
光塩基発生剤、感光性ポリイミド樹脂組成物、レリーフパターンの製造方法並びに物品
【課題】露光後ベーク時の加熱条件を従来より緩和しても、露光部のポリイミド前駆体のイミド化を十分に促進する塩基を発生する塩基発生剤、及び、現像時間を短縮でき、アルコール等を添加しない塩基性水溶液を現像液として用いることが可能で、露光部と未露光部の溶解性コントラストが高くなる感光性ポリイミド樹脂組成物を提供する。
【解決手段】発生する塩基が共有結合を用いて潜在化されていて、電磁波の照射により塩基を発生する塩基発生剤であって、水酸基を有する塩基を発生する光塩基発生剤、並びに、ポリイミド前駆体と、前記本発明に係る光塩基発生剤とを含有する、ネガ型感光性ポリイミド樹脂組成物である。
【解決手段】発生する塩基が共有結合を用いて潜在化されていて、電磁波の照射により塩基を発生する塩基発生剤であって、水酸基を有する塩基を発生する光塩基発生剤、並びに、ポリイミド前駆体と、前記本発明に係る光塩基発生剤とを含有する、ネガ型感光性ポリイミド樹脂組成物である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、前駆体型感光性ポリイミド用光塩基発生剤、ネガ型感光性ポリイミド樹脂組成物、当該感光性ポリイミド樹脂組成物を用いたレリーフパターンの製造方法、及び、当該感光性ポリイミド樹脂組成物を用いて作製した物品に関するものである。
【背景技術】
【0002】
感光性ポリイミド樹脂組成物は、例えば、電子部品、光学製品、光学部品の成形材料、層形成材料又は接着剤などに用いられ、特に、電磁波によるパターニング工程を経て形成される製品又は部材に好適に利用されてきている。
高分子材料であるポリイミドは、耐熱性、寸法安定性、絶縁特性といった性能が有機物の中でもトップクラスの性能を示すため、電子部品の絶縁材料等へ広く適用され、半導体素子の中のチップコーティング膜や、フレキシブルプリント配線板の基材などとして盛んに利用されてきている。
【0003】
一般にポリイミドは溶媒への溶解性に乏しく、加工が困難なため、ポリイミドを所望の形状にパターニングする方法として、溶媒溶解性に優れるポリイミド前駆体の状態で露光と現像によるパターニングを行い、その後、熱処理等によりイミド化を行いポリイミドのパターンを得るという方法がある。
【0004】
ポリイミド前駆体を利用して、パターンを形成する手段として、種々の方法が提案されている。その代表的なものは以下の二つである。
(1)ポリイミド前駆体にはパターン形成能力がなく、ポリイミド前駆体上に感光性樹脂をレジスト層として設けることによりパターンを形成する方法。
(2)ポリイミド前駆体自身に感光性部位を結合や配位させて導入し、その作用により、パターンを形成する方法。または、ポリイミド前駆体に感光性成分を混合し樹脂組成物とし、その感光性成分の作用でパターンを形成する方法。
【0005】
上記(2)を用いるパターニング手法の代表的なものとしては、(i)ポリイミド前駆体のポリアミック酸に電磁波の露光前は溶解抑止剤として作用し、露光後はカルボン酸を形成し溶解促進剤となるナフトキノンジアジド誘導体を混合し、露光部と未露光部の現像液に対する溶解速度のコントラストを大きくすることでパターン形成を行い、その後にイミド化を行い、ポリイミドパターンを得る手法や(特許文献1)、(ii)ポリイミド前駆体にエステル結合またはイオン結合を介してメタクリロイル基を導入し、そこに光ラジカル発生剤を添加し、露光部を架橋させることで露光部と未露光部の現像液に対する溶解速度のコントラストを大きくすることでパターン形成を行い、その後にイミド化を行い、ポリイミドパターンを得る手法などが実用化されている(特許文献2)。
【0006】
(2)の手法は、(1)の方法と比べ、レジスト層が必要ないため大幅にプロセスを簡略化させることができるが、(i)の方法では、溶解性コントラストを高めるためにナフトキノンジアジド誘導体の添加量を増加させると、ポリイミド本来の物性が得られなくなるという問題があった。また(ii)の方法では、ポリイミド前駆体の構造が制約されてしまうという問題があった。
【0007】
この他のパターニング手法としては、(iii)ポリイミド前駆体であるポリアミック酸に、光塩基発生剤を混合し、露光後加熱することで露光によって発生した塩基の作用によって環化を進行させ、現像液に対する溶解性を低下させることで、露光部と未露光部の現像液に対する溶解速度のコントラストを大きくすることでパターン形成を行い、その後にイミド化を行い、ポリイミドパターンを得る手法が報告されている(特許文献3)。
【0008】
非特許文献1には、アミンの光反応性保護基として、o−ヒドロキシ−トランス−桂皮酸を用いることが開示されている。また、特許文献4には、o−ヒドロキシ−トランス−桂皮酸アミドを光塩基発生剤として用い、当該光塩基発生剤と塩基反応性樹脂とを含む感光性ポリイミド樹脂組成物が開示されている。更に、本発明者らも、o−ヒドロキシ−トランス−桂皮酸アミド誘導体を光環化型の光塩基発生剤として用い、当該光塩基発生剤とポリイミド前駆体とを含む感光性ポリイミド樹脂組成物を特許文献5に開示している。また、特許文献6及び7には、カルバメート型の光塩基発生剤を用いた感光性ポリイミド樹脂組成物が開示されている。更に、特許文献8及び9には、オキシム型の光塩基発生剤を用いた感光性ポリイミド樹脂組成物が開示されている。
【0009】
光塩基発生剤を用いた感光性ポリイミド樹脂組成物は、ポリイミド前駆体に、光塩基発生剤を一定比率混合するだけで感光性ポリイミド前駆体を得ることができるため、樹脂組成物を製造するプロセスが簡便である。また、種々の構造のポリイミド前駆体に適用できるため汎用性が高いという利点がある。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】特開昭52−13315号公報
【特許文献2】特開昭54−145794号公報
【特許文献3】特開平8−227154号公報
【特許文献4】特開2009−80452号公報
【特許文献5】国際公開第2009/123122号パンフレット
【特許文献6】特開2006−189591号公報
【特許文献7】特開2008−247747号公報
【特許文献8】特開2007−249013号公報
【特許文献9】特開2008−003581号公報
【非特許文献】
【0011】
【非特許文献1】Chem. Pharm. Bull. 1997, 45(4) p.715-718
【発明の概要】
【発明が解決しようとする課題】
【0012】
しかし、上記のような光塩基発生剤を用いた感光性ポリイミド樹脂組成物は、露光後の加熱工程において露光部のみならず未露光部においてもポリイミド前駆体のイミド化が進行することから、現像性の改善が課題であった。すなわち、未露光部のイミド化も一部進行することにより、未露光部の現像液への溶解性が悪くなるため、現像時間を長くしなければならないという問題があった。また、現像液として、塩基性水溶液だけでは十分に溶解しないため、例えばアルコール等の有機溶剤を添加した塩基性水溶液を用いる必要があった。環境負荷の低減から、アルコール等の有機溶剤を添加しない塩基性水溶液を現像液として用いることが市場で求められている。
更に、未露光部においてポリイミド前駆体のイミド化が進行しすぎた場合、露光部と未露光部の現像液に対する溶解速度のコントラストが不十分となり、レリーフパターンの形状が悪化する恐れがある。
【0013】
露光後ベーク時の加熱温度を低くしたり、加熱時間を短くするなど、加熱条件を緩和することにより、未露光部におけるポリイミド前駆体のイミド化の進行を抑制することができる。しかし、加熱条件を緩和すると、露光部においてもイミド化の進行は抑制されるため、露光部と未露光部の溶解性コントラストは向上しない。そこで、本発明者らは、露光後ベーク時の加熱条件を従来より緩和しても、露光部においてポリイミド前駆体のイミド化を十分に促進する触媒能が高い塩基を発生する光塩基発生剤を得ることに着目した。
【0014】
すなわち本発明は、上記実情を鑑みてなされたものであり、露光後ベーク時の加熱条件を従来より緩和しても、露光部のポリイミド前駆体のイミド化を十分に促進する塩基を発生する光塩基発生剤を提供することを目的とする。また、本発明は、当該光塩基発生剤を用いることにより、現像時間を短縮でき、アルコール等を添加しない塩基性水溶液を現像液として用いることが可能となったり、露光部と未露光部の溶解性コントラストが高くなる、感光性ポリイミド樹脂組成物を提供することを目的とする。
【課題を解決するための手段】
【0015】
本発明に係る前駆体型感光性ポリイミド用光塩基発生剤は、発生する塩基が共有結合を用いて潜在化されていて、電磁波の照射により塩基を発生する塩基発生剤であって、水酸基を有する塩基を発生することを特徴とする。
【0016】
本発明に係る前駆体型感光性ポリイミド用塩基発生剤は、電磁波の照射により水酸基を有する塩基を発生することにより、ポリイミド前駆体のイミド化促進効果が高く、露光後ベーク時の加熱条件を従来より緩和しても、露光部のポリイミド前駆体のイミド化を十分に促進することが可能である。
【0017】
本発明に係る塩基発生剤においては、沸点が150℃以上かつ融点が200℃以下の水酸基を有する塩基を発生することが、ポリイミド前駆体のイミド化促進効果が高くなる点から好ましい。
【0018】
本発明に係る光塩基発生剤においては、下記化学式(1−1)、下記化学式(1−2)又は下記化学式(1−3)で表されること光塩基発生剤であることが好ましい。
【0019】
【化1】

(式(1−1)中、R1及びR2は、それぞれ独立に、水素原子又は有機基であり、同一であっても異なっていても良い。R1及びR2の少なくとも1つは有機基である。R1及びR2は、それらが結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。但し、R1及びR2のいずれかの有機基が水酸基を有する。R3及びR4はそれぞれ独立に、水素原子、ハロゲン原子、水酸基、メルカプト基、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、又は有機基であり、同一であっても異なっていても良い。R5、R6、R7及びR8は、それぞれ独立に、水素原子、ハロゲン原子、水酸基、メルカプト基、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、アミノ基、アンモニオ基又は有機基であり、同一であっても異なっていても良い。R5、R6、R7及びR8は、それらの2つ以上が結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。R9は、水素原子、或いは、加熱及び/又は電磁波の照射により脱保護可能な保護基である。)
【0020】
【化2】
(式(1−2)中、R1及びR2は、それぞれ独立に、水素原子又は有機基であり、同一であっても異なっていても良い。R1及びR2の少なくとも1つは有機基である。R1及びR2は、それらが結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。但し、R1及びR2のいずれかの有機基が水酸基を有する。R10及びR10’はそれぞれ独立に水素原子、ハロゲン原子、水酸基、メルカプト基、ニトロ基、シリル基、シラノール基、又は有機基である。R11、R12、R13、R14及びR15は、それぞれ独立に、水素原子、ハロゲン原子、水酸基、メルカプト基、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、アミノ基、アンモニオ基又は有機基であり、同一であっても異なっていても良い。R11、R12、R13、R14及びR15は、それらの2つ以上が結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。)
【0021】
【化3】
(式(1−3)中、R17は、水酸基を有する有機基で置換されたアミノ基又は水酸基を有する有機基、R18及びR19は、それぞれ独立に、水素原子、ハロゲン原子、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、アミノ基、アンモニオ基又は有機基であり、同一であっても異なっていても良いが、R18及びR19の少なくとも1つはヘテロ原子を有していても良い芳香族化合物である。R17、R18及びR19は、それらの2つ以上が結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。)
【0022】
本発明に係る光塩基発生剤においては、発生する塩基が脂肪族アミンであることが、塩基性が強く、イミド化を促進する触媒能が高いため、より少量の添加で、より低い温度での最終生成物への反応が可能となる点から好ましい。
【0023】
本発明に係る光塩基発生剤においては、発生する塩基が酸性基を含まないことが、発生した塩基のイミド化を促進する触媒能を発揮させる点から好ましい。
【0024】
本発明に係るネガ型感光性ポリイミド樹脂組成物は、ポリイミド前駆体と、前記本発明に係る光塩基発生剤とを含有することを特徴とする。
本発明に係るネガ型感光性ポリイミド樹脂組成物は、前記本発明に係る光塩基発生剤を用いることにより、露光後ベーク時の加熱条件を従来より緩和しても、露光部は十分にイミド化が促進され、未露光部は緩和された加熱条件のためにポリイミド前駆体のイミド化の進行を抑制することができる。また、塩基に導入されている水酸基の効果で、未露光部の現像液への溶解が促進される。これらにより、未露光部の現像液への溶解性が向上するため、未露光部を除去する現像時間を短縮でき、また、アルコール等を添加しない塩基性水溶液を現像液として用いることが可能になる。更に、露光部と未露光部の溶解性コントラストが高くなるため、レリーフパターンの形状が良好になる。
【0025】
本発明に係るネガ型感光性ポリイミド樹脂組成物においては、前記光塩基発生剤が発生する塩基が、150〜200℃のいずれかの温度において、当該塩基100重量部に対して、前記ポリイミド前駆体を10重量部以上溶解する塩基であることが、塩基の触媒能を向上させる点から好ましい。
【0026】
本発明に係るネガ型感光性ポリイミド樹脂組成物は、塗料、印刷インキ、シール剤、又は接着剤、或いは、表示装置、半導体装置、電子部品、微小電気機械システム、光造形物、光学部材又は建築材料の形成材料として好適に用いられる。
【0027】
本発明は、上記本発明に係るネガ型感光性ポリイミド樹脂組成物を用いるレリーフパターンの製造方法を提供する。
本発明に係るレリーフパターンの製造方法は、上記ネガ型感光性ポリイミド樹脂組成物を用いて塗膜又は成形体を形成し、当該塗膜又は成形体を、所定パターン状に電磁波を照射し、照射後又は照射と同時に加熱し、前記照射部位の溶解性を変化させた後、現像することを特徴とする。
【0028】
また、本発明は、上記ネガ型感光性ポリイミド樹脂組成物又はその硬化物により少なくとも一部が形成されている、印刷物、塗料、シール剤、接着剤、表示装置、半導体装置、電子部品、微小電気機械システム、光造形物、光学部材又は建築材料のいずれかの物品を提供する。
【発明の効果】
【0029】
本発明によれば、露光後ベーク時の加熱条件を従来より緩和しても、露光部のポリイミド前駆体のイミド化を十分に促進する光塩基発生剤を提供することができる。
本発明に係るネガ型感光性ポリイミド樹脂組成物は、現像時間を短縮でき、アルコール等を添加しない塩基性水溶液を現像液として用いることが可能となったり、露光部と未露光部の溶解性コントラストを高くすることができる。
【図面の簡単な説明】
【0030】
【図1】試験例16、試験例17、及び比較試験例11における、ポリイミド前駆体に対する塩基の添加量とイミド化率との関係を示すグラフである。
【図2】実施例1、実施例2、及び比較例1の感光性ポリイミド樹脂組成物の露光量と残膜率との関係を示す感度曲線である。
【図3】実施例3、及び比較例2の感光性ポリイミド樹脂組成物の露光量と残膜率との関係を示す感度曲線である。
【発明を実施するための形態】
【0031】
以下、本発明について詳しく説明する。
なお、本発明において(メタ)アクリロイルとは、アクリロイル及び/又はメタクリロイルを意味し、(メタ)アクリルとは、アクリル及び/又はメタクリルを意味し、(メタ)アクリレートとは、アクリレート及び/又はメタクリレートを意味する。
また、本発明において、電磁波とは、波長を特定した場合を除き、可視及び非可視領域の波長の電磁波だけでなく、電子線のような粒子線、及び、電磁波と粒子線を総称する放射線又は電離放射線が含まれる。本明細書では、電磁波の照射を露光ともいう。なお、波長365nm、405nm、436nmの電磁波をそれぞれ、i線、h線、g線とも表記することがある。
また、本発明において、沸点及び融点は、圧力1atm(101325Pa)下での沸点及び融点をいう。
また、本発明において、有機基とは、少なくとも1つの炭素原子を含む官能基の総称を表す。
【0032】
<光塩基発生剤>
本発明に係る前駆体型感光性ポリイミド用光塩基発生剤は、発生する塩基が共有結合を用いて潜在化されていて、電磁波の照射により塩基を発生する光塩基発生剤であって、水酸基を有する塩基を発生することを特徴とする。なおここで、前駆体型感光性ポリイミドとは、ポリイミド前駆体を含む感光性ポリイミド樹脂組成物のことをいう。
本発明に係る前駆体型感光性ポリイミド用塩基発生剤は、電磁波の照射により水酸基を有する塩基(塩基性物質)を発生することにより、露光後ベーク時の加熱条件を従来より緩和しても、露光部のポリイミド前駆体のイミド化を十分に促進することが可能である。
ここで、露光部のポリイミド前駆体のイミド化を十分に促進する程度とは、露光部のポリイミド前駆体の現像液に対する溶解速度が未露光部に対して十分遅くなる程度であり、例えば用いられる現像液に対して露光部のポリイミド前駆体の溶解速度が25℃で10nm/s以下程度に遅くなることを目安とすることができる。また、例えば現像液として0.1〜5.0重量%のテトラメチルアンモニウムヒドロキシド(TMAH)水溶液を用いる場合には、露光部のポリイミド前駆体のイミド化率が45〜60%程度であることが好ましい。
【0033】
露光後ベーク(Post Exposure Bake:PEB)時の加熱条件を従来よりも緩和する、すなわち、加熱温度を低減したり、加熱時間を短くすることにより、未露光部のポリイミド前駆体はイミド化が抑制される。また、塩基に導入されている水酸基の効果で、未露光部の現像液への溶解が促進される。これらの相乗効果により、本発明に係る光塩基発生剤を前駆体型感光性ポリイミド樹脂組成物に用いると、未露光部の現像液への溶解性が向上し、未露光部を除去する現像時間を短縮でき、また、アルコール等を添加しない塩基性水溶液を現像液として用いることが可能になる。更に、露光部は十分にイミド化される一方で、未露光部は現像液への溶解性が高いことから、露光部と未露光部の溶解性コントラストが高くなるため、レリーフパターンの形状が良好になる。
【0034】
本発明に係る光塩基発生剤は、発生する塩基が共有結合を用いて潜在化されているものである。発生する塩基が共有結合を用いて潜在化されている光塩基発生剤とは、発生する塩基部分の窒素原子と隣接する原子とが共有結合を有しており、電磁波の照射により当該共有結合が切断されて塩基が発生する光塩基発生剤である。このような構造の光塩基発生剤は中性にすることができるため、溶剤溶解性が良好であり、ポットライフが向上する。また、ポリイミド前駆体と組み合わせて用いられる成分との相溶性が良好である場合が多いというメリットがある。
【0035】
発生する塩基が共有結合を用いて潜在化されている構造を有する光塩基発生剤としては、例えば、上記特許文献5及び6で開示されたような桂皮酸アミド構造を有する塩基発生剤、上記特許文献7及び8で開示されたようなカルバメート構造を有する塩基発生剤、上記特許文献9及び10で開示されたようなオキシム構造、カルバモイルオキシム構造を有する塩基発生剤等が挙げられるが、これらに限定されず、その他にも公知の光塩基発生剤の構造を用いることができる。
【0036】
本発明に係る光塩基発生剤は、水酸基を有する塩基を発生するものである。発生する塩基が水酸基を有することにより、露光後ベーク温度の条件を、従来よりも低温としたり、加熱時間を短くしても、後述するポリイミド前駆体を効率よくイミド化することができる。アミノ基と水酸基を1分子内に有する化合物は、ポリイミドやポリイミド前駆体との親和性が高く、塗膜中でのポリイミド前駆体のイミド化過程において、よりイミド化が進んだ状態まで、擬似的な溶媒兼、触媒としてイミド化を促進するものとして機能するためと推測される。更に、発生する塩基が水酸基を有することにより、未露光部の塩基性水溶液に対する溶解性が向上する傾向がある。これは、光塩基発生剤中に潜在化された塩基に結合された水酸基が、ポリイミド前駆体と相互作用することにより、塩基性水溶液への溶解性を向上させるためと推測される。なお、水酸基は1つ以上有していればよく、発生する塩基の構造は特に限定されない。フェノール性水酸基は弱い酸性基となる点から、水酸基としては、アルコール性水酸基であることが好ましい。なお、アルコール性水酸基とは、脂肪族炭化水素骨格に直接結合する水酸基を意味し、フェノール性水酸基は、芳香族炭化水素骨格に直接結合する水酸基を意味する。
【0037】
また、発生する塩基は、沸点が150℃以上かつ融点が200℃以下の水酸基を有する塩基であることが、ポリイミド前駆体のイミド化促進効果が高くなる点から好ましい。
一般的な構造のポリイミド前駆体がイミド化に要する加熱温度は、140℃以上であり、通常、露光後加熱温度は140℃〜210℃の範囲内で選択される場合が多い。
発生する塩基の沸点が150℃以上であると、露光後加熱工程における当該塩基の揮発が抑えられ、発生した塩基をイミド化促進触媒として効率よく利用することができる。中でも沸点は160℃以上であることが好ましい。また、融点が200℃以下であると、露光後加熱工程において当該塩基が液体であり、固体の場合に比べ、樹脂組成物中を拡散しやすいか、又は、擬似的に溶媒として機能し、ポリイミド前駆体の分子鎖の自由度を向上させることができる。そのため、少量の塩基であっても効率的にポリイミド前駆体のイミド化を促進することができると推測される。中でも融点は190℃以下であることが好ましい。
【0038】
本発明に係る光塩基発生剤は発生する塩基が共有結合を用いて潜在化されていることから、発生する塩基としては、1級アミン、2級アミン、又は複素環式化合物が挙げられる。またアミンには、それぞれ、脂肪族アミン及び芳香族アミンがある。なお、アミンのうち、アミンの窒素原子に少なくとも1つの芳香族基が直接結合している場合を芳香族アミンといい、それ以外の場合は脂肪族アミンに含まれる。また、ここでの複素環式化合物は、塩基が環状構造を有し且つ芳香族性を有しているものをいう。芳香族複素環式化合物ではない、非芳香族複素環式化合物は、ここでは脂環式アミンとして脂肪族アミンに含まれる。
【0039】
更に、発生する塩基は、アミド結合を形成可能なNH基を1つだけ有するモノアミン等の塩基だけでなく、ジアミン、トリアミン、テトラアミン等のアミド結合を形成可能なNH基を2つ以上有する塩基であってもよい。中でも、アミド結合を形成可能なNH基を1つ有するものであることが、塩基の発生効率、溶剤溶解性、合成上の精製の点から好ましい。
以下に発生する塩基の一例を示すが、これに限定されるものではない。
【0040】
脂肪族1級アミンとしては、エタノールアミン、3−アミノ−1−プロパノール、1−アミノ−2−プロパノール、2−アミノ−1−プロパノール、4−アミノ−1−ブタノール、2−アミノ−1−ブタノール、1−アミノ−2−ブタノール、3−アミノ−2,2−ジメチル−1−プロパノール、4−アミノ−2−メチル−1−ブタノール、バリノール、3−アミノ−1,2−プロパンジオール、2−アミノ−1,3−プロパンジオール、チラミン、ノルエフェドリン、2−アミノ−1−フェニル−1,3−プロパンジオール、2−アミノシクロヘキサノール、4−アミノシクロヘキサノール、4−アミノシクロヘキサンエタノール、4−(2−アミノエチル)シクロヘキサノール等が挙げられる。
【0041】
脂肪族2級アミンとしては、N−メチルエタノールアミン、3−(メチルアミノ)−1−プロパノール、3−(イソプロピルアミノ)プロパノール、N−シクロヘキシルエタノールアミン、α−[2−(メチルアミノ)エチル]ベンジルアルコール、ジエタノールアミン、ジイソプロパノールアミン、3−ピロリジノール、2−ピロリジンメタノール、4−ヒドロキシピペリジン、3−ヒドロキシピペリジン、4−ヒドロキシ−4−フェニルピペリジン、4−(3−ヒドロキシフェニル)ピペリジン、4−ピペリジンメタノール、3−ピペリジンメタノール、2−ピペリジンメタノール、4−ピペリジンエタノール、2−ピペリジンエタノール、2−(4−ピペリジル)−2−プロパノール等が挙げられ、中でも脂環式アミンが好ましい。
【0042】
芳香族1級アミンとしては、4−アミノフェノール、3−アミノフェノール、2−アミノフェノール、4−アミノベンジルアルコール、3−アミノベンジルアルコール、2−アミノベンジルアルコール、2−(4−アミノフェニル)エタノール等が挙げられる。
【0043】
芳香族2級アミンとしては、2−アニリノエタノール、3−アニリノ−1−プロパノール、4−ヒドロキシジフェニルアミン、3−ヒドロキシジフェニルアミン、3,3’−ジヒドロキシジフェニルアミン、4−ベンジルアミノフェノール等が挙げられる。また、アミド結合を形成可能なNH基を有する芳香族複素環式化合物としては、塩基性の点から分子内にイミノ結合(−N=C(−R)−、−C(=NR)−、ここでRは水素原子又は有機基)を有することが好ましく、イミダゾール、プリン、トリアゾールに水酸基が置換された誘導体が挙げられる。これらの中でも、塩基の中の塩基性部位がすべて中性に潜在化されている光塩基発生剤が好ましい。
【0044】
ポリイミド前駆体から最終生成物への反応に対する反応開始温度を低下させる触媒作用は、塩基性の大きい塩基の方が触媒としての効果が大きく、より少量の添加で、より低い温度での最終生成物への反応が可能となる。一般に芳香族アミンよりも脂肪族アミンの方が塩基性が強いため、その触媒能が高い。従って、本発明に係る光塩基発生剤においては、発生する塩基が脂肪族アミンであることが好ましい。また、発生する塩基が脂肪族アミンである場合には、イミド化終了後に、速やかに揮発しやすく、同じ加熱条件によってイミド化された場合、塩基性芳香族へテロ環化合物やアミジン類に比べ、塗膜中への残存が少ない点からも好ましい。
【0045】
また、本発明で発生する塩基が、2級アミン及び/又は複素環式化合物である場合には、光塩基発生剤としての感度が高くなる点から好ましい。これは、2級アミンや複素環式化合物を用いることで、アミド結合部位の活性水素がなくなり、このことにより、電子密度が変化し、異性化の感度が向上するからではないかと推定される。
【0046】
さらに、本発明に係る光塩基発生剤から発生する塩基においては、酸性基を含まないことが、発生した塩基の触媒能を低下させない点から好ましい。但し、フェノール性水酸基のような弱い酸性基を含む塩基を発生する光塩基発生剤は、塩基の触媒能を低下させないことから、本発明の光塩基発生剤において用いられる場合がある。酸性基としては、カルボキシル基、スルホ基、及びリン酸基等の強い酸性基を含まないことが好ましい。
【0047】
本発明に係る光塩基発生剤においては、下記化学式(1−1)、下記化学式(1−2)又は下記化学式(1−3)で表される光塩基発生剤であることが、塩基の存在下での加熱によって最終生成物への反応が促進される様々なポリイミド前駆体に対して利用可能である点から好ましい。中でも、優れた感度を有し、ポリイミド前駆体の種類を問わず、形状が良好なパターンを得ることができる点から、下記化学式(1−1)で表される光塩基発生剤であることが好ましい。
【0048】
【化4】
(式(1−1)中、R1及びR2は、それぞれ独立に、水素原子又は有機基であり、同一であっても異なっていても良い。R1及びR2の少なくとも1つは有機基である。R1及びR2は、それらが結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。但し、R1及びR2のいずれかの有機基が水酸基を有する。R3及びR4はそれぞれ独立に、水素原子、ハロゲン原子、水酸基、メルカプト基、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、又は有機基であり、同一であっても異なっていても良い。R5、R6、R7及びR8は、それぞれ独立に、水素原子、ハロゲン原子、水酸基、メルカプト基、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、アミノ基、アンモニオ基又は有機基であり、同一であっても異なっていても良い。R5、R6、R7及びR8は、それらの2つ以上が結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。R9は、水素原子、或いは、加熱及び/又は電磁波の照射により脱保護可能な保護基である。)
【0049】
【化5】
(式(1−2)中、R1及びR2は、それぞれ独立に、水素原子又は有機基であり、同一であっても異なっていても良い。R1及びR2の少なくとも1つは有機基である。R1及びR2は、それらが結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。但し、R1及びR2のいずれかの有機基が水酸基を有する。R10及びR10’はそれぞれ独立に水素原子、ハロゲン原子、水酸基、メルカプト基、ニトロ基、シリル基、シラノール基、又は有機基である。R11、R12、R13、R14及びR15は、それぞれ独立に、水素原子、ハロゲン原子、水酸基、メルカプト基、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、アミノ基、アンモニオ基又は有機基であり、同一であっても異なっていても良い。R11、R12、R13、R14及びR15は、それらの2つ以上が結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。)
【0050】
【化6】
(式(1−3)中、R17は、水酸基を有する有機基で置換されたアミノ基又は水酸基を有する有機基、R18及びR19は、それぞれ独立に、水素原子、ハロゲン原子、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、アミノ基、アンモニオ基又は有機基であり、同一であっても異なっていても良いが、R18及びR19の少なくとも1つはヘテロ原子を有していても良い芳香族化合物である。R17、R18及びR19は、それらの2つ以上が結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。)
【0051】
上記化学式(1−1)で表される光塩基発生剤は、電磁波が照射されることにより、下記式で示されるように、式(1−1)で中の(−CR4=CR3−C(=O)−)部分がトランス体からシス体へと異性化する。さらに加熱及び/又は電磁波の照射によって、R9が保護基である場合は保護基R9が脱保護されると共に環化し、塩基(NHR1R2)を生成する。式(1−1)で表される光塩基発生剤は電磁波の照射のみでも塩基を発生するが、適宜加熱することにより塩基の発生が促進される。
【0052】
【化7】
【0053】
また、上記化学式(1−2)で表される光塩基発生剤は、電磁波を照射するとカルバメート結合の光脱炭酸反応によって結合が開裂し、塩基(NHR1R2)を発生させる。
更に、上記化学式(1−3)で表される光塩基発生剤は、オキシムエステル誘導体であって、下記反応式に従って、電磁波の吸収により分子内解裂反応を起こし、アミンまたはヒドラジンを発生させる。
【0054】
【化8】
【0055】
上記化学式(1−1)及び化学式(1−2)において、R1及びR2は、それぞれ、独立に水素原子又は有機基であるが、R1及びR2のうち少なくとも1つは有機基である。また、NHR1R2は、塩基であるが、R1及びR2は、それぞれ、アミノ基を含まない有機基であることが好ましい。R1及びR2に、アミノ基が含まれてしまうと、光塩基発生剤自体が塩基となり、ポリイミド前駆体の反応を促進してしまい、露光部と未露光部での溶解性コントラストの差が小さくなってしまう恐れがある。但し、例えば、R1及びR2の有機基中に存在する芳香環にアミノ基が結合している場合のように、電磁波の照射と加熱後に発生する塩基との塩基性と差が生じる場合には、R1及びR2の有機基にアミノ基を含まれていても用いることができる場合もある。
有機基としては、例えば、飽和又は不飽和アルキル基、飽和又は不飽和シクロアルキル基、アリール基、アラルキル基、及び飽和又は不飽和ハロゲン化アルキル基等が挙げられる。これらの有機基は、当該有機基中にヘテロ原子等の炭化水素基以外の結合や置換基を含んでよく、これらは、直鎖状でも分岐状でも環状でも良い。
有機基としては、置換基を含んで良く、不飽和結合を含んで良く、ヘテロ原子の結合を含んで良い、直鎖、分岐又は環状の炭化水素基が好ましい。
R1及びR2における有機基は、通常、1価の有機基であるが、後述する環状構造を形成する場合や、生成するNHR1R2がジアミン等のアミド結合を形成可能なNH基を2つ以上有する塩基の場合等には、2価以上の有機基となり得る。
R1及びR2のいずれかの有機基が水酸基を有する。有機基中に水酸基を1つ以上有すれば有機基内のどこに存在していてもよい。R1及びR2が環状構造を形成する場合には、当該環状構造内に水酸基を1つ以上有すればよい。R1及びR2のいずれかの有機基が水酸基を有することにより、光塩基発生剤は水酸基を有する塩基を発生する。塩基が水酸基を有することにより、触媒能が向上し、露光後ベーク時の加熱条件を緩和することができ、例えばより低温であっても後述するポリイミド前駆体を効率よくイミド化することができる。
【0056】
また、R1及びR2は、それらが結合して環状構造になっていても良い。
環状構造は、飽和又は不飽和の脂環式炭化水素、複素環、及び縮合環、並びに当該脂環式炭化水素、複素環、及び縮合環よりなる群から選ばれる2種以上が組み合されてなる構造であっても良い。
【0057】
前記R1及びR2の有機基中の炭化水素基以外の結合としては、本発明の効果が損なわれない限り、特に限定されず、エーテル結合、チオエーテル結合、カルボニル結合、チオカルボニル結合、エステル結合、アミド結合、ウレタン結合、イミノ結合(−N=C(−R)−、−C(=NR)−、ここでRは水素原子又は有機基)、カーボネート結合、スルホニル結合、スルフィニル結合、アゾ結合等が挙げられる。耐熱性の点から、有機基中の炭化水素基以外の結合としては、エーテル結合、チオエーテル結合、カルボニル結合、チオカルボニル結合、エステル結合、アミド結合、ウレタン結合、イミノ結合(−N=C(−R)−、−C(=NR)−:ここでRは水素原子又は有機基)、カーボネート結合、スルホニル結合、スルフィニル結合が好ましい。
【0058】
前記R1及びR2の有機基中の炭化水素基以外の置換基、すなわち、有機基に包含される置換基において炭化水素基とは異なる置換基、及び炭化水素基に置換されていても良い置換基としては、本発明の効果が損なわれない限り、特に限定されず、ハロゲン原子、メルカプト基(−SH)、スルフィド基(−SR、ここでRは有機基)、シアノ基(−CN)、イソシアノ基(−NC)、シアナト基(−OCN)、イソシアナト基(−NCO)、チオシアナト基(−SCN)、イソチオシアナト基(−NCS)、アルキルエーテル基及びアリールエーテル基(−OR、ここでRは有機基)、アルコキシカルボニル基及びアリールオキシカルボニル基(−COOR、ここでRは有機基)、アルキルスルフィニル基及びアリールスルフィニル基(−SOR、ここでRは有機基)、アルキルスルホニル基及びアリールスルホニル基(−SO2R、ここでRは有機基)、アルコキシスルホニル基及びアリールオキシスルホニル基(−SO3R、ここでRは有機基)、カルバモイル基(−CONRR’、ここでR、R’は有機基)、チオカルバモイル基(−CSNRR’、ここでR、R’は有機基)、スルファモイル基(−SO3NRR’、ここでR、R’は有機基)、ニトロ基(−NO2)、ニトロソ基(−NO)、アシル基(−COR、ここでRは有機基)、アシルオキシ基(−OCOR、ここでRは有機基)、アシルアミノ基(−NHCOR、ここでRは有機基)、カーバメート基(−NHCOOR、ここでRは有機基)、ホスフィノ基(−PR2)、シリル基(−SiR3、ここでRは有機基)、アルコキシシリル基及びアリールオキシシリル基(−Si(OR)XR’Y、ここでR、R’は有機基。Xは1から3の整数、Yは0から2の整数で、X+Y=3。)、アミノ基(−NH2, −NHR, −NRR’:ここで、R及びR’はそれぞれ独立に炭化水素基)等が挙げられる。上記置換基に含まれる水素原子は、炭化水素基によって置換されていても良い。また、上記置換基に含まれる炭化水素基は、直鎖、分岐、及び環状のいずれでも良い。
