
低置換度ヒドロキシプロピルセルロース粉末及び固形製剤
【課題】結合性が高く、流動性が良好で崩壊性に優れる低置換度ヒドロキシプロピルセルロース粉末を提供する。
【解決手段】平均粒子径が10〜100μmであり、かつBET法で測定した比表面積が1.0m2/g以上であり、圧縮圧50MPaで圧縮成型した時の弾性回復率が7%以下である低置換度ヒドロキシプロピルセルロース粉末を提供する。
【解決手段】平均粒子径が10〜100μmであり、かつBET法で測定した比表面積が1.0m2/g以上であり、圧縮圧50MPaで圧縮成型した時の弾性回復率が7%以下である低置換度ヒドロキシプロピルセルロース粉末を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、医薬品又は食品分野等において製剤を製造する際に崩壊性又は結合性を付与するために添加する低置換度ヒドロキシプロピルセルロース関するものである。特に結合性、崩壊性に優れたものに関するものである。
【背景技術】
【0002】
医薬品又は食品分野等の固形製剤において、主薬のみで作製された製剤では、薬物を投与しても十分な崩壊性が得られず、薬効が十分に発揮されない場合や結合性が劣るため錠剤や顆粒剤とした際にその形状を保つことができない等の問題がある。このような場合に低置換度ヒドロキシプロピルセルロース、カルボキシメチルセルロースのカルシウム塩、架橋カルボキシメチルセルロースナトリウム、架橋ポリビニルピロリドン、カルボキシメチルスターチ等の崩壊剤を添加することにより崩壊性を改善することができる。また、結合性を改良するためには結晶セルロースを添加したり、水溶性の結合剤を添加することにより改善することができる。低置換度ヒドロキシプロピルセルロースは上記の崩壊性と結合性を合わせ持つユニークな添加剤として知られている。
【0003】
低置換度ヒドロキシプロピルセルロースは非イオン性であるため、イオン性の薬物等との反応による変質が起きにくい等の利点を有する。低置換度ヒドロキシプロピルセルロースを医薬品の添加剤として使用しうることは特許文献1〜2に記載されている。
【0004】
特許文献1〜4及び特許文献7に記載されているように、アルカリセルロースの調製方法としてシート状のパルプを苛性ソーダ水溶液に浸せき後、圧搾して製造する方法があるが、この方法においては一旦シート状のパルプを苛性ソーダ水溶液に浸せきすると過剰量の苛性ソーダ水溶液がパルプに吸液され、圧搾しても過剰の水又は苛性ソーダを含むアルカリセルロースしか製造できず、副反応への選択率が上昇し、主反応であるエーテル化反応の効率は低いものであった。
【0005】
従来の低置換度ヒドロキシプロピルセルロースは、特許文献3に記載されているように、アルカリセルロースとプロピレンオキサイドを反応させることによって得ることができる。エーテル化反応においては、主反応としてはセルロースのエーテル化があり、副反応としてはプロピレンオキサイドと水との反応によりプロピレングリコールを生成する反応がある。このエーテル化反応に利用されるプロピレンオキサイドの量は、反応触媒として利用される苛性ソーダ水溶液の量により変化し、アルカリセルロース中の水又は苛性ソーダが多すぎると反応効率が低下する問題があった。
【0006】
一方、粉末状のパルプに苛性ソーダ水溶液を滴下又は噴霧して混合することにより製造する場合、アルカリセルロース中の水又は苛性ソーダ量を任意に調製できる利点がある。
また、特許文献3に記載されているように、粗反応生成物中に残存するアルカリを中和する際に酸を含む熱水中に反応生成物を投入して粗反応生成物を溶解させる方法を用いると、得られた低置換度ヒドロキシプロピルセルロースは原料パルプと同様に繊維状の形態となり洗浄性に優れ、精製が容易であるが、その反面粉砕性に劣り流動性の優れた粉体を得ることができない。
【0007】
特許文献2には、エーテル化反応終了後に、反応触媒として用いられたアルカリを水中で部分的に中和し、低置換度ヒドロキシプロピルセルロースを一部溶解させて、繊維質部分を制御する方法が記載されている。この方法では残存するアルカリ量を多くすると、繊維状形態が溶解により減少し、得られた粉砕物の嵩密度は高く、流動性が良好になるが、精製時に洗浄性が低下してしまう問題があることが記載されている。
【0008】
特許文献3では、洗浄脱水物の含水率を70〜90質量%とすることにより、粉砕性が向上し、42%以下の圧縮度と48o以下の安息角を有する低置換度ヒドロキシプロピルセルロース粉体を得ることができることが記載されている。また、粉砕はハンマーミル等の衝撃型粉砕機によって行われることが記載されている。
しかし、衝撃粉砕機では粉砕原料の影響を受けやすく、粉砕原料の形状により、製品の粉体物性が決定される。つまり、特許文献2にあるように部分中和量を多くしたり、溶解工程を経ず、粗反応生成物に残存する苛性ソーダに対して当量の酸を含む水又は熱水中に反応生成物を投入し中和を行うことにより得られた繊維状形態の原料を用いて衝撃粉砕機で粉砕した場合、得られた粉砕物は繊維状粒子が多く含まれる流動性の低い粉体しか得られなかった。また、短時間で高エネルギーを与えて粉砕するため、処理量が低く、生産性の低い方法であった。
また、上記のような方法で製造された低置換度ヒドロキシプロピルセルロースは粉体の流動性を高めるために部分中和量を少なくして溶解させることにより、繊維質部分が少なくなり結合性が低下してしまう問題があった。また、結合性を高めるために部分中和量を多くして繊維質部分を多くすると流動性が低下する問題もあった。
特許文献3の洗浄脱水物の含水率を70〜90質量%に制御して得られたものは繊維状形態の粒子と球状粒子の混在する粉体である。この繊維状形態の粒子の影響を受け流動性は不十分で直接打錠法で製錠する場合、錠剤の質量偏差が大きくなることがあった。更に、球状粒子の結合性が低いため、この混在物である粉体の結合性は不十分であった。
【0009】
特許文献4においては繊維状粒子を完全溶解させることにより、流動性の高い低置換度ヒドロキシプロピルセルロースを得ることができることが記載されているが、この方法で得られた粉体の結合性は低いものであった。
更に、特許文献2において、粉砕を衝撃粉砕に換えてボールミルで行うと結合性に劣ることが記載されている。また、ボールミルで粉砕されたものは崩壊剤として重要な特性である膨潤特性に劣るものであった。
【0010】
特許文献5においては、パルプを粉末化する際に竪型ローラーミルを用いることにより、粉末化パルプの嵩密度を上昇させ、水に溶解した時の未溶解繊維分が少ない水溶性セルロースエーテルについて記載されている。
しかし、水溶性セルロースエーテルは水不溶性の低置換度ヒドロキシプロピルセルロースの製造において必須の中和工程を含んでいない点で異なり、かつ、特許文献5では低置換度ヒドロキシプロピルセルロースの重要な特性である結合性及び崩壊性に関して何ら記載されていない。
【0011】
特許文献6において竪型ローラーミルを用いてセルロースエーテルを製造する方法が記載されているが、これも同様に水溶性セルロースエーテルに関するもので水不溶性である低置換度ヒドロキシプロピルセルロースに関するものではない。
【0012】
特許文献7には安息角45o以下で膨潤するときの体積増加率100%以上の低置換度ヒドロキシプロピルセルロースに関する記載があるが、この低置換度ヒドロキシプロピルセルロースの結合性は低いものであった。
【特許文献1】特公昭48−38858号公報
【特許文献2】特公昭57−53100号公報
【特許文献3】特開平10−305084号公報
【特許文献4】特開平11−322802号公報
【特許文献5】特開2001−9316号公報
【特許文献6】特開平9−76233号公報
【特許文献7】特開平7−324101号公報
【発明の開示】
【発明が解決しようとする課題】
【0013】
本発明は、上記従来技術の欠点を解消するためになされたものであり、結合性が高く、流動性が良好で崩壊性に優れる低置換度ヒドロキシプロピルセルロース粉末を提供することを目的とする。
【課題を解決するための手段】
【0014】
本発明者らは、上記目的を達成するため鋭意検討した結果、低置換度ヒドロキシプロピルセルロースの製造において、粉末化したパルプに苛性ソーダ水溶液を添加混合し、無水セルロースに対する苛性ソーダの質量比が0.1〜0.3であるアルカリセルロースを製造し、次いでエーテル化反応を行った後、溶解工程を経るか又は経ずに苛性ソーダを中和し、洗浄乾燥後、粉砕工程において圧密摩砕することにより、結合性が高く、流動性が良好で崩壊性に優れる低置換度ヒドロキシプロピルセルロース粉末を得ることを見出した。
【0015】
本発明は、平均粒子径が10〜100μmであり、かつBET法で測定した比表面積が1.0m2/g以上である低置換度ヒドロキシプロピルセルロース粉末を提供する。低置換度ヒドロキシプロピルセルロース粉末は、好ましくは、圧縮圧50MPaで圧縮成型した時の弾性回復率が7%以下である。低置換度ヒドロキシプロピルセルロース粉末は、好ましくは、吸水時の膨潤体積増加率が300%以上で、膨潤体積増加速度が100%/分以上である。低置換度ヒドロキシプロピルセルロース粉末は、好ましくは、安息角が42o以下である。
さらに、本発明は、この低置換度ヒドロキシプロピルセルロース粉末を用いた固形製剤を提供する。
本明細書は、無水グルコース単位あたりの置換モル数が0.05〜1.0である水不溶性で吸水膨潤性を有する低置換度ヒドロキシプロピルセルロース粉末の製造方法であって、(1)粉末化したパルプに無水セルロースに対する苛性ソーダの質量比が0.1〜0.3となる量で苛性ソーダ水溶液を添加混合してアルカリセルロースを製造する工程と、(2)得られたアルカリセルロースのエーテル化反応を行って粗反応物を得る工程と、(3)得られた粗反応物中に含有される苛性ソーダを中和する工程と、(4)その後の洗浄・脱水工程と、(5)乾燥工程と、(6)圧密摩砕を行う粉砕工程とを含んでなる低置換度ヒドロキシプロピルセルロース粉末の製造方法を記載する。また、無水グルコース単位あたりの置換モル数が0.05〜1.0である水不溶性で吸水膨潤性を有する低置換度ヒドロキシプロピルセルロース粉末の製造方法であって、(1)粉末化したパルプに無水セルロースに対する苛性ソーダの質量比が0.1〜0.3となる量で苛性ソーダ水溶液を添加混合してアルカリセルロースを製造する工程と、(2)得られたアルカリセルロースのエーテル化反応を行って粗反応物を得る工程と、(3)粗反応物の一部又は全部を溶解する溶解工程を経ることなく、得られた粗反応物中に含有される苛性ソーダを中和する工程と、(4)その後の洗浄・脱水工程と、(5)乾燥工程と、(6)圧密摩砕を行う粉砕工程とを含んでなる低置換度ヒドロキシプロピルセルロース粉末の製造方法を記載する。
上記洗浄・脱水工程は、好ましくは、洗浄するとともに含水率を65質量%以下とする脱水を行う工程である。
【発明の効果】
【0016】
本発明の低置換度ヒドロキシプロピルセルロース粉末は、原料パルプ由来の繊維状形態にも拘わらず流動性が高く、結合性に優れ、更に膨潤特性に優れるものである。
また、本発明の低置換度ヒドロキシプロピルセルロース粉末は、結合性及び崩壊性に優れるため、錠剤中の添加量を削減でき錠剤のサイズを小さくできる利点も有する。
【図面の簡単な説明】
【0017】
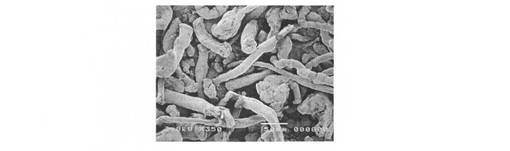
【図1】実施例1で得られた粉体の電子顕微鏡写真を示す。
【図2】比較例4で得られた粉体の電子顕微鏡写真を示す。
【図3】比較例5で得られた粉体の電子顕微鏡写真を示す。
【図4】比較例6で得られた粉体の電子顕微鏡写真を示す。
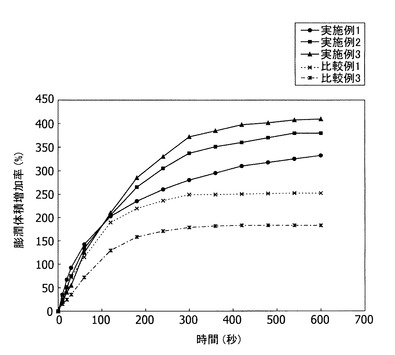
【図5】実施例1〜3と比較例1と3で得られた粉体の時間の経過に伴う膨潤体積増加率を示す。
【発明を実施するための最良の形態】
【0018】
本発明の方法について以下に説明する。まず、原料として使用される粉末状のパルプは何れの粉砕方式を用いても良い。その平均粒子径は、好ましくは60〜300μmである。60μm未満の粉末状パルプを調製するには工業的に非効率的であり、300μmを超えると苛性ソーダ水溶液との混合性に劣る恐れがある。
【0019】
アルカリセルロースを製造する工程は、好ましくは、上記粉末状のパルプに苛性ソーダ水溶液を滴下又は噴霧して混合することにより行う。この際、苛性ソーダはエーテル化反応の触媒として作用する。アルカリセルロースの製造は、好ましくは、内部撹拌型の反応機内で混合を行い、引き続いてエーテル化反応を行うか、他の混合機内で調製したアルカリセルロースを反応機内に仕込んでエーテル化反応を行うか何れの方法を用いても良い。
【0020】
また、アルカリセルロース中の苛性ソーダ量は、反応効率への影響のみではなく、最終製品の膨潤特性及び結合性に影響を与えることが解った。アルカリセルロース中の最適な苛性ソーダ量は、無水セルロース(パルプ中の水分を除いたものをいう。)に対する苛性ソーダの質量比で0.1〜0.3である。0.1未満では膨潤特性の特に吸水膨潤時の体積増加率が低くなり、崩壊性が低下し、結合性も低下する場合がある。また、0.3を超えると後述の吸水時の膨潤体積増加率及び膨潤体積増加速度も低くなり、結合性も低下する場合がある。
苛性ソーダは、好ましくは20〜40質量%の水溶液として添加される。
【0021】
次のエーテル化反応を行う工程は、アルカリセルロースを反応機内に仕込み、窒素置換後、エーテル化剤としてプロピレンオキサイドを反応機内に仕込み反応を行う。プロピレンオキサイドの仕込み比は、好ましくは、無水グルコース単位1モルに対して0.1〜1.0モル程度である。反応温度は40〜80℃程度、反応時間は1〜5時間程度である。
【0022】
なお、エーテル化反応を行う工程の後、必要に応じて溶解工程を行うことができる。溶解工程は、エーテル化反応後の粗反応物の一部又は全部を水又は熱水に溶解することにより行われる。水又は熱水の使用量は、粗反応物の溶解量によって異なるが、粗反応物の全部を溶解させるときの水の量は、通常、粗反応物中の低置換度ヒドロキシプロピルセルロースに対して質量比で0.5〜10である。
後述の洗浄・脱水工程における負荷及び低置換度セルロースエーテル結合性の更なる向上を考慮すると、この溶解工程を行わない方がより好ましい。
【0023】
次に行われる中和工程は、触媒として使用した苛性ソーダが反応生成物に残存するため、好ましくは、その苛性ソーダに対して当量の酸を含む水又は熱水中に粗反応生成物を投入し中和を行う。また、反応生成物に当量の酸を含む水又は熱水を加えて中和を行っても良い。
使用する酸は、塩酸、硫酸、硝酸等の鉱酸やギ酸、酢酸等の有機酸が挙げられる。
【0024】
次の洗浄・脱水工程では、得られた中和物を、好ましくは水又は熱水を用いて洗浄しながら、好ましくは遠心分離、減圧濾過、加圧濾過等から選ばれる方法で脱水を行う。得られた脱水物ケーキ中の低置換度ヒドロキシプロピルセルロースは原料パルプの形態と同様に繊維状ものとなる。溶解工程を経た脱水物では置換モル数にもよるが、脱水率が概ね70〜90質量%であり、溶解工程を経ない脱水率は通常65質量%以下となり、その後の乾燥工程での負荷が低減でき生産性が向上する。更に、溶解工程が無いため工程を簡略化できる利点を有する。
また、製品の結合性の観点から繊維状形態のものを使用して粉砕した方が得られた製品の比表面積が高く、結合性の高いものが得られる。
【0025】
上記で得られた脱水物を乾燥する乾燥工程は、好ましくは、流動層乾燥機、ドラムドライヤー等の乾燥機を用いて60〜120℃にて行うことができる。
【0026】
粉砕工程は、上記方法で得られた乾燥物を圧密摩砕することにより行われる。
