
二特異的融合タンパク質
【課題】生体内及び生体外診断及び治療に有用な、生物学的に活性な二特異的融合タンパク質を提供する。
【解決手段】二特異的融合タンパク質であって、(1)標的を結合することができる第1結合ドメイン、(2)標的を結合することができる第2結合ドメイン、ここで各ドメインは異なる標的を結合することができ、及び(3)第1結合ドメインと第2結合ドメインを連結する、親水性アミノ酸と少なくとも1のペンタマー:EEAKKを含有するリンカーを含む、二特異的融合タンパク質;溶解性二特異的融合タンパク質であって、B7抗原と結合する第1結合ドメイン及びCD40抗原と結合する第2結合ドメインを含み、該第1結合ドメインと第2結合ドメインが、親水性アミノ酸と少なくとも1のペンタマー:EEAKKとを含有するリンカーによって連結されている、溶解性二特異的融合タンパク質。
【解決手段】二特異的融合タンパク質であって、(1)標的を結合することができる第1結合ドメイン、(2)標的を結合することができる第2結合ドメイン、ここで各ドメインは異なる標的を結合することができ、及び(3)第1結合ドメインと第2結合ドメインを連結する、親水性アミノ酸と少なくとも1のペンタマー:EEAKKを含有するリンカーを含む、二特異的融合タンパク質;溶解性二特異的融合タンパク質であって、B7抗原と結合する第1結合ドメイン及びCD40抗原と結合する第2結合ドメインを含み、該第1結合ドメインと第2結合ドメインが、親水性アミノ酸と少なくとも1のペンタマー:EEAKKとを含有するリンカーによって連結されている、溶解性二特異的融合タンパク質。
【発明の詳細な説明】
【技術分野】
【0001】
本出願は、1993年2月1日に出願された米国特許出願第08/013,420号の一部継続出願であり、この明細書を本発明に参考として引用する。
本明細書において、種々の文献が言及される。本発明が関係している技術を更に詳細に述べるために、これらの文献の開示をすべて本明細書に参考として引用する。
本発明は、二特異的融合タンパク質をコードする発現ベクター及び哺乳動物細胞における生物学的に活性な二特異的融合タンパク質の生産方法に関する。
【背景技術】
【0002】
血清から抗体を得るような伝統的な抗体手法又はハイブリドーマ手法に伴う課題のために、所望の結合特性とエフェクター機能をセットした抗体又は抗体誘導体分子(例えば、二特異的融合タンパク質)を設計、操作及び生産する遺伝子工学がますます頻繁に用いられてきている。
ヒト抗体を産生する安定なハイブリドーマの生産で困難に直面したことが、生体内抗体産生及び慣用の試験管内手法を克服するように設計された代替方法の開発をもたらした(Mayforth R.D., Quintans, J.(1990) Current Concepts: Designer and catalytic antibodies. New Eng. J. Med. 323:173-178; Waldmann, T.A.(1991) Monoclonal antibodies in diagnosis and therapy. Science 252:1657-1662; Winter, G., Milstein, C.(1991) Man-made Antibodies. Nature 349:293-299; Morrison, S.L.(1992) In Vitro antibodeis: strategies for production and application. Ann. Rev. Immunol. 10:239-266 )。
【0003】
治療用として、異なる標的抗原に対して2つの全抗体の結合特異性を結合する最初の試みは、化学的に結合した『異種複合体』分子を用いるものであった(Staerz, U.D., Kanagawa, O., Bevan, M.J.(1985) Hybrid antibodies can target sites for attack by T cells. Nature 314:628-631; Perez, P., Hoffman, R.W., Shaw, S., Blurstone, J.A., Segal, D.M.(1985) Specific targeting of cytotoxic T cells by anti-T3 linked to anti-target antibody. Nature 316:354-356; Liu, M.A., Kranz, D.M., Kurnick, J.T., Boyle, L.A., Levy, R., Eisen, H.N.(1985) Heteroantibody duplexes target cells for lysis by cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. USA 82:8648-8652; Jung, G., Ledbetter, J.A., Muller-Eberhard, H.J.(1987) Induction of cytotoxicity in resting human T lymphocytes bound to tumor cells by antibody heteroconjugates. Proc. Natl. Acad. Sci. USA 84:4611-4615; Emmrich, F., Rieber, P., Kurrie, R., Eichmann, K.(1988) Selective stimulation of human T lymphocyte subsets by heteroconjugates of antibodies to the T cell receptor and to subset-specific differentiation antigens. Eur. J. Immunol. 18:645-648; Ledbetter, J.A., June, C.H., Rabinovitch, P.S., Grossmann, A., Tsu, T.T., Imboden, J.B.(1988) Signal transduction through CD4 proximity to the CD3/T cell receptor. Eur. J. Immunol. 18:525-532)。
【0004】
これらの試みは、抗標的細胞抗体に化学的に結合したマウス又はヒトCD3T細胞表面レセプターに特定されたモノクローナル抗体が細胞障害性Tリンパ球(CTL)による標的細胞の溶解の引き金となり、CTLの主要な組織不適合性複合体制限を克服したことを証明した。
二特異的抗体はハイブリッドハイブリドーマから異種ハイブリドーマ法により産生され、異種複合体に見られるものと同様の特性を試験管内で示した(Milstein, C., Cuello, A.C.(1983) Hybrid hybridomas and their use in immunohistochemistry. Nature 305:537-540; Ataerz, U.D., Bevan, M.J.(1986) Hybrid hybridoma producing a bispecific monoclonal antibody that can focus effector cell activity. Proc. Natl. Acad. Sci. USA 83:1453-1457; Clark, M.R., Waldmann, H.(1987) T-cell killing of target cells induced by hybrid antibodies: comparison of two bispecific monoclonal antibodies. J. Natl. Cancer Inst. 79:1393-1401; Lanzavecchia, A., Scheidegger, D.(1987) The use of hybrid hybridomas to target cytotoxic T lymphocytes. Eur. J. Immunol.17:105-111; Gilliland, L.K., Clark, M.R.,Waldmann, H.(1988) Universal bispecific antibody for targeting tumour cells for destruction by cytotoxic T cells. Proc. Natl. Acad. Sci. USA 85:7719-7723 )。しかしながら、この抗体は細胞融合から産生されたものである。
【0005】
異種複合体又は細胞融合からの二特異的抗体を用いて得られた有望な結果にもかかわらず、いくつかの要因のために大量の治療適用には非実用的であった。
それらの要因としては、(1)多量の異種複合体の急速な生体内クリアランス、(2)両方の種類の分子を作成するために要する骨の折れる徹底的な方法、(3)同種複合体又は一特異的抗体にはない徹底的精製の必要及び(4)低収量が挙げられる。
一般的には、異種複合体又は二特異的抗体を用いることに関連する方法には、免疫グロブリン重(H)鎖及び/又は軽(L)鎖をコードする配列を連結しない2つの異なる特異性を用いた共発現方法を含むので、ランダムH−L結合及び/又はランダム(HL)−(HL)結合の課題があり、正しい生産物が少量しか得られず且つ困難な精製計画を招いている。過度の一特異的又は非特異的タンパク質分子がある場合、精製が煩わしくなり確認が困難になることがある。
これらの課題を取り除くための努力として、単鎖二特異的又は二機能性抗体を作成するために遺伝子工学が用いられている(Haber等, 1990; Wels, W.,Harwerth, I.M.,Zwickl, M.,Hardman, N.,Groner B.,Hynes, N.E.(1992) Construction, bacterial expression and characterization of a bifunctional single-chain antibody phosphatase fusion protein targeted to the human ERBB-2 receptor. Biotechnology 10:1128-1132; A. Traunecker等(1991) EMBO Journal10(12):3655-3659)。しかしながら、この努力は有望なものではなかった。
【0006】
単鎖二特異的又は二機能性抗体が細菌系において産生されている。しかしながら、この融合タンパク質は不活性形態で生産された(Haber等,1990)。更に、生産されたこの融合タンパク質は、結合親和性及び/又は結合活性の低下を示すか又は所望の生産物を回収するために複雑な単離及び精製方法を要している(Haber等, 1990; Wels等, 1992a)。
単鎖一価抗体及び単鎖二機能性抗体が述べられている(Wels 等, 1992b)。これらの抗体分子は、そのサイズを最小にし且つその機能修飾を可能にするために遺伝子操作したものである。更に、この二機能性抗体は、アルカリ性ホスファターゼ細菌遺伝子の3′をscFv 遺伝子に結合したという点でのみ二機能性である。これらの二機能性抗体は単一の結合ドメイン(例えば、VL +VH ) を含み、アルカリ性ホスファターゼ遺伝子をその標的に結合した抗体を検出するために単にマーカーとして用いたものである。
FvCD3及びCD4配列を含む Janusin分子が述べられている(A. Traunecker等 “Bispecific single chain molecules (Janusins) target cytotoxic lymphocytes on HIV infected cells, EMBO Journal 10(12):3655-3659 )。 Janusin構築物は、構築物のアミノ末端にCD4分子の一部及び構築物のカルボキシ末端にCD3の結合ドメイン(即ち、VL +VH ) を含んでいる。 Janusin分子は、CD3可変部をCD4分子部分から分離するらせんペプチドリンカーを含んでいない。更に、 Janusin分子は、多量体又は集合体にしばしば見られる。集合体形成を避けるために、追加の精製をしばしば必要とする。
【発明の開示】
【発明が解決しようとする課題】
【0007】
抗体産生に関する上記課題の観点から本発明が求められている。現在、抗体手法に伴う課題、即ち、大量の特異的抗体を得ることの難しさが継続している。歴史的には、抗体は血清又はマウス由来のハイブリドーマから得られていた。しかしながら、血清はたいてい量が限られ品質が変化しうるものであった。更に、マウス由来の抗体は、非マウス体にしばしば有害な免疫応答を起こす傾向があることから、ヒト治療に対する有用性が制限されている。
抗体手法を個々に悩ます課題、たいていは実質量の機能性タンパク質分子の生産に伴う課題を克服するために、生物学的に活性な融合タンパク質の発現を促進する新規な発現ベクターを本明細書に記載する。
【課題を解決するための手段】
【0008】
本発明は、二特異的融合タンパク質を提供する。
ここではさらに、一特異的又は二特異的融合タンパク質をコードする発現ベクターを提供する。
1実施態様においては、発現ベクターは一特異的融合タンパク質をコードし(例えば、図16)、このベクターは細胞表面抗原のような標的を結合することができる第1結合ドメインをコードするDNA配列を含む組換え体一特異的単鎖カセットを含む。
もう1つの実施態様においては、発現ベクターは二特異的融合タンパク質をコードし、このベクターは標的を結合することができる第1結合ドメインをコードするDNA配列及び標的を結合することができる第2結合ドメインをコードするDNA配列を含む組換え体二特異的単鎖カセットを含み、各ドメインは異種標的を結合することができる。
本発明は、また、哺乳動物細胞における生物学的に活性な一特異的又は二特異的融合タンパク質の生産方法を提供する。本方法は、(a)哺乳動物細胞に本発明の組換え体発現ベクターを移入し、(b)段階(a)で移入した哺乳動物細胞を培養し、(c)培養した哺乳動物細胞によって産生された生物学的に活性な二特異的融合タンパク質を回収することを含む。
【発明を実施するための最良の形態】
【0009】
定義
本明細書で用いられる語句の意味を下記に説明する。
本明細書で用いられる『二特異的融合タンパク質』は、少なくとも2種の異なる標的を別々に又は同時に特異的に認識及び結合する免疫学的に反応する任意の分子を意味し、単鎖として表される。
本明細書で用いられる『一特異的融合タンパク質』は、標的を特異的に認識及び結合し、2本の免疫グロブリン重鎖、2本の免疫グロブリン軽鎖又は免疫グロブリン重鎖及び/又は軽鎖及び/又はそのいずれかの部分を含む複合体である免疫学的に反応する任意の分子を意味し、単鎖として表される。
本明細書で用いられる『発現ベクター』は、(1)所望の遺伝子又はDNA配列を発現させるのに必要なプロモーター及び他の配列(例えば、リーダー配列)及び(2)所望の配列又はDNA配列を含む核酸分子を意味する。場合によっては、核酸分子は遺伝子転写物の安定性を高めるポリA及び/又は遺伝子の転写を増強するエンハンサー配列を含んでもよく、これにより遺伝子の発現が影響される。
本明細書で用いられる『結合ドメイン』は、標的の全結合領域又はその任意部分を認識及び結合する結合部位を意味する。具体例としては、(1)抗体の単一可変部(VL 又はVH )、(2)2種以上の可変部(例えば、VL +VH ;VL +VL ;又はVH +VH )又はその相補的決定領域(CDR)又は(3)抗原(例えば、白血球抗原)又はその一部があるが、これらに限定されない。
【0010】
本明細書で用いられる『分子垂れはし』は、本明細書で記載される融合タンパク質の検出及び精製を容易にすることができる分子をコードする任意のDNA配列を包含する。
本明細書で用いられる『二特異的単鎖カセット』は、標的を結合することができる第1結合ドメインをコードするDNA配列及び標的を結合することができる第2結合ドメインをコードするDNA配列を包含し、各ドメインは別々に又は同時に異種標的を結合することができ、DNA配列でコードされた両ドメインは同一カセット上にある。第1及び/又は第2結合ドメインは、2個の可変部(VL +VH 、VH +VL 、VL +VL 又はVH +VH )又は1個の可変部(VL 又はVH )であってもよい。また、第1及び/又は第2結合ドメインは、抗原又はその一部であってもよい。抗原の適切な例としては白血球抗原があるが、これに限定されない。第1結合ドメインは発現タンパク質のアミノ末端に又はその方向に位置し、一方第2結合ドメインは発現タンパク質のカルボキシ末端に又はその方向に位置する(二特異的単鎖DNAカセットのDNA配列の5′又は3′端に各々対応する)。
本明細書で用いられる『単鎖カセット』は、その標的の少なくとも一部を認識及び結合することがきるタンパク質をコードする配列を意味する。そのタンパク質は、複数の標的を認識及び結合するために複数部位を有してもよい。
【0011】
本明細書で記載した本発明を完全に理解するために、更に下記の説明を述べる。
A.ベクター
本明細書に記載される発現ベクターは、結合ドメインをコードするDNA配列(即ち、そのコーディング配列)又は調節配列、即ち、プロモーター又は発現プラスミド内で所望の遺伝子を発現させるのに必要な他の配列(例えば、リーダー配列)を全部又は部分的に置き換えることにより修飾することができる。
修飾した発現ベクター内の置換されたDNA配列は、任意の抗体又は他のレセプターの可変部をコードしてもよい。例えば、DNA配列は、BR96抗原、CD3、L6、CD28、CTLA4又はB7を認識及び結合する抗体の可変部をコードしてもよい。更に、DNA配列は、他の細胞表面抗原に結合することができる可変部をコードしてもよい。また、結合ドメインは、白血球抗原のような抗原の全部又は一部をコードしてもよい。具体的な置換配列を挿入するかについての主要な問題は、置換された配列が所望の結合ドメインを発現することができるように『フレーム内』に位置するかどうかである。
【0012】
置換された配列は、ポリメラーゼ連鎖反応(PCR)法でクローン化される。発現ベクターに挿入することができ、順次真核細胞を形質転換することによりDNA配列を発現することができる多数のDNA配列を作製するために、PCRが用いられる。特異的配列の増幅を達成する他のクローン化法、例えば、リガーゼ連鎖反応(LCR)を用いることもできる。
発現ベクターのプロモーターは、発現に用いられる細胞の種類又は挿入されるDNA配列により、他のプロモーターに容易に置き換えられる。プロモーターの適切な例としては、サイトメガロウイルス(CMV)、トリ骨髄芽球症ウイルス(AMV)及びモロニーマウス白血病ウイルス(MMLV)が挙げられる。
未変性の単量体としての抗体は、1分子当たり2本の相同な重鎖及び2本の相同な軽鎖を含む4連鎖の高分子である。各鎖は、可変(V)部と定常(C)部からなる。軽鎖の可変部(VL )は、可変部(V)と結合部(J)遺伝子でコードされ、重鎖の可変部(VH )は可変部(V)と介在多様性領域(D)を有する結合部(J)遺伝子でコードされる。VL +JL 又はVH +DH +JH 配列でコードされた各可変部フラグメント(VL 又はVH )は、約100個のアミノ酸で構成される。これらの配列内には、抗体の結合部位を作るアミノ酸を含むと思われる相補性決定領域(CDR)と呼ばれる3個の超可変部が含まれる。CDRは、骨格領域(FR)と呼ばれる可変性の極めて低い4個の領域に散在する。
【0013】
抗体の抗原結合部位は、典型的には、VL 及びVH 領域ポリペプチドをそれらのβプリーツシート構造に結合することにより形成され、CDR領域又はその近傍には鎖の間にループを含んでいる。時には、VL +VL 対又はVH +VH (例えば、G17−2軽鎖単量体)又はVL 又はVH 単独で抗原を結合することがある。
従って、『結合ドメイン』は、次の(a)免疫グロブリン(IgG、IgM又は他の免疫グロブリン)のVL +VH 領域、(b)免疫グロブリン(IgG、IgM又は他の免疫グロブリン)のVL +VL 領域、(c)免疫グロブリン(IgG、IgM又は他の免疫グロブリン)のVH +VH 領域、(d)免疫グロブリン(IgG、IgM又は他の免疫グロブリン)の単一VL 領域又は(e)免疫グロブリン(IgG、IgM又は他の免疫グロブリン)の単一VH 領域の1種又は組合わせを含む。
本発明による発現ベクターは、抗体配列を組込んでいるベクターに限定されない。抗原及びレセプターのような他の種類のタンパク質の結合ドメインをコードする配列を用いることもできる。即ち、ベクターは、複数の異なった標的を別々に又はほとんどは同時に結合することができる二特異的融合タンパク質をコードする。
【0014】
本発明の1実施態様においては、組換え体単鎖カセットは、異なった特異性を各々有する複数の(1)可変部及び/又は(2)抗原又はその一部をコードする複合DNA配列を含む。このカッセトにおいて、可変部をコードする配列は、(a)別の可変部又は(b)抗原をコードするDNAにリンカーで結合されることが好ましく、これらはすべて縦列に配置される。典型的には、そのようなリンカーはらせん構造である。らせんペプチドリンカーは、タンパク質分子を適切に折りたたむことができる。更に、らせんペプチドリンカーは、分子の溶解性を高めることができる。単一可変部単独で(VL 又はVH )発現するのと異なり、単鎖タンパク質として2個の可変部(VL +VH ;VL +VL ;VH +VH )は、個々の可変部を短いリンカー、例えば、(Gly4 Ser)3リンカーで連結するとを必要とする。
全領域の短いリンカーは、VL +VH (FV ) フラグメントを有する免疫グロブリン鎖を結合するために用いられる(Huston, J.S., Levinson, D., Mudgett-Hunter, M., Tai, M.S., Novotny, J., Margolies, M.N., Ridge, R.J., Bruccoleri, R.E., Haber, E., Crea, R., Oppermann, H.(1988) Protein engineering of antibody binding sites: recovery of specific activity in an antidigoxin single-chain Fv analogue produced in Escherichia coli. Proc. Natl. Acad. Sci. USA 85:5879-5883; Pluckthoen, 1991)。このリンカーは、タンパク質の折りたたみでは受動体である。一般に、このリンカーは親水性及び可撓性である。
【0015】
慣用的には、FV フラグメントを有する免疫グロブリン鎖の連結には数種の手段、即ち、化学的架橋(Glockshuber等, 1991);ジスルフィド結合による自然架橋(Glockshuber等, 1990a); ジスルフィド結合のない自然結合; 及び遺伝的にコードされたペプチドリンカーによる結合がある(Bird, R.E., Hardman, K.D., Jacobson, J.W., Johnson, S., Kaufman, B.M., Lee, S.M., Lee, T., Pope, S.H., Riordan, G.S., Whitlow, M.(1988) Single-chain antigen-binding proteins. Science 242:423-427; Huston 等, 1988a)。
単鎖カセットは、複数のリンカーを含んでもよい。リンカー数について主要な制約は、本発明のベクターでコードされた機能性融合タンパク質をなお発現することができるその数である。
本発明のらせんペプチドをコードするリンカー(配列番号10、11、12)は、その誘導体分子を作製するために修飾される、即ち、分子内のアミノ酸置換により修飾される。このような誘導体分子は、らせんペプチドリンカーの機能性を保持する。即ち、この置換を有する分子は、本発明の新規な発現ベクターによってコードされた生物学的に活性なタンパク質生産物をなお発現することができる。
【0016】
これらのアミノ酸置換としては、『保存的』として当該技術において既知のアミノ酸置換があるが、必ずしもこれに限定されない。
例えば、タンパク質においてしばしば『保存的アミノ酸置換』と称するある種アミノ酸置換をタンパク質の構造又は機能を変えずに行うことができることは、タンパク質化学の十分に確立された原理である。このような置換としては、疎水性アミノ酸のいずれかをイソロイシン(I)、バリン(V)及びロイシン(L)のいずれかに;グルタミン酸(E)をアスパラギン酸(D)に又はその逆に;アスパラギン(N)をグルタミン(Q)に又はその逆に;及びトレオニン(T)をセリン(S)に又はその逆に置換することが挙げられる。
他の置換も個々のアミノ酸の環境及びタンパク質の三次構造におけるその役割によっては保存的とみなすことができる。例えば、アラニンとバリン(V)のように、グリシン(G)とアラニン(A)もしばしば交換される。
【0017】
相対的に疎水性のメチオニン(M)は、ロイシン及びイソロイシンとしばしば交換され、時にはバリンと交換される。リシン(K)及びアルギニン(R)は、アミノ酸残基の重要な特徴がその電荷であり、これらの2つのアミノ酸残基のpKの違いが重要でない場所ではしばしば交換される。また、他の置換も個々の環境において『保存的』とみなすことができる。
軽鎖及び/又は重鎖配列を有する抗体可変部及び非抗体結合ドメインを含む融合タンパク質をコードするベクターは、本発明に包含される。第1及び/又は第2結合ドメインは、抗体の可変部であってもよい。また、第1及び/又は第2結合ドメインは、抗原又はその一部であってもよい。抗原の一部は、その部分が認識され、認識後これに分子が結合することができるものを包含することが好ましい。例えば、トランスメンブランタンパク質抗原においては、好ましい部分は細胞外部分である。しかしながら、他の部分も本発明に包含される。
適切な抗原及びレセプターの例としては、CD及び非CD分子があるが、これらに限定されない。
【0018】
CD分子としては、CD1、CD2、CD3/TcR、CD4、CD5、CD6、CD7、CD8、CD9、CD10、CD11、CD12、CD13、CD14、CD15、CD16、CDw17、CD18、CD19、CD20、CD21、CD22、CD23、CD24、CD25、CD26、CD27、CD28、CD29、CD30、CD31、CD32、CD33、CD34、CD35、CD36、CD37、CD38、CD39、CD40、CD41、CD42a、b、CD43、CD44、CD45、CD46、CD47、CD48、CD49、CDw50、CD51、CDw52、CD53、CD54、CD55、CD56、CD57、CD58、CD59、CDw60、CD61、CD62、CD63、CD64、CDw65、CD66、CD67、CD68、CD69、CDw70、CD71、CD72、CD73、CD74、CDw75、CD76、CDw78が挙げられるが、これらに限定されない。
非CD分子としては、B7、B7(2)、CTLA4、BR96、GP39、LFA−3、ICAM−2及びインターロイキン(IL)1−8が挙げられるが、これらに限定されない。
【0019】
例えば、CD28抗原は、ほとんどの成熟ヒトT細胞(Damle等(1983) J.Immunol. 131:2296-2300)に見られる免疫グロブリンスーパーファミリーのホモ二量体糖タンパク質である(Aruffo, A., Seed, B.(1987) Molecular cloning of a CD28 cDNA by a high-efficiency COS cell expression system. Proc. Natl. Acad. Sci. USA 84:8573-8577)。CD28抗原と反応するモノクローナル抗体(mAb)は、種々のポリクローナル刺激により起こされるT細胞応答を増加することができる。相同分子、CTLA4は、マウス細胞溶解T細胞cDNAライブラリーの微分スクリーニングにより同定されている (Brunet等(1987) Nature 328:267-270)。
本発明の発現ベクターは、多特異的融合タンパク質をコードすることができるベクター、即ち、多数の標的と反応することができる分子を包含する。例えば、本発現ベクターは、単鎖に発現することができる三特異性融合タンパク質、即ち、3種の標的を認識及び結合する融合タンパク質をコードしてもよい。また、融合タンパク質は、4種の標的を認識及び結合することができる。
