
併用薬剤を送達するための組成物
【課題】治療薬の相乗的又は相加的な組合わせを送達する、改良された送達のための組成物及び方法を提供する。
【解決手段】抗腫瘍薬などの、2種類またはそれ以上の薬剤の非拮抗的な組合せが安定的に会合した送達媒体を含む組成物、及び薬剤の組合せを含有する該組成物の投与による非拮抗的な作用を有する達成システム。
【解決手段】抗腫瘍薬などの、2種類またはそれ以上の薬剤の非拮抗的な組合せが安定的に会合した送達媒体を含む組成物、及び薬剤の組合せを含有する該組成物の投与による非拮抗的な作用を有する達成システム。
【発明の詳細な説明】
【技術分野】
【0001】
関連出願の相互参照
本出願は、米国特許法第119条(e)項(35 U.S.C. §119(e))に基づき、2001年10月3日に提出された米国仮特許出願第60/326,671号;2001年12月17日に提出された第60/341,529号;2002年2月15日に提出された第60/356,759号;2002年4月23日に提出されたカナダ仮特許出願第2,383,259号;2002年8月7日に提出された米国仮特許出願第60/401,984号および2002年9月6日に提出された第60/408,733号の恩典を主張する。これらの出願の内容は参照として本明細書に組み入れられる。
【0002】
技術分野
本発明は、治療薬の相乗的または相加的な組合せを送達する、改良された送達のための組成物および方法に関する。より詳細には、本発明は、送達媒体を含む製剤を提供することによって、薬剤を意図した標的に対して送達する場合に、相乗的または相加的な比を維持することを保証する送達システムに関する。
【背景技術】
【0003】
背景技術
癌、AIDS、感染症、免疫疾患および心血管疾患といった命にかかわる多くの疾患は、数多くの分子機構による影響を受ける。この複雑性のために、単一の薬剤を用いて治癒を達成しようとした場合の奏功性には限界がある。このため、薬剤の併用が、疾患との戦い、特に癌の治療にしばしば用いられている。投与した薬剤の数と急性リンパ球性白血病などの癌の治癒率との間には強い相関があるように思われる(Freiら、Clin. Cancer Res. (1998) 4: 2027-2037(非特許文献1))。ドキソルビシン、シクロホスファミド、ビンクリスチン、メトトレキサート、ロイコボリン(救済用)およびシタラビンの組合せ(ACOMLA)、またはシクロホスファミド、ドキソルビシン、ビンクリスチン、プレドニゾンおよびブレオマイシンの組合せ(CHOP-b)を用いた臨床試験では、組織球性リンパ腫の治療にこれらが用いられて奏功している(Toddら、J. Clin. Oncol. (1984) 2: 986-993(非特許文献2))。
【0004】
薬剤併用の効果は、それらの供給される比によって相乗作用がもたらされる場合に強化される。また、薬剤の相乗的組合せが、必要投与量の減少によって毒性を低下させること、癌の治癒率を高めること(Barriereら、Pharmacotherapy (1992) 12: 397-402(非特許文献3);Schimpff, Support Care Cancer (1993) 1: 5-18(非特許文献4))および微生物の多剤耐性株の蔓延を抑えること(Shlaesら、Clin. Infect. Dis. (1993) 17:S527-S536(非特許文献5))も示されている。作用機序の異なる複数の薬剤を選択することにより、生化学経路における複数の部位を攻撃し、それによって相乗作用を得ることができる(ShahおよびSchwartz、Clin. Cancer Res. (2001) 7: 2168-2181(非特許文献6))。L-カナバニンと5-フルオロウラシル(5-FU)との併用などは、ラット結腸腫瘍モデルにおいて各薬剤の単独での効果の合計よりも強い抗腫瘍活性を示すことが報告されている(Swaffarら、Anti-Cancer Drugs (1995) 6: 586-593(非特許文献7))。シスプラチンとエトポシドは、ヒト小細胞肺癌細胞株SBC-3の増殖の抑制に関して相乗作用を示す(Kanzawaら、Int. J. Cancer (1997) 71(3): 311-319(非特許文献8))。
【0005】
相乗作用に関するこのほかの報告には、以下に関するものがある。
ビンブラスチンと組換えインターフェロンβ(Kueblerら、J. Interferon Res. (1990) 10: 281-291(非特許文献9));
シスプラチンとカルボプラチン(Kobayashiら、Nippon Chiryo Gakkai Shi (1990) 25: 2684-2692(非特許文献10));
エチルデスヒドロキシ-スパルソマイシンとシスプラチンまたはシトシンアラビノシド(AraC)またはメトトレキサートまたは5-FUまたはビンクリスチン(Hofsら、Anti Cancer Drugs (1994) 5: 35-42(非特許文献11));
全トランスレチノイン酸と酪酸またはトリブチリン(Chenら、Chin. Med. Engl. (1999) 112: 352-355(非特許文献12));および
シスプラチンとパクリタキセル(Engblomら、Br. J. Cancer (1999) 79: 286-292(非特許文献13))。
【0006】
以上の試験では、成分の比が相乗作用にとって重要であることが認められた。例えば、5-フルオロウラシルおよびL-カナバニンはモル比が1:1では相乗的であるが、比が5:1では拮抗性であることが見いだされた;シスプラチンおよびカルボプラチンは血中濃度曲線下面積(AUC)比が13:1では相乗作用を示したが、19:5では拮抗作用を示した。
【0007】
相乗的な相互作用を示すものの、相互作用が組合せ比(combination ratio)に依存することが記載されていない、薬剤のその他の組合せもある。このリストは非常に広範囲にわたり、インビボ試験も時に含まれるが、主としてインビトロ培養の報告によって構成される。
【0008】
多数の報告に加えて、さまざまな組合せが臨床の場で有効であることが示されている。これらを以下の表に記した。

a FDA:米国食品医薬品局
【0009】
加えて、ある種の他の組合せは、非拮抗的な併用効果もしくは臨床的有効性を示す可能性があることを文献中のさまざまな報告から推論され、または地域の研究グループによって標準的な医療行為として受容されうる。これらには以下のものがある。
【0010】
相乗的な薬剤の組合せを用いることには前述のような利点があるものの、その治療的使用を制限するさまざまな欠点もある。例えば、相乗作用はしばしば、薬剤曝露の期間および投与の順序といった種々の要因に依存する(BonnerおよびKozelsky、Cancer Chemother. Pharmacol. (1990) 39: 109-112(非特許文献14))。エチルデスヒドロキシ-スパルソマイシンをシスプラチンと併用した試験では、この相乗作用が組合せ比、投与期間および投与の順序によって影響されることが示されている(Hofsら、前記(非特許文献11))。
【0011】
このため、相乗作用を薬剤の併用によって発揮させるためには、これらの薬剤が、規定された比を示す量で存在する必要があることが知られている。事実、同じ薬剤の組合せが、ある比では拮抗的であり、別の比では相乗的であり、さらに別の比では相加的であることもある。拮抗的作用を回避し、薬剤が少なくとも相加的であるようにすることが望ましい。本発明は、個々の比で得られる結果が濃度にも依存することを認識している。比の中にはある濃度では拮抗的であり、別の濃度では非拮抗的なものがある。本発明は、これらの薬剤が対象内部の標的部位に到達した場合に所望の比または投与された比を維持する製剤中にある形で送達することにより、および、標的での濃度は投与したものとは異なる可能性があるという理由から、所望の濃度の範囲で顕著に非拮抗的である比を選択することにより、成分の比が相乗的または相加的な範囲にあることを保証する。
【0012】
PCT公報・国際公開公報第00/51641号(特許文献1)は、相乗的であると言われている抗ウイルス薬の組合せを投与することを記載している。相乗的な比を決定するためにインビトロ試験が用いられた。しかし、この比をインビボで維持すると考えられる投与様式に関する教示はない。実際には、この刊行物は、成分を逐次的に投与しても同時に投与してもよいと記載している。
【0013】
PCT公報・国際公開公報第01/15733号(特許文献2)は、自己免疫疾患の治療のための、相乗的であると想定される組成物を記載している。この場合も、製剤化の方法は、送達後にこの比が維持されることを保証していない。
【0014】
Daoudら、Cancer Chemother. Pharmacol. (1991) 28: 370-376(非特許文献15)は、ヒト卵巣癌細胞に対するシスプラチンおよびリポソーム封入バリノマイシンの相乗的な細胞傷害作用を記載している。この論文は、遊離状態にあるシスプラチンとリポソーム中に封入されたバリノマイシンとをヒト卵巣腫瘍由来細胞株CaOV-3の培養物の処置に用いるインビトロアッセイ法を記載している。著者らは相乗作用および拮抗作用が示される濃度範囲を決定した。リポソーム封入はバリノマイシンを溶解させるために用いられた。実験はインビトロで行われているため、インビボ送達は関係しない。
【0015】
2001年4月10日にDaoudに対して発行された米国特許第6,214,821号(特許文献3)は、トポイソメラーゼI阻害物質およびスタウロスポリンを含む薬学的組成物を記載している。その特許請求の範囲は、スタウロスポリンに、トポイソメラーゼI阻害物質により誘導されるS期停止を無効化する能力、および、正常なp53機能を欠くヒト乳癌細胞に対するその細胞傷害性を増強させる能力があるという発見に基づくように思われる。具体的な医薬製剤は提唱されていない。
【0016】
Fountainらに対する米国特許第5,000,958号(特許文献4)は、インビボでの治療効果の増強を発揮すると言われている、リポソーム中に封入された抗菌薬の混合物を記載している。抗菌薬の適した比は、インビトロでの相乗作用を経験的に調べる併用効果試験によって決定される。ある濃度範囲にわたる相乗的な比の保証に関する考察は行われていない。
【0017】
Schiffelersら、J. Pharmacol. Exp. Therapeutic (2001) 298: 369-375(非特許文献16)は、リポソーム中に共封入されたゲンタマイシンおよびセフタジジムのインビボでの相乗的な相互作用を記載している。望ましい比は、Fountain(前記(特許文献4))のものに類似した併用効果試験を用いて決定されたが、ある濃度範囲にわたって相乗作用が維持される比の決定に関する考察はなされていない。
【0018】
本発明は、第1に、治療薬の決定された相乗的または相加的な比を、それらが内部に投与された製剤の薬物動態を制御することによって維持しうることを認識しており、第2に、標的組織に到達する薬剤混合物における成分の濃度はそれが投与されたものと同じではない可能性があるため、ある濃度範囲にわたって非拮抗的な比が示される必要があることを認識している。相乗作用または相加性を維持するという問題は、治療薬をリポソームなどの送達媒体中に封入した(すなわち、安定的に会合させた)場合には送達媒体が薬物動態を決定し、そのため封入された複数の薬剤が同じように振る舞うという認識により、および、ある濃度範囲にわたって顕著に相乗的/相加的である比を選択することにより、解決される。
【先行技術文献】
【特許文献】
【0019】
【特許文献1】国際公開公報第00/51641号
【特許文献2】国際公開公報第01/15733号
【特許文献3】米国特許第6,214,821号
【特許文献4】米国特許第5,000,958号
【非特許文献】
【0020】
【非特許文献1】Freiら、Clin. Cancer Res. (1998) 4: 2027-2037
【非特許文献2】Toddら、J. Clin. Oncol. (1984) 2: 986-993
【非特許文献3】Barriereら、Pharmacotherapy (1992) 12: 397-402
【非特許文献4】Schimpff, Support Care Cancer (1993) 1: 5-18
【非特許文献5】Shlaesら、Clin. Infect. Dis. (1993) 17:S527-S536
【非特許文献6】ShahおよびSchwartz、Clin. Cancer Res. (2001) 7: 2168-2181
【非特許文献7】Swaffarら、Anti-Cancer Drugs (1995) 6: 586-593
【非特許文献8】Kanzawaら、Int. J. Cancer (1997) 71(3): 311-319
【非特許文献9】Kueblerら、J. Interferon Res. (1990) 10: 281-291
【非特許文献10】Kobayashiら、Nippon Chiryo Gakkai Shi (1990) 25: 2684-2692
【非特許文献11】Hofsら、Anti Cancer Drugs (1994) 5: 35-42
【非特許文献12】Chenら、Chin. Med. Engl. (1999) 112: 352-355
【非特許文献13】Engblomら、Br. J. Cancer (1999) 79: 286-292
【非特許文献14】BonnerおよびKozelsky、Cancer Chemother. Pharmacol. (1990) 39: 109-112
【非特許文献15】Daoudら、Cancer Chemother. Pharmacol. (1991) 28: 370-376
【非特許文献16】Schiffelersら、J. Pharmacol. Exp. Therapeutic (2001) 298: 369-375
【発明の概要】
【0021】
発明の開示
本発明は、非拮抗的な比の複数の治療薬、好ましくは抗腫瘍薬を、2種類またはそれ以上の薬剤を封入する送達媒体組成物を用いて投与するための方法であって、薬剤がある濃度範囲にわたって相乗的または相加的(すなわち、非拮抗的)な比で媒体中に存在するような方法に関する。封入の前に、組合せにおける治療薬の比は、組合せが所望の濃度範囲にわたって相乗作用または相加作用を示すように選択される。送達媒体中への封入により、2種類またはそれ以上の薬剤を協調的な様式で罹患部に送達することが可能になり、それによって薬剤が罹患部に非拮抗的な比で存在することが保証される。この結果は、薬剤を送達媒体中に共封入した場合にも、または罹患部で非拮抗的な比が維持されるように投与される送達媒体中に別個に封入した場合にも達成されると考えられる。組成物の薬物動態(PK)は、協調的な送達が得られるように送達媒体それ自体によって制御される(送達システムのPKが類似しているという条件で)。
【0022】
したがって、1つの面において、本発明は、媒体組成物中に封入された2種類またはそれ以上の薬剤を、所望の濃度範囲にわたって相乗的または相加的である比で含む、非経口的投与のための送達媒体組成物を提供する。本送達媒体組成物は、薬剤を送達媒体組成物中にこれらの比で封入することを含む工程によって調製される。薬剤の非拮抗的な比は、妥当な細胞培養物または無細胞系に対する薬剤の生物活性または作用をある濃度範囲にわたって評価することにより、および、1つの態様においては、「組合せ指数(combination index)」(CI)を決定するためのアルゴリズムを適用することにより、決定される。以下にさらに説明するように、認知されたアルゴリズムを用いて、それぞれの濃度レベルでの組合せ指数を算出することができる。ある濃度範囲にわたってCIが相乗作用または相加作用を表す比を選択する。広い濃度範囲にわたってCIが相乗的であることが好ましい。好ましい薬剤は抗腫瘍薬である。所望の濃度範囲にわたって非拮抗的な効果を維持する薬剤の比が決定される任意の方法を用いうる。
【0023】
より詳細には、本発明は、送達媒体を含む組成物であって、前記送達媒体の内部に少なくとも第1の治療薬および第2の治療薬が、細胞の1%超が影響を受ける(影響率(fa)>0.01)濃度範囲の少なくとも5%にわたって、培養下の妥当な細胞もしくは無細胞系に対して非拮抗的な生物作用を示すような第1の薬剤と第2の薬剤とのモル比で封入されている組成物、または、送達媒体を含む組成物であって、前記送達媒体の内部に少なくとも第1の治療薬および第2の治療薬が、妥当な細胞に対して非拮抗的な細胞傷害作用もしくは細胞分裂抑制作用を示すような第1の薬剤と第2の薬剤とのモル比で封入されており、前記薬剤が抗腫瘍薬である組成物に関する。「妥当な(relevant)」細胞とは、本出願者らによれば、所望の生物作用を調べるのに適した少なくとも1つの細胞培養物または細胞系のことを指している。例えば、薬剤が抗腫瘍薬である場合、「妥当な」細胞は、米国国立癌研究所(National Cancer Institute;(NCI)/米国国立衛生研究所(National Institutes of Health;NIH)の開発中治療薬プログラム(Developmental Therapeutics Program)(DTP)により、その抗癌薬開発プログラムにおいて有用と同定された細胞系であると考えられる。現在、DTPスクリーニングには60種類のヒト腫瘍細胞株が用いられている。このような細胞株の少なくとも1つに対して所望の活性が示される必要があると考えられる。
【0024】
もう1つの面において、本発明は、相乗的または相加的な比にある2種類またはそれ以上の治療薬を、本発明の組成物を投与することによって所望の標的に送達するための方法を対象とする。
【0025】
もう1つの面において、本発明は、ある濃度範囲にわたって非拮抗的である比の少なくとも2つの治療薬を含む送達媒体である、送達媒体を含む治療的組成物を調製するための方法であって、少なくとも2つの治療薬の一団(panel)を提供すること(この際、この一団は少なくとも1つ、しかし好ましくは複数の比の前記薬剤を含む)、ある濃度範囲にわたって、妥当な細胞培養物または無細胞系に対して生物作用を発揮する一団のメンバーの能力を試験すること、その比が、適した濃度範囲にわたって前記細胞培養物または無細胞系に対して相乗的または相加的な作用をもたらすような一団のメンバーを選択すること、および、一団のうち適格なメンバーを表す比の薬剤を薬物送達媒体中に封入する(すなわち、安定的に会合させる)こと、を含む方法を対象とする。
【0026】
以下にさらに説明するように、好ましい態様においては、上記の方法による適切な組合せの設計に際して、非拮抗的な比は、妥当な細胞培養物または無細胞アッセイ系による評価で、細胞の1%またはそれ以上に影響を及ぼす(fa>0.01)、好ましくは細胞の20〜80%に影響を及ぼす(fa=0.2〜0.8)用量または濃度の、少なくとも5%の範囲にわたって組合せ指数(CI)が1.1以下であるものとして選択される。
【図面の簡単な説明】
【0027】
【図1】製剤中に含める治療薬の適切な比を決定するための本発明の方法の概要を示した概略図である。
【図2A】組合せおよび相乗作用のデータを提示するための方法を例示する。
【図2B】組合せおよび相乗作用のデータを提示するための方法を例示する。
【図2C】組合せおよび相乗作用のデータを提示するための方法を例示する。
【図2D】組合せおよび相乗作用のデータを提示するための方法を例示する。
【図2E】組合せおよび相乗作用のデータを提示するための方法を例示する。
【図3】図3Aはイリノテカン:5-FUに関する、モル比1:10(黒い四角)および1:1(黒丸)での、影響を受けたHT29細胞の率(fa)の関数としての組合せ指数(CI)のグラフである。図3Bはエトポシド:カルボプラチンに関する、モル比1:10(黒い菱形)および10:1(黒い四角)での、影響を受けたMCF-7細胞の率(fa)の関数としてのCIのグラフである。
【図4】シスプラチン:エデルフォシン(edelfosine)に関する、モル比10:1(黒い三角)および1:1(黒丸)での、影響を受けたH460細胞の率(fa)の関数としてのCIのグラフである。
【図5A】H460細胞における、モル比10:1、1:1および1:10でのカルボプラチン:ダウノルビシンの関数としてのCI最大値のグラフである。挿入図は、MCF-7細胞における、有効量(ED)値50、75および90、モル比10:1および1:1でのカルボプラチン:ダウノルビシンに関するCIのヒストグラムである。
【図5B】カルボプラチン:ダウノルビシンに関する、モル比1:10(黒い三角)、1:1(黒い四角)および10:1(黒丸)での、影響を受けたH460細胞の率(fa)の関数としてのCIのグラフである。挿入図は、H460細胞における、ED値50、75および90での、モル比1:10、1:1および10:1のカルボプラチン:ダウノルビシンに関するCIのヒストグラムである。
【図6】薬剤を単一のリポソーム(DSPC/DSPG、80:20mol%)中に非拮抗的な比(10:1)で調合した場合の、静脈内投与後の時間の関数としての、カルボプラチン(白丸)およびダウノルビシン(黒丸)の血漿中濃度(nmol/mL)のグラフである。
【図7】図7Aは薬剤を単一のリポソーム(DSPC/DSPG、80:20mol%)中に10:1(黒丸)、5:1(白丸)および1:1(黒い三角)という3種類の比で調合した場合の、静脈内投与後の時間の関数としての、カルボプラチン:ダウノルビシンのモル比のグラフである。図7Bは静脈内投与後の時間の関数として再プロットした、図7Aにおける1:1カルボプラチン:ダウノルビシンのデータのグラフである。
【図8】薬剤を非拮抗的なモル比(10:1)で単一のリポソーム(DSPC/スフィンゴミエリン/DSPE-PEG2000、90:5:5mol%)中に調合した場合の、静脈内投与後の時間の関数としての、カルボプラチン(黒丸)およびダウノルビシン(白丸)の血漿中濃度(nmol/mL)のグラフである。
【図9】ヒトH460非小細胞肺腫瘍を有するマウスに投与した、カルボプラチンおよびダウノルビシンの混合物(黒い逆三角)、カルボプラチンおよびダウノルビシンを単一のリポソーム中に調合したもの(白い逆三角)、または生理食塩水対照(黒丸)の活性を比較したグラフである。カルボプラチンおよびダウノルビシンはDSPC/DSPG(80:20mol%)リポソーム中にモル比1:1で調合した。矢印は薬剤を投与した日を示す。
【図10】ヒトH460非小細胞肺腫瘍を有するマウスに投与した、カルボプラチンおよびダウノルビシンの混合物(黒い三角)、カルボプラチンおよびダウノルビシンを単一のリポソーム中に調合したもの(白い三角)、または生理食塩水対照(黒丸)の活性を比較したグラフである。カルボプラチンおよびダウノルビシンはDSPC/SM/DSPE-PEG2000(90:5:5mol%)リポソーム中にモル比10:1で調合した。x軸に沿った矢印は矢印は投薬スケジュールを表す。
【図11】図11Aはシスプラチン:ダウノルビシンに関する、モル比1:1(黒い四角)および10:1(黒丸)での、影響を受けたH460細胞の率(fa)の関数としてのCIのグラフである。図11BはH460細胞に対する、モル比10:1、1:1および1:10でのシスプラチン:ダウノルビシンの関数としてのCI最大値のグラフである。
【図12】薬剤を非拮抗的なモル比(10:1)で単一のリポソーム(DMPC/Chol、55:45mol%)中に調合した場合の、静脈内投与後の時間の関数としての、シスプラチン(白丸)およびダウノルビシン(黒丸)の血漿中濃度(μmol/mL)のグラフである。
【図13】薬剤を非拮抗的なモル比(10:1)で2つの別個のリポソーム(シスプラチンに関してはDMPC/Chol、55:45mol%、ダウノルビシンに関してはDSPC/DSPE-PEG2000、95:5mol%)中に調合した場合の、静脈内投与後の時間の関数としての、シスプラチン(黒丸)およびダウノルビシン(白丸)の血漿中濃度(μmol/mL)のグラフである。
【図14】ヒトH460非小細胞肺腫瘍を有するマウスに投与した、シスプラチンおよびダウノルビシンの混合物(黒い逆三角)、シスプラチンおよびダウノルビシンを別個のリポソーム中に調合したもの(白い逆三角)、または生理食塩水対照(黒丸)の活性を比較したグラフである。シスプラチンはDMPC/Chol(55:45mol%)リポソーム中に調合し、ダウノルビシンはDSPC/DSPE-PEG2000(95:5mol%)リポソーム中に調合した上で、非拮抗的なモル比(10:1)で投与した。矢印は薬剤を投与した日を示す。
【図15】薬剤を単一のリポソーム(DMPC/Chol、55:45mol%)中に拮抗的なモル比1:1で調合した場合の、静脈内投与後の種々の時点での、血漿中に残留したシスプラチン(黒丸)およびダウノルビシン(白丸)の濃度(nmol/mL)を示したグラフである。挿入図は、投与後の種々の時点でのシスプラチン:ダウノルビシンのモル比を示す。
【図16】ヒトH460非小細胞肺腫瘍を有するマウスに投与した、シスプラチンおよびダウノルビシンの混合物(黒い三角)、シスプラチンおよびダウノルビシンを単一のリポソーム中に調合したもの(白い三角)、または生理食塩水対照(黒丸)の活性を比較したグラフである。薬剤はDMPC/Chol(55:45mol%)リポソーム中に拮抗的なモル比(1:1)で調合した。矢印は薬剤を投与した日を示す。
【図17】図17Aはシスプラチン:トポテカンに関する、モル比1:1(黒丸)および10:1(白丸)での、影響を受けたH460細胞の率(fa)の関数としてのCIのグラフである。図17BはH460細胞に対する、シスプラチン:トポテカンのモル比の関数としてのCI最大値のグラフである。
【図18】薬剤を別個のリポソーム(シスプラチンに関してはDMPC/Chol、55:45mol%、トポテカンに関してはDSPC/Chol、55:45mol%)中に調合した場合の、静脈内投与後の種々の時点での、血漿中に残留したシスプラチン(黒丸)およびトポテカン(白丸)の濃度(μmol/mL)を示したグラフである。挿入図は、投与後の種々の時点でのシスプラチンとトポテカンとのモル比を示す。
【図19】ヒトH460非小細胞肺腫瘍を有するマウスに投与した、シスプラチンおよびトポテカンの混合物(黒い三角)、シスプラチンおよびトポテカンを別個のリポソーム中に調合したもの(白い三角)、または生理食塩水対照(黒丸)の活性を比較したグラフである。シスプラチンはDMPC/Chol(55:45mol%)リポソーム中に調合し、トポテカンはDSPC/Chol(55:45mol%)リポソーム中に調合した上で、非拮抗的なモル比(10:1)で投与した。矢印は薬剤を投与した日を示す。
【図20】図20Aはシスプラチン:イリノテカンに関する、モル比1:1(黒い四角)、10:1(黒丸)、1:5(黒い三角)および1:10(黒い菱形)での、影響を受けたH460細胞の率(fa)の関数としてのCIのグラフである。図20BはH460細胞に対する、シスプラチン:イリノテカンのモル比の関数としてのCI最大値のグラフである。
【図21】薬剤を単一のリポソーム(DSPC/DSPG、80:20mol%)中に同時装填した場合の、静脈内投与後の種々の時点での、血漿中に残留したシスプラチン(黒丸)およびイリノテカン(白丸)の濃度(nmol/mL)を示したグラフである。
【図22】薬剤を別個のリポソーム(シスプラチンに関してはDMPC/Chol、55:45mol%、イリノテカンに関してはDSPC/DSPE-PEG2000、95:5mol%)中に調合した場合の、静脈内投与後の種々の時点での、血漿中に残留したシスプラチン(黒丸)およびイリノテカン(白丸)の濃度(nmol/mL)を示したグラフである。
【図23】ヒトH460非小細胞肺腫瘍を有するマウスに投与した、シスプラチンおよびイリノテカンの混合物(黒い四角)、シスプラチンおよびイリノテカンを別個のリポソーム中に調合して種々の用量で投与したもの(白抜き記号)、または生理食塩水対照(黒丸)の活性を比較したグラフである。DMPC/Chol(55:45mol%)リポソーム中に調合したシスプラチンおよびDSPC/DSPE-PEG2000(95:5mol%)リポソーム中に調合したイリノテカンを非拮抗的なモル比(1:5)で投与した。矢印は薬剤を投与した日を示す。
【図24】ビノレルビンをPOPS(黒い逆三角)、DPPS(黒い三角)、DLPS(黒丸)、DSPS(黒い菱形)またはDOPS(黒い四角)と、ビノレルビン:PSのモル比1:1で併用した場合の、影響を受けたH460細胞の率(fa)の関数としてのCIのグラフである。
【図25】図25AはSCID/rag2マウスに対する、遊離ビノレルビン(黒丸)、またはSM/Chol/DPPS/DSPE-PEG2000、35:45:10:10mol%リポソーム中にビノレルビン:PSのモル比1:1で封入したもの(白丸)を静脈内投与した後の時間の関数としてのビノレルビン血漿中濃度のグラフである。図25Bは図25Aのデータを用いた、SCID/rag2マウスに対する、遊離ビノレルビン(黒色の棒グラフ)、またはSM/Chol/DPPS/DSPE-PEG2000、35:45:10:10mol%中に封入したもの(灰色の棒グラフ)を静脈内投与した後の血漿中濃度曲線下面積(AUC)を示したヒストグラムである。
【図26】H460非小細胞肺腫瘍を有するマウスに投与した、遊離ビノレルビン(白丸)、ビノレルビンをDSPC/Chol/DPPS/DSPE-PEG2000、35:45:10:10mol%リポソーム中に封入したもの(黒い逆三角)、ビノレルビンをSM/Chol/DPPS/DSPE-PEG2000、35:45:10:10mol%リポソーム中に封入したもの(白い三角)、または生理食塩水対照(黒丸)の活性を比較したグラフである。ビノレルビンおよびホスファチジルセリン(DPPS)は非拮抗的なモル比(1:1)で調合した。矢印は薬剤を投与した日を示す。
【図27】H460非小細胞肺腫瘍を有するマウスに投与した、生理食塩水対照(黒丸);遊離ビノレルビン(白丸);ビノレルビンを以下のリポソーム中に封入したもの:SM/Chol/DPPS/DSPE-PEG2000、35:45:10:10(黒い逆三角)、DAPC/Chol/DPPS/DSPE-PEG2000、35:45:10:10mol%(白い三角)およびDSPC/Chol/DSPS/DSPE-PEG2000、35:45:10:10mol%(黒い四角)の効果を示す。ビノレルビンおよびホスファチジルセリン(DPPSまたはDSPS)は非拮抗的なモル比(1:1)で調合した。矢印は薬剤を投与した日を示す。
【図28】P388マウス白血病を有するマウスの生存率に対する、生理食塩水対照(白い三角);遊離ビノレルビン(黒丸);およびビノレルビンをSM/Chol/DPPS/DSPE-PEG2000、35:45:10:10mol%リポソーム中に封入したもの(黒い逆三角)の効果を示す。ビノレルビンおよびホスファチジルセリンは非拮抗的なモル比(1:1)で調合した。x軸に沿った矢印は薬剤を投与した日を示す。
【図29】FUDR:CPT-11の10:1(黒い四角);5:1(黒丸);1:1(黒い三角);1:5(黒い逆三角);および1:10(白丸)という種々の比での組合せによって影響されたHT-29細胞の率の関数としてプロットしたCIを示す。
【図30】静脈内投与後の時間の関数としてのFUDR(黒丸)およびCPT-11(白丸)の血漿中濃度のグラフである。
【図31】腫瘍体積と、生理食塩水対照(黒丸)、CPT-11/FUDR混合物注射(白い逆三角)およびCPT-11/FUDRのリポソーム製剤(黒い逆三角)の腫瘍細胞接種後の時間との関係に関するグラフである。
【発明を実施するための形態】
【0028】
発明の実施の態様1
本発明の方法は、インビトロで所望の濃度範囲にわたって非拮抗的である治療薬の比を決定すること、および、その比が所望の作用部位で確実に維持されると考えられる様式でこの非拮抗的な比を提供することを含む。相乗的な比または相加的な比は、2種類またはそれ以上の治療薬の少なくとも1つの比を妥当な細胞培養物または無細胞系に対してある濃度範囲にわたってインビトロで試験することによって得られた結果に対して、標準的な分析ツールを適用することによって決定される。例えば、個々の薬剤および種々の組合せを、細胞培養物または無細胞系に対する生物作用、例えば細胞死を引き起こすこと、または細胞増殖を抑制することに関して、種々の濃度レベルで試験する。あらかじめ設定した比の濃度レベルを細胞生存率に対してプロットして相関を得て、それを既知かつ確立された数学的手法によって操作して「組合せ指数(combination index)」(CI)を算出することができる。この数学的手法は、CIが1(すなわち0.9〜1.1)であれば薬剤の相加作用を表し、CT>1(すなわち>1.1)であれば拮抗作用を表し、CIが<1(すなわち<0.9)であれば相乗作用を表すというものである。
【0029】
1 略号
以下の略号を用いる。
PE:ホスファチジルエタノールアミン;PS:ホスファチジルセリン;DPPS:ジパルミトイルホスファチジルセリン;DSPS:ジアステロイルホスファチジルセリン;DLPS:ジラウロイルホスファチジルセリン;DOPS:ジオレオイルホスファチジルセリン;POPS:パルミトイルオレオイルホスファチジルセリン;PC:ホスファチジルコリン;SM:スフィンゴミエリン;PG:ホスファチジルグリセロール;PI:ホスファチジルイノシトール;PA:ホスファチジン酸;DSPC:ジアステロイルホスファチジルコリン;DMPC:ジミリストイルホスファチジルコリン;DSPG:ジアステロイルホスファチジルグリセロール;DSPE:ジアステロイルホスファチジルエタノールアミン;Chol:コレステロール;CHまたはCHE:コレステリルヘキサデシルエーテル;
PEG:ポリエチレングリコール;DSPE-PEG:ジアステロイルホスファチジルエタノールアミン-N-[ポリエチレングリコール];PEGの後に数字が続く場合、その数字はダルトン単位で表現したPEGの分子量である;DSPE-PEG2000:ジアステロイルホスファチジルエタノールアミン-N-[ポリエチレングリコール 2000];
SUV:小型単ラメラ小胞;LUV:大型単ラメラ小胞;MLV:多重ラメラ小胞;
MTT:3-(4,5-ジメチルチアゾール-2-イル)-2,5-ジフェニル-2Hテトラゾリウムブロミド;DMSO:ジメチルスルホキシド;OD:光学密度;OGP:N-オクチルβ-D-グルコピラノシド;EDTA:エチレンジアミン四酢酸;HEPES:N-[2-ヒドロキシルエチル]-ピペラジン-N-[2-エタンスルホン酸];HBS:HEPES緩衝生理食塩水(20mM HEPES、150mM NaCl、pH 7.4);SHE:300mMスクロース、20mM HEPES、30mM EDTA;ED50、ED75およびED90:培養下にある細胞の50、75および90%に影響を及ぼすために必要な有効量;LD50:培養下にある細胞の50%の死滅を引き起こすために必要な用量;CI:組合せ指数;CI maxまたはCI最大値:異なる比の薬剤に関してCI値に最も大きな差が観察される単一のfa値(0.2〜0.8の間)について得られたCI値;fa:影響率;TEA:トリエタノールアミン;
FDA:米国食品医薬品局;NCI:米国国立癌研究所。
【0030】
一般的なアプローチの1つを図1に示している。示されているように、薬剤AおよびBを個別に、および2種類の比で併用して、以下に述べるMTTアッセイ法によって評価される細胞死または細胞分裂停止を引き起こす能力に関して調べる。最初に、薬剤A、Bの濃度と2種類の組合せ比(Y:ZおよびX:Y)との相関を、未処理対照細胞の生存性を基準とする百分率として算出した細胞傷害性に対してプロットする。予想通り、個別の薬剤および組合せのいずれに対しても細胞の生存性に対して用量依存的な効果がみられる。この相関性が実証されれば、細胞の生存性または影響率(fa)を、CIの算出において濃度の代用物として用いることができる。
【0031】
CI算出の結果は図1にも示されている;この指数は、ChouおよびTalalay、Advance Enz. Regul. (1985) 22: 27-55の手順に従って影響を受けた細胞の率の関数として算出される。この仮説の状況では、薬剤A+Bの第1の比(X:Y)はすべての濃度で非拮抗的であるが、第2の比(Y:Z)での組合せは拮抗的である。したがって、広範囲にわたって濃度に関係なく非拮抗的であると考えられる薬剤A+Bの比(比1)を得ることが可能である。本発明の組成物に含めることが望ましいのはこの比である。
【0032】
本発明者らは、薬剤の組合せのさまざまな比について「CI最大値」を算出することによる、相乗作用に対する比および濃度の影響の代替的な例も考案している。「CI最大値」は、異なる比の薬剤に関してCI値に最も大きな差が観察された単一のfa値(0.2〜0.8の間)について得られたCI値と定義される。これは図2Aおよび2Bに例示されている;示されているように、イリノテカン/カルボプラチン比が1:10である場合、そのCIが残りの比の値と最も大きく異なるのは影響率の値が0.2の場合である。示されているように、fa 0.2におけるこの比に関するCI値は約2.0である。
【0033】
非拮抗的な比のインビトロでの決定は2種類の薬剤のみの組合せに関して例示されているが、3種類またはそれ以上の薬剤の組合せに同じ手法を適用することにより、同様の様式でその濃度範囲にわたるCI値が得られる。
【0034】
このようにして得られた比は、非拮抗的な比が確実に維持されると考えられるリポソームまたは他の粒子状形態の中に薬剤を所定の比で封入することにより、薬学的組成物において維持される。このため、本組成物は、粒子状の送達媒体を含み、かつ所望の比の治療薬を含む。
【0035】
薬剤はその両方が同じ送達媒体中に含まれるように共封入されることが好ましいが、これは必ずしも必要ではない。粒子状担体は類似した薬物動態を共有しうるため、作用物質は、別個に封入された場合も製剤から協調的な送達を受ける。
【0036】
「封入(encapsulation)」とは、送達媒体との安定的な会合のことを意味する。このため、媒体は、インビボで投与された場合に1つまたは複数の薬剤が媒体と安定的に会合している限り、1つまたは複数の薬剤を取り囲む必要はない。したがって、「〜と安定的に会合した」および「〜中に封入された」または「〜とともに封入された」または「〜の中または〜とともに封入された」は、同義の用語であることを意図している。それらは本明細書において互換的に用いられる。安定的な会合は、送達媒体との共有結合、好ましくは切断可能な結合によるもの、非共有結合、および、薬剤を送達媒体の内部に捕捉することなどを含む、さまざまな手段によって生じさせうる。会合は、薬剤が、それが投与された対象における標的部位に送達されるまで、非拮抗的な比で送達媒体と会合したまま保たれる程度に、十分に安定でなければならない。
【0037】
送達媒体には、脂質担体、リポソーム、脂質ミセル、リポタンパク質ミセル、脂質安定化エマルション、シクロデキストリン、ポリマー性ナノ粒子、ポリマー性微粒子、ブロック共重合体ミセル、ポリマー-脂質ハイブリッド系、誘導体化された一本鎖ポリマーなどが含まれうる。リポソームは、「リポソーム:合理的デザイン(Liposomes: Rational Design)」(A.S. Janoffed., Marcel Dekker, Inc., N.Y.)に記載された通りに、または当業者に知られた別の技法によって調製しうる。本発明に用いるためのリポソームを、「低コレステロール」性であるように調製してもよい。このようなリポソームは「コレステロール非含有性」である、または「コレステロールを実質的に含まない」、または「コレステロールを本質的に含まない」。「コレステロール非含有性」という用語は、リポソームに言及して本明細書で用いる場合、リポソームがコレステロールを含まずに調製されていることを意味する。「コレステロールを実質的に含まない」という用語は、リポソームの相転移特性を大きく変化させるには不十分な量(通常はコレステロールが20mol%未満)のコレステロールの存在を許容する。リポソーム中に組み入れられるコレステロールが20mol%未満であれば、リポソームが20mol%を上回るコレステロールを伴って調製された場合には最適な保持がなされない薬剤の保持が可能になる。さらに、コレステロール20mol%未満として調製されたリポソームは相転移温度の幅が狭く、この特性は、封入された薬剤を熱を与えることによって放出するリポソーム(熱感受性リポソーム)の調製に利用することができる。本発明のリポソームが治療的脂質を含んでもよく、これにはエーテル脂質、ホスファチジン酸、ホスホネート、セラミドおよびセラミド類似体、スフィンゴシンおよびスフィンゴシン類似体、ならびにセリン含有脂質が含まれる。また、リポソームを、循環寿命を延長させるために、ポリエチレングリコール-DSPEなどの表面安定化性親水性ポリマー-脂質結合物とともに調製することもできる。担体の循環寿命を延長させるために、ホスファチジルグリセロール(PG)およびホスファチジルイノシトール(PI)などの負に荷電した脂質をリポソーム製剤に添加することもできる。これらの脂質は、表面安定化剤として親水性ポリマー-脂質結合物を置き換えるために用いうる。本発明の態様に、凝集を防止し、それによって担体の血中滞留時間を延長させるためのPGまたはPIを含むコレステロール非含有リポソームを用いてもよい。
【0038】
ミセルは、疎水性コアに存在する溶けにくい薬剤の送達のために利用される、両親媒性脂質またはポリマー成分から構成される自己集合性粒子である。ミセル性送達媒体の調製のための手段にはさまざまなものがあり、当業者によって容易に行われうる。例えば、脂質ミセルは、Perkinsら、Int. J. Pharm. (2000) 200(1): 27-39(参照として本明細書に組み入れられる)に記載された通りに調製しうる。リポタンパク質ミセルは、低密度および高密度リポタンパク質ならびにカイロミクロンを含む、天然または人工のリポタンパク質から調製することができる。脂質安定化エマルションとは、油が充填されたコアが脂質の単層または二重層などの乳化成分によって安定化されたものを含むように調製されたミセルのことである。コアはトリアシルグリセロール(コーン油)などの脂肪酸エステルを含みうる。単層または二重層には、DSPE-PEGなどの親水性ポリマー脂質結合物が含まれる。これらの送達媒体は、ポリマー脂質結合物の存在下における油のホモジネート化によって調製しうる。脂質安定化エマルションに組み入れられる薬剤は一般に水溶性が乏しい。リポタンパク質に類似した特性を示す合成ポリマー類似体、例えば、ステアリン酸エステルまたはポリ(エチレンオキシド)ブロック-ポリ(ヒドロキシエチル-L-アスパルタミド)およびポリ(エチレンオキシド)-ブロック-ポリ(ヒドロキシヘキシル-L-アスパルタミド)のミセルを、本発明の実施に用いることもできる(Lavasanifarら、J. Biomed. Mater. Res. (2000) 52: 831-835)。
【0039】
シクロデキストリンは空洞形成性で水溶性の多糖であり、その空洞内に水に溶けない薬剤を収容することができる。当業者に知られた手順を用いて薬剤をシクロデキストリン中に封入することができる。例えば、Atwoodら編、「包含性化合物(Inclusion Compounds)」第2巻および第3巻、Academic Press、NY (1984);Benderら、「シクロデキストリンの化学(Cyclodextrin Chemistry)」、Springer-Verlag、Berlin (1978);Szeitliら、「シクロデキストリンおよびその包含複合体(Cyclodextrins and Their Inclusion Complexes)」、Akademiai Kiado, Budapest, Hungary (1982)および国際公開公報第00/40962号を参照されたい。
【0040】
ナノ粒子および微粒子は、ポリマー性外殻(ナノカプセル)によって囲まれた、またはポリマー基質の全体に固体もしくは液体が分散した(ナノスフェア)、薬剤の濃縮性コアである。ナノ粒子および微粒子の一般的な調製法は、Soppimathら(J. Control Release (2001) 70(12): 1-20)に記載されており、これは参照として本明細書に組み入れられる。用いうるその他のポリマー性送達媒体には、親水性外殻によって囲まれた疎水性コアを含む薬剤を含むブロック共重合体ミセルが含まれる;これは一般に、疎水性薬剤のための担体として用いられ、Allenら、「コロイドおよび表面B(Colloids and Surface B)、Biointerfaces (1999) Nov 16 (1-4): 3-27に記載された通りに調製することができる。ポリマー-脂質ハイブリッド系は、脂質単層によって囲まれたポリマー性ナノ粒子からなる。ポリマー粒子は疎水性薬剤の組み入れのための積み荷空間(cargo space)として働き、脂質単層は疎水性コアと外部水性環境との間の安定化性境界として働く。ポリマーにはポリカプロラクトンおよびポリ(d,l-ラクチド)などを用いることができ、脂質単層は一般に脂質の混合物から構成される。適した調製方法は、ポリマー性ナノ粒子に関する上記の参考文献にあるものと同様である。誘導体化された一本鎖ポリマーとは、生物活性物質との共有結合によってポリマー-薬剤結合物を形成するために適合化されたポリマーである。ポリアミノ酸、多糖類(デキストリンもしくはデキストランなど)および合成ポリマー(N-(2-ヒドロキシプロピル)メタクリルアミド(HPMA)共重合体など)を含むさまざまなポリマーが、ポリマー-薬剤結合物の合成用に提唱されている。適した調製法は、VeroneseおよびMorpurgo、IL Farmaco (1999) 54(8): 497-516に詳述されており、これは参照として本明細書に組み入れられる。
【0041】
このように、送達媒体は、治療成分の投与された比の一貫した送達が達成されるように提供される。すなわち、比は、組成物を含む媒体中への薬剤の単純な共封入によって維持してもよく、または、非拮抗的な薬剤の比を同様に維持するように媒体が組成物の薬物動態を制御する場合には、薬剤を別個の媒体中に封入することもできる。
【0042】
本発明の組成物は、拮抗的でない複数の抗腫瘍薬の組成物を送達するために用いることが好ましい。以下の詳細な説明は、治療薬の比を決定する様式、および、本発明の送達システム中に所望の比を封入するための方法を示している。
【0043】
簡潔に述べると、1つのシナリオでは、まず個々の薬剤を種々のインビトロアッセイ法またはインビボアッセイ法で別個にスクリーニングして個々の活性を決定する。続いて、薬剤の対を組み合わせて、同じスクリーニング法でアッセイする。この初期スクリーニングでは、薬剤の比は、以前に同定した50%の活性を示す濃度(IC50値)でのモル比である。または、製剤の目的に関する考察に基づき、その他の固定比(一般的にはモル比1:10、1:1および10:1)を選択する。細胞の生存に対する薬剤の効果に基づいて算出した平均値および薬剤の用量をCalcuSynコンピュータプログラムに入力し、出力データを、影響を受けた細胞の率(fa)の関数として組合せ指数(CI)値を定めるために評価する。
【0044】
CalcuSyn法は、抗腫瘍薬、臓器移植用の免疫抑制剤、自家骨髄移植のための白血病細胞の複合的パージング、殺虫薬、生体応答修飾物質、多剤耐性阻害薬、抗菌薬、抗HIV薬、抗ヘルペス薬およびその他の抗ウイルス薬などの種々の薬剤の試験を行うために首尾良く適用されている。
【0045】
図11Aにおけるシスプラチン:ダウノルビシンのモル比1:1での薬剤に類似した相互作用挙動を示す薬剤の組合せは拮抗的であり、追求の対象とならない。fa>0.01であるfa値のかなりの範囲(好ましくは少なくとも約20%)にわたって非拮抗的な相互作用がみられる化合物の組合せ(すなわち、モル比1:1および10:1のイリノテカン:カルボプラチン;図2A)を、非拮抗的な相互作用の強度(すなわち、低いCI値)を高めるとともに相乗作用が観察されるfaの範囲を拡げる最適な比を明確にするために、さまざまな異なる薬剤/薬剤比で、このインビトロスクリーニングアッセイ法において再び評価する。
