
光分解性ヘテロ二価性架橋剤
【課題】(1)リビングラジカル重合によるポリマー伸長が可能であること;(2)固体基板表面の官能基との結合が可能であること;(3)光照射によるパターニングが可能であること;を具備する新規な架橋剤を提供することを目的とする。
【解決手段】本発明の光分解性ヘテロ二価性架橋剤は、リビングラジカル重合の開始基と、反応性基(但し、前記開始基を除く)とが光分解性基を介して連結されてなり、下記化学式1で表される点に特徴を有する。

式中、Iは開始基を表し、Rは反応性基を表し、Dは光分解性基を表し、S1b−S1a及びS2a−S2bはスペーサーを表し、n及びmはそれぞれ独立して0又は1である。
【解決手段】本発明の光分解性ヘテロ二価性架橋剤は、リビングラジカル重合の開始基と、反応性基(但し、前記開始基を除く)とが光分解性基を介して連結されてなり、下記化学式1で表される点に特徴を有する。
式中、Iは開始基を表し、Rは反応性基を表し、Dは光分解性基を表し、S1b−S1a及びS2a−S2bはスペーサーを表し、n及びmはそれぞれ独立して0又は1である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は光分解性ヘテロ二価性架橋剤に関する。
【背景技術】
【0002】
固体基板上の表面化学を制御することは、材料化学の分野において根本的に重要な問題である。例えば、近年、生体膜表面で起こる高度な分子識別機構をセンシングや分離などの技術に応用するために、固体基板上に生体膜を模擬した界面を自在に形成する手法の確立が求められており、この際、固体基板上の表面化学をいかに制御するかが課題となっている。
【0003】
固体基板上の表面化学を制御する方法としては、当該基板の表面を自己組織化単分子膜(Self−Assembled Monolayer:SAM)、ポリマー薄膜、ポリマーブラシなどで修飾する方法がよく知られている。SAMは、分子配向性に優れ、調製が容易なことから広く研究されている。また、ポリマーブラシは、SAMと類似の特性を示すだけでなく、表面被覆率、膜厚、組成、官能基密度、安定性などの点でSAMより優れた特性を示しうる。しかしながら、SAMと比べて調製法がより困難であるという欠点も有する。
【0004】
ポリマーブラシの化学的調製法は、予め合成したポリマーの末端基を基板表面の官能基と反応させることにより基板表面にポリマーを固定させる「grafting to」法と、基板表面に化学的に固定された重合開始基からの重合反応によってポリマーを伸長させる「grafting from」法の二つに大別される。前者の方法では、基板表面へのポリマーの固定化反応が進むにつれ、既に基板表面に固定されたポリマーが障害となって新たなポリマーが基板表面の官能基に到達しにくくなる。よって、基板表面に高濃度の(多くの)ポリマーを固定することが困難であるという問題点を有する。一方、後者の方法では、基板表面に固定された重合開始基から順次モノマーが重合するために、前者の方法で生じるような立体障害の問題が起こりにくく、高濃度のポリマーを固定化することが可能である。
【0005】
当該「grafting from」法に用いる重合方法として、リビングラジカル重合が注目されている。リビングラジカル重合法は、従来のラジカル重合では実現困難であった分子量と分子量分布を制御できる方法であり、適用できるモノマーも広範囲である。
【0006】
近年、このようなリビングラジカル重合を用いるポリマーブラシの調製方法が数多く報告されている。また、リビングラジカル重合を用いたポリマーの伸長により、従来では得られなかった濃厚ポリマーブラシが膜厚を制御した薄膜として得られるようになり、その基礎的研究及び応用研究が盛んに行われている。応用研究の例としては、刺激応答性表面、細胞接着性表面、タンパク質の結合支持体、クロマトグラフィーの支持体、抗バクテリアコート剤、低摩擦表面に関する研究が挙げられる(非特許文献1及び非特許文献2を参照)。
【0007】
さらに、ポリマーブラシのパターニングについての研究もいくつかなされている。パターニングの方法としては、半導体の加工に用いるフォトレジスト及び用いた一般的なフォトリソグラフィー、遠紫外リソグラフィー、マイクロコンタクトプリンティングなどが報告されている(非特許文献1及び非特許文献2を参照)。
【0008】
本発明者は、自己組織化単分子膜の出発原料であるシランカップリング剤や、チオール又はジスルフィド化合物に光切断可能な2−ニトロベンジル誘導体を導入した化合物の合成とそれを用いた感光性SAMの開発に従事してきた。その中で、基板表面と反応するシリル基;アミンと反応する活性カルボナート基;及びチオールと反応するマレイミド基;の3種の反応性基のうちの、2種の反応性基が、光切断可能な2−ニトロベンジル誘導体で連結された光分解性ヘテロ二価性架橋剤を提案している(特許文献1を参照)。当該光分解性ヘテロ二価性架橋剤を用いると、光分解性基で連結されたアミンやチオールと反応するSAMを調製したり、光分解性ブロック共重合体の合成に応用したりすることができる。
【0009】
また、活性カルボナートやマレイミドを末端に持つ光分解性基で連結したジスルフィドを用いると、金や銀などの基板上あるいはそれらの微粒子上に、アミンやチオールと反応するSAMを形成することが可能である(特許文献2を参照)。
【0010】
さらに、最近、ポリエチレングリコールとリビングラジカル重合の開始基とを間に光分解性基を有する化合物の合成が報告されている。そして、当該化合物の開始基にモノマーであるスチレンを反応させ、ポリエチレングリコールとポリスチレンとが光分解性基を介して連結されてなるブロック共重合体を合成している(非特許文献3を参照)。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】特開2007−186471号公報
【特許文献2】特開2007−291005号公報
【非特許文献】
【0012】
【非特許文献1】Wageesha Senaratne,Luisa Andruzzi,Christopher K. Ober Biomacromolecules,2005,6,2427−2448
【非特許文献2】Raphael Barbey,Laurent Lavanant,Dusko Paripovic,Nicolas Schuwer,Caroline Sugnaux,Stefeno Tugulu,Harm−Anton Klok,Chem. Rev.,2009,109,5437−5527
【非特許文献3】Minhyuck Kang,Bongjin Moon,Macromolecules,2009,42,455−458
【発明の概要】
【発明が解決しようとする課題】
【0013】
固体基板上に自在にポリマーブラシを形成するためには、(1)リビングラジカル重合によるポリマー伸長が可能であること;(2)固体基板表面の官能基との結合が可能であること;(3)光照射によるパターニングが可能であること;を具備する架橋剤を固体基板表面に固定化させることが求められる。しかしながら、このような機能を全て満たす架橋剤の合成はこれまでのところ達成されていない。
【0014】
そこで、本発明の目的は、上記(1)〜(3)を具備する新規な架橋剤を提供することを目的とする。
【課題を解決するための手段】
【0015】
本発明の光分解性ヘテロ二価性架橋剤は、リビングラジカル重合の開始基と、反応性基(但し、前記開始基を除く)とが光分解性基を介して連結されてなり、下記化学式1で表される点に特徴を有する。
【0016】
【化1】
【0017】
式中、Iは開始基を表し、Rは反応性基を表し、Dは光分解性基を表し、S1b−S1a及びS2a−S2bはスペーサーを表し、n及びmはそれぞれ独立して0又は1である。
【発明の効果】
【0018】
本発明の光分解性ヘテロ二価性架橋剤は、リビングラジカル重合の開始基を有するため、当該開始基からポリマーの伸長を行うことができる。また、反応性基を有するために、固体基板表面の官能基との結合が可能である。さらに、光分解性基を有するために、光照射によるパターニングが可能である。
【図面の簡単な説明】
【0019】
【図1】チオールカップリング剤(5a)の光照射による1次反応のUVスペクトル変化を表すグラフである。
【図2】チオールカップリング剤(5a)の光照射による2次反応のUVスペクトル変化を表すグラフである。
【図3】シランカップリング剤(3a)の光照射によるUVスペクトル変化を表すグラフである。
【図4】シランカップリング剤(3b)の光照射(0秒)のUVスペクトルを表すグラフである。
【図5】シランカップリング剤(3b)の光照射(0〜280秒)によるUVスペクトル変化を表すグラフである。
【図6】シランカップリング剤(3b)について、ニトロ基由来の吸収のln(Ct/C0)を時間(秒)に対してプロットしたグラフである。
【図7】シランカップリング剤(3b)について、ニトロソ基由来の吸収のln(Ct/C0)を時間(秒)に対してプロットしたグラフである。
【図8】シランカップリング剤(3b)について、ニトロ基由来の吸収のln(Ct/C0)を時間(秒)に対してプロットしたグラフである。
【図9】シランカップリング剤(3b)について、ニトロソ基由来の吸収のln(Ct/C0)を時間(秒)に対してプロットしたグラフである。
【図10】シランカップリング剤(3a)について、ニトロ基由来の吸収のln(Ct/C0)を時間(秒)に対してプロットしたグラフである。
【図11】シランカップリング剤(3a)について、ニトロソ基由来の吸収のln(Ct/C0)を時間(秒)に対してプロットしたグラフである。
【図12】基板1のXPS wideスキャンスペクトルを表すグラフである。
【図13】基板2のXPS wideスキャンスペクトルを表すグラフである。
【図14】基板1のN1s XPSスペクトルを表すグラフである。
【図15】基板2のN1s XPSスペクトルを表すグラフである。
【図16】基板1のBr3d(5/2) XPSスペクトルを表すグラフである。
【図17】基板2のBr3d(5/2) XPSスペクトルを表すグラフである。
【図18】基板1のC1s XPSスペクトルを表すグラフである。
【図19】基板2のC1s XPSスペクトルを表すグラフである。
【図20】シランカップリング剤(3b)について、ln(1−at)を時間(秒)に対してプロットしたグラフである。
【図21】シランカップリング剤(3a)について、ln(1−at)を時間(秒)に対してプロットしたグラフである。
【図22】基板3のXPS wideスキャンスペクトルを表すグラフである。
【図23】基板4のXPS wideスキャンスペクトルを表すグラフである。
【図24】基板3のN1s XPSスペクトルを表すグラフである。
【図25】基板4のN1s XPSスペクトルを表すグラフである。
【図26】基板3のC1s XPSスペクトルを表すグラフである。
【図27】基板4のC1s XPSスペクトルを表すグラフである。
【図28】基板3のBr3d(5/2) XPSスペクトルを表すグラフである。
【図29】基板4のBr3d(5/2) XPSスペクトルを表すグラフである。
【発明を実施するための形態】
【0020】
以下、本発明の好ましい形態を説明するが、本発明の技術的範囲は特許請求の範囲の記載によって定められるべきものであり、以下の形態のみに制限されない。
【0021】
本形態は、リビングラジカル重合の開始基と、反応性基(但し、前記開始基を除く)とが光分解性基を介して連結されてなり、下記化学式1で表される光分解性ヘテロ二価性架橋剤に関する。
【0022】
【化2】
【0023】
式中、Iは開始基を表し、Rは反応性基を表し、Dは光分解性基を表し、S1b−S1a及びS2a−S2bはスペーサーを表し、n及びmはそれぞれ独立して0又は1である。
【0024】
[開始基]
本形態の光分解性ヘテロ二価性架橋剤は、リビングラジカル重合の開始基を有することを特徴とする。リビングラジカル重合は重合過程において、開始反応と成長反応のみからなり、連鎖移動反応及び停止反応などの成長末端を失活させる副反応を伴わない。したがって、重合度は一定のペースで増大し、長さの揃ったポリマーが得られるという特徴を有する。また、モノマーを追加すれば重合反応が再開するため、ブロック重合やポリマーアロイに適する。
【0025】
リビングラジカル重合の開始基は、リビングラジカル重合の成長末端となる構造を有するものであれば特に制限はないが、好ましくは、電子移動ラジカル重合(Atom Transfer Radical Polymerization:ATRP)又はニトロキシドを介したラジカル重合(Nitroxide−mediated radical
polymerization:NMP)の開始基であることが好ましい。
【0026】
具体的に、ATRPの開始基としては、下記化学式3、化学式4、及び化学式5で表される構造を有する開始基が挙げられる。
【0027】
【化3】
【0028】
式中、R2は水素原子又は炭素原子数1〜8のアルキル基を表す。
【0029】
【化4】
【0030】
式中、R4及びR5はそれぞれ独立して水素原子又は炭素原子数1〜8のアルキル基を表し、Xは臭素原子又は塩素原子を表す。
【0031】
【化5】
【0032】
また、NMPの開始剤としては、下記化学式6で表される構造を有する開始基が挙げられる。
【0033】
【化6】
【0034】
上記R2、R4、又はR5で表される炭素原子数1〜8のアルキル基としては、具体的には、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、n−ペンチル基、イソペンチル基、ネオペンチル基、1,2−ジメチルプロピル基、n−ヘキシル基、シクロヘキシル基、1,3−ジメチルブチル基、1−イソプロピルプロピル基、1,2−ジメチルブチル基、n−ヘプチル基、1,4−ジメチルペンチル基、2−メチル−1−イソプロピルプロピル基、1−エチル−3−メチルブチル基、n−オクチル基、2−エチルヘキシル基などが挙げられる。このうち、メチル基であることが好ましい。
【0035】
[反応性基]
反応性基は、本形態の光分解性ヘテロ二価性架橋剤を固体基板に固定化させる場合、固体基板表面の官能基との結合部位となりうる。また、本形態の光分解性ヘテロ二価性架橋剤を用いてブロック共重合体を合成する場合には、ポリマーとの結合部位となりうる。なお、本形態の反応性基の概念には、上述のリビングラジカル重合の開始基となりうる基は含まない。
【0036】
反応性基は、固体基板表面の官能基やポリマーの末端の官能基と結合することができるものであれば特に制限はないが、具体的には、ヒドロキシ基、エチニル基、アジド基、活性エステル基、マレイミド基、加水分解性シリル基(例えば、トリメトキシシリル基、トリエトキシシリル基、トリクロロシリル基、ジエトキシシリル基、ジメトキシシリル基、ジモノクロロシリル基、モノエトキシシリル基、モノメトキシシリル基、モノクロロシリル基など)、アミノ基、カルボキシル基、チオール基、ジスルフィド基、及びスルホ基からなる群から選択される少なくとも1種であることが好ましい。
【0037】
[光分解性基]
光分解性基は、光の照射により開裂する性質を有する官能基であり、様々な構造を有するものが既に知られている。本形態の架橋剤に使用される光分解性基は特に制限はないが、下記化学式2で示される2−ニトロベンジル誘導体骨格を有する二価の光分解性官能基が好ましく用いられる。
【0038】
【化7】
【0039】
式中、R1は水素原子、炭素原子数1〜8のアルキル基、又は置換されたもしくは非置換のフェニル基を表し、R3は水素原子又は炭素原子数1〜8のアルコキシ基を表す。
【0040】
上記R1が炭素原子数1〜8のアルキル基である場合の、当該アルキル基としては、例えば、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、n−ペンチル基、イソペンチル基、ネオペンチル基、1,2−ジメチルプロピル基、n−ヘキシル基、シクロヘキシル基、1,3−ジメチルブチル基、1−イソプロピルプロピル基、1,2−ジメチルブチル基、n−ヘプチル基、1,4−ジメチルペンチル基、2−メチル−1−イソプロピルプロピル基、1−エチル−3−メチルブチル基、n−オクチル基、2−エチルヘキシル基などが挙げられる。
【0041】
上記R1が置換されたもしくは非置換のフェニル基である場合に、当該フェニル基に場合によって存在する置換基としては、例えば、ハロゲン原子、アシル基、アルキル基、フェニル基、アルコキシル基、ハロゲン化アルキル基、ハロゲン化アルコキシル基、ニトロ基、アミノ基、アルキルアミノ基、アルキルカルボニルアミノ基、アリールアミノ基、アリールカルボニルアミノ基、カルボニル基、アルコキシカルボニル基、アルキルアミノカルボニル基、アルコキシスルホニル基、アルキルチオ基、カルバモイル基、アリールオキシカルボニル基、オキシアルキルエーテル基、シアノ基等が例示できるが、これらに限定されるものではない。これらの置換基は、フェニル基に1〜5個置換可能であり、これらの置換基の種類も、複数個置換する場合には同種もしくは異種のいずれであってもよい。上記置換基の具体的な例を以下に示す。
【0042】
上記置換基のうち、ハロゲン原子とは、フッ素原子、塩素原子、臭素原子及びヨウ素原子である。
【0043】
上記置換基のうち、アシル基としては、アセチル基、エチルカルボニル基、プロピルカルボニル基、ブチルカルボニル基、ペンチルカルボニル基、ヘキシルカルボニル基、ベンゾイル基、p−tert−ブチルベンゾイル基等が挙げられる。
【0044】
上記置換基のうち、アルキル基とは、炭素原子数1〜8の直鎖、分岐鎖又は環状のアルキル基である。具体的には、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、n−ペンチル基、イソペンチル基、ネオペンチル基、1,2−ジメチルプロピル基、n−ヘキシル基、シクロヘキシル基、1,3−ジメチルブチル基、1−イソプロピルプロピル基、1,2−ジメチルブチル基、n−ヘプチル基、1,4−ジメチルペンチル基、2−メチル−1−イソプロピルプロピル基、1−エチル−3−メチルブチル基、n−オクチル基、2−エチルヘキシル基等が挙げられる。
【0045】
上記置換基のうち、アルコキシ基は、炭素原子数1〜8の直鎖、分岐鎖又は環状のアルコキシ基である。具体的には、メトキシ基、エトキシ基、n−プロポキシ基、イソプロポキシ基、n−ブトキシ基、イソブトキシ基、sec−ブトキシ基、tert−ブトキシ基、n−ペンチルオキシ基、イソペンチルオキシ基、ネオペンチルオキシ基、1,2−ジメチル−プロポキシ基、n−ヘキシルオキシ基、シクロヘキシルオキシ基、1,3−ジメチルブトキシ基、1−イソプロピルプロポキシ基等が挙げられる。
【0046】
上記置換基のうち、ハロゲン化アルキル基とは、炭素原子数1〜8の直鎖、分岐鎖又は環状のアルキル基の一部がハロゲン化されたものである。具体的には、クロロメチル基、ブロモメチル基、トリフルオロメチル基、クロロエチル基、2,2,2−トリクロロエチル基、ブロモエチル基、クロロプロピル基、ブロモプロピル基等が挙げられる。
【0047】
上記置換基のうち、ハロゲン化アルコキシル基とは、炭素原子数1〜8の直鎖、分岐鎖又は環状のアルコキシル基の一部がハロゲン化されたものである。具体的には、クロロメトキシ基、ブロモメトキシ基、トリフルオロメトキシ基、クロロエトキシ基、2,2,2−トリクロロエトキシ基、ブロモエトキシ基、クロロプロポキシ基、ブロモプロポキシ基等が挙げられる。
【0048】
上記置換基のうち、アルキルアミノ基とは、炭素原子数1〜8のアルキル部位を有するアルキルアミノ基である。具体的には、メチルアミノ基、エチルアミノ基、n−プロピルアミノ基、n−ブチルアミノ基、sec−ブチルアミノ基、n−ペンチルアミノ基、n−ヘキシルアミノ基、n−ヘプチルアミノ基、n−オクチルアミノ基、2−エチルヘキシルアミノ基等が挙げられる。
【0049】
上記置換基のうち、アルコキシカルボニル基とは、アルコキシル基のアルキル基部分にヘテロ原子を含んでもよい炭素原子数1〜8のアルコキシカルボニル、又はヘテロ原子を含んでもよい炭素原子数3〜8の環状アルコキシカルボニルを示す。具体的には、メトキシカルボニル基、エトキシカルボニル基、n−プロポキシカルボニル基、イソプロポキシカルボニル基、n−ブトキシカルボニル基、イソブトキシカルボニル基、sec−ブトキシカルボニル基、tert−ブトキシカルボニル基等が挙げられる。
【0050】
上記R3が炭素原子数1〜8のアルコキシ基である場合の、当該アルコキシ基としては、メトキシ基、エトキシ基、n−プロポキシ基、イソプロポキシ基、n−ブトキシ基、イソブトキシ基、sec−ブトキシ基、tert−ブトキシ基、n−ペンチルオキシ基、イソペンチルオキシ基、ネオペンチルオキシ基、1,2−ジメチル−プロポキシ基、n−ヘキシルオキシ基、シクロヘキシルオキシ基、1,3−ジメチルブトキシ基、1−イソプロピルプロポキシ基等が挙げられる。
【0051】
上記化学式2で示す光分解性基のうち、R1は水素原子またはメチル基であることが好ましく、R3は水素原子またはメトキシ基であることが好ましい。
【0052】
上記化学式2に示す光分解性基は、光照射により、ベンゼン環の1位に結合する炭素原子(ベンジル位の炭素原子)と、その隣の酸素原子、窒素原子、又は硫黄原子(図示せず)との間の結合が開裂する。当該酸素原子、窒素原子、又は硫黄原子は、上記反応性基に含まれる酸素原子、窒素原子、又は硫黄原子であってもよいし、光分解性基と反応性基の間に介在するスペーサーに含まれる酸素原子、窒素原子、又は硫黄原子であってもよい。上記のような二価の光分解性基の一方にリビングラジカル重合の開始基、他方に反応性基を導入することによって、本形態の光分解性ヘテロ二価性架橋剤を合成することができる。この際、光分解性ヘテロ二価性架橋剤は、光照射により、反応性基と、光分解性基との間が開裂するものであってもよいし、開始基と光分解性基との間が開裂するものであってもよい。本明細書では前者の配置を有する光分解性ヘテロ二価性架橋剤について主に説明するが、後者の配置であっても光照射によるパターニングが可能なことはいうまでもない。
【0053】
[スペーサー]
本形態の光分解性ヘテロ二価性架橋剤は、上述の光分解性基と、開始基および/または反応性基との間に、さらにスペーサーを有してもよい。スペーサーの構造は、特に制限はないが、好ましい形態としては、上述の化学式1におけるS1aが、−O−、−C(O)O−、−OC(O)O−、−NH−、−C(O)NH−、−OC(O)NH−、−S−、−C(O)S−、−OC(O)S−からなる群から選択される少なくとも1種であり;S2aが、−O−であり;S1b及びS2bが、それぞれ独立して、アミド結合、イミド結合、エーテル結合、エステル結合、チオエーテル結合、もしくはスルホンアミド結合を有してもよい、炭素原子数1〜20、好ましくは炭素原子数1〜8のアルキレン基からなる群から選択される少なくとも1種である。
【0054】
また、S1b及びS2bに含まれるアルキレン基の具体例としては、メチレン基、エチレン基、プロピレン基、トリメチレン基、テトラメチレン基、ペンタメチレン基、2,2−ジメチルトリメチレン基、ヘキサメチレン基、トリメチルヘキサメチレン基、オクタメチレン基、及びデカメチレン基などが挙げられる。
【0055】
下記化学式8に、本形態の光分解性ヘテロ二価性架橋剤の一例を示す。
【0056】
【化8】
【0057】
上記化学式8では、光分解性基である2−ニトロベンジル誘導体構造を有する官能基の4位側にATRPの開始基が連結され、さらに1位側に反応性基が連結されてなる構造を有する種々の光分解性ヘテロ二価性架橋剤が示されている。化学式8に示すように、反応性基をヒドロキシ基から、活性カルボナート基(スクシンイミジル基)へと変換し、さらに加水分解性シリル基(トリメトキシシリル基)やチオール基やジスルフィド基へと変換することができる。活性カルボナート基はアミノ基と反応するため、化学式8に示すようにアミノ基含有ポリマーを活性カルボナート基と反応させることにより、容易にポリマーを導入することができる。また、加水分解性シリル基はSi基板への固定化に寄与しうる。また、ジスルフィド基は、Au基板への固定化に寄与しうる。なお、チオール基、ジスルフィド基を還元することによりチオール基に変換することができ、また、チオール基又はジスルフィド基を酸化するとスルホ基に変換することができる。なお、これらの反応性基の変換は、従来公知の合成技術を適宜参照することにより容易に行うことができる。
【0058】
また、下記化学式9に、本形態の光分解性ヘテロ二価性架橋剤の他の一例を示す。
【0059】
【化9】
【0060】
上記化学式9に示すように、反応性基は、ヒドロキシ基から、活性カルボナート基、マレイミド基、カルボキシル基、アジド基、ビニル基(アルケン)、又はエチニル基(アルキン)などに変換することができる。なお、これらの反応性基の変換は、従来公知の合成技術を適宜参照することにより容易に行うことができる。
【0061】
本形態の光分解性ヘテロ二価性架橋剤の製造方法としては、後述の実施例で示すような合成ルートが一例として挙げられる。すなわち、光分解性基にスペーサーを導入した後、リビングラジカル重合の開始基を導入し、その後反応性基を導入又は変換する合成方法である。しかしながら、当該合成方法はあくまでも一例に過ぎず、他の合成ルートによってっても本形態の光分解性ヘテロ二価性架橋剤を合成することは可能である。
【0062】
本形態の光分解性ヘテロ二価性架橋剤は、例えば、固体基板上に固定させることにより容易にSAMを形成することができる。すなわち、本形態の光分解性ヘテロ二価性架橋剤の反応性基と基板表面の官能基とを結合させることにより、ポリマーブラシ形成用基板として使用することができる。例えば、上述のように、反応性基を加水分解性シリル基とした光分解性ヘテロ二価性架橋剤(以下、シランカップリング剤とも称する)は、Si基板表面に固定することができる。また、反応性基をチオール基又はジスルフィド基とした場合は、金(Au)基板表面に固定することができる。これらのSAMは細胞パターニングや有機薄膜トランジスタなどへの応用が期待される。
【0063】
また、SAMを形成した後、固体基板に固定された光分解性ヘテロ二価性架橋剤の開始剤からリビングラジカル重合によってポリマーを伸長させ、ポリマーブラシを作製することができる。
【0064】
