
光起電力素子用材料および光起電力素子
【課題】光電変換効率の高い光起電力素子を提供すること。
【解決手段】特定の構造を有する電子供与性有機材料を含む光起電力素子用材料、およびこれを用いた光起電力素子。
【解決手段】特定の構造を有する電子供与性有機材料を含む光起電力素子用材料、およびこれを用いた光起電力素子。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、光起電力素子用材料およびこれを用いた光起電力素子に関する。
【背景技術】
【0002】
太陽電池は環境に優しい電気エネルギー源として、現在深刻さを増すエネルギー問題に対して有力なエネルギー源と注目されている。現在、太陽電池の光起電力素子の半導体素材としては、単結晶シリコン、多結晶シリコン、アモルファスシリコン、化合物半導体などの無機物が使用されている。しかし、無機半導体を用いて製造される太陽電池は、火力発電や原子力発電などの発電方式と比べてコストが高いために、一般家庭に広く普及するには至っていない。コスト高の要因は主として、真空かつ高温下で半導体薄膜を製造するプロセスにある。そこで、製造プロセスの簡略化が期待される半導体素材として、共役系重合体や有機結晶などの有機半導体や有機色素を用いた有機太陽電池が検討されている。
【0003】
しかし、共役系重合体などを用いた有機太陽電池は、従来の無機半導体を用いた太陽電池と比べて光電変換効率が低いことが最大の課題であり、まだ実用化には至っていない。従来の共役系重合体を用いた有機太陽電池の光電変換効率が低いのは、主として、太陽光の吸収効率が低いことや、太陽光によって生成された電子と正孔が分離しにくいエキシトンという束縛状態が形成されることと、キャリア(電子、正孔)を捕獲するトラップが形成されやすいため生成したキャリアがトラップに捕獲されやすく、キャリアの移動度が遅いことなどによる。
【0004】
これまでの有機半導体による光電変換素子は、現在のところ一般的に次のような素子構成に分類することができる。電子供与性有機材料(p型有機半導体)と仕事関数の小さい金属を接合させるショットキー型、電子受容性有機材料(n型有機半導体)と電子供与性有機材料(p型有機半導体)を接合させるヘテロ接合型などである。これらの素子は、接合部の有機層(数分子層程度)のみが光電流生成に寄与するため光電変換効率が低く、その向上が課題となっている。
【0005】
光電変換効率向上の一つの方法として、電子受容性有機材料(n型有機半導体)と電子供与性有機材料(p型有機半導体)を混合し、光電変換に寄与する接合面を増加させたバルクヘテロ接合型(例えば、非特許文献1参照)がある。なかでも、電子供与性有機材料(p型有機半導体)として共役系重合体を用い、電子受容性有機材料としてn型の半導体特性をもつ導電性高分子のほかC60などのフラーレンやフラーレン誘導体を用いた光電変換材料が報告されている(例えば、非特許文献2参照)。
【0006】
また、太陽光スペクトルの広い範囲にわたる放射エネルギーを効率よく吸収し、光電変換効率を向上させるために、主鎖骨格に電子供与性基と電子吸引性基を導入することでバンドギャップを狭めた電子供与性有機材料が報告されている(例えば、非特許文献3参照)。この電子供与性基としてはチオフェン骨格やオリゴチオフェン骨格が、電子吸引性基としてはベンゾチアジアゾール骨格やキノキサリン骨格などが精力的に研究されている(例えば、特許文献1〜2、非特許文献3〜5参照)。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特表2004−534863号公報
【特許文献2】特表2004−500464号公報
【非特許文献】
【0008】
【非特許文献1】J.J.M.Halls、C.A.Walsh、N.C.Greenham、E.A.Marseglla、R.H.Friend、S.C.Moratti、A.B.Holmes著、「ネイチャー(Nature)」、1995年、376号、498頁
【非特許文献2】G.Yu、J.Gao、J.C.Hummelen、F.Wudl、A.J.Heeger著、「サイエンス(Science)」、1995年、270巻、1789頁
【非特許文献3】E.Bundgaard、F.C.Kreb著、「ソーラー エナジー マテリアルズ アンド ソーラー セル(Solar Energy Materials & Solar Cells)」、2007年、91巻、954頁
【非特許文献4】J.Hou、M.−H.Park、S.Zhang、Y.Yao、L.−M.Chen、J.−H.Li、Y.Yang著、「マクロモレキュルズ(Macromolecules)」、2008年、41巻、6012頁
【非特許文献5】S.C.Prince、A.C.Stuart、W.You著、「マクロモレキュルズ(Macromolecules)」、2010年、43巻、4609頁
【発明の概要】
【発明が解決しようとする課題】
【0009】
しかしながら、上述の電子供与性基としてチオフェン骨格やオリゴチオフェン骨格、電子吸引性基としてベンゾチアジアゾール骨格やキノキサリン骨格を用いた狭バンドギャップ電子供与性有機材料(特許文献1〜2、非特許文献3)では主鎖のチオフェン骨格のねじれのために十分なキャリア移動度が保てないと考えた。
【0010】
キャリア移動度を高めるために、平面性の高い電子供与性基であるベンゾジチオフェン骨格と、電子吸引性基であるベンゾチアジアゾール骨格やキノキサリン骨格等を組み合わせた狭バンドギャップポリマーが報告されている(非特許文献4、5)。しかしながら、ベンゾチアジアゾール骨格またはキノキサリン骨格と、ベンゾジチオフェン骨格を直接連結した狭バンドギャップ電子供与性有機材料(非特許文献4)では、ポリマー主鎖骨格にねじれが生じ、キャリアの移動度が阻害されるために十分な変換効率が得られていない。
【0011】
また、ベンゾチアジアゾール骨格とチオフェン骨格、およびベンゾジチオフェン骨格を組み合わせた狭バンドギャップ電子供与性有機材料(非特許文献5)においては、ねじれが解消され、キャリア移動度が向上するが、平面性が高すぎるために有機溶媒に対する溶解性、並びにフラーレン誘導体等に代表される電子受容性材料との相溶性が低下し、結果として十分な光電変換効率が得られていなかった。これは溶解性を保つために、過剰な長鎖アルキル側鎖を狭バンドギャップ電子供与性有機材料に導入する必要があったことが一因である。
【0012】
すなわち、従来の電子供与性有機材料を用いた光起電力素子では、高いキャリア移動度と有機溶媒に対する溶解性、並びにフラーレン誘導体に代表される電子受容性材料との相溶性を両立させることが困難であり、いずれも光電変換効率が低かった。本発明は光電変換効率の高い光起電力素子を提供することを目的とし、高いキャリア移動度と有機溶媒に対する溶解性およびフラーレン誘導体等の電子受容性材料との相溶性を両立させた電子供与性有機材料を提供する。
【課題を解決するための手段】
【0013】
電子吸引性基としてベンゾチアジアゾール骨格やキノキサリン骨格、電子供与基として平面性の高いベンゾジチオフェン骨格を用いた狭バンドギャップ電子供与性有機材料について、様々な構造を検討した結果、ジフェニルキノキサリン等の平面性をくずした構造を導入することによって、高いキャリア移動度を保ったまま、有機溶媒に対する溶解性とフラーレン等の電子受容性材料との相溶性を両立させることができることを見出した。
【0014】
すなわち本発明は、一般式(1)で表される構造を有する電子供与性有機材料を含む光起電力素子用材料、および、これを用いた光起電力素子である。
【0015】
【化1】

【0016】
上記一般式(1)中、R1およびR2は同じでも異なっていてもよく、置換されていてもよいアリール基、または置換されていてもよいヘテロアリール基を表す。このキノキサリン上に配置された隣接するアリール基またはヘテロアリール基によって、主鎖構造の平面性を保持しつつ電子供与性材料の平面性を部分的に崩すことが可能となり、高いキャリア移動度を保ったまま、有機溶媒に対する溶解性と電子受容性材料との相溶性を両立させることができる。R3〜R6は同じでも異なっていてもよく、水素、アルキル基、アルコキシ基、アリール基、ヘテロアリール基、またはハロゲンを表す。XおよびYは2価の連結基を表す。nは2以上1,000以下の範囲を表す。)
【発明の効果】
【0017】
キノキサリン上にアリールまたはヘテロアリール基を配置し、側鎖の平面性を崩すことによって、高いキャリア移動度を保ったまま有機溶媒に対する溶解性およびフラーレン誘導体等の電子受容性材料との相溶性を両立させることが可能となり、光電変換効率を向上させた光起電力素子を提供することができる。
【図面の簡単な説明】
【0018】
【図1】本発明の光起電力素子の一態様を示した模式図。
【図2】本発明の光起電力素子の別の態様を示した模式図。
【発明を実施するための形態】
【0019】
本発明の光起電力素子用材料は、一般式(1)で表される電子供与性有機材料を含む。
【0020】
【化2】
【0021】
上記一般式(1)中、R1およびR2は同じでも異なっていてもよく、置換されていてもよいアリール基、または置換されていてもよいヘテロアリール基を表す。前述のとおり、このキノキサリン上に配置された隣接するアリール基またはヘテロアリール基によって、主鎖構造の平面性を保持しつつ電子供与性材料の平面性を部分的に崩すことが可能となり、高いキャリア移動度を保ったまま、有機溶媒に対する溶解性と電子受容性材料との相溶性を両立させることができる。
【0022】
ここでアリール基とは、例えば、フェニル基、ナフチル基、ビフェニル基、フェナントリル基、アントリル基、ターフェニル基、ピレニル基、フルオレニル基、ペリレニル基などの芳香族炭化水素基を示し、これは無置換でも置換されていてもかまわない。置換される場合の置換基の例としては、下記アルキル基や下記アルコキシ基、下記ヘテロアリール基、ハロゲンが挙げられる。電子供与性有機材料のキャリア移動度を保ちつつ、側鎖構造の平面性を崩すことによって、有機溶媒対する溶解性およびフラーレン誘導体等の電子受容性材料との相溶性を両立させるためには、R1およびR2が共にフェニル基であることが好ましい。
【0023】
また、ヘテロアリール基とは、例えば、チエニル基、フリル基、ピロリル基、イミダゾリル基、ピラゾリル基、オキサゾリル基、ピリジル基、ピラジル基、ピリミジル基、キノリニル基、イソキノリル基、キノキサリル基、アクリジニル基、インドリル基、カルバゾリル基、ベンゾフラン基、ジベンゾフラン基、ベンゾチオフェン基、ジベンゾチオフェン基、ベンゾジチオフェン基、シロール基、ベンゾシロール基、ジベンゾシロール基などの炭素以外の原子を有する複素芳香族環基を示し、これは無置換でも置換されていてもかまわない。置換される場合の置換基の例としては、下記アルキル基、上記アリール基、ハロゲンが挙げられる。ヘテロアリール基の炭素数は、キャリア移動度を保つために2以上8以下が好ましい。
【0024】
上記一般式(1)中、R3〜R6はそれぞれ同じでも異なっていてもよく、水素、アルキル基、アルコキシ基、上記アリール基、上記ヘテロアリール基、ハロゲンの中から選ばれる。R3およびR4は主鎖骨格のねじれを抑えるため体積の小さな置換基が好ましく、水素、フッ素、アルキル基が好ましく用いられる。合成の容易さから水素が特に好ましく用いられる。
【0025】
R5およびR6は電子供与性の溶解性を保つためにアルキル基、アルコキシ基、アルキルアリール基、アルキルヘテロアリール基が好ましく用いられる。電子供与性有機材料の平面性を高めることでキャリア移動度を向上できることから、アルキルチエニル基が特に好ましく用いられる。合成の容易さからはアルコキシ基が特に好ましく用いられる。
【0026】
ここでアルキル基とは、例えば、メチル基、エチル基、プロピル基、ブチル基、ペンチル基、ヘキシル基、ヘプチル基、オクチル基、ノニル基、デシル基、ウンデシル基、ドデシル基のような飽和脂肪族炭化水素基であり、直鎖状であっても分岐状であっても環状であってもよく、無置換でも置換されていてもかまわない。置換される場合の置換基の例としては、下記アルコキシ基、上記アリール基、上記ヘテロアリール基、ハロゲンが挙げられる。アルキル基の炭素数は、有機材料の溶解性をあげてスピンコートなどのウエットプロセスに適用する場合には4以上であることが好ましく、有機材料の十分なキャリア移動度を保つためには12以下であることが好ましい。
【0027】
また、アルコキシ基とは、例えば、メトキシ基、エトキシ基、プロポキシ基、ブトキシ基などのエーテル結合を介した脂肪族炭化水素基を示し、脂肪族炭化水素基は無置換でも置換されていてもかまわない。置換される場合の置換基の例としては、上記アリール基や上記ヘテロアリール基、ハロゲンが挙げられる。アルコキシ基の好ましい炭素数の範囲は、上述のアルキル基の場合と同じである。
【0028】
また、ハロゲンは、フッ素、塩素、臭素、ヨウ素のいずれかである。
【0029】
上記一般式(1)中、XおよびYは同じでも異なっていてもよい2価の連結基を表す。電子供与性有機材料の電子共役系を伸ばしてキャリア移動度を高めるためには、アリーレン基やヘテロアリーレン基が好ましい。主鎖構造平面性を保ち、キャリア移動度を保つためにはチオフェン基、ジチオフェン基、ビチオフェン基が好ましく、チオフェン基が特に好ましく用いられる。
【0030】
上記一般式(1)中、nは重合度を示し、2以上1,000以下の範囲を表す。nを2以上とすることにより、前述のバルクヘテロ接合の薄膜において有効なキャリアパスを形成させることができるために、光電変換効率を高めることができる。また、優れたキャリアパスを形成させるためにはnは5以上であることが好ましく、合成上の容易さから100未満であることが好ましい。重合度は重量平均分子量から求めることができる。重量平均分子量は、GPC(ゲルパーミエーションクロマトグラフィー)を用いて測定し、ポリスチレンの標準試料に換算して求めることができる。
【0031】
上記の一般式(1)で表される構造を有する電子供与性有機材料として、具体的には下記のような構造が挙げられる。ただし、nは2以上1,000以下の範囲を示す。
【0032】
【化3】
【0033】
【化4】
【0034】
【化5】
【0035】
【化6】
【0036】
【化7】
【0037】
なお、一般式(1)で表される構造を有する電子供与性有機材料は、例えば、前記非特許文献4に記載されている方法に類似した手法や、前記非特許文献5に記載されている方法に類似した手法により合成することができる。
【0038】
本発明の光起電力素子用材料は、一般式(1)で表される構造を有する電子供与性有機材料のみからなるものでもよいし、他の電子供与性有機材料を含んでもよい。他の電子供与性有機材料としては、例えば、ポリチオフェン系重合体、ベンゾチアジアゾール−チオフェン系誘導体、ベンゾチアジアゾール−チオフェン系共重合体、ポリ−p−フェニレンビニレン系重合体、ポリ−p−フェニレン系重合体、ポリフルオレン系重合体、ポリピロール系重合体、ポリアニリン系重合体、ポリアセチレン系重合体、ポリチエニレンビニレン系重合体などの共役系重合体や、H2フタロシアニン(H2Pc)、銅フタロシアニン(CuPc)、亜鉛フタロシアニン(ZnPc)等のフタロシアニン誘導体、ポルフィリン誘導体、N,N’−ジフェニル−N,N’−ジ(3−メチルフェニル)−4,4’−ジフェニル−1,1’−ジアミン(TPD)、N,N’−ジナフチル−N,N’−ジフェニル−4,4’−ジフェニル−1,1’−ジアミン(NPD)等のトリアリールアミン誘導体、4,4’−ジ(カルバゾール−9−イル)ビフェニル(CBP)等のカルバゾール誘導体、オリゴチオフェン誘導体(ターチオフェン、クウォーターチオフェン、セキシチオフェン、オクチチオフェンなど)等の低分子有機化合物が挙げられる。
【0039】
一般式(1)で表される構造を有する電子供与性有機材料はp型半導体特性を示す材料であり、本発明の光起電力素子用材料は、より高い光電変換効率を得るために電子受容性有機材料(n型有機半導体)と組み合わせることが好ましい。
【0040】
本発明で用いる電子受容性有機材料とは、n型半導体特性を示す有機材料であり、例えば、1,4,5,8−ナフタレンテトラカルボキシリックジアンハイドライド(NTCDA)、3,4,9,10−ペリレンテトラカルボキシリックジアンハイドライド(PTCDA)、3,4,9,10−ペリレンテトラカルボキシリックビスベンズイミダゾール(PTCBI)、N,N'−ジオクチル−3,4,9,10−ナフチルテトラカルボキシジイミド(PTCDI−C8H)、2−(4−ビフェニリル)−5−(4−t−ブチルフェニル)−1,3,4−オキサジアゾール(PBD)、2,5−ジ(1−ナフチル)−1,3,4−オキサジアゾール(BND)等のオキサゾール誘導体、3−(4−ビフェニリル)−4−フェニル−5−(4−t−ブチルフェニル)−1,2,4−トリアゾール(TAZ)等のトリアゾール誘導体、フェナントロリン誘導体、ホスフィンオキサイド誘導体、フラーレン化合物(C60、C70、C76、C78、C82、C84、C90、C94を始めとする無置換のものと、[6,6]−フェニル C61 ブチリックアシッドメチルエステル([6,6]−PCBM)、[5,6]−フェニル C61 ブチリックアシッドメチルエステル([5,6]−PCBM)、[6,6]−フェニル C61 ブチリックアシッドヘキシルエステル([6,6]−PCBH)、[6,6]−フェニル C61 ブチリックアシッドドデシルエステル([6,6]−PCBD)、フェニル C71 ブチリックアシッドメチルエステル(PC70BM)、フェニル C85 ブチリックアシッドメチルエステル(PC84BM)など)、カーボンナノチューブ(CNT)、ポリ−p−フェニレンビニレン系重合体にシアノ基を導入した誘導体(CN−PPV)などが挙げられる。中でも、フラーレン化合物は電荷分離速度と電子移動速度が速いため、好ましく用いられる。フラーレン化合物の中でも、C70誘導体(上記PC70BMなど)は光吸収特性に優れ、より高い光電変換効率を得られるために、より好ましい。
