
免疫応答のアジュバントとしてのケモカイン
【課題】樹状細胞は、抗原特異的免疫応答において重要な役割を果たす。抗原提示樹状細胞の移動または活性化を促進または阻害することにより、疾患状態(癌および自己免疫疾患を含む)を処置するための材料および方法を提供すること。
【解決手段】本発明は、樹状細胞は、抗原特異的免疫応答において重要な役割を果たす、抗原提示樹状細胞の移動または活性化を促進または阻害することにより、疾患状態(癌および自己免疫疾患を含む)を処置するための材料および方法を提供する。特に、ケモカインは、免疫応答を開始、増幅または調節するために使用される。1つの実施形態において、ケモカインは、樹状細胞を抗原送達部位に誘引するために使用される。抗原送達部位における樹状細胞の数の増加は、より多くの抗原取り込みおよび改変された免疫応答を意味する。
【解決手段】本発明は、樹状細胞は、抗原特異的免疫応答において重要な役割を果たす、抗原提示樹状細胞の移動または活性化を促進または阻害することにより、疾患状態(癌および自己免疫疾患を含む)を処置するための材料および方法を提供する。特に、ケモカインは、免疫応答を開始、増幅または調節するために使用される。1つの実施形態において、ケモカインは、樹状細胞を抗原送達部位に誘引するために使用される。抗原送達部位における樹状細胞の数の増加は、より多くの抗原取り込みおよび改変された免疫応答を意味する。
【発明の詳細な説明】
【技術分野】
【0001】
本出願は、欧州特許出願第0 974 357 A1(1998年7月16日に出願および2000年1月26日公開)の利益を主張する。
【0002】
(発明の分野)
本発明は、癌を含む疾患状態の処置におけるヒトケモカインの使用に関連する。投与されたケモカインは、すべての抗原提示の樹状細胞または樹状細胞の特定のサブセットのいずれかの移動を方向付ける。1つの実施形態において、樹状細胞を活性化するように設計された疾患特異的抗原および/または部分は、ケモカインとともに投与される。
【背景技術】
【0003】
(発明の背景)
樹状細胞(DC)は、抗原の取り込みおよびT細胞へのそれらの提示に従事している。従って、DCは、抗原特異的免疫応答において重要な役割を果たす。
【0004】
DCは、種々のリンパ組織および非リンパ組織において身体の至る所に広範囲に分布した形態学的に類似な細胞型の多様な集団により表わされる(Cauxら、1995、Immunology Today 16:2;Steinman、1991、Ann.Rev.Immunol.9:271−296)。これらの細胞には、脾臓のリンパDC、およびリンパ節、表皮のランゲルハンス細胞、ならびに血液循環内の明瞭でない細胞が挙げられる。DCは、それらの形態学、表面のMHCクラスII発現の高レベルならびにT細胞の結合および同時刺激を媒介するいくつかのアクセサリー分子(B7−1[CD80]およびB7−2[CD86])(Inabaら、1990、Intern.Rev.Immunol.6:197−206;Frendenthalら、1990、Proc.Natl.Acad.Sci.USA 87:7698)、ならびにT細胞、B細胞、単球、およびナチュラルキラー細胞上で発現された特定の他の表面マーカーの非存在に基づいたグループとして集団的に分類される。
【0005】
DCは、骨髄由来であり、血流から組織の至るところまで前駆体として移動し、ここでDCはこの表皮におけるランゲルハンス細胞のような常在の細胞となる。
【0006】
末梢において、病原体侵入後、新鮮なランゲルハンス細胞のような未成熟DCは、抗原を捕獲しプロセシングする炎症部位(Kaplanら、1992、J.Exp.Med.175:1717−1728;McWilliamら、1994、J.Exp.Med.179:1331−1336)に補充される(Inabaら、1986.J.Exp.Med.164:605−613;Streileinら、1989、J.Immunol.143:3925−3933;Romaniら、1989、J.Exp.Med.169:1169−1178;Pureら、1990.J.Exp.Med.172:1459−1469;Schulerら、1985、J.Exp.Med.161:526−546)。
【0007】
次いで、抗原が負荷されたDCは、リンパ管を経由して末梢組織から、リンパ節のT細胞が豊富な領域まで移動し、ここで成熟DCは、相互連結細胞(IDC)と呼ばれる(Austynら、1988、J.Exp.Med.167:646−651;Kupiec−Weglinskiら、1988、J.Exp.Med.167:632−645;Larsenら、1990、J.Exp.Med.172:1483−1494;Fossum、S.1988、Scand.J.Immunol.27:97−105;Macatoniaら、1987、J.Exp.Med.166:1654−1667;Kripkeら、1990.、J.Immunol.145:2833−2838)。この部位で、これらは、未処置のT細胞に対してプロセシングされた抗原を提示し、そして抗原特異的な初期のT細胞応答を発生する(Liuら、1993、J.Exp.Med.177:1299−1307;Sornasseら、1992、J.Exp.Med.175:15−21;Heuflerら、1988、J.Exp.Med.167:700−705)。
【0008】
末梢組織からリンパ器官までのこれらの移動の間、DCは、表現型および機能において劇的な変化を含む成熟過程を経る(Larsenら、1990、J.Exp.Med.172:1483−1494;Streileinら、1990、Immunol.Rev.117:159−184;De Smedtら、1996、J.Exp.Med.184:1413−1424)。特に、可溶性タンパク質を効率的に捕獲およびプロセシングし特異的な記憶およびエフェクターT細胞を活性化するのに有効な、新鮮なランゲルハンス細胞のような未成熟のDCと対照的に、リンパ器官のIDCのような成熟DCは、未処置のT細胞の初回刺激において顕著に効果的であるが抗原の捕獲およびプロセシングにおいては乏しい(Inabaら、1986.J.Exp.Med.164:605−613;Streileinら、1989、J.Immunol.143:3925−3933;Romaniら、1989、J.Exp.Med.169:1169−1178;Pureら、1990、J.Exp.Med.172:1459−1469;Sallustoら、1995、J.Exp.Med.182:389−400;Cellaら、1997、Current Opin.Immunol.9:10−16)。