この圧密摩砕には、ローラーミル、ボールミル、ビーズミル、石臼型粉砕機等の粉砕機が利用できる。ローラミルは、ローラー又はボールが、その回転運動に伴う遠心力や重力荷重により、ミル壁の被粉砕物を圧縮・剪断しながら転がる粉砕機で、石川島播磨重工業社製ISミル、栗本鐵工所社製VXミル、増野製作所社製MSローラーミル等が利用できる。ボールミルは、鋼球、磁性ボール、玉石及びその類似物を粉砕媒体とする粉砕機で、栗本鉄工社製ボールミル、大塚鉄工社製チューブミル、FRITSCH社製遊星ボールミル等が利用できる。ビーズミルはボールミルと類似するが、使われるボールの径が小さく、機器内部が高速回転することにより、ボールの加速度をより高めることができる点で異なり、例えばアシザワ製作所社製のビーズミルが利用できる。石臼型粉砕機は、石臼が狭いクリアランスで高速回転することにより粉体を摩砕することができる機械で、例えば増幸産業社製のセレンディピターが利用できる。
特に金属異物の混入が少なく、設置面積が小さく、生産性の高いローラーミルが好ましい。
【0027】
なお、従来の衝撃粉砕より製造される低置換度ヒドロキシプロピルセルロースの結合性は、繊維状形態の絡み合いにより発現されると言われてきた。そのため、結合性を高めるために繊維状粒子を多くすると流動性は低下してしまった。しかし、本発明の低置換度ヒドロキシプロピルセルロース粉末は圧密摩砕されることにより繊維状形態が消失しているにも拘わらず、驚くべきことに高い結合性を示すものであった。
【0028】
従来技術における製品の粉体物性の調製は、特許文献2及び特許文献3にあるようにエーテル化反応終了後に、反応触媒として用いられた苛性ソーダを水中で部分的に中和し、低置換度ヒドロキシプロピルセルロースを一部溶解させて、繊維状粒子の量を制御することにより行われていた。また、特許文献4では低置換度ヒドロキシプロピルセルロースを完全溶解させることにより、流動性の高い粉体を調製していた。そして、いずれの粉砕も衝撃力を利用したインパクトミル等の衝撃粉砕機を用いることにより行われてきた。
衝撃粉砕機では、粉砕原料の影響を受けやすく、粉砕原料の形状により、製品の粉体物性が決定される。つまり、特許文献2又は特許文献3にあるように部分中和量を多くしたり、本発明の実施の一形態である溶解工程を経ず、反応生成物に残存する苛性ソーダに対して当量の酸を含む水又は熱水中に反応生成物を投入し中和を行うことにより得られた繊維状形態の原料を用いた場合、得られた粉砕物は繊維状粒子が多く含まれる流動性の低い粉体しか得られなかった。
【0029】
本発明によれば、粉砕原料である繊維状形態の粒子は圧密摩砕を繰り返すことにより、原料パルプ由来の繊維状で中空の管状形態が消失することにより、1次粒子を小さくすることができるため、比表面積が増大する。また、原料パルプ由来の繊維状形態が消失し、粒子形状の揃った粉体が得られる。
【0030】
次に、好ましくは、粉砕物を定法に従い篩い分けを行うことにより目的の低置換度ヒドロキシプロピルセルロース粉末を得ることができる。篩いの目開きは、75〜180μm程度のものが使用できる。
本発明の低置換度ヒドロキシプロピルセルロース粉末の平均粒子径は、好ましくは10〜100μm程度であり、より好ましくは20〜60μm程度である。10μm未満では微粉化により凝集性が増し、粉体の流動性が低下する恐れがあり、100μmを超えると薬物との混合性が低下して不均一となる恐れがある。
【0031】
本発明の低置換度ヒドロキシプロピルセルロース粉末の比表面積は1.0m2/g以上が好ましい。これ未満では高い結合性が得られない恐れがある。
一般的に粉体の比表面積が高い程、結合性が高い粉体となることが知られている。比表面積分析は、粉体粒子表面に吸着占有面積の判った分子を液体窒素の温度で吸着させ、その量から試料の比表面積を求める方法であり、不活性気体の低温低湿物理吸着によるBET法を用いることができる。例えば、MICROMERITICS GEMINI 2375(島津製作所社製)を用いて測定できる。
一般的には平均粒子径を小さくすることにより比表面積を増大させることができるが、上記記載のように平均粒子径が小さくなり過ぎると粉体の凝集性が増し、粉体の流動性が低下する恐れがある。本発明においては、圧密摩砕により、粉体の流動性が得られる平均粒子径でありながら高い比表面積を持つ粉体ができる。
【0032】
弾性回復率は、粉体の圧縮成形性を示す指標である。粉体を錠剤径11.3mmであっって、接触面が平面である平杵を用いて、錠剤質量480mg、圧縮圧50MPaで圧縮成型した時の錠剤厚みより、次式から求めることができる。
弾性回復率={(30秒後の錠剤厚み−最小錠剤厚み)/(最小錠剤厚み)}×100
ここで、「最小錠剤厚み」は、下杵が固定された平杵を用いて上杵にて粉体を圧縮した時の最下点、すなわち錠剤が最も圧縮された時の厚みをいい、「30秒後の錠剤厚み」は、上杵が上方に開放されてから30秒後の錠剤厚みをいう。
本発明の低置換度ヒドロキシプロピルセルロース粉末の弾性回復率は、好ましくは7%以下であり、結合剤として汎用されている結晶セルロースと類似の塑性変形体であり、圧縮時に緻密な成型体を形成する粉体であることが解った。特許文献2や特許文献5に記載されている方法で作成した粉体は、弾性回復率が高く、弾性変形体の性質が強い粉体であった。
【0033】
本発明の低置換度ヒドロキシプロピルセルロース粉末の膨潤特性の測定方法として、例えば、低置換度ヒドロキシプロピルセルロース粉末を打錠圧1tで直径15mmの平面を有する錠剤に成型し、そこに水を滴下することにより、錠剤が膨潤するときの膨潤体積増加率と膨潤体積増加速度により評価できる。無水セルロースに対する苛性ソーダの質量比で0.1〜0.3であるアルカリセルロースを用いた場合、膨潤体積増加率は好ましくは300%以上で、膨潤体積増加速度は好ましくは100%/分以上となる。
膨潤体積増加率は、粉体を打錠圧1tで直径15mmの平面を有する錠剤に成型し、その後上杵の代わりに導管を持つ杵を取り付け、この導管を通じて臼に入った状態の錠剤に水を滴下することにより、錠剤が10分間吸水したときの膨潤体積増加率として得られる。水は、1ml/分の速度で10分間滴下する。体積の増加は、錠剤の厚み変化から以下の式により求めることができる。
膨潤体積増加率=(水添加前後の錠剤厚みの差/水添加前の錠剤厚み)×100
なお、上式中、「水添加前後の錠剤厚みの差」は、10分間の水添加後の錠剤厚みから水添加前の錠剤厚みを引いたものである。
本発明の低置換度ヒドロキシプロピルセルロース粉末の膨潤体積増加率は、崩壊剤として重要な特性である膨潤特性の点から、300%以上が好ましい。膨潤体積増加率が300%未満だと製剤化した場合に崩壊時間が延長する場合がある。
膨潤体積増加速度は、上記方法と同様の条件で膨潤体積増加率を測定したとき、水添加開始から30秒後の初期膨潤率を意味し、以下の式から求めることができる。
膨潤体積増加速度=(初期水添加前後の錠剤厚みの差/水添加前の錠剤厚み)×100/0.5
上式中、「初期水添加前後の錠剤厚みの差」は、水添加開始から30秒後の錠剤厚みから水添加前の錠剤厚みを引いたものである。
本発明の低置換度ヒドロキシプロピルセルロース粉末の膨潤体積増加速度は、崩壊剤として重要な特性である膨潤特性の点から、100%/分以上が好ましい。膨潤体積増加速度が100%/分未満であると、製剤化したときに崩壊時間が延長する場合がある。
【0034】
本発明の低置換度ヒドロキシプロピルセルロース粉末は、流動性が高く、粉体の流動性の指標の一つである安息角が好ましくは42o以下の粉体となる。安息角は、試料を平面上に落下させて堆積させた円錐の母線と水平面とのなす角度をいう。例えば、パウダーテスターPT−D型(ホソカワミクロン社製)を用いて直径80mmの金属製円盤状の台の上に75mmの高さより一定の角度になるまで試料を流出させ、堆積している粉体と台との角度を測定することにより算出できる。この角度が小さいほど流動性に優れる粉体と言える。
【0035】
また、結合性の指標として試料粉体を錠剤径11.3mmの平杵、錠剤質量480mg、圧縮圧150MPaで圧縮成型した時の錠剤硬度によって評価することができる。本発明の低置換度ヒドロキシプロピルセルロース粉末を用いた錠剤硬度は、35kgf以上、特に40kgf以上が好ましい。錠剤硬度は、例えば、タブレッティングテスター(三協パウテック社製)を用いて上記の条件で打錠を行うことにより評価できる。なお、試料粉体の水分により得られる錠剤硬度が変化するため、試料粉体の水分を一定にして行うことが好ましい。例えば試料粉体の水分を2〜4質量%に調製して行うことが好ましい。
更に、低置換度ヒドロキシプロピルセルロース粉末のゆるめ嵩密度によって評価することもできる。例えば、パウダーテスター(ホソカワミクロン社製)を用いて100mlの容器に軽く(タップすることなくの意味である)充填した時の質量を測定することにより算出することができる。本発明の低置換度ヒドロキシプロピルセルロース粉末のゆるめ嵩密度は0.3〜0.5g/ml、特に0.35〜0.45g/mlが好ましい。ゆるめ嵩密度が0.3g/ml未満だと粉体の流動性が低下して取り扱い上好ましくない場合があり、0.5g/mlを超えると結合性が低下する場合がある。
【0036】
本発明の低置換度ヒドロキシプロピルセルロース粉末は、錠剤又は顆粒剤等の固形製剤の結合剤又は崩壊剤として使用できる。錠剤は、乾式直接打錠法、湿式撹拌造粒打錠法、流動層造粒打錠法、乾式造粒打錠法等何れの製造方法によっても得ることができる。
ここで、乾式直接打錠法とは、低置換度ヒドロキシプロピルセルロース粉末と薬物、その他の賦形剤、滑沢剤等を乾式混合後、打錠する方法であり、造粒工程がないため製造工程が簡略化でき、生産性の高い方法である。湿式撹拌造粒打錠法とは、低置換度ヒドロキシプロピルセルロース粉末と薬物、その他の賦形剤等を水や水溶性結合剤溶液を用いて高速撹拌造粒装置を用いて造粒した後、乾燥して得られた粉体と滑沢剤とを混合後、打錠する方法であり、薬物の含量均一性が高い方法である。流動層造粒打錠法とは、低置換度ヒドロキシプロピルセルロース粉末と薬物、その他の賦形剤等を水や水溶性結合剤溶液を用いて流動層造粒装置を用いて造粒後、乾燥して得られた粉体と滑沢剤とを混合後、打錠する方法であり、湿式撹拌造粒打錠法と同様に薬物の含量均一性が高い方法である。乾式造粒打錠法とは、低置換度ヒドロキシプロピルセルロース粉末と薬物、その他の賦形剤等を圧縮により造粒し、打錠する方法であり、水や溶剤に対して敏感な薬物について有効な方法である。また、近年盛んに開発が進められている水無し又は少量の水で口腔内で速やかに崩壊する口腔内速崩壊錠にも適応可能である。この方法は嚥下能力の低い老人、小児に有効な剤型である。
更に、本発明の低置換度ヒドロキシプロピルセルロースは、顆粒剤の結合剤、崩壊剤としても使用できる。顆粒剤は、上記の湿式撹拌造粒、流動層造粒、乾式造粒等何れの製造方法によっても得ることができる。
なお、押出造粒により柱状の顆粒剤や押出造粒後の造粒物をマルメライザーにより球形化処理することも可能である。また、糖等からなる真球状の核に低置換度ヒドロキシプロピルセルロース粉末、薬物粉体その他の賦形剤等の混合粉末を散布しながら結合剤溶液を噴霧するレイヤリングを行なうことができる。
【0037】
本発明の低置換度ヒドロキシプロピルセルロース粉末を用いて製剤化する場合に用いられる薬物としては、特に限定されず、中枢神経系薬物、循環器系薬物、呼吸器系薬物、消化器系薬物、抗生物質及び化学療法剤、代謝系薬物、ビタミン系薬物等が挙げられる。
中枢神経系薬物としては、ジアゼパム、イデベノン、アスピリン、イブプロフェン、パラセタモール、ナプロキセン、ピロキシカム、ジクロフェナック、インドメタシン、スリンダック、ロラゼパム、ニトラゼパム、フェニトイン、アセトアミノフェン、エテンザミド、ケトプロフェン等が挙げられる。
循環器系薬物としては、モルシドミン、ビンポセチン、プロプラノロール、メチルドパ、ジピリダモール、フロセミド、トリアムテレン、ニフェジビン、アテノロール、スピロノラクトン、メトプロロール、ピンドロール、カプトプリル、硝酸イソソルビト等が挙げられる。
呼吸器系薬物としては、アンレキサノクス、デキストロメトルファン、テオフィリン、プソイドエフェドリン、サルブタモール、グアイフェネシン等が挙げられる。
消化器系薬物としては、2−{〔3−メチル−4−(2,2,2−トリフルオロエトキシ)−2−ピリジル〕メチルスルフィニル}ペンズイミダゾール及び5−メトキシ−2−〔(4−メトキシ−3,5−ジメチル−2−ピリジル)メチルスルフィニル〕ベンズミダゾール等の抗潰瘍作用を有するベンズイミダゾール系薬物、シメチジン、ラニチジン、パンクレアチン、ビサコジル、5−アミノサリチル酸等が挙げられる。
抗生物質及び化学療法剤としては、セファレキシン、セファクロール、セフラジン、アモキシシリン、ピバンピシリン、バカンピシリン、ジクロキサシリン、エリスロマイシン、エリスロマイシンステアレート、リンコマイシン、ドキシサイクリン、トリメトプリム/スルファメトキサゾール等が挙げられる。
代謝系薬物としては、セラペプターゼ、塩化リゾチーム、アデノシントリフォスフェート、グリベンクラミド、塩化カリウム等が挙げられる。
ビタミン系薬物としては、ビタミンB1、ビタミンB2、ビタミンB6、ビタミンC等が挙げられる。
【実施例】
【0038】
以下、実施例により本発明を詳細に説明するが、本発明は以下の実施例に限定されるものではない。
[実施例1]
806gの粉末状のパルプ(無水換算750g)を10L内部撹拌型反応機に仕込み26%苛性ソーダ303gを反応機に仕込み45℃で30分間混合して、無水セルロースに対する苛性ソーダの質量比が0.105のアルカリセルロースを得た。次に、窒素置換を実施し、そこにプロピレンオキサイドを123g(セルロースに対して0.164質量部)添加して、ジャケット温度60℃で1.5時間反応を行い、無水グルコース単位あたりヒドロキシプロポキシル基置換モル数0.28のヒドキシプロピルセルロース粗反応物1232gを得た。エーテル化効率は61.4%であった。
次に、10L内部撹拌型反応機に50質量%の酢酸236gを添加混合して中和を行った。この中和物をバッチ式遠心分離機を用いて回転数3000rpmの条件で、90℃の熱水にて洗浄、脱水を行った。脱水物の含水率は58.2質量%であった。この脱水物を棚段乾燥機で80℃、一昼夜乾燥を行った。
乾燥物をバッチ式遊星ボールミルFRITSH社製P−5を用いて255rpmで60分間粉砕を実施した。得られた粉砕物を目開き180μmの篩にて篩過して目的の低置換度ヒドロキシプロピルセルロース粉末を得た。この粉体の平均粒子径、比表面積、ゆるめ嵩密度、安息角、弾性回復率、結合性、膨潤体積増加率、膨潤体積増加速度を以下の方法で評価し、その結果を表1に示した。また、得られた粉体の電子顕微鏡写真を図1に示した。図1は、原料パルプ由来の表面が滑らかな繊維状形態が、圧密摩砕により消失し、一定形状で、かつ空隙のある粒子からなる粉体となっていることを示す。
【0039】
平均粒子径は、レーザー回折法粒度分布測定であるHELOS&RODOS(シンパック社製)を用いて測定した。
比表面積は、MICROMERITICS GEMINI 2375(島津製作所社製)を用いて測定した。
ゆるめ嵩密度は、パウダーテスター(ホソカワミクロン社製)を用いて直径5.03cm、高さ5.03cmの100ccの円筒容器(ステンレス製)に試料をJISの24メッシュの篩を通して、上方(23cm)から均一に軽く(タップすることなくの意味である)充填し、上面をすり切って質量を測定することにより算出した。
安息角は、パウダーテスター(ホソカワミクロン社製)を用いて80mmの円盤状の台の上に75mmの高さより流出させ、堆積している粉体と台との角度を測定することにより算出した。
弾性回復率は、試料粉体をタブレッティングテスター(三協パウテック社製)を用いて錠剤径11.3mmの平杵、錠剤質量480mg、圧縮圧50MPaで圧縮成型した時の錠剤厚みより、次式から求めた。