【0046】
このようにして同定された、最適化された非拮抗的な薬剤の組合せは、二剤(dual-agent)組成物として送達媒体中にある製剤に関する組成物を規定し、および/または第3の薬剤との相乗的または相加的な相互作用を決定するための単一の薬剤単位として用いることができる。
【0047】
非拮抗的な比のインビトロでの決定
本発明の組成物を調製するためには、送達媒体中に含まれる薬剤の所望の比をまず決定する必要がある。この比は、相乗作用または相加作用が組合せによってある濃度範囲にわたって示されるものであることが望ましい。このような比は、インビトロの細胞培養物または無細胞系においてさまざまな数学モデルを用いて決定することができる。
【0048】
相乗的または相加的な併用効果を濃度範囲にわたって示す薬剤の比の決定は、以下に述べる実験データのタイプに応じて、種々のアルゴリズムを用いて行うことができる。これらの方法には、アイソボログラム法(Loeweら、Arzneim-Forsch (1953) 3: 285-290;Steelら、Int. J. Radiol. Oncol. Biol. Phys. (1979) 5: 27-55)、分画産物法(Webb, 「酵素および代謝阻害物質(Enzyme and Metabolic Inhibitors)」(1963)第1巻、pp.1-5. New York: Academic Press)、モンテカルロシミュレーション法、CombiTool、ComboStat、ならびにChou, J. Theor. Biol. (1976) 39: 253-76;およびChou, Mol. Pharmacol. (1974) 10: 235-247に記載された式に基づくChou-Talalayのメディアンエフェクト法が含まれる。それに代わるものには、生存率(Zoliら、Int. J. Cancer (1999) 80: 413-416)、対照と比較した顆粒球/マクロファージ-コロニー形成単位の反応率(Pannacciulliら、Anticancer Res. (1999) 19: 409-412)およびその他(Berenbaum、Pharmacol. Rev. (1989) 41: 93-141;Grecoら、Pharmacol Rev. (1995) 47: 331-385)が含まれる。
【0049】
Chou-Talalayのメディアンエフェクト法が好ましい。この分析には、特定の影響faを引き起こす用量が
D=Dm[fa/(1-fa)]1/m
によって与えられる式が用いられ、ここでDは用いる薬剤の用量であり、faはその用量による影響を受けた細胞の率であり、Dmは効力を表す中位効果(median effect)であり、mは用量-効果曲線の形状を表す係数である(一次反応ではmは1である)。
【0050】
この式をさらに操作して、ChouおよびTalalay、Adv. Enzyme Reg. (1984) 22: 27-55;ならびにChouら、「化学療法における相乗作用および拮抗作用(Synergism and Antagonism in Chemotherapy)」、ChouおよびRideout編、Academic Press: New York 1991: 223-244に記載された多剤効果式に基づいて組合せ指数(CI)を計算することができる。この計算用のコンピュータプログラム(CalcuSyn)は、Chou、「マイクロコンピュータによる用量-効果分析:ED50、LD50、相乗作用、拮抗作用、低用量リスク、受容体リガンド結合および酵素反応速度論(Dose-effect analysis with microcomputers: quantitation of ED50, LD50, synergism, antagonism, low-dose risk, receptor ligand binding and enzyme kinetics)」(CalcuSynマニュアルおよびソフトウエア;Cambridge:Biosoft 1987)において見出すことができる。
【0051】
組合せ指数の式は、酵素反応速度論モデルに由来するChou-Talalayの多剤効果式に基づく。ある式は相乗作用および拮抗作用ではなく相加作用のみを決定する。しかし、CalcuSynプログラムによれば、相乗作用は期待される相加作用を上回るものとして、拮抗作用は期待される相加作用を下回るものとして定義される。ChouおよびTalalayは1983年にCI=1の指定を相加作用として提唱しており、このため本発明者らは、2種類の薬剤の多剤効果式から、同じまたは類似の作用機序を有する排反的な薬剤に関して
CI=(D)1/(Dx)1+(D)2/(Dx)2 [式1]
を得ており、さらに、完全に独立した作用機序を有する排反的でない薬剤に関して
CI=(D)1/(Dx)1+(D)2/(Dx)2+(D1)(D2)/(Dx)1(Dx)2 [式2]
を得ている。CI<1、=1および>1はそれぞれ相乗作用、相加作用および拮抗作用を示す。式1または式2は、併用した薬剤1、(D)1および薬剤2、(D)2(分子にあるもの)が、実際の実験でx%を阻害することを指示している。このため、実験的に観察される阻害率x%は、概数ではなくて小数部を有する可能性が高い。式1および2の(D)1および(D)2(分母にあるもの)は、それぞれ薬剤1および薬剤2が単独でx%を阻害する用量である。
【0052】
単純化のために、排反性は通常、3種類以上の薬剤を組合せに用いる場合に仮定される(CalcuSynマニュアルおよびソフトウエア;Cambridge:Biosoft 1987)。
【0053】
基礎をなす実験データは一般に、培養下の細胞または無細胞系を用いてインビトロで決定される。組合せ指数(CI)は、図1に示されているように、上に説明した通りに濃度範囲の代用パラメーターである、影響を受けた細胞の率(fa)の関数としてプロットすることが好ましい。好ましい薬剤の組合せは、fa値のかなりの範囲にわたって相乗作用または相加作用を示すものである。細胞の1%超が影響を受ける濃度範囲、すなわちfaが0.01を上回る範囲の少なくとも5%にわたって相乗作用を示す薬剤の組合せを選択する。全濃度のうちより多くの割合、例えば、faが0.2〜0.8である範囲の5%が、好都合なCIを示すことが好ましい。より好ましくは、この範囲の10%が好都合なCIを示す。さらにより好ましくは、fa範囲の20%、好ましくは50%超、最も好ましくはfaが0.2〜0.8である範囲の少なくとも70%超が組成物において用いられる。fa値のかなりの範囲にわたって相乗作用を示す組合せを、非拮抗的相互作用の強度を高め、相乗作用が観察されるfaの範囲を拡げる最適な比を定めるために、種々の薬剤比で再び評価することができる。
【0054】
細胞が影響を受ける濃度範囲の全体にわたって相乗作用がみられることが望ましいが、多くの場合には、0.2〜0.8のfa範囲における結果の信頼性の方がかなり高い。このため、本発明の組合せによって示される相乗作用は0.01またはそれ以上という広い範囲内に存在することは述べておくが、相乗作用は0.2〜0.8のfa範囲において実証されることが好ましい。
【0055】
最適な組合せ比を、さらに、第3の薬剤との相乗的または相加的な相互作用を決定するための単一の薬剤単位として用いることもできる。加えて、3剤組合せを、第4の薬剤との非拮抗的な相互作用を決定するための単位として用いることもでき、以降も同様である。
【0056】
上記の通り、細胞培養物に対するインビトロ試験は、「妥当な」細胞を用いて行われると考えられる。細胞の選択は、薬剤の意図した治療用途に依存すると考えられる。組成物を本発明の範囲に含めるための基盤を得るために、要求される非拮抗的な作用を示す必要があるのは、ただ1つの妥当な細胞系または細胞培養物のみでよい。
【0057】
例えば、本発明の1つの好ましい態様において、薬剤の組合せは抗癌療法を目的とする。続いて、試験を行う細胞および試験の性質に対して適した選択を行う。詳細には、腫瘍細胞株が適した被験対象であり、細胞死または細胞分裂停止の計測が適切なエンドポイントである。以下にさらに考察するように、他の適応症に適した非拮抗的な薬剤の組合せを見いだそうとする状況下では、他の標的細胞、および細胞傷害性または細胞分裂停止以外の基準を用いうると考えられる。
【0058】
抗腫瘍薬にかかわる決定のための細胞系は、標準的な細胞系収集機関(例えば、NCIまたはATCC)から、学術機関から、または市販の供給元を含む他の組織から入手しうる。好ましい細胞系には、NCI/NIHの開発中治療薬プログラム(Developmental Therapeutics Program)によって同定された細胞系から選択された1つまたは複数が含まれると考えられる。現在、このプログラムによって用いられている腫瘍細胞株スクリーニングでは、白血病、黒色腫ならびに肺癌、結腸癌、脳腫瘍、卵巣癌、乳癌、前立腺癌および腎癌を代表する60種類のヒト腫瘍細胞株が同定されている。所望の濃度範囲にわたって必要な非拮抗的な作用が示される必要があるのは単一の細胞種のみである;しかし、少なくとも2種類の細胞系、より好ましくは3種類の細胞系、より好ましくは5種類の細胞系、より好ましくは10種類の細胞系がこの作用を示すことが好ましい。細胞系は樹立された腫瘍細胞株でもよく、または患者試料から得られた初代培養物でもよい。細胞系は任意の種に由来するものでよいが、好ましい由来は哺乳動物、特にヒトであると考えられる。細胞系は、種々の実験条件下での選択、および/または外因性遺伝物質の付加もしくは欠失によって遺伝的に改変されたものでもよい。細胞系は、ウイルスまたはプラスミドを用いるトランスフェクション法を非制限的に含む任意の遺伝子導入法によるトランスフェクションを受けたものでもよい。改変には、特定のタンパク質もしくはペプチドの発現をコードするcDNA、プロモーターもしくはエンハンサー配列などの調節因子、またはアンチセンスDNAもしくはRNAの導入が含まれうる。遺伝的に操作された組織培養細胞系には、腫瘍抑制遺伝子、すわなちp53、pTENおよびp16などの遺伝子を有する系または有しない系;ならびに、ドミナントネガティブ法、遺伝子挿入法および他の選択方法を用いて作製された系が含まれうる。例えば抗腫瘍薬の試験のために、細胞の生存度を定量するために用いうる好ましい組織培養細胞系には、H460、MCF-7、SF268、HT29、HCT-116、LS 180、B16-F10、A549、Capan膵細胞、CAOV-3、IGROV1、PC3、MX-1およびMDA-MB-231が非制限的に含まれる。
【0059】
1つの好ましい態様においては、所定の効果(fa)とは、細胞培養物に対する細胞傷害物質の適用後の細胞死または細胞分裂停止のことを指す。細胞死または生存度は、例えば、以下の方法を用いて測定しうる。
【0060】
「MTTアッセイ法」が好ましい。
【0061】
癌以外の適応疾患に対しても2種類またはそれ以上の薬剤の非拮抗的な比を決定することができ、この情報を、これらの疾患の治療のために2種類またはそれ以上の薬剤による治療的製剤を調製する目的で用いることができる。インビトロアッセイ法に関しては、薬物の相乗作用を明確化するために計測可能なさまざまなエンドポイントを、そのエンドポイントがその特定の疾患に対して治療的に妥当であるという条件付きで選択することができる。
【0062】
したがって、例えば、当業者は、炎症性疾患の治療のための2種類またはそれ以上の薬剤の非拮抗的な比を、IL-1、IL-18、COX-2、TNFまたはインターフェロンγなどの炎症誘発性サイトカインの抑制をインビトロで測定することによって選択することができる。他の炎症性シグナルには、プロスタグランジンE2およびトロンボキサンB2の阻害が非制限的に含まれる。特に、エンドトキシンを介したマクロファージ活性化は、添加した薬剤または薬剤の組合せの抗炎症作用を測定するのに適したインビトロアッセイ法をもたらし、当技術分野でよく用いられている。この種のアッセイ法では、大量に増殖させたマクロファージを、リポ多糖などのエンドトキシンの添加によって活性化する。活性化を行った上で、マクロファージによるIL-1およびTNFなどのサイトカインの分泌、ならびにCOX-2の活性化を測定する。抗炎症性薬剤の候補を添加し、それらがIL-1、TNFおよびCOX-2を抑制する能力を評価する。1×10-7Mデキサメタゾンによる滴定が陽性対照として一般に用いられる。当業者には、マクロファージ活性化を用いるアッセイ法が薬剤の組合せの広域スクリーニングに適していること、ならびにIL-1、TNFおよびCOX-2の抑制が相乗作用を明確にするのに適したエンドポイントであることが明らかであると考えられる。炎症性シグナルを測定することに加えて、研究者は、白血球機能に対する2種類またはそれ以上の薬剤の効果を計測するためのインビトロモデルの使用を検討することもできる。機能試験には、脱顆粒の阻害、スーパーオキシドの生成および白血球の遊走が非制限的に含まれる。
【0063】
癌と同様に、増殖は、動脈硬化、再狭窄、または血管増殖性を伴う他の心血管疾患の発症においても重要な事象である。このため、当業者は、2種類またはそれ以上の薬剤の非拮抗的な比を、妥当な血管の増殖性細胞集団に対して適用した本明細書に記載の方法によって薬物の相乗作用を評価することにより、見いだすことができる。特に、通常は血管形成術後に生じる冠動脈再狭窄および末梢動脈再狭窄などの再狭窄は、平滑筋および内皮細胞の増殖に起因する(Fuster, Arch Mal Coeur Vaiss (1997) 90 Spec No 6: 41-47)。本明細書に記載の標準的な方法を用いて、当業者は、2種類またはそれ以上の薬剤が内皮細胞または平滑筋細胞の増殖を阻害するように非拮抗的に作用するか否かを評価することができる。これらのアッセイ法は、不死化細胞系を用いて、または好ましくは初代細胞系を用いて行いうる。これらの細胞系は、市販の供給元(例えば、Clonetics、California)から、または新鮮な組織(例えば、臍帯静脈、動脈、脳)から入手可能であるが、細胞増殖を促進する適切な増殖因子の存在下で維持する必要がある。癌細胞に対する2種類またはそれ以上の薬剤の相乗作用を測定するアッセイ法と同様に、この種のアッセイ法には、増殖および遊走の阻害というエンドポイントが非制限的に含まれる。増殖エンドポイントは、本出願書に記載のMTTアッセイ法などの生/死アッセイ法、[3H]-チミジン取込みに依拠した増殖の測定、または他の類似のアッセイ法に依拠しうる。同じく分裂性癌細胞と同様に、内皮細胞および平滑筋細胞の増殖は細胞周期におけるチェックポイントによって調節されており、細胞周期の阻害を測定するアッセイ法は、血管増殖性疾患の治療のために選択された2種類またはそれ以上の薬剤の非拮抗的な比を明確にするために用いることができる。
【0064】
薬剤の非拮抗的な組合せを、微生物またはウイルスの感染に対する活性の点から同定することもできる。抗菌薬の同定における第1の段階として、薬剤に関する最小発育阻止濃度(MIC)を、当業者に知られたマイクロタイター液体希釈法または寒天希釈抗菌アッセイ法によって決定することができる。これらのアッセイ法は、実験の安全性および標準に関する国家委員会(National Committee of Laboratory Safety and Standards)(NCLSS)による規制を受けている。標準的な液体希釈アッセイ法は、「実験医学における抗生物質(Antibiotics in Laboratory Medicine)」、Lorian, V. 第4版、p 52-111, WilliamsおよびWilkins、Baltimore中のAmsterdam (1996) 「液体培地における抗菌物質の感受性試験(Susceptibility testing of Antimicrobials in liquid media)」に発表されている。MICは、感染性微生物のインビトロ増殖を阻止すると考えられる抗生物質の最低濃度と定義される。上記のアッセイ法では、微生物の接種物を、種々の濃度の薬剤を含む増殖培地(例えば、寒天)上の小さなスポットに微生物の接種物を播くことにより(例えば、1スポット当たり104コロニー形成単位[CFU])、MICを決定しうる。または、種々の濃度の薬剤を含む増殖培地の懸濁液に微生物を接種することもできる。さらに、微生物は上記の通りに処理してもよく、または特定細胞集団(すなわち、マクロファージ)における細胞内感染として存在してもよい。後者の場合には、標準的な方法によって培養下で増殖させた哺乳動物細胞を低濃度の微生物に対して短時間曝露させることにより、細胞内微生物感染を行わせる。微生物の細胞内複製を行わせる期間をおいた後に、細胞およびその細胞内微生物を、哺乳動物細胞を用いる細胞傷害性試験に関して記載されているのと同じ方式で薬剤によって処理する。有効濃度で投与した薬剤が微生物の増殖を阻止するのに十分な、適切な期間の後に、細菌増殖を以下を含む種々の手段によって判定することができる:(i)接種スポットの有無(およびサイズ、適宜)の判定;(ii)処理に対して生存し得た微生物の数の算出を可能にするための、寒天増殖プレート上への処理細菌のプレーティングおよび既知の容積の浮遊液の連続希釈;(iii)顕微鏡的(肉眼による)な判定;(iv)対数増殖期にある微生物を薬剤を含む増殖培地中に浮遊させ、接種後の種々の時点で既知の容積を採取し、連続希釈した上で、生存した微生物の算定のために増殖寒天上にプレーティングして得た、時間-死滅曲線;(v)生存している微生物の算定が可能な、当業者に知られた他の分光学的、分析的、インビトロまたはインビボの方法。単一の薬剤または薬剤の組合せが細胞内に存在する感染物を死滅させる有効性は一般に、微生物を放出させるために宿主細胞を界面活性剤(1%Triton X-100に0.1%ドデシル硫酸ナトリウムを加えたもの)で可溶化した後に、生存している微生物の数を算定するために可溶化物を連続希釈して寒天増殖プレートに播くことによって評価される。
【0065】
上記の手段を用いて、有効な薬剤の組合せを、その拮抗活性、相加活性または相乗的活性に関して評価する。具体的には、化合物の対を固定比(これは等モルでも、MIC値の比でも、または他の固定比でもよい)として細菌に適用し、細菌を種々の濃度の化合物対によって処理する。活性は上記の通りに決定する。拮抗作用、相加作用または相乗作用をさまざまな数学的処理、例えばアイソボログラム、CIなどによって決定する。
【0066】
抗ウイルス活性を備えた薬剤または薬剤の組合せに対する広範囲のスクリーニングは、さまざまなインビトロアッセイ法、一般的にはプラーク減数アッセイ法および細胞変性効果(CPE)阻止アッセイ法によって行うことができ、これらは当業者に周知である。これらのアッセイ法は、1つまたは複数の抗ウイルス薬が組織培養下でのウイルス感染の影響を阻止する程度を直接測定することができる。プラーク減数アッセイ法は、明確なプラークを生じるウイルスおよび細胞系の組合せに対して行われる。Michaelisらは、ウイルス数に対するアフィジコリンおよびその誘導体の非拮抗的な抗ウイルス効果を種々のモル比で決定するための、プラーク減数アッセイ法とChou-Talalay法との併用を示した(Michaelisら、Arzneimittelforschung (2002) 52(5): 393-399)。特定のウイルスおよび細胞系の組合せで明確なプラークが生じない場合には、CPE阻止アッセイ法が好ましい。抗ウイルス薬の非拮抗的な組合せを迅速かつ簡便に同定するためのそのほかの方法には、細胞生存度、ウイルス収量アッセイ法およびHIV急性または慢性感染アッセイ法が非制限的に含まれる。細胞生存度は抗ウイルス薬または薬剤の組合せが細胞の生存度を高める能力を測定するために用いられ、前記のMTTアッセイ法などの定量的アッセイ法を用いて行うことができる。または、ウイルス収量アッセイ法および急性HIV感染アッセイ法は、薬剤がウイルス収量を減少させる能力を評価し、抗ウイルス活性の直接測定を可能にする。前記のアッセイ法が抗ウイルス薬剤の組合せを相乗的、相加的または拮抗的な作用に関してインビトロでスクリーニングするのに適していることは当業者には明らかであると考えられ、このため、本発明の範囲に含まれる。
【0067】
好ましい薬剤の組合せ
上記の非拮抗的な作用に関する基準を満たすことが判明した治療薬のさまざまな組合せは、その後、薬物送達媒体の配合物の形態で提供される。「治療薬」とは、単独で、または他の化合物との組合せで、望ましくない状態または疾患に冒された対象に対して望ましい効果を有する、化合物のことである。
【0068】
標的疾患が癌である場合、ある種の治療薬は併用するのに好都合である。その例には以下のものがある。
「シグナル伝達阻害物質」、これは癌細胞が増殖または分裂する原因になるシグナルを妨げる、またはそれを阻止する;
「細胞傷害物質」;
「細胞周期阻害物質」または「細胞周期制御阻害物質」、これは細胞が正常な細胞周期、すなわち細胞の生涯を、それを派生させた有糸分裂から、分裂して娘細胞を生じさせる有糸分裂後の事象へと進行させることを妨げる;
「チェックポイント阻害物質」、これは細胞周期チェックポイント、例えば、S/G2チェックポイント、G2/MチェックポイントおよびGl/Sチェックポイントの正常な機能を妨げる;
「トポイソメラーゼ阻害物質」、これにはカンプトテシンなどがあり、DNAの複製および転写に必要な酵素であるトポイソメラーゼIまたはIIの活性を妨げる;
「受容体チロシンキナーゼ阻害物質」、これは、チロシンキナーゼ活性を有する増殖因子受容体の活性を妨げる;
「アポトーシス誘導物質」、これはプログラム細胞死を促進する;
「代謝拮抗物質」、これにはゲムシタビンまたはヒドロキシ尿素などがあり、必須代謝産物とよく似ているために、それがかかわる生物反応を妨げる;
「テロメラーゼ阻害物質」、これは、テロメア長を延長させて細胞の寿命およびその複製能力を向上させる酵素であるテロメラーゼの活性を妨げる;
「サイクリン依存性キナーゼ阻害物質」、これは、ヒストン、細胞骨格タンパク質、転写因子、腫瘍抑制遺伝子といった細胞タンパク質のリン酸化を介して細胞周期の異なる時期間の主要な段階を制御するサイクリン依存性キナーゼを妨げる;
「DNA損傷物質」;
「DNA修復阻害物質」;
「血管形成抑制物質」、これは腫瘍増殖時に起こる新たな血管の形成または既存の血管の成長を妨げる;および
「ミトコンドリア毒」、これはミトコンドリアの呼吸鎖機能を直接的または間接的に破壊する。
【0069】
腫瘍の治療のために特に好ましい組合せは、上記の臨床的に承認された組合せである。これらの組合せはすでにヒトでの使用が承認されているため、適切な送達を保証するための再製剤化(reformation)が特に重要である。
【0070】
併用しうる好ましい薬剤には、以下のものが含まれる:カルボプラチン、シスプラチン、シクロホスファミド、ドキソルビシン、ダウノルビシン、エピルビシン、マイトマイシンC、ミトキサントロンなどのDNA損傷物質;5-フルオロウラシル(5-FU)またはFUDR、ゲムシタビンおよびメトトレキサートを含むDNA修復阻害物質;カンプトテシン、イリノテカンおよびトポテカンなどのトポイソメラーゼI阻害物質;ブレオマイシン、ドセタキセル、ドキソルビシン、エトポシド、パクリタキセル、ビンブラスチン、ビンクリスチン、ビンデシンおよびビノレルビンなどのS/G2またはG2/Mチェックポイント阻害物質;G1/S初期チェックポイント阻害物質;G2/Mチェックポイント阻害物質;ゲニステイン、トラスツズマブ(trastuzumab)、ZD1839などの受容体チロシンキナーゼ阻害物質;細胞傷害物質;アポトーシス誘導物質および細胞周期制御阻害物質。
【0071】
薬剤の1つまたは複数の作用機序が不明であってもよく、または誤って同定されていてもよい。相乗的または相加的な薬剤の組合せはすべて本発明の範囲に含まれる。新生物の治療のためには、無秩序な細胞増殖につながる複数の機序を阻害する組合せを、本発明に従って用いるために選択することが好ましい。例えば、本発明は、細胞周期における特定の箇所に影響を及ぼし、それによって非拮抗的な作用を生じる組合せを選択することを含む。例えば、DNA損傷を引き起こす薬剤をDNA修復を阻害するもの、例えば代謝拮抗物質などと対にする。本発明はまた、通常であれば細胞増殖をもたらすと考えられる複数の経路を阻止する組合せを選択することも含む。
【0072】
特に好ましい組合せは、DNA損傷物質とDNA修復阻害物質との組合せ、DNA損傷物質とトポイソメラーゼIまたはトポイソメラーゼII阻害物質との組合せ、トポイソメラーゼI阻害物質とS/G2またはG2/Mチェックポイント阻害物質との組合せ、G1/Sチェックポイント阻害物質またはCDK阻害物質とG2/Mチェックポイント阻害物質との組合せ、受容体チロシンキナーゼ阻害物質と細胞傷害物質との組合せ、アポトーシス誘導物質と細胞傷害物質との組合せ、アポトーシス誘導物質と細胞周期制御阻害物質との組合せ、G1/SまたはG2/Mチェックポイント阻害物質と細胞傷害物質との組合せ、トポイソメラーゼIまたはII阻害物質とDNA修復阻害物質との組合せ、トポイソメラーゼIもしくはII阻害物質またはテロメラーゼ阻害物質と細胞周期制御阻害物質との組合せ、トポイソメラーゼI阻害物質とトポイソメラーゼII阻害物質との組合せ、および2種類の細胞傷害物質の組合せである。
【0073】
併用しうる具体的な薬剤には、以下のものが含まれる:シスプラチン(またはカルボプラチン)と5-FU(またはFUDR)、シスプラチン(またはカルボプラチン)とイリノテカン、イリノテカンと5-FU(またはFUDR)、ビノレルビンとシスプラチン(またはカルボプラチン)、メトトレキサートと5-FU(またはFUDR)、イダルビシンとaraC、シスプラチン(またはカルボプラチン)とタキソール、シスプラチン(またはカルボプラチン)とエトポシド、シスプラチン(またはカルボプラチン)とトポテカン、シスプラチン(またはカルボプラチン)とダウノルビシン、シスプラチン(またはカルボプラチン)とドキソルビシン、シスプラチン(またはカルボプラチン)とゲムシタビン、オキサリプラチンと5-FU(またはFUDR)、ゲムシタビンと5-FU(またはFUDR)、アドリアマイシンとビノレルビン、タキソールとドキソルビシン、フラボプリドールとドキソルビシン、UCN01とドキソルビシン、ブレオマイシンとトリクロロペラジン、ビノレルビンとエデルフォシン、ビノレルビンとスフィンゴシン(およびスフィンゴシン類似体)、ビノレルビンとホスファチジルセリン、ビノレルビンとカンプトテシン、シスプラチン(またはカルボプラチン)とスフィンゴシン(およびスフィンゴシン類似体)、スフィンゴシン(およびスフィンゴシン類似体)とダウノルビシン、ならびにスフィンゴシン(およびスフィンゴシン類似体)とドキソルビシン。
【0074】
好ましい組合せには一般に、臨床の場で有効なことがFDAによる認識ですでに示されている上記のもの、および文献報告に基づいてそれが示唆されるものが含まれる。本発明の方法に用いるための候補薬剤はこれらの特定の組合せには限定されないが、上記のものは、適した併用療法として開示されており、このため、本発明の方法および組成物に用いるのに好ましい。
【0075】
脂質の中には、アポトーシスの誘導といった治療的な作用を発揮しうる「治療的脂質」がある。この定義に含まれるものには、エーテル脂質、ホスファチジン酸、ホスホネート、セラミドおよびセラミド類似体、ジヒドロキシセラミド、フィトセラミド、スフィンゴシン、スフィンゴシン類似体、スフィンゴミエリン、セリン含有脂質およびスフィンガニンなどの脂質がある。本明細書において定義される「セリン含有リン脂質」または「セリン含有脂質」などの用語は、一方の端でセリンと共有結合し、他方の端で、エーテル結合、エステル結合またはアミド結合を介して疎水性部分と連結した三炭素骨格と共有結合しているリン酸基を頭部極性基が含む、リン脂質のことである。このクラスに含まれるものには、長さが5〜23炭素原子で種々の程度に飽和している2つの炭化水素鎖が疎水性部分に存在するホスファチジルセリン(PS)などのリン脂質がある。セリン含有リン脂質またはセリン含有脂質に言及する場合の疎水性部分という用語は、長鎖飽和または不飽和脂肪族炭化水素鎖(選択的には1つまたは複数の芳香族、脂環式または複素環式原子団によって置換されたもの)などの非極性基のことを指す。
【0076】
治療的脂質および他の薬剤の組合せを、相乗的または相加作用を達成するために用いることもできる(実施例17〜21を参照)。
【0077】
非拮抗的な併用効果を示す比を決定するためのハイスループットスクリーニング
薬剤の新規な非拮抗的組合せを同定するために、薬剤の化学物質ライブラリーを互いにさまざまな比でスクリーニングすることができる。化学物質ライブラリーは新規な薬剤を含んでも従来の薬剤を含んでもよい。2種類の薬剤の組合せに関するスクリーニングに加えて、3種類または4種類の薬剤の組合せを非拮抗的な併用効果に関してスクリーニングすることもできる。薬物の相乗作用を決定するために用いるデータ解析法は、前記のメディアンエフェクト解析(Median Effect Analysis)であることが好ましい。この方法によれば、薬剤のライブラリーを、個別に、および種々の比で組み合わせて検討する。続いて、ChouおよびTalalayによって開発された前記の方法を用いて組合せ指数を算出する。特定の比で非拮抗的な作用を示す薬剤の組合せを送達媒体中に非拮抗的な比で封入する。
【0078】
ハイスループットスクリーニングシステムは市販されている(例えば、Zymark Corp., Hopkinton, Mass.;Air Technical Industries, Mentor, Ohio;Beckman Instruments, Inc. Fullerton, Calif.;Precision Systems, Inc., Natick, Mass.などを参照されたい)。これらのシステムは通常、すべての試料および試薬のピペット操作、液体の分注、定めた時間のインキュベーション、ならびにアッセイ法に適した検出器によるマイクロプレートの最終的な読み取りを含む、全手順を自動化している。適合性のあるこれらのシステムは、ハイスループットかつ始動が迅速である上に、高度の汎用性およびカスタマイズ性を備えている。この種のシステムの製造者は、種々のハイスループットスクリーニング方法に関する詳細なプロトコールを用意している。
【0079】
非拮抗的な組成物の調製
薬剤の適切な比が上記のようにして決定されれば、1つまたは複数の送達媒体が2種類またはそれ以上の薬剤を封入するように、薬剤をその適切な比で送達媒体組成物中に収める。組成物中のすべての送達媒体が同一である必要はない。組成物中の送達媒体は粒子であり、そのサイズは投与経路に依存し、本発明の薬剤を封入しうる水性溶媒または他の溶媒中に懸濁化されうる。このような媒体には、例えば、前述した、脂質担体、リポソーム、シクロデキストリン、ポリマー性ナノ粒子およびポリマー性微粒子(ナノカプセルおよびナノスフェアを含む)、ブロック共重合体ミセル、脂質安定化エマルション、誘導体化された一本鎖ポリマー、ポリマー脂質ハイブリッド系、脂質ミセル、リポタンパク質ミセルが含まれる。静脈内投与のための送達媒体は一般に直径約4〜6,000nmである。好ましい直径は直径約5〜500nm、より好ましくは直径5〜200nmである。吸入投与、クモ膜下腔内投与、関節内投与、動脈内投与、腹腔内投与または皮下投与のための送達媒体は一般に4μmから50μmを超える範囲である。眼内投与用に設計される送達媒体組成物は一般にそれよりも小さい。
【0080】
治療薬は送達媒体中に「封入」される。「封入」には、前記の通り、薬剤と送達媒体との共有結合性または非共有結合性の会合が含まれる。例えば、これは薬剤と送達媒体の1つもしくは複数の外層との相互作用、または送達媒体内部への封じ込め、送達媒体の異なる部分間で実現される平衡によるものであってよい。例えば、リポソームの場合、薬剤の封入は、脂質成分との共有結合性もしくは非共有結合性の相互作用を介したリポソームの二重層との相互作用による薬剤の会合、またはリポソームの水性内部への封じ込め、または内部水相と二重層との間の平衡によるものであってよい。ポリマーを基盤とする送達媒体の場合、封入は薬剤と線状もしくは非線状ポリマーとの共有結合のことを指す。さらに、非制限的な例には、ポリマー基質全体への薬剤の分散、またはナノカプセル、ブロック共重合体ミセルもしくはポリマー-脂質ハイブリッド系のコアへの薬剤の濃縮が含まれる。「装填(loading)」とは、1つまたは複数の薬剤を送達媒体中に封入する作業のことを指す。
【0081】
所望の組合せの封入は、別個の送達媒体中への封入、または同じ送達媒体の内部への封入のいずれによって行うこともできる。リポソームなどの別個の送達媒体中への封入が望まれる場合には、各リポソームの脂質組成は協調的な薬物動態が可能となる程度に大きく異なってもよい。媒体組成を変更することにより、封入された薬剤の放出速度を、非拮抗的な比の薬剤が腫瘍部位に送達されることが可能となるように一致させることができる。放出速度を変更する手段には、薬剤の保持性を改良するために小胞を形成する脂質のアシル鎖の長さを延長すること、リポソーム膜から突出したPEGなどの表面接枝状親水性ポリマーの交換を制御すること、およびステロールまたはスフィンゴミエリンなどの膜硬化性物質を膜に組み入れることが含まれる。第1および第2の薬剤を特定の薬剤比で投与したい場合、ならびに、第2の薬剤が第1の薬剤のリポソーム組成物(例えば、DMPC/Chol)の内部にほとんど保持されない場合には、アシル鎖長がさらに長い脂質(例えば、DSPC/Chol)リポソーム組成物中に第2の薬剤を封入することによって薬物動態を改良しうることは、当業者には明らかであると考えられる。または、2種類またはそれ以上の薬剤を同じ送達媒体中に封入してもよい。
【0082】
封入のための技法は送達媒体の性質に依存する。例えば、受動的な装填方法および能動的な装填方法の両方を用いてリポソーム中に治療薬を装填することができる。
【0083】
薬剤をリポソーム中に封入するための受動的方法は、リポソームの調製中に薬剤を封入することを含む。この方法では、薬剤は膜に会合させてもよく、封じ込めた水性空間内に封入してもよい。これには、目的の薬剤を含む水相を、反応容器の壁面に付着させた乾燥した小胞形成性脂質の薄膜と接触させる、Banghamら、J. Mol. Biol. (1965) 12: 238に記載された受動的封じ込め法が含まれる。機械的手段による振盪を加えると、脂質の膨潤が起こって多重ラメラ小胞(MLV)が形成されると考えられる。押出処理法を用いて、MLVを大型単ラメラ小胞(LUV)または小型単ラメラ小胞(SUV)へと変換することができる。用いうるもう1つの受動的装填方法には、DeamerおよびBangham、Biochim. Biophys. Acta (1976) 443: 629に記載されたものが含まれる。この方法では、小胞形成性脂質をエーテル中に溶解した上で、まずエーテルを蒸発させて表面に薄膜を形成させ、その後にこの薄膜を封入しようとする水相と接触させる代わりに、エーテル溶液を前記水相中に直接注入した後にエーテルを蒸発させ、それによって薬剤が封入されたリポソームを得る。用いうるさらにもう1つの方法は、SzokaおよびPapahadjopoulos, P.N.A.S. (1978) 75: 4194に記載された逆相蒸発(REV)法であり、この方法では水に溶けない有機溶媒中にある脂質溶液を水性担体相中に乳濁化し、その後に減圧下で有機溶媒を除去する。
【0084】
用いうるその他の受動的封じ込め法には、リポソームに対して連続的な脱水処理および再水和処理、または凍結解凍を行うことが含まれる。脱水は蒸発または凍結乾燥によって行われる。この技法はKirbyら、Biotechnology (1984) 979-984に記載されている。同じく、ShewおよびDeamer(Biochim. et Biophys. Acta (1985) 816: 1-8)は、超音波処理によって調製したリポソームを、封入しようとする溶質を含む水性溶液と混合し、その混合物を回転フラスコ内で窒素中にて乾燥させる方法を記載している。再水和を行うと、溶質のかなりの割合が封入された大型のリポソームが生成される。
【0085】
2種類またはそれ以上の薬剤の受動的封入は、薬剤の数多くの組合せに対して可能である。このアプローチは水性緩衝液中への薬剤の溶解性によって制限され、薬剤の多くの割合が送達システム内部に捕捉されないという制限もある。装填性は、薬剤を脂質試料と同時凍結乾燥すること、および薬剤を溶解するための最少容積で再水和を行うことによって改良されることがある。溶解性は、緩衝液のpHを変更すること、温度を高めること、または緩衝液に対する塩の添加もしくは除去によって改良されることがある。
【0086】
能動的な封入方法を用いてもよい。例えば、金属錯体化またはpH勾配装填法に従ってリポソームに装填することができる。pH勾配装填法では、選択したpHの水相を封入するリポソームを形成させる。次に、水和リポソームを、薬剤または封入しようとする他の作用物質の荷電を除去もしくは低下させるように選択した異なるpHの水性環境に置く。薬剤がリポソームの内部に移動すると、内部のpHのために荷電した薬剤の状態が生じ、そのため薬剤が脂質二重層を透過することが妨げられ、それによって薬剤がリポソーム中に封じ込められる。
【0087】
pH勾配を作り出すためには、最初の外部媒質を、水素イオン濃度が異なる新たな外部媒質によって置き換える。外部媒質の置換は、さまざまな技法により、例えば、新たな媒体によって平衡化したゲル濾過カラム、例えばセファデックス(Sephadex)G-50カラムに脂質小胞調製物を通過させること(以下の実施例で述べる)により、または遠心分離、透析もしくは関連した技法などによって行いうる。内部媒質は外部媒質に対して酸性でも塩基性でもよい。
【0088】
pH勾配を確立した後に、pH勾配によって装填されうる薬剤を混合物に添加し、上記のようにしてリポソーム中への薬剤の封入を行わせる。
【0089】
pH勾配を用いる装填は、米国特許第5,616,341号、第5,736,155号および第5,785,987号に記載された方法に従って行うことができ、これらは参照として本明細書に組み入れられる。好ましいpH勾配装填法は、pH 2〜6の内部緩衝液としてのクエン酸塩および中性外部緩衝液を用いる、クエン酸塩を利用する装填方法である。
【0090】
リポソームの内外にpH勾配を成立させてそれを維持するにはさまざまな方法を用いることができ、これらはすべて参照として本明細書に組み入れられる。これには、リポソーム膜に挿入しうるイオノフォア、およびプロトンの交換において膜を越える輸送イオンを用いることが含まれうる(例えば、米国特許第5,837,282号を参照)。リポソーム膜を越えてプロトンを往復させることができ、このためpH勾配を成立させうる、リポソームの内部に封入された化合物(例えば、米国特許第5,837,282号を参照)を用いることもできる。これらの化合物は、脱プロトン化された場合は中性であってプロトン化されると荷電するイオン化部分を含む。中性脱プロトン化型(これはプロトン化型と平衡状態にある)は、リポソーム膜を通過することができ、このためプロトンがリポソーム内部に残り、それによって内部のpHが低下する原因となる。この種の化合物の例には、塩化メチルアンモニウム、硫酸メチルアンモニウム、硫酸エチレンジアンモニウム(米国特許第5,785,987号を参照)および硫酸アンモニウムが含まれる。塩基性の内部pHを成立させうる内部装填緩衝液を利用することもできる。この場合には、中性型のものがプロトン化され、プロトンがリポソームの内部から外に出されて内部が塩基性となる。この種の化合物の例には酢酸カルシウムがある(米国特許第5,939,096号を参照)。
【0091】
2種類またはそれ以上の薬剤を、同じ能動的装填方法を用いてリポソーム中に装填することもでき、または異なる能動的装填方法を用いてもよい。例えば、金属錯体化装填法を複数の薬剤を能動的に装填するために用いてもよく、またはpH勾配装填法などの別の能動的装填法と組み合わせてもよい。金属を用いる能動的装填法では一般に、金属イオンが受動的に封入されたリポソーム(受動的に装填された治療薬を含む場合も含まない場合もある)を用いる。金属イオンのさまざまな塩が用いられるが、その塩は薬学的に許容され、水性溶液に溶解するという条件が付く。能動的に装填される薬剤は、金属イオンと錯体を形成し、そのように錯体化した場合にリポソーム内部に保持されるが、金属イオンと錯体を形成しない場合にもリポソーム中に装填されうるという能力に基づいて選択される。金属と配位結合を行える薬剤は一般に、アミン、カルボキシル基、エーテル、ケトン、アシル基、アセチレン、オレフィン、チオール、ヒドロキシル基もしくはハロゲン基、または金属イオンに電子を供与することによって金属イオンと錯体を形成しうる他の適した基などの配位結合部位を含む。金属と結合する作用物質の例には、フルオロキノロンなどのキノロン;ナリジクス酸などのキノロン;ドキソルビシン、ダウノルビシンおよびイダルビシンなどのアントラサイクリン;カナマイシンなどのアミノグリコシド;ならびにブレオマイシン、マイトマイシンCおよびテトラサイクリンなどの他の抗生物質;ならびにシクロホスファミド、チオセミカルバゾン、インドメタシンおよびニトロプルシドなどのナイトロジェンマスタード;トポテカン、イリノテカン、ラートテカン(lurtotecan)、9-アミノカンプトテシン、9-ニトロカンプトテシンおよび10-ヒドロキシカンプトテシンなどのカンプトテシン;ならびにエトポシドなどのポドフィロトキシンが非制限的に含まれる。薬剤の取り込みは、薬剤を外部媒質に添加した後に混合物を適した温度でインキュベートすることによって確認しうる。リポソームの組成、内部媒質の温度およびpH、ならびに薬剤の化学的性質に応じて、薬剤の取り込みは数分で起こることも数時間かかることもある。リポソーム内部で薬剤と金属との間に配位結合が生じたか否かを判定する方法には、分光光度分析、および当業者に周知のその他の従来の技法が含まれる。
【0092】
さらに、リポソーム中にイオノフォアを用いずに行った、金属を利用する手順を用いたリポソーム装填効率および保持特性は、用いた金属およびリポソームの脂質組成に依存する。脂質組成および金属を選択することにより、装填特性または保持特性を、所望の装填またはリポソームからの選択した薬剤の放出を達成するように適合させることができる。
【0093】
複数の薬剤を送達媒体に装填するために、受動的および能動的な装填方法を逐次的に組み合わせることもできる。例えば、受動的に封じ込められたシスプラチンなどのプラチナ性薬剤をMnC12の存在下に含むリポソームは、続いて、ドキソルビシンなどのアントラサイクリンをリポソームの内部に封入するために用いることができる。この方法は、受動的封入によってリポソーム中に封入されたさまざまな薬剤に適用しうる可能性が高い。
【0094】
複数の薬剤を封入した送達媒体組成物の治療的用途
これらの送達媒体組成物は、温血動物および鳥類における種々の疾患の治療に用いうる。したがって、本発明の方法および組成物による治療に適した対象には、ヒト、家畜または家庭内動物などの哺乳動物、ニワトリおよびアヒルなどの家畜化された鳥類対象、ならびに研究用の実験動物が含まれる。本発明の組成物の医学的用途の例には、癌の治療、高血圧、不整脈および再狭窄などの心血管疾患の治療、細菌性、ウイルス性、真菌性もしくは寄生生物性の感染症の治療、本発明の組成物のワクチンとしての使用による疾患の治療および/もしくは予防、炎症の治療、または自己免疫疾患の治療が含まれる。
【0095】
1つの態様において、本発明による送達媒体組成物は、新生物の治療に用いられることが好ましい。腫瘍部位への製剤の送達は、リポソームまたは他の粒子状送達システムの投与によって達成される。リポソームの直径は200nm未満であることが好ましい。腫瘍の血管構造は、内皮における開窓またはギャップのため、正常な血管構造よりも一般に漏出性が高い。これにより、直径200nmまたはそれ未満の送達媒体が、不連続的な内皮細胞層、および、その下層にあって腫瘍に血液を供給する血管周囲の基底膜を通過することが可能になる。送達媒体が血管外漏出後に腫瘍部位内に選択的に蓄積されることにより、薬物送達および治療的有効性が向上する。担体が血管外に漏出するため、血中で測定した担体薬剤-薬剤比は、血管外漏出空間における担体薬剤-薬剤比と同程度であると想定することができる。
【0096】
送達媒体組成物の投与
上記の通り、本発明の送達媒体組成物を、ヒトを含む温血動物、ならびに家畜鳥類に投与することができる。ヒトの疾患の治療の場合、有能な医師は、本発明の組成物を、確立されたプロトコールを用いて、用量、投与計画および投与経路の点でいかにして用いるべきかを決定しうると考えられる。本発明の送達媒体組成物中に封入された薬剤が対象の健常組織に及ぼす毒性が低ければ、このような用途に用量漸増法を用いてもよい。
【0097】
本発明の薬学的組成物は、非経口的に、すなわち動脈内、静脈内、腹腔内、皮下または筋肉内に投与することが好ましい。より好ましくは、薬学的組成物をボーラス注射によって静脈内または腹腔内に投与する。例えば、Rahmanら、米国特許第3,993,754号;Sears、米国特許第4,145,410号;Papahadjopoulosら、米国特許第4,235,871号;Schneider、米国特許第4,224,179号;Lenkら、米国特許第4,522,803号;およびFountainら、米国特許第4,588,578号を参照されたい。
【0098】
他の方法では、製剤を組織に直接適用することにより、本発明の医薬製剤を標的組織に接触させることができる。適用は、外用、「観血的(open)」または「閉鎖的(closed)」処置のいずれによって行うこともできる。「局所的(topical)」とは、環境に露出された組織、例えば皮膚、中咽頭、外耳道などに対する医薬製剤の直接適用を意味する。「観血的」処置とは、患者の皮膚を切開し、医薬製剤を適用しよとする下層の組織が直接見えるようにする処置のことである。これは一般に、肺に到達するための開胸術、腹部臓器に到達するための腹式開腹術、または標的組織に対する他の直接的な外科的アプローチなどの外科処置によって行われる。「閉鎖的」処置とは、内部標的組織を直接目にすることはないが、皮膚内の小さな創を通して挿入した器具を介して到達する侵襲的処置のことである。例えば、針洗浄によって製剤を腹膜に投与することができる。同様に、腰椎穿刺時に注入し、その後に脊椎麻酔または脊髄のメトラザミド(metrazamide)造影のために一般に行われているように患者に適切な体位をとらせることにより、医薬製剤を髄膜または脊髄に投与することもできる。または、製剤を内視鏡装置を用いて投与することもできる。
【0099】
本発明の送達媒体を含む薬学的組成物は標準的な技法に従って調製され、水、緩衝液、0.9%生理食塩水、0.3%グリシン、5%デキストロースなどを含んでよく、安定性の向上のためにアルブミン、リポタンパク質、グロブリンなどの糖タンパク質も含みうる。これらの組成物は、従来のよく知られた滅菌法によって滅菌しうる。その結果得られた水性溶液を使用のためにパッケージ化してもよく、無菌条件下で濾過して凍結乾燥した上で凍結乾燥製剤を投与前に滅菌水溶液と混合してもよい。組成物は、生理的条件に近づけるための薬学的に許容される補助物質、例えば、pH調整剤および緩衝剤、張度調整剤など(例えば、酢酸ナトリウム、乳酸ナトリウム、塩化ナトリウム、塩化カリウム、塩化カルシウムなど)を含みうる。さらに、送達媒体懸濁液が、脂質をフリーラジカルおよび保存時の脂質過酸化損傷から保護する脂質保護剤を含んでもよい。α-トコフェロールなどの脂溶性フリーラジカル失活剤、および、フェリオキサミンなどの水溶性鉄特異的キレート剤が適している。
【0100】
医薬製剤における送達媒体の濃度には、重量比で約0.05%未満、通常は約2〜5%または少なくとも約2〜5%から、10〜30%までというように大きな幅があってよく、これは主として、選択した特定の投与様式に従い、液体の容積、粘性などによって選択される。例えば、治療に伴う液体投与量を少なくするには濃度を高めるとよい。または、刺激性の脂質を含む送達媒体は、投与部位での炎症を軽減するために希釈して低濃度にするとよい。診断の目的で投与する送達媒体の量は、用いる特定の標識、診断しようとする疾病状態、および臨床医の判断に依存すると考えられる。
