
二酸化炭素分離装置
【課題】 100℃以上の高温及び加圧条件下において十分な膜性能を発揮可能なCO2促進輸送膜を用いた二酸化炭素分離装置を提供する。
【解決手段】 少なくとも水素と二酸化炭素と水蒸気が含まれる原料ガスをCO2促進輸送膜10の原料側面に100℃以上の供給温度で供給して、CO2促進輸送膜10を透過した二酸化炭素を透過側面から取り出す二酸化炭素分離装置であって、CO2促進輸送膜10が、ポリビニルアルコール−ポリアクリル酸共重合体ゲル膜に炭酸セシウムを添加したゲル層を親水性の多孔膜に担持させて形成される。
【解決手段】 少なくとも水素と二酸化炭素と水蒸気が含まれる原料ガスをCO2促進輸送膜10の原料側面に100℃以上の供給温度で供給して、CO2促進輸送膜10を透過した二酸化炭素を透過側面から取り出す二酸化炭素分離装置であって、CO2促進輸送膜10が、ポリビニルアルコール−ポリアクリル酸共重合体ゲル膜に炭酸セシウムを添加したゲル層を親水性の多孔膜に担持させて形成される。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、所定の主成分ガスに少なくとも二酸化炭素と水蒸気が含まれる原料ガスをCO2促進輸送膜の原料側面に供給して、CO2促進輸送膜を透過した二酸化炭素を透過側面から取り出す二酸化炭素分離装置に関し、特に、水素を主成分とする燃料電池用等の改質ガスに含まれる二酸化炭素を水素に対する高い選択比率で分離する二酸化炭素分離装置に関する。
【背景技術】
【0002】
現在の水素ステーション用改質システムでは、水蒸気改質により炭化水素を水素及び一酸化炭素(CO)に改質し、更に、CO変成反応を用いて一酸化炭素を水蒸気と反応させることにより水素を製造している。
【0003】
しかし、これらの技術はケミカルプラント用の大規模な水素製造プロセスとして開発されたものであり、日産数10万m3以上の大規模なものが普通で、しかも工場内に設置され、高圧・連続運転が前提となっている。これに対し、将来の水素エネルギ社会を支える重要なインフラとして天然ガスや石油からオンサイトで水素を製造し燃料電池自動車等に水素を供給する水素ステーションでは、水素を製造する規模や設置環境、運転パターンにおいて、従来の大規模水素プラントと大きく異なっており、それに起因する問題が多く残されている。
【0004】
性能面の課題としては、水素ステーションの場合、水素需要(具体的には、水素供給対象の燃料電池自動車の数量等)に対応して、頻繁な起動停止や負荷変化に対応する必要がある。特に、改質システム中でもサイズが最も大きく熱容量の大きなCO変成器に対して、起動時間や負荷応答性等の面で改善が必要となっている。
【0005】
また、燃料電池用改質システムの普及促進に必須とされる家庭用システムでのDSS(毎日の起動停止)運転に対しても、起動時間や負荷応答性等において、改質システム中でもCO変成器に課題が多く残されており、特に、CO変成器の小型化、低温度化が最大の課題である。
【0006】
更に、自動車用への適用についても、水蒸気改質方式はサイズや起動時間の点で目標との大きなギャップがあり、自動車業界ではオンボード改質に関しては効率の高い水蒸気改質よりは、寧ろ昇温反応性に優れた部分酸化方式での実用化を目指す傾向がある。しかし、部分酸化方式では、改質器については大幅な小型化が期待できるものの、CO変成器を伴うため、実用化に向けてはCO変成器の小型化が大きな課題となっている。このようにCO変成器の小型化は水素ステーションだけではなく、自動車用を含む燃料電池改質システムに共通の課題と言える。
【0007】
また、効率面から見ても、水蒸気改質を行う際、S/C(スチームと炭素(原料炭化水素)のモル比)の低下が熱効率上望ましいが、CO変成反応の化学平衡上の制約から効率の高い低S/C条件が採用されていなかった。
【0008】
コスト面での課題としては、水素ステーション全体のコストで最大の割合を占めるのがPSA(プレッシャー・スイング・アドソープション)であることから、そのコストダウンに直接繋がる水素濃度を上げる改質方式が望まれていた。改質器の出口ガス組成は水素以外に、10%程度の一酸化炭素及び二酸化炭素が含まれており、CO変成器では一酸化炭素は減少するものの二酸化炭素は増加するため、現状のプロセス(水蒸気改質+CO変成)では、1%程度のメタンとともに、20%程度の二酸化炭素と1%以下の一酸化炭素の残留は避けられず、その精製のために大型・高コストのPSA装置を設置せざるを得なかった。
【0009】
従来のCO変成器において、小型化や起動時間の短縮を阻害する原因として、以下の(化1)に示すCO変成反応の化学平衡上の制約から、多量のCO変成触媒が必要となっていることが挙げられる。一例として、50kWのPAFC(リン酸型燃料電池)用改質システムでは、改質触媒が20L必要であるのに対して、CO変成触媒は77Lと約4倍の触媒が必要となる。このことが、CO変成器の小型化や起動時間の短縮を阻害する大きな要因となっている。なお、記号「⇔」は、可逆反応であることを示している。
【0010】
(化1)
CO + H2O ⇔ CO2 + H2
【0011】
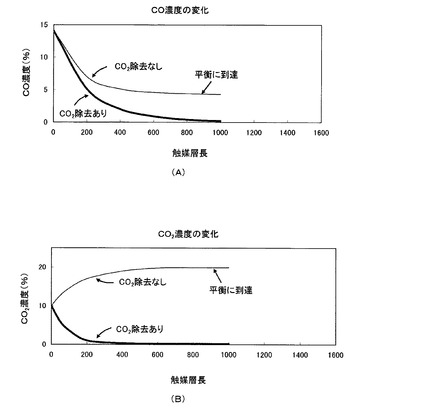
そこで、CO変成器に二酸化炭素を選択的に透過させるCO2促進輸送膜を備え、上記(化1)のCO変成反応で生成された右側の二酸化炭素を効率的にCO変成器外部に除去することで、化学平衡を水素生成側(右側)にシフトさせることができ、同一反応温度において高い転化率が得られる結果、一酸化炭素及び二酸化炭素を平衡の制約による限界を超えて除去することが可能となる。図21及び図22に、この様子を模式的に示す。図22(A)と(B)は、夫々、CO2促進輸送膜を備えている場合と備えていない場合における、CO変成器の触媒層長に対する一酸化炭素及び二酸化炭素の各濃度変化を示している。
【0012】
上記のCO2促進輸送膜を備えたCO変成器(CO2透過型メンブレンリアクター)により、一酸化炭素及び二酸化炭素を平衡の制約による限界を超えて除去することが可能となるため、水素ステーションのPSAの負荷低減及び低S/C化が図れ、水素ステーション全体のコスト低減及び高効率化が図れる。また、CO2促進輸送膜を備えることで、CO変成反応の高速化(高SV化)が図れるため、改質システムの小型化及び起動時間の短縮が図れる。
【0013】
かかるCO2透過型メンブレンリアクターの先行例としては、下記の特許文献1(或いは、同じ発明者による同一内容の特許文献2)に開示されているものがある。
【0014】
該特許文献1、2において提案されている改質システムは、炭化水素、メタノール等の燃料を燃料電池自動車用の水素に車上で改質する際に発生する改質ガスの精製及び水性ガスシフト反応(CO変成反応)に有用なCO2促進輸送膜プロセスを提供するもので、代表的な4種類のプロセスが、同文献に示されている。炭化水素(メタンを含む)を原料とする場合、水性ガスシフター(CO変成器)にCO2促進輸送膜を備えたメンブレンリアクターを用いて二酸化炭素を選択的に除去することにより、一酸化炭素の反応率を高め一酸化炭素濃度を低下させるとともに生成水素の純度を向上させている。また、生成水素中に残留する%オーダーの一酸化炭素及び二酸化炭素はメタネーターで水素と反応させてメタンに変換して濃度を低下させ、燃料電池の被毒等による効率低下を防いでいる。
【0015】
該特許文献1、2では、CO2促進輸送膜として、主としてハロゲン化四級アンモニウム塩((R)4N+X−)を二酸化炭素キャリアとして含むPVA(ポリビニルアルコール)等の親水性ポリマー膜が使用されている。また、該特許文献1、2の実施例6には、二酸化炭素キャリアとしてテトラメチルアンモニウムフルオリド塩50重量%を含む膜厚49μm50重量%のPVA膜とそれを支持する多孔質PTFE(四フッ化エチレン重合体)膜よりなる複合膜で形成されたCO2促進輸送膜の作製方法が開示されており、同実施例7には、混合ガス(25%CO2、75%H2)を全圧3気圧、23℃で処理したときの当該CO2促進輸送膜の膜性能が開示されている。当該膜性能として、CO2パーミアンスRCO2が7.2GPU(=2.4×10−6mol/(m2・s・kPa))、CO2/H2選択性が19となっている。
【0016】
また、下記特許文献3には、CO2促進輸送膜として、炭酸セシウムとアミノ酸とを組み合わせて構成されたCO2吸収剤が開示されている。
【0017】
特許文献3に記載のCO2促進輸送膜の製法は、以下のとおりである。まず、炭酸セシウムの水溶液に市販のアミノ酸を濃度分加えて、よく撹拌し混合水溶液を作製する。その後、ゲルを塗布した多孔PTFE膜(47Φ)のゲル塗布面を、作製した混合溶液に30分以上浸した後、ゆっくり膜を引き上げる。焼結金属の上にシリコーン膜を乗せ(溶液が透過側に漏れるのを防ぐため)その上に47mmΦの上記の含水ゲル膜を乗せ、その上からシリコーンパッキングの入ったセルをかぶせシーリングする。このようにして製造されたCO2促進輸送膜に対して、供給ガスを50cc/分の速度で流し、膜の下側を真空引きし圧力を40torr程度まで下げる。
【0018】
特許文献3の実施例4では、炭酸セシウムと、2,3−ジアミノプロピオン酸塩酸塩をそれぞれ4(mol/kg)のモル濃度で構成したCO2促進輸送膜により、25℃の温度条件下において、CO2透過速度が1.1(10−4cm3(STP)/cm2・s・cmHg)、CO2/N2分離係数が300となっている。なお、CO2パーミアンスRCO2は、圧力差あたりの透過速度で定義されるので、特許文献3の実施例4におけるCO2パーミアンスRCO2は、110GPUと算出されるが、本実施例におけるCO2/H2選択性に関するデータは開示されていない。
【0019】
なお、下記特許文献4には、アルカリ重炭酸塩を添加した酢酸セルロース膜で構成されたCO2分離膜が開示されている。しかし、当該文献4では、CO2/O2選択性についてしか記載されておらず、CO2/H2の選択性についてのデータが開示されていない。更に、開示されたデータは低圧力(0.01気圧程度)の条件下で測定されたものであり、数気圧程度の圧力条件下におけるデータは開示されていない。
【先行技術文献】
【特許文献】
【0020】
【特許文献1】特表2001−511430号公報
【特許文献2】米国特許6579331号明細書
【特許文献3】特開2000−229219号公報
【特許文献4】米国特許第3396510号明細書
【発明の概要】
【発明が解決しようとする課題】
【0021】
CO2促進輸送膜は、基本機能として二酸化炭素を選択的に分離することから、地球温暖化の原因となっている二酸化炭素の吸収或いは除去等を目的とした開発も行われている。しかしながら、CO2促進輸送膜は、CO2透過型メンブレンリアクターへの応用を考えた場合、使用温度、CO2パーミアンス、CO2/H2選択性等に対して、一定以上の性能が要求される。つまり、CO変成反応に供するCO変成触媒の性能が温度とともに低下する傾向にあるため、使用温度は最低でも100℃が必要と考えられる。上記各特許文献1〜3は、いずれも25℃程度の温度条件下で膜性能の測定が行われており、100℃以上の温度条件下においても十分な膜性能を示すCO2促進輸送膜が上記各特許文献によって開示されたということはできない。
【0022】
また、CO2パーミアンス(二酸化炭素透過性の性能指標の一つ)は、CO変成反応の化学平衡を水素生成側(右側)にシフトさせ、一酸化炭素濃度と二酸化炭素濃度を平衡の制約による限界を超えて例えば0.1%程度以下に低減し、且つ、CO変成反応の高速化(高SV化)を図るためには、一定レベル以上(例えば、2×10−5mol/(m2・s・kPa)=60GPU程度以上)が必要と考えられる。しかしながら、上記各特許文献1,2に記載のCO2促進輸送膜のCO2パーミアンスは、10GPUを大きく下回るような値であり、60GPU程度以上のCO2パーミアンスを示すCO2促進輸送膜が上記各特許文献によって開示されたということはできない。また、特許文献3は、CO2/H2選択性は開示されていない上、100℃以上の温度条件でCO2パーミアンスが60GPU以上の能力を示すことは示されていない。特許文献4においても、CO2/H2選択性は開示されていない上、数気圧程度の圧力条件の下でのデータが開示されていない。
【0023】
更に、CO変成反応で生成された水素が二酸化炭素とともにCO2促進輸送膜を通して外部に廃棄されたのでは、当該廃棄ガスから水素を分離回収するというプロセスが必要となる。水素は当然に二酸化炭素より分子サイズが小さいので、二酸化炭素を透過可能な膜は水素も透過できることになるが、膜中の二酸化炭素キャリアによって二酸化炭素のみを選択的に膜の供給側から透過側に向けて輸送可能な促進輸送膜が必要となり、その場合のCO2/H2選択性として90〜100程度以上が必要と考えられる。
【0024】
しかしながら、上記各特許文献1及び2に記載のCO2促進輸送膜は、CO2/H2選択性が19であり、十分な選択性を有しているとは言えない。また、上記特許文献3,4は、CO2/H2選択性が開示されていないため、特許文献3,4によって高いCO2/H2選択性を示すCO2促進輸送膜が開示されたということはできない。
【0025】
ところで、CO2促進輸送膜は、二酸化炭素の促進輸送機能(膜機能)を十分に発揮するには水分が必要である。具体的に説明すると、膜内の二酸化炭素(CO2)と炭酸イオンの反応は、通常以下の(化2)の反応経路式に示す化学反応を示す。これより、膜内の水分が多いほど化学平衡は生成物側(右側)にシフトし、二酸化炭素の透過が促進されることが分かる。
【0026】
(化2)
CO2+CO32−+H2O → 2HCO3−
【0027】
しかし、使用温度が100℃を超える高温になると、膜内の水分が蒸発して膜機能、つまり、二酸化炭素の促進輸送機能が低下する。当該膜機能低下はこれまでの促進輸送膜の常識となっている。一方、高温ほど上記化学反応の速度が大きくなるので、本願の発明者は、加圧下において気相中の水蒸気分圧を増すことにより膜内の水分量を確保することで膜機能が十分に発揮されることを確認した。現時点においては、使用温度が100℃以上を超える高温の温度条件下においても高いCO2分離機能を示す二酸化炭素分離装置は提供されていない。
【0028】
本発明は、上記の問題点に鑑み、100℃以上の高温及び加圧条件下において十分な膜性能を発揮可能なCO2促進輸送膜を用いた二酸化炭素分離装置を提供することを目的とする。
【課題を解決するための手段】
【0029】
上記目的を達成するための本発明に係る二酸化炭素分離装置は、少なくとも水素と二酸化炭素と水蒸気が含まれる原料ガスをCO2促進輸送膜の原料側面に100℃以上の供給温度で供給して、前記CO2促進輸送膜を透過した二酸化炭素を透過側面から取り出す二酸化炭素分離装置であって、前記CO2促進輸送膜が、ポリビニルアルコール−ポリアクリル酸共重合体ゲル膜に炭酸セシウム若しくは重炭酸セシウム若しくは水酸化セシウムからなる添加剤を添加したゲル層を親水性の多孔膜に担持させて形成されることを特徴とする。
【0030】
本発明に係る二酸化炭素分離装置の上記特徴によれば、ポリビニルアルコール−ポリアクリル酸(PVA/PAA)共重合体ゲル膜中に、炭酸セシウム(Cs2CO3)が含まれることから、当該Cs2CO3が透過物質であるPVA/PAAゲル層の二酸化炭素の高濃度側界面から低濃度側界面へと二酸化炭素を輸送する二酸化炭素キャリアとして機能し、100℃以上の高温において90〜100程度以上の対水素選択性(CO2/H2)、及び、2×10−5mol/(m2・s・kPa)(=60GPU)程度以上のCO2パーミアンスを達成可能となる。
【0031】
また、PVA/PAAゲル層を担持する多孔膜が親水性であるので、欠陥の少ないゲル層を安定して作製することができ、高い対水素選択性を維持できる。一般に、多孔膜が疎水性であると、100℃以下においてPVA/PAAゲル膜内の水分が多孔膜内の細孔に侵入して膜性能を低下させるのを防止でき、また、100℃以上においてPVA/PAAゲル膜内の水分が少なくなる状況でも同様の効果が期待できると考えられるため、疎水性の多孔膜の使用が推奨されるが、本発明の二酸化炭素分離装置は、親水性多孔膜を使用することで、以下の理由により欠陥が少なく高い対水素選択性を維持できる。
【0032】
親水性の多孔膜上に、PVA/PAA共重合体とCs2CO3の水溶液からなるキャスト溶液をキャストすると多孔膜の細孔内が液で満たされ、更に、多孔膜の表面にキャスト溶液が塗布される。このキャスト溶液をゲル化すると、多孔膜の表面のみならず細孔内にもゲル層が充填されるので欠陥が生じ難くなり、ゲル層の製膜成功率が高くなる。
【0033】
細孔部分の割合(多孔度)、及び、細孔が膜表面に垂直に真っ直ぐではなく曲がりくねっていること(屈曲率)を考慮すると、細孔内のゲル層はガス透過の大きな抵抗となるので、多孔膜表面のゲル層と比較して透過性は低くなり、ガスパーミアンスは低下する。他方、疎水性の多孔膜上にキャスト溶液をキャストすると多孔膜の細孔内は液で満たされずに多孔膜の表面のみにキャスト溶液が塗布され細孔はガスで満たされるので、疎水性多孔膜上のゲル層におけるガスパーミアンスは、親水性多孔膜と比較して水素及び二酸化炭素の両方において高くなると予想される。
【0034】
しかし、細孔内のゲル層と比較して膜表面のゲル層では微小な欠陥が生じ易く、製膜成功率は低下する。水素は二酸化炭素より分子サイズが非常に小さいので、微小な欠陥個所では二酸化炭素より水素の方が、パーミアンスが著しく大きくなる。なお、欠陥箇所以外では、促進輸送機構で透過する二酸化炭素のパーミアンスは、物理的な溶解、拡散機構で透過する水素のパーミアンスより格段に大きい。
【0035】
結果として、疎水性多孔膜を使用した場合の対水素選択性(CO2/H2)は、親水性多孔膜を使用した場合と比較して低下することになる。従って、実用化の観点からは、CO2促進輸送膜の安定性、耐久性が非常に重要となり、対水素選択性(CO2/H2)の高い親水性多孔膜を使用する方が有利となる。また、親水性多孔膜の使用は、PVA/PAAゲル層に二酸化炭素キャリアとしてCs2CO3を添加することで高いCO2パーミアンスを達成可能であることを前提に実現できるものである。
【0036】
なお、疎水性多孔膜と親水性多孔膜の違いによるガスパーミアンスの差は、キャスト溶液中に予め二酸化炭素キャリアであるCs2CO3を添加せずにゲル化後に含侵させても、細孔内のゲル層がガス透過の大きな抵抗となる点は同じであり、同様に発現するものと推定される。
【0037】
以上より、上記特徴の二酸化炭素分離装置によれば、100℃以上の使用温度、2×10−5mol/(m2・s・kPa)(=60GPU)程度以上のCO2パーミアンス、及び、90〜100程度以上のCO2/H2選択性が実現でき、CO変成器の小型化、起動時間の短縮、及び、高速化(高SV化)が図られる。
【0038】
なお、添加剤として、炭酸セシウムの代わりに水酸化セシウムを添加した場合においても、同様の効果を得ることができる。即ち、水酸化セシウムが添加されたゲル層を含む促進輸送膜をCO2の分離に利用することで、以下の(化3)に示されるような反応が起こり、これによって当該促進輸送膜内に添加されていた水酸化セシウムが炭酸セシウムに転化するためである。
【0039】
(化3)
CO2 + CsOH → CsHCO3
CsHCO3 + CsOH → Cs2CO3 + H2O
【0040】