【0020】
例えば、1実施態様においては、本発明のベクターによってコードされた一特異的融合タンパク質は、図16の単鎖抗体と同じものである。
本発明の1実施態様においては、発現ベクターは、(1)抗体又は細胞表面抗原の第1結合ドメインをコードするDNA配列、(2)抗体又は細胞表面抗原の第2結合ドメインをコードするDNA配列、(3)第1結合ドメインをコードするDNA配列及び第2結合ドメインをコードするDNA配列を連結するらせんペプチドをコードするリンカー及び(4)一又は二特異的融合タンパク質の検出のための分子垂れはしをコードするDNA配列を含む。
分子垂れはしは、抗体、センス又はアンチセンス相補的分子、酵素等の分子垂れはしを認識及び結合する適切な分子により同定される。分子垂れはしの例としては、Fc フラグメント、HIVフラグメント及び赤血球凝集素エピトープ配列HAIが挙げられる (Pati等(1992) Gene 114 (2):285-8)。
好ましい実施態様によれば、発現ベクターは、(1)抗体又は細胞表面抗原の第1結合ドメインをコードするDNA配列、(2)抗体又は細胞表面抗原の第2結合ドメインをコードするDNA配列、(3)第1結合ドメインをコードするDNA配列及び第2結合ドメインをコードするDNA配列を連結するらせんペプチドをコードするリンカーを含み、該ドメインの各々が同種又は異種標的又は抗原を結合することができる組換え体二特異的単鎖DNAカセットを含む。
【0021】
本発明の実験によれば、第1及び/又は第2結合ドメインは細胞表面抗原又は白血球抗原と反応することができる。細胞表面抗原としては、CD3、L6、CD28、CTLA4、CD40又はB7のような分子が挙げられるが、これらに限定されない。即ち、第1及び/又は第2結合ドメインはCD3と反応することができる。また、第1及び/又は第2結合ドメインはL6と反応することもできる。更に、第1及び/又は第2結合ドメインはCD28と反応することもできる。更に、第1及び/又は第2結合ドメインはB7と反応することもできる。更に、第1及び/又は第2結合ドメインはCD40と反応することもできる。
好ましい実施態様においては、発現ベクターはCC9−2と称し、ATCC No.69235 として大腸菌プラスミド内でアメリカンティッシュカルチュアコレクション(ATCC)に寄託されている(CC9−2はpCDM8内にL6VL リーダー配列−CD3sFV −リンカー−L6sFV −免疫グロブリンFC を有する) 。任意のDNA配列が用いられることは当業者に明らかである。適切な配列及び発現のための正しい読み枠のためのカセットとして挿入されるように、用いられるべき可変部又は分子のコーディング配列が既知でなければならないことのみが要求される。
【0022】
更に、任意の抗体の可変部をコードするDNA配列及びリガンドをコードするDNA配列を含む組換え体二特異的単鎖カセットを含む二特異的融合タンパク質をコードする発現ベクターも提供される。そのリガンドの例としては、B7、CTLA4、CD28、CD40、CD3が挙げられるが、これらに限定されない。リガンドの他の例としては、任意の白血球抗原がある。
例えば、本発明は、CD3と反応するドメインをコードするDNA配列及びL6と反応するドメインをコードするDNA配列を含む組換え体二特異的単鎖カセットを含む二特異的融合タンパク質をコードする発現ベクターを提供する。第1又は第2結合ドメインがCD3と反応してもよい。第1又は第2結合ドメインがL6と反応してもよい。
本発明は、更に、B7の少なくとも一部(例えば、細胞外部分)であるドメインをコードするDNA配列及びL6と反応するドメインをコードするDNA配列を含む組換え体二特異的単鎖カセットを含む二特異的融合タンパク質をコードする発現ベクターを提供する。
【0023】
本発明は、更に、CTLA4と反応するドメインをコードするDNA配列及びL6と反応するドメインをコードするDNA配列を含む組換え体二特異的単鎖カセットを含む二特異的融合タンパク質をコードする発現ベクターを提供する。第1又は第2結合ドメインがCTLA4と反応してもよい。第1又は第2結合ドメインがL6と反応してもよい。
また、本発明は、CD28と反応するドメインをコードするDNA配列及びL6と反応するドメインをコードするDNA配列を含む組換え体二特異的単鎖カセットを含む二特異的融合タンパク質をコードする発現ベクターを提供する。第1又は第2結合ドメインがCD28と反応してもよい。第1又は第2結合ドメインがL6と反応してもよい。
例えば、本発明は、CD3と反応するドメインをコードするDNA配列及びB7の細胞外部分をコードするDNA配列を含む組換え体二特異的単鎖カセットを含む二特異的融合タンパク質をコードする発現ベクターを提供する。
【0024】
本発明によれば、発現ベクター及び生物学的に活性な二特異的融合タンパク質の生産方法が提供される。一般に、これらの方法は、(1)抗体又は他の結合タンパク質を合成するB細胞ハイブリドーマ又は他の培養細胞からmRNAを単離し、(2)逆転写によりcDNAを合成し、(3)PCRプライマーとしてアンカー末端のオリゴヌクレオチド又は変性したオリゴヌクレオチドを用いて可変部のような結合ドメインをクローン化し、(4)そのクローン化した可変部の配列を決定し、(5)可変部遺伝子間の遺伝子融合を構築し、(6)その構築された可変部をSalI及びBclIで切断したpUCIgベクター(又は他の適切なベクター)に挿入し、(7)適切な断片配置のクローンをスクリーニングし、そのクローンの配列を決定し、(8)そのクローンをpCDM8又はpiLNXAnのような適切な発現ベクターに移し、(9)DEAE−デキストラン法のような手法でCOS細胞に移入し、(10)その移入された細胞を発現されるべき融合タンパク質に十分な時間、典型的には約72時間インキュベートし、(11)生物学的に活性な二特異的融合タンパク質を生産するためにFITCヤギ抗ヒトIgG及び/又はELISAを用いたファクスでその細胞をスクリーニングすることを含む。
【0025】
B.生物学的に活性な二特異的融合タンパク質の生産方法
生物学的に活性な二特異的融合タンパク質の生産方法を提供する。本方法は、二特異的融合タンパク質を生産するために、本発明の発現ベクターを移入した細胞を培養し、生産されたタンパク質を回収することを含む。
本発明は、更に、哺乳動物細胞における生物学的に活性な二特異的融合タンパク質の生産方法を提供する。本方法は、(a)本発明の発現ベクターを哺乳動物細胞に移入し、(b)段階(a)で移入した哺乳動物細胞を培養し、(c)培養した哺乳動物細胞によって産生された生物学的に活性な二特異的融合タンパク質を回収することを含む。
生物学的に活性な二特異的融合タンパク質の回収方法は、(a)分子垂れはしの存在によって生物学的に活性な二特異的融合タンパク質を同定し、(b)培養した哺乳動物細胞によって産生された生物学的に活性な二特異的融合タンパク質を回収するために、同定した分子垂れはしのある生物学的に活性な二特異的融合タンパク質を分子垂れはしのない分子から分離することを含む。
【0026】
下記実施例においては、哺乳動物由来の細胞が用いられるが、本発明の実施において任意の真核細胞が有効である。例としては、ヒト細胞、例えば、線維芽細胞及びヒツジ、ブタ、マウス、ウシのような他の動物由来の細胞が挙げられる。哺乳動物細胞の具体例としては、COS、HeLa、CHO、DUX、B11、Sp2/0、W138、DHK及びHEPG2細胞が挙げられる。
生物学的に活性な二特異的融合タンパク質が生体内及び生体外診断及び治療に有用な検出マーカー及び治療用薬剤に結合されることは明らかである。その生物学的に活性な二特異的融合タンパク質を結合することができる検出マーカーの中には、酵素、常磁性イオン又は化合物、アビジン−ビオチン特異結合対のもの、蛍光団、発色団、化学発光団、重金属及び放射性同位元素がある。生物学的に活性な二特異的融合タンパク質を結合することができる治療用薬剤の中には、抗腫瘍剤、リンホカイン及び毒素がある。
【0027】
C.本発明の発現ベクターを移入した細胞
発現ベクターの哺乳動物細胞への導入は、リン酸カルシウム移入(Graham, Vander Eb.(1973) Virol. 52:456-467)、DEAE−デキストラン移入 (Lopata, M.A.等,(1984) Nucl. Acids Res. 12:5707) 及び電気穿孔法 (Potter, H.等 (1984) PNAS 81:7161) により行われる。移入方法の選択は、何の種類の移入が行われるかに幾分左右される。電気穿孔法及びCaPO4 移入は、共に、安定に組込まれたDNAを含むセルラインを効率よく生産するために用いられる。電気穿孔法はたいてい浮遊培養を用いて容易に行われ、CaPO4 移入はたいてい粘着細胞を用いて容易に行われる。DEAE−デキストラン移入は安定なセルラインを生産する際には十分に作用しないが、一過性のプロトコールに用いる際にはCaPO4 移入より再現性が高い。他の移入方法も当該技術において既知である。
本明細書に記載される方法で生産された発現ベクター及び新規なタンパク質は上記可変部に限定されず、一般に、任意の単鎖二特異的融合タンパク質の構築、発現及びスクリーニングに適用できる。
【0028】
D.本発明のベクターによってコードされた融合タンパク質の使用
分子垂れはしのない抗L6及び抗CD3単鎖誘導体は、L6又はCD3関連の疾患を治療又は検出するための治療又は診断に有効である。例えば、L6sFvは検出用放射性核種に化学的に結合することができ、また治療用PE40のような毒素に遺伝的に融合することができる。
短いsFvフラグメントの方が腫瘍塊をよりよく浸透することができ且つ腫瘍部位の局在を改良したので、治療用薬剤を高レベルのL6抗原を発現する腫瘍細胞に対して標的とするには少量のL6sFvが効果的である(Colcher, D., Bird, R., Roselli, M., Hardman, K.D., Johnson, S.,Pope, S., Dodd, S.W., Pantoliano, M.W., Milenic, D.E., Schlom, J.(1990) In vivo tumor targeting of a recombinant single-chain antigen-binding protein. J. Nat. Cancer Inst. 82:1191-1197; Yokota, T., Milenic, D.E., Whitlow, M., Schlom, J.(1992) Rapid tumor penetration of a single-chain Fv and comparison with other immunoglobulin forms. Cancer Res. 52:3402-3408 )。
【0029】
CD28の連結反応のように第2シグナルなしのT細胞レセプターシグナルの送達はT細胞アネルギーを招くことがあるので、CD3sFvは生体内免疫抑制の誘導に十分適している。OKT3のF(ab′)2フラグメントが免疫抑制するとともに全抗体OKT3誘導免疫抑制に関与するサイトカイン毒性を著しく低下させることが証明されている(Woodle, E.S., Thistlethwaithe, J.R., Ghobrial, I.A., Jolliffe, L.K., Stuart, F.P., Bluestone, J.A. (1991) OKT3 F(ab′)2 fragments: retention of the immunosuppressive properties of whole antibody with marked reduction in T cell activation and lymphokine release. Transplantation 52:354-360)。
【0030】
おもしろいことに、CD3単鎖単一特異的抗体誘導体が未変性抗体と異なった機能特性を示した。CD3Fv−Ig誘導体は、PLCγ1のチロシンリン酸化を強力に誘発し、未変性抗CD3mAbに比べてPLCγ1とpp35/35との結合を増加させる。この結合は、T細胞を最大限に刺激するように増加することが示されている(Kanner,S.B., Deans,J.P., Ledbetter,J.A.(1992a) Regulation of CD3-induced phospholipase C-γ1 (PLCγ1) tyrosine phosphorylation by CD4 and CD45 receptors. Immunology 75:441-447; Kanner,S.B., Ledbetter,J.A.(1992b) CD45 regulates TCR-induced signalling through tyrosine phosphorylation of phospholipase Cγ1. Biochem. Soc. Trans. 20:178-184 )。更に、T細胞のCD3Fv−Igによる刺激は、活性化を伴う細胞タンパク質のより全体のチロシンリン酸化をもたらした。この分子は未変性mAbより小さいので、CD3−ε上の結合ドメインがより利用され且つTCR/CD3結合チロシンキナーゼとの相互作用を改良することが可能である。
【0031】
動物モデル及びヒトにおける臨床試験において、抗TCR又は抗CD3mAbは標的細胞抗原に架橋する際に標的細胞に対してT細胞応答を強めることが示されている。異種複合又は二特異的mAbは、細胞障害性Tリンパ球又はリンパ球活性化キラー細胞を活性にして悪性標的細胞 (Staerz等, 1986a; Perez等, 1985a; Perez, P., Hoffman, R.W., Titus, J.A., Segal, D.M. (1986) Specific targeting of human peripheral blood T cells by heteroaggregates containing anti-T3 crosslinked to anti-target cell antibodies. J. Exp. Med. 163:166-178; Liu等, 1985a; Staerz 等, 1986b)又はウイルス感染標的細胞(Paya, C.V., McKean, D.J., Segal, D.M., Schoon, R.A., Schowalter, S.D., Leibson, P.J. (1989) Heteroconjugate antibodies enhance cell-mediated anti-herpes simplex virus immunity. J. Immunol. 142:666-671; Zarling, J.M., Moran, P.A., Grosmaire, L.S., McClure, J., shriver, K., Ledbetter, J.A.(1988) Lysis of cells infected with HIV-1 by human lymphocytes targeted with monoclonal antibody heteroconjugates. J. Immunol., 140:2609-2613; voss, L.M., David, C.S., Showalter, S.D., Paya, C.V., Liebson, P.J.(1992) Heteroconjugate antibodies enhance cell-mediated anti-herpes simplex virus immunity in vivo. Int. Immunol. 4:417-420) を破壊する。
【0032】
抗CD3mAbは強力なサイトカイン毒性を生体内で誘導することができる(Abramowicz, D., Schandene, L., Goldman, M., Crusiaux, A., Vereerstraeten, P., De Pauw, L., Wybran, J., Kinnaert, P., Dupont, E., Toussaint, C.(1989) Release of tumor necrosis factor, interleukin-2, and gamma interferon in serum after injection of OKT3 monoclonal antibody in kidney transplant recipients. Transplantation 47:606-608) が、二特異的mAbで治療した患者は、患者のT細胞を試験管内で試薬で前処理してから再投与したので、サイトカイン毒性を生じない(Mezzanzanica, D., Canevari, S., Colnaghi, M.I.(1991) Retargeting of human lymphocytes against human ovarian carcinoma cells by bispecific antibodies: from laboratory to clinic. Int. J. Clin. Lab. Res. 21:159-164; Nitta, T., Sato, K., Yagita, H., Okumura, K., Ishii, S. (1990) Preliminary trial of specific targeting therapy against malignant glioma. Lancet 335:368-371)。マウスモデルにおいて、F(ab′)2フラグメントのような未変性mAbより小さい分子は、一特異的抗腫瘍特異性及び二特異的抗CD3、抗腫瘍特異性の双方に対して腫瘍局在の増加を示している(van Dijk, J., Zegveld, S.T., Fleuren, G.J., Warnaar, S.O.(1991) Localization of monoclonal antibody G250 and bispecific monoclonal antibody CD3/G250 in human renal cell carcinoma xenografts: relative effects of size and affinity. Int. J. Cancer 48:738-743; Nelson, H., Ramsey, P.S., Kerr, L.A., McKean, D.J., Donohue, J.H.(1990) Regional and systemic distribution of anti-tumor x anti-CD3 heteroconjugate antibodies and cultured human peripheral blood lymphocytes in a human colon cancer xenograft. J. Immunol. 145:3507-3515)。
【0033】
CD3−L6FvIg分子は、新規な分子として哺乳動物がん治療、例えば、ヒトがん治療に有効である。この分子は、CD3及び腫瘍抗原L6双方の結合特異性を含み、L6陽性腫瘍細胞の存在下にT細胞増殖を誘導し且つこれらの細胞に対してCTL破壊を特定することが試験管内で示されている。この2つの異なった結合特異性は、ヒトIgG1のヒンジ、CH2及びCH3ドメインに結合して初めは垂れはしとして二特異的分子の確認及び精製のために作用する。この垂れはしは、これ自体垂れはしの付いた二特異的タンパク質の小さな全サイズが未変性IgGの約2/3まで残る相対的に小さなドメイン(約50kDa)である。この垂れはしはヒト由来であるので、この分子の全体の免疫原性は全マウスIgGより著しく低いことが予想される。骨格領域を人化し、また、既知のヒトタンパク質のらせんに似るようにらせんリンカーの溶媒接触部分を構築するような二特異的タンパク質の免疫原性を更に低下させるための追加の修飾も行われる。Ig部分は親和性尾部として作用したが、生体内で二特異的タンパク質の半減期を増加させることもできる。
【0034】
1報告書において、ヒトIgG1定常部に融合したキメラマウス抗結腸直腸がんmAb可変部(Steplewski, Z., Sun, L.K., Sherman, C.W., Ghrayeb, J., Daddona, P., Koprowski, H.(1988) Biological activity of human-mouse IgG1, IgG2, IgG3, and IgG4 chimeric monoclonal antibodies with antitumor activity. proc. Natl. Acad. Sci. USA 85:4852-4856) を転移結腸がんの10人の患者に投与すると、循環時間が6倍増加することが示されている(LoBuglio, A.F., Wheeler, R.H., Trang, J., Haynes, A., Rogers, K., Harvey, E.B., Sun, L., Ghrayeb, J., Khazaeli, M.B. (1989) Mouse/human chimeric monoclonal antibody in man: kinetics and immune response. Proc. Natl. Acad. Sci. USA 86:4220-4224)。
更に、本発明の二特異的融合タンパク質に用いたIg尾部は、CH2ドメインで変異される(残基238でプロリンからセリンに)。この変異は、ヒトIgG1尾部とFcレセプターとの相互作用により仲介されるADCC活性を切断する。これは、生体内でCD3陽性T細胞とFcレセプターをもつ細胞との間の『相互的』破壊を防止するにちがいない(Clark等, 1987a)。最後に、二特異的分子はCOS細胞の一過性移入から発現されるが、タンパク質は安定な移入系で発現されることが可能である。
【0035】
本発明の利点: 本発明は、現在の抗体産生方法に伴う課題を克服する。
二特異的融合タンパク質を生産するに当たり他のもので直面した課題を克服するために、既存のCOS細胞発現系を適応して組換え体二特異的単鎖カセットDNAから機能性単鎖抗体誘導体を分泌することを達成した。単鎖抗体は、ヒトIgG1のFcドメインをヒト抗原(例えば、CD3及びL6)に対するマウス抗体の可変部に融合することにより構築した。Fc領域は、種々の特異性を有する融合タンパク質の同定及び精製に便利な分子垂れはしとして作用した。抗体のこのセグメントによって与えられるエフェクター機能は、ある種の治療に有効であある。
生物学的に活性な分子の回収は、単一結合特異性を有する分子の場合でさえ問題のある細菌発現系と異なり、本発明は、発現した生物学的に活性な融合タンパク質を含む培養上清を生じ、次いで、慣用的なアフィニティークロマトグラフィーで精製されるCOS細胞からの一過性発現を提供する。おもしろいことに、生産された単鎖二特異的融合タンパク質分子は、親抗体と異なった特性を示す。
【0036】
生物学的に活性な分子の回収が単一結合特異性のみを有する分子の場合でさえ問題があることから、これらの二特異的融合タンパク質分子の哺乳動物発現が選ばれた。哺乳動物発現により抗体誘導体の簡便で急速な産生が得られ、分子間の確認、評価及び比較が相対的に短時間で可能なためには重要なことである。
個々のタンパク質ドメインが相互交換可能な組換え体二特異的単鎖カセットDNA上にある遺伝子融合は、新規な組合わせを作り、急速な交換を行い且つ所望の機能を行うのにそれらの効能に対して種々のドメインをスクリーニングするための可能性が生じる。一過性移入系における構築物の発現は、初めの組換え及びスクリーニング段階の場合の他の生産方法より好ましい。このスクリーニングから所望のサブセットの特徴を示す分子だけが、大量生産用の第2発現系にシャトルすること、長いあるいは複雑な作製を排除すること及び作成した大多数の分子の単離操作を必要とする。
【0037】
相互交換可能なDNAカセットを有する組換え体二特異的単鎖DNAカセット、即ち、単鎖可変部又は別のものに交換することができる他の結合ドメインをコードするDNAカセットとしてより大きな分子に構築された抗体結合部位の急速な構築、発現及び分析系を開発することに着手した。第1及び第2結合ドメインの配列は、置換あるいは相互交換することができる。異種の配列は既存のものと置き換えてもよい。
前述の構築物と異なり、第1又は第2結合ドメインのいずれか又は双方に任意の可変部を配置することができるので、この二特異的単鎖カセットは有用である。
抗体構造ドメインの新規な組合わせを構築することにより、例えば、2つの関連のない結合特異性及び所望のエフェクター機能を結合する分子が作製され、治療可能性が改良される。そのような二特異的単鎖抗体誘導体が2つの非相互作用細胞表面レセプターのアダプター分子として作用すると人工レセプター−リガンド対を生じる。
【0038】
ここでのデータは、組換え体二特異的単鎖DNAカセットから発現された二特異的融合タンパク質が、多くの組織培養操作又はCD3に対する毒性一特異的抗体の可能性をなくす精製を必要としないで生体内及び生体外でヒト腫瘍発現L6に対するT細胞による細胞障害性応答を標的とすることができることを示している。
腫瘍細胞結合及びT細胞結合及び活性化に対する特異性を結合する単鎖抗体誘導体は、ヒト疾患に対する単一のモノクローナル抗体に基づく治療より著しい改良を与える。ここに記載した遺伝子融合の設計、構築、発現及び試験の方法は、関連のない分子の機能性ドメイン間の新規な組合わせを単一分子内で共に機能する能力について試験する場合、迅速性、簡便さ及び再現性において著しい利点を与える有用なものである。
下記の実施例において、本発明を具体的に説明する。実施例は本発明を更に理解するために記載されるが、前記特許請求の範囲で記載した本発明を限定するものではない。
【実施例1】
【0039】
材料及び方法:
発現ベクターの修飾:
スタッファーフラグメントを得られた融合タンパク質に対してある機能特性を与えるいくつかの短いスタッファーフラグメントに置き換えることにより、プラスミドpCDM8及びpiLNXAnを修飾した。
哺乳動物発現ベクターは、ベクターの上部に沿って示されている修正と追加が行われた図で表される。融合カセットの発現はCMVプロモーターによって進み、細菌及び哺乳動物細胞における複製はベクターの下部に示された適切な起源によって得られる。ベクターは、適切な宿主細菌株内のp3プラスミドに存在するアンピシリン及びテトラサイクリン遺伝子のナンセンス変異を抑圧するsupF遺伝子を含む。終結及びポリA付加シグナルは、SV40から適切な領域で設けられる。
【0040】
スタッファー領域の5′端のHindIII部位を用いて抗L6抗体(本明細書では抗L6とも呼ばれる)の軽鎖可変部から得られた融合タンパク質を分泌するためのリーダー配列を含むHindIII−SalIカセットを挿入した(図1)。この配列は、HindIII及びSalI付着端突出部分を有する72−mer相補的オリゴヌクレオチドにコードされた。使用したセンスオリゴヌクレオチドはL6VL-LP5/AGC TTA TGG ATT TTC AAG TGC AGA TTT TCA GCT TCC TGC TAA TCA GTG CTT CAG TCA TAA TGT CCA GAG GAG(配列番号1)であり、相補的オリゴヌクレオチドはL6VL-LP3/TCG ACT CCT CTG GAC ATT ATG ACT GAA GCA CTG ATT AGC AGG AAG CTG AAA ATC TGC ACT TGA AAA TCC ATA (配列番号2)であった。センスオリゴヌクレオチドをポリヌクレオチドキナーゼでリン酸化し(Boehringer-Mannheim, Indianapolis, ID) 、連結反応の前に、以前に発表された方法(Sambrook, J., Fritsch, E.F., Maniatis, T.(1989) Molecular cloning - a laboratory manual, second edition. ISBN 0-87969-309-6) に従って補体にアニールした。オリゴヌクレオチドの1本だけをキナーゼして複数の縦列挿入を防止した。
【0041】
更に、スタッファーフラグメントの3′端のXbaI部位を用いてカルボキシル末端のBclI及びXbaI制限部位に隣接した分子垂れはし又は簡単な停止コドンを挿入した(図1)。試験した分子垂れはしは、ヒトIgG1、gp110のV3ループからのヒト免疫不全ウイルス(HIV)ペプチド、FLAGペプチド及びヒトC−κからの定常部ドメインを含んだ。ヒトIgG1配列を、骨髄腫発現ヒト−マウスキメラL6のRNAからの結合逆転写(トリ骨髄芽球症ウイルス; Life Sciences, St. Petersburg, FL)及び/又はPCR反応によりキメラL6トランスフェクトーマのRNAから単離した。ヒンジあるいはCH2領域に適切な変異を含む前進プライマーを用いてPCR反応からFcドメインの各種変異誘導体を構築した。