弾性回復率={(30秒後の錠剤厚み−最小錠剤厚み)/(最小錠剤厚み)}×100
結合性は、タブレッティングテスター(三協パウテック社製)を用いて粉体を錠剤径11.3mmの平杵、錠剤質量480mg、圧縮圧150MPaで圧縮成型した時の錠剤硬度を測定した。
膨潤体積増加率として、粉体を打錠圧1tで直径15mmの平面を有する錠剤に成型し、そこに水を滴下することにより、錠剤が吸水膨潤したときの10分後の体積増加率を測定した。
膨潤体積増加速度は、粉体を打錠圧1tで直径15mmの平面を有する錠剤に成型し、そこに水を滴下することにより、錠剤が膨潤したときの30秒後の体積変化から膨潤体積増加率を求めて算出した。
【0040】
次に、得られた粉体を用いて以下の条件でアスコルビン酸の直打錠を作製した。
錠剤組成 アスコルビン酸造粒末VC−97(BASF武田ビタミン社製) 90質量部
低置換度ヒドロキシプロピルセルロース 10質量部
ステアリン酸マグネシウム 0.5質量部
打錠機 ロータリー打錠機(菊水製作所社製)
錠剤サイズ 直径:8mm、曲面半径:6.5mm、錠剤質量:200mg
打錠圧 本圧:0.5t、予圧:0.2t
打錠速度 40rpm
得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)及び錠剤質量偏差を測定した。その結果を表1に示す。
【0041】
[実施例2]
806gの粉末状のパルプ(無水換算750g)を10L内部撹拌型反応機に仕込み26質量%苛性ソーダ504gを反応機に仕込み45℃で30分間混合して、無水セルロースに対する苛性ソーダの質量比が0.175のアルカリセルロースを得た。次に、窒素置換を実施し、そこにプロピレンオキサイドを143g(セルロースに対して0.190質量部)添加して、ジャケット温度60℃で1.5時間反応を行い、無水グルコース単位あたりヒドロキシプロポキシル基置換モル数0.29のヒドキシプロピルセルロース粗反応物1453gを得た。エーテル化効率は53.6%であった。
次に、10L内部撹拌型反応機に50質量%の酢酸394gを添加し、混合して中和を行った。この中和物をバッチ式遠心分離機を用いて回転数3000rpmの条件で90℃の熱水で洗浄、脱水を行った。脱水物の含水率は60.5質量%であった。この脱水物を棚段乾燥機で80℃、一昼夜乾燥を行った。
乾燥物をバッチ式遊星ボールミルFRITSH社製P−5を用いて255rpmで60分間粉砕を実施した。得られた粉砕物を目開き180μmの篩にて篩過して目的の低置換度ヒドロキシプロピルセルロース粉末を得た。この粉体の平均粒子径、比表面積、ゆるめ嵩密度、安息角、弾性回復率、結合性、膨潤体積増加率、膨潤体積増加速度を実施例1と同様の方法で評価し、その結果を表1に示す。
また、実施例1と同様にアスコルビン酸の直打錠を作製し、得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)及び錠剤質量偏差を測定した。その結果を表1に示した。
【0042】
[実施例3]
806gの粉末状のパルプ(無水換算750g)を10L内部撹拌型反応機に仕込み26質量%苛性ソーダ808gを反応機に仕込み45℃で30分間混合して、無水セルロースに対する苛性ソーダの質量比が0.280のアルカリセルロースを得た。次に、窒素置換を実施し、そこにプロピレンオキサイドを173g(セルロースに対して0.230質量部)添加して、ジャケット温度60℃で1.5時間反応を行い、無水グルコース単位あたりヒドロキシプロポキシル基置換モル数0.30のヒドキシプロピルセルロース粗反応物1787gを得た。エーテル化効率は46.4%であった。
次に、10L内部撹拌型反応機に50質量%の酢酸629gを添加し、混合して中和を行った。この中和物をバッチ式遠心分離機を用いて回転数3000rpmの条件で90℃の熱水で洗浄、脱水を行った。脱水物の含水率は62.2質量%であった。この脱水物を棚段乾燥機で80℃、一昼夜乾燥を行った。
乾燥物をバッチ式遊星ボールミルFRITSH社製P−5を用いて255rpmで60分間粉砕を実施した。得られた粉砕物を目開き180μmの篩にて篩過して目的の低置換度ヒドロキシプロピルセルロース粉末を得た。この粉体の平均粒子径、比表面積、ゆるめ嵩密度、安息角、弾性回復率、結合性、膨潤体積増加率、膨潤体積増加速度を実施例1と同様の方法で評価し、その結果を表1に示す。
また、実施例1と同様にアスコルビン酸の直打錠を作製し、得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)及び錠剤質量偏差を測定した。その結果を表1に示した。
【0043】
[実施例4]
8.6kgの粉末状のパルプ(無水換算8kg)を130L内部撹拌型反応機に仕込み35質量%苛性ソーダ3.5kgを反応機に仕込み45℃で30分間混合して、無水セルロースに対する苛性ソーダの質量比が0.151のアルカリセルロースを得た。次に、窒素置換を実施し、そこにプロピレンオキサイドを1.6kg(セルロースに対して0.2質量部)添加して、ジャケット温度60℃で1.5時間反応を行い、無水グルコース単位あたりヒドロキシプロポキシル基置換モル数0.35のヒドキシプロピルセルロース粗反応物13.7kgを得た。
次に、内部撹拌型反応機に50質量%の酢酸11.8kgを添加し、混合して中和を行った。この中和物をバッチ式減圧濾過機を用いて90℃の熱水で洗浄を行った。脱水物の含水率は59.5質量%であった。この脱水物を流動層乾燥機にて吸気80℃で排気温度60℃になるまで乾燥を行った。エーテル化反応効率は57.6%であった。
乾燥物をローラーミルIHI社製IS−250にて加圧力3MPa、テーブル回転数120rpm、分級回転数300rpm、風量11Nm3/min、供給時間5kg/hrで粉砕、脱水を行った。得られた粉砕物を目開き75μmの篩にて篩過して目的の低置換度ヒドロキシプロピルセルロース粉末を得た。
この粉体の平均粒子径、比表面積、ゆるめ嵩密度、安息角、弾性回復率、結合性、膨潤体積増加率、膨潤体積増加速度を実施例1と同様の方法で評価し、その結果を表1に示す。
また、実施例1と同様にアスコルビン酸の直打錠を作製し、得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)及び錠剤質量偏差を測定した。その結果を表1に示した。
【0044】
[実施例5]
806gの粉末状のパルプ(無水換算750g)を10L内部撹拌型反応機に仕込み26質量%苛性ソーダ303gを反応機に仕込み45℃で30分間混合して、無水セルロースに対する苛性ソーダの質量比が0.105のアルカリセルロースを得た。次に、窒素置換を実施し、そこにプロピレンオキサイドを81.8g(セルロースに対して0.109質量部)添加して、ジャケット温度60℃で1時間反応を行い、無水グルコース単位あたりヒドロキシプロポキシル基置換モル数0.20のヒドキシプロピルセルロース粗反応物1190.8gを得た。エーテル化効率は65.0%であった。
次に、10Lのバッチ式ニーダーに50℃の湯水2250g、氷酢酸59gを張り込み、上記粗反応品1190.8gを全量投入して溶解を行った。完全に溶解するのに20分間要した。その後、50質量%の酢酸118gを20g/分の速度で添加して中和を行った。この中和物をバッチ式遠心分離機を用いて回転数3000rpmの条件で90℃の熱水で洗浄、脱水を行った。脱水物の含水率は62.2質量%であった。この脱水物を棚段乾燥機で80℃、一昼夜乾燥を行った。
乾燥物をバッチ式遊星ボールミルFRITSH社製P−5を用いて255rpmで60分間粉砕を実施した。得られた粉砕物を目開き75μmの篩にて篩過して目的の低置換度ヒドロキシプロピルセルロース粉末を得た。この粉体の平均粒子径、ゆるめ嵩密度、安息角、弾性回復率、結合性、膨潤体積増加率、膨潤体積増加速度を実施例1と同様の方法で評価し、その結果を表1に示す。
また、実施例1と同様にアスコルビン酸の直打錠を作製し、得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)及び錠剤質量偏差を測定した。その結果を表1に示した。
【0045】
[比較例1]
806gの粉末状のパルプ(無水換算750g)を10L内部撹拌型反応機に仕込み13質量%苛性ソーダ300gを反応機に仕込み45℃で30分間混合して、無水セルロースに対する苛性ソーダの質量比が0.052のアルカリセルロースを得た。次に、窒素置換を実施し、そこにプロピレンオキサイドを146g(セルロースに対して0.195質量部)添加して、ジャケット温度60℃で1.5時間反応を行い、無水グルコース単位あたりヒドロキシプロポキシル基置換モル数0.28のヒドキシプロピルセルロース粗反応物1252gを得た。エーテル化効率は50.9%であった。
次に、10L内部撹拌型反応機に50質量%の酢酸117gを添加し、混合して中和を行った。この中和物をバッチ式遠心分離機を用いて回転数3000rpmの条件で90℃の熱水で洗浄、脱水を行った。脱水物の含水率は50.1質量%であった。この脱水物を棚段乾燥機で80℃、一昼夜乾燥を行った。
乾燥物をバッチ式遊星ボールミルFRITSH社製P−5を用いて255rpmで60分間粉砕を実施した。得られた粉砕物を目開き180μmの篩にて篩過して目的の低置換度ヒドロキシプロピルセルロース粉末を得た。この粉体の平均粒子径、比表面積、ゆるめ嵩密度、安息角、弾性回復率、結合性、膨潤体積増加率、膨潤体積増加速度を実施例1と同様の方法で評価し、その結果を表1に示す。
また、実施例1と同様にアスコルビン酸の直打錠を作製し、得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)及び錠剤質量偏差を測定した。その結果を表1に示した。
【0046】
[比較例2]
806gの粉末状のパルプ(無水換算750g)を10L内部撹拌型反応機に仕込み35質量%苛性ソーダ761gを反応機に仕込み45℃で30分間混合して、無水セルロースに対する苛性ソーダの質量比が0.355のアルカリセルロースを得た。次に、窒素置換を実施し、そこにプロピレンオキサイドを161g(セルロースに対して0.214質量部)添加して、ジャケット温度60℃で1.5時間反応を行い、無水グルコース単位あたりヒドロキシプロポキシル基置換モル数0.28のヒドキシプロピルセルロース粗反応物1728gを得た。エーテル化効率は43.5%であった。
次に、10L内部撹拌型反応機に50質量%の酢酸800gを添加し、混合して中和を行った。この中和物をバッチ式遠心分離機を用いて回転数3000rpmの条件で90℃の熱水で洗浄、脱水を行った。脱水物の含水率は65.2質量%であった。この脱水物を棚段乾燥機で80℃、一昼夜乾燥を行った。
乾燥物をバッチ式遊星ボールミルFRITSH社製P−5を用いて255rpmで60分間粉砕を実施した。得られた粉砕物を目開き180μmの篩にて篩過して目的の低置換度ヒドロキシプロピルセルロース粉末を得た。この粉体の平均粒子径、比表面積、ゆるめ嵩密度、安息角、弾性回復率、結合性、膨潤体積増加率、膨潤体積増加速度を実施例1と同様の方法で評価し、その結果を表1に示す。
また、実施例1と同様にアスコルビン酸の直打錠を作製し、得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)及び錠剤質量偏差を測定した。その結果を表1に示した。
【0047】
[比較例3]
806gの粉末状のパルプ(無水換算750g)を10L内部撹拌型反応機に仕込み43質量%苛性ソーダ844gを反応機に仕込み45℃で30分間混合して、無水セルロースに対する苛性ソーダの質量比が0.484のアルカリセルロースを得た。次に、窒素置換を実施し、そこにプロピレンオキサイドを165g(セルロースに対して0.220質量部)添加して、ジャケット温度60℃で1.5時間反応を行い、無水グルコース単位あたりヒドロキシプロポキシル基置換モル数0.27のヒドキシプロピルセルロース粗反応物1815gを得た。エーテル化効率は42.3%であった。
次に、10L内部撹拌型反応機に50質量%の酢酸1088gを添加し、混合して中和を行った。この中和物をバッチ式遠心分離機を用いて回転数3000rpmの条件で90℃の熱水で洗浄、脱水を行った。脱水物の含水率は66.8質量%であった。この脱水物を棚段乾燥機で80℃、一昼夜乾燥を行った。
乾燥物をバッチ式遊星ボールミルFRITSH社製P−5を用いて255rpmで60分間粉砕を実施した。得られた粉砕物を目開き180μmの篩にて篩過して目的の低置換度ヒドロキシプロピルセルロース粉末を得た。この粉体の平均粒子径、比表面積、ゆるめ嵩密度、安息角、弾性回復率、結合性、膨潤体積増加率、膨潤体積増加速度を実施例1と同様の方法で評価し、その結果を表1に示す。
また、実施例1と同様にアスコルビン酸の直打錠を作製し、得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)及び錠剤質量偏差を測定した。その結果を表1に示した。
【0048】
[比較例4]
シート状パルプを43%苛性ソーダ溶液に浸せき後、圧搾して22.2質量%の苛性ソーダを含有し、無水セルロースに対する苛性ソーダの質量比が0.484であるアルカリセルロースを得た。得られたアルカリセルロースを約1cm角のチップ状に裁断し、セルロース換算で350gのアルカリセルロースを5Lの容量の反応機に仕込み、窒素置換を行った。そこにプロピレンオキサイドを79g(セルロースに対して0.226質量部)添加して、ジャケット温度45℃で2時間、65℃で30分間反応を行い、無水グルコース単位あたりヒドロキシプロポキシル基置換モル数0.27のヒドキシプロピルセルロース粗反応物857gを得た。エーテル化効率は42.0%であった。
次に、特許文献2及び特許文献3のように粗反応物の一部を熱水で溶解後、中和を行い(部分中和法)、得られた乾燥物を衝撃粉砕で行った試料を作製した。
5Lのバッチ式ニーダーに50℃の水1750g、氷酢酸52gを張り込み上記粗反応物857gを全量投入して溶解を行った。完全に溶解するのに30分を要した。その後、33%酢酸633gを20g/minの速度で添加して中和析出を行った。
この中和物をバッチ式遠心分離機を用い3000rpmの回転数にて90℃の熱水で洗浄を行った。脱水物の含水率は75.4質量%であった。この脱水物を送風オーブンにて80℃、一昼夜乾燥を行った。
得られた乾燥物を目開き0.3mmのスクリーンを有する高速回転型衝撃粉砕機ビクトリーミルを用いて粉砕を実施し、粉砕物を目開き75μmの篩にて篩過して目的の低置換度ヒドロキシプロピルセルロース粉末を得た。この粉体の平均粒子径、比表面積、ゆるめ嵩密度、安息角、弾性回復率、結合性、膨潤体積増加率、膨潤体積増加速度を実施例1と同様の方法で評価し、その結果を表1に示す。
また、実施例1と同様にアスコルビン酸の直打錠を作製し、得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)及び錠剤質量偏差を測定した。その結果を表1に示した。また、得られた粉体の電子顕微鏡写真を図2に示した。図2は、原料パルプ由来の表面が滑らかな繊維状形態と、溶解工程により生じた球状の緻密な粒子が混在した粉体になっていることを示す。
【0049】
[比較例5]
特許文献4記載の完全溶解後中和を行い、得られた乾燥物を衝撃粉砕で行った試料を作製した。
5Lのバッチ式ニーダーに50℃の水2450gを張り込み比較例4と同様の方法で反応した粗反応物857gを全量投入して溶解(完全溶解)を行った。完全に溶解するのに1時間を要した。その後、33%酢酸793gを20g/minの速度で添加して中和析出を行った。
この中和物をバッチ式遠心分離機を用い3000rpmの回転数にて90℃の熱水で洗浄、脱水を行った。脱水物の含水率は80.1質量%であった。この脱水物を送風オーブンにて80℃、一昼夜乾燥を行った。