【0101】
本発明の薬学的組成物は静脈内に投与されることが好ましい。送達媒体製剤の投与量は、薬剤と脂質との比、ならびに、患者の年齢、体重および状態に基づいた、投与する臨床医の見解に依存すると考えられる。
【0102】
薬学的組成物に加えて、獣医学的な使用に適した製剤を、対象に適した様式で調製して投与することもできる。好ましい獣医学的対象には、哺乳動物種、例えば、非ヒト霊長動物、イヌ、ネコ、ウシ、ウマ、ヒツジ、および家畜化した鳥類が含まれる。対象には実験動物、例えば、特にラット、ウサギ、マウスおよびモルモットも含まれる。
【0103】
インビボでの治療活性の評価
2種類またはそれ以上の薬剤が封入された送達媒体組成物の治療活性は、動物モデルへの投与後に測定することができる。動物モデルは腫瘍を含むことが好ましいが、送達媒体組成物を他の疾患の動物モデルに投与することもできる。マウスおよびラットなどの齧歯類種、例えば、免疫能力があるものおよび免疫不全のものを含む、近交系、非近交系または雑種由来のもの、ならびにノックアウトモデルまたはトランスジェニックモデルを用いることができる。
【0104】
モデルは、細胞浮遊液、胸腺または腫瘍断片として、皮下、静脈内、腹腔内、筋肉内、クモ膜下腔内または同所領域に移植された固形腫瘍または非固形腫瘍からなりうる。腫瘍を、腫瘍形成性/発癌性物質の適用もしくは投与によって成立させてもよく、または適切な遺伝子組換え動物モデルにおいて自然発生させてもよい。腫瘍のタイプは、癌、肉腫、黒色腫、神経膠腫、白血病およびリンパ腫といった、外胚葉、中胚葉または内胚葉由来の腫瘍からなりうる。
【0105】
1つの好ましい態様では、腫瘍のマウスモデルを用いる。免疫不全マウスで増殖させたヒト異種移植固形腫瘍を利用し、規定された遺伝学的性質および増殖性に基づいて選択することができる。これらの実験に用いる腫瘍細胞は遺伝的に操作した上で、または好ましい特性を発現するように選択した上で、マウスに注入することができる。
【0106】
腫瘍が触知可能な(測定可能な)サイズに増殖したところで、送達媒体組成物を投与し(静脈内が好ましい)、腫瘍増殖に対する効果を観測する。意図する治療的処置は、単回のボーラス投与もしくはプッシュ投与、または数日もしくは数週間にわたる多回もしくは連続的な投与からなってよく、経口的、鼻腔内、皮下、静脈内、腹腔内、クモ膜下腔内、腫瘍内経路などの任意の適した経路により、シリンジ、錠剤、液剤およびポンプ(浸透ポンプなど)を用いうる。達成しうる最大抗腫瘍活性を決定するために用量および投与計画の依存性を評価することもできる。
【0107】
腫瘍を含む動物モデルにおける治療活性を決定するためのさまざまな方法を用いうる。これには、固形腫瘍モデル評価法および非固形腫瘍モデル評価法が含まれる。
【0108】
固形腫瘍モデル評価法には、腫瘍体積(質量)、腫瘍重量抑制(TWI%)、腫瘍増殖遅延(T-C)、腫瘍退縮、細胞死の計測およびクローン原性(clonogenic)アッセイ法が含まれる。
【0109】
腫瘍体積の測定値は、直交する長さおよび幅のキャリパーによる測定によって決定される(高さの測定もしばしば行いうる)。腫瘍体積(mL)または質量(g)は、体積=(長さ×幅2/2;または体積=π/6×(長さ×幅×高さ)によって算出される。データを時間に対してプロットする。
【0110】
腫瘍重量抑制(TWI%)は、定められた時点で、投与群の平均腫瘍重量を対照群の平均腫瘍重量によって除算して1を差し引いた上で100を掛けることによって決定される。
【0111】
腫瘍増殖遅延(T-C)は、投与群(T)が任意の決定された腫瘍サイズ(例えば、300mg)に到達するまでの日数の中央値から、対照群が同じ腫瘍サイズに達するまでの日数の中央値を差し引いた値として計測される。
【0112】
投与の結果としての腫瘍退縮を、腫瘍モデルを評価する手段として用いることもできる。結果は腫瘍サイズ(質量)の経時的な減少として表現される。
【0113】
細胞死法による固形腫瘍モデル評価は、腫瘍のすべてが所定のサイズ(例えば、200mg)を超えるまで繰り返し測定することを含みうる。続いて、腫瘍増殖および腫瘍倍加時間を評価する。log10細胞死パラメーターは以下によって算出しうる:
log10細胞死/用量=(T-C)/((3.32)(Td)(投与回数))
log10細胞死(合計)=(T-C)/((3.32(Td))
log10細胞死(正味)=((T-C)-(Rxの期間))/((3.32(Td))
ここで、(T-C)=腫瘍増殖遅延
Td=腫瘍倍加時間である。
【0114】
クローン原性(clonogenicity)アッセイ法は、治療法の有効性を表す。これらのアッセイ法は、切り出し(excision)アッセイ法および固形腫瘍由来の細胞浮遊液の特徴分析を含む。
【0115】
切り出しアッセイ法は、腫瘍から調製した浮遊液におけるどの細胞画分が無限の増殖能を有するか(すなわち、クローン原性があるか)を評価するために用いられる。切り出しアッセイ法には3種類がある。
i)TD50またはエンドポイント希釈アッセイ法:これはインビボの接種材料から採取した細胞が腫瘍を生成するために必要な数を決定する。
ii)インビボコロニーアッセイ法:これは個々の細胞が、例えば肺に、小結節(コロニー)を形成する能力を評価する。
iii)インビトロコロニーアッセイ法、これは個々の細胞が、コロニーが培養皿のプラスチックまたはガラスの表面に形成される場合には液体培地中で、またはコロニーが浮遊液中に形成される寒天などの半流動培地中で、増殖してコロニーとなる能力を調べる。
【0116】
固形腫瘍からの細胞浮遊液の特徴分析は、インビトロおよびインビボでのクローン原性アッセイ法、フローサイトメトリー測定のため、ならびに細胞単位に対して行われるさまざまな生化学的および分子的な分析のために必要とされる。調製は、酵素的、機械的、化学的、それらの組合せ、および界面活性剤などのさまざまな方法による。評価には、細胞収量、細胞形態、腫瘍細胞クローン原性、生化学的または分子的な特徴の保持が含まれる。
【0117】
非固形腫瘍モデル評価法には、寿命の延長(ILS%)、腫瘍増殖遅延(T-C)、長期生存者(治癒)の計測が含まれる。
【0118】
寿命の延長(ILS%)は、対照群または非投与群と比較した投与群の寿命の延長率を計測する。腫瘍増殖遅延(T-C)は、投与(T)群の生存日数の中央値から対照(C)群の生存日数の中央値を差し引いた値を計測する。長期生存者(治癒)は、非投与群または対照群の生存期間の3倍を超えて生存した投与群について計測する。
【0119】
癌に罹患したヒトにおける治療活性を決定する方法は、生存および代用エンドポイントの測定を含む。生存を評価するのに妥当な時点は当該の腫瘍によって決まる。例えば、低悪性度リンパ腫の患者に関する生存率は、診断から5年後または10年後に評価するとよいが、進行性非小細胞肺癌などの悪性度の高い疾患を有する患者の生存は診断から6カ月後または12カ月後に評価するのが最もよい。
【0120】
代用エンドポイントを用いて治療活性を決定する方法には、完全縮小(CR)、部分的縮小(PR)、無増悪生存(PFS)、無増悪期間(TTP)または縮小期間(DOR)、血漿マーカーおよび尿マーカー、酵素阻害ならびに/または受容体の状態、遺伝子発現の変化および生活の質(QOL)の計測が含まれる。
【0121】
完全縮小(complete response)とは、治療している腫瘍の種類にとって適切な期間にわたり、新たな疾患の発生を伴わずに、疾患のすべての既知の部位が消失することを意味する。評価は、以上に述べたようなさまざまな検査に基づく。
【0122】
部分的縮小(partial response)とは、治療している腫瘍の種類にとって適切な期間にわたり、新たな病変の出現を伴わずに、すべての病変の二方向測定値の積の合計が少なくとも50%減少することを意味する。評価は、患者のさまざまな検査(CTスキャン、MRI、超音波、PETスキャン、骨スキャン、身体診察)に基づく。
【0123】
無増悪生存(PFS):患者が生存していて、既存の腫瘍の増殖がなく、新たな腫瘍塊の出現もみられない治療からの期間。PFSは、診断後の所定の時点で生存していて無増悪である期間または患者の割合として表現しうる。
【0124】
無増悪期間(TTP)または縮小期間(DOR)とは、治療時から、既存の腫瘍塊のサイズの増大または新たな腫瘍塊の出現として計測される腫瘍増殖の進行までの期間のことである。
【0125】
血漿マーカーおよび尿マーカーには、以下のマーカーを非制限的に含むマーカーを測定することが含まれる:前立腺特異抗原(PSA)および癌胎児性抗原(CEA)。
【0126】
酵素阻害および/または受容体の状況。チロシンキナーゼ受容体、EGF受容体、PDGF受容体、Her-1およびHer-2受容体を非制限的に含む増殖因子受容体。インテグリン関連キナーゼ、タンパク質キナーゼなどを非制限的に含む酵素。
【0127】
遺伝子発現の変化には、遺伝子発現(ゲノミクス)、およびタンパク質発現の変化(プロテオミクス)の連続解析が含まれる。
【0128】
生活の質(QOL)にはEORTC QLQ-C30採点法などの方法が含まれ、これは5種類の機能的尺度(身体的、役割上、認知的、社会的および情緒的)、3種類の症状尺度(悪心、疼痛および疲労)ならびに全般的な健康状態および生活の質の尺度に関して得られたスコアを評価するものである。この指標からは、癌患者によって多く報告される別の症状(呼吸困難、食欲減退、睡眠障害、便秘および下痢)、さらには疾患およびその治療による認知されている財政的な影響に関する単項目評点も得られる。
【0129】
以下の例は例示を目的として提供するものであり、本発明の範囲を制限するためのものではない。
【実施例】
【0130】
以下の実施例では、細胞傷害性の決定および非拮抗的な作用の評価のために以下の方法を用いている。
【0131】
細胞傷害性アッセイ法
以下の実施例では、標準的なテトラゾリウム比色MTT細胞傷害性アッセイ法のプロトコール(Mosmannら、J. Immunol Methods (1983) 65(1-2):55-63)を、影響を受けた細胞の率に関する読み取り値の決定に用いた。簡潔に述べると、生きた細胞は、テトラゾリウム塩である3-(4,5-ジエチルチアゾイル-2-イル)-2,5ジフェニルテトラゾリウムブロミド(3-(4,5-diethylthiazoyl-2-yl)-2,5-diphenyltetrazolium bromide、MTT)を、分光分析によって読み取れる青色のホルマザンへと還元する。25cm2フラスコ内で増殖させたヒトH460非小細胞肺癌(NSCLC)細胞などの細胞を継代し(継代数<20)、新たなRPMI細胞培養液中に再懸濁した上で、96ウェル細胞培養プレートに、1ウェル当たり細胞1000個(1ウェル中に100μL)の濃度でプレーティングする。続いて細胞を37℃、5%CO2下で24時間インキュベートする。その翌日に、薬剤の系列希釈物を12ウェル細胞培養プレート中に用意する。前もって種々の溶液中に調製しておいた薬剤を新たなRPMI細胞培養液中に希釈する。薬剤を、ラテン方格配置法または「チェッカーボード」希釈法を用いて、単一の薬剤(20μl)および特定の固定比での二剤の組合せ(増分20μL)に関する適切なまたは特定されたウェルに投与する。新たな培地を加えてウェル内の総容積を200μLにする。薬剤の曝露は72時間にわたって行う。
【0132】
薬剤曝露の後に、MTT試薬(1mg/mL、RPMI中)を各ウェルに添加して容積を1ウェル当たり50μLとし、3〜4時間インキュベートする。続いてウェル内容物を吸引し、細胞を破壊して細胞内のホルマザン沈殿物を可溶化するために150μL のジメチルスルホキシド(DMSO)を各ウェルに添加する。96ウェルプレートをプレートシェーカーで振盪させ、波長570nmに設定したマイクロプレート分光光度計で読み取る。光学密度(OD)の読み取り値を記録し、ブランクウェル(培地のみを含む)のOD値を細胞を含む全ウェルから差し引く。薬剤に対する曝露後の細胞生存度は対照ウェル(薬剤に曝露されていない細胞)に対する百分率に基づく。すべてのウェルを3回ずつ検討し、平均値を算出する。
【0133】
薬剤の組合せに関するメディアンエフェクト解析
薬剤の組合せの解析には、ChouおよびTalalayによるメディアンエフェクト原理を基盤とするソフトウエアプログラムCalcuSyn(Biosoft, Ferguson, MO, USA)を用いた。まず、二剤の組合せに関する固定比を、単剤の細胞傷害性プロフィールによるIC50:IC50比から導き出す。続いて、製剤の目的に関する考察に基づき、より妥当な固定比(例えば、10:1〜1:10の範囲;モル比)を選択する。細胞生存性に対する薬剤の効果に基づいて算出した平均値から、用量および各々の分割効果の値をCalcuSynコンピュータプログラムに入力する。するとソフトウエアが、薬剤の組合せが相乗的、相加的または拮抗的のいずれであるかを組合せ指数(CI)値に基づいて算出する。
【0134】
実施例1
用量-効果分析のさまざまな表現
薬剤の量(用量または濃度)とその生物作用との間の関係、ならびに薬剤の組合せの連合効果に関する定量分析は、さまざまなやり方で計測および報告を行うことができる。図2は、このような5通りの方法を、一例としてイリノテカンとカルボプラチンとの組合せを用いて示している。
【0135】
用量-効果分析に関するChouおよびTalalayの理論に基づき、「メディアンエフェクト式」がさまざまな生化学的式の算出に用いられており、これらは当技術分野で広範囲に用いられている。この式の変形により、組合せ指数(CI)の算出に用いられるもののような高次式が生まれている。前述の通り、CIは、複数の薬剤の組合せおよび各組合せの種々の比が拮抗的、相加的または相乗的のいずれであるかを決定するために用いうる。CIプロットは一般に、y軸を表すCIと、x軸にある影響を受けた細胞の割合または影響率(fa)との関係を図示したものである。図2Aは、イリノテカン/カルボプラチンがモル比1:10では拮抗的であり(CI>1.1)、1:1および10:1では相乗作用を有すること(CI<0.9)を示している。
【0136】
本出願者らは、用いた薬剤比に対するCIの依存性を表現する代替的な方法もデザインした。図2Bに示されているように、特定の組合せに関する比特異的な作用における傾向がより適切に図示されるように、最大CI値を各々の比に対してプロットする。CI最大値とは、異なる比の薬剤に関してCI値に最も大きな差が観察される単一のfa値(0.2〜0.8の間)について得られたCI値のことである。
【0137】
個々の比について用いる薬剤の濃度は効果(すなわち、相乗作用または拮抗作用)の決定に一定の役割を果たすため、CIを種々の濃度で測定することも重要と考えられる。これらの濃度は、Chou-Talalayによって「有効量」(ED)とも呼ばれ、インビトロアッセイ法において細胞の指定された比率に影響を及ぼすために必要な薬剤の濃度であり、すなわち、ED50は、対照または非投与細胞集団との比較で細胞の50%に影響を及ぼすために必要な薬剤の濃度のことである。図2Cに示されているように、種々の比の間での濃度-効果における傾向は容易に識別可能である。示されているエラーバーは平均周辺での標準偏差の一つを表しており、これはCalcuSynプログラムによって直接決定される。
【0138】
2種類またはそれ以上の薬剤の間の相乗的相互作用は、陽性の効果を得るために必要な各薬剤の量を減らしうる(「用量減少」としても知られる)という点で有益である。ChouおよびTalalayの「用量減少指数」(DRI)は、所定の効果レベルで、相乗的組合せにおける各薬剤の用量を各薬剤の単独での用量と比較してどの程度減少させうるかの指標である。DRIは、用量減少が治療的有効性を維持したままで宿主に対する毒性の低下につながるような臨床的状況では重要である。図2Dにおけるプロットは、それぞれ単独で90%の細胞死を達成するために必要なイリノテカンおよびカルボプラチンの濃度が、それらを非拮抗的な比で併用した場合に必要な個々の濃度よりも有意に高いことを示している。
【0139】
さらに、前記のデータを、古典的なアイソボログラムとして表現することもできる(図2E)。アイソボログラムには種々のED値で作成しうるという利点があるが、しかし、解釈のために高い効果レベルを選択するほど、その読み取りは困難になる。この理由から、以下の実施例におけるデータは一般に、図2Aおよび2Bに示したプロットのタイプに応じて提示されている。
【0140】
実施例2
CIは濃度に依存する
イリノテカンおよび5-フルオロウラシル(5-FU)のモル比1:1および1:10での組合せ、ならびにエトポシドおよびカルボプラチンのモル比10:1および1:10での組合せという薬剤の組合せを、前の実施例の項で説明した、標準的なテトラゾリウム比色MTT細胞傷害性アッセイ法およびメディアンエフェクト解析を用いて、相加的、相乗的または拮抗的な作用に関して検討した。HT29細胞またはMCF-7細胞を、単一の薬剤ならびに薬剤の所定の比での組合せに対して曝露させた。単一の薬剤および組合せについて、8種類の薬剤濃度を用いた。光学密度の値をMTTアッセイ法によって入手し、これを対照に対する百分率に変換し、平均した上で、影響率の値に変換した。用量および影響率の値をCalcuSynに入力し、図3に示されたCIとfaとの関係をみたグラフを得た。
【0141】
図3Aは、イリノテカンおよび5-FUがモル比1:1では、影響率(用量)による評価で、全範囲範囲にわたって非拮抗的であったことを示している。これに対して、モル比1:10では、同じ2つの薬剤は低濃度では非拮抗的であるが、高濃度では拮抗的であった。図3Bに示されているように、エトポシドおよびカルボプラチンはモル比10:1では全濃度範囲にわたって拮抗的であった。これに対して、モル比1:10では、エトポシドおよびカルボプラチンは低濃度では拮抗的であるが、高濃度では非拮抗的であった。
【0142】
シスプラチンおよびエデルフォシンも、CIをfaに対してプロットすることによって概括されているように、モル比10:1および1:1でH460細胞において異なる併用効果を示した。図4に示されているように、モル比10:1での組合せは、影響率の範囲のうち低濃度側の約50%では非拮抗的であり、高濃度では拮抗的であったが、一方、モル比1:1では全濃度範囲にわたって相乗作用を示した。
【0143】
すなわち、これらの結果は、相乗作用が、薬剤の相互の比だけではなく、それらの濃度にも強く依存することを示している。
【0144】
実施例3
さまざまな二剤の組合せに関するCIの決定
以下の表に示したさまざまな薬剤の組合せを、上記のMTT細胞傷害性アッセイ法プロトコールおよびメディアンエフェクト解析手順を用いて、相加的、相乗的または拮抗的な作用に関して検討した。CIとfaとの関係をみたグラフからの結果を以下に表としてまとめている。非拮抗的な作用が認められたfa範囲のおおよその百分率を、比の後ろのカギ括弧の中に報告している。測定はfa値0.2〜0.8の範囲で行い、CI値1.1未満に該当する曲線の百分率を決定することにより、相乗作用または相加作用(非拮抗的)を示したfa範囲の百分率を算出した。データは、少なくとも1つの実験を3回ずつ行ったものに由来する。
a「相乗的または相加的である百分率%」は、fa値0.2〜0.8の範囲での、Chou-Talalay方法に基づくClと影響率(fa)とのプロット上で、拮抗的な範囲(CI値>1.1は拮抗的である)に該当しないfa範囲の百分率として算出される。CIは用量およびfa値をCalcuSynに入力することによって測定した。
b この比に関するデータセットは「相加的な」範囲にあった(CIが0.9〜1.1の間)。
【0145】
実施例4
カルボプラチンおよびダウノルビシンの相乗作用
相加的、相乗的または拮抗的な作用を計測するための上記の手順を、カルボプラチン/ダウノルビシンをH460細胞にてモル比10:1、1:1および1:10で用い、さらにMCF-7細胞にて比10:1および1:1で用いて再び行った。上記の通りにCI-fa曲線を作成し、続いてfa値0.50、0.75および0.90でのCIを決定することにより(それぞれED50、ED75およびED90でのCI値を得るため)、組合せ指数を各用量について決定した。標準偏差はCalcuSynプログラムによって算出した。図5Aの挿入図に示されているように、カルボプラチンおよびダウノルビシンはモル比10:1では、MCF-7細胞においてED50、ED75およびED90値で相乗的な相互作用を示す。図5Aの挿入図にさらに示されているように、カルボプラチンおよびダウノルビシンは1:1モル比の場合、ED75およびED90では平均CI値による判定で相乗的であるが、ED50では相加的である。H460細胞においては、CI最大値とカルボプラチン/ダウノルビシンのモル比とのプロットから、モル比10:1ではこれらの薬剤は相乗的であるが、モル比1:1では幾分拮抗的な作用が観察されることが判明した。これに対して、比1:10では高度に拮抗的な作用が認められる(図5A)。このデータは図5Bにも、相乗作用に対する濃度の影響をより適切に図示するために、CIと影響を受けたH460細胞の率との関係としてプロットされている。モル比1:1のカルボプラチン/ダウノルビシンは、影響率の値が0.42までの範囲では非拮抗的である。比が10:1だと、fa値のかなりの範囲(0.2より上)で相乗作用が認められ、比1:10はすべてのfa値で拮抗的である。図5Bの挿入図は、H460細胞において比10:1ではED50、75および90で相乗作用(平均CI値による判定)が観察され、比1:1ではED50で相加作用が示されることを示している。比が1:10だと、カルボプラチン/ダウノルビシンはED50、75および90値で高度に拮抗的である。このため、これらの結果に基づき、カルボプラチンおよびダウノルビシンはモル比1:10では、測定したすべてのED値でCI-faプロット内のすべてのfa範囲にわたって拮抗作用が認められるために、これ以上の製剤およびインビボ試験の対象としては選択されないと考えられる。モル比10:1および1:1のカルボプラチン:ダウノルビシンは、これらの比のそれぞれで、薬剤が(細胞の1%超が影響を受ける)fa範囲の少なくとも5%にわたって相乗作用を示しているため、製剤および有効性試験の対象として選択される。
【0146】
実施例5
インビボでのカルボプラチンおよびダウノルビシンの相乗作用の維持
カルボプラチンおよびダウノルビシンを、単一のコレステロール非含有リポソーム中に、モル比10:1、5:1および1:1(カルボプラチン/ダウノルビシン)で共装填(co-load)した。DSPCはクロロホルム中に溶解し、DSPGは微量の14C-CHEとともにクロロホルム/メタノール/水(50:10:1 vol/vol)中に溶解した。これらの溶液をモル比80:20(DSPC/DSPG)で混ぜ合わせた。温度を60℃超に保ちながら、溶媒をN2ガス流によって除去した。続いて脂質薄膜を真空ポンプ内に2分間置き、その後にクロロホルムのみの中に再び溶解した。続いてクロロホルムを上記の通りに除去した。その結果生じた脂質薄膜を残留溶媒を除くために真空下に一晩おき、その後に、カルボプラチンの溶解性を高めるための4%(v/v)DMSOとともに80mg/mLカルボプラチンを含む150mM CuSO4、pH 7.4(pHはトリエタノールアミンで調整)中に再水和した。その結果生じた多重ラメラ小胞(MLVs)を70℃で孔径80および100nmの積み重ねた2枚のフィルターを通して押し出し、合計10回通過させた。この試料を生理食塩水中に入れた上で、接線流透析を用いる交換によって300mMスクロース、20mM HEPES、30mM EDTA、pH 7.4(SHE)中に入れた。モル比10:1、5:1および1:1のカルボプラチン/ダウノルビシンが得られる薬剤-脂質比で、60℃にて5分間インキュベートすることにより、ダウノルビシン(微量の3H-ダウノルビシンを含む)をリポソーム中に装填した。その後に、各試料の緩衝液を接線流によって生理食塩水に交換した。共装填した製剤の調製中の種々の時点での薬剤装填の程度を決定するために、液体シンチレーション計数によってダウノルビシンおよび脂質のレベルを測定した。カルボプラチン濃度は原子吸光分光法によって測定した。共装填製剤におけるモル比10:1、5:1および1:1のカルボプラチン/ダウノルビシンの場合、Balb/cマウスに対して8mg/kgカルボプラチンが静脈内投与され、ダウノルビシンの用量はそれぞれ1.2mg/kg、6mg/kgおよび12mg/kgとなった。指定した時点(各時点当たりマウス3匹)で、血液を心臓穿刺によって採取し、EDTAをコーティングしたマイクロテイナーに入れた。試料を遠心して血漿を慎重に別にチューブに移した。液体シンチレーション計数を用いてダウノルビシンおよび脂質の血漿中濃度を定量した;カルボプラチンの血漿中濃度は原子吸光分光法によって測定した。原子吸光分光法による定量のためには、標準曲線の直線範囲に収まるように試料を0.1%硝酸で希釈した。
【0147】
図6における結果は、薬剤の平均血漿中濃度(+/-標準偏差、SD)を指定時点でプロットしたものであり、存在したカルボプラチンの血漿中モル濃度がダウノルビシンの10倍であることから、カルボプラチンおよびダウノルビシンをモル比10:1で含む共装填リポソーム製剤が、静脈内投与後の薬剤の比を維持したことを示している。図7Aおよび7Bにおける結果は、DSPC/DSPGリポソーム中に調合されたカルボプラチンとダウノルビシンとのモル比10:1、5:1および1:1が、これらの比で調製された製剤の静脈内投与から24時間の経過にわたって(各時点当たりマウス3匹)、血液区画において維持されたことを示している(図7Bは、1:1カルボプラチン/ダウノルビシン製剤の投与後に得られた結果をさらに明確に強調している)。すなわち、これらの結果は、2種類の薬剤のさまざまなモル比での協調的な放出動態を達成しうることを示している。
【0148】
カルボプラチンおよびダウノルビシンをDSPC/SM/DSPE-PEG2000(90:5:5mol%)リポソーム中に、インビボでの薬剤の協調的な放出がこの製剤でも達成されうるか否かを明らかにする目的で、ともに調合した。実施例4で相乗的であると決定されたモル比10:1を選択した。
【0149】
脂質をクロロホルム中に溶解し、クロロホルムをN2ガス下で除去し、試料を真空ポンプ内に一晩置くことにより、脂質薄膜(微量の14C-CHEを含む)を上記の通りに調製した。その結果生じた脂質薄膜を、40mg/mLカルボプラチンを含む150mM CuS04、20mMヒスチジン、pH 7.4(pHはトリエタノールアミンで調整)中で水和させた。MLVを70℃で孔径100nmの2枚の積み重ねたフィルターを通して押し出し、合計10回通過させた。続いて試料を、封入されなかった金属溶液(またはカルボプラチン)を除去するために接線流透析によって300mMスクロース、20mM HEPES、pH 7.4中に入れ換えた。ダウノルビシンの装填(微量レベルの3H-ダウノルビシンを含む)は、カルボプラチン/ダウノルビシンのモル比10:1を達成するための薬剤濃度で60℃にて5分間かけて行った。薬剤装填の程度を決定するために、ダウノルビシンおよび脂質のレベルを液体シンチレーション計数によって測定した;カルボプラチンレベルは原子吸光分光法によって測定した。雄性SCID/rag2マウスに対して、DSPC/SM/DSPE-PEG2000リポソーム中に共装填された組合せで、2.25mg/kgダウノルビシンおよび15mg/kgカルボプラチンを静脈内に投与した。指定された時点(各時点当たりマウス3匹)で心臓穿刺によって血液を採取し、EDTAをコーティングしたマイクロテイナーに入れた。試料を遠心して血漿を慎重に別のチューブに移した。カルボプラチンおよびダウノルビシンの血漿中濃度はそれぞれ原子吸光分光法および液体シンチレーション計数によって測定した。
【0150】
図8に示した結果は、薬剤の平均血漿中濃度(+/-標準偏差、SD)を指定時点でプロットしたものであり、DSPC/SM/DSPE-PEG2000リポソーム中に調合された場合に静脈内投与後に同じ速度で血漿区画から排出されたことを明らかにしている。すなわち、この時間経過中に存在したカルボプラチン(nmol/mL)の血漿中濃度はダウノルビシン(nmol/mL)の概ね10倍であったことから、カルボプラチンおよびダウノルビシンは10:1のモル比に維持された。これらの結果は、種々の製剤を用いて、単一のリポソーム中に共封入された2種類の薬剤の薬物動態を、同様の薬物動態放出プロフィールが達成されるように協調させうることを示している。
【0151】
実施例6
リポソーム性カルボプラチンおよびダウノルビシンの有効性
ダウノルビシンおよびカルボプラチンがモル比1:1(これは実施例4で製剤に関して選択された)で共封入されたDSPC/DSPGリポソーム(80:20mol%)を、脂質薄膜を25mg/mLのカルボプラチンを含む溶液である150mM CuSO4、pH 7.4(pHはトリエタノールアミンで調整)中に水和させた点を除き、実施例5における記載の通りに調製した。さらに、脂質薄膜を乾燥させた後にメタノールまたは水を除くためにクロロホルム中に再び溶解し、続いて溶媒を以前の記載の通りに除去した。
【0152】
実施例26の方法と同じように、まずH460細胞(1×106個)を雌性SCID/rag2マウスの側腹部に皮下接種することにより、有効性試験を行った。腫瘍を約50mg(0.05cm3)のサイズになるまで増殖させ、その時点(第12日)で製剤を尾静脈から注入した。マウス(各群当たりマウス4匹)には4日間隔で3回の注射を行った(q4dスケジュール;第12、16および20日)。腫瘍の増殖はキャリパーによる直接測定によって評価した。マウスに対して、生理食塩水、モル比1:1の遊離性薬剤混合物、またはカルボプラチン/ダウノルビシンのモル比1:1でのリポソーム製剤のいずれかを投与した。遊離物およびリポソーム製剤の投与のいずれに関しても、用量は6.6mg/kgカルボプラチンおよび10mg/kgダウノルビシンとした。リポソーム製剤試料に関する脂質用量は260mg/kg脂質であった。
【0153】
図9に提示した結果(各点は指定日に測定した平均腫瘍サイズ+/-平均の標準誤差(SEM)を表す)は、モル比1:1のリポソーム性カルボプラチンおよびダウノルビシンの投与が、遊離性薬剤混合物および生理食塩水対照との比較で、有効性を高めることを示している。
【0154】
カルボプラチンおよびダウノルビシンをモル比10:1(実施例4で相乗的であることが決定された)で共装填したスフィンゴミエリン含有リポソームにおける有効性も、DSPC/DSPGリポソームで認められた有効性の大きな改善がこの製剤を用いた場合にも得られるか否かを検討するために調べた。リポソームを孔径80nmおよび100nmフィルターを通して10回押し出す処理を行った点を除き、実施例5に概要を述べた手順に従って、カルボプラチンおよびダウノルビシンをDSPC/SM/DSPE-PEG2000(90:5:5mol%)リポソーム中にともに調合した。さらに、ダウノルビシンの装填の前に、接線流透析ではなく固定容積透析により、試料の緩衝液をSHE緩衝液に交換した。実施例26に詳述した通りに、H460腫瘍を有する雌性SCID/rag2マウス(各群当たりマウス4匹)に対して、15mg/kgカルボプラチンおよび2.25mg/kgダウノルビシンを、リポソーム製剤および遊離性薬剤混合物として第14、18および22日に投与した。リポソーム製剤は脂質用量として375mg/kgが投与された。
【0155】
図10に提示した結果(各点は指定日に測定した平均腫瘍サイズ+/-SEMを表す)は、スフィンゴミエリン含有リポソーム中に非拮抗的なモル比である10:1で封入されたリポソーム性カルボプラチンおよびダウノルビシンが、遊離性薬剤および生理食塩水からなる対照との比較で、有効性を大きく高めることを示している。
【0156】
実施例7
シスプラチンおよびダウノルビシンの相乗作用
シスプラチン/ダウノルビシンの組合せを、上記の方法を用いて、相加的、相乗的または拮抗的な作用に関して調べた。その結果を図11にまとめた。図11Aに示されているように、シスプラチン/ダウノルビシンのモル比10:1ではfaの全範囲にわたって相乗作用が観察されたが、モル比1:1ではすべてのfa範囲にわたって拮抗作用が示された。CI最大値(CI max)とシスプラチン-ダウノルビシン比との関係をプロットした図11Bは、組合せ指数に対する2種類の薬剤の組合せ比の依存性をさらに示している。これらの結果は、モル比10:1ではCI max値は相乗的であるが、モル比1:1および1:10ではCI max値は拮抗的であることを示している。
【0157】
実施例8
インビボでのシスプラチンおよびダウノルビシンの相乗作用の維持
シスプラチンおよびダウノルビシンを、DMPC/Chol(55:45mol%)リポソーム中に、実施例7で非拮抗的であることが特定されたモル比10:1で共装填した。
【0158】
150mM CuCl2、20mMヒスチジン(pH 7.4、pHはトリエタノールアミンで調整)に4%(v/v)DMSOを加えたものからなる溶液中に薬剤(40mg/mL)をまず溶解し、その結果生じた溶液をシスプラチンの溶解性を高めるために80℃に加熱することにより、シスプラチンをリポソーム中に受動的に封じ込めた。続いてシスプラチン溶液を80℃で、DMPCおよびコレステロールを微量レベルの14C-CHEとともに含む脂質薄膜に添加した。水和した脂質薄膜を80℃で2枚の100nmフィルターを通して押し出して、リポソームを室温まで冷ました。冷却後に、封入されなかったシスプラチンをペレット化するために試料を卓上遠心器に入れて2000×g、5分間の遠心処理を行い、上清を収集した。過剰な金属イオンの除去は、セファデックスG-50ゲル濾過カラムを通過させてリポソーム画分を収集することによって行った。
【0159】
シスプラチンが装填されたリポソームに、さらにダウノルビシン(微量レベルの3H-ダウノルビシンで標識)を、リポソームを薬剤とともに60℃で15分間インキュベートすることにより、シスプラチン/ダウノルビシンのモル比が10:1となるように装填した。薬剤装填の程度を決定するために、シスプラチンレベルを原子吸光分光法によって測定し、3H-ダウノルビシンおよび脂質のレベルは液体シンチレーション計数によって測定した。
【0160】
協調的な放出がこの製剤によって達成されるか否かを明らかにするために、装填されたリポソームを雄性SCID/rag2マウスの尾静脈に、マウス1匹につき5.0mg/kgのシスプラチンおよび1.0mg/kgのダウノルビシンの用量で注入した。指定された時点(各時点当たりマウス3匹)で血液を心臓穿刺によって採取し、EDTAをコーティングしたマイクロテイナーに入れた。試料を遠心して血漿を慎重に別のチューブに移した。リポソーム性脂質およびダウノルビシンの血漿中濃度はいずれも液体シンチレーション計数によって測定し、シスプラチンレベルは原子吸光分光法によって測定した。
【0161】
図12に示した結果(各点は指定時点に測定した薬剤の平均血漿中濃度+/-SDを表す)は、測定した時点で血漿中濃度(μmol/mL)がモル比10:1に維持されていたことから、ダウノルビシンおよびシスプラチンの協調的な放出が達成されたことを示している。
【0162】
上記の方法によってリポソームにシスプラチンおよびダウノルビシンを共装填することもできるが、薬剤を単一のリポソーム中に装填するためにその他の技法を用いてもよい。代替的な方法では、シスプラチンをクエン酸、pH 4.0とともに受動的に封じ込めた後にダウノルビシンを装填するためにpH勾配を用い、緩衝液交換によって膜の内外にpH勾配を生じさせる。この技法は以下のようにして行うことができる。
【0163】
DSPC/Chol(55:45mol%)からなる脂質薄膜を上記の通りに微量の3H-CHEとともに調製する。シスプラチン粉末を150mM NaClおよび150mMクエン酸(pH 4)に溶解することによってシスプラチン溶液を調製する。シスプラチンの緩衝液への溶解性を最大限に高めるために、溶液を65℃に加熱して脂質薄膜に添加する。その結果生じたMLVを65℃で2枚の孔径100nmフィルターを通して押し出し、合計10回通過させる。続いて、溶液を2000×gで10分間遠心することにより、封入されなかったシスプラチンを製剤から除去する。その結果得られたリポソーム性シスプラチンを含む上清を、封じ込められなかった残留シスプラチンを除去し、二重層の内外にpH勾配を成立させるために、150mM NaClおよび20mM HEPES(pH 7.4)によってあらかじめ平衡化したセファデックスG-50カラムに通過させる。
【0164】
次にダウノルビシンをリポソーム中に装填する。まず熱平衡を達成するためにリポソームを60℃で5分間インキュベートし、続いて薬剤/脂質モル比が0.1:1となるように、ボルテックス処理を行いながらダウノルビシンを脂質製剤に対して添加する。種々の時点での薬剤装填の程度を決定するために、リポソームをOGPとともに溶解し、ダウノルビシンの480nmでの吸光度を測定することによってダウノルビシンの濃度を測定する。製剤のシスプラチン濃度は原子吸光分光法を用いて測定する。脂質濃度は液体シンチレーション計数によって測定する。
【0165】
2種類の薬剤の放出動態を協調させる代替的な手段は、各薬剤を別個の担体中に調合することによって達成される。このことは、シスプラチンをDMPC/コレステロールリポソーム中に調合し、ダウノルビシンをDSPC/DSPE-PEG2000リポソーム中に調合した上で、それらをモル比10:1でマウスに静脈内投与することによって示された。
【0166】
リポソーム性シスプラチンは、シスプラチン(8.5mg/mL)を150mM NaCl、80℃中にまず溶解することによって調製した。この溶液を次に、微量の3H-CHEを含むDMPC/コレステロール(55:45mol%)脂質薄膜に添加して水和させた。その結果生じたMLVを80℃で2枚の孔径100nmフィルターを通して押し出し、その後にリポソームを過剰な金属イオンを除去するための接線流透析によって20mM HEPES、150mM NaCl(pH 7.4)(HBS)中に入れ換えた。押出処理で封入されなかったシスプラチンをペレット化するためにリポソームを遠心した。シスプラチン濃度は原子吸光分光法によって測定し、脂質レベルは液体シンチレーション計数によって測定した。
【0167】
リポソーム性ダウノルビシンは、DSPC/DSPE-PEG2000(95:5mol%)および微量の14C-CHEから構成される脂質薄膜の300mM CuSO4溶液による水和によって調製した。その結果生じたMLVを、重ね合わせた2枚の孔径100nmフィルターに70℃で10回通過させることによって押し出し処理した。押出処理の後に、リポソームを接線流透析によってHBS(pH 7.4)中に入れ換えた。ダウノルビシン(微量レベルの3H-ダウノルビシンを含む)の装填は、最終的な薬剤/脂質重量比が0.1となるようにダウノルビシンを添加することによって開始し、その溶液を60℃に10分間保った。薬剤装填の程度は、3H-ダウノルビシンおよび14C-CHEレベルを計測するための液体シンチレーション計数によって測定した。
【0168】
雄性SCID/rag2マウスに対してリポソーム性シスプラチンを薬剤用量2mg/kgとして、リポソーム性ダウノルビシンを薬剤用量0.375mg/kgとして静脈内注射した。指定された時点(各時点当たりマウス3匹)で血液を心臓穿刺によって採取し、EDTAをコーティングしたマイクロテイナーに入れた。試料を遠心して血漿を慎重に別のチューブに移した。シスプラチンの血漿中濃度は原子吸光分光法によって測定し、ダウノルビシン濃度はシンチレーション計数によって測定した。
【0169】
図13に示した結果(各点は指定時点に測定した薬剤の平均血漿中濃度+/-SDを表す)は、別個のリポソーム中に調合したシスプラチンおよびダウノルビシンが静脈内投与後の種々の時点で10:1のモル比を維持したことを明らかにしている。
【0170】
実施例9
リポソーム性シスプラチンおよびダウノルビシンの有効性
別個のリポソーム中に調合したシスプラチンおよびダウノルビシンの有効性を、実施例26に詳述した通りにSCID/rag2マウス(H460異種移植モデル)において評価した。H460腫瘍を有するマウス(各群当たりマウス4匹)に対して、生理食塩水、または実施例7で非拮抗的であるとインビトロで特定されたモル比10:1のシスプラチン/ダウノルビシンを投与した。シスプラチンおよびダウノルビシンは、押出処理後にDMPC/CholリポソームをHBSに対して透析した点を除き、実施例8の記載の通りに、それぞれDMPC/Chol(55:45mol%)リポソームおよびDSPC/DSPE-PEG2000(95:5mol%)リポソーム中に調合した。薬剤の組合せを投与するマウスに対しては、薬剤を遊離性薬剤混合物(10:1の混合物、モル比)として、またはリポソーム性ダウノルビシンとリポソーム性シスプラチンとの同時投与(リポソーム製剤;モル比10:1)として、第14、17および21日に投与した。遊離物および調合物の投与のいずれに関しても、用量は2.0mg/kgシスプラチンおよび0.375mg/kgダウノルビシンとした。脂質用量は、リポソーム性シスプラチンについては400mg/kgであり、リポソーム性ダウノルビシンについては3.75mg/kgであった。
【0171】
図14はその結果を示しており、ここで各データ点は指定日に測定した平均腫瘍サイズ+/-SEMを表す。生理食塩水対照(黒丸)は腫瘍増殖を抑制しなかった;同様に、遊離混合物(黒い逆三角)も腫瘍増殖に対してわずかな効果しか示さなかった。これに対して、リポソーム製剤(白い三角)は腫瘍増殖を少なくとも32日間にわたって抑制した。
【0172】
実施例10
拮抗的なモル比にある薬剤の組合せのリポソーム性投与の効果
シスプラチンおよびダウノルビシンを、DMPC/Chol(55:45mol%)リポソーム中に、実施例7で拮抗的であることが決定されたモル比1:1で共装填した。シスプラチンは受動的に封じ込め、ダウノルビシンは能動的に封じ込めてシスプラチン/ダウノルビシンのモル比1:1を達成した。単一のリポソーム中への薬剤の装填には、実施例8に概要を示した手順を用いた。
【0173】
DMPC/Cholリポソーム中にある製剤によって協調的な放出が達成されたか否かを明らかにするために、装填したリポソームをBalb/cマウスの尾静脈に2mg/kgシスプラチンおよび3.75mg/kgダウノルビシンの用量で注入した。指定された時点(各時点当たりマウス3匹)で血液を心臓穿刺によって採取し、EDTAをコーティングしたマイクロテイナーに入れた。試料を遠心して血漿を慎重に別のチューブに移した。脂質およびダウノルビシンの血漿中濃度はいずれも液体シンチレーション計数によって測定し、シスプラチン濃度は原子吸光分光法によって測定した。その結果を図15にまとめた(各データ点は指定時点で測定した薬剤の平均血漿中濃度+/-SDを表す)は、ダウノルビシンおよびシスプラチンが血漿から同じ速度で排出され、そのために血漿中濃度(nmol/mL)がモル比1:1に維持されたことを示している(図15の挿入図を参照されたい)。
【0174】
有効性試験は実施例26の記載の通りに行い、H460腫瘍を有する雌性SCJD/rag2マウスに対して2.5mg/kgシスプラチン、4.7mg/kgダウノルビシンを混合物またはリポソーム製剤として、さらに52.83mg/kgの脂質を第11、15および19日に投与した。
【0175】
図16における有効性の結果(各データ点は指定日に測定した平均腫瘍サイズ+/-SEMを表す)は、非拮抗的な比(モル比10:1)の薬剤では腫瘍増殖がかなり抑制されたのに対して(図14参照)、拮抗的な比にあるダウノルビシンおよびシスプラチンの投与が腫瘍増殖の抑制に無効であることを示している。すなわち、これらの結果は、インビトロである濃度範囲にわたって非拮抗的な作用を示す比にある薬剤の組合せを選択することの重要性を強く示している。実際には、図16で用いた薬剤の用量(2.5mg/kgシスプラチンおよび4.7mg/kgダウノルビシン)は、図14で用いた用量よりも高い(2mg/kgシスプラチン、0.375mg/kgダウノルビシン)。
【0176】
実施例11
シスプラチンおよびトポテカンの相乗作用
相乗的、相加的または拮抗的な作用を決定するための上記の手順(実施例1参照)を、シスプラチン/トポテカンをモル比10:1およびモル比1:1の両方で用いて再び行った。図17Aに示されているように、シスプラチン/トポテカンはモル比10:1では、細胞の5%〜99%が影響を受ける(fa=0.05〜fa=0.99)広範囲の用量にわたって非拮抗的な相互作用を示した。これに対して、モル比1:1のシスプラチン/トポテカンは、同じfa範囲にわたって高度に拮抗的であった(図17A)。
【0177】
濃度のこのような影響は、シスプラチン/トポテカンのさまざまなモル比に関するCI最大値の算出からも明らかとなった。図17Bに示されているように、拮抗作用はモル比1:1で最大となるように思われ、非拮抗的な作用はいずれかの薬剤が過剰に存在する場合に明らかである。
【0178】
実施例12
インビボでのシスプラチンおよびトポテカンの相乗作用の維持
シスプラチンおよびトポテカンを、それぞれDMPCICholリポソームおよびDSPC/Cholリポソーム中に調合した上で、実施例11で相乗的であることが特定されたモル比10:1でマウスに静脈内注射した。
【0179】
リポソーム性シスプラチンは、DMPCおよびコレステロール(55:45mol%)からなる脂質薄膜を、150mM NaClおよび8.5mg/mLシスプラチンからなる溶液で水和させることによって調製した。その結果生じたMLVを、重ね合わせた2枚の孔径100nmフィルターに80℃で10回通過させることによって押し出し処理した。押出処理の後に試料を冷却し、沈殿したシスプラチンを遠心分離によって除去した。リポソーム中に封入されなかった残留性の可溶性シスプラチンは、HBSに対する透析によって除去した。封入されなかったシスプラチンの除去後に、薬剤の濃度を原子吸光分光法によって測定した。
【0180】
リポソーム性トポテカンは、DSPCおよびコレステロール(55:45mol%)から構成される脂質薄膜の300mM MnSO4溶液による水和によって調製した。その結果生じたMLVを、重ね合わせた2枚の100nmフィルターに65℃で10回通過させることによって押し出し処理した。押出処理の後に、リポソームをゲル濾過クロマトグラフィーによってSHE緩衝液(300mMスクロース、20mM HEPESおよび30mM EDTA、pH 7.4)中に入れ換えた。トポテカンの装填は、1μgのA23187/脂質μmol(A23187は二重層を介した二価金属イオンとプロトン2個との交換を媒介する陽イオン性イオノフォアである)および最終的なトポテカン/脂質比が0.08(w/w)となるトポテカンを添加することによって開始し、続いて溶液を65℃に15分間保った。トポテカン装填の程度は、封入された薬剤と封入されなかった薬剤をゲル濾過クロマトグラフィーを用いて分離してTriton X-100中に溶解した後に、380nmでの吸光度によって測定した。
【0181】
この製剤を、SCID/rag2雌性マウスに尾静脈から静脈内注射した。リポソーム製剤の用量は5mg/kgシスプラチンおよび0.758mg/kgトポテカンとした。指定された時点(各時点当たりマウス3匹)で血液を心臓穿刺によって採取し、EDTAをコーティングしたマイクロテイナーに入れた。試料を遠心して血漿を慎重に別のチューブに移した。放射標識した脂質の定量には液体シンチレーション計数を用いた。シスプラチンは原子吸光分光法を用いて測定し、トポテカンはリポソームを過剰量の界面活性剤で破壊した後に蛍光分光法(励起380nmおよび発光518nm)によって測定した。
【0182】