なお、本形態の光分解性ヘテロ二価性架橋剤は、ポリマーブラシ以外の用途にも好適に使用されうる。例えば、反応性基にポリマーを導入し、開始基からリビングラジカル重合によってポリマーを伸長させることにより、2種のポリマーが光分解性基で連結したブロック共重合体を得ることもできる。このようなブロック共重合体は、光分解性ポリマーソーム、ナノポーラス材料などへの応用が期待される。
【実施例】
【0065】
[実施例1]光分解性ヘテロ二価性架橋剤の合成
測定機器は以下のものを使用した。
【0066】
【表1】
【0067】
GPCは以下の測定条件で行った。
【0068】
【表2】
【0069】
<1−1>1−(4−(3−(2−ブロモプロパノイルオキシ)プロピルオキシ)−5−メトキシ−2−ニトロフェニル)−エチル N−(11−メルカプトウンデシル)カルバメート(5a)の合成
【0070】
【化10】
【0071】
<1−1−1>3−(4−アセチル−2−メトキシ−5−ニトロフェノキシ)プロパン−1−オールの合成
【0072】
【化11】
【0073】
窒素雰囲気下、50mL二口ナスフラスコに4−ヒドロキシ−5−メトキシ−2−ニトロアセトフェノン1.57g(7.43mmol)、60%水素化ナトリウム0.310g(7.73mmol)、脱水DMF16mLを加え、室温で30分間撹拌した。その後3−ブロモプロパン−1−オール1.03g(7.43mmol)、DMF4mLを加え、80℃で21時間撹拌した。室温まで冷却後、濃縮、水200mLを加え、酢酸エチルで抽出(200mL×4)、洗浄(飽和食塩水100mL×2)、無水硫酸マグネシウムで乾燥、濃縮し、カラムクロマトグラフィー(ヘキサン:酢酸エチル=2:3)、真空乾燥を行い、茶白色固体を得た。
【0074】
【表3】
【0075】
<1−1−2>3−(4−(1−ヒドロキシエチル)−2−メトキシ−5−ニトロフェノキシ)プロパン−1−オールの合成
【0076】
【化12】
【0077】
氷浴上において100mLナスフラスコに3−(4−アセチル−2−メトキシ−5−ニトロフェノキシ)−プロパン−1−オール0.684g(2.54mmol)、メタノール7mL、THF5mL、徐々にNaBH40.194g(5.13mmol)を加え、室温で1時間撹拌した。濃縮、水50mLを加え、抽出(クロロホルム100mL×3)、洗浄(飽和食塩水100mL×2)、無水硫酸マグネシウムで乾燥、濃縮、真空乾燥し、黄色固体を得た。
【0078】
【表4】
【0079】
<1−1−3>3−(4−(1−ヒドロキシエチル)−2−メトキシ−5−ニトロフェノキシ)プロピル 2−ブロモプロピオナート(1a)の合成
【0080】
【化13】
【0081】
50mL二口ナスフラスコに3−(4−(1−ヒドロキシエチル)−2−メトキシ−5−ニトロフェノキシ)−プロパノール0.199g(0.734mmol)、トリエチルアミン0.0743g(0.734mmol)、2‐ブロモプロピオン酸ブロミド0.160g(0.734mmol)、脱水クロロホルム7mLを加え、窒素雰囲気下とし、室温で20時間撹拌した。濃縮、カラムクロマトグラフィー(酢酸エチルのみ)、濃縮、真空乾燥し、黄色固体を得た。
【0082】
【表5】
【0083】
<1−1−4>3−(2−メトキシ−5−ニトロ−4−(1−(N−スクシンイミジルオキシカルボニルオキシ)エチル)−フェノキシ)プロピル 2−ブロモプロピオナート(2a)の合成
【0084】
【化14】
【0085】
50mL二口ナスフラスコに3−(4−(1−ヒドロキシエチル)−2−メトキシ−5−ニトロフェノキシ)−プロピル 2−ブロモプロピオナート0.361g(0.889mmol)、トリエチルアミン0.275g(2.72mmol)、ジ(N−スクシンイミジル)カルボナート0.804g(3.14mmol)、脱水アセトニトリル7mLを加え、窒素雰囲気下とし、室温で20時間撹拌した。濃縮、カラムクロマトグラフィー(酢酸エチルのみ)、濃縮、真空乾燥し、淡黄色固体を得た。
【0086】
【表6】
【0087】
<1−1−5>11−アミノウンデカン−1−チオールの合成
【0088】
【化15】
【0089】
100mLナスフラスコにN−(11−アセチルチオウンデシル)フタルイミド0.556g(1.48mmol)、ヒドラジン一水和物0.231g(4.61mmol)、2−プロパノール20mLを加え、85℃で5時間撹拌し、濃縮、洗浄(クロロホルム)、メンブランろ紙を用いてろ過、H2Oを加えて抽出(クロロホルム50mL×4)、洗浄(飽和食塩水50mL×4)、乾燥、ろ過、カラムクロマトグラフィー(クロロホルムのみ→クロロホルム:メタノール=20:1)、濃縮、真空乾燥し、白色固体を得た。
【0090】
【表7】
【0091】
<1−1−6>1−(4−(3−(2−ブロモプロパノイルオキシ)プロピルオキシ)−5−メトキシ−2−ニトロフェニル)−エチル N−(11−メルカプトウンデシル)カルバメート(5a)の合成
【0092】
【化16】
【0093】
50mL二口ナスフラスコに11−アミノウンデカン−1−チオール0.0451g(0.222mmol)、トリエチルアミン0.0561g(0.554mmol)、3−(2−メトキシ−5−ニトロ−4−(1−N−スクシンイミジルオキシカルボニルオシキエチル)フェノキシ)プロピル 2−ブロモプロピオナート0.237g(0.433mmol)、脱水THF6mLを加え、窒素雰囲気下とし、室温で20時間撹拌した。濃縮、カラムクロマトグラフィー(酢酸エチルのみ)、濃縮、真空乾燥し、黄色粘体を得た。
【0094】
【表8】
【0095】
<1−2>1−(4−(3−(2−ブロモプロパノイルオキシ)プロピルオキシ)−5−メトキシ−2−ニトロフェニル)エチル N−(3−(トリメトキシシリル)プロピル)カルバメート(3a)の合成
<1−2−1>1−(4−(3−(2−ブロモプロパノイルオキシ)プロピルオキシ)−5−メトキシ−2−ニトロフェニル)エチル N−((トリメトキシシリル)プロピル)カルバメート(3a)の合成
【0096】
【化17】
【0097】
窒素雰囲気下で50mL二口ナスフラスコに<1−1−4>で合成した3−(2−メトキシ−5−ニトロ−4−(1−(N−スクシンイミジルオキシカルボニルオキシ)エチル)−フェノキシ)−プロピル 2−ブロモプロピオナート(2a)0.103g(0.189mmol)、無水THF18mL、3−(トリメトキシシリル)プロピルアミン0.104g(0.578mmol)、トリエチルアミン0.168g(1.85mmol)を加え20時間攪拌した。濃縮、洗浄(酢酸エチル)、吸引ろ過、ろ液を濃縮、真空乾燥し、黄色固体0.0835g(粗精製物)を得た。
【0098】
<1−2−2>1−(4−(3−(2−ブロモプロパノイルオキシ)プロピルオキシ)−5−メトキシ−2−ニトロフェニル)エチル N−(3−(トリメトキシシリル)プロピル)カルバメート(3a)の合成
【0099】
【化18】
【0100】
窒素雰囲気下、20mL二口ナスフラスコに<1−1−3>で合成した3−(4−(1−ヒドロキシエチル)−2−メトキシ−5−ニトロフェノキシ)プロピル 2−ブロモプロピオナート(1a)0.151g(0.373mmol)、無水THF0.5mL、パスツールピペットで3−(イソシアナトプロピル)トリメトキシシラン11滴(1滴 0.0121g)、パスツールピペットでジラウリン酸ジブチルスズ(DBTL)2滴を加え、室温で8時間撹拌した。濃縮、真空乾燥、カラムクロマトグラフィー(ジクロロメタンのみ×2[NH2シリカゲル使用])、濃縮、真空乾燥、中圧カラムクロマトグラフィー(ヘキサン:酢酸エチル:テトラメトキシシラン=1:1:0.02[従来シリカゲル使用])、真空乾燥を行い、黄色液体0.0409(0.0669mmol、収率:18%)を得た。
【0101】
【表9】
【0102】
<1−3>1−(4−(3−(2−ブロモ−2−メチルプロパノイルオキシ)プロピルオキシ)−5−メトキシ−2−ニトロフェニル)エチル N−(3−(トリメトキシシリル)プロピル)カルバメート(3b)の合成
<1−3−1>3−(4−(1−ヒドロキシエチル)−2−メトキシ−5−ニトロフェノキシ)プロピル 2−ブロモ−2−メチルプロピオナート(1b)の合成
【0103】
【化19】
【0104】
50mL二口ナスフラスコに<1−1−2>で合成した3−(4−(1−ヒドロキシエチル)−2−メトキシ−5−ニトロフェノキシ)プロパン−1−オール0.5002g(1.8439mmol)を入れ、1時間真空乾燥した。これに窒素雰囲気下で、脱水クロロホルム25mLを加え、トリエチルアミン0.1869g(1.8439mmol)を脱水クロロホルム1.5mLに溶かした溶液、2−ブロモイソ酪酸ブロミド0.5090g(2.2127mmol)を脱水クロロホルム1.5mLに溶かした溶液をパスツールピペットでゆっくり加え、室温で24時間撹拌した。これを濃縮し、H2O100mL、1N HCl0.5mLを加え、抽出(クロロホルム100mL×3)、無水硫酸マグネシウムを加え乾燥させ、ろ過、濃縮し、オープンカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:2)、濃縮、真空乾燥を行い、黄色固体を得た。
【0105】
【表10】
【0106】
<1−3−2>3−(2−メトキシ−5−ニトロ−4−(1−(N−スクシンイミジルオキシカルボニルオキシ)エチル)−フェノキシ)プロピル 2−ブロモ−2−メチルプロピオナート(2b)の合成
【0107】
【化20】
【0108】
50mL二口ナスフラスコに3−(4−(1−ヒドロキシエチル)−2−メトキシ−5−ニトロフェノキシ)プロピル 2−ブロモ−2−メチルプロピオナート0.2853g(0.6789mmol)を入れ、2時間真空乾燥した。これを、窒素雰囲気下で、脱水アセトニトリル1mL、トリエチルアミン0.2061g(2.0367mmol)を脱水アセトニトリル1.5mLに溶かした溶液、ジ(N−スクシンイミジル)カルボナート0.5217g(2.0367mmol)を加え、最後に脱水アセトニトリル4mL加え、室温で24時間撹拌した。これを濃縮し、H2O50mL、抽出(クロロホルム50mL×3)、洗浄(NaHCO3100mL×6)、無水硫酸マグネシウムを加え乾燥させ、ろ過、濃縮し、オープンカラムクロマトグラフィー(カラム直径3.5cm、シリカゲル高さ17cm、ジクロロメタン:酢酸エチル=10:1)を行った。モノと原料の2つのスポットが重なる部分が少量残っていたため、重なって出てきた部分をもう一度回収し、さらにオープンカラムクロマトグラフィー(カラム直径;2.5cm、シリカゲル高さ;17cm、ジクロロメタン:酢酸エチル=10:1)を行い、これを濃縮、真空乾燥し、黄色粘体を得た。
【0109】
【表11】
【0110】
<1−4>ポリ(エチレングリコール)−NH−光分解性基質(4b)の合成
<1−4−1>ポリ(エチレングリコール)−NH−光分解性基質(4b)の合成
【0111】
【化21】
【0112】
50mL二口ナスフラスコに<1−3−2>で合成した3−(2−メトキシ−5−ニトロ−4−(1−(N−スクシンイミジルオキシカルボニルオキシ)エチル)−フェノキシ)プロピル 2−ブロモ−2−メチルプロピオナート(2b)0.1356g(0.2416mmol)、PEG−NH2(数平均分子量2000(PEG2000))0.1757g(0.0805mmol)加え、3時間真空乾燥した。これを窒素雰囲気下で、脱水THF1.5mL、トリエチルアミン0.2445g(2.4160mmol)を加え室温で終夜撹拌した。これを再沈殿(2mLのTHFに溶かし、50mLのジエチルエーテルに滴下し氷浴上1時間撹拌)し、吸引ろ過、真空乾燥し、白色固体を得た。
【0113】
【表12】
【0114】
<1−5>1−(4−(3−(2−ブロモ−2−メチルプロパノイルオキシ)プロピルオキシ)−5−メトキシ−2−ニトロフェニル)エチル N−(3−(トリメトキシシリル)プロピル)カルバメート(3b)の合成
【0115】
【化22】
【0116】
<1−5−1>4−ベンジルオキシ−3−メトキシアセトフェノンの合成
【0117】
【化23】
【0118】
300mLナスフラスコに4−ヒドロキシ−3−メトキシアセトフェノン20.0g(120mol)とアセトン153mL、K2CO3 16.6g(120mol)を入れ、室温で30分間撹拌した。その後、ベンジルブロミド20.7g(120mol)を加え、80℃で4時間還流した。濃縮し、抽出(H2O 200mL、クロロホルム120mL×4回)を行い、有機層を乾燥(無水MgSO4)、ろ過、濃縮し再結晶を2回(第一結晶 ウォーターバス50℃、酢酸エチル5mL;第二結晶 ウォーターバス50℃、酢酸エチル2mL、ヘキサン パスツール2滴)行い、メンブランろ紙を用いて吸引ろ過、真空乾燥し、白色固体を得た。
【0119】
【表13】
【0120】
<1−5−2>4−ベンジルオキシ−5−メトキシ−2−ニトロアセトフェノンの合成
【0121】
【化24】
【0122】
300mLナスフラスコに4−ベンジルオキシ−3−メトキシアセトフェノン18.0g(70.2mmol)と酢酸200mLを入れ、氷浴上で発煙HNO3 23mLを42分間で滴下し、18時間撹拌した。反応溶液を冷水280mLの中に入れ、15分間撹拌し固体を析出させ、メンブランろ紙を用いて吸引ろ過、H2Oで固体を洗浄、再結晶を2回(第一結晶 ウォーターバス52℃、アセトン160mL、ヘキサン3mL;第二結晶 ウォーターバス55℃、アセトン48mL)行い、真空乾燥し、黄色固体を得た。
【0123】
【表14】
【0124】
<1−5−3>4−ヒドロキシ−5−メトキシ−2−ニトロアセトフェノンの合成
【0125】
【化25】
【0126】
300mLナスフラスコに4−ベンジルオキシ−5−メトキシ−2−ニトロアセトフェノン19.1g(63.4mmol)とCF3COOH 200mLを入れ、室温で終夜撹拌した。濃縮し、5% NaHCO3 320mL、2N HClを30mL入れ、抽出(酢酸エチル300mL×1回、200mL×1回、100mL×2回)を行い、有機層を乾燥(無水MgSO4)、ろ過、濃縮、再結晶を2回(第一結晶 ウォーターバス55℃、酢酸エチル80mL、ヘキサンmL;第二結晶 ウォーターバス55℃、酢酸エチル25mL)行い、吸引ろ過、真空乾燥し、黄緑色固体を得た。
【0127】
【表15】
【0128】
<1−5−4>3−(4−アセチル−2−メトキシ−5−ニトロフェノキシ)プロパン−1−オールの合成
【0129】
【化26】
【0130】
窒素雰囲気下で50mL二口ナスフラスコに脱水DMF16mLと60% NaH 0.28g(7.00mmol)、4−ヒドロキシ−5−メトキシ−2−ニトロアセトフェノン1.50g(7.10mmol)を入れ、室温で30分間撹拌した。3−ブロモプロパン−1−オール1.00g(7.19mmol)を加え85℃の油浴上で22時間撹拌した。減圧留去、抽出(H2O 200mL、酢酸エチル200mL×3回、100mL×1回)、洗浄(飽和NaCl水溶液100mL×2回)、有機層を乾燥(無水MgSO4)、ろ過、濃縮、オープンカラムクロマトグラフィーを2回(1回目 ヘキサン:酢酸エチル=1:3;2回目 ヘキサン:酢酸エチル=1:3)行い、真空乾燥し、白色固体を得た。
【0131】
【表16】
【0132】
<1−5−5>3−(4−(1−ヒドロキシエチル)−2−メトキシ−5−ニトロフェノキシ)プロパン−1−オールの合成
【0133】
【化27】
【0134】
100mLナスフラスコに3−(4−アセチル−2−メトキシ−5−ニトロフェノキシ)プロパン−1−オール1.11g(4.12mmol)とメタノール12mL、THF 9mLを入れ、氷浴上でNaBH4 0.33g(8.72mmol)を入れて1時間撹拌した。濃縮、抽出(H2O 80mL、クロロホルム100mL×3回)、洗浄(飽和NaCl水溶液100mL×3回)、乾燥(無水MgSO4)、ろ過、濃縮、真空乾燥し、黄色固体を得た。
【0135】
【表17】
【0136】
<1−5−6>3−(4−(1−ヒドロキシエチル)−2−メトキシ−5−ニトロフェノキシ)プロパン 2−ブロモ−2−メチルプロピオナートの合成
【0137】
【化28】
【0138】
100mL二口ナスフラスコに3−(4−(1−ヒドロキシエチル)−2−メトキシ−5−ニトロフェノキシ)プロパン−1−オール0.58g(2.15mmol)入れ、1時間真空乾燥した。これに水浴上で脱水CHCl3 25mL、メスピペットを用いてトリエチルアミン0.30mL(0.22g、2.17mmol)、脱水CHCl3 10mLに溶かした2−ブロモイソブチリルブロミド0.27mL(0.51g、2.21mmol)を滴下ロートでゆっくり加え、室温で17.5時間撹拌した。濃縮、抽出(H2O 100mL、1N HCl パスツールピペット3滴、クロロホルム100mL×3回)、乾燥(無水MgSO4)、ろ過、濃縮し、オープンカラムクロマトグラフィーを2回(1回目 ヘキサン:酢酸エチル=1:1、2回目 ヘキサン:酢酸エチル=15:1)行い、濃縮、真空乾燥し黄色固体を得た。
【0139】
【表18】
【0140】
<1−5−7>3−(2−メトキシ−5−ニトロ−4−(1−(N−スクシンイミジルオキシカルボニルオキシ)エチル)−フェノキシ)プロピル 2−ブロモ−2−メチルプロピオナートの合成
【0141】
【化29】
【0142】
50mL二口ナスフラスコに3−(4−(1−ヒドロキシエチル)−2−メトキシ−5−ニトロフェノキシ)プロパン 2−ブロモ−2−メチルプロピオナート0.200g(0.476mmol)を入れ、1時間真空乾燥した。窒素雰囲気下で乾燥CH3CN 3mL、トリエチルアミン0.2mL(0.146g、1.44mmol)、ジ(N−スクシンイミジル)カルボナート0.369g(1.44mmol)を加えた。室温で24時間撹拌した。濃縮、抽出(H2O 50mL、CHCl3 50mL×3回)、洗浄(5% NaHCO3水溶液50mL×6回)、乾燥(無水MgSO4)、ろ過、濃縮、オープンカラムクロマトグラフィー(ジクロロメタン:酢酸エチル=10:1)を行い、濃縮、真空乾燥し、黄白色固体を得た。
【0143】
【表19】
【0144】
<1−5−8>1−(4−(3−(2−ブロモ−2−メチルプロパノイルオキシ)プロピルオキシ)−5−メトキシ−2−ニトロフェニル)エチル N−(3−(トリメトキシシシル)プロピル)カルバマートの合成
【0145】
【化30】
【0146】
50mL二口ナスフラスコに3−(2−メトキシ−5−ニトロ−4−(1−(N−スクシンイミジルオキシカルボニルオキシ)エチル)−フェノキシ)プロピル 2−ブロモ−2−メチルプロピオナート0.174g(0.310mmol)を入れ、窒素雰囲気下で脱水THF 6mL、脱水THF 1mLに溶かした3−アミノプロピルトリメトキシシラン0.0586g(0.327mmol)を加え、室温で1時間撹拌した。濃縮、洗浄(酢酸エチルのみ)、濃縮し、中圧カラムクロマトグラフィーを2回(1回目および2回目 ヘキサン:酢酸エチル:アセトン=2:1:1(1.0%テトラメトキシシラン含有))を行い、濃縮、真空乾燥(湯浴中40℃)し、黄色粘体を得た。
【0147】
【表20】
【0148】
<1−6>ポリ(エチレングリコール)−NH−光分解性基質(4c)の合成
【0149】
【化31】
【0150】
<1−6−1>4−ベンジルオキシ−3−メトキシベンズアルデヒドの合成
【0151】
【化32】
【0152】
300mLナスフラスコにバニリン20.0g(131mmol)、アセトン89mL、K2CO3 18.1g(131mmol)を入れ、室温で40分間撹拌した。その後、ベンジルブロミド22.5g(132mmol)加え、80℃で4時間還流した。室温まで放冷後、濃縮し、H2O 200mLを加え、クロロホルム100mL×4回で抽出、無水MgSO4で乾燥、ろ過、濃縮した。再沈殿(アセトン31mLに溶解し、氷浴上のヘキサン310mLに滴下)し、メンブランろ紙を用いて吸引ろ過、真空乾燥を行い、白色固体25.9g(107mmol)得た。この操作で生じたろ液を濃縮、再沈殿(アセトン10mLに溶解し、氷浴上のヘキサン100mLに滴下)し、メンブランろ紙を用いて吸引ろ過、真空乾燥を行い、白色固体4.69g(19.3mmol)得た。
【0153】
【表21】
【0154】
<1−6−2>4−ベンジルオキシ−5−メトキシ−2−ニトロベンズアルデヒドの合成
【0155】
【化33】
【0156】
500mLナスフラスコに4−ベンジルオキシ−3−メトキシベンズアルデヒド15.8g(65.2mmol)、酢酸130mLを入れて撹拌し、氷浴上で発煙HNO3 31mLを65分かけて滴下し、17時間氷浴上で撹拌した。反応溶液を冷水270mLに入れ、15分間撹拌し、固体を析出させた。メンブランろ紙を用いて吸引ろ過、H2Oで固体を洗浄、酢酸エチルに溶かし、無水MgSO4で乾燥させた後、濃縮し、再結晶(ウォーターバス50℃、酢酸エチル150mLで溶かし、室温で静置)、メンブランろ紙を用いて吸引ろ過、真空乾燥を行い、黄色固体9.51g(33.1mmol)を得た。再度再結晶(ウォーターバス50℃、酢酸エチル45mLで溶かし、ヘキサンを5滴加えた)を行い、メンブランろ紙を用いて吸引ろ過、真空乾燥を行い、黄色固体2.34g(8.16mmol)を得た。
【0157】
【表22】
【0158】
<1−6−3>6−ニトロバニリンの合成
【0159】
【化34】
【0160】
200mL二口ナスフラスコに4−ベンジルオキシ−5−メトキシ−2−ニトロベンズアルデヒド9.55g(33.2mmol)、トリフルオロ酢酸61mLを撹拌しながら入れ、60℃で1時間撹拌した。反応溶液を冷ヘキサン180mLの中に入れ、15分間撹拌し、固体を析出させた。メンブランろ紙を用いて吸引ろ過を行い、ヘキサンで固体を洗浄、真空乾燥を行い、再結晶(ウォーターバス58℃、THF 75mLで溶かし、パスツールピペットでヘキサンを5滴加えた)を行った。メンブランろ紙を用いて吸引ろ過、ヘキサンで固体を洗浄、真空乾燥を行い、黄色固体3.89g(19.7mmol)を得た。この操作で生じたろ液を、濃縮し、再結晶(ウォーターバス59℃、酢酸エチル25mLで溶かし、ヘキサンを5滴加えた)を行い、メンブランろ紙を用いて吸引ろ過、真空乾燥を行い、黄色固体0.973g(4.94mmol)得た。
【0161】
【表23】
【0162】
<1−6−4>2−(3−ヨード−1−プロピル)オキサンの合成
【0163】
【化35】
【0164】
窒素雰囲気下、水浴上で20mL二口ナスフラスコに、3−ヨードプロパン−1−オール2.7mL(5.24g、28.2mmol、1.0eq)、脱水ジクロロメタン5mL(6.63g、78.1mmol)、ピリジニウム p−トルエンスルホナート0.270g(1.08mmol、0.04eq)、3,4−ジヒドロ−2H−ピラン2.5mL(2.3g、27.4mmol、1.0eq)を加え、2時間撹拌し、濃縮した。飽和NH4Cl水溶液100mLを加え、抽出(ジクロロメタン100mL×3回)、洗浄(飽和NaCl水溶液100mL×2回)、無水MgSO4で乾燥、ろ過、濃縮、オープンカラムクロマトグラフィー(ヘキサン:酢酸エチル=5:1)を行い、濃縮、真空乾燥を行った。再び、オープンカラムクロマトグラフィー(ヘキサン:酢酸エチル=5:1)を行い、濃縮、真空乾燥を行って、淡橙色液体7.04g(26.1mmol)を得た。
【0165】
【表24】
【0166】
<1−6−5>4−(3−(2−オキサノキシ)プロピル)−5−メトキシ−2−ニトロベンズアルデヒドの合成
【0167】
【化36】
【0168】
窒素雰囲気下、50mL二口ナスフラスコに脱水DMF10mL、K2CO3 0.351g(2.54mmol、0.5eq)、6−ニトロバニリン1.00g(5.08mmol、1.0eq)を入れ室温で30分間撹拌し、2−(3−ヨード−1−プロポキシ)オキサン1.37g(5.08mmol、1.0eq)加え、40℃で7時間、80℃で2時間撹拌後、減圧留去、H2O 100mLを加え、抽出(酢酸エチル100mL×3回)、洗浄(飽和NaCl水溶液100mL×2回)、無水MgSO4で乾燥、ろ過、濃縮を行い黄色固体1.41g(4.17mmol)を得た。
【0169】
【表25】
【0170】
<1−6−6>4−(1−ヒドロキシプロポキシ)−5−メトキシ−2−ニトロベンズアルデヒドの合成
【0171】
【化37】
【0172】
50mLナスフラスコに4−(3−(2−オキサノキシ)プロピル)−5−メトキシ−2−ニトロベンズアルデヒド0.502g(1.48mmol)、テトラヒドロフラン3.0mL、1N HCl 0.15mLを入れ、室温で2時間撹拌した。さらに、1N HClを0.15mL加え、室温で2時間撹拌し、1N HClを0.3mL加え、室温で2時間撹拌した。さらに、1N HClを0.9mL加え、室温で17時間撹拌し、濃縮、オープンカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1)、濃縮、真空乾燥を行い、淡黄色固体を0.270g(1.05mmol)を得た。
【0173】
【表26】
【0174】
<1−6−7>4−(1−ヒドロキシプロポキシ)−5−メトキシ−2−ニトロベンズアルデヒドの合成
【0175】
【化38】
【0176】
窒素雰囲気下、100mL二口ナスフラスコに脱水アセトニトリル32mL、K2CO3 0.0720g(0.521mmol、0.5eq)、6−ニトロバニリン0.270g(1.37mmol、1.3eq)を入れて室温で30分間撹拌し、3−ヨードプロパン−1−オール0.1mL(0.194g、1.04mmol、1.0eq)を加え、室温で1時間、40℃で2時間、80℃で18時間撹拌し、濃縮、H2O 25mLを加え、抽出(酢酸エチル25mL×5回)、洗浄(飽和NH4Cl水溶液25mL×2回)、無水MgSO4で乾燥、ろ過、濃縮、オープンカラムクロマトグラフィー(ジクロロメタン:酢酸エチル=1:1)、濃縮、真空乾燥を行い、黄色固体0.157g(0.615mmol)を得た。
【0177】
【表27】
【0178】
<1−6−8>3−(4−ホルミル−2−メトキシ−5−ニトロフェノキシ)プロピル 2−ブロモ−2−メチルプロピオナートの合成
【0179】
【化39】
【0180】
