
修飾されたADAMディスインテグリンドメインポリペプチドおよびその使用
修飾されたADAM(ディスインテグリンおよびメタロプロテイナーゼ)ポリペプチド(MAP)が提供される。抗血管新生活性および抗腫瘍成長活性のためにMAPを投与するための方法が提供される。本発明の組成物はまた、内皮細胞の機能障害を処置するため、およびインテグリンに関連する状態を診断するためにも有用である。本発明では、MAPとチオレドキシンのN末端セグメントの融合タンパク質およびこれらの融合物をコードする核酸も提供される。

【発明の詳細な説明】
【技術分野】
【0001】
(関連出願への相互参照)
本出願は、2010年2月11日に出願された米国仮出願番号61/303,631号の米国特許法119条(e)項の下での優先権の利益を主張し、上記米国仮出願番号61/303,631号は、全ての図面および表を含めてその全容が、参考として本明細書に援用される。
【0002】
(発明の分野)
本発明は、多機能のプロテアーゼのADAM(ディスインテグリンおよびメタロプロテイナーゼ(A Disintegrin and Metalloproteinase))哺乳動物のファミリーに由来する、遺伝子改変(engineered)されたポリペプチドのクラス、およびそれを作製する方法に関する。本発明は、これらの遺伝子改変されたポリペプチドの、抗血管新生活性および抗腫瘍成長活性についての使用にも関する。本発明は、遺伝子改変されたポリペプチドを、内皮細胞の機能障害およびインテグリンに関連する状態を診断するために投与することにも関する。
【背景技術】
【0003】
(発明の背景)
本発明は、Mineaらによる、発明の名称が「Method of expressing proteins with disulfide bridges」である特許文献1、および2009年11月12日に出願された、発明の名称が「Method of expressing proteins with disulfide bridges with enhanced yields and activity」であるPCT特許出願番号PCT/US09/64256に関連する。そのどちらの内容も、全ての図面も含めて、参照により本明細書に組み込まれる。
【0004】
ADAMは、胚発生のあらゆるステップ(そこでは、細胞増殖、細胞遊走、細胞の特異化、軸索伸長および器官の形態形成を制御する)ならびに成人期における多数の生理的プロセスおよび病理学的プロセス(創傷治癒から種々の炎症プロセスまで、ならびに血管新生および転移から器官の修復および再生まで)に関与する多ドメインの哺乳動物の膜貫通タンパク質または分泌タンパク質のクラスである[非特許文献1〜5(1〜5)]。多数の報告により、ADAMファミリーの多くのタンパク質分解性のメンバーがヒト悪性腫瘍において過剰発現されていると思われることが示されており、これは、これらのプロテアーゼが腫瘍の進行において重要な役割を果たす可能性があることを示している[非特許文献6〜15(6〜15)]。興味深いことに、触媒として活性なADAMの過剰発現は、一般に、がんにおける転帰不良に関連づけられると思われるが、非タンパク質分解性のADAMは、腫瘍インヒビターとしての役割を果たすと思われる。例えば、ヒト神経膠腫におけるADAM22の過剰発現は、腫瘍の成長阻害と相関することが示されている[16]。
【0005】
ADAMタンパク質の構造は、それらのオルソログである、PIII−クラスのヘビ毒メタロプロテイナーゼの構造とよく似ている。PIII−クラスのヘビ毒メタロプロテイナーゼ(SVMP)と同様に、ADAMは、メタロプロテアーゼドメイン、ディスインテグリンドメインおよびシステインリッチドメインを保有する多ドメインのタンパク質である[17、28]。ADAM足場は、いくつかのドメイン間のシステイン残基およびドメイン内のシステイン残基を含有し、とりわけ、ディスインテグリンループと称される構造要素の先端における、ディスインテグリンドメイン内に位置するものが非常に重要である。ドメイン間のシステイン残基は、メタロプロテアーゼとディスインテグリンドメインとの間およびディスインテグリンとシステインリッチドメインとの間のスペーサー領域内に位置する[20]。ADAMおよびPIII−SVMPにおいて見いだされるが、一般に、ヘビ毒ディスインテグリンにおいては見いだされないこれらのシステイン残基は、ADAMおよびPIII−SVMPにおいて、ドメイン間(スペーサー)領域をメタロプロテアーゼドメイン、ディスインテグリンドメインおよびシステインリッチドメインと連結するジスルフィド架橋を形成する[20]。これらのADAM足場におけるスペーサー−ドメインジスルフィド架橋は、3つのドメインを一緒に緊密に折りたたまれた構造にロックする構造要素を示し、これらの多ドメインタンパク質が、細胞外の環境において良好に生存する(すなわち、タンパク質分解性の攻撃などに対する抵抗性が増す)ことを可能にしている。この安定化されたADAM足場は、ジスルフィド結合の遺伝子改変によって新しい折りたたみのPII−クラスのSVMPに自然に進化し、これは、タンパク質分解性の攻撃を伴う機構によって遊離のディスインテグリンを生じる。分子進化の結果として、ADAM足場に特徴的なシステイン残基のいくつか(すなわち、スペーサー−ドメインジスルフィド架橋に関与するシステイン残基)が、異なる残基に変異したか、または欠失した。これらの変異の結果、これらの新しいタンパク質(PIIクラスのSVMP)において、スペーサー領域と、メタロプロテアーゼドメインおよびディスインテグリンドメインとの間にジスルフィド架橋をもはや形成することができなくなり、これにより、それらのドメイン間領域は、タンパク質分解を受けやすくなり、個々のドメインがヘビ毒ディスインテグリンの遊離のドメインとして放出される可能性が与えられる[20]。したがって、PIIヘビ毒ディスインテグリンは、それらが血小板に特異的なインテグリンアルファIIbベータ3(alphaIIbbeta3)との相互作用の親和性が高いことによって、最も強力な天然の血小板凝集インヒビターとなっている、遊離のディスインテグリンドメインポリペプチドのクラスとして明らかになった。ADAM足場のディスインテグリンドメインと対照的に、これらの後期進化のPIIクラスのSVMPは、血液毒性のヘビの毒液において遊離のポリペプチドとして放出され、かつ新規の11アミノ酸ディスインテグリンループを保有する。このループは、このクラスの分子に特徴的な独特の構造要素であり、自然に遺伝子改変されて、細胞外マトリックスタンパク質モチーフの作用を模倣することによって強力な可溶性のインテグリンリガンドとしての機能を果たしている(ECM−模倣物)[19]。このループは、ジスルフィド安定化されたポリペプチドコアから自由に突出し、ループの先端に提示されるトリペプチドモチーフ(通常Arg−Gly−Aspモチーフ)を介してインテグリン受容体と相互作用する[21]。
【0006】
ヒトインテグリンファミリーには24種のメンバーが存在し、そのうちの23種は複雑な発現パターン、組織分布および生理機能を有するが、アルファIIbベータ3インテグリンは、血餅の形成に役立つ、血小板に特異的な受容体である。異常に発現されること、または原形質膜に誤って局在化すること、または不適切に活性化されること、またはこれらの機構の組み合わせのいずれかによって、種々の組織または器官において調節解除された場合に、これらの受容体の多くが異常に機能することが、新形成から炎症性疾患まで、ならびに創傷治癒および組織再生などの複雑な生理応答までにわたる多様な病態に関連づけられている[22〜25]。
【0007】
ヘビ毒ディスインテグリンは、リポソーム処方物で送達される場合、抗がん剤としての治療的な可能性を有する[21、26、27]。それらの天然の生物活性(すなわち、インテグリンアルファIIbベータ3との相互作用の親和性が高いことによる血小板凝集の阻害)に加えて、ヘビ毒ディスインテグリンは、腫瘍関連プロセス、例えば、転移および血管新生などを、これらのプロセスの病因に機構的に関わるインテグリンの定義されたセット(例えば、アルファvベータ3(alphavbeta3)、アルファvベータ5(alphavbeta5)、およびアルファ5ベータ1(alpha5beta1))に関与する能力によって破壊することが示されている[21]。これらの好都合な特質にもかかわらず、これらのポリペプチドは、依然として、それらがヘビ毒に由来することに起因する、潜在的に負の免疫学的特性を保有する。遊離のディスインテグリンは、ヒトまたは任意の他の哺乳動物において存在することは知られていない−これまでに同定された唯一のヒトディスインテグリンは、ADAMファミリータンパク質のメンバーの大きな配列の中に埋もれたサブドメインとして存在する[17]。哺乳動物界において30種を超えるADAMタンパク質が同定されており(ヒトは20種の遺伝子および3種の偽遺伝子を保有する)、それらの全てが、ディスインテグリンドメインを保有する[1]。ヒトにおいて同定された23種のADAM転写物のうち、3種が偽遺伝子由来(ADAM1、ADAM3およびADAM6)であり、それは機能タンパク質に翻訳されない。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】米国特許出願公開第20060246541号明細書
【非特許文献】
【0009】
【非特許文献1】Edwards, D.R., M.M. Handsley,およびC.J. Pennington, The ADAM metalloproteinases. Mol Aspects Med, 2008. 29(5): p. 258−89.
【非特許文献2】Mochizuki, S.およびY. Okada, ADAMs in cancer cell proliferation and progression. Cancer Sci, 2007. 98(5): p. 621−8.
【非特許文献3】Reiss, K., A. Ludwig,およびP. Saftig, Breaking up the tie: disintegrin−like metalloproteinases as regulators of cell migration in inflammation and invasion. Pharmacol Ther, 2006. 111(3): p. 985−1006.
【非特許文献4】Tousseyn, T., et al., (Make) stick and cut loose−−disintegrin metalloproteases in development and disease. Birth Defects Res C Embryo Today, 2006. 78(1): p. 24−46.
【非特許文献5】Arribas, J., J.J. Bech−Serra,およびB. Santiago−Josefat, ADAMs, cell migration and cancer. Cancer Metastasis Rev, 2006. 25(1): p. 57−68.
【非特許文献6】Blanchot−Jossic, F., et al., Up−regulated expression of ADAM17 in human colon carcinoma: co−expression with EGFR in neoplastic and endothelial cells. J Pathol, 2005. 207(2): p. 156−63.
【非特許文献7】Lendeckel, U., et al., Increased expression of ADAM family members in human breast cancer and breast cancer cell lines. J Cancer Res Clin Oncol, 2005. 131(1): p. 41−8.
【非特許文献8】Mazzocca, A., et al., A secreted form of ADAM9 promotes carcinoma invasion through tumor−stromal interactions. Cancer Res, 2005. 65(11): p. 4728−38.
【非特許文献9】McGowan, P.M., et al., ADAM−17 predicts adverse outcome in patients with breast cancer. Ann Oncol, 2008. 19(6): p. 1075−81.
【非特許文献10】McGowan, P.M., et al., ADAM−17 expression in breast cancer correlates with variables of tumor progression. Clin Cancer Res, 2007. 13(8): p. 2335−43.
【非特許文献11】Mitsui, Y., et al., ADAM28 is overexpressed in human breast carcinomas: implications for carcinoma cell proliferation through cleavage of insulin−like growth factor binding protein−3. Cancer Res, 2006. 66(20): p. 9913−20.
【非特許文献12】Najy, A.J., K.C. Day,およびM.L. Day, ADAM15 supports prostate cancer metastasis by modulating tumor cell endothelial cell interaction. Cancer Res, 2008. 68(4): p. 1092−9.
【非特許文献13】O’Shea, C., et al., Expression of ADAM−9 mRNA and protein in human breast cancer. Int J Cancer, 2003. 105(6): p.754−61.
【非特許文献14】Ringel, J., et al., Aberrant expression of a disintegrin and metalloproteinase 17/tumor necrosis factor−alpha converting enzyme increases the malignant potential in human pancreatic ductal adenocarcinoma. Cancer Res, 2006. 66(18): p. 9045−53.
【非特許文献15】Wildeboer, D., et al., Metalloproteinase disintegrins ADAM8 and ADAM19 are highly regulated in human primary brain tumors and their expression levels and activities are associated with invasiveness. J Neuropathol Exp Neurol, 2006. 65(5): p. 516−27.
【発明の概要】
【課題を解決するための手段】
【0010】
(発明の概要)
本明細書では、以下のアミノ酸残基:
a)Cys−Asp−Cys(CDC)モチーフのC末端側の最初のシステイン;
b)その最初のシステインのC末端側の2つの連続したアミノ酸;および
c)トリペプチドモチーフのC末端側のシステイン
において修飾されているADAM由来のポリペプチド(AP)を有する、修飾されたADAM由来のポリペプチド(MAP)であって、アミノ酸残基の修飾が、独立に、
a)欠失;
b)置換;および
c)化学的な修飾
から選択され、APが、(i)Cys−Asp−Cys(CDC)モチーフ、(ii)図1に示されているトリペプチドモチーフを有し、(iii)ADAMのメタロプロテアーゼドメイン、システインリッチドメイン、およびドメイン間のセグメントの全て、または実質的に全てを欠く、ADAM由来のディスインテグリン(distintegrin)様ドメインを有し、ADAMが、ADAM17(CDCモチーフではなくCDPを有するADAM17など)ではない、修飾されたADAM由来のポリペプチド(MAP)の組成物、およびそれに関連する方法が提供される。本明細書では、これらのMAPをコードする核酸が提供される。
【0011】
MAPは、ADAM(ディスインテグリンおよびメタロプロテイナーゼ)から得ることができ、ADAMとしては、ADAM15、ADAM28、ADAM1、ADAM2、ADAM3、ADAM6、ADAM7、ADAM8、ADAM9、ADAM10、ADAM11、ADAM12、ADAM18、ADAM19、ADAM20、ADAM21、ADAM22、ADAM23、ADAM29、ADAM30、ADAM32、ADAM33が挙げられる。これらの対応するMAPは、MAP15、MAP28、MAP1、MAP2、MAP3、MAP6、MAP7、MAP8、MAP9、MAP10、MAP11、MAP12、MAP18、MAP19、MAP20、MAP21、MAP22、MAP23、MAP29、MAP30、MAP32、MAP33であってよい。
【0012】
本明細書では、MAPとチオレドキシンのN末端セグメントの融合タンパク質およびこれらの融合物をコードする核酸も提供される。
【0013】
MAPは、HUVECまたはMDA−MB−435細胞の再構成基底膜を通じた移動を阻害する性質、MDA−MB−435細胞におけるFAKのリン酸化のレベルを上昇させる性質、または培養物におけるHUVECの管形成を阻害する性質を有し得る。
【0014】
本明細書では、MAPを発現させる発現ベクターおよびこれらの発現ベクターを用いて形質転換した原核宿主細胞も提供される。発現ベクターは、誘導性の制御下にあってもよく、例えば、ここで、宿主は、チオレドキシン還元酵素B(trxB)遺伝子および/またはグルタチオン還元酵素(gor)遺伝子に安定な変異も有する。
【0015】
本明細書では、がんに罹患した個体を、有効量の少なくとも1つのMAPを投与することによって処置する方法も提供される。がんは、インテグリンが発現されているがんであってよい。がんは、乳がん、結腸直腸がん、基底細胞がん、腺がん、胃腸がん、結腸がん、肝がん、膀胱がん、膵がん、卵巣がん、子宮頸がん、肺がん、皮膚がん、前立腺がん、腎細胞がん、中枢神経系(CNS)のがん、および白血病であってよい。胃腸がんは、口唇がん、口腔がん、食道がん、小腸がんまたは胃がんであってよい。皮膚がんは、扁平上皮細胞がんまたは基底細胞がんであってよい。
【0016】
本明細書では、インテグリンを発現している細胞を有効量のMAPまたはその融合物と接触させることによって、インテグリンがリガンドと結合することを阻害する方法も提供される。
【0017】
本明細書では、個体におけるがん細胞の存在を、がん細胞を少なくとも1つのMAPと接触させ、少なくとも1つのMAPを検出することによって決定する方法も提供される。MAPは、標識することができ、標識は、陽電子放射型断層撮影法(PET)プローブまたは蛍光プローブであってよい。
【0018】
本明細書では、人工的なECM足場をMAPでコーティングすることによって、人工的なECM足場を調製する方法も提供される。人工的なECM足場は、幹細胞前駆体をさらに含んでよい。人工的なECM足場は、膀胱の足場、食道の足場または肛門の足場であってよい。
【0019】
少なくとも1つのMAPを含む組成物でコーティングしたステントも提供される。
【0020】
(i)Cys−Asp−Cys(CD)モチーフ、(ii)図1に示されているトリペプチドモチーフを有し、(iii)ADAMのメタロプロテアーゼドメイン、システインリッチドメイン、およびドメイン間のセグメントの全て、または実質的に全てを欠き、ADAM由来のディスインテグリン様ドメインを有し、CDCモチーフのC末端側の最初のシステインとドメイン間のジスルフィド結合およびトリペプチドモチーフのC末端側のシステインとドメイン間のジスルフィド結合の破壊をもたらすアミノ酸の修飾をさらに含む、APであるMAPも提供される。
【図面の簡単な説明】
【0021】
【図1A】図1は、23種のヒトADAMタンパク質(AP)のディスインテグリン様ドメインのアミノ酸配列を示す。図1Aは、ヒトADAMタンパク質のディスインテグリン様ドメインの配列アラインメントを示す。横線で消されたアミノ酸残基は、対応するMAPポリペプチドでは除去されるアミノ酸を示す。AP1およびAP17の太字のアミノ酸は、中程度のサイズのヘビ毒ディスインテグリンのシステインパターンを保存するために、対応するMAPでは別のアミノ酸と交換した。図1Bは、APのディスインテグリン様ドメインのディスインテグリンループのそれぞれの先端に提示されるトリペプチドモチーフ(枠で囲まれたアミノ酸残基)を示す。図1Cは、ディスインテグリン様ドメインのディスインテグリンループ(枠で囲まれたアミノ酸残基)を示す。
【図1B】図1は、23種のヒトADAMタンパク質(AP)のディスインテグリン様ドメインのアミノ酸配列を示す。図1Aは、ヒトADAMタンパク質のディスインテグリン様ドメインの配列アラインメントを示す。横線で消されたアミノ酸残基は、対応するMAPポリペプチドでは除去されるアミノ酸を示す。AP1およびAP17の太字のアミノ酸は、中程度のサイズのヘビ毒ディスインテグリンのシステインパターンを保存するために、対応するMAPでは別のアミノ酸と交換した。図1Bは、APのディスインテグリン様ドメインのディスインテグリンループのそれぞれの先端に提示されるトリペプチドモチーフ(枠で囲まれたアミノ酸残基)を示す。図1Cは、ディスインテグリン様ドメインのディスインテグリンループ(枠で囲まれたアミノ酸残基)を示す。
【図1C】図1は、23種のヒトADAMタンパク質(AP)のディスインテグリン様ドメインのアミノ酸配列を示す。図1Aは、ヒトADAMタンパク質のディスインテグリン様ドメインの配列アラインメントを示す。横線で消されたアミノ酸残基は、対応するMAPポリペプチドでは除去されるアミノ酸を示す。AP1およびAP17の太字のアミノ酸は、中程度のサイズのヘビ毒ディスインテグリンのシステインパターンを保存するために、対応するMAPでは別のアミノ酸と交換した。図1Bは、APのディスインテグリン様ドメインのディスインテグリンループのそれぞれの先端に提示されるトリペプチドモチーフ(枠で囲まれたアミノ酸残基)を示す。図1Cは、ディスインテグリン様ドメインのディスインテグリンループ(枠で囲まれたアミノ酸残基)を示す。
【図2A】図2は、MAPポリペプチドのアミノ酸配列を示す。図2Aは、典型的な中程度のサイズのヘビ毒ディスインテグリン(Trimeresurus flavoviridisヘビの毒液から精製した)であるトリメスタチン(trimestatin)の配列に対してアラインメントした、23種のMAPポリペプチドのアミノ酸配列を示す。システイン残基をアラインメントし、それに下線が引かれている。AP1およびAP17におけるアミノ酸残基に対応する、MAP1およびMAP17の交換されたアミノ酸残基がそれぞれ太字で示されている。さらに、AP1、AP3、AP6、AP18、AP21、AP30、およびAP32のN末端残基が、それらの対応するMAPにおいてグリシン残基と交換されている。図2Bは、MAPのディスインテグリン様ドメインのディスインテグリンループのそれぞれの先端に提示され、および中程度のサイズのヘビ毒トリメスタチンによって提示されるトリペプチドモチーフ(枠で囲まれたアミノ酸)を示す。図2Cは、MAPおよびトリメスタチンのディスインテグリンループ(枠で囲まれたアミノ酸)を同定する。
【図2B】図2は、MAPポリペプチドのアミノ酸配列を示す。図2Aは、典型的な中程度のサイズのヘビ毒ディスインテグリン(Trimeresurus flavoviridisヘビの毒液から精製した)であるトリメスタチン(trimestatin)の配列に対してアラインメントした、23種のMAPポリペプチドのアミノ酸配列を示す。システイン残基をアラインメントし、それに下線が引かれている。AP1およびAP17におけるアミノ酸残基に対応する、MAP1およびMAP17の交換されたアミノ酸残基がそれぞれ太字で示されている。さらに、AP1、AP3、AP6、AP18、AP21、AP30、およびAP32のN末端残基が、それらの対応するMAPにおいてグリシン残基と交換されている。図2Bは、MAPのディスインテグリン様ドメインのディスインテグリンループのそれぞれの先端に提示され、および中程度のサイズのヘビ毒トリメスタチンによって提示されるトリペプチドモチーフ(枠で囲まれたアミノ酸)を示す。図2Cは、MAPおよびトリメスタチンのディスインテグリンループ(枠で囲まれたアミノ酸)を同定する。
【図2C】図2は、MAPポリペプチドのアミノ酸配列を示す。図2Aは、典型的な中程度のサイズのヘビ毒ディスインテグリン(Trimeresurus flavoviridisヘビの毒液から精製した)であるトリメスタチン(trimestatin)の配列に対してアラインメントした、23種のMAPポリペプチドのアミノ酸配列を示す。システイン残基をアラインメントし、それに下線が引かれている。AP1およびAP17におけるアミノ酸残基に対応する、MAP1およびMAP17の交換されたアミノ酸残基がそれぞれ太字で示されている。さらに、AP1、AP3、AP6、AP18、AP21、AP30、およびAP32のN末端残基が、それらの対応するMAPにおいてグリシン残基と交換されている。図2Bは、MAPのディスインテグリン様ドメインのディスインテグリンループのそれぞれの先端に提示され、および中程度のサイズのヘビ毒トリメスタチンによって提示されるトリペプチドモチーフ(枠で囲まれたアミノ酸)を示す。図2Cは、MAPおよびトリメスタチンのディスインテグリンループ(枠で囲まれたアミノ酸)を同定する。
【図3】図3は、ヘビ毒ディスインテグリンおよびヒトADAMタンパク質のディスインテグリン様ドメインの配列アラインメントを示す。PIII−クラスのヘビ毒メタロプロテアーゼ(VAP1およびカトロコラスタチン(Catrocollastatin))のディスインテグリン様配列を長いサイズのヘビ毒ディスインテグリン配列(サルモシン(Salmosin)3およびビチスタチン(Bitistatin))、典型的な中程度のサイズのヘビ毒ディスインテグリン(トリメスタチン)およびいくつかのヒトADAMのディスインテグリン様ドメイン(AP7、AP8、AP12、AP19、AP28、およびAP33)とアラインメントした。対応するMAPポリペプチドを生成するために、修飾することができる(例えば、欠失)ディスインテグリン様配列(ヘビ起源およびヒト起源の両方)におけるアミノ酸残基が横線で消されて示されている。これらのディスインテグリンおよびディスインテグリン様配列におけるディスインテグリンループの先端に提示されるトリペプチドアミノ酸モチーフが囲み枠の中に示されている。
【図4】図4は、様々なE.coli宿主(BL21対Origami B)におけるTrx−D9(ネイティブなヒトADAM9ディスインテグリン様ドメイン配列−チオレドキシン融合ポリペプチド)の発現とTrx−MAP9(MAP9−チオレドキシン融合ポリペプチド)の発現とを、SDS−PAGEによって分析して比較している。レーンは、左から右に、PageRulerTMPlus Prestained Protein Ladder(Fermentas、Burlington、ON)、カルベニシリンの下で成長させ、誘導した、Trx−D9で形質転換したBL21(DE3)細胞からの溶解物、最初に3種のAB(カルベニシリン、カナマイシンおよびテトラサイクリン)上に播いたが、カルベニシリンのみの下でさらに増殖させ、誘導した、Trx−D9で形質転換したOrigami B(DE3)細胞からの溶解物、カルベニシリンの下で成長させ、誘導した、Trx−MAP9で形質転換したBL21(DE3)細胞からの溶解物、ならびに、最初に3種のAB(カルベニシリン、カナマイシンおよびテトラサイクリン)上に播いたが、カルベニシリンのみの下でさらに増殖させ、誘導した、Trx−MAP9で形質転換したOrigami B(DE3)細胞からの溶解物を示す。
【図5】図5は、様々なE.coli宿主(BL21対Origami B)におけるTrx−D15(ネイティブなヒトADAM15ディスインテグリン様ドメイン配列−チオレドキシン融合ポリペプチド)の発現とTrx−MAP15(MAP15−チオレドキシン融合ポリペプチド)の発現とを、SDS−PAGEによって分析して比較している。レーンは、左から右に、PageRulerTMPlus Prestained Protein Ladder(Fermentas、Burlington、ON)、カルベニシリンの下で成長させ、誘導した、Trx−D15で形質転換したBL21(DE3)細胞からの溶解物、最初に3種のAB(カルベニシリン、カナマイシンおよびテトラサイクリン)上に播いたが、カルベニシリンのみの下でさらに増殖させ、誘導した、Trx−D15で形質転換したOrigami B(DE3)細胞からの溶解物、カルベニシリンの下で成長させ、誘導した、Trx−MAP15で形質転換したBL21(DE3)細胞からの溶解物、ならびに、最初に3種のAB(カルベニシリン、カナマイシンおよびテトラサイクリン)上に播いたが、カルベニシリンのみの下でさらに増殖させ、誘導した、Trx−MAP15で形質転換したOrigami B(DE3)細胞からの溶解物を示す。
【図6A】図6は、細菌のTrxAの3’側の、pET32a発現ベクターに挿入されたMAPの核酸配列を示す。図6Aは、MAP1、MAP2、MAP3、およびMAP6のDNA配列を示す。図6Bは、MAP7、MAP8、MAP9、およびMAP10の核酸配列を示す。図6Cは、MAP11、MAP12、MAP15、およびMAP17の核酸配列を示す。図6Dは、MAP18、MAP19、MAP20、およびMAP21の核酸配列を示す。図6Eは、MAP22、MAP23、MAP28、およびMAP29の核酸配列を示す。図6Fは、MAP30、MAP32、およびMAP33の核酸配列を示す。
【図6B】図6は、細菌のTrxAの3’側の、pET32a発現ベクターに挿入されたMAPの核酸配列を示す。図6Aは、MAP1、MAP2、MAP3、およびMAP6のDNA配列を示す。図6Bは、MAP7、MAP8、MAP9、およびMAP10の核酸配列を示す。図6Cは、MAP11、MAP12、MAP15、およびMAP17の核酸配列を示す。図6Dは、MAP18、MAP19、MAP20、およびMAP21の核酸配列を示す。図6Eは、MAP22、MAP23、MAP28、およびMAP29の核酸配列を示す。図6Fは、MAP30、MAP32、およびMAP33の核酸配列を示す。
【図6C】図6は、細菌のTrxAの3’側の、pET32a発現ベクターに挿入されたMAPの核酸配列を示す。図6Aは、MAP1、MAP2、MAP3、およびMAP6のDNA配列を示す。図6Bは、MAP7、MAP8、MAP9、およびMAP10の核酸配列を示す。図6Cは、MAP11、MAP12、MAP15、およびMAP17の核酸配列を示す。図6Dは、MAP18、MAP19、MAP20、およびMAP21の核酸配列を示す。図6Eは、MAP22、MAP23、MAP28、およびMAP29の核酸配列を示す。図6Fは、MAP30、MAP32、およびMAP33の核酸配列を示す。
【図6D】図6は、細菌のTrxAの3’側の、pET32a発現ベクターに挿入されたMAPの核酸配列を示す。図6Aは、MAP1、MAP2、MAP3、およびMAP6のDNA配列を示す。図6Bは、MAP7、MAP8、MAP9、およびMAP10の核酸配列を示す。図6Cは、MAP11、MAP12、MAP15、およびMAP17の核酸配列を示す。図6Dは、MAP18、MAP19、MAP20、およびMAP21の核酸配列を示す。図6Eは、MAP22、MAP23、MAP28、およびMAP29の核酸配列を示す。図6Fは、MAP30、MAP32、およびMAP33の核酸配列を示す。
【図6E】図6は、細菌のTrxAの3’側の、pET32a発現ベクターに挿入されたMAPの核酸配列を示す。図6Aは、MAP1、MAP2、MAP3、およびMAP6のDNA配列を示す。図6Bは、MAP7、MAP8、MAP9、およびMAP10の核酸配列を示す。図6Cは、MAP11、MAP12、MAP15、およびMAP17の核酸配列を示す。図6Dは、MAP18、MAP19、MAP20、およびMAP21の核酸配列を示す。図6Eは、MAP22、MAP23、MAP28、およびMAP29の核酸配列を示す。図6Fは、MAP30、MAP32、およびMAP33の核酸配列を示す。
