
代謝安定性鎮痛薬、疼痛薬物療法及び他の物質の合成
【課題】中毒症状又は他の疾患もしくは障害の治療的介入に有用な新規化合物および該化合物を含有する医薬組成物の提供。
【解決手段】下記一般式で表される化合物および該化合物を含有する医薬組成物。

【解決手段】下記一般式で表される化合物および該化合物を含有する医薬組成物。
【発明の詳細な説明】
【技術分野】
【0001】
(関連出願の相互参照)
本出願は、2004年5月28日に出願された、米国特許仮出願第60/575,451号の優先権を主張するものであり、その開示全体は本願明細書に参照として組入れられている。
(発明の技術分野)
本発明は、鎮痛薬受容体と相互作用する医薬物質、これらの物質を調製する方法、並びに痛覚喪失、疼痛、中毒症状及び他の状態に対するそれらの使用に関する。
【背景技術】
【0002】
(発明の背景)
モルヒネ及び他のオピエートは、強力な鎮痛薬として作用する[Foye,
W.O.の著書、「Principles of Medicinal Chemistry」第3版、Lea
& Febiger、フィラデルフィア、1989]。末期患者のため及び癌の疼痛緩和のために麻薬性鎮痛薬の適切な使用を開発及び理解するために、かなりの努力が払われているが、新規薬物療法が非常に必要とされている。モルヒネは、痛覚喪失、呼吸抑制、及び胃腸管輸送の阻害を含む、μオピオイド受容体により媒介された、多くの薬理学的活性を誘起する。[0.
Rayの著書、「Drugs, Society and Human Behavior」第3版、The
C.V. Mosby Co.、セントルイス (1983)を参照されたい]。しかし有害な副作用及び乱用の可能性が、モルヒネの利用可能性及び最適な使用を制限している。更に溶解度及び効能の問題点が、注射可能なモルヒネ量を制限している。重度の疼痛のための長寿命物質の必要性が存在する。高価な静脈内又は硬膜外の投与経路により投与されることが不要である物質の必要性も存在する。呼吸抑制、耐性、尿貯留、便秘、身体的依存、及び/又は中毒を引き起こさない薬物療法も必要である。加えて、存在する疼痛状態の一部は、現在利用可能なオピエートの鎮痛作用に対し抵抗がある。末梢にのみ作用し脳に侵入しない有効な鎮痛薬必要性も存在する。
【0003】
耐性は、オピエート作用に対する低下した感受性と定義され、一般に離脱症状の身体的症状発現により明らかにされる依存性を引き起こす、鎮痛効能の減弱化を示す。[B.
L. Kiefferらの論文、Cell, 108:87-90 (2002)を参照されたい]。耐性は、ほぼ全て痛覚喪失に関連している。耐性は、表面受容体及びオピオイド受容体シグナル伝達の低下により引き起こされると長く考えられてきた。しかし、モルヒネは、効率的なμ受容体内在化を促進しない[J.
L. Whistlerらの論文、Neuron, 23:737-46 (1999)を参照されたい]のに対し、μ-選択的ペプチドであるDAMGO及びアルカロイドフェンタニルなどの他のオピオイドは、そのような内在化を促進する。インビトロモデルは、内在化を誘導する閾値以下の濃度で投与されたDAMGOは、モルヒネの存在下で、μ受容体の内在化を誘導することができることを示している[L.
Heらの論文、Cell, 108:271-82 (2002)を参照されたい]。モルヒネの連続投与後の痛覚喪失は、内在化投与量を下回るDAMGOがラットへ同時投与される場合に、著しく増強される。[L.
Heらの論文、前掲参照]。細胞レベルでのμ受容体内在化とインビボ耐性の間のこの連関は、受容体内在化は、耐性に対する保護を提供し得ることを示唆している。従ってM6Gのどのアナログが受容体内在化に作用するかの決定は、非-中毒性鎮痛薬の開発及び中毒プロセスの我々の理解を深める上での重要な工程である。μ、δ、又はκ受容体のアンタゴニストの開発も、有用な薬物療法を生じる。これらのM6Gアナログ中のN-17メチル基の、他のアルキル、シクロアルキル及びアルケニル基との交換は、オピオイド受容体に薬理学的活性のあるアンタゴニストを提供することができる。このようなアンタゴニストは、薬物中毒、ギャンブル依存症、及びアルコール依存症を含むCNS疾患の治療において有用であろう。一定に帯電し及び4級アミン又はグアナジノ基を含む誘導体の作製も、末梢にのみ作用する鎮痛薬の新規セットを提供する。
【0004】
モルヒネ及び関連したオピエートを体から排除する主要経路は、肝臓における水溶性グルクロニド複合体の形成、それに続く尿中排泄である。モルヒネの場合、3種のグルクロニドが形成される:モルヒネ-6-β-D-グルクロニド、モルヒネ-3-β-D-グルクロニド及びモルヒネ-3,6-ジ-β-D-グルクロニド。モルヒネ-6-β-D-グルクロニド(M6G)は、モルヒネ自身よりも100倍大きい効能を伴う鎮痛薬である。[G. W. Pasternakの論文、Life
Sci., 41:2845-2849 (1987)を参照されたい]。胃酸による消化管内での加水分解に起因したM6Gの低い生体利用効率(11%)は、この化合物からの薬物の開発を著しく制限する。[R. T. Pensonら、Br. J; Clin. Pharmacol., 53:347-354
(2002)を参照されたい]。M6Gをベースにした新規薬物療法の開発は、その鎮痛効能、好ましい副作用プロファイル、及び際だった薬理学的活性のために有望である。[M.H.
Hannaらの論文、Anesthesiology, 102:815-821 (2005)を参照されたい]。
【0005】
概してグルクロニドは、高度に極性のある代謝産物であり、及び血液脳関門(BBB)を通過することができないと考えられている。[G.
W. Pasternakの論文、Clin. Neuropharmacol.,
16:1-18 (1993)を参照されたい]。しかしM6Gは、明らかに予想されるよりもはるかに親油性である。[P. A. Carruptらの論文、J.
Med. Chem., 34:1272-1275 (1991)を参照されたい]。極性表面積(PSA)の計算は、90Å2を上回る分子は、脳に移行しないことを示唆している。[K.
Palmらの論文、J. Pharm. Sci., 85:32-39 (1996a)を参照されたい]。経細胞経路によるBBB通過に関して、分子は、MW<450及びPSA<90Å2を有さねばならない。関連した計算は、経口吸収には、PSA<120Å2が最適であることを示している。[J. Kelderらの論文、Pharmaceutical
Research, 16:1514-1519 (1999)を参照されたい]。ラットにおいて、M6Gは経口投与後に、近位小腸でそれ自身吸収され、M6Gは膜透過可能であることを示している。[R.
Stain-Texierらの論文、Drug
Metab.Dispos., 26:383-387, (1998)を参照されたい]。しかし分子量が450よりも大きいM6Gのアナログは、脳に移行しないと予想され、並びにモルヒネと比べより極性のある性質のために、より好ましいCNS生体分布及び作用特性の開始を有さないと他のものにより予想される。
【発明の開示】
【発明が解決しようとする課題】
【0006】
(発明の簡単な概要)
1つの局面において、本発明は、鎮痛薬受容体に対する薬理学的活性を有する鎮痛薬-関連物質を提供する。典型的態様において、これらの物質は、任意に置換されたアリール、任意に置換されたヘテロアリール、もしくは糖類に連結された鎮痛薬又はその誘導体を含む。好ましい態様において、鎮痛薬又はその誘導体は、オピエート又はオピエート誘導体である。鎮痛薬-関連物質は、その物質が特異的に結合する対応する鎮痛薬受容体のアゴニスト又はアンタゴニストであり得る。
【課題を解決するための手段】
【0007】
本発明の物質は、例えば対象における疼痛の緩和について、又はそのような治療もしくは予防が鎮痛薬受容体の変調(例えば、阻害又は刺激)により影響されやすい疾患又は障害の治療もしくは予防について、有用である。従って別の局面において、本発明は、本願明細書に記した鎮痛薬-関連物質を含有する医薬組成物を提供する。同じく、対象へ有効量の鎮痛薬-関連物質を投与することによる、疼痛の軽減、又は鎮痛薬受容体活性の変調によるそのような治療、もしくは予防に影響されやすい疾患もしくは障害の治療もしくは予防のための方法を提供する。本願明細書に提供された組成物及び方法による治療的介入の影響を受けやすい疾患又は障害は、例えば、鎮痛薬受容体により少なくとも一部、直接又は間接に媒介される中毒又は他のCNS障害を含む。例えば疼痛の治療のためのような、ある種の医薬組成物又は方法の変動において、鎮痛薬-関連物質は、鎮痛薬受容体アゴニストである。別の態様において、例えば薬物中毒、ギャンブル依存症、又はアルコール依存症の治療などのためには、物質は典型的には鎮痛薬受容体アンタゴニストである。
【0008】
脳の鎮痛薬受容体に著しい作用を伴うことなく、末梢において鎮痛薬受容体を変調するために、この物質は、典型的には生理的pHで一定の電荷を有する。このような物質は、例えば、4級アミン、クアニジン(quanidine)などを含むことができ、特に例えば末梢鎮痛薬としての使用に適している。
【0009】
(定義)
特に別に記載しない限りは、本願明細書において使用される全ての技術用語及び科学用語は、本発明が属する技術分野の業者に通常理解されるものと同じ意味を有する。本願明細書に記載されたものに類似したあらゆる方法及び材料を、本発明の実践又は試験に使用することができるが、方法及び材料の例のみが説明されている。本発明の目的のために、以下の用語は、下記のように定義される。
【0010】
用語「1つの(a,an及びthe)」は、本文が別に明確に指摘しない限りは、複数の意味を含む。
用語「アルキル」は、直鎖、分枝鎖、及び環状の基を含む、飽和した脂肪族基を意味し、これらは全て任意に置換されてよい。適当なアルキル基は、メチル、エチルなどを含み、任意に置換されてよい。
用語「アルケニル」は、少なくとも1個の炭素-炭素二重結合を含み、並びに直鎖、分枝鎖、及び環状の基を含む不飽和基を意味し、これらは全て任意に置換されてよい。
【0011】
用語「アルキニル」は、少なくとも1個の炭素-炭素三重結合を含み、並びに直鎖、分枝鎖、及び環状の基を含む不飽和基を意味し、これらは全て任意に置換されてよい。適当なアルキニル基は、任意に置換されてよい、エチニル、プロピニル、ブチニルなどを含む。
用語「アリール」は、共役「π」電子系を有する少なくとも1個の環を有する芳香族基を意味し、炭素環式アリール、二環式アリールを含み、両方とも任意に置換されてよい。
【0012】
用語「アラルキル」は、アリール基で置換されたアルキル基を意味する。適当なアラルキル基は、ベンジルなどを含み、任意に置換されてよい。
用語「ヘテロアリール」は、1、2、3又は4個のO、N、P又はS原子を含み、及び1個又は1個よりも多い環において局在化されない6、10又は14個のπ電子を有する、炭素含有する5-14員の環式不飽和ラジカルを意味する。
【0013】
用語「医薬として許容し得る誘導体」は、例えば医薬として許容し得るエステル、アミド又は塩を含む、薬理学的使用に適している、本願明細書に説明されたような化合物のいずれかの誘導体を意味する。医薬として許容し得るエステル、アミド又は塩は、本発明の化合物及び有機酸又は無機酸の組合せに由来する。
用語「鎮痛薬」は、痛覚喪失を生じること、すなわち麻酔状態又は意識喪失を生じることなく、侵害刺激の認知を変更することにより、疼痛を軽減又は阻害することが可能な化合物を意味する。本発明の好ましい態様において、鎮痛薬はオピエートである。
【0014】
用語「オピエート」は、アヘン又はその任意の誘導体(例えばモルヒネ)、更には合成又は半-合成の麻薬を含む、オピオイド受容体に特異的に結合することが可能な、天然又は合成の物質を意味する。
用語「鎮痛薬-関連物質」は、鎮痛薬の代謝産物、アナログ又は誘導体を意味する。用語「オピエート-関連物質」は、更に本願明細書に説明されたような、オピエートの代謝産物、アナログ、又は誘導体を意味する。本発明に従い、オピエート-関連物質を含む、鎮痛薬-関連物質は、この物質が特異的に結合する1種又は複数の鎮痛薬受容体に関するアゴニスト又はアンタゴニスト活性を有することができる。
【0015】
用語「阻害」は、測定可能な量だけ低下するか、又は全体を妨害することを意味する。
語句「効率的な受容体内在化の促進」は、物質が、その物質が特異的に結合する受容体の細胞内在化を促進することが可能であり、その物質に接触していない対照細胞と比較して、FACSによる対応する受容体の表面染色が80%を超えない、典型的には70%又は60%を超えない、より典型的には50%又は40%を超えないことを意味する。
【0016】
疾患又は障害の「治療する」、「治療」又は「療法」は、本願明細書に説明されたような、本発明の組成物及び方法を使用する、臨床症状又は診断症状のいずれかの軽減又は消失により証明されるような、疾患又は障害の進行の遅延、停止、又は逆行を意味する。
疾患又は障害の「予防する」、「予防」又は「妨害」は、疾患もしくは障害又はその症状の一部もしくは全ての発生又は開始を妨害することを意味する。
【0017】
本願明細書に使用される「中毒」は、実質的に自主規制を超えた物質又は慣習への習慣性の精神的及び生理的依存を特徴とする疾患又は障害を意味する。本願明細書に説明された化合物及び方法を使用する治療の影響を受ける中毒は、物質中毒、例えば、麻薬(例えばモルヒネ、ヘロイン)、アルコール、及びニコチンへの中毒に加え、行動的中毒、例えばギャンブルへの依存症などを含む。
【0018】
本願明細書において使用される用語「対象」は、本発明の組成物が、本願明細書に説明された方法に従い投与される哺乳類患畜を意味する。本発明の方法を使用する治療又は予防が特異的に意図された対象は、ヒトを含む。
用語「治療的に有効な投与計画」は、医薬組成物又はそれらの組合せが、疼痛を軽減するか、又は鎮痛薬受容体の変調による治療に影響を受ける疾患もしくは障害の少なくとも1種の症状もしくは生化学マーカーの顕在化を少なくとも検出可能に妨害、遅延、阻害又は逆行させるのに、十分な量及び回数及び適当な経路で投与されることを意味する。
【0019】
用語「治療的有効量」は、適切な投与計画で投与された場合に、例えば、疼痛の軽減、又は鎮痛薬受容体の変調による治療の影響を受ける疾患もしくは障害の症状もしくは生化学マーカーを妨害、遅延、阻害もしくは逆行するなど、所望の結果を実現するように存在する、本発明の物質又は本発明の物質の他の物質(複数)との組合せの量を意味する。
【発明を実施するための最良の形態】
【0020】
(詳細な説明)
本発明は、鎮痛薬及び鎮痛薬誘導体、特にオピエート-関連化合物に関する。これらの化合物の構造、合成、及び生物学的評価に加え、これらの化合物を含有する医薬組成物並びに例えば疼痛及び物質中毒の治療のためなどのそれらの薬理学的使用が、本願明細書に説明されている。ある態様において、本願明細書に説明された化合物は、モルヒネ代謝産物M6Gに関連している。予想外のことに、M6Gに対する無関係のアナログは、いくつかのインビトロ及びインビボ機能試験において、M6Gよりもより好ましい薬理学的特性を有した。
【0021】
鎮痛薬受容体シグナル伝達及び物質中毒の様々な要素のインビトロ薬理学的及び代謝的試験及び動物モデルは、疼痛及び物質中毒の治療において、ヒトモデルの様々な要素において有効であるであろう薬物療法の開発を予測することができる。それらの両方が最終的な薬物療法開発を促進するための情報を整備するインビトロ及びインビボ要素からの動的医化学フィードバックを用い、例えば、疼痛及び/又は中毒の治療に有用な化合物が同定されている。中毒サイクルの1種又は複数の成分中のこれらの化合物の薬理学的活性は、とりわけ、中毒サイクルの複数の要素が投与された薬理学的物質により対象とされるような併用療法の基本を提供する。
【0022】
本発明の態様を説明する目的で、本願明細書に説明された化学式は、塩、エステル、及びアミドを含む、各化合物の医薬として許容し得る誘導体、更にはラセミ体又は立体化学的に純粋な化合物を含む、置換基の全ての可能性のある立体化学的配置を含むことが理解される。
【0023】
(化合物)
本発明の1つの局面において、鎮痛薬受容体に対する薬理学的活性を有し、並びに例えば疼痛又はそのような受容体の変調を介して治療の影響を受ける、中毒を含む疾患又は障害の治療に有用である化合物が、提供される。本発明の化合物は一般に、下記式Iを有し:
【化1】
【0024】
(式中、Aは、鎮痛薬又はその誘導体であり;Bは、連結基であり、及びCは、任意に置換されたアリール、任意に置換されたヘテロアリール及び糖類からなる群から選択される);及び、それらの全ての医薬として許容し得る誘導体である。好ましい変形において、鎮痛薬Aは、オピエートである。本願明細書に説明された本発明による使用に特に適したオピエートは、例えばナルトレキソン、フェニルピペリジン、ピペリジノール、プロジン、ピペリジルプロピオンアニリド、イソプロジン、プロジリジン、ベンゾモルファン、モルファン、アザビシクラン(azabicyclane)、モルヒナン、プロジン、ジフェニルアミノエチルプロピオネート、メサドン、イソメサドン、プロポキシフェン、デキストロメトロファン、ベンザゾシン-8-オール、ノルビナルトロフィン、ナルトリンドール及びグアニジンナルトリンドールからなる群から選択される。連結基Bは、例えば-S-又は-NH(CO)(CH2)n-であり、ここで添字nは、0〜5の整数である。
【0025】
ある態様において、Cは、ハロゲン、(C1-C5)アルコキシ、ニトロ、又はCO2Rで任意に置換された、アリール又はヘテロアリールであり、ここでRは、H又は(Cl-C5)アルキルである。特定の態様において、Cは、例えば、フェニル、チオフェン、又は糖類である。特に適した糖類は、下記式IIを有するものである:
【化2】
【0026】
(式中、各R8は独立して、H、(C1-C5)アルキルC(O)-、(C7-C10)アラルキルC(O)-、(C7-C10)アラルキル、(C1-C5)アルキル、(C6-C12)アリール及び(C6-C12)アリール(CO)-からなる群から独立して選択され;R9は、CH2OH、CH2O(C1-C5)アルキル、CH2O2C(C1-C5)アルキル、CH2O(C7-C10)アラルキル、CH2O(C6-C12)アリール、CO2H、CO2(C1-C5)アルキル、CO2(C6-C12)アリール、CO2(C7-C10)アラルキルであり;並びに、波線は、分子の残余に結合する点を示す。)。
【0027】
1つの適当な変形において、化合物は、下記式IIIを有する:
【化3】
【0028】
(式中、破線は、単結合、二重結合又は規格化結合(normalized bond)を示し;R1は、H、(C1-C5)アルキルC(O)-、(C7-C10)アラルキルC(O)-、(C7-C10)アラルキル、(C1-C5)アルキル、(C6-C12)アリール、(C6-C12)アリール(CO)-及びシリルであり;R2は、ハロゲン、(C1-C5)アルコキシ、ニトロ及びCO2Rからなる群から選択される置換基により任意に置換された、(C1-C5)アルキル、(C3-C6)シクロアルキル、(C3-C6)シクロアルキル-アルキル、(C5-C7)シクロアルケニル-アルキル、(C6-C12)アリール、(C7-C12)アラルキル、(C6-C12)ヘテロアリール、(C7-C12)ヘテロアラルキル、(C2-C5)アルケニル、(C2-C5)アルキニルであり、ここでRは、H又は(C1-C5)アルキルであり;R3は、H又はOHであり;R4は、Hであり;R5は、Hであるか、又はR6と一緒に、YR7で置換されたヘテロアリール基を形成し;R6は、YR7であるか、又はR5と一緒に、YR7で置換されたヘテロアリール基を形成し;Yは、-S-又は-NH(CO)(CH2)n-であり、ここで添字nは、0〜5の整数であり;並びに、R7は、任意に置換されたアリール、任意に置換されたヘテロアリール又は糖類であり、ここで糖類は、先に説明された式IIを有する。特に適当なR2基は、例えばシクロプロピルメチル、シクロブチルメチル、及びアリルを含む。
【0029】
いくつかの代わりの態様において、化合物は、下記式IVa又はIVbを有する:
【化4】
【0030】
(式中、R1は、H、(C1-C5)アルキルC(O)-、(C7-C10)アラリル、又は(C1-C5)アルキルであり;R2は、(C1-C5)アルキル、C3-C6(シクロアルキル)アルキル、C5-C7(シクロアルケニル)アルキル、C6-C12アリール(C7-C12)アラルキル、トランス(C4-C5)アルケニル、アリル、又はフラン-2-イルアルキルであり;各R8は、独立して、H、(C1-C5)アルキルC(O)、(C7-C10)アラリルC(O)、(C7-C10)アラリル、又は(C1-C5)アルキルであり;並びに、R9は、CH2OH、CH2O2C(C1-C5)アルキル、CH2O(C7-C10)アラリル、CO2H、CO2(C1-C5)アルキル、又はCO2(C7-C10)アラルキルである。)。
【0031】
更に別の変形において、化合物は、下記式Va又はVbを有する:
【化5】
【0032】
(式中、R1は、H、(C1-C5)アルキルC(O)-、(C7-C10)アラリル、又は(C1-C5)アルキルであり;R2は、(C1-C5)アルキル、C3-C6(シクロアルキル)アルキル、C5-C7(シクロアルケニル)アルキル、(C6-C12)アリール(C7-C12)アラルキル、トランス(C4-C5)アルケニル、アリル、又はフラン-2-イルアルキルであり;R8は、独立してH、(C1-C5)アルキルC(O)、(C7-C10)アラリルC(O)、(C7-C10)アラリル、又は(C1-C5)アルキルであり;並びに、R9は、CH2OH、CH2O2C(C1-C5)アルキル、CH2O(C7-C10)アラリル、CO2H、CO2(C1-C5)アルキル、又はCO2(C7-C10)アラルキルであり;及び、添字nは、0〜5の整数である。)。
【0033】
更に別の態様において、化合物は、下記式VIa又はVIbを有する:
【化6】
【0034】
(式中、R1は、H、(Cl-C5)アルキルC(O)-、(C7-C10)アラリル、又は(Cl-C5)アルキルであり;R2は、(Cl-C5)アルキル、C3-C6(シクロアルキル)アルキル、C5-C7(シクロアルケニル)アルキル、(C6-C12)アリール(C7-Cl2)アラルキル、トランス(C4-C5)アルケニル、アリル、又はフラン-2-イルアルキルであり;及び、添字nは、0〜5の整数である。)。
【0035】
特定の態様において、本発明の化合物は、下記式VIIからXXXのいずれか1つを有する:
【化7】
【0036】
(他の態様)
ある態様において、本発明の化合物は、モルヒネ、ジヒドロモルヒネ-6-グルクロニド、コデイン、及び下記式IVを有するジヒドロコデイン-6-グルクロニドスルファーアナログである:
【化8】
【0037】
(式中、Aは、OR、OH、又はOC(0)Rであり;Bは、OC(0)R、OH、又はORであり;Cは、CO2R、CO2H、CH2OR、又はCH2OHであり;Dは、アルキル、シクロアルキル、アルケニル、アルキニル、任意に置換されたアリール、又は任意に置換されたアラルキルであり;並びに、置換基A、B、C、DのRは、アルキル又はアリール基である。)。
【0038】
別の態様において、本発明の化合物は、コデイン、ジヒドロコデイン、モルヒネ、及び下記式Vを有するジヒドロモルヒネアナログを含む:
【化9】
【0039】
(式中、Aは、RO、OH、又はOC(0)Rであり;Bは、OC(0)R、OH、又はORであり;Cは、CO2R、CO2H、CH2OR、又はCH2OHであり;Dは、アルキル、シクロアルキル、アルケニル、アルキニル、任意に置換されたアリール、又は任意に置換されたアラルキルであり;並びに、置換基A、B、C、DのRは、アルキル又はアリール基である。)。
【0040】
更に別の変形において、本発明の化合物は、モルヒネ、ジヒドロモルヒネ、コデイン、及び下記式VIのジヒドロコデインアミドを含む:
【化10】
【0041】
(式中、Aは、RO、OH、又はOC(O)であり、ここでnは0〜5の整数であり;アリールは、フェニル、3,4-ジクロロフェニル、3-メトキシフェニル、4-メトキシフェニル、3-ニトロフェニル、3-カルボメトキシフェニル、3-カルボキシフェニル又はチオフェン-2-イル、又は任意に置換されたアリールもしくはヘテロアリール誘導体であり;Dは、アルキル、シクロアルキル、アルケニル、アルキニル、任意に置換されたアリール、又は任意に置換されたアラルキルであり;並びに、置換基A及びDのRは、アルキル又はアリール基である。)。
【0042】
式IVの化合物は、ジメチルホルムアミド中でのモルヒネの6βトシラート、3-0-アセチルモルヒネ及びコデイン1a-cの、チオ糖ナトリウム塩2a-bのとのSN2反応により合成し(図1参照)、3a-fを生成することができる。出発トシラートの調製については、[L.
H. Welshの論文, J. Org. Chem., 19:1409 (1954);G.
Stork 及び F. Clarkeの論文,J. Am.Chem.Soc.,
78:4619-4624 (1956)を参照されたい]。引き続きエステル保護基は、水酸化ナトリウム水溶液の中での一晩の攪拌により除去され、モルヒネ、コデイン及びそれらの同族のS-β-D-グルクロニド又はS-β-D-グルコース複合体である追加の化合物4a-dを生じる。これらの調製において使用されるチオ糖2a-bは、対応するグリコシルブロミドの、チオ酢酸カリウムとの反応、それに続くチオメトキシドナトリウムによる選択的S-脱アセチル化により作製された(図2参照)。出発グリコシルブロミドの調製については、[G.
N. Bollenbackらの論文、J. Org Chem., 77:3310-3315 (1955)を参照されたい]。関連したS-脱アセチル化については、[Wallaceらの論文、
Tetrahedron Lett., 2693 2694 (1998)を参照されたい]。この合成法は、C-6位置で、イオウ原子を介した、糖類のモルヒネ、コデイン及びそれらのN-17誘導体への結合に関する、一般的合成経路を構成しており、これらの化合物のジヒドロモルヒネ及びジヒドロコデイン誘導体の調製に適用可能である。加えて、モルヒネ、コデイン及び誘導体の6β-トシラートを利用し、前述の全ての化合物のC-6α誘導体の調製が可能になる。
【0043】
式Vの化合物は、モルヒネ、ジヒドロモルヒネ、コデイン、ジヒドロコデイン並びにそれらのN-アルキル及びシクロアルキル誘導体6の6β-フタルイミドから合成することができる(図3参照)。[S.
Makleitらの論文、 Synthetic Commun.,
21:407-412 (1991)を参照されたい]。モルヒネシリーズの化合物に関して、フェノール系ヒドロキシル基は、室温のジメチルホルムアミド中での、tert-ブチルジメチルシリルクロリド又はトリイソプロピルシリルクロリド及びイミダゾールとの反応により、シリルエーテルとして保護される。その後フタルイミド基は、EtOH中でのヒドラジン水和物との加熱により除去され、アミン7を生じる。アミン7は、C-グリコシド8の酸塩化物と反応し、アミド9を生じる。9のR1がシリルエーテルである場合、これは、TBAF処理により除去することができ、フェノール10を生じる。9又は10中の糖保護基は、NaOH水溶液による処理によるか、又は水素化により除去され、化合物11を生じる。これらの合成において使用されるC-グリコシド8は、公知の方法により合成される。[M.
Lewisらの論文、J. Am. Chem. Soc., 104:4976-4978 (1982)]。この合成配列は、C-6位置での、アミド橋を介した、糖類のモルヒネ及びそれらのN-17誘導体への結合の一般的経路を提供し、これらの化合物のジヒドロモルヒネ及びジヒドロコデイン誘導体の調製に適用可能である。
【0044】
式VIの化合物は、式Vの化合物の調製に使用された6β-アミノモルヒネ7cから合成することができる(図4参照)。アミン7cは、アリール及びヘテロアリール酸塩化物で処理され、アミド12a-hを生じる。水性THF中における12a-hのTBAFによる処理は、6β-アミドモルヒネ13a-hを提供する。メチルエステル13eの水酸化ナトリウム水溶液による処理は、カルボン酸14を提供する。この合成配列は、C-6位でアミド橋を介したアリール及びヘテロアリール置換基のモルヒネ、コデイン及びそれらのN-17アルキル誘導体への結合の一般的経路を提供し、これらの化合物のジヒドロモルヒネ及びジヒドロコデイン誘導体の調製に適用可能である。この合成法は、アミン7cのC-6エピマーで出発することにより、前記化合物の全てのC-6a誘導体の調製も提供する。本願明細書に説明された化合物に関する橋頭NのN-メチル置換基の、シクロプロピルメチル又はシクロブチルメチル又は同様の置換基との交換は、典型的には、以下に説明された活性に関する受容体アンタゴニストを供する。このようなアンタゴニストは、薬物中毒、ギャンブル依存症、アルコール依存症、又は他のCNS疾患を拮抗するのに有用である。更に本願明細書に説明された、合成法(代謝が安定した糖類又は他の化学ホモログ又は等配性(isosteric)同等物の結合)の、ナルトレキソン、フェニルピペリジン、ピペリジノール、プロジン、ピペリジンプロピオンアニリド、イソプロジン、プロジリジン、ベンゾモルファン、モルファン、アザビシクラン、モルヒナン、プロジン、ジフェニルアミノエチルプロピオネート、メサドン、イソメサドン、プロポキシフェン、デキストロメトロファン、ベンザゾシン-8-オール、及び全ての関連した立体異性体及びN-置換された誘導体(シクロプロピルメチル、シクロブチルメチル、アリルなど)を含む他の鎮痛薬への適用が、疼痛緩和に加え、中毒及び他の疾患の治療に有用な、薬理学的活性のあるCNS系物質につながるであろう。
【0045】
更に別の変形において、本発明の鎮痛薬-関連物質は、生理的pHで一定電荷(すなわち、その時の100%か、又は実質的に100%イオン化される)を有する。このような変形は、例えば、芳香族基又は糖基からの4級アミン又はグアニジノ(quanidino)部分の除去により、合成することができる。このような修飾は典型的には、脳に移行せず、従って末梢適応に有用である誘導体(例えば脳受容体への著しい作用を伴わずに末梢において疼痛を緩和する、末梢鎮痛薬)を生じる。
【0046】
(医薬組成物及び投与法)
本発明の鎮痛薬-関連物質は、神経系の鎮痛薬受容体シグナル伝達の変調に関連している様々な適用において有用である。例えば本発明の物質は、疼痛(例えば、外科的手技又は癌のような末期疾患に関連した疼痛の緩和)の治療のための鎮痛薬として有用である。これらの物質は、特に例えばCNS疾患又は障害の、鎮痛薬(例えばオピオイド)受容体シグナル伝達の変調による改善の影響を受ける疾患又は障害の治療のためにも有用である。このような疾患又は障害は、例えば様々な中毒を含む。本願明細書に説明された物質を使用する治療の影響を受ける中毒は、例えば麻薬(例えば、モルヒネ、ヘロイン、及び他のオピエート)などの、薬物、ニコチン、及びアルコールに対する中毒に加え、行動中毒(例えばギャンブル依存症)を含む。
【0047】
従って本発明は更に、疼痛の治療に加え、中毒及び他のCNSに関連した障害の治療のための医薬組成物及び方法を提供する。本発明の鎮痛薬-関連物質は、哺乳類、例えばヒト対象へ、単独で、医薬として許容し得る塩もしくはそれらの水和可能な前駆体の形状で、又は化合物が適当な担体もしくは賦形剤(複数)と治療的有効量で混合されている医薬組成物の形状で、送達又は投与することができる。好ましい態様において、対象における薬物中毒の治療のため及び適当な治療的に有効な投与計画で投与される場合は、対象をより低量の薬物に傾向づけるために、十分量の鎮痛薬-関連物質が、インビボにおいて鎮痛薬受容体を阻害するために存在する。
【0048】
本発明の方法で使用される鎮痛薬-関連物質は、この物質を、1種又はそれよりも多い他の医薬として許容し得る成分と共に含有する医薬組成物として投与することができる。医薬組成物は、固形物の形(例えば散剤、顆粒剤、糖衣錠、錠剤又は丸剤など)、半-固形物(例えば、ゲル剤、スラリー、又は軟膏剤など)、液体、又は気体(例えばエアゾール剤又は吸入剤など)の形であり得る。
本発明において使用するために適当な製剤は、例えば、[Remington's Pharmaceutical
Sciences, Mack Publishing Company,フィラデルフィア, PA, 17th ed. (1985)
and Langer, Science, 249:1527-1533 (1990)]に認められる。本願明細書に説明された医薬組成物は、例えば、混合、溶解、造粒、糖衣錠形成、すりつぶし、乳化、カプセル封入、捕獲又は凍結乾燥プロセスなどの、常法により製造することができる。
【0049】
本発明の製剤の調製において、各化合物の医薬として認識された同等物を、代わりに使用することができる。これらの医薬として認められた同等物は、医薬として許容し得るエステル、アミド、もしくは塩、又は医薬として許容し得る酸付加塩であり得る。
医薬として許容し得る塩は、例えば、当該技術分野において周知の方法により調製されるナトリウム、カリウム、リチウム、カルシウム、マグネシウム、バリウム、アンモニウム、及びプロタミン亜鉛塩を含む、製薬産業において通常使用される、無毒の金属、アルカリ土類金属、又はアンモニウム塩である。この用語は、一般に本発明の化合物を、適当な有機酸又は無機酸と反応することにより調製される、無毒の酸付加塩も含む。代表的塩は、例えば塩酸塩、臭化水素酸塩、硫酸塩、亜硫酸塩、酢酸塩、シュウ酸塩、吉草酸塩、オレイン酸塩、ラウリン酸塩、ホウ酸塩、安息香酸塩、乳酸塩、リン酸塩、トシラート、クエン酸塩、マレイン酸塩、フマル酸塩、コハク酸塩、酒石酸塩、及びナプシラートを含む。
【0050】
医薬として許容し得る酸付加塩は、無機酸、例えば塩酸、臭化水素酸、硫酸、硝酸、リン酸などと、並びに有機酸、例えば酢酸、プロピオン酸、グリコール酸、ピルビン酸、シュウ酸、リンゴ酸、マロン酸、コハクサン、マレイン酸、フマル酸、酒石酸、クエン酸、安息香酸、桂皮酸、マンデル酸、メタンスルホン酸、エタンスルホン酸、p-トルエンスルホン酸、サリチル酸などにより形成された、遊離の塩基の生物学的有効性及び特性を保持し、並びに生物学的又はそうでなければ望ましくない塩である。[例えば、Bundgaard編集の著書、Design
of Prodrugs, Elsevier Science Publishers, アムステルダム(1985)を参照]。
【0051】
この鎮痛薬-関連物質は、通常の賦形剤、希釈剤、又は担体と共に製剤し、並びに便利な経口投与のために錠剤へ圧縮し、エリキシル剤又は液剤として製剤し得る。これらの物質は、徐放性剤形などとして製剤することもできる。
脳において鎮痛薬受容体の結合に関連した所望の治療作用を発揮するために、本発明の鎮痛薬-関連物質は、脳細胞及び組織に到達しなければならず;脳血管内皮細胞の微小毛細血管膜を含む、脳血管関門の通過による、それらの血液から脳への通路を必要とする。本発明は、患者の末梢組織(すなわち、中枢神経系組織以外の組織)への鎮痛薬-関連物質の治療的に有効な投薬投与計画を投与する方法を提供する。これは、経口、口腔内、直腸内、非経口、腹腔内、皮内、経皮、皮下、気管内及び筋肉内投与を含む様々な方法で、実現することができる。更にこれらの物質は、デポ剤又は徐放性製剤において、全身様式よりもむしろ局所的に投与することができる(例えば、痛覚喪失に管理された患者)。加えて、これらの物質は、ベシクル内、特にリポソーム内で投与することができる[例えば、Langerの論文, 前掲 Treatの論文,
Liposomes in the Therapy of Infectious Disease and Cancer, Lopez-Berestein and
Fidler編集、Liss, ニューヨーク, pp. 353-365 (1989)を参照されたい]。
【0052】
注射剤に関して、本発明の鎮痛薬-関連物質は、例えば、植物油又は他の同様の油、合成脂肪族系酸グリセリド、高次脂肪族系酸エステル、又はポリエチレングリコールなどの水性又は非水性溶媒中に;並びに望ましいならば、例えば、可溶化剤、等張剤、懸濁化剤、乳化剤、安定化剤及び保存剤などの、通常の添加剤を伴い、それらを溶解、懸濁化、又は乳化することにより、調製物へ製剤することができる。好ましくは注射剤に関して、本発明の化合物は、水溶液、好ましくはハンク液、リンゲル液、又は生理的食塩緩衝液などの、生理的に相溶性のある緩衝液中に製剤することができる。経粘膜的投与に関して、透過されるべき障壁に適した浸透剤が、製剤中で使用される。このような浸透剤は、一般に当該技術分野において公知である。
【0053】
経口投与に関して、鎮痛薬-関連物質は、当該技術分野において周知の医薬として許容し得る担体と組合せ、容易に製剤することができる。このような担体は、治療される対象による経口摂取のために、この化合物を、錠剤、丸剤、糖衣錠、カプセル剤、乳剤、親水性及び疎水性懸濁剤、液剤、ゲル剤、シロップ剤、スラリー、懸濁剤などとして製剤することが可能である。経口使用のための医薬調製は、錠剤又は糖衣錠コアを得るために望ましいならば、適当な助剤の添加後に、この化合物の固形賦形剤との混合、任意に得られる混合物の粉砕、及び顆粒の混合物の加工処理により得ることができる。特に適した賦形剤は、例えば糖(例えば乳糖、ショ糖、マンニトール、又はソルビトール)、セルロース調製物(例えばコーンスターチ、コムギデンプン、コメデンプン、ジャガイモデンプン)、ゼラチン、トラガカントガム、メチルセルロース、ヒドロキシプロピルメチル-セルロース、カルボキシメチルセルロースナトリウム、及び/又はポリビニルピロリドン(PVP)などの、充填剤を含む。望ましいならば、架橋したポリビニルピロリドン、寒天、又はアルギン酸もしくはそれらの塩、例えばアルギン酸ナトリウムのような、崩壊剤を添加することができる。
【0054】
糖衣錠コアは、適当なコーティングと共に提供される。この目的に関して、濃縮された糖溶液を使用し、これは任意にアラビアゴム、タルク、ポリビニルピロリドン、カルボポールゲル、ポリエチレングリコール、二酸化チタン、ラッカー液、及び/又は適当な有機溶媒又は溶媒混合物を含むことができる。色素又は顔料は、活性化合物用量の様々な組合せの同定又は特徴決定のために、錠剤又は糖衣錠コーティングに添加することができる。
【0055】
経口的に使用することができる医薬調製物は、例えば、ゼラチンで形成された押込み式カプセル、更にはゼラチン及び例えばグリセロール又はソルビトールのような可塑剤で製造された封止された軟カプセルを含む。押込み式カプセルは、例えば乳糖などの充填剤、結合剤(例えばデンプン)、及び/又は滑沢剤(例えばタルク又はステアリン酸マグネシウム)、並びに任意に安定化剤と混合された活性成分を含むことができる。軟カプセルにおいて、活性化合物は、例えば脂肪族系油、流動パラフィン、又は液体ポリエチレングリコールなどの、適当な液体中に溶解又は懸濁することができる。
【0056】
口腔内投与に関して、組成物は、通常の様式で製剤された、錠剤又はトローチ剤の形をとることができる。
吸入投与に関して、本発明に従い使用するための化合物は通常、例えばジクロロジフルオロメタン、トリクロロフルオロメタン、ジクロロテトラフルオロエタン、二酸化炭素もしくは他の適当なガスのような、適当な噴射剤の使用を伴う、加圧パックもしくはネブライザーからのエアゾールスプレー調製物、又は噴射剤非含有の乾燥散剤の吸入の形で送達される。加圧されたエアゾールの形において、用量単位は、例えば測定された量を送達するためのバルブを提供することにより決定することができる。吸入器又は通気器において使用するための例えばゼラチンのカプセル及びカートリッジは、この化合物及び、例えば乳糖又はデンプンのような適当な粉末基剤の粉末混合物を含むよう製剤することができる。
【0057】
本発明の鎮痛薬-関連物質は、注射による、例えばボーラス注射又は連続注入による、非経口投与のために製剤することができる。注射用製剤は、添加された保存剤と共に、例えばアンプル内又は反復投与用容器内で、単位剤形で提供される。これらの組成物は、例えば、油ベースのビヒクル又は水性ビヒクル中の懸濁剤、液剤、又は乳剤などのような形状をとることができ、並びに例えば、懸濁剤、安定化剤及び/又は分散剤などの製剤用物質を含むことができる。
【0058】
非経口投与のための医薬製剤は、水溶液型の活性化合物の水溶液を含む。あるいは、活性化合物の懸濁剤は、適当な油性注射用懸濁剤として調製することができる。適当な親油性溶媒又はビヒクルは、脂肪族油分(例えばゴマ油)、合成脂肪酸エステル(例えば、オレイン酸エチル又はトリグリセリド)、又はリポソームを含む。水性注射用懸濁剤は、例えばカルボキシメチルセルロースナトリウム、ソルビトール、又はデキストランなどの、懸濁剤の粘度を増加する物質を含有することができる。任意に懸濁剤は、高度に濃縮された溶液の調製を可能にするために、適当な安定化剤又は化合物の溶解度を増加する物質を含むこともできる。あるいは、活性成分は、使用前に、適当なビヒクル、例えば滅菌パイロジェン非含有水で再構成される散剤の形であり得る。
【0059】
鎮痛薬-関連物質は、例えば、ココアバター、カルボワックス、ポリエチレングリコール又は他のグリセリドなどの、全て体温で溶融するが室温では固形であるような、通常の坐剤基剤を含有する、坐剤又は持続浣腸のような、経直腸組成物中に製剤することができる。
先に説明された製剤に加え、これらの化合物は、デポ剤として製剤することもできる。このような長期作用する製剤は、移植(例えば皮下又は筋肉内)又は筋肉内注射により投与することができる。従って、例えばこれらの化合物は、適当な高分子又は疎水性物質と共に(例えば許容し得る油分中の乳剤)、又はイオン交換樹脂中、又はやや溶けにくい誘導体(例えばやや溶けにくい塩)として、製剤することができる。
【0060】
あるいは、疎水性医薬化合物のための他の送達システムを使用することができる。リポソーム及び乳剤は、疎水性薬物のための送達ビヒクル又は担体の周知の例である。一部の方法において、長期間循環するリポリーム、例えばステルスリポソームを使用することができる。このようなリポソームは一般に、Woodleらの米国特許第5,013,556号に開示されている。
本発明の化合物は、制御された放出手段及び/又は送達装置により投与することができる。ある変形において、ポンプが使用される[例えば、Langerの論文、前掲;Seftonの論文、CRC
Crit. Red: Biomed. End, 14:201 (1987);Buchwaldらの論文、Surgery,
88:507 (1980);Saudekらの論文、N. Engl. J. Med., 321:574
(1989)を参照されたい]。他の態様において、高分子物質が使用される[例えば、Medical
Applications of Controlled j Release, Langer及びWise編集、CRC
Pres., ボカラトン, FL (1974);対照led Drug i Bioovailability,
Drug Product Design and Performa,nce, Smolen及びBull編集、Wiley,
ニューヨーク (1984);Ranger及びPeppasの論文、J.
Macromol. Sci. Rev. Macromol. Chem., 23:61 (1983)参照;同じく、Levyらの論文、Science,
228:190 (1985) Duringら、Ann. Neurol., 25:351 (1989);Howardらの論文、J.
