】
【0097】
(1R,2R)−トランス-2−フェニルシクロプロパン−1−カルボン酸(0.905g、5.6 mmol) を CH2Cl2 (30mL)およびDMF(0.05mL、16.7mmol)の溶液中で塩化オキサリル(0.65mL、7.23 mmol)で0℃窒素静圧下で処理した。反応液を窒素下で1晩室温で撹拌した。減圧下で過剰なジクロロメタンを除去した。
【0098】
酸塩化物[(1R,2R)−トランスー2−フェニルシクロプロパンー1−カルボン酸クロライド]のCH2Cl2 (50mL)溶液 を H−Leu−NH2[L−ロイシンアミド、(S)―2−アミノー4―メチル−n―バレルアミド](0.761g、5.8mmol)およびトリエチレンアミン(1.13g、11.1mmol)のCH2Cl2(60mL)溶液に0℃で滴下して加えた。反応混合物を窒素下で1晩室温で撹拌した。
【0099】
反応混合物を減圧下で蒸発させ、その結果入手した残渣をエチルアセテート/水の混合液に再溶解させた。混合液をエチルアセテート(80mL)および水(50mL)を使用して分液ロートへ移した。混合液を平衡化し水層を除去した。有機層を1.0 M HCl(20 mL)、H2O(90 mL)および塩水(120mL)で連続して洗浄した。次に有機層を無水硫酸マグネシウム上で乾燥させ、濾過し、減圧下で余剰な溶媒を蒸発させた。その結果入手した橙茶色の固体をBiostage SP4システム(カラムSi 40+M 90:10 CH2Cl2: MeOH) を使用して精製し、白色粉末[(1R,2R)−2−フェニルシクロプロパンカルボン酸((S)−1−カルバモイル−3−メチルブチル)−アミド]0.365gを入手した(24%の収率)。GC/MS分析によりこの物質は100%純粋であった。1H−NMR 分光法によって製品の構造と一致するシグナルが得られ98%以上の純度が示された。
【0100】
実施例18 化合物U[2−[1−(4−メトキシフェニル)シクロプロピル]−アセタミド]の調製。
【0101】
【化15】
【0102】
水素化リチウムアルミニウム(0.211mol)を無水エーテル(200mL)中に撹拌した懸濁液を1−(4−メトキシフェニル)−1−シクロプロパンカルボン酸(0.1406mol)で0℃でエーテル100mL中で処理する。次に反応混合物を窒素下で1晩室温で撹拌する。反応混合液を脱イオン水100mLを滴下し冷却する。混合液を濾過し、ケーキ状の固体をジエチルエーテル(1L)で洗浄する。濾過した混合液(エーテルおよび水)を分液ロートへ移す。有機層を水層から分離し塩水で洗浄する。次にエーテル層を硫酸マグネシウム上で乾燥させ、濾過し、室温減圧下で濃縮させる。その結果、[1−(4−メトキシフェニル)シクロプロピル]‐メタノールを入手する。
【0103】
[1−(4−メトキシ−フェニル)シクロプロピル]メタノール(0.074mol)の未希釈液を三臭化リン(0.081mol)を静圧窒素下に0℃で滴下して処理する。反応液を130℃まで加熱し、その温度を6時間維持する。反応混合液を室温まで冷却し橙色の沈殿物を濾過する。橙色の沈殿物をジエチルエーテル200mLで洗浄する。水150mLおよびジエチルエーテル200mLを使用して濾液を分液ロートへ移す。混合液を平衡化し水層をジエチルエーテル200mLでもう1回抽出する。エーテル抽出物およびエーテル洗浄液を合わせて、炭酸水素ナトリウム飽和溶液および食塩水で洗浄する。次にエーテル抽出物を硫酸マグネシウム上で乾燥させ、余剰のジエチルエーテルを30℃減圧下で除去する。この結果、1−(1−ブロモメチル−シクロプロピル)−4−メトキシベンゼンを入手する。この粗物質をさらに精製しないで対応するニトリルへ変換させる。
【0104】
【化16】
【0105】
1−(1−ブロモメチル−シクロプロピル)−4−メトキシベンゼン(60.3mmol)のジメチルスルホキシド(60mL)粗溶液をシアン化ナトリウム(180.7mmol)で処理する。反応混合液を、窒素下で1晩95℃で加熱する。反応液を塩水(150mL)およびクロロホルム(300mL)を使用して分液ロートへ移す。反応混合液を平衡化し水層を除去する。さらに2回クロロホルム(2x300mL)を使って水層を抽出する。有機抽出液を合わせて無水硫酸マグネシウム上で乾燥させ、濾過し、減圧下で濃縮させ[1−(4−メトキシフェニル)シクロプロピル]アセトニトリルを入手する。この粗物質をさらに精製しないで次の段階で(ニトリルの対応するアミドへの加水分解)使用する。[または、本物質から、酸加水分解(例えば、 硫酸を使用)によって対応するカルボン酸を入手できる。]
[1−(4−メトキシ−フェニル)シクロプロピル)アセトニトリル(60.4mmol)のDMSO(75mL)溶液をH2O2(50%w/w)(434mmol)および炭酸カリウム(121mmol)を使って0°Cで処理する。反応混合液を室温で週末中、撹拌する。反応混合液を水(100mL)およびCH2Cl2(200mL)を使用して分液ロートへ移す。反応混合液を平衡化しCH2Cl2層を除去する。水層をさらに2回CH2Cl2(2x300mL)で抽出する。CH2Cl2抽出液を合わせて、水で5回連続して洗浄した後(5x200mL)、塩水(500mL)で洗浄し、無水硫酸マグネシウム上で乾燥させ、濾過し、減圧下で濃縮して、2−[1−(4−メトキシフェニル)−シクロプロピル]―アセトアミドを入手する。
【0106】
実施例19 化合物V[3−(4−クロロフェノキシ)−3−メチルブチル−アミド]の調製。
【0107】
【化17】
【0108】
水酸化リチウムアルミニウム(0.211mol)を無水エーテル(200mL)中で撹拌した懸濁液を2−(4−クロロフェノキシ)−2−メチルプロパン酸(0.1406mol)を使って0℃エーテル100mL中で処理する。反応混合物を窒素下で1晩室温で撹拌する。反応混合液を脱イオン水100mLを滴下し冷却する。混合液を濾過し、ケーキ状固体をジエチルエーテル(1L)で洗浄する。濾過した混合液(エーテルおよび水)を分液ロートへ移す。有機層を水層から分離し塩水で洗浄する。次にエーテル層を硫酸マグネシウム上で乾燥させ、濾過し、室温減圧下で濃縮させる。この結果、2−(4−クロロフェノキシ)−2−メチルプロパン−1−オールを入手する。
【0109】
