
側鎖置換脂環式テトラカルボン酸二無水物及びその製造法
【課題】沸点の低い有機溶媒類に対しても溶解性に優れ、また液晶配向膜の高チルト角発現が可能なポリイミドを与え得るそのモノマーとしての側鎖置換脂環式テトラカルボン酸二無水物及びその製造法の提供。
【解決手段】ビシクロ[3.3.0]オクタン−2,4,6,8−テトラカルボン酸−2:4,6:8−二無水物のビシクロオクタン環に側鎖置換基を導入した下記式[1]で表される化合物及びその製造方法。

(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表す。)
【解決手段】ビシクロ[3.3.0]オクタン−2,4,6,8−テトラカルボン酸−2:4,6:8−二無水物のビシクロオクタン環に側鎖置換基を導入した下記式[1]で表される化合物及びその製造方法。
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表す。)
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、側鎖置換脂環式テトラカルボン酸二無水物及びその製造法に関し、さらに詳述すると、例えば、電子材料用として好適なポリイミドの原料モノマーである側鎖置換脂環式テトラカルボン酸二無水物に関する。
【背景技術】
【0002】
一般に、ポリイミド樹脂はその特長である高い機械的強度、耐熱性、絶縁性、耐溶剤性のために、液晶表示素子や半導体における保護材料、絶縁材料、カラーフィルターなどの電子材料として広く用いられている。また、最近では光導波路用材料等の光通信用材料としての用途も期待されている。
【0003】
近年、この分野の発展は目覚ましく、それに対応して、用いられる材料に対しても益々高度な特性が要求される様になっている。即ち、単に耐熱性、耐溶剤性に優れるだけでなく、用途に応じた性能を多数合わせ有することが期待されている。
【0004】
しかし、特に、全芳香族ポリイミド樹脂においては、濃い琥珀色を呈し着色するため、高い透明性を要求される光学材料用途においては問題が生じてくる。また、全芳香族ポリイミドは有機溶剤に不溶であるため、実際にはその前駆体であるポリアミック酸を熱による脱水閉環によって得る必要がある。
【0005】
透明性を実現する一つの方法として、脂環式テトラカルボン酸二無水物と芳香族ジアミンとの重縮合反応によりポリアミック酸を得て、該当前駆体をイミド化してポリイミドを製造すれば、比較的着色が少なく、高透明性のポリイミドが得られることは知られている(特許文献1、2参照)。
【0006】
近年、下記式[11]
【0007】
【化1】
【0008】
で表されるビシクロ[3.3.0]オクタン−2,4,6,8−テトラカルボン酸−2:4,6:8−二無水物(以下、BODAと略称する。)を使用するポリイミドが、基板への印刷性、密着性に優れ、かつラビング時に基板からの剥離がなく、またラビングによる配向膜への傷がつきにくく、液晶セル駆動時に優れた電圧保持特性が得られる液晶配向処理剤及びそれを用いた液晶配向膜として知られている。(特許文献3参照)。
しかしながら、BODAを酸二無水物として用いたポリイミドは、ジアミンの種類によっては有機溶剤への溶解性が低いという問題点を抱えており、その使用場面に制限があった。
また、ポリアミック酸やポリイミドを溶解させる有機溶媒としては、従来N−メチル−2−ピロリドン(NMP)やγ―ブチロラクトン等のアミド系やラクトン系有機溶媒が多用されている。しかし、これらの溶媒は高沸点のため、液晶配向剤を基板に塗布、焼成して液晶配向膜を作成する際、溶媒を完全に除去するためには高温焼成が必要であった。近年ではプロセス効率の観点から、低温での焼成が望まれて来ており、沸点の低い有機溶媒類に対しても可溶であるポリイミドが望まれていた。
また、液晶配向膜に求められる特性の一つとして、基板面に対する液晶分子の配向傾斜角を任意の値に保つ、いわゆる液晶のプレチルト角制御があり、近年の液晶ディスプレイの大型化、高精細化に伴い、液晶のプレチルト角制御は液晶表示素子において重要な課題となっている。液晶のプレチルト角は、液晶配向膜を構成しているポリイミドの構造を種々選択することで変更、制御出来ることが知られている。
【0009】
ポリイミドの構造によってプレチルト角を制御する技術の中でも、側鎖を有するジアミンをポリイミド原料の一部として用いる方法は、このジアミンの使用割合に応じてプレチルト角が制御できるので、目的のプレチルト角にせしめることが比較的容易であり、プレチルト角を大きくする手段として有用である。液晶のプレチルト角を大きくするジアミンの側鎖構造としては、長鎖のアルキル基又はフルオロアルキル基(例えば特許文献4参照)、環状基又は環状基とアルキル基の組み合わせ(例えば特許文献5参照)、ステロイド骨格(例えば特許文献6参照)などが知られている。
しかし、側鎖を有するジアミンは一般的に溶解性が低く、使用できる割合に限界があった。また、側鎖ジアミンを多く用いすぎると、得られるポリアミック酸やポリイミドを用いた液晶配向剤を基板に均一に塗布することが困難となる問題もあった。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】特開昭60−188427号公報
【特許文献2】特開昭58−208322号公報
【特許文献3】特開平11−249148号公報
【特許文献4】特開平2−282726号公報
【特許文献5】特開平3−179323号公報
【特許文献6】特開平4−281427号公報
【発明の開示】
【発明が解決しようとする課題】
【0011】
本発明は、このような事情に鑑みてなされたものであり、沸点の低い有機溶媒類に対しても溶解性に優れ、また液晶配向膜の高チルト角発現が可能なポリイミドを与えることが期待されるモノマーである、側鎖置換脂環式テトラカルボン酸二無水物及びその製造方法を提供することを目的とする。
【課題を解決するための手段】
【0012】
本発明者らは、上記目的を達成するために鋭意検討を重ねた結果、BODAのビシクロオクタン環に側鎖置換基を導入した原料酸二無水物モノマーの製造方法を確立し、本発明を完成した。得られた酸二無水物モノマーは新規化合物であり、各種有機溶媒に対する溶解性に優れ、また液晶配向膜の高チルト角発現が可能なポリイミドへの誘導が期待される。
【0013】
すなわち、本発明は、
1.下記式[1]で表される化合物。
【0014】
【化2】
【0015】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表す。)
2.前記R1が、メチル基、n−オクチル基及びn−テトラデシル基である1記載の化合物。
3.下記式[2]で表される化合物。
【0016】
【化3】
【0017】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表す。)
4.前記R1が、メチル基、n−オクチル基及びn−テトラデシル基である3記載の化合物。
5.下記式[3]で表される化合物。
【0018】
【化4】
【0019】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表す。)
6.前記R1が、メチル基、n−オクチル基及びn−テトラデシル基である5記載の化合物。
7.下記式[4]で表される化合物。
【0020】
【化5】
【0021】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表し、R2は、炭素数1〜10のアルキル基を表す。)
8.前記R1が、メチル基、n−オクチル基及びn−テトラデシル基である7記載の化合物
9.下記式[5]で表される化合物。
【0022】
【化6】
【0023】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表し、R2は、炭素数1〜10のアルキル基を表す。)
10.前記R1が、メチル基、n−オクチル基及びn−テトラデシル基である9記載の化合物。
11.下記式[6]で表される化合物。
【0024】
【化7】
【0025】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表し、R2は、炭素数1〜10のアルキル基を表す。)
12.前記R1が、メチル基、n−オクチル基及びn−テトラデシル基である11記載の化合物。
13.下記式[9]で表される化合物を塩基の存在下、下記式[10]で表される置換アルキルハライドと反応して、下記式[4]で表される化合物を得、更にこれを加水分解して下記式[3]で表される化合物を得、更に、これを還元して下記式[2]で表される化合物を得た後、脱水剤により下記式[1]で表される化合物を得ることを特徴とする製造方法。
【0026】
【化8】
【0027】
(式中、R2は、炭素数1〜10のアルキル基を表す。)
【0028】
【化9】
【0029】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表し、Xはハロゲン原子を表す。)
【0030】
【化10】
【0031】
(式中、R1及びR2は、前記と同じ意味を表す。)
【0032】
【化11】
【0033】
(式中、R1は、前記と同じ意味を表す。)
【0034】
【化12】
【0035】
(式中、R1は、前記と同じ意味を表す。)
【0036】
【化13】
【0037】
(式中、R1は、前記と同じ意味を表す。)
14.下記式[4]で表される化合物を加水分解して下記式[6]で表される化合物を得ることを特徴とする製造法を提供する。
【0038】
【化14】
【0039】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表し、R2は、炭素数1〜10のアルキル基を表す。)
【0040】
【化15】
【0041】
(式中、R1及びR2は、前記と同じ意味を表す。)
【発明の効果】
【0042】
本発明によれば、各種有機溶媒に対する溶解性に優れ、また液晶配向膜の高チルト角発現が可能なポリアミック酸及びポリイミドへの誘導が期待される側鎖置換テトラカルボン酸二無水物及びその製造法を提供することができる。
【0043】
本発明の側鎖置換BODA酸二無水物を一原料として得られるポリイミドは、例えば、液晶表示素子や半導体における保護材料、絶縁材料などの電子材料、さらに光導波路等の光通信用材料への好適な適用が期待される。
【図面の簡単な説明】
【0044】
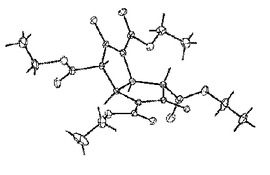
【図1】THOEの単結晶X線構造
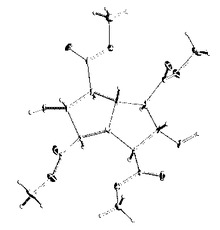
【図2】TMHAの単結晶X線構造
【発明を実施するための最良の形態】
【0045】
以下、本発明についてさらに詳しく説明する。
【0046】
なお、以下において、nはノルマルを、iはイソを、sはセカンダリーを、tはターシャリーを、cはシクロをそれぞれ表す。
【0047】
上記各式において、炭素数1〜10のアルキル基としては、直鎖、分岐、環状のいずれでもよく、その具体例としては、メチル、エチル、n−プロピル、i−プロピル、c−プロピル、n−ブチル、i−ブチル、s−ブチル、t−ブチル、c−ブチル、n−ペンチル、1−メチル−n−ブチル、2−メチル−n−ブチル、3−メチル−n−ブチル、1,1−ジメチル−n−プロピル、c−ペンチル、2−メチル−c−ブチル、n−ヘキシル、1−メチル−n−ペンチル、2−メチル−n−ペンチル、1,1−ジメチル−n−ブチル、1−エチル−n−ブチル、1,1,2−トリメチル−n−プロピル、c−ヘキシル、1−メチル−c−ペンチル、1−エチル−c−ブチル、1,2−ジメチル−c−ブチル、n−ヘプチル、n−オクチル、n−ノニル、n−デシル基等が挙げられる。
【0048】
これらの中でも、得られるポリイミドの有機溶媒に対する溶解性を高めることを考慮すると、R1としては、炭素数1〜20のアルキル基が好ましく、炭素数5〜20のアルキル基がより好ましく、炭素数8〜18のアルキル基がより一層好ましい。
【0049】
上記式[1]で表される2−アルキル−7−アルコキシビシクロ[3.3.01,5]オクタン−2,4,6,8−テトラカルボン酸−2:4,6:8−二無水物(以下、AODAと略記する)の製造法は、下記の一連の反応スキームで表される。
