
人工テンプレートおよび単一プライマー増幅におけるその使用
【課題】抗体の少なくとも一部分をコードする核酸を増幅する方法を提供すること。
【解決手段】上記方法は、a)抗体の少なくとも一部分をコードするテンプレートにプライマーをアニーリングする工程であって、該プライマーは、該テンプレートにアニーリングする第1の部分および該テンプレートにアニーリングしない所定の配列の第2の部分を有する、工程;b)該プライマーの該第1の部分が該テンプレートにアニーリングする位置と該テンプレートの末端との間の該テンプレートの部分に相補的であるポリヌクレオチドを合成する工程であって、該ポリヌクレオチドは、その第1の末端において該プライマーを有し、かつ第2の末端を有する、工程;ならびにc)該テンプレートから、工程(b)において合成された該ポリヌクレオチドを分離する工程などを包含する。
【解決手段】上記方法は、a)抗体の少なくとも一部分をコードするテンプレートにプライマーをアニーリングする工程であって、該プライマーは、該テンプレートにアニーリングする第1の部分および該テンプレートにアニーリングしない所定の配列の第2の部分を有する、工程;b)該プライマーの該第1の部分が該テンプレートにアニーリングする位置と該テンプレートの末端との間の該テンプレートの部分に相補的であるポリヌクレオチドを合成する工程であって、該ポリヌクレオチドは、その第1の末端において該プライマーを有し、かつ第2の末端を有する、工程;ならびにc)該テンプレートから、工程(b)において合成された該ポリヌクレオチドを分離する工程などを包含する。
【発明の詳細な説明】
【技術分野】
【0001】
(関連出願)
本国際出願は、2003年12月15日に出願された米国出願第10/737,252号(これは、2001年9月19日に出願された米国仮出願第60/323,455号に対する優先権を主張する、2002年9月19日に出願された米国出願第10/251,085号の一部継続出願である)に対する優先権を主張する。上述の米国出願および米国仮出願の開示全体は、本明細書中に参考として援用される。
【0002】
(技術分野)
本開示は、標的核酸配列の増幅のために有用な人工テンプレート(engineered template)に関連する。さらに具体的には、その反対端に相補的な配列を含むように操作されたテンプレートが、ネステッド(nested)オリゴヌクレオチド伸長反応(NOER)によって提供される。上記人工テンプレートは、この人工テンプレート内の標的配列を増幅するための単一プライマー増幅(SPA)を可能にする。特に有用な実施形態において、上記人工テンプレート由来の標的配列は、発現ビヒクル中にクローニングされて、例えば、抗体ライブラリーのような、ポリペプチドまたはタンパク質のライブラリーを提供する。
【背景技術】
【0003】
(関連分野の背景)
核酸増幅および増幅生成物の検出のための方法は、核酸配列の検出、同定、定量および配列分析を支援する。核酸増幅は、例えば、抗体のような、関連する遺伝子ライブラリーの構築において重要な工程である。これらのライブラリーは、特異的であり所望する活性を有する抗体に関して、スクリーニングされ得る。核酸分析は、病原体の検出および同定、規定の表現型を引き起こす遺伝子変化の検出、遺伝病(genetic disease)または疾患に対する感受性の診断、発達、疾患および規定の刺激の応答における遺伝子発現の評価、ならびに種々のゲノム計画のために重要である。核酸増幅方法の他の適用としては、稀な細胞の検出、病原体の検出、および悪性疾患における改変された遺伝子発現の検出などが挙げられる。核酸増幅はまた、(例えば、規定の核酸配列の存在の検出のような)定性分析、および(例えば、病原性配列の量の評価、ならびに遺伝子増殖または遺伝子欠失の判定、および正常細胞型から悪性細胞型への細胞形質転換の判定などにおいて有用な)規定の遺伝子配列の定量化のために有用である。核酸配列中の配列変化の検出は、変異遺伝子型の検出、遺伝子分析に関連することとして、薬物耐性を引き起こす変異の検出、薬理ゲノム科学などのために重要である。
【0004】
多くの種類の核酸増幅(例えば、指数関数的増幅、結合線形増幅(linked linear amplification)、ライゲーションベースの増幅、および転写ベースの増幅)が存在する。指数関数的核酸増幅方法の一例は、多数の刊行物において開示される、ポリメラーゼ連鎖反応(PCR)である。例えば、非特許文献1;Mullis K.特許文献1;Mullisら、特許文献2;Erlichら、特許文献3、特許文献4、特許文献5、特許文献6;およびSaiki R.ら、特許文献7を参照のこと。実際に、このポリメラーゼ連鎖反応(PCR)は、最も一般に使用される標的増幅方法である。PCRは、多数の標的配列の複数の二重鎖複製物を生成するために、多サイクルの、変性、2種の異なるオリゴヌクレオチドプライマーの(各々、標的鎖の対向する鎖への)ハイブリダイゼーション、およびヌクレオチドポリメラーゼによるプライマー伸長に基づく。
【0005】
単一のプライマーを利用した複製方法もまた、開示されている。例えば、特許文献8;特許文献9;特許文献10;特許文献11;および特許文献12を参照のこと。このプライマーは、特許文献13において開示されるようなDNA/RNAキメラプライマーであり得る。
【0006】
いくつかの増幅方法は、テンプレート転換オリゴヌクレオチド(template switching oligonucleotide)(TSO)およびブロッキングオリゴヌクレオチド(blocking oligonucleotide)を使用する。例えば、キメラDNAプライマーが利用されたテンプレート転換増幅は、特許文献12;特許文献14;特許文献15において、かつ非特許文献2によって開示される。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】欧州特許第201,184号明細書
【特許文献2】米国特許第4,582,788号明細書
【特許文献3】欧州特許第50,424号明細書
【特許文献4】欧州特許第84,796号明細書
【特許文献5】欧州特許第258,017号明細書
【特許文献6】欧州特許第237,362号明細書
【特許文献7】米国特許第4,683,194号明細書
【特許文献8】米国特許第5,508,178号明細書
【特許文献9】米国特許第5,595,891号明細書
【特許文献10】米国特許第5,683,879号明細書
【特許文献11】米国特許第5,130,238号明細書
【特許文献12】米国特許第5,679,512号明細書
【特許文献13】米国特許第5,744,308号明細書
【特許文献14】米国特許第5,962,272号明細書
【特許文献15】米国特許第6,251,639号明細書
【非特許文献】
【0008】
【非特許文献1】Mullisら、Cold Spring Harbor Symp.Quant.Biol.1986年、第51巻:263−273頁
【非特許文献2】Patelら、Proc.Natl.Acad.Sci.U.S.A.1996年、第93巻:2969−2974頁
【発明の概要】
【発明が解決しようとする課題】
【0009】
しかしながら、既に記載した標的増幅方法は、いくつかの欠点を有する。例えば、転写ベースの増幅方法(例えば、核酸配列ベースの増幅(NASBA)および転写媒介性増幅(TMA))は、プライマーによる増幅生成物の中へのポリメラーゼプロモーター配列の組み込みに対する必要性によって制限され、結果として、この方法は非特異的増幅となる傾向がある。現在の増幅方法の欠点の別の例は、異なる温度で最適な結合を有し得る2回の結合現象の必要性、ならびに天然に存在する配列を含むプライマーの使用である。この要因の組み合わせの結果、ミスプライミング(mis−priming)の可能性が生じ、そして結果として標的配列以外の配列の増幅が生じる。
【0010】
したがって、これらの欠点を克服する核酸増幅方法に対する必要性が存在する。本明細書中で提供される本発明は、この必要性を満たし、そしてさらなる利益を提供する。
【課題を解決するための手段】
【0011】
(要旨)
核酸増幅の新しい方法が、ここ発明され、この方法は、以下の工程:
a)テンプレート核酸配列にプライマーをアニーリングする工程であって、このプライマーは、テンプレートにアニーリングする第1の部分および所定の配列の第2の部分を有する、工程;
b)上記プライマーの第1の部分が上記テンプレートにアニーリングする位置と上記テンプレートの末端との間のテンプレートの部分にアニーリングし、かつ相補的であるポリヌクレオチドを合成する工程であって、このポリヌクレオチドは第1の末端および第2の末端を有し、ここで、その第1の末端は上記プライマーを組み込む、工程;
c)上記テンプレートから、工程(b)において合成されたポリヌクレオチドを分離する工程;
d)工程(b)において合成されたポリヌクレオチドの第2の末端に、ネステッドオリゴヌクレオチドをアニーリングする工程であって、このネステッドオリゴヌクレオチドは、上記ポリヌクレオチドの第2の末端にアニーリングする第1の部分およびプライマーの第2の部分と同一の所定の配列を有する第2の部分を有する工程;
e)工程(b)において合成されたポリヌクレオチドを伸長して、上記所定の配列に相補的なこのポリヌクレオチドの末端部分を提供する、工程;ならびに
f)所定の配列を有する単一のプライマーを使用し、伸長されたポリヌクレオチドを増幅する工程
を包含する。
【0012】
代替的な実施形態において、上記方法は、以下の工程:
a)テンプレート核酸配列に、プライマーおよび境界(boundary)オリゴヌクレオチドをアニーリングする工程であって、このプライマーは、上記テンプレートにアニーリングする第1の部分および所定の配列の第2の部分を有する、工程;
b)上記プライマーの第1の部分がテンプレートにアニーリングする位置と上記境界オリゴヌクレオチドがアニーリングするテンプレートの部分との間のテンプレートの部分にアニーリングし、そしてそれに相補的であるポリヌクレオチドを合成する工程であって、このポリヌクレオチドは第1の末端および第2の末端を有し、ここで、この第1の末端はプライマーを組み込む、工程;
c)上記テンプレートから、工程(b)において合成されたポリヌクレオチドを分離する工程;
d)工程(b)において合成されたポリヌクレオチドの第2の末端に、ネステッドオリゴヌクレオチドをアニーリングする工程であって、このネステッドオリゴヌクレオチドは、上記ポリヌクレオチドの第2の末端にアニーリングする第1の部分および上記プライマーの第2の部分と同一の所定の配列を有する第2の部分を有する、工程;
e)工程(b)において合成されたポリヌクレオチドを伸長して、上記所定の配列に相補的であるポリヌクレオチドの末端部分を提供する工程;ならびに
f)上記所定の配列を有する単一のプライマーを使用し、伸長されたポリヌクレオチドを増幅する工程
を包含する。
【0013】
また、その第1の末端に所定の配列を有し、そしてその他方の末端に、この所定の配列と相補的な配列を有する人工核酸鎖が、それ自身本開示の新しい局面であることが企図される。
【0014】
別の局面において、本開示は、核酸鎖を増幅する新しい方法を提供し、この方法は、その第1の末端に所定の配列を有し、そしてその他方の末端に、この所定の配列に相補的な配列を有する人工核酸鎖を提供する工程;およびポリメラーゼおよびヌクレオチドの存在下において、このヌクレオチドの重合のために適切な条件下で、上記人工核酸鎖と所定の配列を有するプライマーとを接触させる工程を包含する。
【0015】
本明細書中に記載される増幅プロセスおよび人工テンプレートは、適切な発現ベクター中にライゲーションされ得る増幅生成物を調製するために使用され得る。次いで、このベクターは、標的配列によってコードされるポリペプチドまたはタンパク質を生成するために、標準的方法を使用して適切な宿主生物を形質転換するために使用され得る。特に有用な実施形態において、本明細書中に記載される技術は、例えば、抗体ライブラリーのような複雑なライブラリーを構築するための、関連する配列のファミリーを増幅するために使用される。
例えば、本発明は、以下の項目を提供する:
(項目1)
抗体の少なくとも一部分をコードする核酸を増幅する方法であって、以下の工程:
a)抗体の少なくとも一部分をコードするテンプレートにプライマーをアニーリングする工程であって、該プライマーは、該テンプレートにアニーリングする第1の部分および該テンプレートにアニーリングしない所定の配列の第2の部分を有する、工程;
b)該プライマーの該第1の部分が該テンプレートにアニーリングする位置と該テンプレートの末端との間の該テンプレートの部分に相補的であるポリヌクレオチドを合成する工程であって、該ポリヌクレオチドは、その第1の末端において該プライマーを有し、かつ第2の末端を有する、工程;
c)該テンプレートから、工程(b)において合成された該ポリヌクレオチドを分離する工程;
d)工程(b)において合成された該ポリヌクレオチドの該第2の末端に、テンプレートオリゴヌクレオチドをアニーリングする工程であって、該テンプレートオリゴヌクレオチドは、該ポリヌクレオチドの該第2の末端にアニーリングする第1の部分および該プライマーの該第2の部分と同一の所定の配列を有する第2の部分を有する、工程;
e)工程(b)において合成された該ポリヌクレオチドを伸長して、該所定の配列に相補的な該ポリヌクレオチドの末端部分を提供する工程;および
f)該所定の配列を有する単一のプライマーを使用し、該伸長されたポリヌクレオチドを増幅する、工程
を包含する、方法。
(項目2)
項目1に記載の方法であって、ここで、抗体の少なくとも一部分をコードするテンプテートにプライマーをアニーリングする上記工程が、以下:
【化1】

からなる群より選択される配列を含む少なくとも1つのプライマーをアニーリングする工程を包含し、ここで、RはAまたはGであり、KはGまたはTであり、そしてSはCまたはGである、方法。
(項目3)
項目1に記載の方法であって、ここで、抗体の少なくとも一部分をコードするテンプテートにプライマーをアニーリングする上記工程が、IgA抗体の少なくとも一部分をコードするテンプテートにプライマーをアニーリングする工程を包含する、方法。
(項目4)
抗体の少なくとも一部分をコードする核酸を増幅する方法であって、以下の工程:
a)抗体の少なくとも一部分をコードするテンプレートに、プライマーおよび境界オリゴヌクレオチドをアニーリングする工程であって、該プライマーは、該テンプレートにアニーリングする第1の部分および該テンプレートにアニーリングしない所定の配列の第2の部分を有する、工程;
b)該プライマーの該第1の部分が該テンプレートにアニーリングする位置と該境界オリゴヌクレオチドがアニーリングする該テンプレートの部分との間の該テンプレートの部分に相補的であるポリヌクレオチドを合成する工程であって、該ポリヌクレオチドは、その第1の末端において該プライマーを有し、かつ第2の末端を有する、工程;
c)該テンプレートから、工程(b)において合成された該ポリヌクレオチドを分離する工程;
d)工程(b)において合成された該ポリヌクレオチドの該第2の末端に、テンプレートオリゴヌクレオチドをアニーリングする工程であって、該テンプレートオリゴヌクレオチドは、該ポリヌクレオチドの該第2の末端にアニーリングする第1の部分および該プライマーの該第2の部分と同一の所定の配列を有する第2の部分を有する工程;
e)工程(b)において合成された該ポリヌクレオチドを伸長して、該所定の配列に相補的である該ポリヌクレオチドの末端部分を提供する、工程;
f)該所定の配列を有する単一のプライマーを使用し、該伸長されたポリヌクレオチドを増幅する、工程
を包含する、方法。
(項目5)
項目4に記載の方法であって、ここで、抗体の少なくとも一部分をコードするテンプテートにプライマーをアニーリングする上記工程が、以下:
【化2】
【化3】
からなる群より選択される配列を含む少なくとも1つのプライマーをアニーリングする工程を包含し、ここで、RはAまたはGであり、KはGまたはTであり、そしてSはCまたはGである、方法。
(項目6)
項目4に記載の方法であって、ここで、抗体の少なくとも一部分をコードするテンプテートにプライマーをアニーリングする上記工程が、IgA抗体の少なくとも一部分をコードするテンプテートにプライマーをアニーリングする工程を包含する、方法。
(項目7)
抗体ライブラリーを作製する方法であって、以下の工程:
a)IgA抗体の少なくとも一部分をコードする、多種多様なテンプレートの集団を提供する工程;
b)該多種多様なテンプレートの集団と少なくとも1つのプライマーとを接触させる工程であって、該少なくとも1つのプライマーは、該テンプレートにアニーリングする第1の部分および該テンプレートにアニーリングしない所定の配列の第2の部分を有する、工程;
c)該プライマーの該第1の部分が該テンプレートにアニーリングする位置と該テンプレートの末端との間の該テンプレートの部分に相補的である、ポリヌクレオチドを合成する工程であって、該ポリヌクレオチドは、その第1の末端において該プライマーを有し、かつ第2の末端を有する、工程;
d)該テンプレートから、工程(c)において合成された該ポリヌクレオチドを分離する工程;
e)工程(c)において合成された該ポリヌクレオチドの該第2の末端に、少なくとも1つのテンプレートオリゴヌクレオチドをアニーリングする工程であって、該少なくとも1つのテンプレートオリゴヌクレオチドは、該ポリヌクレオチドの該第2の末端にアニーリングする第1の部分および該プライマー該第2の部分と同一の所定の配列を有する第2の部分を有する、工程;
f)工程(c)において合成された該ポリヌクレオチドを伸長して、該所定の配列に相補的である該ポリヌクレオチドの末端部分を提供する工程;および
g)該所定の配列を有する単一のプライマーを使用し、該伸長されたポリヌクレオチドを増幅する工程
を包含する、方法。
(項目8)
項目7に記載の方法であって、ここで、抗体の少なくとも一部分をコードするテンプテートにプライマーをアニーリングする上記工程が、以下:
【化4】
【化5】
からなる群より選択される配列を含む少なくとも1つのプライマーをアニーリングする工程を包含し、ここで、RはAまたはGであり、KはGまたはTであり、そしてSはCまたはGである、方法。
(項目9)
項目7に記載の方法に従って調製された、IgA抗体のライブラリー。
(項目10)
所望の結合特異性を有する抗体を同定する方法であって、以下の工程:
項目7に記載の方法に従ってIgA抗体のライブラリーを調製する工程;および
該ライブラリーをスクリーニングして、所望の結合特異性を有する1つ以上のIgA抗体を同定する工程
を包含する、方法。
(項目11)
項目10に記載の方法に従って同定した、IgA抗体。
【図面の簡単な説明】
【0016】
【図1】テンプレートにアニーリングした、プライマーおよび境界オリゴの概略図。
【図2A】核酸鎖にアニーリングした、制限オリゴの概略図。
【図2B】短縮された5’末端を有するテンプレートにアニーリングした、プライマーの概略図。
【図3】代替の実施形態の概略図であって、ここで、複数ラウンドの重合が実施され、そして、制限オリゴヌクレオチドは、原テンプレートではなく新規に合成された鎖にアニールする。
【図4】新規に合成された核酸鎖にアニーリングした、ネステッドオリゴの概略図。
【図5】本開示に基づいた、人工テンプレートの概略図。
【図6】人工テンプレートの単一プライマー増幅の概略図。
【図7】TMX24CMnptと名づけられたネステッドオリゴの配列を示す。
【図8a】実施例3において生成された、単離されたFabの配列を示す。
【図8b】実施例3において生成された、単離されたFabの配列を示す。
【図8c】実施例3において生成された、単離されたFabの配列を示す。
【図8d】実施例3において生成された、単離されたFabの配列を示す。
【図8e】実施例3において生成された、単離されたFabの配列を示す。
【図9a】実施例5において生成された、単離されたFabの配列を示す。
【図9b】実施例5において生成された、単離されたFabの配列を示す。
【図9c】実施例5において生成された、単離されたFabの配列を示す。
【図9d】実施例5において生成された、単離されたFabの配列を示す。
【発明を実施するための形態】
【0017】
(好ましい実施形態の詳細な説明)
本開示は、標的核酸配列を増幅する方法を提供する。特に有用な実施形態において、上記標的核酸配列は、ポリペプチドまたはタンパク質をコードする遺伝子である。本開示はまた、どのように増幅生成物をクローニングし、かつ適切な発現系において発現させ得るかを記載する。特に有用な実施形態において、本明細書中に記載される技術は、例えば、抗体ライブラリーのような複雑なライブラリーを構築するための、関連する配列のファミリーを増幅するために使用される。
【0018】
上記標的核酸配列は、単一のプライマーのみを含むプロセスを介して指数関数的に増幅される。単一のプライマーを利用する(すなわち、各々異なった配列を有する順方向プライマーおよび逆方向プライマーの両方を使用する必要性が無い)能力は、増幅されるべき標的配列を含む核酸の鎖を操作することによって達成される。操作された核酸の鎖(本明細書中において、時折、「人工テンプレート」という)は、2つのテンプレート、すなわち、以下;
1)増幅されるべき配列を含む、天然核酸または合成核酸(例えば、DNAまたはcDNA)である出発物質
2)ネステッドオリゴヌクレオチド
から調製される。上記出発物質は、原テンプレート(original template)と見なされ得る。上記ネステッドオリゴヌクレオチドは、操作された核酸の鎖の作製の間に、原テンプレートのヌクレオチド配列を伸長するためのテンプレートとして使用される。この操作された核酸の鎖は、一連の操作によって原テンプレートから作製され、その結果その反対端に相補的な配列が存在する。これは、単一のプライマーのみを使用する増幅を可能にする相補的な配列である。
【0019】
精製形態または未精製形態のあらゆる核酸は、増幅されるべき標的核酸配列を含むか、または含むと思われる条件で、本明細書中に記載されるプロセスのための出発物質として利用され得る。したがって、このプロセスにおいて利用される出発物質は、DNAまたはRNA(伝令RNAを含む)であり得、これらのDNAまたはRNAは、一本鎖または二本鎖であり得る。さらに、各々の1つの鎖を含むDNA−RNAハイブリッドが利用され得る。任意のこれらの核酸の混合物がまた利用され得るか、または同じかもしくは異なったプライマーを使用した本明細書における、これまでの増幅反応から生成された核酸が使用され得る。増幅されるべき標的核酸配列は、より大きい分子の一部分(fraction)であり得るか、または個々の分子(discrete molecule)として最初に存在し得る。この出発核酸は、同一であり得るかまたは異なり得る1つより多くの所望の標的核酸配列を含み得る。したがって、本プロセスは、大量の1種類の標的核酸配列の生成のためのみではなく、同一であるかまたは異なった核酸分子上に位置する1つより多くの異なる標的核酸配列を同時に増幅するためにも有用であり得る。
【0020】
核酸は、任意の供給源(例えば:ゲノムライブラリーもしくはcDNAライブラリー、プラスミド、クローニングされたDNAもしくはRNA、または任意の供給源(細菌、酵母、ウイルスおよび高等生物(例えば、植物もしくは動物)が挙げられる)に由来する天然のDNAもしくはRNA)から得られ得る。核酸は、天然に存在し得るか、または全体的もしくは部分的に合成され得る。本発明において使用される核酸を獲得または生成するための技術は、当業者において周知である。核酸が2本の鎖を含む場合、それが原テンプレートとして使用され得る前に、別個の工程かまたはプライマー伸長生成物の合成と同時にかのいずれかとして、その核酸の鎖を分離する必要性がある。さらに、出発物質が第1鎖(first strand)DNAである場合、第2鎖(second strand)DNAは、当業者の範囲内のプロセスによって有利に作製され得、そして原テンプレートとして使用され得、そこから人工テンプレートが作製される。
【0021】
第1鎖cDNAは、本方法のための特に有用な原テンプレートである。DNAテンプレートを生成するための適切な方法は当業者に公知であり、そして当業者によって容易に選択される。好ましい実施形態において、第1鎖cDNAは、オリゴデオキシヌクレオチドプライマーおよび4つのデオキシヌクレオシド三リン酸(dATP、dGTP、dCTP、およびdTTP)の存在下において、任意のRNA出発物質に相補的であるDNAの合成を逆転写酵素が触媒する反応において合成される。この反応は、mRNAの3’末端に対するオリゴ−デオキシヌクレオチドプライマーのアニーリングによって開始され、続いて、上記mRNAヌクレオチド配列と関連する塩基対形成により決定されるように、伸長する鎖の3’末端に対して適切なデオキシヌクレオシドが段階的に添加される。当業者において理解されるように、試料中の全てのmRNAは、mRNAのポリA尾部への、オリゴdTのアニーリングを介した第1鎖cDNAを生成のために使用され得る。
【0022】
一旦原テンプレートが得られると、プライマー20および境界オリゴヌクレオチド30は、原テンプレート10にアニーリングされる(図1を参照のこと)。上記プライマーの3’末端で始まり上記境界オリゴヌクレオチドのおよそ5’末端までの、上記原テンプレートの部分に相補的な核酸の鎖が重合化される。
【0023】