2−(4−クロロフェノキシ)−2−メチルプロパン−1−オール(0.074mol)の未希釈液を三臭化リン(0.081mol)で静圧窒素下0℃で滴下によって処理する。反応液を130℃まで加熱しその温度を6時間維持する。反応混合液を室温へ冷却し橙色の沈殿物を濾過する。橙色の沈殿物をジエチルエーテル200mLで洗浄する。水150mLおよびジエチルエーテル200mLを使用して濾液を分液ロートへ移す。混合液を平衡化し、水層をジエチルエーテル200mLでもう1回抽出する。エーテル抽出液およびエーテル洗浄液を合わせて、炭酸水素ナトリウム飽和溶液および塩水で洗浄する。次にエーテル抽出物を硫酸マグネシウム上で乾燥させ、余剰のジエチルエーテルを30℃、減圧下で除去する。この結果、1−(2−ブロモ−1,1−ジメチルエトキシ)−4−クロロベンゼンを入手する。1−(2−ブロモ−1,1−ジメチルエトキシ)−4−クロロベンゼンの粗生成物をさらに精製しないで対応するニトリルへ変換する。
【0110】
【化18】
【0111】
1−(2−ブロモ−1,1−ジメチルエトキシ)−4−クロロベンゼン(60.3mol)のジメチルスルホキシド(60mL)粗溶液をシアン化ナトリウム(180.7mmol)で処理する。反応混合液を、窒素下で1晩95℃に加熱する。反応混合液を塩水(150mL)およびクロロホルム(300mL)を使用して分液ロートへ移す。反応混合液を平衡化し水層を除去する。水層をさらに2回クロロホルム(2x300mL)を使って抽出する。有機抽出液を合わせて無水硫酸マグネシウム上で乾燥させ、濾過し、減圧下で濃縮させ3−(4−クロロフェノキシ)−3−メチルブチロニトリルを入手する。この粗物質をさらに精製しないで次の段階で(ニトリルの対応するアミドへの加水分解)使用する。[または、本物質から、酸加水分解(例えば、 硫酸を使用)によって対応するカルボン酸を入手できる。]
(3−(4−クロロフェノキシ)−3−メチルブチロニトリル(60.4mmol)のDMSO(75mL)溶液をH2O2(50%w/w)(434mmol)および炭酸カリウム(121mmol)を使って0°Cで処理する。反応混合液を室温で週末の間、撹拌する。反応混合液を水(100mL)およびCH2Cl2(200mL)を使用して分液ロートへ移す。反応混合液を平衡化しCH2Cl2層を除去する。さらに水層を2回CH2Cl2(2x300mL)を使って抽出する。CH2Cl2抽出液を合わせて水で5回連続して洗浄した後(5x200mL)、塩水(500mL)で洗浄し、無水硫酸マグネシウム上で乾燥させ、濾過し、減圧下で濃縮し、3−(4−クロロフェノキシ)−3−メチルブチルアミドを入手する。
【0112】
実施例20 化合物AG[(R)-3−(4−トリフルオロメチルフェニル)―ブチルアミド]の調製。
【0113】
【化19】
【0114】
アセチルアセトナートビス(エチレン)ロジウム(I)(0.3mmol)、(s)−(−)−2,2’―ビス(ジフェニルホスフィノ)−1,1’−ビナフタレン(0.045mmol)、4−(トリフルオロメチル)フェニルボロン酸(2mmol)、K2CO3 (0.5mmol)および ブタ-2-エン酸アミド(1 mmol)を、磁気撹拌棒、セプタム注入口および還流冷却器のある25mL丸底フラスコへ加える。フラスコをアルゴンでフラッシュし、1,4−ジオキサン(3mL)および脱イオン水(0.5mL)で充填する。得られた反応混合液を100℃で16時間攪拌する。(R)−3−(4−トリフルオロメチルフェニル)ブチルアミドをエチルアセテートで抽出し、塩水で洗浄し無水硫酸マグネシウム上で乾燥させる。シリカゲル上のクロマトグラフィーによって所望の生成物が得られる。
【0115】
実施例21 化合物AA[3−(4−トリフルオロメチルフェニル)―ペンタンアミド]の調製。
【0116】
【化20】
【0117】
ビス(トリメチルシリル)アミドリチウム(1.0M、50mL)の冷却溶液に温度を10℃以下に維持しながらトリメチルホスホノアセテート溶液を滴下した。次に、本溶液を室温まで暖め、さらに5分間撹拌し、4’−(トリフルオロメチル)プロピオフェノンのTHF(25mL)溶液を一度に加える。本溶液をゆっくりと50℃に温め、8時間加熱した。本溶液を室温まで冷却し、10%NH4Cl溶液(100mL)で希釈し、エチルアセテート(2x100mL)で抽出した。有機層を合わせて、硫酸マグネシウム上で乾燥させ、濾過し、濾液をヘキサン/エチルアセテートから白色固体が再結晶するまで濃縮し、5.42gの3−(4−トリフルオロメチルフェニル)ペント−2−エン酸メチルエステル中間体を得た(85%の収率)。
【0118】
3−(4−トリフルオロメチルフェニル)ペント−2−エン酸メチルエステルのTHF/MeOH溶液に水酸化ナトリウム水溶液(15mL)を添加した。その結果得られる水溶液を室温で12時間撹拌し、酢酸(3g)を添加した。次に、溶液を濃縮しオイルを入手した。オイルを酢酸エチル(100mL)へ溶解させ水(3x100mL)で洗浄した。酢酸エチル抽出液を合わせて、硫酸マグネシウム上で乾燥させ、濾過し、濃縮させて半固体を入手し、MeOH/THF(2:1,50mL)へ溶解させ10%Pd/Cと水素圧下50psiで24時間振盪した。TLCにより反応が不完全であったことが示された。10%Pd/Cを追加(500mg)し懸濁液をさらに24時間振盪した。懸濁液を濾過し、濾液を濃縮し半固体を入手した(4.97g)。固体をCH2Cl2(30mL)に溶解し、その結果得られた溶液を0℃まで冷却した。この溶液へ塩化オキサリルを加え、9インチ使い捨てピペットを使ってDMFを1滴添加した。この溶液を8時間撹拌し、濃縮して固体とし、追加のCH2Cl2(30mL)に溶解させた。この溶液を再度濃縮して半固体を得、追加のCH2Cl2(50mL)に溶解させ、得られた溶液を、機械的に撹拌させた冷却(5℃)NH4OH (10 mL) 溶液へ約5分間かけて滴下した。次に懸濁液を濃縮し、粘性のある混合液を入手しエチルアセテート(2x100mL)で抽出した。エチルアセテート抽出液を合わせて、硫酸マグネシウム上で乾燥させ、濾過し、濾液を濃縮し、琥珀色の粗固体を入手しCH2Cl2/THFを使用してシリカゲル(50g)へ吸収させた。この固体をシリカゲル(EtOAc/ヘキサン)のクロマトグラフィーで処理し、2.25gの灰色がかった白色の固体を入手した(37%の収率)。この物質はLC/MSにより100%純粋であった。H-NMR により、物質の構造と一致するシグナルが得られ98%以上の純度が示された。
