
修飾PHOS/PSTS細胞周辺リン酸結合タンパク質を持つE.COLI宿主細胞、及び組換えFABを製造する方法
本発明は、野生型において該組換え抗体と共精製する1又はそれ以上のE.coliタンパク質の少なくとも一つの物理的性質を変えるためにE.coli宿主細胞が遺伝的に修飾されていることが特徴である組換え抗体を発現するE.coli宿主細胞を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、組換えタンパク質の発現に使用するE. coli宿主細胞に関するものであり、特に組換え抗体の製造用の改良されたE. coli宿主細胞を提供するものである。
【背景技術】
【0002】
組換えタンパク質の大規模、経済的な精製はますますバイオテクノロジー産業の重要な課題となっている。一般的に、そのタンパク質の遺伝子を含む組換えプラスミドの挿入により目的のタンパク質を製造するように操作した哺乳動物または細菌の細胞系を使用して組換えタンパク質は製造される。タンパク質は直接細胞から周囲の培地に分泌されるか又は細胞内に作られる。後者のタンパク質については、その精製操作の最初の段階は細胞の溶解又は破壊であり、これは機械的切断、浸透圧ショック、又は酵素処理を含む種々の方法により行うことができる。その破壊により細胞の内容物はホモジネートに放出され、そしてさらに非細胞断片を生じ、これは一般的に分別遠心分離又は濾過により除去される。小規模であるが、細胞の自然死によるタンパク質の直接的分泌及びタンパク質生産の過程における細胞内宿主細胞タンパク質の放出に関して同じ問題が生じる。このようにして生産された組換えタンパク質は、混入する宿主細胞タンパク質が毒性又は免疫原性を持つ可能性があるので、それらから分離精製する必要がある。抗体のような治療に必要な組換えタンパク質については高純度であることが必須である。
【0003】
最近まで抗体及び抗体フラグメントは通常哺乳動物細胞において製造されていたが、E. coli, Pichia酵母及び植物のような代替生産システムも使用されてきた。これ等の生産代替方法は特異的抗体生産及び規模、コスト、速さ、資本リスク及び生物学的安全性の間のバランスによって促進されてきた(Humphreys and Glover, 2001, Current Opinion in Drug Discovery and Development, 4, 172-185)。醗酵プラント、操業時間、培地成分及び工程回転率/資本減価償却のより明確なコストに加えて、「下流工程」、すなわち、粗製品の保管、取扱い及び精製に関係する相当なコストが存在する。
【発明の開示】
【発明が解決しようとする課題】
【0004】
大規模な抗体精製は、コストの効率がよくそして物理的耐久性があるので、主として分別沈殿、イオン交換、サイズ排除及び疎水性相互作用クロマトグラフィーに依存している。これ等の方法を使用して精製を行うことができないとするならば、アフィニティークロマトグラフィーのような高価な分析方法をスケールアップするためにかなりの開発が必要になるであろう。混入する宿主タンパク質がpI、大きさ又は疎水性のような組換え抗体の物理的性質に類似している場合にはこの問題が起こりそうである。これ等の混入物を除去するには、大規模に行う場合には極めて好ましくない専門家による精製工程を必要とするであろう。従って、混入する宿主タンパク質が特別に問題である場合には、組換えにより製造される抗体の大規模精製工程を改良しそして単純化するというニーズが存在する。
【課題を解決するための手段】
【0005】
本発明は、組換え抗体を製造するために改良されたE. coli宿主細胞を提供することにより上記問題を解決する。特に、本発明は、該細胞が野生型においては該組換え抗体と共精製する1以上のE. coliタンパク質の少なくとも一つの物理的性質を改変するために遺伝的に修飾されていることを特徴とする組換え抗体を生産するためのE. coli宿主細胞を提供する。
【0006】
従って、組換え抗体と共精製しないように選択したE. coliタンパク質の物理的性質を改変することによりE. coliにおいて生産された抗体の精製工程を改良できることを、われわれは立証することができた。本発明のE. coli宿主細胞を使用した結果として、該細胞を使用して生産された抗体の精製工程を改良することができ、例えば、野生型のE. coliにより生産されたものよりは工程は速やかでありそして/又はより経済的でありうる。したがって、本発明における抗体の精製における改良は、E. coli宿主タンパク質の物理的性質を修飾することにより生じた精製工程に対する何らかの有利な改変であると考えることができる。改良には、限定はしないが、精製の速度の改善、精製コストの減少、または生産された抗体の品質の向上が含まれる。本発明の一態様において、組換え抗体の精製工程は全精製段階の除去により改良され、コスト及び時間の両者の節減を生じる。好ましくは、除去される工程はアフィニティークロマトグラフィー工程、イオン交換工程、サイズ排除工程、又は疎水性相互作用工程である。好ましい態様において、抗体Fab’フラグメントの精製の間に除去される工程は疎水性相互作用工程でありそして改変されるE. coliタンパク質はリン酸結合タンパク質(PhoS/PstS)である。好ましくは該工程の除去は、Fab'のモルベースで、元の精製工程のコストに比較して約15%削減をもたらす。
【0007】
本発明のもう一つの態様において、本発明の宿主細胞において生産された組換え抗体の精製工程は必要なカラムマトリックスの量を減少させることにより改良され、それにより物質のコスト及び操作時間を減少させる。好ましくは通常は組換え抗体と共精製しそして抗体に結合するカラムの能力を減少させる宿主タンパク質の混入数を減少させることによりこれを達成する。好ましい態様において増加するカラムの能力はカチオン交換カラムでありそしてカラムへの結合を防ぐために改変されるE. coli宿主タンパク質はジペプチド結合タンパク質(DppA)、マルトース結合タンパク質(MBP)及びチオレドキシンである。
【0008】
このように、本発明により組換え抗体を製造するための改良されたE.coli宿主細胞が提供される。本発明のE.coli宿主細胞は組換え抗体を生産することができる天然に存在する生物又は突然変異生物でありうる。しかし、好ましくは、宿主生物は組換えDNA技術を使用して組換え抗体の生産をコードする異種DNA配列を導入された生物又は生物の子孫である。本発明に使用するのに適した特別な宿主E. coli株は、これに限定せずに、MC4100,TG1,TG2, DHB4, DH5α,DH1, BL21, XL1Blueを含む。一つの好ましいE.coli宿主はE.coli W3110 (ATCC 27,325)であり、広く組換えタンパク質醗酵に使用されている宿主株である。E. coliにおける外来遺伝子の発現は発現ベクター中に遺伝子のcDNAコピーを挿入することにより行われる。多くの型の発現ベクターが使用可能である。該ベクターは通常マルチクローニング部位(発現カセット)により分離されたプラスミドのDNA複製起点、抗生物質選別マーカー及びプロモーター及び転写ターミネーター及びリボソーム結合部位をコードするDNA配列によって構成されている。
【0009】
用語「野生型」とは、混入する宿主タンパク質が修飾されていない宿主細胞のことである。この宿主細胞の他のタンパク質がこの発明以外の目的で修飾されていることはありうる。
【0010】
遺伝子修飾のために選択されたE. coli宿主タンパク質は、野生型では精製の際に組換え抗体と共精製されることが知られているタンパク質である。用語「共精製」とは同じセットの精製条件の下に他のタンパク質を伴ってあるタンパク質が精製されることである。典型的にこれはクロマトグラフィーのような精製工程の際に組換え発現した抗体に伴って混入したE. coliタンパク質が精製されることである。
【0011】
用語「遺伝子修飾」とは遺伝子配列の1以上の欠失、挿入、置換又は突然変異によりその遺伝子によりコードされたタンパク質の物理的性質に変化を生じることである。好ましくは、これ等の変化はコードされたタンパク質の生理学的または生物学的活性に影響しない。
【0012】
用語「疎水性」とは、水又は有機溶媒中のタンパク質の溶解性及び固体表面及びマトリックスとの相互作用に及ぼす全タンパク質表面上又は局所的部分表面の疎水性及び親水性アミノ酸の総合的影響のことである。
【0013】
用語「pI又は等電点」とはポリペプチドの陽電荷と陰電荷がバランスを取るpHのことである。pIはポリペプチドのアミノ酸残基のネット電荷から計算することができるし、あるいは等電点電気泳動により測定することができる。
【0014】
用語「物理的性質」とは、生理学的又は生物学的活性でなくタンパク質そのものの物理的性質のことである。好ましくは、物理的性質とは大きさ、疎水性および等電点のようなタンパク質の特性である。E. coli宿主タンパク質のいずれの物理的性質が改変されるかは組換え抗体の精製工程及び必要な改良によって規定されるであろう。例えば、ある条件下に特別なイオン交換カラムへの結合を阻止するためにタンパク質の等電点を変更することができる。
【0015】
遺伝子修飾により改変される混入E. coliタンパク質の物理的性質には、限定はしないが、等電点及び/又は大きさ及び/又は疎水性を含めることができる。タンパク質の大きさとはタンパク質の分子量のことである。混入タンパク質の物理的性質の改変はコードするヌクレオチド配列内の特定の配列の付加、欠失、置換又は挿入のいずれかの組合せにより行うことができる。一態様において、タンパク質の物理的性質はN又はC末端の少なくとも一つのアミノ酸の付加又は欠失により改変することができる。一態様において、混入する宿主タンパク質の物理的性質はC末端へアミノ酸タグを付加することにより改変することができる。好ましい態様において改変される物理的性質は等電点でありそしてアミノ酸タグはC末端へ結合したポリアスパラギン酸タグである。一態様において該タグの付加により改変されるE. coliタンパク質はジペプチド結合タンパク質(DppA),マルトース結合タンパク質(MBP)、チオレドキシン及びリン酸結合タンパク質(PhoS/PstS)である。一つの特別な態様においてE. coliリン酸結合タンパク質(PhoS/PstS)のpIは、C末端へ6個のアスパラギン酸残基を含む、ポリアスパラギン酸タグ(ポリD)の付加により7.2から5.1に減少する。
【0016】
物理的性質を改変するために混入するE.coliタンパク質の特定の残基を変更するか又はN又はC末端タグの付加と組み合わせて改変することもまた好ましい。該改変はタンパク質の大きさを変えるための挿入又は欠失又はpI又は疎水性を変えるためのアミノ酸の置換を含むことができる。一態様においてこの残基はタンパク質の表面に局在している。好ましい態様においてPhoSタンパク質の表面残基はタンパク質のpIを減少させるために変更される。好ましくはPhoSタンパク質の機能を維持するためにリン酸結合に関係する残基(Bass, US5,304,472)を避ける。好ましくはタンパク質の表面から突き出ているか又は塩基性残基の大きな群の中又は近くにあるリシン残基が標的にされる。一つの態様において、PhoSタンパク質はC-末端に結合したヘキサポリ-アスパラギン酸タグを持っているが一方で分子の逆の末端における表面残基は置換の標的にされる。好ましくは中性残基を酸性残基に換えるよりも大きなpI変化の可能性を与えるために選択されたリシン残基はグルタミン酸又はアスパラギン酸に置換される。この中における置換突然変異体の命名は文字次いで数字次いで文字からなる。最初の文字は野生型タンパク質におけるアミノ酸を示す。数字はアミノ酸置換が行われるアミノ酸の位置であり、そして二番目の文字は野生型のアミノ酸に代って使用されるアミノ酸を示す。本発明におけるPhoSの好ましい突然変異においてリシン残基(K)275,107,109,110,262,265,266,309,313がグルタミン酸(E)に、単点又は複合点の突然変異として、置換され、さらにリシン(K)318はアスパラギン酸(D)に、単点又は複合点の突然変異として、置換することができる。好ましくは単点置換突然変異はK262E, K265E及びK266Eである。好ましくは、複合点突然変異はK265/266E及びK110/265/266Eである。より好ましくは、全ての突然変異がC-末端に結合したポリアスパラギン酸(ポリD)と複合しておりそして任意にK318D置換も伴っている。好ましい態様において突然変異は少なくとも2単位のpIの減少を生じる。好ましくは本発明の突然変異によりPhoSのpIは7.2から約4及び約5.5の間へ減少する。本発明の一態様において突然変異ポリD K318D,ポリD K265/266E及びポリD K110/265/266Eを使用してE.coliのPhoSタンパク質のpIは7.2から約4.9、約4.8及び約4.5にそれぞれ減少した。
【0017】
好ましくはE.coli宿主タンパク質の全ての遺伝子修飾により必要な精製工程において組換え抗体と共精製されないタンパク質を生じる。好ましくはPhoSタンパク質の変更はイオン交換の際に抗体Fab'フラグメントと共精製されないタンパク質を生じ、そして好ましくは以前必要であった疎水性相互作用工程は最早必要でなくなる。好ましくはPhoSタンパク質の突然変異は全て組換え抗体と同じ塩濃度ではカチオン交換カラムから溶出されないタンパク質を生じる。好ましくは突然変異PhoSタンパク質は100 mMより低いNaClで溶出するが、しかし抗体はpH 4.5において200 mMで溶出する。さらに好ましいのは突然変異PhoSタンパク質は5.0又はそれ以下のpHでは全くカチオン交換カラムに結合しないであろう。本発明の一態様において突然変異PhoSタンパク質は野生型PhoSが結合しないpH 8以上においてアニオン交換カラムに結合するであろう。
【0018】
好ましくは混入する宿主タンパク質の物理的性質の改変はタンパク質の生物活性又は機能に有意な影響を与えない。本発明の一態様において突然変異PhoSタンパク質はPhoS欠損E.coli株、ANCC75(Amemura et al., 1982, Journal of Bacteriology, 152, 692-701)を補充する能力により測定される機能を維持している。好ましくはE.coli宿主タンパク質における突然変異の全ては野生型と比較してE. coliの増殖又は組換え抗体の収量に影響しない。
【0019】
本発明のE. coli宿主細胞において生産される組換え抗体は、いずれかの抗原に結合する免疫グロブリンフラグメント、例えばFv, Fab, Fab'及びF(ab')2フラグメント、及びいずれかのその誘導体、例えば一本鎖Fvフラグメントを含む免疫グロブリン分子のいずれかである。抗体の生産は当業者によく知られておりそして抗体フラグメントは封入体からのリフォールディングにより又は細菌細胞周辺への分泌による機能的発現によりE.coliにおいて定型的に生産されている(Pluckthun and Pack 1997, Immunotechnology, 3, 83-105; Verma et al., 1998, Journal of Immunological Methods, 216, 165-181)。
【0020】
所与抗体の精製工程はE.coliにおける該抗体の発現に続く純粋な組換え抗体を作製するために必要な精製段階の連続である。組換え抗体の精製は当業者によく知られておりそして個々の抗体に対して最小の生成段階を使用して最大の収量及び純度を生じる精製工程を工夫することができる。その例としてはイオン交換クロマトグラフィー、疎水性相互作用クロマトグラフィー、サイズ排除、等電点電気泳動、逆相HPLC, クロマトフォーカシング、SDS-PAGE, 硫酸アンモニウム沈殿、及びアフィニティークロマトグラフィー(例えば、タンパク質A, タンパク質G,又は捕捉試薬としての抗原を使用)を含む1以上の精製段階を使用して組換え抗体を精製することができる。組換え抗体の大量生産に使用される最も一般的な方法は、コスト効率がよくそして物理的に耐久性がよいので、分別沈殿、イオン交換、疎水性相互作用クロマトグラフィー及びサイズ排除クロマトグラフィーである。これ等の方法はその物理的特性、それぞれ等電点、疎水性及び大きさに基づいてタンパク質を分離する。混入するE.coliタンパク質が組換え抗体と類似の物理的性質を持っている場合には、これ等の方法を使用してタンパク質を分離することはできないであろう。このような抗体にはタンパク質-A及びタンパク質-Gアフィニティークロマトグラフィーのような費用のかかる精製段階を追加する必要があるであろう。
【0021】
所与組換え抗体に対する精製工程を検査することにより、組換え抗体と共精製され除去するのが困難である混入E. coliタンパク質が存在するか否かを当業者は速やかに決定することができる。該混入タンパク質の存在は精製工程を工夫した人には既知であり、例えばその除去のためだけに工程に組み込まれた追加の段階がありうる。そのほかには、カラムフラクションのSDS-PAGEゲルはどのタンパク質が組換え抗体と一貫して共精製されているかを明らかにするであろう。
【0022】
精製工程を検査することにより、組換え抗体の精製が、例えばより少ない又は異なる精製段階を使用することにより改良することができるように当業者は該混入タンパク質のどの物理的性質を変えるのが好ましいのか決定することができる。例えば、高価なアフィニティークロマトグラフィーを使用するよりもサイズ排除クロマトグラフィーを使用して組換え抗体から分離できるように混入タンパク質の大きさを減少させたり又は増加させたりすることが適当であろう。そのほかには、イオン交換又は疎水性相互作用クロマトグラフィーにより分離できるように混入タンパク質の等電点又は疎水性を改変することも適当である。所与の精製工程において混入タンパク質の物理的性質の最適変化を当業者は容易に確認することができる。1を超えるE.coli タンパク質の性質及び/又は該タンパク質の1を超える物理的性質を変化させることも好ましい。
【0023】
組換え抗体と共精製されるE.coliタンパク質が選択されそして精製工程を改良するために該タンパク質のどの物理的性質が変えられることが好ましいかが決定されたならば、物理的性質の改変が行われうるような該タンパク質をコードする遺伝子をクローニングする必要がある。これは当業者には定型業務でありそして最初にタンパク質配列をそのタンパク質から得る必要がある。これは、例えば、ウエスタンブロットからのタンパク質のN-末端配列分析により又は該ブロットから作製したトリプシン分解又はCNBrフラグメントの配列分析により行うことができる。これらは全てタンパク質の配列を得るための定型的方法である。該ウエスタンブロットは混入宿主タンパク質を含むカラムフラクションの電気泳動を行ったSDS-PAGEゲルから作製することができる。該タンパク質から得られたアミノ酸配列は次いで、E.coliの全ゲノム配列が一般に公開されているSwissProt又はGenbankのようなデータベースから全タンパク質及びDNA配列を相同性探索により同定するために使用される(Blattner et al., 1997, Science, 277, 1453-1462)。次いで核酸配列に基づくプライマー及び鋳型としてE.coli DNAを使用するPCRのような既知技術を使用して、混入タンパク質をコードする遺伝子をクローニングすることができる。
【0024】
混入するE.coliタンパク質が同定されそして該タンパク質をコードする遺伝子がクローニングされてしまえば、組換え抗体の精製を改良することになる必要なタンパク質の物理的変化を生じるようにその遺伝子を修飾することができる。タンパク質の物理的性質を改変するための方法は当業者によく知られておりそして目的とする結果を達成するために多くの系統的改変が必要となるであろう。タンパク質配列、可能であれば、配列整列及び結晶構造の分析から、改変の処理を受けるタンパク質の領域を同定することは当業者には可能である。選択される領域は物理的性質における意図する変化によるであろう。例えば、タンパク質のpI又は疎水性を変えるためには特別な電荷又は疎水性を持つ表面に露出した残基に焦点を当てる必要があるであろう。大きさを変えるには削除又は修飾することができる特別なドメインに焦点を当てることができる。Rasmol又はWebLab Viewer Liteのようなコンピュータープログラムはタンパク質の結晶構造を見るには有用であり、いずれの残基を変更するかについて賢明にして学識のある選択をすることを可能にする。タンパク質の活性又は構造にとって重要な残基に関するその他の情報は公開情報から入手可能であろう。
【0025】
タンパク質の機能は残っているのが好ましいので、できればタンパク質配列整列及び結晶構造の検査により活性部位領域を避けるようにすべきである。結晶構造が使用できない場合には、タンパク質の機能又は発現を破壊せずに改変できる残基を同定することは、アラニンスキャンニング突然変異誘起(Cunningham and Wells, 1989, Science, 244, 1081-1085)のような当業者既知の方法を使用して試験することができる。
