
二重特異性抗体を精製するための抗体改変方法
【課題】二重特異性抗体を効率的に精製するための抗体可変領域のアミノ酸改変の方法、改変された二重特異性抗体からなる医薬組成物、並びに、二重特異性抗体医薬組成物の製造方法を提供する。
【解決手段】二重特異性抗体を構成する2種類の抗体が、抗体可変領域の表面に存在するアミノ酸を改変することによって、2種類の抗体のH鎖の間に等電点の差異を導入し、等電点の違いを利用して、二重特異性抗体をクロマトグラフィーカラムで効率的に精製する方法。また等電点に差のある抗体の定常領域に各抗原結合部位(重鎖可変領域)を組み込み、これらを共発現させることによって、二重特異性抗体をクロマトグラフィーカラムで効率的に精製する方法。
【解決手段】二重特異性抗体を構成する2種類の抗体が、抗体可変領域の表面に存在するアミノ酸を改変することによって、2種類の抗体のH鎖の間に等電点の差異を導入し、等電点の違いを利用して、二重特異性抗体をクロマトグラフィーカラムで効率的に精製する方法。また等電点に差のある抗体の定常領域に各抗原結合部位(重鎖可変領域)を組み込み、これらを共発現させることによって、二重特異性抗体をクロマトグラフィーカラムで効率的に精製する方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、二重特異性抗体を精製するための抗体改変方法、該二重特異性抗体の分離方法、および該二重特異性抗体を有効成分として含有する医薬組成物等に関する。
【背景技術】
【0002】
抗体は血中での安定性が高く、副作用も少ないことから医薬品として注目されている。その中には二種類の抗原(抗原Aと抗原B)を同時に認識できる二重特異性抗体がある(非特許文献1)。現在、臨床試験が行なわれているMDX-210は、FcγRIを発現しているmonocyte等をHER-2/neuを発現している癌細胞にretargetingするIgG型二重特異性抗体である(非特許文献2)。抗体の製造は、一般的に遺伝子組み換え技術を用いることが多い。具体的には、抗体の蛋白質をコードするDNAをハイブリドーマ、抗体を産生する感作リンパ球等の抗体産生細胞、または抗体遺伝子を提示しているファージライブラリーからクローニングし、適当なベクターに組み込み、これを宿主細胞に導入し産生させる技術である。遺伝子組み換え技術を用いたIgG型二重特異性抗体の製造は、目的の二種類のIgGを構成するH鎖及びL鎖の遺伝子、合計4種の遺伝子を細胞に導入し、共発現により分泌させる。このような発現において、野生型のH鎖及びL鎖の構成遺伝子を発現させた場合には、2種類のH鎖の会合やH鎖とL鎖の会合はランダムに起こるため、目的の二重特異性抗体の比率は極めて少なくなる。具体的には、目的の二重特異性抗体は10種類中1種類のみであり、生産効率は低下してしまう。目的抗体の生産効率の低下は、目的抗体の精製の障害になるばかりでなく、ロット間差などの不均一性を増大させ、生産コストの肥大を招くことになる。
【0003】
二重特異性抗体を開発するための効率的な二重特異性抗体作製方法として、両H鎖に共通するL鎖を取得するための共通L鎖取得技術およびH鎖へテロ会合化のためのKnobs-into-holes技術が報告されている。具体的には、抗原Aと抗原Bを認識する各H鎖に対して、両抗原結合活性を維持することができる共通のL鎖をファージライブラリー(Phage library)等から見出し、さらに、一方のH鎖のCH3領域に存在するアミノ酸側鎖をより大きい側鎖(knob;突起)に置換し、もう一方のH鎖のCH3領域に存在するアミノ酸側鎖をより小さい側鎖(hole;空隙)に置換することにより突起が空隙内に配置されるようにしてH鎖ヘテロダイマーの形成を促進し、目的の二重特異性抗体を効率的に取得することができる(特許文献1、非特許文献3、非特許文献4)。
【0004】
しかしながら、H鎖へテロダイマーのためにKnobs-into-holes技術を用いた場合でも、非特許文献3、非特許文献4に示されたようにKnobs-into-holes技術によって目的のA鎖B鎖へテロダイマーの含有率を最大95%程度まで高めることが可能だが、残りの5%はA鎖ホモダイマー、B鎖ホモダイマーであり不純物となってしまう。医薬品として、二重特異性抗体を開発するためには、共通L鎖(非特許文献3,非特許文献4)を用いた場合に産生される3種類の分子種(A鎖ホモダイマー、B鎖ホモダイマー、A鎖B鎖へテロダイマー)の中から、可能な限り高純度にA鎖B鎖へテロダイマーを精製する必要がある。そのため残りの5%の不純物であるA鎖ホモダイマー、B鎖ホモダイマーを取り除き、A鎖B鎖へテロダイマーを医薬品として開発可能な高純度まで精製する必要がある。共通L鎖を用い、Knobs-into-holes技術を用いない場合は、理論上、A鎖ホモダイマー、A鎖B鎖へテロダイマー、B鎖ホモダイマーが1:2:1で産生され、50%の不純物であるA鎖ホモダイマー、B鎖ホモダイマーを取り除く必要がある。
【0005】
医薬品の製造レベルのクロマトグラフィーによる分離において、A鎖B鎖へテロダイマーとA鎖ホモダイマー、B鎖ホモダイマーを分離する方法がこれまでにいくつか報告されている。A鎖B鎖へテロダイマーを選択的に精製する方法として、非特許文献5にはA鎖にmouse IgG2a、B鎖にrat IgG2bを使用し、mouse IgG2aとrat IgG2bの各H鎖に対するprotein Aへのアフィニティーの違いを利用し、protein Aからの溶出pHをコントロールすることで、A鎖B鎖へテロダイマーを精製する方法が報告されているが、mouseおよびratの定常領域を使用しているため、抗原性の観点から同方法はヒトに対する医薬品への応用は困難である。またこの方法では同じアイソタイプのH鎖からなるA鎖B鎖へテロダイマーを分離することはできないため、利用は限定される。
【0006】
非特許文献6には疎水相互作用クロマトグラフィーを用いたA鎖B鎖へテロダイマーを精製する方法が報告されているが、Anti-CD3 mouse IgG2aとanti-CD19 mouse IgG1からなる目的のA鎖B鎖へテロダイマーが十分にピーク分離しておらず、また、異なるアイソタイプのH鎖を用い、その疎水性の違いを利用して分離していると考えられることから、同じアイソタイプのH鎖からなるA鎖B鎖へテロダイマーを必ずしも分離できるとは限らない。
【0007】
非特許文献7にはThiophilic affinityクロマトグラフィーを用いたA鎖B鎖へテロダイマーを精製する方法が報告されているが、mouse IgG1とrat IgG2aを用い、そのヒンジ領域におけるフリーのシステイン(チオール基)を利用していることから、同じアイソタイプのH鎖からなるA鎖B鎖へテロダイマーの分離には利用できず、またフリーのシステインは保存中のアグリゲーションに関与するため、安定な医薬品製剤の開発には適さない。
【0008】
非特許文献8には抗原を用いたアフィニティークロマトグラフィーが報告されている。しかし、タンパク質あるいはペプチド抗原を用いたアフィニティークロマトグラフィーは、カラムのコストや安定性に課題があるため、アフィニティークロマトグラフィーを用いた医薬品の製造は一般的ではない。また両抗原に結合するA鎖B鎖へテロダイマーの精製のためにはアフィニティークロマトグラフィーを2回実施する必要があり、コストが高くなることが予想される。また抗原の立体構造のみを認識する抗体や低アフィニティで目的の機能を有する抗体が報告されており、このような性質を持つ抗体は抗原を利用したアフィニティクロマトグラフィーを使用することが困難である。従って、アフィニティークロマトグラフィーを用いた二重特異性抗体の精製は汎用的ではないと考えられる。
【0009】
このように二重特異性抗体のA鎖B鎖へテロダイマー精製は限られた範囲内でしか行われておらず、同じH鎖アイソタイプ・定常領域配列からなる二重特異性抗体のA鎖B鎖へテロダイマーを、医薬品として許容されるような高い純度まで精製する方法は報告されていない。二重特異性抗体を構成する2種類の抗体が同じ定常領域配列を有する場合、可変領域配列の違いのみにより、A鎖B鎖へテロダイマーを分離する必要があるが、抗体の可変領域のアミノ酸配列は抗体間で非常にホモロジーが高く(非特許文献9)、可変領域配列の違いだけでA鎖B鎖へテロダイマーを医薬品として許容されるような高い純度まで精製することは困難であった。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】国際公開第96/27011号
【非特許文献】
【0011】
【非特許文献1】Marvin JS,および Zhu Z.著、「Recombinant approaches to IgG-like bispecific antibodies.」、Acta Pharmacol Sin.、2005年Jun、Vol.26(6)、p.649-58.
【非特許文献2】Segal DM ら著、Current Opinion in Immunology、1999年、Vol.11、p.558-562.
【非特許文献3】Merchant AM, 外6名著、「An efficient route to human bispecific IgG.」、Nat Biotechnol.、1998年Jul、Vol.16(7)、p.677-81.
【非特許文献4】Carter P.著、「Bispecific human IgG by design.」、J Immunol Methods. 、2001年Feb 1、Vol.248(1-2)、p.7-15.
【非特許文献5】Lindhofer H, 外3名著、「Preferential species-restricted heavy/light chain pairing in rat/mouse quadromas. Implications for a single-step purification of bispecific antibodies.」、J Immunol.、1995年Jul 1、Vol.155(1)、p.219-25.
【非特許文献6】Manzke O, 外3名著、「Single-step purification of bispecific monoclonal antibodies for immunotherapeutic use by hydrophobic interaction chromatography.」、J Immunol Methods.、1997年Oct 13、Vol.208(1)、p.65-73.
【非特許文献7】Kreutz FT, 外2名著、「Efficient bispecific monoclonal antibody purification using gradient thiophilic affinity chromatography.」、J Chromatogr B Biomed Sci Appl.、1998年Sep 4、Vol.714(2)、p.161-70.
【非特許文献8】Gupta S, およびSuresh M.著、「Affinity chromatography and co-chromatography of bispecific monoclonal antibody immunoconjugates.」、J Biochem Biophys Methods. 、2002年May 31、Vol.51(3)、p.203-16. Review.
【非特許文献9】Carl Branden著、Introduction to Protein Structure 2nd edition, Newton Press.
【発明の概要】
【発明が解決しようとする課題】
【0012】
本発明はこのような状況に鑑みて為されたものであり、その目的は、二重特異性抗体を効率的に精製するための抗体可変領域のアミノ酸改変の方法、改変された二重特異性抗体からなる医薬組成物、並びに、二重特異性抗体医薬組成物の製造方法を提供することにある。また本発明は、重鎖定常領域が改変された二重特異性抗体、改変された二重特異性抗体からなる医薬組成物、並びに、二重特異性抗体医薬組成物の製造方法を提供することにある。
【課題を解決するための手段】
【0013】
本発明者らは、目的物の精製が従来困難であった二重特異性抗体を汎用的なクロマトグラフィーカラムを用いることで効率的に精製できる方法として、抗体可変領域のアミノ酸置換による方法について、鋭意研究を行った。
【0014】
その結果、二重特異性抗体を構成する2種類の抗体が抗体可変領域の表面に存在するアミノ酸を改変することによって、2種類の抗体のH鎖の間に等電点の差異を導入し、等電点の違いを利用して、二重特異性抗体をクロマトグラフィーカラムで効率的に精製する方法を見出した。具体的には、抗体のH鎖において、抗体の機能(活性)を低下させることなく、等電点のみを制御することが可能な改変箇所を見出した。さらに本発明者らは、本発明の方法によって取得された二重特異性抗体が、実際に機能を保持していることを確認した。
【0015】
上述の如く本発明者らは、任意の二重特異性抗体を汎用的なクロマトグラフィーカラムを用いることで効率的に精製できる方法として、抗体可変領域のアミノ酸置換による方法の開発に成功し、本発明を完成させた。
【0016】
また本発明者らは、二重特異性抗体を構成する2種類のH鎖の定常領域に対して、元来等電点に差のある異なるアイソタイプの定常領域をそれぞれのH鎖に用いることで、等電点の違いを利用して、二重特異性抗体をクロマトグラフィーカラムで効率的に精製する方法を見出した。さらに本発明者らは、本発明の方法によって取得された二重特異性抗体が、実際に機能を保持していることも確認した。
【0017】
本発明は、クロマトグラフィーカラムを用いることで効率的に精製するための抗体可変領域のアミノ酸置換方法、および、改変された二重特異性抗体からなる医薬組成物、および、二重特異性抗体医薬組成物の製造方法に関し、さらに重鎖定常領域が改変された二重特異性抗体、および、改変された二重特異性抗体からなる医薬組成物、および、二重特異性抗体医薬組成物の製造方法に関し、より具体的には、
〔1〕 第1のポリペプチドおよび第2のポリペプチドを含む多重特異性抗体の製造方法であって、
(a)第1のポリペプチドと第2のポリペプチドの等電点に差がつくように、第1のポリペプチドのアミノ酸残基をコードする核酸および第2のポリペプチドのアミノ酸残基をコードする核酸の両方またはいずれか一方を改変し、
(b)宿主細胞を該核酸が発現するように培養し、
(c)宿主細胞培養物から多重特異性抗体を回収すること、
を含む多重特異性抗体の製造方法、
〔2〕 工程(a)の改変が、第1のポリペプチドのホモ多量体、第2のポリペプチドのホモ多量体、および第1のポリペプチドと第2のポリペプチドのヘテロ多量体が、標準的なクロマトグラフィーを使用した分析により分離したピークとなるように、核酸を改変することである〔1〕に記載の方法、
〔3〕 前記第1のポリペプチドおよび前記第2のポリペプチドが重鎖可変領域を含む〔1〕に記載の方法、
〔4〕 前記多重特異性抗体が、軽鎖可変領域を含む第3のポリペプチドを含み、前記第1のポリペプチドおよび前記第2のポリペプチドがそれぞれ該第3のポリペプチドと多量体を形成する〔3〕に記載の方法、
〔5〕 前記第1のポリペプチドおよび前記第2のポリペプチドが重鎖定常領域を含む〔1〕〜〔4〕のいずれか1項に記載の方法、
〔6〕 前記第1のポリペプチドおよび第2のポリペプチドに含まれる重鎖定常領域が互いに等電点の異なる重鎖定常領域である〔5〕に記載の方法、
〔7〕 前記等電点の異なる重鎖定常領域がIgG1とIgG4、又は、IgG1とIgG2である〔6〕に記載の方法、
〔8〕 前記多重特異性抗体が、二重特異性抗体である〔1〕に記載の方法、
〔9〕 〔1〕に記載の方法により製造される多重特異性抗体、
〔10〕 第1のポリペプチドおよび第2のポリペプチドを含む多重特異性抗体の精製方法であって、
(a)第1のポリペプチドと第2のポリペプチドの等電点に差がつくように、第1のポリペプチドのアミノ酸残基をコードする核酸および第2のポリペプチドのアミノ酸残基をコードする核酸の両方またはいずれか一方を改変し、
(b)宿主細胞を該核酸が発現するように培養し、
(c)宿主細胞培養物から標準的なクロマトグラフィーにより該多重特異性抗体を精製すること、
を含む多重特異性抗体の精製方法、
〔11〕 工程(a)の改変が、第1のポリペプチドのホモ多量体、第2のポリペプチドのホモ多量体、および第1のポリペプチドと第2のポリペプチドのヘテロ多量体が、標準的なクロマトグラフィーを使用した分析により分離したピークとなるように、核酸を改変することである〔10〕に記載の方法、
〔12〕 前記第1のポリペプチドおよび前記第2のポリペプチドが重鎖可変領域を含む〔10〕に記載の方法、
〔13〕 前記多重特異性抗体が、軽鎖可変領域を含む第3のポリペプチドを含み、前記第1のポリペプチドおよび前記第2のポリペプチドがそれぞれ該第3のポリペプチドと多量体を形成する〔12〕に記載の方法、
〔14〕 前記第1のポリペプチドおよび前記第2のポリペプチドが重鎖定常領域を含む〔10〕〜〔13〕のいずれか1項に記載の方法、
〔15〕 前記第1のポリペプチドおよび第2のポリペプチドに含まれる重鎖定常領域が互いに等電点の異なる重鎖定常領域である〔14〕に記載の方法、
〔16〕 前記等電点の異なる重鎖定常領域がIgG1とIgG4、又は、IgG1とIgG2である〔15〕に記載の方法、
〔17〕 前記多重特異性抗体が、二重特異性抗体である〔10〕に記載の方法、
〔18〕 〔10〕に記載の方法により精製する工程を含む多重特異性抗体の製造方法、
〔19〕 〔18〕に記載の方法により製造される多重特異性抗体、
〔20〕 第1のポリペプチドおよび第2のポリペプチドを含む多重特異性抗体であって、第1のポリペプチドが重鎖可変領域および/または重鎖定常領域を含み、該重鎖可変領域におけるKabatナンバリングによる10位、12位、23位、39位、43位および105位のアミノ酸残基、若しくは、該重鎖定常領域におけるEUナンバリングによる137位、196位、203位、214位、217位、233位、268位、274位、276位、297位、355位、392位、419位、435位のアミノ酸残基から選ばれる、少なくとも1つのアミノ酸残基が電荷を有し、第1のポリペプチドと第2のポリペプチドの等電点が互いに異なる多重特異性抗体、
〔21〕 第2のポリペプチドが重鎖可変領域および/または重鎖定常領域を含み、該重鎖可変領域におけるKabatナンバリングによる10位、12位、23位、39位、43位および105位のアミノ酸残基、若しくは、該重鎖定常領域におけるEUナンバリングによる137位、196位、203位、214位、217位、233位、268位、274位、276位、297位、355位、392位、419位、435位のアミノ酸残基から選ばれる、少なくとも1つのアミノ酸残基が、前記第1のポリペプチドに含まれる重鎖可変領域および/または重鎖定常領域において選ばれる、電荷を有するアミノ酸残基とは反対の電荷を有する、または電荷を有しない〔20〕に記載の多重特異性抗体、
〔22〕 前記電荷を有するアミノ酸残基と当該アミノ酸残基とは反対の電荷を有するアミノ酸残基の組み合わせが、以下の(a)または(b)いずれかの群に含まれるアミノ酸残基からそれぞれ選択される〔20〕に記載の多重特異性抗体:
(a)グルタミン酸(E)、アスパラギン酸(D);
(b)リジン(K)、アルギニン(R)、ヒスチジン(H)、
〔23〕 第1のポリペプチドと第2ポリペプチドの等電点に差があり、第1のポリペプチドのホモ多量体、第2のポリペプチドのホモ多量体、および第1のポリペプチドと第2のポリペプチドのヘテロ多量体が、標準的なクロマトグラフィーを使用した分析により分離したピークとなり得る多重特異性抗体、
〔24〕 前記第1のポリペプチドおよび前記第2のポリペプチドが重鎖可変領域を含む〔23〕に記載の多重特異性抗体、
〔25〕 前記多重特異性抗体が、軽鎖可変領域を含む第3のポリペプチドを含み、前記第1のポリペプチドおよび前記第2のポリペプチドがそれぞれ該第3のポリペプチドと多量体を形成する〔24〕に記載の多重特異性抗体、
〔26〕 前記第1のポリペプチドおよび前記第2のポリペプチドが重鎖定常領域を含む〔23〕〜〔25〕のいずれか1項に記載の多重特異性抗体、
〔27〕 前記第1のポリペプチドおよび第2のポリペプチドに含まれる重鎖定常領域が互いに等電点の異なる重鎖定常領域である〔26〕に記載の多重特異性抗体、
〔28〕 前記等電点の異なる重鎖定常領域がIgG1とIgG4、又は、IgG1とIgG2である〔27〕に記載の多重特異性抗体、
〔29〕 前記多重特異性抗体が、二重特異性抗体である〔23〕に記載の多重特異性抗体、
〔30〕 〔23〕〜〔29〕のいずれか1項に記載の多重特異性抗体および医薬的に許容される担体を含む組成物、
〔31〕 〔23〕〜〔29〕のいずれか1項に記載の多重特異性抗体を構成するポリペプチドをコードする核酸、
〔32〕 〔31〕に記載の核酸を有する宿主細胞、
〔33〕 〔32〕に記載の宿主細胞を培養する工程、細胞培養物からポリペプチドを回収する工程を含む〔23〕〜〔29〕のいずれか1項に記載の多重特異性抗体の製造方法、
〔34〕 第1のポリペプチドの可変領域が以下の(a1)〜(a7)のいずれかに記載のアミノ酸配列からなり、第2のポリペプチドの可変領域が以下の(b1)〜(b3)のいずれかに記載のアミノ酸配列からなり、第3のポリペプチドの可変領域が以下の(c1)または(c2)に記載のアミノ酸配列からなる〔25〕に記載の多重特異性抗体:
(a1)配列番号:7
(a2)配列番号:8
(a3)配列番号:9
(a4)配列番号:10
(a5)配列番号:11
(a6)配列番号:12
(a7)配列番号:13
(b1)配列番号:14
(b2)配列番号:15
(b3)配列番号:16
(c1)配列番号:17
(c2)配列番号:18
〔35〕 第1のポリペプチドの可変領域が配列番号:11に記載のアミノ酸配列からなり、第2のポリペプチドの可変領域が配列番号:16に記載のアミノ酸配列からなり、第3のポリペプチドの可変領域が配列番号:17に記載のアミノ酸配列からなる〔34〕に記載の多重特異性抗体、
〔36〕 第1のポリペプチドの可変領域が配列番号:12に記載のアミノ酸配列からなり、第2のポリペプチドの可変領域が配列番号:16に記載のアミノ酸配列からなり、第3のポリペプチドの可変領域が配列番号:18に記載のアミノ酸配列からなる〔34〕に記載の多重特異性抗体、
〔37〕 第1のポリペプチドおよび第2のポリペプチドがヒトIgG4定常領域を含み、第3のポリペプチドがヒトκ定常領域を含む〔34〕〜〔36〕のいずれか1項に記載の多重特異性抗体、に関する。
【図面の簡単な説明】
【0018】
【図1】ヒト化二重特異性抗体(ヒト化A69(hA69a)/ ヒト化B26 (hB26-F123e4)/ヒト化BBA(hAL-F123j4))の凝固活性について評価した結果を示す図である。評価の結果、キメラ二重特異性抗体と同等以上の凝固活性を示した。
【図2】ヒト化A69-H鎖可変領域(hA69a)とヒト化BBA(hAL-F123j4)およびヒト化hB26-H鎖可変領域(hB26-F123e4)とヒト化BBA(hAL-F123j4)を使用して抗体モデリングを実施した結果を示す図である。表面電荷を変化させることが可能なアミノ酸について側鎖を強調して示した。番号については、Kabatデータベースの配列番号(Kabat EA et al. 1991. Sequences of Proteins of Immunological Interest.NIH)を採用した。
【図3】未改変および可変領域を改変したヒト化A69抗体ホモダイマーと未改変および可変領域を改変したヒト化B26抗体ホモダイマーを用いて等電点電気泳動による分析を実施した結果を示す写真である。分析の結果、改変により等電点が変化していることが確認された。
【図4】可変領域を改変したヒト化A69抗体ホモダイマーを用いて陽イオン交換クロマトグラフィー分析を実施した結果を示す図である。分析の結果、未改変の抗体と比較してピークの移動が確認された。
【図5】可変領域を改変したヒト化B26抗体ホモダイマーを用いて陽イオン交換クロマトグラフィー分析を実施した結果を示す図である。分析の結果、未改変の抗体と比較してピークの移動が確認された。
【図6】可変領域を改変したヒト化二重特異性抗体(H鎖定常領域にknobs-into-holes技術を利用)を用いて凝固活性を評価した結果を示す図である。評価の結果、未改変の抗体と同等の凝固活性を示した。
【図7】可変領域(CDR)を改変したヒト化A69抗体ホモダイマーを用いて等電点電気泳動による分析を実施した結果を示す写真である。分析の結果、未改変の抗体と比較してバンドの移動が確認された。
【図8】可変領域(CDR)を改変したヒト化A69抗体ホモダイマーを用いて抗原であるFactor IXaに対する結合活性を評価した結果を示す図である。評価の結果、未改変の抗体と同等の結合活性を保持していることが示された。
【図9】未改変抗体として、ヒト化A69-H鎖であるhA69a、ヒト化B26-H鎖であるhB26-F123e4とヒト化BBA-L鎖であるhAL-F123j4を用いて作製した未改変のヒト化二重特異性抗体ヒト化の陽イオン交換クロマトグラフィー分析を実施した結果を示す図である。分析の結果、二種類のホモダイマーと二重特異性抗体が分離せず1本のピークとして溶出した。
【図10】ヒト化A69-H鎖の改変体であるhA69-PFとヒト化B26-H鎖の改変体であるhA26-PFとヒト化BBA-L鎖であるhAL-s8を用いて作製したヒト化二重特異性PF抗体の陽イオン交換クロマトグラフィー分析を実施した結果を示す図である。分析の結果、二種類のホモダイマーと二重特異性抗体がそれぞれ分離し、hA69-PF ホモダイマー、ヒト化二重特異性PF抗体、hB26-PF ホモダイマーの順に3本のピークとして溶出した。
【図11】精製したヒト化A69抗体-PFホモダイマーおよびヒト化B26-PF抗体ホモダイマー、ヒト化二重特異性PF抗体を用いて等電点電気泳動による分析を実施した結果を示す写真である。分析の結果、目的の二重特異性抗体が精製できていることが確認された。
【図12】精製したヒト化二重特異性PF抗体(H鎖定常領域は野生型)を用いて凝固活性を評価した結果を示す図である。評価の結果、H鎖定常領域にknobs-into-holes技術を利用した二重特異性抗体(KiH)と同等の凝固活性を示した。
【図13】ヒト化A69抗体ホモダイマーおよびヒト化B26抗体ホモダイマー、ヒト化二重特異性抗体の3種類の抗体が含まれる培養上清から製造用汎用カラムを用いて二重特異性抗体を精製した時のクロマトグラムを示した。
【図14】製造用汎用カラムを用いて精製したヒト化二重特異性抗体(H鎖定常領域は野生型)を用いて凝固活性を評価した結果を示す図である。評価の結果、ヒト化二重特異性PF抗体と同等の凝固活性を示した。
【図15】未改変、IgG2化およびIgG4化ヒト化PM-1抗体を用いて等電点電気泳動による分析を実施した結果を示す写真である。分析の結果、改変により等電点が変化していることが確認された。Aは未改変ヒト化PM-1抗体、BはIgG2化ヒト化PM-1抗体、CはIgG4化ヒト化PM-1抗体を示す。
【図16】未改変、IgG2化およびIgG4化ヒト化PM-1抗体のそれぞれの共発現抗体を用いて等電点電気泳動による分析を実施した結果を示す写真である。分析の結果、各アイソタイプ抗体とアイソタイプハイブリッド抗体がpI差をもって分離することが示された。Aは未改変ヒト化PM-1抗体/IgG2化ヒト化PM-1抗体共発現抗体、Bは未改変ヒト化PM-1抗体/IgG4化ヒト化PM-1抗体共発現抗体、Cはヒト化PM-1抗体精製品(bulk)を示す。
【図17】単独発現させた未改変、IgG2化、IgG4化ヒト化PM-1抗体を用いて陽イオン交換クロマトグラフィー分析を実施した結果を示す図である。分析の結果、未改変の抗体と比較してピークの移動が確認された。
【図18】未改変、IgG2化およびIgG4化ヒト化PM-1抗体のそれぞれの共発現抗体の陽イオン交換クロマトグラフィー分析を実施した結果を示す図である。分析の結果、未改変ヒト化PM-1抗体/IgG2化ヒト化PM-1抗体の組み合わせ、および、未改変ヒト化PM-1抗体/IgG4化ヒト化PM-1抗体の組み合わせにおいて、各アイソタイプのホモダイマー、ヘテロダイマーが3つの主ピークとして観察された。Aは未改変ヒト化PM-1抗体/IgG2化ヒト化PM-1抗体共発現抗体、Bは未改変ヒト化PM-1抗体/IgG4化ヒト化PM-1抗体共発現抗体を示す。
【図19】未改変ヒト化PM-1抗体/IgG4化ヒト化PM-1抗体を共発現させたものから陽イオン交換クロマトグラフィーでホモダイマー、ヘテロダイマーを精製した結果を示す図である。この結果、IgG4化ヒト化PM-1抗体ホモダイマー、未改変ヒト化PM-1/IgG4化ヒト化PM-1ハイブリッド抗体、未改変ヒト化PM-1抗体ホモダイマーの順に3本のピークとして溶出したため、これらを分取した。矢印はおよその分画範囲を示す。
【図20】陽イオン交換クロマトグラフィーで精製した未改変ヒト化PM-1抗体ホモダイマー、未改変ヒト化PM-1/IgG4化ヒト化PM-1ハイブリッド抗体、IgG4化ヒト化PM-1抗体ホモダイマーを用いてリクロマトグラフィーを行った結果を示す図である。この結果、目的のアイソタイプハイブリッド抗体が精製できていることが確認された。
【図21】陽イオン交換クロマトグラフィーで精製した未改変ヒト化PM-1抗体ホモダイマー、未改変/IgG4化ヒト化PM-1ハイブリッド抗体、IgG4化ヒト化PM-1抗体ホモダイマーを用いて等電点電気泳動による分析を実施した結果を示す写真である。分析の結果、目的のアイソタイプハイブリッド抗体が精製できていることが確認された。Aは未改変ヒト化PM-1抗体/IgG4化ヒト化PM-1抗体共発現抗体、Bは未改変ヒト化PM-1抗体分取画分、Cは未改変ヒト化PM-1/IgG4化ヒト化PM-1ハイブリット抗体分取画分、DはIgG4化ヒト化PM-1抗体分取画分を示す。
【図22】陽イオン交換クロマトグラフィーで精製した未改変ヒト化PM-1抗体ホモダイマー、未改変ヒト化PM-1/IgG4化ヒト化PM-1ハイブリッド抗体、IgG4化ヒト化PM-1抗体ホモダイマーを用いてヒトIL-6中和活性を評価した結果を示す図である。評価の結果、いずれの抗体もヒト化PM-1精製抗体と同等の中和活性を示した。AおよびBはヒトgp130発現BaF3細胞株、CおよびDはヒトgp130/ヒトIL-6受容体共発現BaF3細胞株を示す。黒丸(●)はヒト化PM-1抗体精製品(bulk)、白四角(□)は未改変ヒト化PM-1抗体、白三角(△)はIgG4化ヒト化PM-1抗体、×は未改変ヒト化PM-1/IgG4化ヒト化PM-1ハイブリット抗体を示す。
【発明を実施するための形態】
【0019】
まず本発明は、多重特異性抗体を製造するための抗体改変方法を提供する。本発明の製造方法の好ましい態様としては、第1のポリペプチドと第2のポリペプチドの等電点に差がつくように第1のポリペプチドのアミノ酸残基をコードする核酸および第2のポリペプチドのアミノ酸残基をコードする核酸の両方またはいずれか一方を改変することを含む方法である。即ち、第1のポリペプチドと第2のポリペプチドのアミノ酸残基の電荷を変えることによって、ポリペプチドに等電点(pI)の差異を導入し、当該等電点の差異を利用して多重特異性抗体を製造することができる。詳しくは以下の(a)〜(c)の工程を含む製造方法である。
(a)第1のポリペプチドと第2のポリペプチドの等電点に差がつくように、第1のポリペプチドのアミノ酸残基をコードする核酸および第2のポリペプチドのアミノ酸残基をコードする核酸の両方またはいずれか一方を改変し、
(b)宿主細胞を該核酸が発現するように培養し、
(c)宿主細胞培養物から多重特異性抗体を回収すること
【0020】
本発明におけるポリペプチドとは、通常、10アミノ酸程度以上の長さを有するペプチド、およびタンパク質を指す。また、通常、生物由来のポリペプチドであるが、特に限定されず、例えば、人工的に設計された配列からなるポリペプチドであってもよい。また、天然ポリペプチド、あるいは合成ポリペプチド、組換えポリペプチド等のいずれであってもよい。さらに、上記のポリペプチドの断片もまた、本発明のポリペプチドに含まれる。
【0021】
本発明において「ポリペプチドの等電点に差がつく」とは、2種以上のポリペプチドにおいて、表面アミノ酸の電荷の改変を行うことにより、互いの等電点が等しくならないことをいう。等電点の差は、例えば、等電点電気泳動等の手法を用いることにより観察することができる。また本発明においては、当該ポリペプチドの構造や機能(活性)を変化させずに等電点を制御することが好ましい。
【0022】
即ち本発明は、第1のポリペプチドおよび第2のポリペプチドを含む多重特異性抗体の製造方法であって、
(a)第1のポリペプチドと第2のポリペプチドの等電点の差が0.5以上、好ましくは0.7以上、更に好ましくは0.9以上となるように、第1のポリペプチドのアミノ酸残基をコードする核酸および第2のポリペプチドのアミノ酸残基をコードする核酸の両方またはいずれか一方を改変し、
(b)宿主細胞を該核酸が発現するように培養し、
(c)宿主細胞培養物から多重特異性抗体を回収すること、を含む多重特異性抗体の製造方法を提供する。
【0023】
また本発明は、多重特異性抗体を精製するための抗体改変方法を提供する。本発明の精製方法の好ましい態様としては、第1のポリペプチドと第2のポリペプチドの等電点に差がつくように第1のポリペプチドのアミノ酸残基をコードする核酸および第2のポリペプチドのアミノ酸残基をコードする核酸の両方またはいずれか一方を改変することを含む方法である。即ち、第1のポリペプチドと第2のポリペプチドのアミノ酸残基の電荷を変えることによって、ポリペプチドに等電点(pI)の差異を導入し、当該等電点の差異を利用して多重特異性抗体を精製することができる。詳しくは以下の(a)〜(c)の工程を含む精製方法である。
(a)第1のポリペプチドと第2のポリペプチドの等電点に差がつくように、第1のポリペプチドのアミノ酸残基をコードする核酸および第2のポリペプチドのアミノ酸残基をコードする核酸の両方またはいずれか一方を改変し、
(b)宿主細胞を該核酸が発現するように培養し、
(c)宿主細胞培養物から標準的なクロマトグラフィーにより該多重特異性抗体を精製すること
【0024】
なお、上記精製方法により精製する工程を含む多重特異性抗体の製造方法も本発明に含まれる。
【0025】
本発明における核酸は、通常、適当なベクターへクローニング(挿入)され、宿主細胞へ導入される。該ベクターとしては、挿入した核酸を安定に保持するものであれば特に制限されず、例えば宿主に大腸菌を用いるのであれば、クローニング用ベクターとしてはpBluescriptベクター(Stratagene社製)などが好ましいが、市販の種々のベクターを利用することができる。本発明の多重特異性抗体(ポリペプチド)を生産する目的においてベクターを用いる場合には、特に発現ベクターが有用である。発現ベクターとしては、試験管内、大腸菌内、培養細胞内、生物個体内でポリペプチドを発現するベクターであれば特に制限されないが、例えば、試験管内発現であればpBESTベクター(プロメガ社製)、大腸菌であればpETベクター(Invitrogen社製)、培養細胞であればpME18S-FL3ベクター(GenBank Accession No. AB009864)、生物個体であればpME18Sベクター(Mol Cell Biol. 8:466-472(1988))などが好ましい。ベクターへの本発明のDNAの挿入は、常法により、例えば、制限酵素サイトを用いたリガーゼ反応により行うことができる(Current protocols in Molecular Biology edit. Ausubel et al. (1987) Publish. John Wiley & Sons.Section 11.4-11.11)。
【0026】
上記宿主細胞としては特に制限はなく、目的に応じて種々の宿主細胞が用いられる。ポリペプチドを発現させるための細胞としては、例えば、細菌細胞(例:ストレプトコッカス、スタフィロコッカス、大腸菌、ストレプトミセス、枯草菌)、真菌細胞(例:酵母、アスペルギルス)、昆虫細胞(例:ドロソフィラS2、スポドプテラSF9)、動物細胞(例:CHO、COS、HeLa、C127、3T3、BHK、HEK293、Bowes メラノーマ細胞)および植物細胞を例示することができる。宿主細胞へのベクター導入は、例えば、リン酸カルシウム沈殿法、電気パルス穿孔法(Current protocols in Molecular Biology edit. Ausubel et al. (1987) Publish. John Wiley & Sons.Section 9.1-9.9)、リポフェクション法、マイクロインジェクション法などの公知の方法で行うことが可能である。
【0027】
宿主細胞において発現したポリペプチドを小胞体の内腔に、細胞周辺腔に、または細胞外の環境に分泌させるために、適当な分泌シグナルを目的のポリペプチドに組み込むことができる。これらのシグナルは目的のポリペプチドに対して内因性であっても、異種シグナルであってもよい。
【0028】
上記製造方法における多重特異性抗体(ポリペプチド)の回収は、本発明のポリペプチドが培地に分泌される場合は、培地を回収する。本発明のポリペプチドが細胞内に産生される場合は、その細胞をまず溶解し、その後にポリペプチドを回収する。
【0029】
組換え細胞培養物から本発明のポリペプチドを回収し精製するには、硫酸アンモニウムまたはエタノール沈殿、酸抽出、アニオンまたはカチオン交換クロマトグラフィー、ホスホセルロースクロマトグラフィー、疎水性相互作用クロマトグラフィー、アフィニティクロマトグラフィー、ヒドロキシルアパタイトクロマトグラフィーおよびレクチンクロマトグラフィーを含めた公知の方法を用いることができる。
【0030】
また本発明は、本発明の多重特異性抗体、および医薬的に許容される担体を含む組成物(薬剤)に関する。
【0031】
本発明において医薬組成物とは、通常、疾患の治療もしくは予防、あるいは検査・診断のための薬剤を言う。
【0032】
本発明の医薬組成物は、当業者に公知の方法で製剤化することが可能である。例えば、水もしくはそれ以外の薬学的に許容し得る液との無菌性溶液、又は懸濁液剤の注射剤の形で非経口的に使用できる。例えば、薬理学上許容される担体もしくは媒体、具体的には、滅菌水や生理食塩水、植物油、乳化剤、懸濁剤、界面活性剤、安定剤、香味剤、賦形剤、ベヒクル、防腐剤、結合剤などと適宜組み合わせて、一般に認められた製薬実施に要求される単位用量形態で混和することによって製剤化することが考えられる。これら製剤における有効成分量は、指示された範囲の適当な容量が得られるように設定する。
【0033】
注射のための無菌組成物は注射用蒸留水のようなベヒクルを用いて通常の製剤実施に従って処方することができる。
【0034】
注射用の水溶液としては、例えば生理食塩水、ブドウ糖やその他の補助薬(例えばD-ソルビトール、D-マンノース、D-マンニトール、塩化ナトリウム)を含む等張液が挙げられる。適当な溶解補助剤、例えばアルコール(エタノール等)、ポリアルコール(プロピレングリコール、ポリエチレングリコール等)、非イオン性界面活性剤(ポリソルベート80(TM)、HCO-50等)を併用してもよい。
【0035】
油性液としてはゴマ油、大豆油があげられ、溶解補助剤として安息香酸ベンジル及び/またはベンジルアルコールを併用してもよい。また、緩衝剤(例えば、リン酸塩緩衝液及び酢酸ナトリウム緩衝液)、無痛化剤(例えば、塩酸プロカイン)、安定剤(例えば、ベンジルアルコール及びフェノール)、酸化防止剤と配合してもよい。調製された注射液は通常、適当なアンプルに充填する。
【0036】
本発明の医薬組成物は、好ましくは非経口投与により投与される。例えば、注射剤型、経鼻投与剤型、経肺投与剤型、経皮投与型の組成物とすることができる。例えば、静脈内注射、筋肉内注射、腹腔内注射、皮下注射などにより全身または局部的に投与することができる。
投与方法は、患者の年齢、症状により適宜選択することができる。抗体または抗体をコードするポリヌクレオチドを含有する医薬組成物の投与量は、例えば、一回につき体重1kgあたり0.0001mgから1000mgの範囲に設定することが可能である。または、例えば、患者あたり0.001〜100000mgの投与量とすることもできるが、本発明はこれらの数値に必ずしも制限されるものではない。投与量及び投与方法は、患者の体重、年齢、症状などにより変動するが、当業者であればそれらの条件を考慮し適当な投与量及び投与方法を設定することが可能である。
【0037】
また、必要に応じ本発明の多重特異性抗体を、その他の医薬成分と組み合わせて製剤化することもできる。
【0038】
また本発明は、本発明の多重特異性抗体を構成するポリペプチドをコードする核酸を提供する。さらに該核酸を担持するベクターもまた、本発明に含まれる。
【0039】
さらに本発明は、上記核酸を有する宿主細胞を提供する。該宿主細胞は、特に制限されず、例えば、大腸菌や種々の動物細胞などを挙げることができる。宿主細胞は、例えば、本発明の抗体もしくはポリペプチドの製造や発現のための産生系として使用することができる。ポリペプチド製造のための産生系には、in vitroおよびin vivoの産生系がある。in vitroの産生系としては、真核細胞を使用する産生系及び原核細胞を使用する産生系が挙げられる。
【0040】
宿主細胞として使用できる真核細胞として、例えば、動物細胞、植物細胞、真菌細胞が挙げられる。動物細胞としては、哺乳類細胞、例えば、CHO(J. Exp. Med. (1995) 108: 945)、COS、HEK293、3T3、ミエローマ、BHK(baby hamster kidney)、HeLa、Vero等、両生類細胞、例えばアフリカツメガエル卵母細胞(Valle et al., Nature (1981) 291: 338-340)、及び昆虫細胞、例えば、Sf9、Sf21、Tn5が例示される。本発明の抗体の発現においては、CHO-DG44、CHO-DX11B、COS7細胞、HEK293細胞、BHK細胞が好適に用いられる。動物細胞において、大量発現を目的とする場合には特にCHO細胞が好ましい。宿主細胞へのベクターの導入は、例えば、リン酸カルシウム法、DEAEデキストラン法、カチオニックリボソームDOTAP(Boehringer Mannheim製)を用いた方法、エレクトロポレーション法、リポフェクションなどの方法で行うことが可能である。