【0009】
DCの移動(traffic)パターンを制御するシグナルは、複雑であり、十分には理解されていない。
【0010】
TNFαおよびLPSにより提供されたシグナルが、組織から排出リンパ器官まで常在のDCの移動をインビボにおいて誘導することは公知である(De Smedtら、1996、J.Exp.Med.184:1413−1424;MacPhersonら、1995、J.Immunol.154:1317−1322;Roakeら、1995、J.Exp.Med.181:2237−2247;Cumberbatchら、1992、Immunology.75:257−263;Cumberbatchら、1995、Immunology.84:31−35)。
【0011】
ケモカインは、白血球の移動および活性化を調節する低分子量タンパク質である(Oppenheim、1993、Adv.Exp.Med.Biol.351:183−186;Schallら、1994、Curr.Opin.Immunol.6:865−873;Rollins、1997、Blood 90:909−928;Baggioliniら、1994、Adv.Imunol.55:97−179)。これらは、活性化された白血球自身、ならびに炎症刺激に際しての内皮細胞および上皮細胞を含む間質細胞により分泌される(Oppenheim、1993、Adv.Exp.Med.Biol.351:183−186;Schallら、1994、Curr.Opin.Immunol.6:865−873;Rollins、1997、Blood 90:909−928;Baggioliniら、1994、Adv.Immunol.55:97−179)。ケモカインに対する応答は、7つの膜貫通Gタンパク質共役レセプターにより媒介される(Rollins、1997、Blood 90:909−928;Premackら、1996、Nat.Med.2:1174−1178;Murphy、P.M.1994、Ann.Rev.Immunol.12:593−633)。いくつかのケモカイン(例えば、単球走化性タンパク質(MCP)−3、MCP−4、マクロファージ炎症タンパク質(MIP)−1α、MIP−1β、RANTES(発現および分泌された正常なT細胞の活性化に際する制御)、SDF−1、Teck(胸腺発現ケモカイン)およびMDC(マクロファージ由来ケモカイン))は、インビトロにおいてDCを誘引することが報告されている(Sozzaniら、1995、J.Immunol.155:3292−3295;Sozzaniら、1997、J.Immunol.159:1993−2000;Xuら、1996、J.Leukoc.Biol.60:365−371;MacPhersonら、1995、J.Immunol.154:1317−1322;Roakeら、1995、J.Exp.Med.181:2237−2247)。
【0012】
最近では、研究者らは、癌の処置においてDCの活性化を利用することを試みている。動物のモデルにおいて、わずか2×105の抗原パルスDCは、未処置のマウスに注入した場合、免疫を誘導する(Inabaら、1990、Intern.Rev.Immunol.6:197−206)。Flamandら(Eur.J.Immunol、1994、24:605−610)は、B細胞リンパ腫からのイディオタイプ抗原を用いてマウスDCをパルスし、未処置のマウスにそれらを注射した。この処置は、次の腫瘍攻撃からレシピエントマウスを効果的に保護し、そして免疫を持続する状態を確立した。抗原単独の注入、または抗原を用いてパルスされたB細胞は、効果がなく、このことは、抗腫瘍応答を担うDCの独特な特徴であることを示唆する。DCは抗腫瘍免疫を誘導することが可能なだけでなく、DCが、このプロセスの発生に絶対的に必須であると仮定されている(Ostrand−Rosenberg、1994、Current Opinion in Immunol.6:722−727;Grabbeら、1995、Immunol.Today 16:117−120;Huangら、1994、Science 264:961−965)。Huangおよび共同研究者ら(Huangら、1994、Science 264:961−965)は、抗腫瘍免疫を産生することが公知であるB7−1トランスフェクト腫瘍を用いてマウスに接種した。彼らは、MHC適合性APCを有するマウスだけが腫瘍チャレンジを拒絶することが可能であることを実証した。ヒトにおける研究では、DCについての類似の役割を実証している。ペプチド特異的CTLはペプチドパルスDCを使用して、精製CD8+T細胞から容易に誘導されることが報告されているが、ペプチドパルス単球を使用した場合は誘発されていない(Mehta−Damaniら、1994、J.Immunology153:996−1003)。
【0013】
樹状細胞の原発性腫瘍病変への組織学的浸潤が、膀胱癌、肺癌、食道癌、胃癌および鼻咽頭癌の患者における非常に長期化した患者の生存および転移性疾患の発生の減少と関連していることは、臨床的に非常に興味がある。対照的に、比較的乏しい臨床的予後は、DCを伴う散在性の浸潤を示す病変を有する患者で観察され、転移性の病変は、頻繁に、DC浸潤が欠けている(Becker,1993,In Vivo 7:187;Zeidら,1993,Pathology 25:338;Furihatonら,1992,61:409;Tsujitaniら,1990,Cancer 66:2012;Gianniら,1991,Pathol.Res.Pract.187:496;Murphyら,1993,J.Inv.Dermatol.100:3358)。最近、進行したB細胞リンパ腫の患者を、患者自身の腫瘍イディオタイプでパルスしたDCで処置した(Hsuら,1996,Nature Medicine 2(1):52)。このことにより、患者のB細胞リンパ腫において測定可能な減少が生じた。PSM抗原でパルスしたDCを用いた前立腺癌の処置は、Murphyらにより報告されている(The Prostate 1996 29:371)。