【0026】
タンパク質の物理的性質を改変する好ましい方法はタンパク質タグの付加によるものであり、この方法は当業者に広く知られている。現在ではこれは混入宿主タンパク質を変えるよりもむしろ組換えタンパク質の精製を容易にするためにその性質を変えるために使用されている。このようなタグが組換えタンパク質に使用された場合には、タンパク質機能を回復し、溶解性を増し又はタグが抗原性であるので治療のために、それは精製されたタンパク質から除去される必要がある。これにはしばしば困難を生じそしてこの技術を上手に適用するための最大の障害となることがある(Sassenfeld, 1990, Tibtech, 8, 88-93)。本発明においては、タグは組換え抗体自身でなく混入するE. coliタンパク質のpIを変えるために使用されているので該タグの除去は不必要である。タンパク質のpI,疎水性及び大きさは全てアミノ酸タグの付加により変更することができ、その性質は必要とする成果によるであろう。例えば、タンパク質の等電点を変更するのに適するタグはポリ-アルギニンタグ(Sassenfeld and Brewer, 1984, Biotechnology, 2, 76-81; Brewer US 4,532,207; Niederauer et al., 1996, Biotechnology Progress, 10, 237-245; Stempfer et al., 1996, Nature Biotechnology, 14, 329-334)、ポリ-グルタミン酸タグ(Dalbφge et al., 1987, Bio/Technology, 5, 1447-1457; Niederauer et al., 1996)及びbespoke タンパク質ドメインタグ(Graslund et al., 2000, Protein Engineering, 13, 703-709 and Graslund et al., 2002, Journal of Chromatography, 942,157-166)である。疎水性を改変するために使用することができるタグにはポリ-フェニルアラニンタグ(Persson et al., 1988, Analytical Biochemistry, 172, 330-337)及びエラスチン様ポリペプチド(Meyer and Cholkoti, 1999, Nature Biotechnology, 17, 1112-1115)が含まれる。標的タンパク質の大きさ増加させる実行可能な融合相手として使用されてきた多数のタンパク質及びタンパク質のドメインも存在し、それらにはアルカリホスファターゼ(Carrier et al., 1995, Journal of Immunological Methods, 181, 177-186)、β-ガラクトシダーゼ(Nielsen et al., 1988, JIMM 111, 1-9)、マルトース結合タンパク質(di Guan et al., 1988, Gene, 67, 21-30)、GST(グルタチオンS転移酵素、Smith and Johnson 1988, Gene, 67, 31-40)、セルロース結合ドメイン(Ong et al., 1989, Bio/Technology, 7, 604-607)、DsbA(Collins-Racie et al., 1995, Bio/Technology, 13, 982-987)、DsbC(Novagen)、チオレドキシン及びNusA(Novagen)が含まれる。該融合により10〜60 kDaの大きさの増大を生じる。該タグは単独で又は以下に記述する混入タンパク質に対する他の特定の改変と組み合わせて使用することができる。
【0027】
宿主タンパク質の物理的性質を改変するその他の好ましい方法は特定残基の修飾であり、その方法は当業者に良く知られている。例えば、ズブチリシンの静電気的性質はX線結晶構造をガイドとして使用して複数の荷電アミノ酸残基を導入することによりズブチリシンの表面電荷を変えることにより修飾された(Egmond et al., 1996, In Subtilisin Enzymes; Practical Protein Engineering, R Bott and C Betzel eds, 219-228)。Marttila et al., 1998, FEBS Letters, 441, 313-317,は、天然のpI 10.5に対して9.4から4.7の範囲のpIを持つ数個のアビジンの荷電変異体を作製した。この変異体は既知結晶学的データ並びに比較配列整列に基づいてリシン及びアルギニンのような塩基性残基を中性又は酸性アミノ酸と置換することにより作製された。
【0028】
突然変異のためにいずれの表面残基を選択するかは、活性部位に対する残基の位置、溶媒暴露の程度、他の表面残基との潜在的相互作用、関係するアミノ酸R-基の相対的溶媒和能力、アミノ酸のpKa、関係するアミノ酸の相対的疎水性/親水性、関係するR-基の相対的長さを含む構造的及び立体的考慮により支援される。
【0029】
特定の残基の突然変異はオリゴヌクレオチド仲介突然変異誘起(Zoller and Smith, 1982, Nucleic Acid Research, 10, 6487)のような当業者既知の方法により行うことができる。タンパク質の等電点を変更させるには、中性アミノ酸を目的電荷のアミノ酸に換えるか、又は逆の電荷を持つアミノ酸残基に換える必要がある(すなわち、アスパラギン/グルタミン酸の代わりにリシン/アルギニン)。典型的にタンパク質のpIを上昇させるためには、アスパラギン酸及びグルタミン酸のような酸性アミノ酸を置換してリシン及びアルギニンのようなより塩基性のアミノ酸をタンパク質に取り込まなければならない。pIを減少させるには、置換の選択は逆にしなければならない。
【0030】
そのほかには混入するタンパク質の疎水性を減少させることも好ましいであろう、そしてこれはバリン、ロイシン、イソロイシン、フェニルアラニン、トリプトファン、メチオニン及びプロリンのような疎水性残基をより親水性又は極性のある残基、例えばセリン、トレオニン、システイン、チロシン、アスパラギン酸、グルタミン酸、アスパラギン、グルタミン、ヒスチジン、リシン又はアルギニンに置換することにより行うことができる。そのほかに疎水性を増加させるにはアミノ酸置換を逆にしなければならない。
【0031】
タンパク質の発現及び任意の機能に影響することなく削除することができるタンパク質の特別なドメイン又は部分を同定することにより、混入するタンパク質の大きさを減少させることも可能である。これ等はN又はC-末端ドメイン又はインフレーム融合を形成するために削除することができる露出ループである。そのほかにはサイズ排除クロマトグラフィーを使用する分離を改良するために混入するタンパク質の大きさを増加させるために、既に記述したようなアミノ酸タグ又はタンパク質ドメインをタンパク質に融合することができる。
【0032】
混入するタンパク質の発現、活性又は物理的性質に及ぼす置換、欠失、挿入又はタグの正確な影響を予測することは困難であることは、当業者には理解されるであろう。必要な成果を達成するために当業者は一連の種々の変更を試験するであろう。これ等は連続して又は平行して創りそして試験することができる。その効果は以下に記述されるような定型的スクリーニング検定により評価され、必要があればさらに修飾が行われることを当業者は理解するであろう。
【0033】
各変異体を試験するために改変されたタンパク質は非修飾タンパク質を発現しないE.coli宿主細胞において発現されなければならない。これは欠失変異株のようなこの遺伝子を発現しないE.coli宿主を使用することにより又はその遺伝子の発現が抑制される条件下に該宿主を培養することにより達成される。改変された混入タンパク質は該タンパク質の高レベルの発現を生じるプラスミドの方法で導入することができる。そのほかには、変異タンパク質はE.coliゲノムの中に改変遺伝子を直接組み込んで内在性遺伝子を置換することにより試験することができる。
【0034】
各改変タンパク質の発現に成功したことはE.coliの醗酵の後のSDS-PAGE分析により確認することができる。改変タンパク質が適切に発現しない場合がありうるが、その突然変異は廃棄すべきである。変異タンパク質の発現に成功した後、物理的性質の改変はクロマトグラフィー及びゲル電気泳動により評価することができる。例えば、改変が大きさの減少を生じるはずである場合には、サイズ排除カラム及びSDS-PAGEにより試験することができる。改変がpIの変化を生じるはずである場合には、イオン交換クロマトグラフィー及び等電点電気泳動を使用して試験することができる。改変が疎水性の変化を生じるはずである場合には、疎水性相互作用クロマトグラフィー及び溶媒溶解性を使用して試験することができる。組換えタンパク質が存在しなくてもこれ等の方法により改変タンパク質を評価することは当業者には可能である。任意に、試験前に精製組換え抗体を抽出物に添加することにより組換え抗体からの分離を確認することができる。
【0035】
タンパク質の物理的性質が改変されても修飾E.coliタンパク質の生物学的機能は保持されていることが好ましい。突然変異体の機能はタンパク質同定に関わる技術分野において広く知られている種々の方法で試験することができる。例えば、変異タンパク質はその生物学的活性を立証する生物学的検定、例として示すならば、酵素検定により試験することができる。そのほかに、変異タンパク質をコードする遺伝子を目的の遺伝子を欠失したE.coli突然変異体に補充して、次いで生物活性又は細胞増殖を試験することに使用できる。このような突然変異体は既に多数存在するか、又は当業者は容易に創ることができる。
【0036】
上記の方法を使用して、改良精製工程の条件を充たしそして好ましくは機能を維持する少なくとも一つの突然変異体を同定することは当業者には可能であろう。好ましくは、ゲノムに組み込まれた後に突然変異体のいずれかがE.coliの増殖又は組換え抗体の収量に有害な影響を及ぼす場合には、この段階において1を超える突然変異体を選択するであろう。野生型の遺伝子を置換するように選択した突然変異遺伝子をE.coliゲノムの中に組み込むことは当業者既知の方法を使用して行うことができる(Hamilton et al., 1989, Journal of Bacteriology, 171, 4617-4622)。E.coliゲノムDNAに認められる配列に相補的である改変DNA配列は形質転換用のベクターに含まれる。このベクターによるE.coliの形質転換はゲノムの相同組換え及び野生型遺伝子の代わりに改変遺伝子の挿入を生じる。
【0037】
修飾遺伝子配列で野生型遺伝子を置換した後、元の野生型E.coli発現システムにおいて行うように改良E.coli宿主細胞を望ましい組換え抗体遺伝子配列で形質転換する。次いでそのE.coli宿主細胞を元の野生型と同じ条件下に培養しそして細胞の増殖及び組換え抗体の収量を測定しそして野生型と比較する。組換え抗体の収量は使用する組換え抗体用の当業者既知の標準的方法を使用して試験することができる。例えば、抗体Fab'フラグメントはELISAにより定量することができる。好ましくは抗体収量及びE.coli増殖に逆効果は認められてはならない。逆効果が認められるならば選択された他の突然変異の一つを使用すべきでありあるいは新しい突然変異体を作製すべきである。このような突然変異を系統的に又は平行して試験できることは当業者には既知であろう。
【0038】
改変宿主タンパク質が組換え抗体と最早共精製しないことを確認するために、組換え抗体を目的の工程を使用して精製しそして改変宿主タンパク質の存在を例えばSDS-PAGEにより監視すべきである。物理的性質の変化のために改変宿主タンパク質は期待通りに行動するはずでありそして最早組換え抗体とは共精製しないはずである。
【0039】
以下の実施例は説明のために、そして限定するためでなく提供される。
【0040】
実施例
材料と方法
DNA操作
一般的DNA操作には標準的方法を使用した。制限酵素はBoehringer Mannheimから、Precision PlusをStratageneから入手した以外はTaq ポリメラーゼはRocheから入手した。プラスミドの調製はQiagenキットをメーカー説明書に従って使用しておこなった。オリゴヌクレオチドはSigma-Genosys Ltd.,Pampisford, U.K.から入手した。オリゴヌクレオチド及び領域をコードするPCR産物の配列はPRISM Big Dyeサイクル配列分析キット及びGenetic Analyzerソフトウエアを使用するABI PRISM-3100シークエンサーを使用して両鎖の配列分析により確認した。超コンピテントE.coli株XL1Blue MRF' Kan (Stratagene)を全てのDNA操作に使用した。野生型E.coli W3110(ATCC ref.27325)を洗いそして氷冷滅菌10%(v/v)グリセロール中で3回濃縮することにより電気穿孔にコンピテントにし、パルス調節装置を装着したBioRad Gene Pulser を使用して2000V 25 mS及び200Ωにおいて電気穿孔を行った。新しい単一W3110コロニーを100 mlの2xPY培地中37℃でOD600が約0.5〜0.8に達するまで増殖した。その後培養を氷で15分間冷却し、氷冷下試験管中で4℃ 4000 g 10分間遠心分離して細胞ペレットにした。細胞ペレットを再懸濁し、洗い、滅菌脱イオン水で作製した氷冷10%(v/v)グリセロール中で3回ペレットにした。最後のペレット化の後、細胞ペレットを滅菌脱イオン水で作製した氷冷10%(v/v)グリセロールで最終容量10 mlに再懸濁した。細胞を直ちに使用するかまたは液体窒素で凍結して-70℃で保存した。
【0041】
細菌用複合培地及び無リン酸培地
「PhoS培地」を、phoレギュロンを抑制するPO4及びペプチドリッチ培地を供給するためにE.coliの一般的増殖の全て、プラスミド調製及びPhoS発現実験に使用した(「PhoS培地」=1%(w/v)トリプトン、0.5%(w/v)イースト抽出物、0.3%(w/v) KH2PO4, 0.7%(w/v) K2HPO4,0.5%(w/v) NaCl及び0.5%(w/v) DIFCOのcasamino acids)。「無PO4」と定義される培地はNeidhardt et al., 1974 Journal of Bacteriology, 119, 736-747に記述されたMOPS培地にグルコースを0.5%(w/v)、casamino acidsを 0.5%(w/v)、及びチアミンを1 mM添加したものである。組み込み実験に用いられた細胞は2xPY(2%寒天(w/v)、1% フィトーン(w/v)、0.5%イースト抽出物(w/v)、0.5% NaCl(w/v)及び1 M NaOHでpH 7.0にする)を使用して増殖した。必要があれば培地には3%寒天(w/v)、カルベニシリンを200 μg/ml又はクロラムフェニコールを20 μg/ml、IPTGを200 μM及びXPを40 μg/ml添加した。
【0042】
Fab'測定用ELISA
ELISAプレートをPBS中2 μgml-1のHP6045 で4℃において終夜コーティングした(マウスIgG2aモノクロナール抗-ヒトIgG Pan Fd(CH1)であるHP6045をATCCのハイブリドーマHP6045から得た。イムノグロブリンはプロテインA精製により培養上清から回収し、微量のウシIgGをヒツジ抗-ウシIgGカラムで除去した)。dH2Oで4x洗った後、プレート上でサンプルを系統的1/2希釈及び標準を100 μlのサンプル/コンジュゲート緩衝液(100 mM Tris/Cl pH 7, 100 mM NaCl, カゼイン0.2%(w/v)、Tween 20 0.0002%(v/v))中に作製し、そしてプレートを室温で1時間250 r.p.m.において振とうした。dH2Oで4x洗った後、100 μlの表示抗体GD12, HRP結合F(ab')2抗ヒトカッパ鎖(The Binding Site, Birmingham, U.K.)を加え、サンプル/コンジュゲート緩衝液中に1/1000に希釈し、プレートを室温で1時間200 r.p.m.において振とうした。dH2Oで4x洗った後、100 μlの3-3'-5-5’テトラメチルベンジジン(TMB)基質を加え(0.1 M酢酸/クエン酸ナトリウム pH 6、100 μg/ml TMB, H2O20.01%(v/v))、そして自動化プレートリーダーを使用してA630を記録した。細胞周辺抽出物中のFab'の濃度は適当なイソタイプの精製Fab'標準と比較することにより算出した。
【0043】
PhoS遺伝子のクローニング
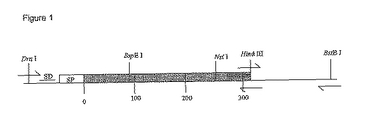
約700 bpの3'-クロモソーム配列を伴う野生型PhoS遺伝子を鋳型としてW3110 E.coliを使用してDra I-BstB I制限断片としてpSK-(Stratagene)中にPCRクローニングした。PhoSのC-末端の改変を容易にする目的で、新規なHind III部位を組み込むためにPhoS遺伝子の終結コドンの3'に隣接する3塩基対の改変を行った(図1の図解参照)。PhoS遺伝子を改変しそしてPhoSタンパク質のクロマトグラフィー挙動で変化を試験した後、最終的な遺伝子構築をE.coli染色体に組換えるためにSal I-BamH IフラグメントとしてpKO3に戻した。PhoS遺伝子及びその周辺のゲノム配列は文献及び公開データーベースで見ることができる(Surin et al., 1984 and Blattner et al., 1997)。
【0044】
突然変異PhoSタンパク質をコードする遺伝子の構築
鋳型としてW3110 を使用し、Taq ポリメラーゼを使用するPCRにより3'ゲノム配列の一部を伴うPhoSコード配列を2部分にクローニングした。これに使用したオリゴヌクレオチドを表1に示す。PCR突然変異誘起を使用して終結コドンの直後の3塩基対を改変してHind III部位をコードした。したがって遺伝子の3'の改変を含むPhoSコード領域はDra I-Hind III制限フラグメントとしてSma I-Hind III制限pSK-プラスミド中にクローニングすることができた。PhoS遺伝子の3'末端に施された改変はポリ(ヘキサ)アスパラギン酸、及びK318D変異を伴うポリ(ヘキサ)アスパラギン酸をコードするコード領域を含む。
【0045】
表面残基の突然変異は図1に示すような有用な制限部位に及ぶオリゴ(表1に示す)を使用するオリゴヌクレオチド指定突然変異誘起PCRを使用して行った。したがってPCR及び制限クローニングを使用することによりこれ等の突然変異を構築して組み合わせることが可能である。
【表1−1】

【表1−2】
【0046】
突然変異PhoSを組み込んだ組換えプラスミドの構築
挿入配列の両側の隣接領域が100%相同であれば、E.coli染色体中への突然変異遺伝子の指定相同組換えの効率は増加する。この隣接領域の長さは通常200〜1000 bpの程度である(Hamilton et al., 1989, Journal of Bacteriology, 171, 4617-4622)。Hind III部位及び元来のBstB I部位を導入するために突然変異改変の3'染色体配列の約700 bpをPCRクローニングした。このHind III-BstB IフラグメントをHind III-Cla I制限pSK-中にクローニングした。
【0047】
pSK-から発現したPhoS突然変異体を全て構築しそして試験した後、目的の最終PhoS遺伝子は全て3'染色体隣接領域の676 bp Hind III-Xho Iフラグメントを同様に制限処理されたPhoS発現プラスミド中に移動することによりその後にクローニングされた上記の3'染色体隣接領域を有した。PhoS組み込みカセットは1852 bp BamH I-Sal I制限フラグメントとして同様に制限処理されたpKO3相同組換え/置換プラスミドに移した(Link et al., 1997, Journal of Bacteriology, 179, 6228-6237)。
【0048】
W3110中のPhoS遺伝子の染色体置換の構築
プラスミドpKO3(Link et al., 1997)をマーカーの無い染色体遺伝子置換を発生させるために使用した。染色体組込みを強制しそして選別するためにこのプラスミドはクロラムフェニコールマーカーを伴うpSC101の温度感受性変異体の複製起点を使用する。レバンスクラーゼをコードするsacB遺伝子はスクロース上で増殖するE.coliにとっては致死的であるので(クロラムフェニコールマーカー及びpSC101起点を一緒に)デインテグレーション及びプラスミドキュアリングを強制しそして選別するために使用する。この方法は既に記述されている(Hamilton et al., 1989, Journal of Bacteriology, 171, 4617-4622及びBlomfield et al., 1991, Molecular Microbiology, 5, 1447-1457)。