【0041】
本発明の光起電力素子用材料において、電子供与性有機材料と電子受容性有機材料の含有比率(重量分率)は特に限定されないが、電子供与性有機材料:電子受容性有機材料の重量分率が、1:99〜99:1の範囲であることが好ましく、より好ましくは10:90〜90:10の範囲であり、さらに好ましくは20:80〜60:40の範囲である。
【0042】
電子供与性有機材料と電子受容性有機材料は混合して用いても積層して用いてもよい。混合方法としては特に限定されるものではないが、所望の比率で溶媒に添加した後、加熱、撹拌、超音波照射などの方法を1種または複数種組み合わせて溶媒中に溶解させる方法が挙げられる。なお、後述するように、光起電力素子用材料が一層の有機半導体層を形成する場合は、上述の含有比率はその一層に含まれる電子供与性有機材料と電子受容性有機材料の含有比率となり、有機半導体層が二層以上の積層構造である場合は、有機半導体層全体における電子供与性有機材料と電子受容性有機材料の含有比率を意味する。
【0043】
光電変換効率をより向上させるためには、キャリアのトラップとなるような不純物は極力除去することが好ましい。本発明では、前述の一般式(1)で表される構造を有する電子供与性有機材料や、電子受容性有機材料の不純物を除去する方法は特に限定されないが、カラムクロマトグラフィー法、再結晶法、昇華法、再沈殿法、ソックスレー抽出法、GPCによる分子量分画法、濾過法、イオン交換法、キレート法等を用いることができる。
【0044】
一般的に低分子有機材料の精製にはカラムクロマトグラフィー法、再結晶法、昇華法が好ましく用いられる。他方、高分子量体の精製には、低分子量成分を除去する場合には再沈殿法やソクスレー抽出法、GPCによる分子量分画法が好ましく用いられ、金属成分を除去する場合には再沈殿法やキレート法、イオン交換法が好ましく用いられる。これらの方法のうち、複数を組み合わせてもよい。
【0045】
次に、本発明の光起電力素子について説明する。本発明の光起電力素子は、少なくとも正極と負極を有し、これらの間に本発明の光起電力素子用材料を含む。図1は本発明の光起電力素子の一例を示す模式図である。図1において符号1は基板、符号2は正極、符号3は本発明の光起電力素子用材料を含む有機半導体層、符号4は負極である。
【0046】
有機半導体層3は本発明の光起電力素子用材料を含む。すなわち、一般式(1)で表される構造を有する電子供与性有機材料を含む。光起電力素子が電子供与性有機材料と電子受容性材料を含む場合、これらの材料は混合されていても積層されていても良いが、混合されていることが好ましい。
【0047】
すなわち、電子供与性有機材料と電子受容性有機材料とを混合することにより光電変換に寄与する接合面を増加させるバルクヘテロ接合型光起電力素子が好ましい。混合されている場合は、一般式(1)で表される構造を有する電子供与性有機材料と電子受容性有機材料が分子レベルで相溶しているか、相分離しているが、ナノメートルのサイズで相分離していることが好ましい。
【0048】
この相分離構造のドメインサイズは特に限定されるものではないが、通常1nm以上50nm以下である。また、電子供与性有機材料と電子受容性有機材料が積層されている場合は、p型半導体特性を示す電子供与性有機材料を有する層が正極側、n型半導体特性を示す電子受容性有機材料を有する層が負極側であることが好ましい。
【0049】
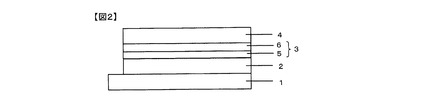
有機半導体層3が積層されている場合の光起電力素子の一例を図2に示す。符号5は一般式(1)で表される構造を有する電子供与性有機材料を有する層、符号6は電子受容性有機材料を有する層である。有機半導体層は5nm〜500nmの厚さが好ましく、より好ましくは30nm〜300nmである。積層されている場合は、本発明の電子供与性有機材料を有する層は上記厚さのうち1nm〜400nmの厚さを有していることが好ましく、より好ましくは15nm〜150nmである。
【0050】
また、有機半導体層3には一般式(1)で表される構造を有する電子供与性有機材料、および電子受容性有機材料以外の電子供与性有機材料(p型有機半導体)を含んでいてもよい。ここで用いる電子供与性有機材料(p型有機半導体)としては、先に電子供与性有機材料の他の化合物として例示したものが挙げられる。
【0051】
本発明の光起電力素子においては、正極2もしくは負極4のいずれかに光透過性を有することが好ましい。電極の光透過性は、有機半導体層3に入射光が到達して起電力が発生する程度であれば、特に限定されるものではない。ここで、本発明における光透過性は、[透過光強度(W/m2)/入射光強度(W/m2)]×100(%)で求められる値である。電極の厚さは光透過性と導電性とを有する範囲であればよく、電極素材によって異なるが20nm〜300nmが好ましい。なお、もう一方の電極は導電性があれば必ずしも光透過性は必要ではなく、厚さも特に限定されない。
【0052】
電極材料としては、一方の電極には仕事関数の大きな導電性素材、もう一方の電極には仕事関数の小さな導電性素材を使用することが好ましい。仕事関数の大きな導電性素材を用いた電極は正極となる。この仕事関数の大きな導電性素材としては金、白金、クロム、ニッケルなどの金属のほか、透明性を有するインジウム、スズ、モリブデンなどの金属酸化物、複合金属酸化物(インジウム錫酸化物(ITO)、インジウム亜鉛酸化物(IZO)など)が好ましく用いられる。ここで、正極2に用いられる導電性素材は、有機半導体層3とオーミック接合するものであることが好ましい。さらに、後述する正孔輸送層を用いた場合においては、正極2に用いられる導電性素材は正孔輸送層とオーミック接合するものであることが好ましい。
【0053】
仕事関数の小さな導電性素材を用いた電極は負極となるが、この仕事関数の小さな導電性素材としては、アルカリ金属やアルカリ土類金属、具体的にはリチウム、マグネシウム、カルシウムなどが使用される。また、錫や銀、アルミニウムも好ましく用いられる。さらに、上記の金属からなる合金や上記の金属の積層体からなる電極も好ましく用いられる。また、負極4と電子輸送層の界面にフッ化リチウムやフッ化セシウムなどの金属フッ化物を導入することで、取り出し電流を向上させることも可能である。ここで、負極4に用いられる導電性素材は、有機半導体層3とオーミック接合するものであることが好ましい。
【0054】
基板1は、光電変換材料の種類や用途に応じて、電極材料や有機半導体層が積層できる基板、例えば、無アルカリガラス、石英ガラス等の無機材料、ポリエステル、ポリカーボネート、ポリオレフィン、ポリアミド、ポリイミド、ポリフェニレンスルフィド、ポリパラキシレン、エポキシ樹脂やフッ素系樹脂等の有機材料から任意の方法によって作製されたフィルムや板が使用可能である。また基板側から光を入射して用いる場合は、上記に示した各基板に80%程度の光透過性を持たせておくことが好ましい。
【0055】
本発明では、正極2と有機半導体層3の間に正孔輸送層を設けてもよい。正孔輸送層を形成する材料としては、ポリチオフェン系重合体、ポリ−p−フェニレンビニレン系重合体、ポリフルオレン系重合体などの導電性高分子や、フタロシアニン誘導体(H2Pc、CuPc、ZnPcなど)、ポルフィリン誘導体などのp型半導体特性を示す低分子有機化合物が好ましく用いられる。特に、ポリチオフェン系重合体であるポリエチレンジオキシチオフェン(PEDOT)やPEDOTにポリスチレンスルホネート(PSS)が添加されたものが好ましく用いられる。正孔輸送層は5nmから600nmの厚さが好ましく、より好ましくは30nmから200nmである。また、光電変換効率をより向上させるために、正孔輸送層をフルオラス化合物(分子内にフッ素原子を一個以上有する有機化合物)により処理することが好ましい。
【0056】
フルオラス化合物としては、例えばベンゾトリフルオリド、ヘキサフルオロベンゼン、1,1,1,3,3,3−ヘキサフルオロ−2−プロパノール、ペルフルオロトルエン、ペルフルオロデカリン、ペルフルオロヘキサン、1H,1H,2H,2H−ヘプタデカフルオロ−1−デカノール(F−デカノール)などが挙げられる。より好ましくはベンゾトリフルオリド、ペルフルオロヘキサン、F−デカノールが用いられる。処理方法としては、正孔輸送層を形成する材料に上述のフルオラス化合物をあらかじめ混合してから正孔輸送層を形成させる方法や、正孔輸送層を形成してから上述のフルオラス化合物を接触させる方法(スピンコート、ディップコート、ブレードコート、蒸着、蒸気処理法など)が挙げられる。
【0057】
また、本発明の光起電力素子は、有機半導体層3と負極4の間に電子輸送層を設けてもよい。電子輸送層を形成する材料として、特に限定されるものではないが、上述の電子受容性有機材料(NTCDA、PTCDA、PTCDI−C8H、オキサゾール誘導体、トリアゾール誘導体、フェナントロリン誘導体、ホスフィンオキサイド誘導体、フラーレン化合物、CNT、CN−PPVなど)のようにn型半導体特性を示す有機材料が好ましく用いられる。電子輸送層は5nm〜600nmの厚さが好ましく、より好ましくは30nm〜200nmである。
【0058】
また、本発明の光起電力素子は、1つ以上の中間電極を介して2層以上の有機半導体層を積層(タンデム化)して直列接合を形成してもよい。例えば、基板/正極/第1の有機半導体層/中間電極/第2の有機半導体層/負極という積層構成を挙げることができる。このように積層することにより、開放電圧を向上させることができる。なお、正極と第1の有機半導体層の間、および、中間電極と第2の有機半導体層の間に上述の正孔輸送層を設けてもよく、第1の有機半導体層と中間電極の間、および、第2の有機半導体層と負極の間に上述の正孔輸送層を設けてもよい。
【0059】
このような積層構成の場合、有機半導体層の少なくとも1層が本発明の光起電力素子用材料を含み、他の層には、短絡電流を低下させないために、一般式(1)で表される構造を有する電子供与性有機材料とはバンドギャップの異なる電子供与性有機材料を含むことが好ましい。
【0060】
このような電子供与性有機材料としては、例えば上述のポリチオフェン系重合体、ポリ−p−フェニレンビニレン系重合体、ポリ−p−フェニレン系重合体、ポリフルオレン系重合体、ポリピロール系重合体、ポリアニリン系重合体、ポリアセチレン系重合体、ポリチエニレンビニレン系重合体、ベンゾチアジアゾール系重合体(例えば、PCPDTBT(poly[2,6−(4,4−bis−(2−ethylhexyl)−4H−cyclopenta[2,1−b;3,4−b’]dithiophene)−alt−4,7−(2,1,3−benzothiadiazole)])や、PSBTBT(poly[(4,4−bis−(2−ethylhexyl)dithieno[3,2−b:2’,3’−d]silole)−2,6−diyl−alt−(2,1,3−benzothiadiazole)−4,7−diyl]))などの共役系重合体や、H2フタロシアニン(H2Pc)、銅フタロシアニン(CuPc)、亜鉛フタロシアニン(ZnPc)等のフタロシアニン誘導体、ポルフィリン誘導体、N,N’−ジフェニル−N,N’−ジ(3−メチルフェニル)−4,4’−ジフェニル−1,1’−ジアミン(TPD)、N,N’−ジナフチル−N,N’−ジフェニル−4,4’−ジフェニル−1,1’−ジアミン(NPD)等のトリアリールアミン誘導体、4,4’−ジ(カルバゾール−9−イル)ビフェニル(CBP)等のカルバゾール誘導体、オリゴチオフェン誘導体(ターチオフェン、クウォーターチオフェン、セキシチオフェン、オクチチオフェンなど)等の低分子有機化合物が挙げられる。
【0061】
また、ここで用いられる中間電極用の素材としては高い導電性を有するものが好ましく、例えば上述の金、白金、クロム、ニッケル、リチウム、マグネシウム、カルシウム、錫、銀、アルミニウムなどの金属や、透明性を有するインジウム、スズ、モリブデンなどの金属酸化物、複合金属酸化物(インジウム錫酸化物(ITO)、インジウム亜鉛酸化物(IZO)など)、上記の金属からなる合金や上記の金属の積層体、ポリエチレンジオキシチオフェン(PEDOT)やPEDOTにポリスチレンスルホネート(PSS)が添加されたもの、などが挙げられる。中間電極は光透過性を有することが好ましいが、光透過性が低い金属のような素材でも膜厚を薄くすることで充分な光透過性を確保できる場合が多い。
【0062】
次に、本発明の光起電力素子の製造方法について例を挙げて説明する。基板上にITOなどの透明電極(この場合正極に相当)をスパッタリング法などにより形成する。次に、一般式(1)で表される構造を有する電子供与性有機材料、および必要により電子受容性有機材料を含む光電変換素子用材料を溶媒に溶解させて溶液を作り、透明電極上に塗布し有機半導体層を形成する。
【0063】
このとき用いられる溶媒は有機溶媒が好ましく、例えば、メタノール、エタノール、ブタノール、トルエン、キシレン、o−クロロフェノール、アセトン、酢酸エチル、エチレングリコール、テトラヒドロフラン、ジクロロメタン、クロロホルム、ジクロロエタン、クロロベンゼン、ジクロロベンゼン、トリクロロベンゼン、クロロナフタレン、ジメチルホルムアミド、ジメチルスルホキシド、N−メチルピロリドン、γ−ブチロラクトンなどが挙げられる。これらを2種以上用いてもよい。
【0064】
また、上述のフルオラス化合物を含有することで光電変換効率をより向上させることができる。常温常圧で液体であるフルオラス化合物(フルオラス溶媒)が好ましく、より好ましくは上述のベンゾトリフルオリド、ペルフルオロヘキサン、F−デカノールが用いられる。フルオラス化合物の含有量は全溶媒量に対して0.01〜20体積%が好ましく、より好ましくは0.1〜2体積%である。また、フルオラス溶媒の含有量は全溶媒中0.01〜30重量%が好ましく、より好ましくは0.1〜4重量%である。
【0065】
一般式(1)で表される構造を有する電子供与性有機材料および電子受容性有機材料を混合して有機半導体層を形成する場合は、一般式(1)で表される構造を有する電子供与性有機材料と電子受容性有機材料を所望の比率で溶媒に添加し、加熱、撹拌、超音波照射などの方法を用いて溶解させ溶液を作り、透明電極上に塗布する。この場合、2種以上の溶媒を混合して用いることで光起電力素子の光電変換効率を向上させることもできる。これは、電子供与性有機材料と電子受容性有機材料がナノレベルで相分離を起こし、電子と正孔の通り道となるキャリアパスが形成されるためと推測される。
【0066】
組み合わせる溶媒は、用いる電子供与性有機材料と電子受容性有機材料の種類によって最適な組み合わせの種類を選択することができる。一般式(1)で表される構造を有する電子供与性有機材料を用いる場合、組み合わせる好ましい溶媒として上述の中でもクロロホルムとクロロベンゼンが挙げられる。この場合、各溶媒の混合体積比率は、クロロホルム:クロロベンゼン=5:95〜95:5の範囲であることが好ましく、さらに好ましくはクロロホルム:クロロベンゼン=10:90〜90:10の範囲である。
【0067】
また、一般式(1)表される構造を有する電子供与性有機材料および電子受容性有機材料を積層して有機半導体層を形成する場合は、例えば電子供与性有機材料の溶液を塗布して電子供与性有機材料を有する層を形成した後に、電子受容性有機材料の溶液を塗布して層を形成する。ここで、電子供与性有機材料および電子受容性有機材料は、分子量が1000以下程度の低分子量体である場合には、蒸着法を用いて層を形成することも可能である。
【0068】
有機半導体層の形成には、スピンコート塗布、ブレードコート塗布、スリットダイコート塗布、スクリーン印刷塗布、バーコーター塗布、鋳型塗布、印刷転写法、浸漬引き上げ法、インクジェット法、スプレー法、真空蒸着法など何れの方法を用いてもよく、膜厚制御や配向制御など、得ようとする有機半導体層特性に応じて形成方法を選択すればよい。例えばスピンコート塗布を行う場合には、一般式(1)で表される構造を有する電子供与性有機材料、および電子受容性有機材料が1〜20g/lの濃度(一般式(1)で表される構造を有する電子供与性有機材料と電子受容性有機材料と溶媒を含む溶液の体積に対する、一般式(1)で表される構造を有する電子供与性有機材料と電子受容性有機材料の重量)であることが好ましく、この濃度にすることで厚さ5〜200nmの均質な有機半導体層を容易に得ることができる。
【0069】
形成した有機半導体層に対して、溶媒を除去するために、減圧下または不活性雰囲気下(窒素やアルゴン雰囲気下)などでアニーリング処理を行ってもよい。アニーリング処理の好ましい温度は40℃〜300℃、より好ましくは50℃〜200℃である。また、アニーリング処理を行うことで、積層した層が界面で互いに浸透して接触する実行面積が増加し、短絡電流を増大させることができる。このアニーリング処理は、負極の形成後に行ってもよい。
【0070】