Neurosurg., 71:105 (1989)を参照されたい]。制御された放出手段及び送達装置は、例えば、米国特許第3,845,770号;第3,916,899号;第3,536,809号;第3,598,123号;及び、第4,008,719号にも開示されている。
【0061】
ある有機溶媒、例えばジメチルスルホキシド(DMSO)も、利用することができるが、通常より大きい毒性を代償とする。加えてこれらの化合物は、例えば治療薬を含有する固形疎水性ポリマーの半透過性マトリックスのような、持続放出システムを使用し送達することができる。様々な種類の持続放出材料が確立されている。持続放出カプセルは、それらの化学性質に応じ、数時間から最大100日を超える期間にわたり化合物を放出する。
【0062】
この医薬組成物は、適当な固相又はゲル相の担体又は賦形剤も含むことができる。このような担体又は賦形剤の例は、炭酸カルシウム、リン酸カルシウム、様々な糖類、デンプン、セルロース誘導体、ゼラチン、及びポリマー、例えばポリエチレングリコールなどを含むが、これらに限定されるものではない。
薬物中毒の治療のために、本発明の化合物は、この物質を薬物-含有製品(例えばニコチン摂取の場合は、タバコ製品、例えば紙巻きタバコなど)へ混入することにより投与することもできる。例えばある態様において、本発明の化合物は、摂取前に、薬物-含有製品へ噴霧又はそうでなければ塗布される。
【0063】
本発明における使用に適した医薬組成物は、活性成分が治療的有効量含まれる組成物を含む。本発明の方法のための治療的有効量は、例えば年齢、体重、全身の健康状態、性別、食事、投与時間及び様式、排泄速度、併用薬、治療担当医の判断、及び治療される特定の苦痛の重症度を含む、様々な要因に応じて決定することができる。活性物質の量は、オピエート-関連物質の比活性、及びその物質が他の治療成分又は予防成分と同時に投与されるかどうかによっても左右される。
【0064】
典型的には、本願明細書に開示された方法に従い治療される対象は、疼痛に苦しむとして確定されるか;鎮痛薬(例えばオピオイド)受容体の変調による治療もしくは予防に影響される疾患もしくは障害と診断されるか;又は、そうではなければ、この化合物を使用する生理的恩恵を受ける対象であると確定される。ある変形において、疼痛に苦しむ対象又は鎮痛薬受容体の変調による治療又は予防に影響される疾患もしくは障害(例えば中毒)と診断された対象は、第二の疾患又は障害には罹患していない。更に一部の態様において、この対象は、治療期間中、生理的及び/又は臨床的作用についてモニタリングされる。例えば疼痛治療に関して、対象は、治療経過を通じて疼痛の相対重症度についてモニタリングすることができ、;又は、鎮痛薬受容体シグナル伝達の変調を介した改善に影響される疾患もしくは障害の治療について、対象は、この疾患もしくは障害に関連した1種又は複数の症状についてモニタリングすることができる。
【実施例】
【0065】
下記実施例は、例証のために提供されているが、請求された発明を制限するものではない。実施例1-32化合物参照番号は、先に使用されたものに対応している。異なる化合物参照名が、実施例33、及び集合的に実施例34-45の各々において使用される。
下記の全般的情報は、実施例1-32に示した化合物の合成及び分析に関して適用される。全ての反応は、特に別に記さない限りは、炉乾燥したガラス製品を用い、周囲温度で磁気攪拌しながら、窒素陽圧下で実施した。大気-及び水分-感受性液体は、シリンジによりゴム隔壁を通して移した。シリカゲル(230-400メッシュ)を、カラムクロマトグラフィーに使用した。DMFを、中間(neutral)アルミナのカラムを通る、濾過により乾燥し、及び使用前に窒素下で活性化された4Å分子篩上に貯蔵した。他の溶媒及び試薬は全て、入手したまま使用した。1H
NMR及び13C NMRスペクトルは、各々、500MHz及び125MHzで記録した。融点は補正しなかった。燃焼分析が特定されない場合は、分析的純度は、D7500積分器(integrator)及びHamilton
PRP-Iステンレス鋼カラム(250mm ×4.6mm内径)を伴う、Hitachi
L74液体クロマトグラフィーを使用する、順相HPLCにより決定した。HPLC移動相:A=55:45:0.01
MeOH/イソプロパノール/HCl04;B=45/55/0.01
MeOH/イソプロパノール/HClO4;C=70:30
CH3CN/MeOH;D=MeOH。
【0066】
更に以下に説明された「I型」、「II型」、及び「III」は一般に、各々、先に説明された式IV、V、及びVIの化合物を意味する。
(実施例1):6β-S-(メチル-2',3'4'-トリ-O-アセチル-β-D-グルコピラノシルウロネート)モルヒネ3a
NaH(46mG, 1.16mmol)の60%分散体を、0℃の2a(424mg,
1.21mmol)のDMF溶液へ添加し、得られた赤色混合物を、10分間攪拌した。こうして形成されたチオールの混合物を、0℃で1b(183mg,
0.416mmol)のDMF(8mL)溶液へ1分間かけてシリンジで添加した。得られた溶液を、室温まで温めながら4時間攪拌した。反応液を、0.5%水性HC(20mL)へ注ぎ、pHを、固形NaHCO3を慎重に添加することにより、9へ上昇させた。この混合物を、CH2Cl2(3
× 50mL)で抽出し、一緒にした有機層を、H2O(10mL)、ブライン(10mL)で洗浄し、Na2SO4上で乾燥し、濾過し、灰色混合物へ濃縮し、フラッシュクロマトグラフィー(15
×2.5cm SiO2, 60:1〜20:1 CH2Cl2/EtOH)により精製し、黄味がかった白色の固形物として3a(144mg,
56%)を得た。3aの分析用試料を、無水EtOHからの再結晶により、白色固形物として得た:Rf=0.10
(20:1 CH2Cl2/MeOH);mp=135℃(分解);1H
NMR δ6.63 (d, J= 7.9 Hz, 1H), 6.50 (d, J= 7.9 Hz, 1H) , 5.81 (ddd, J= 9.1, 5.7,
3.0 Hz, 1H), 5.29-5.21 (m, 2H), 5.05 (d, J= 9.2 Hz, 1H), 5.03 (s, 1H), 4.70 (d,
J= 9.6 Hz, 1H), 4.06 (d, J= 9.6 Hz, 1H), 3.74 (s, 3H), 3.31 (m, 1H), 3.03-2.99
(m, 2H), 2.58 (dd, J= 11.7, 4.0 Hz, 1H), 2.43 (s, 3H), 2.38-2.28 (m, 2H),
2.15-2.09 (m, 2H), 2.05 (s, 3H), 2.02 (s, 3H), 1.76 (dd, J= 12.2, 1.8 Hz, 1H);13C
NMR (CDCl3)δ170.3, 169.6, 169.5, 167.0, 144.7, 138.5, 131.8, 130. 4,
128.5, 126.7, 119.7, 116.8, 93.7, 84.6, 76.3, 73.4, 70.0, 69.4, 59.2, 53.1,
47.2, 45.0, 44.5, 43.1, 39.8, 35.6, 20.9, 20.8, 20.7;C30H36NO11S
[M+H+]のHRMS計算値618.2009、実測値618.2011;3aの平均純度は、tR
=3.47分(移動相A)及びtR =3.98分(移動相B)を生じたことから、分析的HPLCにより、≧99%であることがわかった。
【0067】
(実施例2):3-O-アセチル-6-β-S-(メチル-2',3',4'-トリ-O-アセチル-β-D-グルコピラノシルウロネート)モルヒネ 3b
3aについて説明された手法に従い、NaH(53mg, 1.33mmol)、1a(484mg,
1.38mmol)及び2a(246mg, 0.51mmol)は、黄味がかかった白色粉末として3bを提供した(211mg,
63%):Rf=0.33 (20:1 CH2Cl2/EtOH);mp
194℃(分解);1HNMR (CDC13) δ 6.74 (d, J= 8.2 Hz, 1H), 6.58 (d, J= 8.2
Hz, 1H), 5.83-5.80 (m, 1H), 5.53 (dd, J= 1.5, 10.2 Hz, 1H), 5.29-5.21 (m, 2H),
5.09 (s, 1H), 5.00 (t, 1H), 4.72 (d, J= 10.3 Hz, 1H), 4.06 (d, J= 9.3 Hz, 1H),
3.76-3.73 (4H), 3.33 (m, 1H), 3.07-3.03 (2H), 2.60-2.58 (m, 1H), 2.44 (s, 3H),
2.37-2.20 (s, 3H over m, 2H), 2.15 (dt, J= 3.7, 11.8 Hz, 1H), 2.04 (s, 3H),
2.03 (s, 3H), 2.02 (s, 3H), 1.80 (d, J=11.6 Hz, 1H);13C
NMR (CDCl3) δ170.3, 169.5, 169.4, 168.8, 167.0, 149.0, 133.0, 132.6,
132.0, 131.8, 128.0, 121.8, 119.5, 94. 5, 85.1, 76.2, 73.3, 70.3, 69.4, 59.0,
53.0, 46.9, 45.6, 44.5, 43.2, 40.0, 35.7, 20.93, 20.90, 20.8, 20.7;MS
(ESI) m/z=660 [M+H+];分析(C32H37NO12S)計算値:C
58.26;H 5.65;N 2.12;S
4.86、実測値:C 57.94;H 5.75;N
2.07;S 4.97。
【0068】
(実施例3):3-O-アセチル-6β-S-(2',3',4',5'-テトラ-O-アセチル-β-D-グルコピラノシル)-6β-チオモルヒネ 3c
3aについて説明された手法に従い、チオール2b (500mg, 1.37mmol)、NaH(53mg,
1.32mmol)及びトシラート1b(246mg, 0.51mmol)は、黄味がかかった白色泡状物として3cを生じた(255mg,
74%):Rf=0.20 (20:1 CH2Cl2/MeOH);mp=151.9℃;1H
NMR (CDC13) δ 6.73 (d, J= 8.2 Hz, 1H), 6.58 (d, J= 8.2 Hz, 1H),
5.83-5.79 (m, 1H), 5.50 (dd, J= 1.8, 9.7 Hz, 1H), 5.24-5.17 (m, 2H), 5.04 (t,
J=9.9 Hz, 1H), 4.96 (t, J=9.9 Hz, 1H), 4.69 (d, J= 10.2 Hz, 1H), 4.19 (d, J=
4.2 Hz, 2H), 3.79-3.75 (m, 1H), 3.71 (d, J= 6.0 Hz, 1H), 3.30 (dd, J= 3.3, 5.6:
Hz, 1H), 3.04 (d, J= 18.8 Hz, 1H), 2.57 (dd, J= 4.0, 12.2 Hz, 1H), 2.42 (s,
3H), 2.34-2.29 (m, 2H), 2.14-2.11 (m, 1H), 2.07 (s, 6H), 2.05 (s, 6H), 2.00 (s,
6H), 1.80-1.77 (m, 1H);13C
NMR (CDCl3) δ170.8, 170.1, 169.5, 169.3, 169.5, 148.8, 132.8, 132.1,
131.7, 131.6, 127.9, 121.6, 119.3, 94.4, 85.1, 75.9, 73.8, 70.3, 68.5, 62.4,
58.8, 46.7, 45.8, 44.3, 43.0, 39.7, 35.6, 20.7, 20.6, 20.5;MS
m/z=674 [M+H]+;HRMS m/z計算値674.2271、実測値674.2238;3cの平均純度は、tR
=4.86分(移動相A)及びtR =5.66分(移動相B)を生じたことから、分析的HPLCにより、97.3%であることがわかった。
【0069】
(実施例4):6-βS-(2',3',4',5'-テトラ-O-アセチル-β-D-グルコピラノシル)-6β-チオモルヒネ 3d
この化合物は、3cの調製の副産物として単離した(47mg, 14.6%):Rf=0.08
(20:1 CH2Cl2/MeOH);mp 165.4℃;1H
NMRδ6.63 (d, J=8.2 Hz, 1H), 6.51 (d, J= 8.2 Hz, 1H), 5.79 (ddd, J= 3.6, 5.8,
9.6 Hz, 1H), 5.50 (dd, J= 1.7, 9.6 Hz, 1H), 5.22 (t, J= 9.3 Hz, 1H), 5.09-4.99
(m, 3H), 4.69 (d, J= 10.2 Hz, 1H), 4.27-4.19 (m, 2H), 3.78-3.74 (m, 1H), 3.67
(d, J= 5.8 Hz, 1H), 3.32 (m, 1H), 3.01 (d, J=18.9 Hz, 1H) over bs (1H),
2.61-2.58 (m, 1H), 2.44 (s, 3H), 2.39-2.27 (m, 2H), 2.16-2.11 (m, 2H), 2.08 (s,
3H), 2.06 (s, 3H), 2.05 (s, 3H), 2.03 (s, 3H), 1.78-1.75 (m, 1H);13C
NMRδ171.1, 170.4, 169.69, 169.66, 144.6, 130.3, 131.8, 130.6, 128.3, 126.9,
119.7, 116.7, 94.2, 84.9, 76.1, 74.0, 70.4, 68.7, 62.5, 59.2, 47.2, 45.8, 44.5,
43.2, 39.8, 35.8, 21.0, 20.83, 20.80, 20.6;MS
m/z=632[M+H]+;HRMS m/z C3lH38NO11S
の計算値632.2166、実測値632.2136;3dの平均純度は、tR
=3.98分(移動相A)及びtR =4.40分(移動相B)を生じたことから、分析的HPLCにより、≧98%であることがわかった。
【0070】
(実施例5):6β-S-(メチル-2',3',4'-トリ-O-アセチル-β-D-グルコピラノシルウロネート)コデイン 3e
3aについて説明された一般的手法に従い、1c(231mg, 0.51mmol)、2a(483mg,
1.38mmol)及びNaH(1.33mmol)は、黄味がかかった白色固形物3e(249mg,
77%)を生じた:Rf =0.30, 20:1 CH2Cl2/EtOH);mp=172℃;1H
NMR (CDC13) δ6.64 (d, J= 8.1 Hz, 1H), 6.54 (d, J= 8.1 Hz, 1H), 5.82
(ddd, J= 3.0, 6.0, 9.2 Hz, 1H), 5.53 (dd, J= 1.8, 9.4 Hz, 1H), 5.27-5.22 (m,
2H), 5.14 (s, 1H), 4.99 (t, J= 2.1 Hz, 1H), 4.71 (d, J= 10.3 Hz, 1H), 4.02 (d, J=
7.3 Hz, 1H), 3.83 (s, 3H), 3.79 (d, J= 6.0 Hz, 1H), 3.73 (s, 3H), 3.29 (dd, J=
3.3, 5.8 Hz, 1H), 3.05-3.00 (d, J= 19.0 Hz, 1H over m, 1H), 2.55
(dd, J= 4.3, 12.2 Hz, 1H) , 2.42 (s, 3H), 2.35 (td, J= 3.4, 12.4 Hz, 1H), 2.29
(dd, J= 6.0, 19.0 Hz, 1H), 2.15 (td, J= 5.0, 12.4 Hz, 1H), 2.02 (s, 3H), 2.016
(s, 3H), 2.00 (s, 3H), 1.77 (dd, J= 1.5, 12.3 Hz, 1H);13C
NMR δ 170.2, 169.5, 169.4, 166.9, 146.3, 142.2, 133.1, 130.7, 127.9, 127.7,
119.1, 112.8, 93.6, 85.2, 76.1, 73.4, 70.3, 69.3, 59.1, 56.4, 53.0, 47.0, 45.5,
44.7, 43.3, 40.0, 36.2, 20.9, 20.8, 20.7, 20.6;MS(ESI)
632 [M+H]+;分析(C3lH37NO11S)
C, H, N, S。
【0071】
(実施例6):6-S-(2',3',4',5'-テトラ-O-アセチル-β-D-グルコピラノシル)6β-チオコデイン 3f
3aについて説明された一般的手法に従い、チオール2b (440mg, 1.21mmol)、NaH
(47mg, 1.16mmol)及び1c(203mg, 0.448mmol)は、黄味がかかった白色泡状物として3f(237mg,
82%)を提供した。分析的試料は、沸騰ヘキサンからの再結晶により得た:Rf=0.26
(20:1 CH2Cl2/EtOH);mp=142.5℃;1H
NMR (CDC13) δ6.65 (d, J= 8.2 Hz, 1H), 6.55 (d, J= 8.2 Hz, 1H),
5.85-5.81 (m, 1H), 5.52 (dd, J= 1.9, 9.6 Hz, 1H), 5.22-5.19 (m, 2H), 5.06 (t,
J= 9.8 Hz, 1H), 4.97 (dd, J= 9.5, 10.1 Hz, 1H), 4.69 (d, J= 10.1 Hz, 1H),
4.23-4.16 (m, 2H), 3.83 (s, 3H), 3.76 (d, J= 6.0 Hz, 1H), 3.76-3.71 (m, 1),
3.29 (dd, J= 3.3, 5.8 Hz, 1H), 3.06-3.01 (m, 2H), 2.56 (dd, J= 4.0, 12.2 Hz,
1H), 2.42 (s, 3H), 2.38-2.30 (m, 2H), 2.18-2.15 (m, 1H), 2.12 (s, 3H), 2.09 (s,
6H), 2.00 (s, 3H), 1.99-1.80 (m, 1H);13C
NMR (CDCl3) δ171.0, 170.4, 169.6, 169.5, 146.3, 142.2, 132.9, 130.7,
127.9, 127.6, 119.1, 112.7, 93.8, 85.5, 76.1, 74.0, 70.5, 68.5, 62.5, 59.1,
56.4, 47.0, 46.1, 43.3, 40.0, 20.93, 20.92, 20.8, 20.5;MS
m/z=646 [M+H]+;HRMS C32H40NO11Sの計算値646.2322、実測値646.2304;3fの平均純度は、tR
=5.00分(移動相A)及びtR =5.71分(移動相B)を生じたことから、分析的HPLCにより、≧99%であることがわかった。
【0072】
(実施例7):モルヒネ-6β-S-D-グルクロニド 4a
MeOH 1.6mL中の3b(50.0mg, 0.076mmol)溶液に、5%NaOH水溶液(0.12mL)を添加した。得られた金色の溶液を、室温で18.5時間攪拌し、氷酢酸10滴の添加によりpH5へ酸性とし、濃縮した。得られた白色非晶質固形物を、シリカゲル(16
×1cm, 5:1から1:1 CH3CN/MeOH)上のフラッシュクロマトグラフィー、それに続く100℃(0.25mmHg)で4時間の乾燥により精製し、白色粉末(19.4mg,
54%)を得た:Rf=0.26 (1:1 CH3CN/0.2%AcOHを含むMeOH);mp=>300℃(分解);1H
NMR (D20) δ6.65 (d, J= 8.1 Hz, 1H), 6.61 (d, J= 8.1 Hz, 1H), 5.92
(m, 1H), 5.61 (d, J= 9.6 Hz, 1H), 5.19 (s, 1H), 4.69 (d, J= 9.9 Hz, 1H), 3.87
(d, J= 5.9 Hz, 1H), 3.77 (d, J= 9.0 Hz, 1H), 3.64 (m, 1H), 3.57-3.51 (m, 2H),
3.38 (t, J= 9.2 Hz, 1H), 3.13-3.09 (m, 2H), 2.83 (dd, J= 3.7, 12.3 Hz, 1H),
2.59-2.54 (s, 3H over m, 2H), 2.19 (dt, J= 8.5, 13.2 Hz, 1H), 1.87 (d, J= 12.0
Hz, 1H);13C NMR (D2O) δ 174.6, 144.3, 139.6, 129.8, 128.8, 127.7,
123.3, 119.0, 116.9, 91.7, 85.0, 79.3, 76.1, 71.3, 70.7, 58.0, 45.6, 43.3, 42.6,
40.3, 37.2, 33.0, 19.7;MS (ESI) m/z=478
[M+H]+;分析(C23H27NO8S)計算値:C
57.85;H 5.70;N 2.93;S
6.71、実測値:C 57.75;H 5.95;N
2.73;S 6.58。
【0073】
(実施例8):6β-チオモルヒネ-6β-S-D-グルクロピラノシド 4b
MeOH(6mL)中の3cの褐色溶液(50mg, 0.0742mmol)を、5%NaOH水溶液(0.4mL)に添加した。白色沈殿が、5分以内に形成された。この混合物を、18時間攪拌し、1.5N
AcOH(15滴)の添加により、pHを7に低下した。溶液を、5分間攪拌し、その後飽和NaHCO3 (15滴)を添加し、pHを8.5に低下した。混合物を濃縮し、残渣を、13%NH4OH
3滴添加した水0.5mL中に溶解し、分取的TLC (SiO2,
200:40:5:0.05 CH2Cl2/MeOH/H2O/13% NH4OH)により精製した。1本のUV活性バンドを、SiO2から取り出し、SiO2を4:1
CHCl3/MeOH(100mL)で洗浄した。濾液を濃縮し、残渣を、9:1
CHCl3/MeOH (10mL)を用い綿栓を通して再濾過した。濾液を濃縮し、得られた固形物を、100℃(0.25mmHg)で4時間乾燥し、表題化合物を白色粉末(20mg,
59%)として得た:Rf=0.21;mp=>300℃(dec)
1H NMR (CD3OD) δ6.55 (d, J= 8.0 Hz, 1H), 6.46 (d, J= 8.0
Hz, 1H) , 5.90 (ddd, J= 2.9, 5.8, 9.0 Hz, 1H), 5.52 (dd, J= 1.6, 9.7 Hz, 1H),
5.22 (s, 1H), 4.54 (d, J= 9.8 Hz, 1H), 3.90 (d, J= 11.2 Hz, 1H), 3.77 (d, 1=
6.0 Hz, 1H), 3.70 (dd, J= 5.2, 11.9 Hz, 1H), 3.42 (dd, J= 3.2, 5.6 Hz, 1H),
3.42-3.41 (m, 3H), 3.24 (dd, J= 8.4, 9.6 Hz, 1H), 3.07 (bs, 1H), 3.05 (d, J=
18.7 Hz, 1H), 2.68 (dd, J= 4.0, 12.3 Hz, 1H), 2.49 (s, 3H), 2.49-2.39 (m, 2H),
2.18 (dt, J= 7.9, 12.8 Hz, 1H), 1.81-1.79 (m, 1H);13C
NMR δ 146.3, 140.4, 131.5, 131.4, 130.3, 126.7, 120.4, 117.9, 95.3, 88.4, 82.3,
79.9, 74.8, 71.6, 63.2, 60.7, 48.2, 46.3, 45.3, 43.0, 40.4, 36.7, 30.9, 24.2;MS
m/z=464 [M+H]+;HRMS m/z C23H30NO7Sの計算値
464.1743、実測値464.1727;4bの平均純度は、tR
=3.94分(移動相A)及びtR =4.42分(移動相B)を生じたことから、分析的HPLCにより、97.0%であることがわかった。
【0074】
(実施例9):コデイン6β-S-グルクロニド, 4c
4aの調製について説明された手法に従い、3e(60.4mg, 0.095mmol)、MeOH
(2mL)及び5%NaOH水溶液(0.28mL)は、白色固形物として4c(38mg, 79%)を提供した:Rf=0.08
(1:1 MeCN/0.2%AcOHを含むMeOH);mp=>300℃(分解);1H
NMR (D2O) 6.82 (d, J= 8.2 Hz, 1H), 6.69 (d, J= 8.2 Hz, 1H), 5.83
(ddd, J= 2.8, 6.0, 9.5 Hz, 1H), 5.22 (s, 1H), 4.66 (d, J= 9.9 Hz, 1H), 3.84 (d,
J= 6.0 Hz, 1H), 3.81 (s, 3H), 3.76 (d, J= 6.0 Hz, 1H), 3.57-3.50 (m, 2H), 3.42
(dd, J= 3.3, 5.7 Hz, 1H), 3.40-3.34 (m, 1H), 3.08 (d, J= 19.0 Hz, 1H), 3.01 (m,
1H), 2.60 (dd, J= 4.0, 12.4 Hz, 1H), 2.43-2.37 (s, 3H over m, 1H), 2.30 (td, J=
3.4, 12.6 Hz, 1H) , 2.12 (td, J= 4.8 Hz, 13.0, 1H), 1.75 (d, J= 12.0 Hz, 1H);13C
NMR (D20) δ174.5, 144.4, 140.5, 131.0, 129.5, 127.3, 127.1, 119.0,
112.9, 92.8, 85.1, 79.3, 76.1, 71.3, 70.8, 57.1, 55. 7, 45.1, 43.2, 43.0, 40.5,
37.7, 33.8, 19.3;HRMS [M-H-] C24H28NO8Sの計算値
490.1536、実測値490.1525;4cの平均純度は、tR
=1.62分(移動相C)及びtR =1.46分(移動相D)を生じたことから、分析的HPLCにより、96%であることがわかった。
【0075】
(実施例10):6β-チオコデイン-6β-S-D-グルコース, 4d
エステル3f(120mg, 0.186mmol)を、MeOH (4mL)に溶解し、5%NaOH水溶液(0.6mL)を添加した。この混合物を、室温で18時間攪拌し、その後飽和の水性NH4Cl(1mL)で処理した。混合液を濃縮し、残渣を、分取TLC(SiO2)において、溶離液としてCH2Cl2/MeOH/水/13%濃縮したNH4OH(200:40:5:0.050)を使用し精製した。1本のUV活性バンドを、かみそりの刃でSiO2から取り出し、このSiO2を4:1
CHCl3/MeOH(100mL)で洗浄した。濾液を濃縮し、残渣を、溶離液として9:1
CHCl3/MeOH (10mL)を使用し綿栓を通して再濾過した。濾液を濃縮し、得られた固形物を、100℃(0.25mmHg)で4時間乾燥し、白色粉末として4dを得た(23mg,
26%):Rf=0.17;mp=201.6℃;1H
NMR (CD3OD) δ6. 79 (d, J= 8.3 Hz, 1H), 6.68 (d, J= 8.3 Hz, 1H), 6.02
(ddd, J= 3.0, 5.9, 9.1 Hz, 1H), 5.55 (dd, J= 1.8, 9.8 Hz, 1H), 5.36 (s, 1H),
4.57 (d, J= 9.8 Hz, 1H), 4.10 (dd, J= 3.0, 6.2 Hz, 1H), 3.91 (dd, J= 1.6, 12.1
Hz, 1H), 3.83 (d, J= 6.1 Hz, 1H), 3.81 (s, 3H), 3.70 (dd, J= 5.1, 12.0 Hz, 1H),
3.39-3.23 (複合m, 6H), 3.01-3.00 (m, 1H), 2.98 (s, 3H), 2.90 (dd, J= 6.6, 19.8
Hz, 1H), 2.41 (dt, J= 4.8, 13.8 Hz, 1H), 2.04 (dd, J= 2.7, 14.0 Hz, 1H);13C
NMR (CD30D) δ147.8, 144.5, 131.7, 130.1, 128.9, 125.3, 121.1, 116.2,
94.5, 88.3, 82.3, 79.9, 79.6, 74.8, 71.6, 63.1, 62.3, 57.3, 45.7, 44.3, 41.9,
38.8, 34.8, 23.0.;MS m/z=478 [M+H]+;HRMS
m/z C24H32NO7Sの計算値
478.1899、実測値478.1886;4dの平均純度は、tR
=4.79分(移動相A)及びtR =5.64分(移動相B)を生じたことから、分析的HPLCにより、96.5%であることがわかった。
【0076】
(実施例11):6βメチル-2',3',4'-トリ-O-アセチル-β-D-グルコピラノシルウロネート)アセトアミドコデイン 9b
塩化チオニル(0.2mL, 2.74mmol)を、カルボン酸8a (20.0mg,
0.053mmol)へ添加し、得られた無色の溶液を、窒素下で4時間攪拌した。トルエン(1mL)を添加し、溶液を、45℃の水浴を使い、高圧下での蒸留により濃縮した。得られた残渣を、0℃の氷浴中で冷却した。NEt3を3滴含有するアミン9b(32mg,
0.106mmol)の0℃のCH2Cl2 (2mL)溶液を、シリンジで添加した。得られた溶液を、氷浴を補充することなく、20時間攪拌し、CH2Cl2
(15mL)で希釈し、飽和NaHCO3水溶液(2 ×2mL)及びブライン(2mL)で洗浄した。有機層を乾燥し、濾過し及び濃厚な黄色油状物へ濃縮した。フラッシュクロマトグラフィーによる精製(10
×1cm SiO2, 10:1 CH2Cl2/MeOH)は、表題化合物を白色固形物(10mg,
29%)として得た:Rf=0.17 (10:1 CH2Cl2/MeOH);1H
NMR (CDCl3) δ7.06 (d, J= 6.8 Hz, 1H), 6.666 (d, J= 8.2 Hz, 1H), 6.54
(d, J= 8.2 Hz, 1H), 5.888 (ddd, J= 3.0, 5.8, 9.4 Hz, 1H), 5.60 (dd, J= 1.5, 9.4
Hz, 1H), 5.30 (s, 1H), 5.08 (t, J= 3.0 Hz, 1H), 4.78 (s, 1H), 4.68-4.66 (m,
2H), 4.57 (d, J= 10.4 Hz, 1H), 4.51 (t, J= 6.2 Hz, 1H), 3.86 (s, 3H), 3.82 (s,
3H), 3.30 (m, 1H), 3.03 (d, J= 18.5 Hz, 1H), 2.97 (m, 1H), 2.73 (dd, J= 11.1,
17.0 Hz, 1H), 2.55 (dd, J= 3.9, 11.6 Hz, 1H), 2.43 (s, 3H), 2.37-2.29 (m, 3H),
2.16 (s, 3H), 2.15 (s, 3H), 2.04 (s, 3H), 2.03-1.99 (m, 1H), 1.81-1.79 (m, 1H);MS(ESI)
m/z=657 [M+H]+。
【0077】
(実施例12):6α-(メチル-2',3',4'-トリ-O-アセチル-β-D-グルコピラノシルウロネート)アセトアミドモルヒネ,
10a
化合物9bについて説明された手法に従い、カルボン酸8a (38.5mg, 0.102mmol)、塩化チオニル(0.6mL)及びアミン7a(30mg,
0.075mmol)は、白色固形物として10a(29mg, 51%)を提供した:Rf=0.11
(20:1 CH2Cl2/MeOH);1H
NMR(CDCl3) δ7.01 (d, J = 6.7 Hz, 1H), 6.59 (d, J= 7.7 Hz, 1H), 6.45
(d, J= 7.7 Hz, 1H), 5.88-5.84 (m, 1H), 5.59 (dd, J= 1.6, 9.6 Hz, 1H), 5.08 (t,
J= 3.0 Hz, 1H), 4.72-4.66 (m, 2H), 4.58 (d, J= 16.4 Hz, 1H), 4.49 (t, J= 6.3
Hz, 1H), 3.82 (s, 3H), 3.84-3.72 (m, 2H), 3.29 (m, 1H), 3.01 (d, J= 18.5 Hz,
1H), 2.94 (m, 1H), 2.75-2.69 (dd, J= 11.0, 17.1 Hz, 1H), 2.53 (m, 1H), 2.43 (s,
3H), 2.42-2.29 (m, 2H), 2.19-1.99 (m, 2H), 2.163 (s, 3H), 2.157 (s, 3H) 2.06
(s, 3H), 1.78-1.75 (m, 1H), 0.98 (s, 9H), 0.194 (s, 3H), 0.174 (s, 3H);MS
(ESI) 757 [100, (M+H)+]。中間体シリルエーテル(20mg,0.026mmol)を、THF(1mL)に溶解した。TBAF(0.030mL,
0.030mmol)及び水(0.020ml)を添加し、紫色の溶液を、室温で50分間攪拌し、CH2Cl2
(20mL)で希釈し、NH4Cl (4mL)、ブライン(2mL)及び水(2mL)で洗浄した。有機層を乾燥し(Na2SO4)、濾過し、濃縮し、ピンク色の泡(22mg)を得た:Rf=0.22(10:1
CH2Cl2/MeOH);MS m/z=643
[M+H]+。
【0078】
(実施例13):6β-(α-グルクロノシル)アセトアミドモルヒネ 11a
10a(20mg, 0.031mmol)、5%NaOH水溶液(0.4mL)及びMeOH(2mL)から実施例7について説明された手法により調製し、白色固形物として11a(2.1mg,
18%)を得た:Rf=0.19 (SiO2/0.2%AcOHを含むMeOH);MS
m/z=501 [M-H]-。
(実施例14):6β-(α-D-グルクロノシル)アセトアミドコデイン 11b
【0079】
表題化合物を、実施例7について説明された手法により、白色固形物(13.5mg, 86%)として得た:Rf=0.14
(0.2%AcOHを含むMeOH);1H
NMR (D2O) δ6.97 (d,J=8.4 Hz, 1H), 6.84 (d, J= 8.4 Hz, 1H), 5.89-5.85
(m, 1H), 5.71 (d, J= 12.2 Hz, 1H), 4.94 (s, 1H), 4.62-4.59 (m, 1H), 4.36 (d, J=
5.7 Hz, 1H), 4.22 (m, 1H), 4.03 (d, J= 6.2 Hz, 1H), 3.88 (s, 3H), 3.80-3.73 (m,
2H), 3.35-3.30 (m, 2H), 3.03-2.94 (s, 3H 重なり m,
1H), 2.77-2.71 (m, 1H), 2.59 (dd, J= 3.6, 16.1 Hz, 1H), 2.31 (td, J= 4.7, 13.5
Hz, 1H), 2.06 (m, 1H);MS m/z=515 [M-H]-。
【0080】
(実施例15):6β-(2',3',4',5'-テトラ-O-ベンジル-β-D-グルコピラノシル)-3-O-トリイソプロピルシリルアセトアミドモルヒネ 9c
参照JMM-VI-113-2。SOCl2 (1.5mL)を、カルボン酸8b
(115mg, 0.20mmol)へ添加し、この溶液を、室温で16時間攪拌した。過剰なSOCl2を蒸留により除去し、生じた無色の油状物を、CH2Cl2(2.5mL)中に溶解し、CH2Cl2(1mL)中のアミン7c(43.5mg,
0.10mmol)及びNEt3(0.035mL, 0.25mmol)へ添加した。この溶液を、室温で1時間攪拌し、濃縮及びフラッシュクロマトグラフィーにより精製し(SiO2,
30:1から10:1 EtOAc/MeOH)、白色固形物として9c(70mg,
70%)を提供した:Rf=0.46 (10:1 CH2Cl2/MeOH);1H
NMR (CDCl3) d 7.35-7.10 (20H), 6.59 (d, J= 8.0 Hz, 1H), 6.40 (d, J=
8.0 Hz, 1H), 6.22 (d, J= 6.9 Hz, 1H), 5.71 (ddd, J= 3.0, 6.0, 9.5 Hz, 1H), 5.41
(dd, J= 1.9, 9.5 Hz, 1H), 4.89-4.48 (9H), 4.36 (t, J= 6.3 Hz, 1H), 3.72-3.46
(5H), 3.29 (t, J= 9.0 Hz, 1H), 3.17 (dd, J= 3.0, 5.2 Hz, 1H), 2.96 (d, J= 18.7
Hz, 1H), 2.88 (bs, 1H), 2.72 (dd, 2.7, 15.2 Hz, 1H), 2.50-1.72 (10H),
1.30-1.05(21H)。
【0081】
(実施例16):6β-(2',3',4',5'-テトラ-O-ベンジル-β-D-グルコピラノシル)アセトアミドモルヒネ 10b
JMM-VI-114-2。シリルエーテル9c(60mg, 59.7mmol)を、THF(2mL)中に溶解し、及び水(0.050mL)を添加し、その後TBAF(0.090mL,
0.090mmol, THF中1.0M溶液)を添加した。2.5時間後、1%濃縮したHCl(4mL)を添加し、混合物を2分間攪拌し、水(10mL)で希釈し、固形NaHCO3で塩基性とした。この混合物を、CH2Cl2
(5 ×30mL)で抽出し、乾燥し(Na2SO4)、濾過し濃縮した。フラッシュクロマトグラフィー(24:1から12:1
EtOAc/MeOH)は、白色固形物として10b(42mg, 83%)を提供した:Rf=0.15
(12:1 EtOAc/MeOH);1H NMR (CDC13) δ7.34-7.10 (20H), 6.64 (d, J= 8.2 Hz,
1H), 6.46 (d, J= 8.2 Hz, 1H), 6.34 (d, J= 6.3 Hz, 1H), 5.68 (ddd, J= 2.5, 5.8,
9.5 Hz, 1H), 5.39 (d, J= 9.5 Hz, 1H), 4.89-4.46 (10H), 4.34 (t, J= 6.3 Hz, 1H),
3.72-3.46 (6H), 3.30 (t, J= 9.1 Hz, 1H), 3.18 (bs, 1H), 2.97 (d, J= 18.1 Hz,
1H), 2.94 (bs, 1H), 2.74 (dd, J= 2.5, 15.7 Hz, 1H), 2.55-2.16 (7H), 1.95 (dt,
J= 8.0, 12.6 Hz, 1H), 1.76 (d, J= 11.3 Hz, 1H)。
【0082】
(実施例17):6β-(β-D-グルコピラノシル)アセトアミドジヒドロモルヒネ11c
JMM-VI-116-2。テトラベンジル誘導体10b(16mg, 0.018mmol)を、MeOH(2mL)及び濃HCl(3滴)中に溶解し、その後10%Pd-C(16mg)を添加した。混合物を、24時間水素バルーン(balloom)下で攪拌し、その後セライトを通して濾過し、このセライトをMeOH(10mL)で洗浄した。濾液を濃縮し、残渣を分取TLC(SiO2,
5:4:0.5:0.5 CHCl3/MeOH/H2O/濃NH4OH)で精製し、11cを白色フィルム(2.3mg,
26%)として得た:Rf=0.40;MS
m/z=491 (MH+)。
【0083】
(実施例18):6β-(3-メトキシフェニル)アセトアミドモルヒネ,13a
7c(80mg, 0.182mmol)を、CH2Cl2(4mL)中で溶解し、NEt3(72μL,
0.517mmol)及び3-メトキシフェニルアセチルクロリド(80μL, 0.513mmol)をシリンジにより添加した。得られた淡黄色溶液を、密閉したバイアル中で、室温で乾燥窒素大気下で攪拌した。2時間後、この溶液を濃縮し、残渣を、フラッシュクロマトグラフィー(20:1
CHCl3/MeOH)により精製し、白色泡状物として12a(78mg, 73%)を提供した:Rf
=0.18 (20:1 CH2Cl2/MeOH);MS
m/z: 589(M+H)+。12a(64mg, 0.109mmol)を、5%水性THF(2.2mL)中に溶解した。TBAF(0.18mL,
THF中1.0M溶液)は、シリンジにより添加し、淡黄色溶液を、室温で、乾燥窒素大気下で2時間攪拌した。溶液を濃縮し、1%HCl(2mL)を添加した。混合物を2分間攪拌し、その後、水(20mL)を使い分液ロートに移した。混合物を、固形NaHCO3でアルカリ化し(pH8.5)、CHC13(5×10mL)で抽出した。一緒にしたCHCl3抽出物を、ブライン(4mL)で洗浄し、乾燥し(Na2SO4)、濾過し、濃縮した。フラッシュクロマトグラフィー(20:1
CHCl3/MeOH)は、13aを白色固形物(38mg,
81%)として提供した:mp144℃;Rf =0.14(20:1 CHCl3/MeOH);1H
NMRδ7.24 (J= 7.9 H, 1H), 6.82-6.79 (m, 3H), 6.64 (d, J = 8.2 Hz, 1H), 6.49 (d, J=
8.2 Hz, 1H), 5.67 (ddd, J= 3.6, 6.1, 9.1 Hz, 1H), 5.58-5.53 (m, 2H), 4.65 (s,
1H), 4.37 (t, J= 6.4 Hz, 1H), 3.79 (s, 3H), 3.54 (s, 2H), 3.34 (m, 1H), 3.00
(d, J= 18.6 Hz, 1H), 2.89 (s, 1H), 2.61 (dd, J= 4.0, 12.2 Hz, 1H), 2.43 (s,
3H), 2.38-2.29 (m, 2H), 1.99 (dt, J= 9.6, 12.2 Hz, 1H), 1.76 (dd, 1.9, 12.6 Hz,
1H);13C NMRδ171.1, 160.2, 144.5, 138.9, 136.2, 132.8, 130.3, 129.9,
128.6, 125.8, 121.7, 119.6, 117.1, 115.1, 113.1, 93.0, 59.3, 55.4, 50.5, 47.3,
44.1, 43.9, 43.0, 39.8, 35.5, 20.5;MS m/z =433 [M+H]+、455[M+Na]+;HRMS
m/z 433.2115 [M+H]+;15aの平均純度は、tR
=4.38分(移動相A)及びtR =5.09分(移動相B)を生じたことから、分析的HPLCにより、≧99%であることがわかった。
【0084】
(実施例19):6β-(4-メトキシフェニル)アセトアミドモルヒネ 13b
12aについて説明された一般的手法に従い、7c(78.4mg, 0.178mmol)、NEt3(72μL,
0.517mmol)及び4-メトキシフェニルアセチルクロリド(80μL, 0.523mmol)は、12bを白色泡状物(70mg,
67%)として生じた:Rf=0.11 (20:1 CHCl3/MeOH);MS
m/z:589 (M+H)+, 611 (M+Na)+。参照V-99。13aについて説明された手法に従い、12b(65mg,
0.110mmol)及びTBAF(0.18mL, 0.18mmol)は、13b(29mg, 61%)を白色固形物として生じた:mp
247(dec);Rf=0.14 (20:1 CHCl3/MeOH);1H
NMR δ7.15 (d, J= 8.6 Hz, 2H), 6.86 (d, J= 8.6 Hz, 2H), 6.64 (d, J= 8.1 Hz, 1H),
6.48 (d, J= 8.1 Hz, 1H), 5.67 (ddd, J= 3.0, 5.6, 9.5 Hz, 1H), 5.56 (dd, J= 1.5,
9.5 Hz, 1H), 5.45 (d, J= 6.4 Hz, 1H), 4.65 (s, 1H), 4.36 (t, J= 6.3 Hz, 1H),
3.79 (s, 3H), 3.50 (s, 2H), 3.34 (m, 1H), 3.00 (d, J= 18.6 Hz, 1H), 2.87 (s,
1H), 2.61 (dd, J= 3.8, 11.9 Hz, 1H), 2.44 (s, 3H), 2.38-2.28 (m, 2H), 1.98 (dt,
4.9, 12.5 Hz, 1H) , 1.77 (d, J= 10.7 Hz, 1H);13C
δ171.5, 158.8, 144.4, 138.7, 132.6, 130.4, 129.7, 128.3, 126.5, 125.6, 119.3,
117.0, 114.5, 92.8, 59.1, 55.3, 50.3, 47.0, 43.9, 42.9, 42.7, 39.6, 35.4, 20.2;MS
m/z =433 (M+H)+, 456 (M+Na)+;HRMS
m/z 433.2136 [M+H]+;13bの平均純度は、tR
=4.44分(移動相A)及びtR =5.24分(移動相B)を生じたことから、分析的HPLCにより、>99%であることがわかった。
【0085】
(実施例20):6β-(3-メトキシ)ベンズアミドモルヒネ 13c
12aについて説明された一般的手法に従い、7c(82mg,, 0.185mmol)、NEt3
(76μL, 0.545mmol)及びアニソイルクロリド(71μL, 0.521mmol)は、白色泡状物として12c(88mg,
82%)を生じた;Rf=0.28 (10:1 CH2Cl2/MeOH);MS
m/z:575 (M+H)+。13aについて説明された一般的手法に従い、12c(73mg,,
0.174mmol)及びTBAF(0.210mL, 0.210mmol)は、白色固形物として13c(42.5mg,
58%)を生じた:Rf=0.15 (15:1 dcm/MeOH);mp=212.9℃(dec);1H
NMRδ7.34-7.33 (m, 1H), 7.27-7.23 (m, 2H), 7.02-7.00 (m, 1H), 6.68 (d, J= 8.1
Hz, 1H), 6.51 (d, J= 8.1 Hz, 1H), 6.27 (d, J= 6.8 Hz, 1H), 5.84 (ddd, J= 3.0,
5.8, 9.2 Hz, 1H), 5.68 (dd, J= 1.2 Hz, 9.9 Hz, 1H), 4.85 (s, 1H), 4.59 (t, J=
6.4 Hz, 1H), 3.81 (s, 3H), 3.47 (dd, J= 3.0, 5.0 Hz, 1H), 3.19-3.16 (m, 1H),
3.04 (d, J= 18.5 Hz, 1H), 2.68 (dd, J = 4.0, 12.0 Hz, 1H), 2.49 (s, 3H),
2.43-2.38 (m, 1H), 2.11 (dt, J= 4.8, 12.6 Hz, 1H), 1.80 (d, J= 10.9 Hz, 1H);13C
NMRδ167.5, 160.0, 144.7, 139.1, 135.6, 132.7, 129.9, 129.8, 128.9, 125.5,
119.7, 119.0, 118.2, 117.4, 112.6, 93.0, 59.5, 55.7, 50.9, 47.4, 44.1, 43.0,
39.9, 35.4, 20.6;MS m/z 419 (M+H)+, 441 (M+Na)+;HRMS
m/z = 419.1984 [M+H]+;13cの平均純度は、tR
=4.41分(移動相A)及びtR =5.05分(移動相B)を生じたことから、分析的HPLCにより、99%であることがわかった。
【0086】
(実施例21):6β-ベンズアミドモルヒネ, 13d
12aについて説明された一般的手法に従い、7c(82mg, 0.232mmol)、NEt3(76μL,
0.55mmol)及び塩化ベンジル(61μL, 0.521mmol)は、白色泡状物として12d(79mg,
78%)を生じた;Rf =0.23(20:1 CHCl3/MeOH);MS
m/z: 545 (M+H)+。13aについて説明された一般的手法に従い、12d(65mg,
0.110mmol)及びTBAF(0.18mL, 0.18mmol)は、白色固形物として13d(29mg,
61%)を生じた:Rf=0.15(15:1 CH2Cl2/MeOH);mp=184.4℃(dec);1H
NMRδ7.78 (m, 2H), 7.53-7.50 (m, 1H), 7.42-7.41 (m, 1H), 6.72 (d, J= 8.1 Hz,
1H)), 6.55 (d, J= 8.1 Hz, 1H), 6.18 (d, J= 6.2 Hz, 1H), 5.89 (ddd, J= 3.0, 5.3,
9.5 Hz, 1H), 5.69 (dd, J= 1.5, 9.5 Hz, 1H), 4.86 (s, 1H), 4.64 (t, J= 6.3 Hz,
1H), 3.54 (s, 1H), 3.31 (s, 1H), 3.06 (d, J= 18.7 Hz, 1H), 2.77 (m, 1H), 2.57
(s, 3H), 2.50-2.48 (m, 3H), 2.23-2.19 (m, 1H), 1.86 (dd, J= 2.2, 12.9 Hz, 1H);13C
NMR δ167.4, 144.5, 138.8, 133.9, 132.9, 131.7, 129.8, 128.6, 128.6, 127.0,
125.7, 119.4, 117.1, 92.8, 59.1, 50.6, 47.0, 44.0, 42.9, 39.9, 35.5, 20.5;MS
m/z = 389 [M+H]+, 411(M+Na)+;HRMS
m/z = 389.1867[M+H]+;13dの平均純度は、tR
=4.43分(移動相A)及びtR =5.16分(移動相B)を生じたことから、分析的HPLCにより、99%であることがわかった。
【0087】
(実施例22):6β-(3'-カルボメトキシ)ベンズアミドモルヒネ 13e
12aについて説明された一般的手法に従い、7c(159mg, 0.362mmol)、NEt3(151μL,
1.09mmol)及びモノメチルフタロイルクロリド(217mg, 1.01mmol)は、白色泡状物として12e(122mg,
56%)を生じた;Rf=0.22 (20:1 CHCl3/MeOH);MS
m/z:603 (M+H)+。13aについて説明された一般的手法に従い、12e(102mg,
0.170mmol)及びTBAF(0.47mL, 0.470mmol)は、白色固形物として13eを生じた:Rf=0.14
(30:1 dcm/MeOH);mp=166.0℃(dec);1H
NMR δ8.32 (s, 1H), 8.14 (d, J= 7.8 Hz, 1H), 8.00 (d, J= 7.8 Hz, 1H), 7.49 (t,
J=7.8 Hz, 1H), 6.68 (d, J=8.1 Hz, 1H), 6.52 (d, J=8.1 Hz, 1H), 6.33(d, J=7.0
Hz, 1H), 5.85 (ddd, J=9.4, 5.6, 3.1 Hz, 1H), 5.73 (d, J=9.9, 1H), 4.85 (s, 1H),
4.62 (t, J=6.2 Hz, 1H), 3.93 (s, 3H), 3.42 (dd, J=3.2, 5.2, 1H), 3.10 (bs, 1H),
3.05 (d, J=18.5 Hz, 1H), 2.63 (dd, J=12.1, 4.2 Hz, 1H), 2.46 (s, 3H), 2.40-2.34
(m, 2H), 2.08 (dt, J= 12.5, 4.9 Hz, 1H), 1.81 (d, J=10.8 Hz, 1H);13C
δ166.4, 166.2, 144.3, 138.7, 134.2, 133.0, 132.7, 131.9, 130.5, 129.8, 128.9,
128.3, 127.6, 125.7, 119.5, 117.0, 92.9, 59.1, 52.4, 50.9, 47.1, 44.0, 42.9,
39.9, 35.4, 20.2;MS m/z=447 [M+H]+;HRMS
m/z=447.1930 [M+H]+;13eの平均純度は、tR
=4.39分(移動相A)及びtR =5.06分(移動相B)を生じたことから、分析的HPLCにより、≧99%であることがわかった。
【0088】
(実施例23):6β-(3'-ニトロ)ベンズアミドモルヒネ 13f
12aについて説明された一般的手法に従い、7c(80mg, 0.182mmol)、NEt3(76μL,
0.55mmol)及び3-ニトロベンゾイルクロリド(95mg, 0.51mmol)は、白色泡状物として12f(87mg,
81%)を生じた;Rf=0.17(20:1 CHCl3/MeOH);MS
m/z:590(M+H)+。13aについて説明された一般的手法に従い、12f(73mg,
0.168mmol)及びTBAF(0.200mL, 0.200mmol)は、明黄色固形物として13f(36mg,
49%)を生じた:Rf=0.11(20:1 CHCl3/MeOH);mp=208.3℃(dec);1H NMRδ8.54
(t, J= 1.4 Hz, 1H), 8.26 (dd, J= 1.4, 8.2 Hz, 1H), 8.11 (d, J= 8.0 Hz, 1H),
7.55 (t, J= 8.0 Hz, 1H), 6.85 (d, J= 6.4 Hz, 1H), 6.65 (d, J = 8.1 Hz, 1H),
6.51 (d, J= 8.1 Hz, 1H), 5.84 (ddd, J= 3.0, 5.5, 9.2 Hz, 1H), 5.71 (d, J= 10.3
Hz, 1H), 4.88(s, 1H), 4.63 (t, J= 6.3 Hz, 1H), 3.45 (t, J= 3.2 Hz, 1H), 3.18
(s, 1H), 3.04 (d, J= 18.6 Hz, 1H), 2.68 (dd, J= 3.8, 11.8 Hz, 1H), 2.47 (s,
3H), 2.41-2.34 (m, 2H), 2.15 (dt, J = 7.6, 12.4 Hz, 1H), 1.78 (d, J= 11.5 Hz,
1H);13Cδ165.2, 148.0, 144.4, 138.9, 135.5, 133.3, 132.7, 129.8, 129.7,
128.2, 126.1, 125.4, 122.0, 119.5, 117.3, 92.5, 59.2, 51.1, 47.1, 43.9, 42.8,
39.6, 35.1, 20.3;MS m/z=434 [M+H]+;HRMS
m/z=434.1719 [M+H]+;13fの平均純度は、tR
=4.11分(移動相A)及びtR =4.78分(移動相B)を生じたことから、分析的HPLCにより、>99%であることがわかった。
【0089】
(実施例24):6β-(3',4'-ジクロロ)ベンズアミドモルヒネ,
13g
12aについて説明された一般的手法に従い、7c(81mg, 0.184mmol)、NEt3(77μL,
0.55mmol)及び3,4-ジクロロベンゾイルクロリド(108mg, 0.518mmol)は、白色泡状物として12g(96mg,
85%)を生じた;Rf=0.20(20:1 CH2Cl2/MeOH);MS
m/z:614(M+H)+, 616(M+2+H)+;618(M+4+H)。13aについて説明された一般的手法に従い、12g(81mg,
0.177mmol)及びTBAF(0.22mL, 0.22mmol)は、白色固形物として13g(48mg,
49%)を生じた:Rf=0.17(15:1 CH2Cl2/MeOH);mp=228.5℃(dec);1H
NMRδ7.82 (d, J= 2.1 Hz, 1H), 7.54 (dd, J= 2.1, 8.4 Hz, 1H), 7.39 (d, J= 8.4 Hz,
1H), 6.67 (d, J= 8.0 Hz, 1H), 6.56 (d, J= 6.2 Hz, 1H), 6.52 (d, J= 8.0 Hz, 1H),
5.82 (ddd, J= 2.9, 5.3, 9.5 Hz, 1H), 5.68 (dd, J= 1.2, 9.5 Hz, 1H), 4.83 (s,
1H), 4.58 (t, J= 6.3 Hz, 1H), 3.47 (dd, J= 3.0, 5.3 Hz, 1H), 3.19 (m, 1H), 3.05
(d, J= 18.6 Hz, 1H), 2.70 (dd, J= 3.8, 11.9 Hz, 1H), 2.50 (s, 3H), 2.43-2.37 (m,
2H), 1.77 (d, J= 11.0 Hz, 1H);13C
NMR δ165.4, 144.4, 138.9, 136.1, 133.7, 133.0, 132.5, 130.5, 129. 6, 129.3,
128.4, 126.3, 125.2, 119.5, 117.4, 92.5, 59.2, 52.4, 50.8, 47.2, 43.8, 42.7,
39.5, 35.1, 29.7, 25.8, 20.3, 20.5, 13.6;MS m/z 457 (M+H)+;HRMS
m/z=457.1087 [M+ H+];13gの平均純度は、tR
=3.93分(移動相A)及びtR =4.43分(移動相B)を生じたことから、分析的HPLCにより、>99%であることがわかった。
【0090】
(実施例25):6β-(チオフェン-2'-イル)アセトアミドモルヒネ 13h
12aについて説明された一般的手法に従い、7c(86mg, 0.196mmol)、2-チオフェンアセチルクロリド(0.068mL,
0.552mmol)及びNEt3(0.082mL, 0.588mmol)は、白色泡状物として12h(66mg,
60%)を生じ;Rf=0.23(20:1 CHCl3/MeOH);MS
m/z=565[M+H]+。13aについて説明された一般的手法に従い、12h(56mg,
0.099mmol)及びTBAF(0.120mL, 0.120mmol)は、白色固形物として13h(32mg,
79%)を得た:Rf=0.10(20:1 CHCl3/MeOH);mp=54.4℃;1H NMRδ7.18
(d, J= 4.5 Hz, 1H), 6.94-6.91 (m, 2H), 6.64 (d, J= 8.1 Hz, 1H), 5.82 (d, J= 7.1
Hz, 1H), 5.69 (ddd, J=3.5, 5.5, 9.4 Hz, 1H), 5.59 (dd, J= 1.2, 9.9 Hz. 1H),
4.65 (s, 1H), 4.39 (t, J= 6.4 Hz, 1H), 3.77 (s, 2H), 3.37 (dd, J= 3.3, 5.8 Hz,
1H), 3.01 (d, J= 18.5 Hz, 1H), 2.93 (m, 1H), 2.64 (dd, J= 4.1, 12.1 Hz, 1H),
2.45 (s, 3H), 2.39-2.31 (m, 2H), 2.02 (dt, J= 7.6, 12.6 Hz, 1H), 1.77 (dd, J=
10.8, 1.7 Hz, 1H);13C NMRδ170.2, 144.6, 139.0, 136.0, 132.9, 129.9, 128.4, 127.6,
125.9, 125.6, 119.6, 117.3, 92.9, 59.2, 50.4, 47.3, 44.1, 43.0, 39.7, 37.7,
35.5, 20.5;MS m/z=409[M+H]+, 431(M+Na)+;HRMS
m/z=409.1572[M+H]+;13hの平均純度は、tR
=4.31分(移動相A)及びtR =5.02分(移動相B)を生じたことから、分析的HPLCにより、>99%であることがわかった。
【0091】
(実施例26):6β-(3'-カルボキシ)ベンズアミドモルヒネ 14
参照V-110。13e(31mg, 0.067mmol)を、1:1
THF/水4mLに溶解し、LiOH-H2O(27mg, 0.643nmol)を添加した。無色の溶液を、室温で3.75時間攪拌し、氷酢酸を添加し(15滴)、及びこの溶液を濃縮した。残渣を、SiO2上(5:1から1:1
CHCl3/MeOH)でのフラッシュクロマトグラフィーにより精製した。適当な画分を濃縮し、残渣を10mLの10:1
CHCl3/MeOHと共に攪拌し、紙を通して濾過した。濾液を濃縮し、表題化合物を白色固形物として20mg(69%)提供した:Rf=0.17(1:1
CHCl3/0.2%AcOHを含むMeOH)、mp=217.1℃(dec);1H
NMRδ8.41-8.40 (m, 1H), 8.13-8.12 (m, 1H), 7.90-7.88 (m, 1H), 7.47 (t, J= 7.7
Hz, 1H), 6.62 (d, J= 8.2 Hz, 1H), 6.55 (d, J= 8.2 Hz, 1H), 5.81-5.78 (m, 1H),
5.69 (d, J= 9.9 Hz, 1H), 4.82 (s, 1H), 4.51-4.50 (m, 1H), 3.65 (dd, J= 3.2, 5.4
Hz, 1H), 3.27 (bs, 1H), 3.12 (d, J= 18.9 Hz, 1H), 2.87 (dd, J= 4.0, 12.9 Hz,
1H), 2.64-2.57 (m, 2H), 2.20 (dt, J= 4.8, 12.9 Hz, 1H), 1.81 (dd, J= 2.4, 12.9
Hz, 1H);13C NMRδ180.5, 170.2, 145.6, 141.2, 139.4, 135.5, 133.6, 131.0,
130.9, 130.5, 130.3, 129.5, 129.2, 125.6, 120.7, 118.5, 93.8, 61.3, 52.8, 42.6,
39.9, 35.1, 30.9, 22.1:MS m/z=431
(M-H)-;HRMS m/z C25H25N2O5 の計算値433.1758、実測値433.1749;14の平均純度は、tR
=3.93分(移動相A)及びtR =4.43分(移動相B)を生じたことから、分析的HPLCにより、99%であることがわかった。
【0092】
(実施例27):インビトロ結合アッセイ
A. 方法。細胞膜への結合は、96-ウェルフォーマットで行った。[N. Zaveriらの論文、Eur.
J. Pharmacol., 428:29-36 (2001)を参照されたい]。細胞を、プレートから、ラバーポリスマン(rubber
policeman)でこそぎ落とし、Tris緩衝液中に、Polytronホモジナイザーを使用して均質化し、その後一度遠心し、27,000×gで15分間の追加の遠心により洗浄した。ペレットを、50mM
Tris(pH7.5)中に懸濁し、懸濁液を、μオピオイド受容体、δオピオイド受容体、及びκオピオイド受容体それぞれに結合させるため、[3H]DAMGO、[3H]DPDPE、又は[3H]U69593と共にインキュベーションした。インキュベーションの総容積は、1.0mLであり、試料を、25℃で60〜120分間インキュベーションした。結合反応中のタンパク質の量は、約15μg〜30μgで変動した。ガラスファイバーフィルターを有するTomtec
96ハーベスター(Orange, CT)を用いた濾過により、反応を終結させた。結合した放射能を、Pharmacia
Biotechβ-プレート液体シンチレーションカウンター(ピスカタウェイ,
NJ)で計測し、カウント/分で表した。IC50値は、被験化合物の少なくとも6種の濃度を用いて決定し、Graphpad/Prism
(ISI, サンディエゴ, CA)を用いて計算した。Ki値は、Cheng及びPrusoffの方法を用いて決定した[Y.