【0049】
pKO3組込みプラスミドの使用
1日目 冷却したBioRad電気穿孔キュベット中で100 μlのE.coli細胞を3 μlのpKO3 DNAと混合し次いで2500V,25 μF及び200Ωで電気穿孔した。直ちに900 μlの2xPYを加え、インキュベーター中30℃で1時間250 rpmで振とうすることにより細胞を回復した。細胞を2xPY中で系統的1/10希釈を行いついで100 μlの分割液をクロラムフェニコール20 μg/mlを含み30℃及び43℃に予め加温した2xPY寒天プレート上に注いだ。プレートを終夜30℃及び43℃でインキュベートした。
2日目 30℃で増殖したコロニーの数は電気穿孔の効率の指標を与え、他方43℃で生存し増殖したコロニーは組込みの可能性を示す。43℃プレートのシングルコロニーを取り出し、そして10 mlの2xPYに再懸濁した。シングルコロニーを作るために、この100 μlを5%(w/v)スクロースを含み30℃に予熱した2xPY寒天プレート上に注いだ。プレートを30℃で終夜インキュベートした。
3日目 このコロニーはデインテグレーション及びプラスミドキュアリングを同時に生じた可能性を示している。デインテグレーション及びプラスミドキュアリングが増殖の初期に生じていたならば、多数のコロニー集団はクローンであろう。シングルコロニーを取り出し、そしてクロラムフェニコール20 μg/mlを含むか又は5%(w/v)スクロースを含有する2xPY寒天に接種し、複製した。プレートを終夜30℃でインキュベートした。
4日目 スクロース上で増殖しクロラムフェニコール上で死んだコロニーは染色体置換及びプラスミドキュアリングの可能性を示す。これ等を取り出しそして突然変異特異的オリゴヌクレオチドを使用するPCRによりスクリーニングした。正しい大きさのはっきりとしたPCRバンドを生じるコロニーを削り取り、5%(w/v)スクロースを含有する2xPY寒天上にシングルコロニーを作るためにプレートを終夜30℃でインキュベートした。
5日目 PCR陽性、クロラムフェニコール感受性そしてスクロース耐性E.coliのシングルコロニーをグリセロール保存、化学的コンピテント細胞を作るために使用し、そしてPrecision plusポリメラーゼを使用する直接DNA配列分析のためのPCR産物を作成するための5'及び3'隣接オリゴを使用するPCR反応のPCR鋳型として使用した。
【0050】
PhoS補充検定
ANCC75のようなPhoS株(Amemura et al., 1982, Journal of Bacteriology, 152, 692-701)はそのPO4感受性と捕捉能力が連携していないために構成的にアルカリホスファターゼ(AP)を発現する。PhoS障害をプラスミド発現PhoS遺伝子で補充することが可能であり、この補充を寒天プレート上又は液体培地それぞれでAP活性を検定するための5-ブロモ-4-クロロ-3-インドリルリン酸(XP)又はパラ-ニトロフェニルリン酸(pNPP)のようなアルカリホスファターゼの発色基質を使用して検出することができる。寒天プレートにはイソプロピル β-D-チオガラクトピラノシド(IPTG)200 μM及び抗生物質とともにXPを40μg/ml加え、そして終夜37℃でコロニーを成長させる。液体培地においてAPを検定するには約1.0のOD600の誘導培養の10〜100 μl検体を25μlの0.1% SDS (w/v)及び50 μlのクロロホルムを含む1.5 M Tris. Cl pH 8.0で1 mlにして、5秒間渦巻き混合し、そして30℃で5分間予備インキュベートした。検定は200 μlの15 mM pNPPを加えそして反転により混合して開始し、そして200 μlの1 M KH2PO4を加えそして反転により混合して10分後に停止した。検定は30℃において実施した。マイクロフュージ中で細胞をペレット化して、上清の吸光度を420 nmでブランクと比較して測定した。アルカリホスファターゼ活性の1単位はΔA420OD600-1min-1として定義した。
【0051】
理論的分子量及びpI計算
理論的パラメーターの計算は全てMacVectorソフトウエアを使用して行った。
【0052】
質量分析
Fab”の分子量はMicromass Ultimaトリプル四重極スペクトロメーターを使用して陽イオンエレクトロスプレーイオン化モードにおいて測定した。Fab”検体はTrisを除去するために10 kDa膜のカットオフサイズ(Amicon, U.K.)によるMicrocon濃縮器を使用して10 mM酢酸アンモニウムとの多容量交換により脱塩した。
【0053】
カチオン交換クロマトグラフィー
フラスコを振とうして醗酵細胞ペレットを100 mM Tris.Cl/10 mM EDTA pH 7.4 中に30 OD600/元の培養容量mlにそれぞれ再懸濁し、そして終夜30℃又は60℃で撹拌した。細胞破片を除くために4000 gで10分間遠心分離した後上清に1 M酢酸を加えて精製実験を行う緩衝液のpH(典型的にはpH 4.5, 5.0 又は6.0)にpHを下げ、そして伝導度が≦3.5 mScm-1になるまでdH2Oで希釈した。再度pHをチェックした後、細胞周辺抽出液を20,000 gで10分間遠心分離して澄明とし、次いで0.2 μm膜を通して濾過した。
【0054】
1 ml/minの流速においてFPLC指令ソフトウエア及び10 ml検体注入シリンダーにより稼動するPharmacia P500 FPLCを使用して酢酸ナトリウム(NaAc)緩衝液中での5 ml SPセファロース(Pharmacia)カラム処理を共通して行った。PhoS及びFab'’構築に対しては全て以下の基本的溶出方法を使用した:カラムの平衡は1 mlの平衡緩衝液で行い、9 mlの検体を負荷し、次いで70 ml(カラム容積の約14倍)のNaAc中0〜200 mM NaClグラジエントで溶出し、カラムを7 ml NaAc中1 M NaClで洗い、次いで13 ml 負荷緩衝液(NaAc)で再度平衡した。A280により監視した溶出点の時間及び伝導度を記録しそしてフラクションを手動で採取した。
【0055】
アニオン交換クロマトグラフィー
20 mM Tris.Cl pH 8.0 中の2.5 ml Poros HQカラム(PerSeptive Biosystems)処理を共通して使用した。カチオン交換の検体を使用前に20 mM Tris.Cl pH 8.0に緩衝液を交換した。カラムの平衡は1 mlの洗浄で行い、9 mlの検体を負荷し、次いで10.9 mlの洗浄段階、次いで20 mM Tris.Cl pH 8.0中3.9 mlの1 M NaClで溶出し、そして10 ml負荷緩衝液(20 mM Tris.Cl pH 8.0)で再度平衡した。タンパク質フラクションはA280により監視しそして手動で採取した。
【0056】
SDS-PAGE, イムノブロット及びIEFゲル
SDS-PAGEはMES緩衝液中でSeeBlue2標準を使用してInvitrogenの4〜12% NuPAGEゲルで行った。それをクマシーで染色するか又はタンパク質を1/2xTowbin緩衝液を使用してPVDF膜に移した。IEFゲルはInvitrogenのpH 3〜10 IEF垂直スラブゲルであり、Serva標準pI 3〜10を使用し、クマシーで染色した。脱染色後ゲルをスキャンし及び/又はセルロース膜に挟んで乾燥した。
【0057】
実施例1
宿主タンパク質を改変することにより行うことができる改良を確認するためのFab'抗体フラグメントの精製工程の概要。
E.coliにおいて発現したFab'フラグメントは細胞周辺抽出液から3つのクロマトグラフィー段階を使用して定型的に精製された。第一段階はpH 4.5におけるカチオン交換であり、この間に抗体Fab'フラグメントはカチオン交換カラムに結合した。Fab'フラグメントはカチオン交換から溶出されそしてpH 8.0 においてアニオン交換処理された。Fab'フラグメントはこのpHではアニオン交換に結合せず通過液中に採取された。この段階の間に殆どの残留E.coliタンパク質及びエンドトキシンはアニオン交換カラムに結合し、Fab'調製品から除去された。最後に、SDS-PAGEにより確認されたように、両イオン交換段階において共精製した一つの多量のE.coliタンパク質を除去するために疎水性相互作用クロマトグラフィー(HIC)段階が必要であった。
【0058】
それにより精製工程をスピードアップしそして物質及び労力に関するかなりのコスト節減をもたらすであろうことから、最後のHIC段階を除くことが好ましいことはこの精製工程の概要から明らかであった。この一つの段階を除去することによる推定コスト節減はFab'のモルベースで15%であった。これを達成するためには混入するE.coliタンパク質の物理的性質を、イオン交換でFab'と最早共精製せずに、したがってHIC段階を最早必要としないように改変する必要があるであろう。これは最早カチオン交換カラムに結合しないかあるいはFab'と異なる塩濃度においてカラムから溶出するようにタンパク質のpIを改変することにより達成することができた。この混入タンパク質は後にリン酸結合タンパク質(PhoS/PstS)として同定された(実施例2)。
【0059】
その他の改良の可能性はカチオン交換カラムに結合する全てのタンパク質のSDS-PAGEゲルの検討により同定された。これによりこのカラムに結合した量の多い宿主タンパク質がいくつかあることが明らかにされた。これ等のタンパク質の除去によりカラムのFab'結合能力が有意に増加するのでカラムの大きさを減少しそして精製を速くするのでコスト節減を生じるであろう。これを達成するにはカチオン交換カラムに最早結合しないように混入するタンパク質のpIを改変する必要があろう。これ等の混入タンパク質は後にジペプチド結合タンパク質(DppA)、マルトース結合タンパク質(MBP)、チオレドキシン及び仮に24 kDaタンパク質と呼ぶものと同定された(実施例2)。
【0060】
実施例2
実施例1において選択された混入タンパク質の同定
標準的E.coli W3110醗酵から生産された細胞周辺フラクションを終夜30℃で100 mM Tris.Cl 10 mM EDTA pH 8.0中に抽出した。遠心分離の後上清をH2O及び酢酸でpHが≦4.5そして伝導度が≦3.5 mScm-1になるまで希釈した。これをカチオン交換カラムに正規の方法で負荷した。洗浄した後、結合したタンパク質を溶出した。Fab'及びそのフラグメントは溶出液をプロテインGカラムを2回及びプロテインLを1回通すことにより除去し、次いでFab'の入っていない溶出液を10 kDaカットオフ膜を使用したAmicon撹拌セルで濃縮した。Fab'に関係したペプチドが全て効率よく除去されたことは、抗-カッパ及び抗-CH1イムノブロット、サンドイッチELISA及びHPLCにより示された。クマシー染色4〜12%SDS-PAGEゲルを使用して濃縮検体を分析した。同じゲルをPVDF膜に移し、ポンソーSで染色してバンドの位置を明らかにして清潔なメスで切り取った。タンパク質はN-末端配列分析を行いそしてその結果はSwissProtへ質問するために使用した。結果を以下に示す。
【表2】
【0061】
実施例3
C-末端ポリ-イオン性テイルを持つPhoSタンパク質の創製と試験
pH 4.5におけるカチオン交換精製に対するC-末端ポリ-イオン性テイルの影響
最初にPhoSの3種をクローニングしそしてE.coliにおける発現及びカチオン交換精製を試験した:野生型PhoS(wtPhoS)、C-末端に6個のアスパラギン酸を持つPhoS(PhoS polyD)、及びC-末端に6個のアスパラギン酸そして近くにK318D変異を持つPhoS(PhoS K/D polyD)。大まかにpHを調整した細胞周辺抽出液のクマシー染色SDS-PAGEにより判断して、これ等3種は振とうフラスコ内で全て良く発現した。プラスミド、タンパク質名、分子量、推定pI、測定pI、及びpH 4.5におけるカチオン交換カラムからのNaCl溶出プロフィールの概要を表2に示す。
【0062】
polyDテイルの追加により、野生型タンパク質に比較して塩グラジエントにおいて約20%早く、103 mM NaClにおいて、PhoSタンパク質は溶出した。しかし、pH 4.5 においてSPセファロースカラムに全く結合しないタンパク質の好ましい効果は達成できなかった。追加のK318D表面突然変異(これはC-末端に非常に近い)はカチオン交換効率についてpolyDタグのみよりも利点が多いことを示す証拠が得られた。
【0063】
pH 8.0におけるアニオン交換精製に対するC-末端polyDテイルの影響
細胞周辺抽出液をカチオン交換実験の通過液及び溶出検体の採取と同様に処理しそして緩衝液を20 mM Tris.Cl pH 8.0 に交換した。wt PhoS, PhoS polyD 及びPhoS K/D polyDの検体を小さなPoros HQアニオン交換カラムに流した。結果は図2に示すように、wt PhoSタンパク質はこのpHではカラムに結合しないが、polyD及びK318D polyDの2種のPhoSはともに結合する。したがってこの条件下では、polyDタグの存在により生じたpI変化に加えて、タグ自身はこの条件下に「親和性テイル」として働いているように思われる。
【表3−1】
【表3−2】
【表3−3】
【0064】
実施例4
pH 4.5におけるPhoSのカチオン交換精製に対する表面突然変異及びC-末端polyDテイルの影響
活性部位に結合したPO4を持つPhoSの結晶構造(Luecke and Quiocho, 1990, Nature, 347, 402-406)を分析して突然変異に使用できる表面残基を探した。PhoSのpIを減少させるために、リシン残基(MW 128.17 Da, pKa=10.79)をグルタミン酸(MW 129.12, pKa=4.07)残基に交換した。塩基性残基を酸性残基に交換することにより、中性残基を酸性残基に交換する(約0.07)よりも大きな突然変異当りのpI変化(約0.15のPhoSのpI変化)の可能性を付与する。また、表面に露出したグルタミン酸残基は(リシン残基のように)高度に溶媒和されると思われるので、多くの他の(非荷電)表面残基をグルタミン酸に換える際よりも重大な構造的変動を生じるリスクが少ないであろう。これまでの構造研究又は突然変異研究からPO4結合に重要である又はこれ等の残基に空間的に近い又は実際PhoS分子の中央の中央溝に近いことが分かっている残基を避けることも重要である。タンパク質の表面の非常に遠くに突き出ているリシン残基及び塩基性残基の大きな集団の中に又はその近くに位置するリシンが選択された。
【0065】
塩基性残基の該集団は精製マトリックスと相互作用することができる重要な荷電パッチを形成することができる。したがって1又はそれ以上の酸性残基の戦略的置換は該パッチを「破壊」することができる、すなわち、該領域のネット電荷を有意に変え、それにより精製マトリックスと相互作用する能力が局所的に大きく変化する。最終的に、C-末端は既に、「割れたラグビーボール」のような形のPhoSタンパク質の一つの端に存在するpolyDテイルにより改変されているので、PhoSタンパク質のC-末端の逆の端にある残基を少なくとも見出すことが好ましかった。
【0066】
以下の領域がこれ等の条件に適合すること、及び直線配列中に集めて集団PCR突然変異誘起を容易にすることができることが確認された。
1) 単一突然変異K275Eは窪んだ表面から突き出ておりそしてK272に非常に近接しておりそしてK282に近い。
2)K107, 109及び110のトリプル突然変異、K107はK98及びE155 に近く、単離したK109はPhoSから突き出ており、そしてK110はK109の近傍にあり、D112, D113及びE114に非常に近い。
3) K262, 265及び266のトリプル突然変異、K262はK265/266から離れているがK318 の近傍にあり、他方K265及びK266 は一緒に「V」字を形成しそしてN48 と三角形を形成している。
4) K309, 313及び318のトリプル突然変異は全て、T310及びP319 に隣接する塩基性表面の可能性がある大きな広がった領域に関係している。
【0067】
この4種の突然変異をコードするPhoS遺伝子を構築しそしてE.coliにおける発現及びpH 4.5におけるカチオン交換精製の効率を試験した。
【0068】
単一K275E突然変異(pDPH192)はE.coliにおける耐容性が悪いことが判明しそして/又は細胞周辺抽出液のpHを4.5に調整したときに沈殿された。さらに、細胞から回収したタンパク質のカチオン交換溶出効率はPhoS polyDタンパク質よりも増加することはなかった。
【0069】
K107/109/110Eのトリプル突然変異ではカチオン交換の溶出後にタンパク質を検出しなかった。これはE.coliにおける発現のレベルが(誘導時間経過に示されるように)極めて低かったためばかりでなく細胞周辺抽出液をpH 4.5 に調整したときの沈殿による可能性もある。しかし、この突然変異はPhoS「ラグビーボール」のpolyDテイルの逆の端にある唯一の突然変異であるので、続いてさらに分析するために単一突然変異に分離した。
【0070】
K262/265/266Eにおけるトリプル突然変異(pDPH193)はカチオン交換から33 mM NaClにおいて溶出した。しかしカチオン交換後のPhoS発現/回収のレベルはwt PhoSに比較して減少したので、このトリプル突然変異をやはりさらに分析するために3種の単一突然変異及びダブル突然変異のすべての組み合わせに分離した。
【0071】
K309/313/318E におけるトリプル突然変異(pDPH194)はPhoS polyDに比較してカチオン交換溶出効率を改善し(それぞれ73 mM対83 mM NaCl)そして正常な発現レベルであったが、73 mMにおける溶出はK318D単一変異+polyD(pDPH188)のそれに比較して十分な改善ではなかったので、このトリプル突然変異についてはこれ以上の検討を行わなかった。
【0072】
該組合せが追加の又は相乗効果を生じるか否かを検討するために、K275E及びK318Dの単一突然変異の両者をトリプル変異K262/265/266Eと組み合わせた(それぞれpDPH195及び pDPH194)。しかし、両者ともにK262/265/266Eにおいて既に認められた低いタンパク質発現/回収の欠点があった。
【0073】
K107/109/110Eトリプル突然変異を3種の単一変異に分離することにより、K109E及びK110E単一変異はより多くのタンパク質を生産することができたので、K107E変異がトリプル変異の有害な効果の主な原因であることが示された。K109E (pDPH199)及びK110E(pDPH200)はともにカチオン溶出効率の改善(52〜55 mM NaCl)を示し、そして70 mM NaClにおいてK107E(pDPH198)よりも良かった。そこで、K109E及びK110Eを組み合わせて(pDPH201)相加/相乗効果を試験した。しかし、このダブル突然変異K109/110EはpH 4.5に調整した後発現又は回収ができなかった。これによりK107/109/110が存在するPhoSの領域には3者が寄与する構造上の又は溶媒和の重要な影響があることが示された。タンパク質回収から判断してK110EはK109 Eよりも耐容性が良いように見えたので、K110Eをさらに組合せ研究を行うために選択した。
【0074】
3種の単一突然変異K262, 265, 266Eは全てPhoS polyDのみよりもカチオン交換溶出効率の改善を示した:それぞれ68 mM, 60 mM 及び58 mMそして全て良好なタンパク質発現レベルであった。これらをダブル突然変異に組合わせることにより、K265E(pDPH205)又はK266E (pDPH206)のいずれと組合わせた場合もタンパク質の発現/回収のレベルの減少を認めたので、良好なタンパク質発現レベル(構造/溶解性)を維持するのに重要なのは残基K262であることが示された。さらにこれ等のダブル変異のいずれも単一変異K265E又はK266E以上のカチオン交換溶出効率を改善しなかった。しかしダブル突然変異K265/266E(pDPH207)は良好なタンパク質発現/回収レベルを示し、このタンパク質が38 mM NaClで溶出したので(単一変異のみの60 mM及び58 mMに比較して)、カチオン交換溶出効率への追加効果を示した。
【0075】
さらにK265/266EにK110E及びK318Dを加えて組合わせ突然変異が作られた。K110/265/266E突然変異(pDPH209)はカチオン交換溶出効率の改善が認められ、29 mM NaClにおいて溶出しそして良好なタンパク質発現レベルであった。K265/266E+K318D突然変異(pDPH211)はカチオン交換溶出効率及びタンパク質発現ともにK265/266E変異のみよりも良くなかった。最後にpolyDテイルに加えて4個の突然変異を含む「最大」変異構築:K110/265/266E+K318D(pDPH210)を試験した。しかしこれはタンパク質回収の減少を生じそしてK265/266E又はK110/265/266Eのいずれよりもカチオン交換効率を改善しなかった。