20mL二口ナスフラスコに4−(1−ヒドロキシプロポキシ)−5−メトキシ−2−ニトロベンズアルデヒド0.144g(0.565mmol)を入れ、1時間真空乾燥を行った。これに窒素雰囲気下で、脱水クロロホルム17mLを加え、脱水クロロホルム1mLに溶かしたトリエチルアミン0.0571g(0.564mmol)と、脱水クロロホルム1mLに溶かした2−ブロモイソブチリルブロミド0.161g(0.702mmol)をパスツールピペットでゆっくり加え、室温で24時間撹拌した。濃縮し、H2O 25mLを加え、抽出(クロロホルム25mL×4回)、無水MgSO4で乾燥、濃縮、オープンカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1)、濃縮、真空乾燥を行い、黄色固体0.190g(0.437mmol)を得た。
【0181】
【表28】
【0182】
<1−6−9>3−(4−(1−ヒドロキシメチル)−2−メトキシ−5−ニトロフェノキシ)プロピル 2−ブロモ−2−メチルプロピオナートの合成
【0183】
【化40】
【0184】
氷浴上で50mLナスフラスコに3−(4−ホルミル−2−メトキシ−5−ニトロフェノキシ)プロピル 2−ブロモ−2−メチルプロピオナート0.152g(0.376mmol、1.0eq)、メタノール9mL、テトラヒドロフラン6mLを入れ、NaBH4 0.006g(0.159mmol、0.25eq)をゆっくりと加え、室温で3時間撹拌した。これを濃縮し、H2O 15mLを加え、抽出(クロロホルム25mL×1回、20mL×3回)、洗浄(飽和NaCl水溶液20mL×2回)、無水MgSO4で乾燥、ろ過、濃縮、真空乾燥、オープンカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1)、濃縮、真空乾燥を行い、橙色粘体0.106g(0.261mmol)を得た。
【0185】
【表29】
【0186】
<1−6−10>3−(2−メトキシ−5−ニトロ−4−(1−(N−スクシンイミジルオキシカルボニルオキシ)メチル)−フェノキシ)プロピル 2−ブロモ−2−メチルプロピオナートの合成
【0187】
【化41】
【0188】
50mL二口ナスフラスコに3−(4−(1−ヒドロキシメチル)−2−メトキシ−5−ニトロフェノキシ)プロピル 2−ブロモ−2−メチルプロピオナート0.0629g(0.155mmol)を加え、2.5時間真空乾燥を行った。窒素雰囲気下で、脱水アセトニトリル3mL、1mLの脱水アセトニトリルで溶かしたトリエチルアミン0.0410g(0.405mmol)、ジ(N−スクシンイミジル)カルボナート0.1145g(0.447mmol)さらに脱水アセトニトリル6mLを加え、室温で24時間撹拌を行った。その後、濃縮し、H2O 15mLを加え、抽出(クロロホルム15mL×3回)、洗浄(飽和NaHCO3水溶液25mL×5回)、無水MgSO4で乾燥、濃縮、オープンカラムクロマトグラフィー(ジクロロメタン:酢酸エチル=10:1)、濃縮、真空乾燥を行い、黄色固体0.051g(0.0932mmol)を得た。
【0189】
【表30】
【0190】
<1−6−11>ポリ(エチレングリコール)−NH−光分解性基質(4c)の合成
【0191】
【化42】
【0192】
50mL二口ナスフラスコに3−(2−メトキシ−5−ニトロ−4−(1−(N−スクシンイミジルオキシカルボニルオキシ)メチル)−フェノキシ)プロピル 2−ブロモ−2−メチルプロピオナートを0.077g(0.141mmol、3.0eq)、PEG−NH2(数平均分子量2000(PEG2000))を0.092g(0.046mmol、1.0eq)加え、3時間真空乾燥を行った。その後、窒素雰囲気下で脱水テトラヒドロフラン1mL、トリエチルアミン0.19mL(0.138g、1.36mmol)を加え、室温で49時間攪拌し、濃縮、メンブランろ紙を用いて吸引ろ過、濃縮、再沈殿(THF 1mLに溶かし、10mLのジエチルエーテルに滴下し、氷浴上で1時間攪拌)し、吸引ろ過、真空乾燥を行った。再び、再沈殿(THF 1mLに溶かし、10mLのジエチルエーテルに滴下し、氷浴上で1時間攪拌)し、吸引ろ過、凍結乾燥を行い、白色固体0.0467g(0.0174mmol)得た。
【0193】
【表31】
【0194】
<1−7>1−(5−メトキシ−2−ニトロ−4−(3−トリメトキシシリルプロポキシ)フェニル)エチル 2−ブロモ−2−メチルプロピオナートの合成
【0195】
【化43】
【0196】
<1−7−1>1−(4−アリルオキシ−5−メトキシ−2−ニトロフェニル)エチル 2−ブロモ−2−メチルプロピオナートの合成
【0197】
【化44】
【0198】
窒素雰囲気下で30mL二口ナスフラスコに1−(4−アリルオキシ−5−メトキシ−2−ニトロフェニル)エタノール(特開平2007−186472号公報の段落「0052」〜「0067」に記載の方法により合成した)を0.300g(1.18mmol)、脱水クロロホルム10mL、メスピペットを用いてTEA0.17mL(1.22mmol)、2−ブロモイソブチリルブロミド0.58mL(4.71mmol)入れ、48時間撹拌した。濃縮し、抽出(H2O 50mL、2N HCl3mL、クロロホルム50mL×3回)、洗浄(飽和NaHCO3 50mL×7回)を行い、乾燥(無水MgSO4)、ろ過、濃縮し、カラムクロマトグラフィー(クロロホルムのみ)を2回行い、濃縮、真空乾燥し、黄白色固体を得た。
【0199】
【表32】
【0200】
<1−7−2>1−(5−メトキシ−2−ニトロ−4−(3−トリメトキシシリルプロポキシ)フェニル)エチル 2−ブロモ−2−メチルプロピオナートの合成
【0201】
【化45】
【0202】
20mL二口ナスフラスコに1−(4−アリルオキシ−5−メトキシ−2−ニトロフェニル)エチル 2−ブロモ−2−メチルプロピオナートを0.066g(0.164mmol)入れ、窒素雰囲気下で脱水THF 1mL、1mLの脱水THFに溶かしたトリメトキシシランを0.072g(0.589mmol)、Karstedt触媒7滴を入れ、窒素下にて2時間撹拌した。濃縮し、中圧カラムクロマトグラフィー(ヘキサン:酢酸エチル=2:1(1.0%テトラメトキシシラン含有))し、濃縮、真空乾燥(湯浴中、40℃)し、黄色粘体を得た。
【0203】
【表33】
【0204】
<1−8>2−ニトロ−5−(3−トリメトキシシリルプロポキシ)ベンジル 2−ブロモ−2−メチルプロパノアートの合成
【0205】
【化46】
【0206】
<1−8−1>5−アリルオキシ−2−ニトロベンズアルデヒドの合成
【0207】
【化47】
【0208】
窒素雰囲気下にした300mL二口ナスフラスコに5−ヒドロキシ−2−ニトロベンズアルデヒドを5.00g(29.9mmol)、脱水CH3CNを150mL、K2CO3を7.05g(51.0mmol)入れ、室温で1時間撹拌後、アリルブロミドを4.45g(36.8mmol)入れ、80℃で2時間還流しながら撹拌し、濃縮後、H2O 100mL、2N HClを10mL入れ、抽出(CHCl3 100mL×3回)し、有機層を無水MgSO4で乾燥後、ろ過し、ろ液を濃縮、真空乾燥(湯浴中40℃)し、黄色オイルを得た。
【0209】
【表34】
【0210】
<1−8−2>1−(5−アリルオキシ−2−ニトロフェニル)メタノールの合成
【0211】
【化48】
【0212】
300mLナスフラスコに5−アリルオキシ−2−ニトロベンズアルデヒドを6.08g(29.4mmol)、THFを20mL、メタノールを40mL入れ、氷浴中でNaBH4を3.36g(88.8mmol)撹拌しながら少量ずつ加え、氷浴中で1時間、室温で30分間撹拌し、濃縮後、H2O 100mL、2N HClを20mL加え、抽出(CHCl3 100mL×3回)し、有機層を無水MgSO4で乾燥後、ろ過し、ろ液を濃縮、真空乾燥(湯浴中40℃)し、黄白色固体を得た。
【0213】
【表35】
【0214】
<1−8−3>1−(5−アリルオキシ−2−ニトロ)ベンジル 2−ブロモ−2−メチルプロピオナートの合成
【0215】
【化49】
【0216】
窒素雰囲気下にした200mL二口ナスフラスコに1−(5−アリルオキシ−2−ニトロフェニル)メタノールを1.00g(4.78mmol)、脱水CHCl3を15mL、TEAをメスピペットを用いて0.66mL(0.484g、4.78mmol)、脱水CHCl3 1mLに溶かした2−ブロモイソブチリルブロミドを1.22g(5.31mmol)入れ、16時間撹拌し、濃縮後、H2O 100mL、1N HClを5mL加え、抽出(CHCl3 100mL×3回)し、有機層を無水MgSO4で乾燥後、ろ過し、ろ液を濃縮、カラムクロマトグラフィー(ヘキサン:酢酸エチル=2:1)を行い、濃縮、真空乾燥し、黄色オイルを得た。
【0217】
【表36】
【0218】
<1−8−4>2−ニトロ−5−(3−トリメトキシシリルプロポキシ)ベンジル 2−ブロモ−2−メチルプロパノアートの合成
【0219】
【化50】
【0220】
50mL二口ナスフラスコに1−(5−アリルオキシ−2−ニトロ)ベンジル 2−ブロモ−2−メチルプロピオナートを0.530g(1.48mmol)入れ、1時間真空乾燥した。窒素雰囲気下で脱水THF10mL、ドライTHF 1mLに溶かしたトリメトキシシラン0.592g(4.84mmol)、Karstedt触媒を5滴入れ、3時間撹拌した。濃縮し、中圧カラムクロマトグラフィー(ヘキサン:酢酸エチル=3:1(1.0%テトラメトキシシラン含有))、濃縮、真空乾燥(湯浴中、40℃)し、黄色オイルを得た。
【0221】
【表37】
【0222】
[実施例2]カップリング剤の溶液中における光分解の挙動の追跡
【0223】
【化51】
【0224】
測定機器は以下のものを使用した。
【0225】
【表38】
【0226】
<2−1>チオールカップリング剤(5a)のUV測定
10mLメスフラスコに<1−1>で合成した1−(4−(3−(2−ブロモプロパノイルオキシ)プロピルオキシ)−5−メトキシ−2−ニトロフェニル)−エチル N−(11−メルカプトウンデシル)カルバメートを0.657mg量りとり、THFを用いて0.103mM THF溶液を調製した。熱と300nm以下の波長の光を遮断するために水フィルターとパイレックスガラスフィルター、及び光照射する石英硝子セルを乗せる台を用意した。超高圧水銀灯を起動させ、光量が安定するまで、30分間暖機を行い、照射度計で照度が100mW/cm2となる位置を探した。調製した溶液を石英硝子セルにとり、一定時間光照射し、UVスペクトルを測定した。
【0227】
UV測定の結果を示す。チオールカップリング剤(5a)の光照射前の波長(λ)とモル吸光係数(ε)の値を下記表39に示す。また、比較として既に合成されている1−(4,5−ジメトキシ−2−ニトロフェニル)エチル N−プロピル−カルバメート(A)(下記化学式A)の光照射前の波長(λ)とモル吸光係数(ε)の値を示す。
【0228】
【表39】
【0229】
【化52】
【0230】
化合物(A)と比較するとチオールカップリング剤(5a)のεの値が若干低いことが分かる。これは副生成物であるNHSが混合してしまっているためと考えられる。チオールカップリング剤(2)の1H−NMRより、等モルのNHSが含まれていると考えられるので、厳密にはチオールカップリング剤(5a)のTHF溶液の濃度は0.0875mMである。上記表39における括弧内の数値は0.0875mMの濃度で算出したε値を示す。
【0231】
チオールカップリング剤(5a)の光照射による1次反応と2次反応のUVスペクトル変化をそれぞれ図1お及び図2に示す。溶液中での光分解は、45秒で295nm付近のピークが減少しなくなり、380nm付近のピークが増加しなくなった。この時点を一次反応の終了とし、光分解の終了時間とした。また、420秒で248nm付近のピークが増加しなくなり、284nm付近のピークが減少しなくなった。この時点を二次反応の終了とし、何らかの反応の終了時間とした。化合物(A)において、一次反応は100秒で終了したとされるが、この差異は置換基の違いによるものであると考えた。
【0232】
<2−2>シランカップリング剤(3a)のUV測定
精密天秤用バイアルに<1−2>で合成した1−(4−(3−(2−ブロモプロパノイルオキシ)プロピルオキシ)−5−メトキシ−2−ニトロフェニル)エチル N−((トリメトキシシリル)プロピル)カルバメート(3a)1.223mg(2.00μmol)を精密天秤で量りとり、20mLメスフラスコを使用し、0.1mMエタノール溶液を調製した。エタノールでバックグラウンド測定を行った。調製したサンプルをフタ付き二面セルに入れ、λ>300nmの条件でそれぞれ、0、1、3、5、10、20、30、45、60、75、90、120、150、180、240、300、360、420、540、720、1200、1800、3600秒になるように一定時間光照射し、逐次UV−visスペクトル測定を行った。光照射に際しては石英セルの中心の照度が100mW/cm2なるように照射した。
【0233】
UV測定の結果を下記表40に示す。シランカップリング剤(3a)と化合物(B)(下記化学式B)の光照射前のUVスペクトルのλmaxの値から、シランカップリング剤(3a)と化合物(B)とでほぼ同程度のλmaxが見られることから、シランカップリング剤(3a)と化合物(B)とが類似した分子骨格を持っているといえる。
【0234】
【表40】
【0235】
【化53】
【0236】
シランカップリング剤(3a)の光照射によるUVスペクトル変化を図3に示す。図3より、0〜180secで測定したUVスペクトルのピークの挙動から、220nm、300nm付近のピークの減少、260nm、330nm付近のピークの増大がそれぞれ見られたが、180秒でそれらが見られなくなったことからこの時点で一次反応の終了とし、光分解の終了時間とした。
【0237】
<2−3>シランカップリング剤(3b)のUV測定
(UV サンプルの調整)
精密天秤用バイアルに<1−3>で合成した1−(4−(3−(2−ブロモ−2−メチルプロパノイルオキシ)プロピルオキシ)−5−メトキシ−2−ニトロフェニル)エチル N−(3−(トリメトキシシリル)プロピル)カルバメート(3b)1.248mg(2.00μmol)を精密天秤で量り取り、20mLメスフラスコを用いて、0.1mMエタノール溶液を調製した。
【0238】
(UV−Vis スペクトルの測定)
まず、光源として超高圧水銀灯を用い、光源が安定するまで約30分暖気を行った。照度計で照度が100mW/cm2となる位置を探した。調製した溶液を蓋付き二面石英セルに入れUV−Visスペクトル測定を行った。なお、バックグラウンドはエタノールで測定した。1、3、5、10、15、20、25、30、35、40、45、50、55、60、65、70、75、80、85、90、100、110、120、130、140、150、160、170、180、190、200、210、220、230、240、250、260、270、280、300、360、420、480、600、900、1200、1800、3600秒になるように一定時間光照射(λ>300nm)し、逐次UV−Visスペクトル測定を行った。光照射に関しては石英セルの中心の照度が100mW/cm2になるように照射した。光分解の終了の判断基準は300nm付近のニトロ基由来のスペクトルの減少と、370nm付近のニトロソ基由来のスペクトルの増大がほぼ見られなくなり、等吸収点から外れたスペクトルを示した時点とした。
【0239】
(光反応の反応速度定数)
上記シランカップリング剤(3b)をそれぞれ所定時間光照射したUV−Visスペクトルから反応速度定数の算出を試みた。一次反応終了と判断するまでのニトロ由来の吸収である300nm付近の吸光度Absの値をそれぞれ得た。その後、ランベルト−ベールの式から得られる式に所定時間光照射することにより得られた吸光度At、ニトロ基のモル吸光係数ε1、一次反応終了と判断した時の300nm付近のニトロソ化合物由来の吸収から得たモル吸光係数ε2、初濃度C0を代入し所定時間での前駆体の濃度Ctを求めた。そして一次反応速度式に求めたCtの値、C0を代入し、時間とln(Ct/C0)をプロットし、その近似式の傾きから反応速度定数kを求めた。またニトロソ由来である370nm付近の吸収から所定時間におけるニトロ化合物の濃度を求め、これを一次反応式に代入し、反応速度定数kを求めた。
【0240】
【数1】
【0241】
(結果および考察)
下記表41にシランカップリング剤(3b)および(3a)の極大吸収波長λとモル吸光係数εを示す。
【0242】
【表41】
【0243】
また、図4および5にλ>300nmで光照射を行った際のUV−Visスペクトルの変化を示す。図5より0〜280秒まで照射すると、ニトロ基由来の300nm付近の減少とニトロソ基由来の370nm付近の増大、等吸収点が消失したため、この時点を光分解終了と判断した。280秒で等吸収点が消失したため二次反応が進行するようになったと考えられる。光分解の終了を判断した後も60分まで光照射を続けたところ、スペクトル全体の減少が見られ、二次反応の進行が示唆された。
【0244】
(光反応の反応速度定数)
図6にニトロ基由来の吸収を、図7にニトロソ基由来の吸収の時間(秒)、ln(Ct/C0)についてのプロット示す。シランカップリング剤(3b)の溶液中における光反応の反応速度定数を求めた。このグラフの最小二乗法による近似直線の傾きから求めた反応速度定数kはニトロ基由来では0.0138s-1、ニトロソ基由来では0.0199s-1となった。
【0245】
反応速度定数はニトロ基由来の吸収から得られたものとニトロソ由来の吸収から得られたものから求められるがほぼ同様の値を示し、反応速度定数を求めるにあたり、ニトロ基由来の吸収から求める方法で十分であるということが報告されている。しかし、図6と図7の近似直線からかなりのずれが見られ、一次反応ではない反応も同時に進行しているおそれがあるため、図8にニトロ由来の吸収、図9にニトロソ由来の吸収での100秒までの時間(秒)、ln(Ct/C0)についてのプロット示し、(3b)の溶液中における光反応の反応速度定数を求めた。このグラフの最小二乗法による近似直線の傾きから求めた反応速度定数kはニトロ基由来では0.0129s-1、ニトロソ基由来では0.0160s-1となった。
【0246】
さらにシランカップリング剤(3a)についても図10にニトロ基由来の吸収、図11にはニトロソ基由来の吸収の時間(秒)、ln(Ct/C0)についてのプロット示す。シランカップリング剤(3a)の溶液中における光反応の反応速度定数を求めた。このグラフの最小二乗法による近似直線の傾きから求めた反応速度定数kはニトロ基由来では0.0326s-1、ニトロソ基由来では0.0361s-1となった。
【0247】
表42にシランカップリング剤(3b)と(3a)のニトロ基由来の吸収とニトロソ基由来の吸収の反応速度定数をまとめた。化合物の違いは末端にメチル基がついているかいないかの違いであるため、さほど反応速度に違いがあるとは思われなかったが、実際は、シランカップリング剤(3a)の方が(3b)よりも約2.5倍速いという大きな違いが見られた。
【0248】
【表42】
【0249】
[実施例3]シランカップリング剤を用いて作製したSAMの評価
<3−1>シランカップリング剤(3a)を用いて作製したSAMの評価
シリコンウエハはエナテック社製のVE49149AA(4インチを約1×4cmとしたもの)を用いた。
【0250】
光照射は以下の機器を用いて行った。
【0251】
【表43】
【0252】
測定機器は以下のものを使用した。
【0253】
【表44】
【0254】
下記表45にシランカップリング剤(3a)の量、調製した溶液の濃度、シリコンウエハの大きさと枚数をまとめたものを示す。
【0255】
【表45】
【0256】
<3−1−1>表面修飾条件の検討
【0257】
【化54】
【0258】
<3−1−1−1>シリコンウエハの前処理
50mLナスフラスコに濃H2SO4:30%H2O2=7:3(14mL:6mL)の混合溶液を調製し、シリコンウエハの非鏡面を合わせて入れ、100℃で1時間加熱した。その後、純水約30mLで洗浄し、純水約50mLを入れ超音波洗浄を行った。シリコンウエハを窒素気流で乾燥し、表面修飾に用いた。
【0259】
<3−1−1−2>表面修飾
精密天秤用バイアルにシランカップリング剤(3a)を上記表45のA〜Fに記載の量ずつ量り取り、脱水トルエン10mLで溶解しながら50mLナスフラスコに入れ、シランカップリング剤(3a)の1mM脱水トルエン溶液を調製し、窒素雰囲気下とした。この溶液に前処理済みのシリコンウエハを入れ、室温、50℃、100℃に加熱し、表面修飾を任意の時間で行った。処理後、メタノール約30mLで3回洗浄し、超音波洗浄10分間行い、クロロホルム約30mLで3回洗浄し、クロロホルム約30mLで超音波洗浄を10分間行った。表面を窒素気流で乾燥し、水の静的接触角を測定した。静的接触角を測定後は基板をメタノール、クロロホルムの順で洗浄し、クロロホルム約30mLで超音波洗浄を5分間行い、クロロホルムを入れかえ、さらに超音波洗浄を5分間行い、表面を窒素気流で乾燥し、25mLバイアルに修飾基板を入れ、窒素を充填し、保存した。
【0260】
室温、50℃、100℃にて任意の時間で静的接触角を測定した結果を表46に示す。
【0261】
【表46】
【0262】
室温で表面修飾を行った際は、表面修飾5分で水の静的接触角が67°となり、表面修飾時間3時間で水の静的接触角は72°となりほぼ一定となった。50℃条件下では表面修飾開始5分で水の静的接触角は64°となり、表面修飾30分で水の静的接触角は70°〜73°となり、ほぼ一定となった。100℃条件下では表面修飾開始5分間で水の静的接触角が75°となりそれ以降、接触角は70°〜74°となり、ほぼ一定となった。室温から100℃の水の静的接触角を比較すると、室温条件下よりも加熱条件下のほうが、表面修飾時間が短くても静的接触角が一定の値となることが考えられる。水の静的接触角が一定となったところを表面修飾時間の終了時間とすると、室温では3時間、50℃では30分、100℃では5分間で表面修飾が終了していることが考えられる。トリメトキシシリル基を持つカルボキシレート型のシランカップリング剤と表面修飾時間を比較すると、カルバメート型のシランカップリング剤は表面修飾に要する時間が短く、短時間で表面修飾が行えることが考えられる。これはカルバメート部位のアミノ基とシリコンウエハ表面のシラノールが水素結合し、その引力で基板上に接近することで、トリメトキシシリル基とシラノールの自己縮合反応が促進され、カルボキシレート型のシランカップリング剤よりも、表面に反応する速度が速くなることが要因であると考えられる。
【0263】
<3−1−2>SAMへの光照射による基板上での光分解の挙動の追跡
【0264】
【化55】
【0265】
<3−1−2−1>シリコンウエハの前処理
50mLナスフラスコに濃H2SO4:30%H2O2=7:3(14mL:6mL)の混合溶液を調製し、シリコンウエハを入れ、100℃で1時間加熱した。その後、純水約30mLで洗浄し、純水約50mLを入れ超音波洗浄を行った。シリコンウエハを窒素気流で乾燥し、表面修飾いた。
【0266】
<3−1−2−2>表面修飾
精密天秤用バイアルに2つにシランカップリング剤(3a)を上記表45のa、bに記載した量ずつ量り取り、それぞれ脱水トルエン10mLを加え、溶かしながら50mLナスフラスコに入れ、シランカップリング剤(3a)の1mM脱水トルエン溶液を調製し、窒素雰囲気下とした。この溶液に前処理済みのシリコンウエハを入れ、100℃、1時間加熱した。処理後、メタノール約30mLで3回洗浄し、超音波洗浄を10分間行い、クロロホルム約30mLで3回洗浄し、クロロホルム約30mLで超音波洗浄を10分間行った。表面を窒素気流で乾燥し、水の静的接触角を測定した。静的接触角を測定後は基板をメタノール、クロロホルムの順で洗浄し、クロロホルム約30mLで超音波洗浄を5分間行い、その後、クロロホルムを入れ替え、超音波洗浄を5分間行って、表面を窒素気流で乾燥し、25mLバイアルに修飾基板を入れ、窒素を充填し、保存した。
【0267】
<3−1−2−3>光照射
熱と300nm以上の光を遮断するために水とフィルターとパイレックスガラスフィルター、及び表面修飾したシリコンウエハを置く台を用意した。超高圧水銀灯を起動させ光量が安定するまで60分間暖気した。照射度計で照度が45mW/cm2となる位置を探した。その後、表面修飾したシリコンウエハをチタンピンセットに挟んで置き、任意の時間で光照射を行った。光照射後、シリコンウエハをメタノール、クロロホルムの順で洗い流し、その後クロロホルム約30mLで超音波洗浄10分間行い、窒素気流でシリコンウエハを乾燥させた。
【0268】
<3−1−2−4>水の静的接触角の測定
シランカップリング剤(3a)を用いたSAMへの光照射時間と光照射直後に測定した静的接触角を、協和界面化学株式会社の接触角計の取扱説明書に記載されている「液滴法測定操作」にしたがって測定した。結果を表47に示す。
【0269】
【表47】
【0270】
光照射前の基板では水の静的接触角は73°であった。光照射90秒で静的接触角が42°となり、光照射150秒で静的接触角が安定した。この結果からシランカップリング剤(3a)が基板上で光分解に要する時間は90秒であると判断した。
【0271】
<3−1−3>シランカップリング剤(3a)を用いたSAMに対するXPS測定
<3−1−3−1>測定サンプルの調製
上記3−2項で作製したSAMを測定サンプルとして利用した。調製したシリコンウエハ上のSAMの構造を下記式に示す。
【0272】
【化56】
【0273】
<3−1−3−2>測定対象とする元素と測定条件
XPS測定は表面修飾によるシリコンウエハ上へのシランカップリング剤(3a)の導入、SAMに対する光照射による光分解を確認するために下記表48に示す元素と条件で測定を行った。
【0274】
【表48】
【0275】
測定したスペクトルはmodificationのSiスペクトルを用いてピークシフトの公正、面積による規格化、解析を行った。以下に測定で得られた各サンプルのwideスキャンで得られたスペクトル、N、C、Brのスペクトルの解析結果を示す。
【0276】
図12及び13に、基板1及び基板2のXPS wideスキャンスペクトルをそれぞれ示す。基板1と基板2のスペクトルから400、70eV付近にN、Brに由来するピークが見られた。これは、シリコンウエハ上にシランカップリング剤(3a)が反応、固定化されていることを示している。
【0277】
