
光学活性アミン化合物の製造方法、並びに、ジアステレオマー塩及びその製造方法
【課題】光学活性アミン化合物の実用的な製造方法を提供すること。
【解決手段】式(A−1)又は(A−2)で表されるアミン化合物と式(B−1)又は(B−2)で表される酸性分割剤とを反応させてジアステレオマー塩を形成する工程、得られたジアステレオマー塩の少なくとも一部を析出させ精製ジアステレオマー塩を得る工程、及び、精製ジアステレオマー塩を塩基性水溶液により分解し、式(A−1)又は(A−2)で表される光学活性アミン化合物を得る工程を含むことを特徴とする光学活性アミン化合物の製造方法。

【解決手段】式(A−1)又は(A−2)で表されるアミン化合物と式(B−1)又は(B−2)で表される酸性分割剤とを反応させてジアステレオマー塩を形成する工程、得られたジアステレオマー塩の少なくとも一部を析出させ精製ジアステレオマー塩を得る工程、及び、精製ジアステレオマー塩を塩基性水溶液により分解し、式(A−1)又は(A−2)で表される光学活性アミン化合物を得る工程を含むことを特徴とする光学活性アミン化合物の製造方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、医薬、農薬、電子材料等の原料あるいは中間体として有用な光学活性アミン化合物の製造方法に関する。また、本発明は、前記アミン化合物と酸性分割剤より形成されるジアステレオマー塩、及び、該ジアステレオマー塩の分離方法に関する。
【背景技術】
【0002】
光学活性アミン化合物は、医薬、農薬、電子材料等の原料あるいは中間体などとして、幅広い利用が期待されている。
例えば、3−アミノピペリジン類の光学分割法としては、特許文献1及び2に記載の方法が知られている。
特許文献1には、3−アミノピペリジンに酸性分割剤を作用させ、ジアステレオマー塩を分離し、光学分割を行う方法が記載されている。
また、特許文献2には、キサンチン構造を有する保護基を1位に有する3−アミノピペリジン類に酸性分割剤を作用させ、ジアステレオマー塩を分離し、光学分割を行う方法が記載されている。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】国際公開第2007/75630号パンフレット
【特許文献2】特表2008−519005号公報
【発明の開示】
【発明が解決しようとする課題】
【0004】
本発明の目的は、光学活性アミン化合物の実用的な製造方法を提供することである。
本発明の他の目的は、新規なジアステレオマー塩を提供することである。
また、本発明の他の目的は、光学活性アミン化合物と酸性分割剤とからなるジアステレオマー塩の実用的な製造方法を提供することである。
【課題を解決するための手段】
【0005】
本発明者らは光学活性アミン化合物の実用的製造方法について研究を重ねた結果、特定の酸性分割剤とジアステレオマー塩を生成させることにより、光学分割が可能であることを見出し、本発明を完成させることができた。
前記課題は、以下の手段<1>、<4>及び<7>により解決された。好ましい実施態様である<2>、<3>、<5>、<6>、<8>及び<9>と共に以下に記載する。
<1>下記式(A−1)又は(A−2)で表されるアミン化合物と下記式(B−1)又は(B−2)で表される酸性分割剤とを反応させてジアステレオマー塩を形成する工程、得られたジアステレオマー塩の少なくとも一部を析出させ精製ジアステレオマー塩を得る工程、及び、精製ジアステレオマー塩を塩基性水溶液により分解し、下記式(A−1)又は(A−2)で表される光学活性アミン化合物を得る工程を含むことを特徴とする
光学活性アミン化合物の製造方法、
【0006】
【化1】
(式中、R1及びR2はそれぞれ独立に、水素原子、炭素数1〜10のアルキル基、又は、炭素数6〜10のアリール基を表し、R3は炭素数7〜20のアラルキル基、炭素数2〜20のアルキルオキシカルボニル基、炭素数7〜20のアリールオキシカルボニル基、又は、炭素数8〜20のアラルキルオキシカルボニル基を表し、*は不斉炭素原子であることを表す。)
【0007】
【化2】
(式中、R4及びR9は、酸性官能基を表し、R5〜R8はそれぞれ独立に、アルキル基、アリール基、アラルキル基、アルキルオキシ基、アリールオキシ基、アラルキルオキシ基、アルキルカルボニルオキシ基、アリールカルボニルオキシ基、アラルキルカルボニルオキシ基、又は、ヒドロキシ基を表し、R10は、酸性官能基、又は、アリールアミノカルボニル基を表し、*は不斉炭素原子であることを表す。ただし、R5とR6とは異なる基である。)
<2>前記アミン化合物が、下記式(A−1−1)〜(A−1−3)で表されるアミン化合物である上記<1>に記載の光学活性アミン化合物の製造方法、
【0008】
【化3】
(式中、*は不斉炭素原子であることを表す。)
<3>前記酸性分割剤が、下記式(B−1−1)〜(B−1−4)又は(B−2−1)〜(B−2−6)で表される化合物である上記<1>又は<2>に記載の光学活性アミン化合物の製造方法、
【0009】
【化4】
(式中、*は不斉炭素原子であることを表す。)
<4>下記式(A−1)又は(A−2)で表されるアミン化合物と下記式(B−1)又は(B−2)で表される酸性分割剤とを反応させてジアステレオマー塩を形成する工程、及び、得られたジアステレオマー塩の少なくとも一部を析出させ精製ジアステレオマー塩を得る工程を含むことを特徴とするジアステレオマー塩の製造方法、
【0010】
【化5】
(式中、R1及びR2はそれぞれ独立に、水素原子、炭素数1〜10のアルキル基、又は、炭素数6〜10のアリール基を表し、R3は炭素数7〜20のアラルキル基、炭素数2〜20のアルキルオキシカルボニル基、炭素数7〜20のアリールオキシカルボニル基、又は、炭素数8〜20のアラルキルオキシカルボニル基を表し、*は不斉炭素原子であることを表す。)
【0011】
【化6】
(式中、R4及びR9は、酸性官能基を表し、R5〜R8はそれぞれ独立に、アルキル基、アリール基、アラルキル基、アルキルオキシ基、アリールオキシ基、アラルキルオキシ基、アルキルカルボニルオキシ基、アリールカルボニルオキシ基、アラルキルカルボニルオキシ基、又は、ヒドロキシ基を表し、R10は、酸性官能基、又は、アリールアミノカルボニル基を表し、*は不斉炭素原子であることを表す。ただし、R5とR6とは異なる基である。)
<5>前記アミン化合物が、下記式(A−1−1)〜(A−1−3)で表されるアミン化合物である上記<4>に記載のジアステレオマーの製造方法、
【0012】
【化7】
(式中、*は不斉炭素原子であることを表す。)
<6>前記酸性分割剤が、下記式(B−1−1)〜(B−1−4)又は(B−2−1)〜(B−2−6)で表される化合物である上記<4>又は<5>に記載のジアステレオマーの製造方法、
【0013】
【化8】
(式中、*は不斉炭素原子であることを表す。)
<7>下記式(A−1)又は(A−2)で表され、かつ不斉炭素原子を有するアミン化合物と下記式(B−1)又は(B−2)で表される酸性分割剤とからなることを特徴とするジアステレオマー塩、
【0014】
【化9】
(式中、R1及びR2はそれぞれ独立に、水素原子、炭素数1〜10のアルキル基、又は、炭素数6〜10のアリール基を表し、R3は炭素数7〜20のアラルキル基、炭素数2〜20のアルキルオキシカルボニル基、炭素数7〜20のアリールオキシカルボニル基、又は、炭素数8〜20のアラルキルオキシカルボニル基を表し、*は不斉炭素原子であることを表す。)
【0015】
【化10】
(式中、R4及びR9は、酸性官能基を表し、R5〜R8はそれぞれ独立に、アルキル基、アリール基、アラルキル基、アルキルオキシ基、アリールオキシ基、アラルキルオキシ基、アルキルカルボニルオキシ基、アリールカルボニルオキシ基、アラルキルカルボニルオキシ基、又は、ヒドロキシ基を表し、R10は、酸性官能基、又は、アリールアミノカルボニル基を表し、*は不斉炭素原子であることを表す。ただし、R5とR6とは異なる基である。)
<8>前記アミン化合物が、下記式(A−1−1)〜(A−1−3)で表されるアミン化合物である上記<7>に記載のジアステレオマー塩、
【0016】
【化11】
(式中、*は不斉炭素原子であることを表す。)
<9>前記酸性分割剤が、下記式(B−1−1)〜(B−1−4)又は(B−2−1)〜(B−2−6)で表される化合物である上記<7>又は<8>に記載のジアステレオマー塩。
【0017】
【化12】
(式中、*は不斉炭素原子であることを表す。)
【発明の効果】
【0018】
本発明によれば、光学活性アミン化合物の実用的な製造方法を提供することができた。
また、本発明によれば、新規なジアステレオマー塩を提供することができた。
さらにまた、本発明によれば、光学活性アミン化合物と酸性分割剤とからなるジアステレオマー塩の実用的な製造方法を提供することができた。
【図面の簡単な説明】
【0019】
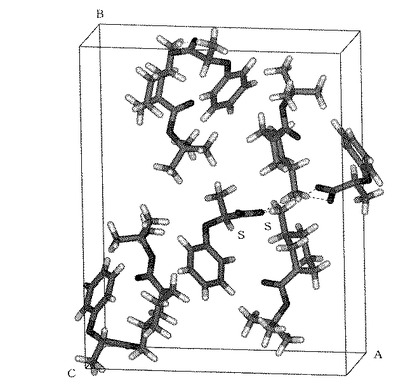
【図1】実施例20で得られた結晶の結晶構造を示す図である。
【発明を実施するための形態】
【0020】
本発明の光学活性アミン化合物の製造方法は、下記式(A−1)又は(A−2)で表されるアミン化合物と下記式(B−1)又は(B−2)で表される酸性分割剤とを反応させてジアステレオマー塩を形成する工程(以下、「形成工程」ともいう。)、得られたジアステレオマー塩の少なくとも一部を析出させ精製ジアステレオマー塩を得る工程(以下、「析出工程」ともいう。)、及び、精製ジアステレオマー塩を塩基性水溶液により分解し、下記式(A−1)又は(A−2)で表される光学活性アミン化合物を得る工程(以下、「単離工程」ともいう。)を含むことを特徴とする。
【0021】
【化13】
(式中、R1及びR2はそれぞれ独立に、水素原子、炭素数1〜10のアルキル基、又は、炭素数6〜10のアリール基を表し、R3は炭素数7〜20のアラルキル基、炭素数2〜20のアルキルオキシカルボニル基、炭素数7〜20のアリールオキシカルボニル基、又は、炭素数8〜20のアラルキルオキシカルボニル基を表し、*は不斉炭素原子であることを表す。)
【0022】
【化14】
(式中、R4及びR9は、酸性官能基を表し、R5〜R8はそれぞれ独立に、アルキル基、アリール基、アラルキル基、アルキルオキシ基、アリールオキシ基、アラルキルオキシ基、アルキルカルボニルオキシ基、アリールカルボニルオキシ基、アラルキルカルボニルオキシ基、又は、ヒドロキシ基を表し、R10は、酸性官能基、又は、アリールアミノカルボニル基を表し、*は不斉炭素原子であることを表す。ただし、R5とR6とは異なる基である。)
【0023】
なお、本発明において、光学活性アミン化合物は、窒素原子において、アンモニウム塩を形成したものも含むものとする。また、光学活性アミン化合物は、塩を形成していないものと、1種又は2種以上の塩を形成したものとの混合物であってもよい。
光学活性アミン化合物の塩における対アニオンは、特に制限はなく、無機アニオンであっても、有機アニオンであってもよく、また、一価アニオンであっても、多価アニオンであってもよい。
本発明の光学活性アミン化合物の製造方法により、光学純度の高い光学活性アミン化合物を、簡便な操作で効率よく製造することができる。
以下、本発明を詳細に説明する。
【0024】
<形成工程>
本発明の光学活性アミン化合物の製造方法は、前記式(A−1)又は(A−2)で表されるアミン化合物と前記式(B−1)又は(B−2)で表される酸性分割剤とを反応させてジアステレオマー塩を形成する工程(形成工程)を含む。