【0076】
このようにして3種のPhoS構築がさらに分析するために選択され、改善されたカチオン交換溶出効率を持つ最小PhoS突然変異であることを示した:PhoS polyD, PhoS K265/266E polyD及びPhoS K110/265/266E polyD。野生型PhoSよりも低い塩濃度で3種の変異体が溶出することによりカチオン交換カラムからのグラジエント又は段階的溶出によりFab'から変異PhoSを分離できるはずである。そのほかに、変異体のpIによってはFab'の結合に影響しない緩衝液のpHに上昇することによりそれらがカチオン交換カラムに結合することを防止することも可能であろう。その他の方法としてはアニオン交換段階により変異PhoSを除去することもできる。変異PhoSタンパク質のpIは後に決定される。
【0077】
実施例5
タンパク質の純度及びpIに及ぼすPhoS突然変異誘起の影響
>95%純度の変異PhoSタンパク質をグラジエント溶出カチオン交換により得た。このタンパク質の純度及びそのC-末端テイルをSDS-PAGE及び質量分析により評価した。MSにより測定した分子量は表2に示す。全て予測分子量と良い一致を示し、いずれもタンパク質は正しく同定されそしてpolyDテイルは完全であることを確認した。図3に示すSDS-PAGEによりこれ等の調製品の純度を確認しそしてpolyDを含むPhoSタンパク質は野生型に比較して若干遅く移動することを示す。IEFゲル(図4)は、wt PhoSは約7.2のpIを有し、一方polyDテイルのみの付加はpIを約5.1に減少することを示している。続くKのE又はDへの表面突然変異の追加によりさらにPhoS K/D polyDの約4.9からPhoS K265/266E polyDの約4.8及びPhoS K110/265/266E+K318D polyDの約4.5への範囲のpIシフトを生じる。
【0078】
実施例6
変異PhoSタンパク質の機能的純度試験:プラスミドから得られたPhoSによるphoS遺伝型の補充
E.coli株ANCC75(Amemura et al., 1982)をphoSの遺伝的背景を提供するために使用した。この細胞はPO4に欠乏しているかのような表現型を永続的に持っているので、phoレギュロンを介するフィードバックのためにPO4が存在しても高レベルのアルカリホスファターゼを誘導する。XPを含む固体培地上での該細胞の殖によりアルカリホスファターゼを高いレベルで持つ又は低いレベルで持つ細胞を区別することができる。該細胞をwt PhoS, PhoS polyD, Phos K265/266E polyD及びPhoS K110/265/266E polyDをコードするプラスミド又は対照プラスミドで形質転換して、変異PhoSタンパク質の全てが、染色体にコードされたPhoSの欠失を補充する能力を有することを確認した。
【0079】
対照として野生型W3110 E.coliを使用して同じ実験を「無リン酸」(PO4が非常に低い)液体培地で繰り返し(図5)、試験した変異PhoSタンパク質は全て低いPO4条件の下に(5時間の点を参照)phoS表現型を100%補充できることそして無リン酸(PO4)条件下にphoS表現型を部分的に補充すること(22,27及び47.5時間の点を参照)が示された。
【0080】
実施例7
変異PhoSタンパク質を発現する安定な組込みE.coli株の作製
3種の変異PhoSタンパク質(polyD, K265/266E polyD及びK110/265/266E polyD)をコードする遺伝子を3’染色体隣接領域とともに染色体置換プラスミドpKO3の中に組込んだ。さらにPhoSのヌルバージョンをシグナルペプチドコード領域に2個のインフレーム終結コドンを持つオリゴヌクレオチドで作製した(それぞれプラスミドpDPH217〜220)。これ等を電気穿孔によりW3110 E.coliに導入し、そして組換え及びプラスミドキュアリングを方法に記述したように試験した。表1に示したスクリーニングオリゴを適当な対立鎖の共通オリゴとともに使用して理論的に組込まれたシングルコロニーの最終的PCRスクリーニングを行い野生型又は変異PhoSが染色体中に組込まれたか否かを試験した。pKO3にコードされたDNAの領域の外側にアニールする2個のオリゴ(Dra I前進及びBstB I逆)を使用し、高信頼度ポリメラーゼ(Precision plus Taq)を使用する染色体DNA(E.coli細胞全体)のPCR、次いでゲル精製PCR産物の直接配列分析を正しい改変が染色体に組込まれたことを確認するために使用した。この結果次の4種のE.coli株が作製された:DPH1はPhoS polyDをコードし、DPH2はPhoS K265/266E polyDをコードし、DPH3はPhoS K110/265/266E polyDをコードし、DPH4はヌルPhoSをコードする。
【0081】
実施例8
DPH3株のFab'の発現及び精製の試験
株DPH3を目的のFab'を発現するプラスミドで形質転換した。標準的な醗酵を実施しそして増殖又は誘導相の間に明らかな欠陥又は問題を認めなかった。醗酵中に採取した検体をTris/EDTA抽出した後通常の方法でELISAにより分析した。図6のデータは、増殖は正常でありそしてペリプラズムにおけるFab'の蓄積は回収時に約380 mg/Lで十分にFab'の正常範囲内であることを示している。遠心分離の後細胞ペレットは硬くそして遠心後容易に再懸濁することができることがわかり、通常これは良好な細胞の表れである。
【0082】
回収培養の50 mlから得たペレットを終夜30℃でTris/EDTA中に抽出し、次いで前記のようにカチオン交換精製を行った。株DPH3の変異PhoS(pI 4.5)がカチオン交換カラムに結合せずそしてFab'フラグメントが結合するようにpHを4.5から5.0に上昇させた。伝導度は3.0 mS/cmであった。検体を5 ml SPセファロースカラムに適用し、原液、通過液及び溶出液検体をクマシー染色SDS-PAGEにより分析した。必要があれば、クマシー染色ゲルで可視化できるように10 kDaカットオフスピンカラムを使用して検体を濃縮した。
【0083】
図7のSDS-PAGEゲルは、このpH及び伝導度の条件下ではDPH3の変異PhoSはSPセファロースカラムに結合しないが、W3110のwt PhoSは結合することを示している。このことは、DPH3の場合には変異PhoSとFab'は異なるフラクションに移行する:それぞれ通過液及び200 mM NaCl溶出液、他方W3110 の場合には両タンパク質はともに200 mM NaCl溶出液中に移行することを意味する。
【0084】
少しでも残留したPhoSはアニオン交換によっても除去されることを確認するために、DPH3及びW3110実験の両者からの通過液及び200 mM NaCl溶出液を濃縮し、脱塩しそして緩衝液を20 mM Tris.Cl pH 8.0に交換しそしてアニオン交換を行った。図8に示すクマシー染色SDS-PAGEゲルは、DPH3については PhoSはアニオン交換カラムに結合するのでFab'から分離されることを示している。しかし、W3110のwt PhoSはこのカラムに結合せずに通過しそしてFab'溶液に混入する。
【0085】
実施例9
株DPH1, DPH2及びDPH3において発現したFab'フラグメントの精製試験
典型的にpH 6におけるイオン交換で精製される実施例8に使用したFab'よりも高いpIを持つFab'を発現するプラスミドで株DPH1, DPH2及びDPH3を形質転換した。標準的醗酵を行いそしてカチオン交換クロマトグラフィーによりFabからPhoS変異体を分離する試験をpH 5, 5.5及び6において前記と同様に行った。
【0086】
pH 5.0
pH 5.0 において行った精製の結果を図9及び10に示す。pH 5においてPhoS polyD(DPH1)はクマシー染色により判断して通過液には現れず(図9、レーン2)、実際Fab'-Bとともにカラムから溶出した(図9、レーン3)。抗-PhoSポリクロナール及び抗-Fdモノクロナールを使用するより感度の高いイムノブロット分析により、Fab'-Bは大量のPhoS polyDとともにカラムに強く結合していることが明らかに示される(図10、レーン3)。しかし通過液フラクション(レーン2)にもPhoS polyDが少しあった。これはPhoS polyDとクロマトグラフィーマトリックスの会合がpH 5.0では弱いことを示すのであろう。
【0087】
これに対して、PhoS K265/266E polyDをコードするDPH2は通過液中に明瞭なクマシー染色PhoSバンドを示した(図9、レーン5)。抗-PhoSイムノブロットは溶出フラクション中に極微量のPhoSしか示さなかった(図10、レーン6)。したがってPhoS K265/266E polyDは実際上pH 5.0 においてSPセファロースには結合しない。
【0088】
最大突然変異PhoS K110/265/266E polyDをコードする株DPH3では、PhoSはやはり通過液中に現れる(図9、レーン8)が、イムノブロットにより溶出フラクション中に残留するPhoSの量(図10、レーン9)はDPH2の場合(図10、レーン6)よりもさらに減少することがを示される。これは実施例8において既に観察したDPH3のクロマトグラフィーにおける性質はDPH2よりも低いpH(pH 約4.7〜5.0)でよく作動するということを支持している。
【0089】
pH 5.0 で負荷された場合に、親のW3110株によりコードされている野生型PhoSの結合性は図9のレーン12に示されている。野生型PhoSの大部分は溶出フラクションにあるが、極微量を通過液中に検出することができる(図10、レーン11及び12)ことを抗-PhoSイムノブロットは示している。(この実験においてイムノブロットにより溶出フラクション中に検出された極微量のPhoSは、高速分析方法に使用されたカラム洗浄の量が少なかったことを反映している可能性があるが、通過液中の微量の野生型PhoSは、カラムへの弱い結合又はカラム/緩衝液フロントアーチファクトを示している可能性もある)。
【0090】
pH 5.5
pH 5.5におけるPhoS精製について3株を分析したところ、DPH1の行動のみがpH 5.0 に比較して大きく変化した。pH 5.5においてDPH1によりコードされたPhoS polyDは今度はクマシー染色ゲルにより通過液中に認められ(図11、レーン2)、それは抗-PhoSブロット(図12、レーン2+3)により確認された。DPH2及びDPH3によりコードされたPhoS変異体はpH 5.5 において通過液中に存在した。W3110によってコードされた野生型PhoSには不明確な行動が認められている。クマシー染色ゲルはpH 5.5において大量のPhoSが通過液中にあることを示している(図11、レーン11)が、他方で抗-PhoSブロットはかなりの量(約40%)がまだ溶出フラクションに残っていることを示している。したがって、このpHにおいて、PhoSは機能的pIの近くにあるに違いなくそして弱く結合している。
【0091】
pH 6.0
3種の変異株全て及び野生型W3110はクマシー染色ゲルにより示されるようにpH 6.0 (図13、レーン2,5,8及び11)においてSPセファロースに結合しないPhoSを生産する。抗-PhoSポリクロナールを使用する分析により、これ等は全て溶出フラクションに同じレベルの微量の検出しうるPhoSがあること(図14、レーン3,6,9,12)が示された。したがって、カチオン交換精製がこのpHで行われるならば、野生型W3110よりも株DPH 1, 2又は3を使用する利点はない。しかし、溶出フラクション中の微量のPhoSにおけるpolyDテイルの存在は、カラムを通過様式で稼動する場合にはC-末端タグは親和性タグとして有効に作動するので、2番目のアニオン交換カラム上でのPhoSとFab'のさらなる分離を非常に容易にすることができる。
【0092】
表3に集めた結果は、50 ml醗酵検体からの生成を試験すると、3種のPhoS突然変異:PhoS polyD, PhoS K265/266E polyD及びPhoS K110/265/266E polyDは全て 系統的に改良された精製プロフィールを持つことを示す。これ等の実験において、SPセファロースカラムに酸性にして適用した時の通過液中にPhoSを認めることができる低いpHによりこれ等の改良は示された。したがって多数の突然変異を持つタンパク質はますます厳密なpHの結合条件においてFab'から分離することができる。この型のPhoSをpH 4.5においてSPセファロースカラムから溶出すためには低いNaCl濃度(W3110, DPH1, 2及び3に対してそれぞれ約103 mM, 83 mM, 38 mM及び29 mM NaCl(表3))が必要であった以前の情報をこれは支持している。
【表4】
【図面の簡単な説明】
【0093】
【図1】PhoS遺伝子のPCRクローニングの図解
【図2】アニオン交換カラム, pH 8.0に結合したPhoSのクマシー染色4〜12% SDS-PAGE。レーン1〜4はwt PhoS, レーン5〜8はPhoS polyD及びレーン9〜12はPhoS K/D polyD。 レーン1,5及び9は原液、レーン2,6及び10は通過液、及びレーン3,4,7,8,11及び12は1 M NaCl溶出段階のそれぞれ初期及び後期フラクション。
【図3】SPセファロースpH 4.5上NaClグラジエントで精製されたPhoSタンパク質のクマシー染色4〜12% SDS-PAGE。レーン1 wt PhoS, レーン2 PhoS polyD, レーン3 PhoS K/D polyD, レーン4 PhoS K265/266E polyD, レーン5 PhoS K110E K318D polyD, レーン6 PhoS K110/265/266E poly D, レーン7 PhoS K110/265/266E K318D polyD, レーン8 PhoS K265/266E K318D polyD.
【図4】SPセファロースpH 4.5上NaClグラジエントで精製されたPhoSタンパク質のクマシー染色pH 3〜10 IEFゲル。レーン1 wt PhoS, レーン2 PhoS polyD, レーン3 PhoS K/D polyD, レーン4 PhoS K265/266E polyD, レーン5 PhoS K110E K318D polyD, レーン6 PhoS K110/265/266E poly D, レーン7 PhoS K110/265/266E K318D polyD, レーン8 PhoS K265/266E K318D polyD.
【図5】phoS及びwt E.coli株におけるアルカリホスファターゼ活性のphoS及びPO4欠乏誘導のwt及び突然変異PhoSタンパク質をコードするプラスミドによる機能的補充。
【図6】DPH3 E.coliの醗酵によるDPH3の増殖及びFab'の生産。
【図7】SPセファロース上pH 5.0 及び3.0 mS/cmにおけるW3110及びDPH3のFab'の精製におけるカチオン交換カラムフラクションのクマシー染色4〜12% SDS-PAGE。レーン1 DPH3通過液、レーン2 DPH3 200 mM NaCl 溶出液、レーン3 W3110通過液、レーン4 W3110 200 mM NaCl 溶出液、レーン5 wt PhoS, レーン6 PhoS polyD, レーン7 Fab'。
【図8】カチオン交換カラムで分離したDPH3又はW3110で生産されたFab'のpH 8.0におけるPoros HQのアニオン交換カラムのフラクション。クマシー染色4〜12% SDS-PAGE。 DPH3カチオン交換の溶出物をアニオン交換カラムに適用して得られた通過液(レーン1)又は溶出液(レーン2)。DPH3カチオン交換の通過液をアニオン交換カラムに適用して得られた通過液(レーン3)又は溶出液(レーン4)。W3110カチオン交換の溶出液をアニオン交換カラムに適用して得られた通過液(レーン5)又は溶出液(レーン6)。W3110カチオン交換の通過液をアニオン交換カラムに適用して得られた通過液(レーン7)又は溶出液(レーン8)。レーン9 wt PhoS,レーン10 PhoS polyD及びレーン11 Fab'。
【図9】Fab'BのpH 5.0におけるカチオン交換精製における株DPH1, DPH2及びDPH3の変異PhoSの行動を比較したクマシー染色4〜12% SDS-PAGEゲル。レーン1 DPH1 原液、レーン2 DPH1 FT, レーン3 DPH1溶出液、レーン4 DPH2原液、レーン5 DPH2 FT、レーン6 DPH2溶出液、レーン7 DPH3原液、レーン8 DPH3 FT、レーン9 DPH3溶出液、レーン10 W3110 原液、レーン11 W3110 FT、レーン12 W3110 溶出液、レーン13 PhoS polyD、レーン14 Fab'。
【図10】Fab'BのpH 5.0におけるカチオン交換精製における株DPH1, DPH2及びDPH3の変異PhoSの行動を比較した4〜12% SDS-PAGEの抗-PhoS及び抗-Fdイムノブロット。
【図11】Fab'BのpH 5.5におけるカチオン交換精製における株DPH1, DPH2及びDPH3の変異PhoSの行動を比較した4〜12% SDS-PAGEのクマシー染色ゲル。
【図12】Fab'BのpH 5.5におけるカチオン交換精製における株DPH1, DPH2及びDPH3の変異PhoSの行動を比較した4〜12% SDS-PAGEの抗-PhoS及び抗-Fdイムノブロット。
【図13】Fab'BのpH 6.0におけるカチオン交換精製における株DPH1, DPH2及びDPH3の変異PhoSの行動を比較した4〜12% SDS-PAGEのクマシー染色ゲル。
【図14】Fab'BのpH 6.0におけるカチオン交換精製における株DPH1, DPH2及びDPH3の変異PhoSの行動を比較した4〜12% SDS-PAGEの抗-PhoS及び抗-Fdイムノブロット。
【配列表】
【技術分野】
【0001】
本発明は、組換えタンパク質の発現に使用するE. coli宿主細胞に関するものであり、特に組換え抗体の製造用の改良されたE. coli宿主細胞を提供するものである。
【背景技術】
【0002】
組換えタンパク質の大規模、経済的な精製はますますバイオテクノロジー産業の重要な課題となっている。一般的に、そのタンパク質の遺伝子を含む組換えプラスミドの挿入により目的のタンパク質を製造するように操作した哺乳動物または細菌の細胞系を使用して組換えタンパク質は製造される。タンパク質は直接細胞から周囲の培地に分泌されるか又は細胞内に作られる。後者のタンパク質については、その精製操作の最初の段階は細胞の溶解又は破壊であり、これは機械的切断、浸透圧ショック、又は酵素処理を含む種々の方法により行うことができる。その破壊により細胞の内容物はホモジネートに放出され、そしてさらに非細胞断片を生じ、これは一般的に分別遠心分離又は濾過により除去される。小規模であるが、細胞の自然死によるタンパク質の直接的分泌及びタンパク質生産の過程における細胞内宿主細胞タンパク質の放出に関して同じ問題が生じる。このようにして生産された組換えタンパク質は、混入する宿主細胞タンパク質が毒性又は免疫原性を持つ可能性があるので、それらから分離精製する必要がある。抗体のような治療に必要な組換えタンパク質については高純度であることが必須である。
【0003】
最近まで抗体及び抗体フラグメントは通常哺乳動物細胞において製造されていたが、E. coli, Pichia酵母及び植物のような代替生産システムも使用されてきた。これ等の生産代替方法は特異的抗体生産及び規模、コスト、速さ、資本リスク及び生物学的安全性の間のバランスによって促進されてきた(Humphreys and Glover, 2001, Current Opinion in Drug Discovery and Development, 4, 172-185)。醗酵プラント、操業時間、培地成分及び工程回転率/資本減価償却のより明確なコストに加えて、「下流工程」、すなわち、粗製品の保管、取扱い及び精製に関係する相当なコストが存在する。
【発明の開示】
【発明が解決しようとする課題】
【0004】
大規模な抗体精製は、コストの効率がよくそして物理的耐久性があるので、主として分別沈殿、イオン交換、サイズ排除及び疎水性相互作用クロマトグラフィーに依存している。これ等の方法を使用して精製を行うことができないとするならば、アフィニティークロマトグラフィーのような高価な分析方法をスケールアップするためにかなりの開発が必要になるであろう。混入する宿主タンパク質がpI、大きさ又は疎水性のような組換え抗体の物理的性質に類似している場合にはこの問題が起こりそうである。これ等の混入物を除去するには、大規模に行う場合には極めて好ましくない専門家による精製工程を必要とするであろう。従って、混入する宿主タンパク質が特別に問題である場合には、組換えにより製造される抗体の大規模精製工程を改良しそして単純化するというニーズが存在する。
【課題を解決するための手段】