Cheng及びW. H. Prusoffの論文、Biochem. Pharmacol.,
22:3099-3108 (1973)を参照されたい]。
【0093】
B.結果。リガンドの親和性及び選択性。表1は、化合物5及び6のKi値を含む。Kiは、被験化合物による受容体への放射性リガンドの結合の阻害を測定するパラメーターである。M6Gのように、試験した各リガンドは、μ選択性であった。I型化合物に関して、最も活性のある化合物に関する親和性の順位は、6b=5a>5d>6aであった。これらの各化合物は、遊離のフェノール性ヒドロキシル基を有した。M6Gと比べて、それらの親和性は、μ受容体で1.5〜2.4倍、δ受容体で2.9〜10.3倍、及びκ受容体で1.4〜369倍増加した。C-5'位にカルボメトキシ基を有する化合物5a及び5bは各々、κ受容体に関して、δ受容体よりも1.7〜2.7倍の選択性を示した。他のリガンドは全て、κ受容体よりもδ受容体に選択性を示した。化合物5aのC-5'位のカルボメトキシ基の、化合物5dのアセトキシメチル基との交換は、κ受容体の親和性を1/270に減少させた。3-アセトキシ基又は3-メチルエーテル基のいずれかを有するμ受容体、δ受容体、及びκ受容体についての残りの化合物の親和性は、M6Gと比較した場合に、有意に減少した。各場合において、糖部分のエステル官能基の加水分解は、リガンドのκ受容体への親和性を大きく減少させる一方で、μ受容体及びδ受容体に対するリガンドの親和性については、明らかに著しく低い効果を有していた。
【0094】
II型化合物のμ結合親和性に関して、遊離のフェノール系ヒドロキシル基及び完全にエステル化されたグルクロン酸部分を有するモルヒネアナログ10(2.80nM)は、最高の親和性を有した。この知見は、I型アナログ3aについて観察されたそれらの結果と一致した。興味深いことに、コデイン誘導体9b(7.43nM)は、I型コデイン化合物よりも有意に高い親和性を示した。化合物11a(14.6nM)及び11b(41.5nM)は、低下した親和性を示した。M6Gとの比較において、化合物10は、μ受容体について4.5倍より高い親和性を示し、化合物9bは、1.7倍高い親和性を示した。II型化合物は、I型化合物よりも有意により選択性であった。μ受容体に関する化合物11bの選択性は、M6Gのものに近いが、これは結合親和性の1/3.2の低下を有した。これらの結果は、モルヒネ又はコデイン核と糖残基の間の炭素橋は、μ受容体選択性に関して重要であることを示唆している。この橋によりもたらされた追加の立体配置的移動度は、これらの分子が、μ受容体と、C-6α糖残基を有するM6Gにより類似して相互作用することは考えられる。
【0095】
化合物14(19.92nM)を除いて、μ受容体でのIII型化合物のKi値は、0.21〜0.59nMであった。これらの化合物は、M6Gよりも、20倍〜64倍より高いμ受容体親和性を有する。III型化合物は、最高の親和性のI型化合物よりも少なくとも9倍高い親和性を有し、最高親和性のII型化合物よりも少なくとも4.7倍高い親和性を有した。これらの結果は、これらのリガンドのC-6置換基は、μ結合ポケットの疎水性領域と相互作用することを示唆している。この知見は更に、化合物14(19.92nM)で観察された親和性の減少により裏付けられる。ほとんどの場合において、II型化合物は、増加したμ対δ選択性及び低下したμ対κ選択性を示した。架橋したNのN-メチル置換基のシクロプロピルメチル又はシクロブチルメチル又は類似の置換基との交換は、典型的には以下に説明された活性に関するこれらの受容体のアンタゴニストを提供する。
【0096】
【表1】
【0097】
(実施例28):Gタンパク質への共役を測定するための[35S]GTPγ-S結合試験
A. 方法。放射標識されたS結合は、基本的に、[J.R. Traynor及びS. R. Nahorskiの論文、Mol.
Pharmacol., 47:848-854(1995)]に説明されたように行った。細胞を組織培養皿から20mM
Hepes、1mM EDTAへはぎ取り、その後500 ×gで10分間遠心した。細胞を、この緩衝液中に再懸濁し、Polytronホモジナイザーを用いてホモジナイズした。ホモジネートを、27,000
×gで15分間遠心し、ペレットを20mM Hepes、10mM MgCl2、100mM
NaCl(pH7.4)を含有する緩衝液A中に再懸濁した。懸濁液を、27,000 ×gで再遠心し、もう一度緩衝液A中に懸濁した。結合アッセイのために、膜(8〜15μgタンパク質)を、[35S]GTPγ-S(50pM)、GDP(10μM)、及び適当な化合物と共に、総容量1.0mLで、25℃で60分間インキュベーションした。試料を、ガラスファイバーフィルター上で濾過し、結合アッセイについて説明されたようにカウントした。統計解析は、プログラムPrismを用いて行った。
【0098】
B. 結果。リガンドの効力及び効能。化合物が会合したGタンパク質のオピオイド受容体介在性の活性化を評価するために、これらの化合物を、[35S]GTPγSアッセイを用いて評価した。これらのデータを、表2にまとめた。このアッセイにおいて、化合物の効力又は受容体親和性は、[35S]GTPγS結合の刺激に関するそのEC50により定義した。アゴニスト効能は、化合物が[35S]GTPγS結合を対照に対し最大刺激する程度として定義される。EC50値は、化合物による[35S]GTPγS結合の50%最大刺激を生じる化合物の濃度を表している。完全アゴニストは、[35S]GTPγS結合を最大程度刺激し、部分アゴニストは、結合のレベルの低下を生じさせる。表2は、ヒトμオピオイド受容体、ヒトδオピオイド受容体、及びヒトκオピオイド受容体での[35S]GTPγS結合の刺激に関する、Emax及びEC50値を提供する。化合物6cを除き全ての化合物は、μ受容体及びδ受容体で、[35S]GTPγS結合を刺激した。これらの化合物は、κ受容体で有効性がはるかに少なかった。この傾向は、M6Gについても観察された。μ受容体で最も有効な化合物は、化合物3b(88.0%)、3d(76.0%)、4b(64.5%)、及び4a(46.6%)であった(表2参照)。これらの各化合物は、遊離のフェノール系ヒドロキシル基を有した。これらの化合物は全て、M6Gよりもより有効であった。同じ一般的傾向が、遊離のフェノール系ヒドロキシルを有する化合物により、δ受容体においても認められ、これは増大した効能を示している。最も有効な化合物は、3b(104.4%)、4a(78.70%),、3e(64.3%)、及び4b(51.5%)であった。アセテート基又はメチル基を伴うフェノール系ヒドロキシル基の保護は、各受容体において効能及び効力を減弱させた。最小刺激を作用する化合物は、所定の受容体でアゴニストと見なすことができる。最大結合よりも少なく誘起したリガンドは、刺激が認められない場合は、部分アゴニスト又はアンタゴニストと見なすことができる。EC50値の全体の順位は、Ki値と相関していた。II型化合物に関して、リガンド10及び9bは、μ受容体において[35S]GTPγS結合の最大刺激を生じ、δ受容体及びκ受容体において、[35S]GTPγS結合の刺激を低下した。III型化合物13a-h及び14に関して、これらの各化合物は、μ受容体、δ受容体及びκ受容体で[35S]GTPγS結合を刺激した。これらの化合物は一般に、δ受容体に対し、μ及びκ受容体でより有効であった。最大刺激を作用する化合物は、所定の受容体でアゴニストとみなされる。最大結合よりも少なく誘起するこれらのリガンドは、刺激が認められない場合には、部分アゴニスト又はアンタゴニストと見なされる。EC50値の全体の順位は、Ki値と相関した。
【0099】
【表2】
【0100】
(実施例29):μ受容体内在化
A. 方法。μ受容体内在化は、[D. E. Keithらの論文、Mol. Pharm.,
53:377-384 (1998)]の方法に従い評価した。一般的用語において、この現象は、表面受容体を、FITC-標識されたFLAG
M1の染色及び蛍光の測定により定量することができるフローサイトメトリー分析を用いて、試験した。内在化を引き起こす効果がある薬物は、対照と比べ低下した染色を生じた。
【0101】
B. 結果。この過程は耐性及び依存の発生において役割を担い得るという理由から、III型化合物を、μ受容体内在化に作用するそれらの能力について評価した(表3参照)。このデータから、ジクロロ化合物13g(47%)及びニトロ化合物13f(59%)は両方とも、μ受容体内在化の誘導において、エトロフィン(61%)よりも有効であることが認められた。他のアミドは、μ受容体の弱い内在化を中程度にする。モルヒネ(93%)は、通常の血漿濃度(1〜50nM)では、μ受容体内在化を引き起こさないことは指摘されなければならない。これらの実験において使用される化合物の濃度は、1μMであった。
【0102】
【表3】
【0103】
(実施例30):化学安定性及び代謝安定性評価
A. 方法。選択された化合物の化学安定性を、pH7.4及びpH2.0で試験した。10μMリン酸カリウム緩衝液の存在下、pH7.4又はpH2.0のいずれかの中の化合物(0.1M)を、5分、1時間、2時間、3時間、24時間、48時間又は72時間後に濃縮した。その後HPLCを用い、出発物質の分解を分析した。選択された化合物の代謝安定性を、ヒト肝臓S9及びミクロソームで試験した。ヒト肝臓プールしたS9(HLS9)の安定性及びマウス肝ミクロソーム(MLM)。アッセイ緩衝液は、最終インキュベーション容積0.25mLについて、3.2mg/mL
HLS9又は1.2mg/mL MLM、0.1Mリン酸カリウム緩衝液(pH7.4)、0.1mM化合物、0.5mM
NADP+、0.5mMグルコース-6-リン酸、5U/mLグルコース-6-リン酸デヒドロゲナーゼ、1mg/mL
DETAPAC、7mM MgCl2を含む。0分、10分、25分、40分又は60分後、1mL
CH2Cl2/IPA(3:1 v:v)の添加により、反応を停止した。遠心後、抽出溶媒(有機相)を、窒素流で蒸発除去し、200μLメタノールに溶解し、激しく攪拌し、遠心し、HPLCバイアル挿入管に移した。HPLCを使用し、所定の時間間隔(0分、10分、25分、40分又は60分)での曲線下面積を、ミクロソーム添加前の曲線下面積と比較することにより、出発物質の分解を分析した。
【0104】
B. 結果。選択型I化合物、及び選択型III化合物は、マウス肝ミクロソーム(MLM)、ヒト肝臓ミクロソームS-9(HLS9)の存在下、pH2及び7.4で評価した(表4参照)。より長い半減期は、所定の条件下で、より大きい安定性と相関している。I型化合物において、M6Gイオウアナログ4aは、HLS9の存在下で、かつpH2及びpH9において、M6Gと同等の安定性を示した。エステル保護基の存在は、I型分子の安定性を低下した。HLS9に対する化合物3aの安定性は、化合物3bよりも大きく(17.86分)、これは3-位のアセテート基は、I型分子において最も不安定なエステル基であることを示唆している。この仮説は、HPLC同時注入により証明された。脱保護された化合物4a-dの安定性において、差異は検出されなかった。III型化合物は、試験した全ての条件下で、I型化合物よりも有意により安定していた。化合物13eにおけるメチルエステル基の存在は、HLS9及びpH7.4に対するこの化合物の安定性を、他のIII型化合物と比べて低下した。評価するための十分量のII型化合物は、得られなかった。II型化合物におけるエステル保護基の存在は、それらをIII型化合物よりもより不安定にした。アミド連結は、S-グリコシド連結に勝る改善された安定性をもたらすと予想される。
【0105】
【表4】
【0106】
(実施例31):モルヒネアナログ3a及び13eの鎮痛薬活性の決定
A. 方法。
1.動物。実験開始時に体重20〜25gの雄のICRマウスを使用した。動物は、標準の実験室条件下で、群で飼育し、12:12時間昼-夜サイクル(08:00点灯)を維持した。動物は、実験実施前、1〜2日間馴化した。
2. 薬物。モルヒネ13e及び3bを、水に溶解した。薬物を、0.1mlの容量で注射した。
【0107】
3.侵害受容試験。侵害受容を、投射熱を使用する痛覚喪失装置(Stoelting)による、掉尾反射アッセイを用いて試験した。この装置は、掉尾反射潜時の自動定量で、動物尾の損傷を防ぐために15-秒カットオフで装備した。試験期間中は、焦点化した光ビームを、動物尾の下側半分に適用し、掉尾反射潜時を記録した。掉尾反射潜時のベースライン値は、各動物の薬物投与前に決定した。基本の掉尾反射潜時は、3.2〜8.0秒(平均5.86±0.16SEM)であった。試験直後に、動物に、被験化合物又はビヒクル対照として生理食塩水を皮下注射した。動物を、注射後30分及び60分に、掉尾反射潜時について試験した。
【0108】
4. データ解析。抗侵害受容を、下記式で定量した:
%抗侵害受容=100*[(試験潜時-ベースライン潜時)/(15-ベースライン潜時)]
動物が15秒カットオフ以前に反応しなかった場合は、動物は、スコア100%に割り当てた。行動の結果は、群間変動としてモルヒネ、13e、及び3bで、並びに反復測定として薬物処置後時間(30分、60分)により、ANOVAを用い、適当ならばStudent
Newman-Keulsポストホック検定により、引き続き解析した。有意水準は、p<0.05とした。
B. 結果
【0109】
1.モルヒネの掉尾反射潜時に対する作用。図5に示したように、モルヒネは、掉尾反射潜時の用量-依存型の増加を生じた。全般的ANOVAは、投与量の有意な作用を示した。[F(3,45)=25.52,
P<0.0001]。従って注射後時間とは無関係に、モルヒネは、掉尾反射潜時に対して同じ作用を生じた。注射後時間横断的に平均化して、モルヒネの3〜30mg/kg投与量は、対照に対し、掉尾反射潜時の有意な増加を生じた(Student
Newman-Keuls, p<0.05)。%MPEに関する用量漸増反応曲線を認め、ここでモルヒネ投与量3mg/kgは、10及び30mg/kg投与量に比べ、より低い鎮痛作用を生じた(Student
Newman-Keuls, p<0.05)。
【0110】
2. 13eの掉尾反射潜時に対する作用。13eの投与は、30分間で明らかであり及び60分間で継続した掉尾反射潜時の増加を生じた(図6参照)。全般的ANOVAは、投与量の有意な作用を示した。[F(5,72)=14.86,
P<0.0001]。注射後時間横断的に平均化して、13eの2つの最高投与量は、対照に対し、掉尾反射潜時の有意な増加を生じた(Student
Newman-Keuls, p<0.05)。13eの投与量30mg/kgの鎮痛作用は、10mg/kg投与量により生じた作用よりも2倍大きかった(Student
Newman-Keuls, P<0.05)。化合物13eは、モルヒネよりもより遅い作用開始を有し、30分よりも60分で有意に増加した効力を有するように見える。その非常に高い親和性(μ受容体でKi=0.23nM)及び[35S]GTPγ結合の刺激に関する強力な完全アゴニスト活性(μでEC50=2.4nM)を考慮し、この化合物は、比較的弱い抗侵害受容活性を有し、ED50値は30分及び60分間で、各々、30.2mg/kg及び12.6mg/kgであった。これは、恐らくED50が約3mg/kgであるモルヒネに比べ短期間の効力が低いであろう。
【0111】
3. 3aの掉尾反射潜時の作用。3aの投与は、化合物の強力な鎮痛薬作用の掉尾反射潜時指標における用量-依存型の増加を生じた(図7参照)。全般的ANOVAは、投与量の有意な作用を示した[F(5,64)=37.26,
P<0.0001]。注射後時間横断的に平均化して、3aの投与量3〜30mg/kgは、対照に対する掉尾反射潜時の有意な増加を生じた(Student
Newman-Keuls, p<0.05)。3aの2つの最高投与量(10及び30mg/kg)は、3mg/kg投与量に対し、より大きい量の痛覚喪失を生じた(Student
Newman-Keuls, p<0.05)。化合物3aは、13eよりも鎮痛薬として有意により強力であり、30及び60分の両方で、約2.5mg/kgのED50を有した。この値は、3aは、モルヒネ(ED50、3mg/kg)よりもわずかにより強力であることも示唆している。3aは、モルヒネよりもわずかに低いインビトロ結合親和性を有するが、ほぼ同じ[35S]GTPγS効力及び%刺激を有し(表1及び2参照)、これは3aの生体利用効率が、モルヒネのそれとほぼ同じであるか又はより大きいことを示唆している。この化合物はオピオイド受容体の各々に関して若干非選択的であるので、この時点で、どの受容体が抗侵害受容活性を媒介するかは定かではない。3aの作用開始は、モルヒネよりもより早いことは明らかである(この活性は、30分時点よりも、60分でより大きい)。
【0112】
(実施例32):ヘロイン依存性及び離脱症状
A. 方法。M6Gアナログのヘロイン依存性動物に対する作用。ヘロイン自己-投与を、依存の最適測定を生じるヘロイン投与量を用いて行った。これまでの研究を基に、0.06mg/kg/0.1ml注入を算出した。安定した反応が確立した後(23時間/日アクセスで約40日間)、陽性対照としての低投与量ナロキソン(0.05〜0.2mg/kg)、及びM6Gアナログ(0、0.01、0.04、0.1mg/kg、皮下注射)及び他のアナログの、ヘロイン摂取の様々な行動測定に対する作用を試験した。4種の投与量を、各化合物について試験した。各M6Gアナログの対象内用量-作用試験を行うことを試みたが、試験は、必要ならば、対象間用量-作用機能も用いた。ナロキソン及びM6Gアナログの12時間にわたる作用を調べるために、Koob研究室が開発したミニポンプを連結したオン/オフゲーティング装置を使用した。一旦安定したヘロイン摂取が達成された後、ラットに、午前6時に浸透圧ミニポンプ(ゲート-オフ位置)を移植することができる。12時間後、ゲーティング装置を、オン位置に回し、ラットを通常23時間セッション時に、ヘロイン自己-投与にアクセスさせた。活動相(午後6時オン午後6時オフ)である暗期間の12時間の期間について、これらのセッション時に、ミニポンプは、ゲート-オンの位置とした。2日後、ミニポンプを除去し、ラテン方陣(Latin
Square)デザインで異なる投与量の被験化合物(すなわち、ナロキソン又はM6Gアナログ)含む別のミニポンプと置き換えた。このサイクルを、3投与量及びビヒクル対照を試験するように繰り返した。必要な多数の薬物及び投与量は、ナロキソン及びM6Gアナログの投与量の対象間デザインで強制することができる。試験される特定の行動は、非依存型パターンの自己投与から、依存型パターンの自己投与への移行を反映することができる、自己投与を介した長期間ヘロイン曝露の試験結果から決定した。個別の獲得軌跡が多項式関数にマッチされると同時に、対象内及び対象間変数を説明する、階層線形モデル(Hiearchical
Linear Modeling)法を使用し、洗練された統計モデル化を行った。
【0113】
B. 結果。ヘロイン自己投与のパラダイムは、現在ラットにおいてよく確立されている[Walkerらの論文、Fur.
J. Pharmacol., 383:115 (1999)を参照されたい]。2,3の試験は、限度のないヘロインアクセスに関連した行動の変化及び依存点までのヘロインの自己-投与の出現を調べた。予備的結果は、ラットは、食物枯渇又は制限のない条件下で、限度のないアクセスで、ヘロインi.v.を容易に自己-投与することを確立した。自己-投与の限度のないアクセスのパターンは、経時的に変更し、依存性の測定を示す動物を生じる。ヒトにおけるヘロイン依存性の動物依存モデルが開発された。延長されたアクセス(投与量0.06mg/kg/0.1ml注入でのヘロイン自己-投与への23時間のアクセス)が与えられた動物は、50〜60日間にわたるヘロインの持続摂取を示した。この期間、自己-注入パターンは、「活動/暗所」相においてのみ認められたものから、自己-投与がほぼ15〜20分毎に生じたパターンへと変化した。2〜3週目から7週目まで、本質的に均等な自己-投与が認められた。各自己-投与回数/日を、約50日間記録した。これらの試験は、ヘロイン投与量、摂取パターン、及び最大薬物摂取の時刻に関する完全な情報を提供した。ヘロイン依存性は、慢性ヘロイン投与からの離脱症状発現として測定することができる[Woolverton及びSchusterの論文、(1983)を参照されたい]。依存性は、依存型ラットにおけるヘロイン除去後の又はヘロインアンタゴニスト投与後の、顕在的禁断徴候として表すことができる。両方とも、身体的徴候及び動機づけ作用を生じる[Weeks及びCollinsの論文、(1964)を参照されたい]。M6Gアナログのヘロイン自己-投与を低下する能力を、自己-投与時及びM6Gアナログの投与後の延長された-アクセスモデルにおいて試験した。
【0114】
M6Gアナログ3a、13e、13g及び13hのヘロイン自己-投与に関する作用を、ヘロインに依存したヒトにおける作用と一致する依存型ラットにおいて試験した。これらの実験の目標は、ヘロイン自己-投与の減少に有効であるM6Gアナログを同定することである。図8-12の試験は、M6Gアナログ13e(0.04mg/kg)、13g(0.2mg/kg)、M6G(0.04mg/kg)、13h(0.01、0.04、0.2mg/kg)は、ヘロイン自己-投与の低下に有効であることを明らかにした。最も有効な化合物は、投与量0.01mg/kgで、ヘロイン摂取を1000μg/kg低下させる13hであった。
【0115】
(実施例33):MG6のC-グリコシドアナログの合成及び生物学的評価
モルヒネ-6-グルクロニド(M6G)は、ラットへ脳室内注射した場合に、鎮痛効力がモルヒネそれ自身の約100倍であるモルヒネの第II相代謝複合体である[Carruptらの論文、J.
Med. Chem., 34, 1272-1275 (1991)を参照されたい]。M6Gは現在、術後疼痛治療のための臨床試験の最終相にある[Gutmanらの米国特許第6,737,518号(2004)]。M6Gの経口生体利用効率は、わずかに11%であり[Personらの論文、Br.
J. Clin. Pharmacol., 53:347-354 (2002)を参照されたい]、またM6Gの代謝安定性の改善は、可能性のある疼痛の薬物療法としてのその有効性を恐らく増大させるであろう。複合糖質のインビボ代謝安定性を改善するための一般的方法は、グリコシド酸素原子の炭素、窒素又はイオウ原子との交換に関連している[Postemgらの論文、
Organic Lett., 5:1721-1723 (2003);Kiefelらの論文、J.
Carbohydr. Chem., 18:937-959 (1999)を参照されたい]。M6Gのグルコシル及びグルクロノシルアナログのライブラリーのデザインにおける戦略は、グリコシド酸素原子がイオウ原子と交換される場合がこれまで利用された[MacDougallらの論文、J.
Med. Chem. 47:5809-5815 (2004)を参照されたい]。M6Gの6-βイオウアナログは、μオピオイド受容体親和性及び機能的効能のわずかな改善を示したが、μ受容体については、δオピオイド受容体及びκオピオイド受容体に対しより少ない選択性を示した。本願明細書において報告したのは、新規μ選択的にアミド結合された炭素グリコシドアナログの14工程の合成経路及び生物学的評価の結果を詳細に説明する、本研究の拡張である。
【0116】
C-β-グリコピラノシルアシルクロリド9は、市販の2,3,4,6-テトラベンジルグリコピラノース4から、5工程の手順により調製した。化合物4を、DMSO/Ac2Oにより酸化させ、対応するラクトン5(94%)を生じた[Kuzuhara,
H.;Fletcher, H.G.の論文、J. Org Chem.,
32:2531-2534 (1967)]。5の-78℃のリチウム酢酸エチルのTHF溶液への添加[Rathke,
M. W.の論文、J. Am. Chem. Soc., 92:3222-3223 (1970)]、それに続く酸性水による後処理は、立体選択的アルドール反応により形成されたヘミケタール6を形成した(91%)
[Lewisらの論文、J. Am. Chem. Soc., 104:4976-4978 (1982)]。アセトニトリル中BF3OEt2の存在下での、0℃での、6のトリエチルシランによる還元[Lewisらの論文、前掲;Tiedemannらの論文、J.
Org Chem., 64:4030-4041 (1999)を参照されたい]は、エチルエステル7(86%)を生じさせた。還流時の1:1
THF/H2O中のLiOHによる、7のエチルエステル基の加水分解及びそれに続く酸性水による後処理は、カルボン酸8(87%)を生じさせた。8の対応するアシルクロリド9への転換は、無希釈のチオニルクロリド中での18時間の攪拌により実現した。2.0当量Et3Nの存在下CH2C12中での、粗酸塩化物9の1.5当量の、3-トリイソプロピルシリル-6-β-アミノモルヒネへの添加[MacDougallらの論文、Bioorg
Med. Chem., 12:5983-5990 (2004)]は、保護されたモルヒネグルコースアナログ10(93%)を生じさせた。10中のトリイソプロピルシリル保護基は、THF中のTBAFの添加により除去され、フェノール11を生じた(90%)。引き続きの酸性メタノール中の活性化された10%Pd/Cによる、11の触媒的水素化は、M6Gのアミド-連結したC-β-グリコピラノシドアナログである化合物3を生じさせた(52%)。HClの非存在下でのPd/Cによる11の水素化分解は、フェナントレン炭素二重結合の還元を生じ、定量的収量の12をもたらしたが、グルコースベンジルエーテル基の水素化分解は生じなかった。これらの条件下で、11の塩基性モルヒナン窒素原子は、恐らくO-脱ベンジル化を阻害した[Czech,
B. P.;Bartsch, R. A.の論文、J. Org Chem.
49:4076-4078 (1984)]。7,8-二重結合の飽和及びグルクロン酸部分のグルコース部分との置換の両方は、鎮痛効力が増大したM6Gアナログを提供することがわかっている[Stachulskiらの論文、Bioorg
Med. Chem. Lett., 13: 1207-1214 (2003)]。従って、3の対応するデヒドロアナログ又はグルクロン酸アナログへの代替経路を開発するよりもむしろ、化合物3を直接評価した。全ての合成化合物に関するスペクトルデータは、割当てられた構造に完全に一致した[新規化合物10、11、12及び3は、1H
NMR、13C NMR及びMSにより特徴決定した。被験化合物3及び11の純度は、HPLCにより決定し、≧98%であることがわかった。]。
【0117】
化合物3及び11のμオピオイド受容体、δオピオイド受容体及びκオピオイド受容体との競合的結合アッセイで得られたIC50値を、実験の項に説明されたように、Ki値へ変換した。被験化合物及び参照物質のKi値は、表5に列記している。結合アッセイにおいて、以下の放射性リガンドを使用した:[3H]DAMGO(μオピオイド受容体アゴニスト);[3H]U69593(κオピオイド受容体アゴニスト);[3H]DPDPE(δオピオイド受容体アゴニスト)。Ki値は、被験化合物3及び11によるこれらの放射性リガンドの受容体への結合の阻害の測定から決定した[Zaveriらの論文、Eur.
J. Pharmacol., 428:29-36 (2001)]。ベンジル誘導体11及び脱保護された同族体3は両方とも、μ受容体選択性であった。M6Gと比べ、化合物11は、μオピオイド受容体に関してM6Gよりも27倍高い効力を有した。化合物11のμ受容体対δ受容体、及びμ受容体対κ受容体への選択性は、各々、10倍及び34倍であった。M6Gと比較して、化合物3は、μオピオイド受容体に関して3.7倍より高い効力を有した。化合物3のμ受容体対δ受容体、及びμ受容体対κ受容体への選択性は、各々、77及び166倍であった。化合物3のμ受容体対δ受容体選択性は、M6Gの値に対しかなり改善されるが、μ受容体対κ受容体はそうではないことは、注目に値する(すなわち、各々12.5倍及び316倍の選択性)。化合物3は、μ受容体に対し、チオ糖2a(2.5倍)及び2b(1.6倍)に比べわずかに大きい効力を有し、並びにδ/μ及びκ/μ受容体選択性比を著しく改善した。化合物3及び11の機能活性は、[35S]
GTPγSアッセイを用いて評価した[Traynor, J.R.及びNahorski S.R.の論文、Modulation,
Mol. Pharmacol. 47:848-854 (1995)]。[35S]GTPγSアッセイは、被験化合物の、μ、δ又はκオピオイド受容体のいずれかに会合したGタンパク質を活性化する能力を測定する。[35S]GTP-γ-S結合を刺激するEmax値を基に、化合物3は、μ受容体及びδ受容体で完全アゴニストであり、及びκ受容体で部分アゴニストであると決定した。化合物11は、δ受容体で完全アゴニストであり、及びμ及びκ受容体で部分アゴニストであると決定した。μ受容体での化合物3の効能(Emax)(75%)は、M6G(45%)、及びチオ糖アナログ2a(46.6%)及び2b(36.0%)よりも、各々、実質的に高い。
【0118】
結論として、C-グリコシド3を、2,3,4,6-テトラ-O-ベンジル-D-グルコース4及び硫酸モルヒネから、14-工程の転換合成により調製した。化合物3は、μオピオイド受容体に対し、M6Gと比べ、3.7倍大きい親和性を示した。化合物3のδ受容体対μ受容体、及びκ受容体対μ受容体に対する選択性の比は、各々、76.7及び166であった。化合物3のδ/μ選択性は、12.5であるM6Gに関する値に比べ、著しく改善された。これらの化合物のインビボ生物学的特性を完全に説明するための、更なる研究が現在進行中である。M6Gの代謝安定性の増加は、より長い寿命の強力なオピオイドアゴニストの新規クラスを提供することができる。
【0119】
【表5】
【0120】
【表6】
【0121】
全般的情報。全ての反応を、特に記さない限りは、炉乾燥したガラス製品を使用し、周囲温度で磁気攪拌しながら、陽圧下で実施した。空気-及び水分-感受性液体を、シリンジによりゴム製隔壁を通して移した。用語ブラインは、塩化ナトリウム飽和溶液を意味する。カラムクロマトグラフィーのために、シリカゲル(230〜400メッシュ)を使用した。DMFを、中間アルミナのカラムを通して乾燥させ、また使用前は、窒素下活性化した4Å分子篩上で貯蔵した。CH2Cl2及びTHFを、使用直前にCaH2から蒸留した。全ての他の溶媒及び試薬は、入手したまま使用した。1H
NMRスペクトルは、300MHz Varian NMRにより、18℃で記録した。化学シフトは、特に別に記載しない限りは、7.26ppmで
CDC13に対するppm(δ)で報告した。高解像度質量分析を、University of
California at Riverside において、Opus V3.1及びDEC
3000 Alpha Stationデータシステムを備えた、VG7070スペクトロメーター上で行った。低解像度質量分析(LRMS)を、ESIを使用するHitachi
M-8000 3DQMS (イオントラップ型)質量分析装置で行った。融点は、補正せずに報告した。分析的純度は、D7500積分器及びHamilton
PRP-Iステンレス鋼カラム(250mm ×4.6mm内径)を備えた、Hitachi
L74液体クロマトグラフィーを使用する、順相HPLCにより決定した。分析的純度を決定するために、移動相A=60/40/0.02
MeOH/2-プロパノール/HClO4 (v:v)又は移動相B=55/45/0.018
MeOH/2-プロパノール/HClO4 (v:v)を使用した。
【0122】
3-O-トリイソプロピルシリル-6-β-(2,3,4,6-テトラ-O-ベンジル-β-D-グルコピラノシル)アセトアミドモルヒネ10。SOC12(2.0mL)を、カルボン酸8(389mg,
0.67mmol)に添加し、この溶液を、室温で18時間攪拌した。過剰なSOCl2を、水浴(60℃)を使用し、高真空下で除去し、及び生じた黄色油状物を、CH2C12(5mL)中に溶解し、CH2Cl2
(5mL)中の3-O-トリイソプロピルシリル-6-β-アミノモルヒネ(196mg, 0.445mmol)及びEt3N(0.12mL,
0.89mmol)へ添加した。この溶液を、室温で1時間攪拌し、濃縮し、フラッシュクロマトグラフィーにより精製し(SiO2,
30:1〜10:1 EtOAc/MeOH)、10を白色固形物として得た(417mg,
93%):Rf=0.46(10:1 CH2Cl2/MeOH);mp=56.6℃;1H
NMR(CDC13)δ7.35-7.10 (20H), 6.59 (d, J=8.0 Hz, 1H), 6.40 (d, J= 8.0
Hz, 1H), 6.22 (d, J= 6.9 Hz, 1H), 5.71 (ddd, J= 3.0, 6.0, 9.5 Hz, 1H), 5.41
(dd, J= 1.9, 9.5 Hz, 1H), 4.89-4.48 (9H), 4.36 (t, J= 6.3 Hz, 1H), 3.72-3.46
(5H), 3.29 (t, J= 9.0 Hz, 1H), 3.17 (dd, J= 3.0, 5.2 Hz, 1H), 2.96 (d, J= 18.7
Hz, 1H), 2.88 (bs, 1H), 2.72 (dd, J= 2.7, 15.2 Hz, 1H), 2.50-1.72 (1OH),
1.30-1.05 (21H)。13C NMR (CDC13)δ170.0, 147.9, 143.6, 138.2, 137.8, 137.6,
132.9, 130.4, 128.7, 128.4, 128.34, 128.32, 128.0, 127.8, 127.76, 127.6, 127.0,
120.8, 118.4, 92.0, 86.8, 80.9, 78.7, 78.1, 77.4, 76.1, 75.7, 75.2, 75.0, 73.3,
68.7, 59.0, 49.3, 46.8, 44.1, 43.1, 40.3, 39.0, 36.4, 20.3, 18.0, 17.99, 12.8,
11.3;MS (EI) m/z 1006 [M+H]+。
【0123】
6-β-(2,2,4,6-テトラ-O-ベンジル-β-D-グルコピラノシル)アセトアミドモルヒネ11。シリルエーテル10(270mg,
0.27mmol)を、THF(4mL)に溶解した。水(0.05mL)及びTBAF(0.4mL,
0.4mmol, THF中1.0M溶液)を添加した。2.5時間後、濃度1%のHCl(4mL)を添加し、この混合物を2分間攪拌し、水(10mL)で希釈し、固形NaHCO3で塩基性とした。この混合物を、CH2C12
(5×30mL)で抽出し、乾燥し(Na2SO4)、濾過し、濃縮した。フラッシュクロマトグラフィー(24:1〜12:1
EtOAc/MeOH)は、11を白色固形物(205mg, 90%)として提供した:Rf=0.15(12:1
EtOAc/MeOH);mp=103.8℃;1H NMRδ7.34-7.10 (20H), 6.64 (d, J= 8.2 Hz, 1H), 6.46 (d, J= 8.2
Hz, 1H), 6.34 (d, J= 6.3 Hz, 1H), 5.68 (ddd, J= 2.5, 5.8, 9.5 Hz, 1H), 5.39 (d,
J= 9.5 Hz, 1H), 4.89-4.46 (10H), 4.34 (t, J= 6.3 Hz, 1H), 3.72-3.46 (6H), 3.30
(t, J= 9.1 Hz, 1H), 3.18 (bs, 1H), 2.97 (d, J= 18.1 Hz, 1H), 2.94 (bs, 1H),
2.74 (dd, J= 2.5, 15.7 Hz, 1H), 2.55-2.16 (7H), 1.95 (dt, J= 8.0, 12.6 Hz, 1H)
, 1.76 (d, J= 11.3 Hz, 1H);13Cδ170.4,
144.2, 143.6, 138.1, 138.0, 137.8, 137.7, 137.6, 132.6, 129.8, 128.38, 128.37,
128.33, 128.3, 128.2, 128.0, 127.8, 127.7, 127.7, 127.65, 127.6, 125.9, 119.1,
116.3, 93.0, 86.8, 80.8, 78.7, 78.1, 77.4, 76.0, 75.7, 75.3, 75.0, 73.3, 68.7,
59.0, 50.0, 47.0, 44.0, 43.0, 39.9, 38.8, 35.9, 20.2;MS
(EI) m/z 850 [M+H]+;HRMS (ESI) C53H57N2O8
についての計算値849.4115、実測値849.4099;11の平均純度は、tR=3.24分(移動相A)及びtR=4.66分(移動相B)を生じたことから、分析的HPLCにより、≧99%であることがわかった。
【0124】
6-β-(2,3,4,6-テトラ-O-ベンジル-β-D-グルコピラノシル)アセトアミド-7,8-ジヒドロモルヒネ12。テトラベンジルエーテル12(35.4mg,
0.042mmol)を、MeOHに溶解し、10%Pd-C(15mg)を添加した。混合物を、水素大気下で24時間攪拌し、その後セライトを通して濾過した。セライトを、MeOH(10mL)及びCH2C12(2mL)で洗浄し、濾液を濃縮し、12を白色固形物(35mg,
100%)として得た:Rf=0.66(200:80:5:0.05 CH2Cl2/MeOH/H2O/13% NH4OH);mp=94.3℃;1H
NMR (CDC13) δ 7.33-7.10 (20H), 6.92 (d, J= 8.4 Hz, 1H), 6.69 (d, J=
8.2 Hz, 1H), 6.56 (d, J= 8.2 Hz, 1H), 4.88-4.29 (9H), 3.73-3.32 (8H), 3.05 (bs,
1H), 2.96 (d, J= 18.4 Hz, 1H), 2.72 (dd, J= 2.7, 15.7 Hz, 1H), 2.49-0.95 (15H);MS
(EI) m/z 852 [M+H]+。
【0125】
ジヒドロモルヒネ-6-β-アセトアミドグルコース3。テトラベンジル誘導体11(41mg,
0.05mmol)を、MeOH(2mL)中に溶解した。濃HCl(13μL)、次に10%Pd-C(40mg)を添加した。この混合物を、水素大気下で、24時間攪拌した。この溶液を、Bio
Rad RG 501-X8混合床樹脂(1g)で中和し、この混合物をセライトを通して濾過した。固形物を、MeOH(30mL)で洗浄し、濾液を濃縮した。残渣を、エチルエーテルで摩砕し(3×2mL)、高真空下で乾燥し、3を白色粉末(12mg,
52%)として得た:Rf=0.4 (SiO2, 5:4:0.5:0.5 CHCl3/MeOH/H2O/濃NH4OH);mp=266.4℃(分解);1H NMRδ6.62
(s, 2H), 4.47 (d, J= 7.8 Hz, 2H), 3.81-3.77 (m, 2H), 3.55-3.43 (m, 4H), 3.00
(t, J= 9.3 Hz, 1H), 2.90 (d, J= 5.4 Hz, 1H), 2.84 (s, 3H), 2.79-2.69 (m, 1H),
2.63 (dd, J= 12.9, 14.4 Hz, 1H), 2.45 (d, J= 11.7 Hz, 1H), 2.30-2.05 (m, 3H),
1.77-1.57 (m, 4H), 1.34-1.24 (2H), 1.00-0.92 (2H);13C
NMR (CD3OD)δ173.9, 143.9, 142.6, 128.5, 122.3, 121.1, 119.4, 92.7,
81.6, 79.5, 77.9, 75.1, 72.0, 63.1, 52.7, 43.1, 41.9, 41.7, 40.3, 34.6, 29.2,
24.6, 21.7; MS (EI) m/z 491 [M+H]+;HRMS
(ESI) C25H35N2O8 の計算値491.2393、実測値491.2375;3の平均純度は、tR
=3.41分(移動相A)及びtR =3.74分(移動相B)を生じたことから、分析的HPLCにより、≧98%であることがわかった。
【0126】
受容体結合。ヒトμオピオイド受容体、ヒトδオピオイド受容体及びヒトκオピオイド受容体を用いて形質移入された細胞由来の膜への結合を、先に説明されたような、96-ウェルフォーマットを使用し行った(新規化合物10、11、12及び3は、1H
NMR、13C NMR及びMSにより特徴付けた。被験化合物3及び11の純度は、HPLCにより決定し、>98%であることがわかった)。細胞を、ラバーポリスマンにより剥ぎ取ることにより、プレートから除去し、Polytronホモジナイザーを用いTris緩衝液中でホモジナイズし、その後1回遠心し、追加の15分間の遠心27,000
×gにより洗浄した。ペレットを、50mM Tris(pH7.5)中に懸濁し、懸濁液を、μオピオイド受容体、δオピオイド受容体又はκオピオイド受容体について、各々、[3H]DAMGO、[3H]DPDPE、又は[3H]U69593と共に、インキュベーションした。総インキュベーション容量は1.0mLであり、及び試料は、25℃で60〜120分間インキュベーションした。結合反応におけるタンパク質の量は、約15μg〜30μgを変動した。この反応は、ガラスファイバーフィルターを備えるTomtec
96収集器(Orange, CT)を使用する濾過により終結させた。結合した放射活性を、Pharmacia
Biotechβ-プレート液体シンチレーションカウンター(ピスカタウェイ,
NJ)によるカウントにより決定し、データをカウント/分で表した。IC50値は、少なくとも6種の被験化合物濃度を用いて決定し、Graphpad/Prism(ISI,
サンディエゴ, CA)を用いて計算した。Ki値は、Cheng及びPrusoffの方法により決定した[Traynor,
J. R.の論文 Mol. Pharmacol., 47:848-854 (1995)]。
【0127】
[35S]GTPγ-S結合。[35S]GTPγ-S結合を、本質的にTraynor及びNahorskiの論文に説明されたように行った[Zaveri,
N.らの論文、Eur. J. Pharmacol., 428:29-36 2001]。μオピエート受容体、δオピエート受容体又はκオピエート受容体を形質移入した細胞を、組織培養皿から20mM
Hepes、1mM EDTAへ剥ぎ取り、その後500 ×gで10分間遠心した。細胞を、この緩衝液中で再懸濁し、Polytronホモジナイザーを使用しホモジナイズした。ホモジネートを、27,000
×gで15分間遠心し、ペレットを20mM Hepes、10mM MgC12、100mM
NaCl(pH7.4)を含有する緩衝液A中に懸濁した。懸濁液を、27,000 ×gで再遠心し、再度緩衝液A中に懸濁した。結合アッセイに関して、膜(8〜15μgタンパク質)を、[35S]GTPγ-S(50pM)、GDP(10μM)、及び被験化合物と共に、総容量1.0mLで25℃で60分間インキュベーションした。試料を、ガラスファイバーフィルター上で濾過し、結合アッセイについて説明されたようにカウントした。プログラムGraphPad
Prismを用い、統計解析を行った。このアッセイにおいて、受容体に関する化合物の効力又は親和性を、[35S]GTPγS結合の刺激に関するそのEC50により決定した。アゴニスト効能は、化合物が対照に対し[35S]GTPγS結合を最大刺激した程度として定義した。EC50値は、[35S]GTPγS結合の50%最大刺激を生じた化合物の濃度を表している。
【0128】
(実施例34):アルコール中毒及びニコチン中毒に対する6-ナルトレキサミド
米国において、アルコール依存症、ニコチン中毒、及びギャンブル依存症は、著しい社会的及び経済的結果を伴う、重篤な精神障害である。疫学的試験は、アルコール依存症は、米国人口の約14%であり、いずれかの時点で肝臓に罹患していることを示した[Regierらの論文、Arch.
Gen. Psychiatry, 50:85-94 (1993)]。毎年米国民100,000名は、アルコール依存症の直接的及び間接的原因で死亡すると推定され[McKinnis及びFoegeの論文、JAMG,,
270:2207-2212 (1993)]、健康及び社会的コストを含む経済的コストは、年間1660億ドルに達している(stagger)(NIDA及びNIAAA、1998)。喫煙は、肺癌に直接関連しており、禁煙は、肺癌を防ぐ主要手段である。アルコール依存症又はニコチンからの離脱症状を治療するための薬物療法の使用、並びにアルコール依存症患者又は喫煙者のリハビリテーションにおける役割は限定されている。多くの薬物療法がテストされ、中毒患者において、アルコール消費を減少又は回避させる。これらの一部は、ジスルフィラム、リチウム、選択的セロトニン再取込みインヒビター、及びアカンプロセートである。ジスルフィラムは、限定的有効性を有することが示されている[Garbuttらの論文、JAMA,,
281:1318-1325 (1999)]。アルコール依存症からの離脱症状を治療するために、薬物療法が使用されるいくつかの方法が存在する。喫煙を止めさせるいくつかの方法が開発されているが、再発率は高い。アルコール依存症に関して、ジスルフィラムのような抑止薬は、アルコールの摂取を喜ばしくないものとする。対照的に、薬物療法は、アルコールの強化(reinforcing)作用を減弱するか、又はアルコールを摂取する衝動もしくは渇望を低下させることにより、アルコール摂取を減少することを試みる。薬理学的物質は、内因性オピオイド、カテコールアミン(特にドパミン、セロトニン)及びグルタミン酸のような興奮性アミノ酸を含む、エタノール又はニコチンの強化又は識別力刺激作用の基礎となるいくつかのニューロトランスミッターシステムに影響するように見える[Kranzlerの論文、Am.
J. Psychiat., 152:391-397 (1995)]。多くの薬物療法が、アルコール又はニコチン依存症の治療において価値があることが示されているという事実にもかかわらず、薬理学的療法(Pharmacotherapy)は大きい作用を有さない。アルコール依存者又は喫煙者の亜群は、ある種の薬物療法に良好に反応することのいくつかの証拠があり、これは治療のマッチングは、薬物療法の効能を増加し得ることを示唆している。しかし戦略的方法、用量及び治療期間に関するかなり基本的情報が、正に入手可能になりつつあり、及びはるかに多い情報が確立されることが必要である。これらの重要な問題点に対処するために、新規方法及び新規薬物療法の開発が明らかに必要である。
【0129】
1994年に、ナルトレキソンは、米国FDAにより、アルコール依存症の治療用に承認された。ナルトレキソンは、アゴニスト活性を伴わず、乱用の可能性もない、純粋なオピオイドμ受容体アンタゴニストである。ナルトレキソンは、問題のない飲酒者の前臨床試験において、強化(すなわち刺激物)作用を低下させ、かつ最初のアルコール消費の望ましくない(すなわち鎮静)特性を増大させることがわかった[Swiftらの論文、Am.
J. Psychiat., 151: 1463-1467 (1994)]。齧歯類及びサルを使用した試験は、オピオイドアンタゴニストであるナロキソン及びナルトレキソンが、自発的消費及びストレスが誘導したアルコール消費の増加を減少させることを示し、このことは、これらの物質が、アルコール消費の強化作用を妨害し得ることを示唆している[O'Brienらの論文、Alcohol,
13:35-39 (1996)]。ナルトレキソンは、より高いレベルの渇望及び貧弱な認知機能を伴うアルコール依存者において最も有益であろう[Jaffeらの論文、J.
Consult. Clin. Psychol., 64:1044-1053 (1996)]。しかし、1つの試験において、治療を受けている患者の15%は、耐え難い悪心を含む有害作用ために、治療を早期終結している[Croopらの論文、Arch.
Gen. Psychiat., 54:1130-1135 (1997)]。ナルトレキソンは、肝臓疾患のアルコール依存症患者での使用を複雑にしかつ治療を混乱させる、用量-依存型肝毒性副作用にも関連している[Masonらの論文、Arch.
Gen. Psychiat., 56:719-724 (1999)]。さらなる欠点は、短い望ましい作用期間及び比較的低い生体利用効率を含み[Wallらの論文、Drug
Metab. Dispos., 9:369-375 (1981)]、恐らくδ受容体及びκ受容体に関する比較的低い親和性が、アルコール飲酒の強化作用を消滅することに関与していると考えられるであろう。ナルトレキソンの有益な作用は、経時的に次第に消滅する。加えてナルトレキソン治療の服薬遵守は様々であり、また愁訴の多い対象においてのみ、プラセボよりも有意により良い活性のある薬物療法であった[Volpicelliらの論文、Arch.
Gen. Psychiat., 54:737-742 (1997)]。まとめると、ナルトレキソンは、アルコール依存症の間で飲酒行動に対し、軽度の作用を生じるように見える。場合によっては、ナルトレキソンの作用は、極めて強固であるが、その効能の証拠には一貫性が少ない。しかしナルトレキソンよりも優れた医薬特性を伴うオピオイドアンタゴニストであるナルメフェンは、アルコール治療薬物療法としてより有望であろう。ナルトレキソンは、禁煙に有用であることが報告されている[O'Malleyら、米国特許第6,541,478号]。
【0130】
オピオイド受容体アンタゴニストは、アルコール-所望行動に対し、明らかに直接作用を有する。例えば、ナロキソンの微量透析による側座核ニューロンへの直接灌流は、アルコール-媒介したドパミン放出を阻害した[Benjaminらの論文、Brain
Res., 621:137-140 (1993)]。オピオイド受容体の遮断によるアルコール消費の減少は、この強化システムにおけるナロキソンの直接作用を示唆している。動物試験は、μ-オピオイド受容体、δ-オピオイド受容体及びκ-オピオイド受容体は、アルコール-誘導した強化に寄与することの証拠を提供した[Herz
(1997);Ulmらの論文、J. Clin. Psychiat., 56:5-14 (1995)]。κ-オピオイド受容体に選択性のあるノルビナルトロフィン(norbinaltorphimine)(norBNI、図1)に関連した、かなりの研究が行われた[Portogheseらの論文、J.