上記原テンプレートにアニーリングした上記プライマー20は、好ましくは上記原テンプレートにアニーリングしない所定の配列の第1の部分22および上記原テンプレートにアニーリングする第2の部分25を含み、そして必要に応じてその第1の部分と第2の部分との間に制限酵素認識部位23を含む。上記プライマーは、増幅されるべき標的配列12に隣接した原テンプレートにアニーリングする。図1に示されるように、上記プライマーは、増幅されるべき標的配列の上流の原テンプレートにアニーリングし得るか、または上記プライマーは、増幅されるべき標的配列12の開始部(beginning)に重なり得ることが企図される。このプライマーの非アニーリング部分22の所定の配列は、もともと上記原テンプレート中に存在するものではなく、そして以下に詳細に記載されるように、増幅処理の間に使用される単一のプライマーがハイブリダイズし得る配列を提供するように選択される。必要に応じて、上記所定の配列は、本明細書中の以下においてより十分に記載されるように、発現ベクター中への人工テンプレート部分の挿入のために有用な制限酵素認識部位を含み得る。
【0024】
原テンプレートにアニーリングした境界オリゴヌクレオチド30は、その核酸の重合を終結させる。核酸重合を終結させることが可能なあらゆるオリゴヌクレオチドが、境界オリゴヌクレオチド30として利用され得る。好ましい実施形態において、上記境界オリゴヌクレオチドは、原テンプレート10にアニーリングする第1の部分35および伸長反応に影響されない第2の部分32を含む。この境界オリゴを、伸長に関する部位としての作用から妨げるための技術は、当業者の範囲内である。一例として、上記境界オリゴ30の部分32は、図1に示されるように、原テンプレート10にアニーリングしないように設計され得る。このような実施形態において、境界オリゴヌクレオチド30はさらなる重合を妨げるが、その3’末端が原テンプレート10にハイブリダイズしないので、核酸合成のためのプライマーとしては機能しない。代替的に、境界オリゴ30の3’末端は、同じ効果を達成するために固定核酸(locked nucleic acid)を含むように設計され得る。固定核酸は、例えば、WO 99/14226において開示され、その内容は本明細書中に参考として援用される。当業者は、境界オリゴの3’末端の伸長が起こらないことを保証する他の方法を構想する。
【0025】
本明細書中に記載されるプライマーおよびオリゴヌクレオチドは、当該分野において周知である、オリゴヌクレオチド合成のための確立された方法を使用して合成され得る。本発明のプライマーを含むオリゴヌクレオチドは、モノマー−モノマー相互作用の規則的パターン(例えば、Watson−Crick塩基対)によって標的ポリヌクレオチドに特異的に結合することが可能な、天然の、もしくは修飾されたモノマーまたは結合体(linkage)の線形オリゴマー(linear oligomer)(例えば、デオキシリボヌクレオチド、リボヌクレオチドなど)を含む。通常、モノマーは、数単量体単位(例えば、3〜4)から数十単量体単位の大きさの範囲内のオリゴヌクレオチドを形成するために、ホスホジエステル結合またはそのアナログによって結合する。プライマーは、代表的には一本鎖であるが、二本鎖にもなり得る。プライマーは、代表的にはデオキシリボ核酸であるが、当該分野において公知である種々の合成的プライマーおよび天然に存在するプライマーは、本開示の方法のために有用であり得る。プライマーは、テンプレートに相補的であり、そのプライマーは合成の開始のための部位として機能するためにハイブリダイズするように設計されるが、そのテンプレートの正確な配列を反映する必要性は無い。このような場合において、テンプレートへのプライマーの特異的ハイブリダイゼーションは、ハイブリダイゼーション条件のストリンジェンシーに依存する。プライマーは、例えば、色素性部分、放射活性部分、または蛍光部分で標識され得、そして検出可能な部分として使用される。
【0026】
核酸の重合は、当業者において公知である方法を使用して達成され得る。重合は、テンプレートの指示に従って遊離ヌクレオチドを順次付加するDNAポリメラーゼを使用し、一般に酵素的に達成される。数種の異なるDNAポリメラーゼが、本プロセスにおける使用のために適切である。ある実施形態において、選択の判定基準としては、エキソヌクレアーゼ活性を欠くこと、または強いエキソヌクレアーゼを有さないDNAポリメラーゼであることが挙げられる。本プロセスにおける使用のための低いエキソヌクレアーゼ活性を有するDNAポリメラーゼは、天然の供給源から単離され得るか、または組み換えDNA技術を介して生成され得る。使用され得るポリメラーゼの例示的な例は、T7 Sequenase v.2.0、エキソヌクレアーゼ活性を欠くDNAポリメラーゼIのKlenowフラグメント、TaqポリメラーゼのKlenowフラグメント、exo.−Pfu DNAポリメラーゼ、Vent.(exo.−)DNAポリメラーゼ、およびDeep Vent.(exo−)DNAポリメラーゼであるが、これらに限定されない。
【0027】
特に有用な実施形態において、出発物質の不必要な部分を消化により取り除くことによって、境界オリゴヌクレオチドの使用が避けられる。図2Aにおいて模式的に示されるこの実施形態において、制限オリゴヌクレオチド70は、あらかじめ選択しておいた位置で出発物質100にアニーリングする。この制限オリゴヌクレオチドは、制限酵素認識部位72を含む出発物質上に、二本鎖部分を提供する。適切な制限酵素認識部位としては、XhoI、SpeI、Nhe1、HindIII、NcoI、XmaI、BglII、BstI、およびPvuIが挙げられるが、これらに限定されない。適切な制限酵素に曝露すると、上記出発物質は消化され、そしてその結果不必要な配列を取り除くために短くなる一方で、原テンプレート110として使用されるものの上で、増幅されるべき所望の標的配列12(またはその一部分)は保存される。一旦原テンプレート110が得られると、プライマー20は、上述の前の実施形態のように標的配列12に隣接して、または重なって原テンプレート110にアニーリングされる(図2Bを参照のこと)。プライマー20の3’末端と原テンプレート110の5’末端との間の原テンプレートの部分に相補的な核酸の鎖40は、重合化される。当業者が理解するように、この実施形態において、制限オリゴヌクレオチドが原テンプレートを生成するために利用される場合、プライマー伸長は短くされた原テンプレート110の5’末端までの全てを進むことを可能にし得るので、境界オリゴヌクレオチドを使用する必要性は無い。
【0028】
一旦重合が完了する(すなわち、伸長する鎖40が境界ヌクレオチド30または短くされた原テンプレート110の5’末端に到達する)と、新規に合成された相補鎖は、物理学的手段、化学的手段または酵素的手段を含む任意の適切な変性方法によって、原テンプレートから分離される。鎖の分離はまた、ヘリカーゼまたは酵素RecAとして公知の酵素のクラスに由来する酵素によって誘導されるRecAは、ヘリカーゼ活性を有し、かつリボATPの存在下においてDNAを変性させることで知られる。ヘリカーゼを用いた、核酸の鎖を分離するための適切な反応条件は、Cold Spring Harbor Symposia on Quantitative Biology、第XLIII巻「DNA:Replication and Recombination」(New York:Cold Spring Harbor Laboratory、1978)、B.Kuhnら、「DNA Helicases」、pp.63−67によって記載され、そしてRecAの使用に関する技術は、C.Radding、Ann.Rev.Genetics、16:405−37(1982)において総説される。
【0029】
したがって、新規に合成された相補鎖は、上記プライマー20によって提供された配列(例えば、所定の配列22、任意の制限酵素認識部位23および上記プライマーのアニーリング部分25)、ならびに、プライマー20が原テンプレート10にアニーリングした位置と、境界オリゴヌクレオチド30にアニーリングした原テンプレート10の部分かまたは原テンプレートの短くされた5’末端のいずれかとの間の原テンプレート10の部分に相補的な、新規に合成された部分45を含む。図4を参照のこと。
【0030】
必要に応じて、続く工程の中で使用するための複数コピーの新規に合成された相補鎖を生成するために、原テンプレートおよびプライマーを使用した複数ラウンドの重合(好ましくは、15ラウンド〜25ラウンド)が実施される。このプロセス中のこの時点での複数コピーの新規に合成された相補鎖の作製は、(増幅の前に全長(entire)の人工テンプレートが生成されるまで待つ代わりに)最終的に生成された人工テンプレート中への標的配列の正確な複製の取り込みを確実にすることを援助する。原テンプレートに基づいた複数ラウンドの重合は、ライブラリーの全てのメンバーのより良い表現が達成されるより大きな可能性を提供すると考えられており、その結果、単一ラウンドの重合と比較して、より大きな多様性を提供する。
【0031】
代替的な実施形態において、新規合成鎖は、上述のようにプライマー20を原テンプレート10にアニーリングし、そしてブロッキングオリゴヌクレオチドの存在も原テンプレートの部分除去のいずれも伴わずに複数ラウンドの重合を実施することによって生成される。この実施形態(これは、図3において図解的に示される)において、プライマーは、全長の新規合成鎖140を提供するために、原テンプレートの全長に沿って伸長される。次に、制限オリゴヌクレオチド170が、上記全長の新規合成鎖にハイブリダイゼーションされる。この制限オリゴヌクレオチドは、上記新規合成鎖上に制限酵素認識部位を含む二本鎖部分を提供する。適切な制限酵素認識部位としては、XhoI、SpeI、Nhel、HindIII、NcoI、XmaI、BglII、BstI、PvuI、XcmI、BsaJI、HpaI、ApaLI、SacI、DraIIIおよびSmaIが挙げられるが、これらに限定されない。適切な制限酵素に曝すことにより、上記新規合成鎖は消化され、そしてその結果短縮される。次いで、ネステッドオリゴヌクレオチド50は、以下により詳細に説明されるように、人工テンプレートの調製を完了させるために、短くされた新規合成鎖142にハイブリダイゼーションされる。
【0032】
人工テンプレートの調製における次の工程は、例えば、図4において示されるように、ネステッドオリゴヌクレオチド50を新規に合成された相補鎖の3’末端にアニーリングさせることに関する。図4において見られるように、ネステッドオリゴヌクレオチド50は、人工テンプレートを完成させるために必要な、さらなる重合のためのテンプレートを提供する。ネステッドオリゴヌクレオチド50は、新規に合成された相補鎖に対してハイブリダイゼーションしないおよび/または新規に合成された相補鎖に対して修飾された塩基を含む部分52を含み、その結果、ネステッドオリゴヌクレオチドがプライマーとして機能することを防いでいる。ネステッドオリゴヌクレオチド50はまた、新規に合成された相補鎖の3’末端にハイブリダイゼーションする部分55を含む。部分55は、図4において示されるように、新規に合成された部分45と重なり合い得るか、または新規に合成された部分45を超えて伸長し得る。ネステッドオリゴヌクレオチド50はまた、必要に応じて、制限酵素認識部位を規定する部分56を含み得る。ネステッドオリゴヌクレオチド50の最終部分58は、プライマー20の部分22と同一な所定の配列を含む。部分55が、新規に合成された相補鎖の初めの3’末端を越えて伸長する地点から、ネステッドオリゴヌクレオチドは、人工テンプレートを形成するためのさらなる重合のためのテンプレートとして機能する。ネステッドオリゴは、(原テンプレートの形成において、その一部分が短縮されていた場合)標的配列の一部分を含み得るか、もしくはポリペプチドまたはタンパク質をコードする遺伝子(またはその一部分)(例えば、1つ以上のCDR領域もしくはフレームワーク領域または抗体の定常領域)を含み得ることが、理解されるはずである。種々の配列を有するネステッドオリゴヌクレオチドの収集物が利用され得ることもまた企図され、その結果、多種多様な生成物のライブラリーをもたらす種々のテンプレートが提供される。このようにして、図4において示されるように、ネステッドオリゴヌクレオチドに相補的なさらなる核酸60を付加することによって、重合は、新規に合成された相補鎖を伸長させる。重合を達成するための技術は、当業者の範囲内にある。前述のように、ある実施形態において、適切なポリメラーゼ(エキソヌクレアーゼ活性を欠く酵素)の選択が好ましくあり得る。
【0033】
一旦重合が完了すると、人工テンプレート120は、例えば、加熱変性のような当業者に周知の技術によって、ネステッドオリゴヌクレオチド50から分離される。結果生じた人工テンプレート120は、初発プライマー20由来である部分、原テンプレートの一部分に相補的な部分45、およびネステッドオリゴヌクレオチドの一部分に相補的な部分65を含む(図5を参照のこと)。注目すべきことに、人工テンプレート120の3’末端は、プライマー20の部分22の所定の配列に相補的な配列を含む、部分68を含む。これは、当業者において公知の技術を使用し、プライマー部分22の所定の配列と同一の配列を有する単一のプライマーを使用して、人工テンプレート120内に含まれる所望の配列を増幅することを可能にする。単一プライマー増幅の間、エキソヌクレアーゼ活性を有するポリメラーゼが存在することは好ましい。なぜならば、このような酵素は「プルーフリーディング」機能を提供することが知られており、かつエキソヌクレアーゼ活性を欠いたポリメラーゼと比較して相対的により高い伸長性(processivity)を有するからである。
【0034】
図6は、新規に合成されたcDNAテンプレートの単一プライマー増幅に関連する工程を図示する。反応混合物中にプライマーが存在する場合、プライマーは、テンプレートに隣り合う配列にハイブリダイゼーションし、そしてこのテンプレートを増幅する。プライマーが存在しない場合は、5’末端の所定の配列とこの所定の配列に相補的な3’末端配列との間で、内部の自己アニーリングが存在すると考えられている。好ましい実施形態において、所定の配列および相補的な所定の配列は、単一プライマー増幅反応の間のミスプライミングを避けるために、より高い温度でアニーリングするように設計され得る。
【0035】
増幅が実施された後、その生成物は、当業者において公知である任意の技術を使用して検出され得る。核酸を検出するために使用される方法の例としては、対立遺伝子特異的オリゴヌクレオチドを用いたハイブリダイゼーション、制限酵素切断、一本鎖立体配座多型(single−stranded conformational polymorphism)(SSCP)、分析ゲル電気泳動、エチジウムブロマイド染色、蛍光共鳴エネルギー移動(fluorescence resonance energy transfer)、ヘアピンFRETアッセイ(essay)、およびTaqManアッセイが挙げられるが、これらに限定されない。
【0036】
一旦人工核酸が所望される回数分増幅されると、その鎖を消化するために制限酵素認識部位23および制限酵素認識部位66または任意の内在性制限酵素認識部位が使用され得、そして標的核酸配列は、適切な発現ベクター中にライゲーションされ得る。次いで、このベクターは、標準の方法を使用して適切な宿主生物を形質転換するために使用され、標的領域によってコードされるポリペプチドまたはタンパク質が生成される。
【0037】
特に有用な実施形態において、本明細書中に記載される方法は、抗体のcDNAを使用し、抗体またはその一部(例えば、可変領域(軽鎖または重鎖のいずれか))をコードする標的配列を増幅するために使用される。このようにして、抗体のライブラリーが増幅され得、そしてスクリーニングされ得る。したがって、例えば、抗体mRNAで開始して、第1鎖cDNAが生成され得、そして原テンプレートを提供するために消化され得る。プライマーは選択された相補性決定領域(CDR)の上流にアニーリングするように設計され得、そしてこの新規合成核酸鎖は、CDRを含む。一例として、上記標的配列が重鎖CDR3である場合、プライマーは、重鎖フレームワーク1(FR1)領域にアニーリングするように設計され得る。当業者は、本開示に基づき、他の上流部位にアニーリングする適切なプライマー、または抗体cDNA内の他に選択された標的を複製する適切なプライマーを、どのようにして設計するかを容易に想定する。
【実施例】
【0038】
以下の実施例は、本発明の例証のために提供されるが、本発明を限定しない。
【0039】
(実施例1)
(IgM重鎖可変遺伝子のレパートリーの増幅)
(第1鎖cDNA合成および修飾)
SuperScript First−Strand Synthesis System for RT−PCR(Invitrogen Life Technologies、Carlsbad、CA)を使用し、基本的にキットの指示書に従って、オリゴdTプライマーを用いて慣習的な第1鎖cDNAを生成するために、ヒト末梢血リンパ球(PBL)mRNAを使用した。この第1鎖cDNA生成物を、QIAGENスピンカラム(QIAGEN、Valencia、CA提供のPCR Purification Kit)を通じて浄化した。制限エンドヌクレアーゼEcoRIによって消化され得る二本鎖DNA領域を生成するために、制限オリゴヌクレオチドを上記第1鎖cDNAに添加した。制限オリゴヌクレオチド(CMEcoRI)の配列は、5’TCC TGT GAG AAT TCC CCG TCG3’(配列番号1)であった。この反応を、第1鎖cDNAおよび0.1μMオリゴヌクレオチドを用いてセットアップした。試料を2分間95℃まで加熱し、次いで特異的アニーリングが起こり得るように64℃で2分間保った。適切な量の10×制限緩衝液H(Roche Diagnostics)を上記試料に添加し、そしてさらに37℃まで冷却した。制限エンドヌクレアーゼEcoRI(New England Biolabs、Beverly MA)を添加し、そして37℃で30分間インキュベートした。この制限酵素を65℃で20分間加熱不活性化し、次いで試料を4℃まで冷却した。
【0040】
(第2鎖線形増幅およびネステッドオリゴ伸長)
プライマー「TMX24VH3a」(最終0.4μM)、dNTP、AmpliTaq酵素およびその10×反応緩衝液(Applied Biosystems、Foster City、CA)に加えて、EcoRIで消化された第1鎖cDNAを、第2鎖cDNA反応における原テンプレートとして使用した。上記プライマーは、所定のTMX24配列、XhoI制限酵素認識部位およびヒト抗体重鎖遺伝子のフレームワーク1領域における第1鎖cDNAにアニーリングする領域を含むように設計した。「TMX24VH3a」の配列は、5’GTG CTG GCC GTT GGA AGA GGA GTG
CTC GAG GAR GTG CAG CTG GTG GAG3’(配列番号2)であり、Rは、塩基Aおよび塩基Gの等モル混合物を意味する。上記試料を95℃で1分間加熱変性し、次いで95℃で5秒間、56℃で10秒間、および68℃で1分間を介する反応を20回サイクルさせた。これは、第2鎖cDNAの線形増幅を可能にする。「TMX24CM0」と命名されたネステッドオリゴを、次いで0.08μMの最終濃度となるように氷上で添加した。「TMX24CM0」の配列は、5’GTG CTG GCC GTT GGA AGA GGA GTG ACT AGT AAT TCT CAC AGG AGA CGA GGG GGA3’(配列番号3)であった。これはその後のクローニング工程において使用されるSpeI制限エンドヌクレアーゼ部位を含む。このネステッドオリゴの3’末端は、逆結合(3’−5’よりもむしろ3’−3’)アデノシンの取り込みにより伸長を防ぐように設計される。上記第2鎖cDNAを、94℃5秒間で加熱変性し、次いで68℃で10秒間および95℃で5秒間の4サイクルアニーリングおよび伸長させることによってネステッドオリゴからさらに伸長し、続いて68℃で30秒間処理し、そして4℃にした。結果生じた第2鎖cDNA、すなわち人工テンプレートを、次いでQIAGENスピンカラム(QIAGEN、Valencia、CA提供のPCR Purification Kit)を使用して洗浄した。この工程は、オリゴヌクレオチドを除去し、そして下流のプロトコールのための容易な緩衝液交換を可能にする。
【0041】
(単一プライマー増幅(SPA))
Advantage2ポリメラーゼミックス(Clontech)およびその10×反応緩衝液、dNTP、ならびに5’GTG CTG GCC GTT GGA AGA GGA GTG3’(配列番号4)の配列を有する単一のプライマー(TMX24)を使用して、上記人工テンプレートを増幅した。この試料を95℃で1分間加熱変性し、次いで95℃で5秒間および68℃で1分間を介する反応を35回サイクルさせた。これ続いて、さらに68℃で3分間反応させ、そして4℃で保持した。
【0042】
(クローニングおよび配列決定)
約450bpの増幅生成物をゲル精製し、次いでXhoIおよびSpeIで消化し、そしてpBluescript KS+(Stratagene)中にクローニングした。個々のクローンを取り出し、そしてそれらのDNA配列を決定した。分析した16クローンの全てがIgM重鎖であり、各々が種々の長さの異なるCDR3配列を有していた。この結果は、この方法によって抗体鎖の多様な集団が増幅されたことを示す(表1を参照のこと)。
【0043】
【表1】
。
【0044】
(実施例2)
VH生成物をベクター中にクローニングするために、ネイティブIgM CH1定常領域が再構成され得、CH1中のEcoRI以外の部位を、上記第1鎖cDNAエンドヌクレアーゼ消化のために利用した。当業者が理解するように、Taqポリメラーゼがこのプロトコールのために使用された場合、新規に合成されたDNA鎖の多くに末端のAが付加される。多様性を最大化させるために、その末端のAの存在をネステッドオリゴヌクレオチドの設計において考慮した。しかしながら、余剰なAの存在は、EcoRI認識部位の減少を招く。IgM定常領域の分析は、他のネイティブ制限酵素認識部位が、この方法のために潜在的に使用され得ることを明らかにした(例えば、DraIII)。CH1ドメイン中のDraIIIネイティブ制限酵素認識部位の使用の結果、上流EcoRI部位が未修飾のまま残存し、そして重鎖レパートリーのクローニングのために使用され得る。重鎖インサートは、XhoIおよびEcoRIによって、EcoRIからCH2ドメインに残存IgM CH1ドメインを有する適切なベクター中にクローニングされる。
【0045】
(第1鎖合成および修飾)
オリゴdTプライマーを用いた慣習的な第1鎖cDNAを生成するために、ヒト末梢血リンパ球(PBL)mRNAを使用した。これを、基本的にキットの指示書に従って、SuperScript First−Strand Synthesis System for RT−PCR(Invitrogen Life Technologies、Carlsbad、CA)を使用して行った。制限エンドヌクレアーゼDraIIIによって消化され得る二本鎖DNA領域を生成するために、制限オリゴヌクレオチドを第1鎖cDNAに添加した。この制限オリゴヌクレオチド(CMDraIII)の配列は、5’GAC GAA CAC GTG GTG TGC AAA G3’(配列番号21)であった。この反応を、第1鎖cDNAおよび1μMオリゴヌクレオチドを用いてセットアップした。試料を2分間95℃まで加熱し、次いで特異的アニーリングが起こり得るように64℃で2分間保持した。適切な量の10×制限緩衝液H(Roche Diagnostics)を、上記試料中に添加し、そしてさらに37℃まで冷却した。制限エンドヌクレアーゼDraIII(New England Biolabs、Beverly
MA)を添加し、そして37℃で30分間インキュベートした。上記制限酵素を65℃で20分間加熱不活性化し、次いでこの試料を4℃まで冷却した。
【0046】
(第2鎖線形増幅およびネステッドオリゴ伸長)
第2鎖cDNA反応において、プライマー「TMX24VH1a」(最終0.4μM)、dNTP、AmpliTaq酵素およびその10×反応緩衝液(Applied Biosystems、Foster City、CA)に加えて、DraIIIで消化された第1鎖cDNAを原テンプレートとして使用した。上記プライマーは、所定のTMX24配列、XhoI制限酵素認識部位およびヒト抗体重鎖遺伝子のフレームワーク1領域における第1鎖cDNAにアニーリングする領域を含むように設計した。「TMX24VH1a」の配列は、5’GTG CTG GCC GTT GGA AGA GGA GTG CTC GAG CAG GTK CAG CTG GTG CAG3’(配列番号22)であり、Kは、塩基Gおよび塩基Tの等モル混合物を意味する。上記試料を94℃で1分間加熱変性し、次いで94℃で5秒間、56℃で10秒間、および68℃で2分間を介する反応を20回サイクルさせた。これは、第2鎖cDNAの線形増幅を可能にする。「TMX24CMnpt」と命名されたネステッドオリゴを、次いで0.2μMの最終濃度となるように氷上で添加した。図7において示されるように、「TMX24CMnpt」(配列番号23)の配列は、オリゴヌクレオチドの伸長を防ぐように設計された、修飾された構造を有する3つの3’末端ヌクレオチドを含む。具体的には、上記ネステッドオリゴは、ホスホチオ酸塩(phosphorthioate)および伸長を防ぎかつエキソヌクレアーゼ活性ならびにエンドヌクレアーゼ活性に対して保護するために設計された2’OMeで修飾された、3つの末端ヌクレオチドを有する。このオリゴの3’末端オリゴヌクレオチドは、ハイブリダイズしない(cの代わりにg)。上記第2鎖cDNAを、94℃1分間で加熱変性し、次いで68℃で2分間アニーリングおよび伸長させてネステッドオリゴからさらに伸長し、続いて4℃にした。結果生じた第2鎖cDNA、または人工テンプレートを、次いでQIAGENスピンカラム(QIAGEN、Valencia、CA提供のPCR Purification Kit)を使用して洗浄した。この工程は、オリゴヌクレオチドを除去し、そして下流のプロトコールのための容易な緩衝液交換を可能にする。この手順を繰り返し、そして、適切なベクターにクローニングされ得る免疫グロブリン生成物のライブラリーを生成するために、VHプライマーパネル(primer panel)(プライマーリストを参照のこと)の残りの部分を伸長した。
