
二成分系現像剤
【課題】耐久時の画像の濃度変動及び高いプロセススピードで間欠印字の画質劣化の低減、キャリア付着やトナー飛散の抑制が可能であり、高い現像性を有する二成分系現像剤の提供。
【解決手段】磁性キャリアとトナーを少なくとも含有する二成分系現像剤であって、トナーは、結着樹脂及びワックスを少なくとも含有するトナー粒子を含有し、磁性キャリアは、磁性コア粒子と樹脂を少なくとも含有し、真比重が2.5g/cm3以上4.2g/cm3以下であり、二成分系現像剤は、前記磁性キャリア100質量部に対して、トナー4質量部以上20質量部以下の割合で含有する二成分系現像剤であり、二成分系現像剤のE100mm/sec(mJ)及びE100mm/sec(mJ)、並びに、前記E10mm/secからE100mm/secを引いたΔE(mJ)が特定の関係をみたすことを特徴とする二成分系現像剤。
【解決手段】磁性キャリアとトナーを少なくとも含有する二成分系現像剤であって、トナーは、結着樹脂及びワックスを少なくとも含有するトナー粒子を含有し、磁性キャリアは、磁性コア粒子と樹脂を少なくとも含有し、真比重が2.5g/cm3以上4.2g/cm3以下であり、二成分系現像剤は、前記磁性キャリア100質量部に対して、トナー4質量部以上20質量部以下の割合で含有する二成分系現像剤であり、二成分系現像剤のE100mm/sec(mJ)及びE100mm/sec(mJ)、並びに、前記E10mm/secからE100mm/secを引いたΔE(mJ)が特定の関係をみたすことを特徴とする二成分系現像剤。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、電子写真法、及び、静電記録法に用いられる二成分系現像剤に関する。
【背景技術】
【0002】
電子写真法において、静電荷像を現像する工程は帯電されたトナーを静電荷像とのクーロン力を利用して静電荷像上に付着させて画像形成する。トナーを用いて静電荷像を現像するための現像剤には、磁性体を樹脂中に分散した磁性トナーを用いる一成分系現像剤と、非磁性トナーと磁性キャリアを混合して用いる二成分系現像剤とに大別される。
特に、高画質を要求されるフルカラー複写機またはフルカラープリンタ等のフルカラー画像形成装置では、後者が好適に用いられている。
フルカラー画像形成装置では、近年、更なる高耐久性が要求されている。そこで、現像剤の流動性を制御することで、低温低湿環境下における劣化かぶりを減少させると共に、ベタ画像の追随性が良好な現像剤が提案されている。具体的には、二成分系現像剤の流速指数25FRI値を0以上2.0以下にコントロールした現像剤である。このような現像剤とすることで、トナー外添剤の埋め込まれにくいトナー表面を有する、あるいはトナー外添剤が埋め込まれてもトナーとしての流動性、帯電性他の特性を有する耐ストレス性の高いトナーを得ることができる。[25FRI=(ブレード回転数10mm/sにおける全エネルギー/100mm/sにおける全エネルギー)](特許文献1)。
しかし、プロセススピード200mm/sec以上の高スピードで間欠印刷した場合、画質が低下し、画像の濃度変動が大きく、改善が必要であった。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】特開2007−183607号公報
【発明の概要】
【発明が解決しようとする課題】
【0004】
本発明は、耐久時の画像の濃度変動及び高いプロセススピードで間欠印字の画質劣化の低減、さらには、キャリア付着やトナー飛散の抑制が可能であり、高い現像性を有する二成分系現像剤の提供を目的とする。
【課題を解決するための手段】
【0005】
発明者らは、鋭意検討を重ねた結果、高いプロセススピードで間欠印刷した場合の画質劣化や、耐久時の画像の濃度変動と、磁性キャリアの真比重と二成分系現像剤のトータルエネルギーの間に相関があることを見出した。
すなわち、磁性キャリアとトナーを少なくとも含有する二成分系現像剤であって、前記トナーは、結着樹脂及びワックスを少なくとも含有するトナー粒子を含有し、前記磁性キャリアは、磁性コア粒子と樹脂を少なくとも含有し、真比重が2.5g/cm3以上4.2g/cm3以下であり、前記二成分系現像剤は、前記磁性キャリア100質量部に対して、トナー4質量部以上20質量部以下の割合で含有する二成分系現像剤であり、前記二成分系現像剤のE100mm/sec(mJ)及びE100mm/sec(mJ)、並びに、前記E10mm/secからE100mm/secを引いたΔE(mJ)が下記式(1)及び(2)をみたすことを特徴とする二成分系現像剤。
式(1):−0.2×E100mm/sec+100≦ΔE≦−0.4×E100mm/sec+200
式(2):80≦E100mm/sec≦360
[前記式(1)及び(2)において、E100mm/sec(mJ)は、回転式プロペラ
型ブレードを備えた粉体流動性分析装置において、プロペラ型ブレードを、前記プロペラ型ブレードの最外縁部の周速を100mm/secで回転させながら、前記二成分系現像剤が満たされた測定容器内の二成分系現像剤粉体層中に垂直に進入させ、前記二成分系現像剤粉体層の底面から100mmの位置から測定を開始し、底面から10mmの位置まで進入させた時に得られる、回転トルクと垂直荷重の総和を表す。また、前記式(1)のΔE(mJ)において、E10mm/sec(mJ)は、回転式プロペラ型ブレードを備えた粉体流動性分析装置において、プロペラ型ブレードを、前記プロペラ型ブレードの最外縁部の周速を10mm/secで回転させながら、前記二成分系現像剤が満たされた測定容器内の二成分系現像剤粉体層中に垂直に進入させ、前記二成分系現像剤粉体層の底面から55mmの位置から測定を開始し、底面から2mmの位置まで進入させた時に得られる、回転トルクと垂直荷重の総和を表す。]
【発明の効果】
【0006】
本発明によれば、耐久時の画像の濃度変動と、間欠印字の画質劣化の低減、さらには、キャリア付着やトナー飛散の抑制および現像性の高い二成分系現像剤を提供することができる。
【図面の簡単な説明】
【0007】
【図1】粉体流動性分析装置の模式図(1−a)、23.5mm径ブレード(1−b)、25mm×25mlスプリット容器(1−c)。
【図2】磁性キャリアの反射電子像を可視化した投影像(2−a)、磁性キャリアの表面状態の模式図(2−b)、磁性キャリアを画像処理して、磁性キャリアを抽出した状態(2−c)、磁性キャリアを画像処理して、磁性キャリア表面上の金属酸化物に由来する部分を抽出した状態(2−d)(図面代用写真)。
【図3】磁性キャリアの反射電子を可視化した投影像(3−a)、投影像を600倍の倍率で可視化した投影図(3−b)、投影図の画像処理の前処理後の様子を示す図(3−c)、磁性キャリアを抽出した状態を示す図(3−d)(図面代用写真)。
【図4】抽出した磁性キャリアから、画像外周部の磁性キャリアを除外した状態を示す図(4−a)、抽出された磁性キャリアから、さらに粒径によって画像処理する粒子を絞り込んだ状態を示す図(4−b)、金属酸化物を抽出した状態の図(4−c)(図面代用写真)。
【図5】表面改質装置の模式図。
【発明を実施するための形態】
【0008】
本発明の二成分系現像剤は、磁性キャリアとトナーを少なくとも含有する二成分系現像剤であって、トナーは、結着樹脂及びワックスを少なくとも含有するトナー粒子を含有し、磁性キャリアは、磁性コア粒子と樹脂を少なくとも含有し、真比重が2.5g/cm3以上4.2g/cm3以下であり、二成分系現像剤は、磁性キャリア100質量部に対して、トナー4質量部以上20質量部以下の割合で含有する二成分系現像剤であり、二成分系現像剤のE100mm/sec(mJ)及びE100mm/sec(mJ)、並びに、E10mm/secからE100mm/secを引いたΔE(mJ)が下記式(1)及び(2)をみたすことを特徴とする。
式(1):−0.2×E100mm/sec+100≦ΔE≦−0.4×E100mm/sec+200
式(2):80≦E100mm/sec≦360
[前記式(1)及び(2)において、E100mm/sec(mJ)は、回転式プロペラ型ブレードを備えた粉体流動性分析装置において、プロペラ型ブレードを、前記プロペラ型ブレードの最外縁部の周速を100mm/secで回転させながら、前記二成分系現像剤が満たされた測定容器内の二成分系現像剤粉体層中に垂直に進入させ、前記二成分系現像剤粉体層の底面から100mmの位置から測定を開始し、底面から10mmの位置まで
進入させた時に得られる、回転トルクと垂直荷重の総和を表す。また、前記式(1)のΔE(mJ)において、E10mm/sec(mJ)は、回転式プロペラ型ブレードを備えた粉体流動性分析装置において、プロペラ型ブレードを、前記プロペラ型ブレードの最外縁部の周速を10mm/secで回転させながら、前記二成分系現像剤が満たされた測定容器内の二成分系現像剤粉体層中に垂直に進入させ、前記二成分系現像剤粉体層の底面から55mmの位置から測定を開始し、底面から2mmの位置まで進入させた時に得られる、回転トルクと垂直荷重の総和を表す。]
ここで、ΔE(mJ)は、現像剤が現像器内で攪拌開始時の低速攪拌されている時に攪拌に必要なエネルギー(E10mm/sec)と、攪拌速度が規定の速度となり高速攪拌されている時に攪拌に必要なエネルギー(E100mm/sec)との差を示している。
このエネルギー差(ΔE)は、−0.2×E100mm/sec+100≦ΔE≦−0.4×E100mm/sec+200の関係を満たすことで、間欠印字の画像劣化及び耐久時の濃度変動の低減を両立させることができる。
そのメカニズムは明確ではないが、発明者らは以下ように推測している。
間欠印刷した場合の画像劣化を抑制するためには、トナーを十分に摩擦帯電させることが必要である。しかし、間欠印刷の場合、印刷されていない時に徐々にトナーの電荷が失われる。この現象は特に高湿環境下で顕著である。しかし、ユーザーの印刷の待ち時間を短くするために、印刷開始時の現像器内で現像剤の攪拌時間は、短いことが好ましい。
短時間に電荷が失われたトナーを再び十分に摩擦帯電させるためには、現像器内での攪拌に必要なエネルギーの高い現像剤にすることが1つの手法として考えられる。しかし、このような現像剤の場合、継続的に現像剤に大きなストレスがかかり、その結果、耐久時に現像剤が劣化し画像濃度が変動する。このため、従来から、間欠印字の画像劣化の防止と、耐久時の濃度変動の防止を両立させることは出来なかった。
発明者らは、上記課題に対し鋭意検討し、現像剤が攪拌され始めた時、すなわち、現像器内で現像剤が低速攪拌されている時の攪拌エネルギーを選択的に高めることで、上記問題を解決した。
間欠印刷時には、低速で攪拌される時間が比較的長い。これは、印刷終了後に現像器が停止し、次に印刷する時に再び現像器が攪拌を開始する。この頻度が高いためである。
それに対し、連続で印刷する場合は現像器が連続で攪拌しており低速で攪拌される時間は比較的短い。このため、耐久時の濃度変動を抑制しつつ、間欠印刷時の画像の濃度変動を抑制するためには、低速攪拌時のエネルギーを選択的に高めることが必要である。
【0009】
この思想から発明者らは、現像剤が現像器内で攪拌開始時の低速攪拌されている時に攪拌に必要なエネルギー(E10mm/sec)と、攪拌速度が規定の速度となり高速攪拌されている時に攪拌に必要なエネルギー(E100mm/sec)の理想的なエネルギーの関係として、以下の関係式を見出した。
−0.2×E100mm/sec+100≦ΔE≦−0.4×E100mm/sec+200
[但し、ΔE(mJ)は、E10mm/secからE100mm/secを引いた値であ
る。]
E10mm/secは、粉体流動性分析装置(図1(1−a))においてプロペラ型ブレードの最外縁部の周速を10mm/secで回転させながら容器内の二成分系現像剤粉体層中に垂直に進入させ、該粉体層の底面から55mmの位置から測定を開始し、底面から2mmの位置まで進入させた時に得られる、回転トルクと垂直荷重の総和(ブレード周速10mm/sec時のトータルエネルギーともいう)で求められる。
一方、E100mm/secは、粉体流動性分析装置(図1(1−a))においてプロペラ型ブレードの最外縁部の周速を100mm/secで回転させながら容器内の二成分系現像剤粉体層中に垂直に進入させ、該粉体層の底面から100mmの位置から測定を開始し、底面から10mmの位置まで進入させた時に得られる、回転トルクと垂直荷重の総和(ブレード周速100mm/sec時のトータルエネルギーともいう)で求められる。
プロペラ型ブレードの最外縁部の周速は低い方が、現像剤が現像器内で低速攪拌されている時の状態をより忠実に再現できる。しかし、10mm/secより小さい周速にするとプロペラの回転が安定しないためか、データの再現性が低くなる。このため、発明者らはデータの信頼性が得られ、かつ、現像機内の状態を忠実に再現できる10mm/secの周速で測定することが必要と考えた。
また、プロペラ型ブレードの最外縁部の周速100mm/secで測定したトータルエネルギー(E100mm/sec)は、攪拌速度が規定の速度となり安定的に攪拌されている時に、攪拌に必要なエネルギーを示している。具体的には、現像器内の現像スリーブと規制ブレードの間で現像剤にかかるエネルギーである。
通常、現像スリーブは現像ドラムに対し、1.2倍以上2.0倍以下のスピードで回転する。このため、プロセススピード200mm/sec以上の本体の場合、現像スリーブは240mm/sec以上のスピードで回転する。
現像剤は、規制ブレードで一部せき止められるため、規制ブレード裏のせき止められた現像剤は、240mm/sec以上のスピードで攪拌されることになる。この時の現像剤にかかるエネルギーは、粉体流動性分析装置においてプロペラ型ブレードの最外縁部の周速を240mm/sec以上で測定することで求められると予想されるが、このような高周速で測定した場合、トータルエネルギーが画質劣化や濃度変動と相関があるとする結果が得られなかった。その理由は明確ではないが以下のように発明者らは推測している。
粉体流動性分析装置で測定する場合、測定容器にフタがないため、プロペラ型ブレードの周速があがればあがるほど、測定容器の上の方に現像剤が逃げてしまう。このように逃げる現像剤の量が多くなってしまうと、現像剤にかかるエネルギーが小さくなってしまう。それに対して、現像器内の場合、密閉系に近い形態のため、大きなエネルギーがかかる。
このため、発明者らは鋭意検討した結果、プロペラ型ブレードの最外縁部の周速100mm/sec以下で測定することで、測定容器の上に逃げる現像剤が少なく、現像剤が逃げた影響を減らすことができ、現像機内の現像剤の状況を再現できることを見出した。
また、100mm/secよりブレード周速が遅くなると、現像剤の攪拌速度が低くなりすぎ、プロセススピード200mm/sec以上の本体の状況を再現できない。
【0010】
E100mm/secとΔEを上記範囲にすることで、高プロセススピードで間欠印刷した場合の画質劣化の低減や、画像の濃度変動を改善することができる。
ΔEが[−0.2×E100mm/sec+100]より小さくなると、間欠で印刷した場合、画像の濃度変動が大きくなる。その理由は明確ではないが、発明者らは低速攪拌時の攪拌に必要なエネルギーが小さく、トナーが十分に電荷を保持しないうちに現像されることに起因すると推測している。
一方、ΔEが[−0.4×E100mm/sec+200]より大きくなると、間欠で印刷した場合、画質が劣化する。その理由は明確ではないが、発明者らは低速攪拌時の攪拌に必要なエネルギーが大きすぎ、低速攪拌時に現像剤が劣化してしまうことに起因すると推測している。
また、E100mm/sec(mJ)を、80≦E100mm/sec≦360、(好ましくは120≦E100mm/sec≦300)の範囲にすることで、トナー飛散防止と耐久時の濃度安定性を向上させることができる。
E100mm/sec(mJ)が80より小さいと現像器でのトナー飛散が多くなり、部材が汚染されてしまう。
一方、E100mm/sec(mJ)が360より大きいと現像器内で現像剤を攪拌する時に大きなエネルギーが必要になる。このため、間欠印刷を有無にかかわらず、現像剤に過度のストレスがかかり、耐久時に画像濃度が変動する。
【0011】
次に、E100mm/sec(mJ)とE10mm/sec(mJ)の具体的な測定方法について述べる。本発明において、E100mm/sec(mJ)及びE10mm/s
ec(mJ)の測定には、回転式ブレードを備えた粉体流動性分析装置(パウダーレオメータFT−4、Freeman Technology社製)(以下、「FT−4」ともいう)を用いた。
上記装置の原理は、粉体サンプル中で回転式ブレードを移動させ、一定のパターンの流れを起こさせる。粉体サンプル中の粒子はブレードが近接すると流動し、通過すると再び静止する。ブレードが粉体中を移動するのに必要としたエネルギーが測定され、この値から、種々の流動性指数が計算される。ブレードはプロペラ型で、回転すると同時に上又は下方向にも運動するので先端はらせんを描くことになる。回転速度と上下運動を変化させることによりブレードのらせん経路の角度や速度を調節することができる。ブレードが粉体層表面に対して右回りのらせん経路に沿って移動するときには粉体を均一に混ぜる作用がある。逆にブレードが粉体層表面に対して左回りのらせん経路に沿って移動するときにはブレードは粉体から抵抗を受けることになる。(図1(1−a))
具体的には、以下の操作により測定を行った。尚、全ての操作において、プロペラ型ブレードは、FT−4測定専用[23.5mm径ブレード(型番:C415)](図1(1−b)参照、以下、「ブレード」と省略)を用いた。
まず、FT−4測定専用[25mm×25mlスプリット容器(型番:2015)](図1(1−c)参照、以下、容器と省略)に23℃/60%RH環境に3日放置されたトナーをすりきり一杯(約30cc)入れることで二成分系現像剤粉体層とした。
(1)コンディショニング操作
(a):ブレードの周速(ブレードの最外縁部の周速)を100(mm/sec)にした。粉体層への垂直方向の進入速度を、移動中のブレードの最外縁部が描く軌跡と粉体層表面とのなす角度(以降、「ブレード軌跡角」)が5(deg)になるスピードにした。粉体層表面に対して時計回り(ブレードの回転により粉体層がほぐされる方向)の回転方向に、粉体層表面から二成分系現像剤粉体層の底面から5mmの位置までブレードを進入させた。その後、ブレードの周速を40(mm/sec)、粉体層への垂直方向の進入速度を、ブレード軌跡角が2(deg)になるスピードで、粉体層表面に対して時計回りの回転方向に、二成分系現像剤粉体層の底面から2mmの位置までブレードを進入させる操作を行った。その後、ブレードの周速を40(mm/sec)、粉体層からの抜き取り速度をブレード軌跡角が5(deg)になるスピードで、粉体層表面に対して時計回りの回転方向に、トナー粉体層の底面から55mmの位置までブレードを移動させ、抜き取りを行った。抜き取りが完了したら、ブレードを時計回り、反時計回りに交互に小さく回転させることでブレードに付着した二成分系現像剤を払い落とした。
(b):一連の上記(1)−(a)の操作を5回行うことで、二成分系現像剤粉体層中に巻き込まれている空気を取り除き、安定した二成分系現像剤粉体層を作った。
・ スプリット操作
上述の容器のスプリット部分(図1(1−c))で二成分系現像剤粉体層をすり切り、粉体層上部の二成分系現像剤を取り除くことで、同じ体積(25ml)の二成分系現像剤粉体層を形成した。
・ 測定操作
(a):上記(1)−(a)と同様の操作を一回行った。
(b):次にブレードの周速を100(mm/sec)、粉体層への垂直方向の進入速度を、ブレード軌跡角が5(deg)になるスピードにした。粉体層表面に対して反時計回り(ブレードの回転により粉体層が押し込まれる方向)の回転方向に、トナー粉体層の底面から5mmの位置までブレードを進入させた。その後、ブレードの周速を40(mm/sec)、粉体層への垂直方向の進入速度を、ブレード軌跡角が2(deg)になるスピードで、粉体層表面に対して時計回りの回転方向に、粉体層の底面から2mmの位置までブレードを進入させる操作を行った。その後、ブレードの周速を40(mm/sec)、粉体層からの垂直方向の抜き取り速度をブレード軌跡角が5(deg)になるスピードで、粉体層表面に対して時計回りの回転方向に、粉体層の底面から55mmの位置までブレードの抜き取りを行った。抜き取りが完了したら、ブレードを時計回り、反時計回りに
交互に小さく回転させることでブレードに付着した二成分系現像剤を払い落とした。
(c):上記、(b)の一連の操作を7回繰り返した。
上記(c)の操作において、7回目のブレードの周速100(mm/sec)で二成分系現像剤粉体層の底面から100mmから10mmの位置までブレードを進入させたときに得られる回転トルクと垂直荷重の総和を、[E100mm/sec]とした。
(d):(3)−(c)と同様に70(mm/sec)、40(mm/sec)、10(mm/sec)に順次周速を落とした測定を行った。周速が10(mm/sec)でトナー粉体層の底面から55mmの位置から測定を開始し、底面から2mmの位置まで進入させた時に得られる回転トルクと垂直荷重の総和を、[E10mm/sec]とした。
そして、E10mm/secからE100mm/secを引いたものをΔE(mJ)とした。
【0012】
上記ΔE及びE100mm/secは、磁性キャリア及びトナーの構造又は物性を制御することで本発明の範囲に調整することが可能である。
磁性キャリアについては、まず、真比重が、2.5g/cm3以上4.2g/cm3以下である。真比重が2.5g/cm3より小さくなると、磁性成分の量が少なくなる。その結果、磁性キャリアが現像スリーブに保持されにくくなり、磁性キャリアがドラム上に付着してしまう。磁性キャリアの真比重が4.2g/cm3より大きくなると、二成分系現像剤の攪拌時の抵抗が大きくなり、E100mm/secが上記範囲より大きくなる。
次に、磁性キャリア表面に、現像剤が現像器内で攪拌開始時の低速攪拌されている時に攪拌に必要なエネルギーに関与する部分と、攪拌速度が規定の速度となり高速攪拌されている時に攪拌に必要なエネルギーに関与する部分の両方を持たせることが好ましい。
具体的には、磁性キャリア表面に、凹凸を存在させ、かつ、凸部と凹部の表面状態を異ならせることが好ましい。
このような磁性キャリアの場合、低速攪拌時は攪拌速度が遅いために、凸部と凹部の両方が攪拌エネルギーに関与する。それに対し、規定の速度での攪拌時には、凹部はほとんど接することがなく、攪拌エネルギーに関与するのは凸部のみになる。
このため、凸部と凹部の表面状態を変えた磁性キャリアの場合、低速攪拌時及び高速攪拌時の攪拌エネルギーをコントロールすることができる。
具体的には凸部に攪拌時の抵抗の少ない金属酸化物部分を存在させ、凹部に攪拌時の抵抗が大きい樹脂部を存在させた磁性キャリアを用いることが好ましい。
これ以外にも、キャリアの粒径、使用している樹脂の種類等によってもE100mm/sec、及びΔEを調整することができる。
また、トナーとしては、トナーの形状(平均円形度)、粒径、粒径分布により、攪拌エネルギーをコントロールすることができる。
なお、上記、どの手法を用いても、E100mm/sec、及びΔEが本発明の範囲に入っていれば、本願発明の効果は発現する。
【0013】
上記E100mm/sec、及びΔEを本発明の範囲に調整するための、磁性キャリアの凹凸度合いとしては、該磁性キャリアの走査型電子顕微鏡(SEM)で撮影された投影像の周囲長をL1とし、包絡線の長さをL2とするとき、包絡度{(L1−L2)/L2}×100が、0.5以上10.0以下が好ましく、1.0以上5.0以下がより好ましい。
そのメカニズムは明確ではないが、発明者らは以下のように推測している。
現像剤が攪拌された時、攪拌速度が低い場合、磁性キャリア粒子の周囲に沿って、他の磁性キャリアやトナーが通過すると思われる。それに対し、高速で攪拌された場合、キャリア粒子の包絡線に近い場所で磁性キャリア粒子やトナーが通過すると思われる。
このため、この周囲長と包絡線がずれるようにキャリアの凹凸度合いをコントロールすることで、低速攪拌及び高速攪拌時の現像剤の攪拌状態を変えることができる。具体的には、包絡度が0.5以上10.0以下であると、E100mm/sec、及びΔEを本発