次に、有機半導体層上にAlなどの金属電極(この場合負極に相当)を真空蒸着法やスパッタ法により形成する。金属電極は、電子輸送層に低分子有機材料を用いて真空蒸着した場合は、引き続き、真空を保持したまま続けて形成することが好ましい。
【0071】
正極と有機半導体層の間に正孔輸送層を設ける場合には、所望のp型有機半導体材料(PEDOTなど)を正極上にスピンコート法、バーコーティング法、ブレードによるキャスト法等で塗布した後、真空恒温槽やホットプレートなどを用いて溶媒を除去し、正孔輸送層を形成する。フタロシアニン誘導体やポルフィリン誘導体などの低分子有機材料を使用する場合には、真空蒸着機を用いた真空蒸着法を適用することも可能である。
【0072】
有機半導体層と負極の間に電子輸送層を設ける場合には、所望のn型有機半導体材料(フラーレン誘導体など)n型無機半導体材料(酸化チタンゲルなど)を有機半導体層上にスピンコート法、バーコーティング法、ブレードによるキャスト法、スプレー法等で塗布した後、真空恒温槽やホットプレートなどを用いて溶媒を除去し、電子輸送層を形成する。フェナントロリン誘導体やC60などの低分子有機材料を使用する場合には、真空蒸着機を用いた真空蒸着法を適用することも可能である。
【0073】
本発明の光起電力素子は、光電変換機能、光整流機能などを利用した種々の光電変換デバイスへの応用が可能である。例えば光電池(太陽電池など)、電子素子(光センサ、光スイッチ、フォトトランジスタなど)、光記録材(光メモリなど)などに有用である。
【実施例】
【0074】
以下、本発明を実施例に基づいてさらに具体的に説明する。なお、本発明は下記実施例に限定されるものではない。また実施例等で用いた化合物のうち、略語を使用しているものについて、以下に示す。
ITO:インジウム錫酸化物
PEDOT:ポリエチレンジオキシチオフェン
PSS:ポリスチレンスルホネート
PC70BM:フェニル C71 ブチリックアシッドメチルエステル
Eg:バンドギャップ
HOMO:最高被占分子軌道
Isc:短絡電流密度
Voc:開放電圧
FF:フィルファクター
η:光電変換効率
なお、1H−NMR測定にはFT−NMR装置((株)日本電子製JEOL JNM−EX270)を用いた。
【0075】
また、平均分子量(数平均分子量、重量平均分子量)はGPC装置(クロロホルムを送液したTOSOH社製、高速GPC装置HLC−8320GPC)を用い、絶対検量線法によって算出した。重合度nは以下の式で算出した。
重合度n=[(重量平均分子量)/(繰り返しユニットの分子量)]
また、光吸収端波長は、ガラス上に約60nmの厚さに形成した薄膜について、日立製作所(株)製のU−3010型分光光度計を用いて測定した薄膜の紫外可視吸収スペクトル(測定波長範囲:300〜900nm)から得た。
【0076】
バンドギャップ(Eg)は下式により、光吸収端波長から算出した。なお、薄膜はクロロホルムを溶媒に用いてスピンコート法により形成した。
Eg(eV)=1240/薄膜の光吸収端波長(nm)
また、最高被占分子軌道(HOMO)準位は、ITOガラス上に約60nmの厚さに形成した薄膜について、表面分析装置(大気中紫外線光電子分光装置AC−2型、理研機器(株)製)を用いて測定した。なお、薄膜はクロロホルムを溶媒に用いてスピンコート法により形成した。
【0077】
合成例1
化合物A−1を式1に示す方法で合成した。なお、合成例1記載の化合物(1−c)はアドバンスドマテリアルズ(Advanced Materials)、2010年、22巻、5240−5244頁に記載されている方法を、化合物(1−g)はジャーナルオブザアメリカンケミカルソサエティ(Journal of the American Chemical Society)、2009年、131巻、7792−7799頁に記載されている方法を参考にして合成した。
【0078】
【化8】
【0079】
化合物(1−a)(アルドリッチ社製)20.0g(68mmol)をエタノール750mlにけん濁させ、メカニカルスターラーを用いて撹拌しているところに、テトラヒドロホウ酸ナトリウム37.8g(1mol)を5回にわけ、1時間かけて0℃で加えた。反応溶混合物を0℃で5時間撹拌した後、室温で12時間放置した。エタノールをおよそ250mlになるまで減圧留去した後、水100mlを0℃でゆっくり加えた。クロロホルム200mlを加えた後、水層および有機層に不溶の固体をろ別した。水層をクロロホルム100mlで3回抽出した後、集めた有機層を無水硫酸マグネシウムで乾燥した。クロロホルムを減圧留去し、化合物(1−b)を薄橙色固体(10.2g、収率56%)として得た。化合物(1−b)はこれ以上精製することなく、以下の反応に用いた。
【0080】
上記化合物(1−b)1.08g(4.07mmol)および化合物(1−c)1.9g(4.07mmol)のエタノール溶液50mlにメタンスルホン酸(東京化成工業(株))1滴を加え、6時間還流した。反応溶液を室温まで冷却した後、炭酸ナトリウムを加えて中和し、溶液をろ過、ついで減圧留去した。シリカゲルカラムクロマトグラフィー(溶離液、ヘキサン:酢酸エチル=10:1)で精製することにより化合物(1−d)を黄色オイル(2.1g、収率73%)として得た。化合物(1−d)の1H−NMRの測定結果を以下に示す。
1H−NMR(270MHz,CDCl3):7.90(s,2H),7.27−7.19(m,6H),6.95−6.88(m,2H),3.69(d,J=5.9Hz,4H),1.67−1.27(m,18H),0.97−0.86(m,12H)ppm。
【0081】
上記化合物(1−d)1.3g(1.85mmol)および2−(tributylstannyl)thiophene(アルドリッチ社製)2.1g(5.5mmol)のトルエン溶液60mlを窒素でバブリングした後、ジクロロビス(トリフェニルホスフィン)パラジウム触媒(東京化成工業(株)製)100mgを加え、8時間還流した。反応溶液を室温まで冷却した後、水50mlを加え、有機層を水で2回、次いで飽和食塩水で洗浄した。無水硫酸マグネシウムで溶媒を乾燥させた後、ろ過し、溶媒を減圧留去した。シリカゲルカラムクロマトグラフィー(溶離液、ヘキサン:酢酸エチル=20:1)で精製することにより化合物(1−e)を黄色固体(1.2g、収率92%)として得た。化合物(1−e)の1H−NMRの測定結果を以下に示す。
1H−NMR(270MHz,CDCl3):8.15(s,2H),7.88(d,J=4.1Hz,2H),7.49(d,J=5.1Hz,2H),7.40(m,2H),7.28−7.24(m,4H),7.17(m,2H),6.93(m,2H),3.78(d,J=5.9Hz,4H),1.71(m,2H),1.61−1.29(m,6H),0.92(t,J=7.0Hz,12H)ppm。
【0082】
上記化合物(1−e)1.2g(1.7mmol)のクロロホルム溶液50mlにN−ブロモスクシンイミド(東京化成工業(株)製)600mg(3.4mmol)を加え、3時間室温で撹拌した。水50mlを加えた後、有機層を水で2回、次いで飽和食塩水で1回洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧留去した。粗生成物をシリカゲルカラムクロマトグラフィー(溶離液、ヘキサン:クロロホルム=3:1)で精製することにより化合物(1−f)を黄色固体(1.1g、収率75%)として得た。化合物(1−f)の1H−NMRの測定結果を以下に示す。
1H−NMR(270MHz,CDCl3):8.06(s,2H),7.54(d,J=4.1Hz,2H),7.49(s,2H),7.21(d,J=8.0Hz,2H),7.13−7.10(m,4H),6.97(d,J=8.0Hz,2H),3.88(d,J=5.1Hz,4H),1.73(m,2H),1.52−1.30(m,16H),0.94(t,J=7.3Hz,12H),ppm。
【0083】
上記化合物(1−f)129mg(0.15mmol)および化合物(1−g)116mg(0.15mmol)をトルエン(和光純薬工業(株)製)10mlに溶解させたところに、トリス(ジベンジリデンアセトン)ジパラジウム(東京化成工業(株)製)8mg、トリス(2−メチルフェニル)ホスフィン(東京化成工業(株)製)を13mg加え、窒素雰囲気下、110℃で12時間撹拌した。次いで、ブロモベンゼン(東京化成工業(株)製)10mgを加え、110℃にて1時間撹拌した。次いで、2−(tributylstannyl)thiophene(アルドリッチ社製)40mgを加え、110℃にてさらに1時間撹拌した。撹拌終了後、反応混合物を室温まで冷却し、メタノール100mlに注いだ。析出した固体をろ取し、メタノール、水、アセトンの順に洗浄した。次いでソックスレー抽出器を用いてアセトン、ヘキサンの順で洗浄した。次に、得られた固体をクロロホルムに溶解させ、セライト(ナカライテスク(株)製)を通して濾過した後、溶媒を減圧留去した。得られた固体を再びクロロホルムに溶解させ、シリカゲルカラム(溶離液:クロロホルム)を通した後に濃縮し、メタノールに再沈澱させ、化合物A−1(40mg)を得た。重量平均分子量は8,800、数平均分子量は6,200、重合度nは7.7であった。また、光吸収端波長は695nm、バンドギャップ(Eg)は1.78eV、最高被占分子軌道(HOMO)準位は−5.03eVであった。
【0084】
合成例2
化合物A−2を式2に示す方法で合成した。なお、合成例2記載の化合物(2−b)はジャーナルオブポリマーサイエンス:パートA(Journal of PolymerScience:PartA)、2010年、48巻、4823−4834頁に記載されている方法を参考にして合成した。
【0085】
【化9】
【0086】
上記化合物(1−b)4.0g(15mmol)およびベンジル(東京化成工業(株)製)3.16g(15mmol)のクロロホルム溶液120mlに濃硫酸1滴を室温で加え、反応溶液を5時間還流した。反応溶液を室温まで冷やした後、5%炭酸ナトリウム水溶液50mlを加え、有機層を水100ml、ついで飽和食塩水100mlで洗浄した。溶媒を無水硫酸マグネシウムで乾燥後、減圧留去した。粗成生成物をメタノールで洗浄し、化合物(2−a)を薄黄色固体(5.2g、収率79%)として得た。化合物(2−a)の1H−NMRの測定結果を以下に示す。
1H−NMR(270MHz,CDCl3):7.91(s,2H),7.65(m,4H),7.38(m,6H)ppm。
【0087】
上記化合物(2−a)1.0g(2.26mmol)、化合物(2−b)(2.2g,6.79mmol)、りん酸三カリウム1.44g(6.79mmol)およびジメチルホルムアミド(ナカライテスク(株)製)30mlに[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(アルドリッチ社製)100mgを加え100℃で5時間撹拌した。反応混合物に酢酸エチル50mlおよび水50mlを加えた後、セライト(ナカライテスク(株)を通してろ過し、有機層を水、次いで飽和食塩水で洗浄した。無水硫酸マグネシウムで乾燥後、溶媒を減圧留去した。粗生成物をシリカゲルカラムクロマトグラフィー(溶離液、ヘキサン:クロロホルム=1:1)で精製することにより化合物(2−c)を橙色固体(1.2g、収率64%)として得た。化合物(2−c)の1H−NMRの測定結果を以下に示す。
1H−NMR(270MHz,CDCl3):8.10(s,2H),7.75(m,6H),7.38(m,6H),7.11(s,2H),2.68(t,J=7.6Hz,4H),1.71(m,4H),1.39−1.29(m,20H),0.88(t,J=7.0Hz,6H)ppm。
【0088】
上記化合物(2−c)1.01g(1.5mmol)のクロロホルム溶液40mlにN−ブロモスクシンイミド(東京化成工業(株)製)533mg(3.0mmol)を加え、3時間室温で撹拌した。水50mlを加えた後、有機層を水で2回、次いで飽和食塩水で1回洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧留去した。粗生成物をシリカゲルカラムクロマトグラフィー(溶離液、クロロホルム)で精製することにより化合物(2−d)を黄色固体(930mg、収率75%)として得た。化合物(2−d)の1H−NMRの測定結果を以下に示す。
1H−NMR(270MHz,CDCl3):8.03(s,2H),7.72−7.68(m,4H),7.52(s,2H),7.42−7.39(m,6H),2.62(t,J=7.3Hz,4H),1.64(m,4H),1.36−1.29(m,20H),0.89(t,J=7.0Hz,6H)ppm。
【0089】
上記化合物(2−d)124mg(0.15mmol)および化合物(1−g)116mg(0.15mmol)をトルエン(和光純薬工業(株)製)8mlに溶解させたところに、トリス(ジベンジリデンアセトン)ジパラジウム(東京化成工業(株)製)8mg、トリス(2−メチルフェニル)ホスフィン(東京化成工業(株)製)を13mg加え、窒素雰囲気下、110℃で12時間撹拌した。次いで、ブロモベンゼン(東京化成工業(株)製)10mgを加え、110℃にて1時間撹拌した。次いで、2−(tributylstannyl)thiophene(アルドリッチ社製)40mgを加え、110℃にてさらに1時間撹拌した。撹拌終了後、反応混合物を室温まで冷却し、メタノール100mlに注いだ。析出した固体をろ取し、メタノール、水、アセトンの順に洗浄した。次いでソックスレー抽出器を用いてアセトン、ヘキサンの順で洗浄した。次に、得られた固体をクロロホルムに溶解させ、セライト(ナカライテスク(株)製)を通して濾過した後、溶媒を減圧留去した。得られた固体を再びクロロホルムに溶解させ、シリカゲルカラム(溶離液:クロロホルム)を通した後に濃縮し、メタノールに再沈澱させ、化合物A−2(110mg)を得た。重量平均分子量は18,600、数平均分子量は13,500、重合度nは16であった。また、光吸収端波長は703nm、バンドギャップ(Eg)は1.76eV、最高被占分子軌道(HOMO)準位は−5.08eVであった。
【0090】
合成例3
化合物A−3を式3に示す方法で合成した。なお、合成例3記載の化合物(3−a)はマクロモレキュルズ(Macromolecules)、2010年、43巻、9779−9786頁に記載されている方法を参考にして合成した。
【0091】
【化10】
【0092】
化合物(2−a)880mg(2.0mmol)および化合物(3−a)1.4g(6.0mmol)のトルエン20ml溶液を窒素でバブリングした後、ジクロロビス(トリフェニルホスフィン)パラジウム触媒(東京化成工業(株)製)80mgを加え、6時間還流した。反応溶液を室温まで冷却した後、水50mlを加え、有機層を水で2回、次いで飽和食塩水で洗浄した。無水硫酸マグネシウムで溶媒を乾燥させた後、ろ過し、溶媒を減圧留去した。シリカゲルカラムクロマトグラフィー(溶離液、ヘキサン:クロロホルム=1:1)で精製することにより化合物(3−b)を黄色固体(1.2g、収率84%)として得た。化合物(3−b)の1H−NMRの測定結果を以下に示す。
1H−NMR(270MHz,CDCl3):8.10(s,2H),7.75(m,6H),7.38(m,6H),7.08(s,2H),2.62(d,J=7.0Hz,4H),1.63(m,1H),1.4−1.2(m,16H),0.92(m,6H)ppm。
【0093】
上記化合物(3−b)1.01g(1.5mmol)のクロロホルム溶液40mlにN−ブロモスクシンイミド(東京化成工業(株)製)533mg(3.0mmol)を加え、3時間室温で撹拌した。水50mlを加えた後、有機層を水で2回、次いで飽和食塩水で1回洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧留去した。粗生成物をシリカゲルカラムクロマトグラフィー(溶離液、クロロホルム)で精製することにより化合物(3−c)を黄色固体(890mg、収率72%)として得た。化合物(3−c)の1H−NMRの測定結果を以下に示す。
1H−NMR(270MHz,CDCl3):8.06(s,2H),7.71(m,4H),7.53(s,2H),7.40(m,6H),2.56(d,J=7.0Hz,4H),1.69(m,2H),1.4−1.3(m,16H),0.9−0.8(m,12H)ppm。
【0094】
上記化合物(3−c)124mg(0.15mmol)および化合物(1−g)116mg(0.15mmol)をトルエン(和光純薬工業(株)製)8mlに溶解させたところに、トリス(ジベンジリデンアセトン)ジパラジウム(東京化成工業(株)製)8mg、トリス(2−メチルフェニル)ホスフィン(東京化成工業(株)製)を13mg加え、窒素雰囲気下、110℃で12時間撹拌した。次いで、ブロモベンゼン(東京化成工業(株)製)10mgを加え、110℃にて1時間撹拌した。次いで、2−(tributylstannyl)thiophene(アルドリッチ社製)40mgを加え、110℃にてさらに1時間撹拌した。撹拌終了後、反応混合物を室温まで冷却し、メタノール100mlに注いだ。析出した固体をろ取し、メタノール、水、アセトンの順に洗浄した。次いでソックスレー抽出器を用いてアセトン、ヘキサンの順で洗浄した。次に、得られた固体をクロロホルムに溶解させ、セライト(ナカライテスク(株)製)を通して濾過した後、溶媒を減圧留去した。得られた固体を再びクロロホルムに溶解させ、シリカゲルカラム(溶離液:クロロホルム)を通した後に濃縮し、メタノールに再沈澱させ、化合物A−3(110mg)を得た。重量平均分子量は42,000、数平均分子量は23,000、重合度nは37であった。また、光吸収端波長は678nm、バンドギャップ(Eg)は1.83eV、最高被占分子軌道(HOMO)準位は−5.15eVであった。