なお、上記(化3)をまとめると、下記(化4)のように表すことができる。即ち、これにより、添加された水酸化セシウムが炭酸セシウムに転化することが示される。
【0041】
(化4)
CO2 + 2CsOH → Cs2CO3 + H2O
【0042】
更に、上記(化3)より、添加剤として、炭酸セシウムの代わりに重炭酸セシウムを添加した場合においても同様の効果を得ることができることが分かる。
【0043】
また、本発明に係る二酸化炭素分離装置は、上記特徴に加えて、前記ゲル層が、ポリビニルアルコール−ポリアクリル酸共重合体ゲル膜と炭酸セシウムの合計重量に対する炭酸セシウムの重量比率が65重量%以上85重量%以下の範囲で構成されることを別の特徴とする。
【0044】
本発明に係る二酸化炭素分離装置の上記特徴によれば、100℃以上の温度条件下で、優れたCO2パーミアンス、並びに優れたCO2/H2選択性の値を実現でき、CO変成器の小型化、起動時間の短縮、及び、高速化(高SV化)が図られる。
【0045】
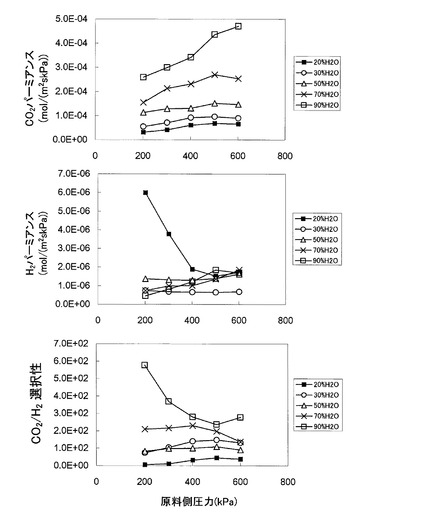
また、本発明に係る二酸化炭素分離装置は、少なくとも水素と二酸化炭素と水蒸気が含まれる原料ガスをCO2促進輸送膜の原料側面に100℃以上の供給温度で供給して、前記CO2促進輸送膜を透過した二酸化炭素を透過側面から取り出す二酸化炭素分離装置であって、前記CO2促進輸送膜が、ポリビニルアルコール−ポリアクリル酸共重合体ゲル膜に炭酸ルビジウム若しくは重炭酸ルビジウム若しくは水酸化ルビジウムからなる添加剤を添加したゲル層を親水性の多孔膜に担持させて形成されることを別の特徴とする。
【0046】
本発明に係る二酸化炭素分離装置の上記特徴によれば、ポリビニルアルコール−ポリアクリル酸(PVA/PAA)共重合体ゲル膜中に、水中への溶解度が比較的高い炭酸塩である炭酸ルビジウム(Rb2CO3)が、透過物質である二酸化炭素をPVA/PAA共重合体ゲル層の二酸化炭素高濃度側界面から低濃度側界面へと輸送する二酸化炭素キャリアとして機能し、100℃以上の高温において90〜100程度以上の対水素選択性(CO2/H2)、及び、2×10−5mol/(m2・s・kPa)(=60GPU)程度以上のCO2パーミアンスを達成可能となる。
【0047】
なお、炭酸ルビジウムの代わりに水酸化ルビジウム若しくは重炭酸ルビジウムを添加した場合においても、同様の効果を得ることができる。これは、炭酸セシウムの代わりに水酸化セシウム若しくは重炭酸セシウムを添加した場合において、炭酸セシウムを添加したときと同様の効果が得られることと同様の理由による。
【0048】
また、本発明に係る二酸化炭素分離装置は、上記特徴に加えて、前記親水性の多孔膜に担持された前記ゲル層が疎水性の多孔膜によって被覆されていることを別の特徴とする。
【0049】
本発明に係る二酸化炭素分離装置の上記特徴によれば、親水性の多孔膜で担持されたゲル層が疎水性の多孔膜によって保護され、使用時におけるCO2促進輸送膜の強度が増す。この結果、CO2促進輸送膜の両側(反応器内外)での圧力差が大きく(例えば、2気圧以上)なっても十分な膜強度を確保できる。更に、ゲル層が疎水性の多孔膜によって被覆されるため、水蒸気が疎水性の多孔膜の膜表面に凝縮しても当該多孔膜が疎水性のために水がはじかれてゲル層内にしみ込むのを防止している。よって、疎水性の多孔膜によって、ゲル層中の二酸化炭素キャリアが水で薄められ、また、薄められた二酸化炭素キャリアがゲル層から流出することを防止できる。
【0050】
また、本発明に係る二酸化炭素分離装置は、上記特徴に加えて、前記ゲル層が、アルデヒド基由来の架橋構造を有することを別の特徴とする。
【0051】
本発明に係るCO2促進輸送膜の上記特徴によれば、ゲル層に形成された架橋構造によって膜内に欠陥が生じにくくなり、この結果、H2パーミアンスが大きく低下する。一方で、CO2パーミアンスは、H2パーミアンスほどの低下を招来しない。これにより、更に高いCO2/H2選択性を示す促進輸送膜を実現することができる。
【0052】
なお、このとき、添加する架橋剤としては、グルタルアルデヒドやホルムアルデヒドを採用することができる。グルタルアルデヒドを添加する場合においては、PVA/PAA共重合体1gに対して0.008〜0.015g程度添加することで、特に高いCO2/H2選択性を示すことができる。
【0053】
また、本発明に係る二酸化炭素分離装置は、上記特徴に加え、前記親水性の多孔膜が100℃以上の耐熱性を備えていることを別の特徴とする。
【0054】
本発明に係る二酸化炭素分離装置の上記特徴によれば、常温から100℃以上に亘る広範な温度範囲での使用が可能となる。具体的には、親水性の多孔膜が100℃以上の耐熱性を備えることで100℃以上の温度領域での使用が可能となる。
【0055】
また、本発明に係るCO2促進輸送膜は、上記特徴に加え、前記ゲル層,並びに前記親水性の多孔膜は、共に軸心を同一にした筒形状であって、一方の膜が、その内側面を他方の膜の外側面と接触させて、前記他方の膜を取り囲むように構成されていることを特徴とする。
【0056】
このとき、前記親水性の多孔膜として、アルミナ等のセラミックス製の膜を利用することができる。
【0057】
また、前記ゲル層を、前記親水性の多孔膜を取り囲むように、前記親水性の多孔膜の外側に形成することができる。
【発明の効果】
【0058】
本発明の構成によれば、100℃以上の高温及び加圧条件下において十分な膜性能を発揮可能なCO2促進輸送膜を用いた二酸化炭素分離装置を実現することができ、また、かかるCO2促進輸送膜を用いた二酸化炭素分離方法を提供することができる。
【図面の簡単な説明】
【0059】
【図1】本発明に係る二酸化炭素分離装置の一実施形態における概略の構成を模式的に示す構成図
【図2】本発明に係るCO2促進輸送膜の一実施形態における構造を模式的に示す断面図
【図3】本発明に係るCO2促進輸送膜の作製方法を示す工程図
【図4】CO2促進輸送膜の比較例サンプルの構造を模式的に示す断面図
【図5】本発明に係るCO2促進輸送膜の膜性能を評価するための実験装置の構成図
【図6】本発明に係るCO2促進輸送膜の親水性多孔膜の使用によるCO2/H2選択性の改善効果を示す図(1)
【図7】本発明に係るCO2促進輸送膜の親水性多孔膜の使用によるCO2/H2選択性の改善効果を示す図(2)
【図8】本発明に係るCO2促進輸送膜のCO2パーミアンスRCO2とCO2/H2選択性の原料ガスの圧力とキャリア濃度に対する依存性を示す図
【図9】本発明に係るCO2促進輸送膜のCO2パーミアンスRCO2とCO2/H2選択性のキャリア濃度に対する依存性を示す図
【図10】本発明に係るCO2促進輸送膜のCO2パーミアンスRCO2とCO2/H2選択性の原料ガスの圧力と使用温度に対する依存性を示す図
【図11】本発明に係るCO2促進輸送膜のCO2パーミアンスRCO2とCO2/H2選択性の使用温度に対する依存性を示す図
【図12】本発明に係るCO2促進輸送膜のCO2パーミアンスRCO2とCO2/H2選択性の原料ガスの圧力と水蒸気モル%に対する依存性を示す図
【図13】本発明に係るCO2促進輸送膜のCO2パーミアンスRCO2とCO2/H2選択性の経時変化を示す図
【図14】本発明に係るCO2促進輸送膜の第2実施形態の実施例1の方法で作製された本発明膜の膜性能を示すグラフ
【図15】本発明に係るCO2促進輸送膜の第2実施形態の実施例2の方法で作製された本発明膜の膜性能を示すグラフ
【図16】本発明に係るCO2促進輸送膜の第2実施形態の実施例3の方法で作製された本発明膜の膜性能を示すグラフ
【図17】本発明に係るCO2促進輸送膜の第2実施形態の実施例1の方法で作製された本発明膜の膜性能の経時変化を示すグラフ
【図18】本発明に係るCO2促進輸送膜の第3実施形態の構造を模式的に示す断面図
【図19】本発明に係るCO2促進輸送膜の第3実施形態のCO2パーミアンス、H2パーミアンス、並びにCO2/H2選択性の原料ガスの圧力に対する依存性を示す図
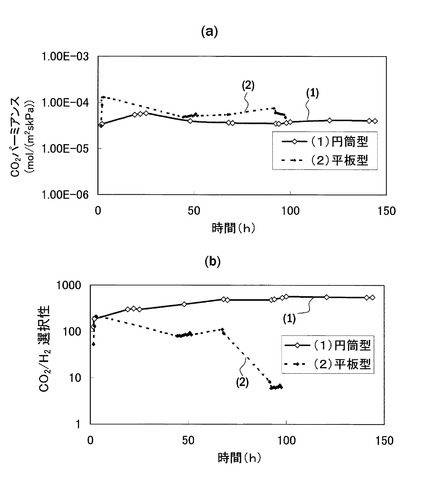
【図20】円筒型と平板型の促進輸送膜における、CO2パーミアンスRCO2とCO2/H2選択性の経時変化の比較図
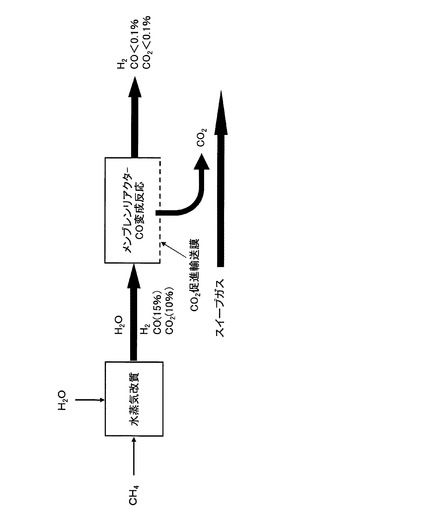
【図21】CO2促進輸送膜を備えたCO変成器における各種ガスの流れを示す図
【図22】CO2促進輸送膜を備えている場合と備えていない場合における、CO変成器の触媒層長に対する一酸化炭素及び二酸化炭素の各濃度変化の比較図
【発明を実施するための形態】
【0060】
本発明に係る二酸化炭素分離装置(以下、適宜「本発明装置」という。)の実施の形態につき、図面に基づいて説明する。
【0061】
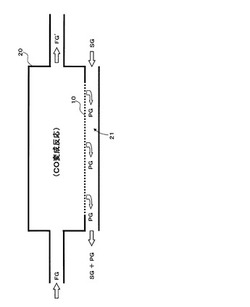
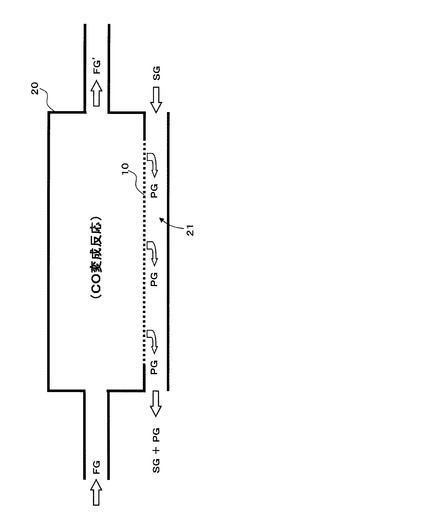
本装置は、図1に示すように、CO2促進輸送膜10を備えたCO変成器(CO2透過型メンブレンリアクター)20を備え、CO2促進輸送膜10によって二酸化炭素が選択的に分離された後の処理済ガスFG’は、排気経路から排気される構成である。
【0062】
CO2透過型メンブレンリアクター20は、例えば、水蒸気改質器(図示せず)で生成された原料ガスFG中の水素以外に含まれる一酸化炭素を、上述の(化1)に示すCO変成反応によって除去する装置であって、CO変成反応で生成される二酸化炭素をCO2促進輸送膜10により選択的にCO変成器外部に除去することで、CO変成反応の化学平衡を水素生成側にシフトさせることができ、同一反応温度において高い転化率で、一酸化炭素及び二酸化炭素を平衡の制約による限界を超えて除去することが可能である。
【0063】
本発明装置では、CO変成反応に供するCO変成触媒の性能が温度とともに低下する傾向にあるため、使用温度は最低でも100℃であると考え、CO2促進輸送膜10の原料側面に供給される原料ガスFGの供給温度は100℃以上となっている。従って、原料ガスFGは、CO2促進輸送膜10より上流側において、CO変成触媒の触媒活性に適した温度に調整された後に、CO変成反応(発熱反応)を経てCO2促進輸送膜10に供給される。なお、CO変成反応後の原料ガス温度を低下させる処理を行ってから原料ガスをCO2促進輸送膜10に供給しても構わない。
【0064】
CO2促進輸送膜10を透過した二酸化炭素を含む透過ガスの分圧を低くして、CO2促進輸送膜10の透過推進力を維持し、透過ガスPGを外部に排出するために、CO2促進輸送膜10の外側面(透過側面)に沿ってスイープガスSGの通路21を設けてあり、当該通路の一方側から空気等のスイープガスSGを送入し、他方側からスイープガスSGとともに透過ガスPGを排出する構造となっている。具体的には、例えば、CO2透過型メンブレンリアクター20を二重管構造として内管内をCO変成器として、内管と外管の間をスイープガスの通路21として構成する。
【0065】
また、原料ガスFGは、上流側の水蒸気改質器から所定の圧力でCO2透過型メンブレンリアクター20に送入される。
【0066】
次に、本発明装置で使用するCO2促進輸送膜10(以下、適宜「本発明膜10」という)について説明する。
【0067】
[本発明膜の第1実施形態]
本発明膜の第1実施形態について、以下に図2〜図13及び表1を参照して説明する。
【0068】
本発明膜は、水分を含むゲル膜内に二酸化炭素キャリアを含有したCO2促進輸送膜であって、100℃以上の使用温度、高い二酸化炭素透過性とCO2/H2選択性を有するCO2透過型メンブレンリアクターへ応用可能なCO2促進輸送膜である。更に、本発明膜は、高いCO2/H2選択性を安定して実現するために、二酸化炭素キャリアを含有したゲル膜を担持する支持膜として、親水性の多孔膜を採用している。
【0069】
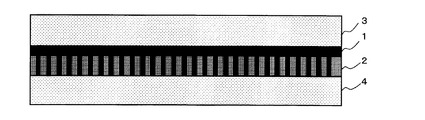
具体的には、本発明膜は、膜材料として、ポリビニルアルコール‐ポリアクリル酸(PVA/PAA)共重合体を使用し、二酸化炭素キャリアとして、炭酸セシウム(Cs2CO3)を使用する。また、本発明膜は、図2に模式的に示すように、二酸化炭素キャリアを含有するPVA/PAAゲル膜1を担持した親水性多孔膜2が、2枚の疎水性多孔膜3,4に挟持される3層構造で構成される。以下、二酸化炭素キャリアを含有するPVA/PAAゲル膜を、二酸化炭素キャリアを含有しないPVA/PAAゲル膜、及び、2枚の疎水性多孔膜を備えた構造の本発明膜と区別するために、適宜「含浸ゲル膜」と略称する。また、この含浸ゲル膜中のPVA/PAAとCs2CO3の全重量を基準として、含浸ゲル膜中において、PVA/PAAは約20〜80重量%の範囲で存在し、Cs2CO3は約20〜80重量%の範囲で存在する。
【0070】
親水性多孔膜は、親水性に加えて、100℃以上の耐熱性、機械的強度、含浸ゲル膜との密着性を有するのが好ましく、更に、多孔度(空隙率)が55%以上で、細孔径は0.1〜1μmの範囲にあるのが好ましい。本実施形態では、これらの条件を備えた親水性多孔膜として、親水性化した四フッ化エチレン重合体(PTFE)多孔膜を使用する。
【0071】
疎水性多孔膜は、疎水性に加えて、100℃以上の耐熱性、機械的強度、含浸ゲル膜との密着性を有するのが好ましく、更に、多孔度(空隙率)が55%以上で、細孔径は0.1〜1μmの範囲にあるのが好ましい。本実施形態では、これらの条件を備えた疎水性多孔膜として、親水性化していない四フッ化エチレン重合体(PTFE)多孔膜を使用する。
【0072】
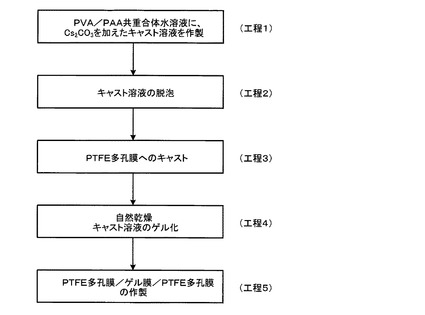
次に、本発明膜の作製方法(本発明方法)の一実施形態について、図3を参照して説明する。
【0073】
まず、PVA/PAA共重合体とCs2CO3を含む水溶液からなるキャスト溶液を作製する(工程1)。より詳細には、PVA/PAA共重合体(例えば、住友精化製の仮称SSゲル)を1g、Cs2CO3を2.33g、サンプル瓶に秤取し、これに水20mlを加えて室温で5日間攪拌して溶解させてキャスト溶液を得る。
【0074】
次に、工程1で得たキャスト溶液中の気泡を除去するために、遠心分離(回転数5000rpmで30分間)を行う(工程2)。
【0075】
次に、工程2で得たキャスト溶液を、親水性PTFE多孔膜(例えば、住友電工製,WPW−020−80、膜厚80μm、細孔径0.2μm、空隙率75%)と疎水性PTFE多孔膜(例えば、住友電工製、フロロポアFP010、膜厚60μm、細孔径0.1μm、空隙率55%)を2枚重ね合わせた層状多孔膜の親水性PTFE多孔膜側の面上に、アプリケータでキャストする(工程3)。なお、後述する実施例のサンプルでのキャスト厚は500μmである。ここで、キャスト溶液は、親水性PTFE多孔膜中の細孔内に浸透するが、疎水性のPTFE多孔膜の境界面で浸透が停止し、層状多孔膜の反対面までキャスト溶液がしみ込まず、層状多孔膜の疎水性PTFE多孔膜側面にはキャスト溶液が存在せず取り扱いが容易となる。
【0076】
次に、キャスト後の親水性PTFE多孔膜を室温で一昼夜自然乾燥させた後、キャスト溶液をゲル化させゲル層を生成する(工程4)。この方法では、工程3において、キャスト溶液を層状多孔膜の親水性PTFE多孔膜側の表面にキャストするため、工程4において、ゲル層は、親水性PTFE多孔膜の表面(キャスト面)に形成されるのみならず細孔内にも充填して形成されるので、欠陥(ピンホール等の微小欠陥)が生じ難くなり、ゲル層の製膜成功率が高くなる。なお、工程4において、自然乾燥させたPTFE多孔膜を、更に、120℃程度の温度で、2時間程度熱架橋するのが望ましい。なお、後述する実施例及び比較例のサンプルでは、何れも熱架橋を行っている。
【0077】
次に、工程4で得た親水性PTFE多孔膜表面のゲル層側に、工程3で用いた層状多孔膜の疎水性PTFE多孔膜と同じ疎水性PTFE多孔膜を重ね、図2に模式的に示すように、疎水性PTFE多孔膜/ゲル層(親水性PTFE多孔膜に担持された含浸ゲル膜)/疎水性PTFE多孔膜よりなる3層構造の本発明膜を得る(工程5)。なお、図2において、含浸ゲル膜1が親水性PTFE多孔膜2の細孔内に充填している様子を模式的に直線状に表示している。
【0078】
以上、工程1〜工程5を経て作製された本発明膜は、後述するようにCO2透過型メンブレンリアクターへ応用可能な膜性能、即ち、使用温度100℃以上、2×10−5mol/(m2・s・kPa)(=60GPU)程度以上のCO2パーミアンス、及び、90〜100程度以上のCO2/H2選択性が実現できる。
【0079】
また、ゲル層を疎水性PTFE多孔膜で挟持した3層構造とすることにより、一方の疎水性PTFE多孔膜は、工程3及び工程4で用いられ、含浸ゲル膜を担持する親水性PTFE多孔膜の支持とキャスト溶液の浸透防止に供せられ、他方の疎水性PTFE多孔膜は、含浸ゲル膜を他方面側から保護するのに用いられる。
【0080】
更に、水蒸気が疎水性PTFE多孔膜の膜表面に凝縮しても当該PTFE多孔膜が疎水性のために水がはじかれて含浸ゲル膜にしみ込むのを防止している。よって、他方のPTFE多孔膜によって、含浸ゲル膜中の二酸化炭素キャリアが水で薄められ、また、薄められた二酸化炭素キャリアが含浸ゲル膜から流出することを防止できる。
【0081】
以下、具体的な実施例の膜性能について説明する。
【0082】
まず、含浸ゲル膜を担持する多孔膜として、親水性PTFE多孔膜を使用した実施例と、疎水性PTFE多孔膜を使用した比較例の各サンプルの膜組成について説明する。
【0083】
実施例のサンプルは、上述の作製方法により作製した。(PVA/PAA:Cs2CO3)の配合比率は、記載の順に、(30重量%:70重量%)となっている。なお、以下では、共重合体重量とキャリア重量の合計重量に対するキャリア重量の比率を「キャリア濃度」と記載する。即ち、上記の例の場合、キャリア濃度は70%である。
【0084】
比較例のサンプルは、上述の作製方法において、親水性PTFE多孔膜と疎水性PTFE多孔膜の層状多孔膜に替えて1層の疎水性PTFE多孔膜を使用して作製された。従って、比較例のサンプルは、図4に模式的に示すように、二酸化炭素キャリアを含有するPVA/PAAゲル膜1が、2枚の疎水性多孔膜3,4の間に挟持される3層構造で構成される。(PVA/PAA:Cs2CO3)の配合比率は、実施例と同じである。
【0085】
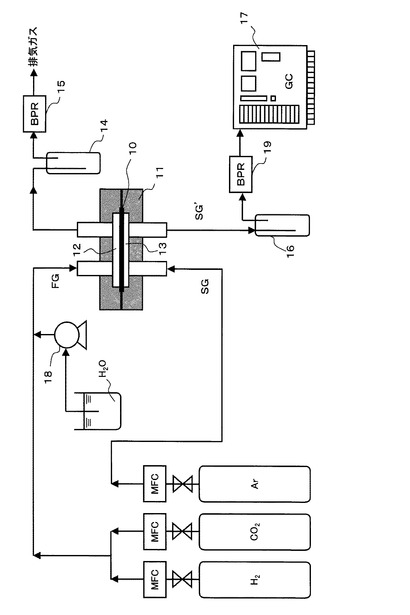
次に、実施例、及び、比較例の各サンプルの膜性能を評価するための実験装置の構成及び実験方法について、図5を参照して説明する。
【0086】
図5に示すように、各サンプル10は、ステンレス製の流通式ガス透過セル11(膜面積:2.88cm2)の原料側室12と透過側室13の間に、2枚のフッ素ゴム製ガスケットをシール材として用いて固定されている。原料ガス(CO2、H2、H2Oからなる混合ガス)FGを、2.24×10−2mol/minの流量で原料側室12に供給し、スイープガス(Arガス)SGを、8.18×10−4mol/minの流量で透過側室13に供給する。原料側室12の圧力は、排気ガスの排出路の途中の冷却トラップ14の下流側に設けられた背圧調整器15で調整される。透過側室13の圧力は大気圧である。透過側室13から排出するスイープガスSG’中の水蒸気を冷却トラップ16で除去した後のガス組成をガスクロマトグラフ17で定量し、これとスイープガスSG中のArの流量よりCO2及びH2のパーミアンス[mol/(m2・s・kPa)]を計算し、その比より、CO2/H2選択性を算出する。なお、冷却トラップ16とガスクロマトグラフ17の間にも背圧調整器19が設けられており、これによって透過側室13の圧力が調整される。