本発明に用いることができる前記式(A−1)又は(A−2)で表されるアミン化合物は、ラセミ体であっても、任意の比率の鏡像異性体混合物であってもよく、鏡像体過剰率(光学純度、enantiomeric excess、ee)が0%以上100%未満であるアミンを用いることができる。
【0025】
前記式(A−1)又は(A−2)中、R1及びR2はそれぞれ独立に、水素原子、炭素数1〜10のアルキル基、又は、炭素数6〜10のアリール基を表し、水素原子、炭素数1〜10のアルキル基、又は、フェニル基であることが好ましく、水素原子であることがより好ましい。
前記式(A−1)又は(A−2)中、R3は炭素数7〜20のアラルキル基、炭素数2〜20のアルキルオキシカルボニル基、炭素数7〜20のアリールオキシカルボニル基、又は、炭素数8〜20のアラルキルオキシカルボニル基を表し、炭素数7〜20のアラルキル基、炭素数2〜20のアルキルオキシカルボニル基、又は、炭素数8〜20のアラルキルオキシカルボニル基であることが好ましく、ベンジル基、t−ブチルオキシカルボニル基、又は、ベンジルオキシカルボニル基であることがより好ましく、t−ブチルオキシカルボニル基であることが特に好ましい。上記態様であると、1回の操作でより光学純度の高いジアステレオマー塩や光学活性アミン化合物を得ることができる。
また、R1〜R3におけるアルキル基やアラルキル基は、直鎖状であっても、分岐や環構造を有していてもよい。
また、前記形成工程で使用するアミン化合物としては、前記式(A−1)で表されるアミン化合物であることが好ましく、下記式(A−1−1)〜(A−1−3)で表されるアミン化合物であることがより好ましく、下記式(A−1−1)で表されるアミン化合物であることが特に好ましい。
【0026】
【化15】
(式中、*は不斉炭素原子であることを表す。)
【0027】
前記式(B−1)におけるR4、並びに、前記式(B−2)におけるR9は、酸性官能基を表し、前記式(B−2)におけるR10は、酸性官能基、又は、アリールアミノカルボニル基を表す。
前記酸性官能基としては、カルボキシル基、リン酸基、及び、スルホン酸基が好ましく例示でき、カルボキシル基がより好ましく例示できる。
前記式(B−2)におけるR10におけるアリールアミノカルボニル基は、そのアリール基上に置換基を有していてもよい。置換基としては、ハロゲン原子、及び、アルキル基が好ましく例示でき、メチル基、フッ素原子、及び、クロロ原子がより好ましく例示できる。
前記式(B−1)又は(B−2)中、R5〜R8はそれぞれ独立に、アルキル基、アリール基、アラルキル基、アルキルオキシ基、アリールオキシ基、アラルキルオキシ基、アルキルカルボニルオキシ基、アリールカルボニルオキシ基、アラルキルカルボニルオキシ基、又は、ヒドロキシ基を表す。
また、R5〜R8のアルキル基、アラルキル基、アルキルオキシ基、アラルキルオキシ基、アルキルカルボニルオキシ基、及び、アラルキルカルボニルオキシ基におけるアルキル基(アルキレン基)は、直鎖状であっても、分岐や環構造を有していてもよい。
また、R5〜R8のアリール基、アラルキル基、アリールオキシ基、アラルキルオキシ基、アリールカルボニルオキシ基、及び、アラルキルカルボニルオキシ基におけるアリール基は、置換基を有していてもよい。置換基としては、ハロゲン原子、及び、アルキル基が好ましく例示でき、メチル基がより好ましく例示できる。
【0028】
前記式(B−1)又は(B−2)で表される酸性分割剤としては、これらの中でも、下記式(B−1−1)〜(B−1−4)又は(B−2−1)〜(B−2−6)で表される化合物であることが好ましく、下記式(B−1−1)又は(B−2−1)で表される化合物であることがより好ましく、下記式(B−1−1)で表される化合物であることが特に好ましい。
【0029】
【化16】
【0030】
また、前記形成工程において使用する前記式(A−1)又は(A−2)で表されるアミン化合物と前記式(B−1)又は(B−2)で表される酸性分割剤との組み合わせとしては、前記式(A−1−1)で表される化合物と前記(B−1−1)で表される化合物との組み合わせ、又は、前記式(A−1−3)で表される化合物と前記(B−2−1)で表される化合物との組み合わせが好ましく、前記式(A−1−1)で表される化合物と前記(B−1−1)で表される化合物との組み合わせが特に好ましい。
【0031】
前記形成工程において得られる本発明のジアステレオマー塩は、前記式(A−1)又は(A−2)で表されるアミン化合物と下前記式(B−1)又は(B−2)で表される酸性分割剤とからなるジアステレオマー塩である。
また、前記ジアステレオマー塩における前記アミン化合物と前記酸性分割剤との結合比は、特に制限はないが、前記アミン化合物1モルに対し、前記酸性分割剤のカルボキシル基が1モルであることが好ましい。
中でも、前記ジアステレオマー塩としては、(前記式(A−1−1)で表される化合物と前記(B−1−1)で表される化合物とからなるジアステレオマー塩、又は、前記式(A−1−3)で表される化合物と前記(B−2−1)で表される化合物とからなるジアステレオマー塩が好ましく例示でき、前記式(A−1−1)で表される化合物と前記(B−1−1)で表される化合物とからなるジアステレオマー塩をより好ましく例示できる。
【0032】
前記形成工程における前記アミン化合物と前記酸性分割剤とのモル混合比(使用比)は、(前記酸性分割剤のモル量):(前記アミン化合物のモル量)=10:1〜1:10であることが好ましく、1.0:1.6〜1.0:2.4であることがより好ましい。
【0033】
前記形成工程におけるジアステレオマー塩の調製は、無溶媒の条件でも可能ではあるが、溶媒を用いて行うことが好ましい。
前記形成工程において用いることができる溶媒は、特に制限はないが、前記酸性分割剤及び前記アミン化合物を溶解させることができ、かつ生成するジアステレオマー塩のうち一方の光学活性体が析出する性質を持つものが操作上好ましい。
前記形成工程において用いることができる溶媒として具体的には、メタノール等のアルコール類、クロロホルム等のハロゲン化炭化水素、トルエン、ヘキサン等の炭化水素類、酢酸エチルなどのエステル類、アセトン等のケトン類、t−ブチルメチルエーテル等のエーテル類、及び、水が例示できる。また、これらの溶媒は、1種単独で使用しても、任意の混合して使用してもよい。これらの中でも溶媒としては、製造コストや環境への配慮の点から、アセトン、2−ブタノン、4−メチル−2−ペンタノンに代表されるケトン類を使用することが好ましい。
また、前記形成工程における溶媒の使用量は、固形分の全重量に対して、1〜20倍量が好ましい。
【0034】
前記形成工程における反応温度は、特に制限はないが、使用した溶媒への基質の溶解度を増加させ、溶媒の使用量を少なくするため、加熱を行うことが好ましい。加熱温度は、特に制限はないが、35〜80℃であることが好ましく、45〜50℃であることがより好ましい。
【0035】
<析出工程>
本発明の光学活性アミン化合物の製造方法は、前記形成工程において得られたジアステレオマー塩の少なくとも一部を析出させ精製ジアステレオマー塩を得る工程(析出工程)を含む。
前記析出工程における析出手段としては、前記形成工程により得られたジアステレオマー塩の少なくとも一部を析出させる手段であれば、特に制限はない。
析出手段としては、例えば、前記形成工程で得られたジアステレオマー塩を含む混合物を静置し、ジアステレオマー塩を析出させることが挙げられるが、中でも、前記形成工程において、溶媒を使用し、かつ加熱を行っている場合、室温以下(30℃以下、好ましくは10〜20℃)に冷却し、静置することが好ましい。
また、析出手段としては、例えば、前記形成工程において、ジアステレオマー塩の良溶媒を使用している場合、ジアステレオマー塩の貧溶媒を添加することにより、析出させてもよい。
これらの中でも、前記形成工程において、溶媒としてt−ブチルメチルエーテルを使用し、加熱を行い、析出工程において、室温以下(30℃以下、好ましくは10〜20℃)に冷却し、ジアステレオマー塩を析出させることが好ましい。
【0036】
前記析出工程において用いることができる溶媒としては、前記形成工程において例示したものを好適に用いることができる。また、溶媒は1種単独で用いても、2種以上を任意の割合で混合して用いてもよい。
【0037】
前記形成工程と前記析出工程とは同時に行っても、逐次行ってもよい。また、形成工程後、一旦ジアステレオマー塩混合物として単離し、析出工程に用いてもよい。
また、操作の簡便性から、前記形成工程及び前記析出工程において同一の溶媒を用いることが好ましく、同一の溶媒として、t−ブチルメチルエーテルを使用することが特に好ましい。
【0038】
前記析出工程において、析出した精製ジアステレオマー塩を溶媒や反応残渣等から分離する方法としては、特に制限はなく、ろ過、乾燥等の公知の方法により行うことができる。
【0039】
前記析出工程では、析出した固体として精製ジアステレオマー塩を得てもよく、析出した固体を除いた溶液から析出した固体とは異なる異性体である精製ジアステレオマー塩を得てもよく、また、析出した固体及び該固体を除いた溶液からそれぞれ精製ジアステレオマー塩を得てもよい。
また、所望の光学純度に応じ、前記析出工程で得られた精製ジアステレオマー塩に対し、さらに析出工程を1回以上繰り返したり、再結晶や再沈殿等の手段を1回以上行い、精製ジアステレオマー塩をさらに精製してもよい。
【0040】
<単離工程>
本発明の光学活性アミン化合物の製造方法は、精製ジアステレオマー塩を塩基性水溶液により分解し、前記式(A−1)又は(A−2)で表される光学活性アミン化合物を得る工程(単離工程)を含む。
前記単離工程においては、精製ジアステレオマー塩を塩基性水溶液により分解し、前記式(A−1)又は(A−2)で表される光学活性アミン化合物を得るが、精製ジアステレオマー塩に有機溶媒及び塩基性水溶液を添加した後、分液し、前記式(A−1)又は(A−2)で表される光学活性アミン化合物を得る手段が好ましく例示できる。
また、上記手段において有機溶媒及び塩基性水溶液の添加は、同時であっても、逐次であってもよく、また、有機溶媒、塩基性水溶液のどちらを先に添加してもよい。これらの溶媒は、1種単独で用いても、2種以上を任意の割合で混合し用いてもよい。
【0041】
前記単離工程に用いることができる有機溶媒は、水と界面を形成し、酸性又は塩基性でも分解せず、かつ前記式(A−1)又は(A−2)で表される光学活性アミン化合物及び酸性分割剤に対して不活性ならば、どのような溶媒でも使用可能である。
前記単離工程に用いることができる有機溶媒として、具体的には、ジクロロメタン等のハロゲン化炭化水素、トルエン、ヘキサン等の炭化水素類、4−メチル−2−ペンタノン等のケトン類、t−ブチルメチルエーテル等のエーテル類が好ましく例示できる。
【0042】
前記単離工程に用いることができる塩基性水溶液は、精製ジアステレオマー塩を前記式(A−1)又は(A−2)で表される光学活性アミン化合物と酸性分割剤とに解離することができる塩基性水溶液であれば、特に制限はなく、アルカリ金属水酸化物又はアルカリ炭酸塩の水溶液であることが好ましく、水酸化ナトリウム又は炭酸ナトリウム水溶液であることがより好ましい。
精製ジアステレオマー塩の分解に使用される塩基(アルカリ化合物)の使用量は、精製ジアステレオマー塩の量に対し、0.5〜10モル当量であることが好ましく、1〜2モル当量であることがより好ましい。