【0095】
合成例4
化合物A−4を式4に示す方法で合成した。なお、合成例4記載の化合物(4−a)はアンゲバンテケミ インターナショナルエディション(Angewandte Chem Internatioal Edition)、2011年、50巻、9697−9702頁に記載されている方法を参考にして合成した。
【0096】
【化11】
【0097】
上記化合物(1−f)129mg(0.15mmol)および化合物(4−a)136mg(0.15mmol)をトルエン(和光純薬工業(株)製)8mlに溶解させたところに、トリス(ジベンジリデンアセトン)ジパラジウム(東京化成工業(株)製)8mg、トリス(2−メチルフェニル)ホスフィン(東京化成工業(株)製)を13mg加え、窒素雰囲気下、110℃で12時間撹拌した。次いで、ブロモベンゼン(東京化成工業(株)製)10mgを加え、110℃にて1時間撹拌した。次いで、2−(tributylstannyl)thiophene(アルドリッチ社製)40mgを加え、110℃にてさらに1時間撹拌した。撹拌終了後、反応混合物を室温まで冷却し、メタノール100mlに注いだ。析出した固体をろ取し、メタノール、水、アセトンの順に洗浄した。次いでソックスレー抽出器を用いてアセトン、ヘキサンの順で洗浄した。得られた固体をクロロベンゼンに溶解させ、メタノールより再沈澱することで、化合物A−4(112mg)を得た。重量平均分子量は18,000、数平均分子量は11,000、重合度nは14であった。また、光吸収端波長は725nm、バンドギャップ(Eg)は1.71eV、最高被占分子軌道(HOMO)準位は−4.92eVであった。
【0098】
合成例5
化合物A−5を式5に示す方法で合成した。
【0099】
【化12】
【0100】
上記化合物(3−c)124mg(0.15mmol)および化合物(4−a)136mg(0.15mmol)をトルエン(和光純薬工業(株)製)8mlに溶解させたところに、トリス(ジベンジリデンアセトン)ジパラジウム(東京化成工業(株)製)8mg、トリス(2−メチルフェニル)ホスフィン(東京化成工業(株)製)を13mg加え、窒素雰囲気下、110℃で12時間撹拌した。次いで、ブロモベンゼン(東京化成工業(株)製)10mgを加え、110℃にて1時間撹拌した。次いで、2−(tributylstannyl)thiophene(アルドリッチ社製)40mgを加え、110℃にてさらに1時間撹拌した。撹拌終了後、反応混合物を室温まで冷却し、メタノール100mlに注いだ。析出した固体をろ取し、メタノール、水、アセトンの順に洗浄した。次いでソックスレー抽出器を用いてアセトン、ヘキサンの順で洗浄した。得られた固体をクロロベンゼンに溶解させ、メタノールより再沈澱することで、化合物A−5(105mg)を得た。重量平均分子量は22,000、数平均分子量は15,000、重合度nは18であった。また、光吸収端波長は708nm、バンドギャップ(Eg)は1.75eV、最高被占分子軌道(HOMO)準位は−5.07eVであった。
【0101】
合成例6
化合物B−1を式6に示す方法で合成した。
【0102】
【化13】
【0103】
化合物(2−a)66mg(0.15mmol)および化合物(1−g)116mg(0.15mmol)をトルエン(和光純薬工業(株)製)8mlに溶解させたところに、トリス(ジベンジリデンアセトン)ジパラジウム(東京化成工業(株)製)8mg、トリス(2−メチルフェニル)ホスフィン(東京化成工業(株)製)を13mg加え、窒素雰囲気下、110℃で12時間撹拌した。次いで、ブロモベンゼン(東京化成工業(株)製)10mgを加え、110℃にて1時間撹拌した。次いで、2−(tributylstannyl)thiophene(アルドリッチ社製)40mgを加え、110℃にてさらに1時間撹拌した。撹拌終了後、反応混合物を室温まで冷却し、メタノール100mlに注いだ。析出した固体をろ取し、メタノール、水、アセトンの順に洗浄した。次いでソックスレー抽出器を用いてアセトン、ヘキサンの順で洗浄した。次に、得られた固体をクロロホルムに溶解させ、セライト(ナカライテスク(株)製)を通して濾過した後、溶媒を減圧留去した。得られた固体を再びクロロホルムに溶解させ、シリカゲルカラム(溶離液:クロロホルム)を通した後に濃縮し、メタノールに再沈澱させ、化合物B−1(80mg)を得た。重量平均分子量は10,200、数平均分子量は9,000、重合度nは14であった。また、光吸収端波長は770nm、バンドギャップ(Eg)は1.61eV、最高被占分子軌道(HOMO)準位は−4.93eVであった。
【0104】
合成例7
化合物B−2を式7に示す方法で合成した。なお、合成例7記載の化合物(7−a)はザジャーナルオブオーガニックケミストリー(The Journal of Organic Chemistry)、2002年、67巻、9073−9076頁に記載されている方法を参考にして合成した。
【0105】
【化14】
【0106】
化合物(7−a)66.6mg(0.15mmol)および化合物(1−g)116mg(0.15mmol)をトルエン(和光純薬工業(株)製)8mlに溶解させたところに、トリス(ジベンジリデンアセトン)ジパラジウム(東京化成工業(株)製)8mg、トリス(2−メチルフェニル)ホスフィン(東京化成工業(株)製)を13mg加え、窒素雰囲気下、110℃で12時間撹拌した。次いで、ブロモベンゼン(東京化成工業(株)製)10mgを加え、110℃にて1時間撹拌した。次いで、2−(tributylstannyl)thiophene(アルドリッチ社製)40mgを加え、110℃にてさらに1時間撹拌した。撹拌終了後、反応混合物を室温まで冷却し、メタノール100mlに注いだ。析出した固体をろ取し、メタノール、水、アセトンの順に洗浄した。次いでソックスレー抽出器を用いてアセトン、ヘキサンの順で洗浄した。次に、得られた固体をクロロホルムに溶解させ、セライト(ナカライテスク(株)製)を通して濾過した後、溶媒を減圧留去した。得られた固体を再びクロロホルムに溶解させ、シリカゲルカラム(溶離液:クロロホルム)を通した後に濃縮し、メタノールに再沈澱させ、化合物B−2(76mg)を得た。重量平均分子量は7,800、数平均分子量は5,600、重合度nは10であった。また、光吸収端波長は915nm、バンドギャップ(Eg)は1.36eV、最高被占分子軌道(HOMO)準位は−4.91eVであった。
【0107】
合成例8
【0108】
【化15】
【0109】
化合物B−3を式8に示す方法で合成した。なお、合成例8記載の化合物(8−a)はマクロモレキュルズ(Macromolecules)、2010年、43巻、811−820頁に記載されている方法を参考にして合成した。
【0110】
化合物(8−a)111mg(0.15mmol)および化合物(1−g)116mg(0.15mmol)をトルエン(和光純薬工業(株)製)8mlに溶解させたところに、トリス(ジベンジリデンアセトン)ジパラジウム(東京化成工業(株)製)8mg、トリス(2−メチルフェニル)ホスフィン(東京化成工業(株)製)を13mg加え、窒素雰囲気下、110℃で12時間撹拌した。次いで、ブロモベンゼン(東京化成工業(株)製)10mgを加え、110℃にて1時間撹拌した。次いで、2−(tributylstannyl)thiophene(アルドリッチ社製)40mgを加え、110℃にてさらに1時間撹拌した。撹拌終了後、反応混合物を室温まで冷却し、メタノール100mlに注いだ。析出した固体をろ取し、メタノール、水、アセトンの順に洗浄した。次いでソックスレー抽出器を用いてアセトン、ヘキサンの順で洗浄した。次に、得られた固体をクロロホルムに溶解させ、セライト(ナカライテスク(株)製)を通して濾過した後、溶媒を減圧留去した。得られた固体を再びクロロホルムに溶解させ、シリカゲルカラム(溶離液:クロロホルム)を通した後に濃縮し、メタノールに再沈澱させ、化合物B−3(90mg)を得た。重量平均分子量は14,100、数平均分子量は11,500、重合度nは14であった。また、光吸収端波長は730nm、バンドギャップ(Eg)は1.70eV、最高被占分子軌道(HOMO)準位は−5.19eVであった。
【0111】
合成例9
化合物B−4を式9に示す方法で合成した。
【0112】
【化16】
【0113】
上記化合物(3−c)124mg(0.15mmol)および化合物(9−a)(アルドリッチ社製)84mg(0.15mmol)をトルエン10mlに溶解させたところに、濃度1M炭酸カリウム水溶液1ml、Aliquat336(アルドリッチ社製)1滴およびテトラキス(トリフェニルホスフィン)パラジウム触媒(東京化成工業(株)製)10mgを加え、窒素雰囲気下、100℃で8時間撹拌した。次いで、ブロモベンゼン(東京化成工業(株)製)30mgを加え、100℃にて1時間撹拌した。次いで、フェニルボロン酸(東京化成工業(株)製)50mgを加え、100℃にてさらに1時間撹拌した。撹拌終了後、反応混合物を室温まで冷却し、メタノール100mlに注いだ。析出した固体をろ取し、メタノール、水、アセトンの順に洗浄した。次いでソックスレー抽出器を用いてアセトン、ヘキサンの順で洗浄した。次に、得られた固体をクロロホルムに溶解させ、セライト(ナカライテスク(株)製)を通して濾過した後、溶媒を減圧留去した。得られた固体を再びクロロホルムに溶解させ、シリカゲルカラム(溶離液:クロロホルム)を通した後に濃縮し、メタノールに再沈殿 させ、化合物B−4(92mg)を得た。重量平均分子量は32,400、数平均分子量は18,400、重合度nは30であった。また、光吸収端波長は600nm、バンドギャップ(Eg)は2.07eV、最高被占分子軌道(HOMO)準位は−5.47であった
実施例1
上記A−1(1mg)とPC70BM(4mg、Solenne社製)をクロロベンゼン0.25mlの入ったサンプル瓶の中に加え、超音波洗浄機((株)井内盛栄堂製US−2(商品名)、出力120W)中で30分間超音波照射することにより溶液Aを得た。
【0114】
スパッタリング法により正極となるITO透明導電層を120nm堆積させたガラス基板を38mm×46mmに切断した後、ITOをフォトリソグラフィー法により38mm×13mmの長方形状にパターニングした。得られた基板をアルカリ洗浄液(フルウチ化学(株)製、“セミコクリーン”EL56(商品名))で10分間超音波洗浄した後、超純水で洗浄した。
【0115】
この基板を30分間UV/オゾン処理した後に、基板上に正孔輸送層となるPEDOT:PSS水溶液(PEDOT0.8重量%、PPS0.5重量%)をスピンコート法により60nmの厚さに成膜した。ホットプレートにより200℃で5分間加熱乾燥した後、上記の溶液AをPEDOT:PSS層上に滴下し、スピンコート法により膜厚100nmの有機半導体層を形成した。その後、有機半導体層が形成された基板と陰極用マスクを真空蒸着装置内に設置して、装置内の真空度が1×10−3Pa以下になるまで再び排気し、抵抗加熱法によって、負極となるアルミニウム層を80nmの厚さに蒸着した。以上のように、ストライプ状のITO層とアルミニウム層が交差する部分の面積が5mm×5mmである光起電力素子を作製した。
【0116】
このようにして作製された光起電力素子の正極と負極をヒューレット・パッカード社製ピコアンメーター/ボルテージソース4140Bに接続して、大気中でITO層側から擬似太陽光(山下電装株式会社製 簡易型ソーラシミュレータ YSS−E40、スペクトル形状:AM1.5、強度:100mW/cm2)を照射し、印加電圧を−1Vから+2Vまで変化させたときの電流値を測定した。この時の短絡電流密度(印加電圧が0Vのときの電流密度の値)は6.49mA/cm2、開放電圧(電流密度が0になるときの印加電圧の値)は0.78V、フィルファクター(FF)は0.61であり、これらの値から算出した光電変換効率は3.09%であった。なお、フィルファクターと光電変換効率は次式により算出した。
フィルファクター=IVmax(mA・V/cm2)/(短絡電流密度(mA/cm2)×開放電圧(V))
(ここで、IVmaxは、印加電圧が0Vから開放電圧値の間で電流密度と印加電圧の積が最大となる点における電流密度と印加電圧の積の値である。)
光電変換効率=[(短絡電流密度(mA/cm2)×開放電圧(V)×フィルファクター)/擬似太陽光強度(100mW/cm2)]×100(%)
以下の実施例と比較例におけるフィルファクターと光電変換効率も全て上式により算出した。
【0117】
実施例2
A−1の代わりに上記A−2を用いた他は実施例1と全く同様にして光起電力素子を作製し、電流−電圧特性を測定した。この時の短絡電流密度は8.23mA/cm2、開放電圧は0.79V、フィルファクター(FF)は0.57であり、これらの値から算出した光電変換効率は3.71%であった。
【0118】
実施例3
A−1の代わりに上記A−3を用いた他は実施例1と全く同様にして光起電力素子を作製し、電流−電圧特性を測定した。この時の短絡電流密度は7.21mA/cm2、開放電圧は0.83V、フィルファクター(FF)は0.53であり、これらの値から算出した光電変換効率は3.17%であった。
【0119】
実施例4
A−1の代わりに上記A−4を用いた他は実施例1と全く同様にして光起電力素子を作製し、電流−電圧特性を測定した。この時の短絡電流密度は9.03mA/cm2、開放電圧は0.71V、フィルファクター(FF)は0.60であり、これらの値から算出した光電変換効率は3.85%であった。
【0120】
実施例5
A−1の代わりに上記A−5を用いた他は実施例1と全く同様にして光起電力素子を作製し、電流−電圧特性を測定した。この時の短絡電流密度は8.62mA/cm2、開放電圧は0.93V、フィルファクター(FF)は0.53であり、これらの値から算出した光電変換効率は4.25%であった。
【0121】
比較例1
A−1の代わりに上記B−1を用いた他は実施例1と全く同様にして光起電力素子を作製し、電流−電圧特性を測定した。この時の短絡電流密度は2.01mA/cm2、開放電圧は0.59V、フィルファクター(FF)は0.31であり、これらの値から算出した光電変換効率は0.37%であった。
【0122】
比較例2
A−1の代わりに上記B−2を用いた他は実施例1と全く同様にして光起電力素子を作製し、電流−電圧特性を測定した。この時の短絡電流密度は4.40mA/cm2、開放電圧は0.52V、フィルファクター(FF)は0.55であり、これらの値から算出した光電変換効率は1.26%であった。
【0123】
比較例3
A−1の代わりに上記B−3を用いた他は実施例1と全く同様にして光起電力素子を作製し、電流−電圧特性を測定した。この時の短絡電流密度は4.14mA/cm2、開放電圧は0.82V、フィルファクター(FF)は0.31であり、これらの値から算出した光電変換効率は1.05%であった。
【0124】
比較例4
A−1の代わりに上記B−4を用いた他は実施例1と全く同様にして光起電力素子を作製し、電流−電圧特性を測定した。この時の短絡電流密度は4.64mA/cm2、開放電圧は1.03V、フィルファクター(FF)は0.38であり、これらの値から算出した光電変換効率は1.82%であった。
【0125】
【表1】
【0126】
表1から明らかなように、一般式(1)で表される構造を有する電子供与性有機材料を用いて作製した光起電力素子(実施例1〜5)は、同様の条件で作製した他の光起電力素子(比較例1〜4)に比べ高い光電変換効率を示した。
【符号の説明】
【0127】
1:基板
2:正極
3:有機半導体層
4:負極
5:電子供与性有機材料を有する層
6:電子受容性有機材料を有する層
【技術分野】
【0001】
本発明は、光起電力素子用材料およびこれを用いた光起電力素子に関する。
【背景技術】
【0002】
太陽電池は環境に優しい電気エネルギー源として、現在深刻さを増すエネルギー問題に対して有力なエネルギー源と注目されている。現在、太陽電池の光起電力素子の半導体素材としては、単結晶シリコン、多結晶シリコン、アモルファスシリコン、化合物半導体などの無機物が使用されている。しかし、無機半導体を用いて製造される太陽電池は、火力発電や原子力発電などの発電方式と比べてコストが高いために、一般家庭に広く普及するには至っていない。コスト高の要因は主として、真空かつ高温下で半導体薄膜を製造するプロセスにある。そこで、製造プロセスの簡略化が期待される半導体素材として、共役系重合体や有機結晶などの有機半導体や有機色素を用いた有機太陽電池が検討されている。
【0003】
しかし、共役系重合体などを用いた有機太陽電池は、従来の無機半導体を用いた太陽電池と比べて光電変換効率が低いことが最大の課題であり、まだ実用化には至っていない。従来の共役系重合体を用いた有機太陽電池の光電変換効率が低いのは、主として、太陽光の吸収効率が低いことや、太陽光によって生成された電子と正孔が分離しにくいエキシトンという束縛状態が形成されることと、キャリア(電子、正孔)を捕獲するトラップが形成されやすいため生成したキャリアがトラップに捕獲されやすく、キャリアの移動度が遅いことなどによる。