【0087】
原料ガスFGは、CO変成器内における原料ガスを模擬するために、CO2、H2、H2Oからなる第1混合ガスを、CO2:5.0%、H2:45%、H2O:50%の混合比率(モル%)に調整した。具体的には、10%CO2と90%H2(モル%)よりなる混合ガス流(25℃での流量:200cm3/min、8.18×10−3mol/min)に水を定量送液ポンプ18で送入し(流量:0.256cm3/min、1.42×10−2mol/min)、100℃以上に加熱して水分を蒸発させて、上記混合比率の混合ガスを調製し、これを原料側室12に供給した。
【0088】
スイープガスSGは、サンプル膜を透過する被測定ガス(CO2、H2)の透過側室側の分圧を低くして、透過推進力を維持するために供給され、被測定ガスと異なるガス種(Arガス)を用いる。具体的には、Arガス(25℃での流量:20cm3/min、8.13×10−4mol/min)を透過側室13に供給した。
【0089】
なお、図示していないが、サンプル膜の使用温度、及び、原料ガスFGとスイープガスSGの温度を一定温度に維持するために、サンプル膜を固定した流通式ガス透過セル11と上記ガスを加熱する予熱コイルを、所定温度に設定したオイル恒温槽内に浸している。
【0090】
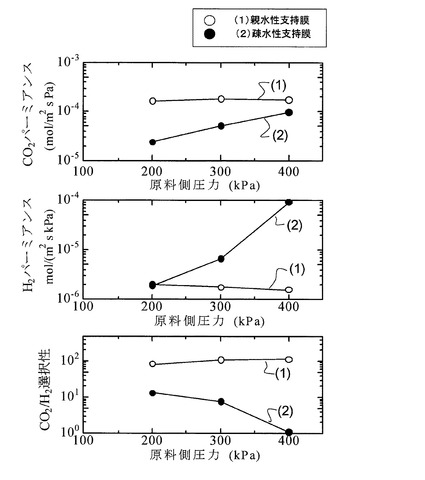
次に、図6及び図7に、(1)実施例と(2)比較例の各サンプルのCO2パーミアンスRCO2、H2パーミアンスRH2、及びCO2/H2選択性を、原料側室12内の原料ガスFGの圧力(グラフ上では「原料側圧力」と記載。以下同様)を200kPa〜400kPaの範囲の加圧状態で測定した結果を示す。なお、図6は測定温度を160℃、図7は測定温度を180℃として、それぞれ測定したものである。また、グラフ上における原料側圧力の値は、原料側室12の圧力を調整するための背圧調整器15が示す圧力値を採用した。
【0091】
図6及び図7より、H2パーミアンスは、疎水性PTFE多孔膜を使用した比較例のサンプルの方が、親水性PTFE多孔膜を使用した実施例のサンプルより、全圧力範囲で高くなっているが、CO2パーミアンス、並びにCO2/H2選択性では、実施例のサンプルの方が、比較例のサンプルより大幅に改善されていることが分かる。これは、親水性膜の場合は、キャスト溶液を膜上にキャストすると、PTFE多孔膜の表面のみならず細孔内にもゲル層が充填されるので欠陥(ピンホール等の微小欠陥)が生じ難くなり、当該微小欠陥を介してガスパーミアンス、特に、H2パーミアンスが上昇するのが抑制されるためと考えられる。これに対し、疎水性膜の場合には、キャスト液が膜の細孔内には侵入せず、その表面に塗布されるので、欠陥が生じやすく、H2パーミアンスが上昇し、これによって選択性が低下するものと考えられる。
【0092】
なお、図6及び図7によれば、測定温度を変更しても、同様の特性が示されていることが分かる。
【0093】
また、上記特許文献1,2に開示されたCO2促進輸送膜では、100℃以上の使用温度、2×10−5mol/(m2・s・kPa)程度以上のCO2パーミアンス、及び、90〜100程度以上のCO2/H2選択性の何れも満足していないのに対して、図6及び図7に示す実施例のサンプルは、全圧力範囲において全ての要件を概ね満足している。また、比較例のサンプルでも、100℃以上の使用温度条件下で、2×10−5mol/(m2・s・kPa)程度以上のCO2パーミアンスを示している。なお、比較例のサンプルでは、300kPa以上でCO2/H2選択性の低下が大きくなることが示唆されている。
【0094】
図6及び図7の結果を勘案すれば、特許文献1、2に開示されたCO2促進輸送膜と比較して、Cs2CO3を含有するPVA/PAAゲル膜を備える本発明膜の方が、100℃以上の高温条件下においてCO2パーミアンスを向上させることができる。そして、特に支持膜を親水性多孔膜とすることで、CO2パーミアンス、及びCO2/H2選択性の値を顕著に向上させることができる。
【0095】
以下では、実施例と同様、親水性PTFE多孔膜によって含浸ゲル膜を担持する構成を有する本発明膜を用いてデータの取得を行った。
【0096】
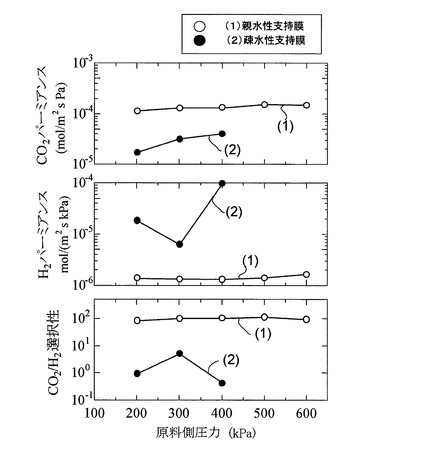
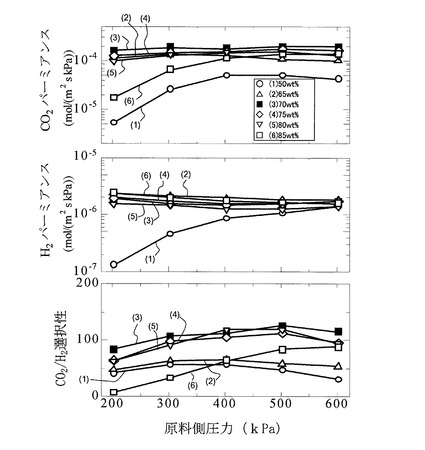
次に、図8に、キャリア濃度を50wt%〜85wt%まで変化させて作製された各サンプルのCO2パーミアンスRCO2、H2パーミアンスRH2、及びCO2/H2選択性を、原料ガスFGの混合比率及び測定温度を図6と同一の条件とし、原料ガスFGの圧力を200kPa〜600kPaの範囲の加圧状態で測定した結果を示す。
【0097】
図8より、160℃の測定温度下において、キャリア濃度70wt%のときにCO2パーミアンスRCO2が最大となり、また、原料ガスFGの圧力が500kPaのときにCO2パーミアンスRCO2が最大となることが分かる。そして、キャリア濃度が80%以下の場合、及びキャリア濃度が85%の場合であって原料ガスFGの圧力が300kPa以上の場合には、いずれも5.0×10−5mol/(m2・s・kPa)以上の高いCO2パーミアンスを示すことが分かる。
【0098】
また、H2パーミアンスRH2は、キャリア濃度が50wt%の場合を除くと、全体的に原料ガスFGの圧力が増加するとともに微減する傾向を示すことが分かる。
【0099】
更に、図8より、キャリア濃度が70wt%以上80wt%以下の場合には、原料ガスFGの圧力が200〜600kPaのいずれの場合においても90〜100程度以上のCO2/H2選択性を示すことが分かる。
【0100】
即ち、図8の結果より、本発明膜によれば、キャリア濃度を調整することにより、使用温度100℃以上(160℃)、2×10−5mol/(m2・s・kPa)(=60GPU)程度以上のCO2パーミアンス、及び、90〜100程度以上のCO2/H2選択性が実現できる。このため、本発明膜をCO2透過型メンブレンリアクターに適用することができる。特に、70wt%〜80wt%の範囲内で全圧力範囲に亘って高いCO2/H2選択性を示すことが示唆されている。
【0101】
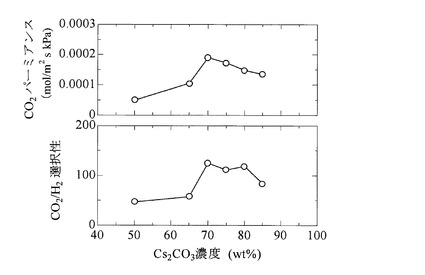
また、図9は、原料ガス圧力を一定(501.3kPa)として、キャリア濃度とCO2パーミアンスRCO2の関係、並びに、キャリア濃度とCO2/H2選択性の関係をそれぞれグラフにしたものである。なお、原料ガスFGの混合比率及び測定温度は図8の場合と同一の条件とした。
【0102】
図9によれば、キャリア濃度が70wt%の場合に、CO2パーミアンス並びにCO2/H2選択性の双方が最も高い値を示すことが分かる。即ち、図9によれば、CO2パーミアンス並びにCO2/H2選択性は、いずれもキャリア濃度に依存することが分かる。特に、本発明膜をCO2促進輸送膜として利用する際には、キャリア濃度を70wt%と設定することで、その能力を最大限発揮させることができる。
【0103】
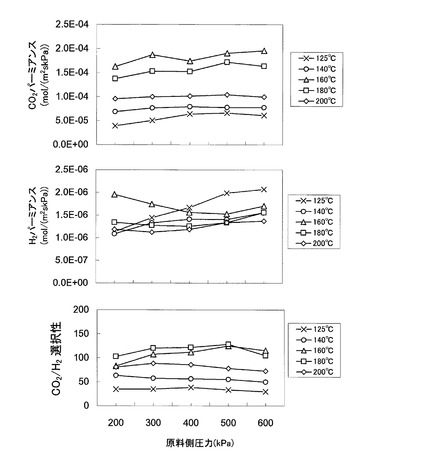
図10は、キャリア濃度を70wt%とし、原料ガスFGの混合比率を図8の場合と同様とした状態の下、測定温度を125℃以上200℃以下の範囲内で変化させたときの、CO2パーミアンスRCO2、H2パーミアンスRH2、及びCO2/H2選択性を、原料側室12内の原料ガスFGの圧力を200kPa〜600kPaの範囲の加圧状態で測定した結果を示す。
【0104】
図10によれば、測定温度が160℃のときが最もCO2パーミアンスRCO2が大きくなっている。また、CO2/H2選択性については、測定温度が160℃並びに180℃におけるCO2/H2選択性が大きく、それより温度が上昇しても低下してもCO2/H2選択性が減少することが分かる。即ち、図10によれば、CO2パーミアンス並びにCO2/H2選択性は、測定温度にも依存することが分かる。特に、本発明膜をCO2促進輸送膜として利用する際には、160℃の温度条件下で本発明膜を設置することで、その能力を最大限発揮させることができることが分かる。これにより、本発明膜によれば、特許文献1、2に開示された従来のCO2促進輸送膜と比較して、十分高い温度条件下(125℃〜200℃)で高いCO2パーミアンスと高いCO2/H2選択性を実現することができ、特に140℃〜180℃で良好な値を実現することが示唆される。
【0105】
なお、本発明膜は、測定温度が200℃の場合であっても、CO2パーミアンスRCO2は1.0×10−4mol/(m2・s・kPa)程度の値を示しており、2×10−5mol/(m2・s・kPa)程度以上のCO2パーミアンスを示すことが分かる。そして、一定温度条件下においては、原料ガスFGの圧力を変化してもCO2パーミアンスの値はあまり変化していないことが分かる。
【0106】
更に、図10によれば、200℃の高温条件下において、CO2/H2選択性は圧力300kPaの下で100に近い値を示していることが分かる。即ち、200℃程度の高温条件下でも、CO2透過型メンブレンリアクターに適用可能なCO2促進輸送膜を実現できることが分かる。
【0107】
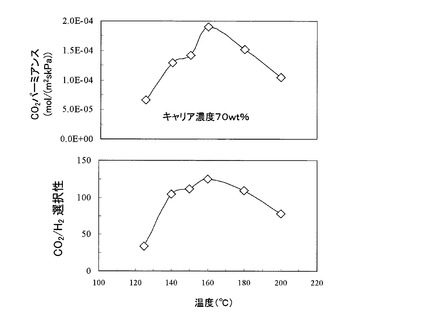
また、図11は、原料ガス圧力を一定(501.3kPa)として、測定温度とCO2パーミアンスRCO2の関係、並びに、測定温度とCO2/H2選択性の関係をそれぞれグラフにしたものである。なお、原料ガスFGの混合比率及び測定温度は図10の場合と同一の条件とした。
【0108】
図11によれば、測定温度が160℃の場合に、CO2パーミアンス並びにCO2/H2選択性の双方が最も高い値を示すことが分かる。即ち、図11によれば、CO2パーミアンス並びにCO2/H2選択性は、いずれも測定温度の高さに依存することが分かる。特に、本発明膜をCO2促進輸送膜として利用する際には、160℃の温度条件下で利用することでその能力を最大限発揮させることができる。
【0109】
図12は、キャリア濃度を70wt%として作製されたサンプルのCO2パーミアンスRCO2、H2パーミアンスRH2、及びCO2/H2選択性を、原料ガスFGの混合比率及び測定温度を図6と同一の条件とし、水蒸気モル%を20%、30%、50%、70%、90%と変化させた場合において、原料ガスFGの圧力を200kPa〜600kPaの範囲の加圧状態で測定した結果を示す。なお、具体的には、CO2、H2、H2Oからなる混合ガスを、CO2のモル%を5%と固定し、H2並びにH2Oのモル%の合計が95%となるように、H2のモル%並びにH2Oのモル%(水蒸気モル%)をそれぞれ変更して測定した。
【0110】
図12によれば、水蒸気モル%が高いほどCO2パーミアンスの値が上昇し、逆に水蒸気モル%が低下するに連れCO2パーミアンスの値も低下することが分かる。そして、水蒸気モル%を30%程度にまで低下させた場合においても、400kPaの原料ガスFGの圧力条件の下で約1×10−4mol/(m2・s・kPa)程度のCO2パーミアンスを示している。
【0111】
なお、H2パーミアンスの値は、水蒸気モル%が20%の場合においては大きく変化しているものの、他の値の場合にはあまり大きな差異が見られない。一方、CO2/H2選択性は、水蒸気モル%が低下するに連れほぼ全体的に低下することが分かる。そして、水蒸気モル%を30%としても、400kPaの原料ガスFGの圧力条件の下で約100程度のCO2/H2選択性を示している。
【0112】
従って、図12のグラフにより、水蒸気モル%を30%以下に低く設定した条件下においても、本発明膜は優れた性能を示し、CO2透過型メンブレンリアクターに適用可能なCO2促進輸送膜を実現できることが分かる。従って、本発明装置は、原料ガスの水蒸気モル%が低い場合においても、CO2分離性能を示すことができる。
【0113】
また、図13は、本発明膜の長期性能を示すグラフ結果である。原料ガスをCO2:5%、H2:45%、H2O:50%の混合比率(モル%)に調整し、原料ガスの圧力を351.03kPa、キャリア濃度を70wt%としたときの、CO2パーミアンスRCO2、並びにCO2/H2選択性の値の経時変化をグラフ化したものである。
【0114】
図13によれば、時間が経過してもCO2パーミアンスRCO2の値は大きな変化を示さず、ほぼ1.6×10−4mol/(m2・s・kPa)程度の値を示した。また、CO2/H2選択性についても、同様に時間が経過しても大きな変化を示さず、ほぼ100程度の値を示した。このように、本発明膜によれば、時間経過に連れて性能が大きく低下するということがなく、長時間にわたって、優れた性能を示すCO2透過型メンブレンリアクターに適用可能なCO2促進輸送膜を実現できることが分かる。
【0115】
また、下記表1は、膜材料を同一(PVA/PAA共重合体)とし、二酸化炭素キャリアとして用いる材料をCs2CO3以外の種々の炭酸塩としたときの、CO2パーミアンス、H2パーミアンス、及びCO2/H2選択性の値を、本発明膜と比較したものである。表1では、本発明膜で用いたCsの炭酸塩以外に、Na、K、Rbの炭酸塩を二酸化炭素キャリアとして利用した場合の上記データを測定した。なお、いずれの場合も、原料ガス圧力を401.33kPa、測定温度を160℃とし、原料ガスをCO2:5.0%、H2:45%、H2O:50%の混合比率(モル%)に調整して各データの測定を行った。また、各膜は、上述した本発明膜の製法と同様の方法により製造した。
【0116】
【表1】

【0117】
表1に示される結果によれば、Na2CO3膜による場合,CO2パーミアンスは非常に低く、そして高いH2パーミアンスを示した。これは、Na2CO3の水中への溶解度が低いことから(表1参照)、キャスト膜を120℃で架橋するときに結晶が生成され、これによって均一な膜が得られなかったためと考えられる。また、K2CO3膜の場合、高いCO2パーミアンスが得られたが、膜に欠陥が生じやすいためにH2パーミアンスも大きくなり、高いCO2/H2選択性は得られなかった。一方、水中への溶解度が高いRb2CO3およびCs2CO3(表1参照)を含む膜では、CO2パーミアンス並びにCO2/H2選択性ともに良好な結果が得られた。
【0118】
以上より、水中への溶解度の高い炭酸塩は、高温においてもCO2のキャリアとして効率よく機能し,それを含有する膜は欠陥が生じにくく、高いCO2透過性及び選択性を示すことが明らかになった。特に、Cs2CO3をキャリアとする本発明膜によれば、高いCO2パーミアンス及び高いCO2/H2選択性を示すCO2促進輸送膜を実現することができる。従って、本発明装置が、かかるCO2促進輸送膜を備えることで、高いCO2分離能力を示すことができる。
【0119】
[本発明膜の第2実施形態]
本発明膜の第2実施形態(以下、適宜「本実施形態」と称する)につき、図14〜図17の各図を参照して説明する。なお、本実施形態は、本発明膜の第1実施形態と比較して本発明膜及び本発明方法の一部構成が異なるのみであるため、同一の構成要素についてはその旨を記載して説明を割愛する。
【0120】
本実施形態では、第1実施形態と比較して、キャスト溶液を作製する工程(上記工程1)の内容が異なる。本実施形態では、第1実施形態の工程1に相当する工程(キャスト溶液作製工程)として以下の3通りの工程を行うものとし、それぞれを実施例1〜3と称する。
【0121】
(実施例1)
まず、PVA/PAA共重合体(例えば、住友精化製の仮称SSゲル)を1gに水20gを加えた後、室温で攪拌してゲルを溶解させる。次に、これにグルタルアルデヒドを0.008〜0.0343g程度加えた後、95℃の温度条件下で15時間攪拌する。そして、これにCs2CO3を2.33g加えて更に室温で攪拌することでキャスト溶液を得る。即ち、実施例1では、ゲル溶解工程、グルタルアルデヒド添加工程、高温下での攪拌工程、Cs2CO3添加工程、室温下での攪拌工程、の順に処理を行うことでキャスト溶液を作製する。
【0122】
(実施例2)
まず、PVA/PAA共重合体を1gに水20gを加えた後、室温で攪拌してゲルを溶解させる。次に、これにCs2CO3を2.33gと、グルタルアルデヒドを0.008〜0.0343g程度加えた後、室温で攪拌して溶解させる。その後、95℃の温度条件下で15時間攪拌することでキャスト溶液を得る。即ち、実施例2では、ゲル溶解工程、グルタルアルデヒド及びCs2CO3添加工程、室温下での攪拌工程、高温下での攪拌工程、の順に処理を行うことでキャスト溶液を作製する。
【0123】
(実施例3)
まず、PVA/PAA共重合体を1gに水20gを加えた後、室温で攪拌してゲルを溶解させる。次に、これにCs2CO3を2.33gと、グルタルアルデヒドを0.008〜0.0343g程度加えた後、室温で攪拌して溶解させることで、キャスト溶液を得る。即ち、実施例3では、ゲル溶解工程、グルタルアルデヒド及びCs2CO3添加工程、室温下での攪拌工程、の順に処理を行うことでキャスト溶液を作製する。
【0124】
なお、実施例1〜3の何れにおいても、キャスト溶液を作製後については、第1実施形態に記載の工程(工程2〜4)と同様の方法を用いてCO2促進輸送膜を得る。即ち、キャスト溶液中の気泡を除去するために遠心分離を行った後、ガラス板上に疎水性PTFE多孔膜(膜厚60μm)と親水性PTFE多孔膜(膜厚80μm)を重ね合わせた層状多孔膜の親水性PTFE多孔膜側の面上に、前述のキャスト溶液をアプリケータを用いて厚さ500μmでキャストする。その後、室温で一昼夜乾燥させる。そして、更にこれを120℃程度の高温条件下で2時間程度保持することで、CO2促進輸送膜を得る。
【0125】
以下、これらの各実施例1〜3の方法で製造された本発明膜の膜性能について説明する。なお、膜組成は、第1実施形態の実施例と同様にキャリア濃度を70wt%とし、膜性能を評価するための実験装置及び実験方法も第1実施形態と同様とする。
【0126】
図14は、実施例1の方法で作製されたキャスト溶液を用いて作製された本発明膜による(a)CO2パーミアンスRCO2、(b)H2パーミアンスRH2、及び(c)CO2/H2選択性を、原料側圧力を200kPa〜600kPaの範囲の加圧状態で測定した結果を示す。なお、図14では、キャスト溶液作製の際に添加されるグルタルアルデヒドの添加量を異ならせて各データの測定を行った。即ち、グルタルアルデヒドの添加量として、(1)0.008g、(2)0.0153g、(3)0g(不添加)の3パターンで実験を行った。なお、グラフ上では、グルタルアルデヒドを「GA」と略記している(以下のグラフも同様)。
【0127】
また、実験条件としては、温度条件を160℃、原料ガスFGをCO2:5.0%、H2:45%、H2O:50%の混合比率(モル%)、原料ガスFGの流量を25℃・1atm下で360cm3/min、透過側の圧力を原料側の圧力から20kPa減、スイープガスSGの流量を25℃・1atm下で40cm3/minとした。なお、この実験条件は、各実施例とも共通である。
【0128】