【0047】
【化6】
上記配列において、RはAおよびGの同じ割合の混合物であり、KはGおよびTの同じ割合の混合物であり、そしてSはCおよびGの同じ割合の混合物である。
【0048】
(単一プライマー増幅(SPA))
上記人工テンプレートを、Advantage 2ポリメラーゼミックス(Clontech)およびその10×反応緩衝液、dNTPならびに5’GTG CTG GCC GTT GGA AGA GGA GTG3’(配列番号4)の配列を有する単一のプライマー(TMX24)を使用して増幅した。その試料を95℃で1分間加熱変性し、次いで95℃で5秒間および68℃で1分間を介する反応を30回サイクルさせた。これに続いてさらに68℃で3分間処理し、そして4℃で保持した。
【0049】
(クローニングおよび配列決定)
約450bpの増幅生成物をゲル精製し、そしてXhoIおよびEcoRIによって消化する。ネイティブEcoRI由来のIgM CH1ドメインの残余部分を含み、CH2ドメインまでかまたはを含んで増幅されたフラグメントをクローニングするために適性のある制限酵素認識部位を含む任意の適切な発現ベクター中に、このインサートをクローニングする。
【0050】
(実施例3)
(B型肝炎陽性ドナー由来のファージミドディスプレイライブラリー(phagmid display library)の構築)
(IgG重鎖およびκ軽鎖についての第1鎖cDNAの合成および修飾)
オリゴdTプライマーを用いた慣習的な第1鎖cDNAを生成するために、B型肝炎ワクチン接種されたドナー由来のヒト末梢血リンパ球(PBL)mRNAを使用した。これは、SuperScript First−Strand Synthesis System for RT−PCR(Invitrogen Life Technologies、Carlsbad、CA)を使用し、基本的にキットの指示書に従って行った。IgGについては制限エンドヌクレアーゼApaLIによって、またはκ軽鎖についてはSacIによって消化され得る二本鎖DNA領域を生成するために、IgGについては制限オリゴヌクレオチド「CGApaLI」を、またはκ軽鎖については制限オリゴヌクレオチド「CKSacI」を第1鎖cDNAに添加した。「CGApaLI」配列は、5’CCA GCG GCG TGC ACA CCT TCC3’(配列番号39)である。「CKSacI」配列は、5’AGG GCC TGA GCT CGC CCG TC3’(配列番号40)である。この反応を、第1鎖cDNA、1μMオリゴヌクレオチド、および適切な量の10×反応緩衝液A(Roche Diagnostics)を用いてセットアップした。この試料を95℃まで2分間加熱し、次いで特異的アニーリングが起こり得るように64℃で2分間保持し、そして37℃まで冷却した。制限エンドヌクレアーゼApaLIまたはSacI(New England Biolabs、Beverly MA)を添加し、そして37℃で30分間インキュベートした。この制限酵素を、SacIに関して65℃で20分間加熱不活性化し、次いで試料を4℃まで冷却した。
【0051】
各々の制限エンドヌクレアーゼによる上記第1鎖cDNAの消化を、当業者において公知である技術を使用し、PCR増幅によって検証した。これらの生成物は、抗体遺伝子のクローニングには使用しなかった。消化された第1鎖cDNAの陽性増幅物を、IgGについて5’VBVH1a内部コントロールプライマーおよび3’CG0内部コントロールプライマーを、およびκについて5’VBVK1aおよび3’CK0内部コントロールプライマーを使用した反応中で観察した。5’VBVH1a/3’CG0プライマーまたは5’VBVK1a/3’CK0プライマーを用いた良好な増幅および5’VBVH1a/3’CG1Zプライマーまたは5’VK1a/3’CK1dx2プライマーを用いたわずかな増幅は、各々の制限エンドヌクレアーゼを用いた第1鎖cDNAテンプレートの首尾良い消化を示す。PCRを検査するチェックのために使用されるプライマーの配列は、以下:
【0052】
【化7】
である。
【0053】
(軽鎖第2鎖線形増幅およびネステッドオリゴ伸長)
フレームワーク1特異的プライマー(最終0.4μM)、dNTP、AmpliTaq酵素およびその10×反応緩衝液(Applied Biosystems、Foster City、CA)を使用した複数の第2鎖cDNA反応をセットアップするために、SacI消化された第1鎖cDNAを、原テンプレートとして使用した。上記プライマーを、κについて所定のTMX24K配列5’GAC GAC CGG CTA CCA AGA GGA GTG3’(配列番号47)、XbaI制限酵素認識部位、およびヒト抗体κ軽鎖遺伝子のフレームワーク1領域中の第1鎖cDNAにアニーリングする領域を含むように設計した。これらのアニーリング配列は、VBaseデータベースプライマー(www.mrc−cpe.cam.ac.uk/imt−doc/public/INTRO.html)由来であり、これらは、公知のヒト抗体の配列に基づいて設計され、そしてκ軽鎖遺伝子の全ヒト抗体レパートリーを網羅すると報告されている。
【0054】
【化8】
【0055】
【化9】
上記配列において、RはAおよびGの同じ割合の混合物であり、MはAおよびCの同じ割合の混合物であり、YはCおよびTの同じ割合の混合物であり、WはAおよびTの同じ割合の混合物であり、そしてSはCおよびGの同じ割合の混合物である。
【0056】
上記試料を94℃で1分間加熱変性し、次いで94℃で5秒間、56℃で10秒間、および68℃で2分間を介する反応を20サイクルさせた。これは、第2鎖DNAの線形増幅を可能にする。κ鎖について「TMX24CKnpt」と命名されたネステッドオリゴを、氷上で0.2μMの最終濃度となるように添加した。「TMX24CKnpt」は、所定の配列TMX24Kを含み、そしてこの配列は、5’GAC GAC CGG CTA CCA AGA GGA GTG CTC GAG CTC AGG CCC TGA TGG GTG ACT TCG CT3’(配列番号61)であった。上記第2鎖cDNAを、94℃で1分間加熱変性させ、次いで68℃で2分間アニーリングおよび伸長させて、ネステッドオリゴからさらに伸長させ、続いて4℃にした。結果生じた第2鎖cDNA(人工テンプレート)を、QIAGENスピンカラム(QIAGEN、Valencia、CA提供のPCR Purification Kit)を使用して洗浄した。この工程は、遊離のオリゴヌクレオチドを除去し、そして下流のプロトコールのための容易な緩衝液交換を可能にする。
【0057】
(軽鎖単一プライマー増幅(SPA))
Advantage 2ポリメラーゼミックス(Clontech)およびその10×反応緩衝液、dNTP、ならびにκ鎖についてのプライマー「TMX24K」を使用し、上記人工テンプレートを増幅した。「TMX24K」についての配列は、5’GAC GAC CGG CTA CCA AGA GGA GTG3’(配列番号62)である。この試料を95℃で1分間加熱変性し、次いで95℃で5秒間および68℃で1分間を介する反応を30サイクルさせた。これに続いてさらに68℃で3分間処理し、そして4℃で保持した。
【0058】
(軽鎖クローニング)
κ増幅生成物をゲル精製し、次いでXbaIおよびSacIによって消化した。そのインサートを、κ軽鎖定常領域の残余部分を含む適切な発現ベクター中にクローニングした。このライゲーションされた生成物を、エレクトロポレーションによってE.coli中に導入し、そして37℃で一晩成長させた。翌朝、軽鎖ライブラリーDNAを回収するためにDNAマキシプレップ(QIAGEN、Valencia、CA)を実施した。次いで、ライブラリーの構築を完了させるために、XhoI/AgeIによる重鎖Fdフラグメントのクローニングのための続く工程において、この軽鎖ライブラリーDNAを使用した。
【0059】
(重鎖第2鎖線形増幅およびネステッドオリゴ伸長)
フレームワーク1特異的プライマー(最終0.4μM)、dNTP、AmpliTaq酵素およびその10×反応緩衝液(Applied Biosystems、Foster City、CA)を使用して複数の第2鎖cDNA反応をセットアップするために、ApaLI消化された第1鎖cDNAを、原テンプレートとして使用した。上記プライマーを、所定のTMX24配列、XhoI制限酵素認識部位、およびヒト抗体重鎖遺伝子のフレームワーク1領域における第1鎖cDNAにアニーリングする領域を含むように設計した。これらのアニーリング配列は、VBaseデータベースプライマー由来であって、これらは公知のヒト抗体の配列に基づいて設計されており、そして重鎖遺伝子の全ヒト抗体レパートリーを網羅すると報告されている。
【0060】
【化10】
【0061】
【化11】
上記配列において、RはAおよびGの同じ割合の混合物であり、KはGおよびTの同じ割合の混合物であり、そしてSはCおよびGの同じ割合の混合物である。
【0062】
上記試料を、94℃で1分間加熱変性し、次いで94℃で5秒間、56℃で10秒間、および68℃で2分間を介する反応を20サイクルさせた。これは、第2鎖cDNAの線形増幅を可能にする。「TMX24CGnpt」と命名されたネステッドオリゴヌクレオチド(配列5’GTG CTG GCC GTT GGA AGA GGA GTG TGT TTG CAC GCC GCT GGT CAG RGC GCC TGA GTT G3’(配列番号77))を、氷上で0.2μMの最終濃度となるように添加した。図1に示されるように、IgMネステッドオリゴについて、オリゴ伸長を防ぐために、3つの3’末端ヌクレオチドを修飾した。上記第2鎖cDNAを、94℃で1分間の加熱変性し、68℃で2分間でアニーリングおよび伸長させてネステッドオリゴからさらに伸長させ、続いて4℃にした。結果生じた第2鎖cDNA(人工テンプレート)を、次いでQIAGENスピンカラム(QIAGEN、Valencia、CA提供のPCR Purification Kit)を使用して洗浄した。この工程は、遊離のオリゴヌクレオチドを除去し、そして下流のプロトコールのための容易な緩衝液交換を可能にする。
【0063】
(単一プライマー増幅(SPA))
Advantage 2ポリメラーゼミックス(Clontech)およびその10×反応緩衝液、dNTP、およびプライマー「TMX24」を使用し、上記人工テンプレートを増幅した。この試料を95℃で1分間加熱変性し、次いで95℃で5秒間および68℃で1分間を介する反応を30サイクルさせた。これに続いて68℃でさらに3分間処理し、そして4℃で保持した。
【0064】
(重鎖クローニングおよびライブラリーの生成)
この増幅された生成物をプールし、次いでゲル精製した。QIAquick PCR purification kit(QIAGEN、Valencia、CA)を用いて、DNAを回収した。このDNAを、XhoI制限酵素およびAgeI制限酵素で順次消化し、次いでゲル精製した。AgeI部位は、ApaLI部位の上流のIgG定常領域のCH1中に、天然に存在する。DNAを、QIAquick Gel extraction Kit(QIAGEN、Valencia、CA)で回収した。
【0065】
軽鎖ライブラリーDNAを、XhoIおよびAgeIで順次消化し、次いでゲル精製した。この軽鎖ライブラリーDNAを、上記重鎖フラグメントとライゲーションした。ライゲーションされたDNAを、反応緩衝液を除去するためにスピンカラム(PCR purification Kit、QIAGEN、Valencia、CA)上に置き、そしてこのDNAを濃縮した。電気的にコンピテントな(electrocompetent)XL−1 Blue細胞(Stratagene)において、最終形質転換を行った。
【0066】
(HBs Agについてのライブラリーのパンニングおよびスクリーニング)
基本的に、Barbas III,CF、Burton,DR、Scott,JK、およびSilverman,GJ(2001)Phage Display:A Laboratory Manual、Cold Spring Harbor Laboratory Press、Cold Spring Harbor、New Yorkに記載されるように、上記ライブラリーを、固定化HBs Agについて4ラウンドパンニングした。パンニングの第2ラウンド、第3ラウンド、および第4ラウンド由来である個々のクローンを、HBs AgについてのELISAによってスクリーニングした。
【0067】
(λ軽鎖についての第1鎖cDNA合成および修飾)
オリゴdTプライマーを用いた慣習的な第1鎖cDNAを生成するために、B型肝炎ワクチン接種されたドナー由来のヒトPBL mRNAを使用した。これを、SuperScript First−Strand Synthesis for RT−PCR(Invitrogen Life Technologies、Carlsbad、CA)を使用し、基本的にキットの指示書に従って行った。制限エンドヌクレアーゼSmaIによって消化され得る二本鎖DNA領域を生成するために、上記第1鎖cDNAに制限オリゴヌクレオチド「CLSmaI」を添加した。「CLSmaI」配列は、5’GAC TTC TAC CCG GGA GCY GTG3’(配列番号78)であって、ここで、YはCおよびTの混合物である。この反応を、第1鎖cDNA、1μMオリゴヌクレオチド、および適切な量の10×反応緩衝液A(Roche Diagnostics)を用いてセットアップした。この試料を95℃まで2分間加熱し、次いで特異的アニーリングが起こり得るように64℃で2分間保持し、そして37℃まで冷却した。制限エンドヌクレアーゼSmaI(New England Biolabs、Beverly MA)を添加し、そして37℃で30分間インキュベートした。この制限酵素を65℃で加熱不活性化し、次いでその試料を4℃まで冷却した。
【0068】
制限エンドヌクレアーゼによる上記第1鎖cDNAの消化を、当業者において公知である技術を使用し、PCR増幅によって検証した。これらの生成物は、抗体遺伝子のクローニングには使用しなかった。5’VBVL1a内部コントロールプライマーおよび3’CL0内部コントロールプライマーを使用した反応において、消化された第1鎖cDNAの陽性増幅物を観察した。5’VBVH1a/3’CL0プライマーを用いた良好な増幅および5’VBVL1a/3’CL2dx2プライマーを用いたわずかな増幅は、SmaIを用いた第1鎖cDNAテンプレートの首尾良い消化を示す。PCRの検査のためのプライマーの配列は、VBVL1a:5’GAC GCG CAC AAC ACG GAG CTC CAG TCT GTG CTG ACT CAG3’(配列番号79)、CL0:5’CCT CAG AGG AGG GYG GGA ACA G3’(配列番号80)およびCL2dx2:5’AGA CAG TGA CGC CGT CTA GAA TTA TGA ACA TTC TGT AGG3’(配列番号81)である。
【0069】
(λ軽鎖第2鎖線形増幅およびネステッドオリゴ伸長)
フレームワーク1特異的プライマー(最終0.4μM)、dNTP、AmpliTaq酵素およびその10×反応緩衝液(Applied Biosystems、Foster City、CA)を使用して複数の第2鎖cDNA反応をセットアップするために、SmaI消化された第1鎖cDNAを、原テンプレートとして使用した。上記プライマーを、所定のTMX24L配列、XbaI部位、およびヒト抗体λ軽鎖遺伝子のフレームワーク領域中の第1鎖cDNAにアニーリングする領域を含むように設計した。これらのアニーリング配列は、VBaseデータベースプライマー(www.mrc−cpe.cam.ac.uk/imt−doc/public/INTRO.html)由来であって、これらは公知のヒト抗体の配列に基づいて設計されており、そしてλ軽鎖遺伝子の全ヒト抗体レパートリーを網羅すると報告されている。λ軽鎖フレームワーク1特異的プライマーは、実施例4におけるものを使用する。
【0070】
上記試料を、94℃で1分間加熱変性し、次いで94℃で5秒間、56℃で10秒間、および68℃で2分間を介する反応を20サイクル行った。これは、上記第2鎖cDNAの線形増幅を可能にする。実施例4において示されるような、「TMX24CLnpt」と命名されたネステッドオリゴヌクレオチドを、氷上で0.2μMの最終濃度となるように添加した。図7に示されるようなIgMネステッドオリゴヌクレオチド「TMX24CMnpt」について、「TMX24CLnpt」の3’末端ヌクレオチドは、オリゴ伸長を防ぐように修飾される。第2鎖cDNAを、94℃で1分間の加熱変性、68℃で2分間でアニーリングおよび伸長させてネステッドオリゴヌクレオチドからさらに伸長し、続いて4℃にした。結果生じた第2鎖cDNA(人工テンプレート)を、PCR Purification Kit(QIAGEN、Valencia、CA)を使用して浄化した。この工程は、遊離のオリゴヌクレオチドを除去し、そして下流のプロトコールのための容易な緩衝液交換を可能にする。
【0071】
(λ軽鎖単一プライマー増幅(SPA))
Advantage 2ポリメラーゼミックス(Clontech)およびその10×反応緩衝液、dNTP、ならびにプライマー「TMX24L」を使用し、上記人工テンプレートを増幅した。「TMX24L」についての所定の配列は、5’GAC GAC CGG CTA CCA AGA GGA CAG3’(配列番号82)である。この試料を95℃で1分間加熱変性し、次いで95℃で5秒間および68℃で1分間を介する反応を30サイクルさせた。これに続いてさらに68℃で3分間処理し、そして4℃で保持した。
【0072】
(λ軽鎖クローニング)
λ軽鎖増幅生成物を、PCR purification kit(QIAGEN、Valencia、CA)によって浄化し、そしてXbaIおよびSacIによって消化した。そのインサートを、gel extraction kit(QIAGEN、Valencia、CA)を使用してゲル精製し、そしてλ軽鎖定常領域の残余部分を含む適切な発現ベクター中にクローニングした。このライゲーションされた生成物を、エレクトロポレーションによってE.coli中に導入し、そして37℃で一晩成長させた。翌朝、λ軽鎖ライブラリーDNAを回収するためにDNAマキシプレップ(QIAGEN、Valencia、CA)を実施した。
【0073】
(重鎖クローニングおよびIgGλライブラリーの生成)
上記で調製したλ軽鎖ライブラリーDNAを、前述されるように、IgGκライブラリーに対して調製した重鎖Fdフラグメントの挿入のために、XhoIおよびAgeIで順次消化した。次いで、ライゲーションされた生成物を、エレクトロポレーションによってE.coli中に導入し、そして37℃で一晩成長させた。翌朝、完全なIgGλライブラリーDNAを回収するために、DNAマキシプレップ(QIAGEN、Valencia、CA)を実施した。
【0074】
(HBs Agについてのライブラリーの選別およびスクリーニング)
IgGκライブラリーについて前述されたように、パンニングおよびスクリーニングを実施した。
【0075】
(DNA配列決定分析および単離されたFabの性質決定)
ELISAスクリーニングによって、HBsAgに対して特異的結合を示し、かつ非特異的タンパク質(オボアルブミン)に対して最低限の結合を示したクローンを、DNA配列決定によって分析した。図8a〜図8eを参照のこと。HBsAgに対して、全部で38の異なるIgGκFab(25重鎖および37軽鎖)および17の異なるIgGλFab(13重鎖および16軽鎖)を、B型肝炎ワクチン接種を受けたドナー由来のPBL mRNAから作製したライブラリーから単離した。
【0076】
(実施例4)
(ヒトPBL mRNAからのファージディスプレイライブラリーの構築)
(軽鎖についての第1鎖cDNA合成および修飾)
ドナーからのヒトPBL mRNAを、オリゴdTプライマーを用いて、基本的にキットの指示書に従って、SuperScript First−Strand Synthesis System for RT−PCR(Invitrogen Life Technologies、Carlsbad、CA)を使用し、慣習的な第1鎖cDNAを生成するために使用した。κ軽鎖反応およびλ軽鎖反応を、別々にセットアップする。κ軽鎖については制限ヌクレアーゼSacIによって、またはλ軽鎖についてはSmaIによって消化され得る二本鎖DNA領域を生成するために、第1鎖cDNAに制限オリゴヌクレオチド「CKSacI」または「CLSmaI」を添加する。複数のλ定常領域(C1、C2、C3、およびC6)がある場合、全ての機能的λ定常ドメイン(C1、C2、C3、およびC6)の中で、SmaI部位が保存されていることに注意することが重要である。「CKSacI」配列は、5’AGG GCC TGA GCT CGC CCG TC3’(配列番号179)であり、「CLSmaI」配列は5’GAC TTC TAC CCG GGA GCY GTG3’(配列番号180)であって、ここで、YはCおよびTの混合物である。この反応を、第1鎖cDNAおよび1μMオリゴヌクレオチドを用いてセットアップする。この試料を95℃まで2分間加熱し、次いで特異的アニーリングが起こり得るように64℃で2分間保持する。適切な量の10×制限緩衝液A(Roche Diagnostics)を上記試料に添加し、そして37℃までさらに冷却する。制限エンドヌクレアーゼSacIまたはSmaI(New England Biolabs、Beverly MA)を添加し、そして37℃で30分間インキュベートする。この制限酵素を、65℃で20分間加熱不活性化し、次いでこの試料を4℃まで冷却する。
【0077】
(軽鎖第2鎖線形増幅およびネステッドオリゴ伸長)
フレームワーク1特異的プライマー(最終0.4μM)、dNTP、AmpliTaq酵素およびその10×反応緩衝液(Applied Biosystems、Foster City、CA)を使用して複数の第2鎖cDNA反応をセットアップするために、SacI消化されたκ第1鎖cDNAまたはSmaI消化されたλ第1鎖cDNAを、原テンプレートとして使用する。上記プライマーを、TMX24K(κについて)配列またはTMX24L(λについて)配列、XbaI制限酵素認識部位、およびヒト抗体κ鎖遺伝子またはヒト抗体λ軽鎖遺伝子のフレームワーク1領域中の第1鎖cDNAにアニーリングする領域を含むように設計する。これらのアニーリング配列は、VBaseデータベースプライマー(www.mrc−cpe.cam.ac.uk/imt−doc/public/INTRO.html)由来であり、これらは公知のヒト抗体の配列に基づいて設計されており、そして軽鎖遺伝子の全ヒト抗体レパートリーを網羅すると報告されている。κ軽鎖フレームワーク1特異的プライマーは、実施例3におけるものを使用する。λ増幅における使用のためのプライマーのリストは、以下を参照のこと。
【0078】
【化12】
上記配列において、RはAおよびGの同じ割合の混合物であり、MはAおよびCの同じ割合の混合物であり、YはCおよびTの同じ割合の混合物であり、WはAおよびTの同じ割合の混合物であり、そしてSはCおよびGの同じ割合の混合物である。
【0079】
上記試料を、94℃で1分間加熱変性し、次いで94℃で5秒間、および56℃で10秒間、および68℃で2分間を介する反応を20サイクル行う。これは、第2鎖cDNAの線形増幅を可能にする。κ鎖について「TMX24CKnpt」と命名されたネステッドオリゴヌクレオチド、またはλ鎖について「TMX24CLnpt」と命名されたネステッドオリゴヌクレオチドを、氷上で0.2μMの最終濃度となるように添加する。このネステッドオリゴヌクレオチドの配列は;「TMX24CKnpt」5’GAC GAC CGG CTA CCA AGA GGA GTG CTC GAG CTC AGG CCC TGA TGG GTG ACT TCG CT3’(配列番号196)および「TMX24CLnpt」5’GAC GAC CGG CTA CCA AGA GGA CAG AAG AGC TCC TGG GTA GAA GTC ACT KAT SAG RCA CAG3’(配列番号197)である。図7に示されるように、IgMネステッドオリゴについて、オリゴ伸長を防げるために3つの3’末端ヌクレオチドを修飾する。上記第2鎖cDNAを、94℃で1分間の加熱変性し、68℃で2分間アニーリングおよび伸長させてネステッドオリゴからさらに伸長し、続いて4℃にする。結果生じた第2鎖cDNA(人工テンプレート)を、QIAGENスピンカラム(QIAGEN、Valencia、CA提供のPCR Purification Kit)を使用して精製する。この工程は、遊離のオリゴヌクレオチドを除去し、そして下流のプロトコールのための容易な緩衝液交換を可能にする。