前記単離工程における有機溶媒及びアルカリ水溶液の使用量は、固形分の全重量に対して、1〜10倍量であることが好ましく、4〜6倍量であることがより好ましい。
【0043】
得られた前記式(A−1)又は(A−2)で表される光学活性アミン化合物は、塩として単離しても、塩を形成していないアミンとして単離してもよい。
前述のような精製ジアステレオマー塩に有機溶媒及び塩基性水溶液を添加した後、分液する手段では、有機層側に前記式(A−1)又は(A−2)で表される光学活性アミン化合物が含まれている。有機層の有機溶媒を公知の方法により留去し、前記式(A−1)又は(A−2)で表される光学活性アミン化合物を回収することができる。また、アンモニウム塩として単離する場合は、分離した有機層、前記式(A−1)又は(A−2)で表される光学活性アミン化合物又はその有機溶媒溶液に、酸を添加し、該アンモニウム塩を得ることもできる。
酸としては、特に制限はなく、無機酸であっても、有機酸であってもよい。中でも、鉱酸を用いることが好ましく、塩酸、硫酸、硝酸を用いることがより好ましい。また、酸は、そのまま添加しても、水溶液として添加してもよい。
【0044】
また、前述のような精製ジアステレオマー塩に有機溶媒及び塩基性水溶液を添加した後、分液する手段では、水層側に光学分割剤が分離される。水層から光学分割剤を回収する方法としては、特に制限はなく、公知の方法を用いることができる。また、回収した光学分割剤を再利用することも可能である。回収した光学分割剤の純度が十分でない場合は、カラムクロマトグラフィーや、再結晶又は蒸留などの公知の方法により精製することができる。
【0045】
また、所望の光学純度の前記式(A−1)又は(A−2)で表される光学活性アミン化合物を得るため、本発明の光学活性アミン化合物の製造方法における形成工程、析出工程及び単離工程を任意の回数繰り返して行ってもよい。
また、本発明の光学活性アミン化合物の製造方法により得られた前記式(A−1)又は(A−2)で表される光学活性アミン化合物は、所望の光学純度に応じ、再結晶や再沈殿等の公知の手段を行い、さらに精製してもよい。
【0046】
本発明における化合物の光学純度の測定方法としては、特に制限はなく、公知の方法を用いることができる。
前記式(A−1)又は(A−2)で表される光学活性アミン化合物については、たとえば、以下の方法により測定することができる。以下に、2つの測定例を示す。
【0047】
〔1−Boc−3−アミノピペリジンの光学純度の測定に好ましい例〕
[分析のための誘導体化]
前記式(A−1)又は(A−2)で表される光学活性アミン化合物(30mg,0.15mmol)をジクロロメタン(2ml)に溶解させ、ここに無水炭酸ナトリウム(35mg,0.33mmol)を懸濁させ、3,5−ジニトロ安息香酸クロライド(35mg,0.15mmol)を添加する。15〜25℃で溶液を12〜16時間撹拌する。
反応溶液に脱イオン水(10ml)を添加して内容物を完溶させ、溶液を静置分液する。
有機層を採り、硫酸マグネシウムで乾燥後、溶媒を減圧留去して測定サンプルである誘導体化化合物を得る。
前記誘導体化化合物を、下記の条件で測定することにより、光学純度を求めることができる。
[光学純度測定条件]
カラム:ダイセル化学工業(株)製キラルセル AD−H 4.6φ×250mm
移動層:ヘキサン:エタノール=25:75(体積比、0.1体積%ジエチルアミン含有する。)
流速:0.7ml/min
カラム温度:40℃
検出器:紫外線(UV) 254nm
【0048】
〔1−ベンジル−3−アミノピペリジンの光学純度の測定に好ましい例〕
[分析のための誘導体化]
前記式(A−1)又は(A−2)で表される光学活性アミン化合物(18.0mg)をアセトニトリル(1ml)に溶解させ、0.4重量%GITC(2,3,4,6−テトラ−O−アセチル−β−D−グルコピラノシル−イソチオシアネート)アセトニトリル溶液(2ml)を添加し、15〜25℃で0.5時間撹拌する。これに0.2重量%エタノールアミンアセトニトリル溶液(1ml)を添加して0.5時間撹拌し、反応液を0.05%リン酸水溶液でメスアップ(40ml)して測定溶液とする。
前記測定溶液を、下記の条件で測定することにより、光学純度を求めることができる。
[測定条件]
カラム:(株)資生堂製カプセルパック C18 SG120 4.6φ×150mm
キャリア:0.03重量%アンモニア水溶液(酢酸にてpHを4に調整した。)/メタノール=62:38(体積比)
検出器:UV 254nm
測定温度:室温
流速:0.5ml/min
【0049】
本発明のジアステレオマー塩は、前述したように、前記式(A−1)又は(A−2)で表される光学活性アミン化合物と前記式(B−1)又は(B−2)で表される酸性分割剤とからなるジアステレオマー塩である。
なお、本発明におけるジアステレオマー塩は、イオン対として形成される塩であっても、プロトン移動のない分子錯体より形成される塩であってもよく、具体的には例えば、前記式(A−1)又は(A−2)で表される光学活性アミン化合物のアミノ基がアンモニウム基となったものと前記式(B−1)又は(B−2)で表される酸性分割剤の酸性官能基のアニオンとの塩であっても、プロトンの移動のない前記式(A−1)又は(A−2)で表される光学活性アミン化合物と前記式(B−1)又は(B−2)で表される酸性分割剤との錯体であってもよく、これらに前記式(A−1)又は(A−2)で表される光学活性アミン化合物及び/又は前記式(B−1)又は(B−2)で表される酸性分割剤がさらに錯形成していてもよい。また、本発明におけるジアステレオマー塩は、その結晶中に溶媒を含むものであってもよい。
【0050】
また、本発明のジアステレオマー塩の製造方法は、前記式(A−1)又は(A−2)で表されるアミン化合物と前記式(B−1)又は(B−2)で表される酸性分割剤とを反応させてジアステレオマー塩を形成する工程(形成工程)、及び、得られたジアステレオマー塩の少なくとも一部を析出させ精製ジアステレオマー塩を得る工程(析出工程)を含むことを特徴とする。
本発明のジアステレオマー塩の製造方法における形成工程及び析出工程はそれぞれ、前述した本発明の光学活性アミン化合物の製造方法における形成工程及び析出工程と同義であり、また、好ましい態様も同様である。
【実施例】
【0051】
以下、実施例により本発明を更に具体的に説明する。なお、本発明はこれらの実施例に限定されるものではない。
【0052】
<3−アミノピペリジン誘導体の光学純度測定方法>
3−アミノピペリジン誘導体の光学純度の測定方法を、1−t−ブトキシカルボニル(Boc)−3−アミノピペリジンの例、及び、1−ベンジル−3−アミノピロリジンの例を、以下に示す。また、1−ベンジルオキシカルボニル(Z)−3−アミノピペリジンについては、1−t−ブトキシカルボニル(Boc)−3−アミノピペリジンの例と同様な方法で行った。
【0053】
〔1−Boc−3−アミノピペリジンの測定方法〕
[分析のための誘導体化]
光学活性1−Boc−3−アミノピペリジン(30mg,0.15mmol)をジクロロメタン(2ml)に溶解させ、ここに無水炭酸ナトリウム(35mg,0.33mmol)を懸濁させ、3,5−ジニトロ安息香酸クロライド(35mg,0.15mmol)を添加した。15〜25℃で溶液を12〜16時間撹拌した。
反応溶液に脱イオン水(10ml)を添加して内容物を完溶させ、溶液を静置分液した。
有機層を採り、硫酸マグネシウムで乾燥後、溶媒を減圧留去して測定サンプルである誘導体化化合物を得た。
前記誘導体化化合物を、下記の条件で測定することにより、光学純度を求めた。
【0054】
[光学純度測定条件]
カラム:ダイセル化学工業(株)製キラルセル AD−H 4.6φ×250mm
移動層:ヘキサン:エタノール=25:75(体積比、0.1体積%のジエチルアミン含有する。)
流速:0.7ml/min
カラム温度:40℃
検出器:UV 254nm
リテンションタイム:(R)体:17min,(S)体:18min
【0055】
〔1−ベンジル−3−アミノピロリジンの測定方法〕
[分析のための誘導体化]
1−ベンジル−3−アミノピロリジン(18.0mg)をアセトニトリル(1ml)に溶解させ、0.4重量%GITC(2,3,4,6−テトラ−O−アセチル−β−D−グルコピラノシル−イソチオシアネート)アセトニトリル溶液(2ml)を添加し、15〜25℃で0.5時間撹拌した。これに0.2重量%エタノールアミンアセトニトリル溶液(1ml)を添加して0.5時間撹拌し、反応液を0.05%リン酸水溶液でメスアップ(40ml)して測定溶液とした。
前記測定溶液を、下記の条件で測定することにより、光学純度を求めた。
[測定条件]
カラム:(株)資生堂製カプセルパック C18 SG120 4.6φ×150mm
キャリア:0.03重量%アンモニア水溶液(酢酸にてpHを4に調整した。)/メタノール=62:38(体積比)
検出器:UV 254nm
測定温度:室温
流速:0.5ml/min
リテンションタイム:(R)体:49min,(S)体:53min
【0056】
(実施例1)
(RS)−1−Boc−3−アミノピペリジン(40.0g,0.200mol),(S)−2−フェノキシプロピオン酸(16.6g,0.100mol),アセトン(850ml)を混合、加熱、還流し内容物を溶解させた。溶液を15〜25℃まで12時間かけて冷却し、更に15〜25℃で12時間撹拌した。生じた結晶を分離して、35〜45℃で減圧乾燥し、粗ジアステレオマー((S)−1−Boc−3−アミノピペリジン/(S)−2−フェノキシプロピオン酸)を得た(収量31.1g,収率85.0%,光学純度63.0%ee)。なお以下に記載される1−Boc−3−アミノピペリジンの絶対配置は、後述する実施例20により求まった絶対構造と、各比旋光度の極性を元にした。
得られた粗ジアステレオマーをアセトンで2回再結晶して、精製ジアステレオマー((S)−1−Boc−3−アミノピペリジン/(S)−2−フェノキシプロピオン酸)を得ることができた(収量22.0g,総収率60.1%,光学純度98.6%ee)。
なお、得られた精製ジアステレオマーは非水滴定法で分析した結果、1−Boc−3−アミノピペリジン:2−フェノキシプロピオン酸の結合比が、1:1であることが判明した。
【0057】
(実施例2)
実施例1で得られた精製ジアステレオマー(20.0g,54.6mmol)にジクロロメタン(66ml)を混合して、溶液の温度を0〜15℃とした。この温度を保ちながら5%水酸化ナトリウム水溶液(66.0g,82.5mmol)を滴下した。15min撹拌後、内容物が完溶したら溶液を静置、分液した。有機層をとり、脱イオン水で洗浄後、硫酸マグネシウムで乾燥した後溶媒を減圧留去して、(S)−1−Boc−3−アミノピペリジンを得た(収量10.4g,精製ジアステレオマーからの回収率95.0%)。
一方、水層は35%塩酸で酸性としたのちジクロロメタンを添加して分割剤((S)−2−フェノキシプロピオン酸)を抽出した。その後硫酸ナトリウムで乾燥後、溶媒を減圧留去することによって、分割剤を定量的に回収した。
上記で得られた(S)−1−Boc−3−アミノピペリジン(5.0g,25.0mmol)をN2気流下で脱水メタノール(50ml)に溶解させ、0〜15℃に冷却した。これに5%塩化水素メタノール溶液(54.5g,75.0mmol)を反応液に温度を保ちながら滴下した。0.5時間撹拌後、溶媒を減圧留去して、(S)−3−アミノピペリジン二塩酸塩を得ることができた(収量4.1g,収率95%,比旋光度[α]D+2.5°,C=1.0 H2O,25℃)。