図14(a)によれば、グルタルアルデヒドを添加した場合、添加しない場合よりもCO2パーミアンスRCO2が微小に低下している。しかしながら、図14(b)に示すように、グルタルアルデヒドを添加した場合、H2パーミアンスRH2が大幅に低下しているため、図14(c)に示すように、グルタルアルデヒドを添加することで添加しない場合と比べてCO2/H2選択性は大きく上昇することが分かる。これは、グルタルアルデヒドを添加したことにより、架橋構造が形成されることで膜の欠陥が生じにくくなったためにH2パーミアンスが大きく低下したことによるものと考えられる。図14(b)及び(c)によれば、グルタルアルデヒドを0.008g添加した場合の方が、0.0153g添加した場合よりもH2パーミアンスが低く、CO2/H2選択性が高いことが分かる。即ち、グルタルアルデヒドの添加量は、多ければ多いほど選択性が高くなるという訳ではなく、実験条件に応じて高い選択性が実現可能な適正な添加量が存在することが示唆される。
【0129】
図15は、実施例2の方法で作製されたキャスト溶液を用いて作製された本発明膜による(a)CO2パーミアンスRCO2、(b)H2パーミアンスRH2、及び(c)CO2/H2選択性を、原料側圧力を200kPa〜600kPaの範囲の加圧状態で測定した結果を示す。なお、図15では、キャスト溶液作製の際に添加されるグルタルアルデヒドの添加量を異ならせて各データの測定を行った。即ち、グルタルアルデヒドの添加量として、(1)0.008g、(2)0.0165g、(3)0g(不添加)の3パターンで実験を行った。その他の実験条件は実施例1の場合と同様である。
【0130】
図15(a)によれば、図14(a)と同様、グルタルアルデヒドを添加した場合、添加しない場合よりもCO2パーミアンスRCO2が微小に低下している。そして、図15(b)によれば、図14(b)と同様、グルタルアルデヒドを添加した場合にH2パーミアンスRH2が大幅に低下しているため、図15(c)に示すように、グルタルアルデヒドを添加することで添加しない場合と比べてCO2/H2選択性は大きく上昇することが分かる。これは、実施例1の場合と同様の理由、即ち、グルタルアルデヒドを添加したことにより、架橋構造が形成されることで膜の欠陥が生じにくくなったためにH2パーミアンスが大きく低下したことによるものと考えられる。図15(b)及び(c)によれば、グルタルアルデヒドを0.008g添加した場合の方が、0.0165g添加した場合よりもH2パーミアンスが低く、CO2/H2選択性が高いことが分かる。即ち、グルタルアルデヒドの添加量は、多ければ多いほど選択性が高くなるという訳ではなく、実験条件に応じて高い選択性が実現可能な適正な添加量が存在することが示唆される。なお、図15(c)より、原料側のガス圧力が高い領域においては、添加するグルタルアルデヒドの添加量による選択性の差分は小さくなっている。
【0131】
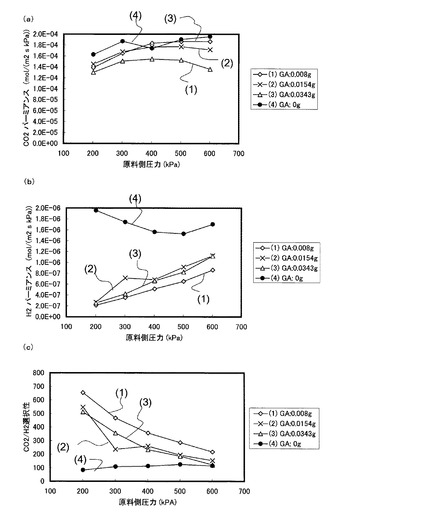
図16は、実施例3の方法で作製されたキャスト溶液を用いて作製された本発明膜による(a)CO2パーミアンスRCO2、(b)H2パーミアンスRH2、及び(c)CO2/H2選択性を、原料側圧力を200kPa〜600kPaの範囲の加圧状態で測定した結果を示す。なお、図15では、キャスト溶液作製の際に添加されるグルタルアルデヒドの添加量を異ならせて各データの測定を行った。即ち、グルタルアルデヒドの添加量として、(1)0.008g、(2)0.0154g、(3)0.0343g、(4)0g(不添加)の4パターンで実験を行った。その他の実験条件は実施例1の場合と同様である。
【0132】
図16(a)によれば、図14(a)と同様、グルタルアルデヒドを添加した場合、添加しない場合よりもCO2パーミアンスRCO2が微小に低下している。そして、図16(b)によれば、図14(b)と同様、グルタルアルデヒドを添加した場合にH2パーミアンスRH2が大幅に低下しているため、図16(c)に示すように、グルタルアルデヒドを添加することで添加しない場合と比べてCO2/H2選択性は大きく上昇することが分かる。これは、実施例1の場合と同様の理由、即ち、グルタルアルデヒドを添加したことにより、架橋構造が形成されることで膜の欠陥が生じにくくなったためにH2パーミアンスが大きく低下したことによるものと考えられる。図16(b)及び(c)によれば、グルタルアルデヒドを0.008g添加した場合の方が、0.0154g添加した場合、並びに0.0343g添加した場合よりもH2パーミアンスが低く、CO2/H2選択性が高いことが分かる。即ち、グルタルアルデヒドの添加量は、多ければ多いほど選択性が高くなるという訳ではなく、実験条件に応じて高い選択性が実現可能な適正な添加量が存在することが示唆される。
【0133】
なお、図16(c)においても、原料側のガス圧力が高い領域においては、添加するグルタルアルデヒドの添加量による選択性の差分は小さくなっている。
【0134】
以上、図14〜図16の各グラフを参照すれば、ゲル膜をグルタルアルデヒドで架橋することにより、グルタルアルデヒド無添加の場合と比較して、CO2の透過性の低下を一定程度に抑制しながらH2の透過性を著しく低下させることが可能となり、これによって高いCO2/H2選択性を示す促進輸送膜を実現することができる。特に、PVA/PAA共重合体を1gに対し、グルタルアルデヒドを0.008〜0.015g程度添加した場合(以下、かかる範囲を「良好範囲」と称する)に、CO2/H2の選択性が著しく向上する。
【0135】
なお、上記実施例1〜3間では膜性能に顕著な差異は見られなかった。即ち、いずれの方法で製膜した場合であっても、グルタルアルデヒドを添加することによるCO2/H2の選択性の向上効果を実現することができる。特に、実施例2及び3においては、グルタルアルデヒドで架橋した場合でもCO2パーミアンスの低下量が抑制された。一方、実施例1においては、原料側のガス圧力を増加させてもH2パーミアンスの増加が一定程度に抑制された。
【0136】
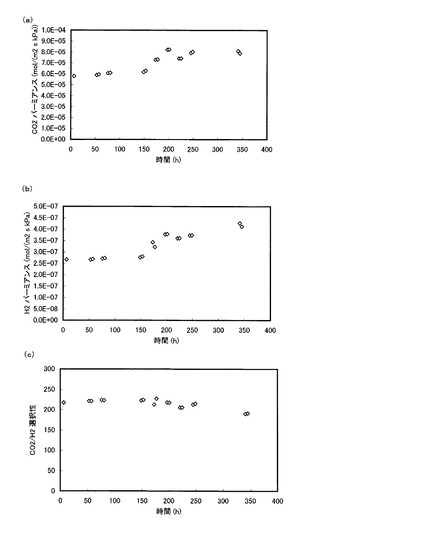
また、図17は、グルタルアルデヒドを添加した場合の長期性能を示すグラフである。具体的には、実施例1の方法によって作製した膜(グルタルアルデヒド添加量:0.0339g)を用いて長期間実験を行ったときの(a)CO2パーミアンスRCO2、(b)H2パーミアンスRH2、(c)CO2/H2選択性の経時変化をそれぞれグラフに表したものである。なお、原料側ガス圧力を401.3kPaとし、その他の実験条件は、図14〜図16と同様とした。
【0137】
実験方法としては、午前10時頃に本発明膜を透過セルにセットして温度を160℃まで上げて原料ガスおよびスイープガスを供給して透過実験を開始し、午後8時頃まで同じ条件で継続した。そして、午後8時頃に供給ガスを停めて温度を室温まで低下させた。再び翌朝の午前10時頃に、透過セルを分解せずにそのまま同じ膜を使用して同様の実験を行った。このような実験を繰り返し2週間続けた結果が図17(a)〜(c)に示されている。
【0138】
図17の実験データは、添加したグルタルアルデヒド量が前記良好範囲よりも若干多いので、CO2パーミアンスは,図14〜図16の値と比較して小さい値となっているが、H2パーミアンスは、時間が経過してもグルタルアルデヒド無添加の場合より著しく小さい値が示されており、CO2/H2選択性も200以上の高い値を維持している。特に、本評価方法のように、スタートアップとシャットダウンを繰返して経時評価を行う場合には、温度の変動(室温〜160℃)や圧力の変動(常圧〜6気圧)を膜に繰返し与えるため、同一温度・同一圧力で実験を継続することで長期性能を評価する場合と比較して、膜に対する負荷が大きくなる。図17によれば、スタートアップ、シャットダウンを繰り返す本評価方法によっても、膜性能が約2週間にわたって安定していることから、グルタルアルデヒド添加によって、膜の安定性が著しく改善されたと言える。
【0139】
なお、本実施形態で添加する材料としてグルタルアルデヒドを採用したが、当該材料の添加工程は、膜に架橋構造を形成するために行われるものであるところ、架橋構造の形成が可能な材料であれば、グルタルアルデヒドに限定されるものではない。アルデヒド基で架橋を形成する場合には、例えば、ホルムアルデヒドも利用可能である。また、二酸化炭素キャリアとして用いる材料をCs2CO3以外の材料(例えばRb2CO3)とした場合でも、同様に添加剤を導入して架橋構造とすることで膜性能を更に高めることが可能である。
【0140】
[本発明膜の第3実施形態]
本発明膜の第3実施形態につき、説明する。なお、本実施形態は、第1及び第2実施形態と比較して本発明膜の形状が異なる。
【0141】
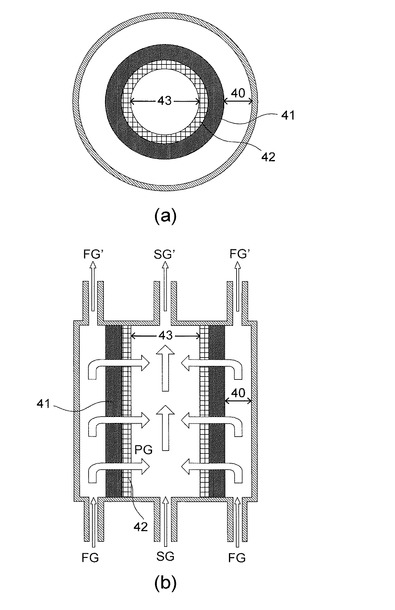
上述の第1及び第2実施形態では、いずれも図2に示すような平板型構造の促進輸送膜を想定して説明を行った。これに対し、本実施形態では、図18に示すような円筒型形状の促進輸送膜を想定している。
【0142】
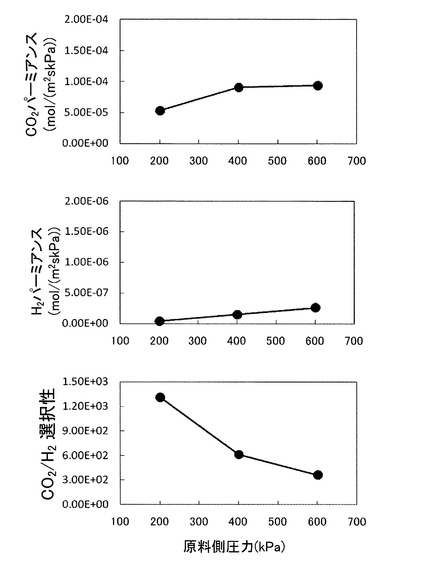
図18は、本実施形態の促進輸送膜の構造を示す概略図である。また、図19は、このような円筒型の形状を示す促進輸送膜を用いたときの、CO2パーミアンス,H2パーミアンス,並びにCO2/H2選択性を示すグラフである。
【0143】
図18において、(a)は水平面に平行に切断したときの断面図,(b)は水平面に鉛直方向に切断したときの断面図である。図18に示される促進輸送膜は、円筒形状の親水性のセラミックス製支持膜42の外周上に、キャリアを含むゲル膜41を担持させた構造である。なお本実施形態では、第1実施形態と同様のキャスト溶液から生成されたゲル膜41を用いた。すなわち、キャリアとしてCs2CO3を用い、熱架橋を施している。
【0144】
また、図18に示すように、ゲル膜41と外枠の間には空間40が設けられており,セラミックス製支持膜42の内側にも空間43が設けられている。
【0145】
膜性能を評価するに際しては、上述の実施形態と同様の原料ガスFGを、空間40内に流入する。一方、空間43内には不活性のスイープガスSGを流入する。空間40内に流入された原料ガスFGのうち、その一部は、キャリアを含むゲル膜41(及び支持膜42)を透過して空間43内に透過ガスPGとして流入する。空間43内には、この透過ガスPGを系外に排出するために不活性のスイープガスSGが流入されており、このスイープガスSGと透過ガスPGが混合された排出ガスSG’が、図5の冷却トラップ16に供給される。パーミアンス及び選択性の算出方法は、第1実施形態と同様である。
【0146】
図19は、促進輸送膜として図18に示す円筒形状の促進輸送膜を用い、測定方法,キャリア濃度,原料ガス圧力を図10のときと同じにして、測定温度を160℃としたときの得られたデータに基づくグラフである。図10の場合と同様、CO2パーミアンス及びCO2/H2選択性ともに高い値を示しており、図18に示すような構造の円筒型の促進輸送膜でも図1に示すような平板型と同様の効果を示すことが分かる。
【0147】
また、図18に示す構造の場合、ゲル膜41が直接原料ガスFGに接触するように、空間40内においてゲル膜41が露出している構成である。すなわち、図2に示す構造と比較して、疎水性膜によってゲル膜41が覆われていない。この疎水性膜は、ゲル膜を安定化させ、経時性能を劣化させにくくする効果を有している。しかし、図20に示すように、円筒型形状の促進輸送膜であれば、疎水性膜で覆わなくても経時性能を向上させる効果を有している。以下にその点を説明する。
【0148】
図20は、平板型と円筒型の促進輸送膜の長期性能を比較したグラフであり、(a)がCO2パーミアンスRCO2、(b)がCO2/H2選択性を示している。いずれのグラフにおいても、(1)が円筒型のデータであり、(2)が平板型のデータである。なお、当該グラフ結果を得るに際しての条件は図13の場合と同一とした。
【0149】
また、図20では、比較例としての平板型の促進輸送膜として、ゲル膜が疎水性膜で覆われていない構造のものを想定している。これは、円筒型形状ではゲル膜の一方の面が原料ガスに曝される状態にあるため、比較の観点から、平板型についてもその条件を共通にする目的である。
【0150】
図20(a)によれば、CO2パーミアンスに関しては、平板型,円筒型共に経時変化はあまり大きくない。これに対し、図20(b)によれば、CO2/H2選択性に関しては、円筒型の促進輸送膜の場合、時間が経過してもその性能はあまり変化がないのに対し、平板型の促進輸送膜の場合、時間経過とともに選択性が低下し、100時間経過時においては最大時の10%程度にまで落ち込んでいる。これにより、ゲル膜を疎水膜で覆わない場合においては、長期性能の観点から見れば円筒型形状の促進膜の方が平板型形状よりも優れていると見ることができる。なお、一方、平板型においても、ゲル膜を疎水膜で覆うことにより、図13や図17に示すように、良好な長期性能を示すことが分かる。
【0151】
なお、本実施形態で用いたセラミックス製支持膜においても、第1実施形態において上述したPTFE多孔膜の場合と同様、100℃以上の耐熱性、機械的強度、含浸ゲル膜との密着性を有するのが好ましい。また、多孔度(空隙率)は40%以上で、細孔径は0.1〜1μmの範囲にあるのが好ましい。
【0152】
また、図18の構成では、セラミックス製支持膜を内側にし、その外側にゲル膜を備えた構造としたが、これとは逆に、支持膜を外側にしてゲル膜をその内側に形成しても良い。また、形状として「円筒形状」と記載したが、これは必ずしも断面が正確な「円」であることを要求するものではなく、楕円形状であっても構わないし、多少の凹凸を有していても構わない。
【0153】
本実施形態によれば、促進膜を円筒形状にすることで、平板型よりも長期性能が向上することが示される。これは、形状を円筒形状にすることで、促進輸送膜が変形しにくくなり、安定化することに由来すると考えられる。一方、平板型の場合は、経時とともに膜が変形して欠陥が生じ、この欠陥からH2が漏れ出すことで選択性が低下するものと考えられる。すなわち、上記実施例では、支持膜としてセラミックス膜を利用したが、この膜は、円筒形状に対する加工が可能であり、且つ、経時と共に変形しにくい材料であれば、セラミックスに限定されるものではない。
【0154】
また、逆に、第1,第2実施形態では支持膜としてPTFE多孔膜を利用としたが、圧力を加えた状態でも割れたりすることなく安定的に平板状態を維持することができれば、PTFE多孔膜に限定されるものではない。
【0155】
[別実施形態]
以下に、別実施形態について説明する。
【0156】
〈1〉上記実施形態では、本発明膜は、PVA/PAA共重合体と二酸化炭素キャリアのCs2CO3を含む水溶液からなるキャスト溶液を、ゲル膜担持用の親水性PTFE多孔膜にキャストした後にゲル化して作製したが、本発明膜は、当該作製方法以外の作製方法で作製しても構わない。例えば、PVA/PAA共重合体ゲル膜に、Cs2CO3水溶液を後から含浸させて作製しても構わない。
【0157】
〈2〉上記第1実施形態において、添加剤として炭酸セシウムをゲル膜に添加してCO2促進輸送膜を製造する場合について説明したが、炭酸セシウムの代わりに水酸化セシウムを用いても同様の効果を得ることができる。これは、水酸化セシウムが添加されたゲル膜をCO2分離に利用することで、上記(化3)に示される反応が生じ、これによって水酸化セシウムが炭酸セシウムに転化するためである。更に、炭酸セシウムの代わりに重炭酸セシウムを用いた場合も、上記(化3)より同様の効果を得ることができることが分かる。
【0158】
なお、添加剤として炭酸ルビジウムをゲル膜に添加してCO2促進輸送膜を製造する場合についても、同様に、炭酸ルビジウムに代えて水酸化ルビジウム若しくは重炭酸ルビジウムを用いることができる。
【0159】
〈3〉上記実施形態では、本発明膜は、疎水性PTFE多孔膜/ゲル層(親水性PTFE多孔膜に担持された含浸ゲル膜)/疎水性PTFE多孔膜よりなる3層構造としたが、本発明膜の支持構造は、必ずしも当該3層構造に限定されない。例えば、疎水性PTFE多孔膜/ゲル層(親水性PTFE多孔膜に担持された含浸ゲル膜)よりなる2層構造でも構わない。
【0160】
〈4〉上記実施形態において例示した、本発明膜の組成における各成分の混合比率、膜の各部の寸法等は、本発明の理解の容易のための例示であり、本発明はそれらの数値のCO2促進輸送膜に限定されるものではない。
【産業上の利用可能性】
【0161】
本発明に係る二酸化炭素分離装置は、二酸化炭素の分離に利用可能であり、特に、水素を主成分とする燃料電池用等の改質ガスに含まれる二酸化炭素を水素に対する高い選択比率で分離可能なCO2促進輸送膜を備えたCO2透過型メンブレンリアクターに有用である。
【符号の説明】
【0162】
1: 二酸化炭素キャリアを含有するPVA/PAAゲル膜(ゲル層)
2: 親水性多孔膜
3、4: 疎水性多孔膜
10: CO2促進輸送膜(サンプル)
11: 流通式ガス透過セル
12: 原料側室
13: 透過側室
14、16: 冷却トラップ
15: 背圧調整器
17: ガスクロマトグラフ
18: 定量送液ポンプ
19: 背圧調整器
20: CO変成器(CO2透過型メンブレンリアクター)
21: スイープガスの通路
40: 空間
41: ゲル膜
42: セラミックス製支持膜
43: 空間
FG: 原料ガス
FG’: 処理済ガス
PG: 透過ガス
SG、SG’: スイープガス
【技術分野】
【0001】
本発明は、所定の主成分ガスに少なくとも二酸化炭素と水蒸気が含まれる原料ガスをCO2促進輸送膜の原料側面に供給して、CO2促進輸送膜を透過した二酸化炭素を透過側面から取り出す二酸化炭素分離装置に関し、特に、水素を主成分とする燃料電池用等の改質ガスに含まれる二酸化炭素を水素に対する高い選択比率で分離する二酸化炭素分離装置に関する。
【背景技術】
【0002】
現在の水素ステーション用改質システムでは、水蒸気改質により炭化水素を水素及び一酸化炭素(CO)に改質し、更に、CO変成反応を用いて一酸化炭素を水蒸気と反応させることにより水素を製造している。
【0003】
しかし、これらの技術はケミカルプラント用の大規模な水素製造プロセスとして開発されたものであり、日産数10万m3以上の大規模なものが普通で、しかも工場内に設置され、高圧・連続運転が前提となっている。これに対し、将来の水素エネルギ社会を支える重要なインフラとして天然ガスや石油からオンサイトで水素を製造し燃料電池自動車等に水素を供給する水素ステーションでは、水素を製造する規模や設置環境、運転パターンにおいて、従来の大規模水素プラントと大きく異なっており、それに起因する問題が多く残されている。
【0004】
性能面の課題としては、水素ステーションの場合、水素需要(具体的には、水素供給対象の燃料電池自動車の数量等)に対応して、頻繁な起動停止や負荷変化に対応する必要がある。特に、改質システム中でもサイズが最も大きく熱容量の大きなCO変成器に対して、起動時間や負荷応答性等の面で改善が必要となっている。
【0005】
また、燃料電池用改質システムの普及促進に必須とされる家庭用システムでのDSS(毎日の起動停止)運転に対しても、起動時間や負荷応答性等において、改質システム中でもCO変成器に課題が多く残されており、特に、CO変成器の小型化、低温度化が最大の課題である。
【0006】