【0080】
(軽鎖単一プライマー増幅(SPA))
Advantage 2ポリメラーゼミックス(Clontech)およびその10×反応緩衝液、dNTP、ならびにκ鎖についてプライマー「TMX24K」を、またはλ鎖について「TMX24L」を使用し、上記人工テンプレートを増幅する。「TMX24K」の配列は5’GAC GAC CGG CTA CCA AGA GGA GTG 3’(配列番号198)であり、そして「TMX24L」の配列は5’GAC GAC CGG CTA CCA AGA GGA CAG3’(配列番号199)である。この試料を95℃で1分間加熱変性し、次いで95℃で5秒間および68℃で1分間を介する反応を30サイクル行う。これに続いてさらに68℃で3分間処理し、そして4℃で保持する。
【0081】
(軽鎖クローニング)
κ増幅生成物およびλ増幅生成物を、PCR purification kit(QUIAGEN)を使用して浄化し、そして別々にゲル精製した。これらの生成物を、XbaIおよびSacIによって消化した。そのインサートを、それぞれの軽鎖定常領域の残余部分を含む適切な発現ベクター中にクローニングした。このライゲーションされた生成物を、エレクトロポレーションによってE.coli中に導入し、そして37℃で一晩成長させた。翌朝、軽鎖ライブラリーDNAを回収するために、DNAマキシプレップを実施した。ライブラリーの構築を完了させるために、この軽鎖ライブラリーDNA調製物を、XhoI/EcoRIによる重鎖Fdフラグメントの挿入のためのクローニングベクターとして使用した。
【0082】
(重鎖についての第1鎖cDNA合成および修飾)
オリゴdTプライマーを用いた慣習的な第1鎖cDNAを生成するために、ヒトPBL mRNAを使用する。これを、SuperScript First−Strand Synthesis System for RT−PCR(Invitrogen Life Technologies、Carlsbad、CA)を使用し、基本的にキットの指示書に従って行う。制限エンドヌクレアーゼDraIIIによって消化され得る二本鎖DNA領域を生成するために、制限オリゴヌクレオチドCMDraIIIを、第1鎖cDNAに添加する。この反応を、第1鎖cDNAおよび1μMオリゴヌクレオチドを用いてセットアップする。試料を2分間95℃まで加熱し、次いで特異的アニーリングが起こり得るように64℃で2分間保持する。適切な量の10×制限緩衝液H(Roche Diagnostics)を上記試料中に添加し、そしてさらに37℃まで冷却する。制限エンドヌクレアーゼDraIII(New England Biolabs、Beverly MA)を添加し、そして37℃で30分間インキュベートし、次いで4℃まで冷却する。
【0083】
DraIIIによる第1鎖cDNAの消化を、PCR増幅によって検証する。増幅生成物は、抗体フラグメントのクローニングには使用されない。2つの異なる緩衝液条件下において、5’VBVH1a内部コントロールプライマーおよび3’CM0内部コントロールプライマーを使用した反応において、消化された第1鎖cDNAの陽性増幅物を観察する。5’VBVH1a/3’CM0プライマーを用いた良好な増幅、および5’VBVH1a/3’CM1プライマーを用いたわずかな増幅は、第1鎖cDNAテンプレートの首尾良いDraIII消化を示す。
【0084】
(重鎖第2鎖線形増幅およびネステッドオリゴ伸長)
フレームワーク1特異的プライマー(最終0.4μM)、dNTP、AmpliTaq酵素およびその10×反応緩衝液(Applied Biosystems、Foster City、CA)を使用して複数の第2鎖cDNA反応をセットアップするために、DraIII消化された第1鎖cDNAを、原テンプレートとして使用する。上記プライマーを、TMX24配列、XbaI制限酵素認識部位、およびヒト抗体重鎖遺伝子のフレームワーク1領域における第1鎖cDNAにアニーリングする領域を含むように設計する。これらのアニーリング配列は、VBaseデータベースプライマー由来であって、これらは公知のヒト抗体の配列に基づいて設計されており、そして実施例3において上述されるように、重鎖遺伝子の全ヒト抗体レパートリーを網羅すると報告されている。重鎖フレームワーク1特異的プライマーは、実施例3において列挙されたものを使用する。
【0085】
上記試料を、94℃で1分間加熱変性し、次いで94℃で5秒間、56℃で10秒間、および68℃で2分間を介する反応を20サイクル行う。これは、第2鎖cDNAの線形増幅を可能にする。「TMX24CMnpt」と命名されたネステッドオリゴヌクレオチド(実施例3において使用したもの)を、氷上で0.2μMの最終濃度となるように添加する。上記第2鎖cDNAを、94℃で1分間の加熱変性し、68℃で2分間のアニーリングおよび伸長によって、ネステッドオリゴからさらに伸長し、続いて4℃にする。結果生じた第2鎖cDNA(人工テンプレート)を、QIAGENスピンカラム(QIAGEN、Valencia、CA提供のPCR Purification Kit)を使用して洗浄する。この工程は、遊離のオリゴヌクレオチドを除去し、そして下流のプロトコールのための容易な緩衝液交換を可能にする。
【0086】
(重鎖単一プライマー増幅(SPA))
Advantage 2ポリメラーゼミックス(Clontech)およびその10×反応緩衝液、dNTP、ならびにプライマー「TMX24」を使用し、上記人工テンプレートを増幅する。この試料を95℃で1分間加熱変性し、次いで95℃で5秒間および68℃で1分間を介する反応を30サイクル行う。これに続いてさらに68℃で3分間処理し、そして4℃で保持する。
【0087】
(重鎖クローニングおよびライブラリーの生成)
約500bpの増幅生成物をゲル精製し、次いでXhoIおよびEcoRIによって消化する。IgM CH1ドメインの残余部分を含む適切な発現ベクター中に、インサートをクローニングする。ライゲーションされたFabライブラリーを含む生成物を、エレクトロポレーションによってE.coli中に導入する。
【0088】
バクテリオファージの表面上にFabライブラリーを生成させるために、サプレッサー株の細胞(例えば、XL1BLUE(Stratagene))を使用する。続いてエレクトロポレーションに続いて、その細胞を37℃で1時間振とうし、次いでカルベニシリンを20μg/mlまで添加する。37℃で1時間の振とうの後、カルベニシリンを50μg/mlまで増やし、37℃にさらに1時間置く。次いで、ファージミド粒子の生成のために必要な全ての成分を提供するため、VCS−M13ヘルパーファージ(Stratagene)を添加し、そして培養物の容積をSB培地で100mlまで増やす。37℃で1時間後、ヘルパーファージDNAを含む細菌を選択するために、70μg/mlまでカナマイシンを添加する。この培養物を、37℃で一晩振とうする。その期間の間、上記細菌は、その表面上にFabを有する新しいファージミド粒子を生成する。翌朝、このファージミド粒子は細菌細胞をスピンアウトすることによって単離され得、次いでこのファージミド粒子を、4% PEG 8000および0.5M NaClを用いて、氷上で30分間上清から沈殿させる。14,300×gの遠沈によって、ファージペレットを沈殿させる。このペレットは、PBS/1% BSA中で再懸濁され得る。この調製物は、細菌性の破片を取り除くために、濾過され得る。結果生じたライブラリーを、4℃で保存する。
【0089】
(実施例5)
(IgEまたは組み換えIgE Fc CH2〜4で免疫されたマウスmRNA由来のファージミドディスプレイライブラリーの構築)
(IgG重鎖およびκ軽鎖についての第1鎖cDNA合成および修飾)
オリゴdTプライマーを用いた慣習的第1鎖cDNAを生成するために、ヒトIgEまたは組み換えヒトIgEで免疫されたマウス由来であるマウス脾臓mRNAを使用した。これを、SuperScript First−Strand Synthesis for RT−PCR(Invitrogen Life Technologies、Carlsbad、CA)を使用し、基本的にキットの指示書に従って行った。IgG1については制限エンドヌクレアーゼXcmI、IgG2aについては制限エンドヌクレアーゼBsaJI、またはκ軽鎖についてはHpaIによって消化され得る二本鎖DNA領域を生成するために、IgG1については制限オリゴヌクレオチド「mCG1XcmI」、IgG2aについては制限オリゴヌクレオチド「mCG2aBsaJI」、またはκ軽鎖については制限オリゴヌクレオチド「mCKHpaI」を、第1鎖cDNAに添加した。「mCG1XcmI」配列は、5’CTAACTCCATGGTGACCCTGGGATG3’(配列番号200)である。「mCG2aBsaJI」配列は、5’CAACTGGCTCCTCGGTGACTCTAG3’(配列番号201)であり、「mCKHpaI」配列は、5’CAGTGAGCAGTTAACATCTGGAGG3’(配列番号202)である。この反応を、第1鎖cDNA、1μMオリゴヌクレオチド、および適切な量の10×NE緩衝液(New England Biolabs、Beverly MA)または10×反応緩衝液A(Roche Diagnostics)を用いてセットアップした。この試料を95℃まで2分間加熱し、次いで特異的アニーリングが起こり得るように64℃で2分間保持し、そしてXcmIおよびHpaIについては37℃まで冷却し、またBsaJIについては60℃まで冷却した。上記制限エンドヌクレアーゼXcmI、制限エンドヌクレアーゼBsaJI、または制限エンドヌクレアーゼHpaI(New England Biolabs、Beverly MA)を添加し、それぞれ、37℃で30分間、60℃で30分間、および37℃で10分間インキュベートした。上記制限酵素を、XcmIについては65℃で20分間、およびBsaJIについては80℃で20分間加熱不活性化し、次いで試料を4℃まで冷却した。
【0090】
各々の制限エンドヌクレアーゼによる上記第1鎖cDNAの消化を、当業者において公知である技術を使用し、PCR増幅によって検証した。これらの生成物は、抗体遺伝子のクローニングには使用しなかった。IgG1について5’TMX24mVHIIB短縮型内部コントロールプライマーおよび3’mCG1内部コントロールプライマーを、IgG2aについて5’TMX24mVKIIB短縮型および3’mCG2の内部コントロールプライマーを、そしてκについて5’TMX24mVKIV短縮型および3’mCK0内部コントロールプライマーを使用した反応において、消化された第1鎖cDNAの陽性増幅物を観察した。5’TMX24mVHIIB短縮型/3’mCG1プライマーもしくは5’TMX24mVHIIB短縮型/3’mCG2aプライマーまたは5’TMX24mVHIV短縮型/3’mCK0プライマーを用いた良好な増幅、および5’TMX24mVHIIB短縮型/3’mCG1Bプライマー、もしくは5’TMX24mVHIIB短縮型/3’mCG2aBプライマーもしくはTMX24mVKIV短縮型/3’mCKBプライマーを用いたわずかな増幅は、各々の制限エンドヌクレアーゼを用いた第1鎖cDNAテンプレートの首尾良い消化を示す。PCRの検査のために使用されるプライマーの配列は、以下:
【0091】
【化13】
【0092】
【化14】
である。
【0093】
(軽鎖第2鎖線形増幅およびネステッドオリゴ伸長)
フレームワーク1特異的プライマー(最終0.4μM)、dNTP、AmpliTaq酵素およびその10×反応緩衝液(Applied Biosystems、Foster City、CA)を使用して複数の第2鎖cDNA反応をセットアップするために、HpaI消化された第1鎖cDNAを、原テンプレートとして使用した。上記プライマーを、TMX24mK配列(κについて)、およびXbaI制限酵素認識部位、ならびにマウス抗体κ軽鎖遺伝子のフレームワーク1領域における第1鎖cDNAにアニーリングする領域を含むように設計した。そしてκ軽鎖遺伝子の全マウス抗体レパートリーを網羅するために、Kabatデータベース(http://immuno.bme.nwu.edu/)由来の公知のマウス抗体の配列に基づいて、これらのアニーリング配列を設計した。
【0094】
【化15】
ここで、RはAおよびGの同じ割合の混合物であり、MはAおよびCの同じ割合の混合物であり、KはGおよびTの同じ割合の混合物であり、WはAおよびTの同じ割合の混合物であり、そしてSはCおよびGの同じ割合の混合物である。
【0095】
上記試料を、94℃で1分間加熱変性し、次いで94℃で5秒間、56℃で10秒間、および68℃で2分間を介する反応を20サイクル行った。これは、第2鎖cDNAの線形増幅を可能にする。κ鎖について「TMX24mCKnoer」と命名されたネステッドオリゴヌクレオチドを、氷上で0.2μMの最終濃度となるように添加した。「TM24CKnpt」の配列は;5’GACGACCGGCTACCAAGAGGAGTGTCCGGATGTTAACTGCTCACTGGATGGTGGGAAGATGG2’OMe[A(ps)U(ps)U(ps)](プロピル)3’(配列番号220)である。上記第2鎖cDNAを、94℃で1分間の加熱変性し、68℃で2分間アニーリングおよび伸長させてネステッドオリゴからさらに伸長し、続いて4℃にした。結果生じた第2鎖cDNA(人工テンプレート)を、QIAGENスピンカラム(QIAGEN、Valencia、CA提供のPCR Purification Kit)を使用して洗浄した。この工程は、遊離のオリゴヌクレオチドを除去し、そして下流のプロトコールのための容易な緩衝液交換を可能にする。
【0096】
(軽鎖単一プライマー増幅)
Advantage 2ポリメラーゼミックス(Clontech)およびその10×反応緩衝液、dNTP、ならびにκ鎖についてのプライマー「TMX24mK」を使用し、上記人工テンプレートを増幅した。「TMX24mK」についての配列は、5’GACGACCGGCTACCAAGAGGAGTG3’(配列番号221)である。この試料を95℃で1分間加熱変性し、次いで95℃で5秒間および68℃で1分間を介する反応を25サイクルさせた。これに続いてさらに68℃で3分間処理し、そして4℃で保持した。
【0097】
(軽鎖クローニング)
κ増幅生成物をゲル精製し、次いでXbaIおよびBspEIによって消化した。そのインサートを、κ軽鎖定常領域の残余部分を含む適切な発現ベクター中にクローニングした。このライゲーションされた生成物を、エレクトロポレーションによってE.coli中に導入し、そして37℃で一晩成長させた。翌朝、軽鎖ライブラリーDNAを回収するためにDNAマキシプレップを実施した。次いで、以下の「(重鎖クローニング)」において記載されるようにライブラリーの構築を完了させるため、XhoI/BlnIによる重鎖Fdフラグメントをクローニングするための後の工程において、この軽鎖ライブラリーDNAを使用した。
【0098】
(重鎖第2鎖線形増幅およびネステッドオリゴ伸長)
フレームワーク1特異的プライマー(最終0.4μM)、dNTP、AmpliTaq酵素およびその10×反応緩衝液(Applied Biosystems、Foster City、CA)を使用して複数の第2鎖cDNA反応をセットアップするために、XcmIおよびBsaJIで消化された第1鎖cDNAを使用した。上記プライマーを、TMX24mH配列、XhoI制限酵素認識部位、およびマウス抗体重鎖遺伝子のフレームワーク1領域における第1鎖cDNAにアニーリングする領域を含むように設計した。そして重鎖遺伝子の全マウス抗体レパートリーを網羅するために、Kabatデータベース(http://immuno.bme.nwu.edu/)由来の公知のマウス抗体の配列に基づいて、これらのアニーリング配列を設計した。
【0099】
【化16】
ここで、RはAおよびGの同じ割合の混合物であり、MはAおよびCの同じ割合の混合物であり、YはCおよびTの同じ割合の混合物であり、そしてSはCおよびGの同じ割合の混合物である。
【0100】
上記試料を94℃で1分間加熱変性し、次いで94℃で5秒間、56℃で10秒間、および68℃で2分間を介する反応を20サイクル行った。これは、第2鎖cDNAの線形増幅を可能にする。IgG1について「TMX24mCG1noer」と命名されたネステッドオリゴおよびIgG2aについて「TMX24mCG2anoer」と命名されたネステッドオリゴを、氷上で0.2μMの最終濃度となるように添加した。「TMX24mCG1noer」の配列は;5’GACGTGGCCGTTGGAAGAGGAGTGCCTAGGGTTACCATGGAGTTAGTTTGGGCAGCAGA2’OMe[U(ps)C(ps)A(ps)](プロピル)3’(配列番号230)であり、そして「TMX24mCG2anoer」の配列は;5’GACGTGGCCGTTGGAAGAGGAGTGCCTAGGGTCATCGAGGAGCCAGTTGTATCTCCACA2’OMe[C(ps)A(ps)U(ps)](プロピル)3’(配列番号231)である。
【0101】
上記第2鎖cDNAを、94℃で1分間の加熱変性、68℃で2分間のアニーリングおよび伸長によって、ネステッドオリゴからさらに伸長し、続いて4℃にした。結果生じた第2鎖cDNA(人工テンプレート)を、QIAGENスピンカラム(QIAGEN、Valencia、CA提供のPCR Purification Kit)を使用して洗浄した。この工程は、遊離のオリゴヌクレオチドを除去し、そして下流のプロトコールのための容易な緩衝液交換を可能にする。
【0102】
(重鎖単一プライマー増幅(SPA))
Advantage 2ポリメラーゼミックス(Clontech)およびその10×反応緩衝液、dNTP、ならびに重鎖についてのプライマー「TMX24mH」を使用し、上記人工テンプレートを増幅した。「TMX24mH」についての配列は、5’GACGTGGCCGTTGGAAGAGGAGTG3’(配列番号232)である。この試料を95℃で1分間加熱変性し、次いで95℃で5秒間および68℃で1分間を介する反応を、IgG1については28サイクル、IgG2aについては30サイクルさせた。これに続いて68℃でさらに3分間処理し、そして4℃で保持した。
【0103】
(重鎖クローニング)
重鎖増幅生成物をゲル精製し、次いでXhoIおよびBlnIによって消化した。そのインサートを、IgG1およびIgG2aについての重鎖定常領域の残余部分を含むκ鎖ライブラリーDNA中にクローニングした。このライゲーションされた生成物を、エレクトロポレーションによってE.coli中に導入し、そして37℃で一晩成長させた。翌朝、IgG1κライブラリーDNAまたはIgG2aκライブラリーDNAを回収するために、DNAマキシプレップを実施した。
【0104】
(組み換えIgE Fc CH2〜4についてのライブラリーのパンニングおよびスクリーニング)
基本的に、Barbas III,CF、Burton,DR、Scott,JK、およびSilverman,GJ(2001)Phage Display:A Laboratory Manual、Cold Spring Harbor Laboratory Press、Cold Spring Harbor、New Yorkに記載されるように、上記ライブラリーを、組み換えIgE Fc CH2〜4について4ラウンドパンニングした。選別の第2ラウンド、第3ラウンド、および第4ラウンド由来の個々のクローンを、組み換えIgE Fc CH2〜4についてのELISAによってスクリーニングした。
【0105】
ELISAによって、IgE Fc CH2〜4に対して特異的結合を示し、かつ非特異的タンパク質(オボアルブミン)に対して最低限の結合を示したクローンを、DNA配列決定によって分析した。IgE Fc CH2〜4に対して、全部で31の異なるFabを、マウスライブラリーから単離した。図9a〜図9dを参照のこと。
【0106】
(実施例6)
(IgA抗体ライブラリーの構築)
(第1鎖cDNA合成および修飾)
ヒト末梢血リンパ球(PBL)mRNAを、オリゴdTプライマーを用いた慣習的な第1鎖cDNAを生成するために使用する。これを、SuperScript II RTcDNA Synthesis Kit(Invitrogen Life Technologies、Carlsbad、CA)を使用し、基本的にその指示書に従って行う。制限エンドヌクレアーゼBsrGIによって消化され得る二本鎖DNA領域を生成するために、第1鎖cDNAに制限オリゴヌクレオチド「CABsrGI」を添加する。「CABsrGI」の配列は、5’TCC GGG GAC CTG TAC ACC ACG AGC AG3’(配列番号279)である。この反応を、第1鎖cDNAおよび0.1μMオリゴヌクレオチドを用いてセットアップする。試料を95℃まで2分間加熱し、次いで特異的アニーリングが起こり得るように64℃で2分間保持する。適切な量の10×制限緩衝液2(New England Biolabs、Beverly MA)を上記試料中に添加し、そしてさらに37℃まで冷却した。制限エンドヌクレアーゼBsrGI(New England Biolabs、Beverly MA)を添加し、そして37℃で30分間インキュベートする。この制限酵素を80℃で20分間加熱不活性化し、次いで試料を4℃まで冷却する。
【0107】
(第2鎖cDNA合成およびネステッドオリゴヌクレオチド伸長反応(NOER))
フレームワーク1特異的プライマー(最終0.4μM)、dNTP、AmpliTaq酵素およびその10×反応緩衝液(Applied Biosystems、Foster City、CA)を使用して複数の第2鎖cDNA反応をセットアップするために、BsrGI消化された第1鎖cDNAを使用する。以下の表A中に列挙されるプライマーは、TMX24配列、XhoI制限酵素認識部位、およびヒト抗体重鎖遺伝子のフレームワーク1領域における第1鎖cDNAにアニーリングする領域を含むように設計された。これらのアニーリング配列は、公知のヒト抗体の配列に基づいて設計され、そして重鎖鎖遺伝子の全長ヒト抗体レパートリーを網羅すると報告されているVbaseデータベースプライマー由来である。
【0108】
【化17】
【0109】
【化18】
上記配列の各々において、RはAまたはGであり、KはGまたはTであり、そしてSはCまたはGである。
【0110】
上記試料を、94℃で1分間加熱変性し、次いで94℃で5秒間、56℃で10秒間、および68℃で2分間を介する反応を20サイクル行う。これは、第2鎖cDNAの線形増幅を可能にする。次いで、伸長オリゴヌクレオチド「TMX24CAnpt」を、氷上で0.2μMの最終濃度となるように添加する。「TMX24CAnpt」の配列は、5’GTG CTG GCC GTT GGA AGA GGA GTG CCT GTA CAG GTC CCC GGA GGC ATC CTC3’(配列番号294)であって、ここで、RはAまたはGである。オリゴ伸長を防ぐために、3つの3’末端ヌクレオチドを修飾する。94℃で1分間の加熱変性し、68℃で2分間での伸長において上記第2鎖cDNAをネステッドオリゴからさらに伸長し、続いて4℃にする。次いで、この第2鎖cDNAを、QIAGENスピンカラム(QIAGEN、Valencia、CA提供のPCR Purification Kit)を使用して洗浄する。この工程は、オリゴヌクレオチドを除去し、そして下流のプロトコールのための容易な緩衝液交換を可能にする。
【0111】
(単一プライマーPCR増幅)
Advantage 2ポリメラーゼミックス(Clontech、Palo Alto、CA)およびその10×反応緩衝液、dNTP、ならびにプライマー「TMX24」を使用し、上記第2鎖cDNAの増幅を実施する。「TMX24」の配列は、5’GTG CTG GCC GTT GGA AGA GGA GTG3’(配列番号295)である。この試料を95℃で1分間加熱変性し、次いで95℃で5秒間および68℃で1分間を介する反応を30サイクルさせる。これに続いて68℃でさらに3分間処理し、そして4℃で保持する。
【0112】
(クローニングおよび配列決定)
約560bpのPCR生成物を、PCR purification kit (QIAGEN、Valencia、CA)を使用して精製し、XhoIおよびBsrGIによって消化し、ゲル精製し、そしてIgA CH1定常領域の残余部を有する適切なFab発現ベクター中にクローニングする。
【0113】
種々の改変が、本明細書中に記載される実施形態を構成し得ると考えられる。したがって、上記は、好ましい実施形態の限定として解釈されるべきではなく、単に、好ましい実施形態の例示として解釈されるべきである。当業者は、本開示の範囲および精神の範囲内で他の改変を構想する。
【技術分野】
【0001】
(関連出願)
本国際出願は、2003年12月15日に出願された米国出願第10/737,252号(これは、2001年9月19日に出願された米国仮出願第60/323,455号に対する優先権を主張する、2002年9月19日に出願された米国出願第10/251,085号の一部継続出願である)に対する優先権を主張する。上述の米国出願および米国仮出願の開示全体は、本明細書中に参考として援用される。
【0002】
(技術分野)
