
使用方法
【課題】被験体における関節リウマチまたは多発性硬化症などの障害を治療する方法は、前記被験体にTh1分化もしくはTh17増殖の調節因子化合物を含む組成物を投与するステップによって実施される。
【解決手段】被験体における関節リウマチを治療する方法であって、前記被験体に、Th1分化もしくはTh17増殖の調節因子化合物を含む組成物を投与するステップを含む方法。
【解決手段】被験体における関節リウマチを治療する方法であって、前記被験体に、Th1分化もしくはTh17増殖の調節因子化合物を含む組成物を投与するステップを含む方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、関節リウマチや多発性硬化症を治療する方法に関する。
【背景技術】
【0002】
抗原に遭遇すると、天然CD4+ヘルパーT前駆(Thp)細胞は、2つの別個のサブセットである1型ヘルパーT(Th1)および2型ヘルパーT(Th2)に分化する。これらの分化したTh細胞は、それらの別個の機能能力および固有のサイトカインプロファイルの両方によって規定される。詳細には、Th1細胞は、インターフェロン−γ、インターロイキン(IL)−2、および腫瘍壊死因子(TNF)−βを産生するが、これらはマクロファージを活性化し、細胞媒介性免疫および食細胞依存性保護応答に関与する。対照的に、Th2細胞は、IL−4、IL−5、IL−6、IL−9、IL−10およびIL−13を産生することが公知であるが、これらは強力な抗体産生、好酸球活性化、および数種のマクロファージ機能の阻害に対して役割を果たすので、食細胞非依存性保護応答を提供する。したがって、Th1およびTh2細胞は、様々な免疫病理学的応答と関連している。
【0003】
さらに、各タイプのTh細胞の発生は、様々なサイトカイン経路によって媒介される。詳細には、IL−4はTh2分化を促進し、同時にTh1発生を遮断することが証明されている。対照的に、IL−12、IL−18およびIFN−γは、Th1細胞の発生に対して重大なサイトカインである。したがって、これらのサイトカイン自体がTh分極を駆動し、Th1とTh2との間の平衡を維持する正および負のフィードバック系を形成する。PEG2は、さらにまたインターフェロンγ(IFN−γ)産生およびT細胞増殖の抑制によってTH1応答を調節することに役割を果たすことも証明されている。
【0004】
Th1細胞は、様々な臓器特異的自己免疫疾患、クローン病、ヘリコバクター・ピロリ誘導性消化性潰瘍、急性腎臓同種移植拒絶反応、および原因不明の反復性流産の病因に関係している。対照的に、アレルゲン特異的Th2応答は、遺伝的に感受性の強い個体におけるアトピー性疾患の原因である。さらに、Th2は、オーメン(Omenn’s syndrome)症候群、特発性肺線維症、および進行性全身性硬化症において優勢である依然として未知の抗原に対して応答する。IL−17(Th−17サイトカインの符号)ノックアウトマウスは、炎症性関節炎の発生に対して顕著な耐性を示す。CIAモデルにおける関節破壊は、抗IL−17中和抗体の投与によって改善することができる。
【0005】
そこで、不均衡なTh1/Th2およびTh17細胞分化に関連する様々な状態を治療する際に有用である新規な治療的処置を開発する高度な満たされていない医療的必要性が残っている。これらの多くの状態のために現在利用できる治療選択肢は不適切である。したがって、Th1/Th2およびTh17のパラダイムは、アレルギー性および自己免疫障害を治療する戦略を開発するための論理的根拠を提供する。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】米国特許第7,189,715号明細書
【非特許文献】
【0007】
【非特許文献1】T−bet regulates IgG class switching and pathogenic auto Ab production,Proc.Natl.Acad.Sci.USA 99(8):5545−50(2002);Imbalance of Th1/Th2 transcription factors in patients with lupus nephritis,Rheumatology(Oxford) 45(8):951−7(2006)
【非特許文献2】Identification of a novel type 1 diabetes susceptibility gene,T−bet,Human Genetics 111(3):177−84(2004);T−bet controls autoaggressive CD8 lymphocyte response in type I diabetes,J.Exp.Med.199(8):1153−62(2004)
【非特許文献3】J.Mol.Med 81(8): 471−80 (2003)
【非特許文献4】Proc.Natl.Acad.Sci.USA 102(5):1596−601(2005)
【発明の概要】
【発明が解決しようとする課題】
【0008】
脂質メディエータであるプロスタグランジンE2(PGE2)は、免疫応答の様々な段階を変調することが証明されている。PGE2が細胞内cAMPの上昇およびIckの不活性化を通してCD4+T細胞活性化を抑制することは周知である。しかし、本明細書に記載したように、EP4受容体によるPGE2刺激は反対の作用も有する可能性があり、つまり、活性化CD4+細胞内でのTh1分化およびIL−17産生を促進することがある。これに一致して、EP4と新規な選択的EP4アンタゴニストまたはPGE2中和抗体いずれかとの拮抗作用は、Th1分化、Th17増殖、ならびに活性化された樹状細胞によるIL−23分泌を抑制する。PGE2によるTh1分化の誘導はPI3Kシグナリングによって媒介されるが、他方IL−17産生の刺激はcAMPシグナリングを必要とする。さらに、EP4アンタゴニストのDBA/1またはC57BL/6マウスへの投与は先天性および適応的免疫応答を抑制し、さらにコラーゲン誘導性関節炎(CIA)および実験的自己免疫脳脊髄炎(EAE)モデルにおける疾患を抑制したので、これはPGE2/EP4シグナリングがこれらの自己免疫病理に極めて重要に関係することを示した。これらの結果は、PGE2/EP4シグナリングの抑制が例えば関節リウマチおよび多発性硬化症などの炎症性自己免疫疾患を改善することに治療的価値を有する可能性があることを示唆している。
【課題を解決するための手段】
【0009】
本発明の第1態様は、被験体における関節リウマチを治療する方法であって、該被験体にTh1分化もしくはTh17増殖の調節因子化合物を含む組成物を投与するステップを含む方法である。一部の実施形態では、調節因子化合物は、式:
【化1】

の化合物、またはその医薬上許容される塩である。
【0010】
本発明のまた別の態様は、関節リウマチを治療するための医薬品の製造における化合物の使用であって、該医薬品は調節因子化合物を含み、該調節因子化合物はTh1分化もしくはTh17増殖の調節因子である使用である。一部の実施形態では、調節因子化合物は、式:
【化2】
の化合物、またはその医薬上許容される塩である。
【0011】
本発明のまた別の態様は、関節リウマチを治療するためのTh1分化もしくはTh17増殖の調節因子化合物である。
【0012】
本発明のまた別の態様は、被験体における多発性硬化症を治療する方法であって、該被験体にTh1分化もしくはTh17増殖の調節因子化合物を含む組成物を投与するステップを含む方法である。一部の実施形態では、調節因子化合物は、式:
【化3】
の化合物、またはその医薬上許容される塩である。
【0013】
本発明のまた別の態様は、多発性硬化症を治療するための医薬品の製造における化合物の使用であって、該医薬品は調節因子化合物を含み、該調節因子化合物はTh1分化もしくはTh17増殖の調節因子である使用である。一部の実施形態では、調節因子化合物は、式:
【化4】
の化合物、またはその医薬上許容される塩である。
【0014】
本発明のまた別の態様は、多発性硬化症を治療するためのTh1分化もしくはTh17増殖の調節因子化合物である。
【0015】
本発明のまた別の態様は、被験体における関節リウマチを治療する方法であって、該被験体にEP4アンタゴニストを含む組成物を投与するステップを含む方法である。一部の実施形態では、EP4アンタゴニストは、式:
【化5】
の化合物、またはその医薬上許容される塩である。
【0016】
本発明のまた別の態様は、関節リウマチを治療するための医薬品の製造における化合物の使用であって、該医薬品はEP4アンタゴニストを含む使用である。一部の実施形態では、EP4アンタゴニストは、式:
【化6】
の化合物、またはその医薬上許容される塩である。
【0017】
本発明のまた別の態様は、関節リウマチを治療するためのEP4アンタゴニストである。一部の実施形態では
【0018】
本発明のまた別の態様は、被験体における多発性硬化症を治療する方法であって、該被験体にEP4アンタゴニストを投与するステップを含む方法である。一部の実施形態では、EP4アンタゴニストは、式:
【化7】
の化合物、またはその医薬上許容される塩である。
【0019】
本発明のまた別の態様は、多発性硬化症を治療するための医薬品の製造における化合物の使用であって、該医薬品はEP4アンタゴニストを含む使用である。一部の実施形態では、EP4アンタゴニストは、式:
【化8】
の化合物、またはその医薬上許容される塩である。
【0020】
本発明のまた別の態様は、多発性硬化症を治療するためのEP4アンタゴニストである。
【発明の効果】
【0021】
本発明の方法によれば、関節リウマチや多発性硬化症を治療することができる。
【0022】
本発明の他の態様については、本明細書において開示し、以下でより詳細に考察する。
【図面の簡単な説明】
【0023】
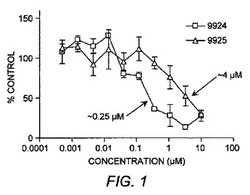
【図1】EP4結合アッセイにおけるER−819924−01およびそのエナンチオマーであるER−819925−01の滴定試験を示す図である。
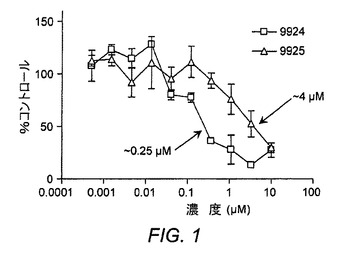
【図2】ER−819924およびER−819762が、SE302細胞におけるPGE2誘導性CRE−PLAPレポーター活性に対する強力な阻害活性を示した図である。どちらの化合物も0.3nM〜10μMの濃度で試験した。
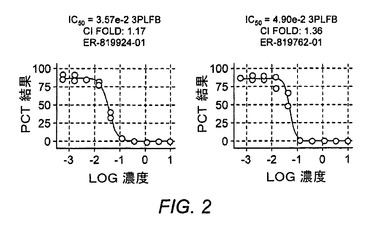
【図3】PGE2/EP4アゴニストが抗CD3/抗CD28を用いて刺激されたCD4+T細胞からのIL−17産生に及ぼす作用を示す図である。
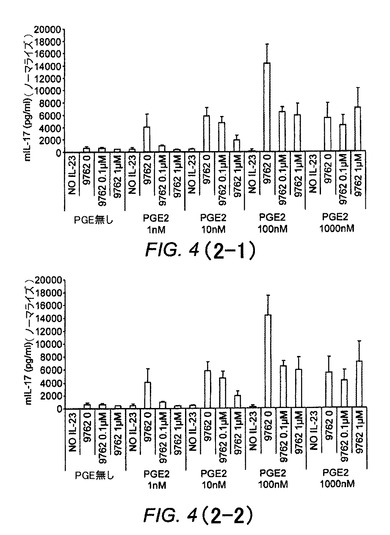
【図4(2−1)】ER−819762が活性化CD4+細胞からのPGE2/PGE1−OH強化IL−17産生を抑制することを示す図である。
【図4(2−2)】ER−819762が活性化CD4+細胞からのPGE2/PGE1−OH強化IL−17産生を抑制することを示す図である。
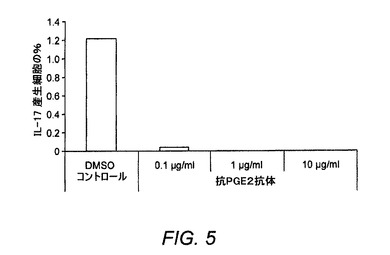
【図5】抗PGE2抗体がIL−23媒介性Th17増殖に及ぼす作用を示す図である。
【図6(2−1)】PGE2/EP4アゴニストがマウスTh1分化に及ぼす作用を示す図である。
【図6(2−2)】PGE2/EP4アゴニストがマウスTh1分化に及ぼす作用を示す図である(IFNg-Th1)。
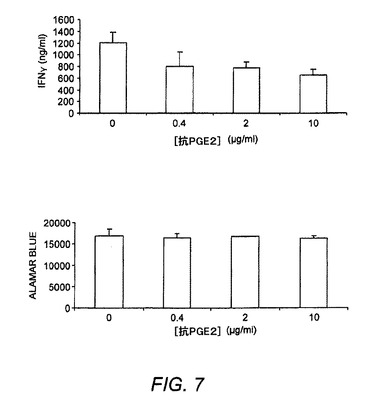
【図7】抗PGE2抗体がマウスTh1分化に及ぼす作用を示す図である。抗PGE2抗体は、Th1分化中に加えられた。
【図8】マウスTh1分化における抗PGE2抗体にER−819762は相加作用を及ぼさないことを示す図である。
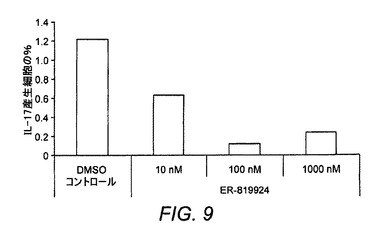
【図9】インビトロでのマウスIL−17増殖の抑制を示す図である。
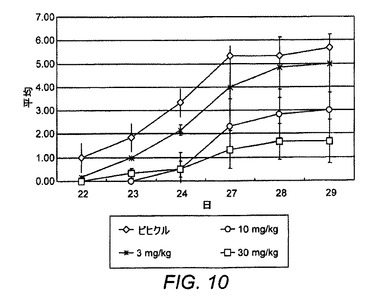
【図10】CIAにおけるER−819924−01の部分的治療評価を示す図である。
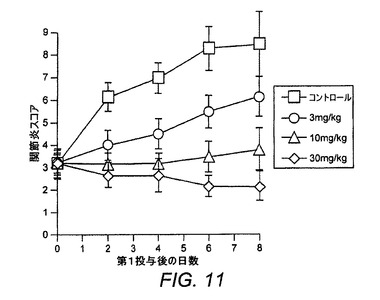
【図11】CIAにおけるER−819924−01の完全治療評価を示す図である。
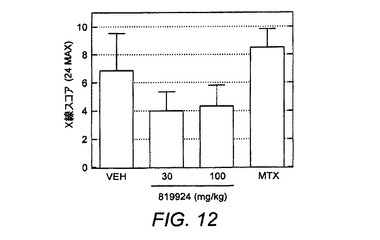
【図12】完全治療的CIA試験からのマウス足のX線分析を示す図である。X線スコアは、骨減少症、骨浸食および新規骨形成の組み合わせの測定値の指標である。
【図13】ヒトCD14+単球を7日間にわたりGM−CSF+IL−4を用いてimDCに分化させ、外因性PGE1−OH(1〜100nM)の存在下もしくは非存在下で、そしてER−819762を用いて、および用いずに、LPS/R−848を用いて再刺激した。
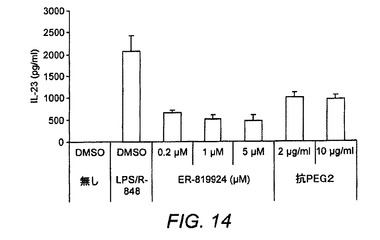
【図14】ヒトCD14+単球を7日間にわたりGM−CSF+IL−4を用いてimDC内に分化させ、外因性PGE1−OH(1〜100nM)の存在下もしくは非存在下で、そしてER−819924もしくは抗PGE2抗体を用いて、および用いずに、LPS/R−848を用いて再刺激した。
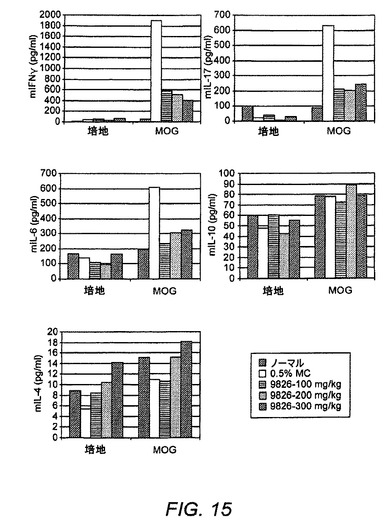
【図15】EAEにおけるTh1/Th17応答のエックスビボ抑制を示す図である。
【発明を実施するための形態】
【0024】
A.定義
本明細書で使用する用語「Th1分化もしくはTh17増殖の調節因子」または「Th1分化もしくはTh17増殖の調節因子化合物」または本明細書で使用する用語「調節因子化合物」は、天然CD4+T細胞のTh1細胞への分化を抑制する、減少させる、または阻害する化合物を意味する。一部の実施形態では、用語「Th1分化もしくはTh17増殖の調節因子」または本明細書で使用する用語「Th1分化もしくはTh17増殖の調節因子化合物」は、IL−17産生CD4+細胞の数または活性化CD4+細胞内でのIL−17産生を抑制する、減少させる、または阻害する化合物を意味する。
【0025】
本明細書で使用する「エナンチオマー的に純粋」は、ステレオマー的に純粋な化合物、または1つのキラル中心を有する化合物の組成物を意味している。
【0026】
本明細書で使用する「ステレオマー的に純粋」は、化合物の1つの立体異性体を含み、その化合物の他方の立体異性体を実際的に含んでいない化合物または組成物を意味している。例えば、1つのキラル中心を有する化合物のステレオマー的に純粋な組成物は、実質的に該化合物の反対エナンチオマーを含んでいない。2つのキラル中心を有する化合物のステレオマー的に純粋な組成物は、該化合物の他方のジアステレオマーを実質的に含んでいない。典型的なステレオマー的に純粋な化合物は、約80重量%超の該化合物の1つの立体異性体および約20重量%未満の該化合物の他方の立体異性体、より好ましくは約90重量%超の該化合物の1つの立体異性体および約10重量%未満の該化合物の他方の立体異性体、一層より好ましくは約95重量%超の該化合物の1つの立体異性体および約5重量%未満の該化合物の他方の立体異性体、および最も好ましくは約97重量%超の該化合物の1つの立体異性体および約3重量%未満の該化合物の他方の立体異性体を含んでいる(例えば、特許文献1)。
【0027】
本明細書で使用する「安定性」は、本明細書に開示した1つ以上の目的のためにそれらの産生、検出、ならびに好ましくはそれらの回収、精製、および使用を許容する条件に受けさせたときに、実質的に変化しない化合物に関する。一部の実施形態では、安定性化合物または化学的に実行可能な化合物は、少なくとも1週間にわたり、水分またはその他の化学的反応性条件の存在下で、40℃以下の温度で維持された場合に実質的に変化させられない化合物である。
【0028】
本明細書で使用する「アルキル」または「アルキル基」は、完全に飽和している直鎖状(すなわち、非分枝状)、分枝状、または環状炭化水素鎖を意味する。所定の実施形態では、アルキル基は、1〜3個の炭素原子を含有している。さらに他の実施形態では、アルキル基は2〜3個の炭素原子を含有し、そしてさらに他の実施形態では、アルキル基は1〜2個の炭素原子を含有している。所定の実施形態では、用語「アルキル」または「アルキル基」は、炭素環としても公知であるシクロアルキル基を意味する。典型的なC1−3アルキル基には、メチル、エチル、プロピル、イソプロピル、およびシクロプロピルが含まれる。
【0029】
本明細書で使用する「アルケニル」または「アルケニル基」は、1つ以上の二重結合を有する直鎖状(すなわち、非分枝状)、分枝状、または環状炭化水素鎖を意味する。所定の実施形態では、アルケニル基は、2〜4個の炭素原子を含有している。さらに他の実施形態では、アルケニル基は3〜4個の炭素原子を含有し、そしてさらに他の実施形態では、アルケニル基は2〜3個の炭素原子を含有している。また別の態様によると、用語「アルケニル」は、2つの二重結合を有する直鎖状炭化水素を意味し、「ジエン」とも呼ばれる。他の実施形態では、用語「アルケニル」または「アルケニル基」は、シクロアルケニル基を意味する。典型的なC2−4アルケニル基には、−CH=CH2、−CH2CH=CH2(アリルとも呼ばれる)、−CH=CHCH3、−CH2CH2CH=CH2、−CH2CH=CHCH3、−CH=CH2CH2CH3、−CH=CH2CH=CH2、およびシクロブテニルが含まれる。
【0030】
本明細書で使用する「アルコキシ」または「アルキルチオ」は、以前に規定したように、アルキル基を意味し、酸素(「アルコキシ」)または硫黄(「アルキルチオ」)原子を通して主要炭素鎖に結合した。
【0031】
本明細書で使用する「メチレン」、「エチレン」、および「プロピレン」は、二価成分であるCH2−、−CH2CH2−、および−CH2CH2CH2−を各々意味する。
【0032】
本明細書で使用する「エテニレン」、「プロペニレン」、および「ブテニレン」は、二価成分である−CH=CH−、−CH=CHCH2−、−CH2CH=CH−、−CH=CHCH2CH2−、−CH2CH=CH2CH2−、および−CH2CH2CH=CH−、を意味するが、このとき各エテニレン基、プロペニレン基、およびブテニレン基はcisまたはtrans配列にあってよい。所定の実施形態では、エテニレン基、プロペニレン基、またはブテニレン基は、trans配列にあってよい。
【0033】
「アルキリデン」は、メチレンのモノまたはジアルキル置換によって形成された二価炭化水素基を意味する。所定の実施形態では、アルキリデン基は、1〜6個の炭素原子を有する。他の実施形態では、アルキリデン基は、2〜6、1〜5、2〜4、または1〜3個の炭素原子を有する。そのような基には、プロピリデン(CH3CH2CH=)、エチリデン(CH3CH=)、およびイソプロピリデン(CH3(CH3)CH=)などが含まれる。
【0034】
「アルケニリデン」は、メチレンのモノまたはジアルケニル置換によって形成された1つ以上の二重結合を有する二価炭化水素基を意味する。所定の実施形態では、アルケニリデン基は、2〜6個の炭素原子を有する。他の実施形態では、アルケニリデン基は、2〜6、2〜5、2〜4、または2〜3個の炭素原子を有する。1つの態様によると、アルケニリデンは、2つの二重結合を有する。典型的なアルケニリデン基には、CH3CH=C=、CH2=CHCH=、CH2=CHCH2CH=、およびCH2=CHCH2CH=CHCH=が含まれる。
【0035】
「C1−6アルキルエステルもしくはアミド」は、C1−6アルキルエステルもしくはC1−6アルキルアミドを意味するが、このとき各C1−6アルキル基は上記に規定したとおりである。そのようなC1−6アルキルエステル基は、式(C1−6アルキル)OC(=O)−または(C1−6アルキル)C(=O)O−の基である。そのようなC1−6アルキルアミド基は、式(C1−6アルキル)NHC(=O)−または(C1−6アルキル)C(=O)NH−の基である。
【0036】
「C2−6アルケニルエステルもしくはアミド」は、C2−6アルケニルエステルもしくはC2−6アルケニルアミドを意味し、ここで、各C2−6アルケニル基が上記に規定されている。そのようなC2−6アルキルエステル基は、式(C2−6アルケニル)OC(=O)−または(C2−6アルケニル)C(=O)O−の基である。そのようなC2−6アルケニルアミド基は、式(C2−6アルケニル)NHC(=O)−または(C2−6アルケニル)C(=O)NH−の基である。