更に、自動車用への適用についても、水蒸気改質方式はサイズや起動時間の点で目標との大きなギャップがあり、自動車業界ではオンボード改質に関しては効率の高い水蒸気改質よりは、寧ろ昇温反応性に優れた部分酸化方式での実用化を目指す傾向がある。しかし、部分酸化方式では、改質器については大幅な小型化が期待できるものの、CO変成器を伴うため、実用化に向けてはCO変成器の小型化が大きな課題となっている。このようにCO変成器の小型化は水素ステーションだけではなく、自動車用を含む燃料電池改質システムに共通の課題と言える。
【0007】
また、効率面から見ても、水蒸気改質を行う際、S/C(スチームと炭素(原料炭化水素)のモル比)の低下が熱効率上望ましいが、CO変成反応の化学平衡上の制約から効率の高い低S/C条件が採用されていなかった。
【0008】
コスト面での課題としては、水素ステーション全体のコストで最大の割合を占めるのがPSA(プレッシャー・スイング・アドソープション)であることから、そのコストダウンに直接繋がる水素濃度を上げる改質方式が望まれていた。改質器の出口ガス組成は水素以外に、10%程度の一酸化炭素及び二酸化炭素が含まれており、CO変成器では一酸化炭素は減少するものの二酸化炭素は増加するため、現状のプロセス(水蒸気改質+CO変成)では、1%程度のメタンとともに、20%程度の二酸化炭素と1%以下の一酸化炭素の残留は避けられず、その精製のために大型・高コストのPSA装置を設置せざるを得なかった。
【0009】
従来のCO変成器において、小型化や起動時間の短縮を阻害する原因として、以下の(化1)に示すCO変成反応の化学平衡上の制約から、多量のCO変成触媒が必要となっていることが挙げられる。一例として、50kWのPAFC(リン酸型燃料電池)用改質システムでは、改質触媒が20L必要であるのに対して、CO変成触媒は77Lと約4倍の触媒が必要となる。このことが、CO変成器の小型化や起動時間の短縮を阻害する大きな要因となっている。なお、記号「⇔」は、可逆反応であることを示している。
【0010】
(化1)
CO + H2O ⇔ CO2 + H2
【0011】
そこで、CO変成器に二酸化炭素を選択的に透過させるCO2促進輸送膜を備え、上記(化1)のCO変成反応で生成された右側の二酸化炭素を効率的にCO変成器外部に除去することで、化学平衡を水素生成側(右側)にシフトさせることができ、同一反応温度において高い転化率が得られる結果、一酸化炭素及び二酸化炭素を平衡の制約による限界を超えて除去することが可能となる。図21及び図22に、この様子を模式的に示す。図22(A)と(B)は、夫々、CO2促進輸送膜を備えている場合と備えていない場合における、CO変成器の触媒層長に対する一酸化炭素及び二酸化炭素の各濃度変化を示している。
【0012】
上記のCO2促進輸送膜を備えたCO変成器(CO2透過型メンブレンリアクター)により、一酸化炭素及び二酸化炭素を平衡の制約による限界を超えて除去することが可能となるため、水素ステーションのPSAの負荷低減及び低S/C化が図れ、水素ステーション全体のコスト低減及び高効率化が図れる。また、CO2促進輸送膜を備えることで、CO変成反応の高速化(高SV化)が図れるため、改質システムの小型化及び起動時間の短縮が図れる。
【0013】
かかるCO2透過型メンブレンリアクターの先行例としては、下記の特許文献1(或いは、同じ発明者による同一内容の特許文献2)に開示されているものがある。
【0014】
該特許文献1、2において提案されている改質システムは、炭化水素、メタノール等の燃料を燃料電池自動車用の水素に車上で改質する際に発生する改質ガスの精製及び水性ガスシフト反応(CO変成反応)に有用なCO2促進輸送膜プロセスを提供するもので、代表的な4種類のプロセスが、同文献に示されている。炭化水素(メタンを含む)を原料とする場合、水性ガスシフター(CO変成器)にCO2促進輸送膜を備えたメンブレンリアクターを用いて二酸化炭素を選択的に除去することにより、一酸化炭素の反応率を高め一酸化炭素濃度を低下させるとともに生成水素の純度を向上させている。また、生成水素中に残留する%オーダーの一酸化炭素及び二酸化炭素はメタネーターで水素と反応させてメタンに変換して濃度を低下させ、燃料電池の被毒等による効率低下を防いでいる。
【0015】
該特許文献1、2では、CO2促進輸送膜として、主としてハロゲン化四級アンモニウム塩((R)4N+X−)を二酸化炭素キャリアとして含むPVA(ポリビニルアルコール)等の親水性ポリマー膜が使用されている。また、該特許文献1、2の実施例6には、二酸化炭素キャリアとしてテトラメチルアンモニウムフルオリド塩50重量%を含む膜厚49μm50重量%のPVA膜とそれを支持する多孔質PTFE(四フッ化エチレン重合体)膜よりなる複合膜で形成されたCO2促進輸送膜の作製方法が開示されており、同実施例7には、混合ガス(25%CO2、75%H2)を全圧3気圧、23℃で処理したときの当該CO2促進輸送膜の膜性能が開示されている。当該膜性能として、CO2パーミアンスRCO2が7.2GPU(=2.4×10−6mol/(m2・s・kPa))、CO2/H2選択性が19となっている。
【0016】
また、下記特許文献3には、CO2促進輸送膜として、炭酸セシウムとアミノ酸とを組み合わせて構成されたCO2吸収剤が開示されている。
【0017】
特許文献3に記載のCO2促進輸送膜の製法は、以下のとおりである。まず、炭酸セシウムの水溶液に市販のアミノ酸を濃度分加えて、よく撹拌し混合水溶液を作製する。その後、ゲルを塗布した多孔PTFE膜(47Φ)のゲル塗布面を、作製した混合溶液に30分以上浸した後、ゆっくり膜を引き上げる。焼結金属の上にシリコーン膜を乗せ(溶液が透過側に漏れるのを防ぐため)その上に47mmΦの上記の含水ゲル膜を乗せ、その上からシリコーンパッキングの入ったセルをかぶせシーリングする。このようにして製造されたCO2促進輸送膜に対して、供給ガスを50cc/分の速度で流し、膜の下側を真空引きし圧力を40torr程度まで下げる。
【0018】
特許文献3の実施例4では、炭酸セシウムと、2,3−ジアミノプロピオン酸塩酸塩をそれぞれ4(mol/kg)のモル濃度で構成したCO2促進輸送膜により、25℃の温度条件下において、CO2透過速度が1.1(10−4cm3(STP)/cm2・s・cmHg)、CO2/N2分離係数が300となっている。なお、CO2パーミアンスRCO2は、圧力差あたりの透過速度で定義されるので、特許文献3の実施例4におけるCO2パーミアンスRCO2は、110GPUと算出されるが、本実施例におけるCO2/H2選択性に関するデータは開示されていない。
【0019】
なお、下記特許文献4には、アルカリ重炭酸塩を添加した酢酸セルロース膜で構成されたCO2分離膜が開示されている。しかし、当該文献4では、CO2/O2選択性についてしか記載されておらず、CO2/H2の選択性についてのデータが開示されていない。更に、開示されたデータは低圧力(0.01気圧程度)の条件下で測定されたものであり、数気圧程度の圧力条件下におけるデータは開示されていない。
【先行技術文献】
【特許文献】
【0020】
【特許文献1】特表2001−511430号公報
【特許文献2】米国特許6579331号明細書
【特許文献3】特開2000−229219号公報
【特許文献4】米国特許第3396510号明細書
【発明の概要】
【発明が解決しようとする課題】
【0021】
CO2促進輸送膜は、基本機能として二酸化炭素を選択的に分離することから、地球温暖化の原因となっている二酸化炭素の吸収或いは除去等を目的とした開発も行われている。しかしながら、CO2促進輸送膜は、CO2透過型メンブレンリアクターへの応用を考えた場合、使用温度、CO2パーミアンス、CO2/H2選択性等に対して、一定以上の性能が要求される。つまり、CO変成反応に供するCO変成触媒の性能が温度とともに低下する傾向にあるため、使用温度は最低でも100℃が必要と考えられる。上記各特許文献1〜3は、いずれも25℃程度の温度条件下で膜性能の測定が行われており、100℃以上の温度条件下においても十分な膜性能を示すCO2促進輸送膜が上記各特許文献によって開示されたということはできない。
【0022】
また、CO2パーミアンス(二酸化炭素透過性の性能指標の一つ)は、CO変成反応の化学平衡を水素生成側(右側)にシフトさせ、一酸化炭素濃度と二酸化炭素濃度を平衡の制約による限界を超えて例えば0.1%程度以下に低減し、且つ、CO変成反応の高速化(高SV化)を図るためには、一定レベル以上(例えば、2×10−5mol/(m2・s・kPa)=60GPU程度以上)が必要と考えられる。しかしながら、上記各特許文献1,2に記載のCO2促進輸送膜のCO2パーミアンスは、10GPUを大きく下回るような値であり、60GPU程度以上のCO2パーミアンスを示すCO2促進輸送膜が上記各特許文献によって開示されたということはできない。また、特許文献3は、CO2/H2選択性は開示されていない上、100℃以上の温度条件でCO2パーミアンスが60GPU以上の能力を示すことは示されていない。特許文献4においても、CO2/H2選択性は開示されていない上、数気圧程度の圧力条件の下でのデータが開示されていない。
【0023】
更に、CO変成反応で生成された水素が二酸化炭素とともにCO2促進輸送膜を通して外部に廃棄されたのでは、当該廃棄ガスから水素を分離回収するというプロセスが必要となる。水素は当然に二酸化炭素より分子サイズが小さいので、二酸化炭素を透過可能な膜は水素も透過できることになるが、膜中の二酸化炭素キャリアによって二酸化炭素のみを選択的に膜の供給側から透過側に向けて輸送可能な促進輸送膜が必要となり、その場合のCO2/H2選択性として90〜100程度以上が必要と考えられる。
【0024】
しかしながら、上記各特許文献1及び2に記載のCO2促進輸送膜は、CO2/H2選択性が19であり、十分な選択性を有しているとは言えない。また、上記特許文献3,4は、CO2/H2選択性が開示されていないため、特許文献3,4によって高いCO2/H2選択性を示すCO2促進輸送膜が開示されたということはできない。
【0025】
ところで、CO2促進輸送膜は、二酸化炭素の促進輸送機能(膜機能)を十分に発揮するには水分が必要である。具体的に説明すると、膜内の二酸化炭素(CO2)と炭酸イオンの反応は、通常以下の(化2)の反応経路式に示す化学反応を示す。これより、膜内の水分が多いほど化学平衡は生成物側(右側)にシフトし、二酸化炭素の透過が促進されることが分かる。
【0026】
(化2)
CO2+CO32−+H2O → 2HCO3−
【0027】
しかし、使用温度が100℃を超える高温になると、膜内の水分が蒸発して膜機能、つまり、二酸化炭素の促進輸送機能が低下する。当該膜機能低下はこれまでの促進輸送膜の常識となっている。一方、高温ほど上記化学反応の速度が大きくなるので、本願の発明者は、加圧下において気相中の水蒸気分圧を増すことにより膜内の水分量を確保することで膜機能が十分に発揮されることを確認した。現時点においては、使用温度が100℃以上を超える高温の温度条件下においても高いCO2分離機能を示す二酸化炭素分離装置は提供されていない。
【0028】
本発明は、上記の問題点に鑑み、100℃以上の高温及び加圧条件下において十分な膜性能を発揮可能なCO2促進輸送膜を用いた二酸化炭素分離装置を提供することを目的とする。
【課題を解決するための手段】
【0029】
上記目的を達成するための本発明に係る二酸化炭素分離装置は、少なくとも水素と二酸化炭素と水蒸気が含まれる原料ガスをCO2促進輸送膜の原料側面に100℃以上の供給温度で供給して、前記CO2促進輸送膜を透過した二酸化炭素を透過側面から取り出す二酸化炭素分離装置であって、前記CO2促進輸送膜が、ポリビニルアルコール−ポリアクリル酸共重合体ゲル膜に炭酸セシウム若しくは重炭酸セシウム若しくは水酸化セシウムからなる添加剤を添加したゲル層を親水性の多孔膜に担持させて形成されることを特徴とする。
【0030】
本発明に係る二酸化炭素分離装置の上記特徴によれば、ポリビニルアルコール−ポリアクリル酸(PVA/PAA)共重合体ゲル膜中に、炭酸セシウム(Cs2CO3)が含まれることから、当該Cs2CO3が透過物質であるPVA/PAAゲル層の二酸化炭素の高濃度側界面から低濃度側界面へと二酸化炭素を輸送する二酸化炭素キャリアとして機能し、100℃以上の高温において90〜100程度以上の対水素選択性(CO2/H2)、及び、2×10−5mol/(m2・s・kPa)(=60GPU)程度以上のCO2パーミアンスを達成可能となる。
【0031】
また、PVA/PAAゲル層を担持する多孔膜が親水性であるので、欠陥の少ないゲル層を安定して作製することができ、高い対水素選択性を維持できる。一般に、多孔膜が疎水性であると、100℃以下においてPVA/PAAゲル膜内の水分が多孔膜内の細孔に侵入して膜性能を低下させるのを防止でき、また、100℃以上においてPVA/PAAゲル膜内の水分が少なくなる状況でも同様の効果が期待できると考えられるため、疎水性の多孔膜の使用が推奨されるが、本発明の二酸化炭素分離装置は、親水性多孔膜を使用することで、以下の理由により欠陥が少なく高い対水素選択性を維持できる。
【0032】
親水性の多孔膜上に、PVA/PAA共重合体とCs2CO3の水溶液からなるキャスト溶液をキャストすると多孔膜の細孔内が液で満たされ、更に、多孔膜の表面にキャスト溶液が塗布される。このキャスト溶液をゲル化すると、多孔膜の表面のみならず細孔内にもゲル層が充填されるので欠陥が生じ難くなり、ゲル層の製膜成功率が高くなる。
【0033】
細孔部分の割合(多孔度)、及び、細孔が膜表面に垂直に真っ直ぐではなく曲がりくねっていること(屈曲率)を考慮すると、細孔内のゲル層はガス透過の大きな抵抗となるので、多孔膜表面のゲル層と比較して透過性は低くなり、ガスパーミアンスは低下する。他方、疎水性の多孔膜上にキャスト溶液をキャストすると多孔膜の細孔内は液で満たされずに多孔膜の表面のみにキャスト溶液が塗布され細孔はガスで満たされるので、疎水性多孔膜上のゲル層におけるガスパーミアンスは、親水性多孔膜と比較して水素及び二酸化炭素の両方において高くなると予想される。
【0034】
しかし、細孔内のゲル層と比較して膜表面のゲル層では微小な欠陥が生じ易く、製膜成功率は低下する。水素は二酸化炭素より分子サイズが非常に小さいので、微小な欠陥個所では二酸化炭素より水素の方が、パーミアンスが著しく大きくなる。なお、欠陥箇所以外では、促進輸送機構で透過する二酸化炭素のパーミアンスは、物理的な溶解、拡散機構で透過する水素のパーミアンスより格段に大きい。
【0035】
結果として、疎水性多孔膜を使用した場合の対水素選択性(CO2/H2)は、親水性多孔膜を使用した場合と比較して低下することになる。従って、実用化の観点からは、CO2促進輸送膜の安定性、耐久性が非常に重要となり、対水素選択性(CO2/H2)の高い親水性多孔膜を使用する方が有利となる。また、親水性多孔膜の使用は、PVA/PAAゲル層に二酸化炭素キャリアとしてCs2CO3を添加することで高いCO2パーミアンスを達成可能であることを前提に実現できるものである。
【0036】
なお、疎水性多孔膜と親水性多孔膜の違いによるガスパーミアンスの差は、キャスト溶液中に予め二酸化炭素キャリアであるCs2CO3を添加せずにゲル化後に含侵させても、細孔内のゲル層がガス透過の大きな抵抗となる点は同じであり、同様に発現するものと推定される。
【0037】
以上より、上記特徴の二酸化炭素分離装置によれば、100℃以上の使用温度、2×10−5mol/(m2・s・kPa)(=60GPU)程度以上のCO2パーミアンス、及び、90〜100程度以上のCO2/H2選択性が実現でき、CO変成器の小型化、起動時間の短縮、及び、高速化(高SV化)が図られる。
【0038】
なお、添加剤として、炭酸セシウムの代わりに水酸化セシウムを添加した場合においても、同様の効果を得ることができる。即ち、水酸化セシウムが添加されたゲル層を含む促進輸送膜をCO2の分離に利用することで、以下の(化3)に示されるような反応が起こり、これによって当該促進輸送膜内に添加されていた水酸化セシウムが炭酸セシウムに転化するためである。
【0039】
(化3)
CO2 + CsOH → CsHCO3
CsHCO3 + CsOH → Cs2CO3 + H2O
【0040】
なお、上記(化3)をまとめると、下記(化4)のように表すことができる。即ち、これにより、添加された水酸化セシウムが炭酸セシウムに転化することが示される。
【0041】
(化4)
CO2 + 2CsOH → Cs2CO3 + H2O
【0042】
更に、上記(化3)より、添加剤として、炭酸セシウムの代わりに重炭酸セシウムを添加した場合においても同様の効果を得ることができることが分かる。
【0043】
また、本発明に係る二酸化炭素分離装置は、上記特徴に加えて、前記ゲル層が、ポリビニルアルコール−ポリアクリル酸共重合体ゲル膜と炭酸セシウムの合計重量に対する炭酸セシウムの重量比率が65重量%以上85重量%以下の範囲で構成されることを別の特徴とする。
【0044】
本発明に係る二酸化炭素分離装置の上記特徴によれば、100℃以上の温度条件下で、優れたCO2パーミアンス、並びに優れたCO2/H2選択性の値を実現でき、CO変成器の小型化、起動時間の短縮、及び、高速化(高SV化)が図られる。
【0045】
また、本発明に係る二酸化炭素分離装置は、少なくとも水素と二酸化炭素と水蒸気が含まれる原料ガスをCO2促進輸送膜の原料側面に100℃以上の供給温度で供給して、前記CO2促進輸送膜を透過した二酸化炭素を透過側面から取り出す二酸化炭素分離装置であって、前記CO2促進輸送膜が、ポリビニルアルコール−ポリアクリル酸共重合体ゲル膜に炭酸ルビジウム若しくは重炭酸ルビジウム若しくは水酸化ルビジウムからなる添加剤を添加したゲル層を親水性の多孔膜に担持させて形成されることを別の特徴とする。
【0046】
本発明に係る二酸化炭素分離装置の上記特徴によれば、ポリビニルアルコール−ポリアクリル酸(PVA/PAA)共重合体ゲル膜中に、水中への溶解度が比較的高い炭酸塩である炭酸ルビジウム(Rb2CO3)が、透過物質である二酸化炭素をPVA/PAA共重合体ゲル層の二酸化炭素高濃度側界面から低濃度側界面へと輸送する二酸化炭素キャリアとして機能し、100℃以上の高温において90〜100程度以上の対水素選択性(CO2/H2)、及び、2×10−5mol/(m2・s・kPa)(=60GPU)程度以上のCO2パーミアンスを達成可能となる。
【0047】
なお、炭酸ルビジウムの代わりに水酸化ルビジウム若しくは重炭酸ルビジウムを添加した場合においても、同様の効果を得ることができる。これは、炭酸セシウムの代わりに水酸化セシウム若しくは重炭酸セシウムを添加した場合において、炭酸セシウムを添加したときと同様の効果が得られることと同様の理由による。
【0048】
また、本発明に係る二酸化炭素分離装置は、上記特徴に加えて、前記親水性の多孔膜に担持された前記ゲル層が疎水性の多孔膜によって被覆されていることを別の特徴とする。
【0049】
本発明に係る二酸化炭素分離装置の上記特徴によれば、親水性の多孔膜で担持されたゲル層が疎水性の多孔膜によって保護され、使用時におけるCO2促進輸送膜の強度が増す。この結果、CO2促進輸送膜の両側(反応器内外)での圧力差が大きく(例えば、2気圧以上)なっても十分な膜強度を確保できる。更に、ゲル層が疎水性の多孔膜によって被覆されるため、水蒸気が疎水性の多孔膜の膜表面に凝縮しても当該多孔膜が疎水性のために水がはじかれてゲル層内にしみ込むのを防止している。よって、疎水性の多孔膜によって、ゲル層中の二酸化炭素キャリアが水で薄められ、また、薄められた二酸化炭素キャリアがゲル層から流出することを防止できる。