得られた乾燥物を目開き0.3mmのスクリーンを有する高速回転型衝撃粉砕機ビクトリーミルを用いて粉砕を実施し、粉砕物を目開き75μmの篩にて篩過して目的の低置換度ヒドロキシプロピルセルロース粉末を得た。この粉体の平均粒子径、ゆるめ嵩密度、安息角、弾性回復率、結合性、膨潤体積増加率、膨潤体積増加速度を実施例1と同様の方法で評価し、その結果を表1に示す。
また、実施例1と同様にアスコルビン酸の直打錠を作製し、得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)及び錠剤質量偏差を測定した。その結果を表1に示した。また、得られた粉体の電子顕微鏡写真を図3に示した。図3は、溶解工程により生じた繊維状粒子が少なく、球状の緻密な粒子からなる粉体になっていることを示す。
【0050】
[比較例6]
目開き0.3mmのスクリーンを有する高速回転型衝撃粉砕機ビクトリーミルを用いた以外は実施例2と同様の方法にて低置換度ヒドロキシプロピルセルロース粉末を得た。この粉体の平均粒子径、比表面積、ゆるめ嵩密度、安息角、弾性回復率、結合性、膨潤体積増加率、膨潤体積増加速度を実施例1と同様の方法で評価し、その結果を表1に示す。
また、実施例1と同様にアスコルビン酸の直打錠を作製し、得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)及び錠剤質量偏差を測定した。その結果を表1に示した。また、得られた粉体の電子顕微鏡写真を図4に示した。図4は、原料パルプ由来の表面が滑らかな繊維状形態からなる粉体になっていることを示す。
【0051】
アルカリセルロース中の無水セルロースに対する苛性ソーダの質量比の膨潤特性に与える影響をみた結果を図5に示す。図5は、横軸に水の添加開始からの測定時間を、縦軸に膨潤体積増加率を示す。
実施例は、比較例に比べ、膨潤体積増加率が大きく、膨潤体積増加速度も大きいことがわかる。
【0052】
【表1】

【0053】
表1から明らかなように、所定の苛性ソーダの質量比を有し、圧密摩砕を用いたものは比表面積が大きく、弾性回復率が小さくなるため、結合性が高くなり、製剤化した場合に錠剤硬度が高い錠剤が得られる。また、膨潤体積増加率及び膨潤体積増加速度が高くなるので、製剤化した場合、崩壊時間が短い製剤が得られた。一方、圧密摩砕を用いても所定の苛性ソーダの質量比を用いない粉体は、比表面積、結合性、崩壊剤として重要な膨潤特性である膨潤体積増加率、錠剤の硬度と崩壊時間に劣ることが比較例1〜3の結果に示されている。また、比較例2〜3の結果では、崩壊剤として重要な膨潤特性である膨潤体積増加速度にも劣っている。所定の苛性ソーダの質量比を用いても、圧密摩砕に換えて衝撃粉砕を用いて得られた粉体は、比表面積、流動性、結合性、錠剤の硬度と崩壊時間に劣り、錠剤質量偏差が大きいことが比較例6の結果に示されている。比較例4は、シート状のパルプを使用しているため、過剰な苛性ソーダを含むアルカリセルロースを用いることとなり、膨潤体積増加速度が劣る。また、衝撃粉砕機を用いているため、比表面積が小さく、かつ弾性回復率が大きく、結合性が低かった。一方、比較例5は、完全溶解により繊維状形態が少ないため、安息角が低く、流動性に優れるが、結合性が低かった。
【0054】
1)口腔内崩壊錠
[実施例6]
実施例1に記載の方法により得られた低置換度ヒドロキシプロピルセルロースを用いて下記条件にて口腔内速崩壊錠を作製した。
錠剤組成 エリスリトール(250μm篩過品) 70質量部
低置換度ヒドロキシプロピルセルロース 30質量部
ステアリン酸マグネシウム 0.5質量部
打錠機 単発打錠機 タブレッティングテスター(三協パウテック社製)
錠剤サイズ 直径:11.3mm、平杵
打錠圧 10、25、50MPa
得られた錠剤の錠剤硬度、口腔内崩壊時間及び服用感を評価した。口腔内崩壊時間は健康な成人4名に口腔内で錠剤を噛まず、軽く口に含んだ状態で錠剤が完全に溶解又は崩壊するまでの時間を測定し、その平均値を算出した。錠剤硬度及び口腔内崩壊時間の結果を表2に示す。
【0055】
[比較例7]
比較例4に記載の方法により得られた低置換度ヒドロキシプロピルセルロースを用いた以外は、実施例6と同様の方法により口腔内速崩壊錠を作製した。得られた錠剤の評価を実施例6と同様の方法にて行い、その結果を表2に示す。
【0056】
【表2】
【0057】
実施例6は、比較例7と比較して高い錠剤硬度を示し、かつ口腔内での崩壊時間が短いものであった。また、実施例6は何れの錠剤もざらつきがなくクリーミーな服用感であったのに対して、比較例7では紙を口に含んだ様であり、服用感が悪いものであった。
【0058】
2)湿式撹拌造粒
[実施例7]
実施例4に記載の方法により得られた低置換度ヒドロキシプロピルセルロースを用いて下記組成の粉体をバーチカルグラニュレーターFM−VG−05(パウレック社製)に仕込み、主軸600rpm、チョッパー1000rpmの回転数にて1分間前混合を実施し、水55gを添加し5分間混合して湿式撹拌造粒を行なった。
錠剤組成 エテンザミド 210g(70質量部)
200#乳糖 60g(20質量部)
低置換度ヒドロキシプロピルセルロース 30g(10質量部)
造粒物を1180μmの目開きの篩にて篩過後、流動層乾燥機にて、吸気60℃、流動エアー量70m3/hrで乾燥を行い、排気温度45℃になるまで乾燥を行った。
得られた顆粒の平均粒子径、ゆるめ嵩密度、固め嵩密度、圧縮度、安息角を表3に示す。
なお、得られた顆粒の平均粒子径、ゆるめ嵩密度、安息角の測定方法は、低置換度ヒドロキシプロピルセルロース粉末における測定方法と同様であり、固め嵩密度及び圧縮度は以下のよう方法により測定した。
すなわち、固め嵩密度とは、ゆるめ嵩密度にタッピングを加えて密充填にした場合の嵩密度をいい、タッピングとは、試料を充填した容器を一定の高さから繰り返し落下させて底部に軽い衝撃を与え、試料を密充填にする操作をいう。従って、固め嵩密度は、ゆるめ嵩密度を測定する際、上面をすり切って秤量した後、更にこの容器の上にキャップ(下記ホソカワミクロン社製パウダーテスターの備品)をはめ、この上縁まで粉体を加えてタップ高さ1.8cmのタッピングを180回行なう。タッピング終了後、キャップを外して容器の上面で粉体をすり切って秤量する。これらの操作は、ホソカワミクロン社製パウダーテスター(PT−D)を使用することにより測定できる。
また、圧縮度とは、かさべりの度合いを示す値であり、以下の式で求められる。
圧縮度(%)=[(固め嵩密度−ゆるめ嵩密度)/固め嵩密度]×100
【0059】
次に、上記顆粒100質量部に対してステアリン酸マグネシウム0.5質量部を加えた後、混合して打錠用の粉体を得た。この粉体を用いて下記条件にて連続打錠を実施した。
打錠機 ロータリー打錠機 (菊水製作所社製)
錠剤サイズ 直径:8mm、曲率半径:6.5mm、錠剤重量:200mg
打錠圧 本圧:1t、予圧:0.2t
打錠速度 40rpm
得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)、日本薬局方摩損度試験に準じた下記方法にて錠剤摩損度、摩損度試験後にキャッピングが発生している錠剤数を測定することによりキャッピング発生率、錠剤質量偏差を評価した。
摩損度試験は、日本薬局方記載の摩損度試験器を用いて20錠を25rpmで16分間試験を行った後の質量減少率を下記の式より算出した。
[{(試験前の錠剤質量)−(試験後の錠剤質量)}/(試験前の錠剤質量)]×100
キャッピング発生率は、上記摩損度試験実施後、錠剤が2層に割れていた錠剤数から、キャッピング発生率を算出した。
錠剤質量偏差は、錠剤20錠の質量を測定することにより、その変動係数(CV値)を算出した。
得られた結果を表4に示す。
【0060】
[比較例8]
比較例4に記載の方法により得られた低置換度ヒドロキシプロピルセルロースを用いた以外は、実施例7と同様の方法により湿式撹拌造粒を実施した。得られた顆粒の平均粒子径、ゆるめ嵩密度、固め嵩密度、圧縮度、安息角を表3に示した。
次に、実施例7と同様の方法にて連続打錠を実施し、その評価を行った。その結果を表4に示す。
【0061】
[比較例9]
低置換度ヒドロキシプロピルセルロースの代わりに微結晶セルロースCeolusPH−101(旭化成ケミカルズ社製)を用いた以外は、実施例7と同様の方法により湿式撹拌造粒を実施した。ただし、微結晶セルロースは低置換度ヒドロキシプロピルセルロースと比較して保水性が低いため、水を36gに減量した。得られた顆粒の平均粒子径、ゆるめ嵩密度、固め嵩密度、圧縮度、安息角を表3に示す。
次に、実施例7と同様の方法にて連続打錠を実施し、その評価を行った。その結果を表4に示す。
【0062】
【表3】
【0063】
実施例7は、比較例8、9と比較してゆるめ嵩密度が高く重質で、圧縮度及び安息角が低いことにより、流動性に優れる顆粒が得られた。
【0064】
【表4】
【0065】
実施例7は、比較例8と比較して錠剤硬度が高く、錠剤質量偏差の低い優れた製剤が得られた。また、微結晶セルロースを用いた比較例9では、キャッピングが発生し、錠剤摩損度が高く、錠剤硬度が低いものであったが、実施例7は、全ての評価項目において優れた製剤であった。
【0066】
3)流動層造粒
[実施例8]
実施例1に記載の方法により得られた低置換度ヒドロキシプロピルセルロースを用いて下記組成の粉体を流動層造粒装置マルチプレックスMP−01(パウレック社製)に仕込み、吸気60℃、流動エアー量50m3/hr、排気温度30〜35℃、ヒドロキシプロピルメチルセルロースTC−5R(信越化学工業社製)5質量%水溶液60gを10g/分の速度で噴霧して造粒を行った。
錠剤組成 アセトアミノフェン 80g(40質量部)
200#乳糖 28g(14質量部)
コーンスターチ 12g( 6質量部)
低置換度ヒドロキシプロピルセルロース 80g(40質量部)
得られた顆粒の平均粒子径、ゆるめ嵩密度、固め嵩密度、圧縮度、安息角を表5に示す。
次に、上記顆粒100質量部に対してステアリン酸マグネシウム0.5質量部を加えた後、混合して打錠用の粉体を得た。この粉体を用いて実施例7と同様に連続打錠を実施した。得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)、錠剤質量偏差を評価した。その結果を表6に示す。
【0067】
[比較例10]
比較例4に記載の方法により得られた低置換度ヒドロキシプロピルセルロースを用いた以外は、実施例8と同様の方法により流動層造粒を実施した。得られた顆粒の平均粒子径、ゆるめ嵩密度、固め嵩密度、圧縮度、安息角を表5に示した。
次に、実施例8と同様の方法にて連続打錠を実施し、その評価を行った。その結果を表6に示す。
【0068】
[比較例11]
比較例5に記載の方法により得られた低置換度ヒドロキシプロピルセルロースを用いた以外は、実施例8と同様の方法により流動層造粒を実施した。得られた顆粒の平均粒子径、ゆるめ嵩密度、固め嵩密度、圧縮度、安息角を表5に示す。
次に、実施例8と同様の方法にて連続打錠を実施し、その評価を行った。その結果を表6に示す。
【0069】
【表5】
【0070】
実施例8は、比較例10、11と比較してゆるめ嵩密度が高く重質で、圧縮度及び安息角が低いことにより、流動性に優れる顆粒が得られた。
【0071】
【表6】
【0072】
実施例8は、比較例10、11と比較して錠剤硬度が高く、錠剤質量偏差の少ない製剤であった。
【0073】
4)押出造粒
[実施例9]
実施例4に記載の方法により得られた低置換度ヒドロキシプロピルセルロースを用いて下記組成の粉体をバーチカルグラニュレーターFM−VG−05(パウレック社製)に仕込み、主軸600rpm、チョッパー1000rpmの回転数にて1分間前混合を実施し、水50gを添加し5分間混合して湿式撹拌造粒を行なった。
錠剤組成 アスピリン 279g(93質量部)
低置換度ヒドロキシプロピルセルロース 15g( 5質量部)
ヒドロキシプロピルメチルセルロースTC−5E 6g( 2質量部)
次に、押出造粒装置ドームグラン(ダルトン社製)1.0mmφのスクリーンを用いて上記湿粉の押出造粒を実施し、固形分に換算した造粒速度を表7に示した。また、得られた押出造粒物をマルメライザー(ダルトン社製)を用いて750rpmで球形化処理を行った。その後、流動層乾燥機にて、吸気60℃、流動エアー量70m3/hrで乾燥を行い、排気温度45℃になるまで乾燥を行った。得られた顆粒の顆粒強度、日本薬局方崩壊試験における崩壊時間(試験液:水)を測定した。その結果を表7に示す。
なお、顆粒強度は、日本薬局方摩損度試験機を用いて、そこに顆粒10gを仕込み直径5mmのガラスビーズ20個を入れ、25rpm、4分間試験を行った後の質量減少率を下記の式より算出した。
[{(試験前の顆粒質量)−(試験後の顆粒質量)}/(試験前の顆粒質量)]×100
【0074】
[比較例12]
比較例4に記載の方法により得られた低置換度ヒドロキシプロピルセルロースを用いた以外は、実施例9と同様の方法により押出造粒を実施し、固形分に換算した造粒速度を表7に示した。得られた湿式造粒物を実施例9と同様の方法にて球形化、乾燥を行った。
得られた顆粒の顆粒強度、日本薬局方崩壊試験における崩壊時間(試験液:水)を測定した。その結果を表7に示す。
【0075】
【表7】
【0076】
実施例9は、比較例12と比較して製粒速度が速く、生産性が高く、得られた顆粒の顆粒強度が高く優れた製剤であった。
【0077】
5)乾式造粒
[実施例10]
実施例4に記載の方法により得られた低置換度ヒドロキシプロピルセルロースを用いて下記組成の粉体をローラーコンパクターMINI(フロイント社製)を用い、ロール圧4MPa、ロール回転数5rpm、スクリュー回転数12rpmで乾式造粒を行った。
錠剤組成 アスコルビン酸 50g(10質量部)
200#乳糖 245g(49質量部)
コーンスターチ 105g(21質量部)
低置換度ヒドロキシプロピルセルロース 100g(20質量部)
ステアリン酸マグネシウム 2.5g(0.5質量部)
造粒物を250μm目開きの篩にて篩過し、この造粒物を下記条件で連続打錠を実施した。
打錠機 ロータリー打錠機 (菊水製作所社製)
錠剤サイズ 直径:8mm、曲率半径:6.5mm、錠剤重量:200mg
打錠圧 本圧:1t、予圧:0.2t
打錠速度 40rpm
得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)を評価した結果を表8に示す。
【0078】
[比較例13]
比較例4に記載の方法により得られた低置換度ヒドロキシプロピルセルロースを用いた以外は、実施例10と同様の方法により乾式造粒を実施し、実施例10と同様の方法にて連続打錠を実施した。得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)を評価した結果を表8に示す。
【0079】
【表8】
【0080】
実施例10は、比較例13と比較して崩壊時間は同等でありながら錠剤硬度が高い製剤であった。
【技術分野】
【0001】
本発明は、医薬品又は食品分野等において製剤を製造する際に崩壊性又は結合性を付与するために添加する低置換度ヒドロキシプロピルセルロース関するものである。特に結合性、崩壊性に優れたものに関するものである。
【背景技術】
【0002】
医薬品又は食品分野等の固形製剤において、主薬のみで作製された製剤では、薬物を投与しても十分な崩壊性が得られず、薬効が十分に発揮されない場合や結合性が劣るため錠剤や顆粒剤とした際にその形状を保つことができない等の問題がある。このような場合に低置換度ヒドロキシプロピルセルロース、カルボキシメチルセルロースのカルシウム塩、架橋カルボキシメチルセルロースナトリウム、架橋ポリビニルピロリドン、カルボキシメチルスターチ等の崩壊剤を添加することにより崩壊性を改善することができる。また、結合性を改良するためには結晶セルロースを添加したり、水溶性の結合剤を添加することにより改善することができる。低置換度ヒドロキシプロピルセルロースは上記の崩壊性と結合性を合わせ持つユニークな添加剤として知られている。
