
予備成形された生分解性ポリマー組成物を使用したデリバリシステムおよび方法
【課題】様々な生分解速度およびデリバリ速度を提供できる、活性薬剤をサブジェクトにデリバリするためのデリバリシステムの提供。
【解決手段】予備成形された、式、I−(−X−LM−G)nで表される生分解ポリマー組成物を使用したデリバリシステム。(式中、Xは二官能性ポリオキシエチレン鎖部分または結合であり、LMは式、−C(O)−、−(CH2)bC(O)−(式中、bは1〜5の整数)であり、−C(O)−(CH2)c−C(O)−(式中、cは2〜10の整数)であり、−C(O)−O−(CH2)d−O−C(O)−(式中、dは2〜10の整数)である)によって表される二官能性結合部分であり、GはN−オキシスクシンイミジルなどの脱離基であり、Iは多求核性化合物から誘導される多官能性結合部分であり、少なくとも部分的に脱溶媒和された架橋されたアルブミンである。
【解決手段】予備成形された、式、I−(−X−LM−G)nで表される生分解ポリマー組成物を使用したデリバリシステム。(式中、Xは二官能性ポリオキシエチレン鎖部分または結合であり、LMは式、−C(O)−、−(CH2)bC(O)−(式中、bは1〜5の整数)であり、−C(O)−(CH2)c−C(O)−(式中、cは2〜10の整数)であり、−C(O)−O−(CH2)d−O−C(O)−(式中、dは2〜10の整数)である)によって表される二官能性結合部分であり、GはN−オキシスクシンイミジルなどの脱離基であり、Iは多求核性化合物から誘導される多官能性結合部分であり、少なくとも部分的に脱溶媒和された架橋されたアルブミンである。
【発明の詳細な説明】
【技術分野】
【0001】
背景
医薬品などの活性薬剤を好ましくは薬剤の徐放のためにサブジェクトにデリバリできるようにする、様々なアプローチが発達している。このようなデリバリシステムは典型的に、サブジェクトの目標領域への薬剤の徐放を可能にしながら、デリバリに先立って環境から薬剤を保護するようにデザインされている。
【0002】
いくつかの従来の徐放システムは、リポソーム、リポ球体、マイクロカプセル、微小粒子、およびナノ粒子などの微細構造、並びにシリンダー、ディスク、および繊維などのマクロ構造に基づく。典型的に医薬品などの活性薬剤がポリマーと混合され、次に所望の形状に形成される。
【0003】
このような従来のシステムの多くは、補綴用途に必要な構造一体性を有する固形移植片を形成するのには使用できない。さらにこのようなシステムの多くは、例えばサブジェクトへの適用時に、医師が活性薬剤(例えば医薬品)を注入できる物品を形成するのには使用できない。またこのようなシステムの多くは、生分解速度、および/またはあらゆる組み込まれた活性薬剤の放出速度に関して容易に制御できないポリマーを含む。
【0004】
アルブミンとポリエチレングリコール(PEG)誘導体が架橋して製造されるヒドロゲル(例えば水性媒体中で膨潤するが、水に溶けないポリマーのクラス)は、可能な医薬品デリバリ用途について既に研究されている(D’Ursoら、Biotech.Tech.、8、71〜76ページ(1994年))。医薬品の制御されたデリバリに対する別のアプローチは、ポリ(乳酸)や、ラクチドとグリコリドの種々のコポリマーなどの合成生体吸収性ポリマーの使用による、マイクロカプセル封入または微小球の形成を伴う。しかし微小球使用の欠点は、外科手術部位表面あるいは負傷または病変組織上に、均一に被覆して保持できないことである。この問題と取り組むために、微小球には非溶剤である溶剤の使用によって作られる第2のポリマー溶液に微小球を懸濁し、次に混合物からフィルムをキャスティングして、微小球含有メンブランが作られている。
【発明の概要】
【発明が解決しようとする課題】
【0005】
したがってその他のデリバリシステム、特に、様々な生分解速度およびデリバリ速度を提供できる、変化させられるポリマー組成物を含むものに対する必要性がある。好ましくは十分な構造一体性を有して取り扱いが容易な、ポリマー組成物を含むデリバリシステムに対する必要性がある。
【課題を解決するための手段】
【0006】
要約
本発明は、活性薬剤をサブジェクトにデリバリするための予備成形されたオブジェクトを提供し、予備成形されたオブジェクトは、式、
I−(−X−LM−G)n
(式中、
Xは二官能性ポリオキシエチレン鎖部分または結合であり、
LMは式、−C(O)−、−(CH2)bC(O)−(式中、bは1〜5の整数である。)、−C(O)−(CH2)c−C(O)−(式中、cは2〜10の整数であり、ラジカルの脂肪族部分は飽和または不飽和でも良い。)、−C(O)−O−(CH2)d−O−C(O)−(式中、dは2〜10の整数であり、あるいは式、−R−C(O)−、−R−C(O)−(CH2)c−C(O)−、または−R−C(O)−O−(CH2)d−O−C(O)−(式中、cは2〜10の整数であり、dは2〜10の整数であり、Rは1〜10個のモノマーラクチド、グリコリド、炭酸トリメチレン、カプロラクトンまたはp−ジオキサノン断片を有するポリマーまたはコポリマーである。)によって表されるオリゴマージラジカルである。)によって表される二官能性結合部分であり、
GはN−オキシスクシンイミジル、N−オキシマレイミジル、N−オキシフタルイミジル、ニトロフェノキシル、N−オキシイミダゾリル、およびトレシルの群より選択される脱離基であり、
Iは多求核性化合物から誘導される多官能性結合部分であり、
nは2〜10の整数であるが、
ただしXが二官能性ポリオキシエチレン鎖部分の場合、−X−I−X−は、式、
−O−(CH2−CH2−O−)a−
(式中、aは20〜300の整数である。)によって表されるジラジカル断片であるPEGである。)
の架橋剤を有する、少なくとも部分的に脱溶媒和された架橋されたアルブミンである。
【0007】
本発明のその他の実施態様としては、その中に組み込まれた再溶媒和していてもしていなくても良い活性薬剤を有する予備成形されたオブジェクトが挙げられ、その中に活性薬剤を有する予備成形されたオブジェクトは、二次生分解性マトリックス内にさらに組み込まれる。その他の実施態様としては、予備成形されたオブジェクトについて上述した化学的性質を有する、二次生分解性マトリックス内にさらに組み込まれた、その中に活性薬剤を有するその他の生分解性ポリマーの予備成形されたオブジェクトが挙げられる。製造方法、および活性薬剤をサブジェクトにデリバリする方法も本発明によって提供される。
【図面の簡単な説明】
【0008】
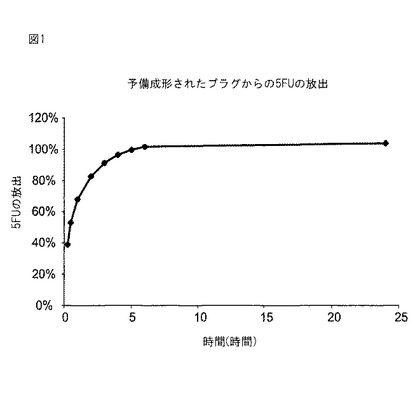
【図1】図1は、PEG−(SS)2架橋剤で架橋されたアルブミンの予備成形されたプラグからの5−フルオロウラシル(5−FU)の放出を示す。
【図2】図2は、PEG−SS2架橋剤との架橋直前にアルブミンに混合された、テトラサイクリンのアルギン酸塩/医薬品複合体からの放出を示す。
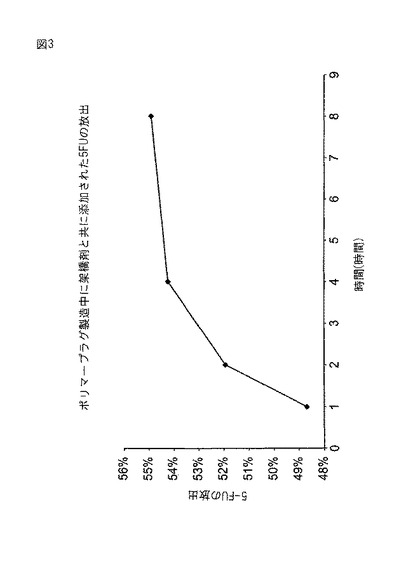
【図3】図3は、プラグ製造中に5−FUが架橋剤と共に添加されたアルブミン/PEG−(SS)2プラグからの5−FUの放出を示す。
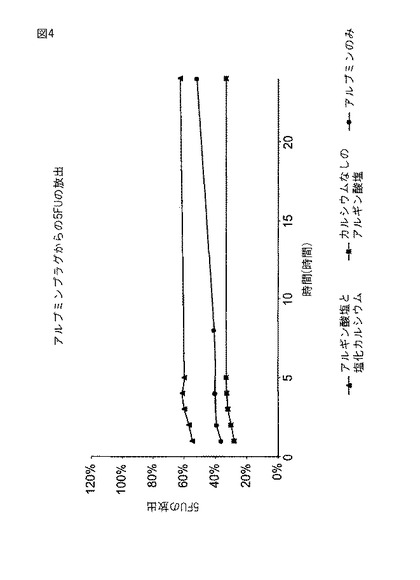
【図4】図4は、塩化カルシウムが添加されたアルブミン/PEG−(SS)2プラグからの5−FUの放出を示す。
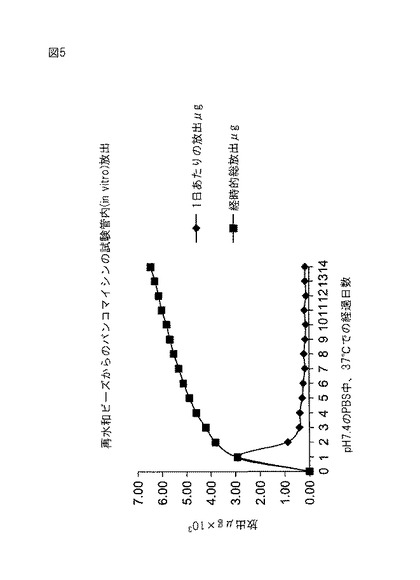
【図5】図5は、再水和されたアルブミン/PEG−(SS)2ビーズからのバンコマイシンの放出を示す。
【図6】図6は、再水和されたアルブミン/PEG−(SS)2ビーズからのバンコマイシンの放出重量%を示す。
【図7】図7は、再水和されたアルブミン/PEG−(SS)2ビーズからのセファゾリン放出を示す。
【図8】図8は、再水和されたアルブミン/PEG−(SS)2ビーズからのセファゾリンの放出重量%を示す。
【図9】図9は、単一ビーズ試験におけるポリマービーズの容積増大%対水取り込み重量%を示す。
【図10】図10は、微小球からのテトラサイクリンの放出プロフィールを示す。
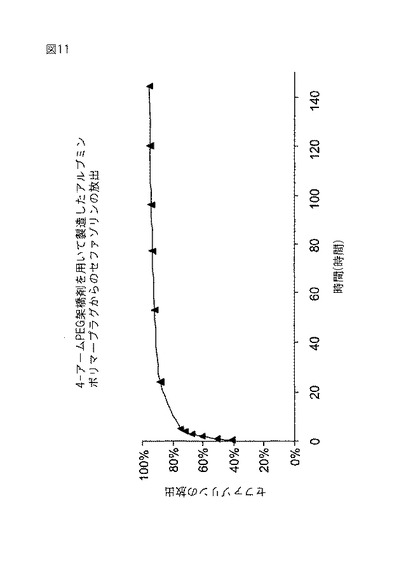
【図11】図11は、アルブミン/4−アーム PEGプラグからのセファゾリンの放出を示す。
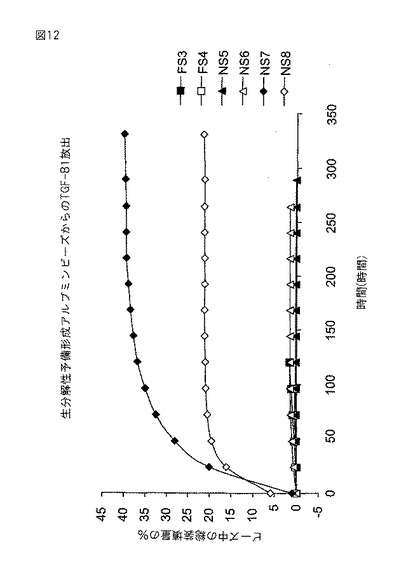
【図12】図12は、種々のビーズから放出されるTGF−β1の百分率を示す。
【発明を実施するための形態】
【0009】
好ましい実施態様の詳細な説明
本発明は、第1の生分解性組成物、および任意に医薬品などの活性薬剤を含む、サブジェクトの目標領域へのデリバリのための予備成形されたオブジェクト、好ましくは予備成形された自立型オブジェクトを提供する。予備成形されたオブジェクトは少なくとも部分的に脱溶媒和させて(例えば少なくとも部分的に脱水して)、引き続いて活性薬剤と組み合わせることができ、あるいは予備成形されたオブジェクトの調製時に活性薬剤を含めることができる。このような予備成形されたオブジェクトは好ましくは、開始材料(例えばタンパク質および架橋剤)を組織に適用してサブジェクト組織存在下で硬化(例えば架橋)させて、組織を結合または封着するのに従来法で使用される生分解性組成物から調製される。しかしここでは好ましいオブジェクトは、同一または同様の組成物から調製されて硬化され(例えば架橋され)、次にサブジェクトの組織に適用される。したがって典型的に予備成形されたオブジェクトと組織の間に結合相互作用はない。
【0010】
本発明の好ましい予備成形されたオブジェクトは、十分な構造一体性を有して、ひとたび形成(例えば硬化)されると、適切な条件下で(例えば脱溶媒和させた場合は水分不在下で、溶媒和させた場合は約4℃の温度で)保管する限りは、それらの全体的形状を維持して自立型である。予備成形されたオブジェクト製造に使用される構成要素の選択次第で、機械的強度、可撓性、硬化速度、および生分解速度の程度が変化できる。本発明の好ましい予備成形されたオブジェクトは、架橋されたタンパク質を含む。それらは典型的に緩衝化された塩基性タンパク質溶液と、多官能性、典型的には二官能性の架橋剤とから調製される。緩衝化されたタンパク質溶液および架橋剤は、典型的に市販される材料を使用して得られるが、これらの材料のほとんどは概して臨床安全性および/または用途の経歴を有することが利点である。
【0011】
本組成物中で使用するのに適切なタンパク質としては、非免疫原性の水溶性タンパク質、好ましくはアルブミン(より好ましくは血清アルブミン、そして最も好ましくはヒト血清アルブミン)が挙げられる。本発明の予備成形されたオブジェクトを調製するのに使用しても良い、好ましい緩衝化されたタンパク質溶液は、約8.0〜約11.0のpHに緩衝化された濃縮水性血清アルブミンを含み、緩衝液濃度は約0.01モル濃度〜約0.25モル濃度の範囲である。適切な緩衝液システムとしては、緩衝液が架橋剤と不利に反応したりそれを変性させたりしない限り、既知の炭酸またはリン酸緩衝液システムなどの生理学的および/または臨床的に許容可能な緩衝液が挙げられる。好ましい緩衝液システムは、pHの値が約9.0〜約10.5で濃度が約0.05〜約0.15モル濃度の範囲の炭酸塩/炭酸水素塩緩衝液システムである。
【0012】
血清アルブミンは既知の分離工程を使用して、血清から容易に分離できる。さらに遺伝子形質転換細胞からアルブミンを製造することが可能である。例えばQuirkらの報告Biotechnology and Applied Biochemistry、11、273〜287ページ(1989年)、KalmanらのNucleic Acids Research、18、6075〜6081ページ(1990年)、SleepらのBiotechnology、8、42〜46ページ(1990年)、およびSijmonsらのBiotechnology、8、217〜221(1990年)を参照されたい。組換えアルブミンを製造する能力は、この方法によって製造されたタンパク質が、ヒト血清から直接分離されたアルブミンを汚染しているかもしれないヒト病原体、ウイルスまたはその他の汚染物質を含まない利点を提供する。
【0013】
本緩衝化された混合物中で使用される場合、血清アルブミンは変性しないことが分かった。使用前にアルブミンが変性されないので、アルブミンがその天然のコイル状立体配座を維持するために、ゲル様の固体を提供する硬化工程中に架橋された後、硬化した予備成形されたオブジェクトは十分な可撓性を維持して、所望ならば組織移植のための適切なマトリックスが提供されると考えられる。
【0014】
本発明中で様々な適切な架橋剤を使用しても良い。架橋剤は多官能性で好ましくは二官能性である。本発明で使用するのに適切な架橋剤としては、式、
I−(−X−LM−G)n
(式中、Xは二官能性ポリオキシエチレン鎖部分または結合であり、LMは二官能性結合部分であり、Gは活性脱離基であり、Iは多求核性化合物(例えばエチレングリコール、ペンタエリスリトール、トリメチロールプロパン、多求核性アミンなど)に由来する多官能性結合部分であり、nは約2〜約10、好ましくは約2〜約4であるが、ただしXが二官能性ポリオキシエチレン鎖部分である場合、−X−I−X−はここで定義したように−PEG−である。)の化合物が挙げられる。
【0015】
好ましい架橋剤は、二官能性であり、式、
G−LM−PEG−LM−G
(式中、−PEG−は、式、
−O−(CH2−CH2−O−)a−
(式中、aは約20〜約300の整数である。)によって表されるジラジカル断片であり、−LM−は、式、−C(O)−、−(CH2)bC(O)−(式中、bは約1〜約5の整数である。)、−C(O)−(CH2)c−C(O)−(式中、cは約2〜約10の整数であり、ラジカルの脂肪族部分は飽和または不飽和でも良い。)、−C(O)−O−(CH2)d−O−C(O)−(式中、dは約2〜約10の整数であり、あるいは式、−R−C(O)−、−R−C(O)−(CH2)c−C(O)−、または−R−C(O)−O−(CH2)d−O−C(O)−(式中、cは約2〜約10の整数であり、dは約2〜約10の整数であり、Rは1〜10個のモノマーラクチド、グリコリド、炭酸トリメチレン、カプロラクトンまたはp−ジオキサノン断片を有するポリマーまたはコポリマーである。)によって表されるオリゴマージラジカルである。)によって表されるジラジカル断片であり、−Gは、スクシンイミジル、マレイミジル、フタルイミジル、ニトロフェニル、イミダゾリル、またはトレシル脱離基などの脱離基である。)を有する。脱離基の文脈で、「スクシンイミジル」とはN−オキシスクシンイミジルを意味し、「マレイミジル」とはN−オキシマレイミジル、「フタルイミジル」とはN−オキシフタルイミジルを意味し、「ニトロフェニル」とは、ニトロフェノキシルを意味し、「イミダゾリル」とはN−オキシイミダゾリルを意味し、「トレシル」とは(CF3−CH2−SO2−O−)を意味する。
【0016】
架橋剤の−PEG−部分は好ましくは市販される化合物から誘導されて、約1,000〜約15,000の量平均分子量を有し、好ましくは約2,000〜約4,000の量平均分子量を有する。これらの化合物は無害であることが実証されており、また分子量が約30,000未満であれば迅速に体から排出されることから、異なるタイプの生物医学的材料中で使用されている。
【0017】
架橋剤の脱離基−G部分は、架橋剤をタンパク質の遊離一級または二級アミン基と反応させ、あるいは化学的に結合させる活性脱離基である。適切な脱離基としては、N−オキシスクシンイミジルと、N−オキシマレイミジルおよびN−オキシフタルイミジルなどのその他のイミドと、N−オキシイミダゾリルなどのヘテロ環式脱離基と、ニトロフェノキシルなどの芳香族脱離基と、あるいはトレシル(CF3−CH2−SO2−O−)などのフッ素化アルキルスルホン脱離基と、が挙げられる。変異原性、発がん性、および催奇形性の研究からは、少量のN−オキシスクシンイミジル基が局所的または全身的毒物学リスクを呈さないことが示唆されるので、この群の脱離基が好ましい。
【0018】
架橋剤は、米国特許番号第4,101,380号または第4,839,345号で報告された手順、1990年4月19日に出願された国際出願整理番号PCT/US90/02133で報告された手順、またはAbuchowskiらによってCancer Biochem.Biophys.、7、175〜186ページ(1984年)で報告された手順などの既知の工程、手順または合成方法を使用して調製しても良い。簡単に述べると、塩基存在下でポリエチレングリコールおよび適切な酸無水物を適切な極性有機溶剤中に溶解し、ポリエチレングリコールジエステル二酸を形成するのに十分な時間還流する。次にジシクロヘキシルカルボジイミドまたはその他の縮合剤の存在下に、適切な極性有機溶剤中で、ジエステル二酸をN−ヒドロキシイミド化合物などの脱離基と反応させて、室温で撹拌して所望の二官能性架橋剤を形成する。