さらに詳細な定性を行うため、基板1、基板2のN、C、Brのnarrowスキャンにより得られたスペクトルを詳細に解析した。図14及び図15に基板1と基板2のN1s XPSスペクトルをそれぞれ示す。
【0278】
基板1では400.0eV、406.1eVにサブピークが見られた。これはそれぞれNH基とNO2基の結合エネルギー由来のサブピークであり、基板上にNH基とNO2基が両方存在していることを示している。
【0279】
表49に各状態の基板のNO2基とNH基の面積比を示す。
【0280】
【表49】
【0281】
基板1の状態ではNO2基とNH基のサブピークの面積比の計算値は[NO2:NH]=1:1となるはずだが実際は[NO2:NH]=1:1.5となっていた。基板1と基板2のピークを比較すると406ev付近に存在するNO2由来のピークがSAMに対して光照射を行って作製した基板2には見られなかった。これはシリコンウエハ上にシランカップリング剤(3a)を修飾して作製したSAMがλ>300nmの光の照射することで光分解し、基板表面がアミンに変換され基板2となっていることを示している。402eV付近に現れるサブピークはSAM作製時特有のコンタミネーションのピークであると考えられる。
【0282】
図16及び図17に基板1及び基板2のBr3d(5/2) XPSスペクトルをそれぞれ示す。また、表50に各基板のカーブフィッティングしたサブピークの結合エネルギー(eV)と帰属を示す。
【0283】
【表50】
【0284】
基板1では75.0、70.8eV付近のそれぞれサブピークが見られ、基板2では70eV付近のピークの消失が見られた。これはSAMに対する光照射により、光分解反応が進行し、SAMの最表面に存在する重合開始剤となるBrが脱離したことが要因で70eV付近が消失したことを示している。また文献A(Kevin Critchley et al.,Langmuir,21,4554−4561(2005))からBrを持つ重合開始剤SAMのBrのピークが71eVに現れることが確認されており、それと比較して、このカップリング剤を用いることによって重合開始剤をシリコンウエハに導入できることが分かった。75eV付近のピークは基板1、基板2に見られることから、コンタミネーション由来のピークであると考えられる。
【0285】
図18及び図19に基板1及び基板2のC1s XPSスペクトルをそれぞれ示す。表51に基板3から得られたC1s XPSスペクトルをカーブフィッティングしたサブピークの結合エネルギー(eV)、表52に基板3の表面構造から予想されるサブピークの面積比とスペクトルから得られたサブピークの面積比を記載したもの、測定したシリコンウエハの表面構造を示す。
【0286】
【表51】
【0287】
【表52】
【0288】
基板1をXPS測定し、得られたスペクトルの波形解析の結果から表51のように帰属できる結合エネルギーのサブピークが存在すると予想できる。これらの結合エネルギーのサブピークの面積比の計算値は表52より[NCOO:COO:CO2CBr、NCO2C、CNCO2:C−O:C−C]=1:1:3:3:15であると考えられたが、実際に波形解析したサブピークの面積比は[NCOO:COO:CO2CBr、NCO2C、CNCO2:C−O:C−C]=1:1:3.8:5.5:9.8となった。298.6eV付近にNCOO、288.8eV付近のCOOのサブピークが存在していることから光分解性基が連結されているカルバメート構造、開始剤であるBrの連結部分であるエステル部位がシリコンウエハ上に存在していることが確認できた。
【0289】
表53に基板2から得られたC1s XPSスペクトルをカーブフィッティングしたサブピークの結合エネルギー(eV)と測定したシリコンウエハの表面構造を示す。
【0290】
【表53】
【0291】
基板2は波形解析の結果から3つのサブピークが存在することが考えられ、NCOO、COO、CO2CBr、NCO2C、CNCO2、C−O結合由来のサブピークが消失し、C−N、C−C結合のサブピークが見られたことから、光照射により、シリコンウエハ上のSAMが光分解し、カルバメート部位、開始剤が存在していたエステル部位が消失、親水性部位のアミン基が導入されたことが考えられる。C−N、C−C結合のサブピークの面積比は[C−N、C−C]=1:1.2であった。しかし、NCOO結合エネルギーに相当する288.9eV付近に※ピークが存在していた。これは文献からシェイク−アップサテラインに由来するピーク、あるいはシリコンウエハ上に残留している洗浄しきれていない切断された光分解性基、光分解しきれていないSAM部分に由来するコンタミネーションピークであるとであると考えられる。
【0292】
<3−2>シランカップリング剤(3b)を用いて作製したSAMの評価
<3−2−1>SAMへの光照射による基板上での光分解の挙動の追跡
【0293】
【化57】
【0294】
<3−2−1−1>シリコンウエハの前処理
2つの50mL太口ナスフラスコにピラニア溶液(Piranha溶液;H2SO4:H2O2=7:3(14mL:6mL)の混合溶液)を調製し、約1cm角のシリコンウエハを鏡面が上向きになるように3枚ずつ、計6枚入れ、100℃で1時間静置した。混合溶液を捨て、純粋で3回洗浄し、純粋を約50mL入れ超音波洗浄を10分間行った。基板を窒素気流で乾燥させ、表面修飾に用いた。
【0295】
<3−2−1−2>表面修飾
2つの50mL太口ナスフラスコにシランカップリング剤(3b)の1.0mMトルエン*溶液(トルエンにモレキュラーシーブを入れて脱水したもの)を入れ、前処理を行った基板を鏡面が上向きになるように3枚ずつ、計6枚入れ、窒素下、100℃にてそれぞれ5、30分、1、2、3、6時間表面修飾を行った。時間毎に基板を取り出し、メタノールで洗浄、試料瓶にて基板をメタノールに浸漬させ超音波洗浄を10分間行った。さらにクロロホルムで洗浄を行い、試料瓶にて基板をクロロホルムに浸漬させ超音波洗浄を10分間行った。基板を窒素気流で乾燥させた。
【0296】
<3−2−1−3>光照射
超高圧水銀灯光量が安定するまで30分間暖気し、表面修飾した基板を大気中で、照度計の測定で約50mW/cm2の位置にチタンピンセットで挟み固定した。λ>300nmの光を5、15、30、50、70、90、120秒の時間で照射した。光照射を行った後、基板をメタノールで洗浄、クロロホルムで洗浄し、さらに試料瓶にて基板をクロロホルムに浸漬させ超音波洗浄を10分間行った。窒素気流で乾燥させた。
【0297】
<3−2−1−4>水の静的接触角の測定
シランカップリング剤(3a)を用いたSAMへの光照射時間と光照射直後に測定した静的接触角を、協和界面化学株式会社の接触角計の取扱説明書に記載されている「液滴法測定操作」にしたがって測定した。1枚のサンプルについて1点の測定を3回行い、その平均値の少数第一位を四捨五入し、その基板の接触角とした。
【0298】
(表面修飾の条件検討)
トルエン溶液で100℃の加熱条件を用いてシランカップリング剤(3b)の表面修飾をした。基板を浸漬する時間を検討し、前処理基板の接触角約0°からの変化を追跡した。下記表54にそれぞれの時間で表面修飾した接触角の値を示す。
【0299】
【表54】
【0300】
表面修飾時間を表54のように振り分けたところ、浸漬開始から30分以降、ほぼ接触角の変化がないことから30分で表面修飾が終了したと考えられる。
【0301】
(光照射による接触角の変化)
表面修飾した基板にそれぞれの時間で光照射を行った。光照射したSAMの水の接触角の値を表55に示す。
【0302】
【表55】
【0303】
表55より光照射前の接触角の値が64°から光照射していくと徐々に接触角が下がっているのがわかる。これは光照射によりニトロソ化合物が脱離し疎水性の減少および、親水性の増大による接触角が変化したものだと考えられる。70秒以降、接触角の値がほぼ変化していないことから70秒で光分解が終了したと考えられる。90秒行った時点を光分解率100%と仮定した時、その時の接触角の値42°をθ∞として、下記のCassieの式を用いて各時間tにおける光分解率αtを求めたものを表57に示す。さらに残存率(1−αt)の自然対数lnをとったものを縦軸、各時間tを横軸にプロットしたものを図20と図21に示す。このグラフの最小二乗法による近似直線の傾きから求めた(3b)の反応速度定数kは0.0301s-1となり(3a)は0.0207s-1となった。
【0304】
【数2】
【0305】
【表56】
【0306】
以上の結果より、SAMの光分解速度は、溶液中の光分解速度とは逆に、(3b)の方が(3a)よりも反応速度が速いという結果となった。
【0307】
[実施例4]SAMを利用して作製したポリマーブラシの評価
シリコンウエハはエナテック社製のVE49149AA(4インチを約1×4cmとしたもの)を用いた。
【0308】
測定機器は以下のものを使用した。
【0309】
【表57】
【0310】
<4−1>Pt−BuA(ポリ(アクリル酸tert−ブチル))ブラシ、PAA(ポリアクリル酸)ブラシの作製
【0311】
【化58】
【0312】
<4−1−1>表面重合に使用するSAM、ポリマーブラシの作製に使用する試薬、条件
表58にポリマーブラシ作製に使用したSAMの作製条件を記載する。SAM作製方法は3−1項で記載した実験方法と同様である。
【0313】
【表58】
【0314】
表59に後述の<4−2−2−2>項で、XPS測定に使用するサンプル作製のためのポリマーブラシ作製時に使用した試薬の量、重合時間、をまとめたものを示す。
【0315】
【表59】
【0316】
<4−2>実験項
<4−2−1>表面処理
3−1項の方法でシランカップリング剤(3a)のSAMを作製し、表面重合に用いた。用いた試薬の量、基板の形状等は表58に示すとおりである。
【0317】
<4−2−2>シランカップリング剤(3a)を用いたポリマーブラシの作製
<4−2−2−1>原料の前処理
100mLナスフラスコにCuBr0.552g(3.85mmol)を量り取り、酢酸約5mLを加え、24時間撹拌後、洗浄(エタノール100mL、ジエチルエーテル100mL)、12時間真空乾燥(湯浴70℃)し、白色固体を得た。ここで得られた白色固体をポリマーブラシ作製に用いた。
【0318】
アクリル酸tert−ブチルは塩基性アルミナカラムで重合禁止剤を除去して使用した。条件を表60に示す。
【0319】
【表60】
【0320】
<4−2−2−2>Pt−BuAブラシの作製[基板3の作製]
50mL二口ナスフラスコに、窒素雰囲気下で、脱水DMF5mL、アニソール5mL、アクリル酸tert−ブチル(t−BuA)10mL、CuBr5.83mg(0.041mmol)、CuBr24.6mg(0.021mmol)、1,4,8,11−アザシクロテトラデカン(Me4Cyclam)10.43mg(0.041mmol)、dnNbpy16.57mg(0.041mmol)を入れ窒素置換し、凍結脱気×3回、室温に戻した後、50mLバイアルに調製した溶液を入れ、50℃で溶液が均一になるまで15分間よく撹拌、窒素置換した。反応溶液に修飾基板を50℃で1時間浸漬させ、静置した。基板を溶液から取り出し、酢酸エチル、THFで洗浄、窒素気流で乾燥させ、基板3を得た。静的接触角測定後、メタノール、クロロホルムの順で洗浄し、表面を窒素気流で乾燥し、25mLバイアルに基板3を入れ、窒素を充填し、保存した。
【0321】
<4−2−2−3>PAAブラシの作製[基板4の作製]
100mLビーカーにメタンスルホン酸100μL、ジクロロメタン10mL入れ基板3を15分間浸漬させ、クロロホルム、純水、メタノール、クロロホルムの順で洗浄、窒素気流で乾燥させ、基板4を得た。静的接触角測定後、メタノール、クロロホルムの順で洗浄し、表面を窒素気流で乾燥し、25mLバイアルに修飾基板を入れ、窒素を充填し、保存した。
【0322】
<4−3>シランカップリング剤(3a)を用いたSAM、シランカップリング剤(3a)を用いたSAMから作製した基板2及び基板3の水の静的接触角
【0323】
【化59】
【0324】
表61に各基板の表面構造を示す。基板1、3、4のSAM時の接触角は72〜75°であった。シリコンウエハ上に形成した3級ATRP開始剤を持つSAMの静的接触角は70〜77°であった)。シランカップリング剤(3a)はATRP開始剤部位が2級であるが、3級ATRP開始剤を持つSAMの静的接触角と類似した値を示した。その後モノマーを重合した基板2、3の接触角はそれぞれ78°、64°であった。この接触角の違いはグラフト密度の異なるポリマー鎖が重合されたことが要因であると考えられる。接触角測定の際、作製されたポリマーブラシが高密度の場合、ポリマーの最表面の性質が最も反映される。しかしグラフト密度が高密度時と比較して、密度が低い(中程度)場合、表面に接触している水がポリマー鎖に浸み込んでいき、接触角が減少する。基板2、3の接触角の変化はこのグラフト密度の変化が要因で現れた変化であることが考えられる。基板2の場合、基板上のSAMからポリマーブラシが密にグラフトされ、ポリマー鎖の最表面(Brとtert−ブトキシカルボニル部位)の性質が水の静的接触角に反映され、SAMの状態よりも接触角が上昇したことが考えられる。基板4ではポリマー鎖のグラフト密度が基板3と比較して低く、ポリマー鎖の間に水が浸み込み、接触角が低下したことが考えられる。基板4の加水分解処理後の水の静的接触角は49°であった。これはPt−BuAブラシ重合後に加水分解を行ったことにより、tert−ブトキシカルボニル基が分解し、カルボキシ基に変換されたことで接触角が低下したことが考えられる。
【0325】
【表61】
【0326】
<4−4>作製したポリマーブラシのXPS測定
<4−4−1>測定試料の調製
測定に使用する各試料は4−2項で示した方法で調製し、基板3、4をとした。
【0327】
<4−4−2>測定対象とする元素と測定条件
このXPS測定は表面修飾によるシリコンウエハ上へのシランカップリング剤(3a)の導入、SAMに対する光照射による光分解、モノマーの重合によるポリマーブラシ作製の有無を確認するために表62に示した元素と条件で測定を行った。
【0328】
【表62】
【0329】
測定したスペクトルは3−3項の基板1のSiスペクトルを用いてピークシフトの公正、面積による規格化、解析を行った。以下に測定で得られた各サンプルのwideスキャンで得られたスペクトル、N、C、Brのスペクトルの解析結果を示す。
【0330】
図22、23に基板3、4のXPS wideスキャンスペクトルそれぞれ示す。文献B(Krzysztof Matyjaszewski et al.,Macromolecules,32,8716−8724(1999))および文献C(Hydeharu Mori et al.,Macromolecules,34,6871−6881(2001))から開始剤を無機材料表面に導入後、重合を行うと、膜厚が増加し、開始剤部位、基板表面は表面全体の深層になる。よってX線を基板に対して90°の角度に照射し、測定したスペクトルでは重合開始剤部位や基板自体のスペクトルは検出されにくくなると考えられる。よって、基板3、4とした基板から実際に得られたスペクトルを見ると、3項で記載した基板1、2と基板3、4の間でSiのピークの強度の変化があまり見られないことから、開始剤表面からモノマーであるt−BuAの重合がわずかしか進行していないことが予想された。さらに詳細な定性を行うため、基板3、4のN、C、Brのnarrowスキャンにより得られたスペクトルを詳細に解析した結果を以下に示す。
【0331】
図24、25に基板3、4のN1s XPSスペクトルをそれぞれ示す。また、表63に各基板のカーブフィッティングしたサブピークの結合エネルギー(eV)と帰属を示す。
【0332】
【表63】
【0333】
モノマーであるアクリル酸tert−ブチル(t−BuA)を重合した後、NO2基由来、NH基由来のピークが基板3、4でそれぞれ400.3、400.0eVと406.1、406.3eVに確認できた。これは重合後、加水分解後も光分解性基である、2−ニトロベンジル誘導体がカルバメート構造で連結されて存在していることを示している。NO2基由来のサブピークはSAMの時と同様だがサブピークの面積比が計算値の[NO2:NH]=1:1ではなく基板3、4の面積比でそれぞれ[NO2:NH]=1:3.9、1:3.5で存在していた。これは重合や加水分解で光分解性基を連結しているカルバメート部位に何らかの影響が及び、光分解性基の部分が脱離しているか、ポリマー鎖が伸展し、膜厚が増加したことでシリコンウエハ底部のNO2基部位が検出されにくくなっていることにより、NO2基由来のサブピークの面積比が減少していることが予想された。基板3に関しては、スペクトルのベースラインが傾いているのはNO2基とNH基部位が基板表面上に形成されている膜の下層にあり、X線を当てて、検出される光電子の量が減少していることが原因であることが推測される。402eV付近に現れるサブピークはここに示した全ての基板に現れていることからSAM作製時特有のコンタミネーションのピークであると考えられる。
【0334】
図26、27に基板3、4のC1s XPSスペクトルをそれぞれ示す。また、表64に基板3から得られたC1s XPSスペクトルをカーブフィッティングしたサブピークの結合エネルギー(eV)、表65に基板3の表面構造から予想されるサブピークの面積比とスペクトルから得られたサブピークの面積比を記載したもの、測定したシリコンウエハの表面構造を示す。
【0335】
【表64】
【0336】
【表65】
【0337】
検出されたピークの波形解析の結果から、SAMと比較してサブピークの構成要素が減少し、CO2R、C−O、CCO2R、C−C結合に由来するサブピークがみられた。これらのサブピークは基板表面にt−BuAが重合した際に得られるPt−BuAブラシの構造に由来する結合エネルギーであり、このXPSスペクトルから基板1を重合開始剤としてt−BuAが重合しPt−BuAブラシの作製に成功したことが考えられる。また表65に示すように、CO2R、C−O、CCO2R、C−Cのサブピークの面積比は[CO2R:C−O:CCO2R:C−C]=1.1:3.2:1.0:4.7となった。Pt−BuAブラシの構造からCO2R、C−O、CCO2R、C−Cのサブピークの面積比の計算値は[CO2R:C−O:CCO2R:C−C]=1.0:1.0:1.0:4.0であり、実際に波形解析して得られたサブピークの面積比とC−O結合部分以外はよく一致していた。C−O結合の面積比が計算値よりも増加した原因としてシリコンウエハ下層部の重合開始剤に由来するサブピークが含まれているあるいは、表面の汚染によるコンタミネーションピークが含まれている可能性があると考えられる。
【0338】
表66に基板4から得られたC1s XPSスペクトルをカーブフィッティングしたサブピークの結合エネルギー(eV)、表67に基板4の表面構造から予想されるサブピークの面積比とスペクトルから得られたサブピークの面積比をまとめたもの、測定したシリコンウエハの表面構造を示す。
【0339】
【表66】
【0340】
【表67】
【0341】
検出されたピークの波形解析の結果から、基板4から得られたすスペクトルの構成要素はCO2H、C−O、CCO2H、C−C結合由来のピークであると考えられる。表67に示したように、サブピークの構成要素の面積比は[CO2H:C−O:CCO2H:C−C]=1.0:3.7:3.2:8.4であった。加水分解により保護基が分解し、完全なPAAブラシが作製されたとき、C1s XPSスペクトルのサブピークにはC−O結合由来のピークは検出されず、ピークの構成要素は[CO2H:CCO2H:C−C]=1:1:2となると考えられる。しかし、基板3のC1s XPSスペクトルから得られたサブピークの面積比と比較すると、作製した基板4ではC−O結合由来のサブピークの面積の相対的な減少が見られため、加水分解により保護基が分解し、カルボン酸が導入されたことは分かった。しかし、加水分解処理後にもC−O結合由来のサブピークが見られた。これは完全にポリマー鎖の保護基の加水分解が進行していないためか、基板表面の前処理により除去できなかったコンタミネーションに由来するC−O結合エネルギーであると推察された。
【0342】
図28、29に基板3、4のBr3d(5/2) XPSスペクトルをそれぞれ示す。また、表68に基板3、4から得られたBr3d(5/2) XPSスペクトルをカーブフィッティングしたサブピークの結合エネルギー(eV)を示す。
【0343】
【表68】
【0344】
基板3、基板1のスペクトルを比較すると基板3では71eV付近の基板1のATRP開始剤部位のBrのピークが消失し、78.9eV付近に新たなピークが出現した。このようなスペクトルが得られたのは、基板3で79eV付近に見られたサブスペクトルは、表面開始剤から重合が進行、基板の最表面の構造が変化したことで、基板1のBrと比較して高結合エネルギー側へシフトしたサブピークが得られたと考えられる。基板3、4のBr3d(5/2) XPSスペクトルを比較するとのスペクトルでは78.9eV付近にピークは見られず75eV付近のピークのみが見られた。これは重合の段階で停止、脱離、転移反応を通じて、ポリマー鎖末端のBrが脱離したこと、加水分解処理により、重合したポリマー鎖が連結されているエステル又は光分解性基が連結されているカルバメート部位が分解を示している可能性がある。
【0345】
<4−5>定量分析
作製した各基板をそれぞれ基板1、2、3、4としたときに、測定対照とした各元素O、N、C、Si、Brが基板上にどのような割合(%)で存在するかを、測定した各元素のピークの面積から算出したものを表69に示す。
【0346】
【表69】
【0347】
基板1、2を比較したとき、光照射により光分解性基とそれに連結されたATRP開始剤が脱離するためO、N、C、Brの割合が変化することが予想された。しかし、基板1、2で算出された各元素の割合を比較すると、基板2でBrが顕著な元素の割合の減少を示していたが、他の元素については、わずかな元素の割合の変化が見られるのみであった。基板3について、基板1と比較するとN、Si、Brについて元素の割合の減少が見られ、Cの割合の増加が見られた。これは重合を行ったことによって基板1から基板3のような構造の変化が起こったことにより、膜厚の増加によるCの増加によって検出されるCの割合が増加したことが要因であることに由来する変化であると考えられる。しかし、基板3、4を比較すると基板3に対して基板4ではCの割合の減少、Siの割合の増加が顕著に見られた。これは加水分解処理により表面に存在するCが減少したことに由来する変化であると考えられる。しかし、Siの割合が増加していることから、表面の膜厚が減少し、加水分解処理により、重合したポリマー鎖が連結されているエステル又は光分解性基が連結されているカルバメート部位の分解がこの結果から予想される。
【技術分野】
【0001】
本発明は光分解性ヘテロ二価性架橋剤に関する。
【背景技術】
【0002】
固体基板上の表面化学を制御することは、材料化学の分野において根本的に重要な問題である。例えば、近年、生体膜表面で起こる高度な分子識別機構をセンシングや分離などの技術に応用するために、固体基板上に生体膜を模擬した界面を自在に形成する手法の確立が求められており、この際、固体基板上の表面化学をいかに制御するかが課題となっている。
【0003】
固体基板上の表面化学を制御する方法としては、当該基板の表面を自己組織化単分子膜(Self−Assembled Monolayer:SAM)、ポリマー薄膜、ポリマーブラシなどで修飾する方法がよく知られている。SAMは、分子配向性に優れ、調製が容易なことから広く研究されている。また、ポリマーブラシは、SAMと類似の特性を示すだけでなく、表面被覆率、膜厚、組成、官能基密度、安定性などの点でSAMより優れた特性を示しうる。しかしながら、SAMと比べて調製法がより困難であるという欠点も有する。
【0004】
ポリマーブラシの化学的調製法は、予め合成したポリマーの末端基を基板表面の官能基と反応させることにより基板表面にポリマーを固定させる「grafting to」法と、基板表面に化学的に固定された重合開始基からの重合反応によってポリマーを伸長させる「grafting from」法の二つに大別される。前者の方法では、基板表面へのポリマーの固定化反応が進むにつれ、既に基板表面に固定されたポリマーが障害となって新たなポリマーが基板表面の官能基に到達しにくくなる。よって、基板表面に高濃度の(多くの)ポリマーを固定することが困難であるという問題点を有する。一方、後者の方法では、基板表面に固定された重合開始基から順次モノマーが重合するために、前者の方法で生じるような立体障害の問題が起こりにくく、高濃度のポリマーを固定化することが可能である。
【0005】
当該「grafting from」法に用いる重合方法として、リビングラジカル重合が注目されている。リビングラジカル重合法は、従来のラジカル重合では実現困難であった分子量と分子量分布を制御できる方法であり、適用できるモノマーも広範囲である。
【0006】
近年、このようなリビングラジカル重合を用いるポリマーブラシの調製方法が数多く報告されている。また、リビングラジカル重合を用いたポリマーの伸長により、従来では得られなかった濃厚ポリマーブラシが膜厚を制御した薄膜として得られるようになり、その基礎的研究及び応用研究が盛んに行われている。応用研究の例としては、刺激応答性表面、細胞接着性表面、タンパク質の結合支持体、クロマトグラフィーの支持体、抗バクテリアコート剤、低摩擦表面に関する研究が挙げられる(非特許文献1及び非特許文献2を参照)。
【0007】
さらに、ポリマーブラシのパターニングについての研究もいくつかなされている。パターニングの方法としては、半導体の加工に用いるフォトレジスト及び用いた一般的なフォトリソグラフィー、遠紫外リソグラフィー、マイクロコンタクトプリンティングなどが報告されている(非特許文献1及び非特許文献2を参照)。
【0008】
本発明者は、自己組織化単分子膜の出発原料であるシランカップリング剤や、チオール又はジスルフィド化合物に光切断可能な2−ニトロベンジル誘導体を導入した化合物の合成とそれを用いた感光性SAMの開発に従事してきた。その中で、基板表面と反応するシリル基;アミンと反応する活性カルボナート基;及びチオールと反応するマレイミド基;の3種の反応性基のうちの、2種の反応性基が、光切断可能な2−ニトロベンジル誘導体で連結された光分解性ヘテロ二価性架橋剤を提案している(特許文献1を参照)。当該光分解性ヘテロ二価性架橋剤を用いると、光分解性基で連結されたアミンやチオールと反応するSAMを調製したり、光分解性ブロック共重合体の合成に応用したりすることができる。