【0119】
実施例22 化合物AW[3−(4−イソプロピルフェニル)ブチルアミド]の調製。
【0120】
【化21】
【0121】
ビス(トリメチルシリル)アミドリチウム(1.0M、56mL)の冷却溶液(0°C)に温度を10℃以下に維持しながらトリメチルホスホノアセテート溶液を滴下した。次に、本溶液を室温まで暖め、さらに5分間撹拌した後、p−イソブチルアセトフェノンのTHF(25mL)溶液をを一度に加えた。この溶液をゆっくりと65℃まで加熱し30時間還流させた。本溶液を室温まで冷却し、10%NH4Cl溶液(100mL)で希釈し、エチルアセテート(2x100mL)で抽出した。有機層を合わせて、硫酸マグネシウム上で乾燥させ、濾過し、濾液を濃縮して白色固体(4.98g)を得、さらにそれをヘキサン/エチルアセテートから再結晶させて、5.93gの3−(4−イソブチルフェニル)ブタ−2−エン酸メチルエステル中間体を得た(85%の収率)。
【0122】
3−(4−イソブチルフェニル)ブタ−2−エン酸メチルエステルのTHF/MeOH溶液に水酸化ナトリウム水溶液(15mL)を加えた。その結果得られる水溶液を室温で12時間撹拌し、酢酸(3g)を添加した。この溶液を濃縮しオイルを入手した。このオイルを酢酸エチル(150mL)へ溶解させ水(3x100mL)で洗浄した。酢酸エチル抽出液を合わせて、硫酸マグネシウム上で乾燥させ、濾過し、濃縮させて固体(4.98g)を入手し、MeOH/THF(2:1,50mL)に溶解させ10%Pd/Cと水素圧下50psiで24時間振盪した。TLCにより反応が不完全であったことが示された。10%Pd/Cを追加(500mg)し懸濁液をさらに24時間振盪した。この懸濁液を濾過し、濾液を濃縮し固体を入手した(4.75g)。この固体をCH2Cl2(30mL)に溶解し、その結果得られた溶液を0℃に冷却した。この溶液に塩化オキサリルを加えた後、9インチ使い捨てピペットを使ってDMFを1滴添加した。この溶液を6時間撹拌し、濃縮して固体を得、追加のCH2Cl2(30mL)に溶解させた。この溶液を再度濃縮して半固体を得、追加のCH2Cl2(50mL)に溶解させ、得られた溶液を、機械的に撹拌しながら冷却した(5℃)NH4OH (10 mL) 溶液へ約5分間かけて滴下した。次にこの懸濁液を濃縮し、固体/水の混合物を入手しエチルアセテート(2x100mL)で抽出した。エチルアセテート抽出液を合わせて、硫酸マグネシウム上で乾燥させ、濾過し、濾液を濃縮し、粗固体を入手しCH2Cl2/THFを使用してシリカゲル(50g)へ吸収させた。この固体をシリカゲル(EtOAc/ヘキサン)を使ったクロマトグラフィーで処理し、2.5gの灰色がかった白色の固体を入手した(40%の収率)。この物質はLC/MSにより100%純粋であった。. H-NMR により、物質の構造と一致するシグナルが得られ98%以上の純度が示された。
【0123】
実施例23 化合物AE[3−(6−メトキシナフタレン−2−シル)―ブチルアミド]の調製。
【0124】
【化22】
【0125】
ビス(トリメチルシリル)アミドリチウム(1.0M、50mL)の冷却溶液にトリメチルホスホノアセテート溶液を温度10℃以下に維持しながら滴下した。次に、この溶液を室温まで温め、さらに10分間撹拌し、2−アセチル−6−メトキシナフタレンのTHF(20mL)溶液を一度に加えた。この溶液をゆっくりと50℃まで温め、14時間加熱した。この溶液を室温に冷却し、10%NH4Cl溶液(100mL)で希釈し、エチルアセテート(2x100mL)で抽出した。有機層を合わせて、硫酸マグネシウム上で乾燥させ、濾過し、濾液を濃縮し白色固体を得、ヘキサン/エチルアセテートから再結晶して、5.88gの3−(6−メトキシナフタレン−2−イル)ブタ−2−エン酸メチルエステル中間体を得た(92%の収率)。
【0126】
3−(6−メトキシナフタレン−2−イル)ブタ−2−エン酸メチルエステルのTHF/MeOH溶液に水酸化ナトリウム水溶液(15mL)を加えた。その結果得られた水溶液を室温で12時間撹拌し、酢酸(3g)を添加した。この溶液を濃縮し固体残渣を入手した。この固体を酢酸エチル(150mL)に溶解させ水(3x100mL)で洗浄した。酢酸エチル抽出液を合わせて、硫酸マグネシウム上で乾燥させ、濾過し、濃縮させて固体を入手し、MeOH/THF(2:1,50mL)に溶解させ10%Pd/Cと水素圧下50psiで24時間振盪した。TLCにより反応が不完全であったことが示された。10%Pd/Cを追加(500mg)し懸濁液をさらに24時間振盪した。この懸濁液を濾過し、濾液を濃縮し半固体を入手した(4.97g)。この固体をCH2Cl2(30mL)に溶解し、その結果得られた溶液を0℃まで冷却した。この溶液へ塩化オキサリルを加えた後、9インチ使い捨てピペットを使ってDMFを一度に加えた。この溶液を6時間撹拌し、濃縮して固体を得、追加のCH2Cl2(30mL)に溶解させた。この溶液を再度濃縮して半固体を得、追加のCH2Cl2(50mL)に溶解させ、得られた溶液を、機械的に撹拌した冷却(5℃)NH4OH (10 mL) 溶液へ約15分間かけて滴下した。次にこの懸濁液を濃縮し、粘性物質と水の混合液を入手しエチルアセテート(2x100mL)で抽出した。エチルアセテート抽出液を合わせて、硫酸マグネシウム上で乾燥させ、濾過し、濾液を濃縮し、粗固体を入手しCH2Cl2/THFを使用してシリカゲル(50g)に吸収させた。この固体をシリカゲル(EtOAc/ヘキサン)を使ったクロマトグラフィーにより処理し、1.87gの灰色がかった白色の固体を入手した(32%の収率)。LC/MSによりこの物質は100%純粋であった。H-NMR により、物質の構造と一致するシグナルが得られ98%以上の純度が示された。
【0127】
実施例24 化合物AX[3−(2−フルオロ−ビフェニル−4−イル)ブチルアミド]の調製。
【0128】
【化23】
【0129】