【0050】
【化16】
【0051】
(式中、R1およびR2は、上記と同じ意味を表す。)
すなわち、第1工程は、テトラアルキル3,7−ジヒドロキシビシクロ[3.3.01,5]オクタン−2,4,6,8−テトラカルボキシレート(TAHA)を塩基の存在下、置換アルキルハライドと反応させて、テトラアルキル−4−アルキル−7−アルコキシビシクロ[3.3.01,5]オクテ−2−エン−2,4,6,8−テトラカルボキシレート(TAAE)を得る。
第2工程は、TAAEを加水分解して4−アルキル−7−アルコキシビシクロ[3.3.01,5]オクテ−2−エン−2,4,6,8−テトラカルボン酸(AECA)を得る。
第3工程は、AECAを還元して4−アルキル−7−アルコキシビシクロ[3.3.01,5]オクタン−2,4,6,8−テトラカルボン酸(AOCA)を得る。
第4工程は、AOCAを脱水剤により2−アルキル−7−アルコキシビシクロ[3.3.01,5]オクタン−2,4,6,8−テトラカルボン酸−2:4,6:8−二無水物(AODA)を得る製造法である。
【0052】
ここで、原料のTAHAは、公知の次の反応スキームで表される製造法で合成される。(Organic Syntheses, 64, 27−38 (1986);Tetrahedron Letters, 49 (2008) 3056−3059)即ち、R2がメチルの場合は、ジメチル−1,3−アセトンジカルボキシレートとグリオギザールを水酸化ナトリウム存在下で反応させて、テトラメチル−3,7−ジソジウムオキシビシクロ[3.3.01,5]オクタ−2,6-ジエン−2,4,6,8−テトラカルボキシレート(TMSO)を得た後、塩酸で酸性にすることによりテトラメチル−3,7−ジヒドロキシビシクロ[3.3.01,5]オクタ−2,6-ジエン−2,4,6,8−テトラカルボキシレート(TMHO)が得られる。続いてこれを還元することによりテトラメチル−3,7−ジヒドロキシビシクロ[3.3.01,5]オクタタン−2,4,6,8−テトラカルボキシレート(TMHA)が合成できる。
【0053】
【化17】
【0054】
また、R2がエチルの場合は、次式で表される製造法で合成される。
【0055】
【化18】
【0056】
即ち、ジエチル−1,3−アセトンジカルボキシレートとグリオギザールから公知の製造法でテトラエチル−3,7−ジヒドロキシビシクロ[3.3.01,5]オクタ−2,6-ジエン−2,4,6,8−テトラカルボキシレート(THOE)が合成される。(特開昭56−127328)続いてこれを還元することによりテトラエチル3,7−ジヒドロキシビシクロ[3.3.01,5]オクタン−2,4,6,8−テトラカルボキシレート(TEHO)が合成できる。
<第1工程>
こうして得られてTAHAを原料として、塩基の存在下、置換アルキルハライドと反応させて、TAAEを得る。
置換アルキルハライドの置換アルキル基(R1)としては、炭素数1〜20のアルキル基及び炭素数2〜20のシアノアルキル基を表し、ハロゲン原子(X)としては、弗素原子、塩素原子、臭素原子及び沃素原子を表す。
具体的には、沃化メタン、沃化エタン、沃化プロパン、沃化ブタン、沃化ペンタン、沃化ヘキサン、沃化ヘプタン、沃化オクタン、沃化ノナン、沃化デカン、臭化ブタン、臭化ペンタン、臭化ヘキサン、臭化ヘプタン、臭化オクタン、臭化ノナン、臭化デカン、臭化ウンデカン、臭化ドデカン、臭化トリデカン、臭化テトラデカン、臭化ペンタデカン、臭化ヘキサデカン、臭化ヘプタデカン、臭化オクタデカン、臭化ノナデカン、臭化エイコサン、BrCH2CN、Br(CH2)2CN、Br(CH2)3CN、Br(CH2)4CN、Br(CH2)5CN、Br(CH2)6CN、Br(CH2)7CN及びBr(CH2)4C(CH3)2CN等が挙げられる。
その使用量は、TAHAに対し、2〜3モル倍が好ましく、2〜2.5モル倍がより好ましい。
【0057】
塩基としては、金属水素化物が好ましく、具体的には、水素化リチウム、水素化ナトリウム及び水素化カリウム等が用いられ、特には水素化ナトリウムが好ましい。
その使用量は、TAHAに対し、2〜3モル倍が好ましく、2〜2.5モル倍がより好ましい。
【0058】
反応溶媒としては、N,N−ジメチルホルムアミド(DMF)、テトラヒドロフラン(THF)及び1,4−ジオキサン等が好ましい。それらの使用量は、2〜20質量倍が好ましく、3〜15質量倍がより好ましい。
【0059】
また、置換アルキルハライドが臭化物の場合は、金属沃化物を加えることもできる。
金属沃化物としては、沃化ナトリウムや沃化カリウムが挙げられる。
その使用量は、TAHAに対し、0.1〜3モル倍が好ましく、0.2〜2.5モル倍がより好ましい。
【0060】
反応温度は、−30〜200℃程度であるが、0〜150℃が好ましい。
【0061】
反応時間は、1〜30時間が好ましく、2〜20時間がより好ましい。
【0062】
反応後は、濃縮して溶媒を除いてから、酢酸エチルを加えてから塩酸水等で酸性にして、有機層を分液した後濃縮することにより油状粗物を得る。これをシリカゲルカラムクロマトグラフィー等で精製することにより目的のTAAEが得られる。
<第2工程>
第2工程の加水分解法としては、通常のエステル化合物からカルボン酸化合物を得る塩基や酸の存在下で行う条件を採用できるが、塩基の存在下で行うことが好ましい。
【0063】
塩基としては、アルカリ金属やアルカリ土類金属の水酸化物等を用いることができ、具体的には、水酸化ナトリウムや水酸化カリウムが経済的で好ましい。その使用量は、TAAEに対し、4〜10モル倍が好ましく、5〜8モル倍がより好ましい。
【0064】
加水分解反応の溶媒としては、水と有機溶媒との混合系が好ましい。有機溶媒としては、例えば、メタノール,エタノール等のアルコール類や、1,4−ジオキサン等が挙げられる。それらの使用量は、水と有機溶媒共に、TAAEに対し、それぞれ1〜10質量倍が好ましく、2〜8質量倍がより好ましい。
【0065】
反応温度は、0〜200℃程度であるが、0〜150℃が好ましい。
【0066】
反応時間は、1〜30時間が好ましく、2〜25時間がより好ましい。
【0067】
反応後は、塩酸水等で酸性にしてから酢酸エチルを加え攪拌すると、結晶が析出し、これを捕集するとAECAの結晶が得られる。また、有機層を濃縮した後、得られた粗物にアセトニトリルを加えて加温溶解後、氷冷すると結晶が析出するので、これを捕集するとAECAの粗結晶が得られる。さらに純度を向上させる場合は、この粗結晶をアセトニトリルからの再結晶を繰り返して純度を上げることができる。
<第3工程>
第3工程の還元法は、二重結合を単結合に変換する種々の一般的還元法が適用できる。例えば、(1)金属および金属塩による還元、(2)金属水素化物による還元、(3)金属水素錯化合物による還元、(4)ジボランおよび置換ボランによる還元、(5)ヒドラジンによる還元、(6)ジイミド還元、(7)リン化合物による還元、(8)電解還元、(9)接触還元等を挙げることができる。
これらの中で、最も実用的な方法は接触還元方法である。本発明で採用できる接触還元法は以下の通りである。触媒金属としては、周期律表第8族のパラジウム、ルテニウム、ロジウム、白金、ニッケル、コバルト及び鉄、又は第1族の銅等が使用できる。これらの金属は単独で、又は他の元素と複合させた多元系で使用される。それらの使用形態は、各金属単身、ラネー型触媒、ケイソウ土、アルミナ、ゼオライト、炭素及びその他の担体に担持させた触媒及び錯体触媒等が挙げられる。
【0068】
具体的には、パラジウム/炭素、ルテニウム/炭素、ロジウム/炭素、白金/炭素、パラジウム/アルミナ、ルテニウム/アルミナ、ロジウム/アルミナ、白金/アルミナ、還元ニッケル、還元コバルト、ラネーニッケル、ラネーコバルト、ラネー銅、酸化銅、銅クロマト、クロロトリス(トリフェニルホスフィン)ロジウム、クロロヒドリドトリス(トリフェニルホスフィン)ルテニウム、ジクロロトリス(トリフェニルホスフィン)ルテニウム及びヒドリドカルボニルトリス(トリフェニルホスフィン)イリジウム等が挙げられる。これらの中で特に好ましいものはパラジウム/炭素及びルテニウム/炭素等である。
【0069】
触媒の使用量は、5%金属担持触媒として基質に対し0.1〜30質量%が、特には、0.5〜20質量%が好ましい。溶媒は、メタノール、エタノール及びプロパノール等に代表されるアルコール類、ジオキサン、テトラヒドロフラン及びジメトキシエタン等に代表されるエーテル類及び酢酸エチル及び酢酸プロピル等に代表されるエステル類等が使用できる。
【0070】
その使用量は、原料に対し1〜50質量倍の範囲が、特には3〜20質量倍の範囲が好ましい。水素圧は常圧から10MPa(100kg/cm2)の範囲が、特には常圧から3MPa(30kg/cm2)の範囲が好ましい。反応温度は、0〜150℃の範囲が、特には10〜100℃の範囲が好ましい。
【0071】
反応は、水素吸収量によって追跡することができ、理論水素量の吸収後サンプリングし1H−NMRで分析し確認することができる。反応後は、濾過により触媒を除いてから、濃縮後、再結晶、又はカラムクロマトグラフィー法で精製することができる。
<第4工程>
脱水法としては、(a)脂肪族カルボン酸無水物法、(b)蟻酸およびp−トルエンスルホン酸法、(c)芳香族炭化水素による共沸法等が挙げられる。
【0072】
これらの中でも、本発明では、操業上簡便であるとともに、目的物がより高収率で得られることから、(a)脂肪族カルボン酸無水物法を用いることが好ましい。
【0073】
脂肪族カルボン酸無水物としては、例えば、無水酢酸、無水プロピオン酸等が挙げられるが、経済性の点から無水酢酸が好ましい。
【0074】
脂肪族カルボン酸無水物の添加量は、原料AOCAに対して2〜30モル倍が好ましく、3〜20モル倍がより好ましい。
【0075】
上記脱水反応は、芳香族炭化水素化合物を共存させて行うことが好ましい。本工程では、反応の進行に伴って反応液が着色し、生成物の結晶も着色し易くなるが、芳香族炭化水素化合物を共存させることで、反応液の着色を軽減できる結果、生成物の着色を抑制することができる。
【0076】
芳香族炭化水素化合物としては、ベンゼン、トルエン、キシレン、エチルベンゼン、キュメン等が挙げられるが、経済性の点からトルエンが好適である。
【0077】
芳香族炭化水素化合物の添加量は、原料AOCAに対し、1〜30質量倍が好ましく、3〜20質量倍がより好ましい。
【0078】
反応温度は、通常50〜150℃程度であるが、反応完結までの時間を短縮することを考慮すると、60〜130℃が好適である。
【0079】
反応時間は、長くなると反応液の着色が強くなることから、10分間〜3時間が好ましく、15分間〜2時間がより好ましい。
【0080】
なお、脱色を目的に活性炭を存在させて反応を行うこともできる。この場合、活性炭の使用量は、原料AOCAに対し、1〜100質量%が好ましく、3〜50質量%がより好ましい。
【0081】
反応の終了は、昇温後、原料AOCAの完全溶解で判断することができる。
【0082】
反応後は、氷冷して撹拌して析出した結晶を、濾過して洗浄し、さらに乾燥して目的のAODAが得られる。
【0083】
以上述べた各工程の反応は、いずれも常圧または加圧下で行うことができ、また回分式でも連続式でもよい。
このようにして得られた側鎖置換テトラカルボン酸二無水物を用いて得られるポリアミック酸をイミド化して出来るポリイミドは、各種有機溶媒に対する溶解性に優れることが期待される。また、この側鎖置換テトラカルボン酸二無水物は、液晶配向膜の高チルト角発現が可能なポリアミック酸及びポリイミドへの誘導が期待できる。
<実施例>
以下に参考例及び実施例を挙げて、本発明を具体的に説明するが、本発明の解釈はこれらに限定されるものではないことはもちろんである。
【0084】
尚、実施例で用いた分析法は以下の通りである。
【0085】
[1] [質量分析(MASS)]
機種:AQ−Tod(JEOL) イオン化法:DART+ 測定範囲:m/z = 100〜1000
[2] [1H NMR]
機種:Varian社製NMR System 400NB(400MHz)
測定溶媒:CDCl3、DMSO−d6
標準物質:tetramethylsilane(TMS)
[3] [融点(m.p.)]