【0050】
また、本発明に係る二酸化炭素分離装置は、上記特徴に加えて、前記ゲル層が、アルデヒド基由来の架橋構造を有することを別の特徴とする。
【0051】
本発明に係るCO2促進輸送膜の上記特徴によれば、ゲル層に形成された架橋構造によって膜内に欠陥が生じにくくなり、この結果、H2パーミアンスが大きく低下する。一方で、CO2パーミアンスは、H2パーミアンスほどの低下を招来しない。これにより、更に高いCO2/H2選択性を示す促進輸送膜を実現することができる。
【0052】
なお、このとき、添加する架橋剤としては、グルタルアルデヒドやホルムアルデヒドを採用することができる。グルタルアルデヒドを添加する場合においては、PVA/PAA共重合体1gに対して0.008〜0.015g程度添加することで、特に高いCO2/H2選択性を示すことができる。
【0053】
また、本発明に係る二酸化炭素分離装置は、上記特徴に加え、前記親水性の多孔膜が100℃以上の耐熱性を備えていることを別の特徴とする。
【0054】
本発明に係る二酸化炭素分離装置の上記特徴によれば、常温から100℃以上に亘る広範な温度範囲での使用が可能となる。具体的には、親水性の多孔膜が100℃以上の耐熱性を備えることで100℃以上の温度領域での使用が可能となる。
【0055】
また、本発明に係るCO2促進輸送膜は、上記特徴に加え、前記ゲル層,並びに前記親水性の多孔膜は、共に軸心を同一にした筒形状であって、一方の膜が、その内側面を他方の膜の外側面と接触させて、前記他方の膜を取り囲むように構成されていることを特徴とする。
【0056】
このとき、前記親水性の多孔膜として、アルミナ等のセラミックス製の膜を利用することができる。
【0057】
また、前記ゲル層を、前記親水性の多孔膜を取り囲むように、前記親水性の多孔膜の外側に形成することができる。
【発明の効果】
【0058】
本発明の構成によれば、100℃以上の高温及び加圧条件下において十分な膜性能を発揮可能なCO2促進輸送膜を用いた二酸化炭素分離装置を実現することができ、また、かかるCO2促進輸送膜を用いた二酸化炭素分離方法を提供することができる。
【図面の簡単な説明】
【0059】
【図1】本発明に係る二酸化炭素分離装置の一実施形態における概略の構成を模式的に示す構成図
【図2】本発明に係るCO2促進輸送膜の一実施形態における構造を模式的に示す断面図
【図3】本発明に係るCO2促進輸送膜の作製方法を示す工程図
【図4】CO2促進輸送膜の比較例サンプルの構造を模式的に示す断面図
【図5】本発明に係るCO2促進輸送膜の膜性能を評価するための実験装置の構成図
【図6】本発明に係るCO2促進輸送膜の親水性多孔膜の使用によるCO2/H2選択性の改善効果を示す図(1)
【図7】本発明に係るCO2促進輸送膜の親水性多孔膜の使用によるCO2/H2選択性の改善効果を示す図(2)
【図8】本発明に係るCO2促進輸送膜のCO2パーミアンスRCO2とCO2/H2選択性の原料ガスの圧力とキャリア濃度に対する依存性を示す図
【図9】本発明に係るCO2促進輸送膜のCO2パーミアンスRCO2とCO2/H2選択性のキャリア濃度に対する依存性を示す図
【図10】本発明に係るCO2促進輸送膜のCO2パーミアンスRCO2とCO2/H2選択性の原料ガスの圧力と使用温度に対する依存性を示す図
【図11】本発明に係るCO2促進輸送膜のCO2パーミアンスRCO2とCO2/H2選択性の使用温度に対する依存性を示す図
【図12】本発明に係るCO2促進輸送膜のCO2パーミアンスRCO2とCO2/H2選択性の原料ガスの圧力と水蒸気モル%に対する依存性を示す図
【図13】本発明に係るCO2促進輸送膜のCO2パーミアンスRCO2とCO2/H2選択性の経時変化を示す図
【図14】本発明に係るCO2促進輸送膜の第2実施形態の実施例1の方法で作製された本発明膜の膜性能を示すグラフ
【図15】本発明に係るCO2促進輸送膜の第2実施形態の実施例2の方法で作製された本発明膜の膜性能を示すグラフ
【図16】本発明に係るCO2促進輸送膜の第2実施形態の実施例3の方法で作製された本発明膜の膜性能を示すグラフ
【図17】本発明に係るCO2促進輸送膜の第2実施形態の実施例1の方法で作製された本発明膜の膜性能の経時変化を示すグラフ
【図18】本発明に係るCO2促進輸送膜の第3実施形態の構造を模式的に示す断面図
【図19】本発明に係るCO2促進輸送膜の第3実施形態のCO2パーミアンス、H2パーミアンス、並びにCO2/H2選択性の原料ガスの圧力に対する依存性を示す図
【図20】円筒型と平板型の促進輸送膜における、CO2パーミアンスRCO2とCO2/H2選択性の経時変化の比較図
【図21】CO2促進輸送膜を備えたCO変成器における各種ガスの流れを示す図
【図22】CO2促進輸送膜を備えている場合と備えていない場合における、CO変成器の触媒層長に対する一酸化炭素及び二酸化炭素の各濃度変化の比較図
【発明を実施するための形態】
【0060】
本発明に係る二酸化炭素分離装置(以下、適宜「本発明装置」という。)の実施の形態につき、図面に基づいて説明する。
【0061】
本装置は、図1に示すように、CO2促進輸送膜10を備えたCO変成器(CO2透過型メンブレンリアクター)20を備え、CO2促進輸送膜10によって二酸化炭素が選択的に分離された後の処理済ガスFG’は、排気経路から排気される構成である。
【0062】
CO2透過型メンブレンリアクター20は、例えば、水蒸気改質器(図示せず)で生成された原料ガスFG中の水素以外に含まれる一酸化炭素を、上述の(化1)に示すCO変成反応によって除去する装置であって、CO変成反応で生成される二酸化炭素をCO2促進輸送膜10により選択的にCO変成器外部に除去することで、CO変成反応の化学平衡を水素生成側にシフトさせることができ、同一反応温度において高い転化率で、一酸化炭素及び二酸化炭素を平衡の制約による限界を超えて除去することが可能である。
【0063】
本発明装置では、CO変成反応に供するCO変成触媒の性能が温度とともに低下する傾向にあるため、使用温度は最低でも100℃であると考え、CO2促進輸送膜10の原料側面に供給される原料ガスFGの供給温度は100℃以上となっている。従って、原料ガスFGは、CO2促進輸送膜10より上流側において、CO変成触媒の触媒活性に適した温度に調整された後に、CO変成反応(発熱反応)を経てCO2促進輸送膜10に供給される。なお、CO変成反応後の原料ガス温度を低下させる処理を行ってから原料ガスをCO2促進輸送膜10に供給しても構わない。
【0064】
CO2促進輸送膜10を透過した二酸化炭素を含む透過ガスの分圧を低くして、CO2促進輸送膜10の透過推進力を維持し、透過ガスPGを外部に排出するために、CO2促進輸送膜10の外側面(透過側面)に沿ってスイープガスSGの通路21を設けてあり、当該通路の一方側から空気等のスイープガスSGを送入し、他方側からスイープガスSGとともに透過ガスPGを排出する構造となっている。具体的には、例えば、CO2透過型メンブレンリアクター20を二重管構造として内管内をCO変成器として、内管と外管の間をスイープガスの通路21として構成する。
【0065】
また、原料ガスFGは、上流側の水蒸気改質器から所定の圧力でCO2透過型メンブレンリアクター20に送入される。
【0066】
次に、本発明装置で使用するCO2促進輸送膜10(以下、適宜「本発明膜10」という)について説明する。
【0067】
[本発明膜の第1実施形態]
本発明膜の第1実施形態について、以下に図2〜図13及び表1を参照して説明する。
【0068】
本発明膜は、水分を含むゲル膜内に二酸化炭素キャリアを含有したCO2促進輸送膜であって、100℃以上の使用温度、高い二酸化炭素透過性とCO2/H2選択性を有するCO2透過型メンブレンリアクターへ応用可能なCO2促進輸送膜である。更に、本発明膜は、高いCO2/H2選択性を安定して実現するために、二酸化炭素キャリアを含有したゲル膜を担持する支持膜として、親水性の多孔膜を採用している。
【0069】
具体的には、本発明膜は、膜材料として、ポリビニルアルコール‐ポリアクリル酸(PVA/PAA)共重合体を使用し、二酸化炭素キャリアとして、炭酸セシウム(Cs2CO3)を使用する。また、本発明膜は、図2に模式的に示すように、二酸化炭素キャリアを含有するPVA/PAAゲル膜1を担持した親水性多孔膜2が、2枚の疎水性多孔膜3,4に挟持される3層構造で構成される。以下、二酸化炭素キャリアを含有するPVA/PAAゲル膜を、二酸化炭素キャリアを含有しないPVA/PAAゲル膜、及び、2枚の疎水性多孔膜を備えた構造の本発明膜と区別するために、適宜「含浸ゲル膜」と略称する。また、この含浸ゲル膜中のPVA/PAAとCs2CO3の全重量を基準として、含浸ゲル膜中において、PVA/PAAは約20〜80重量%の範囲で存在し、Cs2CO3は約20〜80重量%の範囲で存在する。
【0070】
親水性多孔膜は、親水性に加えて、100℃以上の耐熱性、機械的強度、含浸ゲル膜との密着性を有するのが好ましく、更に、多孔度(空隙率)が55%以上で、細孔径は0.1〜1μmの範囲にあるのが好ましい。本実施形態では、これらの条件を備えた親水性多孔膜として、親水性化した四フッ化エチレン重合体(PTFE)多孔膜を使用する。
【0071】
疎水性多孔膜は、疎水性に加えて、100℃以上の耐熱性、機械的強度、含浸ゲル膜との密着性を有するのが好ましく、更に、多孔度(空隙率)が55%以上で、細孔径は0.1〜1μmの範囲にあるのが好ましい。本実施形態では、これらの条件を備えた疎水性多孔膜として、親水性化していない四フッ化エチレン重合体(PTFE)多孔膜を使用する。
【0072】
次に、本発明膜の作製方法(本発明方法)の一実施形態について、図3を参照して説明する。
【0073】
まず、PVA/PAA共重合体とCs2CO3を含む水溶液からなるキャスト溶液を作製する(工程1)。より詳細には、PVA/PAA共重合体(例えば、住友精化製の仮称SSゲル)を1g、Cs2CO3を2.33g、サンプル瓶に秤取し、これに水20mlを加えて室温で5日間攪拌して溶解させてキャスト溶液を得る。
【0074】
次に、工程1で得たキャスト溶液中の気泡を除去するために、遠心分離(回転数5000rpmで30分間)を行う(工程2)。
【0075】
次に、工程2で得たキャスト溶液を、親水性PTFE多孔膜(例えば、住友電工製,WPW−020−80、膜厚80μm、細孔径0.2μm、空隙率75%)と疎水性PTFE多孔膜(例えば、住友電工製、フロロポアFP010、膜厚60μm、細孔径0.1μm、空隙率55%)を2枚重ね合わせた層状多孔膜の親水性PTFE多孔膜側の面上に、アプリケータでキャストする(工程3)。なお、後述する実施例のサンプルでのキャスト厚は500μmである。ここで、キャスト溶液は、親水性PTFE多孔膜中の細孔内に浸透するが、疎水性のPTFE多孔膜の境界面で浸透が停止し、層状多孔膜の反対面までキャスト溶液がしみ込まず、層状多孔膜の疎水性PTFE多孔膜側面にはキャスト溶液が存在せず取り扱いが容易となる。
【0076】
次に、キャスト後の親水性PTFE多孔膜を室温で一昼夜自然乾燥させた後、キャスト溶液をゲル化させゲル層を生成する(工程4)。この方法では、工程3において、キャスト溶液を層状多孔膜の親水性PTFE多孔膜側の表面にキャストするため、工程4において、ゲル層は、親水性PTFE多孔膜の表面(キャスト面)に形成されるのみならず細孔内にも充填して形成されるので、欠陥(ピンホール等の微小欠陥)が生じ難くなり、ゲル層の製膜成功率が高くなる。なお、工程4において、自然乾燥させたPTFE多孔膜を、更に、120℃程度の温度で、2時間程度熱架橋するのが望ましい。なお、後述する実施例及び比較例のサンプルでは、何れも熱架橋を行っている。
【0077】
次に、工程4で得た親水性PTFE多孔膜表面のゲル層側に、工程3で用いた層状多孔膜の疎水性PTFE多孔膜と同じ疎水性PTFE多孔膜を重ね、図2に模式的に示すように、疎水性PTFE多孔膜/ゲル層(親水性PTFE多孔膜に担持された含浸ゲル膜)/疎水性PTFE多孔膜よりなる3層構造の本発明膜を得る(工程5)。なお、図2において、含浸ゲル膜1が親水性PTFE多孔膜2の細孔内に充填している様子を模式的に直線状に表示している。
【0078】
以上、工程1〜工程5を経て作製された本発明膜は、後述するようにCO2透過型メンブレンリアクターへ応用可能な膜性能、即ち、使用温度100℃以上、2×10−5mol/(m2・s・kPa)(=60GPU)程度以上のCO2パーミアンス、及び、90〜100程度以上のCO2/H2選択性が実現できる。
【0079】
また、ゲル層を疎水性PTFE多孔膜で挟持した3層構造とすることにより、一方の疎水性PTFE多孔膜は、工程3及び工程4で用いられ、含浸ゲル膜を担持する親水性PTFE多孔膜の支持とキャスト溶液の浸透防止に供せられ、他方の疎水性PTFE多孔膜は、含浸ゲル膜を他方面側から保護するのに用いられる。
【0080】
更に、水蒸気が疎水性PTFE多孔膜の膜表面に凝縮しても当該PTFE多孔膜が疎水性のために水がはじかれて含浸ゲル膜にしみ込むのを防止している。よって、他方のPTFE多孔膜によって、含浸ゲル膜中の二酸化炭素キャリアが水で薄められ、また、薄められた二酸化炭素キャリアが含浸ゲル膜から流出することを防止できる。
【0081】
以下、具体的な実施例の膜性能について説明する。
【0082】
まず、含浸ゲル膜を担持する多孔膜として、親水性PTFE多孔膜を使用した実施例と、疎水性PTFE多孔膜を使用した比較例の各サンプルの膜組成について説明する。
【0083】
実施例のサンプルは、上述の作製方法により作製した。(PVA/PAA:Cs2CO3)の配合比率は、記載の順に、(30重量%:70重量%)となっている。なお、以下では、共重合体重量とキャリア重量の合計重量に対するキャリア重量の比率を「キャリア濃度」と記載する。即ち、上記の例の場合、キャリア濃度は70%である。
【0084】
比較例のサンプルは、上述の作製方法において、親水性PTFE多孔膜と疎水性PTFE多孔膜の層状多孔膜に替えて1層の疎水性PTFE多孔膜を使用して作製された。従って、比較例のサンプルは、図4に模式的に示すように、二酸化炭素キャリアを含有するPVA/PAAゲル膜1が、2枚の疎水性多孔膜3,4の間に挟持される3層構造で構成される。(PVA/PAA:Cs2CO3)の配合比率は、実施例と同じである。
【0085】
次に、実施例、及び、比較例の各サンプルの膜性能を評価するための実験装置の構成及び実験方法について、図5を参照して説明する。
【0086】
図5に示すように、各サンプル10は、ステンレス製の流通式ガス透過セル11(膜面積:2.88cm2)の原料側室12と透過側室13の間に、2枚のフッ素ゴム製ガスケットをシール材として用いて固定されている。原料ガス(CO2、H2、H2Oからなる混合ガス)FGを、2.24×10−2mol/minの流量で原料側室12に供給し、スイープガス(Arガス)SGを、8.18×10−4mol/minの流量で透過側室13に供給する。原料側室12の圧力は、排気ガスの排出路の途中の冷却トラップ14の下流側に設けられた背圧調整器15で調整される。透過側室13の圧力は大気圧である。透過側室13から排出するスイープガスSG’中の水蒸気を冷却トラップ16で除去した後のガス組成をガスクロマトグラフ17で定量し、これとスイープガスSG中のArの流量よりCO2及びH2のパーミアンス[mol/(m2・s・kPa)]を計算し、その比より、CO2/H2選択性を算出する。なお、冷却トラップ16とガスクロマトグラフ17の間にも背圧調整器19が設けられており、これによって透過側室13の圧力が調整される。
【0087】
原料ガスFGは、CO変成器内における原料ガスを模擬するために、CO2、H2、H2Oからなる第1混合ガスを、CO2:5.0%、H2:45%、H2O:50%の混合比率(モル%)に調整した。具体的には、10%CO2と90%H2(モル%)よりなる混合ガス流(25℃での流量:200cm3/min、8.18×10−3mol/min)に水を定量送液ポンプ18で送入し(流量:0.256cm3/min、1.42×10−2mol/min)、100℃以上に加熱して水分を蒸発させて、上記混合比率の混合ガスを調製し、これを原料側室12に供給した。
【0088】
スイープガスSGは、サンプル膜を透過する被測定ガス(CO2、H2)の透過側室側の分圧を低くして、透過推進力を維持するために供給され、被測定ガスと異なるガス種(Arガス)を用いる。具体的には、Arガス(25℃での流量:20cm3/min、8.13×10−4mol/min)を透過側室13に供給した。
【0089】
なお、図示していないが、サンプル膜の使用温度、及び、原料ガスFGとスイープガスSGの温度を一定温度に維持するために、サンプル膜を固定した流通式ガス透過セル11と上記ガスを加熱する予熱コイルを、所定温度に設定したオイル恒温槽内に浸している。
【0090】
次に、図6及び図7に、(1)実施例と(2)比較例の各サンプルのCO2パーミアンスRCO2、H2パーミアンスRH2、及びCO2/H2選択性を、原料側室12内の原料ガスFGの圧力(グラフ上では「原料側圧力」と記載。以下同様)を200kPa〜400kPaの範囲の加圧状態で測定した結果を示す。なお、図6は測定温度を160℃、図7は測定温度を180℃として、それぞれ測定したものである。また、グラフ上における原料側圧力の値は、原料側室12の圧力を調整するための背圧調整器15が示す圧力値を採用した。
【0091】
図6及び図7より、H2パーミアンスは、疎水性PTFE多孔膜を使用した比較例のサンプルの方が、親水性PTFE多孔膜を使用した実施例のサンプルより、全圧力範囲で高くなっているが、CO2パーミアンス、並びにCO2/H2選択性では、実施例のサンプルの方が、比較例のサンプルより大幅に改善されていることが分かる。これは、親水性膜の場合は、キャスト溶液を膜上にキャストすると、PTFE多孔膜の表面のみならず細孔内にもゲル層が充填されるので欠陥(ピンホール等の微小欠陥)が生じ難くなり、当該微小欠陥を介してガスパーミアンス、特に、H2パーミアンスが上昇するのが抑制されるためと考えられる。これに対し、疎水性膜の場合には、キャスト液が膜の細孔内には侵入せず、その表面に塗布されるので、欠陥が生じやすく、H2パーミアンスが上昇し、これによって選択性が低下するものと考えられる。
【0092】
なお、図6及び図7によれば、測定温度を変更しても、同様の特性が示されていることが分かる。
【0093】
また、上記特許文献1,2に開示されたCO2促進輸送膜では、100℃以上の使用温度、2×10−5mol/(m2・s・kPa)程度以上のCO2パーミアンス、及び、90〜100程度以上のCO2/H2選択性の何れも満足していないのに対して、図6及び図7に示す実施例のサンプルは、全圧力範囲において全ての要件を概ね満足している。また、比較例のサンプルでも、100℃以上の使用温度条件下で、2×10−5mol/(m2・s・kPa)程度以上のCO2パーミアンスを示している。なお、比較例のサンプルでは、300kPa以上でCO2/H2選択性の低下が大きくなることが示唆されている。
【0094】
図6及び図7の結果を勘案すれば、特許文献1、2に開示されたCO2促進輸送膜と比較して、Cs2CO3を含有するPVA/PAAゲル膜を備える本発明膜の方が、100℃以上の高温条件下においてCO2パーミアンスを向上させることができる。そして、特に支持膜を親水性多孔膜とすることで、CO2パーミアンス、及びCO2/H2選択性の値を顕著に向上させることができる。
