
光重合開始剤、光硬化性組成物、パターン形成方法、カラーフィルタ、液晶表示装置、及び光重合開始剤の製造方法
【課題】装置の汚染が無く、解像度、現像性、深部硬化性及び基板との密着性が良好となる光硬化性組成物及び該組成物を用いたカラーフィルターの提供。
【解決手段】下記一般式で表される光重合開始剤。

(式中、R1〜R11は、水素原子、ハロゲン原子、アルキル基、アルケニル基、シクロアルキル基、シクロアルケニル基、ヒドロキシル基、アルコキシ基、アルケニルオキシ基、アルカノイル基、アルケノイル基、アリール基又はヘテロ環基を示す。)
【解決手段】下記一般式で表される光重合開始剤。
(式中、R1〜R11は、水素原子、ハロゲン原子、アルキル基、アルケニル基、シクロアルキル基、シクロアルケニル基、ヒドロキシル基、アルコキシ基、アルケニルオキシ基、アルカノイル基、アルケノイル基、アリール基又はヘテロ環基を示す。)
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、光重合開始剤、光硬化性組成物、該組成物を用いたパターン形成方法、該組成物を用いたカラーフィルタ、該カラーフィルタを有する液晶表示装置、及び光重合開始剤の製造方法に関する。さらに詳しくは、光硬化性組成物中のエチレン性不飽和結合を有する化合物、エーテル結合及び/又はエステル結合を有する25℃で液体の化合物との相溶性、及び溶剤(前記のエーテル結合及び/又はエステル結合を有する25℃で液体の化合物を含む。)に対する溶解性が高く、光(特に波長450nm以下の短波長の光)に対する感度が高く、露光時の光により発生する分解物による重合物の汚染や装置の汚染が無い光重合開始剤であって、解像度、現像性、深部硬化性及び基板との密着性が良好となる光硬化性組成物を提供する光重合開始剤、該光重合開始剤を含有する光硬化性組成物、該光硬化性組成物を用いたパターン形成方法、該光硬化性組成物を用いたカラーフィルタ、該カラーフィルタを有する液晶表示装置、及び前記光重合開始剤の製造方法に関する。
【背景技術】
【0002】
光硬化性樹脂組成物は、例えばバインダー樹脂や重合性モノマー及び光重合開始剤を含有するものであり、光(粒子線等の放射線を含む電磁波)を照射することにより重合硬化させることができるので、光硬化性インキ、感光性印刷版、カラーフィルタ、各種フォトレジスト等に用いられる。光硬化性樹脂組成物を硬化させる光としては、その取り扱いのし易さや感度等の点から、450nm以下の波長の光(紫外線)が用いられる場合が多く、その光源としては波長365nm、405nm、436nmの波長に強い発光を有する高圧水銀灯や、KrF及びArF等のエキシマレーザーが利用されている。また、より微細なパターン形成に用いられる場合に、電子線やEUV(極端紫外光:Extreme Ultra Violet)等の波長の短い電磁波や放射線の適用が検討されている。
光による硬化は、熱硬化に比べて省エネルギーであることや、フォトマスクを介して照射を行うことにより所望のパターンで硬化できることなどから、種々の用途分野において需要が高く、特に生産性が向上し、かつ光重合開始剤の添加量を削減し得る高感度の光重合性開始剤に対して、需要が高まっている。
また、顔料分散法を用いたカラーフィルタは、通常、分散剤等により顔料を分散してなる顔料分散液に光硬化性組成物をガラス基板に塗布して乾燥後、フォトマスクを用いて露光し、現像を行うことによって着色パターンを形成し、加熱することによりパターンを固着して画素を形成する。これらの工程を、各色ごとに繰り返してカラーフィルタを形成する。このようなカラーフィルタの画像形成に用いられる光硬化性組成物には、十分な解像性、基板との密着性及び低現像残渣等の特性が求められる。
さらに近年では、色濃度が高い画素やそれ自体が遮光層として機能する光学濃度の高いブラックマトリックス用レジストが利用されており、光硬化性組成物中の着色顔料やカーボンブラック等の黒色顔料の含有量が高くなり、光硬化性組成物の光透過性が下がる傾向にある。その結果、露光光源からの光が、光硬化性樹脂塗膜の深部まで到達せず、解像性、基板との密着性及び現像性等が悪化するという問題があり、生産性や、カラーフィルタに要求される精度、さらには信頼性が低下する。
また、カラーフィルタが形成されるガラス基板は年々大型化しており、大面積露光が行われているため、露光照度が小さくなる傾向があることや、更なる生産性向上のために露光時間の短縮が求められていることから、より高感度の光重合開始剤が必要とされている。
そこで、高感度及び高解像度等を達成し得る光硬化性組成物を得る方法として、光重合開始剤としてオキシムエステル化合物を用いることが提案されている(特許文献1〜5参照)。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】特許第3860170号明細書
【特許文献2】特開2006−036750号公報
【特許文献3】特許第3992725号明細書
【特許文献4】特開2010−037542号公報
【特許文献5】韓国公開特許第2009−0046108号公報
【発明の概要】
【発明が解決しようとする課題】
【0004】
特許文献1〜5に記載の光重合開始剤では、感度、解像度、現像性、及び相溶性や溶解性、特に光硬化性組成物中のエチレン性不飽和結合を有する化合物(特に(メタ)アクリロイルオキシ基を有する化合物)との相溶性や溶剤に対する溶解性が不十分であり、使用条件に制限があるため、さらなる改善の余地がある。また、特許文献1〜5に記載の光重合開始剤では、露光時の光により発生する分解物がマスクに付着し、その結果、焼付け時のパターン形成不良を起こし、収率の低下を招くことがあった。そこで、発生した分解物が、重合物や装置等を汚染しない光重合開始剤が望まれている。
【0005】
よって、本発明の課題は、光硬化性組成物中のエチレン性不飽和結合を有する化合物(特に(メタ)アクリロイルオキシ基を有する化合物)、エーテル結合及び/又はエステル結合を有する25℃で液体の化合物との相溶性、及び溶剤(前記のエーテル結合及び/又はエステル結合を有する25℃で液体の化合物を含む。)に対する溶解性が高く、光(特に波長450nm以下の短波長の光)に対する感度が高く、露光時の光により発生する分解物による重合物の汚染や装置の汚染が無い光重合開始剤であって、解像度、現像性、深部硬化性及び基板との密着性が良好となる光硬化性組成物を提供し得る光重合開始剤を提供することである。また、該光重合開始剤を含有する光硬化性組成物、該光硬化性組成物を用いたパターン形成方法、該光硬化性組成物を用いたカラーフィルタ及び該カラーフィルタを有する液晶表示装置を提供することである。さらには、前記光重合開始剤を低コストで簡便に製造する方法を提供することである。
【課題を解決するための手段】
【0006】
本発明者は、前記課題を達成するために鋭意研究を重ねた結果、後述する一般式(I−a)で表される特定構造の光重合開始剤であれば、前記課題を解決し得ることを見出した。
すなわち、本発明は、下記[1]〜[17]に関する。
[1]下記一般式(I−a)で表される光重合開始剤。
【0007】
【化1】
【0008】
(式中、R1〜R11は、それぞれ独立に、水素原子、ハロゲン原子、置換もしくは無置換の炭素数1〜20のアルキル基、置換もしくは無置換の炭素数2〜20のアルケニル基、置換もしくは無置換の環形成原子数3〜10のシクロアルキル基、置換もしくは無置換の炭素数4〜20のシクロアルケニル基、ヒドロキシル基、置換もしくは無置換の炭素数1〜20のアルコキシ基、置換もしくは無置換の炭素数2〜20のアルケニルオキシ基、置換もしくは無置換の炭素数1〜20のアルカノイル基、置換もしくは無置換の炭素数2〜20のアルケノイル基、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数3〜14のヘテロ環基を示す。R3は、R4又はR5と一緒になって環を形成していてもよい。R4は、R5と一緒になって環を形成していてもよい。
また、Arは、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数5〜14のヘテロアリール基を示す。
nは、1〜10の整数を示す。nが2〜10の整数の場合、複数のR4及びR5は、それぞれ同一でも異なっていてもよい。)
[2]前記一般式(I−a)中、R1、R2及びR3が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、nが2であり、複数のR4及びR5が、それぞれ独立に、水素原子又は置換もしくは無置換の炭素数1〜20のアルキル基であり、Arが、置換もしくは無置換の環形成炭素数6〜14のアリール基である、上記[1]に記載の光重合開始剤。
[3]前記一般式(I−a)中、R1、R2及びR3が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、nが2であり、複数のR4及びR5が、それぞれ独立に、水素原子又は置換もしくは無置換の炭素数1〜20のアルキル基であり、Arが、置換もしくは無置換の環形成原子数5〜14のヘテロアリール基である、上記[1]に記載の光重合開始剤。
[4]分子量が515以下である、上記[2]又は[3]に記載の光重合開始剤。
【0009】
[5]前記一般式(I−a)中、R1及びR2が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、R3が置換もしくは無置換の炭素数3〜18のシクロアルキル基であり、nが2であり、複数のR4及びR5が、それぞれ独立に水素原子もしくは置換もしくは無置換の炭素数1〜20のアルキル基である、上記[1]に記載の光重合開始剤。
[6]前記一般式(I−a)中、R1及びR2が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、R3が環形成原子数3〜6の環状エーテル基で置換された炭素数1〜20のアルキル基であり、nが2であり、複数のR4及びR5が、それぞれ独立に水素原子もしくは置換もしくは無置換の炭素数1〜20のアルキル基である、上記[1]に記載の光重合開始剤。
[7]分子量が550以下である、上記[5]又は[6]に記載の光重合開始剤。
[8]バインダー樹脂及び/又はエチレン性不飽和結合を有する化合物と、上記[1]〜[7]のいずれかに記載の光重合開始剤を含有する光硬化性組成物。
[9]光重合開始剤の含有量が、光硬化性組成物の固形分に対して2〜50質量%である、上記[8]に記載の光硬化性組成物。
[10]さらに色材を含有する、上記[8]又は[9]に記載の光硬化性組成物。
[11]さらに、エーテル結合及び/又はエステル結合を有する25℃で液体の化合物を光重合開始剤1質量部に対して5〜100質量部含有する、上記[8]〜[10]のいずれかに記載の光硬化性組成物。
[12]エチレン性不飽和結合を有する化合物として、エーテル結合及び/又はエステル結合を持つエチレン性不飽和結合を有する化合物を少なくとも1種用い、溶剤を光硬化性組成物全量の10質量%以下のみ含有する、上記[8]〜[10]のいずれかに記載の光硬化性組成物。
[13]カラーフィルタ用である、上記[10]〜[12]のいずれかに記載の光硬化性組成物。
[14]上記[10]〜[12]のいずれかに記載の光硬化性組成物を用いたカラーフィルタ。
[15]上記[14]に記載のカラーフィルタと対向基板と、前記カラーフィルタと前記対向基板との間に形成された液晶層とを有する液晶表示装置。
[16]上記[10]〜[12]のいずれかに記載の光硬化性組成物を基板に塗布し、乾燥した後、フォトマスクを用いて露光し、次いで現像を行うことによる、パターン形成方法。
[17]下記一般式(1)
【0010】
【化2】
【0011】
(式中、R6〜R11は、それぞれ独立に、水素原子、ハロゲン原子、置換もしくは無置換の炭素数1〜20のアルキル基、置換もしくは無置換の炭素数2〜20のアルケニル基、置換もしくは無置換の環形成原子数3〜10のシクロアルキル基、置換もしくは無置換の炭素数4〜20のシクロアルケニル基、ヒドロキシル基、置換もしくは無置換の炭素数1〜20のアルコキシ基、置換もしくは無置換の炭素数2〜20のアルケニルオキシ基、置換もしくは無置換の炭素数1〜20のアルカノイル基、置換もしくは無置換の炭素数2〜20のアルケノイル基、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数3〜14のヘテロ環基を示す。)
で表されるカルバゾール誘導体と、下記一般式(2a)
【0012】
【化3】
【0013】
(式中、R3、R4’及びR5’は、それぞれ独立に、水素原子、ハロゲン原子、置換もしくは無置換の炭素数1〜20のアルキル基、置換もしくは無置換の炭素数2〜20のアルケニル基、置換もしくは無置換の環形成原子数3〜10のシクロアルキル基、置換もしくは無置換の炭素数4〜20のシクロアルケニル基、ヒドロキシル基、置換もしくは無置換の炭素数1〜20のアルコキシ基、置換もしくは無置換の炭素数2〜20のアルケニルオキシ基、置換もしくは無置換の炭素数1〜20のアルカノイル基、置換もしくは無置換の炭素数2〜20のアルケノイル基、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数3〜14のヘテロ環基を示す。R3は、R5’と一緒になって環を形成していてもよい。)
で表されるアクリレート誘導体とを塩基の存在下に反応させることにより、下記一般式(3a’)
【0014】
【化4】
【0015】
(式中、R3、R4’、R5’、R6〜R11は、前記定義の通りである。)
で表されるカルボニルアルキル基導入体を得、得られたカルボニルアルキル基導入体と下記一般式(4)、(5)
【0016】
【化5】
【0017】
(式中、Xは、ハロゲン原子又は−OC(=O)Arを示し、X’は、ハロゲン原子又は−OC(=O)R2を示す。
Arは、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数5〜14のヘテロアリール基を示す。
R2は、水素原子、ハロゲン原子、置換もしくは無置換の炭素数1〜20のアルキル基、置換もしくは無置換の炭素数2〜20のアルケニル基、置換もしくは無置換の環形成原子数3〜10のシクロアルキル基、置換もしくは無置換の炭素数4〜20のシクロアルケニル基、ヒドロキシル基、置換もしくは無置換の炭素数1〜20のアルコキシ基、置換もしくは無置換の炭素数2〜20のアルケニルオキシ基、置換もしくは無置換の炭素数1〜20のアルカノイル基、置換もしくは無置換の炭素数2〜20のアルケノイル基、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数3〜14のヘテロ環基を示す。)
で表される2つのアシル化剤をルイス酸の存在下に反応させることにより、下記一般式(6a’)
【0018】
【化6】
【0019】
(式中、R2、R3、R4’、R5’、R6〜R11及びArは、前記定義の通りである。)
で表されるジケトン体を得、得られたジケトン体とヒドロキシルアミンを反応させることにより、下記一般式(7a’)
【0020】
【化7】
【0021】
(式中、R2、R3、R4’、R5’、R6〜R11及びArは、前記定義の通りである。)
で表されるオキシム体を得、得られたオキシム体と下記一般式(8)
【0022】
【化8】
【0023】
(式中、Yは、ハロゲン原子又は−OC(=O)R1を示す。
また、R1は、水素原子、ハロゲン原子、置換もしくは無置換の炭素数1〜20のアルキル基、置換もしくは無置換の炭素数2〜20のアルケニル基、置換もしくは無置換の環形成原子数3〜10のシクロアルキル基、置換もしくは無置換の炭素数4〜20のシクロアルケニル基、ヒドロキシル基、置換もしくは無置換の炭素数1〜20のアルコキシ基、置換もしくは無置換の炭素数2〜20のアルケニルオキシ基、置換もしくは無置換の炭素数1〜20のアルカノイル基、置換もしくは無置換の炭素数2〜20のアルケノイル基、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数3〜14のヘテロ環基を示す。)
で表されるエステル化剤とを反応させることによる、下記一般式(I−a1)
【0024】
【化9】
【0025】
(式中、R1〜R3、R4’、R5’、R6〜R11及びArは、前記定義の通りである。)
で表される光重合開始剤の製造方法。
【発明の効果】
【0026】
本発明の光重合開始剤は、光(特に450nm以下の短波長の光であって、例えば波長365nmや波長405nmの光)に対する感度が非常に高いため、薄膜化することが可能であり、低コストで高品質のパターンを形成することができる。さらに、エチレン性不飽和二重結合を有する化合物、エーテル結合及び/又はエステル結合を有する25℃で液体の化合物との相溶性、及び溶剤(前記のエーテル結合及び/又はエステル結合を有する25℃で液体の化合物を含む。)に対する溶解性が高いため、無溶剤の光硬化性組成物を調製することもでき、また、カラーフィルタ用の光硬化性組成物においては、光重合開始剤の添加量の選択幅が広く、結果として、感度、解像度、現像性及び深部硬化性が向上する。
従来、光重合開始剤に種々の置換基を付与することにより分子量を高めながら溶剤への溶解性が高められてきたが、本発明の光重合開始剤は、比較的分子量が小さく、光硬化性組成物への添加量を低減できるうえ、従来の光重合開始剤よりもエチレン性不飽和二重結合を有する化合物、エーテル結合及び/又はエステル結合を有する25℃で液体の化合物との相溶性、及び溶剤(前記のエーテル結合及び/又はエステル結合を有する25℃で液体の化合物を含む。)に対する溶解性が高く、感度、解像度及び現像性、並びに基板との密着性に優れる。
【図面の簡単な説明】
【0027】
【図1】本発明の液晶表示装置を示す模式図である。
【発明を実施するための形態】
【0028】
[光重合開始剤]
本発明の光重合開始剤は、下記一般式(I)で表される。
【化10】
(式中、R1〜R11は、それぞれ独立に、水素原子、ハロゲン原子、置換もしくは無置換の炭素数1〜20のアルキル基、置換もしくは無置換の炭素数2〜20のアルケニル基、置換もしくは無置換の環形成原子数3〜10のシクロアルキル基、置換もしくは無置換の炭素数4〜20のシクロアルケニル基、ヒドロキシル基、置換もしくは無置換の炭素数1〜20のアルコキシ基、置換もしくは無置換の炭素数2〜20のアルケニルオキシ基、置換もしくは無置換の炭素数1〜20のアルカノイル基、置換もしくは無置換の炭素数2〜20のアルケノイル基、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数3〜14のヘテロ環基を示す。R3は、R4又はR5と一緒になって環を形成していてもよい。R4は、R5と一緒になって環を形成していてもよい。
また、Arは、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数5〜14のヘテロアリール基を示す。
Wは、単結合又は酸素原子を示す。Zは、単結合、酸素原子又は>NR3’(R3’は、置換もしくは無置換の炭素数1〜20のアルキル基を示すか、又はR3’はR3とつながって、窒素原子と共に環を形成している。)を示す。
nは、1〜10の整数を示す。nが2〜10の整数の場合、複数のR4及びR5は、それぞれ同一でも異なっていてもよい。)
【0029】
また、一般式(I)で表される本発明の光重合開始剤の中でも、本発明の効果の観点から、下記一般式(I−a)で表される光重合開始剤(W:単結合、Z:酸素原子の場合に相当する。)及び下記一般式(I−b)で表される光重合開始剤(nが1の場合に相当する。)が好ましい。
【0030】
【化11】
【0031】
上記式中、R1〜R11は、それぞれ独立に、水素原子、ハロゲン原子、置換もしくは無置換の炭素数1〜20のアルキル基、置換もしくは無置換の炭素数2〜20のアルケニル基、置換もしくは無置換の環形成原子数3〜10のシクロアルキル基、置換もしくは無置換の炭素数4〜20のシクロアルケニル基、ヒドロキシル基、置換もしくは無置換の炭素数1〜20のアルコキシ基、置換もしくは無置換の炭素数2〜20のアルケニルオキシ基、置換もしくは無置換の炭素数1〜20のアルカノイル基、炭素数2〜20のアルケノイル基、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数3〜14のヘテロ環基を示す。なお、一般式(I−a)においては、R3は、R4又はR5と一緒になって環を形成していてもよく、また、R4は、R5と一緒になって環を形成していてもよい。
また、Arは、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数5〜14のヘテロアリール基を示す。
一般式(I−b)中のWは、単結合又は酸素原子を示す。また、一般式(I−b)中のZは、単結合、酸素原子又は>NR3’(R3’は、置換もしくは無置換の炭素数1〜20のアルキル基を示すか、又はR3’はR3とつながって、窒素原子と共に環を形成している。)を示す。
一般式(I−a)中のnは、1〜10の整数を示す。nが2〜10の整数の場合、複数のR4及びR5は、それぞれ同一でも異なっていてもよい。
【0032】
(一般式(I)、(I−a)及び(I−b)中の各基について)
nとしては、低分子量とする観点、エチレン性不飽和二重結合を有する化合物との相溶性(以下、単に相溶性と称する。)や溶剤への溶解性(以下、単に溶解性と称する。)の観点及び製造容易性の観点から、2〜8の整数が好ましく、2〜4の整数がより好ましく、2がさらに好ましい。
R1〜R11がそれぞれ独立して示すハロゲン原子としては、例えばフッ素原子、塩素原子、臭素原子、ヨウ素原子等が挙げられる。
R1〜R11がそれぞれ独立して示す炭素数1〜20のアルキル基としては、直鎖状でも分岐鎖状でもよく、例えばメチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、t−ブチル基、n−ペンチル基、イソペンチル基、n−ヘキシル基、n−オクチル基、2−エチル−n−オクチル基、n−デシル基、n−ドデシル基等が挙げられる。これらの中でも、低分子量とする観点、相溶性や溶解性の観点及び製造容易性の観点から、炭素数1〜10のアルキル基が好ましく、炭素数1〜5のアルキル基がより好ましい。
R1〜R11がそれぞれ独立して示す炭素数2〜20のアルケニル基としては、直鎖状でも分岐鎖状でもよく、例えばビニル基、アリル基、7−オクテニル基等が挙げられる。これらの中でも、低分子量とする観点、相溶性や溶解性の観点及び製造容易性の観点から、炭素数2〜10のアルケニル基が好ましく、炭素数2〜6のアルケニル基がより好ましい。
R1〜R11がそれぞれ独立して示す環形成原子数3〜10のシクロアルキル基としては、シクロプロピル基、シクロペンチル基、シクロヘキシル基、シクロオクチル基等が挙げられる。これらの中でも、低分子量とする観点、相溶性や溶解性の観点及び製造容易性の観点から、環形成原子数3〜6のシクロアルキル基が好ましい。
R1〜R11がそれぞれ独立して示す炭素数4〜20のシクロアルケニル基としては、シクロペンテニル基、シクロヘキセニル基、シクロヘキサジエニル基、シクロオクテニル基等が挙げられる。これらの中でも、低分子量とする観点、相溶性や溶解性の観点及び製造容易性の観点から、炭素数4〜10のシクロアルケニル基が好ましく、炭素数4〜6のシクロアルケニル基がより好ましい。
【0033】
R1〜R11がそれぞれ独立して示す炭素数1〜20のアルコキシ基としては、アルキル基部位が、前記した炭素数1〜20のアルキル基であるものが挙げられる。これらの中でも、低分子量とする観点、相溶性や溶解性の観点及び製造容易性の観点から、炭素数1〜10のアルコキシ基が好ましく、炭素数1〜5のアルコキシ基がより好ましい。
R1〜R11がそれぞれ独立して示す炭素数2〜20のアルケニルオキシ基としては、アルケニル基部位が、前記した炭素数2〜20のアルケニル基であるものが挙げられる。これらの中でも、低分子量とする観点、相溶性や溶解性の観点及び製造容易性の観点から、炭素数2〜10のアルケニルオキシ基が好ましく、炭素数2〜6のアルケニルオキシ基がより好ましい。
R1〜R11がそれぞれ独立して示す炭素数1〜20のアルカノイル基としては、直鎖状でも分岐鎖状でもよく、例えばメタノイル基、エタノイル基、n−プロパノイル基、イソプロパノイル基、n−ブタノイル基、t−ブタノイル基、n−ヘキサノイル基、n−オクタノイル基、n−デカノイル基、n−ドデカノイル基等が挙げられる。これらの中でも、低分子量とする観点、相溶性や溶解性の観点及び製造容易性の観点から、炭素数1〜10のアルカノイル基が好ましく、炭素数1〜5のアルカノイル基がより好ましい。
R1〜R11がそれぞれ独立して示す炭素数2〜20のアルケノイル基としては、直鎖状でも分岐鎖状でもよく、例えばエテノイル基、n−プロペノイル基、イソプロペノイル基、n−ブテノイル基、t−ブテノイル基、n−ヘキセノイル基、n−オクテノイル基、n−デセノイル基、n−ドデセノイル基等が挙げられる。これらの中でも、低分子量とする観点、相溶性や溶解性の観点及び製造容易性の観点から、炭素数2〜10のアルケノイル基が好ましく、炭素数2〜6のアルケノイル基がより好ましい。
【0034】
R1〜R11がそれぞれ独立して示す環形成炭素数6〜14のアリール基としては、例えばフェニル基、ナフチル基、アントリル基が挙げられる。
R1〜R11がそれぞれ独立して示す環形成原子数3〜14のヘテロ環基としては、例えば2−フラニル基、2−チオフェニル基、2−ピリジニル基、下記式(A)で表される基(以下、置換基(A)と称する。)
【化12】
等の環形成原子数5〜14の不飽和ヘテロ環基;2−テトラヒドロフリル基、3−テトラヒドロフリル基、ピロリジニル基、ピペリジニル基、2,2,6,6−トリメチルピペリジン−4−イル基
等の環形成原子数3〜10の飽和ヘテロ環基が挙げられる。これらの中でも、低分子量とする観点、相溶性や溶解性の観点及び製造容易性の観点から、環形成原子数5〜6の不飽和ヘテロ環基、環形成原子数3〜6の飽和ヘテロ環基が好ましい。
【0035】
また、前記した「R3とR4又はR5が一緒になって環を形成する」というのは、n=2、Wが単結合及びZが酸素原子である場合を例に挙げると下記式で説明され、右側に例示した環が具体例として挙げられる。
【化13】
【0036】
R4とR5が一緒になって形成する環としては、例えば、シクロペンチル環、シクロオクチル環等の環形成炭素数3〜10(好ましくは3〜6)の環が挙げられる。
【0037】
なお、R1〜R11がそれぞれ独立して示す、前記アルキル基、アルケニル基、シクロアルキル基、シクロアルケニル基、アルコキシ基、アルケニルオキシ基、アルカノイル基、アルケノイル基、アリール基及びヘテロ環基は、置換基を有していてもよい。
R1〜R11がそれぞれ独立して示すアルキル基、アルケニル基の置換基としては、ヒドロキシル基;カルボキシル基;メトキシ基、エトキシ基、プロポキシ基等の直鎖状又は分岐鎖状の炭素数1〜18(好ましくは1〜10、より好ましくは1〜5)のアルコキシ基;直鎖状又は分岐鎖状の炭素数2〜18(好ましくは2〜10、より好ましくは2〜6)のアルケニルオキシ基;直鎖状又は分岐鎖状の炭素数2〜18(好ましくは2〜10、より好ましくは2〜6)のアルケニルチオ基;直鎖状又は分岐鎖状の炭素数1〜18(好ましくは1〜10、より好ましくは1〜5)のアルキルチオ基;炭素数5〜18(好ましくは5〜10)のシクロアルケニル基;シクロプロピル基、シクロペンチル基、シクロヘキシル基等の炭素数3〜18(好ましくは3〜10、より好ましくは3〜6)のシクロアルキル基;フェノキシ基等の環形成炭素数6〜10のアリールオキシ基;フッ素原子、塩素原子、臭素原子、ヨウ素原子等のハロゲン原子;酸素原子(=O);硫黄原子(=S);シアノ基;ニトロ基;トリメチルシリル基等のトリアルキルシリル基;トリメトキシシリル基等のトリアルコキシシリル基;フェニル基、ナフチル基、アントリル基等の環形成炭素数6〜14(好ましくは6〜10)のアリール基;−COR12;−OCOR13;−NR14R15;−NHCOR16;−NHCOOR17;−CONR18R19;−COOR20;−SO3NR21R22;−SO3R23;エポキシ基やテトラヒドロフラニル基等の環形成原子数3〜6の環状エーテル基、2−チエニル基、2−ピリジル基、フリル基、チアゾリル基、ベンゾチアゾリル基、モルホリノ基、前記置換基(A)等の飽和もしくは不飽和の環形成原子数3〜10(好ましくは3〜6)のヘテロ環基等が挙げられる。
上記一般式中、R12〜R23は、それぞれ独立に、炭素数1〜20の直鎖状又は分岐鎖状のアルキル基、環形成炭素数6〜14のアリール基を示す。該アルキル基、アリール基としては、R1〜R11の場合と同じものが挙げられる。
【0038】
R1〜R11がそれぞれ独立して示すアルコキシ基、アルケニルオキシ基、アルカノイル基、アルケノイル基の置換基としては、ヒドロキシル基;カルボキシル基;メトキシ基、エトキシ基、プロポキシ基等の直鎖状又は分岐鎖状の炭素数1〜18(好ましくは1〜10、より好ましくは1〜5)のアルコキシ基;直鎖状又は分岐鎖状の炭素数2〜18(好ましくは2〜10、より好ましくは2〜6)のアルケニルオキシ基;直鎖状又は分岐鎖状の炭素数2〜18(好ましくは2〜10、より好ましくは2〜6)のアルケニルチオ基;直鎖状又は分岐鎖状の炭素数1〜18(好ましくは1〜10、より好ましくは1〜5)のアルキルチオ基;炭素数5〜18(好ましくは5〜10)のシクロアルケニル基;シクロプロピル基、シクロペンチル基、シクロヘキシル基等の炭素数3〜18(好ましくは3〜10、より好ましくは3〜6)のシクロアルキル基;フェノキシ基等の環形成炭素数6〜10のアリールオキシ基;フッ素原子、塩素原子、臭素原子、ヨウ素原子等のハロゲン原子;酸素原子(=O);硫黄原子(=S);シアノ基;ニトロ基;トリメチルシリル基等のトリアルキルシリル基;トリメトキシシリル基等のトリアルコキシシリル基;フェニル基、ナフチル基、アントリル基等の環形成炭素数6〜14(好ましくは6〜10)のアリール基;−COR12;−OCOR13;−NR14R15;−NHCOR16;−NHCOOR17;−CONR18R19;−COOR20;−SO3NR21R22;−SO3R23;エポキシ基やテトラヒドロフラニル基等の環形成原子数3〜6の環状エーテル基、2−チエニル基、2−ピリジル基、フリル基、チアゾリル基、ベンゾチアゾリル基、モルホリノ基、前記置換基(A)等の飽和もしくは不飽和の環形成原子数3〜10(好ましくは3〜6)のヘテロ環基等が挙げられる(式中、R12〜R23は、前記定義の通りである。)。
【0039】
R1〜R11がそれぞれ独立して示すシクロアルキル基、シクロアルケニル基の置換基としては、ヒドロキシル基;カルボキシル基;メチル基、エチル基等の直鎖又は分岐状の炭素数1〜18(好ましくは1〜10、より好ましくは1〜5)のアルキル基;メトキシ基、エトキシ基、プロポキシ基等の直鎖状又は分岐鎖状の炭素数1〜18(好ましくは1〜10、より好ましくは1〜5)のアルコキシ基;直鎖状又は分岐鎖状の炭素数2〜18(好ましくは2〜10、より好ましくは2〜6)のアルケニルオキシ基;直鎖状又は分岐鎖状の炭素数2〜18(好ましくは2〜10、より好ましくは2〜6)のアルケニルチオ基;ビニル基、アリル基等の炭素数2〜18(好ましくは2〜10、より好ましくは2〜6)の直鎖又は分岐状のアルケニル基;直鎖状又は分岐鎖状の炭素数1〜18(好ましくは1〜10、より好ましくは1〜5)のアルキルチオ基;炭素数5〜18(好ましくは5〜10)のシクロアルケニル基;フェノキシ基等の環形成炭素数6〜10のアリールオキシ基;フッ素原子、塩素原子、臭素原子、ヨウ素原子等のハロゲン原子;酸素原子(=O);硫黄原子(=S);シアノ基;ニトロ基;トリメチルシリル基等のトリアルキルシリル基;トリメトキシシリル基等のトリアルコキシシリル基;フェニル基、ナフチル基、アントリル基等の環形成炭素数6〜14(好ましくは6〜10)のアリール基;−COR12;−OCOR13;−NR14R15;−NHCOR16;−NHCOOR17;−CONR18R19;−COOR20;−SO3NR21R22;−SO3R23;エポキシ基やテトラヒドロフラニル基等の環形成原子数3〜6の環状エーテル基、2−チエニル基、2−ピリジル基、フリル基、チアゾリル基、ベンゾチアゾリル基、モルホリノ基、前記置換基(A)等の飽和もしくは不飽和の環形成原子数3〜10(好ましくは3〜6)のヘテロ環基等が挙げられる(式中、R12〜R23は、前記定義の通りである。)。
【0040】
R1〜R11がそれぞれ独立して示すアリール基、ヘテロ環基の置換基としては、ヒドロキシル基;カルボキシル基;メチル基、エチル基等の直鎖又は分岐状の炭素数1〜18(好ましくは1〜10、より好ましくは1〜5)のアルキル基;メトキシ基、エトキシ基、プロポキシ基等の直鎖状又は分岐鎖状の炭素数1〜18(好ましくは1〜10、より好ましくは1〜5)のアルコキシ基;直鎖状又は分岐鎖状の炭素数2〜18(好ましくは2〜10、より好ましくは2〜6)のアルケニルオキシ基;直鎖状又は分岐鎖状の炭素数2〜18(好ましくは2〜10、より好ましくは2〜6)のアルケニルチオ基;直鎖状又は分岐鎖状の炭素数1〜18(好ましくは1〜10、より好ましくは1〜5)のアルキルチオ基;ビニル基、アリル基等の直鎖状又は分岐鎖状の炭素数2〜18(好ましくは2〜10、より好ましくは2〜6)のアルケニル基;炭素数5〜18(好ましくは5〜10)のシクロアルケニル基;シクロプロピル基、シクロペンチル基、シクロヘキシル基等の炭素数3〜18(好ましくは3〜10、より好ましくは3〜6)のシクロアルキル基;フェノキシ基等の環形成炭素数6〜10のアリールオキシ基;フッ素原子、塩素原子、臭素原子、ヨウ素原子等のハロゲン原子;シアノ基;ニトロ基;トリメチルシリル基等のトリアルキルシリル基;トリメトキシシリル基等のトリアルコキシシリル基;−COR12;−OCOR13;−NR14R15;−NHCOR16;−NHCOOR17;−CONR18R19;−COOR20;−SO3NR21R22;−SO3R23;エポキシ基やテトラヒドロフラニル基等の環形成原子数3〜6の環状エーテル基、2−チエニル基、2−ピリジル基、フリル基、チアゾリル基、ベンゾチアゾリル基、モルホリノ基、前記置換基(A)等の飽和もしくは不飽和の環形成原子数3〜10(好ましくは3〜6)のヘテロ環基等が挙げられる(式中、R12〜R23は、前記定義の通りである。)。
【0041】
また、Arが示す環形成炭素数6〜14のアリール基としては、例えばフェニル基、ナフチル基、アントリル基、クリセニル基、フェナントレニル基、アズレニル基、アセナフチレニル基等が挙げられる。これらの中でも、低分子量とする観点、相溶性や溶解性の観点及び製造容易性の観点から、環形成炭素数6〜10のアリール基が好ましく、フェニル基がさらに好ましい。Arが示す環形成原子数5〜14のヘテロアリール基としては、例えば2−フラニル基、2−チオフェニル基、2−ピリジニル基等が挙げられる。これらの中でも、低分子量とする観点、相溶性や溶解性の観点及び製造容易性の観点から、環形成原子数5〜6のへテロアリール基が好ましく、2−フラニル基、2−チオフェニル基がより好ましい。
これらのアリール基及びヘテロアリール基は置換基を有していてもよい。該置換基としては、前記したR1〜R11がそれぞれ独立して示すアリール基の置換基と同じものが挙げられ、それらの中でも、炭素数1〜18(好ましくは1〜10、より好ましくは1〜5)のアルキル基が好ましく、メチル基がより好ましい。