Med. Chem., 31:1344-1347 (1987)]。norBNIのSARは、広範に調べられており、2つのナルトレキソンで誘導したファーマコフォアを含む、二価のリガンドである。ただ1つのnorBNIファーマコフォアが、κアンタゴニスト活性に必要であり、及び受容体-結合したリガンドの第二のファーマコフォアにおける塩基性基(N-17位の)は、選択性を付与する「アドレス」として作用すると考えられる。分子の受容体への配向のための固い足場の明らかな必要性の観点において、ナルトリンドール(NTI、図15参照)を含む代わりの構造が開発されている。NTIに関連したこのインドロモルヒナン構造は、norBNIよりも簡単であり、及びκアンタゴニストは、Glu297アドレスサブサイトと相互作用することのかなりの証拠を提供している[Stevensらの論文、J.
Med. Chem., 43:2759-2769 (2000)]。
【0131】
更にκアンタゴニスト活性に必要なアドレス群の正帯電したアミノ官能基の必要要件は、強力かつ選択的オピオイドκアンタゴニストとして非オピエート-誘導した化合物を説明した最近の報告において証明された。化合物JDTic(図16参照)は、2つの塩基性アミン基の重要性を例示しており:両方は各々、アドレス基及びメッセージ基である。更なる研究は、nor-BNIシリーズについて認められないκ選択性におけるフェノール基の重要性を示した[Thomasらの論文、J.
Med. Chem., 47:1070-1073 (2004)]。キメラκ-μオピオイド受容体を基に、負帯電した残基(すなわちGlu297)は、κ-選択的結合及び機能活性に重要であることは明らかである。高度に活性のあるκ-選択性又は非κ-選択性受容体の選択的アンタゴニストは、中毒、特に例えばニコチン又はアルコールのような、物質中毒の治療に有用であろう。
【0132】
ナルメフェン1及びナルトレキソン2の化学構造を、図17に示す。これらの化合物は、オピオイド過量、中毒及びアルコール依存症の治療に使用される。本試験の医化学的目的は、1及び2の各々代謝上脆弱な6-メチレン又は6-ケト基を、アミド部分と置換することによる、これらの化合物の代謝的に安定なアナログの開発であった。μオピオイド受容体及びκオピオイド受容体の選択性及び効力を増加するために、アミド基の芳香環内の様々な置換パターンが調べられた。この目的に合致するために6-αナルトレキサミド又は6-βナルトレキサミドの新規4工程合成が開発された。水性エタノールの還流時のNaOAc存在下でのナルトレキリン2の塩酸ヒドロキシルアミンとの縮合は、対応するオキシム3(図18参照)を生じた。引き続きの3のオキシム官能基の対応するアミン4への還元は、BH3/THFとの48時間の加熱及び引き続きの水性後処理により実現され、アミン4をジアステレオマーの9:1(β/α)混合物として生じた。ジアステレオマーアミンは、シリカゲル上でのクロマトグラフィーにより分離した。アミン4は、CH2C12中のベンゾトリアゾール-1-イル-オキシ-トリス-(ジメチルアミノ)ホスホニウムヘキサ-フルオロリン酸(BOP)及びジイソプロピルエチルアミンの存在下で、カルボン酸(RCOOH)とカップリングした。あるいは、これらのアミドは、CH2C12中の酸塩化物、トリエチルアミン及び4の反応により形成した。両方の場合において、生成物は、3-位のヒドロキシル基のエステル化により生じる副産物を脱エステル化するために、メタノール中のK2CO3で処理した。BOPカップリング法が、3-位に酸塩化物法よりもより少ないエステル化を生じるが、3-位での若干のエステル化は避けられない。従って、中間体アミドエステルの精製を助けるために、過剰な酸誘導体による反応を実施することはより都合が良いことがわかった。これらの方法は、アミド5及び6a-hを簡便に調製することを可能にした。Pd-C及びHClの存在下での水素化分解によって、化合物5のベンジル基を除去したグルコース複合体6aを生じた。
【0133】
【表7】
【0134】
対応するアゴニストについて観察された効力及び選択性[MacDougallらの論文、J.
Med. Chem., 47:5809-5815 (2004);MacDougallらの論文、Bioorganic
Med. Chem., 12:5983-5990 (2004);MacDougallらの論文、Bioorganic
Med. Chem., 15:583-1586 (2005)]は一般に、これらのアンタゴニストにおいて維持された。アミド6d、6e及び6cは全て、オピオイド受容体に関して高い親和性を有し、かつδ受容体及びκ受容体で部分的又は完全なアゴニスト作用を有した(表8)。細胞膜へのGTPγS結合を基に、全ての化合物は、μ受容体で強力なアンタゴニストである。化合物6aは、μ受容体について高度の親和性を有し、δ及びκ受容体について中等度の親和性を有した。6aは、μ及びκ受容体で強力なアンタゴニストであり、δ受容体で部分的アゴニストである。まとめると、このデータは、μ受容体単独又はμ受容体及びκ受容体のアンタゴニスト作用の機能性の結果による、強力な先例(lead)が存在することを示している。
【0135】
【表8】
【0136】
代謝安定性を調べるために、代謝安定性及び化学安定性を試験するための、HPLC及びLCMSアッセイを開発し、一般にこれらのシリーズの化合物は、代謝及び分解に対し著しく安定していることがわかった。これらのアナログを、C-6位の遮断のために肝臓調製物中で安定しているようにデザインし、かつこれは、観測されたものであった。予備的結果は、6d及び6aは、強力なオピオイドアンタゴニストとして有望であることを示した(表7及び8)。テトラ-O-ベンジル-β-D-グルコピラノシルで保護された化合物5が、合成され、そのインビトロ結合及び機能特性を調べた。化合物5は、μ受容体に関して高い親和性を有し、かつκ受容体に関して中程度の及びδ受容体に関して低い親和性を有した。これらの試験は、グルコースアナログそれら自身を、開発候補として指摘している。
【0137】
このデータは、代謝的に安定なオピオイドアンタゴニストは、インビトロにおいて合成することができ、かつ試験することができることの実行可能性を示している。代謝的に安定なC-6置換されたオピオイドアンタゴニストは、好ましい副作用プロファイルを維持するであろう。インビトロ結果は、κアゴニスト又はアンタゴニスト特性のいずれかを伴う、強力なμアンタゴニストを合成することができることを示している。アルコール消費の減少においてκアンタゴニスト又はアゴニスト作用がより効率的であるかどうかを試験するために、アルコール依存症及びニコチン中毒の動物モデルにおいて先例を試験した。高度に活性のあるκ-選択性又は非-κ-選択性受容体の選択性アンタゴニストは、中毒、特に例えばニコチン又はアルコールなどの物質に対する中毒症状の治療に有用であろう。
【0138】
下記実施例(実施例35-43)において、全ての反応は、特に記さない限りは、炉乾燥したガラス製品を使用し、周囲温度で磁気攪拌しながら、窒素陽圧下で実施した。空気-及び水分-感受性液体を、シリンジによりゴム製隔壁を通して移した。カラムクロマトグラフィーのために、シリカゲル(230〜400メッシュ)を使用した。DMFを、中間アルミナのカラムを通して乾燥させ、また使用前は、窒素下活性化した4Å分子篩上で貯蔵した。全ての他の溶媒及び試薬は、入手したまま使用した。1H
NMRスペクトルは、300MHz Varian NMRにより記録した。化学シフトは、7.26ppmで
CDC13に対するppm(δ)で報告した。1H
NMRスペクトルは、特に記載しない限りは、CDC13で記録した。融点は補正せずに報告した。高解像度質量分析を、University
of California at Riverside において、Opus V3.1及びDEC
3000 Alpha Stationデータシステムを備えた、VG7070スペクトロメーター上で行った。燃焼分析が特定されない場合は、分析純度は、D7500積分器及びHamilton
PRP-Iステンレス鋼カラム(250mm ×4.6mm内径)を備えた、Hitachi
L74液体クロマトグラフィーを使用する、順相HPLCにより決定した。
ナルトレキソンオキシム3を、以下の方法に従い調製した。ナルトレキソン(6.2g, 1.46mmol)、NH2OH-HCl(1.5g)及びNaOAc(2.9g)を、無水EtOH(80mL)及び水(5mL)中に溶解した。混合物を、2.5時間還流加熱し、その後濃縮した。水(20mL)を添加し、混合物を、K2CO3により塩基性とし、CHC13で抽出した。CHCl3抽出物は、ブラインで洗浄し、Na2SO4上で乾燥し、濾過及び濃縮し、白色固形物(5.83g,
90%)とした:Rf =0.65 (5:1:0.2 EtOAc/MeOH/NH4OH);MS(EI)
m/z=357[MH]+。
6-β-ナルトレキサミン及び6-α-ナルトレキサミン、4b、4aを、以下の方法に従い調製した。ナルトレキソンオキシム(5.83g, 16.3mmol)を、THF(40mL)に溶解し、カニューレにより、BH3:THF
(300mL, 300mmol, THF中1M溶液)の10℃溶液へ10分かけて移した。白色沈殿が形成され、その後反応液を2日間かけて還流温度へ加熱するにつれて、ゆっくり溶解した。この溶液を、室温へ冷却し、水(10mL)及び1N
KOH(200mL)をゆっくり慎重に添加した。溶液を、2時間還流加熱した。その後10%HCl(225mL)によりpHを2.5に低下した。この溶液を、2時間還流加熱し、濃縮し、THFを除去し、その後K2CO3で塩基性(pH8-9)とした。混合物を、CHC13(4×150mL)で抽出し、抽出物をNa2SO4上で乾燥し、濾過し濃縮した。得られた油状物を、SiO2上のクロマトグラフィー(26×60cm,
CH3CN/MeOH/NH4OH, 25:5:1による溶離)により精製し、4b(2.14g,
38%)を白色-黄色固形物として得た:Rf=0.20;1H
NMR (300 MHz, CD3ODを2滴含むCDC13)
δ6.61 (d, J = 8.1 Hz, 1H), 6.49 (d, J = 8.1 Hz, 1H), 4.17 (d, J = 7.5 Hz, 1H),
3.39-0.45 (20 H);MS m/z = 343 [MH]+。α-及びβ-ジアステレオマーの混合物からなる追加の物質0.64g(12%)を、単離した。この物質の尾部画分の分取クロマトグラフィー(SiO2,
CH3CN/MeOH/NH4OH, 25:5:1)は、4aの分析的試料を生じた:Rf
=0.16;1H NMRδ6.65 (d, J = 8.1 Hz, 1H), 6.46 (d, 8.1 Hz, 1H), 4.50 (d, J =
3.0 Hz, 1H), 3.34 (dt, J = 3.9, 12.6 Hz, 1H), 3.04 (t, J = 6.6 Hz, 1H), 2.95
(s, 1H), 2.63-0.29 (17H);MS m/z 343 [MH]+。
【0139】
【化11】
【0140】
(実施例35):6-アセトアミド-2,3,4,6-テトラ-O-ベンジル-D-グルコピラノシル-14-ヒドロキシ-17-(シクロプロピルメチル)ノルデスモルヒネ, 5
6-ナルトレキサミン(100mg, 0.292mmol)、カルボン酸(RCOOH, スキーム1)(187mg,
0.321mmol)及びBOP(142mg, 0.321mmol)を、CH2CI2(3mL)中に溶解し、NPr2Et
(0.16mL, 0.92mmol)を添加した。無色の溶液を、室温で90分間攪拌し、濃縮した。残渣を、SiO2
(20:1 EtOAc/MeOH)を通して濾過し、白色固形物を提供した。この物質(240mg)を、MeOH
(10mL)に溶解し、K2CO3 (36mg, 0.26mmol)を添加した。混合物を、115分間攪拌し、飽和NH4Cl(10滴)を添加した。混合物を濃縮し、SiO2
上で吸収し、SiO2クロマトグラフィー(20:1
CHCl3/MeOH)により精製し、7を白色固形物(200mg,
6-ナルトレキサミンナルトレキサミンから75%、2-工程)として得た:Rf=0.11
(20:1 CHCl3/MeOH);MS (EI) m/z=907[M]+;1H
NMR (CDCl3, 重要な共鳴)δ7.31-7.06 (m);6.76
(d), 6.71 (d);6.55 (d);6.54 (d) 4.94-4.38 (m);3.86-3.79
(m);13C NMR (CDCl3)δ170.4, 143.6, 138.1, 137.6, 128.4, 128.4,
128.3, 128.1, 127.9, 127.8, 127.78, 127.7, 127.69, 127.64, 118.9, 93.8, 86.8,
81.0, 78.4, 75.8, 75.7, 75.2, 75.0, 73.2, 70.1, 59.2, 50.9, 30.2, 4.1, 3.9。
【0141】
(実施例36):14-ヒドロキシ-17-(シクロプロピルメチル)ノルデスモルヒネ6-アセトアミドグルコース, 6a
無水MeOH(8mL)を、ベンジル化した誘導体5(180mg, 0.198mmol)及び10%活性化したPd-C(180mg)に添加した。濃HCl(0.015mL)を、シリンジにより添加した。このフラスコを、窒素で掃流し、次に水素バルーン下で21時間攪拌し、セライトを通して濾過し(MeOHで溶離)、濃縮した。得られた白色固形物を、エチルエーテル(無水物、3×2mL)で摩砕し、6aを白色固形物(92mg,
85%)として提供した:Rf=(5:4:0.5:0.5 CHCl3/MeOH/H2O/NH4OH);1H
NMR(CD3OD、重要な共鳴) δ6.72 (s), 4.67 (d);
4.60-4.52 (m);3.99 (d); 3.90-3.48 (m); MS
m/z = 547 [M+H]+。
【0142】
(実施例37):酸塩化物を用いた6-ナルトレキサミンのアミド化の一般的手法。6-(ピリジン-3'-イル)カルボキサミド-14-ヒドロキシ-17-(シクロプロピルメチル)ノルデスモルヒネ, 6b
6-アミノナルトレキサミン(53mg, 0.155mmol)を、CH2Cl2(2mL)中に溶解し、塩化ニコチニル-HCl(40mg,
0.226mmol)及びNEt3(0.085mL, 0.465mmol)を添加した。この溶液を、1.5時間攪拌し、濃縮した。残渣を、SiO2(10:1
CH2Cl2/MeOH)上のクロマトグラフィーにより精製し、アミドエステルを黄色-白色固形物(63mg,
74%)として提供した:Rf=0.11(10:1 CH2Cl2/MeOH);MS
m/z=552。このアミドエステル(53mg, 0.096mmol)を、MeOH(2mL)中に溶解し、K2CO3(46mg,
0.33mmol)を添加した。混合物を、室温で1時間攪拌し、NH4Cl(飽和水溶液、10滴)及びSiO2(2g)を添加した。この混合物を濃縮し、SiO2クロマトグラフィー(10:1
CHCl3/MeOH)により精製し、標題化合物を黄味がかった-白色固形物(27mg,
63%)として得た:Rf=0.31(10:1 CHCl3/MeOH);MS
m/z=447。
(実施例38):BOP試薬及びカルボン酸を用いた6-ナルトレキサミンのアミド化の一般的手法。6-(3'-ジメチルアミノ)ベンズアミド-14-ヒドロキシ-17-(シクロプロピルメチル)ノルデスモルヒネ, 6c
【0143】
CH2Cl2(1.5mL)を、ナルトレキサミン(50mg,
0.146mmol)、3-ジメチルアミノ安息香酸(24mg, 0.14mmol)、BOP試薬(64mg,
0.14mmol)及びPr2EtN(0.081mL, 0.47mmol)に添加した。黄色溶液を、室温で80分間攪拌し、濃縮した。残渣を、クロマトグラフィーSiO2(10:1
EtOAc/MeOH)にかけ、白色固形物(70mg)を提供した。この物質を、MeOH(4mL)に溶解し、K2CO3(30mg,
0.22mmol)を添加した。90分後、この混合物を濃縮した。残渣を、SiO2(10:1 CHCl3/MeOH)上のクロマトグラフィーにより精製し、標題化合物を白色固形物(17.4mg,
ナルトレキサミンから24%):Rf=0.16(10:1 CHCl3/MeOH);MS
m/z=490 [MH]+。
【0144】
(実施例39):6-(3'-メトキシ)ベンズアミド-14-ヒドロキシ-17-(シクロプロピルメチル)ノルデスモルヒネ, 6d
参照JMM-VIII-42。化合物6bについて説明された一般的手法を使用し、6-ナルトレキサミン(90mg,
0.263mmol)、3-アニソイルクロリド(0.09mL, 0.69mmol)、NEt3(0.11mL,
0.83mmol)及びCH2Cl2(2mL)は、中間体アミドエステルを白色泡状物(96mg)として生じた;MS
m/z=611。この物質のMeOH(3mL)中K2CO3(43mg,
0.31mmol)による加水分解、それに続くSiO2クロマトグラフィー(10:1
CH2Cl2/MeOH)は、6dを白色固形物として生じた:57mg(78%);MS
m/z=477。
(実施例40):6-(チオフェン-2'-イル)アセトアミドミド(acetamidomido)-14-ヒドロキシ-17-(シクロプロピルメチル)ノルデスモルヒネ, 6e
【0145】
化合物6bについて説明された一般的手法を使用し、6-ナルトレキサミン-(19mg,
0.055mmol)、2-チオフェンアセチルクロリド(0.014mL, 0.11mmol)、NEt3(0.019mL,
0.14mmol)及びCH2Cl2(2mL)は、中間体アミドエステルを白色泡状物(15mg,
46%)として生じた;Rf=0.12(30:1 CH2Cl2/MeOH);MS
m/z=591[MH+]。この物質(13mg, 0.022mmol)のMeOH(2mL)中K2CO3(2.5mg,
0.0.018mmol)による加水分解、それに続くSiO2クロマトグラフィー(15:1
CH2Cl2/MeOH)は、6eを白色固形物として生じた(10mg,
100%):Rf=0.17(15:1 CH2Cl2/MeOH);MS
m/z=467。
(実施例41):6-(3',5'-ジメトキシ)ベンズアミド-14-ヒドロキシ-17-(シクロプロピルメチル)ノルデスモルヒネ, 6f
【0146】
化合物6cについて説明された一般的手法に従い、4(90mg, 0.263mmol)、3,5-ジメトキシ安息香酸(72mg,
0.395mmol)、BOP(174mg, 0.394mmol)、NPr2Et(0.15mL,
0.84mmol)及びCH2C12(3mL)は、中間体アミドエステルを白色-オレンジ色固形物(186mg)として生じた。この物質のMS分析は、これがアミドエステル(m/z=671)及びアミド(m/z=507)の混合物からなることを示した。この物質のMeOH(4mL)中K2CO3(150mg,
1.4mmol)による脱エステル化、及びSiO2クロマトグラフィー(20:
1 CH2Cl2/MeOH)は、6fを白色固形物として提供した:MS
m/z=507。
【0147】
(実施例42):6-(4'-クロロ)ベンズアミド-14-ヒドロキシ-17-(シクロプロピルメチル)ノルデスモルヒネ, 6g
参照VIII-58。化合物6cについて説明された一般的手法に従い、4(90mg,
0.26mmol)、4-クロロ安息香酸(62mg, 0.39mmol)、BOP(174mg, 0.394mmol)及びNEtPr2(0.15mL,
0.842mmol)、引き続きのSiO2クロマトグラフィー(20:1
EtOAc/MeOH)は、中間体アミドエステルを白色固形物としてもたらした:Rf=0.5
(20:1 EtOAc/MeOH);MS m/z=620。この物質のMeOH(4mL)中K2CO3(72mg,
0.53mmol)による脱エステル化、それに続くSiO2クロマトグラフィー(20:1
CH2Cl2/MeOH)は、6gを白色固形物(76mg,
60%)として提供した:Rf=0.41 (20:1 CH2Cl2/MeOH);MS
m/z=480。
【0148】
(実施例43):6-(3'-ヒドロキシ)ベンズアミド-14-ヒドロキシ-17-(シクロプロピルメチル)ノルデスモルヒネ, 6h
参照JMM-VIII-59、VIII-64。化合物8bについて説明された一般的手法に従い、4(90mg,
0.26mmol)、3-アセトキシ安息香酸(71mg, 0.39mmol)、NEtPr2(0.15mL,
0.84mmol)、BOP(174mg, 0.39mmol)及びCH2C12(2mL)、並びにSiO2クロマトグラフィー(20:1
EtOAc/MeOH)は、中間体アミドエステルを白色泡状物(180mg)として生じた:Rf=0.28(10:1
CHCl3/MeOH);MS m/z=667。この物質のMeOH(2mL)中K2CO3(72mg,
0.53mmol)による脱エステル化、及びSiO2クロマトグラフィー(20:1
CH2Cl2/MeOH)は、8hを白色固形物(76mg,
60%)として生じた:Rf=0.41(20:1 CH2Cl2/MeOH);MS
m/z=480。
【0149】
(実施例44):インビトロ代謝安定性
インビトロ試験における化合物の代謝安定性は、インビボ実験における代謝安定性に関する可能性についての重要な情報をもたらすことができる。オピオイドクラスの化合物の代謝の卓越した経路はC-6位での代謝のためであるので、代謝安定性を、合成化合物の半減期を延長させるため、生体利用効率を増加させるため、並びに薬物動態プロファイル及び副作用プロファイルを改善させるために、化合物中にデザインした。薬物のデザイン及び開発の初期段階におけるADMETの不安定性を克服することにより、より多くの薬物-様化合物を生じた。
【0150】
選択された化合物のインビトロ代謝安定性試験を、プールしたヒト肝臓S9、ラット肝ミクロソーム及びマウス肝ミクロソームの存在下において試験した。これらの試験を、ヒト代謝状況を模倣するように行い、動物肝代謝試験を、インビボ動物試験のモデル試験として行った(下記参照)。アッセイ緩衝液は、3.2mg/mLのヒト肝臓プールしたS9又は1.2mg/mLラットもしくはマウス肝ミクロソーム、0.1Mリン酸カリウム緩衝液(pH7.4)、0.1mM化合物、0.5mM
NADP+、0.5mMグルコース-6-リン酸、5IU/mLグルコース-6-リン酸デヒドロゲナーゼ、1mg/mL
DETAPAC、7mM MgCl2 を含み、最終インキュベーション容量0.25mLであった。0分、10分、25分、45分又は60分後、1mL
CH2Cl2/IPA(3:1 v:v)の添加により、反応を停止させ、完全に混合した。遠心後、有機相を蒸発させ、窒素流れ下で乾燥し、200μメタノールに溶解し、ボルテックスし、遠心した。HPLCを使用し、有機抽出物を、Richard
Scientific(Novato, Ca)から購入したAxxi-Chrom Silica 5μmカラムを用いて、Hitachi
HPLC上で分析した。移動相は以下からなる:55/45/0.018 (MeOH/IPA/HClO4)及び流量1.5mL/分、UV検出254nm。ナルトレキソン、ナルメフェン及び化合物6dの保持時間は、各々、2.7分、2.1分、及び2.6分であった。
【0151】
表9に示したように、ナルトレキソン及びナルメフェンは、試験した肝調製物の存在下で、かなりの代謝不安定性を有した。対照的に、ヒト肝臓S-9及びマウス肝ミクロソームの存在下で、化合物6dは、検出的には代謝されなかった。ラット肝ミクロソームの存在下で、少量の代謝産物が認められたが、これは、ナルトレキソン又はナルメフェンよりも、各々、6倍又は22倍安定していた。まとめると、化合物6dについてかなりの代謝安定性が認められ、化合物6d及び同族体の生体利用効率はかなりの量であると推定される。
【0152】
【表9】
【0153】
(実施例45):インビボ活性
オピエートによる急性予備治療後の身体禁断徴候のアンタゴニスト-突発(precipitate)により規定されたような、急性依存状態の観察が、ヒト、サル、イヌ、ハムスター、マウス及びラットにおいて報告されている。ヒト及びラットの両方において、オピエートは、モルヒネへの単回曝露後のオピエート離脱症状の徴候を突発する。ラットのパラダイムは、ヒトにおける状態に類似している。従ってナロキソン注射前4時間のモルフィン(5.0mg/kg)による単回処置後の、ナロキソン(0.03〜3.0mg/kg)は、離脱症状の身体的徴候を突発した[Schulteisらの論文、PsychoPharmacology,
129:56-65 (1997)]。離脱症状強度の全体的指標には、Gellert及びHoltzmanの論文の重み付きスケールを使用した[J.Pharmacol.
Exper. Ther., 205:536-546 (1978)]。このスケールは、体重減少、回避試みの回数、咆哮(roaring)、禁断の激しい震えの回数、腹部収縮の事例の等級化した徴候、並びに下痢、顔面萎縮/歯の根の合わない震え(chattering)、嚥下-運動(swallowing-movement)、多量の唾液分泌、色素涙、下垂症、異常な姿勢、陰茎つくろい(penile
grooming)/勃起/射精、及び取扱い時の興奮を含むチェックされた徴候(存在又は非存在)からなる。本発明者らの研究に関して、Global身体的徴候(SS)等級付けは、Gellert及びHoltzmanの重み付きスケール(1978)を基に、先の個々の徴候の全てを含んだ。モルヒネ(5.0mg/kg)単回投与の4時間後、化合物6a又は6dの単回投与量を、ラット群に投与した。皮下注射後1分で、離脱症状の身体的徴候が10分間認められた。これらの結果は、図19及び20にまとめている。
【0154】
まとめると、成体雄ラットにおいて、モルヒネ5.0mg/kg投与の4時間後に皮下経路により投与された化合物6a及び6dは、離脱症状の身体的徴候を突発した。化合物6dは、離脱症状の身体的徴候の用量-依存型の増加を引き起こし、ピークは0.3mg/kgであった。0.1mg/kg未満の概算EC50値が認められた。より高い投与量は、この作用を減少した。化合物6aに関して、約1mg/kgの概算EC50は、離脱症状の身体的徴候を突発することがわかった。比較のためのナルメフェンは、約0.6〜1mg/kgのEC50値を与えた、離脱症状の身体的徴候における、(雑音が多いが(albeit
noisy))用量-依存性の増加をもたらした。文献の基準について、ナロキソンの離脱症状の身体的徴候を突発する最低有効量は、0.1〜0.3mg/kgと計算されている[Schulteisらの論文、Psychopharmacology,
129:56-65 (1997)]。結論として、このデータは、化合物6a、6d及び同族体の、強力なオピエート受容体アンタゴニストとしてのインビボ効能を裏付けている。これらの化合物は、比較的迅速に中枢神経系へ移行し(10分以内)、オピエート受容体に対し強力な作用を発揮する。この受容体システムは、アルコール及びニコチンに対する易中毒性を強力に制御するので、6のようなオピエートアンタゴニストは、ニコチン中毒及びアルコール中毒の軽減に有効であるはずである。
【0155】
本願明細書に説明された実施例及び態様は、例証のみを目的とし、かつそれらを考慮し様々な修飾又は変更が、当業者に示唆され、本願明細書及び添付された「特許請求の範囲」の精神及び範囲内に含まれることは理解される。本願明細書に列記された全ての刊行物、特許及び特許出願は、あらゆる目的のためにそれらの全体が本願明細書に参照として組入れられている。
【図面の簡単な説明】
【0156】
【図1】図1は、式IVに示した化合物の合成を示す。
【図2】図2は、チオ糖2a及び2bの合成を示す。
【図3】図3は、式Vの化合物の合成を示す。
【図4】図4は、式VIの化合物の合成を示す。
【図5】図5は、モルヒネの投与により誘導された用量-依存型の痛覚喪失を示す。データは、平均%MPE(±SEM)である。アスタリスクは、ビヒクル対照からの有意差を表す(StudentNewman-Keuls、p<0.05)。プラス印は、3mg/kgモルヒネからの有意差を表す(StudentNewman-Keuls、p<0.05)。
【図6】図6は、2種の高投与量13eで生じた鎮痛作用である。データは、平均%MPE(±SEM)である。アスタリスクは、ビヒクル対照からの有意差を表す(StudentNewman Kenls、p<0.05)。プラス印は、10及び30mg/kg13eの間の有意差を表す(Student Newman-Keuls、p<0.05)。
【図7】図7は、3aの投与により誘導された、用量-依存型の痛覚喪失を示す。データは、平均%MPE(±SEM)である。アスタリスクは、ビヒクル対照からの有意差を表す(StudentNewman Kenls、p<0.05)。プラス印は、3mg/kg 3aからの有意差を表す(StudentNewman-Keuls、p<0.05)。
【図8】図8A-Cは、ヘロイン依存型ラット(ラット#96)におけるヘロイン摂取に対する、オピエートアナログ13eの作用を示す。ラットには、活動/暗所相(午後6時〜午前6時)の15分前(午後5時45分)に、オピエートアナログ13e(0、0.04、及び0.2mg/kg、皮下注射)を、対象内デザインで注射した。データは、暗所相での1時間毎(図8A)、3時間毎(図8B)、及び12時間全体(図8C)についての総ヘロイン摂取(60μg/kg/0.1ml注入)で表した。
【図9】図9A-Cは、ヘロイン依存型ラット(ラット#99)におけるヘロイン摂取に対する、オピエートアナログ3aの作用を示す。ラットには、活動/暗所相(午後6時〜午前6時)の15分前(午後5時45分)に、オピエートアナログ3a(0、0.04、及び0.2mg/kg、皮下注射)を、対象内デザインで注射した。データは、暗所相での1時間毎(図9A)、3時間毎(図9B)、及び12時間全体(図9C)についての総ヘロイン摂取(60μg/kg/0.1ml注入)で表した。
【図10】図10A-Cは、ヘロイン-依存型ラット(ラット#100)におけるヘロイン摂取に対するオピエートアナログ13gの作用を示す。ラットには、活動/暗所相(午後6時〜午前6時)の15分前(午後5時45分)に、オピエートアナログ13g(0mg/kg、0.04 mg/kg、及び0.2 mg/kg、皮下注射)を、対象内デザインで注射した。データは、暗所相での1時間毎(図10A)、3時間毎(図10B)、及び12時間全体(図10C)についての総ヘロイン摂取(60μg/kg/0.1ml注入)で表した。
【図11】図11A-Cは、ヘロイン-依存型ラット(ラット#103)におけるヘロイン摂取に対するM6Gの作用を示す。ラットには、活動/暗所相(午後6時〜午前6時)の15分前(午後5時45分)に、M6G(0mg/kg、0.04 mg/kg、及び0.2mg/kg、皮下注射)を、対象内デザインで注射した。データは、暗所相での1時間毎(図11A)、3時間毎(図11B)、及び12時間全体(図11C)についての総ヘロイン摂取(60μg/kg/0.1ml注入)で表した。
【図12】図12A-Cは、ヘロイン-依存型ラット(n=4)におけるヘロイン摂取に対するオピエートアナログ13hの作用を示す。ラットには、活動/暗所相(午後6時〜午前6時)の15分前(午後5時45分)に、オピエートアナログ13h(0[ラット#96]、0.01[ラット#99]、0.04[ラット#100]、及び0.2mg/kg[ラット#103]、皮下注射)を、対象内デザインで注射した。データは、暗所相での1時間毎(図12A)、3時間毎(図12B)、及び12時間全体(図12C)についての総ヘロイン摂取(60μg/kg/0.1ml注入)で表した。
【図13】図13は、実施例33に説明されたMG6(1)及び2種の代謝安定性アナログ(2及び3)の化学構造を示している。
【図14】図14は、実施例33に示されたMG6のC-グリコシドの調製を示している。(i) CH3S(0)CH3, Ac2O;(ii)LiCH2CO2Et、THFその後HCl、-78℃〜室温;(iii)Et3SiH、BF3OEt2、CH3CN、0℃〜室温;(iv)NaOH、H2O、THF、還流、その後HCl;(v)SOC12;(vi) 3-O-トリイソプロピルシリル-6-β-アミノモルヒネ、Et3N、CH2Cl2;(vii)Bu4NF、THF、H2O;(viii) H2、10%Pd-C、MeOH;(ix)H2、10%Pd-C、MeOH、HCl。
【図15】図15は、norBNI及びナルトリンドール(NTI)の化学構造を示している。
【図16】図16は、JDTic及びグアニジンナルトリンドール(GNTI)の化学構造を示している。
【図17】図17は、ナルメフェン(1)及びナルトレキソン(2)の化学構造を示している。
【図18】図18は、オピオイドアンタゴニストの一般的合成を示している。
【図19】図19は、離脱症状の身体的徴候に対する実施例38の化合物6dの作用を示す。
【図20】図20は、離脱症状の身体的徴候に対する実施例35の化合物6aの作用を示す。
【技術分野】
【0001】
(関連出願の相互参照)
本出願は、2004年5月28日に出願された、米国特許仮出願第60/575,451号の優先権を主張するものであり、その開示全体は本願明細書に参照として組入れられている。
(発明の技術分野)
本発明は、鎮痛薬受容体と相互作用する医薬物質、これらの物質を調製する方法、並びに痛覚喪失、疼痛、中毒症状及び他の状態に対するそれらの使用に関する。
【背景技術】
【0002】
(発明の背景)
モルヒネ及び他のオピエートは、強力な鎮痛薬として作用する[Foye,
W.O.の著書、「Principles of Medicinal Chemistry」第3版、Lea
& Febiger、フィラデルフィア、1989]。末期患者のため及び癌の疼痛緩和のために麻薬性鎮痛薬の適切な使用を開発及び理解するために、かなりの努力が払われているが、新規薬物療法が非常に必要とされている。モルヒネは、痛覚喪失、呼吸抑制、及び胃腸管輸送の阻害を含む、μオピオイド受容体により媒介された、多くの薬理学的活性を誘起する。[0.
Rayの著書、「Drugs, Society and Human Behavior」第3版、The
C.V. Mosby Co.、セントルイス (1983)を参照されたい]。しかし有害な副作用及び乱用の可能性が、モルヒネの利用可能性及び最適な使用を制限している。更に溶解度及び効能の問題点が、注射可能なモルヒネ量を制限している。重度の疼痛のための長寿命物質の必要性が存在する。高価な静脈内又は硬膜外の投与経路により投与されることが不要である物質の必要性も存在する。呼吸抑制、耐性、尿貯留、便秘、身体的依存、及び/又は中毒を引き起こさない薬物療法も必要である。加えて、存在する疼痛状態の一部は、現在利用可能なオピエートの鎮痛作用に対し抵抗がある。末梢にのみ作用し脳に侵入しない有効な鎮痛薬必要性も存在する。
【0003】
耐性は、オピエート作用に対する低下した感受性と定義され、一般に離脱症状の身体的症状発現により明らかにされる依存性を引き起こす、鎮痛効能の減弱化を示す。[B.
L. Kiefferらの論文、Cell, 108:87-90 (2002)を参照されたい]。耐性は、ほぼ全て痛覚喪失に関連している。耐性は、表面受容体及びオピオイド受容体シグナル伝達の低下により引き起こされると長く考えられてきた。しかし、モルヒネは、効率的なμ受容体内在化を促進しない[J.
L. Whistlerらの論文、Neuron, 23:737-46 (1999)を参照されたい]のに対し、μ-選択的ペプチドであるDAMGO及びアルカロイドフェンタニルなどの他のオピオイドは、そのような内在化を促進する。インビトロモデルは、内在化を誘導する閾値以下の濃度で投与されたDAMGOは、モルヒネの存在下で、μ受容体の内在化を誘導することができることを示している[L.
Heらの論文、Cell, 108:271-82 (2002)を参照されたい]。モルヒネの連続投与後の痛覚喪失は、内在化投与量を下回るDAMGOがラットへ同時投与される場合に、著しく増強される。[L.
Heらの論文、前掲参照]。細胞レベルでのμ受容体内在化とインビボ耐性の間のこの連関は、受容体内在化は、耐性に対する保護を提供し得ることを示唆している。従ってM6Gのどのアナログが受容体内在化に作用するかの決定は、非-中毒性鎮痛薬の開発及び中毒プロセスの我々の理解を深める上での重要な工程である。μ、δ、又はκ受容体のアンタゴニストの開発も、有用な薬物療法を生じる。これらのM6Gアナログ中のN-17メチル基の、他のアルキル、シクロアルキル及びアルケニル基との交換は、オピオイド受容体に薬理学的活性のあるアンタゴニストを提供することができる。このようなアンタゴニストは、薬物中毒、ギャンブル依存症、及びアルコール依存症を含むCNS疾患の治療において有用であろう。一定に帯電し及び4級アミン又はグアナジノ基を含む誘導体の作製も、末梢にのみ作用する鎮痛薬の新規セットを提供する。
【0004】
モルヒネ及び関連したオピエートを体から排除する主要経路は、肝臓における水溶性グルクロニド複合体の形成、それに続く尿中排泄である。モルヒネの場合、3種のグルクロニドが形成される:モルヒネ-6-β-D-グルクロニド、モルヒネ-3-β-D-グルクロニド及びモルヒネ-3,6-ジ-β-D-グルクロニド。モルヒネ-6-β-D-グルクロニド(M6G)は、モルヒネ自身よりも100倍大きい効能を伴う鎮痛薬である。[G. W. Pasternakの論文、Life
Sci., 41:2845-2849 (1987)を参照されたい]。胃酸による消化管内での加水分解に起因したM6Gの低い生体利用効率(11%)は、この化合物からの薬物の開発を著しく制限する。[R. T. Pensonら、Br. J; Clin. Pharmacol., 53:347-354
(2002)を参照されたい]。M6Gをベースにした新規薬物療法の開発は、その鎮痛効能、好ましい副作用プロファイル、及び際だった薬理学的活性のために有望である。[M.H.
Hannaらの論文、Anesthesiology, 102:815-821 (2005)を参照されたい]。
【0005】
概してグルクロニドは、高度に極性のある代謝産物であり、及び血液脳関門(BBB)を通過することができないと考えられている。[G.
W. Pasternakの論文、Clin. Neuropharmacol.,
16:1-18 (1993)を参照されたい]。しかしM6Gは、明らかに予想されるよりもはるかに親油性である。[P. A. Carruptらの論文、J.
Med. Chem., 34:1272-1275 (1991)を参照されたい]。極性表面積(PSA)の計算は、90Å2を上回る分子は、脳に移行しないことを示唆している。[K.
Palmらの論文、J. Pharm. Sci., 85:32-39 (1996a)を参照されたい]。経細胞経路によるBBB通過に関して、分子は、MW<450及びPSA<90Å2を有さねばならない。関連した計算は、経口吸収には、PSA<120Å2が最適であることを示している。[J. Kelderらの論文、Pharmaceutical
Research, 16:1514-1519 (1999)を参照されたい]。ラットにおいて、M6Gは経口投与後に、近位小腸でそれ自身吸収され、M6Gは膜透過可能であることを示している。[R.
Stain-Texierらの論文、Drug
Metab.Dispos., 26:383-387, (1998)を参照されたい]。しかし分子量が450よりも大きいM6Gのアナログは、脳に移行しないと予想され、並びにモルヒネと比べより極性のある性質のために、より好ましいCNS生体分布及び作用特性の開始を有さないと他のものにより予想される。
【発明の開示】
【発明が解決しようとする課題】
【0006】
(発明の簡単な概要)
1つの局面において、本発明は、鎮痛薬受容体に対する薬理学的活性を有する鎮痛薬-関連物質を提供する。典型的態様において、これらの物質は、任意に置換されたアリール、任意に置換されたヘテロアリール、もしくは糖類に連結された鎮痛薬又はその誘導体を含む。好ましい態様において、鎮痛薬又はその誘導体は、オピエート又はオピエート誘導体である。鎮痛薬-関連物質は、その物質が特異的に結合する対応する鎮痛薬受容体のアゴニスト又はアンタゴニストであり得る。
【課題を解決するための手段】
【0007】
本発明の物質は、例えば対象における疼痛の緩和について、又はそのような治療もしくは予防が鎮痛薬受容体の変調(例えば、阻害又は刺激)により影響されやすい疾患又は障害の治療もしくは予防について、有用である。従って別の局面において、本発明は、本願明細書に記した鎮痛薬-関連物質を含有する医薬組成物を提供する。同じく、対象へ有効量の鎮痛薬-関連物質を投与することによる、疼痛の軽減、又は鎮痛薬受容体活性の変調によるそのような治療、もしくは予防に影響されやすい疾患もしくは障害の治療もしくは予防のための方法を提供する。本願明細書に提供された組成物及び方法による治療的介入の影響を受けやすい疾患又は障害は、例えば、鎮痛薬受容体により少なくとも一部、直接又は間接に媒介される中毒又は他のCNS障害を含む。例えば疼痛の治療のためのような、ある種の医薬組成物又は方法の変動において、鎮痛薬-関連物質は、鎮痛薬受容体アゴニストである。別の態様において、例えば薬物中毒、ギャンブル依存症、又はアルコール依存症の治療などのためには、物質は典型的には鎮痛薬受容体アンタゴニストである。
【0008】
脳の鎮痛薬受容体に著しい作用を伴うことなく、末梢において鎮痛薬受容体を変調するために、この物質は、典型的には生理的pHで一定の電荷を有する。このような物質は、例えば、4級アミン、クアニジン(quanidine)などを含むことができ、特に例えば末梢鎮痛薬としての使用に適している。
【0009】
(定義)
特に別に記載しない限りは、本願明細書において使用される全ての技術用語及び科学用語は、本発明が属する技術分野の業者に通常理解されるものと同じ意味を有する。本願明細書に記載されたものに類似したあらゆる方法及び材料を、本発明の実践又は試験に使用することができるが、方法及び材料の例のみが説明されている。本発明の目的のために、以下の用語は、下記のように定義される。
【0010】
用語「1つの(a,an及びthe)」は、本文が別に明確に指摘しない限りは、複数の意味を含む。
用語「アルキル」は、直鎖、分枝鎖、及び環状の基を含む、飽和した脂肪族基を意味し、これらは全て任意に置換されてよい。適当なアルキル基は、メチル、エチルなどを含み、任意に置換されてよい。
用語「アルケニル」は、少なくとも1個の炭素-炭素二重結合を含み、並びに直鎖、分枝鎖、及び環状の基を含む不飽和基を意味し、これらは全て任意に置換されてよい。
【0011】
用語「アルキニル」は、少なくとも1個の炭素-炭素三重結合を含み、並びに直鎖、分枝鎖、及び環状の基を含む不飽和基を意味し、これらは全て任意に置換されてよい。適当なアルキニル基は、任意に置換されてよい、エチニル、プロピニル、ブチニルなどを含む。
用語「アリール」は、共役「π」電子系を有する少なくとも1個の環を有する芳香族基を意味し、炭素環式アリール、二環式アリールを含み、両方とも任意に置換されてよい。
【0012】
用語「アラルキル」は、アリール基で置換されたアルキル基を意味する。適当なアラルキル基は、ベンジルなどを含み、任意に置換されてよい。
用語「ヘテロアリール」は、1、2、3又は4個のO、N、P又はS原子を含み、及び1個又は1個よりも多い環において局在化されない6、10又は14個のπ電子を有する、炭素含有する5-14員の環式不飽和ラジカルを意味する。
【0013】
用語「医薬として許容し得る誘導体」は、例えば医薬として許容し得るエステル、アミド又は塩を含む、薬理学的使用に適している、本願明細書に説明されたような化合物のいずれかの誘導体を意味する。医薬として許容し得るエステル、アミド又は塩は、本発明の化合物及び有機酸又は無機酸の組合せに由来する。
用語「鎮痛薬」は、痛覚喪失を生じること、すなわち麻酔状態又は意識喪失を生じることなく、侵害刺激の認知を変更することにより、疼痛を軽減又は阻害することが可能な化合物を意味する。本発明の好ましい態様において、鎮痛薬はオピエートである。
【0014】
用語「オピエート」は、アヘン又はその任意の誘導体(例えばモルヒネ)、更には合成又は半-合成の麻薬を含む、オピオイド受容体に特異的に結合することが可能な、天然又は合成の物質を意味する。
用語「鎮痛薬-関連物質」は、鎮痛薬の代謝産物、アナログ又は誘導体を意味する。用語「オピエート-関連物質」は、更に本願明細書に説明されたような、オピエートの代謝産物、アナログ、又は誘導体を意味する。本発明に従い、オピエート-関連物質を含む、鎮痛薬-関連物質は、この物質が特異的に結合する1種又は複数の鎮痛薬受容体に関するアゴニスト又はアンタゴニスト活性を有することができる。
【0015】
用語「阻害」は、測定可能な量だけ低下するか、又は全体を妨害することを意味する。
語句「効率的な受容体内在化の促進」は、物質が、その物質が特異的に結合する受容体の細胞内在化を促進することが可能であり、その物質に接触していない対照細胞と比較して、FACSによる対応する受容体の表面染色が80%を超えない、典型的には70%又は60%を超えない、より典型的には50%又は40%を超えないことを意味する。
【0016】
疾患又は障害の「治療する」、「治療」又は「療法」は、本願明細書に説明されたような、本発明の組成物及び方法を使用する、臨床症状又は診断症状のいずれかの軽減又は消失により証明されるような、疾患又は障害の進行の遅延、停止、又は逆行を意味する。
疾患又は障害の「予防する」、「予防」又は「妨害」は、疾患もしくは障害又はその症状の一部もしくは全ての発生又は開始を妨害することを意味する。
【0017】
本願明細書に使用される「中毒」は、実質的に自主規制を超えた物質又は慣習への習慣性の精神的及び生理的依存を特徴とする疾患又は障害を意味する。本願明細書に説明された化合物及び方法を使用する治療の影響を受ける中毒は、物質中毒、例えば、麻薬(例えばモルヒネ、ヘロイン)、アルコール、及びニコチンへの中毒に加え、行動的中毒、例えばギャンブルへの依存症などを含む。
【0018】
本願明細書において使用される用語「対象」は、本発明の組成物が、本願明細書に説明された方法に従い投与される哺乳類患畜を意味する。本発明の方法を使用する治療又は予防が特異的に意図された対象は、ヒトを含む。
用語「治療的に有効な投与計画」は、医薬組成物又はそれらの組合せが、疼痛を軽減するか、又は鎮痛薬受容体の変調による治療に影響を受ける疾患もしくは障害の少なくとも1種の症状もしくは生化学マーカーの顕在化を少なくとも検出可能に妨害、遅延、阻害又は逆行させるのに、十分な量及び回数及び適当な経路で投与されることを意味する。
【0019】
用語「治療的有効量」は、適切な投与計画で投与された場合に、例えば、疼痛の軽減、又は鎮痛薬受容体の変調による治療の影響を受ける疾患もしくは障害の症状もしくは生化学マーカーを妨害、遅延、阻害もしくは逆行するなど、所望の結果を実現するように存在する、本発明の物質又は本発明の物質の他の物質(複数)との組合せの量を意味する。
【発明を実施するための最良の形態】
【0020】
(詳細な説明)
本発明は、鎮痛薬及び鎮痛薬誘導体、特にオピエート-関連化合物に関する。これらの化合物の構造、合成、及び生物学的評価に加え、これらの化合物を含有する医薬組成物並びに例えば疼痛及び物質中毒の治療のためなどのそれらの薬理学的使用が、本願明細書に説明されている。ある態様において、本願明細書に説明された化合物は、モルヒネ代謝産物M6Gに関連している。予想外のことに、M6Gに対する無関係のアナログは、いくつかのインビトロ及びインビボ機能試験において、M6Gよりもより好ましい薬理学的特性を有した。
【0021】
鎮痛薬受容体シグナル伝達及び物質中毒の様々な要素のインビトロ薬理学的及び代謝的試験及び動物モデルは、疼痛及び物質中毒の治療において、ヒトモデルの様々な要素において有効であるであろう薬物療法の開発を予測することができる。それらの両方が最終的な薬物療法開発を促進するための情報を整備するインビトロ及びインビボ要素からの動的医化学フィードバックを用い、例えば、疼痛及び/又は中毒の治療に有用な化合物が同定されている。中毒サイクルの1種又は複数の成分中のこれらの化合物の薬理学的活性は、とりわけ、中毒サイクルの複数の要素が投与された薬理学的物質により対象とされるような併用療法の基本を提供する。
【0022】
本発明の態様を説明する目的で、本願明細書に説明された化学式は、塩、エステル、及びアミドを含む、各化合物の医薬として許容し得る誘導体、更にはラセミ体又は立体化学的に純粋な化合物を含む、置換基の全ての可能性のある立体化学的配置を含むことが理解される。
【0023】
(化合物)
本発明の1つの局面において、鎮痛薬受容体に対する薬理学的活性を有し、並びに例えば疼痛又はそのような受容体の変調を介して治療の影響を受ける、中毒を含む疾患又は障害の治療に有用である化合物が、提供される。本発明の化合物は一般に、下記式Iを有し:
【化1】
【0024】
(式中、Aは、鎮痛薬又はその誘導体であり;Bは、連結基であり、及びCは、任意に置換されたアリール、任意に置換されたヘテロアリール及び糖類からなる群から選択される);及び、それらの全ての医薬として許容し得る誘導体である。好ましい変形において、鎮痛薬Aは、オピエートである。本願明細書に説明された本発明による使用に特に適したオピエートは、例えばナルトレキソン、フェニルピペリジン、ピペリジノール、プロジン、ピペリジルプロピオンアニリド、イソプロジン、プロジリジン、ベンゾモルファン、モルファン、アザビシクラン(azabicyclane)、モルヒナン、プロジン、ジフェニルアミノエチルプロピオネート、メサドン、イソメサドン、プロポキシフェン、デキストロメトロファン、ベンザゾシン-8-オール、ノルビナルトロフィン、ナルトリンドール及びグアニジンナルトリンドールからなる群から選択される。連結基Bは、例えば-S-又は-NH(CO)(CH2)n-であり、ここで添字nは、0〜5の整数である。
【0025】
ある態様において、Cは、ハロゲン、(C1-C5)アルコキシ、ニトロ、又はCO2Rで任意に置換された、アリール又はヘテロアリールであり、ここでRは、H又は(Cl-C5)アルキルである。特定の態様において、Cは、例えば、フェニル、チオフェン、又は糖類である。特に適した糖類は、下記式IIを有するものである:
【化2】
【0026】
(式中、各R8は独立して、H、(C1-C5)アルキルC(O)-、(C7-C10)アラルキルC(O)-、(C7-C10)アラルキル、(C1-C5)アルキル、(C6-C12)アリール及び(C6-C12)アリール(CO)-からなる群から独立して選択され;R9は、CH2OH、CH2O(C1-C5)アルキル、CH2O2C(C1-C5)アルキル、CH2O(C7-C10)アラルキル、CH2O(C6-C12)アリール、CO2H、CO2(C1-C5)アルキル、CO2(C6-C12)アリール、CO2(C7-C10)アラルキルであり;並びに、波線は、分子の残余に結合する点を示す。)。
【0027】
1つの適当な変形において、化合物は、下記式IIIを有する:
【化3】
【0028】
(式中、破線は、単結合、二重結合又は規格化結合(normalized bond)を示し;R1は、H、(C1-C5)アルキルC(O)-、(C7-C10)アラルキルC(O)-、(C7-C10)アラルキル、(C1-C5)アルキル、(C6-C12)アリール、(C6-C12)アリール(CO)-及びシリルであり;R2は、ハロゲン、(C1-C5)アルコキシ、ニトロ及びCO2Rからなる群から選択される置換基により任意に置換された、(C1-C5)アルキル、(C3-C6)シクロアルキル、(C3-C6)シクロアルキル-アルキル、(C5-C7)シクロアルケニル-アルキル、(C6-C12)アリール、(C7-C12)アラルキル、(C6-C12)ヘテロアリール、(C7-C12)ヘテロアラルキル、(C2-C5)アルケニル、(C2-C5)アルキニルであり、ここでRは、H又は(C1-C5)アルキルであり;R3は、H又はOHであり;R4は、Hであり;R5は、Hであるか、又はR6と一緒に、YR7で置換されたヘテロアリール基を形成し;R6は、YR7であるか、又はR5と一緒に、YR7で置換されたヘテロアリール基を形成し;Yは、-S-又は-NH(CO)(CH2)n-であり、ここで添字nは、0〜5の整数であり;並びに、R7は、任意に置換されたアリール、任意に置換されたヘテロアリール又は糖類であり、ここで糖類は、先に説明された式IIを有する。特に適当なR2基は、例えばシクロプロピルメチル、シクロブチルメチル、及びアリルを含む。
【0029】
いくつかの代わりの態様において、化合物は、下記式IVa又はIVbを有する:
【化4】
【0030】
(式中、R1は、H、(C1-C5)アルキルC(O)-、(C7-C10)アラリル、又は(C1-C5)アルキルであり;R2は、(C1-C5)アルキル、C3-C6(シクロアルキル)アルキル、C5-C7(シクロアルケニル)アルキル、C6-C12アリール(C7-C12)アラルキル、トランス(C4-C5)アルケニル、アリル、又はフラン-2-イルアルキルであり;各R8は、独立して、H、(C1-C5)アルキルC(O)、(C7-C10)アラリルC(O)、(C7-C10)アラリル、又は(C1-C5)アルキルであり;並びに、R9は、CH2OH、CH2O2C(C1-C5)アルキル、CH2O(C7-C10)アラリル、CO2H、CO2(C1-C5)アルキル、又はCO2(C7-C10)アラルキルである。)。
【0031】
更に別の変形において、化合物は、下記式Va又はVbを有する:
【化5】
【0032】
(式中、R1は、H、(C1-C5)アルキルC(O)-、(C7-C10)アラリル、又は(C1-C5)アルキルであり;R2は、(C1-C5)アルキル、C3-C6(シクロアルキル)アルキル、C5-C7(シクロアルケニル)アルキル、(C6-C12)アリール(C7-C12)アラルキル、トランス(C4-C5)アルケニル、アリル、又はフラン-2-イルアルキルであり;R8は、独立してH、(C1-C5)アルキルC(O)、(C7-C10)アラリルC(O)、(C7-C10)アラリル、又は(C1-C5)アルキルであり;並びに、R9は、CH2OH、CH2O2C(C1-C5)アルキル、CH2O(C7-C10)アラリル、CO2H、CO2(C1-C5)アルキル、又はCO2(C7-C10)アラルキルであり;及び、添字nは、0〜5の整数である。)。
【0033】
更に別の態様において、化合物は、下記式VIa又はVIbを有する:
【化6】
【0034】
(式中、R1は、H、(Cl-C5)アルキルC(O)-、(C7-C10)アラリル、又は(Cl-C5)アルキルであり;R2は、(Cl-C5)アルキル、C3-C6(シクロアルキル)アルキル、C5-C7(シクロアルケニル)アルキル、(C6-C12)アリール(C7-Cl2)アラルキル、トランス(C4-C5)アルケニル、アリル、又はフラン-2-イルアルキルであり;及び、添字nは、0〜5の整数である。)。
【0035】
特定の態様において、本発明の化合物は、下記式VIIからXXXのいずれか1つを有する:
【化7】
【0036】
(他の態様)
ある態様において、本発明の化合物は、モルヒネ、ジヒドロモルヒネ-6-グルクロニド、コデイン、及び下記式IVを有するジヒドロコデイン-6-グルクロニドスルファーアナログである:
【化8】
【0037】
(式中、Aは、OR、OH、又はOC(0)Rであり;Bは、OC(0)R、OH、又はORであり;Cは、CO2R、CO2H、CH2OR、又はCH2OHであり;Dは、アルキル、シクロアルキル、アルケニル、アルキニル、任意に置換されたアリール、又は任意に置換されたアラルキルであり;並びに、置換基A、B、C、DのRは、アルキル又はアリール基である。)。
【0038】
別の態様において、本発明の化合物は、コデイン、ジヒドロコデイン、モルヒネ、及び下記式Vを有するジヒドロモルヒネアナログを含む:
【化9】
【0039】
(式中、Aは、RO、OH、又はOC(0)Rであり;Bは、OC(0)R、OH、又はORであり;Cは、CO2R、CO2H、CH2OR、又はCH2OHであり;Dは、アルキル、シクロアルキル、アルケニル、アルキニル、任意に置換されたアリール、又は任意に置換されたアラルキルであり;並びに、置換基A、B、C、DのRは、アルキル又はアリール基である。)。
【0040】
更に別の変形において、本発明の化合物は、モルヒネ、ジヒドロモルヒネ、コデイン、及び下記式VIのジヒドロコデインアミドを含む:
【化10】
【0041】
(式中、Aは、RO、OH、又はOC(O)であり、ここでnは0〜5の整数であり;アリールは、フェニル、3,4-ジクロロフェニル、3-メトキシフェニル、4-メトキシフェニル、3-ニトロフェニル、3-カルボメトキシフェニル、3-カルボキシフェニル又はチオフェン-2-イル、又は任意に置換されたアリールもしくはヘテロアリール誘導体であり;Dは、アルキル、シクロアルキル、アルケニル、アルキニル、任意に置換されたアリール、又は任意に置換されたアラルキルであり;並びに、置換基A及びDのRは、アルキル又はアリール基である。)。
【0042】
式IVの化合物は、ジメチルホルムアミド中でのモルヒネの6βトシラート、3-0-アセチルモルヒネ及びコデイン1a-cの、チオ糖ナトリウム塩2a-bのとのSN2反応により合成し(図1参照)、3a-fを生成することができる。出発トシラートの調製については、[L.