前記R1及びR2の有機基中の炭化水素基及び水酸基とは異なる置換基としては、ハロゲン原子、メルカプト基(−SH)、スルフィド基(−SR、ここでRは有機基)、シアノ基(−CN)、イソシアノ基(−NC)、シアナト基(−OCN)、イソシアナト基(−NCO)、チオシアナト基(−SCN)、イソチオシアナト基(−NCS)、アルキルエーテル基及びアリールエーテル基(−OR、ここでRは有機基)、アルコキシカルボニル基及びアリールオキシカルボニル基(−COOR、ここでRは有機基)、スルフィニル基(−SOR、ここでRは有機基)、アルキルスルホニル基及びアリールスルホニル基(−SO2R、ここでRは有機基)、アルコキシスルホニル基及びアリールオキシスルホニル基(−SO3R、ここでRは有機基)、カルバモイル基(−CONRR’、ここでR、R’は有機基)、チオカルバモイル基(−CSNRR’、ここでR、R’は有機基)、スルファモイル基(−SO3NRR’、ここでR、R’は有機基)、ニトロ基(−NO2)、ニトロソ基(−NO)、アシル基(−COR、ここでRは有機基)、アシルオキシ基(−OCOR、ここでRは有機基)、アシルアミノ基(−NHCOR、ここでRは有機基)、カーバメート基(−NHCOOR、ここでRは有機基)シリル基(−SiR3、ここでRは有機基)、アルコキシシリル基及びアリールオキシシリル基(−Si(OR)XR’Y、ここでR、R’は有機基。Xは1から3の整数、Yは0から2の整数で、X+Y=3。)が好ましい。
なお、上記炭化水素基以外の結合や上記炭化水素基以外の置換基中の水素原子又は有機基が、水酸基に置換された構造であっても良い。
【0059】
また、発生する塩基の熱物性、及び塩基性度の点から、R1及びR2の有機基は、それぞれ独立に炭素数1〜20が好ましく、更に炭素数1〜12が好ましく、特に炭素数1〜8であることが好ましい。
発生する塩基はNHR1R2であるため、1級アミン、2級アミン、又は複素環式化合物が挙げられる。発生する塩基の好適な例としては、上述したのでここでの説明を省略する。
【0060】
また、化学式(1−1)において、R3及びR4は、それぞれ独立に、水素原子、ハロゲン原子、水酸基、メルカプト基、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、又は有機基であり、同一であっても異なっていても良い。
R3及びR4としては、高感度を達成しやすい点から、いずれも水素原子であることが好ましい。
【0061】
本発明において、特に化学式(1−1)中のR3及びR4のうち少なくとも1つが、水素原子ではなく、上記特定の官能基である場合には、R3及びR4の両方共が水素原子の場合と比べて、本発明の光塩基発生剤は、有機溶剤に対する溶解性を更に向上させたり、ポリイミド前駆体との親和性を向上させることが可能である。例えば、R3及びR4のうち少なくとも1つが、アルキル基やアリール基等の有機基である場合、有機溶剤に対する溶解性が向上する。また、例えばR3及びR4のうち少なくとも1つがフッ素等のハロゲン原子である場合、フッ素等のハロゲン原子を含有するポリイミド前駆体との親和性が向上する。このように、R3及び/又はR4を所望の有機溶剤やポリイミド前駆体に合わせて適宜置換基を導入することにより、所望の有機溶剤に対する溶解性や、所望のポリイミド前駆体との親和性を向上することが可能である。
【0062】
ハロゲン原子、有機基としては、本発明の効果が損なわれない限り、特に制限がなく、後述するR5〜R8、R11〜R15に挙げたものと同様のものを用いることができる。
R3及びR4における有機基は、通常、1価の有機基である。
【0063】
R3及びR4が、置換基を有する場合には少なくとも一方が、メチル基、エチル基、プロピル基等の炭素数1〜20のアルキル基;シクロペンチル基、シクロヘキシル基等の炭素数4〜23のシクロアルキル基;シクロペンテニル基、シクロヘキセニル基等の炭素数4〜23のシクロアルケニル基;フェノキシメチル基、2−フェノキシエチル基、4−フェノキシブチル基等の炭素数7〜26のアリールオキシアルキル基(−ROAr基);ベンジル基、3−フェニルプロピル基等の炭素数7〜20のアラルキル基;シアノメチル基、β−シアノエチル基等のシアノ基をもつ炭素数2〜21のアルキル基;ヒドロキシメチル基等の水酸基をもつ炭素数1〜20のアルキル基、メトキシ基、エトキシ基等の炭素数1〜20のアルキルエーテル基、アセトアミド基、ベンゼンスルホナミド基(C6H5SO2NH2−)等の炭素数2〜21のアミド基、メチルチオ基、エチルチオ基等の炭素数1〜20のアルキルチオ基のようなスルフィド基(−SR基)、アセチル基、ベンゾイル基等の炭素数1〜20のアシル基、メトキシカルボニル基、アセトキシ基等の炭素数2〜21のエステル基(−COOR基及び−OCOR基)、フェニル基、ナフチル基、ビフェニル基、トリル基等の炭素数6〜20のアリール基、電子供与性基及び/又は電子吸引性基が置換した炭素数6〜20のアリール基、電子供与性基及び/又は電子吸引性基が置換したベンジル基、シアノ基であることが好ましい。また、上記のアルキル部分は直鎖でも分岐状でも環状でも良い。
【0064】
また、化学式(1−2)において、R10及びR10’はそれぞれ独立に水素原子、ハロゲン原子、水酸基、メルカプト基、ニトロ基、シリル基、シラノール基、又は有機基であるが、当該有機基としては、後述するR5〜R8、R11〜R15に挙げたものと同様のものを用いることができる。
【0065】
また、上記化学式(1−1)のR5〜R8、並びに、上記化学式(1−2)のR11〜R15は、それぞれ独立に、水素原子、ハロゲン原子、水酸基、メルカプト基、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、アミノ基、アンモニオ基又は有機基であり、同一であっても異なっていても良い。R5〜R8及びR11〜R15は、それぞれ、それらの2つ以上が結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。
【0066】
上記化学式(1−1)のR5〜R8、並びに、上記化学式(1−2)のR11〜R15には、置換基を1つ以上導入することが好ましい。特に上記化学式(1−1)で表される塩基発生剤の場合、カルボニル結合のα位およびβ位に位置するα炭素−β炭素間の二重結合がトランス体からシス体への異性化反応を効率よく進める要因としてはいくつかあり、例えば上記炭素−炭素二重結合周囲の立体障害の大きさ、上記炭素−炭素二重結合周囲に広がる共役鎖の電子状態等が挙げられるが、置換基R5〜R8に、上記のような置換基を少なくとも1つ導入することにより、上記炭素−炭素二重結合周囲の共役鎖が拡張し、塩基発生の感度を向上することができる。また、R5〜R8及びR11〜R15において、上記のような置換基を少なくとも1つ導入することにより、吸収する光の波長を調整することが可能であり、置換基を導入することで所望の波長を吸収させるようにすることもできる。芳香族環の共役鎖を伸ばすような置換基を導入することにより、吸収波長を長波長にシフトすることができる。また、溶解性や組み合わせるポリイミド前駆体との相溶性が向上するようにすることもできる。これにより、組み合わせるポリイミド前駆体の吸収波長も考慮しながら、感光性ポリイミド樹脂組成物の感度を向上させることが可能である。
【0067】
所望の波長に対して吸収波長をシフトさせる為に、どのような置換基を導入したら良いかという指針として、Interpretation of the Ultraviolet Spectra of Natural Products(A.I.Scott 1964)や、有機化合物のスペクトルによる同定法第5版(R.M.Silverstein 1993)に記載の表を参考にすることができる。これらを参考とすることで、化合物の極大吸収波長がどの程度長波長化するかの目安を知ることができる。
【0068】
上記化学式(1−2)のR11は、ニトロ基であることが、上記化学式(1−2)で表される光塩基発生剤の感度が向上する点から好ましい。
【0069】
R5〜R8及びR11〜R15において、ハロゲン原子としては、フッ素、塩素、臭素などが挙げられる。
R5〜R8及びR11〜R15において、有機基としては、例えば、飽和又は不飽和アルキル基、飽和又は不飽和シクロアルキル基、アリール基、アラルキル基、及び飽和又は不飽和ハロゲン化アルキル基等が挙げられる。これらの有機基は、当該有機基中にヘテロ原子等の炭化水素基以外の結合や置換基を含んでよく、これらは、直鎖状でも分岐状でも環状でも良い。R5〜R8及びR11〜R15の有機基中の炭化水素基以外の結合としては、前記R1及びR2の炭化水素基以外の結合と同様のものを用いることができる。また、R5〜R8及びR11〜R15の有機基は、炭化水素基以外の結合を介してベンゼン環に結合してもよい。また、R5〜R8及びR11〜R15の有機基において炭化水素基以外の置換基(有機基に包含される置換基において炭化水素基とは異なる置換基、及び炭化水素基に置換されていても良い置換基)としては、前記R1及びR2の炭化水素基以外の置換基と同様のものを用いることができる。
有機基としては、本発明の効果が損なわれない限り、特に制限がなく、飽和又は不飽和アルキル基、飽和又は不飽和シクロアルキル基、アリール基、アラルキル基、及び飽和又は不飽和ハロゲン化アルキル基、シアノ基、イソシアノ基、シアナト基、イソシアナト基、チオシアナト基、イソチオシアナト基、アルコキシカルボニル基、カルバモイル基、チオカルバモイル基、カルボキシル基、カルボキシラート基、アシル基、アシルオキシ基、ヒドロキシイミノ基、飽和又は不飽和アルキルエーテル基、飽和又は不飽和アルキルチオ基、アリールエーテル基、及びアリールチオ基等が挙げられる。
R5〜R8及びR11〜R15における有機基は、通常、1価の有機基であるが、後述する環状構造を形成する場合等には、2価以上の有機基となり得る。
【0070】
また、R5〜R8及びR11〜R15は、それらのうち2つ以上が結合して環状構造になっていても良い。
環状構造は、飽和又は不飽和の脂環式炭化水素、複素環、及び縮合環、並びに当該脂環式炭化水素、複素環、及び縮合環よりなる群から選ばれる2種以上が組み合されてなる構造であっても良い。例えば、R5〜R8及びR11〜R15のそれぞれは、それらの2つ以上が結合して、R5〜R8及びR11〜R15のそれぞれが結合しているベンゼン環の原子を共有してナフタレン、アントラセン、フェナントレン、インデン等の縮合環を形成していても良い。
【0071】
R5〜R8又はR11〜R15としては、水素原子、ハロゲン原子、水酸基、メルカプト基、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、アミノ基、アンモニオ基、メチル基、エチル基、プロピル基等の炭素数1〜20のアルキル基;シクロペンチル基、シクロヘキシル基等の炭素数4〜23のシクロアルキル基;シクロペンテニル基、シクロヘキセニル基等の炭素数4〜23のシクロアルケニル基;フェノキシメチル基、2−フェノキシエチル基、4−フェノキシブチル基等の炭素数7〜26のアリールオキシアルキル基(−ROAr基);ベンジル基、3−フェニルプロピル基等の炭素数7〜20のアラルキル基;シアノメチル基、β−シアノエチル基等のシアノ基をもつ炭素数2〜21のアルキル基;ヒドロキシメチル基等の水酸基をもつ炭素数1〜20のアルキル基、メトキシ基、エトキシ基、ベンジルオキシ基等の炭素数1〜20のアリール基で置換されていても良いアルキルエーテル基;フェノキシ基、ナフチルオキシ基等のアリールエーテル基;アセトアミド基、ベンゼンスルホナミド基(C6H5SO2NH2−)等の炭素数2〜21のアミド基、メチルチオ基、エチルチオ基等の炭素数1〜20のアルキルチオ基(−SR基)、ベンジルチオ基、ナフチルチオ基等のアリールチオ基;アセチル基、ベンゾイル基等の炭素数1〜20のアシル基;チオアシル基;アセチルチオ基、ベンゾイルチオ基等のアシルチオ基;メトキシカルボニル基、アセトキシ基、ベンジルオキシカルボニル基等の炭素数2〜21のエステル基(−COOR基及び−OCOR基)、フェニル基、ナフチル基、ビフェニル基、トリル基等の炭素数6〜20のアリール基、及び、電子供与性基及び/又は電子吸引性基が置換した炭素数6〜20のアリール基、電子供与性基及び/又は電子吸引性基が置換したベンジル基、シアノ基、カルバモイル基、カルバモイルオキシ基、シアノオキシ基(シアナト基)、シアノチオ基(チオシアナト基)、ホルミル基、であることが好ましい。また、上記のアルキル部分は直鎖でも分岐状でも環状でも良い。
また、R5〜R8又はR11〜R15としては、それらの2つ以上が結合して、R5〜R8又はR11〜R15が結合しているベンゼン環の原子を共有してナフタレン、アントラセン、フェナントレン、インデン等の縮合環を形成している場合も、吸収波長が長波長化する点から好ましい。
【0072】
また、式(1−1)においてR9は、水素原子、或いは、加熱及び/又は電磁波の照射により脱保護可能な保護基である。ここで、“脱保護可能な”とは、−OR9から−OHに変化する可能性があることを表す。R9が水素原子の場合には、本発明に係る光塩基発生剤は、環化することで、フェノール性水酸基を消失し、溶解性が変化し、塩基性水溶液等の場合には溶解性が低下する。これにより、ポリイミド前駆体の最終生成物への反応による溶解性の低下を更に補助する機能を有し、露光部と未露光部の溶解性コントラストを更に大きくすることが可能となる。
【0073】
また、R9が加熱及び/又は電磁波の照射により脱保護可能な保護基である場合、加熱及び/又は電磁波の照射により脱保護されて、水酸基を生成する。加熱及び/又は電磁波の照射により脱保護可能な保護基でフェノール性水酸基を保護することにより、当該保護基を適宜選択することによって、ポリイミド前駆体との相溶性が向上し、組み合わせ可能な化合物の範囲が増える。例えば、フェノール性水酸基と共存することが好ましくないポリイミド前駆体に対しても、感光性ポリイミド樹脂組成物中に共存させて用いることが可能になる。R9は、本発明で用いられる光塩基発生剤において式(1−1)中に存在するアミド基が分解しない条件下で、加熱及び/又は電磁波の照射により脱保護可能なフェノール性水酸基の保護基であれば、特に限定されず用いることができる。例えば、アミド結合は、三臭化ホウ素や三塩化アルミニウム等の強ルイス酸や硫酸、塩酸、硝酸等の強酸等が存在する強酸性下における加熱や、水酸化ナトリウム等の強塩基が存在する強塩基性下における加熱により分解する。従って、このような強酸性又は強塩基性条件下での加熱でしか脱保護されない保護基は、本発明の光塩基発生剤に用いられる保護基としては不適切である。R9は、溶解性や相溶性の向上或いは合成時の反応性の変化などを目的として、当該光塩基発生剤と組み合わせて用いられる化合物の種類や、光塩基発生剤の適用方法や合成方法により適宜選択されるものである。
【0074】
R9としては、シリル基、シラノール基、ホスフィノ基、ホスフィニル基、ホスホノ基、又は有機基から選択することができる。R9における有機基は、通常、1価の有機基である。
【0075】
R9としては、下記式(2−1)〜下記式(2−6)で表わされる有機基よりなる群から選択される1種以上であることが、式(1−1)中に存在するアミド基が分解しない条件下で、加熱及び/又は電磁波の照射により脱保護可能な点から好ましい。
【0076】
【化9】
(式(2−1)中、R30、R31、R32はそれぞれ独立に水素原子、ハロゲン原子、または有機基であり、R33は有機基であり、R30、R31、R32、R33はそれぞれ互いに結合して環状構造を示していても良い。式(2−2)中、R34は、有機基である。式(2−3)中、R35、R36、R37はそれぞれ独立に水素原子、ハロゲン原子、または有機基である。式(2−4)中、R38は、有機基である。式(2−5)中、R39は、置換基を有していても良い芳香環である。式(2−6)中、R40は、有機基である。)
【0077】
上記式(2−1)で表される有機基は、水酸基と、各種ビニルエーテル化合物との反応により得ることができる。式(2−1)で表される有機基は、例えば、各種ビニルエーテル化合物の残基である。
上記式(2−2)で表される有機基は、例えば、水酸基と、カーボネート系保護基の導入試薬(たとえばジ−t−ブチルジカルボナートや、塩化ベンジルオキシカルボニル、N−(9−フルオレニルメトキシカルボニルオキシ)コハク酸イミドなど)との反応により得ることができる。式(2−2)で表される有機基は、例えば、各種カーボネート系保護基の残基である。
上記式(2−3)で表される有機基は、例えば、水酸基と、シリルエーテル系保護基の導入試薬(たとえばクロロトリメチルシラン、tert−ブチルジメチルクロロシラン、tert−ブチルジフェニルクロロシランなど)との反応により得ることができる。式(2−3)で表される有機基は、例えば、シリルエーテル系保護基の残基である。
【0078】
上記式(2−4)で表される有機基は、例えば、水酸基と、酸塩化物または酸無水物との反応により得ることができ、式(2−4)で表される有機基は、酸塩化物または酸無水物の残基である。
上記式(2−5)で表される有機基は、例えば、Williamson反応を用いて、水酸基と、ハロゲン化物(たとえばベンジルクロライドなど)により得ることができ、式(2−5)で表される有機基は、ハロゲン化物の残基である。
上記式(2−6)で表される有機基は、例えば、水酸基と、イソシアネート化合物(たとえばベンジルイソシアネートなど)との反応により得ることができ、式(2−6)で表される有機基は、イソシアネート化合物の残基である。
【0079】
上記化学式(1−1)で表される構造は、幾何異性体が存在するが、トランス体のみを用いることが好ましい。しかし、合成および精製工程および保管時などにおいて幾何異性体であるシス体が混ざる可能性もあり、この場合トランス体とシス体の混合物を用いても良いが、溶解性コントラストを高められる点から、シス体の割合が10%未満であることが好ましい。
【0080】
上記化学式(1−3)中、R17は、水酸基を有する有機基で置換されたアミノ基又は水酸基を有する有機基である。R17が、水酸基を有する有機基である場合、R5〜R8、R11〜R15に挙げた有機基と同様の有機基に適宜水酸基が置換された構造を用いることができる。
上記化学式(1−3)のR17が、水酸基を有する有機基で置換されたアミノ基である場合、下記式(1−3’)で表されるカルバモイルオキシム構造を有し、上述のように、アミン及びヒドラジンを発生し得る。
【0081】
【化10】
(式(1−3’)中、R18及びR19は、化学式(1−3)と同じであり、R20及びR21は、上記化学式(1−1)のR1及びR2と同じである。R18、R19、R20及びR21は、それらの2つ以上が結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。但し、R20及びR21のいずれかの有機基が水酸基を有する。)
【0082】
上記化学式(1−3)及び式(1−3’)中、R18及びR19の少なくとも1つはヘテロ原子を有していても良い芳香族化合物であることが特徴的である。R18及びR19の少なくとも1つに芳香族化合物が置換している状態とは、R18及びR19が結合している炭素に芳香族化合物が直接共有結合で結合している状態のことをいう。なお、ここでの芳香族化合物は、環状不飽和有機化合物をいい、芳香族炭化水素と芳香族複素環式化合物が含まれる。芳香族化合物としては、例えば、フェニル基、ビフェニル基、ターフェニル基、ナフチル基、フルオレン基、アントラニル基、フェナントレニル基、ピレニル基等の芳香族炭化水素基の他、フラニル基、チオフェニル基、ピロリル基、(チオ)キサンテニル基、(チオ)キサントニル基、クマリル基、アントラキノリル基等の複素芳香族化合物等が挙げられるが特に限定されない。これらが有していても良い置換基としては、上述したR5〜R8及びR11〜R15と同様であって良い。R18及びR19の一方が、芳香族化合物以外の有機基である場合も、上述したR5〜R8及びR11〜R15と同様であって良い。
【0083】
上記化学式(1−3)及び式(1−3’)中、R17、R18、R19、R20及びR21は、それらの2つ以上が結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。R18及びR19が結合して環状構造を形成している場合としては、例えば、R18及びR19が結合している炭素と共に、フルオレン環を形成している態様が挙げられる。また、R18及びR17やR18及びR20が結合して環状構造を形成している場合としては、例えば、R18及びR17やR18及びR20が脂肪族炭化水素基及び/又は芳香族炭化水素基で連結されている態様が挙げられる。
発生する塩基が水酸基を有するため、R20及びR21のいずれかの有機基は水酸基を有する。R20及びR21は、上記化学式(1−1)のR1及びR2と同じであるため、ここでの説明を省略する。
以下に本発明の光塩基発生剤の一例を示すが、これらに限定されるものではない。
【0084】
【化11】
【0085】
【化12】
【0086】
【化13】
【0087】
本発明に係る光塩基発生剤は、加熱して初期の重量から5%重量が減少したときの温度(5%重量減少温度)が、60℃以上であることが好ましく、更に100℃以上であることが好ましい。ポリイミド前駆体の場合、塗膜を形成する際にN−メチル−2−ピロリドンなどの高沸点溶媒を用いる必要があるが、このように5%重量減少温度が高い場合には残留溶媒の影響が少なくなるような乾燥条件で塗膜を形成することができる。これにより、残留溶媒の影響による露光部と未露光部での溶解性コントラストの減少を抑制することができる。
本発明において、x%重量減少温度とは、熱重量分析装置を用いて重量減少を測定した時に、サンプルの重量が初期重量からx%減少した時点(すなわち、サンプル重量が初期の(100−x)%となった時点)の温度である。
【0088】
本発明の光塩基発生剤は、複数の従来公知の合成ルートで合成することができる。上記化学式(1−1)で表される光塩基発生剤は、例えば、上記特許文献5や本発明者らによる特願2010−176384を参考に合成することができる。上記化学式(1−1)で表される光塩基発生剤において、フェノール性水酸基における保護基(R9)の導入は、合成途中で導入していても良いし、合成の最後に導入しても良い。また、上記化学式(1−2)で表される光塩基発生剤は、例えば、上記特許文献6及び7を参考に合成することができる。また、上記化学式(1−3)で表される光塩基発生剤は、例えば、上記特許文献8及び9を参考に合成することができる。
【0089】
本発明の光塩基発生剤は、ポリイミド前駆体が最終生成物となるための塩基発生の機能を十分に発揮させるために、露光波長の少なくとも一部に対して吸収を有する必要がある。一般的な露光光源である高圧水銀灯の波長としては、365nm、405nm、436nmがある。このため、本発明で用いられる光塩基発生剤は、少なくとも365nm、405nm、436nmの波長の電磁波のうち少なくとも1つの波長の電磁波に対して吸収を有することが好ましい。このような場合、適用可能なポリイミド前駆体の種類がさらに増える点から好ましい。
【0090】
本発明の光塩基発生剤は、モル吸光係数が、電磁波の波長365nmにおいて100以上、又は405nmにおいて1以上であることが、適用可能なポリイミド前駆体の種類がさらに増える点から好ましい。
【0091】
なお、本発明の光塩基発生剤が前記波長領域に吸収を有することは、当該波長領域に吸収をもたない溶媒(例えば、アセトニトリル)に、本発明の光塩基発生剤を1×10−4mol/L以下の濃度(通常、1×10−4mol/L〜1×10−5mol/L程度。適度な吸収強度となるように、適宜、調節してもよい。)で溶解し、紫外可視分光光度計(例えば、UV−2550(株)島津製作所製))により吸光度を測定することにより明らかにすることができる。
【0092】
本発明の光塩基発生剤は、後述するポリイミド前駆体と組み合わせて感光性ポリイミド樹脂組成物を調製するための前駆体型感光性ポリイミド用光塩基発生剤として好適に用いられる。しかしながら、本発明の光塩基発生剤は、塩基によって又は塩基の存在下での加熱によって最終生成物への反応が促進される他の高分子前駆体と組み合わせて感光性樹脂組成物を調製することもできる。
【0093】
<感光性ポリイミド樹脂組成物>
本発明に係るネガ型感光性ポリイミド樹脂組成物は、ポリイミド前駆体と、前記本発明に係る光塩基発生剤とを含有することを特徴とする。
本発明に係るネガ型感光性ポリイミド樹脂組成物は、露光されると前記光塩基発生剤から塩基が発生し、当該塩基の触媒作用により、露光部は露光後ベーク時におけるポリイミド前駆体のイミド化が未露光部に比べ促進され、より低温でイミド化を進行させることができる。本発明のネガ型感光性ポリイミド樹脂組成物を用いて、パターンを得るには、パターンを残したい場所に露光後、塩基が存在する場所ではイミド化が進行し、塩基の存在していない場所ではイミド化が進行し難い温度で、加熱を行う。これにより、露光部の現像液に対する溶解性が低下し、現像後に露光部分が残る。
【0094】
本発明に係るネガ型感光性ポリイミド樹脂組成物は、前記本発明に係る光塩基発生剤を用いることにより、露光後ベーク時の加熱条件を従来より緩和しても、露光部は十分にイミド化が促進され、未露光部は緩和された加熱条件のためにポリイミド前駆体のイミド化の進行を抑制することができる。また、塩基に導入されている水酸基の効果で、未露光部の現像液への溶解が促進される。緩和された加熱条件と水酸基の溶解促進の相乗効果により、未露光部の現像液への溶解性が向上するため、未露光部を除去する現像時間を短縮でき、また、アルコール等を添加しない塩基性水溶液を現像液として用いることが可能になる。更に、露光部は十分に硬化される一方で、未露光部の現像液への溶解性が高いことから、露光部と未露光部の溶解性コントラストが高くなるため、レリーフパターンの形状が良好になる。
【0095】
以下、本発明に係る感光性ポリイミド樹脂組成物の構成成分を、ポリイミド前駆体、光塩基発生剤、必要に応じて適宜含むことができるその他の成分について順に説明する。
光塩基発生剤及びポリイミド前駆体としては、1種単独で用いても良いし、2種以上混合して用いても良い。
【0096】
(ポリイミド前駆体)
本発明の感光性ポリイミド樹脂組成物に用いるポリイミド前駆体とは、反応により最終的に目的の物性を示すポリイミドとなる物質を意味する。
本発明に用いるポリイミド前駆体は、なんらかの溶媒(有機溶剤、又は水溶液)に可溶なものが好適に用いられる。溶媒(有機溶剤、又は水溶液)に可溶なものであると、ポリイミド前駆体の当該溶媒に対する溶解性を変化させることにより、その可溶な溶媒を現像液として用いて、適宜、有機溶剤、塩基性水溶液、酸性水溶液、又は中性水溶液による現像をすることが可能になる。
ここで、ある溶媒に可溶とは、具体的には、基板上に形成された塗膜の25℃における当該溶媒に対する溶解速度が、10nm/sec以上を目安とする。当該溶解速度は、プロセス適性の観点から30nm/sec以上であることがさらに好ましい。
【0097】
例えば、塩基性水溶液に可溶なものは、具体的には、基板上に形成された塗膜の25℃における0.1重量%のテトラメチルアンモニウムヒドロキシド水溶液に対する溶解速度が、10nm/sec以上である。当該溶解速度は30nm/sec以上であることがさらに好ましい。さらには、より一般的に用いられる現像液である2.38重量%のテトラメチルアンモニウムヒドロキシド水溶液に対する溶解速度が、10nm/sec以上であることが好ましく、30nm/sec以上であることがさらに好ましい。上記定義による溶解速度が10nm/secより小さい場合、現像時間が遅くなり作業性、生産性が悪くなると共に、露光部、未露光部間の溶解性コントラストが得にくくなる。
【0098】
上記溶解速度を測定する具体的手順としては、無アルカリガラス等の基板上に形成されたポリイミド前駆体の塗膜を、25℃に調温され、撹拌された現像液(0.1重量%TMAH水溶液または、2.38重量%TMAH水溶液等の塩基性水溶液、有機溶剤等)に一定時間、浸漬し、蒸留水でリンス後、乾燥させた後で測定した膜厚と、初期膜厚との差を、膜減り量とし、その膜減り量を、現像液に浸漬した時間で割ったものが、25℃における単位時間当たりの溶解速度ということになる。
【0099】
また、露光部と未露光部の間に十分な溶解性コントラストを得るために、感光性ポリイミド樹脂組成物を実際に所定の感光パターン形成プロセスにおいて用いた時に、パターン状露光、及び、加熱工程を行って得られる、現像工程前における未露光部位と露光部位の現像液に対する溶解性の比(未露光部位の現像液に対する単位時間当たりの溶解速度/露光部位の現像液に対する単位時間当たりの溶解速度)が、5以上であることが好ましく、10以上であることが更に好ましい。
単位時間当たりの溶解速度は、上記の方法と同様にして求められ、感光性ポリイミド樹脂組成物の塗膜にパターン露光を行い、露光後の加熱を行った後に、露光部、未露光部の溶解速度を、それぞれ求める。
【0100】
本発明においては、塩基の作用によって最終生成物への反応が促進されるポリイミド前駆体が用いられ、塩基の作用によってポリイミド前駆体の最終生成物への反応温度が、塩基の作用がない場合に比べて低下するようなポリイミド前駆体が用いられる。
本発明に用いられるポリイミド前駆体としては、下記化学式(3)で表される繰り返し単位を有するポリイミド前駆体が好適に用いられる。
【0101】
【化14】
(式(3)中、R31は4価の有機基である。R32は2価の有機基である。R33及びR34は、水素原子、又は有機基である。nは1以上の自然数である。)
【0102】
R33及びR34が有機基である場合としては、例えば、アルキル基、アルケニル基、アルキニル基、アリール基、及び、これらにエーテル結合を含有したCnH2nOCmH2m+1などで表される構造等を挙げることができる。
【0103】
ポリイミド前駆体としては、下記式(4)で表されるようなポリアミック酸が、アルカリ現像性の点から好適に用いられる。
【0104】
【化15】
(式(4)中、R31は4価の有機基である。R32は2価の有機基である。nは1以上の自然数である。)
【0105】
なお、式(3)及び式(4)において、R31の4価は、酸二無水物等から誘導されるテトラカルボン酸残基を示し、R32の2価はジアミン残基を示す。なお、R31の4価は酸と結合するための価数のみを示しているが、他に更なる置換基を有していても良い。同様に、R32の2価はアミンと結合するための価数のみを示しているが、他に更なる置換基を有していても良い。
【0106】
ポリアミック酸は、酸二無水物とジアミンを溶液中で混合するのみで得られるので、1段階の反応で合成することができ、合成が容易で低コストで入手できるので好ましい。
【0107】
副次的な効果として、用いるポリイミド前駆体がポリアミック酸である場合、塩基の触媒効果によりイミド化に要する温度が低くても十分な為、最終キュア温度を300℃未満、更に好ましくは250℃以下まで下げることが可能である。従来のポリアミック酸はイミド化するために最終キュア温度を300℃以上とする必要があった為、用途が制限されていたが、最終キュア温度を下げることが可能になったことによって、より広範囲の用途に適用が可能である。
【0108】
ポリアミック酸は、酸二無水物とジアミンの反応により得られるが、最終的に得られるポリイミドに優れた耐熱性及び寸法安定性を付与する点から、前記化学式(4)において、R31又はR32が芳香族化合物であることが好ましく、R31及びR32が芳香族化合物であることがより好ましい。またこのとき、前記化学式(4)のR31において、当該R31に結合している4つの基((−CO−)2(−COOH)2)は同一の芳香環に結合していても良く、異なる芳香環に結合していても良い。同様に、前記化学式(4)のR32において、当該R32に結合している2つの基((−NH−)2)は同一の芳香環に結合していても良く、異なる芳香環に結合していても良い。
【0109】
また、前記化学式(4)で表されるポリアミック酸は、単一の繰り返し単位からなるものでも、2種以上の繰り返し単位から成るものでもよい。
【0110】
本発明のポリイミド前駆体を製造する方法としては、従来公知の手法を適用することができる。例えば、(1)酸二無水物とジアミンから前駆体であるポリアミド酸を合成する手法。(2)酸二無水物に1価のアルコールやアミノ化合物、エポキシ化合物等を反応させ合成した、エステル酸やアミド酸モノマーのカルボン酸に、ジアミノ化合物やその誘導体を反応させてポリイミド前駆体を合成する手法などが挙げられるがこれに限定されない。
【0111】
本発明のポリイミド前駆体を得るための反応に適用可能な酸二無水物としては、例えば、エチレンテトラカルボン酸二無水物、ブタンテトラカルボン酸二無水物、シクロブタンテトラカルボン酸二無水物、メチルシクロブタンテトラカルボン酸二無水物、シクロペンタンテトラカルボン酸二無水物などの脂肪族テトラカルボン酸二無水物;ピロメリット酸二無水物、3,3’,4,4’−ベンゾフェノンテトラカルボン酸二無水物、2,2’,3,3’−ベンゾフェノンテトラカルボン酸二無水物、2,3’,3,4’−ベンゾフェノンテトラカルボン酸二無水物、3,3’,4,4’−ビフェニルテトラカルボン酸二無水物、2,2’,3,3’−ビフェニルテトラカルボン酸二無水物、2,3’,3,4’−ビフェニルテトラカルボン酸二無水物、2,2’,6,6’−ビフェニルテトラカルボン酸二無水物、2,2−ビス(3,4−ジカルボキシフェニル)プロパン二無水物、2,2−ビス(2,3−ジカルボキシフェニル)プロパン二無水物、ビス(3,4−ジカルボキシフェニル)エーテル二無水物、ビス(3,4−ジカルボキシフェニル)スルホン二無水物、1,1−ビス(2,3−ジカルボキシフェニル)エタン二無水物、ビス(2,3−ジカルボキシフェニル)メタン二無水物、ビス(3,4−ジカルボキシフェニル)メタン二無水物、2,2−ビス(3,4−ジカルボキシフェニル)−1,1,1,3,3,3−ヘキサフルオロプロパン二無水物、2,2−ビス(2,3−ジカルボキシフェニル)−1,1,1,3,3,3−ヘキサフルオロプロパン二無水物、1,3−ビス〔(3,4−ジカルボキシ)ベンゾイル〕ベンゼン二無水物、1,4−ビス〔(3,4−ジカルボキシ)ベンゾイル〕ベンゼン二無水物、2,2−ビス{4−〔4−(1,2−ジカルボキシ)フェノキシ〕フェニル}プロパン二無水物、
【0112】
2,2−ビス{4−〔3−(1,2−ジカルボキシ)フェノキシ〕フェニル}プロパン二無水物、ビス{4−〔4−(1,2−ジカルボキシ)フェノキシ〕フェニル}ケトン二無水物、ビス{4−〔3−(1,2−ジカルボキシ)フェノキシ〕フェニル}ケトン二無水物、4,4’−ビス〔4−(1,2−ジカルボキシ)フェノキシ〕ビフェニル二無水物、4,4’−ビス〔3−(1,2−ジカルボキシ)フェノキシ〕ビフェニル二無水物、ビス{4−〔4−(1,2−ジカルボキシ)フェノキシ〕フェニル}ケトン二無水物、ビス{4−〔3−(1,2−ジカルボキシ)フェノキシ〕フェニル}ケトン二無水物、ビス{4−〔4−(1,2−ジカルボキシ)フェノキシ〕フェニル}スルホン二無水物、ビス{4−〔3−(1,2−ジカルボキシ)フェノキシ〕フェニル}スルホン二無水物、ビス{4−〔4−(1,2−ジカルボキシ)フェノキシ〕フェニル}スルフィド二無水物、ビス{4−〔3−(1,2−ジカルボキシ)フェノキシ〕フェニル}スルフィド二無水物、2,2−ビス{4−〔4−(1,2−ジカルボキシ)フェノキシ〕フェニル}−1,1,1,3,3,3−ヘキサフルオロプロパン二無水物、2,2−ビス{4−〔3−(1,2−ジカルボキシ)フェノキシ〕フェニル}−1,1,1,3,3,3−ヘキサフルオロプロパン二無水物、2,3,6,7−ナフタレンテトラカルボン酸二無水物、1,1,1,3,3,3−ヘキサフルオロ−2,2−ビス(2,3−又は3,4−ジカルボキシフェニル)プロパン二無水物、1,4,5,8−ナフタレンテトラカルボン酸二無水物、1,2,5,6−ナフタレンテトラカルボン酸二無水物、1,2,3,4−ベンゼンテトラカルボン酸二無水物、3,4,9,10−ぺリレンテトラカルボン酸二無水物、2,3,6,7−アントラセンテトラカルボン酸二無水物、1,2,7,8−フェナントレンテトラカルボン酸二無水物、ピリジンテトラカルボン酸二無水物、スルホニルジフタル酸無水物、m−ターフェニル−3,3’,4,4’−テトラカルボン酸二無水物、p−ターフェニル−3,3’,4,4’−テトラカルボン酸二無水物等の芳香族テトラカルボン酸二無水物等が挙げられる。これらは単独あるいは2種以上混合して用いられる。そして、特に好ましく用いられるテトラカルボン酸二無水物としてピロメリット酸二無水物、3,3’,4,4’−ベンゾフェノンテトラカルボン酸二無水物、3,3’,4,4’−ビフェニルテトラカルボン酸二無水物、2,2’,6,6’−ビフェニルテトラカルボン酸二無水物、ビス(3,4−ジカルボキシフェニル)エーテル二無水物、2,2−ビス(3,4−ジカルボキシフェニル)−1,1,1,3,3,3−ヘキサフルオロプロパン二無水物が挙げられる。
【0113】
併用する酸二無水物としてフッ素が導入された酸二無水物や、脂環骨格を有する酸二無水物を用いると、透明性をそれほど損なわずに溶解性や熱膨張率等の物性を調整することが可能である。また、ピロメリット酸無水物、3,3’,4,4’−ビフェニルテトラカルボン酸二無水物、1,4,5,8−ナフタレンテトラカルボン酸二無水物などの剛直な酸二無水物を用いると、最終的に得られるポリイミドの線熱膨張係数が小さくなるが、透明性の向上を阻害する傾向があるので、共重合割合に注意しながら併用してもよい。
【0114】
一方、アミン成分も、1種類のジアミン単独で、または2種類以上のジアミンを併用して用いることができる。用いられるジアミン成分は限定されず、p−フェニレンジアミン、