【0014】
インターロイキン4(IL−4)もしくは腫瘍壊死因子α(TNFα)のいずれかと組み合わせた顆粒球−マクロファージコロニー刺激因子(GM−CSF)に応答して、循環単球またはCD34造血性前駆体から多数のDCをインビトロ増殖させる技術が、最近出てきた(Sallustoら,1994,J.Exp.Med.179:1109−1118;Romaniら,1994,J.Exp.Med.180:83−93:Caux
ら,1992,Nature 360:258)。GM−CSFとIL−4の組み合わせは、末梢血単球を強力なDCへと分化させるように誘導する(KiertscherおよびRoth,1996,J.Leukocyte Biol.59:208−281)。これら2つのサイトカインの組み合わせを用いると、インビトロで末梢血から100倍のDCの収量の増加が達成される。
【0015】
マウスにおいて、種々の群により、腫瘍抗原を負荷したインビトロで生成されたDCは、腫瘍の発生を予防し、より重要なことには、確立された腫瘍の退行を誘導することが示された。黒色腫の患者を、MAGE−1腫瘍抗原由来のペプチドでパルスしたGM−CSF活性化APCを用いて処置する臨床試験を行った(Mehta−Damaniら,1994,J.Immunology 153:996−1003)。免疫する前は、2名の患者に由来する腫瘍浸潤リンパ球は、CD4+が優性であり、それぞれ、特異的腫瘍反応性を欠いていた。対照的に、同じ患者に由来する腫瘍浸潤リンパ球を免疫した後は、CD8+が優性であり、MAGE−1特異的抗腫瘍細胞傷害性が示された。従って、これらの研究から、DCが、免疫応答を刺激する独特かつ強い能力を有することが明らかである。
【発明の開示】
【発明が解決しようとする課題】
【0016】
従って、樹状細胞治療は、疾患(特に、癌)の処置に対する非常に確実な(promising)アプローチを示す。抗原提示樹状細胞を増殖かつ活性化させるだけでなく、治療的に有用かつ予防的に有用であるように、DCの移動を促進するために使用され得る、改善された材料および方法が継続して必要である。
【課題を解決するための手段】
【0017】
(発明の要旨)
本発明は、抗原提示樹状細胞の移動もしくは活性化を促進または阻害することにより疾患状態を処置するための材料および方法を提供することによって、前述の必要性を満たす。現在では、ケモカインは、有用な治療的薬剤であることが発見されている。本発明に従って処置され得る疾患状態としては、寄生生物感染、細菌感染、ウイルス感染、真菌感染、癌、自己免疫疾患、移植片拒絶、およびアレルギーが挙げられる。
【0018】
本発明は、疾患状態を処置する方法を提供し、この方法は、疾患状態の処置が必要な個体に、未成熟樹状細胞の抗原送達部位への移動を増大させるに十分な量のケモカインを投与する工程を包含する。本発明の1つの局面において、ケモカイン(例えば、MCP−1、MCP−2、MCP−3、MCP−4、MIP−1α、MIP−3α、RANTES、SDF−1、Teck、DC tactin−β、6Ckine、MDC、MIP−5またはこれらの組み合わせ)が投与される。本発明の好ましい方法において、疾患関連抗原(例えば、腫瘍関連抗原)は、ケモカインとともに投与される。
【0019】
本発明の別の局面は、疾患状態を処置する方法を提供し、この方法は、疾患状態を処置する必要のある個体に、未成熟樹状細胞の抗原送達部位への移動を減少させるに十分な量のケモカインを投与する工程を包含する。
【0020】
本発明のなお別の局面において、サイトカイン(特に、GM−CSFおよびIL−4)は、ケモカインと組み合わせて、ケモカインの前またはケモカインと同時のいずれかで投与される。GM−CSFおよびIL−4の投与は、前駆体からのDCの生成を刺激し、それにより、抗原を捕捉し、プロセシングするに利用可能なDCの数を増大させる。
【0021】
本発明のなお別の局面は、活性化薬剤(例えば、TNF−α、IFN−α、RANK−LまたはRANKのアゴニスト、およびtoll様レセプターファミリーの分子のアンタゴニスト)は、組織から、排出リンパを通ってリンパ器官へ向かうDCの移動を駆動する成熟シグナルを提供するために投与される。
【0022】
本発明はまた、哺乳動物における免疫応答を増強する方法を提供し、この方法は、ケモカインMCP−4またはMCP−4の生物学的に活性なフラグメントを哺乳動物に投与する工程を包含する。ヒトMCP−4(hMCP−4)は、ヒト血液の樹状細胞に対して活性であり、樹状細胞および樹状細胞前駆体を血液から補充する。好ましい局面において、このケモカインは、組換えケモカインである。最も好ましくは、このケモカインは、例えば、組換えケモカインと抗原の融合タンパク質の形態で、抗原とともに投与される。このような抗原は、腫瘍関連抗原、細菌抗原、ウイルス抗原または真菌抗原であり得る。
【0023】
さらに、本発明は、哺乳動物における免疫応答を増強する方法を提供し、この方法は、ケモカイン6Ckineまたは6Ckineの生物学的に活性なフラグメントを哺乳動物に投与する工程を包含する。ヒト6Ckineは、ヒト血液樹状細胞に対して活性であり、樹状細胞および樹状細胞前駆体を血液から補充する。樹状細胞の補充により、ケモカイン6Ckineは、抗腫瘍薬剤として働き、具体的には、腫瘍血管に対する新脈管形成効果を発揮することが示される。好ましい局面において、このケモカインは、組換えケモカインである。最も好ましくは、このケモカインは、例えば、組換えケモカインと抗原の融合タンパク質の形態で、抗原とともに投与される。このような抗原は、腫瘍関連抗原、細菌抗原、ウイルス抗原または真菌抗原であり得る。
【0024】
本発明のなお別の局面において、サイトカイン(特に、GM−CSFおよびIL−4)は、ケモカインと組み合わせて(ケモカインの前またはケモカインと同時のいずれか)投与される。