【0037】
「治療」、「治療する」および「治療するステップ」は、本明細書に記載した疾患または障害の発生を逆転させる、軽減する、遅延させる、進行を阻害する、または予防することを意味する。一部の実施形態では、治療は、1つ以上の症状が発生した後に投与されてよい。他の実施形態では、治療は、症状の非存在下で投与されてよい。例えば、治療は、症状の発生前に(例えば、症状の履歴に照らして、および/または遺伝的もしくはその他の感受性因子に照らして)感受性のある個体に投与されてよい。治療はさらにまた、症状が消散した後に、例えば症状の再発を予防または遅延させるために継続されてよい。
【0038】
本明細書で使用する「患者」または「被験体(subject)」は、動物被験体、好ましくは哺乳動物被験体(例えば、イヌ、ネコ、ウマ、ウシ、ヒツジ、ヤギ、サルなど)、および特にヒト被験者(男性および女性被験者の両方を含み、さらに新生児、幼児、少年、青年、成人および高齢被験者を含む)を意味する。
【0039】
本明細書で使用する用語「医薬上許容される担体」は、それが一緒に調製される化合物の薬理学的活性を破壊しない、非毒性の担体、アジュバント、またはビヒクルを意味する。本発明の組成物中に使用できる医薬上許容される担体、アジュバントまたはビヒクルには、イオン交換剤、アルミナ、ステアリン酸アルミニウム、レシチン、例えばヒト血清アルブミンなどの血清タンパク質、例えばリン酸塩などの緩衝物質、グリシン、ソルビン酸、ソルビン酸カリウム、飽和植物性脂肪酸の部分グリセリド混合物、水、例えば硫酸プロタミン、リン酸水素二ナトリウム、リン酸水素カリウム、塩化ナトリウム、亜鉛塩などの塩類または電解質類、コロイド状シリカ、三ケイ酸マグネシウム、ポリビニルピロリドン、セルロースをベースとする物質、ポリエチレングリコール、シクロデキストリン類、カルボキシメチルセルロースナトリウム、ポリアクリレート類、ワックス類、ポリエチレン・ポリオキシプロピレンブロックポリマー類、ポリエチレングリコールおよび羊毛脂が含まれるがそれらに限定されない。
【0040】
他に特に指示しない限り、本明細書で使用する化学基または成分を記載するために使用する術語は慣習に従うが、このとき名称は左から右へ読み、分子の残りへの結合点は名称の右側にある。例えば、「(C1−3アルコキシ)C1−3アルキル」基は、アルキル末端で分子の残りに結合している。また別の例には、結合点がエチル末端にあるメトキシエチル、および結合点がアミン末端にあるメチルアミノが含まれる。
【0041】
他に特に指示しない限り、二価基が「−」によって指示される2つの末端結合成分を含む化学式によって記載される場合は、その結合は左から右へ読まれると理解される。
【0042】
他に特に指示しない限り、本明細書に示した構造はさらにまたその構造の全てのエナンチオマー、ジアステレオマー、および幾何学的(または立体配座)形、例えば各不斉中心に対するRおよびS立体配置、(Z)および(E)二重結合異性体、ならびに(Z)および(E)配座異性体を含むことが意図されている。このため、本発明の化合物の単一立体化学的異性体ならびにエナンチオマー、ジアステレオマー、および幾何学的(または立体配座)混合物は、本発明の範囲内に含まれる。他に特に指示しない限り、本発明の化合物の全ての互変異性体形は、本発明の範囲内に含まれる。さらに、他に特に指示しない限り、本明細書に記載した構造は、さらにまた1つ以上の同位体が濃縮された原子の存在に関してのみ相違する化合物を含むことも意図されている。例えば、ジューテリウム(重水素)またはトリチウムによる水素の置換、または13Cもしくは14C濃縮炭素による炭素の置換を除いて、本発明の構造を有する化合物は、本発明の範囲内に含まれる。そのような化合物は、例えば、生物学的アッセイにおける分析ツールまたはプローブとして有用である。
【0043】
B.活性化合物/調節因子化合物/EP4アンタゴニスト
本明細書で記載する本発明の活性化合物(本明細書では「調節因子化合物」および/または「EP4アンタゴニスト」と呼ぶこともある)は、一般に上記で例示したような、または本発明の特定のクラス、サブクラス、および種によって例示されるような1つ以上の置換基と任意で置換されてよい。一般に、用語「置換(された)」は、所定の構造内の水素ラジカルと特定の置換基のラジカルとの置換を意味する。他に特に指示しない限り、置換基は、その基の各置換可能な位置で置換基を有していてよく、任意の所定の構造内での2つ以上の位置が特定の基より選択される2つ以上の置換基と置換されてよい場合は、該置換基は1つ1つの位置で同一であっても相違していてもよい。
【0044】
上述したように、本発明は、式I:
【化9】
のエナンチオマー的に純粋な化合物、もしくは活性な化合物、
またはより詳細には式Iaもしくは式Ib:
【化10】
(式中、
R1はC1−3アルキルであり;
Xは、メチレン、エチレン、プロピレン、エテニレン、プロペニレン、またはブテニレンであり;
R5は、フェニル、ピロリル、ベンズイミダゾリル、オキサゾリル、イソキサゾリル、イミダゾチアゾリル、キノリニル、イソキノリニル、インダゾリル、ピリジニル、イミダゾピリジニル、インドリル、ベンゾトリアゾリル、イミダゾリル、ベンゾフラニル、ベンゾチアジアゾリル、ピリジミジニル、ベンゾピラノニル、チアゾリル、チアジアゾリル、フリル、チエニル、ピラゾリル、キノキサリニル、もしくはナフチルであり、C1−4アルキル、C1−3アルコキシ、ヒドロキシル、C1−3アルキルチオ、シクロプロピル、シクロプロピルメチル、トリフルオロメトキシ、5−メチルイソキサゾリル、ピラゾリル、ベンジルオキシ、アセチル、(シアニル)C1−3アルキル、(フェニル)C2−3アルケニルおよびハロよりなる群から独立して選択された0〜5個の置換基で置換されており;
R8は、H、メチル、エチル、プロピル、(C1−3アルコキシ)C1−3アルキル、(C1−3アルキルチオ)C1−3アルキル、C1−3ヒドロキシアルキル、フェニル、ベンジル、フリル、ピロリル、イミダゾリル、ピラゾリル、ピロリル、イソチアゾリル、イソキサゾリル、ピリジル、およびチエニルであり;
(このとき、R8は、メチル、エチル、ハロ、ヒドロキシル、C1−3アルコキシ、C1−3アルキルチオ、(C1−3アルコキシ)C1−3アルキル、(C1−3アルキルチオ)C1−3アルキル、C1−3ヒドロキシアルキル、(C1−3メルカプトアルキル)フェニル、ベンジル、フリル、イミダゾリル、ピラゾリル、ピロリル、イソチアゾリル、イソキサゾリル、ピリジル、およびチエニルより独立して選択される0〜3個の置換基で置換されており);
Ra、Rb、およびRcの各々は、水素、ヒドロキシル、メトキシ、ベンジルオキシ、フルオロ、クロロ、アミノ、メチルアミノ、ジメチルアミノ、およびフェノキシより独立して選択され;
またはRaおよびRb、ならびにRbおよびRcより選択される1対は、一緒になって−O−(CH2)−O−または−O−CH2−CH2−O−である)
またはその医薬上許容される塩、C1−6アルキルエステルもしくはアミド、またはC2−6アルケニルエステルもしくはアミドを提供する。
【0045】
上記の一部の実施形態では:
R1はC1−2アルキルである;
R5は、フェニル、ピロリル、ベンズイミダゾリル、オキサゾリル、イソキサゾリル、イミダゾチアゾリル、キノリニル、イソキノリニル、インダゾリル、ピリジニル、イミダゾピリジニル、インドリル、ベンゾトリアゾリル、イミダゾリル、ベンゾフラニル、ベンゾチアジアゾリル、ピリジミジニル、ベンゾピラノニル、チアゾリル、チアジアゾリル、フリル、チエニル、ピラゾリル、キノキサリニル、もしくはナフチルであり、およびC1−4アルキル、C1−3アルコキシ、ヒドロキシル、C1−3アルキルチオ、シクロプロピル、シクロプロピルメチル、トリフルオロメトキシ、5−メチルイソキサゾリル、ピラゾリル、ベンジルオキシ、アセチル、(シアニル)C1−3アルキル、(フェニル)C2−3アルケニルおよびハロよりなる群から独立して選択された0〜5個の置換基で置換されている;
R8は、メチル、エチル、またはプロピルである(このときR8は0〜3個のヒドロキシル置換基で置換されている);
Xは、メチレンもしくはエチレンである;および
Ra、Rb、およびRcの各々は、Hおよびメトキシよりなる群から独立して選択される;
またはそれらの医薬上許容される塩、C1−6アルキルエステルもしくはアミド、またはC2−6アルケニルエステルもしくはアミドである。
【0046】
上記の一部の実施形態では:
R1は、メチルである;
R5は、フェニル、ピロリルもしくはピラゾリルであり、それらの各々はメチルで0、1または2回置換される;
R8は、エチルである;
Xは、メチレンである;
RaおよびRcの各々は、メトキシである;および
Rbは、Hである;
またはその医薬上許容される塩である。
【0047】
上記の特定の実施形態では、本化合物は:
【化11】
またはそれらの医薬上許容される塩である。
【0048】
本発明の活性化合物には、上記の医薬上許容される塩類が含まれる。医薬上許容される塩類には、医薬上許容される無機塩および有機酸および塩基に由来する塩類が含まれる。適切な酸性塩類の例には、酢酸塩、アジピン酸塩、アルギン酸塩、アスパラギン酸塩、安息香酸塩、ベンゼンスルホン酸塩、重硫酸塩、酪酸塩、クエン酸塩、樟脳酸塩、樟脳スルホン酸塩、シクロペンタンプロピオン酸塩、ジグルコン酸塩、ドデシル硫酸塩、エタンスルホン酸塩、ギ酸塩、フマル酸塩、グルコヘプタン酸塩、グリセロリン酸塩、グリコール酸塩、ヘミ硫酸塩、ヘプタン酸塩、ヘキサン酸塩、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、2−ヒドロキシエタンスルホン酸塩、乳酸塩、マレイン酸塩、マロン酸塩、メタンスルホン酸塩、2−ナフタレンスルホン酸塩、ニコチン酸塩、硝酸塩、シュウ酸塩、パルモエート(palmoate)、ペクチン酸塩、過硫酸塩、3−フェニルプロピオン酸塩、リン酸塩、ピクリン酸塩、ピバル酸塩、プロピオン酸塩、サリチル酸塩、コハク酸塩、硫酸塩、酒石酸塩、チオシアン酸塩、トシル酸塩およびウンデカン酸塩が含まれる。例えばシュウ酸などのその他の酸は、それら自体は医薬上許容されないが、本発明の化合物およびそれらの医薬上許容される酸付加塩を得る際の中間体として有用な塩類の調製において使用することができる。
【0049】
適切な塩基に由来する塩類には、アルカリ金属(例えば、ナトリウムおよびカリウム)、アルカリ土類金属(例えば、マグネシウム)、アンモニウムおよびN+(C1−4アルキル)4塩類が含まれる。本発明は、さらにまた本明細書に開示した化合物の任意の塩基性窒素含有基の四級化も想定している。そのような四級化によって、水溶性もしくは油溶性または分散性生成物を入手できる。
【0050】
C.医薬調製物
本発明の活性化合物(調節因子化合物および/またはEP4アンタゴニストを含む)は、それらの医薬調製物を提供するために医薬上許容される担体と結合することができる。担体および調製物の特定の選択は、該組成物の予定される特定投与経路に依存する。
【0051】
本発明の組成物は、経口、非経口、吸入スプレー、局所、経直腸、経鼻腔、経口腔、経膣または埋め込みリザーバ投与などのために適合する可能性がある。好ましくは、本組成物は、経口、腹腔内または静脈内投与される。本発明の組成物の無菌注射剤形は、水性または油性懸濁剤であってよい。これらの懸濁剤は、適切な分散剤もしくは湿潤剤および懸濁化剤を使用して、当分野において公知の技術にしたがって調製できる。無菌注射用製剤は、例えば1,3−ブタンジオール中の溶液として、非毒性の非経口的に許容される希釈剤または溶媒中の無菌注射用液剤または懸濁剤であってもよい。特に使用できる許容されるビヒクルおよび溶媒は、水、リンガー(Ringer)溶液および等張塩化ナトリウム溶液である。さらに、無菌の不揮発性油は、慣習的には溶媒または懸濁化剤として使用される。
【0052】
この目的で、合成モノ−またはジ−グリセリド類を含む任意のブランドの不揮発性油を使用できる。例えばオレイン酸などの脂肪酸およびそのグリセリド誘導体は、例えば、特にそれらのポリオキシエチル化形にあるオリーブ油またはヒマシ油などの天然の医薬上許容される油と同様に、注射剤の調製において有用である。これらの油性溶液または懸濁液は、例えばエマルジョンおよび懸濁液を含む医薬上許容される剤形の調製において一般に使用されるカルボキシメチルセルロースまたは類似の分散剤などの長鎖アルコール希釈剤または分散剤をさらに含有していてもよい。例えばTween類、Span類および医薬上許容される固体、液体、またはその他の剤形の製造において一般に使用されるその他の乳化剤またはバイオアベイラビリティ強化剤などの他の一般に使用される界面活性剤類もまた調製のために使用できる。
【0053】
本発明の医薬上許容される組成物は、カプセル剤、錠剤、水性懸濁剤または液剤を含む任意の経口投与に許容される剤形で経口投与することができるが、それらに限定されない。経口使用するための錠剤の場合には、一般に使用される担体には、ラクトースおよびコーンスターチが含まれる。典型的には、例えばステアリン酸マグネシウムなどの潤滑剤もまた加えられる。カプセル形での経口投与のために有用な希釈剤には、ラクトースおよび乾燥コーンスターチが含まれる。経口使用のために水性懸濁剤が必要とされる場合は、有効成分が乳化剤および懸濁化剤と結合される。所望であれば、所定の甘味料、フレーバー剤または着色剤もまた加えることができる。
【0054】
または、本発明の医薬上許容される組成物は、直腸投与のための坐剤の形態で投与することができる。これらは、該物質を室温では固体であるが直腸温度では液体である適切な非刺激性賦形剤と混合する工程によって調製することができるので、このため直腸内で融解して薬物を放出する。そのような材料には、カカオ脂、蜜ろうおよびポリエチレングリコールが含まれる。
【0055】
本発明の医薬上許容される組成物は、特に治療の標的が目、皮膚、または下部消化管の疾患を含む、局所適用によって容易に接近できる領域または器官を含む場合には、局所投与することもできる。適切な局所用調製物は、これらの領域または器官の各々のために容易に調製される。
【0056】
下部消化管に対する局所適用は、直腸坐剤調製物(上記参照)または適切な浣腸調製物で実行することができる。局所用経皮パッチもまた使用できる。
【0057】
局所適用のためには、医薬上許容される組成物は、1つ以上の担体中に懸濁化または溶解した活性成分を含有する適切な軟膏剤に調製することができる。本発明の化合物を局所投与するための担体には、鉱油、流動ワセリン(liquid petrolatum)、白色ワセリン、プロピレングリコール、ポリオキシエチレン、ポリオキシプロピレン化合物、乳化ろうおよび水が含まれるがそれらに限定されない。または、医薬上許容される組成物は、1つ以上の医薬上許容される担体中に懸濁化または溶解した活性成分を含有する適切なローション剤またはクリーム剤に調製することができる。適切な担体には、鉱油、ソルビタンモノステアレート、ポリソルベート60、セチルエステル類ロウ、セテアリルアルコール、2−オクチルドデカノール、ベンジルアルコールおよび水が含まれるがそれらに限定されない。
【0058】
眼科使用のためには、医薬上許容される組成物は、等張性のpH調整無菌食塩液中の微粉化懸濁剤として、または好ましくは、等張性のpH調整無菌食塩液中の液剤として、例えば塩化ベンジルアルコニウムなどの保存料を含めて、または含めずに調製することができる。または、眼科使用のためには、医薬上許容される組成物は、例えばワセリンなどの軟膏剤で調製することができる。
【0059】
本発明の医薬上許容される組成物は、さらにまた鼻腔エーロゾルまたは吸入によって投与することもできる。そのような組成物は、医薬調製物の分野において周知の技術にしたがって調製され、ベンジルアルコールもしくはその他の適切な保存料、バイオアベイラビリティを強化するための吸収促進剤、フルオロカーボン、および/または他の従来型可溶化剤もしくは分散剤を使用して、食塩液中の液剤として調製することができる。
【0060】
最も好ましくは、本発明の医薬上許容される組成物は、経口投与のために調製される。
【0061】
D.被験体および使用方法
本発明の活性化合物(調節因子化合物および/またはEP4アンタゴニストを含む)は、様々な相違する状態を治療するために患者または被験体に、特に以下に罹患している患者または被験体に投与することができる。
(a)関節リウマチ;
(b)多発性硬化症;
(c)全身性エリテマトーデス(例えば、非特許文献1);
(d)1型糖尿病(例えば、非特許文献2);
(e)乾癬(例えば、非特許文献3);および
(f)アテローム硬化症(例えば、非特許文献4)
【0062】
活性化合物は、経口、非経口、吸入スプレー、局所的、経直腸、経鼻腔、経口腔、経膣または埋め込みリザーバなどの任意の適切な経路によって被験体に投与されてもよい。本明細書で使用する用語「非経口」には、皮下、静脈下、筋肉内、関節内、滑膜内、胸骨内、クモ膜下、肝内、病巣内および頭蓋内注射または注入技術が含まれる。好ましくは、本組成物は、経口、腹腔内または静脈内投与される。
【0063】
本活性化合物は、被験体に治療有効量、または治療的に有効な量で投与される。単位製剤形にある組成物を製造するために担体材料と結合できる本発明の化合物の量は、治療される宿主、および特定投与経路に依存して変動する。好ましくは、本組成物は、これらの組成物を摂取する患者に0.01〜100mg/kg(体重)/日の阻害剤を投与できるように調製されなければならない。所定の実施形態では、本発明の組成物は、0.01mg〜50mgの用量を提供する。他の実施形態では、0.1〜25mgまたは5mg〜40mgの用量が提供される。
【0064】
任意の特定患者のための特定の用量および治療レジメンは、使用される特定化合物の活性、年齢、体重、全身状態、性別、食事、投与回数、排出速度、併用薬、ならびに治療担当医師の判断および治療される特定疾患の重症度を含む様々な因子に依存することもまた理解されたい。組成物中の本発明の化合物の量は、さらに本組成物中の特定化合物に依存する。
【0065】
以下では、本明細書に記載した本発明をより完全に理解できるように、実施例を記載する。これらの実施例は単に例示するためであり、決していかなる様式でも本発明を限定すると解釈すべきではないと理解されたい。
【実施例】
【0066】
[実施例1〜41]
化合物の合成
Biotage Corporation社によって供給されるEmrys Liberator機器を用いて、マイクロ波援用反応を実施した。溶媒除去は、Buechiロータリー・エバポレータまたはGenevac遠心式エバポレータのどちらかを使用して実施した。分析および分取クロマトグラフィは、酸性、中性、または塩基性条件下のいずれかで、順相または逆相いずれかのHPLCカラムを用いて、Waters autopurification機器を用いて実施した。化合物はELSDクロマトグラムの面積百分率によって決定して、>90%純粋であると推定された。NMRスペクトルは、Varian 300MHz分光計を用いて記録した。
【0067】
以下では本発明の化合物を調製するための一般的方法および実験について記載する。所定の場合には、例として特定化合物について記載する。しかし、各場合において、一連の本発明の化合物は以下に記載するスキームおよび実験にしたがって調製されたことは理解される。
【0068】
スキーム1
【化12】
ER−811160.上記のスキーム1に図示したように、水(50mL)中のシアン化カリウム(22.5g、0.335M(モル))を5分間かけて、水(90mL)およびメタノール(110mL)中の1−Boc−ピペリドン(32.48g、0.1598M)および炭酸アンモニウム(33.8g、0.351M)の溶液に滴下した。添加の完了直後に、オフホワイトの沈降物が形成し始めた。反応フラスコを密封し、懸濁液を室温で72時間にわたり攪拌した。結果として生じた浅黄色の沈降物を濾過し、少量の水で洗浄すると、無色の固体としてER−811160(37.1g、86%)が得られた。
【0069】
スキーム2
【化13】