m−フェニレンジアミン、o−フェニレンジアミン、3,3’−ジアミノジフェニルエーテル、3,4’−ジアミノジフェニルエーテル、4,4’−ジアミノジフェニルエーテル、3,3’−ジアミノジフェニルスルフィド、3,4’−ジアミノジフェニルスルフィド、4,4’−ジアミノジフェニルスルフィド、3,3’−ジアミノジフェニルスルホン、3,4’−ジアミノジフェニルスルホン、4,4’−ジアミノジフェニルスルホン、3,3’−ジアミノベンゾフェノン、4,4’−ジアミノベンゾフェノン、3,4’−ジアミノベンゾフェノン、3,3’−ジアミノジフェニルメタン、4,4’−ジアミノジフェニルメタン、3,4’−ジアミノジフェニルメタン、2,2−ジ(3−アミノフェニル)プロパン、2,2−ジ(4−アミノフェニル)プロパン、2−(3−アミノフェニル)−2−(4−アミノフェニル)プロパン、2,2−ジ(3−アミノフェニル)−1,1,1,3,3,3−ヘキサフルオロプロパン、2,2−ジ(4−アミノフェニル)−1,1,1,3,3,3−ヘキサフルオロプロパン、2−(3−アミノフェニル)−2−(4−アミノフェニル)−1,1,1,3,3,3−ヘキサフルオロプロパン、1,1−ジ(3−アミノフェニル)−1−フェニルエタン、1,1−ジ(4−アミノフェニル)−1−フェニルエタン、1−(3−アミノフェニル)−1−(4−アミノフェニル)−1−フェニルエタン、1,3−ビス(3−アミノフェノキシ)ベンゼン、1,3−ビス(4−アミノフェノキシ)ベンゼン、1,4−ビス(3−アミノフェノキシ)ベンゼン、1,4−ビス(4−アミノフェノキシ)ベンゼン、1,3−ビス(3−アミノベンゾイル)ベンゼン、1,3−ビス(4−アミノベンゾイル)ベンゼン、1,4−ビス(3−アミノベンゾイル)ベンゼン、1,4−ビス(4−アミノベンゾイル)ベンゼン、1,3−ビス(3−アミノ−α,α−ジメチルベンジル)ベンゼン、1,3−ビス(4−アミノ−α,α−ジメチルベンジル)ベンゼン、1,4−ビス(3−アミノ−α,α−ジメチルベンジル)ベンゼン、1,4−ビス(4−アミノ−α,α−ジメチルベンジル)ベンゼン、1,3−ビス(3−アミノ−α,α−ジトリフルオロメチルベンジル)ベンゼン、1,3−ビス(4−アミノ−α,α−ジトリフルオロメチルベンジル)ベンゼン、1,4−ビス(3−アミノ−α,α−ジトリフルオロメチルベンジル)ベンゼン、1,4−ビス(4−アミノ−α,α−ジトリフルオロメチルベンジル)ベンゼン、2,6−ビス(3−アミノフェノキシ)ベンゾニトリル、2,6−ビス(3−アミノフェノキシ)ピリジン、4,4’−ビス(3−アミノフェノキシ)ビフェニル、4,4’−ビス(4−アミノフェノキシ)ビフェニル、ビス[4−(3−アミノフェノキシ)フェニル]ケトン、ビス[4−(4−アミノフェノキシ)フェニル]ケトン、ビス[4−(3−アミノフェノキシ)フェニル]スルフィド、ビス[4−(4−アミノフェノキシ)フェニル]スルフィド、
【0115】
ビス[4−(3−アミノフェノキシ)フェニル]スルホン、ビス[4−(4−アミノフェノキシ)フェニル]スルホン、ビス[4−(3−アミノフェノキシ)フェニル]エーテル、ビス[4−(4−アミノフェノキシ)フェニル]エーテル、2,2−ビス[4−(3−アミノフェノキシ)フェニル]プロパン、2,2−ビス[4−(4−アミノフェノキシ)フェニル]プロパン、2,2−ビス[3−(3−アミノフェノキシ)フェニル]−1,1,1,3,3,3−ヘキサフルオロプロパン、2,2−ビス[4−(4−アミノフェノキシ)フェニル]−1,1,1,3,3,3−ヘキサフルオロプロパン、1,3−ビス[4−(3−アミノフェノキシ)ベンゾイル]ベンゼン、1,3−ビス[4−(4−アミノフェノキシ)ベンゾイル]ベンゼン、1,4−ビス[4−(3−アミノフェノキシ)ベンゾイル]ベンゼン、1,4−ビス[4−(4−アミノフェノキシ)ベンゾイル]ベンゼン、1,3−ビス[4−(3−アミノフェノキシ)−α,α−ジメチルベンジル]ベンゼン、1,3−ビス[4−(4−アミノフェノキシ)−α,α−ジメチルベンジル]ベンゼン、1,4−ビス[4−(3−アミノフェノキシ)−α,α−ジメチルベンジル]ベンゼン、1,4−ビス[4−(4−アミノフェノキシ)−α,α−ジメチルベンジル]ベンゼン、4,4’−ビス[4−(4−アミノフェノキシ)ベンゾイル]ジフェニルエーテル、4,4’−ビス[4−(4−アミノ−α,α−ジメチルベンジル)フェノキシ]ベンゾフェノン、4,4’−ビス[4−(4−アミノ−α,α−ジメチルベンジル)フェノキシ]ジフェニルスルホン、4,4’−ビス[4−(4−アミノフェノキシ)フェノキシ]ジフェニルスルホン、3,3’−ジアミノ−4,4’−ジフェノキシベンゾフェノン、3,3’−ジアミノ−4,4’−ジビフェノキシベンゾフェノン、3,3’−ジアミノ−4−フェノキシベンゾフェノン、3,3’−ジアミノ−4−ビフェノキシベンゾフェノン、6,6’−ビス(3−アミノフェノキシ)−3,3,3’,3’−テトラメチル−1,1’−スピロビインダン、6,6’−ビス(4−アミノフェノキシ)−3,3,3’,3’−テトラメチル−1,1’−スピロビインダン等の芳香族アミン;
【0116】
1,3−ビス(3−アミノプロピル)テトラメチルジシロキサン、1,3−ビス(4−アミノブチル)テトラメチルジシロキサン、α,ω−ビス(3−アミノプロピル)ポリジメチルシロキサン、α,ω−ビス(3−アミノブチル)ポリジメチルシロキサン、ビス(アミノメチル)エーテル、ビス(2−アミノエチル)エーテル、ビス(3−アミノプロピル)エーテル、ビス(2−アミノメトキシ)エチル]エーテル、ビス[2−(2−アミノエトキシ)エチル]エーテル、ビス[2−(3−アミノプロトキシ)エチル]エーテル、1,2−ビス(アミノメトキシ)エタン、1,2−ビス(2−アミノエトキシ)エタン、1,2−ビス[2−(アミノメトキシ)エトキシ]エタン、1,2−ビス[2−(2−アミノエトキシ)エトキシ]エタン、エチレングリコールビス(3−アミノプロピル)エーテル、ジエチレングリコールビス(3−アミノプロピル)エーテル、トリエチレングリコールビス(3−アミノプロピル)エーテル、エチレンジアミン、1,3−ジアミノプロパン、1,4−ジアミノブタン、1,5−ジアミノペンタン、1,6−ジアミノヘキサン、1,7−ジアミノヘプタン、1,8−ジアミノオクタン、1,9−ジアミノノナン、1,10−ジアミノデカン、1,11−ジアミノウンデカン、1,12−ジアミノドデカン等の脂肪族アミン;
【0117】
1,2−ジアミノシクロヘキサン、1,3−ジアミノシクロヘキサン、1,4−ジアミノシクロヘキサン、1,2−ジ(2−アミノエチル)シクロヘキサン、1,3−ジ(2−アミノエチル)シクロヘキサン、1,4−ジ(2−アミノエチル)シクロヘキサン、ビス(4−アミノシクロへキシル)メタン、2,6−ビス(アミノメチル)ビシクロ[2.2.1]ヘプタン、2,5−ビス(アミノメチル)ビシクロ[2.2.1]ヘプタン等の脂環式ジアミンが挙げられる。グアナミン類としては、アセトグアナミン、ベンゾグアナミンなどを挙げることができ、また、上記ジアミンの芳香環上水素原子の一部若しくは全てをフルオロ基、メチル基、メトキシ基、トリフルオロメチル基、又はトリフルオロメトキシ基から選ばれた置換基で置換したジアミンも使用することができる。
【0118】
さらに目的に応じ、架橋点となるエチニル基、ベンゾシクロブテン−4’−イル基、ビニル基、アリル基、シアノ基、イソシアナト基、及びイソプロペニル基のいずれか1種又は2種以上を、上記ジアミンの芳香環上水素原子の一部若しくは全てに置換基として導入しても使用することができる。
【0119】
ジアミンは、目的の物性によって選択することができ、p−フェニレンジアミンなどの剛直なジアミンを用いれば、最終的に得られるポリイミドは低膨張率となる。剛直なジアミンとしては、同一の芳香環に2つアミノ基が結合しているジアミンとして、p−フェニレンジアミン、m−フェニレンジアミン、1,4−ジアミノナフタレン、1,5−ジアミノナフタレン、2、6−ジアミノナフタレン、2,7−ジアミノナフタレン、1,4―ジアミノアントラセンなどが挙げられる。
【0120】
さらに、2つ以上の芳香族環が単結合により結合し、2つ以上のアミノ基がそれぞれ別々の芳香族環上に直接又は置換基の一部として結合しているジアミンが挙げられ、例えば、下記式(5)により表されるものがある。具体例としては、ベンジジン等が挙げられる。
【0121】
【化16】
(化学式(5)中、aは1以上の自然数、アミノ基はベンゼン環同士の結合に対して、メタ位または、パラ位に結合する。)
【0122】
さらに、上記式(5)において、他のベンゼン環との結合に関与せず、ベンゼン環上のアミノ基が置換していない位置に置換基を有するジアミンも用いることができる。これら置換基は、有機基であるがそれらは互いに結合していてもよい。
具体例としては、2,2’−ジメチル−4,4’−ジアミノビフェニル、2,2’−ジトリフルオロメチル−4,4’−ジアミノビフェニル、3,3’−ジクロロ−4,4’−ジアミノビフェニル、3,3’−ジメトキシ−4,4’−ジアミノビフェニル、3,3’−ジメチル−4,4’−ジアミノビフェニル等が挙げられる。
【0123】
最終的に得られるポリイミドを光導波路、光回路部品として用いる場合には、芳香環の置換基としてフッ素を導入すると1μm以下の波長の電磁波に対しての透過率を向上させることができる。
【0124】
一方、ジアミンとして、1,3−ビス(3−アミノプロピル)テトラメチルジシロキサンなどのシロキサン骨格を有するジアミンを用いると、最終的に得られるポリイミドの弾性率が低下し、ガラス転移温度を低下させることができる。
ここで、選択されるジアミンは耐熱性の観点より芳香族ジアミンが好ましいが、目的の物性に応じてジアミンの全体の60モル%、好ましくは40モル%を超えない範囲で、脂肪族ジアミンやシロキサン系ジアミン等の芳香族以外のジアミンを用いても良い。
【0125】
一方、ポリイミド前駆体を合成するには、例えば、アミン成分として4,4’−ジアミノジフェニルエーテルをN−メチルピロリドンなどの有機極性溶媒に溶解させた溶液を冷却しながら、そこへ等モルの3,3’,4,4’−ビフェニルテトラカルボン酸二無水物を徐々に加え撹拌し、ポリイミド前駆体溶液を得ることができる。
このようにして合成されるポリイミド前駆体は、最終的に得られるポリイミドに耐熱性及び寸法安定性を求める場合には、芳香族酸成分及び/又は芳香族アミン成分の共重合割合ができるだけ大きいことが好ましい。具体的には、イミド構造の繰り返し単位を構成する酸成分に占める芳香族酸成分の割合が50モル%以上、特に70モル%以上であることが好ましく、イミド構造の繰り返し単位を構成するアミン成分に占める芳香族アミン成分の割合が40モル%以上、特に60モル%以上であることが好ましく、全芳香族ポリイミドであることが特に好ましい。
【0126】
本発明に用いられるポリイミド前駆体においては、感光性ポリイミド樹脂組成物とした際の感度を高め、マスクパターンを正確に再現するパターン形状を得るために、1μmの膜厚のときに、露光波長に対して少なくとも5%以上の透過率を示すことが好ましく、15%以上の透過率を示すことが更に好ましい。
露光波長に対してポリイミド前駆体の透過率が高いということは、それだけ、電磁波のロスが少ないということであり、高感度の感光性ポリイミド樹脂組成物を得ることができる。
【0127】
また、一般的な露光光源である高圧水銀灯を用いて露光を行う場合には、少なくとも436nm、405nm、365nmの波長の電磁波のうち1つの波長の電磁波に対する透過率が、厚み1μmのフィルムに成膜した時で好ましくは5%以上、更に好ましくは15%、特に好ましくは50%以上である。
【0128】
本発明に用いられるポリイミド前駆体の重量平均分子量は、その用途にもよるが、3,000〜1,000,000の範囲であることが好ましく、5,000〜500,000の範囲であることがさらに好ましく、10,000〜500,000の範囲であることがさらに好ましい。重量平均分子量が3,000未満であると、塗膜又はフィルムとした場合に十分な強度が得られにくい。また、加熱処理等を施しポリイミド等の高分子とした際の膜の強度も低くなる。一方、重量平均分子量が1,000,000を超えると粘度が上昇し、溶解性も低下しやすく、表面が平滑で膜厚が均一な塗膜又はフィルムが得られにくい。
【0129】
ここで用いている分子量とは、ゲル浸透クロマトグラフィー(GPC)によるポリスチレン換算の値のことをいい、ポリイミド前駆体そのものの分子量でも良いし、無水酢酸等で化学的イミド化処理を行った後のものでも良い。
【0130】
なお、ポリイミド前駆体合成時における溶媒は、極性溶媒が望ましく、代表的なものとして、N−メチル−2−ピロリドン、N−アセチル−2−ピロリドン、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N,N−ジエチルホルムアミド、N,N−ジエチルアセトアミド、N,N−ジメチルメトキシアセトアミド、ジメチルスルホキシド、ヘキサメチルフォスホアミド、ピリジン、ジメチルスルホン、テトラメチレンスルホン、ジメチルテトラメチレンスルホン、ジエチレングリコールジメチルエーテル、シクロペンタノン、γ−ブチロラクトン、α−アセチル−γ−ブチロラクトン等があり、これらの溶媒は単独であるいは2種類以上を組み合わせて用いられる。この他にも溶媒として組合せて用いられるものとしてベンゼン、ベンゾニトリル、1,4−ジオキサン、テトラヒドロフラン、ブチロラクトン、キシレン、トルエン、シクロヘキサノン等の非極性溶媒が挙げられ、これらの溶媒は、原料の分散媒、反応調節剤、あるいは生成物からの溶媒の揮散調節剤、皮膜平滑剤などとして使用される。
【0131】
(光塩基発生剤)
本発明に係るネガ型感光性ポリイミド樹脂組成物に用いられる光塩基発生剤は、前記本発明に係る光塩基発生剤と同様のものとすることができる。中でも、光塩基発生剤が発生する塩基が、150〜200℃のいずれかの温度において、当該塩基100重量部に対して、前記ポリイミド前駆体を10重量部以上溶解する塩基であることが好ましい。発生する塩基が前記ポリイミド前駆体を10重量部以上溶解する場合、触媒である塩基とポリイミド前駆体の親和性が向上し、少量の塩基であっても前記ポリイミド前駆体を効率的にイミド化することができるからである。
【0132】
本発明に係る光塩基発生剤は、少量の添加で硬化が可能となり、感光性ポリイミド樹脂組成物の固形分全体に対し、通常、0.1〜30重量%、好ましくは0.5〜20重量%の範囲内で含有させることが好ましい。なお、本発明において固形分は、上述した溶媒以外のもの全てであり、室温で液体のモノマー等も含まれる。
【0133】
<その他の成分>
本発明に係る感光性ポリイミド樹脂組成物は、前記ポリイミド前駆体と、前記光塩基発生剤と、溶媒の単純な混合物であってもよいが、さらに、光又は熱硬化性成分、ポリイミド前駆体以外の非重合性バインダー樹脂、その他の成分を配合して、感光性ポリイミド樹脂組成物を調製してもよい。また、本発明の光塩基発生剤の補助的な役割として、光によって酸又は塩基を発生させる他の感光性成分を加えても良い。また、光塩基発生剤から発生した少量の塩基の作用によって、分解や転位反応して塩基を発生させる塩基増殖剤を併用しても良いし、増感剤を加えてもよい。
【0134】
感光性ポリイミド樹脂組成物を溶解、分散又は希釈する溶剤としては、各種の汎用溶剤を用いることが出来る。また、前駆体としてポリアミド酸を用いる場合には、ポリアミド酸の合成反応により得られた溶液をそのまま用い、そこに必要に応じて他の成分を混合しても良い。
【0135】
使用可能な汎用溶剤としては、例えば、ジエチルエーテル、テトラヒドロフラン、ジオキサン、エチレングリコールジメチルエーテル、エチレングリコールジエチルエーテル、プロピレングリコールジメチルエーテル、プロピレングリコールジエチルエーテル、ジエチレングリコールジメチルエーテル等のエーテル類;エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル、プロピレングリコールモノメチルエーテル、プロピレングリコールモノエチルエーテル、ジエチレングリコールモノメチルエーテル、ジエチレングリコールモノエチルエーテル等のグリコールモノエーテル類(いわゆるセロソルブ類);メチルエチルケトン、アセトン、メチルイソブチルケトン、シクロペンタノン、シクロヘキサノンなどのケトン類;酢酸エチル、酢酸ブチル、酢酸n−プロピル、酢酸i−プロピル、酢酸n−ブチル、酢酸i−ブチル、前記グリコールモノエーテル類の酢酸エステル(例えば、メチルセロソルブアセテート、エチルセロソルブアセテート)、プロピレングリコールモノメチルエーテルアセテート、プロピレングリコールモノエチルエーテルアセテート、蓚酸ジメチル、乳酸メチル、乳酸エチル等のエステル類;エタノール、プロパノール、ブタノール、ヘキサノール、シクロヘキサノール、エチレングリコール、ジエチレングリコール、グリセリン等のアルコール類;塩化メチレン、1,1−ジクロロエタン、1,2−ジクロロエチレン、1−クロロプロパン、1−クロロブタン、1−クロロペンタン、クロロベンゼン、ブロムベンゼン、o−ジクロロベンゼン、m−ジクロロベンゼン等のハロゲン化炭化水素類;N,N−ジメチルホルムアミド、N,N−ジエチルホルムアミド、N,N−ジメチルアセトアミド、N,N−ジエチルアセトアミド、N,N−ジメチルメトキシアセトアミド等のアミド類;N−メチル−2−ピロリドン、N−アセチル−2−ピロリドンなどのピロリドン類;γ−ブチロラクトン、α−アセチル−γ−ブチロラクトン等のラクトン類;ジメチルスルホキシドなどのスルホキシド類、ジメチルスルホン、テトラメチレンスルホン、ジメチルテトラメチレンスルホンなどのスルホン類、ヘキサメチルフォスホアミド等のリン酸アミド類、その他の有機極性溶媒類等が挙げられ、更には、ベンゼン、トルエン、キシレン、ピリジン等の芳香族炭化水素類、及び、その他の有機非極性溶媒類等も挙げられる。これらの溶媒は単独若しくは組み合わせて用いられる。
【0136】
中でも、プロピレングリコールモノメチルエーテル、メチルエチルケトン、シクロペンタノン、シクロヘキサノン、酢酸エチル、プロピレングリコールモノメチルエーテルアセテート、N,N−ジメチルアセトアミド、N−メチル−2−ピロリドン、γ−ブチロラクトン等の極性溶媒、トルエン等の芳香族炭化水素類、及び、これらの溶媒からなる混合溶媒が好適なものとして挙げられる。
【0137】
光硬化性成分としては、エチレン性不飽和結合を1つ又は2つ以上有する化合物を用いることができ、例えば、アミド系モノマー、(メタ)アクリレートモノマー、ウレタン(メタ)アクリレートオリゴマー、ポリエステル(メタ)アクリレートオリゴマー、エポキシ(メタ)アクリレート、及びヒドロキシル基含有(メタ)アクリレート、スチレン等の芳香族ビニル化合物を挙げることができる。また、ポリイミド前駆体が、ポリアミック酸等のカルボン酸成分を構造内に有する場合には、3級アミノ基を有するエチレン性不飽和結合含有化合物を用いると、ポリイミド前駆体のカルボン酸とイオン結合を形成し、感光性ポリイミド樹脂組成物としたときの露光部、未露光部の溶解速度のコントラストが大きくなる。
【0138】
このようなエチレン性不飽和結合を有する光硬化性化合物を用いる場合には、さらに光ラジカル発生剤を添加してもよい。光ラジカル発生剤としては、例えば、ベンゾイン、ベンゾインメチルエーテル、ベンゾインエチルエーテル及びベンゾインイソプロピルエーテル等のベンゾインとそのアルキルエーテル;アセトフェノン、2,2−ジメトキシ−2−フェニルアセトフェノン、2,2−ジエトキシ−2−フェニルアセトフェノン、1,1−ジクロロアセトフェノン、1−ヒドロキシアセトフェノン、1−ヒドロキシシクロヘキシルフェニルケトン及び2−メチル−1−[4−(メチルチオ)フェニル]−2−モルフォリノ−プロパン−1−オン等のアセトフェノン;2−メチルアントラキノン、2−エチルアントラキノン、2−ターシャリ−ブチルアントラキノン、1−クロロアントラキノン及び2−アミルアントラキノン等のアントラキノン;2,4−ジメチルチオキサントン、2,4−ジエチルチオキサントン、2−クロロチオキサントン及び2,4−ジイソピルチオキサントン等のチオキサントン;アセトフェノンジメチルケタール及びベンジルジメチルケタール等のケタール;2,4,6−トリメチルベンゾイルジフェニルホスフィンオキシド等のモノアシルホスフィンオキシドあるいはビスアシルホスフィンオキシド類;ベンゾフェノン等のベンゾフェノン類;並びにキサントン類等が挙げられる。
【0139】
光によって酸を発生させる化合物としては、1,2−ベンゾキノンジアジドあるいは1,2−ナフトキノンジアジド構造を有する感光性ジアゾキノン化合物があり、米国特許明細書第2,772,972号、第2,797,213号、第3,669,658号に提案されている。また、トリアジンやその誘導体、スルホン酸オキシムエステル化合物、スルホン酸ヨードニウム塩、スルホン酸スルフォニウム塩等、公知の光酸発生剤を用いることができる。光によって塩基を発生させる化合物としては、例えば2,6−ジメチル−3,5−ジシアノ−4−(2’−ニトロフェニル)−1,4−ジヒドロピリジン、2,6−ジメチル−3,5−ジアセチル−4−(2’−ニトロフェニル)−1,4−ジヒドロピリジン、2,6−ジメチル−3,5−ジアセチル−4−(2’,4’−ジニトロフェニル)−1,4−ジヒドロピリジンなどが例示できる。
【0140】
塩基増殖剤としては、例えば、9−フルオレニルメチルカルバメート結合を有する化合物、1,1−ジメチル−2−シアノメチルカルバメート結合((CN)CH2C(CH3)2OC(O)NR2)を有する化合物、パラニトロベンジルカルバメート結合を有する化合物、2,4−ジクロロベンジルカルバメート結合を有する化合物、その他にも特開2000−330270号公報の段落0010〜段落0032に記載されているウレタン系化合物や、特開2008−250111号公報の段落0033〜段落0060に記載されているウレタン系化合物等が挙げられる。
【0141】
高分子を透過する波長の電磁波のエネルギーを光塩基発生剤が充分利用できる様にし、感度を向上させたい場合に、増感剤の添加が効果を発揮する場合がある。
特に、ポリイミド前駆体の吸収が360nm以上の波長にもある場合には、増感剤の添加による効果が大きい。増感剤と呼ばれる化合物の具体例としては、チオキサントン及び、ジエチルチオキサントンなどのその誘導体、クマリン系及び、その誘導体、ケトクマリン及び、その誘導体、ケトビスクマリン、及びその誘導体、シクロペンタノン及び、その誘導体、シクロヘキサノン及び、その誘導体、チオピリリウム塩及び、その誘導体、チオキサンテン系、キサンテン系及び、その誘導体などが挙げられる。
【0142】
クマリン、ケトクマリン及び、その誘導体の具体例としては、3,3’−カルボニルビスクマリン、3,3’−カルボニルビス(5,7−ジメトキシクマリン)、3,3’−カルボニルビス(7−アセトキシクマリン)等が挙げられる。チオキサントン及び、その誘導体の具体例としては、ジエチルチオキサントン、イソプロピルチオキサントンなどが挙げられる。さらに他にはベンゾフェノン、アセトフェノン、フェナントレン、2−ニトロフルオレン、5−ニトロアセナフテン、ベンゾキノン、2−エチルアントラキノン、2−ターシャリーブチルアントラキノン、1,2−ベンズアンスラキノン、1,2−ナフトキノン、などが挙げられる。
これらは、光塩基発生剤との組み合わせによって、特に優れた効果を発揮する為、塩基発生剤の構造によって最適な増感作用を示す増感剤が適宜選択される。
【0143】
また、本発明に係る樹脂組成物に加工特性や各種機能性を付与するために、その他に様々な有機又は無機の低分子又は高分子化合物を配合してもよい。例えば、染料、界面活性剤、レベリング剤、可塑剤、微粒子等を用いることができる。微粒子には、ポリスチレン、ポリテトラフルオロエチレン等の有機微粒子、コロイダルシリカ、カーボン、層状珪酸塩等の無機微粒子等が含まれ、それらは多孔質や中空構造であってもよい。また、その機能又は形態としては顔料、フィラー、繊維等がある。
【0144】
また、その他の溶剤以外の任意成分の配合割合は、感光性ポリイミド樹脂組成物の固形分全体に対し、0.1重量%〜95重量%の範囲が好ましい。0.1重量%未満だと、添加物を添加した効果が発揮されにくく、95重量%を超えると、最終的に得られる樹脂硬化物の特性が最終生成物に反映されにくい。
【0145】
本発明に係る感光性ポリイミド樹脂組成物は、印刷インキ、塗料、シール剤、接着剤、電子材料、光回路部品、成形材料、レジスト材料、建築材料、光造形、光学部材等、樹脂材料が用いられる公知の全ての分野、製品に利用できる。塗料、シール剤、接着剤のように、全面露光して用いる用途にも、永久膜や剥離膜などパターンを形成する用途にも、いずれにも好適に用いることができる。
【0146】
本発明に係る感光性ポリイミド樹脂組成物は、耐熱性、寸法安定性、絶縁性等の特性が有効とされる広範な分野、製品、例えば、塗料、印刷インキ、シール剤、又は接着剤、或いは、表示装置、半導体装置、電子部品、微小電気機械システム(Micro Electro Mechanical System(MEMS))、光造形物、光学部材又は建築材料の形成材料として好適に用いられる。例えば具体的には、電子部品の形成材料としては、封止材料、層形成材料として、プリント配線基板、層間絶縁膜、配線被覆膜等に用いることができる。また、表示装置の形成材料としては、層形成材料や画像形成材料として、カラーフィルター、フレキシブルディスプレイ用フィルム、レジスト材料、配向膜等に用いることができる。また、半導体装置の形成材料としては、レジスト材料、バッファーコート膜のような層形成材料等に用いることができる。また、光学部品の形成材料としては、光学材料や層形成材料として、ホログラム、光導波路、光回路、光回路部品、反射防止膜等に用いることができる。また、建築材料としては、塗料、コーティング剤等に用いることができる。また、光造形物の材料としても用いることができる。
【0147】
上記の様な特徴を有することから、本発明に係る感光性ポリイミド樹脂組成物は、パターン形成用材料としても用いることが可能である。特に、感光性ポリイミド樹脂組成物をパターン形成用材料(レジスト)として用いた場合、それによって形成されたパターンは、ポリイミドからなる永久膜として耐熱性や絶縁性を付与する成分として機能し、例えば、カラーフィルター、フレキシブルディスプレー用フィルム、電子部品、半導体装置、層間絶縁膜、配線被覆膜、光回路、光回路部品、反射防止膜、その他の光学部材又は電子部材を形成するのに適している。
【0148】
また、本発明においては、本発明に係るネガ型感光性ポリイミド樹脂組成物又はその熱硬化物により少なくとも一部分が形成されている、印刷物、塗料、シール剤、接着剤、表示装置、半導体装置、電子部品、微小電気機械システム、光造形物、光学部材又は建築材料いずれかの物品が提供される。
【0149】
<レリーフパターンの製造方法>
本発明に係るレリーフパターンの製造方法は、前記本発明に係るネガ型感光性ポリイミド樹脂組成物からなる塗膜又は成形体を形成し、当該塗膜又は成形体を、所定パターン状に電磁波を照射し、照射後又は照射と同時に加熱し、前記照射部位の溶解性を変化させた後、現像することを特徴とする。
【0150】
上記レリーフパターンの製造方法においては、ポリイミド前駆体と、本発明に係る光塩基発生剤とを組み合わせて用いることにより、感光性ポリイミド樹脂組成物からなる塗膜又は成形体の表面を現像液から保護するためのレジスト膜を用いずに、現像を行うパターン形成が可能である。
【0151】
本発明に係る感光性ポリイミド樹脂組成物を何らかの支持体上に塗布するなどして塗膜を形成したり、適した成型方法で成形体を形成し、当該塗膜又は成形体を、所定のパターン状に電磁波を照射し、照射後又は照射と同時に加熱することにより、露光部においてのみ、本発明に係る光塩基発生剤が塩基を発生する。塩基は、露光部のポリイミド前駆体のイミド化を促進する触媒として作用する。本発明の光塩基発生剤を用いることにより、前記加熱条件を従来よりも緩和することができ、感光性ポリイミド樹脂組成物の未露光部のイミド化を従来よりも抑制できるため、現像時間を短縮でき、アルコール等を添加しない塩基性水溶液を現像液として用いることが可能となったり、露光部と未露光部の溶解性コントラストを高くすることができる。
【0152】
ポリイミド前駆体は、塩基の触媒作用によって熱硬化温度が低下する。このような場合、先ず、本発明に係る感光性ポリイミド樹脂組成物の塗膜又は成形体上のパターンを残したい部分を露光する。露光後又は露光と同時に加熱すると、露光部には、塩基が発生し、その部分の熱硬化温度が選択的に低下する。露光後又は露光と同時に、露光部は熱硬化するが未露光部は熱硬化しない処理温度で加熱し、露光部のみ硬化させる。塩基を発生させる加熱工程と、露光部のみ硬化させる反応を行うための加熱工程(露光後ベイク)は、同一の工程としても良いし、別の工程にしても良い。
次に、所定の現像液(有機溶剤や塩基性水溶液等)で未露光部を溶解して熱硬化物からなるパターンを形成する。このパターンを、更に必要に応じ加熱して熱硬化を完結させる。以上の工程によって、通常ネガ型の所望の2次元樹脂パターン(一般的な平面パターン)又は3次元樹脂パターン(立体的に成形された形状)が得られる。
【0153】
本発明の感光性ポリイミド樹脂組成物は、プロピレングリコールモノメチルエーテル、メチルエチルケトン、シクロペンタノン、シクロヘキサノン、酢酸エチル、プロピレングリコールモノメチルエーテルアセテート、N,N−ジメチルアセトアミド、N−メチル−2−ピロリドン、γ−ブチロラクトン等の極性溶媒、トルエン等の芳香族炭化水素類、及び、これらの溶媒からなる混合溶媒に溶解後、浸漬法、スプレー法、フレキソ印刷法、グラビア印刷法、スクリーン印刷法、スピンコート法、ディスペンス法などによって、シリコンウエハ、金属基板、セラミック基板、樹脂フィルムなどの基材表面に塗布し、加熱して溶剤の大部分を除くことにより、基材表面に粘着性のない塗膜を与えることができる。塗膜の厚みには特に制限はないが、0.5〜50μmであることが好ましく、感度および現像速度面から1.0〜20μmであることがより望ましい。塗布した塗膜の乾燥条件としては、例えば、80〜100℃、1分〜20分が挙げられる。
【0154】
この塗膜に、所定のパターンを有するマスクを通して、電磁波を照射しパターン状に露光後を行い、加熱後、膜の未露光部分を、適切な現像液で現像して除去することにより、所望のパターン化された膜を得ることができる。
【0155】
露光工程に用いられる露光方法や露光装置は特に限定されることなく、密着露光でも間接露光でも良く、g線ステッパ、i線ステッパ、超高圧水銀灯を用いるコンタクト/プロキシミティ露光機、ミラープロジェクション露光機、又はその他の紫外線、可視光線、X線、電子線などを照射可能な投影機や線源を使用することができる。
【0156】
例えば上記式(1−1)で表される光塩基発生剤など保護基を有する場合であって、電磁波照射のみによって保護基の脱保護を行う場合、塩基を発生するために照射する電磁波によって脱保護しても良いし、脱保護のための電磁波と、塩基を発生させるための電磁波とで波長を変更しても良い。例えば、長波長の電磁波を照射して脱保護を行い、その後短波長の電磁波で塩基を発生させるための異性化を行うなどが挙げられる。これらの場合の電磁波の照射量は、電磁波によっても異なり、特に限定されず、適宜調整される。
【0157】
露光前又は露光後又は露光と同時に加熱し、保護基を脱保護させて塩基を発生させるための加熱温度としては、組み合わせるポリイミド前駆体や目的により適宜選択され、特に限定されない。感光性ポリイミド樹脂組成物が置かれた環境の温度(例えば、室温)による加熱であっても良く、その場合、徐々に塩基が発生する。また、電磁波の照射時に副生される熱によっても塩基が発生するため、電磁波の照射時に副生される熱により実質的に加熱が同時に行われても良い。反応速度を高くし、効率よく塩基を発生させる点から、加熱温度としては、30℃以上であることが好ましく、更に好ましくは60℃以上、より更に好ましくは100℃以上、特に好ましくは120℃以上である。しかしながら、組み合わせて用いられるポリイミド前駆体によっては、例えば60℃以上の加熱で未露光部についても硬化するものもあるので、好適な加熱温度は、上記に限定されない。また、本発明に係る光塩基発生剤の塩基発生以外の分解を防ぐために、300℃以下で加熱することが好ましく、更に200℃以下で加熱することが好ましい。
なお、例えば上記式(1−1)で表される光塩基発生剤など保護基を有する場合、露光前に加熱を行い脱保護してもよい。保護基等の種類によっては保護基等を導入することで、吸収波長が短波長化するなどして光塩基発生剤の感度が悪くなることがある。このような場合、電磁波照射前の加熱により予め保護基等を脱保護し、電磁波を照射することにより、電磁波照射時の感度を向上させることができる。
なお、保護基の脱保護条件は、組成物中で共存する成分により変化し得る。例えば、他の光酸発生剤や光塩基発生剤が含まれる場合、光照射によって発生した酸・塩基の影響で、露光後の加熱温度が変化する場合がある。
当該電磁波照射前の保護基脱保護のための加熱は、塗膜の乾燥工程であっても良いし、他の加熱工程であっても良い。この場合、加熱温度としては、脱保護が可能な温度を適宜選択すればよいが、50℃〜180℃が好ましく、時間は10秒以上60分以下が好ましい。
また、加熱を行う際には、低温で保護基の脱保護を行い、より高温で塩基を発生させるようにしても良い。
【0158】
前記式(1−1)で表される光塩基発生剤は電磁波の照射のみでも塩基を発生するが、適宜加熱することにより塩基の発生が促進される。従って、効率的に塩基を発生させるために、前記式(1−1)で表される光塩基発生剤を用いる際には、電磁波照射(露光)後又は電磁波照射と同時に加熱を行うことにより塩基を発生する。露光と加熱を交互に行ってもよい。最も効率が良い方法は、露光と同時に加熱する方法である。
【0159】
本発明に係る感光性ポリイミド樹脂組成物の塗膜は、露光部のみ硬化させる反応を行うために、露光工程と現像工程の間に、露光後ベイク(Post Exposure Bake:PEB)を行うことが好ましい。当該PEBは、電磁波の照射により発生した塩基の作用により、塩基が存在する部位と、未照射で塩基が存在しない部位とでイミド化率等の硬化反応の反応率が異なるようになる温度で行うことが好ましく、用いる光塩基発生剤の種類によって適宜選択されるものである。好ましい熱処理の温度の範囲は、通常140℃〜210℃程度であり、より好ましくは150℃〜200℃である。熱処理温度が140℃より低いと、イミド化の効率が悪く、現実的なプロセス条件で露光部、未露光部のイミド化率の差を生ずることが難しくなる。一方、熱処理温度が210℃を超えると、塩基が存在していない未露光部でもイミド化が進行する恐れがあり、露光部と未露光部の溶解性の差を生じ難い。
また、本発明の光塩基発生剤は、発生する塩基が150〜200℃の少なくとも一部の温度で液体であるため、発生する塩基が液体となる温度を適宜選択することが、特に好ましい。発生する塩基が液体である場合、樹脂組成物中を拡散しやすいか、又は、樹脂組成物中のポリイミド前駆体を溶解することができるため、少量の塩基であってもポリイミド前駆体を効率的にイミド化することができるためである。
この熱処理は、公知の方法であればどの方法でもよく、具体的に例示すると、空気、又は窒素雰囲気下の循環オーブン、又はホットプレートによる加熱等が挙げられるが、特に限定されない。
【0160】
(現像液)
現像工程に用いられる現像液としては、前記照射部位の溶解性が変化する溶剤を現像液として用いれば、特に限定されず、塩基性水溶液、有機溶剤など、用いられるポリイミド前駆体に合わせて適宜選択することが可能である。環境への負荷を低減させる点から、現像液としては、有機溶剤の含有量が少ないことが好ましく、具体的には、有機溶剤の含有量が20重量%以下の塩基性水溶液が好ましく、有機溶剤の含有量が10重量%以下の塩基性水溶液が更に好ましい。理想的には、実質的にアルコール等の有機溶剤を含まない塩基性水溶液が最も好ましい。
【0161】
塩基性水溶液としては、特に限定されないが、例えば、濃度が、0.01重量%〜10重量%、好ましくは、0.05重量%〜5重量%のテトラメチルアンモニウムヒドロキシド(TMAH)水溶液の他、ジエタノールアミン、ジエチルアミノエタノール、水酸化ナトリウム、水酸化カリウム、炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム、炭酸水素カリウム、トリエチルアミン、ジエチルアミン、メチルアミン、ジメチルアミン、酢酸ジメチルアミノエチル、ジメチルアミノエタノール、ジメチルアミノエチルメタクリレート、シクロヘキシルアミン、エチレンジアミン、ヘキサメチレンジアミン、テトラメチルアンモニウムなどの水溶液等が挙げられる。
溶質は、1種類でも2種類以上でも良く、全体の重量の50%以上、さらに好ましくは70%以上、水が含まれていれば有機溶剤等を含んでいても良い。
【0162】
また、有機溶剤としては、特に限定されないが、N−メチル−2−ピロリドン、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、ジメチルスルホキシド、γ−ブチロラクロン、ジメチルアクリルアミドなどの極性溶媒、メタノール、エタノール、イソプロパノールなどのアルコール類、酢酸エチル、プロピレングリコールモノメチルエーテルアセテートなどのエステル類、シクロペンタノン、シクロヘキサノン、イソブチルケトン、メチルイソブチルケトンなどのケトン類、その他テトラヒドロフラン、クロロホルム、アセトニトリルなどを、単独であるいは2種類以上を組み合わせて添加してもよい。現像後は水または貧溶媒にて洗浄を行う。この場合においてもエタノール、イソプロピルアルコールなどのアルコール類、乳酸エチル、プロピレングリコールモノメチルエーテルアセテートなどのエステル類などを水に加えても良い。
【0163】
現像後は必要に応じて水または貧溶媒でリンスを行い、80〜100℃で乾燥しパターンを安定なものとする。このレリーフパターンを、耐熱性のあるものとするために180〜500℃、好ましくは200〜350℃の温度で数十分から数時間加熱することによりパターン化された高耐熱性樹脂層が形成される。
【実施例】
【0164】
以下、本発明について実施例を示して具体的に説明する。これらの記載により本発明を制限するものではない。尚、実施例中、部は特に特定しない限り重量部を表す。製造された塩基発生剤の構造は1H NMRによって確認した。