【0019】
代案としては、ポリエチレングリコールおよび適切な塩化ジカルボン酸またはビスクロロホルメートを、混合酸塩化物ポリエチレングリコールエステルまたは混合クロロホルメートポリエチレングリコールエステルを形成するのに十分な時間、適切な極性有機溶剤中に溶解しても良い。次に適切な極性有機溶剤中で、混合エステルをN−ヒドロキシイミド化合物などの化合物と反応させて、所望の二官能性架橋剤を形成するのに十分な時間、高温で撹拌しても良い。
【0020】
予備成形されたオブジェクトは、タンパク質などの材料を架橋する従来の方法、および押出し技術、噴霧乾燥技術、およびエマルジョン重合技術をはじめとする、架橋されたポリマーをビーズまたは微小球などの形状に形成する従来の方法を使用して調製しても良い。このような方法は、ポリマー化学技術分野の当業者には周知である。
【0021】
異なるpH値を有する緩衝液を使用してタンパク質のpHを変更することで、本組成物の硬化時間を目的に合わせても良い。例えば緩衝液のpHを変化させることでアルブミンのpHを変更し、それによって硬化速度時間を約10秒から約10分未満に変化させることが可能である。簡単に述べると、濃縮水性血清アルブミンをより高いpHで架橋剤と混合することで、最速硬化時間が提供される。より高濃度のタンパク質および架橋剤が、比較的より強力な硬化マトリックスを提供することも分かった。しかし混合物が濃くなりすぎ、粘度が高くなりすぎると、得られる硬化マトリックスは弱くなって球状になる。さらに架橋剤濃度が高すぎると、得られる硬化マトリックスは、水またはその他の流体存在下において、マトリックス強度が低くなる程度にまで膨潤するかもしれない。
【0022】
本発明の予備成形されたオブジェクトを形成するのに使用される組成物については、米国特許番号第5,583,114号(Barrowsら)で述べられている。しかしそこでは、組成物は直接組織に適用されて現場で(in situ)硬化されて、組織との結合相互作用が得られる。これらは例えば、現行の方法で標準的に必要な縫合糸数を排除または大きく削減したり、再建外科手術中に皮膚移植片を付着して組織皮弁または自由皮弁を配置したり、歯周部の外科手術において歯肉皮弁を閉じるのに使用される。これらの全ての用途において、組成物は、2つの隣接する生体組織層の間に事実上はさまれた、硬化接着剤の薄層を形成する。代案としては、組成物は、例えば肺外科手術に関連した空気漏れを防止するために、あるいはその他の外科手術手順において出血を阻害または防止するための密封材として使用されることが開示されている。このようにして使用する場合、下にある組織を比較的厚い接着剤層で被覆しても良い。
【0023】
対照的にここで述べる組成物は、サブジェクトに接触させるのに先立ってオブジェクトに成形される。したがって「予備成形された」という用語は、サブジェクトをオブジェクトに接触させるのに先立って、ポリマー組成物に形態または形状を提供することを指す。好ましくは予備成形されたオブジェクトは、自立型の三次元形状を有する。したがってこのように予備成形されたオブジェクトは、薄膜または薄層を含まない。しかし例えば形状が、裏材料上のシート材料または層の形状でパッチを形成する場合、予備成形されたオブジェクトは2mmを越える厚さを有する。本発明の予備成形されたオブジェクトは、マイクロカプセル、微小粒子、ナノ粒子などの微細構造、ならびにビーズまたはその他のボール形のオブジェクト、シリンダー、ディスク、繊維、シート、プラグ、リボン、ウェッジなどのマクロ構造の形態であることができる。好ましい構造は、微小球およびビーズである。典型的にビーズは約1mmを越える直径を有し、微小球は約1μm〜約1000μmの直径を有する。
【0024】
予備成形されたオブジェクトは、形成後に少なくとも部分的に脱溶媒和することができ、好ましくは実質的に完全に脱溶媒和される。ここで「脱溶媒和された」とは、タンパク質と架橋剤との反応に続いて、調製中に使用された溶剤が予備成形されたオブジェクトから除去されることを意味する。典型的にこれは、最初の形成時に予備成形されたオブジェクト中に組み込まれた水の少なくとも一部が除去されるために、予備成形されたオブジェクトが少なくとも部分的に脱水されることを意味する。このような予備成形されたオブジェクトの調製には、水の他に、または水に加えてその他の溶剤も使用できる。注目に値すべきは本発明の好ましい予備成形されたオブジェクトが、脱溶媒和および再溶媒和に際してそれらの形状を維持することである。
【0025】
予備成形されたオブジェクトを少なくとも部分的に脱溶媒和した後は、例えば大気中の湿気による再溶媒和を起こさない環境に、それらを貯蔵することが望ましい。取り扱いが容易で、医師に提供されて使用時に活性薬剤で再溶媒和できることから、このようなオブジェクトが望ましい。
【0026】
脱溶媒和されているか否かに関わらず、予備成形されたオブジェクトは活性薬剤を含むことができる。本発明の予備成形されたオブジェクトの好ましい実施態様は、その中に組み込まれた活性薬剤を有する。ここでの用法で「活性薬剤」とは、それが化学的、薬理学的、物理的、または生物学的に関わらず、所望の効果を生じることができるものである。したがってその中に組み込まれた1つ以上の活性薬剤を有する、得られた予備成形されたオブジェクトは、医薬品、薬物、またはその他の活性薬剤のためのデリバリシステムとして機能できる。予備成形されたオブジェクトは、例えばサブジェクトの体腔または組織空隙に移植する際などに組織と接触させて、あるいは例えば皮膚上などサブジェクトの体外に配置できる。
【0027】
活性薬剤は、例えばタンパク質と架橋剤の反応中など、またはそれに引き続いて、予備成形されたオブジェクトの形成中に添加できる。予備成形されたオブジェクトに形成中に活性薬剤を組み込む場合、活性薬剤が架橋剤またはタンパク質のどちらとも反応しないことが好ましい。特定の好ましい実施態様では、活性薬剤である液体または活性薬剤を含む液体を有する、予備成形され少なくとも部分的に脱溶媒和されたオブジェクトと活性薬剤とを組み合わせて、その中に組み込まれた活性薬剤を有する再溶媒和され予備成形されたオブジェクトを形成する。次にこの再溶媒和され予備成形されたオブジェクトは、活性薬剤のデリバリのためにサブジェクトと接触させて配置できる。予備成形されたオブジェクトの形成中、またはそれに続いて、それらに活性薬剤を組み込む特定方法を以下の実施例で開示する。
【0028】
本発明の一態様では、これは抗菌剤などの活性薬剤を含有する水性組成物で、複数のボール形の予備成形されたオブジェクトを再溶媒和して、膿瘍または創傷清拭した骨空洞などの組織空隙に、このような複数のオブジェクトを詰めることを伴う。したがって特定の一実施態様では、骨髄炎の治療において、抗菌剤を含有する再溶媒和され予備成形されたオブジェクトが使用できる。骨髄炎は骨とその骨髄の感染症であり、骨の病変形成が帰結する。病因は通常ブドウ球菌である。これは治療および根絶が困難な感染症である。現行の慣行は、長期的な予防的抗生物質と複数の外科的壊死組織切除である。壊死組織切除手術に続いて、骨髄炎組織の除去によって生じた空所に、生分解性ではないポリメタクリル酸メチル骨セメントビーズを詰める。このようなビーズは立証された抗生物質担体であるが、それらは生体内(in vivo)で分解せず、組織不適合反応に帰結することもあるために、それらは理想的とは言えない。したがって1つ以上の抗生物質を含有する本発明の予備成形されたオブジェクトは、骨髄炎の治療に使用できる。
【0029】
注目に値すべきは、予備成形されたオブジェクトの生分解速度が、タンパク質、架橋剤、および調製条件を選択することで、所望の用途について目的に合わせられることである。ここでの用法では「生分解」とは、材料の完全性の低下をもたらす可溶化、加水分解、酵素、および生物学的存在の作用をはじめとする様々な機序によって、予備成形されたオブジェクトがより単純な中間体または最終産物に転換することを意味する。必ずしもそうではないが、ポリマー分子はより小さい断片に壊れることができる。このような生分解の範囲には、予備成形されたオブジェクトの生体再吸収、生体吸収または生体侵食が含まれる。典型的に、本発明予備成形されたオブジェクトは、約2日間〜約60日間の間に分解するように調合できる。
【0030】
活性薬剤の放出は、典型的にポリマーの侵食、およびポリマーからの活性薬剤の拡散の組み合わせによって起きる。拡散成分はフィックの法則に従うかも従わないかもしれず、他方、侵食寄与は分解の動態学に従う。活性薬剤の放出速度は、数時間程度から数ヶ月、典型的に約60日間程度までの範囲になるように、目的に合わせることができる。
【0031】
活性薬剤は、例えば治療的または予防的であっても良い、生理的、薬理学的、または生物学的効果を提供する多種多様の薬剤から選択できる。例としては、細胞増殖を向上させ、組織再生を向上させ、血管形成および血管新生を向上させ、神経刺激を向上させ、骨成長を向上させ、感染症(例えば細菌性またはウイルス性)および/または炎症を阻害(例えば防止、低下、または逆転)し、ガン細胞増殖を阻害し(例えば既存のガン状態を治療する、または前ガン状態からガン状態への転換を防止する)、免疫反応を調節し、創傷治癒を促進し、あるいは組織軟化および加湿を促進できる物質またはその代謝前駆体が挙げられる。このような物質としては、テトラサイクリン、バンコマイシン、およびセファロスポリンなどの抗菌剤と、血小板由来増殖因子、トランスフォーミング増殖因子β、上皮性増殖因子、および繊維芽細胞増殖因子などの増殖因子と、5−フルオロウラシル、マイトマイシン、メトトレキセート、ドキソルビシン、およびシスプラチンなどの抗ガン剤(例えば抗有糸分裂薬)と、リドカイン、ブピバカイン、テトラカイン、プロカイン、およびプリロカインなどの局所麻酔薬と、防腐薬(例えばクロルヘキシジン)と、ホルモン(例えばステロイド、インスリン)と、抗ウイルス薬剤と、麻薬拮抗薬と、免疫反応調節剤と、目薬(例えばアトロピン、ピロカルピン、およびチモロール)と、ワクチンと、および美容医薬品と、が挙げられるが、これに限定されるものではない。その他の具体例は、米国特許番号第第5,733,563号(Fortierら)および第5,759,563号(Yeweyら)に列挙されている。本発明の予備成形されたオブジェクトに、このような1つ以上の活性薬剤を組み込んでも良い。
【0032】
好ましくはその中に組み込まれた活性薬剤を有する本発明の予備成形されたオブジェクトは、二次生分解性マトリックスに組み込むことができる。この二次生分解性マトリックスは、液体(例えば液体中の微小球)としてサブジェクトに適用しても良く、あるいは成形オブジェクト(例えば成形オブジェクト内の微小球などの予備成形された成形オブジェクト)に形成できる。このようにして予備成形されたオブジェクトは、サブジェクトへのデリバリのためにマトリックス内に組み込むことができる。二次生分解性マトリックスは、好ましくは上述され、米国特許番号第5,583,114号(Barrowsら)で述べられるような予備成形されたオブジェクトと同様の化学的性質であることができ、あるいはそれは別の生分解性ポリマーであることができる。同様に予備成形されたオブジェクトは、上述の架橋されたタンパク質を含み、あるいは別の生分解性ポリマーでも良い。マトリックスまたは予備成形されたオブジェクトのいずれかのための適切な生分解性ポリマーの例としては、Drug Delivery Systems、V.V.Ranade編、CRC Press,Inc.、BocaRaton、FL、78〜81ページ、1996年、およびBiodegradable Polymers as Drug Delivery Systems、M.Chasin編、Drugs and the Pharmaceutical Sciences Series、第45巻、Marcel Dekker,Inc.NY、1990年で開示されるものが挙げられる。二次生分解性マトリックスの好ましい例としては、ポリ(ホスホエステル)、ポリ(α−ヒドロキシ酸)、親水アクリレートおよびメタアクリレートポリマー、ヒドロキシプロリンポリエステル、ポリ無水物(例えばポリ(ラクチド−コ−グリコリド))、ポリカプロラクトン、ポリ(オルトエステル)、ポリホスファゲン、ポリ(アミノ酸)、多糖類、およびそれらのコポリマーが挙げられる。予備成形されたオブジェクトの好ましい例としては、ポリ(α−ヒドロキシ酸)(例えばポリ(グリコール酸)、ポリ(DL−乳酸)、およびポリ(L−乳酸))、ポリ(アミノ酸)、ポリ(酸無水物)、ポリ(オルトエステル)、ポリ(ホスファジン)、ポリ(ホスホエステル)、ポリラクトン(例えばポリ(ε−カプロラクトン)、ポリ(δ−バレロラクトン)、およびポリ(γ−ブチロラクトン)が挙げられる。予備成形されたオブジェクトは、当業者には既知の技術を使用して、二次生分解性マトリックスに組み込むことができる。例えば米国特許番号第5,583,114号(Barrowsら)で開示される組織密封材を二次生分解性マトリックスとして使用して、その中に組み込まれた予備成形されたオブジェクトをサブジェクトにデリバリできる。
【0033】
以下の実施例によって本発明の目的および利点をさらに実証するが、これらの実施例で言及される特定材料およびその量、ならびにその他の条件と詳細は、本発明を不当に制限するものではない。
【実施例】
【0034】
実施例
アルブミン溶液の調製
アルブミンは、Baxter Healthcare(イリノイ州ディアフィールド)から25%濃度のヒト血清アルブミンとして得た。それを米国特許番号第5,583,114号(Barrowsら)で述べられるような透析(手順A)で処理し、あるいはカットオフ分子量50,000の680cm2ポリスルホン繊維モジュール(Spectrum Laboratories,Inc.から入手できる)を装着した、接線フロー分離のためのMINIKROS Sampler Lab Unit(カリフォルニア州ランチョドミンゲスのSpectrum Laboratories,Inc.から入手できる)を使用して、連続ダイアフィルトレーションによって処理した(手順B)。pH10の0.1M炭酸/炭酸水素塩緩衝液溶液を使用して、Barrows法(米国特許番号第5,582,114号)(工程A)から得られたアルブミンを23%溶液に希釈した。ダイアフィルトレーション(工程B)から得られたアルブミンの場合は、pH8.9〜9.1の0.075M炭酸/炭酸水素塩緩衝液を用いてダイアフィルトレーションを実施した。ダイアフィルトレーション装置を使用して、サンプルを29%〜30%に濃縮した。アルブミンの最終濃度を標準ビウレット滴定によって求めた。
【0035】
ポリエチレングリコールジスクシンイミジルスクシネート(PEG−SS)2架橋剤の合成
30ガロンのガラス内張反応器に、10.9kgのポリエチレングリコール(分子量3400)、760gの無水コハク酸、および4.4kgのトルエンを装填した。装填物を窒素ガスシール下で110℃で6時間混合した。次に反応混合物を80℃に冷却した。13.0kgの無水エタノールを撹拌しながら添加した。それ以上の加熱は行わなかった。温度が25℃に冷めるまで混合物を撹拌した。43.8kgのメチルtert−ブチルエーテル(MtBE)を冷却混合物に撹拌しながら添加して、窒素ガス体下で一晩混合した。反応混合物を遠心分離した。反応器を同量のMtBEでさらに4回すすいだ。このすすぎ水を使用して遠心分離ケークを洗浄した。ブレンダー乾燥機内で、ポリエチレングリコールジスクシネートのケークを真空下、30℃でおよそ19.5時間乾燥した。10.9kgのポリエチレングリコールジスクシネートが回収された。
【0036】
30ガロンガラス内張反応器に、5.6kgのポリエチレングリコールジスクシネート、540gのN−ヒドロキシスクシンイミド、84gの4−ジメチルアミノピリジン、および8.2kgのアセトニトリルを装填した。全固形物が溶解するまで、装填物を室温で1時間混合した。1.3kgの1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミドを添加した。混合物を窒素ガスシール下で、25℃で6時間撹拌した。次に39.6kgの無水エタノールを反応混合物に撹拌しながら添加した。混合物を75ガロンポータブルガラス内張反応器に移した。80.0kgのMtBEを撹拌しながら添加した。冷却ジャケットを使用して混合物を一晩撹拌した。翌朝、反応混合物は5〜7℃であった。反応混合物を遠心分離して、反応フラスコを同量のMtBEで4回すすいだ。このすすぎ水を使用して遠心分離ケークを洗浄した。遠心分離した濡れたケークを40kg無水エタノールと共に30ガロンガラス内張反応器に加えた。反応器を窒素ガス体下、周囲温度でおよそ1時間混合した。混合物を遠心分離して、反応器を40kgの無水エタノールですすぎ、このすすぎ水を使用して遠心分離ケークを洗浄した。次に反応器を40kgのMtBEですすぎ、このこのすすぎ水を使用して遠心分離ケークを洗浄した。ケークのおよそ半分をタンブル乾燥機に移して、真空下、30℃でおよそ18時間乾燥した。液体窒素冷却を使用して、2.7kgの乾燥生成物を粉砕した。ケークの半分からそれぞれ2.55kgのポリエチレングリコールジスクシンイミジルスクシネート、PEG−(SS)2が回収された。