【0041】
植物細胞としては、例えば、ニコチアナ・タバカム(Nicotiana tabacum)由来の細胞およびウキクサ(Lemna minor)が蛋白質生産系として知られており、この細胞をカルス培養する方法により本発明の抗体を産生させることができる。真菌細胞としては、酵母、例えば、サッカロミセス(Saccharomyces)属の細胞(サッカロミセス・セレビシエ(Saccharomyces cerevisiae)、サッカロミセス・ポンベ(Saccharomyces pombe)等)、及び糸状菌、例えば、アスペルギルス(Aspergillus)属の細胞(アスペルギルス・ニガー(Aspergillus niger)等)を用いた蛋白質発現系が公知であり、本発明の抗体産生の宿主として利用できる。
【0042】
原核細胞を使用する場合、細菌細胞を用いる産生系がある。細菌細胞としては、上述の大腸菌(E. coli)に加えて、枯草菌を用いた産生系が知られており、本発明の抗体産生に利用できる。
【0043】
本発明の宿主細胞を用いて抗体を産生する場合、本発明の抗体をコードするポリヌクレオチドを含む発現ベクターにより形質転換された宿主細胞の培養を行い、ポリヌクレオチドを発現させればよい。培養は、公知の方法に従って行うことができる。例えば、動物細胞を宿主とした場合、培養液として、例えば、DMEM、MEM、RPMI1640、IMDMを使用することができる。その際、FBS、牛胎児血清(FCS)等の血清補液を併用しても、無血清培養により細胞を培養してもよい。培養時のpHは、約6〜8とするのが好ましい。培養は、通常、約30〜40℃で約15〜200時間行い、必要に応じて培地の交換、通気、攪拌を加える。
【0044】
一方、in vivoでポリペプチドを産生させる系としては、例えば、動物を使用する産生系や植物を使用する産生系が挙げられる。これらの動物又は植物に目的とするポリヌクレオチドを導入し、動物又は植物の体内でポリペプチドを産生させ、回収する。本発明における「宿主」とは、これらの動物、植物を包含する。
【0045】
動物を使用する場合、哺乳類動物、昆虫を用いる産生系がある。哺乳類動物としては、ヤギ、ブタ、ヒツジ、マウス、ウシ等を用いることができる(Vicki Glaser, SPECTRUM Biotechnology Applications (1993))。また、哺乳類動物を用いる場合、トランスジェニック動物を用いることができる。
【0046】
例えば、本発明の抗体をコードするポリヌクレオチドを、ヤギβカゼインのような乳汁中に固有に産生されるポリペプチドをコードする遺伝子との融合遺伝子として調製する。次いで、この融合遺伝子を含むポリヌクレオチド断片をヤギの胚へ注入し、この胚を雌のヤギへ移植する。胚を受容したヤギから生まれるトランスジェニックヤギ又はその子孫が産生する乳汁から、目的の抗体を得ることができる。トランスジェニックヤギから産生される抗体を含む乳汁量を増加させるために、適宜ホルモンをトランスジェニックヤギに投与してもよい(Ebert et al., Bio/Technology (1994) 12: 699-702)。
【0047】
また、本発明の抗体を産生させる昆虫としては、例えばカイコを用いることができる。カイコを用いる場合、目的の抗体をコードするポリヌクレオチドを挿入したバキュロウィルスをカイコに感染させることにより、このカイコの体液から目的の抗体を得ることができる(Susumu et al., Nature (1985) 315: 592-4)。
【0048】
さらに、植物を本発明の抗体産生に使用する場合、例えばタバコを用いることができる。タバコを用いる場合、目的とする抗体をコードするポリヌクレオチドを植物発現用ベクター、例えばpMON 530に挿入し、このベクターをアグロバクテリウム・ツメファシエンス(Agrobacterium tumefaciens)のようなバクテリアに導入する。このバクテリアをタバコ、例えば、ニコチアナ・タバカム(Nicotiana tabacum)に感染させ、本タバコの葉より所望の抗体を得ることができる(Ma et al., Eur. J. Immunol. (1994) 24: 131-8)。また、同様のバクテリアをウキクサ(Lemna minor)に感染させ、クローン化した後にウキクサの細胞より所望の抗体を得ることができる(Cox KM et al. Nat. Biotechnol. 2006 Dec;24(12):1591-1597)。
【0049】
このようにして得られた抗体は、宿主細胞内または細胞外(培地、乳汁など)から単離し、実質的に純粋で均一な抗体として精製することができる。抗体の分離、精製は、通常のポリペプチドの精製で使用されている分離、精製方法を使用すればよく、何ら限定されるものではない。例えば、クロマトグラフィーカラム、フィルター、限外濾過、塩析、溶媒沈殿、溶媒抽出、蒸留、免疫沈降、SDS-ポリアクリルアミドゲル電気泳動、等電点電気泳動法、透析、再結晶等を適宜選択、組み合わせて抗体を分離、精製することができる。
【0050】
クロマトグラフィーとしては、例えばアフィニティクロマトグラフィー、イオン交換クロマトグラフィー、疎水性クロマトグラフィー、ゲル濾過、逆相クロマトグラフィー、吸着クロマトグラフィー等が挙げられる(Strategies for Protein Purification and Characterization: A Laboratory Course Manual. Ed Daniel R. Marshak et al.(1996) Cold Spring Harbor Laboratory Press)。これらのクロマトグラフィーは、液相クロマトグラフィー、例えばHPLC、FPLC等の液相クロマトグラフィーを用いて行うことができる。アフィニティクロマトグラフィーに用いるカラムとしては、プロテインAカラム、プロテインGカラムが挙げられる。例えば、プロテインAを用いたカラムとして、Hyper D, POROS, Sepharose F. F. (Pharmacia製)等が挙げられる。
【0051】
必要に応じ、抗体の精製前又は精製後に適当なタンパク質修飾酵素を作用させることにより、任意に修飾を加えたり部分的にペプチドを除去することもできる。タンパク質修飾酵素としては、例えば、トリプシン、キモトリプシン、リシルエンドペプチダーゼ、プロテインキナーゼ、グルコシダーゼなどが用いられる。
【0052】
上述のように本発明の宿主細胞を培養し、該細胞培養物からポリペプチドを回収する工程を含む、本発明の多重特異性抗体の製造方法もまた、本発明の好ましい態様の一つである。
【0053】
本発明における「多重特異性抗体」とは、少なくとも2種類の異なる抗原に対して特異的に結合することができる抗体である。本発明の製造方法または精製方法によって得られる好ましい多重特異性抗体として、2つの抗原に対して特異的に結合することができる二重特異性抗体(bispecific antibody;BsAb)(二種特異性抗体と呼ばれる場合もある)を挙げることができる。
【0054】
本発明において、「異なる抗原」とは必ずしも抗原自体が異なる必要はなく、抗原決定基が異なる場合等も本発明の「異なる抗原」に含まれる。従って、例えば、単一分子内の異なる抗原決定基も本発明の異なる抗原に含まれ、このような単一分子内の異なる抗原決定基を各々認識する2つの抗体は、本発明において異なる抗原を認識する抗体として扱われる。
【0055】
本発明における多重特異性抗体は、2種以上の異なる抗原に対して特異性を有する抗体もしくは抗体断片からなる分子である。
【0056】
本発明の上記方法における「核酸の改変」とは、第1のポリペプチドと第2のポリペプチドを標準的なクロマトグラフィーを使用した分析により分離したピークが得られるように、核酸を改変することが含まれる。
【0057】
本発明の方法における、「核酸を改変する」とは、本発明における「改変」によって導入されるアミノ酸残基に対応するように核酸を改変することを言う。より具体的には、元(改変前)のアミノ酸残基をコードする核酸について、改変によって導入されるアミノ酸残基をコードする核酸へ改変することを言う。
【0058】
通常、目的のアミノ酸残基をコードするコドンとなるように、元の核酸に対して、少なくとも1塩基を挿入、欠失または置換するような遺伝子操作もしくは変異処理を行うことを意味する。即ち、元のアミノ酸残基をコードするコドンは、改変によって導入されるアミノ酸残基をコードするコドンによって置換される。このような核酸の改変は、当業者においては公知の技術、例えば、部位特異的変異誘発法、PCR変異導入法等を用いて、適宜実施することが可能である。
【0059】
本発明における改変位置は、例えば、(1)ポリペプチドに表面にあるアミノ酸残基、(2)可変領域、好ましくはFR領域にあるアミノ酸残基、(3)定常領域にあるアミノ酸残基、を挙げることができる。
【0060】
「ポリペプチドの表面にあるアミノ酸」とは、その側鎖が溶媒分子(通常は水分子)に接し得るアミノ酸であり、必ずしも側鎖の全てが溶媒分子に接する必要はなく、側鎖の一部でも溶媒分子に接する場合は、そのアミノ酸は表面にあるアミノ酸である。当業者であれば、市販のソフトウェアを用いたホモロジーモデリング等により、ポリペプチドや抗体のホモロジーモデルを作製することができ、それにより適切な残基を表面にあるアミノ酸として選択することができる。
【0061】
当業者であれば、抗体可変領域における表面アミノ酸はホモロジーモデリング等により作製されたホモロジーモデルにより適宜選択することが可能であるが、例えばH鎖FR領域においてはH1, H3, H5, H8, H10, H12, H13, H15, H16, H19, H23, H25, H26, H39, H42, H43, H44, H46, H68, H71, H72, H73, H75, H76, H81, H82b, H83, H85, H86, H105, H108, H110, H112が表面アミノ酸として例示することができるが、本発明はこれらに限定されることはない。またH鎖CDR領域に関しては、同様にしてホモロジーモデルにより表面アミノ酸を選択することが可能であり、例えばH97はほとんどの抗体で表面に露出している。L鎖FR領域においてはL1, L3, L7, L8, L9, L11, L12, L16, L17, L18, L20, L22, L38, L39, L41, L42, L43, L45, L46, L49, L57, L60, L63, L65, L66, L68, L69, L70, L74, L76, L77, L79, L80, L81, L85, L100, L103, L105, L106, L107, L108が表面アミノ酸として例示することができるが、本発明はこれらに限定されることはない。またL鎖CDR領域に関しては、同様にしてホモロジーモデルにより表面アミノ酸を選択することが可能である。
【0062】
本発明において可変領域にあるアミノ酸残基とは、重鎖可変領域(VH)または軽鎖可変領域(VL)にあるアミノ酸残基が含まれるが、好ましくは、フレームワーク領域(FR)にあるアミノ酸残基である。
【0063】
本発明におけるCDR以外のFR領域において表面に露出するアミノ酸としては、例えば、H10、H12、H23、H39、H43、H105を例示することができるが、これらに制限されない。
【0064】
本発明において、核酸を改変するポリペプチドは、好ましくは、第1のポリペプチドのホモ多量体、第2のポリペプチドのホモ多量体、および第1のポリペプチドと第2のポリペプチドのヘテロ多量体である。例えば、下記実施例に記載のように、第1のポリペプチドのホモ多量体としてヒト化A69-H鎖とヒト化BBA-L鎖のホモダイマー、第2のポリペプチドのホモ多量体としてヒト化B26-H鎖とヒト化BBA-L鎖のホモダイマー、および第1のポリペプチドと第2のポリペプチドのヘテロ多量体としてヒト化A69-H鎖およびヒト化B26-H鎖とヒト化BBA-L鎖のヘテロダイマーを挙げることができるが、これらに制限されない。
【0065】
本発明における標準的なクロマトグラフィーとしては、陽イオン交換クロマトグラフィー、陰イオン交換クロマトグラフィー、疎水クロマトグラフィー、ハイドロキシアパタイトクロマトグラフィー、疎水電荷相互作用クロマトグラフィー、クロマトフォーカシング等が挙げられる。
【0066】
本発明の上記方法において、第1のポリペプチドおよび第2のポリペプチドは、重鎖可変領域(VH)を含んでいることが好ましい。該可変領域には、例えば相補性決定領域(CDR)、フレームワーク領域(FR)が含まれていてもよい。
【0067】
本発明の方法において改変に供されるアミノ酸残基の数は、特に制限されないが、例えば、抗体の可変領域を改変する場合、抗原との結合活性を低下させないために、また抗原性を上げないために、目的のポリペプチドの分離を達成するための必要最低限のアミノ酸残基を改変することが好ましい。
【0068】
また好ましくは、抗原性を上げないために、改変後のアミノ酸配列がヒト配列であることが好ましいが本発明はこれに限定されることはない。さらに、改変後のFR(FR1、FR2、FR3、FR4)が各FRとしてヒト配列になるように、等電点が変化するように導入した改変以外の箇所に変異を導入してもよい。このようにして各FRをヒト配列に置き換える方法は非特許文献(Ono K et al., Mol Immunol. 1999 Apr;36(6):387-395.)で報告されている。また、各FRの等電点を変化させるために、等電点が変化する他のヒトFRに改変してもよい(例えばFR3を等電点が低下する他のヒトFRと交換してもよい)。このようなヒト化方法は非特許文献(Dall'Acqua WF., Methods. 2005 May;36(1):43-60.)で報告されている。
また、僅かな表面電荷の改変において目的のポリペプチドの分離が達成できない場合に、表面電荷の改変とポリペプチドの分離の評価を繰り返し行うことで、所望の多重特異性抗体を取得することが可能である。
【0069】
さらに本発明の上記方法においては、多重特異性抗体が、軽鎖可変領域を含む第3のポリペプチドを含んでいることが好ましい。そして、第1のポリペプチドおよび第2のポリペプチドがそれぞれ第3のポリペプチドと多量体を形成していることが好ましい。
【0070】
さらに本発明の上記方法において、第1のポリペプチドおよび第2のポリペプチドは、重鎖定常領域を含んでいることが好ましい。重鎖定常領域としては、第1のポリペプチドと第2のポリペプチドにpI差が生じるものがより好ましい。そのような重鎖定常領域としては、pI差を有する抗体の重鎖定常領域が挙げられ、元来pIに差のあるIgG1、IgG2、IgG3、IgG4の重鎖定常領域を用いて第1と第2のポリペプチドにpI差を導入することもできるし、第1と第2のポリペプチド中の重鎖定常領域における、これらアイソタイプ間の等電点の違いに起因するアミノ酸のみ、あるいはそれらの等電点には影響しない隣接するアミノ酸を同時に改変することにより非野生型ヒト定常領域を作製し、2つの定常領域にpI差を導入することもできる。定常領域にpI差を導入するための改変箇所としては、例えばH鎖定常領域のEUナンバリングで、H鎖137番目、196番目、203番目、214番目、217番目、233番目、268番目、274番目、276番目、297番目、355番目、392番目、419番目、435番目が挙げられる。
また、重鎖定常領域の糖鎖を除去することによりpI差が生じることから、糖鎖付加部位の297番目もpI鎖を導入するための改変箇所として挙げられる。
また本発明には、上記第1のポリペプチドおよび第2のポリペプチドが重鎖定常領域を含む方法に対して、上述の第1のポリペプチドおよび第2のポリペプチドが重鎖可変領域を含む方法、および/または前記多重特異性抗体が軽鎖可変領域を含む第3のポリペプチドを含み、前記第1のポリペプチドおよび前記第2のポリペプチドがそれぞれ該第3のポリペプチドと多量体を形成する方法とを組み合わせた方法も含まれる。
【0071】
さらに上記方法によって製造される多重特異性抗体も本発明に含まれる。
【0072】
さらに本発明によって提供される多重特異性抗体における第1のポリペプチドが重鎖可変領域および/または重鎖定常領域を含んでいる場合には、上記「等電点に差がつくよう」にするために、例えば該重鎖可変領域におけるKabatナンバリングによる10位、12位、23位、39位、43位および105位のアミノ酸残基、若しくは、該重鎖定常領域におけるEUナンバリングによる137位、196位、203位、214位、217位、233位、268位、274位、276位、297位、355位、392位、419位、435位のアミノ酸残基から選ばれる、少なくとも1つのアミノ酸残基が電荷を有するようにする態様を挙げることができる。上記のナンバリングで示された第1のポリペプチドのアミノ酸残基のうち、当該電荷を有するアミノ酸残基以外のアミノ酸残基は、第1のポリペプチドと第2のポリペプチドの等電点に差がついていれば、当該電荷を有するアミノ酸残基と同種の電荷であっても良いし、電荷を有していなくても、反対の電荷であっても良い。
【0073】
本発明の上記多重特異性抗体は、好ましくは、第2のポリペプチドが、第1のポリペプチドの電荷を有するアミノ酸残基とは反対の電荷を有する、または電荷を有しないことを特徴とする。詳しくは、第2のポリペプチドが重鎖可変領域および/または重鎖定常領域を含み、該重鎖可変領域におけるKabatナンバリングによる10位、12位、23位、39位、43位および105位のアミノ酸残基、若しくは、該重鎖定常領域におけるEUナンバリングによる137位、196位、203位、214位、217位、233位、268位、274位、276位、297位、355位、392位、419位、435位のアミノ酸残基から選ばれる、少なくとも1つのアミノ酸残基が、前記第1のポリペプチドに含まれる重鎖可変領域および/または重鎖定常領域において選ばれる、電荷を有するようにするアミノ酸残基とは反対の電荷を有する、または電荷を有しない、多重特異性抗体である。上記のナンバリングで示された第2のポリペプチドのアミノ酸残基のうち、当該電荷を有するアミノ酸残基以外のアミノ酸残基は、第1のポリペプチドと第2のポリペプチドの等電点に差がついていれば、当該電荷を有するアミノ酸残基と同種の電荷であっても良いし、電荷を有していなくても、反対の電荷であっても良い。
等電点を低下させるためには、例えば、137位はIgG2またはIgG4の配列、196位はIgG1またはIgG2またはIgG4の配列、203位はIgG2またはIgG4の配列、214位はIgG2の配列、217位はIgG1またはIgG3またはIgG4の配列、233位はIgG1またはIgG3またはIgG4の配列、268位はIgG4の配列、274位はIgG2またはIgG3またはIgG4の配列、276位はIgG1またはIgG2またはIgG4の配列、355位はIgG4の配列、392位はIgG3の配列、419位はIgG4の配列、435位はIgG1またはIgG2またはIgG4の配列、を適用することが望ましい。また、等電点を上昇させるためには、例えば、137位はIgG1またはIgG3の配列、196位はIgG3の配列、203位はIgG1またはIgG3の配列、214位はIgG1またはIgG3またはIgG4の配列、217位はIgG2の配列、233位はIgG2の配列、268位はIgG1またはIgG2またはIgG3の配列、274位はIgG1の配列、276位はIgG3の配列、355位はIgG1またはIgG2またはIgG3の配列、392位はIgG1またはIgG2またはIgG4の配列、419位はIgG1またはIgG2またはIgG3の配列、435位はIgG3の配列、を適用することが望ましい。
これらの配列の適用は、両H鎖に十分な等電点の差が付くようであればよく、必ずしも全ての配列を適用する必要はない。
【0074】
アミノ酸の中には、電荷を帯びたアミノ酸が知られている。一般的に正の電荷を帯びたアミノ酸(正電荷アミノ酸)としては、リジン(K)、アルギニン(R)、ヒスチジン(H)が知られている。負の電荷を帯びたアミノ酸(負電荷アミノ酸)としては、アスパラギン酸(D)、グルタミン酸(E)等が知られている。
【0075】
上記「電荷を有するアミノ酸残基」は、好ましくは、以下の(a)または(b)いずれかの群に含まれるアミノ酸残基から適宜選択されるが、特に制限されない。
(a)グルタミン酸(E)、アスパラギン酸(D)
(b)リジン(K)、アルギニン(R)、ヒスチジン(H)
【0076】
上記抗体において、「同種の電荷を有する」とは、例えば、重鎖可変領域における上記Kabatナンバリングによるアミノ酸残基、若しくは重鎖定常領域における上記EUナンバリングによるアミノ酸残基のいずれもが、上記(a)または(b)のいずれか1の群に含まれるアミノ酸残基を有することを意味する。
【0077】
また、「反対の電荷を有する」とは、例えば、重鎖可変領域および/または重鎖定常領域を有する第2のポリペプチドにおける上記Kabatナンバリング若しくは上記EUナンバリングによるアミノ酸残基のなかの少なくとも1つのアミノ酸残基が、第1のポリペプチドに含まれる重鎖可変領域および/または重鎖定常領域における対応する位置のアミノ酸残基であって、上記(a)または(b)のいずれか1の群に含まれるアミノ酸残基を有する場合に、残りのアミノ酸残基が異なる群に含まれるアミノ酸残基を有することを意味する。
【0078】
即ち本発明においては、前記同種の電荷を有するアミノ酸残基が、上記(a)または(b)のいずれかの群に含まれるアミノ酸残基から選択される多重特異性抗体を提供する。
【0079】
なお、元の(改変前の)アミノ酸残基が既に電荷を有する場合、電荷を有さないアミノ酸残基となるように改変することも本発明の好ましい態様の一つである。
【0080】
本発明においては、第1のポリペプチドと第2のポリペプチドの等電点(pI)に差が出るように、アミノ酸残基が改変されることが好ましい。また、改変によって導入されるアミノ酸残基が複数の場合、これらアミノ酸残基の中に電荷を持たないアミノ酸残基が少数程度含まれていてもよい。
【0081】
さらに本発明は、第1のポリペプチドの可変領域が以下の(a1)〜(a7)のいずれかに記載のアミノ酸配列からなり、第2のポリペプチドの可変領域が以下の(b1)〜(b3)のいずれかに記載のアミノ酸配列からなり、第3のポリペプチドの可変領域が以下の(c1)または(c2)に記載のアミノ酸配列からなる、多重特異性抗体を提供する。
(a1)配列番号:7
(a2)配列番号:8
(a3)配列番号:9
(a4)配列番号:10
(a5)配列番号:11
(a6)配列番号:12
(a7)配列番号:13
(b1)配列番号:14
(b2)配列番号:15
(b3)配列番号:16
(c1)配列番号:17
(c2)配列番号:18
なお上記のアミノ酸配列は、本発明において改変に供するアミノ酸をより具体的に例示するためのものであり、可変領域がこれらのアミノ酸である場合に限定されない。
【0082】
上記多重特異性抗体の好ましい態様の一つとして、例えば、第1のポリペプチドの可変領域が配列番号:11に記載のアミノ酸配列からなり、第2のポリペプチドの可変領域が配列番号:16に記載のアミノ酸配列からなり、第3のポリペプチドの可変領域が配列番号:17に記載のアミノ酸配列からなる多重特異性抗体を挙げることができる。
さらに別の好ましい態様の一つとして、例えば、第1のポリペプチドの可変領域が配列番号:12に記載のアミノ酸配列からなり、第2のポリペプチドの可変領域が配列番号:16に記載のアミノ酸配列からなり、第3のポリペプチドの可変領域が配列番号:18に記載のアミノ酸配列からなる多重特異性抗体を挙げることができる。
【0083】
さらに上記多重特異性抗体の好ましい態様の一つとして、第1のポリペプチドおよび第2のポリペプチドがヒトIgG4定常領域を含み、第3のポリペプチドがヒトκ定常領域を含んでいる多重特異性抗体を挙げることができる。
【0084】
本発明において、「抗体」という用語は最も広い意味で使用され、所望の生物学的活性を示す限り、モノクローナル抗体、ポリクローナル抗体、抗体変異体(キメラ抗体、ヒト化抗体、低分子化抗体(抗体断片も含む)、多重特異性抗体等)が含まれる。本発明においては、これら抗体の取得(作成)の際に、好適に本発明の抗体改変方法を用いることができる。
【0085】
本発明における「抗体」には、上述のようにアミノ酸残基の電荷を改変した抗体に対して、さらにアミノ酸の置換、欠失、付加及び/若しくは挿入等により、アミノ酸配列が改変された抗体が含まれる。また、アミノ酸の置換、欠失、付加及び/若しくは挿入、またはキメラ化やヒト化等により、アミノ酸配列が改変された抗体に対して、さらに、アミノ酸残基の電荷が改変された抗体が含まれる。すなわち、マウス抗体をヒト化する工程と同時に改変してもよく、あるいは、ヒト化抗体をさらに改変することであってもよい。
【0086】
アミノ酸の置換、欠失、付加及び/又は挿入、並びにヒト化、キメラ化などのアミノ酸配列の改変は、当業者に公知の方法により行うことが可能である。同様に、本発明における抗体を組換え抗体として作製する際に利用する抗体の可変領域及び定常領域も、アミノ酸の置換、欠失、付加及び/若しくは挿入、またはキメラ化やヒト化等によりそのアミノ酸配列を改変してもよい。
【0087】
本発明における抗体はマウス抗体、ヒト抗体、ラット抗体、ウサギ抗体、ヤギ抗体、ラクダ抗体など、どのような動物由来の抗体でもよい。さらに、例えば、キメラ抗体、中でもヒト化抗体などのアミノ酸配列を置換した改変抗体でもよい。また、各種分子を結合させた抗体修飾物、抗体断片、低分子抗体などいかなる抗体であってもよい。
【0088】
「キメラ抗体」とは、異なる動物由来の配列を組合わせて作製される抗体である。例えば、マウス抗体の重鎖、軽鎖の可変(V)領域とヒト抗体の重鎖、軽鎖の定常(C)領域からなる抗体を例示することができる。キメラ抗体の作製は公知であり、例えば、抗体V領域をコードするDNAをヒト抗体C領域をコードするDNAと連結し、これを発現ベクターに組み込んで宿主に導入し産生させることによりキメラ抗体を得ることができる。
【0089】
「ヒト化抗体」とは、再構成(reshaped)ヒト抗体とも称される、ヒト以外の哺乳動物由来の抗体、例えばマウス抗体の相補性決定領域(CDR;complementarity determining region)をヒト抗体のCDRへ移植したものである。CDRを同定するための方法は公知である(Kabat et al., Sequence of Proteins of Immunological Interest (1987), National Institute of Health, Bethesda, Md.; Chothia et al., Nature (1989) 342: 877)。また、その一般的な遺伝子組換え手法も公知である(欧州特許出願公開番号EP 125023号公報、WO 96/02576 号公報参照)。そこで公知の方法により、例えば、マウス抗体のCDRを決定し、該CDRとヒト抗体のフレームワーク領域(framework region;FR)とが連結された抗体をコードするDNAを取得し、ヒト化抗体を通常の発現ベクターを用いた系により産生することができる。このようなDNAは、CDR及びFR両方の末端領域にオーバーラップする部分を有するように作製した数個のオリゴヌクレオチドをプライマーとして用いてPCR法により合成することができる(WO98/13388号公報に記載の方法を参照)。CDRを介して連結されるヒト抗体のFRは、CDRが良好な抗原結合部位を形成するように選択される。必要に応じ、再構成ヒト抗体のCDRが適切な抗原結合部位を形成するように、抗体の可変領域におけるFRのアミノ酸を改変してもよい(Sato et al., Cancer Res. (1993) 53: 851-6)。改変できるFR中のアミノ酸残基には、抗原に直接、非共有結合により結合する部分(Amit et al., Science (1986) 233: 747-53)、CDR構造に影響または作用する部分(Chothia et al., J. Mol. Biol. (1987) 196: 901-17)及びVH-VL相互作用に関連する部分(EP239400号特許公報)が含まれる。
【0090】
本発明における抗体がキメラ抗体またはヒト化抗体である場合には、これらの抗体のC領域は,好ましくはヒト抗体由来のものが使用される。例えばH鎖では、Cγ1、Cγ2、Cγ3、Cγ4を、L鎖ではCκ、Cλを使用することができる。また、抗体またはその産生の安定性を改善するために、ヒト抗体C領域を必要に応じ修飾してもよい。本発明におけるキメラ抗体は、好ましくはヒト以外の哺乳動物由来抗体の可変領域とヒト抗体由来の定常領域とからなる。一方、ヒト化抗体は、好ましくはヒト以外の哺乳動物由来抗体のCDRと、ヒト抗体由来のFRおよびC領域とからなる。ヒト抗体由来の定常領域は、IgG(IgG1、IgG2、IgG3、IgG4)、IgM、IgA、IgD及びIgE等のアイソタイプごとに固有のアミノ酸配列を有する。本発明におけるヒト化抗体に用いられる定常領域は、どのアイソタイプに属する抗体の定常領域であってもよい。好ましくは、ヒトIgGの定常領域が用いられるが、これに限定されるものではない。また、ヒト化抗体に利用されるヒト抗体由来のFRも特に限定されず、どのアイソタイプに属する抗体のものであってもよい。
【0091】
本発明におけるキメラ抗体及びヒト化抗体の可変領域及び定常領域は、元の抗体の結合特異性を示す限り、欠失、置換、挿入及び/または付加等により改変されていてもよい。
【0092】
ヒト由来の配列を利用したキメラ抗体及びヒト化抗体は、ヒト体内における抗原性が低下しているため、治療目的などでヒトに投与する場合に有用と考えられる。
【0093】
また、低分子化抗体は、体内動態の性質の面からも、大腸菌、植物細胞等を用いて低コストで製造できる点からも抗体として有用である。
【0094】
抗体断片は低分子化抗体の一種である。また、低分子化抗体は、抗体断片をその構造の一部とする抗体も含む。本発明における低分子化抗体は、抗原への結合能を有していれば特にその構造、製造法等は限定されない。低分子化抗体の中には、全長抗体よりも高い活性を有する抗体も存在する(Orita et al., Blood(2005) 105: 562-566)。本明細書において、「抗体断片」とは、全長抗体(whole antibody、例えばwhole IgG等)の一部分であれば特に限定されないが、重鎖可変領域(VH)又は軽鎖可変領域(VL)を含んでいることが好ましい。好ましい抗体断片の例としては、例えば、Fab、F(ab')2、Fab'、Fvなどを挙げることができる。抗体断片中の、VHまたはVLのアミノ酸配列は、置換、欠失、付加及び/又は挿入により改変されていてもよい。さらに抗原への結合能を保持する限り、VH及びVLの一部を欠損させてもよい。例えば、前述の抗体断片のうち「Fv」は、完全な抗原認識部位と結合部位を含む最小の抗体断片である。「Fv」は、1つのVHおよび1つのVLが非共有結合により強く結合したダイマー(VH-VLダイマー)である。各可変領域の3つの相補鎖決定領域(complementarity determining region;CDR)によって、VH-VLダイマーの表面に抗原結合部位を形成する。6つのCDRが抗体に抗原結合部位を付与している。しかしながら、1つの可変領域(または、抗原に特異的な3つのCDRのみを含むFvの半分)であっても、全結合部位よりも親和性は低いが、抗原を認識し、結合する能力を有する。従って、このようなFvより小さい分子も本発明における抗体断片に含まれる。又、抗体断片の可変領域はキメラ化やヒト化されていてもよい。
【0095】
低分子化抗体は、VHとVLの両方を含んでいることが好ましい。低分子化抗体の例としては、Fab、Fab'、F(ab')2及びFv等の抗体断片、並びに、抗体断片を利用して作製され得るscFv(シングルチェインFv)(Huston et al., Proc. Natl. Acad. Sci. USA (1988) 85: 5879-83; Pluckthun「The Pharmacology of Monoclonal Antibodies」Vol.113, Resenburg 及び Moore編, Springer Verlag, New York, pp.269-315, (1994))、Diabody(Holliger et al., Proc. Natl. Acad. Sci. USA (1993) 90: 6444-8; EP404097号; WO93/11161号; Johnson et al., Method in Enzymology (1991) 203: 88-98; Holliger et al., Protein Engineering (1996) 9: 299-305; Perisic et al., Structure (1994) 2: 1217-26; John et al., Protein Engineering (1999) 12(7): 597-604; Atwell et al., Mol.Immunol. (1996) 33: 1301-12)、sc(Fv)2(Hudson et al、J Immunol. Methods (1999) 231: 177-89 ; Orita et al., Blood(2005) 105: 562-566)、Triabody(Journal of Immunological Methods (1999) 231: 177-89)、及びTandem Diabody(Cancer Research (2000) 60: 4336-41)等を挙げることができる。
【0096】
抗体断片は、抗体を酵素、例えばパパイン、ペプシン等のプロテアーゼにより処理して得ることができる(Morimoto et al., J. Biochem. Biophys. Methods (1992) 24: 107-17; Brennan et al., Science (1985) 229: 81参照)。また、該抗体断片のアミノ酸配列を基に、遺伝子組換えにより製造することもできる。
【0097】
抗体断片を改変した構造を有する低分子化抗体は、酵素処理若しくは遺伝子組換えにより得られた抗体断片を利用して構築することができる。又は、低分子化抗体全体をコードする遺伝子を構築し、これを発現ベクターに導入した後、適当な宿主細胞で発現させることもできる(例えば、Co et al., J. Immunol. (1994) 152: 2968-76; Better and Horwitz, Methods Enzymol. (1989) 178: 476-96; Pluckthun and Skerra, Methods Enzymol. (1989) 178: 497-515; Lamoyi, Methods Enzymol. (1986) 121: 652-63; Rousseaux et al., Methods Enzymol. (1986) 121: 663-9; Bird and Walker, Trends Biotechnol. (1991) 9: 132-7参照)。
【0098】
また、上記「scFv」は、2つの可変領域を、必要に応じリンカー等を介して、結合させた一本鎖ポリペプチドである。scFvに含まれる2つの可変領域は、通常、1つのVHと1つのVLであるが、2つのVH又は2つのVLであってもよい。一般にscFvポリペプチドは、VH及びVLドメインの間にリンカーを含み、それにより抗原結合のために必要なVH及びVLの対部分が形成される。通常、同じ分子内でVH及びVLの間で対部分を形成させるために、一般に、VH及びVLを連結するリンカーを10アミノ酸以上の長さのぺプチドリンカーとする。しかしながら、本発明におけるscFvのリンカーは、scFvの形成を妨げない限り、このようなペプチドリンカーに限定されるものではない。scFvの総説として、Pluckthun『The Pharmacology of Monoclonal Antibody』Vol.113(Rosenburg and Moore ed., Springer Verlag, NY, pp.269-315 (1994))を参照することができる。
【0099】
また、「ダイアボディ(diabody; Db)」は、遺伝子融合により構築された二価(bivalent)の抗体断片を指す(P.Holliger et al., Proc.Natl.Acad.Sci.USA 90: 6444-6448 (1993)、EP404,097号、WO93/11161号等)。ダイアボディは、2本のポリペプチド鎖から構成されるダイマーであり、ポリペプチド鎖は各々、同じ鎖中で軽鎖可変領域(VL)及び重鎖可変領域(VH)が、互いに結合できない位に短い、例えば、5残基程度のリンカーにより結合されている。同一ポリペプチド鎖上にコードされるVLとVHとは、その間のリンカーが短いため単鎖V領域フラグメントを形成することが出来ず二量体を形成するため、ダイアボディは2つの抗原結合部位を有することとなる。このとき2つの異なるエピトープ(a、b)に対するVLとVHをVLa-VHbとVLb-VHaの組合わせで5残基程度のリンカーで結んだものを同時に発現させると二重特異性Dbとして分泌される。
【0100】
Diabodyは、2分子のscFvを含むことから、4つの可変領域を含み、その結果、2つの抗原結合部位を持つこととなる。ダイマーを形成させないscFvの場合と異なり、Diabodyの形成を目的とする場合、通常、各scFv分子内のVH及びVL間を結ぶリンカーは、ペプチドリンカーとする場合には、5アミノ酸前後のものとする。しかしながら、Diabodyを形成するscFvのリンカーは、scFvの発現を妨げず、Diabodyの形成を妨げない限り、このようなペプチドリンカーに限定されない。
【0101】
本発明においてさらに好ましくは、多重特異性抗体として二重特異性抗体を挙げることができる。
なお、上記「二重特異性抗体」は、例えば、重鎖可変領域および軽鎖可変領域が1本鎖として連結した構造の抗体(例えば、sc(Fv)2)であってもよい。また重鎖可変領域(VH)および軽鎖可変領域(VL)が連結したscFv(あるいはsc(Fv)2)をFc領域(CH1ドメインを欠いた定常領域)と結合した抗体様分子(例えば、scFv-Fc)であってもよい。scFv-Fcからなる多重特異性抗体は第1のポリペプチドがVH1-linker-VL1-Fcであり、第2のポリペプチドがVH2-linker-VL2-Fcからなる(scFv)2-Fc型の構造をもつ。あるいはsingle domain antibodyをFc領域と結合させた抗体様分子であってもよい(Curr Opin Drug Discov Devel. 2006 , 9(2), 184-93)。
【0102】
本発明の方法における変異導入前の抗体(本明細書においては、単に「本発明の抗体」と記載する場合あり)のH鎖又はL鎖をコードする遺伝子は既知の配列を用いることも可能であり、又、当業者に公知の方法で取得することもできる。例えば、抗体ライブラリーから取得することも可能であるし、モノクローナル抗体を産生するハイブリドーマから抗体をコードする遺伝子をクローニングして取得することも可能である。
【0103】
抗体ライブラリーについては既に多くの抗体ライブラリーが公知になっており、又、抗体ライブラリーの作製方法も公知であるので、当業者は適宜抗体ライブラリーを入手することが可能である。例えば、抗体ファージライブラリーについては、Clackson et al., Nature 1991, 352: 624-8、Marks et al., J. Mol. Biol. 1991, 222: 581-97、Waterhouses et al., Nucleic Acids Res. 1993, 21: 2265-6、Griffiths et al., EMBO J. 1994, 13: 3245-60、Vaughan et al., Nature Biotechnology 1996, 14: 309-14、及び特表平20−504970号公報等の文献を参照することができる。その他、真核細胞をライブラリーとする方法(WO95/15393号パンフレット)やリボソーム提示法等の公知の方法を用いることが可能である。さらに、ヒト抗体ライブラリーを用いて、パンニングによりヒト抗体を取得する技術も知られている。例えば、ヒト抗体の可変領域を一本鎖抗体(scFv)としてファージディスプレイ法によりファージの表面に発現させ、抗原に結合するファージを選択することができる。選択されたファージの遺伝子を解析すれば、抗原に結合するヒト抗体の可変領域をコードするDNA配列を決定することができる。抗原に結合するscFvのDNA配列が明らかになれば、当該配列を元に適当な発現ベクターを作製し、ヒト抗体を取得することができる。これらの方法は既に周知であり、WO92/01047、WO92/20791、WO93/06213、WO93/11236、WO93/19172、WO95/01438、WO95/15388を参考にすることができる。
【0104】
ハイブリドーマから抗体をコードする遺伝子を取得する方法は、基本的には公知技術を使用し、所望の抗原または所望の抗原を発現する細胞を感作抗原として使用して、これを通常の免疫方法にしたがって免疫し、得られる免疫細胞を通常の細胞融合法によって公知の親細胞と融合させ、通常のスクリーニング法により、モノクローナルな抗体産生細胞(ハイブリドーマ)をスクリーニングし、得られたハイブリドーマのmRNAから逆転写酵素を用いて抗体の可変領域(V領域)のcDNAを合成し、これを所望の抗体定常領域(C領域)をコードするDNAと連結することにより得ることができる。
【0105】