【0025】
最後の局面において、本発明は、MCP−4またはMCP−4の生物学的に活性な部分および抗原、ならびに6Ckineまたは6Ckineの生物学的に活性な部分および抗原を含む融合タンパク質を提供する。これらの融合タンパク質は、プラスミド、ウイルスベクターの形態で、または組換えベクターの形態で哺乳動物に投与され得る。
本発明は以下を提供する。
(1)疾患状態の処置のための医薬品の製造における、樹状細胞の移動を方向付け得るケモカインの使用。
(2)項目1に記載の使用であって、前記ケモカインが、MCP−1、MCP−2、MCP−3、MCP−4、MIP−1α、MIP−1β、MIP−3α、RANTES、SDF−1、Teck、DCtactin−β、6Ckine/SLC、LEC、MDC、およびMIP−5からなる群より選択される、使用。
(3)項目1に記載の使用であって、前記ケモカインが、樹状細胞の抗原送達部位への移動を方向付け得る、使用。
(4)項目1に記載の使用であって、前記ケモカインが、樹状細胞のリンパ器官への移動を方向付け得る、使用。
(5)項目1に記載の使用であって、前記疾患状態が、細菌感染、ウイルス感染、真菌感染、寄生生物感染または癌である、使用。
(6)項目1に記載の使用であって、前記疾患状態が、自己免疫疾患、組織拒絶、またはアレルギーである、使用。
(7)項目5に記載の使用であって、前記疾患状態が、黒色腫、乳癌、すい臓癌、結腸癌、肺癌、神経膠腫、肝細胞癌、子宮内膜癌、胃癌、腸癌、腎臓癌、前立腺癌、甲状腺癌、卵巣癌、精巣癌、肝臓癌、頭部および頚部の癌、結腸直腸癌、食道癌、胃癌、眼の癌、膀胱癌、神経膠芽細胞種、および転移性癌腫からなる群より選択される癌である、使用。
(8)項目3に記載の使用であって、前記樹状細胞が、未成熟樹状細胞である、使用。
(9)項目8に記載の使用であって、前記ケモカインが、MCP−1、MCP−2、MCP−3、MCP−4、MIP−1B、MDC、MIP−3α、MIP−1α、RANTESおよびMIP−5からなる群より選択される、使用。
(10)項目4に記載の使用であって、前記ケモカインが、MIP−3βである、使用。
(11)項目3に記載の使用であって、少なくとも1つの疾患関連抗原の使用をさらに包含する、使用。
(12)項目11に記載の使用であって、前記抗原が、腫瘍関連抗原である、使用。
(13)項目11に記載の使用であって、前記抗原が、細菌抗原、ウイルス抗原、または真菌抗原である、使用。
(14)項目12に記載の使用であって、前記腫瘍関連抗原が、Melan−A、チロシナーゼ、p97、β−HCG、GalNAc、MAGE−1、MAGE−2、MAGE−3、MAGE−4、MAGE−12、MART−1、MUC1、MUC2、MUC3、MUC4、MUC18、CEA、DDC、黒色腫抗原gp75、Hker8、高分子量黒色腫抗原、K19、Tyr1およびTyr2、pMel 17遺伝子ファミリーのメンバー、c−Met、PSA、PSM、α−フェトプロテイン、甲状腺ペルオキシダーゼ、gp100、NY−ESO−1、テロメラーゼならびにp53からなる群より選択される、使用。
(15)項目14に記載の使用であって、前記癌が前立腺癌であり、かつ前記腫瘍関連抗原が、PSAおよび/またはPSMである、使用。
(16)項目14に記載の使用であって、前記疾患状態が黒色腫であり、かつ前記腫瘍関連抗原がMelan−A、gp100またはチロシナーゼである、使用。
(17)項目1に記載の使用であって、活性化薬剤の使用をさらに包含する、使用。
(18)項目15に記載の使用であって、前記活性化薬剤が、TNFα、RP−105、抗CD40抗体および非メチル化CpGモチーフを含む核酸、またはtoll様レセプターのリガンドから選択される、使用。
(19)項目1に記載の使用であって、ケモカインとともに、GM−CSFおよびIL−4の組み合わせの使用をさらに包含する、使用。
(20)項目1に記載の使用であって、前記ケモカインが、皮内投与、筋肉内投与、皮下投与、局所投与、またはベクターの形態で投与される、使用。
(21)哺乳動物における免疫応答を増強する方法であって、該方法は、ケモカインMCP−4またはケモカインMCP−4の生物学的に活性な画分を該哺乳動物に投与する工程を包含する、方法。
(22)項目21に記載の方法であって、前記ケモカインが、組換えケモカインである、方法。
(23)項目21に記載の方法であって、前記ケモカインがヒトケモカインである、方法。
(24)項目21に記載の方法であって、該方法は、前記ケモカインの徐放を可能にする物質を送達部位に投与する工程をさらに包含する、方法。
(25)項目21に記載の方法であって、該方法は、前記ケモカインとともに抗原を投与する工程をさらに包含する、方法。
(26)項目25に記載の方法であって、MCP−4および抗原を含む融合タンパク質が、前記哺乳動物に投与される、方法。
(27)項目25に記載の方法であって、前記抗原が、腫瘍関連抗原である、方法。
(28)項目26に記載の方法であって、前記抗原が、腫瘍関連抗原である、方法。
(29)項目25に記載の方法であって、前記抗原が、細菌抗原、ウイルス抗原、または真菌抗原である、方法。
(30)項目26に記載の方法であって、前記抗原が、細菌抗原、ウイルス抗原または真菌抗原である、方法。
(31)項目25に記載の方法であって、前記腫瘍関連抗原が、Melan−A、チロシナーゼ、p97、β−HCG、GalNAc、MAGE−1、MAGE−2、MAGE−3、MAGE−4、MAGE−12、MART−1、MUC1、MUC2、MUC3、MUC4、MUC18、CEA、DDC、黒色腫抗原gp75、Hker8、高分子量黒色腫抗原
、K19、Tyr1およびTyr2、pMel 17遺伝子ファミリーのメンバー、c−Met、PSA、PSM、α−フェトプロテイン、甲状腺ペルオキシダーゼ、gp100、p53ならびにテロメラーゼからなる群より選択される、方法。