【0009】
また、活性カルボナートやマレイミドを末端に持つ光分解性基で連結したジスルフィドを用いると、金や銀などの基板上あるいはそれらの微粒子上に、アミンやチオールと反応するSAMを形成することが可能である(特許文献2を参照)。
【0010】
さらに、最近、ポリエチレングリコールとリビングラジカル重合の開始基とを間に光分解性基を有する化合物の合成が報告されている。そして、当該化合物の開始基にモノマーであるスチレンを反応させ、ポリエチレングリコールとポリスチレンとが光分解性基を介して連結されてなるブロック共重合体を合成している(非特許文献3を参照)。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】特開2007−186471号公報
【特許文献2】特開2007−291005号公報
【非特許文献】
【0012】
【非特許文献1】Wageesha Senaratne,Luisa Andruzzi,Christopher K. Ober Biomacromolecules,2005,6,2427−2448
【非特許文献2】Raphael Barbey,Laurent Lavanant,Dusko Paripovic,Nicolas Schuwer,Caroline Sugnaux,Stefeno Tugulu,Harm−Anton Klok,Chem. Rev.,2009,109,5437−5527
【非特許文献3】Minhyuck Kang,Bongjin Moon,Macromolecules,2009,42,455−458
【発明の概要】
【発明が解決しようとする課題】
【0013】
固体基板上に自在にポリマーブラシを形成するためには、(1)リビングラジカル重合によるポリマー伸長が可能であること;(2)固体基板表面の官能基との結合が可能であること;(3)光照射によるパターニングが可能であること;を具備する架橋剤を固体基板表面に固定化させることが求められる。しかしながら、このような機能を全て満たす架橋剤の合成はこれまでのところ達成されていない。
【0014】
そこで、本発明の目的は、上記(1)〜(3)を具備する新規な架橋剤を提供することを目的とする。
【課題を解決するための手段】
【0015】
本発明の光分解性ヘテロ二価性架橋剤は、リビングラジカル重合の開始基と、反応性基(但し、前記開始基を除く)とが光分解性基を介して連結されてなり、下記化学式1で表される点に特徴を有する。
【0016】
【化1】
【0017】
式中、Iは開始基を表し、Rは反応性基を表し、Dは光分解性基を表し、S1b−S1a及びS2a−S2bはスペーサーを表し、n及びmはそれぞれ独立して0又は1である。
【発明の効果】
【0018】
本発明の光分解性ヘテロ二価性架橋剤は、リビングラジカル重合の開始基を有するため、当該開始基からポリマーの伸長を行うことができる。また、反応性基を有するために、固体基板表面の官能基との結合が可能である。さらに、光分解性基を有するために、光照射によるパターニングが可能である。
【図面の簡単な説明】
【0019】
【図1】チオールカップリング剤(5a)の光照射による1次反応のUVスペクトル変化を表すグラフである。
【図2】チオールカップリング剤(5a)の光照射による2次反応のUVスペクトル変化を表すグラフである。
【図3】シランカップリング剤(3a)の光照射によるUVスペクトル変化を表すグラフである。
【図4】シランカップリング剤(3b)の光照射(0秒)のUVスペクトルを表すグラフである。
【図5】シランカップリング剤(3b)の光照射(0〜280秒)によるUVスペクトル変化を表すグラフである。
【図6】シランカップリング剤(3b)について、ニトロ基由来の吸収のln(Ct/C0)を時間(秒)に対してプロットしたグラフである。
【図7】シランカップリング剤(3b)について、ニトロソ基由来の吸収のln(Ct/C0)を時間(秒)に対してプロットしたグラフである。
【図8】シランカップリング剤(3b)について、ニトロ基由来の吸収のln(Ct/C0)を時間(秒)に対してプロットしたグラフである。
【図9】シランカップリング剤(3b)について、ニトロソ基由来の吸収のln(Ct/C0)を時間(秒)に対してプロットしたグラフである。
【図10】シランカップリング剤(3a)について、ニトロ基由来の吸収のln(Ct/C0)を時間(秒)に対してプロットしたグラフである。
【図11】シランカップリング剤(3a)について、ニトロソ基由来の吸収のln(Ct/C0)を時間(秒)に対してプロットしたグラフである。
【図12】基板1のXPS wideスキャンスペクトルを表すグラフである。
【図13】基板2のXPS wideスキャンスペクトルを表すグラフである。
【図14】基板1のN1s XPSスペクトルを表すグラフである。
【図15】基板2のN1s XPSスペクトルを表すグラフである。
【図16】基板1のBr3d(5/2) XPSスペクトルを表すグラフである。
【図17】基板2のBr3d(5/2) XPSスペクトルを表すグラフである。
【図18】基板1のC1s XPSスペクトルを表すグラフである。
【図19】基板2のC1s XPSスペクトルを表すグラフである。
【図20】シランカップリング剤(3b)について、ln(1−at)を時間(秒)に対してプロットしたグラフである。
【図21】シランカップリング剤(3a)について、ln(1−at)を時間(秒)に対してプロットしたグラフである。
【図22】基板3のXPS wideスキャンスペクトルを表すグラフである。
【図23】基板4のXPS wideスキャンスペクトルを表すグラフである。
【図24】基板3のN1s XPSスペクトルを表すグラフである。
【図25】基板4のN1s XPSスペクトルを表すグラフである。
【図26】基板3のC1s XPSスペクトルを表すグラフである。
【図27】基板4のC1s XPSスペクトルを表すグラフである。
【図28】基板3のBr3d(5/2) XPSスペクトルを表すグラフである。
【図29】基板4のBr3d(5/2) XPSスペクトルを表すグラフである。
【発明を実施するための形態】
【0020】
以下、本発明の好ましい形態を説明するが、本発明の技術的範囲は特許請求の範囲の記載によって定められるべきものであり、以下の形態のみに制限されない。
【0021】
本形態は、リビングラジカル重合の開始基と、反応性基(但し、前記開始基を除く)とが光分解性基を介して連結されてなり、下記化学式1で表される光分解性ヘテロ二価性架橋剤に関する。
【0022】
【化2】
【0023】
式中、Iは開始基を表し、Rは反応性基を表し、Dは光分解性基を表し、S1b−S1a及びS2a−S2bはスペーサーを表し、n及びmはそれぞれ独立して0又は1である。
【0024】
[開始基]
本形態の光分解性ヘテロ二価性架橋剤は、リビングラジカル重合の開始基を有することを特徴とする。リビングラジカル重合は重合過程において、開始反応と成長反応のみからなり、連鎖移動反応及び停止反応などの成長末端を失活させる副反応を伴わない。したがって、重合度は一定のペースで増大し、長さの揃ったポリマーが得られるという特徴を有する。また、モノマーを追加すれば重合反応が再開するため、ブロック重合やポリマーアロイに適する。
【0025】
リビングラジカル重合の開始基は、リビングラジカル重合の成長末端となる構造を有するものであれば特に制限はないが、好ましくは、電子移動ラジカル重合(Atom Transfer Radical Polymerization:ATRP)又はニトロキシドを介したラジカル重合(Nitroxide−mediated radical
polymerization:NMP)の開始基であることが好ましい。
【0026】
具体的に、ATRPの開始基としては、下記化学式3、化学式4、及び化学式5で表される構造を有する開始基が挙げられる。
【0027】
【化3】
【0028】
式中、R2は水素原子又は炭素原子数1〜8のアルキル基を表す。
【0029】
【化4】
【0030】
式中、R4及びR5はそれぞれ独立して水素原子又は炭素原子数1〜8のアルキル基を表し、Xは臭素原子又は塩素原子を表す。
【0031】
【化5】
【0032】
また、NMPの開始剤としては、下記化学式6で表される構造を有する開始基が挙げられる。
【0033】
【化6】
【0034】
上記R2、R4、又はR5で表される炭素原子数1〜8のアルキル基としては、具体的には、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、n−ペンチル基、イソペンチル基、ネオペンチル基、1,2−ジメチルプロピル基、n−ヘキシル基、シクロヘキシル基、1,3−ジメチルブチル基、1−イソプロピルプロピル基、1,2−ジメチルブチル基、n−ヘプチル基、1,4−ジメチルペンチル基、2−メチル−1−イソプロピルプロピル基、1−エチル−3−メチルブチル基、n−オクチル基、2−エチルヘキシル基などが挙げられる。このうち、メチル基であることが好ましい。
【0035】
[反応性基]
反応性基は、本形態の光分解性ヘテロ二価性架橋剤を固体基板に固定化させる場合、固体基板表面の官能基との結合部位となりうる。また、本形態の光分解性ヘテロ二価性架橋剤を用いてブロック共重合体を合成する場合には、ポリマーとの結合部位となりうる。なお、本形態の反応性基の概念には、上述のリビングラジカル重合の開始基となりうる基は含まない。
【0036】
反応性基は、固体基板表面の官能基やポリマーの末端の官能基と結合することができるものであれば特に制限はないが、具体的には、ヒドロキシ基、エチニル基、アジド基、活性エステル基、マレイミド基、加水分解性シリル基(例えば、トリメトキシシリル基、トリエトキシシリル基、トリクロロシリル基、ジエトキシシリル基、ジメトキシシリル基、ジモノクロロシリル基、モノエトキシシリル基、モノメトキシシリル基、モノクロロシリル基など)、アミノ基、カルボキシル基、チオール基、ジスルフィド基、及びスルホ基からなる群から選択される少なくとも1種であることが好ましい。
【0037】
[光分解性基]
光分解性基は、光の照射により開裂する性質を有する官能基であり、様々な構造を有するものが既に知られている。本形態の架橋剤に使用される光分解性基は特に制限はないが、下記化学式2で示される2−ニトロベンジル誘導体骨格を有する二価の光分解性官能基が好ましく用いられる。
【0038】
【化7】
【0039】
式中、R1は水素原子、炭素原子数1〜8のアルキル基、又は置換されたもしくは非置換のフェニル基を表し、R3は水素原子又は炭素原子数1〜8のアルコキシ基を表す。
【0040】
上記R1が炭素原子数1〜8のアルキル基である場合の、当該アルキル基としては、例えば、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、n−ペンチル基、イソペンチル基、ネオペンチル基、1,2−ジメチルプロピル基、n−ヘキシル基、シクロヘキシル基、1,3−ジメチルブチル基、1−イソプロピルプロピル基、1,2−ジメチルブチル基、n−ヘプチル基、1,4−ジメチルペンチル基、2−メチル−1−イソプロピルプロピル基、1−エチル−3−メチルブチル基、n−オクチル基、2−エチルヘキシル基などが挙げられる。
【0041】
上記R1が置換されたもしくは非置換のフェニル基である場合に、当該フェニル基に場合によって存在する置換基としては、例えば、ハロゲン原子、アシル基、アルキル基、フェニル基、アルコキシル基、ハロゲン化アルキル基、ハロゲン化アルコキシル基、ニトロ基、アミノ基、アルキルアミノ基、アルキルカルボニルアミノ基、アリールアミノ基、アリールカルボニルアミノ基、カルボニル基、アルコキシカルボニル基、アルキルアミノカルボニル基、アルコキシスルホニル基、アルキルチオ基、カルバモイル基、アリールオキシカルボニル基、オキシアルキルエーテル基、シアノ基等が例示できるが、これらに限定されるものではない。これらの置換基は、フェニル基に1〜5個置換可能であり、これらの置換基の種類も、複数個置換する場合には同種もしくは異種のいずれであってもよい。上記置換基の具体的な例を以下に示す。
【0042】
上記置換基のうち、ハロゲン原子とは、フッ素原子、塩素原子、臭素原子及びヨウ素原子である。
【0043】
上記置換基のうち、アシル基としては、アセチル基、エチルカルボニル基、プロピルカルボニル基、ブチルカルボニル基、ペンチルカルボニル基、ヘキシルカルボニル基、ベンゾイル基、p−tert−ブチルベンゾイル基等が挙げられる。
【0044】
上記置換基のうち、アルキル基とは、炭素原子数1〜8の直鎖、分岐鎖又は環状のアルキル基である。具体的には、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、n−ペンチル基、イソペンチル基、ネオペンチル基、1,2−ジメチルプロピル基、n−ヘキシル基、シクロヘキシル基、1,3−ジメチルブチル基、1−イソプロピルプロピル基、1,2−ジメチルブチル基、n−ヘプチル基、1,4−ジメチルペンチル基、2−メチル−1−イソプロピルプロピル基、1−エチル−3−メチルブチル基、n−オクチル基、2−エチルヘキシル基等が挙げられる。
【0045】
上記置換基のうち、アルコキシ基は、炭素原子数1〜8の直鎖、分岐鎖又は環状のアルコキシ基である。具体的には、メトキシ基、エトキシ基、n−プロポキシ基、イソプロポキシ基、n−ブトキシ基、イソブトキシ基、sec−ブトキシ基、tert−ブトキシ基、n−ペンチルオキシ基、イソペンチルオキシ基、ネオペンチルオキシ基、1,2−ジメチル−プロポキシ基、n−ヘキシルオキシ基、シクロヘキシルオキシ基、1,3−ジメチルブトキシ基、1−イソプロピルプロポキシ基等が挙げられる。
【0046】
上記置換基のうち、ハロゲン化アルキル基とは、炭素原子数1〜8の直鎖、分岐鎖又は環状のアルキル基の一部がハロゲン化されたものである。具体的には、クロロメチル基、ブロモメチル基、トリフルオロメチル基、クロロエチル基、2,2,2−トリクロロエチル基、ブロモエチル基、クロロプロピル基、ブロモプロピル基等が挙げられる。
【0047】
上記置換基のうち、ハロゲン化アルコキシル基とは、炭素原子数1〜8の直鎖、分岐鎖又は環状のアルコキシル基の一部がハロゲン化されたものである。具体的には、クロロメトキシ基、ブロモメトキシ基、トリフルオロメトキシ基、クロロエトキシ基、2,2,2−トリクロロエトキシ基、ブロモエトキシ基、クロロプロポキシ基、ブロモプロポキシ基等が挙げられる。
【0048】
上記置換基のうち、アルキルアミノ基とは、炭素原子数1〜8のアルキル部位を有するアルキルアミノ基である。具体的には、メチルアミノ基、エチルアミノ基、n−プロピルアミノ基、n−ブチルアミノ基、sec−ブチルアミノ基、n−ペンチルアミノ基、n−ヘキシルアミノ基、n−ヘプチルアミノ基、n−オクチルアミノ基、2−エチルヘキシルアミノ基等が挙げられる。
【0049】
上記置換基のうち、アルコキシカルボニル基とは、アルコキシル基のアルキル基部分にヘテロ原子を含んでもよい炭素原子数1〜8のアルコキシカルボニル、又はヘテロ原子を含んでもよい炭素原子数3〜8の環状アルコキシカルボニルを示す。具体的には、メトキシカルボニル基、エトキシカルボニル基、n−プロポキシカルボニル基、イソプロポキシカルボニル基、n−ブトキシカルボニル基、イソブトキシカルボニル基、sec−ブトキシカルボニル基、tert−ブトキシカルボニル基等が挙げられる。
【0050】
上記R3が炭素原子数1〜8のアルコキシ基である場合の、当該アルコキシ基としては、メトキシ基、エトキシ基、n−プロポキシ基、イソプロポキシ基、n−ブトキシ基、イソブトキシ基、sec−ブトキシ基、tert−ブトキシ基、n−ペンチルオキシ基、イソペンチルオキシ基、ネオペンチルオキシ基、1,2−ジメチル−プロポキシ基、n−ヘキシルオキシ基、シクロヘキシルオキシ基、1,3−ジメチルブトキシ基、1−イソプロピルプロポキシ基等が挙げられる。
【0051】
上記化学式2で示す光分解性基のうち、R1は水素原子またはメチル基であることが好ましく、R3は水素原子またはメトキシ基であることが好ましい。
【0052】
上記化学式2に示す光分解性基は、光照射により、ベンゼン環の1位に結合する炭素原子(ベンジル位の炭素原子)と、その隣の酸素原子、窒素原子、又は硫黄原子(図示せず)との間の結合が開裂する。当該酸素原子、窒素原子、又は硫黄原子は、上記反応性基に含まれる酸素原子、窒素原子、又は硫黄原子であってもよいし、光分解性基と反応性基の間に介在するスペーサーに含まれる酸素原子、窒素原子、又は硫黄原子であってもよい。上記のような二価の光分解性基の一方にリビングラジカル重合の開始基、他方に反応性基を導入することによって、本形態の光分解性ヘテロ二価性架橋剤を合成することができる。この際、光分解性ヘテロ二価性架橋剤は、光照射により、反応性基と、光分解性基との間が開裂するものであってもよいし、開始基と光分解性基との間が開裂するものであってもよい。本明細書では前者の配置を有する光分解性ヘテロ二価性架橋剤について主に説明するが、後者の配置であっても光照射によるパターニングが可能なことはいうまでもない。
【0053】
[スペーサー]
本形態の光分解性ヘテロ二価性架橋剤は、上述の光分解性基と、開始基および/または反応性基との間に、さらにスペーサーを有してもよい。スペーサーの構造は、特に制限はないが、好ましい形態としては、上述の化学式1におけるS1aが、−O−、−C(O)O−、−OC(O)O−、−NH−、−C(O)NH−、−OC(O)NH−、−S−、−C(O)S−、−OC(O)S−からなる群から選択される少なくとも1種であり;S2aが、−O−であり;S1b及びS2bが、それぞれ独立して、アミド結合、イミド結合、エーテル結合、エステル結合、チオエーテル結合、もしくはスルホンアミド結合を有してもよい、炭素原子数1〜20、好ましくは炭素原子数1〜8のアルキレン基からなる群から選択される少なくとも1種である。
【0054】
また、S1b及びS2bに含まれるアルキレン基の具体例としては、メチレン基、エチレン基、プロピレン基、トリメチレン基、テトラメチレン基、ペンタメチレン基、2,2−ジメチルトリメチレン基、ヘキサメチレン基、トリメチルヘキサメチレン基、オクタメチレン基、及びデカメチレン基などが挙げられる。
【0055】
下記化学式8に、本形態の光分解性ヘテロ二価性架橋剤の一例を示す。
【0056】
【化8】
【0057】
上記化学式8では、光分解性基である2−ニトロベンジル誘導体構造を有する官能基の4位側にATRPの開始基が連結され、さらに1位側に反応性基が連結されてなる構造を有する種々の光分解性ヘテロ二価性架橋剤が示されている。化学式8に示すように、反応性基をヒドロキシ基から、活性カルボナート基(スクシンイミジル基)へと変換し、さらに加水分解性シリル基(トリメトキシシリル基)やチオール基やジスルフィド基へと変換することができる。活性カルボナート基はアミノ基と反応するため、化学式8に示すようにアミノ基含有ポリマーを活性カルボナート基と反応させることにより、容易にポリマーを導入することができる。また、加水分解性シリル基はSi基板への固定化に寄与しうる。また、ジスルフィド基は、Au基板への固定化に寄与しうる。なお、チオール基、ジスルフィド基を還元することによりチオール基に変換することができ、また、チオール基又はジスルフィド基を酸化するとスルホ基に変換することができる。なお、これらの反応性基の変換は、従来公知の合成技術を適宜参照することにより容易に行うことができる。
【0058】
また、下記化学式9に、本形態の光分解性ヘテロ二価性架橋剤の他の一例を示す。
【0059】
【化9】
【0060】
上記化学式9に示すように、反応性基は、ヒドロキシ基から、活性カルボナート基、マレイミド基、カルボキシル基、アジド基、ビニル基(アルケン)、又はエチニル基(アルキン)などに変換することができる。なお、これらの反応性基の変換は、従来公知の合成技術を適宜参照することにより容易に行うことができる。
【0061】
本形態の光分解性ヘテロ二価性架橋剤の製造方法としては、後述の実施例で示すような合成ルートが一例として挙げられる。すなわち、光分解性基にスペーサーを導入した後、リビングラジカル重合の開始基を導入し、その後反応性基を導入又は変換する合成方法である。しかしながら、当該合成方法はあくまでも一例に過ぎず、他の合成ルートによってっても本形態の光分解性ヘテロ二価性架橋剤を合成することは可能である。
【0062】
本形態の光分解性ヘテロ二価性架橋剤は、例えば、固体基板上に固定させることにより容易にSAMを形成することができる。すなわち、本形態の光分解性ヘテロ二価性架橋剤の反応性基と基板表面の官能基とを結合させることにより、ポリマーブラシ形成用基板として使用することができる。例えば、上述のように、反応性基を加水分解性シリル基とした光分解性ヘテロ二価性架橋剤(以下、シランカップリング剤とも称する)は、Si基板表面に固定することができる。また、反応性基をチオール基又はジスルフィド基とした場合は、金(Au)基板表面に固定することができる。これらのSAMは細胞パターニングや有機薄膜トランジスタなどへの応用が期待される。
【0063】
また、SAMを形成した後、固体基板に固定された光分解性ヘテロ二価性架橋剤の開始剤からリビングラジカル重合によってポリマーを伸長させ、ポリマーブラシを作製することができる。
【0064】
なお、本形態の光分解性ヘテロ二価性架橋剤は、ポリマーブラシ以外の用途にも好適に使用されうる。例えば、反応性基にポリマーを導入し、開始基からリビングラジカル重合によってポリマーを伸長させることにより、2種のポリマーが光分解性基で連結したブロック共重合体を得ることもできる。このようなブロック共重合体は、光分解性ポリマーソーム、ナノポーラス材料などへの応用が期待される。
【実施例】
【0065】
[実施例1]光分解性ヘテロ二価性架橋剤の合成
測定機器は以下のものを使用した。
【0066】
【表1】
【0067】
GPCは以下の測定条件で行った。
【0068】
【表2】
【0069】
<1−1>1−(4−(3−(2−ブロモプロパノイルオキシ)プロピルオキシ)−5−メトキシ−2−ニトロフェニル)−エチル N−(11−メルカプトウンデシル)カルバメート(5a)の合成
【0070】
【化10】
【0071】
<1−1−1>3−(4−アセチル−2−メトキシ−5−ニトロフェノキシ)プロパン−1−オールの合成
【0072】
【化11】
【0073】
窒素雰囲気下、50mL二口ナスフラスコに4−ヒドロキシ−5−メトキシ−2−ニトロアセトフェノン1.57g(7.43mmol)、60%水素化ナトリウム0.310g(7.73mmol)、脱水DMF16mLを加え、室温で30分間撹拌した。その後3−ブロモプロパン−1−オール1.03g(7.43mmol)、DMF4mLを加え、80℃で21時間撹拌した。室温まで冷却後、濃縮、水200mLを加え、酢酸エチルで抽出(200mL×4)、洗浄(飽和食塩水100mL×2)、無水硫酸マグネシウムで乾燥、濃縮し、カラムクロマトグラフィー(ヘキサン:酢酸エチル=2:3)、真空乾燥を行い、茶白色固体を得た。
【0074】
【表3】
【0075】
<1−1−2>3−(4−(1−ヒドロキシエチル)−2−メトキシ−5−ニトロフェノキシ)プロパン−1−オールの合成
【0076】
【化12】
【0077】
氷浴上において100mLナスフラスコに3−(4−アセチル−2−メトキシ−5−ニトロフェノキシ)−プロパン−1−オール0.684g(2.54mmol)、メタノール7mL、THF5mL、徐々にNaBH40.194g(5.13mmol)を加え、室温で1時間撹拌した。濃縮、水50mLを加え、抽出(クロロホルム100mL×3)、洗浄(飽和食塩水100mL×2)、無水硫酸マグネシウムで乾燥、濃縮、真空乾燥し、黄色固体を得た。
【0078】
【表4】
【0079】
<1−1−3>3−(4−(1−ヒドロキシエチル)−2−メトキシ−5−ニトロフェノキシ)プロピル 2−ブロモプロピオナート(1a)の合成
【0080】
【化13】
【0081】
50mL二口ナスフラスコに3−(4−(1−ヒドロキシエチル)−2−メトキシ−5−ニトロフェノキシ)−プロパノール0.199g(0.734mmol)、トリエチルアミン0.0743g(0.734mmol)、2‐ブロモプロピオン酸ブロミド0.160g(0.734mmol)、脱水クロロホルム7mLを加え、窒素雰囲気下とし、室温で20時間撹拌した。濃縮、カラムクロマトグラフィー(酢酸エチルのみ)、濃縮、真空乾燥し、黄色固体を得た。
【0082】
【表5】
【0083】
<1−1−4>3−(2−メトキシ−5−ニトロ−4−(1−(N−スクシンイミジルオキシカルボニルオキシ)エチル)−フェノキシ)プロピル 2−ブロモプロピオナート(2a)の合成
【0084】
【化14】
【0085】
50mL二口ナスフラスコに3−(4−(1−ヒドロキシエチル)−2−メトキシ−5−ニトロフェノキシ)−プロピル 2−ブロモプロピオナート0.361g(0.889mmol)、トリエチルアミン0.275g(2.72mmol)、ジ(N−スクシンイミジル)カルボナート0.804g(3.14mmol)、脱水アセトニトリル7mLを加え、窒素雰囲気下とし、室温で20時間撹拌した。濃縮、カラムクロマトグラフィー(酢酸エチルのみ)、濃縮、真空乾燥し、淡黄色固体を得た。
【0086】
【表6】
【0087】
<1−1−5>11−アミノウンデカン−1−チオールの合成
【0088】
【化15】
【0089】
100mLナスフラスコにN−(11−アセチルチオウンデシル)フタルイミド0.556g(1.48mmol)、ヒドラジン一水和物0.231g(4.61mmol)、2−プロパノール20mLを加え、85℃で5時間撹拌し、濃縮、洗浄(クロロホルム)、メンブランろ紙を用いてろ過、H2Oを加えて抽出(クロロホルム50mL×4)、洗浄(飽和食塩水50mL×4)、乾燥、ろ過、カラムクロマトグラフィー(クロロホルムのみ→クロロホルム:メタノール=20:1)、濃縮、真空乾燥し、白色固体を得た。
【0090】
【表7】
【0091】
<1−1−6>1−(4−(3−(2−ブロモプロパノイルオキシ)プロピルオキシ)−5−メトキシ−2−ニトロフェニル)−エチル N−(11−メルカプトウンデシル)カルバメート(5a)の合成