【0037】
実施例1
予備成形されたアルブミンポリマープラグからの5−フルオロウラシル(5FU)の放出
シリンジバレルを切断して開放円筒容器を形成した改造5mlポリシリンジ内で、29%アルブミン溶液(テキサス州セギーンのAmerican Biological Technologyから入手できる、あるいは上述したダイアフィルトレーション(手順B)によって得られる)0.5mlと、136mg/mlのPEG−(SS)2架橋剤0.5mlとを混合して、2つのポリマープラグを予備成形した。架橋完了後(15分間)、プラグをシリンジから除去して押し出した。ポリマープラグを真空下で24時間乾燥させた。各プラグが全部で187.6μg、または165.5μgの5FUを含有するように、プラグを2mlの5FU溶液(1.04mg/ml、ミズーリ州セントルイスのSigma Chemical Co.)で戻した。各プラグを別々のバイアルに入れて、pH7.4のリン酸緩衝食塩水5mlをサンプル時間0(t0)時に添加した。バイアルを室温に保持して絶え間なく振盪した。各サンプル時間毎にバイアルの全内容物を取り出して、5mlの新鮮なPBSと入れ替えた。サンプルは0.25時間、0.5時間、1時間、2時間、3時間、4時間、5時間、6時間、および24時間目に採取した。5FUの分析は、BernatchezらのInt.J.Pharm.、106、161〜166ページ(1994年)の方法に従って、高圧液体クロマトグラフィーによって実施した。図1のデータは、5時間の間に5FUが放出されたことを示す。
【0038】
実施例2
バイオポリマープラグからのテトラサイクリンの放出
アルギン酸(30mg、ミズーリ州セントルイスのSigma Chemical Co.)を1.01mg/mlテトラサイクリン(ミズーリ州セントルイスのSigmaChemicalCo.)1mlに添加した。CloropHast pHストリップ(イリノイ州イタスカのFischerCompany)を使用して、1NのNaOHで混合物のpHを6.8にして、次にDRIERITE(オハイオ州ジーニアのW.A.Hammond Drierite Co.)と共に真空デシケーターに入れて24時間乾燥した。得られたルギン酸塩/テトラサイクリンフィルムを粉砕して30%アルブミン溶液(手順B)中に混合し、直ちに5mlポリシリンジ型に入れて等容積のPEG−(SS)2架橋剤(136mg/ml)で架橋した。各プラグを別々のバイアルに入れて、pH7.4のリン酸緩衝食塩水(PBS)5mlをサンプル時間0(t0)時に添加した。バイアルを室温に保持して絶え間なく振盪した。各サンプル時間毎にバイアルの全内容物を取り出して、5mlの新鮮なPBSと入れ替えた。サンプルを1時間、2時間、3時間、5時間、および24時間目に採取した。可変波長検出器を有するHewlett Packard(カリフォルニア州パロアルト)HPLCモデル1190、およびSupelco Co.(ペンシルベニア州ベルフォンテ)からの逆相LC−8−DBカラム(15.0cm×4.6mm、粒度5μm)でサンプルを分析した。HPLC分析のために使用した移動相は、比率が45:45:10:1の0.05Mリン酸アンモニウム緩衝液(pH6.8)、メタノール、アセトニトリル、およびトリエチルアミンから構成された。移動相の流速は1.0ml/分であり、カラム温度は25℃に保った。テトラサイクリンは280nmで検出された。
【0039】
図2のデータは、5時間の間に約70%のテトラサイクリンが放出されたことを示す。収集テトラサイクリン量は、テトラサイクリンの分解による損失について補正しなかった。
【0040】
実施例3
架橋剤およびアルブミンと混合された5FUから製造されるプラグからの5−フルオロウラシルの放出
136mgのPEG−(SS)2架橋剤を1.04mg/mlの5−フルオロウラシル溶液1mlに溶解した。次にこの溶液をあらかじめ5mlの型に入れた1mlの30%アルブミン溶液(実施例2参照)中に混合した。得られたヒドロゲルプラグを15分後に型から取り出して、3つの等しい寸法の小片に切断した(厚さおよそ0.5cm)。次に各小片をバイアルに入れて、4mlのPBS緩衝液(pH7.4)をサンプル時間0(t0)時に添加した。バイアルを室温に保持して絶え間なく振盪した。各サンプル時間毎にバイアルの全内容物を取り出して、4mlの新鮮なPBSと入れ替えた。サンプルは1時間、2時間、3時間、4時間、および8時間目に採取した。5−フルオロウラシルを実施例1に述べるようにして分析した。
【0041】
図3のデータは、8時間の間に約55%の5−フルオロウラシルが放出されたことを示す。残る5FUは、恐らくポリマー中に架橋されており、ポリマーが分解すれば放出される。
【0042】
実施例4
カルシウム無添加のアルギン酸を含有するアルブミンプラグからの5−フルオロウラシルの放出
1.04mg/mlの5−フルオロウラシル1mlを30mgのアルギン酸と混合した。混合物のpHを6.8にして、次にDRIERITEと共に真空デシケーター内に入れて24時間乾燥させた。得られたアルギン酸塩/5FUフィルムを粉砕し、30%アルブミン溶液(実施例2参照)中に混合し、直ちに5mlシリンジ型に入れて等容積のPEG−(SS)2架橋剤(136mg/ml)で架橋した。各プラグを別々のバイアルに入れて、pH7.4のリン酸緩衝食塩水(PBS)5mlをサンプル時間0(t0)時に添加した。バイアルを室温に保持して絶え間なく振盪した。各サンプル時間毎にバイアルの全内容物を取り出して、5mlの新鮮なPBSと入れ替えた。サンプルを1時間、2時間、3時間、4時間、5時間、および24時間目に採取した。
【0043】
5−FUを実施例1に述べるようにして分析した。図4のデータは、5時間の間に約40%の5FUが放出されたことを示す。
【0044】
実施例5
カルシウム添加アルギン酸を含有するアルブミンプラグからの5−フルオロウラシルの放出
1.04mg/mlの5−フルオロウラシル1mlを30mgのアルギン酸と混合した。混合物のpHを6.8にして、150μlの飽和塩化カルシウム/水溶液を添加してアルギン酸塩をゲル化した。ゲルを3個のほぼ同じ容積に分割し、次にDRIERITEと共に真空デシケーター内に入れて24時間乾燥させた。得られたアルギン酸塩/Ca/5FUフィルムを粉砕し、30%アルブミン溶液(実施例2参照)中に混合し、直ちに5mlシリンジ型に入れて等容積のPEG−(SS)2架橋剤(136mg/ml)で架橋した。各プラグを別々のバイアルに入れて、pH7.4のリン酸緩衝食塩水(PBS)5mlをサンプル時間0(t0)時に添加した。バイアルを室温に保持して絶え間なく振盪した。各サンプル時間毎にバイアルの全内容物を取り出して、5mlの新鮮なPBSと入れ替えた。サンプルを1時間、2時間、3時間、4時間、5時間、および24時間目に採取した。
【0045】
5−FUを実施例1に述べるようにして分析した。図4のデータは、5時間の間に約60%の5FUが放出されたことを示す。
【0046】
実施例6
生分解性ビーズの製造方法
二液システムからビーズを調製した。パートAは、等張(0.075M)炭酸緩衝液(pH9)中の29%アルブミン滅菌溶液(実施例1参照)であった。パートBは、使用直前に滅菌水溶液で戻したPEG−(SS)2の260mg/ml溶液であった。静止ミクシングヘッドに接続したデュアルシリンジシステムによって、溶液AおよびBを等容積で混合した。8mm径の孔を有する6インチ×4インチ×1インチのテフロン金型内に液体を注入して、ビーズを調製した。注入した混合物を8mm径の孔内で10分間硬化した。ビーズをテフロン金型から10分後に取り出した。ビーズは2つの異なるやり方で使用できる。一例では水和したビーズをテフロン金型から取り出して、直接使用できる。別の例では、ビーズを金型から取り出した後に脱水できる。乾燥は、真空下または空気中、またはその他の適切な様式で実施できる。球状のビーズ以外にも様々な形状寸法が製造できることに留意すべきである。
【0047】
実施例7
バンコマイシンの試験管内(in vitro)放出試験
実施例6から製造された2個の脱水されたビーズを4mlバイアルに入れた。バンコマイシン(60mg、イリノイ州ノースシカゴのAbbott Laboratories)を2mlの滅菌水に溶解してバイアルに入れた。ビーズを室温で24時間浸漬した。24時間の浸漬後にビーズを取り出して、pH7.4のリン酸緩衝食塩水5mlと共に20mlガラス瓶に入れた。バンコマイシン放出速度は定温水浴内で37℃で測定した。緩衝液を24時間毎に最高3週間まで交換した。UV分光光度計(DU640、カリフォルニア州フラートンのBeckman)によって、これらのサンプルをバンコマイシンについて分析した。バンコマイシンは282nmで検出された。結果を図5および6に示す。
【0048】
実施例8
セファゾリンの試験管内(in vitro)放出
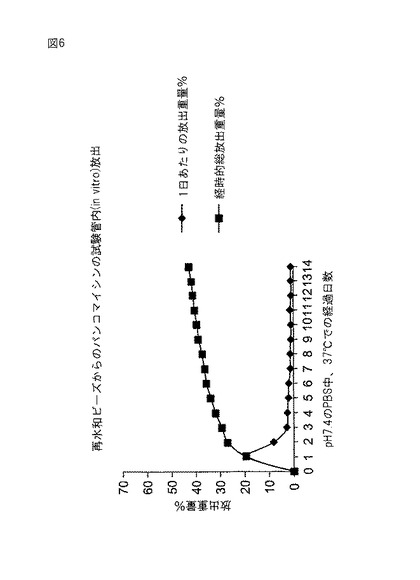
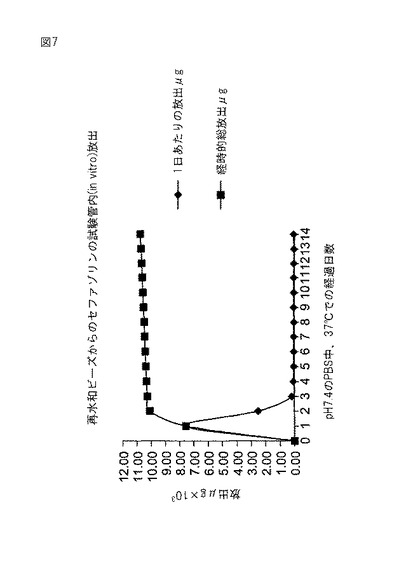
実施例6から作られた2個の脱水されたビーズを4mlバイアルに入れた。セファゾリン(60mg、ニュージャージー州チェリーヒルのMarsam Pharmaceuticals,Inc.)を2mlの滅菌水に溶解してバイアルに入れた。ビーズを室温で24時間浸漬した。24時間の浸漬後にビーズを取り出して、pH7.4のリン酸緩衝食塩水5mlと共に20mlガラス瓶に入れた。セファゾリン放出速度は定温水浴内で37℃で測定した。緩衝液を24時間毎に最高3週間まで交換した。UV−分光光度計(Beckman,DU640,カリフォルニア州フラートン)によって、これらのサンプルをセファゾリンについて分析した。セファゾリンは272nmで吸光する。結果を図7および8に示す。
【0049】
実施例9
架橋程度および溶剤容積の関数としての再水和されたビーズの水取り込み
PEG−(SS)2のいくつかの架橋剤調合物を使用したこと以外は、実施例6と同様にしてビーズを製造した。標準の密封材(NS)調合物からできたビーズは、1mlの水中の130mgのPEG−(SS)2と、pH9.0の炭酸緩衝液中の1mlの29%アルブミンとを混合して製造した。架橋剤濃度が半分(NS*1/2)のビーズでは、ビーズは1mlの水中に65mgのPEG−(SS)2、pH9.0の炭酸緩衝液中に1mlの29%アルブミンを含有した。架橋剤濃度が2倍(NS*2)のビーズでは、ビーズは1mlの水中に260mgのPEG−(SS)2、pH9.0の炭酸緩衝液中に1mlの29%アルブミンを含有した。架橋剤濃度が3倍(NS*3)のビーズでは、ビーズは1mlの水中に390mgのPEG−(SS)2、pH9.0の炭酸緩衝液中に1mlの29%アルブミンを含有した。各調合物からの3個のビーズを20mlのあらかじめ秤量したバイアルに入れた。3mlの水を秤量してバイアルに加えた。6mlの水を使用して同じ試験を繰り返した。ビーズを取り出してバイアル中の水を秤量し、ビーズの水取り込みを2、4、6、8、および24時間目に測定した。結果を表1に示す。
【0050】
【表1】

【0051】
実施例10
ビーズの直径および架橋程度の関数としての水和されたビーズの水取り込み
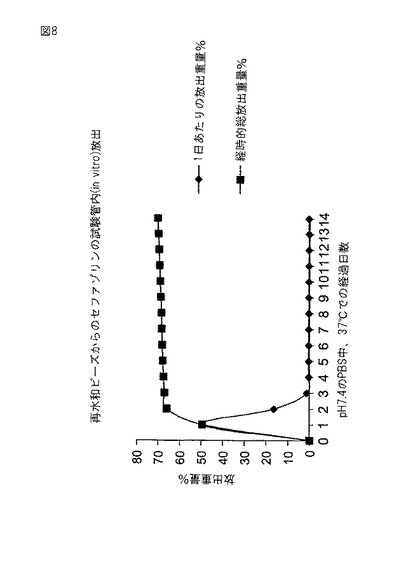
ビーズは実施例6と同様にして製造した。標準、2倍、3倍の架橋剤濃度のビーズは、実施例9で述べたようにして調製した。各調合物からの3個のビーズを20mlのあらかじめ秤量したバイアルに入れた。6mlの水を秤量して、バイアルに加えた。ビーズをバイアルから取り出してバイアル内に残留する水を秤量し、ビーズの水取り込みおよびビーズ径を2、4、6、8、および24時間目に測定した。1個のビーズと2mlの水で試験を繰り返した。ビーズ径はScherr−Tumico Optical Comparator(S.T.Industries)を使用して測定した。複数のビーズについての結果を表2に、単一ビーズについての結果を表3に示す。表4は単一ビーズ試験と複数ビーズ試験における、乾燥ビーズ容積と比較した容積増大%を乾燥ビーズの再水和に伴う時間の関数として示す。図9はビーズ1個の試験の水取り込みに関するこのデータを示す。ビーズ3個の試験は同様の結果をもたらした。
【0052】
【表2】
【0053】
【表3】
【0054】
【表4】
【0055】
実施例11
架橋されたアルブミン微小球の調製
米国特許番号第5,508,060号に従って、種々の修正を加えて、油中水型エマルジョン法によって微小球を調製した。米国特許番号第5,583,114号(Barrowsら)の実施例8で教示されるようにしてヒト血清アルブミン(HSA)溶液を使用し、ウシ血清アルブミン(BSA)Fraction Vは、Sigma Chemical Co.(ミズーリ州セントルイス)から購入した。PEG−(SS)2を架橋剤として使用した。概して、電動の3枚羽根プロペラタイプ撹拌機(Motomatic、米国のElectro−Craft)を使用して、ピーナツ油(Nabisco Foods,Inc.、ニュージャージー州イーストハノーバー)を所定速度で撹拌した。絶え間なく撹拌しながら油浴中に皮下注射針で滴下して、アルブミン溶液と混合した架橋剤の水性溶液を添加した。別の変法では、架橋剤溶液を油に添加してからアルブミン溶液を滴下して加えた。アルブミンと架橋剤との容積比は1:1であり、使用した架橋剤濃度は138mg/mlおよび276mg/mlであった。油と水との比率はおよそ100:1であり、300〜500mlの範囲の油を使用した。所定時間の撹拌後、微小球を収集して酢酸エチルで洗浄し、0.44μmまたは1.1μmのセルロースまたはナイロンフィルターを通して濾過した。
【0056】
実施例11A
以下の修正と共に実施例11の手順を使用して微小球を調製した。ヒト血清アルブミン(HSA)はpH9.4で27%濃度で使用した。PEG−(SS)2溶液は138mg/mlの濃度であった。PEG−(SS)2溶液を油に添加して、次にアルブミン溶液を27ゲージの針を通して滴下して撹拌中の油に添加した。3枚羽根プロペラタイプ撹拌機を使用して、油を1000rpmで15分間撹拌した。撹拌停止後、溶液を85℃に加熱して、実施例11と同様に微小球を濾過して洗浄した。溶液の加熱中に媒体温度が約45℃に達すると、架橋された粒子が凝集しはじめ、混合物から白色フレークが形成した。凝集した粒子を除去して微小球を収集した。この工程から回収された微小球の典型的な画像分析は、平均径が4.22μmで、1.0μm〜15μmの範囲内の粒度分布を有した。
【0057】
実施例11B
撹拌速度が1300rpmで撹拌後に溶液を加熱しなかったこと以外は、実施例11Aで使用したのと同一手順によって微小球を製造した。この工程から回収された微小球の典型的な画像分析は、平均径が3.05μmで、1.0μm〜10μmの範囲内の粒度分布を有した。
【0058】
実施例11C
PEG−(SS)2架橋剤濃度が276mg/ml、撹拌速度が1300rpmで撹拌時間が20分であり、撹拌後に溶液を加熱しなかったこと以外は、実施例11Aで使用したのと同一手順によって微小球を製造した。この工程から回収された微小球の典型的な画像分析は、平均径が5.45μmで、0.5μm〜16μmの範囲内の粒度分布を有した。これらの微小球を実施例12の医薬品装填試験に使用した。
【0059】
実施例11D
pH9.17で15%の血清アルブミン(BSA)溶液を使用し、撹拌速度が1500rpmで撹拌時間が20分であり、撹拌後に溶液を加熱しなかったこと以外は、実施例11Aで使用したのと同一手順によって微小球を製造した。この工程から回収された微小球の典型的な画像分析は、平均径が2.42μmで、1.0μm〜6.0μmの範囲内の粒度分布を有した。
【0060】
実施例12
テトラサイクリンを用いた現場での(in situ)微小球への医薬品装填
2mlのHSA溶液と、等容積の油浴中に医薬品を含有する架橋剤溶液とを混合して、実施例11Cの方法に従って塩酸テトラサイクリンの存在下で微小球を調製した。簡単に述べると、552mgのPEG−(SS)2架橋剤を2mlのテトラサイクリン溶液(10mg/ml)に添加して、架橋剤溶液を調製した。医薬品を含有するPEG−(SS)2溶液を1300rpmで撹拌しながら油に添加した。水性溶液が安定したエマルジョンを形成するのに十分な時間をおいてから、2mlのヒト血清アルブミン(27%、pH9.4)を滴下して添加しエマルジョンを20分間撹拌した。酢酸エチルで洗浄後、粒子は良好に分離した微粉末状を呈した。画像分析からは、粒子が良好に分離して小型であることが示された。