明の範囲にコントロールすることができる。包絡度が0.5未満だと、凹凸が小さいために高速攪拌時に攪拌エネルギーが大きくなってしまう。一方、包絡度が10.0を越えると、凸凹が大きいため、磁性キャリアとしての耐久性が低下し、印刷時にキャリアが破損し、キャリア付着が発生する場合がある。
【0014】
本発明に用いられる磁性キャリアについて詳細に述べる。
本発明の磁性キャリアは、金属酸化物を含有する磁性コア粒子と樹脂とを少なくとも含有する磁性キャリアである。前記磁性キャリアは、走査型電子顕微鏡を用い、加速電圧が2.0kVのときに得られる反射電子の投影像において、金属酸化物に由来する部分の面積が、磁性キャリアの投影像面積に対して、0.5面積%以上8.0面積%以下であり、6.672μm2以上の金属酸化物に由来する部分の面積が、金属酸化物に由来する部分の総面積に対して、10面積%以下であることが、上記ΔEを本発明で規定する範囲に調節するために好ましい。
この金属酸化物に由来する部分は、樹脂部に比べ、トナーに対し付着力が低く、現像剤の攪拌エネルギーを下げることができると発明者らは考えている。
加速電圧が2.0kVのときに得られる反射電子の投影像において、金属酸化物に由来する部分の面積が、磁性キャリアの投影像面積に対して、0.5面積%より小さい場合は、磁性キャリア表面に存在する金属酸化物に由来する部分の面積が小さいために、高速攪拌時に現像剤の攪拌エネルギーを下げることが出来ず、現像剤が劣化し、耐久時に画質が低下する傾向にある。
一方、磁性キャリアの投影像面積に対して、8.0面積%より大きい場合は、金属酸化物に由来する部分が多いため、低速攪拌時の攪拌エネルギーが低くなり、その結果、間欠印刷時に画像濃度が変動する傾向にある。
また、発明者らは、200pixel(5.560μm2)、220pixel(6.116μm2)、240pixel(6.672μm2)・・・と、20pixel毎に、金属露出部分の面積比率と、電荷のリークの関係を検討した。その結果、金属酸化物に由来する部分の面積が6.672μm2以上の部位において、電荷のリークが顕著に発生することを見出した。6.672μm2以上とは、走査型電子顕微鏡による反射電子の投影像の観察において、240pixelの大きさである。(1pixelの面積は0.0278μm2)
発明者らはその理由を以下のように推察している。
金属酸化物に由来する部分の回りには被覆している樹脂が存在している。その樹脂はある程度の厚みをもっており、その厚みにより金属酸化物に由来する部分の接触が抑制され、リークが生じにくい。それに対し、金属酸化物に由来する部分が6.672μm2以上大きさだと、周囲にある樹脂の影響をうけずに接触に寄与する部分が存在できる。このため、キャリア同士が接触したときに、当該部分から電荷がリークすると考えている。
この面積を金属酸化物に由来する部分の総面積に対して、10面積%以下にすることで、長期使用時の帯電量低下を抑制し、画像濃度の変動を抑制することができる。
【0015】
なお、上記走査型電子顕微鏡を用い、所定の加速電圧下で撮影された反射電子の投影像上で金属酸化物に由来する部分とは、主に反射電子を可視化した像(図2(2−a)参照)において、輝度の高い(画像上白く、明るく見える)部分として観察される部分のことを指している。走査型電子顕微鏡は、加速した電子線を試料へ照射し、試料から放出されてくる二次電子や反射電子を検出することで、試料の表面や組成情報を可視化する装置である。走査型電子顕微鏡観察において、放出されてくる反射電子の量は、重元素ほど多いことが知られている。例えば、有機化合物と鉄が平面上に分布している試料であれば、鉄からの反射電子の放出量が多いため、鉄部分が画像上では明るく(輝度が高い、白く)見える。一方、軽元素から構成される有機化合物からの反射電子量は多くはないため、画像上では暗く(輝度が低く、黒く)見えることになる。
上記磁性キャリアの表面には、有機化合物である樹脂部分と金属酸化物である磁性コア
部分とが磁性キャリアの表面に分布している。磁性キャリアの反射電子像においては、金属酸化物部分が明るく、樹脂部分が暗く、画像上で大きなコントラストの差をもった投影像として得られる。図2(2−b)は、図2(2−a)の磁性キャリア表面の金属酸化物部分と樹脂部分の分布を模式的に示したもので、白い部分が、金属酸化物に由来する部分であり、黒い部分が、樹脂部分に相当する。本発明においては、まず、図2(2−a)の磁性キャリアの投影像から磁性キャリアを抽出し、磁性キャリアの投影像面積をもとめる。図2(2−c)の白く抜けている部分が、図2(2−a)の投影像から磁性キャリア部分として抽出された部分を示している。続いて、図2(2−a)の投影像から金属酸化物部分を抽出し(図2(2−d)参照)、図2(2−d)において、白く抜けている箇所が金属酸化物部分を表している。磁性キャリアの投影像面積と金属酸化物部分の面積を画像処理によりそれぞれ求める。次に、磁性キャリアの投影像面積に占める金属酸化物部分の面積の割合と、金属酸化物部分の面積分布を算出する。走査型電子顕微鏡による観察条件、撮影条件、画像手順の詳細は、後述する。また、実際に、白く光る部分が金属酸化物であることは、電子顕微鏡に付属の元素分析装置で確認することもできる。
【0016】
磁性キャリア表面における磁性コア部分の存在状態(すなわち、金属酸化物に由来する部分の面積)をコントロールする方法は、磁性コア粒子の含有する被覆樹脂の組成、被覆樹脂の量、被覆方法など種々の方法で調整することができる。また、樹脂で被覆時に、凹部に選択的に樹脂を導入することが好ましい。
具体的には、固形分濃度の異なる被覆樹脂溶液で複数回にわけて被覆処理するなどによって、磁性コア部分の存在状態をコントロールすることが好ましい。
【0017】
上記磁性コア粒子としては、例えば、表面酸化又は未酸化の鉄、リチウム、カルシウム、マグネシウム、ニッケル、銅、亜鉛、コバルト、マンガン、クロム、希土類の如き金属粒子、それらの合金粒子、酸化物粒子及びフェライト等が好適に例示できる。
また、上記磁性キャリアは、磁性コア粒子へ樹脂を充填した磁性キャリア、及び/又は、磁性コア粒子の表面を樹脂で被覆した被覆磁性キャリアであることが好ましい。樹脂の被覆方法としては、樹脂の如き被覆材を溶剤中に溶解もしくは懸濁せしめて調製した塗布液を磁性コア粒子表面に付着せしめる方法、磁性コア粒子と被覆材とを粉体で混合する方法等、従来公知の方法が適用できる。
また、上記磁性コア粒子は、下記に述べる、多孔質磁性コア粒子であることが好ましく、多孔質フェライト粒子であることがより好ましい。
ここで、上記磁性キャリアとして、多孔質磁性コア粒子中に樹脂を含有させたものが、包絡度を0.5以上10以下となるような凹凸を磁性キャリア表面に構成できるために、特に好ましい。
多孔質磁性コア粒子は、以下のような工程で製造することができる。
多孔質磁性コア粒子の材質としては、以下のものが挙げられる。1)表面が酸化された鉄粉、2)未酸化の鉄粉、3)リチウム、カルシウム、マグネシウム、ニッケル、銅、亜鉛、コバルト、マンガン、クロム及び希土類元素の如き金属粒子、4)鉄、リチウム、カルシウム、マグネシウム、ニッケル、銅、亜鉛、コバルト、マンガン、クロム及び希土類元素の如き金属の合金粒子、又はこれらの元素を含む酸化物粒子、5)マグネタイト粒子又はフェライト粒子。
フェライト粒子とは次式で表される焼結体である。
(M12O)u(M2O)v(M32O3)w(M4O2)x(M52O5)y(Fe2O3)z
(式中、M1は1価、M2は2価、M3は3価、M4は4価、M5は5価の金属であり、u+v+w+x+y+z=1.0とした時に、u、v、w、x及びyは、それぞれ0≦(u,v,w,x,y)≦0.8であり、zは、0.2<z<1.0である。)
また、上記式中において、M1〜M5としては、少なくともLi、Fe、Zn、Ni、Mn、Mg、Co、Cu、Ba、Sr、Ca、Si、V、Bi、In、Ta、Zr、B、
Mo、Na、Sn、Ti、Cr、Al、Sc、Y、La、Ce、Pr、Nd、Sm、Eu、Gd、Tb、Dy、Ho、Er、Tm、Yb、Luからなる群から選ばれる1種類以上の金属元素を表す。
例えば、磁性のLi系フェライト(例えば、(Li2O)a(Fe2O3)b(0.0<a<0.4、0.6≦b<1.0、a+b=1))、Mn系フェライト(例えば、(MnO)a(Fe2O3)b(0.0<a<0.5、0.5≦b<1.0、a+b=1))、Mn−Mg系フェライト(例えば、(MnO)a(MgO)b(Fe2O3)c(0.0<a<0.5、0.0<b<0.5、0.5≦c<1.0、a+b+c=1))、Mn−Mg−Sr系フェライト(例えば、(MnO)a(MgO)b(SrO)c(Fe2O3)d(0.0<a<0.5、0.0<b<0.5、0.0<c<0.5、0.5≦d<1.0、a+b+c+d=1)、Cu−Zn系フェライト(例えば、(CuO)a(ZnO)b(Fe2O3)c(0.0<a<0.5、0.0<b<0.5、0.5≦c<1.0、a+b+c=1)がある。なお、上記フェライトは主元素を示し、それ以外の微量金属を含有するものも含んでいる。
結晶の成長速度のコントロールの容易性の観点から、Mn元素を含有する、Mn系フェライト、Mn−Mg系フェライト、Mn−Mg−Sr系フェライトが好ましい。
【0018】
以下に、多孔質磁性キャリア粒子としてフェライト粒子を用いる場合の製造工程を詳細に説明する。
<工程1(秤量・混合工程)>
上記フェライトの原料を、秤量し、混合する。
フェライト原料としては、例えば以下のものが挙げられる。Li、Fe、Zn、Ni、Mn、Mg、Co、Cu、Ba、Sr、Y、Ca、Si、V、Bi、In、Ta、Zr、B、Mo、Na、Sn、Ti、Cr、Al、希土類金属の金属粒子、酸化物、水酸化物、シュウ酸塩、炭酸塩。
混合する装置としては、例えば以下のものが挙げられる。ボールミル、遊星ミル、ジオットミル、振動ミル。特にボールミルが混合性の観点から好ましい。
具体的には、ボールミル中に、秤量したフェライト原料、ボールを入れ、0.1時間以上20.0時間以下、粉砕・混合する。
<工程2(仮焼成工程)>
粉砕・混合したフェライト原料を、大気中で焼成温度700℃以上1000℃以下の範囲で、0.5時間以上5.0時間以下仮焼成し、フェライト化する。焼成には、例えば以下の炉が用いられる。バーナー式焼却炉、ロータリー式焼却炉、電気炉。
<工程3(粉砕工程)>
工程2で作製した仮焼フェライトを粉砕機で粉砕する。
粉砕機としては、所望の粒径が得られれば特に限定されない。例えば以下のものがあげられる。クラッシャーやハンマーミル、ボールミル、ビーズミル、遊星ミル、ジオットミル。
フェライト粉砕品を所望の粒径にするために、例えば、ボールミルやビーズミルでは用いるボールやビーズの素材、粒径、運転時間を制御することが好ましい。具体的には、仮焼フェライトスラリーの粒径を小さくするためには、比重の重いボールを用いたり、粉砕時間を長くすればよい。また、仮焼フェライトの粒度分布を広くするためには、比重の重いボールやビーズを用い、粉砕時間を短くすることで得ることができる。また、粒径の異なる複数の仮焼フェライトを混合することでも分布の広い仮焼フェライトを得ることができる。
また、ボールミルやビーズミルは、乾式より湿式の法が、粉砕品がミルの中で舞い上がることがなく粉砕効率が高い。このため、乾式より湿式の方がより好ましい。
<工程4(造粒工程)>
仮焼フェライトの粉砕品に対し、水、バインダーと、必要に応じて、細孔調整剤を加える。細孔調整剤としては、発泡剤や樹脂微粒子が挙げられる。発泡剤として、例えば、炭
酸水素ナトリウム、炭酸水素カリウム、炭酸水素リチウム、炭酸水素アンモニウム、炭酸ナトリウム、炭酸カリウム、炭酸リチウム、炭酸アンモニウムが挙げられる。樹脂微粒子として、例えば、ポリエステル、ポリスチレン、スチレン−ビニルトルエン共重合体、スチレン−ビニルナフタリン共重合体、スチレン−アクリル酸エステル共重合体、スチレン−メタクリル酸エステル共重合体、スチレン−α−クロルメタクリル酸メチル共重合体、スチレン−アクリロニトリル共重合体、スチレン−ビニルメチルケトン共重合体、スチレン−ブタジエン共重合体、スチレン−イソプレン共重合体、スチレン−アクリロニトリル−インデン共重合体の如きスチレン共重合体;ポリ塩化ビニル、フェノール樹脂、変性フェノール樹脂、マレイン樹脂、アクリル樹脂、メタクリル樹脂、ポリ酢酸ビニル、シリコーン樹脂;脂肪族多価アルコール、脂肪族ジカルボン酸、芳香族ジカルボン酸、芳香族ジアルコール類及びジフェノール類から選択されるモノマーを構造単位として有するポリエステル樹脂;ポリウレタン樹脂、ポリアミド樹脂、ポリビニルブチラール、テルペン樹脂、クマロンインデン樹脂、石油樹脂、ポリエステルユニットとビニル系重合体ユニットを有しているハイブリッド樹脂の微粒子が挙げられる。
上記バインダーとしては、例えば、ポリビニルアルコールが用いられる。
工程3において、湿式で粉砕した場合は、フェライトスラリー中に含まれている水も考慮し、バインダーと必要に応じて細孔調整剤を加えることが好ましい。
得られたフェライトスラリーを、噴霧乾燥機を用い、100℃以上200℃以下の加温雰囲気下で、乾燥・造粒する。噴霧乾燥機としては、所望の多孔質磁性コア粒子の粒径が得られれば特に限定されない。例えば、スプレードライヤーが使用できる。
<工程5(本焼成工程)>
次に、造粒品を800℃以上1400℃以下で1時間以上24時間以下焼成する。
焼成温度を上げ、焼成時間を長くすることで、多孔質磁性コア粒子の焼成が進み、その結果、細孔径は小さく、かつ、細孔の数も減る。また、焼成する雰囲気をコントロールすることで、ブレークダウンする電界強度を好ましい範囲にコントロールすることができる。例えば、酸素濃度を低くしたり、還元雰囲気(水素存在下)にしたりすることで、多孔質磁性コア粒子のブレークダウン電界強度を下げることができる。
<工程6(選別工程)>
以上の様に焼成した粒子を解砕した後に、必要に応じて、分級や篩で篩分して粗大粒子や微粒子を除去してもよい。
磁性コア粒子の体積分布基準50%粒径(D50)は、18.0μm以上68.0μm以下であることが、画像へのキャリア付着とガサツキの抑制のためより望ましい。
【0019】
上述のように磁性キャリアは、多孔質磁性コア粒子の空隙の少なくとも一部に、樹脂が充填された磁性キャリアであることが好ましい。
多孔質磁性コア粒子は、内部の空隙容積によっては物理的強度が低くなることがあり、磁性キャリアとしての物理的強度を高めるために、多孔質磁性コア粒子の空隙の少なくとも一部に樹脂の充填を行うことが好ましい。多孔質磁性コア粒子に充填される樹脂の量としては、多孔質磁性コア粒子に対して6質量%以上25質量%以下であることが好ましい。磁性キャリア毎の樹脂含有量にバラつきが少なければ、内部空隙内の一部にのみ樹脂が充填されていても、多孔質磁性コア粒子の表面近傍の空隙にのみ樹脂が充填され内部に空隙が残っていても、内部空隙が完全に樹脂で充填されていてもよい。
多孔質磁性コア粒子の空隙に、樹脂を充填する方法としては、特に限定されないが、浸漬法、スプレー法、ハケ塗り法、及び流動床の如き塗布方法により多孔質磁性コア粒子を樹脂溶液に含浸させ、その後、溶剤を揮発させる方法が挙げられる。特に、磁性キャリアの表面に金属酸化物由来の部分を残すためには、樹脂と溶剤を混合した樹脂溶液を多孔質磁性コア粒子の空隙へ充填させる方法が好ましい。多孔質磁性コア粒子の空隙に樹脂を充填させる方法としては、樹脂を溶剤に希釈し、これを多孔質磁性コア粒子の空隙に添加する方法が採用できる。ここで用いられる溶剤は、樹脂を溶解できるものであればよい。有機溶剤に可溶な樹脂である場合は、有機溶剤として、トルエン、キシレン、セルソルブブ
チルアセテート、メチルエチルケトン、メチルイソブチルケトン、メタノールが挙げられる。また、水溶性の樹脂またはエマルジョンタイプの樹脂である場合には、溶剤として水を用いればよい。
上記多孔質磁性コア粒子の空隙に充填する樹脂としては特に限定されず、熱可塑性樹脂、熱硬化性樹脂のどちらを用いてもかまわない。多孔質磁性コア粒子に対する親和性が高いものであることが好ましく、親和性が高い樹脂を用いた場合には、多孔質磁性コア粒子の空隙への樹脂の充填時に、同時に多孔質磁性コア粒子表面も樹脂で覆うことが容易になる。
上記充填する樹脂として、熱可塑性樹脂としては、以下のものが挙げられる。ポリスチレン、ポリメチルメタクリレート、スチレン−アクリル樹脂;スチレン−ブタジエン共重合体、エチレン−酢酸ビニル共重合体、ポリ塩化ビニル、ポリ酢酸ビニル、ポリフッ化ビニリデン樹脂、フルオロカーボン樹脂、パーフルオロカーボン樹脂、ポリビニルピロリドン、石油樹脂、ノボラック樹脂、飽和アルキルポリエステル樹脂、ポリエチレンテレフタレート、ポリブチレンテレフタレート、ポリアリレート、ポリアミド樹脂、ポリアセタール樹脂、ポリカーボネート樹脂、ポリエーテルスルホン樹脂、ポリスルホン樹脂、ポリフェニレンサルファイド樹脂、ポリエーテルケトン樹脂。
また、上記熱硬化性樹脂としては、以下のものが挙げられる。フェノール樹脂、変性フェノール樹脂、マレイン樹脂、アルキド樹脂、エポキシ樹脂、無水マレイン酸とテレフタル酸と多価アルコールとの重縮合によって得られる不飽和ポリエステル、尿素樹脂、メラミン樹脂、尿素−メラミン樹脂、キシレン樹脂、トルエン樹脂、グアナミン樹脂、メラミン−グアナミン樹脂、アセトグアナミン樹脂、グリプタール樹脂、フラン樹脂、シリコーン樹脂、ポリイミド、ポリアミドイミド樹脂、ポリエーテルイミド樹脂、ポリウレタン樹脂。
上記樹脂の中で、熱硬化性樹脂が、磁性キャリアの強度を高めることができるため好ましい。中でもシリコーン樹脂が、現像剤のΔE(攪拌エネルギー差)を本発明の範囲にコントロールしやすいため、より好ましい。
上記シリコーン樹脂の具体例として、以下のものが挙げられる。ストレートシリコーン樹脂では、信越化学社製のKR271、KR255、KR152、東レ・ダウコーニング社製のSR2400、SR2405、SR2410、SR2411。変性シリコーン樹脂では、信越化学社製のKR206(アルキッド変性)、KR5208(アクリル変性)、ES1001N(エポキシ変性)、KR305(ウレタン変性)、東レ・ダウコーニング社製のSR2115(エポキシ変性)、SR2110(アルキッド変性)。
上記樹脂溶液における樹脂固形分の量は、好ましくは1質量%以上50質量%以下であり、より好ましくは1質量%以上30質量%以下である。50質量%より樹脂量の多い樹脂溶液を用いると粘度が高いため多孔質磁性コア粒子の空隙に樹脂溶液が均一に浸透しにくい。また、1質量%未満であると樹脂量が少なく、多孔質磁性コア粒子への樹脂の付着力が低くなる場合がある。
【0020】
多孔質磁性コア粒子へ樹脂を充填しただけでも、磁性キャリアとして用いることも可能である。その場合には、トナーへの帯電付与性を高めるために、予め、樹脂溶液中に荷電制御剤、荷電制御樹脂等などを含有した状態で充填することが好ましい。
また、本発明に用いられる磁性キャリアは、多孔質磁性コア粒子の空隙に樹脂を充填した後、磁性キャリアの表面を樹脂で被覆することが、磁性キャリア表面に金属酸化物に由来する部分の面積や面積分布を調整するうえで、より好ましい。
磁性キャリアの表面を樹脂により被覆することにより、金属酸化物由来の部分の割合や面積をより精密に制御することができるようになるため好ましい。また、磁性キャリア表面からのトナーの離型性、磁性キャリア表面のトナーや外添剤の汚染性、トナーへの帯電付与能や磁性キャリア抵抗を制御する意味からも、磁性キャリア表面を樹脂により被覆することが好ましい。
磁性コア粒子の表面を樹脂で被覆する方法としては、特に限定されないが、浸漬法、ス
プレー法、ハケ塗り法、乾式法、及び流動床の如き塗布方法により被覆する方法が挙げられる。中でも、磁性コア粒子を適度に表面に露出させることができる浸漬法がより好ましい。被覆する樹脂の量としては、磁性コア粒子100質量部に対し、0.1質量部以上5.0質量部以下であることが、金属酸化物部分を表面に適度に露出させることができ、好ましい。被覆する樹脂は、充填に用いた樹脂を用いることができる。
【0021】
本発明に用いられるトナーは、結着樹脂及びワックスを少なくとも含有するトナー粒子を含有するトナーである。上記結着樹脂としては、特に限定されず、従来公知の結着樹脂を用いることができる。具体的には、以下のものが挙げられる。ポリエステル、ポリスチレン;ポリ−p−クロルスチレン、ポリビニルトルエンの如きスチレン誘導体の重合体;スチレン−p−クロルスチレン共重合体、スチレン−ビニルトルエン共重合体、スチレン−ビニルナフタリン共重合体、スチレン−アクリル酸エステル共重合体、スチレン−メタクリル酸エステル共重合体、スチレン−α−クロルメタクリル酸メチル共重合体、スチレン−アクリロニトリル共重合体、スチレン−ビニルメチルケトン共重合体、スチレン−ブタジエン共重合体、スチレン−イソプレン共重合体、スチレン−アクリロニトリル−インデン共重合体の如きスチレン共重合体;ポリ塩化ビニル、フェノール樹脂、変性フェノール樹脂、マレイン樹脂、アクリル樹脂、メタクリル樹脂、ポリ酢酸ビニル、シリコーン樹脂;脂肪族多価アルコール、脂肪族ジカルボン酸、芳香族ジカルボン酸、芳香族ジアルコール類及びジフェノール類から選択されるモノマーを構造単位として有するポリエステル樹脂;ポリウレタン樹脂、ポリアミド樹脂、ポリビニルブチラール、テルペン樹脂、クマロンインデン樹脂、石油樹脂、ポリエステルユニットとビニル系重合体ユニットを有しているハイブリッド樹脂。また、上記ワックスも、特に限定されず、従来公知のワックスを用いることができる。具体的には、以下のものが挙げられる。低分子量ポリエチレン、低分子量ポリプロピレン、アルキレン共重合体、マイクロクリスタリンワックス、パラフィンワックス、フィッシャー・トロプシュワックスの如き炭化水素系ワックス;酸化ポリエチレンワックスの如き炭化水素系ワックスの酸化物又はそれらのブロック共重合物;カルナバワックス、ベヘン酸ベヘニルエステルワックス、モンタン酸エステルワックスの如き脂肪酸エステルを主成分とするワックス類;脱酸カルナバワックスの如き脂肪酸エステル類を一部又は全部を脱酸化したもの。さらに、本発明に用いられるトナーは、必要に応じて、従来公知の着色剤、荷電制御剤、無機微粉体等を含有させることもできる。
一方、上記トナー粒子及びトナーの製造方法についても、特に限定されず、従来公知の製造方法を用いることができる。
本発明に用いられるトナーは、トナーの画像処理解像度512×512画素(1画素あたり0.37μm×0.37μm)のフロー式粒子像測定装置によって計測された前記トナーの円相当径1.98μm以上200.00μm以下の粒子を、0.200以上1.000以下の円形度の範囲において800分割して解析された平均円形度が、0.950以上0.990以下であり、円相当径0.50μm以上200.00μm以下の全粒子に対する、0.50μm以上1.98μm以下の粒子が25.0個数%以下であることが、現像性を向上させつつ耐久時の画像劣化の抑制することができ、好ましい。
平均円形度が、0.950より小さい場合、トナーと磁性キャリアの付着面積が大きくなり、その結果、トナーとキャリアの付着力が上がり現像性が低下する場合がある。
一方、平均円形度が、0.990より大きい場合、トナーと磁性キャリアの付着力が小さくなりすぎトナー飛散が悪化する場合がある。
【0022】
また、本発明に用いられるトナーが、上記円相当径0.50μm以上1.98μm以下の粒子が25.0個数%より大きい場合、耐久時に0.50μm以上1.98μm以下の粒子が磁性キャリアの金属酸化物部分に付着し、高速攪拌時の攪拌エネルギーが大きくなり、耐久時に濃度が変動する場合がある。
トナーの平均円形度及び円相当径0.50μm以上1.98μm以下の粒子の個数%を上記範囲にコントロールする手法としては、分級する方法、または、熱風を用いてトナー
粒子の表面を改質処理する方法が挙げられる。
具体的には、慣性分級方式のエルボージェット(日鉄鉱業社製)、遠心力分級方式のターボプレックス(ホソカワミクロン社製)、TSPセパレータ(ホソカワミクロン社製)、ファカルティ(ホソカワミクロン社製)の如き分級機や篩分機を用い、0.50μm以上1.98μm以下の粒子を取り除くことができる。
また、熱風を用いたトナー粒子の表面改質処理を行いつつ、平均円形度をコントロールすることもできる。さらに、熱風を用いたトナー粒子の表面改質処理の場合、0.50μm以上1.98μm以下の粒子が、表面改質処理時にトナー表面にしっかりと保持される。その結果、0.50μm以上1.98μm以下の粒子を25.0個数%以下にコントロールすることも可能である。