1H NMR測定:日本電子(株)製、JEOL JNM−LA400WB
手動露光機:大日本科研製、MA−1100
吸光度測定:(株)島津製作所製、紫外可視分光光度計UV−2550
塗膜の加熱:アズワン(株)製、HOT PLATE EC−1200(本実施例中、ホットプレートと記載することがある)
【0165】
(合成例1:ポリイミド前駆体1の合成)
4,4’−ジアミノジフェニルエーテル(ODA)20.0g(100mmol)を500mlのセパラブルフラスコに投入し、181gの脱水されたN−メチル−2−ピロリドン(NMP)に溶解させ窒素気流下、オイルバスによって液温が50℃になるように熱電対でモニターし加熱しながら撹拌した。それらが完全に溶解したことを確認した後、そこへ、少しずつ30分かけて3,3’、4,4’−ビフェニルテトラカルボン酸二無水物(BPDA)27.4g(93.1mmol)を添加し、添加終了後、50℃で5時間撹拌した。その後、フタル酸無水物を0.900g(6.08mmol)添加した。その後室温まで冷却し、下記式で表わされる繰り返し単位を有するポリイミド前駆体1溶液を得た。
【0166】
【化17】
【0167】
(合成例2:ポリイミド前駆体2の合成)
2,2’−ビス(トリフルオロメチル)−4,4’−ジアミノビフェニル(TFMB)16.0g(50.0mmol)と2,2’−ジメチル−4,4’−ジアミノビフェニル(mTBHG)10.6g(50.0mmol)とを500mlのセパラブルフラスコに投入し、200gの脱水されたN−メチル−2−ピロリドン(NMP)に溶解させ、窒素気流下、オイルバスによって液温が50℃になるように熱電対でモニターし加熱しながら撹拌した。それらが完全に溶解したことを確認した後、そこへ、少しずつ30分かけて3,3’,4,4’−ビフェニルテトラカルボン酸二無水物(BPDA)27.3g(93.1mmol)を添加し、添加終了後、50℃で5時間撹拌した。その後、フタル酸無水物を0.900g(6.08mmol)添加した。その後室温まで冷却し、下記式で表わされる繰り返し単位を有するポリイミド前駆体2溶液を得た。
【0168】
【化18】
【0169】
(合成例3:ポリイミド前駆体3の合成)
窒素気流下1000mlのセパラブルフラスコに投入に脱水されたN−メチル−2−ピロリドン(NMP)85.5gを投入し、オイルバスによって液温が50℃になるように熱電対でモニターし加熱しながら撹拌した。3,3’,4,4’−ビフェニルテトラカルボン酸二無水物(BPDA)13.0g(44.2mmol)を投入し、その後、1,3−ビス(3−アミノプロピル)テトラメチルジシロキサン5.14g(20.7mmol)を投入した。60分後に溶解を確認後、脱水されたN−メチル−2−ピロリドン(NMP)401.7gを投入し、4,4’−ジアミノジフェニルエーテル(ODA)37.3g(186mmol)を溶解させた。
それらが完全に溶解したことを確認した後、3,3’,4,4’−ビフェニルテトラカルボン酸二無水物(BPDA)46.7g(159mmol)を添加し50℃で5時間撹拌した。その後、フタル酸無水物を0.994g(6.71mmol)添加し、50℃で1時間撹拌し末端封止した。その後室温まで冷却し、下記式で表わされる繰り返し単位を有するポリイミド前駆体3溶液を得た。(式中のRはR1またはR2で表される有機基であり、R1とR2の比率は、R1:R2=90:10となる。)
【0170】
【化19】
【0171】
[試験例:各塩基の触媒能の評価1]
本発明に係る光塩基発生剤から発生する塩基のイミド化を促進する触媒能の評価を行った。
(1)ポリイミド前駆体樹脂組成物溶液の調製、及び塗膜の作製
上記合成例1で得られたポリイミド前駆体1溶液に、表1に記載の塩基を、上記ポリイミド前駆体1溶液の固形分1gに対してそれぞれ0.6mmolの割合で添加し、試験例1〜15及び比較試験例1〜8のポリイミド前駆体樹脂組成物溶液を調製した。
また、上記合成例1で得られたポリイミド前駆体1溶液に、ピペリジンとシクロヘキサノールを、上記ポリイミド前駆体1溶液の固形分1gに対してそれぞれ0.6mmol添加したサンプルを、比較試験例9のポリイミド前駆体樹脂組成物溶液とし、ポリイミド前駆体1溶液そのものを、ブランクとして比較試験例10のポリイミド前駆体樹脂組成物溶液とした。また、塩基としてイソニペコチン酸を用いて、試験例1と同様にしてポリイミド前駆体樹脂組成物溶液を調製したところ、ポリイミド前駆体1溶液にイソニペコチン酸が溶解しなかったため、イミド化率の測定はできなかった。
得られた各ポリイミド前駆体樹脂組成物溶液をクロムめっきされたガラス(50mm×50mm)上に、乾燥後11±1μmとなるようスピンコートし、100℃のホットプレート上で15分乾燥させてそれぞれ対応する塗膜を作製した。
【0172】
(2)イミド化率の測定
イミド化率は、各塗膜について、ホットプレートにより150℃、160℃、170℃でそれぞれ10分間加熱し、得られた各塗膜のIRスペクトルから求めた。
具体的にはFT/IR−6100typeA(日本分光)と付属品名 ATR PRO470−Hのアタッチメントをもちい、下記の条件で測定し、得られたそれぞれのスペクトルを1480−1495cm−1のピークを基準に規格化し、1750−1800cm−1のイミドカルボニルピークの強度比から算出した。
算出の基準として、前記合成例1で得られたポリイミド前駆体1溶液をクロムめっきされたガラス(50mm×50mm)上に、乾燥後11±1μmとなるようスピンコートし、100℃のホットプレート上で15分乾燥した塗膜をイミド化率0%、そのサンプルを、窒素気流下、室温から350℃まで毎分10℃ずつ昇温し、350℃で1時間加熱した後の塗膜をイミド化率100%としてイミド化率を算出した。結果は表1の通りであった。
ホットプレートにより160℃で加熱した際、イミド化率が60%以上であれば、塩基の触媒能は高いと判断される。
なお、表1中の沸点は東京化成工業(株)の2010年度版の試薬カタログ(TCI Fine Chemicals 2010-2011 No.40)、シグマ アルドリッチ ジャパン(株)の2010年度版の試薬カタログ(ALDRICH Chemistry 2009-2010)及び、関東化学(株)の2010年度版のカタログ(The Index of Laboratory Chemicals 2010 No.26)を参照した。減圧状態での沸点が記載されているものに関しては、東京化成工業(株)の2010年度版の試薬カタログ(TCI Fine Chemicals 2010-2011 No.40)内に掲載されている沸点換算表を元に常圧1atm(=101325Pa=760mmHg)での沸点に換算した。
<測定条件>
光源:標準光源
検出器:TGS
積算回数:16
分解:4cm−1
ゼロフィリング:On
アポダイゼーション:Cosine
ゲイン:Auto(32)
アパーチャー:Auto(7.1mm)
スキャンスピード:Auto(2mm/sec)
フィルタ:Auto(10000Hz)
【0173】
【表1】
【0174】
表1の結果から、沸点が150℃以上かつ融点が200℃以下の水酸基を有する塩基は、ポリイミド前駆体のイミド化率を向上させる触媒能が高いことがわかる。
【0175】
[試験例:各塩基の触媒能の評価2]
上記合成例1で得られたポリイミド前駆体1溶液に、上記各塩基の触媒能の評価1で触媒能が高いと評価された、沸点が150℃以上かつ融点が200℃以下の水酸基を有する塩基である4−ピペリジンメタノールを、上記ポリイミド前駆体1溶液の固形分1gに対して表2に示す割合でそれぞれ添加し、試験例16のポリイミド前駆体樹脂組成物溶液を4つの濃度で調製した。
また、上記試験例16において、4−ピペリジンメタノールの代わりに、沸点が150℃以上かつ融点が200℃以下の水酸基を有する塩基である4−ヒドロキシピペリジンを用いた以外は、試験例16と同様にして試験例17のポリイミド前駆体樹脂組成物溶液を4つの濃度で調製した。
また、上記試験例16において、4−ピペリジンメタノールの代わりに、水酸基を有しないピペリジンを用いた以外は、試験例16と同様にして比較試験例11のポリイミド前駆体樹脂組成物溶液を4つの濃度で調製した。
【0176】
【表2】
【0177】
得られた試験例16〜17及び比較試験例11の各濃度のポリイミド前駆体樹脂組成物溶液を、それぞれクロムめっきされたガラス(50mm×50mm)上に、乾燥後11±1μmとなるようスピンコートし、100℃のホットプレート上で15分乾燥させてそれぞれ対応する塗膜を作製した。
各塗膜について、ホットプレートにより160℃で10分間加熱した。その時の各塗膜のイミド化率を、IRスペクトルから上記各塩基の触媒能の評価1と同様の方法で求めた。
各塩基の、ポリイミド前駆体1溶液の固形分1gに対する添加量(mmol/g)とイミド化率の関係を図1に示す。
【0178】
図1の結果から、4−ピペリジンメタノール及び4−ヒドロキシピペリジンを添加した塗膜は、ピペリジンを添加した塗膜に比べて少ない添加量でも高いイミド化率を達成できることが明らかとなった。このことから、沸点が150℃以上かつ融点が200℃以下の水酸基を有する塩基は触媒能が高く、少ない添加量でポリイミド前駆体のイミド化を十分に促進できることがわかる。
【0179】
(製造例1:光塩基発生剤1の合成)
100mLフラスコ中、炭酸カリウム2.00gをメタノール15mLに加えた。50mLフラスコ中、エトキシカルボニルメチル(トリフェニル)ホスホニウムブロミド(東京化成工業(株)製)2.67g(6.2mmol)、2−ヒドロキシ−4−メトキシベンズアルデヒド(東京化成工業(株)製)945mg(6.2mmol)をメタノール10mLに溶解し、よく撹拌した炭酸カリウム溶液にゆっくり滴下した。3時間撹拌した後、TLCにより反応の終了を確認したうえでろ過を行って炭酸カリウムを除き、減圧濃縮した。濃縮後、1Nの水酸化ナトリウム水溶液を50mL加え1時間撹拌した。反応終了後、ろ過によりトリフェニルホスフィンオキシドを除いた後、濃塩酸を滴下し反応液を酸性にした。沈殿物をろ過により集め、少量のクロロホルムにより洗浄することで2−ヒドロキシ−4−メトキシ桂皮酸を1.00g得た。続いて、100mLフラスコ中、2−ヒドロキシ−4−メトキシ桂皮酸1.00g(5.1mmol)を脱水テトラヒドロキシフラン10mLに溶解し、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩(東京化成工業(株)製)1.18g(6.2mmol)を加えた。30分後、4−ピペリジンメタノール(東京化成工業(株)製)712mg(6.2mmol)を加えた。反応終了後、反応溶液を濃縮し、水に溶解した。クロロホルムで抽出した後、1N塩酸、飽和食塩水で洗浄した。その後、シリカゲルカラムクマトグラフィー(展開溶媒:クロロホルム/メタノール=100/1〜10/1、体積比)により精製することにより、下記式で表される製造例1の光塩基発生剤1を100mg得た。
【0180】
【化20】
【0181】
(製造例2:光塩基発生剤の合成)
製造例1において、4−ピペリジンメタノール(東京化成工業(株)製)712mg(6.2mmol)の代わりに、4−ヒドロキシピペリジン(東京化成工業(株)製)625mg(6.2mmol)を用いた以外は、製造例1と同様にして、下記式で表される製造例2の光塩基発生剤2を83mg得た。
【0182】
【化21】
【0183】
(比較製造例1:比較光塩基発生剤1の合成)
実施例1において、4−ピペリジンメタノール(東京化成工業(株)製)712mg(6.2mmol)の代わりに、ピペリジン(東京化成工業(株)製)0.61ml(6.2mmol)を用いた以外は、製造例1と同様にして、下記式で表される比較製造例1の比較光塩基発生剤1を128mg得た。
【0184】
【化22】
【0185】
[評価:光塩基発生剤の塩基発生能]
製造例1、2及び比較製造例1で得られた光塩基発生剤についてそれぞれ1mgの試料を3つ用意し、それぞれを石英製NMR管中で重アセトニトリルに溶解させた。i線を20%透過するフィルタと高圧水銀灯を用いて、1本には400mJ/cm2で光照射を行い、他の1本には800mJ/cm2で光照射を行った。残り1本には光照射を行わなかった。各サンプルの1H NMRを測定し、異性化の割合を求めた。
【0186】
製造例1、2及び比較製造例1で得られた光塩基発生剤は、400mJ/cm2の照射および800mJ/cm2の照射により異性化した。異性化させたサンプルを160℃に加熱すると、異性化した化合物のほぼ100%が環化し、それにともない塩基が発生し、表3に示す塩基発生率(%)となった。
【0187】
【表3】
【0188】
(実施例1:感光性ポリイミド樹脂組成物1の調製)
合成例2で得られたポリイミド前駆体2溶液の固形分100重量部に対し、製造例1で得られた光塩基発生剤1を15重量部添加し、実施例1の感光性ポリイミド樹脂組成物1を調製した。
なお、光塩基発生剤1から発生する塩基(4−ピペリジンメタノール)は、160℃で、当該塩基100重量部に対して、合成例2で得られたポリイミド前駆体(固形分)を10重量部以上溶解した。
【0189】
(実施例2:感光性ポリイミド樹脂組成物2の調製)
実施例1において製造例1で得られた光塩基発生剤1の代わりに、製造例2で得られた光塩基発生剤2を用いた以外は、実施例1と同様にして、実施例2の感光性ポリイミド樹脂組成物2を調製した。
なお、光塩基発生剤2から発生する塩基(4−ヒドロキシピペリジン)は、165℃で、当該塩基100重量部に対して、合成例2で得られたポリイミド前駆体(固形分)を10重量部以上溶解した。
【0190】
(実施例3:感光性ポリイミド樹脂組成物3の調製)
合成例1で得られたポリイミド前駆体1溶液の固形分100重量部に対し、製造例1で得られた光塩基発生剤1を22.3重量部添加し、実施例3の感光性ポリイミド樹脂組成物3を調製した。
なお、光塩基発生剤1から発生する塩基(4−ピペリジンメタノール)は、160℃で、当該塩基100重量部に対して、合成例1で得られたポリイミド前駆体(固形分)を10重量部以上溶解した。
【0191】
(実施例4:感光性ポリイミド樹脂組成物4の調製)
合成例3で得られたポリイミド前駆体3溶液の固形分100重量部に対し、製造例1で得られた光塩基発生剤1を22.3重量部添加し、実施例4の感光性ポリイミド樹脂組成物4を調製した。
なお、光塩基発生剤1から発生する塩基(4−ピペリジンメタノール)は、160℃で、当該塩基100重量部に対して、合成例3で得られたポリイミド前駆体(固形分)を10重量部以上溶解した。
【0192】
(比較例1:比較感光性ポリイミド樹脂組成物1の調製)
実施例1において製造例1で得られた光塩基発生剤の代わりに、比較製造例1で得られた比較光塩基発生剤1を用いた以外は実施例1と同様にして、比較例1の比較感光性ポリイミド樹脂組成物1を調製した。
なお、比較光塩基発生剤1から発生する塩基(ピペリジン)は、沸点が106℃であるため、150℃〜200℃のいずれかの温度における当該塩基に対するポリイミド前駆体の溶解を試みることはできなかった。
【0193】
(比較例2:比較感光性ポリイミド樹脂組成物2の調製)
合成例1で得られたポリイミド前駆体1溶液の固形分100重量部に対し、比較製造例1で得られた比較光塩基発生剤1を20重量部添加し、比較例2の比較感光性ポリイミド樹脂組成物2を調製した。
なお、比較光塩基発生剤1から発生する塩基(ピペリジン)は、沸点が106℃であるため、150℃〜200℃のいずれかの温度における当該塩基に対するポリイミド前駆体の溶解を試みることはできなかった。
【0194】
(比較例3:比較感光性ポリイミド樹脂組成物3の調製)
合成例3で得られたポリイミド前駆体3溶液の固形分100重量部に対し、比較製造例1で得られた比較光塩基発生剤1を20重量部添加し、比較例3の比較感光性ポリイミド樹脂組成物3を調製した。
なお、比較光塩基発生剤1から発生する塩基(ピペリジン)は、沸点が106℃であるため、150℃〜200℃のいずれかの温度における当該塩基に対するポリイミド前駆体の溶解を試みることはできなかった。
【0195】
[感光性ポリイミド樹脂組成物の評価:現像性評価1]
(1)塗膜の作製
実施例1、実施例2、及び比較例1の感光性ポリイミド樹脂組成物を、それぞれ、クロムめっきされたガラス(50mm×50mm)上に乾燥後17±1μmとなるようにスピンコートし、100℃のホットプレート上で15分乾燥させて実施例1、実施例2、及び比較例1の感光性ポリイミド樹脂組成物の塗膜をそれぞれ2枚をセットとして作製した。
上記の通り作製した塗膜のうち1枚を、露光部の評価を行う塗膜とし、手動露光機を用いて高圧水銀灯により、2J/cm2露光を行った。残りの一枚を未露光部の評価を行う塗膜として、露光を行わなかった。その後、各塗膜をホットプレートにより下表4〜6に示す温度(PEB温度)でそれぞれ10分間加熱し、現像性評価サンプルを得た。各現像性評価サンプルにおいては、露光部の評価を行う塗膜1枚と未露光部の評価を行う塗膜1枚の2枚をセットにして評価に用いた。
【0196】
(2)現像性評価
それぞれの塗膜について、50℃のテトラメチルアンモニウムハイドロオキサイド2.38重量%水溶液で現像処理し、600秒後までの塗膜の溶解速度を測定した。実施例1、実施例2、及び比較例1の感光性ポリイミド樹脂組成物の評価結果はそれぞれ表4〜6の通りであった。
現像性を評価する方法としては、各塗膜の露光部と未露光部の時間あたりの膜減り量(溶解速度)を算出し、比較を行った。未露光部の塗膜の溶解速度に対する露光部の塗膜の溶解速度の比(溶解性コントラスト)の値が大きいほど、残膜率が高くなると評価できる。
なお、比較例1の未露光部は、現像液に殆ど溶解せず、膨潤した後、1200秒後に塗膜がガラスから剥れた。
【0197】
【表4】
【0198】
【表5】
【0199】
【表6】
【0200】
評価の結果、実施例1、実施例2はいずれのPEB温度でも比較例1より溶解性コントラストの値が高いことがわかった。実施例1、実施例2の感光性ポリイミド樹脂組成物は、環境負荷の低減から、アルコール等の有機溶剤を添加しない塩基性水溶液を現像液として用いた場合でもより短時間で残膜率の高い良好な形状のパターンが形成できると推測できる。一方、比較例1の感光性ポリイミド樹脂組成物は、アルコール等の有機溶剤を添加しない塩基性水溶液を現像液として用いると、良好なレリーフパターンが得られないことが示唆された。
また、上記の結果から、本発明の感光性ポリイミド樹脂組成物は、露光後ベーク時の加熱条件を従来より緩和しても、ポリイミド前駆体のイミド化を十分に促進でき、更に、塩基に導入されている水酸基の効果で、未露光部の現像液への溶解が促進されることが示唆された。水酸基を有する塩基は、従来現像液に添加していたアルコールの代替として機能し、ポリイミド前駆体の溶解を促進する溶解促進剤としても機能しているのではないかと推測される。
【0201】
[感光性ポリイミド樹脂組成物の評価:現像性評価2]
(1)塗膜の作製
実施例3、及び比較例2の感光性ポリイミド樹脂組成物を、それぞれ、クロムめっきされたガラス(50mm×50mm)上に乾燥後2μmとなるようにスピンコートし、120℃のホットプレート上で15分乾燥させて実施例3、及び比較例2の感光性ポリイミド樹脂組成物の塗膜をそれぞれ2枚をセットとして作製した。
上記の通り作製した塗膜のうち1枚を、露光部の評価を行う塗膜とし、手動露光機を用いて高圧水銀灯により、500mJ/cm2露光を行った。残りの一枚を未露光部の評価を行う塗膜として、露光を行わなかった。その後、各塗膜をホットプレートにより160℃でそれぞれ2分間加熱し、現像性評価サンプルを得た。各現像性評価サンプルにおいては、露光部の評価を行う塗膜1枚と未露光部の評価を行う塗膜1枚の2枚をセットにして評価に用いた。
【0202】
(2)現像性評価
それぞれの塗膜について、23℃のテトラメチルアンモニウムハイドロオキサイド2.38重量%水溶液で現像処理し、塗膜の溶解速度を測定した。現像性は、上記現像性評価1と同様の方法で評価した。
実施例3、及び比較例2の感光性ポリイミド樹脂組成物の評価結果はそれぞれ表7の通りであった。
【0203】
【表7】
【0204】
評価の結果、実施例3は比較例2と比べて未露光部の溶解速度が速く、溶解性コントラストの値が高いことがわかった。
【0205】
[感光性ポリイミド樹脂組成物の評価:現像性評価3]
(1)塗膜の作製
実施例4、及び比較例3の感光性ポリイミド樹脂組成物を、それぞれ、クロムめっきされたガラス(50mm×50mm)上に乾燥後2μmとなるようにスピンコートし、120℃のホットプレート上で15分乾燥させて実施例4、及び比較例3の感光性ポリイミド樹脂組成物の塗膜をそれぞれ2枚をセットとして作製した。
上記の通り作製した塗膜のうち1枚を、露光部の評価を行う塗膜とし、手動露光機を用いて高圧水銀灯により、500mJ/cm2露光を行った。残りの一枚を未露光部の評価を行う塗膜として、露光を行わなかった。その後、各塗膜をホットプレートにより160℃でそれぞれ2分間加熱し、現像性評価サンプルを得た。各現像性評価サンプルにおいては、露光部の評価を行う塗膜1枚と未露光部の評価を行う塗膜1枚の2枚をセットにして評価に用いた。
【0206】
(2)現像性評価
それぞれの塗膜について、23℃のテトラメチルアンモニウムハイドロオキサイド2.38重量%水溶液で現像処理し、塗膜の溶解速度を測定した。現像性は、上記現像性評価1と同様の方法で評価した。
実施例4、及び比較例3の感光性ポリイミド樹脂組成物の評価結果はそれぞれ表8の通りであった。
【0207】
【表8】
【0208】
評価の結果、実施例4は比較例3と比べて未露光部の溶解速度が速く、溶解性コントラストの値が高いことがわかった。
【0209】
現像性評価2〜3の結果から、本発明の光塩基発生剤は、添加するポリイミド前駆体の組成を変えても、露光部のイミド化を十分に促進させ、一方、未露光部は塩基発生剤に導入されている水酸基の効果で現像液への溶解が促進されるために、より短時間で残膜率の高い良好な形状のパターンが形成できることが示唆された。
【0210】
[感光性ポリイミド樹脂組成物の評価:塗膜の感度とγ値の評価1]
実施例1、実施例2、及び比較例1の感光性ポリイミド樹脂組成物を、それぞれ、クロムめっきされたガラス(50mm×50mm)上に乾燥後18±1μmとなるようにスピンコートし、100℃のホットプレート上で15分乾燥させて、各塗膜を得た。
【0211】
作成した塗膜に対し、手動露光機を用いて、高圧水銀灯により全面露光を行った。実施例1の塗膜に対しては、0、25、50、75、100、150、300、400、500、1000mJ/cm2でそれぞれ全面露光を行い、実施例2の塗膜に対しては、0、50、75、100、200、300、500、1000mJ/cm2でそれぞれ全面露光を行った。比較例1の塗膜に対しては、0、25、75、100、200、300、400、500、1000mJ/cm2でそれぞれ全面露光を行った。その後、各塗膜をホットプレートにより実施例1の塗膜に対しては160℃で、実施例2の塗膜に対しては165℃で、比較例1の塗膜に対しては165℃でそれぞれ10分間加熱し、これらの塗膜をそれぞれ、テトラメチルアンモニウムハイドロオキサイド2.5重量%水溶液とイソプロパノールを8:2で混合した溶液に実施例1の塗膜に対しては40℃で5分間、実施例2の塗膜に対しては30℃で10分間、比較例1の塗膜に対しては40℃で6分間現像処理し、基板上の残存膜厚を測定した。結果を、表9に示す。また、露光量と規格化残膜率の関係を表す感度曲線を図2に示す。なお、図中の規格化残膜率は、(現像後膜厚×100)/(現像後の最高膜厚)とした。
また、表中の感度とは、感度曲線から求めた規格化残膜率が約1となる時の露光量である。表中のγ値はコントラストを表す指標であり、値が大きいほどコントラストが高くテーパー角の大きい形状のパターンを得ることができる。γ値は以下の式により求めた。
γ=1/{log(規格化残膜率が約1となる時の露光量)−log(規格化残膜率が0となる時の露光量)}
なお、実施例2、比較例1については規格化残膜率が0となる露光量が正確に測れなかったので、規格化残膜率が最小となる露光量(実施例2は50mJ、比較例1は25mJ)−1mJを仮に規格化残膜率が0となる時の露光量としてγ値を算出した。
【0212】
【表9】
【0213】
UV照射量の増加とともに、残膜率が上昇していることから、UV照射および、加熱によって、塩基発生剤がアミド結合を形成可能なNH基を1つ有する2級アミンを発生し、イミド化が進行していることが示された。本発明の実施例1では100mJ/cm2で、実施例2では75mJ/cm2で規格化残膜率が約1となった。それに対して、比較例1では100mJ/cm2で規格化残膜率が約1となった。このことから、本発明の感光性ポリイミド樹脂組成物は、比較感光性ポリイミド樹脂組成物と同程度の露光量でイミド化が進行することが示された。
一方、γ値は、値が大きいほど溶解性コントラストが高くテーパー角の大きい形状のパターンを得ることができる。本発明の実施例1ではγ値が8.0で、実施例2ではγ値が5.4であったのに対し、比較例1ではγ値が1.6であった。このことから、本発明の感光性ポリイミド樹脂組成物は、比較感光性ポリイミド樹脂組成物と比べて溶解性コントラストが高く、より矩形に近い形状のパターンを得ることができることが示された。
【0214】
[感光性ポリイミド樹脂組成物の評価:塗膜の感度とγ値の評価2]
実施例3、及び比較例2の感光性ポリイミド樹脂組成物を、それぞれ、クロムめっきされたガラス(50mm×50mm)上に乾燥後2μmとなるようにスピンコートし、120℃のホットプレート上で15分乾燥させて、各塗膜を得た。
【0215】
作成した塗膜に対し、手動露光機を用いて、適宜露光量を選んで高圧水銀灯により全面露光を行った。その後、各塗膜をホットプレートにより160℃でそれぞれ2分間加熱した。これらの塗膜をそれぞれ、23℃のテトラメチルアンモニウムハイドロオキサイド2.38重量%水溶液で、実施例3の塗膜に対しては90秒間、比較例2の塗膜に対しては150秒間現像処理し、基板上の残存膜厚を測定した。その結果を表10に示す。また、露光量と規格化残膜率の関係を表す感度曲線を図3に示す。なお、図中の規格化残膜率、表中の感度及びγ値は、上記塗膜の感度とγ値の評価1と同様である。
【0216】
【表10】
【0217】
UV照射量の増加とともに、残膜率が上昇していることから、UV照射および、加熱によって、塩基発生剤がアミド結合を形成可能なNH基を1つ有する2級アミンを発生し、イミド化が進行していることが示された。本発明の実施例3では118mJ/cm2で、比較例2では82mJ/cm2で規格化残膜率が1となった。
一方、γ値は、値が大きいほど溶解性コントラストが高くテーパー角の大きい形状のパターンを得ることができる。本発明の実施例3ではγ値が1.8であったのに対し、比較例2ではγ値が1.1であった。このことから、本発明の感光性ポリイミド樹脂組成物は、比較感光性ポリイミド樹脂組成物と比べて溶解性コントラストが高く、より矩形に近い形状のパターンを得ることができることが示された。
【0218】
[感光性ポリイミド樹脂組成物の評価:パターン形成能評価1]
実施例1、実施例2、及び比較例1の感光性ポリイミド樹脂組成物を、それぞれ、クロムめっきされたガラス(50mm×50mm)上に乾燥後18±1μmとなるようにスピンコートし、100℃のホットプレート上で15分乾燥させて、各塗膜を得た。
【0219】
作成した塗膜に対し、手動露光機を用いて、高圧水銀灯により細線評価用マスクを介して、200mJ/cm2露光を行った。続いて各塗膜を、実施例2及び比較例1の塗膜は165℃、実施例1の塗膜は160℃でそれぞれ5分間加熱した。
これらのサンプルの現像は、環境への負荷を低減させるため、アルコールを含まない、テトラメチルアンモニウムハイドロオキサイド2.38重量%水溶液を用い、200mlのビーカーでマグネチックスターラーによって攪拌させ、湯浴によって50℃に加熱した状態で、サンプルを浸漬させることにより行った。
通常、現像液からアルコールを除去することにより、環境負荷が低減される一方、未露光部の現像速度が低下し、現像時間が長くなる。
【0220】
パターン形成後、塗膜を窒素雰囲気下、350℃、1時間熱処理し(昇温速度10℃/分、自然放冷)後硬化させた。その結果、実施例1及び実施例2の塗膜については、表11に示すような細線パターンを得た。比較例1の塗膜については、現像液に1時間以上浸漬しても未露光部が残存しており、パターンを形成できなかった。この結果より、本発明の感光性ポリイミド樹脂組成物は、良好なパターンを形成できることが明らかとなった。
【0221】
【表11】
【0222】
<パターン形状の評価基準>
◎:パターン断面のテーパー角が55度超過
○:パターン断面のテーパー角が30〜55度
【0223】
比較例1の比較感光性ポリイミド樹脂組成物の場合、未露光部がゲル状になる、又は膨張するなどの現象が見られ、現像液に溶解しにくかった。一方、実施例1及び実施例2の感光性ポリイミド樹脂組成物のように発生塩基が水酸基を有する場合、はっ水性の高いフッ素含有ポリイミド前駆体を用いた場合でも、未露光部における前記のような現象は見られなかった。これは発生塩基の水酸基の効果によって未露光部のアルカリ水溶液に対する親和性が向上したためと考えられる。さらに実施例1の感光性ポリイミド樹脂組成物を用いる場合はPEB温度を160℃に下げることができるため、未露光部のイミド化を抑えられ、現像時間を短縮することができた。
【0224】
[感光性ポリイミド樹脂組成物の評価:パターン形成能評価2]
実施例3、及び比較例2の感光性ポリイミド樹脂組成物を、それぞれ、クロムめっきされたガラス(50mm×50mm)上に乾燥後2μmとなるようにスピンコートし、120℃のホットプレート上で15分乾燥させて、各塗膜を得た。
作成した塗膜に対し、手動露光機を用いて、高圧水銀灯により細線評価用マスクを介して、200mJ/cm2露光を行った。続いて各塗膜を、160℃で2分間加熱した。
これらのサンプルの現像は、環境への負荷を低減させるため、アルコールを含まない、テトラメチルアンモニウムハイドロオキサイド2.38重量%水溶液を用いて90秒間処理を行った。
その結果、実施例3の塗膜については、現像後膜厚1.60μm、残膜率90%の細線パターンの形成を確認した。
一方、比較例2の塗膜については、90秒間の現像処理ではまだ未露光部が残存しており、パターンを形成できなかった。
パターン形成能評価2の結果より、実施例3の感光性ポリイミド樹脂組成物は、比較例2と比べて短時間で良好なパターンを形成できることが明らかとなった。
【0225】
[感光性ポリイミド樹脂組成物の評価:パターン形成能評価3]
実施例4、及び比較例3の感光性ポリイミド樹脂組成物を、それぞれ、クロムめっきされたガラス(50mm×50mm)上に乾燥後2μmとなるようにスピンコートし、120℃のホットプレート上で15分乾燥させて、各塗膜を得た。
作成した塗膜に対し、手動露光機を用いて、高圧水銀灯により細線評価用マスクを介して、200mJ/cm2露光を行った。続いて各塗膜を、160℃で2分間加熱した。
これらのサンプルの現像は、環境への負荷を低減させるため、アルコールを含まない、テトラメチルアンモニウムハイドロオキサイド2.38重量%水溶液を用いて60秒間処理を行った。
その結果、実施例4の塗膜については、現像後膜厚1.48μm、残膜率81%の細線パターンの形成を確認した。
比較例3の塗膜については、60秒間の現像処理ではまだ未露光部が残存しており、パターンを形成できなかった。
パターン形成能評価3の結果より、実施例4の感光性ポリイミド樹脂組成物は、比較例3と比べて短時間で良好なパターンを形成できることが明らかとなった。
【0226】
以上の結果から、水酸基を有する塩基は、イミド化の触媒能が高く、このような塩基を発生する光塩基発生剤を用いた本発明の感光性ポリイミド樹脂組成物は、露光後ベーク時の加熱条件を従来より緩和しても、ポリイミド前駆体のイミド化を十分に促進でき、更に、塩基に導入されている水酸基の効果で、未露光部の現像液への溶解が促進されることが明らかとなった。そして、その結果、本発明の感光性ポリイミド樹脂組成物は、現像時間を短縮でき、アルコール等を添加しない塩基性水溶液を現像液として用いることが可能で、露光部と未露光部の溶解性コントラストを高くすることができ、より解像度の高いパターンを形成できたり、レリーフパターンの形状が良好になるという効果が得られることが明らかとなった。
【技術分野】
【0001】
本発明は、前駆体型感光性ポリイミド用光塩基発生剤、ネガ型感光性ポリイミド樹脂組成物、当該感光性ポリイミド樹脂組成物を用いたレリーフパターンの製造方法、及び、当該感光性ポリイミド樹脂組成物を用いて作製した物品に関するものである。
【背景技術】
【0002】
感光性ポリイミド樹脂組成物は、例えば、電子部品、光学製品、光学部品の成形材料、層形成材料又は接着剤などに用いられ、特に、電磁波によるパターニング工程を経て形成される製品又は部材に好適に利用されてきている。
高分子材料であるポリイミドは、耐熱性、寸法安定性、絶縁特性といった性能が有機物の中でもトップクラスの性能を示すため、電子部品の絶縁材料等へ広く適用され、半導体素子の中のチップコーティング膜や、フレキシブルプリント配線板の基材などとして盛んに利用されてきている。
【0003】
一般にポリイミドは溶媒への溶解性に乏しく、加工が困難なため、ポリイミドを所望の形状にパターニングする方法として、溶媒溶解性に優れるポリイミド前駆体の状態で露光と現像によるパターニングを行い、その後、熱処理等によりイミド化を行いポリイミドのパターンを得るという方法がある。
【0004】
ポリイミド前駆体を利用して、パターンを形成する手段として、種々の方法が提案されている。その代表的なものは以下の二つである。
(1)ポリイミド前駆体にはパターン形成能力がなく、ポリイミド前駆体上に感光性樹脂をレジスト層として設けることによりパターンを形成する方法。
(2)ポリイミド前駆体自身に感光性部位を結合や配位させて導入し、その作用により、パターンを形成する方法。または、ポリイミド前駆体に感光性成分を混合し樹脂組成物とし、その感光性成分の作用でパターンを形成する方法。
【0005】
上記(2)を用いるパターニング手法の代表的なものとしては、(i)ポリイミド前駆体のポリアミック酸に電磁波の露光前は溶解抑止剤として作用し、露光後はカルボン酸を形成し溶解促進剤となるナフトキノンジアジド誘導体を混合し、露光部と未露光部の現像液に対する溶解速度のコントラストを大きくすることでパターン形成を行い、その後にイミド化を行い、ポリイミドパターンを得る手法や(特許文献1)、(ii)ポリイミド前駆体にエステル結合またはイオン結合を介してメタクリロイル基を導入し、そこに光ラジカル発生剤を添加し、露光部を架橋させることで露光部と未露光部の現像液に対する溶解速度のコントラストを大きくすることでパターン形成を行い、その後にイミド化を行い、ポリイミドパターンを得る手法などが実用化されている(特許文献2)。
【0006】
(2)の手法は、(1)の方法と比べ、レジスト層が必要ないため大幅にプロセスを簡略化させることができるが、(i)の方法では、溶解性コントラストを高めるためにナフトキノンジアジド誘導体の添加量を増加させると、ポリイミド本来の物性が得られなくなるという問題があった。また(ii)の方法では、ポリイミド前駆体の構造が制約されてしまうという問題があった。
【0007】
この他のパターニング手法としては、(iii)ポリイミド前駆体であるポリアミック酸に、光塩基発生剤を混合し、露光後加熱することで露光によって発生した塩基の作用によって環化を進行させ、現像液に対する溶解性を低下させることで、露光部と未露光部の現像液に対する溶解速度のコントラストを大きくすることでパターン形成を行い、その後にイミド化を行い、ポリイミドパターンを得る手法が報告されている(特許文献3)。
【0008】
非特許文献1には、アミンの光反応性保護基として、o−ヒドロキシ−トランス−桂皮酸を用いることが開示されている。また、特許文献4には、o−ヒドロキシ−トランス−桂皮酸アミドを光塩基発生剤として用い、当該光塩基発生剤と塩基反応性樹脂とを含む感光性ポリイミド樹脂組成物が開示されている。更に、本発明者らも、o−ヒドロキシ−トランス−桂皮酸アミド誘導体を光環化型の光塩基発生剤として用い、当該光塩基発生剤とポリイミド前駆体とを含む感光性ポリイミド樹脂組成物を特許文献5に開示している。また、特許文献6及び7には、カルバメート型の光塩基発生剤を用いた感光性ポリイミド樹脂組成物が開示されている。更に、特許文献8及び9には、オキシム型の光塩基発生剤を用いた感光性ポリイミド樹脂組成物が開示されている。
【0009】
光塩基発生剤を用いた感光性ポリイミド樹脂組成物は、ポリイミド前駆体に、光塩基発生剤を一定比率混合するだけで感光性ポリイミド前駆体を得ることができるため、樹脂組成物を製造するプロセスが簡便である。また、種々の構造のポリイミド前駆体に適用できるため汎用性が高いという利点がある。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】特開昭52−13315号公報
【特許文献2】特開昭54−145794号公報
【特許文献3】特開平8−227154号公報
【特許文献4】特開2009−80452号公報
【特許文献5】国際公開第2009/123122号パンフレット
【特許文献6】特開2006−189591号公報
【特許文献7】特開2008−247747号公報
【特許文献8】特開2007−249013号公報
【特許文献9】特開2008−003581号公報
【非特許文献】
【0011】