【0061】
実施例13
試験管中での(in vitro)医薬品放出評価
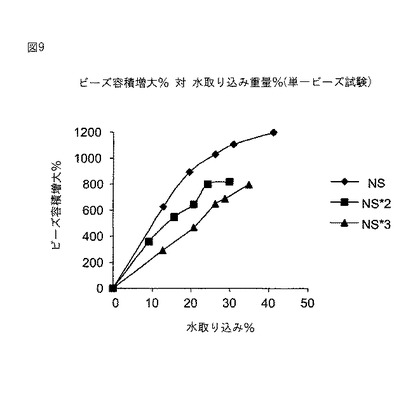
微小球(実施例11A参照)を拡張放出試験器(BIO−DIS、ニュージャージー州エジソンのVanKel)の容器に入れて、試験管中での(in vitro)医薬品放出の試験を実施した。1分あたり浸漬3回の撹拌速度、初期遅延l5秒で、100mlのリン酸緩衝食塩水(PBS、pH7.4)を含有する200mlガラスビーカー内に容器を浸漬した。放出試験中、水浴の温度は電子的に37℃に保った。所定時間経過後、1mlのサンプルを取り出して、新鮮な溶液をPBSレザバーに添加した。0.22m COSTARシリンジフィルター(ニューヨーク州コーニングのCorning)を通してサンプル溶液を濾過し、あらゆる微粒子を除去した。塩化テトラサイクリンの定量は、HPLC法を使用して分光測定UV検出により実施した。使用した装置は、注入器(ShimadzuモデルSil−9A、メリーランド州コロンビアのShimadzu)、溶剤デリバリシステム(Waters 625LC、マサチューセッツ州ミルフォードのMillipore Corporation)、マルチ波長検出器(Waters 490E、マサチューセッツ州ミルフォードのMillipore Corporation)、および積分器(Waters Millennium、マサチューセッツミルフォードのMillipore Corporation)から構成された。医薬品を含有する溶液(20μl〜150μl、直線検出範囲に入るように調節済み)をBeckman逆相C−18カラム(5μm×4.6mm×25cm、米国のBeckman)に注入した。移動相は0.1%のトリフルオロ酢酸を含有する、水とアセトニトリルの80/20混合物であった。流速は1.0ml/分であり、使用したUV波長は275nmであった。医薬品濃度は、医薬品ピークの下の面積と標準曲線から得られたものとを比較して測定した。各サンプルを少なくとも2回分析し、平均放出医薬品総量を計算した。
【0062】
図10は、3つの異なる大きさ範囲の微小球からの塩酸テトラサイクリンの放出プロフィールを示す。分粒は較差沈降法によって実施した。3つの標本全てで初期のバースト放出が観察された。しかしバースト効果に対する顕著な粒度の影響があった。微小球からの初期の医薬品流量は粒度が増大するにつれて低下し、初期のバースト相が主に微小球表面積に左右されることが示された。
【0063】
実施例14
4−アーム PEG架橋剤を用いて製造したアルブミンポリマープラグからのセファゾリンの放出
30%アルブミン溶液(手順B)0.5mlと、110mg/mlのポリ(エチレングリコール)−スクシネート架橋剤の4−アーム−N−ヒドロキシスクシンイミジルエステル(アラバマ州ハンツビルのShearwater Polymers,Inc.)0.5mlから、2個のポリマープラグを予備成形した。架橋剤を添加してシリンダー状プラグを形成する前に、およそ10mgのナトリウムセファゾリン(ミズーリ州セントルイスのSigma Chemical Co.)をアルブミン中に溶解した。各形成プラグを別々のバイアルに入れて、pH7.4のリン酸緩衝食塩水(PBS)8mlをサンプル時間(t0)時に添加した。バイアルを室温に保持して絶え間なく振盪した。各サンプル時間毎にバイアルの全内容物を取り出して、8mlの新鮮なPBSと入れ替えた。サンプルを0.25時間、0.5時間、1時間、2時間、3時間、4時間、5時間、および24時間目に採取した。それ以降は144時間目まで、およそ24時間毎にサンプルを採取した。図11のデータは、最初の5時間でおよそ67%のセファゾリンが、144時間で全セファゾリンの95.6%が放出されることを示す。
【0064】
LiangらのJ.Chromatog.B.、656、45〜465ページ(1994年)の方法に従って、高圧液体クロマトグラフィーによってセファゾリンを分析した。カラムはMICROBONDAPAKの商品名の下に市販されるWaters C−18(3.9×300mm)カラムであり、カラム温度を25℃に保った。移動相は0.02Mのリン酸ナトリウム緩衝液(pH=5.0)およびメタノール(77:23)から構成された。検出に使用した波長は270nmであり、カラム流速は1.0ml/分であった。
【0065】
実施例15
生分解性の予備成形されたアルブミンビーズからのTGF−β1放出
以下のバリエーションで実施例6で述べた方法に従って、ビーズを調製した。実施例6で述べたのと全く同じようにして2個のビーズを調製し、それらの水和した形態(ビーズNS1およびNS2)で使用した。実施例6で述べたのと全く同じようにして2個のビーズを調製し、脱水して水中で再水和した(ビーズNS3およびNS4)。実施例6のパートBに3.75μg/mlヒト組換えトランスフォーミング増殖因子β1(TGF−β1、メリーランド州ロックビルのGibco BRL)20μlを加えて2個のビーズを調製し、ビーズあたり25ngのTGF−β1の最終濃度を得て、それらの水和した形態で使用した(ビーズNS5およびNS6)。実施例6で述べたのと全く同じようにして2個のビーズを調製し、脱水して50ng/mlのTGF−β1溶液2ml中で4時間再水和した(4時間で25%の取り込みが見られたので100ngの内25ngがNS7およびNS8の各ビーズに組み込まれた)。等張(0.075M)炭酸緩衝液(pH8.66)中の23%の滅菌アルブミン溶液(手順B)から成る実施例6のパートA、およびPEG(SS)2の65mg/ml溶液から成るパートB(実施例1参照)から2個のビーズを調製し、滅菌水中で使用直前に戻した(ビーズFS1およびFS2)。等張(0.075M)炭酸緩衝液(pH8.66)中の23%の滅菌アルブミン溶液(手順B)から成る実施例6のパートA、およびPEG(SS)2の65mg/ml溶液から成るパートB(実施例1参照)から2個のビーズを調製し、20μlの3.75μg/mlヒト組換えTGF−β1を添加した滅菌水中で使用直前に戻した(ビーズFS3およびFS4)。
【0066】
12ウェルペトリプレート(ペンシルベニア州ピッツバーグのFisher Scientific)の個々のウェル内で、10%ウシ胎児血清(バージニア州マナッサスのAmerican Type Culture Collection)およびペニシリン/ストレプトマイシン100U/ml(メリーランド州ロックビルのGibco BRL)を含有する4mlの修正Eagle’s Minimum Essential Cell Culture Medium(バージニア州マナッサスのAmerican Type Culture Collection)に全てのビーズを入れた。試験の実施期間中、プレートを細胞培養器(37℃、5%のCO2)に入れた。サンプルを経時的に(t=0から開始)採取して、ビーズが完全に分解するまで各時点で培養液の全容積を置き換えた。各時点の培養液はクリオバイアルに収集して、−20℃で貯蔵した。採取時点は、NSビーズでは0時間、24時間、48時間、72時間、96時間、120時間、144時間、168時間、192時間、216時間、240時間、264時間、289時間、および330時間であり、FSビーズでは、0時間、24時間、48時間、72時間、96時間、および120時間であった。
【0067】
次にTGF−β1濃度を求めるための酵素結合抗体免疫吸着アッセイ(ELISA)(ヒトTGF−β1用QUANTIKINE、ミネソタ州ミネアポリスのR & D Systems)で、製造元の取扱説明書に従って培養液の全サンプルをアッセイした。測定されたTGF−β1は活性形態で販売され、培養中の細胞が生成したものではなかったので、サンプルは活性化を必要としなかった。ビーズの製造で使用したのと同一供給元のTGF−β1を用いて別に用量反応曲線を生成し、この後者の用量反応曲線に従って結果を計算した。
【0068】
図12の結果は、ビーズ(NS7、NS8)の再水和に使用される溶液に増殖因子を組み込むことで、組み込まれた増殖因子の40%までが2〜6日中に放出されることを示す。架橋剤(NS5、NS6、FS3、FS4)へのTGF−β1の組み込みは、ビーズからの顕著な量のTGF放出をもたらさなかった。対照ビーズ(TGF−β1を含まないNS1、NS2、NS3、NS4、FS1、FS2)を試験したところ、アッセイを妨害しなかった。
【0069】
前出の記述が例証のみを目的とするものであり、発明の範囲を逸脱することなく様々な実施態様が想定されるできることは、当業者によって容易に理解される。
【技術分野】
【0001】
背景
医薬品などの活性薬剤を好ましくは薬剤の徐放のためにサブジェクトにデリバリできるようにする、様々なアプローチが発達している。このようなデリバリシステムは典型的に、サブジェクトの目標領域への薬剤の徐放を可能にしながら、デリバリに先立って環境から薬剤を保護するようにデザインされている。
【0002】
いくつかの従来の徐放システムは、リポソーム、リポ球体、マイクロカプセル、微小粒子、およびナノ粒子などの微細構造、並びにシリンダー、ディスク、および繊維などのマクロ構造に基づく。典型的に医薬品などの活性薬剤がポリマーと混合され、次に所望の形状に形成される。
【0003】
このような従来のシステムの多くは、補綴用途に必要な構造一体性を有する固形移植片を形成するのには使用できない。さらにこのようなシステムの多くは、例えばサブジェクトへの適用時に、医師が活性薬剤(例えば医薬品)を注入できる物品を形成するのには使用できない。またこのようなシステムの多くは、生分解速度、および/またはあらゆる組み込まれた活性薬剤の放出速度に関して容易に制御できないポリマーを含む。
【0004】
アルブミンとポリエチレングリコール(PEG)誘導体が架橋して製造されるヒドロゲル(例えば水性媒体中で膨潤するが、水に溶けないポリマーのクラス)は、可能な医薬品デリバリ用途について既に研究されている(D’Ursoら、Biotech.Tech.、8、71〜76ページ(1994年))。医薬品の制御されたデリバリに対する別のアプローチは、ポリ(乳酸)や、ラクチドとグリコリドの種々のコポリマーなどの合成生体吸収性ポリマーの使用による、マイクロカプセル封入または微小球の形成を伴う。しかし微小球使用の欠点は、外科手術部位表面あるいは負傷または病変組織上に、均一に被覆して保持できないことである。この問題と取り組むために、微小球には非溶剤である溶剤の使用によって作られる第2のポリマー溶液に微小球を懸濁し、次に混合物からフィルムをキャスティングして、微小球含有メンブランが作られている。
【発明の概要】
【発明が解決しようとする課題】
【0005】
したがってその他のデリバリシステム、特に、様々な生分解速度およびデリバリ速度を提供できる、変化させられるポリマー組成物を含むものに対する必要性がある。好ましくは十分な構造一体性を有して取り扱いが容易な、ポリマー組成物を含むデリバリシステムに対する必要性がある。
【課題を解決するための手段】
【0006】
要約
本発明は、活性薬剤をサブジェクトにデリバリするための予備成形されたオブジェクトを提供し、予備成形されたオブジェクトは、式、
I−(−X−LM−G)n
(式中、
Xは二官能性ポリオキシエチレン鎖部分または結合であり、
LMは式、−C(O)−、−(CH2)bC(O)−(式中、bは1〜5の整数である。)、−C(O)−(CH2)c−C(O)−(式中、cは2〜10の整数であり、ラジカルの脂肪族部分は飽和または不飽和でも良い。)、−C(O)−O−(CH2)d−O−C(O)−(式中、dは2〜10の整数であり、あるいは式、−R−C(O)−、−R−C(O)−(CH2)c−C(O)−、または−R−C(O)−O−(CH2)d−O−C(O)−(式中、cは2〜10の整数であり、dは2〜10の整数であり、Rは1〜10個のモノマーラクチド、グリコリド、炭酸トリメチレン、カプロラクトンまたはp−ジオキサノン断片を有するポリマーまたはコポリマーである。)によって表されるオリゴマージラジカルである。)によって表される二官能性結合部分であり、
GはN−オキシスクシンイミジル、N−オキシマレイミジル、N−オキシフタルイミジル、ニトロフェノキシル、N−オキシイミダゾリル、およびトレシルの群より選択される脱離基であり、
Iは多求核性化合物から誘導される多官能性結合部分であり、
nは2〜10の整数であるが、
ただしXが二官能性ポリオキシエチレン鎖部分の場合、−X−I−X−は、式、
−O−(CH2−CH2−O−)a−
(式中、aは20〜300の整数である。)によって表されるジラジカル断片であるPEGである。)
の架橋剤を有する、少なくとも部分的に脱溶媒和された架橋されたアルブミンである。
【0007】
本発明のその他の実施態様としては、その中に組み込まれた再溶媒和していてもしていなくても良い活性薬剤を有する予備成形されたオブジェクトが挙げられ、その中に活性薬剤を有する予備成形されたオブジェクトは、二次生分解性マトリックス内にさらに組み込まれる。その他の実施態様としては、予備成形されたオブジェクトについて上述した化学的性質を有する、二次生分解性マトリックス内にさらに組み込まれた、その中に活性薬剤を有するその他の生分解性ポリマーの予備成形されたオブジェクトが挙げられる。製造方法、および活性薬剤をサブジェクトにデリバリする方法も本発明によって提供される。
【図面の簡単な説明】
【0008】
【図1】図1は、PEG−(SS)2架橋剤で架橋されたアルブミンの予備成形されたプラグからの5−フルオロウラシル(5−FU)の放出を示す。
【図2】図2は、PEG−SS2架橋剤との架橋直前にアルブミンに混合された、テトラサイクリンのアルギン酸塩/医薬品複合体からの放出を示す。
【図3】図3は、プラグ製造中に5−FUが架橋剤と共に添加されたアルブミン/PEG−(SS)2プラグからの5−FUの放出を示す。
【図4】図4は、塩化カルシウムが添加されたアルブミン/PEG−(SS)2プラグからの5−FUの放出を示す。
【図5】図5は、再水和されたアルブミン/PEG−(SS)2ビーズからのバンコマイシンの放出を示す。
【図6】図6は、再水和されたアルブミン/PEG−(SS)2ビーズからのバンコマイシンの放出重量%を示す。
【図7】図7は、再水和されたアルブミン/PEG−(SS)2ビーズからのセファゾリン放出を示す。
【図8】図8は、再水和されたアルブミン/PEG−(SS)2ビーズからのセファゾリンの放出重量%を示す。
【図9】図9は、単一ビーズ試験におけるポリマービーズの容積増大%対水取り込み重量%を示す。
【図10】図10は、微小球からのテトラサイクリンの放出プロフィールを示す。
【図11】図11は、アルブミン/4−アーム PEGプラグからのセファゾリンの放出を示す。
【図12】図12は、種々のビーズから放出されるTGF−β1の百分率を示す。
【発明を実施するための形態】
【0009】
好ましい実施態様の詳細な説明
本発明は、第1の生分解性組成物、および任意に医薬品などの活性薬剤を含む、サブジェクトの目標領域へのデリバリのための予備成形されたオブジェクト、好ましくは予備成形された自立型オブジェクトを提供する。予備成形されたオブジェクトは少なくとも部分的に脱溶媒和させて(例えば少なくとも部分的に脱水して)、引き続いて活性薬剤と組み合わせることができ、あるいは予備成形されたオブジェクトの調製時に活性薬剤を含めることができる。このような予備成形されたオブジェクトは好ましくは、開始材料(例えばタンパク質および架橋剤)を組織に適用してサブジェクト組織存在下で硬化(例えば架橋)させて、組織を結合または封着するのに従来法で使用される生分解性組成物から調製される。しかしここでは好ましいオブジェクトは、同一または同様の組成物から調製されて硬化され(例えば架橋され)、次にサブジェクトの組織に適用される。したがって典型的に予備成形されたオブジェクトと組織の間に結合相互作用はない。
【0010】
本発明の好ましい予備成形されたオブジェクトは、十分な構造一体性を有して、ひとたび形成(例えば硬化)されると、適切な条件下で(例えば脱溶媒和させた場合は水分不在下で、溶媒和させた場合は約4℃の温度で)保管する限りは、それらの全体的形状を維持して自立型である。予備成形されたオブジェクト製造に使用される構成要素の選択次第で、機械的強度、可撓性、硬化速度、および生分解速度の程度が変化できる。本発明の好ましい予備成形されたオブジェクトは、架橋されたタンパク質を含む。それらは典型的に緩衝化された塩基性タンパク質溶液と、多官能性、典型的には二官能性の架橋剤とから調製される。緩衝化されたタンパク質溶液および架橋剤は、典型的に市販される材料を使用して得られるが、これらの材料のほとんどは概して臨床安全性および/または用途の経歴を有することが利点である。
【0011】