【0004】
これまでの有機半導体による光電変換素子は、現在のところ一般的に次のような素子構成に分類することができる。電子供与性有機材料(p型有機半導体)と仕事関数の小さい金属を接合させるショットキー型、電子受容性有機材料(n型有機半導体)と電子供与性有機材料(p型有機半導体)を接合させるヘテロ接合型などである。これらの素子は、接合部の有機層(数分子層程度)のみが光電流生成に寄与するため光電変換効率が低く、その向上が課題となっている。
【0005】
光電変換効率向上の一つの方法として、電子受容性有機材料(n型有機半導体)と電子供与性有機材料(p型有機半導体)を混合し、光電変換に寄与する接合面を増加させたバルクヘテロ接合型(例えば、非特許文献1参照)がある。なかでも、電子供与性有機材料(p型有機半導体)として共役系重合体を用い、電子受容性有機材料としてn型の半導体特性をもつ導電性高分子のほかC60などのフラーレンやフラーレン誘導体を用いた光電変換材料が報告されている(例えば、非特許文献2参照)。
【0006】
また、太陽光スペクトルの広い範囲にわたる放射エネルギーを効率よく吸収し、光電変換効率を向上させるために、主鎖骨格に電子供与性基と電子吸引性基を導入することでバンドギャップを狭めた電子供与性有機材料が報告されている(例えば、非特許文献3参照)。この電子供与性基としてはチオフェン骨格やオリゴチオフェン骨格が、電子吸引性基としてはベンゾチアジアゾール骨格やキノキサリン骨格などが精力的に研究されている(例えば、特許文献1〜2、非特許文献3〜5参照)。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特表2004−534863号公報
【特許文献2】特表2004−500464号公報
【非特許文献】
【0008】
【非特許文献1】J.J.M.Halls、C.A.Walsh、N.C.Greenham、E.A.Marseglla、R.H.Friend、S.C.Moratti、A.B.Holmes著、「ネイチャー(Nature)」、1995年、376号、498頁
【非特許文献2】G.Yu、J.Gao、J.C.Hummelen、F.Wudl、A.J.Heeger著、「サイエンス(Science)」、1995年、270巻、1789頁
【非特許文献3】E.Bundgaard、F.C.Kreb著、「ソーラー エナジー マテリアルズ アンド ソーラー セル(Solar Energy Materials & Solar Cells)」、2007年、91巻、954頁
【非特許文献4】J.Hou、M.−H.Park、S.Zhang、Y.Yao、L.−M.Chen、J.−H.Li、Y.Yang著、「マクロモレキュルズ(Macromolecules)」、2008年、41巻、6012頁
【非特許文献5】S.C.Prince、A.C.Stuart、W.You著、「マクロモレキュルズ(Macromolecules)」、2010年、43巻、4609頁
【発明の概要】
【発明が解決しようとする課題】
【0009】
しかしながら、上述の電子供与性基としてチオフェン骨格やオリゴチオフェン骨格、電子吸引性基としてベンゾチアジアゾール骨格やキノキサリン骨格を用いた狭バンドギャップ電子供与性有機材料(特許文献1〜2、非特許文献3)では主鎖のチオフェン骨格のねじれのために十分なキャリア移動度が保てないと考えた。
【0010】
キャリア移動度を高めるために、平面性の高い電子供与性基であるベンゾジチオフェン骨格と、電子吸引性基であるベンゾチアジアゾール骨格やキノキサリン骨格等を組み合わせた狭バンドギャップポリマーが報告されている(非特許文献4、5)。しかしながら、ベンゾチアジアゾール骨格またはキノキサリン骨格と、ベンゾジチオフェン骨格を直接連結した狭バンドギャップ電子供与性有機材料(非特許文献4)では、ポリマー主鎖骨格にねじれが生じ、キャリアの移動度が阻害されるために十分な変換効率が得られていない。
【0011】
また、ベンゾチアジアゾール骨格とチオフェン骨格、およびベンゾジチオフェン骨格を組み合わせた狭バンドギャップ電子供与性有機材料(非特許文献5)においては、ねじれが解消され、キャリア移動度が向上するが、平面性が高すぎるために有機溶媒に対する溶解性、並びにフラーレン誘導体等に代表される電子受容性材料との相溶性が低下し、結果として十分な光電変換効率が得られていなかった。これは溶解性を保つために、過剰な長鎖アルキル側鎖を狭バンドギャップ電子供与性有機材料に導入する必要があったことが一因である。
【0012】
すなわち、従来の電子供与性有機材料を用いた光起電力素子では、高いキャリア移動度と有機溶媒に対する溶解性、並びにフラーレン誘導体に代表される電子受容性材料との相溶性を両立させることが困難であり、いずれも光電変換効率が低かった。本発明は光電変換効率の高い光起電力素子を提供することを目的とし、高いキャリア移動度と有機溶媒に対する溶解性およびフラーレン誘導体等の電子受容性材料との相溶性を両立させた電子供与性有機材料を提供する。
【課題を解決するための手段】
【0013】
電子吸引性基としてベンゾチアジアゾール骨格やキノキサリン骨格、電子供与基として平面性の高いベンゾジチオフェン骨格を用いた狭バンドギャップ電子供与性有機材料について、様々な構造を検討した結果、ジフェニルキノキサリン等の平面性をくずした構造を導入することによって、高いキャリア移動度を保ったまま、有機溶媒に対する溶解性とフラーレン等の電子受容性材料との相溶性を両立させることができることを見出した。
【0014】
すなわち本発明は、一般式(1)で表される構造を有する電子供与性有機材料を含む光起電力素子用材料、および、これを用いた光起電力素子である。
【0015】
【化1】
【0016】
上記一般式(1)中、R1およびR2は同じでも異なっていてもよく、置換されていてもよいアリール基、または置換されていてもよいヘテロアリール基を表す。このキノキサリン上に配置された隣接するアリール基またはヘテロアリール基によって、主鎖構造の平面性を保持しつつ電子供与性材料の平面性を部分的に崩すことが可能となり、高いキャリア移動度を保ったまま、有機溶媒に対する溶解性と電子受容性材料との相溶性を両立させることができる。R3〜R6は同じでも異なっていてもよく、水素、アルキル基、アルコキシ基、アリール基、ヘテロアリール基、またはハロゲンを表す。XおよびYは2価の連結基を表す。nは2以上1,000以下の範囲を表す。)
【発明の効果】
【0017】
キノキサリン上にアリールまたはヘテロアリール基を配置し、側鎖の平面性を崩すことによって、高いキャリア移動度を保ったまま有機溶媒に対する溶解性およびフラーレン誘導体等の電子受容性材料との相溶性を両立させることが可能となり、光電変換効率を向上させた光起電力素子を提供することができる。
【図面の簡単な説明】
【0018】
【図1】本発明の光起電力素子の一態様を示した模式図。
【図2】本発明の光起電力素子の別の態様を示した模式図。
【発明を実施するための形態】
【0019】
本発明の光起電力素子用材料は、一般式(1)で表される電子供与性有機材料を含む。
【0020】
【化2】
【0021】
上記一般式(1)中、R1およびR2は同じでも異なっていてもよく、置換されていてもよいアリール基、または置換されていてもよいヘテロアリール基を表す。前述のとおり、このキノキサリン上に配置された隣接するアリール基またはヘテロアリール基によって、主鎖構造の平面性を保持しつつ電子供与性材料の平面性を部分的に崩すことが可能となり、高いキャリア移動度を保ったまま、有機溶媒に対する溶解性と電子受容性材料との相溶性を両立させることができる。
【0022】
ここでアリール基とは、例えば、フェニル基、ナフチル基、ビフェニル基、フェナントリル基、アントリル基、ターフェニル基、ピレニル基、フルオレニル基、ペリレニル基などの芳香族炭化水素基を示し、これは無置換でも置換されていてもかまわない。置換される場合の置換基の例としては、下記アルキル基や下記アルコキシ基、下記ヘテロアリール基、ハロゲンが挙げられる。電子供与性有機材料のキャリア移動度を保ちつつ、側鎖構造の平面性を崩すことによって、有機溶媒対する溶解性およびフラーレン誘導体等の電子受容性材料との相溶性を両立させるためには、R1およびR2が共にフェニル基であることが好ましい。
【0023】
また、ヘテロアリール基とは、例えば、チエニル基、フリル基、ピロリル基、イミダゾリル基、ピラゾリル基、オキサゾリル基、ピリジル基、ピラジル基、ピリミジル基、キノリニル基、イソキノリル基、キノキサリル基、アクリジニル基、インドリル基、カルバゾリル基、ベンゾフラン基、ジベンゾフラン基、ベンゾチオフェン基、ジベンゾチオフェン基、ベンゾジチオフェン基、シロール基、ベンゾシロール基、ジベンゾシロール基などの炭素以外の原子を有する複素芳香族環基を示し、これは無置換でも置換されていてもかまわない。置換される場合の置換基の例としては、下記アルキル基、上記アリール基、ハロゲンが挙げられる。ヘテロアリール基の炭素数は、キャリア移動度を保つために2以上8以下が好ましい。
【0024】
上記一般式(1)中、R3〜R6はそれぞれ同じでも異なっていてもよく、水素、アルキル基、アルコキシ基、上記アリール基、上記ヘテロアリール基、ハロゲンの中から選ばれる。R3およびR4は主鎖骨格のねじれを抑えるため体積の小さな置換基が好ましく、水素、フッ素、アルキル基が好ましく用いられる。合成の容易さから水素が特に好ましく用いられる。
【0025】
R5およびR6は電子供与性の溶解性を保つためにアルキル基、アルコキシ基、アルキルアリール基、アルキルヘテロアリール基が好ましく用いられる。電子供与性有機材料の平面性を高めることでキャリア移動度を向上できることから、アルキルチエニル基が特に好ましく用いられる。合成の容易さからはアルコキシ基が特に好ましく用いられる。
【0026】
ここでアルキル基とは、例えば、メチル基、エチル基、プロピル基、ブチル基、ペンチル基、ヘキシル基、ヘプチル基、オクチル基、ノニル基、デシル基、ウンデシル基、ドデシル基のような飽和脂肪族炭化水素基であり、直鎖状であっても分岐状であっても環状であってもよく、無置換でも置換されていてもかまわない。置換される場合の置換基の例としては、下記アルコキシ基、上記アリール基、上記ヘテロアリール基、ハロゲンが挙げられる。アルキル基の炭素数は、有機材料の溶解性をあげてスピンコートなどのウエットプロセスに適用する場合には4以上であることが好ましく、有機材料の十分なキャリア移動度を保つためには12以下であることが好ましい。
【0027】
また、アルコキシ基とは、例えば、メトキシ基、エトキシ基、プロポキシ基、ブトキシ基などのエーテル結合を介した脂肪族炭化水素基を示し、脂肪族炭化水素基は無置換でも置換されていてもかまわない。置換される場合の置換基の例としては、上記アリール基や上記ヘテロアリール基、ハロゲンが挙げられる。アルコキシ基の好ましい炭素数の範囲は、上述のアルキル基の場合と同じである。
【0028】
また、ハロゲンは、フッ素、塩素、臭素、ヨウ素のいずれかである。
【0029】
上記一般式(1)中、XおよびYは同じでも異なっていてもよい2価の連結基を表す。電子供与性有機材料の電子共役系を伸ばしてキャリア移動度を高めるためには、アリーレン基やヘテロアリーレン基が好ましい。主鎖構造平面性を保ち、キャリア移動度を保つためにはチオフェン基、ジチオフェン基、ビチオフェン基が好ましく、チオフェン基が特に好ましく用いられる。
【0030】
上記一般式(1)中、nは重合度を示し、2以上1,000以下の範囲を表す。nを2以上とすることにより、前述のバルクヘテロ接合の薄膜において有効なキャリアパスを形成させることができるために、光電変換効率を高めることができる。また、優れたキャリアパスを形成させるためにはnは5以上であることが好ましく、合成上の容易さから100未満であることが好ましい。重合度は重量平均分子量から求めることができる。重量平均分子量は、GPC(ゲルパーミエーションクロマトグラフィー)を用いて測定し、ポリスチレンの標準試料に換算して求めることができる。
【0031】
上記の一般式(1)で表される構造を有する電子供与性有機材料として、具体的には下記のような構造が挙げられる。ただし、nは2以上1,000以下の範囲を示す。
【0032】
【化3】
【0033】
【化4】
【0034】
【化5】
【0035】
【化6】
【0036】
【化7】
【0037】
なお、一般式(1)で表される構造を有する電子供与性有機材料は、例えば、前記非特許文献4に記載されている方法に類似した手法や、前記非特許文献5に記載されている方法に類似した手法により合成することができる。
【0038】
本発明の光起電力素子用材料は、一般式(1)で表される構造を有する電子供与性有機材料のみからなるものでもよいし、他の電子供与性有機材料を含んでもよい。他の電子供与性有機材料としては、例えば、ポリチオフェン系重合体、ベンゾチアジアゾール−チオフェン系誘導体、ベンゾチアジアゾール−チオフェン系共重合体、ポリ−p−フェニレンビニレン系重合体、ポリ−p−フェニレン系重合体、ポリフルオレン系重合体、ポリピロール系重合体、ポリアニリン系重合体、ポリアセチレン系重合体、ポリチエニレンビニレン系重合体などの共役系重合体や、H2フタロシアニン(H2Pc)、銅フタロシアニン(CuPc)、亜鉛フタロシアニン(ZnPc)等のフタロシアニン誘導体、ポルフィリン誘導体、N,N’−ジフェニル−N,N’−ジ(3−メチルフェニル)−4,4’−ジフェニル−1,1’−ジアミン(TPD)、N,N’−ジナフチル−N,N’−ジフェニル−4,4’−ジフェニル−1,1’−ジアミン(NPD)等のトリアリールアミン誘導体、4,4’−ジ(カルバゾール−9−イル)ビフェニル(CBP)等のカルバゾール誘導体、オリゴチオフェン誘導体(ターチオフェン、クウォーターチオフェン、セキシチオフェン、オクチチオフェンなど)等の低分子有機化合物が挙げられる。
【0039】
一般式(1)で表される構造を有する電子供与性有機材料はp型半導体特性を示す材料であり、本発明の光起電力素子用材料は、より高い光電変換効率を得るために電子受容性有機材料(n型有機半導体)と組み合わせることが好ましい。
【0040】
本発明で用いる電子受容性有機材料とは、n型半導体特性を示す有機材料であり、例えば、1,4,5,8−ナフタレンテトラカルボキシリックジアンハイドライド(NTCDA)、3,4,9,10−ペリレンテトラカルボキシリックジアンハイドライド(PTCDA)、3,4,9,10−ペリレンテトラカルボキシリックビスベンズイミダゾール(PTCBI)、N,N'−ジオクチル−3,4,9,10−ナフチルテトラカルボキシジイミド(PTCDI−C8H)、2−(4−ビフェニリル)−5−(4−t−ブチルフェニル)−1,3,4−オキサジアゾール(PBD)、2,5−ジ(1−ナフチル)−1,3,4−オキサジアゾール(BND)等のオキサゾール誘導体、3−(4−ビフェニリル)−4−フェニル−5−(4−t−ブチルフェニル)−1,2,4−トリアゾール(TAZ)等のトリアゾール誘導体、フェナントロリン誘導体、ホスフィンオキサイド誘導体、フラーレン化合物(C60、C70、C76、C78、C82、C84、C90、C94を始めとする無置換のものと、[6,6]−フェニル C61 ブチリックアシッドメチルエステル([6,6]−PCBM)、[5,6]−フェニル C61 ブチリックアシッドメチルエステル([5,6]−PCBM)、[6,6]−フェニル C61 ブチリックアシッドヘキシルエステル([6,6]−PCBH)、[6,6]−フェニル C61 ブチリックアシッドドデシルエステル([6,6]−PCBD)、フェニル C71 ブチリックアシッドメチルエステル(PC70BM)、フェニル C85 ブチリックアシッドメチルエステル(PC84BM)など)、カーボンナノチューブ(CNT)、ポリ−p−フェニレンビニレン系重合体にシアノ基を導入した誘導体(CN−PPV)などが挙げられる。中でも、フラーレン化合物は電荷分離速度と電子移動速度が速いため、好ましく用いられる。フラーレン化合物の中でも、C70誘導体(上記PC70BMなど)は光吸収特性に優れ、より高い光電変換効率を得られるために、より好ましい。
【0041】
本発明の光起電力素子用材料において、電子供与性有機材料と電子受容性有機材料の含有比率(重量分率)は特に限定されないが、電子供与性有機材料:電子受容性有機材料の重量分率が、1:99〜99:1の範囲であることが好ましく、より好ましくは10:90〜90:10の範囲であり、さらに好ましくは20:80〜60:40の範囲である。
【0042】
電子供与性有機材料と電子受容性有機材料は混合して用いても積層して用いてもよい。混合方法としては特に限定されるものではないが、所望の比率で溶媒に添加した後、加熱、撹拌、超音波照射などの方法を1種または複数種組み合わせて溶媒中に溶解させる方法が挙げられる。なお、後述するように、光起電力素子用材料が一層の有機半導体層を形成する場合は、上述の含有比率はその一層に含まれる電子供与性有機材料と電子受容性有機材料の含有比率となり、有機半導体層が二層以上の積層構造である場合は、有機半導体層全体における電子供与性有機材料と電子受容性有機材料の含有比率を意味する。
【0043】
光電変換効率をより向上させるためには、キャリアのトラップとなるような不純物は極力除去することが好ましい。本発明では、前述の一般式(1)で表される構造を有する電子供与性有機材料や、電子受容性有機材料の不純物を除去する方法は特に限定されないが、カラムクロマトグラフィー法、再結晶法、昇華法、再沈殿法、ソックスレー抽出法、GPCによる分子量分画法、濾過法、イオン交換法、キレート法等を用いることができる。