機種:微量融点測定装置(MP−S3)(ヤナコ機器開発研究所社製)
[4] [単結晶X線解析]
装置:SMART APEX2 Ultra(Bruker) X線:Cu−Kα 温度:−100℃
サンプル中から測定可能と思われる結晶を選び出して測定した。
(参考例1)
【0086】
【化19】
【0087】
攪拌羽付き3Lの四つ口反応フラスコに水2L、リン酸水素二ナトリウム8水和物を140g及びクエン酸一水和物を34g仕込み、25℃で機械攪拌下に1,3−ジエチル−アセトンジカルボキシレート127g(0.628mol)と39%グリオキザール水溶液45g(0.302mol)を滴下した。その後、25℃で50時間攪拌を継続し反応を終了させた。
続いて、フラスコ内壁に付着した橙色固形物と水溶液をデカンテーションにより分離した。
この橙色固形物が付着したフラスコ内に水100mlを加えて洗浄した。この操作を2回繰り返した。続いて酢酸エチル500gを加えて橙色固形物を溶解させた後、水100mlを加えて洗浄した。得られた有機層に再び水100mlとn−ヘプタン50mlを加え水洗を繰り返した。この有機層を減圧濃縮すると黄色油状物119gが得られた。
この黄色油状物にn−ヘプタン/酢酸エチル=1/1(v/v)90gを加えて60℃で溶解後、氷冷した。析出した結晶をろ過、n−ヘプタン/酢酸エチル=9/1(v/v)で洗浄後50℃で減圧乾燥すると肌色結晶85.2g(収率67%)が得られた。
この結晶は、単結晶X線構造解析(図1に示す)から目的のテトラエチル−3,7−ジハイドロオキシビシクロ[3.3.01,5]オクタ−2,6−ジエン−2,4,6,8−テトラカルボキシレート(THOE)であることを確認した。
(参考例2)
【0088】
【化20】
【0089】
攪拌羽付き1Lの四つ口反応フラスコにメタノール460mlと水酸化ナトリウム25.6g(0.64mol)を仕込み、60℃で機械攪拌下に溶解させた後、氷冷し5℃とした。続いて攪拌しながら1,3−ジメチル−アセトンジカルボキシレート109.2g(0.627mol)を内温4〜12℃で20分かけて滴下した。しだいに高粘度の黄色スラリー状になったので、90℃油浴で加温すると内温60℃溶解し均一溶液になった。続いて39%グリオキザール水溶液52.7g(0.354mol)を滴下開始すると発熱し、65℃で還流した。20分で滴下を終了し、更に10分間還流させた後、25℃まで冷却し5時間攪拌を継続し反応を終了させた。
続いて、生成した固形物をろ過後、200mlのメタノールで5回洗浄した後減圧乾燥するとTMHO・2Na塩の淡肌色結晶83.5g(収率64.3%)が得られた。
【0090】
この結晶79.6g(0.235mol)に水312gを加えて氷冷攪拌下10℃としたところに、35%塩酸水47gを水320gに溶解した溶液を滴下し酸性にすると、黄色スラリーが白色スラリーになった。続いてろ過後100mlの水で2回洗浄した後減圧乾燥すると固体128.7gが得られた。この固体にクロロホルム90gと水30gを加えて60℃で溶解した後クロロホルム層を分液して濃縮すると、赤色油状物55.9gが得られた。酢酸エチル100gを加えて加温溶解した後n−ヘプタンを加えてからやや濃縮するとスラリー化したので、そのまま氷冷一夜静置させた。続いてろ過後酢酸エチル/n−ヘプタン=1/3(V/V)で洗浄した後減圧乾燥すると淡肌色結晶45.4g(収率52.2%)(m.p.94〜95℃)が得られた。
この結晶は、MASS及び1H−NMRから目的のテトラメチル−3,7−ジハイドロオキシビシクロ[3.3.01,5]オクタ−2,6−ジエン−2,4,6,8−テトラカルボキシレート(TMHO)であることを確認した。
(参考例3)
【0091】
【化21】
【0092】
100ml四つ口反応フラスコに参考例2で得られたTMHO3.7g(10mmol)とメタノール30gを仕込み、−40℃に冷却攪拌下にNaBH40.822gを10分かけて分割添加した。その後一時間かけて20℃に昇温し、20℃で2時間攪拌した。
続いて、氷冷し5℃で35%塩酸水2gを水10gに溶解した溶液を滴下し酸性にした後、濃縮してメタノールを留去してから酢酸エチルを加えて有機層を濃縮するとゼリー状物4.35gが得られた。更にシリカゲルカラムクロマトグラフィーにより精製すると留分1としてゼリー状物1.54g(収率41%)と留分2として白色結晶1.12g(収率30%)(m.p.130〜133℃)が得られた。ゼリー状物は、MASS及び1H−NMRから目的のテトラメチル−3,7−ジハイドロオキシビシクロ[3.3.01,5]オクタン−2,4,6,8−テトラカルボキシレート(TMHA)であることを確認した。
又、留分2の白色結晶はX線構造解析からオールシス−テトラメチル3,7−ジハイドロオキシビシクロ[3.3.01,5]オクタン−2,4,6,8−テトラカルボキシレート(all cis−TMHA)であることを確認した。
MASS(ESI+) : 375.2([M+H]+), 343.2, 325.2, 307.2
1H NMR (CDCl3, ppm) : 2.77〜2.82(m, 4H), 2.85〜2.91(m, 4H), 3.72 (s, 12H), 4.53 (s, 2H)
【0093】
【表1】
【0094】
(参考例4)
【0095】
【化22】
【0096】
100ml四つ口反応フラスコに参考例1で得られたTHOE4.26g(10mmol)、酢酸38.3g(9質量倍)、水3.83g(0.9質量倍)及び3%Pt/カーボン(H2O 60.1wt%含)4.07gを仕込み、窒素置換後更に水素ガスを充填した風船で置換後、大気圧の水素雰囲気下で、20℃で7時間後17℃で16時間攪拌した。
反応後、ろ過により触媒を除去し、ろ液を濃縮・減圧乾燥すると黄色油状物が得られた。この黄色油状物に5%炭酸ナトリウム水溶液と酢酸エチルを加えて溶解させた後、有機層を水洗してから濃縮すると黄色油状物3.37gが得られた。
この黄色油状物をシリカゲルカラムクロマトグラフィーにより精製すると黄色油状物2.72g(収率64%)が得られた
この油状物は、MASS及び1H−NMRから目的のテトラエチル−3,7−ジヒドロキシビシクロ[3.3.01,5]オクタン−2,4,6,8−テトラカルボキシレート(TDHO)であることを確認した。
(実施例1)
【0097】
【化23】
【0098】
100ml四つ口反応フラスコに参考例3で得られたTMHA7.18g(19mmol)とN,N−ジメチルホルムアミド(DMF)57gを仕込み、氷冷攪拌下にNaH(純度55%)2.02g(42mmol)を添加した。その後24℃まで昇温し、20分間攪拌してから、再び氷冷し5℃で1−ブロモテトラデカン10.6g(38mmol)を滴下した。その後、25℃で1時間攪拌した後、40℃で4時間攪拌した。
続いて濃縮して残渣に、酢酸エチルと水を加えて氷冷下に35%塩酸8.8gを滴下し酸性にした。有機層を濃縮すると油状物13.3gが得られた。更にシリカゲルカラムクロマトグラフィーにより2回精製すると黄色油状物1.89g(収率13%)が得られた。この黄色油状物は、MASS及び1H−NMRからテトラメチル−4−テトラデシル−7−テトラデシルオキシビシクロ[3.3.01,5]オクテ−2−エン−2,4,6,8−テトラカルボキシレート(TMTE)であることを確認した。
MASS(ESI+) : 748.8([M+])
1H−NMR(CDCl3, ppm) : 0.882(t, J=6.4Hz, 6H), 1.100〜1.898(m, 50H), 2.550(t, J=10.2Hz, 1H), 2.659(t, J=10.0Hz, 1H), 3.081(t, J=10.0Hz, 1H), 3.284〜3.397(m, 2H), 3.648〜3.813(m, 13H), 4.061(t, J=9.2Hz, 1H), 6.769 (s, 1H)
(実施例2)
【0099】
【化24】
【0100】
100ml四つ口反応フラスコに実施例1で得られたTMTE1.79g(2.30mmol)、メタノール20g及び水酸化ナトリウム1.67g(41mmol)を水8gに溶解した水溶液を仕込み、90℃油浴で加温還流下に、8時間攪拌した。
続いて濃縮して残渣に、酢酸エチルと水を加えてから氷冷し、35%塩酸水溶液4gを滴下し酸性にしてから、20℃で一夜攪拌すると結晶が析出した。この結晶をろ過後酢酸エチルで洗浄した後減圧乾燥すると白色結晶1.0g(収率60%)が得られた。
この結晶は、MASS及び1H−NMRから目的の4−テトラデシル−7−テトラデシルオキシビシクロ[3.3.01,5]オクテ−2−エン−2,4,6,8−テトラカルボン酸(TECA)であることを確認した。
m.p.228−230℃
MASS(ESI−) : 691.7([M−H])−, 646.8
1H−NMR(CDCl3, ppm) : 0.815(t, J=3.4Hz, 6H), 1.197〜1.732(m, 50H), 2.372(t, J=10.0Hz, 1H), 2.452(t, J=8.8Hz, 1H), 2.808(t, J=9.2Hz, 1H), 3.284〜3.397(m, 2H), 3.478(t, J=7.4Hz, 1H), 3.891(t, J=8.8Hz, 1H), 6.518 (s, 1H), 12.430(brs, 4H)
(実施例3)
【0101】
【化25】
【0102】
200ml四つ口反応フラスコに実施例2で得られたTECA9.20g(13.2mmol)、1,4−ジオキサン136g及び5%Pd/カーボン(H2O:55.34wt%含)3.8gを仕込み、窒素置換後水素ガスを充填した風船で置換後、40℃の大気圧の水素雰囲気下で、50時間攪拌した。
反応後、ろ過により触媒を除去し、ろ液を濃縮・減圧乾燥すると白色固体9.09gが得られた。この固体にアセトニトリルを加えて加温するとスラリー化した。氷冷後ろ過・アセトニトリル洗浄してから減圧乾燥すると白色結晶7.68g(収率83.7%)が得られた。
この結晶は、MASS及び1H−NMRから目的の4−テトラデシル−7−テトラデシルオキシビシクロ[3.3.01,5]オクタン−2,4,6,8−テトラカルボン酸(TOCA)であることを確認した。
m.p.195−200℃
MASS(ESI−):693.4([M−H]−)
1H−NMR:0.835(t, J=18.8Hz, 6H), 0.951〜1.554(m, 52H), 1.681〜1.784(m, 2H), 2.010(t, J=13.6Hz, 0.5H), 2.210(t, J=9.6Hz, 0.5H), 2.317〜2.452(m, 1H), 2.473〜2.623(m, 1H), 2.749〜3.059(m, 2H), 3.820(t, J=9.6Hz, 0.5H), 4.060(t, J=8.0Hz, 0.5H), 12.290(brs, 4H)
(実施例4)
【0103】
【化26】
【0104】
100ml四つ口反応フラスコに実施例3で得られたTOCA7.67g(11.0mmol)、トルエン77g及び無水酢酸11.3g(110mmol)を仕込み、110℃油浴で15分間攪拌した。
反応後、濃縮・減圧乾燥すると淡灰色ガム7.57gが得られた。続いてこの淡灰色ガム4.12g(6.2mmol)に、酢酸エチル40g、無水酢酸2.3g(22mmol)及び活性炭2.3gを加えて85℃油浴で1時間攪拌した。更に、ろ過後ろ液を濃縮すると油状物3.46gが得られた。更に、120〜130℃で1時間減圧乾燥すると、淡黄色固体3.14gが得られた。
黄色固体は、MASS及び1H−NMRから目的の4−テトラデシル−7−テトラデシルオキシビシクロ[3.3.01,5]オクタン−2,4,6,8−テトラカルボン酸−2,4:6,8−二無水物(TODA)であることを確認した。
MASS(ESI−):657.4([M−H]−)
1H−NMR:(CDCl3, ppm) : 0.879(t, J=6.8Hz, 6H), 1.254〜1.748(m, 52H), 1.890〜2.182(m, 1H), 2.400〜2.668(m, 2H), 2.871〜3.064(m, 1H), 3.193〜3.500(m, 2H), 4.053(t, J=6.8Hz, 1H), 4.417〜4.494(m, 1H)
(実施例5)
【0105】
【化27】
【0106】
100ml四つ口反応フラスコに参考例2で得られたTMHA5.80g(15.4mmol)とN,N−ジメチルホルムアミド(DMF)60gを仕込み、氷冷攪拌下にNaH(純度55%)1.47g(34mmol)を添加した。その後24℃まで昇温し、60分間攪拌してから、再び氷冷し5℃で1−ヨードオクタン7.77g(32mmol)を滴下した。その後、25℃で1時間攪拌した後、60℃で7時間攪拌した。
続いて濃縮して残渣に、酢酸エチルと水を加えて氷冷下に濃塩酸3.4gを滴下し酸性にした。有機層を濃縮すると油状物8.0gが得られた。更にシリカゲルカラムクロマトグラフィーにより2回精製すると黄色油状物3.15g(収率34%)が得られた。この黄色油状物は、MASS及び1H−NMRからテトラメチル−4−オクチル−7−オクチルオキシビシクロ[3.3.01,5]オクテ−2−エン−2,4,6,8−テトラカルボキシレート(TMOE)であることを確認した。