本組成物中で使用するのに適切なタンパク質としては、非免疫原性の水溶性タンパク質、好ましくはアルブミン(より好ましくは血清アルブミン、そして最も好ましくはヒト血清アルブミン)が挙げられる。本発明の予備成形されたオブジェクトを調製するのに使用しても良い、好ましい緩衝化されたタンパク質溶液は、約8.0〜約11.0のpHに緩衝化された濃縮水性血清アルブミンを含み、緩衝液濃度は約0.01モル濃度〜約0.25モル濃度の範囲である。適切な緩衝液システムとしては、緩衝液が架橋剤と不利に反応したりそれを変性させたりしない限り、既知の炭酸またはリン酸緩衝液システムなどの生理学的および/または臨床的に許容可能な緩衝液が挙げられる。好ましい緩衝液システムは、pHの値が約9.0〜約10.5で濃度が約0.05〜約0.15モル濃度の範囲の炭酸塩/炭酸水素塩緩衝液システムである。
【0012】
血清アルブミンは既知の分離工程を使用して、血清から容易に分離できる。さらに遺伝子形質転換細胞からアルブミンを製造することが可能である。例えばQuirkらの報告Biotechnology and Applied Biochemistry、11、273〜287ページ(1989年)、KalmanらのNucleic Acids Research、18、6075〜6081ページ(1990年)、SleepらのBiotechnology、8、42〜46ページ(1990年)、およびSijmonsらのBiotechnology、8、217〜221(1990年)を参照されたい。組換えアルブミンを製造する能力は、この方法によって製造されたタンパク質が、ヒト血清から直接分離されたアルブミンを汚染しているかもしれないヒト病原体、ウイルスまたはその他の汚染物質を含まない利点を提供する。
【0013】
本緩衝化された混合物中で使用される場合、血清アルブミンは変性しないことが分かった。使用前にアルブミンが変性されないので、アルブミンがその天然のコイル状立体配座を維持するために、ゲル様の固体を提供する硬化工程中に架橋された後、硬化した予備成形されたオブジェクトは十分な可撓性を維持して、所望ならば組織移植のための適切なマトリックスが提供されると考えられる。
【0014】
本発明中で様々な適切な架橋剤を使用しても良い。架橋剤は多官能性で好ましくは二官能性である。本発明で使用するのに適切な架橋剤としては、式、
I−(−X−LM−G)n
(式中、Xは二官能性ポリオキシエチレン鎖部分または結合であり、LMは二官能性結合部分であり、Gは活性脱離基であり、Iは多求核性化合物(例えばエチレングリコール、ペンタエリスリトール、トリメチロールプロパン、多求核性アミンなど)に由来する多官能性結合部分であり、nは約2〜約10、好ましくは約2〜約4であるが、ただしXが二官能性ポリオキシエチレン鎖部分である場合、−X−I−X−はここで定義したように−PEG−である。)の化合物が挙げられる。
【0015】
好ましい架橋剤は、二官能性であり、式、
G−LM−PEG−LM−G
(式中、−PEG−は、式、
−O−(CH2−CH2−O−)a−
(式中、aは約20〜約300の整数である。)によって表されるジラジカル断片であり、−LM−は、式、−C(O)−、−(CH2)bC(O)−(式中、bは約1〜約5の整数である。)、−C(O)−(CH2)c−C(O)−(式中、cは約2〜約10の整数であり、ラジカルの脂肪族部分は飽和または不飽和でも良い。)、−C(O)−O−(CH2)d−O−C(O)−(式中、dは約2〜約10の整数であり、あるいは式、−R−C(O)−、−R−C(O)−(CH2)c−C(O)−、または−R−C(O)−O−(CH2)d−O−C(O)−(式中、cは約2〜約10の整数であり、dは約2〜約10の整数であり、Rは1〜10個のモノマーラクチド、グリコリド、炭酸トリメチレン、カプロラクトンまたはp−ジオキサノン断片を有するポリマーまたはコポリマーである。)によって表されるオリゴマージラジカルである。)によって表されるジラジカル断片であり、−Gは、スクシンイミジル、マレイミジル、フタルイミジル、ニトロフェニル、イミダゾリル、またはトレシル脱離基などの脱離基である。)を有する。脱離基の文脈で、「スクシンイミジル」とはN−オキシスクシンイミジルを意味し、「マレイミジル」とはN−オキシマレイミジル、「フタルイミジル」とはN−オキシフタルイミジルを意味し、「ニトロフェニル」とは、ニトロフェノキシルを意味し、「イミダゾリル」とはN−オキシイミダゾリルを意味し、「トレシル」とは(CF3−CH2−SO2−O−)を意味する。
【0016】
架橋剤の−PEG−部分は好ましくは市販される化合物から誘導されて、約1,000〜約15,000の量平均分子量を有し、好ましくは約2,000〜約4,000の量平均分子量を有する。これらの化合物は無害であることが実証されており、また分子量が約30,000未満であれば迅速に体から排出されることから、異なるタイプの生物医学的材料中で使用されている。
【0017】
架橋剤の脱離基−G部分は、架橋剤をタンパク質の遊離一級または二級アミン基と反応させ、あるいは化学的に結合させる活性脱離基である。適切な脱離基としては、N−オキシスクシンイミジルと、N−オキシマレイミジルおよびN−オキシフタルイミジルなどのその他のイミドと、N−オキシイミダゾリルなどのヘテロ環式脱離基と、ニトロフェノキシルなどの芳香族脱離基と、あるいはトレシル(CF3−CH2−SO2−O−)などのフッ素化アルキルスルホン脱離基と、が挙げられる。変異原性、発がん性、および催奇形性の研究からは、少量のN−オキシスクシンイミジル基が局所的または全身的毒物学リスクを呈さないことが示唆されるので、この群の脱離基が好ましい。
【0018】
架橋剤は、米国特許番号第4,101,380号または第4,839,345号で報告された手順、1990年4月19日に出願された国際出願整理番号PCT/US90/02133で報告された手順、またはAbuchowskiらによってCancer Biochem.Biophys.、7、175〜186ページ(1984年)で報告された手順などの既知の工程、手順または合成方法を使用して調製しても良い。簡単に述べると、塩基存在下でポリエチレングリコールおよび適切な酸無水物を適切な極性有機溶剤中に溶解し、ポリエチレングリコールジエステル二酸を形成するのに十分な時間還流する。次にジシクロヘキシルカルボジイミドまたはその他の縮合剤の存在下に、適切な極性有機溶剤中で、ジエステル二酸をN−ヒドロキシイミド化合物などの脱離基と反応させて、室温で撹拌して所望の二官能性架橋剤を形成する。
【0019】
代案としては、ポリエチレングリコールおよび適切な塩化ジカルボン酸またはビスクロロホルメートを、混合酸塩化物ポリエチレングリコールエステルまたは混合クロロホルメートポリエチレングリコールエステルを形成するのに十分な時間、適切な極性有機溶剤中に溶解しても良い。次に適切な極性有機溶剤中で、混合エステルをN−ヒドロキシイミド化合物などの化合物と反応させて、所望の二官能性架橋剤を形成するのに十分な時間、高温で撹拌しても良い。
【0020】
予備成形されたオブジェクトは、タンパク質などの材料を架橋する従来の方法、および押出し技術、噴霧乾燥技術、およびエマルジョン重合技術をはじめとする、架橋されたポリマーをビーズまたは微小球などの形状に形成する従来の方法を使用して調製しても良い。このような方法は、ポリマー化学技術分野の当業者には周知である。
【0021】
異なるpH値を有する緩衝液を使用してタンパク質のpHを変更することで、本組成物の硬化時間を目的に合わせても良い。例えば緩衝液のpHを変化させることでアルブミンのpHを変更し、それによって硬化速度時間を約10秒から約10分未満に変化させることが可能である。簡単に述べると、濃縮水性血清アルブミンをより高いpHで架橋剤と混合することで、最速硬化時間が提供される。より高濃度のタンパク質および架橋剤が、比較的より強力な硬化マトリックスを提供することも分かった。しかし混合物が濃くなりすぎ、粘度が高くなりすぎると、得られる硬化マトリックスは弱くなって球状になる。さらに架橋剤濃度が高すぎると、得られる硬化マトリックスは、水またはその他の流体存在下において、マトリックス強度が低くなる程度にまで膨潤するかもしれない。
【0022】
本発明の予備成形されたオブジェクトを形成するのに使用される組成物については、米国特許番号第5,583,114号(Barrowsら)で述べられている。しかしそこでは、組成物は直接組織に適用されて現場で(in situ)硬化されて、組織との結合相互作用が得られる。これらは例えば、現行の方法で標準的に必要な縫合糸数を排除または大きく削減したり、再建外科手術中に皮膚移植片を付着して組織皮弁または自由皮弁を配置したり、歯周部の外科手術において歯肉皮弁を閉じるのに使用される。これらの全ての用途において、組成物は、2つの隣接する生体組織層の間に事実上はさまれた、硬化接着剤の薄層を形成する。代案としては、組成物は、例えば肺外科手術に関連した空気漏れを防止するために、あるいはその他の外科手術手順において出血を阻害または防止するための密封材として使用されることが開示されている。このようにして使用する場合、下にある組織を比較的厚い接着剤層で被覆しても良い。
【0023】
対照的にここで述べる組成物は、サブジェクトに接触させるのに先立ってオブジェクトに成形される。したがって「予備成形された」という用語は、サブジェクトをオブジェクトに接触させるのに先立って、ポリマー組成物に形態または形状を提供することを指す。好ましくは予備成形されたオブジェクトは、自立型の三次元形状を有する。したがってこのように予備成形されたオブジェクトは、薄膜または薄層を含まない。しかし例えば形状が、裏材料上のシート材料または層の形状でパッチを形成する場合、予備成形されたオブジェクトは2mmを越える厚さを有する。本発明の予備成形されたオブジェクトは、マイクロカプセル、微小粒子、ナノ粒子などの微細構造、ならびにビーズまたはその他のボール形のオブジェクト、シリンダー、ディスク、繊維、シート、プラグ、リボン、ウェッジなどのマクロ構造の形態であることができる。好ましい構造は、微小球およびビーズである。典型的にビーズは約1mmを越える直径を有し、微小球は約1μm〜約1000μmの直径を有する。
【0024】
予備成形されたオブジェクトは、形成後に少なくとも部分的に脱溶媒和することができ、好ましくは実質的に完全に脱溶媒和される。ここで「脱溶媒和された」とは、タンパク質と架橋剤との反応に続いて、調製中に使用された溶剤が予備成形されたオブジェクトから除去されることを意味する。典型的にこれは、最初の形成時に予備成形されたオブジェクト中に組み込まれた水の少なくとも一部が除去されるために、予備成形されたオブジェクトが少なくとも部分的に脱水されることを意味する。このような予備成形されたオブジェクトの調製には、水の他に、または水に加えてその他の溶剤も使用できる。注目に値すべきは本発明の好ましい予備成形されたオブジェクトが、脱溶媒和および再溶媒和に際してそれらの形状を維持することである。
【0025】
予備成形されたオブジェクトを少なくとも部分的に脱溶媒和した後は、例えば大気中の湿気による再溶媒和を起こさない環境に、それらを貯蔵することが望ましい。取り扱いが容易で、医師に提供されて使用時に活性薬剤で再溶媒和できることから、このようなオブジェクトが望ましい。
【0026】
脱溶媒和されているか否かに関わらず、予備成形されたオブジェクトは活性薬剤を含むことができる。本発明の予備成形されたオブジェクトの好ましい実施態様は、その中に組み込まれた活性薬剤を有する。ここでの用法で「活性薬剤」とは、それが化学的、薬理学的、物理的、または生物学的に関わらず、所望の効果を生じることができるものである。したがってその中に組み込まれた1つ以上の活性薬剤を有する、得られた予備成形されたオブジェクトは、医薬品、薬物、またはその他の活性薬剤のためのデリバリシステムとして機能できる。予備成形されたオブジェクトは、例えばサブジェクトの体腔または組織空隙に移植する際などに組織と接触させて、あるいは例えば皮膚上などサブジェクトの体外に配置できる。
【0027】
活性薬剤は、例えばタンパク質と架橋剤の反応中など、またはそれに引き続いて、予備成形されたオブジェクトの形成中に添加できる。予備成形されたオブジェクトに形成中に活性薬剤を組み込む場合、活性薬剤が架橋剤またはタンパク質のどちらとも反応しないことが好ましい。特定の好ましい実施態様では、活性薬剤である液体または活性薬剤を含む液体を有する、予備成形され少なくとも部分的に脱溶媒和されたオブジェクトと活性薬剤とを組み合わせて、その中に組み込まれた活性薬剤を有する再溶媒和され予備成形されたオブジェクトを形成する。次にこの再溶媒和され予備成形されたオブジェクトは、活性薬剤のデリバリのためにサブジェクトと接触させて配置できる。予備成形されたオブジェクトの形成中、またはそれに続いて、それらに活性薬剤を組み込む特定方法を以下の実施例で開示する。
【0028】
本発明の一態様では、これは抗菌剤などの活性薬剤を含有する水性組成物で、複数のボール形の予備成形されたオブジェクトを再溶媒和して、膿瘍または創傷清拭した骨空洞などの組織空隙に、このような複数のオブジェクトを詰めることを伴う。したがって特定の一実施態様では、骨髄炎の治療において、抗菌剤を含有する再溶媒和され予備成形されたオブジェクトが使用できる。骨髄炎は骨とその骨髄の感染症であり、骨の病変形成が帰結する。病因は通常ブドウ球菌である。これは治療および根絶が困難な感染症である。現行の慣行は、長期的な予防的抗生物質と複数の外科的壊死組織切除である。壊死組織切除手術に続いて、骨髄炎組織の除去によって生じた空所に、生分解性ではないポリメタクリル酸メチル骨セメントビーズを詰める。このようなビーズは立証された抗生物質担体であるが、それらは生体内(in vivo)で分解せず、組織不適合反応に帰結することもあるために、それらは理想的とは言えない。したがって1つ以上の抗生物質を含有する本発明の予備成形されたオブジェクトは、骨髄炎の治療に使用できる。
【0029】
注目に値すべきは、予備成形されたオブジェクトの生分解速度が、タンパク質、架橋剤、および調製条件を選択することで、所望の用途について目的に合わせられることである。ここでの用法では「生分解」とは、材料の完全性の低下をもたらす可溶化、加水分解、酵素、および生物学的存在の作用をはじめとする様々な機序によって、予備成形されたオブジェクトがより単純な中間体または最終産物に転換することを意味する。必ずしもそうではないが、ポリマー分子はより小さい断片に壊れることができる。このような生分解の範囲には、予備成形されたオブジェクトの生体再吸収、生体吸収または生体侵食が含まれる。典型的に、本発明予備成形されたオブジェクトは、約2日間〜約60日間の間に分解するように調合できる。
【0030】
活性薬剤の放出は、典型的にポリマーの侵食、およびポリマーからの活性薬剤の拡散の組み合わせによって起きる。拡散成分はフィックの法則に従うかも従わないかもしれず、他方、侵食寄与は分解の動態学に従う。活性薬剤の放出速度は、数時間程度から数ヶ月、典型的に約60日間程度までの範囲になるように、目的に合わせることができる。
【0031】
活性薬剤は、例えば治療的または予防的であっても良い、生理的、薬理学的、または生物学的効果を提供する多種多様の薬剤から選択できる。例としては、細胞増殖を向上させ、組織再生を向上させ、血管形成および血管新生を向上させ、神経刺激を向上させ、骨成長を向上させ、感染症(例えば細菌性またはウイルス性)および/または炎症を阻害(例えば防止、低下、または逆転)し、ガン細胞増殖を阻害し(例えば既存のガン状態を治療する、または前ガン状態からガン状態への転換を防止する)、免疫反応を調節し、創傷治癒を促進し、あるいは組織軟化および加湿を促進できる物質またはその代謝前駆体が挙げられる。このような物質としては、テトラサイクリン、バンコマイシン、およびセファロスポリンなどの抗菌剤と、血小板由来増殖因子、トランスフォーミング増殖因子β、上皮性増殖因子、および繊維芽細胞増殖因子などの増殖因子と、5−フルオロウラシル、マイトマイシン、メトトレキセート、ドキソルビシン、およびシスプラチンなどの抗ガン剤(例えば抗有糸分裂薬)と、リドカイン、ブピバカイン、テトラカイン、プロカイン、およびプリロカインなどの局所麻酔薬と、防腐薬(例えばクロルヘキシジン)と、ホルモン(例えばステロイド、インスリン)と、抗ウイルス薬剤と、麻薬拮抗薬と、免疫反応調節剤と、目薬(例えばアトロピン、ピロカルピン、およびチモロール)と、ワクチンと、および美容医薬品と、が挙げられるが、これに限定されるものではない。その他の具体例は、米国特許番号第第5,733,563号(Fortierら)および第5,759,563号(Yeweyら)に列挙されている。本発明の予備成形されたオブジェクトに、このような1つ以上の活性薬剤を組み込んでも良い。
【0032】