なお、該置換基がアルキル基又はアルケニル基である場合、Arと共に縮合環を形成していてもよく、例えばフルオレン環、インデン環等を形成していてもよい。
【0042】
Zが表す>NR3’中のR3’は、前述の通り、置換もしくは無置換の炭素数1〜20のアルキル基を示すか、又はR3’はR3とつながって、窒素原子と共に環を形成している。
該炭素数1〜20のアルキル基としては、R1〜R11の場合と同じものが挙げられ、好ましいものも同じものが挙げられ、メチル基が特に好ましい。
また、R3’はR3とつながって、窒素原子と共に形成する環の具体例としては、モルホリン環、ピロリジン環、ピペリジン環、ピペコリン環、ピペラジン環などが挙げられる。これらの中でも、モルホリン環が好ましい。
WとZの組み合わせとしては、Wが単結合の場合、Zは酸素原子又は>Nr3’が好ましく、Wが酸素原子の場合、Zは単結合又は酸素原子が好ましい。
【0043】
さらに、一般式(I−a)において、(a)R1、R2及びR3が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、nが2であり、複数のR4及びR5が、それぞれ独立に、水素原子又は置換もしくは無置換の炭素数1〜20のアルキル基であり、Arが、置換もしくは無置換の環形成炭素数6〜14のアリール基である光重合開始剤、(b)R1、R2及びR3が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、nが2であり、複数のR4及びR5が、それぞれ独立に、水素原子又は置換もしくは無置換の炭素数1〜20のアルキル基であり、Arが、置換もしくは無置換の環形成炭素数5〜14のヘテロアリール基である光重合開始剤、(c)R1及びR2が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、R3が置換もしくは無置換の炭素数3〜18のシクロアルキル基であり、nが2であり、複数のR4及びR5が、それぞれ独立に、水素原子又は置換もしくは無置換の炭素数1〜20のアルキル基である光重合開始剤、(d)R1及びR2が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、R3が環形成原子数3〜6の環状エーテル基で置換された炭素数1〜20のアルキル基であり、nが2であり、複数のR4及びR5が、それぞれ独立に、水素原子又は置換もしくは無置換の炭素数1〜20のアルキル基である光重合開始剤も好ましい。さらに、後述する光硬化性組成物における光重合開始剤の含有量の低減の観点から、上記(a)及び(b)の場合は、分子量が515以下であることが好ましく、500以下であることがより好ましく、上記(c)及び(d)の場合は、分子量が550以下であることが好ましい。
さらに、本発明の効果の観点、及び光重合開始剤の分解物による重合物の汚染や装置の汚染を低減する観点から、上記の好ましい光重合開始剤において、R4〜R11がそれぞれ独立に、水素原子又は炭素数1〜20のアルキル基であることがより好ましい。なお、各基の好ましいものは、前記した通りである。
以下に、本発明の光重合開始剤(I−a)の具体例を示すが、特にこれらに制限されるものではない。
【0044】
【化14】
【0045】
【化15】
【0046】
【化16】
【0047】
【化17】
【0048】
さらに、一般式(I−b)において、(e)R1及びR3が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、Wが酸素原子であり、Zが単結合又は酸素原子であり、Arが、置換もしくは無置換の環形成炭素数6〜14のアリール基である光重合開始剤、(f)R1及びR3が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、R2が、−OCOR13(R13は、前記定義の通りである。)又は−COOR20(R20は、炭素数1〜20のアルキル基又は環形成炭素数6〜14のアリール基を示す。)で表されるエステル基を有する炭素数1〜20のアルキル基であり、Wが酸素原子であり、Zが単結合又は酸素原子であり、Arが、置換もしくは無置換の環形成炭素数6〜14のアリール基である光重合開始剤、(g)R1及びR3が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、Wが単結合であり、Zが>NR3’(R3’は、前記定義のとおりである。)であり、Arが、置換もしくは無置換の環形成炭素数6〜14のアリール基である光重合開始剤、(h)R1及びR3が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、R2が、−OCOR13(R13は、前記定義の通りである。)又は−COOR20(R20は、炭素数1〜20のアルキル基又は環形成炭素数6〜14のアリール基を示す。)で表されるエステル基を有する炭素数1〜20のアルキル基であり、Wが単結合であり、Zが>NR3’(R3’は、前記定義のとおりである。)であり、Arが、置換もしくは無置換の環形成炭素数6〜14のアリール基である光重合開始剤、(i)R1が、置換もしくは無置換の炭素数1〜20のアルキル基であり、R3が水素原子であり、Wが単結合であり、Zが酸素原子であり、Arが、置換もしくは無置換の環形成炭素数6〜14のアリール基である光重合開始剤も好ましい。
なお、上記(g)の光重合開始剤においては、>NR3’中のR3’はR3とつながって、窒素原子と共に環を形成しているのも好ましく、モルホリン環を形成しているのがより好ましい。
さらに、本発明の効果の観点、及び光重合開始剤の分解物による重合物の汚染や装置の汚染を低減する観点から、上記の好ましい光重合開始剤において、R4〜R11がそれぞれ独立に、水素原子又は炭素数1〜20のアルキル基であることがより好ましい。なお、各基の好ましいものは、前記した通りである。
以下に、本発明の光重合開始剤(I−b)の具体例を示すが、特にこれらに制限されるものではない。
【0049】
【化18】
【0050】
【化19】
【0051】
なお、本発明の光重合開始剤としては、前述の通り一般式(I−a)においてn=2であることが好ましく、具体的には下記一般式(I−a1)で表される光重合開始剤が好ましい。
【化20】
上記式(I−a1)中、R1〜R3、R6〜R11及びArは、前記定義の通りである。R4’及びR5’は、それぞれR4及びR5の定義と同じであり、好ましいものも同じである。
【0052】
[光重合開始剤の製造方法]
本発明の光重合開始剤(I)の製造方法に特に制限は無いが、例えば、以下の光重合開始剤(I−a)の製造方法や光重合開始剤(I−b)の製造方法によって容易に製造することができる。
(1.光重合開始剤(I−a)の製造方法)
本発明の光重合開始剤(I−a)の製造方法に特に制限は無いが、例えば、下記工程1〜工程4によって、容易に製造することができる。
【0053】
(工程1)
工程1としては、例えば、前記カルバゾール誘導体(1)とZ(CR4R5)nCOOR3(Zは、フッ素原子、塩素原子、臭素原子、ヨウ素原子等のハロゲン原子を示す。また、R3、R4及びR5は、前記定義の通りである。)で表されるハロゲン化物とを、塩基の存在下に反応させる工程が挙げられる。該工程により、下記一般式(3a)で表されるカルボニルアルキル基導入体が得られる。
【化21】
(式中、R3〜R11は、前記定義の通りであり、好ましいものも同じである。)
前記塩基としては、n−ブチルリチウム、t−ブチルリチウム、水酸化ナトリウム、水酸化カリウム等が挙げられる。反応温度は、通常、好ましくは0〜200℃であり、反応時間は通常、好ましくは2〜100時間である。
【0054】
なお、一般式(I−a)においてn=2の光重合開始剤を製造する場合、工程1は、以下の工程であると製造が容易であり、好ましい。
下記一般式(1)
【化22】
(式中、R6〜R11は、前記定義の通りであり、好ましいものも同じである。)
で表されるカルバゾール誘導体[以下、カルバゾール誘導体(1)と称する。]と、下記一般式(2a)
【0055】
【化23】
(式中、R3、R4’及びR5’は、前記定義の通りであり、好ましいものも同じである。)
で表されるアクリレート誘導体[以下、アクリレート誘導体(2a)と称する。]とを塩基の存在下に反応させることにより、下記一般式(3a’)
【0056】
【化24】
(式中、R3、R4’、R5’及びR6〜R11は、前記定義の通りであり、好ましいものも同じである。)
で表されるカルボニルアルキル基導入体[以下、カルボニルアルキル基導入体(3a’)と称する。]を得る工程。
以下、n=2の光重合開始剤を製造する場合に好ましいとするこの工程1について、詳細に説明する。
【0057】
工程1で使用するアクリレート誘導体(2a)の使用量に特に制限は無いが、カルバゾール誘導体(1)1モルに対して、好ましくは0.5〜2モル、より好ましくは0.8〜1.2モルであり、未反応物を低減する観点から、ほぼ等量で反応させることがさらに好ましい。
工程1は、塩基の存在下に実施する。該塩基としては、Michael付加反応に使用し得る塩基であればよく、有機塩基、無機塩基のいずれも用いることができる。有機塩基としては、例えばピリジン、ジアザビシクロウンデセン(DBU)、ジアザビシクロノネン(DBN)等が挙げられる。無機塩基としては、例えば炭酸ナトリウム、炭酸カリウム等のアルカリ金属炭酸塩;炭酸マグネシウム、炭酸カルシウム等のアルカリ土類金属炭酸塩;炭酸水素ナトリウム、炭酸水素カリウム等のアルカリ金属炭酸水素塩;水酸化ナトリウム、水酸化カリウム等のアルカリ金属水酸化物;水酸化マグネシウム、水酸化カルシウム等のアルカリ土類金属水酸化物等が挙げられる。これらの中でも、アルカリ金属炭酸塩、アルカリ土類金属炭酸塩が好ましく、アルカリ金属炭酸塩がより好ましく、炭酸カリウムがさらに好ましい。
該塩基の使用量に特に制限は無いが、反応効率及び製造コストの観点から、カルバゾール誘導体(1)1モルに対して、好ましくは0.5〜2モル、より好ましくは0.8〜1.2モルであり、ほぼ等量で使用することがさらに好ましい。
工程1は、溶媒の存在下に実施することが好ましい。該溶媒としては、カルバゾール誘導体(1)、アクリレート誘導体(2a)及び塩基を溶解し得る溶媒を適宜選択すればよい。具体的には、ジメチルホルムアミド(DMF)、ジメチスルホキシド(DMSO)、塩化メチレン等が挙げられる。
【0058】
工程1の反応温度に特に制限は無いが、通常、好ましくは10〜50℃、より好ましくは15〜40℃、さらに好ましくは15〜30℃で実施する。反応時間は、カルバゾール誘導体(1)、アクリレート誘導体(2a)及び塩基の種類や使用量並びに反応温度等によっても異なるが、通常、2時間〜48時間程度である。
工程1の実施形態に特に制限は無く、例えばカルバゾール誘導体(1)、アクリレート誘導体(2a)及び塩基を全て溶媒中に添加し、好ましくは前記温度で攪拌することにより、カルボニルアルキル基導入体(3a’)を得ることができる。
【0059】
反応終了後、得られた反応混合液から、抽出等の通常の有機化合物の分離手段によって、カルボニルアルキル基導入体(3a)や(3a’)を得ることができる。適宜、蒸留、カラムクロマトグラフィー、再結晶等の、通常の有機化合物の精製手段によって精製することにより、カルボニルアルキル基導入体(3a)や(3a’)の純度を高めることもできる。
【0060】
以下、前記工程1で得られるカルボニルアルキル基導入体(3a)又は(3a’)を用いた工程2〜4について説明するが、便宜上、n=2に相当するカルボニルアルキル基導入体(3a’)については省略して記載する。
(工程2)
工程2は、工程1で得られたカルボニルアルキル基導入体(3a)と下記一般式(4)、(5)
【化25】
(式中、Xは、ハロゲン原子又は−OC(=O)Arを示し、X’は、ハロゲン原子又は−OC(=O)R2を示す。Ar及びR2は、前記定義の通りであり、好ましいものも同じである。)
で表される2つのアシル化剤[以下、それぞれアシル化剤(4)、アシル化剤(5)と称する。]とをルイス酸の存在下に反応させることにより、下記一般式(6a)
【0061】
【化26】
(式中、R2〜R11及びArは、前記定義の通りであり、好ましいものも同じである。)
で表されるジケトン体[以下、ジケトン体(6a)と称する。]を得る工程である。
【0062】
上記式中、X、X’がそれぞれ独立に示すハロゲン原子としては、フッ素原子、塩素原子、臭素原子、ヨウ素原子等が挙げられ、塩素原子が好ましい。
工程2において、カルボニルアルキル基導入体(3a)と、アシル化剤(4)及び(5)との反応は、目的とするジケトン体(6a)の収率の観点から、カルボニルアルキル基導入体(3a)とアシル化剤(4)とを反応させた後、次いでアシル化剤(5)を反応させることが好ましい。
アシル化剤(4)及び(5)の使用量は、カルボニルアルキル基導入体(3a)にそれぞれのアシル基を1つずつ導入する観点から、カルボニルアルキル基導入体(3a)1モルに対して、それぞれ好ましくは0.8〜1.3モル、より好ましくは1〜1.3モルであり、未反応物を低減する観点から、ほぼ等量で反応させることがさらに好ましい。特に、カルボニルアルキル基導入体(3a)とアシル化剤(4)とを反応させた後、次いでアシル化剤(5)を反応させる場合には、アシル化剤(5)の使用量は、前記範囲を超えても何ら問題ないが、あまり過剰であってもそれに見合う収率が得られるわけではなく、いたずらに生産コストがかさむことになり得る。
工程2は、ルイス酸の存在下に実施する。ルイス酸としては、塩化アルミニウム、三フッ化ホウ素ジエチルエーテル錯体が好ましい。ルイス酸の使用量は、ジケトン体(6a)の収率の観点から、アシル化剤(4)又は(5)1モルに対して、通常、好ましくは0.8〜2.5モル、より好ましくは1〜2モルである。
【0063】
工程2は、溶媒の存在下に実施することが好ましい。該溶媒としては、通常のFriedel−Craftsアシル化反応に使用し得る溶媒であれば特に制限は無い。具体的には、ジクロロメタン、ニトロベンゼン、アセトン、アセトニトリル等が挙げられる。
工程2の反応温度は、反応開始時は−50〜5℃(好ましくは−10〜5℃)とし、反応が進行するにしたがって、徐々に室温(15〜25℃程度)へ戻すことが好ましい。反応時間は、カルボニルアルキル基導入体(3a)、アシル化剤(4)及び(5)の種類及び使用量、並びに反応温度によっても異なるが、通常、好ましくは1〜30時間である。なお、アシル化剤(4)及び(5)の両方がハロゲン化アシル又は酸無水物である場合、カルボニルアルキル基導入体(3a)とアシル化剤(4)とを反応させた後、生成物を単離することなく引き続きアシル化剤(5)と反応させることもできる。その場合、カルボニルアルキル基導入体(3a)とアシル化剤(4)との反応時間は、30分〜5時間程度(好ましくは30分〜3時間)とし、一方、カルボニルアルキル基導入体(3a)とアシル化剤(5)との反応時間を、30分〜24時間程度(好ましくは30分〜18時間)と長めに設けて十分に反応させることが好ましい。
【0064】
収率の観点から、工程2の好ましい実施形態を以下に2通り説明する。
[1]氷浴で冷却しながらカルボニルアルキル基導入体(3a)及びアシル化剤(4)を適宜溶媒中で混合し、その混合溶液へルイス酸をゆっくり(好ましくは5分〜1時間、より好ましくは10分〜40分かけて)添加し、添加終了後、室温へ戻して一定時間(30分〜5時間程度)攪拌を続ける。好ましくは、抽出及び洗浄等の通常の有機化合物の分離手段を行うことによって、一旦、生成物を分離取得する。
こうして得られた生成物を適宜溶媒に溶解し、氷浴で冷却しながらアシル化剤(5)を添加し、そこへルイス酸をゆっくり(好ましくは5分〜1時間、より好ましくは10分〜40分かけて)添加し、添加終了後、室温へ戻して一定時間(30分〜24時間程度)攪拌を続けることにより、ジケトン体(6a)を得ることができる。
[2]氷浴で冷却しながらカルボニルアルキル基導入体(3a)を適宜溶媒と混合してから、ルイス酸をゆっくり(好ましくは5分〜1時間、より好ましくは10分〜40分かけて)添加し、得られた混合溶液へアシル化剤(4)をゆっくり(好ましくは5分〜1時間、より好ましくは10分〜40分かけて)添加した後、室温へ戻して一定時間(30分〜5時間程度)攪拌を続ける。再び氷浴で冷却しながらルイス酸をゆっくり(好ましくは5分〜1時間、より好ましくは10分〜40分かけて)添加し、得られた混合溶液へアシル化剤(5)をゆっくり(好ましくは5分〜1時間、より好ましくは10分〜40分かけて)添加した後、室温へ戻して一定時間以上(好ましくは30分〜24時間程度)攪拌を続けることにより、ジケトン体(6a)を得ることができる。
なお、反応終了後、得られた反応混合液から、抽出等の通常の有機化合物の分離手段によって、ジケトン体(6a)を得ることができる。適宜、蒸留、カラムクロマトグラフィー、再結晶等の、通常の有機化合物の精製手段によって精製することにより、ジケトン体(6a)の純度を高めることもできる。
【0065】
(工程3)
工程3は、工程2で得られたジケトン体(6a)とヒドロキシルアミンを反応させることにより、下記一般式(7a)
【化27】
(式中、R2〜R11及びArは、前記定義の通りであり、好ましいものも同じである。)
で表されるオキシム体[以下、オキシム体(7a)と称する。]を得る工程である。
【0066】
工程3においては、ヒドロキシルアミン供給源としては、特に制限されるものではないが、塩化ヒドロキシルアミンが好ましい。該塩化ヒドロキシルアミンは、例えば水中で酢酸ナトリウム、等と反応させることにより、ヒドロキシルアミンの水溶液を得ることができる。
ヒドロキシルアミン(塩化ヒドロキシルアミン)の使用量は、ジケトン体(6a)1モルに対して、好ましくは0.8〜2モル、より好ましくは1〜1.5モル、さらに好ましくは1〜1.3モルであり、未反応物を低減する観点から、ほぼ等量で反応させることがさらに好ましい。
工程3は、溶媒の存在下に実施することが好ましい。溶媒としては、水溶性有機溶媒が好ましく、該水溶性有機溶媒としては、例えばメタノール、エタノール等のアルコール類、ジメチルホルムアミド(DMF)等が好ましい。
【0067】
工程3の反応温度に特に制限は無いが、オキシム体(7a)の収率の観点からは、通常、好ましくは40〜160℃、より好ましくは50〜140℃、さらに好ましくは70〜110℃である。反応時間は、ジケトン体(6a)の種類及び使用量、並びに反応温度によっても異なるが、通常、好ましくは2〜20時間、より好ましくは4〜12時間である。
工程3の実施形態に特に制限は無く、例えば、塩化ヒドロキシルアミンと酢酸ナトリウムを水中で混合してヒドロキシルアミンの水溶液を得ておき、そこへジケトン体(6a)及び溶媒を添加して、好ましくは前記温度範囲で攪拌することにより、オキシム体(7a)を得ることができる。
反応終了後、得られた反応混合液から、抽出等の通常の有機化合物の分離手段によって、オキシム体(7a)を得ることができる。適宜、蒸留、カラムクロマトグラフィー、再結晶等の、通常の有機化合物の精製手段によって精製することにより、オキシム体(7a)の純度を高めることもできる。
【0068】
(工程4)
工程4は、工程3で得られたオキシム体(7a)と下記一般式(8)
【化28】
(式中、Yは、ハロゲン原子又は−OC(=O)R1を示す。R1は、前記定義の通りであり、好ましいものも同じである。)
で表されるエステル化剤[以下、エステル化剤(8)と称する。]とを反応させることにより、下記一般式(I−a)
【0069】
【化29】
(式中、R1〜R11及びArは、前記定義の通りである。)
で表される光重合開始剤[以下、光重合開始剤(I−a)と称する。]を得る工程である。
【0070】
上記式中、Yが示すハロゲン原子としては、フッ素原子、塩素原子、臭素原子、ヨウ素原子等が挙げられ、塩素原子が好ましい。
工程4で使用するエステル化剤(8)の使用量に特に制限は無いが、オキシム体(7a)1モルに対して、好ましくは0.5〜2モル、より好ましくは0.8〜1.2モルであり、未反応物を低減する観点から、ほぼ等量で反応させることがさらに好ましい。
工程4は、反応を促進するために、塩基の存在下に実施してもよい。塩基としては、有機塩基や無機塩基が挙げられる。有機塩基としては、例えばトリエチルアミン、トリブチルアミン等のアミン類;ピリジン等の含窒素複素環式芳香族化合物等が挙げられる。無機塩基としては、例えば炭酸ナトリウム等のアルカリ金属炭酸塩;炭酸マグネシウム等のアルカリ土類金属炭酸塩;水酸化ナトリウム等のアルカリ金属水酸化物;水酸化マグネシウム等のアルカリ土類金属水酸化物等が挙げられる。これらの中でも、有機塩基が好ましく、アミン類、含窒素複素環式芳香族化合物がより好ましく、反応効率及び製造コストの観点から、トリエチルアミン、ピリジンがさらに好ましい。
塩基を使用する場合、その使用量は、光重合開始剤(I−a)の収率及び製造コストの観点から、オキシム体(7a)1モルに対して、好ましくは1〜5モル、より好ましくは1.5〜3モルである。
【0071】
工程4は、溶媒の存在下に実施することが好ましい。溶媒としては、t−ブチルメチルエーテル、エチルメチルエーテル、シクロペンチルメチルエーテル、テトラヒドロフラン(THF)等のエーテル類が好ましく挙げられる。
工程4の反応温度は、反応開始時は−50〜5℃(好ましくは−10〜5℃)とし、反応が進行するにしたがって、徐々に室温(15〜25℃程度)へ戻すことが好ましい。反応時間は、オキシム体(7a)、エステル化剤(8)の種類及び使用量、並びに反応温度によっても異なるが、通常、好ましくは0.5〜10時間、より好ましくは1〜5時間である。
工程4の実施形態に特に制限は無く、例えばオキシム体(7a)及びエステル化剤(8)を適宜溶媒中で混合し、そこへ塩基を滴下し、滴下終了後、反応液の温度を徐々に室温へ戻して攪拌を続けることにより、本発明の光重合開始剤(I−a)を得ることができる。
反応終了後、得られた反応混合液から、抽出等の通常の有機化合物の分離手段によって、光重合開始剤(I−a)を得ることができる。適宜、蒸留、カラムクロマトグラフィー、再結晶等の、通常の有機化合物の精製手段によって精製することにより、光重合開始剤(I−a)の純度を高めることもできる。
【0072】
こうして得られる本発明の光重合開始剤(I−a)は、カルバゾール骨格中の窒素原子に、エステル基を含有する特殊な置換基が付いている。そのため、光硬化性組成物中のエチレン性不飽和結合を有する化合物(特に(メタ)アクリロイルオキシ基を有する化合物)、エーテル結合及び/又はエステル結合を有する25℃で液体の化合物との相溶性、及び溶剤(前記のエーテル結合及び/又はエステル結合を有する25℃で液体の化合物を含む。)に対する溶解性が高く、光(特に波長450nm以下の短波長の光)に対する感度が高く、解像度及び現像性、ひいては深部硬化性に優れ、さらに基板との密着性が良好になるものと考えられる。また、該光重合開始剤(I−a)は、露光時の光により発生する分解物による重合物の汚染や装置の汚染が無い。
なお、特定化合物や溶剤に対する光重合開始剤(I−a)の相溶性や溶解性の高さは、感度、解像度及び現像性、ひいては深部硬化性の向上に寄与しているものと考えられ、カラーフィルタ用途においては重要な要素であると言える。特にカラーフィルタ用途においては、光重合開始剤が、エーテル結合及び/又はエステル結合を有する溶剤(特に、プロピレングリコールモノエチルエーテルアセテート等)100質量部に対して、5質量部以上溶解することが好ましく、10質量部以上溶解することがさらに好ましく、本発明の光重合開始剤(I−a)はかかる条件を満たしており、カラーフィルタ用光硬化性組成物に含有させる光重合開始剤、さらには高遮光性のブラックマトリックス用光硬化性組成物に含有させる光重合開始剤として適している。
【0073】
(1.光重合開始剤(I−a)の製造方法)
本発明の光重合開始剤(I−b)の製造方法に特に制限は無いが、例えば、下記工程1〜工程4によって、容易に製造することができる。
【0074】
(工程1)
工程1は、W及びZによって製造方法が異なるため、以下に場合分けして説明する。
<Wが単結合であり、Zが>NR3’である場合>
下記一般式(1)
【化30】
(式中、R6〜R11は、前記定義の通りであり、好ましいものも同じである。)
で表されるカルバゾール誘導体[以下、カルバゾール誘導体(1)と称する。]と、下記一般式(2b)
【0075】
【化31】
(式中、R3、R3’、R4及びR5は、前記定義の通りであり、好ましいものも同じである。)
で表されるアクリルアミド誘導体[以下、アクリルアミド誘導体(2b)と称する。]とを塩基の存在下に反応させることにより、下記一般式(3b)
【0076】
【化32】
(式中、R3、R3’、R4〜R11は、前記定義の通りであり、好ましいものも同じである。)
で表されるアミド基導入体[以下、アミド基導入体(3b)と称する。]を得る工程である。
【0077】
工程1で使用するアクリルアミド誘導体(2b)の使用量に特に制限は無いが、カルバゾール誘導体(1)1モルに対して、好ましくは0.5〜2モル、より好ましくは0.8〜1.2モルであり、未反応物を低減する観点から、ほぼ等量で反応させることがさらに好ましい。
工程1は、塩基の存在下に実施する。該塩基としては、Michael付加反応に使用し得る塩基を用いることができ、特に制限は無く、有機塩基、無機塩基のいずれも用いることができる。有機塩基としては、例えばピリジン、ジアザビシクロウンデセン(DBU)、ジアザビシクロノネン(DBN)等が挙げられ、求核性の低い塩基が好ましく、DBU、DBNがより好ましい。無機塩基としては、例えば、炭酸ナトリウム、炭酸カリウム等のアルカリ金属炭酸塩;炭酸マグネシウム、炭酸カルシウム等のアルカリ土類金属炭酸塩;炭酸水素ナトリウム、炭酸水素カリウム等のアルカリ金属炭酸水素塩;水酸化ナトリウム、水酸化カリウム等のアルカリ金属水酸化物;水酸化マグネシウム、水酸化カルシウム等のアルカリ土類金属水酸化物等が挙げられる。これらの中でも、アルカリ金属炭酸塩、アルカリ土類金属炭酸塩が好ましく、アルカリ金属炭酸塩がより好ましく、炭酸カリウムがさらに好ましい。
該塩基の使用量に特に制限は無いが、反応効率及び製造コストの観点から、カルバゾール誘導体(1)1モルに対して、好ましくは0.5〜2モル、より好ましくは0.8〜1.2モルであり、ほぼ等量で使用することがさらに好ましい。
工程1は、溶媒の存在下に実施することが好ましい。該溶媒としては、カルバゾール誘導体(1)、アクリルアミド誘導体(2b)及び塩基を溶解し得る溶媒を適宜選択すればよい。具体的には、ジメチルホルムアミド(DMF)、ジメチスルホキシド(DMSO)、塩化メチレン等が挙げられる。
【0078】
工程1の反応温度に特に制限は無いが、通常、好ましくは10〜50℃、より好ましくは15〜40℃、さらに好ましくは15〜30℃で実施する。反応圧力に特に制限は無いが、常圧下に実施することが好ましい。反応時間は、カルバゾール誘導体(1)、アクリルアミド誘導体(2b)及び塩基の種類や使用量並びに反応温度や反応圧力等によっても異なるが、通常、好ましくは2時間〜48時間程度である。
工程1の実施形態に特に制限は無く、例えばカルバゾール誘導体(1)、アクリルアミド誘導体(2b)及び塩基を全て溶媒中に添加し、好ましくは前記温度で攪拌することにより、アミド基導入体(3b)を製造することができる。
反応終了後、得られた反応混合液から、抽出等の通常の有機化合物の分離手段によって、アミド基導入体(3b)を得ることができる。適宜、蒸留、カラムクロマトグラフィー、再結晶等の、通常の有機化合物の精製手段によって精製することにより、アミド基導入体(3b)の純度を高めることもできる。
【0079】
−別法−
アミド基導入体(3b)の製造には、次の方法を採用することもできる。
前記カルバゾール誘導体(1)と下記一般式(2b’)
【化33】
(式中、R4及びR5は前記定義の通りである。R24は、炭素数1〜5のアルキル基を示す。)
で表されるカルボン酸基導入剤[以下、カルボン酸基導入剤(2b’)と称する。]とを塩基の存在下に反応(第1反応)させて得られる下記一般式
【化34】
(式中、R4〜R11は、前記定義の通りであり、好ましいものも同じである。)
で表されるカルボン酸基含有カルバゾール誘導体を、例えば塩化チオニルの存在下にHNR3R3’(R3及びR3’は、前記定義の通りであり、好ましいものも同じである。)で表されるアミンと反応(第2反応)させることにより、アミド基導入体(3b)を得る方法である。該アミド基導入体(3b)としては、市販品を用いることもできる。
なお、R24が示す炭素数1〜5のアルキル基としては、例えばメチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、t−ブチル基等が挙げられる。これらの中でも、メチル基、エチル基が好ましい。
【0080】
前記第1反応で使用する塩基としては特に制限は無く、有機塩基、無機塩基のいずれも用いることができる。有機塩基としては、例えばピリジン、ジアザビシクロウンデセン(DBU)、ジアザビシクロノネン(DBN)等が挙げられ、求核性の低い塩基が好ましく、DBU、DBNがより好ましい。無機塩基としては、例えば、炭酸ナトリウム、炭酸カリウム等のアルカリ金属炭酸塩;炭酸マグネシウム、炭酸カルシウム等のアルカリ土類金属炭酸塩;炭酸水素ナトリウム、炭酸水素カリウム等のアルカリ金属炭酸水素塩;水酸化ナトリウム、水酸化カリウム等のアルカリ金属水酸化物;水酸化マグネシウム、水酸化カルシウム等のアルカリ土類金属水酸化物等が挙げられる。これらの中でも、塩基としては、無機塩基が好ましく、アルカリ金属水酸化物がより好ましい。
該塩基の使用量に特に制限は無いが、反応効率及び製造コストの観点から、カルバゾール誘導体(1)1モルに対して、好ましくは0.5〜2モル、より好ましくは0.8〜1.2モルであり、ほぼ等量で使用することがさらに好ましい。
第1反応は、溶媒の存在下に実施することが好ましい。溶媒としては、カルバゾール誘導体(1)、カルボン酸基導入剤(2b’)及び前記塩基を溶解し得る溶媒を適宜選択すればよい。具体的には、ジメチルホルムアミド(DMF)、ジメチスルホキシド(DMSO)、塩化メチレン等が挙げられ、DMFが好ましい。
【0081】
第1反応の反応温度に特に制限は無いが、通常、好ましくは10〜50℃、より好ましくは15〜40℃、さらに好ましくは15〜30℃で実施する。第1反応の反応圧力に特に制限は無いが、常圧下に実施することが好ましい。反応時間は、カルバゾール誘導体(1)及び塩基の種類や使用量、及びカルボン酸基導入剤(2b’)の使用量並びに反応温度及び反応圧力等によっても異なるが、通常、好ましくは2時間〜48時間程度である。
第1反応の実施形態に特に制限は無く、例えばカルバゾール誘導体(1)、カルボン酸基導入剤(2b’)及び塩基を全て溶媒中に添加し、好ましくは前記温度で攪拌することにより、カルボン酸基含有カルバゾール誘導体を製造することができる。
第1反応終了後、抽出等の通常の有機化合物の分離手段によって、前記カルボン酸基含有カルバゾール誘導体を得ることができる。適宜、蒸留、カラムクロマトグラフィー、再結晶等の、通常の有機化合物の精製手段によって精製することにより、該カルボン酸基含有カルバゾール誘導体の純度を高めることもできる。
【0082】
第2反応で使用する塩化チオニルの使用量は、第1反応で使用するカルバゾール誘導体(1)1モルに対して、好ましくは1〜10モル程度使用すればよく、より好ましくは2〜5モル程度である。
第2反応で使用するHNR3R3’で表されるアミンの使用量は、第1反応で使用するカルバゾール誘導体(1)1モルに対して、好ましくは1〜10モル程度使用すればよく、より好ましくは2〜7モル程度である。
第2反応は、溶媒の存在下に実施することが好ましい。溶媒としては、前記カルボン酸基含有カルバゾール誘導体、前記アミンを溶解し得る溶媒であれば特に制限は無い。例えば、ジクロロメタン、ニトロベンゼン、アセトン、アセトニトリル等が挙げられ、アセトニトリルが好ましい。
【0083】
第2反応の反応温度に特に制限は無いが、通常、好ましくは10〜50℃、より好ましくは15〜40℃、さらに好ましくは15〜30℃で実施する。第2反応の反応圧力に特に制限は無いが、常圧下に実施することが好ましい。反応時間は、前記カルボン酸基含有カルバゾール誘導体、前記アミン及び塩基の種類や使用量並びに反応温度及び反応圧力等によっても異なるが、通常、好ましくは1時間〜10時間程度である。
第2反応の実施形態に特に制限は無いが、例えば前記カルボン酸基含有カルバゾール誘導体と前記アミンとを溶媒中に添加し、そこへ塩化チオニルを滴下し、滴下終了後、好ましくは前記温度で攪拌することにより、アミド基導入体(3b)を製造することができる。
反応終了後、得られた反応混合液から、抽出等の通常の有機化合物の分離手段によって、アミド基導入体(3b)を得ることができる。適宜、蒸留、カラムクロマトグラフィー、再結晶等の、通常の有機化合物の精製手段によって精製することにより、アミド基導入体(3b)の純度を高めることもできる。
【0084】
<Wが酸素原子であり、Zが単結合である場合>
前記カルバゾール誘導体(1)と炭酸エチレンを塩基の存在下に反応(第1反応)させて得られる、下記一般式
【0085】
【化35】
(式中、R6〜R11は、前記定義の通りであり、好ましいものも同じである。)
【0086】
で表されるヒドロキシル基含有カルバゾール誘導体を、塩基の存在下にR3COY(R3は、前記定義の通りであり、好ましいものも同じである。また、Yは、ハロゲン原子を示す。)で表されるアシルハライドと反応(第2反応)させることにより、下記一般式(3b’)
【化36】
(式中、R3〜R11は、前記定義の通りであり、好ましいものも同じである。)
で表されるエステル基導入体[以下、エステル基導入体(3b’)と称する。]を得る方法が好ましい。
【0087】
前記第1反応で使用する炭酸エチレンの使用量は、カルバゾール誘導体(1)1モルに対して、好ましくは0.7〜15モル、より好ましくは1〜10モルである。
第1反応で使用する塩基としては、有機塩基が好ましく、例えばトリエチルアミン、トリブチルアミン等のアミン類;ピリジン等の含窒素複素環式芳香族化合物等が挙げられる。これらの中でも、アミン類が好ましく、トリエチルアミンがより好ましい。該塩基の使用量に特に制限は無いが、反応効率及び製造コストの観点から、カルバゾール誘導体(1)1モルに対して、好ましくは0.5〜2モル、より好ましくは0.8〜1.2モルであり、ほぼ等量で使用することがさらに好ましい。
第1反応は、溶媒の存在下に実施することが好ましい。溶媒としては、カルバゾール誘導体(1)や炭酸エチレン、並びに前記塩基を溶解し得る溶媒を適宜選択すればよい。具体的には、ジメチルホルムアミド(DMF)、ジメチスルホキシド(DMSO)、塩化メチレン等が挙げられ、DMFが好ましい。
【0088】
第1反応の反応温度に特に制限は無いが、通常、好ましくは60〜130℃、より好ましくは80〜120℃、さらに好ましくは90〜110℃で実施する。第1反応の反応圧力に特に制限は無いが、常圧下に実施することが好ましい。反応時間は、カルバゾール誘導体(1)及び塩基の種類や使用量、炭酸エチレンの使用量、並びに反応温度及び反応圧力等によっても異なるが、通常、好ましくは1時間〜10時間程度である。
第1反応の実施形態に特に制限は無く、例えばカルバゾール誘導体(1)、炭酸エチレン及び塩基を全て溶媒中に添加し、好ましくは前記温度で攪拌することにより、前記ヒドロキシル基含有カルバゾール誘導体を製造することができる。