【0042】
2つの異なった結合特異性、ヒトL6腫瘍抗原及びヒトCD3−εの可変部を挿入及び発現することにより、修飾発現ベクターを試験した。単鎖抗体誘導体は、融合される分子垂れはしによって、種々の結合活性及び親和性を有する抗原を結合した。CH1及びCκドメインなしにもかかわらず、ヒトIgG1のFcドメインが未変性抗体の結合特性を再現する点で最も満足すべき垂れはしであった。他の各種垂れはしを構築及び試験したが、Cκ(Traunecker, A., Lanzavecchia, A.M., Karjalainen, K.(1991) Bispecific single chain molecules(Janusins)target cytotoxic lymphocytes on HIV infected cells. EMBO J. 10:3655-3659) 及びFLAGペプチドは共に確実に機能しなかった。ヒト免疫不全ウイルスによってコードされたgp110のV3ループからのペプチドRKSIRIQRGPGRAFVTIGKI(配列番号3)も親和性尾部として用い、ペプチド特異的mAb110.3によって認識された。この垂れはしは融合されるFvによって可変結果を生じ、L6に融合される際には適切に機能せず、CD3に融合される際には満足に機能した。
【0043】
ペプチド29(RKSIRIQRGPGRAFVTIGKI)(配列番号3)は、HIVのgp110のV3ループセグメントに相当する(HIVペプチド)。付着末端突出部分を有する2本の76−mer相補的オリゴヌクレオチドをアニールして、分子垂れはしを作った。センスオリゴヌクレオチドはBclI突出部分、V3ループ配列及び停止コドンHIVSTOP5 GA TCA AGA TCC GCG GAA ATC GAT TAG AAT CCA GAG AGG CCC TGG GCG CGC CTT CGT TAC GAT CGG CAA GAT CTA GT(配列番号4)を含み、相補的プライマーはXbal突出部分HIVSTOP3/CTA GAC TAG ATC TTG CCG ATC GTA ACG AAG GCG CGC CCA GGG CCT CTC TGG ATT CTA ATC GAT TTC CGC GGA TCT T(配列番号5)を含んだ。センスオリゴヌクレオチドをリン酸化し、非リン酸化逆プライマーにアニールした後、上記BclI−XbaI消化ベクターに連結した。
アミノ末端垂れはしとして有用なIBI製のFLAGペプチドが垂れはしの付いたタンパク質のカルボキシル末端に配置した際になお作用するかを試験した。次の51−mer配列:FLAG5/GAT CAA GAC TAC AAG GAC GAC GAT GAC AAG TGA GCG GCC GCG AAT TCG TC(配列番号6)及びFLAG3/CTA GAG ACG AAT TCG CGG CCG CTC ACT TGT CAT CGT CGT CCT TGT AGT CTT(配列番号6)を含む相補的オリゴヌクレオチドを設計した。これらの配列をキナーゼし、アニールし、抗体結合ドメインに対するベクター末端に連結した。ヒトCκ配列を上記のようにキメラL6RNAから逆転写及びPCRにより得た。Cκを単離するための前向きプライマーはL6CK5BCL/GGT GCT CTG ATC ACT GTG GCT GCA CCA TCT GTC TTC ATC (配列番号8)であり、逆向きプラマーはL6CK3XBA/CCT CCT CAT TCT AGA CTA ACA CTC TCC CCT GTT GAA GCT (配列番号9)であった。
【0044】
CD3とL6結合ドメインの間に組換え体二特異的単鎖カセットを作るために用いられるらせんペプチドリンカーをGATC付着端突出部分を有する78−mer相補的オリゴヌクレオチドにコードした。センスオリゴヌクレオチドは、Fvlink1/GAT CAA TCC AAC TCT GAA GAA GCA AAG AAA GAG GAG GCC AAA AAG GAG GAA GCC AAG AAT CTA ACA GCC TCG AGA GC (配列番号10)であり、アンチセンスオリゴヌクレオチドはFvlink2/GAT CGC TCT CGA GGC TGT TAG ATT TCT TGG CTT CCT CCT TTT TGG CCT CCT CTT TCT TTG CTT CTT CAG AGT TGG ATT(配列番号11)である。コードされたペプチドは親水性であり、分子の溶解性を高める荷電したアミノ酸(DQSNSEEAKKEEAKKEEAKKSNSLESL)(配列番号12)に富んでいる。配列モチーフ (EEAKK)n は、荷電したアミノ酸に富んでいるために特に親水性である。
PCR反応(100μl の全反応量中)は、20μモル各dNTP、50−100ピコモルプライマー、1−10ng鋳型及びTaqポリメラーゼ (Stratagene又はBoehringer-Mannheim)を含むTaqポリメラーゼバッファー(Stratagene, Torrey Pines, CA 又はBoehringer-Mannheim)中で行った。反応は、パーキン−エルマーセタスターミナルサイクラーを用いて、典型的には94℃で1分、55℃で1分及び72℃で1分の工程からなる30サイクルプログラムで行った。連結産物をMC1061/p3に形質転換し、適切な挿入プラスミドのコロニーをスクリーンした。陽性クローンをDNA配列決定及びミニ移入により検定した。
【0045】
V領域の単離:
キメラL6トランスフェクトーマのRNAをNP−40急速溶菌法を用いて単離し、重鎖及び軽鎖双方の全長DNAをL6及びヒト定常部として発表されている配列と同じプライマーを用いて増幅した(Hieter, P.S., Maz, E.E., Seidman, J.G., Maizel, J.V. Jr., Leder, P.(1980) Cloned human and mouse kappa immunoglobulin constant and J region genes conserve homology in functional segments. Cell 22:197-207; Liu 等(1987) Proc. Natl. Acad. Sci. USA 84:3438-3442) が、制限部位をクローン化のために結合した。これらの全長cDNAを鋳型として第2PCR反応に用いて可変部をサブクローン化した。1実施例においては、可変部のサブフラグメントを、特異的プライマーよりむしろランダム六量体を用いて作ったcDNAからPCRによりクローン化した。
【0046】
G19−4細胞由来RNAをNP−40急速溶菌法を用いて抽出した。抗CD3ハイブリドーマG19−4のVL 及びVH 配列を確立されたPCR法を用いて増幅した(Orlandi, R., Gussow, D.H., Jones, P.T., Winter, G.(1989) Cloning immunoglobulin variable regions for expression by the Polymerase Chain Reaction. Proc. Nat. Acad. Sci. 86:3833-3837)。AMV逆転写酵素(Life Sciences) 及び重鎖又は軽鎖の定常部に相補的なプライマーを用いて第1鎖cDNA合成を行った。第1鎖cDNA産物を末端トランスフェラーゼ(Stratagene, Torrey Pines, California)を用いてポリGにつないだ。次いで、100ピコモルの各プライマー及び第1鎖合成からの1−2μl の精製したG末端のcDNA遺伝子の完全なcDNAを用いてPCRを行った。ANCTAIL前向きプライマーはナンセンスDNA配列及びアンカー配列に相補的なポリC配列を含み、ANC−ER前向きプライマーはEcoRI部位及びアンカー部位の上流のナンセンス配列を含み、MHγC及びMCK−3逆向きプライマーは各々重鎖及び軽鎖の定常部に相補的な第1鎖プライマー内の配列を含んだ。プライマー配列は次の通りであった。
ANCTAIL: 5′-GCATGTGCAAGTCCGATGAGTCCCCCCCCCCCCCC-3′ 配列番号13、
ANC-ER: 5′-ACGTCGAGAATTCGCATGTGCAAGTCCGATGAGTCC -3′ 配列番号14、
MHγC: 5′-A(TC)CTCCACACACAGG(AG)(AG)CCAGTGGATAGAC-3′ 配列番号15、
MCK-1: 5′-CTTCCACTTGACATTGATGTCTTTG-3′ 配列番号16、
MCK-3: 5′-CAAGAAGCACACGACTGAGGCA-3 ′ 配列番号17
【0047】
免疫染色及びファクス分析:
Jurkat細胞、抹消血リンパ球及び/又はL6陽性腫瘍細胞(H2981又はH3639)を間接免疫染色により解析した。H2981細胞をトリプシン−EDTA(GIBCO-BRL) 中でインキュベートしてフラスコから剥がした。細胞を種々の濃度の単鎖抗体又は未変性親抗体と結合バッファー(GIBCO-BRL) 中4℃で40分間インキュベートした。第1段階後細胞を洗浄し、FITC結合第2段階試薬と4℃で更に30−40分間インキュベートした。第2段階抗体は、次のものの1種とした:マウスmAbの場合ヤギ抗マウスIg、単一Ig融合物の場合ヤギ抗ヒトIgG(Tago, Inc., Burlingame, CA)、HIVペプチド融合物の場合gp110のV3ループに特定されたFITC−110.3及び全種類のL6含有構築物の場合L6抗体に対するFITC−αイディオタイプ(1B)。40対数アンプリファイアを備えたファクスIVセルソーター(Becton Dickenson and Co., Mountain View, CA) で蛍光を分析した。
2種の抗体を全量10μg/mlの抗体まで種々の割合で共に混合した後に細胞を加えることにより、競合アッセイを行った。2種の抗体のうち1種をFITC、通常はキメラ又はマウスL6又は未変性G19−4で標識した。阻害アッセイの場合には、第2FITC結合抗体を加える30分前に、第1抗体を加えた。
【0048】
融合タンパク質の細胞培養、移入及び精製:
前記発現プラスミドをCOS細胞に移入した(Linsley, P.S., Brady, W., Grosmaire, L., Aruffo, A., Damle, N.K., Ledbetter, J.A.(1991) Binding of the B cell activation antigen B7 to CD28 costimulates T cell proliferation and interleukin 2 mRNA accumulation. J. Exp Med. 173:721-730; Aruffo and Seed, 1987a)。プラスミドDNAを全量12ml/150mmプレート中1μg/mlで移入培養液に加えた。移入したCOS細胞の3採取物からの使用済み無血清培養液をプールし、Ig、HIV又はSTOP分子垂れはしを含む融合タンパク質を精製するために使用した。細胞デブリを低速遠心により除去し、上清を0.2μm フィルターでときどきろ過した後精製した。Ig融合移入物の培養液を0.05M クエン酸ナトリウム、pH8.0(Linsley等, 1991a)で平衡化した固定化プロテインA (Repligen Corp., Cambridge, MA)のカラムに加えた。500mlの上清に対して1mlの充填床容量プロテインAを用いた。培養液を2回用いた後、カラムを100mMリン酸ナトリウム、pH8.0で洗浄し、結合タンパク質を0.05M クエン酸ナトリウム、pH3で溶離した。画分を1/5容量1M トリスpH8.0を含む試験管に集めた。A230 吸収物質のピークを含む画分をプールし、使用前にPBSで透析した。ローリー法に基づくバイオラドタンパク質アッセイキットを用いてタンパク質濃度を求めた。
【0049】
他の分子垂れはしを含む融合タンパク質の場合、ファーマシア社の説明書に従いCNBr活性化セファロース4Bを用いて適当な抗体(抗L6イディオタイプmAb13B又はgp110のV3ループに特定された抗HIVmAb110.3)を固定化して、アフィニティーカラムを作った。親和性基質は約5mgのmAb/ml床容量を含み、典型的なカラムサイズ1×5cm(4ml)とした。試料をpH7に調整し、予め0.1M クエン酸、pH2.2で洗浄し、PBS、pH7.2中で平衡化してある適切なイムノアフィニティーカラムに加えた。カラムをPBSで十分洗浄し、結合物質を0.1M クエン酸塩pH3.0で溶離し、次いで直ちにトリスで中和した。精製した抗体誘導体を最後にPBSに透析し、滅菌ろ過した。
【0050】
細胞接着アッセイ:
10mMEDTAの存在下に実質的に(Linsley, P.S., Clark, E.A., Ledbetter, J.A. (1990) T-cell antigen CD28 mediates adhesion with B cells by interacting with activation antigen B7/BB1. Proc. Natl. Acad. Sci. USA 87:5031-5035)に記載されているように接着アッセイを行った。Jurkat細胞をまず51Crで標識し、抗体刺激とインキュベートし、洗浄し、H2981腫瘍細胞とインキュベートし、顕微鏡的に試験した。H2981に対する非特異的結合を防止するために、Jurkat及びH2981単層を無関係な抗体とインキュベートしてFcレセプターを飽和した後、CD3Ig(本明細書ではCD3FvIgとも呼ぶ)、L6Ig(本明細書ではL6FvIgとも呼ぶ)又はCD3−L6Ig(本明細書ではCD3−L6FvIgとも呼ぶ)抗体誘導体を加えた。接着反応が完了した後、単層を氷冷RPMI培養液で5回洗浄し、0.5M NaOHを加えて可溶化し、γカウンターで放射能を測定した。全結合放射能(cpm) を標識細胞の比活性(cpm/細胞)で割って、結合した細胞数を算出した(図13)。
【0051】
図13において、H2981腫瘍細胞の単層を105 細胞/ウェルの密度で48ウェルプレートに播種し、0.1%パラホルムアルデヒドに23℃で20分間固定し、洗浄し、完全RPMI+10%FBSで阻止し、単独で又は指定した抗体と37℃で1時間予備インキュベートした。抗CD3(G19−4)及び抗L6mAbの異種結合体を陽性対照として用いた。指示した場合を除いてすべての抗体を20μg で用いた。Jurkat細胞を51Crで標識し、10mMEDTA中で予備インキュベートし、腫瘍細胞及び抗体に加えた。プレートを1000rpm で2分間攪拌することにより接着を開始した。反応物を37℃で45分間インキュベートし、氷冷RPMIで5回洗浄し、0.5N NaOHに溶解した。遊離した数をγ計数により求めた。データは結合した細胞数を示した(×103)。3回の測定平均と標準偏差(誤差のすじ)が示されている。
【0052】
SDS−PAGE及びウェスタンブロット法:
4%スタッカーを用いて直線勾配6−15%を生じるアクリルアミドゲルを225ボルトで3時間又は8mAmpで一晩行った。ウェスタンセミドライ移動装置(Ellard Instruments, Seattle, WA) を用いてニトロセルロース膜にゲルを130mAmpで1時間ブロットして免疫反応で検出した。ブロットを1%無脂肪乳、PBS中0.05%NP−40 (BLOTTO、即ち、阻止バッファー) で1−2時間阻止した。第1抗体をアルカリ性ホスファターゼ結合ヤギ抗ヒトIgG (Boehringer-Mannheim)とBLOTTO中1:1500の希釈度で又は非Ig融合物、即ち、Ig垂れはしのないSfV の検出用の適切な希釈度の非結合マウス又はキメラ抗体又は抗イディオタイプ抗体とインキュベートした。いずれの場合にもブロットをBLOTTOで3回洗浄し、第2工程を必要とする場合にはアルカリ性ホスファターゼ結合ヤギ抗マウス又は抗ヒトIgGとインキュベートした。ブロットをウェスタンブルー(Promega, Madison, WI)で展開し、反応を蒸留水で停止した。
【0053】
抗p−tyrとの免疫沈降及びウェスタンブロット法:
JurkatT細胞を刺激しない(0)か又は指定した濃度の未変性G19−4Mab(Ledbetter, J.A., Norris, N.A., Grossmann, A., Grosmaire, L.S., June, C.H., Ucklin, F.M., Cosand, W.L., Rabinovitch P.S.(1989) Enhancd transmembrane signalling activity of monoclonal antibody heteroconjugates suggest molecular interactions between receptors on the T cell surface. Mol. Immunol. 26:137-145)又はCD3Fv−Igで刺激し、ホスファターゼ及びプロテアーゼインヒビター(1mMオルトバナジン酸ナトリウム、1mMPMSF、2mMEGTA、0.5%アプロチニン及び10μg/mlロイペプチン)を含む修飾RIPAバッファー(Kanner, S.B., Reynolds, A.B., Parsons, J.T.(1989) Immunoaffinity purification of tyrosine-phosphorylated cellular proteins. J. Immunol. Methods 120:115-124) に溶解した。細胞溶解物を透明にし(14000rpm で10分)、ウサギ抗p−tyrあるいはPLCγ1抗血清で免疫沈降させた。免疫複合体をプロテインA−セファロースビーズ(Pharmacia, Piscataway, NJ) で回収し、洗浄した。タンパク質をSDS−PAGE(8%)で分離し、PVDF Immobilon(Millipore, Bedford, MA)に4℃で2時間移した。イムノブロットを阻止した後、阻止バッファー中0.5μg/mlのアフィニティー精製ウサギ抗p−tyrを加えた。1μCi/ml の高比活性 125I標識プロテインA(ICN Biomedicals, Costa Mesa, CA) 及びオートラジオグラフィーを用いて検出した。
【0054】
増殖アッセイ:
抹消血リンパ球をリンパ球分離培養液(Organon Teknika,Durham, NC)で希釈及び遠心して単離した。リンパ球を無血清RPMI1640で数回洗浄し、細胞濃度を10%FCSを含むRPMIで106 細胞/mlに調整した。細胞を96ウェル、平底プレートで培養した(0.2ml容量中5×104 細胞/ウェル)。3日培養の最後の6−8時間に1μCi/ml の[3H]チミジン取込みによ、3回の試料について増殖を測定した。PBLを1μg/mlPHA(Wellcome, Charlotte, NC) と培養し、PHAのない培養液で1日静止することにより、PHA活性化T細胞を調製した。
H2981腫瘍細胞を10,000ラドで照射した後、増殖アッセイに使用した。細胞を、融合タンパク質に前結合し洗浄した後PBLとインキュベートする(『前結合』試料)かあるいは3日培養で溶液に融合タンパク質と含めた(『溶液』試料)。前結合試料の洗浄は非結合タンパク質を除去するので、腫瘍細胞に結合したタンパク質のみがアッセイでPBLの刺激を与える。PBLの力価が照射細胞に対して次のように測定された:(2:1)、5:1、125:1、(625:1)、ここで最初の数は腫瘍細胞の減少数と比べて存在したPBL(5×104細胞/ウェル)の相対数を意味する。
【0055】
細胞毒性アッセイ:
H2981腫瘍細胞を[51Cr]と2時間インキュベートし後、融合タンパク質(0.1μg/ml〜10μg/ml)及びPBL(各種エフェクター:標的比10:1〜100:1で)とインキュベートした。細胞を全量0.2ml中10%FCSを含むRPMI中で5時間培養した後、計数した。γカウンターでクロミウム放出を測定して腫瘍細胞を標的とした細胞毒性を測定した。
【0056】
COS細胞は組換え体二特異的単鎖カセットDNAからの抗体を発現することができる: 急速検出、精製及び確認を容易にする抗体分子の一過性哺乳動物発現系を開発することに着手した。種々の特異性及びドメインの交換を有する分子に適応させて作成、試験及び異種単鎖二特異的抗体との間の比較を簡易化するのに、系が十分有用であることは重要なことであった。表面レセプターの細胞外ドメインとヒト免疫グロブリンIgG1の重鎖との間の融合タンパク質を作ることにより可溶性細胞表面レセプターを発現するCOS細胞一過性発現系を従来の研究者らは満足して用いている(Aruffo and Seed, 1987b; Linsley等, 1991b)。
本系は、分子を分泌し且つ培養上清から活性形態として容易に回収することができることから、細菌発現系に対して魅力のある代替品として選ばれた。最初の実験は、COS細胞一過性発現系がゲノム配列よりむしろcDNAを用いてそのままの機能性IgG分子を発現及び分泌することができるかを調べた。抗腫瘍抗原L6特異性をコードする全長κ及びγcDNAカセットをpCDM8に連結し、挿入ベクターをDEAE−デキストラン法によりCOS細胞に共移入した。培養上清がL6陽性腫瘍細胞に対して結合活性をもつ100−500 ng/mlのタンパク質レベルを含むことを見出し、COS細胞が組換え体二特異的単鎖カセットDNAから未変性抗体を構築し分泌することができることを示した。
【0057】
哺乳動物発現ベクターの適応:
そのような分子のCOS細胞発現能が証明されると、個々のタンパク質ドメインをコードする相互交換可能なカセットを用いた単鎖抗体分子を発現する発現ベクターpCDM8及びpiLNXAnを修飾した。ベクターpCDM8及びpiLNXは、調節領域の下流に位置するポリリンカー/スタッファー領域に挿入された遺伝子の発現を達成するために、サイトメガロウイルスあるいはAMVプロモーター及びエンハンサーを用いる。ポリリンカー内の可変部挿入部位に隣接する2つの短いcDNAカセットを含むように、この領域を作り変えた。図1は、ベクターの修飾を図で示したものであり、ベクターの修飾及び発現した単鎖分子の可変部の配置の模式図である。L6κ軽鎖可変部からのリーダーペプチドを含むHindIII−SaII断片をポリリンカーの5′端に挿入してこれに融合した分子の分泌を達成した。ヒトIgG1のヒンジ、CH2及びCH3ドメインをコードするBcII−XbaI、即ち、分子垂れしをポリリンカーの3′端のフレーム内に融合して種々の特異性を有する分子の検出及び精製を容易にする。重鎖及び軽鎖の可変部をコードする単鎖抗体カセットDNAを短いペプチドリンカーによって相互に結合し、これらの2つの短いフランキングカセットの間に単一のオープンリーディングフレームを作るようにSaII−BcII断片(他の適合性末端もときどき用いた)として挿入した。これにより、既存の哺乳動物発現ベクターを適応して任意の特異性を有する単鎖抗体分子(SCA)をコードするcDNAカセットの有効な発現が得られた。本系は、異種分子垂れはし、リンカー及び結合特異性をコードするカセットが可溶性機能分子の発現における相対的有効性に匹敵するように急速発現、精製、スクリーニング及び融合タンパク質の変更が可能である。
【0058】
単鎖一特異的L6FV −Ig、CD3FV 及び二特異的CD3−L6Ig抗体誘導体の構築及び発現:
2つの異なる結合特異性をモデルとして用いて適応した単鎖抗体発現系を試験した。L6腫瘍抗原及びCD3T細胞表面レセプターに特定された抗体の重鎖及び軽鎖の可変部を材料及び方法で記載したように単離した。
抗L6及びG19−4の可変部をオーバーラップ拡張PCRを用いて単一のコーディング領域に融合してVL のカルボキシル末端とVH のアミノ末端の間に(Gly4Ser)3リンカーを作った。
図2は、G19−4の軽鎖及び重鎖可変部を(Gly4Ser)3と融合して作った単のオープンリーディングフレームを示すものである。
L6軽鎖可変部のリーダーペプチド(点描領域)、CD3−L6抗原結合ドメイン(陰のない領域)及びヒトIgG1Fcドメイン(陰のある領域)をコードするcDNA構築物を材料及び方法で記載したように構築した(図2)。
【0059】
図2において、スタッファーフラグメントを除き且つ単鎖抗体分子の発現のためのアダプター配列に置き換えることにより、哺乳動物発現ベクターpCDM8を修飾した。発現分子の分泌を得るために、抗L6の軽鎖可変部のリーダーペプチドをコードするHindIII−SalI断片を挿入した。融合構築物の検出、精製及び確認を容易にするために、ヒトIgG1のFcドメインをBclI−XbaI部分として下流に含めた。HIV由来のV3ループの一部をコードする短い分子垂れはし配列もCD3構築物に用いた。リーダー配列と垂れはしの間に、VL とVH との間の融合カセットを挿入し、2つのドメインを(Gly4Ser)3アミ酸リンカーで分けた。
第1カセットとFcドメインとの間のBclI部位に第2VL −VH カセットを挿入することにより、二特異的分子を構築することができた。立体障害を防止し溶解性を改良するために、27アミノ酸らせんペプチドリンカーをコードするオリゴヌクレオチドで2つの結合特異性を相互に離した。
表示した配列は各ドメイン間の連結を示し、構築で導入されたアミノ酸は太文字体で示されている。
【0060】
可変部融合カセットを修飾発現ベクターに挿入して図2に示した遺伝子融合を作り、COS細胞に移入した。無血清使用済培養液を集め、タンパク質をプロテインAアフィニティークロマトグラフィーで精製した。還元又は非還元SDS−PAGEにかけたタンパク質のウェスタンブロットをアルカリ性ホスファターゼ結合ヤギ抗ヒトIgGで図3に示したようにプローブした。パネルA及びBは、L6Fv−Ig及びCD3Fv−Ig融合タンパク質が還元及び非還元両条件下でMr 55,000を有する単一種として移動したことを示し、このおよそのサイズはこれらの単鎖抗体誘導体が予想される。同様に、CD3−L6FvIg二特異的分子がこれらの両条件下でMr 〜94,000を有する単一種として移動した。比較すると、ヒトヒンジ−CH2−CH3の野生型配列に融合したマウス可変部からなるキメラL6mAb又はL6野生型二量体(WTD) は、還元及び変性の程度により著しい移動度差を示し、これらの分子の重鎖定常部がジスルフィド結合で結合して二量体を形成したことを示した。時折、非還元SDS−PAGEのウェスタンブロットがMr >100,000で移動する一特異的分子又はMr >200,000で移動する二特異的分子の場合に極めてわずかなバンドを示した。
【0061】
抗L6軽鎖シグナルペプチド、抗体結合特異性をコードする可変部融合及びヒトIgG1Fcドメインを含む単一のオープンリーディングフレームを作るように、VL −VH 融合カセットを適応したベクターに挿入した。短いらせんペプチドリンカーを介したCD3及びL6融合カセットを融合することにより、CD3−L6二特異的融合カセットを作り、一特異的構築物の場合のように挿入した。発現系の最初の試験に使用した分子垂れはしは、鎖内ジスルフィド結合を減じるか又は排除するためにヒンジジスルフィドをセリンに変えたヒトFcの変異誘導体とした。これらの単鎖構築物をCOS細胞に個別に移入し、融合タンパク質を固定化プロテインAによるアフィニティークロマトグラフィーで培養上清から精製した。
精製タンパク質の収量は、典型的には、L6Ig融合タンパク質の場合約2mg/リットル、CD3Igの場合約10mg/リットル及びCD3−L6Ig二特異的分子の場合約0.5mg/リットルであった。非還元SDS−PAGEにかけアルカリ性ホスファターゼ結合ヤギ抗ヒトIgGでプローブしたタンパク質のウェスタンブロットを図3に示す。Mr 55,000で移動するCD3Ig及びL6Igレーンに単一種が見られ、およそのサイズがこれらの単鎖抗体誘導体として予想されるが、陰性対照レーンには見られない(図3)。CD3−L6Ig二特異的分子は、Mr 約95,000−98,000で移動し、より高い分子量バンドがMr >200,000に見られる(図3)。
【0062】
L6Ig、CD3Ig及びCD3−L6Ig融合タンパク質の結合活性:
本発明の単鎖抗体誘導体の機能活性を調べ且つこの分子が抗原特異結合することができることを証明するために、まずFcドメイン融合タンパク質の細胞発現標的抗原に対する結合を試験した。ヒト腫瘍細胞系H2981は、高レベルのL6標的抗原を発現するが、CD3又はBR96を発現せず、ヒトJurkatセルラインはCD3を発現するが、L6又はBR96を発現しない。第2段階試薬としてFITC結合ヤギ抗ヒトIgを用いて蛍光活性化セルソーター分析により、結合を検出した。図3に示されるように、融合タンパク質は未変性親抗体(即ち、未変性抗L6及びG19−4抗体)と同様の細胞発現標的抗原に特異的に結合するが、検出可能レベルの抗原のない細胞には結合しない。CD3−L6Ig二特異的融合タンパク質は、Jurkat及びH2981細胞の双方に結合し、これらの分子が1種以上の特異性を有することを間接的に示した。ヤギ抗ヒトIgG又はL6結合特異性に特定されたFITC結合抗イディオタイプ抗体を用いて検出が得られるかでも同様の結果が見られた。
【0063】
10μg/mlの抗体誘導体をH2981腫瘍細胞(L6陽性)、Jurkat細胞(CD3陽性)又はH3396腫瘍細胞(BR96陽性)とインキュベートした(図4)。細胞を洗浄し、第2段階試薬としてFITC結合ヤギ抗ヒトIgGとインキュベートした。次いで、合計10,000個の染色細胞をファクスで分析した(図4)。図3は、L6Ig、CD3Ig及びCD3−L6Igが細胞発現標的抗原に結合することを示している。
融合タンパク質の生物学的特性を調べるために、個々の分子の予想又は所望の特性に基づいて種々の機能アッセイを用いた。従って、各単鎖分子の結果が別々に存在し、L6Ig単鎖抗体誘導体から始める。
【0064】
L6融合タンパク質の結合活性に関する種々の分子垂れはし配列の影響:
各種分子垂れはし配列を材料及び方法で記載したように構築し、L6結合ドメインに融合し、これらの領域を用いてプロテインAに対する結合標的又はそれらを標的とした特異抗体として発現融合構築物を検出及び精製することができるかを求めた。L6結合ドメインに直接融合した場合、Cκ、HIVペプチド及びFLAGペプチドはすべて信頼できる分子垂れはしとして機能しなかった。これら3種のペプチドはすべて、認識可能なL6を発現する移入細胞をもたらさなかった。検出が分子垂れはしによらずL6抗イディオタイプ抗体を利用する場合でさえ、H2981腫瘍細胞に対する結合アッセイにおいて機能性融合タンパク質を検出できなかった。
【0065】
L6誘導体によるエフェクター機能に関する種々のFcドメインの影響:
分子垂れはしのないL6結合ドメインの単純sFv(sFv)及び異種Fcドメイン変異体に結合した各種−Ig融合物を含む他のL6誘導体を構築した。図5及び図6に示されるように、ヒンジ及び/又はCH2ドメインに導入された変異に基づく呼称を各Fc構築物に示した。野生型二量体 (WTD)は、すべてのFcドメイン配列に対して野生型であり、この領域内のアミノ酸残基に対して未変性抗体と同一である。単量体構築物は配列置換、即ち、システイン残基を含み、ヒンジジスルフィドをセリンに変異する (HS1)。単量体1(mut1としても知られる)(HS2)は、IgG1エフェクター機能を仲介するに当たり重要な領域であるCH2ドメイン内の残基238においてプロリンをセリンに置き換わることを含む。単量体(HS3) は、このCH2領域内の数個の残基(234−238)に対して変異する。二量体mut1(DM1) はFcドメインのヒンジ領域に野生型配列を含むが、残基234−238をコードするCH2配列の変異体でもある。
【0066】
L6のSfvは、L6VL −VH 融合カセットの後に他の分子垂れはし配列よりむしろ停止コドンを含んでいる。これらの分子の各々を構築し、COS細胞に移入し、プロテインAセファロースあるいは固定化L6抗イディオタイプ抗体カラムを用いて精製タンパク質を発現した。飽和及び阻害結合分析において、融合タンパク質を未変性キメラL6抗体及びFab′誘導体と比較した(図7−8)。L6Fc変異体は未変性抗体と極めて類似した飽和曲線を生じる(図7)が、Sfv融合タンパク質は十分に結合しなかった(図6)。増加量の試験抗体を腫瘍細胞とインキュベートした後、FITC結合キメラL6を加えることにより阻害試験を行った。再度、すべての−Ig融合物及びキメラ抗体に対して同様の曲線が見られ(図8)、Sfv及びFab′は未変性抗体の結合を阻害する能力が減少した(図7)。未変性抗体の結合と拮抗又は阻害しないにもかかわらず、Sfv融合タンパク質はこの実験において化学的に調製されたL6Fab分子よりわずかに良好であった。
【0067】
ヒトIgG1に関与するエフェクター機能を仲介するこれらの分子の相対的能力を、抗体による細胞の細胞毒性(ADCC)又は補体による細胞毒性(CDC)アッセイにより測定し、特異的溶解を抗体濃度の関数としてプロットした。ヒンジ領域に欠失のある変異体はすべてADCC仲介能に影響しなかったが、CH2ドメイン変異体はいずれもこの過程を仲介しなかった。驚くべきことに、キメラL6はCDCをかなり良好に仲介するが、L6誘導体はいずれも腫瘍標的の補体媒介性溶解を刺激することができたことを発見した。
ヒト腫瘍抗原L6に特定されたマウス抗体の可変部をCOS細胞で発現したヒトIgG1のFcドメインの各種誘導体に融合し、融合タンパク質をアフィニティークロマトグラフィーで精製した(図17−20)。
構築した各Fcドメインが示され、配列置換は下線を引いたアミノ酸で示される。各構築物の精製タンパク質は、飽和及び阻害アッセイにおいてキメラL6に匹敵した。更に、H2981腫瘍細胞を51Crで2時間標識し、洗浄し、これに10倍連続希釈の抗体を含むIMDM+10%FCS及びヒトPBL(ADCC)あるいはウサギ補体(CDC)に加えることにより、これらの分子のADCC及びCDCのような正常なIgG1エフェクター機能を仲介する能力を測定した。検定物を4.5時間インキュベートし、攪拌し、遊離した数をγカウンターで測定した。数値は、3回の培養の平均を示した(SEM<10%)。
【0068】
CD3単鎖抗体は未変性抗体と量的に異なる他は質的に同様な生物活性を示す:
−Ig融合物、−HIVペプチド融合タンパク質、VL −VH sFv(垂れはしなし)及びCD3−L6Fv−Ig二特異的分子を含む各種CD3融合タンパク質を構築した。
HIVペプチドは、CD3に融合した場合信頼できる分子垂れはしとして作用した。3単鎖融合タンパク質の検出及び精製を可能にした。垂れはしを付けた分子を固定化プロテインA(−Ig融合物)又は110.3抗体(HIV融合物)を用いてアフィニティークロマトグラフィーで精製した。単純Sfvはろ過した上清溶液として用い、G19−4抗体のJurkat細胞に対する結合を阻害する上清の能力を測定することにより近似の濃縮物を評価した。これらの変質した分子のCD3T細胞レセプター複合体に対する結合によって生じた細胞応答を調べた。各分子をindo−1の入った末梢血リンパ球又はT細胞に結合し、細胞内カルシウムの移動をフローサイトメトリーによりモニターした。図9−12に示されるように、未変性G19−4抗体の等価濃縮物と比べるとIg及びHIV融合タンパク質の双方が膜貫通シグナル活性を高めた。単純Sfvはカルシウムシグナルを生じるが、未変性抗体に見られるように強くも長くもなかった。
【0069】
一及び二特異的誘導体の連結配列を含む抗CD3可変部の配列を図21に示す。Ig融合タンパク質は固定化プロテインAを用いたアフィニティークロマトグラフィーで精製し、Sfvはろ過上清として用いた。既知濃度の親G19−4MabのJurkat細胞に対する結合を遮断する移入上清の能力を測定することにより、Sfvの濃度を推定した。CD3Fv−Ig分子のシグナル活性を分析するために、PLCγ1のチロシンリン酸化誘導能を試験した。JurkatT細胞をCD3Fv−Ig又は未変性G19−4Mabで刺激し、細胞溶解物のタンパク質をSDS−PAGEで分離し、PDVF膜に移し、抗p−tyr又は抗PLCγ1でプローブした。驚くべきことに、我々は、CD3Fv−IgがPLCγ1を含む全細胞溶解物中の細胞タンパク質のチロシンリン酸化を強力に誘導し且つ多量のpp35/36リンタンパク質がPLCγ1と結合していることを見出した(図22−23)。更に、CD3Fv−Ig及びCD3Sfvの双方がPBL中でカルシウムフラックスを誘導し、未変性G19−4Mabの等価濃度に見られるものより程度が高かった。
【0070】
図21においては、抗CD3MabG19−4の重鎖及び軽鎖の可変部がPCRでクローン化されている。これらのcDNAカセットから構築した遺伝子融合のヌクレオチド及びタンパク質配列を(Gly4Ser)3リンカーと共に太文字体で示。VL −VH 融合カセットのアミノ末端をSalI部位でL6軽鎖可変部リーダーペプチドに融合し、カルボキシ末端をBclI部位でFcドメインのヒンジ領域に直接融合するか又は短い『らせん』ペプチドリンカーに融合して二特異的CD3−L6FvIg抗体誘導体を構築した。L6の可変部は、図1及び2に示されるように、『らせん』リンカーの反対の端にフレーム内で融合した。
図22−23においては、示されているように、Jurkat細胞(107/試料)5、20又は80μg/mlの未変性抗CD3MabG19−4又は1、5又は20μg/mlのCD3Fv−Igで1分又は3分間刺激した。細胞溶解物のタンパク質をウサギ抗p−tyr(パネルA)又はPLCγ1抗血清(パネルB)で免疫沈降させ、プロテインAセファロースで回収し、これをSDS−PAGEで分離し、ニトロセルロースに移し、精製したウサギ抗p−tyr及び125I標識プロテンAで検出した。
【0071】
図9−12は、CD3Fv融合誘導体が異なったレベルの膜貫通シグナル活性を示す線グラフである。図9−10は、CD3FvIgが末梢血T細胞内で細胞内カルシウムを移動することを示すものである。フローサイトメトリー及びカルシウム結合染料を用いて、CD3FvIg(−・−・−)、未変性CD3Mab( ---- )又は非T細胞結合対照L6FvIg( .... )による刺激に伴う細胞内遊離カルシウムの濃度をモニターした。各刺激(2μg)をindo−1の入った充填T細胞に1分の時点(矢印)130nm=静止細胞で及び%応答細胞のように加えた。
図11−12は、CD3FvIgで前処理すると次のクロスリンクしたCD2の刺激に対して末梢血T細胞の応答を脱感作することを示している。indo−1の入った細胞を1μg のCD3FvIg、2μg の未変性CD3mAb又はL6FvIg(非T細胞結合対照)と37℃で15分間インキュベートした。ビオチニル化抗CD2mAb(5μg )を1分の時点で加え、5分の時点でアビジン(20μg )によってクロスリンクした。次いで、クロスリンクしたCD2に対する応答を5分間モニターした。データは、([Ca2+ ])130nm=静止細胞(ネルA)及び%応答細胞(パネルB)として示されている。
【0072】
種々の濃度のCD3抗体誘導体を用いた刺激に応答する末梢血リンパ球及び精製T細胞の増殖アッセイにおいて、3誘導体すべてがT細胞を活性化することを見出した。1μg/mlの抗体処理において測定した増殖応答を表1に示す。増殖応答の同様のパターンが0.2及び5μg/mlにおいても見られた。データは、CD3誘導体がマウス抗体の未変性G19−4(CD3Fv−Igと比較)あるいはFabフラグメント(sFvと比較)で見られたものと同様のT細胞内増殖応答をもたらすことを示している。未変性G19−4で見られるものよりPBLの強い増殖を生じるPMAとの相乗作用で作用するCD3Fv−Igを除き、遺伝子操作した分子は精製T細胞のわずかに強い増殖レベル及びPBLのわずかに弱いレベルを刺激した。
2種の垂れはしを付けたCD3融合タンパク質が未変性活性より強い膜貫通シグナル活性を示したので、これらの分子がT細胞増殖にどのように影響するかを求めた。末梢血リンパ球及び精製T細胞を種々の濃度の抗体誘導体と72時間インキュベートし、トリチウムを入れたチミジンで6時間標識した後、回収し、計数した。表1に1μg/mlにおける抗体処理の結果を示す。増殖応答の同様のパターンが0.2及び5μg/mlにも見られた。
【0073】
【表1】

【0074】
細胞を1μCi/ウェル[3H]チミジンで6時間パルス標識して72時間培養し後増殖を測定した。T細胞を2種のプラスチック粘着で単核細胞及びB細胞から離し、次いでナイロンウールカラムに通過させることにより精製した。
データは、ここで試験した条件下、CD3誘導体の結合がマウス抗体のG19−4(Ig融合)あるいはFabフラグメント(HIV及びSTOP融合)で見られるのと同様のT細胞の増殖応答をもたらすことを示している。遺伝子操作した分子は、精製T細胞のわずかに強い増殖レベル及びPBLのわずかに弱いレベルを刺激する傾向があったが、ほとんどの場合、それらの差はわずかである。このパターンの唯一の例外は、CD3sFvIg及びCD3HIV構築物がPMAと相乗作用して生じた未変性G19−4に比べて強い増殖応答である。
【0075】
CD3−L6二特異的融合タンパク質はJurkat細胞とH2981腫瘍細胞との間の接着を仲介する。
以前に開発された細胞接着(Linsley等, 1990a)を用いてCD3−L6二特異的単分子がCD3又はL6発現細胞に同時に結合できるかを求めた。まずJurkat細胞を抗CD3又はCD3−L6FvIgとインキュベートした。細胞を洗浄し、L6FvIgあり又はなしで予備インキュベートしたH2981粘着細胞に加えた。H2981単層(図13)の顕微鏡試験は、CD3及びL6レセプターがリガンドで遮断されない場合だけを除いて、CD3−L6FvIgタンパク質がEDTAの存在下にJurkat細胞とH2981腫瘍細胞との間の接着を仲介したことを示した。Jurkat細胞を51Crで前標識し、次いで融合タンパク質及びH2981腫瘍細胞とインキュベートした。CD3−L6二特異的融合タンパク質の存在下に標識していない単層に結合したカウント数は、連結していないCD3及びL6抗体に前結合した細胞より非常に多かった。
CD3−L6Ig二特異的融合タンパク質は、H2981腫瘍細胞に対するT細胞の細胞毒性を標的とする(図14)。H2981腫瘍細胞を51Crで2時間標識し、標的に対するエフェクター比10:1及び100:1で抗体刺激及びPBLとインキュベートした(図14)。クロミウムの遊離を材料及び方法で記載したように測定し、3回の実験から各抗体誘導体に特異的な溶解を表にした(SEM<12%)(図14)。阻害アッセイの場合、CD3又はL6Ig単一特異性誘導体を適切な種類の細胞とインキュベートした後、CD3−L6Ig二特異的分子を加えた(図14)。いずれも単一特異性分子と予備インキュベートすると細胞毒性の二特異的構築物による刺激を阻害することができなかった(図14)。
【0076】
CD3−L6二特異的融合タンパク質はH2981腫瘍細胞に対するT細胞の細胞毒性を標的とする。
次に、これら2つの抗原結合ドメインを単分子に結合することの生物学的結果を求めた。腫瘍細胞とT細胞との間の接着を促進する遺伝子操作した二機能性分子がレセプター結合に対する応答の種類又は程度を変えると推論された。
IgG媒介性エフェクター機能から背景原因を排除するために、CH2内のプロリンからセリンへの変異及びヒンジ領域内のいくつかのシステインからセリンへの置換の結果としてこれらの機能を仲介しないFc単量体の変異体に分子の抗原結合部分を結合した。溶解の場合の標的としてH2981腫瘍細胞を用いて標準細胞毒性アッセイを行った。腫瘍に対する細胞毒性を標的とするために静止又は未変性細胞を活性化することができる場合には、エフェクター細胞として静止PBLを用いて求めた(図14)。
【0077】
CD3−L6FvIg分子は、標的に対するエフェクター比10:1において30%の特異的溶解を仲介し、100:1の比では特異的溶解が71%に上昇する。CD3FvIg及びL6FvIgを一緒に混合したもの又は両分子単独の場合の特異的溶解レベルは、E:T10:1で3〜5%及びE:T100:1で9〜20%である。破壊レベルをわずかに減少したが、CD3FvIg及びL6FvIg抗体誘導体はCD3−L6融合タンパク質により仲介された標的細胞毒性を完全に遮断しなかった。PHA活性化T細胞芽球も本アッセイにおいてエフェクターとして用いたが、高レベルの背景破壊を示した。
CD3−L6Ig二特異的融合タンパク質は、H2981腫瘍細胞に結合すると高レベルのT細胞増殖を刺激する(図15)。PBLを単離し、必要とするT細胞増殖刺激物質及び照射H2981腫瘍細胞を存在させて培養した。刺激物質を1又は10μg/mlのいずれかの溶液として加えた。前結合実験の場合、H2981腫瘍細胞に照射し、10μg/mlの抗体誘導体と氷上で1時間インキュベートし、数回洗浄して非結合抗体を除去し、種々の割合で5×104 細胞/ウェルのPBLに加えた。3日培養した後、[3H]チミジンの取込みにより6時間増殖を定した。各処理について4回の培養により数値を求めた(SEM<15%)。
【0078】
CD3−L6二特異的融合タンパク質は試験管内で高レベルのT細胞増殖を刺激する。
CD3T細胞表面レセプター(CD3/TCR複合体)をCD3−L6FvIg融合タンパク質で誘発するとT細胞増殖を刺激するかを調べた。照射H2981腫瘍細胞とインキュベートした静止PBL及び刺激(融合タンパク質)の溶解したものを用いて、増殖アッセイを行った。
また、刺激タンパク質を照射腫瘍細胞に前結合し、非結合タンパク質を数回洗浄して除去した後、アッセイに含め、CD3によってT細胞の刺激に寄与するものからL6抗原に結合できない分子を排除した。H2981腫瘍細胞はFcドメインで変異した抗体誘導体にさえ非特異的に結合する傾向があったので、アッセイからこの非特異的背景の原因を排除するために、不適当な抗体を細胞とインキュベートした後に問題の刺激を加えた。図15は、溶解及び前結合両増殖実験の結果を示すものである。
【0079】
増殖レベルは10μg/mlのCD3−L6FvIg二特異的融合タンパク質の存在下で著しく増大し、有為レベルの増殖が1μg/mlでさえも見られた。L6FvIgを腫瘍細胞に前結合した後に二特異的分子を加えると、被覆腫瘍細胞によって誘導されるこのT細胞増殖の刺激が排除された。CD3IG融合タンパク質は、腫瘍細胞のあり又はなしに依存しない溶液中に存在すると著しいレベルの増殖を刺激することができた。背景レベルの増殖のみがこれらの条件下で見られるので、これらの分子が腫瘍細胞前結合アッセイの洗浄段階で除去されたことは明らかである。これらの結果は、H2981腫瘍細胞に結合した際に、T細胞の細胞毒性及び刺激T細胞増殖を標的とするCD3−L6二特異的融合タンパク質の能力を示すものである。
【図面の簡単な説明】
【0080】
【図1】種々の分子垂れはし又は単純な停止コドンを含む組換え体一特異的単鎖カセットを含むpCDM8発現ベクターの図である。
【図2】組換え体二特異的単鎖カセットを含むpCDM8発現ベクターの図である。
【図3】単鎖抗L6、抗CD3及び抗CD3−L6二特異的融合タンパク質分子のウェスタンブロット写真である。COS細胞移入からの使用済み無血清培養液を採取し、プロテインAアフィニティークロマトグラフィーで精製した。タンパク質を増量バッファーに再浮遊し、非還元(A)又は還元(B)条件下SDS−PAGE勾配ゲル(5−16%)電気泳動にかけ、ニトロセルロースにブロットし、アルカリ性ホスファターゼ結合抗ヒトIgGで検出した。パネルA:レーン1=キメラL6mAb(0.5μg);レーン2=抗L6WTD(0.5μg);レーン3=CD3−L6FvIg二特異的(0.5μg);レーン4=L6FvIg(0.6μg);及びレーン5=CD3Fv−Ig(0.4μg)。パネルB:レーン1=CD3Fv−Ig(0.4μg);レーン2=L6FvIg(0.6μg);レーン3=CD3−L6FvIg二特異的(0.5μg);レーン4=抗L6WTD(0.5μg);及びレーン5=キメラL6mAb(0.5μg)。
【図4】細胞発現標的抗原に結合するL6FvIg、CD3FvIg、CD3−L6FvIg及びBR96を示すファクスプロットである。
【図5】L6FvがヒトIgG1Fcドメインの各種異種変異誘導体に融合したことを示す。ヒンジ及び/又はCH2ドメインに導入された各変異体及び配列変化を示す。更に、各構築物のCDC及びADCC仲介能を示す。
【0081】
【図6】累進的に低くした濃度(3倍連続希釈)のH2981腫瘍細胞とインキュベートしたL6誘導体又はキメラL6の飽和分析の線グラフであり、第2段階試薬としてL6に対するFITC結合抗イディオタイプを用いて結合を検出した。これらのデータから飽和結合曲線を作り、蛍光強度を抗体濃度の関数としてプロットした。記号の説明:キメラL6(白ダイヤモンド形)、WTD(白四角)、DM1(白三角)、HS1(+記号)、HS2(『X』記号)及びHS3(白丸)
【図7】ファクス分析の前に連続希釈の各抗体誘導体と30分間及び1μg/mlのFITC結合L6と30分間インキュベートしたH2981腫瘍細胞の阻害分析の線グラフである。蛍光強度を各分子の抗体濃度の関数としてプロットした。記号の説明:キメラL6(白ダイヤモンド形)、WTD(白四角)、DM1(白三角)、HS1(+記号)、HS2(『X』記号)及びHS3(白丸)。
【図8】H2981腫瘍細胞を51Crで2時間標識し、洗浄し、10倍連続希釈の抗体誘導体を含むIMDM/10%FBS及びエフェクター細胞としてヒトPBLに100:1の標的に対するエフェクター比で加えた。分析物を4.5時間インキュベートし、攪拌し、100μl の遊離数をγカウンターを用いて測定した。次式を用いて破壊%を算出した:破壊%=〔(平均cpm−平均自然遊離)/(平均最大遊離−平均自然遊離)〕×100。数値は3回の培養の平均を示す(SEM<10%)。
【図9】CD3FvIgが抹消血T細胞の細胞内カルシウムを移動し、CD3FvIgとの前処理が次のクロスリンクしたCD2の刺激に対する抹消血T細胞の応答を脱感作することを示す線グラフである。
【図10】CD3FvIgが抹消血T細胞の細胞内カルシウムを移動し、CD3FvIgとの前処理が次のクロスリンクしたCD2の刺激に対する抹消血T細胞の応答を脱感作することを示す線グラフである。
【0082】
【図11】CD3FvIgが抹消血T細胞の細胞内カルシウムを移動し、CD3FvIgとの前処理が次のクロスリンクしたCD2の刺激に対する抹消血T細胞の応答を脱感作することを示す線グラフである。
【図12】CD3FvIgが抹消血T細胞の細胞内カルシウムを移動し、CD3FvIgとの前処理が次のクロスリンクしたCD2の刺激に対する抹消血T細胞の応答を脱感作することを示す線グラフである。
【図13】二特異的分子CD3−L6IgがH2981とJurkat細胞との間の接着を仲介するのでCD3及びL6発現細胞に同時に結合することができることを示す棒グラフである。
【図14】CD3−L6FvIg二特異的融合タンパク質がH2981腫瘍細胞に対するT細胞の細胞毒性を標的とすることを示す棒グラフである。
【図15】H2981腫瘍細胞に結合した際にCD3−L6FvIg二特異的タンパク質が高レベルのT細胞増殖を刺激することを示す棒グラフである。
【0083】
【図16】融合タンパク質として一特異的抗体可変部の発現のための発現ベクターpCDM8をヒトIgG1のFcドメインで修飾することを示す図式である。
【図17】黒い線で示したリンカー配列及び斜線で示した各機能性ドメインを含むL6Fv−Ig誘導体構造の図である。
【図18】キメラL6、WTD、DM1、HS1、HS2及びHS3のFc構築物及びそれらの配列の比較及びADCC又はCDC活性の有無を示す。L6FvはヒトIgG1のFcドメインの各種異種変異誘導体に融合した。ヒンジ及び/又はCH2ドメインに導入された配列変化を下線を引いたアミノ酸で示し、構築物同定を左に示す。
【図19】51Crで2時間標識し、洗浄し、10倍連続希釈の抗体誘導体を含むIMDM+10%FBS及びエフェクター細胞としてヒトPBLに100:1の標的に対するエフェクター比で加えたH2981腫瘍細胞のADCC特性の線グラフである。分析物を4.5時間インキュベートし、攪拌し、100μl の上清の遊離数を測定した。破壊%を〔(平均cpm−平均自然遊離)/(平均最大遊離−平均自然遊離)〕×100として算出した。数値は3回の培養の平均を示す(SEM<10%)。記号の説明:キメラL6(白ダイヤモンド形)、WTD(白四角)、DM1(白三角)、HS1(+記号)、HS2(『X』記号)及びHS3(白丸)。
【図20】PBLの代わりに補体を用いた以外は図19と同様に補体破壊を測定するために行ったアッセイの線グラフである。記号の説明:キメラL6(白ダイヤモンド形)、WTD(白四角)、DM1(白三角)、HS1(+記号)、HS2(『X』記号)及びHS3(白丸)。
【0084】
【図21】L6VL リーダー、CD3FV (V L−V H)及びFvlinkらせんリンカーのアミノ酸及び核酸配列である。
【図22】CD3FvIgが強力なチロシンリン酸化及びT細胞内のPLCγ1の活性化を刺激し、PLCγ1とpp35/36との結合を誘導することを示すPAGEゲルである。
【図23】CD3FvIgが強力なチロシンリン酸化及びT細胞内のPLCγ1の活性化を刺激し、PLCγ1とpp35/36との結合を誘導することを示すPAGEゲルである。
【技術分野】
【0001】
本出願は、1993年2月1日に出願された米国特許出願第08/013,420号の一部継続出願であり、この明細書を本発明に参考として引用する。
本明細書において、種々の文献が言及される。本発明が関係している技術を更に詳細に述べるために、これらの文献の開示をすべて本明細書に参考として引用する。
本発明は、二特異的融合タンパク質をコードする発現ベクター及び哺乳動物細胞における生物学的に活性な二特異的融合タンパク質の生産方法に関する。
【背景技術】
【0002】
血清から抗体を得るような伝統的な抗体手法又はハイブリドーマ手法に伴う課題のために、所望の結合特性とエフェクター機能をセットした抗体又は抗体誘導体分子(例えば、二特異的融合タンパク質)を設計、操作及び生産する遺伝子工学がますます頻繁に用いられてきている。
ヒト抗体を産生する安定なハイブリドーマの生産で困難に直面したことが、生体内抗体産生及び慣用の試験管内手法を克服するように設計された代替方法の開発をもたらした(Mayforth R.D., Quintans, J.(1990) Current Concepts: Designer and catalytic antibodies. New Eng. J. Med. 323:173-178; Waldmann, T.A.(1991) Monoclonal antibodies in diagnosis and therapy. Science 252:1657-1662; Winter, G., Milstein, C.(1991) Man-made Antibodies. Nature 349:293-299; Morrison, S.L.(1992) In Vitro antibodeis: strategies for production and application. Ann. Rev. Immunol. 10:239-266 )。
【0003】
治療用として、異なる標的抗原に対して2つの全抗体の結合特異性を結合する最初の試みは、化学的に結合した『異種複合体』分子を用いるものであった(Staerz, U.D., Kanagawa, O., Bevan, M.J.(1985) Hybrid antibodies can target sites for attack by T cells. Nature 314:628-631; Perez, P., Hoffman, R.W., Shaw, S., Blurstone, J.A., Segal, D.M.(1985) Specific targeting of cytotoxic T cells by anti-T3 linked to anti-target antibody. Nature 316:354-356; Liu, M.A., Kranz, D.M., Kurnick, J.T., Boyle, L.A., Levy, R., Eisen, H.N.(1985) Heteroantibody duplexes target cells for lysis by cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. USA 82:8648-8652; Jung, G., Ledbetter, J.A., Muller-Eberhard, H.J.(1987) Induction of cytotoxicity in resting human T lymphocytes bound to tumor cells by antibody heteroconjugates. Proc. Natl. Acad. Sci. USA 84:4611-4615; Emmrich, F., Rieber, P., Kurrie, R., Eichmann, K.(1988) Selective stimulation of human T lymphocyte subsets by heteroconjugates of antibodies to the T cell receptor and to subset-specific differentiation antigens. Eur. J. Immunol. 18:645-648; Ledbetter, J.A., June, C.H., Rabinovitch, P.S., Grossmann, A., Tsu, T.T., Imboden, J.B.(1988) Signal transduction through CD4 proximity to the CD3/T cell receptor. Eur. J. Immunol. 18:525-532)。