好ましくはその中に組み込まれた活性薬剤を有する本発明の予備成形されたオブジェクトは、二次生分解性マトリックスに組み込むことができる。この二次生分解性マトリックスは、液体(例えば液体中の微小球)としてサブジェクトに適用しても良く、あるいは成形オブジェクト(例えば成形オブジェクト内の微小球などの予備成形された成形オブジェクト)に形成できる。このようにして予備成形されたオブジェクトは、サブジェクトへのデリバリのためにマトリックス内に組み込むことができる。二次生分解性マトリックスは、好ましくは上述され、米国特許番号第5,583,114号(Barrowsら)で述べられるような予備成形されたオブジェクトと同様の化学的性質であることができ、あるいはそれは別の生分解性ポリマーであることができる。同様に予備成形されたオブジェクトは、上述の架橋されたタンパク質を含み、あるいは別の生分解性ポリマーでも良い。マトリックスまたは予備成形されたオブジェクトのいずれかのための適切な生分解性ポリマーの例としては、Drug Delivery Systems、V.V.Ranade編、CRC Press,Inc.、BocaRaton、FL、78〜81ページ、1996年、およびBiodegradable Polymers as Drug Delivery Systems、M.Chasin編、Drugs and the Pharmaceutical Sciences Series、第45巻、Marcel Dekker,Inc.NY、1990年で開示されるものが挙げられる。二次生分解性マトリックスの好ましい例としては、ポリ(ホスホエステル)、ポリ(α−ヒドロキシ酸)、親水アクリレートおよびメタアクリレートポリマー、ヒドロキシプロリンポリエステル、ポリ無水物(例えばポリ(ラクチド−コ−グリコリド))、ポリカプロラクトン、ポリ(オルトエステル)、ポリホスファゲン、ポリ(アミノ酸)、多糖類、およびそれらのコポリマーが挙げられる。予備成形されたオブジェクトの好ましい例としては、ポリ(α−ヒドロキシ酸)(例えばポリ(グリコール酸)、ポリ(DL−乳酸)、およびポリ(L−乳酸))、ポリ(アミノ酸)、ポリ(酸無水物)、ポリ(オルトエステル)、ポリ(ホスファジン)、ポリ(ホスホエステル)、ポリラクトン(例えばポリ(ε−カプロラクトン)、ポリ(δ−バレロラクトン)、およびポリ(γ−ブチロラクトン)が挙げられる。予備成形されたオブジェクトは、当業者には既知の技術を使用して、二次生分解性マトリックスに組み込むことができる。例えば米国特許番号第5,583,114号(Barrowsら)で開示される組織密封材を二次生分解性マトリックスとして使用して、その中に組み込まれた予備成形されたオブジェクトをサブジェクトにデリバリできる。
【0033】
以下の実施例によって本発明の目的および利点をさらに実証するが、これらの実施例で言及される特定材料およびその量、ならびにその他の条件と詳細は、本発明を不当に制限するものではない。
【実施例】
【0034】
実施例
アルブミン溶液の調製
アルブミンは、Baxter Healthcare(イリノイ州ディアフィールド)から25%濃度のヒト血清アルブミンとして得た。それを米国特許番号第5,583,114号(Barrowsら)で述べられるような透析(手順A)で処理し、あるいはカットオフ分子量50,000の680cm2ポリスルホン繊維モジュール(Spectrum Laboratories,Inc.から入手できる)を装着した、接線フロー分離のためのMINIKROS Sampler Lab Unit(カリフォルニア州ランチョドミンゲスのSpectrum Laboratories,Inc.から入手できる)を使用して、連続ダイアフィルトレーションによって処理した(手順B)。pH10の0.1M炭酸/炭酸水素塩緩衝液溶液を使用して、Barrows法(米国特許番号第5,582,114号)(工程A)から得られたアルブミンを23%溶液に希釈した。ダイアフィルトレーション(工程B)から得られたアルブミンの場合は、pH8.9〜9.1の0.075M炭酸/炭酸水素塩緩衝液を用いてダイアフィルトレーションを実施した。ダイアフィルトレーション装置を使用して、サンプルを29%〜30%に濃縮した。アルブミンの最終濃度を標準ビウレット滴定によって求めた。
【0035】
ポリエチレングリコールジスクシンイミジルスクシネート(PEG−SS)2架橋剤の合成
30ガロンのガラス内張反応器に、10.9kgのポリエチレングリコール(分子量3400)、760gの無水コハク酸、および4.4kgのトルエンを装填した。装填物を窒素ガスシール下で110℃で6時間混合した。次に反応混合物を80℃に冷却した。13.0kgの無水エタノールを撹拌しながら添加した。それ以上の加熱は行わなかった。温度が25℃に冷めるまで混合物を撹拌した。43.8kgのメチルtert−ブチルエーテル(MtBE)を冷却混合物に撹拌しながら添加して、窒素ガス体下で一晩混合した。反応混合物を遠心分離した。反応器を同量のMtBEでさらに4回すすいだ。このすすぎ水を使用して遠心分離ケークを洗浄した。ブレンダー乾燥機内で、ポリエチレングリコールジスクシネートのケークを真空下、30℃でおよそ19.5時間乾燥した。10.9kgのポリエチレングリコールジスクシネートが回収された。
【0036】
30ガロンガラス内張反応器に、5.6kgのポリエチレングリコールジスクシネート、540gのN−ヒドロキシスクシンイミド、84gの4−ジメチルアミノピリジン、および8.2kgのアセトニトリルを装填した。全固形物が溶解するまで、装填物を室温で1時間混合した。1.3kgの1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミドを添加した。混合物を窒素ガスシール下で、25℃で6時間撹拌した。次に39.6kgの無水エタノールを反応混合物に撹拌しながら添加した。混合物を75ガロンポータブルガラス内張反応器に移した。80.0kgのMtBEを撹拌しながら添加した。冷却ジャケットを使用して混合物を一晩撹拌した。翌朝、反応混合物は5〜7℃であった。反応混合物を遠心分離して、反応フラスコを同量のMtBEで4回すすいだ。このすすぎ水を使用して遠心分離ケークを洗浄した。遠心分離した濡れたケークを40kg無水エタノールと共に30ガロンガラス内張反応器に加えた。反応器を窒素ガス体下、周囲温度でおよそ1時間混合した。混合物を遠心分離して、反応器を40kgの無水エタノールですすぎ、このすすぎ水を使用して遠心分離ケークを洗浄した。次に反応器を40kgのMtBEですすぎ、このこのすすぎ水を使用して遠心分離ケークを洗浄した。ケークのおよそ半分をタンブル乾燥機に移して、真空下、30℃でおよそ18時間乾燥した。液体窒素冷却を使用して、2.7kgの乾燥生成物を粉砕した。ケークの半分からそれぞれ2.55kgのポリエチレングリコールジスクシンイミジルスクシネート、PEG−(SS)2が回収された。
【0037】
実施例1
予備成形されたアルブミンポリマープラグからの5−フルオロウラシル(5FU)の放出
シリンジバレルを切断して開放円筒容器を形成した改造5mlポリシリンジ内で、29%アルブミン溶液(テキサス州セギーンのAmerican Biological Technologyから入手できる、あるいは上述したダイアフィルトレーション(手順B)によって得られる)0.5mlと、136mg/mlのPEG−(SS)2架橋剤0.5mlとを混合して、2つのポリマープラグを予備成形した。架橋完了後(15分間)、プラグをシリンジから除去して押し出した。ポリマープラグを真空下で24時間乾燥させた。各プラグが全部で187.6μg、または165.5μgの5FUを含有するように、プラグを2mlの5FU溶液(1.04mg/ml、ミズーリ州セントルイスのSigma Chemical Co.)で戻した。各プラグを別々のバイアルに入れて、pH7.4のリン酸緩衝食塩水5mlをサンプル時間0(t0)時に添加した。バイアルを室温に保持して絶え間なく振盪した。各サンプル時間毎にバイアルの全内容物を取り出して、5mlの新鮮なPBSと入れ替えた。サンプルは0.25時間、0.5時間、1時間、2時間、3時間、4時間、5時間、6時間、および24時間目に採取した。5FUの分析は、BernatchezらのInt.J.Pharm.、106、161〜166ページ(1994年)の方法に従って、高圧液体クロマトグラフィーによって実施した。図1のデータは、5時間の間に5FUが放出されたことを示す。
【0038】
実施例2
バイオポリマープラグからのテトラサイクリンの放出
アルギン酸(30mg、ミズーリ州セントルイスのSigma Chemical Co.)を1.01mg/mlテトラサイクリン(ミズーリ州セントルイスのSigmaChemicalCo.)1mlに添加した。CloropHast pHストリップ(イリノイ州イタスカのFischerCompany)を使用して、1NのNaOHで混合物のpHを6.8にして、次にDRIERITE(オハイオ州ジーニアのW.A.Hammond Drierite Co.)と共に真空デシケーターに入れて24時間乾燥した。得られたルギン酸塩/テトラサイクリンフィルムを粉砕して30%アルブミン溶液(手順B)中に混合し、直ちに5mlポリシリンジ型に入れて等容積のPEG−(SS)2架橋剤(136mg/ml)で架橋した。各プラグを別々のバイアルに入れて、pH7.4のリン酸緩衝食塩水(PBS)5mlをサンプル時間0(t0)時に添加した。バイアルを室温に保持して絶え間なく振盪した。各サンプル時間毎にバイアルの全内容物を取り出して、5mlの新鮮なPBSと入れ替えた。サンプルを1時間、2時間、3時間、5時間、および24時間目に採取した。可変波長検出器を有するHewlett Packard(カリフォルニア州パロアルト)HPLCモデル1190、およびSupelco Co.(ペンシルベニア州ベルフォンテ)からの逆相LC−8−DBカラム(15.0cm×4.6mm、粒度5μm)でサンプルを分析した。HPLC分析のために使用した移動相は、比率が45:45:10:1の0.05Mリン酸アンモニウム緩衝液(pH6.8)、メタノール、アセトニトリル、およびトリエチルアミンから構成された。移動相の流速は1.0ml/分であり、カラム温度は25℃に保った。テトラサイクリンは280nmで検出された。
【0039】
図2のデータは、5時間の間に約70%のテトラサイクリンが放出されたことを示す。収集テトラサイクリン量は、テトラサイクリンの分解による損失について補正しなかった。
【0040】
実施例3
架橋剤およびアルブミンと混合された5FUから製造されるプラグからの5−フルオロウラシルの放出
136mgのPEG−(SS)2架橋剤を1.04mg/mlの5−フルオロウラシル溶液1mlに溶解した。次にこの溶液をあらかじめ5mlの型に入れた1mlの30%アルブミン溶液(実施例2参照)中に混合した。得られたヒドロゲルプラグを15分後に型から取り出して、3つの等しい寸法の小片に切断した(厚さおよそ0.5cm)。次に各小片をバイアルに入れて、4mlのPBS緩衝液(pH7.4)をサンプル時間0(t0)時に添加した。バイアルを室温に保持して絶え間なく振盪した。各サンプル時間毎にバイアルの全内容物を取り出して、4mlの新鮮なPBSと入れ替えた。サンプルは1時間、2時間、3時間、4時間、および8時間目に採取した。5−フルオロウラシルを実施例1に述べるようにして分析した。
【0041】
図3のデータは、8時間の間に約55%の5−フルオロウラシルが放出されたことを示す。残る5FUは、恐らくポリマー中に架橋されており、ポリマーが分解すれば放出される。
【0042】
実施例4
カルシウム無添加のアルギン酸を含有するアルブミンプラグからの5−フルオロウラシルの放出
1.04mg/mlの5−フルオロウラシル1mlを30mgのアルギン酸と混合した。混合物のpHを6.8にして、次にDRIERITEと共に真空デシケーター内に入れて24時間乾燥させた。得られたアルギン酸塩/5FUフィルムを粉砕し、30%アルブミン溶液(実施例2参照)中に混合し、直ちに5mlシリンジ型に入れて等容積のPEG−(SS)2架橋剤(136mg/ml)で架橋した。各プラグを別々のバイアルに入れて、pH7.4のリン酸緩衝食塩水(PBS)5mlをサンプル時間0(t0)時に添加した。バイアルを室温に保持して絶え間なく振盪した。各サンプル時間毎にバイアルの全内容物を取り出して、5mlの新鮮なPBSと入れ替えた。サンプルを1時間、2時間、3時間、4時間、5時間、および24時間目に採取した。
【0043】
5−FUを実施例1に述べるようにして分析した。図4のデータは、5時間の間に約40%の5FUが放出されたことを示す。
【0044】
実施例5
カルシウム添加アルギン酸を含有するアルブミンプラグからの5−フルオロウラシルの放出
1.04mg/mlの5−フルオロウラシル1mlを30mgのアルギン酸と混合した。混合物のpHを6.8にして、150μlの飽和塩化カルシウム/水溶液を添加してアルギン酸塩をゲル化した。ゲルを3個のほぼ同じ容積に分割し、次にDRIERITEと共に真空デシケーター内に入れて24時間乾燥させた。得られたアルギン酸塩/Ca/5FUフィルムを粉砕し、30%アルブミン溶液(実施例2参照)中に混合し、直ちに5mlシリンジ型に入れて等容積のPEG−(SS)2架橋剤(136mg/ml)で架橋した。各プラグを別々のバイアルに入れて、pH7.4のリン酸緩衝食塩水(PBS)5mlをサンプル時間0(t0)時に添加した。バイアルを室温に保持して絶え間なく振盪した。各サンプル時間毎にバイアルの全内容物を取り出して、5mlの新鮮なPBSと入れ替えた。サンプルを1時間、2時間、3時間、4時間、5時間、および24時間目に採取した。
【0045】
5−FUを実施例1に述べるようにして分析した。図4のデータは、5時間の間に約60%の5FUが放出されたことを示す。
【0046】
実施例6
生分解性ビーズの製造方法
二液システムからビーズを調製した。パートAは、等張(0.075M)炭酸緩衝液(pH9)中の29%アルブミン滅菌溶液(実施例1参照)であった。パートBは、使用直前に滅菌水溶液で戻したPEG−(SS)2の260mg/ml溶液であった。静止ミクシングヘッドに接続したデュアルシリンジシステムによって、溶液AおよびBを等容積で混合した。8mm径の孔を有する6インチ×4インチ×1インチのテフロン金型内に液体を注入して、ビーズを調製した。注入した混合物を8mm径の孔内で10分間硬化した。ビーズをテフロン金型から10分後に取り出した。ビーズは2つの異なるやり方で使用できる。一例では水和したビーズをテフロン金型から取り出して、直接使用できる。別の例では、ビーズを金型から取り出した後に脱水できる。乾燥は、真空下または空気中、またはその他の適切な様式で実施できる。球状のビーズ以外にも様々な形状寸法が製造できることに留意すべきである。
【0047】
実施例7
バンコマイシンの試験管内(in vitro)放出試験
実施例6から製造された2個の脱水されたビーズを4mlバイアルに入れた。バンコマイシン(60mg、イリノイ州ノースシカゴのAbbott Laboratories)を2mlの滅菌水に溶解してバイアルに入れた。ビーズを室温で24時間浸漬した。24時間の浸漬後にビーズを取り出して、pH7.4のリン酸緩衝食塩水5mlと共に20mlガラス瓶に入れた。バンコマイシン放出速度は定温水浴内で37℃で測定した。緩衝液を24時間毎に最高3週間まで交換した。UV分光光度計(DU640、カリフォルニア州フラートンのBeckman)によって、これらのサンプルをバンコマイシンについて分析した。バンコマイシンは282nmで検出された。結果を図5および6に示す。
【0048】
実施例8
セファゾリンの試験管内(in vitro)放出
実施例6から作られた2個の脱水されたビーズを4mlバイアルに入れた。セファゾリン(60mg、ニュージャージー州チェリーヒルのMarsam Pharmaceuticals,Inc.)を2mlの滅菌水に溶解してバイアルに入れた。ビーズを室温で24時間浸漬した。24時間の浸漬後にビーズを取り出して、pH7.4のリン酸緩衝食塩水5mlと共に20mlガラス瓶に入れた。セファゾリン放出速度は定温水浴内で37℃で測定した。緩衝液を24時間毎に最高3週間まで交換した。UV−分光光度計(Beckman,DU640,カリフォルニア州フラートン)によって、これらのサンプルをセファゾリンについて分析した。セファゾリンは272nmで吸光する。結果を図7および8に示す。
【0049】
実施例9
架橋程度および溶剤容積の関数としての再水和されたビーズの水取り込み