ER−818039.上記のスキーム2に図示したように、アセトン(555mL)中のER−811160(30.0g、0.111M)、3,5−ジメトキシベンジルブロミド(30.9g、0.134M)、および炭酸カリウム(18.5g、0.134M)の懸濁液を一晩還流させながら加熱した。反応溶液を室温へ冷却し、濾過し、インバキュオで濃縮した。橙色の粗残留物を最小量のMTBE(250mL)中に溶解させた。少量(50mL)のヘキサンを加え、生成物を無色の固体として(約2時間かけて)沈殿させ、これを真空濾過によって単離した。フィルターケーキを少量のMTBEで洗浄し、インバキュオで乾燥させると、無色の固体としてER−818039(39.6g、85%)が得られた。
【0070】
スキーム3
【化14】
ER−823143−01.上記のスキーム3に図示したように、ER−818039(2.15g、0.00512M)を含有する1口丸底フラスコに1,4−ジオキサン(3.8mL、0.049M)中の4N HClの溶液を緩徐に加えた。出発材料は20分間かけて緩徐に溶解し、30分後に無色の沈降物が形成された。次にMTBE(3mL)を加えた。2時間後、反応液を濾過し、MTBEを用いて洗浄すると、無色の固体としてER−823143−01(1.81g、99%)が得られた。
【0071】
スキーム4
【化15】
ER−817098:上記のスキーム4に図示したように、窒素雰囲気下で1,2−ジメトキシエタン(0.5mL、0.004M)中のER−823143−01(41.5mg、0.000117M)および4Åモレキュラーシーブの懸濁液に、3,5−ジメトキシベンズアルデヒド(21.3mg、0.000128M)、その後にトリエチルアミン(16.2μL、0.000117M)を加えた。この反応液を1時間にわたり攪拌した。トリアセトキシホウ化水素ナトリウム(34.6mg、0.000163M)を加え、この反応液を一晩攪拌した。シリカゲル・フラッシュ・クロマトグラフィーは、無色の固体としてER−817098(45.3mg、83%)を産生した。
【0072】
スキーム5
【化16】
ER−817116:上記のスキーム5に図示したように、N−メチルピロリジノン(1.0mL、0.010M)中のER−817098−00(50.0mg、0.000106M)および1−ブロモ−2−メトキシエタン(15.6μL、0.000160M)の溶液にテトラヒドロフラン(0.16mL)中の1.0Mリチウムヘキサメチルジシラジド溶液を加えた。温度を80℃へ上昇させ、反応混合液を一晩攪拌した。反応混合液を室温に冷却し、水を用いて急冷し、次にMTBEを用いて数回抽出した。MTBE抽出物を結合し、水(2×)および食塩液(1×)を用いて洗浄した。有機層は硫酸マグネシウムの上方に通して乾燥させ、濾過し、インバキュオで濃縮した。フラッシュ・クロマトグラフィーは、無色油としてER−817116(32.2mg、58%)を生じさせた。
【0073】
スキーム6
【化17】
ER−817118:上記のスキーム6に図示したように、N,N−ジメチルホルムアミド(15mL)中のER−817098(2.85g、0.00607M)の溶液に水素化ナトリウム(364mg、0.00910M)、次にヨードエタン(758μL、0.00910M)を加えた。この反応混合液を一晩攪拌した。水を極めて緩徐に加え、反応混合液はMTBEを用いて数回抽出した。MTBE抽出物を結合し、水(2×)および食塩液(1×)を用いて洗浄した。有機層は硫酸マグネシウムの上方に通して乾燥させ、濾過し、インバキュオで濃縮した。溶離液として酢酸エチルを用いるフラッシュ・クロマトグラフィーは、無色油としてER−817118(2.89g、96%)を生じさせた。
【0074】
スキーム7
【化18】
ER−823914:上記のスキーム7に図示したように、−78℃でテトラヒドロフラン(30.0mL、0.370M)中のER−823143−01(5.03g、0.0141M)の溶液に、エーテル(71mL)中の1.0Mの臭化アリルマグネシウムを緩徐に加えた。この反応混合液を室温へ加温し、一晩攪拌した。反応混合液を−78℃へ冷却し、トリフルオロ酢酸(21.8mL、0.283M)を滴下して処理し、次に少量の残留物量へインバキュオで濃縮した。残留TFAを中和するためにトリエチルアミンを加え、次にこの混合物を乾燥するまでインバキュオで濃縮した。残留した赤色油をメタノール(138mL、3.41M)中に溶解させ、ジ−tert−ブチルジカーボネート(3.34g、0.0148M)、次にトリエチルアミン(2.38mL、0.0169M)で処理し、室温で一晩攪拌した。反応混合液をインバキュオで濃縮し、フラッシュ・クロマトグラフィー(溶離液:酢酸エチル中の50%ヘキサン)によって精製すると、無色の固体としてER−823914(3.25g、52%)が得られた。
【0075】
スキーム8
【化19】
ER−823915:上記のスキーム8に図示したように、N,N−ジメチルホルムアミド(12.4mL、0.160M)中のER−823914(2.20g、0.00496M)の溶液に水素化ナトリウム(298mg、0.00744M)、次にヨードエタン(607μL、0.00744M)を加えた。反応混合液を一晩攪拌し、水を用いて急冷し、次にMTBEを用いて数回抽出した。MTBE抽出物を結合し、水および食塩液を用いて洗浄した。有機層は硫酸マグネシウムの上方に通して乾燥させ、濾過し、インバキュオで濃縮した。フラッシュ・クロマトグラフィー(溶離液:酢酸エチル中の40%ヘキサン)は、無色の泡としてER■823915(0.80g、34%)を生じさせた。
【0076】
スキーム9
【化20】
ER−823917−01:上記のスキーム9に図示したように、ER−823915(799.2mg、0.001695M)を1,4−ジオキサン(10mL)中の4M塩化水素の溶液中に溶解させた。反応混合液を一晩攪拌し、次にインバキュオで濃縮すると、橙色の固体としてER−823917−01(0.69g、定量的)が得られた。
【0077】
スキーム10
【化21】
ER−824184およびER−824185:上記のスキーム10に図示したように、アセトニトリル(1mL)中のER−823915(200mg)の溶液をCHIRALPAK(登録商標)AS−H SFCカラム(30mm×250mm、粒径:5ミクロン)上に注入し、95:5のn−ヘプタン:i−プロパノールを用いて40mL/分の流量で溶出した。溶出フラクションは、290nmに設定した波長を用いてUV検出器を使用して検出した。第1溶出フラクションを単離し、インバキュオで回転式蒸発によって濃縮するとER−824184が得られた;第2溶出フラクションを単離し、インバキュオで回転式蒸発によって濃縮するとER−824185が得られた。
【0078】
スキーム11
【化22】
ER−824188−01:上記のスキーム11に図示したように、ER−824184(25.33g、0.05371M)を1,4−ジオキサン(135mL)中の4M塩化水素の溶液中に溶解させた。反応混合液を一晩攪拌し、次にインバキュオで濃縮すると、橙色の固体としてER−824188−01(21.9g、定量的)が得られた。ER−824188−01の単結晶X線回折分析は、立体中心の絶対配置がスキーム11に図示したようにSであることを証明した。
【0079】
スキーム12
【化23】
ER−824280−01:上記のスキーム12に図示したように、ER−824185(457.2mg、0.0009695M)を1,4−ジオキサン(2.5mL)中の4M塩化水素の溶液中に溶解させた。反応混合液を一晩攪拌し、次にインバキュオで濃縮すると、橙色の固体としてER−824280−01(383.2mg、97%)が得られた。ER−824188−01のMosherアミド誘導体の単結晶X線回折分析は、立体中心の絶対配置がスキーム11に図示したようにRであることを証明した。
【0080】
スキーム13
【化24】
ER−819924:上記のスキーム13に図示したように、ER−824188−01(62.4mg、0.000153M)およびN−メチルピロール−2−カルバルデヒド(0.000229M)をN,N−ジメチルホルムアミド(0.62mL)中に溶解/懸濁させた。30分間にわたり攪拌した後、トリアセトキシホウ化水素ナトリウム(47.8mg、0.000214M)を加えた。反応混合液を一晩攪拌し、次に逆相クロマトグラフィによって精製すると、油としてER−819924(71.1mg、83.4%)が得られた。
【0081】
スキーム14
【化25】
ER−819925:上記のスキーム14に図示したように、ER−824280−01(59.5mg、0.000146M)およびN−メチルピロール−2−カルバルデヒド(0.000219M)をN,N’−ジメチルホルムアミド(0.60mL)中に溶解/懸濁させた。30分間にわたり攪拌した後、トリアセトキシホウ化水素ナトリウム(45.6mg、0.000204M)を加えた。反応混合液を一晩攪拌し、次に逆相クロマトグラフィによって精製すると、油としてER−819925(51.9mg、76.6%)が得られた。
【0082】
スキーム15
【化26】
ER−819762:上記のスキーム15に図示したように、N,N−ジメチルホルムアミド(50mL)中のER−824188−01(5.7g、0.0140M)、1,8−ジアザビシクロ[5.4.0]ウンデク−7−エン(4.4mL、0.029M)および3,5−ジメチルベンジルブロミド(4.7g、0.024M)の溶液を97℃で一晩加熱した。水系後処理およびフラッシュ・クロマトグラフィーによる精製によって、無色の固体としてER−819762(4.86g、71%)が得られた。
【0083】
スキーム16
【化27】
ER−819762−01:上記のスキーム16に図示したように、水(11mL)中のER−819762(4.77g、0.00974M)、アセトニトリル(10mL)および1M HClの溶液を室温でおよそ5分間攪拌した。この溶液を濃縮すると、凍結乾燥後の無色の結晶固体としてER−819762−01(5.1g、定量的)が得られた。ER−819762−01の単結晶X線回折分析は、立体中心の絶対配置がスキーム16に図示したようにSであることを証明した。
【0084】
スキーム17
【化28】
ER−819763:上記のスキーム17に図示したように、N−メチルピロリジノン(669mL)中のER−824280−01(66.9g、0.1640M)、1,8−ジアザビシクロ[5.4.0]ウンデク−7−エン(54mL、0.361M)および3,5−ジメチルベンジルクロリド(42.4g、0.213M)の溶液を2時間にわたり72℃で加熱した。冷却後、水を加えると、所望の生成物が沈降した。濾過および真空下での乾燥は、無色の固体としてER−819763(74.4g、92%)を生じさせた。
【0085】
スキーム18
【化29】
ER−824102:上記のスキーム18に図示したように、室温にあるN,N−ジメチルホルムアミド(25mL)中のER−823143−01(4.00g、0.0112M)の溶液にα−ブロモメシチレン(3.13g、0.0157M)、次にDBU(4.37mL、0.0292M)を加えた。1時間にわたり攪拌した後、反応液を半飽和NH4Cl水溶液で急冷し、酢酸エチルを用いて希釈し、1時間攪拌すると2つの透明層が得られた。有機層を分離し、水層は酢酸エチルにより抽出した(2×)。結合抽出物はNa2SO4の上方に通して乾燥させ、濾過し、インバキュオで濃縮した。MTBEからの結晶化により、無色の固体としてER−824102(4.30g、87%)が得られた。
【0086】
スキーム19
【化30】
ER−819929:上記のスキーム19に図示したように、−65℃のテトラヒドロフラン(35mL)中のER−824102(3.72g、0.0085M)の溶液に、−50℃未満の内部温度を維持しながら10分間かけてエーテル(25.5mL、0.0255M)中の1.0M臭化アリルマグネシウムを加えた。反応混合液を0℃へ加温させた。0℃で3時間置いた後、反応液を飽和NH4Cl水溶液で急冷し、酢酸エチルおよび水を用いて希釈し、10分間攪拌すると2つの透明層が得られた。有機層を分離し、水層は酢酸エチルにより抽出した。結合抽出物を水、食塩液で洗浄し、Na2SO4の上方に通して乾燥させ、濾過し、インバキュオで濃縮すると無色の固体として粗生成物のER−819929(4.15g、定量的)が得られたので、これをそれ以上精製せずに次の工程に使用した。
【0087】
スキーム20
【化31】
ER−819930:上記のスキーム20に図示したように、トリフルオロ酢酸(0.5mL)中のER−819929(37mg、0.000077M)の溶液を室温で16時間にわたり攪拌した。暗赤褐色の反応混合液をEtOAc(5mL)で希釈し、飽和NaHCO3(5mL、注意:ガス発生)を用いて中和した。2層の混合液を10分間攪拌すると、ほぼ無色の2つの透明層が得られた。2つの有機層を分離し、水層はEtOAcで抽出した。結合有機抽出物はNa2SO4の上方に通して乾燥させ、濾過し、インバキュオで濃縮した。1:1ヘプタン−EtOAc、1:3ヘプタン−EtOAc、100%EtOAcを用いて溶出させるフラッシュ・クロマトグラフィーによって精製すると、無色の固体としてER−819930(26mg、73%)が得られた。
【0088】
スキーム21
【化32】
ER−820006およびER−820007:上記のスキーム21に図示したように、DMF(1.5mL)中のER−819930(110mg、0.000238M)および臭化メタリル(72μL、0.000715M)の溶液にテトラヒドロフラン中の1.0Mリチウムヘキサメチルジシラジド溶液(0.52mL、0.00052M)を加えた。室温で18時間攪拌した後、反応混合液をMTBEで希釈し、半飽和NH4Cl水溶液で急冷した。水層を分離し、MTBEで抽出した。結合抽出物はNa2SO4の上方に通して乾燥させ、濾過し、インバキュオで濃縮した。3:2ヘプタン−EtOAc、1:1ヘプタン−EtOAcを用いて溶出させるフラッシュ・クロマトグラフィーによって精製すると、無色油としてラセミ生成物(68mg、55%)が得られた。ラセミ生成物(55mg)は、ヘプタン−イソプロパノール(9:1)を用いて溶出させるChiralpak ASカラム上でのキラルHPLCにかけると、第1溶出エナンチオマーであるER−820006(21mg、38%、[a]D=+83.7°(c=0.35、CHCl3)および第2溶出エナンチオマーであるER−820007(23mg、42%、[a]D=−74.2°(c=0.38、CHCl3)が得られた。絶対立体化学は、エナンチオマーのER−819762/ER−819763対を用いて旋光度およびキラルHPLC保持時間における共通点に基づいて暫定的に指定した。
【0089】
スキーム22
【化33】
ER−819786およびER−819787:上記のスキーム22に図示したように、攪拌棒を装備した5mLマイクロ波反応器バイアルに、ER−819930(110mg、0.000238M)、DMF(1.5mL)、2−(2−ブロモエトキシ)テトラヒドロ−2H−ピラン(108μL、0.000715M)およびテトラヒドロフラン(520μL、0.00052M)中の1.00Mのリチウムヘキサメチルジシラジドを装填した。反応器バイアルを、200℃で15分間にわたりマイクロ波反応器にかけた。さらに2−(2−ブロモメトキシ)テトラヒドロ−2H−ピラン(108μL、0.000715M)およびテトラヒドロフラン中の1.00Mのリチウムヘキサメチルジシラジド(520μL、0.00052M)を加え、反応混合液をマイクロ波照射によってさらに15分間、200℃で加熱した。分取的逆相HPLCによる精製によって、無色のガラス質の油としてラセミ生成物(25mg、21%)が得られた。ラセミ生成物(17mg)は、ヘプタン−イソプロパノール(9:1)を用いて溶出させるChiralpac ASカラム上でのキラルHPLCにかけると、第1溶出エナンチオマーであるER−819786(7.2mg、42%、[α]D=+72.0°(c=0.1、CHCl3)および第2溶出エナンチオマーであるER−819787(7.5mg、44%、[α]D=−73.0°(c=0.1、CHCl3)が得られた。絶対立体化学は、エナンチオマーのER−819762/ER−819763対を用いて旋光度およびキラルHPLC保持時間における共通点に基づいて暫定的に指定した。
【0090】
スキーム23
【化34】
ER−819993およびER−819994:上記のスキーム23に図示したように、攪拌棒を装備した5mLマイクロ波反応器バイアルに、ER−819930(110mg、0.000238M)、DMF(1.5mL)、((4S)−2,2−ジメチル−1,3−ジオキソラン−4−イル)メチル4−メチルベンゼンスルホネート(205mg、0.000715mol)およびテトラヒドロフラン(520μL、0.00052M)中の1.00Mのリチウムヘキサメチルジシラジドを装填した。反応器バイアルを、200℃で15分間にわたりマイクロ波照射によって加熱した。さらに((4S)−2,2−ジメチル−1,3−ジオキソラン−4−イル)メチル4−メチルベンゼンスルホネート(157mg、0.000548M)およびテトラヒドロフラン中の1.00Mのリチウムヘキサメチルジシラジド(477μL、0.000477M)を加え、反応混合液をマイクロ波照射によってさらに15分間、200℃で加熱した。分取的逆相HPLCによる精製によって、ジアステレオマーの1:1混合物としてアセトニドであるER−819993(40mg、30%)およびジオール材料(18mg、14%)が得られた。ヘプタン−イソプロパノール(9:1)を用いて溶出させるChiralpac ASカラム上でのキラルHPLCによるジアステレオマージオールの分離によって、第1溶出ジアステレオマーであるER−819788(5.0mg)および第2溶出ジアステレオマーであるER−819789(5.2mg)が得られた。絶対立体化学は、エナンチオマーのER−819762/ER−819763対を用いてキラルHPLC保持時間における共通点に基づいて暫定的に指定した。
【0091】
スキーム24
【化35】
ER−81990:上記のスキーム24に図示したように、塩化メチレン(500μL)中のER−824220−00(51.8mg、0.000139M)、トリエチルアミン(97μL、0.00070M)、4−ジメチルアミノピリジン(3.4mg、0.000028M)および(R)−(−)−α−メトキシ−α−トリフルオロメチルフェニルアセチルクロリド(0.052mL、0.00028M)の溶液を室温で5時間にわたり攪拌した。フラッシュ・クロマトグラフィーによる精製後に、酢酸エチル/ヘプタン/ペンタンから結晶化させると、結晶としてER−819990(49.2mg、60%)が得られた。
【0092】
以下のセクションおよび以下の表1〜2に例示するが上記で明確には図示していない化合物は、スキーム13および/またはスキーム15に一致する一般方法を使用して合成できる。塩酸塩形で例示した化合物については、それらは対応する遊離塩基にスキーム16に記載した一般条件を受けさせることによって調製することができる。
【0093】
表1.式Iの典型的化合物についての分析データ
【表1A】
【表1B】
【表1C】
【表1D】
【表1E】
【表1F】
【表1G】
【表1H】
【表1I】
【表1J】
【表1K】
【表1L】
【表1M】
【表1N】
【表1O】
【表1P】
【表1Q】
【表1R】
【表1S】
【表1T】
【0094】
分析方法:
方法A1
溶媒A:水中の0.2% Et3N
溶媒B:アセトニトリル中の0.2% Et3N
流量:2.0mL/分
線形勾配:
【表1U】
方法C1
移動相:エタノール中の0.1% Et2NH
流量:1.0mL/分
均一濃度
【0095】
[実施例42〜126]
インビトロ生物学的活性
HEKT−bet−lucアッセイ:本アッセイは、ルシフェラーゼレポーターを駆動するヒトT−betおよびT−box応答エレメントを発現する遺伝子組換えHEK細胞内のT−bet依存性レポーター(ルシフェラーゼ)活性を測定する。HEKT−bet細胞は2×104/ウエルで96ウエルプレート内で平板培養し、化合物を24時間にわたり細胞培養中へ加えた。ルシフェラーゼ活性は、50μLのSteady−Glo試薬(Promega社)を加えることによって測定し、サンプルをVictor Vリーダー(PerkinElmer社)で読み取った。化合物の活性は、化合物処理サンプルを非化合物処理ビヒクル対照と比較することによって決定した。IC50値は、試験化合物の非存在下でのルシフェラーゼの量に対応する最大値および最大阻害で得られた試験化合物値に対応する最小値を利用して計算した。