冷却した(0℃)2−(2−フルオロ−ビフェニル−4−イル)プロピオン酸のTHF溶液にイソブチルクロロホルメートを加え、TEAを滴下した。その結果得られた白色のスラリーを1時間撹拌しTHF(50mL)で希釈して濾過した。濾過したケーキ状固体を追加のTHF(50mL)で洗浄し、濾液を回転式蒸発器で約50mLまで濃縮した。濃縮された濾液を−20℃で撹拌し、 NaBH4 水溶液(20mL)を15分間かけて滴下した。その結果得られた懸濁液を0℃で2時間撹拌し、水(200mL)で希釈し、エチルアセテート(2x100mL)で抽出した。エチルアセテート層を合わせて、1.0N HCl溶液(100mL)で洗浄し5%炭酸水素塩溶液で洗浄した(100mL)。このエチルアセテート溶液を濃縮しオイル状の残渣である2−(2−フルオロービフェニル−4−イル)プロパノールを入手した(4.47g、95%の収率)。
【0130】
冷却した(0℃)2−(2−フルオロ−ビフェニル−4−イル)プロパノールのCH2Cl2溶液にメタンスルホニルクロライドを加え、TEAを滴下により添加した。その結果得られた白色のスラリーを1時間撹拌し水(200mL)で希釈した。この懸濁液をCH2Cl2 (2 x 100 mL)で抽出した。CH2Cl2層を合わせて、水(2x100mL)および5%NH4OH (100mL)を加えた。CH2Cl2 層を追加の水(200mL)で洗浄し、硫酸マグネシウム上で乾燥させた。CH2Cl2層を濃縮しオイル状の残渣を入手し無水DMF(50mL)に溶解させた。この溶液をNaCNで処理し60℃で14時間撹拌した。TLCにより1個の主要な、より極性の低い溶出生成物(メシレートに比較して)と複数の微量の極性が主要生成物およびメシレートに比較して、より低い溶出生成物が示された。反応を室温まで冷却し水(100mL)で希釈した。この溶液をエチルアセテート(2x100mL)で抽出し、硫酸マグネシウム上で乾燥させ濃縮しオイル状残渣を入手し、シリカゲルを使ったクロマトグラフィー(90% Hex、10%EtOAc)により3−(2−フルオロ−ビフェニル−4−イル)ブチロニトリルをオイルとして入手したが、これはゆっくりと室温で固化した(2.05g、49%の収率)。
【0131】
3−(2−フルオロ−ビフェニル−4−イル)ブチロニトリルのtert-ブチルアルコール溶液に水酸化カリウムの微粉末1.87gを添加した。その結果得られた懸濁液を撹拌し、70℃まで温め、2.5時間加熱し室温まで冷却した。反応懸濁液を1.0N HCl溶液(100mL)で希釈しエチルアセテート(2x100mL)で抽出した。エチルアセテート抽出液を合わせて5%炭酸水素塩溶液(100mL)で、続いて水(100mL)で洗浄した。次に有機物を硫酸マグネシウム上で乾燥させ濃縮して白色固体を得、EtOAc/Hexで数回再結晶させ白色の剥片状のプリズム結晶を入手した(1.62g、75%収率)。
【0132】
実施例25 化合物AY[3−(4−モルホリン−4−イルーフェニル)―ブチルアミド]の調製。
【0133】
【化24】
【0134】
冷却(0℃)したカリウム tert-ブトキシド(1.0M、37.1mL)溶液に温度を25℃以下に維持しながらトリメチルホスホノアセテート溶液を滴下した。次に、この溶液を室温まで温め、さらに5分間撹拌した後、4―モルホリノアセトフェノンのTHF(20mL)溶液を一度に加えた。この溶液をゆっくりと60℃まで温め、36時間加熱した。この溶液を室温まで冷却し、1.0N HCl溶液(100mL)で希釈し、エチルアセテート(2x100mL)で抽出した。有機層を合わせて、硫酸マグネシウム上で乾燥させ、濾過し、濾液を濃縮して白色固体を得、ヘキサン/エチルアセテートから再結晶させて、3.33gの3−(4−モルホリノフェニル)ブタ−2−エン酸メチルエステル中間体を得た(69.9%の収率)。
【0135】
3−(4−モルホリノフェニル)ブタ−2−エン酸メチルエステルのTHF/MeOH溶液(1:1)に水酸化ナトリウム水溶液(15mL)を加えたた。その結果得られた溶液を室温で15時間撹拌し、酢酸(3g)を加えた。この溶液のpHは、6.5であった。この溶液を濃縮しオイルを入手した。オイルを酢酸エチル(150mL)に溶解させ水(3x100mL)で洗浄した。酢酸エチル抽出液を合わせて、硫酸マグネシウム上で乾燥させ、濾過し、濃縮して不定形の固体を入手し、MeOH(50mL)に溶解させ10%Pd/Cを加えて水素圧下50psiで8時間振盪した。TLCにより反応が完了したことが示された。この懸濁液を濾過し、濾液を濃縮し半固体を入手した(2.77g)。この半固体をCH2Cl2(30mL)に溶解させ、その結果得られた溶液を0℃に冷却した。この溶液に塩化オキサリルを加えた後、9インチ使い捨てピペットを使ってDMFを1滴添加した。この溶液を4時間撹拌し、濃縮して固体を得、追加のCH2Cl2(30mL)に溶解させた。この溶液を再度濃縮して半固体を得、、追加のCH2Cl2(50mL)に溶解させ、得られた溶液を、機械的に撹拌させた冷却(5℃)NH4OH (15mL) 溶液へ約5分間かけて滴下した。次にこの溶液を濃縮し、固体/水の混合物を入手しエチルアセテート(2x100mL)で抽出した。エチルアセテート抽出液を合わせて、硫酸マグネシウム上で乾燥させ、濾過し、濾液を濃縮し、粗固体を入手しCH2Cl2/THFを使用してシリカゲル(50g)に吸収させた。この固体をシリカゲル(EtOAc/ヘキサン)のクロマトグラフィーで処理し、2.1gのベージュ色の平板状固体を入手した(46%の収率)。LC/MSにより、この物質は100%純粋であった。. H-NMR で測定し、物質の構造と一致するシグナルが得られ98%以上の純度が示された。
【0136】
実施例26 化合物Q−1[1R,2R]-2−フェニルシクロプロパンーカルボン酸―((S)−1―カルバモイル−プロピル)アミド]の調製。
【0137】
【化25】
【0138】
トランスー2−フェニルシクロプロパンカルボニルクロライドのCH2Cl2 (20mL)溶液をL−2−アミノブタンアミドヒドロクロライド(1.61g、11.6mmol)およびトリエチレンアミン(3.36g、33.2mmol)のCH2Cl2 (60mL)溶液中に、0℃で滴下した。反応混合物を窒素下で1晩室温で撹拌した。
【0139】