第1反応終了後、得られた反応混合液から、抽出等の通常の有機化合物の分離手段によって、前記ヒドロキシル基含有カルバゾール誘導体を得ることができる。適宜、蒸留、カラムクロマトグラフィー、再結晶等の、通常の有機化合物の精製手段によって精製することにより、該ヒドロキシル基含有カルバゾール誘導体の純度を高めることもできる。
【0089】
第2反応で使用するR3COYで表されるアシルハライドとしては、アシルクロリド、アシルヨージド等が挙げられ、アシルクロリドが好ましい。
該アシルハライドの使用量は、第1反応で使用するカルバゾール誘導体(1)1モルに対して、好ましくは0.7〜3モル程度、より好ましくは1〜2モルであり、ほぼ等量で使用することがさらに好ましい。
第2反応で使用する塩基としては、例えばトリエチルアミン、トリブチルアミン、ジイソプロピルエチルアミン等のアミン類が好ましい。塩基の使用量は、前記アシルハライド1モルに対して、好ましくは0.7〜3モル程度、より好ましくは1〜2モルであり、ほぼ等量で使用することがさらに好ましい。
第2反応は、溶媒の存在下に実施することが好ましい。溶媒としては、通常のアシル化反応に使用し得る溶媒を使用でき、反応を阻害しない限り特に制限は無い。具体的には、ジクロロメタン、ニトロベンゼン、アセトン、アセトニトリル等が挙げられ、アセトニトリルが好ましい。
【0090】
第2反応の反応温度に特に制限は無いが、通常、好ましくは10〜50℃、より好ましくは15〜40℃、さらに好ましくは15〜30℃で実施する。第2反応の反応圧力に特に制限は無いが、常圧下に実施することが好ましい。反応時間は、前記ヒドロキシル基含有カルバゾール誘導体、前記アシルハライド及び塩基の種類や使用量並びに反応温度及び反応圧力等によっても異なるが、通常、好ましくは1時間〜10時間程度である。
第2反応の実施形態に特に制限は無いが、例えば前記ヒドロキシル基含有カルバゾール誘導体と前記アシルハライドと塩基とを全て溶媒中に添加し、好ましくは前記温度で攪拌することにより、エステル基導入体(3b’)を製造することができる。
反応終了後、得られた反応混合液から、抽出等の通常の有機化合物の分離手段によって、エステル基導入体(3b’)を得ることができる。適宜、蒸留、カラムクロマトグラフィー、再結晶等の、通常の有機化合物の精製手段によって精製することにより、エステル基導入体(3b’)の純度を高めることもできる。
【0091】
(工程2)
工程2は、工程1で得られたアミド基導入体(3b)又はエステル基導入体(3b’)[以下、便宜上、これらをまとめて、官能基導入体(3b)と称する。]と、下記一般式(4)、(5)
【化37】
(式中、Xは、ハロゲン原子又は−OC(=O)Arを示し、X’は、ハロゲン原子又は−OC(=O)R2を示す。Ar及びR2は、前記定義の通りであり、好ましいものも同じである。)
で表される2つのアシル化剤[以下、それぞれアシル化剤(4)、アシル化剤(5)と称する。]とをルイス酸の存在下に反応させることにより、下記一般式(6b)
【0092】
【化38】
(式中、R2〜R11、Ar、W及びZは、前記定義の通りであり、好ましいものも同じである。)
で表されるジケトン体[以下、ジケトン体(6b)と称する。]を得る工程である。
【0093】
上記式中、X、X’がそれぞれ独立に示すハロゲン原子としては、フッ素原子、塩素原子、臭素原子、ヨウ素原子等が挙げられ、塩素原子が好ましい。
工程2において、官能基導入体(3b)と、アシル化剤(4)及び(5)との反応は、目的とするジケトン体(6b)の収率の観点から、官能基導入体(3b)とアシル化剤(4)とを反応させた後、次いでアシル化剤(5)を反応させることが好ましい。
アシル化剤(4)及び(5)の使用量は、官能基導入体(3b)にそれぞれのアシル基を1つずつ導入する観点から、官能基導入体(3b)1モルに対して、それぞれ好ましくは0.8〜1.3モル、より好ましくは1〜1.3モルであり、未反応物を低減する観点から、ほぼ等量で反応させることがさらに好ましい。特に、官能基導入体(3b)とアシル化剤(4)とを反応させた後、次いでアシル化剤(5)を反応させる場合には、アシル化剤(5)の使用量は、前記範囲を超えても何ら問題ないが、あまり過剰であってもそれに見合う収率が得られるわけではなく、いたずらに生産コストがかさむことになり得る。
工程2は、ルイス酸の存在下に実施する。ルイス酸としては、塩化アルミニウム、三フッ化ホウ素ジエチルエーテル錯体が好ましい。ルイス酸の使用量は、ジケトン体(6b)の収率の観点から、アシル化剤(4)又は(5)1モルに対して、通常、好ましくは0.8〜2.5モル、より好ましくは1〜2モルである。
【0094】
工程2は、溶媒の存在下に実施することが好ましい。該溶媒としては、通常のFriedel−Craftsアシル化反応に使用し得る溶媒であれば特に制限は無い。具体的には、ジクロロメタン、ニトロベンゼン、アセトン、アセトニトリル等が挙げられる。
工程2の反応温度は、反応開始時は−50〜5℃(好ましくは−10〜5℃)とし、反応が進行するにしたがって、徐々に室温(15〜25℃程度)へ戻すことが好ましい。反応時間は、官能基導入体(3b)、アシル化剤(4)及び(5)の種類及び使用量、並びに反応温度によっても異なるが、通常、好ましくは1〜30時間である。なお、アシル化剤(4)及び(5)の両方がハロゲン化アシル又は酸無水物である場合、官能基導入体(3b)とアシル化剤(4)とを反応させた後、生成物を単離することなく引き続きアシル化剤(5)と反応させることもできる。その場合、官能基導入体(3b)とアシル化剤(4)との反応時間は、30分〜5時間程度(好ましくは30分〜3時間)とし、一方、官能基導入体(3b)とアシル化剤(5)との反応時間を、30分〜24時間程度(好ましくは30分〜18時間)と長めに設けて十分に反応させることが好ましい。
【0095】
収率の観点から、工程2の好ましい実施形態の1つを説明する。例えば、氷浴で冷却しながら官能基導入体(3b)を適宜溶媒と混合し、その混合溶液へルイス酸をゆっくり(好ましくは5分〜1時間、より好ましくは10分〜40分かけて)添加し、添加終了後、アシル化剤(4)をゆっくり(好ましくは5分〜1時間、より好ましくは10分〜40分かけて)添加し、室温へ戻して一定時間(30分〜5時間程度)攪拌を続ける。再び、氷浴で冷却しながらアシル化剤(5)を添加し、そこへルイス酸をゆっくり(好ましくは5分〜1時間、より好ましくは10分〜40分かけて)添加し、添加終了後、室温へ戻して一定時間(30分〜24時間程度)攪拌を続けることにより、ジケトン体(6b)を得ることができる。
反応終了後、得られた反応混合液から、抽出等の通常の有機化合物の分離手段によって、ジケトン体(6b)を得ることができる。適宜、蒸留、カラムクロマトグラフィー、再結晶等の、通常の有機化合物の精製手段によって精製することにより、ジケトン体(6b)の純度を高めることもできる。
【0096】
(工程3)
工程3は、工程2で得られたジケトン体(6b)とヒドロキシルアミンを反応させることにより、下記一般式(7b)
【化39】
(式中、R2〜R11、Ar、W及びZは、前記定義の通りであり、好ましいものも同じである。)
で表されるオキシム体[以下、オキシム体(7b)と称する。]を得る工程である。
【0097】
工程3においては、ヒドロキシルアミン供給源としては、特に制限されるものではないが、塩化ヒドロキシルアミンが好ましい。該塩化ヒドロキシルアミンは、例えば水中で酢酸ナトリウム、等と反応させることにより、ヒドロキシルアミンの水溶液を得ることができる。
ヒドロキシルアミン(塩化ヒドロキシルアミン)の使用量は、ジケトン体(6b)1モルに対して、好ましくは0.8〜2モル、より好ましくは1〜1.5モル、さらに好ましくは1〜1.3モルであり、未反応物を低減する観点から、ほぼ等量で反応させることがさらに好ましい。
工程3は、溶媒の存在下に実施することが好ましい。溶媒としては、水溶性有機溶媒が好ましく、該水溶性有機溶媒としては、例えばメタノール、エタノール等のアルコール類、ジメチルホルムアミド(DMF)等が好ましい。
【0098】
工程3の反応温度に特に制限は無いが、オキシム体(7b)の収率の観点からは、通常、好ましくは40〜160℃、より好ましくは50〜140℃、さらに好ましくは70〜110℃である。反応時間は、ジケトン体(6b)の種類及び使用量、並びに反応温度によっても異なるが、通常、好ましくは0.5〜20時間、より好ましくは1〜12時間である。
工程3の実施形態に特に制限は無く、例えば、塩化ヒドロキシルアミンと酢酸ナトリウムを水中で混合してヒドロキシルアミンの水溶液を得ておき、そこへジケトン体(6b)及び溶媒を添加して、好ましくは前記温度範囲で攪拌することにより、オキシム体(7b)を得ることができる。
反応終了後、得られた反応混合液から、抽出等の通常の有機化合物の分離手段によって、オキシム体(7b)を得ることができる。適宜、蒸留、カラムクロマトグラフィー、再結晶等の、通常の有機化合物の精製手段によって精製することにより、オキシム体(7b)の純度を高めることもできる。
【0099】
(工程4)
工程4は、工程3で得られたオキシム体(7b)と下記一般式(8)
【化40】
(式中、Yは、ハロゲン原子又は−OC(=O)R1を示す。R1は、前記定義の通りであり、好ましいものも同じである。)
で表されるエステル化剤[以下、エステル化剤(8)と称する。]とを反応させることにより、下記一般式(I−b)
【0100】
【化41】
(式中、R1〜R11、Ar、W及びZは、前記定義の通りである。)
で表される光重合開始剤[以下、光重合開始剤(I−b)と称する。]を得る工程である。
【0101】
上記式中、Yが示すハロゲン原子としては、フッ素原子、塩素原子、臭素原子、ヨウ素原子等が挙げられ、塩素原子が好ましい。
工程4で使用するエステル化剤(8)の使用量に特に制限は無いが、オキシム体(7b)1モルに対して、好ましくは0.5〜2モル、より好ましくは0.8〜1.2モルであり、未反応物を低減する観点から、ほぼ等量で反応させることがさらに好ましい。
工程4は、反応を促進するために、塩基の存在下に実施してもよい。塩基としては、有機塩基や無機塩基が挙げられる。有機塩基としては、例えばトリエチルアミン、トリブチルアミン等のアミン類;ピリジン等の含窒素複素環式芳香族化合物等が挙げられる。無機塩基としては、例えば炭酸ナトリウム等のアルカリ金属炭酸塩;炭酸マグネシウム等のアルカリ土類金属炭酸塩;水酸化ナトリウム等のアルカリ金属水酸化物;水酸化マグネシウム等のアルカリ土類金属水酸化物等が挙げられる。これらの中でも、有機塩基が好ましく、アミン類、含窒素複素環式芳香族化合物がより好ましく、反応効率及び製造コストの観点から、トリエチルアミン、ピリジンがさらに好ましい。
工程4にて塩基を使用する場合、その使用量は、光重合開始剤(I−b)の収率及び製造コストの観点から、オキシム体(7b)1モルに対して、好ましくは1〜5モル、より好ましくは1.5〜3モルである。
【0102】
工程4は、溶媒の存在下に実施することが好ましい。溶媒としては、t−ブチルメチルエーテル、エチルメチルエーテル、シクロペンチルメチルエーテル、テトラヒドロフラン(THF)等のエーテル類が好ましく挙げられる。
工程4の反応温度は、反応開始時は−50〜5℃(好ましくは−10〜5℃)とし、反応が進行するにしたがって、徐々に室温(15〜25℃程度)へ戻すことが好ましい。反応時間は、オキシム体(7b)、エステル化剤(8)の種類及び使用量、並びに反応温度によっても異なるが、通常、好ましくは0.5〜10時間、より好ましくは1〜5時間である。
工程4の実施形態に特に制限は無く、例えばオキシム体(7b)及びエステル化剤(8)を適宜溶媒中で混合し、そこへ塩基を滴下し、滴下終了後、反応液の温度を徐々に室温へ戻して攪拌を続けることにより、本発明の光重合開始剤(I−b)を得ることができる。
反応終了後、得られた反応混合液から、抽出等の通常の有機化合物の分離手段によって、光重合開始剤(I−b)を得ることができる。適宜、蒸留、カラムクロマトグラフィー、再結晶等の、通常の有機化合物の精製手段によって精製することにより、光重合開始剤(I−b)の純度を高めることもできる。
【0103】
こうして得られる本発明の光重合開始剤(I−b)は、カルバゾール骨格中の窒素原子に、エステル基又はアミド結合(若しくは環を形成したアミド結合)を含有する特殊な置換基が付いている。そのため、光硬化性組成物中のエチレン性不飽和結合を有する化合物(特に(メタ)アクリロイルオキシ基を有する化合物)やエーテル結合及び/又はエステル結合を有する25℃で液体の化合物との相溶性が高く、かつ溶剤(前記のエーテル結合及び/又はエステル結合を有する25℃で液体の化合物を含む。)に対する溶解性が高く、光(特に波長450nm以下の短波長の光)に対する感度が高く、解像度及び現像性、ひいては深部硬化性に優れ、さらに基板との密着性が良好になるものと考えられる。また、該光重合開始剤(I−b)は、露光時の光により発生する分解物による重合物の汚染や装置の汚染が無い。
なお、特定化合物や溶剤に対する光重合開始剤(I−b)の相溶性や溶解性の高さは、感度、解像度及び現像性、ひいては深部硬化性の向上に寄与しているものと考えられ、カラーフィルタ用途においては重要な要素であると言える。特にカラーフィルタ用途においては、光重合開始剤が、エーテル結合及び/又はエステル結合を有する溶剤(特に、プロピレングリコールモノエチルエーテルアセテート等)100質量部に対して、5質量部以上溶解することが好ましく、10質量部以上溶解することがさらに好ましく、本発明の光重合開始剤(I−b)はかかる条件を満たしており、カラーフィルタ用光硬化性組成物に含有させる光重合開始剤、さらには高遮光性のブラックマトリックス用光硬化性組成物に含有させる光重合開始剤として適している。
【0104】
次に、本発明の光重合開始剤(I)を含有する光硬化性組成物、該光硬化性組成物を用いたカラーフィルタ、パターン形成方法及び液晶表示装置について順に説明する。
[光硬化性組成物]
本発明の光硬化性組成物は、エチレン性不飽和結合を有する化合物及び/又はバインダー樹脂と、前記光重合開始剤(I)を1種以上含有するものである。本発明の光硬化性組成物には、さらに、色材、そして必要に応じて、前記光重合開始剤(I)以外の光重合開始剤、分散剤、多官能モノマー、単官能モノマー、増感剤及び溶剤等を含有してもよく、こうして得られる光硬化性組成物は、カラーフィルタ用途に有用である。なお、多官能モノマー及び単官能モノマーは、官能基がエチレン性不飽和結合を有していれば、エチレン性不飽和結合を有する化合物にもなり得る。
本発明の光硬化性組成物において、光重合開始剤(I)の含有量は、感度、解像度及び現像性の観点から、光硬化性組成物の固形分に対して、好ましくは2〜50質量%、より好ましくは2〜30質量%、さらに好ましくは4〜15質量%である。
また、本発明の光硬化性組成物が、前記光重合開始剤(I)以外の光重合開始剤を含有する場合、前記光重合開始剤(I)以外の光重合開始剤の含有量は、本発明の効果を著しく阻害しない限り特に制限は無いが、光重合開始剤(I)100質量部に対して、好ましくは100質量部以下、より好ましくは50質量部以下、より好ましくは20質量部以下、さらに好ましくは10質量部以下、特に好ましくは5質量部以下である。
【0105】
(エチレン性不飽和結合を有する化合物)
エチレン性不飽和結合を有する化合物は、光重合開始剤(I)の存在下に光照射されることにより、重合反応を起こし、光硬化性組成物において、バインダーとなり得る化合物であり、形成された画素中で結着剤としての役割を果たし得る。エチレン性不飽和結合を有する化合物は、本発明の光硬化性組成物中に、好ましくは5〜60質量%、より好ましくは10〜40質量%含有させる。この範囲であると、形成された画素と基板との密着性が良好となる傾向にある。なお、エチレン性不飽和結合を有する化合物は、後述するバインダー樹脂と併用してもよい。併用する場合、その合計含有量が前記範囲内であると好ましい。
エチレン性不飽和結合を有する化合物の具体例としては、(メタ)アクリル酸、クロトン酸、α−クロルアクリル酸等の不飽和モノカルボン酸類;マレイン酸、フマル酸、イタコン酸等の不飽和ジカルボン酸又はその無水物類;トリメリット酸、ピロメリット酸等の3価以上の不飽和多価カルボン酸又はその無水物類;スチレン、α−メチルスチレン、クロロスチレン、メトキシスチレン、ジビニルベンゼン、ビニルトルエン等の芳香族ビニル化合物;メチル(メタ)アクリレート、エチル(メタ)アクリレート、n−プロピル(メタ)アクリレート、イソプロピル(メタ)アクリレート、n−ブチル(メタ)アクリレート、s−ブチル(メタ)アクリレート、t−ブチル(メタ)アクリレート、2−エチルヘキシル(メタ)アクリレート、ラウリル(メタ)アクリレート、シクロヘキシル(メタ)アクリレート、ヒドロキシエチル(メタ)アクリレート、フェノキシエチル(メタ)アクリレート、メトキシエチル(メタ)アクリレート、ポリ(プロピルオキシ)プロピル(メタ)アクリレート、ビニル(メタ)アクリレート、エチレングリコールジ(メタ)アクリレート、ポリエチレングリコールジ(メタ)アクリレート、プロピレングリコールジ(メタ)アクリレート、ポリプロピレングリコールジ(メタ)アクリレート、1,6−ヘキサンジオールジ(メタ)アクリレート、トリメチロールプロパントリ(メタ)アクリレート、アリル(メタ)アクリレート、ベンジル(メタ)アクリレート等の、(多価)アルコールをα,β−不飽和カルボン酸でエステル化した化合物;2−アミノエチル(メタ)アクリレート、2−ジメチルアミノエチル(メタ)アクリレート、2−アミノプロピル(メタ)アクリレート、2−ジメチルアミノプロピル(メタ)アクリレート等の不飽和カルボン酸アミノアルキルエステル類;グリシジル(メタ)アクリレート等の不飽和カルボン酸グリシジルエステル類;酢酸ビニル、プロピオン酸ビニル、酪酸ビニル、安息香酸ビニル等のカルボン酸ビニルエステル類;ビニルメチルエーテル、ビニルエチルエーテル、アリルグリシジルエーテル、イソブチルビニルエーテル等の不飽和エーテル類;(メタ)アクリロニトリル、α−クロロアクリロニトリル、シアン化ビニリデン等のシアン化ビニル化合物;(メタ)アクリルアミド、α−クロロアクリルアミド、N−2−ヒドロキシエチル(メタ)アクリルアミド等の不飽和アミド類;マレイミド、N−フェニルマレイミド、N−シクロヘキシルマレイミド等の不飽和イミド類;1,3−ブタジエン、イソプレン、クロロプレン等の脂肪族共役ジエン類等が挙げられる。これらの中でも、本発明の光重合開始剤(I)との相溶性の観点から、(メタ)アクリロイルオキシ基を有する化合物が好ましい。
エチレン性不飽和結合を有する化合物は、1種を単独で使用してもよいし、2種以上を併用してもよい。
【0106】
なお、エチレン性不飽和結合を有する化合物が、エーテル結合及び/又はエステル結合(好ましくはエーテル結合及びエステル結合)を持つエチレン性不飽和結合を有する化合物である場合、本発明の光重合開始剤(I)との相溶性により一層優れるため、本発明の光硬化性組成物において、後述する溶剤の量を低減する又は無溶剤とすることができるため、そのような化合物を用いることも好ましい。エチレン性不飽和結合を有する化合物として、エーテル結合及び/又はエステル結合を持つエチレン性不飽和結合を有する化合物を少なくとも1種用いる場合、溶剤の量は、光硬化性組成物全量に対して10質量%以下のみでよく、さらには5質量%以下、さらには無溶剤とすることもできる。
【0107】
(バインダー樹脂)
バインダー樹脂は、形成された画素中で結着剤としての役割を果たすものであり、基板と密着性が良好なものであれば、特に限定されるものではない。このようなバインダー樹脂は、本発明の光硬化性組成物の固形分中に、好ましくは5〜60質量%、より好ましくは10〜40質量%含有させる。この範囲であると、形成された画素と基板との密着性が良好となる傾向にある。また、バインダー樹脂の重量平均分子量としては、形成された画素と基板との密着性の観点から、好ましくは3,000〜100,000、より好ましくは5,000〜50,000である。なお、本明細書において、重量平均分子量は、ゲル浸透クロマトグラフィー(GPC)で測定したポリスチレン換算の値である。
なお、前述の通り、バインダー樹脂は、前記エチレン性不飽和結合を有する化合物と併用してもよい。
【0108】
上記バインダー樹脂としては、一般的なカラーフィルタ用光硬化性組成物に用いられるバインダー樹脂と同様のものを用いることができる。具体的には、エチレン−酢酸ビニル共重合体、エチレン−塩化ビニル共重合体、ポリスチレン、アクリロニトリル−スチレン共重合体、ABS樹脂、ポリメタクリル酸樹脂、エチレンメタクリル酸樹脂、ポリ塩化ビニル樹脂、塩素化塩化ビニル、ポリビニルアルコール、ポリエチレンテレフタレート、ポリブチレンテレフタレート、ポリカーボネート、ポリビニルアセタール、ポリエーテルエーテルケトン、ポリエーテルサルフォン、ポリフェニレンサルファイド、ポリアリレート、ポリビニルブチラール、エポキシ樹脂、フェノキシ樹脂、ポリイミド樹脂、ポリアミドイミド樹脂、ポリアミック酸樹脂、ポリエーテルイミド樹脂、フェノール樹脂、ユリア樹脂等が挙げられる。さらに、重合可能なモノマーであるメチル(メタ)アクリレート、エチル(メタ)アクリレート、n−プロピル(メタ)アクリレート、イソプロピル(メタ)アクリレート、s−ブチル(メタ)アクリレート、イソブチル(メタ)アクリレート、t−ブチル(メタ)アクリレート、n−ペンチル(メタ)アクリレート、n−ヘキシル(メタ)アクリレート、2−エチルヘキシル(メタ)アクリレート、n−オクチル(メタ)アクリレート、n−デシル(メタ)アクリレート、スチレン、α−メチルスチレン、N−ビニル−2−ピロリドン、グリシジル(メタ)アクリレート、シクロヘキシル(メタ)アクリレート、ジシクロペンタニル(メタ)アクリレート、ジシクロペンタニルオキシエチル(メタ)アクリレート、フェニル(メタ)アクリレート、ベンジル(メタ)アクリレートの1種以上と、アクリル酸、メタクリル酸、アクリル酸の2量体(例えば、東亜合成化学(株)製、商品名「M−5600」)、イタコン酸、クロトン酸、マレイン酸、フマル酸、ビニル酢酸、これらの酸無水物等の1種以上とを反応させて得られるポリマー又はコポリマー等が挙げられる。また、該コポリマーにグリシジル基又は水酸基を有するエチレン性不飽和化合物を付加させたポリマー等も挙げられる。
【0109】
特に好ましいバインダー樹脂としては、カルボキシル基等の酸性官能基を有するアルカリ可溶性樹脂、例えば、アルカリ可溶性アクリル系樹脂を挙げることができる。カルボキシル基を有するアルカリ可溶性樹脂としては、カルボキシル基含有不飽和単量体と他の共重合可能なエチレン性不飽和単量体の共重合体が好ましく、さらに分子内にエポキシ基とエチレン性不飽和基とを併せ持つ化合物、例えばグリシジル(メタ)アクリレート等を付加させ、側鎖にエチレン性不飽和基を導入したものが好ましい。
上記カルボキシル基含有不飽和単量体としては、例えば、(メタ)アクリル酸、クロトン酸、マレイン酸、無水マレイン酸、コハク酸モノ[2−(メタ)アクリロイロキシエチル]、フタル酸モノ[2−(メタ)アクリロイロキシエチル]、ω−カルボキシポリカプロラクトンモノ(メタ)アクリレート等が好ましく、(メタ)アクリル酸が特に好ましい。なお、カルボキシル基含有不飽和単量体は、1種を単独で使用してもよいし、2種以上を併用してもよい。
上記エチレン性不飽和単量体としては、メチル(メタ)アクリレート、n−ブチル(メタ)アクリレート、シクロヘキシル(メタ)アクリレート、イソボルニル(メタ)アクリレート、ジシクロペンタニル(メタ)アクリレート、アリル(メタ)アクリレート、スチレン、α−メチルスチレン、ベンジル(メタ)アクリレート、ヒドロキシエチル(メタ)アクリレート、2−ジメチルアミノエチル(メタ)アクリレート、2−エチルヘキシル(メタ)アクリレート、2−ヒドロキシル(メタ)エチルアクリレート、ジシクロペンテニル(メタ)アクリレート等、及びこれらのマクロモノマー類;N−メチルマレイミド、N−シクロヘキシルマレイミド、N−ベンジルマレイミド、N−フェニルマレイミド、N−メチルフェニルマレイミド等のN置換マレイミド類等を挙げることができる。なお、エチレン性不飽和単量体は、1種を単独で使用してもよいし、2種以上を併用してもよい。
【0110】
(色材)
本発明の光硬化性組成物には、さらに色材を含有させて着色光硬化性組成物としてもよい。該色材としては、顔料、染料、天然色素等が挙げられる。色材は、1種を単独で使用してもよいし、2種以上を併用してもよい。色材は、色再現性、硬化性及び現像性の観点から、本発明の光硬化性組成物の固形分中に、好ましくは5〜60質量%、より好ましくは5〜50質量%含有させる。
顔料としては、有機顔料、無機顔料があるが、発色性及び耐熱性の観点から有機顔料が好ましい。有機顔料としては、例えばカラーインデックス(C.I.;The Society of Dyers and Colourists 発行)においてピグメント(Pigment)に分類されている化合物(C.I.ピグメントブルー、C.I.ピグメントバイオレット、C.I.ピグメントグリーン、C.I.ピグメントレッド、C.I.ピグメントイエロー、C.I.ピグメントオレンジ等)を挙げることができる。
なお、色材は、以下の分散剤によって、より均一に分散されていることが好ましい。
【0111】
(分散剤)
分散剤としては、上記色材を均一に分散することができるものであればよく、公知の分散剤を使用することができる。具体的には、変性ポリウレタン、変性ポリアクリレート、変性ポリエステル、変性ポリアミド等の高分子分散剤、リン酸エステル、アルキルアミン、ポリオキシエチレンアルキルフェニルエーテル等の界面活性剤や顔料誘導体を挙げることができる。これらの中でも、高分子分散剤が好ましく、具体的な市販品の商品名としては、EFKA−4046、EFKA−4047、EFKAポリマー10、EFKAポリマー400、EFKAポリマー401、EFKAポリマー4300、EFKAポリマー4330(以上、チバ・スペシャルティ・ケミカルズ(株)製)、Disperbyk111、Disperbyk161、Disperbyk165、Disperbyk167、Disperbyk182、Disperbyk2000、Disperbyk2001(以上、ビックケミー・ジャパン(株)製)、SOLSPERSE24000、SOLSPERSE27000、SOLSPERSE28000(以上、ルーブリゾール社製)、アジスパー(登録商標)PB821、PB822(味の素ファインテクノ(株)製)等が挙げられる。
【0112】
本発明の光硬化性組成物に分散剤を含有させる場合、その含有量としては、上述の色材を均一に分散することができるものであれば特に限定されるものではないが、色材の分散性、光硬化性組成物の現像性、及び未露光箇所での現像残渣の低減の観点から、光硬化性組成物の固形分全量に対して、好ましくは0.5〜30質量%、より好ましくは1〜20質量%である。
なお、色材(特に顔料)と分散剤は、色材の分散量及び分散性の観点から、後述の溶剤中で混合することにより、分散液としてから使用するのが好ましい。
【0113】
(多官能モノマー)
本発明の光硬化性組成物に適宜含有させてもよい多官能モノマーとしては、上記バインダー樹脂と相溶性のある(メタ)アクリロイルオキシ基を有するモノマー、具体的には二官能(メタ)アクリレート及び三官能以上の(メタ)アクリレート等が好ましく挙げられる。多官能モノマーは1種を単独で使用してもよいし、2種以上を併用してもよい。
二官能(メタ)アクリレートとしては、例えば、1,4−ブタンジオール(メタ)アクリレート、ネオペンチルグリコールジ(メタ)アクリレート、1,6−へキサンジオールジ(メタ)アクリレート、ポリプロピレングリコールジ(メタ)アクリレート、トリエチレングリコールジ(メタ)アクリレート、ポリエチレングリコールジ(メタ)アクリレート、ビスフェノールAジ(メタ)アクリレート等が挙げられる。
また、三官能以上の(メタ)アクリレートとしては、例えば、トリメチロールプロパントリ(メタ)アクリレート、トリメチロールエタントリ(メタ)アクリレート、ペンタエリスリトールトリ(メタ)アクリレート、ペンタエリスリトールテトラ(メタ)アクリレート、ジペンタエリスリトールテトラ(メタ)アクリレート、ジペンタエリスリトールヘキサ(メタ)アクリレート(DPHA)、ジペンタエリスリトールペンタ(メタ)アクリレートや、カルボン酸変性ジペンタエリスリトールペンタ(メタ)アクリレート等のカルボキシル基含有多官能(メタ)アクリレートが挙げられる。
【0114】
本発明の光硬化性組成物に多官能モノマーを含有させる場合、その含有量は、光硬化性組成物の固形分全量に対して、好ましくは3〜50質量%である。この範囲であれば、充分に光硬化が進行するため、アルカリ現像時に露光部分が溶出する恐れがなくなり、且つアルカリ現像性が良好である。
【0115】
(単官能モノマー)
本発明の光硬化性組成物に適宜含有させてもよい単官能モノマーとしては(メタ)アクリレート類が好ましく、例えば、アリル(メタ)アクリレート、ベンジル(メタ)アクリレート、ブトキシエチル(メタ)アクリレート、ブトキシエチレングリコール(メタ)アクリレート、シクロヘキシル(メタ)アクリレート、ジシクロペンタニル(メタ)アクリレート、2−エチルヘキシル(メタ)アクリレート、グリセロール(メタ)アクリレート、グリシジル(メタ)アクリレート、2−ヒドロキシエチル(メタ)アクリレート、2−ヒドロキシプロピル(メタ)アクリレート、イソボニル(メタ)アクリレート、イソデキシル(メタ)アクリレート、イソオクチル(メタ)アクリレート、ラウリル(メタ)アクリレート、2−メトキシエチル(メタ)アクリレート、メトキシエチレングリコール(メタ)アクリレート、フェノキシエチル(メタ)アクリレート、ステアリル(メタ)アクリレート等が挙げられる。
【0116】
(増感剤)
さらに、本発明の光硬化性組成物には、必要に応じて増感剤を含有させてもよい。増感剤は、本発明の光硬化性組成物を基板上に塗布した後、露光した際に、照射されたエネルギーを吸収し、その吸収したエネルギーを上記本発明の光重合開始剤の反応開始に寄与させる役割を担う物質である。
増感剤としては、具体的には、4,4'−ビス(ジエチルアミノ)ベンゾフェノン、4,4'−ビス(ジメチルアミノ)ベンゾフェノン、4−メチル−4'−ジエチルアミノベンゾフェノン、4−メトキシ−4'−ジエチルアミノベンゾフェノン、4,4'−ビス(ジプロピルアミノ)ベンゾフェノン、4,4'−ビス(ジイソプロピルアミノ)ベンゾフェノン等が挙げられる。これらは、1種を単独で使用してもよいし、2種以上を併用してもよい。
本発明の光硬化性組成物に増感剤を含有させる場合、その含有量としては、光硬化性組成物の固形分全量に対して、好ましくは1〜10質量%である。
【0117】
(溶剤)
本発明の光硬化性組成物には、色材を分散させるために溶剤を含有していてもよい。該溶剤としては、一般的なカラーフィルタ用光硬化性組成物に用いられるものであれば特に限定されるものではない。具体的には、エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル、プロピレングリコールモノメチルエーテル、ジエチレングリコールモノエチルエーテル、ジエチレングリコールジメチルエーテル、ジエチレングリコールジエチルエーテル、ジプロピレングリコールジメチルエーテル等の(モノ又はポリ)アルキレングリコール(モノ又はポリ)アルキルエーテル類、及びこれらのアセテート類;プロピレングリコールジアセテート、1,3−ブチレングリコールジアセテート等のジアセテート類;テトラヒドロフラン等のエーテル類;メチルエチルケトン、シクロヘキサノン、2−ヘプタノン等のケトン類;2−ヒドロキシプロピオン酸メチル、3−ヒドロキシプロピオン酸エチル、酢酸エチル、n−ブチルアセテート、イソブチルアセテート、酪酸イソブチル、酪酸n−ブチル、3−メトキシプロピオン酸エチル、3−エトキシプロピオン酸エチル、3−メトキシブチルアセテート、乳酸エチル、シクロヘキサノールアセテート等のエステル類;トルエン、キシレン等の芳香族炭化水素類等を挙げることができる。溶剤は、1種を単独で使用してもよいし、2種以上を併用してもよい。
【0118】
これらの中でも、相溶性、溶解性、顔料分散性及び塗布性の観点からは、エーテル結合及び/又はエステル結合を有する25℃で液体の化合物、具体的にはプロピレングリコールモノメチルエーテルアセテート、3−メトキシブチルアセテート、エチレングリコールモノメチルエーテル、ジエチレングリコールジエチルエーテルが好ましく、エーテル結合及びエステル結合を有する25℃で液体の化合物がより好ましい。
本発明の光硬化性組成物に溶剤を含有させる場合、その含有量としては、光硬化性組成物全量中、好ましくは50〜90質量%である。通常、この範囲であれば、顔料分散性が良好であり、レジスト塗布特性(面内均一性)を良好なものとすることができる。なお、本発明の光重合開始剤は、エーテル結合及び/又はエステル結合を有する25℃で液体の化合物との相溶性に優れているため、上記溶剤がエーテル結合及び/又はエステル結合を有する25℃で液体の化合物であれば、光硬化性組成物における溶剤の含有量を大幅に低減することもできる。その場合、本発明の光硬化性組成物中におけるエーテル結合及び/又はエステル結合を有する25℃で液体の化合物の含有量は、光重合開始剤(I)1質量部に対して好ましくは5〜100質量部の範囲で任意に選択し得るため、必要に応じて、光重合開始剤(I)1質量部に対して5〜20質量部程度、さらには5〜10質量部程度の少ない量とすることも可能である。
【0119】
(その他の成分)
本発明の光硬化性組成物には、さらに必要に応じて、無機充填剤;密着促進剤;凝集防止剤;重合停止剤;連鎖移動剤;レベリング剤;可塑剤;消泡剤;シランカップリング剤;紫外線吸収剤等を含有させてもよい。
【0120】
[カラーフィルタ]
カラーフィルタの製造方法には、色材を含有する本発明の光硬化性組成物を用い、フォトリソグラフィー法により、孔部を有する着色層(パターン)を形成する方法を利用することが好ましい。この方法であれば、例えば上記露光の際に、エネルギーの回折等が生じた場合であっても、その影響を受けにくいものとすることができる。従って、着色層に微細なパターン状に孔部が形成された、高精細なカラーフィルタとすることが可能となる。
【0121】
(パターン形成方法)
パターン形成方法として、本発明の光硬化性組成物を用いて、フォトリソグラフィー法により行う方法を説明する。その方法については、着色層に孔部を形成することが可能な方法であれば特に限定されるものではない。例えば、本発明の光硬化性組成物を基板に塗布し、乾燥した後、開口部と遮蔽部とが設けられたフォトマスク等を用いて、エネルギーを照射することにより露光し、次いで現像を行うことによりパターンを形成する。なお、これを複数回繰り返すことにより、例えば赤色、緑色、青色それぞれのパターンを基板上に形成し、次いで、酸化インジウムスズ(ITO)等の透明電極膜を形成することにより、カラーフィルタを製造することができる。
前記光硬化性組成物の塗布方法に特に制限はなく、例えばスピンコート法やスプレーコート、ディップコート、ロールコート、ビードコート、バーコート等の公知の塗布方法に利用できる。塗布後の乾燥は、通常、好ましくは50〜150℃で15秒〜10分行う。
露光の際に用いられる光源としては、一般的に着色層の形成の際に用いられている光源と同様とすることができるが、超高圧水銀ランプ、低圧水銀ランプ、メタルハライドランプ等、紫外部に高輝線を有するランプが好ましい。これにより、上記光硬化性組成物の特性をより効果的に発揮させることが可能となる。