より具体的には、特に以下の例示に限定される訳ではないが、上記のH鎖及びL鎖をコードする抗体遺伝子を得るための感作抗原は、免疫原性を有する完全抗原と、免疫原性を示さないハプテン等を含む不完全抗原の両方を含む。例えば、目的タンパク質の全長タンパク質、又は部分ペプチドなどを用いることができる。その他、多糖類、核酸、脂質等から構成される物質が抗原となり得ることが知られており、本発明の抗体の抗原は特に限定されるものではない。抗原の調製は、当業者に公知の方法により行うことができ、例えば、バキュロウィルスを用いた方法(例えば、WO98/46777など)などに準じて行うことができる。ハイブリドーマの作製は、たとえば、ミルステインらの方法(G. Kohler and C. Milstein, Methods Enzymol. 1981, 73: 3-46)等に準じて行うことができる。抗原の免疫原性が低い場合には、アルブミン等の免疫原性を有する巨大分子と結合させ、免疫を行えばよい。また、必要に応じ抗原を他の分子と結合させることにより可溶性抗原とすることもできる。受容体のような膜貫通分子を抗原として用いる場合、受容体の細胞外領域部分を断片として用いたり、膜貫通分子を細胞表面上に発現する細胞を免疫原として使用することも可能である。
【0106】
抗体産生細胞は、上述の適当な感作抗原を用いて動物を免疫化することにより得ることができる。または、抗体を産生し得るリンパ球をin vitroで免疫化して抗体産生細胞とすることもできる。免疫化する動物としては、各種哺乳動物を使用できるが、ゲッ歯目、ウサギ目、霊長目の動物が一般的に用いられる。マウス、ラット、ハムスター等のゲッ歯目、ウサギ等のウサギ目、カニクイザル、アカゲザル、マントヒヒ、チンパンジー等のサル等の霊長目の動物を例示することができる。その他、ヒト抗体遺伝子のレパートリーを有するトランスジェニック動物も知られており、このような動物を使用することによりヒト抗体を得ることもできる(WO96/34096; Mendez et al., Nat. Genet. 1997, 15: 146-56参照)。このようなトランスジェニック動物の使用に代えて、例えば、ヒトリンパ球をin vitroで所望の抗原または所望の抗原を発現する細胞で感作し、感作リンパ球をヒトミエローマ細胞、例えばU266と融合させることにより、抗原への結合活性を有する所望のヒト抗体を得ることもできる(特公平1-59878号公報参照)。また、ヒト抗体遺伝子の全てのレパートリーを有するトランスジェニック動物を所望の抗原で免疫することで所望のヒト抗体を取得することができる(WO93/12227、WO92/03918、WO94/02602、WO96/34096、WO96/33735参照)。
【0107】
動物の免疫化は、例えば、感作抗原をPhosphate-Buffered Saline(PBS)または生理食塩水等で適宜希釈、懸濁し、必要に応じてアジュバントを混合して乳化した後、動物の腹腔内または皮下に注射することにより行われる。その後、好ましくは、フロイント不完全アジュバントに混合した感作抗原を4〜21日毎に数回投与する。抗体の産生の確認は、動物の血清中の目的とする抗体力価を慣用の方法により測定することにより行われ得る。
【0108】
ハイブリドーマは、所望の抗原で免疫化した動物またはリンパ球より得られた抗体産生細胞を、慣用の融合剤(例えば、ポリエチレングリコール)を使用してミエローマ細胞と融合して作成することができる(Goding, Monoclonal Antibodies: Principles and Practice, Academic Press, 1986, 59-103)。必要に応じハイブリドーマ細胞を培養・増殖し、免疫沈降、放射免疫分析(RIA)、酵素結合免疫吸着分析(ELISA)等の公知の分析法により該ハイブリドーマより産生される抗体の結合特異性を測定する。その後、必要に応じ、目的とする特異性、親和性または活性が測定された抗体を産生するハイブリドーマを限界希釈法等の手法によりサブクローニングすることもできる。
【0109】
続いて、選択された抗体をコードする遺伝子をハイブリドーマまたは抗体産生細胞(感作リンパ球等)から、抗体に特異的に結合し得るプローブ(例えば、抗体定常領域をコードする配列に相補的なオリゴヌクレオチド等)を用いてクローニングすることができる。また、mRNAからRT-PCRによりクローニングすることも可能である。免疫グロブリンは、IgA、IgD、IgE、IgG及びIgMの5つの異なるクラスに分類される。さらに、これらのクラスは幾つかのサブクラス(アイソタイプ)(例えば、IgG-1、IgG-2、IgG-3、及びIgG-4;IgA-1及びIgA-2等)に分けられる。本発明において抗体の製造に使用するH鎖及びL鎖は、これらいずれのクラス及びアイソタイプに属する抗体に由来するものであってもよく、特に限定されないが、IgGは特に好ましいものである。
【0110】
ここで、H鎖及びL鎖をコードする遺伝子を遺伝子工学的手法により改変することも可能である。例えば、マウス抗体、ラット抗体、ウサギ抗体、ハムスター抗体、ヒツジ抗体、ラクダ抗体等の抗体について、ヒトに対する異種抗原性を低下させること等を目的として人為的に改変した遺伝子組換え型抗体、例えば、キメラ抗体、ヒト化抗体等を適宜作製することができる。キメラ抗体は、ヒト以外の哺乳動物、例えば、マウス抗体のH鎖、L鎖の可変領域とヒト抗体のH鎖、L鎖の定常領域からなる抗体であり、マウス抗体の可変領域をコードするDNAをヒト抗体の定常領域をコードするDNAと連結し、これを発現ベクターに組み込んで宿主に導入し産生させることにより得ることができる。ヒト化抗体は、再構成(reshaped)ヒト抗体とも称され、ヒト以外の哺乳動物、たとえばマウス抗体の相補性決定領域(CDR; complementary determining region) を連結するように設計したDNA配列を、末端部にオーバーラップする部分を有するように作製した数個のオリゴヌクレオチドからPCR法により合成する。得られたDNAをヒト抗体定常領域をコードするDNAと連結し、次いで発現ベクターに組み込んで、これを宿主に導入し産生させることにより得られる(EP239400; WO96/02576参照)。CDRを介して連結されるヒト抗体のFRは、相補性決定領域が良好な抗原結合部位を形成するものが選択される。必要に応じ、再構成ヒト抗体の相補性決定領域が適切な抗原結合部位を形成するように抗体の可変領域のフレームワーク領域のアミノ酸を置換してもよい(K. Sato et al., Cancer Res. 1993, 53: 851-856)。
【0111】
上述のヒト化以外に、例えば、抗原との結合性等の抗体の生物学的特性を改善するために改変を行うことも考えられる。本発明における改変は、部位特異的突然変異(例えば、Kunkel (1985) Proc. Natl. Acad. Sci. USA 82: 488参照)、PCR変異、カセット変異等の方法により行うことができる。一般に、生物学的特性の改善された抗体変異体は70%以上、より好ましくは80%以上、さらに好ましくは90%以上(例えば、95%以上、97%、98%、99%等)のアミノ酸配列相同性及び/または類似性を元となった抗体の可変領域のアミノ酸配列に対して有する。本明細書において、配列の相同性及び/または類似性は、配列相同性が最大の値を取るように必要に応じ配列を整列化、及びギャップ導入した後、元となった抗体残基と相同(同じ残基)または類似(一般的なアミノ酸の側鎖の特性に基き同じグループに分類されるアミノ酸残基)するアミノ酸残基の割合として定義される。通常、天然のアミノ酸残基は、その側鎖の性質に基づいて(1)疎水性:アラニン、イソロイシン、バリン、メチオニン及びロイシン;(2)中性親水性:アスパラギン、グルタミン、システイン、スレオニン及びセリン;(3)酸性:アスパラギン酸及びグルタミン酸;(4)塩基性:アルギニン、ヒスチジン及びリジン;(5)鎖の配向に影響する残基:グリシンおよびプロリン;ならびに(6)芳香族性:チロシン、トリプトファン及びフェニルアラニンのグループに分類される。
【0112】
通常、H鎖及びL鎖の可変領域中に存在する全部で6つの相補性決定領域(超可変部;CDR)が相互作用し、抗体の抗原結合部位を形成している。このうち1つの可変領域であっても全結合部位を含むものよりは低い親和性となるものの、抗原を認識し、結合する能力があることが知られている。従って、本発明のH鎖及びL鎖をコードする抗体遺伝子は、該遺伝子によりコードされるポリペプチドが所望の抗原との結合性を維持していればよく、H鎖及びL鎖の各々の抗原結合部位を含む断片部分をコードしていればよい。
本発明の方法によって、上述のように、例えば、所望の、実際に活性を保持する、二重特異性抗体を効率的に取得することができる。
【0113】
重鎖可変領域は、上述のように、通常3つのCDR領域と4つのFR領域によって構成されている。本発明の好ましい態様において「改変」に供するアミノ酸残基としては、例えば、CDR領域あるいはFR領域に位置するアミノ酸残基の中から適宜選択することができる。一般的にCDR領域のアミノ酸残基の改変は、抗原に対する結合能を低下させる場合がある。従って、本発明において「改変」に供するアミノ酸残基としては、特に限定されるものではないが、FR領域に位置するアミノ酸残基の中から適宜選択することが好ましい。
【0114】
また、ヒトもしくはマウス等の生物において、抗体の可変領域のFRとして利用可能な配列を、当業者であれば、公共のデータベース等を利用して適宜取得することができる。より具体的には、後述の実施例に記載の手段にて、FR領域のアミノ酸配列情報を取得することが可能である。
なお本明細書において引用されたすべての先行技術文献は、参照として本明細書に組み入れられる。
【実施例】
【0115】
以下、本発明を実施例により具体的に説明するが、本発明はこれら実施例に制限されるものではない。
【0116】
〔実施例1〕ハイブリッドL鎖を持つ二重特異性抗体のヒト化
特願2005-112514において血液凝固時間の短縮効果が最も高かった抗FactorIXa抗体 A69-VH、抗FactorX抗体 B26-VH、ハイブリッドL鎖(BBA)の組み合わせから成る二重特異性抗体について、以下のようにヒト化を実施した。
【0117】
1−1.ヒト抗体の相同性検索
一般公開されているKabat Database (ftp://ftp.ebi.ac.uk/pub/databases/kabat/) およびIMGT Database (http://imgt.cines.fr/)よりヒト抗体アミノ酸配列データを入手し、構築したデータベースを用いてマウスA69-H鎖可変領域(アミノ酸配列:配列番号:19)、マウスB26-H鎖可変領域(アミノ酸配列:配列番号:20)、マウスBBA-L鎖可変領域(アミノ酸配列:配列番号:21)に分けてホモロジー検索を行った。その結果、以下に示すヒト抗体配列と高い相同性を持つことが確認されたことから、ヒト化抗体のフレームワーク領域(以下、FR)に使用することにした。
【0118】
(1)A69-H鎖可変領域:KABATID-000064 (Kabat Database)
(Kippsら、J Clin Invest. 1991;87:2087-2096)
(2)B26-H鎖可変領域:EMBL Accession No. AB063872(IMGT Database)
(Unpublished data)
(3)BBA-L鎖可変領域:KABATID-024300 (Kabat Database)
(Welschofら、J Immunol Method. 1995;179:203-214)
【0119】
(1)-(3)のヒト抗体のFRに各マウス抗体の相補性抗原決定領域(以下、CDR)を移植したヒト化抗体を作製した。
【0120】
また、NCBIより一般公開されている相同性検索Web site(http://www.ncbi. nlm.nih.gov/BLAST/)を使用して、(4)-(6)のヒト抗体に相同性の高いヒト抗体の分泌シグナル配列を検索した。検索により得られた以下に示す分泌シグナル配列を使用した。
(4)A69-H鎖可変領域:GenBank Accession No. AF062257
(5)B26-H鎖可変領域:GenBank Accession No. AAC18248
(6)BBA-L鎖可変領域:GenBank Accession No. AAA59100
【0121】
1−2.ヒト化抗体遺伝子発現ベクターの構築
分泌シグナル配列から抗体可変領域にいたるアミノ酸配列をコードする塩基配列において、50 base程度の合成オリゴDNAを3'末端側が約20 base程度ハイブリダイズするように交互に12本作製した。合成オリゴDNAは5'末端側にヒト配列、3'末端側にマウス配列をコードするか、または全塩基がヒト配列をコードするように設計した。さらに、抗体可変領域遺伝子の5'末端にアニ−ルし、XhoI切断配列を有するプライマーと抗体可変領域遺伝子の3'末端にアニ−ルし、SfiI切断配列を有し且つイントロン配列の5'末端配列をコードするプライマーを作製した。
【0122】
2.5μMに調製した合成オリゴDNAを各1μLで混合し、1x TaKaRa Ex Taq Buffer, 0.4 mM dNTPs, 0.5 units TaKaRa Ex Taq(全て宝酒造)を加え、反応液48μLになるように調製した。94℃ 5分保温した後に、94℃ 2分、55℃ 2分、72℃ 2分からなる反応を2サイクル行い、各合成オリゴDNAのアッセンブルおよび伸長反応を実施した。次に、抗体遺伝子の5'末端および3'末端にアニ−ルするプライマー(各10μM)を1μL添加し、94℃ 30秒、55℃ 30秒、72℃ 1分からなる反応を35サイクル行い、72℃ 5分反応させ、抗体可変領域遺伝子を増幅した。PCR後、反応液全量を1 %アガローズゲル電気泳動に供した。目的のサイズ(約400 bp)の増幅断片をQIAquick Gel Extraction Kit(QIAGEN)を用いて、添付説明書記載の方法で精製し、滅菌水30μlで溶出した。該断片をpGEM-T Easy Vector Systems (Promega)を用いて、添付説明書記載の方法でクローニングを行った。各DNA断片の塩基配列は、BigDye Terminator Cycle Sequencing Kit(Applied Biosystems)を用い、DNAシークエンサーABI PRISM 3730xL DNA SequencerまたはABI PRISM 3700 DNA Sequencer(Applied Biosystems)にて、添付説明書記載の方法に従い決定した。
【0123】
正しいヒト化抗体可変領域遺伝子配列であることが確認されたH鎖可変領域断片挿入プラスミドをXhoIおよびSfiIで、L鎖可変領域断片挿入プラスミドをEcoRIで消化した後に、反応液を1 %アガローズゲル電気泳動に供した。目的のサイズ(約400 bp)のDNA断片をQIAquick Gel Extraction Kit(QIAGEN)を用いて、添付説明書記載の方法で精製し、滅菌水30μlで溶出した。その後、以下のようにして動物細胞用発現ベクターを作製した。H鎖がヘテロな組み合わせであるIgG4を優先的に発現させるために、IgG1のknobs-into-hole技術(Merchant AM ら、Nature Biotechnology、1998年、Vol.16、p.677-681)を参考にIgG4のCH3部分へのアミノ酸置換体を用いた。さらにH鎖のダイマー形成促進のためにヒンジにもアミノ酸置換(-ppcpScp- →-ppcpPcp-)を導入した。ニワトリβアクチンプロモーターを有するpCAGGS(Niwa ら、Gene、1991年、Vol. 108、p.193-199)にY349C、T366Wに置換した定常領域遺伝子を組み込んだ発現ベクターにヒト化A69 H鎖可変領域抗体遺伝子断片を挿入し、ヒト化A69H鎖発現ベクターを作製した。また、pCAGGSにE356C、T366S、L368A、Y407Vに置換した定常領域遺伝子を組み込んだ発現ベクターにヒト化B26 H鎖可変領域抗体遺伝子断片を挿入し、ヒト化B26H鎖発現ベクターを作製した。また、pCAGGSに野生型の抗体L鎖定常領域が挿入されたプラスミド(pCAG-gκDNA)をEcoRIで消化し、ヒト化BBA L鎖可変領域抗体遺伝子断片を挿入した発現ベクターを作製した。連結反応はRapid DNA Ligation Kit (Roche Diagnostics)を用い、大腸菌DH5α株 (東洋紡績)を形質転換した。
【0124】
1−3.ヒト化二重特異性抗体の発現
ヒト化二重特異性抗体の発現は、以下の方法を用いて行った。ヒト胎児腎癌細胞由来ヒト化二重特異性抗体の発現は、実施例1−2に記載した方法か以下の方法を用いて行った。ヒト胎児腎癌細胞由来HEK293H株(Invitrogen)を10 % Fetal Bovine Serum (Invitrogen)を含むDMEM培地(Invitrogen)へ懸濁し、5〜6 × 105個 /mLの細胞密度で接着細胞用ディッシュ(直径10 cm, CORNING)の各ディッシュへ10 mLずつ蒔きこみCO2インキュベーター(37℃、5 % CO2)内で一昼夜培養した後に、培地を吸引除去し、1 %のFetal Bovine Serum(Invitrogen)を含むCHO-S-SFM-II(Invitrogen)培地6.9 mLを添加した。1−2で調製したプラスミドDNA混合液(合計13.8μg)を1μg/mL Polyethylenimine (Polysciences Inc.) 20.7μLとCHO-S-SFMII培地 690μLと混合して室温10分間静置したものを各ディッシュの細胞へ投入し、4〜5時間、CO2インキュベーター(37℃にて5 % CO2)内でインキュベートした。その後、1 %のFetal Bovine Serum(Invitrogen)を含むCHO-S-SFM-II(Invitrogen)培地6.9 mLを添加して、3日間 CO2インキュベーター内で培養した。培養上清を回収した後、遠心分離(約2000 g、5分間、室温)して細胞を除去し、さらに0.22μmフィルターMILLEX(R)-GV(Millipore)を通して滅菌した。該サンプルは使用するまで4℃で保存した。
【0125】
1−4.ヒト化二重特異性抗体の精製
実施例1−2に記載の方法で得られた培養上清に100μLのrProtein A SepharoseTM Fast Flow(Amersham Biosciences)を添加し、4℃で4時間以上転倒混和した。その溶液を0.22μmのフィルターカップUltrafree(R)-MC(Millipore)に移し、0.01 % Tween(R) 20を含むTBS 500μLにて3回洗浄後、rProtein A SepharoseTM樹脂に100μLの 0.01 % Tween(R) 20を含む50 mM 酢酸ナトリウム水溶液, pH 3.3に懸濁して2分間静置したのち、抗体を溶出させた。直ちに、6.7μLの1.5 M Tris-HCl , pH 7.8を加えて中和した。
【0126】
1−5.ヒト化二重特異性抗体の濃度定量
以下に示すとおり、2種類の方法で測定した。
Goat anti-human IgG(Biosource International)をcoating bufferにて1μg/mLに調製し、Nunc-Immuno plate(Nunc)に固相化した。Diluent buffer(D.B.)にてブロッキング処理した後、D.B.を用いて適当に希釈した培養上清サンプルを添加した。また、抗体濃度算出のためのスタンダードとして、2000 ng/mLから3倍系列でD.B.にて11段階希釈したヒトIgG4(ヒト型化抗TF抗体、WO 99/51743参照)を同様に添加した。3回洗浄したのち、Goat anti-human IgG, alkaline phosphatase(Biosource International)を反応させた。5回洗浄したのち、Sigma 104(R) phosphatase substrate(Sigma-Aldrich)を基質として発色させ、吸光度リーダーModel 3550(Bio-Rad Laboratories)により、参照波長655 nmとして405 nmの吸光度を測定した。Microplate Manager III(Bio-Rad Laboretories)ソフトウェアを用いて、スタンダードの検量線から培養上清中のヒトIgG濃度を算出した。
【0127】
また、Biacore1000(BIACORE)を使用し、ProteinAを固定化したSensor Chip CM5(BIACORE)を用いて定量した。具体的にはメーカーのプロトコールに従い、活性化したセンサーチップに10 mM 酢酸ナトリウム水溶液(pH 4.0, BIACORE)で50μg/mLに希釈したProteinA(SIGMA)溶液を5μL/分で30分間反応させ、その後ブロッキング操作を実施してProteinA固定化センサーチップを作製した。このセンサーチップを用いて、Biacore 1000(BIACORE)を使用して培養上清および精製品の濃度を測定した。センサーチップの固定および濃度測定にはHBS-EP Buffer(BIACORE)を使用した。また、濃度測定時の標準品として4000 ng/mLから2倍系列でHBS-EP Bufferにて6段階希釈したヒト化IgG4抗体(ヒト型化抗TF抗体、WO 99/51743参照)を使用した。
【0128】
1−6.ヒト化二重特異性抗体の血液凝固活性評価
血友病A血液の凝固能を二重特異性抗体が是正するか明らかにするために、Factor VIII欠乏血漿を用いた活性化部分トロンボプラスチン時間(APTT)に対する同抗体の影響を検討した。様々な濃度の抗体溶液 50μL、Factor VIII欠乏血漿(Biomerieux)50μL及びAPTT試薬(Dade Behring)50μLの混合液を37℃で3分間加温した。凝固反応は20 mMのCaCl2(Dade Behring)50μLを同混合液に加えることにより開始させた。CR-A(Amelung)が接続されたKC10A(Amelung)により凝固するまでの時間を測定した。
【0129】
Factor VIII欠乏血漿の凝固時間を0 %、正常血漿の凝固時間を100 %としたときに作製される検量線を用いて、二重特異性抗体を添加した際の凝固時間から二重特異性抗体のFactor VIII様活性(%)を算出した。
【0130】
1−7.血液凝固活性を保持したヒト化二重特異性抗体の取得
上述した血液凝固活性評価において、血液凝固能が低下したヒト化二重特異性抗体について、活性上昇を目指してヒト抗体FRのアミノ酸を改変した。具体的には、QuikChange Site-Directed Mutagenesis Kit (Stratagene)を用いて、添付説明書記載の方法でヒト化抗体可変領域に変異を導入した。目的のヒト化抗体可変領域遺伝子配列であることが確認されたH鎖可変領域断片挿入プラスミドをXhoIおよびSfiIで、L鎖可変領域断片挿入プラスミドをEcoRIで消化した後に、反応液を1 %アガローズゲル電気泳動に供した。目的のサイズ(約400 bp)のDNA断片をQIAquick Gel Extraction Kit(QIAGEN)を用いて、添付説明書記載の方法で精製し、滅菌水30μlで溶出した。その後、実施例1−2に示す方法で、動物細胞用発現プラスミドを作製した。実施例1−3、1−4、1−5に示す方法でヒト化二重特異性抗体を調製し、実施例1−6に示す方法で血液凝固活性を評価した。
【0131】
FR配列のアミノ酸改変および血液凝固能の評価を繰り返すことでキメラ二重特異性抗体(A69/B26/BBA)と同等の活性を有するヒト化二重特異性抗体(ヒト化A69(hA69a)/ ヒト化B26 (hB26-F123e4)/ヒト化BBA(hAL-F123j4))を取得した(図1)。各抗体可変領域配列を以下の配列番号に示した。
【0132】
(1) ヒト化A69抗体VH(hA69a) 配列番号:1(塩基配列)、配列番号:2(アミノ酸配列)
(2) ヒト化B26抗体VH(hB26-F123e4) 配列番号:3(塩基配列)、配列番号:4(アミノ酸配列)
(3) ヒト化BBA抗体VL(hAL-F123j4) 配列番号:5(塩基配列)、配列番号:6(アミノ酸配列)
【0133】
〔実施例2〕二重特異性抗体の分離に向けた可変領域のアミノ酸改変箇所の選定
二重特異性抗体を調製する際の発現において、2種類のH鎖と1種類のL鎖を使用すると、ヒト化A69-H鎖とヒト化BBA-L鎖のホモダイマー、ヒト化B26-H鎖とヒト化BBA-L鎖のホモダイマー、ヒト化A69-H鎖およびヒト化B26-H鎖とヒト化BBA-L鎖のヘテロダイマーの3種類の抗体が発現する。この3種類の抗体を分離し、二重特異性抗体のみ精製することを目的として、ヒト化A69 H鎖可変領域の等電点を下降させ、ヒト化B26 H鎖可変領域の等電点を上昇させるアミノ酸改変を行った。
【0134】
はじめにヒト化A69抗体とヒト化B26抗体の可変領域表面に露出するアミノ酸残基を確認するために、MOEソフトウェア(Chemical Computing Group Inc.)を用いて、ホモロジーモデリングによりヒト化A69抗体およびヒト化B26抗体の抗体Fv領域モデルを作製した。モデルを図2に示した。本モデルの詳細な解析により、CDR以外のFR配列においては表面に露出するアミノ酸の中で、H10、H12、H23、H39、H43、H105(Kabatナンバリング、Kabat EA et al. 1991. Sequences of Proteins of Immunological Interest.NIH)が、活性を低下させること無く、等電点を変化させることができる候補になると考えられた。
【0135】
〔実施例3〕ヒト化二重特異性抗体の可変領域アミノ酸の改変
実施例2において選定された箇所について改変抗体を作製するためのアミノ酸改変を行った。具体的には、QuikChange Site-Directed Mutagenesis Kit (Stratagene)を用いて、添付説明書記載の方法で作製したヒト化A69抗体H鎖可変領域(hA69a、塩基配列番号:1)およびヒト化B26抗体H鎖可変領域(hB26-F123e4、塩基配列番号:3)に変異を導入した。目的のヒト化抗体可変領域遺伝子配列であることが確認されたH鎖可変領域断片挿入プラスミドをXhoIおよびSfiIで消化した後に、反応液を1 %アガローズゲル電気泳動に供した。目的のサイズ(約400 bp)のDNA断片をQIAquick Gel Extraction Kit(QIAGEN)を用いて添付説明書記載の方法で精製し、滅菌水30μlで溶出した。実施例1−2に示す方法で、調製したDNA断片をknobs-into-hole技術を参考に定常領域アミノ酸を置換した発現プラスミドおよび野生型定常領域をもつ発現プラスミドに挿入し、H鎖発現ベクターを作製した。その後、実施例1−3、1−4、1−5に示す方法でヒト化二重特異性抗体を調製した。改変したヒト化抗体の可変領域配列を以下の表1に記載される配列番号に示した。
【0136】
【表1】

【0137】
〔実施例4〕改変したヒト化抗体の等電点電気泳動による分析
可変領域のアミノ酸改変による表面電荷の変化について評価するために、改変抗体の調製および等電点電気泳動による分析を実施した。
【0138】
ヒト化BBA-L鎖(hAL-F123j4)発現ベクターとヒト化A69-H鎖を改変したhA69-p18、hA69-p8、hA69-p17、hA69-p16および未改変のhA69aの各H鎖発現ベクターを組み合わせて同時に発現させることによりhA69a、hA69-p18、hA69-p8、hA69-p17、hA69-p16の5種類のホモダイマーで構成される抗体を調製した。同様に、ヒト化BBA-L鎖発現ベクターとヒト化B26-H鎖を改変したhB26-p19、hB26-p15および未改変のhB26-F123e4の各H鎖発現ベクターを組み合わせて同時に発現させることによりhB26-F123e4、hB26-p19、hB26-p15の3種類のホモダイマーで構成される抗体を調製した。等電点電気泳動は以下のとおり行った。Phastsystem Cassette (AmerchamBioscience社製) を用いて以下の膨潤液で30 minほどPhast-Gel Dry IEF (AmerchamBioscience社製)ゲルを膨潤させた。
【0139】
20% Glycerol 0.95 mL
ミリQ水 0.95 mL
Bio-Lyte 7/9 (BioRad社製) 10μL
Bio-Lyte3/10 (BioRad社製) 10μL
Pharmalyte 8-10.5 for IEF (AmerchamBioscience社製) 80μL
【0140】
膨潤したゲルを用いてPhastSystem(AmerchamBioscience社製)により以下のプログラムで電気泳動を行った。サンプルはStep 2でゲルに添加した。pIマーカーとして、Calibration Kit for pI(AmerchamBioscience社製)を使用した。
【0141】
Step 1: 2000 V 2.5 mA 3.5 W 15℃ 75 Vh
Step 2: 200 V 2.5 mA 3.5 W 15℃ 15 Vh
Step 3: 2000 V 2.5 mA 3.5 W 15℃ 410 Vh
【0142】
泳動後のゲルは20 % TCAで固定した後、Silver staining Kit, protein(AmerchamBioscience社製)を用い、キットに添付されているプロトコールに従い銀染色を行った。染色後、pIマーカーの既知等電点からサンプルの等電点を算出した。
【0143】
未改変および改変したヒト化A69抗体のホモダイマー、ヒト化B26抗体のホモダイマーの分析結果を図3に示した。表面電荷の改変により等電点電気泳動においてバンドの移動が観察された。pIマーカーを参考に推測した各抗体の等電点は、未改変のhA69aホモダイマーが約8.8であるのに対して、改変したhA69-p18が約8.4、hA69-p17が約8.2、hA69-p8が約8.2、hA69-p16が約8.1であり、改変により最大約0.7の等電点の差を付与することができた。ヒト化B26ホモダイマーも同様に、未改変のhB26-F123e4が約9.1であるのに対して、改変したhB26-p19が約9.3、hB26-p15が約9.4であり、改変により最大約0.3の等電点の差を付与することができた。本検討において選択した可変領域のH12、H23、H39、H43、H105の表面アミノ酸を電荷的に改変することによって等電点を変化させることが可能であることが示された。
【0144】
〔実施例5〕改変したヒト化抗体の陽イオン交換クロマトグラフィー分析
実施例4において作製した改変抗体を用いて以下の方法で陽イオン交換クロマトグラフィーによる分析を行い、改変による両抗体の分離に及ぼす影響を評価した。陽イオン交換クロマトグラフィー分析条件は以下のとおりであり、ヒト化A69抗体のホモダイマー、ヒト化B26抗体のホモダイマーの保持時間を算出した。
【0145】
カラム:ProPac WCX-10, 4×250 mm, (Dionex)
移動相:A: 10 mmol/L NaH2PO4/Na2HPO4, pH 6.25
B: 10 mmol/L NaH2PO4/Na2HPO4, 500 mmol/L NaCl, pH 6.25
流速:1.0 mL/min
グラジエント:10 %B(5 min)→(40 min)→60 %B→(5 min)→100 %B (5 min)
検出:220 nm
【0146】
未改変および改変した5種のヒト化A69抗体のホモダイマーの分析結果を図4に、未改変および改変した3種のヒト化B26抗体のホモダイマーの分析結果を図5に示した。未改変ヒト化A69抗体のホモダイマーとヒト化B26抗体のホモダイマーの保持時間はともに25 min前後であり、両ホモダイマーの分離、まして目的の二重特異性抗体の分離は出来ない。未改変抗体の等電点を低下させる改変を行ったヒト化A69抗体は未改変の抗体と比較してピークの移動が観察され、改変の数に伴い保持時間は約22.4 min、約21.2 min、約20.2 minと短くなった。可変領域の等電点を上昇させる改変を行ったヒト化B26抗体も未改変の抗体と比較してピークの移動が観察され、改変の数に伴い保持時間は約28.4 min、約29.4 minと長くなった。本検討において選択した可変領域のH12、H23、H39、H43、H105の表面アミノ酸の電荷的に改変することによって、2種類の抗体の表面電荷が変化し、それにより保持時間を変化させることが可能であることが示された。
【0147】
実施例4において測定された等電点によると、未改変のhA69aホモダイマーと未改変のhB26-F123e4ホモダイマーはpIに0.3の差があるが両者の保持時時間はともに25min前後であり分離できなかったが(図9)、未改変のhA69aホモダイマーとhB26-p19はpIに0.5の差が付与され、その結果両者は保持時間約2.6minの差で分離されており、またhA69-p18とhB26ホモダイマーはpIに0.7の差が付与され、その結果両者は保持時間約3.4minの差で分離されており、最大でhA69-p16とhB26-p15はpIに1.3の差が付与され、その結果保持時間約9.2 minの差で分離された。このように、改変により2つのホモダイマーの分離が初めて可能になった。
【0148】
〔実施例6〕改変したヒト化二重特異性抗体の凝固活性評価
実施例4、実施例5の分析により表面電荷の変化が観察されたことを受けて、改変した2種類のヒト化抗体H鎖(hA69-p8、hB26-p15)とヒト化L鎖(hAL-F123j4)を発現させてヒト化二重特異性抗体を調製した。H鎖発現ベクターには、ヘテロダイマーを効率的に促進させるために、knobs-into-holes技術を利用したIgG4定常領域が組み込まれた発現ベクターを使用した。調製したヒト化二重特異性抗体を用いて、以下に示す方法に従って凝固活性を評価した。
【0149】
血友病A血液の凝固能を二重特異性抗体が是正するか明らかにするために、Factor VIII欠乏血漿を用いた活性化部分トロンボプラスチン時間(APTT)に対する同抗体の影響を検討した。様々な濃度の抗体溶液 50μL、Factor VIII欠乏血漿(Biomerieux)50μL及びAPTT試薬(Dade Behring)50μLの混合液を37℃で3分間加温した。凝固反応は20 mMのCaCl2(Dade Behring)50μLを同混合液に加えることにより開始させた。CR-A(Amelung)が接続されたKC10A(Amelung)により凝固するまでの時間を測定した。
【0150】
Factor VIII欠乏血漿の凝固時間を0 %、正常血漿の凝固時間を100 %としたときに作製される検量線を用いて、二重特異性抗体を添加した際の凝固時間から二重特異性抗体のFactor VIII様活性(%)を算出した。
【0151】
活性評価結果を図6に示した。可変領域を改変したヒト化二重特異性抗体は、未改変のヒト化二重特異性抗体と同等の凝固活性を示したことから、本実施例における可変領域の改変は抗体の活性には影響がないことが示された。
【0152】
〔実施例7〕CDRを改変したヒト化抗体の作製と評価
実施例2で作製したヒト化A69抗体のモデルを解析した結果、H97は表面に露出するアミノ酸であることが確認された。表1に示した抗体においてヒト化A69-H鎖であるhA69-N97RはCDR3に存在する97番目のアスパラギンをアルギニンに改変した配列を持つ。実施例1−2の方法に従ってhA69-N97Rをもつ発現ベクターを作製し、ヒト化BBA-L鎖であるhAL-F123j4とともに発現し、改変抗体を調製した。この抗体の表面電荷の変化を評価するために、実施例4の方法に従って等電点電気泳動を行った。図7に示すとおり、未改変の抗体(hA69a / hAL-F123j4)の等電点は8.9であるのに対して、改変抗体(hA69-N97R / hAL-F123j4)は9.1であり、CDRのアミノ酸置換においても表面電荷の変化が観察された。
【0153】
また改変抗体の機能を評価するために、以下の方法で抗原であるFactor IXaに対する結合活性を評価した。Coating buffer(100 mM sodium bicarbonate, pH 9.6, 0.02 % sodium azide)で1μg/mLに希釈したFactor IXaβ(Enzyme Research Labratories)を、Nunc-Immuno plate(Nunc-ImmunoTM 96 MicroWellTM plates MaxiSorpTM(Nalge Nunc International))に100μL/wellで分注後、4℃で一晩インキュベーションした。Tween(R) 20を含むPBS(-)で3回洗浄後、diluent buffer (50 mM Tris-HCl, pH 8.1, 1 % bovine serum albumin, 1mM MgCl2, 0.15 M NaCl, 0.05 % Tween(R) 20, 0.02 % sodium azide)でplateを室温で2時間blockingした。Bufferを除去後、diluent bufferで希釈した精製抗体を100μL/well添加し、室温で1時間インキュベーションした。Plateを3回洗浄後、diluent bufferで1/4000希釈したアルカリホスファターゼ標識ヤギ抗マウスIgG(BIOSOURCE)を100μL/well添加し、室温で1時間インキュベーションした。Plateを5回洗浄後、発色基質(Sigma)を100μL/well添加し、室温で30分インキュベーションした。405 nm(対照655 nm)における吸光度をMicroplate Reader Model 3550(Bio-Rad Laboratories)で測定した。その結果、図8に示すとおり、表面電荷を変化させるためにCDRを改変した抗体は改変前の抗体と同等の結合活性を示した。このように表面電荷を改変する際に、改変する箇所は実施例5に示したFRのみでなく、CDRであっても構わないことが示された。
【0154】
〔実施例8〕ヒト化二重特異性PF抗体の作製と評価
未改変の抗体として、表1に示した抗体においてヒト化A69-H鎖であるhA69a、ヒト化B26-H鎖であるhB26-F123e4とヒト化BBA-L鎖であるhAL-F123j4(配列番号:5)を用いて、未改変のヒト化二重特異性抗体を作製した。改変抗体として、表1に示した抗体においてヒト化A69-H鎖の改変体であるhA69-PFLとヒト化B26-H鎖の改変体であるhB26-PFとヒト化BBA-L鎖であるhAL-s8(配列番号:17)を用いて、ヒト化二重特異性PF抗体を作製した。H鎖は野生型の定常領域をもつ発現ベクターを使用して、実施例1−2に示すとおりに発現ベクターを構築し、実施例1−3、実施例1−4、実施例1−5の方法に従って抗体を調製した。この2種類のホモダイマーと二重特異性抗体を含む混合溶液を用いて、実施例5に示す方法で陽イオン交換クロマトグラフィー分析を実施した。
【0155】
未改変のヒト化二重特異性抗体とヒト化二重特異性PF抗体分析結果を図9、図10に示した。その結果、未改変のヒト化二重特異性抗体においては、二種類のホモダイマーと二重特異性抗体が分離せず1本のピークとして溶出したのに対して、ヒト化二重特異性PF抗体は、二種類のホモダイマーと目的の二重特異性抗体がそれぞれ分離し、hA69-PF ホモダイマー、ヒト化二重特異性PF抗体、hB26-PF ホモダイマーの順に3本のピークとして溶出した。陽イオン交換クロマトグラフィー分析の際に3種類のピークを分取することで、二種類のホモダイマーとヒト化二重特異性PF抗体を精製した。この画分をAmicon Ultra, MWCO 10000 (Millipore)による濃縮後、20 mM sodium acetate, 150 mM NaCl, pH 6.0に対して一晩冷所で透析を行い、濃度測定を行った。
【0156】
各抗体を精製した後に、実施例4に示す方法に従って等電点電気泳動を行った。図11に示すとおり、陽イオン交換クロマトグラフィー分析を行う前の抗体は3本のバンドが存在するが、陽イオン交換クロマトグラフィーにより各抗体が精製できることが確認された。ヒト化A69-PF抗体のホモダイマー、ヒト化二重特異性PF抗体、ヒト化B26-PF抗体のホモダイマーの等電点は約7.9、約8.6、約9.2であり、ヒト化A69-PF抗体のホモダイマーのヒト化二重特異性PF抗体等電点の差は約0.7であり、ヒト化B26-PF抗体のホモダイマーのヒト化二重特異性PF抗体等電点の差は約0.6であることが確認された。
【0157】
つづいて、実施例6に示す方法に従って精製した二重特異性PF抗体の凝固活性を評価した。前述したknobs-into-holes技術を利用したIgG4定常領域を用いて発現させたキメラ二重特異性抗体、可変領域を改変していないhA69a(配列番号:2)、hB26-F123e4(配列番号:4)、hAL-F123j4(配列番号:6)から成る二重特異性抗体、精製した二重特異性PF抗体と同じ可変領域をもち、knobs-into-holes技術を利用したIgG4定常領域を用いている二重特異性抗体の3種類の抗体と凝固活性を比較した。評価結果を図12に示した。Knobs-into-holes技術を利用したIgG4定常領域をもつ二重特異性PF抗体と野生型定常領域をもち陽イオン交換クロマトグラフィーにより精製した二重特異性PF抗体の凝固活性は同等であり、本実施例のH10、H12、H23、H39、H43、H105の可変領域改変により活性に影響することなく高純度にて二重特異性抗体が精製できることが示された。
【0158】
〔実施例8〕ヒト化二重特異性抗体発現細胞株の樹立
改変したヒト化二重特異性抗体を調製するために、以下のようにして抗体発現細胞株を樹立した。
【0159】