【0004】
これらの試みは、抗標的細胞抗体に化学的に結合したマウス又はヒトCD3T細胞表面レセプターに特定されたモノクローナル抗体が細胞障害性Tリンパ球(CTL)による標的細胞の溶解の引き金となり、CTLの主要な組織不適合性複合体制限を克服したことを証明した。
二特異的抗体はハイブリッドハイブリドーマから異種ハイブリドーマ法により産生され、異種複合体に見られるものと同様の特性を試験管内で示した(Milstein, C., Cuello, A.C.(1983) Hybrid hybridomas and their use in immunohistochemistry. Nature 305:537-540; Ataerz, U.D., Bevan, M.J.(1986) Hybrid hybridoma producing a bispecific monoclonal antibody that can focus effector cell activity. Proc. Natl. Acad. Sci. USA 83:1453-1457; Clark, M.R., Waldmann, H.(1987) T-cell killing of target cells induced by hybrid antibodies: comparison of two bispecific monoclonal antibodies. J. Natl. Cancer Inst. 79:1393-1401; Lanzavecchia, A., Scheidegger, D.(1987) The use of hybrid hybridomas to target cytotoxic T lymphocytes. Eur. J. Immunol.17:105-111; Gilliland, L.K., Clark, M.R.,Waldmann, H.(1988) Universal bispecific antibody for targeting tumour cells for destruction by cytotoxic T cells. Proc. Natl. Acad. Sci. USA 85:7719-7723 )。しかしながら、この抗体は細胞融合から産生されたものである。
【0005】
異種複合体又は細胞融合からの二特異的抗体を用いて得られた有望な結果にもかかわらず、いくつかの要因のために大量の治療適用には非実用的であった。
それらの要因としては、(1)多量の異種複合体の急速な生体内クリアランス、(2)両方の種類の分子を作成するために要する骨の折れる徹底的な方法、(3)同種複合体又は一特異的抗体にはない徹底的精製の必要及び(4)低収量が挙げられる。
一般的には、異種複合体又は二特異的抗体を用いることに関連する方法には、免疫グロブリン重(H)鎖及び/又は軽(L)鎖をコードする配列を連結しない2つの異なる特異性を用いた共発現方法を含むので、ランダムH−L結合及び/又はランダム(HL)−(HL)結合の課題があり、正しい生産物が少量しか得られず且つ困難な精製計画を招いている。過度の一特異的又は非特異的タンパク質分子がある場合、精製が煩わしくなり確認が困難になることがある。
これらの課題を取り除くための努力として、単鎖二特異的又は二機能性抗体を作成するために遺伝子工学が用いられている(Haber等, 1990; Wels, W.,Harwerth, I.M.,Zwickl, M.,Hardman, N.,Groner B.,Hynes, N.E.(1992) Construction, bacterial expression and characterization of a bifunctional single-chain antibody phosphatase fusion protein targeted to the human ERBB-2 receptor. Biotechnology 10:1128-1132; A. Traunecker等(1991) EMBO Journal10(12):3655-3659)。しかしながら、この努力は有望なものではなかった。
【0006】
単鎖二特異的又は二機能性抗体が細菌系において産生されている。しかしながら、この融合タンパク質は不活性形態で生産された(Haber等,1990)。更に、生産されたこの融合タンパク質は、結合親和性及び/又は結合活性の低下を示すか又は所望の生産物を回収するために複雑な単離及び精製方法を要している(Haber等, 1990; Wels等, 1992a)。
単鎖一価抗体及び単鎖二機能性抗体が述べられている(Wels 等, 1992b)。これらの抗体分子は、そのサイズを最小にし且つその機能修飾を可能にするために遺伝子操作したものである。更に、この二機能性抗体は、アルカリ性ホスファターゼ細菌遺伝子の3′をscFv 遺伝子に結合したという点でのみ二機能性である。これらの二機能性抗体は単一の結合ドメイン(例えば、VL +VH ) を含み、アルカリ性ホスファターゼ遺伝子をその標的に結合した抗体を検出するために単にマーカーとして用いたものである。
FvCD3及びCD4配列を含む Janusin分子が述べられている(A. Traunecker等 “Bispecific single chain molecules (Janusins) target cytotoxic lymphocytes on HIV infected cells, EMBO Journal 10(12):3655-3659 )。 Janusin構築物は、構築物のアミノ末端にCD4分子の一部及び構築物のカルボキシ末端にCD3の結合ドメイン(即ち、VL +VH ) を含んでいる。 Janusin分子は、CD3可変部をCD4分子部分から分離するらせんペプチドリンカーを含んでいない。更に、 Janusin分子は、多量体又は集合体にしばしば見られる。集合体形成を避けるために、追加の精製をしばしば必要とする。
【発明の開示】
【発明が解決しようとする課題】
【0007】
抗体産生に関する上記課題の観点から本発明が求められている。現在、抗体手法に伴う課題、即ち、大量の特異的抗体を得ることの難しさが継続している。歴史的には、抗体は血清又はマウス由来のハイブリドーマから得られていた。しかしながら、血清はたいてい量が限られ品質が変化しうるものであった。更に、マウス由来の抗体は、非マウス体にしばしば有害な免疫応答を起こす傾向があることから、ヒト治療に対する有用性が制限されている。
抗体手法を個々に悩ます課題、たいていは実質量の機能性タンパク質分子の生産に伴う課題を克服するために、生物学的に活性な融合タンパク質の発現を促進する新規な発現ベクターを本明細書に記載する。
【課題を解決するための手段】
【0008】
本発明は、二特異的融合タンパク質を提供する。
ここではさらに、一特異的又は二特異的融合タンパク質をコードする発現ベクターを提供する。
1実施態様においては、発現ベクターは一特異的融合タンパク質をコードし(例えば、図16)、このベクターは細胞表面抗原のような標的を結合することができる第1結合ドメインをコードするDNA配列を含む組換え体一特異的単鎖カセットを含む。
もう1つの実施態様においては、発現ベクターは二特異的融合タンパク質をコードし、このベクターは標的を結合することができる第1結合ドメインをコードするDNA配列及び標的を結合することができる第2結合ドメインをコードするDNA配列を含む組換え体二特異的単鎖カセットを含み、各ドメインは異種標的を結合することができる。
本発明は、また、哺乳動物細胞における生物学的に活性な一特異的又は二特異的融合タンパク質の生産方法を提供する。本方法は、(a)哺乳動物細胞に本発明の組換え体発現ベクターを移入し、(b)段階(a)で移入した哺乳動物細胞を培養し、(c)培養した哺乳動物細胞によって産生された生物学的に活性な二特異的融合タンパク質を回収することを含む。
【発明を実施するための最良の形態】
【0009】
定義
本明細書で用いられる語句の意味を下記に説明する。
本明細書で用いられる『二特異的融合タンパク質』は、少なくとも2種の異なる標的を別々に又は同時に特異的に認識及び結合する免疫学的に反応する任意の分子を意味し、単鎖として表される。
本明細書で用いられる『一特異的融合タンパク質』は、標的を特異的に認識及び結合し、2本の免疫グロブリン重鎖、2本の免疫グロブリン軽鎖又は免疫グロブリン重鎖及び/又は軽鎖及び/又はそのいずれかの部分を含む複合体である免疫学的に反応する任意の分子を意味し、単鎖として表される。
本明細書で用いられる『発現ベクター』は、(1)所望の遺伝子又はDNA配列を発現させるのに必要なプロモーター及び他の配列(例えば、リーダー配列)及び(2)所望の配列又はDNA配列を含む核酸分子を意味する。場合によっては、核酸分子は遺伝子転写物の安定性を高めるポリA及び/又は遺伝子の転写を増強するエンハンサー配列を含んでもよく、これにより遺伝子の発現が影響される。
本明細書で用いられる『結合ドメイン』は、標的の全結合領域又はその任意部分を認識及び結合する結合部位を意味する。具体例としては、(1)抗体の単一可変部(VL 又はVH )、(2)2種以上の可変部(例えば、VL +VH ;VL +VL ;又はVH +VH )又はその相補的決定領域(CDR)又は(3)抗原(例えば、白血球抗原)又はその一部があるが、これらに限定されない。
【0010】
本明細書で用いられる『分子垂れはし』は、本明細書で記載される融合タンパク質の検出及び精製を容易にすることができる分子をコードする任意のDNA配列を包含する。
本明細書で用いられる『二特異的単鎖カセット』は、標的を結合することができる第1結合ドメインをコードするDNA配列及び標的を結合することができる第2結合ドメインをコードするDNA配列を包含し、各ドメインは別々に又は同時に異種標的を結合することができ、DNA配列でコードされた両ドメインは同一カセット上にある。第1及び/又は第2結合ドメインは、2個の可変部(VL +VH 、VH +VL 、VL +VL 又はVH +VH )又は1個の可変部(VL 又はVH )であってもよい。また、第1及び/又は第2結合ドメインは、抗原又はその一部であってもよい。抗原の適切な例としては白血球抗原があるが、これに限定されない。第1結合ドメインは発現タンパク質のアミノ末端に又はその方向に位置し、一方第2結合ドメインは発現タンパク質のカルボキシ末端に又はその方向に位置する(二特異的単鎖DNAカセットのDNA配列の5′又は3′端に各々対応する)。
本明細書で用いられる『単鎖カセット』は、その標的の少なくとも一部を認識及び結合することがきるタンパク質をコードする配列を意味する。そのタンパク質は、複数の標的を認識及び結合するために複数部位を有してもよい。
【0011】
本明細書で記載した本発明を完全に理解するために、更に下記の説明を述べる。
A.ベクター
本明細書に記載される発現ベクターは、結合ドメインをコードするDNA配列(即ち、そのコーディング配列)又は調節配列、即ち、プロモーター又は発現プラスミド内で所望の遺伝子を発現させるのに必要な他の配列(例えば、リーダー配列)を全部又は部分的に置き換えることにより修飾することができる。
修飾した発現ベクター内の置換されたDNA配列は、任意の抗体又は他のレセプターの可変部をコードしてもよい。例えば、DNA配列は、BR96抗原、CD3、L6、CD28、CTLA4又はB7を認識及び結合する抗体の可変部をコードしてもよい。更に、DNA配列は、他の細胞表面抗原に結合することができる可変部をコードしてもよい。また、結合ドメインは、白血球抗原のような抗原の全部又は一部をコードしてもよい。具体的な置換配列を挿入するかについての主要な問題は、置換された配列が所望の結合ドメインを発現することができるように『フレーム内』に位置するかどうかである。
【0012】
置換された配列は、ポリメラーゼ連鎖反応(PCR)法でクローン化される。発現ベクターに挿入することができ、順次真核細胞を形質転換することによりDNA配列を発現することができる多数のDNA配列を作製するために、PCRが用いられる。特異的配列の増幅を達成する他のクローン化法、例えば、リガーゼ連鎖反応(LCR)を用いることもできる。
発現ベクターのプロモーターは、発現に用いられる細胞の種類又は挿入されるDNA配列により、他のプロモーターに容易に置き換えられる。プロモーターの適切な例としては、サイトメガロウイルス(CMV)、トリ骨髄芽球症ウイルス(AMV)及びモロニーマウス白血病ウイルス(MMLV)が挙げられる。
未変性の単量体としての抗体は、1分子当たり2本の相同な重鎖及び2本の相同な軽鎖を含む4連鎖の高分子である。各鎖は、可変(V)部と定常(C)部からなる。軽鎖の可変部(VL )は、可変部(V)と結合部(J)遺伝子でコードされ、重鎖の可変部(VH )は可変部(V)と介在多様性領域(D)を有する結合部(J)遺伝子でコードされる。VL +JL 又はVH +DH +JH 配列でコードされた各可変部フラグメント(VL 又はVH )は、約100個のアミノ酸で構成される。これらの配列内には、抗体の結合部位を作るアミノ酸を含むと思われる相補性決定領域(CDR)と呼ばれる3個の超可変部が含まれる。CDRは、骨格領域(FR)と呼ばれる可変性の極めて低い4個の領域に散在する。
【0013】
抗体の抗原結合部位は、典型的には、VL 及びVH 領域ポリペプチドをそれらのβプリーツシート構造に結合することにより形成され、CDR領域又はその近傍には鎖の間にループを含んでいる。時には、VL +VL 対又はVH +VH (例えば、G17−2軽鎖単量体)又はVL 又はVH 単独で抗原を結合することがある。
従って、『結合ドメイン』は、次の(a)免疫グロブリン(IgG、IgM又は他の免疫グロブリン)のVL +VH 領域、(b)免疫グロブリン(IgG、IgM又は他の免疫グロブリン)のVL +VL 領域、(c)免疫グロブリン(IgG、IgM又は他の免疫グロブリン)のVH +VH 領域、(d)免疫グロブリン(IgG、IgM又は他の免疫グロブリン)の単一VL 領域又は(e)免疫グロブリン(IgG、IgM又は他の免疫グロブリン)の単一VH 領域の1種又は組合わせを含む。
本発明による発現ベクターは、抗体配列を組込んでいるベクターに限定されない。抗原及びレセプターのような他の種類のタンパク質の結合ドメインをコードする配列を用いることもできる。即ち、ベクターは、複数の異なった標的を別々に又はほとんどは同時に結合することができる二特異的融合タンパク質をコードする。
【0014】
本発明の1実施態様においては、組換え体単鎖カセットは、異なった特異性を各々有する複数の(1)可変部及び/又は(2)抗原又はその一部をコードする複合DNA配列を含む。このカッセトにおいて、可変部をコードする配列は、(a)別の可変部又は(b)抗原をコードするDNAにリンカーで結合されることが好ましく、これらはすべて縦列に配置される。典型的には、そのようなリンカーはらせん構造である。らせんペプチドリンカーは、タンパク質分子を適切に折りたたむことができる。更に、らせんペプチドリンカーは、分子の溶解性を高めることができる。単一可変部単独で(VL 又はVH )発現するのと異なり、単鎖タンパク質として2個の可変部(VL +VH ;VL +VL ;VH +VH )は、個々の可変部を短いリンカー、例えば、(Gly4 Ser)3リンカーで連結するとを必要とする。
全領域の短いリンカーは、VL +VH (FV ) フラグメントを有する免疫グロブリン鎖を結合するために用いられる(Huston, J.S., Levinson, D., Mudgett-Hunter, M., Tai, M.S., Novotny, J., Margolies, M.N., Ridge, R.J., Bruccoleri, R.E., Haber, E., Crea, R., Oppermann, H.(1988) Protein engineering of antibody binding sites: recovery of specific activity in an antidigoxin single-chain Fv analogue produced in Escherichia coli. Proc. Natl. Acad. Sci. USA 85:5879-5883; Pluckthoen, 1991)。このリンカーは、タンパク質の折りたたみでは受動体である。一般に、このリンカーは親水性及び可撓性である。
【0015】
慣用的には、FV フラグメントを有する免疫グロブリン鎖の連結には数種の手段、即ち、化学的架橋(Glockshuber等, 1991);ジスルフィド結合による自然架橋(Glockshuber等, 1990a); ジスルフィド結合のない自然結合; 及び遺伝的にコードされたペプチドリンカーによる結合がある(Bird, R.E., Hardman, K.D., Jacobson, J.W., Johnson, S., Kaufman, B.M., Lee, S.M., Lee, T., Pope, S.H., Riordan, G.S., Whitlow, M.(1988) Single-chain antigen-binding proteins. Science 242:423-427; Huston 等, 1988a)。
単鎖カセットは、複数のリンカーを含んでもよい。リンカー数について主要な制約は、本発明のベクターでコードされた機能性融合タンパク質をなお発現することができるその数である。
本発明のらせんペプチドをコードするリンカー(配列番号10、11、12)は、その誘導体分子を作製するために修飾される、即ち、分子内のアミノ酸置換により修飾される。このような誘導体分子は、らせんペプチドリンカーの機能性を保持する。即ち、この置換を有する分子は、本発明の新規な発現ベクターによってコードされた生物学的に活性なタンパク質生産物をなお発現することができる。
【0016】
これらのアミノ酸置換としては、『保存的』として当該技術において既知のアミノ酸置換があるが、必ずしもこれに限定されない。
例えば、タンパク質においてしばしば『保存的アミノ酸置換』と称するある種アミノ酸置換をタンパク質の構造又は機能を変えずに行うことができることは、タンパク質化学の十分に確立された原理である。このような置換としては、疎水性アミノ酸のいずれかをイソロイシン(I)、バリン(V)及びロイシン(L)のいずれかに;グルタミン酸(E)をアスパラギン酸(D)に又はその逆に;アスパラギン(N)をグルタミン(Q)に又はその逆に;及びトレオニン(T)をセリン(S)に又はその逆に置換することが挙げられる。
他の置換も個々のアミノ酸の環境及びタンパク質の三次構造におけるその役割によっては保存的とみなすことができる。例えば、アラニンとバリン(V)のように、グリシン(G)とアラニン(A)もしばしば交換される。
【0017】
相対的に疎水性のメチオニン(M)は、ロイシン及びイソロイシンとしばしば交換され、時にはバリンと交換される。リシン(K)及びアルギニン(R)は、アミノ酸残基の重要な特徴がその電荷であり、これらの2つのアミノ酸残基のpKの違いが重要でない場所ではしばしば交換される。また、他の置換も個々の環境において『保存的』とみなすことができる。
軽鎖及び/又は重鎖配列を有する抗体可変部及び非抗体結合ドメインを含む融合タンパク質をコードするベクターは、本発明に包含される。第1及び/又は第2結合ドメインは、抗体の可変部であってもよい。また、第1及び/又は第2結合ドメインは、抗原又はその一部であってもよい。抗原の一部は、その部分が認識され、認識後これに分子が結合することができるものを包含することが好ましい。例えば、トランスメンブランタンパク質抗原においては、好ましい部分は細胞外部分である。しかしながら、他の部分も本発明に包含される。
適切な抗原及びレセプターの例としては、CD及び非CD分子があるが、これらに限定されない。
【0018】
CD分子としては、CD1、CD2、CD3/TcR、CD4、CD5、CD6、CD7、CD8、CD9、CD10、CD11、CD12、CD13、CD14、CD15、CD16、CDw17、CD18、CD19、CD20、CD21、CD22、CD23、CD24、CD25、CD26、CD27、CD28、CD29、CD30、CD31、CD32、CD33、CD34、CD35、CD36、CD37、CD38、CD39、CD40、CD41、CD42a、b、CD43、CD44、CD45、CD46、CD47、CD48、CD49、CDw50、CD51、CDw52、CD53、CD54、CD55、CD56、CD57、CD58、CD59、CDw60、CD61、CD62、CD63、CD64、CDw65、CD66、CD67、CD68、CD69、CDw70、CD71、CD72、CD73、CD74、CDw75、CD76、CDw78が挙げられるが、これらに限定されない。
非CD分子としては、B7、B7(2)、CTLA4、BR96、GP39、LFA−3、ICAM−2及びインターロイキン(IL)1−8が挙げられるが、これらに限定されない。
【0019】
例えば、CD28抗原は、ほとんどの成熟ヒトT細胞(Damle等(1983) J.Immunol. 131:2296-2300)に見られる免疫グロブリンスーパーファミリーのホモ二量体糖タンパク質である(Aruffo, A., Seed, B.(1987) Molecular cloning of a CD28 cDNA by a high-efficiency COS cell expression system. Proc. Natl. Acad. Sci. USA 84:8573-8577)。CD28抗原と反応するモノクローナル抗体(mAb)は、種々のポリクローナル刺激により起こされるT細胞応答を増加することができる。相同分子、CTLA4は、マウス細胞溶解T細胞cDNAライブラリーの微分スクリーニングにより同定されている (Brunet等(1987) Nature 328:267-270)。
本発明の発現ベクターは、多特異的融合タンパク質をコードすることができるベクター、即ち、多数の標的と反応することができる分子を包含する。例えば、本発現ベクターは、単鎖に発現することができる三特異性融合タンパク質、即ち、3種の標的を認識及び結合する融合タンパク質をコードしてもよい。また、融合タンパク質は、4種の標的を認識及び結合することができる。
【0020】
例えば、1実施態様においては、本発明のベクターによってコードされた一特異的融合タンパク質は、図16の単鎖抗体と同じものである。
本発明の1実施態様においては、発現ベクターは、(1)抗体又は細胞表面抗原の第1結合ドメインをコードするDNA配列、(2)抗体又は細胞表面抗原の第2結合ドメインをコードするDNA配列、(3)第1結合ドメインをコードするDNA配列及び第2結合ドメインをコードするDNA配列を連結するらせんペプチドをコードするリンカー及び(4)一又は二特異的融合タンパク質の検出のための分子垂れはしをコードするDNA配列を含む。
分子垂れはしは、抗体、センス又はアンチセンス相補的分子、酵素等の分子垂れはしを認識及び結合する適切な分子により同定される。分子垂れはしの例としては、Fc フラグメント、HIVフラグメント及び赤血球凝集素エピトープ配列HAIが挙げられる (Pati等(1992) Gene 114 (2):285-8)。
好ましい実施態様によれば、発現ベクターは、(1)抗体又は細胞表面抗原の第1結合ドメインをコードするDNA配列、(2)抗体又は細胞表面抗原の第2結合ドメインをコードするDNA配列、(3)第1結合ドメインをコードするDNA配列及び第2結合ドメインをコードするDNA配列を連結するらせんペプチドをコードするリンカーを含み、該ドメインの各々が同種又は異種標的又は抗原を結合することができる組換え体二特異的単鎖DNAカセットを含む。
【0021】
本発明の実験によれば、第1及び/又は第2結合ドメインは細胞表面抗原又は白血球抗原と反応することができる。細胞表面抗原としては、CD3、L6、CD28、CTLA4、CD40又はB7のような分子が挙げられるが、これらに限定されない。即ち、第1及び/又は第2結合ドメインはCD3と反応することができる。また、第1及び/又は第2結合ドメインはL6と反応することもできる。更に、第1及び/又は第2結合ドメインはCD28と反応することもできる。更に、第1及び/又は第2結合ドメインはB7と反応することもできる。更に、第1及び/又は第2結合ドメインはCD40と反応することもできる。
好ましい実施態様においては、発現ベクターはCC9−2と称し、ATCC No.69235 として大腸菌プラスミド内でアメリカンティッシュカルチュアコレクション(ATCC)に寄託されている(CC9−2はpCDM8内にL6VL リーダー配列−CD3sFV −リンカー−L6sFV −免疫グロブリンFC を有する) 。任意のDNA配列が用いられることは当業者に明らかである。適切な配列及び発現のための正しい読み枠のためのカセットとして挿入されるように、用いられるべき可変部又は分子のコーディング配列が既知でなければならないことのみが要求される。
【0022】
更に、任意の抗体の可変部をコードするDNA配列及びリガンドをコードするDNA配列を含む組換え体二特異的単鎖カセットを含む二特異的融合タンパク質をコードする発現ベクターも提供される。そのリガンドの例としては、B7、CTLA4、CD28、CD40、CD3が挙げられるが、これらに限定されない。リガンドの他の例としては、任意の白血球抗原がある。
例えば、本発明は、CD3と反応するドメインをコードするDNA配列及びL6と反応するドメインをコードするDNA配列を含む組換え体二特異的単鎖カセットを含む二特異的融合タンパク質をコードする発現ベクターを提供する。第1又は第2結合ドメインがCD3と反応してもよい。第1又は第2結合ドメインがL6と反応してもよい。
本発明は、更に、B7の少なくとも一部(例えば、細胞外部分)であるドメインをコードするDNA配列及びL6と反応するドメインをコードするDNA配列を含む組換え体二特異的単鎖カセットを含む二特異的融合タンパク質をコードする発現ベクターを提供する。
【0023】
本発明は、更に、CTLA4と反応するドメインをコードするDNA配列及びL6と反応するドメインをコードするDNA配列を含む組換え体二特異的単鎖カセットを含む二特異的融合タンパク質をコードする発現ベクターを提供する。第1又は第2結合ドメインがCTLA4と反応してもよい。第1又は第2結合ドメインがL6と反応してもよい。
また、本発明は、CD28と反応するドメインをコードするDNA配列及びL6と反応するドメインをコードするDNA配列を含む組換え体二特異的単鎖カセットを含む二特異的融合タンパク質をコードする発現ベクターを提供する。第1又は第2結合ドメインがCD28と反応してもよい。第1又は第2結合ドメインがL6と反応してもよい。
例えば、本発明は、CD3と反応するドメインをコードするDNA配列及びB7の細胞外部分をコードするDNA配列を含む組換え体二特異的単鎖カセットを含む二特異的融合タンパク質をコードする発現ベクターを提供する。
【0024】