また、前記光硬化性組成物を現像する方法としては、不要部分の光硬化性組成物を除去することが可能であれば、その方法等は特に限定されるものではなく、一般的なカラーフィルタの製造の際に行われる現像方法と同様とすることができる。ここで、例えば、現像液の種類、現像液の濃度、現像液や洗浄水の圧力等を最適化することにより、より微細なパターン状に孔部を形成することが可能となる。
【0122】
なお、本工程に用いられる基板としては、一般的なカラーフィルタに用いられる基板(透明基板)と同様のものを用いることができる。具体的には、石英ガラス、無アルカリガラス、合成石英板等の可撓性のない透明なリジッド材、あるいは、透明樹脂フィルム、光学用樹脂板等の可撓性を有する透明なフレキシブル材が挙げられる。
基板の厚みは、特に限定されるものではないが、本発明のカラーフィルタの用途に応じて、例えば100μm〜1mm程度のものを使用することができる。
カラーフィルタの製造においては、例えば、遮光部を形成する遮光部形成工程、上記着色層上にオーバーコート層を形成する工程等、必要に応じて適宜、他の工程を有していてもよい。
【0123】
[液晶表示装置]
本発明の液晶表示装置は、前記した本発明のカラーフィルタと対向基板と、前記カラーフィルタと前記対向基板との間に形成された液晶層とを有するものであり、その構成は特に制限されるものではなく、公知のカラーフィルタが用いられた液晶表示装置と同じ構成をとることができる。
例えば、図1に示すように、本発明の液晶表示装置40は、得られたカラーフィルタ10(表示側基板)と、TFTアレイ基板(液晶駆動側基板)を有する対向基板20を対向させ、両基板の内面側周縁部をシール剤により接合すると、両基板は所定距離のセルギャップを保持した状態で貼り合わされる。そして、基板間の間隙部に液晶を満たして密封し、液晶層30とすることにより、液晶パネルに属するアクティブマトリックス方式のカラー液晶表示装置が得られる。なお、ここで、本発明のカラーフィルタ10は透明基板1、遮光部2及び着色層3を有する。
液晶表示装置の駆動方式に特に制限はなく、一般的に液晶表示装置に用いられている駆動方式を採用することができる。例えば、TN方式、IPS方式、OCB方式及びMVA方式等が挙げられる。また、液晶層を構成する液晶としては、液晶表示装置の駆動方式等に応じて、誘電異方性の異なる各種液晶及びこれらの混合物を用いることができる。
【実施例】
【0124】
次に、本発明を実施例により、さらに詳細に説明するが、本発明は、これらの例によって何ら限定されるものではない。
【0125】
<実施例1>
(工程1)
【化42】
カルバゾール6.8g(40mmol)とエチルアクリレート4.0g(40mmol)及び炭酸カリウム5.5g(40mmol)をジメチルホルムアミド(DMF)に懸濁させ、室温で24時間攪拌した。反応終了後、水を添加してからさらに30分攪拌し、その後、酢酸エチルで抽出、水で洗浄し、硫酸マグネシウムで乾燥させ、溶媒を留去、乾燥することにより、淡黄色液体の上記化合物(i)9.6g(収率93%)を得た。該化合物(i)の1H−NMR測定結果を以下に示す。
【0126】
1H−NMR(400MHz,CDCl3,TMS,ppm)δ:1.15(t,3H)、2.835(t,2H)、4.08(q,2H)、4.64(t,3H)、7.215−7.254(m,2H)、7.45−7.47(m,4H)、8.08(d,2H)
【0127】
(工程2)
【化43】
化合物(i)2.7g(10mmol)をジクロロメタン10mlに溶解させ、氷浴で冷却しながら塩化アルミニウム2.7g(20mmol)を20分かけて添加した。得られた混合溶液へベンゾイルクロリド1.4g(11mmol)を20分かけて滴下した。その後、徐々に室温に戻してから、1時間攪拌した。再び氷浴で冷却しながら、塩化アルミニウム2.7g(20mmol)を20分かけて添加し、そこへアセチルクロリド1.2g(15mmol)を20分かけて滴下した。その後、徐々に室温に戻し一晩攪拌した。反応終了後、氷水に反応溶液を投入し、酢酸エチルで抽出した後、水、飽和炭酸水素ナトリウム水溶液及び飽和食塩水で洗浄し、硫酸マグネシウムで乾燥してから溶媒を留去した。
シリカゲルカラムクロマトグラフィー(溶出液;ジクロロメタン→ノルマルヘキサン:酢酸エチル=2:1)により生成物を単離し、淡黄色固体の化合物(ii)2.8g(収率70%)を得た。該化合物(ii)の1H−NMR測定結果を以下に示す。
【0128】
1H−NMR(400MHz,CDCl3,TMS,ppm)δ:1.16(t,3H)、2.725(s,3H)、2.91(t,2H)、4.08(q,2H)、4.73(t,2H)、7.525−7.86(m,5H)、8.10(dd,2H)、8.18(dd,1H)、8.20(dd,1H)、8.625(d,1H)、8.725(d,1H)
【0129】
(工程3)
【化44】
塩化ヒドロキシルアミン0.12g(1.7mmol)及び酢酸ナトリウム0.17g(2.0mmol)を水1.4mlに溶解させ、そこへ化合物(ii)0.60g(1.58mmol)及びエタノール11mlを添加し、7時間還流させた。
反応終了後、反応溶液の溶媒を留去し、得られた固体を水で洗浄した後、THFに溶解させ、硫酸マグネシウムで乾燥し、溶媒を留去することにより、淡黄色液体の化合物(iii)0.60g(収率89%)を得た。該化合物(iii)の1H−NMR測定結果を以下に示す。
【0130】
1H−NMR(400MHz,CDCl3,TMS,ppm)δ:1.16(t,3H)、2.41(s,3H)、2.89(t,2H)、4.09(q,2H)、4.69(t,2H)、7.475−7.63(m,5H)、7.85(dd,1H)、8.045(dd,2H)、8.07(dd,1H)、8.325(d,1H)、8.60(d,1H)
【0131】
(工程4)
【化45】
化合物(iii)0.60g(1.4mmol)をt−ブチルメチルエーテル7mlに溶解させ、アセチルクロリド0.11g(1.4mmol)添加し、氷浴で冷却しながらトリエチルアミン0.28ml(2.0mmol)を滴下した。徐々に室温に戻しながら3時間攪拌した。反応終了後、氷水に反応混合液を投入し、酢酸エチルで抽出した後、水、飽和炭酸水素ナトリウム水溶液、飽和食塩水で洗浄し、硫酸マグネシウムで乾燥してから溶媒を留去した。シリカゲルカラムクロマトグラフィー(溶出液;ジクロロメタン→ノルマルヘキサン:酢酸エチル=2:1)により生成物を単離し、淡黄色固体の化合物(iv)0.30g(収率70%)を得た。該化合物(iv)の物性を以下に示す。
【0132】
1H−NMR(400MHz,CDCl3,TMS,ppm)δ:1.16(t,3H)、2.23(s,3H)、2.51(s,3H)、2.90(t,2H)、4.09(q,2H)、4.71(t,2H)、7.51−7.62(m,5H)、7.75(dd,2H)、7.99(dd,1H)、8.09(dd,1H)、8.46(d,1H)、8.59(d,1H)
UVスペクトル(移動層;アセトニトリル):λmax=260、292、333nm
熱分解温度(窒素ガス雰囲気下、1.5mgのサンプルを昇温速度10℃/分で加熱し、5%質量減少した時の温度):224.5℃
分子量:470.52
【0133】
<実施例2>
実施例1の工程1において、エチルアクリレート(40mmol)の代わりにイソブチルアクリレート(40mmol)を用い、工程2において、ベンゾイルクロリド(11mmol)の代わりに2−メチルベンゾイルクロリド(11mmol)を用いたこと以外は実施例1と同様に操作を行い、下記の淡黄色固体の化合物(v)を得た。該化合物(v)の物性を以下に示す。
【0134】
【化46】
1H−NMR(400MHz,CDCl3,TMS,ppm)δ:0.81(d、6H)、1.805(m、1H)、2.29(s、3H)、2.35(s、3H)、2.50(s、3H)、2.90(t、2H)、3.81(d、2H)、4.70(t、2H)、7.28−7.53(m、6H)、7.97(dd、1H)、8.10(dd、1H)、8.42(d、1H)、8.51(d、1H)
UVスペクトル(移動層;アセトニトリル):λmax=260、295.5、334nm
熱分解温度(窒素ガス雰囲気下、1.5mgのサンプルを昇温速度10℃/分で加熱し、5%質量減少した時の温度):236℃
分子量:512.6
【0135】
<実施例3>
実施例1の工程1において、エチルアクリレート(40mmol)の代わりに2−メトキシエチルアクリレート(40mmol)を用い、工程2において、ベンゾイルクロリド(11mmol)の代わりに2−メチルベンゾイルクロリド(11mmol)を用いたこと以外は実施例1と同様に操作を行い、下記の淡黄色固体の化合物(vi)を得た。該化合物(vi)の物性を以下に示す。
【0136】
【化47】
1H−NMR(400MHz,CDCl3,TMS,ppm)δ:2.29(s、3H)、2.35(s、3H)、2.50(s、3H)、2.95(t、2H)、3.32(s、3H)、3.50(t、2H)、4.19(t、2H)、4.70(t、2H)、7.30−7.54(m、6H)、7.97(dd、1H)、8.10(dd、1H)、8.42(d、1H)、8.51(d、1H)
UVスペクトル(移動層;アセトニトリル):λmax=258.5、296、334.5nm
熱分解温度(窒素ガス雰囲気下、1.5mgのサンプルを昇温速度10℃/分で加熱し、5%質量減少した時の温度):243℃
分子量:514.57
【0137】
<参考例4>
(工程1)
【化48】
カルバゾール5.0g(30mmol)をジメチルホルムアミド(DMF)6mlに溶解させ、炭酸エチレン13.2g(150mmol)とジアザビシクロウンデセン(DBU)5.5ml(037mmol)を添加し、100℃で3時間加熱還流した。冷却後、水を投入し、酢酸エチルで抽出した後、水洗してから溶媒を留去した。
次に、得られた濃縮物をアセトニトリル15mlで希釈し、そこへアセチルクロライド2g(23mmol)及びトリエチルアミン4ml(29mmol)を添加し、室温で3時間攪拌した。反応終了後、水を投入し、酢酸エチルで抽出した後、水で洗浄してから硫酸マグネシウムで乾燥させ、溶媒を留去することにより、淡黄色液体の上記化合物(vii)6.0g(収率80%)を得た。
【0138】
(工程2)
【化49】
化合物(vii)2.5g(9.9mmol)をジクロロメタン10mlに溶解させ、氷浴で冷却しながら塩化アルミニウム1.5gを20分かけて添加した。その後、ベンゾイルクロリド1.4g(10.0mmol)を滴下し、徐々に室温に戻してから1時間攪拌した。
再び氷浴で冷却しながら塩化アルミニウム1.5gを20分かけて添加、そこへアセチルクロリド0.93g(11.8mmol)を添加し、徐々に室温に戻してから一時間攪拌した。
反応終了後、氷水に反応溶液を投入し、酢酸エチルで抽出した後、水、飽和炭酸水素ナトリウム水溶液、及び飽和食塩水で洗浄し、硫酸マグネシウムで乾燥してから溶媒を留去した。
シリカゲルカラムクロマトグラフィー(溶出液;ジクロロメタン→ノルマルヘキサン:酢酸エチル=2:1)により化合物を単離し、淡黄色固体の化合物(viii)2.8g(収率70%)を得た。
【0139】
(工程3)
【化50】
塩化ヒドロキシルアミン0.38g(5.4mmol)、酢酸ナトリウム0.54g(6.4mmol)を水4.5mlに溶解させ、そこへ前記化合物(viii)2.0g(5.0mmol)及びエタノール35mlを添加し、2時間還流させた。
反応終了後、反応溶液の溶媒を留去し、得られた固体を水で洗浄した後、THFに溶解させ、硫酸マグネシウムで乾燥し、溶媒を留去することにより、淡黄色液体の化合物(ix)1.8g(収率89%)を得た。
【0140】
(工程4)
【化51】
化合物(ix)0.8g(2.0mmol)をt−ブチルメチルエーテル10mlに溶解させ、アセチルクロライド0.16g(2.0mmol)を添加し、氷浴で冷却しながらトリエチルアミン0.4ml(2.9mmol)を滴下した。徐々に室温に戻しながら3時間攪拌した。
反応終了後、氷水に反応溶液を投入し、酢酸エチルで抽出した後、水、飽和炭酸水素ナトリウム水溶液、及び飽和食塩水で洗浄し、硫酸マグネシウムで乾燥してから溶媒を留去した。
シリカゲルカラムクロマトグラフィー(溶出液;ジクロロメタン→ノルマルヘキサン:酢酸エチル=2:1)により化合物を単離し、下記物性の淡黄色固体の化合物(x)0.6g(収率70%)を得た。
【0141】
(化合物(x)の諸物性)
1H−NMR(400MHz,CDCl3,TMS,ppm)δ:1.93(s,3H)、2.50(s,3H)、2.85(s,3H)、4.31(t,2H)、4.40(t,2H)、7.50−7.62(m,5H)、7.75(dd,2H)、7.99(dd,1H)、8.09(dd,1H)、8.46(d,1H)、8.59(d,1H)
UVスペクトル(移動層;アセトニトリル):λmax=260、296、334nm
熱分解温度(窒素ガス雰囲気下、1.5mgのサンプルを昇温速度10℃/分で加熱し、5%質量減少した時の温度):225℃
分子量:456.49
【0142】
<参考例5>
(工程1)
【化52】
9−カルバゾール−9−プロピオン酸0.96g(40mmol)をアセトニトリル10mlで溶解した後、モルホリン18g(210mmol)を添加した。得られた混合液へ塩化チオニル14g(120mmol)を滴下し、室温で3時間攪拌した。
反応終了後、水を添加してから30分攪拌した。その後、酢酸エチルで抽出し、水で洗浄した後、硫酸マグネシウムで乾燥してから溶媒を留去することにより、淡黄色液体の化合物(xi)10g(収率81%)を得た。
【0143】
(工程2〜4)
【化53】
【0144】
上記化学反応式のとおり、実施例5の工程2〜4では、実施例4の工程2〜4において、工程2で化合物(vii)の代わりに化合物(xi)を用いたこと以外は同様の操作を採用することにより、下記物性の淡黄色固体の化合物(xii)を得た。
【0145】
(化合物(xii)の諸物性)
1H−NMR(400MHz,CDCl3,TMS,ppm)δ:2.50(s,3H)、2.80(t,2H)2.85(s,3H)、2.90(br,2H)、3.00(br,2H)、3.35(br,2H)、3.46(br,2H)4.72(t,2H)、7.51−7.62(m,5H)、7.75(dd,2H)、7.99(dd,1H)、8.09(dd,1H)、8.46(d,1H)、8.59(d,1H)
UVスペクトル(移動層;アセトニトリル):λmax=260,292,335nm
熱分解温度(窒素ガス雰囲気下、1.5mgのサンプルを昇温速度10℃/分で加熱し、5%質量減少した時の温度):234℃
分子量:511.57
【0146】
<試験例1〜3、参考試験例4及び5、比較試験例1及び2>相溶性及び感度試験
三官能アクリレート「M−305」(商品名、東亞合成(株)製)100質量部に、実施例1〜3又は参考例4〜5で得た光重合開始剤(化合物(iv)〜(vi)、(x)及び(xii);試験例1〜3及び参考試験例4及び5用)、下記「IRGACURE OXE02」(チバ・スペシャルティ・ケミカルズ(株)製、比較試験例1用。以下、「OXE02」と略称することがある。)又は下記「N−1919」((株)ADEKA製、比較試験例2用)を5質量部添加した。ここで、目視により、得られた混合物中の固体の有無を確認し、エチレン性不飽和結合を有する化合物(特に(メタ)アクリロイルオキシ基を有する化合物)との相溶性の指標とした。結果を表1に示す。次いで、前記混合物をクロロホルムで溶解することにより、光硬化性組成物を調製した。
得られた光硬化性組成物を各々、クロムをスパッタしたガラス基板上にスピンコートし塗布した。
こうして得られた塗布膜にUVを露光しながら、赤外分光装置で810cm-1のピークの減少量を経時的に記録し、二重結合の消失がどの程度進行しているか(反応率)を確認し、感度の指標とした。結果を表1に示す。
【0147】
【化54】
【0148】
【表1】
【0149】
表1より、本発明の光重合開始剤は、エチレン性不飽和二重結合を有する化合物、特に多官能(メタ)アクリレートとの相溶性が高く、無溶剤の光重合開始剤を調製することも可能である。同時に、本発明の光重合開始剤を含有する光硬化性組成物は、従来の光重合開始剤を含有する光硬化性組成物に比べて、より高感度であることが分かる。
【0150】
<試験例6〜8、参考試験例9及び10、比較試験例3及び4>溶剤に対する溶解性試験
プロピレングリコールモノエチルエーテルアセテート(PGMEA)、メタノール、3−メトキシ−3−メチル−1−ブチルアセテート(MMBA)、メチルイソブチルケトン(MIBK)、クロロホルムそれぞれ100質量部に対する光重合開始剤の溶解量を調査し、下記評価基準に従って溶解性を評価した。結果を表2に示す。
A:10質量部以上溶解した。
B:5質量部以上、10質量部未満の範囲で溶解した。
C:2質量部以上、5質量部未満の範囲で溶解した。
D:2質量部未満しか溶解しなかった。
ここで、「溶解した」とは、目視により固体が見えない状態を指す。
【0151】
【表2】
【0152】
表2より、従来の光重合開始剤に比べ、本発明の光重合開始剤は、各種溶剤、特にエーテル結合及び/又はエステル結合を有する25℃で液体の化合物に対する溶解性に優れていることがわかる。
また、比較試験例4で使用した光重合開始剤「N−1919」は、比較試験例3で使用した光重合開始剤「OXE02」に比べ、溶解性が高くなっている。これは、「N−1919」は、「OXE02」の構造に置換基を付与することによって溶解性を高めたものだからである。試験例6〜10で使用した化合物(iv)〜(vi)、(x)及び(xii)は、この「N−1919」よりも溶剤への溶解性が高いのみならず、分子量も小さくなっており、光硬化性組成物中における含有量を低減することができ、より一層優れた光重合開始剤である。
【0153】
<製造例1>バインダー樹脂の製造
ベンジルメタクリレート30質量部、スチレン38質量部、メタクリル酸18質量部及びt−ブチルパーオキシ−2−エチルヘキサノエート(日油(株)製の「パーブチルO」)10質量部の混合液を、プロピレングリコールモノエチルエーテルアセテート150質量部を入れた重合槽中へ、窒素気流下、100℃で3時間かけて滴下した。滴下終了後、さらに100℃で3時間加熱し、重合体溶液を得た。
得られた重合体溶液に、グリシジルメタクリレート14質量部、トリエチルアミン0.2質量部及びp−メトキシフェノール0.05質量部を添加し、110℃で10時間加熱することによりバインダー樹脂を調製した。
得られたバインダー樹脂の固形分濃度は38質量%、酸価は75mgKOH/g、重量平均分子量は10,000であった。
【0154】
<製造例2>青色顔料分散液の調製
【表3】
表3に記載の各成分を各配合量で混合し、直径0.3mmジルコニアビーズを用いペイントシェーカー(浅田鉄工(株)製)で3時間分散させ、青色顔料分散液(固形分濃度:21質量%)を調製した。
【0155】
<製造例3>赤色顔料分散液の調製
【表4】
表4に記載の各成分を各配合量で混合し、直径0.3mmジルコニアビーズを用いペイントシェーカー(浅田鉄工(株)製)で3時間分散させ、赤色顔料分散液(固形分濃度:21質量%)を調製した。
【0156】
<製造例4>緑色顔料分散液の調製
【表5】
表5に記載の各成分を各配合量で混合し、直径0.3mmジルコニアビーズを用いペイントシェーカー(浅田鉄工(株)製)で3時間分散させ、緑色顔料分散液(固形分濃度:21質量%)を調製した。
【0157】
以下の実施例において、感度、解像度、現像性及び密着性の評価を以下の通りに実施した。
(感度)
露光工程において、露光量を100mJ/cm2又は200mJ/cm2とし、光が照射された領域の現像処理及びポストベーク後の膜厚が、露光前の膜厚100%に対して95%以上であった場合に「膜厚が十分」であるとし、下記評価基準に従って評価した。
A:露光量100mJ/cm2であれば、十分な膜厚が得られた。
B:露光量100mJ/cm2では不十分だが、露光量200mJ/cm2であれば十分な膜厚が得られた。
なお、膜厚は、触針式膜厚測定器「SURFCORDER ET4000A」((株)小坂研究所製)を用いて測定した。
(解像度)
下記評価基準に従って、解像度を評価した。
a:露光現像時において、線幅10μm未満でも良好にパターンを得られた。
b:線幅15〜20μmであれば良好なパターンを得られた。
(現像性)
露光時において、未露光部(光が照射されなかった部分)の残渣を観察し、下記評価基準に従って、現像性を評価した。
○:残渣を全く確認できなかった。
×:残渣を確認できた。
(密着性)
現像後に流されずに密着していた独立パターンの線幅を調査し、下記評価基準に従って、密着性を評価した。
◎:線幅を5μm以下としても、良好な孤立パターンが得られた。
○:線幅6〜10μmとすれば、良好な孤立パターンを得られた。
【0158】
<実施例6>青色光硬化性樹脂組成物の調製及びパターン形成
【表6】
表6に記載の各成分を各配合量で混合し、ディゾルバーで1時間攪拌し、青色光硬化性樹脂組成物を調製した。
得られた青色光硬化性樹脂組成物をガラス基板上にスピンコートし、80℃で3分間加熱することにより、青色光硬化樹脂組成物の塗膜を形成した。得られた塗膜を、所定線幅のマスクを通して、塗膜側から光源として高圧水銀灯にて露光した後、100倍に希釈したディスパーズH(ヘンケル社製)を用いてスプレー現像を行い、現像終了後、230℃で30分ポストベークし、パターンを形成した。
感度、解像度、現像性及び密着性の評価結果を表9に示す。
【0159】
<比較例1、2>青色光硬化性樹脂組成物の調製及びパターン形成
実施例6において、実施例1で得た化合物(iv)の代わりに、前記「IRGACURE OXE02」(チバ・スペシャルティ・ケミカルズ(株)製)又は前記「N−1919」((株)ADEKA製)を用いたこと以外は同様にして青色光硬化性樹脂組成物を調製し、パターンを形成した。
感度、解像度、現像性及び密着性の評価結果を表9に示す。
【0160】
<実施例7>赤色光硬化性樹脂組成物の調製及びパターン形成
【表7】
表7に記載の各成分を各配合量で混合し、ディゾルバーで1時間攪拌し、赤色光硬化性樹脂組成物を調製した。
得られた赤色光硬化性樹脂組成物について、実施例6と同様の操作を行うことにより、パターンを形成した。
感度、解像度、現像性及び密着性の評価結果を表9に示す。
【0161】
<比較例3、4>赤色光硬化性樹脂組成物の調製及びパターン形成
実施例7において、実施例1で得た化合物(iv)の代わりに、前記「IRGACURE OXE02」(チバ・スペシャルティ・ケミカルズ(株)製)又は前記「N−1919」((株)ADEKA製)を用いたこと以外は同様にして赤色光硬化性樹脂組成物を調製し、パターンを形成した。
感度、解像度、現像性及び密着性の評価結果を表9に示す。
【0162】
<実施例8>緑色光硬化性樹脂組成物の調製及びパターン形成
【表8】
表8に記載の各成分を各配合量で混合し、ディゾルバーで1時間攪拌し、緑色光硬化性樹脂組成物を調製した。
得られた緑色光硬化性樹脂組成物について、実施例6と同様の操作を行うことにより、パターンを形成した。
感度、解像度、現像性及び密着性の評価結果を表9に示す。
【0163】
<比較例5、6>緑色光硬化性樹脂組成物の調製及びパターン形成
実施例8において、実施例1で得た化合物(iv)の代わりに、前記「IRGACURE OXE02」(チバ・スペシャルティ・ケミカルズ(株)製)又は前記「N−1919」((株)ADEKA製)を用いたこと以外は同様にして緑色光硬化性樹脂組成物を調製し、パターンを形成した。
感度、解像度、現像性及び密着性の評価結果を表9に示す。
【0164】
<実施例9〜11>
実施例6〜8それぞれにおいて、実施例1で得た化合物(iv)の代わりに、実施例2で得た化合物(v)を用いたこと以外は同様にして、青色光硬化性樹脂組成物、赤色光硬化性樹脂組成物及び緑色光硬化性樹脂組成物を調製し、パターンを形成した。
感度、解像度、現像性及び密着性の評価結果を表9に示す。
【0165】
<実施例12〜14>
実施例6〜8それぞれにおいて、実施例1で得た化合物(iv)の代わりに、実施例3で得た化合物(vi)を用いたこと以外は同様にして、青色光硬化性樹脂組成物、赤色光硬化性樹脂組成物及び緑色光硬化性樹脂組成物を調製し、パターンを形成した。
感度、解像度、現像性及び密着性の評価結果を表9に示す。
【0166】
<参考例15〜17>
実施例6〜8それぞれにおいて、実施例1で得た化合物(iv)の代わりに、参考例4で得た化合物(x)を用いたこと以外は同様にして、青色光硬化性樹脂組成物、赤色光硬化性樹脂組成物及び緑色光硬化性樹脂組成物を調製し、パターンを形成した。
感度、解像度、現像性及び密着性の評価結果を表9に示す。
【0167】
<参考例18〜20>
実施例6〜8それぞれにおいて、実施例1で得た化合物(iv)の代わりに、参考例5で得た化合物(xii)を用いたこと以外は同様にして、青色光硬化性樹脂組成物、赤色光硬化性樹脂組成物及び緑色光硬化性樹脂組成物を調製し、パターンを形成した。
感度、解像度、現像性及び密着性の評価結果を表9に示す。
【0168】
【表9】
【0169】
表9より、本発明の光重合開始剤を含有する光硬化性組成物は、従来の光重合開始剤を含有する光硬化性組成物と同等の現像性を有し、また、感度、解像度及び密着性については、従来の光重合開始剤を含有する光硬化性組成物より優れていることがわかる。このことより、本発明の光重合開始剤を含有する光硬化性組成物は、深部硬化性に優れていると言える。
【産業上の利用可能性】
【0170】
本発明の光重合開始剤は、光(特に波長450nm以下の短波長の光)に対する感度が高いため、薄膜化することが可能であり、低コストで高品質のパターンを形成することができること、さらに、エチレン性不飽和結合を有する化合物(特に(メタ)アクリロイルオキシ基を有する化合物)との相溶性や溶剤に対する溶解性が高いことより、カラーフィルタ用光硬化性組成物に含有させる光重合開始剤、さらには高遮光性のブラックマトリックス用光硬化性組成物に含有させる光重合開始剤として有用である。
【符号の説明】
【0171】
1 透明基板
2 遮光部
3 着色層
10 カラーフィルタ
20 対向基板
30 液晶層
40 液晶表示装置
【技術分野】
【0001】
本発明は、光重合開始剤、光硬化性組成物、該組成物を用いたパターン形成方法、該組成物を用いたカラーフィルタ、該カラーフィルタを有する液晶表示装置、及び光重合開始剤の製造方法に関する。さらに詳しくは、光硬化性組成物中のエチレン性不飽和結合を有する化合物、エーテル結合及び/又はエステル結合を有する25℃で液体の化合物との相溶性、及び溶剤(前記のエーテル結合及び/又はエステル結合を有する25℃で液体の化合物を含む。)に対する溶解性が高く、光(特に波長450nm以下の短波長の光)に対する感度が高く、露光時の光により発生する分解物による重合物の汚染や装置の汚染が無い光重合開始剤であって、解像度、現像性、深部硬化性及び基板との密着性が良好となる光硬化性組成物を提供する光重合開始剤、該光重合開始剤を含有する光硬化性組成物、該光硬化性組成物を用いたパターン形成方法、該光硬化性組成物を用いたカラーフィルタ、該カラーフィルタを有する液晶表示装置、及び前記光重合開始剤の製造方法に関する。
【背景技術】
【0002】
光硬化性樹脂組成物は、例えばバインダー樹脂や重合性モノマー及び光重合開始剤を含有するものであり、光(粒子線等の放射線を含む電磁波)を照射することにより重合硬化させることができるので、光硬化性インキ、感光性印刷版、カラーフィルタ、各種フォトレジスト等に用いられる。光硬化性樹脂組成物を硬化させる光としては、その取り扱いのし易さや感度等の点から、450nm以下の波長の光(紫外線)が用いられる場合が多く、その光源としては波長365nm、405nm、436nmの波長に強い発光を有する高圧水銀灯や、KrF及びArF等のエキシマレーザーが利用されている。また、より微細なパターン形成に用いられる場合に、電子線やEUV(極端紫外光:Extreme Ultra Violet)等の波長の短い電磁波や放射線の適用が検討されている。
光による硬化は、熱硬化に比べて省エネルギーであることや、フォトマスクを介して照射を行うことにより所望のパターンで硬化できることなどから、種々の用途分野において需要が高く、特に生産性が向上し、かつ光重合開始剤の添加量を削減し得る高感度の光重合性開始剤に対して、需要が高まっている。
また、顔料分散法を用いたカラーフィルタは、通常、分散剤等により顔料を分散してなる顔料分散液に光硬化性組成物をガラス基板に塗布して乾燥後、フォトマスクを用いて露光し、現像を行うことによって着色パターンを形成し、加熱することによりパターンを固着して画素を形成する。これらの工程を、各色ごとに繰り返してカラーフィルタを形成する。このようなカラーフィルタの画像形成に用いられる光硬化性組成物には、十分な解像性、基板との密着性及び低現像残渣等の特性が求められる。
さらに近年では、色濃度が高い画素やそれ自体が遮光層として機能する光学濃度の高いブラックマトリックス用レジストが利用されており、光硬化性組成物中の着色顔料やカーボンブラック等の黒色顔料の含有量が高くなり、光硬化性組成物の光透過性が下がる傾向にある。その結果、露光光源からの光が、光硬化性樹脂塗膜の深部まで到達せず、解像性、基板との密着性及び現像性等が悪化するという問題があり、生産性や、カラーフィルタに要求される精度、さらには信頼性が低下する。
また、カラーフィルタが形成されるガラス基板は年々大型化しており、大面積露光が行われているため、露光照度が小さくなる傾向があることや、更なる生産性向上のために露光時間の短縮が求められていることから、より高感度の光重合開始剤が必要とされている。
そこで、高感度及び高解像度等を達成し得る光硬化性組成物を得る方法として、光重合開始剤としてオキシムエステル化合物を用いることが提案されている(特許文献1〜5参照)。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】特許第3860170号明細書
【特許文献2】特開2006−036750号公報
【特許文献3】特許第3992725号明細書
【特許文献4】特開2010−037542号公報
【特許文献5】韓国公開特許第2009−0046108号公報
【発明の概要】
【発明が解決しようとする課題】
【0004】
特許文献1〜5に記載の光重合開始剤では、感度、解像度、現像性、及び相溶性や溶解性、特に光硬化性組成物中のエチレン性不飽和結合を有する化合物(特に(メタ)アクリロイルオキシ基を有する化合物)との相溶性や溶剤に対する溶解性が不十分であり、使用条件に制限があるため、さらなる改善の余地がある。また、特許文献1〜5に記載の光重合開始剤では、露光時の光により発生する分解物がマスクに付着し、その結果、焼付け時のパターン形成不良を起こし、収率の低下を招くことがあった。そこで、発生した分解物が、重合物や装置等を汚染しない光重合開始剤が望まれている。
【0005】
よって、本発明の課題は、光硬化性組成物中のエチレン性不飽和結合を有する化合物(特に(メタ)アクリロイルオキシ基を有する化合物)、エーテル結合及び/又はエステル結合を有する25℃で液体の化合物との相溶性、及び溶剤(前記のエーテル結合及び/又はエステル結合を有する25℃で液体の化合物を含む。)に対する溶解性が高く、光(特に波長450nm以下の短波長の光)に対する感度が高く、露光時の光により発生する分解物による重合物の汚染や装置の汚染が無い光重合開始剤であって、解像度、現像性、深部硬化性及び基板との密着性が良好となる光硬化性組成物を提供し得る光重合開始剤を提供することである。また、該光重合開始剤を含有する光硬化性組成物、該光硬化性組成物を用いたパターン形成方法、該光硬化性組成物を用いたカラーフィルタ及び該カラーフィルタを有する液晶表示装置を提供することである。さらには、前記光重合開始剤を低コストで簡便に製造する方法を提供することである。
【課題を解決するための手段】
【0006】
本発明者は、前記課題を達成するために鋭意研究を重ねた結果、後述する一般式(I−a)で表される特定構造の光重合開始剤であれば、前記課題を解決し得ることを見出した。
すなわち、本発明は、下記[1]〜[17]に関する。
[1]下記一般式(I−a)で表される光重合開始剤。
【0007】
【化1】
【0008】
(式中、R1〜R11は、それぞれ独立に、水素原子、ハロゲン原子、置換もしくは無置換の炭素数1〜20のアルキル基、置換もしくは無置換の炭素数2〜20のアルケニル基、置換もしくは無置換の環形成原子数3〜10のシクロアルキル基、置換もしくは無置換の炭素数4〜20のシクロアルケニル基、ヒドロキシル基、置換もしくは無置換の炭素数1〜20のアルコキシ基、置換もしくは無置換の炭素数2〜20のアルケニルオキシ基、置換もしくは無置換の炭素数1〜20のアルカノイル基、置換もしくは無置換の炭素数2〜20のアルケノイル基、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数3〜14のヘテロ環基を示す。R3は、R4又はR5と一緒になって環を形成していてもよい。R4は、R5と一緒になって環を形成していてもよい。
また、Arは、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数5〜14のヘテロアリール基を示す。
nは、1〜10の整数を示す。nが2〜10の整数の場合、複数のR4及びR5は、それぞれ同一でも異なっていてもよい。)
[2]前記一般式(I−a)中、R1、R2及びR3が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、nが2であり、複数のR4及びR5が、それぞれ独立に、水素原子又は置換もしくは無置換の炭素数1〜20のアルキル基であり、Arが、置換もしくは無置換の環形成炭素数6〜14のアリール基である、上記[1]に記載の光重合開始剤。
[3]前記一般式(I−a)中、R1、R2及びR3が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、nが2であり、複数のR4及びR5が、それぞれ独立に、水素原子又は置換もしくは無置換の炭素数1〜20のアルキル基であり、Arが、置換もしくは無置換の環形成原子数5〜14のヘテロアリール基である、上記[1]に記載の光重合開始剤。
[4]分子量が515以下である、上記[2]又は[3]に記載の光重合開始剤。
【0009】
[5]前記一般式(I−a)中、R1及びR2が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、R3が置換もしくは無置換の炭素数3〜18のシクロアルキル基であり、nが2であり、複数のR4及びR5が、それぞれ独立に水素原子もしくは置換もしくは無置換の炭素数1〜20のアルキル基である、上記[1]に記載の光重合開始剤。
[6]前記一般式(I−a)中、R1及びR2が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、R3が環形成原子数3〜6の環状エーテル基で置換された炭素数1〜20のアルキル基であり、nが2であり、複数のR4及びR5が、それぞれ独立に水素原子もしくは置換もしくは無置換の炭素数1〜20のアルキル基である、上記[1]に記載の光重合開始剤。
[7]分子量が550以下である、上記[5]又は[6]に記載の光重合開始剤。