上記熱風を用いてトナー粒子の表面を改質処理する方法としては、熱風でトナー粒子の表面処理をする方法であれば、特に限定されない。例えば、ハイブリタイゼーションシステム(奈良機械製作所製)、メカノフージョンシステム(ホソカワミクロン社製)、ファカルティ(ホソカワミクロン社製)、メテオレインボー MR Type(日本ニューマチック社製)などの装置を用いる方法が挙げられる。ここで、表面処理装置を用いて熱風により表面処理を行い、続いて分級をする方法、若しくは、予め分級したものを、表面処理装置を用いて熱風により表面処理を行う方法のいずれの方法を用いてもよい。
上記表面処理装置を用いた、熱風でトナー粒子の表面を改質処理する方法の概略を、図5を用いて説明するが、これに限定されるものではない。具体的には、従来公知の方法でトナー粒子を得た後、上記表面処理装置に供給する。そして、トナー粒子供給口(100)から供給されたトナー粒子(114)は、高圧エア供給ノズル(115)から噴射されるインジェクションエアにより加速され、その下方にある気流噴射部材(102)へ向かう。気流噴射部材(102)からは拡散エアが噴射され、この拡散エアによりトナー粒子が外側方向へ拡散する。この時、インジェクションエアの流量と拡散エアの流量とを調節することにより、トナーの拡散状態をコントロールすることができる。また、トナー粒子の融着防止を目的として、トナー粒子供給口(100)の外周、表面処理装置外周及び移送配管(116)の外周には冷却ジャケット(106)が設けられている。尚、該冷却ジャケットには冷却水(好ましくはエチレングリコール等の不凍液)を通水することが好ましい。一方、拡散エアにより拡散したトナー粒子は、熱風供給口(101)から供給された熱風により、トナー粒子の表面が処理される。この時、熱風の温度は100℃以上、450℃以下であることが好ましく、100℃以上、400℃以下であることがより好ましい。熱風の温度が100℃未満の場合にはトナー粒子表面の表面面粗さにばらつきが生じる場合がある。また、450℃を超える場合には溶融状態が進みすぎる事でトナー粒子同士の合一が進み、トナー粒子の粗大化や融着が生じる場合がある。
熱風により表面が処理されたトナー粒子は、装置上部外周に設けた冷風供給口(103)から供給される冷風により冷却される。この時、装置内の温度分布の制御、トナー粒子の表面状態をコントロールする目的で、装置の本体側面に設けた第二の冷風供給口(104)から冷風を導入しても良い。第二の冷風供給口(104)の出口はスリット形状、ルーバー形状、多孔板形状、メッシュ形状等を用いる事ができ、導入方向は中心方向へ水平、装置壁面に沿う方向が、目的に応じて選択可能である。
この時、上記冷風供給口内及び第二の冷風供給口内の温度は−50℃以上、10℃以下であることが好ましく、−40℃以上、8℃以下であることがより好ましい。また、上記冷風は除湿された冷風であることが好ましい。具体的には、絶対水分量が5g/m3以下であることが好ましい。更に好ましくは、3g/m3以下である。
これらの冷風温度が−50℃未満の場合には装置内の温度が下がりすぎてしまい、本来の目的である熱による処理が十分に為されず、トナー粒子の球形化ができない場合がある。また、10℃を超える場合には、装置内における熱風ゾーンの制御が不十分になり、表面処理時にワックスを過度にトナー表面に偏析させることがある。
その後、冷却されたトナー粒子は、ブロワーで吸引され、移送配管(116)を通じて、サイクロン等で回収される。
【0023】
本発明の二成分系現像剤は、画像の濃度変動の抑制と画質低下の抑制の観点から、磁性キャリア100質量部に対して、トナーが4質量部以上20質量部以下の割合で含有する二成分系現像剤である。好ましくは、磁性キャリア100質量部に対して、トナーが4質量部以上18質量部以下の割合で含有する二成分系現像剤である。トナーが4質量部より少ないと、画像面積比率が高い画像を連続で印刷した場合に、現像器内のトナーが不足し画像濃度が低下する場合がある。一方、トナーが20質量部より多いと、現像器でのトナー飛散が多くなり、部材を飛散したトナーで汚染する。上記二成分系現像剤において、磁性キャリア100質量部に対するトナー比率は、二成分系現像剤作製時のトナーと磁性キャリアの比率を変えることにより任意にコントロールすることができる。
【0024】
<磁性キャリア等の真密度の測定方法>
磁性キャリアの真比重は、乾式自動密度計アキュピック1330(島津製作所社製)を用いて測定する。まず、23℃、50%RHの環境に24時間放置したサンプル試料を5g精秤し、測定用セル(10cm3)に入れ、本体試料室に挿入する。測定は、試料サンプル質量を本体に入力し測定をスタートさせることにより自動測定できる。
自動測定における測定条件は、20.000psig(2.392×102kPa)で調整されたヘリウムガスを用い、試料室内を10回パージした後、試料室内の圧力変化が0.005psig/min(3.447×10−2kPa/min)になる状態を平衡状態とし、平衡状態になるまで繰り返しヘリウムガスをパージする。平衡状態の時の本体試料室の圧力を測定する。その平衡状態に達した時の圧力変化により試料サンプル体積が算出できる(ボイルの法則)。試料サンプルの体積が算出できることにより、以下の式で試料サンプルの真比重が計算できる。
試料サンプルの真比重(g/cm3)= 試料サンプル質量(g)/試料サンプル体積
(cm3)
この自動測定により5回繰り返し測定した値の平均値を磁性キャリア及び磁性コア粒子の真比重(g/cm3)とする。
【0025】
<トナーの平均円形度、及び、円相当径0.50μm以上1.98μm以下の粒子の個数%の測定方法>
上記トナーの平均円形度は、画像処理解像度512×512画素(1画素あたり0.37μm×0.37μm)のフロー式粒子像測定装置「FPIA−3000型」(シスメックス社製)を用い、校正作業時の測定・解析条件で測定する。
具体的な測定方法は、以下の通りである。まず、ガラス製の容器中に予め不純固形物などを除去したイオン交換水約20mlを入れる。この中に分散剤として「コンタミノンN」(非イオン界面活性剤、陰イオン界面活性剤、有機ビルダーからなるpH7の精密測定器洗浄用中性洗剤の10質量%水溶液、和光純薬工業社製)をイオン交換水で約3質量倍に希釈した希釈液を約0.2ml加える。更に測定試料を約0.02g加え、超音波分散器を用いて2分間分散処理を行い、測定用の分散液とする。その際、分散液の温度が10℃以上40℃以下となる様に適宜冷却する。超音波分散器としては、発振周波数50kHz、電気的出力150Wの卓上型の超音波洗浄器分散器(例えば「VS−150」(ヴェルヴォクリーア社製))を用い、水槽内には所定量のイオン交換水を入れ、この水槽中に前記コンタミノンNを約2ml添加する。
測定には、標準対物レンズ(10倍)を搭載した前記フロー式粒子像分析装置を用い、シース液にはパーティクルシース「PSE−900A」(シスメックス社製)を使用する。
標準対物レンズ(10倍)を用いることで、画像処理解像度512×512画素(1画素あたり0.37μm×0.37μm)となる。
前記手順に従い調整した分散液を前記フロー式粒子像分析装置に導入し、HPF測定モードで、トータルカウントモードにて3000個のトナー粒子を計測する。そして、粒子解
析時の2値化閾値を85%とし、解析粒子径を円相当径1.98μm以上200.00μm未満に限定し、0.200以上1.000以下の円形度の範囲において800分割して解析し、トナー粒子の平均円形度を求める。
測定にあたっては、測定開始前に標準ラテックス粒子(例えば、Duke Scientific社製の「RESEARCH AND TEST PARTICLES Latex Microsphere Suspensions 5200A」をイオン交換水で希釈)を用いて自動焦点調整を行う。その後、測定開始から2時間毎に焦点調整を実施することが好ましい。
なお、本願実施例では、シスメックス社による校正作業が行われた、シスメックス社が発行する校正証明書の発行を受けたフロー式粒子像分析装置を使用した。解析粒子径を円相当径1.98μm以上、200.00μm未満に限定した以外は、校正証明を受けた時の測定及び解析条件で測定を行う。
また、この時、円相当径0.50μm以上200.00μm以下の全粒子に対する、0.50μm以上1.98μm以下の粒子の割合を個数%として求める。
【0026】
<磁性キャリア表面の金属酸化物に由来する部分の面積%の算出方法>
磁性キャリア表面の金属酸化物に由来する部分の面積%は、走査電子顕微鏡による反射電子の投影像の観察と、続く画像処理により求めることができる。
磁性キャリア表面の金属酸化物に由来する部分の面積%は、走査型電子顕微鏡(SEM)、S−4800(日立製作所社製)を用いて算出する。金属酸化物に由来する部分の面積は、加速電圧が2.0kVのときに得られる反射電子を可視化した像の画像処理から算出される。走査型電子顕微鏡観察において、試料から放出されてくる反射電子の量は、重元素ほど多いことが知られている。本発明に用いられる磁性キャリア表面のように、樹脂部分と磁性コアに由来する金属酸化物部分が存在する試料では、金属酸化物部分が明るく(輝度が高い、白く)、樹脂部分は暗く(輝度が低く、黒く)見えるため、それぞれ大きなコントラスト差を持った画像が得られる。
具体的には、走査型電子顕微鏡観察用の試料台上にカーボンテープで磁性キャリアを一層になるように固定し、白金による蒸着は行わずに、以下の条件にて、走査型電子顕微鏡S−4800(日立製作所社製)で観察する。また、フラッシング操作を行ってから観察を行う。
SignalName=SE(U,LA80)
AcceleratingVoltage=2000Volt
EmissionCurrent=10000nA
WorkingDistance=6000um
LensMode=High
Condencer1=5
ScanSpeed=Slow4(40秒)
Magnification=600
DataSize=1280x960
ColorMode=Grayscale
反射電子の投影像は、走査型電子顕微鏡S−4800の制御ソフト上で「コントラスト5、ブライトネス−5」に明るさを調整し、キャプチャスピード/積算枚数「Slow4を40秒」、画像サイズ1280×960pixelsの8bitの256階調グレースケール画像として得る(図3(3−a))。画像上のスケールから、1pixelの長さは0.1667μm、1pixelの面積は0.0278μm2となる。
続いて、得られた反射電子の投影像を用いて、磁性キャリア50個について金属酸化物に由来する部分の面積%を算出する。解析する磁性キャリア50個の選択方法の詳細は後述する。金属酸化物に由来する部分の面積%の算出には、画像処理ソフトImage−Pro Plus5.1J(MediaCybernetics社製)を使用する。
まず、図3(3−b)の画像下部の文字列は画像処理に不必要であり、不要な部分を削
除し1280×895のサイズに切り出す(図3(3−c))。次に、磁性キャリアの部分を抽出し、抽出された磁性キャリア部分のサイズをカウントした。具体的には、まず、解析する磁性キャリアを抽出するため、磁性キャリアと背景部分を分離する。Image−Pro Plus5.1Jの「測定」−「カウント/サイズ」を選択する。「カウント/サイズ」の「輝度レンジ選択」で、輝度レンジを50〜255の範囲に設定して、背景として写りこんでいる輝度の低いカーボンテープ部分を除外し、磁性キャリアの抽出を行う(図3(3−d))。カーボンテープ以外の方法で磁性キャリアを固定した際には、必ずしも背景が輝度の低い領域とならない、あるいは、部分的に磁性キャリアと同じような輝度となる可能性は皆無ではない。しかし、磁性キャリアと背景の境界については、反射電子の投影像から容易に区別できる。抽出を行う際、「カウント/サイズ」の抽出オプションで、4連結を選択し、平滑度5を入力、「穴埋める」にチェックを入れ、画像の全ての境界(外周)上に位置する粒子や他の粒子と重なっている粒子については、計算から除外するものとする。次に「カウント/サイズ」の測定項目で、面積とフェレ径(平均)を選択し、面積の選別レンジを最小300pixel、最大10000000pixelとする(図4(4−a))。また、フェレ径(平均)は、後述する磁性キャリアの体積分布基準50%粒径(D50)の測定値の±25%径の範囲になるよう選別レンジを設定し、画像解析する磁性キャリアを抽出する(図4(4−b))。抽出された磁性キャリアに由来する部分の大きさ(pixel数)を(ja)、それぞれの抽出部分の和(Σja=Ja)、抽出された部分の数(Jc)を求める。抽出された磁性キャリアの数Jcが、Jc=50となるまで、別視野の磁性キャリア投影像について同様の操作を繰り返す。
次に、選択した粒子に対して金属酸化物に由来する部分を抽出した。Image−Pro Plus5.1Jの「カウント/サイズ」の「輝度レンジ選択」で、輝度レンジを140〜255の範囲に設定して、磁性キャリア上の輝度の高い部分の抽出を行う。面積の選別レンジは最小10pixel、最大10000pixelとし、磁性キャリア表面の金属酸化物に由来する部分を抽出する(図4(4−c))。さらに、上記磁性キャリア部分の抽出時と同様に、画像外周に位置する粒子や50%粒径(D50)の測定値の±25%径の範囲から逸脱したものは、計算から除外した。金属酸化物に由来の抽出部分の大きさ(pixel数)(ma)、それぞれの抽出部分の和(Ma)として、下式に従い計算した。
式: 金属酸化物に由来する部分の面積%=Ma/Ja×100
【0027】
<6.672μm2以上の金属酸化物に由来する部分の面積%の算出方法>
6.672μm2以上の金属酸化物に由来する部分の面積の、金属酸化物に由来する部分の総面積に対する面積%は、走査型電子顕微鏡による反射電子の投影像の観察と画像処理、続く統計処理により求めることができる。金属酸化物に由来する部分の面積%を求めるのと同様にして、磁性キャリアの観察を行い、画像から磁性キャリア中の金属酸化物に由来する部分の抽出を行った。抽出された金属酸化物に由来する部分を大きさ20pixel毎のチャンネルに分布させた。1pixelの面積は0.0278μm2として面積換算を行い、金属酸化物に由来する部分の総面積に対して、6.672μm2以上に分布する割合を算出した。
【0028】
<磁性キャリアの包絡度の測定方法>
磁性キャリアの断面加工には、集束イオンビーム加工観察装置(FIB)、FB−2100(日立ハイテクノロジーズ社製)を用いる。FIB用試料台(金属メッシュ)上にカーボンペーストを塗り、その上に磁性キャリアを1粒子ずつ独立して存在するように少量固着させ、導電膜として白金蒸着することで試料を作製する。試料をFIB装置にセットし、加速電圧40kV、Gaイオン源を用いて、粗加工し(ビーム電流39nA)、続いて仕上げ加工(ビーム電流7nA)を行い、試料断面を削り出す。
尚、試料とする磁性キャリアは、各試料の最大径をDmaxとしたときに、D50×0.9≦Dmax≦D50×1.1である磁性キャリアを対象とする。さらに、各試料の固
着面に対して平行な方向における、最大長を含む平面の位置を、固着面からの距離hとする(例えば、半径rの完全な球体の場合、h=rとなる)。固着面より垂直な方向に、固着面からの距離0.9×h以上1.1×h以下の範囲において、断面を削り出す。
断面加工した試料は、そのまま走査型電子顕微鏡(SEM)観察に適用することができる。走査型電子顕微鏡観察において、試料から放出されてくる反射電子の量は、重元素ほど多いことが知られている。例えば、有機化合物と鉄のような金属が平面状に分布している試料であれば、鉄からの反射電子の放出量がより多く検出されるため、鉄部分が画像上では明るく(輝度が高い、白く)見えることになる。一方、軽元素化合物から構成される有機化合物からの反射電子量は少ないため、画像上では暗く(輝度が低く、黒く)見えることになる。磁性キャリアの断面観察においては、磁性コア部領域に由来する金属酸化物部が明るく(輝度が高い、白く)、磁性コア部以外の領域は、暗く(輝度が低く、黒く)見えるため、それぞれ大きなコントラスト差を持った画像が得られる。具体的には、走査型電子顕微鏡(SEM)、S−4800(日立ハイテクノロジーズ社製)を用いて、以下の条件にて観察する。尚、フラッシング操作を行ってから観察する。
SignalName=SE(U,LA100)
AcceleratingVoltage=5000Volt
EmissionCurrent=10000nA
WorkingDistance=4000um
LensMode=High
Condencer1=3
ScanSpeed=Slow4(40sec)
Magnification=1500
DataSize=1280x960
ColorMode=Grayscale
SpecimenBias=0V
尚、反射電子の投影像は、上記条件のほか、走査電子顕微鏡S−4800の制御ソフト上で「コントラスト5、ブライトネス−5」に明るさを調整し、磁性体観察モードはOFFとし、256階調のグレースケール画像として得る。
次に、得られた投影像を用いて、画像処理ソフトImage−Pro Plus5.1J(MediaCybernetics社製)を使用し、磁性キャリア50個について、包絡線L2及び周囲長L1を求め、下記式より包絡度を求める。
式: 包絡度={(L1−L2)/L2}×100
【0029】
<トナーの重量平均粒子径(D4)の測定>
トナーの重量平均粒径(D4)は、100μmのアパーチャーチューブを備えた細孔電気抵抗法による精密粒度分布測定装置「コールター・カウンター Multisizer
3」(登録商標、ベックマン・コールター社製)と、測定条件設定及び測定データ解析をするための付属の専用ソフト「ベックマン・コールター Multisizer 3 Version3.51」(ベックマン・コールター社製)を用いて、実効測定チャンネル数2万5千チャンネルで測定し、測定データの解析を行い、算出する。
測定に使用する電解水溶液は、特級塩化ナトリウムをイオン交換水に溶解して濃度が約1質量%となるようにしたもの、例えば、「ISOTON II」(ベックマン・コールター社製)が使用できる。
尚、測定、解析を行う前に、以下のように前記専用ソフトの設定を行う。
前記専用ソフトの「標準測定方法(SOM)を変更画面」において、コントロールモードの総カウント数を50000粒子に設定し、測定回数を1回、Kd値は「標準粒子10.0μm」(ベックマン・コールター社製)を用いて得られた値を設定する。閾値/ノイズレベルの測定ボタンを押すことで、閾値とノイズレベルを自動設定する。また、カレントを1600μAに、ゲインを2に、電解液をISOTON IIに設定し、測定後のアパーチャーチューブのフラッシュにチェックを入れる。
専用ソフトの「パルスから粒径への変換設定画面」において、ビン間隔を対数粒径に、粒径ビンを256粒径ビンに、粒径範囲を2μm以上60μm以下に設定する。
具体的な測定法は以下の通りである。
(1)Multisizer 3専用のガラス製250ml丸底ビーカーに前記電解水溶液約200mlを入れ、サンプルスタンドにセットし、スターラーロッドの撹拌を反時計回りで24回転/秒にて行う。そして、解析ソフトの「アパーチャーのフラッシュ」機能により、アパーチャーチューブ内の汚れと気泡を除去しておく。
(2)ガラス製の100ml平底ビーカーに前記電解水溶液約30mlを入れ、この中に分散剤として「コンタミノンN」(非イオン界面活性剤、陰イオン界面活性剤、有機ビルダーからなるpH7の精密測定器洗浄用中性洗剤の10質量%水溶液、和光純薬工業社製)をイオン交換水で3質量倍に希釈した希釈液を約0.3ml加える。
(3)発振周波数50kHzの発振器2個を、位相を180度ずらした状態で内蔵し、電気的出力120Wの超音波分散器「Ultrasonic Dispension System Tetora150」(日科機バイオス社製)の水槽内に所定量のイオン交換水を入れ、この水槽中に前記コンタミノンNを約2ml添加する。
(4)前記(2)のビーカーを前記超音波分散器のビーカー固定穴にセットし、超音波分散器を作動させる。そして、ビーカー内の電解水溶液の液面の共振状態が最大となるようにビーカーの高さ位置を調整する。
(5)前記(4)のビーカー内の電解水溶液に超音波を照射した状態で、トナー約10mgを少量ずつ前記電解水溶液に添加し、分散させる。そして、さらに60秒間超音波分散処理を継続する。尚、超音波分散にあたっては、水槽の水温が10℃以上40℃以下となる様に適宜調節する。
(6)サンプルスタンド内に設置した前記(1)の丸底ビーカーに、ピペットを用いてトナーを分散した前記(5)の電解質水溶液を滴下し、測定濃度が約5%となるように調整する。そして、測定粒子数が50000個になるまで測定を行う。
(7)測定データを装置付属の前記専用ソフトにて解析を行い、重量平均粒径(D4)を算出する。尚、専用ソフトでグラフ/体積%と設定したときの、分析/体積統計値(算術平均)画面の「平均径」が重量平均粒径(D4)である。
【0030】
<樹脂のピーク分子量(Mp)、数平均分子量(Mn)、重量平均分子量(Mw)の測定方法>
樹脂の分子量分布は、ゲルパーミエーションクロマトグラフィー(GPC)により、以下のようにして測定する。
まず、室温で24時間かけて、樹脂をテトラヒドロフラン(THF)に溶解する。そして、得られた溶液を、ポア径が0.2μmの耐溶剤性メンブランフィルター「マエショリディスク」(東ソー社製)で濾過してサンプル溶液を得る。尚、サンプル溶液は、THFに可溶な成分の濃度が約0.8質量%となるように調整する。このサンプル溶液を用いて、以下の条件で測定する。
装置 :HLC8120 GPC(検出器:RI)(東ソー社製)
カラム:Shodex KF−801、802、803、804、805、806、
807の7連(昭和電工社製)
溶離液:テトラヒドロフラン(THF)
流速 :1.0ml/min
オーブン温度:40.0℃
試料注入量:0.10ml
試料の分子量の算出にあたっては、標準ポリスチレン樹脂(例えば、商品名「TSKスタンダード ポリスチレン F−850、F−450、F−288、F−128、F−80、F−40、F−20、F−10、F−4、F−2、F−1、A−5000、A−2500、A−1000、A−500」、東ソ−社製)を用いて作成した分子量校正曲線を使用する。
【0031】
<ワックスの最大吸熱温度、樹脂のガラス転移温度(Tg)の測定方法>
ワックスの最大吸熱ピークを示す温度は、示差走査熱量分析装置「Q1000」(TA
Instruments社製)を用いてASTM D3418−82に準じて測定する。
装置検出部の温度補正はインジウムと亜鉛の融点を用い、熱量の補正についてはインジウムの融解熱を用いる。具体的には、ワックスを約10mg精秤し、これをアルミニウム製のパンの中に入れ、リファレンスとして空のアルミニウム製のパンを用い、測定温度範囲30乃至200℃の間で、昇温速度10℃/minで測定を行う。尚、測定においては、一度200℃まで昇温させ、続いて30℃まで降温し、その後に再度昇温を行う。この2度目の昇温過程での温度30乃至200℃の範囲におけるDSC曲線の最大の吸熱ピークを示す温度を、最大吸熱温度とする。
また、樹脂のガラス転移温度(Tg)は、ワックス測定時と同様に、樹脂を約10mg精秤し測定を行う。温度40℃乃至100℃の範囲の比熱変化において、比熱変化が出る前と出た後のベースラインの中間点の線とDSC曲線との交点を、結着樹脂のガラス転移温度(Tg)とする。
【0032】
<樹脂の軟化点の測定方法>
樹脂の軟化点の測定は、定荷重押し出し方式の細管式レオメータ「流動特性評価装置 フローテスターCFT−500D」(島津製作所社製)を用い、装置付属のマニュアルに従って行う。本装置では、測定試料の上部からピストンによって一定荷重を加えつつ、シリンダに充填した測定試料を昇温させて溶融し、シリンダ底部のダイから溶融された測定試料を押し出し、この際のピストン降下量と温度との関係を示す流動曲線を得ることができる。
本発明においては、「流動特性評価装置 フローテスターCFT−500D」に付属のマニュアルに記載の「1/2法における溶融温度」を軟化点とする。尚、1/2法における溶融温度とは、次のようにして算出されたものである。まず、流出が終了した時点におけるピストンの降下量Smaxと、流出が開始した時点におけるピストンの降下量Sminとの差の1/2を求める(これをXとする。X=(Smax−Smin)/2)。そして、流動曲線においてピストンの降下量がXとSminの和であるときの流動曲線の温度が、1/2法における溶融温度である。