【0003】
低置換度ヒドロキシプロピルセルロースは非イオン性であるため、イオン性の薬物等との反応による変質が起きにくい等の利点を有する。低置換度ヒドロキシプロピルセルロースを医薬品の添加剤として使用しうることは特許文献1〜2に記載されている。
【0004】
特許文献1〜4及び特許文献7に記載されているように、アルカリセルロースの調製方法としてシート状のパルプを苛性ソーダ水溶液に浸せき後、圧搾して製造する方法があるが、この方法においては一旦シート状のパルプを苛性ソーダ水溶液に浸せきすると過剰量の苛性ソーダ水溶液がパルプに吸液され、圧搾しても過剰の水又は苛性ソーダを含むアルカリセルロースしか製造できず、副反応への選択率が上昇し、主反応であるエーテル化反応の効率は低いものであった。
【0005】
従来の低置換度ヒドロキシプロピルセルロースは、特許文献3に記載されているように、アルカリセルロースとプロピレンオキサイドを反応させることによって得ることができる。エーテル化反応においては、主反応としてはセルロースのエーテル化があり、副反応としてはプロピレンオキサイドと水との反応によりプロピレングリコールを生成する反応がある。このエーテル化反応に利用されるプロピレンオキサイドの量は、反応触媒として利用される苛性ソーダ水溶液の量により変化し、アルカリセルロース中の水又は苛性ソーダが多すぎると反応効率が低下する問題があった。
【0006】
一方、粉末状のパルプに苛性ソーダ水溶液を滴下又は噴霧して混合することにより製造する場合、アルカリセルロース中の水又は苛性ソーダ量を任意に調製できる利点がある。
また、特許文献3に記載されているように、粗反応生成物中に残存するアルカリを中和する際に酸を含む熱水中に反応生成物を投入して粗反応生成物を溶解させる方法を用いると、得られた低置換度ヒドロキシプロピルセルロースは原料パルプと同様に繊維状の形態となり洗浄性に優れ、精製が容易であるが、その反面粉砕性に劣り流動性の優れた粉体を得ることができない。
【0007】
特許文献2には、エーテル化反応終了後に、反応触媒として用いられたアルカリを水中で部分的に中和し、低置換度ヒドロキシプロピルセルロースを一部溶解させて、繊維質部分を制御する方法が記載されている。この方法では残存するアルカリ量を多くすると、繊維状形態が溶解により減少し、得られた粉砕物の嵩密度は高く、流動性が良好になるが、精製時に洗浄性が低下してしまう問題があることが記載されている。
【0008】
特許文献3では、洗浄脱水物の含水率を70〜90質量%とすることにより、粉砕性が向上し、42%以下の圧縮度と48o以下の安息角を有する低置換度ヒドロキシプロピルセルロース粉体を得ることができることが記載されている。また、粉砕はハンマーミル等の衝撃型粉砕機によって行われることが記載されている。
しかし、衝撃粉砕機では粉砕原料の影響を受けやすく、粉砕原料の形状により、製品の粉体物性が決定される。つまり、特許文献2にあるように部分中和量を多くしたり、溶解工程を経ず、粗反応生成物に残存する苛性ソーダに対して当量の酸を含む水又は熱水中に反応生成物を投入し中和を行うことにより得られた繊維状形態の原料を用いて衝撃粉砕機で粉砕した場合、得られた粉砕物は繊維状粒子が多く含まれる流動性の低い粉体しか得られなかった。また、短時間で高エネルギーを与えて粉砕するため、処理量が低く、生産性の低い方法であった。
また、上記のような方法で製造された低置換度ヒドロキシプロピルセルロースは粉体の流動性を高めるために部分中和量を少なくして溶解させることにより、繊維質部分が少なくなり結合性が低下してしまう問題があった。また、結合性を高めるために部分中和量を多くして繊維質部分を多くすると流動性が低下する問題もあった。
特許文献3の洗浄脱水物の含水率を70〜90質量%に制御して得られたものは繊維状形態の粒子と球状粒子の混在する粉体である。この繊維状形態の粒子の影響を受け流動性は不十分で直接打錠法で製錠する場合、錠剤の質量偏差が大きくなることがあった。更に、球状粒子の結合性が低いため、この混在物である粉体の結合性は不十分であった。
【0009】
特許文献4においては繊維状粒子を完全溶解させることにより、流動性の高い低置換度ヒドロキシプロピルセルロースを得ることができることが記載されているが、この方法で得られた粉体の結合性は低いものであった。
更に、特許文献2において、粉砕を衝撃粉砕に換えてボールミルで行うと結合性に劣ることが記載されている。また、ボールミルで粉砕されたものは崩壊剤として重要な特性である膨潤特性に劣るものであった。
【0010】
特許文献5においては、パルプを粉末化する際に竪型ローラーミルを用いることにより、粉末化パルプの嵩密度を上昇させ、水に溶解した時の未溶解繊維分が少ない水溶性セルロースエーテルについて記載されている。
しかし、水溶性セルロースエーテルは水不溶性の低置換度ヒドロキシプロピルセルロースの製造において必須の中和工程を含んでいない点で異なり、かつ、特許文献5では低置換度ヒドロキシプロピルセルロースの重要な特性である結合性及び崩壊性に関して何ら記載されていない。
【0011】
特許文献6において竪型ローラーミルを用いてセルロースエーテルを製造する方法が記載されているが、これも同様に水溶性セルロースエーテルに関するもので水不溶性である低置換度ヒドロキシプロピルセルロースに関するものではない。
【0012】
特許文献7には安息角45o以下で膨潤するときの体積増加率100%以上の低置換度ヒドロキシプロピルセルロースに関する記載があるが、この低置換度ヒドロキシプロピルセルロースの結合性は低いものであった。
【特許文献1】特公昭48−38858号公報
【特許文献2】特公昭57−53100号公報
【特許文献3】特開平10−305084号公報
【特許文献4】特開平11−322802号公報
【特許文献5】特開2001−9316号公報
【特許文献6】特開平9−76233号公報
【特許文献7】特開平7−324101号公報
【発明の開示】
【発明が解決しようとする課題】
【0013】
本発明は、上記従来技術の欠点を解消するためになされたものであり、結合性が高く、流動性が良好で崩壊性に優れる低置換度ヒドロキシプロピルセルロース粉末を提供することを目的とする。
【課題を解決するための手段】
【0014】
本発明者らは、上記目的を達成するため鋭意検討した結果、低置換度ヒドロキシプロピルセルロースの製造において、粉末化したパルプに苛性ソーダ水溶液を添加混合し、無水セルロースに対する苛性ソーダの質量比が0.1〜0.3であるアルカリセルロースを製造し、次いでエーテル化反応を行った後、溶解工程を経るか又は経ずに苛性ソーダを中和し、洗浄乾燥後、粉砕工程において圧密摩砕することにより、結合性が高く、流動性が良好で崩壊性に優れる低置換度ヒドロキシプロピルセルロース粉末を得ることを見出した。
【0015】
本発明は、平均粒子径が10〜100μmであり、かつBET法で測定した比表面積が1.0m2/g以上である低置換度ヒドロキシプロピルセルロース粉末を提供する。低置換度ヒドロキシプロピルセルロース粉末は、好ましくは、圧縮圧50MPaで圧縮成型した時の弾性回復率が7%以下である。低置換度ヒドロキシプロピルセルロース粉末は、好ましくは、吸水時の膨潤体積増加率が300%以上で、膨潤体積増加速度が100%/分以上である。低置換度ヒドロキシプロピルセルロース粉末は、好ましくは、安息角が42o以下である。
さらに、本発明は、この低置換度ヒドロキシプロピルセルロース粉末を用いた固形製剤を提供する。
本明細書は、無水グルコース単位あたりの置換モル数が0.05〜1.0である水不溶性で吸水膨潤性を有する低置換度ヒドロキシプロピルセルロース粉末の製造方法であって、(1)粉末化したパルプに無水セルロースに対する苛性ソーダの質量比が0.1〜0.3となる量で苛性ソーダ水溶液を添加混合してアルカリセルロースを製造する工程と、(2)得られたアルカリセルロースのエーテル化反応を行って粗反応物を得る工程と、(3)得られた粗反応物中に含有される苛性ソーダを中和する工程と、(4)その後の洗浄・脱水工程と、(5)乾燥工程と、(6)圧密摩砕を行う粉砕工程とを含んでなる低置換度ヒドロキシプロピルセルロース粉末の製造方法を記載する。また、無水グルコース単位あたりの置換モル数が0.05〜1.0である水不溶性で吸水膨潤性を有する低置換度ヒドロキシプロピルセルロース粉末の製造方法であって、(1)粉末化したパルプに無水セルロースに対する苛性ソーダの質量比が0.1〜0.3となる量で苛性ソーダ水溶液を添加混合してアルカリセルロースを製造する工程と、(2)得られたアルカリセルロースのエーテル化反応を行って粗反応物を得る工程と、(3)粗反応物の一部又は全部を溶解する溶解工程を経ることなく、得られた粗反応物中に含有される苛性ソーダを中和する工程と、(4)その後の洗浄・脱水工程と、(5)乾燥工程と、(6)圧密摩砕を行う粉砕工程とを含んでなる低置換度ヒドロキシプロピルセルロース粉末の製造方法を記載する。
上記洗浄・脱水工程は、好ましくは、洗浄するとともに含水率を65質量%以下とする脱水を行う工程である。
【発明の効果】
【0016】
本発明の低置換度ヒドロキシプロピルセルロース粉末は、原料パルプ由来の繊維状形態にも拘わらず流動性が高く、結合性に優れ、更に膨潤特性に優れるものである。
また、本発明の低置換度ヒドロキシプロピルセルロース粉末は、結合性及び崩壊性に優れるため、錠剤中の添加量を削減でき錠剤のサイズを小さくできる利点も有する。
【図面の簡単な説明】
【0017】
【図1】実施例1で得られた粉体の電子顕微鏡写真を示す。
【図2】比較例4で得られた粉体の電子顕微鏡写真を示す。
【図3】比較例5で得られた粉体の電子顕微鏡写真を示す。
【図4】比較例6で得られた粉体の電子顕微鏡写真を示す。
【図5】実施例1〜3と比較例1と3で得られた粉体の時間の経過に伴う膨潤体積増加率を示す。
【発明を実施するための最良の形態】
【0018】
本発明の方法について以下に説明する。まず、原料として使用される粉末状のパルプは何れの粉砕方式を用いても良い。その平均粒子径は、好ましくは60〜300μmである。60μm未満の粉末状パルプを調製するには工業的に非効率的であり、300μmを超えると苛性ソーダ水溶液との混合性に劣る恐れがある。
【0019】
アルカリセルロースを製造する工程は、好ましくは、上記粉末状のパルプに苛性ソーダ水溶液を滴下又は噴霧して混合することにより行う。この際、苛性ソーダはエーテル化反応の触媒として作用する。アルカリセルロースの製造は、好ましくは、内部撹拌型の反応機内で混合を行い、引き続いてエーテル化反応を行うか、他の混合機内で調製したアルカリセルロースを反応機内に仕込んでエーテル化反応を行うか何れの方法を用いても良い。
【0020】
また、アルカリセルロース中の苛性ソーダ量は、反応効率への影響のみではなく、最終製品の膨潤特性及び結合性に影響を与えることが解った。アルカリセルロース中の最適な苛性ソーダ量は、無水セルロース(パルプ中の水分を除いたものをいう。)に対する苛性ソーダの質量比で0.1〜0.3である。0.1未満では膨潤特性の特に吸水膨潤時の体積増加率が低くなり、崩壊性が低下し、結合性も低下する場合がある。また、0.3を超えると後述の吸水時の膨潤体積増加率及び膨潤体積増加速度も低くなり、結合性も低下する場合がある。
苛性ソーダは、好ましくは20〜40質量%の水溶液として添加される。
【0021】
次のエーテル化反応を行う工程は、アルカリセルロースを反応機内に仕込み、窒素置換後、エーテル化剤としてプロピレンオキサイドを反応機内に仕込み反応を行う。プロピレンオキサイドの仕込み比は、好ましくは、無水グルコース単位1モルに対して0.1〜1.0モル程度である。反応温度は40〜80℃程度、反応時間は1〜5時間程度である。
【0022】
なお、エーテル化反応を行う工程の後、必要に応じて溶解工程を行うことができる。溶解工程は、エーテル化反応後の粗反応物の一部又は全部を水又は熱水に溶解することにより行われる。水又は熱水の使用量は、粗反応物の溶解量によって異なるが、粗反応物の全部を溶解させるときの水の量は、通常、粗反応物中の低置換度ヒドロキシプロピルセルロースに対して質量比で0.5〜10である。
後述の洗浄・脱水工程における負荷及び低置換度セルロースエーテル結合性の更なる向上を考慮すると、この溶解工程を行わない方がより好ましい。
【0023】
次に行われる中和工程は、触媒として使用した苛性ソーダが反応生成物に残存するため、好ましくは、その苛性ソーダに対して当量の酸を含む水又は熱水中に粗反応生成物を投入し中和を行う。また、反応生成物に当量の酸を含む水又は熱水を加えて中和を行っても良い。
使用する酸は、塩酸、硫酸、硝酸等の鉱酸やギ酸、酢酸等の有機酸が挙げられる。
【0024】
次の洗浄・脱水工程では、得られた中和物を、好ましくは水又は熱水を用いて洗浄しながら、好ましくは遠心分離、減圧濾過、加圧濾過等から選ばれる方法で脱水を行う。得られた脱水物ケーキ中の低置換度ヒドロキシプロピルセルロースは原料パルプの形態と同様に繊維状ものとなる。溶解工程を経た脱水物では置換モル数にもよるが、脱水率が概ね70〜90質量%であり、溶解工程を経ない脱水率は通常65質量%以下となり、その後の乾燥工程での負荷が低減でき生産性が向上する。更に、溶解工程が無いため工程を簡略化できる利点を有する。
また、製品の結合性の観点から繊維状形態のものを使用して粉砕した方が得られた製品の比表面積が高く、結合性の高いものが得られる。
【0025】
上記で得られた脱水物を乾燥する乾燥工程は、好ましくは、流動層乾燥機、ドラムドライヤー等の乾燥機を用いて60〜120℃にて行うことができる。
【0026】
粉砕工程は、上記方法で得られた乾燥物を圧密摩砕することにより行われる。
この圧密摩砕には、ローラーミル、ボールミル、ビーズミル、石臼型粉砕機等の粉砕機が利用できる。ローラミルは、ローラー又はボールが、その回転運動に伴う遠心力や重力荷重により、ミル壁の被粉砕物を圧縮・剪断しながら転がる粉砕機で、石川島播磨重工業社製ISミル、栗本鐵工所社製VXミル、増野製作所社製MSローラーミル等が利用できる。ボールミルは、鋼球、磁性ボール、玉石及びその類似物を粉砕媒体とする粉砕機で、栗本鉄工社製ボールミル、大塚鉄工社製チューブミル、FRITSCH社製遊星ボールミル等が利用できる。ビーズミルはボールミルと類似するが、使われるボールの径が小さく、機器内部が高速回転することにより、ボールの加速度をより高めることができる点で異なり、例えばアシザワ製作所社製のビーズミルが利用できる。石臼型粉砕機は、石臼が狭いクリアランスで高速回転することにより粉体を摩砕することができる機械で、例えば増幸産業社製のセレンディピターが利用できる。
特に金属異物の混入が少なく、設置面積が小さく、生産性の高いローラーミルが好ましい。
【0027】
なお、従来の衝撃粉砕より製造される低置換度ヒドロキシプロピルセルロースの結合性は、繊維状形態の絡み合いにより発現されると言われてきた。そのため、結合性を高めるために繊維状粒子を多くすると流動性は低下してしまった。しかし、本発明の低置換度ヒドロキシプロピルセルロース粉末は圧密摩砕されることにより繊維状形態が消失しているにも拘わらず、驚くべきことに高い結合性を示すものであった。
【0028】
従来技術における製品の粉体物性の調製は、特許文献2及び特許文献3にあるようにエーテル化反応終了後に、反応触媒として用いられた苛性ソーダを水中で部分的に中和し、低置換度ヒドロキシプロピルセルロースを一部溶解させて、繊維状粒子の量を制御することにより行われていた。また、特許文献4では低置換度ヒドロキシプロピルセルロースを完全溶解させることにより、流動性の高い粉体を調製していた。そして、いずれの粉砕も衝撃力を利用したインパクトミル等の衝撃粉砕機を用いることにより行われてきた。