【図6F】図6は、細菌のTrxAの3’側の、pET32a発現ベクターに挿入されたMAPの核酸配列を示す。図6Aは、MAP1、MAP2、MAP3、およびMAP6のDNA配列を示す。図6Bは、MAP7、MAP8、MAP9、およびMAP10の核酸配列を示す。図6Cは、MAP11、MAP12、MAP15、およびMAP17の核酸配列を示す。図6Dは、MAP18、MAP19、MAP20、およびMAP21の核酸配列を示す。図6Eは、MAP22、MAP23、MAP28、およびMAP29の核酸配列を示す。図6Fは、MAP30、MAP32、およびMAP33の核酸配列を示す。
【図7A】図7は、MAP構築物をpET32a発現ベクターにクローニングするために使用するオリゴヌクレオチドプライマー配列を示す。図7Aは、MAP1、MAP2、MAP3、MAP6、MAP7、MAP8、MAP9、MAP10、MAP11、MAP12、MAP15、MAP17、およびMAP18をクローニングするために使用するオリゴヌクレオチドプライマーを示す。図7Bは、MAP19、MAP20、MAP21、MAP22、MAP23、MAP28、MAP29、MAP30、MAP32、およびMAP33をクローニングするために使用するオリゴヌクレオチドプライマーを示す。
【図7B】図7は、MAP構築物をpET32a発現ベクターにクローニングするために使用するオリゴヌクレオチドプライマー配列を示す。図7Aは、MAP1、MAP2、MAP3、MAP6、MAP7、MAP8、MAP9、MAP10、MAP11、MAP12、MAP15、MAP17、およびMAP18をクローニングするために使用するオリゴヌクレオチドプライマーを示す。図7Bは、MAP19、MAP20、MAP21、MAP22、MAP23、MAP28、MAP29、MAP30、MAP32、およびMAP33をクローニングするために使用するオリゴヌクレオチドプライマーを示す。
【図8A】図8は、E.coli Origami B(DE3)において発現させた全てのTrxA−MAP構築物のアミノ酸配列を示す。TrxA(CGPC)の活性部位はイタリック体で示されており、ディスインテグリンループの先端のトリペプチドモチーフには下線が引かれており、TEV切断認識部位は囲み枠で強調されており、TrxAと種々のMAP構築物との間のリンカー領域は太字のイタリック体で示されている。AP1およびAP17内の残基を交換するために導入された、対応するMAP1およびMAP17内のアミノ酸残基は、太字の二重下線で強調されている。
【図8B】図8は、E.coli Origami B(DE3)において発現させた全てのTrxA−MAP構築物のアミノ酸配列を示す。TrxA(CGPC)の活性部位はイタリック体で示されており、ディスインテグリンループの先端のトリペプチドモチーフには下線が引かれており、TEV切断認識部位は囲み枠で強調されており、TrxAと種々のMAP構築物との間のリンカー領域は太字のイタリック体で示されている。AP1およびAP17内の残基を交換するために導入された、対応するMAP1およびMAP17内のアミノ酸残基は、太字の二重下線で強調されている。
【図8C】図8は、E.coli Origami B(DE3)において発現させた全てのTrxA−MAP構築物のアミノ酸配列を示す。TrxA(CGPC)の活性部位はイタリック体で示されており、ディスインテグリンループの先端のトリペプチドモチーフには下線が引かれており、TEV切断認識部位は囲み枠で強調されており、TrxAと種々のMAP構築物との間のリンカー領域は太字のイタリック体で示されている。AP1およびAP17内の残基を交換するために導入された、対応するMAP1およびMAP17内のアミノ酸残基は、太字の二重下線で強調されている。
【図8D】図8は、E.coli Origami B(DE3)において発現させた全てのTrxA−MAP構築物のアミノ酸配列を示す。TrxA(CGPC)の活性部位はイタリック体で示されており、ディスインテグリンループの先端のトリペプチドモチーフには下線が引かれており、TEV切断認識部位は囲み枠で強調されており、TrxAと種々のMAP構築物との間のリンカー領域は太字のイタリック体で示されている。AP1およびAP17内の残基を交換するために導入された、対応するMAP1およびMAP17内のアミノ酸残基は、太字の二重下線で強調されている。
【図8E】図8は、E.coli Origami B(DE3)において発現させた全てのTrxA−MAP構築物のアミノ酸配列を示す。TrxA(CGPC)の活性部位はイタリック体で示されており、ディスインテグリンループの先端のトリペプチドモチーフには下線が引かれており、TEV切断認識部位は囲み枠で強調されており、TrxAと種々のMAP構築物との間のリンカー領域は太字のイタリック体で示されている。AP1およびAP17内の残基を交換するために導入された、対応するMAP1およびMAP17内のアミノ酸残基は、太字の二重下線で強調されている。
【図8F】図8は、E.coli Origami B(DE3)において発現させた全てのTrxA−MAP構築物のアミノ酸配列を示す。TrxA(CGPC)の活性部位はイタリック体で示されており、ディスインテグリンループの先端のトリペプチドモチーフには下線が引かれており、TEV切断認識部位は囲み枠で強調されており、TrxAと種々のMAP構築物との間のリンカー領域は太字のイタリック体で示されている。AP1およびAP17内の残基を交換するために導入された、対応するMAP1およびMAP17内のアミノ酸残基は、太字の二重下線で強調されている。
【図8G】図8は、E.coli Origami B(DE3)において発現させた全てのTrxA−MAP構築物のアミノ酸配列を示す。TrxA(CGPC)の活性部位はイタリック体で示されており、ディスインテグリンループの先端のトリペプチドモチーフには下線が引かれており、TEV切断認識部位は囲み枠で強調されており、TrxAと種々のMAP構築物との間のリンカー領域は太字のイタリック体で示されている。AP1およびAP17内の残基を交換するために導入された、対応するMAP1およびMAP17内のアミノ酸残基は、太字の二重下線で強調されている。
【図8H】図8は、E.coli Origami B(DE3)において発現させた全てのTrxA−MAP構築物のアミノ酸配列を示す。TrxA(CGPC)の活性部位はイタリック体で示されており、ディスインテグリンループの先端のトリペプチドモチーフには下線が引かれており、TEV切断認識部位は囲み枠で強調されており、TrxAと種々のMAP構築物との間のリンカー領域は太字のイタリック体で示されている。AP1およびAP17内の残基を交換するために導入された、対応するMAP1およびMAP17内のアミノ酸残基は、太字の二重下線で強調されている。
【図9】図9は、SDS−PAGEによって分析した、Origami B宿主におけるTrx−MAP2(MAP2−チオレドキシン融合ポリペプチド)、Trx−MAP7(MAP7−チオレドキシン融合ポリペプチド)、Trx−MAP8(MAP8−チオレドキシン融合ポリペプチド)、およびTrx−MAP9(MAP9−チオレドキシン融合ポリペプチド)の発現レベルを示す。レーンは、左から右に、PageRulerTMPlus Prestained Protein Ladder(Fermentas、Burlington、ON)、Trx−MAP2で形質転換したOrigami B(DE3)細胞からの溶解物、Trx−MAP7で形質転換したOrigami B(DE3)細胞からの溶解物、Trx−MAP8で形質転換したOrigami B(DE3)細胞からの溶解物、およびTrx−MAP9で形質転換したOrigami B(DE3)からの溶解物を示す。形質転換体は全て、最初に3種の抗生物質(カルベニシリン、カナマイシンおよびテトラサイクリン)の上に播いたが、カルベニシリンのみの下でさらに増殖させ、誘導した。
【図10】図10は、SDS−PAGEによって分析した、Origami B宿主におけるTrx−MAP10(MAP10−チオレドキシン融合ポリペプチド)、Trx−MAP12(MAP12−チオレドキシン融合ポリペプチド)、Trx−MAP15(MAP15−チオレドキシン融合ポリペプチド)、およびTrx−MAP17(MAP17−チオレドキシン融合ポリペプチド)の発現レベルを示す。レーンは、左から右に、PageRulerTMPlus Prestained Protein Ladder(Fermentas、Burlington、ON)、Trx−MAP10で形質転換したOrigami B(DE3)細胞からの溶解物、Trx−MAP12で形質転換したOrigami B(DE3)細胞からの溶解物、Trx−MAP15で形質転換したOrigami B(DE3)細胞からの溶解物、およびTrx−MAP17で形質転換したOrigami B(DE3)からの溶解物を示す。形質転換体は全て、最初に3種の抗生物質(カルベニシリン、カナマイシンおよびテトラサイクリン)の上に播いたが、カルベニシリンのみの下でさらに増殖させ、誘導した。
【図11】図11は、SDS−PAGEによって分析した、Origami B宿主におけるTrx−MAP19(MAP19−チオレドキシン融合ポリペプチド)、Trx−MAP23(MAP23−チオレドキシン融合ポリペプチド)、Trx−MAP28(MAP28−チオレドキシン融合ポリペプチド)、およびTrx−MAP33(MAP33−チオレドキシン融合ポリペプチド)の発現レベルを示す。レーンは、左から右に、PageRulerTMPlus Prestained Protein Ladder(Fermentas、Burlington、ON)、Trx−MAP19で形質転換したOrigami B(DE3)細胞からの溶解物、Trx−MAP23で形質転換したOrigami B(DE3)細胞からの溶解物、Trx−MAP28で形質転換したOrigami B(DE3)細胞からの溶解物、およびTrx−MAP33で形質転換したOrigami B(DE3)からの溶解物を示す。形質転換体は全て、最初に3種の抗生物質(カルベニシリン、カナマイシンおよびテトラサイクリン)の上に播いたが、カルベニシリンのみの下でさらに増殖させ、誘導した。
【図12】図12は、図に示されている種々のTrx−MAPの、MDA−MB−231細胞(ヒト乳がん)への結合の、フローサイトメトリーによる検出を示す。
【図13】図13は、図に示されている種々のTrx−MAPの、MDA−MB−231細胞(ヒト乳がん)の骨ホーミングサブクローンへの結合の、フローサイトメトリーによる検出を示す。
【図14】図14は、図に示されている種々のTrx−MAPの、MDA−MB−231細胞(ヒト乳がん)の脳ホーミングサブクローンへの結合の、フローサイトメトリーによる検出を示す。
【図15】図15は、図に示されている種々のTrx−MAPの、ジャーカット細胞(ヒトT細胞白血病細胞株)への結合の、フローサイトメトリーによる検出を示す。
【図16】図16は、MAP9およびMAP15の、HUVEC(ヒト臍帯静脈内皮細胞)への結合、MDA−MB−435(ヒト乳がん)への結合、MDA−MB−231(ヒト乳がん)への結合、および多形神経膠芽腫がん幹細胞株(GBM−CSC)への結合の、フローサイトメトリーによる検出を示す。
【図17】図17は、10nMのMAP9またはMAP15の存在下でのHUVECの管形成アッセイを示す。パネルA−無処置の対照;パネルB−100μMのスラミン;パネルC−10nMのMAP9;パネルD−10nMのMAP15。細胞をCalcein AMで染色し、共焦点顕微鏡を使用してイメージングした。イメージは全て、同じ倍率で取得した(スケールバー=50μm)。
【図18】図18は、他の抗がん処置(アバスチンおよびドセタキセル)と比較した、MDA−MB−231異種移植における、MAP15およびMAP15のリポソーム処方物による処置による腫瘍の成長阻害(パネルA)および生存(パネルB)を示す。
【図19】図19は、MDA−MB−231異種移植における、他の抗がん処置(アバスチンおよびドセタキセル)と比較した、MAP15のリポソーム処方物による処置および他の抗がん処置との組み合わせにおける、微小血管の阻害を、腫瘍血管新生アッセイの顕微鏡写真(パネルA)において、およびランダムな顕微鏡写真(パネルB)から微小血管の密度を定量化することによって示している。
【図20】図20は、ADAM1転写物に対応する核酸配列を示す。
【図21】図21は、ADAM2転写物に対応する核酸配列を示す。
【図22】図22は、ADAM3転写物、バリアント(variant)1に対応する核酸配列を示す。
【図23】図23は、ADAM6転写物に対応する核酸配列を示す。
【図24】図24は、ADAM7転写物に対応する核酸配列を示す。
【図25A】図25は、ADAM8転写物、バリアント1に対応する核酸配列を示す。
【図25B】図25は、ADAM8転写物、バリアント1に対応する核酸配列を示す。
【図26A】図26は、ADAM9転写物、バリアント1に対応する核酸配列を示す。
【図26B】図26は、ADAM9転写物、バリアント1に対応する核酸配列を示す。
【図27A】図27は、ADAM10転写物に対応する核酸配列を示す。
【図27B】図27は、ADAM10転写物に対応する核酸配列を示す。
【図28A】図28は、ADAM11転写物に対応する核酸配列を示す。
【図28B】図28は、ADAM11転写物に対応する核酸配列を示す。
【図29A】図29は、ADAM12転写物、バリアント1に対応する核酸配列を示す。
【図29B】図29は、ADAM12転写物、バリアント1に対応する核酸配列を示す。
【図30】図30は、ADAM15転写物、バリアント6に対応する核酸配列を示す。
【図31A】図31は、ADAM17転写物に対応する核酸配列を示す。
【図31B】図31は、ADAM17転写物に対応する核酸配列を示す。
【図32】図32は、ADAM18転写物、バリアント1に対応する核酸配列を示す。
【図33A】図33は、ADAM19転写物に対応する核酸配列を示す。
【図33B】図33は、ADAM19転写物に対応する核酸配列を示す。
【図33C】図33は、ADAM19転写物に対応する核酸配列を示す。
【図34】図34は、ADAM20転写物に対応する核酸配列を示す。
【図35】図35は、ADAM21転写物に対応する核酸配列を示す。
【図36A】図36は、ADAM22転写物、バリアント1に対応する核酸配列を示す。
【図36B】図36は、ADAM22転写物、バリアント1に対応する核酸配列を示す。
【図36C】図36は、ADAM22転写物、バリアント1に対応する核酸配列を示す。
【図36D】図36は、ADAM22転写物、バリアント1に対応する核酸配列を示す。
【図37】図37は、ADAM23転写物に対応する核酸配列を示す。
【図38A】図38は、ADAM28転写物、バリアント1に対応する核酸配列を示す。
【図38B】図38は、ADAM28転写物、バリアント1に対応する核酸配列を示す。
【図39A】図39は、ADAM29転写物、バリアント1に対応する核酸配列を示す。
【図39B】図39は、ADAM29転写物、バリアント1に対応する核酸配列を示す。
【図40】図40は、ADAM30転写物に対応する核酸配列を示す。
【図41】図41は、ADAM32転写物に対応する核酸配列を示す。
【図42A】図42は、ADAM33転写物、バリアント1に対応する核酸配列を示す。
【図42B】図42は、ADAM33転写物、バリアント1に対応する核酸配列を示す。
【図43A】図43は、ADAM2、ADAM7、ADAM8、ADAM9、ADAM10、ADAM11、ADAM12、ADAM15、ADAM17、ADAM18、ADAM19、ADAM20、ADAM21、ADAM22、ADAM23、ADAM28、ADAM29、ADAM30、ADAM32、およびADAM33ポリペプチドのアミノ酸配列を示す。
【図43B】図43は、ADAM2、ADAM7、ADAM8、ADAM9、ADAM10、ADAM11、ADAM12、ADAM15、ADAM17、ADAM18、ADAM19、ADAM20、ADAM21、ADAM22、ADAM23、ADAM28、ADAM29、ADAM30、ADAM32、およびADAM33ポリペプチドのアミノ酸配列を示す。
【図43C】図43は、ADAM2、ADAM7、ADAM8、ADAM9、ADAM10、ADAM11、ADAM12、ADAM15、ADAM17、ADAM18、ADAM19、ADAM20、ADAM21、ADAM22、ADAM23、ADAM28、ADAM29、ADAM30、ADAM32、およびADAM33ポリペプチドのアミノ酸配列を示す。
【図43D】図43は、ADAM2、ADAM7、ADAM8、ADAM9、ADAM10、ADAM11、ADAM12、ADAM15、ADAM17、ADAM18、ADAM19、ADAM20、ADAM21、ADAM22、ADAM23、ADAM28、ADAM29、ADAM30、ADAM32、およびADAM33ポリペプチドのアミノ酸配列を示す。
【図43E】図43は、ADAM2、ADAM7、ADAM8、ADAM9、ADAM10、ADAM11、ADAM12、ADAM15、ADAM17、ADAM18、ADAM19、ADAM20、ADAM21、ADAM22、ADAM23、ADAM28、ADAM29、ADAM30、ADAM32、およびADAM33ポリペプチドのアミノ酸配列を示す。
【図43F】図43は、ADAM2、ADAM7、ADAM8、ADAM9、ADAM10、ADAM11、ADAM12、ADAM15、ADAM17、ADAM18、ADAM19、ADAM20、ADAM21、ADAM22、ADAM23、ADAM28、ADAM29、ADAM30、ADAM32、およびADAM33ポリペプチドのアミノ酸配列を示す。
【図43G】図43は、ADAM2、ADAM7、ADAM8、ADAM9、ADAM10、ADAM11、ADAM12、ADAM15、ADAM17、ADAM18、ADAM19、ADAM20、ADAM21、ADAM22、ADAM23、ADAM28、ADAM29、ADAM30、ADAM32、およびADAM33ポリペプチドのアミノ酸配列を示す。
【発明を実施するための形態】
【0022】
(発明の詳細な説明)
本明細書では、MAP(修飾されたADAM由来のポリペプチド)と称される独特な設計ポリペプチドのクラス、およびそれをコードする核酸が提供される。本明細書で使用される場合、MAPとは、ADAMタンパク質のネイティブなディスインテグリンドメインの修飾された形態を指す。ADAMをコードする核酸およびアミノ酸の配列の一覧については付属物を参照されたい。本明細書で使用される場合、本明細書では「AP」(「ADAM由来ポリペプチド」)と称することができる「ADAMタンパク質のディスインテグリンドメイン」は、そのメタロプロテアーゼドメイン、システインリッチドメインおよびドメイン間のセグメントの全て、または実質的に全てを欠く、ADAMのディスインテグリンドメインである。実質的に全てを欠くとは、残りのアミノ酸配列が、そのドメインの機能をもはや保持しないことを意味する。図1にAPの例が示されている。APのN末端は、CDCモチーフからN末端側に3位のアミノ酸残基で始まり、CDCのN末端側の最初のシステインの直前までである。APのC末端は、CDCモチーフから12番目のシステイン残基のC末端側に10位のアミノ酸残基で終わり、前記12番目のシステイン残基のC末端側の次のシステインの直前までである。例外が2つある:(1)AP1のC末端は、CDCモチーフから13番目のシステイン残基のC末端側の10位のアミノ酸残基で終わり、前記13番目のシステイン残基のC末端側の最初のシステインの直前までであり、(2)ADAM17は、CDCモチーフではなくCDPモチーフを有し、対応するAP(AP17)の末端が示されている。
【0023】
「MAP」は、APの「修飾された」形態であり、修飾は、本明細書に記載の有益な性質を実現するためのAPのアミノ酸配列の変更(複数可)を伴う。したがって、MAPは、通常APに存在する配列および対応するADAMポリペプチドの配列と比較して、修飾された配列を有する。本明細書で使用される場合、「修飾された」とは、アミノ酸が欠失していることか、置換されていることか、または化学修飾されていることを意味し、ある実施形態では、修飾により、ドメイン間のジスルフィド結合の破壊がもたらされる。図2に例示的なMAPが示されている。典型的な中程度のサイズのヘビ毒ディスインテグリンであるトリメスタチンとアラインメントしたMAP配列が示されている。全てのMAP構築物を、中程度のサイズのヘビ毒ディスインテグリンにならってモデリングし、それらの配列を、これらのネイティブなヘビ毒分子と同様に折りたたまれるように修飾した。MAP(MAP17以外)は、対応するAPのCDCモチーフのC末端側の最初のシステインおよび前記システインのC末端側の2つのアミノ酸ならびにトリペプチドモチーフのC末端側のシステインが欠失するように構築した。あるいは、ジスルフィド結合の形成を妨げるために、システイン残基を代替のアミノ酸で置換することができ、またはシステインアミノ酸残基を化学修飾することができる。アミノ酸置換は、保存的であり得、例えば、APのCDCモチーフのC末端側の最初のシステインをセリン残基で置換することができ、システインのC末端側のアミノ酸残基を荷電アミノ酸で置換することができ、または、トリペプチドモチーフのC末端側のシステインを荷電アミノ酸で置換することができる。そのような変異による手法およびアミノ酸残基の化学的な修飾は当技術分野で周知である。化学的な修飾に関して、例としては、ジスルフィド結合の形成を妨げるために、アルキル化剤を使用してシステイン残基と反応させることがある。MAP10、MAP17、MAP18およびMAP32以外、MAPは、ヘビ毒ディスインテグリンのネイティブなループと同様の11アミノ酸のディスインテグリンループを提示する。MAP10は、10アミノ酸のインテグリンループを提示し、MAP17、MAP18、およびMAP32は、12アミノ酸のディスインテグリンループを提示する。
【0024】
MAPを、活性な可溶性のジスルフィドリッチポリペプチドの生成およびこれらの生成物についての高い発現収率の両方を支持する細菌系において、独立した生物活性のある分子として発現させ、さらに精製することができる。理論に縛られることを望むものではないが、ネイティブなAPから、MAPを、それらのネイティブなADAMのコンフォメーションではなく、ヘビ毒ディスインテグリンの折りたたみをとり得るように設計した。MAPを、Origami B(DE3)E.coli株において高収率で発現させ、哺乳動物細胞の表面受容体クラス(インテグリン)と、ネイティブなヘビ毒ディスインテグリンと同様の様式で相互作用することができる安定かつ活性な遊離のポリペプチドとしてさらに精製することができる。MAPは、MAPが由来する、ADAMポリペプチド由来のAPまたはディスインテグリンドメインの活性の特性であるシグナル伝達性質のいくつかも保持し得る。例えば、保持される特性としては、ADAMディスインテグリンドメインの、古典的なディスインテグリンループの外側に位置するアミノ酸残基を利用することによってインテグリン受容体に関与すると推定される能力に関連するシグナル伝達特質を挙げることができる。ADAMの細胞機能は周知である[1〜5、34]。
【0025】
理論に縛られることを望むものではないが、典型的な中程度のサイズのヘビ毒ディスインテグリン(例えば、トリメスタチン、キストリン(Kistrin)、フラボリジン(Flavoridin)など)を生じさせるPII−クラスのSVMPは、上流のスペーサー領域とディスインテグリンドメインとの間の重要なジスルフィド架橋を形成することができず、したがって、ディスインテグリンドメインが開始する位置のすぐN末端側に位置する残基においてタンパク質分解性の攻撃が起こり、その結果、放出された中程度のサイズのディスインテグリンは、上流のスペーサー領域の部分を含有しない完全なディスインテグリンドメインであると考えられている。対照的に、長いサイズのヘビ毒ディスインテグリン(例えば、ビチスタチン、サルモシン3など)を生じさせるPII−クラスのSVMPは、メタロプロテアーゼドメインと下流のスペーサー領域との間の重要なジスルフィド架橋を形成することができず、したがって、スペーサー領域のさらにN末端においてタンパク質分解性の攻撃が起こり、スペーサー領域の一部がN末端で遊離のディスインテグリンドメインに付着したより長いディスインテグリンが放出されると考えられている(図3における種々のディスインテグリンおよびディスインテグリンドメインの配列アラインメントを参照されたい)。さらに、PII−SVMPが、より多くの変異および/または欠失を含有する場合、同じスペーサー領域においてだけではなく、ディスインテグリンドメインのN末端部分においてもジスルフィド架橋を形成することができず、ヘビ毒ディスインテグリンのより短いバリアントが放出されるとも考えられている(例えば、コントルトロスタチン(Contortrostatin)と同様に二量体化する部分的に短縮されたディスインテグリンドメイン、または、まれな、エキスタチンまたはエリストスタチン(Eristostatin)と同様に非常に短縮されたポリペプチドのいずれか)。さらに、ほとんど全ての場合において、遊離のディスインテグリンドメインは、それらの分子のC末端側の半分において、ヘビ毒ディスインテグリンの特徴である保存された11アミノ酸のディスインテグリンループを提示すると考えられている。
【0026】
ヒトゲノムにおいて同定された23種の異なるADAM転写物(そのうち3種は通常はタンパク質産物に翻訳されない偽遺伝子である)を、ヘビ毒ディスインテグリンの折りたたみ構造をとる本明細書に記載のコードされたMAPを生み出すための基礎として使用した。
【0027】
いくつかのADAM転写物は、いくつものアイソフォームをコードする。それにもかかわらず、異なるADAMのアイソフォームの内側のディスインテグリンドメインの配列は保存されており、したがって、ADAMタンパク質のヒトファミリー内には、23種のみの異なるディスインテグリンドメインが存在する。組換えによって作出した場合、本発明のMAPは、定義されたインテグリンセットと、高い親和性様式で相互作用することができる。この性質により、これらの変異体ポリペプチドが、臨床的な使用および治療的な使用のための広範なスペクトルのインテグリンリガンドになる。
【0028】
他のヒトADAMメンバーの転写物と同様に、非機能性の転写物は、組換え系において人工的に翻訳された場合に、新規の生物学的機能を有する活性なポリペプチドを生成し得る完全なディスインテグリン配列を含有する。ヒトADAMのディスインテグリンドメインは、76個から86個のアミノ酸を有し(ADAM1のディスインテグリンドメインが最も短く、一方、ADAM10のディスインテグリンドメインが最も長い)、例外が2つあり(ADAM1およびADAM17)、これらは全てADAM足場の標準的なシステイン残基を14個含有する(下記のヒトADAMのアラインメントされた配列を参照されたい)。図1を参照されたい。その大部分がそれらのディスインテグリンループの先端にRGDトリペプチドモチーフを含有する、血小板凝集インヒビターとして機能するように進化したヘビ毒ディスインテグリンとは異なり、ADAMのディスインテグリンループは、それらの先端に非常に異なるトリペプチドモチーフを提示し、したがって、より広範な範囲のインテグリンに、それらのヘビ毒対応物とは異なる様式で関与することが予測される。実際、APのそれぞれは、インテグリン受容体の定義された独特のセットに結合し、したがって、独特の様式でシグナル伝達をすると考えられている(ディスインテグリンループの差異を例示しているADAMおよびヘビ毒ディスインテグリンの配列アラインメントについて図3を参照されたい)。理論に縛られることを望むものではないが、2種以上のMAPの組み合わせを使用して、特定の細胞型または細胞型についての疾患状態に特徴的な「インテグリンサイン(integrin signature)」を決定することができると考えられている。インテグリンサインとは、その細胞型またはその細胞型についての疾患状態に独特である細胞の表面上に存在するインテグリンの組み合わせを意味する。
【0029】
ヒトADAM15のディスインテグリンドメインは、そのディスインテグリンループ内にRGDトリペプチドモチーフを含有し、これは、ヒトADAM15が心臓血管系において重要な調節の役割を果たすという仮説を支持する。このADAM15のRGDトリペプチドモチーフは、図1のAP15に示されている。
【0030】
23種の公知のヒトADAMのメンバーの全てのAP部分のそれぞれについて、MAPを生成した。ヒトADAMディスインテグリンドメイン配列を、上記の理論的根拠に従って修飾した。修飾は、通常、ネイティブなADAMタンパク質におけるドメイン間−ディスインテグリンドメインのジスルフィド架橋の形成に関与するADAMディスインテグリンドメイン内の残基(システイン残基を2つ含むものにおいて)を除去することを含む。理論に縛られることを望むものではないが、これらのジスルフィド架橋の見かけの機能は、ディスインテグリンループをADAM内に緊密にパックしたままにして、インテグリン受容体が利用できないようにすることである。これらのジスルフィド架橋の形成に関与する残基を、例えば、欠失などによって修飾することにより、これらのMAPが、ヘビ毒ディスインテグリンの標準的な11アミノ酸のループの移動性およびディスインテグリンの折りたたみ特性を獲得する。長いサイズのヘビ毒ディスインテグリンおよび中程度のサイズのヘビ毒ディスインテグリンとアラインメントした場合、ならびにPIII−クラスのSVMPとアラインメントした場合、ヒトADAMの23種のメンバーの中で、6種のメンバーが上記のスキームに完全に適合する(ADAM7、ADAM8、ADAM12、ADAM19、ADAM28およびADAM33)(ヘビ毒ディスインテグリンおよびヒトADAMディスインテグリンドメインのアラインメントについては図3を参照されたい)。それにもかかわらず、これらの修飾を導入することにより、4種のADAM(10、17、18および32)の例外を伴い、ヒトADAMのメンバーの全てが、11アミノ酸のディスインテグリンループを提示するMAPに変換された。4種の例外に関しては、3種(ADAM17、ADAM18およびADAM32)は、わずかに長い、12アミノ酸のループを提示するMAPに変換され、一方1種のメンバー(ADAM10)は、わずかに短い、10アミノ酸のディスインテグリンループを保有するMAPに変換された(MAP配列アラインメントについては図2のAP10を参照されたい)。さらに、2種のAP(ADAM1およびADAM17)の場合では、ヘビ毒ディスインテグリンドメインのシステインパターン特性を回復させるために、各配列内の1つの追加的なネイティブな残基をアルギニン残基(MAP1を生成するため)またはシステイン残基(MAP17を生成するため)のいずれかと交換した(MAP配列アラインメントについては図2を参照されたい)。
【0031】
本明細書で使用される場合、「ドメイン間領域」または「スペーサー領域」とは、ADAMのメタロプロテアーゼとディスインテグリンドメインとの間のポリペプチド部分(「MDドメイン間領域」)、およびディスインテグリンドメインとシステインリッチドメインとの間のポリペプチド部分(「DCドメイン間領域」)のそれぞれを意味し、MDドメイン間領域は、APのN末端側の少なくとも10アミノ酸残基で始まり、DCドメイン間領域は、APのC末端側の少なくとも10アミノ酸残基で始まる。各ドメイン間の長さは5〜15アミノ酸である。
【0032】
23種のMAP全てのDNA配列を、新規に合成し、pET32a発現ベクター[30]に、細菌チオレドキシンA(TrxA)の下流にクローニングした。2009年11月12日に出願された、名称が「Method of expressing proteins with disulfide bridges with enhanced yields and activity」であるPCT特許出願第PCT/US09/64256に記載の通り、Origami B(DE3)細菌株にMAPを産生させた。この出願では、キメラのヘビ毒ディスインテグリンビクロスタチン(Vicrostatin(VCN))をOrigami B(DE3)/pET32a系において発現させることを実施形態として含む、米国特許出願公開第20060246541号に開示されている発現系に対する改善が記載されている。改善された方法を使用して、正しく折りたたまれた活性なMAPの量を増加させた。これは、Origami B細胞を選択性の低い環境において成長させ、したがって、異種組換え型のタンパク質の産生を誘導する間に、より最適なレドックス環境を示すVCN形質転換体の生成および増大を可能にすることによって実現された。他のE.coli株とは異なり、Origami Bは、E.coliにおける2つの主要な酸化還元経路の制御に決定的に関与する2つの重要な遺伝子、チオレドキシン還元酵素(trxB)およびグルタチオン還元酵素(gor)に変異を保有させることによって、この細菌の細胞質内の微小環境が、タンパク質におけるジスルフィド架橋の形成に対する触媒状態である、より酸化的なレドックス状態に人工的にシフトしているという点で独特である[18、29]。