この反応混合物を減圧下で蒸発させ、その結果入手した残渣をエチルアセテート/水混合液に再溶解させた。この混合液をエチルアセテート(80mL)および水(50mL)を使用して分液ロートへ移した。混合液を平衡化し水層を除去した。有機層をHCl 1.0 M(20 mL)、 H2O (90 mL)および塩水(120mL)で連続して洗浄した。この有機層を無水硫酸マグネシウム上で乾燥、濾過し、減圧下で余剰な溶媒を蒸発させた。その結果入手した橙茶色の固体をBiostage SP4システム(カラムSi 40+M 90:10 CH2Cl2/ MeOH)を使用して精製し、白色粉末0.365gを入手した(24%の収率)。GC/MSによりこの物質は100%純粋であった。. 1H-NMRで測定し、物質の構造と一致するシグナルが得られ98%以上の純度が示された。
【0140】
図4A-4Oは、合成有機化学文献から引用した様々な化合物および主要中間体(図解 1−15)の合成の追加の実施例を示し、それらから当業者は本発明のさまざまな追加化合物の調製について想像することが可能である。
【0141】
実施例27 齧歯類の癲癇モデルによる抗痙攣性の生物学的活性の実証。
本発明の多様な化合物の抗痙攣作用は、多種の齧歯類(マウスおよびラット)癲癇モデルにおいてin vivoで証明された。動物実験をWhite ら、「抗癲癇薬の発見と非臨床試験による開発」(Discovery and preclinical development of antiepileptic drugs), in Antiepileptic Drugs, 5th ed., Levy et al.(Eds.), Lippincott Williams and Wilkins, Philadelphia, PA, 2002 (968 pp.), pp. 36-48 の記載に従って実施し、参照により その全体が本明細書に組み込まれる。化合物A、G、H、IおよびFの結果を以下の表2および3に要約して示す。
【0142】
【表2】
【0143】
【表3】
【0144】
実施例28 ラットの癲癇重積状態モデルによる抗痙攣性生物学的活性の実証
本発明のさまざまな化合物の抗痙攣活性はまた、2つのラットの癲癇重積状態モデルにおいてin vivoで証明された。米国国立衛生研究所(NIH)の米国国立神経疾患脳卒中研究所(NINDS)の抗痙攣剤スクリーニングプログラム(ASP)で開発されたプロトコルに従って動物実験を実施した。表4に結果の要約を示す。
【0145】
【表4】
【0146】
実施例29 In Vitroアッセイ(LDHおよび細胞増殖)における無毒性の実証。
化合物A、I、HおよびFについてStem Cell Innovations, Inc. (Houston, TX) のヒト肝細胞に基づくアッセイACTIVTox(R)(C3A肝細胞を使用)で試験を実施した。 具体的には、試験化合物を多種類の濃度で、乳酸脱水素酵素LDHの放出を測るLDH放出アッセイ(細胞死の指標)で試験した。
【0147】
試験化合物の100μMという濃度は、生理学的状況下で予測される肝細胞への暴露量よりも非常に高い。従って100μM量の標準試験濃度(比較の目的で)を使用して、陰性対照群の吸光度に対する試験化合物存在下での吸光度(LDH放出レベルの測定)の比は、「平均倍数」(平均倍数=平均吸光度/陰性対照の平均吸光度)値として知られている。1.75以下のAVFは、LDH放出アッセイでは試験化合物は、細胞毒性または肝毒性作用が無いことを意味する。
【0148】
ACTIVToxデータ(以下の表5を参照)は、化合物A、I、HおよびFには、生理学的濃度( < 100 μM)では細胞毒性または肝毒性がないことを示す。
【0149】
【表5】
【0150】
化合物AおよびIはまたACTIVTox 細胞増殖アッセイを使って試験し、各々平均倍数値が1.15および1.25であった100μM濃度では増殖中の細胞に対する毒性がないことが示された。
【0151】
本発明は、その真意や本質的な特徴から逸脱することなくその他の具体的な態様で実施されうる。本明細書に記載される実施態様は、あらゆる面で例証的なものでありこれらに限定されるものではない。従って本発明の範囲は、前述の説明によってよりもむしろ、付記された特許請求の範囲によって示される。本特許請求に意味するものと、それに同等なものの範囲内の全ての変更は、本特許請求に組み込まれる。本明細書に引用される参照は、その全体を本明細書に具体的な参照として組み込まれる。
【特許請求の範囲】
【請求項1】
式Iを有するCNS活性化合物であって、
【化1】
式中、Arは、任意に置換されたフェニル、任意に置換されたナフチル、任意に置換されたテトラヒドロナフチル、任意に置換されたインダンまたは任意に置換されたヘテロサイクリックアリルであり、ここでArには最高5個までの置換基が任意に存在して良く、各置換基は独立して、水素、アルキル、シクロアルキル、ハロゲン、アルコキシ、チオアルキル、スルホキシアルキル、スルホニルアルキル、アルキレンジオキシ、ハロアルキル、ハロアルコキシ、OH、CH2OH、CONH2、CN、アセトキシ、N(アルキル)2、ベンジル、ベンジロキシ、α,α-ジメチルベンジル、NO2、CHO、CH3CH(OH)、アセチル、OCH2COOHおよび任意に置換された芳香環系からなる群から選択され、
該任意に置換された芳香環系は、フェニル、フェノキシ、およびヘテロサイクリックアリルからなる群から選択され、ここで5個までの置換基が任意に芳香環上にあってもよく、各置換基は水素、アルキル、シクロアルキル、ハロゲン、アルコキシ、チオアルキル、スルホキシアルキル、 スルホニルアルキル、アルキレンジオキシ、ハロアルキル、ハロアルコキシ、OH、CH2OH、CONH2、CN、アセトキシ、N(アルキル)2、NO2、CHO、CH3CH(OH)、アセチル、およびOCH2COOHからなる群から独立して選択され