【0058】
(実施例3)
実施例1で得られた粗ジアステレオマー分離母液を減圧留去して、これにジクロロメタン(350ml)を添加した。溶液を0〜15℃に冷却して、温度を保ちながら5%水酸化ナトリウム水溶液(50ml)を滴下した。内容物が溶解したことが確認できたら、溶液を静置、分液した(水層から(S)−2−フェノキシプロピオン酸を回収した。)。
有機層を脱イオン水で洗浄後、硫酸マグネシウムで乾燥したのち、溶媒を減圧留去して粗(R)−1−Boc−3−アミノピペリジンを得た(回収量23.0g,光学純度50.0%ee,(R)体含有量17.7g,0.0883mol)。
これとアセトン(750ml),(R)−2−フェノキシプロピオン酸(14.7g,0.0883mol)を混合して溶液を加熱還流した。以下、実施例1と同様の操作を行い、粗ジアステレオマー((R)−1−Boc−3−アミノピペリジン/(R)−2−フェノキシプロピオン酸)を得ることができた(収量31.0g,(RS)体からの収率84.6%,光学純度64.0%ee)。
以下、実施例1と同様に精製ジアステレオマー((R)−1−Boc−3−アミノピペリジン/(R)−2−フェノキシプロピオン酸)を得ることができた(収量23.0g,総収率62.7%,光学純度98.2%ee)。
なお、得られた精製ジアステレオマーは非水滴定法で分析した結果、1−Boc−3−アミノピペリジン:2−フェノキシプロピオン酸の結合比が、1:1であることが判明した。
【0059】
(実施例4)
実施例3で得られた精製ジアステレオマー(20.0g,54.6mmol)にジクロロメタン(66ml)を混合して、溶液の温度を0〜15℃とした。以下、実施例2と同様の操作を行い、(R)−1−Boc−3−アミノピペリジンを得た(収量10.7g,精製ジアステレオマーからの回収率97.0%)。
上記で得られた(R)−1−Boc−3−アミノピペリジン(5.0g,25.0mmol)をN2気流下で脱水メタノール(50ml)に溶解させ、0〜15℃に冷却した。以下、実施例2と同様な操作を行い、(R)−3−アミノピペリジン二塩酸塩を得ることができた(収量4.1g,収率95%,比旋光度[α]D−2.8°,C=1.0 H2O,25℃)。
【0060】
(実施例5)
(RS)−1−Boc−3−アミノピペリジン(40.0g,0.200mol),(R)−2−フェノキシプロピオン酸(16.6g,0.100mol),アセトン(850ml)を混合、加熱、還流し内容物を溶解させた。以下、実施例1と同様な操作を行い、精製ジアステレオマー((R)−1−Boc−3−アミノピペリジン/(R)−2−フェノキシプロピオン酸)を得ることができた(収量22.6g,総収率61.7%,光学純度98.8%ee)。
【0061】
(実施例6〜19)
実施例6〜19については(RS)−3−アミノピペリジン誘導体(基質)、酸性分割剤、及び、基質/酸性分割剤の混合比を、下記表1に示すように変更した以外は実施例1と同様な操作を行い、粗ジアステレオマーを得た。結果を表1示す。
なお、実施例16〜19において、収率が100%を越えているが、収率は所望するジアステレオマー塩の理論生成量を基準としているためである。
【0062】
【表1】
【0063】
(実施例20)
実施例1の過程で得られた精製ジアステレオマー((S)−1−Boc−3−アミノピペリジン/(S)−2−フェノキシプロピオン酸)において、1−Boc−3−アミノピペリジンの絶対構造を確定するために、複合塩のX線単結晶構造解析を行った。
まず溶媒として含水アセトンに試料を溶解させ、自然蒸発法により、サイズ0.4×0.4×0.2mmの柱状の単結晶を得ることができた。得られた結晶学的データ及び結晶構造を表2及び図1に示す。
光学分割剤(S)−2−フェノキシプロピオン酸の絶対配置を用いて複合塩の手系を決定し、その結果、1−Boc−3−アミノピペリジンの絶対配置は、(S)体であることが明らかになった。
【0064】
【表2】
【0065】
(実施例21)
(RS)−1−ベンジル−3−アミノピロリジン(37.0g,0.209mol),(R,R)−2’−メチルタートラニル酸(50.0g,0.209mol),メタノール(260ml)を混合、加熱して内容物を溶解させたのち、反応液を15−25℃まで一夜冷却した。生じた結晶を分離して、粗ジアステレオマー体を得た((R)−1−ベンジル−3−アミノピロリジン/(R,R)−2’−メチルタートラニル酸,結合比=1:1,非水滴定法,収量66.8g,収率153%,光学純度65%ee)。
粗ジアステレオマー体を、メタノールを用いて2回再結晶し、精製ジアステレオマー塩を得た(収量30.8g,収率70.8%,光学純度98%ee)。
精製ジアステレオマー塩(30.8g,0.0740mol),5%水酸化カリウム水溶液(100g,0.089mol),トルエン(100ml)を混合し、静置、分液した。水層をさらにトルエン(100ml×2)で洗浄した(水層から分割剤を回収した。)。
有機層をあわせ、硫酸ナトリウムで乾燥後、溶媒を減圧留去し、(R)−1−ベンジル−3−アミノピロリジンを得た(収量12.4g,精製ジアステレオマー塩からの収率95.5%)。
【0066】
(実施例22)
(R,R)−2’−メチルタートラニル酸の代わりに(S,S)−2’−メチルタートラニル酸を用いて、実施例21と同様な操作を行い、粗ジアステレオマー塩を得た((S)−1−ベンジル−3−アミノピロリジン/(S,S)−2’−メチルタートラニル酸,結合比=1:1,非水滴定法,収量67.9g,収率156%,光学純度63%ee)。
以下、実施例21と同様な操作を行い、精製ジアステレオマー塩(収量31.3g,収率72.0%,光学純度99%ee),(S)−1−ベンジル−3−アミノピロリジン(収量12.8g,精製ジアステレオマー塩からの収率96.3%)を得た。
【0067】
(実施例23〜28)
実施例23〜28については、酸性分割剤を下記表3のように変更した以外は、実施例21と同様の操作を実施して、粗ジアステレオマー体を得ることができた。
【0068】
なお、前記実施例21、22、24において、収率が100%を越えているが、収率は所望するジアステレオマー塩の理論生成量を基準としているためである。
【0069】
【表3】
【産業上の利用可能性】
【0070】
本発明で記載されている光学活性環状アミノ化合物誘導体は、医薬品中間体原料として、注目されている。特に3−アミノピペリジン誘導体は、次期ブロックバスター候補として重要であり、これらの化合物の製造が容易にできる本発明の産業上の利用可能性は極めて高いということができる。
【技術分野】
【0001】
本発明は、医薬、農薬、電子材料等の原料あるいは中間体として有用な光学活性アミン化合物の製造方法に関する。また、本発明は、前記アミン化合物と酸性分割剤より形成されるジアステレオマー塩、及び、該ジアステレオマー塩の分離方法に関する。
【背景技術】
【0002】
光学活性アミン化合物は、医薬、農薬、電子材料等の原料あるいは中間体などとして、幅広い利用が期待されている。
例えば、3−アミノピペリジン類の光学分割法としては、特許文献1及び2に記載の方法が知られている。
特許文献1には、3−アミノピペリジンに酸性分割剤を作用させ、ジアステレオマー塩を分離し、光学分割を行う方法が記載されている。
また、特許文献2には、キサンチン構造を有する保護基を1位に有する3−アミノピペリジン類に酸性分割剤を作用させ、ジアステレオマー塩を分離し、光学分割を行う方法が記載されている。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】国際公開第2007/75630号パンフレット
【特許文献2】特表2008−519005号公報
【発明の開示】
【発明が解決しようとする課題】
【0004】
本発明の目的は、光学活性アミン化合物の実用的な製造方法を提供することである。
本発明の他の目的は、新規なジアステレオマー塩を提供することである。
また、本発明の他の目的は、光学活性アミン化合物と酸性分割剤とからなるジアステレオマー塩の実用的な製造方法を提供することである。
【課題を解決するための手段】
【0005】
本発明者らは光学活性アミン化合物の実用的製造方法について研究を重ねた結果、特定の酸性分割剤とジアステレオマー塩を生成させることにより、光学分割が可能であることを見出し、本発明を完成させることができた。
前記課題は、以下の手段<1>、<4>及び<7>により解決された。好ましい実施態様である<2>、<3>、<5>、<6>、<8>及び<9>と共に以下に記載する。
<1>下記式(A−1)又は(A−2)で表されるアミン化合物と下記式(B−1)又は(B−2)で表される酸性分割剤とを反応させてジアステレオマー塩を形成する工程、得られたジアステレオマー塩の少なくとも一部を析出させ精製ジアステレオマー塩を得る工程、及び、精製ジアステレオマー塩を塩基性水溶液により分解し、下記式(A−1)又は(A−2)で表される光学活性アミン化合物を得る工程を含むことを特徴とする
光学活性アミン化合物の製造方法、
【0006】
【化1】
(式中、R1及びR2はそれぞれ独立に、水素原子、炭素数1〜10のアルキル基、又は、炭素数6〜10のアリール基を表し、R3は炭素数7〜20のアラルキル基、炭素数2〜20のアルキルオキシカルボニル基、炭素数7〜20のアリールオキシカルボニル基、又は、炭素数8〜20のアラルキルオキシカルボニル基を表し、*は不斉炭素原子であることを表す。)
【0007】
【化2】
(式中、R4及びR9は、酸性官能基を表し、R5〜R8はそれぞれ独立に、アルキル基、アリール基、アラルキル基、アルキルオキシ基、アリールオキシ基、アラルキルオキシ基、アルキルカルボニルオキシ基、アリールカルボニルオキシ基、アラルキルカルボニルオキシ基、又は、ヒドロキシ基を表し、R10は、酸性官能基、又は、アリールアミノカルボニル基を表し、*は不斉炭素原子であることを表す。ただし、R5とR6とは異なる基である。)
<2>前記アミン化合物が、下記式(A−1−1)〜(A−1−3)で表されるアミン化合物である上記<1>に記載の光学活性アミン化合物の製造方法、
【0008】
【化3】
(式中、*は不斉炭素原子であることを表す。)
<3>前記酸性分割剤が、下記式(B−1−1)〜(B−1−4)又は(B−2−1)〜(B−2−6)で表される化合物である上記<1>又は<2>に記載の光学活性アミン化合物の製造方法、
【0009】
【化4】
(式中、*は不斉炭素原子であることを表す。)
<4>下記式(A−1)又は(A−2)で表されるアミン化合物と下記式(B−1)又は(B−2)で表される酸性分割剤とを反応させてジアステレオマー塩を形成する工程、及び、得られたジアステレオマー塩の少なくとも一部を析出させ精製ジアステレオマー塩を得る工程を含むことを特徴とするジアステレオマー塩の製造方法、
【0010】
【化5】
(式中、R1及びR2はそれぞれ独立に、水素原子、炭素数1〜10のアルキル基、又は、炭素数6〜10のアリール基を表し、R3は炭素数7〜20のアラルキル基、炭素数2〜20のアルキルオキシカルボニル基、炭素数7〜20のアリールオキシカルボニル基、又は、炭素数8〜20のアラルキルオキシカルボニル基を表し、*は不斉炭素原子であることを表す。)
【0011】
【化6】
(式中、R4及びR9は、酸性官能基を表し、R5〜R8はそれぞれ独立に、アルキル基、アリール基、アラルキル基、アルキルオキシ基、アリールオキシ基、アラルキルオキシ基、アルキルカルボニルオキシ基、アリールカルボニルオキシ基、アラルキルカルボニルオキシ基、又は、ヒドロキシ基を表し、R10は、酸性官能基、又は、アリールアミノカルボニル基を表し、*は不斉炭素原子であることを表す。ただし、R5とR6とは異なる基である。)