【0033】
Origami B株は、野生型E.coli株を用いて得られる成長速度およびバイオマス収率と同様の成長速度およびバイオマス収率を有し、これにより、Origami B株が、VCNのような、発現させることが難しい組換えタンパク質についての魅力的かつスケーラブルな産生の代替になる。この株は、BL21のlacZY変異体からも得られる。BL21のlacY1欠失変異体(元のTuner株)により、培養物中の全ての細胞によるタンパク質の発現のレベルを調整可能にすることができる。lac透過酵素(lacY1)変異により、IPTG(ラクトース誘導体)を集団内の全ての細胞に均一に入れることが可能になり、これにより、制御された、より均一な誘導がもたらされる。IPTGの濃度を調整することにより、標的タンパク質の発現を最適化することができ、有意に低いレベルのIPTGで理論的に最大のレベルを実現することができる。したがって、Origami Bは、BL21(ompTおよびlonプロテアーゼを欠損している)宿主、Tuner(lacZY変異体)宿主およびOrigami(trxB/gor変異体)宿主の望ましい特性を1つの株内で併せ持つ。上記の通り、チオレドキシン還元酵素(trxB)とグルタチオン還元酵素(gor)の両方の変異により、細胞質におけるジスルフィド結合の形成が大きく促進される[29]。
【0034】
Origami B株により、BL21のような細胞質内環境が還元性であるE.coli株を超える明らかな利点が提供されるが(図4および5に、株間の発現レベルの比較が示されている)、単にOrigami B株およびpET32a発現ベクターを使用することでは、可溶性かつ/または活性な産物が生成されることは自動的には保証されない。Origami Bにおけるジスルフィドリッチポリペプチドの生成は、配列依存的であると思われる。例えば、MAP(例えば、MAP9およびMAP15)は、同じ系および産生技法を使用したという事実にもかかわらず、Origami Bにおいて、それらの対応するヒトADAM9およびADAM15のAPバージョンと比較して有意に高い発現収率で発現させることができる(図4および5)。したがって、APを修飾してMAPにした結果、Origami B細胞における発現収率がより高いディスインテグリンドメインを有するポリペプチドがもたらされ得る。
【0035】
さらに、ADAM9およびADAM15の発現したディスインテグリンドメイン(AP)をTEVプロテアーゼ処理およびRP−HPLC精製を伴うプロセスで精製した後、収集される遊離のポリペプチドは、不安定であり、凍結乾燥した粉末から再構成した後に溶液から沈殿するようである。対照的に、同じ精製ステップを用いることによって生成した対応するMAPポリペプチドは、凍結乾燥した後に水で再構成した際に、より可溶性かつ安定であるようである。
【0036】
本発明のMAPは、実質的に単離または実質的に精製されるように調製する。本明細書で使用される場合、MAPに関して、「実質的に精製された」(または単離された)という用語は、絶対的な純度を必要としない。その代わりに、MAPは、好ましくは50%超純粋であるか、より好ましくは少なくとも75%純粋であるか、および最も好ましくは少なくとも95%純粋であるか、少なくとも99%純粋であるか、最も好ましくは100%純粋であるという指標を表す。MAPは、本明細書に記載の通り、合成的に調製することができ、または組換え発現によって調製することができる。
【0037】
「実質的に」という用語は、本明細書で使用される場合、別段の指定のない限り、プラス10%またはマイナス10%を意味する。
【0038】
MAPを含有する薬学的組成物は、最低でも、所望の効果(例えば、がんの成長を阻害するか、またはがんの転移を妨げるかもしくは阻害する)を実現するために有効な量のMAPを含むべきであり、また、緩衝剤、塩、および/または適切なキャリアもしくは賦形剤を含む。一般に、これらの組成物中、MAPは、1日当たり約0.01mg/kg〜約50mg/kg、好ましくは1日当たり約0.1mg/kg〜約5.0mg/kg、および最も好ましくは1日当たり約0.1mg/kg〜約0.5mg/kgをもたらすために十分な量で存在する。
【0039】
MAPは、それを被験体の体内(例えば、血流内)に相当量で送達するために適した、種々のこれまでに公知の手段によって投与することができる。現在、適切な液体ビヒクルまたは賦形剤中のMAPを静脈内投与することが、好ましい投与経路であると考えられている。MAPは水溶性であり、したがって、適切な水溶液(例えば、リン酸緩衝生理食塩水)において有効に投与することができる。あるいは、MAPは、経口的に(適切な結合剤材料または賦形剤材料を用いて処方した錠剤またはカプセル剤の形態で、あるいは水性または油性の懸濁物、溶液、乳剤、シロップ剤またはエリキシル剤の形態で)、または非経口的な懸濁物として投与することができる。MAPは、動脈内または管内に送達することもでき、または意図された作用部位に局所的に導入することができる。当技術分野で周知の通り、アジュバントまたは賦形剤、例えば、局所麻酔薬、保存料、緩衝剤、潤滑剤、湿潤剤、着色料、矯味矯臭薬、充填剤および希釈剤などを、任意のこれらの処方物に適切に含めることができる。
【0040】
MAPは、リポソームによって送達することができる。リポソーム送達は、当技術分野で周知であり、ディスインテグリンの送達について記載されている。例えば、Swensonらは、乳がんの療法のための、リポソームに入れたコントルトロスタチンの静脈内送達の使用について記載している[26]。
【0041】
MAP、例えば、MAP9などは血管新生促進(pro−angiogenic)効果を示す(以下を参照されたい)。理論に縛られることを望まないが、これは、ADAMファミリーのメルトリンクラスのメンバー(ADAM9、ADAM12、およびADAM19)のより一般的な特性を示し得る。メルトリンは、筋肉細胞の分化誘導の過程において発現され、そこで、細胞融合および他のプロセスに関与し、新生血管の安定化において重要な役割を果たし得ることが最初に記載された間葉系のADAMである[33]。したがって、ADAMタンパク質のメルトリンクラスに由来する組換え型のポリペプチドを、疾患、例えば、冠動脈疾患(CAD)または他の虚血性の状態などにおいて、新しい毛細管網目構造の形成を刺激し、側副毛細血管の成長を維持するために治療的に使用することができる。したがって、例えば、MAPは、ステントを利用するCADおよび関連疾患の療法において使用するために、ステントをコーティングすること、またはステントに化学的にカップリングさせることができる。
【0042】
MAPを、薬物、毒素、放射性核種などと直接または間接的に結合体化し、これらの結合体を診断または治療への適用ために使用することもできる。例えば、MAPを使用して、関連のインテグリンを発現している組織または器官を同定または処置することができる。MAPまたはその生物活性のある断片もしくは部分を、検出可能な分子または細胞傷害性分子とカップリングさせ、1つまたは複数の関連のインテグリンを発現している細胞、組織または器官を有する哺乳動物に送達することができる。
【0043】
適切な検出可能な分子を、MAPに直接または間接的に付着させることができ、その適切な検出可能な分子としては、放射性核種、酵素、基質、補因子、インヒビター、蛍光マーカー、化学発光マーカー、磁気粒子などが挙げられる。適切な細胞傷害性分子を、ポリペプチドまたは抗体に直接または間接的に付着させることができ、その適切な細胞傷害性分子としては、細菌毒素または植物毒素(例えば、ジフテリア毒素、シュードモナス外毒素、リシン、アブリンなど)、ならびに治療的な放射性核種、例えば、ヨウ素−131、レニウム−188またはイットリウム−90などが挙げられる(ポリペプチドまたは抗体に直接付着させるか、または、例えば、キレート化部分によって間接的に付着させる)。MAPは、細胞傷害性薬物、例えば、アドリアマイシンなどと結合体化することもできる。検出可能な分子または細胞傷害性分子を間接的に付着させるために、検出可能な分子または細胞傷害性分子を、補体/抗補体対のメンバーと結合体化することができ、ここで、他方のメンバーはポリペプチドまたは抗体部分に結合する(ビオチン/ストレプトアビジンなど)。
【0044】
開示された組成物は、内皮細胞の機能障害に関連する疾患を処置するために、単独で、または組み合わせで使用することができる。ある実施形態では、開示された組成物は、がん処置のために有用である。がんは上皮起源のものであってよい。がんは、乳がん、結腸直腸がん、基底細胞がん、腺がん、胃腸がん、例えば、口唇がん、口腔がん、食道がん、小腸がんおよび胃がんなど、結腸がん、肝がん、膀胱がん、膵がん、卵巣がん、子宮頸がん、肺がん、および皮膚がん、例えば、扁平上皮細胞がんおよび基底細胞がんなど、前立腺がん、腎細胞がん、中枢神経系(CNS)のがん、白血病ならびに全身の上皮細胞に影響を及ぼす他の公知のがんであってよい。理論に縛られることを望むものではないが、がんの多くの形態において、ADAM(膜に繋ぎ止められた形態および/または分泌された形態)とそれらのインテグリン対受容体(integrin counter−receptor)との間に、がんが進行するために機構的に重要なプロセスである病原性のクロストークが存在する。これらの病理学的な相互作用は、がんにおける、腫瘍の進行における以下のいくつかの重要なインテグリン駆動ステップを実行するプロセスにおいて、高度に遊走性の細胞(がん細胞、炎症細胞または内皮細胞)によって組み立てられる多タンパク質複合体(すなわち、インバドソーム(invadosome))にインテグリンを介してADAMのメタロプロテイナーゼをリクルートする働きをする:重要な支持的な役割を果たすことが示されている炎症細胞による腫瘍間質の浸潤、腫瘍血管新生に関連する遊走性のステップ(ここで、既存の血管由来の内皮細胞は、新生血管を組み立てるためにリクルートされる)、および、最後に、転移に関連する遊走性かつ侵襲的な事象。これらの病理学的プロセスをMAPによって破壊することにより、悪性腫瘍が縮小する。それらの、抗血管新生効果、抗炎症効果および抗転移効果の潜在性に加えて、広範なスペクトルの抗インテグリンMAPは、腫瘍の分化にも影響を与える可能性がある。がん幹細胞に正しい分化シグナルを送る能力は、がん療法の主要な目標の1つのままである(療法の過程において、より安定かつ分化した表現型を獲得させることによる)。例えば、Yuanらにより、ヒト多形神経膠芽腫(脳がんの非常に侵攻性の形態)から単離した幹細胞特性を有する細胞集団を操作して、in vitroで分化させ、より良性の特徴をとるようにすることができることが示された[32]。可溶性のインテグリンリガンド(例えば、単一のMAPまたはMAPの組み合わせ)によって分化シグナルがもたらされ、種々のヒト悪性腫瘍のがん幹細胞を、それらの分化系列に完全にコミットさせ(commit)、分化したままになるように誘導することができる。
【0045】
本発明の組成物は、候補化合物を、MAP特異的結合について、MAPの存在下および非存在下で、前記候補化合物のインテグリンへの相対的な結合を比較することによってスクリーニングするためにも使用することができ、ここで、前記MAPの存在下でインテグリンの結合が減少することは、前記MAP特異的インテグリン結合性分子がMAP特異的であることを示す。
【0046】
本発明の組成物は、がん細胞によって提示される特異的な「インテグリンサイン」を利用することによって、がんを早期検出するために使用することができる。「インテグリンサイン」または「インテグリンプロファイル」は、細胞の表面で発現される1つまたは複数のインテグリンである。例えば、がんおよび肉腫は、腫瘍の分化および器官への局在化の状態に基づいて、特異的な「インテグリンサイン」を提示し得る。インテグリンサインに基づいて種々のがんの種類を同定し、診断するために、MAPに特異的な結合を、in vivoにおけるがんイメージング剤(診断的なイメージング)および/またはex vivoにおいて腫瘍検体を染色するための分子ツール(診断的な病理学的方法)として用いることができる。後者の場合、がん検体を様々なMAPで染色することにより、種々の腫瘍の起源および悪性度分類に応じた特徴的な染色パターンを導くことができる。本発明の組成物は、がんを病期分類するため、がんの進行または療法をモニターするために使用することができる。がんは上皮起源のものであってよい。がんは、乳がん、結腸直腸がん、基底細胞がん、腺がん、胃腸がん、例えば、口唇がん、口腔がん、食道がん、小腸がんおよび胃がんなど、結腸がん、肝がん、膀胱がん、膵がん、卵巣がん、子宮頸がん、肺がん、および皮膚がん、例えば、扁平上皮細胞がんおよび基底細胞がんなど、前立腺がん、腎細胞がん、ならびに全身の上皮細胞に影響を及ぼす他の公知のがんであってよい。MAPは、公知のインテグリン親和性を有するMAPが、特定の固形腫瘍のそれぞれの特異的なインテグリンの発現に基づいて、原発腫瘍、転移性の病巣、ならびに腫瘍の新脈管構造(neovasculature)に異なって結合する独特の能力を活用するために、PET(陽電子放出断層撮影法)プローブと一緒に使用することができる。例えば、MAPは、N−スクシンイミジル−4−18F−フルオロベンゾエート(18F−SFB)を最適化されたフッ素化反応条件下で用いることによって、アミノ基を通じて18Fで標識することができ、または、金属キレート化剤であるDOTA/NOTAと結合体化することによって64Cu/68Gaで標識することができる。さらに、蛍光標識したMAPは、in vivoで、およびex vivoで(生検検体上で)、腫瘍インテグリンの発現プロファイル分析のために使用することができる。
【0047】
本発明の組成物は、再生医療における分子ツールとして利用することもできる。例えば、人工的なECM足場(器官の型)をMAPまたはMAPの組み合わせでコーティングすることにより、幹細胞前駆体を誘導してこれらの足場に配置し、望ましい上皮の層および間葉系の層に分化させることができる。例えば、そのような1つまたは複数のMAPを含有する組成物でコーティングした膀胱の足場を使用して、幹細胞前駆体を尿路上皮および筋層の両方にコミットするように向かわせることができる。他の例としては、そのような1つまたは複数のMAPを含有する組成物でコーティングした食道の足場および直腸の足場を使用して、幹細胞前駆体を関連する組織層にコミットするように向かわせることができることが挙げられる。本発明の組成物は、in vitroで胚性幹細胞の成長を支持し、その幹細胞性を維持すること、または同じ系において、これらの全能性細胞がそれらの分化系列にコミットすることをガイドすること、ができる定義された組織培養プレートのコーティングを生み出すためにも使用することができる。
【0048】
本発明の組成物は、種々の虚血性の状態において、毛細血管の側副成長を支持するための新規の薬物、例えば、薬物溶出ステントなどとして使用することができる。例えば、MAPは、放射性核種、例えば、ベータ線放射性核種などと結合体化して、再狭窄を低下させることができる。そのような治療的手法は、放射性の療法を施す臨床医への危険が少ない。
【実施例】
【0049】
(実施例1)
MAPの調製および精製
図6は、pET32a発現ベクターにクローニングした合成のMAPのDNA配列の一覧を示す。図7は、MAPをpET32aベクターにクローニングするために利用した、対応するオリゴヌクレオチドプライマーの一覧を示す。図8は、Origami B(DE3)において発現させた全てのTrxA−MAP構築物のアミノ酸配列を示す。TrxAの活性部位およびディスインテグリンループの先端のトリペプチドモチーフに下線が引かれており、TEV切断部位が囲み枠で強調されており、TrxAと種々のMAP構築物との間のリンカー領域が黒い太字のイタリック体で示されている。MAP1およびMAP17のネイティブな残基と交換するために導入した新しい残基が、太字の二重下線で強調されている。
【0050】
細菌細胞および試薬。Origami B(DE3)E.coli株および細菌チオレドキシンA遺伝子(trxA)を保有するpET32a発現ベクターは、Novagen(San Diego、CA)から購入した。23種のMAP全てのDNA配列は、Epoch Biolabs,Inc.(Sugar Land、TX)によって新規に合成され、プラスミドに挿入された。APのDNA配列は、HUVEC(PromoCell GmbH、Heidelberg、Germany)、MDA−MB−435(ATCC、Manassas、VA)、MDA−MB−231(ATCC、Manassas、VA)、およびジャーカット(ATCC、Manassas、VA)を含めたいくつかの哺乳動物の細胞系から作製したcDNAライブラリーからPCR増幅した。APのDNA配列およびMAPのDNA配列をpET32a発現ベクターにさらにクローニングするために使用したオリゴヌクレオチドプライマーは、Operon Biotechnologies,Inc.(Huntsville、AL)によって合成された。APのDNA配列およびMAPのDNA配列をpET32a発現ベクターにクローニングするために使用した制限酵素およびリガーゼは全て、New England Biolabs,Inc.(Ipswich、MA)から購入した。組換えTEVプロテアーゼは、Invitrogen(Carlsbad、CA)から購入した。
【0051】
MAP発現ベクターの構築および組換え産生。pET32a発現ベクターに、TrxAの下流においてクローニングした合成のMAPのDNA配列が図6A〜Fに列挙されている。MAPをクローニングするために使用したオリゴヌクレオチドプライマーが図7A〜Bに列挙されている。生成した、TrxA遺伝子の下流にMAPのDNA配列がクローニングされたpET32aプラスミドを、最初にDH5α E.coliにおいて増幅し、精製し、配列決定した後、Origami B(DE3)E.coliに移入した。次いで、各MAP構築物について形質転換された細胞を、カルベニシリン(50μg/mL)、テトラサイクリン(12.5μg/mL)、およびカナマイシン(15μg/mL)を補充したLB寒天に播き、37℃で一晩成長させた。これらのプレートから、形質転換されたOrigami Bの個々のコロニー由来の各MAP構築物について、これらのコロニーを、カルベニシリン(50μg/mL)を含有するLB培地に移すことによって、複数の培養物を確立した。これらの最初の培養物を一晩成長させ、さらに、より大きな容積のカルベニシリン(50μg/mL)を含有するLB培地に接種するために使用し、それをシェーカーインキュベーター内、37℃、250rpmで、OD600が0.6〜1に到達するまで成長させた。この時点で、個々のMAP培養物由来の細胞を、1mMのIPTGを用いて誘導し、さらに4〜5時間、37℃、250rpmでインキュベートした。誘導期間の最後に、個々のMAP培養物由来の細胞を、4000×gでペレットにし、マイクロフルダイザー(Microfluidics M−110L、Microfluidics、Newton、MA)で溶解した。マイクロフルダイザーの作動条件は、適用圧力が14,000〜18,000psiであること、細菌のスラリー流速が1分当たり300〜400mlであること、およびスラリーを、プロセッサを複数回通過させることを含んだ。個々のMAP培養物から処理された溶解物由来の不溶性の細胞の破片を、遠心分離によって(40,000×g)除去し、各MAP培養物について、Trx−MAPを含有する可溶性の材料を収集した。次いで、収集した可溶性の溶解物中の発現された融合タンパク質(すなわち、Trx−MAP)を、組換えTEVプロテアーゼと一緒に室温で一晩インキュベートすることによって、タンパク質分解し、それによって、SDS−PAGEによってモニターされた通り、個々のMAPが、そのTrxA融合パートナーから効率的に切断された。タンパク質分解が完了したら、タンパク質分解された溶解物を、0.22μmのフィルターを通過させ、2回蒸留したH2Oにおいて1:100に希釈し、50,000MWCOのカートリッジ(Biomax50、Millipore)を通して限外濾過し、次いで、接線流限外濾過デバイス(Labscale TFF system、Millipore)を使用して5,000MWCOのカートリッジ(Biomax5、Millipore)に対して再濃縮した。
【0052】
MAPについて上記されているのと同じ手順を用いて、APをpET32aにクローニングし、Origami Bに導入して形質転換し、発現させた。
【0053】
組換えMAPの精製。MAPを、高速液体クロマトグラフィー(HPLC)手順を、ヘビ毒ディスインテグリンに対して以前確立されたプロトコールに従って用いることによって、濾過された溶解物から精製した[2]。ネイティブなCNを精製するために以前使用された標準の溶出条件を用いて、C18逆相HPLCによって精製を実施した[2]。上記の通り処理した個々の濾過された溶解物を、Vydac C18カラム(218TP54、Temecula、CA)にローディングした。カラムを、0.1%のTFAを含有する水溶液で10分間リンスし(1分当たり5ml)、その後、80%のアセトニトリルおよび0.1%のTFAを含有する移動相において、直線勾配(0〜100%)で150分にわたって溶出した。MAPは、35〜40%のアセトニトリルで溶出し始めた。
【0054】
MAPの発現分析。振とうフラスコ中、37℃で一晩成長させたE.coli形質転換体を、1mMのIPTG中、37℃、250rpmで5時間誘導した。誘導期間の最後に、細胞を4,000×gでペレットにし、複数の凍結融解サイクルによって溶解し、さらに40,000×gで遠心分離して不溶性の細胞破片を除去した。種々のE.coli宿主由来の可溶性の細胞溶解物5μlを、還元条件下で、プレキャスト4〜20%のNuSep iGel(NuSep Inc.、Lawrenceville、GA)にローディングし、次いでクーマシー(Coomassie)染色した。
【0055】
得られたAP9およびAP15の発現収率は、それらの対応するMAP(MAP9およびMAP15)と比較して低かった(図4および5)。MAP9およびMAP15はどちらも、Origami B(DE3)において、細菌培養物1リットル当たりのHPLC精製されたタンパク質が200mgから350mgであるバッチ間の発現収率で生成した。これらの高収率の精製組換えMAPは、誘導ステップの最後に、ペレットにした細菌の形質転換体を、マイクロフルダイザーを用いて溶解することによって実現された。同様の作出技法により、Origami B(DE3)系において組換えMAPを同様の発現収率で生成した(図9〜11)。対照的に、同様にジスルフィド架橋の形成を支持する、密接に関連する細菌宿主におけるMAPの発現によっては(例えば、trxB変異のみを有するK12誘導体であるAD494(DE3)、または、まれなコドンの使用について最適化されたOrigami B誘導体であるRosetta−gami B(DE3))、Origami B(DE3)における産生に対する追加的な利点は提供されないと思われた。
【0056】
(実施例2)
MAPの異なる細胞結合
上記の方法を用いてOrigami B(DE3)において産生されたTrx−MAPを、複数の細胞株への異なる結合について、フローサイトメトリーによって分析した(図12〜15)。それは、ヒト乳がん株(MDA−MB−231)、および異なるインテグリンプロファイルに基づく、異なる器官に対する向性を有するこの株の2つの転移性のサブクローン−骨−ホーミングサブクローン(MDA−MB−231BONE)および脳−ホーミングサブクローン(MDA−MB−231BRAIN)を含んだ。MDA−MB−231株の2種のサブクローンは、最初にそれらを単離した研究者からの寄贈品であった[31]。細胞を、示されているTrx−MAPと一緒にインキュベートし、対応する抗Trxポリクローナル抗血清(Sigma−Aldrich,Inc.、St.Louis、MO)を用いて調べた。結合した分子を、抗ウサギFITC−標識した抗体を用いてさらに検出した。FITCで標識した二次抗体のみ、または抗Trx抗血清および二次抗体のいずれかと一緒にインキュベートした細胞を対照として使用した。様々な系列の免疫細胞で発現され、免疫応答に関与することが以前示された、ADAM8およびADAM28に対応するTrx−MAP8およびTrx−MAP28は、フローサイトメトリー分析によって示された通り、ジャーカット細胞(T細胞白血病細胞株)に優先的に結合する(図15)。さらに、2つのHPLCによって精製したMAP(MAP9およびMAP15)をFITCで標識し、がん細胞株(MDA−MB−231、MDA−MB−435)、ヒト神経膠芽腫(GBM−CSC)から単離されたがん幹細胞集団[32]、ならびにヒト臍帯静脈内皮細胞(HUVEC)への直接的な結合についてフローサイトメトリーによって分析した。フローサイトメトリーのデータ(図16)は、精製されたMAP9およびMAP15が、全く異なるインテグリンプロファイルを示すことが予測される、異なる細胞株に非常によく結合することを示している。
【0057】
(実施例3)
MAPの抗血管新生効果
次いで、in vitro HUVEC管形成アッセイを用いて、MAPを血管新生活性について試験した。HUVEC細胞を、10nMのMAP9またはMAP15のいずれかの存在下で「内皮細胞管形成」プレート(BD Biosciences)に播いた。公知の管形成インヒビター(スラミン)を陰性対照として使用した。図17を参照されたい:パネルA−無処置の対照;パネルB−100μMのスラミン;パネルC−10nMのMAP9;パネルD−10nMのMAP15。細胞をCalcein AMで染色し、共焦点顕微鏡を使用してイメージングした。イメージは全て、同じ倍率で取得した(スケールバー=50μm)。このアッセイにおいて、MAP15は有意な抗血管新生活性を示した。このアッセイにおいて、MAP9は、無処置の対照と比較したときに、増加した数の管の形成を導くことによって血管新生促進効果を有すると思われた。このMAP9の血管新生促進効果は、MAP9のリポソーム処方物をMDA−MB−231異種移植の動物モデルにおいて静脈内に投与すると、腫瘍の成長が促進されること(すなわち、無処置の対照と比較して、速い腫瘍の成長、大きな腫瘍および生存の減少)を示すin vivoにおける観察によって支持される。
【0058】
図18Aは、MDA−MB−231モデルにおける、様々な処置によって誘導される腫瘍の成長の阻害を示す。同所性に接種したヌードマウス(乳房の脂肪パッド;完全なマトリゲル(Matrigel)中、マウス1匹当たり2.5×106個のMDA−MB−231細胞)について、処置を開始する前に、触知できる腫瘍を成長させた(矢印によって示される)。動物の群(n=10)に、LMAP15を1回の注射当たり100μgのMAP15の用量当量で週2回静脈内に投与する処置、または、アバスチン(1回の注射当たり400μg;およそ20μg/gr)を1週間に1回静脈内に投与する処置、または、ドセタキセル(DTX、160μg)を1週間に1回腹腔内に投与する処置、またはこれらの作用剤の組み合わせの処置を行った。対照群には、空のリポソームのみを与えた。対照群と比較して、全ての処置群において腫瘍の成長に有意な遅延が観察された。統計分析を、ダネットの事後多重比較検定を用いたANOVAを使用して行った(*は、P<0.001を示す)。図18Bは、動物の生存データを示す。処置群は、対照群と比較して生存の増加を示した(対照の動物は全て7週目までに死亡した)。LMAP15で処置した群またはLMAP15およびアバスチンで処置した群のいずれかの生存が最も高かった。この動物モデルからのin vivoにおける効力データは、LMAP15が、アバスチンまたはドセタキセルのいずれとも同様の抗腫瘍の効力を示すことを示している。リポソームMAP15単独で処置した動物は、同じ異種移植モデルにおいてアバスチンもしくはドセタキセルのいずれかで処置した動物、または組み合わせ療法で処置した動物と比較して、良好な生存率(すなわち、試験終了時にまだ生存している動物の数)を示した。
【0059】
(実施例4)
LMAP15の抗血管新生効果
単独でまたは組み合わせて投与されたLMAP15の抗血管新生効果を評価するために、MDA−MB−231試験における各群からの腫瘍を、死亡した動物または屠殺した動物から解剖し、摘出し、その後、微小血管の密度について免疫組織学的検査によって分析した。摘出した腫瘍をTissue−Tek O.C.T(「Optimal Cutting Temperature」化合物、Sakura Finetek USA)に包埋し、次いで、ドライアイス中で凍結させ、5ミクロンの切片に切り、アセトン中に固定し、染色するまで4℃で貯蔵した。CD31を染色するために、アセトンで固定したスライドをPBSで洗浄し、5%ヤギ血清を含有するPBSにおいてブロッキングし、次いで、PBS中に1:50に希釈したラットポリクローナル抗CD31抗体(BD Biosciences、San Diego、CA)200μlと一緒に室温で一晩インキュベートし、製造者のプロトコールに従ってスライドに適用した。この後、PBS中で複数回洗浄し(1回の洗浄当たり7分間)、1:100に希釈したビオチン化ヤギ抗ラット二次抗体200μlを加え、室温で45分間適用した。PBSでさらに3回洗浄した後、2mlのPBS中1滴に希釈したアビジン結合複合体(Vector Laboratories、Burlingame、CA)200μlを各スライドに室温で30分間適用し、その後、抗体で染色した微小血管を可視化するために、3−アミノ−9−エチルカルバゾール色素原を加えた。PBSでさらに3回洗浄した後、スライドを、Myers Hematoxylinを用いて対比染色し、その後マウントした。CD31で染色された微小血管を定量化するために、スライドを、「ランダム視野(random field)」分析[35、36]に供した。イメージを、顕微鏡に取り付けたOlympus E20Nデジタルカメラ(Olympus America、Melville、NY)を使用して、200×で、各スライドにおいてランダム視野から盲検的に取り込んだ。各群について、4つの腫瘍を染色し、40のランダム視野を分析した。各ランダム視野についてCD31陽性領域を、「SimplePCI」高度イメージングソフトウェア(C−Imaging Systems、Cranberry Township、PA)を使用して、染色された全面積の%としてコンピュータで計算し、次いで、各群について平均した。偏りを排除するために、ランダム視野のイメージを取り込み、その後、取り込んだイメージの処理および分析を盲検的に行った。LMAP15は、単独療法として、または異なる作用機構を有する他の抗血管新生薬(例えば、アバスチン)もしくは化学療法薬(例えば、ドセタキセル)と組み合わせて投与されると、この異種移植モデルにおいて微小血管の密度を有意に低下させることが示された(図19)。
【0060】
(実施例5)
転移性乳がんモデル
MAPの抗侵襲薬/抗転移薬としての治療効果を評価するために、自然発生的な転移性乳がんのいくつかの動物モデルにおいて光学的なルシフェラーゼイメージング手法(in vivo生物発光)を使用する。以下の細胞株に、アデノウイルス形質導入系を安定に感染させた:ルシフェラーゼおよび緑色蛍光タンパク質(GFP)レポーター遺伝子の両方を伴う、ヒト乳がん細胞株(MDA−MB−231、トリプル陰性細胞株)および2種のマウス乳がん株(4T1、HER2陰性株、およびD2F2、HER2陽性株)。ヒト異種移植MDA−MB−231モデルでは、完全なマトリゲルに懸濁させた細胞2×106個の接種材料をヌードマウスの乳房の脂肪パッドに注射し、形成された腫瘍が触知できるまで成長させた(移植のおよそ2週間後)。以下の処置群(1群当たり動物5匹)を形成する:対照群1(処置または操作を受けない動物)、対照群2(空のリポソームを静脈内に与えた動物)、対照群3(PBSを静脈内に与えた動物)、処置群1(アバスチンを静脈内に与えた動物)、処置群2(ドセタキセルを腹腔内に与えた動物)、処置群3(静脈内へのアバスチンと腹腔内へのドセタキセルの組み合わせを与えた動物)、処置群4(MAP15を静脈内に与えた動物)、処置群5(MAP15のリポソーム処方物を静脈内に与えた動物)、処置群6(静脈内へのMAP15、静脈内へのアバスチンおよび腹腔内へのドセタキセルの組み合わせを与えた動物)、および処置群7(静脈内へのMAP15のリポソーム処方物、静脈内へのアバスチンおよび腹腔内へのドセタキセルの組み合わせを与えた動物)。処置は、以下の通り施す:アバスチンおよびドセタキセルは、以前に文献において、このモデルにおいて原発腫瘍および転移病巣に対して効果的であると報告された最大の投与量を用いて週に1回投与し、MAP15は、1回の注射当たりポリペプチド100μgの用量で1日おきに投与し、リポソームMAP15は、1回の注射当たりポリペプチド100μgの用量当量で週2回投与する。