H. Welshの論文, J. Org. Chem., 19:1409 (1954);G.
Stork 及び F. Clarkeの論文,J. Am.Chem.Soc.,
78:4619-4624 (1956)を参照されたい]。引き続きエステル保護基は、水酸化ナトリウム水溶液の中での一晩の攪拌により除去され、モルヒネ、コデイン及びそれらの同族のS-β-D-グルクロニド又はS-β-D-グルコース複合体である追加の化合物4a-dを生じる。これらの調製において使用されるチオ糖2a-bは、対応するグリコシルブロミドの、チオ酢酸カリウムとの反応、それに続くチオメトキシドナトリウムによる選択的S-脱アセチル化により作製された(図2参照)。出発グリコシルブロミドの調製については、[G.
N. Bollenbackらの論文、J. Org Chem., 77:3310-3315 (1955)を参照されたい]。関連したS-脱アセチル化については、[Wallaceらの論文、
Tetrahedron Lett., 2693 2694 (1998)を参照されたい]。この合成法は、C-6位置で、イオウ原子を介した、糖類のモルヒネ、コデイン及びそれらのN-17誘導体への結合に関する、一般的合成経路を構成しており、これらの化合物のジヒドロモルヒネ及びジヒドロコデイン誘導体の調製に適用可能である。加えて、モルヒネ、コデイン及び誘導体の6β-トシラートを利用し、前述の全ての化合物のC-6α誘導体の調製が可能になる。
【0043】
式Vの化合物は、モルヒネ、ジヒドロモルヒネ、コデイン、ジヒドロコデイン並びにそれらのN-アルキル及びシクロアルキル誘導体6の6β-フタルイミドから合成することができる(図3参照)。[S.
Makleitらの論文、 Synthetic Commun.,
21:407-412 (1991)を参照されたい]。モルヒネシリーズの化合物に関して、フェノール系ヒドロキシル基は、室温のジメチルホルムアミド中での、tert-ブチルジメチルシリルクロリド又はトリイソプロピルシリルクロリド及びイミダゾールとの反応により、シリルエーテルとして保護される。その後フタルイミド基は、EtOH中でのヒドラジン水和物との加熱により除去され、アミン7を生じる。アミン7は、C-グリコシド8の酸塩化物と反応し、アミド9を生じる。9のR1がシリルエーテルである場合、これは、TBAF処理により除去することができ、フェノール10を生じる。9又は10中の糖保護基は、NaOH水溶液による処理によるか、又は水素化により除去され、化合物11を生じる。これらの合成において使用されるC-グリコシド8は、公知の方法により合成される。[M.
Lewisらの論文、J. Am. Chem. Soc., 104:4976-4978 (1982)]。この合成配列は、C-6位置での、アミド橋を介した、糖類のモルヒネ及びそれらのN-17誘導体への結合の一般的経路を提供し、これらの化合物のジヒドロモルヒネ及びジヒドロコデイン誘導体の調製に適用可能である。
【0044】
式VIの化合物は、式Vの化合物の調製に使用された6β-アミノモルヒネ7cから合成することができる(図4参照)。アミン7cは、アリール及びヘテロアリール酸塩化物で処理され、アミド12a-hを生じる。水性THF中における12a-hのTBAFによる処理は、6β-アミドモルヒネ13a-hを提供する。メチルエステル13eの水酸化ナトリウム水溶液による処理は、カルボン酸14を提供する。この合成配列は、C-6位でアミド橋を介したアリール及びヘテロアリール置換基のモルヒネ、コデイン及びそれらのN-17アルキル誘導体への結合の一般的経路を提供し、これらの化合物のジヒドロモルヒネ及びジヒドロコデイン誘導体の調製に適用可能である。この合成法は、アミン7cのC-6エピマーで出発することにより、前記化合物の全てのC-6a誘導体の調製も提供する。本願明細書に説明された化合物に関する橋頭NのN-メチル置換基の、シクロプロピルメチル又はシクロブチルメチル又は同様の置換基との交換は、典型的には、以下に説明された活性に関する受容体アンタゴニストを供する。このようなアンタゴニストは、薬物中毒、ギャンブル依存症、アルコール依存症、又は他のCNS疾患を拮抗するのに有用である。更に本願明細書に説明された、合成法(代謝が安定した糖類又は他の化学ホモログ又は等配性(isosteric)同等物の結合)の、ナルトレキソン、フェニルピペリジン、ピペリジノール、プロジン、ピペリジンプロピオンアニリド、イソプロジン、プロジリジン、ベンゾモルファン、モルファン、アザビシクラン、モルヒナン、プロジン、ジフェニルアミノエチルプロピオネート、メサドン、イソメサドン、プロポキシフェン、デキストロメトロファン、ベンザゾシン-8-オール、及び全ての関連した立体異性体及びN-置換された誘導体(シクロプロピルメチル、シクロブチルメチル、アリルなど)を含む他の鎮痛薬への適用が、疼痛緩和に加え、中毒及び他の疾患の治療に有用な、薬理学的活性のあるCNS系物質につながるであろう。
【0045】
更に別の変形において、本発明の鎮痛薬-関連物質は、生理的pHで一定電荷(すなわち、その時の100%か、又は実質的に100%イオン化される)を有する。このような変形は、例えば、芳香族基又は糖基からの4級アミン又はグアニジノ(quanidino)部分の除去により、合成することができる。このような修飾は典型的には、脳に移行せず、従って末梢適応に有用である誘導体(例えば脳受容体への著しい作用を伴わずに末梢において疼痛を緩和する、末梢鎮痛薬)を生じる。
【0046】
(医薬組成物及び投与法)
本発明の鎮痛薬-関連物質は、神経系の鎮痛薬受容体シグナル伝達の変調に関連している様々な適用において有用である。例えば本発明の物質は、疼痛(例えば、外科的手技又は癌のような末期疾患に関連した疼痛の緩和)の治療のための鎮痛薬として有用である。これらの物質は、特に例えばCNS疾患又は障害の、鎮痛薬(例えばオピオイド)受容体シグナル伝達の変調による改善の影響を受ける疾患又は障害の治療のためにも有用である。このような疾患又は障害は、例えば様々な中毒を含む。本願明細書に説明された物質を使用する治療の影響を受ける中毒は、例えば麻薬(例えば、モルヒネ、ヘロイン、及び他のオピエート)などの、薬物、ニコチン、及びアルコールに対する中毒に加え、行動中毒(例えばギャンブル依存症)を含む。
【0047】
従って本発明は更に、疼痛の治療に加え、中毒及び他のCNSに関連した障害の治療のための医薬組成物及び方法を提供する。本発明の鎮痛薬-関連物質は、哺乳類、例えばヒト対象へ、単独で、医薬として許容し得る塩もしくはそれらの水和可能な前駆体の形状で、又は化合物が適当な担体もしくは賦形剤(複数)と治療的有効量で混合されている医薬組成物の形状で、送達又は投与することができる。好ましい態様において、対象における薬物中毒の治療のため及び適当な治療的に有効な投与計画で投与される場合は、対象をより低量の薬物に傾向づけるために、十分量の鎮痛薬-関連物質が、インビボにおいて鎮痛薬受容体を阻害するために存在する。
【0048】
本発明の方法で使用される鎮痛薬-関連物質は、この物質を、1種又はそれよりも多い他の医薬として許容し得る成分と共に含有する医薬組成物として投与することができる。医薬組成物は、固形物の形(例えば散剤、顆粒剤、糖衣錠、錠剤又は丸剤など)、半-固形物(例えば、ゲル剤、スラリー、又は軟膏剤など)、液体、又は気体(例えばエアゾール剤又は吸入剤など)の形であり得る。
本発明において使用するために適当な製剤は、例えば、[Remington's Pharmaceutical
Sciences, Mack Publishing Company,フィラデルフィア, PA, 17th ed. (1985)
and Langer, Science, 249:1527-1533 (1990)]に認められる。本願明細書に説明された医薬組成物は、例えば、混合、溶解、造粒、糖衣錠形成、すりつぶし、乳化、カプセル封入、捕獲又は凍結乾燥プロセスなどの、常法により製造することができる。
【0049】
本発明の製剤の調製において、各化合物の医薬として認識された同等物を、代わりに使用することができる。これらの医薬として認められた同等物は、医薬として許容し得るエステル、アミド、もしくは塩、又は医薬として許容し得る酸付加塩であり得る。
医薬として許容し得る塩は、例えば、当該技術分野において周知の方法により調製されるナトリウム、カリウム、リチウム、カルシウム、マグネシウム、バリウム、アンモニウム、及びプロタミン亜鉛塩を含む、製薬産業において通常使用される、無毒の金属、アルカリ土類金属、又はアンモニウム塩である。この用語は、一般に本発明の化合物を、適当な有機酸又は無機酸と反応することにより調製される、無毒の酸付加塩も含む。代表的塩は、例えば塩酸塩、臭化水素酸塩、硫酸塩、亜硫酸塩、酢酸塩、シュウ酸塩、吉草酸塩、オレイン酸塩、ラウリン酸塩、ホウ酸塩、安息香酸塩、乳酸塩、リン酸塩、トシラート、クエン酸塩、マレイン酸塩、フマル酸塩、コハク酸塩、酒石酸塩、及びナプシラートを含む。
【0050】
医薬として許容し得る酸付加塩は、無機酸、例えば塩酸、臭化水素酸、硫酸、硝酸、リン酸などと、並びに有機酸、例えば酢酸、プロピオン酸、グリコール酸、ピルビン酸、シュウ酸、リンゴ酸、マロン酸、コハクサン、マレイン酸、フマル酸、酒石酸、クエン酸、安息香酸、桂皮酸、マンデル酸、メタンスルホン酸、エタンスルホン酸、p-トルエンスルホン酸、サリチル酸などにより形成された、遊離の塩基の生物学的有効性及び特性を保持し、並びに生物学的又はそうでなければ望ましくない塩である。[例えば、Bundgaard編集の著書、Design
of Prodrugs, Elsevier Science Publishers, アムステルダム(1985)を参照]。
【0051】
この鎮痛薬-関連物質は、通常の賦形剤、希釈剤、又は担体と共に製剤し、並びに便利な経口投与のために錠剤へ圧縮し、エリキシル剤又は液剤として製剤し得る。これらの物質は、徐放性剤形などとして製剤することもできる。
脳において鎮痛薬受容体の結合に関連した所望の治療作用を発揮するために、本発明の鎮痛薬-関連物質は、脳細胞及び組織に到達しなければならず;脳血管内皮細胞の微小毛細血管膜を含む、脳血管関門の通過による、それらの血液から脳への通路を必要とする。本発明は、患者の末梢組織(すなわち、中枢神経系組織以外の組織)への鎮痛薬-関連物質の治療的に有効な投薬投与計画を投与する方法を提供する。これは、経口、口腔内、直腸内、非経口、腹腔内、皮内、経皮、皮下、気管内及び筋肉内投与を含む様々な方法で、実現することができる。更にこれらの物質は、デポ剤又は徐放性製剤において、全身様式よりもむしろ局所的に投与することができる(例えば、痛覚喪失に管理された患者)。加えて、これらの物質は、ベシクル内、特にリポソーム内で投与することができる[例えば、Langerの論文, 前掲 Treatの論文,
Liposomes in the Therapy of Infectious Disease and Cancer, Lopez-Berestein and
Fidler編集、Liss, ニューヨーク, pp. 353-365 (1989)を参照されたい]。
【0052】
注射剤に関して、本発明の鎮痛薬-関連物質は、例えば、植物油又は他の同様の油、合成脂肪族系酸グリセリド、高次脂肪族系酸エステル、又はポリエチレングリコールなどの水性又は非水性溶媒中に;並びに望ましいならば、例えば、可溶化剤、等張剤、懸濁化剤、乳化剤、安定化剤及び保存剤などの、通常の添加剤を伴い、それらを溶解、懸濁化、又は乳化することにより、調製物へ製剤することができる。好ましくは注射剤に関して、本発明の化合物は、水溶液、好ましくはハンク液、リンゲル液、又は生理的食塩緩衝液などの、生理的に相溶性のある緩衝液中に製剤することができる。経粘膜的投与に関して、透過されるべき障壁に適した浸透剤が、製剤中で使用される。このような浸透剤は、一般に当該技術分野において公知である。
【0053】
経口投与に関して、鎮痛薬-関連物質は、当該技術分野において周知の医薬として許容し得る担体と組合せ、容易に製剤することができる。このような担体は、治療される対象による経口摂取のために、この化合物を、錠剤、丸剤、糖衣錠、カプセル剤、乳剤、親水性及び疎水性懸濁剤、液剤、ゲル剤、シロップ剤、スラリー、懸濁剤などとして製剤することが可能である。経口使用のための医薬調製は、錠剤又は糖衣錠コアを得るために望ましいならば、適当な助剤の添加後に、この化合物の固形賦形剤との混合、任意に得られる混合物の粉砕、及び顆粒の混合物の加工処理により得ることができる。特に適した賦形剤は、例えば糖(例えば乳糖、ショ糖、マンニトール、又はソルビトール)、セルロース調製物(例えばコーンスターチ、コムギデンプン、コメデンプン、ジャガイモデンプン)、ゼラチン、トラガカントガム、メチルセルロース、ヒドロキシプロピルメチル-セルロース、カルボキシメチルセルロースナトリウム、及び/又はポリビニルピロリドン(PVP)などの、充填剤を含む。望ましいならば、架橋したポリビニルピロリドン、寒天、又はアルギン酸もしくはそれらの塩、例えばアルギン酸ナトリウムのような、崩壊剤を添加することができる。
【0054】
糖衣錠コアは、適当なコーティングと共に提供される。この目的に関して、濃縮された糖溶液を使用し、これは任意にアラビアゴム、タルク、ポリビニルピロリドン、カルボポールゲル、ポリエチレングリコール、二酸化チタン、ラッカー液、及び/又は適当な有機溶媒又は溶媒混合物を含むことができる。色素又は顔料は、活性化合物用量の様々な組合せの同定又は特徴決定のために、錠剤又は糖衣錠コーティングに添加することができる。
【0055】
経口的に使用することができる医薬調製物は、例えば、ゼラチンで形成された押込み式カプセル、更にはゼラチン及び例えばグリセロール又はソルビトールのような可塑剤で製造された封止された軟カプセルを含む。押込み式カプセルは、例えば乳糖などの充填剤、結合剤(例えばデンプン)、及び/又は滑沢剤(例えばタルク又はステアリン酸マグネシウム)、並びに任意に安定化剤と混合された活性成分を含むことができる。軟カプセルにおいて、活性化合物は、例えば脂肪族系油、流動パラフィン、又は液体ポリエチレングリコールなどの、適当な液体中に溶解又は懸濁することができる。
【0056】
口腔内投与に関して、組成物は、通常の様式で製剤された、錠剤又はトローチ剤の形をとることができる。
吸入投与に関して、本発明に従い使用するための化合物は通常、例えばジクロロジフルオロメタン、トリクロロフルオロメタン、ジクロロテトラフルオロエタン、二酸化炭素もしくは他の適当なガスのような、適当な噴射剤の使用を伴う、加圧パックもしくはネブライザーからのエアゾールスプレー調製物、又は噴射剤非含有の乾燥散剤の吸入の形で送達される。加圧されたエアゾールの形において、用量単位は、例えば測定された量を送達するためのバルブを提供することにより決定することができる。吸入器又は通気器において使用するための例えばゼラチンのカプセル及びカートリッジは、この化合物及び、例えば乳糖又はデンプンのような適当な粉末基剤の粉末混合物を含むよう製剤することができる。
【0057】
本発明の鎮痛薬-関連物質は、注射による、例えばボーラス注射又は連続注入による、非経口投与のために製剤することができる。注射用製剤は、添加された保存剤と共に、例えばアンプル内又は反復投与用容器内で、単位剤形で提供される。これらの組成物は、例えば、油ベースのビヒクル又は水性ビヒクル中の懸濁剤、液剤、又は乳剤などのような形状をとることができ、並びに例えば、懸濁剤、安定化剤及び/又は分散剤などの製剤用物質を含むことができる。
【0058】
非経口投与のための医薬製剤は、水溶液型の活性化合物の水溶液を含む。あるいは、活性化合物の懸濁剤は、適当な油性注射用懸濁剤として調製することができる。適当な親油性溶媒又はビヒクルは、脂肪族油分(例えばゴマ油)、合成脂肪酸エステル(例えば、オレイン酸エチル又はトリグリセリド)、又はリポソームを含む。水性注射用懸濁剤は、例えばカルボキシメチルセルロースナトリウム、ソルビトール、又はデキストランなどの、懸濁剤の粘度を増加する物質を含有することができる。任意に懸濁剤は、高度に濃縮された溶液の調製を可能にするために、適当な安定化剤又は化合物の溶解度を増加する物質を含むこともできる。あるいは、活性成分は、使用前に、適当なビヒクル、例えば滅菌パイロジェン非含有水で再構成される散剤の形であり得る。
【0059】
鎮痛薬-関連物質は、例えば、ココアバター、カルボワックス、ポリエチレングリコール又は他のグリセリドなどの、全て体温で溶融するが室温では固形であるような、通常の坐剤基剤を含有する、坐剤又は持続浣腸のような、経直腸組成物中に製剤することができる。
先に説明された製剤に加え、これらの化合物は、デポ剤として製剤することもできる。このような長期作用する製剤は、移植(例えば皮下又は筋肉内)又は筋肉内注射により投与することができる。従って、例えばこれらの化合物は、適当な高分子又は疎水性物質と共に(例えば許容し得る油分中の乳剤)、又はイオン交換樹脂中、又はやや溶けにくい誘導体(例えばやや溶けにくい塩)として、製剤することができる。
【0060】
あるいは、疎水性医薬化合物のための他の送達システムを使用することができる。リポソーム及び乳剤は、疎水性薬物のための送達ビヒクル又は担体の周知の例である。一部の方法において、長期間循環するリポリーム、例えばステルスリポソームを使用することができる。このようなリポソームは一般に、Woodleらの米国特許第5,013,556号に開示されている。
本発明の化合物は、制御された放出手段及び/又は送達装置により投与することができる。ある変形において、ポンプが使用される[例えば、Langerの論文、前掲;Seftonの論文、CRC
Crit. Red: Biomed. End, 14:201 (1987);Buchwaldらの論文、Surgery,
88:507 (1980);Saudekらの論文、N. Engl. J. Med., 321:574
(1989)を参照されたい]。他の態様において、高分子物質が使用される[例えば、Medical
Applications of Controlled j Release, Langer及びWise編集、CRC
Pres., ボカラトン, FL (1974);対照led Drug i Bioovailability,
Drug Product Design and Performa,nce, Smolen及びBull編集、Wiley,
ニューヨーク (1984);Ranger及びPeppasの論文、J.
Macromol. Sci. Rev. Macromol. Chem., 23:61 (1983)参照;同じく、Levyらの論文、Science,
228:190 (1985) Duringら、Ann. Neurol., 25:351 (1989);Howardらの論文、J.
Neurosurg., 71:105 (1989)を参照されたい]。制御された放出手段及び送達装置は、例えば、米国特許第3,845,770号;第3,916,899号;第3,536,809号;第3,598,123号;及び、第4,008,719号にも開示されている。
【0061】
ある有機溶媒、例えばジメチルスルホキシド(DMSO)も、利用することができるが、通常より大きい毒性を代償とする。加えてこれらの化合物は、例えば治療薬を含有する固形疎水性ポリマーの半透過性マトリックスのような、持続放出システムを使用し送達することができる。様々な種類の持続放出材料が確立されている。持続放出カプセルは、それらの化学性質に応じ、数時間から最大100日を超える期間にわたり化合物を放出する。
【0062】
この医薬組成物は、適当な固相又はゲル相の担体又は賦形剤も含むことができる。このような担体又は賦形剤の例は、炭酸カルシウム、リン酸カルシウム、様々な糖類、デンプン、セルロース誘導体、ゼラチン、及びポリマー、例えばポリエチレングリコールなどを含むが、これらに限定されるものではない。
薬物中毒の治療のために、本発明の化合物は、この物質を薬物-含有製品(例えばニコチン摂取の場合は、タバコ製品、例えば紙巻きタバコなど)へ混入することにより投与することもできる。例えばある態様において、本発明の化合物は、摂取前に、薬物-含有製品へ噴霧又はそうでなければ塗布される。
【0063】
本発明における使用に適した医薬組成物は、活性成分が治療的有効量含まれる組成物を含む。本発明の方法のための治療的有効量は、例えば年齢、体重、全身の健康状態、性別、食事、投与時間及び様式、排泄速度、併用薬、治療担当医の判断、及び治療される特定の苦痛の重症度を含む、様々な要因に応じて決定することができる。活性物質の量は、オピエート-関連物質の比活性、及びその物質が他の治療成分又は予防成分と同時に投与されるかどうかによっても左右される。
【0064】
典型的には、本願明細書に開示された方法に従い治療される対象は、疼痛に苦しむとして確定されるか;鎮痛薬(例えばオピオイド)受容体の変調による治療もしくは予防に影響される疾患もしくは障害と診断されるか;又は、そうではなければ、この化合物を使用する生理的恩恵を受ける対象であると確定される。ある変形において、疼痛に苦しむ対象又は鎮痛薬受容体の変調による治療又は予防に影響される疾患もしくは障害(例えば中毒)と診断された対象は、第二の疾患又は障害には罹患していない。更に一部の態様において、この対象は、治療期間中、生理的及び/又は臨床的作用についてモニタリングされる。例えば疼痛治療に関して、対象は、治療経過を通じて疼痛の相対重症度についてモニタリングすることができ、;又は、鎮痛薬受容体シグナル伝達の変調を介した改善に影響される疾患もしくは障害の治療について、対象は、この疾患もしくは障害に関連した1種又は複数の症状についてモニタリングすることができる。
【実施例】
【0065】
下記実施例は、例証のために提供されているが、請求された発明を制限するものではない。実施例1-32化合物参照番号は、先に使用されたものに対応している。異なる化合物参照名が、実施例33、及び集合的に実施例34-45の各々において使用される。
下記の全般的情報は、実施例1-32に示した化合物の合成及び分析に関して適用される。全ての反応は、特に別に記さない限りは、炉乾燥したガラス製品を用い、周囲温度で磁気攪拌しながら、窒素陽圧下で実施した。大気-及び水分-感受性液体は、シリンジによりゴム隔壁を通して移した。シリカゲル(230-400メッシュ)を、カラムクロマトグラフィーに使用した。DMFを、中間(neutral)アルミナのカラムを通る、濾過により乾燥し、及び使用前に窒素下で活性化された4Å分子篩上に貯蔵した。他の溶媒及び試薬は全て、入手したまま使用した。1H
NMR及び13C NMRスペクトルは、各々、500MHz及び125MHzで記録した。融点は補正しなかった。燃焼分析が特定されない場合は、分析的純度は、D7500積分器(integrator)及びHamilton
PRP-Iステンレス鋼カラム(250mm ×4.6mm内径)を伴う、Hitachi
L74液体クロマトグラフィーを使用する、順相HPLCにより決定した。HPLC移動相:A=55:45:0.01
MeOH/イソプロパノール/HCl04;B=45/55/0.01
MeOH/イソプロパノール/HClO4;C=70:30
CH3CN/MeOH;D=MeOH。
【0066】
更に以下に説明された「I型」、「II型」、及び「III」は一般に、各々、先に説明された式IV、V、及びVIの化合物を意味する。
(実施例1):6β-S-(メチル-2',3'4'-トリ-O-アセチル-β-D-グルコピラノシルウロネート)モルヒネ3a
NaH(46mG, 1.16mmol)の60%分散体を、0℃の2a(424mg,
1.21mmol)のDMF溶液へ添加し、得られた赤色混合物を、10分間攪拌した。こうして形成されたチオールの混合物を、0℃で1b(183mg,
0.416mmol)のDMF(8mL)溶液へ1分間かけてシリンジで添加した。得られた溶液を、室温まで温めながら4時間攪拌した。反応液を、0.5%水性HC(20mL)へ注ぎ、pHを、固形NaHCO3を慎重に添加することにより、9へ上昇させた。この混合物を、CH2Cl2(3
× 50mL)で抽出し、一緒にした有機層を、H2O(10mL)、ブライン(10mL)で洗浄し、Na2SO4上で乾燥し、濾過し、灰色混合物へ濃縮し、フラッシュクロマトグラフィー(15
×2.5cm SiO2, 60:1〜20:1 CH2Cl2/EtOH)により精製し、黄味がかった白色の固形物として3a(144mg,
56%)を得た。3aの分析用試料を、無水EtOHからの再結晶により、白色固形物として得た:Rf=0.10
(20:1 CH2Cl2/MeOH);mp=135℃(分解);1H
NMR δ6.63 (d, J= 7.9 Hz, 1H), 6.50 (d, J= 7.9 Hz, 1H) , 5.81 (ddd, J= 9.1, 5.7,
3.0 Hz, 1H), 5.29-5.21 (m, 2H), 5.05 (d, J= 9.2 Hz, 1H), 5.03 (s, 1H), 4.70 (d,
J= 9.6 Hz, 1H), 4.06 (d, J= 9.6 Hz, 1H), 3.74 (s, 3H), 3.31 (m, 1H), 3.03-2.99
(m, 2H), 2.58 (dd, J= 11.7, 4.0 Hz, 1H), 2.43 (s, 3H), 2.38-2.28 (m, 2H),
2.15-2.09 (m, 2H), 2.05 (s, 3H), 2.02 (s, 3H), 1.76 (dd, J= 12.2, 1.8 Hz, 1H);13C
NMR (CDCl3)δ170.3, 169.6, 169.5, 167.0, 144.7, 138.5, 131.8, 130. 4,
128.5, 126.7, 119.7, 116.8, 93.7, 84.6, 76.3, 73.4, 70.0, 69.4, 59.2, 53.1,
47.2, 45.0, 44.5, 43.1, 39.8, 35.6, 20.9, 20.8, 20.7;C30H36NO11S
[M+H+]のHRMS計算値618.2009、実測値618.2011;3aの平均純度は、tR
=3.47分(移動相A)及びtR =3.98分(移動相B)を生じたことから、分析的HPLCにより、≧99%であることがわかった。
【0067】
(実施例2):3-O-アセチル-6-β-S-(メチル-2',3',4'-トリ-O-アセチル-β-D-グルコピラノシルウロネート)モルヒネ 3b
3aについて説明された手法に従い、NaH(53mg, 1.33mmol)、1a(484mg,
1.38mmol)及び2a(246mg, 0.51mmol)は、黄味がかかった白色粉末として3bを提供した(211mg,
63%):Rf=0.33 (20:1 CH2Cl2/EtOH);mp
194℃(分解);1HNMR (CDC13) δ 6.74 (d, J= 8.2 Hz, 1H), 6.58 (d, J= 8.2
Hz, 1H), 5.83-5.80 (m, 1H), 5.53 (dd, J= 1.5, 10.2 Hz, 1H), 5.29-5.21 (m, 2H),
5.09 (s, 1H), 5.00 (t, 1H), 4.72 (d, J= 10.3 Hz, 1H), 4.06 (d, J= 9.3 Hz, 1H),
3.76-3.73 (4H), 3.33 (m, 1H), 3.07-3.03 (2H), 2.60-2.58 (m, 1H), 2.44 (s, 3H),
2.37-2.20 (s, 3H over m, 2H), 2.15 (dt, J= 3.7, 11.8 Hz, 1H), 2.04 (s, 3H),
2.03 (s, 3H), 2.02 (s, 3H), 1.80 (d, J=11.6 Hz, 1H);13C
NMR (CDCl3) δ170.3, 169.5, 169.4, 168.8, 167.0, 149.0, 133.0, 132.6,
132.0, 131.8, 128.0, 121.8, 119.5, 94. 5, 85.1, 76.2, 73.3, 70.3, 69.4, 59.0,
53.0, 46.9, 45.6, 44.5, 43.2, 40.0, 35.7, 20.93, 20.90, 20.8, 20.7;MS
(ESI) m/z=660 [M+H+];分析(C32H37NO12S)計算値:C
58.26;H 5.65;N 2.12;S
4.86、実測値:C 57.94;H 5.75;N
2.07;S 4.97。
【0068】
(実施例3):3-O-アセチル-6β-S-(2',3',4',5'-テトラ-O-アセチル-β-D-グルコピラノシル)-6β-チオモルヒネ 3c
3aについて説明された手法に従い、チオール2b (500mg, 1.37mmol)、NaH(53mg,
1.32mmol)及びトシラート1b(246mg, 0.51mmol)は、黄味がかかった白色泡状物として3cを生じた(255mg,
74%):Rf=0.20 (20:1 CH2Cl2/MeOH);mp=151.9℃;1H
NMR (CDC13) δ 6.73 (d, J= 8.2 Hz, 1H), 6.58 (d, J= 8.2 Hz, 1H),
5.83-5.79 (m, 1H), 5.50 (dd, J= 1.8, 9.7 Hz, 1H), 5.24-5.17 (m, 2H), 5.04 (t,
J=9.9 Hz, 1H), 4.96 (t, J=9.9 Hz, 1H), 4.69 (d, J= 10.2 Hz, 1H), 4.19 (d, J=
4.2 Hz, 2H), 3.79-3.75 (m, 1H), 3.71 (d, J= 6.0 Hz, 1H), 3.30 (dd, J= 3.3, 5.6:
Hz, 1H), 3.04 (d, J= 18.8 Hz, 1H), 2.57 (dd, J= 4.0, 12.2 Hz, 1H), 2.42 (s,
3H), 2.34-2.29 (m, 2H), 2.14-2.11 (m, 1H), 2.07 (s, 6H), 2.05 (s, 6H), 2.00 (s,
6H), 1.80-1.77 (m, 1H);13C
NMR (CDCl3) δ170.8, 170.1, 169.5, 169.3, 169.5, 148.8, 132.8, 132.1,
131.7, 131.6, 127.9, 121.6, 119.3, 94.4, 85.1, 75.9, 73.8, 70.3, 68.5, 62.4,
58.8, 46.7, 45.8, 44.3, 43.0, 39.7, 35.6, 20.7, 20.6, 20.5;MS
m/z=674 [M+H]+;HRMS m/z計算値674.2271、実測値674.2238;3cの平均純度は、tR
=4.86分(移動相A)及びtR =5.66分(移動相B)を生じたことから、分析的HPLCにより、97.3%であることがわかった。
【0069】
(実施例4):6-βS-(2',3',4',5'-テトラ-O-アセチル-β-D-グルコピラノシル)-6β-チオモルヒネ 3d
この化合物は、3cの調製の副産物として単離した(47mg, 14.6%):Rf=0.08
(20:1 CH2Cl2/MeOH);mp 165.4℃;1H
NMRδ6.63 (d, J=8.2 Hz, 1H), 6.51 (d, J= 8.2 Hz, 1H), 5.79 (ddd, J= 3.6, 5.8,
9.6 Hz, 1H), 5.50 (dd, J= 1.7, 9.6 Hz, 1H), 5.22 (t, J= 9.3 Hz, 1H), 5.09-4.99
(m, 3H), 4.69 (d, J= 10.2 Hz, 1H), 4.27-4.19 (m, 2H), 3.78-3.74 (m, 1H), 3.67
(d, J= 5.8 Hz, 1H), 3.32 (m, 1H), 3.01 (d, J=18.9 Hz, 1H) over bs (1H),
2.61-2.58 (m, 1H), 2.44 (s, 3H), 2.39-2.27 (m, 2H), 2.16-2.11 (m, 2H), 2.08 (s,
3H), 2.06 (s, 3H), 2.05 (s, 3H), 2.03 (s, 3H), 1.78-1.75 (m, 1H);13C
NMRδ171.1, 170.4, 169.69, 169.66, 144.6, 130.3, 131.8, 130.6, 128.3, 126.9,
119.7, 116.7, 94.2, 84.9, 76.1, 74.0, 70.4, 68.7, 62.5, 59.2, 47.2, 45.8, 44.5,
43.2, 39.8, 35.8, 21.0, 20.83, 20.80, 20.6;MS
m/z=632[M+H]+;HRMS m/z C3lH38NO11S
の計算値632.2166、実測値632.2136;3dの平均純度は、tR
=3.98分(移動相A)及びtR =4.40分(移動相B)を生じたことから、分析的HPLCにより、≧98%であることがわかった。
【0070】
(実施例5):6β-S-(メチル-2',3',4'-トリ-O-アセチル-β-D-グルコピラノシルウロネート)コデイン 3e
3aについて説明された一般的手法に従い、1c(231mg, 0.51mmol)、2a(483mg,
1.38mmol)及びNaH(1.33mmol)は、黄味がかかった白色固形物3e(249mg,
77%)を生じた:Rf =0.30, 20:1 CH2Cl2/EtOH);mp=172℃;1H
NMR (CDC13) δ6.64 (d, J= 8.1 Hz, 1H), 6.54 (d, J= 8.1 Hz, 1H), 5.82
(ddd, J= 3.0, 6.0, 9.2 Hz, 1H), 5.53 (dd, J= 1.8, 9.4 Hz, 1H), 5.27-5.22 (m,
2H), 5.14 (s, 1H), 4.99 (t, J= 2.1 Hz, 1H), 4.71 (d, J= 10.3 Hz, 1H), 4.02 (d, J=
7.3 Hz, 1H), 3.83 (s, 3H), 3.79 (d, J= 6.0 Hz, 1H), 3.73 (s, 3H), 3.29 (dd, J=
3.3, 5.8 Hz, 1H), 3.05-3.00 (d, J= 19.0 Hz, 1H over m, 1H), 2.55
(dd, J= 4.3, 12.2 Hz, 1H) , 2.42 (s, 3H), 2.35 (td, J= 3.4, 12.4 Hz, 1H), 2.29
(dd, J= 6.0, 19.0 Hz, 1H), 2.15 (td, J= 5.0, 12.4 Hz, 1H), 2.02 (s, 3H), 2.016
(s, 3H), 2.00 (s, 3H), 1.77 (dd, J= 1.5, 12.3 Hz, 1H);13C
NMR δ 170.2, 169.5, 169.4, 166.9, 146.3, 142.2, 133.1, 130.7, 127.9, 127.7,
119.1, 112.8, 93.6, 85.2, 76.1, 73.4, 70.3, 69.3, 59.1, 56.4, 53.0, 47.0, 45.5,
44.7, 43.3, 40.0, 36.2, 20.9, 20.8, 20.7, 20.6;MS(ESI)
632 [M+H]+;分析(C3lH37NO11S)
C, H, N, S。
【0071】
(実施例6):6-S-(2',3',4',5'-テトラ-O-アセチル-β-D-グルコピラノシル)6β-チオコデイン 3f
3aについて説明された一般的手法に従い、チオール2b (440mg, 1.21mmol)、NaH
(47mg, 1.16mmol)及び1c(203mg, 0.448mmol)は、黄味がかかった白色泡状物として3f(237mg,
82%)を提供した。分析的試料は、沸騰ヘキサンからの再結晶により得た:Rf=0.26
(20:1 CH2Cl2/EtOH);mp=142.5℃;1H
NMR (CDC13) δ6.65 (d, J= 8.2 Hz, 1H), 6.55 (d, J= 8.2 Hz, 1H),
5.85-5.81 (m, 1H), 5.52 (dd, J= 1.9, 9.6 Hz, 1H), 5.22-5.19 (m, 2H), 5.06 (t,
J= 9.8 Hz, 1H), 4.97 (dd, J= 9.5, 10.1 Hz, 1H), 4.69 (d, J= 10.1 Hz, 1H),
4.23-4.16 (m, 2H), 3.83 (s, 3H), 3.76 (d, J= 6.0 Hz, 1H), 3.76-3.71 (m, 1),
3.29 (dd, J= 3.3, 5.8 Hz, 1H), 3.06-3.01 (m, 2H), 2.56 (dd, J= 4.0, 12.2 Hz,
1H), 2.42 (s, 3H), 2.38-2.30 (m, 2H), 2.18-2.15 (m, 1H), 2.12 (s, 3H), 2.09 (s,
6H), 2.00 (s, 3H), 1.99-1.80 (m, 1H);13C
NMR (CDCl3) δ171.0, 170.4, 169.6, 169.5, 146.3, 142.2, 132.9, 130.7,
127.9, 127.6, 119.1, 112.7, 93.8, 85.5, 76.1, 74.0, 70.5, 68.5, 62.5, 59.1,
56.4, 47.0, 46.1, 43.3, 40.0, 20.93, 20.92, 20.8, 20.5;MS
m/z=646 [M+H]+;HRMS C32H40NO11Sの計算値646.2322、実測値646.2304;3fの平均純度は、tR
=5.00分(移動相A)及びtR =5.71分(移動相B)を生じたことから、分析的HPLCにより、≧99%であることがわかった。
【0072】
(実施例7):モルヒネ-6β-S-D-グルクロニド 4a
MeOH 1.6mL中の3b(50.0mg, 0.076mmol)溶液に、5%NaOH水溶液(0.12mL)を添加した。得られた金色の溶液を、室温で18.5時間攪拌し、氷酢酸10滴の添加によりpH5へ酸性とし、濃縮した。得られた白色非晶質固形物を、シリカゲル(16
×1cm, 5:1から1:1 CH3CN/MeOH)上のフラッシュクロマトグラフィー、それに続く100℃(0.25mmHg)で4時間の乾燥により精製し、白色粉末(19.4mg,
54%)を得た:Rf=0.26 (1:1 CH3CN/0.2%AcOHを含むMeOH);mp=>300℃(分解);1H
NMR (D20) δ6.65 (d, J= 8.1 Hz, 1H), 6.61 (d, J= 8.1 Hz, 1H), 5.92
(m, 1H), 5.61 (d, J= 9.6 Hz, 1H), 5.19 (s, 1H), 4.69 (d, J= 9.9 Hz, 1H), 3.87
(d, J= 5.9 Hz, 1H), 3.77 (d, J= 9.0 Hz, 1H), 3.64 (m, 1H), 3.57-3.51 (m, 2H),
3.38 (t, J= 9.2 Hz, 1H), 3.13-3.09 (m, 2H), 2.83 (dd, J= 3.7, 12.3 Hz, 1H),
2.59-2.54 (s, 3H over m, 2H), 2.19 (dt, J= 8.5, 13.2 Hz, 1H), 1.87 (d, J= 12.0
Hz, 1H);13C NMR (D2O) δ 174.6, 144.3, 139.6, 129.8, 128.8, 127.7,
123.3, 119.0, 116.9, 91.7, 85.0, 79.3, 76.1, 71.3, 70.7, 58.0, 45.6, 43.3, 42.6,
40.3, 37.2, 33.0, 19.7;MS (ESI) m/z=478
[M+H]+;分析(C23H27NO8S)計算値:C
57.85;H 5.70;N 2.93;S
6.71、実測値:C 57.75;H 5.95;N
2.73;S 6.58。
【0073】
(実施例8):6β-チオモルヒネ-6β-S-D-グルクロピラノシド 4b
MeOH(6mL)中の3cの褐色溶液(50mg, 0.0742mmol)を、5%NaOH水溶液(0.4mL)に添加した。白色沈殿が、5分以内に形成された。この混合物を、18時間攪拌し、1.5N
AcOH(15滴)の添加により、pHを7に低下した。溶液を、5分間攪拌し、その後飽和NaHCO3 (15滴)を添加し、pHを8.5に低下した。混合物を濃縮し、残渣を、13%NH4OH
3滴添加した水0.5mL中に溶解し、分取的TLC (SiO2,
200:40:5:0.05 CH2Cl2/MeOH/H2O/13% NH4OH)により精製した。1本のUV活性バンドを、SiO2から取り出し、SiO2を4:1
CHCl3/MeOH(100mL)で洗浄した。濾液を濃縮し、残渣を、9:1
CHCl3/MeOH (10mL)を用い綿栓を通して再濾過した。濾液を濃縮し、得られた固形物を、100℃(0.25mmHg)で4時間乾燥し、表題化合物を白色粉末(20mg,
59%)として得た:Rf=0.21;mp=>300℃(dec)
1H NMR (CD3OD) δ6.55 (d, J= 8.0 Hz, 1H), 6.46 (d, J= 8.0
Hz, 1H) , 5.90 (ddd, J= 2.9, 5.8, 9.0 Hz, 1H), 5.52 (dd, J= 1.6, 9.7 Hz, 1H),
5.22 (s, 1H), 4.54 (d, J= 9.8 Hz, 1H), 3.90 (d, J= 11.2 Hz, 1H), 3.77 (d, 1=
6.0 Hz, 1H), 3.70 (dd, J= 5.2, 11.9 Hz, 1H), 3.42 (dd, J= 3.2, 5.6 Hz, 1H),
3.42-3.41 (m, 3H), 3.24 (dd, J= 8.4, 9.6 Hz, 1H), 3.07 (bs, 1H), 3.05 (d, J=
18.7 Hz, 1H), 2.68 (dd, J= 4.0, 12.3 Hz, 1H), 2.49 (s, 3H), 2.49-2.39 (m, 2H),
2.18 (dt, J= 7.9, 12.8 Hz, 1H), 1.81-1.79 (m, 1H);13C
NMR δ 146.3, 140.4, 131.5, 131.4, 130.3, 126.7, 120.4, 117.9, 95.3, 88.4, 82.3,
79.9, 74.8, 71.6, 63.2, 60.7, 48.2, 46.3, 45.3, 43.0, 40.4, 36.7, 30.9, 24.2;MS
m/z=464 [M+H]+;HRMS m/z C23H30NO7Sの計算値
464.1743、実測値464.1727;4bの平均純度は、tR
=3.94分(移動相A)及びtR =4.42分(移動相B)を生じたことから、分析的HPLCにより、97.0%であることがわかった。
【0074】
(実施例9):コデイン6β-S-グルクロニド, 4c
4aの調製について説明された手法に従い、3e(60.4mg, 0.095mmol)、MeOH
(2mL)及び5%NaOH水溶液(0.28mL)は、白色固形物として4c(38mg, 79%)を提供した:Rf=0.08
(1:1 MeCN/0.2%AcOHを含むMeOH);mp=>300℃(分解);1H
NMR (D2O) 6.82 (d, J= 8.2 Hz, 1H), 6.69 (d, J= 8.2 Hz, 1H), 5.83
(ddd, J= 2.8, 6.0, 9.5 Hz, 1H), 5.22 (s, 1H), 4.66 (d, J= 9.9 Hz, 1H), 3.84 (d,
J= 6.0 Hz, 1H), 3.81 (s, 3H), 3.76 (d, J= 6.0 Hz, 1H), 3.57-3.50 (m, 2H), 3.42
(dd, J= 3.3, 5.7 Hz, 1H), 3.40-3.34 (m, 1H), 3.08 (d, J= 19.0 Hz, 1H), 3.01 (m,
1H), 2.60 (dd, J= 4.0, 12.4 Hz, 1H), 2.43-2.37 (s, 3H over m, 1H), 2.30 (td, J=
3.4, 12.6 Hz, 1H) , 2.12 (td, J= 4.8 Hz, 13.0, 1H), 1.75 (d, J= 12.0 Hz, 1H);13C
NMR (D20) δ174.5, 144.4, 140.5, 131.0, 129.5, 127.3, 127.1, 119.0,
112.9, 92.8, 85.1, 79.3, 76.1, 71.3, 70.8, 57.1, 55. 7, 45.1, 43.2, 43.0, 40.5,
37.7, 33.8, 19.3;HRMS [M-H-] C24H28NO8Sの計算値
490.1536、実測値490.1525;4cの平均純度は、tR
=1.62分(移動相C)及びtR =1.46分(移動相D)を生じたことから、分析的HPLCにより、96%であることがわかった。
【0075】
(実施例10):6β-チオコデイン-6β-S-D-グルコース, 4d
エステル3f(120mg, 0.186mmol)を、MeOH (4mL)に溶解し、5%NaOH水溶液(0.6mL)を添加した。この混合物を、室温で18時間攪拌し、その後飽和の水性NH4Cl(1mL)で処理した。混合液を濃縮し、残渣を、分取TLC(SiO2)において、溶離液としてCH2Cl2/MeOH/水/13%濃縮したNH4OH(200:40:5:0.050)を使用し精製した。1本のUV活性バンドを、かみそりの刃でSiO2から取り出し、このSiO2を4:1
CHCl3/MeOH(100mL)で洗浄した。濾液を濃縮し、残渣を、溶離液として9:1
CHCl3/MeOH (10mL)を使用し綿栓を通して再濾過した。濾液を濃縮し、得られた固形物を、100℃(0.25mmHg)で4時間乾燥し、白色粉末として4dを得た(23mg,
26%):Rf=0.17;mp=201.6℃;1H
NMR (CD3OD) δ6. 79 (d, J= 8.3 Hz, 1H), 6.68 (d, J= 8.3 Hz, 1H), 6.02
(ddd, J= 3.0, 5.9, 9.1 Hz, 1H), 5.55 (dd, J= 1.8, 9.8 Hz, 1H), 5.36 (s, 1H),
4.57 (d, J= 9.8 Hz, 1H), 4.10 (dd, J= 3.0, 6.2 Hz, 1H), 3.91 (dd, J= 1.6, 12.1
Hz, 1H), 3.83 (d, J= 6.1 Hz, 1H), 3.81 (s, 3H), 3.70 (dd, J= 5.1, 12.0 Hz, 1H),
3.39-3.23 (複合m, 6H), 3.01-3.00 (m, 1H), 2.98 (s, 3H), 2.90 (dd, J= 6.6, 19.8
Hz, 1H), 2.41 (dt, J= 4.8, 13.8 Hz, 1H), 2.04 (dd, J= 2.7, 14.0 Hz, 1H);13C
NMR (CD30D) δ147.8, 144.5, 131.7, 130.1, 128.9, 125.3, 121.1, 116.2,
94.5, 88.3, 82.3, 79.9, 79.6, 74.8, 71.6, 63.1, 62.3, 57.3, 45.7, 44.3, 41.9,
38.8, 34.8, 23.0.;MS m/z=478 [M+H]+;HRMS
m/z C24H32NO7Sの計算値
478.1899、実測値478.1886;4dの平均純度は、tR
=4.79分(移動相A)及びtR =5.64分(移動相B)を生じたことから、分析的HPLCにより、96.5%であることがわかった。
【0076】
(実施例11):6βメチル-2',3',4'-トリ-O-アセチル-β-D-グルコピラノシルウロネート)アセトアミドコデイン 9b
塩化チオニル(0.2mL, 2.74mmol)を、カルボン酸8a (20.0mg,
0.053mmol)へ添加し、得られた無色の溶液を、窒素下で4時間攪拌した。トルエン(1mL)を添加し、溶液を、45℃の水浴を使い、高圧下での蒸留により濃縮した。得られた残渣を、0℃の氷浴中で冷却した。NEt3を3滴含有するアミン9b(32mg,
0.106mmol)の0℃のCH2Cl2 (2mL)溶液を、シリンジで添加した。得られた溶液を、氷浴を補充することなく、20時間攪拌し、CH2Cl2
(15mL)で希釈し、飽和NaHCO3水溶液(2 ×2mL)及びブライン(2mL)で洗浄した。有機層を乾燥し、濾過し及び濃厚な黄色油状物へ濃縮した。フラッシュクロマトグラフィーによる精製(10
×1cm SiO2, 10:1 CH2Cl2/MeOH)は、表題化合物を白色固形物(10mg,
29%)として得た:Rf=0.17 (10:1 CH2Cl2/MeOH);1H
NMR (CDCl3) δ7.06 (d, J= 6.8 Hz, 1H), 6.666 (d, J= 8.2 Hz, 1H), 6.54
(d, J= 8.2 Hz, 1H), 5.888 (ddd, J= 3.0, 5.8, 9.4 Hz, 1H), 5.60 (dd, J= 1.5, 9.4
Hz, 1H), 5.30 (s, 1H), 5.08 (t, J= 3.0 Hz, 1H), 4.78 (s, 1H), 4.68-4.66 (m,
2H), 4.57 (d, J= 10.4 Hz, 1H), 4.51 (t, J= 6.2 Hz, 1H), 3.86 (s, 3H), 3.82 (s,
3H), 3.30 (m, 1H), 3.03 (d, J= 18.5 Hz, 1H), 2.97 (m, 1H), 2.73 (dd, J= 11.1,
17.0 Hz, 1H), 2.55 (dd, J= 3.9, 11.6 Hz, 1H), 2.43 (s, 3H), 2.37-2.29 (m, 3H),
2.16 (s, 3H), 2.15 (s, 3H), 2.04 (s, 3H), 2.03-1.99 (m, 1H), 1.81-1.79 (m, 1H);MS(ESI)
m/z=657 [M+H]+。
【0077】
(実施例12):6α-(メチル-2',3',4'-トリ-O-アセチル-β-D-グルコピラノシルウロネート)アセトアミドモルヒネ,
10a
化合物9bについて説明された手法に従い、カルボン酸8a (38.5mg, 0.102mmol)、塩化チオニル(0.6mL)及びアミン7a(30mg,
0.075mmol)は、白色固形物として10a(29mg, 51%)を提供した:Rf=0.11
(20:1 CH2Cl2/MeOH);1H
NMR(CDCl3) δ7.01 (d, J = 6.7 Hz, 1H), 6.59 (d, J= 7.7 Hz, 1H), 6.45
(d, J= 7.7 Hz, 1H), 5.88-5.84 (m, 1H), 5.59 (dd, J= 1.6, 9.6 Hz, 1H), 5.08 (t,
J= 3.0 Hz, 1H), 4.72-4.66 (m, 2H), 4.58 (d, J= 16.4 Hz, 1H), 4.49 (t, J= 6.3
Hz, 1H), 3.82 (s, 3H), 3.84-3.72 (m, 2H), 3.29 (m, 1H), 3.01 (d, J= 18.5 Hz,
1H), 2.94 (m, 1H), 2.75-2.69 (dd, J= 11.0, 17.1 Hz, 1H), 2.53 (m, 1H), 2.43 (s,
3H), 2.42-2.29 (m, 2H), 2.19-1.99 (m, 2H), 2.163 (s, 3H), 2.157 (s, 3H) 2.06
(s, 3H), 1.78-1.75 (m, 1H), 0.98 (s, 9H), 0.194 (s, 3H), 0.174 (s, 3H);MS
(ESI) 757 [100, (M+H)+]。中間体シリルエーテル(20mg,0.026mmol)を、THF(1mL)に溶解した。TBAF(0.030mL,
0.030mmol)及び水(0.020ml)を添加し、紫色の溶液を、室温で50分間攪拌し、CH2Cl2