R1およびR2は各々独立して、H、任意に置換されたアルキル、任意に置換されたシクロアルキル、または任意に置換されたCW2フェニルであり、ここで各Wは両方のWがエチルでないという条件で、H、メチルおよびエチルからからなる群から独立して選択され、さらにここで、フェニル基またはシクロアルキル基には5個までの置換基が任意に含まれ、各置換基は独立してハロゲン、アルコキシ、チオアルキル、スルホキシアルキル、スルホニルアルキル、ハロアルキル、ハロアルコキシ、CONH2、CN、アセトキシ、N(アルキル)2、NO2およびアセチルからなる群から選択され、R1 または R2の一方がHの場合には、他方のR1またはR2は、(CH2)2SO3Hまたは CHZCOOHであり、ここでZはH、CH3、CH(CH3)2、CH2C6H5、CH2CH(CH3)2およびCH(CH3)CH2CH3からなる群の一つであり、またはR1およびR2が共にシクロアルキル基であり、
R3は、ヒドロキシ、アルキル、シクロアルキルのうちの一つであるか、またはR4も共に、R3またはR4の一方がOHの場合には他方のR3またはR4がエチルでないという条件で、シクロアルキルであり、
R4がアルキル、シクロアルキルのうちのひとつであり、またはR3も共にシクロアルキルであり、そして
Xが、無置換、メチレン、ケトン、CHOH、酸素、NR1、硫黄、スルホンまたはスルホキシドのうちの1つである化合物。
【請求項2】
式IIを有するCNS活性化合物であって、
【化2】
式中、R1およびR2は各々独立して、少なくともH、アルキル、シクロアルキル、CW2フェニル置換基うちの一つであり、ここで各Wは、両方のWがエチルで無いという条件で、H、メチルおよびエチルからなる群から独立して選択され、そしてここでフェニル基またはシクロアルキル基には5個までの置換基が含まれても良く、各置換基は独立してハロゲン、アルコキシ、チオアルキル、スルホキシアルキル、スルホニルアルキル、ハロアルキル、ハロアルコキシ、CONH2、CN、アセトキシ、N(アルキル)2、NO2 およびアセチルからなる群から選択され、R1またはR2の一方がHの場合には、他方のR1 または R2は、(CH2)2SO3HまたはCHZCOOHであり、ここでZはH、CH3、 CH(CH3)2、CH2C6H5、CH2CH(CH3)2およびCH(CH3)CH2CH3からなる群のうちの1つであるか、またはR1およびR2は共にシクロアルキル基であり、
R4およびR5は、各々独立して任意に置換されたフェニルまたは任意に置換されたヘテロサイクリックアリル基であり、ここで5個までの置換基を含んでも良く、各置換基はハロゲン、アルコキシ、チオアルキル、スルホキシアルキル、 スルホニルアルキル、ハロアルコキシ、CH2OH、CONH2、CN、アセトキシ、N(アルキル)2、NO2、アセチルおよびOCH2COOHからなる群から独立して選択され、
Yは無置換またはメチレンのいずれかである化合物。
【請求項3】
式1〜9のうちの一つを有するCNS活性化合物であって
【化3】
式中、R1は、H、CH3、C2H5、(CH2)2SO3H、またはCHZCOOHのうちの一つであり、
Zは、H、CH3、CH(CH3)2、CH2C6H5、CH2CH(CH3)2またはCH(CH3)CH2CH3のうちの一つであり、
R2およびR3は、独立してHまたはCH3のうちの一つであり、
R4は、H、CH3、OH、またはOCH3のうちの一つであり、
R5は、H、Cl、F、CF3、CN、C1-C5 アルキル、C1-C5アルコキシ、OCF3または CONR1R2のうちの1つであり、
n=1〜5、
Q=O、NR2、C=O、S、SO、またはSO2であり、そして
X = O、NR2、または無置換、C=O、S、SO、またはSO2である化合物。
【請求項4】
式A〜BA:
【化4】
【化5】
【化6】
【化7】
【化8】
【化9】
のうちの一つを有するCNS活性化合物。
【請求項5】
CNS活性を調節する医薬組成物であって、該組成物が、
医薬上許容できる担体および
前記担体と結合させた請求項1〜4のうちの一つに記載されたCNSに活性のある化合物を含む医薬組成物。
【請求項6】
さらに医薬品添加物を含む請求項5に記載の医薬組成物。
【請求項7】
請求項5に記載の医薬組成物であって、CNS活性化合物のCNS活性調節の治療有効量を含む医薬組成物。
【請求項8】
請求項7に記載の医薬組成物であって、
患者に抗痙攣作用を提供する、
患者の痙攣を治療および/または予防する、
患者のてんかん発作を治療および/または予防する、
患者の痙性を治療および/または予防する、
患者の感情障害を治療および/または予防する、
患者の双極性感情障害を治療および/または予防する、
患者の慢性頭痛を治療および/または予防する、
患者の群発性頭痛を治療および/または予防する、
患者の片頭痛を治療および/または予防する、
患者の静止不能症候群を治療および/または予防する、
患者の神経因性疼痛を治療および/または予防する、あるいは
患者の運動障害を治療および/または予防する、
ための治療の少なくとも一つに充分な治療有効量を含む医薬組成物。
【請求項9】
CNS活性を調節する方法であって
請求項1〜4のいずれか一つに記載のCNS活性化合物を患者に投与することを含む方法。
【請求項10】
CNS活性化合物が医薬上許容できる担体に結合された、請求項9に記載の方法。
【請求項11】
CNS活性化合物の、患者のCNS活性を調節する治療有効量が患者に投与される請求項9に記載の方法。
【請求項12】
請求項11に記載の方法であって、
患者に抗痙攣作用を提供する、
患者の痙攣を治療および/または予防する、
患者の癲癇発作を治療および/または予防する、
患者の痙性を治療および/または予防する、
患者の感情障害を治療および/または予防する、
患者の双極性感情障害を治療および/または予防する、
患者の慢性頭痛を治療および/または予防する、
患者の群発性頭痛を治療および/または予防する、
患者の片頭痛を治療および/または予防する、
患者の静止不能症候群を治療および/または予防する、
患者の神経因性疼痛を治療および/または予防する、あるいは
患者の運動障害を治療および/または予防する、
治療のうちの少なくとも一つについて十分な治療有効量が患者に投与される請求項11に記載の方法。
【請求項13】
請求項11に記載の方法であって、前記の治療有効量が不安、抑鬱、不眠、片頭痛、統合失調症、パーキンソン病、痙性、アルツハイマー病、双極性障害、慢性または神経因性疼痛、脳卒中、慢性神経変性疾患、ハンチントン病、脳損傷、脊髄損傷または癲癇重積状態の少なくとも1つの治療および/または予防に十分である方法。
【請求項14】
請求項11に記載の方法であって、前記の治療有効量が化学的対応策として十分な方法。