【0061】
MDA−MB−231モデルと同様に、2つのマウス乳がんモデル(4T1モデルおよびD2F2モデル)においてもMAP15の抗侵襲薬/抗転移薬としての効力を決定する。これらのモデルにおいて、PBS中の細胞5×105個の接種材料(4T1またはD2F2)を免疫応答性のBALB/cマウスの乳房の脂肪パッドに注射し、腫瘍が触知できるようになるまで成長させた(移植後およそ1〜1.5週間)。腫瘍が触知できるようになったら、以下の処置群(1群当たり動物5匹)について処置を開始する:対照群1(処置または操作を受けない動物)、対照群2(空のリポソームを静脈内に与えた動物)、対照群3(PBSを静脈内に与えた動物)、処置群1(アバスチンを静脈内に与えた動物)、処置群2(ラパチニブを静脈内に与えた動物)、処置群3(ハーセプチンを静脈内に与えた動物)、処置群4(アバスチン、ラパチニブおよびハーセプチンの組み合わせを静脈内に与えた動物)、処置群5(MAP15を静脈内に与えた動物)、処置群6(MAP15のリポソーム処方物を静脈内に与えた動物)、処置群7(MAP15、アバスチン、ラパチニブ、およびハーセプチンの組み合わせを静脈内に与えた動物)、および処置群8(MAP15のリポソーム処方物、アバスチン、ラパチニブ、およびハーセプチンの組み合わせを静脈内に与えた動物)。処置は、以下の通り施す:アバスチン、ラパチニブおよびハーセプチンは、以前に文献において、これらのモデルにおいて原発腫瘍および転移病巣に対して効果的であると報告された最大の投与量を用いて週に1回投与し、MAP15は、1回の注射当たりポリペプチド100μgの用量で1日おきに投与し、リポソームMAP15は、1回の注射当たりポリペプチド100μgの用量当量で週2回投与する。
【0062】
処置を開始する前、および処置を開始した後の、乳房の脂肪パッドおよび遠位の部位における腫瘍の成長を、全てのモデルにおいて、週に1回のXenogen生物発光イメージングによってモニターする。原発腫瘍サイズも、カリパスによって測定し、体積を式:1×w2×0.5に基づいて算出する。週に1回の生物発光(ルシフェラーゼ)イメージングのために、マウスに、ルシフェリン溶液(PBS中15mg/mLまたは30mg/kg、5〜50mg/kgの用量)を腹腔内経路によって注射し、約5〜15分間、麻酔していない動物において分布させる。次いで、マウスを、妨げられることなく動物を目視検査し、それらの呼吸の状況をモニターすることを可能にする、透明なPlexiglas麻酔箱(2〜4%のイソフルラン(isofluorane))に置く。この設定では、麻酔を箱に供給する麻酔送達管を、Xenogen IVIS 100イメージング計器のイメージングチャンバーの内側に位置する麻酔多岐管に同じ濃度の麻酔が送達されるように分離する。マウスを完全に麻酔した後、マウスを麻酔箱からイメージングチャンバー内の多岐管に取り付けた麻酔ノーズコーン(nose cone)に移し、扉を閉め、Xenogen計器を使用してイメージを取得する。イメージングの時間は、実験に応じて、面(背面/腹面)当たり1〜5分である。動物を背面から腹面に回転させる際(逆もまた同じ)、苦痛または生命力の変化の任意の徴候についてモニターする。取得したイメージを、対照および処置した腫瘍の部位におけるルシフェラーゼ活性のレベルを比較することによって、および各処置群における腫瘍からの発光に関連する曲線下分布面積を比較することによって評価する。上記の手法を用いることによって、原発腫瘍および生物発光の転移病巣に対するMAP15の効力を決定する。動物の体重も週2回測定し、動物を、ストレス応答または倦怠感の任意の徴候について視覚的に観察する。障害の重篤な徴候を示した動物は実験から排除する。
【0063】
これらの試験の終わりに(MDA−MB−231モデルにおける処置の8週間後、および4T1モデルおよびD2F2モデルにおける処置の3週間後)、これらの腫瘍モデルにおいて原発腫瘍の成長を阻害すること、転移病巣の数およびサイズまたはその両方を低下させること、および動物の生存を延長することにおいて、MAP15が、単独で、試験した単独療法のいずれか(アバスチンまたはドセタキセルまたはラパチニブまたはハーセプチンのいずれか)と同じくらい効果的であるか、またはそれよりも効果的であるかの決定を行う。さらに、MDA−MB−231モデルにおいてアバスチンとドセタキセルと組み合わせて、または4T1モデルにおいてアバスチンとラパチニブと組み合わせて、またはD2F2モデルにおいてアバスチンとラパチニブとハーセプチンと組み合わせて投与すると、MAP15は、これらの腫瘍モデルにおいて原発腫瘍の成長を阻害すること、転移病巣の数およびサイズまたはその両方を低下させること、および生存を延長することにおいて、任意の単独療法または他の組み合わせよりも効果的であることが証明された。
【0064】
(付属物)
ADAMの核酸配列およびアミノ酸配列
ADAMのRNA転写物に対応する核酸配列が図20〜42に提供されている。対応するADAMポリペプチドのアミノ酸配列が図43に提供されている。
【0065】
特に定義されていなければ、本明細書において使用される全ての技術用語および科学用語は、本発明が属する分野の当業者に一般に理解されているものと同じ意味を有する。
【0066】
本明細書に例示的に記載されている本発明は、本明細書において具体的に開示されていない、任意の要素、限定が存在しないところで適切に実施することができる。したがって、例えば、「含む(comprising)」、「含む(including)」、「含有する(containing)」などの用語は、限定されることなく、広く読み取られるべきである。さらに、本明細書で使用される用語および表現は、説明する用語として使用され、限定するものではなく、また、そのような用語および表現の使用において、示され、記載されている特徴の等価物またはその部分のいずれも排除するものではなく、特許請求の範囲に記載された本発明の範囲内で種々の改変が可能であることが理解される。
【0067】
したがって、本発明は、好ましい実施形態および任意選択の特徴によって具体的に開示されているが、当業者は、本明細書の開示の中で具体化される本発明の改変、改善および変形を行うことができること、およびそのような改変、改善および変形は、本発明の範囲内であるとみなされることが理解されるべきである。本明細書で提供される材料、方法および実施例は、好ましい実施形態を表し、例示的であり、本発明の範囲を限定するものではない。
【0068】
本明細書では、本発明が広範かつ一般的に記載されている。包括的な開示の範囲に入る狭義の種および下位のグループ分けのそれぞれも本発明の一部を形成する。これは、削除される材料が本明細書において具体的に列挙されているか否かにかかわらず、類概念から任意の対象物を取り除く但し書きまたは否定的な限定を伴う本発明の包括的な説明を包含する。
【0069】
本明細書において言及されている全ての刊行物、特許出願、特許、および他の参考文献は、全ての式および図面を含めて、それぞれが、個別に参照により組み込まれる場合と同じ程度に、明白にその全体が参照により本明細書に組み込まれる。矛盾する場合は、定義を含めた本明細書が優先する。
【0070】
他の実施形態は、以下の特許請求の範囲内に記載されている。
【0071】
【数1】
【0072】
【数2】
【0073】
【数3】
【0074】
【数4】
【技術分野】
【0001】
(関連出願への相互参照)
本出願は、2010年2月11日に出願された米国仮出願番号61/303,631号の米国特許法119条(e)項の下での優先権の利益を主張し、上記米国仮出願番号61/303,631号は、全ての図面および表を含めてその全容が、参考として本明細書に援用される。
【0002】
(発明の分野)
本発明は、多機能のプロテアーゼのADAM(ディスインテグリンおよびメタロプロテイナーゼ(A Disintegrin and Metalloproteinase))哺乳動物のファミリーに由来する、遺伝子改変(engineered)されたポリペプチドのクラス、およびそれを作製する方法に関する。本発明は、これらの遺伝子改変されたポリペプチドの、抗血管新生活性および抗腫瘍成長活性についての使用にも関する。本発明は、遺伝子改変されたポリペプチドを、内皮細胞の機能障害およびインテグリンに関連する状態を診断するために投与することにも関する。
【背景技術】
【0003】
(発明の背景)
本発明は、Mineaらによる、発明の名称が「Method of expressing proteins with disulfide bridges」である特許文献1、および2009年11月12日に出願された、発明の名称が「Method of expressing proteins with disulfide bridges with enhanced yields and activity」であるPCT特許出願番号PCT/US09/64256に関連する。そのどちらの内容も、全ての図面も含めて、参照により本明細書に組み込まれる。
【0004】
ADAMは、胚発生のあらゆるステップ(そこでは、細胞増殖、細胞遊走、細胞の特異化、軸索伸長および器官の形態形成を制御する)ならびに成人期における多数の生理的プロセスおよび病理学的プロセス(創傷治癒から種々の炎症プロセスまで、ならびに血管新生および転移から器官の修復および再生まで)に関与する多ドメインの哺乳動物の膜貫通タンパク質または分泌タンパク質のクラスである[非特許文献1〜5(1〜5)]。多数の報告により、ADAMファミリーの多くのタンパク質分解性のメンバーがヒト悪性腫瘍において過剰発現されていると思われることが示されており、これは、これらのプロテアーゼが腫瘍の進行において重要な役割を果たす可能性があることを示している[非特許文献6〜15(6〜15)]。興味深いことに、触媒として活性なADAMの過剰発現は、一般に、がんにおける転帰不良に関連づけられると思われるが、非タンパク質分解性のADAMは、腫瘍インヒビターとしての役割を果たすと思われる。例えば、ヒト神経膠腫におけるADAM22の過剰発現は、腫瘍の成長阻害と相関することが示されている[16]。
【0005】
ADAMタンパク質の構造は、それらのオルソログである、PIII−クラスのヘビ毒メタロプロテイナーゼの構造とよく似ている。PIII−クラスのヘビ毒メタロプロテイナーゼ(SVMP)と同様に、ADAMは、メタロプロテアーゼドメイン、ディスインテグリンドメインおよびシステインリッチドメインを保有する多ドメインのタンパク質である[17、28]。ADAM足場は、いくつかのドメイン間のシステイン残基およびドメイン内のシステイン残基を含有し、とりわけ、ディスインテグリンループと称される構造要素の先端における、ディスインテグリンドメイン内に位置するものが非常に重要である。ドメイン間のシステイン残基は、メタロプロテアーゼとディスインテグリンドメインとの間およびディスインテグリンとシステインリッチドメインとの間のスペーサー領域内に位置する[20]。ADAMおよびPIII−SVMPにおいて見いだされるが、一般に、ヘビ毒ディスインテグリンにおいては見いだされないこれらのシステイン残基は、ADAMおよびPIII−SVMPにおいて、ドメイン間(スペーサー)領域をメタロプロテアーゼドメイン、ディスインテグリンドメインおよびシステインリッチドメインと連結するジスルフィド架橋を形成する[20]。これらのADAM足場におけるスペーサー−ドメインジスルフィド架橋は、3つのドメインを一緒に緊密に折りたたまれた構造にロックする構造要素を示し、これらの多ドメインタンパク質が、細胞外の環境において良好に生存する(すなわち、タンパク質分解性の攻撃などに対する抵抗性が増す)ことを可能にしている。この安定化されたADAM足場は、ジスルフィド結合の遺伝子改変によって新しい折りたたみのPII−クラスのSVMPに自然に進化し、これは、タンパク質分解性の攻撃を伴う機構によって遊離のディスインテグリンを生じる。分子進化の結果として、ADAM足場に特徴的なシステイン残基のいくつか(すなわち、スペーサー−ドメインジスルフィド架橋に関与するシステイン残基)が、異なる残基に変異したか、または欠失した。これらの変異の結果、これらの新しいタンパク質(PIIクラスのSVMP)において、スペーサー領域と、メタロプロテアーゼドメインおよびディスインテグリンドメインとの間にジスルフィド架橋をもはや形成することができなくなり、これにより、それらのドメイン間領域は、タンパク質分解を受けやすくなり、個々のドメインがヘビ毒ディスインテグリンの遊離のドメインとして放出される可能性が与えられる[20]。したがって、PIIヘビ毒ディスインテグリンは、それらが血小板に特異的なインテグリンアルファIIbベータ3(alphaIIbbeta3)との相互作用の親和性が高いことによって、最も強力な天然の血小板凝集インヒビターとなっている、遊離のディスインテグリンドメインポリペプチドのクラスとして明らかになった。ADAM足場のディスインテグリンドメインと対照的に、これらの後期進化のPIIクラスのSVMPは、血液毒性のヘビの毒液において遊離のポリペプチドとして放出され、かつ新規の11アミノ酸ディスインテグリンループを保有する。このループは、このクラスの分子に特徴的な独特の構造要素であり、自然に遺伝子改変されて、細胞外マトリックスタンパク質モチーフの作用を模倣することによって強力な可溶性のインテグリンリガンドとしての機能を果たしている(ECM−模倣物)[19]。このループは、ジスルフィド安定化されたポリペプチドコアから自由に突出し、ループの先端に提示されるトリペプチドモチーフ(通常Arg−Gly−Aspモチーフ)を介してインテグリン受容体と相互作用する[21]。
【0006】
ヒトインテグリンファミリーには24種のメンバーが存在し、そのうちの23種は複雑な発現パターン、組織分布および生理機能を有するが、アルファIIbベータ3インテグリンは、血餅の形成に役立つ、血小板に特異的な受容体である。異常に発現されること、または原形質膜に誤って局在化すること、または不適切に活性化されること、またはこれらの機構の組み合わせのいずれかによって、種々の組織または器官において調節解除された場合に、これらの受容体の多くが異常に機能することが、新形成から炎症性疾患まで、ならびに創傷治癒および組織再生などの複雑な生理応答までにわたる多様な病態に関連づけられている[22〜25]。
【0007】
ヘビ毒ディスインテグリンは、リポソーム処方物で送達される場合、抗がん剤としての治療的な可能性を有する[21、26、27]。それらの天然の生物活性(すなわち、インテグリンアルファIIbベータ3との相互作用の親和性が高いことによる血小板凝集の阻害)に加えて、ヘビ毒ディスインテグリンは、腫瘍関連プロセス、例えば、転移および血管新生などを、これらのプロセスの病因に機構的に関わるインテグリンの定義されたセット(例えば、アルファvベータ3(alphavbeta3)、アルファvベータ5(alphavbeta5)、およびアルファ5ベータ1(alpha5beta1))に関与する能力によって破壊することが示されている[21]。これらの好都合な特質にもかかわらず、これらのポリペプチドは、依然として、それらがヘビ毒に由来することに起因する、潜在的に負の免疫学的特性を保有する。遊離のディスインテグリンは、ヒトまたは任意の他の哺乳動物において存在することは知られていない−これまでに同定された唯一のヒトディスインテグリンは、ADAMファミリータンパク質のメンバーの大きな配列の中に埋もれたサブドメインとして存在する[17]。哺乳動物界において30種を超えるADAMタンパク質が同定されており(ヒトは20種の遺伝子および3種の偽遺伝子を保有する)、それらの全てが、ディスインテグリンドメインを保有する[1]。ヒトにおいて同定された23種のADAM転写物のうち、3種が偽遺伝子由来(ADAM1、ADAM3およびADAM6)であり、それは機能タンパク質に翻訳されない。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】米国特許出願公開第20060246541号明細書
【非特許文献】
【0009】
【非特許文献1】Edwards, D.R., M.M. Handsley,およびC.J. Pennington, The ADAM metalloproteinases. Mol Aspects Med, 2008. 29(5): p. 258−89.
【非特許文献2】Mochizuki, S.およびY. Okada, ADAMs in cancer cell proliferation and progression. Cancer Sci, 2007. 98(5): p. 621−8.
【非特許文献3】Reiss, K., A. Ludwig,およびP. Saftig, Breaking up the tie: disintegrin−like metalloproteinases as regulators of cell migration in inflammation and invasion. Pharmacol Ther, 2006. 111(3): p. 985−1006.
【非特許文献4】Tousseyn, T., et al., (Make) stick and cut loose−−disintegrin metalloproteases in development and disease. Birth Defects Res C Embryo Today, 2006. 78(1): p. 24−46.
【非特許文献5】Arribas, J., J.J. Bech−Serra,およびB. Santiago−Josefat, ADAMs, cell migration and cancer. Cancer Metastasis Rev, 2006. 25(1): p. 57−68.
【非特許文献6】Blanchot−Jossic, F., et al., Up−regulated expression of ADAM17 in human colon carcinoma: co−expression with EGFR in neoplastic and endothelial cells. J Pathol, 2005. 207(2): p. 156−63.
【非特許文献7】Lendeckel, U., et al., Increased expression of ADAM family members in human breast cancer and breast cancer cell lines. J Cancer Res Clin Oncol, 2005. 131(1): p. 41−8.
【非特許文献8】Mazzocca, A., et al., A secreted form of ADAM9 promotes carcinoma invasion through tumor−stromal interactions. Cancer Res, 2005. 65(11): p. 4728−38.
【非特許文献9】McGowan, P.M., et al., ADAM−17 predicts adverse outcome in patients with breast cancer. Ann Oncol, 2008. 19(6): p. 1075−81.
【非特許文献10】McGowan, P.M., et al., ADAM−17 expression in breast cancer correlates with variables of tumor progression. Clin Cancer Res, 2007. 13(8): p. 2335−43.
【非特許文献11】Mitsui, Y., et al., ADAM28 is overexpressed in human breast carcinomas: implications for carcinoma cell proliferation through cleavage of insulin−like growth factor binding protein−3. Cancer Res, 2006. 66(20): p. 9913−20.
【非特許文献12】Najy, A.J., K.C. Day,およびM.L. Day, ADAM15 supports prostate cancer metastasis by modulating tumor cell endothelial cell interaction. Cancer Res, 2008. 68(4): p. 1092−9.
【非特許文献13】O’Shea, C., et al., Expression of ADAM−9 mRNA and protein in human breast cancer. Int J Cancer, 2003. 105(6): p.754−61.
【非特許文献14】Ringel, J., et al., Aberrant expression of a disintegrin and metalloproteinase 17/tumor necrosis factor−alpha converting enzyme increases the malignant potential in human pancreatic ductal adenocarcinoma. Cancer Res, 2006. 66(18): p. 9045−53.
【非特許文献15】Wildeboer, D., et al., Metalloproteinase disintegrins ADAM8 and ADAM19 are highly regulated in human primary brain tumors and their expression levels and activities are associated with invasiveness. J Neuropathol Exp Neurol, 2006. 65(5): p. 516−27.
【発明の概要】
【課題を解決するための手段】
【0010】
(発明の概要)
本明細書では、以下のアミノ酸残基:
a)Cys−Asp−Cys(CDC)モチーフのC末端側の最初のシステイン;
b)その最初のシステインのC末端側の2つの連続したアミノ酸;および
c)トリペプチドモチーフのC末端側のシステイン
において修飾されているADAM由来のポリペプチド(AP)を有する、修飾されたADAM由来のポリペプチド(MAP)であって、アミノ酸残基の修飾が、独立に、
a)欠失;
b)置換;および
c)化学的な修飾
から選択され、APが、(i)Cys−Asp−Cys(CDC)モチーフ、(ii)図1に示されているトリペプチドモチーフを有し、(iii)ADAMのメタロプロテアーゼドメイン、システインリッチドメイン、およびドメイン間のセグメントの全て、または実質的に全てを欠く、ADAM由来のディスインテグリン(distintegrin)様ドメインを有し、ADAMが、ADAM17(CDCモチーフではなくCDPを有するADAM17など)ではない、修飾されたADAM由来のポリペプチド(MAP)の組成物、およびそれに関連する方法が提供される。本明細書では、これらのMAPをコードする核酸が提供される。
【0011】
MAPは、ADAM(ディスインテグリンおよびメタロプロテイナーゼ)から得ることができ、ADAMとしては、ADAM15、ADAM28、ADAM1、ADAM2、ADAM3、ADAM6、ADAM7、ADAM8、ADAM9、ADAM10、ADAM11、ADAM12、ADAM18、ADAM19、ADAM20、ADAM21、ADAM22、ADAM23、ADAM29、ADAM30、ADAM32、ADAM33が挙げられる。これらの対応するMAPは、MAP15、MAP28、MAP1、MAP2、MAP3、MAP6、MAP7、MAP8、MAP9、MAP10、MAP11、MAP12、MAP18、MAP19、MAP20、MAP21、MAP22、MAP23、MAP29、MAP30、MAP32、MAP33であってよい。
【0012】
本明細書では、MAPとチオレドキシンのN末端セグメントの融合タンパク質およびこれらの融合物をコードする核酸も提供される。
【0013】
MAPは、HUVECまたはMDA−MB−435細胞の再構成基底膜を通じた移動を阻害する性質、MDA−MB−435細胞におけるFAKのリン酸化のレベルを上昇させる性質、または培養物におけるHUVECの管形成を阻害する性質を有し得る。
【0014】
本明細書では、MAPを発現させる発現ベクターおよびこれらの発現ベクターを用いて形質転換した原核宿主細胞も提供される。発現ベクターは、誘導性の制御下にあってもよく、例えば、ここで、宿主は、チオレドキシン還元酵素B(trxB)遺伝子および/またはグルタチオン還元酵素(gor)遺伝子に安定な変異も有する。
【0015】
本明細書では、がんに罹患した個体を、有効量の少なくとも1つのMAPを投与することによって処置する方法も提供される。がんは、インテグリンが発現されているがんであってよい。がんは、乳がん、結腸直腸がん、基底細胞がん、腺がん、胃腸がん、結腸がん、肝がん、膀胱がん、膵がん、卵巣がん、子宮頸がん、肺がん、皮膚がん、前立腺がん、腎細胞がん、中枢神経系(CNS)のがん、および白血病であってよい。胃腸がんは、口唇がん、口腔がん、食道がん、小腸がんまたは胃がんであってよい。皮膚がんは、扁平上皮細胞がんまたは基底細胞がんであってよい。
【0016】
本明細書では、インテグリンを発現している細胞を有効量のMAPまたはその融合物と接触させることによって、インテグリンがリガンドと結合することを阻害する方法も提供される。
【0017】
本明細書では、個体におけるがん細胞の存在を、がん細胞を少なくとも1つのMAPと接触させ、少なくとも1つのMAPを検出することによって決定する方法も提供される。MAPは、標識することができ、標識は、陽電子放射型断層撮影法(PET)プローブまたは蛍光プローブであってよい。
【0018】
本明細書では、人工的なECM足場をMAPでコーティングすることによって、人工的なECM足場を調製する方法も提供される。人工的なECM足場は、幹細胞前駆体をさらに含んでよい。人工的なECM足場は、膀胱の足場、食道の足場または肛門の足場であってよい。
【0019】
少なくとも1つのMAPを含む組成物でコーティングしたステントも提供される。
【0020】
(i)Cys−Asp−Cys(CD)モチーフ、(ii)図1に示されているトリペプチドモチーフを有し、(iii)ADAMのメタロプロテアーゼドメイン、システインリッチドメイン、およびドメイン間のセグメントの全て、または実質的に全てを欠き、ADAM由来のディスインテグリン様ドメインを有し、CDCモチーフのC末端側の最初のシステインとドメイン間のジスルフィド結合およびトリペプチドモチーフのC末端側のシステインとドメイン間のジスルフィド結合の破壊をもたらすアミノ酸の修飾をさらに含む、APであるMAPも提供される。
【図面の簡単な説明】
【0021】
【図1A】図1は、23種のヒトADAMタンパク質(AP)のディスインテグリン様ドメインのアミノ酸配列を示す。図1Aは、ヒトADAMタンパク質のディスインテグリン様ドメインの配列アラインメントを示す。横線で消されたアミノ酸残基は、対応するMAPポリペプチドでは除去されるアミノ酸を示す。AP1およびAP17の太字のアミノ酸は、中程度のサイズのヘビ毒ディスインテグリンのシステインパターンを保存するために、対応するMAPでは別のアミノ酸と交換した。図1Bは、APのディスインテグリン様ドメインのディスインテグリンループのそれぞれの先端に提示されるトリペプチドモチーフ(枠で囲まれたアミノ酸残基)を示す。図1Cは、ディスインテグリン様ドメインのディスインテグリンループ(枠で囲まれたアミノ酸残基)を示す。
【図1B】図1は、23種のヒトADAMタンパク質(AP)のディスインテグリン様ドメインのアミノ酸配列を示す。図1Aは、ヒトADAMタンパク質のディスインテグリン様ドメインの配列アラインメントを示す。横線で消されたアミノ酸残基は、対応するMAPポリペプチドでは除去されるアミノ酸を示す。AP1およびAP17の太字のアミノ酸は、中程度のサイズのヘビ毒ディスインテグリンのシステインパターンを保存するために、対応するMAPでは別のアミノ酸と交換した。図1Bは、APのディスインテグリン様ドメインのディスインテグリンループのそれぞれの先端に提示されるトリペプチドモチーフ(枠で囲まれたアミノ酸残基)を示す。図1Cは、ディスインテグリン様ドメインのディスインテグリンループ(枠で囲まれたアミノ酸残基)を示す。
【図1C】図1は、23種のヒトADAMタンパク質(AP)のディスインテグリン様ドメインのアミノ酸配列を示す。図1Aは、ヒトADAMタンパク質のディスインテグリン様ドメインの配列アラインメントを示す。横線で消されたアミノ酸残基は、対応するMAPポリペプチドでは除去されるアミノ酸を示す。AP1およびAP17の太字のアミノ酸は、中程度のサイズのヘビ毒ディスインテグリンのシステインパターンを保存するために、対応するMAPでは別のアミノ酸と交換した。図1Bは、APのディスインテグリン様ドメインのディスインテグリンループのそれぞれの先端に提示されるトリペプチドモチーフ(枠で囲まれたアミノ酸残基)を示す。図1Cは、ディスインテグリン様ドメインのディスインテグリンループ(枠で囲まれたアミノ酸残基)を示す。
【図2A】図2は、MAPポリペプチドのアミノ酸配列を示す。図2Aは、典型的な中程度のサイズのヘビ毒ディスインテグリン(Trimeresurus flavoviridisヘビの毒液から精製した)であるトリメスタチン(trimestatin)の配列に対してアラインメントした、23種のMAPポリペプチドのアミノ酸配列を示す。システイン残基をアラインメントし、それに下線が引かれている。AP1およびAP17におけるアミノ酸残基に対応する、MAP1およびMAP17の交換されたアミノ酸残基がそれぞれ太字で示されている。さらに、AP1、AP3、AP6、AP18、AP21、AP30、およびAP32のN末端残基が、それらの対応するMAPにおいてグリシン残基と交換されている。図2Bは、MAPのディスインテグリン様ドメインのディスインテグリンループのそれぞれの先端に提示され、および中程度のサイズのヘビ毒トリメスタチンによって提示されるトリペプチドモチーフ(枠で囲まれたアミノ酸)を示す。図2Cは、MAPおよびトリメスタチンのディスインテグリンループ(枠で囲まれたアミノ酸)を同定する。
【図2B】図2は、MAPポリペプチドのアミノ酸配列を示す。図2Aは、典型的な中程度のサイズのヘビ毒ディスインテグリン(Trimeresurus flavoviridisヘビの毒液から精製した)であるトリメスタチン(trimestatin)の配列に対してアラインメントした、23種のMAPポリペプチドのアミノ酸配列を示す。システイン残基をアラインメントし、それに下線が引かれている。AP1およびAP17におけるアミノ酸残基に対応する、MAP1およびMAP17の交換されたアミノ酸残基がそれぞれ太字で示されている。さらに、AP1、AP3、AP6、AP18、AP21、AP30、およびAP32のN末端残基が、それらの対応するMAPにおいてグリシン残基と交換されている。図2Bは、MAPのディスインテグリン様ドメインのディスインテグリンループのそれぞれの先端に提示され、および中程度のサイズのヘビ毒トリメスタチンによって提示されるトリペプチドモチーフ(枠で囲まれたアミノ酸)を示す。図2Cは、MAPおよびトリメスタチンのディスインテグリンループ(枠で囲まれたアミノ酸)を同定する。
【図2C】図2は、MAPポリペプチドのアミノ酸配列を示す。図2Aは、典型的な中程度のサイズのヘビ毒ディスインテグリン(Trimeresurus flavoviridisヘビの毒液から精製した)であるトリメスタチン(trimestatin)の配列に対してアラインメントした、23種のMAPポリペプチドのアミノ酸配列を示す。システイン残基をアラインメントし、それに下線が引かれている。AP1およびAP17におけるアミノ酸残基に対応する、MAP1およびMAP17の交換されたアミノ酸残基がそれぞれ太字で示されている。さらに、AP1、AP3、AP6、AP18、AP21、AP30、およびAP32のN末端残基が、それらの対応するMAPにおいてグリシン残基と交換されている。図2Bは、MAPのディスインテグリン様ドメインのディスインテグリンループのそれぞれの先端に提示され、および中程度のサイズのヘビ毒トリメスタチンによって提示されるトリペプチドモチーフ(枠で囲まれたアミノ酸)を示す。図2Cは、MAPおよびトリメスタチンのディスインテグリンループ(枠で囲まれたアミノ酸)を同定する。