MASS(ESI+) : 581.4([M+]), 549.4
1H−NMR(CDCl3, ppm) : 0.846(t, J=1.5Hz, 6H), 1.066〜1.898(m, 26H), 2.680(t, J=8.4Hz, 1H), 3.098(t, J=10.0Hz, 1H), 3.268〜3.397(m, 2H), 3.641(s, 3H), 3.692(s, 3H), 3.699(s, 3H), 3.742(s, 3H), 3.675(t, J=10.0Hz, 1H), 4.079(t, J=9.6Hz, 1H), 6.792(s, 1H)
(実施例6)
【0107】
【化28】
【0108】
100ml四つ口反応フラスコに実施例5で得られたTMOE5.10g(8.50mmol)、メタノール31g及び水酸化ナトリウム3.40g(85mmol)を水10gに溶解した水溶液を仕込み、20〜24℃で、19時間攪拌した。
続いて、濃縮して残渣に酢酸エチルと水を加えてから氷冷して、35%塩酸水溶液8.5gを滴下し酸性とした。この有機層を水洗してから濃縮すると肌色の固体5.59gが得られた。この固体に酢酸エチルを加えて加温溶解した後、やや濃縮してから氷冷すると結晶が析出した。この結晶をろ過・圧乾燥すると白色結晶1.68g(収率36.5%)が得られた。
この結晶は、MASS及び1H−NMRから4−オクチル−7−オクチルオキシビシクロ[3.3.01,5]オクテ−2−エン−2,4,6,8−テトラカルボン酸−4−メチルエステル(OECM)であることを確認した。
m.p.169−171℃
MASS(ESI+) : 539.2([M+H])+, 536.2, 521.3
MASS(ESI−) : 537.4([M−H]−),479.8
1H−NMR(CDCl3, ppm) : 0.830〜0.855(m, 6H), 1.040〜1.808(m, 26H), 2.283(t, J=10.0Hz, 1H), 2.416(t, J=10.0Hz, 1H), 2.854(t, J=9.6Hz, 1H), 3.295〜3.388(m, 2H), 3.503〜3.589(m, 1H), 3.581 (s, 3H), 3.936(t, J=8.8Hz, 1H), 6.559(d, J=2.2Hz, 1H), 12.464(brs, 3H)
(実施例7)
【0109】
【化29】
【0110】
100ml四つ口反応フラスコに実施例6で得られたOECM2.46g(4.56mmol)、メタノール25g及び水酸化ナトリウム1.10g(27mmol)を水5.5gに溶解した水溶液を仕込み、内温72℃(油浴90℃)で加温還流下に、3時間攪拌した。
続いて濃縮して残渣に、酢酸エチルと水を加えてから氷冷してから、35%塩酸水溶液4gを滴下して酸性にしてから、有機層を分液し、水洗の後濃縮・減圧乾燥すると白色結晶2.31g(収率96%)が得られた。
この結晶は、MASS及び1H−NMRから目的の4−オクチル−7−オクチルオキシビシクロ[3.3.01,5]オクテ−2−エン−2,4,6,8−テトラカルボン酸(OECA)であることを確認した。
m.p.230−240℃
MASS(ESI+):525.4([M+H]+),507.4
1H−NMR(CDCl3, ppm) : 0.830〜0.855(m, 6H), 1.040〜1.808(m, 26H), 2.283(t, J=10.0Hz, 1H), 2.416(t, J=10.0Hz, 1H), 2.854(t, J=9.6Hz, 1H), 3.295〜3.388(m, 2H), 3.503〜3.589(m, 1H), 3.581 (s, 3H), 3.936(t, J=8.8Hz, 1H), 6.559(d, J=2.2Hz, 1H), 12.464(brs, 4H)
(実施例8)
【0111】
【化30】
【0112】
100ml四つ口反応フラスコに実施例5で得られたTMOE12.8g(22mmol)、メタノール128g及び水酸化ナトリウム5.28g(132mmol)を水26gに溶解した水溶液を仕込み、内温68℃(油浴90℃)で還流させながら、2時間攪拌した。
続いて、濃縮して残渣に酢酸エチルと水を加えてから氷冷して、35%塩酸水溶液13.2gを滴下し酸性とした。この有機層を水洗してから濃縮すると暗赤色のペースト11.8gが得られた。このペーストにアセトニトリルを加えて加温溶解した後、やや濃縮してから氷冷すると結晶が析出した。この結晶をろ過後アセトニトリルで2回洗浄してから減圧乾燥すると肌色結晶2.78gが得られた。この結晶をアセトニトリルで再結晶化させて減圧乾燥すると白色結晶2.34g(収率20.3%)が得られた。
この結晶は、実施例7で得られた1H−NMRと一致したことから4−オクチル−7−オクチルオキシビシクロ[3.3.01,5]オクテ−2−エン−2,4,6,8−テトラカルボン酸(OECA)であることを確認した。
m.p.240−245℃
(実施例9)
【0113】
【化31】
【0114】
100ml四つ口反応フラスコに実施例8を繰り返して得られたOECA3.31g(4.76mmol)、1,4−ジオキサン33g及び5%Pd/カーボン(H2O 55.34wt%含)1.48gを仕込み、窒素置換後更に水素ガスを充填した風船で置換後、25℃の大気圧の水素雰囲気下で、45時間攪拌した。
反応後、ろ過により触媒を除去し、ろ液を濃縮・減圧乾燥すると白色結晶1.34g(収率58%)が得られた。
この結晶は、MASS及び1H−NMRから目的の4−オクチル−7−オクチルオキシビシクロ[3.3.01,5]オクタン−2,4,6,8−テトラカルボン酸(OOCA)であることを確認した。
m.p.255−257℃
MASS(ESI−):525.5([M-H]−)
1H−NMR:(CDCl3, ppm) : 0.833〜0.863(m,6H),1.211〜1.555(m,26H), 1.666〜1.771(m,1H),1.973〜2.090(m,1H), 2.212(t, J=9.88Hz, 1H), 2.407(t, J=9.88Hz,1H), 2.507(t, J=13.7Hz, 1H), 2.821〜2.978(m, 1H), 3.025(t, J=9.49Hz, 1H), 3.339(t, J=6.36Hz, 2H), 3.817(t, J=9.68Hz, 1H), 12.296(brs, 4H)
(実施例10)
【0115】
【化32】
【0116】
100ml四つ口反応フラスコに実施例9で得られたOOCA1.28g(2.43mmol)、トルエン12.8g及び無水酢酸2.47g(24.3mmol)を仕込み、120℃油浴で20分間攪拌した。
反応後、濃縮・減圧乾燥すると無色透明な油状物1.37gが得られた。続いてヘキサンを加えて加温してから氷冷すると2層になった。この上澄み液をデカンテーションにより除去してから同じ操作を繰り返した。残った下層の油状物を減圧乾燥すると1.20g(収率100%)の無色透明な油状物が得られた。
油状物は、MASS及び1H−NMRから目的の4−オクチル−7−オクチルオキシビシクロ[3.3.01,5]オクタン−2,4,6,8−テトラカルボン酸−2,4:6,8−二無水物(OODA)であることを確認した。
MASS(ESI+):490.4([M・]+)
1H−NMR:(CDCl3, ppm) : 0.834〜0.899(m, 6H), 1.245〜1.553(m, 26H), 1.950(t, J=10.2Hz, 1H), 2.108(t, J=12.0Hz, 1H), 2.226〜2.321(m, 2H), 2.430(d, J=12.8Hz, 1H), 2.536〜2.652(m, 1H), 2.909〜2.984(m, 1H), 3.201〜3.300(m, 1H), 3.400〜3.559(m, 1H), 3.676〜3.750(m, 1H)
(実施例11)
【0117】
【化33】
【0118】
200ml四つ口反応フラスコに参考例4で得られたTDHO9.23g(21.4mmol)とN,N−ジメチルホルムアミド(DMF)90gを仕込み、氷冷攪拌下にNaH(純度55%)2.80g(64mmol)を添加した。その後25℃まで昇温し、60分間攪拌してから、再び氷冷し15℃で1−ヨードメタン9.10g(64mmol)を滴下した。その後、25℃で40分攪拌した後、40℃で6時間攪拌した。
続いて濃縮して残渣に、酢酸エチルと水を加えて氷冷下に35%塩酸6.4gを滴下し酸性にした。有機層を濃縮すると油状物9.8gが得られた。無色の油状物が上層に浮遊したので、ヘプタンを加えて洗浄すると、油状物6.5gが得られた。更にシリカゲルカラムクロマトグラフィーにより精製すると黄色油状物5.3g(Y54%)が得られた。この黄色油状物は、MASS及び1H−NMRからテトラエチル−4−メチル−7−メトキシビシクロ[3.3.01,5]オクテ−2−エン−2,4,6,8−テトラカルボキシレート(TMME)であることを確認した。
MASS(ESI+):441.17([M+H]+, 100), 427.2 (26)
1H−NMR:(CDCl3, ppm) : 1.19〜1.42(m, 12H), 2.65〜3.10(m, 4H), 3.31〜3.40(m, 3H), 3.67〜3.80(m, 3H), 4.04〜4.37(m, 9H), 6.70(s, 1H)
(実施例12)
【0119】
【化34】
【0120】
50ml四つ口反応フラスコに実施例11で得られたTMME1.95g(4.2mmol)、エタノール13g及び水酸化ナトリウム2.55g(64mmol)を水14gに溶解した水溶液を仕込み、油浴90℃で還流させながら、21時間攪拌した。
続いて、濃縮して残渣に酢酸エチルと水を加えてから氷冷して、35%塩酸水溶液6.4gを滴下し酸性にするとスラリーになった。ろ過により得られたケーキを酢酸エチルと水で洗浄後減圧乾燥すると白色結晶1.08g(収率78%)(m.p.235−236℃)
が得られた。
また、ろ液を分液し有機層を水洗してから濃縮すると固体1.1gが得られた。この固体に酢酸エチルを加えて加温するとスラリーになった。氷冷後ろ過・酢酸エチル洗浄・減圧乾燥すると白色結晶0.26g(収率18%)(m.p.245−248℃)が得られた。
この結晶は、MASS及び1H−NMRから4−メチル−7−メトキシビシクロ[3.3.01,5]オクテ−2−エン−2,4,6,8−テトラカルボン酸(MMCA)であることを確認した。
MASS(ESI+):329.06([M+H]+, 55), 311.05 (33), 302.10 (16), 285.08 (100), 207.05 (78)
1H−NMR:(DMSO−d6, ppm) : 1.26(s, 3H), 2.49(t, J=9.2Hz, 1H), 2.57(t, J=9.6Hz, 1H), 2.84(t, J=9.6Hz, 1H), 3.25(s, 3H), 3.58(d, J1=2.0Hz, J2=8.8Hz, 1H), 3.92(t, J=8.8Hz, 1H), 6.50(s, 1H), 12.54(brs, 4H)
【技術分野】
【0001】
本発明は、側鎖置換脂環式テトラカルボン酸二無水物及びその製造法に関し、さらに詳述すると、例えば、電子材料用として好適なポリイミドの原料モノマーである側鎖置換脂環式テトラカルボン酸二無水物に関する。
【背景技術】
【0002】
一般に、ポリイミド樹脂はその特長である高い機械的強度、耐熱性、絶縁性、耐溶剤性のために、液晶表示素子や半導体における保護材料、絶縁材料、カラーフィルターなどの電子材料として広く用いられている。また、最近では光導波路用材料等の光通信用材料としての用途も期待されている。
【0003】
近年、この分野の発展は目覚ましく、それに対応して、用いられる材料に対しても益々高度な特性が要求される様になっている。即ち、単に耐熱性、耐溶剤性に優れるだけでなく、用途に応じた性能を多数合わせ有することが期待されている。
【0004】
しかし、特に、全芳香族ポリイミド樹脂においては、濃い琥珀色を呈し着色するため、高い透明性を要求される光学材料用途においては問題が生じてくる。また、全芳香族ポリイミドは有機溶剤に不溶であるため、実際にはその前駆体であるポリアミック酸を熱による脱水閉環によって得る必要がある。
【0005】
透明性を実現する一つの方法として、脂環式テトラカルボン酸二無水物と芳香族ジアミンとの重縮合反応によりポリアミック酸を得て、該当前駆体をイミド化してポリイミドを製造すれば、比較的着色が少なく、高透明性のポリイミドが得られることは知られている(特許文献1、2参照)。
【0006】
近年、下記式[11]
【0007】
【化1】
【0008】
で表されるビシクロ[3.3.0]オクタン−2,4,6,8−テトラカルボン酸−2:4,6:8−二無水物(以下、BODAと略称する。)を使用するポリイミドが、基板への印刷性、密着性に優れ、かつラビング時に基板からの剥離がなく、またラビングによる配向膜への傷がつきにくく、液晶セル駆動時に優れた電圧保持特性が得られる液晶配向処理剤及びそれを用いた液晶配向膜として知られている。(特許文献3参照)。