<5>前記アミン化合物が、下記式(A−1−1)〜(A−1−3)で表されるアミン化合物である上記<4>に記載のジアステレオマーの製造方法、
【0012】
【化7】
(式中、*は不斉炭素原子であることを表す。)
<6>前記酸性分割剤が、下記式(B−1−1)〜(B−1−4)又は(B−2−1)〜(B−2−6)で表される化合物である上記<4>又は<5>に記載のジアステレオマーの製造方法、
【0013】
【化8】
(式中、*は不斉炭素原子であることを表す。)
<7>下記式(A−1)又は(A−2)で表され、かつ不斉炭素原子を有するアミン化合物と下記式(B−1)又は(B−2)で表される酸性分割剤とからなることを特徴とするジアステレオマー塩、
【0014】
【化9】
(式中、R1及びR2はそれぞれ独立に、水素原子、炭素数1〜10のアルキル基、又は、炭素数6〜10のアリール基を表し、R3は炭素数7〜20のアラルキル基、炭素数2〜20のアルキルオキシカルボニル基、炭素数7〜20のアリールオキシカルボニル基、又は、炭素数8〜20のアラルキルオキシカルボニル基を表し、*は不斉炭素原子であることを表す。)
【0015】
【化10】
(式中、R4及びR9は、酸性官能基を表し、R5〜R8はそれぞれ独立に、アルキル基、アリール基、アラルキル基、アルキルオキシ基、アリールオキシ基、アラルキルオキシ基、アルキルカルボニルオキシ基、アリールカルボニルオキシ基、アラルキルカルボニルオキシ基、又は、ヒドロキシ基を表し、R10は、酸性官能基、又は、アリールアミノカルボニル基を表し、*は不斉炭素原子であることを表す。ただし、R5とR6とは異なる基である。)
<8>前記アミン化合物が、下記式(A−1−1)〜(A−1−3)で表されるアミン化合物である上記<7>に記載のジアステレオマー塩、
【0016】
【化11】
(式中、*は不斉炭素原子であることを表す。)
<9>前記酸性分割剤が、下記式(B−1−1)〜(B−1−4)又は(B−2−1)〜(B−2−6)で表される化合物である上記<7>又は<8>に記載のジアステレオマー塩。
【0017】
【化12】
(式中、*は不斉炭素原子であることを表す。)
【発明の効果】
【0018】
本発明によれば、光学活性アミン化合物の実用的な製造方法を提供することができた。
また、本発明によれば、新規なジアステレオマー塩を提供することができた。
さらにまた、本発明によれば、光学活性アミン化合物と酸性分割剤とからなるジアステレオマー塩の実用的な製造方法を提供することができた。
【図面の簡単な説明】
【0019】
【図1】実施例20で得られた結晶の結晶構造を示す図である。
【発明を実施するための形態】
【0020】
本発明の光学活性アミン化合物の製造方法は、下記式(A−1)又は(A−2)で表されるアミン化合物と下記式(B−1)又は(B−2)で表される酸性分割剤とを反応させてジアステレオマー塩を形成する工程(以下、「形成工程」ともいう。)、得られたジアステレオマー塩の少なくとも一部を析出させ精製ジアステレオマー塩を得る工程(以下、「析出工程」ともいう。)、及び、精製ジアステレオマー塩を塩基性水溶液により分解し、下記式(A−1)又は(A−2)で表される光学活性アミン化合物を得る工程(以下、「単離工程」ともいう。)を含むことを特徴とする。
【0021】
【化13】
(式中、R1及びR2はそれぞれ独立に、水素原子、炭素数1〜10のアルキル基、又は、炭素数6〜10のアリール基を表し、R3は炭素数7〜20のアラルキル基、炭素数2〜20のアルキルオキシカルボニル基、炭素数7〜20のアリールオキシカルボニル基、又は、炭素数8〜20のアラルキルオキシカルボニル基を表し、*は不斉炭素原子であることを表す。)
【0022】
【化14】
(式中、R4及びR9は、酸性官能基を表し、R5〜R8はそれぞれ独立に、アルキル基、アリール基、アラルキル基、アルキルオキシ基、アリールオキシ基、アラルキルオキシ基、アルキルカルボニルオキシ基、アリールカルボニルオキシ基、アラルキルカルボニルオキシ基、又は、ヒドロキシ基を表し、R10は、酸性官能基、又は、アリールアミノカルボニル基を表し、*は不斉炭素原子であることを表す。ただし、R5とR6とは異なる基である。)
【0023】
なお、本発明において、光学活性アミン化合物は、窒素原子において、アンモニウム塩を形成したものも含むものとする。また、光学活性アミン化合物は、塩を形成していないものと、1種又は2種以上の塩を形成したものとの混合物であってもよい。
光学活性アミン化合物の塩における対アニオンは、特に制限はなく、無機アニオンであっても、有機アニオンであってもよく、また、一価アニオンであっても、多価アニオンであってもよい。
本発明の光学活性アミン化合物の製造方法により、光学純度の高い光学活性アミン化合物を、簡便な操作で効率よく製造することができる。
以下、本発明を詳細に説明する。
【0024】
<形成工程>
本発明の光学活性アミン化合物の製造方法は、前記式(A−1)又は(A−2)で表されるアミン化合物と前記式(B−1)又は(B−2)で表される酸性分割剤とを反応させてジアステレオマー塩を形成する工程(形成工程)を含む。
本発明に用いることができる前記式(A−1)又は(A−2)で表されるアミン化合物は、ラセミ体であっても、任意の比率の鏡像異性体混合物であってもよく、鏡像体過剰率(光学純度、enantiomeric excess、ee)が0%以上100%未満であるアミンを用いることができる。
【0025】
前記式(A−1)又は(A−2)中、R1及びR2はそれぞれ独立に、水素原子、炭素数1〜10のアルキル基、又は、炭素数6〜10のアリール基を表し、水素原子、炭素数1〜10のアルキル基、又は、フェニル基であることが好ましく、水素原子であることがより好ましい。
前記式(A−1)又は(A−2)中、R3は炭素数7〜20のアラルキル基、炭素数2〜20のアルキルオキシカルボニル基、炭素数7〜20のアリールオキシカルボニル基、又は、炭素数8〜20のアラルキルオキシカルボニル基を表し、炭素数7〜20のアラルキル基、炭素数2〜20のアルキルオキシカルボニル基、又は、炭素数8〜20のアラルキルオキシカルボニル基であることが好ましく、ベンジル基、t−ブチルオキシカルボニル基、又は、ベンジルオキシカルボニル基であることがより好ましく、t−ブチルオキシカルボニル基であることが特に好ましい。上記態様であると、1回の操作でより光学純度の高いジアステレオマー塩や光学活性アミン化合物を得ることができる。
また、R1〜R3におけるアルキル基やアラルキル基は、直鎖状であっても、分岐や環構造を有していてもよい。
また、前記形成工程で使用するアミン化合物としては、前記式(A−1)で表されるアミン化合物であることが好ましく、下記式(A−1−1)〜(A−1−3)で表されるアミン化合物であることがより好ましく、下記式(A−1−1)で表されるアミン化合物であることが特に好ましい。
【0026】
【化15】
(式中、*は不斉炭素原子であることを表す。)
【0027】
前記式(B−1)におけるR4、並びに、前記式(B−2)におけるR9は、酸性官能基を表し、前記式(B−2)におけるR10は、酸性官能基、又は、アリールアミノカルボニル基を表す。
前記酸性官能基としては、カルボキシル基、リン酸基、及び、スルホン酸基が好ましく例示でき、カルボキシル基がより好ましく例示できる。
前記式(B−2)におけるR10におけるアリールアミノカルボニル基は、そのアリール基上に置換基を有していてもよい。置換基としては、ハロゲン原子、及び、アルキル基が好ましく例示でき、メチル基、フッ素原子、及び、クロロ原子がより好ましく例示できる。
前記式(B−1)又は(B−2)中、R5〜R8はそれぞれ独立に、アルキル基、アリール基、アラルキル基、アルキルオキシ基、アリールオキシ基、アラルキルオキシ基、アルキルカルボニルオキシ基、アリールカルボニルオキシ基、アラルキルカルボニルオキシ基、又は、ヒドロキシ基を表す。
また、R5〜R8のアルキル基、アラルキル基、アルキルオキシ基、アラルキルオキシ基、アルキルカルボニルオキシ基、及び、アラルキルカルボニルオキシ基におけるアルキル基(アルキレン基)は、直鎖状であっても、分岐や環構造を有していてもよい。
また、R5〜R8のアリール基、アラルキル基、アリールオキシ基、アラルキルオキシ基、アリールカルボニルオキシ基、及び、アラルキルカルボニルオキシ基におけるアリール基は、置換基を有していてもよい。置換基としては、ハロゲン原子、及び、アルキル基が好ましく例示でき、メチル基がより好ましく例示できる。
【0028】
前記式(B−1)又は(B−2)で表される酸性分割剤としては、これらの中でも、下記式(B−1−1)〜(B−1−4)又は(B−2−1)〜(B−2−6)で表される化合物であることが好ましく、下記式(B−1−1)又は(B−2−1)で表される化合物であることがより好ましく、下記式(B−1−1)で表される化合物であることが特に好ましい。
【0029】
【化16】
【0030】
また、前記形成工程において使用する前記式(A−1)又は(A−2)で表されるアミン化合物と前記式(B−1)又は(B−2)で表される酸性分割剤との組み合わせとしては、前記式(A−1−1)で表される化合物と前記(B−1−1)で表される化合物との組み合わせ、又は、前記式(A−1−3)で表される化合物と前記(B−2−1)で表される化合物との組み合わせが好ましく、前記式(A−1−1)で表される化合物と前記(B−1−1)で表される化合物との組み合わせが特に好ましい。
【0031】
前記形成工程において得られる本発明のジアステレオマー塩は、前記式(A−1)又は(A−2)で表されるアミン化合物と下前記式(B−1)又は(B−2)で表される酸性分割剤とからなるジアステレオマー塩である。
また、前記ジアステレオマー塩における前記アミン化合物と前記酸性分割剤との結合比は、特に制限はないが、前記アミン化合物1モルに対し、前記酸性分割剤のカルボキシル基が1モルであることが好ましい。
中でも、前記ジアステレオマー塩としては、(前記式(A−1−1)で表される化合物と前記(B−1−1)で表される化合物とからなるジアステレオマー塩、又は、前記式(A−1−3)で表される化合物と前記(B−2−1)で表される化合物とからなるジアステレオマー塩が好ましく例示でき、前記式(A−1−1)で表される化合物と前記(B−1−1)で表される化合物とからなるジアステレオマー塩をより好ましく例示できる。
【0032】
前記形成工程における前記アミン化合物と前記酸性分割剤とのモル混合比(使用比)は、(前記酸性分割剤のモル量):(前記アミン化合物のモル量)=10:1〜1:10であることが好ましく、1.0:1.6〜1.0:2.4であることがより好ましい。
【0033】
前記形成工程におけるジアステレオマー塩の調製は、無溶媒の条件でも可能ではあるが、溶媒を用いて行うことが好ましい。
前記形成工程において用いることができる溶媒は、特に制限はないが、前記酸性分割剤及び前記アミン化合物を溶解させることができ、かつ生成するジアステレオマー塩のうち一方の光学活性体が析出する性質を持つものが操作上好ましい。
前記形成工程において用いることができる溶媒として具体的には、メタノール等のアルコール類、クロロホルム等のハロゲン化炭化水素、トルエン、ヘキサン等の炭化水素類、酢酸エチルなどのエステル類、アセトン等のケトン類、t−ブチルメチルエーテル等のエーテル類、及び、水が例示できる。また、これらの溶媒は、1種単独で使用しても、任意の混合して使用してもよい。これらの中でも溶媒としては、製造コストや環境への配慮の点から、アセトン、2−ブタノン、4−メチル−2−ペンタノンに代表されるケトン類を使用することが好ましい。