PEG−(SS)2のいくつかの架橋剤調合物を使用したこと以外は、実施例6と同様にしてビーズを製造した。標準の密封材(NS)調合物からできたビーズは、1mlの水中の130mgのPEG−(SS)2と、pH9.0の炭酸緩衝液中の1mlの29%アルブミンとを混合して製造した。架橋剤濃度が半分(NS*1/2)のビーズでは、ビーズは1mlの水中に65mgのPEG−(SS)2、pH9.0の炭酸緩衝液中に1mlの29%アルブミンを含有した。架橋剤濃度が2倍(NS*2)のビーズでは、ビーズは1mlの水中に260mgのPEG−(SS)2、pH9.0の炭酸緩衝液中に1mlの29%アルブミンを含有した。架橋剤濃度が3倍(NS*3)のビーズでは、ビーズは1mlの水中に390mgのPEG−(SS)2、pH9.0の炭酸緩衝液中に1mlの29%アルブミンを含有した。各調合物からの3個のビーズを20mlのあらかじめ秤量したバイアルに入れた。3mlの水を秤量してバイアルに加えた。6mlの水を使用して同じ試験を繰り返した。ビーズを取り出してバイアル中の水を秤量し、ビーズの水取り込みを2、4、6、8、および24時間目に測定した。結果を表1に示す。
【0050】
【表1】
【0051】
実施例10
ビーズの直径および架橋程度の関数としての水和されたビーズの水取り込み
ビーズは実施例6と同様にして製造した。標準、2倍、3倍の架橋剤濃度のビーズは、実施例9で述べたようにして調製した。各調合物からの3個のビーズを20mlのあらかじめ秤量したバイアルに入れた。6mlの水を秤量して、バイアルに加えた。ビーズをバイアルから取り出してバイアル内に残留する水を秤量し、ビーズの水取り込みおよびビーズ径を2、4、6、8、および24時間目に測定した。1個のビーズと2mlの水で試験を繰り返した。ビーズ径はScherr−Tumico Optical Comparator(S.T.Industries)を使用して測定した。複数のビーズについての結果を表2に、単一ビーズについての結果を表3に示す。表4は単一ビーズ試験と複数ビーズ試験における、乾燥ビーズ容積と比較した容積増大%を乾燥ビーズの再水和に伴う時間の関数として示す。図9はビーズ1個の試験の水取り込みに関するこのデータを示す。ビーズ3個の試験は同様の結果をもたらした。
【0052】
【表2】
【0053】
【表3】
【0054】
【表4】
【0055】
実施例11
架橋されたアルブミン微小球の調製
米国特許番号第5,508,060号に従って、種々の修正を加えて、油中水型エマルジョン法によって微小球を調製した。米国特許番号第5,583,114号(Barrowsら)の実施例8で教示されるようにしてヒト血清アルブミン(HSA)溶液を使用し、ウシ血清アルブミン(BSA)Fraction Vは、Sigma Chemical Co.(ミズーリ州セントルイス)から購入した。PEG−(SS)2を架橋剤として使用した。概して、電動の3枚羽根プロペラタイプ撹拌機(Motomatic、米国のElectro−Craft)を使用して、ピーナツ油(Nabisco Foods,Inc.、ニュージャージー州イーストハノーバー)を所定速度で撹拌した。絶え間なく撹拌しながら油浴中に皮下注射針で滴下して、アルブミン溶液と混合した架橋剤の水性溶液を添加した。別の変法では、架橋剤溶液を油に添加してからアルブミン溶液を滴下して加えた。アルブミンと架橋剤との容積比は1:1であり、使用した架橋剤濃度は138mg/mlおよび276mg/mlであった。油と水との比率はおよそ100:1であり、300〜500mlの範囲の油を使用した。所定時間の撹拌後、微小球を収集して酢酸エチルで洗浄し、0.44μmまたは1.1μmのセルロースまたはナイロンフィルターを通して濾過した。
【0056】
実施例11A
以下の修正と共に実施例11の手順を使用して微小球を調製した。ヒト血清アルブミン(HSA)はpH9.4で27%濃度で使用した。PEG−(SS)2溶液は138mg/mlの濃度であった。PEG−(SS)2溶液を油に添加して、次にアルブミン溶液を27ゲージの針を通して滴下して撹拌中の油に添加した。3枚羽根プロペラタイプ撹拌機を使用して、油を1000rpmで15分間撹拌した。撹拌停止後、溶液を85℃に加熱して、実施例11と同様に微小球を濾過して洗浄した。溶液の加熱中に媒体温度が約45℃に達すると、架橋された粒子が凝集しはじめ、混合物から白色フレークが形成した。凝集した粒子を除去して微小球を収集した。この工程から回収された微小球の典型的な画像分析は、平均径が4.22μmで、1.0μm〜15μmの範囲内の粒度分布を有した。
【0057】
実施例11B
撹拌速度が1300rpmで撹拌後に溶液を加熱しなかったこと以外は、実施例11Aで使用したのと同一手順によって微小球を製造した。この工程から回収された微小球の典型的な画像分析は、平均径が3.05μmで、1.0μm〜10μmの範囲内の粒度分布を有した。
【0058】
実施例11C
PEG−(SS)2架橋剤濃度が276mg/ml、撹拌速度が1300rpmで撹拌時間が20分であり、撹拌後に溶液を加熱しなかったこと以外は、実施例11Aで使用したのと同一手順によって微小球を製造した。この工程から回収された微小球の典型的な画像分析は、平均径が5.45μmで、0.5μm〜16μmの範囲内の粒度分布を有した。これらの微小球を実施例12の医薬品装填試験に使用した。
【0059】
実施例11D
pH9.17で15%の血清アルブミン(BSA)溶液を使用し、撹拌速度が1500rpmで撹拌時間が20分であり、撹拌後に溶液を加熱しなかったこと以外は、実施例11Aで使用したのと同一手順によって微小球を製造した。この工程から回収された微小球の典型的な画像分析は、平均径が2.42μmで、1.0μm〜6.0μmの範囲内の粒度分布を有した。
【0060】
実施例12
テトラサイクリンを用いた現場での(in situ)微小球への医薬品装填
2mlのHSA溶液と、等容積の油浴中に医薬品を含有する架橋剤溶液とを混合して、実施例11Cの方法に従って塩酸テトラサイクリンの存在下で微小球を調製した。簡単に述べると、552mgのPEG−(SS)2架橋剤を2mlのテトラサイクリン溶液(10mg/ml)に添加して、架橋剤溶液を調製した。医薬品を含有するPEG−(SS)2溶液を1300rpmで撹拌しながら油に添加した。水性溶液が安定したエマルジョンを形成するのに十分な時間をおいてから、2mlのヒト血清アルブミン(27%、pH9.4)を滴下して添加しエマルジョンを20分間撹拌した。酢酸エチルで洗浄後、粒子は良好に分離した微粉末状を呈した。画像分析からは、粒子が良好に分離して小型であることが示された。
【0061】
実施例13
試験管中での(in vitro)医薬品放出評価
微小球(実施例11A参照)を拡張放出試験器(BIO−DIS、ニュージャージー州エジソンのVanKel)の容器に入れて、試験管中での(in vitro)医薬品放出の試験を実施した。1分あたり浸漬3回の撹拌速度、初期遅延l5秒で、100mlのリン酸緩衝食塩水(PBS、pH7.4)を含有する200mlガラスビーカー内に容器を浸漬した。放出試験中、水浴の温度は電子的に37℃に保った。所定時間経過後、1mlのサンプルを取り出して、新鮮な溶液をPBSレザバーに添加した。0.22m COSTARシリンジフィルター(ニューヨーク州コーニングのCorning)を通してサンプル溶液を濾過し、あらゆる微粒子を除去した。塩化テトラサイクリンの定量は、HPLC法を使用して分光測定UV検出により実施した。使用した装置は、注入器(ShimadzuモデルSil−9A、メリーランド州コロンビアのShimadzu)、溶剤デリバリシステム(Waters 625LC、マサチューセッツ州ミルフォードのMillipore Corporation)、マルチ波長検出器(Waters 490E、マサチューセッツ州ミルフォードのMillipore Corporation)、および積分器(Waters Millennium、マサチューセッツミルフォードのMillipore Corporation)から構成された。医薬品を含有する溶液(20μl〜150μl、直線検出範囲に入るように調節済み)をBeckman逆相C−18カラム(5μm×4.6mm×25cm、米国のBeckman)に注入した。移動相は0.1%のトリフルオロ酢酸を含有する、水とアセトニトリルの80/20混合物であった。流速は1.0ml/分であり、使用したUV波長は275nmであった。医薬品濃度は、医薬品ピークの下の面積と標準曲線から得られたものとを比較して測定した。各サンプルを少なくとも2回分析し、平均放出医薬品総量を計算した。
【0062】
図10は、3つの異なる大きさ範囲の微小球からの塩酸テトラサイクリンの放出プロフィールを示す。分粒は較差沈降法によって実施した。3つの標本全てで初期のバースト放出が観察された。しかしバースト効果に対する顕著な粒度の影響があった。微小球からの初期の医薬品流量は粒度が増大するにつれて低下し、初期のバースト相が主に微小球表面積に左右されることが示された。
【0063】
実施例14
4−アーム PEG架橋剤を用いて製造したアルブミンポリマープラグからのセファゾリンの放出
30%アルブミン溶液(手順B)0.5mlと、110mg/mlのポリ(エチレングリコール)−スクシネート架橋剤の4−アーム−N−ヒドロキシスクシンイミジルエステル(アラバマ州ハンツビルのShearwater Polymers,Inc.)0.5mlから、2個のポリマープラグを予備成形した。架橋剤を添加してシリンダー状プラグを形成する前に、およそ10mgのナトリウムセファゾリン(ミズーリ州セントルイスのSigma Chemical Co.)をアルブミン中に溶解した。各形成プラグを別々のバイアルに入れて、pH7.4のリン酸緩衝食塩水(PBS)8mlをサンプル時間(t0)時に添加した。バイアルを室温に保持して絶え間なく振盪した。各サンプル時間毎にバイアルの全内容物を取り出して、8mlの新鮮なPBSと入れ替えた。サンプルを0.25時間、0.5時間、1時間、2時間、3時間、4時間、5時間、および24時間目に採取した。それ以降は144時間目まで、およそ24時間毎にサンプルを採取した。図11のデータは、最初の5時間でおよそ67%のセファゾリンが、144時間で全セファゾリンの95.6%が放出されることを示す。
【0064】
LiangらのJ.Chromatog.B.、656、45〜465ページ(1994年)の方法に従って、高圧液体クロマトグラフィーによってセファゾリンを分析した。カラムはMICROBONDAPAKの商品名の下に市販されるWaters C−18(3.9×300mm)カラムであり、カラム温度を25℃に保った。移動相は0.02Mのリン酸ナトリウム緩衝液(pH=5.0)およびメタノール(77:23)から構成された。検出に使用した波長は270nmであり、カラム流速は1.0ml/分であった。
【0065】
実施例15
生分解性の予備成形されたアルブミンビーズからのTGF−β1放出
以下のバリエーションで実施例6で述べた方法に従って、ビーズを調製した。実施例6で述べたのと全く同じようにして2個のビーズを調製し、それらの水和した形態(ビーズNS1およびNS2)で使用した。実施例6で述べたのと全く同じようにして2個のビーズを調製し、脱水して水中で再水和した(ビーズNS3およびNS4)。実施例6のパートBに3.75μg/mlヒト組換えトランスフォーミング増殖因子β1(TGF−β1、メリーランド州ロックビルのGibco BRL)20μlを加えて2個のビーズを調製し、ビーズあたり25ngのTGF−β1の最終濃度を得て、それらの水和した形態で使用した(ビーズNS5およびNS6)。実施例6で述べたのと全く同じようにして2個のビーズを調製し、脱水して50ng/mlのTGF−β1溶液2ml中で4時間再水和した(4時間で25%の取り込みが見られたので100ngの内25ngがNS7およびNS8の各ビーズに組み込まれた)。等張(0.075M)炭酸緩衝液(pH8.66)中の23%の滅菌アルブミン溶液(手順B)から成る実施例6のパートA、およびPEG(SS)2の65mg/ml溶液から成るパートB(実施例1参照)から2個のビーズを調製し、滅菌水中で使用直前に戻した(ビーズFS1およびFS2)。等張(0.075M)炭酸緩衝液(pH8.66)中の23%の滅菌アルブミン溶液(手順B)から成る実施例6のパートA、およびPEG(SS)2の65mg/ml溶液から成るパートB(実施例1参照)から2個のビーズを調製し、20μlの3.75μg/mlヒト組換えTGF−β1を添加した滅菌水中で使用直前に戻した(ビーズFS3およびFS4)。
【0066】
12ウェルペトリプレート(ペンシルベニア州ピッツバーグのFisher Scientific)の個々のウェル内で、10%ウシ胎児血清(バージニア州マナッサスのAmerican Type Culture Collection)およびペニシリン/ストレプトマイシン100U/ml(メリーランド州ロックビルのGibco BRL)を含有する4mlの修正Eagle’s Minimum Essential Cell Culture Medium(バージニア州マナッサスのAmerican Type Culture Collection)に全てのビーズを入れた。試験の実施期間中、プレートを細胞培養器(37℃、5%のCO2)に入れた。サンプルを経時的に(t=0から開始)採取して、ビーズが完全に分解するまで各時点で培養液の全容積を置き換えた。各時点の培養液はクリオバイアルに収集して、−20℃で貯蔵した。採取時点は、NSビーズでは0時間、24時間、48時間、72時間、96時間、120時間、144時間、168時間、192時間、216時間、240時間、264時間、289時間、および330時間であり、FSビーズでは、0時間、24時間、48時間、72時間、96時間、および120時間であった。
【0067】
次にTGF−β1濃度を求めるための酵素結合抗体免疫吸着アッセイ(ELISA)(ヒトTGF−β1用QUANTIKINE、ミネソタ州ミネアポリスのR & D Systems)で、製造元の取扱説明書に従って培養液の全サンプルをアッセイした。測定されたTGF−β1は活性形態で販売され、培養中の細胞が生成したものではなかったので、サンプルは活性化を必要としなかった。ビーズの製造で使用したのと同一供給元のTGF−β1を用いて別に用量反応曲線を生成し、この後者の用量反応曲線に従って結果を計算した。
【0068】
図12の結果は、ビーズ(NS7、NS8)の再水和に使用される溶液に増殖因子を組み込むことで、組み込まれた増殖因子の40%までが2〜6日中に放出されることを示す。架橋剤(NS5、NS6、FS3、FS4)へのTGF−β1の組み込みは、ビーズからの顕著な量のTGF放出をもたらさなかった。対照ビーズ(TGF−β1を含まないNS1、NS2、NS3、NS4、FS1、FS2)を試験したところ、アッセイを妨害しなかった。
【0069】
前出の記述が例証のみを目的とするものであり、発明の範囲を逸脱することなく様々な実施態様が想定されるできることは、当業者によって容易に理解される。
【特許請求の範囲】
【請求項1】
式、
I−(−X−LM−G)n
(式中、
Xは二官能性ポリオキシエチレン鎖部分または結合であり、
LMは式、−C(O)−、−(CH2)bC(O)−(式中、bは1〜5の整数である。)、−C(O)−(CH2)c−C(O)−(式中、cは2〜10の整数であり、ラジカルの脂肪族部分は飽和または不飽和でも良い。)、−C(O)−O−(CH2)d−O−C(O)−(式中、dは2〜10の整数であり、あるいは式、−R−C(O)−、−R−C(O)−(CH2)c−C(O)−、または−R−C(O)−O−(CH2)d−O−C(O)−(式中、cは2〜10の整数であり、dは2〜10の整数であり、Rは1〜10個のモノマーラクチド、グリコリド、炭酸トリメチレン、カプロラクトンまたはp−ジオキサノン断片を有するポリマーまたはコポリマーである。)によって表されるオリゴマージラジカルである。)によって表される二官能性結合部分であり、
GはN−オキシスクシンイミジル、N−オキシマレイミジル、N−オキシフタルイミジル、ニトロフェノキシル、N−オキシイミダゾリル、およびトレシルの群より選択される脱離基であり、
Iは多求核性化合物から誘導される多官能性結合部分であり、
nは2〜10の整数であるが、
ただしXが二官能性ポリオキシエチレン鎖部分の場合、−X−I−X−は、式、
−O−(CH2−CH2−O−)a−
(式中、aは20〜300の整数である。)によって表されるジラジカル断片であるPEGである。)
の架橋剤を有する、少なくとも部分的に脱溶媒和された架橋されたアルブミンである、活性薬剤をサブジェクトにデリバリするための予備成形されたオブジェクト。
【請求項2】
架橋されたタンパク質が架橋された血清アルブミンを含む、請求項1に記載の予備成形されたオブジェクト。
【請求項3】
架橋された血清アルブミンが架橋されたヒト血清アルブミンを含む、請求項2に記載の予備成形されたオブジェクト。
【請求項4】
GがN−オキシスクシンイミジルである、請求項1に記載の予備成形されたオブジェクト。
【請求項5】
組み込まれた活性薬剤をさらに含む、請求項1に記載の予備成形されたオブジェクト。
【請求項6】
活性薬剤が、抗菌剤、増殖因子、抗ガン剤、局所麻酔薬、防腐薬、ホルモン、抗ウイルス剤、麻薬拮抗薬、免疫反応調節剤、目薬、ワクチン、それらの代謝前駆体、およびそれらの混合物から選択される、請求項1に記載の予備成形されたオブジェクト。
【請求項7】