(20mL)で希釈し、NH4Cl (4mL)、ブライン(2mL)及び水(2mL)で洗浄した。有機層を乾燥し(Na2SO4)、濾過し、濃縮し、ピンク色の泡(22mg)を得た:Rf=0.22(10:1
CH2Cl2/MeOH);MS m/z=643
[M+H]+。
【0078】
(実施例13):6β-(α-グルクロノシル)アセトアミドモルヒネ 11a
10a(20mg, 0.031mmol)、5%NaOH水溶液(0.4mL)及びMeOH(2mL)から実施例7について説明された手法により調製し、白色固形物として11a(2.1mg,
18%)を得た:Rf=0.19 (SiO2/0.2%AcOHを含むMeOH);MS
m/z=501 [M-H]-。
(実施例14):6β-(α-D-グルクロノシル)アセトアミドコデイン 11b
【0079】
表題化合物を、実施例7について説明された手法により、白色固形物(13.5mg, 86%)として得た:Rf=0.14
(0.2%AcOHを含むMeOH);1H
NMR (D2O) δ6.97 (d,J=8.4 Hz, 1H), 6.84 (d, J= 8.4 Hz, 1H), 5.89-5.85
(m, 1H), 5.71 (d, J= 12.2 Hz, 1H), 4.94 (s, 1H), 4.62-4.59 (m, 1H), 4.36 (d, J=
5.7 Hz, 1H), 4.22 (m, 1H), 4.03 (d, J= 6.2 Hz, 1H), 3.88 (s, 3H), 3.80-3.73 (m,
2H), 3.35-3.30 (m, 2H), 3.03-2.94 (s, 3H 重なり m,
1H), 2.77-2.71 (m, 1H), 2.59 (dd, J= 3.6, 16.1 Hz, 1H), 2.31 (td, J= 4.7, 13.5
Hz, 1H), 2.06 (m, 1H);MS m/z=515 [M-H]-。
【0080】
(実施例15):6β-(2',3',4',5'-テトラ-O-ベンジル-β-D-グルコピラノシル)-3-O-トリイソプロピルシリルアセトアミドモルヒネ 9c
参照JMM-VI-113-2。SOCl2 (1.5mL)を、カルボン酸8b
(115mg, 0.20mmol)へ添加し、この溶液を、室温で16時間攪拌した。過剰なSOCl2を蒸留により除去し、生じた無色の油状物を、CH2Cl2(2.5mL)中に溶解し、CH2Cl2(1mL)中のアミン7c(43.5mg,
0.10mmol)及びNEt3(0.035mL, 0.25mmol)へ添加した。この溶液を、室温で1時間攪拌し、濃縮及びフラッシュクロマトグラフィーにより精製し(SiO2,
30:1から10:1 EtOAc/MeOH)、白色固形物として9c(70mg,
70%)を提供した:Rf=0.46 (10:1 CH2Cl2/MeOH);1H
NMR (CDCl3) d 7.35-7.10 (20H), 6.59 (d, J= 8.0 Hz, 1H), 6.40 (d, J=
8.0 Hz, 1H), 6.22 (d, J= 6.9 Hz, 1H), 5.71 (ddd, J= 3.0, 6.0, 9.5 Hz, 1H), 5.41
(dd, J= 1.9, 9.5 Hz, 1H), 4.89-4.48 (9H), 4.36 (t, J= 6.3 Hz, 1H), 3.72-3.46
(5H), 3.29 (t, J= 9.0 Hz, 1H), 3.17 (dd, J= 3.0, 5.2 Hz, 1H), 2.96 (d, J= 18.7
Hz, 1H), 2.88 (bs, 1H), 2.72 (dd, 2.7, 15.2 Hz, 1H), 2.50-1.72 (10H),
1.30-1.05(21H)。
【0081】
(実施例16):6β-(2',3',4',5'-テトラ-O-ベンジル-β-D-グルコピラノシル)アセトアミドモルヒネ 10b
JMM-VI-114-2。シリルエーテル9c(60mg, 59.7mmol)を、THF(2mL)中に溶解し、及び水(0.050mL)を添加し、その後TBAF(0.090mL,
0.090mmol, THF中1.0M溶液)を添加した。2.5時間後、1%濃縮したHCl(4mL)を添加し、混合物を2分間攪拌し、水(10mL)で希釈し、固形NaHCO3で塩基性とした。この混合物を、CH2Cl2
(5 ×30mL)で抽出し、乾燥し(Na2SO4)、濾過し濃縮した。フラッシュクロマトグラフィー(24:1から12:1
EtOAc/MeOH)は、白色固形物として10b(42mg, 83%)を提供した:Rf=0.15
(12:1 EtOAc/MeOH);1H NMR (CDC13) δ7.34-7.10 (20H), 6.64 (d, J= 8.2 Hz,
1H), 6.46 (d, J= 8.2 Hz, 1H), 6.34 (d, J= 6.3 Hz, 1H), 5.68 (ddd, J= 2.5, 5.8,
9.5 Hz, 1H), 5.39 (d, J= 9.5 Hz, 1H), 4.89-4.46 (10H), 4.34 (t, J= 6.3 Hz, 1H),
3.72-3.46 (6H), 3.30 (t, J= 9.1 Hz, 1H), 3.18 (bs, 1H), 2.97 (d, J= 18.1 Hz,
1H), 2.94 (bs, 1H), 2.74 (dd, J= 2.5, 15.7 Hz, 1H), 2.55-2.16 (7H), 1.95 (dt,
J= 8.0, 12.6 Hz, 1H), 1.76 (d, J= 11.3 Hz, 1H)。
【0082】
(実施例17):6β-(β-D-グルコピラノシル)アセトアミドジヒドロモルヒネ11c
JMM-VI-116-2。テトラベンジル誘導体10b(16mg, 0.018mmol)を、MeOH(2mL)及び濃HCl(3滴)中に溶解し、その後10%Pd-C(16mg)を添加した。混合物を、24時間水素バルーン(balloom)下で攪拌し、その後セライトを通して濾過し、このセライトをMeOH(10mL)で洗浄した。濾液を濃縮し、残渣を分取TLC(SiO2,
5:4:0.5:0.5 CHCl3/MeOH/H2O/濃NH4OH)で精製し、11cを白色フィルム(2.3mg,
26%)として得た:Rf=0.40;MS
m/z=491 (MH+)。
【0083】
(実施例18):6β-(3-メトキシフェニル)アセトアミドモルヒネ,13a
7c(80mg, 0.182mmol)を、CH2Cl2(4mL)中で溶解し、NEt3(72μL,
0.517mmol)及び3-メトキシフェニルアセチルクロリド(80μL, 0.513mmol)をシリンジにより添加した。得られた淡黄色溶液を、密閉したバイアル中で、室温で乾燥窒素大気下で攪拌した。2時間後、この溶液を濃縮し、残渣を、フラッシュクロマトグラフィー(20:1
CHCl3/MeOH)により精製し、白色泡状物として12a(78mg, 73%)を提供した:Rf
=0.18 (20:1 CH2Cl2/MeOH);MS
m/z: 589(M+H)+。12a(64mg, 0.109mmol)を、5%水性THF(2.2mL)中に溶解した。TBAF(0.18mL,
THF中1.0M溶液)は、シリンジにより添加し、淡黄色溶液を、室温で、乾燥窒素大気下で2時間攪拌した。溶液を濃縮し、1%HCl(2mL)を添加した。混合物を2分間攪拌し、その後、水(20mL)を使い分液ロートに移した。混合物を、固形NaHCO3でアルカリ化し(pH8.5)、CHC13(5×10mL)で抽出した。一緒にしたCHCl3抽出物を、ブライン(4mL)で洗浄し、乾燥し(Na2SO4)、濾過し、濃縮した。フラッシュクロマトグラフィー(20:1
CHCl3/MeOH)は、13aを白色固形物(38mg,
81%)として提供した:mp144℃;Rf =0.14(20:1 CHCl3/MeOH);1H
NMRδ7.24 (J= 7.9 H, 1H), 6.82-6.79 (m, 3H), 6.64 (d, J = 8.2 Hz, 1H), 6.49 (d, J=
8.2 Hz, 1H), 5.67 (ddd, J= 3.6, 6.1, 9.1 Hz, 1H), 5.58-5.53 (m, 2H), 4.65 (s,
1H), 4.37 (t, J= 6.4 Hz, 1H), 3.79 (s, 3H), 3.54 (s, 2H), 3.34 (m, 1H), 3.00
(d, J= 18.6 Hz, 1H), 2.89 (s, 1H), 2.61 (dd, J= 4.0, 12.2 Hz, 1H), 2.43 (s,
3H), 2.38-2.29 (m, 2H), 1.99 (dt, J= 9.6, 12.2 Hz, 1H), 1.76 (dd, 1.9, 12.6 Hz,
1H);13C NMRδ171.1, 160.2, 144.5, 138.9, 136.2, 132.8, 130.3, 129.9,
128.6, 125.8, 121.7, 119.6, 117.1, 115.1, 113.1, 93.0, 59.3, 55.4, 50.5, 47.3,
44.1, 43.9, 43.0, 39.8, 35.5, 20.5;MS m/z =433 [M+H]+、455[M+Na]+;HRMS
m/z 433.2115 [M+H]+;15aの平均純度は、tR
=4.38分(移動相A)及びtR =5.09分(移動相B)を生じたことから、分析的HPLCにより、≧99%であることがわかった。
【0084】
(実施例19):6β-(4-メトキシフェニル)アセトアミドモルヒネ 13b
12aについて説明された一般的手法に従い、7c(78.4mg, 0.178mmol)、NEt3(72μL,
0.517mmol)及び4-メトキシフェニルアセチルクロリド(80μL, 0.523mmol)は、12bを白色泡状物(70mg,
67%)として生じた:Rf=0.11 (20:1 CHCl3/MeOH);MS
m/z:589 (M+H)+, 611 (M+Na)+。参照V-99。13aについて説明された手法に従い、12b(65mg,
0.110mmol)及びTBAF(0.18mL, 0.18mmol)は、13b(29mg, 61%)を白色固形物として生じた:mp
247(dec);Rf=0.14 (20:1 CHCl3/MeOH);1H
NMR δ7.15 (d, J= 8.6 Hz, 2H), 6.86 (d, J= 8.6 Hz, 2H), 6.64 (d, J= 8.1 Hz, 1H),
6.48 (d, J= 8.1 Hz, 1H), 5.67 (ddd, J= 3.0, 5.6, 9.5 Hz, 1H), 5.56 (dd, J= 1.5,
9.5 Hz, 1H), 5.45 (d, J= 6.4 Hz, 1H), 4.65 (s, 1H), 4.36 (t, J= 6.3 Hz, 1H),
3.79 (s, 3H), 3.50 (s, 2H), 3.34 (m, 1H), 3.00 (d, J= 18.6 Hz, 1H), 2.87 (s,
1H), 2.61 (dd, J= 3.8, 11.9 Hz, 1H), 2.44 (s, 3H), 2.38-2.28 (m, 2H), 1.98 (dt,
4.9, 12.5 Hz, 1H) , 1.77 (d, J= 10.7 Hz, 1H);13C
δ171.5, 158.8, 144.4, 138.7, 132.6, 130.4, 129.7, 128.3, 126.5, 125.6, 119.3,
117.0, 114.5, 92.8, 59.1, 55.3, 50.3, 47.0, 43.9, 42.9, 42.7, 39.6, 35.4, 20.2;MS
m/z =433 (M+H)+, 456 (M+Na)+;HRMS
m/z 433.2136 [M+H]+;13bの平均純度は、tR
=4.44分(移動相A)及びtR =5.24分(移動相B)を生じたことから、分析的HPLCにより、>99%であることがわかった。
【0085】
(実施例20):6β-(3-メトキシ)ベンズアミドモルヒネ 13c
12aについて説明された一般的手法に従い、7c(82mg,, 0.185mmol)、NEt3
(76μL, 0.545mmol)及びアニソイルクロリド(71μL, 0.521mmol)は、白色泡状物として12c(88mg,
82%)を生じた;Rf=0.28 (10:1 CH2Cl2/MeOH);MS
m/z:575 (M+H)+。13aについて説明された一般的手法に従い、12c(73mg,,
0.174mmol)及びTBAF(0.210mL, 0.210mmol)は、白色固形物として13c(42.5mg,
58%)を生じた:Rf=0.15 (15:1 dcm/MeOH);mp=212.9℃(dec);1H
NMRδ7.34-7.33 (m, 1H), 7.27-7.23 (m, 2H), 7.02-7.00 (m, 1H), 6.68 (d, J= 8.1
Hz, 1H), 6.51 (d, J= 8.1 Hz, 1H), 6.27 (d, J= 6.8 Hz, 1H), 5.84 (ddd, J= 3.0,
5.8, 9.2 Hz, 1H), 5.68 (dd, J= 1.2 Hz, 9.9 Hz, 1H), 4.85 (s, 1H), 4.59 (t, J=
6.4 Hz, 1H), 3.81 (s, 3H), 3.47 (dd, J= 3.0, 5.0 Hz, 1H), 3.19-3.16 (m, 1H),
3.04 (d, J= 18.5 Hz, 1H), 2.68 (dd, J = 4.0, 12.0 Hz, 1H), 2.49 (s, 3H),
2.43-2.38 (m, 1H), 2.11 (dt, J= 4.8, 12.6 Hz, 1H), 1.80 (d, J= 10.9 Hz, 1H);13C
NMRδ167.5, 160.0, 144.7, 139.1, 135.6, 132.7, 129.9, 129.8, 128.9, 125.5,
119.7, 119.0, 118.2, 117.4, 112.6, 93.0, 59.5, 55.7, 50.9, 47.4, 44.1, 43.0,
39.9, 35.4, 20.6;MS m/z 419 (M+H)+, 441 (M+Na)+;HRMS
m/z = 419.1984 [M+H]+;13cの平均純度は、tR
=4.41分(移動相A)及びtR =5.05分(移動相B)を生じたことから、分析的HPLCにより、99%であることがわかった。
【0086】
(実施例21):6β-ベンズアミドモルヒネ, 13d
12aについて説明された一般的手法に従い、7c(82mg, 0.232mmol)、NEt3(76μL,
0.55mmol)及び塩化ベンジル(61μL, 0.521mmol)は、白色泡状物として12d(79mg,
78%)を生じた;Rf =0.23(20:1 CHCl3/MeOH);MS
m/z: 545 (M+H)+。13aについて説明された一般的手法に従い、12d(65mg,
0.110mmol)及びTBAF(0.18mL, 0.18mmol)は、白色固形物として13d(29mg,
61%)を生じた:Rf=0.15(15:1 CH2Cl2/MeOH);mp=184.4℃(dec);1H
NMRδ7.78 (m, 2H), 7.53-7.50 (m, 1H), 7.42-7.41 (m, 1H), 6.72 (d, J= 8.1 Hz,
1H)), 6.55 (d, J= 8.1 Hz, 1H), 6.18 (d, J= 6.2 Hz, 1H), 5.89 (ddd, J= 3.0, 5.3,
9.5 Hz, 1H), 5.69 (dd, J= 1.5, 9.5 Hz, 1H), 4.86 (s, 1H), 4.64 (t, J= 6.3 Hz,
1H), 3.54 (s, 1H), 3.31 (s, 1H), 3.06 (d, J= 18.7 Hz, 1H), 2.77 (m, 1H), 2.57
(s, 3H), 2.50-2.48 (m, 3H), 2.23-2.19 (m, 1H), 1.86 (dd, J= 2.2, 12.9 Hz, 1H);13C
NMR δ167.4, 144.5, 138.8, 133.9, 132.9, 131.7, 129.8, 128.6, 128.6, 127.0,
125.7, 119.4, 117.1, 92.8, 59.1, 50.6, 47.0, 44.0, 42.9, 39.9, 35.5, 20.5;MS
m/z = 389 [M+H]+, 411(M+Na)+;HRMS
m/z = 389.1867[M+H]+;13dの平均純度は、tR
=4.43分(移動相A)及びtR =5.16分(移動相B)を生じたことから、分析的HPLCにより、99%であることがわかった。
【0087】
(実施例22):6β-(3'-カルボメトキシ)ベンズアミドモルヒネ 13e
12aについて説明された一般的手法に従い、7c(159mg, 0.362mmol)、NEt3(151μL,
1.09mmol)及びモノメチルフタロイルクロリド(217mg, 1.01mmol)は、白色泡状物として12e(122mg,
56%)を生じた;Rf=0.22 (20:1 CHCl3/MeOH);MS
m/z:603 (M+H)+。13aについて説明された一般的手法に従い、12e(102mg,
0.170mmol)及びTBAF(0.47mL, 0.470mmol)は、白色固形物として13eを生じた:Rf=0.14
(30:1 dcm/MeOH);mp=166.0℃(dec);1H
NMR δ8.32 (s, 1H), 8.14 (d, J= 7.8 Hz, 1H), 8.00 (d, J= 7.8 Hz, 1H), 7.49 (t,
J=7.8 Hz, 1H), 6.68 (d, J=8.1 Hz, 1H), 6.52 (d, J=8.1 Hz, 1H), 6.33(d, J=7.0
Hz, 1H), 5.85 (ddd, J=9.4, 5.6, 3.1 Hz, 1H), 5.73 (d, J=9.9, 1H), 4.85 (s, 1H),
4.62 (t, J=6.2 Hz, 1H), 3.93 (s, 3H), 3.42 (dd, J=3.2, 5.2, 1H), 3.10 (bs, 1H),
3.05 (d, J=18.5 Hz, 1H), 2.63 (dd, J=12.1, 4.2 Hz, 1H), 2.46 (s, 3H), 2.40-2.34
(m, 2H), 2.08 (dt, J= 12.5, 4.9 Hz, 1H), 1.81 (d, J=10.8 Hz, 1H);13C
δ166.4, 166.2, 144.3, 138.7, 134.2, 133.0, 132.7, 131.9, 130.5, 129.8, 128.9,
128.3, 127.6, 125.7, 119.5, 117.0, 92.9, 59.1, 52.4, 50.9, 47.1, 44.0, 42.9,
39.9, 35.4, 20.2;MS m/z=447 [M+H]+;HRMS
m/z=447.1930 [M+H]+;13eの平均純度は、tR
=4.39分(移動相A)及びtR =5.06分(移動相B)を生じたことから、分析的HPLCにより、≧99%であることがわかった。
【0088】
(実施例23):6β-(3'-ニトロ)ベンズアミドモルヒネ 13f
12aについて説明された一般的手法に従い、7c(80mg, 0.182mmol)、NEt3(76μL,
0.55mmol)及び3-ニトロベンゾイルクロリド(95mg, 0.51mmol)は、白色泡状物として12f(87mg,
81%)を生じた;Rf=0.17(20:1 CHCl3/MeOH);MS
m/z:590(M+H)+。13aについて説明された一般的手法に従い、12f(73mg,
0.168mmol)及びTBAF(0.200mL, 0.200mmol)は、明黄色固形物として13f(36mg,
49%)を生じた:Rf=0.11(20:1 CHCl3/MeOH);mp=208.3℃(dec);1H NMRδ8.54
(t, J= 1.4 Hz, 1H), 8.26 (dd, J= 1.4, 8.2 Hz, 1H), 8.11 (d, J= 8.0 Hz, 1H),
7.55 (t, J= 8.0 Hz, 1H), 6.85 (d, J= 6.4 Hz, 1H), 6.65 (d, J = 8.1 Hz, 1H),
6.51 (d, J= 8.1 Hz, 1H), 5.84 (ddd, J= 3.0, 5.5, 9.2 Hz, 1H), 5.71 (d, J= 10.3
Hz, 1H), 4.88(s, 1H), 4.63 (t, J= 6.3 Hz, 1H), 3.45 (t, J= 3.2 Hz, 1H), 3.18
(s, 1H), 3.04 (d, J= 18.6 Hz, 1H), 2.68 (dd, J= 3.8, 11.8 Hz, 1H), 2.47 (s,
3H), 2.41-2.34 (m, 2H), 2.15 (dt, J = 7.6, 12.4 Hz, 1H), 1.78 (d, J= 11.5 Hz,
1H);13Cδ165.2, 148.0, 144.4, 138.9, 135.5, 133.3, 132.7, 129.8, 129.7,
128.2, 126.1, 125.4, 122.0, 119.5, 117.3, 92.5, 59.2, 51.1, 47.1, 43.9, 42.8,
39.6, 35.1, 20.3;MS m/z=434 [M+H]+;HRMS
m/z=434.1719 [M+H]+;13fの平均純度は、tR
=4.11分(移動相A)及びtR =4.78分(移動相B)を生じたことから、分析的HPLCにより、>99%であることがわかった。
【0089】
(実施例24):6β-(3',4'-ジクロロ)ベンズアミドモルヒネ,
13g
12aについて説明された一般的手法に従い、7c(81mg, 0.184mmol)、NEt3(77μL,
0.55mmol)及び3,4-ジクロロベンゾイルクロリド(108mg, 0.518mmol)は、白色泡状物として12g(96mg,
85%)を生じた;Rf=0.20(20:1 CH2Cl2/MeOH);MS
m/z:614(M+H)+, 616(M+2+H)+;618(M+4+H)。13aについて説明された一般的手法に従い、12g(81mg,
0.177mmol)及びTBAF(0.22mL, 0.22mmol)は、白色固形物として13g(48mg,
49%)を生じた:Rf=0.17(15:1 CH2Cl2/MeOH);mp=228.5℃(dec);1H
NMRδ7.82 (d, J= 2.1 Hz, 1H), 7.54 (dd, J= 2.1, 8.4 Hz, 1H), 7.39 (d, J= 8.4 Hz,
1H), 6.67 (d, J= 8.0 Hz, 1H), 6.56 (d, J= 6.2 Hz, 1H), 6.52 (d, J= 8.0 Hz, 1H),
5.82 (ddd, J= 2.9, 5.3, 9.5 Hz, 1H), 5.68 (dd, J= 1.2, 9.5 Hz, 1H), 4.83 (s,
1H), 4.58 (t, J= 6.3 Hz, 1H), 3.47 (dd, J= 3.0, 5.3 Hz, 1H), 3.19 (m, 1H), 3.05
(d, J= 18.6 Hz, 1H), 2.70 (dd, J= 3.8, 11.9 Hz, 1H), 2.50 (s, 3H), 2.43-2.37 (m,
2H), 1.77 (d, J= 11.0 Hz, 1H);13C
NMR δ165.4, 144.4, 138.9, 136.1, 133.7, 133.0, 132.5, 130.5, 129. 6, 129.3,
128.4, 126.3, 125.2, 119.5, 117.4, 92.5, 59.2, 52.4, 50.8, 47.2, 43.8, 42.7,
39.5, 35.1, 29.7, 25.8, 20.3, 20.5, 13.6;MS m/z 457 (M+H)+;HRMS
m/z=457.1087 [M+ H+];13gの平均純度は、tR
=3.93分(移動相A)及びtR =4.43分(移動相B)を生じたことから、分析的HPLCにより、>99%であることがわかった。
【0090】
(実施例25):6β-(チオフェン-2'-イル)アセトアミドモルヒネ 13h
12aについて説明された一般的手法に従い、7c(86mg, 0.196mmol)、2-チオフェンアセチルクロリド(0.068mL,
0.552mmol)及びNEt3(0.082mL, 0.588mmol)は、白色泡状物として12h(66mg,
60%)を生じ;Rf=0.23(20:1 CHCl3/MeOH);MS
m/z=565[M+H]+。13aについて説明された一般的手法に従い、12h(56mg,
0.099mmol)及びTBAF(0.120mL, 0.120mmol)は、白色固形物として13h(32mg,
79%)を得た:Rf=0.10(20:1 CHCl3/MeOH);mp=54.4℃;1H NMRδ7.18
(d, J= 4.5 Hz, 1H), 6.94-6.91 (m, 2H), 6.64 (d, J= 8.1 Hz, 1H), 5.82 (d, J= 7.1
Hz, 1H), 5.69 (ddd, J=3.5, 5.5, 9.4 Hz, 1H), 5.59 (dd, J= 1.2, 9.9 Hz. 1H),
4.65 (s, 1H), 4.39 (t, J= 6.4 Hz, 1H), 3.77 (s, 2H), 3.37 (dd, J= 3.3, 5.8 Hz,
1H), 3.01 (d, J= 18.5 Hz, 1H), 2.93 (m, 1H), 2.64 (dd, J= 4.1, 12.1 Hz, 1H),
2.45 (s, 3H), 2.39-2.31 (m, 2H), 2.02 (dt, J= 7.6, 12.6 Hz, 1H), 1.77 (dd, J=
10.8, 1.7 Hz, 1H);13C NMRδ170.2, 144.6, 139.0, 136.0, 132.9, 129.9, 128.4, 127.6,
125.9, 125.6, 119.6, 117.3, 92.9, 59.2, 50.4, 47.3, 44.1, 43.0, 39.7, 37.7,
35.5, 20.5;MS m/z=409[M+H]+, 431(M+Na)+;HRMS
m/z=409.1572[M+H]+;13hの平均純度は、tR
=4.31分(移動相A)及びtR =5.02分(移動相B)を生じたことから、分析的HPLCにより、>99%であることがわかった。
【0091】
(実施例26):6β-(3'-カルボキシ)ベンズアミドモルヒネ 14
参照V-110。13e(31mg, 0.067mmol)を、1:1
THF/水4mLに溶解し、LiOH-H2O(27mg, 0.643nmol)を添加した。無色の溶液を、室温で3.75時間攪拌し、氷酢酸を添加し(15滴)、及びこの溶液を濃縮した。残渣を、SiO2上(5:1から1:1
CHCl3/MeOH)でのフラッシュクロマトグラフィーにより精製した。適当な画分を濃縮し、残渣を10mLの10:1
CHCl3/MeOHと共に攪拌し、紙を通して濾過した。濾液を濃縮し、表題化合物を白色固形物として20mg(69%)提供した:Rf=0.17(1:1
CHCl3/0.2%AcOHを含むMeOH)、mp=217.1℃(dec);1H
NMRδ8.41-8.40 (m, 1H), 8.13-8.12 (m, 1H), 7.90-7.88 (m, 1H), 7.47 (t, J= 7.7
Hz, 1H), 6.62 (d, J= 8.2 Hz, 1H), 6.55 (d, J= 8.2 Hz, 1H), 5.81-5.78 (m, 1H),
5.69 (d, J= 9.9 Hz, 1H), 4.82 (s, 1H), 4.51-4.50 (m, 1H), 3.65 (dd, J= 3.2, 5.4
Hz, 1H), 3.27 (bs, 1H), 3.12 (d, J= 18.9 Hz, 1H), 2.87 (dd, J= 4.0, 12.9 Hz,
1H), 2.64-2.57 (m, 2H), 2.20 (dt, J= 4.8, 12.9 Hz, 1H), 1.81 (dd, J= 2.4, 12.9
Hz, 1H);13C NMRδ180.5, 170.2, 145.6, 141.2, 139.4, 135.5, 133.6, 131.0,
130.9, 130.5, 130.3, 129.5, 129.2, 125.6, 120.7, 118.5, 93.8, 61.3, 52.8, 42.6,
39.9, 35.1, 30.9, 22.1:MS m/z=431
(M-H)-;HRMS m/z C25H25N2O5 の計算値433.1758、実測値433.1749;14の平均純度は、tR
=3.93分(移動相A)及びtR =4.43分(移動相B)を生じたことから、分析的HPLCにより、99%であることがわかった。
【0092】
(実施例27):インビトロ結合アッセイ
A. 方法。細胞膜への結合は、96-ウェルフォーマットで行った。[N. Zaveriらの論文、Eur.
J. Pharmacol., 428:29-36 (2001)を参照されたい]。細胞を、プレートから、ラバーポリスマン(rubber
policeman)でこそぎ落とし、Tris緩衝液中に、Polytronホモジナイザーを使用して均質化し、その後一度遠心し、27,000×gで15分間の追加の遠心により洗浄した。ペレットを、50mM
Tris(pH7.5)中に懸濁し、懸濁液を、μオピオイド受容体、δオピオイド受容体、及びκオピオイド受容体それぞれに結合させるため、[3H]DAMGO、[3H]DPDPE、又は[3H]U69593と共にインキュベーションした。インキュベーションの総容積は、1.0mLであり、試料を、25℃で60〜120分間インキュベーションした。結合反応中のタンパク質の量は、約15μg〜30μgで変動した。ガラスファイバーフィルターを有するTomtec
96ハーベスター(Orange, CT)を用いた濾過により、反応を終結させた。結合した放射能を、Pharmacia
Biotechβ-プレート液体シンチレーションカウンター(ピスカタウェイ,
NJ)で計測し、カウント/分で表した。IC50値は、被験化合物の少なくとも6種の濃度を用いて決定し、Graphpad/Prism
(ISI, サンディエゴ, CA)を用いて計算した。Ki値は、Cheng及びPrusoffの方法を用いて決定した[Y.
Cheng及びW. H. Prusoffの論文、Biochem. Pharmacol.,
22:3099-3108 (1973)を参照されたい]。
【0093】
B.結果。リガンドの親和性及び選択性。表1は、化合物5及び6のKi値を含む。Kiは、被験化合物による受容体への放射性リガンドの結合の阻害を測定するパラメーターである。M6Gのように、試験した各リガンドは、μ選択性であった。I型化合物に関して、最も活性のある化合物に関する親和性の順位は、6b=5a>5d>6aであった。これらの各化合物は、遊離のフェノール性ヒドロキシル基を有した。M6Gと比べて、それらの親和性は、μ受容体で1.5〜2.4倍、δ受容体で2.9〜10.3倍、及びκ受容体で1.4〜369倍増加した。C-5'位にカルボメトキシ基を有する化合物5a及び5bは各々、κ受容体に関して、δ受容体よりも1.7〜2.7倍の選択性を示した。他のリガンドは全て、κ受容体よりもδ受容体に選択性を示した。化合物5aのC-5'位のカルボメトキシ基の、化合物5dのアセトキシメチル基との交換は、κ受容体の親和性を1/270に減少させた。3-アセトキシ基又は3-メチルエーテル基のいずれかを有するμ受容体、δ受容体、及びκ受容体についての残りの化合物の親和性は、M6Gと比較した場合に、有意に減少した。各場合において、糖部分のエステル官能基の加水分解は、リガンドのκ受容体への親和性を大きく減少させる一方で、μ受容体及びδ受容体に対するリガンドの親和性については、明らかに著しく低い効果を有していた。
【0094】
II型化合物のμ結合親和性に関して、遊離のフェノール系ヒドロキシル基及び完全にエステル化されたグルクロン酸部分を有するモルヒネアナログ10(2.80nM)は、最高の親和性を有した。この知見は、I型アナログ3aについて観察されたそれらの結果と一致した。興味深いことに、コデイン誘導体9b(7.43nM)は、I型コデイン化合物よりも有意に高い親和性を示した。化合物11a(14.6nM)及び11b(41.5nM)は、低下した親和性を示した。M6Gとの比較において、化合物10は、μ受容体について4.5倍より高い親和性を示し、化合物9bは、1.7倍高い親和性を示した。II型化合物は、I型化合物よりも有意により選択性であった。μ受容体に関する化合物11bの選択性は、M6Gのものに近いが、これは結合親和性の1/3.2の低下を有した。これらの結果は、モルヒネ又はコデイン核と糖残基の間の炭素橋は、μ受容体選択性に関して重要であることを示唆している。この橋によりもたらされた追加の立体配置的移動度は、これらの分子が、μ受容体と、C-6α糖残基を有するM6Gにより類似して相互作用することは考えられる。
【0095】
化合物14(19.92nM)を除いて、μ受容体でのIII型化合物のKi値は、0.21〜0.59nMであった。これらの化合物は、M6Gよりも、20倍〜64倍より高いμ受容体親和性を有する。III型化合物は、最高の親和性のI型化合物よりも少なくとも9倍高い親和性を有し、最高親和性のII型化合物よりも少なくとも4.7倍高い親和性を有した。これらの結果は、これらのリガンドのC-6置換基は、μ結合ポケットの疎水性領域と相互作用することを示唆している。この知見は更に、化合物14(19.92nM)で観察された親和性の減少により裏付けられる。ほとんどの場合において、II型化合物は、増加したμ対δ選択性及び低下したμ対κ選択性を示した。架橋したNのN-メチル置換基のシクロプロピルメチル又はシクロブチルメチル又は類似の置換基との交換は、典型的には以下に説明された活性に関するこれらの受容体のアンタゴニストを提供する。
【0096】
【表1】
【0097】
(実施例28):Gタンパク質への共役を測定するための[35S]GTPγ-S結合試験
A. 方法。放射標識されたS結合は、基本的に、[J.R. Traynor及びS. R. Nahorskiの論文、Mol.
Pharmacol., 47:848-854(1995)]に説明されたように行った。細胞を組織培養皿から20mM
Hepes、1mM EDTAへはぎ取り、その後500 ×gで10分間遠心した。細胞を、この緩衝液中に再懸濁し、Polytronホモジナイザーを用いてホモジナイズした。ホモジネートを、27,000
×gで15分間遠心し、ペレットを20mM Hepes、10mM MgCl2、100mM
NaCl(pH7.4)を含有する緩衝液A中に再懸濁した。懸濁液を、27,000 ×gで再遠心し、もう一度緩衝液A中に懸濁した。結合アッセイのために、膜(8〜15μgタンパク質)を、[35S]GTPγ-S(50pM)、GDP(10μM)、及び適当な化合物と共に、総容量1.0mLで、25℃で60分間インキュベーションした。試料を、ガラスファイバーフィルター上で濾過し、結合アッセイについて説明されたようにカウントした。統計解析は、プログラムPrismを用いて行った。
【0098】
B. 結果。リガンドの効力及び効能。化合物が会合したGタンパク質のオピオイド受容体介在性の活性化を評価するために、これらの化合物を、[35S]GTPγSアッセイを用いて評価した。これらのデータを、表2にまとめた。このアッセイにおいて、化合物の効力又は受容体親和性は、[35S]GTPγS結合の刺激に関するそのEC50により定義した。アゴニスト効能は、化合物が[35S]GTPγS結合を対照に対し最大刺激する程度として定義される。EC50値は、化合物による[35S]GTPγS結合の50%最大刺激を生じる化合物の濃度を表している。完全アゴニストは、[35S]GTPγS結合を最大程度刺激し、部分アゴニストは、結合のレベルの低下を生じさせる。表2は、ヒトμオピオイド受容体、ヒトδオピオイド受容体、及びヒトκオピオイド受容体での[35S]GTPγS結合の刺激に関する、Emax及びEC50値を提供する。化合物6cを除き全ての化合物は、μ受容体及びδ受容体で、[35S]GTPγS結合を刺激した。これらの化合物は、κ受容体で有効性がはるかに少なかった。この傾向は、M6Gについても観察された。μ受容体で最も有効な化合物は、化合物3b(88.0%)、3d(76.0%)、4b(64.5%)、及び4a(46.6%)であった(表2参照)。これらの各化合物は、遊離のフェノール系ヒドロキシル基を有した。これらの化合物は全て、M6Gよりもより有効であった。同じ一般的傾向が、遊離のフェノール系ヒドロキシルを有する化合物により、δ受容体においても認められ、これは増大した効能を示している。最も有効な化合物は、3b(104.4%)、4a(78.70%),、3e(64.3%)、及び4b(51.5%)であった。アセテート基又はメチル基を伴うフェノール系ヒドロキシル基の保護は、各受容体において効能及び効力を減弱させた。最小刺激を作用する化合物は、所定の受容体でアゴニストと見なすことができる。最大結合よりも少なく誘起したリガンドは、刺激が認められない場合は、部分アゴニスト又はアンタゴニストと見なすことができる。EC50値の全体の順位は、Ki値と相関していた。II型化合物に関して、リガンド10及び9bは、μ受容体において[35S]GTPγS結合の最大刺激を生じ、δ受容体及びκ受容体において、[35S]GTPγS結合の刺激を低下した。III型化合物13a-h及び14に関して、これらの各化合物は、μ受容体、δ受容体及びκ受容体で[35S]GTPγS結合を刺激した。これらの化合物は一般に、δ受容体に対し、μ及びκ受容体でより有効であった。最大刺激を作用する化合物は、所定の受容体でアゴニストとみなされる。最大結合よりも少なく誘起するこれらのリガンドは、刺激が認められない場合には、部分アゴニスト又はアンタゴニストと見なされる。EC50値の全体の順位は、Ki値と相関した。
【0099】
【表2】
【0100】
(実施例29):μ受容体内在化
A. 方法。μ受容体内在化は、[D. E. Keithらの論文、Mol. Pharm.,
53:377-384 (1998)]の方法に従い評価した。一般的用語において、この現象は、表面受容体を、FITC-標識されたFLAG
M1の染色及び蛍光の測定により定量することができるフローサイトメトリー分析を用いて、試験した。内在化を引き起こす効果がある薬物は、対照と比べ低下した染色を生じた。
【0101】
B. 結果。この過程は耐性及び依存の発生において役割を担い得るという理由から、III型化合物を、μ受容体内在化に作用するそれらの能力について評価した(表3参照)。このデータから、ジクロロ化合物13g(47%)及びニトロ化合物13f(59%)は両方とも、μ受容体内在化の誘導において、エトロフィン(61%)よりも有効であることが認められた。他のアミドは、μ受容体の弱い内在化を中程度にする。モルヒネ(93%)は、通常の血漿濃度(1〜50nM)では、μ受容体内在化を引き起こさないことは指摘されなければならない。これらの実験において使用される化合物の濃度は、1μMであった。
【0102】
【表3】
【0103】
(実施例30):化学安定性及び代謝安定性評価
A. 方法。選択された化合物の化学安定性を、pH7.4及びpH2.0で試験した。10μMリン酸カリウム緩衝液の存在下、pH7.4又はpH2.0のいずれかの中の化合物(0.1M)を、5分、1時間、2時間、3時間、24時間、48時間又は72時間後に濃縮した。その後HPLCを用い、出発物質の分解を分析した。選択された化合物の代謝安定性を、ヒト肝臓S9及びミクロソームで試験した。ヒト肝臓プールしたS9(HLS9)の安定性及びマウス肝ミクロソーム(MLM)。アッセイ緩衝液は、最終インキュベーション容積0.25mLについて、3.2mg/mL
HLS9又は1.2mg/mL MLM、0.1Mリン酸カリウム緩衝液(pH7.4)、0.1mM化合物、0.5mM
NADP+、0.5mMグルコース-6-リン酸、5U/mLグルコース-6-リン酸デヒドロゲナーゼ、1mg/mL
DETAPAC、7mM MgCl2を含む。0分、10分、25分、40分又は60分後、1mL
CH2Cl2/IPA(3:1 v:v)の添加により、反応を停止した。遠心後、抽出溶媒(有機相)を、窒素流で蒸発除去し、200μLメタノールに溶解し、激しく攪拌し、遠心し、HPLCバイアル挿入管に移した。HPLCを使用し、所定の時間間隔(0分、10分、25分、40分又は60分)での曲線下面積を、ミクロソーム添加前の曲線下面積と比較することにより、出発物質の分解を分析した。
【0104】
B. 結果。選択型I化合物、及び選択型III化合物は、マウス肝ミクロソーム(MLM)、ヒト肝臓ミクロソームS-9(HLS9)の存在下、pH2及び7.4で評価した(表4参照)。より長い半減期は、所定の条件下で、より大きい安定性と相関している。I型化合物において、M6Gイオウアナログ4aは、HLS9の存在下で、かつpH2及びpH9において、M6Gと同等の安定性を示した。エステル保護基の存在は、I型分子の安定性を低下した。HLS9に対する化合物3aの安定性は、化合物3bよりも大きく(17.86分)、これは3-位のアセテート基は、I型分子において最も不安定なエステル基であることを示唆している。この仮説は、HPLC同時注入により証明された。脱保護された化合物4a-dの安定性において、差異は検出されなかった。III型化合物は、試験した全ての条件下で、I型化合物よりも有意により安定していた。化合物13eにおけるメチルエステル基の存在は、HLS9及びpH7.4に対するこの化合物の安定性を、他のIII型化合物と比べて低下した。評価するための十分量のII型化合物は、得られなかった。II型化合物におけるエステル保護基の存在は、それらをIII型化合物よりもより不安定にした。アミド連結は、S-グリコシド連結に勝る改善された安定性をもたらすと予想される。
【0105】
【表4】
【0106】
(実施例31):モルヒネアナログ3a及び13eの鎮痛薬活性の決定
A. 方法。
1.動物。実験開始時に体重20〜25gの雄のICRマウスを使用した。動物は、標準の実験室条件下で、群で飼育し、12:12時間昼-夜サイクル(08:00点灯)を維持した。動物は、実験実施前、1〜2日間馴化した。
2. 薬物。モルヒネ13e及び3bを、水に溶解した。薬物を、0.1mlの容量で注射した。
【0107】
3.侵害受容試験。侵害受容を、投射熱を使用する痛覚喪失装置(Stoelting)による、掉尾反射アッセイを用いて試験した。この装置は、掉尾反射潜時の自動定量で、動物尾の損傷を防ぐために15-秒カットオフで装備した。試験期間中は、焦点化した光ビームを、動物尾の下側半分に適用し、掉尾反射潜時を記録した。掉尾反射潜時のベースライン値は、各動物の薬物投与前に決定した。基本の掉尾反射潜時は、3.2〜8.0秒(平均5.86±0.16SEM)であった。試験直後に、動物に、被験化合物又はビヒクル対照として生理食塩水を皮下注射した。動物を、注射後30分及び60分に、掉尾反射潜時について試験した。
【0108】
4. データ解析。抗侵害受容を、下記式で定量した:
%抗侵害受容=100*[(試験潜時-ベースライン潜時)/(15-ベースライン潜時)]
動物が15秒カットオフ以前に反応しなかった場合は、動物は、スコア100%に割り当てた。行動の結果は、群間変動としてモルヒネ、13e、及び3bで、並びに反復測定として薬物処置後時間(30分、60分)により、ANOVAを用い、適当ならばStudent
Newman-Keulsポストホック検定により、引き続き解析した。有意水準は、p<0.05とした。
B. 結果
【0109】
1.モルヒネの掉尾反射潜時に対する作用。図5に示したように、モルヒネは、掉尾反射潜時の用量-依存型の増加を生じた。全般的ANOVAは、投与量の有意な作用を示した。[F(3,45)=25.52,
P<0.0001]。従って注射後時間とは無関係に、モルヒネは、掉尾反射潜時に対して同じ作用を生じた。注射後時間横断的に平均化して、モルヒネの3〜30mg/kg投与量は、対照に対し、掉尾反射潜時の有意な増加を生じた(Student
Newman-Keuls, p<0.05)。%MPEに関する用量漸増反応曲線を認め、ここでモルヒネ投与量3mg/kgは、10及び30mg/kg投与量に比べ、より低い鎮痛作用を生じた(Student
Newman-Keuls, p<0.05)。
【0110】
2. 13eの掉尾反射潜時に対する作用。13eの投与は、30分間で明らかであり及び60分間で継続した掉尾反射潜時の増加を生じた(図6参照)。全般的ANOVAは、投与量の有意な作用を示した。[F(5,72)=14.86,
P<0.0001]。注射後時間横断的に平均化して、13eの2つの最高投与量は、対照に対し、掉尾反射潜時の有意な増加を生じた(Student
Newman-Keuls, p<0.05)。13eの投与量30mg/kgの鎮痛作用は、10mg/kg投与量により生じた作用よりも2倍大きかった(Student
Newman-Keuls, P<0.05)。化合物13eは、モルヒネよりもより遅い作用開始を有し、30分よりも60分で有意に増加した効力を有するように見える。その非常に高い親和性(μ受容体でKi=0.23nM)及び[35S]GTPγ結合の刺激に関する強力な完全アゴニスト活性(μでEC50=2.4nM)を考慮し、この化合物は、比較的弱い抗侵害受容活性を有し、ED50値は30分及び60分間で、各々、30.2mg/kg及び12.6mg/kgであった。これは、恐らくED50が約3mg/kgであるモルヒネに比べ短期間の効力が低いであろう。
【0111】
3. 3aの掉尾反射潜時の作用。3aの投与は、化合物の強力な鎮痛薬作用の掉尾反射潜時指標における用量-依存型の増加を生じた(図7参照)。全般的ANOVAは、投与量の有意な作用を示した[F(5,64)=37.26,
P<0.0001]。注射後時間横断的に平均化して、3aの投与量3〜30mg/kgは、対照に対する掉尾反射潜時の有意な増加を生じた(Student
Newman-Keuls, p<0.05)。3aの2つの最高投与量(10及び30mg/kg)は、3mg/kg投与量に対し、より大きい量の痛覚喪失を生じた(Student
Newman-Keuls, p<0.05)。化合物3aは、13eよりも鎮痛薬として有意により強力であり、30及び60分の両方で、約2.5mg/kgのED50を有した。この値は、3aは、モルヒネ(ED50、3mg/kg)よりもわずかにより強力であることも示唆している。3aは、モルヒネよりもわずかに低いインビトロ結合親和性を有するが、ほぼ同じ[35S]GTPγS効力及び%刺激を有し(表1及び2参照)、これは3aの生体利用効率が、モルヒネのそれとほぼ同じであるか又はより大きいことを示唆している。この化合物はオピオイド受容体の各々に関して若干非選択的であるので、この時点で、どの受容体が抗侵害受容活性を媒介するかは定かではない。3aの作用開始は、モルヒネよりもより早いことは明らかである(この活性は、30分時点よりも、60分でより大きい)。
【0112】
(実施例32):ヘロイン依存性及び離脱症状
A. 方法。M6Gアナログのヘロイン依存性動物に対する作用。ヘロイン自己-投与を、依存の最適測定を生じるヘロイン投与量を用いて行った。これまでの研究を基に、0.06mg/kg/0.1ml注入を算出した。安定した反応が確立した後(23時間/日アクセスで約40日間)、陽性対照としての低投与量ナロキソン(0.05〜0.2mg/kg)、及びM6Gアナログ(0、0.01、0.04、0.1mg/kg、皮下注射)及び他のアナログの、ヘロイン摂取の様々な行動測定に対する作用を試験した。4種の投与量を、各化合物について試験した。各M6Gアナログの対象内用量-作用試験を行うことを試みたが、試験は、必要ならば、対象間用量-作用機能も用いた。ナロキソン及びM6Gアナログの12時間にわたる作用を調べるために、Koob研究室が開発したミニポンプを連結したオン/オフゲーティング装置を使用した。一旦安定したヘロイン摂取が達成された後、ラットに、午前6時に浸透圧ミニポンプ(ゲート-オフ位置)を移植することができる。12時間後、ゲーティング装置を、オン位置に回し、ラットを通常23時間セッション時に、ヘロイン自己-投与にアクセスさせた。活動相(午後6時オン午後6時オフ)である暗期間の12時間の期間について、これらのセッション時に、ミニポンプは、ゲート-オンの位置とした。2日後、ミニポンプを除去し、ラテン方陣(Latin
Square)デザインで異なる投与量の被験化合物(すなわち、ナロキソン又はM6Gアナログ)含む別のミニポンプと置き換えた。このサイクルを、3投与量及びビヒクル対照を試験するように繰り返した。必要な多数の薬物及び投与量は、ナロキソン及びM6Gアナログの投与量の対象間デザインで強制することができる。試験される特定の行動は、非依存型パターンの自己投与から、依存型パターンの自己投与への移行を反映することができる、自己投与を介した長期間ヘロイン曝露の試験結果から決定した。個別の獲得軌跡が多項式関数にマッチされると同時に、対象内及び対象間変数を説明する、階層線形モデル(Hiearchical
Linear Modeling)法を使用し、洗練された統計モデル化を行った。
【0113】
B. 結果。ヘロイン自己投与のパラダイムは、現在ラットにおいてよく確立されている[Walkerらの論文、Fur.
J. Pharmacol., 383:115 (1999)を参照されたい]。2,3の試験は、限度のないヘロインアクセスに関連した行動の変化及び依存点までのヘロインの自己-投与の出現を調べた。予備的結果は、ラットは、食物枯渇又は制限のない条件下で、限度のないアクセスで、ヘロインi.v.を容易に自己-投与することを確立した。自己-投与の限度のないアクセスのパターンは、経時的に変更し、依存性の測定を示す動物を生じる。ヒトにおけるヘロイン依存性の動物依存モデルが開発された。延長されたアクセス(投与量0.06mg/kg/0.1ml注入でのヘロイン自己-投与への23時間のアクセス)が与えられた動物は、50〜60日間にわたるヘロインの持続摂取を示した。この期間、自己-注入パターンは、「活動/暗所」相においてのみ認められたものから、自己-投与がほぼ15〜20分毎に生じたパターンへと変化した。2〜3週目から7週目まで、本質的に均等な自己-投与が認められた。各自己-投与回数/日を、約50日間記録した。これらの試験は、ヘロイン投与量、摂取パターン、及び最大薬物摂取の時刻に関する完全な情報を提供した。ヘロイン依存性は、慢性ヘロイン投与からの離脱症状発現として測定することができる[Woolverton及びSchusterの論文、(1983)を参照されたい]。依存性は、依存型ラットにおけるヘロイン除去後の又はヘロインアンタゴニスト投与後の、顕在的禁断徴候として表すことができる。両方とも、身体的徴候及び動機づけ作用を生じる[Weeks及びCollinsの論文、(1964)を参照されたい]。M6Gアナログのヘロイン自己-投与を低下する能力を、自己-投与時及びM6Gアナログの投与後の延長された-アクセスモデルにおいて試験した。
【0114】
M6Gアナログ3a、13e、13g及び13hのヘロイン自己-投与に関する作用を、ヘロインに依存したヒトにおける作用と一致する依存型ラットにおいて試験した。これらの実験の目標は、ヘロイン自己-投与の減少に有効であるM6Gアナログを同定することである。図8-12の試験は、M6Gアナログ13e(0.04mg/kg)、13g(0.2mg/kg)、M6G(0.04mg/kg)、13h(0.01、0.04、0.2mg/kg)は、ヘロイン自己-投与の低下に有効であることを明らかにした。最も有効な化合物は、投与量0.01mg/kgで、ヘロイン摂取を1000μg/kg低下させる13hであった。
【0115】
(実施例33):MG6のC-グリコシドアナログの合成及び生物学的評価
モルヒネ-6-グルクロニド(M6G)は、ラットへ脳室内注射した場合に、鎮痛効力がモルヒネそれ自身の約100倍であるモルヒネの第II相代謝複合体である[Carruptらの論文、J.