【0044】
一般的に低分子有機材料の精製にはカラムクロマトグラフィー法、再結晶法、昇華法が好ましく用いられる。他方、高分子量体の精製には、低分子量成分を除去する場合には再沈殿法やソクスレー抽出法、GPCによる分子量分画法が好ましく用いられ、金属成分を除去する場合には再沈殿法やキレート法、イオン交換法が好ましく用いられる。これらの方法のうち、複数を組み合わせてもよい。
【0045】
次に、本発明の光起電力素子について説明する。本発明の光起電力素子は、少なくとも正極と負極を有し、これらの間に本発明の光起電力素子用材料を含む。図1は本発明の光起電力素子の一例を示す模式図である。図1において符号1は基板、符号2は正極、符号3は本発明の光起電力素子用材料を含む有機半導体層、符号4は負極である。
【0046】
有機半導体層3は本発明の光起電力素子用材料を含む。すなわち、一般式(1)で表される構造を有する電子供与性有機材料を含む。光起電力素子が電子供与性有機材料と電子受容性材料を含む場合、これらの材料は混合されていても積層されていても良いが、混合されていることが好ましい。
【0047】
すなわち、電子供与性有機材料と電子受容性有機材料とを混合することにより光電変換に寄与する接合面を増加させるバルクヘテロ接合型光起電力素子が好ましい。混合されている場合は、一般式(1)で表される構造を有する電子供与性有機材料と電子受容性有機材料が分子レベルで相溶しているか、相分離しているが、ナノメートルのサイズで相分離していることが好ましい。
【0048】
この相分離構造のドメインサイズは特に限定されるものではないが、通常1nm以上50nm以下である。また、電子供与性有機材料と電子受容性有機材料が積層されている場合は、p型半導体特性を示す電子供与性有機材料を有する層が正極側、n型半導体特性を示す電子受容性有機材料を有する層が負極側であることが好ましい。
【0049】
有機半導体層3が積層されている場合の光起電力素子の一例を図2に示す。符号5は一般式(1)で表される構造を有する電子供与性有機材料を有する層、符号6は電子受容性有機材料を有する層である。有機半導体層は5nm〜500nmの厚さが好ましく、より好ましくは30nm〜300nmである。積層されている場合は、本発明の電子供与性有機材料を有する層は上記厚さのうち1nm〜400nmの厚さを有していることが好ましく、より好ましくは15nm〜150nmである。
【0050】
また、有機半導体層3には一般式(1)で表される構造を有する電子供与性有機材料、および電子受容性有機材料以外の電子供与性有機材料(p型有機半導体)を含んでいてもよい。ここで用いる電子供与性有機材料(p型有機半導体)としては、先に電子供与性有機材料の他の化合物として例示したものが挙げられる。
【0051】
本発明の光起電力素子においては、正極2もしくは負極4のいずれかに光透過性を有することが好ましい。電極の光透過性は、有機半導体層3に入射光が到達して起電力が発生する程度であれば、特に限定されるものではない。ここで、本発明における光透過性は、[透過光強度(W/m2)/入射光強度(W/m2)]×100(%)で求められる値である。電極の厚さは光透過性と導電性とを有する範囲であればよく、電極素材によって異なるが20nm〜300nmが好ましい。なお、もう一方の電極は導電性があれば必ずしも光透過性は必要ではなく、厚さも特に限定されない。
【0052】
電極材料としては、一方の電極には仕事関数の大きな導電性素材、もう一方の電極には仕事関数の小さな導電性素材を使用することが好ましい。仕事関数の大きな導電性素材を用いた電極は正極となる。この仕事関数の大きな導電性素材としては金、白金、クロム、ニッケルなどの金属のほか、透明性を有するインジウム、スズ、モリブデンなどの金属酸化物、複合金属酸化物(インジウム錫酸化物(ITO)、インジウム亜鉛酸化物(IZO)など)が好ましく用いられる。ここで、正極2に用いられる導電性素材は、有機半導体層3とオーミック接合するものであることが好ましい。さらに、後述する正孔輸送層を用いた場合においては、正極2に用いられる導電性素材は正孔輸送層とオーミック接合するものであることが好ましい。
【0053】
仕事関数の小さな導電性素材を用いた電極は負極となるが、この仕事関数の小さな導電性素材としては、アルカリ金属やアルカリ土類金属、具体的にはリチウム、マグネシウム、カルシウムなどが使用される。また、錫や銀、アルミニウムも好ましく用いられる。さらに、上記の金属からなる合金や上記の金属の積層体からなる電極も好ましく用いられる。また、負極4と電子輸送層の界面にフッ化リチウムやフッ化セシウムなどの金属フッ化物を導入することで、取り出し電流を向上させることも可能である。ここで、負極4に用いられる導電性素材は、有機半導体層3とオーミック接合するものであることが好ましい。
【0054】
基板1は、光電変換材料の種類や用途に応じて、電極材料や有機半導体層が積層できる基板、例えば、無アルカリガラス、石英ガラス等の無機材料、ポリエステル、ポリカーボネート、ポリオレフィン、ポリアミド、ポリイミド、ポリフェニレンスルフィド、ポリパラキシレン、エポキシ樹脂やフッ素系樹脂等の有機材料から任意の方法によって作製されたフィルムや板が使用可能である。また基板側から光を入射して用いる場合は、上記に示した各基板に80%程度の光透過性を持たせておくことが好ましい。
【0055】
本発明では、正極2と有機半導体層3の間に正孔輸送層を設けてもよい。正孔輸送層を形成する材料としては、ポリチオフェン系重合体、ポリ−p−フェニレンビニレン系重合体、ポリフルオレン系重合体などの導電性高分子や、フタロシアニン誘導体(H2Pc、CuPc、ZnPcなど)、ポルフィリン誘導体などのp型半導体特性を示す低分子有機化合物が好ましく用いられる。特に、ポリチオフェン系重合体であるポリエチレンジオキシチオフェン(PEDOT)やPEDOTにポリスチレンスルホネート(PSS)が添加されたものが好ましく用いられる。正孔輸送層は5nmから600nmの厚さが好ましく、より好ましくは30nmから200nmである。また、光電変換効率をより向上させるために、正孔輸送層をフルオラス化合物(分子内にフッ素原子を一個以上有する有機化合物)により処理することが好ましい。
【0056】
フルオラス化合物としては、例えばベンゾトリフルオリド、ヘキサフルオロベンゼン、1,1,1,3,3,3−ヘキサフルオロ−2−プロパノール、ペルフルオロトルエン、ペルフルオロデカリン、ペルフルオロヘキサン、1H,1H,2H,2H−ヘプタデカフルオロ−1−デカノール(F−デカノール)などが挙げられる。より好ましくはベンゾトリフルオリド、ペルフルオロヘキサン、F−デカノールが用いられる。処理方法としては、正孔輸送層を形成する材料に上述のフルオラス化合物をあらかじめ混合してから正孔輸送層を形成させる方法や、正孔輸送層を形成してから上述のフルオラス化合物を接触させる方法(スピンコート、ディップコート、ブレードコート、蒸着、蒸気処理法など)が挙げられる。
【0057】
また、本発明の光起電力素子は、有機半導体層3と負極4の間に電子輸送層を設けてもよい。電子輸送層を形成する材料として、特に限定されるものではないが、上述の電子受容性有機材料(NTCDA、PTCDA、PTCDI−C8H、オキサゾール誘導体、トリアゾール誘導体、フェナントロリン誘導体、ホスフィンオキサイド誘導体、フラーレン化合物、CNT、CN−PPVなど)のようにn型半導体特性を示す有機材料が好ましく用いられる。電子輸送層は5nm〜600nmの厚さが好ましく、より好ましくは30nm〜200nmである。
【0058】
また、本発明の光起電力素子は、1つ以上の中間電極を介して2層以上の有機半導体層を積層(タンデム化)して直列接合を形成してもよい。例えば、基板/正極/第1の有機半導体層/中間電極/第2の有機半導体層/負極という積層構成を挙げることができる。このように積層することにより、開放電圧を向上させることができる。なお、正極と第1の有機半導体層の間、および、中間電極と第2の有機半導体層の間に上述の正孔輸送層を設けてもよく、第1の有機半導体層と中間電極の間、および、第2の有機半導体層と負極の間に上述の正孔輸送層を設けてもよい。
【0059】
このような積層構成の場合、有機半導体層の少なくとも1層が本発明の光起電力素子用材料を含み、他の層には、短絡電流を低下させないために、一般式(1)で表される構造を有する電子供与性有機材料とはバンドギャップの異なる電子供与性有機材料を含むことが好ましい。
【0060】
このような電子供与性有機材料としては、例えば上述のポリチオフェン系重合体、ポリ−p−フェニレンビニレン系重合体、ポリ−p−フェニレン系重合体、ポリフルオレン系重合体、ポリピロール系重合体、ポリアニリン系重合体、ポリアセチレン系重合体、ポリチエニレンビニレン系重合体、ベンゾチアジアゾール系重合体(例えば、PCPDTBT(poly[2,6−(4,4−bis−(2−ethylhexyl)−4H−cyclopenta[2,1−b;3,4−b’]dithiophene)−alt−4,7−(2,1,3−benzothiadiazole)])や、PSBTBT(poly[(4,4−bis−(2−ethylhexyl)dithieno[3,2−b:2’,3’−d]silole)−2,6−diyl−alt−(2,1,3−benzothiadiazole)−4,7−diyl]))などの共役系重合体や、H2フタロシアニン(H2Pc)、銅フタロシアニン(CuPc)、亜鉛フタロシアニン(ZnPc)等のフタロシアニン誘導体、ポルフィリン誘導体、N,N’−ジフェニル−N,N’−ジ(3−メチルフェニル)−4,4’−ジフェニル−1,1’−ジアミン(TPD)、N,N’−ジナフチル−N,N’−ジフェニル−4,4’−ジフェニル−1,1’−ジアミン(NPD)等のトリアリールアミン誘導体、4,4’−ジ(カルバゾール−9−イル)ビフェニル(CBP)等のカルバゾール誘導体、オリゴチオフェン誘導体(ターチオフェン、クウォーターチオフェン、セキシチオフェン、オクチチオフェンなど)等の低分子有機化合物が挙げられる。
【0061】
また、ここで用いられる中間電極用の素材としては高い導電性を有するものが好ましく、例えば上述の金、白金、クロム、ニッケル、リチウム、マグネシウム、カルシウム、錫、銀、アルミニウムなどの金属や、透明性を有するインジウム、スズ、モリブデンなどの金属酸化物、複合金属酸化物(インジウム錫酸化物(ITO)、インジウム亜鉛酸化物(IZO)など)、上記の金属からなる合金や上記の金属の積層体、ポリエチレンジオキシチオフェン(PEDOT)やPEDOTにポリスチレンスルホネート(PSS)が添加されたもの、などが挙げられる。中間電極は光透過性を有することが好ましいが、光透過性が低い金属のような素材でも膜厚を薄くすることで充分な光透過性を確保できる場合が多い。
【0062】
次に、本発明の光起電力素子の製造方法について例を挙げて説明する。基板上にITOなどの透明電極(この場合正極に相当)をスパッタリング法などにより形成する。次に、一般式(1)で表される構造を有する電子供与性有機材料、および必要により電子受容性有機材料を含む光電変換素子用材料を溶媒に溶解させて溶液を作り、透明電極上に塗布し有機半導体層を形成する。
【0063】
このとき用いられる溶媒は有機溶媒が好ましく、例えば、メタノール、エタノール、ブタノール、トルエン、キシレン、o−クロロフェノール、アセトン、酢酸エチル、エチレングリコール、テトラヒドロフラン、ジクロロメタン、クロロホルム、ジクロロエタン、クロロベンゼン、ジクロロベンゼン、トリクロロベンゼン、クロロナフタレン、ジメチルホルムアミド、ジメチルスルホキシド、N−メチルピロリドン、γ−ブチロラクトンなどが挙げられる。これらを2種以上用いてもよい。
【0064】
また、上述のフルオラス化合物を含有することで光電変換効率をより向上させることができる。常温常圧で液体であるフルオラス化合物(フルオラス溶媒)が好ましく、より好ましくは上述のベンゾトリフルオリド、ペルフルオロヘキサン、F−デカノールが用いられる。フルオラス化合物の含有量は全溶媒量に対して0.01〜20体積%が好ましく、より好ましくは0.1〜2体積%である。また、フルオラス溶媒の含有量は全溶媒中0.01〜30重量%が好ましく、より好ましくは0.1〜4重量%である。
【0065】
一般式(1)で表される構造を有する電子供与性有機材料および電子受容性有機材料を混合して有機半導体層を形成する場合は、一般式(1)で表される構造を有する電子供与性有機材料と電子受容性有機材料を所望の比率で溶媒に添加し、加熱、撹拌、超音波照射などの方法を用いて溶解させ溶液を作り、透明電極上に塗布する。この場合、2種以上の溶媒を混合して用いることで光起電力素子の光電変換効率を向上させることもできる。これは、電子供与性有機材料と電子受容性有機材料がナノレベルで相分離を起こし、電子と正孔の通り道となるキャリアパスが形成されるためと推測される。
【0066】
組み合わせる溶媒は、用いる電子供与性有機材料と電子受容性有機材料の種類によって最適な組み合わせの種類を選択することができる。一般式(1)で表される構造を有する電子供与性有機材料を用いる場合、組み合わせる好ましい溶媒として上述の中でもクロロホルムとクロロベンゼンが挙げられる。この場合、各溶媒の混合体積比率は、クロロホルム:クロロベンゼン=5:95〜95:5の範囲であることが好ましく、さらに好ましくはクロロホルム:クロロベンゼン=10:90〜90:10の範囲である。
【0067】
また、一般式(1)表される構造を有する電子供与性有機材料および電子受容性有機材料を積層して有機半導体層を形成する場合は、例えば電子供与性有機材料の溶液を塗布して電子供与性有機材料を有する層を形成した後に、電子受容性有機材料の溶液を塗布して層を形成する。ここで、電子供与性有機材料および電子受容性有機材料は、分子量が1000以下程度の低分子量体である場合には、蒸着法を用いて層を形成することも可能である。
【0068】
有機半導体層の形成には、スピンコート塗布、ブレードコート塗布、スリットダイコート塗布、スクリーン印刷塗布、バーコーター塗布、鋳型塗布、印刷転写法、浸漬引き上げ法、インクジェット法、スプレー法、真空蒸着法など何れの方法を用いてもよく、膜厚制御や配向制御など、得ようとする有機半導体層特性に応じて形成方法を選択すればよい。例えばスピンコート塗布を行う場合には、一般式(1)で表される構造を有する電子供与性有機材料、および電子受容性有機材料が1〜20g/lの濃度(一般式(1)で表される構造を有する電子供与性有機材料と電子受容性有機材料と溶媒を含む溶液の体積に対する、一般式(1)で表される構造を有する電子供与性有機材料と電子受容性有機材料の重量)であることが好ましく、この濃度にすることで厚さ5〜200nmの均質な有機半導体層を容易に得ることができる。
【0069】
形成した有機半導体層に対して、溶媒を除去するために、減圧下または不活性雰囲気下(窒素やアルゴン雰囲気下)などでアニーリング処理を行ってもよい。アニーリング処理の好ましい温度は40℃〜300℃、より好ましくは50℃〜200℃である。また、アニーリング処理を行うことで、積層した層が界面で互いに浸透して接触する実行面積が増加し、短絡電流を増大させることができる。このアニーリング処理は、負極の形成後に行ってもよい。
【0070】
次に、有機半導体層上にAlなどの金属電極(この場合負極に相当)を真空蒸着法やスパッタ法により形成する。金属電極は、電子輸送層に低分子有機材料を用いて真空蒸着した場合は、引き続き、真空を保持したまま続けて形成することが好ましい。
【0071】
正極と有機半導体層の間に正孔輸送層を設ける場合には、所望のp型有機半導体材料(PEDOTなど)を正極上にスピンコート法、バーコーティング法、ブレードによるキャスト法等で塗布した後、真空恒温槽やホットプレートなどを用いて溶媒を除去し、正孔輸送層を形成する。フタロシアニン誘導体やポルフィリン誘導体などの低分子有機材料を使用する場合には、真空蒸着機を用いた真空蒸着法を適用することも可能である。
【0072】
有機半導体層と負極の間に電子輸送層を設ける場合には、所望のn型有機半導体材料(フラーレン誘導体など)n型無機半導体材料(酸化チタンゲルなど)を有機半導体層上にスピンコート法、バーコーティング法、ブレードによるキャスト法、スプレー法等で塗布した後、真空恒温槽やホットプレートなどを用いて溶媒を除去し、電子輸送層を形成する。フェナントロリン誘導体やC60などの低分子有機材料を使用する場合には、真空蒸着機を用いた真空蒸着法を適用することも可能である。
【0073】
本発明の光起電力素子は、光電変換機能、光整流機能などを利用した種々の光電変換デバイスへの応用が可能である。例えば光電池(太陽電池など)、電子素子(光センサ、光スイッチ、フォトトランジスタなど)、光記録材(光メモリなど)などに有用である。
【実施例】
【0074】
以下、本発明を実施例に基づいてさらに具体的に説明する。なお、本発明は下記実施例に限定されるものではない。また実施例等で用いた化合物のうち、略語を使用しているものについて、以下に示す。
ITO:インジウム錫酸化物
PEDOT:ポリエチレンジオキシチオフェン
PSS:ポリスチレンスルホネート
PC70BM:フェニル C71 ブチリックアシッドメチルエステル
Eg:バンドギャップ