また、前記形成工程における溶媒の使用量は、固形分の全重量に対して、1〜20倍量が好ましい。
【0034】
前記形成工程における反応温度は、特に制限はないが、使用した溶媒への基質の溶解度を増加させ、溶媒の使用量を少なくするため、加熱を行うことが好ましい。加熱温度は、特に制限はないが、35〜80℃であることが好ましく、45〜50℃であることがより好ましい。
【0035】
<析出工程>
本発明の光学活性アミン化合物の製造方法は、前記形成工程において得られたジアステレオマー塩の少なくとも一部を析出させ精製ジアステレオマー塩を得る工程(析出工程)を含む。
前記析出工程における析出手段としては、前記形成工程により得られたジアステレオマー塩の少なくとも一部を析出させる手段であれば、特に制限はない。
析出手段としては、例えば、前記形成工程で得られたジアステレオマー塩を含む混合物を静置し、ジアステレオマー塩を析出させることが挙げられるが、中でも、前記形成工程において、溶媒を使用し、かつ加熱を行っている場合、室温以下(30℃以下、好ましくは10〜20℃)に冷却し、静置することが好ましい。
また、析出手段としては、例えば、前記形成工程において、ジアステレオマー塩の良溶媒を使用している場合、ジアステレオマー塩の貧溶媒を添加することにより、析出させてもよい。
これらの中でも、前記形成工程において、溶媒としてt−ブチルメチルエーテルを使用し、加熱を行い、析出工程において、室温以下(30℃以下、好ましくは10〜20℃)に冷却し、ジアステレオマー塩を析出させることが好ましい。
【0036】
前記析出工程において用いることができる溶媒としては、前記形成工程において例示したものを好適に用いることができる。また、溶媒は1種単独で用いても、2種以上を任意の割合で混合して用いてもよい。
【0037】
前記形成工程と前記析出工程とは同時に行っても、逐次行ってもよい。また、形成工程後、一旦ジアステレオマー塩混合物として単離し、析出工程に用いてもよい。
また、操作の簡便性から、前記形成工程及び前記析出工程において同一の溶媒を用いることが好ましく、同一の溶媒として、t−ブチルメチルエーテルを使用することが特に好ましい。
【0038】
前記析出工程において、析出した精製ジアステレオマー塩を溶媒や反応残渣等から分離する方法としては、特に制限はなく、ろ過、乾燥等の公知の方法により行うことができる。
【0039】
前記析出工程では、析出した固体として精製ジアステレオマー塩を得てもよく、析出した固体を除いた溶液から析出した固体とは異なる異性体である精製ジアステレオマー塩を得てもよく、また、析出した固体及び該固体を除いた溶液からそれぞれ精製ジアステレオマー塩を得てもよい。
また、所望の光学純度に応じ、前記析出工程で得られた精製ジアステレオマー塩に対し、さらに析出工程を1回以上繰り返したり、再結晶や再沈殿等の手段を1回以上行い、精製ジアステレオマー塩をさらに精製してもよい。
【0040】
<単離工程>
本発明の光学活性アミン化合物の製造方法は、精製ジアステレオマー塩を塩基性水溶液により分解し、前記式(A−1)又は(A−2)で表される光学活性アミン化合物を得る工程(単離工程)を含む。
前記単離工程においては、精製ジアステレオマー塩を塩基性水溶液により分解し、前記式(A−1)又は(A−2)で表される光学活性アミン化合物を得るが、精製ジアステレオマー塩に有機溶媒及び塩基性水溶液を添加した後、分液し、前記式(A−1)又は(A−2)で表される光学活性アミン化合物を得る手段が好ましく例示できる。
また、上記手段において有機溶媒及び塩基性水溶液の添加は、同時であっても、逐次であってもよく、また、有機溶媒、塩基性水溶液のどちらを先に添加してもよい。これらの溶媒は、1種単独で用いても、2種以上を任意の割合で混合し用いてもよい。
【0041】
前記単離工程に用いることができる有機溶媒は、水と界面を形成し、酸性又は塩基性でも分解せず、かつ前記式(A−1)又は(A−2)で表される光学活性アミン化合物及び酸性分割剤に対して不活性ならば、どのような溶媒でも使用可能である。
前記単離工程に用いることができる有機溶媒として、具体的には、ジクロロメタン等のハロゲン化炭化水素、トルエン、ヘキサン等の炭化水素類、4−メチル−2−ペンタノン等のケトン類、t−ブチルメチルエーテル等のエーテル類が好ましく例示できる。
【0042】
前記単離工程に用いることができる塩基性水溶液は、精製ジアステレオマー塩を前記式(A−1)又は(A−2)で表される光学活性アミン化合物と酸性分割剤とに解離することができる塩基性水溶液であれば、特に制限はなく、アルカリ金属水酸化物又はアルカリ炭酸塩の水溶液であることが好ましく、水酸化ナトリウム又は炭酸ナトリウム水溶液であることがより好ましい。
精製ジアステレオマー塩の分解に使用される塩基(アルカリ化合物)の使用量は、精製ジアステレオマー塩の量に対し、0.5〜10モル当量であることが好ましく、1〜2モル当量であることがより好ましい。
前記単離工程における有機溶媒及びアルカリ水溶液の使用量は、固形分の全重量に対して、1〜10倍量であることが好ましく、4〜6倍量であることがより好ましい。
【0043】
得られた前記式(A−1)又は(A−2)で表される光学活性アミン化合物は、塩として単離しても、塩を形成していないアミンとして単離してもよい。
前述のような精製ジアステレオマー塩に有機溶媒及び塩基性水溶液を添加した後、分液する手段では、有機層側に前記式(A−1)又は(A−2)で表される光学活性アミン化合物が含まれている。有機層の有機溶媒を公知の方法により留去し、前記式(A−1)又は(A−2)で表される光学活性アミン化合物を回収することができる。また、アンモニウム塩として単離する場合は、分離した有機層、前記式(A−1)又は(A−2)で表される光学活性アミン化合物又はその有機溶媒溶液に、酸を添加し、該アンモニウム塩を得ることもできる。
酸としては、特に制限はなく、無機酸であっても、有機酸であってもよい。中でも、鉱酸を用いることが好ましく、塩酸、硫酸、硝酸を用いることがより好ましい。また、酸は、そのまま添加しても、水溶液として添加してもよい。
【0044】
また、前述のような精製ジアステレオマー塩に有機溶媒及び塩基性水溶液を添加した後、分液する手段では、水層側に光学分割剤が分離される。水層から光学分割剤を回収する方法としては、特に制限はなく、公知の方法を用いることができる。また、回収した光学分割剤を再利用することも可能である。回収した光学分割剤の純度が十分でない場合は、カラムクロマトグラフィーや、再結晶又は蒸留などの公知の方法により精製することができる。
【0045】
また、所望の光学純度の前記式(A−1)又は(A−2)で表される光学活性アミン化合物を得るため、本発明の光学活性アミン化合物の製造方法における形成工程、析出工程及び単離工程を任意の回数繰り返して行ってもよい。
また、本発明の光学活性アミン化合物の製造方法により得られた前記式(A−1)又は(A−2)で表される光学活性アミン化合物は、所望の光学純度に応じ、再結晶や再沈殿等の公知の手段を行い、さらに精製してもよい。
【0046】
本発明における化合物の光学純度の測定方法としては、特に制限はなく、公知の方法を用いることができる。
前記式(A−1)又は(A−2)で表される光学活性アミン化合物については、たとえば、以下の方法により測定することができる。以下に、2つの測定例を示す。
【0047】
〔1−Boc−3−アミノピペリジンの光学純度の測定に好ましい例〕
[分析のための誘導体化]
前記式(A−1)又は(A−2)で表される光学活性アミン化合物(30mg,0.15mmol)をジクロロメタン(2ml)に溶解させ、ここに無水炭酸ナトリウム(35mg,0.33mmol)を懸濁させ、3,5−ジニトロ安息香酸クロライド(35mg,0.15mmol)を添加する。15〜25℃で溶液を12〜16時間撹拌する。
反応溶液に脱イオン水(10ml)を添加して内容物を完溶させ、溶液を静置分液する。
有機層を採り、硫酸マグネシウムで乾燥後、溶媒を減圧留去して測定サンプルである誘導体化化合物を得る。
前記誘導体化化合物を、下記の条件で測定することにより、光学純度を求めることができる。
[光学純度測定条件]
カラム:ダイセル化学工業(株)製キラルセル AD−H 4.6φ×250mm
移動層:ヘキサン:エタノール=25:75(体積比、0.1体積%ジエチルアミン含有する。)
流速:0.7ml/min
カラム温度:40℃
検出器:紫外線(UV) 254nm
【0048】
〔1−ベンジル−3−アミノピペリジンの光学純度の測定に好ましい例〕
[分析のための誘導体化]
前記式(A−1)又は(A−2)で表される光学活性アミン化合物(18.0mg)をアセトニトリル(1ml)に溶解させ、0.4重量%GITC(2,3,4,6−テトラ−O−アセチル−β−D−グルコピラノシル−イソチオシアネート)アセトニトリル溶液(2ml)を添加し、15〜25℃で0.5時間撹拌する。これに0.2重量%エタノールアミンアセトニトリル溶液(1ml)を添加して0.5時間撹拌し、反応液を0.05%リン酸水溶液でメスアップ(40ml)して測定溶液とする。
前記測定溶液を、下記の条件で測定することにより、光学純度を求めることができる。
[測定条件]
カラム:(株)資生堂製カプセルパック C18 SG120 4.6φ×150mm
キャリア:0.03重量%アンモニア水溶液(酢酸にてpHを4に調整した。)/メタノール=62:38(体積比)
検出器:UV 254nm
測定温度:室温
流速:0.5ml/min
【0049】
本発明のジアステレオマー塩は、前述したように、前記式(A−1)又は(A−2)で表される光学活性アミン化合物と前記式(B−1)又は(B−2)で表される酸性分割剤とからなるジアステレオマー塩である。
なお、本発明におけるジアステレオマー塩は、イオン対として形成される塩であっても、プロトン移動のない分子錯体より形成される塩であってもよく、具体的には例えば、前記式(A−1)又は(A−2)で表される光学活性アミン化合物のアミノ基がアンモニウム基となったものと前記式(B−1)又は(B−2)で表される酸性分割剤の酸性官能基のアニオンとの塩であっても、プロトンの移動のない前記式(A−1)又は(A−2)で表される光学活性アミン化合物と前記式(B−1)又は(B−2)で表される酸性分割剤との錯体であってもよく、これらに前記式(A−1)又は(A−2)で表される光学活性アミン化合物及び/又は前記式(B−1)又は(B−2)で表される酸性分割剤がさらに錯形成していてもよい。また、本発明におけるジアステレオマー塩は、その結晶中に溶媒を含むものであってもよい。
【0050】
また、本発明のジアステレオマー塩の製造方法は、前記式(A−1)又は(A−2)で表されるアミン化合物と前記式(B−1)又は(B−2)で表される酸性分割剤とを反応させてジアステレオマー塩を形成する工程(形成工程)、及び、得られたジアステレオマー塩の少なくとも一部を析出させ精製ジアステレオマー塩を得る工程(析出工程)を含むことを特徴とする。
本発明のジアステレオマー塩の製造方法における形成工程及び析出工程はそれぞれ、前述した本発明の光学活性アミン化合物の製造方法における形成工程及び析出工程と同義であり、また、好ましい態様も同様である。
【実施例】
【0051】