本発明によれば、発現ベクター及び生物学的に活性な二特異的融合タンパク質の生産方法が提供される。一般に、これらの方法は、(1)抗体又は他の結合タンパク質を合成するB細胞ハイブリドーマ又は他の培養細胞からmRNAを単離し、(2)逆転写によりcDNAを合成し、(3)PCRプライマーとしてアンカー末端のオリゴヌクレオチド又は変性したオリゴヌクレオチドを用いて可変部のような結合ドメインをクローン化し、(4)そのクローン化した可変部の配列を決定し、(5)可変部遺伝子間の遺伝子融合を構築し、(6)その構築された可変部をSalI及びBclIで切断したpUCIgベクター(又は他の適切なベクター)に挿入し、(7)適切な断片配置のクローンをスクリーニングし、そのクローンの配列を決定し、(8)そのクローンをpCDM8又はpiLNXAnのような適切な発現ベクターに移し、(9)DEAE−デキストラン法のような手法でCOS細胞に移入し、(10)その移入された細胞を発現されるべき融合タンパク質に十分な時間、典型的には約72時間インキュベートし、(11)生物学的に活性な二特異的融合タンパク質を生産するためにFITCヤギ抗ヒトIgG及び/又はELISAを用いたファクスでその細胞をスクリーニングすることを含む。
【0025】
B.生物学的に活性な二特異的融合タンパク質の生産方法
生物学的に活性な二特異的融合タンパク質の生産方法を提供する。本方法は、二特異的融合タンパク質を生産するために、本発明の発現ベクターを移入した細胞を培養し、生産されたタンパク質を回収することを含む。
本発明は、更に、哺乳動物細胞における生物学的に活性な二特異的融合タンパク質の生産方法を提供する。本方法は、(a)本発明の発現ベクターを哺乳動物細胞に移入し、(b)段階(a)で移入した哺乳動物細胞を培養し、(c)培養した哺乳動物細胞によって産生された生物学的に活性な二特異的融合タンパク質を回収することを含む。
生物学的に活性な二特異的融合タンパク質の回収方法は、(a)分子垂れはしの存在によって生物学的に活性な二特異的融合タンパク質を同定し、(b)培養した哺乳動物細胞によって産生された生物学的に活性な二特異的融合タンパク質を回収するために、同定した分子垂れはしのある生物学的に活性な二特異的融合タンパク質を分子垂れはしのない分子から分離することを含む。
【0026】
下記実施例においては、哺乳動物由来の細胞が用いられるが、本発明の実施において任意の真核細胞が有効である。例としては、ヒト細胞、例えば、線維芽細胞及びヒツジ、ブタ、マウス、ウシのような他の動物由来の細胞が挙げられる。哺乳動物細胞の具体例としては、COS、HeLa、CHO、DUX、B11、Sp2/0、W138、DHK及びHEPG2細胞が挙げられる。
生物学的に活性な二特異的融合タンパク質が生体内及び生体外診断及び治療に有用な検出マーカー及び治療用薬剤に結合されることは明らかである。その生物学的に活性な二特異的融合タンパク質を結合することができる検出マーカーの中には、酵素、常磁性イオン又は化合物、アビジン−ビオチン特異結合対のもの、蛍光団、発色団、化学発光団、重金属及び放射性同位元素がある。生物学的に活性な二特異的融合タンパク質を結合することができる治療用薬剤の中には、抗腫瘍剤、リンホカイン及び毒素がある。
【0027】
C.本発明の発現ベクターを移入した細胞
発現ベクターの哺乳動物細胞への導入は、リン酸カルシウム移入(Graham, Vander Eb.(1973) Virol. 52:456-467)、DEAE−デキストラン移入 (Lopata, M.A.等,(1984) Nucl. Acids Res. 12:5707) 及び電気穿孔法 (Potter, H.等 (1984) PNAS 81:7161) により行われる。移入方法の選択は、何の種類の移入が行われるかに幾分左右される。電気穿孔法及びCaPO4 移入は、共に、安定に組込まれたDNAを含むセルラインを効率よく生産するために用いられる。電気穿孔法はたいてい浮遊培養を用いて容易に行われ、CaPO4 移入はたいてい粘着細胞を用いて容易に行われる。DEAE−デキストラン移入は安定なセルラインを生産する際には十分に作用しないが、一過性のプロトコールに用いる際にはCaPO4 移入より再現性が高い。他の移入方法も当該技術において既知である。
本明細書に記載される方法で生産された発現ベクター及び新規なタンパク質は上記可変部に限定されず、一般に、任意の単鎖二特異的融合タンパク質の構築、発現及びスクリーニングに適用できる。
【0028】
D.本発明のベクターによってコードされた融合タンパク質の使用
分子垂れはしのない抗L6及び抗CD3単鎖誘導体は、L6又はCD3関連の疾患を治療又は検出するための治療又は診断に有効である。例えば、L6sFvは検出用放射性核種に化学的に結合することができ、また治療用PE40のような毒素に遺伝的に融合することができる。
短いsFvフラグメントの方が腫瘍塊をよりよく浸透することができ且つ腫瘍部位の局在を改良したので、治療用薬剤を高レベルのL6抗原を発現する腫瘍細胞に対して標的とするには少量のL6sFvが効果的である(Colcher, D., Bird, R., Roselli, M., Hardman, K.D., Johnson, S.,Pope, S., Dodd, S.W., Pantoliano, M.W., Milenic, D.E., Schlom, J.(1990) In vivo tumor targeting of a recombinant single-chain antigen-binding protein. J. Nat. Cancer Inst. 82:1191-1197; Yokota, T., Milenic, D.E., Whitlow, M., Schlom, J.(1992) Rapid tumor penetration of a single-chain Fv and comparison with other immunoglobulin forms. Cancer Res. 52:3402-3408 )。
【0029】
CD28の連結反応のように第2シグナルなしのT細胞レセプターシグナルの送達はT細胞アネルギーを招くことがあるので、CD3sFvは生体内免疫抑制の誘導に十分適している。OKT3のF(ab′)2フラグメントが免疫抑制するとともに全抗体OKT3誘導免疫抑制に関与するサイトカイン毒性を著しく低下させることが証明されている(Woodle, E.S., Thistlethwaithe, J.R., Ghobrial, I.A., Jolliffe, L.K., Stuart, F.P., Bluestone, J.A. (1991) OKT3 F(ab′)2 fragments: retention of the immunosuppressive properties of whole antibody with marked reduction in T cell activation and lymphokine release. Transplantation 52:354-360)。
【0030】
おもしろいことに、CD3単鎖単一特異的抗体誘導体が未変性抗体と異なった機能特性を示した。CD3Fv−Ig誘導体は、PLCγ1のチロシンリン酸化を強力に誘発し、未変性抗CD3mAbに比べてPLCγ1とpp35/35との結合を増加させる。この結合は、T細胞を最大限に刺激するように増加することが示されている(Kanner,S.B., Deans,J.P., Ledbetter,J.A.(1992a) Regulation of CD3-induced phospholipase C-γ1 (PLCγ1) tyrosine phosphorylation by CD4 and CD45 receptors. Immunology 75:441-447; Kanner,S.B., Ledbetter,J.A.(1992b) CD45 regulates TCR-induced signalling through tyrosine phosphorylation of phospholipase Cγ1. Biochem. Soc. Trans. 20:178-184 )。更に、T細胞のCD3Fv−Igによる刺激は、活性化を伴う細胞タンパク質のより全体のチロシンリン酸化をもたらした。この分子は未変性mAbより小さいので、CD3−ε上の結合ドメインがより利用され且つTCR/CD3結合チロシンキナーゼとの相互作用を改良することが可能である。
【0031】
動物モデル及びヒトにおける臨床試験において、抗TCR又は抗CD3mAbは標的細胞抗原に架橋する際に標的細胞に対してT細胞応答を強めることが示されている。異種複合又は二特異的mAbは、細胞障害性Tリンパ球又はリンパ球活性化キラー細胞を活性にして悪性標的細胞 (Staerz等, 1986a; Perez等, 1985a; Perez, P., Hoffman, R.W., Titus, J.A., Segal, D.M. (1986) Specific targeting of human peripheral blood T cells by heteroaggregates containing anti-T3 crosslinked to anti-target cell antibodies. J. Exp. Med. 163:166-178; Liu等, 1985a; Staerz 等, 1986b)又はウイルス感染標的細胞(Paya, C.V., McKean, D.J., Segal, D.M., Schoon, R.A., Schowalter, S.D., Leibson, P.J. (1989) Heteroconjugate antibodies enhance cell-mediated anti-herpes simplex virus immunity. J. Immunol. 142:666-671; Zarling, J.M., Moran, P.A., Grosmaire, L.S., McClure, J., shriver, K., Ledbetter, J.A.(1988) Lysis of cells infected with HIV-1 by human lymphocytes targeted with monoclonal antibody heteroconjugates. J. Immunol., 140:2609-2613; voss, L.M., David, C.S., Showalter, S.D., Paya, C.V., Liebson, P.J.(1992) Heteroconjugate antibodies enhance cell-mediated anti-herpes simplex virus immunity in vivo. Int. Immunol. 4:417-420) を破壊する。
【0032】
抗CD3mAbは強力なサイトカイン毒性を生体内で誘導することができる(Abramowicz, D., Schandene, L., Goldman, M., Crusiaux, A., Vereerstraeten, P., De Pauw, L., Wybran, J., Kinnaert, P., Dupont, E., Toussaint, C.(1989) Release of tumor necrosis factor, interleukin-2, and gamma interferon in serum after injection of OKT3 monoclonal antibody in kidney transplant recipients. Transplantation 47:606-608) が、二特異的mAbで治療した患者は、患者のT細胞を試験管内で試薬で前処理してから再投与したので、サイトカイン毒性を生じない(Mezzanzanica, D., Canevari, S., Colnaghi, M.I.(1991) Retargeting of human lymphocytes against human ovarian carcinoma cells by bispecific antibodies: from laboratory to clinic. Int. J. Clin. Lab. Res. 21:159-164; Nitta, T., Sato, K., Yagita, H., Okumura, K., Ishii, S. (1990) Preliminary trial of specific targeting therapy against malignant glioma. Lancet 335:368-371)。マウスモデルにおいて、F(ab′)2フラグメントのような未変性mAbより小さい分子は、一特異的抗腫瘍特異性及び二特異的抗CD3、抗腫瘍特異性の双方に対して腫瘍局在の増加を示している(van Dijk, J., Zegveld, S.T., Fleuren, G.J., Warnaar, S.O.(1991) Localization of monoclonal antibody G250 and bispecific monoclonal antibody CD3/G250 in human renal cell carcinoma xenografts: relative effects of size and affinity. Int. J. Cancer 48:738-743; Nelson, H., Ramsey, P.S., Kerr, L.A., McKean, D.J., Donohue, J.H.(1990) Regional and systemic distribution of anti-tumor x anti-CD3 heteroconjugate antibodies and cultured human peripheral blood lymphocytes in a human colon cancer xenograft. J. Immunol. 145:3507-3515)。
【0033】
CD3−L6FvIg分子は、新規な分子として哺乳動物がん治療、例えば、ヒトがん治療に有効である。この分子は、CD3及び腫瘍抗原L6双方の結合特異性を含み、L6陽性腫瘍細胞の存在下にT細胞増殖を誘導し且つこれらの細胞に対してCTL破壊を特定することが試験管内で示されている。この2つの異なった結合特異性は、ヒトIgG1のヒンジ、CH2及びCH3ドメインに結合して初めは垂れはしとして二特異的分子の確認及び精製のために作用する。この垂れはしは、これ自体垂れはしの付いた二特異的タンパク質の小さな全サイズが未変性IgGの約2/3まで残る相対的に小さなドメイン(約50kDa)である。この垂れはしはヒト由来であるので、この分子の全体の免疫原性は全マウスIgGより著しく低いことが予想される。骨格領域を人化し、また、既知のヒトタンパク質のらせんに似るようにらせんリンカーの溶媒接触部分を構築するような二特異的タンパク質の免疫原性を更に低下させるための追加の修飾も行われる。Ig部分は親和性尾部として作用したが、生体内で二特異的タンパク質の半減期を増加させることもできる。
【0034】
1報告書において、ヒトIgG1定常部に融合したキメラマウス抗結腸直腸がんmAb可変部(Steplewski, Z., Sun, L.K., Sherman, C.W., Ghrayeb, J., Daddona, P., Koprowski, H.(1988) Biological activity of human-mouse IgG1, IgG2, IgG3, and IgG4 chimeric monoclonal antibodies with antitumor activity. proc. Natl. Acad. Sci. USA 85:4852-4856) を転移結腸がんの10人の患者に投与すると、循環時間が6倍増加することが示されている(LoBuglio, A.F., Wheeler, R.H., Trang, J., Haynes, A., Rogers, K., Harvey, E.B., Sun, L., Ghrayeb, J., Khazaeli, M.B. (1989) Mouse/human chimeric monoclonal antibody in man: kinetics and immune response. Proc. Natl. Acad. Sci. USA 86:4220-4224)。
更に、本発明の二特異的融合タンパク質に用いたIg尾部は、CH2ドメインで変異される(残基238でプロリンからセリンに)。この変異は、ヒトIgG1尾部とFcレセプターとの相互作用により仲介されるADCC活性を切断する。これは、生体内でCD3陽性T細胞とFcレセプターをもつ細胞との間の『相互的』破壊を防止するにちがいない(Clark等, 1987a)。最後に、二特異的分子はCOS細胞の一過性移入から発現されるが、タンパク質は安定な移入系で発現されることが可能である。
【0035】
本発明の利点: 本発明は、現在の抗体産生方法に伴う課題を克服する。
二特異的融合タンパク質を生産するに当たり他のもので直面した課題を克服するために、既存のCOS細胞発現系を適応して組換え体二特異的単鎖カセットDNAから機能性単鎖抗体誘導体を分泌することを達成した。単鎖抗体は、ヒトIgG1のFcドメインをヒト抗原(例えば、CD3及びL6)に対するマウス抗体の可変部に融合することにより構築した。Fc領域は、種々の特異性を有する融合タンパク質の同定及び精製に便利な分子垂れはしとして作用した。抗体のこのセグメントによって与えられるエフェクター機能は、ある種の治療に有効であある。
生物学的に活性な分子の回収は、単一結合特異性を有する分子の場合でさえ問題のある細菌発現系と異なり、本発明は、発現した生物学的に活性な融合タンパク質を含む培養上清を生じ、次いで、慣用的なアフィニティークロマトグラフィーで精製されるCOS細胞からの一過性発現を提供する。おもしろいことに、生産された単鎖二特異的融合タンパク質分子は、親抗体と異なった特性を示す。
【0036】
生物学的に活性な分子の回収が単一結合特異性のみを有する分子の場合でさえ問題があることから、これらの二特異的融合タンパク質分子の哺乳動物発現が選ばれた。哺乳動物発現により抗体誘導体の簡便で急速な産生が得られ、分子間の確認、評価及び比較が相対的に短時間で可能なためには重要なことである。
個々のタンパク質ドメインが相互交換可能な組換え体二特異的単鎖カセットDNA上にある遺伝子融合は、新規な組合わせを作り、急速な交換を行い且つ所望の機能を行うのにそれらの効能に対して種々のドメインをスクリーニングするための可能性が生じる。一過性移入系における構築物の発現は、初めの組換え及びスクリーニング段階の場合の他の生産方法より好ましい。このスクリーニングから所望のサブセットの特徴を示す分子だけが、大量生産用の第2発現系にシャトルすること、長いあるいは複雑な作製を排除すること及び作成した大多数の分子の単離操作を必要とする。
【0037】
相互交換可能なDNAカセットを有する組換え体二特異的単鎖DNAカセット、即ち、単鎖可変部又は別のものに交換することができる他の結合ドメインをコードするDNAカセットとしてより大きな分子に構築された抗体結合部位の急速な構築、発現及び分析系を開発することに着手した。第1及び第2結合ドメインの配列は、置換あるいは相互交換することができる。異種の配列は既存のものと置き換えてもよい。
前述の構築物と異なり、第1又は第2結合ドメインのいずれか又は双方に任意の可変部を配置することができるので、この二特異的単鎖カセットは有用である。
抗体構造ドメインの新規な組合わせを構築することにより、例えば、2つの関連のない結合特異性及び所望のエフェクター機能を結合する分子が作製され、治療可能性が改良される。そのような二特異的単鎖抗体誘導体が2つの非相互作用細胞表面レセプターのアダプター分子として作用すると人工レセプター−リガンド対を生じる。
【0038】
ここでのデータは、組換え体二特異的単鎖DNAカセットから発現された二特異的融合タンパク質が、多くの組織培養操作又はCD3に対する毒性一特異的抗体の可能性をなくす精製を必要としないで生体内及び生体外でヒト腫瘍発現L6に対するT細胞による細胞障害性応答を標的とすることができることを示している。
腫瘍細胞結合及びT細胞結合及び活性化に対する特異性を結合する単鎖抗体誘導体は、ヒト疾患に対する単一のモノクローナル抗体に基づく治療より著しい改良を与える。ここに記載した遺伝子融合の設計、構築、発現及び試験の方法は、関連のない分子の機能性ドメイン間の新規な組合わせを単一分子内で共に機能する能力について試験する場合、迅速性、簡便さ及び再現性において著しい利点を与える有用なものである。
下記の実施例において、本発明を具体的に説明する。実施例は本発明を更に理解するために記載されるが、前記特許請求の範囲で記載した本発明を限定するものではない。
【実施例1】
【0039】
材料及び方法:
発現ベクターの修飾:
スタッファーフラグメントを得られた融合タンパク質に対してある機能特性を与えるいくつかの短いスタッファーフラグメントに置き換えることにより、プラスミドpCDM8及びpiLNXAnを修飾した。
哺乳動物発現ベクターは、ベクターの上部に沿って示されている修正と追加が行われた図で表される。融合カセットの発現はCMVプロモーターによって進み、細菌及び哺乳動物細胞における複製はベクターの下部に示された適切な起源によって得られる。ベクターは、適切な宿主細菌株内のp3プラスミドに存在するアンピシリン及びテトラサイクリン遺伝子のナンセンス変異を抑圧するsupF遺伝子を含む。終結及びポリA付加シグナルは、SV40から適切な領域で設けられる。
【0040】
スタッファー領域の5′端のHindIII部位を用いて抗L6抗体(本明細書では抗L6とも呼ばれる)の軽鎖可変部から得られた融合タンパク質を分泌するためのリーダー配列を含むHindIII−SalIカセットを挿入した(図1)。この配列は、HindIII及びSalI付着端突出部分を有する72−mer相補的オリゴヌクレオチドにコードされた。使用したセンスオリゴヌクレオチドはL6VL-LP5/AGC TTA TGG ATT TTC AAG TGC AGA TTT TCA GCT TCC TGC TAA TCA GTG CTT CAG TCA TAA TGT CCA GAG GAG(配列番号1)であり、相補的オリゴヌクレオチドはL6VL-LP3/TCG ACT CCT CTG GAC ATT ATG ACT GAA GCA CTG ATT AGC AGG AAG CTG AAA ATC TGC ACT TGA AAA TCC ATA (配列番号2)であった。センスオリゴヌクレオチドをポリヌクレオチドキナーゼでリン酸化し(Boehringer-Mannheim, Indianapolis, ID) 、連結反応の前に、以前に発表された方法(Sambrook, J., Fritsch, E.F., Maniatis, T.(1989) Molecular cloning - a laboratory manual, second edition. ISBN 0-87969-309-6) に従って補体にアニールした。オリゴヌクレオチドの1本だけをキナーゼして複数の縦列挿入を防止した。
【0041】
更に、スタッファーフラグメントの3′端のXbaI部位を用いてカルボキシル末端のBclI及びXbaI制限部位に隣接した分子垂れはし又は簡単な停止コドンを挿入した(図1)。試験した分子垂れはしは、ヒトIgG1、gp110のV3ループからのヒト免疫不全ウイルス(HIV)ペプチド、FLAGペプチド及びヒトC−κからの定常部ドメインを含んだ。ヒトIgG1配列を、骨髄腫発現ヒト−マウスキメラL6のRNAからの結合逆転写(トリ骨髄芽球症ウイルス; Life Sciences, St. Petersburg, FL)及び/又はPCR反応によりキメラL6トランスフェクトーマのRNAから単離した。ヒンジあるいはCH2領域に適切な変異を含む前進プライマーを用いてPCR反応からFcドメインの各種変異誘導体を構築した。
【0042】
2つの異なった結合特異性、ヒトL6腫瘍抗原及びヒトCD3−εの可変部を挿入及び発現することにより、修飾発現ベクターを試験した。単鎖抗体誘導体は、融合される分子垂れはしによって、種々の結合活性及び親和性を有する抗原を結合した。CH1及びCκドメインなしにもかかわらず、ヒトIgG1のFcドメインが未変性抗体の結合特性を再現する点で最も満足すべき垂れはしであった。他の各種垂れはしを構築及び試験したが、Cκ(Traunecker, A., Lanzavecchia, A.M., Karjalainen, K.(1991) Bispecific single chain molecules(Janusins)target cytotoxic lymphocytes on HIV infected cells. EMBO J. 10:3655-3659) 及びFLAGペプチドは共に確実に機能しなかった。ヒト免疫不全ウイルスによってコードされたgp110のV3ループからのペプチドRKSIRIQRGPGRAFVTIGKI(配列番号3)も親和性尾部として用い、ペプチド特異的mAb110.3によって認識された。この垂れはしは融合されるFvによって可変結果を生じ、L6に融合される際には適切に機能せず、CD3に融合される際には満足に機能した。
【0043】
ペプチド29(RKSIRIQRGPGRAFVTIGKI)(配列番号3)は、HIVのgp110のV3ループセグメントに相当する(HIVペプチド)。付着末端突出部分を有する2本の76−mer相補的オリゴヌクレオチドをアニールして、分子垂れはしを作った。センスオリゴヌクレオチドはBclI突出部分、V3ループ配列及び停止コドンHIVSTOP5 GA TCA AGA TCC GCG GAA ATC GAT TAG AAT CCA GAG AGG CCC TGG GCG CGC CTT CGT TAC GAT CGG CAA GAT CTA GT(配列番号4)を含み、相補的プライマーはXbal突出部分HIVSTOP3/CTA GAC TAG ATC TTG CCG ATC GTA ACG AAG GCG CGC CCA GGG CCT CTC TGG ATT CTA ATC GAT TTC CGC GGA TCT T(配列番号5)を含んだ。センスオリゴヌクレオチドをリン酸化し、非リン酸化逆プライマーにアニールした後、上記BclI−XbaI消化ベクターに連結した。
アミノ末端垂れはしとして有用なIBI製のFLAGペプチドが垂れはしの付いたタンパク質のカルボキシル末端に配置した際になお作用するかを試験した。次の51−mer配列:FLAG5/GAT CAA GAC TAC AAG GAC GAC GAT GAC AAG TGA GCG GCC GCG AAT TCG TC(配列番号6)及びFLAG3/CTA GAG ACG AAT TCG CGG CCG CTC ACT TGT CAT CGT CGT CCT TGT AGT CTT(配列番号6)を含む相補的オリゴヌクレオチドを設計した。これらの配列をキナーゼし、アニールし、抗体結合ドメインに対するベクター末端に連結した。ヒトCκ配列を上記のようにキメラL6RNAから逆転写及びPCRにより得た。Cκを単離するための前向きプライマーはL6CK5BCL/GGT GCT CTG ATC ACT GTG GCT GCA CCA TCT GTC TTC ATC (配列番号8)であり、逆向きプラマーはL6CK3XBA/CCT CCT CAT TCT AGA CTA ACA CTC TCC CCT GTT GAA GCT (配列番号9)であった。
【0044】
CD3とL6結合ドメインの間に組換え体二特異的単鎖カセットを作るために用いられるらせんペプチドリンカーをGATC付着端突出部分を有する78−mer相補的オリゴヌクレオチドにコードした。センスオリゴヌクレオチドは、Fvlink1/GAT CAA TCC AAC TCT GAA GAA GCA AAG AAA GAG GAG GCC AAA AAG GAG GAA GCC AAG AAT CTA ACA GCC TCG AGA GC (配列番号10)であり、アンチセンスオリゴヌクレオチドはFvlink2/GAT CGC TCT CGA GGC TGT TAG ATT TCT TGG CTT CCT CCT TTT TGG CCT CCT CTT TCT TTG CTT CTT CAG AGT TGG ATT(配列番号11)である。コードされたペプチドは親水性であり、分子の溶解性を高める荷電したアミノ酸(DQSNSEEAKKEEAKKEEAKKSNSLESL)(配列番号12)に富んでいる。配列モチーフ (EEAKK)n は、荷電したアミノ酸に富んでいるために特に親水性である。