測定試料には、約1.0gの樹脂を、25℃の環境下で、錠剤成型圧縮機(例えば、NT−100H、エヌピーエーシステム社製)を用いて約10MPaで、約60秒間圧縮成型し、直径約8mmの円柱状としたものを用いる。また、CFT−500Dの測定条件は、以下の通りである。
試験モード:昇温法
開始温度:40℃
到達温度:200℃
測定間隔:1.0℃
昇温速度:4.0℃/min
ピストン断面積:1.000cm2
試験荷重(ピストン荷重):10.0kgf(0.9807MPa)
予熱時間:300秒
ダイの穴の直径:1.0mm
ダイの長さ:1.0mm
【0033】
<磁性キャリア等の体積分布基準の50%粒径(D50)の測定方法>
粒度分布測定は、レーザー回折・散乱方式の粒度分布測定装置「マイクロトラックMT3300EX」(日機装社製)にて測定を行った。測定には、乾式測定用の試料供給機「ワンショットドライ型サンプルコンディショナーTurbotrac」(日機装社製)を
装着して行った。Turbotracの供給条件として、真空源として集塵機を用い、風量約33リットル/sec、圧力約17kPaとした。制御は、ソフトウエア上で自動的に行う。粒径は体積分布基準の累積値である50%粒径(D50)を求める。制御及び解析は付属ソフト(バージョン10.3.3−202D)を用いて行う。測定条件は、以下の通りである。
SetZero時間:10秒
測定時間 :10秒
測定回数 :1回
粒子屈折率 :1.81
粒子形状 :非球形
測定上限 :1408μm
測定下限 :0.243μm
測定環境 :常温常湿環境(23℃、50%RH)
【実施例】
【0034】
以下、本発明の具体的実施例について説明するが、本発明はこれらの実施例に限定されるものではない。尚、以下の配合における部数は特に説明が無い場合は質量部である。
【0035】
〔樹脂の製造例1〕
冷却管、攪拌機、及び、窒素導入管のついた反応槽中に、以下の材料を秤量した。
テレフタル酸 22.6質量部無水トリメリット酸 1.7質量部ポリオキシプロピレン(2.2)−2,2−ビス(4−ヒドロキシフェニル)プロパン
75.6質量部チタニウムジヒドロキシビス(トリエタノールアミネート) 0.1質量部
その後、200℃に加熱し、窒素を導入しながら生成する水を除去しながら10時間反応させ、その後、10mmHgに減圧し1時間反応させ、樹脂1を合成した。GPCで求めた樹脂1の分子量は、重量平均分子量(Mw)6200、数平均分子量(Mn)2500であり、ピーク分子量(Mp)2900、ガラス転移点は55℃、軟化点は93℃であった。
【0036】
〔樹脂の製造例2〕
冷却管、攪拌機、及び、窒素導入管のついた反応槽中に、以下の材料を秤量した。
テレフタル酸 17.2質量部ポリオキシエチレン(2.2)−2,2−ビス(4−ヒドロキシフェニル)プロパン
76.6質量部チタニウムジヒドロキシビス(トリエタノールアミネート) 0.1質量部
その後、220℃に加熱し、窒素を導入しながら生成する水を除去しながら10時間反応させた。さらに、無水トリメリット酸6.1質量部を加え、180℃に加熱し、2時間反応させ樹脂2を合成した。GPCで求めた樹脂2の分子量は、重量平均分子量(Mw)86000、数平均分子量(Mn)6000、ピーク分子量(Mp)12800、ガラス転移点は62℃、軟化点は132℃であった。
【0037】
〔トナー製造例1〕
樹脂1 50.0質量部樹脂2 50.0質量部フィッシャー・トロプシュワックス(最大吸熱温度:75℃) 7.5質量部C.I.ピグメントブルー15:3 5.0質量部
上記材料をヘンシェルミキサー(FM−75型、三井三池化工機(株)製)でよく混合した後、温度130℃に設定した二軸混練機(PCM−30型、池貝鉄工(株)製)にて
混練した。得られた混練物を冷却し、ハンマーミルにて1mm以下に粗粉砕し、粗砕物を得た。得られた粗砕物を、高圧気体を用いた衝突式気流粉砕機を用いて微粉砕した。
次に、得られた微粉砕物を、図5に示す表面改質装置により表面改質を行った。表面改質時の条件は、原料供給速度は2.0kg/hr、熱風流量4.5m3/min、熱風の吐出温度は230℃、冷風温度0℃、冷風流量3.0m3/minであった。次に、コアンダ効果を利用した風力分級機(エルボジェットラボEJ−L3、日鉄鉱業社製)で分級し、微粉及び粗粉を同時に分級除去、トナー粒子1を得た。
得られたトナー粒子1 100.0質量部に、無機微粒子として、個数平均粒径が40nmであり、i−ブチルトリメトキシシランで処理された酸化チタン微粉体1.0質量部、一次平均粒子径16nmであり、ヘキサメチルジシラザン20質量%で表面処理した疎水性シリカ微粒子0.5質量部を添加し、個数平均粒径が200nmであり、ヘキサメチルジシラザンで処理されたゾルゲル法シリカ微粉体を1.0質量部を外添混合し、トナー1を得た。
【0038】
〔トナー製造例2〕
トナー製造例1のうち、表面改質時の条件の熱風流量を4.5m3/minから4.0m3/min、熱風の吐出温度は230℃から210℃に変更した以外は、トナー製造例1と同様にして、トナー2を得た。
【0039】
〔トナー製造例3〕
トナー製造例1のうち、表面改質時の条件の熱風流量を4.5m3/minから5.0m3/min、熱風の吐出温度は230℃から250℃に変更した以外は、トナー製造例1と同様にして、トナー3を得た。
【0040】
〔トナー製造例4〕
トナー製造例1のうち、表面改質処理を行なわない以外は、トナー製造例1と同様にして、トナー4を得た。
【0041】
トナー1乃至4の物性を表1にしめす。
【0042】
【表1】

【0043】
〔多孔質磁性コアの製造例1〕
<工程1(秤量・混合工程)>
Fe2O3 59.7質量%
MnCO3 34.4質量%
Mg(OH)2 4.8質量%
SrCO3 1.1質量%
となるようにフェライト原材料を秤量した。その後、ジルコニア(φ10mm)のボールを用いた乾式ボールミルで2時間粉砕・混合した。
<工程2(仮焼成工程)>
粉砕・混合した後、バーナー式焼成炉を用い、大気中で950℃で2時間焼成し、仮焼
フェライトを作製した。
フェライトの組成は、下記の通り。
(MnO)a(MgO)b(SrO)c(Fe2O3)d
上記式において、a=0.39、b=0.11、c=0.01、d=0.49
<工程3(粉砕工程)>
クラッシャーで0.5mm程度に粉砕した後に、ジルコニア(φ10mm)のボールを用い、仮焼フェライト100質量部に対し、水を30質量部加え、湿式ボールミルで2時間粉砕した。そのスラリーを、ジルコニアのビーズ(φ1.0mm)を用いた湿式ビーズミルで3時間粉砕し、フェライトスラリー(仮焼フェライト微粉砕品)を得た。
得られた仮焼フェライト微粉砕品の、体積基準の50%粒径(D50)は1.7μmであった。
<工程4(造粒工程)>
フェライトスラリーに、バインダーとして仮焼フェライト100質量部に対してポリビニルアルコール2.0質量部を添加し、スプレードライヤー(製造元:大川原化工機)で、約36μmの球状粒子に造粒した。
<工程5(本焼成工程)>
焼成雰囲気をコントロールするために、電気炉にて窒素雰囲気下(酸素濃度0.01体積%以下)で、1150℃で4時間焼成した。
<工程6(選別工程)>
凝集した粒子を解砕した後に、目開き250μmの篩で篩分して粗大粒子を除去し、多孔質磁性コア1を得た。
【0044】
<多孔質磁性コアの製造例2>
多孔質磁性コア製造例1のうち、工程1でフェライト原材料の比率を、
Fe2O3 62.8質量%
MnCO3 31.2質量%
Mg(OH)2 6.0質量%
に変更した。フェライトの組成は、下記の通り。
(MnO)a(MgO)b(Fe2O3)c
上記式において、a=0.45、b=0.15、c=0.40
工程3のクラッシャーでの粉砕粒度を0.5mm程度から0.3mm程度に変更し、湿式ボールミルのボールをジルコニア(φ10mm)からステンレス(φ10mm)に、湿式ビーズミルのビーズをジルコニア(φ1.0mm)からステンレス(φ1.0mm)に、粉砕時間を3時間から2時間に変更した。
得られた仮焼フェライト微粉砕品は、D50=0.8μmであった。
工程4のポリビニルアルコールの添加量を2質量部から1.0質量部に変更した。
工程5の焼成温度を1150℃から1200℃に、焼成雰囲気を窒素雰囲気下(酸素濃度0.01体積%以下)から窒素雰囲気下(酸素濃度1.0体積%)に、焼成時間を4時間から8時間に変更した。上記以外は、多孔質磁性コア製造例1と同様にして、多孔質磁性コア2を得た。
【0045】
<多孔質磁性コアの製造例3>
多孔質磁性コア製造例1のうち、工程3の湿式ビーズミルの粉砕時間を2時間から3時間に変更した。得られた仮焼フェライト微粉砕品は、D50=1.3μmであった。また、工程5の焼成温度を1150℃から1050℃に変更した。上記以外は、多孔質磁性コア製造例1と同様にして、多孔質磁性コア3を得た
【0046】
<多孔質磁性コアの製造例4>
多孔質磁性コア製造例1のうち、工程1でフェライト原材料の比率を、
Fe2O3 62.4質量%
MnCO3 30.5質量%
Mg(OH)2 6.4質量%
SrCO3 0.7質量%
に変更した。フェライトの組成は、下記の通り。
(MnO)a(MgO)b(SrO)c(Fe2O3)d
上記式において、a=0.34、b=0.14、c=0.01、d=0.51
工程3のクラッシャーでの粉砕粒度を0.5mm程度から1.0mm程度に変更し、湿式ボールミルのボールをジルコニア(φ10mm)からアルミナ(φ10mm)に、粉砕時間を2時間から1時間に変更した。湿式ビーズミルのビーズをジルコニア(φ1.0mm)からアルミナ(φ1.0mm)に変更した。得られた仮焼フェライト微粉砕品は、D50=4.8μmであった。
工程4のポリビニルアルコールの添加量を2.0質量部から5.0質量部に変更した。
工程5の焼成温度を1150℃から1000℃に、焼成雰囲気を窒素雰囲気下(酸素濃度0.01体積%以下)から窒素雰囲気下(酸素濃度0.5体積%)に変更した。上記以外は、多孔質磁性コア製造例1と同様にして、多孔質磁性コア4を得た。
【0047】
<多孔質磁性コアの製造例5>
多孔質磁性コア製造例1のうち、工程1でフェライト原材料の比率を、
Fe2O3 62.8質量%
MnCO3 31.2質量%
Mg(OH)2 6.0質量%
に変更した。フェライトの組成は、下記の通り。
(MnO)a(MgO)b(Fe2O3)c
上記式において、a=0.45、b=0.15、c=0.40
工程3のクラッシャーでの粉砕粒度を0.5mm程度から0.3mm程度に変更し、湿式ボールミルのボールをジルコニア(φ10mm)からステンレス(φ10mm)に、湿式ビーズミルのビーズをジルコニア(φ1.0mm)からステンレス(φ1.0mm)に、粉砕時間を3時間から2時間に変更した。
得られた仮焼フェライト微粉砕品は、D50=0.8μmであった。
工程4のポリビニルアルコールの添加量を2.0質量部から1.0質量部に変更した。
工程5の焼成温度を1150℃から1200℃に、焼成雰囲気を窒素雰囲気下(酸素濃度0.01体積%以下)から窒素雰囲気下(酸素濃度1.0体積%)に、焼成時間を4時間から8時間に変更した。上記以外は、多孔質磁性コア製造例1と同様にして、多孔質磁性コア5を得た。
【0048】
<多孔質磁性コアの製造例6>
多孔質磁性コア製造例4のうち、工程3の湿式ビーズミルの粉砕時間を3時間から2時間に変更した。得られた仮焼フェライト微粉砕品は、D50=5.6μmであった。
工程4のポリビニルアルコールの添加量を5.0質量部から10.0質量部に変更した。
工程5の焼成時間を4時間から2時間に、焼成雰囲気を窒素雰囲気下(酸素濃度0.5体積%)から窒素雰囲気下(酸素濃度1.0体積%)に変更した。上記以外は、多孔質磁性コア製造例4と同様にして、多孔質磁性コア6を得た。
【0049】
<多孔質磁性コアの製造例7>
多孔質磁性コア製造例4のうち、工程3のクラッシャーでの粉砕粒度を1.0mm程度から0.3mm程度に変更し、湿式ボールミルのボールをアルミナ(φ10mm)からステンレス(φ10mm)に、粉砕時間を1時間から3時間に変更した。湿式ビーズミルのビーズをアルミナ(φ1.0mm)からステンレス(φ1.0mm)、粉砕時間を3時間から2時間に変更した。得られた仮焼フェライト微粉砕品は、D50=0.4μmであっ
た。
工程4のポリビニルアルコールの添加量を5.0質量部から1.0質量部に変更した。
工程5の焼成温度を1000℃から1250℃に変更した。上記以外は、多孔質磁性コア製造例4と同様にして、多孔質磁性コア7を得た。
【0050】
<磁性コアの製造例1>
マグネタイト微粒子(球形、個数平均粒径250nm、磁化の強さ65Am2/kg、残留磁化4.2Am2/kg、保磁力4.4kA/m、500V/cmにおける比抵抗3.3×105Ω・cm)と、シラン系カップリング剤(3−(2−アミノエチルアミノプロピル)トリメトキシシラン)(マグネタイト微粒子の質量に対して3.0質量%の量)とを、容器に導入した。そして、該容器内において温度100℃以上で高速混合撹拌して、マグネタイト微粒子を表面処理した。
・フェノール 10質量部
・ホルムアルデヒド溶液(ホルムアルデヒド37質量%水溶液) 16質量部
・表面処理した上記マグネタイト微粒子 84質量部
上記材料を反応釜に導入し、温度40℃にしてよく混合した。
その後、撹拌しながら平均昇温速度3℃/分で、温度85℃に加熱し、28質量%アンモニア水4質量部および水25質量部を反応釜に加えた。温度85℃にて保持し、3時間重合反応させて硬化させた。このときの撹拌翼の周速は1.8m/秒とした。
重合反応させた後、温度30℃まで冷却して水を添加した。上澄み液を除去して得られた沈殿物を水洗し、さらに風乾した。得られた風乾物を、減圧下(5hPa以下)にて、温度60℃で乾燥して、樹脂中に磁性体が分散された磁性コア1を得た。
【0051】
<磁性コアの製造例2>
工程1:
Fe2O3 71.0質量%
CuO 12.5質量%
ZuO 16.5質量%
となるようにフェライト原材料を秤量した。その後、ジルコニア(φ10mm)のボールを用いた乾式ボールミルで2時間粉砕・混合した。
工程2:
粉砕・混合した後、大気中で950℃で2時間焼成し、仮焼フェライトを作製した。
フェライトの組成は、下記の通り。
(CuO)a(ZnO)b(Fe2O3)c
上記式において、a=0.20、b=0.25、c=0.55
工程3:
クラッシャーで0.5mm程度に粉砕した後に、ステンレスのボール(φ10mm)を用い、仮焼フェライト100質量部に対し水を30質量部加え、湿式ボールミルで6時間粉砕した。得られた仮焼フェライト微粉砕品は、D50=0.5μm、D90=2.8μmであった。
工程4:
フェライトスラリーに、バインダーとして仮焼フェライト100質量部に対しポリビニルアルコール2.0質量部を添加し、スプレードライヤー(製造元:大川原化工機)で球状粒子に造粒した。
工程5:
大気中で1300℃で4時間焼成した。
工程6:
凝集した粒子を解砕した後に、目開き250μmの篩で篩分して粗大粒子を除去し、磁性コア2を得た。
【0052】
<樹脂液の調製>
樹脂液1
ストレートシリコーン(SR2411 東レ・ダウコーニング) 20.0質量部
γ−アミノプロピルトリエトキシシラン 0.5質量部
トルエン 79.5質量部
以上を、ボールミル(ソーダガラスボール φ10mm)を用いて1時間混合し、樹脂液1を得た。
【0053】
樹脂液2
ポリメチルメタクリレート重合体(Mw=66,000) 5.0質量部ボントロンP51(オリエント化学工業株式会社) 2.0質量部
トルエン 93.0質量部
以上を、ビーズミル(ジルコニアビーズ φ1.0mm)を用いて3時間分散混合し、樹脂液2を得た。
【0054】
<磁性キャリア1の製造例>
工程1(樹脂充填工程):
多孔質磁性コア1 100.0質量部を万能攪拌混合機(ダルトン社製)に入れ、減圧下、80℃に加熱しながら撹拌する。続いて、樹脂液1を多孔質磁性コア1 100質量部に対し樹脂成分として8質量部になるように添加し2時間加熱を続け、溶剤を除去した。得られた試料をジュリアミキサー(徳寿工作所社製)に移し、窒素雰囲気下に200℃で2時間熱処理して、開口70μmのメッシュで分級して、充填コア1を得た。
工程2(樹脂コート工程):
充填コア1 100.0質量部をナウタミキサ(ホソカワミクロン社製)に投入し、さらに、樹脂液2を樹脂成分として0.5質量部になるようにナウタミキサに投入した。減圧下で70℃に加熱し、1.7S−1(100rpm)で混合し、4時間かけて溶媒除去及び塗布操作を行った。その後、得られた試料をジュリアミキサー(徳寿工作所社製)に移し、窒素雰囲気下、温度200℃で2時間熱処理した後、開口70μmのメッシュで分級して磁性キャリア1を得た。
【0055】
<磁性キャリア2〜13の製造例>
磁性キャリア1の製造例において、コア粒子、充填樹脂量、コート樹脂量を表2のように変え、磁性キャリア2乃至13を得た。得られたキャリア1乃至13の物性を表3に示す。
【0056】
【表2】
【0057】
【表3】
【0058】
〔実施例1乃至22及び比較例1乃至12〕
作製した磁性キャリア及びトナーを用い、磁性キャリアとトナーの組み合わせ、及び、磁性キャリア100質量部に対するトナー比率を表4の記載に従い、二成分系現像剤を作製した。二成分系現像剤は、V型混合機で5分間混合した。また、得られた二成分系現像剤のE100mm/sec、E10mm/sec、ΔE、25FRI(E10mm/sec/E100mm/sec)の数値を表5に示す。
【0059】
【表4】
【0060】
【表5】
【0061】
<二成分系現像剤の評価>
画像形成装置として、キヤノン製カラー複写機imageRUNNER iR C3580を用い、下記評価が可能なように当該複写機を改造した。シアン位置の現像器に上記現像剤を入れ、評価を行った。現像条件としては、現像ドラムに対する現像スリーブ周速を1.5倍、プロセススピード 245mm/secとなるように改造した。そして、現像スリーブには、周波数2.0kHz、Vpp1.3kVの交流電圧と直流電圧VDCを印加した。直流電圧VDCはFFH画像(ベタ部)のトナーの紙上への載り量が0.5mg/cm2となるように調整した。
初期のキャリア付着、トナー飛散、ガサツキの評価を行った。
また、画像面積比率25%で50000枚の連続印刷後、画像濃度安定性、ガサツキの評価を行った。
上記評価を、温度23℃/湿度50%RHの環境と、温度30℃/湿度80%RHの2つの環境で行った。
なおFFH画像とは、256階調を16進数で表示した値であり、00Hを1階調目(
白地部)、FFHを256階調目(ベタ部)とする。
印刷環境 温度23℃/湿度50%RH(以下「N/N」)
温度30℃/湿度80%RH(以下「H/H」
紙 カラー複写機・プリンタ用普通紙 CS−814(A4、81.4g/m2)
(キヤノンマーケティングジャパン株式会社より販売)
【0062】
<キャリア付着>
00H画像を印刷し、感光ドラム上の一部分を透明な粘着テープを密着させてサンプリングし、1cm×1cm中の感光ドラム上に付着していた磁性キャリアの個数をカウントし、1cm2当りの付着キャリア粒子の個数を算出した。
A:3個以下 (優秀)
B:4個以上10個以下 (良い)
C:11個以上20個以下(本発明において許容レベル)
D:21個以上 (本発明において許容できない)
【0063】
<トナー飛散>
現像器、及び、本体内のトナーの汚れ具合を目視にて確認した。
A:トナーの飛散がほとんどない (優秀)
B:トナーの飛散がわずかに確認される (良い)
C:トナーの飛散が確認される (本発明において許容レベル)
D:ひどいトナー飛散が確認される (本発明において許容できない)
【0064】
<ガサツキ>
画像比率5%のFFH画像を、10枚印刷→10分放置→10枚印刷を繰り返し、1000枚印刷した。
その後、1画素を1ドットで形成するドット画像(FFH画像)を作成した。紙上の1ドットあたりの面積が、20000μm2以上25000μm2以下となるように、レーザービームのスポット径を調整した。デジタルマイクロスコープVHX−500(レンズワイドレンジズームレンズVH−Z100・キーエンス社製)を用い、ドット1000個の面積を測定した。ドット面積の個数平均(S)とドット面積の標準偏差(σ)を算出し、ガサツキを下記式により算出した。
ガサツキ指数(I)=σ/S×100
A:Iが4.0未満 (優秀)
B:Iが4.0以上6.0未満 (良好)
C:Iが6.0以上8.0未満 (本発明において許容レベル)
D:Iが8.0以上 (本発明において許容できない)
【0065】
<画像濃度安定性>
耐久評価を行う前に、20mm×20mmのFFH画像を1枚印刷した。X−Riteカラー反射濃度計(500シリーズ:X−Rite社製)を使用し、FFH画像部分の画像濃度を測定した。耐久後に、同じ画像を1枚印刷し、画像濃度を測定した。初期及び耐久後の画像濃度の差を以下の基準で評価とした。
(評価基準)
A:0.00以上0.05未満 (優秀)
B:0.05以上0.10未満 (良い)
C:0.10以上0.20未満 (本発明において許容レベル)
D:0.20以上 (本発明において許容できない)
【0066】
【表6】
【0067】
【表7】
【技術分野】
【0001】
本発明は、電子写真法、及び、静電記録法に用いられる二成分系現像剤に関する。
【背景技術】
【0002】
電子写真法において、静電荷像を現像する工程は帯電されたトナーを静電荷像とのクーロン力を利用して静電荷像上に付着させて画像形成する。トナーを用いて静電荷像を現像するための現像剤には、磁性体を樹脂中に分散した磁性トナーを用いる一成分系現像剤と、非磁性トナーと磁性キャリアを混合して用いる二成分系現像剤とに大別される。
特に、高画質を要求されるフルカラー複写機またはフルカラープリンタ等のフルカラー画像形成装置では、後者が好適に用いられている。
フルカラー画像形成装置では、近年、更なる高耐久性が要求されている。そこで、現像剤の流動性を制御することで、低温低湿環境下における劣化かぶりを減少させると共に、ベタ画像の追随性が良好な現像剤が提案されている。具体的には、二成分系現像剤の流速指数25FRI値を0以上2.0以下にコントロールした現像剤である。このような現像剤とすることで、トナー外添剤の埋め込まれにくいトナー表面を有する、あるいはトナー外添剤が埋め込まれてもトナーとしての流動性、帯電性他の特性を有する耐ストレス性の高いトナーを得ることができる。[25FRI=(ブレード回転数10mm/sにおける全エネルギー/100mm/sにおける全エネルギー)](特許文献1)。
しかし、プロセススピード200mm/sec以上の高スピードで間欠印刷した場合、画質が低下し、画像の濃度変動が大きく、改善が必要であった。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】特開2007−183607号公報
【発明の概要】
【発明が解決しようとする課題】
【0004】
本発明は、耐久時の画像の濃度変動及び高いプロセススピードで間欠印字の画質劣化の低減、さらには、キャリア付着やトナー飛散の抑制が可能であり、高い現像性を有する二成分系現像剤の提供を目的とする。
【課題を解決するための手段】
【0005】
発明者らは、鋭意検討を重ねた結果、高いプロセススピードで間欠印刷した場合の画質劣化や、耐久時の画像の濃度変動と、磁性キャリアの真比重と二成分系現像剤のトータルエネルギーの間に相関があることを見出した。
すなわち、磁性キャリアとトナーを少なくとも含有する二成分系現像剤であって、前記トナーは、結着樹脂及びワックスを少なくとも含有するトナー粒子を含有し、前記磁性キャリアは、磁性コア粒子と樹脂を少なくとも含有し、真比重が2.5g/cm3以上4.2g/cm3以下であり、前記二成分系現像剤は、前記磁性キャリア100質量部に対して、トナー4質量部以上20質量部以下の割合で含有する二成分系現像剤であり、前記二成分系現像剤のE100mm/sec(mJ)及びE100mm/sec(mJ)、並びに、前記E10mm/secからE100mm/secを引いたΔE(mJ)が下記式(1)及び(2)をみたすことを特徴とする二成分系現像剤。