(32)項目26に記載の方法であって、前記腫瘍関連抗原が、Melan−A、チロシナーゼ、p97、β−HCG、GalNAc、MAGE−1、MAGE−2、MAGE−3、MAGE−4、MAGE−12、MART−1、MUC1、MUC2、MUC3、MUC4、MUC18、CEA、DDC、黒色腫抗原gp75、Hker8、高分子量黒色腫抗原、K19、Tyr1およびTyr2、pMel 17遺伝子ファミリーのメンバー、c−Met、PSA、PSM、α−フェトプロテイン、甲状腺ペルオキシダーゼ、gp100、p53ならびにテロメラーゼからなる群より選択される、方法。
(33)項目25に記載の方法であって、GM−CSFおよびIL−4の組み合わせを投与する工程をさらに包含する、方法。
(34)項目26に記載の方法であって、GM−CSFおよびIL−4の組み合わせを投与する工程をさらに包含する、方法。
(35)項目21に記載の方法であって、前記ケモカインとともに活性化薬剤を投与する工程をさらに包含する、方法。
(36)項目21に記載の方法であって、前記ケモカインが、皮内投与、筋肉内投与、皮下投与、局所投与、またはベクターの形態で投与される、方法。
(37)MCP−4および抗原を含む、融合タンパク質。
(38)項目37に記載の融合タンパク質であって、前記抗原が腫瘍関連抗原である、融合タンパク質。
(39)項目38に記載の融合タンパク質であって、前記腫瘍関連抗原が、Melan−A、チロシナーゼ、p97、β−HCG、GalNAc、MAGE−1、MAGE−2、MAGE−3、MAGE−4、MAGE−12、MART−1、MUC1、MUC2、MUC3、MUC4、MUC18、CEA、DDC、黒色腫抗原gp75、Hker8、高分子量黒色腫抗原、K19、Tyr1およびTyr2、pMel 17遺伝子ファミリーのメンバー、c−Met、PSA、PSM、α−フェトプロテイン、甲状腺ペルオキシダーゼ、gp100、p53ならびにテロメラーゼからなる群より選択される、融合タンパク質。
(40)項目38に記載の融合タンパク質であって、前記抗原が、細菌抗原、ウイルス抗原または真菌抗原である、融合タンパク質。
(41)項目37に記載の融合タンパク質を含む、プラスミド。
(42)項目39に記載のプラスミドであって、樹状細胞に特に適したプロモーター配列をさらに含む、プラスミド。
(43)項目37に記載の融合タンパク質を含む、ウイルスベクター。
(44)哺乳動物における免疫応答を増強する方法であって、該方法は、ケモカイン6Ckineまたはケモカイン6Ckineの生物学的に活性な画分を該哺乳動物に投与する工程を包含する、方法。
(45)項目44に記載の方法であって、前記ケモカインが、組換えケモカインである、方法。
(46)項目44に記載の方法であって、前記ケモカインがヒトケモカインである、方法。
(47)項目44に記載の方法であって、該方法は、前記ケモカインの徐放を可能にする物質を送達部位に投与する工程をさらに包含する、方法。
(48)項目44に記載の方法であって、該方法は、前記ケモカインとともに抗原を投与する工程をさらに包含する、方法。
(49)項目48に記載の方法であって、6Ckineおよび抗原を含む融合タンパク質が、前記哺乳動物に投与される、方法。
(50)項目48に記載の方法であって、前記抗原が、腫瘍関連抗原である、方法。
(51)項目49に記載の方法であって、前記抗原が、腫瘍関連抗原である、方法。
(52)項目48に記載の方法であって、前記抗原が、細菌抗原、ウイルス抗原、または真菌抗原である、方法。
(53)項目49に記載の方法であって、前記抗原が、細菌抗原、ウイルス抗原、または真菌抗原である、方法。
(54)項目48に記載の方法であって、前記腫瘍関連抗原が、Melan−A、チロシナーゼ、p97、β−HCG、GalNAc、MAGE−1、MAGE−2、MAGE−3、MAGE−4、MAGE−12、MART−1、MUC1、MUC2、MUC3、MUC4、MUC18、CEA、DDC、黒色腫抗原gp75、Hker8、高分子量黒色腫抗原、K19、Tyr1およびTyr2、pMel 17遺伝子ファミリーのメンバー、c−Met、PSA、PSM、α−フェトプロテイン、甲状腺ペルオキシダーゼ、gp100、p53ならびにテロメラーゼならびにC26結腸癌からなる群より選択される、方法。
(55)項目49に記載の方法であって、前記腫瘍関連抗原が、Melan−A、チロシナーゼ、p97、β−HCG、GalNAc、MAGE−1、MAGE−2、MAGE−3、MAGE−4、MAGE−12、MART−1、MUC1、MUC2、MUC3、MUC4、MUC18、CEA、DDC、黒色腫抗原gp75、Hker8、高分子量黒色腫抗原、K19、Tyr1およびTyr2、pMel 17遺伝子ファミリーのメンバー、c−Met、PSA、PSM、α−フェトプロテイン、甲状腺ペルオキシダーゼ、gp100、p53ならびにテロメラーゼならびにC26結腸癌からなる群より選択される、方法。
(56)項目48に記載の方法であって、GM−CSFおよびIL−4の組み合わせを投与する工程をさらに包含する、方法。
(57)項目49に記載の方法であって、GM−CSFおよびIL−4の組み合わせを投与する工程をさらに包含する、方法。
(58)項目44に記載の方法であって、前記ケモカインとともに活性化薬剤を投与する工程をさらに包含する、方法。
(59)項目44に記載の方法であって、前記ケモカインが、皮内投与、筋肉内投与、皮下投与
、局所投与、またはベクターの形態で投与される、方法。
(60)6Ckineおよび抗原を含む、融合タンパク質。
(61)項目60に記載の融合タンパク質であって、前記抗原が腫瘍関連抗原である、融合タンパク質。
(62)項目61に記載の融合タンパク質であって、前記腫瘍関連抗原が、Melan−A、チロシナーゼ、p97、β−HCG、GalNAc、MAGE−1、MAGE−2、MAGE−3、MAGE−4、MAGE−12、MART−1、MUC1、MUC2、MUC3、MUC4、MUC18、CEA、DDC、黒色腫抗原gp75、Hker8、高分子量黒色腫抗原、K19、Tyr1およびTyr2、pMel 17遺伝子ファミリーのメンバー、c−Met、PSA、PSM、α−フェトプロテイン、甲状腺ペルオキシダーゼ、gp100、p53ならびにテロメラーゼからなる群より選択される、融合タンパク質。
(63)項目61に記載の融合タンパク質であって、前記抗原が、細菌抗原、ウイルス抗原または真菌抗原である、融合タンパク質。