PCR反応(100μl の全反応量中)は、20μモル各dNTP、50−100ピコモルプライマー、1−10ng鋳型及びTaqポリメラーゼ (Stratagene又はBoehringer-Mannheim)を含むTaqポリメラーゼバッファー(Stratagene, Torrey Pines, CA 又はBoehringer-Mannheim)中で行った。反応は、パーキン−エルマーセタスターミナルサイクラーを用いて、典型的には94℃で1分、55℃で1分及び72℃で1分の工程からなる30サイクルプログラムで行った。連結産物をMC1061/p3に形質転換し、適切な挿入プラスミドのコロニーをスクリーンした。陽性クローンをDNA配列決定及びミニ移入により検定した。
【0045】
V領域の単離:
キメラL6トランスフェクトーマのRNAをNP−40急速溶菌法を用いて単離し、重鎖及び軽鎖双方の全長DNAをL6及びヒト定常部として発表されている配列と同じプライマーを用いて増幅した(Hieter, P.S., Maz, E.E., Seidman, J.G., Maizel, J.V. Jr., Leder, P.(1980) Cloned human and mouse kappa immunoglobulin constant and J region genes conserve homology in functional segments. Cell 22:197-207; Liu 等(1987) Proc. Natl. Acad. Sci. USA 84:3438-3442) が、制限部位をクローン化のために結合した。これらの全長cDNAを鋳型として第2PCR反応に用いて可変部をサブクローン化した。1実施例においては、可変部のサブフラグメントを、特異的プライマーよりむしろランダム六量体を用いて作ったcDNAからPCRによりクローン化した。
【0046】
G19−4細胞由来RNAをNP−40急速溶菌法を用いて抽出した。抗CD3ハイブリドーマG19−4のVL 及びVH 配列を確立されたPCR法を用いて増幅した(Orlandi, R., Gussow, D.H., Jones, P.T., Winter, G.(1989) Cloning immunoglobulin variable regions for expression by the Polymerase Chain Reaction. Proc. Nat. Acad. Sci. 86:3833-3837)。AMV逆転写酵素(Life Sciences) 及び重鎖又は軽鎖の定常部に相補的なプライマーを用いて第1鎖cDNA合成を行った。第1鎖cDNA産物を末端トランスフェラーゼ(Stratagene, Torrey Pines, California)を用いてポリGにつないだ。次いで、100ピコモルの各プライマー及び第1鎖合成からの1−2μl の精製したG末端のcDNA遺伝子の完全なcDNAを用いてPCRを行った。ANCTAIL前向きプライマーはナンセンスDNA配列及びアンカー配列に相補的なポリC配列を含み、ANC−ER前向きプライマーはEcoRI部位及びアンカー部位の上流のナンセンス配列を含み、MHγC及びMCK−3逆向きプライマーは各々重鎖及び軽鎖の定常部に相補的な第1鎖プライマー内の配列を含んだ。プライマー配列は次の通りであった。
ANCTAIL: 5′-GCATGTGCAAGTCCGATGAGTCCCCCCCCCCCCCC-3′ 配列番号13、
ANC-ER: 5′-ACGTCGAGAATTCGCATGTGCAAGTCCGATGAGTCC -3′ 配列番号14、
MHγC: 5′-A(TC)CTCCACACACAGG(AG)(AG)CCAGTGGATAGAC-3′ 配列番号15、
MCK-1: 5′-CTTCCACTTGACATTGATGTCTTTG-3′ 配列番号16、
MCK-3: 5′-CAAGAAGCACACGACTGAGGCA-3 ′ 配列番号17
【0047】
免疫染色及びファクス分析:
Jurkat細胞、抹消血リンパ球及び/又はL6陽性腫瘍細胞(H2981又はH3639)を間接免疫染色により解析した。H2981細胞をトリプシン−EDTA(GIBCO-BRL) 中でインキュベートしてフラスコから剥がした。細胞を種々の濃度の単鎖抗体又は未変性親抗体と結合バッファー(GIBCO-BRL) 中4℃で40分間インキュベートした。第1段階後細胞を洗浄し、FITC結合第2段階試薬と4℃で更に30−40分間インキュベートした。第2段階抗体は、次のものの1種とした:マウスmAbの場合ヤギ抗マウスIg、単一Ig融合物の場合ヤギ抗ヒトIgG(Tago, Inc., Burlingame, CA)、HIVペプチド融合物の場合gp110のV3ループに特定されたFITC−110.3及び全種類のL6含有構築物の場合L6抗体に対するFITC−αイディオタイプ(1B)。40対数アンプリファイアを備えたファクスIVセルソーター(Becton Dickenson and Co., Mountain View, CA) で蛍光を分析した。
2種の抗体を全量10μg/mlの抗体まで種々の割合で共に混合した後に細胞を加えることにより、競合アッセイを行った。2種の抗体のうち1種をFITC、通常はキメラ又はマウスL6又は未変性G19−4で標識した。阻害アッセイの場合には、第2FITC結合抗体を加える30分前に、第1抗体を加えた。
【0048】
融合タンパク質の細胞培養、移入及び精製:
前記発現プラスミドをCOS細胞に移入した(Linsley, P.S., Brady, W., Grosmaire, L., Aruffo, A., Damle, N.K., Ledbetter, J.A.(1991) Binding of the B cell activation antigen B7 to CD28 costimulates T cell proliferation and interleukin 2 mRNA accumulation. J. Exp Med. 173:721-730; Aruffo and Seed, 1987a)。プラスミドDNAを全量12ml/150mmプレート中1μg/mlで移入培養液に加えた。移入したCOS細胞の3採取物からの使用済み無血清培養液をプールし、Ig、HIV又はSTOP分子垂れはしを含む融合タンパク質を精製するために使用した。細胞デブリを低速遠心により除去し、上清を0.2μm フィルターでときどきろ過した後精製した。Ig融合移入物の培養液を0.05M クエン酸ナトリウム、pH8.0(Linsley等, 1991a)で平衡化した固定化プロテインA (Repligen Corp., Cambridge, MA)のカラムに加えた。500mlの上清に対して1mlの充填床容量プロテインAを用いた。培養液を2回用いた後、カラムを100mMリン酸ナトリウム、pH8.0で洗浄し、結合タンパク質を0.05M クエン酸ナトリウム、pH3で溶離した。画分を1/5容量1M トリスpH8.0を含む試験管に集めた。A230 吸収物質のピークを含む画分をプールし、使用前にPBSで透析した。ローリー法に基づくバイオラドタンパク質アッセイキットを用いてタンパク質濃度を求めた。
【0049】
他の分子垂れはしを含む融合タンパク質の場合、ファーマシア社の説明書に従いCNBr活性化セファロース4Bを用いて適当な抗体(抗L6イディオタイプmAb13B又はgp110のV3ループに特定された抗HIVmAb110.3)を固定化して、アフィニティーカラムを作った。親和性基質は約5mgのmAb/ml床容量を含み、典型的なカラムサイズ1×5cm(4ml)とした。試料をpH7に調整し、予め0.1M クエン酸、pH2.2で洗浄し、PBS、pH7.2中で平衡化してある適切なイムノアフィニティーカラムに加えた。カラムをPBSで十分洗浄し、結合物質を0.1M クエン酸塩pH3.0で溶離し、次いで直ちにトリスで中和した。精製した抗体誘導体を最後にPBSに透析し、滅菌ろ過した。
【0050】
細胞接着アッセイ:
10mMEDTAの存在下に実質的に(Linsley, P.S., Clark, E.A., Ledbetter, J.A. (1990) T-cell antigen CD28 mediates adhesion with B cells by interacting with activation antigen B7/BB1. Proc. Natl. Acad. Sci. USA 87:5031-5035)に記載されているように接着アッセイを行った。Jurkat細胞をまず51Crで標識し、抗体刺激とインキュベートし、洗浄し、H2981腫瘍細胞とインキュベートし、顕微鏡的に試験した。H2981に対する非特異的結合を防止するために、Jurkat及びH2981単層を無関係な抗体とインキュベートしてFcレセプターを飽和した後、CD3Ig(本明細書ではCD3FvIgとも呼ぶ)、L6Ig(本明細書ではL6FvIgとも呼ぶ)又はCD3−L6Ig(本明細書ではCD3−L6FvIgとも呼ぶ)抗体誘導体を加えた。接着反応が完了した後、単層を氷冷RPMI培養液で5回洗浄し、0.5M NaOHを加えて可溶化し、γカウンターで放射能を測定した。全結合放射能(cpm) を標識細胞の比活性(cpm/細胞)で割って、結合した細胞数を算出した(図13)。
【0051】
図13において、H2981腫瘍細胞の単層を105 細胞/ウェルの密度で48ウェルプレートに播種し、0.1%パラホルムアルデヒドに23℃で20分間固定し、洗浄し、完全RPMI+10%FBSで阻止し、単独で又は指定した抗体と37℃で1時間予備インキュベートした。抗CD3(G19−4)及び抗L6mAbの異種結合体を陽性対照として用いた。指示した場合を除いてすべての抗体を20μg で用いた。Jurkat細胞を51Crで標識し、10mMEDTA中で予備インキュベートし、腫瘍細胞及び抗体に加えた。プレートを1000rpm で2分間攪拌することにより接着を開始した。反応物を37℃で45分間インキュベートし、氷冷RPMIで5回洗浄し、0.5N NaOHに溶解した。遊離した数をγ計数により求めた。データは結合した細胞数を示した(×103)。3回の測定平均と標準偏差(誤差のすじ)が示されている。
【0052】
SDS−PAGE及びウェスタンブロット法:
4%スタッカーを用いて直線勾配6−15%を生じるアクリルアミドゲルを225ボルトで3時間又は8mAmpで一晩行った。ウェスタンセミドライ移動装置(Ellard Instruments, Seattle, WA) を用いてニトロセルロース膜にゲルを130mAmpで1時間ブロットして免疫反応で検出した。ブロットを1%無脂肪乳、PBS中0.05%NP−40 (BLOTTO、即ち、阻止バッファー) で1−2時間阻止した。第1抗体をアルカリ性ホスファターゼ結合ヤギ抗ヒトIgG (Boehringer-Mannheim)とBLOTTO中1:1500の希釈度で又は非Ig融合物、即ち、Ig垂れはしのないSfV の検出用の適切な希釈度の非結合マウス又はキメラ抗体又は抗イディオタイプ抗体とインキュベートした。いずれの場合にもブロットをBLOTTOで3回洗浄し、第2工程を必要とする場合にはアルカリ性ホスファターゼ結合ヤギ抗マウス又は抗ヒトIgGとインキュベートした。ブロットをウェスタンブルー(Promega, Madison, WI)で展開し、反応を蒸留水で停止した。
【0053】
抗p−tyrとの免疫沈降及びウェスタンブロット法:
JurkatT細胞を刺激しない(0)か又は指定した濃度の未変性G19−4Mab(Ledbetter, J.A., Norris, N.A., Grossmann, A., Grosmaire, L.S., June, C.H., Ucklin, F.M., Cosand, W.L., Rabinovitch P.S.(1989) Enhancd transmembrane signalling activity of monoclonal antibody heteroconjugates suggest molecular interactions between receptors on the T cell surface. Mol. Immunol. 26:137-145)又はCD3Fv−Igで刺激し、ホスファターゼ及びプロテアーゼインヒビター(1mMオルトバナジン酸ナトリウム、1mMPMSF、2mMEGTA、0.5%アプロチニン及び10μg/mlロイペプチン)を含む修飾RIPAバッファー(Kanner, S.B., Reynolds, A.B., Parsons, J.T.(1989) Immunoaffinity purification of tyrosine-phosphorylated cellular proteins. J. Immunol. Methods 120:115-124) に溶解した。細胞溶解物を透明にし(14000rpm で10分)、ウサギ抗p−tyrあるいはPLCγ1抗血清で免疫沈降させた。免疫複合体をプロテインA−セファロースビーズ(Pharmacia, Piscataway, NJ) で回収し、洗浄した。タンパク質をSDS−PAGE(8%)で分離し、PVDF Immobilon(Millipore, Bedford, MA)に4℃で2時間移した。イムノブロットを阻止した後、阻止バッファー中0.5μg/mlのアフィニティー精製ウサギ抗p−tyrを加えた。1μCi/ml の高比活性 125I標識プロテインA(ICN Biomedicals, Costa Mesa, CA) 及びオートラジオグラフィーを用いて検出した。
【0054】
増殖アッセイ:
抹消血リンパ球をリンパ球分離培養液(Organon Teknika,Durham, NC)で希釈及び遠心して単離した。リンパ球を無血清RPMI1640で数回洗浄し、細胞濃度を10%FCSを含むRPMIで106 細胞/mlに調整した。細胞を96ウェル、平底プレートで培養した(0.2ml容量中5×104 細胞/ウェル)。3日培養の最後の6−8時間に1μCi/ml の[3H]チミジン取込みによ、3回の試料について増殖を測定した。PBLを1μg/mlPHA(Wellcome, Charlotte, NC) と培養し、PHAのない培養液で1日静止することにより、PHA活性化T細胞を調製した。
H2981腫瘍細胞を10,000ラドで照射した後、増殖アッセイに使用した。細胞を、融合タンパク質に前結合し洗浄した後PBLとインキュベートする(『前結合』試料)かあるいは3日培養で溶液に融合タンパク質と含めた(『溶液』試料)。前結合試料の洗浄は非結合タンパク質を除去するので、腫瘍細胞に結合したタンパク質のみがアッセイでPBLの刺激を与える。PBLの力価が照射細胞に対して次のように測定された:(2:1)、5:1、125:1、(625:1)、ここで最初の数は腫瘍細胞の減少数と比べて存在したPBL(5×104細胞/ウェル)の相対数を意味する。
【0055】
細胞毒性アッセイ:
H2981腫瘍細胞を[51Cr]と2時間インキュベートし後、融合タンパク質(0.1μg/ml〜10μg/ml)及びPBL(各種エフェクター:標的比10:1〜100:1で)とインキュベートした。細胞を全量0.2ml中10%FCSを含むRPMI中で5時間培養した後、計数した。γカウンターでクロミウム放出を測定して腫瘍細胞を標的とした細胞毒性を測定した。
【0056】
COS細胞は組換え体二特異的単鎖カセットDNAからの抗体を発現することができる: 急速検出、精製及び確認を容易にする抗体分子の一過性哺乳動物発現系を開発することに着手した。種々の特異性及びドメインの交換を有する分子に適応させて作成、試験及び異種単鎖二特異的抗体との間の比較を簡易化するのに、系が十分有用であることは重要なことであった。表面レセプターの細胞外ドメインとヒト免疫グロブリンIgG1の重鎖との間の融合タンパク質を作ることにより可溶性細胞表面レセプターを発現するCOS細胞一過性発現系を従来の研究者らは満足して用いている(Aruffo and Seed, 1987b; Linsley等, 1991b)。
本系は、分子を分泌し且つ培養上清から活性形態として容易に回収することができることから、細菌発現系に対して魅力のある代替品として選ばれた。最初の実験は、COS細胞一過性発現系がゲノム配列よりむしろcDNAを用いてそのままの機能性IgG分子を発現及び分泌することができるかを調べた。抗腫瘍抗原L6特異性をコードする全長κ及びγcDNAカセットをpCDM8に連結し、挿入ベクターをDEAE−デキストラン法によりCOS細胞に共移入した。培養上清がL6陽性腫瘍細胞に対して結合活性をもつ100−500 ng/mlのタンパク質レベルを含むことを見出し、COS細胞が組換え体二特異的単鎖カセットDNAから未変性抗体を構築し分泌することができることを示した。
【0057】
哺乳動物発現ベクターの適応:
そのような分子のCOS細胞発現能が証明されると、個々のタンパク質ドメインをコードする相互交換可能なカセットを用いた単鎖抗体分子を発現する発現ベクターpCDM8及びpiLNXAnを修飾した。ベクターpCDM8及びpiLNXは、調節領域の下流に位置するポリリンカー/スタッファー領域に挿入された遺伝子の発現を達成するために、サイトメガロウイルスあるいはAMVプロモーター及びエンハンサーを用いる。ポリリンカー内の可変部挿入部位に隣接する2つの短いcDNAカセットを含むように、この領域を作り変えた。図1は、ベクターの修飾を図で示したものであり、ベクターの修飾及び発現した単鎖分子の可変部の配置の模式図である。L6κ軽鎖可変部からのリーダーペプチドを含むHindIII−SaII断片をポリリンカーの5′端に挿入してこれに融合した分子の分泌を達成した。ヒトIgG1のヒンジ、CH2及びCH3ドメインをコードするBcII−XbaI、即ち、分子垂れしをポリリンカーの3′端のフレーム内に融合して種々の特異性を有する分子の検出及び精製を容易にする。重鎖及び軽鎖の可変部をコードする単鎖抗体カセットDNAを短いペプチドリンカーによって相互に結合し、これらの2つの短いフランキングカセットの間に単一のオープンリーディングフレームを作るようにSaII−BcII断片(他の適合性末端もときどき用いた)として挿入した。これにより、既存の哺乳動物発現ベクターを適応して任意の特異性を有する単鎖抗体分子(SCA)をコードするcDNAカセットの有効な発現が得られた。本系は、異種分子垂れはし、リンカー及び結合特異性をコードするカセットが可溶性機能分子の発現における相対的有効性に匹敵するように急速発現、精製、スクリーニング及び融合タンパク質の変更が可能である。
【0058】
単鎖一特異的L6FV −Ig、CD3FV 及び二特異的CD3−L6Ig抗体誘導体の構築及び発現:
2つの異なる結合特異性をモデルとして用いて適応した単鎖抗体発現系を試験した。L6腫瘍抗原及びCD3T細胞表面レセプターに特定された抗体の重鎖及び軽鎖の可変部を材料及び方法で記載したように単離した。
抗L6及びG19−4の可変部をオーバーラップ拡張PCRを用いて単一のコーディング領域に融合してVL のカルボキシル末端とVH のアミノ末端の間に(Gly4Ser)3リンカーを作った。
図2は、G19−4の軽鎖及び重鎖可変部を(Gly4Ser)3と融合して作った単のオープンリーディングフレームを示すものである。
L6軽鎖可変部のリーダーペプチド(点描領域)、CD3−L6抗原結合ドメイン(陰のない領域)及びヒトIgG1Fcドメイン(陰のある領域)をコードするcDNA構築物を材料及び方法で記載したように構築した(図2)。
【0059】
図2において、スタッファーフラグメントを除き且つ単鎖抗体分子の発現のためのアダプター配列に置き換えることにより、哺乳動物発現ベクターpCDM8を修飾した。発現分子の分泌を得るために、抗L6の軽鎖可変部のリーダーペプチドをコードするHindIII−SalI断片を挿入した。融合構築物の検出、精製及び確認を容易にするために、ヒトIgG1のFcドメインをBclI−XbaI部分として下流に含めた。HIV由来のV3ループの一部をコードする短い分子垂れはし配列もCD3構築物に用いた。リーダー配列と垂れはしの間に、VL とVH との間の融合カセットを挿入し、2つのドメインを(Gly4Ser)3アミ酸リンカーで分けた。
第1カセットとFcドメインとの間のBclI部位に第2VL −VH カセットを挿入することにより、二特異的分子を構築することができた。立体障害を防止し溶解性を改良するために、27アミノ酸らせんペプチドリンカーをコードするオリゴヌクレオチドで2つの結合特異性を相互に離した。
表示した配列は各ドメイン間の連結を示し、構築で導入されたアミノ酸は太文字体で示されている。
【0060】
可変部融合カセットを修飾発現ベクターに挿入して図2に示した遺伝子融合を作り、COS細胞に移入した。無血清使用済培養液を集め、タンパク質をプロテインAアフィニティークロマトグラフィーで精製した。還元又は非還元SDS−PAGEにかけたタンパク質のウェスタンブロットをアルカリ性ホスファターゼ結合ヤギ抗ヒトIgGで図3に示したようにプローブした。パネルA及びBは、L6Fv−Ig及びCD3Fv−Ig融合タンパク質が還元及び非還元両条件下でMr 55,000を有する単一種として移動したことを示し、このおよそのサイズはこれらの単鎖抗体誘導体が予想される。同様に、CD3−L6FvIg二特異的分子がこれらの両条件下でMr 〜94,000を有する単一種として移動した。比較すると、ヒトヒンジ−CH2−CH3の野生型配列に融合したマウス可変部からなるキメラL6mAb又はL6野生型二量体(WTD) は、還元及び変性の程度により著しい移動度差を示し、これらの分子の重鎖定常部がジスルフィド結合で結合して二量体を形成したことを示した。時折、非還元SDS−PAGEのウェスタンブロットがMr >100,000で移動する一特異的分子又はMr >200,000で移動する二特異的分子の場合に極めてわずかなバンドを示した。
【0061】
抗L6軽鎖シグナルペプチド、抗体結合特異性をコードする可変部融合及びヒトIgG1Fcドメインを含む単一のオープンリーディングフレームを作るように、VL −VH 融合カセットを適応したベクターに挿入した。短いらせんペプチドリンカーを介したCD3及びL6融合カセットを融合することにより、CD3−L6二特異的融合カセットを作り、一特異的構築物の場合のように挿入した。発現系の最初の試験に使用した分子垂れはしは、鎖内ジスルフィド結合を減じるか又は排除するためにヒンジジスルフィドをセリンに変えたヒトFcの変異誘導体とした。これらの単鎖構築物をCOS細胞に個別に移入し、融合タンパク質を固定化プロテインAによるアフィニティークロマトグラフィーで培養上清から精製した。
精製タンパク質の収量は、典型的には、L6Ig融合タンパク質の場合約2mg/リットル、CD3Igの場合約10mg/リットル及びCD3−L6Ig二特異的分子の場合約0.5mg/リットルであった。非還元SDS−PAGEにかけアルカリ性ホスファターゼ結合ヤギ抗ヒトIgGでプローブしたタンパク質のウェスタンブロットを図3に示す。Mr 55,000で移動するCD3Ig及びL6Igレーンに単一種が見られ、およそのサイズがこれらの単鎖抗体誘導体として予想されるが、陰性対照レーンには見られない(図3)。CD3−L6Ig二特異的分子は、Mr 約95,000−98,000で移動し、より高い分子量バンドがMr >200,000に見られる(図3)。
【0062】
L6Ig、CD3Ig及びCD3−L6Ig融合タンパク質の結合活性:
本発明の単鎖抗体誘導体の機能活性を調べ且つこの分子が抗原特異結合することができることを証明するために、まずFcドメイン融合タンパク質の細胞発現標的抗原に対する結合を試験した。ヒト腫瘍細胞系H2981は、高レベルのL6標的抗原を発現するが、CD3又はBR96を発現せず、ヒトJurkatセルラインはCD3を発現するが、L6又はBR96を発現しない。第2段階試薬としてFITC結合ヤギ抗ヒトIgを用いて蛍光活性化セルソーター分析により、結合を検出した。図3に示されるように、融合タンパク質は未変性親抗体(即ち、未変性抗L6及びG19−4抗体)と同様の細胞発現標的抗原に特異的に結合するが、検出可能レベルの抗原のない細胞には結合しない。CD3−L6Ig二特異的融合タンパク質は、Jurkat及びH2981細胞の双方に結合し、これらの分子が1種以上の特異性を有することを間接的に示した。ヤギ抗ヒトIgG又はL6結合特異性に特定されたFITC結合抗イディオタイプ抗体を用いて検出が得られるかでも同様の結果が見られた。
【0063】
10μg/mlの抗体誘導体をH2981腫瘍細胞(L6陽性)、Jurkat細胞(CD3陽性)又はH3396腫瘍細胞(BR96陽性)とインキュベートした(図4)。細胞を洗浄し、第2段階試薬としてFITC結合ヤギ抗ヒトIgGとインキュベートした。次いで、合計10,000個の染色細胞をファクスで分析した(図4)。図3は、L6Ig、CD3Ig及びCD3−L6Igが細胞発現標的抗原に結合することを示している。
融合タンパク質の生物学的特性を調べるために、個々の分子の予想又は所望の特性に基づいて種々の機能アッセイを用いた。従って、各単鎖分子の結果が別々に存在し、L6Ig単鎖抗体誘導体から始める。
【0064】
L6融合タンパク質の結合活性に関する種々の分子垂れはし配列の影響:
各種分子垂れはし配列を材料及び方法で記載したように構築し、L6結合ドメインに融合し、これらの領域を用いてプロテインAに対する結合標的又はそれらを標的とした特異抗体として発現融合構築物を検出及び精製することができるかを求めた。L6結合ドメインに直接融合した場合、Cκ、HIVペプチド及びFLAGペプチドはすべて信頼できる分子垂れはしとして機能しなかった。これら3種のペプチドはすべて、認識可能なL6を発現する移入細胞をもたらさなかった。検出が分子垂れはしによらずL6抗イディオタイプ抗体を利用する場合でさえ、H2981腫瘍細胞に対する結合アッセイにおいて機能性融合タンパク質を検出できなかった。
【0065】
L6誘導体によるエフェクター機能に関する種々のFcドメインの影響:
分子垂れはしのないL6結合ドメインの単純sFv(sFv)及び異種Fcドメイン変異体に結合した各種−Ig融合物を含む他のL6誘導体を構築した。図5及び図6に示されるように、ヒンジ及び/又はCH2ドメインに導入された変異に基づく呼称を各Fc構築物に示した。野生型二量体 (WTD)は、すべてのFcドメイン配列に対して野生型であり、この領域内のアミノ酸残基に対して未変性抗体と同一である。単量体構築物は配列置換、即ち、システイン残基を含み、ヒンジジスルフィドをセリンに変異する (HS1)。単量体1(mut1としても知られる)(HS2)は、IgG1エフェクター機能を仲介するに当たり重要な領域であるCH2ドメイン内の残基238においてプロリンをセリンに置き換わることを含む。単量体(HS3) は、このCH2領域内の数個の残基(234−238)に対して変異する。二量体mut1(DM1) はFcドメインのヒンジ領域に野生型配列を含むが、残基234−238をコードするCH2配列の変異体でもある。
【0066】
L6のSfvは、L6VL −VH 融合カセットの後に他の分子垂れはし配列よりむしろ停止コドンを含んでいる。これらの分子の各々を構築し、COS細胞に移入し、プロテインAセファロースあるいは固定化L6抗イディオタイプ抗体カラムを用いて精製タンパク質を発現した。飽和及び阻害結合分析において、融合タンパク質を未変性キメラL6抗体及びFab′誘導体と比較した(図7−8)。L6Fc変異体は未変性抗体と極めて類似した飽和曲線を生じる(図7)が、Sfv融合タンパク質は十分に結合しなかった(図6)。増加量の試験抗体を腫瘍細胞とインキュベートした後、FITC結合キメラL6を加えることにより阻害試験を行った。再度、すべての−Ig融合物及びキメラ抗体に対して同様の曲線が見られ(図8)、Sfv及びFab′は未変性抗体の結合を阻害する能力が減少した(図7)。未変性抗体の結合と拮抗又は阻害しないにもかかわらず、Sfv融合タンパク質はこの実験において化学的に調製されたL6Fab分子よりわずかに良好であった。
【0067】
ヒトIgG1に関与するエフェクター機能を仲介するこれらの分子の相対的能力を、抗体による細胞の細胞毒性(ADCC)又は補体による細胞毒性(CDC)アッセイにより測定し、特異的溶解を抗体濃度の関数としてプロットした。ヒンジ領域に欠失のある変異体はすべてADCC仲介能に影響しなかったが、CH2ドメイン変異体はいずれもこの過程を仲介しなかった。驚くべきことに、キメラL6はCDCをかなり良好に仲介するが、L6誘導体はいずれも腫瘍標的の補体媒介性溶解を刺激することができたことを発見した。