【非特許文献1】Chem. Pharm. Bull. 1997, 45(4) p.715-718
【発明の概要】
【発明が解決しようとする課題】
【0012】
しかし、上記のような光塩基発生剤を用いた感光性ポリイミド樹脂組成物は、露光後の加熱工程において露光部のみならず未露光部においてもポリイミド前駆体のイミド化が進行することから、現像性の改善が課題であった。すなわち、未露光部のイミド化も一部進行することにより、未露光部の現像液への溶解性が悪くなるため、現像時間を長くしなければならないという問題があった。また、現像液として、塩基性水溶液だけでは十分に溶解しないため、例えばアルコール等の有機溶剤を添加した塩基性水溶液を用いる必要があった。環境負荷の低減から、アルコール等の有機溶剤を添加しない塩基性水溶液を現像液として用いることが市場で求められている。
更に、未露光部においてポリイミド前駆体のイミド化が進行しすぎた場合、露光部と未露光部の現像液に対する溶解速度のコントラストが不十分となり、レリーフパターンの形状が悪化する恐れがある。
【0013】
露光後ベーク時の加熱温度を低くしたり、加熱時間を短くするなど、加熱条件を緩和することにより、未露光部におけるポリイミド前駆体のイミド化の進行を抑制することができる。しかし、加熱条件を緩和すると、露光部においてもイミド化の進行は抑制されるため、露光部と未露光部の溶解性コントラストは向上しない。そこで、本発明者らは、露光後ベーク時の加熱条件を従来より緩和しても、露光部においてポリイミド前駆体のイミド化を十分に促進する触媒能が高い塩基を発生する光塩基発生剤を得ることに着目した。
【0014】
すなわち本発明は、上記実情を鑑みてなされたものであり、露光後ベーク時の加熱条件を従来より緩和しても、露光部のポリイミド前駆体のイミド化を十分に促進する塩基を発生する光塩基発生剤を提供することを目的とする。また、本発明は、当該光塩基発生剤を用いることにより、現像時間を短縮でき、アルコール等を添加しない塩基性水溶液を現像液として用いることが可能となったり、露光部と未露光部の溶解性コントラストが高くなる、感光性ポリイミド樹脂組成物を提供することを目的とする。
【課題を解決するための手段】
【0015】
本発明に係る前駆体型感光性ポリイミド用光塩基発生剤は、発生する塩基が共有結合を用いて潜在化されていて、電磁波の照射により塩基を発生する塩基発生剤であって、水酸基を有する塩基を発生することを特徴とする。
【0016】
本発明に係る前駆体型感光性ポリイミド用塩基発生剤は、電磁波の照射により水酸基を有する塩基を発生することにより、ポリイミド前駆体のイミド化促進効果が高く、露光後ベーク時の加熱条件を従来より緩和しても、露光部のポリイミド前駆体のイミド化を十分に促進することが可能である。
【0017】
本発明に係る塩基発生剤においては、沸点が150℃以上かつ融点が200℃以下の水酸基を有する塩基を発生することが、ポリイミド前駆体のイミド化促進効果が高くなる点から好ましい。
【0018】
本発明に係る光塩基発生剤においては、下記化学式(1−1)、下記化学式(1−2)又は下記化学式(1−3)で表されること光塩基発生剤であることが好ましい。
【0019】
【化1】
(式(1−1)中、R1及びR2は、それぞれ独立に、水素原子又は有機基であり、同一であっても異なっていても良い。R1及びR2の少なくとも1つは有機基である。R1及びR2は、それらが結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。但し、R1及びR2のいずれかの有機基が水酸基を有する。R3及びR4はそれぞれ独立に、水素原子、ハロゲン原子、水酸基、メルカプト基、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、又は有機基であり、同一であっても異なっていても良い。R5、R6、R7及びR8は、それぞれ独立に、水素原子、ハロゲン原子、水酸基、メルカプト基、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、アミノ基、アンモニオ基又は有機基であり、同一であっても異なっていても良い。R5、R6、R7及びR8は、それらの2つ以上が結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。R9は、水素原子、或いは、加熱及び/又は電磁波の照射により脱保護可能な保護基である。)
【0020】
【化2】
(式(1−2)中、R1及びR2は、それぞれ独立に、水素原子又は有機基であり、同一であっても異なっていても良い。R1及びR2の少なくとも1つは有機基である。R1及びR2は、それらが結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。但し、R1及びR2のいずれかの有機基が水酸基を有する。R10及びR10’はそれぞれ独立に水素原子、ハロゲン原子、水酸基、メルカプト基、ニトロ基、シリル基、シラノール基、又は有機基である。R11、R12、R13、R14及びR15は、それぞれ独立に、水素原子、ハロゲン原子、水酸基、メルカプト基、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、アミノ基、アンモニオ基又は有機基であり、同一であっても異なっていても良い。R11、R12、R13、R14及びR15は、それらの2つ以上が結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。)
【0021】
【化3】
(式(1−3)中、R17は、水酸基を有する有機基で置換されたアミノ基又は水酸基を有する有機基、R18及びR19は、それぞれ独立に、水素原子、ハロゲン原子、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、アミノ基、アンモニオ基又は有機基であり、同一であっても異なっていても良いが、R18及びR19の少なくとも1つはヘテロ原子を有していても良い芳香族化合物である。R17、R18及びR19は、それらの2つ以上が結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。)
【0022】
本発明に係る光塩基発生剤においては、発生する塩基が脂肪族アミンであることが、塩基性が強く、イミド化を促進する触媒能が高いため、より少量の添加で、より低い温度での最終生成物への反応が可能となる点から好ましい。
【0023】
本発明に係る光塩基発生剤においては、発生する塩基が酸性基を含まないことが、発生した塩基のイミド化を促進する触媒能を発揮させる点から好ましい。
【0024】
本発明に係るネガ型感光性ポリイミド樹脂組成物は、ポリイミド前駆体と、前記本発明に係る光塩基発生剤とを含有することを特徴とする。
本発明に係るネガ型感光性ポリイミド樹脂組成物は、前記本発明に係る光塩基発生剤を用いることにより、露光後ベーク時の加熱条件を従来より緩和しても、露光部は十分にイミド化が促進され、未露光部は緩和された加熱条件のためにポリイミド前駆体のイミド化の進行を抑制することができる。また、塩基に導入されている水酸基の効果で、未露光部の現像液への溶解が促進される。これらにより、未露光部の現像液への溶解性が向上するため、未露光部を除去する現像時間を短縮でき、また、アルコール等を添加しない塩基性水溶液を現像液として用いることが可能になる。更に、露光部と未露光部の溶解性コントラストが高くなるため、レリーフパターンの形状が良好になる。
【0025】
本発明に係るネガ型感光性ポリイミド樹脂組成物においては、前記光塩基発生剤が発生する塩基が、150〜200℃のいずれかの温度において、当該塩基100重量部に対して、前記ポリイミド前駆体を10重量部以上溶解する塩基であることが、塩基の触媒能を向上させる点から好ましい。
【0026】
本発明に係るネガ型感光性ポリイミド樹脂組成物は、塗料、印刷インキ、シール剤、又は接着剤、或いは、表示装置、半導体装置、電子部品、微小電気機械システム、光造形物、光学部材又は建築材料の形成材料として好適に用いられる。
【0027】
本発明は、上記本発明に係るネガ型感光性ポリイミド樹脂組成物を用いるレリーフパターンの製造方法を提供する。
本発明に係るレリーフパターンの製造方法は、上記ネガ型感光性ポリイミド樹脂組成物を用いて塗膜又は成形体を形成し、当該塗膜又は成形体を、所定パターン状に電磁波を照射し、照射後又は照射と同時に加熱し、前記照射部位の溶解性を変化させた後、現像することを特徴とする。
【0028】
また、本発明は、上記ネガ型感光性ポリイミド樹脂組成物又はその硬化物により少なくとも一部が形成されている、印刷物、塗料、シール剤、接着剤、表示装置、半導体装置、電子部品、微小電気機械システム、光造形物、光学部材又は建築材料のいずれかの物品を提供する。
【発明の効果】
【0029】
本発明によれば、露光後ベーク時の加熱条件を従来より緩和しても、露光部のポリイミド前駆体のイミド化を十分に促進する光塩基発生剤を提供することができる。
本発明に係るネガ型感光性ポリイミド樹脂組成物は、現像時間を短縮でき、アルコール等を添加しない塩基性水溶液を現像液として用いることが可能となったり、露光部と未露光部の溶解性コントラストを高くすることができる。
【図面の簡単な説明】
【0030】
【図1】試験例16、試験例17、及び比較試験例11における、ポリイミド前駆体に対する塩基の添加量とイミド化率との関係を示すグラフである。
【図2】実施例1、実施例2、及び比較例1の感光性ポリイミド樹脂組成物の露光量と残膜率との関係を示す感度曲線である。
【図3】実施例3、及び比較例2の感光性ポリイミド樹脂組成物の露光量と残膜率との関係を示す感度曲線である。
【発明を実施するための形態】
【0031】
以下、本発明について詳しく説明する。
なお、本発明において(メタ)アクリロイルとは、アクリロイル及び/又はメタクリロイルを意味し、(メタ)アクリルとは、アクリル及び/又はメタクリルを意味し、(メタ)アクリレートとは、アクリレート及び/又はメタクリレートを意味する。
また、本発明において、電磁波とは、波長を特定した場合を除き、可視及び非可視領域の波長の電磁波だけでなく、電子線のような粒子線、及び、電磁波と粒子線を総称する放射線又は電離放射線が含まれる。本明細書では、電磁波の照射を露光ともいう。なお、波長365nm、405nm、436nmの電磁波をそれぞれ、i線、h線、g線とも表記することがある。
また、本発明において、沸点及び融点は、圧力1atm(101325Pa)下での沸点及び融点をいう。
また、本発明において、有機基とは、少なくとも1つの炭素原子を含む官能基の総称を表す。
【0032】
<光塩基発生剤>
本発明に係る前駆体型感光性ポリイミド用光塩基発生剤は、発生する塩基が共有結合を用いて潜在化されていて、電磁波の照射により塩基を発生する光塩基発生剤であって、水酸基を有する塩基を発生することを特徴とする。なおここで、前駆体型感光性ポリイミドとは、ポリイミド前駆体を含む感光性ポリイミド樹脂組成物のことをいう。
本発明に係る前駆体型感光性ポリイミド用塩基発生剤は、電磁波の照射により水酸基を有する塩基(塩基性物質)を発生することにより、露光後ベーク時の加熱条件を従来より緩和しても、露光部のポリイミド前駆体のイミド化を十分に促進することが可能である。
ここで、露光部のポリイミド前駆体のイミド化を十分に促進する程度とは、露光部のポリイミド前駆体の現像液に対する溶解速度が未露光部に対して十分遅くなる程度であり、例えば用いられる現像液に対して露光部のポリイミド前駆体の溶解速度が25℃で10nm/s以下程度に遅くなることを目安とすることができる。また、例えば現像液として0.1〜5.0重量%のテトラメチルアンモニウムヒドロキシド(TMAH)水溶液を用いる場合には、露光部のポリイミド前駆体のイミド化率が45〜60%程度であることが好ましい。
【0033】
露光後ベーク(Post Exposure Bake:PEB)時の加熱条件を従来よりも緩和する、すなわち、加熱温度を低減したり、加熱時間を短くすることにより、未露光部のポリイミド前駆体はイミド化が抑制される。また、塩基に導入されている水酸基の効果で、未露光部の現像液への溶解が促進される。これらの相乗効果により、本発明に係る光塩基発生剤を前駆体型感光性ポリイミド樹脂組成物に用いると、未露光部の現像液への溶解性が向上し、未露光部を除去する現像時間を短縮でき、また、アルコール等を添加しない塩基性水溶液を現像液として用いることが可能になる。更に、露光部は十分にイミド化される一方で、未露光部は現像液への溶解性が高いことから、露光部と未露光部の溶解性コントラストが高くなるため、レリーフパターンの形状が良好になる。
【0034】
本発明に係る光塩基発生剤は、発生する塩基が共有結合を用いて潜在化されているものである。発生する塩基が共有結合を用いて潜在化されている光塩基発生剤とは、発生する塩基部分の窒素原子と隣接する原子とが共有結合を有しており、電磁波の照射により当該共有結合が切断されて塩基が発生する光塩基発生剤である。このような構造の光塩基発生剤は中性にすることができるため、溶剤溶解性が良好であり、ポットライフが向上する。また、ポリイミド前駆体と組み合わせて用いられる成分との相溶性が良好である場合が多いというメリットがある。
【0035】
発生する塩基が共有結合を用いて潜在化されている構造を有する光塩基発生剤としては、例えば、上記特許文献5及び6で開示されたような桂皮酸アミド構造を有する塩基発生剤、上記特許文献7及び8で開示されたようなカルバメート構造を有する塩基発生剤、上記特許文献9及び10で開示されたようなオキシム構造、カルバモイルオキシム構造を有する塩基発生剤等が挙げられるが、これらに限定されず、その他にも公知の光塩基発生剤の構造を用いることができる。
【0036】
本発明に係る光塩基発生剤は、水酸基を有する塩基を発生するものである。発生する塩基が水酸基を有することにより、露光後ベーク温度の条件を、従来よりも低温としたり、加熱時間を短くしても、後述するポリイミド前駆体を効率よくイミド化することができる。アミノ基と水酸基を1分子内に有する化合物は、ポリイミドやポリイミド前駆体との親和性が高く、塗膜中でのポリイミド前駆体のイミド化過程において、よりイミド化が進んだ状態まで、擬似的な溶媒兼、触媒としてイミド化を促進するものとして機能するためと推測される。更に、発生する塩基が水酸基を有することにより、未露光部の塩基性水溶液に対する溶解性が向上する傾向がある。これは、光塩基発生剤中に潜在化された塩基に結合された水酸基が、ポリイミド前駆体と相互作用することにより、塩基性水溶液への溶解性を向上させるためと推測される。なお、水酸基は1つ以上有していればよく、発生する塩基の構造は特に限定されない。フェノール性水酸基は弱い酸性基となる点から、水酸基としては、アルコール性水酸基であることが好ましい。なお、アルコール性水酸基とは、脂肪族炭化水素骨格に直接結合する水酸基を意味し、フェノール性水酸基は、芳香族炭化水素骨格に直接結合する水酸基を意味する。
【0037】
また、発生する塩基は、沸点が150℃以上かつ融点が200℃以下の水酸基を有する塩基であることが、ポリイミド前駆体のイミド化促進効果が高くなる点から好ましい。
一般的な構造のポリイミド前駆体がイミド化に要する加熱温度は、140℃以上であり、通常、露光後加熱温度は140℃〜210℃の範囲内で選択される場合が多い。
発生する塩基の沸点が150℃以上であると、露光後加熱工程における当該塩基の揮発が抑えられ、発生した塩基をイミド化促進触媒として効率よく利用することができる。中でも沸点は160℃以上であることが好ましい。また、融点が200℃以下であると、露光後加熱工程において当該塩基が液体であり、固体の場合に比べ、樹脂組成物中を拡散しやすいか、又は、擬似的に溶媒として機能し、ポリイミド前駆体の分子鎖の自由度を向上させることができる。そのため、少量の塩基であっても効率的にポリイミド前駆体のイミド化を促進することができると推測される。中でも融点は190℃以下であることが好ましい。
【0038】
本発明に係る光塩基発生剤は発生する塩基が共有結合を用いて潜在化されていることから、発生する塩基としては、1級アミン、2級アミン、又は複素環式化合物が挙げられる。またアミンには、それぞれ、脂肪族アミン及び芳香族アミンがある。なお、アミンのうち、アミンの窒素原子に少なくとも1つの芳香族基が直接結合している場合を芳香族アミンといい、それ以外の場合は脂肪族アミンに含まれる。また、ここでの複素環式化合物は、塩基が環状構造を有し且つ芳香族性を有しているものをいう。芳香族複素環式化合物ではない、非芳香族複素環式化合物は、ここでは脂環式アミンとして脂肪族アミンに含まれる。
【0039】
更に、発生する塩基は、アミド結合を形成可能なNH基を1つだけ有するモノアミン等の塩基だけでなく、ジアミン、トリアミン、テトラアミン等のアミド結合を形成可能なNH基を2つ以上有する塩基であってもよい。中でも、アミド結合を形成可能なNH基を1つ有するものであることが、塩基の発生効率、溶剤溶解性、合成上の精製の点から好ましい。
以下に発生する塩基の一例を示すが、これに限定されるものではない。
【0040】
脂肪族1級アミンとしては、エタノールアミン、3−アミノ−1−プロパノール、1−アミノ−2−プロパノール、2−アミノ−1−プロパノール、4−アミノ−1−ブタノール、2−アミノ−1−ブタノール、1−アミノ−2−ブタノール、3−アミノ−2,2−ジメチル−1−プロパノール、4−アミノ−2−メチル−1−ブタノール、バリノール、3−アミノ−1,2−プロパンジオール、2−アミノ−1,3−プロパンジオール、チラミン、ノルエフェドリン、2−アミノ−1−フェニル−1,3−プロパンジオール、2−アミノシクロヘキサノール、4−アミノシクロヘキサノール、4−アミノシクロヘキサンエタノール、4−(2−アミノエチル)シクロヘキサノール等が挙げられる。
【0041】
脂肪族2級アミンとしては、N−メチルエタノールアミン、3−(メチルアミノ)−1−プロパノール、3−(イソプロピルアミノ)プロパノール、N−シクロヘキシルエタノールアミン、α−[2−(メチルアミノ)エチル]ベンジルアルコール、ジエタノールアミン、ジイソプロパノールアミン、3−ピロリジノール、2−ピロリジンメタノール、4−ヒドロキシピペリジン、3−ヒドロキシピペリジン、4−ヒドロキシ−4−フェニルピペリジン、4−(3−ヒドロキシフェニル)ピペリジン、4−ピペリジンメタノール、3−ピペリジンメタノール、2−ピペリジンメタノール、4−ピペリジンエタノール、2−ピペリジンエタノール、2−(4−ピペリジル)−2−プロパノール等が挙げられ、中でも脂環式アミンが好ましい。
【0042】
芳香族1級アミンとしては、4−アミノフェノール、3−アミノフェノール、2−アミノフェノール、4−アミノベンジルアルコール、3−アミノベンジルアルコール、2−アミノベンジルアルコール、2−(4−アミノフェニル)エタノール等が挙げられる。
【0043】
芳香族2級アミンとしては、2−アニリノエタノール、3−アニリノ−1−プロパノール、4−ヒドロキシジフェニルアミン、3−ヒドロキシジフェニルアミン、3,3’−ジヒドロキシジフェニルアミン、4−ベンジルアミノフェノール等が挙げられる。また、アミド結合を形成可能なNH基を有する芳香族複素環式化合物としては、塩基性の点から分子内にイミノ結合(−N=C(−R)−、−C(=NR)−、ここでRは水素原子又は有機基)を有することが好ましく、イミダゾール、プリン、トリアゾールに水酸基が置換された誘導体が挙げられる。これらの中でも、塩基の中の塩基性部位がすべて中性に潜在化されている光塩基発生剤が好ましい。
【0044】
ポリイミド前駆体から最終生成物への反応に対する反応開始温度を低下させる触媒作用は、塩基性の大きい塩基の方が触媒としての効果が大きく、より少量の添加で、より低い温度での最終生成物への反応が可能となる。一般に芳香族アミンよりも脂肪族アミンの方が塩基性が強いため、その触媒能が高い。従って、本発明に係る光塩基発生剤においては、発生する塩基が脂肪族アミンであることが好ましい。また、発生する塩基が脂肪族アミンである場合には、イミド化終了後に、速やかに揮発しやすく、同じ加熱条件によってイミド化された場合、塩基性芳香族へテロ環化合物やアミジン類に比べ、塗膜中への残存が少ない点からも好ましい。
【0045】
また、本発明で発生する塩基が、2級アミン及び/又は複素環式化合物である場合には、光塩基発生剤としての感度が高くなる点から好ましい。これは、2級アミンや複素環式化合物を用いることで、アミド結合部位の活性水素がなくなり、このことにより、電子密度が変化し、異性化の感度が向上するからではないかと推定される。
【0046】
さらに、本発明に係る光塩基発生剤から発生する塩基においては、酸性基を含まないことが、発生した塩基の触媒能を低下させない点から好ましい。但し、フェノール性水酸基のような弱い酸性基を含む塩基を発生する光塩基発生剤は、塩基の触媒能を低下させないことから、本発明の光塩基発生剤において用いられる場合がある。酸性基としては、カルボキシル基、スルホ基、及びリン酸基等の強い酸性基を含まないことが好ましい。
【0047】
本発明に係る光塩基発生剤においては、下記化学式(1−1)、下記化学式(1−2)又は下記化学式(1−3)で表される光塩基発生剤であることが、塩基の存在下での加熱によって最終生成物への反応が促進される様々なポリイミド前駆体に対して利用可能である点から好ましい。中でも、優れた感度を有し、ポリイミド前駆体の種類を問わず、形状が良好なパターンを得ることができる点から、下記化学式(1−1)で表される光塩基発生剤であることが好ましい。
【0048】
【化4】
(式(1−1)中、R1及びR2は、それぞれ独立に、水素原子又は有機基であり、同一であっても異なっていても良い。R1及びR2の少なくとも1つは有機基である。R1及びR2は、それらが結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。但し、R1及びR2のいずれかの有機基が水酸基を有する。R3及びR4はそれぞれ独立に、水素原子、ハロゲン原子、水酸基、メルカプト基、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、又は有機基であり、同一であっても異なっていても良い。R5、R6、R7及びR8は、それぞれ独立に、水素原子、ハロゲン原子、水酸基、メルカプト基、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、アミノ基、アンモニオ基又は有機基であり、同一であっても異なっていても良い。R5、R6、R7及びR8は、それらの2つ以上が結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。R9は、水素原子、或いは、加熱及び/又は電磁波の照射により脱保護可能な保護基である。)
【0049】
【化5】
(式(1−2)中、R1及びR2は、それぞれ独立に、水素原子又は有機基であり、同一であっても異なっていても良い。R1及びR2の少なくとも1つは有機基である。R1及びR2は、それらが結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。但し、R1及びR2のいずれかの有機基が水酸基を有する。R10及びR10’はそれぞれ独立に水素原子、ハロゲン原子、水酸基、メルカプト基、ニトロ基、シリル基、シラノール基、又は有機基である。R11、R12、R13、R14及びR15は、それぞれ独立に、水素原子、ハロゲン原子、水酸基、メルカプト基、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、アミノ基、アンモニオ基又は有機基であり、同一であっても異なっていても良い。R11、R12、R13、R14及びR15は、それらの2つ以上が結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。)
【0050】
【化6】
(式(1−3)中、R17は、水酸基を有する有機基で置換されたアミノ基又は水酸基を有する有機基、R18及びR19は、それぞれ独立に、水素原子、ハロゲン原子、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、アミノ基、アンモニオ基又は有機基であり、同一であっても異なっていても良いが、R18及びR19の少なくとも1つはヘテロ原子を有していても良い芳香族化合物である。R17、R18及びR19は、それらの2つ以上が結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。)
【0051】
上記化学式(1−1)で表される光塩基発生剤は、電磁波が照射されることにより、下記式で示されるように、式(1−1)で中の(−CR4=CR3−C(=O)−)部分がトランス体からシス体へと異性化する。さらに加熱及び/又は電磁波の照射によって、R9が保護基である場合は保護基R9が脱保護されると共に環化し、塩基(NHR1R2)を生成する。式(1−1)で表される光塩基発生剤は電磁波の照射のみでも塩基を発生するが、適宜加熱することにより塩基の発生が促進される。
【0052】
【化7】
【0053】
また、上記化学式(1−2)で表される光塩基発生剤は、電磁波を照射するとカルバメート結合の光脱炭酸反応によって結合が開裂し、塩基(NHR1R2)を発生させる。
更に、上記化学式(1−3)で表される光塩基発生剤は、オキシムエステル誘導体であって、下記反応式に従って、電磁波の吸収により分子内解裂反応を起こし、アミンまたはヒドラジンを発生させる。
【0054】
【化8】
【0055】
上記化学式(1−1)及び化学式(1−2)において、R1及びR2は、それぞれ、独立に水素原子又は有機基であるが、R1及びR2のうち少なくとも1つは有機基である。また、NHR1R2は、塩基であるが、R1及びR2は、それぞれ、アミノ基を含まない有機基であることが好ましい。R1及びR2に、アミノ基が含まれてしまうと、光塩基発生剤自体が塩基となり、ポリイミド前駆体の反応を促進してしまい、露光部と未露光部での溶解性コントラストの差が小さくなってしまう恐れがある。但し、例えば、R1及びR2の有機基中に存在する芳香環にアミノ基が結合している場合のように、電磁波の照射と加熱後に発生する塩基との塩基性と差が生じる場合には、R1及びR2の有機基にアミノ基を含まれていても用いることができる場合もある。
有機基としては、例えば、飽和又は不飽和アルキル基、飽和又は不飽和シクロアルキル基、アリール基、アラルキル基、及び飽和又は不飽和ハロゲン化アルキル基等が挙げられる。これらの有機基は、当該有機基中にヘテロ原子等の炭化水素基以外の結合や置換基を含んでよく、これらは、直鎖状でも分岐状でも環状でも良い。
有機基としては、置換基を含んで良く、不飽和結合を含んで良く、ヘテロ原子の結合を含んで良い、直鎖、分岐又は環状の炭化水素基が好ましい。
R1及びR2における有機基は、通常、1価の有機基であるが、後述する環状構造を形成する場合や、生成するNHR1R2がジアミン等のアミド結合を形成可能なNH基を2つ以上有する塩基の場合等には、2価以上の有機基となり得る。
R1及びR2のいずれかの有機基が水酸基を有する。有機基中に水酸基を1つ以上有すれば有機基内のどこに存在していてもよい。R1及びR2が環状構造を形成する場合には、当該環状構造内に水酸基を1つ以上有すればよい。R1及びR2のいずれかの有機基が水酸基を有することにより、光塩基発生剤は水酸基を有する塩基を発生する。塩基が水酸基を有することにより、触媒能が向上し、露光後ベーク時の加熱条件を緩和することができ、例えばより低温であっても後述するポリイミド前駆体を効率よくイミド化することができる。
【0056】
また、R1及びR2は、それらが結合して環状構造になっていても良い。
環状構造は、飽和又は不飽和の脂環式炭化水素、複素環、及び縮合環、並びに当該脂環式炭化水素、複素環、及び縮合環よりなる群から選ばれる2種以上が組み合されてなる構造であっても良い。
【0057】
前記R1及びR2の有機基中の炭化水素基以外の結合としては、本発明の効果が損なわれない限り、特に限定されず、エーテル結合、チオエーテル結合、カルボニル結合、チオカルボニル結合、エステル結合、アミド結合、ウレタン結合、イミノ結合(−N=C(−R)−、−C(=NR)−、ここでRは水素原子又は有機基)、カーボネート結合、スルホニル結合、スルフィニル結合、アゾ結合等が挙げられる。耐熱性の点から、有機基中の炭化水素基以外の結合としては、エーテル結合、チオエーテル結合、カルボニル結合、チオカルボニル結合、エステル結合、アミド結合、ウレタン結合、イミノ結合(−N=C(−R)−、−C(=NR)−:ここでRは水素原子又は有機基)、カーボネート結合、スルホニル結合、スルフィニル結合が好ましい。
【0058】
前記R1及びR2の有機基中の炭化水素基以外の置換基、すなわち、有機基に包含される置換基において炭化水素基とは異なる置換基、及び炭化水素基に置換されていても良い置換基としては、本発明の効果が損なわれない限り、特に限定されず、ハロゲン原子、メルカプト基(−SH)、スルフィド基(−SR、ここでRは有機基)、シアノ基(−CN)、イソシアノ基(−NC)、シアナト基(−OCN)、イソシアナト基(−NCO)、チオシアナト基(−SCN)、イソチオシアナト基(−NCS)、アルキルエーテル基及びアリールエーテル基(−OR、ここでRは有機基)、アルコキシカルボニル基及びアリールオキシカルボニル基(−COOR、ここでRは有機基)、アルキルスルフィニル基及びアリールスルフィニル基(−SOR、ここでRは有機基)、アルキルスルホニル基及びアリールスルホニル基(−SO2R、ここでRは有機基)、アルコキシスルホニル基及びアリールオキシスルホニル基(−SO3R、ここでRは有機基)、カルバモイル基(−CONRR’、ここでR、R’は有機基)、チオカルバモイル基(−CSNRR’、ここでR、R’は有機基)、スルファモイル基(−SO3NRR’、ここでR、R’は有機基)、ニトロ基(−NO2)、ニトロソ基(−NO)、アシル基(−COR、ここでRは有機基)、アシルオキシ基(−OCOR、ここでRは有機基)、アシルアミノ基(−NHCOR、ここでRは有機基)、カーバメート基(−NHCOOR、ここでRは有機基)、ホスフィノ基(−PR2)、シリル基(−SiR3、ここでRは有機基)、アルコキシシリル基及びアリールオキシシリル基(−Si(OR)XR’Y、ここでR、R’は有機基。Xは1から3の整数、Yは0から2の整数で、X+Y=3。)、アミノ基(−NH2, −NHR, −NRR’:ここで、R及びR’はそれぞれ独立に炭化水素基)等が挙げられる。上記置換基に含まれる水素原子は、炭化水素基によって置換されていても良い。また、上記置換基に含まれる炭化水素基は、直鎖、分岐、及び環状のいずれでも良い。
前記R1及びR2の有機基中の炭化水素基及び水酸基とは異なる置換基としては、ハロゲン原子、メルカプト基(−SH)、スルフィド基(−SR、ここでRは有機基)、シアノ基(−CN)、イソシアノ基(−NC)、シアナト基(−OCN)、イソシアナト基(−NCO)、チオシアナト基(−SCN)、イソチオシアナト基(−NCS)、アルキルエーテル基及びアリールエーテル基(−OR、ここでRは有機基)、アルコキシカルボニル基及びアリールオキシカルボニル基(−COOR、ここでRは有機基)、スルフィニル基(−SOR、ここでRは有機基)、アルキルスルホニル基及びアリールスルホニル基(−SO2R、ここでRは有機基)、アルコキシスルホニル基及びアリールオキシスルホニル基(−SO3R、ここでRは有機基)、カルバモイル基(−CONRR’、ここでR、R’は有機基)、チオカルバモイル基(−CSNRR’、ここでR、R’は有機基)、スルファモイル基(−SO3NRR’、ここでR、R’は有機基)、ニトロ基(−NO2)、ニトロソ基(−NO)、アシル基(−COR、ここでRは有機基)、アシルオキシ基(−OCOR、ここでRは有機基)、アシルアミノ基(−NHCOR、ここでRは有機基)、カーバメート基(−NHCOOR、ここでRは有機基)シリル基(−SiR3、ここでRは有機基)、アルコキシシリル基及びアリールオキシシリル基(−Si(OR)XR’Y、ここでR、R’は有機基。Xは1から3の整数、Yは0から2の整数で、X+Y=3。)が好ましい。
なお、上記炭化水素基以外の結合や上記炭化水素基以外の置換基中の水素原子又は有機基が、水酸基に置換された構造であっても良い。
【0059】
また、発生する塩基の熱物性、及び塩基性度の点から、R1及びR2の有機基は、それぞれ独立に炭素数1〜20が好ましく、更に炭素数1〜12が好ましく、特に炭素数1〜8であることが好ましい。
発生する塩基はNHR1R2であるため、1級アミン、2級アミン、又は複素環式化合物が挙げられる。発生する塩基の好適な例としては、上述したのでここでの説明を省略する。
【0060】
また、化学式(1−1)において、R3及びR4は、それぞれ独立に、水素原子、ハロゲン原子、水酸基、メルカプト基、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、又は有機基であり、同一であっても異なっていても良い。
R3及びR4としては、高感度を達成しやすい点から、いずれも水素原子であることが好ましい。
【0061】
本発明において、特に化学式(1−1)中のR3及びR4のうち少なくとも1つが、水素原子ではなく、上記特定の官能基である場合には、R3及びR4の両方共が水素原子の場合と比べて、本発明の光塩基発生剤は、有機溶剤に対する溶解性を更に向上させたり、ポリイミド前駆体との親和性を向上させることが可能である。例えば、R3及びR4のうち少なくとも1つが、アルキル基やアリール基等の有機基である場合、有機溶剤に対する溶解性が向上する。また、例えばR3及びR4のうち少なくとも1つがフッ素等のハロゲン原子である場合、フッ素等のハロゲン原子を含有するポリイミド前駆体との親和性が向上する。このように、R3及び/又はR4を所望の有機溶剤やポリイミド前駆体に合わせて適宜置換基を導入することにより、所望の有機溶剤に対する溶解性や、所望のポリイミド前駆体との親和性を向上することが可能である。
【0062】
ハロゲン原子、有機基としては、本発明の効果が損なわれない限り、特に制限がなく、後述するR5〜R8、R11〜R15に挙げたものと同様のものを用いることができる。
R3及びR4における有機基は、通常、1価の有機基である。
【0063】
R3及びR4が、置換基を有する場合には少なくとも一方が、メチル基、エチル基、プロピル基等の炭素数1〜20のアルキル基;シクロペンチル基、シクロヘキシル基等の炭素数4〜23のシクロアルキル基;シクロペンテニル基、シクロヘキセニル基等の炭素数4〜23のシクロアルケニル基;フェノキシメチル基、2−フェノキシエチル基、4−フェノキシブチル基等の炭素数7〜26のアリールオキシアルキル基(−ROAr基);ベンジル基、3−フェニルプロピル基等の炭素数7〜20のアラルキル基;シアノメチル基、β−シアノエチル基等のシアノ基をもつ炭素数2〜21のアルキル基;ヒドロキシメチル基等の水酸基をもつ炭素数1〜20のアルキル基、メトキシ基、エトキシ基等の炭素数1〜20のアルキルエーテル基、アセトアミド基、ベンゼンスルホナミド基(C6H5SO2NH2−)等の炭素数2〜21のアミド基、メチルチオ基、エチルチオ基等の炭素数1〜20のアルキルチオ基のようなスルフィド基(−SR基)、アセチル基、ベンゾイル基等の炭素数1〜20のアシル基、メトキシカルボニル基、アセトキシ基等の炭素数2〜21のエステル基(−COOR基及び−OCOR基)、フェニル基、ナフチル基、ビフェニル基、トリル基等の炭素数6〜20のアリール基、電子供与性基及び/又は電子吸引性基が置換した炭素数6〜20のアリール基、電子供与性基及び/又は電子吸引性基が置換したベンジル基、シアノ基であることが好ましい。また、上記のアルキル部分は直鎖でも分岐状でも環状でも良い。
【0064】
また、化学式(1−2)において、R10及びR10’はそれぞれ独立に水素原子、ハロゲン原子、水酸基、メルカプト基、ニトロ基、シリル基、シラノール基、又は有機基であるが、当該有機基としては、後述するR5〜R8、R11〜R15に挙げたものと同様のものを用いることができる。
【0065】
また、上記化学式(1−1)のR5〜R8、並びに、上記化学式(1−2)のR11〜R15は、それぞれ独立に、水素原子、ハロゲン原子、水酸基、メルカプト基、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、アミノ基、アンモニオ基又は有機基であり、同一であっても異なっていても良い。R5〜R8及びR11〜R15は、それぞれ、それらの2つ以上が結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。
【0066】
上記化学式(1−1)のR5〜R8、並びに、上記化学式(1−2)のR11〜R15には、置換基を1つ以上導入することが好ましい。特に上記化学式(1−1)で表される塩基発生剤の場合、カルボニル結合のα位およびβ位に位置するα炭素−β炭素間の二重結合がトランス体からシス体への異性化反応を効率よく進める要因としてはいくつかあり、例えば上記炭素−炭素二重結合周囲の立体障害の大きさ、上記炭素−炭素二重結合周囲に広がる共役鎖の電子状態等が挙げられるが、置換基R5〜R8に、上記のような置換基を少なくとも1つ導入することにより、上記炭素−炭素二重結合周囲の共役鎖が拡張し、塩基発生の感度を向上することができる。また、R5〜R8及びR11〜R15において、上記のような置換基を少なくとも1つ導入することにより、吸収する光の波長を調整することが可能であり、置換基を導入することで所望の波長を吸収させるようにすることもできる。芳香族環の共役鎖を伸ばすような置換基を導入することにより、吸収波長を長波長にシフトすることができる。また、溶解性や組み合わせるポリイミド前駆体との相溶性が向上するようにすることもできる。これにより、組み合わせるポリイミド前駆体の吸収波長も考慮しながら、感光性ポリイミド樹脂組成物の感度を向上させることが可能である。