本開示は、標的核酸配列の増幅のために有用な人工テンプレート(engineered template)に関連する。さらに具体的には、その反対端に相補的な配列を含むように操作されたテンプレートが、ネステッド(nested)オリゴヌクレオチド伸長反応(NOER)によって提供される。上記人工テンプレートは、この人工テンプレート内の標的配列を増幅するための単一プライマー増幅(SPA)を可能にする。特に有用な実施形態において、上記人工テンプレート由来の標的配列は、発現ビヒクル中にクローニングされて、例えば、抗体ライブラリーのような、ポリペプチドまたはタンパク質のライブラリーを提供する。
【背景技術】
【0003】
(関連分野の背景)
核酸増幅および増幅生成物の検出のための方法は、核酸配列の検出、同定、定量および配列分析を支援する。核酸増幅は、例えば、抗体のような、関連する遺伝子ライブラリーの構築において重要な工程である。これらのライブラリーは、特異的であり所望する活性を有する抗体に関して、スクリーニングされ得る。核酸分析は、病原体の検出および同定、規定の表現型を引き起こす遺伝子変化の検出、遺伝病(genetic disease)または疾患に対する感受性の診断、発達、疾患および規定の刺激の応答における遺伝子発現の評価、ならびに種々のゲノム計画のために重要である。核酸増幅方法の他の適用としては、稀な細胞の検出、病原体の検出、および悪性疾患における改変された遺伝子発現の検出などが挙げられる。核酸増幅はまた、(例えば、規定の核酸配列の存在の検出のような)定性分析、および(例えば、病原性配列の量の評価、ならびに遺伝子増殖または遺伝子欠失の判定、および正常細胞型から悪性細胞型への細胞形質転換の判定などにおいて有用な)規定の遺伝子配列の定量化のために有用である。核酸配列中の配列変化の検出は、変異遺伝子型の検出、遺伝子分析に関連することとして、薬物耐性を引き起こす変異の検出、薬理ゲノム科学などのために重要である。
【0004】
多くの種類の核酸増幅(例えば、指数関数的増幅、結合線形増幅(linked linear amplification)、ライゲーションベースの増幅、および転写ベースの増幅)が存在する。指数関数的核酸増幅方法の一例は、多数の刊行物において開示される、ポリメラーゼ連鎖反応(PCR)である。例えば、非特許文献1;Mullis K.特許文献1;Mullisら、特許文献2;Erlichら、特許文献3、特許文献4、特許文献5、特許文献6;およびSaiki R.ら、特許文献7を参照のこと。実際に、このポリメラーゼ連鎖反応(PCR)は、最も一般に使用される標的増幅方法である。PCRは、多数の標的配列の複数の二重鎖複製物を生成するために、多サイクルの、変性、2種の異なるオリゴヌクレオチドプライマーの(各々、標的鎖の対向する鎖への)ハイブリダイゼーション、およびヌクレオチドポリメラーゼによるプライマー伸長に基づく。
【0005】
単一のプライマーを利用した複製方法もまた、開示されている。例えば、特許文献8;特許文献9;特許文献10;特許文献11;および特許文献12を参照のこと。このプライマーは、特許文献13において開示されるようなDNA/RNAキメラプライマーであり得る。
【0006】
いくつかの増幅方法は、テンプレート転換オリゴヌクレオチド(template switching oligonucleotide)(TSO)およびブロッキングオリゴヌクレオチド(blocking oligonucleotide)を使用する。例えば、キメラDNAプライマーが利用されたテンプレート転換増幅は、特許文献12;特許文献14;特許文献15において、かつ非特許文献2によって開示される。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】欧州特許第201,184号明細書
【特許文献2】米国特許第4,582,788号明細書
【特許文献3】欧州特許第50,424号明細書
【特許文献4】欧州特許第84,796号明細書
【特許文献5】欧州特許第258,017号明細書
【特許文献6】欧州特許第237,362号明細書
【特許文献7】米国特許第4,683,194号明細書
【特許文献8】米国特許第5,508,178号明細書
【特許文献9】米国特許第5,595,891号明細書
【特許文献10】米国特許第5,683,879号明細書
【特許文献11】米国特許第5,130,238号明細書
【特許文献12】米国特許第5,679,512号明細書
【特許文献13】米国特許第5,744,308号明細書
【特許文献14】米国特許第5,962,272号明細書
【特許文献15】米国特許第6,251,639号明細書
【非特許文献】
【0008】
【非特許文献1】Mullisら、Cold Spring Harbor Symp.Quant.Biol.1986年、第51巻:263−273頁
【非特許文献2】Patelら、Proc.Natl.Acad.Sci.U.S.A.1996年、第93巻:2969−2974頁
【発明の概要】
【発明が解決しようとする課題】
【0009】
しかしながら、既に記載した標的増幅方法は、いくつかの欠点を有する。例えば、転写ベースの増幅方法(例えば、核酸配列ベースの増幅(NASBA)および転写媒介性増幅(TMA))は、プライマーによる増幅生成物の中へのポリメラーゼプロモーター配列の組み込みに対する必要性によって制限され、結果として、この方法は非特異的増幅となる傾向がある。現在の増幅方法の欠点の別の例は、異なる温度で最適な結合を有し得る2回の結合現象の必要性、ならびに天然に存在する配列を含むプライマーの使用である。この要因の組み合わせの結果、ミスプライミング(mis−priming)の可能性が生じ、そして結果として標的配列以外の配列の増幅が生じる。
【0010】
したがって、これらの欠点を克服する核酸増幅方法に対する必要性が存在する。本明細書中で提供される本発明は、この必要性を満たし、そしてさらなる利益を提供する。
【課題を解決するための手段】
【0011】
(要旨)
核酸増幅の新しい方法が、ここ発明され、この方法は、以下の工程:
a)テンプレート核酸配列にプライマーをアニーリングする工程であって、このプライマーは、テンプレートにアニーリングする第1の部分および所定の配列の第2の部分を有する、工程;
b)上記プライマーの第1の部分が上記テンプレートにアニーリングする位置と上記テンプレートの末端との間のテンプレートの部分にアニーリングし、かつ相補的であるポリヌクレオチドを合成する工程であって、このポリヌクレオチドは第1の末端および第2の末端を有し、ここで、その第1の末端は上記プライマーを組み込む、工程;
c)上記テンプレートから、工程(b)において合成されたポリヌクレオチドを分離する工程;
d)工程(b)において合成されたポリヌクレオチドの第2の末端に、ネステッドオリゴヌクレオチドをアニーリングする工程であって、このネステッドオリゴヌクレオチドは、上記ポリヌクレオチドの第2の末端にアニーリングする第1の部分およびプライマーの第2の部分と同一の所定の配列を有する第2の部分を有する工程;
e)工程(b)において合成されたポリヌクレオチドを伸長して、上記所定の配列に相補的なこのポリヌクレオチドの末端部分を提供する、工程;ならびに
f)所定の配列を有する単一のプライマーを使用し、伸長されたポリヌクレオチドを増幅する工程
を包含する。
【0012】
代替的な実施形態において、上記方法は、以下の工程:
a)テンプレート核酸配列に、プライマーおよび境界(boundary)オリゴヌクレオチドをアニーリングする工程であって、このプライマーは、上記テンプレートにアニーリングする第1の部分および所定の配列の第2の部分を有する、工程;
b)上記プライマーの第1の部分がテンプレートにアニーリングする位置と上記境界オリゴヌクレオチドがアニーリングするテンプレートの部分との間のテンプレートの部分にアニーリングし、そしてそれに相補的であるポリヌクレオチドを合成する工程であって、このポリヌクレオチドは第1の末端および第2の末端を有し、ここで、この第1の末端はプライマーを組み込む、工程;
c)上記テンプレートから、工程(b)において合成されたポリヌクレオチドを分離する工程;
d)工程(b)において合成されたポリヌクレオチドの第2の末端に、ネステッドオリゴヌクレオチドをアニーリングする工程であって、このネステッドオリゴヌクレオチドは、上記ポリヌクレオチドの第2の末端にアニーリングする第1の部分および上記プライマーの第2の部分と同一の所定の配列を有する第2の部分を有する、工程;
e)工程(b)において合成されたポリヌクレオチドを伸長して、上記所定の配列に相補的であるポリヌクレオチドの末端部分を提供する工程;ならびに
f)上記所定の配列を有する単一のプライマーを使用し、伸長されたポリヌクレオチドを増幅する工程
を包含する。
【0013】
また、その第1の末端に所定の配列を有し、そしてその他方の末端に、この所定の配列と相補的な配列を有する人工核酸鎖が、それ自身本開示の新しい局面であることが企図される。
【0014】
別の局面において、本開示は、核酸鎖を増幅する新しい方法を提供し、この方法は、その第1の末端に所定の配列を有し、そしてその他方の末端に、この所定の配列に相補的な配列を有する人工核酸鎖を提供する工程;およびポリメラーゼおよびヌクレオチドの存在下において、このヌクレオチドの重合のために適切な条件下で、上記人工核酸鎖と所定の配列を有するプライマーとを接触させる工程を包含する。
【0015】
本明細書中に記載される増幅プロセスおよび人工テンプレートは、適切な発現ベクター中にライゲーションされ得る増幅生成物を調製するために使用され得る。次いで、このベクターは、標的配列によってコードされるポリペプチドまたはタンパク質を生成するために、標準的方法を使用して適切な宿主生物を形質転換するために使用され得る。特に有用な実施形態において、本明細書中に記載される技術は、例えば、抗体ライブラリーのような複雑なライブラリーを構築するための、関連する配列のファミリーを増幅するために使用される。
例えば、本発明は、以下の項目を提供する:
(項目1)
抗体の少なくとも一部分をコードする核酸を増幅する方法であって、以下の工程:
a)抗体の少なくとも一部分をコードするテンプレートにプライマーをアニーリングする工程であって、該プライマーは、該テンプレートにアニーリングする第1の部分および該テンプレートにアニーリングしない所定の配列の第2の部分を有する、工程;
b)該プライマーの該第1の部分が該テンプレートにアニーリングする位置と該テンプレートの末端との間の該テンプレートの部分に相補的であるポリヌクレオチドを合成する工程であって、該ポリヌクレオチドは、その第1の末端において該プライマーを有し、かつ第2の末端を有する、工程;
c)該テンプレートから、工程(b)において合成された該ポリヌクレオチドを分離する工程;
d)工程(b)において合成された該ポリヌクレオチドの該第2の末端に、テンプレートオリゴヌクレオチドをアニーリングする工程であって、該テンプレートオリゴヌクレオチドは、該ポリヌクレオチドの該第2の末端にアニーリングする第1の部分および該プライマーの該第2の部分と同一の所定の配列を有する第2の部分を有する、工程;
e)工程(b)において合成された該ポリヌクレオチドを伸長して、該所定の配列に相補的な該ポリヌクレオチドの末端部分を提供する工程;および
f)該所定の配列を有する単一のプライマーを使用し、該伸長されたポリヌクレオチドを増幅する、工程
を包含する、方法。
(項目2)
項目1に記載の方法であって、ここで、抗体の少なくとも一部分をコードするテンプテートにプライマーをアニーリングする上記工程が、以下:
【化1】
からなる群より選択される配列を含む少なくとも1つのプライマーをアニーリングする工程を包含し、ここで、RはAまたはGであり、KはGまたはTであり、そしてSはCまたはGである、方法。
(項目3)
項目1に記載の方法であって、ここで、抗体の少なくとも一部分をコードするテンプテートにプライマーをアニーリングする上記工程が、IgA抗体の少なくとも一部分をコードするテンプテートにプライマーをアニーリングする工程を包含する、方法。
(項目4)
抗体の少なくとも一部分をコードする核酸を増幅する方法であって、以下の工程:
a)抗体の少なくとも一部分をコードするテンプレートに、プライマーおよび境界オリゴヌクレオチドをアニーリングする工程であって、該プライマーは、該テンプレートにアニーリングする第1の部分および該テンプレートにアニーリングしない所定の配列の第2の部分を有する、工程;
b)該プライマーの該第1の部分が該テンプレートにアニーリングする位置と該境界オリゴヌクレオチドがアニーリングする該テンプレートの部分との間の該テンプレートの部分に相補的であるポリヌクレオチドを合成する工程であって、該ポリヌクレオチドは、その第1の末端において該プライマーを有し、かつ第2の末端を有する、工程;
c)該テンプレートから、工程(b)において合成された該ポリヌクレオチドを分離する工程;
d)工程(b)において合成された該ポリヌクレオチドの該第2の末端に、テンプレートオリゴヌクレオチドをアニーリングする工程であって、該テンプレートオリゴヌクレオチドは、該ポリヌクレオチドの該第2の末端にアニーリングする第1の部分および該プライマーの該第2の部分と同一の所定の配列を有する第2の部分を有する工程;
e)工程(b)において合成された該ポリヌクレオチドを伸長して、該所定の配列に相補的である該ポリヌクレオチドの末端部分を提供する、工程;
f)該所定の配列を有する単一のプライマーを使用し、該伸長されたポリヌクレオチドを増幅する、工程
を包含する、方法。
(項目5)
項目4に記載の方法であって、ここで、抗体の少なくとも一部分をコードするテンプテートにプライマーをアニーリングする上記工程が、以下:
【化2】
【化3】
からなる群より選択される配列を含む少なくとも1つのプライマーをアニーリングする工程を包含し、ここで、RはAまたはGであり、KはGまたはTであり、そしてSはCまたはGである、方法。
(項目6)
項目4に記載の方法であって、ここで、抗体の少なくとも一部分をコードするテンプテートにプライマーをアニーリングする上記工程が、IgA抗体の少なくとも一部分をコードするテンプテートにプライマーをアニーリングする工程を包含する、方法。
(項目7)
抗体ライブラリーを作製する方法であって、以下の工程:
a)IgA抗体の少なくとも一部分をコードする、多種多様なテンプレートの集団を提供する工程;
b)該多種多様なテンプレートの集団と少なくとも1つのプライマーとを接触させる工程であって、該少なくとも1つのプライマーは、該テンプレートにアニーリングする第1の部分および該テンプレートにアニーリングしない所定の配列の第2の部分を有する、工程;
c)該プライマーの該第1の部分が該テンプレートにアニーリングする位置と該テンプレートの末端との間の該テンプレートの部分に相補的である、ポリヌクレオチドを合成する工程であって、該ポリヌクレオチドは、その第1の末端において該プライマーを有し、かつ第2の末端を有する、工程;
d)該テンプレートから、工程(c)において合成された該ポリヌクレオチドを分離する工程;
e)工程(c)において合成された該ポリヌクレオチドの該第2の末端に、少なくとも1つのテンプレートオリゴヌクレオチドをアニーリングする工程であって、該少なくとも1つのテンプレートオリゴヌクレオチドは、該ポリヌクレオチドの該第2の末端にアニーリングする第1の部分および該プライマー該第2の部分と同一の所定の配列を有する第2の部分を有する、工程;
f)工程(c)において合成された該ポリヌクレオチドを伸長して、該所定の配列に相補的である該ポリヌクレオチドの末端部分を提供する工程;および
g)該所定の配列を有する単一のプライマーを使用し、該伸長されたポリヌクレオチドを増幅する工程
を包含する、方法。
(項目8)
項目7に記載の方法であって、ここで、抗体の少なくとも一部分をコードするテンプテートにプライマーをアニーリングする上記工程が、以下:
【化4】
【化5】
からなる群より選択される配列を含む少なくとも1つのプライマーをアニーリングする工程を包含し、ここで、RはAまたはGであり、KはGまたはTであり、そしてSはCまたはGである、方法。
(項目9)
項目7に記載の方法に従って調製された、IgA抗体のライブラリー。
(項目10)
所望の結合特異性を有する抗体を同定する方法であって、以下の工程:
項目7に記載の方法に従ってIgA抗体のライブラリーを調製する工程;および
該ライブラリーをスクリーニングして、所望の結合特異性を有する1つ以上のIgA抗体を同定する工程
を包含する、方法。
(項目11)
項目10に記載の方法に従って同定した、IgA抗体。
【図面の簡単な説明】
【0016】
【図1】テンプレートにアニーリングした、プライマーおよび境界オリゴの概略図。
【図2A】核酸鎖にアニーリングした、制限オリゴの概略図。
【図2B】短縮された5’末端を有するテンプレートにアニーリングした、プライマーの概略図。
【図3】代替の実施形態の概略図であって、ここで、複数ラウンドの重合が実施され、そして、制限オリゴヌクレオチドは、原テンプレートではなく新規に合成された鎖にアニールする。
【図4】新規に合成された核酸鎖にアニーリングした、ネステッドオリゴの概略図。
【図5】本開示に基づいた、人工テンプレートの概略図。
【図6】人工テンプレートの単一プライマー増幅の概略図。
【図7】TMX24CMnptと名づけられたネステッドオリゴの配列を示す。
【図8a】実施例3において生成された、単離されたFabの配列を示す。
【図8b】実施例3において生成された、単離されたFabの配列を示す。
【図8c】実施例3において生成された、単離されたFabの配列を示す。
【図8d】実施例3において生成された、単離されたFabの配列を示す。
【図8e】実施例3において生成された、単離されたFabの配列を示す。
【図9a】実施例5において生成された、単離されたFabの配列を示す。
【図9b】実施例5において生成された、単離されたFabの配列を示す。
【図9c】実施例5において生成された、単離されたFabの配列を示す。
【図9d】実施例5において生成された、単離されたFabの配列を示す。
【発明を実施するための形態】
【0017】
(好ましい実施形態の詳細な説明)
本開示は、標的核酸配列を増幅する方法を提供する。特に有用な実施形態において、上記標的核酸配列は、ポリペプチドまたはタンパク質をコードする遺伝子である。本開示はまた、どのように増幅生成物をクローニングし、かつ適切な発現系において発現させ得るかを記載する。特に有用な実施形態において、本明細書中に記載される技術は、例えば、抗体ライブラリーのような複雑なライブラリーを構築するための、関連する配列のファミリーを増幅するために使用される。
【0018】
上記標的核酸配列は、単一のプライマーのみを含むプロセスを介して指数関数的に増幅される。単一のプライマーを利用する(すなわち、各々異なった配列を有する順方向プライマーおよび逆方向プライマーの両方を使用する必要性が無い)能力は、増幅されるべき標的配列を含む核酸の鎖を操作することによって達成される。操作された核酸の鎖(本明細書中において、時折、「人工テンプレート」という)は、2つのテンプレート、すなわち、以下;
1)増幅されるべき配列を含む、天然核酸または合成核酸(例えば、DNAまたはcDNA)である出発物質
2)ネステッドオリゴヌクレオチド
から調製される。上記出発物質は、原テンプレート(original template)と見なされ得る。上記ネステッドオリゴヌクレオチドは、操作された核酸の鎖の作製の間に、原テンプレートのヌクレオチド配列を伸長するためのテンプレートとして使用される。この操作された核酸の鎖は、一連の操作によって原テンプレートから作製され、その結果その反対端に相補的な配列が存在する。これは、単一のプライマーのみを使用する増幅を可能にする相補的な配列である。
【0019】
精製形態または未精製形態のあらゆる核酸は、増幅されるべき標的核酸配列を含むか、または含むと思われる条件で、本明細書中に記載されるプロセスのための出発物質として利用され得る。したがって、このプロセスにおいて利用される出発物質は、DNAまたはRNA(伝令RNAを含む)であり得、これらのDNAまたはRNAは、一本鎖または二本鎖であり得る。さらに、各々の1つの鎖を含むDNA−RNAハイブリッドが利用され得る。任意のこれらの核酸の混合物がまた利用され得るか、または同じかもしくは異なったプライマーを使用した本明細書における、これまでの増幅反応から生成された核酸が使用され得る。増幅されるべき標的核酸配列は、より大きい分子の一部分(fraction)であり得るか、または個々の分子(discrete molecule)として最初に存在し得る。この出発核酸は、同一であり得るかまたは異なり得る1つより多くの所望の標的核酸配列を含み得る。したがって、本プロセスは、大量の1種類の標的核酸配列の生成のためのみではなく、同一であるかまたは異なった核酸分子上に位置する1つより多くの異なる標的核酸配列を同時に増幅するためにも有用であり得る。
【0020】
核酸は、任意の供給源(例えば:ゲノムライブラリーもしくはcDNAライブラリー、プラスミド、クローニングされたDNAもしくはRNA、または任意の供給源(細菌、酵母、ウイルスおよび高等生物(例えば、植物もしくは動物)が挙げられる)に由来する天然のDNAもしくはRNA)から得られ得る。核酸は、天然に存在し得るか、または全体的もしくは部分的に合成され得る。本発明において使用される核酸を獲得または生成するための技術は、当業者において周知である。核酸が2本の鎖を含む場合、それが原テンプレートとして使用され得る前に、別個の工程かまたはプライマー伸長生成物の合成と同時にかのいずれかとして、その核酸の鎖を分離する必要性がある。さらに、出発物質が第1鎖(first strand)DNAである場合、第2鎖(second strand)DNAは、当業者の範囲内のプロセスによって有利に作製され得、そして原テンプレートとして使用され得、そこから人工テンプレートが作製される。
【0021】
第1鎖cDNAは、本方法のための特に有用な原テンプレートである。DNAテンプレートを生成するための適切な方法は当業者に公知であり、そして当業者によって容易に選択される。好ましい実施形態において、第1鎖cDNAは、オリゴデオキシヌクレオチドプライマーおよび4つのデオキシヌクレオシド三リン酸(dATP、dGTP、dCTP、およびdTTP)の存在下において、任意のRNA出発物質に相補的であるDNAの合成を逆転写酵素が触媒する反応において合成される。この反応は、mRNAの3’末端に対するオリゴ−デオキシヌクレオチドプライマーのアニーリングによって開始され、続いて、上記mRNAヌクレオチド配列と関連する塩基対形成により決定されるように、伸長する鎖の3’末端に対して適切なデオキシヌクレオシドが段階的に添加される。当業者において理解されるように、試料中の全てのmRNAは、mRNAのポリA尾部への、オリゴdTのアニーリングを介した第1鎖cDNAを生成のために使用され得る。
【0022】
一旦原テンプレートが得られると、プライマー20および境界オリゴヌクレオチド30は、原テンプレート10にアニーリングされる(図1を参照のこと)。上記プライマーの3’末端で始まり上記境界オリゴヌクレオチドのおよそ5’末端までの、上記原テンプレートの部分に相補的な核酸の鎖が重合化される。
【0023】
上記原テンプレートにアニーリングした上記プライマー20は、好ましくは上記原テンプレートにアニーリングしない所定の配列の第1の部分22および上記原テンプレートにアニーリングする第2の部分25を含み、そして必要に応じてその第1の部分と第2の部分との間に制限酵素認識部位23を含む。上記プライマーは、増幅されるべき標的配列12に隣接した原テンプレートにアニーリングする。図1に示されるように、上記プライマーは、増幅されるべき標的配列の上流の原テンプレートにアニーリングし得るか、または上記プライマーは、増幅されるべき標的配列12の開始部(beginning)に重なり得ることが企図される。このプライマーの非アニーリング部分22の所定の配列は、もともと上記原テンプレート中に存在するものではなく、そして以下に詳細に記載されるように、増幅処理の間に使用される単一のプライマーがハイブリダイズし得る配列を提供するように選択される。必要に応じて、上記所定の配列は、本明細書中の以下においてより十分に記載されるように、発現ベクター中への人工テンプレート部分の挿入のために有用な制限酵素認識部位を含み得る。
【0024】
原テンプレートにアニーリングした境界オリゴヌクレオチド30は、その核酸の重合を終結させる。核酸重合を終結させることが可能なあらゆるオリゴヌクレオチドが、境界オリゴヌクレオチド30として利用され得る。好ましい実施形態において、上記境界オリゴヌクレオチドは、原テンプレート10にアニーリングする第1の部分35および伸長反応に影響されない第2の部分32を含む。この境界オリゴを、伸長に関する部位としての作用から妨げるための技術は、当業者の範囲内である。一例として、上記境界オリゴ30の部分32は、図1に示されるように、原テンプレート10にアニーリングしないように設計され得る。このような実施形態において、境界オリゴヌクレオチド30はさらなる重合を妨げるが、その3’末端が原テンプレート10にハイブリダイズしないので、核酸合成のためのプライマーとしては機能しない。代替的に、境界オリゴ30の3’末端は、同じ効果を達成するために固定核酸(locked nucleic acid)を含むように設計され得る。固定核酸は、例えば、WO 99/14226において開示され、その内容は本明細書中に参考として援用される。当業者は、境界オリゴの3’末端の伸長が起こらないことを保証する他の方法を構想する。
【0025】