【図3】図3は、ヘビ毒ディスインテグリンおよびヒトADAMタンパク質のディスインテグリン様ドメインの配列アラインメントを示す。PIII−クラスのヘビ毒メタロプロテアーゼ(VAP1およびカトロコラスタチン(Catrocollastatin))のディスインテグリン様配列を長いサイズのヘビ毒ディスインテグリン配列(サルモシン(Salmosin)3およびビチスタチン(Bitistatin))、典型的な中程度のサイズのヘビ毒ディスインテグリン(トリメスタチン)およびいくつかのヒトADAMのディスインテグリン様ドメイン(AP7、AP8、AP12、AP19、AP28、およびAP33)とアラインメントした。対応するMAPポリペプチドを生成するために、修飾することができる(例えば、欠失)ディスインテグリン様配列(ヘビ起源およびヒト起源の両方)におけるアミノ酸残基が横線で消されて示されている。これらのディスインテグリンおよびディスインテグリン様配列におけるディスインテグリンループの先端に提示されるトリペプチドアミノ酸モチーフが囲み枠の中に示されている。
【図4】図4は、様々なE.coli宿主(BL21対Origami B)におけるTrx−D9(ネイティブなヒトADAM9ディスインテグリン様ドメイン配列−チオレドキシン融合ポリペプチド)の発現とTrx−MAP9(MAP9−チオレドキシン融合ポリペプチド)の発現とを、SDS−PAGEによって分析して比較している。レーンは、左から右に、PageRulerTMPlus Prestained Protein Ladder(Fermentas、Burlington、ON)、カルベニシリンの下で成長させ、誘導した、Trx−D9で形質転換したBL21(DE3)細胞からの溶解物、最初に3種のAB(カルベニシリン、カナマイシンおよびテトラサイクリン)上に播いたが、カルベニシリンのみの下でさらに増殖させ、誘導した、Trx−D9で形質転換したOrigami B(DE3)細胞からの溶解物、カルベニシリンの下で成長させ、誘導した、Trx−MAP9で形質転換したBL21(DE3)細胞からの溶解物、ならびに、最初に3種のAB(カルベニシリン、カナマイシンおよびテトラサイクリン)上に播いたが、カルベニシリンのみの下でさらに増殖させ、誘導した、Trx−MAP9で形質転換したOrigami B(DE3)細胞からの溶解物を示す。
【図5】図5は、様々なE.coli宿主(BL21対Origami B)におけるTrx−D15(ネイティブなヒトADAM15ディスインテグリン様ドメイン配列−チオレドキシン融合ポリペプチド)の発現とTrx−MAP15(MAP15−チオレドキシン融合ポリペプチド)の発現とを、SDS−PAGEによって分析して比較している。レーンは、左から右に、PageRulerTMPlus Prestained Protein Ladder(Fermentas、Burlington、ON)、カルベニシリンの下で成長させ、誘導した、Trx−D15で形質転換したBL21(DE3)細胞からの溶解物、最初に3種のAB(カルベニシリン、カナマイシンおよびテトラサイクリン)上に播いたが、カルベニシリンのみの下でさらに増殖させ、誘導した、Trx−D15で形質転換したOrigami B(DE3)細胞からの溶解物、カルベニシリンの下で成長させ、誘導した、Trx−MAP15で形質転換したBL21(DE3)細胞からの溶解物、ならびに、最初に3種のAB(カルベニシリン、カナマイシンおよびテトラサイクリン)上に播いたが、カルベニシリンのみの下でさらに増殖させ、誘導した、Trx−MAP15で形質転換したOrigami B(DE3)細胞からの溶解物を示す。
【図6A】図6は、細菌のTrxAの3’側の、pET32a発現ベクターに挿入されたMAPの核酸配列を示す。図6Aは、MAP1、MAP2、MAP3、およびMAP6のDNA配列を示す。図6Bは、MAP7、MAP8、MAP9、およびMAP10の核酸配列を示す。図6Cは、MAP11、MAP12、MAP15、およびMAP17の核酸配列を示す。図6Dは、MAP18、MAP19、MAP20、およびMAP21の核酸配列を示す。図6Eは、MAP22、MAP23、MAP28、およびMAP29の核酸配列を示す。図6Fは、MAP30、MAP32、およびMAP33の核酸配列を示す。
【図6B】図6は、細菌のTrxAの3’側の、pET32a発現ベクターに挿入されたMAPの核酸配列を示す。図6Aは、MAP1、MAP2、MAP3、およびMAP6のDNA配列を示す。図6Bは、MAP7、MAP8、MAP9、およびMAP10の核酸配列を示す。図6Cは、MAP11、MAP12、MAP15、およびMAP17の核酸配列を示す。図6Dは、MAP18、MAP19、MAP20、およびMAP21の核酸配列を示す。図6Eは、MAP22、MAP23、MAP28、およびMAP29の核酸配列を示す。図6Fは、MAP30、MAP32、およびMAP33の核酸配列を示す。
【図6C】図6は、細菌のTrxAの3’側の、pET32a発現ベクターに挿入されたMAPの核酸配列を示す。図6Aは、MAP1、MAP2、MAP3、およびMAP6のDNA配列を示す。図6Bは、MAP7、MAP8、MAP9、およびMAP10の核酸配列を示す。図6Cは、MAP11、MAP12、MAP15、およびMAP17の核酸配列を示す。図6Dは、MAP18、MAP19、MAP20、およびMAP21の核酸配列を示す。図6Eは、MAP22、MAP23、MAP28、およびMAP29の核酸配列を示す。図6Fは、MAP30、MAP32、およびMAP33の核酸配列を示す。
【図6D】図6は、細菌のTrxAの3’側の、pET32a発現ベクターに挿入されたMAPの核酸配列を示す。図6Aは、MAP1、MAP2、MAP3、およびMAP6のDNA配列を示す。図6Bは、MAP7、MAP8、MAP9、およびMAP10の核酸配列を示す。図6Cは、MAP11、MAP12、MAP15、およびMAP17の核酸配列を示す。図6Dは、MAP18、MAP19、MAP20、およびMAP21の核酸配列を示す。図6Eは、MAP22、MAP23、MAP28、およびMAP29の核酸配列を示す。図6Fは、MAP30、MAP32、およびMAP33の核酸配列を示す。
【図6E】図6は、細菌のTrxAの3’側の、pET32a発現ベクターに挿入されたMAPの核酸配列を示す。図6Aは、MAP1、MAP2、MAP3、およびMAP6のDNA配列を示す。図6Bは、MAP7、MAP8、MAP9、およびMAP10の核酸配列を示す。図6Cは、MAP11、MAP12、MAP15、およびMAP17の核酸配列を示す。図6Dは、MAP18、MAP19、MAP20、およびMAP21の核酸配列を示す。図6Eは、MAP22、MAP23、MAP28、およびMAP29の核酸配列を示す。図6Fは、MAP30、MAP32、およびMAP33の核酸配列を示す。
【図6F】図6は、細菌のTrxAの3’側の、pET32a発現ベクターに挿入されたMAPの核酸配列を示す。図6Aは、MAP1、MAP2、MAP3、およびMAP6のDNA配列を示す。図6Bは、MAP7、MAP8、MAP9、およびMAP10の核酸配列を示す。図6Cは、MAP11、MAP12、MAP15、およびMAP17の核酸配列を示す。図6Dは、MAP18、MAP19、MAP20、およびMAP21の核酸配列を示す。図6Eは、MAP22、MAP23、MAP28、およびMAP29の核酸配列を示す。図6Fは、MAP30、MAP32、およびMAP33の核酸配列を示す。
【図7A】図7は、MAP構築物をpET32a発現ベクターにクローニングするために使用するオリゴヌクレオチドプライマー配列を示す。図7Aは、MAP1、MAP2、MAP3、MAP6、MAP7、MAP8、MAP9、MAP10、MAP11、MAP12、MAP15、MAP17、およびMAP18をクローニングするために使用するオリゴヌクレオチドプライマーを示す。図7Bは、MAP19、MAP20、MAP21、MAP22、MAP23、MAP28、MAP29、MAP30、MAP32、およびMAP33をクローニングするために使用するオリゴヌクレオチドプライマーを示す。
【図7B】図7は、MAP構築物をpET32a発現ベクターにクローニングするために使用するオリゴヌクレオチドプライマー配列を示す。図7Aは、MAP1、MAP2、MAP3、MAP6、MAP7、MAP8、MAP9、MAP10、MAP11、MAP12、MAP15、MAP17、およびMAP18をクローニングするために使用するオリゴヌクレオチドプライマーを示す。図7Bは、MAP19、MAP20、MAP21、MAP22、MAP23、MAP28、MAP29、MAP30、MAP32、およびMAP33をクローニングするために使用するオリゴヌクレオチドプライマーを示す。
【図8A】図8は、E.coli Origami B(DE3)において発現させた全てのTrxA−MAP構築物のアミノ酸配列を示す。TrxA(CGPC)の活性部位はイタリック体で示されており、ディスインテグリンループの先端のトリペプチドモチーフには下線が引かれており、TEV切断認識部位は囲み枠で強調されており、TrxAと種々のMAP構築物との間のリンカー領域は太字のイタリック体で示されている。AP1およびAP17内の残基を交換するために導入された、対応するMAP1およびMAP17内のアミノ酸残基は、太字の二重下線で強調されている。
【図8B】図8は、E.coli Origami B(DE3)において発現させた全てのTrxA−MAP構築物のアミノ酸配列を示す。TrxA(CGPC)の活性部位はイタリック体で示されており、ディスインテグリンループの先端のトリペプチドモチーフには下線が引かれており、TEV切断認識部位は囲み枠で強調されており、TrxAと種々のMAP構築物との間のリンカー領域は太字のイタリック体で示されている。AP1およびAP17内の残基を交換するために導入された、対応するMAP1およびMAP17内のアミノ酸残基は、太字の二重下線で強調されている。
【図8C】図8は、E.coli Origami B(DE3)において発現させた全てのTrxA−MAP構築物のアミノ酸配列を示す。TrxA(CGPC)の活性部位はイタリック体で示されており、ディスインテグリンループの先端のトリペプチドモチーフには下線が引かれており、TEV切断認識部位は囲み枠で強調されており、TrxAと種々のMAP構築物との間のリンカー領域は太字のイタリック体で示されている。AP1およびAP17内の残基を交換するために導入された、対応するMAP1およびMAP17内のアミノ酸残基は、太字の二重下線で強調されている。
【図8D】図8は、E.coli Origami B(DE3)において発現させた全てのTrxA−MAP構築物のアミノ酸配列を示す。TrxA(CGPC)の活性部位はイタリック体で示されており、ディスインテグリンループの先端のトリペプチドモチーフには下線が引かれており、TEV切断認識部位は囲み枠で強調されており、TrxAと種々のMAP構築物との間のリンカー領域は太字のイタリック体で示されている。AP1およびAP17内の残基を交換するために導入された、対応するMAP1およびMAP17内のアミノ酸残基は、太字の二重下線で強調されている。
【図8E】図8は、E.coli Origami B(DE3)において発現させた全てのTrxA−MAP構築物のアミノ酸配列を示す。TrxA(CGPC)の活性部位はイタリック体で示されており、ディスインテグリンループの先端のトリペプチドモチーフには下線が引かれており、TEV切断認識部位は囲み枠で強調されており、TrxAと種々のMAP構築物との間のリンカー領域は太字のイタリック体で示されている。AP1およびAP17内の残基を交換するために導入された、対応するMAP1およびMAP17内のアミノ酸残基は、太字の二重下線で強調されている。
【図8F】図8は、E.coli Origami B(DE3)において発現させた全てのTrxA−MAP構築物のアミノ酸配列を示す。TrxA(CGPC)の活性部位はイタリック体で示されており、ディスインテグリンループの先端のトリペプチドモチーフには下線が引かれており、TEV切断認識部位は囲み枠で強調されており、TrxAと種々のMAP構築物との間のリンカー領域は太字のイタリック体で示されている。AP1およびAP17内の残基を交換するために導入された、対応するMAP1およびMAP17内のアミノ酸残基は、太字の二重下線で強調されている。
【図8G】図8は、E.coli Origami B(DE3)において発現させた全てのTrxA−MAP構築物のアミノ酸配列を示す。TrxA(CGPC)の活性部位はイタリック体で示されており、ディスインテグリンループの先端のトリペプチドモチーフには下線が引かれており、TEV切断認識部位は囲み枠で強調されており、TrxAと種々のMAP構築物との間のリンカー領域は太字のイタリック体で示されている。AP1およびAP17内の残基を交換するために導入された、対応するMAP1およびMAP17内のアミノ酸残基は、太字の二重下線で強調されている。
【図8H】図8は、E.coli Origami B(DE3)において発現させた全てのTrxA−MAP構築物のアミノ酸配列を示す。TrxA(CGPC)の活性部位はイタリック体で示されており、ディスインテグリンループの先端のトリペプチドモチーフには下線が引かれており、TEV切断認識部位は囲み枠で強調されており、TrxAと種々のMAP構築物との間のリンカー領域は太字のイタリック体で示されている。AP1およびAP17内の残基を交換するために導入された、対応するMAP1およびMAP17内のアミノ酸残基は、太字の二重下線で強調されている。
【図9】図9は、SDS−PAGEによって分析した、Origami B宿主におけるTrx−MAP2(MAP2−チオレドキシン融合ポリペプチド)、Trx−MAP7(MAP7−チオレドキシン融合ポリペプチド)、Trx−MAP8(MAP8−チオレドキシン融合ポリペプチド)、およびTrx−MAP9(MAP9−チオレドキシン融合ポリペプチド)の発現レベルを示す。レーンは、左から右に、PageRulerTMPlus Prestained Protein Ladder(Fermentas、Burlington、ON)、Trx−MAP2で形質転換したOrigami B(DE3)細胞からの溶解物、Trx−MAP7で形質転換したOrigami B(DE3)細胞からの溶解物、Trx−MAP8で形質転換したOrigami B(DE3)細胞からの溶解物、およびTrx−MAP9で形質転換したOrigami B(DE3)からの溶解物を示す。形質転換体は全て、最初に3種の抗生物質(カルベニシリン、カナマイシンおよびテトラサイクリン)の上に播いたが、カルベニシリンのみの下でさらに増殖させ、誘導した。
【図10】図10は、SDS−PAGEによって分析した、Origami B宿主におけるTrx−MAP10(MAP10−チオレドキシン融合ポリペプチド)、Trx−MAP12(MAP12−チオレドキシン融合ポリペプチド)、Trx−MAP15(MAP15−チオレドキシン融合ポリペプチド)、およびTrx−MAP17(MAP17−チオレドキシン融合ポリペプチド)の発現レベルを示す。レーンは、左から右に、PageRulerTMPlus Prestained Protein Ladder(Fermentas、Burlington、ON)、Trx−MAP10で形質転換したOrigami B(DE3)細胞からの溶解物、Trx−MAP12で形質転換したOrigami B(DE3)細胞からの溶解物、Trx−MAP15で形質転換したOrigami B(DE3)細胞からの溶解物、およびTrx−MAP17で形質転換したOrigami B(DE3)からの溶解物を示す。形質転換体は全て、最初に3種の抗生物質(カルベニシリン、カナマイシンおよびテトラサイクリン)の上に播いたが、カルベニシリンのみの下でさらに増殖させ、誘導した。
【図11】図11は、SDS−PAGEによって分析した、Origami B宿主におけるTrx−MAP19(MAP19−チオレドキシン融合ポリペプチド)、Trx−MAP23(MAP23−チオレドキシン融合ポリペプチド)、Trx−MAP28(MAP28−チオレドキシン融合ポリペプチド)、およびTrx−MAP33(MAP33−チオレドキシン融合ポリペプチド)の発現レベルを示す。レーンは、左から右に、PageRulerTMPlus Prestained Protein Ladder(Fermentas、Burlington、ON)、Trx−MAP19で形質転換したOrigami B(DE3)細胞からの溶解物、Trx−MAP23で形質転換したOrigami B(DE3)細胞からの溶解物、Trx−MAP28で形質転換したOrigami B(DE3)細胞からの溶解物、およびTrx−MAP33で形質転換したOrigami B(DE3)からの溶解物を示す。形質転換体は全て、最初に3種の抗生物質(カルベニシリン、カナマイシンおよびテトラサイクリン)の上に播いたが、カルベニシリンのみの下でさらに増殖させ、誘導した。
【図12】図12は、図に示されている種々のTrx−MAPの、MDA−MB−231細胞(ヒト乳がん)への結合の、フローサイトメトリーによる検出を示す。
【図13】図13は、図に示されている種々のTrx−MAPの、MDA−MB−231細胞(ヒト乳がん)の骨ホーミングサブクローンへの結合の、フローサイトメトリーによる検出を示す。
【図14】図14は、図に示されている種々のTrx−MAPの、MDA−MB−231細胞(ヒト乳がん)の脳ホーミングサブクローンへの結合の、フローサイトメトリーによる検出を示す。
【図15】図15は、図に示されている種々のTrx−MAPの、ジャーカット細胞(ヒトT細胞白血病細胞株)への結合の、フローサイトメトリーによる検出を示す。
【図16】図16は、MAP9およびMAP15の、HUVEC(ヒト臍帯静脈内皮細胞)への結合、MDA−MB−435(ヒト乳がん)への結合、MDA−MB−231(ヒト乳がん)への結合、および多形神経膠芽腫がん幹細胞株(GBM−CSC)への結合の、フローサイトメトリーによる検出を示す。
【図17】図17は、10nMのMAP9またはMAP15の存在下でのHUVECの管形成アッセイを示す。パネルA−無処置の対照;パネルB−100μMのスラミン;パネルC−10nMのMAP9;パネルD−10nMのMAP15。細胞をCalcein AMで染色し、共焦点顕微鏡を使用してイメージングした。イメージは全て、同じ倍率で取得した(スケールバー=50μm)。
【図18】図18は、他の抗がん処置(アバスチンおよびドセタキセル)と比較した、MDA−MB−231異種移植における、MAP15およびMAP15のリポソーム処方物による処置による腫瘍の成長阻害(パネルA)および生存(パネルB)を示す。
【図19】図19は、MDA−MB−231異種移植における、他の抗がん処置(アバスチンおよびドセタキセル)と比較した、MAP15のリポソーム処方物による処置および他の抗がん処置との組み合わせにおける、微小血管の阻害を、腫瘍血管新生アッセイの顕微鏡写真(パネルA)において、およびランダムな顕微鏡写真(パネルB)から微小血管の密度を定量化することによって示している。
【図20】図20は、ADAM1転写物に対応する核酸配列を示す。
【図21】図21は、ADAM2転写物に対応する核酸配列を示す。
【図22】図22は、ADAM3転写物、バリアント(variant)1に対応する核酸配列を示す。
【図23】図23は、ADAM6転写物に対応する核酸配列を示す。
【図24】図24は、ADAM7転写物に対応する核酸配列を示す。
【図25A】図25は、ADAM8転写物、バリアント1に対応する核酸配列を示す。
【図25B】図25は、ADAM8転写物、バリアント1に対応する核酸配列を示す。
【図26A】図26は、ADAM9転写物、バリアント1に対応する核酸配列を示す。
【図26B】図26は、ADAM9転写物、バリアント1に対応する核酸配列を示す。
【図27A】図27は、ADAM10転写物に対応する核酸配列を示す。
【図27B】図27は、ADAM10転写物に対応する核酸配列を示す。
【図28A】図28は、ADAM11転写物に対応する核酸配列を示す。
【図28B】図28は、ADAM11転写物に対応する核酸配列を示す。
【図29A】図29は、ADAM12転写物、バリアント1に対応する核酸配列を示す。
【図29B】図29は、ADAM12転写物、バリアント1に対応する核酸配列を示す。
【図30】図30は、ADAM15転写物、バリアント6に対応する核酸配列を示す。
【図31A】図31は、ADAM17転写物に対応する核酸配列を示す。
【図31B】図31は、ADAM17転写物に対応する核酸配列を示す。
【図32】図32は、ADAM18転写物、バリアント1に対応する核酸配列を示す。
【図33A】図33は、ADAM19転写物に対応する核酸配列を示す。
【図33B】図33は、ADAM19転写物に対応する核酸配列を示す。
【図33C】図33は、ADAM19転写物に対応する核酸配列を示す。
【図34】図34は、ADAM20転写物に対応する核酸配列を示す。
【図35】図35は、ADAM21転写物に対応する核酸配列を示す。
【図36A】図36は、ADAM22転写物、バリアント1に対応する核酸配列を示す。
【図36B】図36は、ADAM22転写物、バリアント1に対応する核酸配列を示す。
【図36C】図36は、ADAM22転写物、バリアント1に対応する核酸配列を示す。
【図36D】図36は、ADAM22転写物、バリアント1に対応する核酸配列を示す。
【図37】図37は、ADAM23転写物に対応する核酸配列を示す。
【図38A】図38は、ADAM28転写物、バリアント1に対応する核酸配列を示す。
【図38B】図38は、ADAM28転写物、バリアント1に対応する核酸配列を示す。
【図39A】図39は、ADAM29転写物、バリアント1に対応する核酸配列を示す。
【図39B】図39は、ADAM29転写物、バリアント1に対応する核酸配列を示す。
【図40】図40は、ADAM30転写物に対応する核酸配列を示す。
【図41】図41は、ADAM32転写物に対応する核酸配列を示す。
【図42A】図42は、ADAM33転写物、バリアント1に対応する核酸配列を示す。
【図42B】図42は、ADAM33転写物、バリアント1に対応する核酸配列を示す。
【図43A】図43は、ADAM2、ADAM7、ADAM8、ADAM9、ADAM10、ADAM11、ADAM12、ADAM15、ADAM17、ADAM18、ADAM19、ADAM20、ADAM21、ADAM22、ADAM23、ADAM28、ADAM29、ADAM30、ADAM32、およびADAM33ポリペプチドのアミノ酸配列を示す。
【図43B】図43は、ADAM2、ADAM7、ADAM8、ADAM9、ADAM10、ADAM11、ADAM12、ADAM15、ADAM17、ADAM18、ADAM19、ADAM20、ADAM21、ADAM22、ADAM23、ADAM28、ADAM29、ADAM30、ADAM32、およびADAM33ポリペプチドのアミノ酸配列を示す。
【図43C】図43は、ADAM2、ADAM7、ADAM8、ADAM9、ADAM10、ADAM11、ADAM12、ADAM15、ADAM17、ADAM18、ADAM19、ADAM20、ADAM21、ADAM22、ADAM23、ADAM28、ADAM29、ADAM30、ADAM32、およびADAM33ポリペプチドのアミノ酸配列を示す。
【図43D】図43は、ADAM2、ADAM7、ADAM8、ADAM9、ADAM10、ADAM11、ADAM12、ADAM15、ADAM17、ADAM18、ADAM19、ADAM20、ADAM21、ADAM22、ADAM23、ADAM28、ADAM29、ADAM30、ADAM32、およびADAM33ポリペプチドのアミノ酸配列を示す。
【図43E】図43は、ADAM2、ADAM7、ADAM8、ADAM9、ADAM10、ADAM11、ADAM12、ADAM15、ADAM17、ADAM18、ADAM19、ADAM20、ADAM21、ADAM22、ADAM23、ADAM28、ADAM29、ADAM30、ADAM32、およびADAM33ポリペプチドのアミノ酸配列を示す。
【図43F】図43は、ADAM2、ADAM7、ADAM8、ADAM9、ADAM10、ADAM11、ADAM12、ADAM15、ADAM17、ADAM18、ADAM19、ADAM20、ADAM21、ADAM22、ADAM23、ADAM28、ADAM29、ADAM30、ADAM32、およびADAM33ポリペプチドのアミノ酸配列を示す。
【図43G】図43は、ADAM2、ADAM7、ADAM8、ADAM9、ADAM10、ADAM11、ADAM12、ADAM15、ADAM17、ADAM18、ADAM19、ADAM20、ADAM21、ADAM22、ADAM23、ADAM28、ADAM29、ADAM30、ADAM32、およびADAM33ポリペプチドのアミノ酸配列を示す。
【発明を実施するための形態】
【0022】
(発明の詳細な説明)
本明細書では、MAP(修飾されたADAM由来のポリペプチド)と称される独特な設計ポリペプチドのクラス、およびそれをコードする核酸が提供される。本明細書で使用される場合、MAPとは、ADAMタンパク質のネイティブなディスインテグリンドメインの修飾された形態を指す。ADAMをコードする核酸およびアミノ酸の配列の一覧については付属物を参照されたい。本明細書で使用される場合、本明細書では「AP」(「ADAM由来ポリペプチド」)と称することができる「ADAMタンパク質のディスインテグリンドメイン」は、そのメタロプロテアーゼドメイン、システインリッチドメインおよびドメイン間のセグメントの全て、または実質的に全てを欠く、ADAMのディスインテグリンドメインである。実質的に全てを欠くとは、残りのアミノ酸配列が、そのドメインの機能をもはや保持しないことを意味する。図1にAPの例が示されている。APのN末端は、CDCモチーフからN末端側に3位のアミノ酸残基で始まり、CDCのN末端側の最初のシステインの直前までである。APのC末端は、CDCモチーフから12番目のシステイン残基のC末端側に10位のアミノ酸残基で終わり、前記12番目のシステイン残基のC末端側の次のシステインの直前までである。例外が2つある:(1)AP1のC末端は、CDCモチーフから13番目のシステイン残基のC末端側の10位のアミノ酸残基で終わり、前記13番目のシステイン残基のC末端側の最初のシステインの直前までであり、(2)ADAM17は、CDCモチーフではなくCDPモチーフを有し、対応するAP(AP17)の末端が示されている。
【0023】
「MAP」は、APの「修飾された」形態であり、修飾は、本明細書に記載の有益な性質を実現するためのAPのアミノ酸配列の変更(複数可)を伴う。したがって、MAPは、通常APに存在する配列および対応するADAMポリペプチドの配列と比較して、修飾された配列を有する。本明細書で使用される場合、「修飾された」とは、アミノ酸が欠失していることか、置換されていることか、または化学修飾されていることを意味し、ある実施形態では、修飾により、ドメイン間のジスルフィド結合の破壊がもたらされる。図2に例示的なMAPが示されている。典型的な中程度のサイズのヘビ毒ディスインテグリンであるトリメスタチンとアラインメントしたMAP配列が示されている。全てのMAP構築物を、中程度のサイズのヘビ毒ディスインテグリンにならってモデリングし、それらの配列を、これらのネイティブなヘビ毒分子と同様に折りたたまれるように修飾した。MAP(MAP17以外)は、対応するAPのCDCモチーフのC末端側の最初のシステインおよび前記システインのC末端側の2つのアミノ酸ならびにトリペプチドモチーフのC末端側のシステインが欠失するように構築した。あるいは、ジスルフィド結合の形成を妨げるために、システイン残基を代替のアミノ酸で置換することができ、またはシステインアミノ酸残基を化学修飾することができる。アミノ酸置換は、保存的であり得、例えば、APのCDCモチーフのC末端側の最初のシステインをセリン残基で置換することができ、システインのC末端側のアミノ酸残基を荷電アミノ酸で置換することができ、または、トリペプチドモチーフのC末端側のシステインを荷電アミノ酸で置換することができる。そのような変異による手法およびアミノ酸残基の化学的な修飾は当技術分野で周知である。化学的な修飾に関して、例としては、ジスルフィド結合の形成を妨げるために、アルキル化剤を使用してシステイン残基と反応させることがある。MAP10、MAP17、MAP18およびMAP32以外、MAPは、ヘビ毒ディスインテグリンのネイティブなループと同様の11アミノ酸のディスインテグリンループを提示する。MAP10は、10アミノ酸のインテグリンループを提示し、MAP17、MAP18、およびMAP32は、12アミノ酸のディスインテグリンループを提示する。
【0024】
MAPを、活性な可溶性のジスルフィドリッチポリペプチドの生成およびこれらの生成物についての高い発現収率の両方を支持する細菌系において、独立した生物活性のある分子として発現させ、さらに精製することができる。理論に縛られることを望むものではないが、ネイティブなAPから、MAPを、それらのネイティブなADAMのコンフォメーションではなく、ヘビ毒ディスインテグリンの折りたたみをとり得るように設計した。MAPを、Origami B(DE3)E.coli株において高収率で発現させ、哺乳動物細胞の表面受容体クラス(インテグリン)と、ネイティブなヘビ毒ディスインテグリンと同様の様式で相互作用することができる安定かつ活性な遊離のポリペプチドとしてさらに精製することができる。MAPは、MAPが由来する、ADAMポリペプチド由来のAPまたはディスインテグリンドメインの活性の特性であるシグナル伝達性質のいくつかも保持し得る。例えば、保持される特性としては、ADAMディスインテグリンドメインの、古典的なディスインテグリンループの外側に位置するアミノ酸残基を利用することによってインテグリン受容体に関与すると推定される能力に関連するシグナル伝達特質を挙げることができる。ADAMの細胞機能は周知である[1〜5、34]。
【0025】
理論に縛られることを望むものではないが、典型的な中程度のサイズのヘビ毒ディスインテグリン(例えば、トリメスタチン、キストリン(Kistrin)、フラボリジン(Flavoridin)など)を生じさせるPII−クラスのSVMPは、上流のスペーサー領域とディスインテグリンドメインとの間の重要なジスルフィド架橋を形成することができず、したがって、ディスインテグリンドメインが開始する位置のすぐN末端側に位置する残基においてタンパク質分解性の攻撃が起こり、その結果、放出された中程度のサイズのディスインテグリンは、上流のスペーサー領域の部分を含有しない完全なディスインテグリンドメインであると考えられている。対照的に、長いサイズのヘビ毒ディスインテグリン(例えば、ビチスタチン、サルモシン3など)を生じさせるPII−クラスのSVMPは、メタロプロテアーゼドメインと下流のスペーサー領域との間の重要なジスルフィド架橋を形成することができず、したがって、スペーサー領域のさらにN末端においてタンパク質分解性の攻撃が起こり、スペーサー領域の一部がN末端で遊離のディスインテグリンドメインに付着したより長いディスインテグリンが放出されると考えられている(図3における種々のディスインテグリンおよびディスインテグリンドメインの配列アラインメントを参照されたい)。さらに、PII−SVMPが、より多くの変異および/または欠失を含有する場合、同じスペーサー領域においてだけではなく、ディスインテグリンドメインのN末端部分においてもジスルフィド架橋を形成することができず、ヘビ毒ディスインテグリンのより短いバリアントが放出されるとも考えられている(例えば、コントルトロスタチン(Contortrostatin)と同様に二量体化する部分的に短縮されたディスインテグリンドメイン、または、まれな、エキスタチンまたはエリストスタチン(Eristostatin)と同様に非常に短縮されたポリペプチドのいずれか)。さらに、ほとんど全ての場合において、遊離のディスインテグリンドメインは、それらの分子のC末端側の半分において、ヘビ毒ディスインテグリンの特徴である保存された11アミノ酸のディスインテグリンループを提示すると考えられている。
【0026】
ヒトゲノムにおいて同定された23種の異なるADAM転写物(そのうち3種は通常はタンパク質産物に翻訳されない偽遺伝子である)を、ヘビ毒ディスインテグリンの折りたたみ構造をとる本明細書に記載のコードされたMAPを生み出すための基礎として使用した。
【0027】
いくつかのADAM転写物は、いくつものアイソフォームをコードする。それにもかかわらず、異なるADAMのアイソフォームの内側のディスインテグリンドメインの配列は保存されており、したがって、ADAMタンパク質のヒトファミリー内には、23種のみの異なるディスインテグリンドメインが存在する。組換えによって作出した場合、本発明のMAPは、定義されたインテグリンセットと、高い親和性様式で相互作用することができる。この性質により、これらの変異体ポリペプチドが、臨床的な使用および治療的な使用のための広範なスペクトルのインテグリンリガンドになる。