【0067】
所望の波長に対して吸収波長をシフトさせる為に、どのような置換基を導入したら良いかという指針として、Interpretation of the Ultraviolet Spectra of Natural Products(A.I.Scott 1964)や、有機化合物のスペクトルによる同定法第5版(R.M.Silverstein 1993)に記載の表を参考にすることができる。これらを参考とすることで、化合物の極大吸収波長がどの程度長波長化するかの目安を知ることができる。
【0068】
上記化学式(1−2)のR11は、ニトロ基であることが、上記化学式(1−2)で表される光塩基発生剤の感度が向上する点から好ましい。
【0069】
R5〜R8及びR11〜R15において、ハロゲン原子としては、フッ素、塩素、臭素などが挙げられる。
R5〜R8及びR11〜R15において、有機基としては、例えば、飽和又は不飽和アルキル基、飽和又は不飽和シクロアルキル基、アリール基、アラルキル基、及び飽和又は不飽和ハロゲン化アルキル基等が挙げられる。これらの有機基は、当該有機基中にヘテロ原子等の炭化水素基以外の結合や置換基を含んでよく、これらは、直鎖状でも分岐状でも環状でも良い。R5〜R8及びR11〜R15の有機基中の炭化水素基以外の結合としては、前記R1及びR2の炭化水素基以外の結合と同様のものを用いることができる。また、R5〜R8及びR11〜R15の有機基は、炭化水素基以外の結合を介してベンゼン環に結合してもよい。また、R5〜R8及びR11〜R15の有機基において炭化水素基以外の置換基(有機基に包含される置換基において炭化水素基とは異なる置換基、及び炭化水素基に置換されていても良い置換基)としては、前記R1及びR2の炭化水素基以外の置換基と同様のものを用いることができる。
有機基としては、本発明の効果が損なわれない限り、特に制限がなく、飽和又は不飽和アルキル基、飽和又は不飽和シクロアルキル基、アリール基、アラルキル基、及び飽和又は不飽和ハロゲン化アルキル基、シアノ基、イソシアノ基、シアナト基、イソシアナト基、チオシアナト基、イソチオシアナト基、アルコキシカルボニル基、カルバモイル基、チオカルバモイル基、カルボキシル基、カルボキシラート基、アシル基、アシルオキシ基、ヒドロキシイミノ基、飽和又は不飽和アルキルエーテル基、飽和又は不飽和アルキルチオ基、アリールエーテル基、及びアリールチオ基等が挙げられる。
R5〜R8及びR11〜R15における有機基は、通常、1価の有機基であるが、後述する環状構造を形成する場合等には、2価以上の有機基となり得る。
【0070】
また、R5〜R8及びR11〜R15は、それらのうち2つ以上が結合して環状構造になっていても良い。
環状構造は、飽和又は不飽和の脂環式炭化水素、複素環、及び縮合環、並びに当該脂環式炭化水素、複素環、及び縮合環よりなる群から選ばれる2種以上が組み合されてなる構造であっても良い。例えば、R5〜R8及びR11〜R15のそれぞれは、それらの2つ以上が結合して、R5〜R8及びR11〜R15のそれぞれが結合しているベンゼン環の原子を共有してナフタレン、アントラセン、フェナントレン、インデン等の縮合環を形成していても良い。
【0071】
R5〜R8又はR11〜R15としては、水素原子、ハロゲン原子、水酸基、メルカプト基、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、アミノ基、アンモニオ基、メチル基、エチル基、プロピル基等の炭素数1〜20のアルキル基;シクロペンチル基、シクロヘキシル基等の炭素数4〜23のシクロアルキル基;シクロペンテニル基、シクロヘキセニル基等の炭素数4〜23のシクロアルケニル基;フェノキシメチル基、2−フェノキシエチル基、4−フェノキシブチル基等の炭素数7〜26のアリールオキシアルキル基(−ROAr基);ベンジル基、3−フェニルプロピル基等の炭素数7〜20のアラルキル基;シアノメチル基、β−シアノエチル基等のシアノ基をもつ炭素数2〜21のアルキル基;ヒドロキシメチル基等の水酸基をもつ炭素数1〜20のアルキル基、メトキシ基、エトキシ基、ベンジルオキシ基等の炭素数1〜20のアリール基で置換されていても良いアルキルエーテル基;フェノキシ基、ナフチルオキシ基等のアリールエーテル基;アセトアミド基、ベンゼンスルホナミド基(C6H5SO2NH2−)等の炭素数2〜21のアミド基、メチルチオ基、エチルチオ基等の炭素数1〜20のアルキルチオ基(−SR基)、ベンジルチオ基、ナフチルチオ基等のアリールチオ基;アセチル基、ベンゾイル基等の炭素数1〜20のアシル基;チオアシル基;アセチルチオ基、ベンゾイルチオ基等のアシルチオ基;メトキシカルボニル基、アセトキシ基、ベンジルオキシカルボニル基等の炭素数2〜21のエステル基(−COOR基及び−OCOR基)、フェニル基、ナフチル基、ビフェニル基、トリル基等の炭素数6〜20のアリール基、及び、電子供与性基及び/又は電子吸引性基が置換した炭素数6〜20のアリール基、電子供与性基及び/又は電子吸引性基が置換したベンジル基、シアノ基、カルバモイル基、カルバモイルオキシ基、シアノオキシ基(シアナト基)、シアノチオ基(チオシアナト基)、ホルミル基、であることが好ましい。また、上記のアルキル部分は直鎖でも分岐状でも環状でも良い。
また、R5〜R8又はR11〜R15としては、それらの2つ以上が結合して、R5〜R8又はR11〜R15が結合しているベンゼン環の原子を共有してナフタレン、アントラセン、フェナントレン、インデン等の縮合環を形成している場合も、吸収波長が長波長化する点から好ましい。
【0072】
また、式(1−1)においてR9は、水素原子、或いは、加熱及び/又は電磁波の照射により脱保護可能な保護基である。ここで、“脱保護可能な”とは、−OR9から−OHに変化する可能性があることを表す。R9が水素原子の場合には、本発明に係る光塩基発生剤は、環化することで、フェノール性水酸基を消失し、溶解性が変化し、塩基性水溶液等の場合には溶解性が低下する。これにより、ポリイミド前駆体の最終生成物への反応による溶解性の低下を更に補助する機能を有し、露光部と未露光部の溶解性コントラストを更に大きくすることが可能となる。
【0073】
また、R9が加熱及び/又は電磁波の照射により脱保護可能な保護基である場合、加熱及び/又は電磁波の照射により脱保護されて、水酸基を生成する。加熱及び/又は電磁波の照射により脱保護可能な保護基でフェノール性水酸基を保護することにより、当該保護基を適宜選択することによって、ポリイミド前駆体との相溶性が向上し、組み合わせ可能な化合物の範囲が増える。例えば、フェノール性水酸基と共存することが好ましくないポリイミド前駆体に対しても、感光性ポリイミド樹脂組成物中に共存させて用いることが可能になる。R9は、本発明で用いられる光塩基発生剤において式(1−1)中に存在するアミド基が分解しない条件下で、加熱及び/又は電磁波の照射により脱保護可能なフェノール性水酸基の保護基であれば、特に限定されず用いることができる。例えば、アミド結合は、三臭化ホウ素や三塩化アルミニウム等の強ルイス酸や硫酸、塩酸、硝酸等の強酸等が存在する強酸性下における加熱や、水酸化ナトリウム等の強塩基が存在する強塩基性下における加熱により分解する。従って、このような強酸性又は強塩基性条件下での加熱でしか脱保護されない保護基は、本発明の光塩基発生剤に用いられる保護基としては不適切である。R9は、溶解性や相溶性の向上或いは合成時の反応性の変化などを目的として、当該光塩基発生剤と組み合わせて用いられる化合物の種類や、光塩基発生剤の適用方法や合成方法により適宜選択されるものである。
【0074】
R9としては、シリル基、シラノール基、ホスフィノ基、ホスフィニル基、ホスホノ基、又は有機基から選択することができる。R9における有機基は、通常、1価の有機基である。
【0075】
R9としては、下記式(2−1)〜下記式(2−6)で表わされる有機基よりなる群から選択される1種以上であることが、式(1−1)中に存在するアミド基が分解しない条件下で、加熱及び/又は電磁波の照射により脱保護可能な点から好ましい。
【0076】
【化9】
(式(2−1)中、R30、R31、R32はそれぞれ独立に水素原子、ハロゲン原子、または有機基であり、R33は有機基であり、R30、R31、R32、R33はそれぞれ互いに結合して環状構造を示していても良い。式(2−2)中、R34は、有機基である。式(2−3)中、R35、R36、R37はそれぞれ独立に水素原子、ハロゲン原子、または有機基である。式(2−4)中、R38は、有機基である。式(2−5)中、R39は、置換基を有していても良い芳香環である。式(2−6)中、R40は、有機基である。)
【0077】
上記式(2−1)で表される有機基は、水酸基と、各種ビニルエーテル化合物との反応により得ることができる。式(2−1)で表される有機基は、例えば、各種ビニルエーテル化合物の残基である。
上記式(2−2)で表される有機基は、例えば、水酸基と、カーボネート系保護基の導入試薬(たとえばジ−t−ブチルジカルボナートや、塩化ベンジルオキシカルボニル、N−(9−フルオレニルメトキシカルボニルオキシ)コハク酸イミドなど)との反応により得ることができる。式(2−2)で表される有機基は、例えば、各種カーボネート系保護基の残基である。
上記式(2−3)で表される有機基は、例えば、水酸基と、シリルエーテル系保護基の導入試薬(たとえばクロロトリメチルシラン、tert−ブチルジメチルクロロシラン、tert−ブチルジフェニルクロロシランなど)との反応により得ることができる。式(2−3)で表される有機基は、例えば、シリルエーテル系保護基の残基である。
【0078】
上記式(2−4)で表される有機基は、例えば、水酸基と、酸塩化物または酸無水物との反応により得ることができ、式(2−4)で表される有機基は、酸塩化物または酸無水物の残基である。
上記式(2−5)で表される有機基は、例えば、Williamson反応を用いて、水酸基と、ハロゲン化物(たとえばベンジルクロライドなど)により得ることができ、式(2−5)で表される有機基は、ハロゲン化物の残基である。
上記式(2−6)で表される有機基は、例えば、水酸基と、イソシアネート化合物(たとえばベンジルイソシアネートなど)との反応により得ることができ、式(2−6)で表される有機基は、イソシアネート化合物の残基である。
【0079】
上記化学式(1−1)で表される構造は、幾何異性体が存在するが、トランス体のみを用いることが好ましい。しかし、合成および精製工程および保管時などにおいて幾何異性体であるシス体が混ざる可能性もあり、この場合トランス体とシス体の混合物を用いても良いが、溶解性コントラストを高められる点から、シス体の割合が10%未満であることが好ましい。
【0080】
上記化学式(1−3)中、R17は、水酸基を有する有機基で置換されたアミノ基又は水酸基を有する有機基である。R17が、水酸基を有する有機基である場合、R5〜R8、R11〜R15に挙げた有機基と同様の有機基に適宜水酸基が置換された構造を用いることができる。
上記化学式(1−3)のR17が、水酸基を有する有機基で置換されたアミノ基である場合、下記式(1−3’)で表されるカルバモイルオキシム構造を有し、上述のように、アミン及びヒドラジンを発生し得る。
【0081】
【化10】
(式(1−3’)中、R18及びR19は、化学式(1−3)と同じであり、R20及びR21は、上記化学式(1−1)のR1及びR2と同じである。R18、R19、R20及びR21は、それらの2つ以上が結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。但し、R20及びR21のいずれかの有機基が水酸基を有する。)
【0082】
上記化学式(1−3)及び式(1−3’)中、R18及びR19の少なくとも1つはヘテロ原子を有していても良い芳香族化合物であることが特徴的である。R18及びR19の少なくとも1つに芳香族化合物が置換している状態とは、R18及びR19が結合している炭素に芳香族化合物が直接共有結合で結合している状態のことをいう。なお、ここでの芳香族化合物は、環状不飽和有機化合物をいい、芳香族炭化水素と芳香族複素環式化合物が含まれる。芳香族化合物としては、例えば、フェニル基、ビフェニル基、ターフェニル基、ナフチル基、フルオレン基、アントラニル基、フェナントレニル基、ピレニル基等の芳香族炭化水素基の他、フラニル基、チオフェニル基、ピロリル基、(チオ)キサンテニル基、(チオ)キサントニル基、クマリル基、アントラキノリル基等の複素芳香族化合物等が挙げられるが特に限定されない。これらが有していても良い置換基としては、上述したR5〜R8及びR11〜R15と同様であって良い。R18及びR19の一方が、芳香族化合物以外の有機基である場合も、上述したR5〜R8及びR11〜R15と同様であって良い。
【0083】
上記化学式(1−3)及び式(1−3’)中、R17、R18、R19、R20及びR21は、それらの2つ以上が結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。R18及びR19が結合して環状構造を形成している場合としては、例えば、R18及びR19が結合している炭素と共に、フルオレン環を形成している態様が挙げられる。また、R18及びR17やR18及びR20が結合して環状構造を形成している場合としては、例えば、R18及びR17やR18及びR20が脂肪族炭化水素基及び/又は芳香族炭化水素基で連結されている態様が挙げられる。
発生する塩基が水酸基を有するため、R20及びR21のいずれかの有機基は水酸基を有する。R20及びR21は、上記化学式(1−1)のR1及びR2と同じであるため、ここでの説明を省略する。
以下に本発明の光塩基発生剤の一例を示すが、これらに限定されるものではない。
【0084】
【化11】
【0085】
【化12】
【0086】
【化13】
【0087】
本発明に係る光塩基発生剤は、加熱して初期の重量から5%重量が減少したときの温度(5%重量減少温度)が、60℃以上であることが好ましく、更に100℃以上であることが好ましい。ポリイミド前駆体の場合、塗膜を形成する際にN−メチル−2−ピロリドンなどの高沸点溶媒を用いる必要があるが、このように5%重量減少温度が高い場合には残留溶媒の影響が少なくなるような乾燥条件で塗膜を形成することができる。これにより、残留溶媒の影響による露光部と未露光部での溶解性コントラストの減少を抑制することができる。
本発明において、x%重量減少温度とは、熱重量分析装置を用いて重量減少を測定した時に、サンプルの重量が初期重量からx%減少した時点(すなわち、サンプル重量が初期の(100−x)%となった時点)の温度である。
【0088】
本発明の光塩基発生剤は、複数の従来公知の合成ルートで合成することができる。上記化学式(1−1)で表される光塩基発生剤は、例えば、上記特許文献5や本発明者らによる特願2010−176384を参考に合成することができる。上記化学式(1−1)で表される光塩基発生剤において、フェノール性水酸基における保護基(R9)の導入は、合成途中で導入していても良いし、合成の最後に導入しても良い。また、上記化学式(1−2)で表される光塩基発生剤は、例えば、上記特許文献6及び7を参考に合成することができる。また、上記化学式(1−3)で表される光塩基発生剤は、例えば、上記特許文献8及び9を参考に合成することができる。
【0089】
本発明の光塩基発生剤は、ポリイミド前駆体が最終生成物となるための塩基発生の機能を十分に発揮させるために、露光波長の少なくとも一部に対して吸収を有する必要がある。一般的な露光光源である高圧水銀灯の波長としては、365nm、405nm、436nmがある。このため、本発明で用いられる光塩基発生剤は、少なくとも365nm、405nm、436nmの波長の電磁波のうち少なくとも1つの波長の電磁波に対して吸収を有することが好ましい。このような場合、適用可能なポリイミド前駆体の種類がさらに増える点から好ましい。
【0090】
本発明の光塩基発生剤は、モル吸光係数が、電磁波の波長365nmにおいて100以上、又は405nmにおいて1以上であることが、適用可能なポリイミド前駆体の種類がさらに増える点から好ましい。
【0091】
なお、本発明の光塩基発生剤が前記波長領域に吸収を有することは、当該波長領域に吸収をもたない溶媒(例えば、アセトニトリル)に、本発明の光塩基発生剤を1×10−4mol/L以下の濃度(通常、1×10−4mol/L〜1×10−5mol/L程度。適度な吸収強度となるように、適宜、調節してもよい。)で溶解し、紫外可視分光光度計(例えば、UV−2550(株)島津製作所製))により吸光度を測定することにより明らかにすることができる。
【0092】
本発明の光塩基発生剤は、後述するポリイミド前駆体と組み合わせて感光性ポリイミド樹脂組成物を調製するための前駆体型感光性ポリイミド用光塩基発生剤として好適に用いられる。しかしながら、本発明の光塩基発生剤は、塩基によって又は塩基の存在下での加熱によって最終生成物への反応が促進される他の高分子前駆体と組み合わせて感光性樹脂組成物を調製することもできる。
【0093】
<感光性ポリイミド樹脂組成物>
本発明に係るネガ型感光性ポリイミド樹脂組成物は、ポリイミド前駆体と、前記本発明に係る光塩基発生剤とを含有することを特徴とする。
本発明に係るネガ型感光性ポリイミド樹脂組成物は、露光されると前記光塩基発生剤から塩基が発生し、当該塩基の触媒作用により、露光部は露光後ベーク時におけるポリイミド前駆体のイミド化が未露光部に比べ促進され、より低温でイミド化を進行させることができる。本発明のネガ型感光性ポリイミド樹脂組成物を用いて、パターンを得るには、パターンを残したい場所に露光後、塩基が存在する場所ではイミド化が進行し、塩基の存在していない場所ではイミド化が進行し難い温度で、加熱を行う。これにより、露光部の現像液に対する溶解性が低下し、現像後に露光部分が残る。
【0094】
本発明に係るネガ型感光性ポリイミド樹脂組成物は、前記本発明に係る光塩基発生剤を用いることにより、露光後ベーク時の加熱条件を従来より緩和しても、露光部は十分にイミド化が促進され、未露光部は緩和された加熱条件のためにポリイミド前駆体のイミド化の進行を抑制することができる。また、塩基に導入されている水酸基の効果で、未露光部の現像液への溶解が促進される。緩和された加熱条件と水酸基の溶解促進の相乗効果により、未露光部の現像液への溶解性が向上するため、未露光部を除去する現像時間を短縮でき、また、アルコール等を添加しない塩基性水溶液を現像液として用いることが可能になる。更に、露光部は十分に硬化される一方で、未露光部の現像液への溶解性が高いことから、露光部と未露光部の溶解性コントラストが高くなるため、レリーフパターンの形状が良好になる。
【0095】
以下、本発明に係る感光性ポリイミド樹脂組成物の構成成分を、ポリイミド前駆体、光塩基発生剤、必要に応じて適宜含むことができるその他の成分について順に説明する。
光塩基発生剤及びポリイミド前駆体としては、1種単独で用いても良いし、2種以上混合して用いても良い。
【0096】
(ポリイミド前駆体)
本発明の感光性ポリイミド樹脂組成物に用いるポリイミド前駆体とは、反応により最終的に目的の物性を示すポリイミドとなる物質を意味する。
本発明に用いるポリイミド前駆体は、なんらかの溶媒(有機溶剤、又は水溶液)に可溶なものが好適に用いられる。溶媒(有機溶剤、又は水溶液)に可溶なものであると、ポリイミド前駆体の当該溶媒に対する溶解性を変化させることにより、その可溶な溶媒を現像液として用いて、適宜、有機溶剤、塩基性水溶液、酸性水溶液、又は中性水溶液による現像をすることが可能になる。
ここで、ある溶媒に可溶とは、具体的には、基板上に形成された塗膜の25℃における当該溶媒に対する溶解速度が、10nm/sec以上を目安とする。当該溶解速度は、プロセス適性の観点から30nm/sec以上であることがさらに好ましい。
【0097】
例えば、塩基性水溶液に可溶なものは、具体的には、基板上に形成された塗膜の25℃における0.1重量%のテトラメチルアンモニウムヒドロキシド水溶液に対する溶解速度が、10nm/sec以上である。当該溶解速度は30nm/sec以上であることがさらに好ましい。さらには、より一般的に用いられる現像液である2.38重量%のテトラメチルアンモニウムヒドロキシド水溶液に対する溶解速度が、10nm/sec以上であることが好ましく、30nm/sec以上であることがさらに好ましい。上記定義による溶解速度が10nm/secより小さい場合、現像時間が遅くなり作業性、生産性が悪くなると共に、露光部、未露光部間の溶解性コントラストが得にくくなる。
【0098】
上記溶解速度を測定する具体的手順としては、無アルカリガラス等の基板上に形成されたポリイミド前駆体の塗膜を、25℃に調温され、撹拌された現像液(0.1重量%TMAH水溶液または、2.38重量%TMAH水溶液等の塩基性水溶液、有機溶剤等)に一定時間、浸漬し、蒸留水でリンス後、乾燥させた後で測定した膜厚と、初期膜厚との差を、膜減り量とし、その膜減り量を、現像液に浸漬した時間で割ったものが、25℃における単位時間当たりの溶解速度ということになる。
【0099】
また、露光部と未露光部の間に十分な溶解性コントラストを得るために、感光性ポリイミド樹脂組成物を実際に所定の感光パターン形成プロセスにおいて用いた時に、パターン状露光、及び、加熱工程を行って得られる、現像工程前における未露光部位と露光部位の現像液に対する溶解性の比(未露光部位の現像液に対する単位時間当たりの溶解速度/露光部位の現像液に対する単位時間当たりの溶解速度)が、5以上であることが好ましく、10以上であることが更に好ましい。
単位時間当たりの溶解速度は、上記の方法と同様にして求められ、感光性ポリイミド樹脂組成物の塗膜にパターン露光を行い、露光後の加熱を行った後に、露光部、未露光部の溶解速度を、それぞれ求める。
【0100】
本発明においては、塩基の作用によって最終生成物への反応が促進されるポリイミド前駆体が用いられ、塩基の作用によってポリイミド前駆体の最終生成物への反応温度が、塩基の作用がない場合に比べて低下するようなポリイミド前駆体が用いられる。
本発明に用いられるポリイミド前駆体としては、下記化学式(3)で表される繰り返し単位を有するポリイミド前駆体が好適に用いられる。
【0101】
【化14】
(式(3)中、R31は4価の有機基である。R32は2価の有機基である。R33及びR34は、水素原子、又は有機基である。nは1以上の自然数である。)
【0102】
R33及びR34が有機基である場合としては、例えば、アルキル基、アルケニル基、アルキニル基、アリール基、及び、これらにエーテル結合を含有したCnH2nOCmH2m+1などで表される構造等を挙げることができる。
【0103】
ポリイミド前駆体としては、下記式(4)で表されるようなポリアミック酸が、アルカリ現像性の点から好適に用いられる。
【0104】
【化15】
(式(4)中、R31は4価の有機基である。R32は2価の有機基である。nは1以上の自然数である。)
【0105】
なお、式(3)及び式(4)において、R31の4価は、酸二無水物等から誘導されるテトラカルボン酸残基を示し、R32の2価はジアミン残基を示す。なお、R31の4価は酸と結合するための価数のみを示しているが、他に更なる置換基を有していても良い。同様に、R32の2価はアミンと結合するための価数のみを示しているが、他に更なる置換基を有していても良い。
【0106】
ポリアミック酸は、酸二無水物とジアミンを溶液中で混合するのみで得られるので、1段階の反応で合成することができ、合成が容易で低コストで入手できるので好ましい。
【0107】
副次的な効果として、用いるポリイミド前駆体がポリアミック酸である場合、塩基の触媒効果によりイミド化に要する温度が低くても十分な為、最終キュア温度を300℃未満、更に好ましくは250℃以下まで下げることが可能である。従来のポリアミック酸はイミド化するために最終キュア温度を300℃以上とする必要があった為、用途が制限されていたが、最終キュア温度を下げることが可能になったことによって、より広範囲の用途に適用が可能である。
【0108】
ポリアミック酸は、酸二無水物とジアミンの反応により得られるが、最終的に得られるポリイミドに優れた耐熱性及び寸法安定性を付与する点から、前記化学式(4)において、R31又はR32が芳香族化合物であることが好ましく、R31及びR32が芳香族化合物であることがより好ましい。またこのとき、前記化学式(4)のR31において、当該R31に結合している4つの基((−CO−)2(−COOH)2)は同一の芳香環に結合していても良く、異なる芳香環に結合していても良い。同様に、前記化学式(4)のR32において、当該R32に結合している2つの基((−NH−)2)は同一の芳香環に結合していても良く、異なる芳香環に結合していても良い。
【0109】
また、前記化学式(4)で表されるポリアミック酸は、単一の繰り返し単位からなるものでも、2種以上の繰り返し単位から成るものでもよい。
【0110】
本発明のポリイミド前駆体を製造する方法としては、従来公知の手法を適用することができる。例えば、(1)酸二無水物とジアミンから前駆体であるポリアミド酸を合成する手法。(2)酸二無水物に1価のアルコールやアミノ化合物、エポキシ化合物等を反応させ合成した、エステル酸やアミド酸モノマーのカルボン酸に、ジアミノ化合物やその誘導体を反応させてポリイミド前駆体を合成する手法などが挙げられるがこれに限定されない。
【0111】
本発明のポリイミド前駆体を得るための反応に適用可能な酸二無水物としては、例えば、エチレンテトラカルボン酸二無水物、ブタンテトラカルボン酸二無水物、シクロブタンテトラカルボン酸二無水物、メチルシクロブタンテトラカルボン酸二無水物、シクロペンタンテトラカルボン酸二無水物などの脂肪族テトラカルボン酸二無水物;ピロメリット酸二無水物、3,3’,4,4’−ベンゾフェノンテトラカルボン酸二無水物、2,2’,3,3’−ベンゾフェノンテトラカルボン酸二無水物、2,3’,3,4’−ベンゾフェノンテトラカルボン酸二無水物、3,3’,4,4’−ビフェニルテトラカルボン酸二無水物、2,2’,3,3’−ビフェニルテトラカルボン酸二無水物、2,3’,3,4’−ビフェニルテトラカルボン酸二無水物、2,2’,6,6’−ビフェニルテトラカルボン酸二無水物、2,2−ビス(3,4−ジカルボキシフェニル)プロパン二無水物、2,2−ビス(2,3−ジカルボキシフェニル)プロパン二無水物、ビス(3,4−ジカルボキシフェニル)エーテル二無水物、ビス(3,4−ジカルボキシフェニル)スルホン二無水物、1,1−ビス(2,3−ジカルボキシフェニル)エタン二無水物、ビス(2,3−ジカルボキシフェニル)メタン二無水物、ビス(3,4−ジカルボキシフェニル)メタン二無水物、2,2−ビス(3,4−ジカルボキシフェニル)−1,1,1,3,3,3−ヘキサフルオロプロパン二無水物、2,2−ビス(2,3−ジカルボキシフェニル)−1,1,1,3,3,3−ヘキサフルオロプロパン二無水物、1,3−ビス〔(3,4−ジカルボキシ)ベンゾイル〕ベンゼン二無水物、1,4−ビス〔(3,4−ジカルボキシ)ベンゾイル〕ベンゼン二無水物、2,2−ビス{4−〔4−(1,2−ジカルボキシ)フェノキシ〕フェニル}プロパン二無水物、
【0112】
2,2−ビス{4−〔3−(1,2−ジカルボキシ)フェノキシ〕フェニル}プロパン二無水物、ビス{4−〔4−(1,2−ジカルボキシ)フェノキシ〕フェニル}ケトン二無水物、ビス{4−〔3−(1,2−ジカルボキシ)フェノキシ〕フェニル}ケトン二無水物、4,4’−ビス〔4−(1,2−ジカルボキシ)フェノキシ〕ビフェニル二無水物、4,4’−ビス〔3−(1,2−ジカルボキシ)フェノキシ〕ビフェニル二無水物、ビス{4−〔4−(1,2−ジカルボキシ)フェノキシ〕フェニル}ケトン二無水物、ビス{4−〔3−(1,2−ジカルボキシ)フェノキシ〕フェニル}ケトン二無水物、ビス{4−〔4−(1,2−ジカルボキシ)フェノキシ〕フェニル}スルホン二無水物、ビス{4−〔3−(1,2−ジカルボキシ)フェノキシ〕フェニル}スルホン二無水物、ビス{4−〔4−(1,2−ジカルボキシ)フェノキシ〕フェニル}スルフィド二無水物、ビス{4−〔3−(1,2−ジカルボキシ)フェノキシ〕フェニル}スルフィド二無水物、2,2−ビス{4−〔4−(1,2−ジカルボキシ)フェノキシ〕フェニル}−1,1,1,3,3,3−ヘキサフルオロプロパン二無水物、2,2−ビス{4−〔3−(1,2−ジカルボキシ)フェノキシ〕フェニル}−1,1,1,3,3,3−ヘキサフルオロプロパン二無水物、2,3,6,7−ナフタレンテトラカルボン酸二無水物、1,1,1,3,3,3−ヘキサフルオロ−2,2−ビス(2,3−又は3,4−ジカルボキシフェニル)プロパン二無水物、1,4,5,8−ナフタレンテトラカルボン酸二無水物、1,2,5,6−ナフタレンテトラカルボン酸二無水物、1,2,3,4−ベンゼンテトラカルボン酸二無水物、3,4,9,10−ぺリレンテトラカルボン酸二無水物、2,3,6,7−アントラセンテトラカルボン酸二無水物、1,2,7,8−フェナントレンテトラカルボン酸二無水物、ピリジンテトラカルボン酸二無水物、スルホニルジフタル酸無水物、m−ターフェニル−3,3’,4,4’−テトラカルボン酸二無水物、p−ターフェニル−3,3’,4,4’−テトラカルボン酸二無水物等の芳香族テトラカルボン酸二無水物等が挙げられる。これらは単独あるいは2種以上混合して用いられる。そして、特に好ましく用いられるテトラカルボン酸二無水物としてピロメリット酸二無水物、3,3’,4,4’−ベンゾフェノンテトラカルボン酸二無水物、3,3’,4,4’−ビフェニルテトラカルボン酸二無水物、2,2’,6,6’−ビフェニルテトラカルボン酸二無水物、ビス(3,4−ジカルボキシフェニル)エーテル二無水物、2,2−ビス(3,4−ジカルボキシフェニル)−1,1,1,3,3,3−ヘキサフルオロプロパン二無水物が挙げられる。
【0113】
併用する酸二無水物としてフッ素が導入された酸二無水物や、脂環骨格を有する酸二無水物を用いると、透明性をそれほど損なわずに溶解性や熱膨張率等の物性を調整することが可能である。また、ピロメリット酸無水物、3,3’,4,4’−ビフェニルテトラカルボン酸二無水物、1,4,5,8−ナフタレンテトラカルボン酸二無水物などの剛直な酸二無水物を用いると、最終的に得られるポリイミドの線熱膨張係数が小さくなるが、透明性の向上を阻害する傾向があるので、共重合割合に注意しながら併用してもよい。
【0114】
一方、アミン成分も、1種類のジアミン単独で、または2種類以上のジアミンを併用して用いることができる。用いられるジアミン成分は限定されず、p−フェニレンジアミン、
m−フェニレンジアミン、o−フェニレンジアミン、3,3’−ジアミノジフェニルエーテル、3,4’−ジアミノジフェニルエーテル、4,4’−ジアミノジフェニルエーテル、3,3’−ジアミノジフェニルスルフィド、3,4’−ジアミノジフェニルスルフィド、4,4’−ジアミノジフェニルスルフィド、3,3’−ジアミノジフェニルスルホン、3,4’−ジアミノジフェニルスルホン、4,4’−ジアミノジフェニルスルホン、3,3’−ジアミノベンゾフェノン、4,4’−ジアミノベンゾフェノン、3,4’−ジアミノベンゾフェノン、3,3’−ジアミノジフェニルメタン、4,4’−ジアミノジフェニルメタン、3,4’−ジアミノジフェニルメタン、2,2−ジ(3−アミノフェニル)プロパン、2,2−ジ(4−アミノフェニル)プロパン、2−(3−アミノフェニル)−2−(4−アミノフェニル)プロパン、2,2−ジ(3−アミノフェニル)−1,1,1,3,3,3−ヘキサフルオロプロパン、2,2−ジ(4−アミノフェニル)−1,1,1,3,3,3−ヘキサフルオロプロパン、2−(3−アミノフェニル)−2−(4−アミノフェニル)−1,1,1,3,3,3−ヘキサフルオロプロパン、1,1−ジ(3−アミノフェニル)−1−フェニルエタン、1,1−ジ(4−アミノフェニル)−1−フェニルエタン、1−(3−アミノフェニル)−1−(4−アミノフェニル)−1−フェニルエタン、1,3−ビス(3−アミノフェノキシ)ベンゼン、1,3−ビス(4−アミノフェノキシ)ベンゼン、1,4−ビス(3−アミノフェノキシ)ベンゼン、1,4−ビス(4−アミノフェノキシ)ベンゼン、1,3−ビス(3−アミノベンゾイル)ベンゼン、1,3−ビス(4−アミノベンゾイル)ベンゼン、1,4−ビス(3−アミノベンゾイル)ベンゼン、1,4−ビス(4−アミノベンゾイル)ベンゼン、1,3−ビス(3−アミノ−α,α−ジメチルベンジル)ベンゼン、1,3−ビス(4−アミノ−α,α−ジメチルベンジル)ベンゼン、1,4−ビス(3−アミノ−α,α−ジメチルベンジル)ベンゼン、1,4−ビス(4−アミノ−α,α−ジメチルベンジル)ベンゼン、1,3−ビス(3−アミノ−α,α−ジトリフルオロメチルベンジル)ベンゼン、1,3−ビス(4−アミノ−α,α−ジトリフルオロメチルベンジル)ベンゼン、1,4−ビス(3−アミノ−α,α−ジトリフルオロメチルベンジル)ベンゼン、1,4−ビス(4−アミノ−α,α−ジトリフルオロメチルベンジル)ベンゼン、2,6−ビス(3−アミノフェノキシ)ベンゾニトリル、2,6−ビス(3−アミノフェノキシ)ピリジン、4,4’−ビス(3−アミノフェノキシ)ビフェニル、4,4’−ビス(4−アミノフェノキシ)ビフェニル、ビス[4−(3−アミノフェノキシ)フェニル]ケトン、ビス[4−(4−アミノフェノキシ)フェニル]ケトン、ビス[4−(3−アミノフェノキシ)フェニル]スルフィド、ビス[4−(4−アミノフェノキシ)フェニル]スルフィド、
【0115】
ビス[4−(3−アミノフェノキシ)フェニル]スルホン、ビス[4−(4−アミノフェノキシ)フェニル]スルホン、ビス[4−(3−アミノフェノキシ)フェニル]エーテル、ビス[4−(4−アミノフェノキシ)フェニル]エーテル、2,2−ビス[4−(3−アミノフェノキシ)フェニル]プロパン、2,2−ビス[4−(4−アミノフェノキシ)フェニル]プロパン、2,2−ビス[3−(3−アミノフェノキシ)フェニル]−1,1,1,3,3,3−ヘキサフルオロプロパン、2,2−ビス[4−(4−アミノフェノキシ)フェニル]−1,1,1,3,3,3−ヘキサフルオロプロパン、1,3−ビス[4−(3−アミノフェノキシ)ベンゾイル]ベンゼン、1,3−ビス[4−(4−アミノフェノキシ)ベンゾイル]ベンゼン、1,4−ビス[4−(3−アミノフェノキシ)ベンゾイル]ベンゼン、1,4−ビス[4−(4−アミノフェノキシ)ベンゾイル]ベンゼン、1,3−ビス[4−(3−アミノフェノキシ)−α,α−ジメチルベンジル]ベンゼン、1,3−ビス[4−(4−アミノフェノキシ)−α,α−ジメチルベンジル]ベンゼン、1,4−ビス[4−(3−アミノフェノキシ)−α,α−ジメチルベンジル]ベンゼン、1,4−ビス[4−(4−アミノフェノキシ)−α,α−ジメチルベンジル]ベンゼン、4,4’−ビス[4−(4−アミノフェノキシ)ベンゾイル]ジフェニルエーテル、4,4’−ビス[4−(4−アミノ−α,α−ジメチルベンジル)フェノキシ]ベンゾフェノン、4,4’−ビス[4−(4−アミノ−α,α−ジメチルベンジル)フェノキシ]ジフェニルスルホン、4,4’−ビス[4−(4−アミノフェノキシ)フェノキシ]ジフェニルスルホン、3,3’−ジアミノ−4,4’−ジフェノキシベンゾフェノン、3,3’−ジアミノ−4,4’−ジビフェノキシベンゾフェノン、3,3’−ジアミノ−4−フェノキシベンゾフェノン、3,3’−ジアミノ−4−ビフェノキシベンゾフェノン、6,6’−ビス(3−アミノフェノキシ)−3,3,3’,3’−テトラメチル−1,1’−スピロビインダン、6,6’−ビス(4−アミノフェノキシ)−3,3,3’,3’−テトラメチル−1,1’−スピロビインダン等の芳香族アミン;
【0116】
1,3−ビス(3−アミノプロピル)テトラメチルジシロキサン、1,3−ビス(4−アミノブチル)テトラメチルジシロキサン、α,ω−ビス(3−アミノプロピル)ポリジメチルシロキサン、α,ω−ビス(3−アミノブチル)ポリジメチルシロキサン、ビス(アミノメチル)エーテル、ビス(2−アミノエチル)エーテル、ビス(3−アミノプロピル)エーテル、ビス(2−アミノメトキシ)エチル]エーテル、ビス[2−(2−アミノエトキシ)エチル]エーテル、ビス[2−(3−アミノプロトキシ)エチル]エーテル、1,2−ビス(アミノメトキシ)エタン、1,2−ビス(2−アミノエトキシ)エタン、1,2−ビス[2−(アミノメトキシ)エトキシ]エタン、1,2−ビス[2−(2−アミノエトキシ)エトキシ]エタン、エチレングリコールビス(3−アミノプロピル)エーテル、ジエチレングリコールビス(3−アミノプロピル)エーテル、トリエチレングリコールビス(3−アミノプロピル)エーテル、エチレンジアミン、1,3−ジアミノプロパン、1,4−ジアミノブタン、1,5−ジアミノペンタン、1,6−ジアミノヘキサン、1,7−ジアミノヘプタン、1,8−ジアミノオクタン、1,9−ジアミノノナン、1,10−ジアミノデカン、1,11−ジアミノウンデカン、1,12−ジアミノドデカン等の脂肪族アミン;
【0117】
1,2−ジアミノシクロヘキサン、1,3−ジアミノシクロヘキサン、1,4−ジアミノシクロヘキサン、1,2−ジ(2−アミノエチル)シクロヘキサン、1,3−ジ(2−アミノエチル)シクロヘキサン、1,4−ジ(2−アミノエチル)シクロヘキサン、ビス(4−アミノシクロへキシル)メタン、2,6−ビス(アミノメチル)ビシクロ[2.2.1]ヘプタン、2,5−ビス(アミノメチル)ビシクロ[2.2.1]ヘプタン等の脂環式ジアミンが挙げられる。グアナミン類としては、アセトグアナミン、ベンゾグアナミンなどを挙げることができ、また、上記ジアミンの芳香環上水素原子の一部若しくは全てをフルオロ基、メチル基、メトキシ基、トリフルオロメチル基、又はトリフルオロメトキシ基から選ばれた置換基で置換したジアミンも使用することができる。