[8]バインダー樹脂及び/又はエチレン性不飽和結合を有する化合物と、上記[1]〜[7]のいずれかに記載の光重合開始剤を含有する光硬化性組成物。
[9]光重合開始剤の含有量が、光硬化性組成物の固形分に対して2〜50質量%である、上記[8]に記載の光硬化性組成物。
[10]さらに色材を含有する、上記[8]又は[9]に記載の光硬化性組成物。
[11]さらに、エーテル結合及び/又はエステル結合を有する25℃で液体の化合物を光重合開始剤1質量部に対して5〜100質量部含有する、上記[8]〜[10]のいずれかに記載の光硬化性組成物。
[12]エチレン性不飽和結合を有する化合物として、エーテル結合及び/又はエステル結合を持つエチレン性不飽和結合を有する化合物を少なくとも1種用い、溶剤を光硬化性組成物全量の10質量%以下のみ含有する、上記[8]〜[10]のいずれかに記載の光硬化性組成物。
[13]カラーフィルタ用である、上記[10]〜[12]のいずれかに記載の光硬化性組成物。
[14]上記[10]〜[12]のいずれかに記載の光硬化性組成物を用いたカラーフィルタ。
[15]上記[14]に記載のカラーフィルタと対向基板と、前記カラーフィルタと前記対向基板との間に形成された液晶層とを有する液晶表示装置。
[16]上記[10]〜[12]のいずれかに記載の光硬化性組成物を基板に塗布し、乾燥した後、フォトマスクを用いて露光し、次いで現像を行うことによる、パターン形成方法。
[17]下記一般式(1)
【0010】
【化2】
【0011】
(式中、R6〜R11は、それぞれ独立に、水素原子、ハロゲン原子、置換もしくは無置換の炭素数1〜20のアルキル基、置換もしくは無置換の炭素数2〜20のアルケニル基、置換もしくは無置換の環形成原子数3〜10のシクロアルキル基、置換もしくは無置換の炭素数4〜20のシクロアルケニル基、ヒドロキシル基、置換もしくは無置換の炭素数1〜20のアルコキシ基、置換もしくは無置換の炭素数2〜20のアルケニルオキシ基、置換もしくは無置換の炭素数1〜20のアルカノイル基、置換もしくは無置換の炭素数2〜20のアルケノイル基、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数3〜14のヘテロ環基を示す。)
で表されるカルバゾール誘導体と、下記一般式(2a)
【0012】
【化3】
【0013】
(式中、R3、R4’及びR5’は、それぞれ独立に、水素原子、ハロゲン原子、置換もしくは無置換の炭素数1〜20のアルキル基、置換もしくは無置換の炭素数2〜20のアルケニル基、置換もしくは無置換の環形成原子数3〜10のシクロアルキル基、置換もしくは無置換の炭素数4〜20のシクロアルケニル基、ヒドロキシル基、置換もしくは無置換の炭素数1〜20のアルコキシ基、置換もしくは無置換の炭素数2〜20のアルケニルオキシ基、置換もしくは無置換の炭素数1〜20のアルカノイル基、置換もしくは無置換の炭素数2〜20のアルケノイル基、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数3〜14のヘテロ環基を示す。R3は、R5’と一緒になって環を形成していてもよい。)
で表されるアクリレート誘導体とを塩基の存在下に反応させることにより、下記一般式(3a’)
【0014】
【化4】
【0015】
(式中、R3、R4’、R5’、R6〜R11は、前記定義の通りである。)
で表されるカルボニルアルキル基導入体を得、得られたカルボニルアルキル基導入体と下記一般式(4)、(5)
【0016】
【化5】
【0017】
(式中、Xは、ハロゲン原子又は−OC(=O)Arを示し、X’は、ハロゲン原子又は−OC(=O)R2を示す。
Arは、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数5〜14のヘテロアリール基を示す。
R2は、水素原子、ハロゲン原子、置換もしくは無置換の炭素数1〜20のアルキル基、置換もしくは無置換の炭素数2〜20のアルケニル基、置換もしくは無置換の環形成原子数3〜10のシクロアルキル基、置換もしくは無置換の炭素数4〜20のシクロアルケニル基、ヒドロキシル基、置換もしくは無置換の炭素数1〜20のアルコキシ基、置換もしくは無置換の炭素数2〜20のアルケニルオキシ基、置換もしくは無置換の炭素数1〜20のアルカノイル基、置換もしくは無置換の炭素数2〜20のアルケノイル基、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数3〜14のヘテロ環基を示す。)
で表される2つのアシル化剤をルイス酸の存在下に反応させることにより、下記一般式(6a’)
【0018】
【化6】
【0019】
(式中、R2、R3、R4’、R5’、R6〜R11及びArは、前記定義の通りである。)
で表されるジケトン体を得、得られたジケトン体とヒドロキシルアミンを反応させることにより、下記一般式(7a’)
【0020】
【化7】
【0021】
(式中、R2、R3、R4’、R5’、R6〜R11及びArは、前記定義の通りである。)
で表されるオキシム体を得、得られたオキシム体と下記一般式(8)
【0022】
【化8】
【0023】
(式中、Yは、ハロゲン原子又は−OC(=O)R1を示す。
また、R1は、水素原子、ハロゲン原子、置換もしくは無置換の炭素数1〜20のアルキル基、置換もしくは無置換の炭素数2〜20のアルケニル基、置換もしくは無置換の環形成原子数3〜10のシクロアルキル基、置換もしくは無置換の炭素数4〜20のシクロアルケニル基、ヒドロキシル基、置換もしくは無置換の炭素数1〜20のアルコキシ基、置換もしくは無置換の炭素数2〜20のアルケニルオキシ基、置換もしくは無置換の炭素数1〜20のアルカノイル基、置換もしくは無置換の炭素数2〜20のアルケノイル基、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数3〜14のヘテロ環基を示す。)
で表されるエステル化剤とを反応させることによる、下記一般式(I−a1)
【0024】
【化9】
【0025】
(式中、R1〜R3、R4’、R5’、R6〜R11及びArは、前記定義の通りである。)
で表される光重合開始剤の製造方法。
【発明の効果】
【0026】
本発明の光重合開始剤は、光(特に450nm以下の短波長の光であって、例えば波長365nmや波長405nmの光)に対する感度が非常に高いため、薄膜化することが可能であり、低コストで高品質のパターンを形成することができる。さらに、エチレン性不飽和二重結合を有する化合物、エーテル結合及び/又はエステル結合を有する25℃で液体の化合物との相溶性、及び溶剤(前記のエーテル結合及び/又はエステル結合を有する25℃で液体の化合物を含む。)に対する溶解性が高いため、無溶剤の光硬化性組成物を調製することもでき、また、カラーフィルタ用の光硬化性組成物においては、光重合開始剤の添加量の選択幅が広く、結果として、感度、解像度、現像性及び深部硬化性が向上する。
従来、光重合開始剤に種々の置換基を付与することにより分子量を高めながら溶剤への溶解性が高められてきたが、本発明の光重合開始剤は、比較的分子量が小さく、光硬化性組成物への添加量を低減できるうえ、従来の光重合開始剤よりもエチレン性不飽和二重結合を有する化合物、エーテル結合及び/又はエステル結合を有する25℃で液体の化合物との相溶性、及び溶剤(前記のエーテル結合及び/又はエステル結合を有する25℃で液体の化合物を含む。)に対する溶解性が高く、感度、解像度及び現像性、並びに基板との密着性に優れる。
【図面の簡単な説明】
【0027】
【図1】本発明の液晶表示装置を示す模式図である。
【発明を実施するための形態】
【0028】
[光重合開始剤]
本発明の光重合開始剤は、下記一般式(I)で表される。
【化10】
(式中、R1〜R11は、それぞれ独立に、水素原子、ハロゲン原子、置換もしくは無置換の炭素数1〜20のアルキル基、置換もしくは無置換の炭素数2〜20のアルケニル基、置換もしくは無置換の環形成原子数3〜10のシクロアルキル基、置換もしくは無置換の炭素数4〜20のシクロアルケニル基、ヒドロキシル基、置換もしくは無置換の炭素数1〜20のアルコキシ基、置換もしくは無置換の炭素数2〜20のアルケニルオキシ基、置換もしくは無置換の炭素数1〜20のアルカノイル基、置換もしくは無置換の炭素数2〜20のアルケノイル基、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数3〜14のヘテロ環基を示す。R3は、R4又はR5と一緒になって環を形成していてもよい。R4は、R5と一緒になって環を形成していてもよい。
また、Arは、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数5〜14のヘテロアリール基を示す。
Wは、単結合又は酸素原子を示す。Zは、単結合、酸素原子又は>NR3’(R3’は、置換もしくは無置換の炭素数1〜20のアルキル基を示すか、又はR3’はR3とつながって、窒素原子と共に環を形成している。)を示す。
nは、1〜10の整数を示す。nが2〜10の整数の場合、複数のR4及びR5は、それぞれ同一でも異なっていてもよい。)
【0029】
また、一般式(I)で表される本発明の光重合開始剤の中でも、本発明の効果の観点から、下記一般式(I−a)で表される光重合開始剤(W:単結合、Z:酸素原子の場合に相当する。)及び下記一般式(I−b)で表される光重合開始剤(nが1の場合に相当する。)が好ましい。
【0030】
【化11】
【0031】
上記式中、R1〜R11は、それぞれ独立に、水素原子、ハロゲン原子、置換もしくは無置換の炭素数1〜20のアルキル基、置換もしくは無置換の炭素数2〜20のアルケニル基、置換もしくは無置換の環形成原子数3〜10のシクロアルキル基、置換もしくは無置換の炭素数4〜20のシクロアルケニル基、ヒドロキシル基、置換もしくは無置換の炭素数1〜20のアルコキシ基、置換もしくは無置換の炭素数2〜20のアルケニルオキシ基、置換もしくは無置換の炭素数1〜20のアルカノイル基、炭素数2〜20のアルケノイル基、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数3〜14のヘテロ環基を示す。なお、一般式(I−a)においては、R3は、R4又はR5と一緒になって環を形成していてもよく、また、R4は、R5と一緒になって環を形成していてもよい。
また、Arは、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数5〜14のヘテロアリール基を示す。
一般式(I−b)中のWは、単結合又は酸素原子を示す。また、一般式(I−b)中のZは、単結合、酸素原子又は>NR3’(R3’は、置換もしくは無置換の炭素数1〜20のアルキル基を示すか、又はR3’はR3とつながって、窒素原子と共に環を形成している。)を示す。
一般式(I−a)中のnは、1〜10の整数を示す。nが2〜10の整数の場合、複数のR4及びR5は、それぞれ同一でも異なっていてもよい。
【0032】
(一般式(I)、(I−a)及び(I−b)中の各基について)
nとしては、低分子量とする観点、エチレン性不飽和二重結合を有する化合物との相溶性(以下、単に相溶性と称する。)や溶剤への溶解性(以下、単に溶解性と称する。)の観点及び製造容易性の観点から、2〜8の整数が好ましく、2〜4の整数がより好ましく、2がさらに好ましい。
R1〜R11がそれぞれ独立して示すハロゲン原子としては、例えばフッ素原子、塩素原子、臭素原子、ヨウ素原子等が挙げられる。
R1〜R11がそれぞれ独立して示す炭素数1〜20のアルキル基としては、直鎖状でも分岐鎖状でもよく、例えばメチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、t−ブチル基、n−ペンチル基、イソペンチル基、n−ヘキシル基、n−オクチル基、2−エチル−n−オクチル基、n−デシル基、n−ドデシル基等が挙げられる。これらの中でも、低分子量とする観点、相溶性や溶解性の観点及び製造容易性の観点から、炭素数1〜10のアルキル基が好ましく、炭素数1〜5のアルキル基がより好ましい。
R1〜R11がそれぞれ独立して示す炭素数2〜20のアルケニル基としては、直鎖状でも分岐鎖状でもよく、例えばビニル基、アリル基、7−オクテニル基等が挙げられる。これらの中でも、低分子量とする観点、相溶性や溶解性の観点及び製造容易性の観点から、炭素数2〜10のアルケニル基が好ましく、炭素数2〜6のアルケニル基がより好ましい。
R1〜R11がそれぞれ独立して示す環形成原子数3〜10のシクロアルキル基としては、シクロプロピル基、シクロペンチル基、シクロヘキシル基、シクロオクチル基等が挙げられる。これらの中でも、低分子量とする観点、相溶性や溶解性の観点及び製造容易性の観点から、環形成原子数3〜6のシクロアルキル基が好ましい。
R1〜R11がそれぞれ独立して示す炭素数4〜20のシクロアルケニル基としては、シクロペンテニル基、シクロヘキセニル基、シクロヘキサジエニル基、シクロオクテニル基等が挙げられる。これらの中でも、低分子量とする観点、相溶性や溶解性の観点及び製造容易性の観点から、炭素数4〜10のシクロアルケニル基が好ましく、炭素数4〜6のシクロアルケニル基がより好ましい。
【0033】
R1〜R11がそれぞれ独立して示す炭素数1〜20のアルコキシ基としては、アルキル基部位が、前記した炭素数1〜20のアルキル基であるものが挙げられる。これらの中でも、低分子量とする観点、相溶性や溶解性の観点及び製造容易性の観点から、炭素数1〜10のアルコキシ基が好ましく、炭素数1〜5のアルコキシ基がより好ましい。
R1〜R11がそれぞれ独立して示す炭素数2〜20のアルケニルオキシ基としては、アルケニル基部位が、前記した炭素数2〜20のアルケニル基であるものが挙げられる。これらの中でも、低分子量とする観点、相溶性や溶解性の観点及び製造容易性の観点から、炭素数2〜10のアルケニルオキシ基が好ましく、炭素数2〜6のアルケニルオキシ基がより好ましい。
R1〜R11がそれぞれ独立して示す炭素数1〜20のアルカノイル基としては、直鎖状でも分岐鎖状でもよく、例えばメタノイル基、エタノイル基、n−プロパノイル基、イソプロパノイル基、n−ブタノイル基、t−ブタノイル基、n−ヘキサノイル基、n−オクタノイル基、n−デカノイル基、n−ドデカノイル基等が挙げられる。これらの中でも、低分子量とする観点、相溶性や溶解性の観点及び製造容易性の観点から、炭素数1〜10のアルカノイル基が好ましく、炭素数1〜5のアルカノイル基がより好ましい。
R1〜R11がそれぞれ独立して示す炭素数2〜20のアルケノイル基としては、直鎖状でも分岐鎖状でもよく、例えばエテノイル基、n−プロペノイル基、イソプロペノイル基、n−ブテノイル基、t−ブテノイル基、n−ヘキセノイル基、n−オクテノイル基、n−デセノイル基、n−ドデセノイル基等が挙げられる。これらの中でも、低分子量とする観点、相溶性や溶解性の観点及び製造容易性の観点から、炭素数2〜10のアルケノイル基が好ましく、炭素数2〜6のアルケノイル基がより好ましい。
【0034】
R1〜R11がそれぞれ独立して示す環形成炭素数6〜14のアリール基としては、例えばフェニル基、ナフチル基、アントリル基が挙げられる。
R1〜R11がそれぞれ独立して示す環形成原子数3〜14のヘテロ環基としては、例えば2−フラニル基、2−チオフェニル基、2−ピリジニル基、下記式(A)で表される基(以下、置換基(A)と称する。)
【化12】
等の環形成原子数5〜14の不飽和ヘテロ環基;2−テトラヒドロフリル基、3−テトラヒドロフリル基、ピロリジニル基、ピペリジニル基、2,2,6,6−トリメチルピペリジン−4−イル基
等の環形成原子数3〜10の飽和ヘテロ環基が挙げられる。これらの中でも、低分子量とする観点、相溶性や溶解性の観点及び製造容易性の観点から、環形成原子数5〜6の不飽和ヘテロ環基、環形成原子数3〜6の飽和ヘテロ環基が好ましい。
【0035】
また、前記した「R3とR4又はR5が一緒になって環を形成する」というのは、n=2、Wが単結合及びZが酸素原子である場合を例に挙げると下記式で説明され、右側に例示した環が具体例として挙げられる。
【化13】
【0036】
R4とR5が一緒になって形成する環としては、例えば、シクロペンチル環、シクロオクチル環等の環形成炭素数3〜10(好ましくは3〜6)の環が挙げられる。
【0037】
なお、R1〜R11がそれぞれ独立して示す、前記アルキル基、アルケニル基、シクロアルキル基、シクロアルケニル基、アルコキシ基、アルケニルオキシ基、アルカノイル基、アルケノイル基、アリール基及びヘテロ環基は、置換基を有していてもよい。
R1〜R11がそれぞれ独立して示すアルキル基、アルケニル基の置換基としては、ヒドロキシル基;カルボキシル基;メトキシ基、エトキシ基、プロポキシ基等の直鎖状又は分岐鎖状の炭素数1〜18(好ましくは1〜10、より好ましくは1〜5)のアルコキシ基;直鎖状又は分岐鎖状の炭素数2〜18(好ましくは2〜10、より好ましくは2〜6)のアルケニルオキシ基;直鎖状又は分岐鎖状の炭素数2〜18(好ましくは2〜10、より好ましくは2〜6)のアルケニルチオ基;直鎖状又は分岐鎖状の炭素数1〜18(好ましくは1〜10、より好ましくは1〜5)のアルキルチオ基;炭素数5〜18(好ましくは5〜10)のシクロアルケニル基;シクロプロピル基、シクロペンチル基、シクロヘキシル基等の炭素数3〜18(好ましくは3〜10、より好ましくは3〜6)のシクロアルキル基;フェノキシ基等の環形成炭素数6〜10のアリールオキシ基;フッ素原子、塩素原子、臭素原子、ヨウ素原子等のハロゲン原子;酸素原子(=O);硫黄原子(=S);シアノ基;ニトロ基;トリメチルシリル基等のトリアルキルシリル基;トリメトキシシリル基等のトリアルコキシシリル基;フェニル基、ナフチル基、アントリル基等の環形成炭素数6〜14(好ましくは6〜10)のアリール基;−COR12;−OCOR13;−NR14R15;−NHCOR16;−NHCOOR17;−CONR18R19;−COOR20;−SO3NR21R22;−SO3R23;エポキシ基やテトラヒドロフラニル基等の環形成原子数3〜6の環状エーテル基、2−チエニル基、2−ピリジル基、フリル基、チアゾリル基、ベンゾチアゾリル基、モルホリノ基、前記置換基(A)等の飽和もしくは不飽和の環形成原子数3〜10(好ましくは3〜6)のヘテロ環基等が挙げられる。
上記一般式中、R12〜R23は、それぞれ独立に、炭素数1〜20の直鎖状又は分岐鎖状のアルキル基、環形成炭素数6〜14のアリール基を示す。該アルキル基、アリール基としては、R1〜R11の場合と同じものが挙げられる。
【0038】
R1〜R11がそれぞれ独立して示すアルコキシ基、アルケニルオキシ基、アルカノイル基、アルケノイル基の置換基としては、ヒドロキシル基;カルボキシル基;メトキシ基、エトキシ基、プロポキシ基等の直鎖状又は分岐鎖状の炭素数1〜18(好ましくは1〜10、より好ましくは1〜5)のアルコキシ基;直鎖状又は分岐鎖状の炭素数2〜18(好ましくは2〜10、より好ましくは2〜6)のアルケニルオキシ基;直鎖状又は分岐鎖状の炭素数2〜18(好ましくは2〜10、より好ましくは2〜6)のアルケニルチオ基;直鎖状又は分岐鎖状の炭素数1〜18(好ましくは1〜10、より好ましくは1〜5)のアルキルチオ基;炭素数5〜18(好ましくは5〜10)のシクロアルケニル基;シクロプロピル基、シクロペンチル基、シクロヘキシル基等の炭素数3〜18(好ましくは3〜10、より好ましくは3〜6)のシクロアルキル基;フェノキシ基等の環形成炭素数6〜10のアリールオキシ基;フッ素原子、塩素原子、臭素原子、ヨウ素原子等のハロゲン原子;酸素原子(=O);硫黄原子(=S);シアノ基;ニトロ基;トリメチルシリル基等のトリアルキルシリル基;トリメトキシシリル基等のトリアルコキシシリル基;フェニル基、ナフチル基、アントリル基等の環形成炭素数6〜14(好ましくは6〜10)のアリール基;−COR12;−OCOR13;−NR14R15;−NHCOR16;−NHCOOR17;−CONR18R19;−COOR20;−SO3NR21R22;−SO3R23;エポキシ基やテトラヒドロフラニル基等の環形成原子数3〜6の環状エーテル基、2−チエニル基、2−ピリジル基、フリル基、チアゾリル基、ベンゾチアゾリル基、モルホリノ基、前記置換基(A)等の飽和もしくは不飽和の環形成原子数3〜10(好ましくは3〜6)のヘテロ環基等が挙げられる(式中、R12〜R23は、前記定義の通りである。)。
【0039】
R1〜R11がそれぞれ独立して示すシクロアルキル基、シクロアルケニル基の置換基としては、ヒドロキシル基;カルボキシル基;メチル基、エチル基等の直鎖又は分岐状の炭素数1〜18(好ましくは1〜10、より好ましくは1〜5)のアルキル基;メトキシ基、エトキシ基、プロポキシ基等の直鎖状又は分岐鎖状の炭素数1〜18(好ましくは1〜10、より好ましくは1〜5)のアルコキシ基;直鎖状又は分岐鎖状の炭素数2〜18(好ましくは2〜10、より好ましくは2〜6)のアルケニルオキシ基;直鎖状又は分岐鎖状の炭素数2〜18(好ましくは2〜10、より好ましくは2〜6)のアルケニルチオ基;ビニル基、アリル基等の炭素数2〜18(好ましくは2〜10、より好ましくは2〜6)の直鎖又は分岐状のアルケニル基;直鎖状又は分岐鎖状の炭素数1〜18(好ましくは1〜10、より好ましくは1〜5)のアルキルチオ基;炭素数5〜18(好ましくは5〜10)のシクロアルケニル基;フェノキシ基等の環形成炭素数6〜10のアリールオキシ基;フッ素原子、塩素原子、臭素原子、ヨウ素原子等のハロゲン原子;酸素原子(=O);硫黄原子(=S);シアノ基;ニトロ基;トリメチルシリル基等のトリアルキルシリル基;トリメトキシシリル基等のトリアルコキシシリル基;フェニル基、ナフチル基、アントリル基等の環形成炭素数6〜14(好ましくは6〜10)のアリール基;−COR12;−OCOR13;−NR14R15;−NHCOR16;−NHCOOR17;−CONR18R19;−COOR20;−SO3NR21R22;−SO3R23;エポキシ基やテトラヒドロフラニル基等の環形成原子数3〜6の環状エーテル基、2−チエニル基、2−ピリジル基、フリル基、チアゾリル基、ベンゾチアゾリル基、モルホリノ基、前記置換基(A)等の飽和もしくは不飽和の環形成原子数3〜10(好ましくは3〜6)のヘテロ環基等が挙げられる(式中、R12〜R23は、前記定義の通りである。)。
【0040】
R1〜R11がそれぞれ独立して示すアリール基、ヘテロ環基の置換基としては、ヒドロキシル基;カルボキシル基;メチル基、エチル基等の直鎖又は分岐状の炭素数1〜18(好ましくは1〜10、より好ましくは1〜5)のアルキル基;メトキシ基、エトキシ基、プロポキシ基等の直鎖状又は分岐鎖状の炭素数1〜18(好ましくは1〜10、より好ましくは1〜5)のアルコキシ基;直鎖状又は分岐鎖状の炭素数2〜18(好ましくは2〜10、より好ましくは2〜6)のアルケニルオキシ基;直鎖状又は分岐鎖状の炭素数2〜18(好ましくは2〜10、より好ましくは2〜6)のアルケニルチオ基;直鎖状又は分岐鎖状の炭素数1〜18(好ましくは1〜10、より好ましくは1〜5)のアルキルチオ基;ビニル基、アリル基等の直鎖状又は分岐鎖状の炭素数2〜18(好ましくは2〜10、より好ましくは2〜6)のアルケニル基;炭素数5〜18(好ましくは5〜10)のシクロアルケニル基;シクロプロピル基、シクロペンチル基、シクロヘキシル基等の炭素数3〜18(好ましくは3〜10、より好ましくは3〜6)のシクロアルキル基;フェノキシ基等の環形成炭素数6〜10のアリールオキシ基;フッ素原子、塩素原子、臭素原子、ヨウ素原子等のハロゲン原子;シアノ基;ニトロ基;トリメチルシリル基等のトリアルキルシリル基;トリメトキシシリル基等のトリアルコキシシリル基;−COR12;−OCOR13;−NR14R15;−NHCOR16;−NHCOOR17;−CONR18R19;−COOR20;−SO3NR21R22;−SO3R23;エポキシ基やテトラヒドロフラニル基等の環形成原子数3〜6の環状エーテル基、2−チエニル基、2−ピリジル基、フリル基、チアゾリル基、ベンゾチアゾリル基、モルホリノ基、前記置換基(A)等の飽和もしくは不飽和の環形成原子数3〜10(好ましくは3〜6)のヘテロ環基等が挙げられる(式中、R12〜R23は、前記定義の通りである。)。
【0041】
また、Arが示す環形成炭素数6〜14のアリール基としては、例えばフェニル基、ナフチル基、アントリル基、クリセニル基、フェナントレニル基、アズレニル基、アセナフチレニル基等が挙げられる。これらの中でも、低分子量とする観点、相溶性や溶解性の観点及び製造容易性の観点から、環形成炭素数6〜10のアリール基が好ましく、フェニル基がさらに好ましい。Arが示す環形成原子数5〜14のヘテロアリール基としては、例えば2−フラニル基、2−チオフェニル基、2−ピリジニル基等が挙げられる。これらの中でも、低分子量とする観点、相溶性や溶解性の観点及び製造容易性の観点から、環形成原子数5〜6のへテロアリール基が好ましく、2−フラニル基、2−チオフェニル基がより好ましい。
これらのアリール基及びヘテロアリール基は置換基を有していてもよい。該置換基としては、前記したR1〜R11がそれぞれ独立して示すアリール基の置換基と同じものが挙げられ、それらの中でも、炭素数1〜18(好ましくは1〜10、より好ましくは1〜5)のアルキル基が好ましく、メチル基がより好ましい。
なお、該置換基がアルキル基又はアルケニル基である場合、Arと共に縮合環を形成していてもよく、例えばフルオレン環、インデン環等を形成していてもよい。
【0042】
Zが表す>NR3’中のR3’は、前述の通り、置換もしくは無置換の炭素数1〜20のアルキル基を示すか、又はR3’はR3とつながって、窒素原子と共に環を形成している。
該炭素数1〜20のアルキル基としては、R1〜R11の場合と同じものが挙げられ、好ましいものも同じものが挙げられ、メチル基が特に好ましい。
また、R3’はR3とつながって、窒素原子と共に形成する環の具体例としては、モルホリン環、ピロリジン環、ピペリジン環、ピペコリン環、ピペラジン環などが挙げられる。これらの中でも、モルホリン環が好ましい。
WとZの組み合わせとしては、Wが単結合の場合、Zは酸素原子又は>Nr3’が好ましく、Wが酸素原子の場合、Zは単結合又は酸素原子が好ましい。
【0043】
さらに、一般式(I−a)において、(a)R1、R2及びR3が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、nが2であり、複数のR4及びR5が、それぞれ独立に、水素原子又は置換もしくは無置換の炭素数1〜20のアルキル基であり、Arが、置換もしくは無置換の環形成炭素数6〜14のアリール基である光重合開始剤、(b)R1、R2及びR3が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、nが2であり、複数のR4及びR5が、それぞれ独立に、水素原子又は置換もしくは無置換の炭素数1〜20のアルキル基であり、Arが、置換もしくは無置換の環形成炭素数5〜14のヘテロアリール基である光重合開始剤、(c)R1及びR2が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、R3が置換もしくは無置換の炭素数3〜18のシクロアルキル基であり、nが2であり、複数のR4及びR5が、それぞれ独立に、水素原子又は置換もしくは無置換の炭素数1〜20のアルキル基である光重合開始剤、(d)R1及びR2が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、R3が環形成原子数3〜6の環状エーテル基で置換された炭素数1〜20のアルキル基であり、nが2であり、複数のR4及びR5が、それぞれ独立に、水素原子又は置換もしくは無置換の炭素数1〜20のアルキル基である光重合開始剤も好ましい。さらに、後述する光硬化性組成物における光重合開始剤の含有量の低減の観点から、上記(a)及び(b)の場合は、分子量が515以下であることが好ましく、500以下であることがより好ましく、上記(c)及び(d)の場合は、分子量が550以下であることが好ましい。
さらに、本発明の効果の観点、及び光重合開始剤の分解物による重合物の汚染や装置の汚染を低減する観点から、上記の好ましい光重合開始剤において、R4〜R11がそれぞれ独立に、水素原子又は炭素数1〜20のアルキル基であることがより好ましい。なお、各基の好ましいものは、前記した通りである。
以下に、本発明の光重合開始剤(I−a)の具体例を示すが、特にこれらに制限されるものではない。
【0044】
【化14】
【0045】
【化15】
【0046】
【化16】
【0047】
【化17】
【0048】
さらに、一般式(I−b)において、(e)R1及びR3が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、Wが酸素原子であり、Zが単結合又は酸素原子であり、Arが、置換もしくは無置換の環形成炭素数6〜14のアリール基である光重合開始剤、(f)R1及びR3が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、R2が、−OCOR13(R13は、前記定義の通りである。)又は−COOR20(R20は、炭素数1〜20のアルキル基又は環形成炭素数6〜14のアリール基を示す。)で表されるエステル基を有する炭素数1〜20のアルキル基であり、Wが酸素原子であり、Zが単結合又は酸素原子であり、Arが、置換もしくは無置換の環形成炭素数6〜14のアリール基である光重合開始剤、(g)R1及びR3が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、Wが単結合であり、Zが>NR3’(R3’は、前記定義のとおりである。)であり、Arが、置換もしくは無置換の環形成炭素数6〜14のアリール基である光重合開始剤、(h)R1及びR3が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、R2が、−OCOR13(R13は、前記定義の通りである。)又は−COOR20(R20は、炭素数1〜20のアルキル基又は環形成炭素数6〜14のアリール基を示す。)で表されるエステル基を有する炭素数1〜20のアルキル基であり、Wが単結合であり、Zが>NR3’(R3’は、前記定義のとおりである。)であり、Arが、置換もしくは無置換の環形成炭素数6〜14のアリール基である光重合開始剤、(i)R1が、置換もしくは無置換の炭素数1〜20のアルキル基であり、R3が水素原子であり、Wが単結合であり、Zが酸素原子であり、Arが、置換もしくは無置換の環形成炭素数6〜14のアリール基である光重合開始剤も好ましい。
なお、上記(g)の光重合開始剤においては、>NR3’中のR3’はR3とつながって、窒素原子と共に環を形成しているのも好ましく、モルホリン環を形成しているのがより好ましい。
さらに、本発明の効果の観点、及び光重合開始剤の分解物による重合物の汚染や装置の汚染を低減する観点から、上記の好ましい光重合開始剤において、R4〜R11がそれぞれ独立に、水素原子又は炭素数1〜20のアルキル基であることがより好ましい。なお、各基の好ましいものは、前記した通りである。
以下に、本発明の光重合開始剤(I−b)の具体例を示すが、特にこれらに制限されるものではない。
【0049】
【化18】
【0050】
【化19】
【0051】
なお、本発明の光重合開始剤としては、前述の通り一般式(I−a)においてn=2であることが好ましく、具体的には下記一般式(I−a1)で表される光重合開始剤が好ましい。
【化20】
上記式(I−a1)中、R1〜R3、R6〜R11及びArは、前記定義の通りである。R4’及びR5’は、それぞれR4及びR5の定義と同じであり、好ましいものも同じである。
【0052】
[光重合開始剤の製造方法]
本発明の光重合開始剤(I)の製造方法に特に制限は無いが、例えば、以下の光重合開始剤(I−a)の製造方法や光重合開始剤(I−b)の製造方法によって容易に製造することができる。
(1.光重合開始剤(I−a)の製造方法)
本発明の光重合開始剤(I−a)の製造方法に特に制限は無いが、例えば、下記工程1〜工程4によって、容易に製造することができる。
【0053】
(工程1)
工程1としては、例えば、前記カルバゾール誘導体(1)とZ(CR4R5)nCOOR3(Zは、フッ素原子、塩素原子、臭素原子、ヨウ素原子等のハロゲン原子を示す。また、R3、R4及びR5は、前記定義の通りである。)で表されるハロゲン化物とを、塩基の存在下に反応させる工程が挙げられる。該工程により、下記一般式(3a)で表されるカルボニルアルキル基導入体が得られる。
【化21】
(式中、R3〜R11は、前記定義の通りであり、好ましいものも同じである。)
前記塩基としては、n−ブチルリチウム、t−ブチルリチウム、水酸化ナトリウム、水酸化カリウム等が挙げられる。反応温度は、通常、好ましくは0〜200℃であり、反応時間は通常、好ましくは2〜100時間である。
【0054】
なお、一般式(I−a)においてn=2の光重合開始剤を製造する場合、工程1は、以下の工程であると製造が容易であり、好ましい。
下記一般式(1)
【化22】
(式中、R6〜R11は、前記定義の通りであり、好ましいものも同じである。)
で表されるカルバゾール誘導体[以下、カルバゾール誘導体(1)と称する。]と、下記一般式(2a)
【0055】
【化23】
(式中、R3、R4’及びR5’は、前記定義の通りであり、好ましいものも同じである。)
で表されるアクリレート誘導体[以下、アクリレート誘導体(2a)と称する。]とを塩基の存在下に反応させることにより、下記一般式(3a’)
【0056】
【化24】
(式中、R3、R4’、R5’及びR6〜R11は、前記定義の通りであり、好ましいものも同じである。)
で表されるカルボニルアルキル基導入体[以下、カルボニルアルキル基導入体(3a’)と称する。]を得る工程。
以下、n=2の光重合開始剤を製造する場合に好ましいとするこの工程1について、詳細に説明する。
【0057】
工程1で使用するアクリレート誘導体(2a)の使用量に特に制限は無いが、カルバゾール誘導体(1)1モルに対して、好ましくは0.5〜2モル、より好ましくは0.8〜1.2モルであり、未反応物を低減する観点から、ほぼ等量で反応させることがさらに好ましい。
工程1は、塩基の存在下に実施する。該塩基としては、Michael付加反応に使用し得る塩基であればよく、有機塩基、無機塩基のいずれも用いることができる。有機塩基としては、例えばピリジン、ジアザビシクロウンデセン(DBU)、ジアザビシクロノネン(DBN)等が挙げられる。無機塩基としては、例えば炭酸ナトリウム、炭酸カリウム等のアルカリ金属炭酸塩;炭酸マグネシウム、炭酸カルシウム等のアルカリ土類金属炭酸塩;炭酸水素ナトリウム、炭酸水素カリウム等のアルカリ金属炭酸水素塩;水酸化ナトリウム、水酸化カリウム等のアルカリ金属水酸化物;水酸化マグネシウム、水酸化カルシウム等のアルカリ土類金属水酸化物等が挙げられる。これらの中でも、アルカリ金属炭酸塩、アルカリ土類金属炭酸塩が好ましく、アルカリ金属炭酸塩がより好ましく、炭酸カリウムがさらに好ましい。
該塩基の使用量に特に制限は無いが、反応効率及び製造コストの観点から、カルバゾール誘導体(1)1モルに対して、好ましくは0.5〜2モル、より好ましくは0.8〜1.2モルであり、ほぼ等量で使用することがさらに好ましい。