(64)項目60に記載の融合タンパク質を含む、プラスミド。
(65)項目64に記載のプラスミドであって、樹状細胞に特に適したプロモーター配列をさらに含む、プラスミド。
(66)項目60に記載の融合タンパク質を含む、ウイルスベクター。
【0026】
(発明の詳細な説明)
本明細書中で引用されるすべての参考文献は、その全体が、参考として援用される。
【0027】
インビボでのDC移動を誘導するシグナルと、それらのケモカインに対する応答との間の関係は、これまで公知ではなかった。本発明者らは、DCにより発現されるケモカインレセプターのパターンは、それらの成熟段階に従って変化し、ケモカインがDCサブセットの移動を駆動するために使用され得、それにより、免疫応答の開始を制御し得ることを発見した。ケモカインは、本発明に従って、未成熟DCサブセットを抗原送達部位に選択的に誘引するアジュバントとして使用され得る。自己免疫疾患、組織拒絶またはアレルギーの状況において、本発明は、例えば、CCR6アゴニストおよびアンタゴニスト、CCR7アゴニストおよびアンタゴニスト、ならびにCCR2アゴニストおよびアンタゴニストの発生を介して、DCの移動を妨害することにより、DC機能をブロックする方法を提供する。
【0028】
免疫系に抗原を提示するDCのサブセットに依存して、応答は劇的に変化する。DCは、寛容を誘導し得る。胸腺の髄質において見出されたDCは、自己選択的胸腺細胞を発生させるネガティブ選択において役割を果たし得る(Brockerら,1997,J.Exp.Med.185(3):541−550)。DCはまた、自己反応性末梢T細胞に寛容させる(Kurtsら,1997,J.Exp.Med.186(2):239−245;Adlerら,1998,J.Exp.Med.187(10):1555−1564)。マウスDCの特定のサブセット(おそらく、リンパ起源の)は、免疫寛容を誘導することが提唱された(Ardavin,1993,Nature 362(6422):761−763)。さらに、リンパDCに対する候補ヒト対応物(DC−2)(Grouardら,1997,J.Exp.Med.185(6):1101−1111)がIL−12を分泌できないという最近の説明は、この亜集団による提示の後に、免疫応答がTH−2型に向かって偏向され得ることを示唆する。
【0029】
目的が免疫応答を減少させることである場合、DCに寛容させること(自己免疫、アレルギー)が増大されるか、または応答の質がDC−2を特異的に増大させることにより改変される(TH2より大きなTH1、すなわち、アレルギーにおいて)。
【0030】
本発明において使用するためのケモカインは、DCの制限されたサブセット、特に、未成熟DCに対して活性な、身体の天然のタンパク質である。これらのケモカインのいくつか(MIP−3α、Teck、MDCおよびMCP−4、ならびに6Ckineが挙げられるが、これらに限定されない)が、本発明者らにより同定された。
【0031】
本発明を実施する際に使用されるケモカインは、天然産物に対して同一のアミノ酸配列を有する組換えタンパク質、または天然産物に由来し得るが、その本来の走化性特性を保持しつつその薬物動態特性を変化させる改変を含む組換えタンパク質であり得る。ケモカインの送達の様式は、注射(皮内、筋肉内および皮下が挙げられる)または局所的(例えば、軟膏またはパッチ)によってであり得る。
【0032】
ケモカインはまた、ベクター(例えば、ウイルスベクター)(例えば、アデノウイルス、ポックスウイルス、レトロウイルス、レンチウイルス)、または操作されたプラスミドDNAによって核酸配列として送達され得る。
【0033】
本明細書中で使用される用語「ケモカイン」は、走化性因子を含み得る。走化性因子は、低分子化合物(これは、未成熟DCにより発現されるケモカインレセプターの選択的アゴニストである)であり得る。CCR6(ケモカインMIP−3αの天然のレセプター)は、このようなレセプターの例である。
【0034】
本発明の特に好ましい実施形態において、ケモカインは、疾患関連抗原とともに投与される。この抗原は、任意の分子部分であり得、これに対して、免疫応答の増大または低下が調べられる。これは、ヒトまたは動物において疾患を引き起こすことが公知の生物(例えば、細菌、ウイルス、寄生生物(例えば、Leishmania)、および真菌)に由来する抗原を含む。これはまた、腫瘍により発現される抗原(腫瘍関連抗原)および植物抗原(アレルゲン)を含む。
【0035】
本発明において使用するための腫瘍関連抗原としては、以下が挙げられるが、これらに限定されない:Melan−A、チロシナーゼ、p97、β−HCG、GalNAc、MAGE−1、MAGE−2、MAGE−3、MAGE−4、MAGE−12、MART−1、MUC1、MUC2、MUC3、MUC4、MUC18、CEA、DDC、黒色腫抗原gp75、HKer 8、高分子量黒色腫抗原、K19、Tyr1およびTyr2、pMel 17遺伝子ファミリーのメンバー、c−Met、PSA、PSM、αフェトプロテイン、甲状腺ペルオキシダーゼ、gp100、NY−ESO−1、テロメラーゼ、ならびにp53。この列挙は、本発明の実施において使用され得る抗原の型を網羅することを意図するのではなく、単に例示するに過ぎない。
【0036】
抗原の異なる組み合わせが使用され得、これは、異なる人種グループ、性別、地理的分布、および疾患の病期に従って最適な機能を示す。本発明の1つの実施形態において、少なくとも2つ以上の異なる抗原が、ケモカインの投与とともに投与される。
【0037】
抗原は、ケモカインと同じ部位および同じ時間に、または48時間を越えない遅延の後に送達または投与され得る。同時投与または組み合わせた投与は、本明細書中で使用される場合、ケモカインおよび抗原が、被験体に、一般的な処置スケジュールの過程の間に、(a)時間内に同時または(b)異なる時間のいずれかに投与される。後者の場合において、2つの化合物は、意図された効果を達成するために十分に近い時間内に投与される。抗原は、タンパク質、または1または数個のペプチドの形態、あるいは送達ベクターに含まれる核酸配列の形態にあり得る。
【0038】