ヒトIgG4の野生型H鎖定常領域遺伝子を鋳型にしてH鎖定常領域のN末端側の2アミノ酸(Ala-Ser)をコードする塩基配列がNheI認識配列(GCTAGC)になるように設計した5'末端側プライマーと3'末端側にアニーリングし、かつNotI認識部位を持つように設計したプライマーを用いてH鎖定常領域をPCR増幅し、pBluescriptKS+ベクター(東洋紡)をNheI, NotI(ともに宝酒造)で消化したベクターと連結したpBCH4(IgG4定常領域遺伝子を含む)を作製した。表1に示すヒト化A69-H鎖抗体(hA69-KQ)およびヒト化B26-H鎖抗体(hB26-PF)のH鎖可変領域の5'末端側塩基配列に相補的でコザック配列(CCACC)およびEcoRI認識配列を有するプライマーとNheI認識配列を有する3'末端側塩基配列にプライマーを用いてPCRを行い、得られたPCR産物をEcoRI, NheI(ともに宝酒造)で消化、同様にEcoRI, NheIで消化したpBCH4に挿入して可変領域と定常領域を連結した。作製したヒト化A69-H鎖抗体ベクターをEcoRI, NotI(ともに宝酒造)で消化し、同様にEcoRI, NotIで消化した動物細胞用発現ベクターpCXND3にクローニングした。
【0160】
本ベクターpCXND3の構築の流れについて、以下に述べる。DHFR-ΔE-rVH-PM1-f(WO92/19759参照)の抗体H鎖遺伝子とベクターを分割するために、制限酵素EcoRI, SmaI部位で消化し、ベクター側のみ回収した後に、EcoRI-NotI -BamHI adaptor(宝酒造)をクローニングした。このベクターをpCHOIと命名した。pCHOIのDHFR遺伝子発現部位をpCXN(Niwaら、Gene 1991;108:193-200)の制限酵素HindIII部位にクローニングしたベクターをpCXND3と命名した。また、作製したヒト化B26-H鎖抗体ベクターをEcoRI, NotI(ともに宝酒造)で消化し、同様にEcoRI, NotIで消化した動物細胞用発現ベクターpCXZD1にクローニングした。pCXZD1ベクターはpCXND3ベクターのネオマイシン耐性遺伝子をゼオシン耐性遺伝子に置き換えた発現ベクターである。また、ヒト化BBA-L鎖抗体(hAL-AQ、配列番号:18)のL鎖可変領域の5'末端側塩基配列に相補的でコザック配列を有する合成オリゴヌクレオチドおよびBsiWI部位を有する3'末端側塩基配列に相補的な合成オリゴヌクレオチドを用いてPCRを行い、得られたPCR産物をヒトkappa鎖定常領域がpBluescriptKS+ベクターに挿入されているpBCLベクターにクローニングした。BsiWI部位により、ヒトL鎖可変領域と定常領域が連結している。作製されたL鎖遺伝子断片を発現ベクターpUCAGにクローニングした。本ベクターpUCAGは、pCXN(Niwaら、Gene 1991;108:193-200)を制限酵素BamHIで消化して得られる2.6 kbpの断片をpUC19ベクター(東洋紡)の制限酵素BamHI部位に連結し、クローニングしたベクターである。L鎖をpUCAGにクローニングしたベクターを制限酵素BamHIで消化し、ハイグロマイシン耐性遺伝子を含む発現ベクターpHygDHFR-4bにクローニングした。作製した3種類の発現ベクターを制限酵素で直鎖上にしたのちに、CHO-DG44細胞に遺伝子導入して抗体発現細胞株を樹立した。
【0161】
安定発現細胞株の作製は次のようにして行った。GenePulserII(Bio-Rad)を用いたエレクトロポレーション法により遺伝子導入した。各抗体発現ベクターとPBSに懸濁したCHO細胞(1×107細胞/mL)の0.75 mLを混合したものを氷上で10分間冷却し、キュベットに移した後に1.5 kV、25μFDの容量にてパルスを与えた。室温にて10分間の回復期間の後、エレクトロポレーション処理された細胞を、HT supplement(Invitrogen)を1倍濃度で含むCHO-S-SFMII培地(Invitrogen)40 mLに懸濁した。同様の培地で10倍希釈液を作製し、96ウェル培養用プレートに100μL/wellで分注した。CO2インキュベーター(5 % CO2)で24時間培養後、Geneticin(Invitrogen)を0.5 mg/mL、Zeocin(Invitrogen)を0.6 mg/mL、HygromycinB(Invitrogen)を0.4 mg/mLになるように、添加して2週間培養した。薬剤耐性を示す形質転換細胞のコロニーを順次拡大培養し、樹立した高産生細胞株を用いて大量培養を行い、培養上清を得た。
【0162】
〔実施例9〕ヒト化二重特異性抗体の製造用汎用カラムによる分離精製
実施例8で得られた培養上清から以下の方法で二重特異性抗体を精製した。培養上清を平衡化バッファー (20 mmol/L Sodium Phosphate buffer, 1 mol/L NaCl)で平衡化したrProtein A Sepharose Fast Flowカラム (Amersham Biosciences、50 mmI.D.×9.9 cmH. = 194.3 mL-resin)に添加し、洗浄用バッファー1(20 mmol/L Sodium Phosphate buffer, 1 mol/L NaCl, pH7.0)、洗浄用バッファー2(50 mmol/L Sodium Acetate buffer, pH 6.0)で洗浄した後に100 mmol/L Acetic acidを用いて溶出した。溶出後に直ちに20 mmol/L Sodium Acetate buffer, pH6.0で3倍希釈した。
【0163】
得られた精製溶液をSolvent A (20 mmol/L sodium Acetate buffer, pH 6.0)で平衡化した製造用汎用カラムであるSP TOYOPEARL 650Mカラム (東ソー、26 mmI.D.×22.3 cmH. = 118.3 mL-resin)に添加した。以下に示すような溶液およびGradientで抗体の表面電荷の差を用いた分離を行った。
【0164】
Solvent A : 20 mmol/L Sodium Acetate buffer, pH6.0
Solvent B : 20 mmol/L Sodium Acetate buffer, 1 mol/L NaCl, pH6.0
Flow rate : 10 mL/min (113 cm/h)溶出時のみ5.3 mL/min (60 cm/h)
Gradient : 0→15%B Step wise 3 Column Volume (CV)通液
15→22 %B gradient 2.5 CV
22→30 %B gradient 6 CV
30→100 %B Step wise 3 CV通液
【0165】
溶出の結果、図13に示すような3本のピークが検出され、製造用汎用カラムを使用した場合にも二重特異性抗体が分離精製できることが示された。
【0166】
〔実施例10〕改変したヒト化二重特異性抗体の活性評価
実施例9で調製したヒト化二重特異性抗体について、実施例6に示す方法に従って凝固活性を評価した。評価結果を図14に示した。実施例8で調製したヒト化二重特異性PF抗体と比較して実施例9で精製したヒト化二重特異性抗体の凝固活性は同等であった。hA69-PFLとhA69-KQのように可変領域のアミノ酸配列が若干異なっていても、製造用汎用カラムを使用して精製した抗体であっても活性には影響がないことが示された。
【0167】
以上のことから、二重特異性抗体を調製する際に、H鎖可変領域の改変により構造や抗体の機能(活性)を変えることなく表面電荷を変化させることで、目的のヒト化二重特異性抗体と二種のホモダイマーを形成する抗体とを分離精製できることが見出された。本方法を用いることで製造用汎用カラムにおいても二重特異性抗体が分離精製可能であることが示されたことから、二重特異性抗体からなる医薬品の製造方法として有用である。
【0168】
〔実施例11〕アイソタイプハイブリッド抗体の作成
11−1.ヒトIgG2抗体H鎖定常領域遺伝子のクローニング
ヒトIgG2抗体H鎖定常領域の遺伝子をクローニングするため、以下の操作を行った。
cDNA断片増幅のために50μLの反応液(各1μLの20μM K62プライマー(5' cac cgt ctc ctc agc ctc cac caa 3'/配列番号:22)、K63プライマー(5' gtg gca ctc att tac ccg gag aca 3'/配列番号:23)、5μLのMTC Multiple Tissue cDNA Panels(peripheral leukocytes) (Clontech)、4μLの5×Prime STAR Buffer、4μLの2.5 mM dNTPs、1μLのPrimeSTAR HS DNA Polymerase (以上TaKaRa))を調製し、PCRに供した。PCRは、サーマルサイクラーGeneAmp PCR system 9700(Parkin Elmer)を用いて、98℃で2分間加熱後、98℃ 10秒、60℃ 5秒、72℃ 2 minからなる反応を30 cycle行い、最後に72℃で10 min加熱した。PCR後、反応液を1% アガローズゲル電気泳動に供した。目的のサイズ(約1000 bp)の増幅断片をQIAqucick Gel Extractio Kit(QIAGEN)にて添付説明書記載の方法に従って精製し、滅菌水50μLで溶出した後、増幅断片の末端にA(Adenosine)を付加するためr-Taq処理をおこなった。r-Taq処理は、得られた増幅断片を10μLのrTaq反応液(1μLの10×rTaq反応溶液、1μLの2.5 mM dNTPs、1μLのrTaq、7μLの上記増幅断片)を72℃で30 min保温した。r-Taq処理した断片をpCR2.1-TOPO vector(Invitrogen)へクローニングし、塩基配列を決定した。各DNA断片の塩基配列は、BigDye Terminator 3.1 Cycle Sequencing Kit(Applied Biosystems)を用い、DNAシークエンサーABI PRISM 3730xL Genetic Analyzer(Applied Biosystems)にて、添付説明書記載の方法に従い決定した。
【0169】
決定した塩基配列をAccession.No.BX640623と比較し、翻訳したアミノ酸配列が異なっている塩基はPCR増幅時に挿入された変異と考え、Quick Change Site-Directed Mutagenesis Kit(Stratagene)を用いてアミノ酸置換を行い、BX640623のアミノ酸配列と同じ配列になるように改変した。Quick Change Site-Directed Mutagenesis Kit(Stratagene)は添付説明書記載の方法に従って行った。さらに、ヒトIgG2-H鎖定常領域遺伝子と目的の可変領域遺伝子を連結するために、ヒトIgG2-H鎖定常領域の最初の2アミノ酸(AlaSer)が制限酵素NheI認識配列(GCTAGC)になるように変異させた。本試験に使用したヒトIgG2-H鎖定常領域の塩基配列およびアミノ酸配列を、それぞれ配列番号:24および配列番号:25に示した。
【0170】
11−2.アイソタイプ置換抗体の発現ベクター構築
ヒト化PM-1抗体のH鎖可変領域とヒトIgG1、ヒトIgG2、ヒトIgG4の各種H鎖定常領域を連結した抗体発現ベクターを以下のように作製した。
非特許文献(Sato K et al, Cancer Research 1993, 53: 851-856)に示されているヒト化抗ヒトインターロイキン6受容体抗体(ヒト化PM-1抗体)のH鎖可変領域の5'末端側塩基配列に相補的でコザック配列を有する合成オリゴヌクレオチドおよび制限酵素NheI認識配列を有し、3'末端側塩基配列に相補的な合成オリゴヌクレオチドを用いてPCRを行い、得られたPCR産物をヒトIgG1-H鎖定常領域(Sato K et al, Cancer Research 1993, 53: 851-856 参照)がpBluescript KS+ベクター(TOYOBO)に挿入されているpB-CHベクターにクローニングした。H鎖可変領域と定常領域が連結したH鎖遺伝子断片をニワトリβアクチンプロモーターにより発現が制御されるpCAGGSベクター(Niwa et al.1991 Gene, 108: 193-199)に挿入した。PCR増幅したヒト化PM-1抗体のH鎖可変領域遺伝子をヒトIgG4定常領域遺伝子(WO 99/51743参照)および実施例11−1で作製したヒトIgG2-H鎖遺伝子の5'末端のNheIと連結してpCAGGSベクターに挿入した。各種H鎖発現ベクターは、NheI配列によりヒト化PM-1抗体のH鎖可変領域とヒトH鎖定常領域が連結してH鎖を発現する。
【0171】
同様にヒト化PM-1抗体のL鎖可変領域の5'末端側塩基配列に相補的でコザック配列を有する合成オリゴヌクレオチドおよび制限酵素BsiWI認識配列を有する3'末端側塩基配列に相補的な合成オリゴヌクレオチドを用いてPCRを行い、得られたPCR産物をヒトkappa鎖定常領域がpBluescript KS+ベクター(TOYOBO)に挿入されているpB-CLベクターにクローニングした。L鎖可変領域と定常領域が連結したL鎖遺伝子断片をニワトリβアクチンプロモーターにより発現が制御されるpCAGGSベクターに挿入した。BsiWI配列によりヒト化PM-1抗体のL鎖可変領域とヒトkappa鎖定常領域が連結してL鎖を発現する。
【0172】
11−3.アイソタイプハイブリッド抗体の発現
アイソタイプハイブリッド抗体はヒトIgG1、ヒトIgG2、ヒトIgG4の各種定常領域を有するヒト化PM-1抗体H鎖発現ベクターをそれぞれ二種類ずつ組み合わせ、ヒト化PM-1抗体L鎖発現ベクターとともに発現用細胞で共発現させることにより可能となる。各抗体の発現は、実施例4−2に記載した方法か以下の方法を用いて行った。ヒト胎児腎癌細胞由来HEK293H株(Invitrogen)を10% Fetal Bovine Serum (Invitrogen)を含むDMEM培地(Invitrogen)へ懸濁し、5〜6 × 105個 /mLの細胞密度で接着細胞用ディッシュ(直径10 cm, CORNING)の各ディッシュへ10 mLずつ蒔きこみCO2インキュベーター(37℃、5% CO2)内で一昼夜培養した後に、培地を吸引除去し、CHO-S-SFM-II(Invitrogen)培地6.9 mLを添加した。11−2で調製したプラスミドDNAを用いて以下のように各アイソタイプ抗体発現用混合液とハイブリッド抗体発現用の混合液(合計13.8μg)を調製した。
(1)L鎖発現ベクター6.9μg、IgG1-H鎖発現ベクター6.9μg
(2)L鎖発現ベクター6.9μg、IgG2-H鎖発現ベクター6.9μg
(3)L鎖発現ベクター6.9μg、IgG4-H鎖発現ベクター6.9μg
(4) L鎖発現ベクター6.9μg、IgG1-H鎖発現ベクター3.45μg、IgG2-H鎖発現ベクター3.45μg
(5) L鎖発現ベクター6.9μg、IgG2-H鎖発現ベクター3.45μg、IgG4-H鎖発現ベクター3.45μg
(6) L鎖発現ベクター6.9μg、IgG1-H鎖発現ベクター3.45μg、IgG4-H鎖発現ベクター3.45μg
【0173】
混合液それぞれを1μg/mL Polyethylenimine (Polysciences Inc.) 20.7μLおよびCHO-S-SFMII培地 690μLと混合して室温10分間静置したものを各ディッシュの細胞へ投入し、4〜5時間、CO2インキュベーター(37℃にて5% CO2)内でインキュベートした。その後、CHO-S-SFM-II(Invitrogen)培地6.9 mLを添加して、3日間 CO2インキュベーター内で培養した。培養上清を回収した後、遠心分離(約2000 g、5分間、室温)して細胞を除去し、さらに0.22μmフィルターMILLEX(R)-GV(Millipore)を通して滅菌した。該サンプルは使用するまで4℃で保存した。
【0174】
11−4.アイソタイプハイブリッド抗体の精製
実施例11−3に記載の方法で得られた培養上清に100μLのrProtein A SepharoseTM Fast Flow(Amersham Biosciences)を添加し、4℃で4時間以上転倒混和した。その溶液を0.22μmのフィルターカップUltrafree(R)-MC(Millipore)に移し、TBS 500μLにて3回洗浄後、rProtein A SepharoseTM樹脂に100μLの50 mM 酢酸ナトリウム水溶液、pH 3.0に懸濁して2分間静置したのち、抗体を溶出させた。直ちに、6.7μLの1.5M Tris-HCl、150 mM NaCl、pH 8.0を加えて中和した。得られた抗体溶液は活性測定用としてPBSに、また、DSC測定用として150 mM NaClを含む20 mM 酢酸緩衝液、pH 6.0に透析することで緩衝液を置換した。精製されたヒトIgG1のH鎖定常領域を有する抗体は「未改変ヒト化抗PM-1抗体」、ヒトIgG2のH鎖定常領域を有する抗体は「IgG2化ヒト化抗PM-1抗体」、ヒトIgG4のH鎖定常領域を有する抗体は「IgG4化ヒト化抗PM-1抗体」と以下に記載する。
【0175】
11−5.アイソタイプハイブリッド抗体の濃度定量
11−4で得た抗体を含む溶液2μLをND-1000 Spectrophotometer(NanoDrop)、あるいは50μLを分光光度計DU-600(BECKMAN)に供し、280 nmでの吸光度を測定した。得られた値から以下の式を用いて抗体濃度を算出した。ブランクにはPBSまたは150 mM NaClを含む20 mM 酢酸緩衝液、pH6.0を使用した。
【0176】
抗体濃度(mg/mL)=吸光度×希釈倍率÷14.6×10
【0177】
〔実施例12〕アイソタイプハイブリッド抗体の分析
12−1.アイソタイプハイブリッド抗体の等電点電気泳動による分析
定常領域の置換による表面電荷の変化について評価するために、等電点電気泳動による分析を実施した。
【0178】
等電点電気泳動は以下のとおり行った。Phastsystem Cassette(AmerchamBioscience社製)を用いて以下の膨潤液で30 minほどPhast-Gel Dry IEF(AmerchamBioscience社製)ゲルを膨潤させた。
【0179】
20% Glycerol 1.5 mL
Pharmalyte 8-10.5 for IEF(AmerchamBioscience社製) 100μL
【0180】
膨潤したゲルを用いてPhastSystem(AmerchamBioscience社製)により以下のプログラムで電気泳動を行った。サンプルはStep 2でゲルに添加した。pIマーカーとして、Calibration Kit for pI(AmerchamBioscience社製)を使用した。
【0181】
Step 1: 2000 V 2.5 mA 3.5 W 15℃ 75 Vh
Step 2: 200 V 2.5 mA 3.5 W 15℃ 15 Vh
Step 3: 2000 V 2.5 mA 3.5 W 15℃ 410 Vh
【0182】
泳動後のゲルは20% TCAで固定した後、Silver staining Kit、protein(AmerchamBioscience社製)を用い、キットに添付されているプロトコールに従い銀染色を行った。染色後、pIマーカーの既知等電点からサンプルの等電点を算出した。
【0183】
未改変、IgG2化およびIgG4化ヒト化PM-1抗体の分析結果を図15に示す。アイソタイプ置換により等電点電気泳動においてバンドの移動が観察された。pIマーカーを参考に推測した各抗体の等電点は未改変ヒト化PM-1抗体が約9.3であるのに対して、IgG2化ヒト化PM-1抗体が約8.9、IgG4化ヒト化PM-1抗体が約8.7であり、置換により最大約0.6の等電点の差を付与することができた。本検討において抗体アイソタイプの定常領域を置換することによって等電点を変化させることが可能であることが示された。
【0184】
次に、未改変、IgG2化ヒト化PM-1抗体および未改変、IgG4化ヒト化PM-1抗体の共発現抗体分析結果を図16に示す。これより、いずれの組み合わせにおいても各アイソタイプのホモダイマー、ヘテロダイマーが3つの主バンドとして観察され、pIマーカーを参考に推測した各アイソタイプハイブリット抗体の等電点は、未改変ヒト化PM-1/IgG2化ヒトPM-1のハイブリッド抗体が9.2、未改変ヒト化PM-1/IgG4化ヒトPM-1のハイブリッド抗体が9.0であった。本検討において各アイソタイプ抗体の発現ベクターを組み合わせて共発現させることで、アイソタイプハイブリッド抗体を作成することが可能であり、それらが等電点の差をもって分離することが示された。
【0185】
12−2.アイソタイプハイブリッド抗体の陽イオン交換クロマトグラフィー分析
実施例11において作製したアイソタイプハイブリッド抗体を用いて以下の方法で陽イオン交換クロマトグラフィーによる分析を行い、アイソタイプ置換が分離に及ぼす影響を評価した。陽イオン交換クロマトグラフィー分析条件は以下のとおりであり、未改変ヒト化PM-1抗体、IgG2化ヒト化PM-1抗体、IgG4化ヒト化PM-1抗体、および未改変ヒト化PM-1抗体/IgG2化ヒト化PM-1抗体のハイブリッド抗体、未改変ヒト化PM-1抗体/IgG4化ヒト化PM-1抗体のハイブリッド抗体の保持時間を算出した。
【0186】
カラム:ProPac WCX-10, 4×250 mm(Dionex)
移動相:A: 25 mmol/L MES/NaOH, pH 6.1
B: 25 mmol/L MES/NaOH, 250 mmol/L NaCl, pH 6.1
流速:0.5 mL/min
グラジエント:25 %B(5 min)→(105 min)→67 %B→(1 min)→100 %B (5 min)
検出:280 nm
【0187】
単独発現させた未改変、IgG2化およびIgG4化ヒト化PM-1抗体の分析結果を図17に示す。未改変ヒト化PM-1抗体、IgG2化ヒト化PM-1抗体およびIgG4化ヒト化PM-1抗体の保持時間はそれぞれ約60.2 min、30.5 minおよび30.3 minであり、アイソタイプ置換によって30分弱保持時間が変化した。一方、等電点電気泳動からpI差が認められたIgG2化ヒト化PM-1抗体とIgG4化ヒト化PM-1抗体の保持時間はほぼ同じであった。次に、未改変、IgG2化および未改変、IgG4化ヒト化PM-1抗体の共発現抗体分析結果を図18に示した。未改変ヒト化PM-1抗体/IgG2化ヒト化PM-1抗体の組み合わせ、および、未改変ヒト化PM-1抗体/IgG4化ヒト化PM-1抗体の組み合わせにおいて、各アイソタイプのホモダイマー、ヘテロダイマーが3つの主ピークとして観察された。保持時間は未改変ヒト化PM-1/IgG2化ヒトPM-1のハイブリッド抗体が約43.8 min、未改変ヒト化PM-1/IgG4化ヒトPM-1のハイブリッド抗体が約45.1 minであり、それぞれのホモダイマーとは10分以上の保持時間差で分離した。本検討において、各アイソタイプ抗体の発現ベクターを組み合わせて共発現させることでアイソタイプハイブリッド抗体を作成することが可能であり、それらがイオン交換クロマトグラフィーをもって分離可能であることが示された。
【0188】
〔実施例13〕アイソタイプハイブリッド抗体の陽イオン交換クロマトグラフィーによる分離精製
実施例11で得られた抗体溶液をAmicon-Ultra4(Amicon)で濃縮後EasySep(トミー精工)に封入し、5 mMクエン酸緩衝液(pH 6.5)に対し透析することで緩衝液を置換後、以下の条件でアイソタイプハイブリッド抗体を精製した。
【0189】
カラム:Poly CAT A, 4.6×100 mm, 粒子径 3μm, 孔径150 nm(Poly LC)
移動相:A: 25 mmol/L MES/NaOH, pH 6.1
B: 25 mmol/L MES/NaOH, 250 mmol/L 酢酸ナトリウム, pH 6.1
流速:1.0 mL/min
グラジエント:35 %B(5 min)→(54 min)→65 %B→(1 min)→100 %B (5 min)
検出:280 nm
【0190】
一回につき約100 - 200μgを注入し、未改変ヒト化PM-1抗体、未改変ヒト化PM-1/ IgG4化ヒト化PM-1アイソタイプハイブリット抗体、IgG4化ヒト化PM-1抗体ピークを分取した。分取時のクロマトグラムを図19に示す。複数回のピーク分取画分をそれぞれ混合してAmicon-Ultra4(Amicon)で濃縮後EasySep(トミー精工)に封入し、活性測定用としてPBSに、また、DSC測定用として150 mM NaClを含む20 mM 酢酸緩衝液、pH 6.0に透析することで緩衝液を置換した。分取ピークを上記同様の条件で再分析した結果を図20に示す。これより、アイソタイプハイブリット抗体がイオン交換クロマトグラフ法で分取精製可能であることが示された。
【0191】
本技術は、共通のH鎖可変領域を持つ抗体をpI値の異なるアイソタイプの定常領域を利用して分離することが出来たことから、pI差のない異なるH鎖可変領域であってもpI値の異なるアイソタイプのH鎖定常領域と連結することにより二重特異性抗体をイオン交換クロマトグラフィーで分離することが可能である。また、異なるH鎖可変領域を持つ場合、実施例9に示した可変領域への変異導入技術と組み合わせることで更に分子間のpI差を増大し、分離精製をより容易にすることができる。H鎖可変領域への変異導入が困難な場合、これらを天然に存在するIgGアイソタイプ配列に変換することで、抗原性を考慮することなく二重特異性抗体のイオン交換クロマトグラフィーによる分離精製が可能となる。
【0192】
〔実施例14〕アイソタイプハイブリッド抗体分取精製品の等電点電気泳動
分取品の純度を評価するために、等電点電気泳動による分析を実施した。
【0193】
等電点電気泳動は以下のとおり行った。Phastsystem Cassette(AmerchamBioscience社製)を用いて以下の膨潤液で30 minほどPhast-Gel Dry IEF(AmerchamBioscience社製)ゲルを膨潤させた。
【0194】
ミリQ水 1.5 mL
Pharmalyte 5-8 for IEF(AmerchamBioscience社製) 50μL
Pharmalyte 8-10.5 for IEF(AmerchamBioscience社製) 50μL
【0195】
膨潤したゲルを用いてPhastSystem(AmerchamBioscience社製)により以下のプログラムで電気泳動を行った。サンプルはStep 2でゲルに添加した。pIマーカーとして、Calibration Kit for pI(AmerchamBioscience社製)を使用した。
【0196】
Step 1: 2000 V 2.5 mA 3.5 W 15℃ 75 Vh
Step 2: 200 V 2.5 mA 3.5 W 15℃ 15 Vh
Step 3: 2000 V 2.5 mA 3.5 W 15℃ 410 Vh
【0197】
泳動後のゲルは20% TCAで固定した後、Silver staining Kit、protein(AmerchamBioscience社製)を用い、キットに添付されているプロトコールに従い銀染色を行った。
【0198】
アイソタイプハイブリッド抗体分取精製品の分析結果を図21に示す。イオン交換クロマトグラフィーにより、各アイソタイプのホモダイマーをほとんど含まずに精製できることが示された。
【0199】
〔実施例15〕アイソタイプハイブリッド抗体分取精製品の活性評価
15−1.ヒトgp130発現BaF3細胞株、ヒトgp130/ヒトIL-6受容体共発現BaF3細胞株の樹立
IL-6依存増殖性を示す細胞株を得るために、以下に示すとおり、ヒトgp130を発現したBaF3細胞株の樹立を行った。
全長ヒトgp130 cDNA (Hibiら、Cell 1990;63:1149-1157 (GenBank # NM_002184))をPCRにより増幅し、pCHOI (Hirataら、FEBS Letter 1994;356:244-248)のDHFR遺伝子発現部位を除去し、Zeocin耐性遺伝子発現部位を挿入した発現ベクターpCOS2Zeoにクローニングし、pCOS2Zeo/gp130を構築した。
【0200】
10μgのpCOS2Zeo/gp130をPBSに懸濁したBaF3細胞(0.8 x 107cells)に混合し、Gene Pulser(Bio-Rad)を用いて0.33 kV, 950μFDの容量でパルスを加えた。エレクトロポーレーション処理により遺伝子導入したBaF3細胞を0.2 ng/mLのmouse interleukin-3(Peprotech)、10% Fetal Bovine Serum(以下FBS、HyClone)を含むRPMI1640培地(Invitrogen)で一昼夜培養し、100 ng/mLのhuman interleukin-6(R&D)、100 ng/mL のhuman interleukin-6 soluble receptor(R&D systems)および10% FBSを含むRPMI1640培地を加えて選抜し、ヒトgp130発現BaF3細胞株(以下、BaF3/gp130)を樹立した。
【0201】
15−2.アイソタイプハイブリッド抗体分取精製品のヒトIL-6中和活性評価
IL-6依存性増殖を示すBaF3/gp130を用いて、以下に示すとおり、IL-6中和活性を評価した。精製した未改変ヒト化PM-1抗体、未改変/IgG4化ヒト化PM-1アイソタイプハイブリッド抗体およびIgG4化ヒト化PM-1抗体を10μg/mLになるように10% FBSを含むRPMI1640に希釈した。この溶液を用いて希釈公比3、合計7系列の希釈液を調製し、96well-plate(CORNING)の各wellに50μLずつ分注した。次に、BaF3/gp130を10% FBS(HyClone)を含むRPMI1640培地で3回洗浄した後に、5 x 104cells/mLとなるように60 ng/mLのhuman interleukin-6(TORAY)、60 ng/mL の可溶性ヒトIL-6受容体(自社調製品)および10% FBSを含むRPMI1640培地に懸濁し、各wellに50μLずつ混合した後、抗体サンプルを分注した。ヒト可溶性IL-6受容体は以下に示す方法で調製した。ヒト可溶性IL-6受容体(Yamasakiら、Science 1988;241:825-828(GenBank # X12830))の1番目から344番目のアミノ酸をコードする遺伝子をCHO細胞に導入後に培養上清から精製して調製した。37℃、5% CO2条件下で、72時間培養し、PBSで2倍に希釈したWST-8試薬(Cell Counting Kit-8、株式会社同仁化学研究所)を20μL/wellで加え、直後にSUNRISE CLASSIC(TECAN)を用いて450 nmの吸光度(参照波長620 nm)を測定した。2時間培養した後に、再度450 nmの吸光度(参照波長620 nm)を測定し、2時間の吸光度変化を指標にIL-6中和活性を評価した。
【0202】
その結果、図22に示すとおり、分取精製した未改変ヒト化PM-1抗体、未改変/IgG4化ヒト化PM-1アイソタイプハイブリッド抗体、およびIgG4化ヒト化PM-1抗体はヒト化PM-1抗体精製品(bulk)と中和活性が同等であった。以上より、アイソタイプハイブリット抗体は本来の抗原結合能を失わず、中和抗体としての機能を有することが示された。
【産業上の利用可能性】
【0203】
本発明の方法においては、アミノ酸の置換数が少数でよく、その構造・機能(活性)を変化させることなく等電点を制御させることが可能であり、それにより二重特異性抗体を汎用的なクロマトグラフィーカラムを用いることで効率的に、且つ、医薬品として開発可能な高純度まで精製することが可能になるため、医薬品として二重特異性抗体を開発する上で非常に有用性が高い。
本発明の方法を用いることにより、実際に活性を保持する二重特異性抗体の効率的な取得が可能である。
【技術分野】
【0001】
本発明は、二重特異性抗体を精製するための抗体改変方法、該二重特異性抗体の分離方法、および該二重特異性抗体を有効成分として含有する医薬組成物等に関する。
【背景技術】
【0002】
抗体は血中での安定性が高く、副作用も少ないことから医薬品として注目されている。その中には二種類の抗原(抗原Aと抗原B)を同時に認識できる二重特異性抗体がある(非特許文献1)。現在、臨床試験が行なわれているMDX-210は、FcγRIを発現しているmonocyte等をHER-2/neuを発現している癌細胞にretargetingするIgG型二重特異性抗体である(非特許文献2)。抗体の製造は、一般的に遺伝子組み換え技術を用いることが多い。具体的には、抗体の蛋白質をコードするDNAをハイブリドーマ、抗体を産生する感作リンパ球等の抗体産生細胞、または抗体遺伝子を提示しているファージライブラリーからクローニングし、適当なベクターに組み込み、これを宿主細胞に導入し産生させる技術である。遺伝子組み換え技術を用いたIgG型二重特異性抗体の製造は、目的の二種類のIgGを構成するH鎖及びL鎖の遺伝子、合計4種の遺伝子を細胞に導入し、共発現により分泌させる。このような発現において、野生型のH鎖及びL鎖の構成遺伝子を発現させた場合には、2種類のH鎖の会合やH鎖とL鎖の会合はランダムに起こるため、目的の二重特異性抗体の比率は極めて少なくなる。具体的には、目的の二重特異性抗体は10種類中1種類のみであり、生産効率は低下してしまう。目的抗体の生産効率の低下は、目的抗体の精製の障害になるばかりでなく、ロット間差などの不均一性を増大させ、生産コストの肥大を招くことになる。
【0003】
二重特異性抗体を開発するための効率的な二重特異性抗体作製方法として、両H鎖に共通するL鎖を取得するための共通L鎖取得技術およびH鎖へテロ会合化のためのKnobs-into-holes技術が報告されている。具体的には、抗原Aと抗原Bを認識する各H鎖に対して、両抗原結合活性を維持することができる共通のL鎖をファージライブラリー(Phage library)等から見出し、さらに、一方のH鎖のCH3領域に存在するアミノ酸側鎖をより大きい側鎖(knob;突起)に置換し、もう一方のH鎖のCH3領域に存在するアミノ酸側鎖をより小さい側鎖(hole;空隙)に置換することにより突起が空隙内に配置されるようにしてH鎖ヘテロダイマーの形成を促進し、目的の二重特異性抗体を効率的に取得することができる(特許文献1、非特許文献3、非特許文献4)。
【0004】
しかしながら、H鎖へテロダイマーのためにKnobs-into-holes技術を用いた場合でも、非特許文献3、非特許文献4に示されたようにKnobs-into-holes技術によって目的のA鎖B鎖へテロダイマーの含有率を最大95%程度まで高めることが可能だが、残りの5%はA鎖ホモダイマー、B鎖ホモダイマーであり不純物となってしまう。医薬品として、二重特異性抗体を開発するためには、共通L鎖(非特許文献3,非特許文献4)を用いた場合に産生される3種類の分子種(A鎖ホモダイマー、B鎖ホモダイマー、A鎖B鎖へテロダイマー)の中から、可能な限り高純度にA鎖B鎖へテロダイマーを精製する必要がある。そのため残りの5%の不純物であるA鎖ホモダイマー、B鎖ホモダイマーを取り除き、A鎖B鎖へテロダイマーを医薬品として開発可能な高純度まで精製する必要がある。共通L鎖を用い、Knobs-into-holes技術を用いない場合は、理論上、A鎖ホモダイマー、A鎖B鎖へテロダイマー、B鎖ホモダイマーが1:2:1で産生され、50%の不純物であるA鎖ホモダイマー、B鎖ホモダイマーを取り除く必要がある。
【0005】
医薬品の製造レベルのクロマトグラフィーによる分離において、A鎖B鎖へテロダイマーとA鎖ホモダイマー、B鎖ホモダイマーを分離する方法がこれまでにいくつか報告されている。A鎖B鎖へテロダイマーを選択的に精製する方法として、非特許文献5にはA鎖にmouse IgG2a、B鎖にrat IgG2bを使用し、mouse IgG2aとrat IgG2bの各H鎖に対するprotein Aへのアフィニティーの違いを利用し、protein Aからの溶出pHをコントロールすることで、A鎖B鎖へテロダイマーを精製する方法が報告されているが、mouseおよびratの定常領域を使用しているため、抗原性の観点から同方法はヒトに対する医薬品への応用は困難である。またこの方法では同じアイソタイプのH鎖からなるA鎖B鎖へテロダイマーを分離することはできないため、利用は限定される。
【0006】
非特許文献6には疎水相互作用クロマトグラフィーを用いたA鎖B鎖へテロダイマーを精製する方法が報告されているが、Anti-CD3 mouse IgG2aとanti-CD19 mouse IgG1からなる目的のA鎖B鎖へテロダイマーが十分にピーク分離しておらず、また、異なるアイソタイプのH鎖を用い、その疎水性の違いを利用して分離していると考えられることから、同じアイソタイプのH鎖からなるA鎖B鎖へテロダイマーを必ずしも分離できるとは限らない。
【0007】
非特許文献7にはThiophilic affinityクロマトグラフィーを用いたA鎖B鎖へテロダイマーを精製する方法が報告されているが、mouse IgG1とrat IgG2aを用い、そのヒンジ領域におけるフリーのシステイン(チオール基)を利用していることから、同じアイソタイプのH鎖からなるA鎖B鎖へテロダイマーの分離には利用できず、またフリーのシステインは保存中のアグリゲーションに関与するため、安定な医薬品製剤の開発には適さない。
【0008】
非特許文献8には抗原を用いたアフィニティークロマトグラフィーが報告されている。しかし、タンパク質あるいはペプチド抗原を用いたアフィニティークロマトグラフィーは、カラムのコストや安定性に課題があるため、アフィニティークロマトグラフィーを用いた医薬品の製造は一般的ではない。また両抗原に結合するA鎖B鎖へテロダイマーの精製のためにはアフィニティークロマトグラフィーを2回実施する必要があり、コストが高くなることが予想される。また抗原の立体構造のみを認識する抗体や低アフィニティで目的の機能を有する抗体が報告されており、このような性質を持つ抗体は抗原を利用したアフィニティクロマトグラフィーを使用することが困難である。従って、アフィニティークロマトグラフィーを用いた二重特異性抗体の精製は汎用的ではないと考えられる。
【0009】
このように二重特異性抗体のA鎖B鎖へテロダイマー精製は限られた範囲内でしか行われておらず、同じH鎖アイソタイプ・定常領域配列からなる二重特異性抗体のA鎖B鎖へテロダイマーを、医薬品として許容されるような高い純度まで精製する方法は報告されていない。二重特異性抗体を構成する2種類の抗体が同じ定常領域配列を有する場合、可変領域配列の違いのみにより、A鎖B鎖へテロダイマーを分離する必要があるが、抗体の可変領域のアミノ酸配列は抗体間で非常にホモロジーが高く(非特許文献9)、可変領域配列の違いだけでA鎖B鎖へテロダイマーを医薬品として許容されるような高い純度まで精製することは困難であった。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】国際公開第96/27011号
【非特許文献】
【0011】
【非特許文献1】Marvin JS,および Zhu Z.著、「Recombinant approaches to IgG-like bispecific antibodies.」、Acta Pharmacol Sin.、2005年Jun、Vol.26(6)、p.649-58.
【非特許文献2】Segal DM ら著、Current Opinion in Immunology、1999年、Vol.11、p.558-562.
【非特許文献3】Merchant AM, 外6名著、「An efficient route to human bispecific IgG.」、Nat Biotechnol.、1998年Jul、Vol.16(7)、p.677-81.
【非特許文献4】Carter P.著、「Bispecific human IgG by design.」、J Immunol Methods. 、2001年Feb 1、Vol.248(1-2)、p.7-15.
【非特許文献5】Lindhofer H, 外3名著、「Preferential species-restricted heavy/light chain pairing in rat/mouse quadromas. Implications for a single-step purification of bispecific antibodies.」、J Immunol.、1995年Jul 1、Vol.155(1)、p.219-25.
【非特許文献6】Manzke O, 外3名著、「Single-step purification of bispecific monoclonal antibodies for immunotherapeutic use by hydrophobic interaction chromatography.」、J Immunol Methods.、1997年Oct 13、Vol.208(1)、p.65-73.
【非特許文献7】Kreutz FT, 外2名著、「Efficient bispecific monoclonal antibody purification using gradient thiophilic affinity chromatography.」、J Chromatogr B Biomed Sci Appl.、1998年Sep 4、Vol.714(2)、p.161-70.
【非特許文献8】Gupta S, およびSuresh M.著、「Affinity chromatography and co-chromatography of bispecific monoclonal antibody immunoconjugates.」、J Biochem Biophys Methods. 、2002年May 31、Vol.51(3)、p.203-16. Review.
【非特許文献9】Carl Branden著、Introduction to Protein Structure 2nd edition, Newton Press.