【0028】
他のヒトADAMメンバーの転写物と同様に、非機能性の転写物は、組換え系において人工的に翻訳された場合に、新規の生物学的機能を有する活性なポリペプチドを生成し得る完全なディスインテグリン配列を含有する。ヒトADAMのディスインテグリンドメインは、76個から86個のアミノ酸を有し(ADAM1のディスインテグリンドメインが最も短く、一方、ADAM10のディスインテグリンドメインが最も長い)、例外が2つあり(ADAM1およびADAM17)、これらは全てADAM足場の標準的なシステイン残基を14個含有する(下記のヒトADAMのアラインメントされた配列を参照されたい)。図1を参照されたい。その大部分がそれらのディスインテグリンループの先端にRGDトリペプチドモチーフを含有する、血小板凝集インヒビターとして機能するように進化したヘビ毒ディスインテグリンとは異なり、ADAMのディスインテグリンループは、それらの先端に非常に異なるトリペプチドモチーフを提示し、したがって、より広範な範囲のインテグリンに、それらのヘビ毒対応物とは異なる様式で関与することが予測される。実際、APのそれぞれは、インテグリン受容体の定義された独特のセットに結合し、したがって、独特の様式でシグナル伝達をすると考えられている(ディスインテグリンループの差異を例示しているADAMおよびヘビ毒ディスインテグリンの配列アラインメントについて図3を参照されたい)。理論に縛られることを望むものではないが、2種以上のMAPの組み合わせを使用して、特定の細胞型または細胞型についての疾患状態に特徴的な「インテグリンサイン(integrin signature)」を決定することができると考えられている。インテグリンサインとは、その細胞型またはその細胞型についての疾患状態に独特である細胞の表面上に存在するインテグリンの組み合わせを意味する。
【0029】
ヒトADAM15のディスインテグリンドメインは、そのディスインテグリンループ内にRGDトリペプチドモチーフを含有し、これは、ヒトADAM15が心臓血管系において重要な調節の役割を果たすという仮説を支持する。このADAM15のRGDトリペプチドモチーフは、図1のAP15に示されている。
【0030】
23種の公知のヒトADAMのメンバーの全てのAP部分のそれぞれについて、MAPを生成した。ヒトADAMディスインテグリンドメイン配列を、上記の理論的根拠に従って修飾した。修飾は、通常、ネイティブなADAMタンパク質におけるドメイン間−ディスインテグリンドメインのジスルフィド架橋の形成に関与するADAMディスインテグリンドメイン内の残基(システイン残基を2つ含むものにおいて)を除去することを含む。理論に縛られることを望むものではないが、これらのジスルフィド架橋の見かけの機能は、ディスインテグリンループをADAM内に緊密にパックしたままにして、インテグリン受容体が利用できないようにすることである。これらのジスルフィド架橋の形成に関与する残基を、例えば、欠失などによって修飾することにより、これらのMAPが、ヘビ毒ディスインテグリンの標準的な11アミノ酸のループの移動性およびディスインテグリンの折りたたみ特性を獲得する。長いサイズのヘビ毒ディスインテグリンおよび中程度のサイズのヘビ毒ディスインテグリンとアラインメントした場合、ならびにPIII−クラスのSVMPとアラインメントした場合、ヒトADAMの23種のメンバーの中で、6種のメンバーが上記のスキームに完全に適合する(ADAM7、ADAM8、ADAM12、ADAM19、ADAM28およびADAM33)(ヘビ毒ディスインテグリンおよびヒトADAMディスインテグリンドメインのアラインメントについては図3を参照されたい)。それにもかかわらず、これらの修飾を導入することにより、4種のADAM(10、17、18および32)の例外を伴い、ヒトADAMのメンバーの全てが、11アミノ酸のディスインテグリンループを提示するMAPに変換された。4種の例外に関しては、3種(ADAM17、ADAM18およびADAM32)は、わずかに長い、12アミノ酸のループを提示するMAPに変換され、一方1種のメンバー(ADAM10)は、わずかに短い、10アミノ酸のディスインテグリンループを保有するMAPに変換された(MAP配列アラインメントについては図2のAP10を参照されたい)。さらに、2種のAP(ADAM1およびADAM17)の場合では、ヘビ毒ディスインテグリンドメインのシステインパターン特性を回復させるために、各配列内の1つの追加的なネイティブな残基をアルギニン残基(MAP1を生成するため)またはシステイン残基(MAP17を生成するため)のいずれかと交換した(MAP配列アラインメントについては図2を参照されたい)。
【0031】
本明細書で使用される場合、「ドメイン間領域」または「スペーサー領域」とは、ADAMのメタロプロテアーゼとディスインテグリンドメインとの間のポリペプチド部分(「MDドメイン間領域」)、およびディスインテグリンドメインとシステインリッチドメインとの間のポリペプチド部分(「DCドメイン間領域」)のそれぞれを意味し、MDドメイン間領域は、APのN末端側の少なくとも10アミノ酸残基で始まり、DCドメイン間領域は、APのC末端側の少なくとも10アミノ酸残基で始まる。各ドメイン間の長さは5〜15アミノ酸である。
【0032】
23種のMAP全てのDNA配列を、新規に合成し、pET32a発現ベクター[30]に、細菌チオレドキシンA(TrxA)の下流にクローニングした。2009年11月12日に出願された、名称が「Method of expressing proteins with disulfide bridges with enhanced yields and activity」であるPCT特許出願第PCT/US09/64256に記載の通り、Origami B(DE3)細菌株にMAPを産生させた。この出願では、キメラのヘビ毒ディスインテグリンビクロスタチン(Vicrostatin(VCN))をOrigami B(DE3)/pET32a系において発現させることを実施形態として含む、米国特許出願公開第20060246541号に開示されている発現系に対する改善が記載されている。改善された方法を使用して、正しく折りたたまれた活性なMAPの量を増加させた。これは、Origami B細胞を選択性の低い環境において成長させ、したがって、異種組換え型のタンパク質の産生を誘導する間に、より最適なレドックス環境を示すVCN形質転換体の生成および増大を可能にすることによって実現された。他のE.coli株とは異なり、Origami Bは、E.coliにおける2つの主要な酸化還元経路の制御に決定的に関与する2つの重要な遺伝子、チオレドキシン還元酵素(trxB)およびグルタチオン還元酵素(gor)に変異を保有させることによって、この細菌の細胞質内の微小環境が、タンパク質におけるジスルフィド架橋の形成に対する触媒状態である、より酸化的なレドックス状態に人工的にシフトしているという点で独特である[18、29]。
【0033】
Origami B株は、野生型E.coli株を用いて得られる成長速度およびバイオマス収率と同様の成長速度およびバイオマス収率を有し、これにより、Origami B株が、VCNのような、発現させることが難しい組換えタンパク質についての魅力的かつスケーラブルな産生の代替になる。この株は、BL21のlacZY変異体からも得られる。BL21のlacY1欠失変異体(元のTuner株)により、培養物中の全ての細胞によるタンパク質の発現のレベルを調整可能にすることができる。lac透過酵素(lacY1)変異により、IPTG(ラクトース誘導体)を集団内の全ての細胞に均一に入れることが可能になり、これにより、制御された、より均一な誘導がもたらされる。IPTGの濃度を調整することにより、標的タンパク質の発現を最適化することができ、有意に低いレベルのIPTGで理論的に最大のレベルを実現することができる。したがって、Origami Bは、BL21(ompTおよびlonプロテアーゼを欠損している)宿主、Tuner(lacZY変異体)宿主およびOrigami(trxB/gor変異体)宿主の望ましい特性を1つの株内で併せ持つ。上記の通り、チオレドキシン還元酵素(trxB)とグルタチオン還元酵素(gor)の両方の変異により、細胞質におけるジスルフィド結合の形成が大きく促進される[29]。
【0034】
Origami B株により、BL21のような細胞質内環境が還元性であるE.coli株を超える明らかな利点が提供されるが(図4および5に、株間の発現レベルの比較が示されている)、単にOrigami B株およびpET32a発現ベクターを使用することでは、可溶性かつ/または活性な産物が生成されることは自動的には保証されない。Origami Bにおけるジスルフィドリッチポリペプチドの生成は、配列依存的であると思われる。例えば、MAP(例えば、MAP9およびMAP15)は、同じ系および産生技法を使用したという事実にもかかわらず、Origami Bにおいて、それらの対応するヒトADAM9およびADAM15のAPバージョンと比較して有意に高い発現収率で発現させることができる(図4および5)。したがって、APを修飾してMAPにした結果、Origami B細胞における発現収率がより高いディスインテグリンドメインを有するポリペプチドがもたらされ得る。
【0035】
さらに、ADAM9およびADAM15の発現したディスインテグリンドメイン(AP)をTEVプロテアーゼ処理およびRP−HPLC精製を伴うプロセスで精製した後、収集される遊離のポリペプチドは、不安定であり、凍結乾燥した粉末から再構成した後に溶液から沈殿するようである。対照的に、同じ精製ステップを用いることによって生成した対応するMAPポリペプチドは、凍結乾燥した後に水で再構成した際に、より可溶性かつ安定であるようである。
【0036】
本発明のMAPは、実質的に単離または実質的に精製されるように調製する。本明細書で使用される場合、MAPに関して、「実質的に精製された」(または単離された)という用語は、絶対的な純度を必要としない。その代わりに、MAPは、好ましくは50%超純粋であるか、より好ましくは少なくとも75%純粋であるか、および最も好ましくは少なくとも95%純粋であるか、少なくとも99%純粋であるか、最も好ましくは100%純粋であるという指標を表す。MAPは、本明細書に記載の通り、合成的に調製することができ、または組換え発現によって調製することができる。
【0037】
「実質的に」という用語は、本明細書で使用される場合、別段の指定のない限り、プラス10%またはマイナス10%を意味する。
【0038】
MAPを含有する薬学的組成物は、最低でも、所望の効果(例えば、がんの成長を阻害するか、またはがんの転移を妨げるかもしくは阻害する)を実現するために有効な量のMAPを含むべきであり、また、緩衝剤、塩、および/または適切なキャリアもしくは賦形剤を含む。一般に、これらの組成物中、MAPは、1日当たり約0.01mg/kg〜約50mg/kg、好ましくは1日当たり約0.1mg/kg〜約5.0mg/kg、および最も好ましくは1日当たり約0.1mg/kg〜約0.5mg/kgをもたらすために十分な量で存在する。
【0039】
MAPは、それを被験体の体内(例えば、血流内)に相当量で送達するために適した、種々のこれまでに公知の手段によって投与することができる。現在、適切な液体ビヒクルまたは賦形剤中のMAPを静脈内投与することが、好ましい投与経路であると考えられている。MAPは水溶性であり、したがって、適切な水溶液(例えば、リン酸緩衝生理食塩水)において有効に投与することができる。あるいは、MAPは、経口的に(適切な結合剤材料または賦形剤材料を用いて処方した錠剤またはカプセル剤の形態で、あるいは水性または油性の懸濁物、溶液、乳剤、シロップ剤またはエリキシル剤の形態で)、または非経口的な懸濁物として投与することができる。MAPは、動脈内または管内に送達することもでき、または意図された作用部位に局所的に導入することができる。当技術分野で周知の通り、アジュバントまたは賦形剤、例えば、局所麻酔薬、保存料、緩衝剤、潤滑剤、湿潤剤、着色料、矯味矯臭薬、充填剤および希釈剤などを、任意のこれらの処方物に適切に含めることができる。
【0040】
MAPは、リポソームによって送達することができる。リポソーム送達は、当技術分野で周知であり、ディスインテグリンの送達について記載されている。例えば、Swensonらは、乳がんの療法のための、リポソームに入れたコントルトロスタチンの静脈内送達の使用について記載している[26]。
【0041】
MAP、例えば、MAP9などは血管新生促進(pro−angiogenic)効果を示す(以下を参照されたい)。理論に縛られることを望まないが、これは、ADAMファミリーのメルトリンクラスのメンバー(ADAM9、ADAM12、およびADAM19)のより一般的な特性を示し得る。メルトリンは、筋肉細胞の分化誘導の過程において発現され、そこで、細胞融合および他のプロセスに関与し、新生血管の安定化において重要な役割を果たし得ることが最初に記載された間葉系のADAMである[33]。したがって、ADAMタンパク質のメルトリンクラスに由来する組換え型のポリペプチドを、疾患、例えば、冠動脈疾患(CAD)または他の虚血性の状態などにおいて、新しい毛細管網目構造の形成を刺激し、側副毛細血管の成長を維持するために治療的に使用することができる。したがって、例えば、MAPは、ステントを利用するCADおよび関連疾患の療法において使用するために、ステントをコーティングすること、またはステントに化学的にカップリングさせることができる。
【0042】
MAPを、薬物、毒素、放射性核種などと直接または間接的に結合体化し、これらの結合体を診断または治療への適用ために使用することもできる。例えば、MAPを使用して、関連のインテグリンを発現している組織または器官を同定または処置することができる。MAPまたはその生物活性のある断片もしくは部分を、検出可能な分子または細胞傷害性分子とカップリングさせ、1つまたは複数の関連のインテグリンを発現している細胞、組織または器官を有する哺乳動物に送達することができる。
【0043】
適切な検出可能な分子を、MAPに直接または間接的に付着させることができ、その適切な検出可能な分子としては、放射性核種、酵素、基質、補因子、インヒビター、蛍光マーカー、化学発光マーカー、磁気粒子などが挙げられる。適切な細胞傷害性分子を、ポリペプチドまたは抗体に直接または間接的に付着させることができ、その適切な細胞傷害性分子としては、細菌毒素または植物毒素(例えば、ジフテリア毒素、シュードモナス外毒素、リシン、アブリンなど)、ならびに治療的な放射性核種、例えば、ヨウ素−131、レニウム−188またはイットリウム−90などが挙げられる(ポリペプチドまたは抗体に直接付着させるか、または、例えば、キレート化部分によって間接的に付着させる)。MAPは、細胞傷害性薬物、例えば、アドリアマイシンなどと結合体化することもできる。検出可能な分子または細胞傷害性分子を間接的に付着させるために、検出可能な分子または細胞傷害性分子を、補体/抗補体対のメンバーと結合体化することができ、ここで、他方のメンバーはポリペプチドまたは抗体部分に結合する(ビオチン/ストレプトアビジンなど)。
【0044】
開示された組成物は、内皮細胞の機能障害に関連する疾患を処置するために、単独で、または組み合わせで使用することができる。ある実施形態では、開示された組成物は、がん処置のために有用である。がんは上皮起源のものであってよい。がんは、乳がん、結腸直腸がん、基底細胞がん、腺がん、胃腸がん、例えば、口唇がん、口腔がん、食道がん、小腸がんおよび胃がんなど、結腸がん、肝がん、膀胱がん、膵がん、卵巣がん、子宮頸がん、肺がん、および皮膚がん、例えば、扁平上皮細胞がんおよび基底細胞がんなど、前立腺がん、腎細胞がん、中枢神経系(CNS)のがん、白血病ならびに全身の上皮細胞に影響を及ぼす他の公知のがんであってよい。理論に縛られることを望むものではないが、がんの多くの形態において、ADAM(膜に繋ぎ止められた形態および/または分泌された形態)とそれらのインテグリン対受容体(integrin counter−receptor)との間に、がんが進行するために機構的に重要なプロセスである病原性のクロストークが存在する。これらの病理学的な相互作用は、がんにおける、腫瘍の進行における以下のいくつかの重要なインテグリン駆動ステップを実行するプロセスにおいて、高度に遊走性の細胞(がん細胞、炎症細胞または内皮細胞)によって組み立てられる多タンパク質複合体(すなわち、インバドソーム(invadosome))にインテグリンを介してADAMのメタロプロテイナーゼをリクルートする働きをする:重要な支持的な役割を果たすことが示されている炎症細胞による腫瘍間質の浸潤、腫瘍血管新生に関連する遊走性のステップ(ここで、既存の血管由来の内皮細胞は、新生血管を組み立てるためにリクルートされる)、および、最後に、転移に関連する遊走性かつ侵襲的な事象。これらの病理学的プロセスをMAPによって破壊することにより、悪性腫瘍が縮小する。それらの、抗血管新生効果、抗炎症効果および抗転移効果の潜在性に加えて、広範なスペクトルの抗インテグリンMAPは、腫瘍の分化にも影響を与える可能性がある。がん幹細胞に正しい分化シグナルを送る能力は、がん療法の主要な目標の1つのままである(療法の過程において、より安定かつ分化した表現型を獲得させることによる)。例えば、Yuanらにより、ヒト多形神経膠芽腫(脳がんの非常に侵攻性の形態)から単離した幹細胞特性を有する細胞集団を操作して、in vitroで分化させ、より良性の特徴をとるようにすることができることが示された[32]。可溶性のインテグリンリガンド(例えば、単一のMAPまたはMAPの組み合わせ)によって分化シグナルがもたらされ、種々のヒト悪性腫瘍のがん幹細胞を、それらの分化系列に完全にコミットさせ(commit)、分化したままになるように誘導することができる。
【0045】
本発明の組成物は、候補化合物を、MAP特異的結合について、MAPの存在下および非存在下で、前記候補化合物のインテグリンへの相対的な結合を比較することによってスクリーニングするためにも使用することができ、ここで、前記MAPの存在下でインテグリンの結合が減少することは、前記MAP特異的インテグリン結合性分子がMAP特異的であることを示す。
【0046】
本発明の組成物は、がん細胞によって提示される特異的な「インテグリンサイン」を利用することによって、がんを早期検出するために使用することができる。「インテグリンサイン」または「インテグリンプロファイル」は、細胞の表面で発現される1つまたは複数のインテグリンである。例えば、がんおよび肉腫は、腫瘍の分化および器官への局在化の状態に基づいて、特異的な「インテグリンサイン」を提示し得る。インテグリンサインに基づいて種々のがんの種類を同定し、診断するために、MAPに特異的な結合を、in vivoにおけるがんイメージング剤(診断的なイメージング)および/またはex vivoにおいて腫瘍検体を染色するための分子ツール(診断的な病理学的方法)として用いることができる。後者の場合、がん検体を様々なMAPで染色することにより、種々の腫瘍の起源および悪性度分類に応じた特徴的な染色パターンを導くことができる。本発明の組成物は、がんを病期分類するため、がんの進行または療法をモニターするために使用することができる。がんは上皮起源のものであってよい。がんは、乳がん、結腸直腸がん、基底細胞がん、腺がん、胃腸がん、例えば、口唇がん、口腔がん、食道がん、小腸がんおよび胃がんなど、結腸がん、肝がん、膀胱がん、膵がん、卵巣がん、子宮頸がん、肺がん、および皮膚がん、例えば、扁平上皮細胞がんおよび基底細胞がんなど、前立腺がん、腎細胞がん、ならびに全身の上皮細胞に影響を及ぼす他の公知のがんであってよい。MAPは、公知のインテグリン親和性を有するMAPが、特定の固形腫瘍のそれぞれの特異的なインテグリンの発現に基づいて、原発腫瘍、転移性の病巣、ならびに腫瘍の新脈管構造(neovasculature)に異なって結合する独特の能力を活用するために、PET(陽電子放出断層撮影法)プローブと一緒に使用することができる。例えば、MAPは、N−スクシンイミジル−4−18F−フルオロベンゾエート(18F−SFB)を最適化されたフッ素化反応条件下で用いることによって、アミノ基を通じて18Fで標識することができ、または、金属キレート化剤であるDOTA/NOTAと結合体化することによって64Cu/68Gaで標識することができる。さらに、蛍光標識したMAPは、in vivoで、およびex vivoで(生検検体上で)、腫瘍インテグリンの発現プロファイル分析のために使用することができる。
【0047】
本発明の組成物は、再生医療における分子ツールとして利用することもできる。例えば、人工的なECM足場(器官の型)をMAPまたはMAPの組み合わせでコーティングすることにより、幹細胞前駆体を誘導してこれらの足場に配置し、望ましい上皮の層および間葉系の層に分化させることができる。例えば、そのような1つまたは複数のMAPを含有する組成物でコーティングした膀胱の足場を使用して、幹細胞前駆体を尿路上皮および筋層の両方にコミットするように向かわせることができる。他の例としては、そのような1つまたは複数のMAPを含有する組成物でコーティングした食道の足場および直腸の足場を使用して、幹細胞前駆体を関連する組織層にコミットするように向かわせることができることが挙げられる。本発明の組成物は、in vitroで胚性幹細胞の成長を支持し、その幹細胞性を維持すること、または同じ系において、これらの全能性細胞がそれらの分化系列にコミットすることをガイドすること、ができる定義された組織培養プレートのコーティングを生み出すためにも使用することができる。
【0048】
本発明の組成物は、種々の虚血性の状態において、毛細血管の側副成長を支持するための新規の薬物、例えば、薬物溶出ステントなどとして使用することができる。例えば、MAPは、放射性核種、例えば、ベータ線放射性核種などと結合体化して、再狭窄を低下させることができる。そのような治療的手法は、放射性の療法を施す臨床医への危険が少ない。
【実施例】
【0049】
(実施例1)
MAPの調製および精製
図6は、pET32a発現ベクターにクローニングした合成のMAPのDNA配列の一覧を示す。図7は、MAPをpET32aベクターにクローニングするために利用した、対応するオリゴヌクレオチドプライマーの一覧を示す。図8は、Origami B(DE3)において発現させた全てのTrxA−MAP構築物のアミノ酸配列を示す。TrxAの活性部位およびディスインテグリンループの先端のトリペプチドモチーフに下線が引かれており、TEV切断部位が囲み枠で強調されており、TrxAと種々のMAP構築物との間のリンカー領域が黒い太字のイタリック体で示されている。MAP1およびMAP17のネイティブな残基と交換するために導入した新しい残基が、太字の二重下線で強調されている。
【0050】
細菌細胞および試薬。Origami B(DE3)E.coli株および細菌チオレドキシンA遺伝子(trxA)を保有するpET32a発現ベクターは、Novagen(San Diego、CA)から購入した。23種のMAP全てのDNA配列は、Epoch Biolabs,Inc.(Sugar Land、TX)によって新規に合成され、プラスミドに挿入された。APのDNA配列は、HUVEC(PromoCell GmbH、Heidelberg、Germany)、MDA−MB−435(ATCC、Manassas、VA)、MDA−MB−231(ATCC、Manassas、VA)、およびジャーカット(ATCC、Manassas、VA)を含めたいくつかの哺乳動物の細胞系から作製したcDNAライブラリーからPCR増幅した。APのDNA配列およびMAPのDNA配列をpET32a発現ベクターにさらにクローニングするために使用したオリゴヌクレオチドプライマーは、Operon Biotechnologies,Inc.(Huntsville、AL)によって合成された。APのDNA配列およびMAPのDNA配列をpET32a発現ベクターにクローニングするために使用した制限酵素およびリガーゼは全て、New England Biolabs,Inc.(Ipswich、MA)から購入した。組換えTEVプロテアーゼは、Invitrogen(Carlsbad、CA)から購入した。
【0051】
MAP発現ベクターの構築および組換え産生。pET32a発現ベクターに、TrxAの下流においてクローニングした合成のMAPのDNA配列が図6A〜Fに列挙されている。MAPをクローニングするために使用したオリゴヌクレオチドプライマーが図7A〜Bに列挙されている。生成した、TrxA遺伝子の下流にMAPのDNA配列がクローニングされたpET32aプラスミドを、最初にDH5α E.coliにおいて増幅し、精製し、配列決定した後、Origami B(DE3)E.coliに移入した。次いで、各MAP構築物について形質転換された細胞を、カルベニシリン(50μg/mL)、テトラサイクリン(12.5μg/mL)、およびカナマイシン(15μg/mL)を補充したLB寒天に播き、37℃で一晩成長させた。これらのプレートから、形質転換されたOrigami Bの個々のコロニー由来の各MAP構築物について、これらのコロニーを、カルベニシリン(50μg/mL)を含有するLB培地に移すことによって、複数の培養物を確立した。これらの最初の培養物を一晩成長させ、さらに、より大きな容積のカルベニシリン(50μg/mL)を含有するLB培地に接種するために使用し、それをシェーカーインキュベーター内、37℃、250rpmで、OD600が0.6〜1に到達するまで成長させた。この時点で、個々のMAP培養物由来の細胞を、1mMのIPTGを用いて誘導し、さらに4〜5時間、37℃、250rpmでインキュベートした。誘導期間の最後に、個々のMAP培養物由来の細胞を、4000×gでペレットにし、マイクロフルダイザー(Microfluidics M−110L、Microfluidics、Newton、MA)で溶解した。マイクロフルダイザーの作動条件は、適用圧力が14,000〜18,000psiであること、細菌のスラリー流速が1分当たり300〜400mlであること、およびスラリーを、プロセッサを複数回通過させることを含んだ。個々のMAP培養物から処理された溶解物由来の不溶性の細胞の破片を、遠心分離によって(40,000×g)除去し、各MAP培養物について、Trx−MAPを含有する可溶性の材料を収集した。次いで、収集した可溶性の溶解物中の発現された融合タンパク質(すなわち、Trx−MAP)を、組換えTEVプロテアーゼと一緒に室温で一晩インキュベートすることによって、タンパク質分解し、それによって、SDS−PAGEによってモニターされた通り、個々のMAPが、そのTrxA融合パートナーから効率的に切断された。タンパク質分解が完了したら、タンパク質分解された溶解物を、0.22μmのフィルターを通過させ、2回蒸留したH2Oにおいて1:100に希釈し、50,000MWCOのカートリッジ(Biomax50、Millipore)を通して限外濾過し、次いで、接線流限外濾過デバイス(Labscale TFF system、Millipore)を使用して5,000MWCOのカートリッジ(Biomax5、Millipore)に対して再濃縮した。
【0052】
MAPについて上記されているのと同じ手順を用いて、APをpET32aにクローニングし、Origami Bに導入して形質転換し、発現させた。
【0053】
組換えMAPの精製。MAPを、高速液体クロマトグラフィー(HPLC)手順を、ヘビ毒ディスインテグリンに対して以前確立されたプロトコールに従って用いることによって、濾過された溶解物から精製した[2]。ネイティブなCNを精製するために以前使用された標準の溶出条件を用いて、C18逆相HPLCによって精製を実施した[2]。上記の通り処理した個々の濾過された溶解物を、Vydac C18カラム(218TP54、Temecula、CA)にローディングした。カラムを、0.1%のTFAを含有する水溶液で10分間リンスし(1分当たり5ml)、その後、80%のアセトニトリルおよび0.1%のTFAを含有する移動相において、直線勾配(0〜100%)で150分にわたって溶出した。MAPは、35〜40%のアセトニトリルで溶出し始めた。
【0054】
MAPの発現分析。振とうフラスコ中、37℃で一晩成長させたE.coli形質転換体を、1mMのIPTG中、37℃、250rpmで5時間誘導した。誘導期間の最後に、細胞を4,000×gでペレットにし、複数の凍結融解サイクルによって溶解し、さらに40,000×gで遠心分離して不溶性の細胞破片を除去した。種々のE.coli宿主由来の可溶性の細胞溶解物5μlを、還元条件下で、プレキャスト4〜20%のNuSep iGel(NuSep Inc.、Lawrenceville、GA)にローディングし、次いでクーマシー(Coomassie)染色した。
【0055】
得られたAP9およびAP15の発現収率は、それらの対応するMAP(MAP9およびMAP15)と比較して低かった(図4および5)。MAP9およびMAP15はどちらも、Origami B(DE3)において、細菌培養物1リットル当たりのHPLC精製されたタンパク質が200mgから350mgであるバッチ間の発現収率で生成した。これらの高収率の精製組換えMAPは、誘導ステップの最後に、ペレットにした細菌の形質転換体を、マイクロフルダイザーを用いて溶解することによって実現された。同様の作出技法により、Origami B(DE3)系において組換えMAPを同様の発現収率で生成した(図9〜11)。対照的に、同様にジスルフィド架橋の形成を支持する、密接に関連する細菌宿主におけるMAPの発現によっては(例えば、trxB変異のみを有するK12誘導体であるAD494(DE3)、または、まれなコドンの使用について最適化されたOrigami B誘導体であるRosetta−gami B(DE3))、Origami B(DE3)における産生に対する追加的な利点は提供されないと思われた。
【0056】
(実施例2)
MAPの異なる細胞結合
上記の方法を用いてOrigami B(DE3)において産生されたTrx−MAPを、複数の細胞株への異なる結合について、フローサイトメトリーによって分析した(図12〜15)。それは、ヒト乳がん株(MDA−MB−231)、および異なるインテグリンプロファイルに基づく、異なる器官に対する向性を有するこの株の2つの転移性のサブクローン−骨−ホーミングサブクローン(MDA−MB−231BONE)および脳−ホーミングサブクローン(MDA−MB−231BRAIN)を含んだ。MDA−MB−231株の2種のサブクローンは、最初にそれらを単離した研究者からの寄贈品であった[31]。細胞を、示されているTrx−MAPと一緒にインキュベートし、対応する抗Trxポリクローナル抗血清(Sigma−Aldrich,Inc.、St.Louis、MO)を用いて調べた。結合した分子を、抗ウサギFITC−標識した抗体を用いてさらに検出した。FITCで標識した二次抗体のみ、または抗Trx抗血清および二次抗体のいずれかと一緒にインキュベートした細胞を対照として使用した。様々な系列の免疫細胞で発現され、免疫応答に関与することが以前示された、ADAM8およびADAM28に対応するTrx−MAP8およびTrx−MAP28は、フローサイトメトリー分析によって示された通り、ジャーカット細胞(T細胞白血病細胞株)に優先的に結合する(図15)。さらに、2つのHPLCによって精製したMAP(MAP9およびMAP15)をFITCで標識し、がん細胞株(MDA−MB−231、MDA−MB−435)、ヒト神経膠芽腫(GBM−CSC)から単離されたがん幹細胞集団[32]、ならびにヒト臍帯静脈内皮細胞(HUVEC)への直接的な結合についてフローサイトメトリーによって分析した。フローサイトメトリーのデータ(図16)は、精製されたMAP9およびMAP15が、全く異なるインテグリンプロファイルを示すことが予測される、異なる細胞株に非常によく結合することを示している。
【0057】
(実施例3)
MAPの抗血管新生効果
次いで、in vitro HUVEC管形成アッセイを用いて、MAPを血管新生活性について試験した。HUVEC細胞を、10nMのMAP9またはMAP15のいずれかの存在下で「内皮細胞管形成」プレート(BD Biosciences)に播いた。公知の管形成インヒビター(スラミン)を陰性対照として使用した。図17を参照されたい:パネルA−無処置の対照;パネルB−100μMのスラミン;パネルC−10nMのMAP9;パネルD−10nMのMAP15。細胞をCalcein AMで染色し、共焦点顕微鏡を使用してイメージングした。イメージは全て、同じ倍率で取得した(スケールバー=50μm)。このアッセイにおいて、MAP15は有意な抗血管新生活性を示した。このアッセイにおいて、MAP9は、無処置の対照と比較したときに、増加した数の管の形成を導くことによって血管新生促進効果を有すると思われた。このMAP9の血管新生促進効果は、MAP9のリポソーム処方物をMDA−MB−231異種移植の動物モデルにおいて静脈内に投与すると、腫瘍の成長が促進されること(すなわち、無処置の対照と比較して、速い腫瘍の成長、大きな腫瘍および生存の減少)を示すin vivoにおける観察によって支持される。