Med. Chem., 34, 1272-1275 (1991)を参照されたい]。M6Gは現在、術後疼痛治療のための臨床試験の最終相にある[Gutmanらの米国特許第6,737,518号(2004)]。M6Gの経口生体利用効率は、わずかに11%であり[Personらの論文、Br.
J. Clin. Pharmacol., 53:347-354 (2002)を参照されたい]、またM6Gの代謝安定性の改善は、可能性のある疼痛の薬物療法としてのその有効性を恐らく増大させるであろう。複合糖質のインビボ代謝安定性を改善するための一般的方法は、グリコシド酸素原子の炭素、窒素又はイオウ原子との交換に関連している[Postemgらの論文、
Organic Lett., 5:1721-1723 (2003);Kiefelらの論文、J.
Carbohydr. Chem., 18:937-959 (1999)を参照されたい]。M6Gのグルコシル及びグルクロノシルアナログのライブラリーのデザインにおける戦略は、グリコシド酸素原子がイオウ原子と交換される場合がこれまで利用された[MacDougallらの論文、J.
Med. Chem. 47:5809-5815 (2004)を参照されたい]。M6Gの6-βイオウアナログは、μオピオイド受容体親和性及び機能的効能のわずかな改善を示したが、μ受容体については、δオピオイド受容体及びκオピオイド受容体に対しより少ない選択性を示した。本願明細書において報告したのは、新規μ選択的にアミド結合された炭素グリコシドアナログの14工程の合成経路及び生物学的評価の結果を詳細に説明する、本研究の拡張である。
【0116】
C-β-グリコピラノシルアシルクロリド9は、市販の2,3,4,6-テトラベンジルグリコピラノース4から、5工程の手順により調製した。化合物4を、DMSO/Ac2Oにより酸化させ、対応するラクトン5(94%)を生じた[Kuzuhara,
H.;Fletcher, H.G.の論文、J. Org Chem.,
32:2531-2534 (1967)]。5の-78℃のリチウム酢酸エチルのTHF溶液への添加[Rathke,
M. W.の論文、J. Am. Chem. Soc., 92:3222-3223 (1970)]、それに続く酸性水による後処理は、立体選択的アルドール反応により形成されたヘミケタール6を形成した(91%)
[Lewisらの論文、J. Am. Chem. Soc., 104:4976-4978 (1982)]。アセトニトリル中BF3OEt2の存在下での、0℃での、6のトリエチルシランによる還元[Lewisらの論文、前掲;Tiedemannらの論文、J.
Org Chem., 64:4030-4041 (1999)を参照されたい]は、エチルエステル7(86%)を生じさせた。還流時の1:1
THF/H2O中のLiOHによる、7のエチルエステル基の加水分解及びそれに続く酸性水による後処理は、カルボン酸8(87%)を生じさせた。8の対応するアシルクロリド9への転換は、無希釈のチオニルクロリド中での18時間の攪拌により実現した。2.0当量Et3Nの存在下CH2C12中での、粗酸塩化物9の1.5当量の、3-トリイソプロピルシリル-6-β-アミノモルヒネへの添加[MacDougallらの論文、Bioorg
Med. Chem., 12:5983-5990 (2004)]は、保護されたモルヒネグルコースアナログ10(93%)を生じさせた。10中のトリイソプロピルシリル保護基は、THF中のTBAFの添加により除去され、フェノール11を生じた(90%)。引き続きの酸性メタノール中の活性化された10%Pd/Cによる、11の触媒的水素化は、M6Gのアミド-連結したC-β-グリコピラノシドアナログである化合物3を生じさせた(52%)。HClの非存在下でのPd/Cによる11の水素化分解は、フェナントレン炭素二重結合の還元を生じ、定量的収量の12をもたらしたが、グルコースベンジルエーテル基の水素化分解は生じなかった。これらの条件下で、11の塩基性モルヒナン窒素原子は、恐らくO-脱ベンジル化を阻害した[Czech,
B. P.;Bartsch, R. A.の論文、J. Org Chem.
49:4076-4078 (1984)]。7,8-二重結合の飽和及びグルクロン酸部分のグルコース部分との置換の両方は、鎮痛効力が増大したM6Gアナログを提供することがわかっている[Stachulskiらの論文、Bioorg
Med. Chem. Lett., 13: 1207-1214 (2003)]。従って、3の対応するデヒドロアナログ又はグルクロン酸アナログへの代替経路を開発するよりもむしろ、化合物3を直接評価した。全ての合成化合物に関するスペクトルデータは、割当てられた構造に完全に一致した[新規化合物10、11、12及び3は、1H
NMR、13C NMR及びMSにより特徴決定した。被験化合物3及び11の純度は、HPLCにより決定し、≧98%であることがわかった。]。
【0117】
化合物3及び11のμオピオイド受容体、δオピオイド受容体及びκオピオイド受容体との競合的結合アッセイで得られたIC50値を、実験の項に説明されたように、Ki値へ変換した。被験化合物及び参照物質のKi値は、表5に列記している。結合アッセイにおいて、以下の放射性リガンドを使用した:[3H]DAMGO(μオピオイド受容体アゴニスト);[3H]U69593(κオピオイド受容体アゴニスト);[3H]DPDPE(δオピオイド受容体アゴニスト)。Ki値は、被験化合物3及び11によるこれらの放射性リガンドの受容体への結合の阻害の測定から決定した[Zaveriらの論文、Eur.
J. Pharmacol., 428:29-36 (2001)]。ベンジル誘導体11及び脱保護された同族体3は両方とも、μ受容体選択性であった。M6Gと比べ、化合物11は、μオピオイド受容体に関してM6Gよりも27倍高い効力を有した。化合物11のμ受容体対δ受容体、及びμ受容体対κ受容体への選択性は、各々、10倍及び34倍であった。M6Gと比較して、化合物3は、μオピオイド受容体に関して3.7倍より高い効力を有した。化合物3のμ受容体対δ受容体、及びμ受容体対κ受容体への選択性は、各々、77及び166倍であった。化合物3のμ受容体対δ受容体選択性は、M6Gの値に対しかなり改善されるが、μ受容体対κ受容体はそうではないことは、注目に値する(すなわち、各々12.5倍及び316倍の選択性)。化合物3は、μ受容体に対し、チオ糖2a(2.5倍)及び2b(1.6倍)に比べわずかに大きい効力を有し、並びにδ/μ及びκ/μ受容体選択性比を著しく改善した。化合物3及び11の機能活性は、[35S]
GTPγSアッセイを用いて評価した[Traynor, J.R.及びNahorski S.R.の論文、Modulation,
Mol. Pharmacol. 47:848-854 (1995)]。[35S]GTPγSアッセイは、被験化合物の、μ、δ又はκオピオイド受容体のいずれかに会合したGタンパク質を活性化する能力を測定する。[35S]GTP-γ-S結合を刺激するEmax値を基に、化合物3は、μ受容体及びδ受容体で完全アゴニストであり、及びκ受容体で部分アゴニストであると決定した。化合物11は、δ受容体で完全アゴニストであり、及びμ及びκ受容体で部分アゴニストであると決定した。μ受容体での化合物3の効能(Emax)(75%)は、M6G(45%)、及びチオ糖アナログ2a(46.6%)及び2b(36.0%)よりも、各々、実質的に高い。
【0118】
結論として、C-グリコシド3を、2,3,4,6-テトラ-O-ベンジル-D-グルコース4及び硫酸モルヒネから、14-工程の転換合成により調製した。化合物3は、μオピオイド受容体に対し、M6Gと比べ、3.7倍大きい親和性を示した。化合物3のδ受容体対μ受容体、及びκ受容体対μ受容体に対する選択性の比は、各々、76.7及び166であった。化合物3のδ/μ選択性は、12.5であるM6Gに関する値に比べ、著しく改善された。これらの化合物のインビボ生物学的特性を完全に説明するための、更なる研究が現在進行中である。M6Gの代謝安定性の増加は、より長い寿命の強力なオピオイドアゴニストの新規クラスを提供することができる。
【0119】
【表5】
【0120】
【表6】
【0121】
全般的情報。全ての反応を、特に記さない限りは、炉乾燥したガラス製品を使用し、周囲温度で磁気攪拌しながら、陽圧下で実施した。空気-及び水分-感受性液体を、シリンジによりゴム製隔壁を通して移した。用語ブラインは、塩化ナトリウム飽和溶液を意味する。カラムクロマトグラフィーのために、シリカゲル(230〜400メッシュ)を使用した。DMFを、中間アルミナのカラムを通して乾燥させ、また使用前は、窒素下活性化した4Å分子篩上で貯蔵した。CH2Cl2及びTHFを、使用直前にCaH2から蒸留した。全ての他の溶媒及び試薬は、入手したまま使用した。1H
NMRスペクトルは、300MHz Varian NMRにより、18℃で記録した。化学シフトは、特に別に記載しない限りは、7.26ppmで
CDC13に対するppm(δ)で報告した。高解像度質量分析を、University of
California at Riverside において、Opus V3.1及びDEC
3000 Alpha Stationデータシステムを備えた、VG7070スペクトロメーター上で行った。低解像度質量分析(LRMS)を、ESIを使用するHitachi
M-8000 3DQMS (イオントラップ型)質量分析装置で行った。融点は、補正せずに報告した。分析的純度は、D7500積分器及びHamilton
PRP-Iステンレス鋼カラム(250mm ×4.6mm内径)を備えた、Hitachi
L74液体クロマトグラフィーを使用する、順相HPLCにより決定した。分析的純度を決定するために、移動相A=60/40/0.02
MeOH/2-プロパノール/HClO4 (v:v)又は移動相B=55/45/0.018
MeOH/2-プロパノール/HClO4 (v:v)を使用した。
【0122】
3-O-トリイソプロピルシリル-6-β-(2,3,4,6-テトラ-O-ベンジル-β-D-グルコピラノシル)アセトアミドモルヒネ10。SOC12(2.0mL)を、カルボン酸8(389mg,
0.67mmol)に添加し、この溶液を、室温で18時間攪拌した。過剰なSOCl2を、水浴(60℃)を使用し、高真空下で除去し、及び生じた黄色油状物を、CH2C12(5mL)中に溶解し、CH2Cl2
(5mL)中の3-O-トリイソプロピルシリル-6-β-アミノモルヒネ(196mg, 0.445mmol)及びEt3N(0.12mL,
0.89mmol)へ添加した。この溶液を、室温で1時間攪拌し、濃縮し、フラッシュクロマトグラフィーにより精製し(SiO2,
30:1〜10:1 EtOAc/MeOH)、10を白色固形物として得た(417mg,
93%):Rf=0.46(10:1 CH2Cl2/MeOH);mp=56.6℃;1H
NMR(CDC13)δ7.35-7.10 (20H), 6.59 (d, J=8.0 Hz, 1H), 6.40 (d, J= 8.0
Hz, 1H), 6.22 (d, J= 6.9 Hz, 1H), 5.71 (ddd, J= 3.0, 6.0, 9.5 Hz, 1H), 5.41
(dd, J= 1.9, 9.5 Hz, 1H), 4.89-4.48 (9H), 4.36 (t, J= 6.3 Hz, 1H), 3.72-3.46
(5H), 3.29 (t, J= 9.0 Hz, 1H), 3.17 (dd, J= 3.0, 5.2 Hz, 1H), 2.96 (d, J= 18.7
Hz, 1H), 2.88 (bs, 1H), 2.72 (dd, J= 2.7, 15.2 Hz, 1H), 2.50-1.72 (1OH),
1.30-1.05 (21H)。13C NMR (CDC13)δ170.0, 147.9, 143.6, 138.2, 137.8, 137.6,
132.9, 130.4, 128.7, 128.4, 128.34, 128.32, 128.0, 127.8, 127.76, 127.6, 127.0,
120.8, 118.4, 92.0, 86.8, 80.9, 78.7, 78.1, 77.4, 76.1, 75.7, 75.2, 75.0, 73.3,
68.7, 59.0, 49.3, 46.8, 44.1, 43.1, 40.3, 39.0, 36.4, 20.3, 18.0, 17.99, 12.8,
11.3;MS (EI) m/z 1006 [M+H]+。
【0123】
6-β-(2,2,4,6-テトラ-O-ベンジル-β-D-グルコピラノシル)アセトアミドモルヒネ11。シリルエーテル10(270mg,
0.27mmol)を、THF(4mL)に溶解した。水(0.05mL)及びTBAF(0.4mL,
0.4mmol, THF中1.0M溶液)を添加した。2.5時間後、濃度1%のHCl(4mL)を添加し、この混合物を2分間攪拌し、水(10mL)で希釈し、固形NaHCO3で塩基性とした。この混合物を、CH2C12
(5×30mL)で抽出し、乾燥し(Na2SO4)、濾過し、濃縮した。フラッシュクロマトグラフィー(24:1〜12:1
EtOAc/MeOH)は、11を白色固形物(205mg, 90%)として提供した:Rf=0.15(12:1
EtOAc/MeOH);mp=103.8℃;1H NMRδ7.34-7.10 (20H), 6.64 (d, J= 8.2 Hz, 1H), 6.46 (d, J= 8.2
Hz, 1H), 6.34 (d, J= 6.3 Hz, 1H), 5.68 (ddd, J= 2.5, 5.8, 9.5 Hz, 1H), 5.39 (d,
J= 9.5 Hz, 1H), 4.89-4.46 (10H), 4.34 (t, J= 6.3 Hz, 1H), 3.72-3.46 (6H), 3.30
(t, J= 9.1 Hz, 1H), 3.18 (bs, 1H), 2.97 (d, J= 18.1 Hz, 1H), 2.94 (bs, 1H),
2.74 (dd, J= 2.5, 15.7 Hz, 1H), 2.55-2.16 (7H), 1.95 (dt, J= 8.0, 12.6 Hz, 1H)
, 1.76 (d, J= 11.3 Hz, 1H);13Cδ170.4,
144.2, 143.6, 138.1, 138.0, 137.8, 137.7, 137.6, 132.6, 129.8, 128.38, 128.37,
128.33, 128.3, 128.2, 128.0, 127.8, 127.7, 127.7, 127.65, 127.6, 125.9, 119.1,
116.3, 93.0, 86.8, 80.8, 78.7, 78.1, 77.4, 76.0, 75.7, 75.3, 75.0, 73.3, 68.7,
59.0, 50.0, 47.0, 44.0, 43.0, 39.9, 38.8, 35.9, 20.2;MS
(EI) m/z 850 [M+H]+;HRMS (ESI) C53H57N2O8
についての計算値849.4115、実測値849.4099;11の平均純度は、tR=3.24分(移動相A)及びtR=4.66分(移動相B)を生じたことから、分析的HPLCにより、≧99%であることがわかった。
【0124】
6-β-(2,3,4,6-テトラ-O-ベンジル-β-D-グルコピラノシル)アセトアミド-7,8-ジヒドロモルヒネ12。テトラベンジルエーテル12(35.4mg,
0.042mmol)を、MeOHに溶解し、10%Pd-C(15mg)を添加した。混合物を、水素大気下で24時間攪拌し、その後セライトを通して濾過した。セライトを、MeOH(10mL)及びCH2C12(2mL)で洗浄し、濾液を濃縮し、12を白色固形物(35mg,
100%)として得た:Rf=0.66(200:80:5:0.05 CH2Cl2/MeOH/H2O/13% NH4OH);mp=94.3℃;1H
NMR (CDC13) δ 7.33-7.10 (20H), 6.92 (d, J= 8.4 Hz, 1H), 6.69 (d, J=
8.2 Hz, 1H), 6.56 (d, J= 8.2 Hz, 1H), 4.88-4.29 (9H), 3.73-3.32 (8H), 3.05 (bs,
1H), 2.96 (d, J= 18.4 Hz, 1H), 2.72 (dd, J= 2.7, 15.7 Hz, 1H), 2.49-0.95 (15H);MS
(EI) m/z 852 [M+H]+。
【0125】
ジヒドロモルヒネ-6-β-アセトアミドグルコース3。テトラベンジル誘導体11(41mg,
0.05mmol)を、MeOH(2mL)中に溶解した。濃HCl(13μL)、次に10%Pd-C(40mg)を添加した。この混合物を、水素大気下で、24時間攪拌した。この溶液を、Bio
Rad RG 501-X8混合床樹脂(1g)で中和し、この混合物をセライトを通して濾過した。固形物を、MeOH(30mL)で洗浄し、濾液を濃縮した。残渣を、エチルエーテルで摩砕し(3×2mL)、高真空下で乾燥し、3を白色粉末(12mg,
52%)として得た:Rf=0.4 (SiO2, 5:4:0.5:0.5 CHCl3/MeOH/H2O/濃NH4OH);mp=266.4℃(分解);1H NMRδ6.62
(s, 2H), 4.47 (d, J= 7.8 Hz, 2H), 3.81-3.77 (m, 2H), 3.55-3.43 (m, 4H), 3.00
(t, J= 9.3 Hz, 1H), 2.90 (d, J= 5.4 Hz, 1H), 2.84 (s, 3H), 2.79-2.69 (m, 1H),
2.63 (dd, J= 12.9, 14.4 Hz, 1H), 2.45 (d, J= 11.7 Hz, 1H), 2.30-2.05 (m, 3H),
1.77-1.57 (m, 4H), 1.34-1.24 (2H), 1.00-0.92 (2H);13C
NMR (CD3OD)δ173.9, 143.9, 142.6, 128.5, 122.3, 121.1, 119.4, 92.7,
81.6, 79.5, 77.9, 75.1, 72.0, 63.1, 52.7, 43.1, 41.9, 41.7, 40.3, 34.6, 29.2,
24.6, 21.7; MS (EI) m/z 491 [M+H]+;HRMS
(ESI) C25H35N2O8 の計算値491.2393、実測値491.2375;3の平均純度は、tR
=3.41分(移動相A)及びtR =3.74分(移動相B)を生じたことから、分析的HPLCにより、≧98%であることがわかった。
【0126】
受容体結合。ヒトμオピオイド受容体、ヒトδオピオイド受容体及びヒトκオピオイド受容体を用いて形質移入された細胞由来の膜への結合を、先に説明されたような、96-ウェルフォーマットを使用し行った(新規化合物10、11、12及び3は、1H
NMR、13C NMR及びMSにより特徴付けた。被験化合物3及び11の純度は、HPLCにより決定し、>98%であることがわかった)。細胞を、ラバーポリスマンにより剥ぎ取ることにより、プレートから除去し、Polytronホモジナイザーを用いTris緩衝液中でホモジナイズし、その後1回遠心し、追加の15分間の遠心27,000
×gにより洗浄した。ペレットを、50mM Tris(pH7.5)中に懸濁し、懸濁液を、μオピオイド受容体、δオピオイド受容体又はκオピオイド受容体について、各々、[3H]DAMGO、[3H]DPDPE、又は[3H]U69593と共に、インキュベーションした。総インキュベーション容量は1.0mLであり、及び試料は、25℃で60〜120分間インキュベーションした。結合反応におけるタンパク質の量は、約15μg〜30μgを変動した。この反応は、ガラスファイバーフィルターを備えるTomtec
96収集器(Orange, CT)を使用する濾過により終結させた。結合した放射活性を、Pharmacia
Biotechβ-プレート液体シンチレーションカウンター(ピスカタウェイ,
NJ)によるカウントにより決定し、データをカウント/分で表した。IC50値は、少なくとも6種の被験化合物濃度を用いて決定し、Graphpad/Prism(ISI,
サンディエゴ, CA)を用いて計算した。Ki値は、Cheng及びPrusoffの方法により決定した[Traynor,
J. R.の論文 Mol. Pharmacol., 47:848-854 (1995)]。
【0127】
[35S]GTPγ-S結合。[35S]GTPγ-S結合を、本質的にTraynor及びNahorskiの論文に説明されたように行った[Zaveri,
N.らの論文、Eur. J. Pharmacol., 428:29-36 2001]。μオピエート受容体、δオピエート受容体又はκオピエート受容体を形質移入した細胞を、組織培養皿から20mM
Hepes、1mM EDTAへ剥ぎ取り、その後500 ×gで10分間遠心した。細胞を、この緩衝液中で再懸濁し、Polytronホモジナイザーを使用しホモジナイズした。ホモジネートを、27,000
×gで15分間遠心し、ペレットを20mM Hepes、10mM MgC12、100mM
NaCl(pH7.4)を含有する緩衝液A中に懸濁した。懸濁液を、27,000 ×gで再遠心し、再度緩衝液A中に懸濁した。結合アッセイに関して、膜(8〜15μgタンパク質)を、[35S]GTPγ-S(50pM)、GDP(10μM)、及び被験化合物と共に、総容量1.0mLで25℃で60分間インキュベーションした。試料を、ガラスファイバーフィルター上で濾過し、結合アッセイについて説明されたようにカウントした。プログラムGraphPad
Prismを用い、統計解析を行った。このアッセイにおいて、受容体に関する化合物の効力又は親和性を、[35S]GTPγS結合の刺激に関するそのEC50により決定した。アゴニスト効能は、化合物が対照に対し[35S]GTPγS結合を最大刺激した程度として定義した。EC50値は、[35S]GTPγS結合の50%最大刺激を生じた化合物の濃度を表している。
【0128】
(実施例34):アルコール中毒及びニコチン中毒に対する6-ナルトレキサミド
米国において、アルコール依存症、ニコチン中毒、及びギャンブル依存症は、著しい社会的及び経済的結果を伴う、重篤な精神障害である。疫学的試験は、アルコール依存症は、米国人口の約14%であり、いずれかの時点で肝臓に罹患していることを示した[Regierらの論文、Arch.
Gen. Psychiatry, 50:85-94 (1993)]。毎年米国民100,000名は、アルコール依存症の直接的及び間接的原因で死亡すると推定され[McKinnis及びFoegeの論文、JAMG,,
270:2207-2212 (1993)]、健康及び社会的コストを含む経済的コストは、年間1660億ドルに達している(stagger)(NIDA及びNIAAA、1998)。喫煙は、肺癌に直接関連しており、禁煙は、肺癌を防ぐ主要手段である。アルコール依存症又はニコチンからの離脱症状を治療するための薬物療法の使用、並びにアルコール依存症患者又は喫煙者のリハビリテーションにおける役割は限定されている。多くの薬物療法がテストされ、中毒患者において、アルコール消費を減少又は回避させる。これらの一部は、ジスルフィラム、リチウム、選択的セロトニン再取込みインヒビター、及びアカンプロセートである。ジスルフィラムは、限定的有効性を有することが示されている[Garbuttらの論文、JAMA,,
281:1318-1325 (1999)]。アルコール依存症からの離脱症状を治療するために、薬物療法が使用されるいくつかの方法が存在する。喫煙を止めさせるいくつかの方法が開発されているが、再発率は高い。アルコール依存症に関して、ジスルフィラムのような抑止薬は、アルコールの摂取を喜ばしくないものとする。対照的に、薬物療法は、アルコールの強化(reinforcing)作用を減弱するか、又はアルコールを摂取する衝動もしくは渇望を低下させることにより、アルコール摂取を減少することを試みる。薬理学的物質は、内因性オピオイド、カテコールアミン(特にドパミン、セロトニン)及びグルタミン酸のような興奮性アミノ酸を含む、エタノール又はニコチンの強化又は識別力刺激作用の基礎となるいくつかのニューロトランスミッターシステムに影響するように見える[Kranzlerの論文、Am.
J. Psychiat., 152:391-397 (1995)]。多くの薬物療法が、アルコール又はニコチン依存症の治療において価値があることが示されているという事実にもかかわらず、薬理学的療法(Pharmacotherapy)は大きい作用を有さない。アルコール依存者又は喫煙者の亜群は、ある種の薬物療法に良好に反応することのいくつかの証拠があり、これは治療のマッチングは、薬物療法の効能を増加し得ることを示唆している。しかし戦略的方法、用量及び治療期間に関するかなり基本的情報が、正に入手可能になりつつあり、及びはるかに多い情報が確立されることが必要である。これらの重要な問題点に対処するために、新規方法及び新規薬物療法の開発が明らかに必要である。
【0129】
1994年に、ナルトレキソンは、米国FDAにより、アルコール依存症の治療用に承認された。ナルトレキソンは、アゴニスト活性を伴わず、乱用の可能性もない、純粋なオピオイドμ受容体アンタゴニストである。ナルトレキソンは、問題のない飲酒者の前臨床試験において、強化(すなわち刺激物)作用を低下させ、かつ最初のアルコール消費の望ましくない(すなわち鎮静)特性を増大させることがわかった[Swiftらの論文、Am.
J. Psychiat., 151: 1463-1467 (1994)]。齧歯類及びサルを使用した試験は、オピオイドアンタゴニストであるナロキソン及びナルトレキソンが、自発的消費及びストレスが誘導したアルコール消費の増加を減少させることを示し、このことは、これらの物質が、アルコール消費の強化作用を妨害し得ることを示唆している[O'Brienらの論文、Alcohol,
13:35-39 (1996)]。ナルトレキソンは、より高いレベルの渇望及び貧弱な認知機能を伴うアルコール依存者において最も有益であろう[Jaffeらの論文、J.
Consult. Clin. Psychol., 64:1044-1053 (1996)]。しかし、1つの試験において、治療を受けている患者の15%は、耐え難い悪心を含む有害作用ために、治療を早期終結している[Croopらの論文、Arch.
Gen. Psychiat., 54:1130-1135 (1997)]。ナルトレキソンは、肝臓疾患のアルコール依存症患者での使用を複雑にしかつ治療を混乱させる、用量-依存型肝毒性副作用にも関連している[Masonらの論文、Arch.
Gen. Psychiat., 56:719-724 (1999)]。さらなる欠点は、短い望ましい作用期間及び比較的低い生体利用効率を含み[Wallらの論文、Drug
Metab. Dispos., 9:369-375 (1981)]、恐らくδ受容体及びκ受容体に関する比較的低い親和性が、アルコール飲酒の強化作用を消滅することに関与していると考えられるであろう。ナルトレキソンの有益な作用は、経時的に次第に消滅する。加えてナルトレキソン治療の服薬遵守は様々であり、また愁訴の多い対象においてのみ、プラセボよりも有意により良い活性のある薬物療法であった[Volpicelliらの論文、Arch.
Gen. Psychiat., 54:737-742 (1997)]。まとめると、ナルトレキソンは、アルコール依存症の間で飲酒行動に対し、軽度の作用を生じるように見える。場合によっては、ナルトレキソンの作用は、極めて強固であるが、その効能の証拠には一貫性が少ない。しかしナルトレキソンよりも優れた医薬特性を伴うオピオイドアンタゴニストであるナルメフェンは、アルコール治療薬物療法としてより有望であろう。ナルトレキソンは、禁煙に有用であることが報告されている[O'Malleyら、米国特許第6,541,478号]。
【0130】
オピオイド受容体アンタゴニストは、アルコール-所望行動に対し、明らかに直接作用を有する。例えば、ナロキソンの微量透析による側座核ニューロンへの直接灌流は、アルコール-媒介したドパミン放出を阻害した[Benjaminらの論文、Brain
Res., 621:137-140 (1993)]。オピオイド受容体の遮断によるアルコール消費の減少は、この強化システムにおけるナロキソンの直接作用を示唆している。動物試験は、μ-オピオイド受容体、δ-オピオイド受容体及びκ-オピオイド受容体は、アルコール-誘導した強化に寄与することの証拠を提供した[Herz
(1997);Ulmらの論文、J. Clin. Psychiat., 56:5-14 (1995)]。κ-オピオイド受容体に選択性のあるノルビナルトロフィン(norbinaltorphimine)(norBNI、図1)に関連した、かなりの研究が行われた[Portogheseらの論文、J.
Med. Chem., 31:1344-1347 (1987)]。norBNIのSARは、広範に調べられており、2つのナルトレキソンで誘導したファーマコフォアを含む、二価のリガンドである。ただ1つのnorBNIファーマコフォアが、κアンタゴニスト活性に必要であり、及び受容体-結合したリガンドの第二のファーマコフォアにおける塩基性基(N-17位の)は、選択性を付与する「アドレス」として作用すると考えられる。分子の受容体への配向のための固い足場の明らかな必要性の観点において、ナルトリンドール(NTI、図15参照)を含む代わりの構造が開発されている。NTIに関連したこのインドロモルヒナン構造は、norBNIよりも簡単であり、及びκアンタゴニストは、Glu297アドレスサブサイトと相互作用することのかなりの証拠を提供している[Stevensらの論文、J.
Med. Chem., 43:2759-2769 (2000)]。
【0131】
更にκアンタゴニスト活性に必要なアドレス群の正帯電したアミノ官能基の必要要件は、強力かつ選択的オピオイドκアンタゴニストとして非オピエート-誘導した化合物を説明した最近の報告において証明された。化合物JDTic(図16参照)は、2つの塩基性アミン基の重要性を例示しており:両方は各々、アドレス基及びメッセージ基である。更なる研究は、nor-BNIシリーズについて認められないκ選択性におけるフェノール基の重要性を示した[Thomasらの論文、J.
Med. Chem., 47:1070-1073 (2004)]。キメラκ-μオピオイド受容体を基に、負帯電した残基(すなわちGlu297)は、κ-選択的結合及び機能活性に重要であることは明らかである。高度に活性のあるκ-選択性又は非κ-選択性受容体の選択的アンタゴニストは、中毒、特に例えばニコチン又はアルコールのような、物質中毒の治療に有用であろう。
【0132】
ナルメフェン1及びナルトレキソン2の化学構造を、図17に示す。これらの化合物は、オピオイド過量、中毒及びアルコール依存症の治療に使用される。本試験の医化学的目的は、1及び2の各々代謝上脆弱な6-メチレン又は6-ケト基を、アミド部分と置換することによる、これらの化合物の代謝的に安定なアナログの開発であった。μオピオイド受容体及びκオピオイド受容体の選択性及び効力を増加するために、アミド基の芳香環内の様々な置換パターンが調べられた。この目的に合致するために6-αナルトレキサミド又は6-βナルトレキサミドの新規4工程合成が開発された。水性エタノールの還流時のNaOAc存在下でのナルトレキリン2の塩酸ヒドロキシルアミンとの縮合は、対応するオキシム3(図18参照)を生じた。引き続きの3のオキシム官能基の対応するアミン4への還元は、BH3/THFとの48時間の加熱及び引き続きの水性後処理により実現され、アミン4をジアステレオマーの9:1(β/α)混合物として生じた。ジアステレオマーアミンは、シリカゲル上でのクロマトグラフィーにより分離した。アミン4は、CH2C12中のベンゾトリアゾール-1-イル-オキシ-トリス-(ジメチルアミノ)ホスホニウムヘキサ-フルオロリン酸(BOP)及びジイソプロピルエチルアミンの存在下で、カルボン酸(RCOOH)とカップリングした。あるいは、これらのアミドは、CH2C12中の酸塩化物、トリエチルアミン及び4の反応により形成した。両方の場合において、生成物は、3-位のヒドロキシル基のエステル化により生じる副産物を脱エステル化するために、メタノール中のK2CO3で処理した。BOPカップリング法が、3-位に酸塩化物法よりもより少ないエステル化を生じるが、3-位での若干のエステル化は避けられない。従って、中間体アミドエステルの精製を助けるために、過剰な酸誘導体による反応を実施することはより都合が良いことがわかった。これらの方法は、アミド5及び6a-hを簡便に調製することを可能にした。Pd-C及びHClの存在下での水素化分解によって、化合物5のベンジル基を除去したグルコース複合体6aを生じた。
【0133】
【表7】
【0134】
対応するアゴニストについて観察された効力及び選択性[MacDougallらの論文、J.
Med. Chem., 47:5809-5815 (2004);MacDougallらの論文、Bioorganic
Med. Chem., 12:5983-5990 (2004);MacDougallらの論文、Bioorganic
Med. Chem., 15:583-1586 (2005)]は一般に、これらのアンタゴニストにおいて維持された。アミド6d、6e及び6cは全て、オピオイド受容体に関して高い親和性を有し、かつδ受容体及びκ受容体で部分的又は完全なアゴニスト作用を有した(表8)。細胞膜へのGTPγS結合を基に、全ての化合物は、μ受容体で強力なアンタゴニストである。化合物6aは、μ受容体について高度の親和性を有し、δ及びκ受容体について中等度の親和性を有した。6aは、μ及びκ受容体で強力なアンタゴニストであり、δ受容体で部分的アゴニストである。まとめると、このデータは、μ受容体単独又はμ受容体及びκ受容体のアンタゴニスト作用の機能性の結果による、強力な先例(lead)が存在することを示している。
【0135】
【表8】
【0136】
代謝安定性を調べるために、代謝安定性及び化学安定性を試験するための、HPLC及びLCMSアッセイを開発し、一般にこれらのシリーズの化合物は、代謝及び分解に対し著しく安定していることがわかった。これらのアナログを、C-6位の遮断のために肝臓調製物中で安定しているようにデザインし、かつこれは、観測されたものであった。予備的結果は、6d及び6aは、強力なオピオイドアンタゴニストとして有望であることを示した(表7及び8)。テトラ-O-ベンジル-β-D-グルコピラノシルで保護された化合物5が、合成され、そのインビトロ結合及び機能特性を調べた。化合物5は、μ受容体に関して高い親和性を有し、かつκ受容体に関して中程度の及びδ受容体に関して低い親和性を有した。これらの試験は、グルコースアナログそれら自身を、開発候補として指摘している。
【0137】
このデータは、代謝的に安定なオピオイドアンタゴニストは、インビトロにおいて合成することができ、かつ試験することができることの実行可能性を示している。代謝的に安定なC-6置換されたオピオイドアンタゴニストは、好ましい副作用プロファイルを維持するであろう。インビトロ結果は、κアゴニスト又はアンタゴニスト特性のいずれかを伴う、強力なμアンタゴニストを合成することができることを示している。アルコール消費の減少においてκアンタゴニスト又はアゴニスト作用がより効率的であるかどうかを試験するために、アルコール依存症及びニコチン中毒の動物モデルにおいて先例を試験した。高度に活性のあるκ-選択性又は非-κ-選択性受容体の選択性アンタゴニストは、中毒、特に例えばニコチン又はアルコールなどの物質に対する中毒症状の治療に有用であろう。
【0138】
下記実施例(実施例35-43)において、全ての反応は、特に記さない限りは、炉乾燥したガラス製品を使用し、周囲温度で磁気攪拌しながら、窒素陽圧下で実施した。空気-及び水分-感受性液体を、シリンジによりゴム製隔壁を通して移した。カラムクロマトグラフィーのために、シリカゲル(230〜400メッシュ)を使用した。DMFを、中間アルミナのカラムを通して乾燥させ、また使用前は、窒素下活性化した4Å分子篩上で貯蔵した。全ての他の溶媒及び試薬は、入手したまま使用した。1H
NMRスペクトルは、300MHz Varian NMRにより記録した。化学シフトは、7.26ppmで
CDC13に対するppm(δ)で報告した。1H
NMRスペクトルは、特に記載しない限りは、CDC13で記録した。融点は補正せずに報告した。高解像度質量分析を、University
of California at Riverside において、Opus V3.1及びDEC
3000 Alpha Stationデータシステムを備えた、VG7070スペクトロメーター上で行った。燃焼分析が特定されない場合は、分析純度は、D7500積分器及びHamilton
PRP-Iステンレス鋼カラム(250mm ×4.6mm内径)を備えた、Hitachi
L74液体クロマトグラフィーを使用する、順相HPLCにより決定した。
ナルトレキソンオキシム3を、以下の方法に従い調製した。ナルトレキソン(6.2g, 1.46mmol)、NH2OH-HCl(1.5g)及びNaOAc(2.9g)を、無水EtOH(80mL)及び水(5mL)中に溶解した。混合物を、2.5時間還流加熱し、その後濃縮した。水(20mL)を添加し、混合物を、K2CO3により塩基性とし、CHC13で抽出した。CHCl3抽出物は、ブラインで洗浄し、Na2SO4上で乾燥し、濾過及び濃縮し、白色固形物(5.83g,
90%)とした:Rf =0.65 (5:1:0.2 EtOAc/MeOH/NH4OH);MS(EI)
m/z=357[MH]+。
6-β-ナルトレキサミン及び6-α-ナルトレキサミン、4b、4aを、以下の方法に従い調製した。ナルトレキソンオキシム(5.83g, 16.3mmol)を、THF(40mL)に溶解し、カニューレにより、BH3:THF
(300mL, 300mmol, THF中1M溶液)の10℃溶液へ10分かけて移した。白色沈殿が形成され、その後反応液を2日間かけて還流温度へ加熱するにつれて、ゆっくり溶解した。この溶液を、室温へ冷却し、水(10mL)及び1N
KOH(200mL)をゆっくり慎重に添加した。溶液を、2時間還流加熱した。その後10%HCl(225mL)によりpHを2.5に低下した。この溶液を、2時間還流加熱し、濃縮し、THFを除去し、その後K2CO3で塩基性(pH8-9)とした。混合物を、CHC13(4×150mL)で抽出し、抽出物をNa2SO4上で乾燥し、濾過し濃縮した。得られた油状物を、SiO2上のクロマトグラフィー(26×60cm,
CH3CN/MeOH/NH4OH, 25:5:1による溶離)により精製し、4b(2.14g,
38%)を白色-黄色固形物として得た:Rf=0.20;1H
NMR (300 MHz, CD3ODを2滴含むCDC13)
δ6.61 (d, J = 8.1 Hz, 1H), 6.49 (d, J = 8.1 Hz, 1H), 4.17 (d, J = 7.5 Hz, 1H),
3.39-0.45 (20 H);MS m/z = 343 [MH]+。α-及びβ-ジアステレオマーの混合物からなる追加の物質0.64g(12%)を、単離した。この物質の尾部画分の分取クロマトグラフィー(SiO2,
CH3CN/MeOH/NH4OH, 25:5:1)は、4aの分析的試料を生じた:Rf
=0.16;1H NMRδ6.65 (d, J = 8.1 Hz, 1H), 6.46 (d, 8.1 Hz, 1H), 4.50 (d, J =
3.0 Hz, 1H), 3.34 (dt, J = 3.9, 12.6 Hz, 1H), 3.04 (t, J = 6.6 Hz, 1H), 2.95
(s, 1H), 2.63-0.29 (17H);MS m/z 343 [MH]+。
【0139】
【化11】
【0140】
(実施例35):6-アセトアミド-2,3,4,6-テトラ-O-ベンジル-D-グルコピラノシル-14-ヒドロキシ-17-(シクロプロピルメチル)ノルデスモルヒネ, 5
6-ナルトレキサミン(100mg, 0.292mmol)、カルボン酸(RCOOH, スキーム1)(187mg,
0.321mmol)及びBOP(142mg, 0.321mmol)を、CH2CI2(3mL)中に溶解し、NPr2Et
(0.16mL, 0.92mmol)を添加した。無色の溶液を、室温で90分間攪拌し、濃縮した。残渣を、SiO2
(20:1 EtOAc/MeOH)を通して濾過し、白色固形物を提供した。この物質(240mg)を、MeOH
(10mL)に溶解し、K2CO3 (36mg, 0.26mmol)を添加した。混合物を、115分間攪拌し、飽和NH4Cl(10滴)を添加した。混合物を濃縮し、SiO2
上で吸収し、SiO2クロマトグラフィー(20:1
CHCl3/MeOH)により精製し、7を白色固形物(200mg,
6-ナルトレキサミンナルトレキサミンから75%、2-工程)として得た:Rf=0.11
(20:1 CHCl3/MeOH);MS (EI) m/z=907[M]+;1H
NMR (CDCl3, 重要な共鳴)δ7.31-7.06 (m);6.76
(d), 6.71 (d);6.55 (d);6.54 (d) 4.94-4.38 (m);3.86-3.79
(m);13C NMR (CDCl3)δ170.4, 143.6, 138.1, 137.6, 128.4, 128.4,
128.3, 128.1, 127.9, 127.8, 127.78, 127.7, 127.69, 127.64, 118.9, 93.8, 86.8,
81.0, 78.4, 75.8, 75.7, 75.2, 75.0, 73.2, 70.1, 59.2, 50.9, 30.2, 4.1, 3.9。
【0141】
(実施例36):14-ヒドロキシ-17-(シクロプロピルメチル)ノルデスモルヒネ6-アセトアミドグルコース, 6a
無水MeOH(8mL)を、ベンジル化した誘導体5(180mg, 0.198mmol)及び10%活性化したPd-C(180mg)に添加した。濃HCl(0.015mL)を、シリンジにより添加した。このフラスコを、窒素で掃流し、次に水素バルーン下で21時間攪拌し、セライトを通して濾過し(MeOHで溶離)、濃縮した。得られた白色固形物を、エチルエーテル(無水物、3×2mL)で摩砕し、6aを白色固形物(92mg,
85%)として提供した:Rf=(5:4:0.5:0.5 CHCl3/MeOH/H2O/NH4OH);1H
NMR(CD3OD、重要な共鳴) δ6.72 (s), 4.67 (d);
4.60-4.52 (m);3.99 (d); 3.90-3.48 (m); MS
m/z = 547 [M+H]+。
【0142】
(実施例37):酸塩化物を用いた6-ナルトレキサミンのアミド化の一般的手法。6-(ピリジン-3'-イル)カルボキサミド-14-ヒドロキシ-17-(シクロプロピルメチル)ノルデスモルヒネ, 6b
6-アミノナルトレキサミン(53mg, 0.155mmol)を、CH2Cl2(2mL)中に溶解し、塩化ニコチニル-HCl(40mg,
0.226mmol)及びNEt3(0.085mL, 0.465mmol)を添加した。この溶液を、1.5時間攪拌し、濃縮した。残渣を、SiO2(10:1
CH2Cl2/MeOH)上のクロマトグラフィーにより精製し、アミドエステルを黄色-白色固形物(63mg,
74%)として提供した:Rf=0.11(10:1 CH2Cl2/MeOH);MS
m/z=552。このアミドエステル(53mg, 0.096mmol)を、MeOH(2mL)中に溶解し、K2CO3(46mg,
0.33mmol)を添加した。混合物を、室温で1時間攪拌し、NH4Cl(飽和水溶液、10滴)及びSiO2(2g)を添加した。この混合物を濃縮し、SiO2クロマトグラフィー(10:1
CHCl3/MeOH)により精製し、標題化合物を黄味がかった-白色固形物(27mg,
63%)として得た:Rf=0.31(10:1 CHCl3/MeOH);MS
m/z=447。
(実施例38):BOP試薬及びカルボン酸を用いた6-ナルトレキサミンのアミド化の一般的手法。6-(3'-ジメチルアミノ)ベンズアミド-14-ヒドロキシ-17-(シクロプロピルメチル)ノルデスモルヒネ, 6c
【0143】
CH2Cl2(1.5mL)を、ナルトレキサミン(50mg,
0.146mmol)、3-ジメチルアミノ安息香酸(24mg, 0.14mmol)、BOP試薬(64mg,
0.14mmol)及びPr2EtN(0.081mL, 0.47mmol)に添加した。黄色溶液を、室温で80分間攪拌し、濃縮した。残渣を、クロマトグラフィーSiO2(10:1
EtOAc/MeOH)にかけ、白色固形物(70mg)を提供した。この物質を、MeOH(4mL)に溶解し、K2CO3(30mg,
0.22mmol)を添加した。90分後、この混合物を濃縮した。残渣を、SiO2(10:1 CHCl3/MeOH)上のクロマトグラフィーにより精製し、標題化合物を白色固形物(17.4mg,
ナルトレキサミンから24%):Rf=0.16(10:1 CHCl3/MeOH);MS
m/z=490 [MH]+。
【0144】
(実施例39):6-(3'-メトキシ)ベンズアミド-14-ヒドロキシ-17-(シクロプロピルメチル)ノルデスモルヒネ, 6d
参照JMM-VIII-42。化合物6bについて説明された一般的手法を使用し、6-ナルトレキサミン(90mg,
0.263mmol)、3-アニソイルクロリド(0.09mL, 0.69mmol)、NEt3(0.11mL,
0.83mmol)及びCH2Cl2(2mL)は、中間体アミドエステルを白色泡状物(96mg)として生じた;MS
m/z=611。この物質のMeOH(3mL)中K2CO3(43mg,
0.31mmol)による加水分解、それに続くSiO2クロマトグラフィー(10:1
CH2Cl2/MeOH)は、6dを白色固形物として生じた:57mg(78%);MS
m/z=477。
(実施例40):6-(チオフェン-2'-イル)アセトアミドミド(acetamidomido)-14-ヒドロキシ-17-(シクロプロピルメチル)ノルデスモルヒネ, 6e
【0145】
化合物6bについて説明された一般的手法を使用し、6-ナルトレキサミン-(19mg,
0.055mmol)、2-チオフェンアセチルクロリド(0.014mL, 0.11mmol)、NEt3(0.019mL,
0.14mmol)及びCH2Cl2(2mL)は、中間体アミドエステルを白色泡状物(15mg,
46%)として生じた;Rf=0.12(30:1 CH2Cl2/MeOH);MS
m/z=591[MH+]。この物質(13mg, 0.022mmol)のMeOH(2mL)中K2CO3(2.5mg,
0.0.018mmol)による加水分解、それに続くSiO2クロマトグラフィー(15:1
CH2Cl2/MeOH)は、6eを白色固形物として生じた(10mg,
100%):Rf=0.17(15:1 CH2Cl2/MeOH);MS
m/z=467。
(実施例41):6-(3',5'-ジメトキシ)ベンズアミド-14-ヒドロキシ-17-(シクロプロピルメチル)ノルデスモルヒネ, 6f
【0146】
化合物6cについて説明された一般的手法に従い、4(90mg, 0.263mmol)、3,5-ジメトキシ安息香酸(72mg,
0.395mmol)、BOP(174mg, 0.394mmol)、NPr2Et(0.15mL,
0.84mmol)及びCH2C12(3mL)は、中間体アミドエステルを白色-オレンジ色固形物(186mg)として生じた。この物質のMS分析は、これがアミドエステル(m/z=671)及びアミド(m/z=507)の混合物からなることを示した。この物質のMeOH(4mL)中K2CO3(150mg,
1.4mmol)による脱エステル化、及びSiO2クロマトグラフィー(20:
1 CH2Cl2/MeOH)は、6fを白色固形物として提供した:MS
m/z=507。
【0147】
(実施例42):6-(4'-クロロ)ベンズアミド-14-ヒドロキシ-17-(シクロプロピルメチル)ノルデスモルヒネ, 6g
参照VIII-58。化合物6cについて説明された一般的手法に従い、4(90mg,
0.26mmol)、4-クロロ安息香酸(62mg, 0.39mmol)、BOP(174mg, 0.394mmol)及びNEtPr2(0.15mL,
0.842mmol)、引き続きのSiO2クロマトグラフィー(20:1
EtOAc/MeOH)は、中間体アミドエステルを白色固形物としてもたらした:Rf=0.5
(20:1 EtOAc/MeOH);MS m/z=620。この物質のMeOH(4mL)中K2CO3(72mg,
0.53mmol)による脱エステル化、それに続くSiO2クロマトグラフィー(20:1
CH2Cl2/MeOH)は、6gを白色固形物(76mg,
60%)として提供した:Rf=0.41 (20:1 CH2Cl2/MeOH);MS
m/z=480。
【0148】
(実施例43):6-(3'-ヒドロキシ)ベンズアミド-14-ヒドロキシ-17-(シクロプロピルメチル)ノルデスモルヒネ, 6h
参照JMM-VIII-59、VIII-64。化合物8bについて説明された一般的手法に従い、4(90mg,
0.26mmol)、3-アセトキシ安息香酸(71mg, 0.39mmol)、NEtPr2(0.15mL,
0.84mmol)、BOP(174mg, 0.39mmol)及びCH2C12(2mL)、並びにSiO2クロマトグラフィー(20:1
EtOAc/MeOH)は、中間体アミドエステルを白色泡状物(180mg)として生じた:Rf=0.28(10:1
CHCl3/MeOH);MS m/z=667。この物質のMeOH(2mL)中K2CO3(72mg,
0.53mmol)による脱エステル化、及びSiO2クロマトグラフィー(20:1
CH2Cl2/MeOH)は、8hを白色固形物(76mg,
60%)として生じた:Rf=0.41(20:1 CH2Cl2/MeOH);MS
m/z=480。
【0149】
(実施例44):インビトロ代謝安定性
インビトロ試験における化合物の代謝安定性は、インビボ実験における代謝安定性に関する可能性についての重要な情報をもたらすことができる。オピオイドクラスの化合物の代謝の卓越した経路はC-6位での代謝のためであるので、代謝安定性を、合成化合物の半減期を延長させるため、生体利用効率を増加させるため、並びに薬物動態プロファイル及び副作用プロファイルを改善させるために、化合物中にデザインした。薬物のデザイン及び開発の初期段階におけるADMETの不安定性を克服することにより、より多くの薬物-様化合物を生じた。
【0150】
選択された化合物のインビトロ代謝安定性試験を、プールしたヒト肝臓S9、ラット肝ミクロソーム及びマウス肝ミクロソームの存在下において試験した。これらの試験を、ヒト代謝状況を模倣するように行い、動物肝代謝試験を、インビボ動物試験のモデル試験として行った(下記参照)。アッセイ緩衝液は、3.2mg/mLのヒト肝臓プールしたS9又は1.2mg/mLラットもしくはマウス肝ミクロソーム、0.1Mリン酸カリウム緩衝液(pH7.4)、0.1mM化合物、0.5mM
NADP+、0.5mMグルコース-6-リン酸、5IU/mLグルコース-6-リン酸デヒドロゲナーゼ、1mg/mL
DETAPAC、7mM MgCl2 を含み、最終インキュベーション容量0.25mLであった。0分、10分、25分、45分又は60分後、1mL
CH2Cl2/IPA(3:1 v:v)の添加により、反応を停止させ、完全に混合した。遠心後、有機相を蒸発させ、窒素流れ下で乾燥し、200μメタノールに溶解し、ボルテックスし、遠心した。HPLCを使用し、有機抽出物を、Richard
Scientific(Novato, Ca)から購入したAxxi-Chrom Silica 5μmカラムを用いて、Hitachi
HPLC上で分析した。移動相は以下からなる:55/45/0.018 (MeOH/IPA/HClO4)及び流量1.5mL/分、UV検出254nm。ナルトレキソン、ナルメフェン及び化合物6dの保持時間は、各々、2.7分、2.1分、及び2.6分であった。
【0151】
表9に示したように、ナルトレキソン及びナルメフェンは、試験した肝調製物の存在下で、かなりの代謝不安定性を有した。対照的に、ヒト肝臓S-9及びマウス肝ミクロソームの存在下で、化合物6dは、検出的には代謝されなかった。ラット肝ミクロソームの存在下で、少量の代謝産物が認められたが、これは、ナルトレキソン又はナルメフェンよりも、各々、6倍又は22倍安定していた。まとめると、化合物6dについてかなりの代謝安定性が認められ、化合物6d及び同族体の生体利用効率はかなりの量であると推定される。
【0152】
【表9】
【0153】
(実施例45):インビボ活性
オピエートによる急性予備治療後の身体禁断徴候のアンタゴニスト-突発(precipitate)により規定されたような、急性依存状態の観察が、ヒト、サル、イヌ、ハムスター、マウス及びラットにおいて報告されている。ヒト及びラットの両方において、オピエートは、モルヒネへの単回曝露後のオピエート離脱症状の徴候を突発する。ラットのパラダイムは、ヒトにおける状態に類似している。従ってナロキソン注射前4時間のモルフィン(5.0mg/kg)による単回処置後の、ナロキソン(0.03〜3.0mg/kg)は、離脱症状の身体的徴候を突発した[Schulteisらの論文、PsychoPharmacology,
129:56-65 (1997)]。離脱症状強度の全体的指標には、Gellert及びHoltzmanの論文の重み付きスケールを使用した[J.Pharmacol.