図18(各データ点は指定時点で測定した薬剤の平均血漿中濃度+/-SDを表す)は、上記のリポソーム中にある状態で送達した場合に静脈内投与後の種々の時点でシスプラチンの血漿中濃度はトポテカンの概ね10倍であったことから、シスプラチンおよびトポテカンの血漿中濃度がモル比10:1に維持されたことを示している。これらの結果は、2種類のリポソーム中に封入された2種類の薬剤の薬剤保持特性およびリポソーム排出特性を、協調的な薬剤排出速度が実現されるように協調させうることを示している。図18の挿入図は、静脈内投与後に血漿中に存在するシスプラチン-トポテカンのモル比(+/-SD)が、時間経過に伴ってほとんど変化しないことを示している。
【0183】
非拮抗的な比が確実にインビボで維持されるように、シスプラチンおよびトポテカンを単一のリポソーム中に調合することもできる。これは、シスプラチンの受動的封じ込めの後にイオノフォアを介したトポテカンの装填を行うことによって実施しうる。シスプラチン粉末を150mM MnCl2溶液中に溶解することにより、シスプラチン溶液をまず調製する。シスプラチンのMnCl2溶液への溶解性を最大限に高めるために、この溶液を65℃に加熱する。DSPC/Chol(55:45mol%)から構成され、微量の3H-CHEを含む脂質薄膜をシスプラチン/MnCl2溶液によって水和させる。その結果生じたMLVを65℃で2枚の100nmフィルターを通して押し出し、合計10回通過させる。続いて、製剤を室温まで冷却し、溶液を2000×gで遠心することによって不溶性シスプラチンを製剤から除去する。その結果得られた、リポソーム性シスプラチンおよび可溶性であるが封入されなかったシスプラチンを含む上清を、SHE緩衝液、300mMスクロース、20mM HEPESおよび30mM EDTA(pH 7.4)に対して室温で一晩透析する。
【0184】
その後に、イオノフォアを介したプロトン勾配を用いて、トポテカンをリポソーム中に装填する。薬剤の取り込みは薬剤-脂質重量比0.08:1(w/w)で行わせる。二価陽イオンイオノフォアA23187(1μgイオノフォア/脂質μmol)をリポソームに添加した後に、A23187の二重層への取り込みを促すために混合物を60℃で15分間インキュベートする。その後にトポテカンを添加し、薬剤の取り込みを促すために混合物を60℃で60分間インキュベートする。試料を300mMスクロースに対して透析することにより、封入されなかったトポテカンおよびA23187を調製物から除去する。トポテカン装填の程度は380nmでの吸光度を測定することによって定量する。シスプラチンレベルは原子吸光分光法によって測定し、脂質レベルは液体シンチレーション計数によって測定する。
【0185】
実施例13
リポソーム性シスプラチンおよびトポテカンの有効性
別個のリポソーム中に装填されたシスプラチンおよびトポテカンの有効性を、2種類の薬剤を別個のリポソーム中に調合した上で、それらの製剤を、実施例11で非拮抗的であることが特定されたモル比10:1で投与することによって調べた。リポソーム性シスプラチンは、実施例12の手順の記載の通りにDMPC/Chol(55:45mol%)リポソーム中に受動的に封じ込めた。トポテカンは、トポテカンの装填は最終的なトポテカン/脂質重量比が0.1(w/w)となるように行った点を除き、実施例12の通りにDSPC/Chol(55:45mol%)中に調合した。装填後に外部緩衝液をHBSに交換した。
【0186】
有効性試験は実施例26に詳述した通りに行い、H460腫瘍を有する雌性SCID/rag2マウス(各群当たりマウス4匹)に対して、生理食塩水(対照)、シスプラチン/トポテカンの遊離性混合物またはリポソーム性混合物を、実施例11で非拮抗的として特定されたモル比10:1で静脈内に投与した(第13、17、21日に)。遊離物およびリポソーム製剤のいずれの投与に関しても、用量は1.6mg/kgシスプラチンおよび0.25mg/kgトポテカンとした。脂質用量はシスプラチン製剤については250mg/kgであり、トポテカン製剤ではそれよりも2.5mg/kg多かった。
【0187】
図19はその結果を示している(各データ点は、指定日に測定した平均腫瘍サイズ+/-SEMを表す)。生理食塩水対照(黒丸)およびシスプラチン/トポテカンの10:1混合物(黒い三角)は、腫瘍体積の増加を停止させる効果がなかった。しかし、シスプラチン/トポテカン10:1リポソーム製剤(白い三角)は、腫瘍体積の増加を少なくとも35日間にわたって阻止した。
【0188】
実施例14
シスプラチンおよびイリノテカンの相乗作用
シスプラチンおよびイリノテカンのモル比1:1、10:1、1:5および1:10での組合せを、上記の方法に従って相乗作用、相加作用または拮抗作用に関して調べた(実施例1参照)。図20Aにまとめた結果は、モル比10:1、1:5および1:10ではfa値の全範囲にわたって非拮抗的であるが、1:1の比はfa値のかなりの範囲にわたって拮抗的であったことを示している。図20Bはさらに、組合せ指数の最大値をシスプラチン-イリノテカンのモル比に対してプロットすることによって概括されているように、併用効果の性質に対する比の依存性を示している。
【0189】
実施例15
インビボでのシスプラチンおよびイリノテカンの相乗作用の維持
6.0mg/mLシスプラチンを含む225mM銅(75mM CuCl2、150mM CuSO4、トリエタノールアミン(TEA)、pH 6.8)中に脂質薄膜を再水和させた点を除き、実施例5の記載の通りに調製したDSPC/DSPG(80:20mol%)リポソーム中に、シスプラチンおよびイリノテカンを共装填した。押出処理および封入されなかった薬剤の除去の後のリポソーム性シスプラチンの濃度はシスプラチン0.025モル/脂質1モルであった。その結果生じたリポソームをSHE、pH 6.8に対して一晩透析した。続いてイリノテカンを調製物に添加し、リポソームを45℃で1.5時間インキュベートした。HPLCによる評価では、添加したイリノテカンの60%がリポソームに装填された。続いてリポソームの緩衝液を接線流によって0.9%生理食塩水に交換した。接線流の後に、リポソームは最初のシスプラチンおよびイリノテカンの約80%を保持していた。それぞれ原子吸光分光法およびHPLC分析によって評価したシスプラチンおよびイリノテカンの分析により、最終的な調製物がシスプラチン-イリノテカンをモル比1:3で有することが示された。SCID/rag2マウスに対して2mg/kgシスプラチンおよび38.6mg/kgイリノテカンを静脈内投与した。指定された時点(各時点当たりマウス3匹)で血液を心臓穿刺によって採取し、EDTAをコーティングしたマイクロテイナーに入れた。試料を遠心して血漿を慎重に別のチューブに移した。イリノテカンおよびシスプラチンの血漿中濃度はそれぞれHPLCおよび原子吸光分光法によって測定した。
【0190】
図21における結果(各データ点は指定時点で測定した薬剤の平均血漿中濃度+/-SDを表す)は、DSPC/DSPGリポソーム中に共装填されたシスプラチンおよびイリノテカンを含む製剤の静脈内注射後の薬剤排出速度は同程度であり、非拮抗的な薬剤モル比が投与後24時間の経過にわたって維持されたことを示している。
【0191】
インビボでのリポソーム性シスプラチンおよびイリノテカンの協調的な放出も、この2種類の薬剤を別個の送達媒体中に調合した上で薬剤をモル比1:5(シスプラチン/イリノテカン)で投与することによって達成された。
【0192】
リポソーム性シスプラチンは、上記の受動的な装填法に従って調製した。DMPC/Chol(55:45mol%)からなる脂質薄膜を、8.5mg/mLシスプラチンを含む150mM NaCl溶液によって水和させた後に、上記の通りの押出処理を行った。上記の通りに遠心分離後の上清中のリポソームを収集した後に、接線流透析によってHBSに交換した。
【0193】
リポソーム性イリノテカンは、DSPC/DSPE-PEG2000(95:5mol%)からなる脂質薄膜を、150mM CuCl2、20mMヒスチジン、pH 6.8(pHはTEAで調整)からなる溶液によって水和させることによって調製した。その結果生じたMLVを65℃で重ね合わせた2枚の孔径100nmフィルターを通して押し出し、接線流によて緩衝液をHBSに交換した。押出処理を行ったリポソームに対して、イリノテカンを薬剤脂質重量比が0.1:1となるように60℃で1分間かけて装填した。イリノテカンの装填の程度は、Triton X-100中への溶解後に370nmでの吸光度によって測定した;脂質レベルは液体シンチレーション計数によって測定した。
【0194】
リポソーム性シスプラチンを雄性SCID/rag2マウスに対して薬剤用量2.0mg/kgとして投与し、リポソーム性イリノテカンをマウスに対して20mg/kgで投与した。指定された時点(各時点当たりマウス3匹)で血液を心臓穿刺によって採取し、EDTAをコーティングしたマイクロテイナーに入れた。試料を遠心して血漿を慎重に別のチューブに移した。イリノテカンの血漿中濃度はHPLCによって測定し、シスプラチン濃度は原子吸光分光法によって測定した。
【0195】
これらのリポソーム製剤中にある状態でこの相乗的な比(モル比1:5)でともに投与されたシスプラチンおよびイリノテカンは、種々の時点でイリノテカンの血漿中濃度(nmol/mL)がシスプラチンの概ね5倍(nmol/mL)であったことから示されるように、1:5というこの比を静脈内投与後にも維持した(図22)。
【0196】
実施例16
リポソーム性シスプラチンおよびイリノテカンの有効性
別個のリポソーム中に調合されたリポソーム性シスプラチンおよびイリノテカンに対して有効性試験を行った。実施例15に詳述した通りに、シスプラチンはDMPC/Chol(55:45mol%)リポソーム中に受動的に封じ込め、イリノテカンはDSPC/DSPE-PEG2000(95:5mol%)リポソーム中に装填した。実施例26に記載の方法に従って、リポソーム性シスプラチンおよびイリノテカンを、実施例14で非拮抗的であることが決定されたモル比1:5で、H460腫瘍を有するSCID/rag2マウスに対して同時投与した。リポソーム性シスプラチンおよびイリノテカンの投与(第14、18および22日に各群当たりマウス4匹に対して)は、非拮抗的なモル比1:5で以下の用量を用いて行った:1mg/kgシスプラチン、10mg/kgイリノテカンおよび130mg/kg脂質(白い四角);2.5mg/kgシスプラチン、25mg/kgイリノテカンおよび175mg/kg脂質(白い三角);または5mg/kgシスプラチン、50mg/kgイリノテカンおよび250mg/kg脂質(白い逆三角)。遊離性シスプラチン/イリノテカンは、モル比1:5を反映する1mg/kgシスプラチンおよび10mg/kgイリノテカンの用量で投与した(黒い四角)。
【0197】
図23(各データ点は指定日に測定した平均腫瘍サイズ+/-SEMを表す)は、遊離性薬剤混合物および生理食塩水を投与したマウスと比較して、リポソーム製剤の場合には腫瘍増殖が大きく抑制されたことを示している。
【0198】
実施例17
薬剤および脂質の組合せの相乗作用
ビノレルビンを、脂質二重層に組み入れられたPOPS(黒い逆三角)、DPPS(黒い三角)、DLPS(黒丸)、DSPS(黒い菱形)またはDOPS(黒い四角)といった種々の治療的脂質候補とともにモル比1:1で含む組合せを、上記の方法を用いて相加的、相乗的または拮抗的な作用に関して検討した(実施例1参照)。
【0199】
図24における結果は、ビノレルビンと被験脂質とのすべての組合せが、fa値のかなりの範囲にわたってH460細胞に対して相乗作用を示すことを示している。特に、ビノレルビンとDLPS、DSPSおよびDOPSとの組合せはfa値の大半で相乗作用を示し、fa=0.2〜fa=0.8の範囲で最も顕著であった。
【0200】
実施例18
リポソーム性ビノレルビンおよびホスファチジルセリンの薬物動態
以下の通りに、SM/Chol/DPPS/DSPE-PEG2000(35:45:10:10mol%)からなるリポソームを調製して、ビノレルビンを装填した。
【0201】
脂質をクロロホルム中に100mg/mLの濃度で溶解した上で、適当な量を混ぜ合わせた。これに対する例外はDPPSであり、これはCHCl3/メタノール/H2O/クエン酸緩衝液(20:10.5:1:1 v/v)を用いて25mg/mLの濃度で溶解した。製剤化工程の全体を通じて脂質を追跡するために、微量の放射性脂質3H-CHEをこの時点で添加した。ごくわずかな溶媒が残るまでクロロホルムをN2ガス流によって除去した。その結果得られた脂質薄膜を真空下に一晩置き、残留性溶媒を除去した。脂質薄膜をクエン酸緩衝液(300mM、pH 4.0)中に再水和させ、その結果生じたMLVを65℃で2枚の孔径100nmフィルターを通して押し出し、合計10回通過させた。
【0202】
外部緩衝液のpHを0.2M Na2HPO4を用いて徐々に高めるpH勾配装填法により、ビノレルビンをこれらの製剤中に装填した。既知の量のリポソームを対応する量のビノレルビン(薬剤/脂質重量比0.1(w/w))と混ぜ合わせ、60℃で15分間インキュベートした。pH勾配を成立させるために、0.2M Na2HPO4をクエン酸緩衝液の10倍の容積として添加した。実施例17で非拮抗的として特定されたビノレルビン/ホスファチジルセリンのモル比が達成されるように、ビノレルビンをリポソーム中に装填した。
【0203】
ビノレルビン装填リポソームの可溶化には界面活性剤OGPを用いた;薬剤濃度は270nmでの吸光度によって測定し、脂質の定量には液体シンチレーション計数を用いた。
【0204】
その結果得られたビノレルビン装填リポソームおよび遊離ビノレルビンを、SCID/rag2マウスに対して薬剤用量10mg/kgとして静脈内投与した。指定された時点(各時点当たりマウス3匹)で血液を心臓穿刺によって採取し、EDTAをコーティングしたマイクロテイナーに入れた。試料を遠心して血漿を慎重に別のチューブに移した。シンチレーション計数を用いて、残留性3H-CHEリポソームマーカーに関して血液を分析した。ビノレルビンの血漿中濃度はHPLCによりアッセイした。
【0205】
図25Aおよび25Bは、ビノレルビンを封入したSM/Chol/DPPS/DSPE-PEG2000リポソームが、遊離ビノレルビンの投与と比較して、薬剤の血漿中濃度を大きく高めることを示している。遊離ビノレルビンの平均血中濃度曲線下面積(AUC)は0.112μg/mLであったが、リポソーム中に製剤化すると125.3μg/mLに増加し、平均AUCは1120倍に増加した。
【0206】
実施例19
H460ヒト肺癌モデルにおけるリポソーム性ホスファチジルセリンおよびビノレルビンの有効性
実施例18の記載の通りに、DSPC/Chol/DSPS/DSPE-PEG2000(35:45:10:10mol%)リポソーム、SM/Chol/DPPS/DSPE-PEG2000(35:45:10:10mol%)リポソームおよびDAPC/Chol/DPPS/DSPE-PEG2000(35:45:10:10mol%)リポソームを調製し、ビノレルビンを装填した。ホスファチジルセリンおよびビノレルビンはリポソーム中に非拮抗的なモル比(1:1)で存在させた。有効性試験は、実施例26に記載した通り、H460ヒト肺癌モデルを用いて行った。
【0207】
図26は、DSPC/Chol/DPPS/DSPE-PEG2000およびSM/Chol/DPPS/DSPE-PEG2000からなり、ビノレルビンが封入されたリポソームの静脈内投与を受けたH460腫瘍保有マウス(各群当たりマウス4匹)について示しており、この投与により、遊離ビノレルビンおよび生理食塩水の投与後に観察された速度と比較して腫瘍増殖速度が低下した。腫瘍細胞の接種から13日、17日および21日後に、遊離ビノレルビンを5mg/kgで投与し、リポソーム性ビノレルビンは薬剤については5mg/kg、脂質については50mg/kgの用量で投与した。
【0208】
図27(各データ点は指定日に測定した平均腫瘍サイズ+/-SEMを表す)は、SM/Chol/DPPS/DSPE-PEG2000;DAPC/Chol/DPPS/DSPE-PEG2000およびDSPC/Chol/DSPS/DSPE-PEG2000からなり、ビノレルビンを封入したリポソームが、遊離ビノレルビンおよび生理食塩水と比較して、時間経過に伴う腫瘍体積の減少を示したことを示している。腫瘍保有マウス(1群当たり4匹)に対してビノレルビンを用量5mg/kgで投与し(遊離性およびリポソーム性)、リポソーム群に対しては脂質を用量50mg/kgで投与した。マウスには第13、17および21日に静脈内投与を行った。
【0209】
実施例20
マウス白血病モデルにおけるリポソーム性ホスファチジルセリンおよびビノレルビンの有効性
リポソームを孔径100nmフィルターと80nmフィルターを重ねたものを通して押し出した点を除き、実施例18の記載の通りに、SM/Chol/DPPS/DSPE-PEG2000(35:45:10:10mol%)からなるリポソームを調製し、ビノレルビンを装填した。
【0210】
実施例27の記載の通りに、P388/wt細胞をBDF-1マウスの腹腔内に接種した。その後に、BDF-1雌性マウスに以下のいずれかを腹腔内投与した:生理食塩水;遊離ビノレルビン(10mg/kg)、およびビノレルビンが装填されたSM/Chol/DPPS/DSPE-PEG2000リポソーム(10mg/kgビノレルビンおよび100mg/kg脂質)。遊離性およびリポソーム性ビノレルビンの腹腔内投与を第1日に一投与群当たりマウス4匹に行った。
【0211】
図28に示した生存曲線は、SM/Chol/DPPS/DSPE-PEG2000からなるリポソーム中に封入したビノレルビンの投与が、遊離ビノレルビンおよび生理食塩水の投与と比較して、BDF-1マウスの生存率の大幅な増加をもたらしたことを示している。
【0212】
実施例21
スフィンゴシンとドキソルビシンとの共調合
ホスファチジルセリンのほかに、他の治療的脂質をリポソーム膜に組み入れることもできる。例えば、スフィンゴシンおよびスフィンゴシン類似体はリポソーム中に調合するのに適合した脂質であり、水性内部に封入した治療薬(例えば、ドキソルビシン)とともに調合することができる。このような薬学的組成物(スフィンゴシン)の調製は以下のようにして行いうる。
【0213】
スフィンゴシンの典型的なリポソーム製剤は、DSPC/Chol/スフィンゴシン(45:45:10mol%)から構成される。以前の実施例に詳述した通りに脂質薄膜を調製する。脂質薄膜をクエン酸緩衝液(300mM、pH 4)中に再水和させ、その結果生じたMLVを65℃で2枚の100nmフィルターを通して押し出し、合計10回通過させる。続いて、pH勾配を成立させるためにHBS(pH 7.4)で平衡化したセファデックスG-50カラムを通過させることによる、リポソームの外部緩衝液の交換によるpH勾配装填法を用いて、ドキソルビシンをこれらの製剤に装填する。
【0214】
続いて、リポソームおよびドキソルビシン溶液をともに60℃でインキュベートし、装填を行わせる。種々の時点での装填の程度を決定するために、100μLの試料を1mL セファデックスG-50スパンカラムにかけ、その後に遠心処理を行う。脂質の定量に液体シンチレーション計数を用い、ドキソルビシンの定量には480nmでの吸光度を用いて、スパンカラムの溶出液の薬剤-脂質比を導き出す。薬剤のアッセイのためには、リポソームをTriton X-100中でインキュベートした後に吸光度の読み取りを行う。
【0215】
実施例22
フロクスウリジン(FUDR)およびイリノテカン(CPT-11)の相乗作用
相加的、相乗的または拮抗的な作用を計測するための上記の手順を、HT 29細胞においてFUDR/CPT-11をモル比10:1、5:1、1:1、1:5および1:10で用いて再び行った。上記の通りにCI-fa曲線を作成することにより、組合せ指数を各用量に関して決定した。影響を受けたHT-29細胞の率に対してCIをプロットした図29におけるデータは、相乗作用に対する濃度の影響を明らかに示している。比が5:1または1:1の場合は、影響率の値の全範囲(0.2〜0.8)にわたって相乗作用が観察されるが、比10:1では0.76未満のfa値で非拮抗的であり、FUDR/CPT-11のモル比が1:5であれば0.62未満のfa値で非拮抗的である。比1:10はfa値の実質的な範囲(50%超)にわたって拮抗的である。これらの結果に基づき、この比がfa値の有意な範囲(細胞の1%超が影響を受ける範囲の少なくとも20%)にわたって相乗作用を示したことから、FUDR:CPT-11のモル比1:1を製剤化および有効性試験の対象として選択した。5:1および10:1の比で調製した製剤も、fa値の実質的な範囲にわたって所定の非拮抗的な比を示すという必要条件を満たすと考えられる。
【0216】
実施例23
インビボでのFUDRおよびCPT-11の相乗作用の維持
FUDRおよびCPT-11をDSPC/DSPG/Chol(70:20:10mol%)リポソーム中に、実施例Aにおいて相乗的であることが特定されたモル比1:1で調合した。DSPCおよびコレステロールをクロロホルム中に溶解し、DSPGをクロロホルム/メタノール/水(16/18/1)中に溶解することによって脂質薄膜を調製した。これらの溶液を所定のモル比が得られるように混ぜ合わせ、リポソーム脂質標識として微量の14C-CHEを添加した。溶媒の除去後に、生成された脂質薄膜を、250mM CuSO4および25mg/mLのFUDR(微量の3H-FUDRを含む)からなる溶液によって70℃で水和させた。その結果生じたMLVを、70℃で、重ね合わせた2枚の孔径100nmフィルターに10回通過させることにより、押し出し処理を行った。その後に、リポソームの緩衝液を接線流透析によってSHE、pH 7.4に交換し、それにより、封入されなかったFUDRおよびCuSO4を除去した。
【0217】
FUDRとCPT-11とのモル比が1:1となるように、CPT-11をこれらのリポソームに添加した。試料を50℃で5分間インキュベートすることにより、CPT-11のリポソームへの装填を促した。装填の後に、EDTAまたは封入されなかった薬剤を除去するために接線流透析によって試料をHBS、pH 7.4中に入れ換えた。CPT-11装填の程度はHPLCを用いて計測した。FUDRおよび脂質のレベルは液体シンチレーションを用いて測定した。
【0218】
これらの製剤をBalb/c雌性マウスに尾静脈を介して静脈内注射した。リポソーム製剤の用量はFUDR 8.38mg/kgおよびCPT11 20mg/kgであった。指定された時点(各時点当たりマウス3匹)で血液を心臓穿刺によって採取し、EDTAをコーティングしたマイクロテイナーに入れた。試料を遠心して血漿を別のチューブに移した。血漿中の放射標識脂質およびFUDRの定量には液体シンチレーション計数を用いた。CPT-11の血漿中濃度はHPLCを用いて定量した。
【0219】
図30は、上記のリポソーム中にある状態で送達した場合に静脈内投与後の種々の時点でFUDRの血漿中濃度がCPT-11とほぼ等しかったことから、FUDRおよびCPT-11の血漿中濃度がモル比1:1に維持されたことを示している。各データ点は指定時点で測定した薬剤の平均血漿中濃度(薬剤nmol/血漿mL)+/-標準偏差を表す。
【0220】
実施例24
リポソーム性FUDRおよびCPT-11の有効性
薬剤装填後にリポソーム外部緩衝液を0.9%NaClに交換した点を除いて、実施例Bの記載の通りに、FUDRおよびイリノテカンをモル比1:1で共封入したDSPC/DSPG/Chol(70:20:10mol%)リポソームを調製した。
【0221】
実施例26の方法を用いて、側腹部に2×106個のHT-29細胞を皮下接種した雌性SCID/rag2マウスにおける有効性試験を行った。腫瘍をサイズが180mg(0.18cm3)と計測されるまで増殖させ、その時点(第21日)で指定の製剤を注入した。腫瘍の増殖はキャリパーによる直接測定によって評価した。マウスに対して、生理食塩水、モル比1:1の遊離性薬剤混合物、またはFUDR/CPT-11のモル比1:1でのリポソーム製剤のいずれかを単回投与(矢印)した。遊離物およびリポソーム製剤の投与のいずれに関しても、用量は9.25mg/kg FUDRおよび25mg/kg CPT-11とした。リポソーム製剤試料に関する脂質用量は278mg/kgであった。
【0222】
図31に提示した結果は、単一のリポソーム中にモル比1:1で封入されたFUDRおよびCPT-11が、遊離性薬剤混合物または生理食塩水のいずれかを注入したマウスと比較して、治療活性を有意に改善することを示している。各データ点は平均腫瘍サイズ+/-平均の標準誤差(SEM)である。
【0223】
実施例25
さまざまな3剤の組合せに関するCIの決定
トポテカン、シスプラチン、HB5-5A(エデルフォシン類似体)およびスフィンゴシンを含む組合せを、標準的なテトラゾリウム比色MTT細胞傷害性アッセイ法(実施例の項の細胞傷害性アッセイ法を参照)を用いて、相加的、相乗的または拮抗的な作用に関して検討した。以前の実施例に記載したメディアンエフェクト解析を用いて併用効果を算出した。CTとfaとの関係を示すグラフを以前の実施例の記載の通りに作成し、fa値0.50、0.75および0.90に対応するCI値(ED50、75および90によって表される)を以下の表に報告した。
a 組合せ指数(CI)は、用量-効果分析に関するChou-Talalay理論に基づく相乗作用(CI<0.9)または相加作用(CIが0.9〜1.1の間)の決定に用いられる。各値はCalcuSynソフトウエアを用いて算出される。
b ED50、ED75、ED90は、測定される応答のそれぞれ50、75または90%が影響を受ける薬剤の用量のことを指す。
【0224】
実施例26
腫瘍モデルの作製、細胞の調製および皮下固形腫瘍の移植法
H460ヒト非小細胞肺癌細胞は、NCIのDCTC腫瘍レポジトリ(Tumor Repository)から入手しうる。細胞は組織培養下で最大20回の継代にわたって維持される。20回の継代後に、液体窒素中で保存していた凍結貯蔵物から新たな細胞を増殖させる。培養細胞が集密度80〜90%に達した時点で、それらをハンクス平衡塩類溶液ですすぎ洗い、付着細胞を0.25%トリプシン溶液によって取り出す。細胞を血球計算器で算定し、培地で希釈して濃度を20×106細胞/mLにする。
【0225】
各マウスの背部の約2cm×2cmの領域を電気バリカンを用いて剃毛する。第0日に、28g針を用いて、マウスに1×106個の腫瘍細胞、容積50μLを皮下接種する(接種1回/匹)。
【0226】
腫瘍が約0.50〜0.100cm3の所定のサイズに達した時点で、投与の1日前または投与日(〜第10日から第14日)に、すべての腫瘍を計測する。適切な腫瘍サイズを選択した後に、小さすぎる腫瘍と大きすぎる腫瘍を除外し、腫瘍をランダム分布させて(n=4)、各群の平均腫瘍体積を決定する。
【0227】
マウスを、生理食塩水対照、媒体対照、陽性対照および被験物質の種々の希釈物などの対照群および投与群からなる適切な投与群に配分する。
【0228】
投与群は以下の通りとする。
a 単回投与または4〜7日毎の投与などの代替的な投薬計画も考慮しうる。
【0229】
マウスに対して、個々のマウスの体重に基づき、処方用量(10μL/g、指定に応じて)をマウスに投与するために必要な容積の試料を静脈内注射する。
【0230】
腫瘍増殖の測定値はバーニアキャリパーを用いて計測し、測定は投与日から開始する。腫瘍の長さの測定値(mm)は最長方向の軸から求め、幅の測定(mm)はこの軸に対して直交するようにする。長さおよび幅の測定値から、式(L×W2/2)/1000を用いて腫瘍体積(cm3)を算出する。腫瘍測定時にマウスの体重を記録する。
【0231】
個々のマウスの体重を、有効性試験の間、最終投与から14日間にわたり、さまざまな日(一般には月曜、水曜および金曜というように1日おきにする)に記録する。
【0232】
全マウスを、前投与期間および投与期間にわたって、生死および病状に関して少なくとも1日1回観察し、必要と思われる場合はそれ以上観察する。詳細には、病的状態の徴候は、体重減少、食欲変化、体毛の荒れ、身づくろいの減少、行動変化(歩行変化など)、傾眠および顕著なストレスの発現に基づく。重篤な中毒または腫瘍に伴う病的状態が認められた場合には、マウスを安楽死させ(CO2による窒息)、中毒の他の徴候を評価するために剖検を行う。瀕死状態のマウスは人道的な理由から屠殺する必要があり、屠殺に関する判断は動物飼育技術者(Animal Care Technician)および試験責任者(Study Director)/監督者(Manager)の裁量に委ねられる。これらの所見のいずれかおよびすべてを生データとして記録し、死亡時はその翌日として記録する。
【0233】
データは表または図の形式として以下の通りに提示する。
1.投与開始前およびグループ分けの後の各群に関する個々のマウスの腫瘍体積。
2.時間の関数としての各群の平均体重。
3.時間の関数としての各群の平均腫瘍体積。
4.図および表を含む生データの作成、これには腫瘍増殖と時間との関係、腫瘍増殖阻止および腫瘍増殖遅延が含まれる。
5.異常または顕著な観察所見のまとめ。
【0234】
実施例27
腫瘍モデルの作成、細胞の調製および腹腔内腫瘍の移植方法
マウスを体重別にグループ分けする。25g針を用いて、BDF-1マウス(n=4)の腹腔内に1×106個のP388細胞、容積500μLを移植する(第0日)。ATCC癌細胞貯蔵部門(ATCC tumor repository)から入手したP388細胞をBDF-1マウスの腹水として維持し、新たなマウスに週1回継代する。マウスを安楽死させ、20g針を用いて腹壁を介して腹水細胞を採取する。実験に用いる細胞は継代回数3〜20回のものを用いる。マウス体内での20回の継代後には、液体窒素中の凍結貯蔵物から新たな細胞を増殖させてマウスに接種する。実験用の細胞はハンクス平衡塩類溶液ですすぎ洗い、血球計算器で算定した上で、HBSSで希釈して濃度を2×106個/mLとする。
【0235】
試験群のグループ分けは、全マウスに腫瘍細胞を投与した後にランダムに行う。必要なグループ分けは固形腫瘍試験に対して行われるものと同様である(実施例26参照)。
【0236】
マウスに対して、個々のマウスの体重に基づき、処方用量(10μL/g、指定に応じて)をマウスに投与するために必要な容積の試料を静脈内または腹腔内に注射する。腹腔内腫瘍の場合には、腫瘍細胞の接種から1日後に投与を開始する。
【0237】
マウスの健康状態を毎日詳細に観察する。病的状態および病態の進行の徴候については実施例26に記載した通りに詳細に観察する。マウスの体重を検査時に測定する。
【0238】
マウスを屠殺した後には、腫瘍量および/または臓器外観の生理的に観察される変化の程度を評価するために肉眼的剖検を行う。所見は記録する。
【0239】
群の体重を、有効性試験の間、最終投与から14日間にわたって月曜から金曜まで記録する。
【0240】
全マウスを、前投与期間および投与期間にわたって生死および病状に関して少なくとも1日1回観察し、必要と思われる場合はそれ以上観察する。詳細には、病的状態の徴候は、体重減少、食欲変化、体毛の荒れ、身づくろいの減少、行動変化(歩行変化など)、傾眠および顕著なストレスの発現に基づく。重篤な中毒または腫瘍に伴う病的状態が認められた場合には、マウスを安楽死させ(CO2による窒息)、中毒の他の徴候を評価するために剖検を行う。瀕死状態のマウスは人道的な理由から屠殺する必要があり、屠殺に関する判断は動物飼育技術者(Animal Care Technician)および試験責任者(Study Director)/監督者(Manager)の裁量に委ねられる。これらの所見を生データとして記録し、死亡時はその翌日として記録する。
【0241】
データは表または図の形式として提示し、これには時間の関数としての各群の平均体重および寿命の延長が含まれる。
【技術分野】
【0001】
関連出願の相互参照
本出願は、米国特許法第119条(e)項(35 U.S.C. §119(e))に基づき、2001年10月3日に提出された米国仮特許出願第60/326,671号;2001年12月17日に提出された第60/341,529号;2002年2月15日に提出された第60/356,759号;2002年4月23日に提出されたカナダ仮特許出願第2,383,259号;2002年8月7日に提出された米国仮特許出願第60/401,984号および2002年9月6日に提出された第60/408,733号の恩典を主張する。これらの出願の内容は参照として本明細書に組み入れられる。
【0002】
技術分野
本発明は、治療薬の相乗的または相加的な組合せを送達する、改良された送達のための組成物および方法に関する。より詳細には、本発明は、送達媒体を含む製剤を提供することによって、薬剤を意図した標的に対して送達する場合に、相乗的または相加的な比を維持することを保証する送達システムに関する。
【背景技術】
【0003】
背景技術
癌、AIDS、感染症、免疫疾患および心血管疾患といった命にかかわる多くの疾患は、数多くの分子機構による影響を受ける。この複雑性のために、単一の薬剤を用いて治癒を達成しようとした場合の奏功性には限界がある。このため、薬剤の併用が、疾患との戦い、特に癌の治療にしばしば用いられている。投与した薬剤の数と急性リンパ球性白血病などの癌の治癒率との間には強い相関があるように思われる(Freiら、Clin. Cancer Res. (1998) 4: 2027-2037(非特許文献1))。ドキソルビシン、シクロホスファミド、ビンクリスチン、メトトレキサート、ロイコボリン(救済用)およびシタラビンの組合せ(ACOMLA)、またはシクロホスファミド、ドキソルビシン、ビンクリスチン、プレドニゾンおよびブレオマイシンの組合せ(CHOP-b)を用いた臨床試験では、組織球性リンパ腫の治療にこれらが用いられて奏功している(Toddら、J. Clin. Oncol. (1984) 2: 986-993(非特許文献2))。
【0004】
薬剤併用の効果は、それらの供給される比によって相乗作用がもたらされる場合に強化される。また、薬剤の相乗的組合せが、必要投与量の減少によって毒性を低下させること、癌の治癒率を高めること(Barriereら、Pharmacotherapy (1992) 12: 397-402(非特許文献3);Schimpff, Support Care Cancer (1993) 1: 5-18(非特許文献4))および微生物の多剤耐性株の蔓延を抑えること(Shlaesら、Clin. Infect. Dis. (1993) 17:S527-S536(非特許文献5))も示されている。作用機序の異なる複数の薬剤を選択することにより、生化学経路における複数の部位を攻撃し、それによって相乗作用を得ることができる(ShahおよびSchwartz、Clin. Cancer Res. (2001) 7: 2168-2181(非特許文献6))。L-カナバニンと5-フルオロウラシル(5-FU)との併用などは、ラット結腸腫瘍モデルにおいて各薬剤の単独での効果の合計よりも強い抗腫瘍活性を示すことが報告されている(Swaffarら、Anti-Cancer Drugs (1995) 6: 586-593(非特許文献7))。シスプラチンとエトポシドは、ヒト小細胞肺癌細胞株SBC-3の増殖の抑制に関して相乗作用を示す(Kanzawaら、Int. J. Cancer (1997) 71(3): 311-319(非特許文献8))。
【0005】
相乗作用に関するこのほかの報告には、以下に関するものがある。
ビンブラスチンと組換えインターフェロンβ(Kueblerら、J. Interferon Res. (1990) 10: 281-291(非特許文献9));
シスプラチンとカルボプラチン(Kobayashiら、Nippon Chiryo Gakkai Shi (1990) 25: 2684-2692(非特許文献10));
エチルデスヒドロキシ-スパルソマイシンとシスプラチンまたはシトシンアラビノシド(AraC)またはメトトレキサートまたは5-FUまたはビンクリスチン(Hofsら、Anti Cancer Drugs (1994) 5: 35-42(非特許文献11));
全トランスレチノイン酸と酪酸またはトリブチリン(Chenら、Chin. Med. Engl. (1999) 112: 352-355(非特許文献12));および
シスプラチンとパクリタキセル(Engblomら、Br. J. Cancer (1999) 79: 286-292(非特許文献13))。
【0006】
以上の試験では、成分の比が相乗作用にとって重要であることが認められた。例えば、5-フルオロウラシルおよびL-カナバニンはモル比が1:1では相乗的であるが、比が5:1では拮抗性であることが見いだされた;シスプラチンおよびカルボプラチンは血中濃度曲線下面積(AUC)比が13:1では相乗作用を示したが、19:5では拮抗作用を示した。
【0007】
相乗的な相互作用を示すものの、相互作用が組合せ比(combination ratio)に依存することが記載されていない、薬剤のその他の組合せもある。このリストは非常に広範囲にわたり、インビボ試験も時に含まれるが、主としてインビトロ培養の報告によって構成される。
【0008】
多数の報告に加えて、さまざまな組合せが臨床の場で有効であることが示されている。これらを以下の表に記した。
a FDA:米国食品医薬品局
【0009】
加えて、ある種の他の組合せは、非拮抗的な併用効果もしくは臨床的有効性を示す可能性があることを文献中のさまざまな報告から推論され、または地域の研究グループによって標準的な医療行為として受容されうる。これらには以下のものがある。
【0010】
相乗的な薬剤の組合せを用いることには前述のような利点があるものの、その治療的使用を制限するさまざまな欠点もある。例えば、相乗作用はしばしば、薬剤曝露の期間および投与の順序といった種々の要因に依存する(BonnerおよびKozelsky、Cancer Chemother. Pharmacol. (1990) 39: 109-112(非特許文献14))。エチルデスヒドロキシ-スパルソマイシンをシスプラチンと併用した試験では、この相乗作用が組合せ比、投与期間および投与の順序によって影響されることが示されている(Hofsら、前記(非特許文献11))。
【0011】
このため、相乗作用を薬剤の併用によって発揮させるためには、これらの薬剤が、規定された比を示す量で存在する必要があることが知られている。事実、同じ薬剤の組合せが、ある比では拮抗的であり、別の比では相乗的であり、さらに別の比では相加的であることもある。拮抗的作用を回避し、薬剤が少なくとも相加的であるようにすることが望ましい。本発明は、個々の比で得られる結果が濃度にも依存することを認識している。比の中にはある濃度では拮抗的であり、別の濃度では非拮抗的なものがある。本発明は、これらの薬剤が対象内部の標的部位に到達した場合に所望の比または投与された比を維持する製剤中にある形で送達することにより、および、標的での濃度は投与したものとは異なる可能性があるという理由から、所望の濃度の範囲で顕著に非拮抗的である比を選択することにより、成分の比が相乗的または相加的な範囲にあることを保証する。
【0012】
PCT公報・国際公開公報第00/51641号(特許文献1)は、相乗的であると言われている抗ウイルス薬の組合せを投与することを記載している。相乗的な比を決定するためにインビトロ試験が用いられた。しかし、この比をインビボで維持すると考えられる投与様式に関する教示はない。実際には、この刊行物は、成分を逐次的に投与しても同時に投与してもよいと記載している。
【0013】
PCT公報・国際公開公報第01/15733号(特許文献2)は、自己免疫疾患の治療のための、相乗的であると想定される組成物を記載している。この場合も、製剤化の方法は、送達後にこの比が維持されることを保証していない。
【0014】
Daoudら、Cancer Chemother. Pharmacol. (1991) 28: 370-376(非特許文献15)は、ヒト卵巣癌細胞に対するシスプラチンおよびリポソーム封入バリノマイシンの相乗的な細胞傷害作用を記載している。この論文は、遊離状態にあるシスプラチンとリポソーム中に封入されたバリノマイシンとをヒト卵巣腫瘍由来細胞株CaOV-3の培養物の処置に用いるインビトロアッセイ法を記載している。著者らは相乗作用および拮抗作用が示される濃度範囲を決定した。リポソーム封入はバリノマイシンを溶解させるために用いられた。実験はインビトロで行われているため、インビボ送達は関係しない。
【0015】
2001年4月10日にDaoudに対して発行された米国特許第6,214,821号(特許文献3)は、トポイソメラーゼI阻害物質およびスタウロスポリンを含む薬学的組成物を記載している。その特許請求の範囲は、スタウロスポリンに、トポイソメラーゼI阻害物質により誘導されるS期停止を無効化する能力、および、正常なp53機能を欠くヒト乳癌細胞に対するその細胞傷害性を増強させる能力があるという発見に基づくように思われる。具体的な医薬製剤は提唱されていない。
【0016】
Fountainらに対する米国特許第5,000,958号(特許文献4)は、インビボでの治療効果の増強を発揮すると言われている、リポソーム中に封入された抗菌薬の混合物を記載している。抗菌薬の適した比は、インビトロでの相乗作用を経験的に調べる併用効果試験によって決定される。ある濃度範囲にわたる相乗的な比の保証に関する考察は行われていない。
【0017】
Schiffelersら、J. Pharmacol. Exp. Therapeutic (2001) 298: 369-375(非特許文献16)は、リポソーム中に共封入されたゲンタマイシンおよびセフタジジムのインビボでの相乗的な相互作用を記載している。望ましい比は、Fountain(前記(特許文献4))のものに類似した併用効果試験を用いて決定されたが、ある濃度範囲にわたって相乗作用が維持される比の決定に関する考察はなされていない。
【0018】
本発明は、第1に、治療薬の決定された相乗的または相加的な比を、それらが内部に投与された製剤の薬物動態を制御することによって維持しうることを認識しており、第2に、標的組織に到達する薬剤混合物における成分の濃度はそれが投与されたものと同じではない可能性があるため、ある濃度範囲にわたって非拮抗的な比が示される必要があることを認識している。相乗作用または相加性を維持するという問題は、治療薬をリポソームなどの送達媒体中に封入した(すなわち、安定的に会合させた)場合には送達媒体が薬物動態を決定し、そのため封入された複数の薬剤が同じように振る舞うという認識により、および、ある濃度範囲にわたって顕著に相乗的/相加的である比を選択することにより、解決される。
【先行技術文献】
【特許文献】
【0019】
【特許文献1】国際公開公報第00/51641号
【特許文献2】国際公開公報第01/15733号
【特許文献3】米国特許第6,214,821号
【特許文献4】米国特許第5,000,958号
【非特許文献】
【0020】
【非特許文献1】Freiら、Clin. Cancer Res. (1998) 4: 2027-2037
【非特許文献2】Toddら、J. Clin. Oncol. (1984) 2: 986-993
【非特許文献3】Barriereら、Pharmacotherapy (1992) 12: 397-402
【非特許文献4】Schimpff, Support Care Cancer (1993) 1: 5-18
【非特許文献5】Shlaesら、Clin. Infect. Dis. (1993) 17:S527-S536
【非特許文献6】ShahおよびSchwartz、Clin. Cancer Res. (2001) 7: 2168-2181
【非特許文献7】Swaffarら、Anti-Cancer Drugs (1995) 6: 586-593
【非特許文献8】Kanzawaら、Int. J. Cancer (1997) 71(3): 311-319
【非特許文献9】Kueblerら、J. Interferon Res. (1990) 10: 281-291
【非特許文献10】Kobayashiら、Nippon Chiryo Gakkai Shi (1990) 25: 2684-2692
【非特許文献11】Hofsら、Anti Cancer Drugs (1994) 5: 35-42
【非特許文献12】Chenら、Chin. Med. Engl. (1999) 112: 352-355
【非特許文献13】Engblomら、Br. J. Cancer (1999) 79: 286-292
【非特許文献14】BonnerおよびKozelsky、Cancer Chemother. Pharmacol. (1990) 39: 109-112
【非特許文献15】Daoudら、Cancer Chemother. Pharmacol. (1991) 28: 370-376
【非特許文献16】Schiffelersら、J. Pharmacol. Exp. Therapeutic (2001) 298: 369-375
【発明の概要】
【0021】
発明の開示
本発明は、非拮抗的な比の複数の治療薬、好ましくは抗腫瘍薬を、2種類またはそれ以上の薬剤を封入する送達媒体組成物を用いて投与するための方法であって、薬剤がある濃度範囲にわたって相乗的または相加的(すなわち、非拮抗的)な比で媒体中に存在するような方法に関する。封入の前に、組合せにおける治療薬の比は、組合せが所望の濃度範囲にわたって相乗作用または相加作用を示すように選択される。送達媒体中への封入により、2種類またはそれ以上の薬剤を協調的な様式で罹患部に送達することが可能になり、それによって薬剤が罹患部に非拮抗的な比で存在することが保証される。この結果は、薬剤を送達媒体中に共封入した場合にも、または罹患部で非拮抗的な比が維持されるように投与される送達媒体中に別個に封入した場合にも達成されると考えられる。組成物の薬物動態(PK)は、協調的な送達が得られるように送達媒体それ自体によって制御される(送達システムのPKが類似しているという条件で)。
【0022】
したがって、1つの面において、本発明は、媒体組成物中に封入された2種類またはそれ以上の薬剤を、所望の濃度範囲にわたって相乗的または相加的である比で含む、非経口的投与のための送達媒体組成物を提供する。本送達媒体組成物は、薬剤を送達媒体組成物中にこれらの比で封入することを含む工程によって調製される。薬剤の非拮抗的な比は、妥当な細胞培養物または無細胞系に対する薬剤の生物活性または作用をある濃度範囲にわたって評価することにより、および、1つの態様においては、「組合せ指数(combination index)」(CI)を決定するためのアルゴリズムを適用することにより、決定される。以下にさらに説明するように、認知されたアルゴリズムを用いて、それぞれの濃度レベルでの組合せ指数を算出することができる。ある濃度範囲にわたってCIが相乗作用または相加作用を表す比を選択する。広い濃度範囲にわたってCIが相乗的であることが好ましい。好ましい薬剤は抗腫瘍薬である。所望の濃度範囲にわたって非拮抗的な効果を維持する薬剤の比が決定される任意の方法を用いうる。
【0023】
より詳細には、本発明は、送達媒体を含む組成物であって、前記送達媒体の内部に少なくとも第1の治療薬および第2の治療薬が、細胞の1%超が影響を受ける(影響率(fa)>0.01)濃度範囲の少なくとも5%にわたって、培養下の妥当な細胞もしくは無細胞系に対して非拮抗的な生物作用を示すような第1の薬剤と第2の薬剤とのモル比で封入されている組成物、または、送達媒体を含む組成物であって、前記送達媒体の内部に少なくとも第1の治療薬および第2の治療薬が、妥当な細胞に対して非拮抗的な細胞傷害作用もしくは細胞分裂抑制作用を示すような第1の薬剤と第2の薬剤とのモル比で封入されており、前記薬剤が抗腫瘍薬である組成物に関する。「妥当な(relevant)」細胞とは、本出願者らによれば、所望の生物作用を調べるのに適した少なくとも1つの細胞培養物または細胞系のことを指している。例えば、薬剤が抗腫瘍薬である場合、「妥当な」細胞は、米国国立癌研究所(National Cancer Institute;(NCI)/米国国立衛生研究所(National Institutes of Health;NIH)の開発中治療薬プログラム(Developmental Therapeutics Program)(DTP)により、その抗癌薬開発プログラムにおいて有用と同定された細胞系であると考えられる。現在、DTPスクリーニングには60種類のヒト腫瘍細胞株が用いられている。このような細胞株の少なくとも1つに対して所望の活性が示される必要があると考えられる。