本明細書中に記載されるプライマーおよびオリゴヌクレオチドは、当該分野において周知である、オリゴヌクレオチド合成のための確立された方法を使用して合成され得る。本発明のプライマーを含むオリゴヌクレオチドは、モノマー−モノマー相互作用の規則的パターン(例えば、Watson−Crick塩基対)によって標的ポリヌクレオチドに特異的に結合することが可能な、天然の、もしくは修飾されたモノマーまたは結合体(linkage)の線形オリゴマー(linear oligomer)(例えば、デオキシリボヌクレオチド、リボヌクレオチドなど)を含む。通常、モノマーは、数単量体単位(例えば、3〜4)から数十単量体単位の大きさの範囲内のオリゴヌクレオチドを形成するために、ホスホジエステル結合またはそのアナログによって結合する。プライマーは、代表的には一本鎖であるが、二本鎖にもなり得る。プライマーは、代表的にはデオキシリボ核酸であるが、当該分野において公知である種々の合成的プライマーおよび天然に存在するプライマーは、本開示の方法のために有用であり得る。プライマーは、テンプレートに相補的であり、そのプライマーは合成の開始のための部位として機能するためにハイブリダイズするように設計されるが、そのテンプレートの正確な配列を反映する必要性は無い。このような場合において、テンプレートへのプライマーの特異的ハイブリダイゼーションは、ハイブリダイゼーション条件のストリンジェンシーに依存する。プライマーは、例えば、色素性部分、放射活性部分、または蛍光部分で標識され得、そして検出可能な部分として使用される。
【0026】
核酸の重合は、当業者において公知である方法を使用して達成され得る。重合は、テンプレートの指示に従って遊離ヌクレオチドを順次付加するDNAポリメラーゼを使用し、一般に酵素的に達成される。数種の異なるDNAポリメラーゼが、本プロセスにおける使用のために適切である。ある実施形態において、選択の判定基準としては、エキソヌクレアーゼ活性を欠くこと、または強いエキソヌクレアーゼを有さないDNAポリメラーゼであることが挙げられる。本プロセスにおける使用のための低いエキソヌクレアーゼ活性を有するDNAポリメラーゼは、天然の供給源から単離され得るか、または組み換えDNA技術を介して生成され得る。使用され得るポリメラーゼの例示的な例は、T7 Sequenase v.2.0、エキソヌクレアーゼ活性を欠くDNAポリメラーゼIのKlenowフラグメント、TaqポリメラーゼのKlenowフラグメント、exo.−Pfu DNAポリメラーゼ、Vent.(exo.−)DNAポリメラーゼ、およびDeep Vent.(exo−)DNAポリメラーゼであるが、これらに限定されない。
【0027】
特に有用な実施形態において、出発物質の不必要な部分を消化により取り除くことによって、境界オリゴヌクレオチドの使用が避けられる。図2Aにおいて模式的に示されるこの実施形態において、制限オリゴヌクレオチド70は、あらかじめ選択しておいた位置で出発物質100にアニーリングする。この制限オリゴヌクレオチドは、制限酵素認識部位72を含む出発物質上に、二本鎖部分を提供する。適切な制限酵素認識部位としては、XhoI、SpeI、Nhe1、HindIII、NcoI、XmaI、BglII、BstI、およびPvuIが挙げられるが、これらに限定されない。適切な制限酵素に曝露すると、上記出発物質は消化され、そしてその結果不必要な配列を取り除くために短くなる一方で、原テンプレート110として使用されるものの上で、増幅されるべき所望の標的配列12(またはその一部分)は保存される。一旦原テンプレート110が得られると、プライマー20は、上述の前の実施形態のように標的配列12に隣接して、または重なって原テンプレート110にアニーリングされる(図2Bを参照のこと)。プライマー20の3’末端と原テンプレート110の5’末端との間の原テンプレートの部分に相補的な核酸の鎖40は、重合化される。当業者が理解するように、この実施形態において、制限オリゴヌクレオチドが原テンプレートを生成するために利用される場合、プライマー伸長は短くされた原テンプレート110の5’末端までの全てを進むことを可能にし得るので、境界オリゴヌクレオチドを使用する必要性は無い。
【0028】
一旦重合が完了する(すなわち、伸長する鎖40が境界ヌクレオチド30または短くされた原テンプレート110の5’末端に到達する)と、新規に合成された相補鎖は、物理学的手段、化学的手段または酵素的手段を含む任意の適切な変性方法によって、原テンプレートから分離される。鎖の分離はまた、ヘリカーゼまたは酵素RecAとして公知の酵素のクラスに由来する酵素によって誘導されるRecAは、ヘリカーゼ活性を有し、かつリボATPの存在下においてDNAを変性させることで知られる。ヘリカーゼを用いた、核酸の鎖を分離するための適切な反応条件は、Cold Spring Harbor Symposia on Quantitative Biology、第XLIII巻「DNA:Replication and Recombination」(New York:Cold Spring Harbor Laboratory、1978)、B.Kuhnら、「DNA Helicases」、pp.63−67によって記載され、そしてRecAの使用に関する技術は、C.Radding、Ann.Rev.Genetics、16:405−37(1982)において総説される。
【0029】
したがって、新規に合成された相補鎖は、上記プライマー20によって提供された配列(例えば、所定の配列22、任意の制限酵素認識部位23および上記プライマーのアニーリング部分25)、ならびに、プライマー20が原テンプレート10にアニーリングした位置と、境界オリゴヌクレオチド30にアニーリングした原テンプレート10の部分かまたは原テンプレートの短くされた5’末端のいずれかとの間の原テンプレート10の部分に相補的な、新規に合成された部分45を含む。図4を参照のこと。
【0030】
必要に応じて、続く工程の中で使用するための複数コピーの新規に合成された相補鎖を生成するために、原テンプレートおよびプライマーを使用した複数ラウンドの重合(好ましくは、15ラウンド〜25ラウンド)が実施される。このプロセス中のこの時点での複数コピーの新規に合成された相補鎖の作製は、(増幅の前に全長(entire)の人工テンプレートが生成されるまで待つ代わりに)最終的に生成された人工テンプレート中への標的配列の正確な複製の取り込みを確実にすることを援助する。原テンプレートに基づいた複数ラウンドの重合は、ライブラリーの全てのメンバーのより良い表現が達成されるより大きな可能性を提供すると考えられており、その結果、単一ラウンドの重合と比較して、より大きな多様性を提供する。
【0031】
代替的な実施形態において、新規合成鎖は、上述のようにプライマー20を原テンプレート10にアニーリングし、そしてブロッキングオリゴヌクレオチドの存在も原テンプレートの部分除去のいずれも伴わずに複数ラウンドの重合を実施することによって生成される。この実施形態(これは、図3において図解的に示される)において、プライマーは、全長の新規合成鎖140を提供するために、原テンプレートの全長に沿って伸長される。次に、制限オリゴヌクレオチド170が、上記全長の新規合成鎖にハイブリダイゼーションされる。この制限オリゴヌクレオチドは、上記新規合成鎖上に制限酵素認識部位を含む二本鎖部分を提供する。適切な制限酵素認識部位としては、XhoI、SpeI、Nhel、HindIII、NcoI、XmaI、BglII、BstI、PvuI、XcmI、BsaJI、HpaI、ApaLI、SacI、DraIIIおよびSmaIが挙げられるが、これらに限定されない。適切な制限酵素に曝すことにより、上記新規合成鎖は消化され、そしてその結果短縮される。次いで、ネステッドオリゴヌクレオチド50は、以下により詳細に説明されるように、人工テンプレートの調製を完了させるために、短くされた新規合成鎖142にハイブリダイゼーションされる。
【0032】
人工テンプレートの調製における次の工程は、例えば、図4において示されるように、ネステッドオリゴヌクレオチド50を新規に合成された相補鎖の3’末端にアニーリングさせることに関する。図4において見られるように、ネステッドオリゴヌクレオチド50は、人工テンプレートを完成させるために必要な、さらなる重合のためのテンプレートを提供する。ネステッドオリゴヌクレオチド50は、新規に合成された相補鎖に対してハイブリダイゼーションしないおよび/または新規に合成された相補鎖に対して修飾された塩基を含む部分52を含み、その結果、ネステッドオリゴヌクレオチドがプライマーとして機能することを防いでいる。ネステッドオリゴヌクレオチド50はまた、新規に合成された相補鎖の3’末端にハイブリダイゼーションする部分55を含む。部分55は、図4において示されるように、新規に合成された部分45と重なり合い得るか、または新規に合成された部分45を超えて伸長し得る。ネステッドオリゴヌクレオチド50はまた、必要に応じて、制限酵素認識部位を規定する部分56を含み得る。ネステッドオリゴヌクレオチド50の最終部分58は、プライマー20の部分22と同一な所定の配列を含む。部分55が、新規に合成された相補鎖の初めの3’末端を越えて伸長する地点から、ネステッドオリゴヌクレオチドは、人工テンプレートを形成するためのさらなる重合のためのテンプレートとして機能する。ネステッドオリゴは、(原テンプレートの形成において、その一部分が短縮されていた場合)標的配列の一部分を含み得るか、もしくはポリペプチドまたはタンパク質をコードする遺伝子(またはその一部分)(例えば、1つ以上のCDR領域もしくはフレームワーク領域または抗体の定常領域)を含み得ることが、理解されるはずである。種々の配列を有するネステッドオリゴヌクレオチドの収集物が利用され得ることもまた企図され、その結果、多種多様な生成物のライブラリーをもたらす種々のテンプレートが提供される。このようにして、図4において示されるように、ネステッドオリゴヌクレオチドに相補的なさらなる核酸60を付加することによって、重合は、新規に合成された相補鎖を伸長させる。重合を達成するための技術は、当業者の範囲内にある。前述のように、ある実施形態において、適切なポリメラーゼ(エキソヌクレアーゼ活性を欠く酵素)の選択が好ましくあり得る。
【0033】
一旦重合が完了すると、人工テンプレート120は、例えば、加熱変性のような当業者に周知の技術によって、ネステッドオリゴヌクレオチド50から分離される。結果生じた人工テンプレート120は、初発プライマー20由来である部分、原テンプレートの一部分に相補的な部分45、およびネステッドオリゴヌクレオチドの一部分に相補的な部分65を含む(図5を参照のこと)。注目すべきことに、人工テンプレート120の3’末端は、プライマー20の部分22の所定の配列に相補的な配列を含む、部分68を含む。これは、当業者において公知の技術を使用し、プライマー部分22の所定の配列と同一の配列を有する単一のプライマーを使用して、人工テンプレート120内に含まれる所望の配列を増幅することを可能にする。単一プライマー増幅の間、エキソヌクレアーゼ活性を有するポリメラーゼが存在することは好ましい。なぜならば、このような酵素は「プルーフリーディング」機能を提供することが知られており、かつエキソヌクレアーゼ活性を欠いたポリメラーゼと比較して相対的により高い伸長性(processivity)を有するからである。
【0034】
図6は、新規に合成されたcDNAテンプレートの単一プライマー増幅に関連する工程を図示する。反応混合物中にプライマーが存在する場合、プライマーは、テンプレートに隣り合う配列にハイブリダイゼーションし、そしてこのテンプレートを増幅する。プライマーが存在しない場合は、5’末端の所定の配列とこの所定の配列に相補的な3’末端配列との間で、内部の自己アニーリングが存在すると考えられている。好ましい実施形態において、所定の配列および相補的な所定の配列は、単一プライマー増幅反応の間のミスプライミングを避けるために、より高い温度でアニーリングするように設計され得る。
【0035】
増幅が実施された後、その生成物は、当業者において公知である任意の技術を使用して検出され得る。核酸を検出するために使用される方法の例としては、対立遺伝子特異的オリゴヌクレオチドを用いたハイブリダイゼーション、制限酵素切断、一本鎖立体配座多型(single−stranded conformational polymorphism)(SSCP)、分析ゲル電気泳動、エチジウムブロマイド染色、蛍光共鳴エネルギー移動(fluorescence resonance energy transfer)、ヘアピンFRETアッセイ(essay)、およびTaqManアッセイが挙げられるが、これらに限定されない。
【0036】
一旦人工核酸が所望される回数分増幅されると、その鎖を消化するために制限酵素認識部位23および制限酵素認識部位66または任意の内在性制限酵素認識部位が使用され得、そして標的核酸配列は、適切な発現ベクター中にライゲーションされ得る。次いで、このベクターは、標準の方法を使用して適切な宿主生物を形質転換するために使用され、標的領域によってコードされるポリペプチドまたはタンパク質が生成される。
【0037】
特に有用な実施形態において、本明細書中に記載される方法は、抗体のcDNAを使用し、抗体またはその一部(例えば、可変領域(軽鎖または重鎖のいずれか))をコードする標的配列を増幅するために使用される。このようにして、抗体のライブラリーが増幅され得、そしてスクリーニングされ得る。したがって、例えば、抗体mRNAで開始して、第1鎖cDNAが生成され得、そして原テンプレートを提供するために消化され得る。プライマーは選択された相補性決定領域(CDR)の上流にアニーリングするように設計され得、そしてこの新規合成核酸鎖は、CDRを含む。一例として、上記標的配列が重鎖CDR3である場合、プライマーは、重鎖フレームワーク1(FR1)領域にアニーリングするように設計され得る。当業者は、本開示に基づき、他の上流部位にアニーリングする適切なプライマー、または抗体cDNA内の他に選択された標的を複製する適切なプライマーを、どのようにして設計するかを容易に想定する。
【実施例】
【0038】
以下の実施例は、本発明の例証のために提供されるが、本発明を限定しない。
【0039】
(実施例1)
(IgM重鎖可変遺伝子のレパートリーの増幅)
(第1鎖cDNA合成および修飾)
SuperScript First−Strand Synthesis System for RT−PCR(Invitrogen Life Technologies、Carlsbad、CA)を使用し、基本的にキットの指示書に従って、オリゴdTプライマーを用いて慣習的な第1鎖cDNAを生成するために、ヒト末梢血リンパ球(PBL)mRNAを使用した。この第1鎖cDNA生成物を、QIAGENスピンカラム(QIAGEN、Valencia、CA提供のPCR Purification Kit)を通じて浄化した。制限エンドヌクレアーゼEcoRIによって消化され得る二本鎖DNA領域を生成するために、制限オリゴヌクレオチドを上記第1鎖cDNAに添加した。制限オリゴヌクレオチド(CMEcoRI)の配列は、5’TCC TGT GAG AAT TCC CCG TCG3’(配列番号1)であった。この反応を、第1鎖cDNAおよび0.1μMオリゴヌクレオチドを用いてセットアップした。試料を2分間95℃まで加熱し、次いで特異的アニーリングが起こり得るように64℃で2分間保った。適切な量の10×制限緩衝液H(Roche Diagnostics)を上記試料に添加し、そしてさらに37℃まで冷却した。制限エンドヌクレアーゼEcoRI(New England Biolabs、Beverly MA)を添加し、そして37℃で30分間インキュベートした。この制限酵素を65℃で20分間加熱不活性化し、次いで試料を4℃まで冷却した。
【0040】
(第2鎖線形増幅およびネステッドオリゴ伸長)
プライマー「TMX24VH3a」(最終0.4μM)、dNTP、AmpliTaq酵素およびその10×反応緩衝液(Applied Biosystems、Foster City、CA)に加えて、EcoRIで消化された第1鎖cDNAを、第2鎖cDNA反応における原テンプレートとして使用した。上記プライマーは、所定のTMX24配列、XhoI制限酵素認識部位およびヒト抗体重鎖遺伝子のフレームワーク1領域における第1鎖cDNAにアニーリングする領域を含むように設計した。「TMX24VH3a」の配列は、5’GTG CTG GCC GTT GGA AGA GGA GTG
CTC GAG GAR GTG CAG CTG GTG GAG3’(配列番号2)であり、Rは、塩基Aおよび塩基Gの等モル混合物を意味する。上記試料を95℃で1分間加熱変性し、次いで95℃で5秒間、56℃で10秒間、および68℃で1分間を介する反応を20回サイクルさせた。これは、第2鎖cDNAの線形増幅を可能にする。「TMX24CM0」と命名されたネステッドオリゴを、次いで0.08μMの最終濃度となるように氷上で添加した。「TMX24CM0」の配列は、5’GTG CTG GCC GTT GGA AGA GGA GTG ACT AGT AAT TCT CAC AGG AGA CGA GGG GGA3’(配列番号3)であった。これはその後のクローニング工程において使用されるSpeI制限エンドヌクレアーゼ部位を含む。このネステッドオリゴの3’末端は、逆結合(3’−5’よりもむしろ3’−3’)アデノシンの取り込みにより伸長を防ぐように設計される。上記第2鎖cDNAを、94℃5秒間で加熱変性し、次いで68℃で10秒間および95℃で5秒間の4サイクルアニーリングおよび伸長させることによってネステッドオリゴからさらに伸長し、続いて68℃で30秒間処理し、そして4℃にした。結果生じた第2鎖cDNA、すなわち人工テンプレートを、次いでQIAGENスピンカラム(QIAGEN、Valencia、CA提供のPCR Purification Kit)を使用して洗浄した。この工程は、オリゴヌクレオチドを除去し、そして下流のプロトコールのための容易な緩衝液交換を可能にする。
【0041】
(単一プライマー増幅(SPA))
Advantage2ポリメラーゼミックス(Clontech)およびその10×反応緩衝液、dNTP、ならびに5’GTG CTG GCC GTT GGA AGA GGA GTG3’(配列番号4)の配列を有する単一のプライマー(TMX24)を使用して、上記人工テンプレートを増幅した。この試料を95℃で1分間加熱変性し、次いで95℃で5秒間および68℃で1分間を介する反応を35回サイクルさせた。これ続いて、さらに68℃で3分間反応させ、そして4℃で保持した。
【0042】
(クローニングおよび配列決定)
約450bpの増幅生成物をゲル精製し、次いでXhoIおよびSpeIで消化し、そしてpBluescript KS+(Stratagene)中にクローニングした。個々のクローンを取り出し、そしてそれらのDNA配列を決定した。分析した16クローンの全てがIgM重鎖であり、各々が種々の長さの異なるCDR3配列を有していた。この結果は、この方法によって抗体鎖の多様な集団が増幅されたことを示す(表1を参照のこと)。
【0043】
【表1】
。
【0044】
(実施例2)
VH生成物をベクター中にクローニングするために、ネイティブIgM CH1定常領域が再構成され得、CH1中のEcoRI以外の部位を、上記第1鎖cDNAエンドヌクレアーゼ消化のために利用した。当業者が理解するように、Taqポリメラーゼがこのプロトコールのために使用された場合、新規に合成されたDNA鎖の多くに末端のAが付加される。多様性を最大化させるために、その末端のAの存在をネステッドオリゴヌクレオチドの設計において考慮した。しかしながら、余剰なAの存在は、EcoRI認識部位の減少を招く。IgM定常領域の分析は、他のネイティブ制限酵素認識部位が、この方法のために潜在的に使用され得ることを明らかにした(例えば、DraIII)。CH1ドメイン中のDraIIIネイティブ制限酵素認識部位の使用の結果、上流EcoRI部位が未修飾のまま残存し、そして重鎖レパートリーのクローニングのために使用され得る。重鎖インサートは、XhoIおよびEcoRIによって、EcoRIからCH2ドメインに残存IgM CH1ドメインを有する適切なベクター中にクローニングされる。
【0045】
(第1鎖合成および修飾)
オリゴdTプライマーを用いた慣習的な第1鎖cDNAを生成するために、ヒト末梢血リンパ球(PBL)mRNAを使用した。これを、基本的にキットの指示書に従って、SuperScript First−Strand Synthesis System for RT−PCR(Invitrogen Life Technologies、Carlsbad、CA)を使用して行った。制限エンドヌクレアーゼDraIIIによって消化され得る二本鎖DNA領域を生成するために、制限オリゴヌクレオチドを第1鎖cDNAに添加した。この制限オリゴヌクレオチド(CMDraIII)の配列は、5’GAC GAA CAC GTG GTG TGC AAA G3’(配列番号21)であった。この反応を、第1鎖cDNAおよび1μMオリゴヌクレオチドを用いてセットアップした。試料を2分間95℃まで加熱し、次いで特異的アニーリングが起こり得るように64℃で2分間保持した。適切な量の10×制限緩衝液H(Roche Diagnostics)を、上記試料中に添加し、そしてさらに37℃まで冷却した。制限エンドヌクレアーゼDraIII(New England Biolabs、Beverly
MA)を添加し、そして37℃で30分間インキュベートした。上記制限酵素を65℃で20分間加熱不活性化し、次いでこの試料を4℃まで冷却した。
【0046】
(第2鎖線形増幅およびネステッドオリゴ伸長)
第2鎖cDNA反応において、プライマー「TMX24VH1a」(最終0.4μM)、dNTP、AmpliTaq酵素およびその10×反応緩衝液(Applied Biosystems、Foster City、CA)に加えて、DraIIIで消化された第1鎖cDNAを原テンプレートとして使用した。上記プライマーは、所定のTMX24配列、XhoI制限酵素認識部位およびヒト抗体重鎖遺伝子のフレームワーク1領域における第1鎖cDNAにアニーリングする領域を含むように設計した。「TMX24VH1a」の配列は、5’GTG CTG GCC GTT GGA AGA GGA GTG CTC GAG CAG GTK CAG CTG GTG CAG3’(配列番号22)であり、Kは、塩基Gおよび塩基Tの等モル混合物を意味する。上記試料を94℃で1分間加熱変性し、次いで94℃で5秒間、56℃で10秒間、および68℃で2分間を介する反応を20回サイクルさせた。これは、第2鎖cDNAの線形増幅を可能にする。「TMX24CMnpt」と命名されたネステッドオリゴを、次いで0.2μMの最終濃度となるように氷上で添加した。図7において示されるように、「TMX24CMnpt」(配列番号23)の配列は、オリゴヌクレオチドの伸長を防ぐように設計された、修飾された構造を有する3つの3’末端ヌクレオチドを含む。具体的には、上記ネステッドオリゴは、ホスホチオ酸塩(phosphorthioate)および伸長を防ぎかつエキソヌクレアーゼ活性ならびにエンドヌクレアーゼ活性に対して保護するために設計された2’OMeで修飾された、3つの末端ヌクレオチドを有する。このオリゴの3’末端オリゴヌクレオチドは、ハイブリダイズしない(cの代わりにg)。上記第2鎖cDNAを、94℃1分間で加熱変性し、次いで68℃で2分間アニーリングおよび伸長させてネステッドオリゴからさらに伸長し、続いて4℃にした。結果生じた第2鎖cDNA、または人工テンプレートを、次いでQIAGENスピンカラム(QIAGEN、Valencia、CA提供のPCR Purification Kit)を使用して洗浄した。この工程は、オリゴヌクレオチドを除去し、そして下流のプロトコールのための容易な緩衝液交換を可能にする。この手順を繰り返し、そして、適切なベクターにクローニングされ得る免疫グロブリン生成物のライブラリーを生成するために、VHプライマーパネル(primer panel)(プライマーリストを参照のこと)の残りの部分を伸長した。
【0047】
【化6】
上記配列において、RはAおよびGの同じ割合の混合物であり、KはGおよびTの同じ割合の混合物であり、そしてSはCおよびGの同じ割合の混合物である。
【0048】
(単一プライマー増幅(SPA))
上記人工テンプレートを、Advantage 2ポリメラーゼミックス(Clontech)およびその10×反応緩衝液、dNTPならびに5’GTG CTG GCC GTT GGA AGA GGA GTG3’(配列番号4)の配列を有する単一のプライマー(TMX24)を使用して増幅した。その試料を95℃で1分間加熱変性し、次いで95℃で5秒間および68℃で1分間を介する反応を30回サイクルさせた。これに続いてさらに68℃で3分間処理し、そして4℃で保持した。
【0049】
(クローニングおよび配列決定)
約450bpの増幅生成物をゲル精製し、そしてXhoIおよびEcoRIによって消化する。ネイティブEcoRI由来のIgM CH1ドメインの残余部分を含み、CH2ドメインまでかまたはを含んで増幅されたフラグメントをクローニングするために適性のある制限酵素認識部位を含む任意の適切な発現ベクター中に、このインサートをクローニングする。
【0050】
(実施例3)
(B型肝炎陽性ドナー由来のファージミドディスプレイライブラリー(phagmid display library)の構築)