【発明の概要】
【発明が解決しようとする課題】
【0012】
本発明はこのような状況に鑑みて為されたものであり、その目的は、二重特異性抗体を効率的に精製するための抗体可変領域のアミノ酸改変の方法、改変された二重特異性抗体からなる医薬組成物、並びに、二重特異性抗体医薬組成物の製造方法を提供することにある。また本発明は、重鎖定常領域が改変された二重特異性抗体、改変された二重特異性抗体からなる医薬組成物、並びに、二重特異性抗体医薬組成物の製造方法を提供することにある。
【課題を解決するための手段】
【0013】
本発明者らは、目的物の精製が従来困難であった二重特異性抗体を汎用的なクロマトグラフィーカラムを用いることで効率的に精製できる方法として、抗体可変領域のアミノ酸置換による方法について、鋭意研究を行った。
【0014】
その結果、二重特異性抗体を構成する2種類の抗体が抗体可変領域の表面に存在するアミノ酸を改変することによって、2種類の抗体のH鎖の間に等電点の差異を導入し、等電点の違いを利用して、二重特異性抗体をクロマトグラフィーカラムで効率的に精製する方法を見出した。具体的には、抗体のH鎖において、抗体の機能(活性)を低下させることなく、等電点のみを制御することが可能な改変箇所を見出した。さらに本発明者らは、本発明の方法によって取得された二重特異性抗体が、実際に機能を保持していることを確認した。
【0015】
上述の如く本発明者らは、任意の二重特異性抗体を汎用的なクロマトグラフィーカラムを用いることで効率的に精製できる方法として、抗体可変領域のアミノ酸置換による方法の開発に成功し、本発明を完成させた。
【0016】
また本発明者らは、二重特異性抗体を構成する2種類のH鎖の定常領域に対して、元来等電点に差のある異なるアイソタイプの定常領域をそれぞれのH鎖に用いることで、等電点の違いを利用して、二重特異性抗体をクロマトグラフィーカラムで効率的に精製する方法を見出した。さらに本発明者らは、本発明の方法によって取得された二重特異性抗体が、実際に機能を保持していることも確認した。
【0017】
本発明は、クロマトグラフィーカラムを用いることで効率的に精製するための抗体可変領域のアミノ酸置換方法、および、改変された二重特異性抗体からなる医薬組成物、および、二重特異性抗体医薬組成物の製造方法に関し、さらに重鎖定常領域が改変された二重特異性抗体、および、改変された二重特異性抗体からなる医薬組成物、および、二重特異性抗体医薬組成物の製造方法に関し、より具体的には、
〔1〕 第1のポリペプチドおよび第2のポリペプチドを含む多重特異性抗体の製造方法であって、
(a)第1のポリペプチドと第2のポリペプチドの等電点に差がつくように、第1のポリペプチドのアミノ酸残基をコードする核酸および第2のポリペプチドのアミノ酸残基をコードする核酸の両方またはいずれか一方を改変し、
(b)宿主細胞を該核酸が発現するように培養し、
(c)宿主細胞培養物から多重特異性抗体を回収すること、
を含む多重特異性抗体の製造方法、
〔2〕 工程(a)の改変が、第1のポリペプチドのホモ多量体、第2のポリペプチドのホモ多量体、および第1のポリペプチドと第2のポリペプチドのヘテロ多量体が、標準的なクロマトグラフィーを使用した分析により分離したピークとなるように、核酸を改変することである〔1〕に記載の方法、
〔3〕 前記第1のポリペプチドおよび前記第2のポリペプチドが重鎖可変領域を含む〔1〕に記載の方法、
〔4〕 前記多重特異性抗体が、軽鎖可変領域を含む第3のポリペプチドを含み、前記第1のポリペプチドおよび前記第2のポリペプチドがそれぞれ該第3のポリペプチドと多量体を形成する〔3〕に記載の方法、
〔5〕 前記第1のポリペプチドおよび前記第2のポリペプチドが重鎖定常領域を含む〔1〕〜〔4〕のいずれか1項に記載の方法、
〔6〕 前記第1のポリペプチドおよび第2のポリペプチドに含まれる重鎖定常領域が互いに等電点の異なる重鎖定常領域である〔5〕に記載の方法、
〔7〕 前記等電点の異なる重鎖定常領域がIgG1とIgG4、又は、IgG1とIgG2である〔6〕に記載の方法、
〔8〕 前記多重特異性抗体が、二重特異性抗体である〔1〕に記載の方法、
〔9〕 〔1〕に記載の方法により製造される多重特異性抗体、
〔10〕 第1のポリペプチドおよび第2のポリペプチドを含む多重特異性抗体の精製方法であって、
(a)第1のポリペプチドと第2のポリペプチドの等電点に差がつくように、第1のポリペプチドのアミノ酸残基をコードする核酸および第2のポリペプチドのアミノ酸残基をコードする核酸の両方またはいずれか一方を改変し、
(b)宿主細胞を該核酸が発現するように培養し、
(c)宿主細胞培養物から標準的なクロマトグラフィーにより該多重特異性抗体を精製すること、
を含む多重特異性抗体の精製方法、
〔11〕 工程(a)の改変が、第1のポリペプチドのホモ多量体、第2のポリペプチドのホモ多量体、および第1のポリペプチドと第2のポリペプチドのヘテロ多量体が、標準的なクロマトグラフィーを使用した分析により分離したピークとなるように、核酸を改変することである〔10〕に記載の方法、
〔12〕 前記第1のポリペプチドおよび前記第2のポリペプチドが重鎖可変領域を含む〔10〕に記載の方法、
〔13〕 前記多重特異性抗体が、軽鎖可変領域を含む第3のポリペプチドを含み、前記第1のポリペプチドおよび前記第2のポリペプチドがそれぞれ該第3のポリペプチドと多量体を形成する〔12〕に記載の方法、
〔14〕 前記第1のポリペプチドおよび前記第2のポリペプチドが重鎖定常領域を含む〔10〕〜〔13〕のいずれか1項に記載の方法、
〔15〕 前記第1のポリペプチドおよび第2のポリペプチドに含まれる重鎖定常領域が互いに等電点の異なる重鎖定常領域である〔14〕に記載の方法、
〔16〕 前記等電点の異なる重鎖定常領域がIgG1とIgG4、又は、IgG1とIgG2である〔15〕に記載の方法、
〔17〕 前記多重特異性抗体が、二重特異性抗体である〔10〕に記載の方法、
〔18〕 〔10〕に記載の方法により精製する工程を含む多重特異性抗体の製造方法、
〔19〕 〔18〕に記載の方法により製造される多重特異性抗体、
〔20〕 第1のポリペプチドおよび第2のポリペプチドを含む多重特異性抗体であって、第1のポリペプチドが重鎖可変領域および/または重鎖定常領域を含み、該重鎖可変領域におけるKabatナンバリングによる10位、12位、23位、39位、43位および105位のアミノ酸残基、若しくは、該重鎖定常領域におけるEUナンバリングによる137位、196位、203位、214位、217位、233位、268位、274位、276位、297位、355位、392位、419位、435位のアミノ酸残基から選ばれる、少なくとも1つのアミノ酸残基が電荷を有し、第1のポリペプチドと第2のポリペプチドの等電点が互いに異なる多重特異性抗体、
〔21〕 第2のポリペプチドが重鎖可変領域および/または重鎖定常領域を含み、該重鎖可変領域におけるKabatナンバリングによる10位、12位、23位、39位、43位および105位のアミノ酸残基、若しくは、該重鎖定常領域におけるEUナンバリングによる137位、196位、203位、214位、217位、233位、268位、274位、276位、297位、355位、392位、419位、435位のアミノ酸残基から選ばれる、少なくとも1つのアミノ酸残基が、前記第1のポリペプチドに含まれる重鎖可変領域および/または重鎖定常領域において選ばれる、電荷を有するアミノ酸残基とは反対の電荷を有する、または電荷を有しない〔20〕に記載の多重特異性抗体、
〔22〕 前記電荷を有するアミノ酸残基と当該アミノ酸残基とは反対の電荷を有するアミノ酸残基の組み合わせが、以下の(a)または(b)いずれかの群に含まれるアミノ酸残基からそれぞれ選択される〔20〕に記載の多重特異性抗体:
(a)グルタミン酸(E)、アスパラギン酸(D);
(b)リジン(K)、アルギニン(R)、ヒスチジン(H)、
〔23〕 第1のポリペプチドと第2ポリペプチドの等電点に差があり、第1のポリペプチドのホモ多量体、第2のポリペプチドのホモ多量体、および第1のポリペプチドと第2のポリペプチドのヘテロ多量体が、標準的なクロマトグラフィーを使用した分析により分離したピークとなり得る多重特異性抗体、
〔24〕 前記第1のポリペプチドおよび前記第2のポリペプチドが重鎖可変領域を含む〔23〕に記載の多重特異性抗体、
〔25〕 前記多重特異性抗体が、軽鎖可変領域を含む第3のポリペプチドを含み、前記第1のポリペプチドおよび前記第2のポリペプチドがそれぞれ該第3のポリペプチドと多量体を形成する〔24〕に記載の多重特異性抗体、
〔26〕 前記第1のポリペプチドおよび前記第2のポリペプチドが重鎖定常領域を含む〔23〕〜〔25〕のいずれか1項に記載の多重特異性抗体、
〔27〕 前記第1のポリペプチドおよび第2のポリペプチドに含まれる重鎖定常領域が互いに等電点の異なる重鎖定常領域である〔26〕に記載の多重特異性抗体、
〔28〕 前記等電点の異なる重鎖定常領域がIgG1とIgG4、又は、IgG1とIgG2である〔27〕に記載の多重特異性抗体、
〔29〕 前記多重特異性抗体が、二重特異性抗体である〔23〕に記載の多重特異性抗体、
〔30〕 〔23〕〜〔29〕のいずれか1項に記載の多重特異性抗体および医薬的に許容される担体を含む組成物、
〔31〕 〔23〕〜〔29〕のいずれか1項に記載の多重特異性抗体を構成するポリペプチドをコードする核酸、
〔32〕 〔31〕に記載の核酸を有する宿主細胞、
〔33〕 〔32〕に記載の宿主細胞を培養する工程、細胞培養物からポリペプチドを回収する工程を含む〔23〕〜〔29〕のいずれか1項に記載の多重特異性抗体の製造方法、
〔34〕 第1のポリペプチドの可変領域が以下の(a1)〜(a7)のいずれかに記載のアミノ酸配列からなり、第2のポリペプチドの可変領域が以下の(b1)〜(b3)のいずれかに記載のアミノ酸配列からなり、第3のポリペプチドの可変領域が以下の(c1)または(c2)に記載のアミノ酸配列からなる〔25〕に記載の多重特異性抗体:
(a1)配列番号:7
(a2)配列番号:8
(a3)配列番号:9
(a4)配列番号:10
(a5)配列番号:11
(a6)配列番号:12
(a7)配列番号:13
(b1)配列番号:14
(b2)配列番号:15
(b3)配列番号:16
(c1)配列番号:17
(c2)配列番号:18
〔35〕 第1のポリペプチドの可変領域が配列番号:11に記載のアミノ酸配列からなり、第2のポリペプチドの可変領域が配列番号:16に記載のアミノ酸配列からなり、第3のポリペプチドの可変領域が配列番号:17に記載のアミノ酸配列からなる〔34〕に記載の多重特異性抗体、
〔36〕 第1のポリペプチドの可変領域が配列番号:12に記載のアミノ酸配列からなり、第2のポリペプチドの可変領域が配列番号:16に記載のアミノ酸配列からなり、第3のポリペプチドの可変領域が配列番号:18に記載のアミノ酸配列からなる〔34〕に記載の多重特異性抗体、
〔37〕 第1のポリペプチドおよび第2のポリペプチドがヒトIgG4定常領域を含み、第3のポリペプチドがヒトκ定常領域を含む〔34〕〜〔36〕のいずれか1項に記載の多重特異性抗体、に関する。
【図面の簡単な説明】
【0018】
【図1】ヒト化二重特異性抗体(ヒト化A69(hA69a)/ ヒト化B26 (hB26-F123e4)/ヒト化BBA(hAL-F123j4))の凝固活性について評価した結果を示す図である。評価の結果、キメラ二重特異性抗体と同等以上の凝固活性を示した。
【図2】ヒト化A69-H鎖可変領域(hA69a)とヒト化BBA(hAL-F123j4)およびヒト化hB26-H鎖可変領域(hB26-F123e4)とヒト化BBA(hAL-F123j4)を使用して抗体モデリングを実施した結果を示す図である。表面電荷を変化させることが可能なアミノ酸について側鎖を強調して示した。番号については、Kabatデータベースの配列番号(Kabat EA et al. 1991. Sequences of Proteins of Immunological Interest.NIH)を採用した。
【図3】未改変および可変領域を改変したヒト化A69抗体ホモダイマーと未改変および可変領域を改変したヒト化B26抗体ホモダイマーを用いて等電点電気泳動による分析を実施した結果を示す写真である。分析の結果、改変により等電点が変化していることが確認された。
【図4】可変領域を改変したヒト化A69抗体ホモダイマーを用いて陽イオン交換クロマトグラフィー分析を実施した結果を示す図である。分析の結果、未改変の抗体と比較してピークの移動が確認された。
【図5】可変領域を改変したヒト化B26抗体ホモダイマーを用いて陽イオン交換クロマトグラフィー分析を実施した結果を示す図である。分析の結果、未改変の抗体と比較してピークの移動が確認された。
【図6】可変領域を改変したヒト化二重特異性抗体(H鎖定常領域にknobs-into-holes技術を利用)を用いて凝固活性を評価した結果を示す図である。評価の結果、未改変の抗体と同等の凝固活性を示した。
【図7】可変領域(CDR)を改変したヒト化A69抗体ホモダイマーを用いて等電点電気泳動による分析を実施した結果を示す写真である。分析の結果、未改変の抗体と比較してバンドの移動が確認された。
【図8】可変領域(CDR)を改変したヒト化A69抗体ホモダイマーを用いて抗原であるFactor IXaに対する結合活性を評価した結果を示す図である。評価の結果、未改変の抗体と同等の結合活性を保持していることが示された。
【図9】未改変抗体として、ヒト化A69-H鎖であるhA69a、ヒト化B26-H鎖であるhB26-F123e4とヒト化BBA-L鎖であるhAL-F123j4を用いて作製した未改変のヒト化二重特異性抗体ヒト化の陽イオン交換クロマトグラフィー分析を実施した結果を示す図である。分析の結果、二種類のホモダイマーと二重特異性抗体が分離せず1本のピークとして溶出した。
【図10】ヒト化A69-H鎖の改変体であるhA69-PFとヒト化B26-H鎖の改変体であるhA26-PFとヒト化BBA-L鎖であるhAL-s8を用いて作製したヒト化二重特異性PF抗体の陽イオン交換クロマトグラフィー分析を実施した結果を示す図である。分析の結果、二種類のホモダイマーと二重特異性抗体がそれぞれ分離し、hA69-PF ホモダイマー、ヒト化二重特異性PF抗体、hB26-PF ホモダイマーの順に3本のピークとして溶出した。
【図11】精製したヒト化A69抗体-PFホモダイマーおよびヒト化B26-PF抗体ホモダイマー、ヒト化二重特異性PF抗体を用いて等電点電気泳動による分析を実施した結果を示す写真である。分析の結果、目的の二重特異性抗体が精製できていることが確認された。
【図12】精製したヒト化二重特異性PF抗体(H鎖定常領域は野生型)を用いて凝固活性を評価した結果を示す図である。評価の結果、H鎖定常領域にknobs-into-holes技術を利用した二重特異性抗体(KiH)と同等の凝固活性を示した。
【図13】ヒト化A69抗体ホモダイマーおよびヒト化B26抗体ホモダイマー、ヒト化二重特異性抗体の3種類の抗体が含まれる培養上清から製造用汎用カラムを用いて二重特異性抗体を精製した時のクロマトグラムを示した。
【図14】製造用汎用カラムを用いて精製したヒト化二重特異性抗体(H鎖定常領域は野生型)を用いて凝固活性を評価した結果を示す図である。評価の結果、ヒト化二重特異性PF抗体と同等の凝固活性を示した。
【図15】未改変、IgG2化およびIgG4化ヒト化PM-1抗体を用いて等電点電気泳動による分析を実施した結果を示す写真である。分析の結果、改変により等電点が変化していることが確認された。Aは未改変ヒト化PM-1抗体、BはIgG2化ヒト化PM-1抗体、CはIgG4化ヒト化PM-1抗体を示す。
【図16】未改変、IgG2化およびIgG4化ヒト化PM-1抗体のそれぞれの共発現抗体を用いて等電点電気泳動による分析を実施した結果を示す写真である。分析の結果、各アイソタイプ抗体とアイソタイプハイブリッド抗体がpI差をもって分離することが示された。Aは未改変ヒト化PM-1抗体/IgG2化ヒト化PM-1抗体共発現抗体、Bは未改変ヒト化PM-1抗体/IgG4化ヒト化PM-1抗体共発現抗体、Cはヒト化PM-1抗体精製品(bulk)を示す。
【図17】単独発現させた未改変、IgG2化、IgG4化ヒト化PM-1抗体を用いて陽イオン交換クロマトグラフィー分析を実施した結果を示す図である。分析の結果、未改変の抗体と比較してピークの移動が確認された。
【図18】未改変、IgG2化およびIgG4化ヒト化PM-1抗体のそれぞれの共発現抗体の陽イオン交換クロマトグラフィー分析を実施した結果を示す図である。分析の結果、未改変ヒト化PM-1抗体/IgG2化ヒト化PM-1抗体の組み合わせ、および、未改変ヒト化PM-1抗体/IgG4化ヒト化PM-1抗体の組み合わせにおいて、各アイソタイプのホモダイマー、ヘテロダイマーが3つの主ピークとして観察された。Aは未改変ヒト化PM-1抗体/IgG2化ヒト化PM-1抗体共発現抗体、Bは未改変ヒト化PM-1抗体/IgG4化ヒト化PM-1抗体共発現抗体を示す。
【図19】未改変ヒト化PM-1抗体/IgG4化ヒト化PM-1抗体を共発現させたものから陽イオン交換クロマトグラフィーでホモダイマー、ヘテロダイマーを精製した結果を示す図である。この結果、IgG4化ヒト化PM-1抗体ホモダイマー、未改変ヒト化PM-1/IgG4化ヒト化PM-1ハイブリッド抗体、未改変ヒト化PM-1抗体ホモダイマーの順に3本のピークとして溶出したため、これらを分取した。矢印はおよその分画範囲を示す。
【図20】陽イオン交換クロマトグラフィーで精製した未改変ヒト化PM-1抗体ホモダイマー、未改変ヒト化PM-1/IgG4化ヒト化PM-1ハイブリッド抗体、IgG4化ヒト化PM-1抗体ホモダイマーを用いてリクロマトグラフィーを行った結果を示す図である。この結果、目的のアイソタイプハイブリッド抗体が精製できていることが確認された。
【図21】陽イオン交換クロマトグラフィーで精製した未改変ヒト化PM-1抗体ホモダイマー、未改変/IgG4化ヒト化PM-1ハイブリッド抗体、IgG4化ヒト化PM-1抗体ホモダイマーを用いて等電点電気泳動による分析を実施した結果を示す写真である。分析の結果、目的のアイソタイプハイブリッド抗体が精製できていることが確認された。Aは未改変ヒト化PM-1抗体/IgG4化ヒト化PM-1抗体共発現抗体、Bは未改変ヒト化PM-1抗体分取画分、Cは未改変ヒト化PM-1/IgG4化ヒト化PM-1ハイブリット抗体分取画分、DはIgG4化ヒト化PM-1抗体分取画分を示す。
【図22】陽イオン交換クロマトグラフィーで精製した未改変ヒト化PM-1抗体ホモダイマー、未改変ヒト化PM-1/IgG4化ヒト化PM-1ハイブリッド抗体、IgG4化ヒト化PM-1抗体ホモダイマーを用いてヒトIL-6中和活性を評価した結果を示す図である。評価の結果、いずれの抗体もヒト化PM-1精製抗体と同等の中和活性を示した。AおよびBはヒトgp130発現BaF3細胞株、CおよびDはヒトgp130/ヒトIL-6受容体共発現BaF3細胞株を示す。黒丸(●)はヒト化PM-1抗体精製品(bulk)、白四角(□)は未改変ヒト化PM-1抗体、白三角(△)はIgG4化ヒト化PM-1抗体、×は未改変ヒト化PM-1/IgG4化ヒト化PM-1ハイブリット抗体を示す。
【発明を実施するための形態】
【0019】
まず本発明は、多重特異性抗体を製造するための抗体改変方法を提供する。本発明の製造方法の好ましい態様としては、第1のポリペプチドと第2のポリペプチドの等電点に差がつくように第1のポリペプチドのアミノ酸残基をコードする核酸および第2のポリペプチドのアミノ酸残基をコードする核酸の両方またはいずれか一方を改変することを含む方法である。即ち、第1のポリペプチドと第2のポリペプチドのアミノ酸残基の電荷を変えることによって、ポリペプチドに等電点(pI)の差異を導入し、当該等電点の差異を利用して多重特異性抗体を製造することができる。詳しくは以下の(a)〜(c)の工程を含む製造方法である。
(a)第1のポリペプチドと第2のポリペプチドの等電点に差がつくように、第1のポリペプチドのアミノ酸残基をコードする核酸および第2のポリペプチドのアミノ酸残基をコードする核酸の両方またはいずれか一方を改変し、
(b)宿主細胞を該核酸が発現するように培養し、
(c)宿主細胞培養物から多重特異性抗体を回収すること
【0020】
本発明におけるポリペプチドとは、通常、10アミノ酸程度以上の長さを有するペプチド、およびタンパク質を指す。また、通常、生物由来のポリペプチドであるが、特に限定されず、例えば、人工的に設計された配列からなるポリペプチドであってもよい。また、天然ポリペプチド、あるいは合成ポリペプチド、組換えポリペプチド等のいずれであってもよい。さらに、上記のポリペプチドの断片もまた、本発明のポリペプチドに含まれる。
【0021】
本発明において「ポリペプチドの等電点に差がつく」とは、2種以上のポリペプチドにおいて、表面アミノ酸の電荷の改変を行うことにより、互いの等電点が等しくならないことをいう。等電点の差は、例えば、等電点電気泳動等の手法を用いることにより観察することができる。また本発明においては、当該ポリペプチドの構造や機能(活性)を変化させずに等電点を制御することが好ましい。
【0022】
即ち本発明は、第1のポリペプチドおよび第2のポリペプチドを含む多重特異性抗体の製造方法であって、
(a)第1のポリペプチドと第2のポリペプチドの等電点の差が0.5以上、好ましくは0.7以上、更に好ましくは0.9以上となるように、第1のポリペプチドのアミノ酸残基をコードする核酸および第2のポリペプチドのアミノ酸残基をコードする核酸の両方またはいずれか一方を改変し、
(b)宿主細胞を該核酸が発現するように培養し、
(c)宿主細胞培養物から多重特異性抗体を回収すること、を含む多重特異性抗体の製造方法を提供する。
【0023】
また本発明は、多重特異性抗体を精製するための抗体改変方法を提供する。本発明の精製方法の好ましい態様としては、第1のポリペプチドと第2のポリペプチドの等電点に差がつくように第1のポリペプチドのアミノ酸残基をコードする核酸および第2のポリペプチドのアミノ酸残基をコードする核酸の両方またはいずれか一方を改変することを含む方法である。即ち、第1のポリペプチドと第2のポリペプチドのアミノ酸残基の電荷を変えることによって、ポリペプチドに等電点(pI)の差異を導入し、当該等電点の差異を利用して多重特異性抗体を精製することができる。詳しくは以下の(a)〜(c)の工程を含む精製方法である。
(a)第1のポリペプチドと第2のポリペプチドの等電点に差がつくように、第1のポリペプチドのアミノ酸残基をコードする核酸および第2のポリペプチドのアミノ酸残基をコードする核酸の両方またはいずれか一方を改変し、
(b)宿主細胞を該核酸が発現するように培養し、
(c)宿主細胞培養物から標準的なクロマトグラフィーにより該多重特異性抗体を精製すること
【0024】
なお、上記精製方法により精製する工程を含む多重特異性抗体の製造方法も本発明に含まれる。
【0025】
本発明における核酸は、通常、適当なベクターへクローニング(挿入)され、宿主細胞へ導入される。該ベクターとしては、挿入した核酸を安定に保持するものであれば特に制限されず、例えば宿主に大腸菌を用いるのであれば、クローニング用ベクターとしてはpBluescriptベクター(Stratagene社製)などが好ましいが、市販の種々のベクターを利用することができる。本発明の多重特異性抗体(ポリペプチド)を生産する目的においてベクターを用いる場合には、特に発現ベクターが有用である。発現ベクターとしては、試験管内、大腸菌内、培養細胞内、生物個体内でポリペプチドを発現するベクターであれば特に制限されないが、例えば、試験管内発現であればpBESTベクター(プロメガ社製)、大腸菌であればpETベクター(Invitrogen社製)、培養細胞であればpME18S-FL3ベクター(GenBank Accession No. AB009864)、生物個体であればpME18Sベクター(Mol Cell Biol. 8:466-472(1988))などが好ましい。ベクターへの本発明のDNAの挿入は、常法により、例えば、制限酵素サイトを用いたリガーゼ反応により行うことができる(Current protocols in Molecular Biology edit. Ausubel et al. (1987) Publish. John Wiley & Sons.Section 11.4-11.11)。
【0026】
上記宿主細胞としては特に制限はなく、目的に応じて種々の宿主細胞が用いられる。ポリペプチドを発現させるための細胞としては、例えば、細菌細胞(例:ストレプトコッカス、スタフィロコッカス、大腸菌、ストレプトミセス、枯草菌)、真菌細胞(例:酵母、アスペルギルス)、昆虫細胞(例:ドロソフィラS2、スポドプテラSF9)、動物細胞(例:CHO、COS、HeLa、C127、3T3、BHK、HEK293、Bowes メラノーマ細胞)および植物細胞を例示することができる。宿主細胞へのベクター導入は、例えば、リン酸カルシウム沈殿法、電気パルス穿孔法(Current protocols in Molecular Biology edit. Ausubel et al. (1987) Publish. John Wiley & Sons.Section 9.1-9.9)、リポフェクション法、マイクロインジェクション法などの公知の方法で行うことが可能である。
【0027】
宿主細胞において発現したポリペプチドを小胞体の内腔に、細胞周辺腔に、または細胞外の環境に分泌させるために、適当な分泌シグナルを目的のポリペプチドに組み込むことができる。これらのシグナルは目的のポリペプチドに対して内因性であっても、異種シグナルであってもよい。
【0028】
上記製造方法における多重特異性抗体(ポリペプチド)の回収は、本発明のポリペプチドが培地に分泌される場合は、培地を回収する。本発明のポリペプチドが細胞内に産生される場合は、その細胞をまず溶解し、その後にポリペプチドを回収する。
【0029】
組換え細胞培養物から本発明のポリペプチドを回収し精製するには、硫酸アンモニウムまたはエタノール沈殿、酸抽出、アニオンまたはカチオン交換クロマトグラフィー、ホスホセルロースクロマトグラフィー、疎水性相互作用クロマトグラフィー、アフィニティクロマトグラフィー、ヒドロキシルアパタイトクロマトグラフィーおよびレクチンクロマトグラフィーを含めた公知の方法を用いることができる。
【0030】
また本発明は、本発明の多重特異性抗体、および医薬的に許容される担体を含む組成物(薬剤)に関する。
【0031】
本発明において医薬組成物とは、通常、疾患の治療もしくは予防、あるいは検査・診断のための薬剤を言う。
【0032】
本発明の医薬組成物は、当業者に公知の方法で製剤化することが可能である。例えば、水もしくはそれ以外の薬学的に許容し得る液との無菌性溶液、又は懸濁液剤の注射剤の形で非経口的に使用できる。例えば、薬理学上許容される担体もしくは媒体、具体的には、滅菌水や生理食塩水、植物油、乳化剤、懸濁剤、界面活性剤、安定剤、香味剤、賦形剤、ベヒクル、防腐剤、結合剤などと適宜組み合わせて、一般に認められた製薬実施に要求される単位用量形態で混和することによって製剤化することが考えられる。これら製剤における有効成分量は、指示された範囲の適当な容量が得られるように設定する。
【0033】
注射のための無菌組成物は注射用蒸留水のようなベヒクルを用いて通常の製剤実施に従って処方することができる。
【0034】
注射用の水溶液としては、例えば生理食塩水、ブドウ糖やその他の補助薬(例えばD-ソルビトール、D-マンノース、D-マンニトール、塩化ナトリウム)を含む等張液が挙げられる。適当な溶解補助剤、例えばアルコール(エタノール等)、ポリアルコール(プロピレングリコール、ポリエチレングリコール等)、非イオン性界面活性剤(ポリソルベート80(TM)、HCO-50等)を併用してもよい。
【0035】
油性液としてはゴマ油、大豆油があげられ、溶解補助剤として安息香酸ベンジル及び/またはベンジルアルコールを併用してもよい。また、緩衝剤(例えば、リン酸塩緩衝液及び酢酸ナトリウム緩衝液)、無痛化剤(例えば、塩酸プロカイン)、安定剤(例えば、ベンジルアルコール及びフェノール)、酸化防止剤と配合してもよい。調製された注射液は通常、適当なアンプルに充填する。
【0036】
本発明の医薬組成物は、好ましくは非経口投与により投与される。例えば、注射剤型、経鼻投与剤型、経肺投与剤型、経皮投与型の組成物とすることができる。例えば、静脈内注射、筋肉内注射、腹腔内注射、皮下注射などにより全身または局部的に投与することができる。
投与方法は、患者の年齢、症状により適宜選択することができる。抗体または抗体をコードするポリヌクレオチドを含有する医薬組成物の投与量は、例えば、一回につき体重1kgあたり0.0001mgから1000mgの範囲に設定することが可能である。または、例えば、患者あたり0.001〜100000mgの投与量とすることもできるが、本発明はこれらの数値に必ずしも制限されるものではない。投与量及び投与方法は、患者の体重、年齢、症状などにより変動するが、当業者であればそれらの条件を考慮し適当な投与量及び投与方法を設定することが可能である。
【0037】
また、必要に応じ本発明の多重特異性抗体を、その他の医薬成分と組み合わせて製剤化することもできる。
【0038】
また本発明は、本発明の多重特異性抗体を構成するポリペプチドをコードする核酸を提供する。さらに該核酸を担持するベクターもまた、本発明に含まれる。
【0039】
さらに本発明は、上記核酸を有する宿主細胞を提供する。該宿主細胞は、特に制限されず、例えば、大腸菌や種々の動物細胞などを挙げることができる。宿主細胞は、例えば、本発明の抗体もしくはポリペプチドの製造や発現のための産生系として使用することができる。ポリペプチド製造のための産生系には、in vitroおよびin vivoの産生系がある。in vitroの産生系としては、真核細胞を使用する産生系及び原核細胞を使用する産生系が挙げられる。
【0040】
宿主細胞として使用できる真核細胞として、例えば、動物細胞、植物細胞、真菌細胞が挙げられる。動物細胞としては、哺乳類細胞、例えば、CHO(J. Exp. Med. (1995) 108: 945)、COS、HEK293、3T3、ミエローマ、BHK(baby hamster kidney)、HeLa、Vero等、両生類細胞、例えばアフリカツメガエル卵母細胞(Valle et al., Nature (1981) 291: 338-340)、及び昆虫細胞、例えば、Sf9、Sf21、Tn5が例示される。本発明の抗体の発現においては、CHO-DG44、CHO-DX11B、COS7細胞、HEK293細胞、BHK細胞が好適に用いられる。動物細胞において、大量発現を目的とする場合には特にCHO細胞が好ましい。宿主細胞へのベクターの導入は、例えば、リン酸カルシウム法、DEAEデキストラン法、カチオニックリボソームDOTAP(Boehringer Mannheim製)を用いた方法、エレクトロポレーション法、リポフェクションなどの方法で行うことが可能である。
【0041】
植物細胞としては、例えば、ニコチアナ・タバカム(Nicotiana tabacum)由来の細胞およびウキクサ(Lemna minor)が蛋白質生産系として知られており、この細胞をカルス培養する方法により本発明の抗体を産生させることができる。真菌細胞としては、酵母、例えば、サッカロミセス(Saccharomyces)属の細胞(サッカロミセス・セレビシエ(Saccharomyces cerevisiae)、サッカロミセス・ポンベ(Saccharomyces pombe)等)、及び糸状菌、例えば、アスペルギルス(Aspergillus)属の細胞(アスペルギルス・ニガー(Aspergillus niger)等)を用いた蛋白質発現系が公知であり、本発明の抗体産生の宿主として利用できる。
【0042】
原核細胞を使用する場合、細菌細胞を用いる産生系がある。細菌細胞としては、上述の大腸菌(E. coli)に加えて、枯草菌を用いた産生系が知られており、本発明の抗体産生に利用できる。
【0043】
本発明の宿主細胞を用いて抗体を産生する場合、本発明の抗体をコードするポリヌクレオチドを含む発現ベクターにより形質転換された宿主細胞の培養を行い、ポリヌクレオチドを発現させればよい。培養は、公知の方法に従って行うことができる。例えば、動物細胞を宿主とした場合、培養液として、例えば、DMEM、MEM、RPMI1640、IMDMを使用することができる。その際、FBS、牛胎児血清(FCS)等の血清補液を併用しても、無血清培養により細胞を培養してもよい。培養時のpHは、約6〜8とするのが好ましい。培養は、通常、約30〜40℃で約15〜200時間行い、必要に応じて培地の交換、通気、攪拌を加える。
【0044】
一方、in vivoでポリペプチドを産生させる系としては、例えば、動物を使用する産生系や植物を使用する産生系が挙げられる。これらの動物又は植物に目的とするポリヌクレオチドを導入し、動物又は植物の体内でポリペプチドを産生させ、回収する。本発明における「宿主」とは、これらの動物、植物を包含する。
【0045】
動物を使用する場合、哺乳類動物、昆虫を用いる産生系がある。哺乳類動物としては、ヤギ、ブタ、ヒツジ、マウス、ウシ等を用いることができる(Vicki Glaser, SPECTRUM Biotechnology Applications (1993))。また、哺乳類動物を用いる場合、トランスジェニック動物を用いることができる。
【0046】
例えば、本発明の抗体をコードするポリヌクレオチドを、ヤギβカゼインのような乳汁中に固有に産生されるポリペプチドをコードする遺伝子との融合遺伝子として調製する。次いで、この融合遺伝子を含むポリヌクレオチド断片をヤギの胚へ注入し、この胚を雌のヤギへ移植する。胚を受容したヤギから生まれるトランスジェニックヤギ又はその子孫が産生する乳汁から、目的の抗体を得ることができる。トランスジェニックヤギから産生される抗体を含む乳汁量を増加させるために、適宜ホルモンをトランスジェニックヤギに投与してもよい(Ebert et al., Bio/Technology (1994) 12: 699-702)。
【0047】
また、本発明の抗体を産生させる昆虫としては、例えばカイコを用いることができる。カイコを用いる場合、目的の抗体をコードするポリヌクレオチドを挿入したバキュロウィルスをカイコに感染させることにより、このカイコの体液から目的の抗体を得ることができる(Susumu et al., Nature (1985) 315: 592-4)。
【0048】
さらに、植物を本発明の抗体産生に使用する場合、例えばタバコを用いることができる。タバコを用いる場合、目的とする抗体をコードするポリヌクレオチドを植物発現用ベクター、例えばpMON 530に挿入し、このベクターをアグロバクテリウム・ツメファシエンス(Agrobacterium tumefaciens)のようなバクテリアに導入する。このバクテリアをタバコ、例えば、ニコチアナ・タバカム(Nicotiana tabacum)に感染させ、本タバコの葉より所望の抗体を得ることができる(Ma et al., Eur. J. Immunol. (1994) 24: 131-8)。また、同様のバクテリアをウキクサ(Lemna minor)に感染させ、クローン化した後にウキクサの細胞より所望の抗体を得ることができる(Cox KM et al. Nat. Biotechnol. 2006 Dec;24(12):1591-1597)。
【0049】
このようにして得られた抗体は、宿主細胞内または細胞外(培地、乳汁など)から単離し、実質的に純粋で均一な抗体として精製することができる。抗体の分離、精製は、通常のポリペプチドの精製で使用されている分離、精製方法を使用すればよく、何ら限定されるものではない。例えば、クロマトグラフィーカラム、フィルター、限外濾過、塩析、溶媒沈殿、溶媒抽出、蒸留、免疫沈降、SDS-ポリアクリルアミドゲル電気泳動、等電点電気泳動法、透析、再結晶等を適宜選択、組み合わせて抗体を分離、精製することができる。
【0050】
クロマトグラフィーとしては、例えばアフィニティクロマトグラフィー、イオン交換クロマトグラフィー、疎水性クロマトグラフィー、ゲル濾過、逆相クロマトグラフィー、吸着クロマトグラフィー等が挙げられる(Strategies for Protein Purification and Characterization: A Laboratory Course Manual. Ed Daniel R. Marshak et al.(1996) Cold Spring Harbor Laboratory Press)。これらのクロマトグラフィーは、液相クロマトグラフィー、例えばHPLC、FPLC等の液相クロマトグラフィーを用いて行うことができる。アフィニティクロマトグラフィーに用いるカラムとしては、プロテインAカラム、プロテインGカラムが挙げられる。例えば、プロテインAを用いたカラムとして、Hyper D, POROS, Sepharose F. F. (Pharmacia製)等が挙げられる。
【0051】
必要に応じ、抗体の精製前又は精製後に適当なタンパク質修飾酵素を作用させることにより、任意に修飾を加えたり部分的にペプチドを除去することもできる。タンパク質修飾酵素としては、例えば、トリプシン、キモトリプシン、リシルエンドペプチダーゼ、プロテインキナーゼ、グルコシダーゼなどが用いられる。
【0052】
上述のように本発明の宿主細胞を培養し、該細胞培養物からポリペプチドを回収する工程を含む、本発明の多重特異性抗体の製造方法もまた、本発明の好ましい態様の一つである。
【0053】
本発明における「多重特異性抗体」とは、少なくとも2種類の異なる抗原に対して特異的に結合することができる抗体である。本発明の製造方法または精製方法によって得られる好ましい多重特異性抗体として、2つの抗原に対して特異的に結合することができる二重特異性抗体(bispecific antibody;BsAb)(二種特異性抗体と呼ばれる場合もある)を挙げることができる。
【0054】
本発明において、「異なる抗原」とは必ずしも抗原自体が異なる必要はなく、抗原決定基が異なる場合等も本発明の「異なる抗原」に含まれる。従って、例えば、単一分子内の異なる抗原決定基も本発明の異なる抗原に含まれ、このような単一分子内の異なる抗原決定基を各々認識する2つの抗体は、本発明において異なる抗原を認識する抗体として扱われる。
【0055】
本発明における多重特異性抗体は、2種以上の異なる抗原に対して特異性を有する抗体もしくは抗体断片からなる分子である。
【0056】
本発明の上記方法における「核酸の改変」とは、第1のポリペプチドと第2のポリペプチドを標準的なクロマトグラフィーを使用した分析により分離したピークが得られるように、核酸を改変することが含まれる。
【0057】
本発明の方法における、「核酸を改変する」とは、本発明における「改変」によって導入されるアミノ酸残基に対応するように核酸を改変することを言う。より具体的には、元(改変前)のアミノ酸残基をコードする核酸について、改変によって導入されるアミノ酸残基をコードする核酸へ改変することを言う。
【0058】
通常、目的のアミノ酸残基をコードするコドンとなるように、元の核酸に対して、少なくとも1塩基を挿入、欠失または置換するような遺伝子操作もしくは変異処理を行うことを意味する。即ち、元のアミノ酸残基をコードするコドンは、改変によって導入されるアミノ酸残基をコードするコドンによって置換される。このような核酸の改変は、当業者においては公知の技術、例えば、部位特異的変異誘発法、PCR変異導入法等を用いて、適宜実施することが可能である。
【0059】
本発明における改変位置は、例えば、(1)ポリペプチドに表面にあるアミノ酸残基、(2)可変領域、好ましくはFR領域にあるアミノ酸残基、(3)定常領域にあるアミノ酸残基、を挙げることができる。
【0060】
「ポリペプチドの表面にあるアミノ酸」とは、その側鎖が溶媒分子(通常は水分子)に接し得るアミノ酸であり、必ずしも側鎖の全てが溶媒分子に接する必要はなく、側鎖の一部でも溶媒分子に接する場合は、そのアミノ酸は表面にあるアミノ酸である。当業者であれば、市販のソフトウェアを用いたホモロジーモデリング等により、ポリペプチドや抗体のホモロジーモデルを作製することができ、それにより適切な残基を表面にあるアミノ酸として選択することができる。
【0061】
当業者であれば、抗体可変領域における表面アミノ酸はホモロジーモデリング等により作製されたホモロジーモデルにより適宜選択することが可能であるが、例えばH鎖FR領域においてはH1, H3, H5, H8, H10, H12, H13, H15, H16, H19, H23, H25, H26, H39, H42, H43, H44, H46, H68, H71, H72, H73, H75, H76, H81, H82b, H83, H85, H86, H105, H108, H110, H112が表面アミノ酸として例示することができるが、本発明はこれらに限定されることはない。またH鎖CDR領域に関しては、同様にしてホモロジーモデルにより表面アミノ酸を選択することが可能であり、例えばH97はほとんどの抗体で表面に露出している。L鎖FR領域においてはL1, L3, L7, L8, L9, L11, L12, L16, L17, L18, L20, L22, L38, L39, L41, L42, L43, L45, L46, L49, L57, L60, L63, L65, L66, L68, L69, L70, L74, L76, L77, L79, L80, L81, L85, L100, L103, L105, L106, L107, L108が表面アミノ酸として例示することができるが、本発明はこれらに限定されることはない。またL鎖CDR領域に関しては、同様にしてホモロジーモデルにより表面アミノ酸を選択することが可能である。
【0062】