【0095】
以下では、実施例と同様、親水性PTFE多孔膜によって含浸ゲル膜を担持する構成を有する本発明膜を用いてデータの取得を行った。
【0096】
次に、図8に、キャリア濃度を50wt%〜85wt%まで変化させて作製された各サンプルのCO2パーミアンスRCO2、H2パーミアンスRH2、及びCO2/H2選択性を、原料ガスFGの混合比率及び測定温度を図6と同一の条件とし、原料ガスFGの圧力を200kPa〜600kPaの範囲の加圧状態で測定した結果を示す。
【0097】
図8より、160℃の測定温度下において、キャリア濃度70wt%のときにCO2パーミアンスRCO2が最大となり、また、原料ガスFGの圧力が500kPaのときにCO2パーミアンスRCO2が最大となることが分かる。そして、キャリア濃度が80%以下の場合、及びキャリア濃度が85%の場合であって原料ガスFGの圧力が300kPa以上の場合には、いずれも5.0×10−5mol/(m2・s・kPa)以上の高いCO2パーミアンスを示すことが分かる。
【0098】
また、H2パーミアンスRH2は、キャリア濃度が50wt%の場合を除くと、全体的に原料ガスFGの圧力が増加するとともに微減する傾向を示すことが分かる。
【0099】
更に、図8より、キャリア濃度が70wt%以上80wt%以下の場合には、原料ガスFGの圧力が200〜600kPaのいずれの場合においても90〜100程度以上のCO2/H2選択性を示すことが分かる。
【0100】
即ち、図8の結果より、本発明膜によれば、キャリア濃度を調整することにより、使用温度100℃以上(160℃)、2×10−5mol/(m2・s・kPa)(=60GPU)程度以上のCO2パーミアンス、及び、90〜100程度以上のCO2/H2選択性が実現できる。このため、本発明膜をCO2透過型メンブレンリアクターに適用することができる。特に、70wt%〜80wt%の範囲内で全圧力範囲に亘って高いCO2/H2選択性を示すことが示唆されている。
【0101】
また、図9は、原料ガス圧力を一定(501.3kPa)として、キャリア濃度とCO2パーミアンスRCO2の関係、並びに、キャリア濃度とCO2/H2選択性の関係をそれぞれグラフにしたものである。なお、原料ガスFGの混合比率及び測定温度は図8の場合と同一の条件とした。
【0102】
図9によれば、キャリア濃度が70wt%の場合に、CO2パーミアンス並びにCO2/H2選択性の双方が最も高い値を示すことが分かる。即ち、図9によれば、CO2パーミアンス並びにCO2/H2選択性は、いずれもキャリア濃度に依存することが分かる。特に、本発明膜をCO2促進輸送膜として利用する際には、キャリア濃度を70wt%と設定することで、その能力を最大限発揮させることができる。
【0103】
図10は、キャリア濃度を70wt%とし、原料ガスFGの混合比率を図8の場合と同様とした状態の下、測定温度を125℃以上200℃以下の範囲内で変化させたときの、CO2パーミアンスRCO2、H2パーミアンスRH2、及びCO2/H2選択性を、原料側室12内の原料ガスFGの圧力を200kPa〜600kPaの範囲の加圧状態で測定した結果を示す。
【0104】
図10によれば、測定温度が160℃のときが最もCO2パーミアンスRCO2が大きくなっている。また、CO2/H2選択性については、測定温度が160℃並びに180℃におけるCO2/H2選択性が大きく、それより温度が上昇しても低下してもCO2/H2選択性が減少することが分かる。即ち、図10によれば、CO2パーミアンス並びにCO2/H2選択性は、測定温度にも依存することが分かる。特に、本発明膜をCO2促進輸送膜として利用する際には、160℃の温度条件下で本発明膜を設置することで、その能力を最大限発揮させることができることが分かる。これにより、本発明膜によれば、特許文献1、2に開示された従来のCO2促進輸送膜と比較して、十分高い温度条件下(125℃〜200℃)で高いCO2パーミアンスと高いCO2/H2選択性を実現することができ、特に140℃〜180℃で良好な値を実現することが示唆される。
【0105】
なお、本発明膜は、測定温度が200℃の場合であっても、CO2パーミアンスRCO2は1.0×10−4mol/(m2・s・kPa)程度の値を示しており、2×10−5mol/(m2・s・kPa)程度以上のCO2パーミアンスを示すことが分かる。そして、一定温度条件下においては、原料ガスFGの圧力を変化してもCO2パーミアンスの値はあまり変化していないことが分かる。
【0106】
更に、図10によれば、200℃の高温条件下において、CO2/H2選択性は圧力300kPaの下で100に近い値を示していることが分かる。即ち、200℃程度の高温条件下でも、CO2透過型メンブレンリアクターに適用可能なCO2促進輸送膜を実現できることが分かる。
【0107】
また、図11は、原料ガス圧力を一定(501.3kPa)として、測定温度とCO2パーミアンスRCO2の関係、並びに、測定温度とCO2/H2選択性の関係をそれぞれグラフにしたものである。なお、原料ガスFGの混合比率及び測定温度は図10の場合と同一の条件とした。
【0108】
図11によれば、測定温度が160℃の場合に、CO2パーミアンス並びにCO2/H2選択性の双方が最も高い値を示すことが分かる。即ち、図11によれば、CO2パーミアンス並びにCO2/H2選択性は、いずれも測定温度の高さに依存することが分かる。特に、本発明膜をCO2促進輸送膜として利用する際には、160℃の温度条件下で利用することでその能力を最大限発揮させることができる。
【0109】
図12は、キャリア濃度を70wt%として作製されたサンプルのCO2パーミアンスRCO2、H2パーミアンスRH2、及びCO2/H2選択性を、原料ガスFGの混合比率及び測定温度を図6と同一の条件とし、水蒸気モル%を20%、30%、50%、70%、90%と変化させた場合において、原料ガスFGの圧力を200kPa〜600kPaの範囲の加圧状態で測定した結果を示す。なお、具体的には、CO2、H2、H2Oからなる混合ガスを、CO2のモル%を5%と固定し、H2並びにH2Oのモル%の合計が95%となるように、H2のモル%並びにH2Oのモル%(水蒸気モル%)をそれぞれ変更して測定した。
【0110】
図12によれば、水蒸気モル%が高いほどCO2パーミアンスの値が上昇し、逆に水蒸気モル%が低下するに連れCO2パーミアンスの値も低下することが分かる。そして、水蒸気モル%を30%程度にまで低下させた場合においても、400kPaの原料ガスFGの圧力条件の下で約1×10−4mol/(m2・s・kPa)程度のCO2パーミアンスを示している。
【0111】
なお、H2パーミアンスの値は、水蒸気モル%が20%の場合においては大きく変化しているものの、他の値の場合にはあまり大きな差異が見られない。一方、CO2/H2選択性は、水蒸気モル%が低下するに連れほぼ全体的に低下することが分かる。そして、水蒸気モル%を30%としても、400kPaの原料ガスFGの圧力条件の下で約100程度のCO2/H2選択性を示している。
【0112】
従って、図12のグラフにより、水蒸気モル%を30%以下に低く設定した条件下においても、本発明膜は優れた性能を示し、CO2透過型メンブレンリアクターに適用可能なCO2促進輸送膜を実現できることが分かる。従って、本発明装置は、原料ガスの水蒸気モル%が低い場合においても、CO2分離性能を示すことができる。
【0113】
また、図13は、本発明膜の長期性能を示すグラフ結果である。原料ガスをCO2:5%、H2:45%、H2O:50%の混合比率(モル%)に調整し、原料ガスの圧力を351.03kPa、キャリア濃度を70wt%としたときの、CO2パーミアンスRCO2、並びにCO2/H2選択性の値の経時変化をグラフ化したものである。
【0114】
図13によれば、時間が経過してもCO2パーミアンスRCO2の値は大きな変化を示さず、ほぼ1.6×10−4mol/(m2・s・kPa)程度の値を示した。また、CO2/H2選択性についても、同様に時間が経過しても大きな変化を示さず、ほぼ100程度の値を示した。このように、本発明膜によれば、時間経過に連れて性能が大きく低下するということがなく、長時間にわたって、優れた性能を示すCO2透過型メンブレンリアクターに適用可能なCO2促進輸送膜を実現できることが分かる。
【0115】
また、下記表1は、膜材料を同一(PVA/PAA共重合体)とし、二酸化炭素キャリアとして用いる材料をCs2CO3以外の種々の炭酸塩としたときの、CO2パーミアンス、H2パーミアンス、及びCO2/H2選択性の値を、本発明膜と比較したものである。表1では、本発明膜で用いたCsの炭酸塩以外に、Na、K、Rbの炭酸塩を二酸化炭素キャリアとして利用した場合の上記データを測定した。なお、いずれの場合も、原料ガス圧力を401.33kPa、測定温度を160℃とし、原料ガスをCO2:5.0%、H2:45%、H2O:50%の混合比率(モル%)に調整して各データの測定を行った。また、各膜は、上述した本発明膜の製法と同様の方法により製造した。
【0116】
【表1】
【0117】
表1に示される結果によれば、Na2CO3膜による場合,CO2パーミアンスは非常に低く、そして高いH2パーミアンスを示した。これは、Na2CO3の水中への溶解度が低いことから(表1参照)、キャスト膜を120℃で架橋するときに結晶が生成され、これによって均一な膜が得られなかったためと考えられる。また、K2CO3膜の場合、高いCO2パーミアンスが得られたが、膜に欠陥が生じやすいためにH2パーミアンスも大きくなり、高いCO2/H2選択性は得られなかった。一方、水中への溶解度が高いRb2CO3およびCs2CO3(表1参照)を含む膜では、CO2パーミアンス並びにCO2/H2選択性ともに良好な結果が得られた。
【0118】
以上より、水中への溶解度の高い炭酸塩は、高温においてもCO2のキャリアとして効率よく機能し,それを含有する膜は欠陥が生じにくく、高いCO2透過性及び選択性を示すことが明らかになった。特に、Cs2CO3をキャリアとする本発明膜によれば、高いCO2パーミアンス及び高いCO2/H2選択性を示すCO2促進輸送膜を実現することができる。従って、本発明装置が、かかるCO2促進輸送膜を備えることで、高いCO2分離能力を示すことができる。
【0119】
[本発明膜の第2実施形態]
本発明膜の第2実施形態(以下、適宜「本実施形態」と称する)につき、図14〜図17の各図を参照して説明する。なお、本実施形態は、本発明膜の第1実施形態と比較して本発明膜及び本発明方法の一部構成が異なるのみであるため、同一の構成要素についてはその旨を記載して説明を割愛する。
【0120】
本実施形態では、第1実施形態と比較して、キャスト溶液を作製する工程(上記工程1)の内容が異なる。本実施形態では、第1実施形態の工程1に相当する工程(キャスト溶液作製工程)として以下の3通りの工程を行うものとし、それぞれを実施例1〜3と称する。
【0121】
(実施例1)
まず、PVA/PAA共重合体(例えば、住友精化製の仮称SSゲル)を1gに水20gを加えた後、室温で攪拌してゲルを溶解させる。次に、これにグルタルアルデヒドを0.008〜0.0343g程度加えた後、95℃の温度条件下で15時間攪拌する。そして、これにCs2CO3を2.33g加えて更に室温で攪拌することでキャスト溶液を得る。即ち、実施例1では、ゲル溶解工程、グルタルアルデヒド添加工程、高温下での攪拌工程、Cs2CO3添加工程、室温下での攪拌工程、の順に処理を行うことでキャスト溶液を作製する。
【0122】
(実施例2)
まず、PVA/PAA共重合体を1gに水20gを加えた後、室温で攪拌してゲルを溶解させる。次に、これにCs2CO3を2.33gと、グルタルアルデヒドを0.008〜0.0343g程度加えた後、室温で攪拌して溶解させる。その後、95℃の温度条件下で15時間攪拌することでキャスト溶液を得る。即ち、実施例2では、ゲル溶解工程、グルタルアルデヒド及びCs2CO3添加工程、室温下での攪拌工程、高温下での攪拌工程、の順に処理を行うことでキャスト溶液を作製する。
【0123】
(実施例3)
まず、PVA/PAA共重合体を1gに水20gを加えた後、室温で攪拌してゲルを溶解させる。次に、これにCs2CO3を2.33gと、グルタルアルデヒドを0.008〜0.0343g程度加えた後、室温で攪拌して溶解させることで、キャスト溶液を得る。即ち、実施例3では、ゲル溶解工程、グルタルアルデヒド及びCs2CO3添加工程、室温下での攪拌工程、の順に処理を行うことでキャスト溶液を作製する。
【0124】
なお、実施例1〜3の何れにおいても、キャスト溶液を作製後については、第1実施形態に記載の工程(工程2〜4)と同様の方法を用いてCO2促進輸送膜を得る。即ち、キャスト溶液中の気泡を除去するために遠心分離を行った後、ガラス板上に疎水性PTFE多孔膜(膜厚60μm)と親水性PTFE多孔膜(膜厚80μm)を重ね合わせた層状多孔膜の親水性PTFE多孔膜側の面上に、前述のキャスト溶液をアプリケータを用いて厚さ500μmでキャストする。その後、室温で一昼夜乾燥させる。そして、更にこれを120℃程度の高温条件下で2時間程度保持することで、CO2促進輸送膜を得る。
【0125】
以下、これらの各実施例1〜3の方法で製造された本発明膜の膜性能について説明する。なお、膜組成は、第1実施形態の実施例と同様にキャリア濃度を70wt%とし、膜性能を評価するための実験装置及び実験方法も第1実施形態と同様とする。
【0126】
図14は、実施例1の方法で作製されたキャスト溶液を用いて作製された本発明膜による(a)CO2パーミアンスRCO2、(b)H2パーミアンスRH2、及び(c)CO2/H2選択性を、原料側圧力を200kPa〜600kPaの範囲の加圧状態で測定した結果を示す。なお、図14では、キャスト溶液作製の際に添加されるグルタルアルデヒドの添加量を異ならせて各データの測定を行った。即ち、グルタルアルデヒドの添加量として、(1)0.008g、(2)0.0153g、(3)0g(不添加)の3パターンで実験を行った。なお、グラフ上では、グルタルアルデヒドを「GA」と略記している(以下のグラフも同様)。
【0127】
また、実験条件としては、温度条件を160℃、原料ガスFGをCO2:5.0%、H2:45%、H2O:50%の混合比率(モル%)、原料ガスFGの流量を25℃・1atm下で360cm3/min、透過側の圧力を原料側の圧力から20kPa減、スイープガスSGの流量を25℃・1atm下で40cm3/minとした。なお、この実験条件は、各実施例とも共通である。
【0128】
図14(a)によれば、グルタルアルデヒドを添加した場合、添加しない場合よりもCO2パーミアンスRCO2が微小に低下している。しかしながら、図14(b)に示すように、グルタルアルデヒドを添加した場合、H2パーミアンスRH2が大幅に低下しているため、図14(c)に示すように、グルタルアルデヒドを添加することで添加しない場合と比べてCO2/H2選択性は大きく上昇することが分かる。これは、グルタルアルデヒドを添加したことにより、架橋構造が形成されることで膜の欠陥が生じにくくなったためにH2パーミアンスが大きく低下したことによるものと考えられる。図14(b)及び(c)によれば、グルタルアルデヒドを0.008g添加した場合の方が、0.0153g添加した場合よりもH2パーミアンスが低く、CO2/H2選択性が高いことが分かる。即ち、グルタルアルデヒドの添加量は、多ければ多いほど選択性が高くなるという訳ではなく、実験条件に応じて高い選択性が実現可能な適正な添加量が存在することが示唆される。
【0129】
図15は、実施例2の方法で作製されたキャスト溶液を用いて作製された本発明膜による(a)CO2パーミアンスRCO2、(b)H2パーミアンスRH2、及び(c)CO2/H2選択性を、原料側圧力を200kPa〜600kPaの範囲の加圧状態で測定した結果を示す。なお、図15では、キャスト溶液作製の際に添加されるグルタルアルデヒドの添加量を異ならせて各データの測定を行った。即ち、グルタルアルデヒドの添加量として、(1)0.008g、(2)0.0165g、(3)0g(不添加)の3パターンで実験を行った。その他の実験条件は実施例1の場合と同様である。
【0130】
図15(a)によれば、図14(a)と同様、グルタルアルデヒドを添加した場合、添加しない場合よりもCO2パーミアンスRCO2が微小に低下している。そして、図15(b)によれば、図14(b)と同様、グルタルアルデヒドを添加した場合にH2パーミアンスRH2が大幅に低下しているため、図15(c)に示すように、グルタルアルデヒドを添加することで添加しない場合と比べてCO2/H2選択性は大きく上昇することが分かる。これは、実施例1の場合と同様の理由、即ち、グルタルアルデヒドを添加したことにより、架橋構造が形成されることで膜の欠陥が生じにくくなったためにH2パーミアンスが大きく低下したことによるものと考えられる。図15(b)及び(c)によれば、グルタルアルデヒドを0.008g添加した場合の方が、0.0165g添加した場合よりもH2パーミアンスが低く、CO2/H2選択性が高いことが分かる。即ち、グルタルアルデヒドの添加量は、多ければ多いほど選択性が高くなるという訳ではなく、実験条件に応じて高い選択性が実現可能な適正な添加量が存在することが示唆される。なお、図15(c)より、原料側のガス圧力が高い領域においては、添加するグルタルアルデヒドの添加量による選択性の差分は小さくなっている。
【0131】
図16は、実施例3の方法で作製されたキャスト溶液を用いて作製された本発明膜による(a)CO2パーミアンスRCO2、(b)H2パーミアンスRH2、及び(c)CO2/H2選択性を、原料側圧力を200kPa〜600kPaの範囲の加圧状態で測定した結果を示す。なお、図15では、キャスト溶液作製の際に添加されるグルタルアルデヒドの添加量を異ならせて各データの測定を行った。即ち、グルタルアルデヒドの添加量として、(1)0.008g、(2)0.0154g、(3)0.0343g、(4)0g(不添加)の4パターンで実験を行った。その他の実験条件は実施例1の場合と同様である。
【0132】
図16(a)によれば、図14(a)と同様、グルタルアルデヒドを添加した場合、添加しない場合よりもCO2パーミアンスRCO2が微小に低下している。そして、図16(b)によれば、図14(b)と同様、グルタルアルデヒドを添加した場合にH2パーミアンスRH2が大幅に低下しているため、図16(c)に示すように、グルタルアルデヒドを添加することで添加しない場合と比べてCO2/H2選択性は大きく上昇することが分かる。これは、実施例1の場合と同様の理由、即ち、グルタルアルデヒドを添加したことにより、架橋構造が形成されることで膜の欠陥が生じにくくなったためにH2パーミアンスが大きく低下したことによるものと考えられる。図16(b)及び(c)によれば、グルタルアルデヒドを0.008g添加した場合の方が、0.0154g添加した場合、並びに0.0343g添加した場合よりもH2パーミアンスが低く、CO2/H2選択性が高いことが分かる。即ち、グルタルアルデヒドの添加量は、多ければ多いほど選択性が高くなるという訳ではなく、実験条件に応じて高い選択性が実現可能な適正な添加量が存在することが示唆される。
【0133】