【0096】
正規化HEKT−bet IC50値の決定:化合物は、マイクロタイタープレート内でアッセイした。各プレートは、ER−819544である参照化合物を含んでいた。特定化合物についての非正規化IC50値を同一マイクロタイタープレート内で参照化合物について決定したIC50値で割ると、相対効力値が得られた。次に相対効力値に参照化合物の確定効力を掛けると、正規化HEKT−bet IC50値が得られた。本アッセイでは、ER−819544の確定効力は0.035mMであった。本明細書に提供したIC50値は、正規化法を使用して入手した。
【0097】
本発明の典型的な化合物は、上記に記載したHEKT−bet−lucアッセイにおいて上述した方法にしたがってアッセイした。以下の表2は、上記に記載した正規化HEKT−bet−lucアッセイによって決定した指示量(μM)までのIC50を有する本発明の典型的な化合物を記載している。
【0098】
表2.典型的な化合物のIC50値
【表2A】
【表2B】
【表2C】
【表2D】
【表2E】
【表2F】
【表2G】
【表2H】
【表2I】
【表2J】
【表2K】
【表2L】
【表2M】
【表2N】
【表2O】
【表2P】
【表2Q】
【表2R】
【表2S】
【表2T】
【表2U】
【表2V】
【表2W】
【表2X】
【表2Y】
【表2Z】
【0099】
[実施例126]
インビボ生物学的活性:能動免疫
CIAにおける関節炎発生の抑制。DBA1/Jマウスを第0日にbCII/CFAで免疫し、その後第21日にbCII/IFAで追加免疫した。関節炎の発生を試験経過にわたって監視した。関節炎のスコアは、以下の通りである:0=正常な足、スコア1=1〜2本の指が炎症を起こしている足、スコア2=3本または1〜2本の指+手関節または足関節が炎症を起こしている、スコア3=手+3本以上の指が炎症を起こしている;およびスコア4=多数の指(3〜4)+重大な手関節または足関節の炎症。
【0100】
(A)化合物の部分的治療評価。活性化合物は、コラーゲンIIに対する抗体の誘導後第20日から、しかし疾患発生の前に指示した用量で1日1回の経口投与によって与えた。(B)化合物の完全治療評価。活性化合物は、疾患が発生した後(第2免疫後第7日から)投与した。(C)完全治療CIA試験からのマウス足についてのX線分析。X線スコアは、骨減少症、骨浸食および新規骨形成の組み合わせの測定値の指標である。(D)代表的なX線写真。
【0101】
データは、以下の表3に示した。一般に、これらのデータは好都合にこのモデルにおけるメトトレキセートの活性を比較する。
【0102】
[実施例127]
インビボ生物学的活性:受動免疫
CAIAにおける関節炎発生の抑制。BALB/cマウスに第0日に1mgの抗タイプIIコラーゲン抗体をi.v.(静脈内)注射し、3日後に25μgのLPSを活性化合物とともにi.p.(腹腔内)注射し、メトトレキセート(MTX)は第0日〜第7日にわたり1日1回PO投与した。関節炎スコアおよび体重は、試験の全経過にわたって監視した。
【0103】
データは、以下の表3に示した。一般に、これらのデータは好都合にもこのモデルにおけるにおいて特に活性ではないメトトレキセートと比較する。
【0104】
【表3】
【0105】
[実施例128〜134]
材料および方法
マウスおよび試薬。BALB/cおよびDO11.10マウスは、Jackson Laboratory社から購入した。C57BL/6およびDBA/1マウスは、Charles River Laboratories社から購入した。マウスIL−2、IL−12、IL−23およびヒトGM−CSFは、R & D systems社から購入した。ヒトIL−4およびGM−CSFは、Peprotec社からである。抗CD3(クローン145−2C11)、抗CD28(クローン37.51)、抗IL−4(クローン11B11)、抗IFNγ(クローンXMG12)およびPE−抗マウスIL−17(クローンTC11−18H10)は、Pharmingen社から購入した。抗TCR(クローンH57−597)は、eBioscience社から購入した。OVAペプチドおよびマイトマイシンCは、Sigma社から購入した。PGE2、PGE1−アルコールおよび抗PGE2は、Cayman Chemicals社から購入した。LPSおよびR−848は、InVivoGen社からである。CD14+細胞単離キットは、MiltenyiBiotec社からである。CD4+T細胞単離キットは、MiltenyiBiotec社またはStemCell Technologies社からである。IFNγ ELISAキットはPIERCE社からである;IL−4 ELISAキットは、R&D systems社からである;IL−23 ELISAキットは、eBioscience社からである。Alamar blue試薬は、Biosource International社からである。Celltiter−glo試薬は、Promega社からである。
【0106】
放射性リガンドEP4結合。放射性リガンドEP4結合アッセイは、EP4発現性膜標本からの放射標識PGE2の置換を測定する。放射性リガンドEP4結合アッセイキットは、Millipore社から購入し、このアッセイは、製造業者の取扱説明書にしたがって実施した。
【0107】
CRE−PLAPレポーターアッセイ。内因性EP4を発現するSE302細胞は、一晩にわたりER−819762の存在下または非存在下でPGE2を用いて刺激し、PLAP活性を測定した。
【0108】
インビトロT細胞アッセイ。天然CD4+T細胞は、製造業者によって記載されたようにRobosepによってBALB/cまたはDO11.10マウスのいずれかの脾臓から精製した。BALB/cマウスについては、1×105のCD4+T細胞を、中性条件下(1μg/mLの平板結合抗CD3+1μg/mLの可溶性抗CD28+10ng/mLのマウスIL−2)またはTh1促進条件下(中性+5ng/mLのマウスIL−12+10μg/mLの抗IL−4)またはTh2分化条件下(中性+10ng/mLのマウスIL−4+10μg/mLの抗IL−4)のいずれかで、10%の正常ウシ胎児血清(Hyclone)または活性炭処理FBS(Hyclone社)(外因性PGE2またはEP4アゴニストが培養に加えられた場合)のいずれかを含有する100μLの完全RPMI培地(Cellgro社)中で967ウエルプレート内で3〜6日間培養した。培養上清中のIFNγまたはIL−4はELISAによって検出した。細胞ペレットを使用して、製造業者の取扱説明書にしたがってAlamar blueまたはCellTiter−Glo試薬のいずれかを用いて細胞増殖を測定した。DO11.10マウスに対して、BALB/cマウス由来のマイトマイシンC処置脾臓細胞を抗原提示細胞として使用し、5:1の比率(100μL培地中の5×105のマイトマイシンC処置脾臓細胞+100μL培地中の1×105 CD4+T細胞)で天然CD4+T細胞と共培養し、上述したように中性、Th1またはTh2促進条件下のいずれかでOVAペプチド(0.3ng/mL)を用いて刺激した。EP4アゴニスト、アンタゴニストまたは抗PGE2抗体をTh細胞分化中に加えた。
【0109】
EP4アゴニスト/アンタゴニストがIL−17産生に及ぼす作用を試験するために、C57BL/6マウス由来の全CD4+T細胞を3〜5日間にわたり指示した濃度のIL−23(10ng/mL)またはEP4アゴニスト/アンタゴニストの存在下または非存在下において平板結合抗CD3(2μg/mL)+可溶性抗CD28(2μg/mL)により活性化した。培養上清をIL−17 ELISAによって分析し、細胞ペレットを使用してCellTite−Glo試薬を用いた細胞増殖を測定した。
【0110】
IL−23誘導性Th17増殖。CD4+T細胞をC57BL/6マウスから単離し、5日間にわたり、IL−23(30ng/mL)を伴う、または伴わない、抗TCR(1μg/mL平板結合)および抗I−CD28(2μg/mL、可溶性)を用いて活性化した。IL−17産生細胞は、製造業者(BD社)によって記載されたようにIL−17細胞内染色によって分析した。
【0111】
インビトロヒト単球由来DCアッセイ。CD14+細胞はMiltenyi CD14マイクロビーズを用いてヒトPBMCから精製し、10%木炭処理したFBSを含有する完全RPMI培地中でヒトGM−CSF(500U/mL)+ヒトIL−4(500U/mL)を用いて8日間にわたり分化させた。未結合imDC(未成熟樹状細胞)は、24時間にわたり指示した濃度にあるEP4アゴニスト/アンタゴニストまたは抗PGE2抗体の添加を伴って、または伴わずに、LPS(10ng/mL)+R−848(2.5μg/mL)を用いて刺激した。培養上清中のIL−23は、ELISA(eBioscience社)によって測定した。
【0112】
コラーゲン誘導性関節炎モデル。雄性DBA/1マウスをFreundの完全アジュバント中で乳化した150μgのウシII型コラーゲンを含有する、0.1mLエマルジョンを用いて尾の基部で皮内免疫した。初回免疫の3週間後、全マウスにFreundの不完全アジュバント中に乳化させたウシII型コラーゲンを用いて追加免疫した。各マウスの足における関節炎の症状の重症度は、公知の技術にしたがったWood et al.の方法によって判定した。
【0113】
PLP誘導性EAEモデル。SJLマウスに、Freundの完全アジュバント中に乳化させた35μgのPLP139−151を含有する0.1mLエマルジョンを皮下注射した。1×109/200μLの百日咳菌を各マウスへ第0日および第2日に注射した。EAEスコアは、公知の技術にしたがって評価した。
【0114】
エックスビボLNまたは脾臓細胞試験。単一懸濁細胞は、bCIIを用いた一次免疫の後第15日マウスまたはEAE試験終了時に排液LNまたは脾臓から調製し、bCII(50μg/mL)、MOG35−55(12.5μg/mL)またはPBS/培地のいずれかを用いて48〜72時間にわたり刺激した。培養上清中のサイトカイン産生をELISAによって分析し、細胞増殖はCellTiter−Glo試薬のいずれかによって測定した。「ミックス・アンド・マッチ(mix & match)」リンパ球反応のためには、排液LN由来の精製CD4+T細胞を1:5の比率でAPC(マイトマイシンC処理全LN細胞)と共培養し、上述したように刺激して分析した。
【0115】
[実施例128]
選択的EP4受容体アンタゴニストとして同定されたER−819924
ER−819924−01は、競合的放射性リガンド結合アッセイ(MDS Pharma社による)においてEP4へ選択的に結合するが他のEP/プロスタノイド/ロイコトリエン受容体には結合しないことが見いだされた(表4および図1)。さらに、1μMでのER−819924−01は、FLIPR機能的スクリーンにおいてEP4受容体に対してのみ活性を示し、他の132GPCRのいずれに対しても活性を示さなかった(Millipore社が外部委託したデータ)(表5)。ER−819924−01はさらに、PGE2に対する強力な阻害活性がSE302細胞においてCRE−PLAP受容体活性を誘導することも証明した(28nMのIC50値を伴う)(図2)。
【0116】
表4:プロスタノイド/ロイコトリエン受容体に対するER−819924−01の競合的リガンド結合アッセイ
【表4】
【0117】
表5:132 GPCRに対するER−819924−01のFLIPRスクリーン。アゴニストモードおよびアンタゴニストモードの両方を測定したが、表にはアンタゴニスト活性だけを示した。
【表5】
【0118】
[実施例129]
活性化マウスCD4+T細胞中でのIL−17産生にPGE2/EP4が及ぼす作用
EP4がさらにTh17応答においても役割を果たすかどうかを調査するために、PGE2またはEP4アゴニストによるEP4活性化が活性化マウスCD4+T細胞内でのIL−17産生に及ぼす作用を試験した。全CD4+T細胞は、3〜5日間にわたり、PGE2またはEP4アゴニストPGE1−OH(0.1nM〜1,000nM)の存在下または非存在下でIL−23を伴う、または伴わずに、抗CD3/抗CD28により刺激した。培養上清中のIL−17はELISAによって測定し、細胞増殖はCellTiter−gloによって測定した。結果は、EP4アゴニストが抗CD3/抗CD28刺激CD4+T細胞中の細胞増殖を抑制しながら、IL−17産生を増強することを証明した(図3)。同様に高度に選択的なEP4アンタゴニストであるまた別のBOAT化合物であるER−819762−01は、この系における活性を抑制した(図4)。さらに、抗PGE2抗体は、天然CD4+T細胞がIL−23の存在下で抗CD3/抗CD28を用いて活性化されるとIL−17産生細胞の数を減少させた(図5)。これらをまとめると、これらの結果は、EP4拮抗作用がTh17応答を抑制できるという仮説を裏付けた。
【0119】
[実施例130]
マウスTh1分化においてPGE2/EP4が果たす役割
EP4活性化がTh1分化において役割を果たすかどうかを調査するために、PGE2またはEP4アゴニストおよび抗PGE2抗体が及ぼす作用をマウスTh1分化アッセイにおいて試験した。天然CD4T細胞は、3日間にわたりTh1促進剤を用いて分化した。培養上清中のTh1サイトカインIFNγはELISAによって測定した。細胞増殖は、Alamar blueアッセイによって測定した。PGE2またはEP4アゴニストPGE1−OHをTh1分化中に加えた。結果は、PGE2およびEP4アゴニスト(PGE1−OH)はTh1分化を強化するが(図6)、他方PGE2に対する抗体が分化Th1細胞におけるIFN−γ産生を部分的に抑制することを証明したが(図7)、これはPGE2/EP4がTh1分化を調節することにある役割を果たすことを示している。さらに、ER−819762は抗PGE2抗体に活性を加えなかったが(図8)、これはER−819762によるTh1分化の抑制が主としてEP4に対する活性に起因することを示唆していた。
【0120】
[実施例131]
インビトロでのマウスIL−17増殖の抑制
ER−819924−01がIL−23誘導性Th17増殖に及ぼす作用をインビトロで試験した(図9)。マウスCD4+T細胞は、5日間にわたりER−819924−01または抗PGE2抗体の存在下または非存在下において抗TCR/抗CD28モノクロール抗体およびIL−23(30ng/mL)とともに培養した。IL−17産生T細胞は、細胞内染色のFACSによって分析した。ER−819924−01は、約10〜100nMのIC50値を備えるIL−23誘導性Th17産生細胞の数を減少させた。
【0121】
[実施例132]
CIAにおける関節炎発生の抑制
ER−819924−01の抗関節炎作用を評価するために、マウスCIAモデルにおいて幾つかの試験を実施した。最初にER−819924−01は、化合物が第20日から毎日経口投与される(すなわち、病原性抗II型コラーゲン抗体の誘導後で、関節炎発生の前に)部分治療投与レジメンにおいて試験した。関節炎スコア(炎症を起こした足の炎症反応指数として測定した)および体重を2週間にわたって監視した。部分治療投与条件下で、ER−819924−01は、約10mg/kgのED50を備えて関節炎の発生を効果的に抑制した(図10)。本発明者らは、さらに化合物が疾患の誘導後に投与される完全治療投与レジメンにおいてER−819924−01について試験した。完全治療投与条件下で、ER−819924−01は、約10mg/kgの用量でさらに関節炎の発生を効果的に防止した(図11)。さらに、ER−819924−01は、別個の試験において30mg/kgの用量でX線スコアを有意に改善した(図12)。
【0122】
[実施例133]
活性化ヒトMoDC(単球由来樹状細胞)における最適なIL−23産生のためには、PGE2/EP4シグナリングが必要とされ、化合物はこの活性を抑制する
ヒトCD14+単球を7日間にわたりGM−CSF+IL−4を用いてimDCに分化させ、外因性PGE1−OH(1〜100nM)の存在下または非存在下において、そしてER−819762(図13)もしくはER−819924または抗PGE2抗体(図14)を用いて、または用いずに、LPS/R−848を用いて24時間にわたり再刺激した。培養上清中のIL−23分泌は、ELISAによって測定した。表6は、ヒトMoDC IL−23分泌アッセイにおける代表的化合物のIC50値を示している。
【0123】
表6:活性化ヒトMoDCにおけるPGE1−OH(10nM)強化IL−23産生を阻害する際の代表的な化合物のIC50値。
【表6】
【0124】
[実施例134]
EAEにおけるTh1/Th17反応のエックスビボ抑制
排液リンパ節は、治療的MOG EAE試験の終了時にマウスから採取した。単一細胞懸濁液を調製し、48時間にわたりMOGまたは培地のいずれかを用いて刺激した。上清は、Search Light複数サイトカイン分析(PIERCE Technology社製)のために収集した。データは、図15に示した。
【0125】
本発明の多数の実施形態について記載してきたが、本発明の化合物および方法を利用する他の実施形態を提供するために基本的実施例を変化させられ得ることは明白である。このため本発明の範囲は、例として提示してきた特定実施形態によってではなく添付の特許請求項によって規定されることは理解される。
【産業上の利用可能性】
【0126】
本発明にかかる方法は、関節リウマチや多発性硬化症の治療に有効である。
【技術分野】
【0001】
本発明は、関節リウマチや多発性硬化症を治療する方法に関する。
【背景技術】
【0002】
抗原に遭遇すると、天然CD4+ヘルパーT前駆(Thp)細胞は、2つの別個のサブセットである1型ヘルパーT(Th1)および2型ヘルパーT(Th2)に分化する。これらの分化したTh細胞は、それらの別個の機能能力および固有のサイトカインプロファイルの両方によって規定される。詳細には、Th1細胞は、インターフェロン−γ、インターロイキン(IL)−2、および腫瘍壊死因子(TNF)−βを産生するが、これらはマクロファージを活性化し、細胞媒介性免疫および食細胞依存性保護応答に関与する。対照的に、Th2細胞は、IL−4、IL−5、IL−6、IL−9、IL−10およびIL−13を産生することが公知であるが、これらは強力な抗体産生、好酸球活性化、および数種のマクロファージ機能の阻害に対して役割を果たすので、食細胞非依存性保護応答を提供する。したがって、Th1およびTh2細胞は、様々な免疫病理学的応答と関連している。
【0003】
さらに、各タイプのTh細胞の発生は、様々なサイトカイン経路によって媒介される。詳細には、IL−4はTh2分化を促進し、同時にTh1発生を遮断することが証明されている。対照的に、IL−12、IL−18およびIFN−γは、Th1細胞の発生に対して重大なサイトカインである。したがって、これらのサイトカイン自体がTh分極を駆動し、Th1とTh2との間の平衡を維持する正および負のフィードバック系を形成する。PEG2は、さらにまたインターフェロンγ(IFN−γ)産生およびT細胞増殖の抑制によってTH1応答を調節することに役割を果たすことも証明されている。
【0004】
Th1細胞は、様々な臓器特異的自己免疫疾患、クローン病、ヘリコバクター・ピロリ誘導性消化性潰瘍、急性腎臓同種移植拒絶反応、および原因不明の反復性流産の病因に関係している。対照的に、アレルゲン特異的Th2応答は、遺伝的に感受性の強い個体におけるアトピー性疾患の原因である。さらに、Th2は、オーメン(Omenn’s syndrome)症候群、特発性肺線維症、および進行性全身性硬化症において優勢である依然として未知の抗原に対して応答する。IL−17(Th−17サイトカインの符号)ノックアウトマウスは、炎症性関節炎の発生に対して顕著な耐性を示す。CIAモデルにおける関節破壊は、抗IL−17中和抗体の投与によって改善することができる。
【0005】
そこで、不均衡なTh1/Th2およびTh17細胞分化に関連する様々な状態を治療する際に有用である新規な治療的処置を開発する高度な満たされていない医療的必要性が残っている。これらの多くの状態のために現在利用できる治療選択肢は不適切である。したがって、Th1/Th2およびTh17のパラダイムは、アレルギー性および自己免疫障害を治療する戦略を開発するための論理的根拠を提供する。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】米国特許第7,189,715号明細書