【0024】
もう1つの面において、本発明は、相乗的または相加的な比にある2種類またはそれ以上の治療薬を、本発明の組成物を投与することによって所望の標的に送達するための方法を対象とする。
【0025】
もう1つの面において、本発明は、ある濃度範囲にわたって非拮抗的である比の少なくとも2つの治療薬を含む送達媒体である、送達媒体を含む治療的組成物を調製するための方法であって、少なくとも2つの治療薬の一団(panel)を提供すること(この際、この一団は少なくとも1つ、しかし好ましくは複数の比の前記薬剤を含む)、ある濃度範囲にわたって、妥当な細胞培養物または無細胞系に対して生物作用を発揮する一団のメンバーの能力を試験すること、その比が、適した濃度範囲にわたって前記細胞培養物または無細胞系に対して相乗的または相加的な作用をもたらすような一団のメンバーを選択すること、および、一団のうち適格なメンバーを表す比の薬剤を薬物送達媒体中に封入する(すなわち、安定的に会合させる)こと、を含む方法を対象とする。
【0026】
以下にさらに説明するように、好ましい態様においては、上記の方法による適切な組合せの設計に際して、非拮抗的な比は、妥当な細胞培養物または無細胞アッセイ系による評価で、細胞の1%またはそれ以上に影響を及ぼす(fa>0.01)、好ましくは細胞の20〜80%に影響を及ぼす(fa=0.2〜0.8)用量または濃度の、少なくとも5%の範囲にわたって組合せ指数(CI)が1.1以下であるものとして選択される。
【図面の簡単な説明】
【0027】
【図1】製剤中に含める治療薬の適切な比を決定するための本発明の方法の概要を示した概略図である。
【図2A】組合せおよび相乗作用のデータを提示するための方法を例示する。
【図2B】組合せおよび相乗作用のデータを提示するための方法を例示する。
【図2C】組合せおよび相乗作用のデータを提示するための方法を例示する。
【図2D】組合せおよび相乗作用のデータを提示するための方法を例示する。
【図2E】組合せおよび相乗作用のデータを提示するための方法を例示する。
【図3】図3Aはイリノテカン:5-FUに関する、モル比1:10(黒い四角)および1:1(黒丸)での、影響を受けたHT29細胞の率(fa)の関数としての組合せ指数(CI)のグラフである。図3Bはエトポシド:カルボプラチンに関する、モル比1:10(黒い菱形)および10:1(黒い四角)での、影響を受けたMCF-7細胞の率(fa)の関数としてのCIのグラフである。
【図4】シスプラチン:エデルフォシン(edelfosine)に関する、モル比10:1(黒い三角)および1:1(黒丸)での、影響を受けたH460細胞の率(fa)の関数としてのCIのグラフである。
【図5A】H460細胞における、モル比10:1、1:1および1:10でのカルボプラチン:ダウノルビシンの関数としてのCI最大値のグラフである。挿入図は、MCF-7細胞における、有効量(ED)値50、75および90、モル比10:1および1:1でのカルボプラチン:ダウノルビシンに関するCIのヒストグラムである。
【図5B】カルボプラチン:ダウノルビシンに関する、モル比1:10(黒い三角)、1:1(黒い四角)および10:1(黒丸)での、影響を受けたH460細胞の率(fa)の関数としてのCIのグラフである。挿入図は、H460細胞における、ED値50、75および90での、モル比1:10、1:1および10:1のカルボプラチン:ダウノルビシンに関するCIのヒストグラムである。
【図6】薬剤を単一のリポソーム(DSPC/DSPG、80:20mol%)中に非拮抗的な比(10:1)で調合した場合の、静脈内投与後の時間の関数としての、カルボプラチン(白丸)およびダウノルビシン(黒丸)の血漿中濃度(nmol/mL)のグラフである。
【図7】図7Aは薬剤を単一のリポソーム(DSPC/DSPG、80:20mol%)中に10:1(黒丸)、5:1(白丸)および1:1(黒い三角)という3種類の比で調合した場合の、静脈内投与後の時間の関数としての、カルボプラチン:ダウノルビシンのモル比のグラフである。図7Bは静脈内投与後の時間の関数として再プロットした、図7Aにおける1:1カルボプラチン:ダウノルビシンのデータのグラフである。
【図8】薬剤を非拮抗的なモル比(10:1)で単一のリポソーム(DSPC/スフィンゴミエリン/DSPE-PEG2000、90:5:5mol%)中に調合した場合の、静脈内投与後の時間の関数としての、カルボプラチン(黒丸)およびダウノルビシン(白丸)の血漿中濃度(nmol/mL)のグラフである。
【図9】ヒトH460非小細胞肺腫瘍を有するマウスに投与した、カルボプラチンおよびダウノルビシンの混合物(黒い逆三角)、カルボプラチンおよびダウノルビシンを単一のリポソーム中に調合したもの(白い逆三角)、または生理食塩水対照(黒丸)の活性を比較したグラフである。カルボプラチンおよびダウノルビシンはDSPC/DSPG(80:20mol%)リポソーム中にモル比1:1で調合した。矢印は薬剤を投与した日を示す。
【図10】ヒトH460非小細胞肺腫瘍を有するマウスに投与した、カルボプラチンおよびダウノルビシンの混合物(黒い三角)、カルボプラチンおよびダウノルビシンを単一のリポソーム中に調合したもの(白い三角)、または生理食塩水対照(黒丸)の活性を比較したグラフである。カルボプラチンおよびダウノルビシンはDSPC/SM/DSPE-PEG2000(90:5:5mol%)リポソーム中にモル比10:1で調合した。x軸に沿った矢印は矢印は投薬スケジュールを表す。
【図11】図11Aはシスプラチン:ダウノルビシンに関する、モル比1:1(黒い四角)および10:1(黒丸)での、影響を受けたH460細胞の率(fa)の関数としてのCIのグラフである。図11BはH460細胞に対する、モル比10:1、1:1および1:10でのシスプラチン:ダウノルビシンの関数としてのCI最大値のグラフである。
【図12】薬剤を非拮抗的なモル比(10:1)で単一のリポソーム(DMPC/Chol、55:45mol%)中に調合した場合の、静脈内投与後の時間の関数としての、シスプラチン(白丸)およびダウノルビシン(黒丸)の血漿中濃度(μmol/mL)のグラフである。
【図13】薬剤を非拮抗的なモル比(10:1)で2つの別個のリポソーム(シスプラチンに関してはDMPC/Chol、55:45mol%、ダウノルビシンに関してはDSPC/DSPE-PEG2000、95:5mol%)中に調合した場合の、静脈内投与後の時間の関数としての、シスプラチン(黒丸)およびダウノルビシン(白丸)の血漿中濃度(μmol/mL)のグラフである。
【図14】ヒトH460非小細胞肺腫瘍を有するマウスに投与した、シスプラチンおよびダウノルビシンの混合物(黒い逆三角)、シスプラチンおよびダウノルビシンを別個のリポソーム中に調合したもの(白い逆三角)、または生理食塩水対照(黒丸)の活性を比較したグラフである。シスプラチンはDMPC/Chol(55:45mol%)リポソーム中に調合し、ダウノルビシンはDSPC/DSPE-PEG2000(95:5mol%)リポソーム中に調合した上で、非拮抗的なモル比(10:1)で投与した。矢印は薬剤を投与した日を示す。
【図15】薬剤を単一のリポソーム(DMPC/Chol、55:45mol%)中に拮抗的なモル比1:1で調合した場合の、静脈内投与後の種々の時点での、血漿中に残留したシスプラチン(黒丸)およびダウノルビシン(白丸)の濃度(nmol/mL)を示したグラフである。挿入図は、投与後の種々の時点でのシスプラチン:ダウノルビシンのモル比を示す。
【図16】ヒトH460非小細胞肺腫瘍を有するマウスに投与した、シスプラチンおよびダウノルビシンの混合物(黒い三角)、シスプラチンおよびダウノルビシンを単一のリポソーム中に調合したもの(白い三角)、または生理食塩水対照(黒丸)の活性を比較したグラフである。薬剤はDMPC/Chol(55:45mol%)リポソーム中に拮抗的なモル比(1:1)で調合した。矢印は薬剤を投与した日を示す。
【図17】図17Aはシスプラチン:トポテカンに関する、モル比1:1(黒丸)および10:1(白丸)での、影響を受けたH460細胞の率(fa)の関数としてのCIのグラフである。図17BはH460細胞に対する、シスプラチン:トポテカンのモル比の関数としてのCI最大値のグラフである。
【図18】薬剤を別個のリポソーム(シスプラチンに関してはDMPC/Chol、55:45mol%、トポテカンに関してはDSPC/Chol、55:45mol%)中に調合した場合の、静脈内投与後の種々の時点での、血漿中に残留したシスプラチン(黒丸)およびトポテカン(白丸)の濃度(μmol/mL)を示したグラフである。挿入図は、投与後の種々の時点でのシスプラチンとトポテカンとのモル比を示す。
【図19】ヒトH460非小細胞肺腫瘍を有するマウスに投与した、シスプラチンおよびトポテカンの混合物(黒い三角)、シスプラチンおよびトポテカンを別個のリポソーム中に調合したもの(白い三角)、または生理食塩水対照(黒丸)の活性を比較したグラフである。シスプラチンはDMPC/Chol(55:45mol%)リポソーム中に調合し、トポテカンはDSPC/Chol(55:45mol%)リポソーム中に調合した上で、非拮抗的なモル比(10:1)で投与した。矢印は薬剤を投与した日を示す。
【図20】図20Aはシスプラチン:イリノテカンに関する、モル比1:1(黒い四角)、10:1(黒丸)、1:5(黒い三角)および1:10(黒い菱形)での、影響を受けたH460細胞の率(fa)の関数としてのCIのグラフである。図20BはH460細胞に対する、シスプラチン:イリノテカンのモル比の関数としてのCI最大値のグラフである。
【図21】薬剤を単一のリポソーム(DSPC/DSPG、80:20mol%)中に同時装填した場合の、静脈内投与後の種々の時点での、血漿中に残留したシスプラチン(黒丸)およびイリノテカン(白丸)の濃度(nmol/mL)を示したグラフである。
【図22】薬剤を別個のリポソーム(シスプラチンに関してはDMPC/Chol、55:45mol%、イリノテカンに関してはDSPC/DSPE-PEG2000、95:5mol%)中に調合した場合の、静脈内投与後の種々の時点での、血漿中に残留したシスプラチン(黒丸)およびイリノテカン(白丸)の濃度(nmol/mL)を示したグラフである。
【図23】ヒトH460非小細胞肺腫瘍を有するマウスに投与した、シスプラチンおよびイリノテカンの混合物(黒い四角)、シスプラチンおよびイリノテカンを別個のリポソーム中に調合して種々の用量で投与したもの(白抜き記号)、または生理食塩水対照(黒丸)の活性を比較したグラフである。DMPC/Chol(55:45mol%)リポソーム中に調合したシスプラチンおよびDSPC/DSPE-PEG2000(95:5mol%)リポソーム中に調合したイリノテカンを非拮抗的なモル比(1:5)で投与した。矢印は薬剤を投与した日を示す。
【図24】ビノレルビンをPOPS(黒い逆三角)、DPPS(黒い三角)、DLPS(黒丸)、DSPS(黒い菱形)またはDOPS(黒い四角)と、ビノレルビン:PSのモル比1:1で併用した場合の、影響を受けたH460細胞の率(fa)の関数としてのCIのグラフである。
【図25】図25AはSCID/rag2マウスに対する、遊離ビノレルビン(黒丸)、またはSM/Chol/DPPS/DSPE-PEG2000、35:45:10:10mol%リポソーム中にビノレルビン:PSのモル比1:1で封入したもの(白丸)を静脈内投与した後の時間の関数としてのビノレルビン血漿中濃度のグラフである。図25Bは図25Aのデータを用いた、SCID/rag2マウスに対する、遊離ビノレルビン(黒色の棒グラフ)、またはSM/Chol/DPPS/DSPE-PEG2000、35:45:10:10mol%中に封入したもの(灰色の棒グラフ)を静脈内投与した後の血漿中濃度曲線下面積(AUC)を示したヒストグラムである。
【図26】H460非小細胞肺腫瘍を有するマウスに投与した、遊離ビノレルビン(白丸)、ビノレルビンをDSPC/Chol/DPPS/DSPE-PEG2000、35:45:10:10mol%リポソーム中に封入したもの(黒い逆三角)、ビノレルビンをSM/Chol/DPPS/DSPE-PEG2000、35:45:10:10mol%リポソーム中に封入したもの(白い三角)、または生理食塩水対照(黒丸)の活性を比較したグラフである。ビノレルビンおよびホスファチジルセリン(DPPS)は非拮抗的なモル比(1:1)で調合した。矢印は薬剤を投与した日を示す。
【図27】H460非小細胞肺腫瘍を有するマウスに投与した、生理食塩水対照(黒丸);遊離ビノレルビン(白丸);ビノレルビンを以下のリポソーム中に封入したもの:SM/Chol/DPPS/DSPE-PEG2000、35:45:10:10(黒い逆三角)、DAPC/Chol/DPPS/DSPE-PEG2000、35:45:10:10mol%(白い三角)およびDSPC/Chol/DSPS/DSPE-PEG2000、35:45:10:10mol%(黒い四角)の効果を示す。ビノレルビンおよびホスファチジルセリン(DPPSまたはDSPS)は非拮抗的なモル比(1:1)で調合した。矢印は薬剤を投与した日を示す。
【図28】P388マウス白血病を有するマウスの生存率に対する、生理食塩水対照(白い三角);遊離ビノレルビン(黒丸);およびビノレルビンをSM/Chol/DPPS/DSPE-PEG2000、35:45:10:10mol%リポソーム中に封入したもの(黒い逆三角)の効果を示す。ビノレルビンおよびホスファチジルセリンは非拮抗的なモル比(1:1)で調合した。x軸に沿った矢印は薬剤を投与した日を示す。
【図29】FUDR:CPT-11の10:1(黒い四角);5:1(黒丸);1:1(黒い三角);1:5(黒い逆三角);および1:10(白丸)という種々の比での組合せによって影響されたHT-29細胞の率の関数としてプロットしたCIを示す。
【図30】静脈内投与後の時間の関数としてのFUDR(黒丸)およびCPT-11(白丸)の血漿中濃度のグラフである。
【図31】腫瘍体積と、生理食塩水対照(黒丸)、CPT-11/FUDR混合物注射(白い逆三角)およびCPT-11/FUDRのリポソーム製剤(黒い逆三角)の腫瘍細胞接種後の時間との関係に関するグラフである。
【発明を実施するための形態】
【0028】
発明の実施の態様1
本発明の方法は、インビトロで所望の濃度範囲にわたって非拮抗的である治療薬の比を決定すること、および、その比が所望の作用部位で確実に維持されると考えられる様式でこの非拮抗的な比を提供することを含む。相乗的な比または相加的な比は、2種類またはそれ以上の治療薬の少なくとも1つの比を妥当な細胞培養物または無細胞系に対してある濃度範囲にわたってインビトロで試験することによって得られた結果に対して、標準的な分析ツールを適用することによって決定される。例えば、個々の薬剤および種々の組合せを、細胞培養物または無細胞系に対する生物作用、例えば細胞死を引き起こすこと、または細胞増殖を抑制することに関して、種々の濃度レベルで試験する。あらかじめ設定した比の濃度レベルを細胞生存率に対してプロットして相関を得て、それを既知かつ確立された数学的手法によって操作して「組合せ指数(combination index)」(CI)を算出することができる。この数学的手法は、CIが1(すなわち0.9〜1.1)であれば薬剤の相加作用を表し、CT>1(すなわち>1.1)であれば拮抗作用を表し、CIが<1(すなわち<0.9)であれば相乗作用を表すというものである。
【0029】
1 略号
以下の略号を用いる。
PE:ホスファチジルエタノールアミン;PS:ホスファチジルセリン;DPPS:ジパルミトイルホスファチジルセリン;DSPS:ジアステロイルホスファチジルセリン;DLPS:ジラウロイルホスファチジルセリン;DOPS:ジオレオイルホスファチジルセリン;POPS:パルミトイルオレオイルホスファチジルセリン;PC:ホスファチジルコリン;SM:スフィンゴミエリン;PG:ホスファチジルグリセロール;PI:ホスファチジルイノシトール;PA:ホスファチジン酸;DSPC:ジアステロイルホスファチジルコリン;DMPC:ジミリストイルホスファチジルコリン;DSPG:ジアステロイルホスファチジルグリセロール;DSPE:ジアステロイルホスファチジルエタノールアミン;Chol:コレステロール;CHまたはCHE:コレステリルヘキサデシルエーテル;
PEG:ポリエチレングリコール;DSPE-PEG:ジアステロイルホスファチジルエタノールアミン-N-[ポリエチレングリコール];PEGの後に数字が続く場合、その数字はダルトン単位で表現したPEGの分子量である;DSPE-PEG2000:ジアステロイルホスファチジルエタノールアミン-N-[ポリエチレングリコール 2000];
SUV:小型単ラメラ小胞;LUV:大型単ラメラ小胞;MLV:多重ラメラ小胞;
MTT:3-(4,5-ジメチルチアゾール-2-イル)-2,5-ジフェニル-2Hテトラゾリウムブロミド;DMSO:ジメチルスルホキシド;OD:光学密度;OGP:N-オクチルβ-D-グルコピラノシド;EDTA:エチレンジアミン四酢酸;HEPES:N-[2-ヒドロキシルエチル]-ピペラジン-N-[2-エタンスルホン酸];HBS:HEPES緩衝生理食塩水(20mM HEPES、150mM NaCl、pH 7.4);SHE:300mMスクロース、20mM HEPES、30mM EDTA;ED50、ED75およびED90:培養下にある細胞の50、75および90%に影響を及ぼすために必要な有効量;LD50:培養下にある細胞の50%の死滅を引き起こすために必要な用量;CI:組合せ指数;CI maxまたはCI最大値:異なる比の薬剤に関してCI値に最も大きな差が観察される単一のfa値(0.2〜0.8の間)について得られたCI値;fa:影響率;TEA:トリエタノールアミン;
FDA:米国食品医薬品局;NCI:米国国立癌研究所。
【0030】
一般的なアプローチの1つを図1に示している。示されているように、薬剤AおよびBを個別に、および2種類の比で併用して、以下に述べるMTTアッセイ法によって評価される細胞死または細胞分裂停止を引き起こす能力に関して調べる。最初に、薬剤A、Bの濃度と2種類の組合せ比(Y:ZおよびX:Y)との相関を、未処理対照細胞の生存性を基準とする百分率として算出した細胞傷害性に対してプロットする。予想通り、個別の薬剤および組合せのいずれに対しても細胞の生存性に対して用量依存的な効果がみられる。この相関性が実証されれば、細胞の生存性または影響率(fa)を、CIの算出において濃度の代用物として用いることができる。
【0031】
CI算出の結果は図1にも示されている;この指数は、ChouおよびTalalay、Advance Enz. Regul. (1985) 22: 27-55の手順に従って影響を受けた細胞の率の関数として算出される。この仮説の状況では、薬剤A+Bの第1の比(X:Y)はすべての濃度で非拮抗的であるが、第2の比(Y:Z)での組合せは拮抗的である。したがって、広範囲にわたって濃度に関係なく非拮抗的であると考えられる薬剤A+Bの比(比1)を得ることが可能である。本発明の組成物に含めることが望ましいのはこの比である。
【0032】
本発明者らは、薬剤の組合せのさまざまな比について「CI最大値」を算出することによる、相乗作用に対する比および濃度の影響の代替的な例も考案している。「CI最大値」は、異なる比の薬剤に関してCI値に最も大きな差が観察された単一のfa値(0.2〜0.8の間)について得られたCI値と定義される。これは図2Aおよび2Bに例示されている;示されているように、イリノテカン/カルボプラチン比が1:10である場合、そのCIが残りの比の値と最も大きく異なるのは影響率の値が0.2の場合である。示されているように、fa 0.2におけるこの比に関するCI値は約2.0である。
【0033】
非拮抗的な比のインビトロでの決定は2種類の薬剤のみの組合せに関して例示されているが、3種類またはそれ以上の薬剤の組合せに同じ手法を適用することにより、同様の様式でその濃度範囲にわたるCI値が得られる。
【0034】
このようにして得られた比は、非拮抗的な比が確実に維持されると考えられるリポソームまたは他の粒子状形態の中に薬剤を所定の比で封入することにより、薬学的組成物において維持される。このため、本組成物は、粒子状の送達媒体を含み、かつ所望の比の治療薬を含む。
【0035】
薬剤はその両方が同じ送達媒体中に含まれるように共封入されることが好ましいが、これは必ずしも必要ではない。粒子状担体は類似した薬物動態を共有しうるため、作用物質は、別個に封入された場合も製剤から協調的な送達を受ける。
【0036】
「封入(encapsulation)」とは、送達媒体との安定的な会合のことを意味する。このため、媒体は、インビボで投与された場合に1つまたは複数の薬剤が媒体と安定的に会合している限り、1つまたは複数の薬剤を取り囲む必要はない。したがって、「〜と安定的に会合した」および「〜中に封入された」または「〜とともに封入された」または「〜の中または〜とともに封入された」は、同義の用語であることを意図している。それらは本明細書において互換的に用いられる。安定的な会合は、送達媒体との共有結合、好ましくは切断可能な結合によるもの、非共有結合、および、薬剤を送達媒体の内部に捕捉することなどを含む、さまざまな手段によって生じさせうる。会合は、薬剤が、それが投与された対象における標的部位に送達されるまで、非拮抗的な比で送達媒体と会合したまま保たれる程度に、十分に安定でなければならない。
【0037】
送達媒体には、脂質担体、リポソーム、脂質ミセル、リポタンパク質ミセル、脂質安定化エマルション、シクロデキストリン、ポリマー性ナノ粒子、ポリマー性微粒子、ブロック共重合体ミセル、ポリマー-脂質ハイブリッド系、誘導体化された一本鎖ポリマーなどが含まれうる。リポソームは、「リポソーム:合理的デザイン(Liposomes: Rational Design)」(A.S. Janoffed., Marcel Dekker, Inc., N.Y.)に記載された通りに、または当業者に知られた別の技法によって調製しうる。本発明に用いるためのリポソームを、「低コレステロール」性であるように調製してもよい。このようなリポソームは「コレステロール非含有性」である、または「コレステロールを実質的に含まない」、または「コレステロールを本質的に含まない」。「コレステロール非含有性」という用語は、リポソームに言及して本明細書で用いる場合、リポソームがコレステロールを含まずに調製されていることを意味する。「コレステロールを実質的に含まない」という用語は、リポソームの相転移特性を大きく変化させるには不十分な量(通常はコレステロールが20mol%未満)のコレステロールの存在を許容する。リポソーム中に組み入れられるコレステロールが20mol%未満であれば、リポソームが20mol%を上回るコレステロールを伴って調製された場合には最適な保持がなされない薬剤の保持が可能になる。さらに、コレステロール20mol%未満として調製されたリポソームは相転移温度の幅が狭く、この特性は、封入された薬剤を熱を与えることによって放出するリポソーム(熱感受性リポソーム)の調製に利用することができる。本発明のリポソームが治療的脂質を含んでもよく、これにはエーテル脂質、ホスファチジン酸、ホスホネート、セラミドおよびセラミド類似体、スフィンゴシンおよびスフィンゴシン類似体、ならびにセリン含有脂質が含まれる。また、リポソームを、循環寿命を延長させるために、ポリエチレングリコール-DSPEなどの表面安定化性親水性ポリマー-脂質結合物とともに調製することもできる。担体の循環寿命を延長させるために、ホスファチジルグリセロール(PG)およびホスファチジルイノシトール(PI)などの負に荷電した脂質をリポソーム製剤に添加することもできる。これらの脂質は、表面安定化剤として親水性ポリマー-脂質結合物を置き換えるために用いうる。本発明の態様に、凝集を防止し、それによって担体の血中滞留時間を延長させるためのPGまたはPIを含むコレステロール非含有リポソームを用いてもよい。
【0038】
ミセルは、疎水性コアに存在する溶けにくい薬剤の送達のために利用される、両親媒性脂質またはポリマー成分から構成される自己集合性粒子である。ミセル性送達媒体の調製のための手段にはさまざまなものがあり、当業者によって容易に行われうる。例えば、脂質ミセルは、Perkinsら、Int. J. Pharm. (2000) 200(1): 27-39(参照として本明細書に組み入れられる)に記載された通りに調製しうる。リポタンパク質ミセルは、低密度および高密度リポタンパク質ならびにカイロミクロンを含む、天然または人工のリポタンパク質から調製することができる。脂質安定化エマルションとは、油が充填されたコアが脂質の単層または二重層などの乳化成分によって安定化されたものを含むように調製されたミセルのことである。コアはトリアシルグリセロール(コーン油)などの脂肪酸エステルを含みうる。単層または二重層には、DSPE-PEGなどの親水性ポリマー脂質結合物が含まれる。これらの送達媒体は、ポリマー脂質結合物の存在下における油のホモジネート化によって調製しうる。脂質安定化エマルションに組み入れられる薬剤は一般に水溶性が乏しい。リポタンパク質に類似した特性を示す合成ポリマー類似体、例えば、ステアリン酸エステルまたはポリ(エチレンオキシド)ブロック-ポリ(ヒドロキシエチル-L-アスパルタミド)およびポリ(エチレンオキシド)-ブロック-ポリ(ヒドロキシヘキシル-L-アスパルタミド)のミセルを、本発明の実施に用いることもできる(Lavasanifarら、J. Biomed. Mater. Res. (2000) 52: 831-835)。
【0039】
シクロデキストリンは空洞形成性で水溶性の多糖であり、その空洞内に水に溶けない薬剤を収容することができる。当業者に知られた手順を用いて薬剤をシクロデキストリン中に封入することができる。例えば、Atwoodら編、「包含性化合物(Inclusion Compounds)」第2巻および第3巻、Academic Press、NY (1984);Benderら、「シクロデキストリンの化学(Cyclodextrin Chemistry)」、Springer-Verlag、Berlin (1978);Szeitliら、「シクロデキストリンおよびその包含複合体(Cyclodextrins and Their Inclusion Complexes)」、Akademiai Kiado, Budapest, Hungary (1982)および国際公開公報第00/40962号を参照されたい。
【0040】
ナノ粒子および微粒子は、ポリマー性外殻(ナノカプセル)によって囲まれた、またはポリマー基質の全体に固体もしくは液体が分散した(ナノスフェア)、薬剤の濃縮性コアである。ナノ粒子および微粒子の一般的な調製法は、Soppimathら(J. Control Release (2001) 70(12): 1-20)に記載されており、これは参照として本明細書に組み入れられる。用いうるその他のポリマー性送達媒体には、親水性外殻によって囲まれた疎水性コアを含む薬剤を含むブロック共重合体ミセルが含まれる;これは一般に、疎水性薬剤のための担体として用いられ、Allenら、「コロイドおよび表面B(Colloids and Surface B)、Biointerfaces (1999) Nov 16 (1-4): 3-27に記載された通りに調製することができる。ポリマー-脂質ハイブリッド系は、脂質単層によって囲まれたポリマー性ナノ粒子からなる。ポリマー粒子は疎水性薬剤の組み入れのための積み荷空間(cargo space)として働き、脂質単層は疎水性コアと外部水性環境との間の安定化性境界として働く。ポリマーにはポリカプロラクトンおよびポリ(d,l-ラクチド)などを用いることができ、脂質単層は一般に脂質の混合物から構成される。適した調製方法は、ポリマー性ナノ粒子に関する上記の参考文献にあるものと同様である。誘導体化された一本鎖ポリマーとは、生物活性物質との共有結合によってポリマー-薬剤結合物を形成するために適合化されたポリマーである。ポリアミノ酸、多糖類(デキストリンもしくはデキストランなど)および合成ポリマー(N-(2-ヒドロキシプロピル)メタクリルアミド(HPMA)共重合体など)を含むさまざまなポリマーが、ポリマー-薬剤結合物の合成用に提唱されている。適した調製法は、VeroneseおよびMorpurgo、IL Farmaco (1999) 54(8): 497-516に詳述されており、これは参照として本明細書に組み入れられる。
【0041】
このように、送達媒体は、治療成分の投与された比の一貫した送達が達成されるように提供される。すなわち、比は、組成物を含む媒体中への薬剤の単純な共封入によって維持してもよく、または、非拮抗的な薬剤の比を同様に維持するように媒体が組成物の薬物動態を制御する場合には、薬剤を別個の媒体中に封入することもできる。
【0042】
本発明の組成物は、拮抗的でない複数の抗腫瘍薬の組成物を送達するために用いることが好ましい。以下の詳細な説明は、治療薬の比を決定する様式、および、本発明の送達システム中に所望の比を封入するための方法を示している。
【0043】
簡潔に述べると、1つのシナリオでは、まず個々の薬剤を種々のインビトロアッセイ法またはインビボアッセイ法で別個にスクリーニングして個々の活性を決定する。続いて、薬剤の対を組み合わせて、同じスクリーニング法でアッセイする。この初期スクリーニングでは、薬剤の比は、以前に同定した50%の活性を示す濃度(IC50値)でのモル比である。または、製剤の目的に関する考察に基づき、その他の固定比(一般的にはモル比1:10、1:1および10:1)を選択する。細胞の生存に対する薬剤の効果に基づいて算出した平均値および薬剤の用量をCalcuSynコンピュータプログラムに入力し、出力データを、影響を受けた細胞の率(fa)の関数として組合せ指数(CI)値を定めるために評価する。
【0044】
CalcuSyn法は、抗腫瘍薬、臓器移植用の免疫抑制剤、自家骨髄移植のための白血病細胞の複合的パージング、殺虫薬、生体応答修飾物質、多剤耐性阻害薬、抗菌薬、抗HIV薬、抗ヘルペス薬およびその他の抗ウイルス薬などの種々の薬剤の試験を行うために首尾良く適用されている。
【0045】
図11Aにおけるシスプラチン:ダウノルビシンのモル比1:1での薬剤に類似した相互作用挙動を示す薬剤の組合せは拮抗的であり、追求の対象とならない。fa>0.01であるfa値のかなりの範囲(好ましくは少なくとも約20%)にわたって非拮抗的な相互作用がみられる化合物の組合せ(すなわち、モル比1:1および10:1のイリノテカン:カルボプラチン;図2A)を、非拮抗的な相互作用の強度(すなわち、低いCI値)を高めるとともに相乗作用が観察されるfaの範囲を拡げる最適な比を明確にするために、さまざまな異なる薬剤/薬剤比で、このインビトロスクリーニングアッセイ法において再び評価する。
【0046】
このようにして同定された、最適化された非拮抗的な薬剤の組合せは、二剤(dual-agent)組成物として送達媒体中にある製剤に関する組成物を規定し、および/または第3の薬剤との相乗的または相加的な相互作用を決定するための単一の薬剤単位として用いることができる。
【0047】
非拮抗的な比のインビトロでの決定
本発明の組成物を調製するためには、送達媒体中に含まれる薬剤の所望の比をまず決定する必要がある。この比は、相乗作用または相加作用が組合せによってある濃度範囲にわたって示されるものであることが望ましい。このような比は、インビトロの細胞培養物または無細胞系においてさまざまな数学モデルを用いて決定することができる。
【0048】
相乗的または相加的な併用効果を濃度範囲にわたって示す薬剤の比の決定は、以下に述べる実験データのタイプに応じて、種々のアルゴリズムを用いて行うことができる。これらの方法には、アイソボログラム法(Loeweら、Arzneim-Forsch (1953) 3: 285-290;Steelら、Int. J. Radiol. Oncol. Biol. Phys. (1979) 5: 27-55)、分画産物法(Webb, 「酵素および代謝阻害物質(Enzyme and Metabolic Inhibitors)」(1963)第1巻、pp.1-5. New York: Academic Press)、モンテカルロシミュレーション法、CombiTool、ComboStat、ならびにChou, J. Theor. Biol. (1976) 39: 253-76;およびChou, Mol. Pharmacol. (1974) 10: 235-247に記載された式に基づくChou-Talalayのメディアンエフェクト法が含まれる。それに代わるものには、生存率(Zoliら、Int. J. Cancer (1999) 80: 413-416)、対照と比較した顆粒球/マクロファージ-コロニー形成単位の反応率(Pannacciulliら、Anticancer Res. (1999) 19: 409-412)およびその他(Berenbaum、Pharmacol. Rev. (1989) 41: 93-141;Grecoら、Pharmacol Rev. (1995) 47: 331-385)が含まれる。
【0049】
Chou-Talalayのメディアンエフェクト法が好ましい。この分析には、特定の影響faを引き起こす用量が
D=Dm[fa/(1-fa)]1/m
によって与えられる式が用いられ、ここでDは用いる薬剤の用量であり、faはその用量による影響を受けた細胞の率であり、Dmは効力を表す中位効果(median effect)であり、mは用量-効果曲線の形状を表す係数である(一次反応ではmは1である)。
【0050】
この式をさらに操作して、ChouおよびTalalay、Adv. Enzyme Reg. (1984) 22: 27-55;ならびにChouら、「化学療法における相乗作用および拮抗作用(Synergism and Antagonism in Chemotherapy)」、ChouおよびRideout編、Academic Press: New York 1991: 223-244に記載された多剤効果式に基づいて組合せ指数(CI)を計算することができる。この計算用のコンピュータプログラム(CalcuSyn)は、Chou、「マイクロコンピュータによる用量-効果分析:ED50、LD50、相乗作用、拮抗作用、低用量リスク、受容体リガンド結合および酵素反応速度論(Dose-effect analysis with microcomputers: quantitation of ED50, LD50, synergism, antagonism, low-dose risk, receptor ligand binding and enzyme kinetics)」(CalcuSynマニュアルおよびソフトウエア;Cambridge:Biosoft 1987)において見出すことができる。
【0051】
組合せ指数の式は、酵素反応速度論モデルに由来するChou-Talalayの多剤効果式に基づく。ある式は相乗作用および拮抗作用ではなく相加作用のみを決定する。しかし、CalcuSynプログラムによれば、相乗作用は期待される相加作用を上回るものとして、拮抗作用は期待される相加作用を下回るものとして定義される。ChouおよびTalalayは1983年にCI=1の指定を相加作用として提唱しており、このため本発明者らは、2種類の薬剤の多剤効果式から、同じまたは類似の作用機序を有する排反的な薬剤に関して
CI=(D)1/(Dx)1+(D)2/(Dx)2 [式1]
を得ており、さらに、完全に独立した作用機序を有する排反的でない薬剤に関して
CI=(D)1/(Dx)1+(D)2/(Dx)2+(D1)(D2)/(Dx)1(Dx)2 [式2]
を得ている。CI<1、=1および>1はそれぞれ相乗作用、相加作用および拮抗作用を示す。式1または式2は、併用した薬剤1、(D)1および薬剤2、(D)2(分子にあるもの)が、実際の実験でx%を阻害することを指示している。このため、実験的に観察される阻害率x%は、概数ではなくて小数部を有する可能性が高い。式1および2の(D)1および(D)2(分母にあるもの)は、それぞれ薬剤1および薬剤2が単独でx%を阻害する用量である。
【0052】
単純化のために、排反性は通常、3種類以上の薬剤を組合せに用いる場合に仮定される(CalcuSynマニュアルおよびソフトウエア;Cambridge:Biosoft 1987)。
【0053】
基礎をなす実験データは一般に、培養下の細胞または無細胞系を用いてインビトロで決定される。組合せ指数(CI)は、図1に示されているように、上に説明した通りに濃度範囲の代用パラメーターである、影響を受けた細胞の率(fa)の関数としてプロットすることが好ましい。好ましい薬剤の組合せは、fa値のかなりの範囲にわたって相乗作用または相加作用を示すものである。細胞の1%超が影響を受ける濃度範囲、すなわちfaが0.01を上回る範囲の少なくとも5%にわたって相乗作用を示す薬剤の組合せを選択する。全濃度のうちより多くの割合、例えば、faが0.2〜0.8である範囲の5%が、好都合なCIを示すことが好ましい。より好ましくは、この範囲の10%が好都合なCIを示す。さらにより好ましくは、fa範囲の20%、好ましくは50%超、最も好ましくはfaが0.2〜0.8である範囲の少なくとも70%超が組成物において用いられる。fa値のかなりの範囲にわたって相乗作用を示す組合せを、非拮抗的相互作用の強度を高め、相乗作用が観察されるfaの範囲を拡げる最適な比を定めるために、種々の薬剤比で再び評価することができる。
【0054】
細胞が影響を受ける濃度範囲の全体にわたって相乗作用がみられることが望ましいが、多くの場合には、0.2〜0.8のfa範囲における結果の信頼性の方がかなり高い。このため、本発明の組合せによって示される相乗作用は0.01またはそれ以上という広い範囲内に存在することは述べておくが、相乗作用は0.2〜0.8のfa範囲において実証されることが好ましい。
【0055】
最適な組合せ比を、さらに、第3の薬剤との相乗的または相加的な相互作用を決定するための単一の薬剤単位として用いることもできる。加えて、3剤組合せを、第4の薬剤との非拮抗的な相互作用を決定するための単位として用いることもでき、以降も同様である。
【0056】
上記の通り、細胞培養物に対するインビトロ試験は、「妥当な」細胞を用いて行われると考えられる。細胞の選択は、薬剤の意図した治療用途に依存すると考えられる。組成物を本発明の範囲に含めるための基盤を得るために、要求される非拮抗的な作用を示す必要があるのは、ただ1つの妥当な細胞系または細胞培養物のみでよい。
【0057】
例えば、本発明の1つの好ましい態様において、薬剤の組合せは抗癌療法を目的とする。続いて、試験を行う細胞および試験の性質に対して適した選択を行う。詳細には、腫瘍細胞株が適した被験対象であり、細胞死または細胞分裂停止の計測が適切なエンドポイントである。以下にさらに考察するように、他の適応症に適した非拮抗的な薬剤の組合せを見いだそうとする状況下では、他の標的細胞、および細胞傷害性または細胞分裂停止以外の基準を用いうると考えられる。
【0058】
抗腫瘍薬にかかわる決定のための細胞系は、標準的な細胞系収集機関(例えば、NCIまたはATCC)から、学術機関から、または市販の供給元を含む他の組織から入手しうる。好ましい細胞系には、NCI/NIHの開発中治療薬プログラム(Developmental Therapeutics Program)によって同定された細胞系から選択された1つまたは複数が含まれると考えられる。現在、このプログラムによって用いられている腫瘍細胞株スクリーニングでは、白血病、黒色腫ならびに肺癌、結腸癌、脳腫瘍、卵巣癌、乳癌、前立腺癌および腎癌を代表する60種類のヒト腫瘍細胞株が同定されている。所望の濃度範囲にわたって必要な非拮抗的な作用が示される必要があるのは単一の細胞種のみである;しかし、少なくとも2種類の細胞系、より好ましくは3種類の細胞系、より好ましくは5種類の細胞系、より好ましくは10種類の細胞系がこの作用を示すことが好ましい。細胞系は樹立された腫瘍細胞株でもよく、または患者試料から得られた初代培養物でもよい。細胞系は任意の種に由来するものでよいが、好ましい由来は哺乳動物、特にヒトであると考えられる。細胞系は、種々の実験条件下での選択、および/または外因性遺伝物質の付加もしくは欠失によって遺伝的に改変されたものでもよい。細胞系は、ウイルスまたはプラスミドを用いるトランスフェクション法を非制限的に含む任意の遺伝子導入法によるトランスフェクションを受けたものでもよい。改変には、特定のタンパク質もしくはペプチドの発現をコードするcDNA、プロモーターもしくはエンハンサー配列などの調節因子、またはアンチセンスDNAもしくはRNAの導入が含まれうる。遺伝的に操作された組織培養細胞系には、腫瘍抑制遺伝子、すわなちp53、pTENおよびp16などの遺伝子を有する系または有しない系;ならびに、ドミナントネガティブ法、遺伝子挿入法および他の選択方法を用いて作製された系が含まれうる。例えば抗腫瘍薬の試験のために、細胞の生存度を定量するために用いうる好ましい組織培養細胞系には、H460、MCF-7、SF268、HT29、HCT-116、LS 180、B16-F10、A549、Capan膵細胞、CAOV-3、IGROV1、PC3、MX-1およびMDA-MB-231が非制限的に含まれる。
【0059】
1つの好ましい態様においては、所定の効果(fa)とは、細胞培養物に対する細胞傷害物質の適用後の細胞死または細胞分裂停止のことを指す。細胞死または生存度は、例えば、以下の方法を用いて測定しうる。
【0060】
「MTTアッセイ法」が好ましい。
【0061】
癌以外の適応疾患に対しても2種類またはそれ以上の薬剤の非拮抗的な比を決定することができ、この情報を、これらの疾患の治療のために2種類またはそれ以上の薬剤による治療的製剤を調製する目的で用いることができる。インビトロアッセイ法に関しては、薬物の相乗作用を明確化するために計測可能なさまざまなエンドポイントを、そのエンドポイントがその特定の疾患に対して治療的に妥当であるという条件付きで選択することができる。
【0062】
したがって、例えば、当業者は、炎症性疾患の治療のための2種類またはそれ以上の薬剤の非拮抗的な比を、IL-1、IL-18、COX-2、TNFまたはインターフェロンγなどの炎症誘発性サイトカインの抑制をインビトロで測定することによって選択することができる。他の炎症性シグナルには、プロスタグランジンE2およびトロンボキサンB2の阻害が非制限的に含まれる。特に、エンドトキシンを介したマクロファージ活性化は、添加した薬剤または薬剤の組合せの抗炎症作用を測定するのに適したインビトロアッセイ法をもたらし、当技術分野でよく用いられている。この種のアッセイ法では、大量に増殖させたマクロファージを、リポ多糖などのエンドトキシンの添加によって活性化する。活性化を行った上で、マクロファージによるIL-1およびTNFなどのサイトカインの分泌、ならびにCOX-2の活性化を測定する。抗炎症性薬剤の候補を添加し、それらがIL-1、TNFおよびCOX-2を抑制する能力を評価する。1×10-7Mデキサメタゾンによる滴定が陽性対照として一般に用いられる。当業者には、マクロファージ活性化を用いるアッセイ法が薬剤の組合せの広域スクリーニングに適していること、ならびにIL-1、TNFおよびCOX-2の抑制が相乗作用を明確にするのに適したエンドポイントであることが明らかであると考えられる。炎症性シグナルを測定することに加えて、研究者は、白血球機能に対する2種類またはそれ以上の薬剤の効果を計測するためのインビトロモデルの使用を検討することもできる。機能試験には、脱顆粒の阻害、スーパーオキシドの生成および白血球の遊走が非制限的に含まれる。
【0063】
癌と同様に、増殖は、動脈硬化、再狭窄、または血管増殖性を伴う他の心血管疾患の発症においても重要な事象である。このため、当業者は、2種類またはそれ以上の薬剤の非拮抗的な比を、妥当な血管の増殖性細胞集団に対して適用した本明細書に記載の方法によって薬物の相乗作用を評価することにより、見いだすことができる。特に、通常は血管形成術後に生じる冠動脈再狭窄および末梢動脈再狭窄などの再狭窄は、平滑筋および内皮細胞の増殖に起因する(Fuster, Arch Mal Coeur Vaiss (1997) 90 Spec No 6: 41-47)。本明細書に記載の標準的な方法を用いて、当業者は、2種類またはそれ以上の薬剤が内皮細胞または平滑筋細胞の増殖を阻害するように非拮抗的に作用するか否かを評価することができる。これらのアッセイ法は、不死化細胞系を用いて、または好ましくは初代細胞系を用いて行いうる。これらの細胞系は、市販の供給元(例えば、Clonetics、California)から、または新鮮な組織(例えば、臍帯静脈、動脈、脳)から入手可能であるが、細胞増殖を促進する適切な増殖因子の存在下で維持する必要がある。癌細胞に対する2種類またはそれ以上の薬剤の相乗作用を測定するアッセイ法と同様に、この種のアッセイ法には、増殖および遊走の阻害というエンドポイントが非制限的に含まれる。増殖エンドポイントは、本出願書に記載のMTTアッセイ法などの生/死アッセイ法、[3H]-チミジン取込みに依拠した増殖の測定、または他の類似のアッセイ法に依拠しうる。同じく分裂性癌細胞と同様に、内皮細胞および平滑筋細胞の増殖は細胞周期におけるチェックポイントによって調節されており、細胞周期の阻害を測定するアッセイ法は、血管増殖性疾患の治療のために選択された2種類またはそれ以上の薬剤の非拮抗的な比を明確にするために用いることができる。
【0064】
薬剤の非拮抗的な組合せを、微生物またはウイルスの感染に対する活性の点から同定することもできる。抗菌薬の同定における第1の段階として、薬剤に関する最小発育阻止濃度(MIC)を、当業者に知られたマイクロタイター液体希釈法または寒天希釈抗菌アッセイ法によって決定することができる。これらのアッセイ法は、実験の安全性および標準に関する国家委員会(National Committee of Laboratory Safety and Standards)(NCLSS)による規制を受けている。標準的な液体希釈アッセイ法は、「実験医学における抗生物質(Antibiotics in Laboratory Medicine)」、Lorian, V. 第4版、p 52-111, WilliamsおよびWilkins、Baltimore中のAmsterdam (1996) 「液体培地における抗菌物質の感受性試験(Susceptibility testing of Antimicrobials in liquid media)」に発表されている。MICは、感染性微生物のインビトロ増殖を阻止すると考えられる抗生物質の最低濃度と定義される。上記のアッセイ法では、微生物の接種物を、種々の濃度の薬剤を含む増殖培地(例えば、寒天)上の小さなスポットに微生物の接種物を播くことにより(例えば、1スポット当たり104コロニー形成単位[CFU])、MICを決定しうる。または、種々の濃度の薬剤を含む増殖培地の懸濁液に微生物を接種することもできる。さらに、微生物は上記の通りに処理してもよく、または特定細胞集団(すなわち、マクロファージ)における細胞内感染として存在してもよい。後者の場合には、標準的な方法によって培養下で増殖させた哺乳動物細胞を低濃度の微生物に対して短時間曝露させることにより、細胞内微生物感染を行わせる。微生物の細胞内複製を行わせる期間をおいた後に、細胞およびその細胞内微生物を、哺乳動物細胞を用いる細胞傷害性試験に関して記載されているのと同じ方式で薬剤によって処理する。有効濃度で投与した薬剤が微生物の増殖を阻止するのに十分な、適切な期間の後に、細菌増殖を以下を含む種々の手段によって判定することができる:(i)接種スポットの有無(およびサイズ、適宜)の判定;(ii)処理に対して生存し得た微生物の数の算出を可能にするための、寒天増殖プレート上への処理細菌のプレーティングおよび既知の容積の浮遊液の連続希釈;(iii)顕微鏡的(肉眼による)な判定;(iv)対数増殖期にある微生物を薬剤を含む増殖培地中に浮遊させ、接種後の種々の時点で既知の容積を採取し、連続希釈した上で、生存した微生物の算定のために増殖寒天上にプレーティングして得た、時間-死滅曲線;(v)生存している微生物の算定が可能な、当業者に知られた他の分光学的、分析的、インビトロまたはインビボの方法。単一の薬剤または薬剤の組合せが細胞内に存在する感染物を死滅させる有効性は一般に、微生物を放出させるために宿主細胞を界面活性剤(1%Triton X-100に0.1%ドデシル硫酸ナトリウムを加えたもの)で可溶化した後に、生存している微生物の数を算定するために可溶化物を連続希釈して寒天増殖プレートに播くことによって評価される。