原発性癌および転移制癌が、ともに、本発明に従って処置され得る。処置され得る癌の型としては、以下が挙げられるが、これらに限定されない:黒色腫、乳癌、膵臓癌、結腸癌、肺癌、神経膠腫、肝細胞癌、子宮内膜癌、胃癌、腸癌、腎臓癌、前立腺癌、甲状腺癌、卵巣癌、精巣癌、肝臓癌、頭部および頚部の癌、結腸直腸癌、食道癌、胃癌、眼の癌、膀胱癌、神経膠芽細胞種、および転移性癌。用語「癌(腫)」とは、呼吸器系癌、胃腸管系癌、尿生殖器系癌、前立腺癌、内分泌系癌、および黒色腫を含む、上皮組織または内分泌組織の悪性腫瘍をいう。転移性は、本明細書中でこの用語が使用される場合、原発性腫瘍(局所的リンパ節を含む)から離れた部位への腫瘍の拡がりとして規定される。
【0039】
DCの成熟を達成、誘導または刺激するように設計された部分は、有利に投与され得る。このような薬剤は、組織からリンパ節への移動を促進する成熟シグナルを提供する。この部分は、例えば、TNF−αもしくはRP−105のような身体の天然産物、またはDC上の特異的構造を認識するアゴニスト抗体(例えば、抗CD−40抗体)、または別の物質であり得る。活性化物質は、非メチル化CpGモチーフ、またはDCを刺激することが公知のtoll様レセプターのアゴニストを含む核酸の配列であり得る。ケモカインおよび/または抗原がプラスミドベクターにより送達される本発明の実施形態において、これらの核酸配列は、ベクターの一部であり得る。
【0040】
GM−CSFおよびIL−4は、有利には、ケモカインおよび/または抗原と組み合わせて投与され得る。GM−CSFおよびIL−4の投与組み合わせは、前駆体からDCの生成を刺激する。GM−CSFおよびIL−4は、循環未成熟DCの数を増大させる目的で投与され得る。これは、ケモカインの引き続く注射により、局所的に補充され得る。このプロトコルは、GM−CSFおよびIL−4を使用して、少なくとも5〜7日間の全身的な前処置を含む。別の方法は、GM−CSFおよびIL−4の局所的投与により、同じ部位に送達される抗原を拾い上げ得る、DC前駆体(単球)の未成熟DCへの局所的分化に都合がよい。
【0041】
一般に、ケモカインおよび/または抗原および/または活性化薬剤および/またはサイトカインは、薬学的キャリア中に有効量のケモカインおよび/または抗原および/または活性化薬剤および/またはサイトカインを含む薬学的組成物として投与される。これらの試薬は、生理学的に刺激のない安定剤および賦形剤とともに、例えば、従来の薬学的に受容可能なキャリアまたは賦形剤(例えば、免疫原性アジュバント)中に、さらなる活性成分または不活性成分を用いた治療的用途のために組み合わせられ得る。薬学的キャリアは、患者に本発明の組成物を送達するために適切な任意の適合性の、非毒性物質であり得る。
【0042】
有効な治療に必要な試薬の量は、多くの異なる要因(投与の手段、標的部位、患者の身体的状態、および投与されている他の医薬品が挙げられる)に依存する。従って、処置投薬量は、安全性および有効性を最適化するために力価測定されるべきである。特定の癌を処置するための有効用量を試験する動物は、ヒト投薬量のさらなる予測指標を提供する。種々の考慮事項が、例えば、Gilmanら(編)(1990)Goodman and
Gilman’s:The Pharmacological Bases of Therapeutics,第8版,Pergamon Press;およびRemington’s Pharmaceutical Sciences,第17版(1990),Mack Publishing Co.,Easton,PAに記載される。投与のための方法は、例えば、静脈内投与、腹腔内投与、または筋肉内投与、経皮的拡散などについて、そこでおよび以下に議論される。薬学的に受容可能なキャリアとしては、水、生理食塩水、緩衝液、および例えば、the Merck Index,Merck&Co.,Rahway,New Jerseyに記載される他の化合物が挙げられる。徐放処方物、または徐放装置は、連続的な投与のために使用され得る。
【0043】
ケモカインおよび/または抗原および/または活性化薬剤についての投薬量範囲は、通常は、適切なキャリアとともに、1mM濃度未満、代表的には、約10μM濃度未満、通常は、約100nM未満、好ましくは、約10pM(ピコモル濃度)未満、および最も好ましくは、約1fM(フェムトモル濃度)未満の量にあると予測される。一般に、処置は、化合物の最適な用量未満のより少量の投薬量を用いて開始される。その後、投薬量は、その環境下での最適な効果が達成されるまで、少量ずつ増大される。特定の状況のための適切な用量および投薬レジメンの決定は、当該分野の技術範囲内である。
【0044】
本願発明の実施におけるGM−CSFおよびIL−4の好ましい生物学的に活性な用量は、循環CD14+/CD13+前駆体細胞の数;DC前駆体および成熟DCの表面上での抗原提示分子の発現;T細胞への抗原提示活性;および/または成熟DC機能と一致した抗原依存性T細胞応答の刺激の最大の増加を誘導する投薬組み合わせである。本発明の実施において、皮下投与するために使用されるIL−4の量は、代表的には、約0.05〜約8.0μg/kg/日、好ましくは、0.25〜6.0μg/kg/日、最も好ましくは、0.50〜4.0μg/kg/日の範囲にある。皮下投与に使用されるGM−CSFの量は、代表的には、約0.25μg/kg/日〜約10.0μg/kg/日、好ましくは、約1.0〜8.0μg/kg/日、最も好ましくは、2.5〜5.0μg/kg/日の範囲にある。特定の患者のための有効量は、上記のパラメーターの1以上における有意な変化を測定することにより確立される。
【0045】