式(1):−0.2×E100mm/sec+100≦ΔE≦−0.4×E100mm/sec+200
式(2):80≦E100mm/sec≦360
[前記式(1)及び(2)において、E100mm/sec(mJ)は、回転式プロペラ
型ブレードを備えた粉体流動性分析装置において、プロペラ型ブレードを、前記プロペラ型ブレードの最外縁部の周速を100mm/secで回転させながら、前記二成分系現像剤が満たされた測定容器内の二成分系現像剤粉体層中に垂直に進入させ、前記二成分系現像剤粉体層の底面から100mmの位置から測定を開始し、底面から10mmの位置まで進入させた時に得られる、回転トルクと垂直荷重の総和を表す。また、前記式(1)のΔE(mJ)において、E10mm/sec(mJ)は、回転式プロペラ型ブレードを備えた粉体流動性分析装置において、プロペラ型ブレードを、前記プロペラ型ブレードの最外縁部の周速を10mm/secで回転させながら、前記二成分系現像剤が満たされた測定容器内の二成分系現像剤粉体層中に垂直に進入させ、前記二成分系現像剤粉体層の底面から55mmの位置から測定を開始し、底面から2mmの位置まで進入させた時に得られる、回転トルクと垂直荷重の総和を表す。]
【発明の効果】
【0006】
本発明によれば、耐久時の画像の濃度変動と、間欠印字の画質劣化の低減、さらには、キャリア付着やトナー飛散の抑制および現像性の高い二成分系現像剤を提供することができる。
【図面の簡単な説明】
【0007】
【図1】粉体流動性分析装置の模式図(1−a)、23.5mm径ブレード(1−b)、25mm×25mlスプリット容器(1−c)。
【図2】磁性キャリアの反射電子像を可視化した投影像(2−a)、磁性キャリアの表面状態の模式図(2−b)、磁性キャリアを画像処理して、磁性キャリアを抽出した状態(2−c)、磁性キャリアを画像処理して、磁性キャリア表面上の金属酸化物に由来する部分を抽出した状態(2−d)(図面代用写真)。
【図3】磁性キャリアの反射電子を可視化した投影像(3−a)、投影像を600倍の倍率で可視化した投影図(3−b)、投影図の画像処理の前処理後の様子を示す図(3−c)、磁性キャリアを抽出した状態を示す図(3−d)(図面代用写真)。
【図4】抽出した磁性キャリアから、画像外周部の磁性キャリアを除外した状態を示す図(4−a)、抽出された磁性キャリアから、さらに粒径によって画像処理する粒子を絞り込んだ状態を示す図(4−b)、金属酸化物を抽出した状態の図(4−c)(図面代用写真)。
【図5】表面改質装置の模式図。
【発明を実施するための形態】
【0008】
本発明の二成分系現像剤は、磁性キャリアとトナーを少なくとも含有する二成分系現像剤であって、トナーは、結着樹脂及びワックスを少なくとも含有するトナー粒子を含有し、磁性キャリアは、磁性コア粒子と樹脂を少なくとも含有し、真比重が2.5g/cm3以上4.2g/cm3以下であり、二成分系現像剤は、磁性キャリア100質量部に対して、トナー4質量部以上20質量部以下の割合で含有する二成分系現像剤であり、二成分系現像剤のE100mm/sec(mJ)及びE100mm/sec(mJ)、並びに、E10mm/secからE100mm/secを引いたΔE(mJ)が下記式(1)及び(2)をみたすことを特徴とする。
式(1):−0.2×E100mm/sec+100≦ΔE≦−0.4×E100mm/sec+200
式(2):80≦E100mm/sec≦360
[前記式(1)及び(2)において、E100mm/sec(mJ)は、回転式プロペラ型ブレードを備えた粉体流動性分析装置において、プロペラ型ブレードを、前記プロペラ型ブレードの最外縁部の周速を100mm/secで回転させながら、前記二成分系現像剤が満たされた測定容器内の二成分系現像剤粉体層中に垂直に進入させ、前記二成分系現像剤粉体層の底面から100mmの位置から測定を開始し、底面から10mmの位置まで
進入させた時に得られる、回転トルクと垂直荷重の総和を表す。また、前記式(1)のΔE(mJ)において、E10mm/sec(mJ)は、回転式プロペラ型ブレードを備えた粉体流動性分析装置において、プロペラ型ブレードを、前記プロペラ型ブレードの最外縁部の周速を10mm/secで回転させながら、前記二成分系現像剤が満たされた測定容器内の二成分系現像剤粉体層中に垂直に進入させ、前記二成分系現像剤粉体層の底面から55mmの位置から測定を開始し、底面から2mmの位置まで進入させた時に得られる、回転トルクと垂直荷重の総和を表す。]
ここで、ΔE(mJ)は、現像剤が現像器内で攪拌開始時の低速攪拌されている時に攪拌に必要なエネルギー(E10mm/sec)と、攪拌速度が規定の速度となり高速攪拌されている時に攪拌に必要なエネルギー(E100mm/sec)との差を示している。
このエネルギー差(ΔE)は、−0.2×E100mm/sec+100≦ΔE≦−0.4×E100mm/sec+200の関係を満たすことで、間欠印字の画像劣化及び耐久時の濃度変動の低減を両立させることができる。
そのメカニズムは明確ではないが、発明者らは以下ように推測している。
間欠印刷した場合の画像劣化を抑制するためには、トナーを十分に摩擦帯電させることが必要である。しかし、間欠印刷の場合、印刷されていない時に徐々にトナーの電荷が失われる。この現象は特に高湿環境下で顕著である。しかし、ユーザーの印刷の待ち時間を短くするために、印刷開始時の現像器内で現像剤の攪拌時間は、短いことが好ましい。
短時間に電荷が失われたトナーを再び十分に摩擦帯電させるためには、現像器内での攪拌に必要なエネルギーの高い現像剤にすることが1つの手法として考えられる。しかし、このような現像剤の場合、継続的に現像剤に大きなストレスがかかり、その結果、耐久時に現像剤が劣化し画像濃度が変動する。このため、従来から、間欠印字の画像劣化の防止と、耐久時の濃度変動の防止を両立させることは出来なかった。
発明者らは、上記課題に対し鋭意検討し、現像剤が攪拌され始めた時、すなわち、現像器内で現像剤が低速攪拌されている時の攪拌エネルギーを選択的に高めることで、上記問題を解決した。
間欠印刷時には、低速で攪拌される時間が比較的長い。これは、印刷終了後に現像器が停止し、次に印刷する時に再び現像器が攪拌を開始する。この頻度が高いためである。
それに対し、連続で印刷する場合は現像器が連続で攪拌しており低速で攪拌される時間は比較的短い。このため、耐久時の濃度変動を抑制しつつ、間欠印刷時の画像の濃度変動を抑制するためには、低速攪拌時のエネルギーを選択的に高めることが必要である。
【0009】
この思想から発明者らは、現像剤が現像器内で攪拌開始時の低速攪拌されている時に攪拌に必要なエネルギー(E10mm/sec)と、攪拌速度が規定の速度となり高速攪拌されている時に攪拌に必要なエネルギー(E100mm/sec)の理想的なエネルギーの関係として、以下の関係式を見出した。
−0.2×E100mm/sec+100≦ΔE≦−0.4×E100mm/sec+200
[但し、ΔE(mJ)は、E10mm/secからE100mm/secを引いた値であ
る。]
E10mm/secは、粉体流動性分析装置(図1(1−a))においてプロペラ型ブレードの最外縁部の周速を10mm/secで回転させながら容器内の二成分系現像剤粉体層中に垂直に進入させ、該粉体層の底面から55mmの位置から測定を開始し、底面から2mmの位置まで進入させた時に得られる、回転トルクと垂直荷重の総和(ブレード周速10mm/sec時のトータルエネルギーともいう)で求められる。
一方、E100mm/secは、粉体流動性分析装置(図1(1−a))においてプロペラ型ブレードの最外縁部の周速を100mm/secで回転させながら容器内の二成分系現像剤粉体層中に垂直に進入させ、該粉体層の底面から100mmの位置から測定を開始し、底面から10mmの位置まで進入させた時に得られる、回転トルクと垂直荷重の総和(ブレード周速100mm/sec時のトータルエネルギーともいう)で求められる。
プロペラ型ブレードの最外縁部の周速は低い方が、現像剤が現像器内で低速攪拌されている時の状態をより忠実に再現できる。しかし、10mm/secより小さい周速にするとプロペラの回転が安定しないためか、データの再現性が低くなる。このため、発明者らはデータの信頼性が得られ、かつ、現像機内の状態を忠実に再現できる10mm/secの周速で測定することが必要と考えた。
また、プロペラ型ブレードの最外縁部の周速100mm/secで測定したトータルエネルギー(E100mm/sec)は、攪拌速度が規定の速度となり安定的に攪拌されている時に、攪拌に必要なエネルギーを示している。具体的には、現像器内の現像スリーブと規制ブレードの間で現像剤にかかるエネルギーである。
通常、現像スリーブは現像ドラムに対し、1.2倍以上2.0倍以下のスピードで回転する。このため、プロセススピード200mm/sec以上の本体の場合、現像スリーブは240mm/sec以上のスピードで回転する。
現像剤は、規制ブレードで一部せき止められるため、規制ブレード裏のせき止められた現像剤は、240mm/sec以上のスピードで攪拌されることになる。この時の現像剤にかかるエネルギーは、粉体流動性分析装置においてプロペラ型ブレードの最外縁部の周速を240mm/sec以上で測定することで求められると予想されるが、このような高周速で測定した場合、トータルエネルギーが画質劣化や濃度変動と相関があるとする結果が得られなかった。その理由は明確ではないが以下のように発明者らは推測している。
粉体流動性分析装置で測定する場合、測定容器にフタがないため、プロペラ型ブレードの周速があがればあがるほど、測定容器の上の方に現像剤が逃げてしまう。このように逃げる現像剤の量が多くなってしまうと、現像剤にかかるエネルギーが小さくなってしまう。それに対して、現像器内の場合、密閉系に近い形態のため、大きなエネルギーがかかる。
このため、発明者らは鋭意検討した結果、プロペラ型ブレードの最外縁部の周速100mm/sec以下で測定することで、測定容器の上に逃げる現像剤が少なく、現像剤が逃げた影響を減らすことができ、現像機内の現像剤の状況を再現できることを見出した。
また、100mm/secよりブレード周速が遅くなると、現像剤の攪拌速度が低くなりすぎ、プロセススピード200mm/sec以上の本体の状況を再現できない。
【0010】
E100mm/secとΔEを上記範囲にすることで、高プロセススピードで間欠印刷した場合の画質劣化の低減や、画像の濃度変動を改善することができる。
ΔEが[−0.2×E100mm/sec+100]より小さくなると、間欠で印刷した場合、画像の濃度変動が大きくなる。その理由は明確ではないが、発明者らは低速攪拌時の攪拌に必要なエネルギーが小さく、トナーが十分に電荷を保持しないうちに現像されることに起因すると推測している。
一方、ΔEが[−0.4×E100mm/sec+200]より大きくなると、間欠で印刷した場合、画質が劣化する。その理由は明確ではないが、発明者らは低速攪拌時の攪拌に必要なエネルギーが大きすぎ、低速攪拌時に現像剤が劣化してしまうことに起因すると推測している。
また、E100mm/sec(mJ)を、80≦E100mm/sec≦360、(好ましくは120≦E100mm/sec≦300)の範囲にすることで、トナー飛散防止と耐久時の濃度安定性を向上させることができる。
E100mm/sec(mJ)が80より小さいと現像器でのトナー飛散が多くなり、部材が汚染されてしまう。
一方、E100mm/sec(mJ)が360より大きいと現像器内で現像剤を攪拌する時に大きなエネルギーが必要になる。このため、間欠印刷を有無にかかわらず、現像剤に過度のストレスがかかり、耐久時に画像濃度が変動する。
【0011】
次に、E100mm/sec(mJ)とE10mm/sec(mJ)の具体的な測定方法について述べる。本発明において、E100mm/sec(mJ)及びE10mm/s
ec(mJ)の測定には、回転式ブレードを備えた粉体流動性分析装置(パウダーレオメータFT−4、Freeman Technology社製)(以下、「FT−4」ともいう)を用いた。
上記装置の原理は、粉体サンプル中で回転式ブレードを移動させ、一定のパターンの流れを起こさせる。粉体サンプル中の粒子はブレードが近接すると流動し、通過すると再び静止する。ブレードが粉体中を移動するのに必要としたエネルギーが測定され、この値から、種々の流動性指数が計算される。ブレードはプロペラ型で、回転すると同時に上又は下方向にも運動するので先端はらせんを描くことになる。回転速度と上下運動を変化させることによりブレードのらせん経路の角度や速度を調節することができる。ブレードが粉体層表面に対して右回りのらせん経路に沿って移動するときには粉体を均一に混ぜる作用がある。逆にブレードが粉体層表面に対して左回りのらせん経路に沿って移動するときにはブレードは粉体から抵抗を受けることになる。(図1(1−a))
具体的には、以下の操作により測定を行った。尚、全ての操作において、プロペラ型ブレードは、FT−4測定専用[23.5mm径ブレード(型番:C415)](図1(1−b)参照、以下、「ブレード」と省略)を用いた。
まず、FT−4測定専用[25mm×25mlスプリット容器(型番:2015)](図1(1−c)参照、以下、容器と省略)に23℃/60%RH環境に3日放置されたトナーをすりきり一杯(約30cc)入れることで二成分系現像剤粉体層とした。
(1)コンディショニング操作
(a):ブレードの周速(ブレードの最外縁部の周速)を100(mm/sec)にした。粉体層への垂直方向の進入速度を、移動中のブレードの最外縁部が描く軌跡と粉体層表面とのなす角度(以降、「ブレード軌跡角」)が5(deg)になるスピードにした。粉体層表面に対して時計回り(ブレードの回転により粉体層がほぐされる方向)の回転方向に、粉体層表面から二成分系現像剤粉体層の底面から5mmの位置までブレードを進入させた。その後、ブレードの周速を40(mm/sec)、粉体層への垂直方向の進入速度を、ブレード軌跡角が2(deg)になるスピードで、粉体層表面に対して時計回りの回転方向に、二成分系現像剤粉体層の底面から2mmの位置までブレードを進入させる操作を行った。その後、ブレードの周速を40(mm/sec)、粉体層からの抜き取り速度をブレード軌跡角が5(deg)になるスピードで、粉体層表面に対して時計回りの回転方向に、トナー粉体層の底面から55mmの位置までブレードを移動させ、抜き取りを行った。抜き取りが完了したら、ブレードを時計回り、反時計回りに交互に小さく回転させることでブレードに付着した二成分系現像剤を払い落とした。
(b):一連の上記(1)−(a)の操作を5回行うことで、二成分系現像剤粉体層中に巻き込まれている空気を取り除き、安定した二成分系現像剤粉体層を作った。
・ スプリット操作
上述の容器のスプリット部分(図1(1−c))で二成分系現像剤粉体層をすり切り、粉体層上部の二成分系現像剤を取り除くことで、同じ体積(25ml)の二成分系現像剤粉体層を形成した。
・ 測定操作
(a):上記(1)−(a)と同様の操作を一回行った。
(b):次にブレードの周速を100(mm/sec)、粉体層への垂直方向の進入速度を、ブレード軌跡角が5(deg)になるスピードにした。粉体層表面に対して反時計回り(ブレードの回転により粉体層が押し込まれる方向)の回転方向に、トナー粉体層の底面から5mmの位置までブレードを進入させた。その後、ブレードの周速を40(mm/sec)、粉体層への垂直方向の進入速度を、ブレード軌跡角が2(deg)になるスピードで、粉体層表面に対して時計回りの回転方向に、粉体層の底面から2mmの位置までブレードを進入させる操作を行った。その後、ブレードの周速を40(mm/sec)、粉体層からの垂直方向の抜き取り速度をブレード軌跡角が5(deg)になるスピードで、粉体層表面に対して時計回りの回転方向に、粉体層の底面から55mmの位置までブレードの抜き取りを行った。抜き取りが完了したら、ブレードを時計回り、反時計回りに
交互に小さく回転させることでブレードに付着した二成分系現像剤を払い落とした。
(c):上記、(b)の一連の操作を7回繰り返した。
上記(c)の操作において、7回目のブレードの周速100(mm/sec)で二成分系現像剤粉体層の底面から100mmから10mmの位置までブレードを進入させたときに得られる回転トルクと垂直荷重の総和を、[E100mm/sec]とした。
(d):(3)−(c)と同様に70(mm/sec)、40(mm/sec)、10(mm/sec)に順次周速を落とした測定を行った。周速が10(mm/sec)でトナー粉体層の底面から55mmの位置から測定を開始し、底面から2mmの位置まで進入させた時に得られる回転トルクと垂直荷重の総和を、[E10mm/sec]とした。
そして、E10mm/secからE100mm/secを引いたものをΔE(mJ)とした。
【0012】
上記ΔE及びE100mm/secは、磁性キャリア及びトナーの構造又は物性を制御することで本発明の範囲に調整することが可能である。
磁性キャリアについては、まず、真比重が、2.5g/cm3以上4.2g/cm3以下である。真比重が2.5g/cm3より小さくなると、磁性成分の量が少なくなる。その結果、磁性キャリアが現像スリーブに保持されにくくなり、磁性キャリアがドラム上に付着してしまう。磁性キャリアの真比重が4.2g/cm3より大きくなると、二成分系現像剤の攪拌時の抵抗が大きくなり、E100mm/secが上記範囲より大きくなる。
次に、磁性キャリア表面に、現像剤が現像器内で攪拌開始時の低速攪拌されている時に攪拌に必要なエネルギーに関与する部分と、攪拌速度が規定の速度となり高速攪拌されている時に攪拌に必要なエネルギーに関与する部分の両方を持たせることが好ましい。
具体的には、磁性キャリア表面に、凹凸を存在させ、かつ、凸部と凹部の表面状態を異ならせることが好ましい。
このような磁性キャリアの場合、低速攪拌時は攪拌速度が遅いために、凸部と凹部の両方が攪拌エネルギーに関与する。それに対し、規定の速度での攪拌時には、凹部はほとんど接することがなく、攪拌エネルギーに関与するのは凸部のみになる。
このため、凸部と凹部の表面状態を変えた磁性キャリアの場合、低速攪拌時及び高速攪拌時の攪拌エネルギーをコントロールすることができる。
具体的には凸部に攪拌時の抵抗の少ない金属酸化物部分を存在させ、凹部に攪拌時の抵抗が大きい樹脂部を存在させた磁性キャリアを用いることが好ましい。
これ以外にも、キャリアの粒径、使用している樹脂の種類等によってもE100mm/sec、及びΔEを調整することができる。
また、トナーとしては、トナーの形状(平均円形度)、粒径、粒径分布により、攪拌エネルギーをコントロールすることができる。
なお、上記、どの手法を用いても、E100mm/sec、及びΔEが本発明の範囲に入っていれば、本願発明の効果は発現する。
【0013】
上記E100mm/sec、及びΔEを本発明の範囲に調整するための、磁性キャリアの凹凸度合いとしては、該磁性キャリアの走査型電子顕微鏡(SEM)で撮影された投影像の周囲長をL1とし、包絡線の長さをL2とするとき、包絡度{(L1−L2)/L2}×100が、0.5以上10.0以下が好ましく、1.0以上5.0以下がより好ましい。
そのメカニズムは明確ではないが、発明者らは以下のように推測している。
現像剤が攪拌された時、攪拌速度が低い場合、磁性キャリア粒子の周囲に沿って、他の磁性キャリアやトナーが通過すると思われる。それに対し、高速で攪拌された場合、キャリア粒子の包絡線に近い場所で磁性キャリア粒子やトナーが通過すると思われる。
このため、この周囲長と包絡線がずれるようにキャリアの凹凸度合いをコントロールすることで、低速攪拌及び高速攪拌時の現像剤の攪拌状態を変えることができる。具体的には、包絡度が0.5以上10.0以下であると、E100mm/sec、及びΔEを本発
明の範囲にコントロールすることができる。包絡度が0.5未満だと、凹凸が小さいために高速攪拌時に攪拌エネルギーが大きくなってしまう。一方、包絡度が10.0を越えると、凸凹が大きいため、磁性キャリアとしての耐久性が低下し、印刷時にキャリアが破損し、キャリア付着が発生する場合がある。
【0014】
本発明に用いられる磁性キャリアについて詳細に述べる。
本発明の磁性キャリアは、金属酸化物を含有する磁性コア粒子と樹脂とを少なくとも含有する磁性キャリアである。前記磁性キャリアは、走査型電子顕微鏡を用い、加速電圧が2.0kVのときに得られる反射電子の投影像において、金属酸化物に由来する部分の面積が、磁性キャリアの投影像面積に対して、0.5面積%以上8.0面積%以下であり、6.672μm2以上の金属酸化物に由来する部分の面積が、金属酸化物に由来する部分の総面積に対して、10面積%以下であることが、上記ΔEを本発明で規定する範囲に調節するために好ましい。
この金属酸化物に由来する部分は、樹脂部に比べ、トナーに対し付着力が低く、現像剤の攪拌エネルギーを下げることができると発明者らは考えている。
加速電圧が2.0kVのときに得られる反射電子の投影像において、金属酸化物に由来する部分の面積が、磁性キャリアの投影像面積に対して、0.5面積%より小さい場合は、磁性キャリア表面に存在する金属酸化物に由来する部分の面積が小さいために、高速攪拌時に現像剤の攪拌エネルギーを下げることが出来ず、現像剤が劣化し、耐久時に画質が低下する傾向にある。
一方、磁性キャリアの投影像面積に対して、8.0面積%より大きい場合は、金属酸化物に由来する部分が多いため、低速攪拌時の攪拌エネルギーが低くなり、その結果、間欠印刷時に画像濃度が変動する傾向にある。
また、発明者らは、200pixel(5.560μm2)、220pixel(6.116μm2)、240pixel(6.672μm2)・・・と、20pixel毎に、金属露出部分の面積比率と、電荷のリークの関係を検討した。その結果、金属酸化物に由来する部分の面積が6.672μm2以上の部位において、電荷のリークが顕著に発生することを見出した。6.672μm2以上とは、走査型電子顕微鏡による反射電子の投影像の観察において、240pixelの大きさである。(1pixelの面積は0.0278μm2)
発明者らはその理由を以下のように推察している。
金属酸化物に由来する部分の回りには被覆している樹脂が存在している。その樹脂はある程度の厚みをもっており、その厚みにより金属酸化物に由来する部分の接触が抑制され、リークが生じにくい。それに対し、金属酸化物に由来する部分が6.672μm2以上大きさだと、周囲にある樹脂の影響をうけずに接触に寄与する部分が存在できる。このため、キャリア同士が接触したときに、当該部分から電荷がリークすると考えている。
この面積を金属酸化物に由来する部分の総面積に対して、10面積%以下にすることで、長期使用時の帯電量低下を抑制し、画像濃度の変動を抑制することができる。
【0015】
なお、上記走査型電子顕微鏡を用い、所定の加速電圧下で撮影された反射電子の投影像上で金属酸化物に由来する部分とは、主に反射電子を可視化した像(図2(2−a)参照)において、輝度の高い(画像上白く、明るく見える)部分として観察される部分のことを指している。走査型電子顕微鏡は、加速した電子線を試料へ照射し、試料から放出されてくる二次電子や反射電子を検出することで、試料の表面や組成情報を可視化する装置である。走査型電子顕微鏡観察において、放出されてくる反射電子の量は、重元素ほど多いことが知られている。例えば、有機化合物と鉄が平面上に分布している試料であれば、鉄からの反射電子の放出量が多いため、鉄部分が画像上では明るく(輝度が高い、白く)見える。一方、軽元素から構成される有機化合物からの反射電子量は多くはないため、画像上では暗く(輝度が低く、黒く)見えることになる。
上記磁性キャリアの表面には、有機化合物である樹脂部分と金属酸化物である磁性コア
部分とが磁性キャリアの表面に分布している。磁性キャリアの反射電子像においては、金属酸化物部分が明るく、樹脂部分が暗く、画像上で大きなコントラストの差をもった投影像として得られる。図2(2−b)は、図2(2−a)の磁性キャリア表面の金属酸化物部分と樹脂部分の分布を模式的に示したもので、白い部分が、金属酸化物に由来する部分であり、黒い部分が、樹脂部分に相当する。本発明においては、まず、図2(2−a)の磁性キャリアの投影像から磁性キャリアを抽出し、磁性キャリアの投影像面積をもとめる。図2(2−c)の白く抜けている部分が、図2(2−a)の投影像から磁性キャリア部分として抽出された部分を示している。続いて、図2(2−a)の投影像から金属酸化物部分を抽出し(図2(2−d)参照)、図2(2−d)において、白く抜けている箇所が金属酸化物部分を表している。磁性キャリアの投影像面積と金属酸化物部分の面積を画像処理によりそれぞれ求める。次に、磁性キャリアの投影像面積に占める金属酸化物部分の面積の割合と、金属酸化物部分の面積分布を算出する。走査型電子顕微鏡による観察条件、撮影条件、画像手順の詳細は、後述する。また、実際に、白く光る部分が金属酸化物であることは、電子顕微鏡に付属の元素分析装置で確認することもできる。