【0065】
上記の手段を用いて、有効な薬剤の組合せを、その拮抗活性、相加活性または相乗的活性に関して評価する。具体的には、化合物の対を固定比(これは等モルでも、MIC値の比でも、または他の固定比でもよい)として細菌に適用し、細菌を種々の濃度の化合物対によって処理する。活性は上記の通りに決定する。拮抗作用、相加作用または相乗作用をさまざまな数学的処理、例えばアイソボログラム、CIなどによって決定する。
【0066】
抗ウイルス活性を備えた薬剤または薬剤の組合せに対する広範囲のスクリーニングは、さまざまなインビトロアッセイ法、一般的にはプラーク減数アッセイ法および細胞変性効果(CPE)阻止アッセイ法によって行うことができ、これらは当業者に周知である。これらのアッセイ法は、1つまたは複数の抗ウイルス薬が組織培養下でのウイルス感染の影響を阻止する程度を直接測定することができる。プラーク減数アッセイ法は、明確なプラークを生じるウイルスおよび細胞系の組合せに対して行われる。Michaelisらは、ウイルス数に対するアフィジコリンおよびその誘導体の非拮抗的な抗ウイルス効果を種々のモル比で決定するための、プラーク減数アッセイ法とChou-Talalay法との併用を示した(Michaelisら、Arzneimittelforschung (2002) 52(5): 393-399)。特定のウイルスおよび細胞系の組合せで明確なプラークが生じない場合には、CPE阻止アッセイ法が好ましい。抗ウイルス薬の非拮抗的な組合せを迅速かつ簡便に同定するためのそのほかの方法には、細胞生存度、ウイルス収量アッセイ法およびHIV急性または慢性感染アッセイ法が非制限的に含まれる。細胞生存度は抗ウイルス薬または薬剤の組合せが細胞の生存度を高める能力を測定するために用いられ、前記のMTTアッセイ法などの定量的アッセイ法を用いて行うことができる。または、ウイルス収量アッセイ法および急性HIV感染アッセイ法は、薬剤がウイルス収量を減少させる能力を評価し、抗ウイルス活性の直接測定を可能にする。前記のアッセイ法が抗ウイルス薬剤の組合せを相乗的、相加的または拮抗的な作用に関してインビトロでスクリーニングするのに適していることは当業者には明らかであると考えられ、このため、本発明の範囲に含まれる。
【0067】
好ましい薬剤の組合せ
上記の非拮抗的な作用に関する基準を満たすことが判明した治療薬のさまざまな組合せは、その後、薬物送達媒体の配合物の形態で提供される。「治療薬」とは、単独で、または他の化合物との組合せで、望ましくない状態または疾患に冒された対象に対して望ましい効果を有する、化合物のことである。
【0068】
標的疾患が癌である場合、ある種の治療薬は併用するのに好都合である。その例には以下のものがある。
「シグナル伝達阻害物質」、これは癌細胞が増殖または分裂する原因になるシグナルを妨げる、またはそれを阻止する;
「細胞傷害物質」;
「細胞周期阻害物質」または「細胞周期制御阻害物質」、これは細胞が正常な細胞周期、すなわち細胞の生涯を、それを派生させた有糸分裂から、分裂して娘細胞を生じさせる有糸分裂後の事象へと進行させることを妨げる;
「チェックポイント阻害物質」、これは細胞周期チェックポイント、例えば、S/G2チェックポイント、G2/MチェックポイントおよびGl/Sチェックポイントの正常な機能を妨げる;
「トポイソメラーゼ阻害物質」、これにはカンプトテシンなどがあり、DNAの複製および転写に必要な酵素であるトポイソメラーゼIまたはIIの活性を妨げる;
「受容体チロシンキナーゼ阻害物質」、これは、チロシンキナーゼ活性を有する増殖因子受容体の活性を妨げる;
「アポトーシス誘導物質」、これはプログラム細胞死を促進する;
「代謝拮抗物質」、これにはゲムシタビンまたはヒドロキシ尿素などがあり、必須代謝産物とよく似ているために、それがかかわる生物反応を妨げる;
「テロメラーゼ阻害物質」、これは、テロメア長を延長させて細胞の寿命およびその複製能力を向上させる酵素であるテロメラーゼの活性を妨げる;
「サイクリン依存性キナーゼ阻害物質」、これは、ヒストン、細胞骨格タンパク質、転写因子、腫瘍抑制遺伝子といった細胞タンパク質のリン酸化を介して細胞周期の異なる時期間の主要な段階を制御するサイクリン依存性キナーゼを妨げる;
「DNA損傷物質」;
「DNA修復阻害物質」;
「血管形成抑制物質」、これは腫瘍増殖時に起こる新たな血管の形成または既存の血管の成長を妨げる;および
「ミトコンドリア毒」、これはミトコンドリアの呼吸鎖機能を直接的または間接的に破壊する。
【0069】
腫瘍の治療のために特に好ましい組合せは、上記の臨床的に承認された組合せである。これらの組合せはすでにヒトでの使用が承認されているため、適切な送達を保証するための再製剤化(reformation)が特に重要である。
【0070】
併用しうる好ましい薬剤には、以下のものが含まれる:カルボプラチン、シスプラチン、シクロホスファミド、ドキソルビシン、ダウノルビシン、エピルビシン、マイトマイシンC、ミトキサントロンなどのDNA損傷物質;5-フルオロウラシル(5-FU)またはFUDR、ゲムシタビンおよびメトトレキサートを含むDNA修復阻害物質;カンプトテシン、イリノテカンおよびトポテカンなどのトポイソメラーゼI阻害物質;ブレオマイシン、ドセタキセル、ドキソルビシン、エトポシド、パクリタキセル、ビンブラスチン、ビンクリスチン、ビンデシンおよびビノレルビンなどのS/G2またはG2/Mチェックポイント阻害物質;G1/S初期チェックポイント阻害物質;G2/Mチェックポイント阻害物質;ゲニステイン、トラスツズマブ(trastuzumab)、ZD1839などの受容体チロシンキナーゼ阻害物質;細胞傷害物質;アポトーシス誘導物質および細胞周期制御阻害物質。
【0071】
薬剤の1つまたは複数の作用機序が不明であってもよく、または誤って同定されていてもよい。相乗的または相加的な薬剤の組合せはすべて本発明の範囲に含まれる。新生物の治療のためには、無秩序な細胞増殖につながる複数の機序を阻害する組合せを、本発明に従って用いるために選択することが好ましい。例えば、本発明は、細胞周期における特定の箇所に影響を及ぼし、それによって非拮抗的な作用を生じる組合せを選択することを含む。例えば、DNA損傷を引き起こす薬剤をDNA修復を阻害するもの、例えば代謝拮抗物質などと対にする。本発明はまた、通常であれば細胞増殖をもたらすと考えられる複数の経路を阻止する組合せを選択することも含む。
【0072】
特に好ましい組合せは、DNA損傷物質とDNA修復阻害物質との組合せ、DNA損傷物質とトポイソメラーゼIまたはトポイソメラーゼII阻害物質との組合せ、トポイソメラーゼI阻害物質とS/G2またはG2/Mチェックポイント阻害物質との組合せ、G1/Sチェックポイント阻害物質またはCDK阻害物質とG2/Mチェックポイント阻害物質との組合せ、受容体チロシンキナーゼ阻害物質と細胞傷害物質との組合せ、アポトーシス誘導物質と細胞傷害物質との組合せ、アポトーシス誘導物質と細胞周期制御阻害物質との組合せ、G1/SまたはG2/Mチェックポイント阻害物質と細胞傷害物質との組合せ、トポイソメラーゼIまたはII阻害物質とDNA修復阻害物質との組合せ、トポイソメラーゼIもしくはII阻害物質またはテロメラーゼ阻害物質と細胞周期制御阻害物質との組合せ、トポイソメラーゼI阻害物質とトポイソメラーゼII阻害物質との組合せ、および2種類の細胞傷害物質の組合せである。
【0073】
併用しうる具体的な薬剤には、以下のものが含まれる:シスプラチン(またはカルボプラチン)と5-FU(またはFUDR)、シスプラチン(またはカルボプラチン)とイリノテカン、イリノテカンと5-FU(またはFUDR)、ビノレルビンとシスプラチン(またはカルボプラチン)、メトトレキサートと5-FU(またはFUDR)、イダルビシンとaraC、シスプラチン(またはカルボプラチン)とタキソール、シスプラチン(またはカルボプラチン)とエトポシド、シスプラチン(またはカルボプラチン)とトポテカン、シスプラチン(またはカルボプラチン)とダウノルビシン、シスプラチン(またはカルボプラチン)とドキソルビシン、シスプラチン(またはカルボプラチン)とゲムシタビン、オキサリプラチンと5-FU(またはFUDR)、ゲムシタビンと5-FU(またはFUDR)、アドリアマイシンとビノレルビン、タキソールとドキソルビシン、フラボプリドールとドキソルビシン、UCN01とドキソルビシン、ブレオマイシンとトリクロロペラジン、ビノレルビンとエデルフォシン、ビノレルビンとスフィンゴシン(およびスフィンゴシン類似体)、ビノレルビンとホスファチジルセリン、ビノレルビンとカンプトテシン、シスプラチン(またはカルボプラチン)とスフィンゴシン(およびスフィンゴシン類似体)、スフィンゴシン(およびスフィンゴシン類似体)とダウノルビシン、ならびにスフィンゴシン(およびスフィンゴシン類似体)とドキソルビシン。
【0074】
好ましい組合せには一般に、臨床の場で有効なことがFDAによる認識ですでに示されている上記のもの、および文献報告に基づいてそれが示唆されるものが含まれる。本発明の方法に用いるための候補薬剤はこれらの特定の組合せには限定されないが、上記のものは、適した併用療法として開示されており、このため、本発明の方法および組成物に用いるのに好ましい。
【0075】
脂質の中には、アポトーシスの誘導といった治療的な作用を発揮しうる「治療的脂質」がある。この定義に含まれるものには、エーテル脂質、ホスファチジン酸、ホスホネート、セラミドおよびセラミド類似体、ジヒドロキシセラミド、フィトセラミド、スフィンゴシン、スフィンゴシン類似体、スフィンゴミエリン、セリン含有脂質およびスフィンガニンなどの脂質がある。本明細書において定義される「セリン含有リン脂質」または「セリン含有脂質」などの用語は、一方の端でセリンと共有結合し、他方の端で、エーテル結合、エステル結合またはアミド結合を介して疎水性部分と連結した三炭素骨格と共有結合しているリン酸基を頭部極性基が含む、リン脂質のことである。このクラスに含まれるものには、長さが5〜23炭素原子で種々の程度に飽和している2つの炭化水素鎖が疎水性部分に存在するホスファチジルセリン(PS)などのリン脂質がある。セリン含有リン脂質またはセリン含有脂質に言及する場合の疎水性部分という用語は、長鎖飽和または不飽和脂肪族炭化水素鎖(選択的には1つまたは複数の芳香族、脂環式または複素環式原子団によって置換されたもの)などの非極性基のことを指す。
【0076】
治療的脂質および他の薬剤の組合せを、相乗的または相加作用を達成するために用いることもできる(実施例17〜21を参照)。
【0077】
非拮抗的な併用効果を示す比を決定するためのハイスループットスクリーニング
薬剤の新規な非拮抗的組合せを同定するために、薬剤の化学物質ライブラリーを互いにさまざまな比でスクリーニングすることができる。化学物質ライブラリーは新規な薬剤を含んでも従来の薬剤を含んでもよい。2種類の薬剤の組合せに関するスクリーニングに加えて、3種類または4種類の薬剤の組合せを非拮抗的な併用効果に関してスクリーニングすることもできる。薬物の相乗作用を決定するために用いるデータ解析法は、前記のメディアンエフェクト解析(Median Effect Analysis)であることが好ましい。この方法によれば、薬剤のライブラリーを、個別に、および種々の比で組み合わせて検討する。続いて、ChouおよびTalalayによって開発された前記の方法を用いて組合せ指数を算出する。特定の比で非拮抗的な作用を示す薬剤の組合せを送達媒体中に非拮抗的な比で封入する。
【0078】
ハイスループットスクリーニングシステムは市販されている(例えば、Zymark Corp., Hopkinton, Mass.;Air Technical Industries, Mentor, Ohio;Beckman Instruments, Inc. Fullerton, Calif.;Precision Systems, Inc., Natick, Mass.などを参照されたい)。これらのシステムは通常、すべての試料および試薬のピペット操作、液体の分注、定めた時間のインキュベーション、ならびにアッセイ法に適した検出器によるマイクロプレートの最終的な読み取りを含む、全手順を自動化している。適合性のあるこれらのシステムは、ハイスループットかつ始動が迅速である上に、高度の汎用性およびカスタマイズ性を備えている。この種のシステムの製造者は、種々のハイスループットスクリーニング方法に関する詳細なプロトコールを用意している。
【0079】
非拮抗的な組成物の調製
薬剤の適切な比が上記のようにして決定されれば、1つまたは複数の送達媒体が2種類またはそれ以上の薬剤を封入するように、薬剤をその適切な比で送達媒体組成物中に収める。組成物中のすべての送達媒体が同一である必要はない。組成物中の送達媒体は粒子であり、そのサイズは投与経路に依存し、本発明の薬剤を封入しうる水性溶媒または他の溶媒中に懸濁化されうる。このような媒体には、例えば、前述した、脂質担体、リポソーム、シクロデキストリン、ポリマー性ナノ粒子およびポリマー性微粒子(ナノカプセルおよびナノスフェアを含む)、ブロック共重合体ミセル、脂質安定化エマルション、誘導体化された一本鎖ポリマー、ポリマー脂質ハイブリッド系、脂質ミセル、リポタンパク質ミセルが含まれる。静脈内投与のための送達媒体は一般に直径約4〜6,000nmである。好ましい直径は直径約5〜500nm、より好ましくは直径5〜200nmである。吸入投与、クモ膜下腔内投与、関節内投与、動脈内投与、腹腔内投与または皮下投与のための送達媒体は一般に4μmから50μmを超える範囲である。眼内投与用に設計される送達媒体組成物は一般にそれよりも小さい。
【0080】
治療薬は送達媒体中に「封入」される。「封入」には、前記の通り、薬剤と送達媒体との共有結合性または非共有結合性の会合が含まれる。例えば、これは薬剤と送達媒体の1つもしくは複数の外層との相互作用、または送達媒体内部への封じ込め、送達媒体の異なる部分間で実現される平衡によるものであってよい。例えば、リポソームの場合、薬剤の封入は、脂質成分との共有結合性もしくは非共有結合性の相互作用を介したリポソームの二重層との相互作用による薬剤の会合、またはリポソームの水性内部への封じ込め、または内部水相と二重層との間の平衡によるものであってよい。ポリマーを基盤とする送達媒体の場合、封入は薬剤と線状もしくは非線状ポリマーとの共有結合のことを指す。さらに、非制限的な例には、ポリマー基質全体への薬剤の分散、またはナノカプセル、ブロック共重合体ミセルもしくはポリマー-脂質ハイブリッド系のコアへの薬剤の濃縮が含まれる。「装填(loading)」とは、1つまたは複数の薬剤を送達媒体中に封入する作業のことを指す。
【0081】
所望の組合せの封入は、別個の送達媒体中への封入、または同じ送達媒体の内部への封入のいずれによって行うこともできる。リポソームなどの別個の送達媒体中への封入が望まれる場合には、各リポソームの脂質組成は協調的な薬物動態が可能となる程度に大きく異なってもよい。媒体組成を変更することにより、封入された薬剤の放出速度を、非拮抗的な比の薬剤が腫瘍部位に送達されることが可能となるように一致させることができる。放出速度を変更する手段には、薬剤の保持性を改良するために小胞を形成する脂質のアシル鎖の長さを延長すること、リポソーム膜から突出したPEGなどの表面接枝状親水性ポリマーの交換を制御すること、およびステロールまたはスフィンゴミエリンなどの膜硬化性物質を膜に組み入れることが含まれる。第1および第2の薬剤を特定の薬剤比で投与したい場合、ならびに、第2の薬剤が第1の薬剤のリポソーム組成物(例えば、DMPC/Chol)の内部にほとんど保持されない場合には、アシル鎖長がさらに長い脂質(例えば、DSPC/Chol)リポソーム組成物中に第2の薬剤を封入することによって薬物動態を改良しうることは、当業者には明らかであると考えられる。または、2種類またはそれ以上の薬剤を同じ送達媒体中に封入してもよい。
【0082】
封入のための技法は送達媒体の性質に依存する。例えば、受動的な装填方法および能動的な装填方法の両方を用いてリポソーム中に治療薬を装填することができる。
【0083】
薬剤をリポソーム中に封入するための受動的方法は、リポソームの調製中に薬剤を封入することを含む。この方法では、薬剤は膜に会合させてもよく、封じ込めた水性空間内に封入してもよい。これには、目的の薬剤を含む水相を、反応容器の壁面に付着させた乾燥した小胞形成性脂質の薄膜と接触させる、Banghamら、J. Mol. Biol. (1965) 12: 238に記載された受動的封じ込め法が含まれる。機械的手段による振盪を加えると、脂質の膨潤が起こって多重ラメラ小胞(MLV)が形成されると考えられる。押出処理法を用いて、MLVを大型単ラメラ小胞(LUV)または小型単ラメラ小胞(SUV)へと変換することができる。用いうるもう1つの受動的装填方法には、DeamerおよびBangham、Biochim. Biophys. Acta (1976) 443: 629に記載されたものが含まれる。この方法では、小胞形成性脂質をエーテル中に溶解した上で、まずエーテルを蒸発させて表面に薄膜を形成させ、その後にこの薄膜を封入しようとする水相と接触させる代わりに、エーテル溶液を前記水相中に直接注入した後にエーテルを蒸発させ、それによって薬剤が封入されたリポソームを得る。用いうるさらにもう1つの方法は、SzokaおよびPapahadjopoulos, P.N.A.S. (1978) 75: 4194に記載された逆相蒸発(REV)法であり、この方法では水に溶けない有機溶媒中にある脂質溶液を水性担体相中に乳濁化し、その後に減圧下で有機溶媒を除去する。
【0084】
用いうるその他の受動的封じ込め法には、リポソームに対して連続的な脱水処理および再水和処理、または凍結解凍を行うことが含まれる。脱水は蒸発または凍結乾燥によって行われる。この技法はKirbyら、Biotechnology (1984) 979-984に記載されている。同じく、ShewおよびDeamer(Biochim. et Biophys. Acta (1985) 816: 1-8)は、超音波処理によって調製したリポソームを、封入しようとする溶質を含む水性溶液と混合し、その混合物を回転フラスコ内で窒素中にて乾燥させる方法を記載している。再水和を行うと、溶質のかなりの割合が封入された大型のリポソームが生成される。
【0085】
2種類またはそれ以上の薬剤の受動的封入は、薬剤の数多くの組合せに対して可能である。このアプローチは水性緩衝液中への薬剤の溶解性によって制限され、薬剤の多くの割合が送達システム内部に捕捉されないという制限もある。装填性は、薬剤を脂質試料と同時凍結乾燥すること、および薬剤を溶解するための最少容積で再水和を行うことによって改良されることがある。溶解性は、緩衝液のpHを変更すること、温度を高めること、または緩衝液に対する塩の添加もしくは除去によって改良されることがある。
【0086】
能動的な封入方法を用いてもよい。例えば、金属錯体化またはpH勾配装填法に従ってリポソームに装填することができる。pH勾配装填法では、選択したpHの水相を封入するリポソームを形成させる。次に、水和リポソームを、薬剤または封入しようとする他の作用物質の荷電を除去もしくは低下させるように選択した異なるpHの水性環境に置く。薬剤がリポソームの内部に移動すると、内部のpHのために荷電した薬剤の状態が生じ、そのため薬剤が脂質二重層を透過することが妨げられ、それによって薬剤がリポソーム中に封じ込められる。
【0087】
pH勾配を作り出すためには、最初の外部媒質を、水素イオン濃度が異なる新たな外部媒質によって置き換える。外部媒質の置換は、さまざまな技法により、例えば、新たな媒体によって平衡化したゲル濾過カラム、例えばセファデックス(Sephadex)G-50カラムに脂質小胞調製物を通過させること(以下の実施例で述べる)により、または遠心分離、透析もしくは関連した技法などによって行いうる。内部媒質は外部媒質に対して酸性でも塩基性でもよい。
【0088】
pH勾配を確立した後に、pH勾配によって装填されうる薬剤を混合物に添加し、上記のようにしてリポソーム中への薬剤の封入を行わせる。
【0089】
pH勾配を用いる装填は、米国特許第5,616,341号、第5,736,155号および第5,785,987号に記載された方法に従って行うことができ、これらは参照として本明細書に組み入れられる。好ましいpH勾配装填法は、pH 2〜6の内部緩衝液としてのクエン酸塩および中性外部緩衝液を用いる、クエン酸塩を利用する装填方法である。
【0090】
リポソームの内外にpH勾配を成立させてそれを維持するにはさまざまな方法を用いることができ、これらはすべて参照として本明細書に組み入れられる。これには、リポソーム膜に挿入しうるイオノフォア、およびプロトンの交換において膜を越える輸送イオンを用いることが含まれうる(例えば、米国特許第5,837,282号を参照)。リポソーム膜を越えてプロトンを往復させることができ、このためpH勾配を成立させうる、リポソームの内部に封入された化合物(例えば、米国特許第5,837,282号を参照)を用いることもできる。これらの化合物は、脱プロトン化された場合は中性であってプロトン化されると荷電するイオン化部分を含む。中性脱プロトン化型(これはプロトン化型と平衡状態にある)は、リポソーム膜を通過することができ、このためプロトンがリポソーム内部に残り、それによって内部のpHが低下する原因となる。この種の化合物の例には、塩化メチルアンモニウム、硫酸メチルアンモニウム、硫酸エチレンジアンモニウム(米国特許第5,785,987号を参照)および硫酸アンモニウムが含まれる。塩基性の内部pHを成立させうる内部装填緩衝液を利用することもできる。この場合には、中性型のものがプロトン化され、プロトンがリポソームの内部から外に出されて内部が塩基性となる。この種の化合物の例には酢酸カルシウムがある(米国特許第5,939,096号を参照)。
【0091】
2種類またはそれ以上の薬剤を、同じ能動的装填方法を用いてリポソーム中に装填することもでき、または異なる能動的装填方法を用いてもよい。例えば、金属錯体化装填法を複数の薬剤を能動的に装填するために用いてもよく、またはpH勾配装填法などの別の能動的装填法と組み合わせてもよい。金属を用いる能動的装填法では一般に、金属イオンが受動的に封入されたリポソーム(受動的に装填された治療薬を含む場合も含まない場合もある)を用いる。金属イオンのさまざまな塩が用いられるが、その塩は薬学的に許容され、水性溶液に溶解するという条件が付く。能動的に装填される薬剤は、金属イオンと錯体を形成し、そのように錯体化した場合にリポソーム内部に保持されるが、金属イオンと錯体を形成しない場合にもリポソーム中に装填されうるという能力に基づいて選択される。金属と配位結合を行える薬剤は一般に、アミン、カルボキシル基、エーテル、ケトン、アシル基、アセチレン、オレフィン、チオール、ヒドロキシル基もしくはハロゲン基、または金属イオンに電子を供与することによって金属イオンと錯体を形成しうる他の適した基などの配位結合部位を含む。金属と結合する作用物質の例には、フルオロキノロンなどのキノロン;ナリジクス酸などのキノロン;ドキソルビシン、ダウノルビシンおよびイダルビシンなどのアントラサイクリン;カナマイシンなどのアミノグリコシド;ならびにブレオマイシン、マイトマイシンCおよびテトラサイクリンなどの他の抗生物質;ならびにシクロホスファミド、チオセミカルバゾン、インドメタシンおよびニトロプルシドなどのナイトロジェンマスタード;トポテカン、イリノテカン、ラートテカン(lurtotecan)、9-アミノカンプトテシン、9-ニトロカンプトテシンおよび10-ヒドロキシカンプトテシンなどのカンプトテシン;ならびにエトポシドなどのポドフィロトキシンが非制限的に含まれる。薬剤の取り込みは、薬剤を外部媒質に添加した後に混合物を適した温度でインキュベートすることによって確認しうる。リポソームの組成、内部媒質の温度およびpH、ならびに薬剤の化学的性質に応じて、薬剤の取り込みは数分で起こることも数時間かかることもある。リポソーム内部で薬剤と金属との間に配位結合が生じたか否かを判定する方法には、分光光度分析、および当業者に周知のその他の従来の技法が含まれる。
【0092】
さらに、リポソーム中にイオノフォアを用いずに行った、金属を利用する手順を用いたリポソーム装填効率および保持特性は、用いた金属およびリポソームの脂質組成に依存する。脂質組成および金属を選択することにより、装填特性または保持特性を、所望の装填またはリポソームからの選択した薬剤の放出を達成するように適合させることができる。
【0093】
複数の薬剤を送達媒体に装填するために、受動的および能動的な装填方法を逐次的に組み合わせることもできる。例えば、受動的に封じ込められたシスプラチンなどのプラチナ性薬剤をMnC12の存在下に含むリポソームは、続いて、ドキソルビシンなどのアントラサイクリンをリポソームの内部に封入するために用いることができる。この方法は、受動的封入によってリポソーム中に封入されたさまざまな薬剤に適用しうる可能性が高い。
【0094】
複数の薬剤を封入した送達媒体組成物の治療的用途
これらの送達媒体組成物は、温血動物および鳥類における種々の疾患の治療に用いうる。したがって、本発明の方法および組成物による治療に適した対象には、ヒト、家畜または家庭内動物などの哺乳動物、ニワトリおよびアヒルなどの家畜化された鳥類対象、ならびに研究用の実験動物が含まれる。本発明の組成物の医学的用途の例には、癌の治療、高血圧、不整脈および再狭窄などの心血管疾患の治療、細菌性、ウイルス性、真菌性もしくは寄生生物性の感染症の治療、本発明の組成物のワクチンとしての使用による疾患の治療および/もしくは予防、炎症の治療、または自己免疫疾患の治療が含まれる。
【0095】
1つの態様において、本発明による送達媒体組成物は、新生物の治療に用いられることが好ましい。腫瘍部位への製剤の送達は、リポソームまたは他の粒子状送達システムの投与によって達成される。リポソームの直径は200nm未満であることが好ましい。腫瘍の血管構造は、内皮における開窓またはギャップのため、正常な血管構造よりも一般に漏出性が高い。これにより、直径200nmまたはそれ未満の送達媒体が、不連続的な内皮細胞層、および、その下層にあって腫瘍に血液を供給する血管周囲の基底膜を通過することが可能になる。送達媒体が血管外漏出後に腫瘍部位内に選択的に蓄積されることにより、薬物送達および治療的有効性が向上する。担体が血管外に漏出するため、血中で測定した担体薬剤-薬剤比は、血管外漏出空間における担体薬剤-薬剤比と同程度であると想定することができる。
【0096】
送達媒体組成物の投与
上記の通り、本発明の送達媒体組成物を、ヒトを含む温血動物、ならびに家畜鳥類に投与することができる。ヒトの疾患の治療の場合、有能な医師は、本発明の組成物を、確立されたプロトコールを用いて、用量、投与計画および投与経路の点でいかにして用いるべきかを決定しうると考えられる。本発明の送達媒体組成物中に封入された薬剤が対象の健常組織に及ぼす毒性が低ければ、このような用途に用量漸増法を用いてもよい。
【0097】
本発明の薬学的組成物は、非経口的に、すなわち動脈内、静脈内、腹腔内、皮下または筋肉内に投与することが好ましい。より好ましくは、薬学的組成物をボーラス注射によって静脈内または腹腔内に投与する。例えば、Rahmanら、米国特許第3,993,754号;Sears、米国特許第4,145,410号;Papahadjopoulosら、米国特許第4,235,871号;Schneider、米国特許第4,224,179号;Lenkら、米国特許第4,522,803号;およびFountainら、米国特許第4,588,578号を参照されたい。
【0098】
他の方法では、製剤を組織に直接適用することにより、本発明の医薬製剤を標的組織に接触させることができる。適用は、外用、「観血的(open)」または「閉鎖的(closed)」処置のいずれによって行うこともできる。「局所的(topical)」とは、環境に露出された組織、例えば皮膚、中咽頭、外耳道などに対する医薬製剤の直接適用を意味する。「観血的」処置とは、患者の皮膚を切開し、医薬製剤を適用しよとする下層の組織が直接見えるようにする処置のことである。これは一般に、肺に到達するための開胸術、腹部臓器に到達するための腹式開腹術、または標的組織に対する他の直接的な外科的アプローチなどの外科処置によって行われる。「閉鎖的」処置とは、内部標的組織を直接目にすることはないが、皮膚内の小さな創を通して挿入した器具を介して到達する侵襲的処置のことである。例えば、針洗浄によって製剤を腹膜に投与することができる。同様に、腰椎穿刺時に注入し、その後に脊椎麻酔または脊髄のメトラザミド(metrazamide)造影のために一般に行われているように患者に適切な体位をとらせることにより、医薬製剤を髄膜または脊髄に投与することもできる。または、製剤を内視鏡装置を用いて投与することもできる。
【0099】
本発明の送達媒体を含む薬学的組成物は標準的な技法に従って調製され、水、緩衝液、0.9%生理食塩水、0.3%グリシン、5%デキストロースなどを含んでよく、安定性の向上のためにアルブミン、リポタンパク質、グロブリンなどの糖タンパク質も含みうる。これらの組成物は、従来のよく知られた滅菌法によって滅菌しうる。その結果得られた水性溶液を使用のためにパッケージ化してもよく、無菌条件下で濾過して凍結乾燥した上で凍結乾燥製剤を投与前に滅菌水溶液と混合してもよい。組成物は、生理的条件に近づけるための薬学的に許容される補助物質、例えば、pH調整剤および緩衝剤、張度調整剤など(例えば、酢酸ナトリウム、乳酸ナトリウム、塩化ナトリウム、塩化カリウム、塩化カルシウムなど)を含みうる。さらに、送達媒体懸濁液が、脂質をフリーラジカルおよび保存時の脂質過酸化損傷から保護する脂質保護剤を含んでもよい。α-トコフェロールなどの脂溶性フリーラジカル失活剤、および、フェリオキサミンなどの水溶性鉄特異的キレート剤が適している。
【0100】
医薬製剤における送達媒体の濃度には、重量比で約0.05%未満、通常は約2〜5%または少なくとも約2〜5%から、10〜30%までというように大きな幅があってよく、これは主として、選択した特定の投与様式に従い、液体の容積、粘性などによって選択される。例えば、治療に伴う液体投与量を少なくするには濃度を高めるとよい。または、刺激性の脂質を含む送達媒体は、投与部位での炎症を軽減するために希釈して低濃度にするとよい。診断の目的で投与する送達媒体の量は、用いる特定の標識、診断しようとする疾病状態、および臨床医の判断に依存すると考えられる。
【0101】
本発明の薬学的組成物は静脈内に投与されることが好ましい。送達媒体製剤の投与量は、薬剤と脂質との比、ならびに、患者の年齢、体重および状態に基づいた、投与する臨床医の見解に依存すると考えられる。
【0102】
薬学的組成物に加えて、獣医学的な使用に適した製剤を、対象に適した様式で調製して投与することもできる。好ましい獣医学的対象には、哺乳動物種、例えば、非ヒト霊長動物、イヌ、ネコ、ウシ、ウマ、ヒツジ、および家畜化した鳥類が含まれる。対象には実験動物、例えば、特にラット、ウサギ、マウスおよびモルモットも含まれる。
【0103】
インビボでの治療活性の評価
2種類またはそれ以上の薬剤が封入された送達媒体組成物の治療活性は、動物モデルへの投与後に測定することができる。動物モデルは腫瘍を含むことが好ましいが、送達媒体組成物を他の疾患の動物モデルに投与することもできる。マウスおよびラットなどの齧歯類種、例えば、免疫能力があるものおよび免疫不全のものを含む、近交系、非近交系または雑種由来のもの、ならびにノックアウトモデルまたはトランスジェニックモデルを用いることができる。
【0104】
モデルは、細胞浮遊液、胸腺または腫瘍断片として、皮下、静脈内、腹腔内、筋肉内、クモ膜下腔内または同所領域に移植された固形腫瘍または非固形腫瘍からなりうる。腫瘍を、腫瘍形成性/発癌性物質の適用もしくは投与によって成立させてもよく、または適切な遺伝子組換え動物モデルにおいて自然発生させてもよい。腫瘍のタイプは、癌、肉腫、黒色腫、神経膠腫、白血病およびリンパ腫といった、外胚葉、中胚葉または内胚葉由来の腫瘍からなりうる。
【0105】
1つの好ましい態様では、腫瘍のマウスモデルを用いる。免疫不全マウスで増殖させたヒト異種移植固形腫瘍を利用し、規定された遺伝学的性質および増殖性に基づいて選択することができる。これらの実験に用いる腫瘍細胞は遺伝的に操作した上で、または好ましい特性を発現するように選択した上で、マウスに注入することができる。
【0106】
腫瘍が触知可能な(測定可能な)サイズに増殖したところで、送達媒体組成物を投与し(静脈内が好ましい)、腫瘍増殖に対する効果を観測する。意図する治療的処置は、単回のボーラス投与もしくはプッシュ投与、または数日もしくは数週間にわたる多回もしくは連続的な投与からなってよく、経口的、鼻腔内、皮下、静脈内、腹腔内、クモ膜下腔内、腫瘍内経路などの任意の適した経路により、シリンジ、錠剤、液剤およびポンプ(浸透ポンプなど)を用いうる。達成しうる最大抗腫瘍活性を決定するために用量および投与計画の依存性を評価することもできる。
【0107】
腫瘍を含む動物モデルにおける治療活性を決定するためのさまざまな方法を用いうる。これには、固形腫瘍モデル評価法および非固形腫瘍モデル評価法が含まれる。
【0108】
固形腫瘍モデル評価法には、腫瘍体積(質量)、腫瘍重量抑制(TWI%)、腫瘍増殖遅延(T-C)、腫瘍退縮、細胞死の計測およびクローン原性(clonogenic)アッセイ法が含まれる。
【0109】
腫瘍体積の測定値は、直交する長さおよび幅のキャリパーによる測定によって決定される(高さの測定もしばしば行いうる)。腫瘍体積(mL)または質量(g)は、体積=(長さ×幅2/2;または体積=π/6×(長さ×幅×高さ)によって算出される。データを時間に対してプロットする。
【0110】
腫瘍重量抑制(TWI%)は、定められた時点で、投与群の平均腫瘍重量を対照群の平均腫瘍重量によって除算して1を差し引いた上で100を掛けることによって決定される。
【0111】
腫瘍増殖遅延(T-C)は、投与群(T)が任意の決定された腫瘍サイズ(例えば、300mg)に到達するまでの日数の中央値から、対照群が同じ腫瘍サイズに達するまでの日数の中央値を差し引いた値として計測される。
【0112】
投与の結果としての腫瘍退縮を、腫瘍モデルを評価する手段として用いることもできる。結果は腫瘍サイズ(質量)の経時的な減少として表現される。
【0113】
細胞死法による固形腫瘍モデル評価は、腫瘍のすべてが所定のサイズ(例えば、200mg)を超えるまで繰り返し測定することを含みうる。続いて、腫瘍増殖および腫瘍倍加時間を評価する。log10細胞死パラメーターは以下によって算出しうる:
log10細胞死/用量=(T-C)/((3.32)(Td)(投与回数))
log10細胞死(合計)=(T-C)/((3.32(Td))
log10細胞死(正味)=((T-C)-(Rxの期間))/((3.32(Td))
ここで、(T-C)=腫瘍増殖遅延
Td=腫瘍倍加時間である。
【0114】
クローン原性(clonogenicity)アッセイ法は、治療法の有効性を表す。これらのアッセイ法は、切り出し(excision)アッセイ法および固形腫瘍由来の細胞浮遊液の特徴分析を含む。
【0115】
切り出しアッセイ法は、腫瘍から調製した浮遊液におけるどの細胞画分が無限の増殖能を有するか(すなわち、クローン原性があるか)を評価するために用いられる。切り出しアッセイ法には3種類がある。
i)TD50またはエンドポイント希釈アッセイ法:これはインビボの接種材料から採取した細胞が腫瘍を生成するために必要な数を決定する。
ii)インビボコロニーアッセイ法:これは個々の細胞が、例えば肺に、小結節(コロニー)を形成する能力を評価する。
iii)インビトロコロニーアッセイ法、これは個々の細胞が、コロニーが培養皿のプラスチックまたはガラスの表面に形成される場合には液体培地中で、またはコロニーが浮遊液中に形成される寒天などの半流動培地中で、増殖してコロニーとなる能力を調べる。
【0116】
固形腫瘍からの細胞浮遊液の特徴分析は、インビトロおよびインビボでのクローン原性アッセイ法、フローサイトメトリー測定のため、ならびに細胞単位に対して行われるさまざまな生化学的および分子的な分析のために必要とされる。調製は、酵素的、機械的、化学的、それらの組合せ、および界面活性剤などのさまざまな方法による。評価には、細胞収量、細胞形態、腫瘍細胞クローン原性、生化学的または分子的な特徴の保持が含まれる。
【0117】
非固形腫瘍モデル評価法には、寿命の延長(ILS%)、腫瘍増殖遅延(T-C)、長期生存者(治癒)の計測が含まれる。
【0118】
寿命の延長(ILS%)は、対照群または非投与群と比較した投与群の寿命の延長率を計測する。腫瘍増殖遅延(T-C)は、投与(T)群の生存日数の中央値から対照(C)群の生存日数の中央値を差し引いた値を計測する。長期生存者(治癒)は、非投与群または対照群の生存期間の3倍を超えて生存した投与群について計測する。
【0119】
癌に罹患したヒトにおける治療活性を決定する方法は、生存および代用エンドポイントの測定を含む。生存を評価するのに妥当な時点は当該の腫瘍によって決まる。例えば、低悪性度リンパ腫の患者に関する生存率は、診断から5年後または10年後に評価するとよいが、進行性非小細胞肺癌などの悪性度の高い疾患を有する患者の生存は診断から6カ月後または12カ月後に評価するのが最もよい。
【0120】
代用エンドポイントを用いて治療活性を決定する方法には、完全縮小(CR)、部分的縮小(PR)、無増悪生存(PFS)、無増悪期間(TTP)または縮小期間(DOR)、血漿マーカーおよび尿マーカー、酵素阻害ならびに/または受容体の状態、遺伝子発現の変化および生活の質(QOL)の計測が含まれる。
【0121】
完全縮小(complete response)とは、治療している腫瘍の種類にとって適切な期間にわたり、新たな疾患の発生を伴わずに、疾患のすべての既知の部位が消失することを意味する。評価は、以上に述べたようなさまざまな検査に基づく。
【0122】
部分的縮小(partial response)とは、治療している腫瘍の種類にとって適切な期間にわたり、新たな病変の出現を伴わずに、すべての病変の二方向測定値の積の合計が少なくとも50%減少することを意味する。評価は、患者のさまざまな検査(CTスキャン、MRI、超音波、PETスキャン、骨スキャン、身体診察)に基づく。
【0123】
無増悪生存(PFS):患者が生存していて、既存の腫瘍の増殖がなく、新たな腫瘍塊の出現もみられない治療からの期間。PFSは、診断後の所定の時点で生存していて無増悪である期間または患者の割合として表現しうる。
【0124】
無増悪期間(TTP)または縮小期間(DOR)とは、治療時から、既存の腫瘍塊のサイズの増大または新たな腫瘍塊の出現として計測される腫瘍増殖の進行までの期間のことである。
【0125】
血漿マーカーおよび尿マーカーには、以下のマーカーを非制限的に含むマーカーを測定することが含まれる:前立腺特異抗原(PSA)および癌胎児性抗原(CEA)。
【0126】
酵素阻害および/または受容体の状況。チロシンキナーゼ受容体、EGF受容体、PDGF受容体、Her-1およびHer-2受容体を非制限的に含む増殖因子受容体。インテグリン関連キナーゼ、タンパク質キナーゼなどを非制限的に含む酵素。
【0127】
遺伝子発現の変化には、遺伝子発現(ゲノミクス)、およびタンパク質発現の変化(プロテオミクス)の連続解析が含まれる。
【0128】
生活の質(QOL)にはEORTC QLQ-C30採点法などの方法が含まれ、これは5種類の機能的尺度(身体的、役割上、認知的、社会的および情緒的)、3種類の症状尺度(悪心、疼痛および疲労)ならびに全般的な健康状態および生活の質の尺度に関して得られたスコアを評価するものである。この指標からは、癌患者によって多く報告される別の症状(呼吸困難、食欲減退、睡眠障害、便秘および下痢)、さらには疾患およびその治療による認知されている財政的な影響に関する単項目評点も得られる。
【0129】
以下の例は例示を目的として提供するものであり、本発明の範囲を制限するためのものではない。
【実施例】
【0130】
以下の実施例では、細胞傷害性の決定および非拮抗的な作用の評価のために以下の方法を用いている。
【0131】
細胞傷害性アッセイ法
以下の実施例では、標準的なテトラゾリウム比色MTT細胞傷害性アッセイ法のプロトコール(Mosmannら、J. Immunol Methods (1983) 65(1-2):55-63)を、影響を受けた細胞の率に関する読み取り値の決定に用いた。簡潔に述べると、生きた細胞は、テトラゾリウム塩である3-(4,5-ジエチルチアゾイル-2-イル)-2,5ジフェニルテトラゾリウムブロミド(3-(4,5-diethylthiazoyl-2-yl)-2,5-diphenyltetrazolium bromide、MTT)を、分光分析によって読み取れる青色のホルマザンへと還元する。25cm2フラスコ内で増殖させたヒトH460非小細胞肺癌(NSCLC)細胞などの細胞を継代し(継代数<20)、新たなRPMI細胞培養液中に再懸濁した上で、96ウェル細胞培養プレートに、1ウェル当たり細胞1000個(1ウェル中に100μL)の濃度でプレーティングする。続いて細胞を37℃、5%CO2下で24時間インキュベートする。その翌日に、薬剤の系列希釈物を12ウェル細胞培養プレート中に用意する。前もって種々の溶液中に調製しておいた薬剤を新たなRPMI細胞培養液中に希釈する。薬剤を、ラテン方格配置法または「チェッカーボード」希釈法を用いて、単一の薬剤(20μl)および特定の固定比での二剤の組合せ(増分20μL)に関する適切なまたは特定されたウェルに投与する。新たな培地を加えてウェル内の総容積を200μLにする。薬剤の曝露は72時間にわたって行う。
【0132】
薬剤曝露の後に、MTT試薬(1mg/mL、RPMI中)を各ウェルに添加して容積を1ウェル当たり50μLとし、3〜4時間インキュベートする。続いてウェル内容物を吸引し、細胞を破壊して細胞内のホルマザン沈殿物を可溶化するために150μL のジメチルスルホキシド(DMSO)を各ウェルに添加する。96ウェルプレートをプレートシェーカーで振盪させ、波長570nmに設定したマイクロプレート分光光度計で読み取る。光学密度(OD)の読み取り値を記録し、ブランクウェル(培地のみを含む)のOD値を細胞を含む全ウェルから差し引く。薬剤に対する曝露後の細胞生存度は対照ウェル(薬剤に曝露されていない細胞)に対する百分率に基づく。すべてのウェルを3回ずつ検討し、平均値を算出する。
【0133】
薬剤の組合せに関するメディアンエフェクト解析
薬剤の組合せの解析には、ChouおよびTalalayによるメディアンエフェクト原理を基盤とするソフトウエアプログラムCalcuSyn(Biosoft, Ferguson, MO, USA)を用いた。まず、二剤の組合せに関する固定比を、単剤の細胞傷害性プロフィールによるIC50:IC50比から導き出す。続いて、製剤の目的に関する考察に基づき、より妥当な固定比(例えば、10:1〜1:10の範囲;モル比)を選択する。細胞生存性に対する薬剤の効果に基づいて算出した平均値から、用量および各々の分割効果の値をCalcuSynコンピュータプログラムに入力する。するとソフトウエアが、薬剤の組合せが相乗的、相加的または拮抗的のいずれであるかを組合せ指数(CI)値に基づいて算出する。
【0134】
実施例1
用量-効果分析のさまざまな表現
薬剤の量(用量または濃度)とその生物作用との間の関係、ならびに薬剤の組合せの連合効果に関する定量分析は、さまざまなやり方で計測および報告を行うことができる。図2は、このような5通りの方法を、一例としてイリノテカンとカルボプラチンとの組合せを用いて示している。
【0135】
用量-効果分析に関するChouおよびTalalayの理論に基づき、「メディアンエフェクト式」がさまざまな生化学的式の算出に用いられており、これらは当技術分野で広範囲に用いられている。この式の変形により、組合せ指数(CI)の算出に用いられるもののような高次式が生まれている。前述の通り、CIは、複数の薬剤の組合せおよび各組合せの種々の比が拮抗的、相加的または相乗的のいずれであるかを決定するために用いうる。CIプロットは一般に、y軸を表すCIと、x軸にある影響を受けた細胞の割合または影響率(fa)との関係を図示したものである。図2Aは、イリノテカン/カルボプラチンがモル比1:10では拮抗的であり(CI>1.1)、1:1および10:1では相乗作用を有すること(CI<0.9)を示している。
【0136】
本出願者らは、用いた薬剤比に対するCIの依存性を表現する代替的な方法もデザインした。図2Bに示されているように、特定の組合せに関する比特異的な作用における傾向がより適切に図示されるように、最大CI値を各々の比に対してプロットする。CI最大値とは、異なる比の薬剤に関してCI値に最も大きな差が観察される単一のfa値(0.2〜0.8の間)について得られたCI値のことである。
【0137】
個々の比について用いる薬剤の濃度は効果(すなわち、相乗作用または拮抗作用)の決定に一定の役割を果たすため、CIを種々の濃度で測定することも重要と考えられる。これらの濃度は、Chou-Talalayによって「有効量」(ED)とも呼ばれ、インビトロアッセイ法において細胞の指定された比率に影響を及ぼすために必要な薬剤の濃度であり、すなわち、ED50は、対照または非投与細胞集団との比較で細胞の50%に影響を及ぼすために必要な薬剤の濃度のことである。図2Cに示されているように、種々の比の間での濃度-効果における傾向は容易に識別可能である。示されているエラーバーは平均周辺での標準偏差の一つを表しており、これはCalcuSynプログラムによって直接決定される。
【0138】
2種類またはそれ以上の薬剤の間の相乗的相互作用は、陽性の効果を得るために必要な各薬剤の量を減らしうる(「用量減少」としても知られる)という点で有益である。ChouおよびTalalayの「用量減少指数」(DRI)は、所定の効果レベルで、相乗的組合せにおける各薬剤の用量を各薬剤の単独での用量と比較してどの程度減少させうるかの指標である。DRIは、用量減少が治療的有効性を維持したままで宿主に対する毒性の低下につながるような臨床的状況では重要である。図2Dにおけるプロットは、それぞれ単独で90%の細胞死を達成するために必要なイリノテカンおよびカルボプラチンの濃度が、それらを非拮抗的な比で併用した場合に必要な個々の濃度よりも有意に高いことを示している。
【0139】
さらに、前記のデータを、古典的なアイソボログラムとして表現することもできる(図2E)。アイソボログラムには種々のED値で作成しうるという利点があるが、しかし、解釈のために高い効果レベルを選択するほど、その読み取りは困難になる。この理由から、以下の実施例におけるデータは一般に、図2Aおよび2Bに示したプロットのタイプに応じて提示されている。
【0140】
実施例2
CIは濃度に依存する
イリノテカンおよび5-フルオロウラシル(5-FU)のモル比1:1および1:10での組合せ、ならびにエトポシドおよびカルボプラチンのモル比10:1および1:10での組合せという薬剤の組合せを、前の実施例の項で説明した、標準的なテトラゾリウム比色MTT細胞傷害性アッセイ法およびメディアンエフェクト解析を用いて、相加的、相乗的または拮抗的な作用に関して検討した。HT29細胞またはMCF-7細胞を、単一の薬剤ならびに薬剤の所定の比での組合せに対して曝露させた。単一の薬剤および組合せについて、8種類の薬剤濃度を用いた。光学密度の値をMTTアッセイ法によって入手し、これを対照に対する百分率に変換し、平均した上で、影響率の値に変換した。用量および影響率の値をCalcuSynに入力し、図3に示されたCIとfaとの関係をみたグラフを得た。