【0092】
【化16】
【0093】
50mL二口ナスフラスコに11−アミノウンデカン−1−チオール0.0451g(0.222mmol)、トリエチルアミン0.0561g(0.554mmol)、3−(2−メトキシ−5−ニトロ−4−(1−N−スクシンイミジルオキシカルボニルオシキエチル)フェノキシ)プロピル 2−ブロモプロピオナート0.237g(0.433mmol)、脱水THF6mLを加え、窒素雰囲気下とし、室温で20時間撹拌した。濃縮、カラムクロマトグラフィー(酢酸エチルのみ)、濃縮、真空乾燥し、黄色粘体を得た。
【0094】
【表8】
【0095】
<1−2>1−(4−(3−(2−ブロモプロパノイルオキシ)プロピルオキシ)−5−メトキシ−2−ニトロフェニル)エチル N−(3−(トリメトキシシリル)プロピル)カルバメート(3a)の合成
<1−2−1>1−(4−(3−(2−ブロモプロパノイルオキシ)プロピルオキシ)−5−メトキシ−2−ニトロフェニル)エチル N−((トリメトキシシリル)プロピル)カルバメート(3a)の合成
【0096】
【化17】
【0097】
窒素雰囲気下で50mL二口ナスフラスコに<1−1−4>で合成した3−(2−メトキシ−5−ニトロ−4−(1−(N−スクシンイミジルオキシカルボニルオキシ)エチル)−フェノキシ)−プロピル 2−ブロモプロピオナート(2a)0.103g(0.189mmol)、無水THF18mL、3−(トリメトキシシリル)プロピルアミン0.104g(0.578mmol)、トリエチルアミン0.168g(1.85mmol)を加え20時間攪拌した。濃縮、洗浄(酢酸エチル)、吸引ろ過、ろ液を濃縮、真空乾燥し、黄色固体0.0835g(粗精製物)を得た。
【0098】
<1−2−2>1−(4−(3−(2−ブロモプロパノイルオキシ)プロピルオキシ)−5−メトキシ−2−ニトロフェニル)エチル N−(3−(トリメトキシシリル)プロピル)カルバメート(3a)の合成
【0099】
【化18】
【0100】
窒素雰囲気下、20mL二口ナスフラスコに<1−1−3>で合成した3−(4−(1−ヒドロキシエチル)−2−メトキシ−5−ニトロフェノキシ)プロピル 2−ブロモプロピオナート(1a)0.151g(0.373mmol)、無水THF0.5mL、パスツールピペットで3−(イソシアナトプロピル)トリメトキシシラン11滴(1滴 0.0121g)、パスツールピペットでジラウリン酸ジブチルスズ(DBTL)2滴を加え、室温で8時間撹拌した。濃縮、真空乾燥、カラムクロマトグラフィー(ジクロロメタンのみ×2[NH2シリカゲル使用])、濃縮、真空乾燥、中圧カラムクロマトグラフィー(ヘキサン:酢酸エチル:テトラメトキシシラン=1:1:0.02[従来シリカゲル使用])、真空乾燥を行い、黄色液体0.0409(0.0669mmol、収率:18%)を得た。
【0101】
【表9】
【0102】
<1−3>1−(4−(3−(2−ブロモ−2−メチルプロパノイルオキシ)プロピルオキシ)−5−メトキシ−2−ニトロフェニル)エチル N−(3−(トリメトキシシリル)プロピル)カルバメート(3b)の合成
<1−3−1>3−(4−(1−ヒドロキシエチル)−2−メトキシ−5−ニトロフェノキシ)プロピル 2−ブロモ−2−メチルプロピオナート(1b)の合成
【0103】
【化19】
【0104】
50mL二口ナスフラスコに<1−1−2>で合成した3−(4−(1−ヒドロキシエチル)−2−メトキシ−5−ニトロフェノキシ)プロパン−1−オール0.5002g(1.8439mmol)を入れ、1時間真空乾燥した。これに窒素雰囲気下で、脱水クロロホルム25mLを加え、トリエチルアミン0.1869g(1.8439mmol)を脱水クロロホルム1.5mLに溶かした溶液、2−ブロモイソ酪酸ブロミド0.5090g(2.2127mmol)を脱水クロロホルム1.5mLに溶かした溶液をパスツールピペットでゆっくり加え、室温で24時間撹拌した。これを濃縮し、H2O100mL、1N HCl0.5mLを加え、抽出(クロロホルム100mL×3)、無水硫酸マグネシウムを加え乾燥させ、ろ過、濃縮し、オープンカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:2)、濃縮、真空乾燥を行い、黄色固体を得た。
【0105】
【表10】
【0106】
<1−3−2>3−(2−メトキシ−5−ニトロ−4−(1−(N−スクシンイミジルオキシカルボニルオキシ)エチル)−フェノキシ)プロピル 2−ブロモ−2−メチルプロピオナート(2b)の合成
【0107】
【化20】
【0108】
50mL二口ナスフラスコに3−(4−(1−ヒドロキシエチル)−2−メトキシ−5−ニトロフェノキシ)プロピル 2−ブロモ−2−メチルプロピオナート0.2853g(0.6789mmol)を入れ、2時間真空乾燥した。これを、窒素雰囲気下で、脱水アセトニトリル1mL、トリエチルアミン0.2061g(2.0367mmol)を脱水アセトニトリル1.5mLに溶かした溶液、ジ(N−スクシンイミジル)カルボナート0.5217g(2.0367mmol)を加え、最後に脱水アセトニトリル4mL加え、室温で24時間撹拌した。これを濃縮し、H2O50mL、抽出(クロロホルム50mL×3)、洗浄(NaHCO3100mL×6)、無水硫酸マグネシウムを加え乾燥させ、ろ過、濃縮し、オープンカラムクロマトグラフィー(カラム直径3.5cm、シリカゲル高さ17cm、ジクロロメタン:酢酸エチル=10:1)を行った。モノと原料の2つのスポットが重なる部分が少量残っていたため、重なって出てきた部分をもう一度回収し、さらにオープンカラムクロマトグラフィー(カラム直径;2.5cm、シリカゲル高さ;17cm、ジクロロメタン:酢酸エチル=10:1)を行い、これを濃縮、真空乾燥し、黄色粘体を得た。
【0109】
【表11】
【0110】
<1−4>ポリ(エチレングリコール)−NH−光分解性基質(4b)の合成
<1−4−1>ポリ(エチレングリコール)−NH−光分解性基質(4b)の合成
【0111】
【化21】
【0112】
50mL二口ナスフラスコに<1−3−2>で合成した3−(2−メトキシ−5−ニトロ−4−(1−(N−スクシンイミジルオキシカルボニルオキシ)エチル)−フェノキシ)プロピル 2−ブロモ−2−メチルプロピオナート(2b)0.1356g(0.2416mmol)、PEG−NH2(数平均分子量2000(PEG2000))0.1757g(0.0805mmol)加え、3時間真空乾燥した。これを窒素雰囲気下で、脱水THF1.5mL、トリエチルアミン0.2445g(2.4160mmol)を加え室温で終夜撹拌した。これを再沈殿(2mLのTHFに溶かし、50mLのジエチルエーテルに滴下し氷浴上1時間撹拌)し、吸引ろ過、真空乾燥し、白色固体を得た。
【0113】
【表12】
【0114】
<1−5>1−(4−(3−(2−ブロモ−2−メチルプロパノイルオキシ)プロピルオキシ)−5−メトキシ−2−ニトロフェニル)エチル N−(3−(トリメトキシシリル)プロピル)カルバメート(3b)の合成
【0115】
【化22】
【0116】
<1−5−1>4−ベンジルオキシ−3−メトキシアセトフェノンの合成
【0117】
【化23】
【0118】
300mLナスフラスコに4−ヒドロキシ−3−メトキシアセトフェノン20.0g(120mol)とアセトン153mL、K2CO3 16.6g(120mol)を入れ、室温で30分間撹拌した。その後、ベンジルブロミド20.7g(120mol)を加え、80℃で4時間還流した。濃縮し、抽出(H2O 200mL、クロロホルム120mL×4回)を行い、有機層を乾燥(無水MgSO4)、ろ過、濃縮し再結晶を2回(第一結晶 ウォーターバス50℃、酢酸エチル5mL;第二結晶 ウォーターバス50℃、酢酸エチル2mL、ヘキサン パスツール2滴)行い、メンブランろ紙を用いて吸引ろ過、真空乾燥し、白色固体を得た。
【0119】
【表13】
【0120】
<1−5−2>4−ベンジルオキシ−5−メトキシ−2−ニトロアセトフェノンの合成
【0121】
【化24】
【0122】
300mLナスフラスコに4−ベンジルオキシ−3−メトキシアセトフェノン18.0g(70.2mmol)と酢酸200mLを入れ、氷浴上で発煙HNO3 23mLを42分間で滴下し、18時間撹拌した。反応溶液を冷水280mLの中に入れ、15分間撹拌し固体を析出させ、メンブランろ紙を用いて吸引ろ過、H2Oで固体を洗浄、再結晶を2回(第一結晶 ウォーターバス52℃、アセトン160mL、ヘキサン3mL;第二結晶 ウォーターバス55℃、アセトン48mL)行い、真空乾燥し、黄色固体を得た。
【0123】
【表14】
【0124】
<1−5−3>4−ヒドロキシ−5−メトキシ−2−ニトロアセトフェノンの合成
【0125】
【化25】
【0126】
300mLナスフラスコに4−ベンジルオキシ−5−メトキシ−2−ニトロアセトフェノン19.1g(63.4mmol)とCF3COOH 200mLを入れ、室温で終夜撹拌した。濃縮し、5% NaHCO3 320mL、2N HClを30mL入れ、抽出(酢酸エチル300mL×1回、200mL×1回、100mL×2回)を行い、有機層を乾燥(無水MgSO4)、ろ過、濃縮、再結晶を2回(第一結晶 ウォーターバス55℃、酢酸エチル80mL、ヘキサンmL;第二結晶 ウォーターバス55℃、酢酸エチル25mL)行い、吸引ろ過、真空乾燥し、黄緑色固体を得た。
【0127】
【表15】
【0128】
<1−5−4>3−(4−アセチル−2−メトキシ−5−ニトロフェノキシ)プロパン−1−オールの合成
【0129】
【化26】
【0130】
窒素雰囲気下で50mL二口ナスフラスコに脱水DMF16mLと60% NaH 0.28g(7.00mmol)、4−ヒドロキシ−5−メトキシ−2−ニトロアセトフェノン1.50g(7.10mmol)を入れ、室温で30分間撹拌した。3−ブロモプロパン−1−オール1.00g(7.19mmol)を加え85℃の油浴上で22時間撹拌した。減圧留去、抽出(H2O 200mL、酢酸エチル200mL×3回、100mL×1回)、洗浄(飽和NaCl水溶液100mL×2回)、有機層を乾燥(無水MgSO4)、ろ過、濃縮、オープンカラムクロマトグラフィーを2回(1回目 ヘキサン:酢酸エチル=1:3;2回目 ヘキサン:酢酸エチル=1:3)行い、真空乾燥し、白色固体を得た。
【0131】
【表16】
【0132】
<1−5−5>3−(4−(1−ヒドロキシエチル)−2−メトキシ−5−ニトロフェノキシ)プロパン−1−オールの合成
【0133】
【化27】
【0134】
100mLナスフラスコに3−(4−アセチル−2−メトキシ−5−ニトロフェノキシ)プロパン−1−オール1.11g(4.12mmol)とメタノール12mL、THF 9mLを入れ、氷浴上でNaBH4 0.33g(8.72mmol)を入れて1時間撹拌した。濃縮、抽出(H2O 80mL、クロロホルム100mL×3回)、洗浄(飽和NaCl水溶液100mL×3回)、乾燥(無水MgSO4)、ろ過、濃縮、真空乾燥し、黄色固体を得た。
【0135】
【表17】
【0136】
<1−5−6>3−(4−(1−ヒドロキシエチル)−2−メトキシ−5−ニトロフェノキシ)プロパン 2−ブロモ−2−メチルプロピオナートの合成
【0137】
【化28】
【0138】
100mL二口ナスフラスコに3−(4−(1−ヒドロキシエチル)−2−メトキシ−5−ニトロフェノキシ)プロパン−1−オール0.58g(2.15mmol)入れ、1時間真空乾燥した。これに水浴上で脱水CHCl3 25mL、メスピペットを用いてトリエチルアミン0.30mL(0.22g、2.17mmol)、脱水CHCl3 10mLに溶かした2−ブロモイソブチリルブロミド0.27mL(0.51g、2.21mmol)を滴下ロートでゆっくり加え、室温で17.5時間撹拌した。濃縮、抽出(H2O 100mL、1N HCl パスツールピペット3滴、クロロホルム100mL×3回)、乾燥(無水MgSO4)、ろ過、濃縮し、オープンカラムクロマトグラフィーを2回(1回目 ヘキサン:酢酸エチル=1:1、2回目 ヘキサン:酢酸エチル=15:1)行い、濃縮、真空乾燥し黄色固体を得た。
【0139】
【表18】
【0140】
<1−5−7>3−(2−メトキシ−5−ニトロ−4−(1−(N−スクシンイミジルオキシカルボニルオキシ)エチル)−フェノキシ)プロピル 2−ブロモ−2−メチルプロピオナートの合成
【0141】
【化29】
【0142】
50mL二口ナスフラスコに3−(4−(1−ヒドロキシエチル)−2−メトキシ−5−ニトロフェノキシ)プロパン 2−ブロモ−2−メチルプロピオナート0.200g(0.476mmol)を入れ、1時間真空乾燥した。窒素雰囲気下で乾燥CH3CN 3mL、トリエチルアミン0.2mL(0.146g、1.44mmol)、ジ(N−スクシンイミジル)カルボナート0.369g(1.44mmol)を加えた。室温で24時間撹拌した。濃縮、抽出(H2O 50mL、CHCl3 50mL×3回)、洗浄(5% NaHCO3水溶液50mL×6回)、乾燥(無水MgSO4)、ろ過、濃縮、オープンカラムクロマトグラフィー(ジクロロメタン:酢酸エチル=10:1)を行い、濃縮、真空乾燥し、黄白色固体を得た。
【0143】
【表19】
【0144】
<1−5−8>1−(4−(3−(2−ブロモ−2−メチルプロパノイルオキシ)プロピルオキシ)−5−メトキシ−2−ニトロフェニル)エチル N−(3−(トリメトキシシシル)プロピル)カルバマートの合成
【0145】
【化30】
【0146】
50mL二口ナスフラスコに3−(2−メトキシ−5−ニトロ−4−(1−(N−スクシンイミジルオキシカルボニルオキシ)エチル)−フェノキシ)プロピル 2−ブロモ−2−メチルプロピオナート0.174g(0.310mmol)を入れ、窒素雰囲気下で脱水THF 6mL、脱水THF 1mLに溶かした3−アミノプロピルトリメトキシシラン0.0586g(0.327mmol)を加え、室温で1時間撹拌した。濃縮、洗浄(酢酸エチルのみ)、濃縮し、中圧カラムクロマトグラフィーを2回(1回目および2回目 ヘキサン:酢酸エチル:アセトン=2:1:1(1.0%テトラメトキシシラン含有))を行い、濃縮、真空乾燥(湯浴中40℃)し、黄色粘体を得た。
【0147】
【表20】
【0148】
<1−6>ポリ(エチレングリコール)−NH−光分解性基質(4c)の合成
【0149】
【化31】
【0150】
<1−6−1>4−ベンジルオキシ−3−メトキシベンズアルデヒドの合成
【0151】
【化32】
【0152】
300mLナスフラスコにバニリン20.0g(131mmol)、アセトン89mL、K2CO3 18.1g(131mmol)を入れ、室温で40分間撹拌した。その後、ベンジルブロミド22.5g(132mmol)加え、80℃で4時間還流した。室温まで放冷後、濃縮し、H2O 200mLを加え、クロロホルム100mL×4回で抽出、無水MgSO4で乾燥、ろ過、濃縮した。再沈殿(アセトン31mLに溶解し、氷浴上のヘキサン310mLに滴下)し、メンブランろ紙を用いて吸引ろ過、真空乾燥を行い、白色固体25.9g(107mmol)得た。この操作で生じたろ液を濃縮、再沈殿(アセトン10mLに溶解し、氷浴上のヘキサン100mLに滴下)し、メンブランろ紙を用いて吸引ろ過、真空乾燥を行い、白色固体4.69g(19.3mmol)得た。
【0153】
【表21】
【0154】
<1−6−2>4−ベンジルオキシ−5−メトキシ−2−ニトロベンズアルデヒドの合成
【0155】
【化33】
【0156】
500mLナスフラスコに4−ベンジルオキシ−3−メトキシベンズアルデヒド15.8g(65.2mmol)、酢酸130mLを入れて撹拌し、氷浴上で発煙HNO3 31mLを65分かけて滴下し、17時間氷浴上で撹拌した。反応溶液を冷水270mLに入れ、15分間撹拌し、固体を析出させた。メンブランろ紙を用いて吸引ろ過、H2Oで固体を洗浄、酢酸エチルに溶かし、無水MgSO4で乾燥させた後、濃縮し、再結晶(ウォーターバス50℃、酢酸エチル150mLで溶かし、室温で静置)、メンブランろ紙を用いて吸引ろ過、真空乾燥を行い、黄色固体9.51g(33.1mmol)を得た。再度再結晶(ウォーターバス50℃、酢酸エチル45mLで溶かし、ヘキサンを5滴加えた)を行い、メンブランろ紙を用いて吸引ろ過、真空乾燥を行い、黄色固体2.34g(8.16mmol)を得た。
【0157】
【表22】
【0158】
<1−6−3>6−ニトロバニリンの合成
【0159】
【化34】
【0160】
200mL二口ナスフラスコに4−ベンジルオキシ−5−メトキシ−2−ニトロベンズアルデヒド9.55g(33.2mmol)、トリフルオロ酢酸61mLを撹拌しながら入れ、60℃で1時間撹拌した。反応溶液を冷ヘキサン180mLの中に入れ、15分間撹拌し、固体を析出させた。メンブランろ紙を用いて吸引ろ過を行い、ヘキサンで固体を洗浄、真空乾燥を行い、再結晶(ウォーターバス58℃、THF 75mLで溶かし、パスツールピペットでヘキサンを5滴加えた)を行った。メンブランろ紙を用いて吸引ろ過、ヘキサンで固体を洗浄、真空乾燥を行い、黄色固体3.89g(19.7mmol)を得た。この操作で生じたろ液を、濃縮し、再結晶(ウォーターバス59℃、酢酸エチル25mLで溶かし、ヘキサンを5滴加えた)を行い、メンブランろ紙を用いて吸引ろ過、真空乾燥を行い、黄色固体0.973g(4.94mmol)得た。
【0161】
【表23】
【0162】
<1−6−4>2−(3−ヨード−1−プロピル)オキサンの合成
【0163】
【化35】
【0164】
窒素雰囲気下、水浴上で20mL二口ナスフラスコに、3−ヨードプロパン−1−オール2.7mL(5.24g、28.2mmol、1.0eq)、脱水ジクロロメタン5mL(6.63g、78.1mmol)、ピリジニウム p−トルエンスルホナート0.270g(1.08mmol、0.04eq)、3,4−ジヒドロ−2H−ピラン2.5mL(2.3g、27.4mmol、1.0eq)を加え、2時間撹拌し、濃縮した。飽和NH4Cl水溶液100mLを加え、抽出(ジクロロメタン100mL×3回)、洗浄(飽和NaCl水溶液100mL×2回)、無水MgSO4で乾燥、ろ過、濃縮、オープンカラムクロマトグラフィー(ヘキサン:酢酸エチル=5:1)を行い、濃縮、真空乾燥を行った。再び、オープンカラムクロマトグラフィー(ヘキサン:酢酸エチル=5:1)を行い、濃縮、真空乾燥を行って、淡橙色液体7.04g(26.1mmol)を得た。
【0165】
【表24】
【0166】
<1−6−5>4−(3−(2−オキサノキシ)プロピル)−5−メトキシ−2−ニトロベンズアルデヒドの合成
【0167】
【化36】
【0168】
窒素雰囲気下、50mL二口ナスフラスコに脱水DMF10mL、K2CO3 0.351g(2.54mmol、0.5eq)、6−ニトロバニリン1.00g(5.08mmol、1.0eq)を入れ室温で30分間撹拌し、2−(3−ヨード−1−プロポキシ)オキサン1.37g(5.08mmol、1.0eq)加え、40℃で7時間、80℃で2時間撹拌後、減圧留去、H2O 100mLを加え、抽出(酢酸エチル100mL×3回)、洗浄(飽和NaCl水溶液100mL×2回)、無水MgSO4で乾燥、ろ過、濃縮を行い黄色固体1.41g(4.17mmol)を得た。
【0169】
【表25】
【0170】
<1−6−6>4−(1−ヒドロキシプロポキシ)−5−メトキシ−2−ニトロベンズアルデヒドの合成
【0171】
【化37】
【0172】
50mLナスフラスコに4−(3−(2−オキサノキシ)プロピル)−5−メトキシ−2−ニトロベンズアルデヒド0.502g(1.48mmol)、テトラヒドロフラン3.0mL、1N HCl 0.15mLを入れ、室温で2時間撹拌した。さらに、1N HClを0.15mL加え、室温で2時間撹拌し、1N HClを0.3mL加え、室温で2時間撹拌した。さらに、1N HClを0.9mL加え、室温で17時間撹拌し、濃縮、オープンカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1)、濃縮、真空乾燥を行い、淡黄色固体を0.270g(1.05mmol)を得た。
【0173】
【表26】
【0174】
<1−6−7>4−(1−ヒドロキシプロポキシ)−5−メトキシ−2−ニトロベンズアルデヒドの合成
【0175】
【化38】
【0176】
窒素雰囲気下、100mL二口ナスフラスコに脱水アセトニトリル32mL、K2CO3 0.0720g(0.521mmol、0.5eq)、6−ニトロバニリン0.270g(1.37mmol、1.3eq)を入れて室温で30分間撹拌し、3−ヨードプロパン−1−オール0.1mL(0.194g、1.04mmol、1.0eq)を加え、室温で1時間、40℃で2時間、80℃で18時間撹拌し、濃縮、H2O 25mLを加え、抽出(酢酸エチル25mL×5回)、洗浄(飽和NH4Cl水溶液25mL×2回)、無水MgSO4で乾燥、ろ過、濃縮、オープンカラムクロマトグラフィー(ジクロロメタン:酢酸エチル=1:1)、濃縮、真空乾燥を行い、黄色固体0.157g(0.615mmol)を得た。
【0177】
【表27】
【0178】
<1−6−8>3−(4−ホルミル−2−メトキシ−5−ニトロフェノキシ)プロピル 2−ブロモ−2−メチルプロピオナートの合成
【0179】
【化39】
【0180】
20mL二口ナスフラスコに4−(1−ヒドロキシプロポキシ)−5−メトキシ−2−ニトロベンズアルデヒド0.144g(0.565mmol)を入れ、1時間真空乾燥を行った。これに窒素雰囲気下で、脱水クロロホルム17mLを加え、脱水クロロホルム1mLに溶かしたトリエチルアミン0.0571g(0.564mmol)と、脱水クロロホルム1mLに溶かした2−ブロモイソブチリルブロミド0.161g(0.702mmol)をパスツールピペットでゆっくり加え、室温で24時間撹拌した。濃縮し、H2O 25mLを加え、抽出(クロロホルム25mL×4回)、無水MgSO4で乾燥、濃縮、オープンカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1)、濃縮、真空乾燥を行い、黄色固体0.190g(0.437mmol)を得た。
【0181】
【表28】
【0182】
<1−6−9>3−(4−(1−ヒドロキシメチル)−2−メトキシ−5−ニトロフェノキシ)プロピル 2−ブロモ−2−メチルプロピオナートの合成
【0183】
【化40】
【0184】
氷浴上で50mLナスフラスコに3−(4−ホルミル−2−メトキシ−5−ニトロフェノキシ)プロピル 2−ブロモ−2−メチルプロピオナート0.152g(0.376mmol、1.0eq)、メタノール9mL、テトラヒドロフラン6mLを入れ、NaBH4 0.006g(0.159mmol、0.25eq)をゆっくりと加え、室温で3時間撹拌した。これを濃縮し、H2O 15mLを加え、抽出(クロロホルム25mL×1回、20mL×3回)、洗浄(飽和NaCl水溶液20mL×2回)、無水MgSO4で乾燥、ろ過、濃縮、真空乾燥、オープンカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1)、濃縮、真空乾燥を行い、橙色粘体0.106g(0.261mmol)を得た。
【0185】
【表29】
【0186】
<1−6−10>3−(2−メトキシ−5−ニトロ−4−(1−(N−スクシンイミジルオキシカルボニルオキシ)メチル)−フェノキシ)プロピル 2−ブロモ−2−メチルプロピオナートの合成
【0187】
【化41】
【0188】
50mL二口ナスフラスコに3−(4−(1−ヒドロキシメチル)−2−メトキシ−5−ニトロフェノキシ)プロピル 2−ブロモ−2−メチルプロピオナート0.0629g(0.155mmol)を加え、2.5時間真空乾燥を行った。窒素雰囲気下で、脱水アセトニトリル3mL、1mLの脱水アセトニトリルで溶かしたトリエチルアミン0.0410g(0.405mmol)、ジ(N−スクシンイミジル)カルボナート0.1145g(0.447mmol)さらに脱水アセトニトリル6mLを加え、室温で24時間撹拌を行った。その後、濃縮し、H2O 15mLを加え、抽出(クロロホルム15mL×3回)、洗浄(飽和NaHCO3水溶液25mL×5回)、無水MgSO4で乾燥、濃縮、オープンカラムクロマトグラフィー(ジクロロメタン:酢酸エチル=10:1)、濃縮、真空乾燥を行い、黄色固体0.051g(0.0932mmol)を得た。
【0189】
【表30】
【0190】
<1−6−11>ポリ(エチレングリコール)−NH−光分解性基質(4c)の合成
【0191】
【化42】
【0192】
50mL二口ナスフラスコに3−(2−メトキシ−5−ニトロ−4−(1−(N−スクシンイミジルオキシカルボニルオキシ)メチル)−フェノキシ)プロピル 2−ブロモ−2−メチルプロピオナートを0.077g(0.141mmol、3.0eq)、PEG−NH2(数平均分子量2000(PEG2000))を0.092g(0.046mmol、1.0eq)加え、3時間真空乾燥を行った。その後、窒素雰囲気下で脱水テトラヒドロフラン1mL、トリエチルアミン0.19mL(0.138g、1.36mmol)を加え、室温で49時間攪拌し、濃縮、メンブランろ紙を用いて吸引ろ過、濃縮、再沈殿(THF 1mLに溶かし、10mLのジエチルエーテルに滴下し、氷浴上で1時間攪拌)し、吸引ろ過、真空乾燥を行った。再び、再沈殿(THF 1mLに溶かし、10mLのジエチルエーテルに滴下し、氷浴上で1時間攪拌)し、吸引ろ過、凍結乾燥を行い、白色固体0.0467g(0.0174mmol)得た。
【0193】
【表31】
【0194】
<1−7>1−(5−メトキシ−2−ニトロ−4−(3−トリメトキシシリルプロポキシ)フェニル)エチル 2−ブロモ−2−メチルプロピオナートの合成
【0195】
【化43】
【0196】
<1−7−1>1−(4−アリルオキシ−5−メトキシ−2−ニトロフェニル)エチル 2−ブロモ−2−メチルプロピオナートの合成
【0197】
【化44】
【0198】
窒素雰囲気下で30mL二口ナスフラスコに1−(4−アリルオキシ−5−メトキシ−2−ニトロフェニル)エタノール(特開平2007−186472号公報の段落「0052」〜「0067」に記載の方法により合成した)を0.300g(1.18mmol)、脱水クロロホルム10mL、メスピペットを用いてTEA0.17mL(1.22mmol)、2−ブロモイソブチリルブロミド0.58mL(4.71mmol)入れ、48時間撹拌した。濃縮し、抽出(H2O 50mL、2N HCl3mL、クロロホルム50mL×3回)、洗浄(飽和NaHCO3 50mL×7回)を行い、乾燥(無水MgSO4)、ろ過、濃縮し、カラムクロマトグラフィー(クロロホルムのみ)を2回行い、濃縮、真空乾燥し、黄白色固体を得た。