衝撃粉砕機では、粉砕原料の影響を受けやすく、粉砕原料の形状により、製品の粉体物性が決定される。つまり、特許文献2又は特許文献3にあるように部分中和量を多くしたり、本発明の実施の一形態である溶解工程を経ず、反応生成物に残存する苛性ソーダに対して当量の酸を含む水又は熱水中に反応生成物を投入し中和を行うことにより得られた繊維状形態の原料を用いた場合、得られた粉砕物は繊維状粒子が多く含まれる流動性の低い粉体しか得られなかった。
【0029】
本発明によれば、粉砕原料である繊維状形態の粒子は圧密摩砕を繰り返すことにより、原料パルプ由来の繊維状で中空の管状形態が消失することにより、1次粒子を小さくすることができるため、比表面積が増大する。また、原料パルプ由来の繊維状形態が消失し、粒子形状の揃った粉体が得られる。
【0030】
次に、好ましくは、粉砕物を定法に従い篩い分けを行うことにより目的の低置換度ヒドロキシプロピルセルロース粉末を得ることができる。篩いの目開きは、75〜180μm程度のものが使用できる。
本発明の低置換度ヒドロキシプロピルセルロース粉末の平均粒子径は、好ましくは10〜100μm程度であり、より好ましくは20〜60μm程度である。10μm未満では微粉化により凝集性が増し、粉体の流動性が低下する恐れがあり、100μmを超えると薬物との混合性が低下して不均一となる恐れがある。
【0031】
本発明の低置換度ヒドロキシプロピルセルロース粉末の比表面積は1.0m2/g以上が好ましい。これ未満では高い結合性が得られない恐れがある。
一般的に粉体の比表面積が高い程、結合性が高い粉体となることが知られている。比表面積分析は、粉体粒子表面に吸着占有面積の判った分子を液体窒素の温度で吸着させ、その量から試料の比表面積を求める方法であり、不活性気体の低温低湿物理吸着によるBET法を用いることができる。例えば、MICROMERITICS GEMINI 2375(島津製作所社製)を用いて測定できる。
一般的には平均粒子径を小さくすることにより比表面積を増大させることができるが、上記記載のように平均粒子径が小さくなり過ぎると粉体の凝集性が増し、粉体の流動性が低下する恐れがある。本発明においては、圧密摩砕により、粉体の流動性が得られる平均粒子径でありながら高い比表面積を持つ粉体ができる。
【0032】
弾性回復率は、粉体の圧縮成形性を示す指標である。粉体を錠剤径11.3mmであっって、接触面が平面である平杵を用いて、錠剤質量480mg、圧縮圧50MPaで圧縮成型した時の錠剤厚みより、次式から求めることができる。
弾性回復率={(30秒後の錠剤厚み−最小錠剤厚み)/(最小錠剤厚み)}×100
ここで、「最小錠剤厚み」は、下杵が固定された平杵を用いて上杵にて粉体を圧縮した時の最下点、すなわち錠剤が最も圧縮された時の厚みをいい、「30秒後の錠剤厚み」は、上杵が上方に開放されてから30秒後の錠剤厚みをいう。
本発明の低置換度ヒドロキシプロピルセルロース粉末の弾性回復率は、好ましくは7%以下であり、結合剤として汎用されている結晶セルロースと類似の塑性変形体であり、圧縮時に緻密な成型体を形成する粉体であることが解った。特許文献2や特許文献5に記載されている方法で作成した粉体は、弾性回復率が高く、弾性変形体の性質が強い粉体であった。
【0033】
本発明の低置換度ヒドロキシプロピルセルロース粉末の膨潤特性の測定方法として、例えば、低置換度ヒドロキシプロピルセルロース粉末を打錠圧1tで直径15mmの平面を有する錠剤に成型し、そこに水を滴下することにより、錠剤が膨潤するときの膨潤体積増加率と膨潤体積増加速度により評価できる。無水セルロースに対する苛性ソーダの質量比で0.1〜0.3であるアルカリセルロースを用いた場合、膨潤体積増加率は好ましくは300%以上で、膨潤体積増加速度は好ましくは100%/分以上となる。
膨潤体積増加率は、粉体を打錠圧1tで直径15mmの平面を有する錠剤に成型し、その後上杵の代わりに導管を持つ杵を取り付け、この導管を通じて臼に入った状態の錠剤に水を滴下することにより、錠剤が10分間吸水したときの膨潤体積増加率として得られる。水は、1ml/分の速度で10分間滴下する。体積の増加は、錠剤の厚み変化から以下の式により求めることができる。
膨潤体積増加率=(水添加前後の錠剤厚みの差/水添加前の錠剤厚み)×100
なお、上式中、「水添加前後の錠剤厚みの差」は、10分間の水添加後の錠剤厚みから水添加前の錠剤厚みを引いたものである。
本発明の低置換度ヒドロキシプロピルセルロース粉末の膨潤体積増加率は、崩壊剤として重要な特性である膨潤特性の点から、300%以上が好ましい。膨潤体積増加率が300%未満だと製剤化した場合に崩壊時間が延長する場合がある。
膨潤体積増加速度は、上記方法と同様の条件で膨潤体積増加率を測定したとき、水添加開始から30秒後の初期膨潤率を意味し、以下の式から求めることができる。
膨潤体積増加速度=(初期水添加前後の錠剤厚みの差/水添加前の錠剤厚み)×100/0.5
上式中、「初期水添加前後の錠剤厚みの差」は、水添加開始から30秒後の錠剤厚みから水添加前の錠剤厚みを引いたものである。
本発明の低置換度ヒドロキシプロピルセルロース粉末の膨潤体積増加速度は、崩壊剤として重要な特性である膨潤特性の点から、100%/分以上が好ましい。膨潤体積増加速度が100%/分未満であると、製剤化したときに崩壊時間が延長する場合がある。
【0034】
本発明の低置換度ヒドロキシプロピルセルロース粉末は、流動性が高く、粉体の流動性の指標の一つである安息角が好ましくは42o以下の粉体となる。安息角は、試料を平面上に落下させて堆積させた円錐の母線と水平面とのなす角度をいう。例えば、パウダーテスターPT−D型(ホソカワミクロン社製)を用いて直径80mmの金属製円盤状の台の上に75mmの高さより一定の角度になるまで試料を流出させ、堆積している粉体と台との角度を測定することにより算出できる。この角度が小さいほど流動性に優れる粉体と言える。
【0035】
また、結合性の指標として試料粉体を錠剤径11.3mmの平杵、錠剤質量480mg、圧縮圧150MPaで圧縮成型した時の錠剤硬度によって評価することができる。本発明の低置換度ヒドロキシプロピルセルロース粉末を用いた錠剤硬度は、35kgf以上、特に40kgf以上が好ましい。錠剤硬度は、例えば、タブレッティングテスター(三協パウテック社製)を用いて上記の条件で打錠を行うことにより評価できる。なお、試料粉体の水分により得られる錠剤硬度が変化するため、試料粉体の水分を一定にして行うことが好ましい。例えば試料粉体の水分を2〜4質量%に調製して行うことが好ましい。
更に、低置換度ヒドロキシプロピルセルロース粉末のゆるめ嵩密度によって評価することもできる。例えば、パウダーテスター(ホソカワミクロン社製)を用いて100mlの容器に軽く(タップすることなくの意味である)充填した時の質量を測定することにより算出することができる。本発明の低置換度ヒドロキシプロピルセルロース粉末のゆるめ嵩密度は0.3〜0.5g/ml、特に0.35〜0.45g/mlが好ましい。ゆるめ嵩密度が0.3g/ml未満だと粉体の流動性が低下して取り扱い上好ましくない場合があり、0.5g/mlを超えると結合性が低下する場合がある。
【0036】
本発明の低置換度ヒドロキシプロピルセルロース粉末は、錠剤又は顆粒剤等の固形製剤の結合剤又は崩壊剤として使用できる。錠剤は、乾式直接打錠法、湿式撹拌造粒打錠法、流動層造粒打錠法、乾式造粒打錠法等何れの製造方法によっても得ることができる。
ここで、乾式直接打錠法とは、低置換度ヒドロキシプロピルセルロース粉末と薬物、その他の賦形剤、滑沢剤等を乾式混合後、打錠する方法であり、造粒工程がないため製造工程が簡略化でき、生産性の高い方法である。湿式撹拌造粒打錠法とは、低置換度ヒドロキシプロピルセルロース粉末と薬物、その他の賦形剤等を水や水溶性結合剤溶液を用いて高速撹拌造粒装置を用いて造粒した後、乾燥して得られた粉体と滑沢剤とを混合後、打錠する方法であり、薬物の含量均一性が高い方法である。流動層造粒打錠法とは、低置換度ヒドロキシプロピルセルロース粉末と薬物、その他の賦形剤等を水や水溶性結合剤溶液を用いて流動層造粒装置を用いて造粒後、乾燥して得られた粉体と滑沢剤とを混合後、打錠する方法であり、湿式撹拌造粒打錠法と同様に薬物の含量均一性が高い方法である。乾式造粒打錠法とは、低置換度ヒドロキシプロピルセルロース粉末と薬物、その他の賦形剤等を圧縮により造粒し、打錠する方法であり、水や溶剤に対して敏感な薬物について有効な方法である。また、近年盛んに開発が進められている水無し又は少量の水で口腔内で速やかに崩壊する口腔内速崩壊錠にも適応可能である。この方法は嚥下能力の低い老人、小児に有効な剤型である。
更に、本発明の低置換度ヒドロキシプロピルセルロースは、顆粒剤の結合剤、崩壊剤としても使用できる。顆粒剤は、上記の湿式撹拌造粒、流動層造粒、乾式造粒等何れの製造方法によっても得ることができる。
なお、押出造粒により柱状の顆粒剤や押出造粒後の造粒物をマルメライザーにより球形化処理することも可能である。また、糖等からなる真球状の核に低置換度ヒドロキシプロピルセルロース粉末、薬物粉体その他の賦形剤等の混合粉末を散布しながら結合剤溶液を噴霧するレイヤリングを行なうことができる。
【0037】
本発明の低置換度ヒドロキシプロピルセルロース粉末を用いて製剤化する場合に用いられる薬物としては、特に限定されず、中枢神経系薬物、循環器系薬物、呼吸器系薬物、消化器系薬物、抗生物質及び化学療法剤、代謝系薬物、ビタミン系薬物等が挙げられる。
中枢神経系薬物としては、ジアゼパム、イデベノン、アスピリン、イブプロフェン、パラセタモール、ナプロキセン、ピロキシカム、ジクロフェナック、インドメタシン、スリンダック、ロラゼパム、ニトラゼパム、フェニトイン、アセトアミノフェン、エテンザミド、ケトプロフェン等が挙げられる。
循環器系薬物としては、モルシドミン、ビンポセチン、プロプラノロール、メチルドパ、ジピリダモール、フロセミド、トリアムテレン、ニフェジビン、アテノロール、スピロノラクトン、メトプロロール、ピンドロール、カプトプリル、硝酸イソソルビト等が挙げられる。
呼吸器系薬物としては、アンレキサノクス、デキストロメトルファン、テオフィリン、プソイドエフェドリン、サルブタモール、グアイフェネシン等が挙げられる。
消化器系薬物としては、2−{〔3−メチル−4−(2,2,2−トリフルオロエトキシ)−2−ピリジル〕メチルスルフィニル}ペンズイミダゾール及び5−メトキシ−2−〔(4−メトキシ−3,5−ジメチル−2−ピリジル)メチルスルフィニル〕ベンズミダゾール等の抗潰瘍作用を有するベンズイミダゾール系薬物、シメチジン、ラニチジン、パンクレアチン、ビサコジル、5−アミノサリチル酸等が挙げられる。
抗生物質及び化学療法剤としては、セファレキシン、セファクロール、セフラジン、アモキシシリン、ピバンピシリン、バカンピシリン、ジクロキサシリン、エリスロマイシン、エリスロマイシンステアレート、リンコマイシン、ドキシサイクリン、トリメトプリム/スルファメトキサゾール等が挙げられる。
代謝系薬物としては、セラペプターゼ、塩化リゾチーム、アデノシントリフォスフェート、グリベンクラミド、塩化カリウム等が挙げられる。
ビタミン系薬物としては、ビタミンB1、ビタミンB2、ビタミンB6、ビタミンC等が挙げられる。
【実施例】
【0038】
以下、実施例により本発明を詳細に説明するが、本発明は以下の実施例に限定されるものではない。
[実施例1]
806gの粉末状のパルプ(無水換算750g)を10L内部撹拌型反応機に仕込み26%苛性ソーダ303gを反応機に仕込み45℃で30分間混合して、無水セルロースに対する苛性ソーダの質量比が0.105のアルカリセルロースを得た。次に、窒素置換を実施し、そこにプロピレンオキサイドを123g(セルロースに対して0.164質量部)添加して、ジャケット温度60℃で1.5時間反応を行い、無水グルコース単位あたりヒドロキシプロポキシル基置換モル数0.28のヒドキシプロピルセルロース粗反応物1232gを得た。エーテル化効率は61.4%であった。
次に、10L内部撹拌型反応機に50質量%の酢酸236gを添加混合して中和を行った。この中和物をバッチ式遠心分離機を用いて回転数3000rpmの条件で、90℃の熱水にて洗浄、脱水を行った。脱水物の含水率は58.2質量%であった。この脱水物を棚段乾燥機で80℃、一昼夜乾燥を行った。
乾燥物をバッチ式遊星ボールミルFRITSH社製P−5を用いて255rpmで60分間粉砕を実施した。得られた粉砕物を目開き180μmの篩にて篩過して目的の低置換度ヒドロキシプロピルセルロース粉末を得た。この粉体の平均粒子径、比表面積、ゆるめ嵩密度、安息角、弾性回復率、結合性、膨潤体積増加率、膨潤体積増加速度を以下の方法で評価し、その結果を表1に示した。また、得られた粉体の電子顕微鏡写真を図1に示した。図1は、原料パルプ由来の表面が滑らかな繊維状形態が、圧密摩砕により消失し、一定形状で、かつ空隙のある粒子からなる粉体となっていることを示す。
【0039】
平均粒子径は、レーザー回折法粒度分布測定であるHELOS&RODOS(シンパック社製)を用いて測定した。
比表面積は、MICROMERITICS GEMINI 2375(島津製作所社製)を用いて測定した。
ゆるめ嵩密度は、パウダーテスター(ホソカワミクロン社製)を用いて直径5.03cm、高さ5.03cmの100ccの円筒容器(ステンレス製)に試料をJISの24メッシュの篩を通して、上方(23cm)から均一に軽く(タップすることなくの意味である)充填し、上面をすり切って質量を測定することにより算出した。
安息角は、パウダーテスター(ホソカワミクロン社製)を用いて80mmの円盤状の台の上に75mmの高さより流出させ、堆積している粉体と台との角度を測定することにより算出した。
弾性回復率は、試料粉体をタブレッティングテスター(三協パウテック社製)を用いて錠剤径11.3mmの平杵、錠剤質量480mg、圧縮圧50MPaで圧縮成型した時の錠剤厚みより、次式から求めた。
弾性回復率={(30秒後の錠剤厚み−最小錠剤厚み)/(最小錠剤厚み)}×100
結合性は、タブレッティングテスター(三協パウテック社製)を用いて粉体を錠剤径11.3mmの平杵、錠剤質量480mg、圧縮圧150MPaで圧縮成型した時の錠剤硬度を測定した。
膨潤体積増加率として、粉体を打錠圧1tで直径15mmの平面を有する錠剤に成型し、そこに水を滴下することにより、錠剤が吸水膨潤したときの10分後の体積増加率を測定した。
膨潤体積増加速度は、粉体を打錠圧1tで直径15mmの平面を有する錠剤に成型し、そこに水を滴下することにより、錠剤が膨潤したときの30秒後の体積変化から膨潤体積増加率を求めて算出した。
【0040】
次に、得られた粉体を用いて以下の条件でアスコルビン酸の直打錠を作製した。
錠剤組成 アスコルビン酸造粒末VC−97(BASF武田ビタミン社製) 90質量部
低置換度ヒドロキシプロピルセルロース 10質量部
ステアリン酸マグネシウム 0.5質量部
打錠機 ロータリー打錠機(菊水製作所社製)
錠剤サイズ 直径:8mm、曲面半径:6.5mm、錠剤質量:200mg
打錠圧 本圧:0.5t、予圧:0.2t
打錠速度 40rpm
得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)及び錠剤質量偏差を測定した。その結果を表1に示す。
【0041】
[実施例2]