以下、実施例により本発明を更に具体的に説明する。なお、本発明はこれらの実施例に限定されるものではない。
【0052】
<3−アミノピペリジン誘導体の光学純度測定方法>
3−アミノピペリジン誘導体の光学純度の測定方法を、1−t−ブトキシカルボニル(Boc)−3−アミノピペリジンの例、及び、1−ベンジル−3−アミノピロリジンの例を、以下に示す。また、1−ベンジルオキシカルボニル(Z)−3−アミノピペリジンについては、1−t−ブトキシカルボニル(Boc)−3−アミノピペリジンの例と同様な方法で行った。
【0053】
〔1−Boc−3−アミノピペリジンの測定方法〕
[分析のための誘導体化]
光学活性1−Boc−3−アミノピペリジン(30mg,0.15mmol)をジクロロメタン(2ml)に溶解させ、ここに無水炭酸ナトリウム(35mg,0.33mmol)を懸濁させ、3,5−ジニトロ安息香酸クロライド(35mg,0.15mmol)を添加した。15〜25℃で溶液を12〜16時間撹拌した。
反応溶液に脱イオン水(10ml)を添加して内容物を完溶させ、溶液を静置分液した。
有機層を採り、硫酸マグネシウムで乾燥後、溶媒を減圧留去して測定サンプルである誘導体化化合物を得た。
前記誘導体化化合物を、下記の条件で測定することにより、光学純度を求めた。
【0054】
[光学純度測定条件]
カラム:ダイセル化学工業(株)製キラルセル AD−H 4.6φ×250mm
移動層:ヘキサン:エタノール=25:75(体積比、0.1体積%のジエチルアミン含有する。)
流速:0.7ml/min
カラム温度:40℃
検出器:UV 254nm
リテンションタイム:(R)体:17min,(S)体:18min
【0055】
〔1−ベンジル−3−アミノピロリジンの測定方法〕
[分析のための誘導体化]
1−ベンジル−3−アミノピロリジン(18.0mg)をアセトニトリル(1ml)に溶解させ、0.4重量%GITC(2,3,4,6−テトラ−O−アセチル−β−D−グルコピラノシル−イソチオシアネート)アセトニトリル溶液(2ml)を添加し、15〜25℃で0.5時間撹拌した。これに0.2重量%エタノールアミンアセトニトリル溶液(1ml)を添加して0.5時間撹拌し、反応液を0.05%リン酸水溶液でメスアップ(40ml)して測定溶液とした。
前記測定溶液を、下記の条件で測定することにより、光学純度を求めた。
[測定条件]
カラム:(株)資生堂製カプセルパック C18 SG120 4.6φ×150mm
キャリア:0.03重量%アンモニア水溶液(酢酸にてpHを4に調整した。)/メタノール=62:38(体積比)
検出器:UV 254nm
測定温度:室温
流速:0.5ml/min
リテンションタイム:(R)体:49min,(S)体:53min
【0056】
(実施例1)
(RS)−1−Boc−3−アミノピペリジン(40.0g,0.200mol),(S)−2−フェノキシプロピオン酸(16.6g,0.100mol),アセトン(850ml)を混合、加熱、還流し内容物を溶解させた。溶液を15〜25℃まで12時間かけて冷却し、更に15〜25℃で12時間撹拌した。生じた結晶を分離して、35〜45℃で減圧乾燥し、粗ジアステレオマー((S)−1−Boc−3−アミノピペリジン/(S)−2−フェノキシプロピオン酸)を得た(収量31.1g,収率85.0%,光学純度63.0%ee)。なお以下に記載される1−Boc−3−アミノピペリジンの絶対配置は、後述する実施例20により求まった絶対構造と、各比旋光度の極性を元にした。
得られた粗ジアステレオマーをアセトンで2回再結晶して、精製ジアステレオマー((S)−1−Boc−3−アミノピペリジン/(S)−2−フェノキシプロピオン酸)を得ることができた(収量22.0g,総収率60.1%,光学純度98.6%ee)。
なお、得られた精製ジアステレオマーは非水滴定法で分析した結果、1−Boc−3−アミノピペリジン:2−フェノキシプロピオン酸の結合比が、1:1であることが判明した。
【0057】
(実施例2)
実施例1で得られた精製ジアステレオマー(20.0g,54.6mmol)にジクロロメタン(66ml)を混合して、溶液の温度を0〜15℃とした。この温度を保ちながら5%水酸化ナトリウム水溶液(66.0g,82.5mmol)を滴下した。15min撹拌後、内容物が完溶したら溶液を静置、分液した。有機層をとり、脱イオン水で洗浄後、硫酸マグネシウムで乾燥した後溶媒を減圧留去して、(S)−1−Boc−3−アミノピペリジンを得た(収量10.4g,精製ジアステレオマーからの回収率95.0%)。
一方、水層は35%塩酸で酸性としたのちジクロロメタンを添加して分割剤((S)−2−フェノキシプロピオン酸)を抽出した。その後硫酸ナトリウムで乾燥後、溶媒を減圧留去することによって、分割剤を定量的に回収した。
上記で得られた(S)−1−Boc−3−アミノピペリジン(5.0g,25.0mmol)をN2気流下で脱水メタノール(50ml)に溶解させ、0〜15℃に冷却した。これに5%塩化水素メタノール溶液(54.5g,75.0mmol)を反応液に温度を保ちながら滴下した。0.5時間撹拌後、溶媒を減圧留去して、(S)−3−アミノピペリジン二塩酸塩を得ることができた(収量4.1g,収率95%,比旋光度[α]D+2.5°,C=1.0 H2O,25℃)。
【0058】
(実施例3)
実施例1で得られた粗ジアステレオマー分離母液を減圧留去して、これにジクロロメタン(350ml)を添加した。溶液を0〜15℃に冷却して、温度を保ちながら5%水酸化ナトリウム水溶液(50ml)を滴下した。内容物が溶解したことが確認できたら、溶液を静置、分液した(水層から(S)−2−フェノキシプロピオン酸を回収した。)。
有機層を脱イオン水で洗浄後、硫酸マグネシウムで乾燥したのち、溶媒を減圧留去して粗(R)−1−Boc−3−アミノピペリジンを得た(回収量23.0g,光学純度50.0%ee,(R)体含有量17.7g,0.0883mol)。
これとアセトン(750ml),(R)−2−フェノキシプロピオン酸(14.7g,0.0883mol)を混合して溶液を加熱還流した。以下、実施例1と同様の操作を行い、粗ジアステレオマー((R)−1−Boc−3−アミノピペリジン/(R)−2−フェノキシプロピオン酸)を得ることができた(収量31.0g,(RS)体からの収率84.6%,光学純度64.0%ee)。
以下、実施例1と同様に精製ジアステレオマー((R)−1−Boc−3−アミノピペリジン/(R)−2−フェノキシプロピオン酸)を得ることができた(収量23.0g,総収率62.7%,光学純度98.2%ee)。
なお、得られた精製ジアステレオマーは非水滴定法で分析した結果、1−Boc−3−アミノピペリジン:2−フェノキシプロピオン酸の結合比が、1:1であることが判明した。
【0059】
(実施例4)
実施例3で得られた精製ジアステレオマー(20.0g,54.6mmol)にジクロロメタン(66ml)を混合して、溶液の温度を0〜15℃とした。以下、実施例2と同様の操作を行い、(R)−1−Boc−3−アミノピペリジンを得た(収量10.7g,精製ジアステレオマーからの回収率97.0%)。
上記で得られた(R)−1−Boc−3−アミノピペリジン(5.0g,25.0mmol)をN2気流下で脱水メタノール(50ml)に溶解させ、0〜15℃に冷却した。以下、実施例2と同様な操作を行い、(R)−3−アミノピペリジン二塩酸塩を得ることができた(収量4.1g,収率95%,比旋光度[α]D−2.8°,C=1.0 H2O,25℃)。
【0060】
(実施例5)
(RS)−1−Boc−3−アミノピペリジン(40.0g,0.200mol),(R)−2−フェノキシプロピオン酸(16.6g,0.100mol),アセトン(850ml)を混合、加熱、還流し内容物を溶解させた。以下、実施例1と同様な操作を行い、精製ジアステレオマー((R)−1−Boc−3−アミノピペリジン/(R)−2−フェノキシプロピオン酸)を得ることができた(収量22.6g,総収率61.7%,光学純度98.8%ee)。
【0061】
(実施例6〜19)
実施例6〜19については(RS)−3−アミノピペリジン誘導体(基質)、酸性分割剤、及び、基質/酸性分割剤の混合比を、下記表1に示すように変更した以外は実施例1と同様な操作を行い、粗ジアステレオマーを得た。結果を表1示す。
なお、実施例16〜19において、収率が100%を越えているが、収率は所望するジアステレオマー塩の理論生成量を基準としているためである。
【0062】
【表1】
【0063】
(実施例20)
実施例1の過程で得られた精製ジアステレオマー((S)−1−Boc−3−アミノピペリジン/(S)−2−フェノキシプロピオン酸)において、1−Boc−3−アミノピペリジンの絶対構造を確定するために、複合塩のX線単結晶構造解析を行った。
まず溶媒として含水アセトンに試料を溶解させ、自然蒸発法により、サイズ0.4×0.4×0.2mmの柱状の単結晶を得ることができた。得られた結晶学的データ及び結晶構造を表2及び図1に示す。
光学分割剤(S)−2−フェノキシプロピオン酸の絶対配置を用いて複合塩の手系を決定し、その結果、1−Boc−3−アミノピペリジンの絶対配置は、(S)体であることが明らかになった。
【0064】
【表2】
【0065】
(実施例21)
(RS)−1−ベンジル−3−アミノピロリジン(37.0g,0.209mol),(R,R)−2’−メチルタートラニル酸(50.0g,0.209mol),メタノール(260ml)を混合、加熱して内容物を溶解させたのち、反応液を15−25℃まで一夜冷却した。生じた結晶を分離して、粗ジアステレオマー体を得た((R)−1−ベンジル−3−アミノピロリジン/(R,R)−2’−メチルタートラニル酸,結合比=1:1,非水滴定法,収量66.8g,収率153%,光学純度65%ee)。
粗ジアステレオマー体を、メタノールを用いて2回再結晶し、精製ジアステレオマー塩を得た(収量30.8g,収率70.8%,光学純度98%ee)。
精製ジアステレオマー塩(30.8g,0.0740mol),5%水酸化カリウム水溶液(100g,0.089mol),トルエン(100ml)を混合し、静置、分液した。水層をさらにトルエン(100ml×2)で洗浄した(水層から分割剤を回収した。)。
有機層をあわせ、硫酸ナトリウムで乾燥後、溶媒を減圧留去し、(R)−1−ベンジル−3−アミノピロリジンを得た(収量12.4g,精製ジアステレオマー塩からの収率95.5%)。
【0066】
(実施例22)
(R,R)−2’−メチルタートラニル酸の代わりに(S,S)−2’−メチルタートラニル酸を用いて、実施例21と同様な操作を行い、粗ジアステレオマー塩を得た((S)−1−ベンジル−3−アミノピロリジン/(S,S)−2’−メチルタートラニル酸,結合比=1:1,非水滴定法,収量67.9g,収率156%,光学純度63%ee)。
以下、実施例21と同様な操作を行い、精製ジアステレオマー塩(収量31.3g,収率72.0%,光学純度99%ee),(S)−1−ベンジル−3−アミノピロリジン(収量12.8g,精製ジアステレオマー塩からの収率96.3%)を得た。
【0067】
(実施例23〜28)
実施例23〜28については、酸性分割剤を下記表3のように変更した以外は、実施例21と同様の操作を実施して、粗ジアステレオマー体を得ることができた。
【0068】
なお、前記実施例21、22、24において、収率が100%を越えているが、収率は所望するジアステレオマー塩の理論生成量を基準としているためである。
【0069】
【表3】
【産業上の利用可能性】
【0070】