【0016】
磁性キャリア表面における磁性コア部分の存在状態(すなわち、金属酸化物に由来する部分の面積)をコントロールする方法は、磁性コア粒子の含有する被覆樹脂の組成、被覆樹脂の量、被覆方法など種々の方法で調整することができる。また、樹脂で被覆時に、凹部に選択的に樹脂を導入することが好ましい。
具体的には、固形分濃度の異なる被覆樹脂溶液で複数回にわけて被覆処理するなどによって、磁性コア部分の存在状態をコントロールすることが好ましい。
【0017】
上記磁性コア粒子としては、例えば、表面酸化又は未酸化の鉄、リチウム、カルシウム、マグネシウム、ニッケル、銅、亜鉛、コバルト、マンガン、クロム、希土類の如き金属粒子、それらの合金粒子、酸化物粒子及びフェライト等が好適に例示できる。
また、上記磁性キャリアは、磁性コア粒子へ樹脂を充填した磁性キャリア、及び/又は、磁性コア粒子の表面を樹脂で被覆した被覆磁性キャリアであることが好ましい。樹脂の被覆方法としては、樹脂の如き被覆材を溶剤中に溶解もしくは懸濁せしめて調製した塗布液を磁性コア粒子表面に付着せしめる方法、磁性コア粒子と被覆材とを粉体で混合する方法等、従来公知の方法が適用できる。
また、上記磁性コア粒子は、下記に述べる、多孔質磁性コア粒子であることが好ましく、多孔質フェライト粒子であることがより好ましい。
ここで、上記磁性キャリアとして、多孔質磁性コア粒子中に樹脂を含有させたものが、包絡度を0.5以上10以下となるような凹凸を磁性キャリア表面に構成できるために、特に好ましい。
多孔質磁性コア粒子は、以下のような工程で製造することができる。
多孔質磁性コア粒子の材質としては、以下のものが挙げられる。1)表面が酸化された鉄粉、2)未酸化の鉄粉、3)リチウム、カルシウム、マグネシウム、ニッケル、銅、亜鉛、コバルト、マンガン、クロム及び希土類元素の如き金属粒子、4)鉄、リチウム、カルシウム、マグネシウム、ニッケル、銅、亜鉛、コバルト、マンガン、クロム及び希土類元素の如き金属の合金粒子、又はこれらの元素を含む酸化物粒子、5)マグネタイト粒子又はフェライト粒子。
フェライト粒子とは次式で表される焼結体である。
(M12O)u(M2O)v(M32O3)w(M4O2)x(M52O5)y(Fe2O3)z
(式中、M1は1価、M2は2価、M3は3価、M4は4価、M5は5価の金属であり、u+v+w+x+y+z=1.0とした時に、u、v、w、x及びyは、それぞれ0≦(u,v,w,x,y)≦0.8であり、zは、0.2<z<1.0である。)
また、上記式中において、M1〜M5としては、少なくともLi、Fe、Zn、Ni、Mn、Mg、Co、Cu、Ba、Sr、Ca、Si、V、Bi、In、Ta、Zr、B、
Mo、Na、Sn、Ti、Cr、Al、Sc、Y、La、Ce、Pr、Nd、Sm、Eu、Gd、Tb、Dy、Ho、Er、Tm、Yb、Luからなる群から選ばれる1種類以上の金属元素を表す。
例えば、磁性のLi系フェライト(例えば、(Li2O)a(Fe2O3)b(0.0<a<0.4、0.6≦b<1.0、a+b=1))、Mn系フェライト(例えば、(MnO)a(Fe2O3)b(0.0<a<0.5、0.5≦b<1.0、a+b=1))、Mn−Mg系フェライト(例えば、(MnO)a(MgO)b(Fe2O3)c(0.0<a<0.5、0.0<b<0.5、0.5≦c<1.0、a+b+c=1))、Mn−Mg−Sr系フェライト(例えば、(MnO)a(MgO)b(SrO)c(Fe2O3)d(0.0<a<0.5、0.0<b<0.5、0.0<c<0.5、0.5≦d<1.0、a+b+c+d=1)、Cu−Zn系フェライト(例えば、(CuO)a(ZnO)b(Fe2O3)c(0.0<a<0.5、0.0<b<0.5、0.5≦c<1.0、a+b+c=1)がある。なお、上記フェライトは主元素を示し、それ以外の微量金属を含有するものも含んでいる。
結晶の成長速度のコントロールの容易性の観点から、Mn元素を含有する、Mn系フェライト、Mn−Mg系フェライト、Mn−Mg−Sr系フェライトが好ましい。
【0018】
以下に、多孔質磁性キャリア粒子としてフェライト粒子を用いる場合の製造工程を詳細に説明する。
<工程1(秤量・混合工程)>
上記フェライトの原料を、秤量し、混合する。
フェライト原料としては、例えば以下のものが挙げられる。Li、Fe、Zn、Ni、Mn、Mg、Co、Cu、Ba、Sr、Y、Ca、Si、V、Bi、In、Ta、Zr、B、Mo、Na、Sn、Ti、Cr、Al、希土類金属の金属粒子、酸化物、水酸化物、シュウ酸塩、炭酸塩。
混合する装置としては、例えば以下のものが挙げられる。ボールミル、遊星ミル、ジオットミル、振動ミル。特にボールミルが混合性の観点から好ましい。
具体的には、ボールミル中に、秤量したフェライト原料、ボールを入れ、0.1時間以上20.0時間以下、粉砕・混合する。
<工程2(仮焼成工程)>
粉砕・混合したフェライト原料を、大気中で焼成温度700℃以上1000℃以下の範囲で、0.5時間以上5.0時間以下仮焼成し、フェライト化する。焼成には、例えば以下の炉が用いられる。バーナー式焼却炉、ロータリー式焼却炉、電気炉。
<工程3(粉砕工程)>
工程2で作製した仮焼フェライトを粉砕機で粉砕する。
粉砕機としては、所望の粒径が得られれば特に限定されない。例えば以下のものがあげられる。クラッシャーやハンマーミル、ボールミル、ビーズミル、遊星ミル、ジオットミル。
フェライト粉砕品を所望の粒径にするために、例えば、ボールミルやビーズミルでは用いるボールやビーズの素材、粒径、運転時間を制御することが好ましい。具体的には、仮焼フェライトスラリーの粒径を小さくするためには、比重の重いボールを用いたり、粉砕時間を長くすればよい。また、仮焼フェライトの粒度分布を広くするためには、比重の重いボールやビーズを用い、粉砕時間を短くすることで得ることができる。また、粒径の異なる複数の仮焼フェライトを混合することでも分布の広い仮焼フェライトを得ることができる。
また、ボールミルやビーズミルは、乾式より湿式の法が、粉砕品がミルの中で舞い上がることがなく粉砕効率が高い。このため、乾式より湿式の方がより好ましい。
<工程4(造粒工程)>
仮焼フェライトの粉砕品に対し、水、バインダーと、必要に応じて、細孔調整剤を加える。細孔調整剤としては、発泡剤や樹脂微粒子が挙げられる。発泡剤として、例えば、炭
酸水素ナトリウム、炭酸水素カリウム、炭酸水素リチウム、炭酸水素アンモニウム、炭酸ナトリウム、炭酸カリウム、炭酸リチウム、炭酸アンモニウムが挙げられる。樹脂微粒子として、例えば、ポリエステル、ポリスチレン、スチレン−ビニルトルエン共重合体、スチレン−ビニルナフタリン共重合体、スチレン−アクリル酸エステル共重合体、スチレン−メタクリル酸エステル共重合体、スチレン−α−クロルメタクリル酸メチル共重合体、スチレン−アクリロニトリル共重合体、スチレン−ビニルメチルケトン共重合体、スチレン−ブタジエン共重合体、スチレン−イソプレン共重合体、スチレン−アクリロニトリル−インデン共重合体の如きスチレン共重合体;ポリ塩化ビニル、フェノール樹脂、変性フェノール樹脂、マレイン樹脂、アクリル樹脂、メタクリル樹脂、ポリ酢酸ビニル、シリコーン樹脂;脂肪族多価アルコール、脂肪族ジカルボン酸、芳香族ジカルボン酸、芳香族ジアルコール類及びジフェノール類から選択されるモノマーを構造単位として有するポリエステル樹脂;ポリウレタン樹脂、ポリアミド樹脂、ポリビニルブチラール、テルペン樹脂、クマロンインデン樹脂、石油樹脂、ポリエステルユニットとビニル系重合体ユニットを有しているハイブリッド樹脂の微粒子が挙げられる。
上記バインダーとしては、例えば、ポリビニルアルコールが用いられる。
工程3において、湿式で粉砕した場合は、フェライトスラリー中に含まれている水も考慮し、バインダーと必要に応じて細孔調整剤を加えることが好ましい。
得られたフェライトスラリーを、噴霧乾燥機を用い、100℃以上200℃以下の加温雰囲気下で、乾燥・造粒する。噴霧乾燥機としては、所望の多孔質磁性コア粒子の粒径が得られれば特に限定されない。例えば、スプレードライヤーが使用できる。
<工程5(本焼成工程)>
次に、造粒品を800℃以上1400℃以下で1時間以上24時間以下焼成する。
焼成温度を上げ、焼成時間を長くすることで、多孔質磁性コア粒子の焼成が進み、その結果、細孔径は小さく、かつ、細孔の数も減る。また、焼成する雰囲気をコントロールすることで、ブレークダウンする電界強度を好ましい範囲にコントロールすることができる。例えば、酸素濃度を低くしたり、還元雰囲気(水素存在下)にしたりすることで、多孔質磁性コア粒子のブレークダウン電界強度を下げることができる。
<工程6(選別工程)>
以上の様に焼成した粒子を解砕した後に、必要に応じて、分級や篩で篩分して粗大粒子や微粒子を除去してもよい。
磁性コア粒子の体積分布基準50%粒径(D50)は、18.0μm以上68.0μm以下であることが、画像へのキャリア付着とガサツキの抑制のためより望ましい。
【0019】
上述のように磁性キャリアは、多孔質磁性コア粒子の空隙の少なくとも一部に、樹脂が充填された磁性キャリアであることが好ましい。
多孔質磁性コア粒子は、内部の空隙容積によっては物理的強度が低くなることがあり、磁性キャリアとしての物理的強度を高めるために、多孔質磁性コア粒子の空隙の少なくとも一部に樹脂の充填を行うことが好ましい。多孔質磁性コア粒子に充填される樹脂の量としては、多孔質磁性コア粒子に対して6質量%以上25質量%以下であることが好ましい。磁性キャリア毎の樹脂含有量にバラつきが少なければ、内部空隙内の一部にのみ樹脂が充填されていても、多孔質磁性コア粒子の表面近傍の空隙にのみ樹脂が充填され内部に空隙が残っていても、内部空隙が完全に樹脂で充填されていてもよい。
多孔質磁性コア粒子の空隙に、樹脂を充填する方法としては、特に限定されないが、浸漬法、スプレー法、ハケ塗り法、及び流動床の如き塗布方法により多孔質磁性コア粒子を樹脂溶液に含浸させ、その後、溶剤を揮発させる方法が挙げられる。特に、磁性キャリアの表面に金属酸化物由来の部分を残すためには、樹脂と溶剤を混合した樹脂溶液を多孔質磁性コア粒子の空隙へ充填させる方法が好ましい。多孔質磁性コア粒子の空隙に樹脂を充填させる方法としては、樹脂を溶剤に希釈し、これを多孔質磁性コア粒子の空隙に添加する方法が採用できる。ここで用いられる溶剤は、樹脂を溶解できるものであればよい。有機溶剤に可溶な樹脂である場合は、有機溶剤として、トルエン、キシレン、セルソルブブ
チルアセテート、メチルエチルケトン、メチルイソブチルケトン、メタノールが挙げられる。また、水溶性の樹脂またはエマルジョンタイプの樹脂である場合には、溶剤として水を用いればよい。
上記多孔質磁性コア粒子の空隙に充填する樹脂としては特に限定されず、熱可塑性樹脂、熱硬化性樹脂のどちらを用いてもかまわない。多孔質磁性コア粒子に対する親和性が高いものであることが好ましく、親和性が高い樹脂を用いた場合には、多孔質磁性コア粒子の空隙への樹脂の充填時に、同時に多孔質磁性コア粒子表面も樹脂で覆うことが容易になる。
上記充填する樹脂として、熱可塑性樹脂としては、以下のものが挙げられる。ポリスチレン、ポリメチルメタクリレート、スチレン−アクリル樹脂;スチレン−ブタジエン共重合体、エチレン−酢酸ビニル共重合体、ポリ塩化ビニル、ポリ酢酸ビニル、ポリフッ化ビニリデン樹脂、フルオロカーボン樹脂、パーフルオロカーボン樹脂、ポリビニルピロリドン、石油樹脂、ノボラック樹脂、飽和アルキルポリエステル樹脂、ポリエチレンテレフタレート、ポリブチレンテレフタレート、ポリアリレート、ポリアミド樹脂、ポリアセタール樹脂、ポリカーボネート樹脂、ポリエーテルスルホン樹脂、ポリスルホン樹脂、ポリフェニレンサルファイド樹脂、ポリエーテルケトン樹脂。
また、上記熱硬化性樹脂としては、以下のものが挙げられる。フェノール樹脂、変性フェノール樹脂、マレイン樹脂、アルキド樹脂、エポキシ樹脂、無水マレイン酸とテレフタル酸と多価アルコールとの重縮合によって得られる不飽和ポリエステル、尿素樹脂、メラミン樹脂、尿素−メラミン樹脂、キシレン樹脂、トルエン樹脂、グアナミン樹脂、メラミン−グアナミン樹脂、アセトグアナミン樹脂、グリプタール樹脂、フラン樹脂、シリコーン樹脂、ポリイミド、ポリアミドイミド樹脂、ポリエーテルイミド樹脂、ポリウレタン樹脂。
上記樹脂の中で、熱硬化性樹脂が、磁性キャリアの強度を高めることができるため好ましい。中でもシリコーン樹脂が、現像剤のΔE(攪拌エネルギー差)を本発明の範囲にコントロールしやすいため、より好ましい。
上記シリコーン樹脂の具体例として、以下のものが挙げられる。ストレートシリコーン樹脂では、信越化学社製のKR271、KR255、KR152、東レ・ダウコーニング社製のSR2400、SR2405、SR2410、SR2411。変性シリコーン樹脂では、信越化学社製のKR206(アルキッド変性)、KR5208(アクリル変性)、ES1001N(エポキシ変性)、KR305(ウレタン変性)、東レ・ダウコーニング社製のSR2115(エポキシ変性)、SR2110(アルキッド変性)。
上記樹脂溶液における樹脂固形分の量は、好ましくは1質量%以上50質量%以下であり、より好ましくは1質量%以上30質量%以下である。50質量%より樹脂量の多い樹脂溶液を用いると粘度が高いため多孔質磁性コア粒子の空隙に樹脂溶液が均一に浸透しにくい。また、1質量%未満であると樹脂量が少なく、多孔質磁性コア粒子への樹脂の付着力が低くなる場合がある。
【0020】
多孔質磁性コア粒子へ樹脂を充填しただけでも、磁性キャリアとして用いることも可能である。その場合には、トナーへの帯電付与性を高めるために、予め、樹脂溶液中に荷電制御剤、荷電制御樹脂等などを含有した状態で充填することが好ましい。
また、本発明に用いられる磁性キャリアは、多孔質磁性コア粒子の空隙に樹脂を充填した後、磁性キャリアの表面を樹脂で被覆することが、磁性キャリア表面に金属酸化物に由来する部分の面積や面積分布を調整するうえで、より好ましい。
磁性キャリアの表面を樹脂により被覆することにより、金属酸化物由来の部分の割合や面積をより精密に制御することができるようになるため好ましい。また、磁性キャリア表面からのトナーの離型性、磁性キャリア表面のトナーや外添剤の汚染性、トナーへの帯電付与能や磁性キャリア抵抗を制御する意味からも、磁性キャリア表面を樹脂により被覆することが好ましい。
磁性コア粒子の表面を樹脂で被覆する方法としては、特に限定されないが、浸漬法、ス
プレー法、ハケ塗り法、乾式法、及び流動床の如き塗布方法により被覆する方法が挙げられる。中でも、磁性コア粒子を適度に表面に露出させることができる浸漬法がより好ましい。被覆する樹脂の量としては、磁性コア粒子100質量部に対し、0.1質量部以上5.0質量部以下であることが、金属酸化物部分を表面に適度に露出させることができ、好ましい。被覆する樹脂は、充填に用いた樹脂を用いることができる。
【0021】
本発明に用いられるトナーは、結着樹脂及びワックスを少なくとも含有するトナー粒子を含有するトナーである。上記結着樹脂としては、特に限定されず、従来公知の結着樹脂を用いることができる。具体的には、以下のものが挙げられる。ポリエステル、ポリスチレン;ポリ−p−クロルスチレン、ポリビニルトルエンの如きスチレン誘導体の重合体;スチレン−p−クロルスチレン共重合体、スチレン−ビニルトルエン共重合体、スチレン−ビニルナフタリン共重合体、スチレン−アクリル酸エステル共重合体、スチレン−メタクリル酸エステル共重合体、スチレン−α−クロルメタクリル酸メチル共重合体、スチレン−アクリロニトリル共重合体、スチレン−ビニルメチルケトン共重合体、スチレン−ブタジエン共重合体、スチレン−イソプレン共重合体、スチレン−アクリロニトリル−インデン共重合体の如きスチレン共重合体;ポリ塩化ビニル、フェノール樹脂、変性フェノール樹脂、マレイン樹脂、アクリル樹脂、メタクリル樹脂、ポリ酢酸ビニル、シリコーン樹脂;脂肪族多価アルコール、脂肪族ジカルボン酸、芳香族ジカルボン酸、芳香族ジアルコール類及びジフェノール類から選択されるモノマーを構造単位として有するポリエステル樹脂;ポリウレタン樹脂、ポリアミド樹脂、ポリビニルブチラール、テルペン樹脂、クマロンインデン樹脂、石油樹脂、ポリエステルユニットとビニル系重合体ユニットを有しているハイブリッド樹脂。また、上記ワックスも、特に限定されず、従来公知のワックスを用いることができる。具体的には、以下のものが挙げられる。低分子量ポリエチレン、低分子量ポリプロピレン、アルキレン共重合体、マイクロクリスタリンワックス、パラフィンワックス、フィッシャー・トロプシュワックスの如き炭化水素系ワックス;酸化ポリエチレンワックスの如き炭化水素系ワックスの酸化物又はそれらのブロック共重合物;カルナバワックス、ベヘン酸ベヘニルエステルワックス、モンタン酸エステルワックスの如き脂肪酸エステルを主成分とするワックス類;脱酸カルナバワックスの如き脂肪酸エステル類を一部又は全部を脱酸化したもの。さらに、本発明に用いられるトナーは、必要に応じて、従来公知の着色剤、荷電制御剤、無機微粉体等を含有させることもできる。
一方、上記トナー粒子及びトナーの製造方法についても、特に限定されず、従来公知の製造方法を用いることができる。
本発明に用いられるトナーは、トナーの画像処理解像度512×512画素(1画素あたり0.37μm×0.37μm)のフロー式粒子像測定装置によって計測された前記トナーの円相当径1.98μm以上200.00μm以下の粒子を、0.200以上1.000以下の円形度の範囲において800分割して解析された平均円形度が、0.950以上0.990以下であり、円相当径0.50μm以上200.00μm以下の全粒子に対する、0.50μm以上1.98μm以下の粒子が25.0個数%以下であることが、現像性を向上させつつ耐久時の画像劣化の抑制することができ、好ましい。
平均円形度が、0.950より小さい場合、トナーと磁性キャリアの付着面積が大きくなり、その結果、トナーとキャリアの付着力が上がり現像性が低下する場合がある。
一方、平均円形度が、0.990より大きい場合、トナーと磁性キャリアの付着力が小さくなりすぎトナー飛散が悪化する場合がある。
【0022】
また、本発明に用いられるトナーが、上記円相当径0.50μm以上1.98μm以下の粒子が25.0個数%より大きい場合、耐久時に0.50μm以上1.98μm以下の粒子が磁性キャリアの金属酸化物部分に付着し、高速攪拌時の攪拌エネルギーが大きくなり、耐久時に濃度が変動する場合がある。
トナーの平均円形度及び円相当径0.50μm以上1.98μm以下の粒子の個数%を上記範囲にコントロールする手法としては、分級する方法、または、熱風を用いてトナー
粒子の表面を改質処理する方法が挙げられる。
具体的には、慣性分級方式のエルボージェット(日鉄鉱業社製)、遠心力分級方式のターボプレックス(ホソカワミクロン社製)、TSPセパレータ(ホソカワミクロン社製)、ファカルティ(ホソカワミクロン社製)の如き分級機や篩分機を用い、0.50μm以上1.98μm以下の粒子を取り除くことができる。
また、熱風を用いたトナー粒子の表面改質処理を行いつつ、平均円形度をコントロールすることもできる。さらに、熱風を用いたトナー粒子の表面改質処理の場合、0.50μm以上1.98μm以下の粒子が、表面改質処理時にトナー表面にしっかりと保持される。その結果、0.50μm以上1.98μm以下の粒子を25.0個数%以下にコントロールすることも可能である。
上記熱風を用いてトナー粒子の表面を改質処理する方法としては、熱風でトナー粒子の表面処理をする方法であれば、特に限定されない。例えば、ハイブリタイゼーションシステム(奈良機械製作所製)、メカノフージョンシステム(ホソカワミクロン社製)、ファカルティ(ホソカワミクロン社製)、メテオレインボー MR Type(日本ニューマチック社製)などの装置を用いる方法が挙げられる。ここで、表面処理装置を用いて熱風により表面処理を行い、続いて分級をする方法、若しくは、予め分級したものを、表面処理装置を用いて熱風により表面処理を行う方法のいずれの方法を用いてもよい。
上記表面処理装置を用いた、熱風でトナー粒子の表面を改質処理する方法の概略を、図5を用いて説明するが、これに限定されるものではない。具体的には、従来公知の方法でトナー粒子を得た後、上記表面処理装置に供給する。そして、トナー粒子供給口(100)から供給されたトナー粒子(114)は、高圧エア供給ノズル(115)から噴射されるインジェクションエアにより加速され、その下方にある気流噴射部材(102)へ向かう。気流噴射部材(102)からは拡散エアが噴射され、この拡散エアによりトナー粒子が外側方向へ拡散する。この時、インジェクションエアの流量と拡散エアの流量とを調節することにより、トナーの拡散状態をコントロールすることができる。また、トナー粒子の融着防止を目的として、トナー粒子供給口(100)の外周、表面処理装置外周及び移送配管(116)の外周には冷却ジャケット(106)が設けられている。尚、該冷却ジャケットには冷却水(好ましくはエチレングリコール等の不凍液)を通水することが好ましい。一方、拡散エアにより拡散したトナー粒子は、熱風供給口(101)から供給された熱風により、トナー粒子の表面が処理される。この時、熱風の温度は100℃以上、450℃以下であることが好ましく、100℃以上、400℃以下であることがより好ましい。熱風の温度が100℃未満の場合にはトナー粒子表面の表面面粗さにばらつきが生じる場合がある。また、450℃を超える場合には溶融状態が進みすぎる事でトナー粒子同士の合一が進み、トナー粒子の粗大化や融着が生じる場合がある。
熱風により表面が処理されたトナー粒子は、装置上部外周に設けた冷風供給口(103)から供給される冷風により冷却される。この時、装置内の温度分布の制御、トナー粒子の表面状態をコントロールする目的で、装置の本体側面に設けた第二の冷風供給口(104)から冷風を導入しても良い。第二の冷風供給口(104)の出口はスリット形状、ルーバー形状、多孔板形状、メッシュ形状等を用いる事ができ、導入方向は中心方向へ水平、装置壁面に沿う方向が、目的に応じて選択可能である。
この時、上記冷風供給口内及び第二の冷風供給口内の温度は−50℃以上、10℃以下であることが好ましく、−40℃以上、8℃以下であることがより好ましい。また、上記冷風は除湿された冷風であることが好ましい。具体的には、絶対水分量が5g/m3以下であることが好ましい。更に好ましくは、3g/m3以下である。
これらの冷風温度が−50℃未満の場合には装置内の温度が下がりすぎてしまい、本来の目的である熱による処理が十分に為されず、トナー粒子の球形化ができない場合がある。また、10℃を超える場合には、装置内における熱風ゾーンの制御が不十分になり、表面処理時にワックスを過度にトナー表面に偏析させることがある。
その後、冷却されたトナー粒子は、ブロワーで吸引され、移送配管(116)を通じて、サイクロン等で回収される。
【0023】
本発明の二成分系現像剤は、画像の濃度変動の抑制と画質低下の抑制の観点から、磁性キャリア100質量部に対して、トナーが4質量部以上20質量部以下の割合で含有する二成分系現像剤である。好ましくは、磁性キャリア100質量部に対して、トナーが4質量部以上18質量部以下の割合で含有する二成分系現像剤である。トナーが4質量部より少ないと、画像面積比率が高い画像を連続で印刷した場合に、現像器内のトナーが不足し画像濃度が低下する場合がある。一方、トナーが20質量部より多いと、現像器でのトナー飛散が多くなり、部材を飛散したトナーで汚染する。上記二成分系現像剤において、磁性キャリア100質量部に対するトナー比率は、二成分系現像剤作製時のトナーと磁性キャリアの比率を変えることにより任意にコントロールすることができる。
【0024】
<磁性キャリア等の真密度の測定方法>