(IgG重鎖およびκ軽鎖についての第1鎖cDNAの合成および修飾)
オリゴdTプライマーを用いた慣習的な第1鎖cDNAを生成するために、B型肝炎ワクチン接種されたドナー由来のヒト末梢血リンパ球(PBL)mRNAを使用した。これは、SuperScript First−Strand Synthesis System for RT−PCR(Invitrogen Life Technologies、Carlsbad、CA)を使用し、基本的にキットの指示書に従って行った。IgGについては制限エンドヌクレアーゼApaLIによって、またはκ軽鎖についてはSacIによって消化され得る二本鎖DNA領域を生成するために、IgGについては制限オリゴヌクレオチド「CGApaLI」を、またはκ軽鎖については制限オリゴヌクレオチド「CKSacI」を第1鎖cDNAに添加した。「CGApaLI」配列は、5’CCA GCG GCG TGC ACA CCT TCC3’(配列番号39)である。「CKSacI」配列は、5’AGG GCC TGA GCT CGC CCG TC3’(配列番号40)である。この反応を、第1鎖cDNA、1μMオリゴヌクレオチド、および適切な量の10×反応緩衝液A(Roche Diagnostics)を用いてセットアップした。この試料を95℃まで2分間加熱し、次いで特異的アニーリングが起こり得るように64℃で2分間保持し、そして37℃まで冷却した。制限エンドヌクレアーゼApaLIまたはSacI(New England Biolabs、Beverly MA)を添加し、そして37℃で30分間インキュベートした。この制限酵素を、SacIに関して65℃で20分間加熱不活性化し、次いで試料を4℃まで冷却した。
【0051】
各々の制限エンドヌクレアーゼによる上記第1鎖cDNAの消化を、当業者において公知である技術を使用し、PCR増幅によって検証した。これらの生成物は、抗体遺伝子のクローニングには使用しなかった。消化された第1鎖cDNAの陽性増幅物を、IgGについて5’VBVH1a内部コントロールプライマーおよび3’CG0内部コントロールプライマーを、およびκについて5’VBVK1aおよび3’CK0内部コントロールプライマーを使用した反応中で観察した。5’VBVH1a/3’CG0プライマーまたは5’VBVK1a/3’CK0プライマーを用いた良好な増幅および5’VBVH1a/3’CG1Zプライマーまたは5’VK1a/3’CK1dx2プライマーを用いたわずかな増幅は、各々の制限エンドヌクレアーゼを用いた第1鎖cDNAテンプレートの首尾良い消化を示す。PCRを検査するチェックのために使用されるプライマーの配列は、以下:
【0052】
【化7】
である。
【0053】
(軽鎖第2鎖線形増幅およびネステッドオリゴ伸長)
フレームワーク1特異的プライマー(最終0.4μM)、dNTP、AmpliTaq酵素およびその10×反応緩衝液(Applied Biosystems、Foster City、CA)を使用した複数の第2鎖cDNA反応をセットアップするために、SacI消化された第1鎖cDNAを、原テンプレートとして使用した。上記プライマーを、κについて所定のTMX24K配列5’GAC GAC CGG CTA CCA AGA GGA GTG3’(配列番号47)、XbaI制限酵素認識部位、およびヒト抗体κ軽鎖遺伝子のフレームワーク1領域中の第1鎖cDNAにアニーリングする領域を含むように設計した。これらのアニーリング配列は、VBaseデータベースプライマー(www.mrc−cpe.cam.ac.uk/imt−doc/public/INTRO.html)由来であり、これらは、公知のヒト抗体の配列に基づいて設計され、そしてκ軽鎖遺伝子の全ヒト抗体レパートリーを網羅すると報告されている。
【0054】
【化8】
【0055】
【化9】
上記配列において、RはAおよびGの同じ割合の混合物であり、MはAおよびCの同じ割合の混合物であり、YはCおよびTの同じ割合の混合物であり、WはAおよびTの同じ割合の混合物であり、そしてSはCおよびGの同じ割合の混合物である。
【0056】
上記試料を94℃で1分間加熱変性し、次いで94℃で5秒間、56℃で10秒間、および68℃で2分間を介する反応を20サイクルさせた。これは、第2鎖DNAの線形増幅を可能にする。κ鎖について「TMX24CKnpt」と命名されたネステッドオリゴを、氷上で0.2μMの最終濃度となるように添加した。「TMX24CKnpt」は、所定の配列TMX24Kを含み、そしてこの配列は、5’GAC GAC CGG CTA CCA AGA GGA GTG CTC GAG CTC AGG CCC TGA TGG GTG ACT TCG CT3’(配列番号61)であった。上記第2鎖cDNAを、94℃で1分間加熱変性させ、次いで68℃で2分間アニーリングおよび伸長させて、ネステッドオリゴからさらに伸長させ、続いて4℃にした。結果生じた第2鎖cDNA(人工テンプレート)を、QIAGENスピンカラム(QIAGEN、Valencia、CA提供のPCR Purification Kit)を使用して洗浄した。この工程は、遊離のオリゴヌクレオチドを除去し、そして下流のプロトコールのための容易な緩衝液交換を可能にする。
【0057】
(軽鎖単一プライマー増幅(SPA))
Advantage 2ポリメラーゼミックス(Clontech)およびその10×反応緩衝液、dNTP、ならびにκ鎖についてのプライマー「TMX24K」を使用し、上記人工テンプレートを増幅した。「TMX24K」についての配列は、5’GAC GAC CGG CTA CCA AGA GGA GTG3’(配列番号62)である。この試料を95℃で1分間加熱変性し、次いで95℃で5秒間および68℃で1分間を介する反応を30サイクルさせた。これに続いてさらに68℃で3分間処理し、そして4℃で保持した。
【0058】
(軽鎖クローニング)
κ増幅生成物をゲル精製し、次いでXbaIおよびSacIによって消化した。そのインサートを、κ軽鎖定常領域の残余部分を含む適切な発現ベクター中にクローニングした。このライゲーションされた生成物を、エレクトロポレーションによってE.coli中に導入し、そして37℃で一晩成長させた。翌朝、軽鎖ライブラリーDNAを回収するためにDNAマキシプレップ(QIAGEN、Valencia、CA)を実施した。次いで、ライブラリーの構築を完了させるために、XhoI/AgeIによる重鎖Fdフラグメントのクローニングのための続く工程において、この軽鎖ライブラリーDNAを使用した。
【0059】
(重鎖第2鎖線形増幅およびネステッドオリゴ伸長)
フレームワーク1特異的プライマー(最終0.4μM)、dNTP、AmpliTaq酵素およびその10×反応緩衝液(Applied Biosystems、Foster City、CA)を使用して複数の第2鎖cDNA反応をセットアップするために、ApaLI消化された第1鎖cDNAを、原テンプレートとして使用した。上記プライマーを、所定のTMX24配列、XhoI制限酵素認識部位、およびヒト抗体重鎖遺伝子のフレームワーク1領域における第1鎖cDNAにアニーリングする領域を含むように設計した。これらのアニーリング配列は、VBaseデータベースプライマー由来であって、これらは公知のヒト抗体の配列に基づいて設計されており、そして重鎖遺伝子の全ヒト抗体レパートリーを網羅すると報告されている。
【0060】
【化10】
【0061】
【化11】
上記配列において、RはAおよびGの同じ割合の混合物であり、KはGおよびTの同じ割合の混合物であり、そしてSはCおよびGの同じ割合の混合物である。
【0062】
上記試料を、94℃で1分間加熱変性し、次いで94℃で5秒間、56℃で10秒間、および68℃で2分間を介する反応を20サイクルさせた。これは、第2鎖cDNAの線形増幅を可能にする。「TMX24CGnpt」と命名されたネステッドオリゴヌクレオチド(配列5’GTG CTG GCC GTT GGA AGA GGA GTG TGT TTG CAC GCC GCT GGT CAG RGC GCC TGA GTT G3’(配列番号77))を、氷上で0.2μMの最終濃度となるように添加した。図1に示されるように、IgMネステッドオリゴについて、オリゴ伸長を防ぐために、3つの3’末端ヌクレオチドを修飾した。上記第2鎖cDNAを、94℃で1分間の加熱変性し、68℃で2分間でアニーリングおよび伸長させてネステッドオリゴからさらに伸長させ、続いて4℃にした。結果生じた第2鎖cDNA(人工テンプレート)を、次いでQIAGENスピンカラム(QIAGEN、Valencia、CA提供のPCR Purification Kit)を使用して洗浄した。この工程は、遊離のオリゴヌクレオチドを除去し、そして下流のプロトコールのための容易な緩衝液交換を可能にする。
【0063】
(単一プライマー増幅(SPA))
Advantage 2ポリメラーゼミックス(Clontech)およびその10×反応緩衝液、dNTP、およびプライマー「TMX24」を使用し、上記人工テンプレートを増幅した。この試料を95℃で1分間加熱変性し、次いで95℃で5秒間および68℃で1分間を介する反応を30サイクルさせた。これに続いて68℃でさらに3分間処理し、そして4℃で保持した。
【0064】
(重鎖クローニングおよびライブラリーの生成)
この増幅された生成物をプールし、次いでゲル精製した。QIAquick PCR purification kit(QIAGEN、Valencia、CA)を用いて、DNAを回収した。このDNAを、XhoI制限酵素およびAgeI制限酵素で順次消化し、次いでゲル精製した。AgeI部位は、ApaLI部位の上流のIgG定常領域のCH1中に、天然に存在する。DNAを、QIAquick Gel extraction Kit(QIAGEN、Valencia、CA)で回収した。
【0065】
軽鎖ライブラリーDNAを、XhoIおよびAgeIで順次消化し、次いでゲル精製した。この軽鎖ライブラリーDNAを、上記重鎖フラグメントとライゲーションした。ライゲーションされたDNAを、反応緩衝液を除去するためにスピンカラム(PCR purification Kit、QIAGEN、Valencia、CA)上に置き、そしてこのDNAを濃縮した。電気的にコンピテントな(electrocompetent)XL−1 Blue細胞(Stratagene)において、最終形質転換を行った。
【0066】
(HBs Agについてのライブラリーのパンニングおよびスクリーニング)
基本的に、Barbas III,CF、Burton,DR、Scott,JK、およびSilverman,GJ(2001)Phage Display:A Laboratory Manual、Cold Spring Harbor Laboratory Press、Cold Spring Harbor、New Yorkに記載されるように、上記ライブラリーを、固定化HBs Agについて4ラウンドパンニングした。パンニングの第2ラウンド、第3ラウンド、および第4ラウンド由来である個々のクローンを、HBs AgについてのELISAによってスクリーニングした。
【0067】
(λ軽鎖についての第1鎖cDNA合成および修飾)
オリゴdTプライマーを用いた慣習的な第1鎖cDNAを生成するために、B型肝炎ワクチン接種されたドナー由来のヒトPBL mRNAを使用した。これを、SuperScript First−Strand Synthesis for RT−PCR(Invitrogen Life Technologies、Carlsbad、CA)を使用し、基本的にキットの指示書に従って行った。制限エンドヌクレアーゼSmaIによって消化され得る二本鎖DNA領域を生成するために、上記第1鎖cDNAに制限オリゴヌクレオチド「CLSmaI」を添加した。「CLSmaI」配列は、5’GAC TTC TAC CCG GGA GCY GTG3’(配列番号78)であって、ここで、YはCおよびTの混合物である。この反応を、第1鎖cDNA、1μMオリゴヌクレオチド、および適切な量の10×反応緩衝液A(Roche Diagnostics)を用いてセットアップした。この試料を95℃まで2分間加熱し、次いで特異的アニーリングが起こり得るように64℃で2分間保持し、そして37℃まで冷却した。制限エンドヌクレアーゼSmaI(New England Biolabs、Beverly MA)を添加し、そして37℃で30分間インキュベートした。この制限酵素を65℃で加熱不活性化し、次いでその試料を4℃まで冷却した。
【0068】
制限エンドヌクレアーゼによる上記第1鎖cDNAの消化を、当業者において公知である技術を使用し、PCR増幅によって検証した。これらの生成物は、抗体遺伝子のクローニングには使用しなかった。5’VBVL1a内部コントロールプライマーおよび3’CL0内部コントロールプライマーを使用した反応において、消化された第1鎖cDNAの陽性増幅物を観察した。5’VBVH1a/3’CL0プライマーを用いた良好な増幅および5’VBVL1a/3’CL2dx2プライマーを用いたわずかな増幅は、SmaIを用いた第1鎖cDNAテンプレートの首尾良い消化を示す。PCRの検査のためのプライマーの配列は、VBVL1a:5’GAC GCG CAC AAC ACG GAG CTC CAG TCT GTG CTG ACT CAG3’(配列番号79)、CL0:5’CCT CAG AGG AGG GYG GGA ACA G3’(配列番号80)およびCL2dx2:5’AGA CAG TGA CGC CGT CTA GAA TTA TGA ACA TTC TGT AGG3’(配列番号81)である。
【0069】
(λ軽鎖第2鎖線形増幅およびネステッドオリゴ伸長)
フレームワーク1特異的プライマー(最終0.4μM)、dNTP、AmpliTaq酵素およびその10×反応緩衝液(Applied Biosystems、Foster City、CA)を使用して複数の第2鎖cDNA反応をセットアップするために、SmaI消化された第1鎖cDNAを、原テンプレートとして使用した。上記プライマーを、所定のTMX24L配列、XbaI部位、およびヒト抗体λ軽鎖遺伝子のフレームワーク領域中の第1鎖cDNAにアニーリングする領域を含むように設計した。これらのアニーリング配列は、VBaseデータベースプライマー(www.mrc−cpe.cam.ac.uk/imt−doc/public/INTRO.html)由来であって、これらは公知のヒト抗体の配列に基づいて設計されており、そしてλ軽鎖遺伝子の全ヒト抗体レパートリーを網羅すると報告されている。λ軽鎖フレームワーク1特異的プライマーは、実施例4におけるものを使用する。
【0070】
上記試料を、94℃で1分間加熱変性し、次いで94℃で5秒間、56℃で10秒間、および68℃で2分間を介する反応を20サイクル行った。これは、上記第2鎖cDNAの線形増幅を可能にする。実施例4において示されるような、「TMX24CLnpt」と命名されたネステッドオリゴヌクレオチドを、氷上で0.2μMの最終濃度となるように添加した。図7に示されるようなIgMネステッドオリゴヌクレオチド「TMX24CMnpt」について、「TMX24CLnpt」の3’末端ヌクレオチドは、オリゴ伸長を防ぐように修飾される。第2鎖cDNAを、94℃で1分間の加熱変性、68℃で2分間でアニーリングおよび伸長させてネステッドオリゴヌクレオチドからさらに伸長し、続いて4℃にした。結果生じた第2鎖cDNA(人工テンプレート)を、PCR Purification Kit(QIAGEN、Valencia、CA)を使用して浄化した。この工程は、遊離のオリゴヌクレオチドを除去し、そして下流のプロトコールのための容易な緩衝液交換を可能にする。
【0071】
(λ軽鎖単一プライマー増幅(SPA))
Advantage 2ポリメラーゼミックス(Clontech)およびその10×反応緩衝液、dNTP、ならびにプライマー「TMX24L」を使用し、上記人工テンプレートを増幅した。「TMX24L」についての所定の配列は、5’GAC GAC CGG CTA CCA AGA GGA CAG3’(配列番号82)である。この試料を95℃で1分間加熱変性し、次いで95℃で5秒間および68℃で1分間を介する反応を30サイクルさせた。これに続いてさらに68℃で3分間処理し、そして4℃で保持した。
【0072】
(λ軽鎖クローニング)
λ軽鎖増幅生成物を、PCR purification kit(QIAGEN、Valencia、CA)によって浄化し、そしてXbaIおよびSacIによって消化した。そのインサートを、gel extraction kit(QIAGEN、Valencia、CA)を使用してゲル精製し、そしてλ軽鎖定常領域の残余部分を含む適切な発現ベクター中にクローニングした。このライゲーションされた生成物を、エレクトロポレーションによってE.coli中に導入し、そして37℃で一晩成長させた。翌朝、λ軽鎖ライブラリーDNAを回収するためにDNAマキシプレップ(QIAGEN、Valencia、CA)を実施した。
【0073】
(重鎖クローニングおよびIgGλライブラリーの生成)
上記で調製したλ軽鎖ライブラリーDNAを、前述されるように、IgGκライブラリーに対して調製した重鎖Fdフラグメントの挿入のために、XhoIおよびAgeIで順次消化した。次いで、ライゲーションされた生成物を、エレクトロポレーションによってE.coli中に導入し、そして37℃で一晩成長させた。翌朝、完全なIgGλライブラリーDNAを回収するために、DNAマキシプレップ(QIAGEN、Valencia、CA)を実施した。
【0074】
(HBs Agについてのライブラリーの選別およびスクリーニング)
IgGκライブラリーについて前述されたように、パンニングおよびスクリーニングを実施した。
【0075】
(DNA配列決定分析および単離されたFabの性質決定)
ELISAスクリーニングによって、HBsAgに対して特異的結合を示し、かつ非特異的タンパク質(オボアルブミン)に対して最低限の結合を示したクローンを、DNA配列決定によって分析した。図8a〜図8eを参照のこと。HBsAgに対して、全部で38の異なるIgGκFab(25重鎖および37軽鎖)および17の異なるIgGλFab(13重鎖および16軽鎖)を、B型肝炎ワクチン接種を受けたドナー由来のPBL mRNAから作製したライブラリーから単離した。
【0076】
(実施例4)
(ヒトPBL mRNAからのファージディスプレイライブラリーの構築)
(軽鎖についての第1鎖cDNA合成および修飾)
ドナーからのヒトPBL mRNAを、オリゴdTプライマーを用いて、基本的にキットの指示書に従って、SuperScript First−Strand Synthesis System for RT−PCR(Invitrogen Life Technologies、Carlsbad、CA)を使用し、慣習的な第1鎖cDNAを生成するために使用した。κ軽鎖反応およびλ軽鎖反応を、別々にセットアップする。κ軽鎖については制限ヌクレアーゼSacIによって、またはλ軽鎖についてはSmaIによって消化され得る二本鎖DNA領域を生成するために、第1鎖cDNAに制限オリゴヌクレオチド「CKSacI」または「CLSmaI」を添加する。複数のλ定常領域(C1、C2、C3、およびC6)がある場合、全ての機能的λ定常ドメイン(C1、C2、C3、およびC6)の中で、SmaI部位が保存されていることに注意することが重要である。「CKSacI」配列は、5’AGG GCC TGA GCT CGC CCG TC3’(配列番号179)であり、「CLSmaI」配列は5’GAC TTC TAC CCG GGA GCY GTG3’(配列番号180)であって、ここで、YはCおよびTの混合物である。この反応を、第1鎖cDNAおよび1μMオリゴヌクレオチドを用いてセットアップする。この試料を95℃まで2分間加熱し、次いで特異的アニーリングが起こり得るように64℃で2分間保持する。適切な量の10×制限緩衝液A(Roche Diagnostics)を上記試料に添加し、そして37℃までさらに冷却する。制限エンドヌクレアーゼSacIまたはSmaI(New England Biolabs、Beverly MA)を添加し、そして37℃で30分間インキュベートする。この制限酵素を、65℃で20分間加熱不活性化し、次いでこの試料を4℃まで冷却する。
【0077】
(軽鎖第2鎖線形増幅およびネステッドオリゴ伸長)
フレームワーク1特異的プライマー(最終0.4μM)、dNTP、AmpliTaq酵素およびその10×反応緩衝液(Applied Biosystems、Foster City、CA)を使用して複数の第2鎖cDNA反応をセットアップするために、SacI消化されたκ第1鎖cDNAまたはSmaI消化されたλ第1鎖cDNAを、原テンプレートとして使用する。上記プライマーを、TMX24K(κについて)配列またはTMX24L(λについて)配列、XbaI制限酵素認識部位、およびヒト抗体κ鎖遺伝子またはヒト抗体λ軽鎖遺伝子のフレームワーク1領域中の第1鎖cDNAにアニーリングする領域を含むように設計する。これらのアニーリング配列は、VBaseデータベースプライマー(www.mrc−cpe.cam.ac.uk/imt−doc/public/INTRO.html)由来であり、これらは公知のヒト抗体の配列に基づいて設計されており、そして軽鎖遺伝子の全ヒト抗体レパートリーを網羅すると報告されている。κ軽鎖フレームワーク1特異的プライマーは、実施例3におけるものを使用する。λ増幅における使用のためのプライマーのリストは、以下を参照のこと。
【0078】
【化12】
上記配列において、RはAおよびGの同じ割合の混合物であり、MはAおよびCの同じ割合の混合物であり、YはCおよびTの同じ割合の混合物であり、WはAおよびTの同じ割合の混合物であり、そしてSはCおよびGの同じ割合の混合物である。
【0079】
上記試料を、94℃で1分間加熱変性し、次いで94℃で5秒間、および56℃で10秒間、および68℃で2分間を介する反応を20サイクル行う。これは、第2鎖cDNAの線形増幅を可能にする。κ鎖について「TMX24CKnpt」と命名されたネステッドオリゴヌクレオチド、またはλ鎖について「TMX24CLnpt」と命名されたネステッドオリゴヌクレオチドを、氷上で0.2μMの最終濃度となるように添加する。このネステッドオリゴヌクレオチドの配列は;「TMX24CKnpt」5’GAC GAC CGG CTA CCA AGA GGA GTG CTC GAG CTC AGG CCC TGA TGG GTG ACT TCG CT3’(配列番号196)および「TMX24CLnpt」5’GAC GAC CGG CTA CCA AGA GGA CAG AAG AGC TCC TGG GTA GAA GTC ACT KAT SAG RCA CAG3’(配列番号197)である。図7に示されるように、IgMネステッドオリゴについて、オリゴ伸長を防げるために3つの3’末端ヌクレオチドを修飾する。上記第2鎖cDNAを、94℃で1分間の加熱変性し、68℃で2分間アニーリングおよび伸長させてネステッドオリゴからさらに伸長し、続いて4℃にする。結果生じた第2鎖cDNA(人工テンプレート)を、QIAGENスピンカラム(QIAGEN、Valencia、CA提供のPCR Purification Kit)を使用して精製する。この工程は、遊離のオリゴヌクレオチドを除去し、そして下流のプロトコールのための容易な緩衝液交換を可能にする。
【0080】
(軽鎖単一プライマー増幅(SPA))
Advantage 2ポリメラーゼミックス(Clontech)およびその10×反応緩衝液、dNTP、ならびにκ鎖についてプライマー「TMX24K」を、またはλ鎖について「TMX24L」を使用し、上記人工テンプレートを増幅する。「TMX24K」の配列は5’GAC GAC CGG CTA CCA AGA GGA GTG 3’(配列番号198)であり、そして「TMX24L」の配列は5’GAC GAC CGG CTA CCA AGA GGA CAG3’(配列番号199)である。この試料を95℃で1分間加熱変性し、次いで95℃で5秒間および68℃で1分間を介する反応を30サイクル行う。これに続いてさらに68℃で3分間処理し、そして4℃で保持する。
【0081】
(軽鎖クローニング)
κ増幅生成物およびλ増幅生成物を、PCR purification kit(QUIAGEN)を使用して浄化し、そして別々にゲル精製した。これらの生成物を、XbaIおよびSacIによって消化した。そのインサートを、それぞれの軽鎖定常領域の残余部分を含む適切な発現ベクター中にクローニングした。このライゲーションされた生成物を、エレクトロポレーションによってE.coli中に導入し、そして37℃で一晩成長させた。翌朝、軽鎖ライブラリーDNAを回収するために、DNAマキシプレップを実施した。ライブラリーの構築を完了させるために、この軽鎖ライブラリーDNA調製物を、XhoI/EcoRIによる重鎖Fdフラグメントの挿入のためのクローニングベクターとして使用した。
【0082】
(重鎖についての第1鎖cDNA合成および修飾)