本発明で記載されている光学活性環状アミノ化合物誘導体は、医薬品中間体原料として、注目されている。特に3−アミノピペリジン誘導体は、次期ブロックバスター候補として重要であり、これらの化合物の製造が容易にできる本発明の産業上の利用可能性は極めて高いということができる。
【特許請求の範囲】
【請求項1】
下記式(A−1)又は(A−2)で表されるアミン化合物と下記式(B−1)又は(B−2)で表される酸性分割剤とを反応させてジアステレオマー塩を形成する工程、
得られたジアステレオマー塩の少なくとも一部を析出させ精製ジアステレオマー塩を得る工程、及び、
精製ジアステレオマー塩を塩基性水溶液により分解し、下記式(A−1)又は(A−2)で表される光学活性アミン化合物を得る工程を含むことを特徴とする
光学活性アミン化合物の製造方法。
【化1】
(式中、R1及びR2はそれぞれ独立に、水素原子、炭素数1〜10のアルキル基、又は、炭素数6〜10のアリール基を表し、R3は炭素数7〜20のアラルキル基、炭素数2〜20のアルキルオキシカルボニル基、炭素数7〜20のアリールオキシカルボニル基、又は、炭素数8〜20のアラルキルオキシカルボニル基を表し、*は不斉炭素原子であることを表す。)
【化2】
(式中、R4及びR9は、酸性官能基を表し、R5〜R8はそれぞれ独立に、アルキル基、アリール基、アラルキル基、アルキルオキシ基、アリールオキシ基、アラルキルオキシ基、アルキルカルボニルオキシ基、アリールカルボニルオキシ基、アラルキルカルボニルオキシ基、又は、ヒドロキシ基を表し、R10は、酸性官能基、又は、アリールアミノカルボニル基を表し、*は不斉炭素原子であることを表す。ただし、R5とR6とは異なる基である。)
【請求項2】
前記アミン化合物が、下記式(A−1−1)〜(A−1−3)で表されるアミン化合物である請求項1に記載の光学活性アミン化合物の製造方法。
【化3】
(式中、*は不斉炭素原子であることを表す。)
【請求項3】
前記酸性分割剤が、下記式(B−1−1)〜(B−1−4)又は(B−2−1)〜(B−2−6)で表される化合物である請求項1又は2に記載の光学活性アミン化合物の製造方法。
【化4】
(式中、*は不斉炭素原子であることを表す。)
【請求項4】
下記式(A−1)又は(A−2)で表されるアミン化合物と下記式(B−1)又は(B−2)で表される酸性分割剤とを反応させてジアステレオマー塩を形成する工程、及び、
得られたジアステレオマー塩の少なくとも一部を析出させ精製ジアステレオマー塩を得る工程を含むことを特徴とする
ジアステレオマー塩の製造方法。
【化5】
(式中、R1及びR2はそれぞれ独立に、水素原子、炭素数1〜10のアルキル基、又は、炭素数6〜10のアリール基を表し、R3は炭素数7〜20のアラルキル基、炭素数2〜20のアルキルオキシカルボニル基、炭素数7〜20のアリールオキシカルボニル基、又は、炭素数8〜20のアラルキルオキシカルボニル基を表し、*は不斉炭素原子であることを表す。)
【化6】
(式中、R4及びR9は、酸性官能基を表し、R5〜R8はそれぞれ独立に、アルキル基、アリール基、アラルキル基、アルキルオキシ基、アリールオキシ基、アラルキルオキシ基、アルキルカルボニルオキシ基、アリールカルボニルオキシ基、アラルキルカルボニルオキシ基、又は、ヒドロキシ基を表し、R10は、酸性官能基、又は、アリールアミノカルボニル基を表し、*は不斉炭素原子であることを表す。ただし、R5とR6とは異なる基である。)
【請求項5】
前記アミン化合物が、下記式(A−1−1)〜(A−1−3)で表されるアミン化合物である請求項4に記載のジアステレオマーの製造方法。
【化7】
(式中、*は不斉炭素原子であることを表す。)
【請求項6】
前記酸性分割剤が、下記式(B−1−1)〜(B−1−4)又は(B−2−1)〜(B−2−6)で表される化合物である請求項4又は5に記載のジアステレオマーの製造方法。
【化8】
(式中、*は不斉炭素原子であることを表す。)
【請求項7】
下記式(A−1)又は(A−2)で表され、かつ不斉炭素原子を有するアミン化合物と
下記式(B−1)又は(B−2)で表される酸性分割剤とからなることを特徴とする
ジアステレオマー塩。
【化9】
(式中、R1及びR2はそれぞれ独立に、水素原子、炭素数1〜10のアルキル基、又は、炭素数6〜10のアリール基を表し、R3は炭素数7〜20のアラルキル基、炭素数2〜20のアルキルオキシカルボニル基、炭素数7〜20のアリールオキシカルボニル基、又は、炭素数8〜20のアラルキルオキシカルボニル基を表し、*は不斉炭素原子であることを表す。)
【化10】
(式中、R4及びR9は、酸性官能基を表し、R5〜R8はそれぞれ独立に、アルキル基、アリール基、アラルキル基、アルキルオキシ基、アリールオキシ基、アラルキルオキシ基、アルキルカルボニルオキシ基、アリールカルボニルオキシ基、アラルキルカルボニルオキシ基、又は、ヒドロキシ基を表し、R10は、酸性官能基、又は、アリールアミノカルボニル基を表し、*は不斉炭素原子であることを表す。ただし、R5とR6とは異なる基である。)
【請求項8】
前記アミン化合物が、下記式(A−1−1)〜(A−1−3)で表されるアミン化合物である請求項7に記載のジアステレオマー塩。
【化11】
(式中、*は不斉炭素原子であることを表す。)
【請求項9】
前記酸性分割剤が、下記式(B−1−1)〜(B−1−4)又は(B−2−1)〜(B−2−6)で表される化合物である請求項7又は8に記載のジアステレオマー塩。
【化12】
(式中、*は不斉炭素原子であることを表す。)
【請求項1】
下記式(A−1)又は(A−2)で表されるアミン化合物と下記式(B−1)又は(B−2)で表される酸性分割剤とを反応させてジアステレオマー塩を形成する工程、
得られたジアステレオマー塩の少なくとも一部を析出させ精製ジアステレオマー塩を得る工程、及び、
精製ジアステレオマー塩を塩基性水溶液により分解し、下記式(A−1)又は(A−2)で表される光学活性アミン化合物を得る工程を含むことを特徴とする
光学活性アミン化合物の製造方法。
【化1】
(式中、R1及びR2はそれぞれ独立に、水素原子、炭素数1〜10のアルキル基、又は、炭素数6〜10のアリール基を表し、R3は炭素数7〜20のアラルキル基、炭素数2〜20のアルキルオキシカルボニル基、炭素数7〜20のアリールオキシカルボニル基、又は、炭素数8〜20のアラルキルオキシカルボニル基を表し、*は不斉炭素原子であることを表す。)
【化2】
(式中、R4及びR9は、酸性官能基を表し、R5〜R8はそれぞれ独立に、アルキル基、アリール基、アラルキル基、アルキルオキシ基、アリールオキシ基、アラルキルオキシ基、アルキルカルボニルオキシ基、アリールカルボニルオキシ基、アラルキルカルボニルオキシ基、又は、ヒドロキシ基を表し、R10は、酸性官能基、又は、アリールアミノカルボニル基を表し、*は不斉炭素原子であることを表す。ただし、R5とR6とは異なる基である。)
【請求項2】
前記アミン化合物が、下記式(A−1−1)〜(A−1−3)で表されるアミン化合物である請求項1に記載の光学活性アミン化合物の製造方法。
【化3】
(式中、*は不斉炭素原子であることを表す。)
【請求項3】
前記酸性分割剤が、下記式(B−1−1)〜(B−1−4)又は(B−2−1)〜(B−2−6)で表される化合物である請求項1又は2に記載の光学活性アミン化合物の製造方法。
【化4】
(式中、*は不斉炭素原子であることを表す。)
【請求項4】
下記式(A−1)又は(A−2)で表されるアミン化合物と下記式(B−1)又は(B−2)で表される酸性分割剤とを反応させてジアステレオマー塩を形成する工程、及び、
得られたジアステレオマー塩の少なくとも一部を析出させ精製ジアステレオマー塩を得る工程を含むことを特徴とする
ジアステレオマー塩の製造方法。
【化5】
(式中、R1及びR2はそれぞれ独立に、水素原子、炭素数1〜10のアルキル基、又は、炭素数6〜10のアリール基を表し、R3は炭素数7〜20のアラルキル基、炭素数2〜20のアルキルオキシカルボニル基、炭素数7〜20のアリールオキシカルボニル基、又は、炭素数8〜20のアラルキルオキシカルボニル基を表し、*は不斉炭素原子であることを表す。)
【化6】
(式中、R4及びR9は、酸性官能基を表し、R5〜R8はそれぞれ独立に、アルキル基、アリール基、アラルキル基、アルキルオキシ基、アリールオキシ基、アラルキルオキシ基、アルキルカルボニルオキシ基、アリールカルボニルオキシ基、アラルキルカルボニルオキシ基、又は、ヒドロキシ基を表し、R10は、酸性官能基、又は、アリールアミノカルボニル基を表し、*は不斉炭素原子であることを表す。ただし、R5とR6とは異なる基である。)
【請求項5】
前記アミン化合物が、下記式(A−1−1)〜(A−1−3)で表されるアミン化合物である請求項4に記載のジアステレオマーの製造方法。
【化7】
(式中、*は不斉炭素原子であることを表す。)
【請求項6】
前記酸性分割剤が、下記式(B−1−1)〜(B−1−4)又は(B−2−1)〜(B−2−6)で表される化合物である請求項4又は5に記載のジアステレオマーの製造方法。
【化8】
(式中、*は不斉炭素原子であることを表す。)
【請求項7】
下記式(A−1)又は(A−2)で表され、かつ不斉炭素原子を有するアミン化合物と
下記式(B−1)又は(B−2)で表される酸性分割剤とからなることを特徴とする
ジアステレオマー塩。
【化9】
(式中、R1及びR2はそれぞれ独立に、水素原子、炭素数1〜10のアルキル基、又は、炭素数6〜10のアリール基を表し、R3は炭素数7〜20のアラルキル基、炭素数2〜20のアルキルオキシカルボニル基、炭素数7〜20のアリールオキシカルボニル基、又は、炭素数8〜20のアラルキルオキシカルボニル基を表し、*は不斉炭素原子であることを表す。)
【化10】
(式中、R4及びR9は、酸性官能基を表し、R5〜R8はそれぞれ独立に、アルキル基、アリール基、アラルキル基、アルキルオキシ基、アリールオキシ基、アラルキルオキシ基、アルキルカルボニルオキシ基、アリールカルボニルオキシ基、アラルキルカルボニルオキシ基、又は、ヒドロキシ基を表し、R10は、酸性官能基、又は、アリールアミノカルボニル基を表し、*は不斉炭素原子であることを表す。ただし、R5とR6とは異なる基である。)
【請求項8】
前記アミン化合物が、下記式(A−1−1)〜(A−1−3)で表されるアミン化合物である請求項7に記載のジアステレオマー塩。
【化11】
(式中、*は不斉炭素原子であることを表す。)
【請求項9】
前記酸性分割剤が、下記式(B−1−1)〜(B−1−4)又は(B−2−1)〜(B−2−6)で表される化合物である請求項7又は8に記載のジアステレオマー塩。
【化12】
(式中、*は不斉炭素原子であることを表す。)
【図1】

【公開番号】特開2011−57619(P2011−57619A)
【公開日】平成23年3月24日(2011.3.24)
【国際特許分類】
【出願番号】特願2009−209169(P2009−209169)
【出願日】平成21年9月10日(2009.9.10)
【出願人】(000125369)学校法人東海大学 (352)
【出願人】(591169386)大東化学株式会社 (11)
【Fターム(参考)】
【公開日】平成23年3月24日(2011.3.24)
【国際特許分類】
【出願日】平成21年9月10日(2009.9.10)
【出願人】(000125369)学校法人東海大学 (352)
【出願人】(591169386)大東化学株式会社 (11)
【Fターム(参考)】
[ Back to top ]