【0199】
【表32】
【0200】
<1−7−2>1−(5−メトキシ−2−ニトロ−4−(3−トリメトキシシリルプロポキシ)フェニル)エチル 2−ブロモ−2−メチルプロピオナートの合成
【0201】
【化45】
【0202】
20mL二口ナスフラスコに1−(4−アリルオキシ−5−メトキシ−2−ニトロフェニル)エチル 2−ブロモ−2−メチルプロピオナートを0.066g(0.164mmol)入れ、窒素雰囲気下で脱水THF 1mL、1mLの脱水THFに溶かしたトリメトキシシランを0.072g(0.589mmol)、Karstedt触媒7滴を入れ、窒素下にて2時間撹拌した。濃縮し、中圧カラムクロマトグラフィー(ヘキサン:酢酸エチル=2:1(1.0%テトラメトキシシラン含有))し、濃縮、真空乾燥(湯浴中、40℃)し、黄色粘体を得た。
【0203】
【表33】
【0204】
<1−8>2−ニトロ−5−(3−トリメトキシシリルプロポキシ)ベンジル 2−ブロモ−2−メチルプロパノアートの合成
【0205】
【化46】
【0206】
<1−8−1>5−アリルオキシ−2−ニトロベンズアルデヒドの合成
【0207】
【化47】
【0208】
窒素雰囲気下にした300mL二口ナスフラスコに5−ヒドロキシ−2−ニトロベンズアルデヒドを5.00g(29.9mmol)、脱水CH3CNを150mL、K2CO3を7.05g(51.0mmol)入れ、室温で1時間撹拌後、アリルブロミドを4.45g(36.8mmol)入れ、80℃で2時間還流しながら撹拌し、濃縮後、H2O 100mL、2N HClを10mL入れ、抽出(CHCl3 100mL×3回)し、有機層を無水MgSO4で乾燥後、ろ過し、ろ液を濃縮、真空乾燥(湯浴中40℃)し、黄色オイルを得た。
【0209】
【表34】
【0210】
<1−8−2>1−(5−アリルオキシ−2−ニトロフェニル)メタノールの合成
【0211】
【化48】
【0212】
300mLナスフラスコに5−アリルオキシ−2−ニトロベンズアルデヒドを6.08g(29.4mmol)、THFを20mL、メタノールを40mL入れ、氷浴中でNaBH4を3.36g(88.8mmol)撹拌しながら少量ずつ加え、氷浴中で1時間、室温で30分間撹拌し、濃縮後、H2O 100mL、2N HClを20mL加え、抽出(CHCl3 100mL×3回)し、有機層を無水MgSO4で乾燥後、ろ過し、ろ液を濃縮、真空乾燥(湯浴中40℃)し、黄白色固体を得た。
【0213】
【表35】
【0214】
<1−8−3>1−(5−アリルオキシ−2−ニトロ)ベンジル 2−ブロモ−2−メチルプロピオナートの合成
【0215】
【化49】
【0216】
窒素雰囲気下にした200mL二口ナスフラスコに1−(5−アリルオキシ−2−ニトロフェニル)メタノールを1.00g(4.78mmol)、脱水CHCl3を15mL、TEAをメスピペットを用いて0.66mL(0.484g、4.78mmol)、脱水CHCl3 1mLに溶かした2−ブロモイソブチリルブロミドを1.22g(5.31mmol)入れ、16時間撹拌し、濃縮後、H2O 100mL、1N HClを5mL加え、抽出(CHCl3 100mL×3回)し、有機層を無水MgSO4で乾燥後、ろ過し、ろ液を濃縮、カラムクロマトグラフィー(ヘキサン:酢酸エチル=2:1)を行い、濃縮、真空乾燥し、黄色オイルを得た。
【0217】
【表36】
【0218】
<1−8−4>2−ニトロ−5−(3−トリメトキシシリルプロポキシ)ベンジル 2−ブロモ−2−メチルプロパノアートの合成
【0219】
【化50】
【0220】
50mL二口ナスフラスコに1−(5−アリルオキシ−2−ニトロ)ベンジル 2−ブロモ−2−メチルプロピオナートを0.530g(1.48mmol)入れ、1時間真空乾燥した。窒素雰囲気下で脱水THF10mL、ドライTHF 1mLに溶かしたトリメトキシシラン0.592g(4.84mmol)、Karstedt触媒を5滴入れ、3時間撹拌した。濃縮し、中圧カラムクロマトグラフィー(ヘキサン:酢酸エチル=3:1(1.0%テトラメトキシシラン含有))、濃縮、真空乾燥(湯浴中、40℃)し、黄色オイルを得た。
【0221】
【表37】
【0222】
[実施例2]カップリング剤の溶液中における光分解の挙動の追跡
【0223】
【化51】
【0224】
測定機器は以下のものを使用した。
【0225】
【表38】
【0226】
<2−1>チオールカップリング剤(5a)のUV測定
10mLメスフラスコに<1−1>で合成した1−(4−(3−(2−ブロモプロパノイルオキシ)プロピルオキシ)−5−メトキシ−2−ニトロフェニル)−エチル N−(11−メルカプトウンデシル)カルバメートを0.657mg量りとり、THFを用いて0.103mM THF溶液を調製した。熱と300nm以下の波長の光を遮断するために水フィルターとパイレックスガラスフィルター、及び光照射する石英硝子セルを乗せる台を用意した。超高圧水銀灯を起動させ、光量が安定するまで、30分間暖機を行い、照射度計で照度が100mW/cm2となる位置を探した。調製した溶液を石英硝子セルにとり、一定時間光照射し、UVスペクトルを測定した。
【0227】
UV測定の結果を示す。チオールカップリング剤(5a)の光照射前の波長(λ)とモル吸光係数(ε)の値を下記表39に示す。また、比較として既に合成されている1−(4,5−ジメトキシ−2−ニトロフェニル)エチル N−プロピル−カルバメート(A)(下記化学式A)の光照射前の波長(λ)とモル吸光係数(ε)の値を示す。
【0228】
【表39】
【0229】
【化52】
【0230】
化合物(A)と比較するとチオールカップリング剤(5a)のεの値が若干低いことが分かる。これは副生成物であるNHSが混合してしまっているためと考えられる。チオールカップリング剤(2)の1H−NMRより、等モルのNHSが含まれていると考えられるので、厳密にはチオールカップリング剤(5a)のTHF溶液の濃度は0.0875mMである。上記表39における括弧内の数値は0.0875mMの濃度で算出したε値を示す。
【0231】
チオールカップリング剤(5a)の光照射による1次反応と2次反応のUVスペクトル変化をそれぞれ図1お及び図2に示す。溶液中での光分解は、45秒で295nm付近のピークが減少しなくなり、380nm付近のピークが増加しなくなった。この時点を一次反応の終了とし、光分解の終了時間とした。また、420秒で248nm付近のピークが増加しなくなり、284nm付近のピークが減少しなくなった。この時点を二次反応の終了とし、何らかの反応の終了時間とした。化合物(A)において、一次反応は100秒で終了したとされるが、この差異は置換基の違いによるものであると考えた。
【0232】
<2−2>シランカップリング剤(3a)のUV測定
精密天秤用バイアルに<1−2>で合成した1−(4−(3−(2−ブロモプロパノイルオキシ)プロピルオキシ)−5−メトキシ−2−ニトロフェニル)エチル N−((トリメトキシシリル)プロピル)カルバメート(3a)1.223mg(2.00μmol)を精密天秤で量りとり、20mLメスフラスコを使用し、0.1mMエタノール溶液を調製した。エタノールでバックグラウンド測定を行った。調製したサンプルをフタ付き二面セルに入れ、λ>300nmの条件でそれぞれ、0、1、3、5、10、20、30、45、60、75、90、120、150、180、240、300、360、420、540、720、1200、1800、3600秒になるように一定時間光照射し、逐次UV−visスペクトル測定を行った。光照射に際しては石英セルの中心の照度が100mW/cm2なるように照射した。
【0233】
UV測定の結果を下記表40に示す。シランカップリング剤(3a)と化合物(B)(下記化学式B)の光照射前のUVスペクトルのλmaxの値から、シランカップリング剤(3a)と化合物(B)とでほぼ同程度のλmaxが見られることから、シランカップリング剤(3a)と化合物(B)とが類似した分子骨格を持っているといえる。
【0234】
【表40】
【0235】
【化53】
【0236】
シランカップリング剤(3a)の光照射によるUVスペクトル変化を図3に示す。図3より、0〜180secで測定したUVスペクトルのピークの挙動から、220nm、300nm付近のピークの減少、260nm、330nm付近のピークの増大がそれぞれ見られたが、180秒でそれらが見られなくなったことからこの時点で一次反応の終了とし、光分解の終了時間とした。
【0237】
<2−3>シランカップリング剤(3b)のUV測定
(UV サンプルの調整)
精密天秤用バイアルに<1−3>で合成した1−(4−(3−(2−ブロモ−2−メチルプロパノイルオキシ)プロピルオキシ)−5−メトキシ−2−ニトロフェニル)エチル N−(3−(トリメトキシシリル)プロピル)カルバメート(3b)1.248mg(2.00μmol)を精密天秤で量り取り、20mLメスフラスコを用いて、0.1mMエタノール溶液を調製した。
【0238】
(UV−Vis スペクトルの測定)
まず、光源として超高圧水銀灯を用い、光源が安定するまで約30分暖気を行った。照度計で照度が100mW/cm2となる位置を探した。調製した溶液を蓋付き二面石英セルに入れUV−Visスペクトル測定を行った。なお、バックグラウンドはエタノールで測定した。1、3、5、10、15、20、25、30、35、40、45、50、55、60、65、70、75、80、85、90、100、110、120、130、140、150、160、170、180、190、200、210、220、230、240、250、260、270、280、300、360、420、480、600、900、1200、1800、3600秒になるように一定時間光照射(λ>300nm)し、逐次UV−Visスペクトル測定を行った。光照射に関しては石英セルの中心の照度が100mW/cm2になるように照射した。光分解の終了の判断基準は300nm付近のニトロ基由来のスペクトルの減少と、370nm付近のニトロソ基由来のスペクトルの増大がほぼ見られなくなり、等吸収点から外れたスペクトルを示した時点とした。
【0239】
(光反応の反応速度定数)
上記シランカップリング剤(3b)をそれぞれ所定時間光照射したUV−Visスペクトルから反応速度定数の算出を試みた。一次反応終了と判断するまでのニトロ由来の吸収である300nm付近の吸光度Absの値をそれぞれ得た。その後、ランベルト−ベールの式から得られる式に所定時間光照射することにより得られた吸光度At、ニトロ基のモル吸光係数ε1、一次反応終了と判断した時の300nm付近のニトロソ化合物由来の吸収から得たモル吸光係数ε2、初濃度C0を代入し所定時間での前駆体の濃度Ctを求めた。そして一次反応速度式に求めたCtの値、C0を代入し、時間とln(Ct/C0)をプロットし、その近似式の傾きから反応速度定数kを求めた。またニトロソ由来である370nm付近の吸収から所定時間におけるニトロ化合物の濃度を求め、これを一次反応式に代入し、反応速度定数kを求めた。
【0240】
【数1】
【0241】
(結果および考察)
下記表41にシランカップリング剤(3b)および(3a)の極大吸収波長λとモル吸光係数εを示す。
【0242】
【表41】
【0243】
また、図4および5にλ>300nmで光照射を行った際のUV−Visスペクトルの変化を示す。図5より0〜280秒まで照射すると、ニトロ基由来の300nm付近の減少とニトロソ基由来の370nm付近の増大、等吸収点が消失したため、この時点を光分解終了と判断した。280秒で等吸収点が消失したため二次反応が進行するようになったと考えられる。光分解の終了を判断した後も60分まで光照射を続けたところ、スペクトル全体の減少が見られ、二次反応の進行が示唆された。
【0244】
(光反応の反応速度定数)
図6にニトロ基由来の吸収を、図7にニトロソ基由来の吸収の時間(秒)、ln(Ct/C0)についてのプロット示す。シランカップリング剤(3b)の溶液中における光反応の反応速度定数を求めた。このグラフの最小二乗法による近似直線の傾きから求めた反応速度定数kはニトロ基由来では0.0138s-1、ニトロソ基由来では0.0199s-1となった。
【0245】
反応速度定数はニトロ基由来の吸収から得られたものとニトロソ由来の吸収から得られたものから求められるがほぼ同様の値を示し、反応速度定数を求めるにあたり、ニトロ基由来の吸収から求める方法で十分であるということが報告されている。しかし、図6と図7の近似直線からかなりのずれが見られ、一次反応ではない反応も同時に進行しているおそれがあるため、図8にニトロ由来の吸収、図9にニトロソ由来の吸収での100秒までの時間(秒)、ln(Ct/C0)についてのプロット示し、(3b)の溶液中における光反応の反応速度定数を求めた。このグラフの最小二乗法による近似直線の傾きから求めた反応速度定数kはニトロ基由来では0.0129s-1、ニトロソ基由来では0.0160s-1となった。
【0246】
さらにシランカップリング剤(3a)についても図10にニトロ基由来の吸収、図11にはニトロソ基由来の吸収の時間(秒)、ln(Ct/C0)についてのプロット示す。シランカップリング剤(3a)の溶液中における光反応の反応速度定数を求めた。このグラフの最小二乗法による近似直線の傾きから求めた反応速度定数kはニトロ基由来では0.0326s-1、ニトロソ基由来では0.0361s-1となった。
【0247】
表42にシランカップリング剤(3b)と(3a)のニトロ基由来の吸収とニトロソ基由来の吸収の反応速度定数をまとめた。化合物の違いは末端にメチル基がついているかいないかの違いであるため、さほど反応速度に違いがあるとは思われなかったが、実際は、シランカップリング剤(3a)の方が(3b)よりも約2.5倍速いという大きな違いが見られた。
【0248】
【表42】
【0249】
[実施例3]シランカップリング剤を用いて作製したSAMの評価
<3−1>シランカップリング剤(3a)を用いて作製したSAMの評価
シリコンウエハはエナテック社製のVE49149AA(4インチを約1×4cmとしたもの)を用いた。
【0250】
光照射は以下の機器を用いて行った。
【0251】
【表43】
【0252】
測定機器は以下のものを使用した。
【0253】
【表44】
【0254】
下記表45にシランカップリング剤(3a)の量、調製した溶液の濃度、シリコンウエハの大きさと枚数をまとめたものを示す。
【0255】
【表45】
【0256】
<3−1−1>表面修飾条件の検討
【0257】
【化54】
【0258】
<3−1−1−1>シリコンウエハの前処理
50mLナスフラスコに濃H2SO4:30%H2O2=7:3(14mL:6mL)の混合溶液を調製し、シリコンウエハの非鏡面を合わせて入れ、100℃で1時間加熱した。その後、純水約30mLで洗浄し、純水約50mLを入れ超音波洗浄を行った。シリコンウエハを窒素気流で乾燥し、表面修飾に用いた。
【0259】
<3−1−1−2>表面修飾
精密天秤用バイアルにシランカップリング剤(3a)を上記表45のA〜Fに記載の量ずつ量り取り、脱水トルエン10mLで溶解しながら50mLナスフラスコに入れ、シランカップリング剤(3a)の1mM脱水トルエン溶液を調製し、窒素雰囲気下とした。この溶液に前処理済みのシリコンウエハを入れ、室温、50℃、100℃に加熱し、表面修飾を任意の時間で行った。処理後、メタノール約30mLで3回洗浄し、超音波洗浄10分間行い、クロロホルム約30mLで3回洗浄し、クロロホルム約30mLで超音波洗浄を10分間行った。表面を窒素気流で乾燥し、水の静的接触角を測定した。静的接触角を測定後は基板をメタノール、クロロホルムの順で洗浄し、クロロホルム約30mLで超音波洗浄を5分間行い、クロロホルムを入れかえ、さらに超音波洗浄を5分間行い、表面を窒素気流で乾燥し、25mLバイアルに修飾基板を入れ、窒素を充填し、保存した。
【0260】
室温、50℃、100℃にて任意の時間で静的接触角を測定した結果を表46に示す。
【0261】
【表46】
【0262】
室温で表面修飾を行った際は、表面修飾5分で水の静的接触角が67°となり、表面修飾時間3時間で水の静的接触角は72°となりほぼ一定となった。50℃条件下では表面修飾開始5分で水の静的接触角は64°となり、表面修飾30分で水の静的接触角は70°〜73°となり、ほぼ一定となった。100℃条件下では表面修飾開始5分間で水の静的接触角が75°となりそれ以降、接触角は70°〜74°となり、ほぼ一定となった。室温から100℃の水の静的接触角を比較すると、室温条件下よりも加熱条件下のほうが、表面修飾時間が短くても静的接触角が一定の値となることが考えられる。水の静的接触角が一定となったところを表面修飾時間の終了時間とすると、室温では3時間、50℃では30分、100℃では5分間で表面修飾が終了していることが考えられる。トリメトキシシリル基を持つカルボキシレート型のシランカップリング剤と表面修飾時間を比較すると、カルバメート型のシランカップリング剤は表面修飾に要する時間が短く、短時間で表面修飾が行えることが考えられる。これはカルバメート部位のアミノ基とシリコンウエハ表面のシラノールが水素結合し、その引力で基板上に接近することで、トリメトキシシリル基とシラノールの自己縮合反応が促進され、カルボキシレート型のシランカップリング剤よりも、表面に反応する速度が速くなることが要因であると考えられる。
【0263】
<3−1−2>SAMへの光照射による基板上での光分解の挙動の追跡
【0264】
【化55】
【0265】
<3−1−2−1>シリコンウエハの前処理
50mLナスフラスコに濃H2SO4:30%H2O2=7:3(14mL:6mL)の混合溶液を調製し、シリコンウエハを入れ、100℃で1時間加熱した。その後、純水約30mLで洗浄し、純水約50mLを入れ超音波洗浄を行った。シリコンウエハを窒素気流で乾燥し、表面修飾いた。
【0266】
<3−1−2−2>表面修飾
精密天秤用バイアルに2つにシランカップリング剤(3a)を上記表45のa、bに記載した量ずつ量り取り、それぞれ脱水トルエン10mLを加え、溶かしながら50mLナスフラスコに入れ、シランカップリング剤(3a)の1mM脱水トルエン溶液を調製し、窒素雰囲気下とした。この溶液に前処理済みのシリコンウエハを入れ、100℃、1時間加熱した。処理後、メタノール約30mLで3回洗浄し、超音波洗浄を10分間行い、クロロホルム約30mLで3回洗浄し、クロロホルム約30mLで超音波洗浄を10分間行った。表面を窒素気流で乾燥し、水の静的接触角を測定した。静的接触角を測定後は基板をメタノール、クロロホルムの順で洗浄し、クロロホルム約30mLで超音波洗浄を5分間行い、その後、クロロホルムを入れ替え、超音波洗浄を5分間行って、表面を窒素気流で乾燥し、25mLバイアルに修飾基板を入れ、窒素を充填し、保存した。
【0267】
<3−1−2−3>光照射
熱と300nm以上の光を遮断するために水とフィルターとパイレックスガラスフィルター、及び表面修飾したシリコンウエハを置く台を用意した。超高圧水銀灯を起動させ光量が安定するまで60分間暖気した。照射度計で照度が45mW/cm2となる位置を探した。その後、表面修飾したシリコンウエハをチタンピンセットに挟んで置き、任意の時間で光照射を行った。光照射後、シリコンウエハをメタノール、クロロホルムの順で洗い流し、その後クロロホルム約30mLで超音波洗浄10分間行い、窒素気流でシリコンウエハを乾燥させた。
【0268】
<3−1−2−4>水の静的接触角の測定
シランカップリング剤(3a)を用いたSAMへの光照射時間と光照射直後に測定した静的接触角を、協和界面化学株式会社の接触角計の取扱説明書に記載されている「液滴法測定操作」にしたがって測定した。結果を表47に示す。
【0269】
【表47】
【0270】
光照射前の基板では水の静的接触角は73°であった。光照射90秒で静的接触角が42°となり、光照射150秒で静的接触角が安定した。この結果からシランカップリング剤(3a)が基板上で光分解に要する時間は90秒であると判断した。
【0271】
<3−1−3>シランカップリング剤(3a)を用いたSAMに対するXPS測定
<3−1−3−1>測定サンプルの調製
上記3−2項で作製したSAMを測定サンプルとして利用した。調製したシリコンウエハ上のSAMの構造を下記式に示す。
【0272】
【化56】
【0273】
<3−1−3−2>測定対象とする元素と測定条件
XPS測定は表面修飾によるシリコンウエハ上へのシランカップリング剤(3a)の導入、SAMに対する光照射による光分解を確認するために下記表48に示す元素と条件で測定を行った。
【0274】
【表48】
【0275】
測定したスペクトルはmodificationのSiスペクトルを用いてピークシフトの公正、面積による規格化、解析を行った。以下に測定で得られた各サンプルのwideスキャンで得られたスペクトル、N、C、Brのスペクトルの解析結果を示す。
【0276】
図12及び13に、基板1及び基板2のXPS wideスキャンスペクトルをそれぞれ示す。基板1と基板2のスペクトルから400、70eV付近にN、Brに由来するピークが見られた。これは、シリコンウエハ上にシランカップリング剤(3a)が反応、固定化されていることを示している。
【0277】
さらに詳細な定性を行うため、基板1、基板2のN、C、Brのnarrowスキャンにより得られたスペクトルを詳細に解析した。図14及び図15に基板1と基板2のN1s XPSスペクトルをそれぞれ示す。
【0278】
基板1では400.0eV、406.1eVにサブピークが見られた。これはそれぞれNH基とNO2基の結合エネルギー由来のサブピークであり、基板上にNH基とNO2基が両方存在していることを示している。
【0279】
表49に各状態の基板のNO2基とNH基の面積比を示す。
【0280】
【表49】
【0281】
基板1の状態ではNO2基とNH基のサブピークの面積比の計算値は[NO2:NH]=1:1となるはずだが実際は[NO2:NH]=1:1.5となっていた。基板1と基板2のピークを比較すると406ev付近に存在するNO2由来のピークがSAMに対して光照射を行って作製した基板2には見られなかった。これはシリコンウエハ上にシランカップリング剤(3a)を修飾して作製したSAMがλ>300nmの光の照射することで光分解し、基板表面がアミンに変換され基板2となっていることを示している。402eV付近に現れるサブピークはSAM作製時特有のコンタミネーションのピークであると考えられる。
【0282】
図16及び図17に基板1及び基板2のBr3d(5/2) XPSスペクトルをそれぞれ示す。また、表50に各基板のカーブフィッティングしたサブピークの結合エネルギー(eV)と帰属を示す。
【0283】
【表50】
【0284】
基板1では75.0、70.8eV付近のそれぞれサブピークが見られ、基板2では70eV付近のピークの消失が見られた。これはSAMに対する光照射により、光分解反応が進行し、SAMの最表面に存在する重合開始剤となるBrが脱離したことが要因で70eV付近が消失したことを示している。また文献A(Kevin Critchley et al.,Langmuir,21,4554−4561(2005))からBrを持つ重合開始剤SAMのBrのピークが71eVに現れることが確認されており、それと比較して、このカップリング剤を用いることによって重合開始剤をシリコンウエハに導入できることが分かった。75eV付近のピークは基板1、基板2に見られることから、コンタミネーション由来のピークであると考えられる。
【0285】
図18及び図19に基板1及び基板2のC1s XPSスペクトルをそれぞれ示す。表51に基板3から得られたC1s XPSスペクトルをカーブフィッティングしたサブピークの結合エネルギー(eV)、表52に基板3の表面構造から予想されるサブピークの面積比とスペクトルから得られたサブピークの面積比を記載したもの、測定したシリコンウエハの表面構造を示す。
【0286】
【表51】
【0287】
【表52】
【0288】
基板1をXPS測定し、得られたスペクトルの波形解析の結果から表51のように帰属できる結合エネルギーのサブピークが存在すると予想できる。これらの結合エネルギーのサブピークの面積比の計算値は表52より[NCOO:COO:CO2CBr、NCO2C、CNCO2:C−O:C−C]=1:1:3:3:15であると考えられたが、実際に波形解析したサブピークの面積比は[NCOO:COO:CO2CBr、NCO2C、CNCO2:C−O:C−C]=1:1:3.8:5.5:9.8となった。298.6eV付近にNCOO、288.8eV付近のCOOのサブピークが存在していることから光分解性基が連結されているカルバメート構造、開始剤であるBrの連結部分であるエステル部位がシリコンウエハ上に存在していることが確認できた。
【0289】
表53に基板2から得られたC1s XPSスペクトルをカーブフィッティングしたサブピークの結合エネルギー(eV)と測定したシリコンウエハの表面構造を示す。
【0290】
【表53】
【0291】
基板2は波形解析の結果から3つのサブピークが存在することが考えられ、NCOO、COO、CO2CBr、NCO2C、CNCO2、C−O結合由来のサブピークが消失し、C−N、C−C結合のサブピークが見られたことから、光照射により、シリコンウエハ上のSAMが光分解し、カルバメート部位、開始剤が存在していたエステル部位が消失、親水性部位のアミン基が導入されたことが考えられる。C−N、C−C結合のサブピークの面積比は[C−N、C−C]=1:1.2であった。しかし、NCOO結合エネルギーに相当する288.9eV付近に※ピークが存在していた。これは文献からシェイク−アップサテラインに由来するピーク、あるいはシリコンウエハ上に残留している洗浄しきれていない切断された光分解性基、光分解しきれていないSAM部分に由来するコンタミネーションピークであるとであると考えられる。
【0292】
<3−2>シランカップリング剤(3b)を用いて作製したSAMの評価
<3−2−1>SAMへの光照射による基板上での光分解の挙動の追跡
【0293】
【化57】