【0118】
さらに目的に応じ、架橋点となるエチニル基、ベンゾシクロブテン−4’−イル基、ビニル基、アリル基、シアノ基、イソシアナト基、及びイソプロペニル基のいずれか1種又は2種以上を、上記ジアミンの芳香環上水素原子の一部若しくは全てに置換基として導入しても使用することができる。
【0119】
ジアミンは、目的の物性によって選択することができ、p−フェニレンジアミンなどの剛直なジアミンを用いれば、最終的に得られるポリイミドは低膨張率となる。剛直なジアミンとしては、同一の芳香環に2つアミノ基が結合しているジアミンとして、p−フェニレンジアミン、m−フェニレンジアミン、1,4−ジアミノナフタレン、1,5−ジアミノナフタレン、2、6−ジアミノナフタレン、2,7−ジアミノナフタレン、1,4―ジアミノアントラセンなどが挙げられる。
【0120】
さらに、2つ以上の芳香族環が単結合により結合し、2つ以上のアミノ基がそれぞれ別々の芳香族環上に直接又は置換基の一部として結合しているジアミンが挙げられ、例えば、下記式(5)により表されるものがある。具体例としては、ベンジジン等が挙げられる。
【0121】
【化16】
(化学式(5)中、aは1以上の自然数、アミノ基はベンゼン環同士の結合に対して、メタ位または、パラ位に結合する。)
【0122】
さらに、上記式(5)において、他のベンゼン環との結合に関与せず、ベンゼン環上のアミノ基が置換していない位置に置換基を有するジアミンも用いることができる。これら置換基は、有機基であるがそれらは互いに結合していてもよい。
具体例としては、2,2’−ジメチル−4,4’−ジアミノビフェニル、2,2’−ジトリフルオロメチル−4,4’−ジアミノビフェニル、3,3’−ジクロロ−4,4’−ジアミノビフェニル、3,3’−ジメトキシ−4,4’−ジアミノビフェニル、3,3’−ジメチル−4,4’−ジアミノビフェニル等が挙げられる。
【0123】
最終的に得られるポリイミドを光導波路、光回路部品として用いる場合には、芳香環の置換基としてフッ素を導入すると1μm以下の波長の電磁波に対しての透過率を向上させることができる。
【0124】
一方、ジアミンとして、1,3−ビス(3−アミノプロピル)テトラメチルジシロキサンなどのシロキサン骨格を有するジアミンを用いると、最終的に得られるポリイミドの弾性率が低下し、ガラス転移温度を低下させることができる。
ここで、選択されるジアミンは耐熱性の観点より芳香族ジアミンが好ましいが、目的の物性に応じてジアミンの全体の60モル%、好ましくは40モル%を超えない範囲で、脂肪族ジアミンやシロキサン系ジアミン等の芳香族以外のジアミンを用いても良い。
【0125】
一方、ポリイミド前駆体を合成するには、例えば、アミン成分として4,4’−ジアミノジフェニルエーテルをN−メチルピロリドンなどの有機極性溶媒に溶解させた溶液を冷却しながら、そこへ等モルの3,3’,4,4’−ビフェニルテトラカルボン酸二無水物を徐々に加え撹拌し、ポリイミド前駆体溶液を得ることができる。
このようにして合成されるポリイミド前駆体は、最終的に得られるポリイミドに耐熱性及び寸法安定性を求める場合には、芳香族酸成分及び/又は芳香族アミン成分の共重合割合ができるだけ大きいことが好ましい。具体的には、イミド構造の繰り返し単位を構成する酸成分に占める芳香族酸成分の割合が50モル%以上、特に70モル%以上であることが好ましく、イミド構造の繰り返し単位を構成するアミン成分に占める芳香族アミン成分の割合が40モル%以上、特に60モル%以上であることが好ましく、全芳香族ポリイミドであることが特に好ましい。
【0126】
本発明に用いられるポリイミド前駆体においては、感光性ポリイミド樹脂組成物とした際の感度を高め、マスクパターンを正確に再現するパターン形状を得るために、1μmの膜厚のときに、露光波長に対して少なくとも5%以上の透過率を示すことが好ましく、15%以上の透過率を示すことが更に好ましい。
露光波長に対してポリイミド前駆体の透過率が高いということは、それだけ、電磁波のロスが少ないということであり、高感度の感光性ポリイミド樹脂組成物を得ることができる。
【0127】
また、一般的な露光光源である高圧水銀灯を用いて露光を行う場合には、少なくとも436nm、405nm、365nmの波長の電磁波のうち1つの波長の電磁波に対する透過率が、厚み1μmのフィルムに成膜した時で好ましくは5%以上、更に好ましくは15%、特に好ましくは50%以上である。
【0128】
本発明に用いられるポリイミド前駆体の重量平均分子量は、その用途にもよるが、3,000〜1,000,000の範囲であることが好ましく、5,000〜500,000の範囲であることがさらに好ましく、10,000〜500,000の範囲であることがさらに好ましい。重量平均分子量が3,000未満であると、塗膜又はフィルムとした場合に十分な強度が得られにくい。また、加熱処理等を施しポリイミド等の高分子とした際の膜の強度も低くなる。一方、重量平均分子量が1,000,000を超えると粘度が上昇し、溶解性も低下しやすく、表面が平滑で膜厚が均一な塗膜又はフィルムが得られにくい。
【0129】
ここで用いている分子量とは、ゲル浸透クロマトグラフィー(GPC)によるポリスチレン換算の値のことをいい、ポリイミド前駆体そのものの分子量でも良いし、無水酢酸等で化学的イミド化処理を行った後のものでも良い。
【0130】
なお、ポリイミド前駆体合成時における溶媒は、極性溶媒が望ましく、代表的なものとして、N−メチル−2−ピロリドン、N−アセチル−2−ピロリドン、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N,N−ジエチルホルムアミド、N,N−ジエチルアセトアミド、N,N−ジメチルメトキシアセトアミド、ジメチルスルホキシド、ヘキサメチルフォスホアミド、ピリジン、ジメチルスルホン、テトラメチレンスルホン、ジメチルテトラメチレンスルホン、ジエチレングリコールジメチルエーテル、シクロペンタノン、γ−ブチロラクトン、α−アセチル−γ−ブチロラクトン等があり、これらの溶媒は単独であるいは2種類以上を組み合わせて用いられる。この他にも溶媒として組合せて用いられるものとしてベンゼン、ベンゾニトリル、1,4−ジオキサン、テトラヒドロフラン、ブチロラクトン、キシレン、トルエン、シクロヘキサノン等の非極性溶媒が挙げられ、これらの溶媒は、原料の分散媒、反応調節剤、あるいは生成物からの溶媒の揮散調節剤、皮膜平滑剤などとして使用される。
【0131】
(光塩基発生剤)
本発明に係るネガ型感光性ポリイミド樹脂組成物に用いられる光塩基発生剤は、前記本発明に係る光塩基発生剤と同様のものとすることができる。中でも、光塩基発生剤が発生する塩基が、150〜200℃のいずれかの温度において、当該塩基100重量部に対して、前記ポリイミド前駆体を10重量部以上溶解する塩基であることが好ましい。発生する塩基が前記ポリイミド前駆体を10重量部以上溶解する場合、触媒である塩基とポリイミド前駆体の親和性が向上し、少量の塩基であっても前記ポリイミド前駆体を効率的にイミド化することができるからである。
【0132】
本発明に係る光塩基発生剤は、少量の添加で硬化が可能となり、感光性ポリイミド樹脂組成物の固形分全体に対し、通常、0.1〜30重量%、好ましくは0.5〜20重量%の範囲内で含有させることが好ましい。なお、本発明において固形分は、上述した溶媒以外のもの全てであり、室温で液体のモノマー等も含まれる。
【0133】
<その他の成分>
本発明に係る感光性ポリイミド樹脂組成物は、前記ポリイミド前駆体と、前記光塩基発生剤と、溶媒の単純な混合物であってもよいが、さらに、光又は熱硬化性成分、ポリイミド前駆体以外の非重合性バインダー樹脂、その他の成分を配合して、感光性ポリイミド樹脂組成物を調製してもよい。また、本発明の光塩基発生剤の補助的な役割として、光によって酸又は塩基を発生させる他の感光性成分を加えても良い。また、光塩基発生剤から発生した少量の塩基の作用によって、分解や転位反応して塩基を発生させる塩基増殖剤を併用しても良いし、増感剤を加えてもよい。
【0134】
感光性ポリイミド樹脂組成物を溶解、分散又は希釈する溶剤としては、各種の汎用溶剤を用いることが出来る。また、前駆体としてポリアミド酸を用いる場合には、ポリアミド酸の合成反応により得られた溶液をそのまま用い、そこに必要に応じて他の成分を混合しても良い。
【0135】
使用可能な汎用溶剤としては、例えば、ジエチルエーテル、テトラヒドロフラン、ジオキサン、エチレングリコールジメチルエーテル、エチレングリコールジエチルエーテル、プロピレングリコールジメチルエーテル、プロピレングリコールジエチルエーテル、ジエチレングリコールジメチルエーテル等のエーテル類;エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル、プロピレングリコールモノメチルエーテル、プロピレングリコールモノエチルエーテル、ジエチレングリコールモノメチルエーテル、ジエチレングリコールモノエチルエーテル等のグリコールモノエーテル類(いわゆるセロソルブ類);メチルエチルケトン、アセトン、メチルイソブチルケトン、シクロペンタノン、シクロヘキサノンなどのケトン類;酢酸エチル、酢酸ブチル、酢酸n−プロピル、酢酸i−プロピル、酢酸n−ブチル、酢酸i−ブチル、前記グリコールモノエーテル類の酢酸エステル(例えば、メチルセロソルブアセテート、エチルセロソルブアセテート)、プロピレングリコールモノメチルエーテルアセテート、プロピレングリコールモノエチルエーテルアセテート、蓚酸ジメチル、乳酸メチル、乳酸エチル等のエステル類;エタノール、プロパノール、ブタノール、ヘキサノール、シクロヘキサノール、エチレングリコール、ジエチレングリコール、グリセリン等のアルコール類;塩化メチレン、1,1−ジクロロエタン、1,2−ジクロロエチレン、1−クロロプロパン、1−クロロブタン、1−クロロペンタン、クロロベンゼン、ブロムベンゼン、o−ジクロロベンゼン、m−ジクロロベンゼン等のハロゲン化炭化水素類;N,N−ジメチルホルムアミド、N,N−ジエチルホルムアミド、N,N−ジメチルアセトアミド、N,N−ジエチルアセトアミド、N,N−ジメチルメトキシアセトアミド等のアミド類;N−メチル−2−ピロリドン、N−アセチル−2−ピロリドンなどのピロリドン類;γ−ブチロラクトン、α−アセチル−γ−ブチロラクトン等のラクトン類;ジメチルスルホキシドなどのスルホキシド類、ジメチルスルホン、テトラメチレンスルホン、ジメチルテトラメチレンスルホンなどのスルホン類、ヘキサメチルフォスホアミド等のリン酸アミド類、その他の有機極性溶媒類等が挙げられ、更には、ベンゼン、トルエン、キシレン、ピリジン等の芳香族炭化水素類、及び、その他の有機非極性溶媒類等も挙げられる。これらの溶媒は単独若しくは組み合わせて用いられる。
【0136】
中でも、プロピレングリコールモノメチルエーテル、メチルエチルケトン、シクロペンタノン、シクロヘキサノン、酢酸エチル、プロピレングリコールモノメチルエーテルアセテート、N,N−ジメチルアセトアミド、N−メチル−2−ピロリドン、γ−ブチロラクトン等の極性溶媒、トルエン等の芳香族炭化水素類、及び、これらの溶媒からなる混合溶媒が好適なものとして挙げられる。
【0137】
光硬化性成分としては、エチレン性不飽和結合を1つ又は2つ以上有する化合物を用いることができ、例えば、アミド系モノマー、(メタ)アクリレートモノマー、ウレタン(メタ)アクリレートオリゴマー、ポリエステル(メタ)アクリレートオリゴマー、エポキシ(メタ)アクリレート、及びヒドロキシル基含有(メタ)アクリレート、スチレン等の芳香族ビニル化合物を挙げることができる。また、ポリイミド前駆体が、ポリアミック酸等のカルボン酸成分を構造内に有する場合には、3級アミノ基を有するエチレン性不飽和結合含有化合物を用いると、ポリイミド前駆体のカルボン酸とイオン結合を形成し、感光性ポリイミド樹脂組成物としたときの露光部、未露光部の溶解速度のコントラストが大きくなる。
【0138】
このようなエチレン性不飽和結合を有する光硬化性化合物を用いる場合には、さらに光ラジカル発生剤を添加してもよい。光ラジカル発生剤としては、例えば、ベンゾイン、ベンゾインメチルエーテル、ベンゾインエチルエーテル及びベンゾインイソプロピルエーテル等のベンゾインとそのアルキルエーテル;アセトフェノン、2,2−ジメトキシ−2−フェニルアセトフェノン、2,2−ジエトキシ−2−フェニルアセトフェノン、1,1−ジクロロアセトフェノン、1−ヒドロキシアセトフェノン、1−ヒドロキシシクロヘキシルフェニルケトン及び2−メチル−1−[4−(メチルチオ)フェニル]−2−モルフォリノ−プロパン−1−オン等のアセトフェノン;2−メチルアントラキノン、2−エチルアントラキノン、2−ターシャリ−ブチルアントラキノン、1−クロロアントラキノン及び2−アミルアントラキノン等のアントラキノン;2,4−ジメチルチオキサントン、2,4−ジエチルチオキサントン、2−クロロチオキサントン及び2,4−ジイソピルチオキサントン等のチオキサントン;アセトフェノンジメチルケタール及びベンジルジメチルケタール等のケタール;2,4,6−トリメチルベンゾイルジフェニルホスフィンオキシド等のモノアシルホスフィンオキシドあるいはビスアシルホスフィンオキシド類;ベンゾフェノン等のベンゾフェノン類;並びにキサントン類等が挙げられる。
【0139】
光によって酸を発生させる化合物としては、1,2−ベンゾキノンジアジドあるいは1,2−ナフトキノンジアジド構造を有する感光性ジアゾキノン化合物があり、米国特許明細書第2,772,972号、第2,797,213号、第3,669,658号に提案されている。また、トリアジンやその誘導体、スルホン酸オキシムエステル化合物、スルホン酸ヨードニウム塩、スルホン酸スルフォニウム塩等、公知の光酸発生剤を用いることができる。光によって塩基を発生させる化合物としては、例えば2,6−ジメチル−3,5−ジシアノ−4−(2’−ニトロフェニル)−1,4−ジヒドロピリジン、2,6−ジメチル−3,5−ジアセチル−4−(2’−ニトロフェニル)−1,4−ジヒドロピリジン、2,6−ジメチル−3,5−ジアセチル−4−(2’,4’−ジニトロフェニル)−1,4−ジヒドロピリジンなどが例示できる。
【0140】
塩基増殖剤としては、例えば、9−フルオレニルメチルカルバメート結合を有する化合物、1,1−ジメチル−2−シアノメチルカルバメート結合((CN)CH2C(CH3)2OC(O)NR2)を有する化合物、パラニトロベンジルカルバメート結合を有する化合物、2,4−ジクロロベンジルカルバメート結合を有する化合物、その他にも特開2000−330270号公報の段落0010〜段落0032に記載されているウレタン系化合物や、特開2008−250111号公報の段落0033〜段落0060に記載されているウレタン系化合物等が挙げられる。
【0141】
高分子を透過する波長の電磁波のエネルギーを光塩基発生剤が充分利用できる様にし、感度を向上させたい場合に、増感剤の添加が効果を発揮する場合がある。
特に、ポリイミド前駆体の吸収が360nm以上の波長にもある場合には、増感剤の添加による効果が大きい。増感剤と呼ばれる化合物の具体例としては、チオキサントン及び、ジエチルチオキサントンなどのその誘導体、クマリン系及び、その誘導体、ケトクマリン及び、その誘導体、ケトビスクマリン、及びその誘導体、シクロペンタノン及び、その誘導体、シクロヘキサノン及び、その誘導体、チオピリリウム塩及び、その誘導体、チオキサンテン系、キサンテン系及び、その誘導体などが挙げられる。
【0142】
クマリン、ケトクマリン及び、その誘導体の具体例としては、3,3’−カルボニルビスクマリン、3,3’−カルボニルビス(5,7−ジメトキシクマリン)、3,3’−カルボニルビス(7−アセトキシクマリン)等が挙げられる。チオキサントン及び、その誘導体の具体例としては、ジエチルチオキサントン、イソプロピルチオキサントンなどが挙げられる。さらに他にはベンゾフェノン、アセトフェノン、フェナントレン、2−ニトロフルオレン、5−ニトロアセナフテン、ベンゾキノン、2−エチルアントラキノン、2−ターシャリーブチルアントラキノン、1,2−ベンズアンスラキノン、1,2−ナフトキノン、などが挙げられる。
これらは、光塩基発生剤との組み合わせによって、特に優れた効果を発揮する為、塩基発生剤の構造によって最適な増感作用を示す増感剤が適宜選択される。
【0143】
また、本発明に係る樹脂組成物に加工特性や各種機能性を付与するために、その他に様々な有機又は無機の低分子又は高分子化合物を配合してもよい。例えば、染料、界面活性剤、レベリング剤、可塑剤、微粒子等を用いることができる。微粒子には、ポリスチレン、ポリテトラフルオロエチレン等の有機微粒子、コロイダルシリカ、カーボン、層状珪酸塩等の無機微粒子等が含まれ、それらは多孔質や中空構造であってもよい。また、その機能又は形態としては顔料、フィラー、繊維等がある。
【0144】
また、その他の溶剤以外の任意成分の配合割合は、感光性ポリイミド樹脂組成物の固形分全体に対し、0.1重量%〜95重量%の範囲が好ましい。0.1重量%未満だと、添加物を添加した効果が発揮されにくく、95重量%を超えると、最終的に得られる樹脂硬化物の特性が最終生成物に反映されにくい。
【0145】
本発明に係る感光性ポリイミド樹脂組成物は、印刷インキ、塗料、シール剤、接着剤、電子材料、光回路部品、成形材料、レジスト材料、建築材料、光造形、光学部材等、樹脂材料が用いられる公知の全ての分野、製品に利用できる。塗料、シール剤、接着剤のように、全面露光して用いる用途にも、永久膜や剥離膜などパターンを形成する用途にも、いずれにも好適に用いることができる。
【0146】
本発明に係る感光性ポリイミド樹脂組成物は、耐熱性、寸法安定性、絶縁性等の特性が有効とされる広範な分野、製品、例えば、塗料、印刷インキ、シール剤、又は接着剤、或いは、表示装置、半導体装置、電子部品、微小電気機械システム(Micro Electro Mechanical System(MEMS))、光造形物、光学部材又は建築材料の形成材料として好適に用いられる。例えば具体的には、電子部品の形成材料としては、封止材料、層形成材料として、プリント配線基板、層間絶縁膜、配線被覆膜等に用いることができる。また、表示装置の形成材料としては、層形成材料や画像形成材料として、カラーフィルター、フレキシブルディスプレイ用フィルム、レジスト材料、配向膜等に用いることができる。また、半導体装置の形成材料としては、レジスト材料、バッファーコート膜のような層形成材料等に用いることができる。また、光学部品の形成材料としては、光学材料や層形成材料として、ホログラム、光導波路、光回路、光回路部品、反射防止膜等に用いることができる。また、建築材料としては、塗料、コーティング剤等に用いることができる。また、光造形物の材料としても用いることができる。
【0147】
上記の様な特徴を有することから、本発明に係る感光性ポリイミド樹脂組成物は、パターン形成用材料としても用いることが可能である。特に、感光性ポリイミド樹脂組成物をパターン形成用材料(レジスト)として用いた場合、それによって形成されたパターンは、ポリイミドからなる永久膜として耐熱性や絶縁性を付与する成分として機能し、例えば、カラーフィルター、フレキシブルディスプレー用フィルム、電子部品、半導体装置、層間絶縁膜、配線被覆膜、光回路、光回路部品、反射防止膜、その他の光学部材又は電子部材を形成するのに適している。
【0148】
また、本発明においては、本発明に係るネガ型感光性ポリイミド樹脂組成物又はその熱硬化物により少なくとも一部分が形成されている、印刷物、塗料、シール剤、接着剤、表示装置、半導体装置、電子部品、微小電気機械システム、光造形物、光学部材又は建築材料いずれかの物品が提供される。
【0149】
<レリーフパターンの製造方法>
本発明に係るレリーフパターンの製造方法は、前記本発明に係るネガ型感光性ポリイミド樹脂組成物からなる塗膜又は成形体を形成し、当該塗膜又は成形体を、所定パターン状に電磁波を照射し、照射後又は照射と同時に加熱し、前記照射部位の溶解性を変化させた後、現像することを特徴とする。
【0150】
上記レリーフパターンの製造方法においては、ポリイミド前駆体と、本発明に係る光塩基発生剤とを組み合わせて用いることにより、感光性ポリイミド樹脂組成物からなる塗膜又は成形体の表面を現像液から保護するためのレジスト膜を用いずに、現像を行うパターン形成が可能である。
【0151】
本発明に係る感光性ポリイミド樹脂組成物を何らかの支持体上に塗布するなどして塗膜を形成したり、適した成型方法で成形体を形成し、当該塗膜又は成形体を、所定のパターン状に電磁波を照射し、照射後又は照射と同時に加熱することにより、露光部においてのみ、本発明に係る光塩基発生剤が塩基を発生する。塩基は、露光部のポリイミド前駆体のイミド化を促進する触媒として作用する。本発明の光塩基発生剤を用いることにより、前記加熱条件を従来よりも緩和することができ、感光性ポリイミド樹脂組成物の未露光部のイミド化を従来よりも抑制できるため、現像時間を短縮でき、アルコール等を添加しない塩基性水溶液を現像液として用いることが可能となったり、露光部と未露光部の溶解性コントラストを高くすることができる。
【0152】
ポリイミド前駆体は、塩基の触媒作用によって熱硬化温度が低下する。このような場合、先ず、本発明に係る感光性ポリイミド樹脂組成物の塗膜又は成形体上のパターンを残したい部分を露光する。露光後又は露光と同時に加熱すると、露光部には、塩基が発生し、その部分の熱硬化温度が選択的に低下する。露光後又は露光と同時に、露光部は熱硬化するが未露光部は熱硬化しない処理温度で加熱し、露光部のみ硬化させる。塩基を発生させる加熱工程と、露光部のみ硬化させる反応を行うための加熱工程(露光後ベイク)は、同一の工程としても良いし、別の工程にしても良い。
次に、所定の現像液(有機溶剤や塩基性水溶液等)で未露光部を溶解して熱硬化物からなるパターンを形成する。このパターンを、更に必要に応じ加熱して熱硬化を完結させる。以上の工程によって、通常ネガ型の所望の2次元樹脂パターン(一般的な平面パターン)又は3次元樹脂パターン(立体的に成形された形状)が得られる。
【0153】
本発明の感光性ポリイミド樹脂組成物は、プロピレングリコールモノメチルエーテル、メチルエチルケトン、シクロペンタノン、シクロヘキサノン、酢酸エチル、プロピレングリコールモノメチルエーテルアセテート、N,N−ジメチルアセトアミド、N−メチル−2−ピロリドン、γ−ブチロラクトン等の極性溶媒、トルエン等の芳香族炭化水素類、及び、これらの溶媒からなる混合溶媒に溶解後、浸漬法、スプレー法、フレキソ印刷法、グラビア印刷法、スクリーン印刷法、スピンコート法、ディスペンス法などによって、シリコンウエハ、金属基板、セラミック基板、樹脂フィルムなどの基材表面に塗布し、加熱して溶剤の大部分を除くことにより、基材表面に粘着性のない塗膜を与えることができる。塗膜の厚みには特に制限はないが、0.5〜50μmであることが好ましく、感度および現像速度面から1.0〜20μmであることがより望ましい。塗布した塗膜の乾燥条件としては、例えば、80〜100℃、1分〜20分が挙げられる。
【0154】
この塗膜に、所定のパターンを有するマスクを通して、電磁波を照射しパターン状に露光後を行い、加熱後、膜の未露光部分を、適切な現像液で現像して除去することにより、所望のパターン化された膜を得ることができる。
【0155】
露光工程に用いられる露光方法や露光装置は特に限定されることなく、密着露光でも間接露光でも良く、g線ステッパ、i線ステッパ、超高圧水銀灯を用いるコンタクト/プロキシミティ露光機、ミラープロジェクション露光機、又はその他の紫外線、可視光線、X線、電子線などを照射可能な投影機や線源を使用することができる。
【0156】
例えば上記式(1−1)で表される光塩基発生剤など保護基を有する場合であって、電磁波照射のみによって保護基の脱保護を行う場合、塩基を発生するために照射する電磁波によって脱保護しても良いし、脱保護のための電磁波と、塩基を発生させるための電磁波とで波長を変更しても良い。例えば、長波長の電磁波を照射して脱保護を行い、その後短波長の電磁波で塩基を発生させるための異性化を行うなどが挙げられる。これらの場合の電磁波の照射量は、電磁波によっても異なり、特に限定されず、適宜調整される。
【0157】
露光前又は露光後又は露光と同時に加熱し、保護基を脱保護させて塩基を発生させるための加熱温度としては、組み合わせるポリイミド前駆体や目的により適宜選択され、特に限定されない。感光性ポリイミド樹脂組成物が置かれた環境の温度(例えば、室温)による加熱であっても良く、その場合、徐々に塩基が発生する。また、電磁波の照射時に副生される熱によっても塩基が発生するため、電磁波の照射時に副生される熱により実質的に加熱が同時に行われても良い。反応速度を高くし、効率よく塩基を発生させる点から、加熱温度としては、30℃以上であることが好ましく、更に好ましくは60℃以上、より更に好ましくは100℃以上、特に好ましくは120℃以上である。しかしながら、組み合わせて用いられるポリイミド前駆体によっては、例えば60℃以上の加熱で未露光部についても硬化するものもあるので、好適な加熱温度は、上記に限定されない。また、本発明に係る光塩基発生剤の塩基発生以外の分解を防ぐために、300℃以下で加熱することが好ましく、更に200℃以下で加熱することが好ましい。
なお、例えば上記式(1−1)で表される光塩基発生剤など保護基を有する場合、露光前に加熱を行い脱保護してもよい。保護基等の種類によっては保護基等を導入することで、吸収波長が短波長化するなどして光塩基発生剤の感度が悪くなることがある。このような場合、電磁波照射前の加熱により予め保護基等を脱保護し、電磁波を照射することにより、電磁波照射時の感度を向上させることができる。
なお、保護基の脱保護条件は、組成物中で共存する成分により変化し得る。例えば、他の光酸発生剤や光塩基発生剤が含まれる場合、光照射によって発生した酸・塩基の影響で、露光後の加熱温度が変化する場合がある。
当該電磁波照射前の保護基脱保護のための加熱は、塗膜の乾燥工程であっても良いし、他の加熱工程であっても良い。この場合、加熱温度としては、脱保護が可能な温度を適宜選択すればよいが、50℃〜180℃が好ましく、時間は10秒以上60分以下が好ましい。
また、加熱を行う際には、低温で保護基の脱保護を行い、より高温で塩基を発生させるようにしても良い。
【0158】
前記式(1−1)で表される光塩基発生剤は電磁波の照射のみでも塩基を発生するが、適宜加熱することにより塩基の発生が促進される。従って、効率的に塩基を発生させるために、前記式(1−1)で表される光塩基発生剤を用いる際には、電磁波照射(露光)後又は電磁波照射と同時に加熱を行うことにより塩基を発生する。露光と加熱を交互に行ってもよい。最も効率が良い方法は、露光と同時に加熱する方法である。
【0159】
本発明に係る感光性ポリイミド樹脂組成物の塗膜は、露光部のみ硬化させる反応を行うために、露光工程と現像工程の間に、露光後ベイク(Post Exposure Bake:PEB)を行うことが好ましい。当該PEBは、電磁波の照射により発生した塩基の作用により、塩基が存在する部位と、未照射で塩基が存在しない部位とでイミド化率等の硬化反応の反応率が異なるようになる温度で行うことが好ましく、用いる光塩基発生剤の種類によって適宜選択されるものである。好ましい熱処理の温度の範囲は、通常140℃〜210℃程度であり、より好ましくは150℃〜200℃である。熱処理温度が140℃より低いと、イミド化の効率が悪く、現実的なプロセス条件で露光部、未露光部のイミド化率の差を生ずることが難しくなる。一方、熱処理温度が210℃を超えると、塩基が存在していない未露光部でもイミド化が進行する恐れがあり、露光部と未露光部の溶解性の差を生じ難い。
また、本発明の光塩基発生剤は、発生する塩基が150〜200℃の少なくとも一部の温度で液体であるため、発生する塩基が液体となる温度を適宜選択することが、特に好ましい。発生する塩基が液体である場合、樹脂組成物中を拡散しやすいか、又は、樹脂組成物中のポリイミド前駆体を溶解することができるため、少量の塩基であってもポリイミド前駆体を効率的にイミド化することができるためである。
この熱処理は、公知の方法であればどの方法でもよく、具体的に例示すると、空気、又は窒素雰囲気下の循環オーブン、又はホットプレートによる加熱等が挙げられるが、特に限定されない。
【0160】
(現像液)
現像工程に用いられる現像液としては、前記照射部位の溶解性が変化する溶剤を現像液として用いれば、特に限定されず、塩基性水溶液、有機溶剤など、用いられるポリイミド前駆体に合わせて適宜選択することが可能である。環境への負荷を低減させる点から、現像液としては、有機溶剤の含有量が少ないことが好ましく、具体的には、有機溶剤の含有量が20重量%以下の塩基性水溶液が好ましく、有機溶剤の含有量が10重量%以下の塩基性水溶液が更に好ましい。理想的には、実質的にアルコール等の有機溶剤を含まない塩基性水溶液が最も好ましい。
【0161】
塩基性水溶液としては、特に限定されないが、例えば、濃度が、0.01重量%〜10重量%、好ましくは、0.05重量%〜5重量%のテトラメチルアンモニウムヒドロキシド(TMAH)水溶液の他、ジエタノールアミン、ジエチルアミノエタノール、水酸化ナトリウム、水酸化カリウム、炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム、炭酸水素カリウム、トリエチルアミン、ジエチルアミン、メチルアミン、ジメチルアミン、酢酸ジメチルアミノエチル、ジメチルアミノエタノール、ジメチルアミノエチルメタクリレート、シクロヘキシルアミン、エチレンジアミン、ヘキサメチレンジアミン、テトラメチルアンモニウムなどの水溶液等が挙げられる。
溶質は、1種類でも2種類以上でも良く、全体の重量の50%以上、さらに好ましくは70%以上、水が含まれていれば有機溶剤等を含んでいても良い。
【0162】
また、有機溶剤としては、特に限定されないが、N−メチル−2−ピロリドン、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、ジメチルスルホキシド、γ−ブチロラクロン、ジメチルアクリルアミドなどの極性溶媒、メタノール、エタノール、イソプロパノールなどのアルコール類、酢酸エチル、プロピレングリコールモノメチルエーテルアセテートなどのエステル類、シクロペンタノン、シクロヘキサノン、イソブチルケトン、メチルイソブチルケトンなどのケトン類、その他テトラヒドロフラン、クロロホルム、アセトニトリルなどを、単独であるいは2種類以上を組み合わせて添加してもよい。現像後は水または貧溶媒にて洗浄を行う。この場合においてもエタノール、イソプロピルアルコールなどのアルコール類、乳酸エチル、プロピレングリコールモノメチルエーテルアセテートなどのエステル類などを水に加えても良い。
【0163】
現像後は必要に応じて水または貧溶媒でリンスを行い、80〜100℃で乾燥しパターンを安定なものとする。このレリーフパターンを、耐熱性のあるものとするために180〜500℃、好ましくは200〜350℃の温度で数十分から数時間加熱することによりパターン化された高耐熱性樹脂層が形成される。
【実施例】
【0164】
以下、本発明について実施例を示して具体的に説明する。これらの記載により本発明を制限するものではない。尚、実施例中、部は特に特定しない限り重量部を表す。製造された塩基発生剤の構造は1H NMRによって確認した。
1H NMR測定:日本電子(株)製、JEOL JNM−LA400WB
手動露光機:大日本科研製、MA−1100
吸光度測定:(株)島津製作所製、紫外可視分光光度計UV−2550
塗膜の加熱:アズワン(株)製、HOT PLATE EC−1200(本実施例中、ホットプレートと記載することがある)
【0165】
(合成例1:ポリイミド前駆体1の合成)
4,4’−ジアミノジフェニルエーテル(ODA)20.0g(100mmol)を500mlのセパラブルフラスコに投入し、181gの脱水されたN−メチル−2−ピロリドン(NMP)に溶解させ窒素気流下、オイルバスによって液温が50℃になるように熱電対でモニターし加熱しながら撹拌した。それらが完全に溶解したことを確認した後、そこへ、少しずつ30分かけて3,3’、4,4’−ビフェニルテトラカルボン酸二無水物(BPDA)27.4g(93.1mmol)を添加し、添加終了後、50℃で5時間撹拌した。その後、フタル酸無水物を0.900g(6.08mmol)添加した。その後室温まで冷却し、下記式で表わされる繰り返し単位を有するポリイミド前駆体1溶液を得た。
【0166】
【化17】
【0167】
(合成例2:ポリイミド前駆体2の合成)
2,2’−ビス(トリフルオロメチル)−4,4’−ジアミノビフェニル(TFMB)16.0g(50.0mmol)と2,2’−ジメチル−4,4’−ジアミノビフェニル(mTBHG)10.6g(50.0mmol)とを500mlのセパラブルフラスコに投入し、200gの脱水されたN−メチル−2−ピロリドン(NMP)に溶解させ、窒素気流下、オイルバスによって液温が50℃になるように熱電対でモニターし加熱しながら撹拌した。それらが完全に溶解したことを確認した後、そこへ、少しずつ30分かけて3,3’,4,4’−ビフェニルテトラカルボン酸二無水物(BPDA)27.3g(93.1mmol)を添加し、添加終了後、50℃で5時間撹拌した。その後、フタル酸無水物を0.900g(6.08mmol)添加した。その後室温まで冷却し、下記式で表わされる繰り返し単位を有するポリイミド前駆体2溶液を得た。
【0168】
【化18】
【0169】
(合成例3:ポリイミド前駆体3の合成)
窒素気流下1000mlのセパラブルフラスコに投入に脱水されたN−メチル−2−ピロリドン(NMP)85.5gを投入し、オイルバスによって液温が50℃になるように熱電対でモニターし加熱しながら撹拌した。3,3’,4,4’−ビフェニルテトラカルボン酸二無水物(BPDA)13.0g(44.2mmol)を投入し、その後、1,3−ビス(3−アミノプロピル)テトラメチルジシロキサン5.14g(20.7mmol)を投入した。60分後に溶解を確認後、脱水されたN−メチル−2−ピロリドン(NMP)401.7gを投入し、4,4’−ジアミノジフェニルエーテル(ODA)37.3g(186mmol)を溶解させた。
それらが完全に溶解したことを確認した後、3,3’,4,4’−ビフェニルテトラカルボン酸二無水物(BPDA)46.7g(159mmol)を添加し50℃で5時間撹拌した。その後、フタル酸無水物を0.994g(6.71mmol)添加し、50℃で1時間撹拌し末端封止した。その後室温まで冷却し、下記式で表わされる繰り返し単位を有するポリイミド前駆体3溶液を得た。(式中のRはR1またはR2で表される有機基であり、R1とR2の比率は、R1:R2=90:10となる。)
【0170】
【化19】
【0171】
[試験例:各塩基の触媒能の評価1]
本発明に係る光塩基発生剤から発生する塩基のイミド化を促進する触媒能の評価を行った。
(1)ポリイミド前駆体樹脂組成物溶液の調製、及び塗膜の作製
上記合成例1で得られたポリイミド前駆体1溶液に、表1に記載の塩基を、上記ポリイミド前駆体1溶液の固形分1gに対してそれぞれ0.6mmolの割合で添加し、試験例1〜15及び比較試験例1〜8のポリイミド前駆体樹脂組成物溶液を調製した。
また、上記合成例1で得られたポリイミド前駆体1溶液に、ピペリジンとシクロヘキサノールを、上記ポリイミド前駆体1溶液の固形分1gに対してそれぞれ0.6mmol添加したサンプルを、比較試験例9のポリイミド前駆体樹脂組成物溶液とし、ポリイミド前駆体1溶液そのものを、ブランクとして比較試験例10のポリイミド前駆体樹脂組成物溶液とした。また、塩基としてイソニペコチン酸を用いて、試験例1と同様にしてポリイミド前駆体樹脂組成物溶液を調製したところ、ポリイミド前駆体1溶液にイソニペコチン酸が溶解しなかったため、イミド化率の測定はできなかった。
得られた各ポリイミド前駆体樹脂組成物溶液をクロムめっきされたガラス(50mm×50mm)上に、乾燥後11±1μmとなるようスピンコートし、100℃のホットプレート上で15分乾燥させてそれぞれ対応する塗膜を作製した。
【0172】
(2)イミド化率の測定
イミド化率は、各塗膜について、ホットプレートにより150℃、160℃、170℃でそれぞれ10分間加熱し、得られた各塗膜のIRスペクトルから求めた。
具体的にはFT/IR−6100typeA(日本分光)と付属品名 ATR PRO470−Hのアタッチメントをもちい、下記の条件で測定し、得られたそれぞれのスペクトルを1480−1495cm−1のピークを基準に規格化し、1750−1800cm−1のイミドカルボニルピークの強度比から算出した。
算出の基準として、前記合成例1で得られたポリイミド前駆体1溶液をクロムめっきされたガラス(50mm×50mm)上に、乾燥後11±1μmとなるようスピンコートし、100℃のホットプレート上で15分乾燥した塗膜をイミド化率0%、そのサンプルを、窒素気流下、室温から350℃まで毎分10℃ずつ昇温し、350℃で1時間加熱した後の塗膜をイミド化率100%としてイミド化率を算出した。結果は表1の通りであった。
ホットプレートにより160℃で加熱した際、イミド化率が60%以上であれば、塩基の触媒能は高いと判断される。