【非特許文献】
【0007】
【非特許文献1】T−bet regulates IgG class switching and pathogenic auto Ab production,Proc.Natl.Acad.Sci.USA 99(8):5545−50(2002);Imbalance of Th1/Th2 transcription factors in patients with lupus nephritis,Rheumatology(Oxford) 45(8):951−7(2006)
【非特許文献2】Identification of a novel type 1 diabetes susceptibility gene,T−bet,Human Genetics 111(3):177−84(2004);T−bet controls autoaggressive CD8 lymphocyte response in type I diabetes,J.Exp.Med.199(8):1153−62(2004)
【非特許文献3】J.Mol.Med 81(8): 471−80 (2003)
【非特許文献4】Proc.Natl.Acad.Sci.USA 102(5):1596−601(2005)
【発明の概要】
【発明が解決しようとする課題】
【0008】
脂質メディエータであるプロスタグランジンE2(PGE2)は、免疫応答の様々な段階を変調することが証明されている。PGE2が細胞内cAMPの上昇およびIckの不活性化を通してCD4+T細胞活性化を抑制することは周知である。しかし、本明細書に記載したように、EP4受容体によるPGE2刺激は反対の作用も有する可能性があり、つまり、活性化CD4+細胞内でのTh1分化およびIL−17産生を促進することがある。これに一致して、EP4と新規な選択的EP4アンタゴニストまたはPGE2中和抗体いずれかとの拮抗作用は、Th1分化、Th17増殖、ならびに活性化された樹状細胞によるIL−23分泌を抑制する。PGE2によるTh1分化の誘導はPI3Kシグナリングによって媒介されるが、他方IL−17産生の刺激はcAMPシグナリングを必要とする。さらに、EP4アンタゴニストのDBA/1またはC57BL/6マウスへの投与は先天性および適応的免疫応答を抑制し、さらにコラーゲン誘導性関節炎(CIA)および実験的自己免疫脳脊髄炎(EAE)モデルにおける疾患を抑制したので、これはPGE2/EP4シグナリングがこれらの自己免疫病理に極めて重要に関係することを示した。これらの結果は、PGE2/EP4シグナリングの抑制が例えば関節リウマチおよび多発性硬化症などの炎症性自己免疫疾患を改善することに治療的価値を有する可能性があることを示唆している。
【課題を解決するための手段】
【0009】
本発明の第1態様は、被験体における関節リウマチを治療する方法であって、該被験体にTh1分化もしくはTh17増殖の調節因子化合物を含む組成物を投与するステップを含む方法である。一部の実施形態では、調節因子化合物は、式:
【化1】
の化合物、またはその医薬上許容される塩である。
【0010】
本発明のまた別の態様は、関節リウマチを治療するための医薬品の製造における化合物の使用であって、該医薬品は調節因子化合物を含み、該調節因子化合物はTh1分化もしくはTh17増殖の調節因子である使用である。一部の実施形態では、調節因子化合物は、式:
【化2】
の化合物、またはその医薬上許容される塩である。
【0011】
本発明のまた別の態様は、関節リウマチを治療するためのTh1分化もしくはTh17増殖の調節因子化合物である。
【0012】
本発明のまた別の態様は、被験体における多発性硬化症を治療する方法であって、該被験体にTh1分化もしくはTh17増殖の調節因子化合物を含む組成物を投与するステップを含む方法である。一部の実施形態では、調節因子化合物は、式:
【化3】
の化合物、またはその医薬上許容される塩である。
【0013】
本発明のまた別の態様は、多発性硬化症を治療するための医薬品の製造における化合物の使用であって、該医薬品は調節因子化合物を含み、該調節因子化合物はTh1分化もしくはTh17増殖の調節因子である使用である。一部の実施形態では、調節因子化合物は、式:
【化4】
の化合物、またはその医薬上許容される塩である。
【0014】
本発明のまた別の態様は、多発性硬化症を治療するためのTh1分化もしくはTh17増殖の調節因子化合物である。
【0015】
本発明のまた別の態様は、被験体における関節リウマチを治療する方法であって、該被験体にEP4アンタゴニストを含む組成物を投与するステップを含む方法である。一部の実施形態では、EP4アンタゴニストは、式:
【化5】
の化合物、またはその医薬上許容される塩である。
【0016】
本発明のまた別の態様は、関節リウマチを治療するための医薬品の製造における化合物の使用であって、該医薬品はEP4アンタゴニストを含む使用である。一部の実施形態では、EP4アンタゴニストは、式:
【化6】
の化合物、またはその医薬上許容される塩である。
【0017】
本発明のまた別の態様は、関節リウマチを治療するためのEP4アンタゴニストである。一部の実施形態では
【0018】
本発明のまた別の態様は、被験体における多発性硬化症を治療する方法であって、該被験体にEP4アンタゴニストを投与するステップを含む方法である。一部の実施形態では、EP4アンタゴニストは、式:
【化7】
の化合物、またはその医薬上許容される塩である。
【0019】
本発明のまた別の態様は、多発性硬化症を治療するための医薬品の製造における化合物の使用であって、該医薬品はEP4アンタゴニストを含む使用である。一部の実施形態では、EP4アンタゴニストは、式:
【化8】
の化合物、またはその医薬上許容される塩である。
【0020】
本発明のまた別の態様は、多発性硬化症を治療するためのEP4アンタゴニストである。
【発明の効果】
【0021】
本発明の方法によれば、関節リウマチや多発性硬化症を治療することができる。
【0022】
本発明の他の態様については、本明細書において開示し、以下でより詳細に考察する。
【図面の簡単な説明】
【0023】
【図1】EP4結合アッセイにおけるER−819924−01およびそのエナンチオマーであるER−819925−01の滴定試験を示す図である。
【図2】ER−819924およびER−819762が、SE302細胞におけるPGE2誘導性CRE−PLAPレポーター活性に対する強力な阻害活性を示した図である。どちらの化合物も0.3nM〜10μMの濃度で試験した。
【図3】PGE2/EP4アゴニストが抗CD3/抗CD28を用いて刺激されたCD4+T細胞からのIL−17産生に及ぼす作用を示す図である。
【図4(2−1)】ER−819762が活性化CD4+細胞からのPGE2/PGE1−OH強化IL−17産生を抑制することを示す図である。
【図4(2−2)】ER−819762が活性化CD4+細胞からのPGE2/PGE1−OH強化IL−17産生を抑制することを示す図である。
【図5】抗PGE2抗体がIL−23媒介性Th17増殖に及ぼす作用を示す図である。
【図6(2−1)】PGE2/EP4アゴニストがマウスTh1分化に及ぼす作用を示す図である。
【図6(2−2)】PGE2/EP4アゴニストがマウスTh1分化に及ぼす作用を示す図である(IFNg-Th1)。
【図7】抗PGE2抗体がマウスTh1分化に及ぼす作用を示す図である。抗PGE2抗体は、Th1分化中に加えられた。
【図8】マウスTh1分化における抗PGE2抗体にER−819762は相加作用を及ぼさないことを示す図である。
【図9】インビトロでのマウスIL−17増殖の抑制を示す図である。
【図10】CIAにおけるER−819924−01の部分的治療評価を示す図である。
【図11】CIAにおけるER−819924−01の完全治療評価を示す図である。
【図12】完全治療的CIA試験からのマウス足のX線分析を示す図である。X線スコアは、骨減少症、骨浸食および新規骨形成の組み合わせの測定値の指標である。
【図13】ヒトCD14+単球を7日間にわたりGM−CSF+IL−4を用いてimDCに分化させ、外因性PGE1−OH(1〜100nM)の存在下もしくは非存在下で、そしてER−819762を用いて、および用いずに、LPS/R−848を用いて再刺激した。
【図14】ヒトCD14+単球を7日間にわたりGM−CSF+IL−4を用いてimDC内に分化させ、外因性PGE1−OH(1〜100nM)の存在下もしくは非存在下で、そしてER−819924もしくは抗PGE2抗体を用いて、および用いずに、LPS/R−848を用いて再刺激した。
【図15】EAEにおけるTh1/Th17応答のエックスビボ抑制を示す図である。
【発明を実施するための形態】
【0024】
A.定義
本明細書で使用する用語「Th1分化もしくはTh17増殖の調節因子」または「Th1分化もしくはTh17増殖の調節因子化合物」または本明細書で使用する用語「調節因子化合物」は、天然CD4+T細胞のTh1細胞への分化を抑制する、減少させる、または阻害する化合物を意味する。一部の実施形態では、用語「Th1分化もしくはTh17増殖の調節因子」または本明細書で使用する用語「Th1分化もしくはTh17増殖の調節因子化合物」は、IL−17産生CD4+細胞の数または活性化CD4+細胞内でのIL−17産生を抑制する、減少させる、または阻害する化合物を意味する。
【0025】
本明細書で使用する「エナンチオマー的に純粋」は、ステレオマー的に純粋な化合物、または1つのキラル中心を有する化合物の組成物を意味している。
【0026】
本明細書で使用する「ステレオマー的に純粋」は、化合物の1つの立体異性体を含み、その化合物の他方の立体異性体を実際的に含んでいない化合物または組成物を意味している。例えば、1つのキラル中心を有する化合物のステレオマー的に純粋な組成物は、実質的に該化合物の反対エナンチオマーを含んでいない。2つのキラル中心を有する化合物のステレオマー的に純粋な組成物は、該化合物の他方のジアステレオマーを実質的に含んでいない。典型的なステレオマー的に純粋な化合物は、約80重量%超の該化合物の1つの立体異性体および約20重量%未満の該化合物の他方の立体異性体、より好ましくは約90重量%超の該化合物の1つの立体異性体および約10重量%未満の該化合物の他方の立体異性体、一層より好ましくは約95重量%超の該化合物の1つの立体異性体および約5重量%未満の該化合物の他方の立体異性体、および最も好ましくは約97重量%超の該化合物の1つの立体異性体および約3重量%未満の該化合物の他方の立体異性体を含んでいる(例えば、特許文献1)。
【0027】
本明細書で使用する「安定性」は、本明細書に開示した1つ以上の目的のためにそれらの産生、検出、ならびに好ましくはそれらの回収、精製、および使用を許容する条件に受けさせたときに、実質的に変化しない化合物に関する。一部の実施形態では、安定性化合物または化学的に実行可能な化合物は、少なくとも1週間にわたり、水分またはその他の化学的反応性条件の存在下で、40℃以下の温度で維持された場合に実質的に変化させられない化合物である。
【0028】
本明細書で使用する「アルキル」または「アルキル基」は、完全に飽和している直鎖状(すなわち、非分枝状)、分枝状、または環状炭化水素鎖を意味する。所定の実施形態では、アルキル基は、1〜3個の炭素原子を含有している。さらに他の実施形態では、アルキル基は2〜3個の炭素原子を含有し、そしてさらに他の実施形態では、アルキル基は1〜2個の炭素原子を含有している。所定の実施形態では、用語「アルキル」または「アルキル基」は、炭素環としても公知であるシクロアルキル基を意味する。典型的なC1−3アルキル基には、メチル、エチル、プロピル、イソプロピル、およびシクロプロピルが含まれる。
【0029】
本明細書で使用する「アルケニル」または「アルケニル基」は、1つ以上の二重結合を有する直鎖状(すなわち、非分枝状)、分枝状、または環状炭化水素鎖を意味する。所定の実施形態では、アルケニル基は、2〜4個の炭素原子を含有している。さらに他の実施形態では、アルケニル基は3〜4個の炭素原子を含有し、そしてさらに他の実施形態では、アルケニル基は2〜3個の炭素原子を含有している。また別の態様によると、用語「アルケニル」は、2つの二重結合を有する直鎖状炭化水素を意味し、「ジエン」とも呼ばれる。他の実施形態では、用語「アルケニル」または「アルケニル基」は、シクロアルケニル基を意味する。典型的なC2−4アルケニル基には、−CH=CH2、−CH2CH=CH2(アリルとも呼ばれる)、−CH=CHCH3、−CH2CH2CH=CH2、−CH2CH=CHCH3、−CH=CH2CH2CH3、−CH=CH2CH=CH2、およびシクロブテニルが含まれる。
【0030】
本明細書で使用する「アルコキシ」または「アルキルチオ」は、以前に規定したように、アルキル基を意味し、酸素(「アルコキシ」)または硫黄(「アルキルチオ」)原子を通して主要炭素鎖に結合した。
【0031】
本明細書で使用する「メチレン」、「エチレン」、および「プロピレン」は、二価成分であるCH2−、−CH2CH2−、および−CH2CH2CH2−を各々意味する。
【0032】
本明細書で使用する「エテニレン」、「プロペニレン」、および「ブテニレン」は、二価成分である−CH=CH−、−CH=CHCH2−、−CH2CH=CH−、−CH=CHCH2CH2−、−CH2CH=CH2CH2−、および−CH2CH2CH=CH−、を意味するが、このとき各エテニレン基、プロペニレン基、およびブテニレン基はcisまたはtrans配列にあってよい。所定の実施形態では、エテニレン基、プロペニレン基、またはブテニレン基は、trans配列にあってよい。
【0033】
「アルキリデン」は、メチレンのモノまたはジアルキル置換によって形成された二価炭化水素基を意味する。所定の実施形態では、アルキリデン基は、1〜6個の炭素原子を有する。他の実施形態では、アルキリデン基は、2〜6、1〜5、2〜4、または1〜3個の炭素原子を有する。そのような基には、プロピリデン(CH3CH2CH=)、エチリデン(CH3CH=)、およびイソプロピリデン(CH3(CH3)CH=)などが含まれる。
【0034】
「アルケニリデン」は、メチレンのモノまたはジアルケニル置換によって形成された1つ以上の二重結合を有する二価炭化水素基を意味する。所定の実施形態では、アルケニリデン基は、2〜6個の炭素原子を有する。他の実施形態では、アルケニリデン基は、2〜6、2〜5、2〜4、または2〜3個の炭素原子を有する。1つの態様によると、アルケニリデンは、2つの二重結合を有する。典型的なアルケニリデン基には、CH3CH=C=、CH2=CHCH=、CH2=CHCH2CH=、およびCH2=CHCH2CH=CHCH=が含まれる。
【0035】
「C1−6アルキルエステルもしくはアミド」は、C1−6アルキルエステルもしくはC1−6アルキルアミドを意味するが、このとき各C1−6アルキル基は上記に規定したとおりである。そのようなC1−6アルキルエステル基は、式(C1−6アルキル)OC(=O)−または(C1−6アルキル)C(=O)O−の基である。そのようなC1−6アルキルアミド基は、式(C1−6アルキル)NHC(=O)−または(C1−6アルキル)C(=O)NH−の基である。
【0036】
「C2−6アルケニルエステルもしくはアミド」は、C2−6アルケニルエステルもしくはC2−6アルケニルアミドを意味し、ここで、各C2−6アルケニル基が上記に規定されている。そのようなC2−6アルキルエステル基は、式(C2−6アルケニル)OC(=O)−または(C2−6アルケニル)C(=O)O−の基である。そのようなC2−6アルケニルアミド基は、式(C2−6アルケニル)NHC(=O)−または(C2−6アルケニル)C(=O)NH−の基である。
【0037】
「治療」、「治療する」および「治療するステップ」は、本明細書に記載した疾患または障害の発生を逆転させる、軽減する、遅延させる、進行を阻害する、または予防することを意味する。一部の実施形態では、治療は、1つ以上の症状が発生した後に投与されてよい。他の実施形態では、治療は、症状の非存在下で投与されてよい。例えば、治療は、症状の発生前に(例えば、症状の履歴に照らして、および/または遺伝的もしくはその他の感受性因子に照らして)感受性のある個体に投与されてよい。治療はさらにまた、症状が消散した後に、例えば症状の再発を予防または遅延させるために継続されてよい。
【0038】
本明細書で使用する「患者」または「被験体(subject)」は、動物被験体、好ましくは哺乳動物被験体(例えば、イヌ、ネコ、ウマ、ウシ、ヒツジ、ヤギ、サルなど)、および特にヒト被験者(男性および女性被験者の両方を含み、さらに新生児、幼児、少年、青年、成人および高齢被験者を含む)を意味する。
【0039】
本明細書で使用する用語「医薬上許容される担体」は、それが一緒に調製される化合物の薬理学的活性を破壊しない、非毒性の担体、アジュバント、またはビヒクルを意味する。本発明の組成物中に使用できる医薬上許容される担体、アジュバントまたはビヒクルには、イオン交換剤、アルミナ、ステアリン酸アルミニウム、レシチン、例えばヒト血清アルブミンなどの血清タンパク質、例えばリン酸塩などの緩衝物質、グリシン、ソルビン酸、ソルビン酸カリウム、飽和植物性脂肪酸の部分グリセリド混合物、水、例えば硫酸プロタミン、リン酸水素二ナトリウム、リン酸水素カリウム、塩化ナトリウム、亜鉛塩などの塩類または電解質類、コロイド状シリカ、三ケイ酸マグネシウム、ポリビニルピロリドン、セルロースをベースとする物質、ポリエチレングリコール、シクロデキストリン類、カルボキシメチルセルロースナトリウム、ポリアクリレート類、ワックス類、ポリエチレン・ポリオキシプロピレンブロックポリマー類、ポリエチレングリコールおよび羊毛脂が含まれるがそれらに限定されない。
【0040】
他に特に指示しない限り、本明細書で使用する化学基または成分を記載するために使用する術語は慣習に従うが、このとき名称は左から右へ読み、分子の残りへの結合点は名称の右側にある。例えば、「(C1−3アルコキシ)C1−3アルキル」基は、アルキル末端で分子の残りに結合している。また別の例には、結合点がエチル末端にあるメトキシエチル、および結合点がアミン末端にあるメチルアミノが含まれる。
【0041】
他に特に指示しない限り、二価基が「−」によって指示される2つの末端結合成分を含む化学式によって記載される場合は、その結合は左から右へ読まれると理解される。
【0042】
他に特に指示しない限り、本明細書に示した構造はさらにまたその構造の全てのエナンチオマー、ジアステレオマー、および幾何学的(または立体配座)形、例えば各不斉中心に対するRおよびS立体配置、(Z)および(E)二重結合異性体、ならびに(Z)および(E)配座異性体を含むことが意図されている。このため、本発明の化合物の単一立体化学的異性体ならびにエナンチオマー、ジアステレオマー、および幾何学的(または立体配座)混合物は、本発明の範囲内に含まれる。他に特に指示しない限り、本発明の化合物の全ての互変異性体形は、本発明の範囲内に含まれる。さらに、他に特に指示しない限り、本明細書に記載した構造は、さらにまた1つ以上の同位体が濃縮された原子の存在に関してのみ相違する化合物を含むことも意図されている。例えば、ジューテリウム(重水素)またはトリチウムによる水素の置換、または13Cもしくは14C濃縮炭素による炭素の置換を除いて、本発明の構造を有する化合物は、本発明の範囲内に含まれる。そのような化合物は、例えば、生物学的アッセイにおける分析ツールまたはプローブとして有用である。
【0043】
B.活性化合物/調節因子化合物/EP4アンタゴニスト
本明細書で記載する本発明の活性化合物(本明細書では「調節因子化合物」および/または「EP4アンタゴニスト」と呼ぶこともある)は、一般に上記で例示したような、または本発明の特定のクラス、サブクラス、および種によって例示されるような1つ以上の置換基と任意で置換されてよい。一般に、用語「置換(された)」は、所定の構造内の水素ラジカルと特定の置換基のラジカルとの置換を意味する。他に特に指示しない限り、置換基は、その基の各置換可能な位置で置換基を有していてよく、任意の所定の構造内での2つ以上の位置が特定の基より選択される2つ以上の置換基と置換されてよい場合は、該置換基は1つ1つの位置で同一であっても相違していてもよい。