HOMO:最高被占分子軌道
Isc:短絡電流密度
Voc:開放電圧
FF:フィルファクター
η:光電変換効率
なお、1H−NMR測定にはFT−NMR装置((株)日本電子製JEOL JNM−EX270)を用いた。
【0075】
また、平均分子量(数平均分子量、重量平均分子量)はGPC装置(クロロホルムを送液したTOSOH社製、高速GPC装置HLC−8320GPC)を用い、絶対検量線法によって算出した。重合度nは以下の式で算出した。
重合度n=[(重量平均分子量)/(繰り返しユニットの分子量)]
また、光吸収端波長は、ガラス上に約60nmの厚さに形成した薄膜について、日立製作所(株)製のU−3010型分光光度計を用いて測定した薄膜の紫外可視吸収スペクトル(測定波長範囲:300〜900nm)から得た。
【0076】
バンドギャップ(Eg)は下式により、光吸収端波長から算出した。なお、薄膜はクロロホルムを溶媒に用いてスピンコート法により形成した。
Eg(eV)=1240/薄膜の光吸収端波長(nm)
また、最高被占分子軌道(HOMO)準位は、ITOガラス上に約60nmの厚さに形成した薄膜について、表面分析装置(大気中紫外線光電子分光装置AC−2型、理研機器(株)製)を用いて測定した。なお、薄膜はクロロホルムを溶媒に用いてスピンコート法により形成した。
【0077】
合成例1
化合物A−1を式1に示す方法で合成した。なお、合成例1記載の化合物(1−c)はアドバンスドマテリアルズ(Advanced Materials)、2010年、22巻、5240−5244頁に記載されている方法を、化合物(1−g)はジャーナルオブザアメリカンケミカルソサエティ(Journal of the American Chemical Society)、2009年、131巻、7792−7799頁に記載されている方法を参考にして合成した。
【0078】
【化8】
【0079】
化合物(1−a)(アルドリッチ社製)20.0g(68mmol)をエタノール750mlにけん濁させ、メカニカルスターラーを用いて撹拌しているところに、テトラヒドロホウ酸ナトリウム37.8g(1mol)を5回にわけ、1時間かけて0℃で加えた。反応溶混合物を0℃で5時間撹拌した後、室温で12時間放置した。エタノールをおよそ250mlになるまで減圧留去した後、水100mlを0℃でゆっくり加えた。クロロホルム200mlを加えた後、水層および有機層に不溶の固体をろ別した。水層をクロロホルム100mlで3回抽出した後、集めた有機層を無水硫酸マグネシウムで乾燥した。クロロホルムを減圧留去し、化合物(1−b)を薄橙色固体(10.2g、収率56%)として得た。化合物(1−b)はこれ以上精製することなく、以下の反応に用いた。
【0080】
上記化合物(1−b)1.08g(4.07mmol)および化合物(1−c)1.9g(4.07mmol)のエタノール溶液50mlにメタンスルホン酸(東京化成工業(株))1滴を加え、6時間還流した。反応溶液を室温まで冷却した後、炭酸ナトリウムを加えて中和し、溶液をろ過、ついで減圧留去した。シリカゲルカラムクロマトグラフィー(溶離液、ヘキサン:酢酸エチル=10:1)で精製することにより化合物(1−d)を黄色オイル(2.1g、収率73%)として得た。化合物(1−d)の1H−NMRの測定結果を以下に示す。
1H−NMR(270MHz,CDCl3):7.90(s,2H),7.27−7.19(m,6H),6.95−6.88(m,2H),3.69(d,J=5.9Hz,4H),1.67−1.27(m,18H),0.97−0.86(m,12H)ppm。
【0081】
上記化合物(1−d)1.3g(1.85mmol)および2−(tributylstannyl)thiophene(アルドリッチ社製)2.1g(5.5mmol)のトルエン溶液60mlを窒素でバブリングした後、ジクロロビス(トリフェニルホスフィン)パラジウム触媒(東京化成工業(株)製)100mgを加え、8時間還流した。反応溶液を室温まで冷却した後、水50mlを加え、有機層を水で2回、次いで飽和食塩水で洗浄した。無水硫酸マグネシウムで溶媒を乾燥させた後、ろ過し、溶媒を減圧留去した。シリカゲルカラムクロマトグラフィー(溶離液、ヘキサン:酢酸エチル=20:1)で精製することにより化合物(1−e)を黄色固体(1.2g、収率92%)として得た。化合物(1−e)の1H−NMRの測定結果を以下に示す。
1H−NMR(270MHz,CDCl3):8.15(s,2H),7.88(d,J=4.1Hz,2H),7.49(d,J=5.1Hz,2H),7.40(m,2H),7.28−7.24(m,4H),7.17(m,2H),6.93(m,2H),3.78(d,J=5.9Hz,4H),1.71(m,2H),1.61−1.29(m,6H),0.92(t,J=7.0Hz,12H)ppm。
【0082】
上記化合物(1−e)1.2g(1.7mmol)のクロロホルム溶液50mlにN−ブロモスクシンイミド(東京化成工業(株)製)600mg(3.4mmol)を加え、3時間室温で撹拌した。水50mlを加えた後、有機層を水で2回、次いで飽和食塩水で1回洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧留去した。粗生成物をシリカゲルカラムクロマトグラフィー(溶離液、ヘキサン:クロロホルム=3:1)で精製することにより化合物(1−f)を黄色固体(1.1g、収率75%)として得た。化合物(1−f)の1H−NMRの測定結果を以下に示す。
1H−NMR(270MHz,CDCl3):8.06(s,2H),7.54(d,J=4.1Hz,2H),7.49(s,2H),7.21(d,J=8.0Hz,2H),7.13−7.10(m,4H),6.97(d,J=8.0Hz,2H),3.88(d,J=5.1Hz,4H),1.73(m,2H),1.52−1.30(m,16H),0.94(t,J=7.3Hz,12H),ppm。
【0083】
上記化合物(1−f)129mg(0.15mmol)および化合物(1−g)116mg(0.15mmol)をトルエン(和光純薬工業(株)製)10mlに溶解させたところに、トリス(ジベンジリデンアセトン)ジパラジウム(東京化成工業(株)製)8mg、トリス(2−メチルフェニル)ホスフィン(東京化成工業(株)製)を13mg加え、窒素雰囲気下、110℃で12時間撹拌した。次いで、ブロモベンゼン(東京化成工業(株)製)10mgを加え、110℃にて1時間撹拌した。次いで、2−(tributylstannyl)thiophene(アルドリッチ社製)40mgを加え、110℃にてさらに1時間撹拌した。撹拌終了後、反応混合物を室温まで冷却し、メタノール100mlに注いだ。析出した固体をろ取し、メタノール、水、アセトンの順に洗浄した。次いでソックスレー抽出器を用いてアセトン、ヘキサンの順で洗浄した。次に、得られた固体をクロロホルムに溶解させ、セライト(ナカライテスク(株)製)を通して濾過した後、溶媒を減圧留去した。得られた固体を再びクロロホルムに溶解させ、シリカゲルカラム(溶離液:クロロホルム)を通した後に濃縮し、メタノールに再沈澱させ、化合物A−1(40mg)を得た。重量平均分子量は8,800、数平均分子量は6,200、重合度nは7.7であった。また、光吸収端波長は695nm、バンドギャップ(Eg)は1.78eV、最高被占分子軌道(HOMO)準位は−5.03eVであった。
【0084】
合成例2
化合物A−2を式2に示す方法で合成した。なお、合成例2記載の化合物(2−b)はジャーナルオブポリマーサイエンス:パートA(Journal of PolymerScience:PartA)、2010年、48巻、4823−4834頁に記載されている方法を参考にして合成した。
【0085】
【化9】
【0086】
上記化合物(1−b)4.0g(15mmol)およびベンジル(東京化成工業(株)製)3.16g(15mmol)のクロロホルム溶液120mlに濃硫酸1滴を室温で加え、反応溶液を5時間還流した。反応溶液を室温まで冷やした後、5%炭酸ナトリウム水溶液50mlを加え、有機層を水100ml、ついで飽和食塩水100mlで洗浄した。溶媒を無水硫酸マグネシウムで乾燥後、減圧留去した。粗成生成物をメタノールで洗浄し、化合物(2−a)を薄黄色固体(5.2g、収率79%)として得た。化合物(2−a)の1H−NMRの測定結果を以下に示す。
1H−NMR(270MHz,CDCl3):7.91(s,2H),7.65(m,4H),7.38(m,6H)ppm。
【0087】
上記化合物(2−a)1.0g(2.26mmol)、化合物(2−b)(2.2g,6.79mmol)、りん酸三カリウム1.44g(6.79mmol)およびジメチルホルムアミド(ナカライテスク(株)製)30mlに[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(アルドリッチ社製)100mgを加え100℃で5時間撹拌した。反応混合物に酢酸エチル50mlおよび水50mlを加えた後、セライト(ナカライテスク(株)を通してろ過し、有機層を水、次いで飽和食塩水で洗浄した。無水硫酸マグネシウムで乾燥後、溶媒を減圧留去した。粗生成物をシリカゲルカラムクロマトグラフィー(溶離液、ヘキサン:クロロホルム=1:1)で精製することにより化合物(2−c)を橙色固体(1.2g、収率64%)として得た。化合物(2−c)の1H−NMRの測定結果を以下に示す。
1H−NMR(270MHz,CDCl3):8.10(s,2H),7.75(m,6H),7.38(m,6H),7.11(s,2H),2.68(t,J=7.6Hz,4H),1.71(m,4H),1.39−1.29(m,20H),0.88(t,J=7.0Hz,6H)ppm。
【0088】
上記化合物(2−c)1.01g(1.5mmol)のクロロホルム溶液40mlにN−ブロモスクシンイミド(東京化成工業(株)製)533mg(3.0mmol)を加え、3時間室温で撹拌した。水50mlを加えた後、有機層を水で2回、次いで飽和食塩水で1回洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧留去した。粗生成物をシリカゲルカラムクロマトグラフィー(溶離液、クロロホルム)で精製することにより化合物(2−d)を黄色固体(930mg、収率75%)として得た。化合物(2−d)の1H−NMRの測定結果を以下に示す。
1H−NMR(270MHz,CDCl3):8.03(s,2H),7.72−7.68(m,4H),7.52(s,2H),7.42−7.39(m,6H),2.62(t,J=7.3Hz,4H),1.64(m,4H),1.36−1.29(m,20H),0.89(t,J=7.0Hz,6H)ppm。
【0089】
上記化合物(2−d)124mg(0.15mmol)および化合物(1−g)116mg(0.15mmol)をトルエン(和光純薬工業(株)製)8mlに溶解させたところに、トリス(ジベンジリデンアセトン)ジパラジウム(東京化成工業(株)製)8mg、トリス(2−メチルフェニル)ホスフィン(東京化成工業(株)製)を13mg加え、窒素雰囲気下、110℃で12時間撹拌した。次いで、ブロモベンゼン(東京化成工業(株)製)10mgを加え、110℃にて1時間撹拌した。次いで、2−(tributylstannyl)thiophene(アルドリッチ社製)40mgを加え、110℃にてさらに1時間撹拌した。撹拌終了後、反応混合物を室温まで冷却し、メタノール100mlに注いだ。析出した固体をろ取し、メタノール、水、アセトンの順に洗浄した。次いでソックスレー抽出器を用いてアセトン、ヘキサンの順で洗浄した。次に、得られた固体をクロロホルムに溶解させ、セライト(ナカライテスク(株)製)を通して濾過した後、溶媒を減圧留去した。得られた固体を再びクロロホルムに溶解させ、シリカゲルカラム(溶離液:クロロホルム)を通した後に濃縮し、メタノールに再沈澱させ、化合物A−2(110mg)を得た。重量平均分子量は18,600、数平均分子量は13,500、重合度nは16であった。また、光吸収端波長は703nm、バンドギャップ(Eg)は1.76eV、最高被占分子軌道(HOMO)準位は−5.08eVであった。
【0090】
合成例3
化合物A−3を式3に示す方法で合成した。なお、合成例3記載の化合物(3−a)はマクロモレキュルズ(Macromolecules)、2010年、43巻、9779−9786頁に記載されている方法を参考にして合成した。
【0091】
【化10】
【0092】
化合物(2−a)880mg(2.0mmol)および化合物(3−a)1.4g(6.0mmol)のトルエン20ml溶液を窒素でバブリングした後、ジクロロビス(トリフェニルホスフィン)パラジウム触媒(東京化成工業(株)製)80mgを加え、6時間還流した。反応溶液を室温まで冷却した後、水50mlを加え、有機層を水で2回、次いで飽和食塩水で洗浄した。無水硫酸マグネシウムで溶媒を乾燥させた後、ろ過し、溶媒を減圧留去した。シリカゲルカラムクロマトグラフィー(溶離液、ヘキサン:クロロホルム=1:1)で精製することにより化合物(3−b)を黄色固体(1.2g、収率84%)として得た。化合物(3−b)の1H−NMRの測定結果を以下に示す。
1H−NMR(270MHz,CDCl3):8.10(s,2H),7.75(m,6H),7.38(m,6H),7.08(s,2H),2.62(d,J=7.0Hz,4H),1.63(m,1H),1.4−1.2(m,16H),0.92(m,6H)ppm。
【0093】
上記化合物(3−b)1.01g(1.5mmol)のクロロホルム溶液40mlにN−ブロモスクシンイミド(東京化成工業(株)製)533mg(3.0mmol)を加え、3時間室温で撹拌した。水50mlを加えた後、有機層を水で2回、次いで飽和食塩水で1回洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧留去した。粗生成物をシリカゲルカラムクロマトグラフィー(溶離液、クロロホルム)で精製することにより化合物(3−c)を黄色固体(890mg、収率72%)として得た。化合物(3−c)の1H−NMRの測定結果を以下に示す。
1H−NMR(270MHz,CDCl3):8.06(s,2H),7.71(m,4H),7.53(s,2H),7.40(m,6H),2.56(d,J=7.0Hz,4H),1.69(m,2H),1.4−1.3(m,16H),0.9−0.8(m,12H)ppm。
【0094】
上記化合物(3−c)124mg(0.15mmol)および化合物(1−g)116mg(0.15mmol)をトルエン(和光純薬工業(株)製)8mlに溶解させたところに、トリス(ジベンジリデンアセトン)ジパラジウム(東京化成工業(株)製)8mg、トリス(2−メチルフェニル)ホスフィン(東京化成工業(株)製)を13mg加え、窒素雰囲気下、110℃で12時間撹拌した。次いで、ブロモベンゼン(東京化成工業(株)製)10mgを加え、110℃にて1時間撹拌した。次いで、2−(tributylstannyl)thiophene(アルドリッチ社製)40mgを加え、110℃にてさらに1時間撹拌した。撹拌終了後、反応混合物を室温まで冷却し、メタノール100mlに注いだ。析出した固体をろ取し、メタノール、水、アセトンの順に洗浄した。次いでソックスレー抽出器を用いてアセトン、ヘキサンの順で洗浄した。次に、得られた固体をクロロホルムに溶解させ、セライト(ナカライテスク(株)製)を通して濾過した後、溶媒を減圧留去した。得られた固体を再びクロロホルムに溶解させ、シリカゲルカラム(溶離液:クロロホルム)を通した後に濃縮し、メタノールに再沈澱させ、化合物A−3(110mg)を得た。重量平均分子量は42,000、数平均分子量は23,000、重合度nは37であった。また、光吸収端波長は678nm、バンドギャップ(Eg)は1.83eV、最高被占分子軌道(HOMO)準位は−5.15eVであった。
【0095】
合成例4
化合物A−4を式4に示す方法で合成した。なお、合成例4記載の化合物(4−a)はアンゲバンテケミ インターナショナルエディション(Angewandte Chem Internatioal Edition)、2011年、50巻、9697−9702頁に記載されている方法を参考にして合成した。
【0096】
【化11】
【0097】
上記化合物(1−f)129mg(0.15mmol)および化合物(4−a)136mg(0.15mmol)をトルエン(和光純薬工業(株)製)8mlに溶解させたところに、トリス(ジベンジリデンアセトン)ジパラジウム(東京化成工業(株)製)8mg、トリス(2−メチルフェニル)ホスフィン(東京化成工業(株)製)を13mg加え、窒素雰囲気下、110℃で12時間撹拌した。次いで、ブロモベンゼン(東京化成工業(株)製)10mgを加え、110℃にて1時間撹拌した。次いで、2−(tributylstannyl)thiophene(アルドリッチ社製)40mgを加え、110℃にてさらに1時間撹拌した。撹拌終了後、反応混合物を室温まで冷却し、メタノール100mlに注いだ。析出した固体をろ取し、メタノール、水、アセトンの順に洗浄した。次いでソックスレー抽出器を用いてアセトン、ヘキサンの順で洗浄した。得られた固体をクロロベンゼンに溶解させ、メタノールより再沈澱することで、化合物A−4(112mg)を得た。重量平均分子量は18,000、数平均分子量は11,000、重合度nは14であった。また、光吸収端波長は725nm、バンドギャップ(Eg)は1.71eV、最高被占分子軌道(HOMO)準位は−4.92eVであった。