工程1は、溶媒の存在下に実施することが好ましい。該溶媒としては、カルバゾール誘導体(1)、アクリレート誘導体(2a)及び塩基を溶解し得る溶媒を適宜選択すればよい。具体的には、ジメチルホルムアミド(DMF)、ジメチスルホキシド(DMSO)、塩化メチレン等が挙げられる。
【0058】
工程1の反応温度に特に制限は無いが、通常、好ましくは10〜50℃、より好ましくは15〜40℃、さらに好ましくは15〜30℃で実施する。反応時間は、カルバゾール誘導体(1)、アクリレート誘導体(2a)及び塩基の種類や使用量並びに反応温度等によっても異なるが、通常、2時間〜48時間程度である。
工程1の実施形態に特に制限は無く、例えばカルバゾール誘導体(1)、アクリレート誘導体(2a)及び塩基を全て溶媒中に添加し、好ましくは前記温度で攪拌することにより、カルボニルアルキル基導入体(3a’)を得ることができる。
【0059】
反応終了後、得られた反応混合液から、抽出等の通常の有機化合物の分離手段によって、カルボニルアルキル基導入体(3a)や(3a’)を得ることができる。適宜、蒸留、カラムクロマトグラフィー、再結晶等の、通常の有機化合物の精製手段によって精製することにより、カルボニルアルキル基導入体(3a)や(3a’)の純度を高めることもできる。
【0060】
以下、前記工程1で得られるカルボニルアルキル基導入体(3a)又は(3a’)を用いた工程2〜4について説明するが、便宜上、n=2に相当するカルボニルアルキル基導入体(3a’)については省略して記載する。
(工程2)
工程2は、工程1で得られたカルボニルアルキル基導入体(3a)と下記一般式(4)、(5)
【化25】
(式中、Xは、ハロゲン原子又は−OC(=O)Arを示し、X’は、ハロゲン原子又は−OC(=O)R2を示す。Ar及びR2は、前記定義の通りであり、好ましいものも同じである。)
で表される2つのアシル化剤[以下、それぞれアシル化剤(4)、アシル化剤(5)と称する。]とをルイス酸の存在下に反応させることにより、下記一般式(6a)
【0061】
【化26】
(式中、R2〜R11及びArは、前記定義の通りであり、好ましいものも同じである。)
で表されるジケトン体[以下、ジケトン体(6a)と称する。]を得る工程である。
【0062】
上記式中、X、X’がそれぞれ独立に示すハロゲン原子としては、フッ素原子、塩素原子、臭素原子、ヨウ素原子等が挙げられ、塩素原子が好ましい。
工程2において、カルボニルアルキル基導入体(3a)と、アシル化剤(4)及び(5)との反応は、目的とするジケトン体(6a)の収率の観点から、カルボニルアルキル基導入体(3a)とアシル化剤(4)とを反応させた後、次いでアシル化剤(5)を反応させることが好ましい。
アシル化剤(4)及び(5)の使用量は、カルボニルアルキル基導入体(3a)にそれぞれのアシル基を1つずつ導入する観点から、カルボニルアルキル基導入体(3a)1モルに対して、それぞれ好ましくは0.8〜1.3モル、より好ましくは1〜1.3モルであり、未反応物を低減する観点から、ほぼ等量で反応させることがさらに好ましい。特に、カルボニルアルキル基導入体(3a)とアシル化剤(4)とを反応させた後、次いでアシル化剤(5)を反応させる場合には、アシル化剤(5)の使用量は、前記範囲を超えても何ら問題ないが、あまり過剰であってもそれに見合う収率が得られるわけではなく、いたずらに生産コストがかさむことになり得る。
工程2は、ルイス酸の存在下に実施する。ルイス酸としては、塩化アルミニウム、三フッ化ホウ素ジエチルエーテル錯体が好ましい。ルイス酸の使用量は、ジケトン体(6a)の収率の観点から、アシル化剤(4)又は(5)1モルに対して、通常、好ましくは0.8〜2.5モル、より好ましくは1〜2モルである。
【0063】
工程2は、溶媒の存在下に実施することが好ましい。該溶媒としては、通常のFriedel−Craftsアシル化反応に使用し得る溶媒であれば特に制限は無い。具体的には、ジクロロメタン、ニトロベンゼン、アセトン、アセトニトリル等が挙げられる。
工程2の反応温度は、反応開始時は−50〜5℃(好ましくは−10〜5℃)とし、反応が進行するにしたがって、徐々に室温(15〜25℃程度)へ戻すことが好ましい。反応時間は、カルボニルアルキル基導入体(3a)、アシル化剤(4)及び(5)の種類及び使用量、並びに反応温度によっても異なるが、通常、好ましくは1〜30時間である。なお、アシル化剤(4)及び(5)の両方がハロゲン化アシル又は酸無水物である場合、カルボニルアルキル基導入体(3a)とアシル化剤(4)とを反応させた後、生成物を単離することなく引き続きアシル化剤(5)と反応させることもできる。その場合、カルボニルアルキル基導入体(3a)とアシル化剤(4)との反応時間は、30分〜5時間程度(好ましくは30分〜3時間)とし、一方、カルボニルアルキル基導入体(3a)とアシル化剤(5)との反応時間を、30分〜24時間程度(好ましくは30分〜18時間)と長めに設けて十分に反応させることが好ましい。
【0064】
収率の観点から、工程2の好ましい実施形態を以下に2通り説明する。
[1]氷浴で冷却しながらカルボニルアルキル基導入体(3a)及びアシル化剤(4)を適宜溶媒中で混合し、その混合溶液へルイス酸をゆっくり(好ましくは5分〜1時間、より好ましくは10分〜40分かけて)添加し、添加終了後、室温へ戻して一定時間(30分〜5時間程度)攪拌を続ける。好ましくは、抽出及び洗浄等の通常の有機化合物の分離手段を行うことによって、一旦、生成物を分離取得する。
こうして得られた生成物を適宜溶媒に溶解し、氷浴で冷却しながらアシル化剤(5)を添加し、そこへルイス酸をゆっくり(好ましくは5分〜1時間、より好ましくは10分〜40分かけて)添加し、添加終了後、室温へ戻して一定時間(30分〜24時間程度)攪拌を続けることにより、ジケトン体(6a)を得ることができる。
[2]氷浴で冷却しながらカルボニルアルキル基導入体(3a)を適宜溶媒と混合してから、ルイス酸をゆっくり(好ましくは5分〜1時間、より好ましくは10分〜40分かけて)添加し、得られた混合溶液へアシル化剤(4)をゆっくり(好ましくは5分〜1時間、より好ましくは10分〜40分かけて)添加した後、室温へ戻して一定時間(30分〜5時間程度)攪拌を続ける。再び氷浴で冷却しながらルイス酸をゆっくり(好ましくは5分〜1時間、より好ましくは10分〜40分かけて)添加し、得られた混合溶液へアシル化剤(5)をゆっくり(好ましくは5分〜1時間、より好ましくは10分〜40分かけて)添加した後、室温へ戻して一定時間以上(好ましくは30分〜24時間程度)攪拌を続けることにより、ジケトン体(6a)を得ることができる。
なお、反応終了後、得られた反応混合液から、抽出等の通常の有機化合物の分離手段によって、ジケトン体(6a)を得ることができる。適宜、蒸留、カラムクロマトグラフィー、再結晶等の、通常の有機化合物の精製手段によって精製することにより、ジケトン体(6a)の純度を高めることもできる。
【0065】
(工程3)
工程3は、工程2で得られたジケトン体(6a)とヒドロキシルアミンを反応させることにより、下記一般式(7a)
【化27】
(式中、R2〜R11及びArは、前記定義の通りであり、好ましいものも同じである。)
で表されるオキシム体[以下、オキシム体(7a)と称する。]を得る工程である。
【0066】
工程3においては、ヒドロキシルアミン供給源としては、特に制限されるものではないが、塩化ヒドロキシルアミンが好ましい。該塩化ヒドロキシルアミンは、例えば水中で酢酸ナトリウム、等と反応させることにより、ヒドロキシルアミンの水溶液を得ることができる。
ヒドロキシルアミン(塩化ヒドロキシルアミン)の使用量は、ジケトン体(6a)1モルに対して、好ましくは0.8〜2モル、より好ましくは1〜1.5モル、さらに好ましくは1〜1.3モルであり、未反応物を低減する観点から、ほぼ等量で反応させることがさらに好ましい。
工程3は、溶媒の存在下に実施することが好ましい。溶媒としては、水溶性有機溶媒が好ましく、該水溶性有機溶媒としては、例えばメタノール、エタノール等のアルコール類、ジメチルホルムアミド(DMF)等が好ましい。
【0067】
工程3の反応温度に特に制限は無いが、オキシム体(7a)の収率の観点からは、通常、好ましくは40〜160℃、より好ましくは50〜140℃、さらに好ましくは70〜110℃である。反応時間は、ジケトン体(6a)の種類及び使用量、並びに反応温度によっても異なるが、通常、好ましくは2〜20時間、より好ましくは4〜12時間である。
工程3の実施形態に特に制限は無く、例えば、塩化ヒドロキシルアミンと酢酸ナトリウムを水中で混合してヒドロキシルアミンの水溶液を得ておき、そこへジケトン体(6a)及び溶媒を添加して、好ましくは前記温度範囲で攪拌することにより、オキシム体(7a)を得ることができる。
反応終了後、得られた反応混合液から、抽出等の通常の有機化合物の分離手段によって、オキシム体(7a)を得ることができる。適宜、蒸留、カラムクロマトグラフィー、再結晶等の、通常の有機化合物の精製手段によって精製することにより、オキシム体(7a)の純度を高めることもできる。
【0068】
(工程4)
工程4は、工程3で得られたオキシム体(7a)と下記一般式(8)
【化28】
(式中、Yは、ハロゲン原子又は−OC(=O)R1を示す。R1は、前記定義の通りであり、好ましいものも同じである。)
で表されるエステル化剤[以下、エステル化剤(8)と称する。]とを反応させることにより、下記一般式(I−a)
【0069】
【化29】
(式中、R1〜R11及びArは、前記定義の通りである。)
で表される光重合開始剤[以下、光重合開始剤(I−a)と称する。]を得る工程である。
【0070】
上記式中、Yが示すハロゲン原子としては、フッ素原子、塩素原子、臭素原子、ヨウ素原子等が挙げられ、塩素原子が好ましい。
工程4で使用するエステル化剤(8)の使用量に特に制限は無いが、オキシム体(7a)1モルに対して、好ましくは0.5〜2モル、より好ましくは0.8〜1.2モルであり、未反応物を低減する観点から、ほぼ等量で反応させることがさらに好ましい。
工程4は、反応を促進するために、塩基の存在下に実施してもよい。塩基としては、有機塩基や無機塩基が挙げられる。有機塩基としては、例えばトリエチルアミン、トリブチルアミン等のアミン類;ピリジン等の含窒素複素環式芳香族化合物等が挙げられる。無機塩基としては、例えば炭酸ナトリウム等のアルカリ金属炭酸塩;炭酸マグネシウム等のアルカリ土類金属炭酸塩;水酸化ナトリウム等のアルカリ金属水酸化物;水酸化マグネシウム等のアルカリ土類金属水酸化物等が挙げられる。これらの中でも、有機塩基が好ましく、アミン類、含窒素複素環式芳香族化合物がより好ましく、反応効率及び製造コストの観点から、トリエチルアミン、ピリジンがさらに好ましい。
塩基を使用する場合、その使用量は、光重合開始剤(I−a)の収率及び製造コストの観点から、オキシム体(7a)1モルに対して、好ましくは1〜5モル、より好ましくは1.5〜3モルである。
【0071】
工程4は、溶媒の存在下に実施することが好ましい。溶媒としては、t−ブチルメチルエーテル、エチルメチルエーテル、シクロペンチルメチルエーテル、テトラヒドロフラン(THF)等のエーテル類が好ましく挙げられる。
工程4の反応温度は、反応開始時は−50〜5℃(好ましくは−10〜5℃)とし、反応が進行するにしたがって、徐々に室温(15〜25℃程度)へ戻すことが好ましい。反応時間は、オキシム体(7a)、エステル化剤(8)の種類及び使用量、並びに反応温度によっても異なるが、通常、好ましくは0.5〜10時間、より好ましくは1〜5時間である。
工程4の実施形態に特に制限は無く、例えばオキシム体(7a)及びエステル化剤(8)を適宜溶媒中で混合し、そこへ塩基を滴下し、滴下終了後、反応液の温度を徐々に室温へ戻して攪拌を続けることにより、本発明の光重合開始剤(I−a)を得ることができる。
反応終了後、得られた反応混合液から、抽出等の通常の有機化合物の分離手段によって、光重合開始剤(I−a)を得ることができる。適宜、蒸留、カラムクロマトグラフィー、再結晶等の、通常の有機化合物の精製手段によって精製することにより、光重合開始剤(I−a)の純度を高めることもできる。
【0072】
こうして得られる本発明の光重合開始剤(I−a)は、カルバゾール骨格中の窒素原子に、エステル基を含有する特殊な置換基が付いている。そのため、光硬化性組成物中のエチレン性不飽和結合を有する化合物(特に(メタ)アクリロイルオキシ基を有する化合物)、エーテル結合及び/又はエステル結合を有する25℃で液体の化合物との相溶性、及び溶剤(前記のエーテル結合及び/又はエステル結合を有する25℃で液体の化合物を含む。)に対する溶解性が高く、光(特に波長450nm以下の短波長の光)に対する感度が高く、解像度及び現像性、ひいては深部硬化性に優れ、さらに基板との密着性が良好になるものと考えられる。また、該光重合開始剤(I−a)は、露光時の光により発生する分解物による重合物の汚染や装置の汚染が無い。
なお、特定化合物や溶剤に対する光重合開始剤(I−a)の相溶性や溶解性の高さは、感度、解像度及び現像性、ひいては深部硬化性の向上に寄与しているものと考えられ、カラーフィルタ用途においては重要な要素であると言える。特にカラーフィルタ用途においては、光重合開始剤が、エーテル結合及び/又はエステル結合を有する溶剤(特に、プロピレングリコールモノエチルエーテルアセテート等)100質量部に対して、5質量部以上溶解することが好ましく、10質量部以上溶解することがさらに好ましく、本発明の光重合開始剤(I−a)はかかる条件を満たしており、カラーフィルタ用光硬化性組成物に含有させる光重合開始剤、さらには高遮光性のブラックマトリックス用光硬化性組成物に含有させる光重合開始剤として適している。
【0073】
(1.光重合開始剤(I−a)の製造方法)
本発明の光重合開始剤(I−b)の製造方法に特に制限は無いが、例えば、下記工程1〜工程4によって、容易に製造することができる。
【0074】
(工程1)
工程1は、W及びZによって製造方法が異なるため、以下に場合分けして説明する。
<Wが単結合であり、Zが>NR3’である場合>
下記一般式(1)
【化30】
(式中、R6〜R11は、前記定義の通りであり、好ましいものも同じである。)
で表されるカルバゾール誘導体[以下、カルバゾール誘導体(1)と称する。]と、下記一般式(2b)
【0075】
【化31】
(式中、R3、R3’、R4及びR5は、前記定義の通りであり、好ましいものも同じである。)
で表されるアクリルアミド誘導体[以下、アクリルアミド誘導体(2b)と称する。]とを塩基の存在下に反応させることにより、下記一般式(3b)
【0076】
【化32】
(式中、R3、R3’、R4〜R11は、前記定義の通りであり、好ましいものも同じである。)
で表されるアミド基導入体[以下、アミド基導入体(3b)と称する。]を得る工程である。
【0077】
工程1で使用するアクリルアミド誘導体(2b)の使用量に特に制限は無いが、カルバゾール誘導体(1)1モルに対して、好ましくは0.5〜2モル、より好ましくは0.8〜1.2モルであり、未反応物を低減する観点から、ほぼ等量で反応させることがさらに好ましい。
工程1は、塩基の存在下に実施する。該塩基としては、Michael付加反応に使用し得る塩基を用いることができ、特に制限は無く、有機塩基、無機塩基のいずれも用いることができる。有機塩基としては、例えばピリジン、ジアザビシクロウンデセン(DBU)、ジアザビシクロノネン(DBN)等が挙げられ、求核性の低い塩基が好ましく、DBU、DBNがより好ましい。無機塩基としては、例えば、炭酸ナトリウム、炭酸カリウム等のアルカリ金属炭酸塩;炭酸マグネシウム、炭酸カルシウム等のアルカリ土類金属炭酸塩;炭酸水素ナトリウム、炭酸水素カリウム等のアルカリ金属炭酸水素塩;水酸化ナトリウム、水酸化カリウム等のアルカリ金属水酸化物;水酸化マグネシウム、水酸化カルシウム等のアルカリ土類金属水酸化物等が挙げられる。これらの中でも、アルカリ金属炭酸塩、アルカリ土類金属炭酸塩が好ましく、アルカリ金属炭酸塩がより好ましく、炭酸カリウムがさらに好ましい。
該塩基の使用量に特に制限は無いが、反応効率及び製造コストの観点から、カルバゾール誘導体(1)1モルに対して、好ましくは0.5〜2モル、より好ましくは0.8〜1.2モルであり、ほぼ等量で使用することがさらに好ましい。
工程1は、溶媒の存在下に実施することが好ましい。該溶媒としては、カルバゾール誘導体(1)、アクリルアミド誘導体(2b)及び塩基を溶解し得る溶媒を適宜選択すればよい。具体的には、ジメチルホルムアミド(DMF)、ジメチスルホキシド(DMSO)、塩化メチレン等が挙げられる。
【0078】
工程1の反応温度に特に制限は無いが、通常、好ましくは10〜50℃、より好ましくは15〜40℃、さらに好ましくは15〜30℃で実施する。反応圧力に特に制限は無いが、常圧下に実施することが好ましい。反応時間は、カルバゾール誘導体(1)、アクリルアミド誘導体(2b)及び塩基の種類や使用量並びに反応温度や反応圧力等によっても異なるが、通常、好ましくは2時間〜48時間程度である。
工程1の実施形態に特に制限は無く、例えばカルバゾール誘導体(1)、アクリルアミド誘導体(2b)及び塩基を全て溶媒中に添加し、好ましくは前記温度で攪拌することにより、アミド基導入体(3b)を製造することができる。
反応終了後、得られた反応混合液から、抽出等の通常の有機化合物の分離手段によって、アミド基導入体(3b)を得ることができる。適宜、蒸留、カラムクロマトグラフィー、再結晶等の、通常の有機化合物の精製手段によって精製することにより、アミド基導入体(3b)の純度を高めることもできる。
【0079】
−別法−
アミド基導入体(3b)の製造には、次の方法を採用することもできる。
前記カルバゾール誘導体(1)と下記一般式(2b’)
【化33】
(式中、R4及びR5は前記定義の通りである。R24は、炭素数1〜5のアルキル基を示す。)
で表されるカルボン酸基導入剤[以下、カルボン酸基導入剤(2b’)と称する。]とを塩基の存在下に反応(第1反応)させて得られる下記一般式
【化34】
(式中、R4〜R11は、前記定義の通りであり、好ましいものも同じである。)
で表されるカルボン酸基含有カルバゾール誘導体を、例えば塩化チオニルの存在下にHNR3R3’(R3及びR3’は、前記定義の通りであり、好ましいものも同じである。)で表されるアミンと反応(第2反応)させることにより、アミド基導入体(3b)を得る方法である。該アミド基導入体(3b)としては、市販品を用いることもできる。
なお、R24が示す炭素数1〜5のアルキル基としては、例えばメチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、t−ブチル基等が挙げられる。これらの中でも、メチル基、エチル基が好ましい。
【0080】
前記第1反応で使用する塩基としては特に制限は無く、有機塩基、無機塩基のいずれも用いることができる。有機塩基としては、例えばピリジン、ジアザビシクロウンデセン(DBU)、ジアザビシクロノネン(DBN)等が挙げられ、求核性の低い塩基が好ましく、DBU、DBNがより好ましい。無機塩基としては、例えば、炭酸ナトリウム、炭酸カリウム等のアルカリ金属炭酸塩;炭酸マグネシウム、炭酸カルシウム等のアルカリ土類金属炭酸塩;炭酸水素ナトリウム、炭酸水素カリウム等のアルカリ金属炭酸水素塩;水酸化ナトリウム、水酸化カリウム等のアルカリ金属水酸化物;水酸化マグネシウム、水酸化カルシウム等のアルカリ土類金属水酸化物等が挙げられる。これらの中でも、塩基としては、無機塩基が好ましく、アルカリ金属水酸化物がより好ましい。
該塩基の使用量に特に制限は無いが、反応効率及び製造コストの観点から、カルバゾール誘導体(1)1モルに対して、好ましくは0.5〜2モル、より好ましくは0.8〜1.2モルであり、ほぼ等量で使用することがさらに好ましい。
第1反応は、溶媒の存在下に実施することが好ましい。溶媒としては、カルバゾール誘導体(1)、カルボン酸基導入剤(2b’)及び前記塩基を溶解し得る溶媒を適宜選択すればよい。具体的には、ジメチルホルムアミド(DMF)、ジメチスルホキシド(DMSO)、塩化メチレン等が挙げられ、DMFが好ましい。
【0081】
第1反応の反応温度に特に制限は無いが、通常、好ましくは10〜50℃、より好ましくは15〜40℃、さらに好ましくは15〜30℃で実施する。第1反応の反応圧力に特に制限は無いが、常圧下に実施することが好ましい。反応時間は、カルバゾール誘導体(1)及び塩基の種類や使用量、及びカルボン酸基導入剤(2b’)の使用量並びに反応温度及び反応圧力等によっても異なるが、通常、好ましくは2時間〜48時間程度である。
第1反応の実施形態に特に制限は無く、例えばカルバゾール誘導体(1)、カルボン酸基導入剤(2b’)及び塩基を全て溶媒中に添加し、好ましくは前記温度で攪拌することにより、カルボン酸基含有カルバゾール誘導体を製造することができる。
第1反応終了後、抽出等の通常の有機化合物の分離手段によって、前記カルボン酸基含有カルバゾール誘導体を得ることができる。適宜、蒸留、カラムクロマトグラフィー、再結晶等の、通常の有機化合物の精製手段によって精製することにより、該カルボン酸基含有カルバゾール誘導体の純度を高めることもできる。
【0082】
第2反応で使用する塩化チオニルの使用量は、第1反応で使用するカルバゾール誘導体(1)1モルに対して、好ましくは1〜10モル程度使用すればよく、より好ましくは2〜5モル程度である。
第2反応で使用するHNR3R3’で表されるアミンの使用量は、第1反応で使用するカルバゾール誘導体(1)1モルに対して、好ましくは1〜10モル程度使用すればよく、より好ましくは2〜7モル程度である。
第2反応は、溶媒の存在下に実施することが好ましい。溶媒としては、前記カルボン酸基含有カルバゾール誘導体、前記アミンを溶解し得る溶媒であれば特に制限は無い。例えば、ジクロロメタン、ニトロベンゼン、アセトン、アセトニトリル等が挙げられ、アセトニトリルが好ましい。
【0083】
第2反応の反応温度に特に制限は無いが、通常、好ましくは10〜50℃、より好ましくは15〜40℃、さらに好ましくは15〜30℃で実施する。第2反応の反応圧力に特に制限は無いが、常圧下に実施することが好ましい。反応時間は、前記カルボン酸基含有カルバゾール誘導体、前記アミン及び塩基の種類や使用量並びに反応温度及び反応圧力等によっても異なるが、通常、好ましくは1時間〜10時間程度である。
第2反応の実施形態に特に制限は無いが、例えば前記カルボン酸基含有カルバゾール誘導体と前記アミンとを溶媒中に添加し、そこへ塩化チオニルを滴下し、滴下終了後、好ましくは前記温度で攪拌することにより、アミド基導入体(3b)を製造することができる。
反応終了後、得られた反応混合液から、抽出等の通常の有機化合物の分離手段によって、アミド基導入体(3b)を得ることができる。適宜、蒸留、カラムクロマトグラフィー、再結晶等の、通常の有機化合物の精製手段によって精製することにより、アミド基導入体(3b)の純度を高めることもできる。
【0084】
<Wが酸素原子であり、Zが単結合である場合>
前記カルバゾール誘導体(1)と炭酸エチレンを塩基の存在下に反応(第1反応)させて得られる、下記一般式
【0085】
【化35】
(式中、R6〜R11は、前記定義の通りであり、好ましいものも同じである。)
【0086】
で表されるヒドロキシル基含有カルバゾール誘導体を、塩基の存在下にR3COY(R3は、前記定義の通りであり、好ましいものも同じである。また、Yは、ハロゲン原子を示す。)で表されるアシルハライドと反応(第2反応)させることにより、下記一般式(3b’)
【化36】
(式中、R3〜R11は、前記定義の通りであり、好ましいものも同じである。)
で表されるエステル基導入体[以下、エステル基導入体(3b’)と称する。]を得る方法が好ましい。
【0087】
前記第1反応で使用する炭酸エチレンの使用量は、カルバゾール誘導体(1)1モルに対して、好ましくは0.7〜15モル、より好ましくは1〜10モルである。
第1反応で使用する塩基としては、有機塩基が好ましく、例えばトリエチルアミン、トリブチルアミン等のアミン類;ピリジン等の含窒素複素環式芳香族化合物等が挙げられる。これらの中でも、アミン類が好ましく、トリエチルアミンがより好ましい。該塩基の使用量に特に制限は無いが、反応効率及び製造コストの観点から、カルバゾール誘導体(1)1モルに対して、好ましくは0.5〜2モル、より好ましくは0.8〜1.2モルであり、ほぼ等量で使用することがさらに好ましい。
第1反応は、溶媒の存在下に実施することが好ましい。溶媒としては、カルバゾール誘導体(1)や炭酸エチレン、並びに前記塩基を溶解し得る溶媒を適宜選択すればよい。具体的には、ジメチルホルムアミド(DMF)、ジメチスルホキシド(DMSO)、塩化メチレン等が挙げられ、DMFが好ましい。
【0088】
第1反応の反応温度に特に制限は無いが、通常、好ましくは60〜130℃、より好ましくは80〜120℃、さらに好ましくは90〜110℃で実施する。第1反応の反応圧力に特に制限は無いが、常圧下に実施することが好ましい。反応時間は、カルバゾール誘導体(1)及び塩基の種類や使用量、炭酸エチレンの使用量、並びに反応温度及び反応圧力等によっても異なるが、通常、好ましくは1時間〜10時間程度である。
第1反応の実施形態に特に制限は無く、例えばカルバゾール誘導体(1)、炭酸エチレン及び塩基を全て溶媒中に添加し、好ましくは前記温度で攪拌することにより、前記ヒドロキシル基含有カルバゾール誘導体を製造することができる。
第1反応終了後、得られた反応混合液から、抽出等の通常の有機化合物の分離手段によって、前記ヒドロキシル基含有カルバゾール誘導体を得ることができる。適宜、蒸留、カラムクロマトグラフィー、再結晶等の、通常の有機化合物の精製手段によって精製することにより、該ヒドロキシル基含有カルバゾール誘導体の純度を高めることもできる。
【0089】
第2反応で使用するR3COYで表されるアシルハライドとしては、アシルクロリド、アシルヨージド等が挙げられ、アシルクロリドが好ましい。
該アシルハライドの使用量は、第1反応で使用するカルバゾール誘導体(1)1モルに対して、好ましくは0.7〜3モル程度、より好ましくは1〜2モルであり、ほぼ等量で使用することがさらに好ましい。
第2反応で使用する塩基としては、例えばトリエチルアミン、トリブチルアミン、ジイソプロピルエチルアミン等のアミン類が好ましい。塩基の使用量は、前記アシルハライド1モルに対して、好ましくは0.7〜3モル程度、より好ましくは1〜2モルであり、ほぼ等量で使用することがさらに好ましい。
第2反応は、溶媒の存在下に実施することが好ましい。溶媒としては、通常のアシル化反応に使用し得る溶媒を使用でき、反応を阻害しない限り特に制限は無い。具体的には、ジクロロメタン、ニトロベンゼン、アセトン、アセトニトリル等が挙げられ、アセトニトリルが好ましい。
【0090】
第2反応の反応温度に特に制限は無いが、通常、好ましくは10〜50℃、より好ましくは15〜40℃、さらに好ましくは15〜30℃で実施する。第2反応の反応圧力に特に制限は無いが、常圧下に実施することが好ましい。反応時間は、前記ヒドロキシル基含有カルバゾール誘導体、前記アシルハライド及び塩基の種類や使用量並びに反応温度及び反応圧力等によっても異なるが、通常、好ましくは1時間〜10時間程度である。
第2反応の実施形態に特に制限は無いが、例えば前記ヒドロキシル基含有カルバゾール誘導体と前記アシルハライドと塩基とを全て溶媒中に添加し、好ましくは前記温度で攪拌することにより、エステル基導入体(3b’)を製造することができる。
反応終了後、得られた反応混合液から、抽出等の通常の有機化合物の分離手段によって、エステル基導入体(3b’)を得ることができる。適宜、蒸留、カラムクロマトグラフィー、再結晶等の、通常の有機化合物の精製手段によって精製することにより、エステル基導入体(3b’)の純度を高めることもできる。
【0091】
(工程2)
工程2は、工程1で得られたアミド基導入体(3b)又はエステル基導入体(3b’)[以下、便宜上、これらをまとめて、官能基導入体(3b)と称する。]と、下記一般式(4)、(5)
【化37】
(式中、Xは、ハロゲン原子又は−OC(=O)Arを示し、X’は、ハロゲン原子又は−OC(=O)R2を示す。Ar及びR2は、前記定義の通りであり、好ましいものも同じである。)
で表される2つのアシル化剤[以下、それぞれアシル化剤(4)、アシル化剤(5)と称する。]とをルイス酸の存在下に反応させることにより、下記一般式(6b)
【0092】
【化38】
(式中、R2〜R11、Ar、W及びZは、前記定義の通りであり、好ましいものも同じである。)
で表されるジケトン体[以下、ジケトン体(6b)と称する。]を得る工程である。
【0093】
上記式中、X、X’がそれぞれ独立に示すハロゲン原子としては、フッ素原子、塩素原子、臭素原子、ヨウ素原子等が挙げられ、塩素原子が好ましい。
工程2において、官能基導入体(3b)と、アシル化剤(4)及び(5)との反応は、目的とするジケトン体(6b)の収率の観点から、官能基導入体(3b)とアシル化剤(4)とを反応させた後、次いでアシル化剤(5)を反応させることが好ましい。
アシル化剤(4)及び(5)の使用量は、官能基導入体(3b)にそれぞれのアシル基を1つずつ導入する観点から、官能基導入体(3b)1モルに対して、それぞれ好ましくは0.8〜1.3モル、より好ましくは1〜1.3モルであり、未反応物を低減する観点から、ほぼ等量で反応させることがさらに好ましい。特に、官能基導入体(3b)とアシル化剤(4)とを反応させた後、次いでアシル化剤(5)を反応させる場合には、アシル化剤(5)の使用量は、前記範囲を超えても何ら問題ないが、あまり過剰であってもそれに見合う収率が得られるわけではなく、いたずらに生産コストがかさむことになり得る。
工程2は、ルイス酸の存在下に実施する。ルイス酸としては、塩化アルミニウム、三フッ化ホウ素ジエチルエーテル錯体が好ましい。ルイス酸の使用量は、ジケトン体(6b)の収率の観点から、アシル化剤(4)又は(5)1モルに対して、通常、好ましくは0.8〜2.5モル、より好ましくは1〜2モルである。
【0094】
工程2は、溶媒の存在下に実施することが好ましい。該溶媒としては、通常のFriedel−Craftsアシル化反応に使用し得る溶媒であれば特に制限は無い。具体的には、ジクロロメタン、ニトロベンゼン、アセトン、アセトニトリル等が挙げられる。
工程2の反応温度は、反応開始時は−50〜5℃(好ましくは−10〜5℃)とし、反応が進行するにしたがって、徐々に室温(15〜25℃程度)へ戻すことが好ましい。反応時間は、官能基導入体(3b)、アシル化剤(4)及び(5)の種類及び使用量、並びに反応温度によっても異なるが、通常、好ましくは1〜30時間である。なお、アシル化剤(4)及び(5)の両方がハロゲン化アシル又は酸無水物である場合、官能基導入体(3b)とアシル化剤(4)とを反応させた後、生成物を単離することなく引き続きアシル化剤(5)と反応させることもできる。その場合、官能基導入体(3b)とアシル化剤(4)との反応時間は、30分〜5時間程度(好ましくは30分〜3時間)とし、一方、官能基導入体(3b)とアシル化剤(5)との反応時間を、30分〜24時間程度(好ましくは30分〜18時間)と長めに設けて十分に反応させることが好ましい。
【0095】
収率の観点から、工程2の好ましい実施形態の1つを説明する。例えば、氷浴で冷却しながら官能基導入体(3b)を適宜溶媒と混合し、その混合溶液へルイス酸をゆっくり(好ましくは5分〜1時間、より好ましくは10分〜40分かけて)添加し、添加終了後、アシル化剤(4)をゆっくり(好ましくは5分〜1時間、より好ましくは10分〜40分かけて)添加し、室温へ戻して一定時間(30分〜5時間程度)攪拌を続ける。再び、氷浴で冷却しながらアシル化剤(5)を添加し、そこへルイス酸をゆっくり(好ましくは5分〜1時間、より好ましくは10分〜40分かけて)添加し、添加終了後、室温へ戻して一定時間(30分〜24時間程度)攪拌を続けることにより、ジケトン体(6b)を得ることができる。
反応終了後、得られた反応混合液から、抽出等の通常の有機化合物の分離手段によって、ジケトン体(6b)を得ることができる。適宜、蒸留、カラムクロマトグラフィー、再結晶等の、通常の有機化合物の精製手段によって精製することにより、ジケトン体(6b)の純度を高めることもできる。
【0096】
(工程3)
工程3は、工程2で得られたジケトン体(6b)とヒドロキシルアミンを反応させることにより、下記一般式(7b)
【化39】
(式中、R2〜R11、Ar、W及びZは、前記定義の通りであり、好ましいものも同じである。)
で表されるオキシム体[以下、オキシム体(7b)と称する。]を得る工程である。
【0097】
工程3においては、ヒドロキシルアミン供給源としては、特に制限されるものではないが、塩化ヒドロキシルアミンが好ましい。該塩化ヒドロキシルアミンは、例えば水中で酢酸ナトリウム、等と反応させることにより、ヒドロキシルアミンの水溶液を得ることができる。
ヒドロキシルアミン(塩化ヒドロキシルアミン)の使用量は、ジケトン体(6b)1モルに対して、好ましくは0.8〜2モル、より好ましくは1〜1.5モル、さらに好ましくは1〜1.3モルであり、未反応物を低減する観点から、ほぼ等量で反応させることがさらに好ましい。
工程3は、溶媒の存在下に実施することが好ましい。溶媒としては、水溶性有機溶媒が好ましく、該水溶性有機溶媒としては、例えばメタノール、エタノール等のアルコール類、ジメチルホルムアミド(DMF)等が好ましい。
【0098】
工程3の反応温度に特に制限は無いが、オキシム体(7b)の収率の観点からは、通常、好ましくは40〜160℃、より好ましくは50〜140℃、さらに好ましくは70〜110℃である。反応時間は、ジケトン体(6b)の種類及び使用量、並びに反応温度によっても異なるが、通常、好ましくは0.5〜20時間、より好ましくは1〜12時間である。
工程3の実施形態に特に制限は無く、例えば、塩化ヒドロキシルアミンと酢酸ナトリウムを水中で混合してヒドロキシルアミンの水溶液を得ておき、そこへジケトン体(6b)及び溶媒を添加して、好ましくは前記温度範囲で攪拌することにより、オキシム体(7b)を得ることができる。
反応終了後、得られた反応混合液から、抽出等の通常の有機化合物の分離手段によって、オキシム体(7b)を得ることができる。適宜、蒸留、カラムクロマトグラフィー、再結晶等の、通常の有機化合物の精製手段によって精製することにより、オキシム体(7b)の純度を高めることもできる。
【0099】
(工程4)
工程4は、工程3で得られたオキシム体(7b)と下記一般式(8)
【化40】
(式中、Yは、ハロゲン原子又は−OC(=O)R1を示す。R1は、前記定義の通りであり、好ましいものも同じである。)
で表されるエステル化剤[以下、エステル化剤(8)と称する。]とを反応させることにより、下記一般式(I−b)
【0100】
【化41】
(式中、R1〜R11、Ar、W及びZは、前記定義の通りである。)
で表される光重合開始剤[以下、光重合開始剤(I−b)と称する。]を得る工程である。
【0101】
上記式中、Yが示すハロゲン原子としては、フッ素原子、塩素原子、臭素原子、ヨウ素原子等が挙げられ、塩素原子が好ましい。
工程4で使用するエステル化剤(8)の使用量に特に制限は無いが、オキシム体(7b)1モルに対して、好ましくは0.5〜2モル、より好ましくは0.8〜1.2モルであり、未反応物を低減する観点から、ほぼ等量で反応させることがさらに好ましい。