806gの粉末状のパルプ(無水換算750g)を10L内部撹拌型反応機に仕込み26質量%苛性ソーダ504gを反応機に仕込み45℃で30分間混合して、無水セルロースに対する苛性ソーダの質量比が0.175のアルカリセルロースを得た。次に、窒素置換を実施し、そこにプロピレンオキサイドを143g(セルロースに対して0.190質量部)添加して、ジャケット温度60℃で1.5時間反応を行い、無水グルコース単位あたりヒドロキシプロポキシル基置換モル数0.29のヒドキシプロピルセルロース粗反応物1453gを得た。エーテル化効率は53.6%であった。
次に、10L内部撹拌型反応機に50質量%の酢酸394gを添加し、混合して中和を行った。この中和物をバッチ式遠心分離機を用いて回転数3000rpmの条件で90℃の熱水で洗浄、脱水を行った。脱水物の含水率は60.5質量%であった。この脱水物を棚段乾燥機で80℃、一昼夜乾燥を行った。
乾燥物をバッチ式遊星ボールミルFRITSH社製P−5を用いて255rpmで60分間粉砕を実施した。得られた粉砕物を目開き180μmの篩にて篩過して目的の低置換度ヒドロキシプロピルセルロース粉末を得た。この粉体の平均粒子径、比表面積、ゆるめ嵩密度、安息角、弾性回復率、結合性、膨潤体積増加率、膨潤体積増加速度を実施例1と同様の方法で評価し、その結果を表1に示す。
また、実施例1と同様にアスコルビン酸の直打錠を作製し、得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)及び錠剤質量偏差を測定した。その結果を表1に示した。
【0042】
[実施例3]
806gの粉末状のパルプ(無水換算750g)を10L内部撹拌型反応機に仕込み26質量%苛性ソーダ808gを反応機に仕込み45℃で30分間混合して、無水セルロースに対する苛性ソーダの質量比が0.280のアルカリセルロースを得た。次に、窒素置換を実施し、そこにプロピレンオキサイドを173g(セルロースに対して0.230質量部)添加して、ジャケット温度60℃で1.5時間反応を行い、無水グルコース単位あたりヒドロキシプロポキシル基置換モル数0.30のヒドキシプロピルセルロース粗反応物1787gを得た。エーテル化効率は46.4%であった。
次に、10L内部撹拌型反応機に50質量%の酢酸629gを添加し、混合して中和を行った。この中和物をバッチ式遠心分離機を用いて回転数3000rpmの条件で90℃の熱水で洗浄、脱水を行った。脱水物の含水率は62.2質量%であった。この脱水物を棚段乾燥機で80℃、一昼夜乾燥を行った。
乾燥物をバッチ式遊星ボールミルFRITSH社製P−5を用いて255rpmで60分間粉砕を実施した。得られた粉砕物を目開き180μmの篩にて篩過して目的の低置換度ヒドロキシプロピルセルロース粉末を得た。この粉体の平均粒子径、比表面積、ゆるめ嵩密度、安息角、弾性回復率、結合性、膨潤体積増加率、膨潤体積増加速度を実施例1と同様の方法で評価し、その結果を表1に示す。
また、実施例1と同様にアスコルビン酸の直打錠を作製し、得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)及び錠剤質量偏差を測定した。その結果を表1に示した。
【0043】
[実施例4]
8.6kgの粉末状のパルプ(無水換算8kg)を130L内部撹拌型反応機に仕込み35質量%苛性ソーダ3.5kgを反応機に仕込み45℃で30分間混合して、無水セルロースに対する苛性ソーダの質量比が0.151のアルカリセルロースを得た。次に、窒素置換を実施し、そこにプロピレンオキサイドを1.6kg(セルロースに対して0.2質量部)添加して、ジャケット温度60℃で1.5時間反応を行い、無水グルコース単位あたりヒドロキシプロポキシル基置換モル数0.35のヒドキシプロピルセルロース粗反応物13.7kgを得た。
次に、内部撹拌型反応機に50質量%の酢酸11.8kgを添加し、混合して中和を行った。この中和物をバッチ式減圧濾過機を用いて90℃の熱水で洗浄を行った。脱水物の含水率は59.5質量%であった。この脱水物を流動層乾燥機にて吸気80℃で排気温度60℃になるまで乾燥を行った。エーテル化反応効率は57.6%であった。
乾燥物をローラーミルIHI社製IS−250にて加圧力3MPa、テーブル回転数120rpm、分級回転数300rpm、風量11Nm3/min、供給時間5kg/hrで粉砕、脱水を行った。得られた粉砕物を目開き75μmの篩にて篩過して目的の低置換度ヒドロキシプロピルセルロース粉末を得た。
この粉体の平均粒子径、比表面積、ゆるめ嵩密度、安息角、弾性回復率、結合性、膨潤体積増加率、膨潤体積増加速度を実施例1と同様の方法で評価し、その結果を表1に示す。
また、実施例1と同様にアスコルビン酸の直打錠を作製し、得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)及び錠剤質量偏差を測定した。その結果を表1に示した。
【0044】
[実施例5]
806gの粉末状のパルプ(無水換算750g)を10L内部撹拌型反応機に仕込み26質量%苛性ソーダ303gを反応機に仕込み45℃で30分間混合して、無水セルロースに対する苛性ソーダの質量比が0.105のアルカリセルロースを得た。次に、窒素置換を実施し、そこにプロピレンオキサイドを81.8g(セルロースに対して0.109質量部)添加して、ジャケット温度60℃で1時間反応を行い、無水グルコース単位あたりヒドロキシプロポキシル基置換モル数0.20のヒドキシプロピルセルロース粗反応物1190.8gを得た。エーテル化効率は65.0%であった。
次に、10Lのバッチ式ニーダーに50℃の湯水2250g、氷酢酸59gを張り込み、上記粗反応品1190.8gを全量投入して溶解を行った。完全に溶解するのに20分間要した。その後、50質量%の酢酸118gを20g/分の速度で添加して中和を行った。この中和物をバッチ式遠心分離機を用いて回転数3000rpmの条件で90℃の熱水で洗浄、脱水を行った。脱水物の含水率は62.2質量%であった。この脱水物を棚段乾燥機で80℃、一昼夜乾燥を行った。
乾燥物をバッチ式遊星ボールミルFRITSH社製P−5を用いて255rpmで60分間粉砕を実施した。得られた粉砕物を目開き75μmの篩にて篩過して目的の低置換度ヒドロキシプロピルセルロース粉末を得た。この粉体の平均粒子径、ゆるめ嵩密度、安息角、弾性回復率、結合性、膨潤体積増加率、膨潤体積増加速度を実施例1と同様の方法で評価し、その結果を表1に示す。
また、実施例1と同様にアスコルビン酸の直打錠を作製し、得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)及び錠剤質量偏差を測定した。その結果を表1に示した。
【0045】
[比較例1]
806gの粉末状のパルプ(無水換算750g)を10L内部撹拌型反応機に仕込み13質量%苛性ソーダ300gを反応機に仕込み45℃で30分間混合して、無水セルロースに対する苛性ソーダの質量比が0.052のアルカリセルロースを得た。次に、窒素置換を実施し、そこにプロピレンオキサイドを146g(セルロースに対して0.195質量部)添加して、ジャケット温度60℃で1.5時間反応を行い、無水グルコース単位あたりヒドロキシプロポキシル基置換モル数0.28のヒドキシプロピルセルロース粗反応物1252gを得た。エーテル化効率は50.9%であった。
次に、10L内部撹拌型反応機に50質量%の酢酸117gを添加し、混合して中和を行った。この中和物をバッチ式遠心分離機を用いて回転数3000rpmの条件で90℃の熱水で洗浄、脱水を行った。脱水物の含水率は50.1質量%であった。この脱水物を棚段乾燥機で80℃、一昼夜乾燥を行った。
乾燥物をバッチ式遊星ボールミルFRITSH社製P−5を用いて255rpmで60分間粉砕を実施した。得られた粉砕物を目開き180μmの篩にて篩過して目的の低置換度ヒドロキシプロピルセルロース粉末を得た。この粉体の平均粒子径、比表面積、ゆるめ嵩密度、安息角、弾性回復率、結合性、膨潤体積増加率、膨潤体積増加速度を実施例1と同様の方法で評価し、その結果を表1に示す。
また、実施例1と同様にアスコルビン酸の直打錠を作製し、得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)及び錠剤質量偏差を測定した。その結果を表1に示した。
【0046】
[比較例2]
806gの粉末状のパルプ(無水換算750g)を10L内部撹拌型反応機に仕込み35質量%苛性ソーダ761gを反応機に仕込み45℃で30分間混合して、無水セルロースに対する苛性ソーダの質量比が0.355のアルカリセルロースを得た。次に、窒素置換を実施し、そこにプロピレンオキサイドを161g(セルロースに対して0.214質量部)添加して、ジャケット温度60℃で1.5時間反応を行い、無水グルコース単位あたりヒドロキシプロポキシル基置換モル数0.28のヒドキシプロピルセルロース粗反応物1728gを得た。エーテル化効率は43.5%であった。
次に、10L内部撹拌型反応機に50質量%の酢酸800gを添加し、混合して中和を行った。この中和物をバッチ式遠心分離機を用いて回転数3000rpmの条件で90℃の熱水で洗浄、脱水を行った。脱水物の含水率は65.2質量%であった。この脱水物を棚段乾燥機で80℃、一昼夜乾燥を行った。
乾燥物をバッチ式遊星ボールミルFRITSH社製P−5を用いて255rpmで60分間粉砕を実施した。得られた粉砕物を目開き180μmの篩にて篩過して目的の低置換度ヒドロキシプロピルセルロース粉末を得た。この粉体の平均粒子径、比表面積、ゆるめ嵩密度、安息角、弾性回復率、結合性、膨潤体積増加率、膨潤体積増加速度を実施例1と同様の方法で評価し、その結果を表1に示す。
また、実施例1と同様にアスコルビン酸の直打錠を作製し、得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)及び錠剤質量偏差を測定した。その結果を表1に示した。
【0047】
[比較例3]
806gの粉末状のパルプ(無水換算750g)を10L内部撹拌型反応機に仕込み43質量%苛性ソーダ844gを反応機に仕込み45℃で30分間混合して、無水セルロースに対する苛性ソーダの質量比が0.484のアルカリセルロースを得た。次に、窒素置換を実施し、そこにプロピレンオキサイドを165g(セルロースに対して0.220質量部)添加して、ジャケット温度60℃で1.5時間反応を行い、無水グルコース単位あたりヒドロキシプロポキシル基置換モル数0.27のヒドキシプロピルセルロース粗反応物1815gを得た。エーテル化効率は42.3%であった。
次に、10L内部撹拌型反応機に50質量%の酢酸1088gを添加し、混合して中和を行った。この中和物をバッチ式遠心分離機を用いて回転数3000rpmの条件で90℃の熱水で洗浄、脱水を行った。脱水物の含水率は66.8質量%であった。この脱水物を棚段乾燥機で80℃、一昼夜乾燥を行った。
乾燥物をバッチ式遊星ボールミルFRITSH社製P−5を用いて255rpmで60分間粉砕を実施した。得られた粉砕物を目開き180μmの篩にて篩過して目的の低置換度ヒドロキシプロピルセルロース粉末を得た。この粉体の平均粒子径、比表面積、ゆるめ嵩密度、安息角、弾性回復率、結合性、膨潤体積増加率、膨潤体積増加速度を実施例1と同様の方法で評価し、その結果を表1に示す。
また、実施例1と同様にアスコルビン酸の直打錠を作製し、得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)及び錠剤質量偏差を測定した。その結果を表1に示した。
【0048】
[比較例4]
シート状パルプを43%苛性ソーダ溶液に浸せき後、圧搾して22.2質量%の苛性ソーダを含有し、無水セルロースに対する苛性ソーダの質量比が0.484であるアルカリセルロースを得た。得られたアルカリセルロースを約1cm角のチップ状に裁断し、セルロース換算で350gのアルカリセルロースを5Lの容量の反応機に仕込み、窒素置換を行った。そこにプロピレンオキサイドを79g(セルロースに対して0.226質量部)添加して、ジャケット温度45℃で2時間、65℃で30分間反応を行い、無水グルコース単位あたりヒドロキシプロポキシル基置換モル数0.27のヒドキシプロピルセルロース粗反応物857gを得た。エーテル化効率は42.0%であった。
次に、特許文献2及び特許文献3のように粗反応物の一部を熱水で溶解後、中和を行い(部分中和法)、得られた乾燥物を衝撃粉砕で行った試料を作製した。
5Lのバッチ式ニーダーに50℃の水1750g、氷酢酸52gを張り込み上記粗反応物857gを全量投入して溶解を行った。完全に溶解するのに30分を要した。その後、33%酢酸633gを20g/minの速度で添加して中和析出を行った。
この中和物をバッチ式遠心分離機を用い3000rpmの回転数にて90℃の熱水で洗浄を行った。脱水物の含水率は75.4質量%であった。この脱水物を送風オーブンにて80℃、一昼夜乾燥を行った。
得られた乾燥物を目開き0.3mmのスクリーンを有する高速回転型衝撃粉砕機ビクトリーミルを用いて粉砕を実施し、粉砕物を目開き75μmの篩にて篩過して目的の低置換度ヒドロキシプロピルセルロース粉末を得た。この粉体の平均粒子径、比表面積、ゆるめ嵩密度、安息角、弾性回復率、結合性、膨潤体積増加率、膨潤体積増加速度を実施例1と同様の方法で評価し、その結果を表1に示す。
また、実施例1と同様にアスコルビン酸の直打錠を作製し、得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)及び錠剤質量偏差を測定した。その結果を表1に示した。また、得られた粉体の電子顕微鏡写真を図2に示した。図2は、原料パルプ由来の表面が滑らかな繊維状形態と、溶解工程により生じた球状の緻密な粒子が混在した粉体になっていることを示す。
【0049】
[比較例5]
特許文献4記載の完全溶解後中和を行い、得られた乾燥物を衝撃粉砕で行った試料を作製した。
5Lのバッチ式ニーダーに50℃の水2450gを張り込み比較例4と同様の方法で反応した粗反応物857gを全量投入して溶解(完全溶解)を行った。完全に溶解するのに1時間を要した。その後、33%酢酸793gを20g/minの速度で添加して中和析出を行った。
この中和物をバッチ式遠心分離機を用い3000rpmの回転数にて90℃の熱水で洗浄、脱水を行った。脱水物の含水率は80.1質量%であった。この脱水物を送風オーブンにて80℃、一昼夜乾燥を行った。
得られた乾燥物を目開き0.3mmのスクリーンを有する高速回転型衝撃粉砕機ビクトリーミルを用いて粉砕を実施し、粉砕物を目開き75μmの篩にて篩過して目的の低置換度ヒドロキシプロピルセルロース粉末を得た。この粉体の平均粒子径、ゆるめ嵩密度、安息角、弾性回復率、結合性、膨潤体積増加率、膨潤体積増加速度を実施例1と同様の方法で評価し、その結果を表1に示す。
また、実施例1と同様にアスコルビン酸の直打錠を作製し、得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)及び錠剤質量偏差を測定した。その結果を表1に示した。また、得られた粉体の電子顕微鏡写真を図3に示した。図3は、溶解工程により生じた繊維状粒子が少なく、球状の緻密な粒子からなる粉体になっていることを示す。
【0050】
[比較例6]
目開き0.3mmのスクリーンを有する高速回転型衝撃粉砕機ビクトリーミルを用いた以外は実施例2と同様の方法にて低置換度ヒドロキシプロピルセルロース粉末を得た。この粉体の平均粒子径、比表面積、ゆるめ嵩密度、安息角、弾性回復率、結合性、膨潤体積増加率、膨潤体積増加速度を実施例1と同様の方法で評価し、その結果を表1に示す。
また、実施例1と同様にアスコルビン酸の直打錠を作製し、得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)及び錠剤質量偏差を測定した。その結果を表1に示した。また、得られた粉体の電子顕微鏡写真を図4に示した。図4は、原料パルプ由来の表面が滑らかな繊維状形態からなる粉体になっていることを示す。