【0005】
本発明は、組換え抗体を製造するために改良されたE. coli宿主細胞を提供することにより上記問題を解決する。特に、本発明は、該細胞が野生型においては該組換え抗体と共精製する1以上のE. coliタンパク質の少なくとも一つの物理的性質を改変するために遺伝的に修飾されていることを特徴とする組換え抗体を生産するためのE. coli宿主細胞を提供する。
【0006】
従って、組換え抗体と共精製しないように選択したE. coliタンパク質の物理的性質を改変することによりE. coliにおいて生産された抗体の精製工程を改良できることを、われわれは立証することができた。本発明のE. coli宿主細胞を使用した結果として、該細胞を使用して生産された抗体の精製工程を改良することができ、例えば、野生型のE. coliにより生産されたものよりは工程は速やかでありそして/又はより経済的でありうる。したがって、本発明における抗体の精製における改良は、E. coli宿主タンパク質の物理的性質を修飾することにより生じた精製工程に対する何らかの有利な改変であると考えることができる。改良には、限定はしないが、精製の速度の改善、精製コストの減少、または生産された抗体の品質の向上が含まれる。本発明の一態様において、組換え抗体の精製工程は全精製段階の除去により改良され、コスト及び時間の両者の節減を生じる。好ましくは、除去される工程はアフィニティークロマトグラフィー工程、イオン交換工程、サイズ排除工程、又は疎水性相互作用工程である。好ましい態様において、抗体Fab’フラグメントの精製の間に除去される工程は疎水性相互作用工程でありそして改変されるE. coliタンパク質はリン酸結合タンパク質(PhoS/PstS)である。好ましくは該工程の除去は、Fab'のモルベースで、元の精製工程のコストに比較して約15%削減をもたらす。
【0007】
本発明のもう一つの態様において、本発明の宿主細胞において生産された組換え抗体の精製工程は必要なカラムマトリックスの量を減少させることにより改良され、それにより物質のコスト及び操作時間を減少させる。好ましくは通常は組換え抗体と共精製しそして抗体に結合するカラムの能力を減少させる宿主タンパク質の混入数を減少させることによりこれを達成する。好ましい態様において増加するカラムの能力はカチオン交換カラムでありそしてカラムへの結合を防ぐために改変されるE. coli宿主タンパク質はジペプチド結合タンパク質(DppA)、マルトース結合タンパク質(MBP)及びチオレドキシンである。
【0008】
このように、本発明により組換え抗体を製造するための改良されたE.coli宿主細胞が提供される。本発明のE.coli宿主細胞は組換え抗体を生産することができる天然に存在する生物又は突然変異生物でありうる。しかし、好ましくは、宿主生物は組換えDNA技術を使用して組換え抗体の生産をコードする異種DNA配列を導入された生物又は生物の子孫である。本発明に使用するのに適した特別な宿主E. coli株は、これに限定せずに、MC4100,TG1,TG2, DHB4, DH5α,DH1, BL21, XL1Blueを含む。一つの好ましいE.coli宿主はE.coli W3110 (ATCC 27,325)であり、広く組換えタンパク質醗酵に使用されている宿主株である。E. coliにおける外来遺伝子の発現は発現ベクター中に遺伝子のcDNAコピーを挿入することにより行われる。多くの型の発現ベクターが使用可能である。該ベクターは通常マルチクローニング部位(発現カセット)により分離されたプラスミドのDNA複製起点、抗生物質選別マーカー及びプロモーター及び転写ターミネーター及びリボソーム結合部位をコードするDNA配列によって構成されている。
【0009】
用語「野生型」とは、混入する宿主タンパク質が修飾されていない宿主細胞のことである。この宿主細胞の他のタンパク質がこの発明以外の目的で修飾されていることはありうる。
【0010】
遺伝子修飾のために選択されたE. coli宿主タンパク質は、野生型では精製の際に組換え抗体と共精製されることが知られているタンパク質である。用語「共精製」とは同じセットの精製条件の下に他のタンパク質を伴ってあるタンパク質が精製されることである。典型的にこれはクロマトグラフィーのような精製工程の際に組換え発現した抗体に伴って混入したE. coliタンパク質が精製されることである。
【0011】
用語「遺伝子修飾」とは遺伝子配列の1以上の欠失、挿入、置換又は突然変異によりその遺伝子によりコードされたタンパク質の物理的性質に変化を生じることである。好ましくは、これ等の変化はコードされたタンパク質の生理学的または生物学的活性に影響しない。
【0012】
用語「疎水性」とは、水又は有機溶媒中のタンパク質の溶解性及び固体表面及びマトリックスとの相互作用に及ぼす全タンパク質表面上又は局所的部分表面の疎水性及び親水性アミノ酸の総合的影響のことである。
【0013】
用語「pI又は等電点」とはポリペプチドの陽電荷と陰電荷がバランスを取るpHのことである。pIはポリペプチドのアミノ酸残基のネット電荷から計算することができるし、あるいは等電点電気泳動により測定することができる。
【0014】
用語「物理的性質」とは、生理学的又は生物学的活性でなくタンパク質そのものの物理的性質のことである。好ましくは、物理的性質とは大きさ、疎水性および等電点のようなタンパク質の特性である。E. coli宿主タンパク質のいずれの物理的性質が改変されるかは組換え抗体の精製工程及び必要な改良によって規定されるであろう。例えば、ある条件下に特別なイオン交換カラムへの結合を阻止するためにタンパク質の等電点を変更することができる。
【0015】
遺伝子修飾により改変される混入E. coliタンパク質の物理的性質には、限定はしないが、等電点及び/又は大きさ及び/又は疎水性を含めることができる。タンパク質の大きさとはタンパク質の分子量のことである。混入タンパク質の物理的性質の改変はコードするヌクレオチド配列内の特定の配列の付加、欠失、置換又は挿入のいずれかの組合せにより行うことができる。一態様において、タンパク質の物理的性質はN又はC末端の少なくとも一つのアミノ酸の付加又は欠失により改変することができる。一態様において、混入する宿主タンパク質の物理的性質はC末端へアミノ酸タグを付加することにより改変することができる。好ましい態様において改変される物理的性質は等電点でありそしてアミノ酸タグはC末端へ結合したポリアスパラギン酸タグである。一態様において該タグの付加により改変されるE. coliタンパク質はジペプチド結合タンパク質(DppA),マルトース結合タンパク質(MBP)、チオレドキシン及びリン酸結合タンパク質(PhoS/PstS)である。一つの特別な態様においてE. coliリン酸結合タンパク質(PhoS/PstS)のpIは、C末端へ6個のアスパラギン酸残基を含む、ポリアスパラギン酸タグ(ポリD)の付加により7.2から5.1に減少する。
【0016】
物理的性質を改変するために混入するE.coliタンパク質の特定の残基を変更するか又はN又はC末端タグの付加と組み合わせて改変することもまた好ましい。該改変はタンパク質の大きさを変えるための挿入又は欠失又はpI又は疎水性を変えるためのアミノ酸の置換を含むことができる。一態様においてこの残基はタンパク質の表面に局在している。好ましい態様においてPhoSタンパク質の表面残基はタンパク質のpIを減少させるために変更される。好ましくはPhoSタンパク質の機能を維持するためにリン酸結合に関係する残基(Bass, US5,304,472)を避ける。好ましくはタンパク質の表面から突き出ているか又は塩基性残基の大きな群の中又は近くにあるリシン残基が標的にされる。一つの態様において、PhoSタンパク質はC-末端に結合したヘキサポリ-アスパラギン酸タグを持っているが一方で分子の逆の末端における表面残基は置換の標的にされる。好ましくは中性残基を酸性残基に換えるよりも大きなpI変化の可能性を与えるために選択されたリシン残基はグルタミン酸又はアスパラギン酸に置換される。この中における置換突然変異体の命名は文字次いで数字次いで文字からなる。最初の文字は野生型タンパク質におけるアミノ酸を示す。数字はアミノ酸置換が行われるアミノ酸の位置であり、そして二番目の文字は野生型のアミノ酸に代って使用されるアミノ酸を示す。本発明におけるPhoSの好ましい突然変異においてリシン残基(K)275,107,109,110,262,265,266,309,313がグルタミン酸(E)に、単点又は複合点の突然変異として、置換され、さらにリシン(K)318はアスパラギン酸(D)に、単点又は複合点の突然変異として、置換することができる。好ましくは単点置換突然変異はK262E, K265E及びK266Eである。好ましくは、複合点突然変異はK265/266E及びK110/265/266Eである。より好ましくは、全ての突然変異がC-末端に結合したポリアスパラギン酸(ポリD)と複合しておりそして任意にK318D置換も伴っている。好ましい態様において突然変異は少なくとも2単位のpIの減少を生じる。好ましくは本発明の突然変異によりPhoSのpIは7.2から約4及び約5.5の間へ減少する。本発明の一態様において突然変異ポリD K318D,ポリD K265/266E及びポリD K110/265/266Eを使用してE.coliのPhoSタンパク質のpIは7.2から約4.9、約4.8及び約4.5にそれぞれ減少した。
【0017】
好ましくはE.coli宿主タンパク質の全ての遺伝子修飾により必要な精製工程において組換え抗体と共精製されないタンパク質を生じる。好ましくはPhoSタンパク質の変更はイオン交換の際に抗体Fab'フラグメントと共精製されないタンパク質を生じ、そして好ましくは以前必要であった疎水性相互作用工程は最早必要でなくなる。好ましくはPhoSタンパク質の突然変異は全て組換え抗体と同じ塩濃度ではカチオン交換カラムから溶出されないタンパク質を生じる。好ましくは突然変異PhoSタンパク質は100 mMより低いNaClで溶出するが、しかし抗体はpH 4.5において200 mMで溶出する。さらに好ましいのは突然変異PhoSタンパク質は5.0又はそれ以下のpHでは全くカチオン交換カラムに結合しないであろう。本発明の一態様において突然変異PhoSタンパク質は野生型PhoSが結合しないpH 8以上においてアニオン交換カラムに結合するであろう。
【0018】
好ましくは混入する宿主タンパク質の物理的性質の改変はタンパク質の生物活性又は機能に有意な影響を与えない。本発明の一態様において突然変異PhoSタンパク質はPhoS欠損E.coli株、ANCC75(Amemura et al., 1982, Journal of Bacteriology, 152, 692-701)を補充する能力により測定される機能を維持している。好ましくはE.coli宿主タンパク質における突然変異の全ては野生型と比較してE. coliの増殖又は組換え抗体の収量に影響しない。
【0019】
本発明のE. coli宿主細胞において生産される組換え抗体は、いずれかの抗原に結合する免疫グロブリンフラグメント、例えばFv, Fab, Fab'及びF(ab')2フラグメント、及びいずれかのその誘導体、例えば一本鎖Fvフラグメントを含む免疫グロブリン分子のいずれかである。抗体の生産は当業者によく知られておりそして抗体フラグメントは封入体からのリフォールディングにより又は細菌細胞周辺への分泌による機能的発現によりE.coliにおいて定型的に生産されている(Pluckthun and Pack 1997, Immunotechnology, 3, 83-105; Verma et al., 1998, Journal of Immunological Methods, 216, 165-181)。
【0020】
所与抗体の精製工程はE.coliにおける該抗体の発現に続く純粋な組換え抗体を作製するために必要な精製段階の連続である。組換え抗体の精製は当業者によく知られておりそして個々の抗体に対して最小の生成段階を使用して最大の収量及び純度を生じる精製工程を工夫することができる。その例としてはイオン交換クロマトグラフィー、疎水性相互作用クロマトグラフィー、サイズ排除、等電点電気泳動、逆相HPLC, クロマトフォーカシング、SDS-PAGE, 硫酸アンモニウム沈殿、及びアフィニティークロマトグラフィー(例えば、タンパク質A, タンパク質G,又は捕捉試薬としての抗原を使用)を含む1以上の精製段階を使用して組換え抗体を精製することができる。組換え抗体の大量生産に使用される最も一般的な方法は、コスト効率がよくそして物理的に耐久性がよいので、分別沈殿、イオン交換、疎水性相互作用クロマトグラフィー及びサイズ排除クロマトグラフィーである。これ等の方法はその物理的特性、それぞれ等電点、疎水性及び大きさに基づいてタンパク質を分離する。混入するE.coliタンパク質が組換え抗体と類似の物理的性質を持っている場合には、これ等の方法を使用してタンパク質を分離することはできないであろう。このような抗体にはタンパク質-A及びタンパク質-Gアフィニティークロマトグラフィーのような費用のかかる精製段階を追加する必要があるであろう。
【0021】
所与組換え抗体に対する精製工程を検査することにより、組換え抗体と共精製され除去するのが困難である混入E. coliタンパク質が存在するか否かを当業者は速やかに決定することができる。該混入タンパク質の存在は精製工程を工夫した人には既知であり、例えばその除去のためだけに工程に組み込まれた追加の段階がありうる。そのほかには、カラムフラクションのSDS-PAGEゲルはどのタンパク質が組換え抗体と一貫して共精製されているかを明らかにするであろう。
【0022】
精製工程を検査することにより、組換え抗体の精製が、例えばより少ない又は異なる精製段階を使用することにより改良することができるように当業者は該混入タンパク質のどの物理的性質を変えるのが好ましいのか決定することができる。例えば、高価なアフィニティークロマトグラフィーを使用するよりもサイズ排除クロマトグラフィーを使用して組換え抗体から分離できるように混入タンパク質の大きさを減少させたり又は増加させたりすることが適当であろう。そのほかには、イオン交換又は疎水性相互作用クロマトグラフィーにより分離できるように混入タンパク質の等電点又は疎水性を改変することも適当である。所与の精製工程において混入タンパク質の物理的性質の最適変化を当業者は容易に確認することができる。1を超えるE.coli タンパク質の性質及び/又は該タンパク質の1を超える物理的性質を変化させることも好ましい。
【0023】
組換え抗体と共精製されるE.coliタンパク質が選択されそして精製工程を改良するために該タンパク質のどの物理的性質が変えられることが好ましいかが決定されたならば、物理的性質の改変が行われうるような該タンパク質をコードする遺伝子をクローニングする必要がある。これは当業者には定型業務でありそして最初にタンパク質配列をそのタンパク質から得る必要がある。これは、例えば、ウエスタンブロットからのタンパク質のN-末端配列分析により又は該ブロットから作製したトリプシン分解又はCNBrフラグメントの配列分析により行うことができる。これらは全てタンパク質の配列を得るための定型的方法である。該ウエスタンブロットは混入宿主タンパク質を含むカラムフラクションの電気泳動を行ったSDS-PAGEゲルから作製することができる。該タンパク質から得られたアミノ酸配列は次いで、E.coliの全ゲノム配列が一般に公開されているSwissProt又はGenbankのようなデータベースから全タンパク質及びDNA配列を相同性探索により同定するために使用される(Blattner et al., 1997, Science, 277, 1453-1462)。次いで核酸配列に基づくプライマー及び鋳型としてE.coli DNAを使用するPCRのような既知技術を使用して、混入タンパク質をコードする遺伝子をクローニングすることができる。
【0024】
混入するE.coliタンパク質が同定されそして該タンパク質をコードする遺伝子がクローニングされてしまえば、組換え抗体の精製を改良することになる必要なタンパク質の物理的変化を生じるようにその遺伝子を修飾することができる。タンパク質の物理的性質を改変するための方法は当業者によく知られておりそして目的とする結果を達成するために多くの系統的改変が必要となるであろう。タンパク質配列、可能であれば、配列整列及び結晶構造の分析から、改変の処理を受けるタンパク質の領域を同定することは当業者には可能である。選択される領域は物理的性質における意図する変化によるであろう。例えば、タンパク質のpI又は疎水性を変えるためには特別な電荷又は疎水性を持つ表面に露出した残基に焦点を当てる必要があるであろう。大きさを変えるには削除又は修飾することができる特別なドメインに焦点を当てることができる。Rasmol又はWebLab Viewer Liteのようなコンピュータープログラムはタンパク質の結晶構造を見るには有用であり、いずれの残基を変更するかについて賢明にして学識のある選択をすることを可能にする。タンパク質の活性又は構造にとって重要な残基に関するその他の情報は公開情報から入手可能であろう。
【0025】
タンパク質の機能は残っているのが好ましいので、できればタンパク質配列整列及び結晶構造の検査により活性部位領域を避けるようにすべきである。結晶構造が使用できない場合には、タンパク質の機能又は発現を破壊せずに改変できる残基を同定することは、アラニンスキャンニング突然変異誘起(Cunningham and Wells, 1989, Science, 244, 1081-1085)のような当業者既知の方法を使用して試験することができる。
【0026】
タンパク質の物理的性質を改変する好ましい方法はタンパク質タグの付加によるものであり、この方法は当業者に広く知られている。現在ではこれは混入宿主タンパク質を変えるよりもむしろ組換えタンパク質の精製を容易にするためにその性質を変えるために使用されている。このようなタグが組換えタンパク質に使用された場合には、タンパク質機能を回復し、溶解性を増し又はタグが抗原性であるので治療のために、それは精製されたタンパク質から除去される必要がある。これにはしばしば困難を生じそしてこの技術を上手に適用するための最大の障害となることがある(Sassenfeld, 1990, Tibtech, 8, 88-93)。本発明においては、タグは組換え抗体自身でなく混入するE. coliタンパク質のpIを変えるために使用されているので該タグの除去は不必要である。タンパク質のpI,疎水性及び大きさは全てアミノ酸タグの付加により変更することができ、その性質は必要とする成果によるであろう。例えば、タンパク質の等電点を変更するのに適するタグはポリ-アルギニンタグ(Sassenfeld and Brewer, 1984, Biotechnology, 2, 76-81; Brewer US 4,532,207; Niederauer et al., 1996, Biotechnology Progress, 10, 237-245; Stempfer et al., 1996, Nature Biotechnology, 14, 329-334)、ポリ-グルタミン酸タグ(Dalbφge et al., 1987, Bio/Technology, 5, 1447-1457; Niederauer et al., 1996)及びbespoke タンパク質ドメインタグ(Graslund et al., 2000, Protein Engineering, 13, 703-709 and Graslund et al., 2002, Journal of Chromatography, 942,157-166)である。疎水性を改変するために使用することができるタグにはポリ-フェニルアラニンタグ(Persson et al., 1988, Analytical Biochemistry, 172, 330-337)及びエラスチン様ポリペプチド(Meyer and Cholkoti, 1999, Nature Biotechnology, 17, 1112-1115)が含まれる。標的タンパク質の大きさ増加させる実行可能な融合相手として使用されてきた多数のタンパク質及びタンパク質のドメインも存在し、それらにはアルカリホスファターゼ(Carrier et al., 1995, Journal of Immunological Methods, 181, 177-186)、β-ガラクトシダーゼ(Nielsen et al., 1988, JIMM 111, 1-9)、マルトース結合タンパク質(di Guan et al., 1988, Gene, 67, 21-30)、GST(グルタチオンS転移酵素、Smith and Johnson 1988, Gene, 67, 31-40)、セルロース結合ドメイン(Ong et al., 1989, Bio/Technology, 7, 604-607)、DsbA(Collins-Racie et al., 1995, Bio/Technology, 13, 982-987)、DsbC(Novagen)、チオレドキシン及びNusA(Novagen)が含まれる。該融合により10〜60 kDaの大きさの増大を生じる。該タグは単独で又は以下に記述する混入タンパク質に対する他の特定の改変と組み合わせて使用することができる。