本発明において可変領域にあるアミノ酸残基とは、重鎖可変領域(VH)または軽鎖可変領域(VL)にあるアミノ酸残基が含まれるが、好ましくは、フレームワーク領域(FR)にあるアミノ酸残基である。
【0063】
本発明におけるCDR以外のFR領域において表面に露出するアミノ酸としては、例えば、H10、H12、H23、H39、H43、H105を例示することができるが、これらに制限されない。
【0064】
本発明において、核酸を改変するポリペプチドは、好ましくは、第1のポリペプチドのホモ多量体、第2のポリペプチドのホモ多量体、および第1のポリペプチドと第2のポリペプチドのヘテロ多量体である。例えば、下記実施例に記載のように、第1のポリペプチドのホモ多量体としてヒト化A69-H鎖とヒト化BBA-L鎖のホモダイマー、第2のポリペプチドのホモ多量体としてヒト化B26-H鎖とヒト化BBA-L鎖のホモダイマー、および第1のポリペプチドと第2のポリペプチドのヘテロ多量体としてヒト化A69-H鎖およびヒト化B26-H鎖とヒト化BBA-L鎖のヘテロダイマーを挙げることができるが、これらに制限されない。
【0065】
本発明における標準的なクロマトグラフィーとしては、陽イオン交換クロマトグラフィー、陰イオン交換クロマトグラフィー、疎水クロマトグラフィー、ハイドロキシアパタイトクロマトグラフィー、疎水電荷相互作用クロマトグラフィー、クロマトフォーカシング等が挙げられる。
【0066】
本発明の上記方法において、第1のポリペプチドおよび第2のポリペプチドは、重鎖可変領域(VH)を含んでいることが好ましい。該可変領域には、例えば相補性決定領域(CDR)、フレームワーク領域(FR)が含まれていてもよい。
【0067】
本発明の方法において改変に供されるアミノ酸残基の数は、特に制限されないが、例えば、抗体の可変領域を改変する場合、抗原との結合活性を低下させないために、また抗原性を上げないために、目的のポリペプチドの分離を達成するための必要最低限のアミノ酸残基を改変することが好ましい。
【0068】
また好ましくは、抗原性を上げないために、改変後のアミノ酸配列がヒト配列であることが好ましいが本発明はこれに限定されることはない。さらに、改変後のFR(FR1、FR2、FR3、FR4)が各FRとしてヒト配列になるように、等電点が変化するように導入した改変以外の箇所に変異を導入してもよい。このようにして各FRをヒト配列に置き換える方法は非特許文献(Ono K et al., Mol Immunol. 1999 Apr;36(6):387-395.)で報告されている。また、各FRの等電点を変化させるために、等電点が変化する他のヒトFRに改変してもよい(例えばFR3を等電点が低下する他のヒトFRと交換してもよい)。このようなヒト化方法は非特許文献(Dall'Acqua WF., Methods. 2005 May;36(1):43-60.)で報告されている。
また、僅かな表面電荷の改変において目的のポリペプチドの分離が達成できない場合に、表面電荷の改変とポリペプチドの分離の評価を繰り返し行うことで、所望の多重特異性抗体を取得することが可能である。
【0069】
さらに本発明の上記方法においては、多重特異性抗体が、軽鎖可変領域を含む第3のポリペプチドを含んでいることが好ましい。そして、第1のポリペプチドおよび第2のポリペプチドがそれぞれ第3のポリペプチドと多量体を形成していることが好ましい。
【0070】
さらに本発明の上記方法において、第1のポリペプチドおよび第2のポリペプチドは、重鎖定常領域を含んでいることが好ましい。重鎖定常領域としては、第1のポリペプチドと第2のポリペプチドにpI差が生じるものがより好ましい。そのような重鎖定常領域としては、pI差を有する抗体の重鎖定常領域が挙げられ、元来pIに差のあるIgG1、IgG2、IgG3、IgG4の重鎖定常領域を用いて第1と第2のポリペプチドにpI差を導入することもできるし、第1と第2のポリペプチド中の重鎖定常領域における、これらアイソタイプ間の等電点の違いに起因するアミノ酸のみ、あるいはそれらの等電点には影響しない隣接するアミノ酸を同時に改変することにより非野生型ヒト定常領域を作製し、2つの定常領域にpI差を導入することもできる。定常領域にpI差を導入するための改変箇所としては、例えばH鎖定常領域のEUナンバリングで、H鎖137番目、196番目、203番目、214番目、217番目、233番目、268番目、274番目、276番目、297番目、355番目、392番目、419番目、435番目が挙げられる。
また、重鎖定常領域の糖鎖を除去することによりpI差が生じることから、糖鎖付加部位の297番目もpI鎖を導入するための改変箇所として挙げられる。
また本発明には、上記第1のポリペプチドおよび第2のポリペプチドが重鎖定常領域を含む方法に対して、上述の第1のポリペプチドおよび第2のポリペプチドが重鎖可変領域を含む方法、および/または前記多重特異性抗体が軽鎖可変領域を含む第3のポリペプチドを含み、前記第1のポリペプチドおよび前記第2のポリペプチドがそれぞれ該第3のポリペプチドと多量体を形成する方法とを組み合わせた方法も含まれる。
【0071】
さらに上記方法によって製造される多重特異性抗体も本発明に含まれる。
【0072】
さらに本発明によって提供される多重特異性抗体における第1のポリペプチドが重鎖可変領域および/または重鎖定常領域を含んでいる場合には、上記「等電点に差がつくよう」にするために、例えば該重鎖可変領域におけるKabatナンバリングによる10位、12位、23位、39位、43位および105位のアミノ酸残基、若しくは、該重鎖定常領域におけるEUナンバリングによる137位、196位、203位、214位、217位、233位、268位、274位、276位、297位、355位、392位、419位、435位のアミノ酸残基から選ばれる、少なくとも1つのアミノ酸残基が電荷を有するようにする態様を挙げることができる。上記のナンバリングで示された第1のポリペプチドのアミノ酸残基のうち、当該電荷を有するアミノ酸残基以外のアミノ酸残基は、第1のポリペプチドと第2のポリペプチドの等電点に差がついていれば、当該電荷を有するアミノ酸残基と同種の電荷であっても良いし、電荷を有していなくても、反対の電荷であっても良い。
【0073】
本発明の上記多重特異性抗体は、好ましくは、第2のポリペプチドが、第1のポリペプチドの電荷を有するアミノ酸残基とは反対の電荷を有する、または電荷を有しないことを特徴とする。詳しくは、第2のポリペプチドが重鎖可変領域および/または重鎖定常領域を含み、該重鎖可変領域におけるKabatナンバリングによる10位、12位、23位、39位、43位および105位のアミノ酸残基、若しくは、該重鎖定常領域におけるEUナンバリングによる137位、196位、203位、214位、217位、233位、268位、274位、276位、297位、355位、392位、419位、435位のアミノ酸残基から選ばれる、少なくとも1つのアミノ酸残基が、前記第1のポリペプチドに含まれる重鎖可変領域および/または重鎖定常領域において選ばれる、電荷を有するようにするアミノ酸残基とは反対の電荷を有する、または電荷を有しない、多重特異性抗体である。上記のナンバリングで示された第2のポリペプチドのアミノ酸残基のうち、当該電荷を有するアミノ酸残基以外のアミノ酸残基は、第1のポリペプチドと第2のポリペプチドの等電点に差がついていれば、当該電荷を有するアミノ酸残基と同種の電荷であっても良いし、電荷を有していなくても、反対の電荷であっても良い。
等電点を低下させるためには、例えば、137位はIgG2またはIgG4の配列、196位はIgG1またはIgG2またはIgG4の配列、203位はIgG2またはIgG4の配列、214位はIgG2の配列、217位はIgG1またはIgG3またはIgG4の配列、233位はIgG1またはIgG3またはIgG4の配列、268位はIgG4の配列、274位はIgG2またはIgG3またはIgG4の配列、276位はIgG1またはIgG2またはIgG4の配列、355位はIgG4の配列、392位はIgG3の配列、419位はIgG4の配列、435位はIgG1またはIgG2またはIgG4の配列、を適用することが望ましい。また、等電点を上昇させるためには、例えば、137位はIgG1またはIgG3の配列、196位はIgG3の配列、203位はIgG1またはIgG3の配列、214位はIgG1またはIgG3またはIgG4の配列、217位はIgG2の配列、233位はIgG2の配列、268位はIgG1またはIgG2またはIgG3の配列、274位はIgG1の配列、276位はIgG3の配列、355位はIgG1またはIgG2またはIgG3の配列、392位はIgG1またはIgG2またはIgG4の配列、419位はIgG1またはIgG2またはIgG3の配列、435位はIgG3の配列、を適用することが望ましい。
これらの配列の適用は、両H鎖に十分な等電点の差が付くようであればよく、必ずしも全ての配列を適用する必要はない。
【0074】
アミノ酸の中には、電荷を帯びたアミノ酸が知られている。一般的に正の電荷を帯びたアミノ酸(正電荷アミノ酸)としては、リジン(K)、アルギニン(R)、ヒスチジン(H)が知られている。負の電荷を帯びたアミノ酸(負電荷アミノ酸)としては、アスパラギン酸(D)、グルタミン酸(E)等が知られている。
【0075】
上記「電荷を有するアミノ酸残基」は、好ましくは、以下の(a)または(b)いずれかの群に含まれるアミノ酸残基から適宜選択されるが、特に制限されない。
(a)グルタミン酸(E)、アスパラギン酸(D)
(b)リジン(K)、アルギニン(R)、ヒスチジン(H)
【0076】
上記抗体において、「同種の電荷を有する」とは、例えば、重鎖可変領域における上記Kabatナンバリングによるアミノ酸残基、若しくは重鎖定常領域における上記EUナンバリングによるアミノ酸残基のいずれもが、上記(a)または(b)のいずれか1の群に含まれるアミノ酸残基を有することを意味する。
【0077】
また、「反対の電荷を有する」とは、例えば、重鎖可変領域および/または重鎖定常領域を有する第2のポリペプチドにおける上記Kabatナンバリング若しくは上記EUナンバリングによるアミノ酸残基のなかの少なくとも1つのアミノ酸残基が、第1のポリペプチドに含まれる重鎖可変領域および/または重鎖定常領域における対応する位置のアミノ酸残基であって、上記(a)または(b)のいずれか1の群に含まれるアミノ酸残基を有する場合に、残りのアミノ酸残基が異なる群に含まれるアミノ酸残基を有することを意味する。
【0078】
即ち本発明においては、前記同種の電荷を有するアミノ酸残基が、上記(a)または(b)のいずれかの群に含まれるアミノ酸残基から選択される多重特異性抗体を提供する。
【0079】
なお、元の(改変前の)アミノ酸残基が既に電荷を有する場合、電荷を有さないアミノ酸残基となるように改変することも本発明の好ましい態様の一つである。
【0080】
本発明においては、第1のポリペプチドと第2のポリペプチドの等電点(pI)に差が出るように、アミノ酸残基が改変されることが好ましい。また、改変によって導入されるアミノ酸残基が複数の場合、これらアミノ酸残基の中に電荷を持たないアミノ酸残基が少数程度含まれていてもよい。
【0081】
さらに本発明は、第1のポリペプチドの可変領域が以下の(a1)〜(a7)のいずれかに記載のアミノ酸配列からなり、第2のポリペプチドの可変領域が以下の(b1)〜(b3)のいずれかに記載のアミノ酸配列からなり、第3のポリペプチドの可変領域が以下の(c1)または(c2)に記載のアミノ酸配列からなる、多重特異性抗体を提供する。
(a1)配列番号:7
(a2)配列番号:8
(a3)配列番号:9
(a4)配列番号:10
(a5)配列番号:11
(a6)配列番号:12
(a7)配列番号:13
(b1)配列番号:14
(b2)配列番号:15
(b3)配列番号:16
(c1)配列番号:17
(c2)配列番号:18
なお上記のアミノ酸配列は、本発明において改変に供するアミノ酸をより具体的に例示するためのものであり、可変領域がこれらのアミノ酸である場合に限定されない。
【0082】
上記多重特異性抗体の好ましい態様の一つとして、例えば、第1のポリペプチドの可変領域が配列番号:11に記載のアミノ酸配列からなり、第2のポリペプチドの可変領域が配列番号:16に記載のアミノ酸配列からなり、第3のポリペプチドの可変領域が配列番号:17に記載のアミノ酸配列からなる多重特異性抗体を挙げることができる。
さらに別の好ましい態様の一つとして、例えば、第1のポリペプチドの可変領域が配列番号:12に記載のアミノ酸配列からなり、第2のポリペプチドの可変領域が配列番号:16に記載のアミノ酸配列からなり、第3のポリペプチドの可変領域が配列番号:18に記載のアミノ酸配列からなる多重特異性抗体を挙げることができる。
【0083】
さらに上記多重特異性抗体の好ましい態様の一つとして、第1のポリペプチドおよび第2のポリペプチドがヒトIgG4定常領域を含み、第3のポリペプチドがヒトκ定常領域を含んでいる多重特異性抗体を挙げることができる。
【0084】
本発明において、「抗体」という用語は最も広い意味で使用され、所望の生物学的活性を示す限り、モノクローナル抗体、ポリクローナル抗体、抗体変異体(キメラ抗体、ヒト化抗体、低分子化抗体(抗体断片も含む)、多重特異性抗体等)が含まれる。本発明においては、これら抗体の取得(作成)の際に、好適に本発明の抗体改変方法を用いることができる。
【0085】
本発明における「抗体」には、上述のようにアミノ酸残基の電荷を改変した抗体に対して、さらにアミノ酸の置換、欠失、付加及び/若しくは挿入等により、アミノ酸配列が改変された抗体が含まれる。また、アミノ酸の置換、欠失、付加及び/若しくは挿入、またはキメラ化やヒト化等により、アミノ酸配列が改変された抗体に対して、さらに、アミノ酸残基の電荷が改変された抗体が含まれる。すなわち、マウス抗体をヒト化する工程と同時に改変してもよく、あるいは、ヒト化抗体をさらに改変することであってもよい。
【0086】
アミノ酸の置換、欠失、付加及び/又は挿入、並びにヒト化、キメラ化などのアミノ酸配列の改変は、当業者に公知の方法により行うことが可能である。同様に、本発明における抗体を組換え抗体として作製する際に利用する抗体の可変領域及び定常領域も、アミノ酸の置換、欠失、付加及び/若しくは挿入、またはキメラ化やヒト化等によりそのアミノ酸配列を改変してもよい。
【0087】
本発明における抗体はマウス抗体、ヒト抗体、ラット抗体、ウサギ抗体、ヤギ抗体、ラクダ抗体など、どのような動物由来の抗体でもよい。さらに、例えば、キメラ抗体、中でもヒト化抗体などのアミノ酸配列を置換した改変抗体でもよい。また、各種分子を結合させた抗体修飾物、抗体断片、低分子抗体などいかなる抗体であってもよい。
【0088】
「キメラ抗体」とは、異なる動物由来の配列を組合わせて作製される抗体である。例えば、マウス抗体の重鎖、軽鎖の可変(V)領域とヒト抗体の重鎖、軽鎖の定常(C)領域からなる抗体を例示することができる。キメラ抗体の作製は公知であり、例えば、抗体V領域をコードするDNAをヒト抗体C領域をコードするDNAと連結し、これを発現ベクターに組み込んで宿主に導入し産生させることによりキメラ抗体を得ることができる。
【0089】
「ヒト化抗体」とは、再構成(reshaped)ヒト抗体とも称される、ヒト以外の哺乳動物由来の抗体、例えばマウス抗体の相補性決定領域(CDR;complementarity determining region)をヒト抗体のCDRへ移植したものである。CDRを同定するための方法は公知である(Kabat et al., Sequence of Proteins of Immunological Interest (1987), National Institute of Health, Bethesda, Md.; Chothia et al., Nature (1989) 342: 877)。また、その一般的な遺伝子組換え手法も公知である(欧州特許出願公開番号EP 125023号公報、WO 96/02576 号公報参照)。そこで公知の方法により、例えば、マウス抗体のCDRを決定し、該CDRとヒト抗体のフレームワーク領域(framework region;FR)とが連結された抗体をコードするDNAを取得し、ヒト化抗体を通常の発現ベクターを用いた系により産生することができる。このようなDNAは、CDR及びFR両方の末端領域にオーバーラップする部分を有するように作製した数個のオリゴヌクレオチドをプライマーとして用いてPCR法により合成することができる(WO98/13388号公報に記載の方法を参照)。CDRを介して連結されるヒト抗体のFRは、CDRが良好な抗原結合部位を形成するように選択される。必要に応じ、再構成ヒト抗体のCDRが適切な抗原結合部位を形成するように、抗体の可変領域におけるFRのアミノ酸を改変してもよい(Sato et al., Cancer Res. (1993) 53: 851-6)。改変できるFR中のアミノ酸残基には、抗原に直接、非共有結合により結合する部分(Amit et al., Science (1986) 233: 747-53)、CDR構造に影響または作用する部分(Chothia et al., J. Mol. Biol. (1987) 196: 901-17)及びVH-VL相互作用に関連する部分(EP239400号特許公報)が含まれる。
【0090】
本発明における抗体がキメラ抗体またはヒト化抗体である場合には、これらの抗体のC領域は,好ましくはヒト抗体由来のものが使用される。例えばH鎖では、Cγ1、Cγ2、Cγ3、Cγ4を、L鎖ではCκ、Cλを使用することができる。また、抗体またはその産生の安定性を改善するために、ヒト抗体C領域を必要に応じ修飾してもよい。本発明におけるキメラ抗体は、好ましくはヒト以外の哺乳動物由来抗体の可変領域とヒト抗体由来の定常領域とからなる。一方、ヒト化抗体は、好ましくはヒト以外の哺乳動物由来抗体のCDRと、ヒト抗体由来のFRおよびC領域とからなる。ヒト抗体由来の定常領域は、IgG(IgG1、IgG2、IgG3、IgG4)、IgM、IgA、IgD及びIgE等のアイソタイプごとに固有のアミノ酸配列を有する。本発明におけるヒト化抗体に用いられる定常領域は、どのアイソタイプに属する抗体の定常領域であってもよい。好ましくは、ヒトIgGの定常領域が用いられるが、これに限定されるものではない。また、ヒト化抗体に利用されるヒト抗体由来のFRも特に限定されず、どのアイソタイプに属する抗体のものであってもよい。
【0091】
本発明におけるキメラ抗体及びヒト化抗体の可変領域及び定常領域は、元の抗体の結合特異性を示す限り、欠失、置換、挿入及び/または付加等により改変されていてもよい。
【0092】
ヒト由来の配列を利用したキメラ抗体及びヒト化抗体は、ヒト体内における抗原性が低下しているため、治療目的などでヒトに投与する場合に有用と考えられる。
【0093】
また、低分子化抗体は、体内動態の性質の面からも、大腸菌、植物細胞等を用いて低コストで製造できる点からも抗体として有用である。
【0094】
抗体断片は低分子化抗体の一種である。また、低分子化抗体は、抗体断片をその構造の一部とする抗体も含む。本発明における低分子化抗体は、抗原への結合能を有していれば特にその構造、製造法等は限定されない。低分子化抗体の中には、全長抗体よりも高い活性を有する抗体も存在する(Orita et al., Blood(2005) 105: 562-566)。本明細書において、「抗体断片」とは、全長抗体(whole antibody、例えばwhole IgG等)の一部分であれば特に限定されないが、重鎖可変領域(VH)又は軽鎖可変領域(VL)を含んでいることが好ましい。好ましい抗体断片の例としては、例えば、Fab、F(ab')2、Fab'、Fvなどを挙げることができる。抗体断片中の、VHまたはVLのアミノ酸配列は、置換、欠失、付加及び/又は挿入により改変されていてもよい。さらに抗原への結合能を保持する限り、VH及びVLの一部を欠損させてもよい。例えば、前述の抗体断片のうち「Fv」は、完全な抗原認識部位と結合部位を含む最小の抗体断片である。「Fv」は、1つのVHおよび1つのVLが非共有結合により強く結合したダイマー(VH-VLダイマー)である。各可変領域の3つの相補鎖決定領域(complementarity determining region;CDR)によって、VH-VLダイマーの表面に抗原結合部位を形成する。6つのCDRが抗体に抗原結合部位を付与している。しかしながら、1つの可変領域(または、抗原に特異的な3つのCDRのみを含むFvの半分)であっても、全結合部位よりも親和性は低いが、抗原を認識し、結合する能力を有する。従って、このようなFvより小さい分子も本発明における抗体断片に含まれる。又、抗体断片の可変領域はキメラ化やヒト化されていてもよい。
【0095】
低分子化抗体は、VHとVLの両方を含んでいることが好ましい。低分子化抗体の例としては、Fab、Fab'、F(ab')2及びFv等の抗体断片、並びに、抗体断片を利用して作製され得るscFv(シングルチェインFv)(Huston et al., Proc. Natl. Acad. Sci. USA (1988) 85: 5879-83; Pluckthun「The Pharmacology of Monoclonal Antibodies」Vol.113, Resenburg 及び Moore編, Springer Verlag, New York, pp.269-315, (1994))、Diabody(Holliger et al., Proc. Natl. Acad. Sci. USA (1993) 90: 6444-8; EP404097号; WO93/11161号; Johnson et al., Method in Enzymology (1991) 203: 88-98; Holliger et al., Protein Engineering (1996) 9: 299-305; Perisic et al., Structure (1994) 2: 1217-26; John et al., Protein Engineering (1999) 12(7): 597-604; Atwell et al., Mol.Immunol. (1996) 33: 1301-12)、sc(Fv)2(Hudson et al、J Immunol. Methods (1999) 231: 177-89 ; Orita et al., Blood(2005) 105: 562-566)、Triabody(Journal of Immunological Methods (1999) 231: 177-89)、及びTandem Diabody(Cancer Research (2000) 60: 4336-41)等を挙げることができる。
【0096】
抗体断片は、抗体を酵素、例えばパパイン、ペプシン等のプロテアーゼにより処理して得ることができる(Morimoto et al., J. Biochem. Biophys. Methods (1992) 24: 107-17; Brennan et al., Science (1985) 229: 81参照)。また、該抗体断片のアミノ酸配列を基に、遺伝子組換えにより製造することもできる。
【0097】
抗体断片を改変した構造を有する低分子化抗体は、酵素処理若しくは遺伝子組換えにより得られた抗体断片を利用して構築することができる。又は、低分子化抗体全体をコードする遺伝子を構築し、これを発現ベクターに導入した後、適当な宿主細胞で発現させることもできる(例えば、Co et al., J. Immunol. (1994) 152: 2968-76; Better and Horwitz, Methods Enzymol. (1989) 178: 476-96; Pluckthun and Skerra, Methods Enzymol. (1989) 178: 497-515; Lamoyi, Methods Enzymol. (1986) 121: 652-63; Rousseaux et al., Methods Enzymol. (1986) 121: 663-9; Bird and Walker, Trends Biotechnol. (1991) 9: 132-7参照)。
【0098】
また、上記「scFv」は、2つの可変領域を、必要に応じリンカー等を介して、結合させた一本鎖ポリペプチドである。scFvに含まれる2つの可変領域は、通常、1つのVHと1つのVLであるが、2つのVH又は2つのVLであってもよい。一般にscFvポリペプチドは、VH及びVLドメインの間にリンカーを含み、それにより抗原結合のために必要なVH及びVLの対部分が形成される。通常、同じ分子内でVH及びVLの間で対部分を形成させるために、一般に、VH及びVLを連結するリンカーを10アミノ酸以上の長さのぺプチドリンカーとする。しかしながら、本発明におけるscFvのリンカーは、scFvの形成を妨げない限り、このようなペプチドリンカーに限定されるものではない。scFvの総説として、Pluckthun『The Pharmacology of Monoclonal Antibody』Vol.113(Rosenburg and Moore ed., Springer Verlag, NY, pp.269-315 (1994))を参照することができる。
【0099】
また、「ダイアボディ(diabody; Db)」は、遺伝子融合により構築された二価(bivalent)の抗体断片を指す(P.Holliger et al., Proc.Natl.Acad.Sci.USA 90: 6444-6448 (1993)、EP404,097号、WO93/11161号等)。ダイアボディは、2本のポリペプチド鎖から構成されるダイマーであり、ポリペプチド鎖は各々、同じ鎖中で軽鎖可変領域(VL)及び重鎖可変領域(VH)が、互いに結合できない位に短い、例えば、5残基程度のリンカーにより結合されている。同一ポリペプチド鎖上にコードされるVLとVHとは、その間のリンカーが短いため単鎖V領域フラグメントを形成することが出来ず二量体を形成するため、ダイアボディは2つの抗原結合部位を有することとなる。このとき2つの異なるエピトープ(a、b)に対するVLとVHをVLa-VHbとVLb-VHaの組合わせで5残基程度のリンカーで結んだものを同時に発現させると二重特異性Dbとして分泌される。
【0100】
Diabodyは、2分子のscFvを含むことから、4つの可変領域を含み、その結果、2つの抗原結合部位を持つこととなる。ダイマーを形成させないscFvの場合と異なり、Diabodyの形成を目的とする場合、通常、各scFv分子内のVH及びVL間を結ぶリンカーは、ペプチドリンカーとする場合には、5アミノ酸前後のものとする。しかしながら、Diabodyを形成するscFvのリンカーは、scFvの発現を妨げず、Diabodyの形成を妨げない限り、このようなペプチドリンカーに限定されない。
【0101】
本発明においてさらに好ましくは、多重特異性抗体として二重特異性抗体を挙げることができる。
なお、上記「二重特異性抗体」は、例えば、重鎖可変領域および軽鎖可変領域が1本鎖として連結した構造の抗体(例えば、sc(Fv)2)であってもよい。また重鎖可変領域(VH)および軽鎖可変領域(VL)が連結したscFv(あるいはsc(Fv)2)をFc領域(CH1ドメインを欠いた定常領域)と結合した抗体様分子(例えば、scFv-Fc)であってもよい。scFv-Fcからなる多重特異性抗体は第1のポリペプチドがVH1-linker-VL1-Fcであり、第2のポリペプチドがVH2-linker-VL2-Fcからなる(scFv)2-Fc型の構造をもつ。あるいはsingle domain antibodyをFc領域と結合させた抗体様分子であってもよい(Curr Opin Drug Discov Devel. 2006 , 9(2), 184-93)。
【0102】
本発明の方法における変異導入前の抗体(本明細書においては、単に「本発明の抗体」と記載する場合あり)のH鎖又はL鎖をコードする遺伝子は既知の配列を用いることも可能であり、又、当業者に公知の方法で取得することもできる。例えば、抗体ライブラリーから取得することも可能であるし、モノクローナル抗体を産生するハイブリドーマから抗体をコードする遺伝子をクローニングして取得することも可能である。
【0103】
抗体ライブラリーについては既に多くの抗体ライブラリーが公知になっており、又、抗体ライブラリーの作製方法も公知であるので、当業者は適宜抗体ライブラリーを入手することが可能である。例えば、抗体ファージライブラリーについては、Clackson et al., Nature 1991, 352: 624-8、Marks et al., J. Mol. Biol. 1991, 222: 581-97、Waterhouses et al., Nucleic Acids Res. 1993, 21: 2265-6、Griffiths et al., EMBO J. 1994, 13: 3245-60、Vaughan et al., Nature Biotechnology 1996, 14: 309-14、及び特表平20−504970号公報等の文献を参照することができる。その他、真核細胞をライブラリーとする方法(WO95/15393号パンフレット)やリボソーム提示法等の公知の方法を用いることが可能である。さらに、ヒト抗体ライブラリーを用いて、パンニングによりヒト抗体を取得する技術も知られている。例えば、ヒト抗体の可変領域を一本鎖抗体(scFv)としてファージディスプレイ法によりファージの表面に発現させ、抗原に結合するファージを選択することができる。選択されたファージの遺伝子を解析すれば、抗原に結合するヒト抗体の可変領域をコードするDNA配列を決定することができる。抗原に結合するscFvのDNA配列が明らかになれば、当該配列を元に適当な発現ベクターを作製し、ヒト抗体を取得することができる。これらの方法は既に周知であり、WO92/01047、WO92/20791、WO93/06213、WO93/11236、WO93/19172、WO95/01438、WO95/15388を参考にすることができる。
【0104】
ハイブリドーマから抗体をコードする遺伝子を取得する方法は、基本的には公知技術を使用し、所望の抗原または所望の抗原を発現する細胞を感作抗原として使用して、これを通常の免疫方法にしたがって免疫し、得られる免疫細胞を通常の細胞融合法によって公知の親細胞と融合させ、通常のスクリーニング法により、モノクローナルな抗体産生細胞(ハイブリドーマ)をスクリーニングし、得られたハイブリドーマのmRNAから逆転写酵素を用いて抗体の可変領域(V領域)のcDNAを合成し、これを所望の抗体定常領域(C領域)をコードするDNAと連結することにより得ることができる。
【0105】
より具体的には、特に以下の例示に限定される訳ではないが、上記のH鎖及びL鎖をコードする抗体遺伝子を得るための感作抗原は、免疫原性を有する完全抗原と、免疫原性を示さないハプテン等を含む不完全抗原の両方を含む。例えば、目的タンパク質の全長タンパク質、又は部分ペプチドなどを用いることができる。その他、多糖類、核酸、脂質等から構成される物質が抗原となり得ることが知られており、本発明の抗体の抗原は特に限定されるものではない。抗原の調製は、当業者に公知の方法により行うことができ、例えば、バキュロウィルスを用いた方法(例えば、WO98/46777など)などに準じて行うことができる。ハイブリドーマの作製は、たとえば、ミルステインらの方法(G. Kohler and C. Milstein, Methods Enzymol. 1981, 73: 3-46)等に準じて行うことができる。抗原の免疫原性が低い場合には、アルブミン等の免疫原性を有する巨大分子と結合させ、免疫を行えばよい。また、必要に応じ抗原を他の分子と結合させることにより可溶性抗原とすることもできる。受容体のような膜貫通分子を抗原として用いる場合、受容体の細胞外領域部分を断片として用いたり、膜貫通分子を細胞表面上に発現する細胞を免疫原として使用することも可能である。
【0106】
抗体産生細胞は、上述の適当な感作抗原を用いて動物を免疫化することにより得ることができる。または、抗体を産生し得るリンパ球をin vitroで免疫化して抗体産生細胞とすることもできる。免疫化する動物としては、各種哺乳動物を使用できるが、ゲッ歯目、ウサギ目、霊長目の動物が一般的に用いられる。マウス、ラット、ハムスター等のゲッ歯目、ウサギ等のウサギ目、カニクイザル、アカゲザル、マントヒヒ、チンパンジー等のサル等の霊長目の動物を例示することができる。その他、ヒト抗体遺伝子のレパートリーを有するトランスジェニック動物も知られており、このような動物を使用することによりヒト抗体を得ることもできる(WO96/34096; Mendez et al., Nat. Genet. 1997, 15: 146-56参照)。このようなトランスジェニック動物の使用に代えて、例えば、ヒトリンパ球をin vitroで所望の抗原または所望の抗原を発現する細胞で感作し、感作リンパ球をヒトミエローマ細胞、例えばU266と融合させることにより、抗原への結合活性を有する所望のヒト抗体を得ることもできる(特公平1-59878号公報参照)。また、ヒト抗体遺伝子の全てのレパートリーを有するトランスジェニック動物を所望の抗原で免疫することで所望のヒト抗体を取得することができる(WO93/12227、WO92/03918、WO94/02602、WO96/34096、WO96/33735参照)。
【0107】
動物の免疫化は、例えば、感作抗原をPhosphate-Buffered Saline(PBS)または生理食塩水等で適宜希釈、懸濁し、必要に応じてアジュバントを混合して乳化した後、動物の腹腔内または皮下に注射することにより行われる。その後、好ましくは、フロイント不完全アジュバントに混合した感作抗原を4〜21日毎に数回投与する。抗体の産生の確認は、動物の血清中の目的とする抗体力価を慣用の方法により測定することにより行われ得る。
【0108】
ハイブリドーマは、所望の抗原で免疫化した動物またはリンパ球より得られた抗体産生細胞を、慣用の融合剤(例えば、ポリエチレングリコール)を使用してミエローマ細胞と融合して作成することができる(Goding, Monoclonal Antibodies: Principles and Practice, Academic Press, 1986, 59-103)。必要に応じハイブリドーマ細胞を培養・増殖し、免疫沈降、放射免疫分析(RIA)、酵素結合免疫吸着分析(ELISA)等の公知の分析法により該ハイブリドーマより産生される抗体の結合特異性を測定する。その後、必要に応じ、目的とする特異性、親和性または活性が測定された抗体を産生するハイブリドーマを限界希釈法等の手法によりサブクローニングすることもできる。
【0109】
続いて、選択された抗体をコードする遺伝子をハイブリドーマまたは抗体産生細胞(感作リンパ球等)から、抗体に特異的に結合し得るプローブ(例えば、抗体定常領域をコードする配列に相補的なオリゴヌクレオチド等)を用いてクローニングすることができる。また、mRNAからRT-PCRによりクローニングすることも可能である。免疫グロブリンは、IgA、IgD、IgE、IgG及びIgMの5つの異なるクラスに分類される。さらに、これらのクラスは幾つかのサブクラス(アイソタイプ)(例えば、IgG-1、IgG-2、IgG-3、及びIgG-4;IgA-1及びIgA-2等)に分けられる。本発明において抗体の製造に使用するH鎖及びL鎖は、これらいずれのクラス及びアイソタイプに属する抗体に由来するものであってもよく、特に限定されないが、IgGは特に好ましいものである。
【0110】
ここで、H鎖及びL鎖をコードする遺伝子を遺伝子工学的手法により改変することも可能である。例えば、マウス抗体、ラット抗体、ウサギ抗体、ハムスター抗体、ヒツジ抗体、ラクダ抗体等の抗体について、ヒトに対する異種抗原性を低下させること等を目的として人為的に改変した遺伝子組換え型抗体、例えば、キメラ抗体、ヒト化抗体等を適宜作製することができる。キメラ抗体は、ヒト以外の哺乳動物、例えば、マウス抗体のH鎖、L鎖の可変領域とヒト抗体のH鎖、L鎖の定常領域からなる抗体であり、マウス抗体の可変領域をコードするDNAをヒト抗体の定常領域をコードするDNAと連結し、これを発現ベクターに組み込んで宿主に導入し産生させることにより得ることができる。ヒト化抗体は、再構成(reshaped)ヒト抗体とも称され、ヒト以外の哺乳動物、たとえばマウス抗体の相補性決定領域(CDR; complementary determining region) を連結するように設計したDNA配列を、末端部にオーバーラップする部分を有するように作製した数個のオリゴヌクレオチドからPCR法により合成する。得られたDNAをヒト抗体定常領域をコードするDNAと連結し、次いで発現ベクターに組み込んで、これを宿主に導入し産生させることにより得られる(EP239400; WO96/02576参照)。CDRを介して連結されるヒト抗体のFRは、相補性決定領域が良好な抗原結合部位を形成するものが選択される。必要に応じ、再構成ヒト抗体の相補性決定領域が適切な抗原結合部位を形成するように抗体の可変領域のフレームワーク領域のアミノ酸を置換してもよい(K. Sato et al., Cancer Res. 1993, 53: 851-856)。
【0111】
上述のヒト化以外に、例えば、抗原との結合性等の抗体の生物学的特性を改善するために改変を行うことも考えられる。本発明における改変は、部位特異的突然変異(例えば、Kunkel (1985) Proc. Natl. Acad. Sci. USA 82: 488参照)、PCR変異、カセット変異等の方法により行うことができる。一般に、生物学的特性の改善された抗体変異体は70%以上、より好ましくは80%以上、さらに好ましくは90%以上(例えば、95%以上、97%、98%、99%等)のアミノ酸配列相同性及び/または類似性を元となった抗体の可変領域のアミノ酸配列に対して有する。本明細書において、配列の相同性及び/または類似性は、配列相同性が最大の値を取るように必要に応じ配列を整列化、及びギャップ導入した後、元となった抗体残基と相同(同じ残基)または類似(一般的なアミノ酸の側鎖の特性に基き同じグループに分類されるアミノ酸残基)するアミノ酸残基の割合として定義される。通常、天然のアミノ酸残基は、その側鎖の性質に基づいて(1)疎水性:アラニン、イソロイシン、バリン、メチオニン及びロイシン;(2)中性親水性:アスパラギン、グルタミン、システイン、スレオニン及びセリン;(3)酸性:アスパラギン酸及びグルタミン酸;(4)塩基性:アルギニン、ヒスチジン及びリジン;(5)鎖の配向に影響する残基:グリシンおよびプロリン;ならびに(6)芳香族性:チロシン、トリプトファン及びフェニルアラニンのグループに分類される。
【0112】
通常、H鎖及びL鎖の可変領域中に存在する全部で6つの相補性決定領域(超可変部;CDR)が相互作用し、抗体の抗原結合部位を形成している。このうち1つの可変領域であっても全結合部位を含むものよりは低い親和性となるものの、抗原を認識し、結合する能力があることが知られている。従って、本発明のH鎖及びL鎖をコードする抗体遺伝子は、該遺伝子によりコードされるポリペプチドが所望の抗原との結合性を維持していればよく、H鎖及びL鎖の各々の抗原結合部位を含む断片部分をコードしていればよい。
本発明の方法によって、上述のように、例えば、所望の、実際に活性を保持する、二重特異性抗体を効率的に取得することができる。
【0113】
重鎖可変領域は、上述のように、通常3つのCDR領域と4つのFR領域によって構成されている。本発明の好ましい態様において「改変」に供するアミノ酸残基としては、例えば、CDR領域あるいはFR領域に位置するアミノ酸残基の中から適宜選択することができる。一般的にCDR領域のアミノ酸残基の改変は、抗原に対する結合能を低下させる場合がある。従って、本発明において「改変」に供するアミノ酸残基としては、特に限定されるものではないが、FR領域に位置するアミノ酸残基の中から適宜選択することが好ましい。
【0114】
また、ヒトもしくはマウス等の生物において、抗体の可変領域のFRとして利用可能な配列を、当業者であれば、公共のデータベース等を利用して適宜取得することができる。より具体的には、後述の実施例に記載の手段にて、FR領域のアミノ酸配列情報を取得することが可能である。
なお本明細書において引用されたすべての先行技術文献は、参照として本明細書に組み入れられる。
【実施例】
【0115】
以下、本発明を実施例により具体的に説明するが、本発明はこれら実施例に制限されるものではない。
【0116】
〔実施例1〕ハイブリッドL鎖を持つ二重特異性抗体のヒト化
特願2005-112514において血液凝固時間の短縮効果が最も高かった抗FactorIXa抗体 A69-VH、抗FactorX抗体 B26-VH、ハイブリッドL鎖(BBA)の組み合わせから成る二重特異性抗体について、以下のようにヒト化を実施した。
【0117】
1−1.ヒト抗体の相同性検索
一般公開されているKabat Database (ftp://ftp.ebi.ac.uk/pub/databases/kabat/) およびIMGT Database (http://imgt.cines.fr/)よりヒト抗体アミノ酸配列データを入手し、構築したデータベースを用いてマウスA69-H鎖可変領域(アミノ酸配列:配列番号:19)、マウスB26-H鎖可変領域(アミノ酸配列:配列番号:20)、マウスBBA-L鎖可変領域(アミノ酸配列:配列番号:21)に分けてホモロジー検索を行った。その結果、以下に示すヒト抗体配列と高い相同性を持つことが確認されたことから、ヒト化抗体のフレームワーク領域(以下、FR)に使用することにした。
【0118】
(1)A69-H鎖可変領域:KABATID-000064 (Kabat Database)
(Kippsら、J Clin Invest. 1991;87:2087-2096)