【0058】
図18Aは、MDA−MB−231モデルにおける、様々な処置によって誘導される腫瘍の成長の阻害を示す。同所性に接種したヌードマウス(乳房の脂肪パッド;完全なマトリゲル(Matrigel)中、マウス1匹当たり2.5×106個のMDA−MB−231細胞)について、処置を開始する前に、触知できる腫瘍を成長させた(矢印によって示される)。動物の群(n=10)に、LMAP15を1回の注射当たり100μgのMAP15の用量当量で週2回静脈内に投与する処置、または、アバスチン(1回の注射当たり400μg;およそ20μg/gr)を1週間に1回静脈内に投与する処置、または、ドセタキセル(DTX、160μg)を1週間に1回腹腔内に投与する処置、またはこれらの作用剤の組み合わせの処置を行った。対照群には、空のリポソームのみを与えた。対照群と比較して、全ての処置群において腫瘍の成長に有意な遅延が観察された。統計分析を、ダネットの事後多重比較検定を用いたANOVAを使用して行った(*は、P<0.001を示す)。図18Bは、動物の生存データを示す。処置群は、対照群と比較して生存の増加を示した(対照の動物は全て7週目までに死亡した)。LMAP15で処置した群またはLMAP15およびアバスチンで処置した群のいずれかの生存が最も高かった。この動物モデルからのin vivoにおける効力データは、LMAP15が、アバスチンまたはドセタキセルのいずれとも同様の抗腫瘍の効力を示すことを示している。リポソームMAP15単独で処置した動物は、同じ異種移植モデルにおいてアバスチンもしくはドセタキセルのいずれかで処置した動物、または組み合わせ療法で処置した動物と比較して、良好な生存率(すなわち、試験終了時にまだ生存している動物の数)を示した。
【0059】
(実施例4)
LMAP15の抗血管新生効果
単独でまたは組み合わせて投与されたLMAP15の抗血管新生効果を評価するために、MDA−MB−231試験における各群からの腫瘍を、死亡した動物または屠殺した動物から解剖し、摘出し、その後、微小血管の密度について免疫組織学的検査によって分析した。摘出した腫瘍をTissue−Tek O.C.T(「Optimal Cutting Temperature」化合物、Sakura Finetek USA)に包埋し、次いで、ドライアイス中で凍結させ、5ミクロンの切片に切り、アセトン中に固定し、染色するまで4℃で貯蔵した。CD31を染色するために、アセトンで固定したスライドをPBSで洗浄し、5%ヤギ血清を含有するPBSにおいてブロッキングし、次いで、PBS中に1:50に希釈したラットポリクローナル抗CD31抗体(BD Biosciences、San Diego、CA)200μlと一緒に室温で一晩インキュベートし、製造者のプロトコールに従ってスライドに適用した。この後、PBS中で複数回洗浄し(1回の洗浄当たり7分間)、1:100に希釈したビオチン化ヤギ抗ラット二次抗体200μlを加え、室温で45分間適用した。PBSでさらに3回洗浄した後、2mlのPBS中1滴に希釈したアビジン結合複合体(Vector Laboratories、Burlingame、CA)200μlを各スライドに室温で30分間適用し、その後、抗体で染色した微小血管を可視化するために、3−アミノ−9−エチルカルバゾール色素原を加えた。PBSでさらに3回洗浄した後、スライドを、Myers Hematoxylinを用いて対比染色し、その後マウントした。CD31で染色された微小血管を定量化するために、スライドを、「ランダム視野(random field)」分析[35、36]に供した。イメージを、顕微鏡に取り付けたOlympus E20Nデジタルカメラ(Olympus America、Melville、NY)を使用して、200×で、各スライドにおいてランダム視野から盲検的に取り込んだ。各群について、4つの腫瘍を染色し、40のランダム視野を分析した。各ランダム視野についてCD31陽性領域を、「SimplePCI」高度イメージングソフトウェア(C−Imaging Systems、Cranberry Township、PA)を使用して、染色された全面積の%としてコンピュータで計算し、次いで、各群について平均した。偏りを排除するために、ランダム視野のイメージを取り込み、その後、取り込んだイメージの処理および分析を盲検的に行った。LMAP15は、単独療法として、または異なる作用機構を有する他の抗血管新生薬(例えば、アバスチン)もしくは化学療法薬(例えば、ドセタキセル)と組み合わせて投与されると、この異種移植モデルにおいて微小血管の密度を有意に低下させることが示された(図19)。
【0060】
(実施例5)
転移性乳がんモデル
MAPの抗侵襲薬/抗転移薬としての治療効果を評価するために、自然発生的な転移性乳がんのいくつかの動物モデルにおいて光学的なルシフェラーゼイメージング手法(in vivo生物発光)を使用する。以下の細胞株に、アデノウイルス形質導入系を安定に感染させた:ルシフェラーゼおよび緑色蛍光タンパク質(GFP)レポーター遺伝子の両方を伴う、ヒト乳がん細胞株(MDA−MB−231、トリプル陰性細胞株)および2種のマウス乳がん株(4T1、HER2陰性株、およびD2F2、HER2陽性株)。ヒト異種移植MDA−MB−231モデルでは、完全なマトリゲルに懸濁させた細胞2×106個の接種材料をヌードマウスの乳房の脂肪パッドに注射し、形成された腫瘍が触知できるまで成長させた(移植のおよそ2週間後)。以下の処置群(1群当たり動物5匹)を形成する:対照群1(処置または操作を受けない動物)、対照群2(空のリポソームを静脈内に与えた動物)、対照群3(PBSを静脈内に与えた動物)、処置群1(アバスチンを静脈内に与えた動物)、処置群2(ドセタキセルを腹腔内に与えた動物)、処置群3(静脈内へのアバスチンと腹腔内へのドセタキセルの組み合わせを与えた動物)、処置群4(MAP15を静脈内に与えた動物)、処置群5(MAP15のリポソーム処方物を静脈内に与えた動物)、処置群6(静脈内へのMAP15、静脈内へのアバスチンおよび腹腔内へのドセタキセルの組み合わせを与えた動物)、および処置群7(静脈内へのMAP15のリポソーム処方物、静脈内へのアバスチンおよび腹腔内へのドセタキセルの組み合わせを与えた動物)。処置は、以下の通り施す:アバスチンおよびドセタキセルは、以前に文献において、このモデルにおいて原発腫瘍および転移病巣に対して効果的であると報告された最大の投与量を用いて週に1回投与し、MAP15は、1回の注射当たりポリペプチド100μgの用量で1日おきに投与し、リポソームMAP15は、1回の注射当たりポリペプチド100μgの用量当量で週2回投与する。
【0061】
MDA−MB−231モデルと同様に、2つのマウス乳がんモデル(4T1モデルおよびD2F2モデル)においてもMAP15の抗侵襲薬/抗転移薬としての効力を決定する。これらのモデルにおいて、PBS中の細胞5×105個の接種材料(4T1またはD2F2)を免疫応答性のBALB/cマウスの乳房の脂肪パッドに注射し、腫瘍が触知できるようになるまで成長させた(移植後およそ1〜1.5週間)。腫瘍が触知できるようになったら、以下の処置群(1群当たり動物5匹)について処置を開始する:対照群1(処置または操作を受けない動物)、対照群2(空のリポソームを静脈内に与えた動物)、対照群3(PBSを静脈内に与えた動物)、処置群1(アバスチンを静脈内に与えた動物)、処置群2(ラパチニブを静脈内に与えた動物)、処置群3(ハーセプチンを静脈内に与えた動物)、処置群4(アバスチン、ラパチニブおよびハーセプチンの組み合わせを静脈内に与えた動物)、処置群5(MAP15を静脈内に与えた動物)、処置群6(MAP15のリポソーム処方物を静脈内に与えた動物)、処置群7(MAP15、アバスチン、ラパチニブ、およびハーセプチンの組み合わせを静脈内に与えた動物)、および処置群8(MAP15のリポソーム処方物、アバスチン、ラパチニブ、およびハーセプチンの組み合わせを静脈内に与えた動物)。処置は、以下の通り施す:アバスチン、ラパチニブおよびハーセプチンは、以前に文献において、これらのモデルにおいて原発腫瘍および転移病巣に対して効果的であると報告された最大の投与量を用いて週に1回投与し、MAP15は、1回の注射当たりポリペプチド100μgの用量で1日おきに投与し、リポソームMAP15は、1回の注射当たりポリペプチド100μgの用量当量で週2回投与する。
【0062】
処置を開始する前、および処置を開始した後の、乳房の脂肪パッドおよび遠位の部位における腫瘍の成長を、全てのモデルにおいて、週に1回のXenogen生物発光イメージングによってモニターする。原発腫瘍サイズも、カリパスによって測定し、体積を式:1×w2×0.5に基づいて算出する。週に1回の生物発光(ルシフェラーゼ)イメージングのために、マウスに、ルシフェリン溶液(PBS中15mg/mLまたは30mg/kg、5〜50mg/kgの用量)を腹腔内経路によって注射し、約5〜15分間、麻酔していない動物において分布させる。次いで、マウスを、妨げられることなく動物を目視検査し、それらの呼吸の状況をモニターすることを可能にする、透明なPlexiglas麻酔箱(2〜4%のイソフルラン(isofluorane))に置く。この設定では、麻酔を箱に供給する麻酔送達管を、Xenogen IVIS 100イメージング計器のイメージングチャンバーの内側に位置する麻酔多岐管に同じ濃度の麻酔が送達されるように分離する。マウスを完全に麻酔した後、マウスを麻酔箱からイメージングチャンバー内の多岐管に取り付けた麻酔ノーズコーン(nose cone)に移し、扉を閉め、Xenogen計器を使用してイメージを取得する。イメージングの時間は、実験に応じて、面(背面/腹面)当たり1〜5分である。動物を背面から腹面に回転させる際(逆もまた同じ)、苦痛または生命力の変化の任意の徴候についてモニターする。取得したイメージを、対照および処置した腫瘍の部位におけるルシフェラーゼ活性のレベルを比較することによって、および各処置群における腫瘍からの発光に関連する曲線下分布面積を比較することによって評価する。上記の手法を用いることによって、原発腫瘍および生物発光の転移病巣に対するMAP15の効力を決定する。動物の体重も週2回測定し、動物を、ストレス応答または倦怠感の任意の徴候について視覚的に観察する。障害の重篤な徴候を示した動物は実験から排除する。
【0063】
これらの試験の終わりに(MDA−MB−231モデルにおける処置の8週間後、および4T1モデルおよびD2F2モデルにおける処置の3週間後)、これらの腫瘍モデルにおいて原発腫瘍の成長を阻害すること、転移病巣の数およびサイズまたはその両方を低下させること、および動物の生存を延長することにおいて、MAP15が、単独で、試験した単独療法のいずれか(アバスチンまたはドセタキセルまたはラパチニブまたはハーセプチンのいずれか)と同じくらい効果的であるか、またはそれよりも効果的であるかの決定を行う。さらに、MDA−MB−231モデルにおいてアバスチンとドセタキセルと組み合わせて、または4T1モデルにおいてアバスチンとラパチニブと組み合わせて、またはD2F2モデルにおいてアバスチンとラパチニブとハーセプチンと組み合わせて投与すると、MAP15は、これらの腫瘍モデルにおいて原発腫瘍の成長を阻害すること、転移病巣の数およびサイズまたはその両方を低下させること、および生存を延長することにおいて、任意の単独療法または他の組み合わせよりも効果的であることが証明された。
【0064】
(付属物)
ADAMの核酸配列およびアミノ酸配列
ADAMのRNA転写物に対応する核酸配列が図20〜42に提供されている。対応するADAMポリペプチドのアミノ酸配列が図43に提供されている。
【0065】
特に定義されていなければ、本明細書において使用される全ての技術用語および科学用語は、本発明が属する分野の当業者に一般に理解されているものと同じ意味を有する。
【0066】
本明細書に例示的に記載されている本発明は、本明細書において具体的に開示されていない、任意の要素、限定が存在しないところで適切に実施することができる。したがって、例えば、「含む(comprising)」、「含む(including)」、「含有する(containing)」などの用語は、限定されることなく、広く読み取られるべきである。さらに、本明細書で使用される用語および表現は、説明する用語として使用され、限定するものではなく、また、そのような用語および表現の使用において、示され、記載されている特徴の等価物またはその部分のいずれも排除するものではなく、特許請求の範囲に記載された本発明の範囲内で種々の改変が可能であることが理解される。
【0067】
したがって、本発明は、好ましい実施形態および任意選択の特徴によって具体的に開示されているが、当業者は、本明細書の開示の中で具体化される本発明の改変、改善および変形を行うことができること、およびそのような改変、改善および変形は、本発明の範囲内であるとみなされることが理解されるべきである。本明細書で提供される材料、方法および実施例は、好ましい実施形態を表し、例示的であり、本発明の範囲を限定するものではない。
【0068】
本明細書では、本発明が広範かつ一般的に記載されている。包括的な開示の範囲に入る狭義の種および下位のグループ分けのそれぞれも本発明の一部を形成する。これは、削除される材料が本明細書において具体的に列挙されているか否かにかかわらず、類概念から任意の対象物を取り除く但し書きまたは否定的な限定を伴う本発明の包括的な説明を包含する。
【0069】
本明細書において言及されている全ての刊行物、特許出願、特許、および他の参考文献は、全ての式および図面を含めて、それぞれが、個別に参照により組み込まれる場合と同じ程度に、明白にその全体が参照により本明細書に組み込まれる。矛盾する場合は、定義を含めた本明細書が優先する。
【0070】
他の実施形態は、以下の特許請求の範囲内に記載されている。
【0071】
【数1】
【0072】
【数2】
【0073】
【数3】
【0074】
【数4】
【特許請求の範囲】
【請求項1】
以下のアミノ酸残基:
a)Cys−Asp−Cys(CDC)モチーフのC末端側の最初のシステイン;
b)該最初のシステインのC末端側の2つの連続したアミノ酸;および
c)トリペプチドモチーフのC末端側のシステイン
において修飾されているADAM由来のポリペプチド(AP)を含む、修飾されたADAM由来のポリペプチド(MAP)であって、該アミノ酸残基の該修飾は、独立に、
a)欠失;
b)置換;および
c)化学的な修飾
からなる群より選択され、ここで、ADAMは「ディスインテグリンおよびメタロプロテイナーゼ」を意味し、該APは、
(i)該Cys−Asp−Cys(CDC)モチーフを含み、
(ii)図1に示されているトリペプチドモチーフを含み、そして
(iii)ADAMのメタロプロテアーゼドメイン、システインリッチドメイン、およびドメイン間のセグメントの全てまたは実質的に全てを欠く
ADAM由来のディスインテグリン様ドメインを含み、該ADAMはADAM17ではない、修飾されたADAM由来のポリペプチド(MAP)。
【請求項2】
前記MAPが、
【化1】
からなる群より選択されるADAMに由来する、請求項1に記載のMAP。
【請求項3】
前記MAPが、
【化2】
からなる群より選択される、請求項1に記載のMAP。
【請求項4】
前記アミノ酸残基の前記修飾が、欠失である、請求項1に記載のMAP。
【請求項5】
チオレドキシンをコードするN末端セグメントと、請求項1に記載の修飾されたADAM由来のポリペプチド(MAP)をコードするC末端セグメントとを含む融合タンパク質。
【請求項6】
請求項1に記載のMAPであって、
a)該MAPは、HUVECまたはMDA−MB−435細胞の、再構成基底膜を通じた移動を阻害することができる;
b)該MAPは、MDA−MB−435細胞におけるFAKのリン酸化のレベルを上昇させることができる;または、
c)該MAPは、培養物中のHUVECの管形成を阻害することができる、
MAP。
【請求項7】
請求項1に記載のMAPをコードする核酸。
【請求項8】
前記MAPをコードする核酸の5’側に、チオレドキシンをコードする核酸配列をさらに含む請求項7に記載の核酸であって、チオレドキシン−MAP融合タンパク質をコードする、核酸。
【請求項9】
請求項7に記載の核酸を含む発現ベクター。
【請求項10】
請求項9に記載の発現ベクターを用いて形質転換した原核宿主細胞であって、該発現ベクターは誘導性の制御下にあり、該宿主はまた、チオレドキシン還元酵素B(trxB)遺伝子および/またはグルタチオン還元酵素(gor)遺伝子に安定な変異を有し、該trxB変異および該gor変異は、成長の間に該宿主細胞内に該発現ベクター、ならびにtrxB変異およびgor変異が維持されるように選択可能である、原核宿主細胞。
【請求項11】
がんに罹患した個体を処置する方法であって、該個体に、有効量の少なくとも1つの請求項1に記載のMAPを投与するステップを含む、方法。
【請求項12】
前記がんが、インテグリンを発現しているがんである、請求項11に記載の方法。
【請求項13】
前記がんが、乳がん、結腸直腸がん、基底細胞がん、腺がん、胃腸がん、結腸がん、肝がん、膀胱がん、膵がん、卵巣がん、子宮頸がん、肺がん、皮膚がん、前立腺がん、腎細胞がん、中枢神経系(CNS)のがん、および白血病からなる群より選択される、請求項12に記載の方法。
【請求項14】
前記胃腸がんが、口唇がん、口腔がん、食道がん、小腸がんおよび胃がんからなる群より選択される、請求項13に記載の方法。
【請求項15】
前記皮膚がんが、扁平上皮細胞がんおよび基底細胞がんからなる群より選択される、請求項13に記載の方法。
【請求項16】
インテグリンがリガンドと結合することを阻害する方法であって、インテグリンを発現する細胞を、有効量の請求項1に記載のMAPと接触させるステップを含む、方法。
【請求項17】
前記MAPが、
【化3】
からなる群より選択される、請求項16に記載の方法。
【請求項18】
前記MAPが、チオレドキシンのN末端セグメントとの融合体をさらに含む、請求項16に記載の方法。
【請求項19】
a)前記MAPは、HUVECまたはMDA−MB−435細胞の、再構成基底膜を通じた移動を阻害することができる;
b)前記MAPは、MDA−MB−435細胞におけるFAKのリン酸化のレベルを上昇させることができる;または、
c)前記MAPは、培養物中のHUVECの管形成を阻害することができる、
請求項16に記載の方法。
【請求項20】
個体におけるがん細胞の存在を決定する方法であって、該がん細胞を、少なくとも1つの請求項1に記載のMAPと接触させるステップと、該少なくとも1つのMAPを検出するステップとを含む、方法。
【請求項21】
前記がんが、インテグリンを発現しているがんである、請求項20に記載の方法。
【請求項22】
前記がんが、乳がん、結腸直腸がん、基底細胞がん、腺がん、胃腸がん、結腸がん、肝がん、膀胱がん、膵がん、卵巣がん、子宮頸がん、肺がん、皮膚がん、前立腺がん、腎細胞がん、中枢神経系(CNS)のがん、および白血病からなる群より選択される、請求項20に記載の方法。
【請求項23】
前記胃腸がんが、口唇がん、口腔がん、食道がん、小腸がんおよび胃がんからなる群より選択される、請求項22に記載の方法。
【請求項24】
前記皮膚がんが、扁平上皮細胞がんおよび基底細胞がんからなる群より選択される、請求項22に記載の方法。
【請求項25】
前記MAPが標識されている、請求項20に記載の方法。
【請求項26】
前記標識が陽電子放射型断層撮影法プローブまたは蛍光プローブである、請求項25に記載の方法。
【請求項27】
人工的なECM足場を調製する方法であって、該人工的なECM足場を、請求項1に記載のMAPでコーティングするステップを含む、方法。
【請求項28】
前記人工的なECM足場に幹細胞前駆体を導入することをさらに含む、請求項27に記載の方法。
【請求項29】
前記人工的なECM足場が、膀胱の足場、食道の足場または肛門の足場を含む、請求項27に記載の方法。
【請求項30】
請求項1に記載のMAPを原核宿主細胞において発現させる方法であって、
a)請求項10に記載の原核宿主細胞を成長させるステップであって、前記発現ベクターは、第1の抗生物質において該発現ベクターを選択可能にする抗生物質耐性遺伝子を有し、前記trxB変異および前記gor変異は、反応器に播種するのに適した十分な数の細胞を得るための該第1の抗生物質および少なくとも1種の追加的な抗生物質の存在下における成長の間に、該宿主細胞内に該発現ベクターならびにtrxB変異およびgor変異が維持されるように該少なくとも1種の追加的な抗生物質において選択可能であり、ここで、該反応器において宿主細胞は成長し、融合タンパク質の発現が誘導される、ステップと;
b)ステップa)の細胞を該反応器に播種し、該細胞を成長させ、該融合タンパク質の発現を誘導するステップであって、該反応器内の該細胞を、該第1の抗生物質の存在下、かつ該少なくとも1種の追加的な抗生物質の非存在下で成長させる、ステップと
を含む、方法。
【請求項31】
前記宿主細胞が、前記trxB遺伝子および前記gor遺伝子の両方の変異体産物を発現する、請求項30に記載の方法。
【請求項32】
前記宿主細胞が、trxB遺伝子およびgor遺伝子の両方について変異体である、請求項31に記載の方法。
【請求項33】
前記trxB遺伝子および前記gor遺伝子が異なる抗生物質において選択可能である、請求項31に記載の方法。
【請求項34】
前記融合タンパク質のチオレドキシン部分が、配列:
【化4】
を有する、請求項30に記載の方法。
【請求項35】
前記宿主が、ompT遺伝子産物またはlon遺伝子産物の任意の1つまたは複数を欠損している、請求項30に記載の方法。
【請求項36】
切断部位をコードする配列が、チオレドキシンをコードする配列とジスルフィドリッチタンパク質をコードする配列との間に位置する、請求項30に記載の方法。
【請求項37】
前記融合タンパク質が、受容体に対するリガンドであるペプチド配列をさらに含む、請求項30に記載の方法。
【請求項38】
前記原核宿主細胞がOrigami株である、請求項30に記載の方法。
【請求項39】
請求項1に記載の組成物でコーティングしたステント。
【請求項40】
ADAM由来のポリペプチド(AP)を含む、修飾されたADAM由来のポリペプチド(MAP)であって、該ADAM由来のポリペプチド(AP)は、
(i)Cys−Asp−Cys(CDC)モチーフを含み、
(ii)図1に示されているトリペプチドモチーフを含み、ならびに
(iii)ADAMのメタロプロテアーゼドメイン、システインリッチドメイン、およびドメイン間のセグメントの全てまたは実質的に全てを欠く
ADAM由来のディスインテグリン様ドメインを含み、かつ該CDCモチーフのC末端側の最初のシステインとドメイン間のジスルフィド結合および該トリペプチドモチーフのC末端側のシステインとドメイン間のジスルフィド結合の破壊をもたらす1つまたは複数のアミノ酸の修飾をさらに含み、ADAMは、「ディスインテグリンおよびメタロプロテイナーゼ」を意味する、修飾されたADAM由来のポリペプチド(MAP)。
【請求項41】
図2に記載の任意のポリペプチド配列によるポリペプチド。
【請求項42】
請求項41に記載のポリペプチドをコードする核酸。
【請求項1】
以下のアミノ酸残基:
a)Cys−Asp−Cys(CDC)モチーフのC末端側の最初のシステイン;
b)該最初のシステインのC末端側の2つの連続したアミノ酸;および
c)トリペプチドモチーフのC末端側のシステイン
において修飾されているADAM由来のポリペプチド(AP)を含む、修飾されたADAM由来のポリペプチド(MAP)であって、該アミノ酸残基の該修飾は、独立に、
a)欠失;
b)置換;および
c)化学的な修飾
からなる群より選択され、ここで、ADAMは「ディスインテグリンおよびメタロプロテイナーゼ」を意味し、該APは、
(i)該Cys−Asp−Cys(CDC)モチーフを含み、
(ii)図1に示されているトリペプチドモチーフを含み、そして
(iii)ADAMのメタロプロテアーゼドメイン、システインリッチドメイン、およびドメイン間のセグメントの全てまたは実質的に全てを欠く
ADAM由来のディスインテグリン様ドメインを含み、該ADAMはADAM17ではない、修飾されたADAM由来のポリペプチド(MAP)。
【請求項2】
前記MAPが、
【化1】
からなる群より選択されるADAMに由来する、請求項1に記載のMAP。
【請求項3】
前記MAPが、
【化2】
からなる群より選択される、請求項1に記載のMAP。
【請求項4】
前記アミノ酸残基の前記修飾が、欠失である、請求項1に記載のMAP。
【請求項5】
チオレドキシンをコードするN末端セグメントと、請求項1に記載の修飾されたADAM由来のポリペプチド(MAP)をコードするC末端セグメントとを含む融合タンパク質。
【請求項6】
請求項1に記載のMAPであって、
a)該MAPは、HUVECまたはMDA−MB−435細胞の、再構成基底膜を通じた移動を阻害することができる;
b)該MAPは、MDA−MB−435細胞におけるFAKのリン酸化のレベルを上昇させることができる;または、
c)該MAPは、培養物中のHUVECの管形成を阻害することができる、
MAP。
【請求項7】
請求項1に記載のMAPをコードする核酸。
【請求項8】
前記MAPをコードする核酸の5’側に、チオレドキシンをコードする核酸配列をさらに含む請求項7に記載の核酸であって、チオレドキシン−MAP融合タンパク質をコードする、核酸。
【請求項9】
請求項7に記載の核酸を含む発現ベクター。
【請求項10】
請求項9に記載の発現ベクターを用いて形質転換した原核宿主細胞であって、該発現ベクターは誘導性の制御下にあり、該宿主はまた、チオレドキシン還元酵素B(trxB)遺伝子および/またはグルタチオン還元酵素(gor)遺伝子に安定な変異を有し、該trxB変異および該gor変異は、成長の間に該宿主細胞内に該発現ベクター、ならびにtrxB変異およびgor変異が維持されるように選択可能である、原核宿主細胞。
【請求項11】
がんに罹患した個体を処置する方法であって、該個体に、有効量の少なくとも1つの請求項1に記載のMAPを投与するステップを含む、方法。
【請求項12】
前記がんが、インテグリンを発現しているがんである、請求項11に記載の方法。
【請求項13】
前記がんが、乳がん、結腸直腸がん、基底細胞がん、腺がん、胃腸がん、結腸がん、肝がん、膀胱がん、膵がん、卵巣がん、子宮頸がん、肺がん、皮膚がん、前立腺がん、腎細胞がん、中枢神経系(CNS)のがん、および白血病からなる群より選択される、請求項12に記載の方法。
【請求項14】
前記胃腸がんが、口唇がん、口腔がん、食道がん、小腸がんおよび胃がんからなる群より選択される、請求項13に記載の方法。
【請求項15】
前記皮膚がんが、扁平上皮細胞がんおよび基底細胞がんからなる群より選択される、請求項13に記載の方法。
【請求項16】
インテグリンがリガンドと結合することを阻害する方法であって、インテグリンを発現する細胞を、有効量の請求項1に記載のMAPと接触させるステップを含む、方法。
【請求項17】
前記MAPが、
【化3】
からなる群より選択される、請求項16に記載の方法。
【請求項18】
前記MAPが、チオレドキシンのN末端セグメントとの融合体をさらに含む、請求項16に記載の方法。
【請求項19】
a)前記MAPは、HUVECまたはMDA−MB−435細胞の、再構成基底膜を通じた移動を阻害することができる;
b)前記MAPは、MDA−MB−435細胞におけるFAKのリン酸化のレベルを上昇させることができる;または、
c)前記MAPは、培養物中のHUVECの管形成を阻害することができる、
請求項16に記載の方法。
【請求項20】
個体におけるがん細胞の存在を決定する方法であって、該がん細胞を、少なくとも1つの請求項1に記載のMAPと接触させるステップと、該少なくとも1つのMAPを検出するステップとを含む、方法。
【請求項21】
前記がんが、インテグリンを発現しているがんである、請求項20に記載の方法。
【請求項22】
前記がんが、乳がん、結腸直腸がん、基底細胞がん、腺がん、胃腸がん、結腸がん、肝がん、膀胱がん、膵がん、卵巣がん、子宮頸がん、肺がん、皮膚がん、前立腺がん、腎細胞がん、中枢神経系(CNS)のがん、および白血病からなる群より選択される、請求項20に記載の方法。
【請求項23】
前記胃腸がんが、口唇がん、口腔がん、食道がん、小腸がんおよび胃がんからなる群より選択される、請求項22に記載の方法。
【請求項24】
前記皮膚がんが、扁平上皮細胞がんおよび基底細胞がんからなる群より選択される、請求項22に記載の方法。
【請求項25】
前記MAPが標識されている、請求項20に記載の方法。
【請求項26】
前記標識が陽電子放射型断層撮影法プローブまたは蛍光プローブである、請求項25に記載の方法。
【請求項27】
人工的なECM足場を調製する方法であって、該人工的なECM足場を、請求項1に記載のMAPでコーティングするステップを含む、方法。
【請求項28】
前記人工的なECM足場に幹細胞前駆体を導入することをさらに含む、請求項27に記載の方法。
【請求項29】
前記人工的なECM足場が、膀胱の足場、食道の足場または肛門の足場を含む、請求項27に記載の方法。
【請求項30】
請求項1に記載のMAPを原核宿主細胞において発現させる方法であって、
a)請求項10に記載の原核宿主細胞を成長させるステップであって、前記発現ベクターは、第1の抗生物質において該発現ベクターを選択可能にする抗生物質耐性遺伝子を有し、前記trxB変異および前記gor変異は、反応器に播種するのに適した十分な数の細胞を得るための該第1の抗生物質および少なくとも1種の追加的な抗生物質の存在下における成長の間に、該宿主細胞内に該発現ベクターならびにtrxB変異およびgor変異が維持されるように該少なくとも1種の追加的な抗生物質において選択可能であり、ここで、該反応器において宿主細胞は成長し、融合タンパク質の発現が誘導される、ステップと;
b)ステップa)の細胞を該反応器に播種し、該細胞を成長させ、該融合タンパク質の発現を誘導するステップであって、該反応器内の該細胞を、該第1の抗生物質の存在下、かつ該少なくとも1種の追加的な抗生物質の非存在下で成長させる、ステップと
を含む、方法。
【請求項31】
前記宿主細胞が、前記trxB遺伝子および前記gor遺伝子の両方の変異体産物を発現する、請求項30に記載の方法。
【請求項32】
前記宿主細胞が、trxB遺伝子およびgor遺伝子の両方について変異体である、請求項31に記載の方法。
【請求項33】
前記trxB遺伝子および前記gor遺伝子が異なる抗生物質において選択可能である、請求項31に記載の方法。
【請求項34】
前記融合タンパク質のチオレドキシン部分が、配列:
【化4】
を有する、請求項30に記載の方法。
【請求項35】
前記宿主が、ompT遺伝子産物またはlon遺伝子産物の任意の1つまたは複数を欠損している、請求項30に記載の方法。
【請求項36】
切断部位をコードする配列が、チオレドキシンをコードする配列とジスルフィドリッチタンパク質をコードする配列との間に位置する、請求項30に記載の方法。
【請求項37】
前記融合タンパク質が、受容体に対するリガンドであるペプチド配列をさらに含む、請求項30に記載の方法。
【請求項38】
前記原核宿主細胞がOrigami株である、請求項30に記載の方法。
【請求項39】
請求項1に記載の組成物でコーティングしたステント。
【請求項40】
ADAM由来のポリペプチド(AP)を含む、修飾されたADAM由来のポリペプチド(MAP)であって、該ADAM由来のポリペプチド(AP)は、
(i)Cys−Asp−Cys(CDC)モチーフを含み、
(ii)図1に示されているトリペプチドモチーフを含み、ならびに
(iii)ADAMのメタロプロテアーゼドメイン、システインリッチドメイン、およびドメイン間のセグメントの全てまたは実質的に全てを欠く
ADAM由来のディスインテグリン様ドメインを含み、かつ該CDCモチーフのC末端側の最初のシステインとドメイン間のジスルフィド結合および該トリペプチドモチーフのC末端側のシステインとドメイン間のジスルフィド結合の破壊をもたらす1つまたは複数のアミノ酸の修飾をさらに含み、ADAMは、「ディスインテグリンおよびメタロプロテイナーゼ」を意味する、修飾されたADAM由来のポリペプチド(MAP)。
【請求項41】
図2に記載の任意のポリペプチド配列によるポリペプチド。
【請求項42】
請求項41に記載のポリペプチドをコードする核酸。
【図1A】