【0027】
宿主タンパク質の物理的性質を改変するその他の好ましい方法は特定残基の修飾であり、その方法は当業者に良く知られている。例えば、ズブチリシンの静電気的性質はX線結晶構造をガイドとして使用して複数の荷電アミノ酸残基を導入することによりズブチリシンの表面電荷を変えることにより修飾された(Egmond et al., 1996, In Subtilisin Enzymes; Practical Protein Engineering, R Bott and C Betzel eds, 219-228)。Marttila et al., 1998, FEBS Letters, 441, 313-317,は、天然のpI 10.5に対して9.4から4.7の範囲のpIを持つ数個のアビジンの荷電変異体を作製した。この変異体は既知結晶学的データ並びに比較配列整列に基づいてリシン及びアルギニンのような塩基性残基を中性又は酸性アミノ酸と置換することにより作製された。
【0028】
突然変異のためにいずれの表面残基を選択するかは、活性部位に対する残基の位置、溶媒暴露の程度、他の表面残基との潜在的相互作用、関係するアミノ酸R-基の相対的溶媒和能力、アミノ酸のpKa、関係するアミノ酸の相対的疎水性/親水性、関係するR-基の相対的長さを含む構造的及び立体的考慮により支援される。
【0029】
特定の残基の突然変異はオリゴヌクレオチド仲介突然変異誘起(Zoller and Smith, 1982, Nucleic Acid Research, 10, 6487)のような当業者既知の方法により行うことができる。タンパク質の等電点を変更させるには、中性アミノ酸を目的電荷のアミノ酸に換えるか、又は逆の電荷を持つアミノ酸残基に換える必要がある(すなわち、アスパラギン/グルタミン酸の代わりにリシン/アルギニン)。典型的にタンパク質のpIを上昇させるためには、アスパラギン酸及びグルタミン酸のような酸性アミノ酸を置換してリシン及びアルギニンのようなより塩基性のアミノ酸をタンパク質に取り込まなければならない。pIを減少させるには、置換の選択は逆にしなければならない。
【0030】
そのほかには混入するタンパク質の疎水性を減少させることも好ましいであろう、そしてこれはバリン、ロイシン、イソロイシン、フェニルアラニン、トリプトファン、メチオニン及びプロリンのような疎水性残基をより親水性又は極性のある残基、例えばセリン、トレオニン、システイン、チロシン、アスパラギン酸、グルタミン酸、アスパラギン、グルタミン、ヒスチジン、リシン又はアルギニンに置換することにより行うことができる。そのほかに疎水性を増加させるにはアミノ酸置換を逆にしなければならない。
【0031】
タンパク質の発現及び任意の機能に影響することなく削除することができるタンパク質の特別なドメイン又は部分を同定することにより、混入するタンパク質の大きさを減少させることも可能である。これ等はN又はC-末端ドメイン又はインフレーム融合を形成するために削除することができる露出ループである。そのほかにはサイズ排除クロマトグラフィーを使用する分離を改良するために混入するタンパク質の大きさを増加させるために、既に記述したようなアミノ酸タグ又はタンパク質ドメインをタンパク質に融合することができる。
【0032】
混入するタンパク質の発現、活性又は物理的性質に及ぼす置換、欠失、挿入又はタグの正確な影響を予測することは困難であることは、当業者には理解されるであろう。必要な成果を達成するために当業者は一連の種々の変更を試験するであろう。これ等は連続して又は平行して創りそして試験することができる。その効果は以下に記述されるような定型的スクリーニング検定により評価され、必要があればさらに修飾が行われることを当業者は理解するであろう。
【0033】
各変異体を試験するために改変されたタンパク質は非修飾タンパク質を発現しないE.coli宿主細胞において発現されなければならない。これは欠失変異株のようなこの遺伝子を発現しないE.coli宿主を使用することにより又はその遺伝子の発現が抑制される条件下に該宿主を培養することにより達成される。改変された混入タンパク質は該タンパク質の高レベルの発現を生じるプラスミドの方法で導入することができる。そのほかには、変異タンパク質はE.coliゲノムの中に改変遺伝子を直接組み込んで内在性遺伝子を置換することにより試験することができる。
【0034】
各改変タンパク質の発現に成功したことはE.coliの醗酵の後のSDS-PAGE分析により確認することができる。改変タンパク質が適切に発現しない場合がありうるが、その突然変異は廃棄すべきである。変異タンパク質の発現に成功した後、物理的性質の改変はクロマトグラフィー及びゲル電気泳動により評価することができる。例えば、改変が大きさの減少を生じるはずである場合には、サイズ排除カラム及びSDS-PAGEにより試験することができる。改変がpIの変化を生じるはずである場合には、イオン交換クロマトグラフィー及び等電点電気泳動を使用して試験することができる。改変が疎水性の変化を生じるはずである場合には、疎水性相互作用クロマトグラフィー及び溶媒溶解性を使用して試験することができる。組換えタンパク質が存在しなくてもこれ等の方法により改変タンパク質を評価することは当業者には可能である。任意に、試験前に精製組換え抗体を抽出物に添加することにより組換え抗体からの分離を確認することができる。
【0035】
タンパク質の物理的性質が改変されても修飾E.coliタンパク質の生物学的機能は保持されていることが好ましい。突然変異体の機能はタンパク質同定に関わる技術分野において広く知られている種々の方法で試験することができる。例えば、変異タンパク質はその生物学的活性を立証する生物学的検定、例として示すならば、酵素検定により試験することができる。そのほかに、変異タンパク質をコードする遺伝子を目的の遺伝子を欠失したE.coli突然変異体に補充して、次いで生物活性又は細胞増殖を試験することに使用できる。このような突然変異体は既に多数存在するか、又は当業者は容易に創ることができる。
【0036】
上記の方法を使用して、改良精製工程の条件を充たしそして好ましくは機能を維持する少なくとも一つの突然変異体を同定することは当業者には可能であろう。好ましくは、ゲノムに組み込まれた後に突然変異体のいずれかがE.coliの増殖又は組換え抗体の収量に有害な影響を及ぼす場合には、この段階において1を超える突然変異体を選択するであろう。野生型の遺伝子を置換するように選択した突然変異遺伝子をE.coliゲノムの中に組み込むことは当業者既知の方法を使用して行うことができる(Hamilton et al., 1989, Journal of Bacteriology, 171, 4617-4622)。E.coliゲノムDNAに認められる配列に相補的である改変DNA配列は形質転換用のベクターに含まれる。このベクターによるE.coliの形質転換はゲノムの相同組換え及び野生型遺伝子の代わりに改変遺伝子の挿入を生じる。
【0037】
修飾遺伝子配列で野生型遺伝子を置換した後、元の野生型E.coli発現システムにおいて行うように改良E.coli宿主細胞を望ましい組換え抗体遺伝子配列で形質転換する。次いでそのE.coli宿主細胞を元の野生型と同じ条件下に培養しそして細胞の増殖及び組換え抗体の収量を測定しそして野生型と比較する。組換え抗体の収量は使用する組換え抗体用の当業者既知の標準的方法を使用して試験することができる。例えば、抗体Fab'フラグメントはELISAにより定量することができる。好ましくは抗体収量及びE.coli増殖に逆効果は認められてはならない。逆効果が認められるならば選択された他の突然変異の一つを使用すべきでありあるいは新しい突然変異体を作製すべきである。このような突然変異を系統的に又は平行して試験できることは当業者には既知であろう。
【0038】
改変宿主タンパク質が組換え抗体と最早共精製しないことを確認するために、組換え抗体を目的の工程を使用して精製しそして改変宿主タンパク質の存在を例えばSDS-PAGEにより監視すべきである。物理的性質の変化のために改変宿主タンパク質は期待通りに行動するはずでありそして最早組換え抗体とは共精製しないはずである。
【0039】
以下の実施例は説明のために、そして限定するためでなく提供される。
【0040】
実施例
材料と方法
DNA操作
一般的DNA操作には標準的方法を使用した。制限酵素はBoehringer Mannheimから、Precision PlusをStratageneから入手した以外はTaq ポリメラーゼはRocheから入手した。プラスミドの調製はQiagenキットをメーカー説明書に従って使用しておこなった。オリゴヌクレオチドはSigma-Genosys Ltd.,Pampisford, U.K.から入手した。オリゴヌクレオチド及び領域をコードするPCR産物の配列はPRISM Big Dyeサイクル配列分析キット及びGenetic Analyzerソフトウエアを使用するABI PRISM-3100シークエンサーを使用して両鎖の配列分析により確認した。超コンピテントE.coli株XL1Blue MRF' Kan (Stratagene)を全てのDNA操作に使用した。野生型E.coli W3110(ATCC ref.27325)を洗いそして氷冷滅菌10%(v/v)グリセロール中で3回濃縮することにより電気穿孔にコンピテントにし、パルス調節装置を装着したBioRad Gene Pulser を使用して2000V 25 mS及び200Ωにおいて電気穿孔を行った。新しい単一W3110コロニーを100 mlの2xPY培地中37℃でOD600が約0.5〜0.8に達するまで増殖した。その後培養を氷で15分間冷却し、氷冷下試験管中で4℃ 4000 g 10分間遠心分離して細胞ペレットにした。細胞ペレットを再懸濁し、洗い、滅菌脱イオン水で作製した氷冷10%(v/v)グリセロール中で3回ペレットにした。最後のペレット化の後、細胞ペレットを滅菌脱イオン水で作製した氷冷10%(v/v)グリセロールで最終容量10 mlに再懸濁した。細胞を直ちに使用するかまたは液体窒素で凍結して-70℃で保存した。
【0041】
細菌用複合培地及び無リン酸培地
「PhoS培地」を、phoレギュロンを抑制するPO4及びペプチドリッチ培地を供給するためにE.coliの一般的増殖の全て、プラスミド調製及びPhoS発現実験に使用した(「PhoS培地」=1%(w/v)トリプトン、0.5%(w/v)イースト抽出物、0.3%(w/v) KH2PO4, 0.7%(w/v) K2HPO4,0.5%(w/v) NaCl及び0.5%(w/v) DIFCOのcasamino acids)。「無PO4」と定義される培地はNeidhardt et al., 1974 Journal of Bacteriology, 119, 736-747に記述されたMOPS培地にグルコースを0.5%(w/v)、casamino acidsを 0.5%(w/v)、及びチアミンを1 mM添加したものである。組み込み実験に用いられた細胞は2xPY(2%寒天(w/v)、1% フィトーン(w/v)、0.5%イースト抽出物(w/v)、0.5% NaCl(w/v)及び1 M NaOHでpH 7.0にする)を使用して増殖した。必要があれば培地には3%寒天(w/v)、カルベニシリンを200 μg/ml又はクロラムフェニコールを20 μg/ml、IPTGを200 μM及びXPを40 μg/ml添加した。
【0042】
Fab'測定用ELISA
ELISAプレートをPBS中2 μgml-1のHP6045 で4℃において終夜コーティングした(マウスIgG2aモノクロナール抗-ヒトIgG Pan Fd(CH1)であるHP6045をATCCのハイブリドーマHP6045から得た。イムノグロブリンはプロテインA精製により培養上清から回収し、微量のウシIgGをヒツジ抗-ウシIgGカラムで除去した)。dH2Oで4x洗った後、プレート上でサンプルを系統的1/2希釈及び標準を100 μlのサンプル/コンジュゲート緩衝液(100 mM Tris/Cl pH 7, 100 mM NaCl, カゼイン0.2%(w/v)、Tween 20 0.0002%(v/v))中に作製し、そしてプレートを室温で1時間250 r.p.m.において振とうした。dH2Oで4x洗った後、100 μlの表示抗体GD12, HRP結合F(ab')2抗ヒトカッパ鎖(The Binding Site, Birmingham, U.K.)を加え、サンプル/コンジュゲート緩衝液中に1/1000に希釈し、プレートを室温で1時間200 r.p.m.において振とうした。dH2Oで4x洗った後、100 μlの3-3'-5-5’テトラメチルベンジジン(TMB)基質を加え(0.1 M酢酸/クエン酸ナトリウム pH 6、100 μg/ml TMB, H2O20.01%(v/v))、そして自動化プレートリーダーを使用してA630を記録した。細胞周辺抽出物中のFab'の濃度は適当なイソタイプの精製Fab'標準と比較することにより算出した。
【0043】
PhoS遺伝子のクローニング
約700 bpの3'-クロモソーム配列を伴う野生型PhoS遺伝子を鋳型としてW3110 E.coliを使用してDra I-BstB I制限断片としてpSK-(Stratagene)中にPCRクローニングした。PhoSのC-末端の改変を容易にする目的で、新規なHind III部位を組み込むためにPhoS遺伝子の終結コドンの3'に隣接する3塩基対の改変を行った(図1の図解参照)。PhoS遺伝子を改変しそしてPhoSタンパク質のクロマトグラフィー挙動で変化を試験した後、最終的な遺伝子構築をE.coli染色体に組換えるためにSal I-BamH IフラグメントとしてpKO3に戻した。PhoS遺伝子及びその周辺のゲノム配列は文献及び公開データーベースで見ることができる(Surin et al., 1984 and Blattner et al., 1997)。
【0044】
突然変異PhoSタンパク質をコードする遺伝子の構築
鋳型としてW3110 を使用し、Taq ポリメラーゼを使用するPCRにより3'ゲノム配列の一部を伴うPhoSコード配列を2部分にクローニングした。これに使用したオリゴヌクレオチドを表1に示す。PCR突然変異誘起を使用して終結コドンの直後の3塩基対を改変してHind III部位をコードした。したがって遺伝子の3'の改変を含むPhoSコード領域はDra I-Hind III制限フラグメントとしてSma I-Hind III制限pSK-プラスミド中にクローニングすることができた。PhoS遺伝子の3'末端に施された改変はポリ(ヘキサ)アスパラギン酸、及びK318D変異を伴うポリ(ヘキサ)アスパラギン酸をコードするコード領域を含む。
【0045】
表面残基の突然変異は図1に示すような有用な制限部位に及ぶオリゴ(表1に示す)を使用するオリゴヌクレオチド指定突然変異誘起PCRを使用して行った。したがってPCR及び制限クローニングを使用することによりこれ等の突然変異を構築して組み合わせることが可能である。
【表1−1】
【表1−2】
【0046】
突然変異PhoSを組み込んだ組換えプラスミドの構築
挿入配列の両側の隣接領域が100%相同であれば、E.coli染色体中への突然変異遺伝子の指定相同組換えの効率は増加する。この隣接領域の長さは通常200〜1000 bpの程度である(Hamilton et al., 1989, Journal of Bacteriology, 171, 4617-4622)。Hind III部位及び元来のBstB I部位を導入するために突然変異改変の3'染色体配列の約700 bpをPCRクローニングした。このHind III-BstB IフラグメントをHind III-Cla I制限pSK-中にクローニングした。
【0047】
pSK-から発現したPhoS突然変異体を全て構築しそして試験した後、目的の最終PhoS遺伝子は全て3'染色体隣接領域の676 bp Hind III-Xho Iフラグメントを同様に制限処理されたPhoS発現プラスミド中に移動することによりその後にクローニングされた上記の3'染色体隣接領域を有した。PhoS組み込みカセットは1852 bp BamH I-Sal I制限フラグメントとして同様に制限処理されたpKO3相同組換え/置換プラスミドに移した(Link et al., 1997, Journal of Bacteriology, 179, 6228-6237)。
【0048】
W3110中のPhoS遺伝子の染色体置換の構築
プラスミドpKO3(Link et al., 1997)をマーカーの無い染色体遺伝子置換を発生させるために使用した。染色体組込みを強制しそして選別するためにこのプラスミドはクロラムフェニコールマーカーを伴うpSC101の温度感受性変異体の複製起点を使用する。レバンスクラーゼをコードするsacB遺伝子はスクロース上で増殖するE.coliにとっては致死的であるので(クロラムフェニコールマーカー及びpSC101起点を一緒に)デインテグレーション及びプラスミドキュアリングを強制しそして選別するために使用する。この方法は既に記述されている(Hamilton et al., 1989, Journal of Bacteriology, 171, 4617-4622及びBlomfield et al., 1991, Molecular Microbiology, 5, 1447-1457)。
【0049】
pKO3組込みプラスミドの使用
1日目 冷却したBioRad電気穿孔キュベット中で100 μlのE.coli細胞を3 μlのpKO3 DNAと混合し次いで2500V,25 μF及び200Ωで電気穿孔した。直ちに900 μlの2xPYを加え、インキュベーター中30℃で1時間250 rpmで振とうすることにより細胞を回復した。細胞を2xPY中で系統的1/10希釈を行いついで100 μlの分割液をクロラムフェニコール20 μg/mlを含み30℃及び43℃に予め加温した2xPY寒天プレート上に注いだ。プレートを終夜30℃及び43℃でインキュベートした。
2日目 30℃で増殖したコロニーの数は電気穿孔の効率の指標を与え、他方43℃で生存し増殖したコロニーは組込みの可能性を示す。43℃プレートのシングルコロニーを取り出し、そして10 mlの2xPYに再懸濁した。シングルコロニーを作るために、この100 μlを5%(w/v)スクロースを含み30℃に予熱した2xPY寒天プレート上に注いだ。プレートを30℃で終夜インキュベートした。
3日目 このコロニーはデインテグレーション及びプラスミドキュアリングを同時に生じた可能性を示している。デインテグレーション及びプラスミドキュアリングが増殖の初期に生じていたならば、多数のコロニー集団はクローンであろう。シングルコロニーを取り出し、そしてクロラムフェニコール20 μg/mlを含むか又は5%(w/v)スクロースを含有する2xPY寒天に接種し、複製した。プレートを終夜30℃でインキュベートした。
4日目 スクロース上で増殖しクロラムフェニコール上で死んだコロニーは染色体置換及びプラスミドキュアリングの可能性を示す。これ等を取り出しそして突然変異特異的オリゴヌクレオチドを使用するPCRによりスクリーニングした。正しい大きさのはっきりとしたPCRバンドを生じるコロニーを削り取り、5%(w/v)スクロースを含有する2xPY寒天上にシングルコロニーを作るためにプレートを終夜30℃でインキュベートした。
5日目 PCR陽性、クロラムフェニコール感受性そしてスクロース耐性E.coliのシングルコロニーをグリセロール保存、化学的コンピテント細胞を作るために使用し、そしてPrecision plusポリメラーゼを使用する直接DNA配列分析のためのPCR産物を作成するための5'及び3'隣接オリゴを使用するPCR反応のPCR鋳型として使用した。
【0050】
PhoS補充検定
ANCC75のようなPhoS株(Amemura et al., 1982, Journal of Bacteriology, 152, 692-701)はそのPO4感受性と捕捉能力が連携していないために構成的にアルカリホスファターゼ(AP)を発現する。PhoS障害をプラスミド発現PhoS遺伝子で補充することが可能であり、この補充を寒天プレート上又は液体培地それぞれでAP活性を検定するための5-ブロモ-4-クロロ-3-インドリルリン酸(XP)又はパラ-ニトロフェニルリン酸(pNPP)のようなアルカリホスファターゼの発色基質を使用して検出することができる。寒天プレートにはイソプロピル β-D-チオガラクトピラノシド(IPTG)200 μM及び抗生物質とともにXPを40μg/ml加え、そして終夜37℃でコロニーを成長させる。液体培地においてAPを検定するには約1.0のOD600の誘導培養の10〜100 μl検体を25μlの0.1% SDS (w/v)及び50 μlのクロロホルムを含む1.5 M Tris. Cl pH 8.0で1 mlにして、5秒間渦巻き混合し、そして30℃で5分間予備インキュベートした。検定は200 μlの15 mM pNPPを加えそして反転により混合して開始し、そして200 μlの1 M KH2PO4を加えそして反転により混合して10分後に停止した。検定は30℃において実施した。マイクロフュージ中で細胞をペレット化して、上清の吸光度を420 nmでブランクと比較して測定した。アルカリホスファターゼ活性の1単位はΔA420OD600-1min-1として定義した。