(2)B26-H鎖可変領域:EMBL Accession No. AB063872(IMGT Database)
(Unpublished data)
(3)BBA-L鎖可変領域:KABATID-024300 (Kabat Database)
(Welschofら、J Immunol Method. 1995;179:203-214)
【0119】
(1)-(3)のヒト抗体のFRに各マウス抗体の相補性抗原決定領域(以下、CDR)を移植したヒト化抗体を作製した。
【0120】
また、NCBIより一般公開されている相同性検索Web site(http://www.ncbi. nlm.nih.gov/BLAST/)を使用して、(4)-(6)のヒト抗体に相同性の高いヒト抗体の分泌シグナル配列を検索した。検索により得られた以下に示す分泌シグナル配列を使用した。
(4)A69-H鎖可変領域:GenBank Accession No. AF062257
(5)B26-H鎖可変領域:GenBank Accession No. AAC18248
(6)BBA-L鎖可変領域:GenBank Accession No. AAA59100
【0121】
1−2.ヒト化抗体遺伝子発現ベクターの構築
分泌シグナル配列から抗体可変領域にいたるアミノ酸配列をコードする塩基配列において、50 base程度の合成オリゴDNAを3'末端側が約20 base程度ハイブリダイズするように交互に12本作製した。合成オリゴDNAは5'末端側にヒト配列、3'末端側にマウス配列をコードするか、または全塩基がヒト配列をコードするように設計した。さらに、抗体可変領域遺伝子の5'末端にアニ−ルし、XhoI切断配列を有するプライマーと抗体可変領域遺伝子の3'末端にアニ−ルし、SfiI切断配列を有し且つイントロン配列の5'末端配列をコードするプライマーを作製した。
【0122】
2.5μMに調製した合成オリゴDNAを各1μLで混合し、1x TaKaRa Ex Taq Buffer, 0.4 mM dNTPs, 0.5 units TaKaRa Ex Taq(全て宝酒造)を加え、反応液48μLになるように調製した。94℃ 5分保温した後に、94℃ 2分、55℃ 2分、72℃ 2分からなる反応を2サイクル行い、各合成オリゴDNAのアッセンブルおよび伸長反応を実施した。次に、抗体遺伝子の5'末端および3'末端にアニ−ルするプライマー(各10μM)を1μL添加し、94℃ 30秒、55℃ 30秒、72℃ 1分からなる反応を35サイクル行い、72℃ 5分反応させ、抗体可変領域遺伝子を増幅した。PCR後、反応液全量を1 %アガローズゲル電気泳動に供した。目的のサイズ(約400 bp)の増幅断片をQIAquick Gel Extraction Kit(QIAGEN)を用いて、添付説明書記載の方法で精製し、滅菌水30μlで溶出した。該断片をpGEM-T Easy Vector Systems (Promega)を用いて、添付説明書記載の方法でクローニングを行った。各DNA断片の塩基配列は、BigDye Terminator Cycle Sequencing Kit(Applied Biosystems)を用い、DNAシークエンサーABI PRISM 3730xL DNA SequencerまたはABI PRISM 3700 DNA Sequencer(Applied Biosystems)にて、添付説明書記載の方法に従い決定した。
【0123】
正しいヒト化抗体可変領域遺伝子配列であることが確認されたH鎖可変領域断片挿入プラスミドをXhoIおよびSfiIで、L鎖可変領域断片挿入プラスミドをEcoRIで消化した後に、反応液を1 %アガローズゲル電気泳動に供した。目的のサイズ(約400 bp)のDNA断片をQIAquick Gel Extraction Kit(QIAGEN)を用いて、添付説明書記載の方法で精製し、滅菌水30μlで溶出した。その後、以下のようにして動物細胞用発現ベクターを作製した。H鎖がヘテロな組み合わせであるIgG4を優先的に発現させるために、IgG1のknobs-into-hole技術(Merchant AM ら、Nature Biotechnology、1998年、Vol.16、p.677-681)を参考にIgG4のCH3部分へのアミノ酸置換体を用いた。さらにH鎖のダイマー形成促進のためにヒンジにもアミノ酸置換(-ppcpScp- →-ppcpPcp-)を導入した。ニワトリβアクチンプロモーターを有するpCAGGS(Niwa ら、Gene、1991年、Vol. 108、p.193-199)にY349C、T366Wに置換した定常領域遺伝子を組み込んだ発現ベクターにヒト化A69 H鎖可変領域抗体遺伝子断片を挿入し、ヒト化A69H鎖発現ベクターを作製した。また、pCAGGSにE356C、T366S、L368A、Y407Vに置換した定常領域遺伝子を組み込んだ発現ベクターにヒト化B26 H鎖可変領域抗体遺伝子断片を挿入し、ヒト化B26H鎖発現ベクターを作製した。また、pCAGGSに野生型の抗体L鎖定常領域が挿入されたプラスミド(pCAG-gκDNA)をEcoRIで消化し、ヒト化BBA L鎖可変領域抗体遺伝子断片を挿入した発現ベクターを作製した。連結反応はRapid DNA Ligation Kit (Roche Diagnostics)を用い、大腸菌DH5α株 (東洋紡績)を形質転換した。
【0124】
1−3.ヒト化二重特異性抗体の発現
ヒト化二重特異性抗体の発現は、以下の方法を用いて行った。ヒト胎児腎癌細胞由来ヒト化二重特異性抗体の発現は、実施例1−2に記載した方法か以下の方法を用いて行った。ヒト胎児腎癌細胞由来HEK293H株(Invitrogen)を10 % Fetal Bovine Serum (Invitrogen)を含むDMEM培地(Invitrogen)へ懸濁し、5〜6 × 105個 /mLの細胞密度で接着細胞用ディッシュ(直径10 cm, CORNING)の各ディッシュへ10 mLずつ蒔きこみCO2インキュベーター(37℃、5 % CO2)内で一昼夜培養した後に、培地を吸引除去し、1 %のFetal Bovine Serum(Invitrogen)を含むCHO-S-SFM-II(Invitrogen)培地6.9 mLを添加した。1−2で調製したプラスミドDNA混合液(合計13.8μg)を1μg/mL Polyethylenimine (Polysciences Inc.) 20.7μLとCHO-S-SFMII培地 690μLと混合して室温10分間静置したものを各ディッシュの細胞へ投入し、4〜5時間、CO2インキュベーター(37℃にて5 % CO2)内でインキュベートした。その後、1 %のFetal Bovine Serum(Invitrogen)を含むCHO-S-SFM-II(Invitrogen)培地6.9 mLを添加して、3日間 CO2インキュベーター内で培養した。培養上清を回収した後、遠心分離(約2000 g、5分間、室温)して細胞を除去し、さらに0.22μmフィルターMILLEX(R)-GV(Millipore)を通して滅菌した。該サンプルは使用するまで4℃で保存した。
【0125】
1−4.ヒト化二重特異性抗体の精製
実施例1−2に記載の方法で得られた培養上清に100μLのrProtein A SepharoseTM Fast Flow(Amersham Biosciences)を添加し、4℃で4時間以上転倒混和した。その溶液を0.22μmのフィルターカップUltrafree(R)-MC(Millipore)に移し、0.01 % Tween(R) 20を含むTBS 500μLにて3回洗浄後、rProtein A SepharoseTM樹脂に100μLの 0.01 % Tween(R) 20を含む50 mM 酢酸ナトリウム水溶液, pH 3.3に懸濁して2分間静置したのち、抗体を溶出させた。直ちに、6.7μLの1.5 M Tris-HCl , pH 7.8を加えて中和した。
【0126】
1−5.ヒト化二重特異性抗体の濃度定量
以下に示すとおり、2種類の方法で測定した。
Goat anti-human IgG(Biosource International)をcoating bufferにて1μg/mLに調製し、Nunc-Immuno plate(Nunc)に固相化した。Diluent buffer(D.B.)にてブロッキング処理した後、D.B.を用いて適当に希釈した培養上清サンプルを添加した。また、抗体濃度算出のためのスタンダードとして、2000 ng/mLから3倍系列でD.B.にて11段階希釈したヒトIgG4(ヒト型化抗TF抗体、WO 99/51743参照)を同様に添加した。3回洗浄したのち、Goat anti-human IgG, alkaline phosphatase(Biosource International)を反応させた。5回洗浄したのち、Sigma 104(R) phosphatase substrate(Sigma-Aldrich)を基質として発色させ、吸光度リーダーModel 3550(Bio-Rad Laboratories)により、参照波長655 nmとして405 nmの吸光度を測定した。Microplate Manager III(Bio-Rad Laboretories)ソフトウェアを用いて、スタンダードの検量線から培養上清中のヒトIgG濃度を算出した。
【0127】
また、Biacore1000(BIACORE)を使用し、ProteinAを固定化したSensor Chip CM5(BIACORE)を用いて定量した。具体的にはメーカーのプロトコールに従い、活性化したセンサーチップに10 mM 酢酸ナトリウム水溶液(pH 4.0, BIACORE)で50μg/mLに希釈したProteinA(SIGMA)溶液を5μL/分で30分間反応させ、その後ブロッキング操作を実施してProteinA固定化センサーチップを作製した。このセンサーチップを用いて、Biacore 1000(BIACORE)を使用して培養上清および精製品の濃度を測定した。センサーチップの固定および濃度測定にはHBS-EP Buffer(BIACORE)を使用した。また、濃度測定時の標準品として4000 ng/mLから2倍系列でHBS-EP Bufferにて6段階希釈したヒト化IgG4抗体(ヒト型化抗TF抗体、WO 99/51743参照)を使用した。
【0128】
1−6.ヒト化二重特異性抗体の血液凝固活性評価
血友病A血液の凝固能を二重特異性抗体が是正するか明らかにするために、Factor VIII欠乏血漿を用いた活性化部分トロンボプラスチン時間(APTT)に対する同抗体の影響を検討した。様々な濃度の抗体溶液 50μL、Factor VIII欠乏血漿(Biomerieux)50μL及びAPTT試薬(Dade Behring)50μLの混合液を37℃で3分間加温した。凝固反応は20 mMのCaCl2(Dade Behring)50μLを同混合液に加えることにより開始させた。CR-A(Amelung)が接続されたKC10A(Amelung)により凝固するまでの時間を測定した。
【0129】
Factor VIII欠乏血漿の凝固時間を0 %、正常血漿の凝固時間を100 %としたときに作製される検量線を用いて、二重特異性抗体を添加した際の凝固時間から二重特異性抗体のFactor VIII様活性(%)を算出した。
【0130】
1−7.血液凝固活性を保持したヒト化二重特異性抗体の取得
上述した血液凝固活性評価において、血液凝固能が低下したヒト化二重特異性抗体について、活性上昇を目指してヒト抗体FRのアミノ酸を改変した。具体的には、QuikChange Site-Directed Mutagenesis Kit (Stratagene)を用いて、添付説明書記載の方法でヒト化抗体可変領域に変異を導入した。目的のヒト化抗体可変領域遺伝子配列であることが確認されたH鎖可変領域断片挿入プラスミドをXhoIおよびSfiIで、L鎖可変領域断片挿入プラスミドをEcoRIで消化した後に、反応液を1 %アガローズゲル電気泳動に供した。目的のサイズ(約400 bp)のDNA断片をQIAquick Gel Extraction Kit(QIAGEN)を用いて、添付説明書記載の方法で精製し、滅菌水30μlで溶出した。その後、実施例1−2に示す方法で、動物細胞用発現プラスミドを作製した。実施例1−3、1−4、1−5に示す方法でヒト化二重特異性抗体を調製し、実施例1−6に示す方法で血液凝固活性を評価した。
【0131】
FR配列のアミノ酸改変および血液凝固能の評価を繰り返すことでキメラ二重特異性抗体(A69/B26/BBA)と同等の活性を有するヒト化二重特異性抗体(ヒト化A69(hA69a)/ ヒト化B26 (hB26-F123e4)/ヒト化BBA(hAL-F123j4))を取得した(図1)。各抗体可変領域配列を以下の配列番号に示した。
【0132】
(1) ヒト化A69抗体VH(hA69a) 配列番号:1(塩基配列)、配列番号:2(アミノ酸配列)
(2) ヒト化B26抗体VH(hB26-F123e4) 配列番号:3(塩基配列)、配列番号:4(アミノ酸配列)
(3) ヒト化BBA抗体VL(hAL-F123j4) 配列番号:5(塩基配列)、配列番号:6(アミノ酸配列)
【0133】
〔実施例2〕二重特異性抗体の分離に向けた可変領域のアミノ酸改変箇所の選定
二重特異性抗体を調製する際の発現において、2種類のH鎖と1種類のL鎖を使用すると、ヒト化A69-H鎖とヒト化BBA-L鎖のホモダイマー、ヒト化B26-H鎖とヒト化BBA-L鎖のホモダイマー、ヒト化A69-H鎖およびヒト化B26-H鎖とヒト化BBA-L鎖のヘテロダイマーの3種類の抗体が発現する。この3種類の抗体を分離し、二重特異性抗体のみ精製することを目的として、ヒト化A69 H鎖可変領域の等電点を下降させ、ヒト化B26 H鎖可変領域の等電点を上昇させるアミノ酸改変を行った。
【0134】
はじめにヒト化A69抗体とヒト化B26抗体の可変領域表面に露出するアミノ酸残基を確認するために、MOEソフトウェア(Chemical Computing Group Inc.)を用いて、ホモロジーモデリングによりヒト化A69抗体およびヒト化B26抗体の抗体Fv領域モデルを作製した。モデルを図2に示した。本モデルの詳細な解析により、CDR以外のFR配列においては表面に露出するアミノ酸の中で、H10、H12、H23、H39、H43、H105(Kabatナンバリング、Kabat EA et al. 1991. Sequences of Proteins of Immunological Interest.NIH)が、活性を低下させること無く、等電点を変化させることができる候補になると考えられた。
【0135】
〔実施例3〕ヒト化二重特異性抗体の可変領域アミノ酸の改変
実施例2において選定された箇所について改変抗体を作製するためのアミノ酸改変を行った。具体的には、QuikChange Site-Directed Mutagenesis Kit (Stratagene)を用いて、添付説明書記載の方法で作製したヒト化A69抗体H鎖可変領域(hA69a、塩基配列番号:1)およびヒト化B26抗体H鎖可変領域(hB26-F123e4、塩基配列番号:3)に変異を導入した。目的のヒト化抗体可変領域遺伝子配列であることが確認されたH鎖可変領域断片挿入プラスミドをXhoIおよびSfiIで消化した後に、反応液を1 %アガローズゲル電気泳動に供した。目的のサイズ(約400 bp)のDNA断片をQIAquick Gel Extraction Kit(QIAGEN)を用いて添付説明書記載の方法で精製し、滅菌水30μlで溶出した。実施例1−2に示す方法で、調製したDNA断片をknobs-into-hole技術を参考に定常領域アミノ酸を置換した発現プラスミドおよび野生型定常領域をもつ発現プラスミドに挿入し、H鎖発現ベクターを作製した。その後、実施例1−3、1−4、1−5に示す方法でヒト化二重特異性抗体を調製した。改変したヒト化抗体の可変領域配列を以下の表1に記載される配列番号に示した。
【0136】
【表1】
【0137】
〔実施例4〕改変したヒト化抗体の等電点電気泳動による分析
可変領域のアミノ酸改変による表面電荷の変化について評価するために、改変抗体の調製および等電点電気泳動による分析を実施した。
【0138】
ヒト化BBA-L鎖(hAL-F123j4)発現ベクターとヒト化A69-H鎖を改変したhA69-p18、hA69-p8、hA69-p17、hA69-p16および未改変のhA69aの各H鎖発現ベクターを組み合わせて同時に発現させることによりhA69a、hA69-p18、hA69-p8、hA69-p17、hA69-p16の5種類のホモダイマーで構成される抗体を調製した。同様に、ヒト化BBA-L鎖発現ベクターとヒト化B26-H鎖を改変したhB26-p19、hB26-p15および未改変のhB26-F123e4の各H鎖発現ベクターを組み合わせて同時に発現させることによりhB26-F123e4、hB26-p19、hB26-p15の3種類のホモダイマーで構成される抗体を調製した。等電点電気泳動は以下のとおり行った。Phastsystem Cassette (AmerchamBioscience社製) を用いて以下の膨潤液で30 minほどPhast-Gel Dry IEF (AmerchamBioscience社製)ゲルを膨潤させた。
【0139】
20% Glycerol 0.95 mL
ミリQ水 0.95 mL
Bio-Lyte 7/9 (BioRad社製) 10μL
Bio-Lyte3/10 (BioRad社製) 10μL
Pharmalyte 8-10.5 for IEF (AmerchamBioscience社製) 80μL
【0140】
膨潤したゲルを用いてPhastSystem(AmerchamBioscience社製)により以下のプログラムで電気泳動を行った。サンプルはStep 2でゲルに添加した。pIマーカーとして、Calibration Kit for pI(AmerchamBioscience社製)を使用した。
【0141】
Step 1: 2000 V 2.5 mA 3.5 W 15℃ 75 Vh
Step 2: 200 V 2.5 mA 3.5 W 15℃ 15 Vh
Step 3: 2000 V 2.5 mA 3.5 W 15℃ 410 Vh
【0142】
泳動後のゲルは20 % TCAで固定した後、Silver staining Kit, protein(AmerchamBioscience社製)を用い、キットに添付されているプロトコールに従い銀染色を行った。染色後、pIマーカーの既知等電点からサンプルの等電点を算出した。
【0143】
未改変および改変したヒト化A69抗体のホモダイマー、ヒト化B26抗体のホモダイマーの分析結果を図3に示した。表面電荷の改変により等電点電気泳動においてバンドの移動が観察された。pIマーカーを参考に推測した各抗体の等電点は、未改変のhA69aホモダイマーが約8.8であるのに対して、改変したhA69-p18が約8.4、hA69-p17が約8.2、hA69-p8が約8.2、hA69-p16が約8.1であり、改変により最大約0.7の等電点の差を付与することができた。ヒト化B26ホモダイマーも同様に、未改変のhB26-F123e4が約9.1であるのに対して、改変したhB26-p19が約9.3、hB26-p15が約9.4であり、改変により最大約0.3の等電点の差を付与することができた。本検討において選択した可変領域のH12、H23、H39、H43、H105の表面アミノ酸を電荷的に改変することによって等電点を変化させることが可能であることが示された。
【0144】
〔実施例5〕改変したヒト化抗体の陽イオン交換クロマトグラフィー分析
実施例4において作製した改変抗体を用いて以下の方法で陽イオン交換クロマトグラフィーによる分析を行い、改変による両抗体の分離に及ぼす影響を評価した。陽イオン交換クロマトグラフィー分析条件は以下のとおりであり、ヒト化A69抗体のホモダイマー、ヒト化B26抗体のホモダイマーの保持時間を算出した。
【0145】
カラム:ProPac WCX-10, 4×250 mm, (Dionex)
移動相:A: 10 mmol/L NaH2PO4/Na2HPO4, pH 6.25
B: 10 mmol/L NaH2PO4/Na2HPO4, 500 mmol/L NaCl, pH 6.25
流速:1.0 mL/min
グラジエント:10 %B(5 min)→(40 min)→60 %B→(5 min)→100 %B (5 min)
検出:220 nm
【0146】
未改変および改変した5種のヒト化A69抗体のホモダイマーの分析結果を図4に、未改変および改変した3種のヒト化B26抗体のホモダイマーの分析結果を図5に示した。未改変ヒト化A69抗体のホモダイマーとヒト化B26抗体のホモダイマーの保持時間はともに25 min前後であり、両ホモダイマーの分離、まして目的の二重特異性抗体の分離は出来ない。未改変抗体の等電点を低下させる改変を行ったヒト化A69抗体は未改変の抗体と比較してピークの移動が観察され、改変の数に伴い保持時間は約22.4 min、約21.2 min、約20.2 minと短くなった。可変領域の等電点を上昇させる改変を行ったヒト化B26抗体も未改変の抗体と比較してピークの移動が観察され、改変の数に伴い保持時間は約28.4 min、約29.4 minと長くなった。本検討において選択した可変領域のH12、H23、H39、H43、H105の表面アミノ酸の電荷的に改変することによって、2種類の抗体の表面電荷が変化し、それにより保持時間を変化させることが可能であることが示された。
【0147】
実施例4において測定された等電点によると、未改変のhA69aホモダイマーと未改変のhB26-F123e4ホモダイマーはpIに0.3の差があるが両者の保持時時間はともに25min前後であり分離できなかったが(図9)、未改変のhA69aホモダイマーとhB26-p19はpIに0.5の差が付与され、その結果両者は保持時間約2.6minの差で分離されており、またhA69-p18とhB26ホモダイマーはpIに0.7の差が付与され、その結果両者は保持時間約3.4minの差で分離されており、最大でhA69-p16とhB26-p15はpIに1.3の差が付与され、その結果保持時間約9.2 minの差で分離された。このように、改変により2つのホモダイマーの分離が初めて可能になった。
【0148】
〔実施例6〕改変したヒト化二重特異性抗体の凝固活性評価
実施例4、実施例5の分析により表面電荷の変化が観察されたことを受けて、改変した2種類のヒト化抗体H鎖(hA69-p8、hB26-p15)とヒト化L鎖(hAL-F123j4)を発現させてヒト化二重特異性抗体を調製した。H鎖発現ベクターには、ヘテロダイマーを効率的に促進させるために、knobs-into-holes技術を利用したIgG4定常領域が組み込まれた発現ベクターを使用した。調製したヒト化二重特異性抗体を用いて、以下に示す方法に従って凝固活性を評価した。
【0149】
血友病A血液の凝固能を二重特異性抗体が是正するか明らかにするために、Factor VIII欠乏血漿を用いた活性化部分トロンボプラスチン時間(APTT)に対する同抗体の影響を検討した。様々な濃度の抗体溶液 50μL、Factor VIII欠乏血漿(Biomerieux)50μL及びAPTT試薬(Dade Behring)50μLの混合液を37℃で3分間加温した。凝固反応は20 mMのCaCl2(Dade Behring)50μLを同混合液に加えることにより開始させた。CR-A(Amelung)が接続されたKC10A(Amelung)により凝固するまでの時間を測定した。
【0150】
Factor VIII欠乏血漿の凝固時間を0 %、正常血漿の凝固時間を100 %としたときに作製される検量線を用いて、二重特異性抗体を添加した際の凝固時間から二重特異性抗体のFactor VIII様活性(%)を算出した。
【0151】
活性評価結果を図6に示した。可変領域を改変したヒト化二重特異性抗体は、未改変のヒト化二重特異性抗体と同等の凝固活性を示したことから、本実施例における可変領域の改変は抗体の活性には影響がないことが示された。
【0152】
〔実施例7〕CDRを改変したヒト化抗体の作製と評価
実施例2で作製したヒト化A69抗体のモデルを解析した結果、H97は表面に露出するアミノ酸であることが確認された。表1に示した抗体においてヒト化A69-H鎖であるhA69-N97RはCDR3に存在する97番目のアスパラギンをアルギニンに改変した配列を持つ。実施例1−2の方法に従ってhA69-N97Rをもつ発現ベクターを作製し、ヒト化BBA-L鎖であるhAL-F123j4とともに発現し、改変抗体を調製した。この抗体の表面電荷の変化を評価するために、実施例4の方法に従って等電点電気泳動を行った。図7に示すとおり、未改変の抗体(hA69a / hAL-F123j4)の等電点は8.9であるのに対して、改変抗体(hA69-N97R / hAL-F123j4)は9.1であり、CDRのアミノ酸置換においても表面電荷の変化が観察された。
【0153】
また改変抗体の機能を評価するために、以下の方法で抗原であるFactor IXaに対する結合活性を評価した。Coating buffer(100 mM sodium bicarbonate, pH 9.6, 0.02 % sodium azide)で1μg/mLに希釈したFactor IXaβ(Enzyme Research Labratories)を、Nunc-Immuno plate(Nunc-ImmunoTM 96 MicroWellTM plates MaxiSorpTM(Nalge Nunc International))に100μL/wellで分注後、4℃で一晩インキュベーションした。Tween(R) 20を含むPBS(-)で3回洗浄後、diluent buffer (50 mM Tris-HCl, pH 8.1, 1 % bovine serum albumin, 1mM MgCl2, 0.15 M NaCl, 0.05 % Tween(R) 20, 0.02 % sodium azide)でplateを室温で2時間blockingした。Bufferを除去後、diluent bufferで希釈した精製抗体を100μL/well添加し、室温で1時間インキュベーションした。Plateを3回洗浄後、diluent bufferで1/4000希釈したアルカリホスファターゼ標識ヤギ抗マウスIgG(BIOSOURCE)を100μL/well添加し、室温で1時間インキュベーションした。Plateを5回洗浄後、発色基質(Sigma)を100μL/well添加し、室温で30分インキュベーションした。405 nm(対照655 nm)における吸光度をMicroplate Reader Model 3550(Bio-Rad Laboratories)で測定した。その結果、図8に示すとおり、表面電荷を変化させるためにCDRを改変した抗体は改変前の抗体と同等の結合活性を示した。このように表面電荷を改変する際に、改変する箇所は実施例5に示したFRのみでなく、CDRであっても構わないことが示された。
【0154】
〔実施例8〕ヒト化二重特異性PF抗体の作製と評価
未改変の抗体として、表1に示した抗体においてヒト化A69-H鎖であるhA69a、ヒト化B26-H鎖であるhB26-F123e4とヒト化BBA-L鎖であるhAL-F123j4(配列番号:5)を用いて、未改変のヒト化二重特異性抗体を作製した。改変抗体として、表1に示した抗体においてヒト化A69-H鎖の改変体であるhA69-PFLとヒト化B26-H鎖の改変体であるhB26-PFとヒト化BBA-L鎖であるhAL-s8(配列番号:17)を用いて、ヒト化二重特異性PF抗体を作製した。H鎖は野生型の定常領域をもつ発現ベクターを使用して、実施例1−2に示すとおりに発現ベクターを構築し、実施例1−3、実施例1−4、実施例1−5の方法に従って抗体を調製した。この2種類のホモダイマーと二重特異性抗体を含む混合溶液を用いて、実施例5に示す方法で陽イオン交換クロマトグラフィー分析を実施した。
【0155】
未改変のヒト化二重特異性抗体とヒト化二重特異性PF抗体分析結果を図9、図10に示した。その結果、未改変のヒト化二重特異性抗体においては、二種類のホモダイマーと二重特異性抗体が分離せず1本のピークとして溶出したのに対して、ヒト化二重特異性PF抗体は、二種類のホモダイマーと目的の二重特異性抗体がそれぞれ分離し、hA69-PF ホモダイマー、ヒト化二重特異性PF抗体、hB26-PF ホモダイマーの順に3本のピークとして溶出した。陽イオン交換クロマトグラフィー分析の際に3種類のピークを分取することで、二種類のホモダイマーとヒト化二重特異性PF抗体を精製した。この画分をAmicon Ultra, MWCO 10000 (Millipore)による濃縮後、20 mM sodium acetate, 150 mM NaCl, pH 6.0に対して一晩冷所で透析を行い、濃度測定を行った。
【0156】
各抗体を精製した後に、実施例4に示す方法に従って等電点電気泳動を行った。図11に示すとおり、陽イオン交換クロマトグラフィー分析を行う前の抗体は3本のバンドが存在するが、陽イオン交換クロマトグラフィーにより各抗体が精製できることが確認された。ヒト化A69-PF抗体のホモダイマー、ヒト化二重特異性PF抗体、ヒト化B26-PF抗体のホモダイマーの等電点は約7.9、約8.6、約9.2であり、ヒト化A69-PF抗体のホモダイマーのヒト化二重特異性PF抗体等電点の差は約0.7であり、ヒト化B26-PF抗体のホモダイマーのヒト化二重特異性PF抗体等電点の差は約0.6であることが確認された。
【0157】
つづいて、実施例6に示す方法に従って精製した二重特異性PF抗体の凝固活性を評価した。前述したknobs-into-holes技術を利用したIgG4定常領域を用いて発現させたキメラ二重特異性抗体、可変領域を改変していないhA69a(配列番号:2)、hB26-F123e4(配列番号:4)、hAL-F123j4(配列番号:6)から成る二重特異性抗体、精製した二重特異性PF抗体と同じ可変領域をもち、knobs-into-holes技術を利用したIgG4定常領域を用いている二重特異性抗体の3種類の抗体と凝固活性を比較した。評価結果を図12に示した。Knobs-into-holes技術を利用したIgG4定常領域をもつ二重特異性PF抗体と野生型定常領域をもち陽イオン交換クロマトグラフィーにより精製した二重特異性PF抗体の凝固活性は同等であり、本実施例のH10、H12、H23、H39、H43、H105の可変領域改変により活性に影響することなく高純度にて二重特異性抗体が精製できることが示された。
【0158】
〔実施例8〕ヒト化二重特異性抗体発現細胞株の樹立
改変したヒト化二重特異性抗体を調製するために、以下のようにして抗体発現細胞株を樹立した。
【0159】
ヒトIgG4の野生型H鎖定常領域遺伝子を鋳型にしてH鎖定常領域のN末端側の2アミノ酸(Ala-Ser)をコードする塩基配列がNheI認識配列(GCTAGC)になるように設計した5'末端側プライマーと3'末端側にアニーリングし、かつNotI認識部位を持つように設計したプライマーを用いてH鎖定常領域をPCR増幅し、pBluescriptKS+ベクター(東洋紡)をNheI, NotI(ともに宝酒造)で消化したベクターと連結したpBCH4(IgG4定常領域遺伝子を含む)を作製した。表1に示すヒト化A69-H鎖抗体(hA69-KQ)およびヒト化B26-H鎖抗体(hB26-PF)のH鎖可変領域の5'末端側塩基配列に相補的でコザック配列(CCACC)およびEcoRI認識配列を有するプライマーとNheI認識配列を有する3'末端側塩基配列にプライマーを用いてPCRを行い、得られたPCR産物をEcoRI, NheI(ともに宝酒造)で消化、同様にEcoRI, NheIで消化したpBCH4に挿入して可変領域と定常領域を連結した。作製したヒト化A69-H鎖抗体ベクターをEcoRI, NotI(ともに宝酒造)で消化し、同様にEcoRI, NotIで消化した動物細胞用発現ベクターpCXND3にクローニングした。
【0160】
本ベクターpCXND3の構築の流れについて、以下に述べる。DHFR-ΔE-rVH-PM1-f(WO92/19759参照)の抗体H鎖遺伝子とベクターを分割するために、制限酵素EcoRI, SmaI部位で消化し、ベクター側のみ回収した後に、EcoRI-NotI -BamHI adaptor(宝酒造)をクローニングした。このベクターをpCHOIと命名した。pCHOIのDHFR遺伝子発現部位をpCXN(Niwaら、Gene 1991;108:193-200)の制限酵素HindIII部位にクローニングしたベクターをpCXND3と命名した。また、作製したヒト化B26-H鎖抗体ベクターをEcoRI, NotI(ともに宝酒造)で消化し、同様にEcoRI, NotIで消化した動物細胞用発現ベクターpCXZD1にクローニングした。pCXZD1ベクターはpCXND3ベクターのネオマイシン耐性遺伝子をゼオシン耐性遺伝子に置き換えた発現ベクターである。また、ヒト化BBA-L鎖抗体(hAL-AQ、配列番号:18)のL鎖可変領域の5'末端側塩基配列に相補的でコザック配列を有する合成オリゴヌクレオチドおよびBsiWI部位を有する3'末端側塩基配列に相補的な合成オリゴヌクレオチドを用いてPCRを行い、得られたPCR産物をヒトkappa鎖定常領域がpBluescriptKS+ベクターに挿入されているpBCLベクターにクローニングした。BsiWI部位により、ヒトL鎖可変領域と定常領域が連結している。作製されたL鎖遺伝子断片を発現ベクターpUCAGにクローニングした。本ベクターpUCAGは、pCXN(Niwaら、Gene 1991;108:193-200)を制限酵素BamHIで消化して得られる2.6 kbpの断片をpUC19ベクター(東洋紡)の制限酵素BamHI部位に連結し、クローニングしたベクターである。L鎖をpUCAGにクローニングしたベクターを制限酵素BamHIで消化し、ハイグロマイシン耐性遺伝子を含む発現ベクターpHygDHFR-4bにクローニングした。作製した3種類の発現ベクターを制限酵素で直鎖上にしたのちに、CHO-DG44細胞に遺伝子導入して抗体発現細胞株を樹立した。
【0161】
安定発現細胞株の作製は次のようにして行った。GenePulserII(Bio-Rad)を用いたエレクトロポレーション法により遺伝子導入した。各抗体発現ベクターとPBSに懸濁したCHO細胞(1×107細胞/mL)の0.75 mLを混合したものを氷上で10分間冷却し、キュベットに移した後に1.5 kV、25μFDの容量にてパルスを与えた。室温にて10分間の回復期間の後、エレクトロポレーション処理された細胞を、HT supplement(Invitrogen)を1倍濃度で含むCHO-S-SFMII培地(Invitrogen)40 mLに懸濁した。同様の培地で10倍希釈液を作製し、96ウェル培養用プレートに100μL/wellで分注した。CO2インキュベーター(5 % CO2)で24時間培養後、Geneticin(Invitrogen)を0.5 mg/mL、Zeocin(Invitrogen)を0.6 mg/mL、HygromycinB(Invitrogen)を0.4 mg/mLになるように、添加して2週間培養した。薬剤耐性を示す形質転換細胞のコロニーを順次拡大培養し、樹立した高産生細胞株を用いて大量培養を行い、培養上清を得た。
【0162】
〔実施例9〕ヒト化二重特異性抗体の製造用汎用カラムによる分離精製
実施例8で得られた培養上清から以下の方法で二重特異性抗体を精製した。培養上清を平衡化バッファー (20 mmol/L Sodium Phosphate buffer, 1 mol/L NaCl)で平衡化したrProtein A Sepharose Fast Flowカラム (Amersham Biosciences、50 mmI.D.×9.9 cmH. = 194.3 mL-resin)に添加し、洗浄用バッファー1(20 mmol/L Sodium Phosphate buffer, 1 mol/L NaCl, pH7.0)、洗浄用バッファー2(50 mmol/L Sodium Acetate buffer, pH 6.0)で洗浄した後に100 mmol/L Acetic acidを用いて溶出した。溶出後に直ちに20 mmol/L Sodium Acetate buffer, pH6.0で3倍希釈した。
【0163】
得られた精製溶液をSolvent A (20 mmol/L sodium Acetate buffer, pH 6.0)で平衡化した製造用汎用カラムであるSP TOYOPEARL 650Mカラム (東ソー、26 mmI.D.×22.3 cmH. = 118.3 mL-resin)に添加した。以下に示すような溶液およびGradientで抗体の表面電荷の差を用いた分離を行った。
【0164】
Solvent A : 20 mmol/L Sodium Acetate buffer, pH6.0
Solvent B : 20 mmol/L Sodium Acetate buffer, 1 mol/L NaCl, pH6.0
Flow rate : 10 mL/min (113 cm/h)溶出時のみ5.3 mL/min (60 cm/h)
Gradient : 0→15%B Step wise 3 Column Volume (CV)通液
15→22 %B gradient 2.5 CV
22→30 %B gradient 6 CV
30→100 %B Step wise 3 CV通液
【0165】
溶出の結果、図13に示すような3本のピークが検出され、製造用汎用カラムを使用した場合にも二重特異性抗体が分離精製できることが示された。
【0166】
〔実施例10〕改変したヒト化二重特異性抗体の活性評価
実施例9で調製したヒト化二重特異性抗体について、実施例6に示す方法に従って凝固活性を評価した。評価結果を図14に示した。実施例8で調製したヒト化二重特異性PF抗体と比較して実施例9で精製したヒト化二重特異性抗体の凝固活性は同等であった。hA69-PFLとhA69-KQのように可変領域のアミノ酸配列が若干異なっていても、製造用汎用カラムを使用して精製した抗体であっても活性には影響がないことが示された。
【0167】
以上のことから、二重特異性抗体を調製する際に、H鎖可変領域の改変により構造や抗体の機能(活性)を変えることなく表面電荷を変化させることで、目的のヒト化二重特異性抗体と二種のホモダイマーを形成する抗体とを分離精製できることが見出された。本方法を用いることで製造用汎用カラムにおいても二重特異性抗体が分離精製可能であることが示されたことから、二重特異性抗体からなる医薬品の製造方法として有用である。
【0168】
〔実施例11〕アイソタイプハイブリッド抗体の作成
11−1.ヒトIgG2抗体H鎖定常領域遺伝子のクローニング
ヒトIgG2抗体H鎖定常領域の遺伝子をクローニングするため、以下の操作を行った。
cDNA断片増幅のために50μLの反応液(各1μLの20μM K62プライマー(5' cac cgt ctc ctc agc ctc cac caa 3'/配列番号:22)、K63プライマー(5' gtg gca ctc att tac ccg gag aca 3'/配列番号:23)、5μLのMTC Multiple Tissue cDNA Panels(peripheral leukocytes) (Clontech)、4μLの5×Prime STAR Buffer、4μLの2.5 mM dNTPs、1μLのPrimeSTAR HS DNA Polymerase (以上TaKaRa))を調製し、PCRに供した。PCRは、サーマルサイクラーGeneAmp PCR system 9700(Parkin Elmer)を用いて、98℃で2分間加熱後、98℃ 10秒、60℃ 5秒、72℃ 2 minからなる反応を30 cycle行い、最後に72℃で10 min加熱した。PCR後、反応液を1% アガローズゲル電気泳動に供した。目的のサイズ(約1000 bp)の増幅断片をQIAqucick Gel Extractio Kit(QIAGEN)にて添付説明書記載の方法に従って精製し、滅菌水50μLで溶出した後、増幅断片の末端にA(Adenosine)を付加するためr-Taq処理をおこなった。r-Taq処理は、得られた増幅断片を10μLのrTaq反応液(1μLの10×rTaq反応溶液、1μLの2.5 mM dNTPs、1μLのrTaq、7μLの上記増幅断片)を72℃で30 min保温した。r-Taq処理した断片をpCR2.1-TOPO vector(Invitrogen)へクローニングし、塩基配列を決定した。各DNA断片の塩基配列は、BigDye Terminator 3.1 Cycle Sequencing Kit(Applied Biosystems)を用い、DNAシークエンサーABI PRISM 3730xL Genetic Analyzer(Applied Biosystems)にて、添付説明書記載の方法に従い決定した。
【0169】
決定した塩基配列をAccession.No.BX640623と比較し、翻訳したアミノ酸配列が異なっている塩基はPCR増幅時に挿入された変異と考え、Quick Change Site-Directed Mutagenesis Kit(Stratagene)を用いてアミノ酸置換を行い、BX640623のアミノ酸配列と同じ配列になるように改変した。Quick Change Site-Directed Mutagenesis Kit(Stratagene)は添付説明書記載の方法に従って行った。さらに、ヒトIgG2-H鎖定常領域遺伝子と目的の可変領域遺伝子を連結するために、ヒトIgG2-H鎖定常領域の最初の2アミノ酸(AlaSer)が制限酵素NheI認識配列(GCTAGC)になるように変異させた。本試験に使用したヒトIgG2-H鎖定常領域の塩基配列およびアミノ酸配列を、それぞれ配列番号:24および配列番号:25に示した。
【0170】
11−2.アイソタイプ置換抗体の発現ベクター構築