【図1B】

【図1C】

【図4】

【図5】

【図6A】

【図6B】

【図6C】

【図6D】

【図6E】

【図6F】

【図7A】

【図7B】

【図8A】

【図8B】

【図8C】

【図8D】

【図8E】

【図8F】

【図8G】

【図8H】

【図9】

【図10】

【図11】
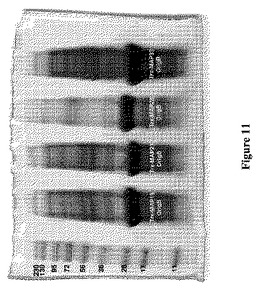
【図17A】

【図17B】

【図17C】

【図17D】

【図20】

【図21】

【図23】

【図24】

【図34】

【図35】

【図37】

【図40】

【図41】

【図43A】

【図43B】

【図43C】

【図43D】

【図43E】

【図43F】

【図43G】

【図2A】

【図2B】

【図2C】
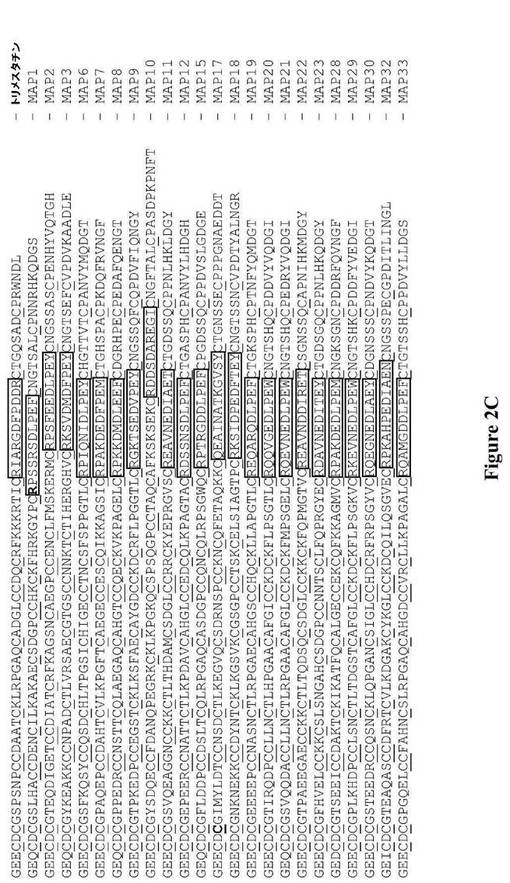
【図3】

【図12】
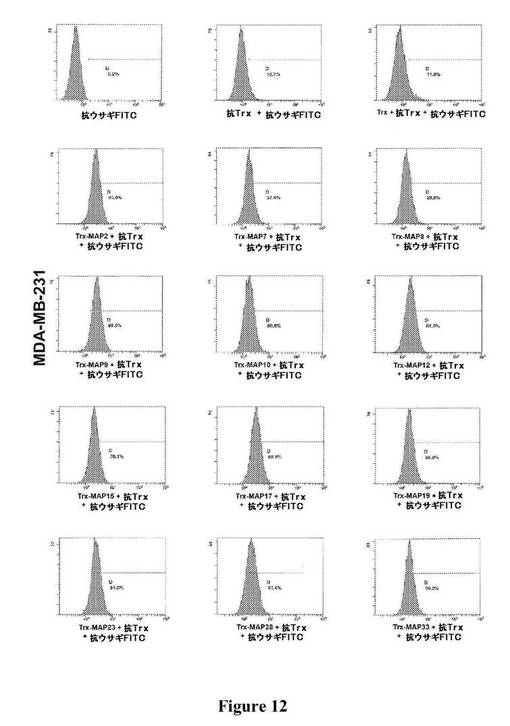
【図13】
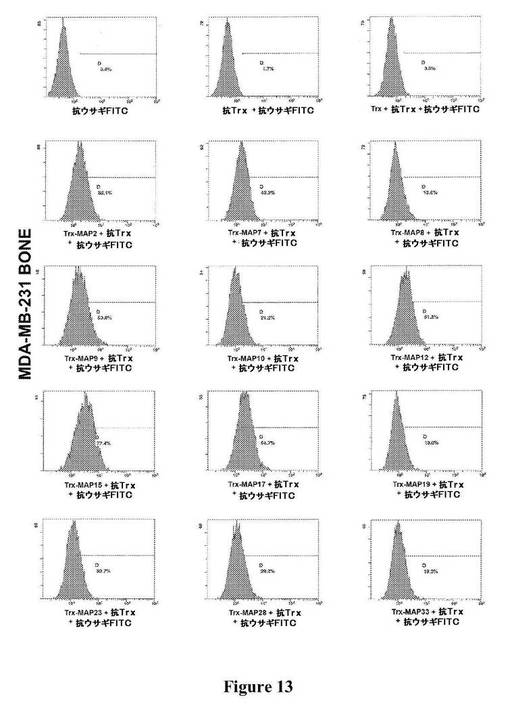
【図14】
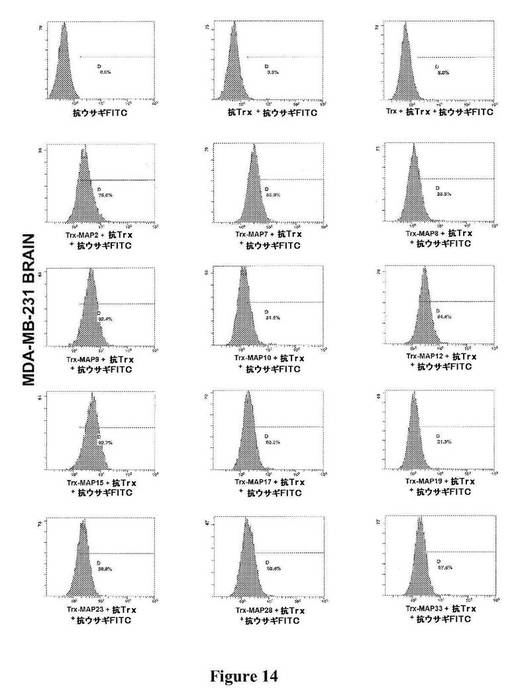
【図15】
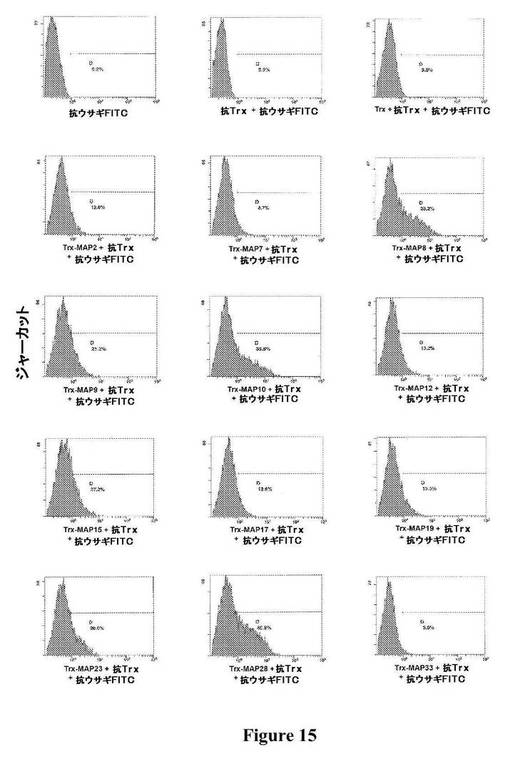
【図16】
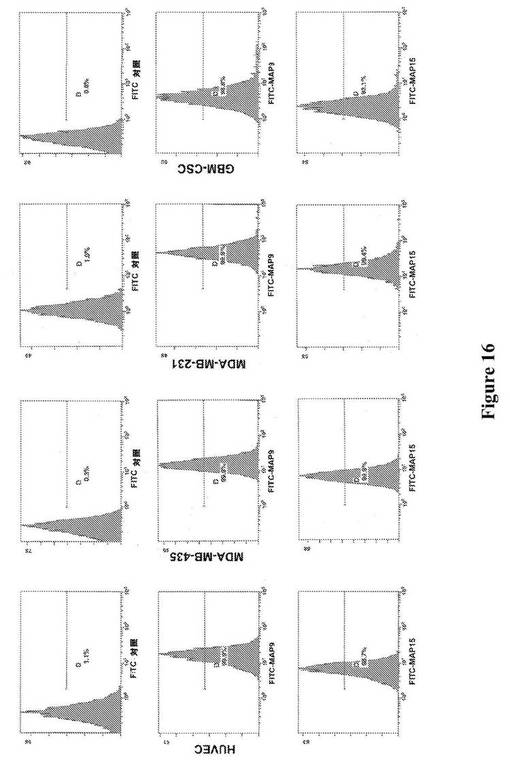
【図18】
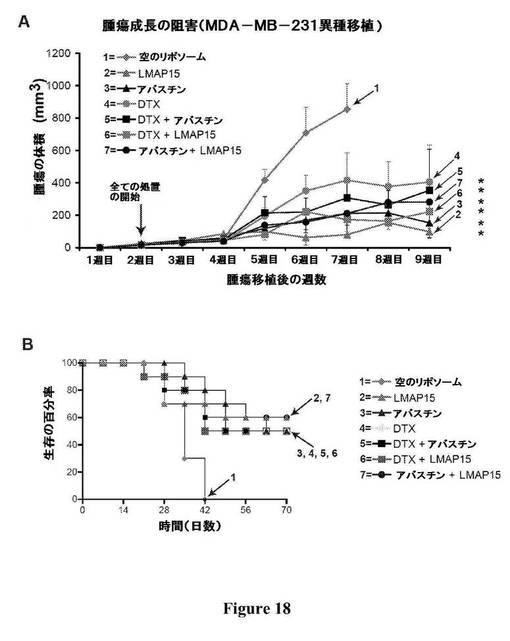
【図19】
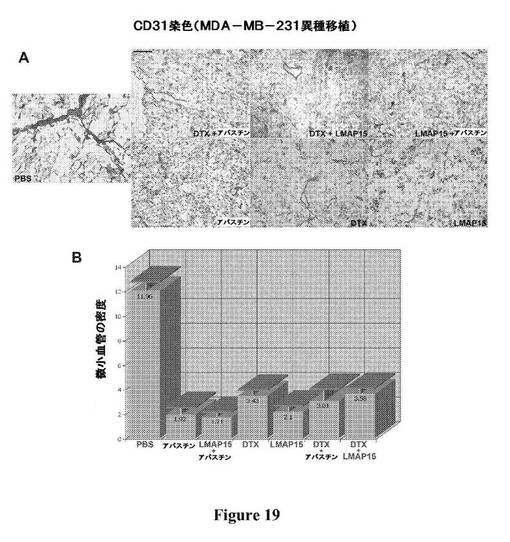
【図22】

【図25A】

【図25B】

【図26A】

【図26B】

【図27A】

【図27B】

【図28A】

【図28B】

【図29A】

【図29B】

【図30】

【図31A】

【図31B】

【図32】

【図33A】

【図33B】

【図33C】

【図36A】

【図36B】

【図36C】

【図36D】

【図38A】

【図38B】

【図39A】

【図39B】

【図42A】

【図42B】

【図1B】
【図1C】
【図4】
【図5】
【図6A】
【図6B】
【図6C】
【図6D】
【図6E】
【図6F】
【図7A】
【図7B】
【図8A】
【図8B】
【図8C】
【図8D】
【図8E】
【図8F】
【図8G】
【図8H】
【図9】
【図10】
【図11】
【図17A】
【図17B】
【図17C】
【図17D】
【図20】
【図21】
【図23】
【図24】
【図34】
【図35】
【図37】
【図40】
【図41】
【図43A】
【図43B】
【図43C】
【図43D】
【図43E】
【図43F】
【図43G】
【図2A】
【図2B】
【図2C】
【図3】
【図12】
【図13】
【図14】
【図15】
【図16】
【図18】
【図19】
【図22】
【図25A】
【図25B】
【図26A】
【図26B】
【図27A】
【図27B】
【図28A】
【図28B】
【図29A】
【図29B】
【図30】
【図31A】
【図31B】
【図32】
【図33A】
【図33B】
【図33C】
【図36A】
【図36B】
【図36C】
【図36D】
【図38A】
【図38B】
【図39A】
【図39B】
【図42A】
【図42B】
【公表番号】特表2013−519374(P2013−519374A)
【公表日】平成25年5月30日(2013.5.30)
【国際特許分類】
【出願番号】特願2012−552964(P2012−552964)
【出願日】平成23年2月9日(2011.2.9)
【国際出願番号】PCT/US2011/024243
【国際公開番号】WO2011/100362
【国際公開日】平成23年8月18日(2011.8.18)
【出願人】(510090748)ユニバーシティー オブ サザン カリフォルニア (5)
【Fターム(参考)】
【公表日】平成25年5月30日(2013.5.30)
【国際特許分類】
【出願日】平成23年2月9日(2011.2.9)
【国際出願番号】PCT/US2011/024243
【国際公開番号】WO2011/100362
【国際公開日】平成23年8月18日(2011.8.18)
【出願人】(510090748)ユニバーシティー オブ サザン カリフォルニア (5)
【Fターム(参考)】
[ Back to top ]