【0141】
図3Aは、イリノテカンおよび5-FUがモル比1:1では、影響率(用量)による評価で、全範囲範囲にわたって非拮抗的であったことを示している。これに対して、モル比1:10では、同じ2つの薬剤は低濃度では非拮抗的であるが、高濃度では拮抗的であった。図3Bに示されているように、エトポシドおよびカルボプラチンはモル比10:1では全濃度範囲にわたって拮抗的であった。これに対して、モル比1:10では、エトポシドおよびカルボプラチンは低濃度では拮抗的であるが、高濃度では非拮抗的であった。
【0142】
シスプラチンおよびエデルフォシンも、CIをfaに対してプロットすることによって概括されているように、モル比10:1および1:1でH460細胞において異なる併用効果を示した。図4に示されているように、モル比10:1での組合せは、影響率の範囲のうち低濃度側の約50%では非拮抗的であり、高濃度では拮抗的であったが、一方、モル比1:1では全濃度範囲にわたって相乗作用を示した。
【0143】
すなわち、これらの結果は、相乗作用が、薬剤の相互の比だけではなく、それらの濃度にも強く依存することを示している。
【0144】
実施例3
さまざまな二剤の組合せに関するCIの決定
以下の表に示したさまざまな薬剤の組合せを、上記のMTT細胞傷害性アッセイ法プロトコールおよびメディアンエフェクト解析手順を用いて、相加的、相乗的または拮抗的な作用に関して検討した。CIとfaとの関係をみたグラフからの結果を以下に表としてまとめている。非拮抗的な作用が認められたfa範囲のおおよその百分率を、比の後ろのカギ括弧の中に報告している。測定はfa値0.2〜0.8の範囲で行い、CI値1.1未満に該当する曲線の百分率を決定することにより、相乗作用または相加作用(非拮抗的)を示したfa範囲の百分率を算出した。データは、少なくとも1つの実験を3回ずつ行ったものに由来する。
a「相乗的または相加的である百分率%」は、fa値0.2〜0.8の範囲での、Chou-Talalay方法に基づくClと影響率(fa)とのプロット上で、拮抗的な範囲(CI値>1.1は拮抗的である)に該当しないfa範囲の百分率として算出される。CIは用量およびfa値をCalcuSynに入力することによって測定した。
b この比に関するデータセットは「相加的な」範囲にあった(CIが0.9〜1.1の間)。
【0145】
実施例4
カルボプラチンおよびダウノルビシンの相乗作用
相加的、相乗的または拮抗的な作用を計測するための上記の手順を、カルボプラチン/ダウノルビシンをH460細胞にてモル比10:1、1:1および1:10で用い、さらにMCF-7細胞にて比10:1および1:1で用いて再び行った。上記の通りにCI-fa曲線を作成し、続いてfa値0.50、0.75および0.90でのCIを決定することにより(それぞれED50、ED75およびED90でのCI値を得るため)、組合せ指数を各用量について決定した。標準偏差はCalcuSynプログラムによって算出した。図5Aの挿入図に示されているように、カルボプラチンおよびダウノルビシンはモル比10:1では、MCF-7細胞においてED50、ED75およびED90値で相乗的な相互作用を示す。図5Aの挿入図にさらに示されているように、カルボプラチンおよびダウノルビシンは1:1モル比の場合、ED75およびED90では平均CI値による判定で相乗的であるが、ED50では相加的である。H460細胞においては、CI最大値とカルボプラチン/ダウノルビシンのモル比とのプロットから、モル比10:1ではこれらの薬剤は相乗的であるが、モル比1:1では幾分拮抗的な作用が観察されることが判明した。これに対して、比1:10では高度に拮抗的な作用が認められる(図5A)。このデータは図5Bにも、相乗作用に対する濃度の影響をより適切に図示するために、CIと影響を受けたH460細胞の率との関係としてプロットされている。モル比1:1のカルボプラチン/ダウノルビシンは、影響率の値が0.42までの範囲では非拮抗的である。比が10:1だと、fa値のかなりの範囲(0.2より上)で相乗作用が認められ、比1:10はすべてのfa値で拮抗的である。図5Bの挿入図は、H460細胞において比10:1ではED50、75および90で相乗作用(平均CI値による判定)が観察され、比1:1ではED50で相加作用が示されることを示している。比が1:10だと、カルボプラチン/ダウノルビシンはED50、75および90値で高度に拮抗的である。このため、これらの結果に基づき、カルボプラチンおよびダウノルビシンはモル比1:10では、測定したすべてのED値でCI-faプロット内のすべてのfa範囲にわたって拮抗作用が認められるために、これ以上の製剤およびインビボ試験の対象としては選択されないと考えられる。モル比10:1および1:1のカルボプラチン:ダウノルビシンは、これらの比のそれぞれで、薬剤が(細胞の1%超が影響を受ける)fa範囲の少なくとも5%にわたって相乗作用を示しているため、製剤および有効性試験の対象として選択される。
【0146】
実施例5
インビボでのカルボプラチンおよびダウノルビシンの相乗作用の維持
カルボプラチンおよびダウノルビシンを、単一のコレステロール非含有リポソーム中に、モル比10:1、5:1および1:1(カルボプラチン/ダウノルビシン)で共装填(co-load)した。DSPCはクロロホルム中に溶解し、DSPGは微量の14C-CHEとともにクロロホルム/メタノール/水(50:10:1 vol/vol)中に溶解した。これらの溶液をモル比80:20(DSPC/DSPG)で混ぜ合わせた。温度を60℃超に保ちながら、溶媒をN2ガス流によって除去した。続いて脂質薄膜を真空ポンプ内に2分間置き、その後にクロロホルムのみの中に再び溶解した。続いてクロロホルムを上記の通りに除去した。その結果生じた脂質薄膜を残留溶媒を除くために真空下に一晩おき、その後に、カルボプラチンの溶解性を高めるための4%(v/v)DMSOとともに80mg/mLカルボプラチンを含む150mM CuSO4、pH 7.4(pHはトリエタノールアミンで調整)中に再水和した。その結果生じた多重ラメラ小胞(MLVs)を70℃で孔径80および100nmの積み重ねた2枚のフィルターを通して押し出し、合計10回通過させた。この試料を生理食塩水中に入れた上で、接線流透析を用いる交換によって300mMスクロース、20mM HEPES、30mM EDTA、pH 7.4(SHE)中に入れた。モル比10:1、5:1および1:1のカルボプラチン/ダウノルビシンが得られる薬剤-脂質比で、60℃にて5分間インキュベートすることにより、ダウノルビシン(微量の3H-ダウノルビシンを含む)をリポソーム中に装填した。その後に、各試料の緩衝液を接線流によって生理食塩水に交換した。共装填した製剤の調製中の種々の時点での薬剤装填の程度を決定するために、液体シンチレーション計数によってダウノルビシンおよび脂質のレベルを測定した。カルボプラチン濃度は原子吸光分光法によって測定した。共装填製剤におけるモル比10:1、5:1および1:1のカルボプラチン/ダウノルビシンの場合、Balb/cマウスに対して8mg/kgカルボプラチンが静脈内投与され、ダウノルビシンの用量はそれぞれ1.2mg/kg、6mg/kgおよび12mg/kgとなった。指定した時点(各時点当たりマウス3匹)で、血液を心臓穿刺によって採取し、EDTAをコーティングしたマイクロテイナーに入れた。試料を遠心して血漿を慎重に別にチューブに移した。液体シンチレーション計数を用いてダウノルビシンおよび脂質の血漿中濃度を定量した;カルボプラチンの血漿中濃度は原子吸光分光法によって測定した。原子吸光分光法による定量のためには、標準曲線の直線範囲に収まるように試料を0.1%硝酸で希釈した。
【0147】
図6における結果は、薬剤の平均血漿中濃度(+/-標準偏差、SD)を指定時点でプロットしたものであり、存在したカルボプラチンの血漿中モル濃度がダウノルビシンの10倍であることから、カルボプラチンおよびダウノルビシンをモル比10:1で含む共装填リポソーム製剤が、静脈内投与後の薬剤の比を維持したことを示している。図7Aおよび7Bにおける結果は、DSPC/DSPGリポソーム中に調合されたカルボプラチンとダウノルビシンとのモル比10:1、5:1および1:1が、これらの比で調製された製剤の静脈内投与から24時間の経過にわたって(各時点当たりマウス3匹)、血液区画において維持されたことを示している(図7Bは、1:1カルボプラチン/ダウノルビシン製剤の投与後に得られた結果をさらに明確に強調している)。すなわち、これらの結果は、2種類の薬剤のさまざまなモル比での協調的な放出動態を達成しうることを示している。
【0148】
カルボプラチンおよびダウノルビシンをDSPC/SM/DSPE-PEG2000(90:5:5mol%)リポソーム中に、インビボでの薬剤の協調的な放出がこの製剤でも達成されうるか否かを明らかにする目的で、ともに調合した。実施例4で相乗的であると決定されたモル比10:1を選択した。
【0149】
脂質をクロロホルム中に溶解し、クロロホルムをN2ガス下で除去し、試料を真空ポンプ内に一晩置くことにより、脂質薄膜(微量の14C-CHEを含む)を上記の通りに調製した。その結果生じた脂質薄膜を、40mg/mLカルボプラチンを含む150mM CuS04、20mMヒスチジン、pH 7.4(pHはトリエタノールアミンで調整)中で水和させた。MLVを70℃で孔径100nmの2枚の積み重ねたフィルターを通して押し出し、合計10回通過させた。続いて試料を、封入されなかった金属溶液(またはカルボプラチン)を除去するために接線流透析によって300mMスクロース、20mM HEPES、pH 7.4中に入れ換えた。ダウノルビシンの装填(微量レベルの3H-ダウノルビシンを含む)は、カルボプラチン/ダウノルビシンのモル比10:1を達成するための薬剤濃度で60℃にて5分間かけて行った。薬剤装填の程度を決定するために、ダウノルビシンおよび脂質のレベルを液体シンチレーション計数によって測定した;カルボプラチンレベルは原子吸光分光法によって測定した。雄性SCID/rag2マウスに対して、DSPC/SM/DSPE-PEG2000リポソーム中に共装填された組合せで、2.25mg/kgダウノルビシンおよび15mg/kgカルボプラチンを静脈内に投与した。指定された時点(各時点当たりマウス3匹)で心臓穿刺によって血液を採取し、EDTAをコーティングしたマイクロテイナーに入れた。試料を遠心して血漿を慎重に別のチューブに移した。カルボプラチンおよびダウノルビシンの血漿中濃度はそれぞれ原子吸光分光法および液体シンチレーション計数によって測定した。
【0150】
図8に示した結果は、薬剤の平均血漿中濃度(+/-標準偏差、SD)を指定時点でプロットしたものであり、DSPC/SM/DSPE-PEG2000リポソーム中に調合された場合に静脈内投与後に同じ速度で血漿区画から排出されたことを明らかにしている。すなわち、この時間経過中に存在したカルボプラチン(nmol/mL)の血漿中濃度はダウノルビシン(nmol/mL)の概ね10倍であったことから、カルボプラチンおよびダウノルビシンは10:1のモル比に維持された。これらの結果は、種々の製剤を用いて、単一のリポソーム中に共封入された2種類の薬剤の薬物動態を、同様の薬物動態放出プロフィールが達成されるように協調させうることを示している。
【0151】
実施例6
リポソーム性カルボプラチンおよびダウノルビシンの有効性
ダウノルビシンおよびカルボプラチンがモル比1:1(これは実施例4で製剤に関して選択された)で共封入されたDSPC/DSPGリポソーム(80:20mol%)を、脂質薄膜を25mg/mLのカルボプラチンを含む溶液である150mM CuSO4、pH 7.4(pHはトリエタノールアミンで調整)中に水和させた点を除き、実施例5における記載の通りに調製した。さらに、脂質薄膜を乾燥させた後にメタノールまたは水を除くためにクロロホルム中に再び溶解し、続いて溶媒を以前の記載の通りに除去した。
【0152】
実施例26の方法と同じように、まずH460細胞(1×106個)を雌性SCID/rag2マウスの側腹部に皮下接種することにより、有効性試験を行った。腫瘍を約50mg(0.05cm3)のサイズになるまで増殖させ、その時点(第12日)で製剤を尾静脈から注入した。マウス(各群当たりマウス4匹)には4日間隔で3回の注射を行った(q4dスケジュール;第12、16および20日)。腫瘍の増殖はキャリパーによる直接測定によって評価した。マウスに対して、生理食塩水、モル比1:1の遊離性薬剤混合物、またはカルボプラチン/ダウノルビシンのモル比1:1でのリポソーム製剤のいずれかを投与した。遊離物およびリポソーム製剤の投与のいずれに関しても、用量は6.6mg/kgカルボプラチンおよび10mg/kgダウノルビシンとした。リポソーム製剤試料に関する脂質用量は260mg/kg脂質であった。
【0153】
図9に提示した結果(各点は指定日に測定した平均腫瘍サイズ+/-平均の標準誤差(SEM)を表す)は、モル比1:1のリポソーム性カルボプラチンおよびダウノルビシンの投与が、遊離性薬剤混合物および生理食塩水対照との比較で、有効性を高めることを示している。
【0154】
カルボプラチンおよびダウノルビシンをモル比10:1(実施例4で相乗的であることが決定された)で共装填したスフィンゴミエリン含有リポソームにおける有効性も、DSPC/DSPGリポソームで認められた有効性の大きな改善がこの製剤を用いた場合にも得られるか否かを検討するために調べた。リポソームを孔径80nmおよび100nmフィルターを通して10回押し出す処理を行った点を除き、実施例5に概要を述べた手順に従って、カルボプラチンおよびダウノルビシンをDSPC/SM/DSPE-PEG2000(90:5:5mol%)リポソーム中にともに調合した。さらに、ダウノルビシンの装填の前に、接線流透析ではなく固定容積透析により、試料の緩衝液をSHE緩衝液に交換した。実施例26に詳述した通りに、H460腫瘍を有する雌性SCID/rag2マウス(各群当たりマウス4匹)に対して、15mg/kgカルボプラチンおよび2.25mg/kgダウノルビシンを、リポソーム製剤および遊離性薬剤混合物として第14、18および22日に投与した。リポソーム製剤は脂質用量として375mg/kgが投与された。
【0155】
図10に提示した結果(各点は指定日に測定した平均腫瘍サイズ+/-SEMを表す)は、スフィンゴミエリン含有リポソーム中に非拮抗的なモル比である10:1で封入されたリポソーム性カルボプラチンおよびダウノルビシンが、遊離性薬剤および生理食塩水からなる対照との比較で、有効性を大きく高めることを示している。
【0156】
実施例7
シスプラチンおよびダウノルビシンの相乗作用
シスプラチン/ダウノルビシンの組合せを、上記の方法を用いて、相加的、相乗的または拮抗的な作用に関して調べた。その結果を図11にまとめた。図11Aに示されているように、シスプラチン/ダウノルビシンのモル比10:1ではfaの全範囲にわたって相乗作用が観察されたが、モル比1:1ではすべてのfa範囲にわたって拮抗作用が示された。CI最大値(CI max)とシスプラチン-ダウノルビシン比との関係をプロットした図11Bは、組合せ指数に対する2種類の薬剤の組合せ比の依存性をさらに示している。これらの結果は、モル比10:1ではCI max値は相乗的であるが、モル比1:1および1:10ではCI max値は拮抗的であることを示している。
【0157】
実施例8
インビボでのシスプラチンおよびダウノルビシンの相乗作用の維持
シスプラチンおよびダウノルビシンを、DMPC/Chol(55:45mol%)リポソーム中に、実施例7で非拮抗的であることが特定されたモル比10:1で共装填した。
【0158】
150mM CuCl2、20mMヒスチジン(pH 7.4、pHはトリエタノールアミンで調整)に4%(v/v)DMSOを加えたものからなる溶液中に薬剤(40mg/mL)をまず溶解し、その結果生じた溶液をシスプラチンの溶解性を高めるために80℃に加熱することにより、シスプラチンをリポソーム中に受動的に封じ込めた。続いてシスプラチン溶液を80℃で、DMPCおよびコレステロールを微量レベルの14C-CHEとともに含む脂質薄膜に添加した。水和した脂質薄膜を80℃で2枚の100nmフィルターを通して押し出して、リポソームを室温まで冷ました。冷却後に、封入されなかったシスプラチンをペレット化するために試料を卓上遠心器に入れて2000×g、5分間の遠心処理を行い、上清を収集した。過剰な金属イオンの除去は、セファデックスG-50ゲル濾過カラムを通過させてリポソーム画分を収集することによって行った。
【0159】
シスプラチンが装填されたリポソームに、さらにダウノルビシン(微量レベルの3H-ダウノルビシンで標識)を、リポソームを薬剤とともに60℃で15分間インキュベートすることにより、シスプラチン/ダウノルビシンのモル比が10:1となるように装填した。薬剤装填の程度を決定するために、シスプラチンレベルを原子吸光分光法によって測定し、3H-ダウノルビシンおよび脂質のレベルは液体シンチレーション計数によって測定した。
【0160】
協調的な放出がこの製剤によって達成されるか否かを明らかにするために、装填されたリポソームを雄性SCID/rag2マウスの尾静脈に、マウス1匹につき5.0mg/kgのシスプラチンおよび1.0mg/kgのダウノルビシンの用量で注入した。指定された時点(各時点当たりマウス3匹)で血液を心臓穿刺によって採取し、EDTAをコーティングしたマイクロテイナーに入れた。試料を遠心して血漿を慎重に別のチューブに移した。リポソーム性脂質およびダウノルビシンの血漿中濃度はいずれも液体シンチレーション計数によって測定し、シスプラチンレベルは原子吸光分光法によって測定した。
【0161】
図12に示した結果(各点は指定時点に測定した薬剤の平均血漿中濃度+/-SDを表す)は、測定した時点で血漿中濃度(μmol/mL)がモル比10:1に維持されていたことから、ダウノルビシンおよびシスプラチンの協調的な放出が達成されたことを示している。
【0162】
上記の方法によってリポソームにシスプラチンおよびダウノルビシンを共装填することもできるが、薬剤を単一のリポソーム中に装填するためにその他の技法を用いてもよい。代替的な方法では、シスプラチンをクエン酸、pH 4.0とともに受動的に封じ込めた後にダウノルビシンを装填するためにpH勾配を用い、緩衝液交換によって膜の内外にpH勾配を生じさせる。この技法は以下のようにして行うことができる。
【0163】
DSPC/Chol(55:45mol%)からなる脂質薄膜を上記の通りに微量の3H-CHEとともに調製する。シスプラチン粉末を150mM NaClおよび150mMクエン酸(pH 4)に溶解することによってシスプラチン溶液を調製する。シスプラチンの緩衝液への溶解性を最大限に高めるために、溶液を65℃に加熱して脂質薄膜に添加する。その結果生じたMLVを65℃で2枚の孔径100nmフィルターを通して押し出し、合計10回通過させる。続いて、溶液を2000×gで10分間遠心することにより、封入されなかったシスプラチンを製剤から除去する。その結果得られたリポソーム性シスプラチンを含む上清を、封じ込められなかった残留シスプラチンを除去し、二重層の内外にpH勾配を成立させるために、150mM NaClおよび20mM HEPES(pH 7.4)によってあらかじめ平衡化したセファデックスG-50カラムに通過させる。
【0164】
次にダウノルビシンをリポソーム中に装填する。まず熱平衡を達成するためにリポソームを60℃で5分間インキュベートし、続いて薬剤/脂質モル比が0.1:1となるように、ボルテックス処理を行いながらダウノルビシンを脂質製剤に対して添加する。種々の時点での薬剤装填の程度を決定するために、リポソームをOGPとともに溶解し、ダウノルビシンの480nmでの吸光度を測定することによってダウノルビシンの濃度を測定する。製剤のシスプラチン濃度は原子吸光分光法を用いて測定する。脂質濃度は液体シンチレーション計数によって測定する。
【0165】
2種類の薬剤の放出動態を協調させる代替的な手段は、各薬剤を別個の担体中に調合することによって達成される。このことは、シスプラチンをDMPC/コレステロールリポソーム中に調合し、ダウノルビシンをDSPC/DSPE-PEG2000リポソーム中に調合した上で、それらをモル比10:1でマウスに静脈内投与することによって示された。
【0166】
リポソーム性シスプラチンは、シスプラチン(8.5mg/mL)を150mM NaCl、80℃中にまず溶解することによって調製した。この溶液を次に、微量の3H-CHEを含むDMPC/コレステロール(55:45mol%)脂質薄膜に添加して水和させた。その結果生じたMLVを80℃で2枚の孔径100nmフィルターを通して押し出し、その後にリポソームを過剰な金属イオンを除去するための接線流透析によって20mM HEPES、150mM NaCl(pH 7.4)(HBS)中に入れ換えた。押出処理で封入されなかったシスプラチンをペレット化するためにリポソームを遠心した。シスプラチン濃度は原子吸光分光法によって測定し、脂質レベルは液体シンチレーション計数によって測定した。
【0167】
リポソーム性ダウノルビシンは、DSPC/DSPE-PEG2000(95:5mol%)および微量の14C-CHEから構成される脂質薄膜の300mM CuSO4溶液による水和によって調製した。その結果生じたMLVを、重ね合わせた2枚の孔径100nmフィルターに70℃で10回通過させることによって押し出し処理した。押出処理の後に、リポソームを接線流透析によってHBS(pH 7.4)中に入れ換えた。ダウノルビシン(微量レベルの3H-ダウノルビシンを含む)の装填は、最終的な薬剤/脂質重量比が0.1となるようにダウノルビシンを添加することによって開始し、その溶液を60℃に10分間保った。薬剤装填の程度は、3H-ダウノルビシンおよび14C-CHEレベルを計測するための液体シンチレーション計数によって測定した。
【0168】
雄性SCID/rag2マウスに対してリポソーム性シスプラチンを薬剤用量2mg/kgとして、リポソーム性ダウノルビシンを薬剤用量0.375mg/kgとして静脈内注射した。指定された時点(各時点当たりマウス3匹)で血液を心臓穿刺によって採取し、EDTAをコーティングしたマイクロテイナーに入れた。試料を遠心して血漿を慎重に別のチューブに移した。シスプラチンの血漿中濃度は原子吸光分光法によって測定し、ダウノルビシン濃度はシンチレーション計数によって測定した。
【0169】
図13に示した結果(各点は指定時点に測定した薬剤の平均血漿中濃度+/-SDを表す)は、別個のリポソーム中に調合したシスプラチンおよびダウノルビシンが静脈内投与後の種々の時点で10:1のモル比を維持したことを明らかにしている。
【0170】
実施例9
リポソーム性シスプラチンおよびダウノルビシンの有効性
別個のリポソーム中に調合したシスプラチンおよびダウノルビシンの有効性を、実施例26に詳述した通りにSCID/rag2マウス(H460異種移植モデル)において評価した。H460腫瘍を有するマウス(各群当たりマウス4匹)に対して、生理食塩水、または実施例7で非拮抗的であるとインビトロで特定されたモル比10:1のシスプラチン/ダウノルビシンを投与した。シスプラチンおよびダウノルビシンは、押出処理後にDMPC/CholリポソームをHBSに対して透析した点を除き、実施例8の記載の通りに、それぞれDMPC/Chol(55:45mol%)リポソームおよびDSPC/DSPE-PEG2000(95:5mol%)リポソーム中に調合した。薬剤の組合せを投与するマウスに対しては、薬剤を遊離性薬剤混合物(10:1の混合物、モル比)として、またはリポソーム性ダウノルビシンとリポソーム性シスプラチンとの同時投与(リポソーム製剤;モル比10:1)として、第14、17および21日に投与した。遊離物および調合物の投与のいずれに関しても、用量は2.0mg/kgシスプラチンおよび0.375mg/kgダウノルビシンとした。脂質用量は、リポソーム性シスプラチンについては400mg/kgであり、リポソーム性ダウノルビシンについては3.75mg/kgであった。
【0171】
図14はその結果を示しており、ここで各データ点は指定日に測定した平均腫瘍サイズ+/-SEMを表す。生理食塩水対照(黒丸)は腫瘍増殖を抑制しなかった;同様に、遊離混合物(黒い逆三角)も腫瘍増殖に対してわずかな効果しか示さなかった。これに対して、リポソーム製剤(白い三角)は腫瘍増殖を少なくとも32日間にわたって抑制した。
【0172】
実施例10
拮抗的なモル比にある薬剤の組合せのリポソーム性投与の効果
シスプラチンおよびダウノルビシンを、DMPC/Chol(55:45mol%)リポソーム中に、実施例7で拮抗的であることが決定されたモル比1:1で共装填した。シスプラチンは受動的に封じ込め、ダウノルビシンは能動的に封じ込めてシスプラチン/ダウノルビシンのモル比1:1を達成した。単一のリポソーム中への薬剤の装填には、実施例8に概要を示した手順を用いた。
【0173】
DMPC/Cholリポソーム中にある製剤によって協調的な放出が達成されたか否かを明らかにするために、装填したリポソームをBalb/cマウスの尾静脈に2mg/kgシスプラチンおよび3.75mg/kgダウノルビシンの用量で注入した。指定された時点(各時点当たりマウス3匹)で血液を心臓穿刺によって採取し、EDTAをコーティングしたマイクロテイナーに入れた。試料を遠心して血漿を慎重に別のチューブに移した。脂質およびダウノルビシンの血漿中濃度はいずれも液体シンチレーション計数によって測定し、シスプラチン濃度は原子吸光分光法によって測定した。その結果を図15にまとめた(各データ点は指定時点で測定した薬剤の平均血漿中濃度+/-SDを表す)は、ダウノルビシンおよびシスプラチンが血漿から同じ速度で排出され、そのために血漿中濃度(nmol/mL)がモル比1:1に維持されたことを示している(図15の挿入図を参照されたい)。
【0174】
有効性試験は実施例26の記載の通りに行い、H460腫瘍を有する雌性SCJD/rag2マウスに対して2.5mg/kgシスプラチン、4.7mg/kgダウノルビシンを混合物またはリポソーム製剤として、さらに52.83mg/kgの脂質を第11、15および19日に投与した。
【0175】
図16における有効性の結果(各データ点は指定日に測定した平均腫瘍サイズ+/-SEMを表す)は、非拮抗的な比(モル比10:1)の薬剤では腫瘍増殖がかなり抑制されたのに対して(図14参照)、拮抗的な比にあるダウノルビシンおよびシスプラチンの投与が腫瘍増殖の抑制に無効であることを示している。すなわち、これらの結果は、インビトロである濃度範囲にわたって非拮抗的な作用を示す比にある薬剤の組合せを選択することの重要性を強く示している。実際には、図16で用いた薬剤の用量(2.5mg/kgシスプラチンおよび4.7mg/kgダウノルビシン)は、図14で用いた用量よりも高い(2mg/kgシスプラチン、0.375mg/kgダウノルビシン)。
【0176】
実施例11
シスプラチンおよびトポテカンの相乗作用
相乗的、相加的または拮抗的な作用を決定するための上記の手順(実施例1参照)を、シスプラチン/トポテカンをモル比10:1およびモル比1:1の両方で用いて再び行った。図17Aに示されているように、シスプラチン/トポテカンはモル比10:1では、細胞の5%〜99%が影響を受ける(fa=0.05〜fa=0.99)広範囲の用量にわたって非拮抗的な相互作用を示した。これに対して、モル比1:1のシスプラチン/トポテカンは、同じfa範囲にわたって高度に拮抗的であった(図17A)。
【0177】
濃度のこのような影響は、シスプラチン/トポテカンのさまざまなモル比に関するCI最大値の算出からも明らかとなった。図17Bに示されているように、拮抗作用はモル比1:1で最大となるように思われ、非拮抗的な作用はいずれかの薬剤が過剰に存在する場合に明らかである。
【0178】
実施例12
インビボでのシスプラチンおよびトポテカンの相乗作用の維持
シスプラチンおよびトポテカンを、それぞれDMPCICholリポソームおよびDSPC/Cholリポソーム中に調合した上で、実施例11で相乗的であることが特定されたモル比10:1でマウスに静脈内注射した。
【0179】
リポソーム性シスプラチンは、DMPCおよびコレステロール(55:45mol%)からなる脂質薄膜を、150mM NaClおよび8.5mg/mLシスプラチンからなる溶液で水和させることによって調製した。その結果生じたMLVを、重ね合わせた2枚の孔径100nmフィルターに80℃で10回通過させることによって押し出し処理した。押出処理の後に試料を冷却し、沈殿したシスプラチンを遠心分離によって除去した。リポソーム中に封入されなかった残留性の可溶性シスプラチンは、HBSに対する透析によって除去した。封入されなかったシスプラチンの除去後に、薬剤の濃度を原子吸光分光法によって測定した。
【0180】
リポソーム性トポテカンは、DSPCおよびコレステロール(55:45mol%)から構成される脂質薄膜の300mM MnSO4溶液による水和によって調製した。その結果生じたMLVを、重ね合わせた2枚の100nmフィルターに65℃で10回通過させることによって押し出し処理した。押出処理の後に、リポソームをゲル濾過クロマトグラフィーによってSHE緩衝液(300mMスクロース、20mM HEPESおよび30mM EDTA、pH 7.4)中に入れ換えた。トポテカンの装填は、1μgのA23187/脂質μmol(A23187は二重層を介した二価金属イオンとプロトン2個との交換を媒介する陽イオン性イオノフォアである)および最終的なトポテカン/脂質比が0.08(w/w)となるトポテカンを添加することによって開始し、続いて溶液を65℃に15分間保った。トポテカン装填の程度は、封入された薬剤と封入されなかった薬剤をゲル濾過クロマトグラフィーを用いて分離してTriton X-100中に溶解した後に、380nmでの吸光度によって測定した。
【0181】
この製剤を、SCID/rag2雌性マウスに尾静脈から静脈内注射した。リポソーム製剤の用量は5mg/kgシスプラチンおよび0.758mg/kgトポテカンとした。指定された時点(各時点当たりマウス3匹)で血液を心臓穿刺によって採取し、EDTAをコーティングしたマイクロテイナーに入れた。試料を遠心して血漿を慎重に別のチューブに移した。放射標識した脂質の定量には液体シンチレーション計数を用いた。シスプラチンは原子吸光分光法を用いて測定し、トポテカンはリポソームを過剰量の界面活性剤で破壊した後に蛍光分光法(励起380nmおよび発光518nm)によって測定した。
【0182】
図18(各データ点は指定時点で測定した薬剤の平均血漿中濃度+/-SDを表す)は、上記のリポソーム中にある状態で送達した場合に静脈内投与後の種々の時点でシスプラチンの血漿中濃度はトポテカンの概ね10倍であったことから、シスプラチンおよびトポテカンの血漿中濃度がモル比10:1に維持されたことを示している。これらの結果は、2種類のリポソーム中に封入された2種類の薬剤の薬剤保持特性およびリポソーム排出特性を、協調的な薬剤排出速度が実現されるように協調させうることを示している。図18の挿入図は、静脈内投与後に血漿中に存在するシスプラチン-トポテカンのモル比(+/-SD)が、時間経過に伴ってほとんど変化しないことを示している。
【0183】
非拮抗的な比が確実にインビボで維持されるように、シスプラチンおよびトポテカンを単一のリポソーム中に調合することもできる。これは、シスプラチンの受動的封じ込めの後にイオノフォアを介したトポテカンの装填を行うことによって実施しうる。シスプラチン粉末を150mM MnCl2溶液中に溶解することにより、シスプラチン溶液をまず調製する。シスプラチンのMnCl2溶液への溶解性を最大限に高めるために、この溶液を65℃に加熱する。DSPC/Chol(55:45mol%)から構成され、微量の3H-CHEを含む脂質薄膜をシスプラチン/MnCl2溶液によって水和させる。その結果生じたMLVを65℃で2枚の100nmフィルターを通して押し出し、合計10回通過させる。続いて、製剤を室温まで冷却し、溶液を2000×gで遠心することによって不溶性シスプラチンを製剤から除去する。その結果得られた、リポソーム性シスプラチンおよび可溶性であるが封入されなかったシスプラチンを含む上清を、SHE緩衝液、300mMスクロース、20mM HEPESおよび30mM EDTA(pH 7.4)に対して室温で一晩透析する。
【0184】
その後に、イオノフォアを介したプロトン勾配を用いて、トポテカンをリポソーム中に装填する。薬剤の取り込みは薬剤-脂質重量比0.08:1(w/w)で行わせる。二価陽イオンイオノフォアA23187(1μgイオノフォア/脂質μmol)をリポソームに添加した後に、A23187の二重層への取り込みを促すために混合物を60℃で15分間インキュベートする。その後にトポテカンを添加し、薬剤の取り込みを促すために混合物を60℃で60分間インキュベートする。試料を300mMスクロースに対して透析することにより、封入されなかったトポテカンおよびA23187を調製物から除去する。トポテカン装填の程度は380nmでの吸光度を測定することによって定量する。シスプラチンレベルは原子吸光分光法によって測定し、脂質レベルは液体シンチレーション計数によって測定する。
【0185】
実施例13
リポソーム性シスプラチンおよびトポテカンの有効性
別個のリポソーム中に装填されたシスプラチンおよびトポテカンの有効性を、2種類の薬剤を別個のリポソーム中に調合した上で、それらの製剤を、実施例11で非拮抗的であることが特定されたモル比10:1で投与することによって調べた。リポソーム性シスプラチンは、実施例12の手順の記載の通りにDMPC/Chol(55:45mol%)リポソーム中に受動的に封じ込めた。トポテカンは、トポテカンの装填は最終的なトポテカン/脂質重量比が0.1(w/w)となるように行った点を除き、実施例12の通りにDSPC/Chol(55:45mol%)中に調合した。装填後に外部緩衝液をHBSに交換した。
【0186】
有効性試験は実施例26に詳述した通りに行い、H460腫瘍を有する雌性SCID/rag2マウス(各群当たりマウス4匹)に対して、生理食塩水(対照)、シスプラチン/トポテカンの遊離性混合物またはリポソーム性混合物を、実施例11で非拮抗的として特定されたモル比10:1で静脈内に投与した(第13、17、21日に)。遊離物およびリポソーム製剤のいずれの投与に関しても、用量は1.6mg/kgシスプラチンおよび0.25mg/kgトポテカンとした。脂質用量はシスプラチン製剤については250mg/kgであり、トポテカン製剤ではそれよりも2.5mg/kg多かった。
【0187】
図19はその結果を示している(各データ点は、指定日に測定した平均腫瘍サイズ+/-SEMを表す)。生理食塩水対照(黒丸)およびシスプラチン/トポテカンの10:1混合物(黒い三角)は、腫瘍体積の増加を停止させる効果がなかった。しかし、シスプラチン/トポテカン10:1リポソーム製剤(白い三角)は、腫瘍体積の増加を少なくとも35日間にわたって阻止した。
【0188】
実施例14
シスプラチンおよびイリノテカンの相乗作用
シスプラチンおよびイリノテカンのモル比1:1、10:1、1:5および1:10での組合せを、上記の方法に従って相乗作用、相加作用または拮抗作用に関して調べた(実施例1参照)。図20Aにまとめた結果は、モル比10:1、1:5および1:10ではfa値の全範囲にわたって非拮抗的であるが、1:1の比はfa値のかなりの範囲にわたって拮抗的であったことを示している。図20Bはさらに、組合せ指数の最大値をシスプラチン-イリノテカンのモル比に対してプロットすることによって概括されているように、併用効果の性質に対する比の依存性を示している。
【0189】
実施例15
インビボでのシスプラチンおよびイリノテカンの相乗作用の維持
6.0mg/mLシスプラチンを含む225mM銅(75mM CuCl2、150mM CuSO4、トリエタノールアミン(TEA)、pH 6.8)中に脂質薄膜を再水和させた点を除き、実施例5の記載の通りに調製したDSPC/DSPG(80:20mol%)リポソーム中に、シスプラチンおよびイリノテカンを共装填した。押出処理および封入されなかった薬剤の除去の後のリポソーム性シスプラチンの濃度はシスプラチン0.025モル/脂質1モルであった。その結果生じたリポソームをSHE、pH 6.8に対して一晩透析した。続いてイリノテカンを調製物に添加し、リポソームを45℃で1.5時間インキュベートした。HPLCによる評価では、添加したイリノテカンの60%がリポソームに装填された。続いてリポソームの緩衝液を接線流によって0.9%生理食塩水に交換した。接線流の後に、リポソームは最初のシスプラチンおよびイリノテカンの約80%を保持していた。それぞれ原子吸光分光法およびHPLC分析によって評価したシスプラチンおよびイリノテカンの分析により、最終的な調製物がシスプラチン-イリノテカンをモル比1:3で有することが示された。SCID/rag2マウスに対して2mg/kgシスプラチンおよび38.6mg/kgイリノテカンを静脈内投与した。指定された時点(各時点当たりマウス3匹)で血液を心臓穿刺によって採取し、EDTAをコーティングしたマイクロテイナーに入れた。試料を遠心して血漿を慎重に別のチューブに移した。イリノテカンおよびシスプラチンの血漿中濃度はそれぞれHPLCおよび原子吸光分光法によって測定した。
【0190】
図21における結果(各データ点は指定時点で測定した薬剤の平均血漿中濃度+/-SDを表す)は、DSPC/DSPGリポソーム中に共装填されたシスプラチンおよびイリノテカンを含む製剤の静脈内注射後の薬剤排出速度は同程度であり、非拮抗的な薬剤モル比が投与後24時間の経過にわたって維持されたことを示している。
【0191】
インビボでのリポソーム性シスプラチンおよびイリノテカンの協調的な放出も、この2種類の薬剤を別個の送達媒体中に調合した上で薬剤をモル比1:5(シスプラチン/イリノテカン)で投与することによって達成された。
【0192】
リポソーム性シスプラチンは、上記の受動的な装填法に従って調製した。DMPC/Chol(55:45mol%)からなる脂質薄膜を、8.5mg/mLシスプラチンを含む150mM NaCl溶液によって水和させた後に、上記の通りの押出処理を行った。上記の通りに遠心分離後の上清中のリポソームを収集した後に、接線流透析によってHBSに交換した。
【0193】
リポソーム性イリノテカンは、DSPC/DSPE-PEG2000(95:5mol%)からなる脂質薄膜を、150mM CuCl2、20mMヒスチジン、pH 6.8(pHはTEAで調整)からなる溶液によって水和させることによって調製した。その結果生じたMLVを65℃で重ね合わせた2枚の孔径100nmフィルターを通して押し出し、接線流によて緩衝液をHBSに交換した。押出処理を行ったリポソームに対して、イリノテカンを薬剤脂質重量比が0.1:1となるように60℃で1分間かけて装填した。イリノテカンの装填の程度は、Triton X-100中への溶解後に370nmでの吸光度によって測定した;脂質レベルは液体シンチレーション計数によって測定した。
【0194】
リポソーム性シスプラチンを雄性SCID/rag2マウスに対して薬剤用量2.0mg/kgとして投与し、リポソーム性イリノテカンをマウスに対して20mg/kgで投与した。指定された時点(各時点当たりマウス3匹)で血液を心臓穿刺によって採取し、EDTAをコーティングしたマイクロテイナーに入れた。試料を遠心して血漿を慎重に別のチューブに移した。イリノテカンの血漿中濃度はHPLCによって測定し、シスプラチン濃度は原子吸光分光法によって測定した。
【0195】
これらのリポソーム製剤中にある状態でこの相乗的な比(モル比1:5)でともに投与されたシスプラチンおよびイリノテカンは、種々の時点でイリノテカンの血漿中濃度(nmol/mL)がシスプラチンの概ね5倍(nmol/mL)であったことから示されるように、1:5というこの比を静脈内投与後にも維持した(図22)。
【0196】
実施例16
リポソーム性シスプラチンおよびイリノテカンの有効性
別個のリポソーム中に調合されたリポソーム性シスプラチンおよびイリノテカンに対して有効性試験を行った。実施例15に詳述した通りに、シスプラチンはDMPC/Chol(55:45mol%)リポソーム中に受動的に封じ込め、イリノテカンはDSPC/DSPE-PEG2000(95:5mol%)リポソーム中に装填した。実施例26に記載の方法に従って、リポソーム性シスプラチンおよびイリノテカンを、実施例14で非拮抗的であることが決定されたモル比1:5で、H460腫瘍を有するSCID/rag2マウスに対して同時投与した。リポソーム性シスプラチンおよびイリノテカンの投与(第14、18および22日に各群当たりマウス4匹に対して)は、非拮抗的なモル比1:5で以下の用量を用いて行った:1mg/kgシスプラチン、10mg/kgイリノテカンおよび130mg/kg脂質(白い四角);2.5mg/kgシスプラチン、25mg/kgイリノテカンおよび175mg/kg脂質(白い三角);または5mg/kgシスプラチン、50mg/kgイリノテカンおよび250mg/kg脂質(白い逆三角)。遊離性シスプラチン/イリノテカンは、モル比1:5を反映する1mg/kgシスプラチンおよび10mg/kgイリノテカンの用量で投与した(黒い四角)。
【0197】
図23(各データ点は指定日に測定した平均腫瘍サイズ+/-SEMを表す)は、遊離性薬剤混合物および生理食塩水を投与したマウスと比較して、リポソーム製剤の場合には腫瘍増殖が大きく抑制されたことを示している。
【0198】
実施例17
薬剤および脂質の組合せの相乗作用
ビノレルビンを、脂質二重層に組み入れられたPOPS(黒い逆三角)、DPPS(黒い三角)、DLPS(黒丸)、DSPS(黒い菱形)またはDOPS(黒い四角)といった種々の治療的脂質候補とともにモル比1:1で含む組合せを、上記の方法を用いて相加的、相乗的または拮抗的な作用に関して検討した(実施例1参照)。
【0199】
図24における結果は、ビノレルビンと被験脂質とのすべての組合せが、fa値のかなりの範囲にわたってH460細胞に対して相乗作用を示すことを示している。特に、ビノレルビンとDLPS、DSPSおよびDOPSとの組合せはfa値の大半で相乗作用を示し、fa=0.2〜fa=0.8の範囲で最も顕著であった。
【0200】
実施例18
リポソーム性ビノレルビンおよびホスファチジルセリンの薬物動態
以下の通りに、SM/Chol/DPPS/DSPE-PEG2000(35:45:10:10mol%)からなるリポソームを調製して、ビノレルビンを装填した。
【0201】
脂質をクロロホルム中に100mg/mLの濃度で溶解した上で、適当な量を混ぜ合わせた。これに対する例外はDPPSであり、これはCHCl3/メタノール/H2O/クエン酸緩衝液(20:10.5:1:1 v/v)を用いて25mg/mLの濃度で溶解した。製剤化工程の全体を通じて脂質を追跡するために、微量の放射性脂質3H-CHEをこの時点で添加した。ごくわずかな溶媒が残るまでクロロホルムをN2ガス流によって除去した。その結果得られた脂質薄膜を真空下に一晩置き、残留性溶媒を除去した。脂質薄膜をクエン酸緩衝液(300mM、pH 4.0)中に再水和させ、その結果生じたMLVを65℃で2枚の孔径100nmフィルターを通して押し出し、合計10回通過させた。
【0202】
外部緩衝液のpHを0.2M Na2HPO4を用いて徐々に高めるpH勾配装填法により、ビノレルビンをこれらの製剤中に装填した。既知の量のリポソームを対応する量のビノレルビン(薬剤/脂質重量比0.1(w/w))と混ぜ合わせ、60℃で15分間インキュベートした。pH勾配を成立させるために、0.2M Na2HPO4をクエン酸緩衝液の10倍の容積として添加した。実施例17で非拮抗的として特定されたビノレルビン/ホスファチジルセリンのモル比が達成されるように、ビノレルビンをリポソーム中に装填した。
【0203】
ビノレルビン装填リポソームの可溶化には界面活性剤OGPを用いた;薬剤濃度は270nmでの吸光度によって測定し、脂質の定量には液体シンチレーション計数を用いた。
【0204】
その結果得られたビノレルビン装填リポソームおよび遊離ビノレルビンを、SCID/rag2マウスに対して薬剤用量10mg/kgとして静脈内投与した。指定された時点(各時点当たりマウス3匹)で血液を心臓穿刺によって採取し、EDTAをコーティングしたマイクロテイナーに入れた。試料を遠心して血漿を慎重に別のチューブに移した。シンチレーション計数を用いて、残留性3H-CHEリポソームマーカーに関して血液を分析した。ビノレルビンの血漿中濃度はHPLCによりアッセイした。
【0205】
図25Aおよび25Bは、ビノレルビンを封入したSM/Chol/DPPS/DSPE-PEG2000リポソームが、遊離ビノレルビンの投与と比較して、薬剤の血漿中濃度を大きく高めることを示している。遊離ビノレルビンの平均血中濃度曲線下面積(AUC)は0.112μg/mLであったが、リポソーム中に製剤化すると125.3μg/mLに増加し、平均AUCは1120倍に増加した。
【0206】
実施例19
H460ヒト肺癌モデルにおけるリポソーム性ホスファチジルセリンおよびビノレルビンの有効性
実施例18の記載の通りに、DSPC/Chol/DSPS/DSPE-PEG2000(35:45:10:10mol%)リポソーム、SM/Chol/DPPS/DSPE-PEG2000(35:45:10:10mol%)リポソームおよびDAPC/Chol/DPPS/DSPE-PEG2000(35:45:10:10mol%)リポソームを調製し、ビノレルビンを装填した。ホスファチジルセリンおよびビノレルビンはリポソーム中に非拮抗的なモル比(1:1)で存在させた。有効性試験は、実施例26に記載した通り、H460ヒト肺癌モデルを用いて行った。
【0207】
図26は、DSPC/Chol/DPPS/DSPE-PEG2000およびSM/Chol/DPPS/DSPE-PEG2000からなり、ビノレルビンが封入されたリポソームの静脈内投与を受けたH460腫瘍保有マウス(各群当たりマウス4匹)について示しており、この投与により、遊離ビノレルビンおよび生理食塩水の投与後に観察された速度と比較して腫瘍増殖速度が低下した。腫瘍細胞の接種から13日、17日および21日後に、遊離ビノレルビンを5mg/kgで投与し、リポソーム性ビノレルビンは薬剤については5mg/kg、脂質については50mg/kgの用量で投与した。
【0208】
図27(各データ点は指定日に測定した平均腫瘍サイズ+/-SEMを表す)は、SM/Chol/DPPS/DSPE-PEG2000;DAPC/Chol/DPPS/DSPE-PEG2000およびDSPC/Chol/DSPS/DSPE-PEG2000からなり、ビノレルビンを封入したリポソームが、遊離ビノレルビンおよび生理食塩水と比較して、時間経過に伴う腫瘍体積の減少を示したことを示している。腫瘍保有マウス(1群当たり4匹)に対してビノレルビンを用量5mg/kgで投与し(遊離性およびリポソーム性)、リポソーム群に対しては脂質を用量50mg/kgで投与した。マウスには第13、17および21日に静脈内投与を行った。
【0209】
実施例20
マウス白血病モデルにおけるリポソーム性ホスファチジルセリンおよびビノレルビンの有効性
リポソームを孔径100nmフィルターと80nmフィルターを重ねたものを通して押し出した点を除き、実施例18の記載の通りに、SM/Chol/DPPS/DSPE-PEG2000(35:45:10:10mol%)からなるリポソームを調製し、ビノレルビンを装填した。
【0210】
実施例27の記載の通りに、P388/wt細胞をBDF-1マウスの腹腔内に接種した。その後に、BDF-1雌性マウスに以下のいずれかを腹腔内投与した:生理食塩水;遊離ビノレルビン(10mg/kg)、およびビノレルビンが装填されたSM/Chol/DPPS/DSPE-PEG2000リポソーム(10mg/kgビノレルビンおよび100mg/kg脂質)。遊離性およびリポソーム性ビノレルビンの腹腔内投与を第1日に一投与群当たりマウス4匹に行った。
【0211】