ヒト化PM-1抗体のH鎖可変領域とヒトIgG1、ヒトIgG2、ヒトIgG4の各種H鎖定常領域を連結した抗体発現ベクターを以下のように作製した。
非特許文献(Sato K et al, Cancer Research 1993, 53: 851-856)に示されているヒト化抗ヒトインターロイキン6受容体抗体(ヒト化PM-1抗体)のH鎖可変領域の5'末端側塩基配列に相補的でコザック配列を有する合成オリゴヌクレオチドおよび制限酵素NheI認識配列を有し、3'末端側塩基配列に相補的な合成オリゴヌクレオチドを用いてPCRを行い、得られたPCR産物をヒトIgG1-H鎖定常領域(Sato K et al, Cancer Research 1993, 53: 851-856 参照)がpBluescript KS+ベクター(TOYOBO)に挿入されているpB-CHベクターにクローニングした。H鎖可変領域と定常領域が連結したH鎖遺伝子断片をニワトリβアクチンプロモーターにより発現が制御されるpCAGGSベクター(Niwa et al.1991 Gene, 108: 193-199)に挿入した。PCR増幅したヒト化PM-1抗体のH鎖可変領域遺伝子をヒトIgG4定常領域遺伝子(WO 99/51743参照)および実施例11−1で作製したヒトIgG2-H鎖遺伝子の5'末端のNheIと連結してpCAGGSベクターに挿入した。各種H鎖発現ベクターは、NheI配列によりヒト化PM-1抗体のH鎖可変領域とヒトH鎖定常領域が連結してH鎖を発現する。
【0171】
同様にヒト化PM-1抗体のL鎖可変領域の5'末端側塩基配列に相補的でコザック配列を有する合成オリゴヌクレオチドおよび制限酵素BsiWI認識配列を有する3'末端側塩基配列に相補的な合成オリゴヌクレオチドを用いてPCRを行い、得られたPCR産物をヒトkappa鎖定常領域がpBluescript KS+ベクター(TOYOBO)に挿入されているpB-CLベクターにクローニングした。L鎖可変領域と定常領域が連結したL鎖遺伝子断片をニワトリβアクチンプロモーターにより発現が制御されるpCAGGSベクターに挿入した。BsiWI配列によりヒト化PM-1抗体のL鎖可変領域とヒトkappa鎖定常領域が連結してL鎖を発現する。
【0172】
11−3.アイソタイプハイブリッド抗体の発現
アイソタイプハイブリッド抗体はヒトIgG1、ヒトIgG2、ヒトIgG4の各種定常領域を有するヒト化PM-1抗体H鎖発現ベクターをそれぞれ二種類ずつ組み合わせ、ヒト化PM-1抗体L鎖発現ベクターとともに発現用細胞で共発現させることにより可能となる。各抗体の発現は、実施例4−2に記載した方法か以下の方法を用いて行った。ヒト胎児腎癌細胞由来HEK293H株(Invitrogen)を10% Fetal Bovine Serum (Invitrogen)を含むDMEM培地(Invitrogen)へ懸濁し、5〜6 × 105個 /mLの細胞密度で接着細胞用ディッシュ(直径10 cm, CORNING)の各ディッシュへ10 mLずつ蒔きこみCO2インキュベーター(37℃、5% CO2)内で一昼夜培養した後に、培地を吸引除去し、CHO-S-SFM-II(Invitrogen)培地6.9 mLを添加した。11−2で調製したプラスミドDNAを用いて以下のように各アイソタイプ抗体発現用混合液とハイブリッド抗体発現用の混合液(合計13.8μg)を調製した。
(1)L鎖発現ベクター6.9μg、IgG1-H鎖発現ベクター6.9μg
(2)L鎖発現ベクター6.9μg、IgG2-H鎖発現ベクター6.9μg
(3)L鎖発現ベクター6.9μg、IgG4-H鎖発現ベクター6.9μg
(4) L鎖発現ベクター6.9μg、IgG1-H鎖発現ベクター3.45μg、IgG2-H鎖発現ベクター3.45μg
(5) L鎖発現ベクター6.9μg、IgG2-H鎖発現ベクター3.45μg、IgG4-H鎖発現ベクター3.45μg
(6) L鎖発現ベクター6.9μg、IgG1-H鎖発現ベクター3.45μg、IgG4-H鎖発現ベクター3.45μg
【0173】
混合液それぞれを1μg/mL Polyethylenimine (Polysciences Inc.) 20.7μLおよびCHO-S-SFMII培地 690μLと混合して室温10分間静置したものを各ディッシュの細胞へ投入し、4〜5時間、CO2インキュベーター(37℃にて5% CO2)内でインキュベートした。その後、CHO-S-SFM-II(Invitrogen)培地6.9 mLを添加して、3日間 CO2インキュベーター内で培養した。培養上清を回収した後、遠心分離(約2000 g、5分間、室温)して細胞を除去し、さらに0.22μmフィルターMILLEX(R)-GV(Millipore)を通して滅菌した。該サンプルは使用するまで4℃で保存した。
【0174】
11−4.アイソタイプハイブリッド抗体の精製
実施例11−3に記載の方法で得られた培養上清に100μLのrProtein A SepharoseTM Fast Flow(Amersham Biosciences)を添加し、4℃で4時間以上転倒混和した。その溶液を0.22μmのフィルターカップUltrafree(R)-MC(Millipore)に移し、TBS 500μLにて3回洗浄後、rProtein A SepharoseTM樹脂に100μLの50 mM 酢酸ナトリウム水溶液、pH 3.0に懸濁して2分間静置したのち、抗体を溶出させた。直ちに、6.7μLの1.5M Tris-HCl、150 mM NaCl、pH 8.0を加えて中和した。得られた抗体溶液は活性測定用としてPBSに、また、DSC測定用として150 mM NaClを含む20 mM 酢酸緩衝液、pH 6.0に透析することで緩衝液を置換した。精製されたヒトIgG1のH鎖定常領域を有する抗体は「未改変ヒト化抗PM-1抗体」、ヒトIgG2のH鎖定常領域を有する抗体は「IgG2化ヒト化抗PM-1抗体」、ヒトIgG4のH鎖定常領域を有する抗体は「IgG4化ヒト化抗PM-1抗体」と以下に記載する。
【0175】
11−5.アイソタイプハイブリッド抗体の濃度定量
11−4で得た抗体を含む溶液2μLをND-1000 Spectrophotometer(NanoDrop)、あるいは50μLを分光光度計DU-600(BECKMAN)に供し、280 nmでの吸光度を測定した。得られた値から以下の式を用いて抗体濃度を算出した。ブランクにはPBSまたは150 mM NaClを含む20 mM 酢酸緩衝液、pH6.0を使用した。
【0176】
抗体濃度(mg/mL)=吸光度×希釈倍率÷14.6×10
【0177】
〔実施例12〕アイソタイプハイブリッド抗体の分析
12−1.アイソタイプハイブリッド抗体の等電点電気泳動による分析
定常領域の置換による表面電荷の変化について評価するために、等電点電気泳動による分析を実施した。
【0178】
等電点電気泳動は以下のとおり行った。Phastsystem Cassette(AmerchamBioscience社製)を用いて以下の膨潤液で30 minほどPhast-Gel Dry IEF(AmerchamBioscience社製)ゲルを膨潤させた。
【0179】
20% Glycerol 1.5 mL
Pharmalyte 8-10.5 for IEF(AmerchamBioscience社製) 100μL
【0180】
膨潤したゲルを用いてPhastSystem(AmerchamBioscience社製)により以下のプログラムで電気泳動を行った。サンプルはStep 2でゲルに添加した。pIマーカーとして、Calibration Kit for pI(AmerchamBioscience社製)を使用した。
【0181】
Step 1: 2000 V 2.5 mA 3.5 W 15℃ 75 Vh
Step 2: 200 V 2.5 mA 3.5 W 15℃ 15 Vh
Step 3: 2000 V 2.5 mA 3.5 W 15℃ 410 Vh
【0182】
泳動後のゲルは20% TCAで固定した後、Silver staining Kit、protein(AmerchamBioscience社製)を用い、キットに添付されているプロトコールに従い銀染色を行った。染色後、pIマーカーの既知等電点からサンプルの等電点を算出した。
【0183】
未改変、IgG2化およびIgG4化ヒト化PM-1抗体の分析結果を図15に示す。アイソタイプ置換により等電点電気泳動においてバンドの移動が観察された。pIマーカーを参考に推測した各抗体の等電点は未改変ヒト化PM-1抗体が約9.3であるのに対して、IgG2化ヒト化PM-1抗体が約8.9、IgG4化ヒト化PM-1抗体が約8.7であり、置換により最大約0.6の等電点の差を付与することができた。本検討において抗体アイソタイプの定常領域を置換することによって等電点を変化させることが可能であることが示された。
【0184】
次に、未改変、IgG2化ヒト化PM-1抗体および未改変、IgG4化ヒト化PM-1抗体の共発現抗体分析結果を図16に示す。これより、いずれの組み合わせにおいても各アイソタイプのホモダイマー、ヘテロダイマーが3つの主バンドとして観察され、pIマーカーを参考に推測した各アイソタイプハイブリット抗体の等電点は、未改変ヒト化PM-1/IgG2化ヒトPM-1のハイブリッド抗体が9.2、未改変ヒト化PM-1/IgG4化ヒトPM-1のハイブリッド抗体が9.0であった。本検討において各アイソタイプ抗体の発現ベクターを組み合わせて共発現させることで、アイソタイプハイブリッド抗体を作成することが可能であり、それらが等電点の差をもって分離することが示された。
【0185】
12−2.アイソタイプハイブリッド抗体の陽イオン交換クロマトグラフィー分析
実施例11において作製したアイソタイプハイブリッド抗体を用いて以下の方法で陽イオン交換クロマトグラフィーによる分析を行い、アイソタイプ置換が分離に及ぼす影響を評価した。陽イオン交換クロマトグラフィー分析条件は以下のとおりであり、未改変ヒト化PM-1抗体、IgG2化ヒト化PM-1抗体、IgG4化ヒト化PM-1抗体、および未改変ヒト化PM-1抗体/IgG2化ヒト化PM-1抗体のハイブリッド抗体、未改変ヒト化PM-1抗体/IgG4化ヒト化PM-1抗体のハイブリッド抗体の保持時間を算出した。
【0186】
カラム:ProPac WCX-10, 4×250 mm(Dionex)
移動相:A: 25 mmol/L MES/NaOH, pH 6.1
B: 25 mmol/L MES/NaOH, 250 mmol/L NaCl, pH 6.1
流速:0.5 mL/min
グラジエント:25 %B(5 min)→(105 min)→67 %B→(1 min)→100 %B (5 min)
検出:280 nm
【0187】
単独発現させた未改変、IgG2化およびIgG4化ヒト化PM-1抗体の分析結果を図17に示す。未改変ヒト化PM-1抗体、IgG2化ヒト化PM-1抗体およびIgG4化ヒト化PM-1抗体の保持時間はそれぞれ約60.2 min、30.5 minおよび30.3 minであり、アイソタイプ置換によって30分弱保持時間が変化した。一方、等電点電気泳動からpI差が認められたIgG2化ヒト化PM-1抗体とIgG4化ヒト化PM-1抗体の保持時間はほぼ同じであった。次に、未改変、IgG2化および未改変、IgG4化ヒト化PM-1抗体の共発現抗体分析結果を図18に示した。未改変ヒト化PM-1抗体/IgG2化ヒト化PM-1抗体の組み合わせ、および、未改変ヒト化PM-1抗体/IgG4化ヒト化PM-1抗体の組み合わせにおいて、各アイソタイプのホモダイマー、ヘテロダイマーが3つの主ピークとして観察された。保持時間は未改変ヒト化PM-1/IgG2化ヒトPM-1のハイブリッド抗体が約43.8 min、未改変ヒト化PM-1/IgG4化ヒトPM-1のハイブリッド抗体が約45.1 minであり、それぞれのホモダイマーとは10分以上の保持時間差で分離した。本検討において、各アイソタイプ抗体の発現ベクターを組み合わせて共発現させることでアイソタイプハイブリッド抗体を作成することが可能であり、それらがイオン交換クロマトグラフィーをもって分離可能であることが示された。
【0188】
〔実施例13〕アイソタイプハイブリッド抗体の陽イオン交換クロマトグラフィーによる分離精製
実施例11で得られた抗体溶液をAmicon-Ultra4(Amicon)で濃縮後EasySep(トミー精工)に封入し、5 mMクエン酸緩衝液(pH 6.5)に対し透析することで緩衝液を置換後、以下の条件でアイソタイプハイブリッド抗体を精製した。
【0189】
カラム:Poly CAT A, 4.6×100 mm, 粒子径 3μm, 孔径150 nm(Poly LC)
移動相:A: 25 mmol/L MES/NaOH, pH 6.1
B: 25 mmol/L MES/NaOH, 250 mmol/L 酢酸ナトリウム, pH 6.1
流速:1.0 mL/min
グラジエント:35 %B(5 min)→(54 min)→65 %B→(1 min)→100 %B (5 min)
検出:280 nm
【0190】
一回につき約100 - 200μgを注入し、未改変ヒト化PM-1抗体、未改変ヒト化PM-1/ IgG4化ヒト化PM-1アイソタイプハイブリット抗体、IgG4化ヒト化PM-1抗体ピークを分取した。分取時のクロマトグラムを図19に示す。複数回のピーク分取画分をそれぞれ混合してAmicon-Ultra4(Amicon)で濃縮後EasySep(トミー精工)に封入し、活性測定用としてPBSに、また、DSC測定用として150 mM NaClを含む20 mM 酢酸緩衝液、pH 6.0に透析することで緩衝液を置換した。分取ピークを上記同様の条件で再分析した結果を図20に示す。これより、アイソタイプハイブリット抗体がイオン交換クロマトグラフ法で分取精製可能であることが示された。
【0191】
本技術は、共通のH鎖可変領域を持つ抗体をpI値の異なるアイソタイプの定常領域を利用して分離することが出来たことから、pI差のない異なるH鎖可変領域であってもpI値の異なるアイソタイプのH鎖定常領域と連結することにより二重特異性抗体をイオン交換クロマトグラフィーで分離することが可能である。また、異なるH鎖可変領域を持つ場合、実施例9に示した可変領域への変異導入技術と組み合わせることで更に分子間のpI差を増大し、分離精製をより容易にすることができる。H鎖可変領域への変異導入が困難な場合、これらを天然に存在するIgGアイソタイプ配列に変換することで、抗原性を考慮することなく二重特異性抗体のイオン交換クロマトグラフィーによる分離精製が可能となる。
【0192】
〔実施例14〕アイソタイプハイブリッド抗体分取精製品の等電点電気泳動
分取品の純度を評価するために、等電点電気泳動による分析を実施した。
【0193】
等電点電気泳動は以下のとおり行った。Phastsystem Cassette(AmerchamBioscience社製)を用いて以下の膨潤液で30 minほどPhast-Gel Dry IEF(AmerchamBioscience社製)ゲルを膨潤させた。
【0194】
ミリQ水 1.5 mL
Pharmalyte 5-8 for IEF(AmerchamBioscience社製) 50μL
Pharmalyte 8-10.5 for IEF(AmerchamBioscience社製) 50μL
【0195】
膨潤したゲルを用いてPhastSystem(AmerchamBioscience社製)により以下のプログラムで電気泳動を行った。サンプルはStep 2でゲルに添加した。pIマーカーとして、Calibration Kit for pI(AmerchamBioscience社製)を使用した。
【0196】
Step 1: 2000 V 2.5 mA 3.5 W 15℃ 75 Vh
Step 2: 200 V 2.5 mA 3.5 W 15℃ 15 Vh
Step 3: 2000 V 2.5 mA 3.5 W 15℃ 410 Vh
【0197】
泳動後のゲルは20% TCAで固定した後、Silver staining Kit、protein(AmerchamBioscience社製)を用い、キットに添付されているプロトコールに従い銀染色を行った。
【0198】
アイソタイプハイブリッド抗体分取精製品の分析結果を図21に示す。イオン交換クロマトグラフィーにより、各アイソタイプのホモダイマーをほとんど含まずに精製できることが示された。
【0199】
〔実施例15〕アイソタイプハイブリッド抗体分取精製品の活性評価
15−1.ヒトgp130発現BaF3細胞株、ヒトgp130/ヒトIL-6受容体共発現BaF3細胞株の樹立
IL-6依存増殖性を示す細胞株を得るために、以下に示すとおり、ヒトgp130を発現したBaF3細胞株の樹立を行った。
全長ヒトgp130 cDNA (Hibiら、Cell 1990;63:1149-1157 (GenBank # NM_002184))をPCRにより増幅し、pCHOI (Hirataら、FEBS Letter 1994;356:244-248)のDHFR遺伝子発現部位を除去し、Zeocin耐性遺伝子発現部位を挿入した発現ベクターpCOS2Zeoにクローニングし、pCOS2Zeo/gp130を構築した。
【0200】
10μgのpCOS2Zeo/gp130をPBSに懸濁したBaF3細胞(0.8 x 107cells)に混合し、Gene Pulser(Bio-Rad)を用いて0.33 kV, 950μFDの容量でパルスを加えた。エレクトロポーレーション処理により遺伝子導入したBaF3細胞を0.2 ng/mLのmouse interleukin-3(Peprotech)、10% Fetal Bovine Serum(以下FBS、HyClone)を含むRPMI1640培地(Invitrogen)で一昼夜培養し、100 ng/mLのhuman interleukin-6(R&D)、100 ng/mL のhuman interleukin-6 soluble receptor(R&D systems)および10% FBSを含むRPMI1640培地を加えて選抜し、ヒトgp130発現BaF3細胞株(以下、BaF3/gp130)を樹立した。
【0201】
15−2.アイソタイプハイブリッド抗体分取精製品のヒトIL-6中和活性評価
IL-6依存性増殖を示すBaF3/gp130を用いて、以下に示すとおり、IL-6中和活性を評価した。精製した未改変ヒト化PM-1抗体、未改変/IgG4化ヒト化PM-1アイソタイプハイブリッド抗体およびIgG4化ヒト化PM-1抗体を10μg/mLになるように10% FBSを含むRPMI1640に希釈した。この溶液を用いて希釈公比3、合計7系列の希釈液を調製し、96well-plate(CORNING)の各wellに50μLずつ分注した。次に、BaF3/gp130を10% FBS(HyClone)を含むRPMI1640培地で3回洗浄した後に、5 x 104cells/mLとなるように60 ng/mLのhuman interleukin-6(TORAY)、60 ng/mL の可溶性ヒトIL-6受容体(自社調製品)および10% FBSを含むRPMI1640培地に懸濁し、各wellに50μLずつ混合した後、抗体サンプルを分注した。ヒト可溶性IL-6受容体は以下に示す方法で調製した。ヒト可溶性IL-6受容体(Yamasakiら、Science 1988;241:825-828(GenBank # X12830))の1番目から344番目のアミノ酸をコードする遺伝子をCHO細胞に導入後に培養上清から精製して調製した。37℃、5% CO2条件下で、72時間培養し、PBSで2倍に希釈したWST-8試薬(Cell Counting Kit-8、株式会社同仁化学研究所)を20μL/wellで加え、直後にSUNRISE CLASSIC(TECAN)を用いて450 nmの吸光度(参照波長620 nm)を測定した。2時間培養した後に、再度450 nmの吸光度(参照波長620 nm)を測定し、2時間の吸光度変化を指標にIL-6中和活性を評価した。
【0202】
その結果、図22に示すとおり、分取精製した未改変ヒト化PM-1抗体、未改変/IgG4化ヒト化PM-1アイソタイプハイブリッド抗体、およびIgG4化ヒト化PM-1抗体はヒト化PM-1抗体精製品(bulk)と中和活性が同等であった。以上より、アイソタイプハイブリット抗体は本来の抗原結合能を失わず、中和抗体としての機能を有することが示された。
【産業上の利用可能性】
【0203】
本発明の方法においては、アミノ酸の置換数が少数でよく、その構造・機能(活性)を変化させることなく等電点を制御させることが可能であり、それにより二重特異性抗体を汎用的なクロマトグラフィーカラムを用いることで効率的に、且つ、医薬品として開発可能な高純度まで精製することが可能になるため、医薬品として二重特異性抗体を開発する上で非常に有用性が高い。
本発明の方法を用いることにより、実際に活性を保持する二重特異性抗体の効率的な取得が可能である。
【特許請求の範囲】
【請求項1】
第1のポリペプチドおよび第2のポリペプチドを含む多重特異性抗体の製造方法であって、
(a)第1のポリペプチドと第2のポリペプチドの等電点に差がつくように、第1のポリペプチドのアミノ酸残基をコードする核酸および第2のポリペプチドのアミノ酸残基をコードする核酸の両方またはいずれか一方を改変し、
(b)宿主細胞を該核酸が発現するように培養し、
(c)宿主細胞培養物から多重特異性抗体を回収すること、
を含む多重特異性抗体の製造方法。
【請求項2】
工程(a)の改変が、第1のポリペプチドのホモ多量体、第2のポリペプチドのホモ多量体、および第1のポリペプチドと第2のポリペプチドのヘテロ多量体が、標準的なクロマトグラフィーを使用した分析により分離したピークとなるように、核酸を改変することである請求項1に記載の方法。
【請求項3】
前記第1のポリペプチドおよび前記第2のポリペプチドが重鎖可変領域を含む請求項1に記載の方法。
【請求項4】
前記多重特異性抗体が、軽鎖可変領域を含む第3のポリペプチドを含み、前記第1のポリペプチドおよび前記第2のポリペプチドがそれぞれ該第3のポリペプチドと多量体を形成する請求項3に記載の方法。
【請求項5】
前記第1のポリペプチドおよび前記第2のポリペプチドが重鎖定常領域を含む請求項1〜4のいずれか1項に記載の方法。
【請求項6】
前記第1のポリペプチドおよび第2のポリペプチドに含まれる重鎖定常領域が互いに等電点の異なる重鎖定常領域である請求項5に記載の方法。
【請求項7】
前記等電点の異なる重鎖定常領域がIgG1とIgG4、又は、IgG1とIgG2である請求項6に記載の方法。
【請求項8】
前記多重特異性抗体が、二重特異性抗体である請求項1に記載の方法。
【請求項9】
請求項1に記載の方法により製造される多重特異性抗体。
【請求項10】
第1のポリペプチドおよび第2のポリペプチドを含む多重特異性抗体の精製方法であって、
(a)第1のポリペプチドと第2のポリペプチドの等電点に差がつくように、第1のポリペプチドのアミノ酸残基をコードする核酸および第2のポリペプチドのアミノ酸残基をコードする核酸の両方またはいずれか一方を改変し、
(b)宿主細胞を該核酸が発現するように培養し、
(c)宿主細胞培養物から標準的なクロマトグラフィーにより該多重特異性抗体を精製すること、
を含む多重特異性抗体の精製方法。
【請求項11】
工程(a)の改変が、第1のポリペプチドのホモ多量体、第2のポリペプチドのホモ多量体、および第1のポリペプチドと第2のポリペプチドのヘテロ多量体が、標準的なクロマトグラフィーを使用した分析により分離したピークとなるように、核酸を改変することである請求項10に記載の方法。
【請求項12】
前記第1のポリペプチドおよび前記第2のポリペプチドが重鎖可変領域を含む請求項10に記載の方法。
【請求項13】
前記多重特異性抗体が、軽鎖可変領域を含む第3のポリペプチドを含み、前記第1のポリペプチドおよび前記第2のポリペプチドがそれぞれ該第3のポリペプチドと多量体を形成する請求項12に記載の方法。
【請求項14】
前記第1のポリペプチドおよび前記第2のポリペプチドが重鎖定常領域を含む請求項10〜13のいずれか1項に記載の方法。
【請求項15】
前記第1のポリペプチドおよび第2のポリペプチドに含まれる重鎖定常領域が互いに等電点の異なる重鎖定常領域である請求項14に記載の方法。
【請求項16】
前記等電点の異なる重鎖定常領域がIgG1とIgG4、又は、IgG1とIgG2である請求項15に記載の方法。
【請求項17】
前記多重特異性抗体が、二重特異性抗体である請求項10に記載の方法。
【請求項18】
請求項10に記載の方法により精製する工程を含む多重特異性抗体の製造方法。
【請求項19】
請求項18に記載の方法により製造される多重特異性抗体。
【請求項20】
第1のポリペプチドおよび第2のポリペプチドを含む多重特異性抗体であって、第1のポリペプチドが重鎖可変領域および/または重鎖定常領域を含み、該重鎖可変領域におけるKabatナンバリングによる10位、12位、23位、39位、43位および105位のアミノ酸残基、若しくは、該重鎖定常領域におけるEUナンバリングによる137位、196位、203位、214位、217位、233位、268位、274位、276位、297位、355位、392位、419位、435位のアミノ酸残基から選ばれる、少なくとも1つのアミノ酸残基が電荷を有し、第1のポリペプチドと第2のポリペプチドの等電点が互いに異なる多重特異性抗体。
【請求項21】
第2のポリペプチドが重鎖可変領域および/または重鎖定常領域を含み、該重鎖可変領域におけるKabatナンバリングによる10位、12位、23位、39位、43位および105位のアミノ酸残基、若しくは、該重鎖定常領域におけるEUナンバリングによる137位、196位、203位、214位、217位、233位、268位、274位、276位、297位、355位、392位、419位、435位のアミノ酸残基から選ばれる、少なくとも1つのアミノ酸残基が、前記第1のポリペプチドに含まれる重鎖可変領域および/または重鎖定常領域ににおいて選ばれる、電荷を有するアミノ酸残基とは反対の電荷を有する、または電荷を有しない請求項20に記載の多重特異性抗体。
【請求項22】
前記電荷を有するアミノ酸残基と当該アミノ酸残基とは反対の電荷を有するアミノ酸残基の組み合わせが、以下の(a)または(b)いずれかの群に含まれるアミノ酸残基からそれぞれ選択される請求項20に記載の多重特異性抗体:
(a)グルタミン酸(E)、アスパラギン酸(D);
(b)リジン(K)、アルギニン(R)、ヒスチジン(H)。
【請求項23】
第1のポリペプチドと第2ポリペプチドの等電点に差があり、第1のポリペプチドのホモ多量体、第2のポリペプチドのホモ多量体、および第1のポリペプチドと第2のポリペプチドのヘテロ多量体が、標準的なクロマトグラフィーを使用した分析により分離したピークとなり得る多重特異性抗体。
【請求項24】
前記第1のポリペプチドおよび前記第2のポリペプチドが重鎖可変領域を含む請求項23に記載の多重特異性抗体。
【請求項25】
前記多重特異性抗体が、軽鎖可変領域を含む第3のポリペプチドを含み、前記第1のポリペプチドおよび前記第2のポリペプチドがそれぞれ該第3のポリペプチドと多量体を形成する請求項24に記載の多重特異性抗体。
【請求項26】
前記第1のポリペプチドおよび前記第2のポリペプチドが重鎖定常領域を含む請求項23〜25のいずれか1項に記載の多重特異性抗体。
【請求項27】
前記第1のポリペプチドおよび第2のポリペプチドに含まれる重鎖定常領域が互いに等電点の異なる重鎖定常領域である請求項26に記載の多重特異性抗体。
【請求項28】
前記等電点の異なる重鎖定常領域がIgG1とIgG4、又は、IgG1とIgG2である請求項27に記載の多重特異性抗体。
【請求項29】
前記多重特異性抗体が、二重特異性抗体である請求項23に記載の多重特異性抗体。
【請求項30】
請求項23〜29のいずれか1項に記載の多重特異性抗体および医薬的に許容される担体を含む組成物。
【請求項31】
請求項23〜29のいずれか1項に記載の多重特異性抗体を構成するポリペプチドをコードする核酸。
【請求項32】
請求項31に記載の核酸を有する宿主細胞。
【請求項33】
請求項32に記載の宿主細胞を培養する工程、細胞培養物からポリペプチドを回収する工程を含む請求項23〜29のいずれか1項に記載の多重特異性抗体の製造方法。
【請求項34】
第1のポリペプチドの可変領域が以下の(a1)〜(a7)のいずれかに記載のアミノ酸配列からなり、第2のポリペプチドの可変領域が以下の(b1)〜(b3)のいずれかに記載のアミノ酸配列からなり、第3のポリペプチドの可変領域が以下の(c1)または(c2)に記載のアミノ酸配列からなる請求項25に記載の多重特異性抗体:
(a1)配列番号:7
(a2)配列番号:8
(a3)配列番号:9
(a4)配列番号:10
(a5)配列番号:11
(a6)配列番号:12
(a7)配列番号:13
(b1)配列番号:14
(b2)配列番号:15
(b3)配列番号:16
(c1)配列番号:17
(c2)配列番号:18
【請求項35】
第1のポリペプチドの可変領域が配列番号:11に記載のアミノ酸配列からなり、第2のポリペプチドの可変領域が配列番号:16に記載のアミノ酸配列からなり、第3のポリペプチドの可変領域が配列番号:17に記載のアミノ酸配列からなる請求項34に記載の多重特異性抗体。
【請求項36】
第1のポリペプチドの可変領域が配列番号:12に記載のアミノ酸配列からなり、第2のポリペプチドの可変領域が配列番号:16に記載のアミノ酸配列からなり、第3のポリペプチドの可変領域が配列番号:18に記載のアミノ酸配列からなる請求項34に記載の多重特異性抗体。
【請求項37】
第1のポリペプチドおよび第2のポリペプチドがヒトIgG4定常領域を含み、第3のポリペプチドがヒトκ定常領域を含む請求項34〜36のいずれか1項に記載の多重特異性抗体。
【請求項1】
第1のポリペプチドおよび第2のポリペプチドを含む多重特異性抗体の製造方法であって、
(a)第1のポリペプチドと第2のポリペプチドの等電点に差がつくように、第1のポリペプチドのアミノ酸残基をコードする核酸および第2のポリペプチドのアミノ酸残基をコードする核酸の両方またはいずれか一方を改変し、
(b)宿主細胞を該核酸が発現するように培養し、
(c)宿主細胞培養物から多重特異性抗体を回収すること、
を含む多重特異性抗体の製造方法。
【請求項2】
工程(a)の改変が、第1のポリペプチドのホモ多量体、第2のポリペプチドのホモ多量体、および第1のポリペプチドと第2のポリペプチドのヘテロ多量体が、標準的なクロマトグラフィーを使用した分析により分離したピークとなるように、核酸を改変することである請求項1に記載の方法。
【請求項3】
前記第1のポリペプチドおよび前記第2のポリペプチドが重鎖可変領域を含む請求項1に記載の方法。
【請求項4】
前記多重特異性抗体が、軽鎖可変領域を含む第3のポリペプチドを含み、前記第1のポリペプチドおよび前記第2のポリペプチドがそれぞれ該第3のポリペプチドと多量体を形成する請求項3に記載の方法。
【請求項5】
前記第1のポリペプチドおよび前記第2のポリペプチドが重鎖定常領域を含む請求項1〜4のいずれか1項に記載の方法。
【請求項6】
前記第1のポリペプチドおよび第2のポリペプチドに含まれる重鎖定常領域が互いに等電点の異なる重鎖定常領域である請求項5に記載の方法。
【請求項7】
前記等電点の異なる重鎖定常領域がIgG1とIgG4、又は、IgG1とIgG2である請求項6に記載の方法。
【請求項8】
前記多重特異性抗体が、二重特異性抗体である請求項1に記載の方法。
【請求項9】
請求項1に記載の方法により製造される多重特異性抗体。
【請求項10】
第1のポリペプチドおよび第2のポリペプチドを含む多重特異性抗体の精製方法であって、
(a)第1のポリペプチドと第2のポリペプチドの等電点に差がつくように、第1のポリペプチドのアミノ酸残基をコードする核酸および第2のポリペプチドのアミノ酸残基をコードする核酸の両方またはいずれか一方を改変し、
(b)宿主細胞を該核酸が発現するように培養し、
(c)宿主細胞培養物から標準的なクロマトグラフィーにより該多重特異性抗体を精製すること、
を含む多重特異性抗体の精製方法。
【請求項11】
工程(a)の改変が、第1のポリペプチドのホモ多量体、第2のポリペプチドのホモ多量体、および第1のポリペプチドと第2のポリペプチドのヘテロ多量体が、標準的なクロマトグラフィーを使用した分析により分離したピークとなるように、核酸を改変することである請求項10に記載の方法。
【請求項12】
前記第1のポリペプチドおよび前記第2のポリペプチドが重鎖可変領域を含む請求項10に記載の方法。
【請求項13】
前記多重特異性抗体が、軽鎖可変領域を含む第3のポリペプチドを含み、前記第1のポリペプチドおよび前記第2のポリペプチドがそれぞれ該第3のポリペプチドと多量体を形成する請求項12に記載の方法。
【請求項14】
前記第1のポリペプチドおよび前記第2のポリペプチドが重鎖定常領域を含む請求項10〜13のいずれか1項に記載の方法。
【請求項15】
前記第1のポリペプチドおよび第2のポリペプチドに含まれる重鎖定常領域が互いに等電点の異なる重鎖定常領域である請求項14に記載の方法。
【請求項16】
前記等電点の異なる重鎖定常領域がIgG1とIgG4、又は、IgG1とIgG2である請求項15に記載の方法。
【請求項17】
前記多重特異性抗体が、二重特異性抗体である請求項10に記載の方法。
【請求項18】
請求項10に記載の方法により精製する工程を含む多重特異性抗体の製造方法。
【請求項19】
請求項18に記載の方法により製造される多重特異性抗体。
【請求項20】
第1のポリペプチドおよび第2のポリペプチドを含む多重特異性抗体であって、第1のポリペプチドが重鎖可変領域および/または重鎖定常領域を含み、該重鎖可変領域におけるKabatナンバリングによる10位、12位、23位、39位、43位および105位のアミノ酸残基、若しくは、該重鎖定常領域におけるEUナンバリングによる137位、196位、203位、214位、217位、233位、268位、274位、276位、297位、355位、392位、419位、435位のアミノ酸残基から選ばれる、少なくとも1つのアミノ酸残基が電荷を有し、第1のポリペプチドと第2のポリペプチドの等電点が互いに異なる多重特異性抗体。
【請求項21】
第2のポリペプチドが重鎖可変領域および/または重鎖定常領域を含み、該重鎖可変領域におけるKabatナンバリングによる10位、12位、23位、39位、43位および105位のアミノ酸残基、若しくは、該重鎖定常領域におけるEUナンバリングによる137位、196位、203位、214位、217位、233位、268位、274位、276位、297位、355位、392位、419位、435位のアミノ酸残基から選ばれる、少なくとも1つのアミノ酸残基が、前記第1のポリペプチドに含まれる重鎖可変領域および/または重鎖定常領域ににおいて選ばれる、電荷を有するアミノ酸残基とは反対の電荷を有する、または電荷を有しない請求項20に記載の多重特異性抗体。
【請求項22】
前記電荷を有するアミノ酸残基と当該アミノ酸残基とは反対の電荷を有するアミノ酸残基の組み合わせが、以下の(a)または(b)いずれかの群に含まれるアミノ酸残基からそれぞれ選択される請求項20に記載の多重特異性抗体:
(a)グルタミン酸(E)、アスパラギン酸(D);
(b)リジン(K)、アルギニン(R)、ヒスチジン(H)。
【請求項23】
第1のポリペプチドと第2ポリペプチドの等電点に差があり、第1のポリペプチドのホモ多量体、第2のポリペプチドのホモ多量体、および第1のポリペプチドと第2のポリペプチドのヘテロ多量体が、標準的なクロマトグラフィーを使用した分析により分離したピークとなり得る多重特異性抗体。
【請求項24】
前記第1のポリペプチドおよび前記第2のポリペプチドが重鎖可変領域を含む請求項23に記載の多重特異性抗体。
【請求項25】
前記多重特異性抗体が、軽鎖可変領域を含む第3のポリペプチドを含み、前記第1のポリペプチドおよび前記第2のポリペプチドがそれぞれ該第3のポリペプチドと多量体を形成する請求項24に記載の多重特異性抗体。
【請求項26】
前記第1のポリペプチドおよび前記第2のポリペプチドが重鎖定常領域を含む請求項23〜25のいずれか1項に記載の多重特異性抗体。
【請求項27】
前記第1のポリペプチドおよび第2のポリペプチドに含まれる重鎖定常領域が互いに等電点の異なる重鎖定常領域である請求項26に記載の多重特異性抗体。
【請求項28】
前記等電点の異なる重鎖定常領域がIgG1とIgG4、又は、IgG1とIgG2である請求項27に記載の多重特異性抗体。
【請求項29】
前記多重特異性抗体が、二重特異性抗体である請求項23に記載の多重特異性抗体。
【請求項30】
請求項23〜29のいずれか1項に記載の多重特異性抗体および医薬的に許容される担体を含む組成物。
【請求項31】
請求項23〜29のいずれか1項に記載の多重特異性抗体を構成するポリペプチドをコードする核酸。
【請求項32】
請求項31に記載の核酸を有する宿主細胞。
【請求項33】
請求項32に記載の宿主細胞を培養する工程、細胞培養物からポリペプチドを回収する工程を含む請求項23〜29のいずれか1項に記載の多重特異性抗体の製造方法。
【請求項34】
第1のポリペプチドの可変領域が以下の(a1)〜(a7)のいずれかに記載のアミノ酸配列からなり、第2のポリペプチドの可変領域が以下の(b1)〜(b3)のいずれかに記載のアミノ酸配列からなり、第3のポリペプチドの可変領域が以下の(c1)または(c2)に記載のアミノ酸配列からなる請求項25に記載の多重特異性抗体:
(a1)配列番号:7
(a2)配列番号:8
(a3)配列番号:9
(a4)配列番号:10
(a5)配列番号:11
(a6)配列番号:12
(a7)配列番号:13
(b1)配列番号:14
(b2)配列番号:15
(b3)配列番号:16
(c1)配列番号:17
(c2)配列番号:18
【請求項35】
第1のポリペプチドの可変領域が配列番号:11に記載のアミノ酸配列からなり、第2のポリペプチドの可変領域が配列番号:16に記載のアミノ酸配列からなり、第3のポリペプチドの可変領域が配列番号:17に記載のアミノ酸配列からなる請求項34に記載の多重特異性抗体。
【請求項36】
第1のポリペプチドの可変領域が配列番号:12に記載のアミノ酸配列からなり、第2のポリペプチドの可変領域が配列番号:16に記載のアミノ酸配列からなり、第3のポリペプチドの可変領域が配列番号:18に記載のアミノ酸配列からなる請求項34に記載の多重特異性抗体。
【請求項37】
第1のポリペプチドおよび第2のポリペプチドがヒトIgG4定常領域を含み、第3のポリペプチドがヒトκ定常領域を含む請求項34〜36のいずれか1項に記載の多重特異性抗体。
【図1】

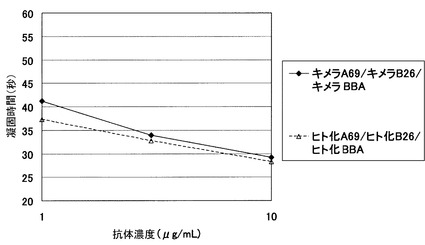
【図2】
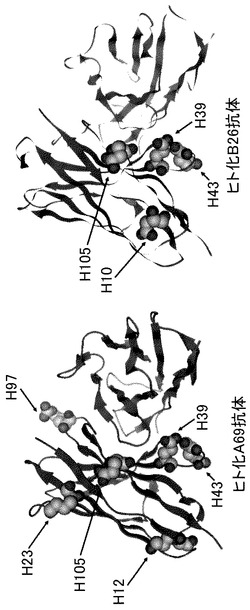
【図3】
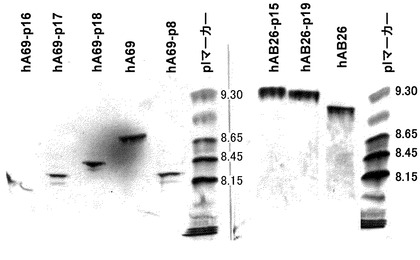
【図4】
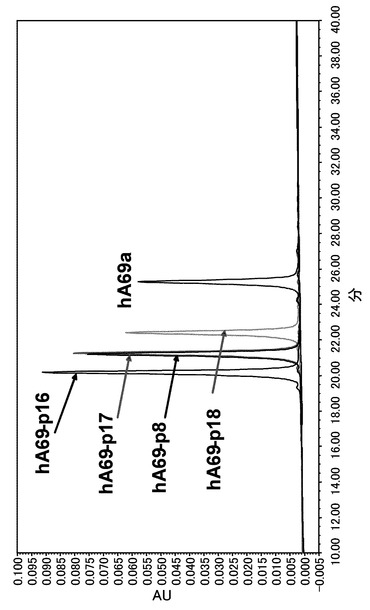
【図5】
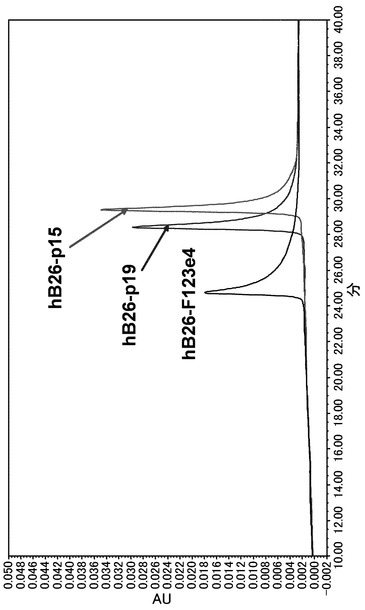
【図6】
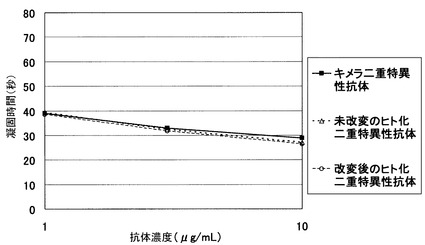
【図7】
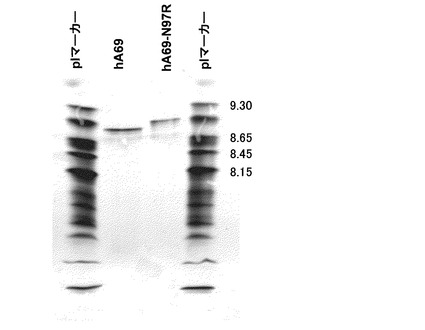
【図8】
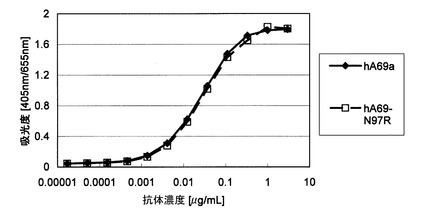
【図9】
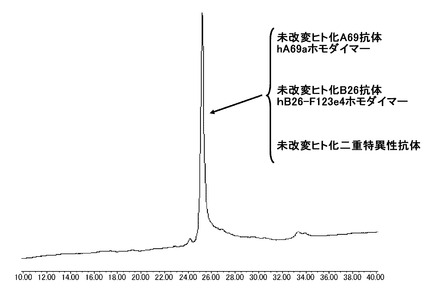
【図10】
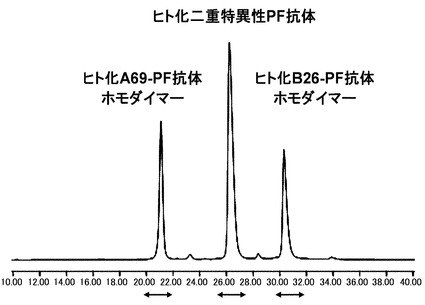
【図11】
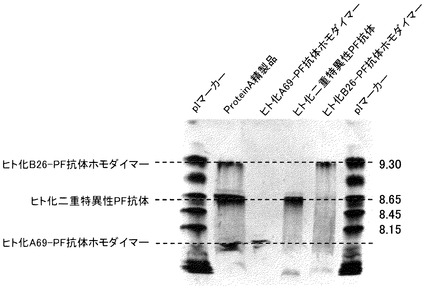
【図12】
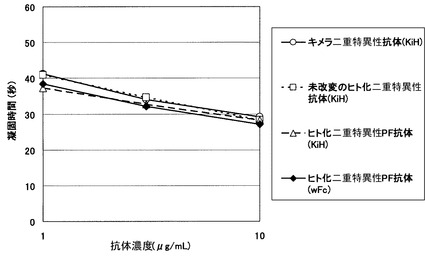
【図13】
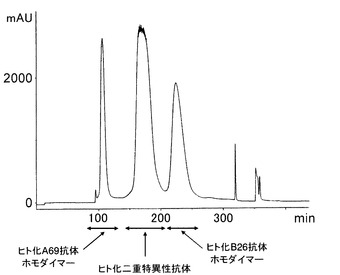
【図14】
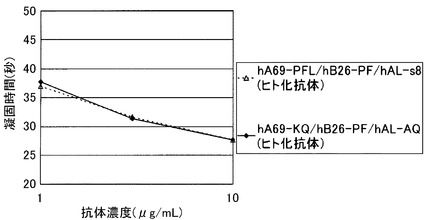
【図15】
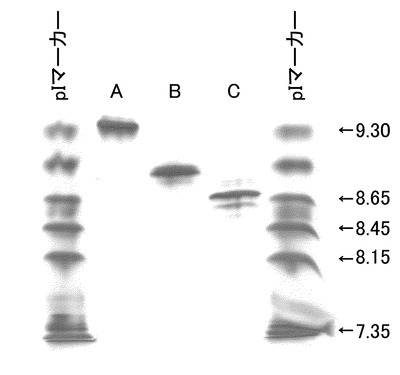
【図16】
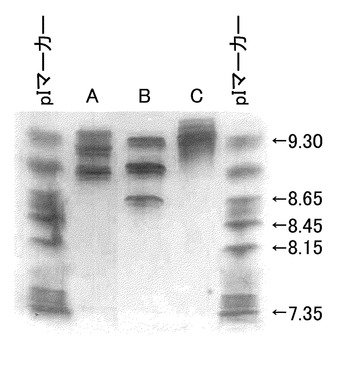
【図17】
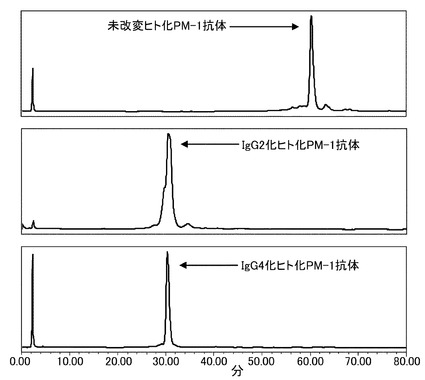
【図18】
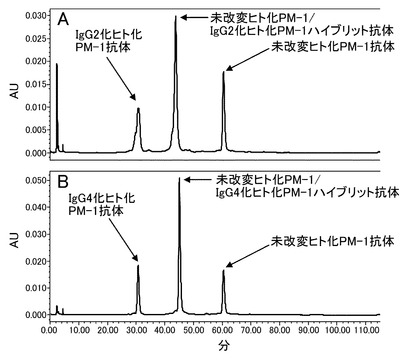
【図19】
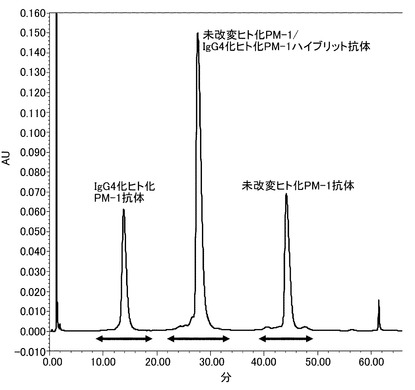
【図20】
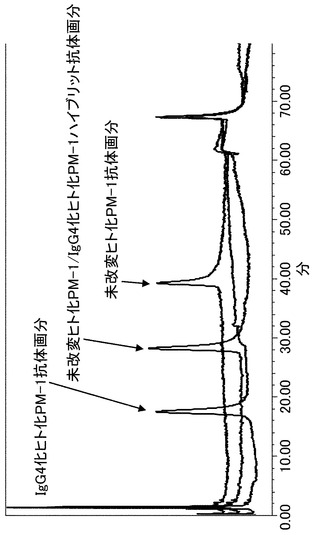
【図21】
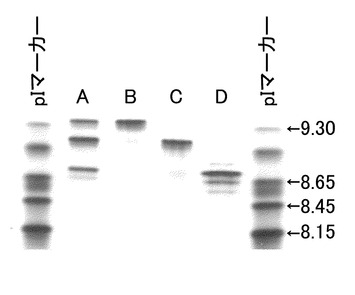
【図22】
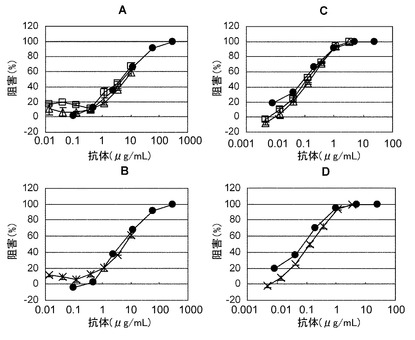
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【公開番号】特開2013−78313(P2013−78313A)
【公開日】平成25年5月2日(2013.5.2)
【国際特許分類】
【出願番号】特願2012−237732(P2012−237732)
【出願日】平成24年10月29日(2012.10.29)
【分割の表示】特願2008−508649(P2008−508649)の分割
【原出願日】平成19年3月30日(2007.3.30)
【出願人】(000003311)中外製薬株式会社 (228)
【Fターム(参考)】
【公開日】平成25年5月2日(2013.5.2)
【国際特許分類】
【出願日】平成24年10月29日(2012.10.29)
【分割の表示】特願2008−508649(P2008−508649)の分割
【原出願日】平成19年3月30日(2007.3.30)
【出願人】(000003311)中外製薬株式会社 (228)
【Fターム(参考)】
[ Back to top ]