【0044】
上述したように、本発明は、式I:
【化9】
のエナンチオマー的に純粋な化合物、もしくは活性な化合物、
またはより詳細には式Iaもしくは式Ib:
【化10】
(式中、
R1はC1−3アルキルであり;
Xは、メチレン、エチレン、プロピレン、エテニレン、プロペニレン、またはブテニレンであり;
R5は、フェニル、ピロリル、ベンズイミダゾリル、オキサゾリル、イソキサゾリル、イミダゾチアゾリル、キノリニル、イソキノリニル、インダゾリル、ピリジニル、イミダゾピリジニル、インドリル、ベンゾトリアゾリル、イミダゾリル、ベンゾフラニル、ベンゾチアジアゾリル、ピリジミジニル、ベンゾピラノニル、チアゾリル、チアジアゾリル、フリル、チエニル、ピラゾリル、キノキサリニル、もしくはナフチルであり、C1−4アルキル、C1−3アルコキシ、ヒドロキシル、C1−3アルキルチオ、シクロプロピル、シクロプロピルメチル、トリフルオロメトキシ、5−メチルイソキサゾリル、ピラゾリル、ベンジルオキシ、アセチル、(シアニル)C1−3アルキル、(フェニル)C2−3アルケニルおよびハロよりなる群から独立して選択された0〜5個の置換基で置換されており;
R8は、H、メチル、エチル、プロピル、(C1−3アルコキシ)C1−3アルキル、(C1−3アルキルチオ)C1−3アルキル、C1−3ヒドロキシアルキル、フェニル、ベンジル、フリル、ピロリル、イミダゾリル、ピラゾリル、ピロリル、イソチアゾリル、イソキサゾリル、ピリジル、およびチエニルであり;
(このとき、R8は、メチル、エチル、ハロ、ヒドロキシル、C1−3アルコキシ、C1−3アルキルチオ、(C1−3アルコキシ)C1−3アルキル、(C1−3アルキルチオ)C1−3アルキル、C1−3ヒドロキシアルキル、(C1−3メルカプトアルキル)フェニル、ベンジル、フリル、イミダゾリル、ピラゾリル、ピロリル、イソチアゾリル、イソキサゾリル、ピリジル、およびチエニルより独立して選択される0〜3個の置換基で置換されており);
Ra、Rb、およびRcの各々は、水素、ヒドロキシル、メトキシ、ベンジルオキシ、フルオロ、クロロ、アミノ、メチルアミノ、ジメチルアミノ、およびフェノキシより独立して選択され;
またはRaおよびRb、ならびにRbおよびRcより選択される1対は、一緒になって−O−(CH2)−O−または−O−CH2−CH2−O−である)
またはその医薬上許容される塩、C1−6アルキルエステルもしくはアミド、またはC2−6アルケニルエステルもしくはアミドを提供する。
【0045】
上記の一部の実施形態では:
R1はC1−2アルキルである;
R5は、フェニル、ピロリル、ベンズイミダゾリル、オキサゾリル、イソキサゾリル、イミダゾチアゾリル、キノリニル、イソキノリニル、インダゾリル、ピリジニル、イミダゾピリジニル、インドリル、ベンゾトリアゾリル、イミダゾリル、ベンゾフラニル、ベンゾチアジアゾリル、ピリジミジニル、ベンゾピラノニル、チアゾリル、チアジアゾリル、フリル、チエニル、ピラゾリル、キノキサリニル、もしくはナフチルであり、およびC1−4アルキル、C1−3アルコキシ、ヒドロキシル、C1−3アルキルチオ、シクロプロピル、シクロプロピルメチル、トリフルオロメトキシ、5−メチルイソキサゾリル、ピラゾリル、ベンジルオキシ、アセチル、(シアニル)C1−3アルキル、(フェニル)C2−3アルケニルおよびハロよりなる群から独立して選択された0〜5個の置換基で置換されている;
R8は、メチル、エチル、またはプロピルである(このときR8は0〜3個のヒドロキシル置換基で置換されている);
Xは、メチレンもしくはエチレンである;および
Ra、Rb、およびRcの各々は、Hおよびメトキシよりなる群から独立して選択される;
またはそれらの医薬上許容される塩、C1−6アルキルエステルもしくはアミド、またはC2−6アルケニルエステルもしくはアミドである。
【0046】
上記の一部の実施形態では:
R1は、メチルである;
R5は、フェニル、ピロリルもしくはピラゾリルであり、それらの各々はメチルで0、1または2回置換される;
R8は、エチルである;
Xは、メチレンである;
RaおよびRcの各々は、メトキシである;および
Rbは、Hである;
またはその医薬上許容される塩である。
【0047】
上記の特定の実施形態では、本化合物は:
【化11】
またはそれらの医薬上許容される塩である。
【0048】
本発明の活性化合物には、上記の医薬上許容される塩類が含まれる。医薬上許容される塩類には、医薬上許容される無機塩および有機酸および塩基に由来する塩類が含まれる。適切な酸性塩類の例には、酢酸塩、アジピン酸塩、アルギン酸塩、アスパラギン酸塩、安息香酸塩、ベンゼンスルホン酸塩、重硫酸塩、酪酸塩、クエン酸塩、樟脳酸塩、樟脳スルホン酸塩、シクロペンタンプロピオン酸塩、ジグルコン酸塩、ドデシル硫酸塩、エタンスルホン酸塩、ギ酸塩、フマル酸塩、グルコヘプタン酸塩、グリセロリン酸塩、グリコール酸塩、ヘミ硫酸塩、ヘプタン酸塩、ヘキサン酸塩、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、2−ヒドロキシエタンスルホン酸塩、乳酸塩、マレイン酸塩、マロン酸塩、メタンスルホン酸塩、2−ナフタレンスルホン酸塩、ニコチン酸塩、硝酸塩、シュウ酸塩、パルモエート(palmoate)、ペクチン酸塩、過硫酸塩、3−フェニルプロピオン酸塩、リン酸塩、ピクリン酸塩、ピバル酸塩、プロピオン酸塩、サリチル酸塩、コハク酸塩、硫酸塩、酒石酸塩、チオシアン酸塩、トシル酸塩およびウンデカン酸塩が含まれる。例えばシュウ酸などのその他の酸は、それら自体は医薬上許容されないが、本発明の化合物およびそれらの医薬上許容される酸付加塩を得る際の中間体として有用な塩類の調製において使用することができる。
【0049】
適切な塩基に由来する塩類には、アルカリ金属(例えば、ナトリウムおよびカリウム)、アルカリ土類金属(例えば、マグネシウム)、アンモニウムおよびN+(C1−4アルキル)4塩類が含まれる。本発明は、さらにまた本明細書に開示した化合物の任意の塩基性窒素含有基の四級化も想定している。そのような四級化によって、水溶性もしくは油溶性または分散性生成物を入手できる。
【0050】
C.医薬調製物
本発明の活性化合物(調節因子化合物および/またはEP4アンタゴニストを含む)は、それらの医薬調製物を提供するために医薬上許容される担体と結合することができる。担体および調製物の特定の選択は、該組成物の予定される特定投与経路に依存する。
【0051】
本発明の組成物は、経口、非経口、吸入スプレー、局所、経直腸、経鼻腔、経口腔、経膣または埋め込みリザーバ投与などのために適合する可能性がある。好ましくは、本組成物は、経口、腹腔内または静脈内投与される。本発明の組成物の無菌注射剤形は、水性または油性懸濁剤であってよい。これらの懸濁剤は、適切な分散剤もしくは湿潤剤および懸濁化剤を使用して、当分野において公知の技術にしたがって調製できる。無菌注射用製剤は、例えば1,3−ブタンジオール中の溶液として、非毒性の非経口的に許容される希釈剤または溶媒中の無菌注射用液剤または懸濁剤であってもよい。特に使用できる許容されるビヒクルおよび溶媒は、水、リンガー(Ringer)溶液および等張塩化ナトリウム溶液である。さらに、無菌の不揮発性油は、慣習的には溶媒または懸濁化剤として使用される。
【0052】
この目的で、合成モノ−またはジ−グリセリド類を含む任意のブランドの不揮発性油を使用できる。例えばオレイン酸などの脂肪酸およびそのグリセリド誘導体は、例えば、特にそれらのポリオキシエチル化形にあるオリーブ油またはヒマシ油などの天然の医薬上許容される油と同様に、注射剤の調製において有用である。これらの油性溶液または懸濁液は、例えばエマルジョンおよび懸濁液を含む医薬上許容される剤形の調製において一般に使用されるカルボキシメチルセルロースまたは類似の分散剤などの長鎖アルコール希釈剤または分散剤をさらに含有していてもよい。例えばTween類、Span類および医薬上許容される固体、液体、またはその他の剤形の製造において一般に使用されるその他の乳化剤またはバイオアベイラビリティ強化剤などの他の一般に使用される界面活性剤類もまた調製のために使用できる。
【0053】
本発明の医薬上許容される組成物は、カプセル剤、錠剤、水性懸濁剤または液剤を含む任意の経口投与に許容される剤形で経口投与することができるが、それらに限定されない。経口使用するための錠剤の場合には、一般に使用される担体には、ラクトースおよびコーンスターチが含まれる。典型的には、例えばステアリン酸マグネシウムなどの潤滑剤もまた加えられる。カプセル形での経口投与のために有用な希釈剤には、ラクトースおよび乾燥コーンスターチが含まれる。経口使用のために水性懸濁剤が必要とされる場合は、有効成分が乳化剤および懸濁化剤と結合される。所望であれば、所定の甘味料、フレーバー剤または着色剤もまた加えることができる。
【0054】
または、本発明の医薬上許容される組成物は、直腸投与のための坐剤の形態で投与することができる。これらは、該物質を室温では固体であるが直腸温度では液体である適切な非刺激性賦形剤と混合する工程によって調製することができるので、このため直腸内で融解して薬物を放出する。そのような材料には、カカオ脂、蜜ろうおよびポリエチレングリコールが含まれる。
【0055】
本発明の医薬上許容される組成物は、特に治療の標的が目、皮膚、または下部消化管の疾患を含む、局所適用によって容易に接近できる領域または器官を含む場合には、局所投与することもできる。適切な局所用調製物は、これらの領域または器官の各々のために容易に調製される。
【0056】
下部消化管に対する局所適用は、直腸坐剤調製物(上記参照)または適切な浣腸調製物で実行することができる。局所用経皮パッチもまた使用できる。
【0057】
局所適用のためには、医薬上許容される組成物は、1つ以上の担体中に懸濁化または溶解した活性成分を含有する適切な軟膏剤に調製することができる。本発明の化合物を局所投与するための担体には、鉱油、流動ワセリン(liquid petrolatum)、白色ワセリン、プロピレングリコール、ポリオキシエチレン、ポリオキシプロピレン化合物、乳化ろうおよび水が含まれるがそれらに限定されない。または、医薬上許容される組成物は、1つ以上の医薬上許容される担体中に懸濁化または溶解した活性成分を含有する適切なローション剤またはクリーム剤に調製することができる。適切な担体には、鉱油、ソルビタンモノステアレート、ポリソルベート60、セチルエステル類ロウ、セテアリルアルコール、2−オクチルドデカノール、ベンジルアルコールおよび水が含まれるがそれらに限定されない。
【0058】
眼科使用のためには、医薬上許容される組成物は、等張性のpH調整無菌食塩液中の微粉化懸濁剤として、または好ましくは、等張性のpH調整無菌食塩液中の液剤として、例えば塩化ベンジルアルコニウムなどの保存料を含めて、または含めずに調製することができる。または、眼科使用のためには、医薬上許容される組成物は、例えばワセリンなどの軟膏剤で調製することができる。
【0059】
本発明の医薬上許容される組成物は、さらにまた鼻腔エーロゾルまたは吸入によって投与することもできる。そのような組成物は、医薬調製物の分野において周知の技術にしたがって調製され、ベンジルアルコールもしくはその他の適切な保存料、バイオアベイラビリティを強化するための吸収促進剤、フルオロカーボン、および/または他の従来型可溶化剤もしくは分散剤を使用して、食塩液中の液剤として調製することができる。
【0060】
最も好ましくは、本発明の医薬上許容される組成物は、経口投与のために調製される。
【0061】
D.被験体および使用方法
本発明の活性化合物(調節因子化合物および/またはEP4アンタゴニストを含む)は、様々な相違する状態を治療するために患者または被験体に、特に以下に罹患している患者または被験体に投与することができる。
(a)関節リウマチ;
(b)多発性硬化症;
(c)全身性エリテマトーデス(例えば、非特許文献1);
(d)1型糖尿病(例えば、非特許文献2);
(e)乾癬(例えば、非特許文献3);および
(f)アテローム硬化症(例えば、非特許文献4)
【0062】
活性化合物は、経口、非経口、吸入スプレー、局所的、経直腸、経鼻腔、経口腔、経膣または埋め込みリザーバなどの任意の適切な経路によって被験体に投与されてもよい。本明細書で使用する用語「非経口」には、皮下、静脈下、筋肉内、関節内、滑膜内、胸骨内、クモ膜下、肝内、病巣内および頭蓋内注射または注入技術が含まれる。好ましくは、本組成物は、経口、腹腔内または静脈内投与される。
【0063】
本活性化合物は、被験体に治療有効量、または治療的に有効な量で投与される。単位製剤形にある組成物を製造するために担体材料と結合できる本発明の化合物の量は、治療される宿主、および特定投与経路に依存して変動する。好ましくは、本組成物は、これらの組成物を摂取する患者に0.01〜100mg/kg(体重)/日の阻害剤を投与できるように調製されなければならない。所定の実施形態では、本発明の組成物は、0.01mg〜50mgの用量を提供する。他の実施形態では、0.1〜25mgまたは5mg〜40mgの用量が提供される。
【0064】
任意の特定患者のための特定の用量および治療レジメンは、使用される特定化合物の活性、年齢、体重、全身状態、性別、食事、投与回数、排出速度、併用薬、ならびに治療担当医師の判断および治療される特定疾患の重症度を含む様々な因子に依存することもまた理解されたい。組成物中の本発明の化合物の量は、さらに本組成物中の特定化合物に依存する。
【0065】
以下では、本明細書に記載した本発明をより完全に理解できるように、実施例を記載する。これらの実施例は単に例示するためであり、決していかなる様式でも本発明を限定すると解釈すべきではないと理解されたい。
【実施例】
【0066】
[実施例1〜41]
化合物の合成
Biotage Corporation社によって供給されるEmrys Liberator機器を用いて、マイクロ波援用反応を実施した。溶媒除去は、Buechiロータリー・エバポレータまたはGenevac遠心式エバポレータのどちらかを使用して実施した。分析および分取クロマトグラフィは、酸性、中性、または塩基性条件下のいずれかで、順相または逆相いずれかのHPLCカラムを用いて、Waters autopurification機器を用いて実施した。化合物はELSDクロマトグラムの面積百分率によって決定して、>90%純粋であると推定された。NMRスペクトルは、Varian 300MHz分光計を用いて記録した。
【0067】
以下では本発明の化合物を調製するための一般的方法および実験について記載する。所定の場合には、例として特定化合物について記載する。しかし、各場合において、一連の本発明の化合物は以下に記載するスキームおよび実験にしたがって調製されたことは理解される。
【0068】
スキーム1
【化12】
ER−811160.上記のスキーム1に図示したように、水(50mL)中のシアン化カリウム(22.5g、0.335M(モル))を5分間かけて、水(90mL)およびメタノール(110mL)中の1−Boc−ピペリドン(32.48g、0.1598M)および炭酸アンモニウム(33.8g、0.351M)の溶液に滴下した。添加の完了直後に、オフホワイトの沈降物が形成し始めた。反応フラスコを密封し、懸濁液を室温で72時間にわたり攪拌した。結果として生じた浅黄色の沈降物を濾過し、少量の水で洗浄すると、無色の固体としてER−811160(37.1g、86%)が得られた。
【0069】
スキーム2
【化13】
ER−818039.上記のスキーム2に図示したように、アセトン(555mL)中のER−811160(30.0g、0.111M)、3,5−ジメトキシベンジルブロミド(30.9g、0.134M)、および炭酸カリウム(18.5g、0.134M)の懸濁液を一晩還流させながら加熱した。反応溶液を室温へ冷却し、濾過し、インバキュオで濃縮した。橙色の粗残留物を最小量のMTBE(250mL)中に溶解させた。少量(50mL)のヘキサンを加え、生成物を無色の固体として(約2時間かけて)沈殿させ、これを真空濾過によって単離した。フィルターケーキを少量のMTBEで洗浄し、インバキュオで乾燥させると、無色の固体としてER−818039(39.6g、85%)が得られた。
【0070】
スキーム3
【化14】
ER−823143−01.上記のスキーム3に図示したように、ER−818039(2.15g、0.00512M)を含有する1口丸底フラスコに1,4−ジオキサン(3.8mL、0.049M)中の4N HClの溶液を緩徐に加えた。出発材料は20分間かけて緩徐に溶解し、30分後に無色の沈降物が形成された。次にMTBE(3mL)を加えた。2時間後、反応液を濾過し、MTBEを用いて洗浄すると、無色の固体としてER−823143−01(1.81g、99%)が得られた。
【0071】
スキーム4
【化15】
ER−817098:上記のスキーム4に図示したように、窒素雰囲気下で1,2−ジメトキシエタン(0.5mL、0.004M)中のER−823143−01(41.5mg、0.000117M)および4Åモレキュラーシーブの懸濁液に、3,5−ジメトキシベンズアルデヒド(21.3mg、0.000128M)、その後にトリエチルアミン(16.2μL、0.000117M)を加えた。この反応液を1時間にわたり攪拌した。トリアセトキシホウ化水素ナトリウム(34.6mg、0.000163M)を加え、この反応液を一晩攪拌した。シリカゲル・フラッシュ・クロマトグラフィーは、無色の固体としてER−817098(45.3mg、83%)を産生した。
【0072】
スキーム5
【化16】
ER−817116:上記のスキーム5に図示したように、N−メチルピロリジノン(1.0mL、0.010M)中のER−817098−00(50.0mg、0.000106M)および1−ブロモ−2−メトキシエタン(15.6μL、0.000160M)の溶液にテトラヒドロフラン(0.16mL)中の1.0Mリチウムヘキサメチルジシラジド溶液を加えた。温度を80℃へ上昇させ、反応混合液を一晩攪拌した。反応混合液を室温に冷却し、水を用いて急冷し、次にMTBEを用いて数回抽出した。MTBE抽出物を結合し、水(2×)および食塩液(1×)を用いて洗浄した。有機層は硫酸マグネシウムの上方に通して乾燥させ、濾過し、インバキュオで濃縮した。フラッシュ・クロマトグラフィーは、無色油としてER−817116(32.2mg、58%)を生じさせた。
【0073】
スキーム6
【化17】
ER−817118:上記のスキーム6に図示したように、N,N−ジメチルホルムアミド(15mL)中のER−817098(2.85g、0.00607M)の溶液に水素化ナトリウム(364mg、0.00910M)、次にヨードエタン(758μL、0.00910M)を加えた。この反応混合液を一晩攪拌した。水を極めて緩徐に加え、反応混合液はMTBEを用いて数回抽出した。MTBE抽出物を結合し、水(2×)および食塩液(1×)を用いて洗浄した。有機層は硫酸マグネシウムの上方に通して乾燥させ、濾過し、インバキュオで濃縮した。溶離液として酢酸エチルを用いるフラッシュ・クロマトグラフィーは、無色油としてER−817118(2.89g、96%)を生じさせた。
【0074】
スキーム7
【化18】
ER−823914:上記のスキーム7に図示したように、−78℃でテトラヒドロフラン(30.0mL、0.370M)中のER−823143−01(5.03g、0.0141M)の溶液に、エーテル(71mL)中の1.0Mの臭化アリルマグネシウムを緩徐に加えた。この反応混合液を室温へ加温し、一晩攪拌した。反応混合液を−78℃へ冷却し、トリフルオロ酢酸(21.8mL、0.283M)を滴下して処理し、次に少量の残留物量へインバキュオで濃縮した。残留TFAを中和するためにトリエチルアミンを加え、次にこの混合物を乾燥するまでインバキュオで濃縮した。残留した赤色油をメタノール(138mL、3.41M)中に溶解させ、ジ−tert−ブチルジカーボネート(3.34g、0.0148M)、次にトリエチルアミン(2.38mL、0.0169M)で処理し、室温で一晩攪拌した。反応混合液をインバキュオで濃縮し、フラッシュ・クロマトグラフィー(溶離液:酢酸エチル中の50%ヘキサン)によって精製すると、無色の固体としてER−823914(3.25g、52%)が得られた。