図28に示した生存曲線は、SM/Chol/DPPS/DSPE-PEG2000からなるリポソーム中に封入したビノレルビンの投与が、遊離ビノレルビンおよび生理食塩水の投与と比較して、BDF-1マウスの生存率の大幅な増加をもたらしたことを示している。
【0212】
実施例21
スフィンゴシンとドキソルビシンとの共調合
ホスファチジルセリンのほかに、他の治療的脂質をリポソーム膜に組み入れることもできる。例えば、スフィンゴシンおよびスフィンゴシン類似体はリポソーム中に調合するのに適合した脂質であり、水性内部に封入した治療薬(例えば、ドキソルビシン)とともに調合することができる。このような薬学的組成物(スフィンゴシン)の調製は以下のようにして行いうる。
【0213】
スフィンゴシンの典型的なリポソーム製剤は、DSPC/Chol/スフィンゴシン(45:45:10mol%)から構成される。以前の実施例に詳述した通りに脂質薄膜を調製する。脂質薄膜をクエン酸緩衝液(300mM、pH 4)中に再水和させ、その結果生じたMLVを65℃で2枚の100nmフィルターを通して押し出し、合計10回通過させる。続いて、pH勾配を成立させるためにHBS(pH 7.4)で平衡化したセファデックスG-50カラムを通過させることによる、リポソームの外部緩衝液の交換によるpH勾配装填法を用いて、ドキソルビシンをこれらの製剤に装填する。
【0214】
続いて、リポソームおよびドキソルビシン溶液をともに60℃でインキュベートし、装填を行わせる。種々の時点での装填の程度を決定するために、100μLの試料を1mL セファデックスG-50スパンカラムにかけ、その後に遠心処理を行う。脂質の定量に液体シンチレーション計数を用い、ドキソルビシンの定量には480nmでの吸光度を用いて、スパンカラムの溶出液の薬剤-脂質比を導き出す。薬剤のアッセイのためには、リポソームをTriton X-100中でインキュベートした後に吸光度の読み取りを行う。
【0215】
実施例22
フロクスウリジン(FUDR)およびイリノテカン(CPT-11)の相乗作用
相加的、相乗的または拮抗的な作用を計測するための上記の手順を、HT 29細胞においてFUDR/CPT-11をモル比10:1、5:1、1:1、1:5および1:10で用いて再び行った。上記の通りにCI-fa曲線を作成することにより、組合せ指数を各用量に関して決定した。影響を受けたHT-29細胞の率に対してCIをプロットした図29におけるデータは、相乗作用に対する濃度の影響を明らかに示している。比が5:1または1:1の場合は、影響率の値の全範囲(0.2〜0.8)にわたって相乗作用が観察されるが、比10:1では0.76未満のfa値で非拮抗的であり、FUDR/CPT-11のモル比が1:5であれば0.62未満のfa値で非拮抗的である。比1:10はfa値の実質的な範囲(50%超)にわたって拮抗的である。これらの結果に基づき、この比がfa値の有意な範囲(細胞の1%超が影響を受ける範囲の少なくとも20%)にわたって相乗作用を示したことから、FUDR:CPT-11のモル比1:1を製剤化および有効性試験の対象として選択した。5:1および10:1の比で調製した製剤も、fa値の実質的な範囲にわたって所定の非拮抗的な比を示すという必要条件を満たすと考えられる。
【0216】
実施例23
インビボでのFUDRおよびCPT-11の相乗作用の維持
FUDRおよびCPT-11をDSPC/DSPG/Chol(70:20:10mol%)リポソーム中に、実施例Aにおいて相乗的であることが特定されたモル比1:1で調合した。DSPCおよびコレステロールをクロロホルム中に溶解し、DSPGをクロロホルム/メタノール/水(16/18/1)中に溶解することによって脂質薄膜を調製した。これらの溶液を所定のモル比が得られるように混ぜ合わせ、リポソーム脂質標識として微量の14C-CHEを添加した。溶媒の除去後に、生成された脂質薄膜を、250mM CuSO4および25mg/mLのFUDR(微量の3H-FUDRを含む)からなる溶液によって70℃で水和させた。その結果生じたMLVを、70℃で、重ね合わせた2枚の孔径100nmフィルターに10回通過させることにより、押し出し処理を行った。その後に、リポソームの緩衝液を接線流透析によってSHE、pH 7.4に交換し、それにより、封入されなかったFUDRおよびCuSO4を除去した。
【0217】
FUDRとCPT-11とのモル比が1:1となるように、CPT-11をこれらのリポソームに添加した。試料を50℃で5分間インキュベートすることにより、CPT-11のリポソームへの装填を促した。装填の後に、EDTAまたは封入されなかった薬剤を除去するために接線流透析によって試料をHBS、pH 7.4中に入れ換えた。CPT-11装填の程度はHPLCを用いて計測した。FUDRおよび脂質のレベルは液体シンチレーションを用いて測定した。
【0218】
これらの製剤をBalb/c雌性マウスに尾静脈を介して静脈内注射した。リポソーム製剤の用量はFUDR 8.38mg/kgおよびCPT11 20mg/kgであった。指定された時点(各時点当たりマウス3匹)で血液を心臓穿刺によって採取し、EDTAをコーティングしたマイクロテイナーに入れた。試料を遠心して血漿を別のチューブに移した。血漿中の放射標識脂質およびFUDRの定量には液体シンチレーション計数を用いた。CPT-11の血漿中濃度はHPLCを用いて定量した。
【0219】
図30は、上記のリポソーム中にある状態で送達した場合に静脈内投与後の種々の時点でFUDRの血漿中濃度がCPT-11とほぼ等しかったことから、FUDRおよびCPT-11の血漿中濃度がモル比1:1に維持されたことを示している。各データ点は指定時点で測定した薬剤の平均血漿中濃度(薬剤nmol/血漿mL)+/-標準偏差を表す。
【0220】
実施例24
リポソーム性FUDRおよびCPT-11の有効性
薬剤装填後にリポソーム外部緩衝液を0.9%NaClに交換した点を除いて、実施例Bの記載の通りに、FUDRおよびイリノテカンをモル比1:1で共封入したDSPC/DSPG/Chol(70:20:10mol%)リポソームを調製した。
【0221】
実施例26の方法を用いて、側腹部に2×106個のHT-29細胞を皮下接種した雌性SCID/rag2マウスにおける有効性試験を行った。腫瘍をサイズが180mg(0.18cm3)と計測されるまで増殖させ、その時点(第21日)で指定の製剤を注入した。腫瘍の増殖はキャリパーによる直接測定によって評価した。マウスに対して、生理食塩水、モル比1:1の遊離性薬剤混合物、またはFUDR/CPT-11のモル比1:1でのリポソーム製剤のいずれかを単回投与(矢印)した。遊離物およびリポソーム製剤の投与のいずれに関しても、用量は9.25mg/kg FUDRおよび25mg/kg CPT-11とした。リポソーム製剤試料に関する脂質用量は278mg/kgであった。
【0222】
図31に提示した結果は、単一のリポソーム中にモル比1:1で封入されたFUDRおよびCPT-11が、遊離性薬剤混合物または生理食塩水のいずれかを注入したマウスと比較して、治療活性を有意に改善することを示している。各データ点は平均腫瘍サイズ+/-平均の標準誤差(SEM)である。
【0223】
実施例25
さまざまな3剤の組合せに関するCIの決定
トポテカン、シスプラチン、HB5-5A(エデルフォシン類似体)およびスフィンゴシンを含む組合せを、標準的なテトラゾリウム比色MTT細胞傷害性アッセイ法(実施例の項の細胞傷害性アッセイ法を参照)を用いて、相加的、相乗的または拮抗的な作用に関して検討した。以前の実施例に記載したメディアンエフェクト解析を用いて併用効果を算出した。CTとfaとの関係を示すグラフを以前の実施例の記載の通りに作成し、fa値0.50、0.75および0.90に対応するCI値(ED50、75および90によって表される)を以下の表に報告した。
a 組合せ指数(CI)は、用量-効果分析に関するChou-Talalay理論に基づく相乗作用(CI<0.9)または相加作用(CIが0.9〜1.1の間)の決定に用いられる。各値はCalcuSynソフトウエアを用いて算出される。
b ED50、ED75、ED90は、測定される応答のそれぞれ50、75または90%が影響を受ける薬剤の用量のことを指す。
【0224】
実施例26
腫瘍モデルの作製、細胞の調製および皮下固形腫瘍の移植法
H460ヒト非小細胞肺癌細胞は、NCIのDCTC腫瘍レポジトリ(Tumor Repository)から入手しうる。細胞は組織培養下で最大20回の継代にわたって維持される。20回の継代後に、液体窒素中で保存していた凍結貯蔵物から新たな細胞を増殖させる。培養細胞が集密度80〜90%に達した時点で、それらをハンクス平衡塩類溶液ですすぎ洗い、付着細胞を0.25%トリプシン溶液によって取り出す。細胞を血球計算器で算定し、培地で希釈して濃度を20×106細胞/mLにする。
【0225】
各マウスの背部の約2cm×2cmの領域を電気バリカンを用いて剃毛する。第0日に、28g針を用いて、マウスに1×106個の腫瘍細胞、容積50μLを皮下接種する(接種1回/匹)。
【0226】
腫瘍が約0.50〜0.100cm3の所定のサイズに達した時点で、投与の1日前または投与日(〜第10日から第14日)に、すべての腫瘍を計測する。適切な腫瘍サイズを選択した後に、小さすぎる腫瘍と大きすぎる腫瘍を除外し、腫瘍をランダム分布させて(n=4)、各群の平均腫瘍体積を決定する。
【0227】
マウスを、生理食塩水対照、媒体対照、陽性対照および被験物質の種々の希釈物などの対照群および投与群からなる適切な投与群に配分する。
【0228】
投与群は以下の通りとする。
a 単回投与または4〜7日毎の投与などの代替的な投薬計画も考慮しうる。
【0229】
マウスに対して、個々のマウスの体重に基づき、処方用量(10μL/g、指定に応じて)をマウスに投与するために必要な容積の試料を静脈内注射する。
【0230】
腫瘍増殖の測定値はバーニアキャリパーを用いて計測し、測定は投与日から開始する。腫瘍の長さの測定値(mm)は最長方向の軸から求め、幅の測定(mm)はこの軸に対して直交するようにする。長さおよび幅の測定値から、式(L×W2/2)/1000を用いて腫瘍体積(cm3)を算出する。腫瘍測定時にマウスの体重を記録する。
【0231】
個々のマウスの体重を、有効性試験の間、最終投与から14日間にわたり、さまざまな日(一般には月曜、水曜および金曜というように1日おきにする)に記録する。
【0232】
全マウスを、前投与期間および投与期間にわたって、生死および病状に関して少なくとも1日1回観察し、必要と思われる場合はそれ以上観察する。詳細には、病的状態の徴候は、体重減少、食欲変化、体毛の荒れ、身づくろいの減少、行動変化(歩行変化など)、傾眠および顕著なストレスの発現に基づく。重篤な中毒または腫瘍に伴う病的状態が認められた場合には、マウスを安楽死させ(CO2による窒息)、中毒の他の徴候を評価するために剖検を行う。瀕死状態のマウスは人道的な理由から屠殺する必要があり、屠殺に関する判断は動物飼育技術者(Animal Care Technician)および試験責任者(Study Director)/監督者(Manager)の裁量に委ねられる。これらの所見のいずれかおよびすべてを生データとして記録し、死亡時はその翌日として記録する。
【0233】
データは表または図の形式として以下の通りに提示する。
1.投与開始前およびグループ分けの後の各群に関する個々のマウスの腫瘍体積。
2.時間の関数としての各群の平均体重。
3.時間の関数としての各群の平均腫瘍体積。
4.図および表を含む生データの作成、これには腫瘍増殖と時間との関係、腫瘍増殖阻止および腫瘍増殖遅延が含まれる。
5.異常または顕著な観察所見のまとめ。
【0234】
実施例27
腫瘍モデルの作成、細胞の調製および腹腔内腫瘍の移植方法
マウスを体重別にグループ分けする。25g針を用いて、BDF-1マウス(n=4)の腹腔内に1×106個のP388細胞、容積500μLを移植する(第0日)。ATCC癌細胞貯蔵部門(ATCC tumor repository)から入手したP388細胞をBDF-1マウスの腹水として維持し、新たなマウスに週1回継代する。マウスを安楽死させ、20g針を用いて腹壁を介して腹水細胞を採取する。実験に用いる細胞は継代回数3〜20回のものを用いる。マウス体内での20回の継代後には、液体窒素中の凍結貯蔵物から新たな細胞を増殖させてマウスに接種する。実験用の細胞はハンクス平衡塩類溶液ですすぎ洗い、血球計算器で算定した上で、HBSSで希釈して濃度を2×106個/mLとする。
【0235】
試験群のグループ分けは、全マウスに腫瘍細胞を投与した後にランダムに行う。必要なグループ分けは固形腫瘍試験に対して行われるものと同様である(実施例26参照)。
【0236】
マウスに対して、個々のマウスの体重に基づき、処方用量(10μL/g、指定に応じて)をマウスに投与するために必要な容積の試料を静脈内または腹腔内に注射する。腹腔内腫瘍の場合には、腫瘍細胞の接種から1日後に投与を開始する。
【0237】
マウスの健康状態を毎日詳細に観察する。病的状態および病態の進行の徴候については実施例26に記載した通りに詳細に観察する。マウスの体重を検査時に測定する。
【0238】
マウスを屠殺した後には、腫瘍量および/または臓器外観の生理的に観察される変化の程度を評価するために肉眼的剖検を行う。所見は記録する。
【0239】
群の体重を、有効性試験の間、最終投与から14日間にわたって月曜から金曜まで記録する。
【0240】
全マウスを、前投与期間および投与期間にわたって生死および病状に関して少なくとも1日1回観察し、必要と思われる場合はそれ以上観察する。詳細には、病的状態の徴候は、体重減少、食欲変化、体毛の荒れ、身づくろいの減少、行動変化(歩行変化など)、傾眠および顕著なストレスの発現に基づく。重篤な中毒または腫瘍に伴う病的状態が認められた場合には、マウスを安楽死させ(CO2による窒息)、中毒の他の徴候を評価するために剖検を行う。瀕死状態のマウスは人道的な理由から屠殺する必要があり、屠殺に関する判断は動物飼育技術者(Animal Care Technician)および試験責任者(Study Director)/監督者(Manager)の裁量に委ねられる。これらの所見を生データとして記録し、死亡時はその翌日として記録する。
【0241】
データは表または図の形式として提示し、これには時間の関数としての各群の平均体重および寿命の延長が含まれる。
【特許請求の範囲】
【請求項1】
送達媒体を含む組成物であって、生物作用に関するインビトロアッセイ法において細胞の1%超が影響を受ける(fa>0.01)濃度範囲の、少なくとも5%にわたって、培養下の妥当な細胞または無細胞系に対する非拮抗的な生物作用が示されるような第1の薬剤対第2の薬剤のモル比で、少なくとも第1の治療薬および第2の治療薬が該送達媒体に安定的に会合している、組成物。
【請求項2】
送達媒体を含む組成物であって、妥当な細胞に対する非拮抗的な細胞傷害作用または細胞分裂抑制作用または生物作用が示されるような第1の薬剤対第2の薬剤のモル比で、少なくとも第1の治療薬および第2の治療薬が該送達媒体に安定的に会合しており、該薬剤が抗腫瘍薬である、組成物。
【請求項3】
細胞傷害性に関するインビトロアッセイ法において妥当な細胞の1%超が影響を受ける(fa>0.01)濃度範囲の、少なくとも5%にわたって非拮抗的な作用が示される、請求項2記載の組成物。
【請求項4】
インビトロアッセイ法において細胞の10〜90%が影響を受ける(fa=0.1〜0.9)濃度範囲の、少なくとも5%にわたって非拮抗的な作用が示される、請求項1記載の組成物。
【請求項5】
インビトロアッセイ法において細胞の20〜80%が影響を受ける(fa=0.2〜0.8)濃度範囲の、少なくとも5%にわたって非拮抗的な作用が示される、請求項4記載の組成物。
【請求項6】
インビトロアッセイ法において細胞の20〜80%が影響を受ける濃度範囲の、少なくとも20%にわたって非拮抗的な作用が示される、請求項5記載の組成物。
【請求項7】
対象に投与した場合に、薬剤を送達媒体と安定的に会合させずに同じ比で投与した場合に得られるよりも高い治療活性がもたらされる、請求項1記載の組成物。
【請求項8】
薬剤が抗腫瘍薬である、請求項1記載の組成物。
【請求項9】
第3の薬剤を含む、請求項1記載の組成物。
【請求項10】
送達媒体の平均径が4.5〜500nmの間である、請求項1記載の組成物。
【請求項11】
媒体の平均径が250nm未満である、請求項10記載の組成物。
【請求項12】
送達媒体が、
リポソーム、および/または
脂質ミセル、および/または
ブロック共重合体ミセル、および/または
微粒子、および/または
ナノ粒子、および/または
ポリマー脂質ハイブリッド系、および/または
誘導体化された一本鎖ポリマー
を含む、請求項1または2記載の組成物。
【請求項13】
第1および第2の薬剤が共封入されている、請求項1記載の組成物。
【請求項14】
インビトロアッセイ法において細胞の10〜90%が影響を受ける(fa=0.1〜0.9)濃度範囲の、少なくとも5%にわたって非拮抗的な作用が示される、請求項13記載の組成物。
【請求項15】
インビトロアッセイ法において細胞の20〜80%が影響を受ける(fa=0.2〜0.8)濃度範囲の、少なくとも5%にわたって非拮抗的な作用が示される、請求項14記載の組成物。
【請求項16】
インビトロアッセイ法において細胞の20〜80%が影響を受ける濃度範囲の、少なくとも20%にわたって非拮抗的な作用が示される、請求項15記載の組成物。
【請求項17】
対象に投与した場合に、薬剤を送達媒体と安定的に会合させずに同じ比で投与した場合に得られるよりも高い治療活性がもたらされる、請求項2記載の組成物。
【請求項18】
薬剤の少なくとも1つが、DNA損傷物質、DNA修復阻害物質、トポイソメラーゼI阻害物質、トポイソメラーゼII阻害物質、細胞チェックポイント阻害物質、CDK阻害物質、受容体チロシンキナーゼ阻害物質、細胞傷害物質、アポトーシス誘導物質、代謝拮抗物質、細胞周期制御阻害物質、治療的脂質、テロメラーゼ阻害物質、血管形成抑制物質、ミトコンドリア毒、シグナル伝達阻害物質および免疫作用物質からなる群より選択される、請求項2記載の組成物。
【請求項19】
第1の薬剤が細胞傷害物質であって第2の薬剤が細胞周期阻害物質である、または
第1の薬剤がDNA損傷物質であって第2の薬剤がDNA修復阻害物質である、または
第1の薬剤がトポイソメラーゼI阻害物質であって第2の薬剤がS/G2チェックポイント阻害物質もしくはG2/Mチェックポイント阻害物質である、または
第1の薬剤がG1/Sチェックポイント阻害物質もしくはサイクリン依存性キナーゼ阻害物質であって第2の薬剤がG2/Mチェックポイント阻害物質である、または
第1の薬剤が受容体キナーゼ阻害物質であって第2の薬剤が細胞傷害物質である、または
第1の薬剤がアポトーシス誘導物質であって第2の薬剤が細胞傷害物質である、または
第1の薬剤がアポトーシス誘導物質であって第2の薬剤が細胞周期制御物質である、または
第1の薬剤がテロメラーゼ阻害物質であって第2の薬剤が細胞周期制御阻害物質である、または
第1および第2の薬剤が代謝拮抗物質である、または
第1および第2の薬剤が細胞傷害物質である、または
第1の薬剤が治療的脂質であって第2の薬剤が細胞傷害物質である、または
第1の薬剤がトポイソメラーゼI阻害物質であって第2の薬剤がDNA修復阻害物質である、または
アポトーシス誘導物質がセリン含有脂質である、
請求項18記載の組成物。
【請求項20】
第1の薬剤がイリノテカンであって第2の薬剤が5-FUもしくはFUDRである、または
第1の薬剤がシスプラチン(もしくはカルボプラチン)であって第2の薬剤が5-FUもしくはFUDRである、または
第1の薬剤がイダルビシンであって第2の薬剤がAraCもしくはFUDRである、または
第1の薬剤がオキサリプラチンであって第2の薬剤が5-FUもしくはFUDRである、または
第1の薬剤がイリノテカンであって第2の薬剤がシスプラチン(もしくはカルボプラチン)である、または
第1の薬剤がゲムシタビンであって第2の薬剤がシスプラチン(もしくはカルボプラチン)である、または
第1の薬剤がメトトレキサートであって第2の薬剤が5-FUもしくはFUDRである、または
第1の薬剤がパクリタキセルであって第2の薬剤がシスプラチン(もしくはカルボプラチン)である、または
第1の薬剤がエトポシドであって第2の薬剤がシスプラチン(もしくはカルボプラチン)である、または
第1の薬剤がドセタキセルもしくはパクリタキセルであって第2の薬剤がドキソルビシンである、または
第1の薬剤がドキソルビシンであって第2の薬剤がビノレルビンである、または
第1の薬剤がカルボプラチンであって第2の薬剤がビノレルビンである、または
第1の薬剤が5-FUもしくはFUDRであって第2の薬剤がゲムシタビンである、
請求項2記載の組成物。
【請求項21】
送達媒体を含む組成物を調製するための方法であって、媒体には、非拮抗的なモル比にある少なくとも第1の治療薬および第2の治療薬が安定的に会合しており、
a)第1および第2の薬剤のモル比を、細胞の1%超が該比の薬剤によって影響を受ける(fa>0.01)濃度範囲の、少なくとも5%にわたり非拮抗的であるよう、生物作用に関する妥当な細胞培養物アッセイ法または無細胞アッセイ法において決定する段階、ならびに
b)段階a)において非拮抗的であると決定されたモル比で、該送達媒体に薬剤を封入する段階
を含む方法。
【請求項22】
薬物送達媒体を含む組成物を調製するための方法であって、該媒体には、非拮抗的なモル比にある少なくとも第1の治療薬および第2の治療薬が安定的に会合しており、
a)妥当な細胞培養物アッセイ法または無細胞アッセイ法において、非拮抗的である該第1および第2の薬剤のモル比を決定する段階、この際、該薬剤は抗腫瘍薬である、および
b)該送達媒体に、段階a)において非拮抗的であると決定されたモル比の薬剤を封入する段階
を含む方法。
【請求項23】
細胞傷害性または細胞増殖抑制(cytostasis)に関するインビトロアッセイ法において細胞の1%〜99%が影響を受ける(fa=0.01〜0.99)濃度範囲の、少なくとも5%にわたって非拮抗的な作用が示される、請求項21記載の方法。
【請求項24】
細胞傷害性または細胞増殖抑制に関するインビトロアッセイ法において細胞の10〜90%が影響を受ける(fa=0.1〜0.9)濃度範囲の、少なくとも5%にわたって非拮抗的な作用が示される、請求項23記載の方法。
【請求項25】
細胞傷害性または細胞増殖抑制に関するインビトロアッセイ法において細胞の20〜80%が影響を受ける(fa=0.2〜0.8)濃度範囲の、少なくとも5%にわたって非拮抗的な作用が示される、請求項24記載の方法。
【請求項26】
細胞傷害性または細胞増殖抑制に関するインビトロアッセイ法において細胞の20〜80%が影響を受ける濃度範囲の、少なくとも20%にわたって相乗作用が示される、請求項25記載の方法。
【請求項27】
細胞傷害性または細胞増殖抑制に関するインビトロアッセイ法において細胞の1%超が影響を受ける(fa>0.01)濃度範囲の、少なくとも5%にわたって非拮抗的な作用が示される、請求項22記載の方法。
【請求項28】
該非拮抗的な作用が、細胞傷害性または細胞増殖抑制に関するインビトロアッセイ法において細胞の10〜90%が細胞影響を受ける(fa=0.1〜0.9)濃度範囲の少なくとも5%にわたって示される、請求項27記載の方法。
【請求項29】
細胞傷害性または細胞増殖抑制に関するインビトロアッセイ法において細胞の20〜80%が影響を受ける(fa=0.2〜0.8)濃度範囲の、少なくとも5%にわたって非拮抗的な作用が示される、請求項28記載の方法。
【請求項30】
細胞傷害性または細胞増殖抑制に関するインビトロアッセイ法において細胞の20〜80%が影響を受ける濃度範囲の、少なくとも20%にわたって相乗作用が示される、請求項29記載の方法。
【請求項31】
決定が、薬剤の少なくとも1つの比を複数の濃度で試験することと、ある濃度範囲にわたって該比に関して相乗的、相加的または拮抗的な作用を算出するためのアルゴリズムを適用することとを用いる、請求項21記載の方法。
【請求項32】
複数の比を試験することを用いる、請求項31記載の方法。
【請求項33】
アルゴリズムがChou-Talalayのメディアンエフェクト法である、請求項31記載の方法。
【請求項34】
薬剤が抗腫瘍薬である、請求項31記載の方法。
【請求項35】
薬剤の少なくとも1つが、DNA損傷物質、DNA修復阻害物質、トポイソメラーゼI阻害物質、トポイソメラーゼII阻害物質、チェックポイント阻害物質、CDK阻害物質、受容体チロシンキナーゼ阻害物質、細胞傷害物質、アポトーシス誘導物質、代謝拮抗物質、細胞周期制御阻害物質、治療的脂質、テロメラーゼ阻害物質、血管形成抑制物質、ミトコンドリア毒、シグナル伝達阻害物質および免疫作用物質からなる群より選択される、請求項22記載の方法。
【請求項36】
第1の薬剤が細胞傷害物質であって第2の薬剤が細胞周期阻害物質である、または
第1の薬剤がDNA損傷物質であって第2の薬剤がDNA修復阻害物質である、または
第1の薬剤がトポイソメラーゼI阻害物質であって第2の薬剤がS/G2チェックポイント阻害物質もしくはG2/Mチェックポイント阻害物質である、または
第1の薬剤がG1/Sチェックポイント阻害物質もしくはサイクリン依存性キナーゼ阻害物質であって第2の薬剤がG2/Mチェックポイント阻害物質である、または
第1の薬剤が受容体キナーゼ阻害物質であって第2の薬剤が細胞傷害物質である、または
第1の薬剤がアポトーシス誘導物質であって第2の薬剤が細胞傷害物質である、または
第1の薬剤がアポトーシス誘導物質であって第2の薬剤が細胞周期制御物質である、または
第1の薬剤がテロメラーゼ阻害物質であって第2の薬剤が細胞周期制御阻害物質である、または
第1および第2の薬剤が代謝拮抗物質である、または
第1および第2の薬剤が細胞傷害物質である、または
第1の薬剤が治療的脂質であって第2の薬剤が細胞傷害物質である、または
第1の薬剤がトポイソメラーゼI阻害物質であって第2の薬剤がDNA修復阻害物質である、または
アポトーシス誘導物質がセリン含有脂質である、
請求項22記載の方法。
【請求項37】
第1の薬剤がイリノテカンであって第2の薬剤が5-FUもしくはFUDRである、または
第1の薬剤がシスプラチンであって第2の薬剤が5-FUもしくはFUDRである、または
第1の薬剤がイダルビシンであって第2の薬剤がAraCである、または
第1の薬剤がオキサリプラチンであって第2の薬剤が5-FUもしくはFUDRである、または
第1の薬剤がイリノテカンであって第2の薬剤がシスプラチン(もしくはカルボプラチン)である、または
第1の薬剤がゲムシタビンであって第2の薬剤がシスプラチン(もしくはカルボプラチン)である、または
第1の薬剤がメトトレキサートであって第2の薬剤が5-FUもしくはFUDRである、または
第1の薬剤がパクリタキセルであって第2の薬剤がシスプラチン(もしくはカルボプラチン)である、または
第1の薬剤がエトポシドであって第2の薬剤がシスプラチン(もしくはカルボプラチン)である、または
第1の薬剤がドセタキセルもしくはパクリタキセルであって第2の薬剤がドキソルビシンである、または
第1の薬剤がアドリアマイシンであって第2の薬剤がビノレルビンである、または
第1の薬剤がカルボプラチンであって第2の薬剤がビノレルビンである、または
第1の薬剤が5-FUもしくはFUDRであって第2の薬剤がゲムシタビンである、
請求項27記載の方法。
【請求項38】
請求項21記載の方法によって調製される組成物。
【請求項39】
請求項22記載の方法によって調製される組成物。
【請求項40】
請求項31記載の方法によって調製される組成物。
【請求項41】
対象における疾病状態を治療するための方法であって、このような治療を必要としている対象に対して、請求項1記載の組成物の治療的有効量を投与する段階を含む方法。
【請求項42】
対象における疾病状態を治療するための方法であって、このような治療を必要としている対象に対して、請求項2記載の組成物の治療的有効量を投与する段階を含む方法。
【請求項43】
対象における疾病状態を治療するための方法であって、このような治療を必要としている対象に対して、請求項38記載の組成物の治療的有効量を投与する段階を含む方法。
【請求項44】
対象における疾病状態を治療するための方法であって、このような治療を必要としている対象に対して、請求項39記載の組成物の治療的有効量を投与する段階を含む方法。
【請求項45】
対象がヒトである、請求項41記載の方法。
【請求項46】
対象が非ヒト哺乳動物または鳥類である、請求項41記載の方法。
【請求項47】
対象がヒトである、請求項42記載の方法。
【請求項48】
対象が非ヒト哺乳動物または鳥類である、請求項42記載の方法。
【請求項1】
送達媒体を含む組成物であって、生物作用に関するインビトロアッセイ法において細胞の1%超が影響を受ける(fa>0.01)濃度範囲の、少なくとも5%にわたって、培養下の妥当な細胞または無細胞系に対する非拮抗的な生物作用が示されるような第1の薬剤対第2の薬剤のモル比で、少なくとも第1の治療薬および第2の治療薬が該送達媒体に安定的に会合している、組成物。
【請求項2】
送達媒体を含む組成物であって、妥当な細胞に対する非拮抗的な細胞傷害作用または細胞分裂抑制作用または生物作用が示されるような第1の薬剤対第2の薬剤のモル比で、少なくとも第1の治療薬および第2の治療薬が該送達媒体に安定的に会合しており、該薬剤が抗腫瘍薬である、組成物。
【請求項3】
細胞傷害性に関するインビトロアッセイ法において妥当な細胞の1%超が影響を受ける(fa>0.01)濃度範囲の、少なくとも5%にわたって非拮抗的な作用が示される、請求項2記載の組成物。
【請求項4】
インビトロアッセイ法において細胞の10〜90%が影響を受ける(fa=0.1〜0.9)濃度範囲の、少なくとも5%にわたって非拮抗的な作用が示される、請求項1記載の組成物。
【請求項5】
インビトロアッセイ法において細胞の20〜80%が影響を受ける(fa=0.2〜0.8)濃度範囲の、少なくとも5%にわたって非拮抗的な作用が示される、請求項4記載の組成物。
【請求項6】
インビトロアッセイ法において細胞の20〜80%が影響を受ける濃度範囲の、少なくとも20%にわたって非拮抗的な作用が示される、請求項5記載の組成物。
【請求項7】
対象に投与した場合に、薬剤を送達媒体と安定的に会合させずに同じ比で投与した場合に得られるよりも高い治療活性がもたらされる、請求項1記載の組成物。
【請求項8】
薬剤が抗腫瘍薬である、請求項1記載の組成物。
【請求項9】
第3の薬剤を含む、請求項1記載の組成物。
【請求項10】
送達媒体の平均径が4.5〜500nmの間である、請求項1記載の組成物。
【請求項11】
媒体の平均径が250nm未満である、請求項10記載の組成物。
【請求項12】
送達媒体が、
リポソーム、および/または
脂質ミセル、および/または
ブロック共重合体ミセル、および/または
微粒子、および/または
ナノ粒子、および/または
ポリマー脂質ハイブリッド系、および/または
誘導体化された一本鎖ポリマー
を含む、請求項1または2記載の組成物。
【請求項13】
第1および第2の薬剤が共封入されている、請求項1記載の組成物。
【請求項14】
インビトロアッセイ法において細胞の10〜90%が影響を受ける(fa=0.1〜0.9)濃度範囲の、少なくとも5%にわたって非拮抗的な作用が示される、請求項13記載の組成物。
【請求項15】
インビトロアッセイ法において細胞の20〜80%が影響を受ける(fa=0.2〜0.8)濃度範囲の、少なくとも5%にわたって非拮抗的な作用が示される、請求項14記載の組成物。
【請求項16】
インビトロアッセイ法において細胞の20〜80%が影響を受ける濃度範囲の、少なくとも20%にわたって非拮抗的な作用が示される、請求項15記載の組成物。
【請求項17】
対象に投与した場合に、薬剤を送達媒体と安定的に会合させずに同じ比で投与した場合に得られるよりも高い治療活性がもたらされる、請求項2記載の組成物。
【請求項18】
薬剤の少なくとも1つが、DNA損傷物質、DNA修復阻害物質、トポイソメラーゼI阻害物質、トポイソメラーゼII阻害物質、細胞チェックポイント阻害物質、CDK阻害物質、受容体チロシンキナーゼ阻害物質、細胞傷害物質、アポトーシス誘導物質、代謝拮抗物質、細胞周期制御阻害物質、治療的脂質、テロメラーゼ阻害物質、血管形成抑制物質、ミトコンドリア毒、シグナル伝達阻害物質および免疫作用物質からなる群より選択される、請求項2記載の組成物。
【請求項19】
第1の薬剤が細胞傷害物質であって第2の薬剤が細胞周期阻害物質である、または
第1の薬剤がDNA損傷物質であって第2の薬剤がDNA修復阻害物質である、または
第1の薬剤がトポイソメラーゼI阻害物質であって第2の薬剤がS/G2チェックポイント阻害物質もしくはG2/Mチェックポイント阻害物質である、または
第1の薬剤がG1/Sチェックポイント阻害物質もしくはサイクリン依存性キナーゼ阻害物質であって第2の薬剤がG2/Mチェックポイント阻害物質である、または
第1の薬剤が受容体キナーゼ阻害物質であって第2の薬剤が細胞傷害物質である、または
第1の薬剤がアポトーシス誘導物質であって第2の薬剤が細胞傷害物質である、または
第1の薬剤がアポトーシス誘導物質であって第2の薬剤が細胞周期制御物質である、または
第1の薬剤がテロメラーゼ阻害物質であって第2の薬剤が細胞周期制御阻害物質である、または
第1および第2の薬剤が代謝拮抗物質である、または
第1および第2の薬剤が細胞傷害物質である、または
第1の薬剤が治療的脂質であって第2の薬剤が細胞傷害物質である、または
第1の薬剤がトポイソメラーゼI阻害物質であって第2の薬剤がDNA修復阻害物質である、または
アポトーシス誘導物質がセリン含有脂質である、
請求項18記載の組成物。
【請求項20】
第1の薬剤がイリノテカンであって第2の薬剤が5-FUもしくはFUDRである、または
第1の薬剤がシスプラチン(もしくはカルボプラチン)であって第2の薬剤が5-FUもしくはFUDRである、または
第1の薬剤がイダルビシンであって第2の薬剤がAraCもしくはFUDRである、または
第1の薬剤がオキサリプラチンであって第2の薬剤が5-FUもしくはFUDRである、または
第1の薬剤がイリノテカンであって第2の薬剤がシスプラチン(もしくはカルボプラチン)である、または
第1の薬剤がゲムシタビンであって第2の薬剤がシスプラチン(もしくはカルボプラチン)である、または
第1の薬剤がメトトレキサートであって第2の薬剤が5-FUもしくはFUDRである、または
第1の薬剤がパクリタキセルであって第2の薬剤がシスプラチン(もしくはカルボプラチン)である、または
第1の薬剤がエトポシドであって第2の薬剤がシスプラチン(もしくはカルボプラチン)である、または
第1の薬剤がドセタキセルもしくはパクリタキセルであって第2の薬剤がドキソルビシンである、または
第1の薬剤がドキソルビシンであって第2の薬剤がビノレルビンである、または
第1の薬剤がカルボプラチンであって第2の薬剤がビノレルビンである、または
第1の薬剤が5-FUもしくはFUDRであって第2の薬剤がゲムシタビンである、
請求項2記載の組成物。
【請求項21】
送達媒体を含む組成物を調製するための方法であって、媒体には、非拮抗的なモル比にある少なくとも第1の治療薬および第2の治療薬が安定的に会合しており、
a)第1および第2の薬剤のモル比を、細胞の1%超が該比の薬剤によって影響を受ける(fa>0.01)濃度範囲の、少なくとも5%にわたり非拮抗的であるよう、生物作用に関する妥当な細胞培養物アッセイ法または無細胞アッセイ法において決定する段階、ならびに
b)段階a)において非拮抗的であると決定されたモル比で、該送達媒体に薬剤を封入する段階
を含む方法。
【請求項22】
薬物送達媒体を含む組成物を調製するための方法であって、該媒体には、非拮抗的なモル比にある少なくとも第1の治療薬および第2の治療薬が安定的に会合しており、
a)妥当な細胞培養物アッセイ法または無細胞アッセイ法において、非拮抗的である該第1および第2の薬剤のモル比を決定する段階、この際、該薬剤は抗腫瘍薬である、および
b)該送達媒体に、段階a)において非拮抗的であると決定されたモル比の薬剤を封入する段階
を含む方法。
【請求項23】
細胞傷害性または細胞増殖抑制(cytostasis)に関するインビトロアッセイ法において細胞の1%〜99%が影響を受ける(fa=0.01〜0.99)濃度範囲の、少なくとも5%にわたって非拮抗的な作用が示される、請求項21記載の方法。
【請求項24】
細胞傷害性または細胞増殖抑制に関するインビトロアッセイ法において細胞の10〜90%が影響を受ける(fa=0.1〜0.9)濃度範囲の、少なくとも5%にわたって非拮抗的な作用が示される、請求項23記載の方法。
【請求項25】
細胞傷害性または細胞増殖抑制に関するインビトロアッセイ法において細胞の20〜80%が影響を受ける(fa=0.2〜0.8)濃度範囲の、少なくとも5%にわたって非拮抗的な作用が示される、請求項24記載の方法。
【請求項26】
細胞傷害性または細胞増殖抑制に関するインビトロアッセイ法において細胞の20〜80%が影響を受ける濃度範囲の、少なくとも20%にわたって相乗作用が示される、請求項25記載の方法。
【請求項27】
細胞傷害性または細胞増殖抑制に関するインビトロアッセイ法において細胞の1%超が影響を受ける(fa>0.01)濃度範囲の、少なくとも5%にわたって非拮抗的な作用が示される、請求項22記載の方法。
【請求項28】
該非拮抗的な作用が、細胞傷害性または細胞増殖抑制に関するインビトロアッセイ法において細胞の10〜90%が細胞影響を受ける(fa=0.1〜0.9)濃度範囲の少なくとも5%にわたって示される、請求項27記載の方法。
【請求項29】
細胞傷害性または細胞増殖抑制に関するインビトロアッセイ法において細胞の20〜80%が影響を受ける(fa=0.2〜0.8)濃度範囲の、少なくとも5%にわたって非拮抗的な作用が示される、請求項28記載の方法。
【請求項30】
細胞傷害性または細胞増殖抑制に関するインビトロアッセイ法において細胞の20〜80%が影響を受ける濃度範囲の、少なくとも20%にわたって相乗作用が示される、請求項29記載の方法。
【請求項31】
決定が、薬剤の少なくとも1つの比を複数の濃度で試験することと、ある濃度範囲にわたって該比に関して相乗的、相加的または拮抗的な作用を算出するためのアルゴリズムを適用することとを用いる、請求項21記載の方法。
【請求項32】
複数の比を試験することを用いる、請求項31記載の方法。
【請求項33】
アルゴリズムがChou-Talalayのメディアンエフェクト法である、請求項31記載の方法。
【請求項34】
薬剤が抗腫瘍薬である、請求項31記載の方法。
【請求項35】
薬剤の少なくとも1つが、DNA損傷物質、DNA修復阻害物質、トポイソメラーゼI阻害物質、トポイソメラーゼII阻害物質、チェックポイント阻害物質、CDK阻害物質、受容体チロシンキナーゼ阻害物質、細胞傷害物質、アポトーシス誘導物質、代謝拮抗物質、細胞周期制御阻害物質、治療的脂質、テロメラーゼ阻害物質、血管形成抑制物質、ミトコンドリア毒、シグナル伝達阻害物質および免疫作用物質からなる群より選択される、請求項22記載の方法。
【請求項36】
第1の薬剤が細胞傷害物質であって第2の薬剤が細胞周期阻害物質である、または
第1の薬剤がDNA損傷物質であって第2の薬剤がDNA修復阻害物質である、または
第1の薬剤がトポイソメラーゼI阻害物質であって第2の薬剤がS/G2チェックポイント阻害物質もしくはG2/Mチェックポイント阻害物質である、または
第1の薬剤がG1/Sチェックポイント阻害物質もしくはサイクリン依存性キナーゼ阻害物質であって第2の薬剤がG2/Mチェックポイント阻害物質である、または
第1の薬剤が受容体キナーゼ阻害物質であって第2の薬剤が細胞傷害物質である、または
第1の薬剤がアポトーシス誘導物質であって第2の薬剤が細胞傷害物質である、または
第1の薬剤がアポトーシス誘導物質であって第2の薬剤が細胞周期制御物質である、または
第1の薬剤がテロメラーゼ阻害物質であって第2の薬剤が細胞周期制御阻害物質である、または
第1および第2の薬剤が代謝拮抗物質である、または
第1および第2の薬剤が細胞傷害物質である、または
第1の薬剤が治療的脂質であって第2の薬剤が細胞傷害物質である、または
第1の薬剤がトポイソメラーゼI阻害物質であって第2の薬剤がDNA修復阻害物質である、または
アポトーシス誘導物質がセリン含有脂質である、
請求項22記載の方法。
【請求項37】
第1の薬剤がイリノテカンであって第2の薬剤が5-FUもしくはFUDRである、または
第1の薬剤がシスプラチンであって第2の薬剤が5-FUもしくはFUDRである、または
第1の薬剤がイダルビシンであって第2の薬剤がAraCである、または
第1の薬剤がオキサリプラチンであって第2の薬剤が5-FUもしくはFUDRである、または
第1の薬剤がイリノテカンであって第2の薬剤がシスプラチン(もしくはカルボプラチン)である、または
第1の薬剤がゲムシタビンであって第2の薬剤がシスプラチン(もしくはカルボプラチン)である、または
第1の薬剤がメトトレキサートであって第2の薬剤が5-FUもしくはFUDRである、または
第1の薬剤がパクリタキセルであって第2の薬剤がシスプラチン(もしくはカルボプラチン)である、または
第1の薬剤がエトポシドであって第2の薬剤がシスプラチン(もしくはカルボプラチン)である、または
第1の薬剤がドセタキセルもしくはパクリタキセルであって第2の薬剤がドキソルビシンである、または
第1の薬剤がアドリアマイシンであって第2の薬剤がビノレルビンである、または
第1の薬剤がカルボプラチンであって第2の薬剤がビノレルビンである、または
第1の薬剤が5-FUもしくはFUDRであって第2の薬剤がゲムシタビンである、
請求項27記載の方法。
【請求項38】
請求項21記載の方法によって調製される組成物。
【請求項39】
請求項22記載の方法によって調製される組成物。
【請求項40】
請求項31記載の方法によって調製される組成物。
【請求項41】
対象における疾病状態を治療するための方法であって、このような治療を必要としている対象に対して、請求項1記載の組成物の治療的有効量を投与する段階を含む方法。
【請求項42】
対象における疾病状態を治療するための方法であって、このような治療を必要としている対象に対して、請求項2記載の組成物の治療的有効量を投与する段階を含む方法。
【請求項43】
対象における疾病状態を治療するための方法であって、このような治療を必要としている対象に対して、請求項38記載の組成物の治療的有効量を投与する段階を含む方法。
【請求項44】
対象における疾病状態を治療するための方法であって、このような治療を必要としている対象に対して、請求項39記載の組成物の治療的有効量を投与する段階を含む方法。
【請求項45】
対象がヒトである、請求項41記載の方法。
【請求項46】
対象が非ヒト哺乳動物または鳥類である、請求項41記載の方法。
【請求項47】
対象がヒトである、請求項42記載の方法。
【請求項48】
対象が非ヒト哺乳動物または鳥類である、請求項42記載の方法。
【図1】

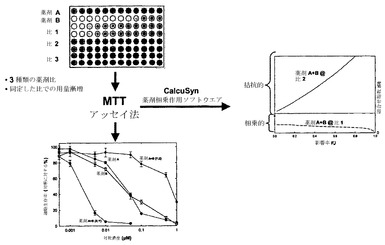
【図2A】
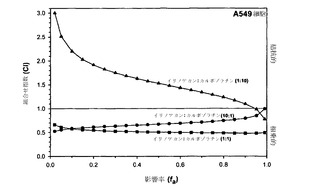
【図2B】
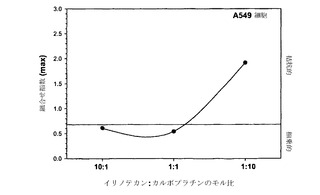
【図2C】
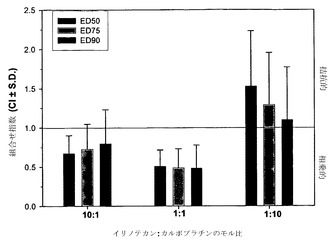
【図2D】
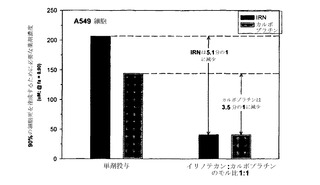
【図2E】
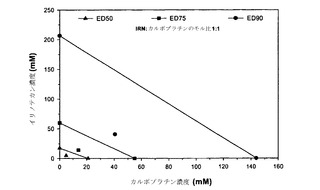
【図3】
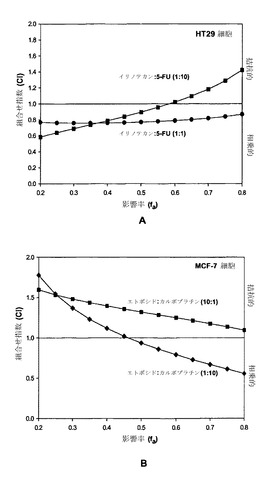
【図4】
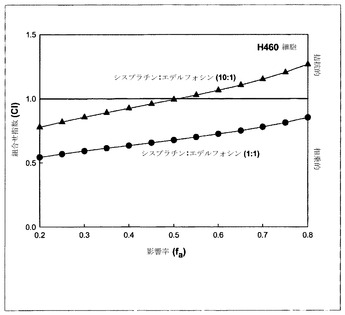
【図5A】
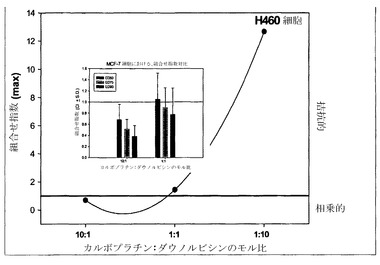
【図5B】
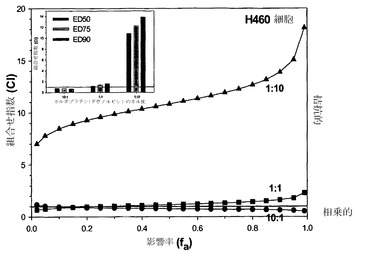
【図6】
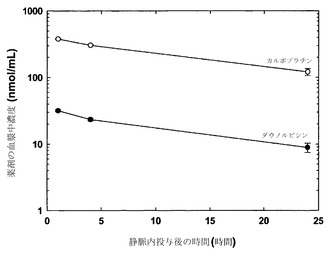
【図7】
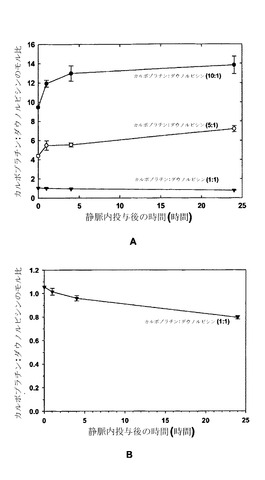
【図8】
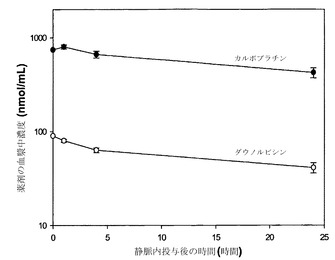
【図9】
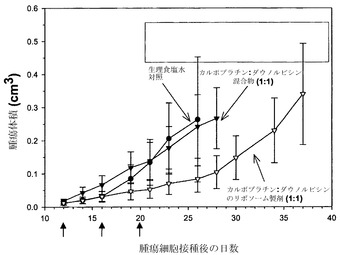
【図10】
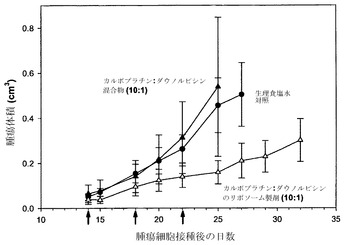
【図11】
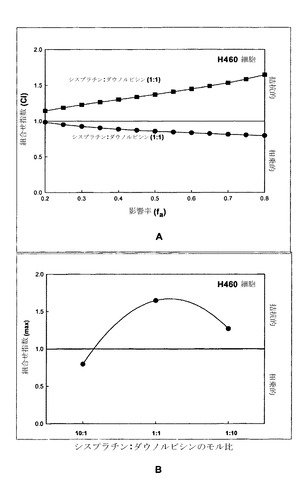
【図12】
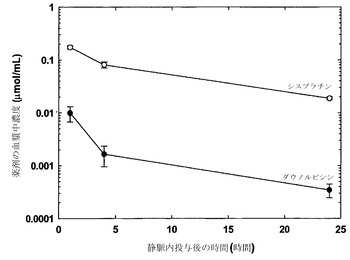
【図13】
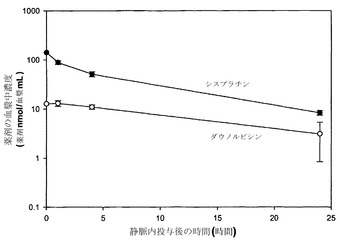
【図14】
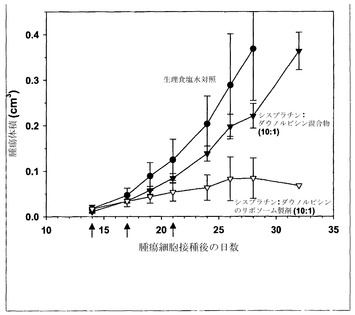
【図15】
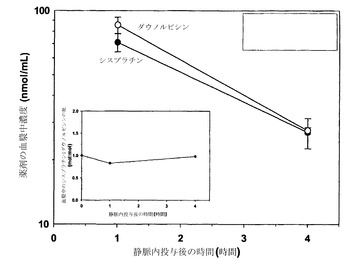
【図16】
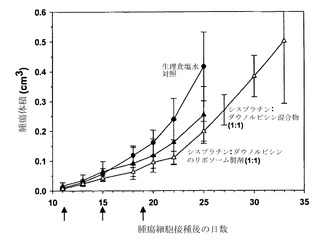
【図17】
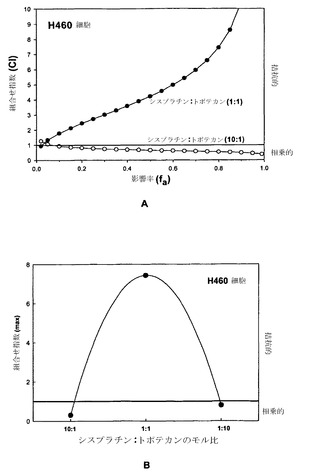
【図18】
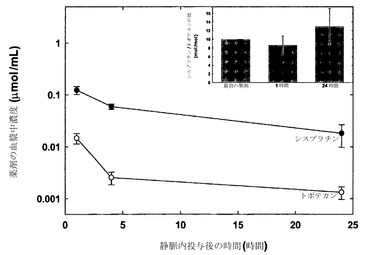
【図19】
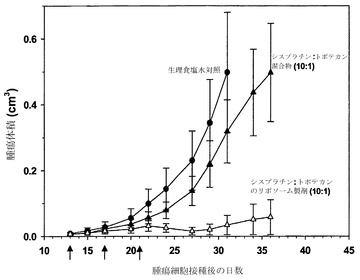
【図20】
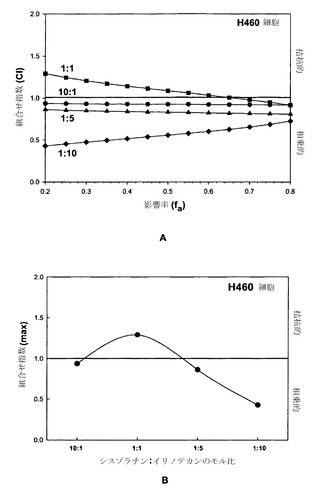
【図21】
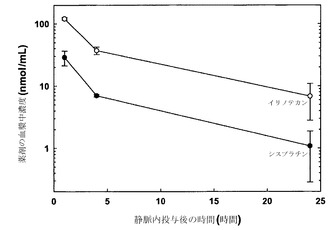
【図22】
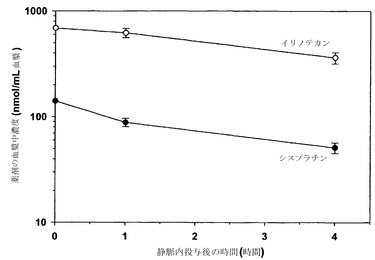
【図23】
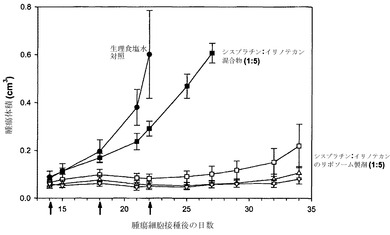
【図24】
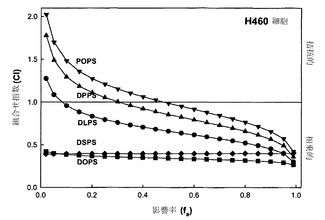
【図25】
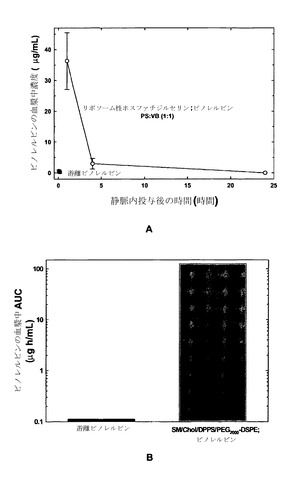
【図26】
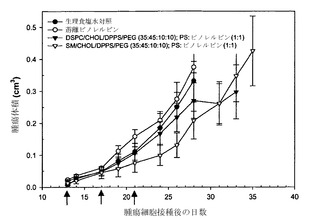
【図27】
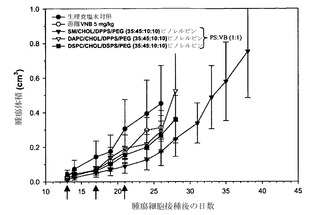
【図28】
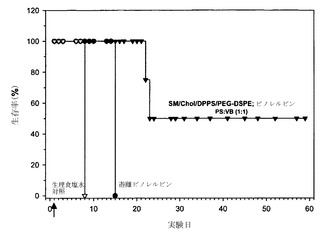
【図29】
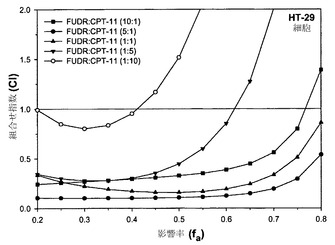
【図30】
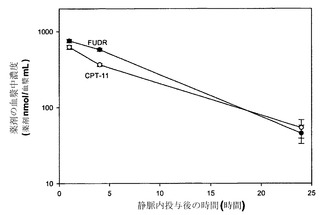
【図31】
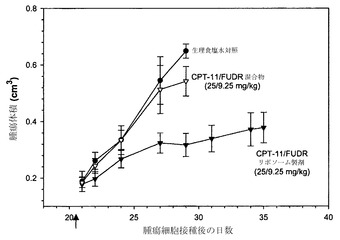
【図2A】
【図2B】
【図2C】
【図2D】
【図2E】
【図3】
【図4】
【図5A】
【図5B】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図31】
【公開番号】特開2010−180210(P2010−180210A)
【公開日】平成22年8月19日(2010.8.19)
【国際特許分類】
【出願番号】特願2010−34701(P2010−34701)
【出願日】平成22年2月19日(2010.2.19)
【分割の表示】特願2003−532029(P2003−532029)の分割
【原出願日】平成14年10月3日(2002.10.3)
【出願人】(507250966)セレーター ファーマシューティカルズ インコーポレイテッド (2)
【Fターム(参考)】
【公開日】平成22年8月19日(2010.8.19)
【国際特許分類】
【出願日】平成22年2月19日(2010.2.19)
【分割の表示】特願2003−532029(P2003−532029)の分割
【原出願日】平成14年10月3日(2002.10.3)
【出願人】(507250966)セレーター ファーマシューティカルズ インコーポレイテッド (2)
【Fターム(参考)】
[ Back to top ]