【0051】
理論的分子量及びpI計算
理論的パラメーターの計算は全てMacVectorソフトウエアを使用して行った。
【0052】
質量分析
Fab”の分子量はMicromass Ultimaトリプル四重極スペクトロメーターを使用して陽イオンエレクトロスプレーイオン化モードにおいて測定した。Fab”検体はTrisを除去するために10 kDa膜のカットオフサイズ(Amicon, U.K.)によるMicrocon濃縮器を使用して10 mM酢酸アンモニウムとの多容量交換により脱塩した。
【0053】
カチオン交換クロマトグラフィー
フラスコを振とうして醗酵細胞ペレットを100 mM Tris.Cl/10 mM EDTA pH 7.4 中に30 OD600/元の培養容量mlにそれぞれ再懸濁し、そして終夜30℃又は60℃で撹拌した。細胞破片を除くために4000 gで10分間遠心分離した後上清に1 M酢酸を加えて精製実験を行う緩衝液のpH(典型的にはpH 4.5, 5.0 又は6.0)にpHを下げ、そして伝導度が≦3.5 mScm-1になるまでdH2Oで希釈した。再度pHをチェックした後、細胞周辺抽出液を20,000 gで10分間遠心分離して澄明とし、次いで0.2 μm膜を通して濾過した。
【0054】
1 ml/minの流速においてFPLC指令ソフトウエア及び10 ml検体注入シリンダーにより稼動するPharmacia P500 FPLCを使用して酢酸ナトリウム(NaAc)緩衝液中での5 ml SPセファロース(Pharmacia)カラム処理を共通して行った。PhoS及びFab'’構築に対しては全て以下の基本的溶出方法を使用した:カラムの平衡は1 mlの平衡緩衝液で行い、9 mlの検体を負荷し、次いで70 ml(カラム容積の約14倍)のNaAc中0〜200 mM NaClグラジエントで溶出し、カラムを7 ml NaAc中1 M NaClで洗い、次いで13 ml 負荷緩衝液(NaAc)で再度平衡した。A280により監視した溶出点の時間及び伝導度を記録しそしてフラクションを手動で採取した。
【0055】
アニオン交換クロマトグラフィー
20 mM Tris.Cl pH 8.0 中の2.5 ml Poros HQカラム(PerSeptive Biosystems)処理を共通して使用した。カチオン交換の検体を使用前に20 mM Tris.Cl pH 8.0に緩衝液を交換した。カラムの平衡は1 mlの洗浄で行い、9 mlの検体を負荷し、次いで10.9 mlの洗浄段階、次いで20 mM Tris.Cl pH 8.0中3.9 mlの1 M NaClで溶出し、そして10 ml負荷緩衝液(20 mM Tris.Cl pH 8.0)で再度平衡した。タンパク質フラクションはA280により監視しそして手動で採取した。
【0056】
SDS-PAGE, イムノブロット及びIEFゲル
SDS-PAGEはMES緩衝液中でSeeBlue2標準を使用してInvitrogenの4〜12% NuPAGEゲルで行った。それをクマシーで染色するか又はタンパク質を1/2xTowbin緩衝液を使用してPVDF膜に移した。IEFゲルはInvitrogenのpH 3〜10 IEF垂直スラブゲルであり、Serva標準pI 3〜10を使用し、クマシーで染色した。脱染色後ゲルをスキャンし及び/又はセルロース膜に挟んで乾燥した。
【0057】
実施例1
宿主タンパク質を改変することにより行うことができる改良を確認するためのFab'抗体フラグメントの精製工程の概要。
E.coliにおいて発現したFab'フラグメントは細胞周辺抽出液から3つのクロマトグラフィー段階を使用して定型的に精製された。第一段階はpH 4.5におけるカチオン交換であり、この間に抗体Fab'フラグメントはカチオン交換カラムに結合した。Fab'フラグメントはカチオン交換から溶出されそしてpH 8.0 においてアニオン交換処理された。Fab'フラグメントはこのpHではアニオン交換に結合せず通過液中に採取された。この段階の間に殆どの残留E.coliタンパク質及びエンドトキシンはアニオン交換カラムに結合し、Fab'調製品から除去された。最後に、SDS-PAGEにより確認されたように、両イオン交換段階において共精製した一つの多量のE.coliタンパク質を除去するために疎水性相互作用クロマトグラフィー(HIC)段階が必要であった。
【0058】
それにより精製工程をスピードアップしそして物質及び労力に関するかなりのコスト節減をもたらすであろうことから、最後のHIC段階を除くことが好ましいことはこの精製工程の概要から明らかであった。この一つの段階を除去することによる推定コスト節減はFab'のモルベースで15%であった。これを達成するためには混入するE.coliタンパク質の物理的性質を、イオン交換でFab'と最早共精製せずに、したがってHIC段階を最早必要としないように改変する必要があるであろう。これは最早カチオン交換カラムに結合しないかあるいはFab'と異なる塩濃度においてカラムから溶出するようにタンパク質のpIを改変することにより達成することができた。この混入タンパク質は後にリン酸結合タンパク質(PhoS/PstS)として同定された(実施例2)。
【0059】
その他の改良の可能性はカチオン交換カラムに結合する全てのタンパク質のSDS-PAGEゲルの検討により同定された。これによりこのカラムに結合した量の多い宿主タンパク質がいくつかあることが明らかにされた。これ等のタンパク質の除去によりカラムのFab'結合能力が有意に増加するのでカラムの大きさを減少しそして精製を速くするのでコスト節減を生じるであろう。これを達成するにはカチオン交換カラムに最早結合しないように混入するタンパク質のpIを改変する必要があろう。これ等の混入タンパク質は後にジペプチド結合タンパク質(DppA)、マルトース結合タンパク質(MBP)、チオレドキシン及び仮に24 kDaタンパク質と呼ぶものと同定された(実施例2)。
【0060】
実施例2
実施例1において選択された混入タンパク質の同定
標準的E.coli W3110醗酵から生産された細胞周辺フラクションを終夜30℃で100 mM Tris.Cl 10 mM EDTA pH 8.0中に抽出した。遠心分離の後上清をH2O及び酢酸でpHが≦4.5そして伝導度が≦3.5 mScm-1になるまで希釈した。これをカチオン交換カラムに正規の方法で負荷した。洗浄した後、結合したタンパク質を溶出した。Fab'及びそのフラグメントは溶出液をプロテインGカラムを2回及びプロテインLを1回通すことにより除去し、次いでFab'の入っていない溶出液を10 kDaカットオフ膜を使用したAmicon撹拌セルで濃縮した。Fab'に関係したペプチドが全て効率よく除去されたことは、抗-カッパ及び抗-CH1イムノブロット、サンドイッチELISA及びHPLCにより示された。クマシー染色4〜12%SDS-PAGEゲルを使用して濃縮検体を分析した。同じゲルをPVDF膜に移し、ポンソーSで染色してバンドの位置を明らかにして清潔なメスで切り取った。タンパク質はN-末端配列分析を行いそしてその結果はSwissProtへ質問するために使用した。結果を以下に示す。
【表2】
【0061】
実施例3
C-末端ポリ-イオン性テイルを持つPhoSタンパク質の創製と試験
pH 4.5におけるカチオン交換精製に対するC-末端ポリ-イオン性テイルの影響
最初にPhoSの3種をクローニングしそしてE.coliにおける発現及びカチオン交換精製を試験した:野生型PhoS(wtPhoS)、C-末端に6個のアスパラギン酸を持つPhoS(PhoS polyD)、及びC-末端に6個のアスパラギン酸そして近くにK318D変異を持つPhoS(PhoS K/D polyD)。大まかにpHを調整した細胞周辺抽出液のクマシー染色SDS-PAGEにより判断して、これ等3種は振とうフラスコ内で全て良く発現した。プラスミド、タンパク質名、分子量、推定pI、測定pI、及びpH 4.5におけるカチオン交換カラムからのNaCl溶出プロフィールの概要を表2に示す。
【0062】
polyDテイルの追加により、野生型タンパク質に比較して塩グラジエントにおいて約20%早く、103 mM NaClにおいて、PhoSタンパク質は溶出した。しかし、pH 4.5 においてSPセファロースカラムに全く結合しないタンパク質の好ましい効果は達成できなかった。追加のK318D表面突然変異(これはC-末端に非常に近い)はカチオン交換効率についてpolyDタグのみよりも利点が多いことを示す証拠が得られた。
【0063】
pH 8.0におけるアニオン交換精製に対するC-末端polyDテイルの影響
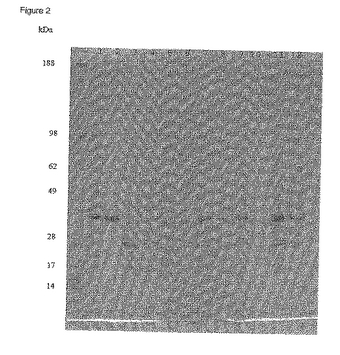
細胞周辺抽出液をカチオン交換実験の通過液及び溶出検体の採取と同様に処理しそして緩衝液を20 mM Tris.Cl pH 8.0 に交換した。wt PhoS, PhoS polyD 及びPhoS K/D polyDの検体を小さなPoros HQアニオン交換カラムに流した。結果は図2に示すように、wt PhoSタンパク質はこのpHではカラムに結合しないが、polyD及びK318D polyDの2種のPhoSはともに結合する。したがってこの条件下では、polyDタグの存在により生じたpI変化に加えて、タグ自身はこの条件下に「親和性テイル」として働いているように思われる。
【表3−1】
【表3−2】
【表3−3】
【0064】
実施例4
pH 4.5におけるPhoSのカチオン交換精製に対する表面突然変異及びC-末端polyDテイルの影響
活性部位に結合したPO4を持つPhoSの結晶構造(Luecke and Quiocho, 1990, Nature, 347, 402-406)を分析して突然変異に使用できる表面残基を探した。PhoSのpIを減少させるために、リシン残基(MW 128.17 Da, pKa=10.79)をグルタミン酸(MW 129.12, pKa=4.07)残基に交換した。塩基性残基を酸性残基に交換することにより、中性残基を酸性残基に交換する(約0.07)よりも大きな突然変異当りのpI変化(約0.15のPhoSのpI変化)の可能性を付与する。また、表面に露出したグルタミン酸残基は(リシン残基のように)高度に溶媒和されると思われるので、多くの他の(非荷電)表面残基をグルタミン酸に換える際よりも重大な構造的変動を生じるリスクが少ないであろう。これまでの構造研究又は突然変異研究からPO4結合に重要である又はこれ等の残基に空間的に近い又は実際PhoS分子の中央の中央溝に近いことが分かっている残基を避けることも重要である。タンパク質の表面の非常に遠くに突き出ているリシン残基及び塩基性残基の大きな集団の中に又はその近くに位置するリシンが選択された。
【0065】
塩基性残基の該集団は精製マトリックスと相互作用することができる重要な荷電パッチを形成することができる。したがって1又はそれ以上の酸性残基の戦略的置換は該パッチを「破壊」することができる、すなわち、該領域のネット電荷を有意に変え、それにより精製マトリックスと相互作用する能力が局所的に大きく変化する。最終的に、C-末端は既に、「割れたラグビーボール」のような形のPhoSタンパク質の一つの端に存在するpolyDテイルにより改変されているので、PhoSタンパク質のC-末端の逆の端にある残基を少なくとも見出すことが好ましかった。
【0066】
以下の領域がこれ等の条件に適合すること、及び直線配列中に集めて集団PCR突然変異誘起を容易にすることができることが確認された。
1) 単一突然変異K275Eは窪んだ表面から突き出ておりそしてK272に非常に近接しておりそしてK282に近い。
2)K107, 109及び110のトリプル突然変異、K107はK98及びE155 に近く、単離したK109はPhoSから突き出ており、そしてK110はK109の近傍にあり、D112, D113及びE114に非常に近い。
3) K262, 265及び266のトリプル突然変異、K262はK265/266から離れているがK318 の近傍にあり、他方K265及びK266 は一緒に「V」字を形成しそしてN48 と三角形を形成している。
4) K309, 313及び318のトリプル突然変異は全て、T310及びP319 に隣接する塩基性表面の可能性がある大きな広がった領域に関係している。
【0067】
この4種の突然変異をコードするPhoS遺伝子を構築しそしてE.coliにおける発現及びpH 4.5におけるカチオン交換精製の効率を試験した。
【0068】
単一K275E突然変異(pDPH192)はE.coliにおける耐容性が悪いことが判明しそして/又は細胞周辺抽出液のpHを4.5に調整したときに沈殿された。さらに、細胞から回収したタンパク質のカチオン交換溶出効率はPhoS polyDタンパク質よりも増加することはなかった。
【0069】
K107/109/110Eのトリプル突然変異ではカチオン交換の溶出後にタンパク質を検出しなかった。これはE.coliにおける発現のレベルが(誘導時間経過に示されるように)極めて低かったためばかりでなく細胞周辺抽出液をpH 4.5 に調整したときの沈殿による可能性もある。しかし、この突然変異はPhoS「ラグビーボール」のpolyDテイルの逆の端にある唯一の突然変異であるので、続いてさらに分析するために単一突然変異に分離した。
【0070】
K262/265/266Eにおけるトリプル突然変異(pDPH193)はカチオン交換から33 mM NaClにおいて溶出した。しかしカチオン交換後のPhoS発現/回収のレベルはwt PhoSに比較して減少したので、このトリプル突然変異をやはりさらに分析するために3種の単一突然変異及びダブル突然変異のすべての組み合わせに分離した。
【0071】
K309/313/318E におけるトリプル突然変異(pDPH194)はPhoS polyDに比較してカチオン交換溶出効率を改善し(それぞれ73 mM対83 mM NaCl)そして正常な発現レベルであったが、73 mMにおける溶出はK318D単一変異+polyD(pDPH188)のそれに比較して十分な改善ではなかったので、このトリプル突然変異についてはこれ以上の検討を行わなかった。
【0072】
該組合せが追加の又は相乗効果を生じるか否かを検討するために、K275E及びK318Dの単一突然変異の両者をトリプル変異K262/265/266Eと組み合わせた(それぞれpDPH195及び pDPH194)。しかし、両者ともにK262/265/266Eにおいて既に認められた低いタンパク質発現/回収の欠点があった。
【0073】
K107/109/110Eトリプル突然変異を3種の単一変異に分離することにより、K109E及びK110E単一変異はより多くのタンパク質を生産することができたので、K107E変異がトリプル変異の有害な効果の主な原因であることが示された。K109E (pDPH199)及びK110E(pDPH200)はともにカチオン溶出効率の改善(52〜55 mM NaCl)を示し、そして70 mM NaClにおいてK107E(pDPH198)よりも良かった。そこで、K109E及びK110Eを組み合わせて(pDPH201)相加/相乗効果を試験した。しかし、このダブル突然変異K109/110EはpH 4.5に調整した後発現又は回収ができなかった。これによりK107/109/110が存在するPhoSの領域には3者が寄与する構造上の又は溶媒和の重要な影響があることが示された。タンパク質回収から判断してK110EはK109 Eよりも耐容性が良いように見えたので、K110Eをさらに組合せ研究を行うために選択した。
【0074】
3種の単一突然変異K262, 265, 266Eは全てPhoS polyDのみよりもカチオン交換溶出効率の改善を示した:それぞれ68 mM, 60 mM 及び58 mMそして全て良好なタンパク質発現レベルであった。これらをダブル突然変異に組合わせることにより、K265E(pDPH205)又はK266E (pDPH206)のいずれと組合わせた場合もタンパク質の発現/回収のレベルの減少を認めたので、良好なタンパク質発現レベル(構造/溶解性)を維持するのに重要なのは残基K262であることが示された。さらにこれ等のダブル変異のいずれも単一変異K265E又はK266E以上のカチオン交換溶出効率を改善しなかった。しかしダブル突然変異K265/266E(pDPH207)は良好なタンパク質発現/回収レベルを示し、このタンパク質が38 mM NaClで溶出したので(単一変異のみの60 mM及び58 mMに比較して)、カチオン交換溶出効率への追加効果を示した。
【0075】
さらにK265/266EにK110E及びK318Dを加えて組合わせ突然変異が作られた。K110/265/266E突然変異(pDPH209)はカチオン交換溶出効率の改善が認められ、29 mM NaClにおいて溶出しそして良好なタンパク質発現レベルであった。K265/266E+K318D突然変異(pDPH211)はカチオン交換溶出効率及びタンパク質発現ともにK265/266E変異のみよりも良くなかった。最後にpolyDテイルに加えて4個の突然変異を含む「最大」変異構築:K110/265/266E+K318D(pDPH210)を試験した。しかしこれはタンパク質回収の減少を生じそしてK265/266E又はK110/265/266Eのいずれよりもカチオン交換効率を改善しなかった。
【0076】
このようにして3種のPhoS構築がさらに分析するために選択され、改善されたカチオン交換溶出効率を持つ最小PhoS突然変異であることを示した:PhoS polyD, PhoS K265/266E polyD及びPhoS K110/265/266E polyD。野生型PhoSよりも低い塩濃度で3種の変異体が溶出することによりカチオン交換カラムからのグラジエント又は段階的溶出によりFab'から変異PhoSを分離できるはずである。そのほかに、変異体のpIによってはFab'の結合に影響しない緩衝液のpHに上昇することによりそれらがカチオン交換カラムに結合することを防止することも可能であろう。その他の方法としてはアニオン交換段階により変異PhoSを除去することもできる。変異PhoSタンパク質のpIは後に決定される。
【0077】
実施例5
タンパク質の純度及びpIに及ぼすPhoS突然変異誘起の影響
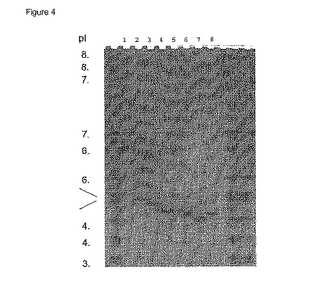
>95%純度の変異PhoSタンパク質をグラジエント溶出カチオン交換により得た。このタンパク質の純度及びそのC-末端テイルをSDS-PAGE及び質量分析により評価した。MSにより測定した分子量は表2に示す。全て予測分子量と良い一致を示し、いずれもタンパク質は正しく同定されそしてpolyDテイルは完全であることを確認した。図3に示すSDS-PAGEによりこれ等の調製品の純度を確認しそしてpolyDを含むPhoSタンパク質は野生型に比較して若干遅く移動することを示す。IEFゲル(図4)は、wt PhoSは約7.2のpIを有し、一方polyDテイルのみの付加はpIを約5.1に減少することを示している。続くKのE又はDへの表面突然変異の追加によりさらにPhoS K/D polyDの約4.9からPhoS K265/266E polyDの約4.8及びPhoS K110/265/266E+K318D polyDの約4.5への範囲のpIシフトを生じる。
【0078】
実施例6
変異PhoSタンパク質の機能的純度試験:プラスミドから得られたPhoSによるphoS遺伝型の補充
E.coli株ANCC75(Amemura et al., 1982)をphoSの遺伝的背景を提供するために使用した。この細胞はPO4に欠乏しているかのような表現型を永続的に持っているので、phoレギュロンを介するフィードバックのためにPO4が存在しても高レベルのアルカリホスファターゼを誘導する。XPを含む固体培地上での該細胞の殖によりアルカリホスファターゼを高いレベルで持つ又は低いレベルで持つ細胞を区別することができる。該細胞をwt PhoS, PhoS polyD, Phos K265/266E polyD及びPhoS K110/265/266E polyDをコードするプラスミド又は対照プラスミドで形質転換して、変異PhoSタンパク質の全てが、染色体にコードされたPhoSの欠失を補充する能力を有することを確認した。
【0079】
対照として野生型W3110 E.coliを使用して同じ実験を「無リン酸」(PO4が非常に低い)液体培地で繰り返し(図5)、試験した変異PhoSタンパク質は全て低いPO4条件の下に(5時間の点を参照)phoS表現型を100%補充できることそして無リン酸(PO4)条件下にphoS表現型を部分的に補充すること(22,27及び47.5時間の点を参照)が示された。
【0080】
実施例7