しかしながら、BODAを酸二無水物として用いたポリイミドは、ジアミンの種類によっては有機溶剤への溶解性が低いという問題点を抱えており、その使用場面に制限があった。
また、ポリアミック酸やポリイミドを溶解させる有機溶媒としては、従来N−メチル−2−ピロリドン(NMP)やγ―ブチロラクトン等のアミド系やラクトン系有機溶媒が多用されている。しかし、これらの溶媒は高沸点のため、液晶配向剤を基板に塗布、焼成して液晶配向膜を作成する際、溶媒を完全に除去するためには高温焼成が必要であった。近年ではプロセス効率の観点から、低温での焼成が望まれて来ており、沸点の低い有機溶媒類に対しても可溶であるポリイミドが望まれていた。
また、液晶配向膜に求められる特性の一つとして、基板面に対する液晶分子の配向傾斜角を任意の値に保つ、いわゆる液晶のプレチルト角制御があり、近年の液晶ディスプレイの大型化、高精細化に伴い、液晶のプレチルト角制御は液晶表示素子において重要な課題となっている。液晶のプレチルト角は、液晶配向膜を構成しているポリイミドの構造を種々選択することで変更、制御出来ることが知られている。
【0009】
ポリイミドの構造によってプレチルト角を制御する技術の中でも、側鎖を有するジアミンをポリイミド原料の一部として用いる方法は、このジアミンの使用割合に応じてプレチルト角が制御できるので、目的のプレチルト角にせしめることが比較的容易であり、プレチルト角を大きくする手段として有用である。液晶のプレチルト角を大きくするジアミンの側鎖構造としては、長鎖のアルキル基又はフルオロアルキル基(例えば特許文献4参照)、環状基又は環状基とアルキル基の組み合わせ(例えば特許文献5参照)、ステロイド骨格(例えば特許文献6参照)などが知られている。
しかし、側鎖を有するジアミンは一般的に溶解性が低く、使用できる割合に限界があった。また、側鎖ジアミンを多く用いすぎると、得られるポリアミック酸やポリイミドを用いた液晶配向剤を基板に均一に塗布することが困難となる問題もあった。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】特開昭60−188427号公報
【特許文献2】特開昭58−208322号公報
【特許文献3】特開平11−249148号公報
【特許文献4】特開平2−282726号公報
【特許文献5】特開平3−179323号公報
【特許文献6】特開平4−281427号公報
【発明の開示】
【発明が解決しようとする課題】
【0011】
本発明は、このような事情に鑑みてなされたものであり、沸点の低い有機溶媒類に対しても溶解性に優れ、また液晶配向膜の高チルト角発現が可能なポリイミドを与えることが期待されるモノマーである、側鎖置換脂環式テトラカルボン酸二無水物及びその製造方法を提供することを目的とする。
【課題を解決するための手段】
【0012】
本発明者らは、上記目的を達成するために鋭意検討を重ねた結果、BODAのビシクロオクタン環に側鎖置換基を導入した原料酸二無水物モノマーの製造方法を確立し、本発明を完成した。得られた酸二無水物モノマーは新規化合物であり、各種有機溶媒に対する溶解性に優れ、また液晶配向膜の高チルト角発現が可能なポリイミドへの誘導が期待される。
【0013】
すなわち、本発明は、
1.下記式[1]で表される化合物。
【0014】
【化2】
【0015】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表す。)
2.前記R1が、メチル基、n−オクチル基及びn−テトラデシル基である1記載の化合物。
3.下記式[2]で表される化合物。
【0016】
【化3】
【0017】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表す。)
4.前記R1が、メチル基、n−オクチル基及びn−テトラデシル基である3記載の化合物。
5.下記式[3]で表される化合物。
【0018】
【化4】
【0019】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表す。)
6.前記R1が、メチル基、n−オクチル基及びn−テトラデシル基である5記載の化合物。
7.下記式[4]で表される化合物。
【0020】
【化5】
【0021】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表し、R2は、炭素数1〜10のアルキル基を表す。)
8.前記R1が、メチル基、n−オクチル基及びn−テトラデシル基である7記載の化合物
9.下記式[5]で表される化合物。
【0022】
【化6】
【0023】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表し、R2は、炭素数1〜10のアルキル基を表す。)
10.前記R1が、メチル基、n−オクチル基及びn−テトラデシル基である9記載の化合物。
11.下記式[6]で表される化合物。
【0024】
【化7】
【0025】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表し、R2は、炭素数1〜10のアルキル基を表す。)
12.前記R1が、メチル基、n−オクチル基及びn−テトラデシル基である11記載の化合物。
13.下記式[9]で表される化合物を塩基の存在下、下記式[10]で表される置換アルキルハライドと反応して、下記式[4]で表される化合物を得、更にこれを加水分解して下記式[3]で表される化合物を得、更に、これを還元して下記式[2]で表される化合物を得た後、脱水剤により下記式[1]で表される化合物を得ることを特徴とする製造方法。
【0026】
【化8】
【0027】
(式中、R2は、炭素数1〜10のアルキル基を表す。)
【0028】
【化9】
【0029】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表し、Xはハロゲン原子を表す。)
【0030】
【化10】
【0031】
(式中、R1及びR2は、前記と同じ意味を表す。)
【0032】
【化11】
【0033】
(式中、R1は、前記と同じ意味を表す。)
【0034】
【化12】
【0035】
(式中、R1は、前記と同じ意味を表す。)
【0036】
【化13】
【0037】
(式中、R1は、前記と同じ意味を表す。)
14.下記式[4]で表される化合物を加水分解して下記式[6]で表される化合物を得ることを特徴とする製造法を提供する。
【0038】
【化14】
【0039】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表し、R2は、炭素数1〜10のアルキル基を表す。)
【0040】
【化15】
【0041】
(式中、R1及びR2は、前記と同じ意味を表す。)
【発明の効果】
【0042】
本発明によれば、各種有機溶媒に対する溶解性に優れ、また液晶配向膜の高チルト角発現が可能なポリアミック酸及びポリイミドへの誘導が期待される側鎖置換テトラカルボン酸二無水物及びその製造法を提供することができる。
【0043】
本発明の側鎖置換BODA酸二無水物を一原料として得られるポリイミドは、例えば、液晶表示素子や半導体における保護材料、絶縁材料などの電子材料、さらに光導波路等の光通信用材料への好適な適用が期待される。
【図面の簡単な説明】
【0044】
【図1】THOEの単結晶X線構造
【図2】TMHAの単結晶X線構造
【発明を実施するための最良の形態】
【0045】
以下、本発明についてさらに詳しく説明する。
【0046】
なお、以下において、nはノルマルを、iはイソを、sはセカンダリーを、tはターシャリーを、cはシクロをそれぞれ表す。
【0047】
上記各式において、炭素数1〜10のアルキル基としては、直鎖、分岐、環状のいずれでもよく、その具体例としては、メチル、エチル、n−プロピル、i−プロピル、c−プロピル、n−ブチル、i−ブチル、s−ブチル、t−ブチル、c−ブチル、n−ペンチル、1−メチル−n−ブチル、2−メチル−n−ブチル、3−メチル−n−ブチル、1,1−ジメチル−n−プロピル、c−ペンチル、2−メチル−c−ブチル、n−ヘキシル、1−メチル−n−ペンチル、2−メチル−n−ペンチル、1,1−ジメチル−n−ブチル、1−エチル−n−ブチル、1,1,2−トリメチル−n−プロピル、c−ヘキシル、1−メチル−c−ペンチル、1−エチル−c−ブチル、1,2−ジメチル−c−ブチル、n−ヘプチル、n−オクチル、n−ノニル、n−デシル基等が挙げられる。
【0048】
これらの中でも、得られるポリイミドの有機溶媒に対する溶解性を高めることを考慮すると、R1としては、炭素数1〜20のアルキル基が好ましく、炭素数5〜20のアルキル基がより好ましく、炭素数8〜18のアルキル基がより一層好ましい。
【0049】
上記式[1]で表される2−アルキル−7−アルコキシビシクロ[3.3.01,5]オクタン−2,4,6,8−テトラカルボン酸−2:4,6:8−二無水物(以下、AODAと略記する)の製造法は、下記の一連の反応スキームで表される。
【0050】
【化16】
【0051】
(式中、R1およびR2は、上記と同じ意味を表す。)
すなわち、第1工程は、テトラアルキル3,7−ジヒドロキシビシクロ[3.3.01,5]オクタン−2,4,6,8−テトラカルボキシレート(TAHA)を塩基の存在下、置換アルキルハライドと反応させて、テトラアルキル−4−アルキル−7−アルコキシビシクロ[3.3.01,5]オクテ−2−エン−2,4,6,8−テトラカルボキシレート(TAAE)を得る。
第2工程は、TAAEを加水分解して4−アルキル−7−アルコキシビシクロ[3.3.01,5]オクテ−2−エン−2,4,6,8−テトラカルボン酸(AECA)を得る。
第3工程は、AECAを還元して4−アルキル−7−アルコキシビシクロ[3.3.01,5]オクタン−2,4,6,8−テトラカルボン酸(AOCA)を得る。
第4工程は、AOCAを脱水剤により2−アルキル−7−アルコキシビシクロ[3.3.01,5]オクタン−2,4,6,8−テトラカルボン酸−2:4,6:8−二無水物(AODA)を得る製造法である。
【0052】
ここで、原料のTAHAは、公知の次の反応スキームで表される製造法で合成される。(Organic Syntheses, 64, 27−38 (1986);Tetrahedron Letters, 49 (2008) 3056−3059)即ち、R2がメチルの場合は、ジメチル−1,3−アセトンジカルボキシレートとグリオギザールを水酸化ナトリウム存在下で反応させて、テトラメチル−3,7−ジソジウムオキシビシクロ[3.3.01,5]オクタ−2,6-ジエン−2,4,6,8−テトラカルボキシレート(TMSO)を得た後、塩酸で酸性にすることによりテトラメチル−3,7−ジヒドロキシビシクロ[3.3.01,5]オクタ−2,6-ジエン−2,4,6,8−テトラカルボキシレート(TMHO)が得られる。続いてこれを還元することによりテトラメチル−3,7−ジヒドロキシビシクロ[3.3.01,5]オクタタン−2,4,6,8−テトラカルボキシレート(TMHA)が合成できる。
【0053】
【化17】
【0054】
また、R2がエチルの場合は、次式で表される製造法で合成される。
【0055】
【化18】
【0056】
即ち、ジエチル−1,3−アセトンジカルボキシレートとグリオギザールから公知の製造法でテトラエチル−3,7−ジヒドロキシビシクロ[3.3.01,5]オクタ−2,6-ジエン−2,4,6,8−テトラカルボキシレート(THOE)が合成される。(特開昭56−127328)続いてこれを還元することによりテトラエチル3,7−ジヒドロキシビシクロ[3.3.01,5]オクタン−2,4,6,8−テトラカルボキシレート(TEHO)が合成できる。
<第1工程>
こうして得られてTAHAを原料として、塩基の存在下、置換アルキルハライドと反応させて、TAAEを得る。
置換アルキルハライドの置換アルキル基(R1)としては、炭素数1〜20のアルキル基及び炭素数2〜20のシアノアルキル基を表し、ハロゲン原子(X)としては、弗素原子、塩素原子、臭素原子及び沃素原子を表す。
具体的には、沃化メタン、沃化エタン、沃化プロパン、沃化ブタン、沃化ペンタン、沃化ヘキサン、沃化ヘプタン、沃化オクタン、沃化ノナン、沃化デカン、臭化ブタン、臭化ペンタン、臭化ヘキサン、臭化ヘプタン、臭化オクタン、臭化ノナン、臭化デカン、臭化ウンデカン、臭化ドデカン、臭化トリデカン、臭化テトラデカン、臭化ペンタデカン、臭化ヘキサデカン、臭化ヘプタデカン、臭化オクタデカン、臭化ノナデカン、臭化エイコサン、BrCH2CN、Br(CH2)2CN、Br(CH2)3CN、Br(CH2)4CN、Br(CH2)5CN、Br(CH2)6CN、Br(CH2)7CN及びBr(CH2)4C(CH3)2CN等が挙げられる。
その使用量は、TAHAに対し、2〜3モル倍が好ましく、2〜2.5モル倍がより好ましい。
【0057】
塩基としては、金属水素化物が好ましく、具体的には、水素化リチウム、水素化ナトリウム及び水素化カリウム等が用いられ、特には水素化ナトリウムが好ましい。
その使用量は、TAHAに対し、2〜3モル倍が好ましく、2〜2.5モル倍がより好ましい。
【0058】