【0294】
<3−2−1−1>シリコンウエハの前処理
2つの50mL太口ナスフラスコにピラニア溶液(Piranha溶液;H2SO4:H2O2=7:3(14mL:6mL)の混合溶液)を調製し、約1cm角のシリコンウエハを鏡面が上向きになるように3枚ずつ、計6枚入れ、100℃で1時間静置した。混合溶液を捨て、純粋で3回洗浄し、純粋を約50mL入れ超音波洗浄を10分間行った。基板を窒素気流で乾燥させ、表面修飾に用いた。
【0295】
<3−2−1−2>表面修飾
2つの50mL太口ナスフラスコにシランカップリング剤(3b)の1.0mMトルエン*溶液(トルエンにモレキュラーシーブを入れて脱水したもの)を入れ、前処理を行った基板を鏡面が上向きになるように3枚ずつ、計6枚入れ、窒素下、100℃にてそれぞれ5、30分、1、2、3、6時間表面修飾を行った。時間毎に基板を取り出し、メタノールで洗浄、試料瓶にて基板をメタノールに浸漬させ超音波洗浄を10分間行った。さらにクロロホルムで洗浄を行い、試料瓶にて基板をクロロホルムに浸漬させ超音波洗浄を10分間行った。基板を窒素気流で乾燥させた。
【0296】
<3−2−1−3>光照射
超高圧水銀灯光量が安定するまで30分間暖気し、表面修飾した基板を大気中で、照度計の測定で約50mW/cm2の位置にチタンピンセットで挟み固定した。λ>300nmの光を5、15、30、50、70、90、120秒の時間で照射した。光照射を行った後、基板をメタノールで洗浄、クロロホルムで洗浄し、さらに試料瓶にて基板をクロロホルムに浸漬させ超音波洗浄を10分間行った。窒素気流で乾燥させた。
【0297】
<3−2−1−4>水の静的接触角の測定
シランカップリング剤(3a)を用いたSAMへの光照射時間と光照射直後に測定した静的接触角を、協和界面化学株式会社の接触角計の取扱説明書に記載されている「液滴法測定操作」にしたがって測定した。1枚のサンプルについて1点の測定を3回行い、その平均値の少数第一位を四捨五入し、その基板の接触角とした。
【0298】
(表面修飾の条件検討)
トルエン溶液で100℃の加熱条件を用いてシランカップリング剤(3b)の表面修飾をした。基板を浸漬する時間を検討し、前処理基板の接触角約0°からの変化を追跡した。下記表54にそれぞれの時間で表面修飾した接触角の値を示す。
【0299】
【表54】
【0300】
表面修飾時間を表54のように振り分けたところ、浸漬開始から30分以降、ほぼ接触角の変化がないことから30分で表面修飾が終了したと考えられる。
【0301】
(光照射による接触角の変化)
表面修飾した基板にそれぞれの時間で光照射を行った。光照射したSAMの水の接触角の値を表55に示す。
【0302】
【表55】
【0303】
表55より光照射前の接触角の値が64°から光照射していくと徐々に接触角が下がっているのがわかる。これは光照射によりニトロソ化合物が脱離し疎水性の減少および、親水性の増大による接触角が変化したものだと考えられる。70秒以降、接触角の値がほぼ変化していないことから70秒で光分解が終了したと考えられる。90秒行った時点を光分解率100%と仮定した時、その時の接触角の値42°をθ∞として、下記のCassieの式を用いて各時間tにおける光分解率αtを求めたものを表57に示す。さらに残存率(1−αt)の自然対数lnをとったものを縦軸、各時間tを横軸にプロットしたものを図20と図21に示す。このグラフの最小二乗法による近似直線の傾きから求めた(3b)の反応速度定数kは0.0301s-1となり(3a)は0.0207s-1となった。
【0304】
【数2】
【0305】
【表56】
【0306】
以上の結果より、SAMの光分解速度は、溶液中の光分解速度とは逆に、(3b)の方が(3a)よりも反応速度が速いという結果となった。
【0307】
[実施例4]SAMを利用して作製したポリマーブラシの評価
シリコンウエハはエナテック社製のVE49149AA(4インチを約1×4cmとしたもの)を用いた。
【0308】
測定機器は以下のものを使用した。
【0309】
【表57】
【0310】
<4−1>Pt−BuA(ポリ(アクリル酸tert−ブチル))ブラシ、PAA(ポリアクリル酸)ブラシの作製
【0311】
【化58】
【0312】
<4−1−1>表面重合に使用するSAM、ポリマーブラシの作製に使用する試薬、条件
表58にポリマーブラシ作製に使用したSAMの作製条件を記載する。SAM作製方法は3−1項で記載した実験方法と同様である。
【0313】
【表58】
【0314】
表59に後述の<4−2−2−2>項で、XPS測定に使用するサンプル作製のためのポリマーブラシ作製時に使用した試薬の量、重合時間、をまとめたものを示す。
【0315】
【表59】
【0316】
<4−2>実験項
<4−2−1>表面処理
3−1項の方法でシランカップリング剤(3a)のSAMを作製し、表面重合に用いた。用いた試薬の量、基板の形状等は表58に示すとおりである。
【0317】
<4−2−2>シランカップリング剤(3a)を用いたポリマーブラシの作製
<4−2−2−1>原料の前処理
100mLナスフラスコにCuBr0.552g(3.85mmol)を量り取り、酢酸約5mLを加え、24時間撹拌後、洗浄(エタノール100mL、ジエチルエーテル100mL)、12時間真空乾燥(湯浴70℃)し、白色固体を得た。ここで得られた白色固体をポリマーブラシ作製に用いた。
【0318】
アクリル酸tert−ブチルは塩基性アルミナカラムで重合禁止剤を除去して使用した。条件を表60に示す。
【0319】
【表60】
【0320】
<4−2−2−2>Pt−BuAブラシの作製[基板3の作製]
50mL二口ナスフラスコに、窒素雰囲気下で、脱水DMF5mL、アニソール5mL、アクリル酸tert−ブチル(t−BuA)10mL、CuBr5.83mg(0.041mmol)、CuBr24.6mg(0.021mmol)、1,4,8,11−アザシクロテトラデカン(Me4Cyclam)10.43mg(0.041mmol)、dnNbpy16.57mg(0.041mmol)を入れ窒素置換し、凍結脱気×3回、室温に戻した後、50mLバイアルに調製した溶液を入れ、50℃で溶液が均一になるまで15分間よく撹拌、窒素置換した。反応溶液に修飾基板を50℃で1時間浸漬させ、静置した。基板を溶液から取り出し、酢酸エチル、THFで洗浄、窒素気流で乾燥させ、基板3を得た。静的接触角測定後、メタノール、クロロホルムの順で洗浄し、表面を窒素気流で乾燥し、25mLバイアルに基板3を入れ、窒素を充填し、保存した。
【0321】
<4−2−2−3>PAAブラシの作製[基板4の作製]
100mLビーカーにメタンスルホン酸100μL、ジクロロメタン10mL入れ基板3を15分間浸漬させ、クロロホルム、純水、メタノール、クロロホルムの順で洗浄、窒素気流で乾燥させ、基板4を得た。静的接触角測定後、メタノール、クロロホルムの順で洗浄し、表面を窒素気流で乾燥し、25mLバイアルに修飾基板を入れ、窒素を充填し、保存した。
【0322】
<4−3>シランカップリング剤(3a)を用いたSAM、シランカップリング剤(3a)を用いたSAMから作製した基板2及び基板3の水の静的接触角
【0323】
【化59】
【0324】
表61に各基板の表面構造を示す。基板1、3、4のSAM時の接触角は72〜75°であった。シリコンウエハ上に形成した3級ATRP開始剤を持つSAMの静的接触角は70〜77°であった)。シランカップリング剤(3a)はATRP開始剤部位が2級であるが、3級ATRP開始剤を持つSAMの静的接触角と類似した値を示した。その後モノマーを重合した基板2、3の接触角はそれぞれ78°、64°であった。この接触角の違いはグラフト密度の異なるポリマー鎖が重合されたことが要因であると考えられる。接触角測定の際、作製されたポリマーブラシが高密度の場合、ポリマーの最表面の性質が最も反映される。しかしグラフト密度が高密度時と比較して、密度が低い(中程度)場合、表面に接触している水がポリマー鎖に浸み込んでいき、接触角が減少する。基板2、3の接触角の変化はこのグラフト密度の変化が要因で現れた変化であることが考えられる。基板2の場合、基板上のSAMからポリマーブラシが密にグラフトされ、ポリマー鎖の最表面(Brとtert−ブトキシカルボニル部位)の性質が水の静的接触角に反映され、SAMの状態よりも接触角が上昇したことが考えられる。基板4ではポリマー鎖のグラフト密度が基板3と比較して低く、ポリマー鎖の間に水が浸み込み、接触角が低下したことが考えられる。基板4の加水分解処理後の水の静的接触角は49°であった。これはPt−BuAブラシ重合後に加水分解を行ったことにより、tert−ブトキシカルボニル基が分解し、カルボキシ基に変換されたことで接触角が低下したことが考えられる。
【0325】
【表61】
【0326】
<4−4>作製したポリマーブラシのXPS測定
<4−4−1>測定試料の調製
測定に使用する各試料は4−2項で示した方法で調製し、基板3、4をとした。
【0327】
<4−4−2>測定対象とする元素と測定条件
このXPS測定は表面修飾によるシリコンウエハ上へのシランカップリング剤(3a)の導入、SAMに対する光照射による光分解、モノマーの重合によるポリマーブラシ作製の有無を確認するために表62に示した元素と条件で測定を行った。
【0328】
【表62】
【0329】
測定したスペクトルは3−3項の基板1のSiスペクトルを用いてピークシフトの公正、面積による規格化、解析を行った。以下に測定で得られた各サンプルのwideスキャンで得られたスペクトル、N、C、Brのスペクトルの解析結果を示す。
【0330】
図22、23に基板3、4のXPS wideスキャンスペクトルそれぞれ示す。文献B(Krzysztof Matyjaszewski et al.,Macromolecules,32,8716−8724(1999))および文献C(Hydeharu Mori et al.,Macromolecules,34,6871−6881(2001))から開始剤を無機材料表面に導入後、重合を行うと、膜厚が増加し、開始剤部位、基板表面は表面全体の深層になる。よってX線を基板に対して90°の角度に照射し、測定したスペクトルでは重合開始剤部位や基板自体のスペクトルは検出されにくくなると考えられる。よって、基板3、4とした基板から実際に得られたスペクトルを見ると、3項で記載した基板1、2と基板3、4の間でSiのピークの強度の変化があまり見られないことから、開始剤表面からモノマーであるt−BuAの重合がわずかしか進行していないことが予想された。さらに詳細な定性を行うため、基板3、4のN、C、Brのnarrowスキャンにより得られたスペクトルを詳細に解析した結果を以下に示す。
【0331】
図24、25に基板3、4のN1s XPSスペクトルをそれぞれ示す。また、表63に各基板のカーブフィッティングしたサブピークの結合エネルギー(eV)と帰属を示す。
【0332】
【表63】
【0333】
モノマーであるアクリル酸tert−ブチル(t−BuA)を重合した後、NO2基由来、NH基由来のピークが基板3、4でそれぞれ400.3、400.0eVと406.1、406.3eVに確認できた。これは重合後、加水分解後も光分解性基である、2−ニトロベンジル誘導体がカルバメート構造で連結されて存在していることを示している。NO2基由来のサブピークはSAMの時と同様だがサブピークの面積比が計算値の[NO2:NH]=1:1ではなく基板3、4の面積比でそれぞれ[NO2:NH]=1:3.9、1:3.5で存在していた。これは重合や加水分解で光分解性基を連結しているカルバメート部位に何らかの影響が及び、光分解性基の部分が脱離しているか、ポリマー鎖が伸展し、膜厚が増加したことでシリコンウエハ底部のNO2基部位が検出されにくくなっていることにより、NO2基由来のサブピークの面積比が減少していることが予想された。基板3に関しては、スペクトルのベースラインが傾いているのはNO2基とNH基部位が基板表面上に形成されている膜の下層にあり、X線を当てて、検出される光電子の量が減少していることが原因であることが推測される。402eV付近に現れるサブピークはここに示した全ての基板に現れていることからSAM作製時特有のコンタミネーションのピークであると考えられる。
【0334】
図26、27に基板3、4のC1s XPSスペクトルをそれぞれ示す。また、表64に基板3から得られたC1s XPSスペクトルをカーブフィッティングしたサブピークの結合エネルギー(eV)、表65に基板3の表面構造から予想されるサブピークの面積比とスペクトルから得られたサブピークの面積比を記載したもの、測定したシリコンウエハの表面構造を示す。
【0335】
【表64】
【0336】
【表65】
【0337】
検出されたピークの波形解析の結果から、SAMと比較してサブピークの構成要素が減少し、CO2R、C−O、CCO2R、C−C結合に由来するサブピークがみられた。これらのサブピークは基板表面にt−BuAが重合した際に得られるPt−BuAブラシの構造に由来する結合エネルギーであり、このXPSスペクトルから基板1を重合開始剤としてt−BuAが重合しPt−BuAブラシの作製に成功したことが考えられる。また表65に示すように、CO2R、C−O、CCO2R、C−Cのサブピークの面積比は[CO2R:C−O:CCO2R:C−C]=1.1:3.2:1.0:4.7となった。Pt−BuAブラシの構造からCO2R、C−O、CCO2R、C−Cのサブピークの面積比の計算値は[CO2R:C−O:CCO2R:C−C]=1.0:1.0:1.0:4.0であり、実際に波形解析して得られたサブピークの面積比とC−O結合部分以外はよく一致していた。C−O結合の面積比が計算値よりも増加した原因としてシリコンウエハ下層部の重合開始剤に由来するサブピークが含まれているあるいは、表面の汚染によるコンタミネーションピークが含まれている可能性があると考えられる。
【0338】
表66に基板4から得られたC1s XPSスペクトルをカーブフィッティングしたサブピークの結合エネルギー(eV)、表67に基板4の表面構造から予想されるサブピークの面積比とスペクトルから得られたサブピークの面積比をまとめたもの、測定したシリコンウエハの表面構造を示す。
【0339】
【表66】
【0340】
【表67】
【0341】
検出されたピークの波形解析の結果から、基板4から得られたすスペクトルの構成要素はCO2H、C−O、CCO2H、C−C結合由来のピークであると考えられる。表67に示したように、サブピークの構成要素の面積比は[CO2H:C−O:CCO2H:C−C]=1.0:3.7:3.2:8.4であった。加水分解により保護基が分解し、完全なPAAブラシが作製されたとき、C1s XPSスペクトルのサブピークにはC−O結合由来のピークは検出されず、ピークの構成要素は[CO2H:CCO2H:C−C]=1:1:2となると考えられる。しかし、基板3のC1s XPSスペクトルから得られたサブピークの面積比と比較すると、作製した基板4ではC−O結合由来のサブピークの面積の相対的な減少が見られため、加水分解により保護基が分解し、カルボン酸が導入されたことは分かった。しかし、加水分解処理後にもC−O結合由来のサブピークが見られた。これは完全にポリマー鎖の保護基の加水分解が進行していないためか、基板表面の前処理により除去できなかったコンタミネーションに由来するC−O結合エネルギーであると推察された。
【0342】
図28、29に基板3、4のBr3d(5/2) XPSスペクトルをそれぞれ示す。また、表68に基板3、4から得られたBr3d(5/2) XPSスペクトルをカーブフィッティングしたサブピークの結合エネルギー(eV)を示す。
【0343】
【表68】
【0344】
基板3、基板1のスペクトルを比較すると基板3では71eV付近の基板1のATRP開始剤部位のBrのピークが消失し、78.9eV付近に新たなピークが出現した。このようなスペクトルが得られたのは、基板3で79eV付近に見られたサブスペクトルは、表面開始剤から重合が進行、基板の最表面の構造が変化したことで、基板1のBrと比較して高結合エネルギー側へシフトしたサブピークが得られたと考えられる。基板3、4のBr3d(5/2) XPSスペクトルを比較するとのスペクトルでは78.9eV付近にピークは見られず75eV付近のピークのみが見られた。これは重合の段階で停止、脱離、転移反応を通じて、ポリマー鎖末端のBrが脱離したこと、加水分解処理により、重合したポリマー鎖が連結されているエステル又は光分解性基が連結されているカルバメート部位が分解を示している可能性がある。
【0345】
<4−5>定量分析
作製した各基板をそれぞれ基板1、2、3、4としたときに、測定対照とした各元素O、N、C、Si、Brが基板上にどのような割合(%)で存在するかを、測定した各元素のピークの面積から算出したものを表69に示す。
【0346】
【表69】
【0347】
基板1、2を比較したとき、光照射により光分解性基とそれに連結されたATRP開始剤が脱離するためO、N、C、Brの割合が変化することが予想された。しかし、基板1、2で算出された各元素の割合を比較すると、基板2でBrが顕著な元素の割合の減少を示していたが、他の元素については、わずかな元素の割合の変化が見られるのみであった。基板3について、基板1と比較するとN、Si、Brについて元素の割合の減少が見られ、Cの割合の増加が見られた。これは重合を行ったことによって基板1から基板3のような構造の変化が起こったことにより、膜厚の増加によるCの増加によって検出されるCの割合が増加したことが要因であることに由来する変化であると考えられる。しかし、基板3、4を比較すると基板3に対して基板4ではCの割合の減少、Siの割合の増加が顕著に見られた。これは加水分解処理により表面に存在するCが減少したことに由来する変化であると考えられる。しかし、Siの割合が増加していることから、表面の膜厚が減少し、加水分解処理により、重合したポリマー鎖が連結されているエステル又は光分解性基が連結されているカルバメート部位の分解がこの結果から予想される。
【特許請求の範囲】
【請求項1】
リビングラジカル重合の開始基と、反応性基(但し、前記開始基を除く)とが光分解性基を介して連結されてなる、下記化学式1で表される光分解性ヘテロ二価性架橋剤。
【化1】
式中、Iは開始基を表し、Rは反応性基を表し、Dは光分解性基を表し、S1b−S1a及びS2a−S2bはスペーサーを表し、n及びmはそれぞれ独立して0又は1である。
【請求項2】
前記光分解性基が下記化学式2で示される基のうちのいずれか1種である、請求項1に記載の光分解性ヘテロ二価性架橋剤。
【化2】
式中、R1は水素原子、炭素原子数1〜8のアルキル基、又は置換されたもしくは非置換のフェニル基を表し、R3は水素原子又は炭素原子数1〜8のアルコキシ基を表す。
【請求項3】
前記開始基が下記化学式3、化学式4、化学式5、又は化学式6で表される、請求項1又は2に記載の光分解性ヘテロ二価性架橋剤;
【化3】
式中、R2は水素原子又はメチル基を表す。
【化4】
式中、R4及びR5はそれぞれ独立して水素原子又は炭素原子数1〜8のアルキル基を表し、Xは臭素原子又は塩素原子を表す。
【化5】
【化6】
【請求項4】
前記反応性基が、ヒドロキシ基、ビニル基、エチニル基、アジド基、活性エステル基、マレイミド基、加水分解性シリル基、アミノ基、カルボキシル基、チオール基、ジスルフィド基、及びスルホ基からなる群から選択される少なくとも1種を含む、請求項1〜3のいずれか1項に記載の光分解性ヘテロ二価性架橋剤。
【請求項5】
前記S1aが、−O−、−C(O)O−、−OC(O)O−、−NH−、−C(O)NH−、−OC(O)NH−、−S−、−C(O)S−、−OC(O)S−からなる群から選択される少なくとも1種を表し、
前記S2aが、−O−を表し、
前記S1b及びS2bが、それぞれ独立して、アミド結合、イミド結合、エーテル結合、エステル結合、チオエーテル結合、もしくはスルホンアミド結合を有してもよい炭素原子数1〜20のアルキレン基からなる群から選択される少なくとも1種を表す、請求項1〜4のいずれか1項に記載の光分解性ヘテロ二価性架橋剤。
【請求項6】
請求項1〜5のいずれか1項に記載の光分解性ヘテロ二価性架橋剤の反応性基と、基板表面の官能基とが結合されてなる、ポリマーブラシ形成用基板。
【請求項1】
リビングラジカル重合の開始基と、反応性基(但し、前記開始基を除く)とが光分解性基を介して連結されてなる、下記化学式1で表される光分解性ヘテロ二価性架橋剤。
【化1】
式中、Iは開始基を表し、Rは反応性基を表し、Dは光分解性基を表し、S1b−S1a及びS2a−S2bはスペーサーを表し、n及びmはそれぞれ独立して0又は1である。
【請求項2】
前記光分解性基が下記化学式2で示される基のうちのいずれか1種である、請求項1に記載の光分解性ヘテロ二価性架橋剤。
【化2】
式中、R1は水素原子、炭素原子数1〜8のアルキル基、又は置換されたもしくは非置換のフェニル基を表し、R3は水素原子又は炭素原子数1〜8のアルコキシ基を表す。
【請求項3】
前記開始基が下記化学式3、化学式4、化学式5、又は化学式6で表される、請求項1又は2に記載の光分解性ヘテロ二価性架橋剤;
【化3】
式中、R2は水素原子又はメチル基を表す。
【化4】
式中、R4及びR5はそれぞれ独立して水素原子又は炭素原子数1〜8のアルキル基を表し、Xは臭素原子又は塩素原子を表す。
【化5】
【化6】
【請求項4】
前記反応性基が、ヒドロキシ基、ビニル基、エチニル基、アジド基、活性エステル基、マレイミド基、加水分解性シリル基、アミノ基、カルボキシル基、チオール基、ジスルフィド基、及びスルホ基からなる群から選択される少なくとも1種を含む、請求項1〜3のいずれか1項に記載の光分解性ヘテロ二価性架橋剤。
【請求項5】
前記S1aが、−O−、−C(O)O−、−OC(O)O−、−NH−、−C(O)NH−、−OC(O)NH−、−S−、−C(O)S−、−OC(O)S−からなる群から選択される少なくとも1種を表し、
前記S2aが、−O−を表し、
前記S1b及びS2bが、それぞれ独立して、アミド結合、イミド結合、エーテル結合、エステル結合、チオエーテル結合、もしくはスルホンアミド結合を有してもよい炭素原子数1〜20のアルキレン基からなる群から選択される少なくとも1種を表す、請求項1〜4のいずれか1項に記載の光分解性ヘテロ二価性架橋剤。
【請求項6】
請求項1〜5のいずれか1項に記載の光分解性ヘテロ二価性架橋剤の反応性基と、基板表面の官能基とが結合されてなる、ポリマーブラシ形成用基板。
【図1】

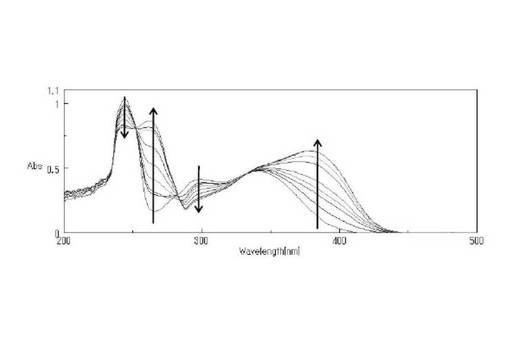
【図2】
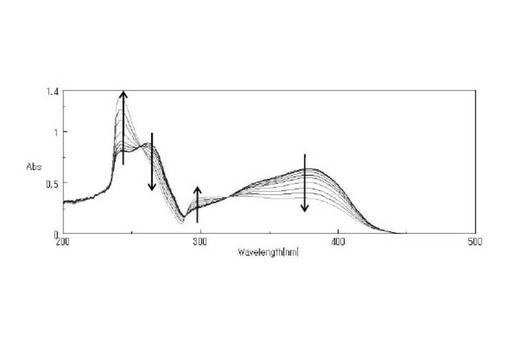
【図3】
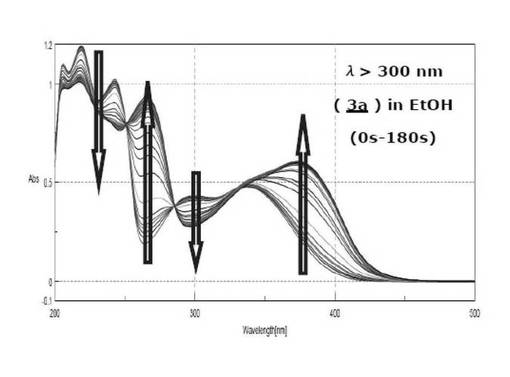
【図4】
【図5】
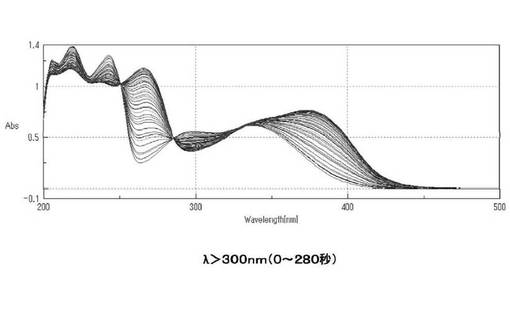
【図6】
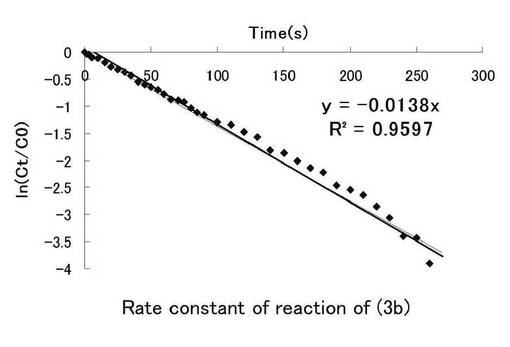
【図7】
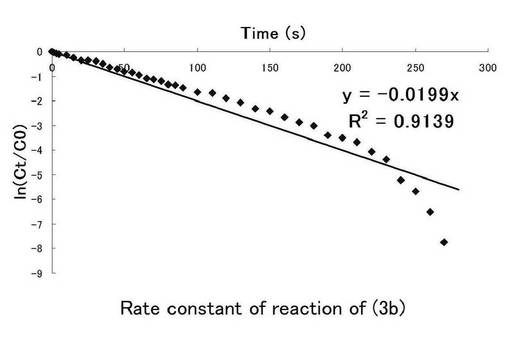
【図8】
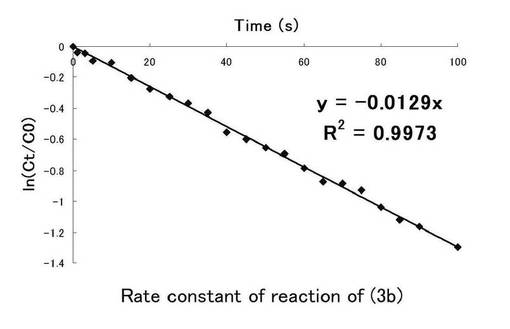
【図9】
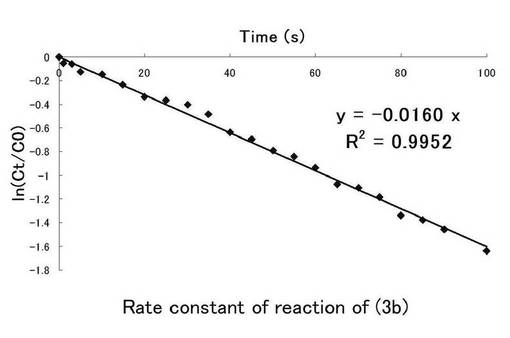
【図10】
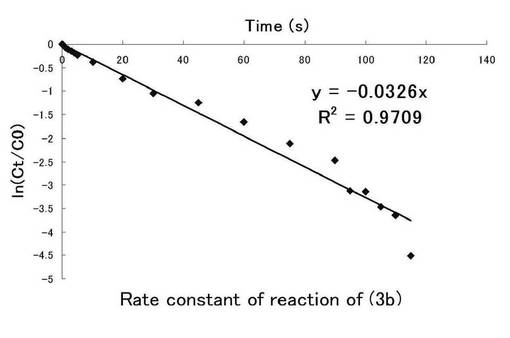
【図11】
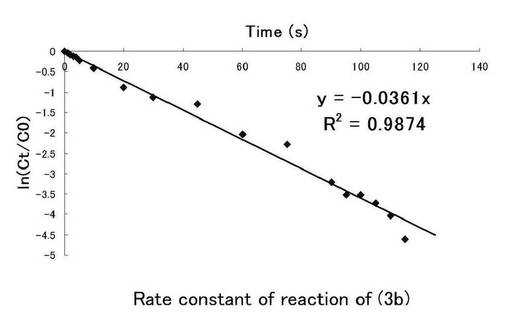
【図12】
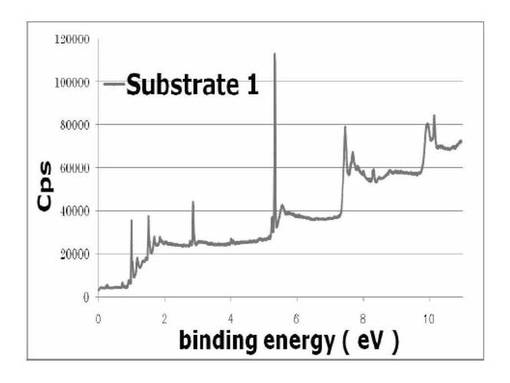
【図13】
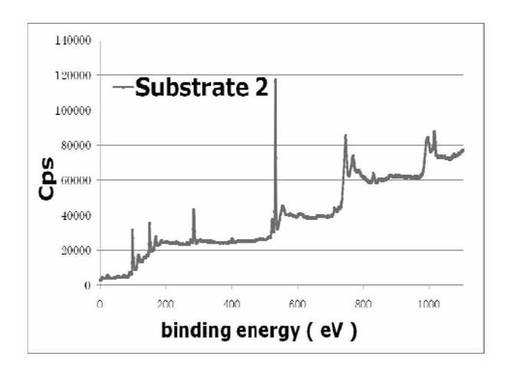
【図14】
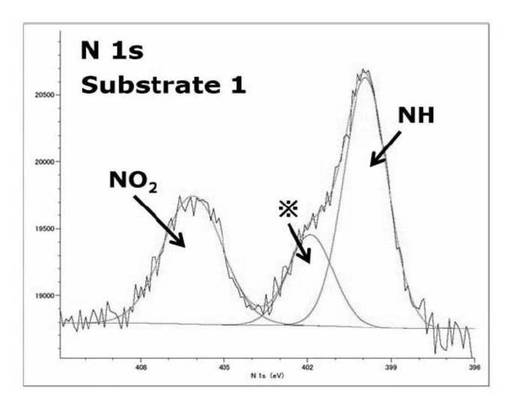
【図15】
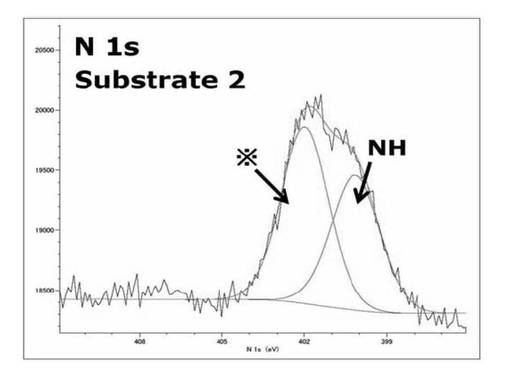
【図16】
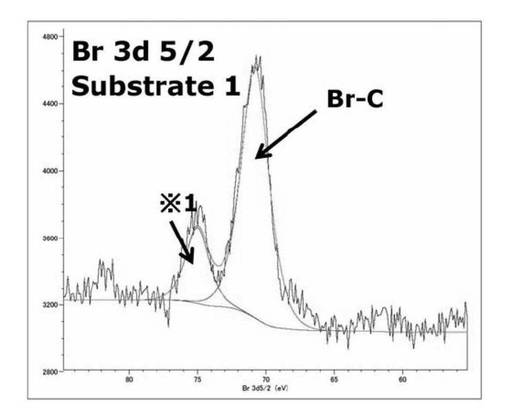
【図17】
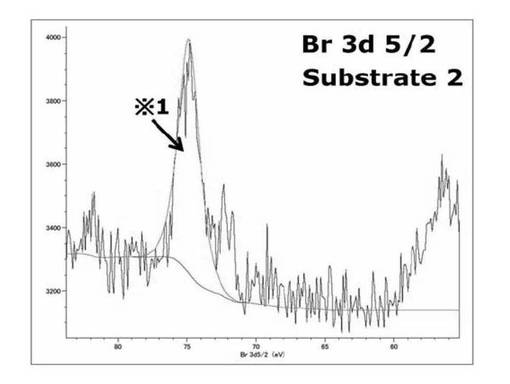
【図18】
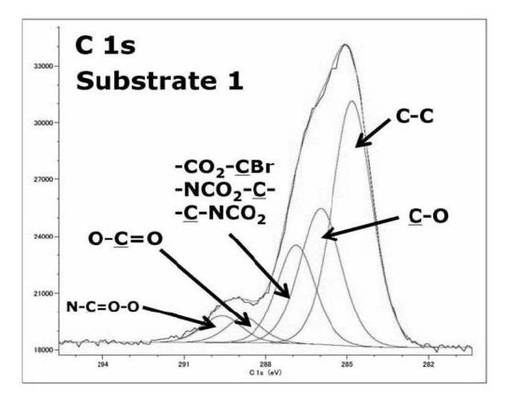
【図19】
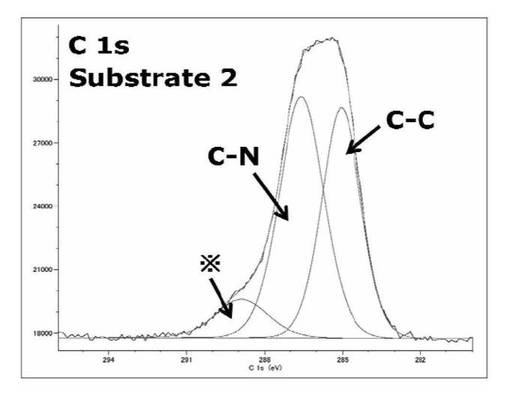
【図20】
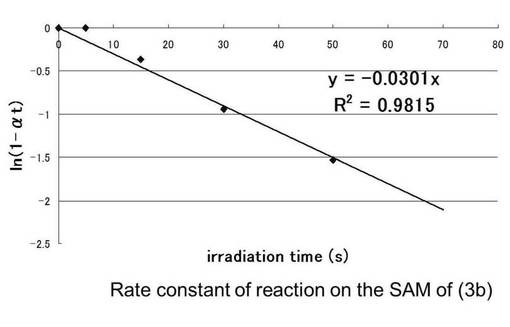
【図21】
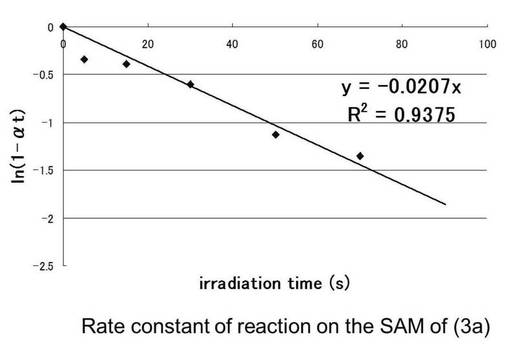
【図22】

【図23】
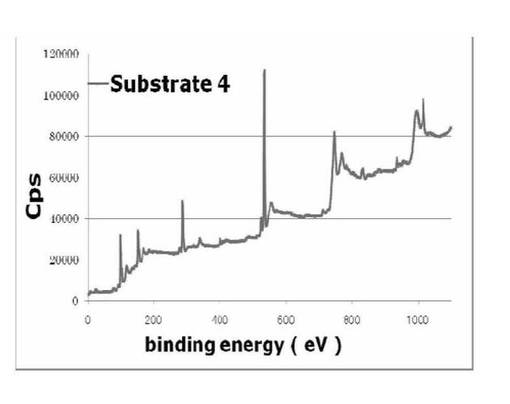
【図24】
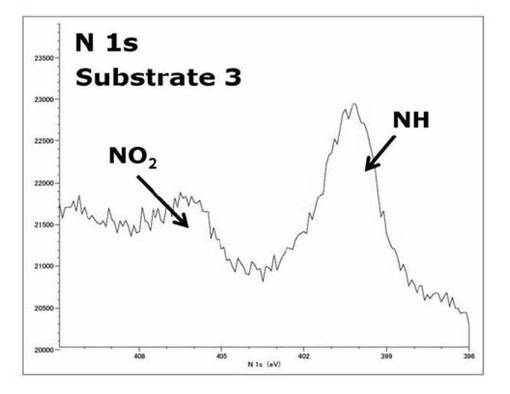
【図25】
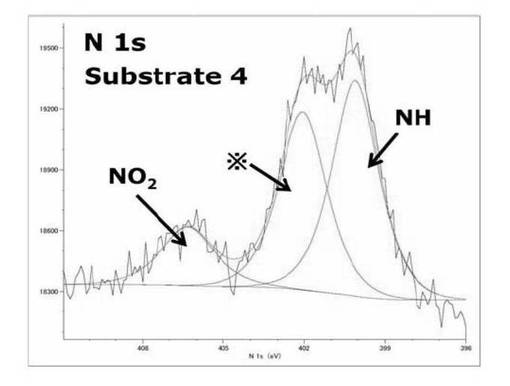
【図26】
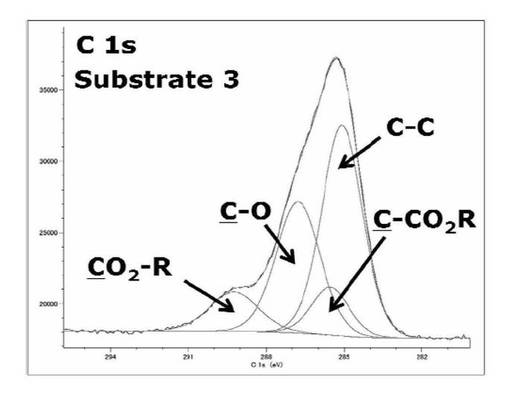
【図27】
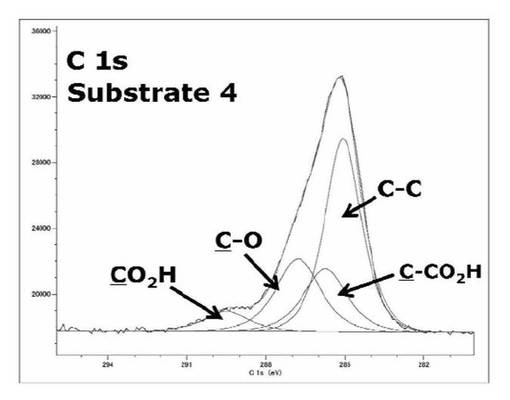
【図28】
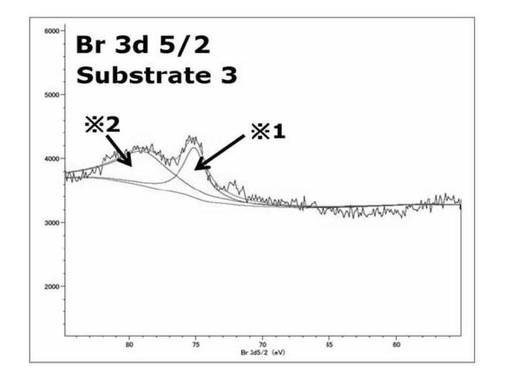
【図29】
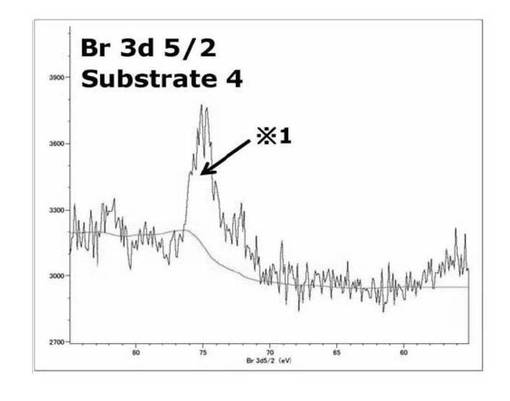
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【公開番号】特開2012−72394(P2012−72394A)
【公開日】平成24年4月12日(2012.4.12)
【国際特許分類】
【出願番号】特願2011−188009(P2011−188009)
【出願日】平成23年8月30日(2011.8.30)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.パイレックス
【出願人】(592218300)学校法人神奈川大学 (243)
【Fターム(参考)】
【公開日】平成24年4月12日(2012.4.12)
【国際特許分類】
【出願日】平成23年8月30日(2011.8.30)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.パイレックス
【出願人】(592218300)学校法人神奈川大学 (243)
【Fターム(参考)】
[ Back to top ]