なお、図16(c)においても、原料側のガス圧力が高い領域においては、添加するグルタルアルデヒドの添加量による選択性の差分は小さくなっている。
【0134】
以上、図14〜図16の各グラフを参照すれば、ゲル膜をグルタルアルデヒドで架橋することにより、グルタルアルデヒド無添加の場合と比較して、CO2の透過性の低下を一定程度に抑制しながらH2の透過性を著しく低下させることが可能となり、これによって高いCO2/H2選択性を示す促進輸送膜を実現することができる。特に、PVA/PAA共重合体を1gに対し、グルタルアルデヒドを0.008〜0.015g程度添加した場合(以下、かかる範囲を「良好範囲」と称する)に、CO2/H2の選択性が著しく向上する。
【0135】
なお、上記実施例1〜3間では膜性能に顕著な差異は見られなかった。即ち、いずれの方法で製膜した場合であっても、グルタルアルデヒドを添加することによるCO2/H2の選択性の向上効果を実現することができる。特に、実施例2及び3においては、グルタルアルデヒドで架橋した場合でもCO2パーミアンスの低下量が抑制された。一方、実施例1においては、原料側のガス圧力を増加させてもH2パーミアンスの増加が一定程度に抑制された。
【0136】
また、図17は、グルタルアルデヒドを添加した場合の長期性能を示すグラフである。具体的には、実施例1の方法によって作製した膜(グルタルアルデヒド添加量:0.0339g)を用いて長期間実験を行ったときの(a)CO2パーミアンスRCO2、(b)H2パーミアンスRH2、(c)CO2/H2選択性の経時変化をそれぞれグラフに表したものである。なお、原料側ガス圧力を401.3kPaとし、その他の実験条件は、図14〜図16と同様とした。
【0137】
実験方法としては、午前10時頃に本発明膜を透過セルにセットして温度を160℃まで上げて原料ガスおよびスイープガスを供給して透過実験を開始し、午後8時頃まで同じ条件で継続した。そして、午後8時頃に供給ガスを停めて温度を室温まで低下させた。再び翌朝の午前10時頃に、透過セルを分解せずにそのまま同じ膜を使用して同様の実験を行った。このような実験を繰り返し2週間続けた結果が図17(a)〜(c)に示されている。
【0138】
図17の実験データは、添加したグルタルアルデヒド量が前記良好範囲よりも若干多いので、CO2パーミアンスは,図14〜図16の値と比較して小さい値となっているが、H2パーミアンスは、時間が経過してもグルタルアルデヒド無添加の場合より著しく小さい値が示されており、CO2/H2選択性も200以上の高い値を維持している。特に、本評価方法のように、スタートアップとシャットダウンを繰返して経時評価を行う場合には、温度の変動(室温〜160℃)や圧力の変動(常圧〜6気圧)を膜に繰返し与えるため、同一温度・同一圧力で実験を継続することで長期性能を評価する場合と比較して、膜に対する負荷が大きくなる。図17によれば、スタートアップ、シャットダウンを繰り返す本評価方法によっても、膜性能が約2週間にわたって安定していることから、グルタルアルデヒド添加によって、膜の安定性が著しく改善されたと言える。
【0139】
なお、本実施形態で添加する材料としてグルタルアルデヒドを採用したが、当該材料の添加工程は、膜に架橋構造を形成するために行われるものであるところ、架橋構造の形成が可能な材料であれば、グルタルアルデヒドに限定されるものではない。アルデヒド基で架橋を形成する場合には、例えば、ホルムアルデヒドも利用可能である。また、二酸化炭素キャリアとして用いる材料をCs2CO3以外の材料(例えばRb2CO3)とした場合でも、同様に添加剤を導入して架橋構造とすることで膜性能を更に高めることが可能である。
【0140】
[本発明膜の第3実施形態]
本発明膜の第3実施形態につき、説明する。なお、本実施形態は、第1及び第2実施形態と比較して本発明膜の形状が異なる。
【0141】
上述の第1及び第2実施形態では、いずれも図2に示すような平板型構造の促進輸送膜を想定して説明を行った。これに対し、本実施形態では、図18に示すような円筒型形状の促進輸送膜を想定している。
【0142】
図18は、本実施形態の促進輸送膜の構造を示す概略図である。また、図19は、このような円筒型の形状を示す促進輸送膜を用いたときの、CO2パーミアンス,H2パーミアンス,並びにCO2/H2選択性を示すグラフである。
【0143】
図18において、(a)は水平面に平行に切断したときの断面図,(b)は水平面に鉛直方向に切断したときの断面図である。図18に示される促進輸送膜は、円筒形状の親水性のセラミックス製支持膜42の外周上に、キャリアを含むゲル膜41を担持させた構造である。なお本実施形態では、第1実施形態と同様のキャスト溶液から生成されたゲル膜41を用いた。すなわち、キャリアとしてCs2CO3を用い、熱架橋を施している。
【0144】
また、図18に示すように、ゲル膜41と外枠の間には空間40が設けられており,セラミックス製支持膜42の内側にも空間43が設けられている。
【0145】
膜性能を評価するに際しては、上述の実施形態と同様の原料ガスFGを、空間40内に流入する。一方、空間43内には不活性のスイープガスSGを流入する。空間40内に流入された原料ガスFGのうち、その一部は、キャリアを含むゲル膜41(及び支持膜42)を透過して空間43内に透過ガスPGとして流入する。空間43内には、この透過ガスPGを系外に排出するために不活性のスイープガスSGが流入されており、このスイープガスSGと透過ガスPGが混合された排出ガスSG’が、図5の冷却トラップ16に供給される。パーミアンス及び選択性の算出方法は、第1実施形態と同様である。
【0146】
図19は、促進輸送膜として図18に示す円筒形状の促進輸送膜を用い、測定方法,キャリア濃度,原料ガス圧力を図10のときと同じにして、測定温度を160℃としたときの得られたデータに基づくグラフである。図10の場合と同様、CO2パーミアンス及びCO2/H2選択性ともに高い値を示しており、図18に示すような構造の円筒型の促進輸送膜でも図1に示すような平板型と同様の効果を示すことが分かる。
【0147】
また、図18に示す構造の場合、ゲル膜41が直接原料ガスFGに接触するように、空間40内においてゲル膜41が露出している構成である。すなわち、図2に示す構造と比較して、疎水性膜によってゲル膜41が覆われていない。この疎水性膜は、ゲル膜を安定化させ、経時性能を劣化させにくくする効果を有している。しかし、図20に示すように、円筒型形状の促進輸送膜であれば、疎水性膜で覆わなくても経時性能を向上させる効果を有している。以下にその点を説明する。
【0148】
図20は、平板型と円筒型の促進輸送膜の長期性能を比較したグラフであり、(a)がCO2パーミアンスRCO2、(b)がCO2/H2選択性を示している。いずれのグラフにおいても、(1)が円筒型のデータであり、(2)が平板型のデータである。なお、当該グラフ結果を得るに際しての条件は図13の場合と同一とした。
【0149】
また、図20では、比較例としての平板型の促進輸送膜として、ゲル膜が疎水性膜で覆われていない構造のものを想定している。これは、円筒型形状ではゲル膜の一方の面が原料ガスに曝される状態にあるため、比較の観点から、平板型についてもその条件を共通にする目的である。
【0150】
図20(a)によれば、CO2パーミアンスに関しては、平板型,円筒型共に経時変化はあまり大きくない。これに対し、図20(b)によれば、CO2/H2選択性に関しては、円筒型の促進輸送膜の場合、時間が経過してもその性能はあまり変化がないのに対し、平板型の促進輸送膜の場合、時間経過とともに選択性が低下し、100時間経過時においては最大時の10%程度にまで落ち込んでいる。これにより、ゲル膜を疎水膜で覆わない場合においては、長期性能の観点から見れば円筒型形状の促進膜の方が平板型形状よりも優れていると見ることができる。なお、一方、平板型においても、ゲル膜を疎水膜で覆うことにより、図13や図17に示すように、良好な長期性能を示すことが分かる。
【0151】
なお、本実施形態で用いたセラミックス製支持膜においても、第1実施形態において上述したPTFE多孔膜の場合と同様、100℃以上の耐熱性、機械的強度、含浸ゲル膜との密着性を有するのが好ましい。また、多孔度(空隙率)は40%以上で、細孔径は0.1〜1μmの範囲にあるのが好ましい。
【0152】
また、図18の構成では、セラミックス製支持膜を内側にし、その外側にゲル膜を備えた構造としたが、これとは逆に、支持膜を外側にしてゲル膜をその内側に形成しても良い。また、形状として「円筒形状」と記載したが、これは必ずしも断面が正確な「円」であることを要求するものではなく、楕円形状であっても構わないし、多少の凹凸を有していても構わない。
【0153】
本実施形態によれば、促進膜を円筒形状にすることで、平板型よりも長期性能が向上することが示される。これは、形状を円筒形状にすることで、促進輸送膜が変形しにくくなり、安定化することに由来すると考えられる。一方、平板型の場合は、経時とともに膜が変形して欠陥が生じ、この欠陥からH2が漏れ出すことで選択性が低下するものと考えられる。すなわち、上記実施例では、支持膜としてセラミックス膜を利用したが、この膜は、円筒形状に対する加工が可能であり、且つ、経時と共に変形しにくい材料であれば、セラミックスに限定されるものではない。
【0154】
また、逆に、第1,第2実施形態では支持膜としてPTFE多孔膜を利用としたが、圧力を加えた状態でも割れたりすることなく安定的に平板状態を維持することができれば、PTFE多孔膜に限定されるものではない。
【0155】
[別実施形態]
以下に、別実施形態について説明する。
【0156】
〈1〉上記実施形態では、本発明膜は、PVA/PAA共重合体と二酸化炭素キャリアのCs2CO3を含む水溶液からなるキャスト溶液を、ゲル膜担持用の親水性PTFE多孔膜にキャストした後にゲル化して作製したが、本発明膜は、当該作製方法以外の作製方法で作製しても構わない。例えば、PVA/PAA共重合体ゲル膜に、Cs2CO3水溶液を後から含浸させて作製しても構わない。
【0157】
〈2〉上記第1実施形態において、添加剤として炭酸セシウムをゲル膜に添加してCO2促進輸送膜を製造する場合について説明したが、炭酸セシウムの代わりに水酸化セシウムを用いても同様の効果を得ることができる。これは、水酸化セシウムが添加されたゲル膜をCO2分離に利用することで、上記(化3)に示される反応が生じ、これによって水酸化セシウムが炭酸セシウムに転化するためである。更に、炭酸セシウムの代わりに重炭酸セシウムを用いた場合も、上記(化3)より同様の効果を得ることができることが分かる。
【0158】
なお、添加剤として炭酸ルビジウムをゲル膜に添加してCO2促進輸送膜を製造する場合についても、同様に、炭酸ルビジウムに代えて水酸化ルビジウム若しくは重炭酸ルビジウムを用いることができる。
【0159】
〈3〉上記実施形態では、本発明膜は、疎水性PTFE多孔膜/ゲル層(親水性PTFE多孔膜に担持された含浸ゲル膜)/疎水性PTFE多孔膜よりなる3層構造としたが、本発明膜の支持構造は、必ずしも当該3層構造に限定されない。例えば、疎水性PTFE多孔膜/ゲル層(親水性PTFE多孔膜に担持された含浸ゲル膜)よりなる2層構造でも構わない。
【0160】
〈4〉上記実施形態において例示した、本発明膜の組成における各成分の混合比率、膜の各部の寸法等は、本発明の理解の容易のための例示であり、本発明はそれらの数値のCO2促進輸送膜に限定されるものではない。
【産業上の利用可能性】
【0161】
本発明に係る二酸化炭素分離装置は、二酸化炭素の分離に利用可能であり、特に、水素を主成分とする燃料電池用等の改質ガスに含まれる二酸化炭素を水素に対する高い選択比率で分離可能なCO2促進輸送膜を備えたCO2透過型メンブレンリアクターに有用である。
【符号の説明】
【0162】
1: 二酸化炭素キャリアを含有するPVA/PAAゲル膜(ゲル層)
2: 親水性多孔膜
3、4: 疎水性多孔膜
10: CO2促進輸送膜(サンプル)
11: 流通式ガス透過セル
12: 原料側室
13: 透過側室
14、16: 冷却トラップ
15: 背圧調整器
17: ガスクロマトグラフ
18: 定量送液ポンプ
19: 背圧調整器
20: CO変成器(CO2透過型メンブレンリアクター)
21: スイープガスの通路
40: 空間
41: ゲル膜
42: セラミックス製支持膜
43: 空間
FG: 原料ガス
FG’: 処理済ガス
PG: 透過ガス
SG、SG’: スイープガス
【特許請求の範囲】
【請求項1】
少なくとも水素と二酸化炭素と水蒸気が含まれる原料ガスをCO2促進輸送膜の原料側面に100℃以上の供給温度で供給して、前記CO2促進輸送膜を透過した二酸化炭素を透過側面から取り出す二酸化炭素分離装置であって、
前記CO2促進輸送膜が、ポリビニルアルコール−ポリアクリル酸共重合体ゲル膜に炭酸セシウム若しくは重炭酸セシウム若しくは水酸化セシウムからなる添加剤を添加したゲル層を親水性の多孔膜に担持させて形成されることを特徴とする二酸化炭素分離装置。
【請求項2】
前記ゲル層が、ポリビニルアルコール−ポリアクリル酸共重合体ゲル膜と炭酸セシウムの合計重量に対する炭酸セシウムの重量比率が65重量%以上85重量%以下の範囲で構成されることを特徴とする請求項1に記載の二酸化炭素分離装置。
【請求項3】
少なくとも水素と二酸化炭素と水蒸気が含まれる原料ガスをCO2促進輸送膜の原料側面に100℃以上の供給温度で供給して、前記CO2促進輸送膜を透過した二酸化炭素を透過側面から取り出す二酸化炭素分離装置であって、
前記CO2促進輸送膜が、ポリビニルアルコール−ポリアクリル酸共重合体ゲル膜に炭酸ルビジウム若しくは重炭酸ルビジウム若しくは水酸化ルビジウムからなる添加剤を添加したゲル層を親水性の多孔膜に担持させて形成されることを特徴とする二酸化炭素分離装置。
【請求項4】
前記親水性の多孔膜に担持された前記ゲル層が疎水性の多孔膜によって被覆されていることを特徴とする請求項1〜3の何れか1項に記載の二酸化炭素分離装置。
【請求項5】
前記ゲル層が、アルデヒド基由来の架橋構造を有することを特徴とする請求項1〜4の何れか1項に記載の二酸化炭素分離装置。
【請求項6】
前記多孔膜が100℃以上の耐熱性を備えていることを特徴とする請求項1〜5の何れか1項に記載の二酸化炭素分離装置。
【請求項7】
前記ゲル層,並びに前記親水性の多孔膜は、共に軸心を同一にした筒形状であって、一方の膜が、その内側面を他方の膜の外側面と接触させて、前記他方の膜を取り囲むように構成されていることを特徴とする請求項1〜3のいずれか1項に記載の二酸化炭素分離装置。
【請求項8】
前記親水性の多孔膜が、セラミックス製の多孔膜であることを特徴とする請求項7に記載の二酸化炭素分離装置。
【請求項9】
前記ゲル層が、前記親水性の多孔膜を取り囲むように、前記親水性の多孔膜の外側に形成されていることを特徴とする請求項7又は8に記載の二酸化炭素分離装置。
【請求項1】
少なくとも水素と二酸化炭素と水蒸気が含まれる原料ガスをCO2促進輸送膜の原料側面に100℃以上の供給温度で供給して、前記CO2促進輸送膜を透過した二酸化炭素を透過側面から取り出す二酸化炭素分離装置であって、
前記CO2促進輸送膜が、ポリビニルアルコール−ポリアクリル酸共重合体ゲル膜に炭酸セシウム若しくは重炭酸セシウム若しくは水酸化セシウムからなる添加剤を添加したゲル層を親水性の多孔膜に担持させて形成されることを特徴とする二酸化炭素分離装置。
【請求項2】
前記ゲル層が、ポリビニルアルコール−ポリアクリル酸共重合体ゲル膜と炭酸セシウムの合計重量に対する炭酸セシウムの重量比率が65重量%以上85重量%以下の範囲で構成されることを特徴とする請求項1に記載の二酸化炭素分離装置。
【請求項3】
少なくとも水素と二酸化炭素と水蒸気が含まれる原料ガスをCO2促進輸送膜の原料側面に100℃以上の供給温度で供給して、前記CO2促進輸送膜を透過した二酸化炭素を透過側面から取り出す二酸化炭素分離装置であって、
前記CO2促進輸送膜が、ポリビニルアルコール−ポリアクリル酸共重合体ゲル膜に炭酸ルビジウム若しくは重炭酸ルビジウム若しくは水酸化ルビジウムからなる添加剤を添加したゲル層を親水性の多孔膜に担持させて形成されることを特徴とする二酸化炭素分離装置。
【請求項4】
前記親水性の多孔膜に担持された前記ゲル層が疎水性の多孔膜によって被覆されていることを特徴とする請求項1〜3の何れか1項に記載の二酸化炭素分離装置。
【請求項5】
前記ゲル層が、アルデヒド基由来の架橋構造を有することを特徴とする請求項1〜4の何れか1項に記載の二酸化炭素分離装置。
【請求項6】
前記多孔膜が100℃以上の耐熱性を備えていることを特徴とする請求項1〜5の何れか1項に記載の二酸化炭素分離装置。
【請求項7】
前記ゲル層,並びに前記親水性の多孔膜は、共に軸心を同一にした筒形状であって、一方の膜が、その内側面を他方の膜の外側面と接触させて、前記他方の膜を取り囲むように構成されていることを特徴とする請求項1〜3のいずれか1項に記載の二酸化炭素分離装置。
【請求項8】
前記親水性の多孔膜が、セラミックス製の多孔膜であることを特徴とする請求項7に記載の二酸化炭素分離装置。
【請求項9】
前記ゲル層が、前記親水性の多孔膜を取り囲むように、前記親水性の多孔膜の外側に形成されていることを特徴とする請求項7又は8に記載の二酸化炭素分離装置。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【公開番号】特開2013−107076(P2013−107076A)
【公開日】平成25年6月6日(2013.6.6)
【国際特許分類】
【出願番号】特願2012−279552(P2012−279552)
【出願日】平成24年12月21日(2012.12.21)
【分割の表示】特願2009−12353(P2009−12353)の分割
【原出願日】平成21年1月22日(2009.1.22)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成20年度独立行政法人新エネルギー・産業技術総合開発機構「水素製造・輸送・貯蔵システム等技術開発/水素製造機器要素技術に関する研究開発/CO2膜分離法を用いた水素製造装置改質システムの開発」委託研究、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(305009898)株式会社ルネッサンス・エナジー・リサーチ (9)
【出願人】(504150450)国立大学法人神戸大学 (421)
【Fターム(参考)】
【公開日】平成25年6月6日(2013.6.6)
【国際特許分類】
【出願日】平成24年12月21日(2012.12.21)
【分割の表示】特願2009−12353(P2009−12353)の分割
【原出願日】平成21年1月22日(2009.1.22)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成20年度独立行政法人新エネルギー・産業技術総合開発機構「水素製造・輸送・貯蔵システム等技術開発/水素製造機器要素技術に関する研究開発/CO2膜分離法を用いた水素製造装置改質システムの開発」委託研究、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(305009898)株式会社ルネッサンス・エナジー・リサーチ (9)
【出願人】(504150450)国立大学法人神戸大学 (421)
【Fターム(参考)】
[ Back to top ]