【0098】
合成例5
化合物A−5を式5に示す方法で合成した。
【0099】
【化12】
【0100】
上記化合物(3−c)124mg(0.15mmol)および化合物(4−a)136mg(0.15mmol)をトルエン(和光純薬工業(株)製)8mlに溶解させたところに、トリス(ジベンジリデンアセトン)ジパラジウム(東京化成工業(株)製)8mg、トリス(2−メチルフェニル)ホスフィン(東京化成工業(株)製)を13mg加え、窒素雰囲気下、110℃で12時間撹拌した。次いで、ブロモベンゼン(東京化成工業(株)製)10mgを加え、110℃にて1時間撹拌した。次いで、2−(tributylstannyl)thiophene(アルドリッチ社製)40mgを加え、110℃にてさらに1時間撹拌した。撹拌終了後、反応混合物を室温まで冷却し、メタノール100mlに注いだ。析出した固体をろ取し、メタノール、水、アセトンの順に洗浄した。次いでソックスレー抽出器を用いてアセトン、ヘキサンの順で洗浄した。得られた固体をクロロベンゼンに溶解させ、メタノールより再沈澱することで、化合物A−5(105mg)を得た。重量平均分子量は22,000、数平均分子量は15,000、重合度nは18であった。また、光吸収端波長は708nm、バンドギャップ(Eg)は1.75eV、最高被占分子軌道(HOMO)準位は−5.07eVであった。
【0101】
合成例6
化合物B−1を式6に示す方法で合成した。
【0102】
【化13】
【0103】
化合物(2−a)66mg(0.15mmol)および化合物(1−g)116mg(0.15mmol)をトルエン(和光純薬工業(株)製)8mlに溶解させたところに、トリス(ジベンジリデンアセトン)ジパラジウム(東京化成工業(株)製)8mg、トリス(2−メチルフェニル)ホスフィン(東京化成工業(株)製)を13mg加え、窒素雰囲気下、110℃で12時間撹拌した。次いで、ブロモベンゼン(東京化成工業(株)製)10mgを加え、110℃にて1時間撹拌した。次いで、2−(tributylstannyl)thiophene(アルドリッチ社製)40mgを加え、110℃にてさらに1時間撹拌した。撹拌終了後、反応混合物を室温まで冷却し、メタノール100mlに注いだ。析出した固体をろ取し、メタノール、水、アセトンの順に洗浄した。次いでソックスレー抽出器を用いてアセトン、ヘキサンの順で洗浄した。次に、得られた固体をクロロホルムに溶解させ、セライト(ナカライテスク(株)製)を通して濾過した後、溶媒を減圧留去した。得られた固体を再びクロロホルムに溶解させ、シリカゲルカラム(溶離液:クロロホルム)を通した後に濃縮し、メタノールに再沈澱させ、化合物B−1(80mg)を得た。重量平均分子量は10,200、数平均分子量は9,000、重合度nは14であった。また、光吸収端波長は770nm、バンドギャップ(Eg)は1.61eV、最高被占分子軌道(HOMO)準位は−4.93eVであった。
【0104】
合成例7
化合物B−2を式7に示す方法で合成した。なお、合成例7記載の化合物(7−a)はザジャーナルオブオーガニックケミストリー(The Journal of Organic Chemistry)、2002年、67巻、9073−9076頁に記載されている方法を参考にして合成した。
【0105】
【化14】
【0106】
化合物(7−a)66.6mg(0.15mmol)および化合物(1−g)116mg(0.15mmol)をトルエン(和光純薬工業(株)製)8mlに溶解させたところに、トリス(ジベンジリデンアセトン)ジパラジウム(東京化成工業(株)製)8mg、トリス(2−メチルフェニル)ホスフィン(東京化成工業(株)製)を13mg加え、窒素雰囲気下、110℃で12時間撹拌した。次いで、ブロモベンゼン(東京化成工業(株)製)10mgを加え、110℃にて1時間撹拌した。次いで、2−(tributylstannyl)thiophene(アルドリッチ社製)40mgを加え、110℃にてさらに1時間撹拌した。撹拌終了後、反応混合物を室温まで冷却し、メタノール100mlに注いだ。析出した固体をろ取し、メタノール、水、アセトンの順に洗浄した。次いでソックスレー抽出器を用いてアセトン、ヘキサンの順で洗浄した。次に、得られた固体をクロロホルムに溶解させ、セライト(ナカライテスク(株)製)を通して濾過した後、溶媒を減圧留去した。得られた固体を再びクロロホルムに溶解させ、シリカゲルカラム(溶離液:クロロホルム)を通した後に濃縮し、メタノールに再沈澱させ、化合物B−2(76mg)を得た。重量平均分子量は7,800、数平均分子量は5,600、重合度nは10であった。また、光吸収端波長は915nm、バンドギャップ(Eg)は1.36eV、最高被占分子軌道(HOMO)準位は−4.91eVであった。
【0107】
合成例8
【0108】
【化15】
【0109】
化合物B−3を式8に示す方法で合成した。なお、合成例8記載の化合物(8−a)はマクロモレキュルズ(Macromolecules)、2010年、43巻、811−820頁に記載されている方法を参考にして合成した。
【0110】
化合物(8−a)111mg(0.15mmol)および化合物(1−g)116mg(0.15mmol)をトルエン(和光純薬工業(株)製)8mlに溶解させたところに、トリス(ジベンジリデンアセトン)ジパラジウム(東京化成工業(株)製)8mg、トリス(2−メチルフェニル)ホスフィン(東京化成工業(株)製)を13mg加え、窒素雰囲気下、110℃で12時間撹拌した。次いで、ブロモベンゼン(東京化成工業(株)製)10mgを加え、110℃にて1時間撹拌した。次いで、2−(tributylstannyl)thiophene(アルドリッチ社製)40mgを加え、110℃にてさらに1時間撹拌した。撹拌終了後、反応混合物を室温まで冷却し、メタノール100mlに注いだ。析出した固体をろ取し、メタノール、水、アセトンの順に洗浄した。次いでソックスレー抽出器を用いてアセトン、ヘキサンの順で洗浄した。次に、得られた固体をクロロホルムに溶解させ、セライト(ナカライテスク(株)製)を通して濾過した後、溶媒を減圧留去した。得られた固体を再びクロロホルムに溶解させ、シリカゲルカラム(溶離液:クロロホルム)を通した後に濃縮し、メタノールに再沈澱させ、化合物B−3(90mg)を得た。重量平均分子量は14,100、数平均分子量は11,500、重合度nは14であった。また、光吸収端波長は730nm、バンドギャップ(Eg)は1.70eV、最高被占分子軌道(HOMO)準位は−5.19eVであった。
【0111】
合成例9
化合物B−4を式9に示す方法で合成した。
【0112】
【化16】
【0113】
上記化合物(3−c)124mg(0.15mmol)および化合物(9−a)(アルドリッチ社製)84mg(0.15mmol)をトルエン10mlに溶解させたところに、濃度1M炭酸カリウム水溶液1ml、Aliquat336(アルドリッチ社製)1滴およびテトラキス(トリフェニルホスフィン)パラジウム触媒(東京化成工業(株)製)10mgを加え、窒素雰囲気下、100℃で8時間撹拌した。次いで、ブロモベンゼン(東京化成工業(株)製)30mgを加え、100℃にて1時間撹拌した。次いで、フェニルボロン酸(東京化成工業(株)製)50mgを加え、100℃にてさらに1時間撹拌した。撹拌終了後、反応混合物を室温まで冷却し、メタノール100mlに注いだ。析出した固体をろ取し、メタノール、水、アセトンの順に洗浄した。次いでソックスレー抽出器を用いてアセトン、ヘキサンの順で洗浄した。次に、得られた固体をクロロホルムに溶解させ、セライト(ナカライテスク(株)製)を通して濾過した後、溶媒を減圧留去した。得られた固体を再びクロロホルムに溶解させ、シリカゲルカラム(溶離液:クロロホルム)を通した後に濃縮し、メタノールに再沈殿 させ、化合物B−4(92mg)を得た。重量平均分子量は32,400、数平均分子量は18,400、重合度nは30であった。また、光吸収端波長は600nm、バンドギャップ(Eg)は2.07eV、最高被占分子軌道(HOMO)準位は−5.47であった
実施例1
上記A−1(1mg)とPC70BM(4mg、Solenne社製)をクロロベンゼン0.25mlの入ったサンプル瓶の中に加え、超音波洗浄機((株)井内盛栄堂製US−2(商品名)、出力120W)中で30分間超音波照射することにより溶液Aを得た。
【0114】
スパッタリング法により正極となるITO透明導電層を120nm堆積させたガラス基板を38mm×46mmに切断した後、ITOをフォトリソグラフィー法により38mm×13mmの長方形状にパターニングした。得られた基板をアルカリ洗浄液(フルウチ化学(株)製、“セミコクリーン”EL56(商品名))で10分間超音波洗浄した後、超純水で洗浄した。
【0115】
この基板を30分間UV/オゾン処理した後に、基板上に正孔輸送層となるPEDOT:PSS水溶液(PEDOT0.8重量%、PPS0.5重量%)をスピンコート法により60nmの厚さに成膜した。ホットプレートにより200℃で5分間加熱乾燥した後、上記の溶液AをPEDOT:PSS層上に滴下し、スピンコート法により膜厚100nmの有機半導体層を形成した。その後、有機半導体層が形成された基板と陰極用マスクを真空蒸着装置内に設置して、装置内の真空度が1×10−3Pa以下になるまで再び排気し、抵抗加熱法によって、負極となるアルミニウム層を80nmの厚さに蒸着した。以上のように、ストライプ状のITO層とアルミニウム層が交差する部分の面積が5mm×5mmである光起電力素子を作製した。
【0116】
このようにして作製された光起電力素子の正極と負極をヒューレット・パッカード社製ピコアンメーター/ボルテージソース4140Bに接続して、大気中でITO層側から擬似太陽光(山下電装株式会社製 簡易型ソーラシミュレータ YSS−E40、スペクトル形状:AM1.5、強度:100mW/cm2)を照射し、印加電圧を−1Vから+2Vまで変化させたときの電流値を測定した。この時の短絡電流密度(印加電圧が0Vのときの電流密度の値)は6.49mA/cm2、開放電圧(電流密度が0になるときの印加電圧の値)は0.78V、フィルファクター(FF)は0.61であり、これらの値から算出した光電変換効率は3.09%であった。なお、フィルファクターと光電変換効率は次式により算出した。
フィルファクター=IVmax(mA・V/cm2)/(短絡電流密度(mA/cm2)×開放電圧(V))
(ここで、IVmaxは、印加電圧が0Vから開放電圧値の間で電流密度と印加電圧の積が最大となる点における電流密度と印加電圧の積の値である。)
光電変換効率=[(短絡電流密度(mA/cm2)×開放電圧(V)×フィルファクター)/擬似太陽光強度(100mW/cm2)]×100(%)
以下の実施例と比較例におけるフィルファクターと光電変換効率も全て上式により算出した。
【0117】
実施例2
A−1の代わりに上記A−2を用いた他は実施例1と全く同様にして光起電力素子を作製し、電流−電圧特性を測定した。この時の短絡電流密度は8.23mA/cm2、開放電圧は0.79V、フィルファクター(FF)は0.57であり、これらの値から算出した光電変換効率は3.71%であった。
【0118】
実施例3
A−1の代わりに上記A−3を用いた他は実施例1と全く同様にして光起電力素子を作製し、電流−電圧特性を測定した。この時の短絡電流密度は7.21mA/cm2、開放電圧は0.83V、フィルファクター(FF)は0.53であり、これらの値から算出した光電変換効率は3.17%であった。
【0119】
実施例4
A−1の代わりに上記A−4を用いた他は実施例1と全く同様にして光起電力素子を作製し、電流−電圧特性を測定した。この時の短絡電流密度は9.03mA/cm2、開放電圧は0.71V、フィルファクター(FF)は0.60であり、これらの値から算出した光電変換効率は3.85%であった。
【0120】
実施例5
A−1の代わりに上記A−5を用いた他は実施例1と全く同様にして光起電力素子を作製し、電流−電圧特性を測定した。この時の短絡電流密度は8.62mA/cm2、開放電圧は0.93V、フィルファクター(FF)は0.53であり、これらの値から算出した光電変換効率は4.25%であった。
【0121】
比較例1
A−1の代わりに上記B−1を用いた他は実施例1と全く同様にして光起電力素子を作製し、電流−電圧特性を測定した。この時の短絡電流密度は2.01mA/cm2、開放電圧は0.59V、フィルファクター(FF)は0.31であり、これらの値から算出した光電変換効率は0.37%であった。
【0122】
比較例2
A−1の代わりに上記B−2を用いた他は実施例1と全く同様にして光起電力素子を作製し、電流−電圧特性を測定した。この時の短絡電流密度は4.40mA/cm2、開放電圧は0.52V、フィルファクター(FF)は0.55であり、これらの値から算出した光電変換効率は1.26%であった。
【0123】
比較例3
A−1の代わりに上記B−3を用いた他は実施例1と全く同様にして光起電力素子を作製し、電流−電圧特性を測定した。この時の短絡電流密度は4.14mA/cm2、開放電圧は0.82V、フィルファクター(FF)は0.31であり、これらの値から算出した光電変換効率は1.05%であった。
【0124】
比較例4
A−1の代わりに上記B−4を用いた他は実施例1と全く同様にして光起電力素子を作製し、電流−電圧特性を測定した。この時の短絡電流密度は4.64mA/cm2、開放電圧は1.03V、フィルファクター(FF)は0.38であり、これらの値から算出した光電変換効率は1.82%であった。
【0125】
【表1】
【0126】
表1から明らかなように、一般式(1)で表される構造を有する電子供与性有機材料を用いて作製した光起電力素子(実施例1〜5)は、同様の条件で作製した他の光起電力素子(比較例1〜4)に比べ高い光電変換効率を示した。
【符号の説明】
【0127】
1:基板
2:正極
3:有機半導体層
4:負極
5:電子供与性有機材料を有する層
6:電子受容性有機材料を有する層
【特許請求の範囲】
【請求項1】
一般式(1)で表される構造を有する電子供与性有機材料を含むことを特徴とする光起電力素子用材料。
【化1】
(上記一般式(1)中、R1およびR2は同じでも異なっていてもよく、置換されていてもよいアリール基、または置換されていてもよいヘテロアリール基を表す。R3〜R6は同じでも異なっていてもよく、水素、アルキル基、アルコキシ基、アリール基、ヘテロアリール基、またはハロゲンを表す。XおよびYは2価の連結基を表す。nは2以上1,000以下の範囲を表す。)
【請求項2】
さらに電子受容性有機材料を含む請求項1に記載の光起電力素子用材料。
【請求項3】
前記電子受容性有機材料がフラーレン化合物である請求項2記載の光起電力素子用材料。
【請求項4】
前記フラーレン化合物がC70誘導体である請求項3記載の光起電力素子用材料。
【請求項5】
少なくとも正極と負極を有する光起電力素子であって、負極と正極の間に請求項1〜4のいずれか記載の光起電力素子用材料を含むことを特徴とする光起電力素子。
【請求項1】
一般式(1)で表される構造を有する電子供与性有機材料を含むことを特徴とする光起電力素子用材料。
【化1】
(上記一般式(1)中、R1およびR2は同じでも異なっていてもよく、置換されていてもよいアリール基、または置換されていてもよいヘテロアリール基を表す。R3〜R6は同じでも異なっていてもよく、水素、アルキル基、アルコキシ基、アリール基、ヘテロアリール基、またはハロゲンを表す。XおよびYは2価の連結基を表す。nは2以上1,000以下の範囲を表す。)
【請求項2】
さらに電子受容性有機材料を含む請求項1に記載の光起電力素子用材料。
【請求項3】
前記電子受容性有機材料がフラーレン化合物である請求項2記載の光起電力素子用材料。
【請求項4】
前記フラーレン化合物がC70誘導体である請求項3記載の光起電力素子用材料。
【請求項5】
少なくとも正極と負極を有する光起電力素子であって、負極と正極の間に請求項1〜4のいずれか記載の光起電力素子用材料を含むことを特徴とする光起電力素子。
【図1】


【図2】
【図2】
【公開番号】特開2013−102148(P2013−102148A)
【公開日】平成25年5月23日(2013.5.23)
【国際特許分類】
【出願番号】特願2012−225804(P2012−225804)
【出願日】平成24年10月11日(2012.10.11)
【国等の委託研究の成果に係る記載事項】(出願人による申告)国等の委託研究の成果に係る特許出願(平成23年度独立行政法人新エネルギー・産業技術総合開発機構最先端研究開発支援プログラム(低炭素社会実現に資する有機系太陽電池の開発)、産業技術力強化法第19条の適用を受ける特許出願。)
【出願人】(000003159)東レ株式会社 (7,677)
【Fターム(参考)】
【公開日】平成25年5月23日(2013.5.23)
【国際特許分類】
【出願日】平成24年10月11日(2012.10.11)
【国等の委託研究の成果に係る記載事項】(出願人による申告)国等の委託研究の成果に係る特許出願(平成23年度独立行政法人新エネルギー・産業技術総合開発機構最先端研究開発支援プログラム(低炭素社会実現に資する有機系太陽電池の開発)、産業技術力強化法第19条の適用を受ける特許出願。)
【出願人】(000003159)東レ株式会社 (7,677)
【Fターム(参考)】
[ Back to top ]