反応溶媒としては、N,N−ジメチルホルムアミド(DMF)、テトラヒドロフラン(THF)及び1,4−ジオキサン等が好ましい。それらの使用量は、2〜20質量倍が好ましく、3〜15質量倍がより好ましい。
【0059】
また、置換アルキルハライドが臭化物の場合は、金属沃化物を加えることもできる。
金属沃化物としては、沃化ナトリウムや沃化カリウムが挙げられる。
その使用量は、TAHAに対し、0.1〜3モル倍が好ましく、0.2〜2.5モル倍がより好ましい。
【0060】
反応温度は、−30〜200℃程度であるが、0〜150℃が好ましい。
【0061】
反応時間は、1〜30時間が好ましく、2〜20時間がより好ましい。
【0062】
反応後は、濃縮して溶媒を除いてから、酢酸エチルを加えてから塩酸水等で酸性にして、有機層を分液した後濃縮することにより油状粗物を得る。これをシリカゲルカラムクロマトグラフィー等で精製することにより目的のTAAEが得られる。
<第2工程>
第2工程の加水分解法としては、通常のエステル化合物からカルボン酸化合物を得る塩基や酸の存在下で行う条件を採用できるが、塩基の存在下で行うことが好ましい。
【0063】
塩基としては、アルカリ金属やアルカリ土類金属の水酸化物等を用いることができ、具体的には、水酸化ナトリウムや水酸化カリウムが経済的で好ましい。その使用量は、TAAEに対し、4〜10モル倍が好ましく、5〜8モル倍がより好ましい。
【0064】
加水分解反応の溶媒としては、水と有機溶媒との混合系が好ましい。有機溶媒としては、例えば、メタノール,エタノール等のアルコール類や、1,4−ジオキサン等が挙げられる。それらの使用量は、水と有機溶媒共に、TAAEに対し、それぞれ1〜10質量倍が好ましく、2〜8質量倍がより好ましい。
【0065】
反応温度は、0〜200℃程度であるが、0〜150℃が好ましい。
【0066】
反応時間は、1〜30時間が好ましく、2〜25時間がより好ましい。
【0067】
反応後は、塩酸水等で酸性にしてから酢酸エチルを加え攪拌すると、結晶が析出し、これを捕集するとAECAの結晶が得られる。また、有機層を濃縮した後、得られた粗物にアセトニトリルを加えて加温溶解後、氷冷すると結晶が析出するので、これを捕集するとAECAの粗結晶が得られる。さらに純度を向上させる場合は、この粗結晶をアセトニトリルからの再結晶を繰り返して純度を上げることができる。
<第3工程>
第3工程の還元法は、二重結合を単結合に変換する種々の一般的還元法が適用できる。例えば、(1)金属および金属塩による還元、(2)金属水素化物による還元、(3)金属水素錯化合物による還元、(4)ジボランおよび置換ボランによる還元、(5)ヒドラジンによる還元、(6)ジイミド還元、(7)リン化合物による還元、(8)電解還元、(9)接触還元等を挙げることができる。
これらの中で、最も実用的な方法は接触還元方法である。本発明で採用できる接触還元法は以下の通りである。触媒金属としては、周期律表第8族のパラジウム、ルテニウム、ロジウム、白金、ニッケル、コバルト及び鉄、又は第1族の銅等が使用できる。これらの金属は単独で、又は他の元素と複合させた多元系で使用される。それらの使用形態は、各金属単身、ラネー型触媒、ケイソウ土、アルミナ、ゼオライト、炭素及びその他の担体に担持させた触媒及び錯体触媒等が挙げられる。
【0068】
具体的には、パラジウム/炭素、ルテニウム/炭素、ロジウム/炭素、白金/炭素、パラジウム/アルミナ、ルテニウム/アルミナ、ロジウム/アルミナ、白金/アルミナ、還元ニッケル、還元コバルト、ラネーニッケル、ラネーコバルト、ラネー銅、酸化銅、銅クロマト、クロロトリス(トリフェニルホスフィン)ロジウム、クロロヒドリドトリス(トリフェニルホスフィン)ルテニウム、ジクロロトリス(トリフェニルホスフィン)ルテニウム及びヒドリドカルボニルトリス(トリフェニルホスフィン)イリジウム等が挙げられる。これらの中で特に好ましいものはパラジウム/炭素及びルテニウム/炭素等である。
【0069】
触媒の使用量は、5%金属担持触媒として基質に対し0.1〜30質量%が、特には、0.5〜20質量%が好ましい。溶媒は、メタノール、エタノール及びプロパノール等に代表されるアルコール類、ジオキサン、テトラヒドロフラン及びジメトキシエタン等に代表されるエーテル類及び酢酸エチル及び酢酸プロピル等に代表されるエステル類等が使用できる。
【0070】
その使用量は、原料に対し1〜50質量倍の範囲が、特には3〜20質量倍の範囲が好ましい。水素圧は常圧から10MPa(100kg/cm2)の範囲が、特には常圧から3MPa(30kg/cm2)の範囲が好ましい。反応温度は、0〜150℃の範囲が、特には10〜100℃の範囲が好ましい。
【0071】
反応は、水素吸収量によって追跡することができ、理論水素量の吸収後サンプリングし1H−NMRで分析し確認することができる。反応後は、濾過により触媒を除いてから、濃縮後、再結晶、又はカラムクロマトグラフィー法で精製することができる。
<第4工程>
脱水法としては、(a)脂肪族カルボン酸無水物法、(b)蟻酸およびp−トルエンスルホン酸法、(c)芳香族炭化水素による共沸法等が挙げられる。
【0072】
これらの中でも、本発明では、操業上簡便であるとともに、目的物がより高収率で得られることから、(a)脂肪族カルボン酸無水物法を用いることが好ましい。
【0073】
脂肪族カルボン酸無水物としては、例えば、無水酢酸、無水プロピオン酸等が挙げられるが、経済性の点から無水酢酸が好ましい。
【0074】
脂肪族カルボン酸無水物の添加量は、原料AOCAに対して2〜30モル倍が好ましく、3〜20モル倍がより好ましい。
【0075】
上記脱水反応は、芳香族炭化水素化合物を共存させて行うことが好ましい。本工程では、反応の進行に伴って反応液が着色し、生成物の結晶も着色し易くなるが、芳香族炭化水素化合物を共存させることで、反応液の着色を軽減できる結果、生成物の着色を抑制することができる。
【0076】
芳香族炭化水素化合物としては、ベンゼン、トルエン、キシレン、エチルベンゼン、キュメン等が挙げられるが、経済性の点からトルエンが好適である。
【0077】
芳香族炭化水素化合物の添加量は、原料AOCAに対し、1〜30質量倍が好ましく、3〜20質量倍がより好ましい。
【0078】
反応温度は、通常50〜150℃程度であるが、反応完結までの時間を短縮することを考慮すると、60〜130℃が好適である。
【0079】
反応時間は、長くなると反応液の着色が強くなることから、10分間〜3時間が好ましく、15分間〜2時間がより好ましい。
【0080】
なお、脱色を目的に活性炭を存在させて反応を行うこともできる。この場合、活性炭の使用量は、原料AOCAに対し、1〜100質量%が好ましく、3〜50質量%がより好ましい。
【0081】
反応の終了は、昇温後、原料AOCAの完全溶解で判断することができる。
【0082】
反応後は、氷冷して撹拌して析出した結晶を、濾過して洗浄し、さらに乾燥して目的のAODAが得られる。
【0083】
以上述べた各工程の反応は、いずれも常圧または加圧下で行うことができ、また回分式でも連続式でもよい。
このようにして得られた側鎖置換テトラカルボン酸二無水物を用いて得られるポリアミック酸をイミド化して出来るポリイミドは、各種有機溶媒に対する溶解性に優れることが期待される。また、この側鎖置換テトラカルボン酸二無水物は、液晶配向膜の高チルト角発現が可能なポリアミック酸及びポリイミドへの誘導が期待できる。
<実施例>
以下に参考例及び実施例を挙げて、本発明を具体的に説明するが、本発明の解釈はこれらに限定されるものではないことはもちろんである。
【0084】
尚、実施例で用いた分析法は以下の通りである。
【0085】
[1] [質量分析(MASS)]
機種:AQ−Tod(JEOL) イオン化法:DART+ 測定範囲:m/z = 100〜1000
[2] [1H NMR]
機種:Varian社製NMR System 400NB(400MHz)
測定溶媒:CDCl3、DMSO−d6
標準物質:tetramethylsilane(TMS)
[3] [融点(m.p.)]
機種:微量融点測定装置(MP−S3)(ヤナコ機器開発研究所社製)
[4] [単結晶X線解析]
装置:SMART APEX2 Ultra(Bruker) X線:Cu−Kα 温度:−100℃
サンプル中から測定可能と思われる結晶を選び出して測定した。
(参考例1)
【0086】
【化19】
【0087】
攪拌羽付き3Lの四つ口反応フラスコに水2L、リン酸水素二ナトリウム8水和物を140g及びクエン酸一水和物を34g仕込み、25℃で機械攪拌下に1,3−ジエチル−アセトンジカルボキシレート127g(0.628mol)と39%グリオキザール水溶液45g(0.302mol)を滴下した。その後、25℃で50時間攪拌を継続し反応を終了させた。
続いて、フラスコ内壁に付着した橙色固形物と水溶液をデカンテーションにより分離した。
この橙色固形物が付着したフラスコ内に水100mlを加えて洗浄した。この操作を2回繰り返した。続いて酢酸エチル500gを加えて橙色固形物を溶解させた後、水100mlを加えて洗浄した。得られた有機層に再び水100mlとn−ヘプタン50mlを加え水洗を繰り返した。この有機層を減圧濃縮すると黄色油状物119gが得られた。
この黄色油状物にn−ヘプタン/酢酸エチル=1/1(v/v)90gを加えて60℃で溶解後、氷冷した。析出した結晶をろ過、n−ヘプタン/酢酸エチル=9/1(v/v)で洗浄後50℃で減圧乾燥すると肌色結晶85.2g(収率67%)が得られた。
この結晶は、単結晶X線構造解析(図1に示す)から目的のテトラエチル−3,7−ジハイドロオキシビシクロ[3.3.01,5]オクタ−2,6−ジエン−2,4,6,8−テトラカルボキシレート(THOE)であることを確認した。
(参考例2)
【0088】
【化20】
【0089】
攪拌羽付き1Lの四つ口反応フラスコにメタノール460mlと水酸化ナトリウム25.6g(0.64mol)を仕込み、60℃で機械攪拌下に溶解させた後、氷冷し5℃とした。続いて攪拌しながら1,3−ジメチル−アセトンジカルボキシレート109.2g(0.627mol)を内温4〜12℃で20分かけて滴下した。しだいに高粘度の黄色スラリー状になったので、90℃油浴で加温すると内温60℃溶解し均一溶液になった。続いて39%グリオキザール水溶液52.7g(0.354mol)を滴下開始すると発熱し、65℃で還流した。20分で滴下を終了し、更に10分間還流させた後、25℃まで冷却し5時間攪拌を継続し反応を終了させた。
続いて、生成した固形物をろ過後、200mlのメタノールで5回洗浄した後減圧乾燥するとTMHO・2Na塩の淡肌色結晶83.5g(収率64.3%)が得られた。
【0090】
この結晶79.6g(0.235mol)に水312gを加えて氷冷攪拌下10℃としたところに、35%塩酸水47gを水320gに溶解した溶液を滴下し酸性にすると、黄色スラリーが白色スラリーになった。続いてろ過後100mlの水で2回洗浄した後減圧乾燥すると固体128.7gが得られた。この固体にクロロホルム90gと水30gを加えて60℃で溶解した後クロロホルム層を分液して濃縮すると、赤色油状物55.9gが得られた。酢酸エチル100gを加えて加温溶解した後n−ヘプタンを加えてからやや濃縮するとスラリー化したので、そのまま氷冷一夜静置させた。続いてろ過後酢酸エチル/n−ヘプタン=1/3(V/V)で洗浄した後減圧乾燥すると淡肌色結晶45.4g(収率52.2%)(m.p.94〜95℃)が得られた。
この結晶は、MASS及び1H−NMRから目的のテトラメチル−3,7−ジハイドロオキシビシクロ[3.3.01,5]オクタ−2,6−ジエン−2,4,6,8−テトラカルボキシレート(TMHO)であることを確認した。
(参考例3)
【0091】
【化21】
【0092】
100ml四つ口反応フラスコに参考例2で得られたTMHO3.7g(10mmol)とメタノール30gを仕込み、−40℃に冷却攪拌下にNaBH40.822gを10分かけて分割添加した。その後一時間かけて20℃に昇温し、20℃で2時間攪拌した。
続いて、氷冷し5℃で35%塩酸水2gを水10gに溶解した溶液を滴下し酸性にした後、濃縮してメタノールを留去してから酢酸エチルを加えて有機層を濃縮するとゼリー状物4.35gが得られた。更にシリカゲルカラムクロマトグラフィーにより精製すると留分1としてゼリー状物1.54g(収率41%)と留分2として白色結晶1.12g(収率30%)(m.p.130〜133℃)が得られた。ゼリー状物は、MASS及び1H−NMRから目的のテトラメチル−3,7−ジハイドロオキシビシクロ[3.3.01,5]オクタン−2,4,6,8−テトラカルボキシレート(TMHA)であることを確認した。
又、留分2の白色結晶はX線構造解析からオールシス−テトラメチル3,7−ジハイドロオキシビシクロ[3.3.01,5]オクタン−2,4,6,8−テトラカルボキシレート(all cis−TMHA)であることを確認した。
MASS(ESI+) : 375.2([M+H]+), 343.2, 325.2, 307.2
1H NMR (CDCl3, ppm) : 2.77〜2.82(m, 4H), 2.85〜2.91(m, 4H), 3.72 (s, 12H), 4.53 (s, 2H)