二次生分解性ポリマーマトリックス内に分散した、請求項1に記載の予備成形されたオブジェクト。
【請求項8】
シート、ボール、ビーズ、球体、プラグ、繊維、リボン、またはウェッジの形態である、請求項1に記載の予備成形されたオブジェクト。
【請求項9】
約1mmを越える直径を有するビーズの形態である、請求項1に記載の予備成形されたオブジェクト。
【請求項10】
約1μmから約1000μmの直径を有する微小球の形態である、請求項1に記載の予備成形されたオブジェクト。
【請求項11】
式、
I−(−X−LM−G)n
(式中、
Xは二官能性ポリオキシエチレン鎖部分または結合であり、
LMは、式、−C(O)−、−(CH2)bC(O)−(式中、bは1〜5の整数である。)−C(O)−(CH2)c−C(O)−(式中、cは2〜10の整数であり、ラジカルの脂肪族部分は飽和でも不飽和でも良い。)、−C(O)−O−(CH2)d−O−C(O)−(式中、dは2〜10の整数であり、あるいは式、−R−C(O)−、−R−C(O)−(CH2)c−C(O)−、または−R−C(O)−O−(CH2)d−O−C(O)−(式中、cは2〜10の整数であり、dは2〜10の整数であり、Rは1〜10個のモノマーラクチド、グリコリド、炭酸トリメチレン、カプロラクトンまたはp−ジオキサノンの断片を有するポリマーまたはコポリマーである。)によって表されるオリゴマージラジカルである。)によって表される二官能性結合部分であり、
GはN−オキシスクシンイミジル、N−オキシマレイミジル、N−オキシフタルイミジル、ニトロフェノキシル、N−オキシイミダゾリル、およびトレシルの群より選択される脱離基であり、
Iは多求核性化合物から誘導される多官能性結合部分であり、
nは2〜10の整数であるが、
ただしXが二官能性ポリオキシエチレン鎖部分の場合、−X−I−X−は、式、
−O−(CH2−CH2−O−)a−
(式中、aは20〜300の整数である。)によって表されるジラジカル断片であるPEGである。)
の架橋剤を有する架橋されたタンパク質内に分散した活性薬剤を含む、活性薬剤をサブジェクトにデリバリするための予備成形されたオブジェクト。
【請求項12】
架橋されたタンパク質が架橋された血清アルブミンを含む、請求項11に記載の予備成形されたオブジェクト。
【請求項13】
架橋された血清アルブミンが架橋されたヒト血清アルブミンを含む、請求項12に記載の予備成形されたオブジェクト。
【請求項14】
GがN−オキシスクシンイミジルである、請求項11に記載の予備成形されたオブジェクト。
【請求項15】
少なくとも部分的に脱水された、請求項11に記載の予備成形されたオブジェクト。
【請求項16】
活性薬剤が抗菌剤、増殖因子、抗ガン剤、局所麻酔薬、防腐薬、ホルモン、抗ウイルス剤、麻薬拮抗薬、免疫反応調節剤、目薬、ワクチン、それらの代謝前駆体、およびそれらの混合物から選択される、請求項11に記載の予備成形されたオブジェクト。
【請求項17】
二次生分解性ポリマーマトリックス中に分散した、請求項11に記載の予備成形されたオブジェクト。
【請求項18】
シート、ボール、ビーズ、球体、プラグ、繊維、リボン、またはウェッジの形態である、請求項11に記載の予備成形されたオブジェクト。
【請求項19】
約1mmを越える直径を有するビーズの形態である、請求項11に記載の予備成形されたオブジェクト。
【請求項20】
直径約1μm〜約1000μmの微小球の形態である、請求項11に記載の予備成形されたオブジェクト。
【請求項21】
少なくとも部分的に脱水されたボール形のオブジェクトである、請求項11に記載の予備成形されたオブジェクト。
【請求項22】
タンパク質を含む第1の水性混合物を提供するステップと、
式、
I−(−X−LM−G)n
(式中、
Xは二官能性ポリオキシエチレン鎖部分または結合であり、
LMは式、−C(O)−、−(CH2)bC(O)−(式中、bは1〜5の整数である。)、−C(O)−(CH2)c−C(O)−(式中、cは2〜10の整数であり、ラジカルの脂肪族部分は飽和または不飽和でも良い。)、−C(O)−O−(CH2)d−O−C(O)−(式中、dは2〜10の整数であり、あるいは式、−R−C(O)−、−R−C(O)−(CH2)c−C(O)−、または−R−C(O)−O−(CH2)d−O−C(O)−(式中、cは2〜10の整数であり、dは2〜10の整数であり、Rは1〜10個のモノマーラクチド、グリコリド、炭酸トリメチレン、カプロラクトンまたはp−ジオキサノン断片を有するポリマーまたはコポリマーである。)によって表されるオリゴマージラジカルである。)によって表される二官能性結合部分であり、
GはN−オキシスクシンイミジル、N−オキシマレイミジル、N−オキシフタルイミジル、ニトロフェノキシル、N−オキシイミダゾリル、およびトレシルの群より選択される脱離基であり、
Iは多求核性化合物から誘導される多官能性結合部分であり、
nは2〜10の整数であるが、
ただし、Xが二官能性ポリオキシエチレン鎖部分である場合、−X−I−X−は、式、
−O−(CH2−CH2−O−)a−
(式中、aは20〜300の整数である。)によって表されるジラジカル断片であるPEGである。)
の架橋剤を含む第2の水性混合物を提供するステップと、
第1および第2の水性混合物を混合して物品を形成するステップと、
物品をタンパク質と架橋剤との架橋に有効な条件に曝すステップと、からなる
薬剤をデリバリするための予備成形されたオブジェクトを形成する方法。
【請求項23】
サブジェクトを請求項5に記載の予備成形されたオブジェクトに接触させるステップを含む、活性薬剤をサブジェクトにデリバリする方法。
【請求項24】
接触させるステップが、予備成形されたオブジェクトをサブジェクトに体内埋植するステップを含む、請求項23に記載の方法。
【請求項25】
接触させるステップが、複数の予備成形されたオブジェクトをサブジェクト内の組織空隙に詰めるステップを含む、請求項23に記載の方法。
【請求項26】
サブジェクトを請求項11に記載の予備成形されたオブジェクトに接触させるステップを含む、活性薬剤をサブジェクトにデリバリする方法。
【請求項27】
接触させるステップが、予備成形されたオブジェクトをサブジェクトに体内埋植するステップを含む、請求項26に記載の方法。
【請求項28】
接触させるステップが、複数の予備成形されたオブジェクトをサブジェクト内の組織空隙に詰めるステップを含む、請求項26に記載の方法。
【請求項1】
式、
I−(−X−LM−G)n
(式中、
Xは二官能性ポリオキシエチレン鎖部分または結合であり、
LMは式、−C(O)−、−(CH2)bC(O)−(式中、bは1〜5の整数である。)、−C(O)−(CH2)c−C(O)−(式中、cは2〜10の整数であり、ラジカルの脂肪族部分は飽和または不飽和でも良い。)、−C(O)−O−(CH2)d−O−C(O)−(式中、dは2〜10の整数であり、あるいは式、−R−C(O)−、−R−C(O)−(CH2)c−C(O)−、または−R−C(O)−O−(CH2)d−O−C(O)−(式中、cは2〜10の整数であり、dは2〜10の整数であり、Rは1〜10個のモノマーラクチド、グリコリド、炭酸トリメチレン、カプロラクトンまたはp−ジオキサノン断片を有するポリマーまたはコポリマーである。)によって表されるオリゴマージラジカルである。)によって表される二官能性結合部分であり、
GはN−オキシスクシンイミジル、N−オキシマレイミジル、N−オキシフタルイミジル、ニトロフェノキシル、N−オキシイミダゾリル、およびトレシルの群より選択される脱離基であり、
Iは多求核性化合物から誘導される多官能性結合部分であり、
nは2〜10の整数であるが、
ただしXが二官能性ポリオキシエチレン鎖部分の場合、−X−I−X−は、式、
−O−(CH2−CH2−O−)a−
(式中、aは20〜300の整数である。)によって表されるジラジカル断片であるPEGである。)
の架橋剤を有する、少なくとも部分的に脱溶媒和された架橋されたアルブミンである、活性薬剤をサブジェクトにデリバリするための予備成形されたオブジェクト。
【請求項2】
架橋されたタンパク質が架橋された血清アルブミンを含む、請求項1に記載の予備成形されたオブジェクト。
【請求項3】
架橋された血清アルブミンが架橋されたヒト血清アルブミンを含む、請求項2に記載の予備成形されたオブジェクト。
【請求項4】
GがN−オキシスクシンイミジルである、請求項1に記載の予備成形されたオブジェクト。
【請求項5】
組み込まれた活性薬剤をさらに含む、請求項1に記載の予備成形されたオブジェクト。
【請求項6】
活性薬剤が、抗菌剤、増殖因子、抗ガン剤、局所麻酔薬、防腐薬、ホルモン、抗ウイルス剤、麻薬拮抗薬、免疫反応調節剤、目薬、ワクチン、それらの代謝前駆体、およびそれらの混合物から選択される、請求項1に記載の予備成形されたオブジェクト。
【請求項7】
二次生分解性ポリマーマトリックス内に分散した、請求項1に記載の予備成形されたオブジェクト。
【請求項8】
シート、ボール、ビーズ、球体、プラグ、繊維、リボン、またはウェッジの形態である、請求項1に記載の予備成形されたオブジェクト。
【請求項9】
約1mmを越える直径を有するビーズの形態である、請求項1に記載の予備成形されたオブジェクト。
【請求項10】
約1μmから約1000μmの直径を有する微小球の形態である、請求項1に記載の予備成形されたオブジェクト。
【請求項11】
式、
I−(−X−LM−G)n
(式中、
Xは二官能性ポリオキシエチレン鎖部分または結合であり、
LMは、式、−C(O)−、−(CH2)bC(O)−(式中、bは1〜5の整数である。)−C(O)−(CH2)c−C(O)−(式中、cは2〜10の整数であり、ラジカルの脂肪族部分は飽和でも不飽和でも良い。)、−C(O)−O−(CH2)d−O−C(O)−(式中、dは2〜10の整数であり、あるいは式、−R−C(O)−、−R−C(O)−(CH2)c−C(O)−、または−R−C(O)−O−(CH2)d−O−C(O)−(式中、cは2〜10の整数であり、dは2〜10の整数であり、Rは1〜10個のモノマーラクチド、グリコリド、炭酸トリメチレン、カプロラクトンまたはp−ジオキサノンの断片を有するポリマーまたはコポリマーである。)によって表されるオリゴマージラジカルである。)によって表される二官能性結合部分であり、
GはN−オキシスクシンイミジル、N−オキシマレイミジル、N−オキシフタルイミジル、ニトロフェノキシル、N−オキシイミダゾリル、およびトレシルの群より選択される脱離基であり、
Iは多求核性化合物から誘導される多官能性結合部分であり、
nは2〜10の整数であるが、
ただしXが二官能性ポリオキシエチレン鎖部分の場合、−X−I−X−は、式、
−O−(CH2−CH2−O−)a−
(式中、aは20〜300の整数である。)によって表されるジラジカル断片であるPEGである。)
の架橋剤を有する架橋されたタンパク質内に分散した活性薬剤を含む、活性薬剤をサブジェクトにデリバリするための予備成形されたオブジェクト。
【請求項12】
架橋されたタンパク質が架橋された血清アルブミンを含む、請求項11に記載の予備成形されたオブジェクト。
【請求項13】
架橋された血清アルブミンが架橋されたヒト血清アルブミンを含む、請求項12に記載の予備成形されたオブジェクト。
【請求項14】
GがN−オキシスクシンイミジルである、請求項11に記載の予備成形されたオブジェクト。
【請求項15】
少なくとも部分的に脱水された、請求項11に記載の予備成形されたオブジェクト。
【請求項16】
活性薬剤が抗菌剤、増殖因子、抗ガン剤、局所麻酔薬、防腐薬、ホルモン、抗ウイルス剤、麻薬拮抗薬、免疫反応調節剤、目薬、ワクチン、それらの代謝前駆体、およびそれらの混合物から選択される、請求項11に記載の予備成形されたオブジェクト。
【請求項17】
二次生分解性ポリマーマトリックス中に分散した、請求項11に記載の予備成形されたオブジェクト。
【請求項18】
シート、ボール、ビーズ、球体、プラグ、繊維、リボン、またはウェッジの形態である、請求項11に記載の予備成形されたオブジェクト。
【請求項19】
約1mmを越える直径を有するビーズの形態である、請求項11に記載の予備成形されたオブジェクト。
【請求項20】
直径約1μm〜約1000μmの微小球の形態である、請求項11に記載の予備成形されたオブジェクト。
【請求項21】
少なくとも部分的に脱水されたボール形のオブジェクトである、請求項11に記載の予備成形されたオブジェクト。
【請求項22】
タンパク質を含む第1の水性混合物を提供するステップと、
式、
I−(−X−LM−G)n
(式中、
Xは二官能性ポリオキシエチレン鎖部分または結合であり、
LMは式、−C(O)−、−(CH2)bC(O)−(式中、bは1〜5の整数である。)、−C(O)−(CH2)c−C(O)−(式中、cは2〜10の整数であり、ラジカルの脂肪族部分は飽和または不飽和でも良い。)、−C(O)−O−(CH2)d−O−C(O)−(式中、dは2〜10の整数であり、あるいは式、−R−C(O)−、−R−C(O)−(CH2)c−C(O)−、または−R−C(O)−O−(CH2)d−O−C(O)−(式中、cは2〜10の整数であり、dは2〜10の整数であり、Rは1〜10個のモノマーラクチド、グリコリド、炭酸トリメチレン、カプロラクトンまたはp−ジオキサノン断片を有するポリマーまたはコポリマーである。)によって表されるオリゴマージラジカルである。)によって表される二官能性結合部分であり、
GはN−オキシスクシンイミジル、N−オキシマレイミジル、N−オキシフタルイミジル、ニトロフェノキシル、N−オキシイミダゾリル、およびトレシルの群より選択される脱離基であり、
Iは多求核性化合物から誘導される多官能性結合部分であり、
nは2〜10の整数であるが、
ただし、Xが二官能性ポリオキシエチレン鎖部分である場合、−X−I−X−は、式、
−O−(CH2−CH2−O−)a−
(式中、aは20〜300の整数である。)によって表されるジラジカル断片であるPEGである。)
の架橋剤を含む第2の水性混合物を提供するステップと、
第1および第2の水性混合物を混合して物品を形成するステップと、
物品をタンパク質と架橋剤との架橋に有効な条件に曝すステップと、からなる
薬剤をデリバリするための予備成形されたオブジェクトを形成する方法。
【請求項23】
サブジェクトを請求項5に記載の予備成形されたオブジェクトに接触させるステップを含む、活性薬剤をサブジェクトにデリバリする方法。
【請求項24】
接触させるステップが、予備成形されたオブジェクトをサブジェクトに体内埋植するステップを含む、請求項23に記載の方法。
【請求項25】
接触させるステップが、複数の予備成形されたオブジェクトをサブジェクト内の組織空隙に詰めるステップを含む、請求項23に記載の方法。
【請求項26】
サブジェクトを請求項11に記載の予備成形されたオブジェクトに接触させるステップを含む、活性薬剤をサブジェクトにデリバリする方法。
【請求項27】
接触させるステップが、予備成形されたオブジェクトをサブジェクトに体内埋植するステップを含む、請求項26に記載の方法。
【請求項28】
接触させるステップが、複数の予備成形されたオブジェクトをサブジェクト内の組織空隙に詰めるステップを含む、請求項26に記載の方法。
【図1】

【図2】

【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】

【図11】
【図12】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【公開番号】特開2013−10774(P2013−10774A)
【公開日】平成25年1月17日(2013.1.17)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−181148(P2012−181148)
【出願日】平成24年8月17日(2012.8.17)
【分割の表示】特願2001−559433(P2001−559433)の分割
【原出願日】平成13年2月16日(2001.2.16)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.テフロン
【出願人】(510103222)ネオメンド,インコーポレイティド (1)
【Fターム(参考)】
【公開日】平成25年1月17日(2013.1.17)
【国際特許分類】
【出願番号】特願2012−181148(P2012−181148)
【出願日】平成24年8月17日(2012.8.17)
【分割の表示】特願2001−559433(P2001−559433)の分割
【原出願日】平成13年2月16日(2001.2.16)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.テフロン
【出願人】(510103222)ネオメンド,インコーポレイティド (1)
【Fターム(参考)】
[ Back to top ]