磁性キャリアの真比重は、乾式自動密度計アキュピック1330(島津製作所社製)を用いて測定する。まず、23℃、50%RHの環境に24時間放置したサンプル試料を5g精秤し、測定用セル(10cm3)に入れ、本体試料室に挿入する。測定は、試料サンプル質量を本体に入力し測定をスタートさせることにより自動測定できる。
自動測定における測定条件は、20.000psig(2.392×102kPa)で調整されたヘリウムガスを用い、試料室内を10回パージした後、試料室内の圧力変化が0.005psig/min(3.447×10−2kPa/min)になる状態を平衡状態とし、平衡状態になるまで繰り返しヘリウムガスをパージする。平衡状態の時の本体試料室の圧力を測定する。その平衡状態に達した時の圧力変化により試料サンプル体積が算出できる(ボイルの法則)。試料サンプルの体積が算出できることにより、以下の式で試料サンプルの真比重が計算できる。
試料サンプルの真比重(g/cm3)= 試料サンプル質量(g)/試料サンプル体積
(cm3)
この自動測定により5回繰り返し測定した値の平均値を磁性キャリア及び磁性コア粒子の真比重(g/cm3)とする。
【0025】
<トナーの平均円形度、及び、円相当径0.50μm以上1.98μm以下の粒子の個数%の測定方法>
上記トナーの平均円形度は、画像処理解像度512×512画素(1画素あたり0.37μm×0.37μm)のフロー式粒子像測定装置「FPIA−3000型」(シスメックス社製)を用い、校正作業時の測定・解析条件で測定する。
具体的な測定方法は、以下の通りである。まず、ガラス製の容器中に予め不純固形物などを除去したイオン交換水約20mlを入れる。この中に分散剤として「コンタミノンN」(非イオン界面活性剤、陰イオン界面活性剤、有機ビルダーからなるpH7の精密測定器洗浄用中性洗剤の10質量%水溶液、和光純薬工業社製)をイオン交換水で約3質量倍に希釈した希釈液を約0.2ml加える。更に測定試料を約0.02g加え、超音波分散器を用いて2分間分散処理を行い、測定用の分散液とする。その際、分散液の温度が10℃以上40℃以下となる様に適宜冷却する。超音波分散器としては、発振周波数50kHz、電気的出力150Wの卓上型の超音波洗浄器分散器(例えば「VS−150」(ヴェルヴォクリーア社製))を用い、水槽内には所定量のイオン交換水を入れ、この水槽中に前記コンタミノンNを約2ml添加する。
測定には、標準対物レンズ(10倍)を搭載した前記フロー式粒子像分析装置を用い、シース液にはパーティクルシース「PSE−900A」(シスメックス社製)を使用する。
標準対物レンズ(10倍)を用いることで、画像処理解像度512×512画素(1画素あたり0.37μm×0.37μm)となる。
前記手順に従い調整した分散液を前記フロー式粒子像分析装置に導入し、HPF測定モードで、トータルカウントモードにて3000個のトナー粒子を計測する。そして、粒子解
析時の2値化閾値を85%とし、解析粒子径を円相当径1.98μm以上200.00μm未満に限定し、0.200以上1.000以下の円形度の範囲において800分割して解析し、トナー粒子の平均円形度を求める。
測定にあたっては、測定開始前に標準ラテックス粒子(例えば、Duke Scientific社製の「RESEARCH AND TEST PARTICLES Latex Microsphere Suspensions 5200A」をイオン交換水で希釈)を用いて自動焦点調整を行う。その後、測定開始から2時間毎に焦点調整を実施することが好ましい。
なお、本願実施例では、シスメックス社による校正作業が行われた、シスメックス社が発行する校正証明書の発行を受けたフロー式粒子像分析装置を使用した。解析粒子径を円相当径1.98μm以上、200.00μm未満に限定した以外は、校正証明を受けた時の測定及び解析条件で測定を行う。
また、この時、円相当径0.50μm以上200.00μm以下の全粒子に対する、0.50μm以上1.98μm以下の粒子の割合を個数%として求める。
【0026】
<磁性キャリア表面の金属酸化物に由来する部分の面積%の算出方法>
磁性キャリア表面の金属酸化物に由来する部分の面積%は、走査電子顕微鏡による反射電子の投影像の観察と、続く画像処理により求めることができる。
磁性キャリア表面の金属酸化物に由来する部分の面積%は、走査型電子顕微鏡(SEM)、S−4800(日立製作所社製)を用いて算出する。金属酸化物に由来する部分の面積は、加速電圧が2.0kVのときに得られる反射電子を可視化した像の画像処理から算出される。走査型電子顕微鏡観察において、試料から放出されてくる反射電子の量は、重元素ほど多いことが知られている。本発明に用いられる磁性キャリア表面のように、樹脂部分と磁性コアに由来する金属酸化物部分が存在する試料では、金属酸化物部分が明るく(輝度が高い、白く)、樹脂部分は暗く(輝度が低く、黒く)見えるため、それぞれ大きなコントラスト差を持った画像が得られる。
具体的には、走査型電子顕微鏡観察用の試料台上にカーボンテープで磁性キャリアを一層になるように固定し、白金による蒸着は行わずに、以下の条件にて、走査型電子顕微鏡S−4800(日立製作所社製)で観察する。また、フラッシング操作を行ってから観察を行う。
SignalName=SE(U,LA80)
AcceleratingVoltage=2000Volt
EmissionCurrent=10000nA
WorkingDistance=6000um
LensMode=High
Condencer1=5
ScanSpeed=Slow4(40秒)
Magnification=600
DataSize=1280x960
ColorMode=Grayscale
反射電子の投影像は、走査型電子顕微鏡S−4800の制御ソフト上で「コントラスト5、ブライトネス−5」に明るさを調整し、キャプチャスピード/積算枚数「Slow4を40秒」、画像サイズ1280×960pixelsの8bitの256階調グレースケール画像として得る(図3(3−a))。画像上のスケールから、1pixelの長さは0.1667μm、1pixelの面積は0.0278μm2となる。
続いて、得られた反射電子の投影像を用いて、磁性キャリア50個について金属酸化物に由来する部分の面積%を算出する。解析する磁性キャリア50個の選択方法の詳細は後述する。金属酸化物に由来する部分の面積%の算出には、画像処理ソフトImage−Pro Plus5.1J(MediaCybernetics社製)を使用する。
まず、図3(3−b)の画像下部の文字列は画像処理に不必要であり、不要な部分を削
除し1280×895のサイズに切り出す(図3(3−c))。次に、磁性キャリアの部分を抽出し、抽出された磁性キャリア部分のサイズをカウントした。具体的には、まず、解析する磁性キャリアを抽出するため、磁性キャリアと背景部分を分離する。Image−Pro Plus5.1Jの「測定」−「カウント/サイズ」を選択する。「カウント/サイズ」の「輝度レンジ選択」で、輝度レンジを50〜255の範囲に設定して、背景として写りこんでいる輝度の低いカーボンテープ部分を除外し、磁性キャリアの抽出を行う(図3(3−d))。カーボンテープ以外の方法で磁性キャリアを固定した際には、必ずしも背景が輝度の低い領域とならない、あるいは、部分的に磁性キャリアと同じような輝度となる可能性は皆無ではない。しかし、磁性キャリアと背景の境界については、反射電子の投影像から容易に区別できる。抽出を行う際、「カウント/サイズ」の抽出オプションで、4連結を選択し、平滑度5を入力、「穴埋める」にチェックを入れ、画像の全ての境界(外周)上に位置する粒子や他の粒子と重なっている粒子については、計算から除外するものとする。次に「カウント/サイズ」の測定項目で、面積とフェレ径(平均)を選択し、面積の選別レンジを最小300pixel、最大10000000pixelとする(図4(4−a))。また、フェレ径(平均)は、後述する磁性キャリアの体積分布基準50%粒径(D50)の測定値の±25%径の範囲になるよう選別レンジを設定し、画像解析する磁性キャリアを抽出する(図4(4−b))。抽出された磁性キャリアに由来する部分の大きさ(pixel数)を(ja)、それぞれの抽出部分の和(Σja=Ja)、抽出された部分の数(Jc)を求める。抽出された磁性キャリアの数Jcが、Jc=50となるまで、別視野の磁性キャリア投影像について同様の操作を繰り返す。
次に、選択した粒子に対して金属酸化物に由来する部分を抽出した。Image−Pro Plus5.1Jの「カウント/サイズ」の「輝度レンジ選択」で、輝度レンジを140〜255の範囲に設定して、磁性キャリア上の輝度の高い部分の抽出を行う。面積の選別レンジは最小10pixel、最大10000pixelとし、磁性キャリア表面の金属酸化物に由来する部分を抽出する(図4(4−c))。さらに、上記磁性キャリア部分の抽出時と同様に、画像外周に位置する粒子や50%粒径(D50)の測定値の±25%径の範囲から逸脱したものは、計算から除外した。金属酸化物に由来の抽出部分の大きさ(pixel数)(ma)、それぞれの抽出部分の和(Ma)として、下式に従い計算した。
式: 金属酸化物に由来する部分の面積%=Ma/Ja×100
【0027】
<6.672μm2以上の金属酸化物に由来する部分の面積%の算出方法>
6.672μm2以上の金属酸化物に由来する部分の面積の、金属酸化物に由来する部分の総面積に対する面積%は、走査型電子顕微鏡による反射電子の投影像の観察と画像処理、続く統計処理により求めることができる。金属酸化物に由来する部分の面積%を求めるのと同様にして、磁性キャリアの観察を行い、画像から磁性キャリア中の金属酸化物に由来する部分の抽出を行った。抽出された金属酸化物に由来する部分を大きさ20pixel毎のチャンネルに分布させた。1pixelの面積は0.0278μm2として面積換算を行い、金属酸化物に由来する部分の総面積に対して、6.672μm2以上に分布する割合を算出した。
【0028】
<磁性キャリアの包絡度の測定方法>
磁性キャリアの断面加工には、集束イオンビーム加工観察装置(FIB)、FB−2100(日立ハイテクノロジーズ社製)を用いる。FIB用試料台(金属メッシュ)上にカーボンペーストを塗り、その上に磁性キャリアを1粒子ずつ独立して存在するように少量固着させ、導電膜として白金蒸着することで試料を作製する。試料をFIB装置にセットし、加速電圧40kV、Gaイオン源を用いて、粗加工し(ビーム電流39nA)、続いて仕上げ加工(ビーム電流7nA)を行い、試料断面を削り出す。
尚、試料とする磁性キャリアは、各試料の最大径をDmaxとしたときに、D50×0.9≦Dmax≦D50×1.1である磁性キャリアを対象とする。さらに、各試料の固
着面に対して平行な方向における、最大長を含む平面の位置を、固着面からの距離hとする(例えば、半径rの完全な球体の場合、h=rとなる)。固着面より垂直な方向に、固着面からの距離0.9×h以上1.1×h以下の範囲において、断面を削り出す。
断面加工した試料は、そのまま走査型電子顕微鏡(SEM)観察に適用することができる。走査型電子顕微鏡観察において、試料から放出されてくる反射電子の量は、重元素ほど多いことが知られている。例えば、有機化合物と鉄のような金属が平面状に分布している試料であれば、鉄からの反射電子の放出量がより多く検出されるため、鉄部分が画像上では明るく(輝度が高い、白く)見えることになる。一方、軽元素化合物から構成される有機化合物からの反射電子量は少ないため、画像上では暗く(輝度が低く、黒く)見えることになる。磁性キャリアの断面観察においては、磁性コア部領域に由来する金属酸化物部が明るく(輝度が高い、白く)、磁性コア部以外の領域は、暗く(輝度が低く、黒く)見えるため、それぞれ大きなコントラスト差を持った画像が得られる。具体的には、走査型電子顕微鏡(SEM)、S−4800(日立ハイテクノロジーズ社製)を用いて、以下の条件にて観察する。尚、フラッシング操作を行ってから観察する。
SignalName=SE(U,LA100)
AcceleratingVoltage=5000Volt
EmissionCurrent=10000nA
WorkingDistance=4000um
LensMode=High
Condencer1=3
ScanSpeed=Slow4(40sec)
Magnification=1500
DataSize=1280x960
ColorMode=Grayscale
SpecimenBias=0V
尚、反射電子の投影像は、上記条件のほか、走査電子顕微鏡S−4800の制御ソフト上で「コントラスト5、ブライトネス−5」に明るさを調整し、磁性体観察モードはOFFとし、256階調のグレースケール画像として得る。
次に、得られた投影像を用いて、画像処理ソフトImage−Pro Plus5.1J(MediaCybernetics社製)を使用し、磁性キャリア50個について、包絡線L2及び周囲長L1を求め、下記式より包絡度を求める。
式: 包絡度={(L1−L2)/L2}×100
【0029】
<トナーの重量平均粒子径(D4)の測定>
トナーの重量平均粒径(D4)は、100μmのアパーチャーチューブを備えた細孔電気抵抗法による精密粒度分布測定装置「コールター・カウンター Multisizer
3」(登録商標、ベックマン・コールター社製)と、測定条件設定及び測定データ解析をするための付属の専用ソフト「ベックマン・コールター Multisizer 3 Version3.51」(ベックマン・コールター社製)を用いて、実効測定チャンネル数2万5千チャンネルで測定し、測定データの解析を行い、算出する。
測定に使用する電解水溶液は、特級塩化ナトリウムをイオン交換水に溶解して濃度が約1質量%となるようにしたもの、例えば、「ISOTON II」(ベックマン・コールター社製)が使用できる。
尚、測定、解析を行う前に、以下のように前記専用ソフトの設定を行う。
前記専用ソフトの「標準測定方法(SOM)を変更画面」において、コントロールモードの総カウント数を50000粒子に設定し、測定回数を1回、Kd値は「標準粒子10.0μm」(ベックマン・コールター社製)を用いて得られた値を設定する。閾値/ノイズレベルの測定ボタンを押すことで、閾値とノイズレベルを自動設定する。また、カレントを1600μAに、ゲインを2に、電解液をISOTON IIに設定し、測定後のアパーチャーチューブのフラッシュにチェックを入れる。
専用ソフトの「パルスから粒径への変換設定画面」において、ビン間隔を対数粒径に、粒径ビンを256粒径ビンに、粒径範囲を2μm以上60μm以下に設定する。
具体的な測定法は以下の通りである。
(1)Multisizer 3専用のガラス製250ml丸底ビーカーに前記電解水溶液約200mlを入れ、サンプルスタンドにセットし、スターラーロッドの撹拌を反時計回りで24回転/秒にて行う。そして、解析ソフトの「アパーチャーのフラッシュ」機能により、アパーチャーチューブ内の汚れと気泡を除去しておく。
(2)ガラス製の100ml平底ビーカーに前記電解水溶液約30mlを入れ、この中に分散剤として「コンタミノンN」(非イオン界面活性剤、陰イオン界面活性剤、有機ビルダーからなるpH7の精密測定器洗浄用中性洗剤の10質量%水溶液、和光純薬工業社製)をイオン交換水で3質量倍に希釈した希釈液を約0.3ml加える。
(3)発振周波数50kHzの発振器2個を、位相を180度ずらした状態で内蔵し、電気的出力120Wの超音波分散器「Ultrasonic Dispension System Tetora150」(日科機バイオス社製)の水槽内に所定量のイオン交換水を入れ、この水槽中に前記コンタミノンNを約2ml添加する。
(4)前記(2)のビーカーを前記超音波分散器のビーカー固定穴にセットし、超音波分散器を作動させる。そして、ビーカー内の電解水溶液の液面の共振状態が最大となるようにビーカーの高さ位置を調整する。
(5)前記(4)のビーカー内の電解水溶液に超音波を照射した状態で、トナー約10mgを少量ずつ前記電解水溶液に添加し、分散させる。そして、さらに60秒間超音波分散処理を継続する。尚、超音波分散にあたっては、水槽の水温が10℃以上40℃以下となる様に適宜調節する。
(6)サンプルスタンド内に設置した前記(1)の丸底ビーカーに、ピペットを用いてトナーを分散した前記(5)の電解質水溶液を滴下し、測定濃度が約5%となるように調整する。そして、測定粒子数が50000個になるまで測定を行う。
(7)測定データを装置付属の前記専用ソフトにて解析を行い、重量平均粒径(D4)を算出する。尚、専用ソフトでグラフ/体積%と設定したときの、分析/体積統計値(算術平均)画面の「平均径」が重量平均粒径(D4)である。
【0030】
<樹脂のピーク分子量(Mp)、数平均分子量(Mn)、重量平均分子量(Mw)の測定方法>
樹脂の分子量分布は、ゲルパーミエーションクロマトグラフィー(GPC)により、以下のようにして測定する。
まず、室温で24時間かけて、樹脂をテトラヒドロフラン(THF)に溶解する。そして、得られた溶液を、ポア径が0.2μmの耐溶剤性メンブランフィルター「マエショリディスク」(東ソー社製)で濾過してサンプル溶液を得る。尚、サンプル溶液は、THFに可溶な成分の濃度が約0.8質量%となるように調整する。このサンプル溶液を用いて、以下の条件で測定する。
装置 :HLC8120 GPC(検出器:RI)(東ソー社製)
カラム:Shodex KF−801、802、803、804、805、806、
807の7連(昭和電工社製)
溶離液:テトラヒドロフラン(THF)
流速 :1.0ml/min
オーブン温度:40.0℃
試料注入量:0.10ml
試料の分子量の算出にあたっては、標準ポリスチレン樹脂(例えば、商品名「TSKスタンダード ポリスチレン F−850、F−450、F−288、F−128、F−80、F−40、F−20、F−10、F−4、F−2、F−1、A−5000、A−2500、A−1000、A−500」、東ソ−社製)を用いて作成した分子量校正曲線を使用する。
【0031】
<ワックスの最大吸熱温度、樹脂のガラス転移温度(Tg)の測定方法>
ワックスの最大吸熱ピークを示す温度は、示差走査熱量分析装置「Q1000」(TA
Instruments社製)を用いてASTM D3418−82に準じて測定する。
装置検出部の温度補正はインジウムと亜鉛の融点を用い、熱量の補正についてはインジウムの融解熱を用いる。具体的には、ワックスを約10mg精秤し、これをアルミニウム製のパンの中に入れ、リファレンスとして空のアルミニウム製のパンを用い、測定温度範囲30乃至200℃の間で、昇温速度10℃/minで測定を行う。尚、測定においては、一度200℃まで昇温させ、続いて30℃まで降温し、その後に再度昇温を行う。この2度目の昇温過程での温度30乃至200℃の範囲におけるDSC曲線の最大の吸熱ピークを示す温度を、最大吸熱温度とする。
また、樹脂のガラス転移温度(Tg)は、ワックス測定時と同様に、樹脂を約10mg精秤し測定を行う。温度40℃乃至100℃の範囲の比熱変化において、比熱変化が出る前と出た後のベースラインの中間点の線とDSC曲線との交点を、結着樹脂のガラス転移温度(Tg)とする。
【0032】
<樹脂の軟化点の測定方法>
樹脂の軟化点の測定は、定荷重押し出し方式の細管式レオメータ「流動特性評価装置 フローテスターCFT−500D」(島津製作所社製)を用い、装置付属のマニュアルに従って行う。本装置では、測定試料の上部からピストンによって一定荷重を加えつつ、シリンダに充填した測定試料を昇温させて溶融し、シリンダ底部のダイから溶融された測定試料を押し出し、この際のピストン降下量と温度との関係を示す流動曲線を得ることができる。
本発明においては、「流動特性評価装置 フローテスターCFT−500D」に付属のマニュアルに記載の「1/2法における溶融温度」を軟化点とする。尚、1/2法における溶融温度とは、次のようにして算出されたものである。まず、流出が終了した時点におけるピストンの降下量Smaxと、流出が開始した時点におけるピストンの降下量Sminとの差の1/2を求める(これをXとする。X=(Smax−Smin)/2)。そして、流動曲線においてピストンの降下量がXとSminの和であるときの流動曲線の温度が、1/2法における溶融温度である。
測定試料には、約1.0gの樹脂を、25℃の環境下で、錠剤成型圧縮機(例えば、NT−100H、エヌピーエーシステム社製)を用いて約10MPaで、約60秒間圧縮成型し、直径約8mmの円柱状としたものを用いる。また、CFT−500Dの測定条件は、以下の通りである。
試験モード:昇温法
開始温度:40℃
到達温度:200℃
測定間隔:1.0℃
昇温速度:4.0℃/min
ピストン断面積:1.000cm2
試験荷重(ピストン荷重):10.0kgf(0.9807MPa)
予熱時間:300秒
ダイの穴の直径:1.0mm
ダイの長さ:1.0mm
【0033】
<磁性キャリア等の体積分布基準の50%粒径(D50)の測定方法>
粒度分布測定は、レーザー回折・散乱方式の粒度分布測定装置「マイクロトラックMT3300EX」(日機装社製)にて測定を行った。測定には、乾式測定用の試料供給機「ワンショットドライ型サンプルコンディショナーTurbotrac」(日機装社製)を
装着して行った。Turbotracの供給条件として、真空源として集塵機を用い、風量約33リットル/sec、圧力約17kPaとした。制御は、ソフトウエア上で自動的に行う。粒径は体積分布基準の累積値である50%粒径(D50)を求める。制御及び解析は付属ソフト(バージョン10.3.3−202D)を用いて行う。測定条件は、以下の通りである。
SetZero時間:10秒
測定時間 :10秒
測定回数 :1回
粒子屈折率 :1.81
粒子形状 :非球形
測定上限 :1408μm
測定下限 :0.243μm
測定環境 :常温常湿環境(23℃、50%RH)
【実施例】
【0034】
以下、本発明の具体的実施例について説明するが、本発明はこれらの実施例に限定されるものではない。尚、以下の配合における部数は特に説明が無い場合は質量部である。
【0035】
〔樹脂の製造例1〕
冷却管、攪拌機、及び、窒素導入管のついた反応槽中に、以下の材料を秤量した。
テレフタル酸 22.6質量部無水トリメリット酸 1.7質量部ポリオキシプロピレン(2.2)−2,2−ビス(4−ヒドロキシフェニル)プロパン
75.6質量部チタニウムジヒドロキシビス(トリエタノールアミネート) 0.1質量部
その後、200℃に加熱し、窒素を導入しながら生成する水を除去しながら10時間反応させ、その後、10mmHgに減圧し1時間反応させ、樹脂1を合成した。GPCで求めた樹脂1の分子量は、重量平均分子量(Mw)6200、数平均分子量(Mn)2500であり、ピーク分子量(Mp)2900、ガラス転移点は55℃、軟化点は93℃であった。
【0036】
〔樹脂の製造例2〕
冷却管、攪拌機、及び、窒素導入管のついた反応槽中に、以下の材料を秤量した。
テレフタル酸 17.2質量部ポリオキシエチレン(2.2)−2,2−ビス(4−ヒドロキシフェニル)プロパン
76.6質量部チタニウムジヒドロキシビス(トリエタノールアミネート) 0.1質量部
その後、220℃に加熱し、窒素を導入しながら生成する水を除去しながら10時間反応させた。さらに、無水トリメリット酸6.1質量部を加え、180℃に加熱し、2時間反応させ樹脂2を合成した。GPCで求めた樹脂2の分子量は、重量平均分子量(Mw)86000、数平均分子量(Mn)6000、ピーク分子量(Mp)12800、ガラス転移点は62℃、軟化点は132℃であった。
【0037】
〔トナー製造例1〕
樹脂1 50.0質量部樹脂2 50.0質量部フィッシャー・トロプシュワックス(最大吸熱温度:75℃) 7.5質量部C.I.ピグメントブルー15:3 5.0質量部
上記材料をヘンシェルミキサー(FM−75型、三井三池化工機(株)製)でよく混合した後、温度130℃に設定した二軸混練機(PCM−30型、池貝鉄工(株)製)にて
混練した。得られた混練物を冷却し、ハンマーミルにて1mm以下に粗粉砕し、粗砕物を得た。得られた粗砕物を、高圧気体を用いた衝突式気流粉砕機を用いて微粉砕した。
次に、得られた微粉砕物を、図5に示す表面改質装置により表面改質を行った。表面改質時の条件は、原料供給速度は2.0kg/hr、熱風流量4.5m3/min、熱風の吐出温度は230℃、冷風温度0℃、冷風流量3.0m3/minであった。次に、コアンダ効果を利用した風力分級機(エルボジェットラボEJ−L3、日鉄鉱業社製)で分級し、微粉及び粗粉を同時に分級除去、トナー粒子1を得た。