工程4は、反応を促進するために、塩基の存在下に実施してもよい。塩基としては、有機塩基や無機塩基が挙げられる。有機塩基としては、例えばトリエチルアミン、トリブチルアミン等のアミン類;ピリジン等の含窒素複素環式芳香族化合物等が挙げられる。無機塩基としては、例えば炭酸ナトリウム等のアルカリ金属炭酸塩;炭酸マグネシウム等のアルカリ土類金属炭酸塩;水酸化ナトリウム等のアルカリ金属水酸化物;水酸化マグネシウム等のアルカリ土類金属水酸化物等が挙げられる。これらの中でも、有機塩基が好ましく、アミン類、含窒素複素環式芳香族化合物がより好ましく、反応効率及び製造コストの観点から、トリエチルアミン、ピリジンがさらに好ましい。
工程4にて塩基を使用する場合、その使用量は、光重合開始剤(I−b)の収率及び製造コストの観点から、オキシム体(7b)1モルに対して、好ましくは1〜5モル、より好ましくは1.5〜3モルである。
【0102】
工程4は、溶媒の存在下に実施することが好ましい。溶媒としては、t−ブチルメチルエーテル、エチルメチルエーテル、シクロペンチルメチルエーテル、テトラヒドロフラン(THF)等のエーテル類が好ましく挙げられる。
工程4の反応温度は、反応開始時は−50〜5℃(好ましくは−10〜5℃)とし、反応が進行するにしたがって、徐々に室温(15〜25℃程度)へ戻すことが好ましい。反応時間は、オキシム体(7b)、エステル化剤(8)の種類及び使用量、並びに反応温度によっても異なるが、通常、好ましくは0.5〜10時間、より好ましくは1〜5時間である。
工程4の実施形態に特に制限は無く、例えばオキシム体(7b)及びエステル化剤(8)を適宜溶媒中で混合し、そこへ塩基を滴下し、滴下終了後、反応液の温度を徐々に室温へ戻して攪拌を続けることにより、本発明の光重合開始剤(I−b)を得ることができる。
反応終了後、得られた反応混合液から、抽出等の通常の有機化合物の分離手段によって、光重合開始剤(I−b)を得ることができる。適宜、蒸留、カラムクロマトグラフィー、再結晶等の、通常の有機化合物の精製手段によって精製することにより、光重合開始剤(I−b)の純度を高めることもできる。
【0103】
こうして得られる本発明の光重合開始剤(I−b)は、カルバゾール骨格中の窒素原子に、エステル基又はアミド結合(若しくは環を形成したアミド結合)を含有する特殊な置換基が付いている。そのため、光硬化性組成物中のエチレン性不飽和結合を有する化合物(特に(メタ)アクリロイルオキシ基を有する化合物)やエーテル結合及び/又はエステル結合を有する25℃で液体の化合物との相溶性が高く、かつ溶剤(前記のエーテル結合及び/又はエステル結合を有する25℃で液体の化合物を含む。)に対する溶解性が高く、光(特に波長450nm以下の短波長の光)に対する感度が高く、解像度及び現像性、ひいては深部硬化性に優れ、さらに基板との密着性が良好になるものと考えられる。また、該光重合開始剤(I−b)は、露光時の光により発生する分解物による重合物の汚染や装置の汚染が無い。
なお、特定化合物や溶剤に対する光重合開始剤(I−b)の相溶性や溶解性の高さは、感度、解像度及び現像性、ひいては深部硬化性の向上に寄与しているものと考えられ、カラーフィルタ用途においては重要な要素であると言える。特にカラーフィルタ用途においては、光重合開始剤が、エーテル結合及び/又はエステル結合を有する溶剤(特に、プロピレングリコールモノエチルエーテルアセテート等)100質量部に対して、5質量部以上溶解することが好ましく、10質量部以上溶解することがさらに好ましく、本発明の光重合開始剤(I−b)はかかる条件を満たしており、カラーフィルタ用光硬化性組成物に含有させる光重合開始剤、さらには高遮光性のブラックマトリックス用光硬化性組成物に含有させる光重合開始剤として適している。
【0104】
次に、本発明の光重合開始剤(I)を含有する光硬化性組成物、該光硬化性組成物を用いたカラーフィルタ、パターン形成方法及び液晶表示装置について順に説明する。
[光硬化性組成物]
本発明の光硬化性組成物は、エチレン性不飽和結合を有する化合物及び/又はバインダー樹脂と、前記光重合開始剤(I)を1種以上含有するものである。本発明の光硬化性組成物には、さらに、色材、そして必要に応じて、前記光重合開始剤(I)以外の光重合開始剤、分散剤、多官能モノマー、単官能モノマー、増感剤及び溶剤等を含有してもよく、こうして得られる光硬化性組成物は、カラーフィルタ用途に有用である。なお、多官能モノマー及び単官能モノマーは、官能基がエチレン性不飽和結合を有していれば、エチレン性不飽和結合を有する化合物にもなり得る。
本発明の光硬化性組成物において、光重合開始剤(I)の含有量は、感度、解像度及び現像性の観点から、光硬化性組成物の固形分に対して、好ましくは2〜50質量%、より好ましくは2〜30質量%、さらに好ましくは4〜15質量%である。
また、本発明の光硬化性組成物が、前記光重合開始剤(I)以外の光重合開始剤を含有する場合、前記光重合開始剤(I)以外の光重合開始剤の含有量は、本発明の効果を著しく阻害しない限り特に制限は無いが、光重合開始剤(I)100質量部に対して、好ましくは100質量部以下、より好ましくは50質量部以下、より好ましくは20質量部以下、さらに好ましくは10質量部以下、特に好ましくは5質量部以下である。
【0105】
(エチレン性不飽和結合を有する化合物)
エチレン性不飽和結合を有する化合物は、光重合開始剤(I)の存在下に光照射されることにより、重合反応を起こし、光硬化性組成物において、バインダーとなり得る化合物であり、形成された画素中で結着剤としての役割を果たし得る。エチレン性不飽和結合を有する化合物は、本発明の光硬化性組成物中に、好ましくは5〜60質量%、より好ましくは10〜40質量%含有させる。この範囲であると、形成された画素と基板との密着性が良好となる傾向にある。なお、エチレン性不飽和結合を有する化合物は、後述するバインダー樹脂と併用してもよい。併用する場合、その合計含有量が前記範囲内であると好ましい。
エチレン性不飽和結合を有する化合物の具体例としては、(メタ)アクリル酸、クロトン酸、α−クロルアクリル酸等の不飽和モノカルボン酸類;マレイン酸、フマル酸、イタコン酸等の不飽和ジカルボン酸又はその無水物類;トリメリット酸、ピロメリット酸等の3価以上の不飽和多価カルボン酸又はその無水物類;スチレン、α−メチルスチレン、クロロスチレン、メトキシスチレン、ジビニルベンゼン、ビニルトルエン等の芳香族ビニル化合物;メチル(メタ)アクリレート、エチル(メタ)アクリレート、n−プロピル(メタ)アクリレート、イソプロピル(メタ)アクリレート、n−ブチル(メタ)アクリレート、s−ブチル(メタ)アクリレート、t−ブチル(メタ)アクリレート、2−エチルヘキシル(メタ)アクリレート、ラウリル(メタ)アクリレート、シクロヘキシル(メタ)アクリレート、ヒドロキシエチル(メタ)アクリレート、フェノキシエチル(メタ)アクリレート、メトキシエチル(メタ)アクリレート、ポリ(プロピルオキシ)プロピル(メタ)アクリレート、ビニル(メタ)アクリレート、エチレングリコールジ(メタ)アクリレート、ポリエチレングリコールジ(メタ)アクリレート、プロピレングリコールジ(メタ)アクリレート、ポリプロピレングリコールジ(メタ)アクリレート、1,6−ヘキサンジオールジ(メタ)アクリレート、トリメチロールプロパントリ(メタ)アクリレート、アリル(メタ)アクリレート、ベンジル(メタ)アクリレート等の、(多価)アルコールをα,β−不飽和カルボン酸でエステル化した化合物;2−アミノエチル(メタ)アクリレート、2−ジメチルアミノエチル(メタ)アクリレート、2−アミノプロピル(メタ)アクリレート、2−ジメチルアミノプロピル(メタ)アクリレート等の不飽和カルボン酸アミノアルキルエステル類;グリシジル(メタ)アクリレート等の不飽和カルボン酸グリシジルエステル類;酢酸ビニル、プロピオン酸ビニル、酪酸ビニル、安息香酸ビニル等のカルボン酸ビニルエステル類;ビニルメチルエーテル、ビニルエチルエーテル、アリルグリシジルエーテル、イソブチルビニルエーテル等の不飽和エーテル類;(メタ)アクリロニトリル、α−クロロアクリロニトリル、シアン化ビニリデン等のシアン化ビニル化合物;(メタ)アクリルアミド、α−クロロアクリルアミド、N−2−ヒドロキシエチル(メタ)アクリルアミド等の不飽和アミド類;マレイミド、N−フェニルマレイミド、N−シクロヘキシルマレイミド等の不飽和イミド類;1,3−ブタジエン、イソプレン、クロロプレン等の脂肪族共役ジエン類等が挙げられる。これらの中でも、本発明の光重合開始剤(I)との相溶性の観点から、(メタ)アクリロイルオキシ基を有する化合物が好ましい。
エチレン性不飽和結合を有する化合物は、1種を単独で使用してもよいし、2種以上を併用してもよい。
【0106】
なお、エチレン性不飽和結合を有する化合物が、エーテル結合及び/又はエステル結合(好ましくはエーテル結合及びエステル結合)を持つエチレン性不飽和結合を有する化合物である場合、本発明の光重合開始剤(I)との相溶性により一層優れるため、本発明の光硬化性組成物において、後述する溶剤の量を低減する又は無溶剤とすることができるため、そのような化合物を用いることも好ましい。エチレン性不飽和結合を有する化合物として、エーテル結合及び/又はエステル結合を持つエチレン性不飽和結合を有する化合物を少なくとも1種用いる場合、溶剤の量は、光硬化性組成物全量に対して10質量%以下のみでよく、さらには5質量%以下、さらには無溶剤とすることもできる。
【0107】
(バインダー樹脂)
バインダー樹脂は、形成された画素中で結着剤としての役割を果たすものであり、基板と密着性が良好なものであれば、特に限定されるものではない。このようなバインダー樹脂は、本発明の光硬化性組成物の固形分中に、好ましくは5〜60質量%、より好ましくは10〜40質量%含有させる。この範囲であると、形成された画素と基板との密着性が良好となる傾向にある。また、バインダー樹脂の重量平均分子量としては、形成された画素と基板との密着性の観点から、好ましくは3,000〜100,000、より好ましくは5,000〜50,000である。なお、本明細書において、重量平均分子量は、ゲル浸透クロマトグラフィー(GPC)で測定したポリスチレン換算の値である。
なお、前述の通り、バインダー樹脂は、前記エチレン性不飽和結合を有する化合物と併用してもよい。
【0108】
上記バインダー樹脂としては、一般的なカラーフィルタ用光硬化性組成物に用いられるバインダー樹脂と同様のものを用いることができる。具体的には、エチレン−酢酸ビニル共重合体、エチレン−塩化ビニル共重合体、ポリスチレン、アクリロニトリル−スチレン共重合体、ABS樹脂、ポリメタクリル酸樹脂、エチレンメタクリル酸樹脂、ポリ塩化ビニル樹脂、塩素化塩化ビニル、ポリビニルアルコール、ポリエチレンテレフタレート、ポリブチレンテレフタレート、ポリカーボネート、ポリビニルアセタール、ポリエーテルエーテルケトン、ポリエーテルサルフォン、ポリフェニレンサルファイド、ポリアリレート、ポリビニルブチラール、エポキシ樹脂、フェノキシ樹脂、ポリイミド樹脂、ポリアミドイミド樹脂、ポリアミック酸樹脂、ポリエーテルイミド樹脂、フェノール樹脂、ユリア樹脂等が挙げられる。さらに、重合可能なモノマーであるメチル(メタ)アクリレート、エチル(メタ)アクリレート、n−プロピル(メタ)アクリレート、イソプロピル(メタ)アクリレート、s−ブチル(メタ)アクリレート、イソブチル(メタ)アクリレート、t−ブチル(メタ)アクリレート、n−ペンチル(メタ)アクリレート、n−ヘキシル(メタ)アクリレート、2−エチルヘキシル(メタ)アクリレート、n−オクチル(メタ)アクリレート、n−デシル(メタ)アクリレート、スチレン、α−メチルスチレン、N−ビニル−2−ピロリドン、グリシジル(メタ)アクリレート、シクロヘキシル(メタ)アクリレート、ジシクロペンタニル(メタ)アクリレート、ジシクロペンタニルオキシエチル(メタ)アクリレート、フェニル(メタ)アクリレート、ベンジル(メタ)アクリレートの1種以上と、アクリル酸、メタクリル酸、アクリル酸の2量体(例えば、東亜合成化学(株)製、商品名「M−5600」)、イタコン酸、クロトン酸、マレイン酸、フマル酸、ビニル酢酸、これらの酸無水物等の1種以上とを反応させて得られるポリマー又はコポリマー等が挙げられる。また、該コポリマーにグリシジル基又は水酸基を有するエチレン性不飽和化合物を付加させたポリマー等も挙げられる。
【0109】
特に好ましいバインダー樹脂としては、カルボキシル基等の酸性官能基を有するアルカリ可溶性樹脂、例えば、アルカリ可溶性アクリル系樹脂を挙げることができる。カルボキシル基を有するアルカリ可溶性樹脂としては、カルボキシル基含有不飽和単量体と他の共重合可能なエチレン性不飽和単量体の共重合体が好ましく、さらに分子内にエポキシ基とエチレン性不飽和基とを併せ持つ化合物、例えばグリシジル(メタ)アクリレート等を付加させ、側鎖にエチレン性不飽和基を導入したものが好ましい。
上記カルボキシル基含有不飽和単量体としては、例えば、(メタ)アクリル酸、クロトン酸、マレイン酸、無水マレイン酸、コハク酸モノ[2−(メタ)アクリロイロキシエチル]、フタル酸モノ[2−(メタ)アクリロイロキシエチル]、ω−カルボキシポリカプロラクトンモノ(メタ)アクリレート等が好ましく、(メタ)アクリル酸が特に好ましい。なお、カルボキシル基含有不飽和単量体は、1種を単独で使用してもよいし、2種以上を併用してもよい。
上記エチレン性不飽和単量体としては、メチル(メタ)アクリレート、n−ブチル(メタ)アクリレート、シクロヘキシル(メタ)アクリレート、イソボルニル(メタ)アクリレート、ジシクロペンタニル(メタ)アクリレート、アリル(メタ)アクリレート、スチレン、α−メチルスチレン、ベンジル(メタ)アクリレート、ヒドロキシエチル(メタ)アクリレート、2−ジメチルアミノエチル(メタ)アクリレート、2−エチルヘキシル(メタ)アクリレート、2−ヒドロキシル(メタ)エチルアクリレート、ジシクロペンテニル(メタ)アクリレート等、及びこれらのマクロモノマー類;N−メチルマレイミド、N−シクロヘキシルマレイミド、N−ベンジルマレイミド、N−フェニルマレイミド、N−メチルフェニルマレイミド等のN置換マレイミド類等を挙げることができる。なお、エチレン性不飽和単量体は、1種を単独で使用してもよいし、2種以上を併用してもよい。
【0110】
(色材)
本発明の光硬化性組成物には、さらに色材を含有させて着色光硬化性組成物としてもよい。該色材としては、顔料、染料、天然色素等が挙げられる。色材は、1種を単独で使用してもよいし、2種以上を併用してもよい。色材は、色再現性、硬化性及び現像性の観点から、本発明の光硬化性組成物の固形分中に、好ましくは5〜60質量%、より好ましくは5〜50質量%含有させる。
顔料としては、有機顔料、無機顔料があるが、発色性及び耐熱性の観点から有機顔料が好ましい。有機顔料としては、例えばカラーインデックス(C.I.;The Society of Dyers and Colourists 発行)においてピグメント(Pigment)に分類されている化合物(C.I.ピグメントブルー、C.I.ピグメントバイオレット、C.I.ピグメントグリーン、C.I.ピグメントレッド、C.I.ピグメントイエロー、C.I.ピグメントオレンジ等)を挙げることができる。
なお、色材は、以下の分散剤によって、より均一に分散されていることが好ましい。
【0111】
(分散剤)
分散剤としては、上記色材を均一に分散することができるものであればよく、公知の分散剤を使用することができる。具体的には、変性ポリウレタン、変性ポリアクリレート、変性ポリエステル、変性ポリアミド等の高分子分散剤、リン酸エステル、アルキルアミン、ポリオキシエチレンアルキルフェニルエーテル等の界面活性剤や顔料誘導体を挙げることができる。これらの中でも、高分子分散剤が好ましく、具体的な市販品の商品名としては、EFKA−4046、EFKA−4047、EFKAポリマー10、EFKAポリマー400、EFKAポリマー401、EFKAポリマー4300、EFKAポリマー4330(以上、チバ・スペシャルティ・ケミカルズ(株)製)、Disperbyk111、Disperbyk161、Disperbyk165、Disperbyk167、Disperbyk182、Disperbyk2000、Disperbyk2001(以上、ビックケミー・ジャパン(株)製)、SOLSPERSE24000、SOLSPERSE27000、SOLSPERSE28000(以上、ルーブリゾール社製)、アジスパー(登録商標)PB821、PB822(味の素ファインテクノ(株)製)等が挙げられる。
【0112】
本発明の光硬化性組成物に分散剤を含有させる場合、その含有量としては、上述の色材を均一に分散することができるものであれば特に限定されるものではないが、色材の分散性、光硬化性組成物の現像性、及び未露光箇所での現像残渣の低減の観点から、光硬化性組成物の固形分全量に対して、好ましくは0.5〜30質量%、より好ましくは1〜20質量%である。
なお、色材(特に顔料)と分散剤は、色材の分散量及び分散性の観点から、後述の溶剤中で混合することにより、分散液としてから使用するのが好ましい。
【0113】
(多官能モノマー)
本発明の光硬化性組成物に適宜含有させてもよい多官能モノマーとしては、上記バインダー樹脂と相溶性のある(メタ)アクリロイルオキシ基を有するモノマー、具体的には二官能(メタ)アクリレート及び三官能以上の(メタ)アクリレート等が好ましく挙げられる。多官能モノマーは1種を単独で使用してもよいし、2種以上を併用してもよい。
二官能(メタ)アクリレートとしては、例えば、1,4−ブタンジオール(メタ)アクリレート、ネオペンチルグリコールジ(メタ)アクリレート、1,6−へキサンジオールジ(メタ)アクリレート、ポリプロピレングリコールジ(メタ)アクリレート、トリエチレングリコールジ(メタ)アクリレート、ポリエチレングリコールジ(メタ)アクリレート、ビスフェノールAジ(メタ)アクリレート等が挙げられる。
また、三官能以上の(メタ)アクリレートとしては、例えば、トリメチロールプロパントリ(メタ)アクリレート、トリメチロールエタントリ(メタ)アクリレート、ペンタエリスリトールトリ(メタ)アクリレート、ペンタエリスリトールテトラ(メタ)アクリレート、ジペンタエリスリトールテトラ(メタ)アクリレート、ジペンタエリスリトールヘキサ(メタ)アクリレート(DPHA)、ジペンタエリスリトールペンタ(メタ)アクリレートや、カルボン酸変性ジペンタエリスリトールペンタ(メタ)アクリレート等のカルボキシル基含有多官能(メタ)アクリレートが挙げられる。
【0114】
本発明の光硬化性組成物に多官能モノマーを含有させる場合、その含有量は、光硬化性組成物の固形分全量に対して、好ましくは3〜50質量%である。この範囲であれば、充分に光硬化が進行するため、アルカリ現像時に露光部分が溶出する恐れがなくなり、且つアルカリ現像性が良好である。
【0115】
(単官能モノマー)
本発明の光硬化性組成物に適宜含有させてもよい単官能モノマーとしては(メタ)アクリレート類が好ましく、例えば、アリル(メタ)アクリレート、ベンジル(メタ)アクリレート、ブトキシエチル(メタ)アクリレート、ブトキシエチレングリコール(メタ)アクリレート、シクロヘキシル(メタ)アクリレート、ジシクロペンタニル(メタ)アクリレート、2−エチルヘキシル(メタ)アクリレート、グリセロール(メタ)アクリレート、グリシジル(メタ)アクリレート、2−ヒドロキシエチル(メタ)アクリレート、2−ヒドロキシプロピル(メタ)アクリレート、イソボニル(メタ)アクリレート、イソデキシル(メタ)アクリレート、イソオクチル(メタ)アクリレート、ラウリル(メタ)アクリレート、2−メトキシエチル(メタ)アクリレート、メトキシエチレングリコール(メタ)アクリレート、フェノキシエチル(メタ)アクリレート、ステアリル(メタ)アクリレート等が挙げられる。
【0116】
(増感剤)
さらに、本発明の光硬化性組成物には、必要に応じて増感剤を含有させてもよい。増感剤は、本発明の光硬化性組成物を基板上に塗布した後、露光した際に、照射されたエネルギーを吸収し、その吸収したエネルギーを上記本発明の光重合開始剤の反応開始に寄与させる役割を担う物質である。
増感剤としては、具体的には、4,4'−ビス(ジエチルアミノ)ベンゾフェノン、4,4'−ビス(ジメチルアミノ)ベンゾフェノン、4−メチル−4'−ジエチルアミノベンゾフェノン、4−メトキシ−4'−ジエチルアミノベンゾフェノン、4,4'−ビス(ジプロピルアミノ)ベンゾフェノン、4,4'−ビス(ジイソプロピルアミノ)ベンゾフェノン等が挙げられる。これらは、1種を単独で使用してもよいし、2種以上を併用してもよい。
本発明の光硬化性組成物に増感剤を含有させる場合、その含有量としては、光硬化性組成物の固形分全量に対して、好ましくは1〜10質量%である。
【0117】
(溶剤)
本発明の光硬化性組成物には、色材を分散させるために溶剤を含有していてもよい。該溶剤としては、一般的なカラーフィルタ用光硬化性組成物に用いられるものであれば特に限定されるものではない。具体的には、エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル、プロピレングリコールモノメチルエーテル、ジエチレングリコールモノエチルエーテル、ジエチレングリコールジメチルエーテル、ジエチレングリコールジエチルエーテル、ジプロピレングリコールジメチルエーテル等の(モノ又はポリ)アルキレングリコール(モノ又はポリ)アルキルエーテル類、及びこれらのアセテート類;プロピレングリコールジアセテート、1,3−ブチレングリコールジアセテート等のジアセテート類;テトラヒドロフラン等のエーテル類;メチルエチルケトン、シクロヘキサノン、2−ヘプタノン等のケトン類;2−ヒドロキシプロピオン酸メチル、3−ヒドロキシプロピオン酸エチル、酢酸エチル、n−ブチルアセテート、イソブチルアセテート、酪酸イソブチル、酪酸n−ブチル、3−メトキシプロピオン酸エチル、3−エトキシプロピオン酸エチル、3−メトキシブチルアセテート、乳酸エチル、シクロヘキサノールアセテート等のエステル類;トルエン、キシレン等の芳香族炭化水素類等を挙げることができる。溶剤は、1種を単独で使用してもよいし、2種以上を併用してもよい。
【0118】
これらの中でも、相溶性、溶解性、顔料分散性及び塗布性の観点からは、エーテル結合及び/又はエステル結合を有する25℃で液体の化合物、具体的にはプロピレングリコールモノメチルエーテルアセテート、3−メトキシブチルアセテート、エチレングリコールモノメチルエーテル、ジエチレングリコールジエチルエーテルが好ましく、エーテル結合及びエステル結合を有する25℃で液体の化合物がより好ましい。
本発明の光硬化性組成物に溶剤を含有させる場合、その含有量としては、光硬化性組成物全量中、好ましくは50〜90質量%である。通常、この範囲であれば、顔料分散性が良好であり、レジスト塗布特性(面内均一性)を良好なものとすることができる。なお、本発明の光重合開始剤は、エーテル結合及び/又はエステル結合を有する25℃で液体の化合物との相溶性に優れているため、上記溶剤がエーテル結合及び/又はエステル結合を有する25℃で液体の化合物であれば、光硬化性組成物における溶剤の含有量を大幅に低減することもできる。その場合、本発明の光硬化性組成物中におけるエーテル結合及び/又はエステル結合を有する25℃で液体の化合物の含有量は、光重合開始剤(I)1質量部に対して好ましくは5〜100質量部の範囲で任意に選択し得るため、必要に応じて、光重合開始剤(I)1質量部に対して5〜20質量部程度、さらには5〜10質量部程度の少ない量とすることも可能である。
【0119】
(その他の成分)
本発明の光硬化性組成物には、さらに必要に応じて、無機充填剤;密着促進剤;凝集防止剤;重合停止剤;連鎖移動剤;レベリング剤;可塑剤;消泡剤;シランカップリング剤;紫外線吸収剤等を含有させてもよい。
【0120】
[カラーフィルタ]
カラーフィルタの製造方法には、色材を含有する本発明の光硬化性組成物を用い、フォトリソグラフィー法により、孔部を有する着色層(パターン)を形成する方法を利用することが好ましい。この方法であれば、例えば上記露光の際に、エネルギーの回折等が生じた場合であっても、その影響を受けにくいものとすることができる。従って、着色層に微細なパターン状に孔部が形成された、高精細なカラーフィルタとすることが可能となる。
【0121】
(パターン形成方法)
パターン形成方法として、本発明の光硬化性組成物を用いて、フォトリソグラフィー法により行う方法を説明する。その方法については、着色層に孔部を形成することが可能な方法であれば特に限定されるものではない。例えば、本発明の光硬化性組成物を基板に塗布し、乾燥した後、開口部と遮蔽部とが設けられたフォトマスク等を用いて、エネルギーを照射することにより露光し、次いで現像を行うことによりパターンを形成する。なお、これを複数回繰り返すことにより、例えば赤色、緑色、青色それぞれのパターンを基板上に形成し、次いで、酸化インジウムスズ(ITO)等の透明電極膜を形成することにより、カラーフィルタを製造することができる。
前記光硬化性組成物の塗布方法に特に制限はなく、例えばスピンコート法やスプレーコート、ディップコート、ロールコート、ビードコート、バーコート等の公知の塗布方法に利用できる。塗布後の乾燥は、通常、好ましくは50〜150℃で15秒〜10分行う。
露光の際に用いられる光源としては、一般的に着色層の形成の際に用いられている光源と同様とすることができるが、超高圧水銀ランプ、低圧水銀ランプ、メタルハライドランプ等、紫外部に高輝線を有するランプが好ましい。これにより、上記光硬化性組成物の特性をより効果的に発揮させることが可能となる。
また、前記光硬化性組成物を現像する方法としては、不要部分の光硬化性組成物を除去することが可能であれば、その方法等は特に限定されるものではなく、一般的なカラーフィルタの製造の際に行われる現像方法と同様とすることができる。ここで、例えば、現像液の種類、現像液の濃度、現像液や洗浄水の圧力等を最適化することにより、より微細なパターン状に孔部を形成することが可能となる。
【0122】
なお、本工程に用いられる基板としては、一般的なカラーフィルタに用いられる基板(透明基板)と同様のものを用いることができる。具体的には、石英ガラス、無アルカリガラス、合成石英板等の可撓性のない透明なリジッド材、あるいは、透明樹脂フィルム、光学用樹脂板等の可撓性を有する透明なフレキシブル材が挙げられる。
基板の厚みは、特に限定されるものではないが、本発明のカラーフィルタの用途に応じて、例えば100μm〜1mm程度のものを使用することができる。
カラーフィルタの製造においては、例えば、遮光部を形成する遮光部形成工程、上記着色層上にオーバーコート層を形成する工程等、必要に応じて適宜、他の工程を有していてもよい。
【0123】
[液晶表示装置]
本発明の液晶表示装置は、前記した本発明のカラーフィルタと対向基板と、前記カラーフィルタと前記対向基板との間に形成された液晶層とを有するものであり、その構成は特に制限されるものではなく、公知のカラーフィルタが用いられた液晶表示装置と同じ構成をとることができる。
例えば、図1に示すように、本発明の液晶表示装置40は、得られたカラーフィルタ10(表示側基板)と、TFTアレイ基板(液晶駆動側基板)を有する対向基板20を対向させ、両基板の内面側周縁部をシール剤により接合すると、両基板は所定距離のセルギャップを保持した状態で貼り合わされる。そして、基板間の間隙部に液晶を満たして密封し、液晶層30とすることにより、液晶パネルに属するアクティブマトリックス方式のカラー液晶表示装置が得られる。なお、ここで、本発明のカラーフィルタ10は透明基板1、遮光部2及び着色層3を有する。
液晶表示装置の駆動方式に特に制限はなく、一般的に液晶表示装置に用いられている駆動方式を採用することができる。例えば、TN方式、IPS方式、OCB方式及びMVA方式等が挙げられる。また、液晶層を構成する液晶としては、液晶表示装置の駆動方式等に応じて、誘電異方性の異なる各種液晶及びこれらの混合物を用いることができる。
【実施例】
【0124】
次に、本発明を実施例により、さらに詳細に説明するが、本発明は、これらの例によって何ら限定されるものではない。
【0125】
<実施例1>
(工程1)
【化42】
カルバゾール6.8g(40mmol)とエチルアクリレート4.0g(40mmol)及び炭酸カリウム5.5g(40mmol)をジメチルホルムアミド(DMF)に懸濁させ、室温で24時間攪拌した。反応終了後、水を添加してからさらに30分攪拌し、その後、酢酸エチルで抽出、水で洗浄し、硫酸マグネシウムで乾燥させ、溶媒を留去、乾燥することにより、淡黄色液体の上記化合物(i)9.6g(収率93%)を得た。該化合物(i)の1H−NMR測定結果を以下に示す。
【0126】
1H−NMR(400MHz,CDCl3,TMS,ppm)δ:1.15(t,3H)、2.835(t,2H)、4.08(q,2H)、4.64(t,3H)、7.215−7.254(m,2H)、7.45−7.47(m,4H)、8.08(d,2H)
【0127】
(工程2)
【化43】
化合物(i)2.7g(10mmol)をジクロロメタン10mlに溶解させ、氷浴で冷却しながら塩化アルミニウム2.7g(20mmol)を20分かけて添加した。得られた混合溶液へベンゾイルクロリド1.4g(11mmol)を20分かけて滴下した。その後、徐々に室温に戻してから、1時間攪拌した。再び氷浴で冷却しながら、塩化アルミニウム2.7g(20mmol)を20分かけて添加し、そこへアセチルクロリド1.2g(15mmol)を20分かけて滴下した。その後、徐々に室温に戻し一晩攪拌した。反応終了後、氷水に反応溶液を投入し、酢酸エチルで抽出した後、水、飽和炭酸水素ナトリウム水溶液及び飽和食塩水で洗浄し、硫酸マグネシウムで乾燥してから溶媒を留去した。
シリカゲルカラムクロマトグラフィー(溶出液;ジクロロメタン→ノルマルヘキサン:酢酸エチル=2:1)により生成物を単離し、淡黄色固体の化合物(ii)2.8g(収率70%)を得た。該化合物(ii)の1H−NMR測定結果を以下に示す。
【0128】
1H−NMR(400MHz,CDCl3,TMS,ppm)δ:1.16(t,3H)、2.725(s,3H)、2.91(t,2H)、4.08(q,2H)、4.73(t,2H)、7.525−7.86(m,5H)、8.10(dd,2H)、8.18(dd,1H)、8.20(dd,1H)、8.625(d,1H)、8.725(d,1H)
【0129】
(工程3)
【化44】
塩化ヒドロキシルアミン0.12g(1.7mmol)及び酢酸ナトリウム0.17g(2.0mmol)を水1.4mlに溶解させ、そこへ化合物(ii)0.60g(1.58mmol)及びエタノール11mlを添加し、7時間還流させた。
反応終了後、反応溶液の溶媒を留去し、得られた固体を水で洗浄した後、THFに溶解させ、硫酸マグネシウムで乾燥し、溶媒を留去することにより、淡黄色液体の化合物(iii)0.60g(収率89%)を得た。該化合物(iii)の1H−NMR測定結果を以下に示す。
【0130】
1H−NMR(400MHz,CDCl3,TMS,ppm)δ:1.16(t,3H)、2.41(s,3H)、2.89(t,2H)、4.09(q,2H)、4.69(t,2H)、7.475−7.63(m,5H)、7.85(dd,1H)、8.045(dd,2H)、8.07(dd,1H)、8.325(d,1H)、8.60(d,1H)
【0131】
(工程4)
【化45】
化合物(iii)0.60g(1.4mmol)をt−ブチルメチルエーテル7mlに溶解させ、アセチルクロリド0.11g(1.4mmol)添加し、氷浴で冷却しながらトリエチルアミン0.28ml(2.0mmol)を滴下した。徐々に室温に戻しながら3時間攪拌した。反応終了後、氷水に反応混合液を投入し、酢酸エチルで抽出した後、水、飽和炭酸水素ナトリウム水溶液、飽和食塩水で洗浄し、硫酸マグネシウムで乾燥してから溶媒を留去した。シリカゲルカラムクロマトグラフィー(溶出液;ジクロロメタン→ノルマルヘキサン:酢酸エチル=2:1)により生成物を単離し、淡黄色固体の化合物(iv)0.30g(収率70%)を得た。該化合物(iv)の物性を以下に示す。
【0132】
1H−NMR(400MHz,CDCl3,TMS,ppm)δ:1.16(t,3H)、2.23(s,3H)、2.51(s,3H)、2.90(t,2H)、4.09(q,2H)、4.71(t,2H)、7.51−7.62(m,5H)、7.75(dd,2H)、7.99(dd,1H)、8.09(dd,1H)、8.46(d,1H)、8.59(d,1H)
UVスペクトル(移動層;アセトニトリル):λmax=260、292、333nm
熱分解温度(窒素ガス雰囲気下、1.5mgのサンプルを昇温速度10℃/分で加熱し、5%質量減少した時の温度):224.5℃
分子量:470.52
【0133】
<実施例2>
実施例1の工程1において、エチルアクリレート(40mmol)の代わりにイソブチルアクリレート(40mmol)を用い、工程2において、ベンゾイルクロリド(11mmol)の代わりに2−メチルベンゾイルクロリド(11mmol)を用いたこと以外は実施例1と同様に操作を行い、下記の淡黄色固体の化合物(v)を得た。該化合物(v)の物性を以下に示す。
【0134】
【化46】
1H−NMR(400MHz,CDCl3,TMS,ppm)δ:0.81(d、6H)、1.805(m、1H)、2.29(s、3H)、2.35(s、3H)、2.50(s、3H)、2.90(t、2H)、3.81(d、2H)、4.70(t、2H)、7.28−7.53(m、6H)、7.97(dd、1H)、8.10(dd、1H)、8.42(d、1H)、8.51(d、1H)
UVスペクトル(移動層;アセトニトリル):λmax=260、295.5、334nm
熱分解温度(窒素ガス雰囲気下、1.5mgのサンプルを昇温速度10℃/分で加熱し、5%質量減少した時の温度):236℃
分子量:512.6
【0135】
<実施例3>
実施例1の工程1において、エチルアクリレート(40mmol)の代わりに2−メトキシエチルアクリレート(40mmol)を用い、工程2において、ベンゾイルクロリド(11mmol)の代わりに2−メチルベンゾイルクロリド(11mmol)を用いたこと以外は実施例1と同様に操作を行い、下記の淡黄色固体の化合物(vi)を得た。該化合物(vi)の物性を以下に示す。
【0136】
【化47】
1H−NMR(400MHz,CDCl3,TMS,ppm)δ:2.29(s、3H)、2.35(s、3H)、2.50(s、3H)、2.95(t、2H)、3.32(s、3H)、3.50(t、2H)、4.19(t、2H)、4.70(t、2H)、7.30−7.54(m、6H)、7.97(dd、1H)、8.10(dd、1H)、8.42(d、1H)、8.51(d、1H)
UVスペクトル(移動層;アセトニトリル):λmax=258.5、296、334.5nm
熱分解温度(窒素ガス雰囲気下、1.5mgのサンプルを昇温速度10℃/分で加熱し、5%質量減少した時の温度):243℃
分子量:514.57
【0137】
<参考例4>
(工程1)
【化48】
カルバゾール5.0g(30mmol)をジメチルホルムアミド(DMF)6mlに溶解させ、炭酸エチレン13.2g(150mmol)とジアザビシクロウンデセン(DBU)5.5ml(037mmol)を添加し、100℃で3時間加熱還流した。冷却後、水を投入し、酢酸エチルで抽出した後、水洗してから溶媒を留去した。
次に、得られた濃縮物をアセトニトリル15mlで希釈し、そこへアセチルクロライド2g(23mmol)及びトリエチルアミン4ml(29mmol)を添加し、室温で3時間攪拌した。反応終了後、水を投入し、酢酸エチルで抽出した後、水で洗浄してから硫酸マグネシウムで乾燥させ、溶媒を留去することにより、淡黄色液体の上記化合物(vii)6.0g(収率80%)を得た。