【請求項15】
CNS活性化合物[3−(4−シアノフェノキシ)ブチルアミド]を製造する方法であって、
【化10】
の化学変化を含む方法。
【請求項16】
請求項15に記載の方法であって、さらに、
塩基性溶液中で4−シアノフェノ−ルを還流しながら加熱する方法と、
β-ブチロラクトンを還流溶液へ添加する方法と、
該溶液を室温に冷却する方法と、
水およびジエチルエ―テルを添加して該溶液の二相混合液を調製する方法と、
形成後にジエチルエーテル相を除去する方法と、
形成後に水相にpH〜2のHCl溶液を添加して酸性化する方法と、
酸性化した水相をエチルアセテートで抽出する方法と、
エチルアセテート層を乾燥させ3−(4−シアノフェノキシ)酪酸を生成する方法と、
有機溶媒で3−(4−シアノフェノキシ)酪酸の溶液を調製する方法と、
塩化オキサリルで該溶液を処理して[3−(4−シアノフェノキシ)ブチリルクロライド]を得る方法と、
3−(4−シアノフェノキシ)ブチリルクロライド溶液へアンモニアガスを発泡により通す方法と、そして
該溶液から[3−(4−シアノフェノキシ)ブチルアミド]を入手する方法
を含む製造方法。
【請求項1】
式Iを有するCNS活性化合物であって、
【化1】
式中、Arは、任意に置換されたフェニル、任意に置換されたナフチル、任意に置換されたテトラヒドロナフチル、任意に置換されたインダンまたは任意に置換されたヘテロサイクリックアリルであり、ここでArには最高5個までの置換基が任意に存在して良く、各置換基は独立して、水素、アルキル、シクロアルキル、ハロゲン、アルコキシ、チオアルキル、スルホキシアルキル、スルホニルアルキル、アルキレンジオキシ、ハロアルキル、ハロアルコキシ、OH、CH2OH、CONH2、CN、アセトキシ、N(アルキル)2、ベンジル、ベンジロキシ、α,α-ジメチルベンジル、NO2、CHO、CH3CH(OH)、アセチル、OCH2COOHおよび任意に置換された芳香環系からなる群から選択され、
該任意に置換された芳香環系は、フェニル、フェノキシ、およびヘテロサイクリックアリルからなる群から選択され、ここで5個までの置換基が任意に芳香環上にあってもよく、各置換基は水素、アルキル、シクロアルキル、ハロゲン、アルコキシ、チオアルキル、スルホキシアルキル、 スルホニルアルキル、アルキレンジオキシ、ハロアルキル、ハロアルコキシ、OH、CH2OH、CONH2、CN、アセトキシ、N(アルキル)2、NO2、CHO、CH3CH(OH)、アセチル、およびOCH2COOHからなる群から独立して選択され
R1およびR2は各々独立して、H、任意に置換されたアルキル、任意に置換されたシクロアルキル、または任意に置換されたCW2フェニルであり、ここで各Wは両方のWがエチルでないという条件で、H、メチルおよびエチルからからなる群から独立して選択され、さらにここで、フェニル基またはシクロアルキル基には5個までの置換基が任意に含まれ、各置換基は独立してハロゲン、アルコキシ、チオアルキル、スルホキシアルキル、スルホニルアルキル、ハロアルキル、ハロアルコキシ、CONH2、CN、アセトキシ、N(アルキル)2、NO2およびアセチルからなる群から選択され、R1 または R2の一方がHの場合には、他方のR1またはR2は、(CH2)2SO3Hまたは CHZCOOHであり、ここでZはH、CH3、CH(CH3)2、CH2C6H5、CH2CH(CH3)2およびCH(CH3)CH2CH3からなる群の一つであり、またはR1およびR2が共にシクロアルキル基であり、
R3は、ヒドロキシ、アルキル、シクロアルキルのうちの一つであるか、またはR4も共に、R3またはR4の一方がOHの場合には他方のR3またはR4がエチルでないという条件で、シクロアルキルであり、
R4がアルキル、シクロアルキルのうちのひとつであり、またはR3も共にシクロアルキルであり、そして
Xが、無置換、メチレン、ケトン、CHOH、酸素、NR1、硫黄、スルホンまたはスルホキシドのうちの1つである化合物。
【請求項2】
式IIを有するCNS活性化合物であって、
【化2】
式中、R1およびR2は各々独立して、少なくともH、アルキル、シクロアルキル、CW2フェニル置換基うちの一つであり、ここで各Wは、両方のWがエチルで無いという条件で、H、メチルおよびエチルからなる群から独立して選択され、そしてここでフェニル基またはシクロアルキル基には5個までの置換基が含まれても良く、各置換基は独立してハロゲン、アルコキシ、チオアルキル、スルホキシアルキル、スルホニルアルキル、ハロアルキル、ハロアルコキシ、CONH2、CN、アセトキシ、N(アルキル)2、NO2 およびアセチルからなる群から選択され、R1またはR2の一方がHの場合には、他方のR1 または R2は、(CH2)2SO3HまたはCHZCOOHであり、ここでZはH、CH3、 CH(CH3)2、CH2C6H5、CH2CH(CH3)2およびCH(CH3)CH2CH3からなる群のうちの1つであるか、またはR1およびR2は共にシクロアルキル基であり、
R4およびR5は、各々独立して任意に置換されたフェニルまたは任意に置換されたヘテロサイクリックアリル基であり、ここで5個までの置換基を含んでも良く、各置換基はハロゲン、アルコキシ、チオアルキル、スルホキシアルキル、 スルホニルアルキル、ハロアルコキシ、CH2OH、CONH2、CN、アセトキシ、N(アルキル)2、NO2、アセチルおよびOCH2COOHからなる群から独立して選択され、
Yは無置換またはメチレンのいずれかである化合物。
【請求項3】
式1〜9のうちの一つを有するCNS活性化合物であって
【化3】
式中、R1は、H、CH3、C2H5、(CH2)2SO3H、またはCHZCOOHのうちの一つであり、
Zは、H、CH3、CH(CH3)2、CH2C6H5、CH2CH(CH3)2またはCH(CH3)CH2CH3のうちの一つであり、
R2およびR3は、独立してHまたはCH3のうちの一つであり、
R4は、H、CH3、OH、またはOCH3のうちの一つであり、
R5は、H、Cl、F、CF3、CN、C1-C5 アルキル、C1-C5アルコキシ、OCF3または CONR1R2のうちの1つであり、
n=1〜5、
Q=O、NR2、C=O、S、SO、またはSO2であり、そして