【0093】
【表1】
【0094】
(参考例4)
【0095】
【化22】
【0096】
100ml四つ口反応フラスコに参考例1で得られたTHOE4.26g(10mmol)、酢酸38.3g(9質量倍)、水3.83g(0.9質量倍)及び3%Pt/カーボン(H2O 60.1wt%含)4.07gを仕込み、窒素置換後更に水素ガスを充填した風船で置換後、大気圧の水素雰囲気下で、20℃で7時間後17℃で16時間攪拌した。
反応後、ろ過により触媒を除去し、ろ液を濃縮・減圧乾燥すると黄色油状物が得られた。この黄色油状物に5%炭酸ナトリウム水溶液と酢酸エチルを加えて溶解させた後、有機層を水洗してから濃縮すると黄色油状物3.37gが得られた。
この黄色油状物をシリカゲルカラムクロマトグラフィーにより精製すると黄色油状物2.72g(収率64%)が得られた
この油状物は、MASS及び1H−NMRから目的のテトラエチル−3,7−ジヒドロキシビシクロ[3.3.01,5]オクタン−2,4,6,8−テトラカルボキシレート(TDHO)であることを確認した。
(実施例1)
【0097】
【化23】
【0098】
100ml四つ口反応フラスコに参考例3で得られたTMHA7.18g(19mmol)とN,N−ジメチルホルムアミド(DMF)57gを仕込み、氷冷攪拌下にNaH(純度55%)2.02g(42mmol)を添加した。その後24℃まで昇温し、20分間攪拌してから、再び氷冷し5℃で1−ブロモテトラデカン10.6g(38mmol)を滴下した。その後、25℃で1時間攪拌した後、40℃で4時間攪拌した。
続いて濃縮して残渣に、酢酸エチルと水を加えて氷冷下に35%塩酸8.8gを滴下し酸性にした。有機層を濃縮すると油状物13.3gが得られた。更にシリカゲルカラムクロマトグラフィーにより2回精製すると黄色油状物1.89g(収率13%)が得られた。この黄色油状物は、MASS及び1H−NMRからテトラメチル−4−テトラデシル−7−テトラデシルオキシビシクロ[3.3.01,5]オクテ−2−エン−2,4,6,8−テトラカルボキシレート(TMTE)であることを確認した。
MASS(ESI+) : 748.8([M+])
1H−NMR(CDCl3, ppm) : 0.882(t, J=6.4Hz, 6H), 1.100〜1.898(m, 50H), 2.550(t, J=10.2Hz, 1H), 2.659(t, J=10.0Hz, 1H), 3.081(t, J=10.0Hz, 1H), 3.284〜3.397(m, 2H), 3.648〜3.813(m, 13H), 4.061(t, J=9.2Hz, 1H), 6.769 (s, 1H)
(実施例2)
【0099】
【化24】
【0100】
100ml四つ口反応フラスコに実施例1で得られたTMTE1.79g(2.30mmol)、メタノール20g及び水酸化ナトリウム1.67g(41mmol)を水8gに溶解した水溶液を仕込み、90℃油浴で加温還流下に、8時間攪拌した。
続いて濃縮して残渣に、酢酸エチルと水を加えてから氷冷し、35%塩酸水溶液4gを滴下し酸性にしてから、20℃で一夜攪拌すると結晶が析出した。この結晶をろ過後酢酸エチルで洗浄した後減圧乾燥すると白色結晶1.0g(収率60%)が得られた。
この結晶は、MASS及び1H−NMRから目的の4−テトラデシル−7−テトラデシルオキシビシクロ[3.3.01,5]オクテ−2−エン−2,4,6,8−テトラカルボン酸(TECA)であることを確認した。
m.p.228−230℃
MASS(ESI−) : 691.7([M−H])−, 646.8
1H−NMR(CDCl3, ppm) : 0.815(t, J=3.4Hz, 6H), 1.197〜1.732(m, 50H), 2.372(t, J=10.0Hz, 1H), 2.452(t, J=8.8Hz, 1H), 2.808(t, J=9.2Hz, 1H), 3.284〜3.397(m, 2H), 3.478(t, J=7.4Hz, 1H), 3.891(t, J=8.8Hz, 1H), 6.518 (s, 1H), 12.430(brs, 4H)
(実施例3)
【0101】
【化25】
【0102】
200ml四つ口反応フラスコに実施例2で得られたTECA9.20g(13.2mmol)、1,4−ジオキサン136g及び5%Pd/カーボン(H2O:55.34wt%含)3.8gを仕込み、窒素置換後水素ガスを充填した風船で置換後、40℃の大気圧の水素雰囲気下で、50時間攪拌した。
反応後、ろ過により触媒を除去し、ろ液を濃縮・減圧乾燥すると白色固体9.09gが得られた。この固体にアセトニトリルを加えて加温するとスラリー化した。氷冷後ろ過・アセトニトリル洗浄してから減圧乾燥すると白色結晶7.68g(収率83.7%)が得られた。
この結晶は、MASS及び1H−NMRから目的の4−テトラデシル−7−テトラデシルオキシビシクロ[3.3.01,5]オクタン−2,4,6,8−テトラカルボン酸(TOCA)であることを確認した。
m.p.195−200℃
MASS(ESI−):693.4([M−H]−)
1H−NMR:0.835(t, J=18.8Hz, 6H), 0.951〜1.554(m, 52H), 1.681〜1.784(m, 2H), 2.010(t, J=13.6Hz, 0.5H), 2.210(t, J=9.6Hz, 0.5H), 2.317〜2.452(m, 1H), 2.473〜2.623(m, 1H), 2.749〜3.059(m, 2H), 3.820(t, J=9.6Hz, 0.5H), 4.060(t, J=8.0Hz, 0.5H), 12.290(brs, 4H)
(実施例4)
【0103】
【化26】
【0104】
100ml四つ口反応フラスコに実施例3で得られたTOCA7.67g(11.0mmol)、トルエン77g及び無水酢酸11.3g(110mmol)を仕込み、110℃油浴で15分間攪拌した。
反応後、濃縮・減圧乾燥すると淡灰色ガム7.57gが得られた。続いてこの淡灰色ガム4.12g(6.2mmol)に、酢酸エチル40g、無水酢酸2.3g(22mmol)及び活性炭2.3gを加えて85℃油浴で1時間攪拌した。更に、ろ過後ろ液を濃縮すると油状物3.46gが得られた。更に、120〜130℃で1時間減圧乾燥すると、淡黄色固体3.14gが得られた。
黄色固体は、MASS及び1H−NMRから目的の4−テトラデシル−7−テトラデシルオキシビシクロ[3.3.01,5]オクタン−2,4,6,8−テトラカルボン酸−2,4:6,8−二無水物(TODA)であることを確認した。
MASS(ESI−):657.4([M−H]−)
1H−NMR:(CDCl3, ppm) : 0.879(t, J=6.8Hz, 6H), 1.254〜1.748(m, 52H), 1.890〜2.182(m, 1H), 2.400〜2.668(m, 2H), 2.871〜3.064(m, 1H), 3.193〜3.500(m, 2H), 4.053(t, J=6.8Hz, 1H), 4.417〜4.494(m, 1H)
(実施例5)
【0105】
【化27】
【0106】
100ml四つ口反応フラスコに参考例2で得られたTMHA5.80g(15.4mmol)とN,N−ジメチルホルムアミド(DMF)60gを仕込み、氷冷攪拌下にNaH(純度55%)1.47g(34mmol)を添加した。その後24℃まで昇温し、60分間攪拌してから、再び氷冷し5℃で1−ヨードオクタン7.77g(32mmol)を滴下した。その後、25℃で1時間攪拌した後、60℃で7時間攪拌した。
続いて濃縮して残渣に、酢酸エチルと水を加えて氷冷下に濃塩酸3.4gを滴下し酸性にした。有機層を濃縮すると油状物8.0gが得られた。更にシリカゲルカラムクロマトグラフィーにより2回精製すると黄色油状物3.15g(収率34%)が得られた。この黄色油状物は、MASS及び1H−NMRからテトラメチル−4−オクチル−7−オクチルオキシビシクロ[3.3.01,5]オクテ−2−エン−2,4,6,8−テトラカルボキシレート(TMOE)であることを確認した。
MASS(ESI+) : 581.4([M+]), 549.4
1H−NMR(CDCl3, ppm) : 0.846(t, J=1.5Hz, 6H), 1.066〜1.898(m, 26H), 2.680(t, J=8.4Hz, 1H), 3.098(t, J=10.0Hz, 1H), 3.268〜3.397(m, 2H), 3.641(s, 3H), 3.692(s, 3H), 3.699(s, 3H), 3.742(s, 3H), 3.675(t, J=10.0Hz, 1H), 4.079(t, J=9.6Hz, 1H), 6.792(s, 1H)
(実施例6)
【0107】
【化28】
【0108】
100ml四つ口反応フラスコに実施例5で得られたTMOE5.10g(8.50mmol)、メタノール31g及び水酸化ナトリウム3.40g(85mmol)を水10gに溶解した水溶液を仕込み、20〜24℃で、19時間攪拌した。
続いて、濃縮して残渣に酢酸エチルと水を加えてから氷冷して、35%塩酸水溶液8.5gを滴下し酸性とした。この有機層を水洗してから濃縮すると肌色の固体5.59gが得られた。この固体に酢酸エチルを加えて加温溶解した後、やや濃縮してから氷冷すると結晶が析出した。この結晶をろ過・圧乾燥すると白色結晶1.68g(収率36.5%)が得られた。
この結晶は、MASS及び1H−NMRから4−オクチル−7−オクチルオキシビシクロ[3.3.01,5]オクテ−2−エン−2,4,6,8−テトラカルボン酸−4−メチルエステル(OECM)であることを確認した。
m.p.169−171℃
MASS(ESI+) : 539.2([M+H])+, 536.2, 521.3
MASS(ESI−) : 537.4([M−H]−),479.8
1H−NMR(CDCl3, ppm) : 0.830〜0.855(m, 6H), 1.040〜1.808(m, 26H), 2.283(t, J=10.0Hz, 1H), 2.416(t, J=10.0Hz, 1H), 2.854(t, J=9.6Hz, 1H), 3.295〜3.388(m, 2H), 3.503〜3.589(m, 1H), 3.581 (s, 3H), 3.936(t, J=8.8Hz, 1H), 6.559(d, J=2.2Hz, 1H), 12.464(brs, 3H)
(実施例7)
【0109】
【化29】
【0110】
100ml四つ口反応フラスコに実施例6で得られたOECM2.46g(4.56mmol)、メタノール25g及び水酸化ナトリウム1.10g(27mmol)を水5.5gに溶解した水溶液を仕込み、内温72℃(油浴90℃)で加温還流下に、3時間攪拌した。
続いて濃縮して残渣に、酢酸エチルと水を加えてから氷冷してから、35%塩酸水溶液4gを滴下して酸性にしてから、有機層を分液し、水洗の後濃縮・減圧乾燥すると白色結晶2.31g(収率96%)が得られた。
この結晶は、MASS及び1H−NMRから目的の4−オクチル−7−オクチルオキシビシクロ[3.3.01,5]オクテ−2−エン−2,4,6,8−テトラカルボン酸(OECA)であることを確認した。
m.p.230−240℃
MASS(ESI+):525.4([M+H]+),507.4
1H−NMR(CDCl3, ppm) : 0.830〜0.855(m, 6H), 1.040〜1.808(m, 26H), 2.283(t, J=10.0Hz, 1H), 2.416(t, J=10.0Hz, 1H), 2.854(t, J=9.6Hz, 1H), 3.295〜3.388(m, 2H), 3.503〜3.589(m, 1H), 3.581 (s, 3H), 3.936(t, J=8.8Hz, 1H), 6.559(d, J=2.2Hz, 1H), 12.464(brs, 4H)
(実施例8)
【0111】
【化30】
【0112】
100ml四つ口反応フラスコに実施例5で得られたTMOE12.8g(22mmol)、メタノール128g及び水酸化ナトリウム5.28g(132mmol)を水26gに溶解した水溶液を仕込み、内温68℃(油浴90℃)で還流させながら、2時間攪拌した。
続いて、濃縮して残渣に酢酸エチルと水を加えてから氷冷して、35%塩酸水溶液13.2gを滴下し酸性とした。この有機層を水洗してから濃縮すると暗赤色のペースト11.8gが得られた。このペーストにアセトニトリルを加えて加温溶解した後、やや濃縮してから氷冷すると結晶が析出した。この結晶をろ過後アセトニトリルで2回洗浄してから減圧乾燥すると肌色結晶2.78gが得られた。この結晶をアセトニトリルで再結晶化させて減圧乾燥すると白色結晶2.34g(収率20.3%)が得られた。
この結晶は、実施例7で得られた1H−NMRと一致したことから4−オクチル−7−オクチルオキシビシクロ[3.3.01,5]オクテ−2−エン−2,4,6,8−テトラカルボン酸(OECA)であることを確認した。
m.p.240−245℃
(実施例9)
【0113】
【化31】
【0114】