【0138】
(工程2)
【化49】
化合物(vii)2.5g(9.9mmol)をジクロロメタン10mlに溶解させ、氷浴で冷却しながら塩化アルミニウム1.5gを20分かけて添加した。その後、ベンゾイルクロリド1.4g(10.0mmol)を滴下し、徐々に室温に戻してから1時間攪拌した。
再び氷浴で冷却しながら塩化アルミニウム1.5gを20分かけて添加、そこへアセチルクロリド0.93g(11.8mmol)を添加し、徐々に室温に戻してから一時間攪拌した。
反応終了後、氷水に反応溶液を投入し、酢酸エチルで抽出した後、水、飽和炭酸水素ナトリウム水溶液、及び飽和食塩水で洗浄し、硫酸マグネシウムで乾燥してから溶媒を留去した。
シリカゲルカラムクロマトグラフィー(溶出液;ジクロロメタン→ノルマルヘキサン:酢酸エチル=2:1)により化合物を単離し、淡黄色固体の化合物(viii)2.8g(収率70%)を得た。
【0139】
(工程3)
【化50】
塩化ヒドロキシルアミン0.38g(5.4mmol)、酢酸ナトリウム0.54g(6.4mmol)を水4.5mlに溶解させ、そこへ前記化合物(viii)2.0g(5.0mmol)及びエタノール35mlを添加し、2時間還流させた。
反応終了後、反応溶液の溶媒を留去し、得られた固体を水で洗浄した後、THFに溶解させ、硫酸マグネシウムで乾燥し、溶媒を留去することにより、淡黄色液体の化合物(ix)1.8g(収率89%)を得た。
【0140】
(工程4)
【化51】
化合物(ix)0.8g(2.0mmol)をt−ブチルメチルエーテル10mlに溶解させ、アセチルクロライド0.16g(2.0mmol)を添加し、氷浴で冷却しながらトリエチルアミン0.4ml(2.9mmol)を滴下した。徐々に室温に戻しながら3時間攪拌した。
反応終了後、氷水に反応溶液を投入し、酢酸エチルで抽出した後、水、飽和炭酸水素ナトリウム水溶液、及び飽和食塩水で洗浄し、硫酸マグネシウムで乾燥してから溶媒を留去した。
シリカゲルカラムクロマトグラフィー(溶出液;ジクロロメタン→ノルマルヘキサン:酢酸エチル=2:1)により化合物を単離し、下記物性の淡黄色固体の化合物(x)0.6g(収率70%)を得た。
【0141】
(化合物(x)の諸物性)
1H−NMR(400MHz,CDCl3,TMS,ppm)δ:1.93(s,3H)、2.50(s,3H)、2.85(s,3H)、4.31(t,2H)、4.40(t,2H)、7.50−7.62(m,5H)、7.75(dd,2H)、7.99(dd,1H)、8.09(dd,1H)、8.46(d,1H)、8.59(d,1H)
UVスペクトル(移動層;アセトニトリル):λmax=260、296、334nm
熱分解温度(窒素ガス雰囲気下、1.5mgのサンプルを昇温速度10℃/分で加熱し、5%質量減少した時の温度):225℃
分子量:456.49
【0142】
<参考例5>
(工程1)
【化52】
9−カルバゾール−9−プロピオン酸0.96g(40mmol)をアセトニトリル10mlで溶解した後、モルホリン18g(210mmol)を添加した。得られた混合液へ塩化チオニル14g(120mmol)を滴下し、室温で3時間攪拌した。
反応終了後、水を添加してから30分攪拌した。その後、酢酸エチルで抽出し、水で洗浄した後、硫酸マグネシウムで乾燥してから溶媒を留去することにより、淡黄色液体の化合物(xi)10g(収率81%)を得た。
【0143】
(工程2〜4)
【化53】
【0144】
上記化学反応式のとおり、実施例5の工程2〜4では、実施例4の工程2〜4において、工程2で化合物(vii)の代わりに化合物(xi)を用いたこと以外は同様の操作を採用することにより、下記物性の淡黄色固体の化合物(xii)を得た。
【0145】
(化合物(xii)の諸物性)
1H−NMR(400MHz,CDCl3,TMS,ppm)δ:2.50(s,3H)、2.80(t,2H)2.85(s,3H)、2.90(br,2H)、3.00(br,2H)、3.35(br,2H)、3.46(br,2H)4.72(t,2H)、7.51−7.62(m,5H)、7.75(dd,2H)、7.99(dd,1H)、8.09(dd,1H)、8.46(d,1H)、8.59(d,1H)
UVスペクトル(移動層;アセトニトリル):λmax=260,292,335nm
熱分解温度(窒素ガス雰囲気下、1.5mgのサンプルを昇温速度10℃/分で加熱し、5%質量減少した時の温度):234℃
分子量:511.57
【0146】
<試験例1〜3、参考試験例4及び5、比較試験例1及び2>相溶性及び感度試験
三官能アクリレート「M−305」(商品名、東亞合成(株)製)100質量部に、実施例1〜3又は参考例4〜5で得た光重合開始剤(化合物(iv)〜(vi)、(x)及び(xii);試験例1〜3及び参考試験例4及び5用)、下記「IRGACURE OXE02」(チバ・スペシャルティ・ケミカルズ(株)製、比較試験例1用。以下、「OXE02」と略称することがある。)又は下記「N−1919」((株)ADEKA製、比較試験例2用)を5質量部添加した。ここで、目視により、得られた混合物中の固体の有無を確認し、エチレン性不飽和結合を有する化合物(特に(メタ)アクリロイルオキシ基を有する化合物)との相溶性の指標とした。結果を表1に示す。次いで、前記混合物をクロロホルムで溶解することにより、光硬化性組成物を調製した。
得られた光硬化性組成物を各々、クロムをスパッタしたガラス基板上にスピンコートし塗布した。
こうして得られた塗布膜にUVを露光しながら、赤外分光装置で810cm-1のピークの減少量を経時的に記録し、二重結合の消失がどの程度進行しているか(反応率)を確認し、感度の指標とした。結果を表1に示す。
【0147】
【化54】
【0148】
【表1】
【0149】
表1より、本発明の光重合開始剤は、エチレン性不飽和二重結合を有する化合物、特に多官能(メタ)アクリレートとの相溶性が高く、無溶剤の光重合開始剤を調製することも可能である。同時に、本発明の光重合開始剤を含有する光硬化性組成物は、従来の光重合開始剤を含有する光硬化性組成物に比べて、より高感度であることが分かる。
【0150】
<試験例6〜8、参考試験例9及び10、比較試験例3及び4>溶剤に対する溶解性試験
プロピレングリコールモノエチルエーテルアセテート(PGMEA)、メタノール、3−メトキシ−3−メチル−1−ブチルアセテート(MMBA)、メチルイソブチルケトン(MIBK)、クロロホルムそれぞれ100質量部に対する光重合開始剤の溶解量を調査し、下記評価基準に従って溶解性を評価した。結果を表2に示す。
A:10質量部以上溶解した。
B:5質量部以上、10質量部未満の範囲で溶解した。
C:2質量部以上、5質量部未満の範囲で溶解した。
D:2質量部未満しか溶解しなかった。
ここで、「溶解した」とは、目視により固体が見えない状態を指す。
【0151】
【表2】
【0152】
表2より、従来の光重合開始剤に比べ、本発明の光重合開始剤は、各種溶剤、特にエーテル結合及び/又はエステル結合を有する25℃で液体の化合物に対する溶解性に優れていることがわかる。
また、比較試験例4で使用した光重合開始剤「N−1919」は、比較試験例3で使用した光重合開始剤「OXE02」に比べ、溶解性が高くなっている。これは、「N−1919」は、「OXE02」の構造に置換基を付与することによって溶解性を高めたものだからである。試験例6〜10で使用した化合物(iv)〜(vi)、(x)及び(xii)は、この「N−1919」よりも溶剤への溶解性が高いのみならず、分子量も小さくなっており、光硬化性組成物中における含有量を低減することができ、より一層優れた光重合開始剤である。
【0153】
<製造例1>バインダー樹脂の製造
ベンジルメタクリレート30質量部、スチレン38質量部、メタクリル酸18質量部及びt−ブチルパーオキシ−2−エチルヘキサノエート(日油(株)製の「パーブチルO」)10質量部の混合液を、プロピレングリコールモノエチルエーテルアセテート150質量部を入れた重合槽中へ、窒素気流下、100℃で3時間かけて滴下した。滴下終了後、さらに100℃で3時間加熱し、重合体溶液を得た。
得られた重合体溶液に、グリシジルメタクリレート14質量部、トリエチルアミン0.2質量部及びp−メトキシフェノール0.05質量部を添加し、110℃で10時間加熱することによりバインダー樹脂を調製した。
得られたバインダー樹脂の固形分濃度は38質量%、酸価は75mgKOH/g、重量平均分子量は10,000であった。
【0154】
<製造例2>青色顔料分散液の調製
【表3】
表3に記載の各成分を各配合量で混合し、直径0.3mmジルコニアビーズを用いペイントシェーカー(浅田鉄工(株)製)で3時間分散させ、青色顔料分散液(固形分濃度:21質量%)を調製した。
【0155】
<製造例3>赤色顔料分散液の調製
【表4】
表4に記載の各成分を各配合量で混合し、直径0.3mmジルコニアビーズを用いペイントシェーカー(浅田鉄工(株)製)で3時間分散させ、赤色顔料分散液(固形分濃度:21質量%)を調製した。
【0156】
<製造例4>緑色顔料分散液の調製
【表5】
表5に記載の各成分を各配合量で混合し、直径0.3mmジルコニアビーズを用いペイントシェーカー(浅田鉄工(株)製)で3時間分散させ、緑色顔料分散液(固形分濃度:21質量%)を調製した。
【0157】
以下の実施例において、感度、解像度、現像性及び密着性の評価を以下の通りに実施した。
(感度)
露光工程において、露光量を100mJ/cm2又は200mJ/cm2とし、光が照射された領域の現像処理及びポストベーク後の膜厚が、露光前の膜厚100%に対して95%以上であった場合に「膜厚が十分」であるとし、下記評価基準に従って評価した。
A:露光量100mJ/cm2であれば、十分な膜厚が得られた。
B:露光量100mJ/cm2では不十分だが、露光量200mJ/cm2であれば十分な膜厚が得られた。
なお、膜厚は、触針式膜厚測定器「SURFCORDER ET4000A」((株)小坂研究所製)を用いて測定した。
(解像度)
下記評価基準に従って、解像度を評価した。
a:露光現像時において、線幅10μm未満でも良好にパターンを得られた。
b:線幅15〜20μmであれば良好なパターンを得られた。
(現像性)
露光時において、未露光部(光が照射されなかった部分)の残渣を観察し、下記評価基準に従って、現像性を評価した。
○:残渣を全く確認できなかった。
×:残渣を確認できた。
(密着性)
現像後に流されずに密着していた独立パターンの線幅を調査し、下記評価基準に従って、密着性を評価した。
◎:線幅を5μm以下としても、良好な孤立パターンが得られた。
○:線幅6〜10μmとすれば、良好な孤立パターンを得られた。
【0158】
<実施例6>青色光硬化性樹脂組成物の調製及びパターン形成
【表6】
表6に記載の各成分を各配合量で混合し、ディゾルバーで1時間攪拌し、青色光硬化性樹脂組成物を調製した。
得られた青色光硬化性樹脂組成物をガラス基板上にスピンコートし、80℃で3分間加熱することにより、青色光硬化樹脂組成物の塗膜を形成した。得られた塗膜を、所定線幅のマスクを通して、塗膜側から光源として高圧水銀灯にて露光した後、100倍に希釈したディスパーズH(ヘンケル社製)を用いてスプレー現像を行い、現像終了後、230℃で30分ポストベークし、パターンを形成した。
感度、解像度、現像性及び密着性の評価結果を表9に示す。
【0159】
<比較例1、2>青色光硬化性樹脂組成物の調製及びパターン形成
実施例6において、実施例1で得た化合物(iv)の代わりに、前記「IRGACURE OXE02」(チバ・スペシャルティ・ケミカルズ(株)製)又は前記「N−1919」((株)ADEKA製)を用いたこと以外は同様にして青色光硬化性樹脂組成物を調製し、パターンを形成した。
感度、解像度、現像性及び密着性の評価結果を表9に示す。
【0160】
<実施例7>赤色光硬化性樹脂組成物の調製及びパターン形成
【表7】
表7に記載の各成分を各配合量で混合し、ディゾルバーで1時間攪拌し、赤色光硬化性樹脂組成物を調製した。
得られた赤色光硬化性樹脂組成物について、実施例6と同様の操作を行うことにより、パターンを形成した。
感度、解像度、現像性及び密着性の評価結果を表9に示す。
【0161】
<比較例3、4>赤色光硬化性樹脂組成物の調製及びパターン形成
実施例7において、実施例1で得た化合物(iv)の代わりに、前記「IRGACURE OXE02」(チバ・スペシャルティ・ケミカルズ(株)製)又は前記「N−1919」((株)ADEKA製)を用いたこと以外は同様にして赤色光硬化性樹脂組成物を調製し、パターンを形成した。
感度、解像度、現像性及び密着性の評価結果を表9に示す。
【0162】
<実施例8>緑色光硬化性樹脂組成物の調製及びパターン形成
【表8】
表8に記載の各成分を各配合量で混合し、ディゾルバーで1時間攪拌し、緑色光硬化性樹脂組成物を調製した。
得られた緑色光硬化性樹脂組成物について、実施例6と同様の操作を行うことにより、パターンを形成した。
感度、解像度、現像性及び密着性の評価結果を表9に示す。
【0163】
<比較例5、6>緑色光硬化性樹脂組成物の調製及びパターン形成
実施例8において、実施例1で得た化合物(iv)の代わりに、前記「IRGACURE OXE02」(チバ・スペシャルティ・ケミカルズ(株)製)又は前記「N−1919」((株)ADEKA製)を用いたこと以外は同様にして緑色光硬化性樹脂組成物を調製し、パターンを形成した。
感度、解像度、現像性及び密着性の評価結果を表9に示す。
【0164】
<実施例9〜11>
実施例6〜8それぞれにおいて、実施例1で得た化合物(iv)の代わりに、実施例2で得た化合物(v)を用いたこと以外は同様にして、青色光硬化性樹脂組成物、赤色光硬化性樹脂組成物及び緑色光硬化性樹脂組成物を調製し、パターンを形成した。
感度、解像度、現像性及び密着性の評価結果を表9に示す。
【0165】
<実施例12〜14>
実施例6〜8それぞれにおいて、実施例1で得た化合物(iv)の代わりに、実施例3で得た化合物(vi)を用いたこと以外は同様にして、青色光硬化性樹脂組成物、赤色光硬化性樹脂組成物及び緑色光硬化性樹脂組成物を調製し、パターンを形成した。
感度、解像度、現像性及び密着性の評価結果を表9に示す。
【0166】
<参考例15〜17>
実施例6〜8それぞれにおいて、実施例1で得た化合物(iv)の代わりに、参考例4で得た化合物(x)を用いたこと以外は同様にして、青色光硬化性樹脂組成物、赤色光硬化性樹脂組成物及び緑色光硬化性樹脂組成物を調製し、パターンを形成した。
感度、解像度、現像性及び密着性の評価結果を表9に示す。
【0167】
<参考例18〜20>
実施例6〜8それぞれにおいて、実施例1で得た化合物(iv)の代わりに、参考例5で得た化合物(xii)を用いたこと以外は同様にして、青色光硬化性樹脂組成物、赤色光硬化性樹脂組成物及び緑色光硬化性樹脂組成物を調製し、パターンを形成した。
感度、解像度、現像性及び密着性の評価結果を表9に示す。
【0168】
【表9】
【0169】
表9より、本発明の光重合開始剤を含有する光硬化性組成物は、従来の光重合開始剤を含有する光硬化性組成物と同等の現像性を有し、また、感度、解像度及び密着性については、従来の光重合開始剤を含有する光硬化性組成物より優れていることがわかる。このことより、本発明の光重合開始剤を含有する光硬化性組成物は、深部硬化性に優れていると言える。
【産業上の利用可能性】
【0170】
本発明の光重合開始剤は、光(特に波長450nm以下の短波長の光)に対する感度が高いため、薄膜化することが可能であり、低コストで高品質のパターンを形成することができること、さらに、エチレン性不飽和結合を有する化合物(特に(メタ)アクリロイルオキシ基を有する化合物)との相溶性や溶剤に対する溶解性が高いことより、カラーフィルタ用光硬化性組成物に含有させる光重合開始剤、さらには高遮光性のブラックマトリックス用光硬化性組成物に含有させる光重合開始剤として有用である。
【符号の説明】
【0171】
1 透明基板
2 遮光部
3 着色層
10 カラーフィルタ
20 対向基板
30 液晶層
40 液晶表示装置
【特許請求の範囲】
【請求項1】
下記一般式(I−a)で表される光重合開始剤。
【化1】
(式中、R1〜R11は、それぞれ独立に、水素原子、ハロゲン原子、置換もしくは無置換の炭素数1〜20のアルキル基、置換もしくは無置換の炭素数2〜20のアルケニル基、置換もしくは無置換の環形成原子数3〜10のシクロアルキル基、置換もしくは無置換の炭素数4〜20のシクロアルケニル基、ヒドロキシル基、置換もしくは無置換の炭素数1〜20のアルコキシ基、置換もしくは無置換の炭素数2〜20のアルケニルオキシ基、置換もしくは無置換の炭素数1〜20のアルカノイル基、置換もしくは無置換の炭素数2〜20のアルケノイル基、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数3〜14のヘテロ環基を示す。R3は、R4又はR5と一緒になって環を形成していてもよい。R4は、R5と一緒になって環を形成していてもよい。
また、Arは、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数5〜14のヘテロアリール基を示す。
nは、1〜10の整数を示す。nが2〜10の整数の場合、複数のR4及びR5は、それぞれ同一でも異なっていてもよい。)
【請求項2】
前記一般式(I−a)中、R1、R2及びR3が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、nが2であり、複数のR4及びR5が、それぞれ独立に、水素原子又は置換もしくは無置換の炭素数1〜20のアルキル基であり、Arが、置換もしくは無置換の環形成炭素数6〜14のアリール基である、請求項1に記載の光重合開始剤。
【請求項3】
前記一般式(I−a)中、R1、R2及びR3が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、nが2であり、複数のR4及びR5が、それぞれ独立に、水素原子又は置換もしくは無置換の炭素数1〜20のアルキル基であり、Arが、置換もしくは無置換の環形成原子数5〜14のヘテロアリール基である、請求項1に記載の光重合開始剤。
【請求項4】
分子量が515以下である、請求項2又は3に記載の光重合開始剤。
【請求項5】
前記一般式(I−a)中、R1及びR2が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、R3が置換もしくは無置換の炭素数3〜18のシクロアルキル基であり、nが2であり、複数のR4及びR5が、それぞれ独立に水素原子もしくは置換もしくは無置換の炭素数1〜20のアルキル基である、請求項1に記載の光重合開始剤。
【請求項6】
前記一般式(I−a)中、R1及びR2が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、R3が環形成原子数3〜6の環状エーテル基で置換された炭素数1〜20のアルキル基であり、nが2であり、複数のR4及びR5が、それぞれ独立に水素原子もしくは置換もしくは無置換の炭素数1〜20のアルキル基である、請求項1に記載の光重合開始剤。
【請求項7】
分子量が550以下である、請求項5又は6に記載の光重合開始剤。
【請求項8】
バインダー樹脂及び/又はエチレン性不飽和結合を有する化合物と、請求項1〜7のいずれかに記載の光重合開始剤を含有する光硬化性組成物。
【請求項9】
光重合開始剤の含有量が、光硬化性組成物の固形分に対して2〜50質量%である、請求項8に記載の光硬化性組成物。
【請求項10】
さらに色材を含有する、請求項8又は9に記載の光硬化性組成物。
【請求項11】
さらに、エーテル結合及び/又はエステル結合を有する25℃で液体の化合物を光重合開始剤1質量部に対して5〜100質量部含有する、請求項8〜10のいずれかに記載の光硬化性組成物。
【請求項12】
エチレン性不飽和結合を有する化合物として、エーテル結合及び/又はエステル結合を持つエチレン性不飽和結合を有する化合物を少なくとも1種用い、溶剤を光硬化性組成物全量の10質量%以下のみ含有する、請求項8〜10のいずれかに記載の光硬化性組成物。
【請求項13】
カラーフィルタ用である、請求項10〜12のいずれかに記載の光硬化性組成物。
【請求項14】
請求項10〜12のいずれかに記載の光硬化性組成物を用いたカラーフィルタ。
【請求項15】
請求項14に記載のカラーフィルタと対向基板と、前記カラーフィルタと前記対向基板との間に形成された液晶層とを有する液晶表示装置。
【請求項16】
請求項10〜12のいずれかに記載の光硬化性組成物を基板に塗布し、乾燥した後、フォトマスクを用いて露光し、次いで現像を行うことによる、パターン形成方法。
【請求項17】
下記一般式(1)
【化2】
(式中、R6〜R11は、それぞれ独立に、水素原子、ハロゲン原子、置換もしくは無置換の炭素数1〜20のアルキル基、置換もしくは無置換の炭素数2〜20のアルケニル基、置換もしくは無置換の環形成原子数3〜10のシクロアルキル基、置換もしくは無置換の炭素数4〜20のシクロアルケニル基、ヒドロキシル基、置換もしくは無置換の炭素数1〜20のアルコキシ基、置換もしくは無置換の炭素数2〜20のアルケニルオキシ基、置換もしくは無置換の炭素数1〜20のアルカノイル基、置換もしくは無置換の炭素数2〜20のアルケノイル基、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数3〜14のヘテロ環基を示す。)
で表されるカルバゾール誘導体と、下記一般式(2a)
【化3】
(式中、R3、R4’及びR5’は、それぞれ独立に、水素原子、ハロゲン原子、置換もしくは無置換の炭素数1〜20のアルキル基、置換もしくは無置換の炭素数2〜20のアルケニル基、置換もしくは無置換の環形成原子数3〜10のシクロアルキル基、置換もしくは無置換の炭素数4〜20のシクロアルケニル基、ヒドロキシル基、置換もしくは無置換の炭素数1〜20のアルコキシ基、置換もしくは無置換の炭素数2〜20のアルケニルオキシ基、置換もしくは無置換の炭素数1〜20のアルカノイル基、置換もしくは無置換の炭素数2〜20のアルケノイル基、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数3〜14のヘテロ環基を示す。R3は、R5'と一緒になって環を形成していてもよい。)
で表されるアクリレート誘導体とを塩基の存在下に反応させることにより、下記一般式(3a’)
【化4】
(式中、R3、R4’、R5’、R6〜R11は、前記定義の通りである。)
で表されるカルボニルアルキル基導入体を得、得られたカルボニルアルキル基導入体と下記一般式(4)、(5)
【化5】
(式中、Xは、ハロゲン原子又は−OC(=O)Arを示し、X’は、ハロゲン原子又は−OC(=O)R2を示す。
Arは、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数5〜14のヘテロアリール基を示す。
R2は、水素原子、ハロゲン原子、置換もしくは無置換の炭素数1〜20のアルキル基、置換もしくは無置換の炭素数2〜20のアルケニル基、置換もしくは無置換の環形成原子数3〜10のシクロアルキル基、置換もしくは無置換の炭素数4〜20のシクロアルケニル基、ヒドロキシル基、置換もしくは無置換の炭素数1〜20のアルコキシ基、置換もしくは無置換の炭素数2〜20のアルケニルオキシ基、置換もしくは無置換の炭素数1〜20のアルカノイル基、置換もしくは無置換の炭素数2〜20のアルケノイル基、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数3〜14のヘテロ環基を示す。)
で表される2つのアシル化剤をルイス酸の存在下に反応させることにより、下記一般式(6a’)
【化6】
(式中、R2、R3、R4’、R5’、R6〜R11及びArは、前記定義の通りである。)
で表されるジケトン体を得、得られたジケトン体とヒドロキシルアミンを反応させることにより、下記一般式(7a’)
【化7】
(式中、R2、R3、R4’、R5’、R6〜R11及びArは、前記定義の通りである。)
で表されるオキシム体を得、得られたオキシム体と下記一般式(8)
【化8】
(式中、Yは、ハロゲン原子又は−OC(=O)R1を示す。
また、R1は、水素原子、ハロゲン原子、置換もしくは無置換の炭素数1〜20のアルキル基、置換もしくは無置換の炭素数2〜20のアルケニル基、置換もしくは無置換の環形成原子数3〜10のシクロアルキル基、置換もしくは無置換の炭素数4〜20のシクロアルケニル基、ヒドロキシル基、置換もしくは無置換の炭素数1〜20のアルコキシ基、置換もしくは無置換の炭素数2〜20のアルケニルオキシ基、置換もしくは無置換の炭素数1〜20のアルカノイル基、置換もしくは無置換の炭素数2〜20のアルケノイル基、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数3〜14のヘテロ環基を示す。)
で表されるエステル化剤とを反応させることによる、下記一般式(I−a1)
【化9】
(式中、R1〜R3、R4’、R5’、R6〜R11及びArは、前記定義の通りである。)
で表される光重合開始剤の製造方法。
【請求項1】
下記一般式(I−a)で表される光重合開始剤。
【化1】
(式中、R1〜R11は、それぞれ独立に、水素原子、ハロゲン原子、置換もしくは無置換の炭素数1〜20のアルキル基、置換もしくは無置換の炭素数2〜20のアルケニル基、置換もしくは無置換の環形成原子数3〜10のシクロアルキル基、置換もしくは無置換の炭素数4〜20のシクロアルケニル基、ヒドロキシル基、置換もしくは無置換の炭素数1〜20のアルコキシ基、置換もしくは無置換の炭素数2〜20のアルケニルオキシ基、置換もしくは無置換の炭素数1〜20のアルカノイル基、置換もしくは無置換の炭素数2〜20のアルケノイル基、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数3〜14のヘテロ環基を示す。R3は、R4又はR5と一緒になって環を形成していてもよい。R4は、R5と一緒になって環を形成していてもよい。
また、Arは、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数5〜14のヘテロアリール基を示す。
nは、1〜10の整数を示す。nが2〜10の整数の場合、複数のR4及びR5は、それぞれ同一でも異なっていてもよい。)
【請求項2】
前記一般式(I−a)中、R1、R2及びR3が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、nが2であり、複数のR4及びR5が、それぞれ独立に、水素原子又は置換もしくは無置換の炭素数1〜20のアルキル基であり、Arが、置換もしくは無置換の環形成炭素数6〜14のアリール基である、請求項1に記載の光重合開始剤。
【請求項3】
前記一般式(I−a)中、R1、R2及びR3が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、nが2であり、複数のR4及びR5が、それぞれ独立に、水素原子又は置換もしくは無置換の炭素数1〜20のアルキル基であり、Arが、置換もしくは無置換の環形成原子数5〜14のヘテロアリール基である、請求項1に記載の光重合開始剤。
【請求項4】
分子量が515以下である、請求項2又は3に記載の光重合開始剤。
【請求項5】
前記一般式(I−a)中、R1及びR2が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、R3が置換もしくは無置換の炭素数3〜18のシクロアルキル基であり、nが2であり、複数のR4及びR5が、それぞれ独立に水素原子もしくは置換もしくは無置換の炭素数1〜20のアルキル基である、請求項1に記載の光重合開始剤。
【請求項6】
前記一般式(I−a)中、R1及びR2が、それぞれ独立に、置換もしくは無置換の炭素数1〜20のアルキル基であり、R3が環形成原子数3〜6の環状エーテル基で置換された炭素数1〜20のアルキル基であり、nが2であり、複数のR4及びR5が、それぞれ独立に水素原子もしくは置換もしくは無置換の炭素数1〜20のアルキル基である、請求項1に記載の光重合開始剤。
【請求項7】
分子量が550以下である、請求項5又は6に記載の光重合開始剤。
【請求項8】
バインダー樹脂及び/又はエチレン性不飽和結合を有する化合物と、請求項1〜7のいずれかに記載の光重合開始剤を含有する光硬化性組成物。
【請求項9】
光重合開始剤の含有量が、光硬化性組成物の固形分に対して2〜50質量%である、請求項8に記載の光硬化性組成物。
【請求項10】
さらに色材を含有する、請求項8又は9に記載の光硬化性組成物。
【請求項11】
さらに、エーテル結合及び/又はエステル結合を有する25℃で液体の化合物を光重合開始剤1質量部に対して5〜100質量部含有する、請求項8〜10のいずれかに記載の光硬化性組成物。
【請求項12】
エチレン性不飽和結合を有する化合物として、エーテル結合及び/又はエステル結合を持つエチレン性不飽和結合を有する化合物を少なくとも1種用い、溶剤を光硬化性組成物全量の10質量%以下のみ含有する、請求項8〜10のいずれかに記載の光硬化性組成物。
【請求項13】
カラーフィルタ用である、請求項10〜12のいずれかに記載の光硬化性組成物。
【請求項14】
請求項10〜12のいずれかに記載の光硬化性組成物を用いたカラーフィルタ。
【請求項15】
請求項14に記載のカラーフィルタと対向基板と、前記カラーフィルタと前記対向基板との間に形成された液晶層とを有する液晶表示装置。
【請求項16】
請求項10〜12のいずれかに記載の光硬化性組成物を基板に塗布し、乾燥した後、フォトマスクを用いて露光し、次いで現像を行うことによる、パターン形成方法。
【請求項17】
下記一般式(1)
【化2】
(式中、R6〜R11は、それぞれ独立に、水素原子、ハロゲン原子、置換もしくは無置換の炭素数1〜20のアルキル基、置換もしくは無置換の炭素数2〜20のアルケニル基、置換もしくは無置換の環形成原子数3〜10のシクロアルキル基、置換もしくは無置換の炭素数4〜20のシクロアルケニル基、ヒドロキシル基、置換もしくは無置換の炭素数1〜20のアルコキシ基、置換もしくは無置換の炭素数2〜20のアルケニルオキシ基、置換もしくは無置換の炭素数1〜20のアルカノイル基、置換もしくは無置換の炭素数2〜20のアルケノイル基、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数3〜14のヘテロ環基を示す。)
で表されるカルバゾール誘導体と、下記一般式(2a)
【化3】
(式中、R3、R4’及びR5’は、それぞれ独立に、水素原子、ハロゲン原子、置換もしくは無置換の炭素数1〜20のアルキル基、置換もしくは無置換の炭素数2〜20のアルケニル基、置換もしくは無置換の環形成原子数3〜10のシクロアルキル基、置換もしくは無置換の炭素数4〜20のシクロアルケニル基、ヒドロキシル基、置換もしくは無置換の炭素数1〜20のアルコキシ基、置換もしくは無置換の炭素数2〜20のアルケニルオキシ基、置換もしくは無置換の炭素数1〜20のアルカノイル基、置換もしくは無置換の炭素数2〜20のアルケノイル基、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数3〜14のヘテロ環基を示す。R3は、R5'と一緒になって環を形成していてもよい。)
で表されるアクリレート誘導体とを塩基の存在下に反応させることにより、下記一般式(3a’)
【化4】
(式中、R3、R4’、R5’、R6〜R11は、前記定義の通りである。)
で表されるカルボニルアルキル基導入体を得、得られたカルボニルアルキル基導入体と下記一般式(4)、(5)
【化5】
(式中、Xは、ハロゲン原子又は−OC(=O)Arを示し、X’は、ハロゲン原子又は−OC(=O)R2を示す。
Arは、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数5〜14のヘテロアリール基を示す。
R2は、水素原子、ハロゲン原子、置換もしくは無置換の炭素数1〜20のアルキル基、置換もしくは無置換の炭素数2〜20のアルケニル基、置換もしくは無置換の環形成原子数3〜10のシクロアルキル基、置換もしくは無置換の炭素数4〜20のシクロアルケニル基、ヒドロキシル基、置換もしくは無置換の炭素数1〜20のアルコキシ基、置換もしくは無置換の炭素数2〜20のアルケニルオキシ基、置換もしくは無置換の炭素数1〜20のアルカノイル基、置換もしくは無置換の炭素数2〜20のアルケノイル基、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数3〜14のヘテロ環基を示す。)
で表される2つのアシル化剤をルイス酸の存在下に反応させることにより、下記一般式(6a’)
【化6】
(式中、R2、R3、R4’、R5’、R6〜R11及びArは、前記定義の通りである。)
で表されるジケトン体を得、得られたジケトン体とヒドロキシルアミンを反応させることにより、下記一般式(7a’)
【化7】
(式中、R2、R3、R4’、R5’、R6〜R11及びArは、前記定義の通りである。)
で表されるオキシム体を得、得られたオキシム体と下記一般式(8)
【化8】
(式中、Yは、ハロゲン原子又は−OC(=O)R1を示す。
また、R1は、水素原子、ハロゲン原子、置換もしくは無置換の炭素数1〜20のアルキル基、置換もしくは無置換の炭素数2〜20のアルケニル基、置換もしくは無置換の環形成原子数3〜10のシクロアルキル基、置換もしくは無置換の炭素数4〜20のシクロアルケニル基、ヒドロキシル基、置換もしくは無置換の炭素数1〜20のアルコキシ基、置換もしくは無置換の炭素数2〜20のアルケニルオキシ基、置換もしくは無置換の炭素数1〜20のアルカノイル基、置換もしくは無置換の炭素数2〜20のアルケノイル基、置換もしくは無置換の環形成炭素数6〜14のアリール基又は置換もしくは無置換の環形成原子数3〜14のヘテロ環基を示す。)
で表されるエステル化剤とを反応させることによる、下記一般式(I−a1)
【化9】
(式中、R1〜R3、R4’、R5’、R6〜R11及びArは、前記定義の通りである。)
で表される光重合開始剤の製造方法。
【図1】

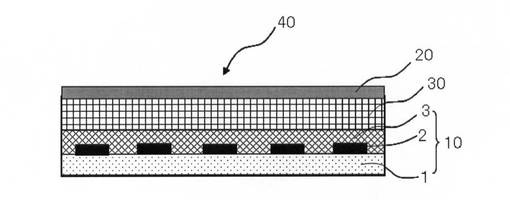
【公開番号】特開2012−251128(P2012−251128A)
【公開日】平成24年12月20日(2012.12.20)
【国際特許分類】
【出願番号】特願2011−272507(P2011−272507)
【出願日】平成23年12月13日(2011.12.13)
【分割の表示】特願2012−501867(P2012−501867)の分割
【原出願日】平成23年2月24日(2011.2.24)
【出願人】(000002897)大日本印刷株式会社 (14,506)
【Fターム(参考)】
【公開日】平成24年12月20日(2012.12.20)
【国際特許分類】
【出願日】平成23年12月13日(2011.12.13)
【分割の表示】特願2012−501867(P2012−501867)の分割
【原出願日】平成23年2月24日(2011.2.24)
【出願人】(000002897)大日本印刷株式会社 (14,506)
【Fターム(参考)】
[ Back to top ]