X = O、NR2、または無置換、C=O、S、SO、またはSO2である化合物。
【請求項4】
式A〜BA:
【化4】
【化5】
【化6】
【化7】
【化8】
【化9】
のうちの一つを有するCNS活性化合物。
【請求項5】
CNS活性を調節する医薬組成物であって、該組成物が、
医薬上許容できる担体および
前記担体と結合させた請求項1〜4のうちの一つに記載されたCNSに活性のある化合物を含む医薬組成物。
【請求項6】
さらに医薬品添加物を含む請求項5に記載の医薬組成物。
【請求項7】
請求項5に記載の医薬組成物であって、CNS活性化合物のCNS活性調節の治療有効量を含む医薬組成物。
【請求項8】
請求項7に記載の医薬組成物であって、
患者に抗痙攣作用を提供する、
患者の痙攣を治療および/または予防する、
患者のてんかん発作を治療および/または予防する、
患者の痙性を治療および/または予防する、
患者の感情障害を治療および/または予防する、
患者の双極性感情障害を治療および/または予防する、
患者の慢性頭痛を治療および/または予防する、
患者の群発性頭痛を治療および/または予防する、
患者の片頭痛を治療および/または予防する、
患者の静止不能症候群を治療および/または予防する、
患者の神経因性疼痛を治療および/または予防する、あるいは
患者の運動障害を治療および/または予防する、
ための治療の少なくとも一つに充分な治療有効量を含む医薬組成物。
【請求項9】
CNS活性を調節する方法であって
請求項1〜4のいずれか一つに記載のCNS活性化合物を患者に投与することを含む方法。
【請求項10】
CNS活性化合物が医薬上許容できる担体に結合された、請求項9に記載の方法。
【請求項11】
CNS活性化合物の、患者のCNS活性を調節する治療有効量が患者に投与される請求項9に記載の方法。
【請求項12】
請求項11に記載の方法であって、
患者に抗痙攣作用を提供する、
患者の痙攣を治療および/または予防する、
患者の癲癇発作を治療および/または予防する、
患者の痙性を治療および/または予防する、
患者の感情障害を治療および/または予防する、
患者の双極性感情障害を治療および/または予防する、
患者の慢性頭痛を治療および/または予防する、
患者の群発性頭痛を治療および/または予防する、
患者の片頭痛を治療および/または予防する、
患者の静止不能症候群を治療および/または予防する、
患者の神経因性疼痛を治療および/または予防する、あるいは
患者の運動障害を治療および/または予防する、
治療のうちの少なくとも一つについて十分な治療有効量が患者に投与される請求項11に記載の方法。
【請求項13】
請求項11に記載の方法であって、前記の治療有効量が不安、抑鬱、不眠、片頭痛、統合失調症、パーキンソン病、痙性、アルツハイマー病、双極性障害、慢性または神経因性疼痛、脳卒中、慢性神経変性疾患、ハンチントン病、脳損傷、脊髄損傷または癲癇重積状態の少なくとも1つの治療および/または予防に十分である方法。
【請求項14】
請求項11に記載の方法であって、前記の治療有効量が化学的対応策として十分な方法。
【請求項15】
CNS活性化合物[3−(4−シアノフェノキシ)ブチルアミド]を製造する方法であって、
【化10】
の化学変化を含む方法。
【請求項16】
請求項15に記載の方法であって、さらに、
塩基性溶液中で4−シアノフェノ−ルを還流しながら加熱する方法と、
β-ブチロラクトンを還流溶液へ添加する方法と、
該溶液を室温に冷却する方法と、
水およびジエチルエ―テルを添加して該溶液の二相混合液を調製する方法と、
形成後にジエチルエーテル相を除去する方法と、
形成後に水相にpH〜2のHCl溶液を添加して酸性化する方法と、
酸性化した水相をエチルアセテートで抽出する方法と、
エチルアセテート層を乾燥させ3−(4−シアノフェノキシ)酪酸を生成する方法と、
有機溶媒で3−(4−シアノフェノキシ)酪酸の溶液を調製する方法と、
塩化オキサリルで該溶液を処理して[3−(4−シアノフェノキシ)ブチリルクロライド]を得る方法と、
3−(4−シアノフェノキシ)ブチリルクロライド溶液へアンモニアガスを発泡により通す方法と、そして
該溶液から[3−(4−シアノフェノキシ)ブチルアミド]を入手する方法
を含む製造方法。
【図1】

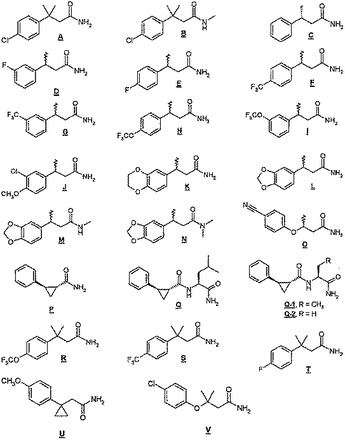
【図2】
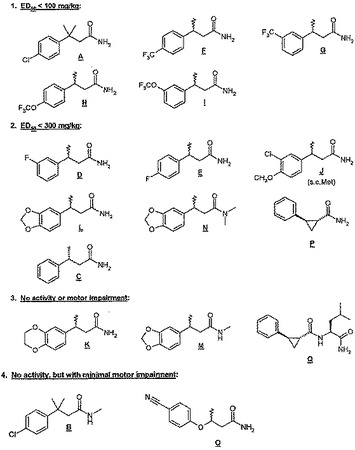
【図3A】
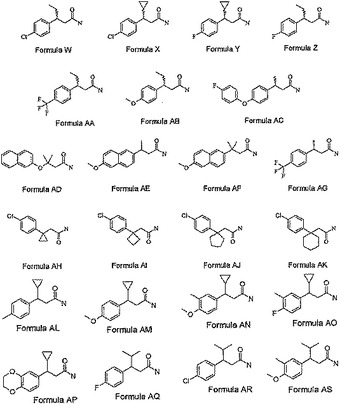
【図3B】
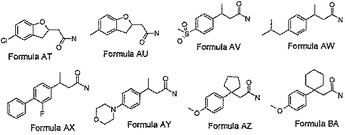
【図4A】
【図4B】
【図4C】

【図4D】
【図4E】
【図4F】
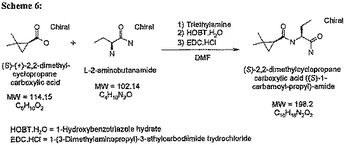
【図4G】

【図4H】
【図4I】
【図4J】
【図4K】
【図4L】
【図4M】
【図4N】
【図4O】

【図2】
【図3A】
【図3B】
【図4A】
【図4B】
【図4C】
【図4D】
【図4E】
【図4F】
【図4G】
【図4H】
【図4I】
【図4J】
【図4K】
【図4L】
【図4M】
【図4N】
【図4O】
【公表番号】特表2011−529022(P2011−529022A)
【公表日】平成23年12月1日(2011.12.1)
【国際特許分類】
【出願番号】特願2011−500935(P2011−500935)
【出願日】平成21年3月18日(2009.3.18)
【国際出願番号】PCT/US2009/037558
【国際公開番号】WO2009/117515
【国際公開日】平成21年9月24日(2009.9.24)
【出願人】(510250847)オーリムメッド・ファルマ・インコーポレーテッド (1)
【Fターム(参考)】
【公表日】平成23年12月1日(2011.12.1)
【国際特許分類】
【出願日】平成21年3月18日(2009.3.18)
【国際出願番号】PCT/US2009/037558
【国際公開番号】WO2009/117515
【国際公開日】平成21年9月24日(2009.9.24)
【出願人】(510250847)オーリムメッド・ファルマ・インコーポレーテッド (1)
【Fターム(参考)】
[ Back to top ]