ヒト腫瘍抗原L6に特定されたマウス抗体の可変部をCOS細胞で発現したヒトIgG1のFcドメインの各種誘導体に融合し、融合タンパク質をアフィニティークロマトグラフィーで精製した(図17−20)。
構築した各Fcドメインが示され、配列置換は下線を引いたアミノ酸で示される。各構築物の精製タンパク質は、飽和及び阻害アッセイにおいてキメラL6に匹敵した。更に、H2981腫瘍細胞を51Crで2時間標識し、洗浄し、これに10倍連続希釈の抗体を含むIMDM+10%FCS及びヒトPBL(ADCC)あるいはウサギ補体(CDC)に加えることにより、これらの分子のADCC及びCDCのような正常なIgG1エフェクター機能を仲介する能力を測定した。検定物を4.5時間インキュベートし、攪拌し、遊離した数をγカウンターで測定した。数値は、3回の培養の平均を示した(SEM<10%)。
【0068】
CD3単鎖抗体は未変性抗体と量的に異なる他は質的に同様な生物活性を示す:
−Ig融合物、−HIVペプチド融合タンパク質、VL −VH sFv(垂れはしなし)及びCD3−L6Fv−Ig二特異的分子を含む各種CD3融合タンパク質を構築した。
HIVペプチドは、CD3に融合した場合信頼できる分子垂れはしとして作用した。3単鎖融合タンパク質の検出及び精製を可能にした。垂れはしを付けた分子を固定化プロテインA(−Ig融合物)又は110.3抗体(HIV融合物)を用いてアフィニティークロマトグラフィーで精製した。単純Sfvはろ過した上清溶液として用い、G19−4抗体のJurkat細胞に対する結合を阻害する上清の能力を測定することにより近似の濃縮物を評価した。これらの変質した分子のCD3T細胞レセプター複合体に対する結合によって生じた細胞応答を調べた。各分子をindo−1の入った末梢血リンパ球又はT細胞に結合し、細胞内カルシウムの移動をフローサイトメトリーによりモニターした。図9−12に示されるように、未変性G19−4抗体の等価濃縮物と比べるとIg及びHIV融合タンパク質の双方が膜貫通シグナル活性を高めた。単純Sfvはカルシウムシグナルを生じるが、未変性抗体に見られるように強くも長くもなかった。
【0069】
一及び二特異的誘導体の連結配列を含む抗CD3可変部の配列を図21に示す。Ig融合タンパク質は固定化プロテインAを用いたアフィニティークロマトグラフィーで精製し、Sfvはろ過上清として用いた。既知濃度の親G19−4MabのJurkat細胞に対する結合を遮断する移入上清の能力を測定することにより、Sfvの濃度を推定した。CD3Fv−Ig分子のシグナル活性を分析するために、PLCγ1のチロシンリン酸化誘導能を試験した。JurkatT細胞をCD3Fv−Ig又は未変性G19−4Mabで刺激し、細胞溶解物のタンパク質をSDS−PAGEで分離し、PDVF膜に移し、抗p−tyr又は抗PLCγ1でプローブした。驚くべきことに、我々は、CD3Fv−IgがPLCγ1を含む全細胞溶解物中の細胞タンパク質のチロシンリン酸化を強力に誘導し且つ多量のpp35/36リンタンパク質がPLCγ1と結合していることを見出した(図22−23)。更に、CD3Fv−Ig及びCD3Sfvの双方がPBL中でカルシウムフラックスを誘導し、未変性G19−4Mabの等価濃度に見られるものより程度が高かった。
【0070】
図21においては、抗CD3MabG19−4の重鎖及び軽鎖の可変部がPCRでクローン化されている。これらのcDNAカセットから構築した遺伝子融合のヌクレオチド及びタンパク質配列を(Gly4Ser)3リンカーと共に太文字体で示。VL −VH 融合カセットのアミノ末端をSalI部位でL6軽鎖可変部リーダーペプチドに融合し、カルボキシ末端をBclI部位でFcドメインのヒンジ領域に直接融合するか又は短い『らせん』ペプチドリンカーに融合して二特異的CD3−L6FvIg抗体誘導体を構築した。L6の可変部は、図1及び2に示されるように、『らせん』リンカーの反対の端にフレーム内で融合した。
図22−23においては、示されているように、Jurkat細胞(107/試料)5、20又は80μg/mlの未変性抗CD3MabG19−4又は1、5又は20μg/mlのCD3Fv−Igで1分又は3分間刺激した。細胞溶解物のタンパク質をウサギ抗p−tyr(パネルA)又はPLCγ1抗血清(パネルB)で免疫沈降させ、プロテインAセファロースで回収し、これをSDS−PAGEで分離し、ニトロセルロースに移し、精製したウサギ抗p−tyr及び125I標識プロテンAで検出した。
【0071】
図9−12は、CD3Fv融合誘導体が異なったレベルの膜貫通シグナル活性を示す線グラフである。図9−10は、CD3FvIgが末梢血T細胞内で細胞内カルシウムを移動することを示すものである。フローサイトメトリー及びカルシウム結合染料を用いて、CD3FvIg(−・−・−)、未変性CD3Mab( ---- )又は非T細胞結合対照L6FvIg( .... )による刺激に伴う細胞内遊離カルシウムの濃度をモニターした。各刺激(2μg)をindo−1の入った充填T細胞に1分の時点(矢印)130nm=静止細胞で及び%応答細胞のように加えた。
図11−12は、CD3FvIgで前処理すると次のクロスリンクしたCD2の刺激に対して末梢血T細胞の応答を脱感作することを示している。indo−1の入った細胞を1μg のCD3FvIg、2μg の未変性CD3mAb又はL6FvIg(非T細胞結合対照)と37℃で15分間インキュベートした。ビオチニル化抗CD2mAb(5μg )を1分の時点で加え、5分の時点でアビジン(20μg )によってクロスリンクした。次いで、クロスリンクしたCD2に対する応答を5分間モニターした。データは、([Ca2+ ])130nm=静止細胞(ネルA)及び%応答細胞(パネルB)として示されている。
【0072】
種々の濃度のCD3抗体誘導体を用いた刺激に応答する末梢血リンパ球及び精製T細胞の増殖アッセイにおいて、3誘導体すべてがT細胞を活性化することを見出した。1μg/mlの抗体処理において測定した増殖応答を表1に示す。増殖応答の同様のパターンが0.2及び5μg/mlにおいても見られた。データは、CD3誘導体がマウス抗体の未変性G19−4(CD3Fv−Igと比較)あるいはFabフラグメント(sFvと比較)で見られたものと同様のT細胞内増殖応答をもたらすことを示している。未変性G19−4で見られるものよりPBLの強い増殖を生じるPMAとの相乗作用で作用するCD3Fv−Igを除き、遺伝子操作した分子は精製T細胞のわずかに強い増殖レベル及びPBLのわずかに弱いレベルを刺激した。
2種の垂れはしを付けたCD3融合タンパク質が未変性活性より強い膜貫通シグナル活性を示したので、これらの分子がT細胞増殖にどのように影響するかを求めた。末梢血リンパ球及び精製T細胞を種々の濃度の抗体誘導体と72時間インキュベートし、トリチウムを入れたチミジンで6時間標識した後、回収し、計数した。表1に1μg/mlにおける抗体処理の結果を示す。増殖応答の同様のパターンが0.2及び5μg/mlにも見られた。
【0073】
【表1】
【0074】
細胞を1μCi/ウェル[3H]チミジンで6時間パルス標識して72時間培養し後増殖を測定した。T細胞を2種のプラスチック粘着で単核細胞及びB細胞から離し、次いでナイロンウールカラムに通過させることにより精製した。
データは、ここで試験した条件下、CD3誘導体の結合がマウス抗体のG19−4(Ig融合)あるいはFabフラグメント(HIV及びSTOP融合)で見られるのと同様のT細胞の増殖応答をもたらすことを示している。遺伝子操作した分子は、精製T細胞のわずかに強い増殖レベル及びPBLのわずかに弱いレベルを刺激する傾向があったが、ほとんどの場合、それらの差はわずかである。このパターンの唯一の例外は、CD3sFvIg及びCD3HIV構築物がPMAと相乗作用して生じた未変性G19−4に比べて強い増殖応答である。
【0075】
CD3−L6二特異的融合タンパク質はJurkat細胞とH2981腫瘍細胞との間の接着を仲介する。
以前に開発された細胞接着(Linsley等, 1990a)を用いてCD3−L6二特異的単分子がCD3又はL6発現細胞に同時に結合できるかを求めた。まずJurkat細胞を抗CD3又はCD3−L6FvIgとインキュベートした。細胞を洗浄し、L6FvIgあり又はなしで予備インキュベートしたH2981粘着細胞に加えた。H2981単層(図13)の顕微鏡試験は、CD3及びL6レセプターがリガンドで遮断されない場合だけを除いて、CD3−L6FvIgタンパク質がEDTAの存在下にJurkat細胞とH2981腫瘍細胞との間の接着を仲介したことを示した。Jurkat細胞を51Crで前標識し、次いで融合タンパク質及びH2981腫瘍細胞とインキュベートした。CD3−L6二特異的融合タンパク質の存在下に標識していない単層に結合したカウント数は、連結していないCD3及びL6抗体に前結合した細胞より非常に多かった。
CD3−L6Ig二特異的融合タンパク質は、H2981腫瘍細胞に対するT細胞の細胞毒性を標的とする(図14)。H2981腫瘍細胞を51Crで2時間標識し、標的に対するエフェクター比10:1及び100:1で抗体刺激及びPBLとインキュベートした(図14)。クロミウムの遊離を材料及び方法で記載したように測定し、3回の実験から各抗体誘導体に特異的な溶解を表にした(SEM<12%)(図14)。阻害アッセイの場合、CD3又はL6Ig単一特異性誘導体を適切な種類の細胞とインキュベートした後、CD3−L6Ig二特異的分子を加えた(図14)。いずれも単一特異性分子と予備インキュベートすると細胞毒性の二特異的構築物による刺激を阻害することができなかった(図14)。
【0076】
CD3−L6二特異的融合タンパク質はH2981腫瘍細胞に対するT細胞の細胞毒性を標的とする。
次に、これら2つの抗原結合ドメインを単分子に結合することの生物学的結果を求めた。腫瘍細胞とT細胞との間の接着を促進する遺伝子操作した二機能性分子がレセプター結合に対する応答の種類又は程度を変えると推論された。
IgG媒介性エフェクター機能から背景原因を排除するために、CH2内のプロリンからセリンへの変異及びヒンジ領域内のいくつかのシステインからセリンへの置換の結果としてこれらの機能を仲介しないFc単量体の変異体に分子の抗原結合部分を結合した。溶解の場合の標的としてH2981腫瘍細胞を用いて標準細胞毒性アッセイを行った。腫瘍に対する細胞毒性を標的とするために静止又は未変性細胞を活性化することができる場合には、エフェクター細胞として静止PBLを用いて求めた(図14)。
【0077】
CD3−L6FvIg分子は、標的に対するエフェクター比10:1において30%の特異的溶解を仲介し、100:1の比では特異的溶解が71%に上昇する。CD3FvIg及びL6FvIgを一緒に混合したもの又は両分子単独の場合の特異的溶解レベルは、E:T10:1で3〜5%及びE:T100:1で9〜20%である。破壊レベルをわずかに減少したが、CD3FvIg及びL6FvIg抗体誘導体はCD3−L6融合タンパク質により仲介された標的細胞毒性を完全に遮断しなかった。PHA活性化T細胞芽球も本アッセイにおいてエフェクターとして用いたが、高レベルの背景破壊を示した。
CD3−L6Ig二特異的融合タンパク質は、H2981腫瘍細胞に結合すると高レベルのT細胞増殖を刺激する(図15)。PBLを単離し、必要とするT細胞増殖刺激物質及び照射H2981腫瘍細胞を存在させて培養した。刺激物質を1又は10μg/mlのいずれかの溶液として加えた。前結合実験の場合、H2981腫瘍細胞に照射し、10μg/mlの抗体誘導体と氷上で1時間インキュベートし、数回洗浄して非結合抗体を除去し、種々の割合で5×104 細胞/ウェルのPBLに加えた。3日培養した後、[3H]チミジンの取込みにより6時間増殖を定した。各処理について4回の培養により数値を求めた(SEM<15%)。
【0078】
CD3−L6二特異的融合タンパク質は試験管内で高レベルのT細胞増殖を刺激する。
CD3T細胞表面レセプター(CD3/TCR複合体)をCD3−L6FvIg融合タンパク質で誘発するとT細胞増殖を刺激するかを調べた。照射H2981腫瘍細胞とインキュベートした静止PBL及び刺激(融合タンパク質)の溶解したものを用いて、増殖アッセイを行った。
また、刺激タンパク質を照射腫瘍細胞に前結合し、非結合タンパク質を数回洗浄して除去した後、アッセイに含め、CD3によってT細胞の刺激に寄与するものからL6抗原に結合できない分子を排除した。H2981腫瘍細胞はFcドメインで変異した抗体誘導体にさえ非特異的に結合する傾向があったので、アッセイからこの非特異的背景の原因を排除するために、不適当な抗体を細胞とインキュベートした後に問題の刺激を加えた。図15は、溶解及び前結合両増殖実験の結果を示すものである。
【0079】
増殖レベルは10μg/mlのCD3−L6FvIg二特異的融合タンパク質の存在下で著しく増大し、有為レベルの増殖が1μg/mlでさえも見られた。L6FvIgを腫瘍細胞に前結合した後に二特異的分子を加えると、被覆腫瘍細胞によって誘導されるこのT細胞増殖の刺激が排除された。CD3IG融合タンパク質は、腫瘍細胞のあり又はなしに依存しない溶液中に存在すると著しいレベルの増殖を刺激することができた。背景レベルの増殖のみがこれらの条件下で見られるので、これらの分子が腫瘍細胞前結合アッセイの洗浄段階で除去されたことは明らかである。これらの結果は、H2981腫瘍細胞に結合した際に、T細胞の細胞毒性及び刺激T細胞増殖を標的とするCD3−L6二特異的融合タンパク質の能力を示すものである。
【図面の簡単な説明】
【0080】
【図1】種々の分子垂れはし又は単純な停止コドンを含む組換え体一特異的単鎖カセットを含むpCDM8発現ベクターの図である。
【図2】組換え体二特異的単鎖カセットを含むpCDM8発現ベクターの図である。
【図3】単鎖抗L6、抗CD3及び抗CD3−L6二特異的融合タンパク質分子のウェスタンブロット写真である。COS細胞移入からの使用済み無血清培養液を採取し、プロテインAアフィニティークロマトグラフィーで精製した。タンパク質を増量バッファーに再浮遊し、非還元(A)又は還元(B)条件下SDS−PAGE勾配ゲル(5−16%)電気泳動にかけ、ニトロセルロースにブロットし、アルカリ性ホスファターゼ結合抗ヒトIgGで検出した。パネルA:レーン1=キメラL6mAb(0.5μg);レーン2=抗L6WTD(0.5μg);レーン3=CD3−L6FvIg二特異的(0.5μg);レーン4=L6FvIg(0.6μg);及びレーン5=CD3Fv−Ig(0.4μg)。パネルB:レーン1=CD3Fv−Ig(0.4μg);レーン2=L6FvIg(0.6μg);レーン3=CD3−L6FvIg二特異的(0.5μg);レーン4=抗L6WTD(0.5μg);及びレーン5=キメラL6mAb(0.5μg)。
【図4】細胞発現標的抗原に結合するL6FvIg、CD3FvIg、CD3−L6FvIg及びBR96を示すファクスプロットである。
【図5】L6FvがヒトIgG1Fcドメインの各種異種変異誘導体に融合したことを示す。ヒンジ及び/又はCH2ドメインに導入された各変異体及び配列変化を示す。更に、各構築物のCDC及びADCC仲介能を示す。
【0081】
【図6】累進的に低くした濃度(3倍連続希釈)のH2981腫瘍細胞とインキュベートしたL6誘導体又はキメラL6の飽和分析の線グラフであり、第2段階試薬としてL6に対するFITC結合抗イディオタイプを用いて結合を検出した。これらのデータから飽和結合曲線を作り、蛍光強度を抗体濃度の関数としてプロットした。記号の説明:キメラL6(白ダイヤモンド形)、WTD(白四角)、DM1(白三角)、HS1(+記号)、HS2(『X』記号)及びHS3(白丸)
【図7】ファクス分析の前に連続希釈の各抗体誘導体と30分間及び1μg/mlのFITC結合L6と30分間インキュベートしたH2981腫瘍細胞の阻害分析の線グラフである。蛍光強度を各分子の抗体濃度の関数としてプロットした。記号の説明:キメラL6(白ダイヤモンド形)、WTD(白四角)、DM1(白三角)、HS1(+記号)、HS2(『X』記号)及びHS3(白丸)。
【図8】H2981腫瘍細胞を51Crで2時間標識し、洗浄し、10倍連続希釈の抗体誘導体を含むIMDM/10%FBS及びエフェクター細胞としてヒトPBLに100:1の標的に対するエフェクター比で加えた。分析物を4.5時間インキュベートし、攪拌し、100μl の遊離数をγカウンターを用いて測定した。次式を用いて破壊%を算出した:破壊%=〔(平均cpm−平均自然遊離)/(平均最大遊離−平均自然遊離)〕×100。数値は3回の培養の平均を示す(SEM<10%)。
【図9】CD3FvIgが抹消血T細胞の細胞内カルシウムを移動し、CD3FvIgとの前処理が次のクロスリンクしたCD2の刺激に対する抹消血T細胞の応答を脱感作することを示す線グラフである。
【図10】CD3FvIgが抹消血T細胞の細胞内カルシウムを移動し、CD3FvIgとの前処理が次のクロスリンクしたCD2の刺激に対する抹消血T細胞の応答を脱感作することを示す線グラフである。
【0082】
【図11】CD3FvIgが抹消血T細胞の細胞内カルシウムを移動し、CD3FvIgとの前処理が次のクロスリンクしたCD2の刺激に対する抹消血T細胞の応答を脱感作することを示す線グラフである。
【図12】CD3FvIgが抹消血T細胞の細胞内カルシウムを移動し、CD3FvIgとの前処理が次のクロスリンクしたCD2の刺激に対する抹消血T細胞の応答を脱感作することを示す線グラフである。
【図13】二特異的分子CD3−L6IgがH2981とJurkat細胞との間の接着を仲介するのでCD3及びL6発現細胞に同時に結合することができることを示す棒グラフである。
【図14】CD3−L6FvIg二特異的融合タンパク質がH2981腫瘍細胞に対するT細胞の細胞毒性を標的とすることを示す棒グラフである。
【図15】H2981腫瘍細胞に結合した際にCD3−L6FvIg二特異的タンパク質が高レベルのT細胞増殖を刺激することを示す棒グラフである。
【0083】
【図16】融合タンパク質として一特異的抗体可変部の発現のための発現ベクターpCDM8をヒトIgG1のFcドメインで修飾することを示す図式である。
【図17】黒い線で示したリンカー配列及び斜線で示した各機能性ドメインを含むL6Fv−Ig誘導体構造の図である。
【図18】キメラL6、WTD、DM1、HS1、HS2及びHS3のFc構築物及びそれらの配列の比較及びADCC又はCDC活性の有無を示す。L6FvはヒトIgG1のFcドメインの各種異種変異誘導体に融合した。ヒンジ及び/又はCH2ドメインに導入された配列変化を下線を引いたアミノ酸で示し、構築物同定を左に示す。
【図19】51Crで2時間標識し、洗浄し、10倍連続希釈の抗体誘導体を含むIMDM+10%FBS及びエフェクター細胞としてヒトPBLに100:1の標的に対するエフェクター比で加えたH2981腫瘍細胞のADCC特性の線グラフである。分析物を4.5時間インキュベートし、攪拌し、100μl の上清の遊離数を測定した。破壊%を〔(平均cpm−平均自然遊離)/(平均最大遊離−平均自然遊離)〕×100として算出した。数値は3回の培養の平均を示す(SEM<10%)。記号の説明:キメラL6(白ダイヤモンド形)、WTD(白四角)、DM1(白三角)、HS1(+記号)、HS2(『X』記号)及びHS3(白丸)。
【図20】PBLの代わりに補体を用いた以外は図19と同様に補体破壊を測定するために行ったアッセイの線グラフである。記号の説明:キメラL6(白ダイヤモンド形)、WTD(白四角)、DM1(白三角)、HS1(+記号)、HS2(『X』記号)及びHS3(白丸)。
【0084】
【図21】L6VL リーダー、CD3FV (V L−V H)及びFvlinkらせんリンカーのアミノ酸及び核酸配列である。
【図22】CD3FvIgが強力なチロシンリン酸化及びT細胞内のPLCγ1の活性化を刺激し、PLCγ1とpp35/36との結合を誘導することを示すPAGEゲルである。
【図23】CD3FvIgが強力なチロシンリン酸化及びT細胞内のPLCγ1の活性化を刺激し、PLCγ1とpp35/36との結合を誘導することを示すPAGEゲルである。
【特許請求の範囲】
【請求項1】
親水性アミノ酸及び少なくとも1のペンタマー:EEAKKを含有する親水性らせんペプチドを含むリンカーで結合している抗CD3/抗L6二特異的融合タンパク質。
【請求項2】
二特異的融合タンパク質であって、(1)標的を結合することができる第1結合ドメイン、(2)標的を結合することができる第2結合ドメイン、ここで各ドメインは異なる標的を結合することができ、及び(3)第1結合ドメインと第2結合ドメインを連結する、親水性アミノ酸と少なくとも1のペンタマー:EEAKKを含有するリンカー
を含む、二特異的融合タンパク質。
【請求項3】
溶解性二特異的融合タンパク質であって、B7抗原と結合する第1結合ドメイン及びCD40抗原と結合する第2結合ドメインを含み、該第1結合ドメインと第2結合ドメインが、親水性アミノ酸と少なくとも1のペンタマー:EEAKKとを含有するリンカーによって連結されている、溶解性二特異的融合タンパク質。
【請求項4】
さらに分子垂れはしを含む、請求項3記載の溶解性二特異的融合タンパク質。
【請求項5】
該分子垂れはしがFcフラグメントである、請求項4記載の溶解性二特異的融合タンパク質。
【請求項6】
該分子垂れはしがHIVフラグメントである、請求項4記載の溶解性二特異的融合タンパク質。
【請求項7】
該分子垂れはしが赤血球凝集素エピトープ配列HAIである、請求項4記載の溶解性二特異的融合タンパク質。
【請求項8】
第1結合ドメインがCTLA4の細胞外部分である、請求項3記載の溶解性二特異的融合タンパク質。
【請求項9】
第1結合ドメインがB7抗原を認識し及び結合する抗体の可変部の部分である、請求項3記載の溶解性二特異的融合タンパク質。
【請求項10】
第1結合ドメインがCD28の細胞外部分である、請求項3記載の溶解性二特異的融合タンパク質。
【請求項11】
第2結合ドメインがCD40抗原を認識し及び結合する抗体の可変部である、請求項3記載の溶解性二特異的融合タンパク質。
【請求項12】
第1結合ドメインがCTLA4の細胞外部分であり、第2結合ドメインが抗CD40モノクローナル抗体の可変部である、請求項3記載の溶解性二特異的融合タンパク質。
【請求項13】
第1結合ドメインがCD28の細胞外部分であり、第2結合ドメインが抗CD40モノクローナル抗体の可変部である、請求項3記載の溶解性二特異的融合タンパク質。
【請求項1】
親水性アミノ酸及び少なくとも1のペンタマー:EEAKKを含有する親水性らせんペプチドを含むリンカーで結合している抗CD3/抗L6二特異的融合タンパク質。
【請求項2】
二特異的融合タンパク質であって、(1)標的を結合することができる第1結合ドメイン、(2)標的を結合することができる第2結合ドメイン、ここで各ドメインは異なる標的を結合することができ、及び(3)第1結合ドメインと第2結合ドメインを連結する、親水性アミノ酸と少なくとも1のペンタマー:EEAKKを含有するリンカー
を含む、二特異的融合タンパク質。
【請求項3】
溶解性二特異的融合タンパク質であって、B7抗原と結合する第1結合ドメイン及びCD40抗原と結合する第2結合ドメインを含み、該第1結合ドメインと第2結合ドメインが、親水性アミノ酸と少なくとも1のペンタマー:EEAKKとを含有するリンカーによって連結されている、溶解性二特異的融合タンパク質。
【請求項4】
さらに分子垂れはしを含む、請求項3記載の溶解性二特異的融合タンパク質。
【請求項5】
該分子垂れはしがFcフラグメントである、請求項4記載の溶解性二特異的融合タンパク質。
【請求項6】
該分子垂れはしがHIVフラグメントである、請求項4記載の溶解性二特異的融合タンパク質。
【請求項7】
該分子垂れはしが赤血球凝集素エピトープ配列HAIである、請求項4記載の溶解性二特異的融合タンパク質。
【請求項8】
第1結合ドメインがCTLA4の細胞外部分である、請求項3記載の溶解性二特異的融合タンパク質。
【請求項9】
第1結合ドメインがB7抗原を認識し及び結合する抗体の可変部の部分である、請求項3記載の溶解性二特異的融合タンパク質。
【請求項10】
第1結合ドメインがCD28の細胞外部分である、請求項3記載の溶解性二特異的融合タンパク質。
【請求項11】
第2結合ドメインがCD40抗原を認識し及び結合する抗体の可変部である、請求項3記載の溶解性二特異的融合タンパク質。
【請求項12】
第1結合ドメインがCTLA4の細胞外部分であり、第2結合ドメインが抗CD40モノクローナル抗体の可変部である、請求項3記載の溶解性二特異的融合タンパク質。
【請求項13】
第1結合ドメインがCD28の細胞外部分であり、第2結合ドメインが抗CD40モノクローナル抗体の可変部である、請求項3記載の溶解性二特異的融合タンパク質。
【図1】

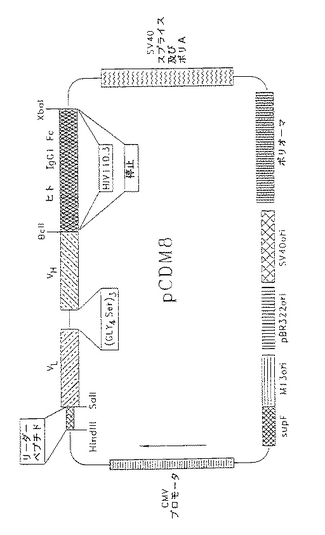
【図2】
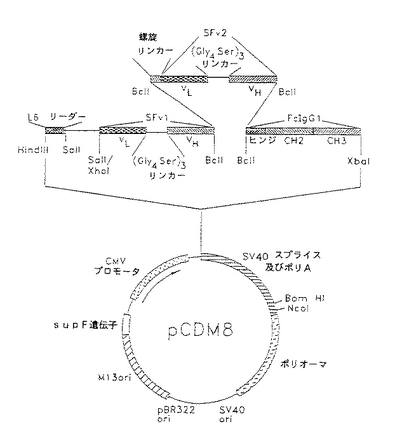
【図3】
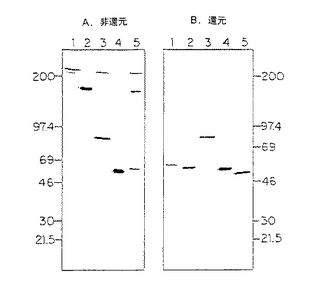
【図4】
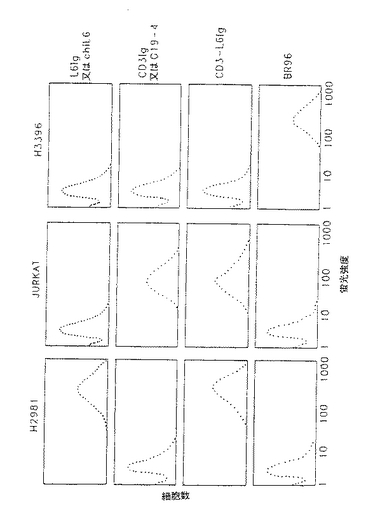
【図5】
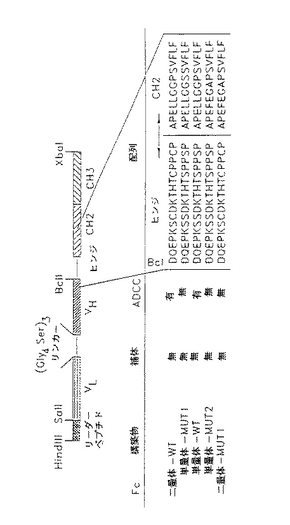
【図6】
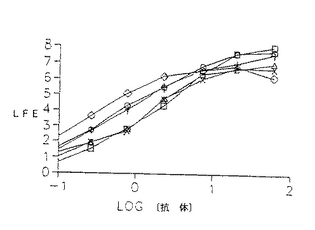
【図7】
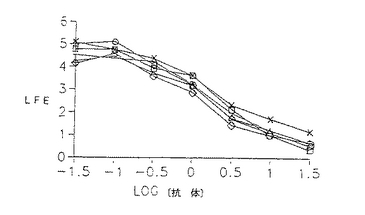
【図8】
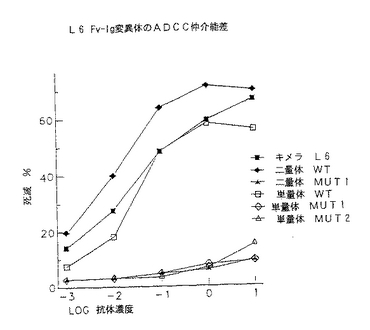
【図9】
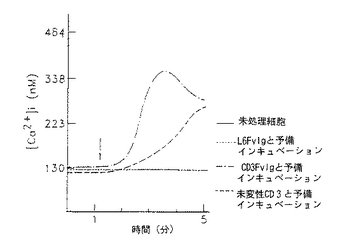
【図10】
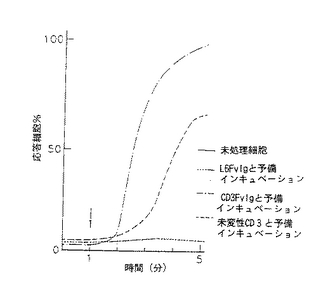
【図11】
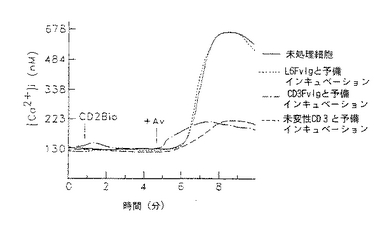
【図12】
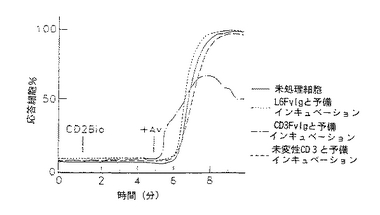
【図13】
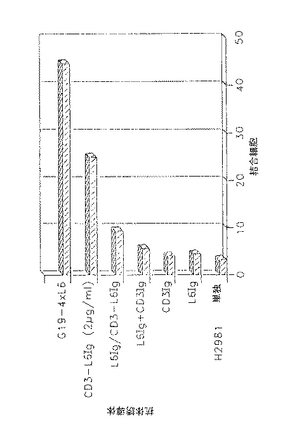
【図14】
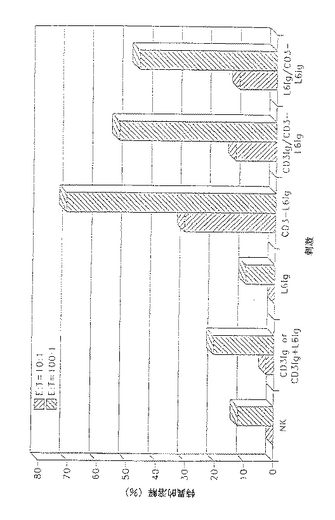
【図15】
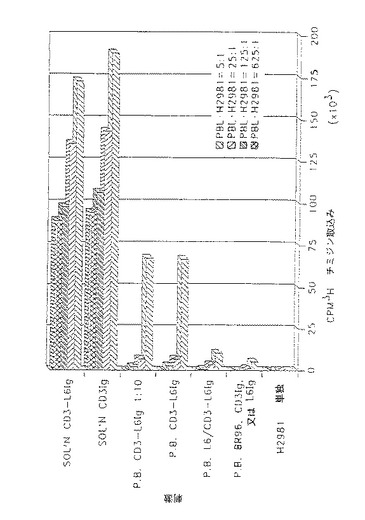
【図16】
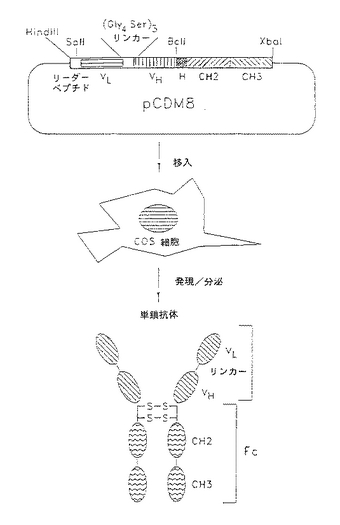
【図17】
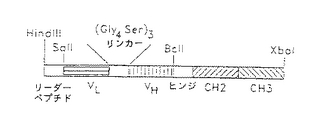
【図18】
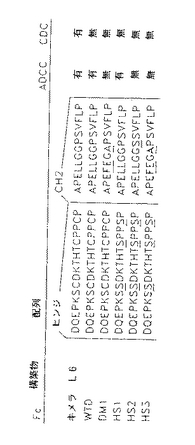
【図19】
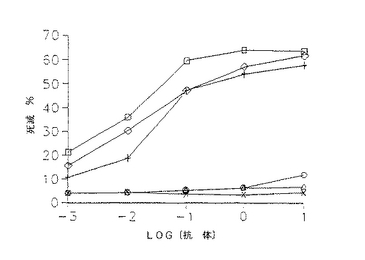
【図20】
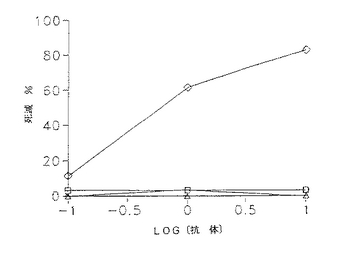
【図21】
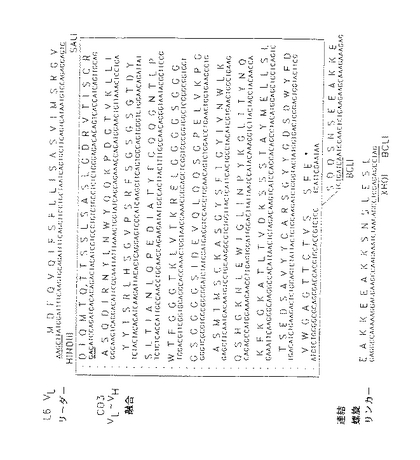
【図22】
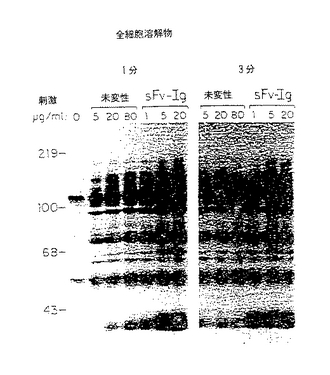
【図23】
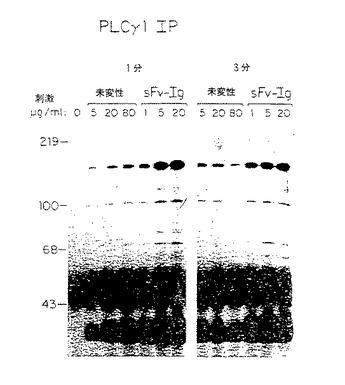
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【公開番号】特開2006−241165(P2006−241165A)
【公開日】平成18年9月14日(2006.9.14)
【国際特許分類】
【出願番号】特願2006−102332(P2006−102332)
【出願日】平成18年4月3日(2006.4.3)
【分割の表示】特願平6−9684の分割
【原出願日】平成6年1月31日(1994.1.31)
【出願人】(391015708)ブリストル−マイヤーズ スクイブ カンパニー (494)
【氏名又は名称原語表記】BRISTOL−MYERS SQUIBB COMPANY
【Fターム(参考)】
【公開日】平成18年9月14日(2006.9.14)
【国際特許分類】
【出願日】平成18年4月3日(2006.4.3)
【分割の表示】特願平6−9684の分割
【原出願日】平成6年1月31日(1994.1.31)
【出願人】(391015708)ブリストル−マイヤーズ スクイブ カンパニー (494)
【氏名又は名称原語表記】BRISTOL−MYERS SQUIBB COMPANY
【Fターム(参考)】
[ Back to top ]