得られたトナー粒子1 100.0質量部に、無機微粒子として、個数平均粒径が40nmであり、i−ブチルトリメトキシシランで処理された酸化チタン微粉体1.0質量部、一次平均粒子径16nmであり、ヘキサメチルジシラザン20質量%で表面処理した疎水性シリカ微粒子0.5質量部を添加し、個数平均粒径が200nmであり、ヘキサメチルジシラザンで処理されたゾルゲル法シリカ微粉体を1.0質量部を外添混合し、トナー1を得た。
【0038】
〔トナー製造例2〕
トナー製造例1のうち、表面改質時の条件の熱風流量を4.5m3/minから4.0m3/min、熱風の吐出温度は230℃から210℃に変更した以外は、トナー製造例1と同様にして、トナー2を得た。
【0039】
〔トナー製造例3〕
トナー製造例1のうち、表面改質時の条件の熱風流量を4.5m3/minから5.0m3/min、熱風の吐出温度は230℃から250℃に変更した以外は、トナー製造例1と同様にして、トナー3を得た。
【0040】
〔トナー製造例4〕
トナー製造例1のうち、表面改質処理を行なわない以外は、トナー製造例1と同様にして、トナー4を得た。
【0041】
トナー1乃至4の物性を表1にしめす。
【0042】
【表1】
【0043】
〔多孔質磁性コアの製造例1〕
<工程1(秤量・混合工程)>
Fe2O3 59.7質量%
MnCO3 34.4質量%
Mg(OH)2 4.8質量%
SrCO3 1.1質量%
となるようにフェライト原材料を秤量した。その後、ジルコニア(φ10mm)のボールを用いた乾式ボールミルで2時間粉砕・混合した。
<工程2(仮焼成工程)>
粉砕・混合した後、バーナー式焼成炉を用い、大気中で950℃で2時間焼成し、仮焼
フェライトを作製した。
フェライトの組成は、下記の通り。
(MnO)a(MgO)b(SrO)c(Fe2O3)d
上記式において、a=0.39、b=0.11、c=0.01、d=0.49
<工程3(粉砕工程)>
クラッシャーで0.5mm程度に粉砕した後に、ジルコニア(φ10mm)のボールを用い、仮焼フェライト100質量部に対し、水を30質量部加え、湿式ボールミルで2時間粉砕した。そのスラリーを、ジルコニアのビーズ(φ1.0mm)を用いた湿式ビーズミルで3時間粉砕し、フェライトスラリー(仮焼フェライト微粉砕品)を得た。
得られた仮焼フェライト微粉砕品の、体積基準の50%粒径(D50)は1.7μmであった。
<工程4(造粒工程)>
フェライトスラリーに、バインダーとして仮焼フェライト100質量部に対してポリビニルアルコール2.0質量部を添加し、スプレードライヤー(製造元:大川原化工機)で、約36μmの球状粒子に造粒した。
<工程5(本焼成工程)>
焼成雰囲気をコントロールするために、電気炉にて窒素雰囲気下(酸素濃度0.01体積%以下)で、1150℃で4時間焼成した。
<工程6(選別工程)>
凝集した粒子を解砕した後に、目開き250μmの篩で篩分して粗大粒子を除去し、多孔質磁性コア1を得た。
【0044】
<多孔質磁性コアの製造例2>
多孔質磁性コア製造例1のうち、工程1でフェライト原材料の比率を、
Fe2O3 62.8質量%
MnCO3 31.2質量%
Mg(OH)2 6.0質量%
に変更した。フェライトの組成は、下記の通り。
(MnO)a(MgO)b(Fe2O3)c
上記式において、a=0.45、b=0.15、c=0.40
工程3のクラッシャーでの粉砕粒度を0.5mm程度から0.3mm程度に変更し、湿式ボールミルのボールをジルコニア(φ10mm)からステンレス(φ10mm)に、湿式ビーズミルのビーズをジルコニア(φ1.0mm)からステンレス(φ1.0mm)に、粉砕時間を3時間から2時間に変更した。
得られた仮焼フェライト微粉砕品は、D50=0.8μmであった。
工程4のポリビニルアルコールの添加量を2質量部から1.0質量部に変更した。
工程5の焼成温度を1150℃から1200℃に、焼成雰囲気を窒素雰囲気下(酸素濃度0.01体積%以下)から窒素雰囲気下(酸素濃度1.0体積%)に、焼成時間を4時間から8時間に変更した。上記以外は、多孔質磁性コア製造例1と同様にして、多孔質磁性コア2を得た。
【0045】
<多孔質磁性コアの製造例3>
多孔質磁性コア製造例1のうち、工程3の湿式ビーズミルの粉砕時間を2時間から3時間に変更した。得られた仮焼フェライト微粉砕品は、D50=1.3μmであった。また、工程5の焼成温度を1150℃から1050℃に変更した。上記以外は、多孔質磁性コア製造例1と同様にして、多孔質磁性コア3を得た
【0046】
<多孔質磁性コアの製造例4>
多孔質磁性コア製造例1のうち、工程1でフェライト原材料の比率を、
Fe2O3 62.4質量%
MnCO3 30.5質量%
Mg(OH)2 6.4質量%
SrCO3 0.7質量%
に変更した。フェライトの組成は、下記の通り。
(MnO)a(MgO)b(SrO)c(Fe2O3)d
上記式において、a=0.34、b=0.14、c=0.01、d=0.51
工程3のクラッシャーでの粉砕粒度を0.5mm程度から1.0mm程度に変更し、湿式ボールミルのボールをジルコニア(φ10mm)からアルミナ(φ10mm)に、粉砕時間を2時間から1時間に変更した。湿式ビーズミルのビーズをジルコニア(φ1.0mm)からアルミナ(φ1.0mm)に変更した。得られた仮焼フェライト微粉砕品は、D50=4.8μmであった。
工程4のポリビニルアルコールの添加量を2.0質量部から5.0質量部に変更した。
工程5の焼成温度を1150℃から1000℃に、焼成雰囲気を窒素雰囲気下(酸素濃度0.01体積%以下)から窒素雰囲気下(酸素濃度0.5体積%)に変更した。上記以外は、多孔質磁性コア製造例1と同様にして、多孔質磁性コア4を得た。
【0047】
<多孔質磁性コアの製造例5>
多孔質磁性コア製造例1のうち、工程1でフェライト原材料の比率を、
Fe2O3 62.8質量%
MnCO3 31.2質量%
Mg(OH)2 6.0質量%
に変更した。フェライトの組成は、下記の通り。
(MnO)a(MgO)b(Fe2O3)c
上記式において、a=0.45、b=0.15、c=0.40
工程3のクラッシャーでの粉砕粒度を0.5mm程度から0.3mm程度に変更し、湿式ボールミルのボールをジルコニア(φ10mm)からステンレス(φ10mm)に、湿式ビーズミルのビーズをジルコニア(φ1.0mm)からステンレス(φ1.0mm)に、粉砕時間を3時間から2時間に変更した。
得られた仮焼フェライト微粉砕品は、D50=0.8μmであった。
工程4のポリビニルアルコールの添加量を2.0質量部から1.0質量部に変更した。
工程5の焼成温度を1150℃から1200℃に、焼成雰囲気を窒素雰囲気下(酸素濃度0.01体積%以下)から窒素雰囲気下(酸素濃度1.0体積%)に、焼成時間を4時間から8時間に変更した。上記以外は、多孔質磁性コア製造例1と同様にして、多孔質磁性コア5を得た。
【0048】
<多孔質磁性コアの製造例6>
多孔質磁性コア製造例4のうち、工程3の湿式ビーズミルの粉砕時間を3時間から2時間に変更した。得られた仮焼フェライト微粉砕品は、D50=5.6μmであった。
工程4のポリビニルアルコールの添加量を5.0質量部から10.0質量部に変更した。
工程5の焼成時間を4時間から2時間に、焼成雰囲気を窒素雰囲気下(酸素濃度0.5体積%)から窒素雰囲気下(酸素濃度1.0体積%)に変更した。上記以外は、多孔質磁性コア製造例4と同様にして、多孔質磁性コア6を得た。
【0049】
<多孔質磁性コアの製造例7>
多孔質磁性コア製造例4のうち、工程3のクラッシャーでの粉砕粒度を1.0mm程度から0.3mm程度に変更し、湿式ボールミルのボールをアルミナ(φ10mm)からステンレス(φ10mm)に、粉砕時間を1時間から3時間に変更した。湿式ビーズミルのビーズをアルミナ(φ1.0mm)からステンレス(φ1.0mm)、粉砕時間を3時間から2時間に変更した。得られた仮焼フェライト微粉砕品は、D50=0.4μmであっ
た。
工程4のポリビニルアルコールの添加量を5.0質量部から1.0質量部に変更した。
工程5の焼成温度を1000℃から1250℃に変更した。上記以外は、多孔質磁性コア製造例4と同様にして、多孔質磁性コア7を得た。
【0050】
<磁性コアの製造例1>
マグネタイト微粒子(球形、個数平均粒径250nm、磁化の強さ65Am2/kg、残留磁化4.2Am2/kg、保磁力4.4kA/m、500V/cmにおける比抵抗3.3×105Ω・cm)と、シラン系カップリング剤(3−(2−アミノエチルアミノプロピル)トリメトキシシラン)(マグネタイト微粒子の質量に対して3.0質量%の量)とを、容器に導入した。そして、該容器内において温度100℃以上で高速混合撹拌して、マグネタイト微粒子を表面処理した。
・フェノール 10質量部
・ホルムアルデヒド溶液(ホルムアルデヒド37質量%水溶液) 16質量部
・表面処理した上記マグネタイト微粒子 84質量部
上記材料を反応釜に導入し、温度40℃にしてよく混合した。
その後、撹拌しながら平均昇温速度3℃/分で、温度85℃に加熱し、28質量%アンモニア水4質量部および水25質量部を反応釜に加えた。温度85℃にて保持し、3時間重合反応させて硬化させた。このときの撹拌翼の周速は1.8m/秒とした。
重合反応させた後、温度30℃まで冷却して水を添加した。上澄み液を除去して得られた沈殿物を水洗し、さらに風乾した。得られた風乾物を、減圧下(5hPa以下)にて、温度60℃で乾燥して、樹脂中に磁性体が分散された磁性コア1を得た。
【0051】
<磁性コアの製造例2>
工程1:
Fe2O3 71.0質量%
CuO 12.5質量%
ZuO 16.5質量%
となるようにフェライト原材料を秤量した。その後、ジルコニア(φ10mm)のボールを用いた乾式ボールミルで2時間粉砕・混合した。
工程2:
粉砕・混合した後、大気中で950℃で2時間焼成し、仮焼フェライトを作製した。
フェライトの組成は、下記の通り。
(CuO)a(ZnO)b(Fe2O3)c
上記式において、a=0.20、b=0.25、c=0.55
工程3:
クラッシャーで0.5mm程度に粉砕した後に、ステンレスのボール(φ10mm)を用い、仮焼フェライト100質量部に対し水を30質量部加え、湿式ボールミルで6時間粉砕した。得られた仮焼フェライト微粉砕品は、D50=0.5μm、D90=2.8μmであった。
工程4:
フェライトスラリーに、バインダーとして仮焼フェライト100質量部に対しポリビニルアルコール2.0質量部を添加し、スプレードライヤー(製造元:大川原化工機)で球状粒子に造粒した。
工程5:
大気中で1300℃で4時間焼成した。
工程6:
凝集した粒子を解砕した後に、目開き250μmの篩で篩分して粗大粒子を除去し、磁性コア2を得た。
【0052】
<樹脂液の調製>
樹脂液1
ストレートシリコーン(SR2411 東レ・ダウコーニング) 20.0質量部
γ−アミノプロピルトリエトキシシラン 0.5質量部
トルエン 79.5質量部
以上を、ボールミル(ソーダガラスボール φ10mm)を用いて1時間混合し、樹脂液1を得た。
【0053】
樹脂液2
ポリメチルメタクリレート重合体(Mw=66,000) 5.0質量部ボントロンP51(オリエント化学工業株式会社) 2.0質量部
トルエン 93.0質量部
以上を、ビーズミル(ジルコニアビーズ φ1.0mm)を用いて3時間分散混合し、樹脂液2を得た。
【0054】
<磁性キャリア1の製造例>
工程1(樹脂充填工程):
多孔質磁性コア1 100.0質量部を万能攪拌混合機(ダルトン社製)に入れ、減圧下、80℃に加熱しながら撹拌する。続いて、樹脂液1を多孔質磁性コア1 100質量部に対し樹脂成分として8質量部になるように添加し2時間加熱を続け、溶剤を除去した。得られた試料をジュリアミキサー(徳寿工作所社製)に移し、窒素雰囲気下に200℃で2時間熱処理して、開口70μmのメッシュで分級して、充填コア1を得た。
工程2(樹脂コート工程):
充填コア1 100.0質量部をナウタミキサ(ホソカワミクロン社製)に投入し、さらに、樹脂液2を樹脂成分として0.5質量部になるようにナウタミキサに投入した。減圧下で70℃に加熱し、1.7S−1(100rpm)で混合し、4時間かけて溶媒除去及び塗布操作を行った。その後、得られた試料をジュリアミキサー(徳寿工作所社製)に移し、窒素雰囲気下、温度200℃で2時間熱処理した後、開口70μmのメッシュで分級して磁性キャリア1を得た。
【0055】
<磁性キャリア2〜13の製造例>
磁性キャリア1の製造例において、コア粒子、充填樹脂量、コート樹脂量を表2のように変え、磁性キャリア2乃至13を得た。得られたキャリア1乃至13の物性を表3に示す。
【0056】
【表2】
【0057】
【表3】
【0058】
〔実施例1乃至22及び比較例1乃至12〕
作製した磁性キャリア及びトナーを用い、磁性キャリアとトナーの組み合わせ、及び、磁性キャリア100質量部に対するトナー比率を表4の記載に従い、二成分系現像剤を作製した。二成分系現像剤は、V型混合機で5分間混合した。また、得られた二成分系現像剤のE100mm/sec、E10mm/sec、ΔE、25FRI(E10mm/sec/E100mm/sec)の数値を表5に示す。
【0059】
【表4】
【0060】
【表5】
【0061】
<二成分系現像剤の評価>
画像形成装置として、キヤノン製カラー複写機imageRUNNER iR C3580を用い、下記評価が可能なように当該複写機を改造した。シアン位置の現像器に上記現像剤を入れ、評価を行った。現像条件としては、現像ドラムに対する現像スリーブ周速を1.5倍、プロセススピード 245mm/secとなるように改造した。そして、現像スリーブには、周波数2.0kHz、Vpp1.3kVの交流電圧と直流電圧VDCを印加した。直流電圧VDCはFFH画像(ベタ部)のトナーの紙上への載り量が0.5mg/cm2となるように調整した。
初期のキャリア付着、トナー飛散、ガサツキの評価を行った。
また、画像面積比率25%で50000枚の連続印刷後、画像濃度安定性、ガサツキの評価を行った。
上記評価を、温度23℃/湿度50%RHの環境と、温度30℃/湿度80%RHの2つの環境で行った。
なおFFH画像とは、256階調を16進数で表示した値であり、00Hを1階調目(
白地部)、FFHを256階調目(ベタ部)とする。
印刷環境 温度23℃/湿度50%RH(以下「N/N」)
温度30℃/湿度80%RH(以下「H/H」
紙 カラー複写機・プリンタ用普通紙 CS−814(A4、81.4g/m2)
(キヤノンマーケティングジャパン株式会社より販売)
【0062】
<キャリア付着>
00H画像を印刷し、感光ドラム上の一部分を透明な粘着テープを密着させてサンプリングし、1cm×1cm中の感光ドラム上に付着していた磁性キャリアの個数をカウントし、1cm2当りの付着キャリア粒子の個数を算出した。
A:3個以下 (優秀)
B:4個以上10個以下 (良い)
C:11個以上20個以下(本発明において許容レベル)
D:21個以上 (本発明において許容できない)
【0063】
<トナー飛散>
現像器、及び、本体内のトナーの汚れ具合を目視にて確認した。
A:トナーの飛散がほとんどない (優秀)
B:トナーの飛散がわずかに確認される (良い)
C:トナーの飛散が確認される (本発明において許容レベル)
D:ひどいトナー飛散が確認される (本発明において許容できない)
【0064】
<ガサツキ>
画像比率5%のFFH画像を、10枚印刷→10分放置→10枚印刷を繰り返し、1000枚印刷した。
その後、1画素を1ドットで形成するドット画像(FFH画像)を作成した。紙上の1ドットあたりの面積が、20000μm2以上25000μm2以下となるように、レーザービームのスポット径を調整した。デジタルマイクロスコープVHX−500(レンズワイドレンジズームレンズVH−Z100・キーエンス社製)を用い、ドット1000個の面積を測定した。ドット面積の個数平均(S)とドット面積の標準偏差(σ)を算出し、ガサツキを下記式により算出した。
ガサツキ指数(I)=σ/S×100
A:Iが4.0未満 (優秀)
B:Iが4.0以上6.0未満 (良好)
C:Iが6.0以上8.0未満 (本発明において許容レベル)
D:Iが8.0以上 (本発明において許容できない)
【0065】
<画像濃度安定性>
耐久評価を行う前に、20mm×20mmのFFH画像を1枚印刷した。X−Riteカラー反射濃度計(500シリーズ:X−Rite社製)を使用し、FFH画像部分の画像濃度を測定した。耐久後に、同じ画像を1枚印刷し、画像濃度を測定した。初期及び耐久後の画像濃度の差を以下の基準で評価とした。
(評価基準)
A:0.00以上0.05未満 (優秀)
B:0.05以上0.10未満 (良い)
C:0.10以上0.20未満 (本発明において許容レベル)
D:0.20以上 (本発明において許容できない)
【0066】
【表6】
【0067】
【表7】
【特許請求の範囲】
【請求項1】
磁性キャリアとトナーを少なくとも含有する二成分系現像剤であって、
前記トナーは、結着樹脂及びワックスを少なくとも含有するトナー粒子を含有し、
前記磁性キャリアは、磁性コア粒子と樹脂を少なくとも含有し、真比重が2.5g/cm3以上4.2g/cm3以下であり、
前記二成分系現像剤は、前記磁性キャリア100質量部に対して、トナー4質量部以上20質量部以下の割合で含有する二成分系現像剤であり、
前記二成分系現像剤のE100mm/sec(mJ)及びE100mm/sec(mJ)、並びに、前記E10mm/secからE100mm/secを引いたΔE(mJ)が下記式(1)及び(2)をみたすことを特徴とする二成分系現像剤。
式(1):−0.2×E100mm/sec+100≦ΔE≦−0.4×E100mm/sec+200
式(2):80≦E100mm/sec≦360
[前記式(1)及び(2)において、E100mm/sec(mJ)は、回転式プロペラ型ブレードを備えた粉体流動性分析装置において、プロペラ型ブレードを、前記プロペラ型ブレードの最外縁部の周速を100mm/secで回転させながら、前記二成分系現像剤が満たされた測定容器内の二成分系現像剤粉体層中に垂直に進入させ、前記二成分系現像剤粉体層の底面から100mmの位置から測定を開始し、底面から10mmの位置まで進入させた時に得られる、回転トルクと垂直荷重の総和を表す。また、前記式(1)のΔE(mJ)において、E10mm/sec(mJ)は、回転式プロペラ型ブレードを備えた粉体流動性分析装置において、プロペラ型ブレードを、前記プロペラ型ブレードの最外縁部の周速を10mm/secで回転させながら、前記二成分系現像剤が満たされた測定容器内の二成分系現像剤粉体層中に垂直に進入させ、前記二成分系現像剤粉体層の底面から55mmの位置から測定を開始し、底面から2mmの位置まで進入させた時に得られる、回転トルクと垂直荷重の総和を表す。]
【請求項2】
前記トナーの画像処理解像度512×512画素(1画素あたり0.37μm×0.37μm)のフロー式粒子像測定装置によって計測された前記トナーの円相当径1.98μm以上200.00μm以下の粒子を、0.200以上1.000以下の円形度の範囲において800分割して解析された平均円形度が、0.950以上0.990以下であり、
円相当径0.50μm以上200.00μm以下の全粒子に対する、0.50μm以上1.98μm以下の粒子が25.0個数%以下であることを特徴とする請求項1に記載の二成分系現像剤。
【請求項3】
前記磁性キャリアは、金属酸化物を含有する磁性コア粒子と樹脂とを少なくとも含有する磁性キャリアであって、前記磁性キャリアは、走査型電子顕微鏡を用い、加速電圧が2.0kVのときに得られる反射電子の投影像において、金属酸化物に由来する部分の面積が、磁性キャリアの投影像面積に対して、0.5面積%以上8.0面積%以下であり、6.672μm2以上の金属酸化物に由来する部分の面積が、金属酸化物に由来する部分の総面積に対して、10面積%以下であることを特徴とする請求項1又は2に記載の二成分系現像剤。
【請求項4】
前記トナー粒子が少なくとも熱風により表面処理されたトナー粒子であることを特徴とする請求項1乃至3のいずれか1項に記載の二成分系現像剤。
【請求項1】
磁性キャリアとトナーを少なくとも含有する二成分系現像剤であって、
前記トナーは、結着樹脂及びワックスを少なくとも含有するトナー粒子を含有し、
前記磁性キャリアは、磁性コア粒子と樹脂を少なくとも含有し、真比重が2.5g/cm3以上4.2g/cm3以下であり、
前記二成分系現像剤は、前記磁性キャリア100質量部に対して、トナー4質量部以上20質量部以下の割合で含有する二成分系現像剤であり、
前記二成分系現像剤のE100mm/sec(mJ)及びE100mm/sec(mJ)、並びに、前記E10mm/secからE100mm/secを引いたΔE(mJ)が下記式(1)及び(2)をみたすことを特徴とする二成分系現像剤。
式(1):−0.2×E100mm/sec+100≦ΔE≦−0.4×E100mm/sec+200
式(2):80≦E100mm/sec≦360
[前記式(1)及び(2)において、E100mm/sec(mJ)は、回転式プロペラ型ブレードを備えた粉体流動性分析装置において、プロペラ型ブレードを、前記プロペラ型ブレードの最外縁部の周速を100mm/secで回転させながら、前記二成分系現像剤が満たされた測定容器内の二成分系現像剤粉体層中に垂直に進入させ、前記二成分系現像剤粉体層の底面から100mmの位置から測定を開始し、底面から10mmの位置まで進入させた時に得られる、回転トルクと垂直荷重の総和を表す。また、前記式(1)のΔE(mJ)において、E10mm/sec(mJ)は、回転式プロペラ型ブレードを備えた粉体流動性分析装置において、プロペラ型ブレードを、前記プロペラ型ブレードの最外縁部の周速を10mm/secで回転させながら、前記二成分系現像剤が満たされた測定容器内の二成分系現像剤粉体層中に垂直に進入させ、前記二成分系現像剤粉体層の底面から55mmの位置から測定を開始し、底面から2mmの位置まで進入させた時に得られる、回転トルクと垂直荷重の総和を表す。]
【請求項2】
前記トナーの画像処理解像度512×512画素(1画素あたり0.37μm×0.37μm)のフロー式粒子像測定装置によって計測された前記トナーの円相当径1.98μm以上200.00μm以下の粒子を、0.200以上1.000以下の円形度の範囲において800分割して解析された平均円形度が、0.950以上0.990以下であり、
円相当径0.50μm以上200.00μm以下の全粒子に対する、0.50μm以上1.98μm以下の粒子が25.0個数%以下であることを特徴とする請求項1に記載の二成分系現像剤。
【請求項3】
前記磁性キャリアは、金属酸化物を含有する磁性コア粒子と樹脂とを少なくとも含有する磁性キャリアであって、前記磁性キャリアは、走査型電子顕微鏡を用い、加速電圧が2.0kVのときに得られる反射電子の投影像において、金属酸化物に由来する部分の面積が、磁性キャリアの投影像面積に対して、0.5面積%以上8.0面積%以下であり、6.672μm2以上の金属酸化物に由来する部分の面積が、金属酸化物に由来する部分の総面積に対して、10面積%以下であることを特徴とする請求項1又は2に記載の二成分系現像剤。
【請求項4】
前記トナー粒子が少なくとも熱風により表面処理されたトナー粒子であることを特徴とする請求項1乃至3のいずれか1項に記載の二成分系現像剤。
【図1】

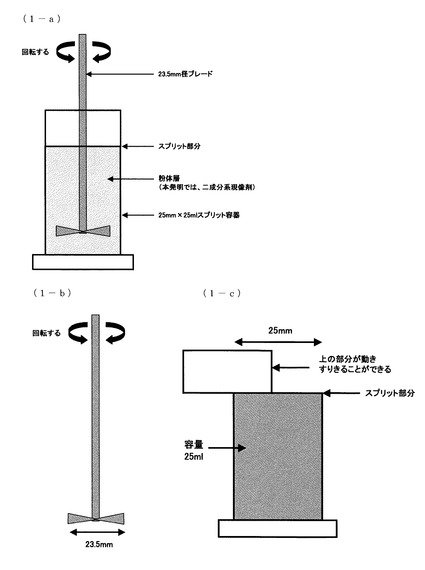
【図2】

【図3】

【図4】

【図5】
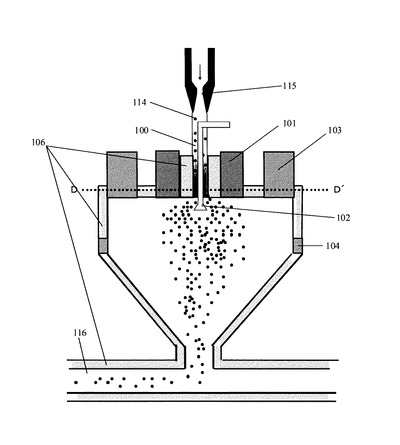
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2011−158742(P2011−158742A)
【公開日】平成23年8月18日(2011.8.18)
【国際特許分類】
【出願番号】特願2010−20974(P2010−20974)
【出願日】平成22年2月2日(2010.2.2)
【出願人】(000001007)キヤノン株式会社 (59,756)
【Fターム(参考)】
【公開日】平成23年8月18日(2011.8.18)
【国際特許分類】
【出願日】平成22年2月2日(2010.2.2)
【出願人】(000001007)キヤノン株式会社 (59,756)
【Fターム(参考)】
[ Back to top ]