100ml四つ口反応フラスコに実施例8を繰り返して得られたOECA3.31g(4.76mmol)、1,4−ジオキサン33g及び5%Pd/カーボン(H2O 55.34wt%含)1.48gを仕込み、窒素置換後更に水素ガスを充填した風船で置換後、25℃の大気圧の水素雰囲気下で、45時間攪拌した。
反応後、ろ過により触媒を除去し、ろ液を濃縮・減圧乾燥すると白色結晶1.34g(収率58%)が得られた。
この結晶は、MASS及び1H−NMRから目的の4−オクチル−7−オクチルオキシビシクロ[3.3.01,5]オクタン−2,4,6,8−テトラカルボン酸(OOCA)であることを確認した。
m.p.255−257℃
MASS(ESI−):525.5([M-H]−)
1H−NMR:(CDCl3, ppm) : 0.833〜0.863(m,6H),1.211〜1.555(m,26H), 1.666〜1.771(m,1H),1.973〜2.090(m,1H), 2.212(t, J=9.88Hz, 1H), 2.407(t, J=9.88Hz,1H), 2.507(t, J=13.7Hz, 1H), 2.821〜2.978(m, 1H), 3.025(t, J=9.49Hz, 1H), 3.339(t, J=6.36Hz, 2H), 3.817(t, J=9.68Hz, 1H), 12.296(brs, 4H)
(実施例10)
【0115】
【化32】
【0116】
100ml四つ口反応フラスコに実施例9で得られたOOCA1.28g(2.43mmol)、トルエン12.8g及び無水酢酸2.47g(24.3mmol)を仕込み、120℃油浴で20分間攪拌した。
反応後、濃縮・減圧乾燥すると無色透明な油状物1.37gが得られた。続いてヘキサンを加えて加温してから氷冷すると2層になった。この上澄み液をデカンテーションにより除去してから同じ操作を繰り返した。残った下層の油状物を減圧乾燥すると1.20g(収率100%)の無色透明な油状物が得られた。
油状物は、MASS及び1H−NMRから目的の4−オクチル−7−オクチルオキシビシクロ[3.3.01,5]オクタン−2,4,6,8−テトラカルボン酸−2,4:6,8−二無水物(OODA)であることを確認した。
MASS(ESI+):490.4([M・]+)
1H−NMR:(CDCl3, ppm) : 0.834〜0.899(m, 6H), 1.245〜1.553(m, 26H), 1.950(t, J=10.2Hz, 1H), 2.108(t, J=12.0Hz, 1H), 2.226〜2.321(m, 2H), 2.430(d, J=12.8Hz, 1H), 2.536〜2.652(m, 1H), 2.909〜2.984(m, 1H), 3.201〜3.300(m, 1H), 3.400〜3.559(m, 1H), 3.676〜3.750(m, 1H)
(実施例11)
【0117】
【化33】
【0118】
200ml四つ口反応フラスコに参考例4で得られたTDHO9.23g(21.4mmol)とN,N−ジメチルホルムアミド(DMF)90gを仕込み、氷冷攪拌下にNaH(純度55%)2.80g(64mmol)を添加した。その後25℃まで昇温し、60分間攪拌してから、再び氷冷し15℃で1−ヨードメタン9.10g(64mmol)を滴下した。その後、25℃で40分攪拌した後、40℃で6時間攪拌した。
続いて濃縮して残渣に、酢酸エチルと水を加えて氷冷下に35%塩酸6.4gを滴下し酸性にした。有機層を濃縮すると油状物9.8gが得られた。無色の油状物が上層に浮遊したので、ヘプタンを加えて洗浄すると、油状物6.5gが得られた。更にシリカゲルカラムクロマトグラフィーにより精製すると黄色油状物5.3g(Y54%)が得られた。この黄色油状物は、MASS及び1H−NMRからテトラエチル−4−メチル−7−メトキシビシクロ[3.3.01,5]オクテ−2−エン−2,4,6,8−テトラカルボキシレート(TMME)であることを確認した。
MASS(ESI+):441.17([M+H]+, 100), 427.2 (26)
1H−NMR:(CDCl3, ppm) : 1.19〜1.42(m, 12H), 2.65〜3.10(m, 4H), 3.31〜3.40(m, 3H), 3.67〜3.80(m, 3H), 4.04〜4.37(m, 9H), 6.70(s, 1H)
(実施例12)
【0119】
【化34】
【0120】
50ml四つ口反応フラスコに実施例11で得られたTMME1.95g(4.2mmol)、エタノール13g及び水酸化ナトリウム2.55g(64mmol)を水14gに溶解した水溶液を仕込み、油浴90℃で還流させながら、21時間攪拌した。
続いて、濃縮して残渣に酢酸エチルと水を加えてから氷冷して、35%塩酸水溶液6.4gを滴下し酸性にするとスラリーになった。ろ過により得られたケーキを酢酸エチルと水で洗浄後減圧乾燥すると白色結晶1.08g(収率78%)(m.p.235−236℃)
が得られた。
また、ろ液を分液し有機層を水洗してから濃縮すると固体1.1gが得られた。この固体に酢酸エチルを加えて加温するとスラリーになった。氷冷後ろ過・酢酸エチル洗浄・減圧乾燥すると白色結晶0.26g(収率18%)(m.p.245−248℃)が得られた。
この結晶は、MASS及び1H−NMRから4−メチル−7−メトキシビシクロ[3.3.01,5]オクテ−2−エン−2,4,6,8−テトラカルボン酸(MMCA)であることを確認した。
MASS(ESI+):329.06([M+H]+, 55), 311.05 (33), 302.10 (16), 285.08 (100), 207.05 (78)
1H−NMR:(DMSO−d6, ppm) : 1.26(s, 3H), 2.49(t, J=9.2Hz, 1H), 2.57(t, J=9.6Hz, 1H), 2.84(t, J=9.6Hz, 1H), 3.25(s, 3H), 3.58(d, J1=2.0Hz, J2=8.8Hz, 1H), 3.92(t, J=8.8Hz, 1H), 6.50(s, 1H), 12.54(brs, 4H)
【特許請求の範囲】
【請求項1】
下記式[1]で表される化合物。
【化1】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表す。)
【請求項2】
前記R1が、メチル基、n−オクチル基及びn−テトラデシル基である請求項1記載の化合物。
【請求項3】
下記式[2]で表される化合物。
【化2】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表す。)
【請求項4】
前記R1が、メチル基、n−オクチル基及びn−テトラデシル基である請求項3記載の化合物。
【請求項5】
下記式[3]で表される化合物。
【化3】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表す。)
【請求項6】
前記R1が、メチル基、n−オクチル基及びn−テトラデシル基である請求項5記載の化合物。
【請求項7】
下記式[4]で表される化合物。
【化4】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表し、R2は、炭素数1〜10のアルキル基を表す。)
【請求項8】
前記R1が、メチル基、n−オクチル基及びn−テトラデシル基である請求項7記載の化合物
【請求項9】
下記式[5]で表される化合物。
【化5】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表し、R2は、炭素数1〜10のアルキル基を表す。)
【請求項10】
前記R1が、メチル基、n−オクチル基及びn−テトラデシル基である請求項9記載の化合物。
【請求項11】
下記式[6]で表される化合物。
【化6】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表し、R2は、炭素数1〜10のアルキル基を表す。)
【請求項12】
前記R1が、メチル基、n−オクチル基及びn−テトラデシル基である請求項11記載の化合物。
【請求項13】
下記式[9]で表される化合物を塩基の存在下、下記式[10]で表される置換アルキルハライドと反応して、下記式[4]で表される化合物を得、更にこれを加水分解して下記式[3]で表される化合物を得、更に、これを還元して下記式[2]で表される化合物を得た後、脱水剤により下記式[1]で表される化合物を得ることを特徴とする製造方法。
【化7】
(式中、R2は、炭素数1〜10のアルキル基を表す。)
【化8】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表し、Xはハロゲン原子を表す。)
【化9】
(式中、R1及びR2は、前記と同じ意味を表す。)
【化10】
(式中、R1は、前記と同じ意味を表す。)
【化11】
(式中、R1は、前記と同じ意味を表す。)
【化12】
(式中、R1は、前記と同じ意味を表す。)
【請求項14】
下記式[4]で表される化合物を加水分解して下記式[6]で表される化合物を得ることを特徴とする製造方法。
【化13】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表し、R2は、炭素数1〜10のアルキル基を表す。)
【化14】
(式中、R1及びR2は、前記と同じ意味を表す。)
【請求項1】
下記式[1]で表される化合物。
【化1】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表す。)
【請求項2】
前記R1が、メチル基、n−オクチル基及びn−テトラデシル基である請求項1記載の化合物。
【請求項3】
下記式[2]で表される化合物。
【化2】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表す。)
【請求項4】
前記R1が、メチル基、n−オクチル基及びn−テトラデシル基である請求項3記載の化合物。
【請求項5】
下記式[3]で表される化合物。
【化3】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表す。)
【請求項6】
前記R1が、メチル基、n−オクチル基及びn−テトラデシル基である請求項5記載の化合物。
【請求項7】
下記式[4]で表される化合物。
【化4】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表し、R2は、炭素数1〜10のアルキル基を表す。)
【請求項8】
前記R1が、メチル基、n−オクチル基及びn−テトラデシル基である請求項7記載の化合物
【請求項9】
下記式[5]で表される化合物。
【化5】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表し、R2は、炭素数1〜10のアルキル基を表す。)
【請求項10】
前記R1が、メチル基、n−オクチル基及びn−テトラデシル基である請求項9記載の化合物。
【請求項11】
下記式[6]で表される化合物。
【化6】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表し、R2は、炭素数1〜10のアルキル基を表す。)
【請求項12】
前記R1が、メチル基、n−オクチル基及びn−テトラデシル基である請求項11記載の化合物。
【請求項13】
下記式[9]で表される化合物を塩基の存在下、下記式[10]で表される置換アルキルハライドと反応して、下記式[4]で表される化合物を得、更にこれを加水分解して下記式[3]で表される化合物を得、更に、これを還元して下記式[2]で表される化合物を得た後、脱水剤により下記式[1]で表される化合物を得ることを特徴とする製造方法。
【化7】
(式中、R2は、炭素数1〜10のアルキル基を表す。)
【化8】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表し、Xはハロゲン原子を表す。)
【化9】
(式中、R1及びR2は、前記と同じ意味を表す。)
【化10】
(式中、R1は、前記と同じ意味を表す。)
【化11】
(式中、R1は、前記と同じ意味を表す。)
【化12】
(式中、R1は、前記と同じ意味を表す。)
【請求項14】
下記式[4]で表される化合物を加水分解して下記式[6]で表される化合物を得ることを特徴とする製造方法。
【化13】
(式中、R1は、炭素数1〜20のアルキル基及びシアノアルキル基を表し、R2は、炭素数1〜10のアルキル基を表す。)
【化14】
(式中、R1及びR2は、前記と同じ意味を表す。)
【図1】

【図2】
【図2】
【公開番号】特開2012−241001(P2012−241001A)
【公開日】平成24年12月10日(2012.12.10)
【国際特許分類】
【出願番号】特願2011−115974(P2011−115974)
【出願日】平成23年5月24日(2011.5.24)
【出願人】(000003986)日産化学工業株式会社 (510)
【Fターム(参考)】
【公開日】平成24年12月10日(2012.12.10)
【国際特許分類】
【出願日】平成23年5月24日(2011.5.24)
【出願人】(000003986)日産化学工業株式会社 (510)
【Fターム(参考)】
[ Back to top ]