【0075】
スキーム8
【化19】
ER−823915:上記のスキーム8に図示したように、N,N−ジメチルホルムアミド(12.4mL、0.160M)中のER−823914(2.20g、0.00496M)の溶液に水素化ナトリウム(298mg、0.00744M)、次にヨードエタン(607μL、0.00744M)を加えた。反応混合液を一晩攪拌し、水を用いて急冷し、次にMTBEを用いて数回抽出した。MTBE抽出物を結合し、水および食塩液を用いて洗浄した。有機層は硫酸マグネシウムの上方に通して乾燥させ、濾過し、インバキュオで濃縮した。フラッシュ・クロマトグラフィー(溶離液:酢酸エチル中の40%ヘキサン)は、無色の泡としてER■823915(0.80g、34%)を生じさせた。
【0076】
スキーム9
【化20】
ER−823917−01:上記のスキーム9に図示したように、ER−823915(799.2mg、0.001695M)を1,4−ジオキサン(10mL)中の4M塩化水素の溶液中に溶解させた。反応混合液を一晩攪拌し、次にインバキュオで濃縮すると、橙色の固体としてER−823917−01(0.69g、定量的)が得られた。
【0077】
スキーム10
【化21】
ER−824184およびER−824185:上記のスキーム10に図示したように、アセトニトリル(1mL)中のER−823915(200mg)の溶液をCHIRALPAK(登録商標)AS−H SFCカラム(30mm×250mm、粒径:5ミクロン)上に注入し、95:5のn−ヘプタン:i−プロパノールを用いて40mL/分の流量で溶出した。溶出フラクションは、290nmに設定した波長を用いてUV検出器を使用して検出した。第1溶出フラクションを単離し、インバキュオで回転式蒸発によって濃縮するとER−824184が得られた;第2溶出フラクションを単離し、インバキュオで回転式蒸発によって濃縮するとER−824185が得られた。
【0078】
スキーム11
【化22】
ER−824188−01:上記のスキーム11に図示したように、ER−824184(25.33g、0.05371M)を1,4−ジオキサン(135mL)中の4M塩化水素の溶液中に溶解させた。反応混合液を一晩攪拌し、次にインバキュオで濃縮すると、橙色の固体としてER−824188−01(21.9g、定量的)が得られた。ER−824188−01の単結晶X線回折分析は、立体中心の絶対配置がスキーム11に図示したようにSであることを証明した。
【0079】
スキーム12
【化23】
ER−824280−01:上記のスキーム12に図示したように、ER−824185(457.2mg、0.0009695M)を1,4−ジオキサン(2.5mL)中の4M塩化水素の溶液中に溶解させた。反応混合液を一晩攪拌し、次にインバキュオで濃縮すると、橙色の固体としてER−824280−01(383.2mg、97%)が得られた。ER−824188−01のMosherアミド誘導体の単結晶X線回折分析は、立体中心の絶対配置がスキーム11に図示したようにRであることを証明した。
【0080】
スキーム13
【化24】
ER−819924:上記のスキーム13に図示したように、ER−824188−01(62.4mg、0.000153M)およびN−メチルピロール−2−カルバルデヒド(0.000229M)をN,N−ジメチルホルムアミド(0.62mL)中に溶解/懸濁させた。30分間にわたり攪拌した後、トリアセトキシホウ化水素ナトリウム(47.8mg、0.000214M)を加えた。反応混合液を一晩攪拌し、次に逆相クロマトグラフィによって精製すると、油としてER−819924(71.1mg、83.4%)が得られた。
【0081】
スキーム14
【化25】
ER−819925:上記のスキーム14に図示したように、ER−824280−01(59.5mg、0.000146M)およびN−メチルピロール−2−カルバルデヒド(0.000219M)をN,N’−ジメチルホルムアミド(0.60mL)中に溶解/懸濁させた。30分間にわたり攪拌した後、トリアセトキシホウ化水素ナトリウム(45.6mg、0.000204M)を加えた。反応混合液を一晩攪拌し、次に逆相クロマトグラフィによって精製すると、油としてER−819925(51.9mg、76.6%)が得られた。
【0082】
スキーム15
【化26】
ER−819762:上記のスキーム15に図示したように、N,N−ジメチルホルムアミド(50mL)中のER−824188−01(5.7g、0.0140M)、1,8−ジアザビシクロ[5.4.0]ウンデク−7−エン(4.4mL、0.029M)および3,5−ジメチルベンジルブロミド(4.7g、0.024M)の溶液を97℃で一晩加熱した。水系後処理およびフラッシュ・クロマトグラフィーによる精製によって、無色の固体としてER−819762(4.86g、71%)が得られた。
【0083】
スキーム16
【化27】
ER−819762−01:上記のスキーム16に図示したように、水(11mL)中のER−819762(4.77g、0.00974M)、アセトニトリル(10mL)および1M HClの溶液を室温でおよそ5分間攪拌した。この溶液を濃縮すると、凍結乾燥後の無色の結晶固体としてER−819762−01(5.1g、定量的)が得られた。ER−819762−01の単結晶X線回折分析は、立体中心の絶対配置がスキーム16に図示したようにSであることを証明した。
【0084】
スキーム17
【化28】
ER−819763:上記のスキーム17に図示したように、N−メチルピロリジノン(669mL)中のER−824280−01(66.9g、0.1640M)、1,8−ジアザビシクロ[5.4.0]ウンデク−7−エン(54mL、0.361M)および3,5−ジメチルベンジルクロリド(42.4g、0.213M)の溶液を2時間にわたり72℃で加熱した。冷却後、水を加えると、所望の生成物が沈降した。濾過および真空下での乾燥は、無色の固体としてER−819763(74.4g、92%)を生じさせた。
【0085】
スキーム18
【化29】
ER−824102:上記のスキーム18に図示したように、室温にあるN,N−ジメチルホルムアミド(25mL)中のER−823143−01(4.00g、0.0112M)の溶液にα−ブロモメシチレン(3.13g、0.0157M)、次にDBU(4.37mL、0.0292M)を加えた。1時間にわたり攪拌した後、反応液を半飽和NH4Cl水溶液で急冷し、酢酸エチルを用いて希釈し、1時間攪拌すると2つの透明層が得られた。有機層を分離し、水層は酢酸エチルにより抽出した(2×)。結合抽出物はNa2SO4の上方に通して乾燥させ、濾過し、インバキュオで濃縮した。MTBEからの結晶化により、無色の固体としてER−824102(4.30g、87%)が得られた。
【0086】
スキーム19
【化30】
ER−819929:上記のスキーム19に図示したように、−65℃のテトラヒドロフラン(35mL)中のER−824102(3.72g、0.0085M)の溶液に、−50℃未満の内部温度を維持しながら10分間かけてエーテル(25.5mL、0.0255M)中の1.0M臭化アリルマグネシウムを加えた。反応混合液を0℃へ加温させた。0℃で3時間置いた後、反応液を飽和NH4Cl水溶液で急冷し、酢酸エチルおよび水を用いて希釈し、10分間攪拌すると2つの透明層が得られた。有機層を分離し、水層は酢酸エチルにより抽出した。結合抽出物を水、食塩液で洗浄し、Na2SO4の上方に通して乾燥させ、濾過し、インバキュオで濃縮すると無色の固体として粗生成物のER−819929(4.15g、定量的)が得られたので、これをそれ以上精製せずに次の工程に使用した。
【0087】
スキーム20
【化31】
ER−819930:上記のスキーム20に図示したように、トリフルオロ酢酸(0.5mL)中のER−819929(37mg、0.000077M)の溶液を室温で16時間にわたり攪拌した。暗赤褐色の反応混合液をEtOAc(5mL)で希釈し、飽和NaHCO3(5mL、注意:ガス発生)を用いて中和した。2層の混合液を10分間攪拌すると、ほぼ無色の2つの透明層が得られた。2つの有機層を分離し、水層はEtOAcで抽出した。結合有機抽出物はNa2SO4の上方に通して乾燥させ、濾過し、インバキュオで濃縮した。1:1ヘプタン−EtOAc、1:3ヘプタン−EtOAc、100%EtOAcを用いて溶出させるフラッシュ・クロマトグラフィーによって精製すると、無色の固体としてER−819930(26mg、73%)が得られた。
【0088】
スキーム21
【化32】
ER−820006およびER−820007:上記のスキーム21に図示したように、DMF(1.5mL)中のER−819930(110mg、0.000238M)および臭化メタリル(72μL、0.000715M)の溶液にテトラヒドロフラン中の1.0Mリチウムヘキサメチルジシラジド溶液(0.52mL、0.00052M)を加えた。室温で18時間攪拌した後、反応混合液をMTBEで希釈し、半飽和NH4Cl水溶液で急冷した。水層を分離し、MTBEで抽出した。結合抽出物はNa2SO4の上方に通して乾燥させ、濾過し、インバキュオで濃縮した。3:2ヘプタン−EtOAc、1:1ヘプタン−EtOAcを用いて溶出させるフラッシュ・クロマトグラフィーによって精製すると、無色油としてラセミ生成物(68mg、55%)が得られた。ラセミ生成物(55mg)は、ヘプタン−イソプロパノール(9:1)を用いて溶出させるChiralpak ASカラム上でのキラルHPLCにかけると、第1溶出エナンチオマーであるER−820006(21mg、38%、[a]D=+83.7°(c=0.35、CHCl3)および第2溶出エナンチオマーであるER−820007(23mg、42%、[a]D=−74.2°(c=0.38、CHCl3)が得られた。絶対立体化学は、エナンチオマーのER−819762/ER−819763対を用いて旋光度およびキラルHPLC保持時間における共通点に基づいて暫定的に指定した。
【0089】
スキーム22
【化33】
ER−819786およびER−819787:上記のスキーム22に図示したように、攪拌棒を装備した5mLマイクロ波反応器バイアルに、ER−819930(110mg、0.000238M)、DMF(1.5mL)、2−(2−ブロモエトキシ)テトラヒドロ−2H−ピラン(108μL、0.000715M)およびテトラヒドロフラン(520μL、0.00052M)中の1.00Mのリチウムヘキサメチルジシラジドを装填した。反応器バイアルを、200℃で15分間にわたりマイクロ波反応器にかけた。さらに2−(2−ブロモメトキシ)テトラヒドロ−2H−ピラン(108μL、0.000715M)およびテトラヒドロフラン中の1.00Mのリチウムヘキサメチルジシラジド(520μL、0.00052M)を加え、反応混合液をマイクロ波照射によってさらに15分間、200℃で加熱した。分取的逆相HPLCによる精製によって、無色のガラス質の油としてラセミ生成物(25mg、21%)が得られた。ラセミ生成物(17mg)は、ヘプタン−イソプロパノール(9:1)を用いて溶出させるChiralpac ASカラム上でのキラルHPLCにかけると、第1溶出エナンチオマーであるER−819786(7.2mg、42%、[α]D=+72.0°(c=0.1、CHCl3)および第2溶出エナンチオマーであるER−819787(7.5mg、44%、[α]D=−73.0°(c=0.1、CHCl3)が得られた。絶対立体化学は、エナンチオマーのER−819762/ER−819763対を用いて旋光度およびキラルHPLC保持時間における共通点に基づいて暫定的に指定した。
【0090】
スキーム23
【化34】
ER−819993およびER−819994:上記のスキーム23に図示したように、攪拌棒を装備した5mLマイクロ波反応器バイアルに、ER−819930(110mg、0.000238M)、DMF(1.5mL)、((4S)−2,2−ジメチル−1,3−ジオキソラン−4−イル)メチル4−メチルベンゼンスルホネート(205mg、0.000715mol)およびテトラヒドロフラン(520μL、0.00052M)中の1.00Mのリチウムヘキサメチルジシラジドを装填した。反応器バイアルを、200℃で15分間にわたりマイクロ波照射によって加熱した。さらに((4S)−2,2−ジメチル−1,3−ジオキソラン−4−イル)メチル4−メチルベンゼンスルホネート(157mg、0.000548M)およびテトラヒドロフラン中の1.00Mのリチウムヘキサメチルジシラジド(477μL、0.000477M)を加え、反応混合液をマイクロ波照射によってさらに15分間、200℃で加熱した。分取的逆相HPLCによる精製によって、ジアステレオマーの1:1混合物としてアセトニドであるER−819993(40mg、30%)およびジオール材料(18mg、14%)が得られた。ヘプタン−イソプロパノール(9:1)を用いて溶出させるChiralpac ASカラム上でのキラルHPLCによるジアステレオマージオールの分離によって、第1溶出ジアステレオマーであるER−819788(5.0mg)および第2溶出ジアステレオマーであるER−819789(5.2mg)が得られた。絶対立体化学は、エナンチオマーのER−819762/ER−819763対を用いてキラルHPLC保持時間における共通点に基づいて暫定的に指定した。
【0091】
スキーム24
【化35】
ER−81990:上記のスキーム24に図示したように、塩化メチレン(500μL)中のER−824220−00(51.8mg、0.000139M)、トリエチルアミン(97μL、0.00070M)、4−ジメチルアミノピリジン(3.4mg、0.000028M)および(R)−(−)−α−メトキシ−α−トリフルオロメチルフェニルアセチルクロリド(0.052mL、0.00028M)の溶液を室温で5時間にわたり攪拌した。フラッシュ・クロマトグラフィーによる精製後に、酢酸エチル/ヘプタン/ペンタンから結晶化させると、結晶としてER−819990(49.2mg、60%)が得られた。
【0092】
以下のセクションおよび以下の表1〜2に例示するが上記で明確には図示していない化合物は、スキーム13および/またはスキーム15に一致する一般方法を使用して合成できる。塩酸塩形で例示した化合物については、それらは対応する遊離塩基にスキーム16に記載した一般条件を受けさせることによって調製することができる。
【0093】
表1.式Iの典型的化合物についての分析データ
【表1A】
【表1B】
【表1C】
【表1D】
【表1E】
【表1F】
【表1G】
【表1H】
【表1I】
【表1J】
【表1K】
【表1L】
【表1M】
【表1N】
【表1O】
【表1P】
【表1Q】
【表1R】
【表1S】
【表1T】
【0094】
分析方法:
方法A1
溶媒A:水中の0.2% Et3N
溶媒B:アセトニトリル中の0.2% Et3N
流量:2.0mL/分
線形勾配:
【表1U】
方法C1
移動相:エタノール中の0.1% Et2NH
流量:1.0mL/分
均一濃度
【0095】
[実施例42〜126]
インビトロ生物学的活性
HEKT−bet−lucアッセイ:本アッセイは、ルシフェラーゼレポーターを駆動するヒトT−betおよびT−box応答エレメントを発現する遺伝子組換えHEK細胞内のT−bet依存性レポーター(ルシフェラーゼ)活性を測定する。HEKT−bet細胞は2×104/ウエルで96ウエルプレート内で平板培養し、化合物を24時間にわたり細胞培養中へ加えた。ルシフェラーゼ活性は、50μLのSteady−Glo試薬(Promega社)を加えることによって測定し、サンプルをVictor Vリーダー(PerkinElmer社)で読み取った。化合物の活性は、化合物処理サンプルを非化合物処理ビヒクル対照と比較することによって決定した。IC50値は、試験化合物の非存在下でのルシフェラーゼの量に対応する最大値および最大阻害で得られた試験化合物値に対応する最小値を利用して計算した。
【0096】
正規化HEKT−bet IC50値の決定:化合物は、マイクロタイタープレート内でアッセイした。各プレートは、ER−819544である参照化合物を含んでいた。特定化合物についての非正規化IC50値を同一マイクロタイタープレート内で参照化合物について決定したIC50値で割ると、相対効力値が得られた。次に相対効力値に参照化合物の確定効力を掛けると、正規化HEKT−bet IC50値が得られた。本アッセイでは、ER−819544の確定効力は0.035mMであった。本明細書に提供したIC50値は、正規化法を使用して入手した。
【0097】
本発明の典型的な化合物は、上記に記載したHEKT−bet−lucアッセイにおいて上述した方法にしたがってアッセイした。以下の表2は、上記に記載した正規化HEKT−bet−lucアッセイによって決定した指示量(μM)までのIC50を有する本発明の典型的な化合物を記載している。
【0098】
表2.典型的な化合物のIC50値
【表2A】
【表2B】
【表2C】
【表2D】
【表2E】
【表2F】
【表2G】
【表2H】
【表2I】
【表2J】
【表2K】
【表2L】
【表2M】
【表2N】
【表2O】
【表2P】
【表2Q】
【表2R】
【表2S】
【表2T】
【表2U】
【表2V】
【表2W】
【表2X】
【表2Y】
【表2Z】
【0099】
[実施例126]
インビボ生物学的活性:能動免疫
CIAにおける関節炎発生の抑制。DBA1/Jマウスを第0日にbCII/CFAで免疫し、その後第21日にbCII/IFAで追加免疫した。関節炎の発生を試験経過にわたって監視した。関節炎のスコアは、以下の通りである:0=正常な足、スコア1=1〜2本の指が炎症を起こしている足、スコア2=3本または1〜2本の指+手関節または足関節が炎症を起こしている、スコア3=手+3本以上の指が炎症を起こしている;およびスコア4=多数の指(3〜4)+重大な手関節または足関節の炎症。
【0100】
(A)化合物の部分的治療評価。活性化合物は、コラーゲンIIに対する抗体の誘導後第20日から、しかし疾患発生の前に指示した用量で1日1回の経口投与によって与えた。(B)化合物の完全治療評価。活性化合物は、疾患が発生した後(第2免疫後第7日から)投与した。(C)完全治療CIA試験からのマウス足についてのX線分析。X線スコアは、骨減少症、骨浸食および新規骨形成の組み合わせの測定値の指標である。(D)代表的なX線写真。
【0101】
データは、以下の表3に示した。一般に、これらのデータは好都合にこのモデルにおけるメトトレキセートの活性を比較する。
【0102】
[実施例127]
インビボ生物学的活性:受動免疫
CAIAにおける関節炎発生の抑制。BALB/cマウスに第0日に1mgの抗タイプIIコラーゲン抗体をi.v.(静脈内)注射し、3日後に25μgのLPSを活性化合物とともにi.p.(腹腔内)注射し、メトトレキセート(MTX)は第0日〜第7日にわたり1日1回PO投与した。関節炎スコアおよび体重は、試験の全経過にわたって監視した。
【0103】
データは、以下の表3に示した。一般に、これらのデータは好都合にもこのモデルにおけるにおいて特に活性ではないメトトレキセートと比較する。
【0104】
【表3】
【0105】
[実施例128〜134]
材料および方法
マウスおよび試薬。BALB/cおよびDO11.10マウスは、Jackson Laboratory社から購入した。C57BL/6およびDBA/1マウスは、Charles River Laboratories社から購入した。マウスIL−2、IL−12、IL−23およびヒトGM−CSFは、R & D systems社から購入した。ヒトIL−4およびGM−CSFは、Peprotec社からである。抗CD3(クローン145−2C11)、抗CD28(クローン37.51)、抗IL−4(クローン11B11)、抗IFNγ(クローンXMG12)およびPE−抗マウスIL−17(クローンTC11−18H10)は、Pharmingen社から購入した。抗TCR(クローンH57−597)は、eBioscience社から購入した。OVAペプチドおよびマイトマイシンCは、Sigma社から購入した。PGE2、PGE1−アルコールおよび抗PGE2は、Cayman Chemicals社から購入した。LPSおよびR−848は、InVivoGen社からである。CD14+細胞単離キットは、MiltenyiBiotec社からである。CD4+T細胞単離キットは、MiltenyiBiotec社またはStemCell Technologies社からである。IFNγ ELISAキットはPIERCE社からである;IL−4 ELISAキットは、R&D systems社からである;IL−23 ELISAキットは、eBioscience社からである。Alamar blue試薬は、Biosource International社からである。Celltiter−glo試薬は、Promega社からである。