変異PhoSタンパク質を発現する安定な組込みE.coli株の作製
3種の変異PhoSタンパク質(polyD, K265/266E polyD及びK110/265/266E polyD)をコードする遺伝子を3’染色体隣接領域とともに染色体置換プラスミドpKO3の中に組込んだ。さらにPhoSのヌルバージョンをシグナルペプチドコード領域に2個のインフレーム終結コドンを持つオリゴヌクレオチドで作製した(それぞれプラスミドpDPH217〜220)。これ等を電気穿孔によりW3110 E.coliに導入し、そして組換え及びプラスミドキュアリングを方法に記述したように試験した。表1に示したスクリーニングオリゴを適当な対立鎖の共通オリゴとともに使用して理論的に組込まれたシングルコロニーの最終的PCRスクリーニングを行い野生型又は変異PhoSが染色体中に組込まれたか否かを試験した。pKO3にコードされたDNAの領域の外側にアニールする2個のオリゴ(Dra I前進及びBstB I逆)を使用し、高信頼度ポリメラーゼ(Precision plus Taq)を使用する染色体DNA(E.coli細胞全体)のPCR、次いでゲル精製PCR産物の直接配列分析を正しい改変が染色体に組込まれたことを確認するために使用した。この結果次の4種のE.coli株が作製された:DPH1はPhoS polyDをコードし、DPH2はPhoS K265/266E polyDをコードし、DPH3はPhoS K110/265/266E polyDをコードし、DPH4はヌルPhoSをコードする。
【0081】
実施例8
DPH3株のFab'の発現及び精製の試験
株DPH3を目的のFab'を発現するプラスミドで形質転換した。標準的な醗酵を実施しそして増殖又は誘導相の間に明らかな欠陥又は問題を認めなかった。醗酵中に採取した検体をTris/EDTA抽出した後通常の方法でELISAにより分析した。図6のデータは、増殖は正常でありそしてペリプラズムにおけるFab'の蓄積は回収時に約380 mg/Lで十分にFab'の正常範囲内であることを示している。遠心分離の後細胞ペレットは硬くそして遠心後容易に再懸濁することができることがわかり、通常これは良好な細胞の表れである。
【0082】
回収培養の50 mlから得たペレットを終夜30℃でTris/EDTA中に抽出し、次いで前記のようにカチオン交換精製を行った。株DPH3の変異PhoS(pI 4.5)がカチオン交換カラムに結合せずそしてFab'フラグメントが結合するようにpHを4.5から5.0に上昇させた。伝導度は3.0 mS/cmであった。検体を5 ml SPセファロースカラムに適用し、原液、通過液及び溶出液検体をクマシー染色SDS-PAGEにより分析した。必要があれば、クマシー染色ゲルで可視化できるように10 kDaカットオフスピンカラムを使用して検体を濃縮した。
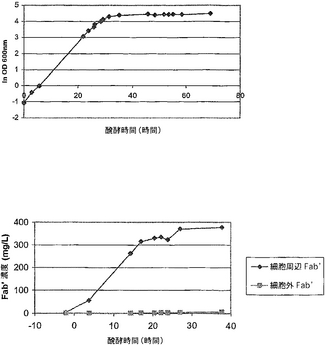
【0083】
図7のSDS-PAGEゲルは、このpH及び伝導度の条件下ではDPH3の変異PhoSはSPセファロースカラムに結合しないが、W3110のwt PhoSは結合することを示している。このことは、DPH3の場合には変異PhoSとFab'は異なるフラクションに移行する:それぞれ通過液及び200 mM NaCl溶出液、他方W3110 の場合には両タンパク質はともに200 mM NaCl溶出液中に移行することを意味する。
【0084】
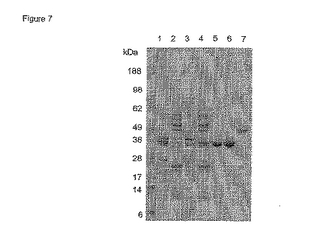
少しでも残留したPhoSはアニオン交換によっても除去されることを確認するために、DPH3及びW3110実験の両者からの通過液及び200 mM NaCl溶出液を濃縮し、脱塩しそして緩衝液を20 mM Tris.Cl pH 8.0に交換しそしてアニオン交換を行った。図8に示すクマシー染色SDS-PAGEゲルは、DPH3については PhoSはアニオン交換カラムに結合するのでFab'から分離されることを示している。しかし、W3110のwt PhoSはこのカラムに結合せずに通過しそしてFab'溶液に混入する。
【0085】
実施例9
株DPH1, DPH2及びDPH3において発現したFab'フラグメントの精製試験
典型的にpH 6におけるイオン交換で精製される実施例8に使用したFab'よりも高いpIを持つFab'を発現するプラスミドで株DPH1, DPH2及びDPH3を形質転換した。標準的醗酵を行いそしてカチオン交換クロマトグラフィーによりFabからPhoS変異体を分離する試験をpH 5, 5.5及び6において前記と同様に行った。
【0086】
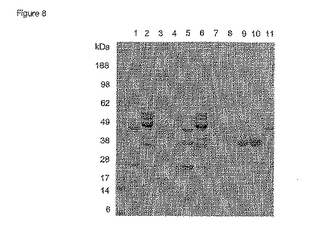
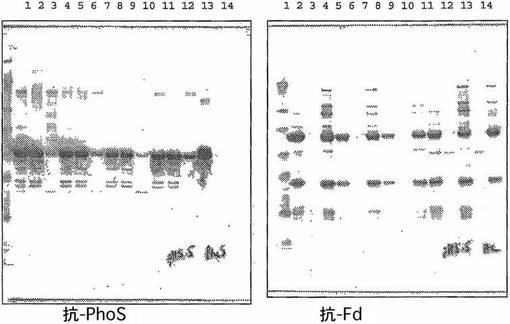
pH 5.0
pH 5.0 において行った精製の結果を図9及び10に示す。pH 5においてPhoS polyD(DPH1)はクマシー染色により判断して通過液には現れず(図9、レーン2)、実際Fab'-Bとともにカラムから溶出した(図9、レーン3)。抗-PhoSポリクロナール及び抗-Fdモノクロナールを使用するより感度の高いイムノブロット分析により、Fab'-Bは大量のPhoS polyDとともにカラムに強く結合していることが明らかに示される(図10、レーン3)。しかし通過液フラクション(レーン2)にもPhoS polyDが少しあった。これはPhoS polyDとクロマトグラフィーマトリックスの会合がpH 5.0では弱いことを示すのであろう。
【0087】
これに対して、PhoS K265/266E polyDをコードするDPH2は通過液中に明瞭なクマシー染色PhoSバンドを示した(図9、レーン5)。抗-PhoSイムノブロットは溶出フラクション中に極微量のPhoSしか示さなかった(図10、レーン6)。したがってPhoS K265/266E polyDは実際上pH 5.0 においてSPセファロースには結合しない。
【0088】
最大突然変異PhoS K110/265/266E polyDをコードする株DPH3では、PhoSはやはり通過液中に現れる(図9、レーン8)が、イムノブロットにより溶出フラクション中に残留するPhoSの量(図10、レーン9)はDPH2の場合(図10、レーン6)よりもさらに減少することがを示される。これは実施例8において既に観察したDPH3のクロマトグラフィーにおける性質はDPH2よりも低いpH(pH 約4.7〜5.0)でよく作動するということを支持している。
【0089】
pH 5.0 で負荷された場合に、親のW3110株によりコードされている野生型PhoSの結合性は図9のレーン12に示されている。野生型PhoSの大部分は溶出フラクションにあるが、極微量を通過液中に検出することができる(図10、レーン11及び12)ことを抗-PhoSイムノブロットは示している。(この実験においてイムノブロットにより溶出フラクション中に検出された極微量のPhoSは、高速分析方法に使用されたカラム洗浄の量が少なかったことを反映している可能性があるが、通過液中の微量の野生型PhoSは、カラムへの弱い結合又はカラム/緩衝液フロントアーチファクトを示している可能性もある)。
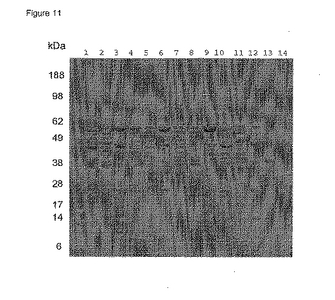
【0090】
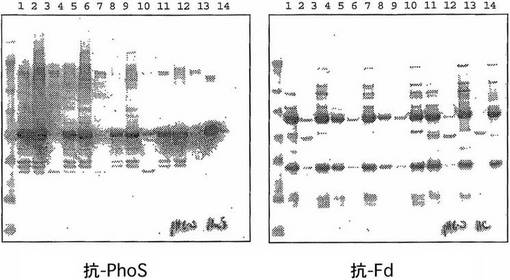
pH 5.5
pH 5.5におけるPhoS精製について3株を分析したところ、DPH1の行動のみがpH 5.0 に比較して大きく変化した。pH 5.5においてDPH1によりコードされたPhoS polyDは今度はクマシー染色ゲルにより通過液中に認められ(図11、レーン2)、それは抗-PhoSブロット(図12、レーン2+3)により確認された。DPH2及びDPH3によりコードされたPhoS変異体はpH 5.5 において通過液中に存在した。W3110によってコードされた野生型PhoSには不明確な行動が認められている。クマシー染色ゲルはpH 5.5において大量のPhoSが通過液中にあることを示している(図11、レーン11)が、他方で抗-PhoSブロットはかなりの量(約40%)がまだ溶出フラクションに残っていることを示している。したがって、このpHにおいて、PhoSは機能的pIの近くにあるに違いなくそして弱く結合している。
【0091】
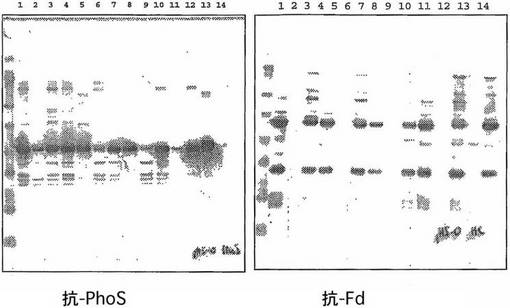
pH 6.0
3種の変異株全て及び野生型W3110はクマシー染色ゲルにより示されるようにpH 6.0 (図13、レーン2,5,8及び11)においてSPセファロースに結合しないPhoSを生産する。抗-PhoSポリクロナールを使用する分析により、これ等は全て溶出フラクションに同じレベルの微量の検出しうるPhoSがあること(図14、レーン3,6,9,12)が示された。したがって、カチオン交換精製がこのpHで行われるならば、野生型W3110よりも株DPH 1, 2又は3を使用する利点はない。しかし、溶出フラクション中の微量のPhoSにおけるpolyDテイルの存在は、カラムを通過様式で稼動する場合にはC-末端タグは親和性タグとして有効に作動するので、2番目のアニオン交換カラム上でのPhoSとFab'のさらなる分離を非常に容易にすることができる。
【0092】
表3に集めた結果は、50 ml醗酵検体からの生成を試験すると、3種のPhoS突然変異:PhoS polyD, PhoS K265/266E polyD及びPhoS K110/265/266E polyDは全て 系統的に改良された精製プロフィールを持つことを示す。これ等の実験において、SPセファロースカラムに酸性にして適用した時の通過液中にPhoSを認めることができる低いpHによりこれ等の改良は示された。したがって多数の突然変異を持つタンパク質はますます厳密なpHの結合条件においてFab'から分離することができる。この型のPhoSをpH 4.5においてSPセファロースカラムから溶出すためには低いNaCl濃度(W3110, DPH1, 2及び3に対してそれぞれ約103 mM, 83 mM, 38 mM及び29 mM NaCl(表3))が必要であった以前の情報をこれは支持している。
【表4】
【図面の簡単な説明】
【0093】
【図1】PhoS遺伝子のPCRクローニングの図解
【図2】アニオン交換カラム, pH 8.0に結合したPhoSのクマシー染色4〜12% SDS-PAGE。レーン1〜4はwt PhoS, レーン5〜8はPhoS polyD及びレーン9〜12はPhoS K/D polyD。 レーン1,5及び9は原液、レーン2,6及び10は通過液、及びレーン3,4,7,8,11及び12は1 M NaCl溶出段階のそれぞれ初期及び後期フラクション。
【図3】SPセファロースpH 4.5上NaClグラジエントで精製されたPhoSタンパク質のクマシー染色4〜12% SDS-PAGE。レーン1 wt PhoS, レーン2 PhoS polyD, レーン3 PhoS K/D polyD, レーン4 PhoS K265/266E polyD, レーン5 PhoS K110E K318D polyD, レーン6 PhoS K110/265/266E poly D, レーン7 PhoS K110/265/266E K318D polyD, レーン8 PhoS K265/266E K318D polyD.
【図4】SPセファロースpH 4.5上NaClグラジエントで精製されたPhoSタンパク質のクマシー染色pH 3〜10 IEFゲル。レーン1 wt PhoS, レーン2 PhoS polyD, レーン3 PhoS K/D polyD, レーン4 PhoS K265/266E polyD, レーン5 PhoS K110E K318D polyD, レーン6 PhoS K110/265/266E poly D, レーン7 PhoS K110/265/266E K318D polyD, レーン8 PhoS K265/266E K318D polyD.
【図5】phoS及びwt E.coli株におけるアルカリホスファターゼ活性のphoS及びPO4欠乏誘導のwt及び突然変異PhoSタンパク質をコードするプラスミドによる機能的補充。
【図6】DPH3 E.coliの醗酵によるDPH3の増殖及びFab'の生産。
【図7】SPセファロース上pH 5.0 及び3.0 mS/cmにおけるW3110及びDPH3のFab'の精製におけるカチオン交換カラムフラクションのクマシー染色4〜12% SDS-PAGE。レーン1 DPH3通過液、レーン2 DPH3 200 mM NaCl 溶出液、レーン3 W3110通過液、レーン4 W3110 200 mM NaCl 溶出液、レーン5 wt PhoS, レーン6 PhoS polyD, レーン7 Fab'。
【図8】カチオン交換カラムで分離したDPH3又はW3110で生産されたFab'のpH 8.0におけるPoros HQのアニオン交換カラムのフラクション。クマシー染色4〜12% SDS-PAGE。 DPH3カチオン交換の溶出物をアニオン交換カラムに適用して得られた通過液(レーン1)又は溶出液(レーン2)。DPH3カチオン交換の通過液をアニオン交換カラムに適用して得られた通過液(レーン3)又は溶出液(レーン4)。W3110カチオン交換の溶出液をアニオン交換カラムに適用して得られた通過液(レーン5)又は溶出液(レーン6)。W3110カチオン交換の通過液をアニオン交換カラムに適用して得られた通過液(レーン7)又は溶出液(レーン8)。レーン9 wt PhoS,レーン10 PhoS polyD及びレーン11 Fab'。
【図9】Fab'BのpH 5.0におけるカチオン交換精製における株DPH1, DPH2及びDPH3の変異PhoSの行動を比較したクマシー染色4〜12% SDS-PAGEゲル。レーン1 DPH1 原液、レーン2 DPH1 FT, レーン3 DPH1溶出液、レーン4 DPH2原液、レーン5 DPH2 FT、レーン6 DPH2溶出液、レーン7 DPH3原液、レーン8 DPH3 FT、レーン9 DPH3溶出液、レーン10 W3110 原液、レーン11 W3110 FT、レーン12 W3110 溶出液、レーン13 PhoS polyD、レーン14 Fab'。
【図10】Fab'BのpH 5.0におけるカチオン交換精製における株DPH1, DPH2及びDPH3の変異PhoSの行動を比較した4〜12% SDS-PAGEの抗-PhoS及び抗-Fdイムノブロット。
【図11】Fab'BのpH 5.5におけるカチオン交換精製における株DPH1, DPH2及びDPH3の変異PhoSの行動を比較した4〜12% SDS-PAGEのクマシー染色ゲル。
【図12】Fab'BのpH 5.5におけるカチオン交換精製における株DPH1, DPH2及びDPH3の変異PhoSの行動を比較した4〜12% SDS-PAGEの抗-PhoS及び抗-Fdイムノブロット。
【図13】Fab'BのpH 6.0におけるカチオン交換精製における株DPH1, DPH2及びDPH3の変異PhoSの行動を比較した4〜12% SDS-PAGEのクマシー染色ゲル。
【図14】Fab'BのpH 6.0におけるカチオン交換精製における株DPH1, DPH2及びDPH3の変異PhoSの行動を比較した4〜12% SDS-PAGEの抗-PhoS及び抗-Fdイムノブロット。
【配列表】
【特許請求の範囲】
【請求項1】
野生型において該組換え抗体と共精製する1又はそれ以上のE.coliタンパク質の少なくとも一つの物理的性質を変えるためにE.coli宿主細胞が遺伝的に修飾されていることが特徴である組換え抗体を発現するE.coli宿主細胞。
【請求項2】
改変されるE.coliタンパク質の物理的性質が等電点、疎水性又は大きさである請求項1に記載の宿主細胞。
【請求項3】
改変されるE.coliタンパク質の物理的性質が等電点である請求項2に記載の宿主細胞。
【請求項4】
改変される宿主タンパク質がリン酸結合タンパク質(PhoS/PstS)、ジペプチド結合タンパク質(DppA)、マルトース結合タンパク質(MBP)又はチオレドキシン1である請求項1から3に記載の宿主細胞。
【請求項5】
改変される宿主タンパク質がリン酸結合タンパク質(PhoS/PstS)である請求項1から3に記載の宿主細胞。
【請求項6】
宿主タンパク質の等電点がC-末端へのポリ-アスパラギン酸タグの付加により改変されている請求項4に記載の宿主細胞。
【請求項7】
リン酸結合タンパク質(PhoS/PstS)の等電点が残基110,265,266又は318のリシンを一つ又はそれ以上グルタミン酸又はアスパラギン酸で置換することにより減少している請求項5に記載の宿主細胞。
【請求項8】
リン酸結合タンパク質(PhoS/PstS)の等電点がC-末端へポリ-アスパラギン酸タグの付加によりさらに減少している請求項7に記載の宿主細胞。
【請求項9】
リン酸結合タンパク質(PhoS/PstS)の等電点が残基265及び266のリシンをグルタミン酸で置換すること及びC-末端へのポリ-アスパラギン酸タグの付加により減少している請求項5に記載の宿主細胞。
【請求項10】
リン酸結合タンパク質(PhoS/PstS)の等電点が残基110,265及び266のリシンをグルタミン酸で置換及びC-末端へのポリ-アスパラギン酸タグの付加により減少している請求項5に記載の宿主細胞。
【請求項11】
組換え抗体がFab又はFab'フラグメントである請求項1から10に記載の宿主細胞。
【請求項12】
請求項1から11に記載の宿主細胞を醗酵することからなる組換え抗体を製造する方法。
【請求項1】
野生型において該組換え抗体と共精製する1又はそれ以上のE.coliタンパク質の少なくとも一つの物理的性質を変えるためにE.coli宿主細胞が遺伝的に修飾されていることが特徴である組換え抗体を発現するE.coli宿主細胞。
【請求項2】
改変されるE.coliタンパク質の物理的性質が等電点、疎水性又は大きさである請求項1に記載の宿主細胞。
【請求項3】
改変されるE.coliタンパク質の物理的性質が等電点である請求項2に記載の宿主細胞。
【請求項4】
改変される宿主タンパク質がリン酸結合タンパク質(PhoS/PstS)、ジペプチド結合タンパク質(DppA)、マルトース結合タンパク質(MBP)又はチオレドキシン1である請求項1から3に記載の宿主細胞。
【請求項5】
改変される宿主タンパク質がリン酸結合タンパク質(PhoS/PstS)である請求項1から3に記載の宿主細胞。
【請求項6】
宿主タンパク質の等電点がC-末端へのポリ-アスパラギン酸タグの付加により改変されている請求項4に記載の宿主細胞。
【請求項7】
リン酸結合タンパク質(PhoS/PstS)の等電点が残基110,265,266又は318のリシンを一つ又はそれ以上グルタミン酸又はアスパラギン酸で置換することにより減少している請求項5に記載の宿主細胞。
【請求項8】
リン酸結合タンパク質(PhoS/PstS)の等電点がC-末端へポリ-アスパラギン酸タグの付加によりさらに減少している請求項7に記載の宿主細胞。
【請求項9】
リン酸結合タンパク質(PhoS/PstS)の等電点が残基265及び266のリシンをグルタミン酸で置換すること及びC-末端へのポリ-アスパラギン酸タグの付加により減少している請求項5に記載の宿主細胞。
【請求項10】
リン酸結合タンパク質(PhoS/PstS)の等電点が残基110,265及び266のリシンをグルタミン酸で置換及びC-末端へのポリ-アスパラギン酸タグの付加により減少している請求項5に記載の宿主細胞。
【請求項11】
組換え抗体がFab又はFab'フラグメントである請求項1から10に記載の宿主細胞。
【請求項12】
請求項1から11に記載の宿主細胞を醗酵することからなる組換え抗体を製造する方法。
【図1】

【図2】
【図3】

【図4】
【図5】
【図6】
【図7】
【図8】
【図9】

【図11】
【図13】
【図10】
【図12】
【図14】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図11】
【図13】
【図10】
【図12】
【図14】
【公表番号】特表2006−502727(P2006−502727A)
【公表日】平成18年1月26日(2006.1.26)
【国際特許分類】
【出願番号】特願2004−544473(P2004−544473)
【出願日】平成15年10月15日(2003.10.15)
【国際出願番号】PCT/GB2003/004474
【国際公開番号】WO2004/035792
【国際公開日】平成16年4月29日(2004.4.29)
【出願人】(501460693)セルテック アール アンド ディ リミテッド (29)
【Fターム(参考)】
【公表日】平成18年1月26日(2006.1.26)
【国際特許分類】
【出願日】平成15年10月15日(2003.10.15)
【国際出願番号】PCT/GB2003/004474
【国際公開番号】WO2004/035792
【国際公開日】平成16年4月29日(2004.4.29)
【出願人】(501460693)セルテック アール アンド ディ リミテッド (29)
【Fターム(参考)】
[ Back to top ]