なお、表1中の沸点は東京化成工業(株)の2010年度版の試薬カタログ(TCI Fine Chemicals 2010-2011 No.40)、シグマ アルドリッチ ジャパン(株)の2010年度版の試薬カタログ(ALDRICH Chemistry 2009-2010)及び、関東化学(株)の2010年度版のカタログ(The Index of Laboratory Chemicals 2010 No.26)を参照した。減圧状態での沸点が記載されているものに関しては、東京化成工業(株)の2010年度版の試薬カタログ(TCI Fine Chemicals 2010-2011 No.40)内に掲載されている沸点換算表を元に常圧1atm(=101325Pa=760mmHg)での沸点に換算した。
<測定条件>
光源:標準光源
検出器:TGS
積算回数:16
分解:4cm−1
ゼロフィリング:On
アポダイゼーション:Cosine
ゲイン:Auto(32)
アパーチャー:Auto(7.1mm)
スキャンスピード:Auto(2mm/sec)
フィルタ:Auto(10000Hz)
【0173】
【表1】
【0174】
表1の結果から、沸点が150℃以上かつ融点が200℃以下の水酸基を有する塩基は、ポリイミド前駆体のイミド化率を向上させる触媒能が高いことがわかる。
【0175】
[試験例:各塩基の触媒能の評価2]
上記合成例1で得られたポリイミド前駆体1溶液に、上記各塩基の触媒能の評価1で触媒能が高いと評価された、沸点が150℃以上かつ融点が200℃以下の水酸基を有する塩基である4−ピペリジンメタノールを、上記ポリイミド前駆体1溶液の固形分1gに対して表2に示す割合でそれぞれ添加し、試験例16のポリイミド前駆体樹脂組成物溶液を4つの濃度で調製した。
また、上記試験例16において、4−ピペリジンメタノールの代わりに、沸点が150℃以上かつ融点が200℃以下の水酸基を有する塩基である4−ヒドロキシピペリジンを用いた以外は、試験例16と同様にして試験例17のポリイミド前駆体樹脂組成物溶液を4つの濃度で調製した。
また、上記試験例16において、4−ピペリジンメタノールの代わりに、水酸基を有しないピペリジンを用いた以外は、試験例16と同様にして比較試験例11のポリイミド前駆体樹脂組成物溶液を4つの濃度で調製した。
【0176】
【表2】
【0177】
得られた試験例16〜17及び比較試験例11の各濃度のポリイミド前駆体樹脂組成物溶液を、それぞれクロムめっきされたガラス(50mm×50mm)上に、乾燥後11±1μmとなるようスピンコートし、100℃のホットプレート上で15分乾燥させてそれぞれ対応する塗膜を作製した。
各塗膜について、ホットプレートにより160℃で10分間加熱した。その時の各塗膜のイミド化率を、IRスペクトルから上記各塩基の触媒能の評価1と同様の方法で求めた。
各塩基の、ポリイミド前駆体1溶液の固形分1gに対する添加量(mmol/g)とイミド化率の関係を図1に示す。
【0178】
図1の結果から、4−ピペリジンメタノール及び4−ヒドロキシピペリジンを添加した塗膜は、ピペリジンを添加した塗膜に比べて少ない添加量でも高いイミド化率を達成できることが明らかとなった。このことから、沸点が150℃以上かつ融点が200℃以下の水酸基を有する塩基は触媒能が高く、少ない添加量でポリイミド前駆体のイミド化を十分に促進できることがわかる。
【0179】
(製造例1:光塩基発生剤1の合成)
100mLフラスコ中、炭酸カリウム2.00gをメタノール15mLに加えた。50mLフラスコ中、エトキシカルボニルメチル(トリフェニル)ホスホニウムブロミド(東京化成工業(株)製)2.67g(6.2mmol)、2−ヒドロキシ−4−メトキシベンズアルデヒド(東京化成工業(株)製)945mg(6.2mmol)をメタノール10mLに溶解し、よく撹拌した炭酸カリウム溶液にゆっくり滴下した。3時間撹拌した後、TLCにより反応の終了を確認したうえでろ過を行って炭酸カリウムを除き、減圧濃縮した。濃縮後、1Nの水酸化ナトリウム水溶液を50mL加え1時間撹拌した。反応終了後、ろ過によりトリフェニルホスフィンオキシドを除いた後、濃塩酸を滴下し反応液を酸性にした。沈殿物をろ過により集め、少量のクロロホルムにより洗浄することで2−ヒドロキシ−4−メトキシ桂皮酸を1.00g得た。続いて、100mLフラスコ中、2−ヒドロキシ−4−メトキシ桂皮酸1.00g(5.1mmol)を脱水テトラヒドロキシフラン10mLに溶解し、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩(東京化成工業(株)製)1.18g(6.2mmol)を加えた。30分後、4−ピペリジンメタノール(東京化成工業(株)製)712mg(6.2mmol)を加えた。反応終了後、反応溶液を濃縮し、水に溶解した。クロロホルムで抽出した後、1N塩酸、飽和食塩水で洗浄した。その後、シリカゲルカラムクマトグラフィー(展開溶媒:クロロホルム/メタノール=100/1〜10/1、体積比)により精製することにより、下記式で表される製造例1の光塩基発生剤1を100mg得た。
【0180】
【化20】
【0181】
(製造例2:光塩基発生剤の合成)
製造例1において、4−ピペリジンメタノール(東京化成工業(株)製)712mg(6.2mmol)の代わりに、4−ヒドロキシピペリジン(東京化成工業(株)製)625mg(6.2mmol)を用いた以外は、製造例1と同様にして、下記式で表される製造例2の光塩基発生剤2を83mg得た。
【0182】
【化21】
【0183】
(比較製造例1:比較光塩基発生剤1の合成)
実施例1において、4−ピペリジンメタノール(東京化成工業(株)製)712mg(6.2mmol)の代わりに、ピペリジン(東京化成工業(株)製)0.61ml(6.2mmol)を用いた以外は、製造例1と同様にして、下記式で表される比較製造例1の比較光塩基発生剤1を128mg得た。
【0184】
【化22】
【0185】
[評価:光塩基発生剤の塩基発生能]
製造例1、2及び比較製造例1で得られた光塩基発生剤についてそれぞれ1mgの試料を3つ用意し、それぞれを石英製NMR管中で重アセトニトリルに溶解させた。i線を20%透過するフィルタと高圧水銀灯を用いて、1本には400mJ/cm2で光照射を行い、他の1本には800mJ/cm2で光照射を行った。残り1本には光照射を行わなかった。各サンプルの1H NMRを測定し、異性化の割合を求めた。
【0186】
製造例1、2及び比較製造例1で得られた光塩基発生剤は、400mJ/cm2の照射および800mJ/cm2の照射により異性化した。異性化させたサンプルを160℃に加熱すると、異性化した化合物のほぼ100%が環化し、それにともない塩基が発生し、表3に示す塩基発生率(%)となった。
【0187】
【表3】
【0188】
(実施例1:感光性ポリイミド樹脂組成物1の調製)
合成例2で得られたポリイミド前駆体2溶液の固形分100重量部に対し、製造例1で得られた光塩基発生剤1を15重量部添加し、実施例1の感光性ポリイミド樹脂組成物1を調製した。
なお、光塩基発生剤1から発生する塩基(4−ピペリジンメタノール)は、160℃で、当該塩基100重量部に対して、合成例2で得られたポリイミド前駆体(固形分)を10重量部以上溶解した。
【0189】
(実施例2:感光性ポリイミド樹脂組成物2の調製)
実施例1において製造例1で得られた光塩基発生剤1の代わりに、製造例2で得られた光塩基発生剤2を用いた以外は、実施例1と同様にして、実施例2の感光性ポリイミド樹脂組成物2を調製した。
なお、光塩基発生剤2から発生する塩基(4−ヒドロキシピペリジン)は、165℃で、当該塩基100重量部に対して、合成例2で得られたポリイミド前駆体(固形分)を10重量部以上溶解した。
【0190】
(実施例3:感光性ポリイミド樹脂組成物3の調製)
合成例1で得られたポリイミド前駆体1溶液の固形分100重量部に対し、製造例1で得られた光塩基発生剤1を22.3重量部添加し、実施例3の感光性ポリイミド樹脂組成物3を調製した。
なお、光塩基発生剤1から発生する塩基(4−ピペリジンメタノール)は、160℃で、当該塩基100重量部に対して、合成例1で得られたポリイミド前駆体(固形分)を10重量部以上溶解した。
【0191】
(実施例4:感光性ポリイミド樹脂組成物4の調製)
合成例3で得られたポリイミド前駆体3溶液の固形分100重量部に対し、製造例1で得られた光塩基発生剤1を22.3重量部添加し、実施例4の感光性ポリイミド樹脂組成物4を調製した。
なお、光塩基発生剤1から発生する塩基(4−ピペリジンメタノール)は、160℃で、当該塩基100重量部に対して、合成例3で得られたポリイミド前駆体(固形分)を10重量部以上溶解した。
【0192】
(比較例1:比較感光性ポリイミド樹脂組成物1の調製)
実施例1において製造例1で得られた光塩基発生剤の代わりに、比較製造例1で得られた比較光塩基発生剤1を用いた以外は実施例1と同様にして、比較例1の比較感光性ポリイミド樹脂組成物1を調製した。
なお、比較光塩基発生剤1から発生する塩基(ピペリジン)は、沸点が106℃であるため、150℃〜200℃のいずれかの温度における当該塩基に対するポリイミド前駆体の溶解を試みることはできなかった。
【0193】
(比較例2:比較感光性ポリイミド樹脂組成物2の調製)
合成例1で得られたポリイミド前駆体1溶液の固形分100重量部に対し、比較製造例1で得られた比較光塩基発生剤1を20重量部添加し、比較例2の比較感光性ポリイミド樹脂組成物2を調製した。
なお、比較光塩基発生剤1から発生する塩基(ピペリジン)は、沸点が106℃であるため、150℃〜200℃のいずれかの温度における当該塩基に対するポリイミド前駆体の溶解を試みることはできなかった。
【0194】
(比較例3:比較感光性ポリイミド樹脂組成物3の調製)
合成例3で得られたポリイミド前駆体3溶液の固形分100重量部に対し、比較製造例1で得られた比較光塩基発生剤1を20重量部添加し、比較例3の比較感光性ポリイミド樹脂組成物3を調製した。
なお、比較光塩基発生剤1から発生する塩基(ピペリジン)は、沸点が106℃であるため、150℃〜200℃のいずれかの温度における当該塩基に対するポリイミド前駆体の溶解を試みることはできなかった。
【0195】
[感光性ポリイミド樹脂組成物の評価:現像性評価1]
(1)塗膜の作製
実施例1、実施例2、及び比較例1の感光性ポリイミド樹脂組成物を、それぞれ、クロムめっきされたガラス(50mm×50mm)上に乾燥後17±1μmとなるようにスピンコートし、100℃のホットプレート上で15分乾燥させて実施例1、実施例2、及び比較例1の感光性ポリイミド樹脂組成物の塗膜をそれぞれ2枚をセットとして作製した。
上記の通り作製した塗膜のうち1枚を、露光部の評価を行う塗膜とし、手動露光機を用いて高圧水銀灯により、2J/cm2露光を行った。残りの一枚を未露光部の評価を行う塗膜として、露光を行わなかった。その後、各塗膜をホットプレートにより下表4〜6に示す温度(PEB温度)でそれぞれ10分間加熱し、現像性評価サンプルを得た。各現像性評価サンプルにおいては、露光部の評価を行う塗膜1枚と未露光部の評価を行う塗膜1枚の2枚をセットにして評価に用いた。
【0196】
(2)現像性評価
それぞれの塗膜について、50℃のテトラメチルアンモニウムハイドロオキサイド2.38重量%水溶液で現像処理し、600秒後までの塗膜の溶解速度を測定した。実施例1、実施例2、及び比較例1の感光性ポリイミド樹脂組成物の評価結果はそれぞれ表4〜6の通りであった。
現像性を評価する方法としては、各塗膜の露光部と未露光部の時間あたりの膜減り量(溶解速度)を算出し、比較を行った。未露光部の塗膜の溶解速度に対する露光部の塗膜の溶解速度の比(溶解性コントラスト)の値が大きいほど、残膜率が高くなると評価できる。
なお、比較例1の未露光部は、現像液に殆ど溶解せず、膨潤した後、1200秒後に塗膜がガラスから剥れた。
【0197】
【表4】
【0198】
【表5】
【0199】
【表6】
【0200】
評価の結果、実施例1、実施例2はいずれのPEB温度でも比較例1より溶解性コントラストの値が高いことがわかった。実施例1、実施例2の感光性ポリイミド樹脂組成物は、環境負荷の低減から、アルコール等の有機溶剤を添加しない塩基性水溶液を現像液として用いた場合でもより短時間で残膜率の高い良好な形状のパターンが形成できると推測できる。一方、比較例1の感光性ポリイミド樹脂組成物は、アルコール等の有機溶剤を添加しない塩基性水溶液を現像液として用いると、良好なレリーフパターンが得られないことが示唆された。
また、上記の結果から、本発明の感光性ポリイミド樹脂組成物は、露光後ベーク時の加熱条件を従来より緩和しても、ポリイミド前駆体のイミド化を十分に促進でき、更に、塩基に導入されている水酸基の効果で、未露光部の現像液への溶解が促進されることが示唆された。水酸基を有する塩基は、従来現像液に添加していたアルコールの代替として機能し、ポリイミド前駆体の溶解を促進する溶解促進剤としても機能しているのではないかと推測される。
【0201】
[感光性ポリイミド樹脂組成物の評価:現像性評価2]
(1)塗膜の作製
実施例3、及び比較例2の感光性ポリイミド樹脂組成物を、それぞれ、クロムめっきされたガラス(50mm×50mm)上に乾燥後2μmとなるようにスピンコートし、120℃のホットプレート上で15分乾燥させて実施例3、及び比較例2の感光性ポリイミド樹脂組成物の塗膜をそれぞれ2枚をセットとして作製した。
上記の通り作製した塗膜のうち1枚を、露光部の評価を行う塗膜とし、手動露光機を用いて高圧水銀灯により、500mJ/cm2露光を行った。残りの一枚を未露光部の評価を行う塗膜として、露光を行わなかった。その後、各塗膜をホットプレートにより160℃でそれぞれ2分間加熱し、現像性評価サンプルを得た。各現像性評価サンプルにおいては、露光部の評価を行う塗膜1枚と未露光部の評価を行う塗膜1枚の2枚をセットにして評価に用いた。
【0202】
(2)現像性評価
それぞれの塗膜について、23℃のテトラメチルアンモニウムハイドロオキサイド2.38重量%水溶液で現像処理し、塗膜の溶解速度を測定した。現像性は、上記現像性評価1と同様の方法で評価した。
実施例3、及び比較例2の感光性ポリイミド樹脂組成物の評価結果はそれぞれ表7の通りであった。
【0203】
【表7】
【0204】
評価の結果、実施例3は比較例2と比べて未露光部の溶解速度が速く、溶解性コントラストの値が高いことがわかった。
【0205】
[感光性ポリイミド樹脂組成物の評価:現像性評価3]
(1)塗膜の作製
実施例4、及び比較例3の感光性ポリイミド樹脂組成物を、それぞれ、クロムめっきされたガラス(50mm×50mm)上に乾燥後2μmとなるようにスピンコートし、120℃のホットプレート上で15分乾燥させて実施例4、及び比較例3の感光性ポリイミド樹脂組成物の塗膜をそれぞれ2枚をセットとして作製した。
上記の通り作製した塗膜のうち1枚を、露光部の評価を行う塗膜とし、手動露光機を用いて高圧水銀灯により、500mJ/cm2露光を行った。残りの一枚を未露光部の評価を行う塗膜として、露光を行わなかった。その後、各塗膜をホットプレートにより160℃でそれぞれ2分間加熱し、現像性評価サンプルを得た。各現像性評価サンプルにおいては、露光部の評価を行う塗膜1枚と未露光部の評価を行う塗膜1枚の2枚をセットにして評価に用いた。
【0206】
(2)現像性評価
それぞれの塗膜について、23℃のテトラメチルアンモニウムハイドロオキサイド2.38重量%水溶液で現像処理し、塗膜の溶解速度を測定した。現像性は、上記現像性評価1と同様の方法で評価した。
実施例4、及び比較例3の感光性ポリイミド樹脂組成物の評価結果はそれぞれ表8の通りであった。
【0207】
【表8】
【0208】
評価の結果、実施例4は比較例3と比べて未露光部の溶解速度が速く、溶解性コントラストの値が高いことがわかった。
【0209】
現像性評価2〜3の結果から、本発明の光塩基発生剤は、添加するポリイミド前駆体の組成を変えても、露光部のイミド化を十分に促進させ、一方、未露光部は塩基発生剤に導入されている水酸基の効果で現像液への溶解が促進されるために、より短時間で残膜率の高い良好な形状のパターンが形成できることが示唆された。
【0210】
[感光性ポリイミド樹脂組成物の評価:塗膜の感度とγ値の評価1]
実施例1、実施例2、及び比較例1の感光性ポリイミド樹脂組成物を、それぞれ、クロムめっきされたガラス(50mm×50mm)上に乾燥後18±1μmとなるようにスピンコートし、100℃のホットプレート上で15分乾燥させて、各塗膜を得た。
【0211】
作成した塗膜に対し、手動露光機を用いて、高圧水銀灯により全面露光を行った。実施例1の塗膜に対しては、0、25、50、75、100、150、300、400、500、1000mJ/cm2でそれぞれ全面露光を行い、実施例2の塗膜に対しては、0、50、75、100、200、300、500、1000mJ/cm2でそれぞれ全面露光を行った。比較例1の塗膜に対しては、0、25、75、100、200、300、400、500、1000mJ/cm2でそれぞれ全面露光を行った。その後、各塗膜をホットプレートにより実施例1の塗膜に対しては160℃で、実施例2の塗膜に対しては165℃で、比較例1の塗膜に対しては165℃でそれぞれ10分間加熱し、これらの塗膜をそれぞれ、テトラメチルアンモニウムハイドロオキサイド2.5重量%水溶液とイソプロパノールを8:2で混合した溶液に実施例1の塗膜に対しては40℃で5分間、実施例2の塗膜に対しては30℃で10分間、比較例1の塗膜に対しては40℃で6分間現像処理し、基板上の残存膜厚を測定した。結果を、表9に示す。また、露光量と規格化残膜率の関係を表す感度曲線を図2に示す。なお、図中の規格化残膜率は、(現像後膜厚×100)/(現像後の最高膜厚)とした。
また、表中の感度とは、感度曲線から求めた規格化残膜率が約1となる時の露光量である。表中のγ値はコントラストを表す指標であり、値が大きいほどコントラストが高くテーパー角の大きい形状のパターンを得ることができる。γ値は以下の式により求めた。
γ=1/{log(規格化残膜率が約1となる時の露光量)−log(規格化残膜率が0となる時の露光量)}
なお、実施例2、比較例1については規格化残膜率が0となる露光量が正確に測れなかったので、規格化残膜率が最小となる露光量(実施例2は50mJ、比較例1は25mJ)−1mJを仮に規格化残膜率が0となる時の露光量としてγ値を算出した。
【0212】
【表9】
【0213】
UV照射量の増加とともに、残膜率が上昇していることから、UV照射および、加熱によって、塩基発生剤がアミド結合を形成可能なNH基を1つ有する2級アミンを発生し、イミド化が進行していることが示された。本発明の実施例1では100mJ/cm2で、実施例2では75mJ/cm2で規格化残膜率が約1となった。それに対して、比較例1では100mJ/cm2で規格化残膜率が約1となった。このことから、本発明の感光性ポリイミド樹脂組成物は、比較感光性ポリイミド樹脂組成物と同程度の露光量でイミド化が進行することが示された。
一方、γ値は、値が大きいほど溶解性コントラストが高くテーパー角の大きい形状のパターンを得ることができる。本発明の実施例1ではγ値が8.0で、実施例2ではγ値が5.4であったのに対し、比較例1ではγ値が1.6であった。このことから、本発明の感光性ポリイミド樹脂組成物は、比較感光性ポリイミド樹脂組成物と比べて溶解性コントラストが高く、より矩形に近い形状のパターンを得ることができることが示された。
【0214】
[感光性ポリイミド樹脂組成物の評価:塗膜の感度とγ値の評価2]
実施例3、及び比較例2の感光性ポリイミド樹脂組成物を、それぞれ、クロムめっきされたガラス(50mm×50mm)上に乾燥後2μmとなるようにスピンコートし、120℃のホットプレート上で15分乾燥させて、各塗膜を得た。
【0215】
作成した塗膜に対し、手動露光機を用いて、適宜露光量を選んで高圧水銀灯により全面露光を行った。その後、各塗膜をホットプレートにより160℃でそれぞれ2分間加熱した。これらの塗膜をそれぞれ、23℃のテトラメチルアンモニウムハイドロオキサイド2.38重量%水溶液で、実施例3の塗膜に対しては90秒間、比較例2の塗膜に対しては150秒間現像処理し、基板上の残存膜厚を測定した。その結果を表10に示す。また、露光量と規格化残膜率の関係を表す感度曲線を図3に示す。なお、図中の規格化残膜率、表中の感度及びγ値は、上記塗膜の感度とγ値の評価1と同様である。
【0216】
【表10】
【0217】
UV照射量の増加とともに、残膜率が上昇していることから、UV照射および、加熱によって、塩基発生剤がアミド結合を形成可能なNH基を1つ有する2級アミンを発生し、イミド化が進行していることが示された。本発明の実施例3では118mJ/cm2で、比較例2では82mJ/cm2で規格化残膜率が1となった。
一方、γ値は、値が大きいほど溶解性コントラストが高くテーパー角の大きい形状のパターンを得ることができる。本発明の実施例3ではγ値が1.8であったのに対し、比較例2ではγ値が1.1であった。このことから、本発明の感光性ポリイミド樹脂組成物は、比較感光性ポリイミド樹脂組成物と比べて溶解性コントラストが高く、より矩形に近い形状のパターンを得ることができることが示された。
【0218】
[感光性ポリイミド樹脂組成物の評価:パターン形成能評価1]
実施例1、実施例2、及び比較例1の感光性ポリイミド樹脂組成物を、それぞれ、クロムめっきされたガラス(50mm×50mm)上に乾燥後18±1μmとなるようにスピンコートし、100℃のホットプレート上で15分乾燥させて、各塗膜を得た。
【0219】
作成した塗膜に対し、手動露光機を用いて、高圧水銀灯により細線評価用マスクを介して、200mJ/cm2露光を行った。続いて各塗膜を、実施例2及び比較例1の塗膜は165℃、実施例1の塗膜は160℃でそれぞれ5分間加熱した。
これらのサンプルの現像は、環境への負荷を低減させるため、アルコールを含まない、テトラメチルアンモニウムハイドロオキサイド2.38重量%水溶液を用い、200mlのビーカーでマグネチックスターラーによって攪拌させ、湯浴によって50℃に加熱した状態で、サンプルを浸漬させることにより行った。
通常、現像液からアルコールを除去することにより、環境負荷が低減される一方、未露光部の現像速度が低下し、現像時間が長くなる。
【0220】
パターン形成後、塗膜を窒素雰囲気下、350℃、1時間熱処理し(昇温速度10℃/分、自然放冷)後硬化させた。その結果、実施例1及び実施例2の塗膜については、表11に示すような細線パターンを得た。比較例1の塗膜については、現像液に1時間以上浸漬しても未露光部が残存しており、パターンを形成できなかった。この結果より、本発明の感光性ポリイミド樹脂組成物は、良好なパターンを形成できることが明らかとなった。
【0221】
【表11】
【0222】
<パターン形状の評価基準>
◎:パターン断面のテーパー角が55度超過
○:パターン断面のテーパー角が30〜55度
【0223】
比較例1の比較感光性ポリイミド樹脂組成物の場合、未露光部がゲル状になる、又は膨張するなどの現象が見られ、現像液に溶解しにくかった。一方、実施例1及び実施例2の感光性ポリイミド樹脂組成物のように発生塩基が水酸基を有する場合、はっ水性の高いフッ素含有ポリイミド前駆体を用いた場合でも、未露光部における前記のような現象は見られなかった。これは発生塩基の水酸基の効果によって未露光部のアルカリ水溶液に対する親和性が向上したためと考えられる。さらに実施例1の感光性ポリイミド樹脂組成物を用いる場合はPEB温度を160℃に下げることができるため、未露光部のイミド化を抑えられ、現像時間を短縮することができた。
【0224】
[感光性ポリイミド樹脂組成物の評価:パターン形成能評価2]
実施例3、及び比較例2の感光性ポリイミド樹脂組成物を、それぞれ、クロムめっきされたガラス(50mm×50mm)上に乾燥後2μmとなるようにスピンコートし、120℃のホットプレート上で15分乾燥させて、各塗膜を得た。
作成した塗膜に対し、手動露光機を用いて、高圧水銀灯により細線評価用マスクを介して、200mJ/cm2露光を行った。続いて各塗膜を、160℃で2分間加熱した。
これらのサンプルの現像は、環境への負荷を低減させるため、アルコールを含まない、テトラメチルアンモニウムハイドロオキサイド2.38重量%水溶液を用いて90秒間処理を行った。
その結果、実施例3の塗膜については、現像後膜厚1.60μm、残膜率90%の細線パターンの形成を確認した。
一方、比較例2の塗膜については、90秒間の現像処理ではまだ未露光部が残存しており、パターンを形成できなかった。
パターン形成能評価2の結果より、実施例3の感光性ポリイミド樹脂組成物は、比較例2と比べて短時間で良好なパターンを形成できることが明らかとなった。
【0225】
[感光性ポリイミド樹脂組成物の評価:パターン形成能評価3]
実施例4、及び比較例3の感光性ポリイミド樹脂組成物を、それぞれ、クロムめっきされたガラス(50mm×50mm)上に乾燥後2μmとなるようにスピンコートし、120℃のホットプレート上で15分乾燥させて、各塗膜を得た。
作成した塗膜に対し、手動露光機を用いて、高圧水銀灯により細線評価用マスクを介して、200mJ/cm2露光を行った。続いて各塗膜を、160℃で2分間加熱した。
これらのサンプルの現像は、環境への負荷を低減させるため、アルコールを含まない、テトラメチルアンモニウムハイドロオキサイド2.38重量%水溶液を用いて60秒間処理を行った。
その結果、実施例4の塗膜については、現像後膜厚1.48μm、残膜率81%の細線パターンの形成を確認した。
比較例3の塗膜については、60秒間の現像処理ではまだ未露光部が残存しており、パターンを形成できなかった。
パターン形成能評価3の結果より、実施例4の感光性ポリイミド樹脂組成物は、比較例3と比べて短時間で良好なパターンを形成できることが明らかとなった。
【0226】
以上の結果から、水酸基を有する塩基は、イミド化の触媒能が高く、このような塩基を発生する光塩基発生剤を用いた本発明の感光性ポリイミド樹脂組成物は、露光後ベーク時の加熱条件を従来より緩和しても、ポリイミド前駆体のイミド化を十分に促進でき、更に、塩基に導入されている水酸基の効果で、未露光部の現像液への溶解が促進されることが明らかとなった。そして、その結果、本発明の感光性ポリイミド樹脂組成物は、現像時間を短縮でき、アルコール等を添加しない塩基性水溶液を現像液として用いることが可能で、露光部と未露光部の溶解性コントラストを高くすることができ、より解像度の高いパターンを形成できたり、レリーフパターンの形状が良好になるという効果が得られることが明らかとなった。
【特許請求の範囲】
【請求項1】
発生する塩基が共有結合を用いて潜在化されていて、電磁波の照射により塩基を発生する塩基発生剤であって、水酸基を有する塩基を発生することを特徴とする、前駆体型感光性ポリイミド用光塩基発生剤。
【請求項2】
沸点が150℃以上かつ融点が200℃以下の水酸基を有する塩基を発生することを特徴とする、請求項1に記載の前駆体型感光性ポリイミド用光塩基発生剤。
【請求項3】
下記化学式(1−1)、下記化学式(1−2)又は下記化学式(1−3)で表されることを特徴とする、請求項1又は2に記載の前駆体型感光性ポリイミド用光塩基発生剤。
【化1】
(式(1−1)中、R1及びR2は、それぞれ独立に、水素原子又は有機基であり、同一であっても異なっていても良い。R1及びR2の少なくとも1つは有機基である。R1及びR2は、それらが結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。但し、R1及びR2のいずれかの有機基が水酸基を有する。R3及びR4はそれぞれ独立に、水素原子、ハロゲン原子、水酸基、メルカプト基、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、又は有機基であり、同一であっても異なっていても良い。R5、R6、R7及びR8は、それぞれ独立に、水素原子、ハロゲン原子、水酸基、メルカプト基、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、アミノ基、アンモニオ基又は有機基であり、同一であっても異なっていても良い。R5、R6、R7及びR8は、それらの2つ以上が結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。R9は、水素原子、或いは、加熱及び/又は電磁波の照射により脱保護可能な保護基である。)
【化2】
(式(1−2)中、R1及びR2は、それぞれ独立に、水素原子又は有機基であり、同一であっても異なっていても良い。R1及びR2の少なくとも1つは有機基である。R1及びR2は、それらが結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。但し、R1及びR2のいずれかの有機基が水酸基を有する。R10及びR10’はそれぞれ独立に水素原子、ハロゲン原子、水酸基、メルカプト基、ニトロ基、シリル基、シラノール基、又は有機基である。R11、R12、R13、R14及びR15は、それぞれ独立に、水素原子、ハロゲン原子、水酸基、メルカプト基、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、アミノ基、アンモニオ基又は有機基であり、同一であっても異なっていても良い。R11、R12、R13、R14及びR15は、それらの2つ以上が結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。)
【化3】
(式(1−3)中、R17は、水酸基を有する有機基で置換されたアミノ基又は水酸基を有する有機基、R18及びR19は、それぞれ独立に、水素原子、ハロゲン原子、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、アミノ基、アンモニオ基又は有機基であり、同一であっても異なっていても良いが、R18及びR19の少なくとも1つはヘテロ原子を有していても良い芳香族化合物である。R17、R18及びR19は、それらの2つ以上が結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。)
【請求項4】
発生する塩基が脂肪族アミンであることを特徴とする、請求項1乃至3のいずれか1項に記載の前駆体型感光性ポリイミド用光塩基発生剤。
【請求項5】
発生する塩基が酸性基を含まないことを特徴とする、請求項1乃至4のいずれか1項に記載の前駆体型感光性ポリイミド用光塩基発生剤。
【請求項6】
ポリイミド前駆体と、前記請求項1乃至5のいずれか1項に記載の光塩基発生剤とを含有する、ネガ型感光性ポリイミド樹脂組成物。
【請求項7】
前記光塩基発生剤が発生する塩基が、150〜200℃のいずれかの温度において、当該塩基100重量部に対して、前記ポリイミド前駆体を10重量部以上溶解する塩基である請求項6に記載のネガ型感光性ポリイミド樹脂組成物。
【請求項8】
塗料、印刷インキ、シール剤、又は接着剤、或いは、表示装置、半導体装置、電子部品、微小電気機械システム、光造形物、光学部材又は建築材料の形成材料として用いられる請求項6又は7に記載のネガ型感光性ポリイミド樹脂組成物。
【請求項9】
前記請求項6乃至8のいずれか1項に記載のネガ型感光性ポリイミド樹脂組成物を用いて塗膜又は成形体を形成し、当該塗膜又は成形体を、所定パターン状に電磁波を照射し、照射後又は照射と同時に加熱し、前記照射部位の溶解性を変化させた後、現像することを特徴とするレリーフパターンの製造方法。
【請求項10】
前記請求項6乃至8のいずれか1項に記載のネガ型感光性ポリイミド樹脂組成物又はその硬化物により少なくとも一部が形成されている、印刷物、塗料、シール剤、接着剤、表示装置、半導体装置、電子部品、微小電気機械システム、光造形物、光学部材又は建築材料のいずれかの物品。
【請求項1】
発生する塩基が共有結合を用いて潜在化されていて、電磁波の照射により塩基を発生する塩基発生剤であって、水酸基を有する塩基を発生することを特徴とする、前駆体型感光性ポリイミド用光塩基発生剤。
【請求項2】
沸点が150℃以上かつ融点が200℃以下の水酸基を有する塩基を発生することを特徴とする、請求項1に記載の前駆体型感光性ポリイミド用光塩基発生剤。
【請求項3】
下記化学式(1−1)、下記化学式(1−2)又は下記化学式(1−3)で表されることを特徴とする、請求項1又は2に記載の前駆体型感光性ポリイミド用光塩基発生剤。
【化1】
(式(1−1)中、R1及びR2は、それぞれ独立に、水素原子又は有機基であり、同一であっても異なっていても良い。R1及びR2の少なくとも1つは有機基である。R1及びR2は、それらが結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。但し、R1及びR2のいずれかの有機基が水酸基を有する。R3及びR4はそれぞれ独立に、水素原子、ハロゲン原子、水酸基、メルカプト基、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、又は有機基であり、同一であっても異なっていても良い。R5、R6、R7及びR8は、それぞれ独立に、水素原子、ハロゲン原子、水酸基、メルカプト基、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、アミノ基、アンモニオ基又は有機基であり、同一であっても異なっていても良い。R5、R6、R7及びR8は、それらの2つ以上が結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。R9は、水素原子、或いは、加熱及び/又は電磁波の照射により脱保護可能な保護基である。)
【化2】
(式(1−2)中、R1及びR2は、それぞれ独立に、水素原子又は有機基であり、同一であっても異なっていても良い。R1及びR2の少なくとも1つは有機基である。R1及びR2は、それらが結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。但し、R1及びR2のいずれかの有機基が水酸基を有する。R10及びR10’はそれぞれ独立に水素原子、ハロゲン原子、水酸基、メルカプト基、ニトロ基、シリル基、シラノール基、又は有機基である。R11、R12、R13、R14及びR15は、それぞれ独立に、水素原子、ハロゲン原子、水酸基、メルカプト基、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、アミノ基、アンモニオ基又は有機基であり、同一であっても異なっていても良い。R11、R12、R13、R14及びR15は、それらの2つ以上が結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。)
【化3】
(式(1−3)中、R17は、水酸基を有する有機基で置換されたアミノ基又は水酸基を有する有機基、R18及びR19は、それぞれ独立に、水素原子、ハロゲン原子、スルフィド基、シリル基、シラノール基、ニトロ基、ニトロソ基、スルフィノ基、スルホ基、スルホナト基、ホスフィノ基、ホスフィニル基、ホスホノ基、ホスホナト基、アミノ基、アンモニオ基又は有機基であり、同一であっても異なっていても良いが、R18及びR19の少なくとも1つはヘテロ原子を有していても良い芳香族化合物である。R17、R18及びR19は、それらの2つ以上が結合して環状構造を形成していても良く、ヘテロ原子の結合を含んでいても良い。)
【請求項4】
発生する塩基が脂肪族アミンであることを特徴とする、請求項1乃至3のいずれか1項に記載の前駆体型感光性ポリイミド用光塩基発生剤。
【請求項5】
発生する塩基が酸性基を含まないことを特徴とする、請求項1乃至4のいずれか1項に記載の前駆体型感光性ポリイミド用光塩基発生剤。
【請求項6】
ポリイミド前駆体と、前記請求項1乃至5のいずれか1項に記載の光塩基発生剤とを含有する、ネガ型感光性ポリイミド樹脂組成物。
【請求項7】
前記光塩基発生剤が発生する塩基が、150〜200℃のいずれかの温度において、当該塩基100重量部に対して、前記ポリイミド前駆体を10重量部以上溶解する塩基である請求項6に記載のネガ型感光性ポリイミド樹脂組成物。
【請求項8】
塗料、印刷インキ、シール剤、又は接着剤、或いは、表示装置、半導体装置、電子部品、微小電気機械システム、光造形物、光学部材又は建築材料の形成材料として用いられる請求項6又は7に記載のネガ型感光性ポリイミド樹脂組成物。
【請求項9】
前記請求項6乃至8のいずれか1項に記載のネガ型感光性ポリイミド樹脂組成物を用いて塗膜又は成形体を形成し、当該塗膜又は成形体を、所定パターン状に電磁波を照射し、照射後又は照射と同時に加熱し、前記照射部位の溶解性を変化させた後、現像することを特徴とするレリーフパターンの製造方法。
【請求項10】
前記請求項6乃至8のいずれか1項に記載のネガ型感光性ポリイミド樹脂組成物又はその硬化物により少なくとも一部が形成されている、印刷物、塗料、シール剤、接着剤、表示装置、半導体装置、電子部品、微小電気機械システム、光造形物、光学部材又は建築材料のいずれかの物品。
【図1】

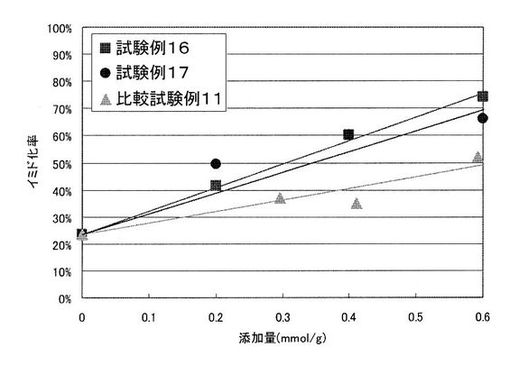
【図2】
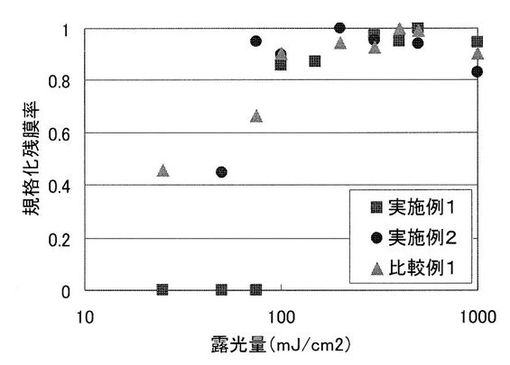
【図3】
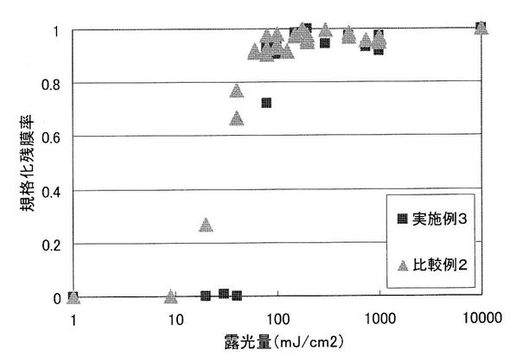
【図2】
【図3】
【公開番号】特開2012−118523(P2012−118523A)
【公開日】平成24年6月21日(2012.6.21)
【国際特許分類】
【出願番号】特願2011−246287(P2011−246287)
【出願日】平成23年11月10日(2011.11.10)
【出願人】(000002897)大日本印刷株式会社 (14,506)
【Fターム(参考)】
【公開日】平成24年6月21日(2012.6.21)
【国際特許分類】
【出願日】平成23年11月10日(2011.11.10)
【出願人】(000002897)大日本印刷株式会社 (14,506)
【Fターム(参考)】
[ Back to top ]