【0106】
放射性リガンドEP4結合。放射性リガンドEP4結合アッセイは、EP4発現性膜標本からの放射標識PGE2の置換を測定する。放射性リガンドEP4結合アッセイキットは、Millipore社から購入し、このアッセイは、製造業者の取扱説明書にしたがって実施した。
【0107】
CRE−PLAPレポーターアッセイ。内因性EP4を発現するSE302細胞は、一晩にわたりER−819762の存在下または非存在下でPGE2を用いて刺激し、PLAP活性を測定した。
【0108】
インビトロT細胞アッセイ。天然CD4+T細胞は、製造業者によって記載されたようにRobosepによってBALB/cまたはDO11.10マウスのいずれかの脾臓から精製した。BALB/cマウスについては、1×105のCD4+T細胞を、中性条件下(1μg/mLの平板結合抗CD3+1μg/mLの可溶性抗CD28+10ng/mLのマウスIL−2)またはTh1促進条件下(中性+5ng/mLのマウスIL−12+10μg/mLの抗IL−4)またはTh2分化条件下(中性+10ng/mLのマウスIL−4+10μg/mLの抗IL−4)のいずれかで、10%の正常ウシ胎児血清(Hyclone)または活性炭処理FBS(Hyclone社)(外因性PGE2またはEP4アゴニストが培養に加えられた場合)のいずれかを含有する100μLの完全RPMI培地(Cellgro社)中で967ウエルプレート内で3〜6日間培養した。培養上清中のIFNγまたはIL−4はELISAによって検出した。細胞ペレットを使用して、製造業者の取扱説明書にしたがってAlamar blueまたはCellTiter−Glo試薬のいずれかを用いて細胞増殖を測定した。DO11.10マウスに対して、BALB/cマウス由来のマイトマイシンC処置脾臓細胞を抗原提示細胞として使用し、5:1の比率(100μL培地中の5×105のマイトマイシンC処置脾臓細胞+100μL培地中の1×105 CD4+T細胞)で天然CD4+T細胞と共培養し、上述したように中性、Th1またはTh2促進条件下のいずれかでOVAペプチド(0.3ng/mL)を用いて刺激した。EP4アゴニスト、アンタゴニストまたは抗PGE2抗体をTh細胞分化中に加えた。
【0109】
EP4アゴニスト/アンタゴニストがIL−17産生に及ぼす作用を試験するために、C57BL/6マウス由来の全CD4+T細胞を3〜5日間にわたり指示した濃度のIL−23(10ng/mL)またはEP4アゴニスト/アンタゴニストの存在下または非存在下において平板結合抗CD3(2μg/mL)+可溶性抗CD28(2μg/mL)により活性化した。培養上清をIL−17 ELISAによって分析し、細胞ペレットを使用してCellTite−Glo試薬を用いた細胞増殖を測定した。
【0110】
IL−23誘導性Th17増殖。CD4+T細胞をC57BL/6マウスから単離し、5日間にわたり、IL−23(30ng/mL)を伴う、または伴わない、抗TCR(1μg/mL平板結合)および抗I−CD28(2μg/mL、可溶性)を用いて活性化した。IL−17産生細胞は、製造業者(BD社)によって記載されたようにIL−17細胞内染色によって分析した。
【0111】
インビトロヒト単球由来DCアッセイ。CD14+細胞はMiltenyi CD14マイクロビーズを用いてヒトPBMCから精製し、10%木炭処理したFBSを含有する完全RPMI培地中でヒトGM−CSF(500U/mL)+ヒトIL−4(500U/mL)を用いて8日間にわたり分化させた。未結合imDC(未成熟樹状細胞)は、24時間にわたり指示した濃度にあるEP4アゴニスト/アンタゴニストまたは抗PGE2抗体の添加を伴って、または伴わずに、LPS(10ng/mL)+R−848(2.5μg/mL)を用いて刺激した。培養上清中のIL−23は、ELISA(eBioscience社)によって測定した。
【0112】
コラーゲン誘導性関節炎モデル。雄性DBA/1マウスをFreundの完全アジュバント中で乳化した150μgのウシII型コラーゲンを含有する、0.1mLエマルジョンを用いて尾の基部で皮内免疫した。初回免疫の3週間後、全マウスにFreundの不完全アジュバント中に乳化させたウシII型コラーゲンを用いて追加免疫した。各マウスの足における関節炎の症状の重症度は、公知の技術にしたがったWood et al.の方法によって判定した。
【0113】
PLP誘導性EAEモデル。SJLマウスに、Freundの完全アジュバント中に乳化させた35μgのPLP139−151を含有する0.1mLエマルジョンを皮下注射した。1×109/200μLの百日咳菌を各マウスへ第0日および第2日に注射した。EAEスコアは、公知の技術にしたがって評価した。
【0114】
エックスビボLNまたは脾臓細胞試験。単一懸濁細胞は、bCIIを用いた一次免疫の後第15日マウスまたはEAE試験終了時に排液LNまたは脾臓から調製し、bCII(50μg/mL)、MOG35−55(12.5μg/mL)またはPBS/培地のいずれかを用いて48〜72時間にわたり刺激した。培養上清中のサイトカイン産生をELISAによって分析し、細胞増殖はCellTiter−Glo試薬のいずれかによって測定した。「ミックス・アンド・マッチ(mix & match)」リンパ球反応のためには、排液LN由来の精製CD4+T細胞を1:5の比率でAPC(マイトマイシンC処理全LN細胞)と共培養し、上述したように刺激して分析した。
【0115】
[実施例128]
選択的EP4受容体アンタゴニストとして同定されたER−819924
ER−819924−01は、競合的放射性リガンド結合アッセイ(MDS Pharma社による)においてEP4へ選択的に結合するが他のEP/プロスタノイド/ロイコトリエン受容体には結合しないことが見いだされた(表4および図1)。さらに、1μMでのER−819924−01は、FLIPR機能的スクリーンにおいてEP4受容体に対してのみ活性を示し、他の132GPCRのいずれに対しても活性を示さなかった(Millipore社が外部委託したデータ)(表5)。ER−819924−01はさらに、PGE2に対する強力な阻害活性がSE302細胞においてCRE−PLAP受容体活性を誘導することも証明した(28nMのIC50値を伴う)(図2)。
【0116】
表4:プロスタノイド/ロイコトリエン受容体に対するER−819924−01の競合的リガンド結合アッセイ
【表4】
【0117】
表5:132 GPCRに対するER−819924−01のFLIPRスクリーン。アゴニストモードおよびアンタゴニストモードの両方を測定したが、表にはアンタゴニスト活性だけを示した。
【表5】
【0118】
[実施例129]
活性化マウスCD4+T細胞中でのIL−17産生にPGE2/EP4が及ぼす作用
EP4がさらにTh17応答においても役割を果たすかどうかを調査するために、PGE2またはEP4アゴニストによるEP4活性化が活性化マウスCD4+T細胞内でのIL−17産生に及ぼす作用を試験した。全CD4+T細胞は、3〜5日間にわたり、PGE2またはEP4アゴニストPGE1−OH(0.1nM〜1,000nM)の存在下または非存在下でIL−23を伴う、または伴わずに、抗CD3/抗CD28により刺激した。培養上清中のIL−17はELISAによって測定し、細胞増殖はCellTiter−gloによって測定した。結果は、EP4アゴニストが抗CD3/抗CD28刺激CD4+T細胞中の細胞増殖を抑制しながら、IL−17産生を増強することを証明した(図3)。同様に高度に選択的なEP4アンタゴニストであるまた別のBOAT化合物であるER−819762−01は、この系における活性を抑制した(図4)。さらに、抗PGE2抗体は、天然CD4+T細胞がIL−23の存在下で抗CD3/抗CD28を用いて活性化されるとIL−17産生細胞の数を減少させた(図5)。これらをまとめると、これらの結果は、EP4拮抗作用がTh17応答を抑制できるという仮説を裏付けた。
【0119】
[実施例130]
マウスTh1分化においてPGE2/EP4が果たす役割
EP4活性化がTh1分化において役割を果たすかどうかを調査するために、PGE2またはEP4アゴニストおよび抗PGE2抗体が及ぼす作用をマウスTh1分化アッセイにおいて試験した。天然CD4T細胞は、3日間にわたりTh1促進剤を用いて分化した。培養上清中のTh1サイトカインIFNγはELISAによって測定した。細胞増殖は、Alamar blueアッセイによって測定した。PGE2またはEP4アゴニストPGE1−OHをTh1分化中に加えた。結果は、PGE2およびEP4アゴニスト(PGE1−OH)はTh1分化を強化するが(図6)、他方PGE2に対する抗体が分化Th1細胞におけるIFN−γ産生を部分的に抑制することを証明したが(図7)、これはPGE2/EP4がTh1分化を調節することにある役割を果たすことを示している。さらに、ER−819762は抗PGE2抗体に活性を加えなかったが(図8)、これはER−819762によるTh1分化の抑制が主としてEP4に対する活性に起因することを示唆していた。
【0120】
[実施例131]
インビトロでのマウスIL−17増殖の抑制
ER−819924−01がIL−23誘導性Th17増殖に及ぼす作用をインビトロで試験した(図9)。マウスCD4+T細胞は、5日間にわたりER−819924−01または抗PGE2抗体の存在下または非存在下において抗TCR/抗CD28モノクロール抗体およびIL−23(30ng/mL)とともに培養した。IL−17産生T細胞は、細胞内染色のFACSによって分析した。ER−819924−01は、約10〜100nMのIC50値を備えるIL−23誘導性Th17産生細胞の数を減少させた。
【0121】
[実施例132]
CIAにおける関節炎発生の抑制
ER−819924−01の抗関節炎作用を評価するために、マウスCIAモデルにおいて幾つかの試験を実施した。最初にER−819924−01は、化合物が第20日から毎日経口投与される(すなわち、病原性抗II型コラーゲン抗体の誘導後で、関節炎発生の前に)部分治療投与レジメンにおいて試験した。関節炎スコア(炎症を起こした足の炎症反応指数として測定した)および体重を2週間にわたって監視した。部分治療投与条件下で、ER−819924−01は、約10mg/kgのED50を備えて関節炎の発生を効果的に抑制した(図10)。本発明者らは、さらに化合物が疾患の誘導後に投与される完全治療投与レジメンにおいてER−819924−01について試験した。完全治療投与条件下で、ER−819924−01は、約10mg/kgの用量でさらに関節炎の発生を効果的に防止した(図11)。さらに、ER−819924−01は、別個の試験において30mg/kgの用量でX線スコアを有意に改善した(図12)。
【0122】
[実施例133]
活性化ヒトMoDC(単球由来樹状細胞)における最適なIL−23産生のためには、PGE2/EP4シグナリングが必要とされ、化合物はこの活性を抑制する
ヒトCD14+単球を7日間にわたりGM−CSF+IL−4を用いてimDCに分化させ、外因性PGE1−OH(1〜100nM)の存在下または非存在下において、そしてER−819762(図13)もしくはER−819924または抗PGE2抗体(図14)を用いて、または用いずに、LPS/R−848を用いて24時間にわたり再刺激した。培養上清中のIL−23分泌は、ELISAによって測定した。表6は、ヒトMoDC IL−23分泌アッセイにおける代表的化合物のIC50値を示している。
【0123】
表6:活性化ヒトMoDCにおけるPGE1−OH(10nM)強化IL−23産生を阻害する際の代表的な化合物のIC50値。
【表6】
【0124】
[実施例134]
EAEにおけるTh1/Th17反応のエックスビボ抑制
排液リンパ節は、治療的MOG EAE試験の終了時にマウスから採取した。単一細胞懸濁液を調製し、48時間にわたりMOGまたは培地のいずれかを用いて刺激した。上清は、Search Light複数サイトカイン分析(PIERCE Technology社製)のために収集した。データは、図15に示した。
【0125】
本発明の多数の実施形態について記載してきたが、本発明の化合物および方法を利用する他の実施形態を提供するために基本的実施例を変化させられ得ることは明白である。このため本発明の範囲は、例として提示してきた特定実施形態によってではなく添付の特許請求項によって規定されることは理解される。
【産業上の利用可能性】
【0126】
本発明にかかる方法は、関節リウマチや多発性硬化症の治療に有効である。
【特許請求の範囲】
【請求項1】
被験体における関節リウマチを治療する方法であって、前記被験体に、Th1分化もしくはTh17増殖の調節因子化合物を含む組成物を投与するステップを含む方法。
【請求項2】
前記調節因子化合物は、式:
【化1】
の化合物、またはその医薬上許容される塩である、請求項1に記載の方法。
【請求項3】
関節リウマチを治療するための医薬品の製造における化合物の使用であって、前記医薬品は調節因子化合物を含み、前記調節因子化合物はTh1分化もしくはTh17増殖の調節因子である使用。
【請求項4】
前記調節因子化合物は、式:
【化2】
の化合物、またはその医薬上許容される塩である、請求項3に記載の方法。
【請求項5】
関節リウマチを治療するためのTh1分化もしくはTh17増殖の調節因子化合物。
【請求項6】
被験体における多発性硬化症を治療する方法であって、前記被験体にTh1分化もしくはTh17増殖の調節因子化合物を含む組成物を投与するステップを含む方法。
【請求項7】
前記調節因子化合物は、式:
【化3】
の化合物、またはその医薬上許容される塩である、請求項6に記載の方法。
【請求項8】
多発性硬化症を治療するための医薬品の製造における化合物の使用であって、前記医薬品は調節因子化合物を含み、前記調節因子化合物はTh1分化もしくはTh17増殖の調節因子である使用。
【請求項9】
前記調節因子化合物は、式:
【化4】
の化合物、またはその医薬上許容される塩である、請求項8に記載の使用。
【請求項10】
多発性硬化症を治療するためのTh1分化もしくはTh17増殖の調節因子化合物。
【請求項11】
被験体における関節リウマチを治療する方法であって、前記被験体にEP4アンタゴニストを含む組成物を投与するステップを含む方法。
【請求項12】
前記EP4アンタゴニストは、式:
【化5】
の化合物、またはその医薬上許容される塩である、請求項11に記載の方法。
【請求項13】
関節リウマチを治療するための医薬品の製造における化合物の使用であって、前記医薬品はEP4アンタゴニストを含む使用。
【請求項14】
前記EP4アンタゴニストは、式:
【化6】
の化合物、またはその医薬上許容される塩である、請求項13に記載の使用。
【請求項15】
関節リウマチを治療するためのEP4アンタゴニスト。
【請求項16】
被験体における多発性硬化症を治療する方法であって、前記被験体にEP4アンタゴニストを投与するステップを含む方法。
【請求項17】
前記EP4アンタゴニストは、式:
【化7】
の化合物、またはその医薬上許容される塩である、請求項16に記載の方法。
【請求項18】
多発性硬化症を治療するための医薬品の製造における化合物の使用であって、前記医薬品はEP4アンタゴニストを含む使用。
【請求項19】
前記EP4アンタゴニストは、式:
【化8】
の化合物、またはその医薬上許容される塩である、請求項18に記載の使用。
【請求項20】
多発性硬化症を治療するためのEP4アンタゴニスト。
【請求項1】
被験体における関節リウマチを治療する方法であって、前記被験体に、Th1分化もしくはTh17増殖の調節因子化合物を含む組成物を投与するステップを含む方法。
【請求項2】
前記調節因子化合物は、式:
【化1】
の化合物、またはその医薬上許容される塩である、請求項1に記載の方法。
【請求項3】
関節リウマチを治療するための医薬品の製造における化合物の使用であって、前記医薬品は調節因子化合物を含み、前記調節因子化合物はTh1分化もしくはTh17増殖の調節因子である使用。
【請求項4】
前記調節因子化合物は、式:
【化2】
の化合物、またはその医薬上許容される塩である、請求項3に記載の方法。
【請求項5】
関節リウマチを治療するためのTh1分化もしくはTh17増殖の調節因子化合物。
【請求項6】
被験体における多発性硬化症を治療する方法であって、前記被験体にTh1分化もしくはTh17増殖の調節因子化合物を含む組成物を投与するステップを含む方法。
【請求項7】
前記調節因子化合物は、式:
【化3】
の化合物、またはその医薬上許容される塩である、請求項6に記載の方法。
【請求項8】
多発性硬化症を治療するための医薬品の製造における化合物の使用であって、前記医薬品は調節因子化合物を含み、前記調節因子化合物はTh1分化もしくはTh17増殖の調節因子である使用。
【請求項9】
前記調節因子化合物は、式:
【化4】
の化合物、またはその医薬上許容される塩である、請求項8に記載の使用。
【請求項10】
多発性硬化症を治療するためのTh1分化もしくはTh17増殖の調節因子化合物。
【請求項11】
被験体における関節リウマチを治療する方法であって、前記被験体にEP4アンタゴニストを含む組成物を投与するステップを含む方法。
【請求項12】
前記EP4アンタゴニストは、式:
【化5】
の化合物、またはその医薬上許容される塩である、請求項11に記載の方法。
【請求項13】
関節リウマチを治療するための医薬品の製造における化合物の使用であって、前記医薬品はEP4アンタゴニストを含む使用。
【請求項14】
前記EP4アンタゴニストは、式:
【化6】
の化合物、またはその医薬上許容される塩である、請求項13に記載の使用。
【請求項15】
関節リウマチを治療するためのEP4アンタゴニスト。
【請求項16】
被験体における多発性硬化症を治療する方法であって、前記被験体にEP4アンタゴニストを投与するステップを含む方法。
【請求項17】
前記EP4アンタゴニストは、式:
【化7】
の化合物、またはその医薬上許容される塩である、請求項16に記載の方法。
【請求項18】
多発性硬化症を治療するための医薬品の製造における化合物の使用であって、前記医薬品はEP4アンタゴニストを含む使用。
【請求項19】
前記EP4アンタゴニストは、式:
【化8】
の化合物、またはその医薬上許容される塩である、請求項18に記載の使用。
【請求項20】
多発性硬化症を治療するためのEP4アンタゴニスト。
【図1】

【図2】
【図3】

【図4】
【図5】
【図6】

【図7】
【図8】

【図9】
【図10】
【図11】
【図12】
【図13】

【図14】
【図15】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【公表番号】特表2011−503182(P2011−503182A)
【公表日】平成23年1月27日(2011.1.27)
【国際特許分類】
【出願番号】特願2010−534035(P2010−534035)
【出願日】平成20年11月13日(2008.11.13)
【国際出願番号】PCT/US2008/012732
【国際公開番号】WO2009/064431
【国際公開日】平成21年5月22日(2009.5.22)
【出願人】(506137147)エーザイ・アール・アンド・ディー・マネジメント株式会社 (215)
【Fターム(参考)】
【公表日】平成23年1月27日(2011.1.27)
【国際特許分類】
【出願日】平成20年11月13日(2008.11.13)
【国際出願番号】PCT/US2008/012732
【国際公開番号】WO2009/064431
【国際公開日】平成21年5月22日(2009.5.22)
【出願人】(506137147)エーザイ・アール・アンド・ディー・マネジメント株式会社 (215)
【Fターム(参考)】
[ Back to top ]