Exper. Ther., 205:536-546 (1978)]。このスケールは、体重減少、回避試みの回数、咆哮(roaring)、禁断の激しい震えの回数、腹部収縮の事例の等級化した徴候、並びに下痢、顔面萎縮/歯の根の合わない震え(chattering)、嚥下-運動(swallowing-movement)、多量の唾液分泌、色素涙、下垂症、異常な姿勢、陰茎つくろい(penile
grooming)/勃起/射精、及び取扱い時の興奮を含むチェックされた徴候(存在又は非存在)からなる。本発明者らの研究に関して、Global身体的徴候(SS)等級付けは、Gellert及びHoltzmanの重み付きスケール(1978)を基に、先の個々の徴候の全てを含んだ。モルヒネ(5.0mg/kg)単回投与の4時間後、化合物6a又は6dの単回投与量を、ラット群に投与した。皮下注射後1分で、離脱症状の身体的徴候が10分間認められた。これらの結果は、図19及び20にまとめている。
【0154】
まとめると、成体雄ラットにおいて、モルヒネ5.0mg/kg投与の4時間後に皮下経路により投与された化合物6a及び6dは、離脱症状の身体的徴候を突発した。化合物6dは、離脱症状の身体的徴候の用量-依存型の増加を引き起こし、ピークは0.3mg/kgであった。0.1mg/kg未満の概算EC50値が認められた。より高い投与量は、この作用を減少した。化合物6aに関して、約1mg/kgの概算EC50は、離脱症状の身体的徴候を突発することがわかった。比較のためのナルメフェンは、約0.6〜1mg/kgのEC50値を与えた、離脱症状の身体的徴候における、(雑音が多いが(albeit
noisy))用量-依存性の増加をもたらした。文献の基準について、ナロキソンの離脱症状の身体的徴候を突発する最低有効量は、0.1〜0.3mg/kgと計算されている[Schulteisらの論文、Psychopharmacology,
129:56-65 (1997)]。結論として、このデータは、化合物6a、6d及び同族体の、強力なオピエート受容体アンタゴニストとしてのインビボ効能を裏付けている。これらの化合物は、比較的迅速に中枢神経系へ移行し(10分以内)、オピエート受容体に対し強力な作用を発揮する。この受容体システムは、アルコール及びニコチンに対する易中毒性を強力に制御するので、6のようなオピエートアンタゴニストは、ニコチン中毒及びアルコール中毒の軽減に有効であるはずである。
【0155】
本願明細書に説明された実施例及び態様は、例証のみを目的とし、かつそれらを考慮し様々な修飾又は変更が、当業者に示唆され、本願明細書及び添付された「特許請求の範囲」の精神及び範囲内に含まれることは理解される。本願明細書に列記された全ての刊行物、特許及び特許出願は、あらゆる目的のためにそれらの全体が本願明細書に参照として組入れられている。
【図面の簡単な説明】
【0156】
【図1】図1は、式IVに示した化合物の合成を示す。
【図2】図2は、チオ糖2a及び2bの合成を示す。
【図3】図3は、式Vの化合物の合成を示す。
【図4】図4は、式VIの化合物の合成を示す。
【図5】図5は、モルヒネの投与により誘導された用量-依存型の痛覚喪失を示す。データは、平均%MPE(±SEM)である。アスタリスクは、ビヒクル対照からの有意差を表す(StudentNewman-Keuls、p<0.05)。プラス印は、3mg/kgモルヒネからの有意差を表す(StudentNewman-Keuls、p<0.05)。
【図6】図6は、2種の高投与量13eで生じた鎮痛作用である。データは、平均%MPE(±SEM)である。アスタリスクは、ビヒクル対照からの有意差を表す(StudentNewman Kenls、p<0.05)。プラス印は、10及び30mg/kg13eの間の有意差を表す(Student Newman-Keuls、p<0.05)。
【図7】図7は、3aの投与により誘導された、用量-依存型の痛覚喪失を示す。データは、平均%MPE(±SEM)である。アスタリスクは、ビヒクル対照からの有意差を表す(StudentNewman Kenls、p<0.05)。プラス印は、3mg/kg 3aからの有意差を表す(StudentNewman-Keuls、p<0.05)。
【図8】図8A-Cは、ヘロイン依存型ラット(ラット#96)におけるヘロイン摂取に対する、オピエートアナログ13eの作用を示す。ラットには、活動/暗所相(午後6時〜午前6時)の15分前(午後5時45分)に、オピエートアナログ13e(0、0.04、及び0.2mg/kg、皮下注射)を、対象内デザインで注射した。データは、暗所相での1時間毎(図8A)、3時間毎(図8B)、及び12時間全体(図8C)についての総ヘロイン摂取(60μg/kg/0.1ml注入)で表した。
【図9】図9A-Cは、ヘロイン依存型ラット(ラット#99)におけるヘロイン摂取に対する、オピエートアナログ3aの作用を示す。ラットには、活動/暗所相(午後6時〜午前6時)の15分前(午後5時45分)に、オピエートアナログ3a(0、0.04、及び0.2mg/kg、皮下注射)を、対象内デザインで注射した。データは、暗所相での1時間毎(図9A)、3時間毎(図9B)、及び12時間全体(図9C)についての総ヘロイン摂取(60μg/kg/0.1ml注入)で表した。
【図10】図10A-Cは、ヘロイン-依存型ラット(ラット#100)におけるヘロイン摂取に対するオピエートアナログ13gの作用を示す。ラットには、活動/暗所相(午後6時〜午前6時)の15分前(午後5時45分)に、オピエートアナログ13g(0mg/kg、0.04 mg/kg、及び0.2 mg/kg、皮下注射)を、対象内デザインで注射した。データは、暗所相での1時間毎(図10A)、3時間毎(図10B)、及び12時間全体(図10C)についての総ヘロイン摂取(60μg/kg/0.1ml注入)で表した。
【図11】図11A-Cは、ヘロイン-依存型ラット(ラット#103)におけるヘロイン摂取に対するM6Gの作用を示す。ラットには、活動/暗所相(午後6時〜午前6時)の15分前(午後5時45分)に、M6G(0mg/kg、0.04 mg/kg、及び0.2mg/kg、皮下注射)を、対象内デザインで注射した。データは、暗所相での1時間毎(図11A)、3時間毎(図11B)、及び12時間全体(図11C)についての総ヘロイン摂取(60μg/kg/0.1ml注入)で表した。
【図12】図12A-Cは、ヘロイン-依存型ラット(n=4)におけるヘロイン摂取に対するオピエートアナログ13hの作用を示す。ラットには、活動/暗所相(午後6時〜午前6時)の15分前(午後5時45分)に、オピエートアナログ13h(0[ラット#96]、0.01[ラット#99]、0.04[ラット#100]、及び0.2mg/kg[ラット#103]、皮下注射)を、対象内デザインで注射した。データは、暗所相での1時間毎(図12A)、3時間毎(図12B)、及び12時間全体(図12C)についての総ヘロイン摂取(60μg/kg/0.1ml注入)で表した。
【図13】図13は、実施例33に説明されたMG6(1)及び2種の代謝安定性アナログ(2及び3)の化学構造を示している。
【図14】図14は、実施例33に示されたMG6のC-グリコシドの調製を示している。(i) CH3S(0)CH3, Ac2O;(ii)LiCH2CO2Et、THFその後HCl、-78℃〜室温;(iii)Et3SiH、BF3OEt2、CH3CN、0℃〜室温;(iv)NaOH、H2O、THF、還流、その後HCl;(v)SOC12;(vi) 3-O-トリイソプロピルシリル-6-β-アミノモルヒネ、Et3N、CH2Cl2;(vii)Bu4NF、THF、H2O;(viii) H2、10%Pd-C、MeOH;(ix)H2、10%Pd-C、MeOH、HCl。
【図15】図15は、norBNI及びナルトリンドール(NTI)の化学構造を示している。
【図16】図16は、JDTic及びグアニジンナルトリンドール(GNTI)の化学構造を示している。
【図17】図17は、ナルメフェン(1)及びナルトレキソン(2)の化学構造を示している。
【図18】図18は、オピオイドアンタゴニストの一般的合成を示している。
【図19】図19は、離脱症状の身体的徴候に対する実施例38の化合物6dの作用を示す。
【図20】図20は、離脱症状の身体的徴候に対する実施例35の化合物6aの作用を示す。
【特許請求の範囲】
【請求項1】
下記式の化合物:
A-B-C
(式中、Aは、鎮痛薬又はその誘導体であり;Bは、連結基であり、かつCは、任意に置換されたアリール、任意に置換されたヘテロアリール及び糖類からなる群から選択される);並びに、それらの全ての医薬として許容し得る誘導体。
【請求項2】
前記鎮痛薬又はその誘導体が、オピエートである、請求項1記載の化合物。
【請求項3】
前記オピエートが、ナルトレキソン、フェニルピペリジン、ピペリジノール、プロジン、ピペリジルプロピオンアニリド、イソプロジン、プロジリジン、ベンゾモルファン、モルファン、アザビシクラン、モルヒナン、プロジン、ジフェニルアミノエチルプロピオネート、メサドン、イソメサドン、プロポキシフェン、デキストロメトロファン、ベンザゾシン-8-オール、ノルビナルトロフィン、ナルトリンドール及びグアニジンナルトリンドールからなる群から選択される、請求項2記載の化合物。
【請求項4】
前記連結基が、-S-及び-NH(CO)(CH2)n-からなる群から選択され;及び、添字nは、0〜5の整数である、請求項1記載の化合物。
【請求項5】
前記連結基が-S-である、請求項1記載の化合物。
【請求項6】
前記連結基が、-NH(CO)(CH2)nであり;かつ、添字nが、0〜5の整数である、請求項1記載の化合物。
【請求項7】
Cが、ハロゲン、(C1-C5)アルコキシ、ニトロ、CO2Rにより任意に置換されたアリールであり、かつRが、H又は(C1-C5)アルキルである、請求項1記載の化合物。
【請求項8】
Cが、フェニルである、請求項1記載の化合物。
【請求項9】
Cが、ハロゲン、(C1-C5)アルコキシ、ニトロ、CO2Rにより任意に置換されたヘテロアリールであり、かつRが、H又は(C1-C5)アルキルである、請求項1記載の化合物。
【請求項10】
Cが、チオフェンである、請求項1記載の化合物。
【請求項11】
Cが、糖類である、請求項1記載の化合物。
【請求項12】
前記糖類が、下記式を有する、請求項1記載の化合物:
【化1】
(式中、各R8は、H、(C1-C5)アルキルC(O)-、(C7-C10)アラルキルC(O)-、(C7-C10)アラルキル、(C1-C5)アルキル、(C6-C12)アリール及び(C6-C12)アリール(CO)-からなる群から独立して選択され;
R9は、CH2OH、CH2O(C1-C5)アルキル、CH2O2C(C1-C5)アルキル、CH2O(C7-C10)アラルキル、CH2O(C6-C12)アリール、CO2H、CO2(C1-C5)アルキル、CO2(C6-C12)アリール、CO2(C7-C10)アラルキルからなる群から選択され;並びに、波線は、残りの分子への結合点を示す。)。
【請求項13】
下記式を有する、請求項1記載の化合物:
【化2】
(式中、破線は、単結合、二重結合又は規格化結合を示し;
R1は、H、(C1-C5)アルキルC(O)-、(C7-C10)アラルキルC(O)-、(C7-C10)アラルキル、(C1-C5)アルキル、(C6-C12)アリール、(C6-C12)アリール(CO)-及びシリルからなる群から選択され;
R2は、ハロゲン、(C1-C5)アルコキシ、ニトロ及びCO2Rからなる群から選択される置換基により任意に置換された、(C1-C5)アルキル、(C3-C6)シクロアルキル、(C3-C6)シクロアルキル-アルキル、(C5-C7)シクロアルケニル-アルキル、(C6-C12)アリール、(C7-C12)アラルキル、(C6-C12)ヘテロアリール、(C7-C12)ヘテロアラルキル、(C2-C5)アルケニル、(C2-C5)アルキニルからなる群から選択され;
Rは、H又は(C1-C5)アルキルであり;
R3は、H及びOHからなる群から選択され;
R4は、Hであり;
R5は、Hであるか、又はR6と共に、YR7で置換されたヘテロアリール基を形成し;
R6は、YR7であるか、又はR5と共に、YR7で置換されたヘテロアリール基を形成し;
Yは、-S-及び-NH(CO)(CH2)n-からなる群から選択され;
添字nは、0〜5の整数であり;
R7は、任意に置換されたアリール、任意に置換されたヘテロアリール及び糖類からなる群から選択され;
ここで糖類は、下記式を有し:
【化3】
各R8は、H、(C1-C5)アルキルC(O)-、(C7-C10)アラルキルC(O)-、(C7-C10)アラルキル、(C1-C5)アルキル、(C6-C12)アリール、(C6-C12)アリール(CO)-及びシリルからなる群から独立して選択され;並びに
R9は、CH2OH、CH2O(C1-C5)アルキル、CH2O2C(C1-C5)アルキル、CH2O(C7-C10)アラルキル、CH2O(C6-C12)アリール、CO2H、CO2(C1-C5)アルキル、CO2(C6-C12)アリール、CO2(C7-C10)アラルキルからなる群から選択される。)。
【請求項14】
下記式を有する、請求項1記載の化合物:
【化4】
(式中、R1は、H、(C1-C5)アルキルC(O)-、(C7-C10)アラリル、及び(C1-C5)アルキルからなる群から独立して選択され;
R2は、(C1-C5)アルキル、C3-C6(シクロアルキル)アルキル、C5-C7(シクロアルケニル)アルキル、C6-C12アリール(C7-C12)アラルキル、トランス(C4-C5)アルケニル、アリル、及びフラン-2-イルアルキルからなる群から選択され;
各R8は、H、(C1-C5)アルキルC(O)、(C7-C10)アラリルC(O)、(C7-C10)アラリル、及び(C1-C5)アルキルからなる群から独立して選択され;並びに
R9は、CH2OH、CH2O2C(C1-C5)アルキル、CH2O(C7-C10)アラリル、CO2H、CO2(C1-C5)アルキル、及びCO2(C7-C10)アラルキルからなる群から選択される。)。
【請求項15】
下記式を有する、請求項1記載の化合物:
【化5】
(式中、R1は、H、(C1-C5)アルキルC(O)-、(C7-C10)アラリル、及び(C1-C5)アルキルからなる群から選択され;
R2は、(C1-C5)アルキル、C3-C6(シクロアルキル)アルキル、C5-C7(シクロアルケニル)アルキル、(C6-C12)アリール(C7-C12)アラルキル、トランス(C4-C5)アルケニル、アリル、及びフラン-2-イルアルキルからなる群から選択され;
各R8は、H、(C1-C5)アルキルC(O)、(C7-C10)アラリルC(O)、(C7-C10)アラリル、及び(C1-C5)アルキルからなる群から独立して選択され;並びに
R9は、CH2OH、CH2O2C(C1-C5)アルキル、CH2O(C7-C10)アラリル、CO2H、CO2(C1-C5)アルキル、及びCO2(C7-C10)アラルキルからなる群から選択される。)。
【請求項16】
下記式を有する、請求項1記載の化合物:
【化6】
(式中、R1は、H、(C1-C5)アルキルC(O)-、(C7-C10)アラリル及び(C1-C5)アルキルからなる群から選択され;
R2は、(C1-C5)アルキル、C3-C6(シクロアルキル)アルキル、C5-C7(シクロアルケニル)アルキル、(C6-C12)アリール(C7-C12)アラルキル、トランス(C4-C5)アルケニル、アリル及びフラン-2-イルアルキルからなる群から選択され;
各R8は、H、(C1-C5)アルキルC(O)、(C7-C10)アラリルC(O)、(C7-C10)アラリル、(C1-C5)アルキルからなる群から独立して選択され、
R9は、CH2OH、CH2O2C(C1-C5)アルキル、CH2O(C7-C10)アラリル、CO2H、CO2(C1-C5)アルキル及びCO2(C7-C10)アラルキルからなる群から選択され;
かつ、添字nは、0〜5の整数である。)。
【請求項17】
下記式を有する、請求項1記載の化合物:
【化7】
(式中、R1は、H、(C1-C5)アルキルC(O)-、(C7-C10)アラリル及び(C1-C5)アルキルからなる群から選択され;
R2は、(C1-C5)アルキル、C3-C6(シクロアルキル)アルキル、C5-C7(シクロアルケニル)アルキル、(C6-C12)アリール(C7-C12)アラルキル、トランス(C4-C5)アルケニル、アリル及びフラン-2-イルアルキルからなる群から選択され;
各R8は、H、(C1-C5)アルキルC(O)、(C7-C10)アラリルC(O)、(C7-C10)アラリル、(C1-C5)アルキルからなる群から独立して選択され、
R9は、CH2OH、CH2O2C(C1-C5)アルキル、CH2O(C7-C10)アラリル、CO2H、CO2(C1-C5)アルキル及びCO2(C7-C10)アラルキルからなる群から選択され;
かつ、添字nは、0〜5の整数である。)。
【請求項18】
下記式を有する、請求項1記載の化合物:
【化8】
(式中、R1は、H、(C1-C5)アルキルC(O)-、(C7-C10)アラリル及び(C1-C5)アルキルからなる群から選択され;
R2は、(C1-C5)アルキル、C3-C6(シクロアルキル)アルキル、C5-C7(シクロアルケニル)アルキル、(C6-C12)アリール(C7-C12)アラルキル、トランス(C4-C5)アルケニル、アリル及びフラン-2-イルアルキルからなる群から選択され;
かつ、添字nは、0〜5の整数である。)。
【請求項19】
下記式を有する、請求項1記載の化合物:
【化9】
(式中、R1が、H、(C1-C5)アルキルC(O)-、(C7-C10)アラリル及び(C1-C5)アルキルからなる群から選択され;
R2が、(C1-C5)アルキル、C3-C6(シクロアルキル)アルキル、C5-C7(シクロアルケニル)アルキル、(C6-C12)アリール(C7-C12)アラルキル、トランス(C4-C5)アルケニル、アリル及びフラン-2-イルアルキルからなる群から選択され;
かつ、添字nは、0〜5の整数である。)。
【請求項20】
R2が、シクロプロピルメチル、シクロブチルメチル、及びアリルからなる群から選択される、請求項13記載の化合物。
【請求項21】
以下からなる群から選択される式を有する、請求項1記載の化合物:
【化10】
【請求項22】
請求項1〜21のいずれか1項記載の化合物;及び
医薬として許容し得る賦形剤又は担体;を含有する、医薬組成物。
【請求項23】
疼痛緩和が必要な対象における疼痛を緩和する方法であって、請求項1〜21のいずれか1項記載の化合物の有効量を、疼痛に苦しむ対象へ投与することを含む、前記方法。
【請求項24】
前記作用物質が、オピオイド受容体の効率的な内在化を促進する、請求項23記載の方法。
【請求項25】
前記方法が、末梢の疼痛を治療し、及び物質が生理的pHで一定の電荷を有する、請求項23記載の方法。
【請求項26】
対象における中毒症状を治療する方法であって、請求項1〜21のいずれか1項記載の化合物の有効量を、中毒に苦しむ対象へ投与することを含む、前記方法。
【請求項27】
前記化合物が、オピオイド受容体アンタゴニストである、請求項26記載の方法。
【請求項28】
前記中毒症状が、物質中毒である、請求項26記載の方法。
【請求項29】
前記物質中毒が、麻薬中毒、アルコール中毒、及びニコチン中毒からなる群から選択される、請求項28記載の方法。
【請求項30】
前記中毒症状が、ギャンブル依存症である、請求項26記載の方法。
【請求項1】
下記式の化合物:
A-B-C
(式中、Aは、鎮痛薬又はその誘導体であり;Bは、連結基であり、かつCは、任意に置換されたアリール、任意に置換されたヘテロアリール及び糖類からなる群から選択される);並びに、それらの全ての医薬として許容し得る誘導体。
【請求項2】
前記鎮痛薬又はその誘導体が、オピエートである、請求項1記載の化合物。
【請求項3】
前記オピエートが、ナルトレキソン、フェニルピペリジン、ピペリジノール、プロジン、ピペリジルプロピオンアニリド、イソプロジン、プロジリジン、ベンゾモルファン、モルファン、アザビシクラン、モルヒナン、プロジン、ジフェニルアミノエチルプロピオネート、メサドン、イソメサドン、プロポキシフェン、デキストロメトロファン、ベンザゾシン-8-オール、ノルビナルトロフィン、ナルトリンドール及びグアニジンナルトリンドールからなる群から選択される、請求項2記載の化合物。
【請求項4】
前記連結基が、-S-及び-NH(CO)(CH2)n-からなる群から選択され;及び、添字nは、0〜5の整数である、請求項1記載の化合物。
【請求項5】
前記連結基が-S-である、請求項1記載の化合物。
【請求項6】
前記連結基が、-NH(CO)(CH2)nであり;かつ、添字nが、0〜5の整数である、請求項1記載の化合物。
【請求項7】
Cが、ハロゲン、(C1-C5)アルコキシ、ニトロ、CO2Rにより任意に置換されたアリールであり、かつRが、H又は(C1-C5)アルキルである、請求項1記載の化合物。
【請求項8】
Cが、フェニルである、請求項1記載の化合物。
【請求項9】
Cが、ハロゲン、(C1-C5)アルコキシ、ニトロ、CO2Rにより任意に置換されたヘテロアリールであり、かつRが、H又は(C1-C5)アルキルである、請求項1記載の化合物。
【請求項10】
Cが、チオフェンである、請求項1記載の化合物。
【請求項11】
Cが、糖類である、請求項1記載の化合物。
【請求項12】
前記糖類が、下記式を有する、請求項1記載の化合物:
【化1】
(式中、各R8は、H、(C1-C5)アルキルC(O)-、(C7-C10)アラルキルC(O)-、(C7-C10)アラルキル、(C1-C5)アルキル、(C6-C12)アリール及び(C6-C12)アリール(CO)-からなる群から独立して選択され;
R9は、CH2OH、CH2O(C1-C5)アルキル、CH2O2C(C1-C5)アルキル、CH2O(C7-C10)アラルキル、CH2O(C6-C12)アリール、CO2H、CO2(C1-C5)アルキル、CO2(C6-C12)アリール、CO2(C7-C10)アラルキルからなる群から選択され;並びに、波線は、残りの分子への結合点を示す。)。
【請求項13】
下記式を有する、請求項1記載の化合物:
【化2】
(式中、破線は、単結合、二重結合又は規格化結合を示し;
R1は、H、(C1-C5)アルキルC(O)-、(C7-C10)アラルキルC(O)-、(C7-C10)アラルキル、(C1-C5)アルキル、(C6-C12)アリール、(C6-C12)アリール(CO)-及びシリルからなる群から選択され;
R2は、ハロゲン、(C1-C5)アルコキシ、ニトロ及びCO2Rからなる群から選択される置換基により任意に置換された、(C1-C5)アルキル、(C3-C6)シクロアルキル、(C3-C6)シクロアルキル-アルキル、(C5-C7)シクロアルケニル-アルキル、(C6-C12)アリール、(C7-C12)アラルキル、(C6-C12)ヘテロアリール、(C7-C12)ヘテロアラルキル、(C2-C5)アルケニル、(C2-C5)アルキニルからなる群から選択され;
Rは、H又は(C1-C5)アルキルであり;
R3は、H及びOHからなる群から選択され;
R4は、Hであり;
R5は、Hであるか、又はR6と共に、YR7で置換されたヘテロアリール基を形成し;
R6は、YR7であるか、又はR5と共に、YR7で置換されたヘテロアリール基を形成し;
Yは、-S-及び-NH(CO)(CH2)n-からなる群から選択され;
添字nは、0〜5の整数であり;
R7は、任意に置換されたアリール、任意に置換されたヘテロアリール及び糖類からなる群から選択され;
ここで糖類は、下記式を有し:
【化3】
各R8は、H、(C1-C5)アルキルC(O)-、(C7-C10)アラルキルC(O)-、(C7-C10)アラルキル、(C1-C5)アルキル、(C6-C12)アリール、(C6-C12)アリール(CO)-及びシリルからなる群から独立して選択され;並びに
R9は、CH2OH、CH2O(C1-C5)アルキル、CH2O2C(C1-C5)アルキル、CH2O(C7-C10)アラルキル、CH2O(C6-C12)アリール、CO2H、CO2(C1-C5)アルキル、CO2(C6-C12)アリール、CO2(C7-C10)アラルキルからなる群から選択される。)。
【請求項14】
下記式を有する、請求項1記載の化合物:
【化4】
(式中、R1は、H、(C1-C5)アルキルC(O)-、(C7-C10)アラリル、及び(C1-C5)アルキルからなる群から独立して選択され;
R2は、(C1-C5)アルキル、C3-C6(シクロアルキル)アルキル、C5-C7(シクロアルケニル)アルキル、C6-C12アリール(C7-C12)アラルキル、トランス(C4-C5)アルケニル、アリル、及びフラン-2-イルアルキルからなる群から選択され;
各R8は、H、(C1-C5)アルキルC(O)、(C7-C10)アラリルC(O)、(C7-C10)アラリル、及び(C1-C5)アルキルからなる群から独立して選択され;並びに
R9は、CH2OH、CH2O2C(C1-C5)アルキル、CH2O(C7-C10)アラリル、CO2H、CO2(C1-C5)アルキル、及びCO2(C7-C10)アラルキルからなる群から選択される。)。
【請求項15】
下記式を有する、請求項1記載の化合物:
【化5】
(式中、R1は、H、(C1-C5)アルキルC(O)-、(C7-C10)アラリル、及び(C1-C5)アルキルからなる群から選択され;
R2は、(C1-C5)アルキル、C3-C6(シクロアルキル)アルキル、C5-C7(シクロアルケニル)アルキル、(C6-C12)アリール(C7-C12)アラルキル、トランス(C4-C5)アルケニル、アリル、及びフラン-2-イルアルキルからなる群から選択され;
各R8は、H、(C1-C5)アルキルC(O)、(C7-C10)アラリルC(O)、(C7-C10)アラリル、及び(C1-C5)アルキルからなる群から独立して選択され;並びに
R9は、CH2OH、CH2O2C(C1-C5)アルキル、CH2O(C7-C10)アラリル、CO2H、CO2(C1-C5)アルキル、及びCO2(C7-C10)アラルキルからなる群から選択される。)。
【請求項16】
下記式を有する、請求項1記載の化合物:
【化6】
(式中、R1は、H、(C1-C5)アルキルC(O)-、(C7-C10)アラリル及び(C1-C5)アルキルからなる群から選択され;
R2は、(C1-C5)アルキル、C3-C6(シクロアルキル)アルキル、C5-C7(シクロアルケニル)アルキル、(C6-C12)アリール(C7-C12)アラルキル、トランス(C4-C5)アルケニル、アリル及びフラン-2-イルアルキルからなる群から選択され;
各R8は、H、(C1-C5)アルキルC(O)、(C7-C10)アラリルC(O)、(C7-C10)アラリル、(C1-C5)アルキルからなる群から独立して選択され、
R9は、CH2OH、CH2O2C(C1-C5)アルキル、CH2O(C7-C10)アラリル、CO2H、CO2(C1-C5)アルキル及びCO2(C7-C10)アラルキルからなる群から選択され;
かつ、添字nは、0〜5の整数である。)。
【請求項17】
下記式を有する、請求項1記載の化合物:
【化7】
(式中、R1は、H、(C1-C5)アルキルC(O)-、(C7-C10)アラリル及び(C1-C5)アルキルからなる群から選択され;
R2は、(C1-C5)アルキル、C3-C6(シクロアルキル)アルキル、C5-C7(シクロアルケニル)アルキル、(C6-C12)アリール(C7-C12)アラルキル、トランス(C4-C5)アルケニル、アリル及びフラン-2-イルアルキルからなる群から選択され;
各R8は、H、(C1-C5)アルキルC(O)、(C7-C10)アラリルC(O)、(C7-C10)アラリル、(C1-C5)アルキルからなる群から独立して選択され、
R9は、CH2OH、CH2O2C(C1-C5)アルキル、CH2O(C7-C10)アラリル、CO2H、CO2(C1-C5)アルキル及びCO2(C7-C10)アラルキルからなる群から選択され;
かつ、添字nは、0〜5の整数である。)。
【請求項18】
下記式を有する、請求項1記載の化合物:
【化8】
(式中、R1は、H、(C1-C5)アルキルC(O)-、(C7-C10)アラリル及び(C1-C5)アルキルからなる群から選択され;
R2は、(C1-C5)アルキル、C3-C6(シクロアルキル)アルキル、C5-C7(シクロアルケニル)アルキル、(C6-C12)アリール(C7-C12)アラルキル、トランス(C4-C5)アルケニル、アリル及びフラン-2-イルアルキルからなる群から選択され;
かつ、添字nは、0〜5の整数である。)。
【請求項19】
下記式を有する、請求項1記載の化合物:
【化9】
(式中、R1が、H、(C1-C5)アルキルC(O)-、(C7-C10)アラリル及び(C1-C5)アルキルからなる群から選択され;
R2が、(C1-C5)アルキル、C3-C6(シクロアルキル)アルキル、C5-C7(シクロアルケニル)アルキル、(C6-C12)アリール(C7-C12)アラルキル、トランス(C4-C5)アルケニル、アリル及びフラン-2-イルアルキルからなる群から選択され;
かつ、添字nは、0〜5の整数である。)。
【請求項20】
R2が、シクロプロピルメチル、シクロブチルメチル、及びアリルからなる群から選択される、請求項13記載の化合物。
【請求項21】
以下からなる群から選択される式を有する、請求項1記載の化合物:
【化10】
【請求項22】
請求項1〜21のいずれか1項記載の化合物;及び
医薬として許容し得る賦形剤又は担体;を含有する、医薬組成物。
【請求項23】
疼痛緩和が必要な対象における疼痛を緩和する方法であって、請求項1〜21のいずれか1項記載の化合物の有効量を、疼痛に苦しむ対象へ投与することを含む、前記方法。
【請求項24】
前記作用物質が、オピオイド受容体の効率的な内在化を促進する、請求項23記載の方法。
【請求項25】
前記方法が、末梢の疼痛を治療し、及び物質が生理的pHで一定の電荷を有する、請求項23記載の方法。
【請求項26】
対象における中毒症状を治療する方法であって、請求項1〜21のいずれか1項記載の化合物の有効量を、中毒に苦しむ対象へ投与することを含む、前記方法。
【請求項27】
前記化合物が、オピオイド受容体アンタゴニストである、請求項26記載の方法。
【請求項28】
前記中毒症状が、物質中毒である、請求項26記載の方法。
【請求項29】
前記物質中毒が、麻薬中毒、アルコール中毒、及びニコチン中毒からなる群から選択される、請求項28記載の方法。
【請求項30】
前記中毒症状が、ギャンブル依存症である、請求項26記載の方法。
【図1】

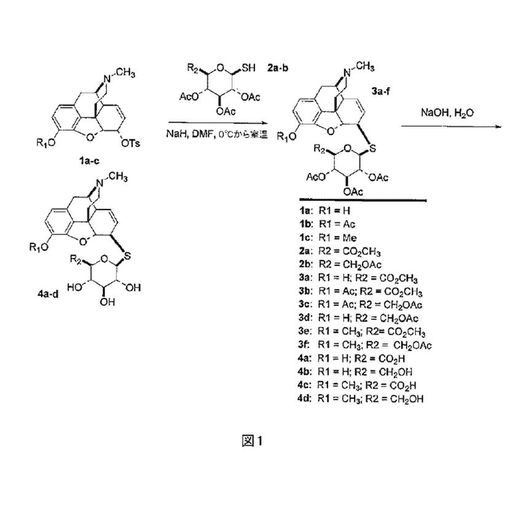
【図2】
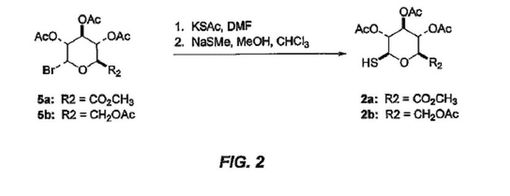
【図3】
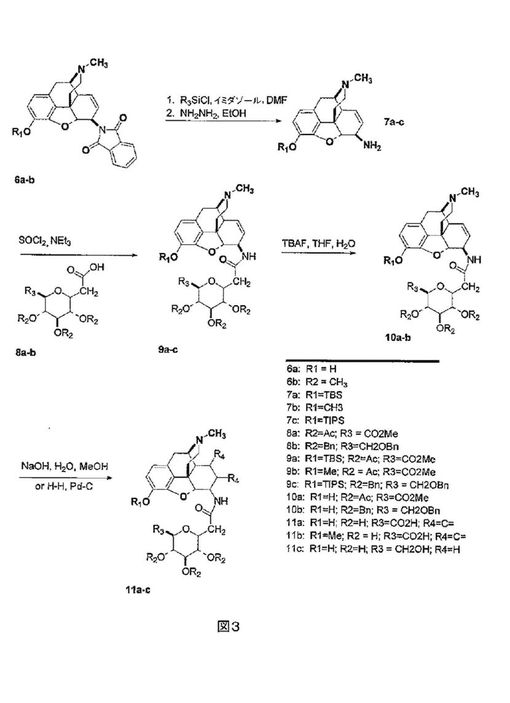
【図4】
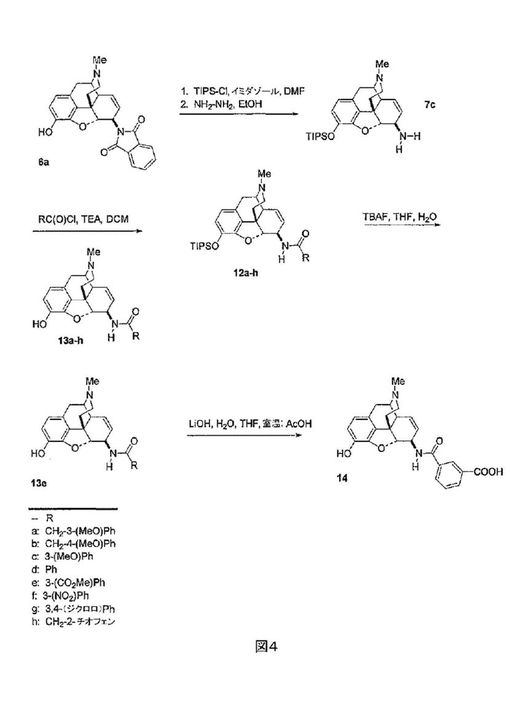
【図5】

【図6】
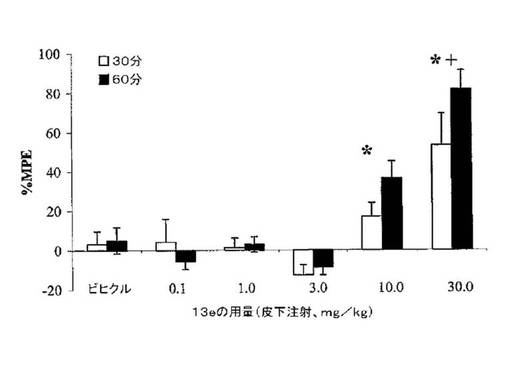
【図7】
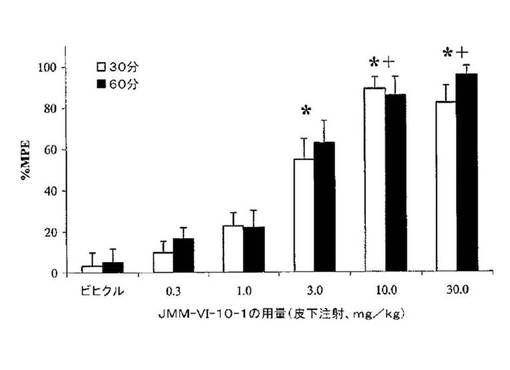
【図8A】

【図8B】
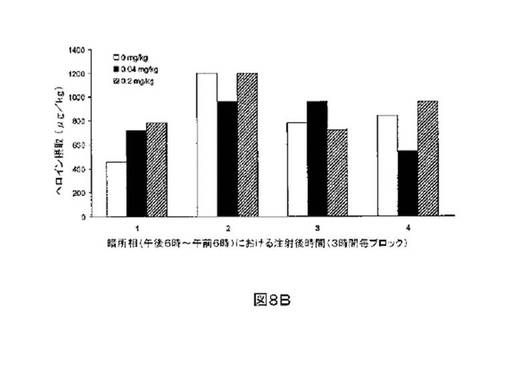
【図8C】

【図9A】
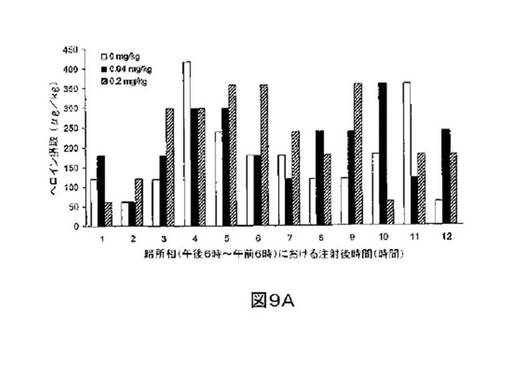
【図9B】

【図9C】
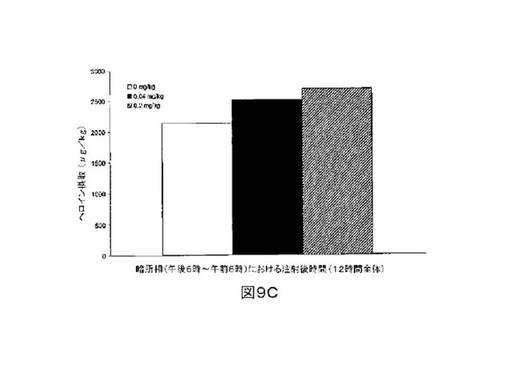
【図10A】
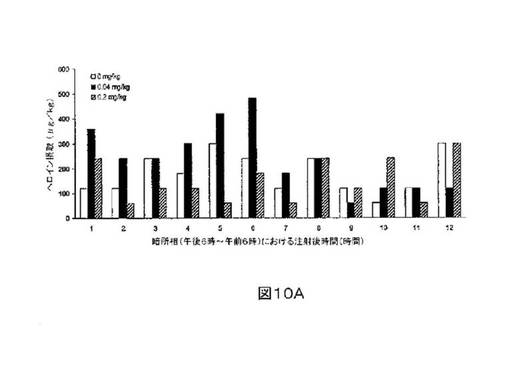
【図10B】

【図10C】
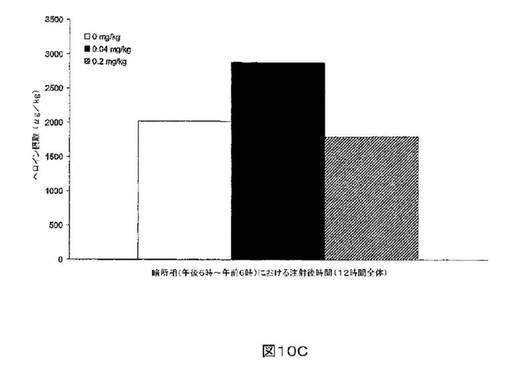
【図11A】
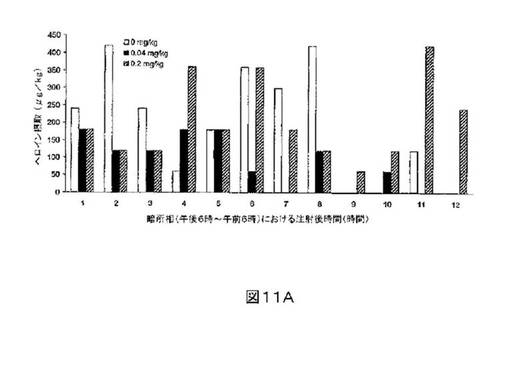
【図11B】

【図11C】
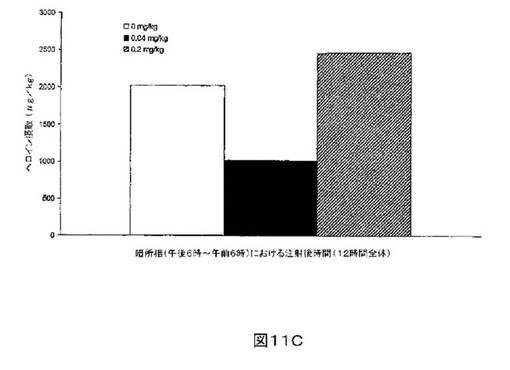
【図12A】
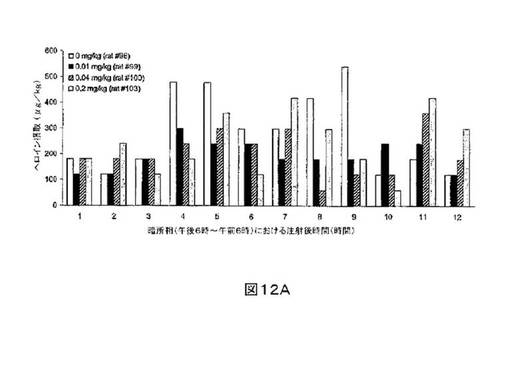
【図12B】
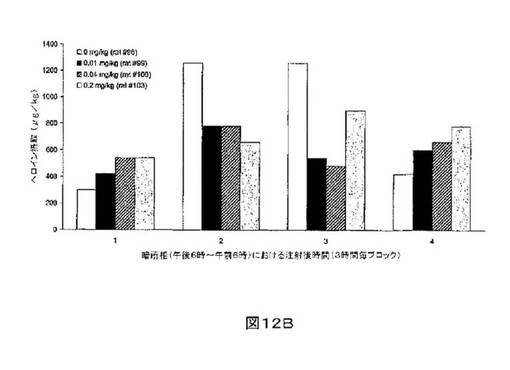
【図12C】
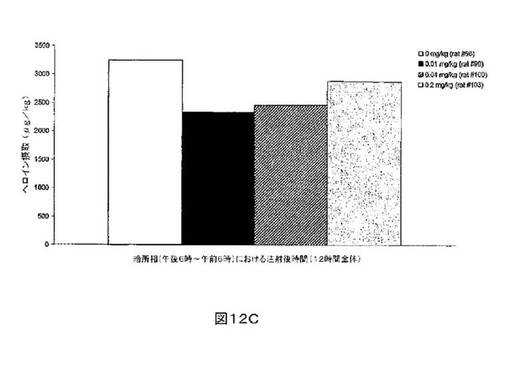
【図13】
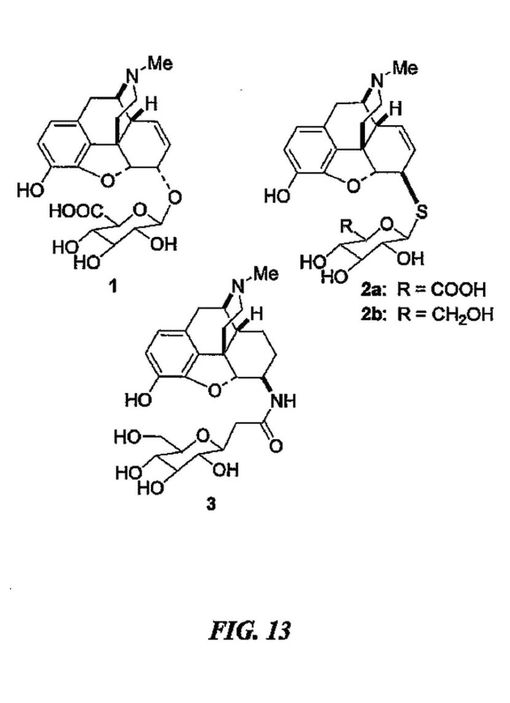
【図14】

【図15】
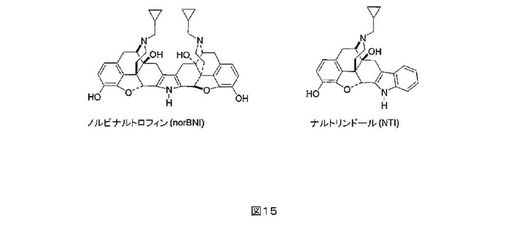
【図16】
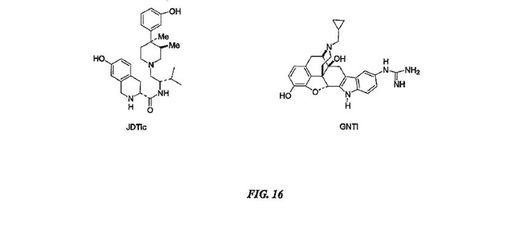
【図17】

【図18】
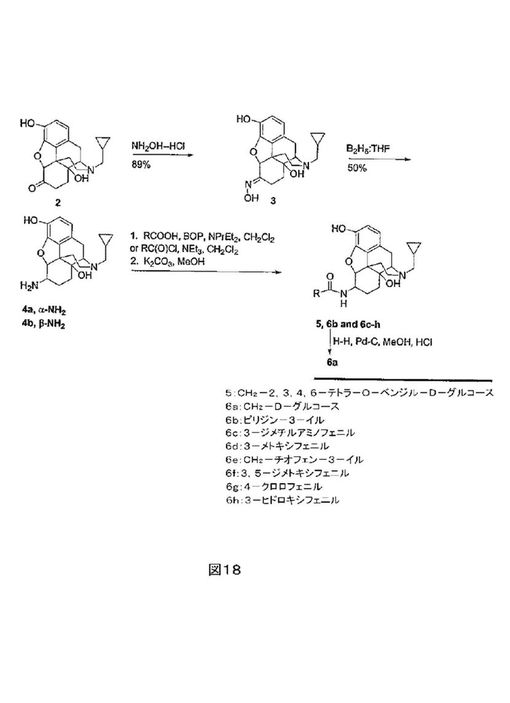
【図19】
【図20】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8A】
【図8B】
【図8C】
【図9A】
【図9B】
【図9C】
【図10A】
【図10B】
【図10C】
【図11A】
【図11B】
【図11C】
【図12A】
【図12B】
【図12C】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【公開番号】特開2012−254984(P2012−254984A)
【公開日】平成24年12月27日(2012.12.27)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−139959(P2012−139959)
【出願日】平成24年6月21日(2012.6.21)
【分割の表示】特願2007−515474(P2007−515474)の分割
【原出願日】平成17年5月31日(2005.5.31)
【出願人】(506392861)ヒューマン バイオモレキュラル リサーチ インスティテュート (5)
【Fターム(参考)】
【公開日】平成24年12月27日(2012.12.27)
【国際特許分類】
【出願番号】特願2012−139959(P2012−139959)
【出願日】平成24年6月21日(2012.6.21)
【分割の表示】特願2007−515474(P2007−515474)の分割
【原出願日】平成17年5月31日(2005.5.31)
【出願人】(506392861)ヒューマン バイオモレキュラル リサーチ インスティテュート (5)
【Fターム(参考)】
[ Back to top ]