オリゴdTプライマーを用いた慣習的な第1鎖cDNAを生成するために、ヒトPBL mRNAを使用する。これを、SuperScript First−Strand Synthesis System for RT−PCR(Invitrogen Life Technologies、Carlsbad、CA)を使用し、基本的にキットの指示書に従って行う。制限エンドヌクレアーゼDraIIIによって消化され得る二本鎖DNA領域を生成するために、制限オリゴヌクレオチドCMDraIIIを、第1鎖cDNAに添加する。この反応を、第1鎖cDNAおよび1μMオリゴヌクレオチドを用いてセットアップする。試料を2分間95℃まで加熱し、次いで特異的アニーリングが起こり得るように64℃で2分間保持する。適切な量の10×制限緩衝液H(Roche Diagnostics)を上記試料中に添加し、そしてさらに37℃まで冷却する。制限エンドヌクレアーゼDraIII(New England Biolabs、Beverly MA)を添加し、そして37℃で30分間インキュベートし、次いで4℃まで冷却する。
【0083】
DraIIIによる第1鎖cDNAの消化を、PCR増幅によって検証する。増幅生成物は、抗体フラグメントのクローニングには使用されない。2つの異なる緩衝液条件下において、5’VBVH1a内部コントロールプライマーおよび3’CM0内部コントロールプライマーを使用した反応において、消化された第1鎖cDNAの陽性増幅物を観察する。5’VBVH1a/3’CM0プライマーを用いた良好な増幅、および5’VBVH1a/3’CM1プライマーを用いたわずかな増幅は、第1鎖cDNAテンプレートの首尾良いDraIII消化を示す。
【0084】
(重鎖第2鎖線形増幅およびネステッドオリゴ伸長)
フレームワーク1特異的プライマー(最終0.4μM)、dNTP、AmpliTaq酵素およびその10×反応緩衝液(Applied Biosystems、Foster City、CA)を使用して複数の第2鎖cDNA反応をセットアップするために、DraIII消化された第1鎖cDNAを、原テンプレートとして使用する。上記プライマーを、TMX24配列、XbaI制限酵素認識部位、およびヒト抗体重鎖遺伝子のフレームワーク1領域における第1鎖cDNAにアニーリングする領域を含むように設計する。これらのアニーリング配列は、VBaseデータベースプライマー由来であって、これらは公知のヒト抗体の配列に基づいて設計されており、そして実施例3において上述されるように、重鎖遺伝子の全ヒト抗体レパートリーを網羅すると報告されている。重鎖フレームワーク1特異的プライマーは、実施例3において列挙されたものを使用する。
【0085】
上記試料を、94℃で1分間加熱変性し、次いで94℃で5秒間、56℃で10秒間、および68℃で2分間を介する反応を20サイクル行う。これは、第2鎖cDNAの線形増幅を可能にする。「TMX24CMnpt」と命名されたネステッドオリゴヌクレオチド(実施例3において使用したもの)を、氷上で0.2μMの最終濃度となるように添加する。上記第2鎖cDNAを、94℃で1分間の加熱変性し、68℃で2分間のアニーリングおよび伸長によって、ネステッドオリゴからさらに伸長し、続いて4℃にする。結果生じた第2鎖cDNA(人工テンプレート)を、QIAGENスピンカラム(QIAGEN、Valencia、CA提供のPCR Purification Kit)を使用して洗浄する。この工程は、遊離のオリゴヌクレオチドを除去し、そして下流のプロトコールのための容易な緩衝液交換を可能にする。
【0086】
(重鎖単一プライマー増幅(SPA))
Advantage 2ポリメラーゼミックス(Clontech)およびその10×反応緩衝液、dNTP、ならびにプライマー「TMX24」を使用し、上記人工テンプレートを増幅する。この試料を95℃で1分間加熱変性し、次いで95℃で5秒間および68℃で1分間を介する反応を30サイクル行う。これに続いてさらに68℃で3分間処理し、そして4℃で保持する。
【0087】
(重鎖クローニングおよびライブラリーの生成)
約500bpの増幅生成物をゲル精製し、次いでXhoIおよびEcoRIによって消化する。IgM CH1ドメインの残余部分を含む適切な発現ベクター中に、インサートをクローニングする。ライゲーションされたFabライブラリーを含む生成物を、エレクトロポレーションによってE.coli中に導入する。
【0088】
バクテリオファージの表面上にFabライブラリーを生成させるために、サプレッサー株の細胞(例えば、XL1BLUE(Stratagene))を使用する。続いてエレクトロポレーションに続いて、その細胞を37℃で1時間振とうし、次いでカルベニシリンを20μg/mlまで添加する。37℃で1時間の振とうの後、カルベニシリンを50μg/mlまで増やし、37℃にさらに1時間置く。次いで、ファージミド粒子の生成のために必要な全ての成分を提供するため、VCS−M13ヘルパーファージ(Stratagene)を添加し、そして培養物の容積をSB培地で100mlまで増やす。37℃で1時間後、ヘルパーファージDNAを含む細菌を選択するために、70μg/mlまでカナマイシンを添加する。この培養物を、37℃で一晩振とうする。その期間の間、上記細菌は、その表面上にFabを有する新しいファージミド粒子を生成する。翌朝、このファージミド粒子は細菌細胞をスピンアウトすることによって単離され得、次いでこのファージミド粒子を、4% PEG 8000および0.5M NaClを用いて、氷上で30分間上清から沈殿させる。14,300×gの遠沈によって、ファージペレットを沈殿させる。このペレットは、PBS/1% BSA中で再懸濁され得る。この調製物は、細菌性の破片を取り除くために、濾過され得る。結果生じたライブラリーを、4℃で保存する。
【0089】
(実施例5)
(IgEまたは組み換えIgE Fc CH2〜4で免疫されたマウスmRNA由来のファージミドディスプレイライブラリーの構築)
(IgG重鎖およびκ軽鎖についての第1鎖cDNA合成および修飾)
オリゴdTプライマーを用いた慣習的第1鎖cDNAを生成するために、ヒトIgEまたは組み換えヒトIgEで免疫されたマウス由来であるマウス脾臓mRNAを使用した。これを、SuperScript First−Strand Synthesis for RT−PCR(Invitrogen Life Technologies、Carlsbad、CA)を使用し、基本的にキットの指示書に従って行った。IgG1については制限エンドヌクレアーゼXcmI、IgG2aについては制限エンドヌクレアーゼBsaJI、またはκ軽鎖についてはHpaIによって消化され得る二本鎖DNA領域を生成するために、IgG1については制限オリゴヌクレオチド「mCG1XcmI」、IgG2aについては制限オリゴヌクレオチド「mCG2aBsaJI」、またはκ軽鎖については制限オリゴヌクレオチド「mCKHpaI」を、第1鎖cDNAに添加した。「mCG1XcmI」配列は、5’CTAACTCCATGGTGACCCTGGGATG3’(配列番号200)である。「mCG2aBsaJI」配列は、5’CAACTGGCTCCTCGGTGACTCTAG3’(配列番号201)であり、「mCKHpaI」配列は、5’CAGTGAGCAGTTAACATCTGGAGG3’(配列番号202)である。この反応を、第1鎖cDNA、1μMオリゴヌクレオチド、および適切な量の10×NE緩衝液(New England Biolabs、Beverly MA)または10×反応緩衝液A(Roche Diagnostics)を用いてセットアップした。この試料を95℃まで2分間加熱し、次いで特異的アニーリングが起こり得るように64℃で2分間保持し、そしてXcmIおよびHpaIについては37℃まで冷却し、またBsaJIについては60℃まで冷却した。上記制限エンドヌクレアーゼXcmI、制限エンドヌクレアーゼBsaJI、または制限エンドヌクレアーゼHpaI(New England Biolabs、Beverly MA)を添加し、それぞれ、37℃で30分間、60℃で30分間、および37℃で10分間インキュベートした。上記制限酵素を、XcmIについては65℃で20分間、およびBsaJIについては80℃で20分間加熱不活性化し、次いで試料を4℃まで冷却した。
【0090】
各々の制限エンドヌクレアーゼによる上記第1鎖cDNAの消化を、当業者において公知である技術を使用し、PCR増幅によって検証した。これらの生成物は、抗体遺伝子のクローニングには使用しなかった。IgG1について5’TMX24mVHIIB短縮型内部コントロールプライマーおよび3’mCG1内部コントロールプライマーを、IgG2aについて5’TMX24mVKIIB短縮型および3’mCG2の内部コントロールプライマーを、そしてκについて5’TMX24mVKIV短縮型および3’mCK0内部コントロールプライマーを使用した反応において、消化された第1鎖cDNAの陽性増幅物を観察した。5’TMX24mVHIIB短縮型/3’mCG1プライマーもしくは5’TMX24mVHIIB短縮型/3’mCG2aプライマーまたは5’TMX24mVHIV短縮型/3’mCK0プライマーを用いた良好な増幅、および5’TMX24mVHIIB短縮型/3’mCG1Bプライマー、もしくは5’TMX24mVHIIB短縮型/3’mCG2aBプライマーもしくはTMX24mVKIV短縮型/3’mCKBプライマーを用いたわずかな増幅は、各々の制限エンドヌクレアーゼを用いた第1鎖cDNAテンプレートの首尾良い消化を示す。PCRの検査のために使用されるプライマーの配列は、以下:
【0091】
【化13】
【0092】
【化14】
である。
【0093】
(軽鎖第2鎖線形増幅およびネステッドオリゴ伸長)
フレームワーク1特異的プライマー(最終0.4μM)、dNTP、AmpliTaq酵素およびその10×反応緩衝液(Applied Biosystems、Foster City、CA)を使用して複数の第2鎖cDNA反応をセットアップするために、HpaI消化された第1鎖cDNAを、原テンプレートとして使用した。上記プライマーを、TMX24mK配列(κについて)、およびXbaI制限酵素認識部位、ならびにマウス抗体κ軽鎖遺伝子のフレームワーク1領域における第1鎖cDNAにアニーリングする領域を含むように設計した。そしてκ軽鎖遺伝子の全マウス抗体レパートリーを網羅するために、Kabatデータベース(http://immuno.bme.nwu.edu/)由来の公知のマウス抗体の配列に基づいて、これらのアニーリング配列を設計した。
【0094】
【化15】
ここで、RはAおよびGの同じ割合の混合物であり、MはAおよびCの同じ割合の混合物であり、KはGおよびTの同じ割合の混合物であり、WはAおよびTの同じ割合の混合物であり、そしてSはCおよびGの同じ割合の混合物である。
【0095】
上記試料を、94℃で1分間加熱変性し、次いで94℃で5秒間、56℃で10秒間、および68℃で2分間を介する反応を20サイクル行った。これは、第2鎖cDNAの線形増幅を可能にする。κ鎖について「TMX24mCKnoer」と命名されたネステッドオリゴヌクレオチドを、氷上で0.2μMの最終濃度となるように添加した。「TM24CKnpt」の配列は;5’GACGACCGGCTACCAAGAGGAGTGTCCGGATGTTAACTGCTCACTGGATGGTGGGAAGATGG2’OMe[A(ps)U(ps)U(ps)](プロピル)3’(配列番号220)である。上記第2鎖cDNAを、94℃で1分間の加熱変性し、68℃で2分間アニーリングおよび伸長させてネステッドオリゴからさらに伸長し、続いて4℃にした。結果生じた第2鎖cDNA(人工テンプレート)を、QIAGENスピンカラム(QIAGEN、Valencia、CA提供のPCR Purification Kit)を使用して洗浄した。この工程は、遊離のオリゴヌクレオチドを除去し、そして下流のプロトコールのための容易な緩衝液交換を可能にする。
【0096】
(軽鎖単一プライマー増幅)
Advantage 2ポリメラーゼミックス(Clontech)およびその10×反応緩衝液、dNTP、ならびにκ鎖についてのプライマー「TMX24mK」を使用し、上記人工テンプレートを増幅した。「TMX24mK」についての配列は、5’GACGACCGGCTACCAAGAGGAGTG3’(配列番号221)である。この試料を95℃で1分間加熱変性し、次いで95℃で5秒間および68℃で1分間を介する反応を25サイクルさせた。これに続いてさらに68℃で3分間処理し、そして4℃で保持した。
【0097】
(軽鎖クローニング)
κ増幅生成物をゲル精製し、次いでXbaIおよびBspEIによって消化した。そのインサートを、κ軽鎖定常領域の残余部分を含む適切な発現ベクター中にクローニングした。このライゲーションされた生成物を、エレクトロポレーションによってE.coli中に導入し、そして37℃で一晩成長させた。翌朝、軽鎖ライブラリーDNAを回収するためにDNAマキシプレップを実施した。次いで、以下の「(重鎖クローニング)」において記載されるようにライブラリーの構築を完了させるため、XhoI/BlnIによる重鎖Fdフラグメントをクローニングするための後の工程において、この軽鎖ライブラリーDNAを使用した。
【0098】
(重鎖第2鎖線形増幅およびネステッドオリゴ伸長)
フレームワーク1特異的プライマー(最終0.4μM)、dNTP、AmpliTaq酵素およびその10×反応緩衝液(Applied Biosystems、Foster City、CA)を使用して複数の第2鎖cDNA反応をセットアップするために、XcmIおよびBsaJIで消化された第1鎖cDNAを使用した。上記プライマーを、TMX24mH配列、XhoI制限酵素認識部位、およびマウス抗体重鎖遺伝子のフレームワーク1領域における第1鎖cDNAにアニーリングする領域を含むように設計した。そして重鎖遺伝子の全マウス抗体レパートリーを網羅するために、Kabatデータベース(http://immuno.bme.nwu.edu/)由来の公知のマウス抗体の配列に基づいて、これらのアニーリング配列を設計した。
【0099】
【化16】
ここで、RはAおよびGの同じ割合の混合物であり、MはAおよびCの同じ割合の混合物であり、YはCおよびTの同じ割合の混合物であり、そしてSはCおよびGの同じ割合の混合物である。
【0100】
上記試料を94℃で1分間加熱変性し、次いで94℃で5秒間、56℃で10秒間、および68℃で2分間を介する反応を20サイクル行った。これは、第2鎖cDNAの線形増幅を可能にする。IgG1について「TMX24mCG1noer」と命名されたネステッドオリゴおよびIgG2aについて「TMX24mCG2anoer」と命名されたネステッドオリゴを、氷上で0.2μMの最終濃度となるように添加した。「TMX24mCG1noer」の配列は;5’GACGTGGCCGTTGGAAGAGGAGTGCCTAGGGTTACCATGGAGTTAGTTTGGGCAGCAGA2’OMe[U(ps)C(ps)A(ps)](プロピル)3’(配列番号230)であり、そして「TMX24mCG2anoer」の配列は;5’GACGTGGCCGTTGGAAGAGGAGTGCCTAGGGTCATCGAGGAGCCAGTTGTATCTCCACA2’OMe[C(ps)A(ps)U(ps)](プロピル)3’(配列番号231)である。
【0101】
上記第2鎖cDNAを、94℃で1分間の加熱変性、68℃で2分間のアニーリングおよび伸長によって、ネステッドオリゴからさらに伸長し、続いて4℃にした。結果生じた第2鎖cDNA(人工テンプレート)を、QIAGENスピンカラム(QIAGEN、Valencia、CA提供のPCR Purification Kit)を使用して洗浄した。この工程は、遊離のオリゴヌクレオチドを除去し、そして下流のプロトコールのための容易な緩衝液交換を可能にする。
【0102】
(重鎖単一プライマー増幅(SPA))
Advantage 2ポリメラーゼミックス(Clontech)およびその10×反応緩衝液、dNTP、ならびに重鎖についてのプライマー「TMX24mH」を使用し、上記人工テンプレートを増幅した。「TMX24mH」についての配列は、5’GACGTGGCCGTTGGAAGAGGAGTG3’(配列番号232)である。この試料を95℃で1分間加熱変性し、次いで95℃で5秒間および68℃で1分間を介する反応を、IgG1については28サイクル、IgG2aについては30サイクルさせた。これに続いて68℃でさらに3分間処理し、そして4℃で保持した。
【0103】
(重鎖クローニング)
重鎖増幅生成物をゲル精製し、次いでXhoIおよびBlnIによって消化した。そのインサートを、IgG1およびIgG2aについての重鎖定常領域の残余部分を含むκ鎖ライブラリーDNA中にクローニングした。このライゲーションされた生成物を、エレクトロポレーションによってE.coli中に導入し、そして37℃で一晩成長させた。翌朝、IgG1κライブラリーDNAまたはIgG2aκライブラリーDNAを回収するために、DNAマキシプレップを実施した。
【0104】
(組み換えIgE Fc CH2〜4についてのライブラリーのパンニングおよびスクリーニング)
基本的に、Barbas III,CF、Burton,DR、Scott,JK、およびSilverman,GJ(2001)Phage Display:A Laboratory Manual、Cold Spring Harbor Laboratory Press、Cold Spring Harbor、New Yorkに記載されるように、上記ライブラリーを、組み換えIgE Fc CH2〜4について4ラウンドパンニングした。選別の第2ラウンド、第3ラウンド、および第4ラウンド由来の個々のクローンを、組み換えIgE Fc CH2〜4についてのELISAによってスクリーニングした。
【0105】
ELISAによって、IgE Fc CH2〜4に対して特異的結合を示し、かつ非特異的タンパク質(オボアルブミン)に対して最低限の結合を示したクローンを、DNA配列決定によって分析した。IgE Fc CH2〜4に対して、全部で31の異なるFabを、マウスライブラリーから単離した。図9a〜図9dを参照のこと。
【0106】
(実施例6)
(IgA抗体ライブラリーの構築)
(第1鎖cDNA合成および修飾)
ヒト末梢血リンパ球(PBL)mRNAを、オリゴdTプライマーを用いた慣習的な第1鎖cDNAを生成するために使用する。これを、SuperScript II RTcDNA Synthesis Kit(Invitrogen Life Technologies、Carlsbad、CA)を使用し、基本的にその指示書に従って行う。制限エンドヌクレアーゼBsrGIによって消化され得る二本鎖DNA領域を生成するために、第1鎖cDNAに制限オリゴヌクレオチド「CABsrGI」を添加する。「CABsrGI」の配列は、5’TCC GGG GAC CTG TAC ACC ACG AGC AG3’(配列番号279)である。この反応を、第1鎖cDNAおよび0.1μMオリゴヌクレオチドを用いてセットアップする。試料を95℃まで2分間加熱し、次いで特異的アニーリングが起こり得るように64℃で2分間保持する。適切な量の10×制限緩衝液2(New England Biolabs、Beverly MA)を上記試料中に添加し、そしてさらに37℃まで冷却した。制限エンドヌクレアーゼBsrGI(New England Biolabs、Beverly MA)を添加し、そして37℃で30分間インキュベートする。この制限酵素を80℃で20分間加熱不活性化し、次いで試料を4℃まで冷却する。
【0107】
(第2鎖cDNA合成およびネステッドオリゴヌクレオチド伸長反応(NOER))
フレームワーク1特異的プライマー(最終0.4μM)、dNTP、AmpliTaq酵素およびその10×反応緩衝液(Applied Biosystems、Foster City、CA)を使用して複数の第2鎖cDNA反応をセットアップするために、BsrGI消化された第1鎖cDNAを使用する。以下の表A中に列挙されるプライマーは、TMX24配列、XhoI制限酵素認識部位、およびヒト抗体重鎖遺伝子のフレームワーク1領域における第1鎖cDNAにアニーリングする領域を含むように設計された。これらのアニーリング配列は、公知のヒト抗体の配列に基づいて設計され、そして重鎖鎖遺伝子の全長ヒト抗体レパートリーを網羅すると報告されているVbaseデータベースプライマー由来である。
【0108】
【化17】
【0109】
【化18】
上記配列の各々において、RはAまたはGであり、KはGまたはTであり、そしてSはCまたはGである。
【0110】
上記試料を、94℃で1分間加熱変性し、次いで94℃で5秒間、56℃で10秒間、および68℃で2分間を介する反応を20サイクル行う。これは、第2鎖cDNAの線形増幅を可能にする。次いで、伸長オリゴヌクレオチド「TMX24CAnpt」を、氷上で0.2μMの最終濃度となるように添加する。「TMX24CAnpt」の配列は、5’GTG CTG GCC GTT GGA AGA GGA GTG CCT GTA CAG GTC CCC GGA GGC ATC CTC3’(配列番号294)であって、ここで、RはAまたはGである。オリゴ伸長を防ぐために、3つの3’末端ヌクレオチドを修飾する。94℃で1分間の加熱変性し、68℃で2分間での伸長において上記第2鎖cDNAをネステッドオリゴからさらに伸長し、続いて4℃にする。次いで、この第2鎖cDNAを、QIAGENスピンカラム(QIAGEN、Valencia、CA提供のPCR Purification Kit)を使用して洗浄する。この工程は、オリゴヌクレオチドを除去し、そして下流のプロトコールのための容易な緩衝液交換を可能にする。
【0111】
(単一プライマーPCR増幅)
Advantage 2ポリメラーゼミックス(Clontech、Palo Alto、CA)およびその10×反応緩衝液、dNTP、ならびにプライマー「TMX24」を使用し、上記第2鎖cDNAの増幅を実施する。「TMX24」の配列は、5’GTG CTG GCC GTT GGA AGA GGA GTG3’(配列番号295)である。この試料を95℃で1分間加熱変性し、次いで95℃で5秒間および68℃で1分間を介する反応を30サイクルさせる。これに続いて68℃でさらに3分間処理し、そして4℃で保持する。
【0112】
(クローニングおよび配列決定)
約560bpのPCR生成物を、PCR purification kit (QIAGEN、Valencia、CA)を使用して精製し、XhoIおよびBsrGIによって消化し、ゲル精製し、そしてIgA CH1定常領域の残余部を有する適切なFab発現ベクター中にクローニングする。
【0113】
種々の改変が、本明細書中に記載される実施形態を構成し得ると考えられる。したがって、上記は、好ましい実施形態の限定として解釈されるべきではなく、単に、好ましい実施形態の例示として解釈されるべきである。当業者は、本開示の範囲および精神の範囲内で他の改変を構想する。
【特許請求の範囲】
【請求項1】
本明細書に記載の方法。
【請求項1】
本明細書に記載の方法。
【図1】

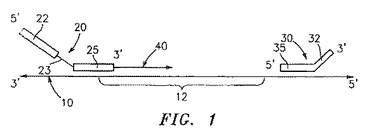
【図2A】
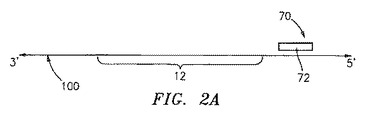
【図2B】
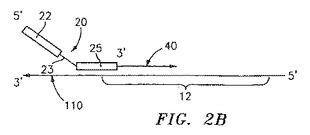
【図3】

【図4】
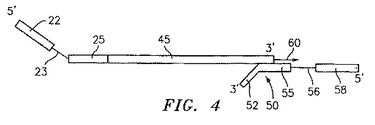
【図5】
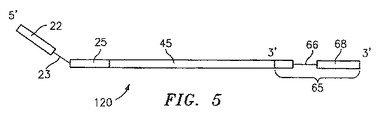
【図6】
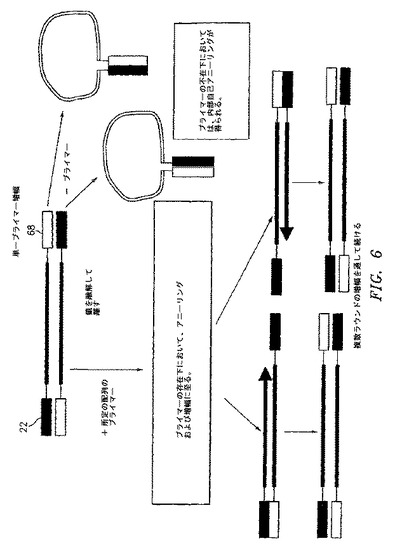
【図7】
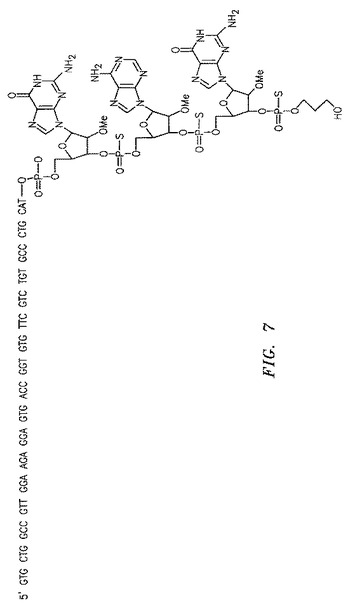
【図8a】

【図8b】
【図8c】
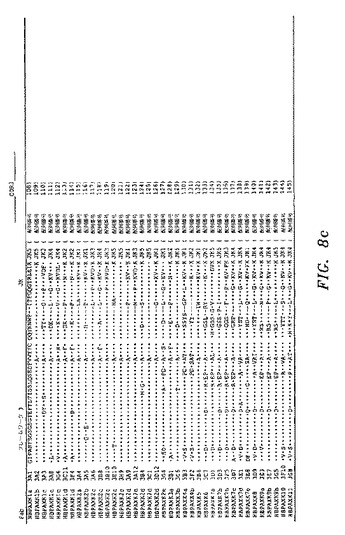
【図8d】
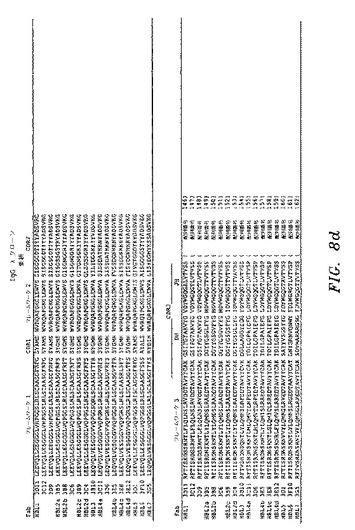
【図8e】
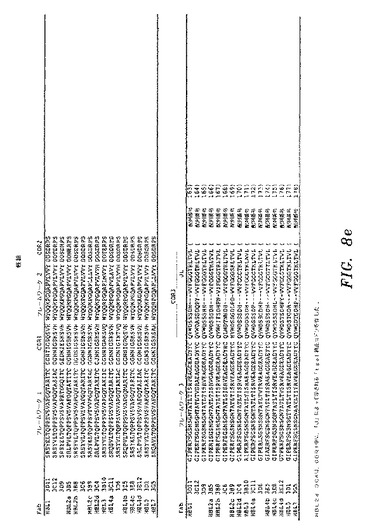
【図9a】
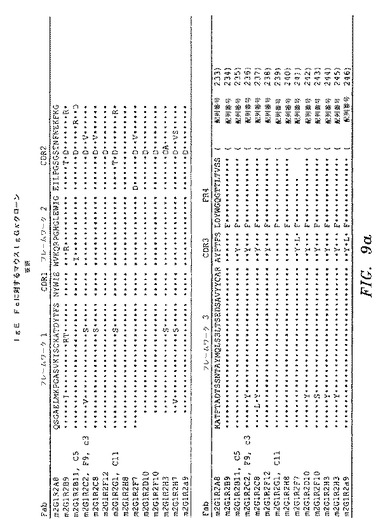
【図9b】
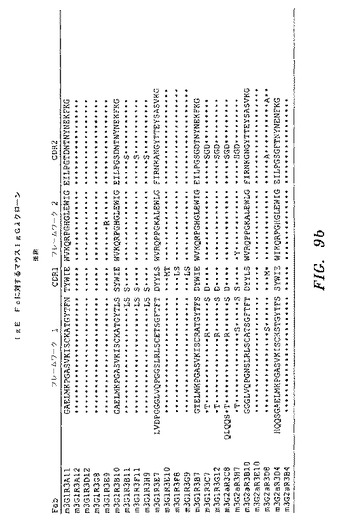
【図9c】
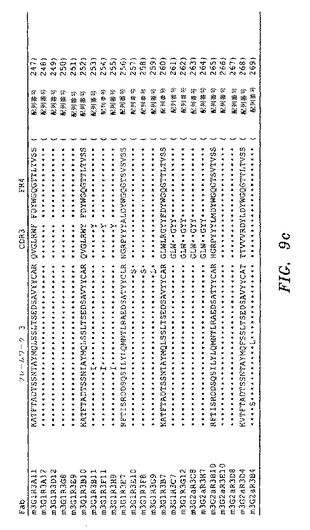
【図9d】

【図2A】
【図2B】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8a】
【図8b】
【図8c】
【図8d】
【図8e】
【図9a】
【図9b】
【図9c】
【図9d】
【公開番号】特開2012−157371(P2012−157371A)
【公開日】平成24年8月23日(2012.8.23)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−122101(P2012−122101)
【出願日】平成24年5月29日(2012.5.29)
【分割の表示】特願2006−545814(P2006−545814)の分割
【原出願日】平成16年12月15日(2004.12.15)
【出願人】(503102674)アレクシオン ファーマシューティカルズ, インコーポレイテッド (51)
【Fターム(参考)】
【公開日】平成24年8月23日(2012.8.23)
【国際特許分類】
【出願番号】特願2012−122101(P2012−122101)
【出願日】平成24年5月29日(2012.5.29)
【分割の表示】特願2006−545814(P2006−545814)の分割
【原出願日】平成16年12月15日(2004.12.15)
【出願人】(503102674)アレクシオン ファーマシューティカルズ, インコーポレイテッド (51)
【Fターム(参考)】
[ Back to top ]