【0051】
アルカリセルロース中の無水セルロースに対する苛性ソーダの質量比の膨潤特性に与える影響をみた結果を図5に示す。図5は、横軸に水の添加開始からの測定時間を、縦軸に膨潤体積増加率を示す。
実施例は、比較例に比べ、膨潤体積増加率が大きく、膨潤体積増加速度も大きいことがわかる。
【0052】
【表1】
【0053】
表1から明らかなように、所定の苛性ソーダの質量比を有し、圧密摩砕を用いたものは比表面積が大きく、弾性回復率が小さくなるため、結合性が高くなり、製剤化した場合に錠剤硬度が高い錠剤が得られる。また、膨潤体積増加率及び膨潤体積増加速度が高くなるので、製剤化した場合、崩壊時間が短い製剤が得られた。一方、圧密摩砕を用いても所定の苛性ソーダの質量比を用いない粉体は、比表面積、結合性、崩壊剤として重要な膨潤特性である膨潤体積増加率、錠剤の硬度と崩壊時間に劣ることが比較例1〜3の結果に示されている。また、比較例2〜3の結果では、崩壊剤として重要な膨潤特性である膨潤体積増加速度にも劣っている。所定の苛性ソーダの質量比を用いても、圧密摩砕に換えて衝撃粉砕を用いて得られた粉体は、比表面積、流動性、結合性、錠剤の硬度と崩壊時間に劣り、錠剤質量偏差が大きいことが比較例6の結果に示されている。比較例4は、シート状のパルプを使用しているため、過剰な苛性ソーダを含むアルカリセルロースを用いることとなり、膨潤体積増加速度が劣る。また、衝撃粉砕機を用いているため、比表面積が小さく、かつ弾性回復率が大きく、結合性が低かった。一方、比較例5は、完全溶解により繊維状形態が少ないため、安息角が低く、流動性に優れるが、結合性が低かった。
【0054】
1)口腔内崩壊錠
[実施例6]
実施例1に記載の方法により得られた低置換度ヒドロキシプロピルセルロースを用いて下記条件にて口腔内速崩壊錠を作製した。
錠剤組成 エリスリトール(250μm篩過品) 70質量部
低置換度ヒドロキシプロピルセルロース 30質量部
ステアリン酸マグネシウム 0.5質量部
打錠機 単発打錠機 タブレッティングテスター(三協パウテック社製)
錠剤サイズ 直径:11.3mm、平杵
打錠圧 10、25、50MPa
得られた錠剤の錠剤硬度、口腔内崩壊時間及び服用感を評価した。口腔内崩壊時間は健康な成人4名に口腔内で錠剤を噛まず、軽く口に含んだ状態で錠剤が完全に溶解又は崩壊するまでの時間を測定し、その平均値を算出した。錠剤硬度及び口腔内崩壊時間の結果を表2に示す。
【0055】
[比較例7]
比較例4に記載の方法により得られた低置換度ヒドロキシプロピルセルロースを用いた以外は、実施例6と同様の方法により口腔内速崩壊錠を作製した。得られた錠剤の評価を実施例6と同様の方法にて行い、その結果を表2に示す。
【0056】
【表2】
【0057】
実施例6は、比較例7と比較して高い錠剤硬度を示し、かつ口腔内での崩壊時間が短いものであった。また、実施例6は何れの錠剤もざらつきがなくクリーミーな服用感であったのに対して、比較例7では紙を口に含んだ様であり、服用感が悪いものであった。
【0058】
2)湿式撹拌造粒
[実施例7]
実施例4に記載の方法により得られた低置換度ヒドロキシプロピルセルロースを用いて下記組成の粉体をバーチカルグラニュレーターFM−VG−05(パウレック社製)に仕込み、主軸600rpm、チョッパー1000rpmの回転数にて1分間前混合を実施し、水55gを添加し5分間混合して湿式撹拌造粒を行なった。
錠剤組成 エテンザミド 210g(70質量部)
200#乳糖 60g(20質量部)
低置換度ヒドロキシプロピルセルロース 30g(10質量部)
造粒物を1180μmの目開きの篩にて篩過後、流動層乾燥機にて、吸気60℃、流動エアー量70m3/hrで乾燥を行い、排気温度45℃になるまで乾燥を行った。
得られた顆粒の平均粒子径、ゆるめ嵩密度、固め嵩密度、圧縮度、安息角を表3に示す。
なお、得られた顆粒の平均粒子径、ゆるめ嵩密度、安息角の測定方法は、低置換度ヒドロキシプロピルセルロース粉末における測定方法と同様であり、固め嵩密度及び圧縮度は以下のよう方法により測定した。
すなわち、固め嵩密度とは、ゆるめ嵩密度にタッピングを加えて密充填にした場合の嵩密度をいい、タッピングとは、試料を充填した容器を一定の高さから繰り返し落下させて底部に軽い衝撃を与え、試料を密充填にする操作をいう。従って、固め嵩密度は、ゆるめ嵩密度を測定する際、上面をすり切って秤量した後、更にこの容器の上にキャップ(下記ホソカワミクロン社製パウダーテスターの備品)をはめ、この上縁まで粉体を加えてタップ高さ1.8cmのタッピングを180回行なう。タッピング終了後、キャップを外して容器の上面で粉体をすり切って秤量する。これらの操作は、ホソカワミクロン社製パウダーテスター(PT−D)を使用することにより測定できる。
また、圧縮度とは、かさべりの度合いを示す値であり、以下の式で求められる。
圧縮度(%)=[(固め嵩密度−ゆるめ嵩密度)/固め嵩密度]×100
【0059】
次に、上記顆粒100質量部に対してステアリン酸マグネシウム0.5質量部を加えた後、混合して打錠用の粉体を得た。この粉体を用いて下記条件にて連続打錠を実施した。
打錠機 ロータリー打錠機 (菊水製作所社製)
錠剤サイズ 直径:8mm、曲率半径:6.5mm、錠剤重量:200mg
打錠圧 本圧:1t、予圧:0.2t
打錠速度 40rpm
得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)、日本薬局方摩損度試験に準じた下記方法にて錠剤摩損度、摩損度試験後にキャッピングが発生している錠剤数を測定することによりキャッピング発生率、錠剤質量偏差を評価した。
摩損度試験は、日本薬局方記載の摩損度試験器を用いて20錠を25rpmで16分間試験を行った後の質量減少率を下記の式より算出した。
[{(試験前の錠剤質量)−(試験後の錠剤質量)}/(試験前の錠剤質量)]×100
キャッピング発生率は、上記摩損度試験実施後、錠剤が2層に割れていた錠剤数から、キャッピング発生率を算出した。
錠剤質量偏差は、錠剤20錠の質量を測定することにより、その変動係数(CV値)を算出した。
得られた結果を表4に示す。
【0060】
[比較例8]
比較例4に記載の方法により得られた低置換度ヒドロキシプロピルセルロースを用いた以外は、実施例7と同様の方法により湿式撹拌造粒を実施した。得られた顆粒の平均粒子径、ゆるめ嵩密度、固め嵩密度、圧縮度、安息角を表3に示した。
次に、実施例7と同様の方法にて連続打錠を実施し、その評価を行った。その結果を表4に示す。
【0061】
[比較例9]
低置換度ヒドロキシプロピルセルロースの代わりに微結晶セルロースCeolusPH−101(旭化成ケミカルズ社製)を用いた以外は、実施例7と同様の方法により湿式撹拌造粒を実施した。ただし、微結晶セルロースは低置換度ヒドロキシプロピルセルロースと比較して保水性が低いため、水を36gに減量した。得られた顆粒の平均粒子径、ゆるめ嵩密度、固め嵩密度、圧縮度、安息角を表3に示す。
次に、実施例7と同様の方法にて連続打錠を実施し、その評価を行った。その結果を表4に示す。
【0062】
【表3】
【0063】
実施例7は、比較例8、9と比較してゆるめ嵩密度が高く重質で、圧縮度及び安息角が低いことにより、流動性に優れる顆粒が得られた。
【0064】
【表4】
【0065】
実施例7は、比較例8と比較して錠剤硬度が高く、錠剤質量偏差の低い優れた製剤が得られた。また、微結晶セルロースを用いた比較例9では、キャッピングが発生し、錠剤摩損度が高く、錠剤硬度が低いものであったが、実施例7は、全ての評価項目において優れた製剤であった。
【0066】
3)流動層造粒
[実施例8]
実施例1に記載の方法により得られた低置換度ヒドロキシプロピルセルロースを用いて下記組成の粉体を流動層造粒装置マルチプレックスMP−01(パウレック社製)に仕込み、吸気60℃、流動エアー量50m3/hr、排気温度30〜35℃、ヒドロキシプロピルメチルセルロースTC−5R(信越化学工業社製)5質量%水溶液60gを10g/分の速度で噴霧して造粒を行った。
錠剤組成 アセトアミノフェン 80g(40質量部)
200#乳糖 28g(14質量部)
コーンスターチ 12g( 6質量部)
低置換度ヒドロキシプロピルセルロース 80g(40質量部)
得られた顆粒の平均粒子径、ゆるめ嵩密度、固め嵩密度、圧縮度、安息角を表5に示す。
次に、上記顆粒100質量部に対してステアリン酸マグネシウム0.5質量部を加えた後、混合して打錠用の粉体を得た。この粉体を用いて実施例7と同様に連続打錠を実施した。得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)、錠剤質量偏差を評価した。その結果を表6に示す。
【0067】
[比較例10]
比較例4に記載の方法により得られた低置換度ヒドロキシプロピルセルロースを用いた以外は、実施例8と同様の方法により流動層造粒を実施した。得られた顆粒の平均粒子径、ゆるめ嵩密度、固め嵩密度、圧縮度、安息角を表5に示した。
次に、実施例8と同様の方法にて連続打錠を実施し、その評価を行った。その結果を表6に示す。
【0068】
[比較例11]
比較例5に記載の方法により得られた低置換度ヒドロキシプロピルセルロースを用いた以外は、実施例8と同様の方法により流動層造粒を実施した。得られた顆粒の平均粒子径、ゆるめ嵩密度、固め嵩密度、圧縮度、安息角を表5に示す。
次に、実施例8と同様の方法にて連続打錠を実施し、その評価を行った。その結果を表6に示す。
【0069】
【表5】
【0070】
実施例8は、比較例10、11と比較してゆるめ嵩密度が高く重質で、圧縮度及び安息角が低いことにより、流動性に優れる顆粒が得られた。
【0071】
【表6】
【0072】
実施例8は、比較例10、11と比較して錠剤硬度が高く、錠剤質量偏差の少ない製剤であった。
【0073】
4)押出造粒
[実施例9]
実施例4に記載の方法により得られた低置換度ヒドロキシプロピルセルロースを用いて下記組成の粉体をバーチカルグラニュレーターFM−VG−05(パウレック社製)に仕込み、主軸600rpm、チョッパー1000rpmの回転数にて1分間前混合を実施し、水50gを添加し5分間混合して湿式撹拌造粒を行なった。
錠剤組成 アスピリン 279g(93質量部)
低置換度ヒドロキシプロピルセルロース 15g( 5質量部)
ヒドロキシプロピルメチルセルロースTC−5E 6g( 2質量部)
次に、押出造粒装置ドームグラン(ダルトン社製)1.0mmφのスクリーンを用いて上記湿粉の押出造粒を実施し、固形分に換算した造粒速度を表7に示した。また、得られた押出造粒物をマルメライザー(ダルトン社製)を用いて750rpmで球形化処理を行った。その後、流動層乾燥機にて、吸気60℃、流動エアー量70m3/hrで乾燥を行い、排気温度45℃になるまで乾燥を行った。得られた顆粒の顆粒強度、日本薬局方崩壊試験における崩壊時間(試験液:水)を測定した。その結果を表7に示す。
なお、顆粒強度は、日本薬局方摩損度試験機を用いて、そこに顆粒10gを仕込み直径5mmのガラスビーズ20個を入れ、25rpm、4分間試験を行った後の質量減少率を下記の式より算出した。
[{(試験前の顆粒質量)−(試験後の顆粒質量)}/(試験前の顆粒質量)]×100
【0074】
[比較例12]
比較例4に記載の方法により得られた低置換度ヒドロキシプロピルセルロースを用いた以外は、実施例9と同様の方法により押出造粒を実施し、固形分に換算した造粒速度を表7に示した。得られた湿式造粒物を実施例9と同様の方法にて球形化、乾燥を行った。
得られた顆粒の顆粒強度、日本薬局方崩壊試験における崩壊時間(試験液:水)を測定した。その結果を表7に示す。
【0075】
【表7】
【0076】
実施例9は、比較例12と比較して製粒速度が速く、生産性が高く、得られた顆粒の顆粒強度が高く優れた製剤であった。
【0077】
5)乾式造粒
[実施例10]
実施例4に記載の方法により得られた低置換度ヒドロキシプロピルセルロースを用いて下記組成の粉体をローラーコンパクターMINI(フロイント社製)を用い、ロール圧4MPa、ロール回転数5rpm、スクリュー回転数12rpmで乾式造粒を行った。
錠剤組成 アスコルビン酸 50g(10質量部)
200#乳糖 245g(49質量部)
コーンスターチ 105g(21質量部)
低置換度ヒドロキシプロピルセルロース 100g(20質量部)
ステアリン酸マグネシウム 2.5g(0.5質量部)
造粒物を250μm目開きの篩にて篩過し、この造粒物を下記条件で連続打錠を実施した。
打錠機 ロータリー打錠機 (菊水製作所社製)
錠剤サイズ 直径:8mm、曲率半径:6.5mm、錠剤重量:200mg
打錠圧 本圧:1t、予圧:0.2t
打錠速度 40rpm
得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)を評価した結果を表8に示す。
【0078】
[比較例13]
比較例4に記載の方法により得られた低置換度ヒドロキシプロピルセルロースを用いた以外は、実施例10と同様の方法により乾式造粒を実施し、実施例10と同様の方法にて連続打錠を実施した。得られた錠剤の錠剤硬度、日本薬局方崩壊試験における崩壊時間(試験液:水)を評価した結果を表8に示す。
【0079】
【表8】
【0080】
実施例10は、比較例13と比較して崩壊時間は同等でありながら錠剤硬度が高い製剤であった。
【特許請求の範囲】
【請求項1】
平均粒子径が10〜100μmであり、かつBET法で測定した比表面積が1.0m2/g以上であり、圧縮圧50MPaで圧縮成型した時の弾性回復率が7%以下である低置換度ヒドロキシプロピルセルロース粉末。
【請求項2】
吸水時の膨潤体積増加率が300%以上で、膨潤体積増加速度が100%/分以上である請求項1に記載の低置換度ヒドロキシプロピルセルロース粉末。
【請求項3】
安息角が42o以下である請求項2又は請求項3に記載の低置換度ヒドロキシプロピルセルロース粉末。
【請求項4】
請求項1〜3のいずれかに記載の低置換度ヒドロキシプロピルセルロース粉末を用いた固形製剤。
【請求項1】
平均粒子径が10〜100μmであり、かつBET法で測定した比表面積が1.0m2/g以上であり、圧縮圧50MPaで圧縮成型した時の弾性回復率が7%以下である低置換度ヒドロキシプロピルセルロース粉末。
【請求項2】
吸水時の膨潤体積増加率が300%以上で、膨潤体積増加速度が100%/分以上である請求項1に記載の低置換度ヒドロキシプロピルセルロース粉末。
【請求項3】
安息角が42o以下である請求項2又は請求項3に記載の低置換度ヒドロキシプロピルセルロース粉末。
【請求項4】
請求項1〜3のいずれかに記載の低置換度ヒドロキシプロピルセルロース粉末を用いた固形製剤。
【図5】

【図1】

【図2】
【図3】

【図4】

【図1】
【図2】
【図3】
【図4】
【公開番号】特開2012−211340(P2012−211340A)
【公開日】平成24年11月1日(2012.11.1)
【国際特許分類】
【出願番号】特願2012−169614(P2012−169614)
【出願日】平成24年7月31日(2012.7.31)
【分割の表示】特願2007−206387(P2007−206387)の分割
【原出願日】平成19年8月8日(2007.8.8)
【出願人】(000002060)信越化学工業株式会社 (3,361)
【Fターム(参考)】
【公開日】平成24年11月1日(2012.11.1)
【国際特許分類】
【出願日】平成24年7月31日(2012.7.31)
【分割の表示】特願2007−206387(P2007−206387)の分割
【原出願日】平成19年8月8日(2007.8.8)
【出願人】(000002060)信越化学工業株式会社 (3,361)
【Fターム(参考)】
[ Back to top ]