ケモカインMCP−4またはMCP−4の生物学的に活性なフラグメントの投与が、用量依存性様式においてインビボでマウスの樹状細胞の補充を促進し、血液から単離されたヒト樹状細胞に対しても活性であることが見出された。生物学的に活性なフラグメントは、測定可能な免疫応答を刺激するに十分なMCP−4分子の部分を意味する。この応答は、血清中の免疫グロブリンレベルの抗原特異的刺激の増強(代表的には、B細胞応答として公知)として測定され得る。さらに、MCP−4の生物学的に活性なフラグメントは、免疫グロブリンの特定のクラス(例えば、T細胞における増大を要するIgG2a)の生成を刺激する。さらに、MCP−4の生物学的に活性なフラグメントは、抗原特異的抗腫瘍応答を増強する。増強した応答は、抗原を発現する腫瘍を使用したチャレンジの後のより遅延した腫瘍増殖またはより遅延した腫瘍指標により測定され得る。増強した免疫応答はまた、リンパ球の規定された集団(血液、脾臓、リンパ節、腫瘍)の抗原特異的細胞傷害性応答の分析により測定され得る。当然のことながら、CCR2アゴニスト(例えば、薬物発見スクリーニングにより見出される)である低分子はまた、抗原特異的抗腫瘍応答を増強することが認識される。根本的理由は、全てのMCP(1〜4)が天然のCCR2アゴニストであり、引き続き人工的な低分子アゴニストが同じ効果を有し得ることである。多くの現在の治療剤は、有機化学合成により得られる低分子である。
【0046】
好ましい実施形態は、組換えhMCP−4タンパク質単独または送達部位における徐放を可能にする物質(デポー);hMCP−4またはhMCP−4の画分および抗原からなる融合タンパク質(9アミノ酸より大きなペプチドまたはタンパク質); hMCP−4またはhMCP−4の画分(抗原を伴うかまたは伴わない)をコードするDNAまたはウイルスベクター(9アミノ酸より大きなペプチドまたはタンパク質)、あるいは送達ベクターに含まれる核酸配列と組み合わせられた組換えhMCP−4タンパク質からなるが、これらに限定されない。
【0047】
ヒトMCP−4(hMCP−4)は、ケモカインのCCファミリーに属する。その配列は、1996年に最初に公開された(Uguccioniら,1996,Monocyte Chemotactic Protein 4(MCP−4),A Novel Structural and Functional Analogue of MCP−3およびEotaxin,J.Exp.Med.183:2379−2394)。ヒトMCP−4は、75アミノ酸残基からなる8.6kDaのペプチドである(図3)。これはまた、CK−β−10、SCY−A13およびNCC−1(Swiss−Prot登録番号Q99616)として公知であり、新たなケモカイン命名においてCCL13と再度名づけられた(Zlotnikら,2000,Chemokines:A New Classification System and Their Role In Immunity,Immunity,12:121−127)。
【0048】
6Ckineは、ケモカインのCCファミリーに属する(Hedrickら,1997,J.Immunol.159:1589−1593)。CK−β−9、exodus−2およびSLC(ヒトタンパク質についてはSwiss−Prot登録番号000585
)として公知であり、CCL21と再度名づけた。ヒト6Ckine(h6Ckine)は、ケモカインCCR7に結合する一方、マウス6Ckine(m6Ckine)は、CCR7およびCXCR3レセプターに結合するが、親和性は低い(Jenhら,1999,J.Immunol.162:3765−3769)。マウス6Ckineは、マウスにおける腫瘍に注射された場合、抗腫瘍効果を有することが示されている(Sharmaら,2000,J.Immunol.164:4558−4563)。
【0049】
6Ckine様MIP−3βおよびMCP−4は、成熟DCの移動を誘導する。興味深いことに、6CkineならびにMIP−3βは、成熟の後に全てのヒトDC集団(CD1a+ランゲルハンス細胞、CD14+間質DC、単球由来DC、循環血液CD11c+DC、単球、および循環血液CD11c−プラズマ細胞様DCを含む)の移動を誘導し得る。6Ckineに対する応答は、いくつかのDCアクチベーター(CD40−L、TNF−α、およびLPSを含む)により誘導される成熟の後に観察される。MIP−3βの場合において見られるように、CCR7は、6Ckineを介してDC活性化の間に上方調節され、おそらく6Ckineに対する応答を説明する。
【0050】
従って、ケモカインh6Ckineは、癌処置において使用され得ることが提唱されている。好ましい実施形態は、以下からなるが、これらに限定されない:組換えh6Ckineタンパク質単独または送達部位におけるその徐放を可能にする物質(腫瘍部位におけるデポー)と組み合わせた組換えh6Ckineタンパク質;融合タンパク質またはh6Ckineもしくはh6Ckineの画分および腫瘍への構築物の送達を可能にする標的化部分(例えば、抗体または抗体のフラグメント、タンパク質リガンド、10アミノ酸よ
り大きなペプチド)からなる、化学的連結により作製された構築物;h6Ckineもし
くはh6Ckineの画分(上記の標的化部分を有するかまたは有さない)をコードするDNAもしくはウイルスベクター(例えば、アデノウイルス)。
【実施例】
【0051】
本発明は、以下の非限定的な実施例により例示され得る。以下の実施例は、以下の材料および方法を参照することによって容易に理解され得る。
【0052】
(造血因子、試薬および細胞株) 組換えGM−CSF(比活性:2.106U/mg
,Schering−Plough Research Institute,Kenilworth,NJ)を、飽和濃度の100ng/mlにて使用した。組換えヒトTNFα(比活性:2×107U/mg,Genzyme,Boston,MA)を、最適濃度の2.5ng/mにて使用した。組換えヒトSCF(比活性:4×105U/mg,R&D Abington,UK)を、25ng/mlの最適濃度にて使用した。組換えヒトIL−4(比活性:2.107U/mg,Schering−Plough Research Institute,Kenilworth,NJ)を、50U/mlの飽和濃度にて使用した。組換えヒトケモカインMIP−1α(比活性:2×105U/mg,9×1012U/M)、RANTES(比活性:1×104U/mg,8×1010U/M)、MIP−3α(比活性:4×105U/mg,3×1012U/M)およびMIP−3β(比活性:1×104U/mg,9×1010U/M)を、R&D(Abington,UK)から得た。LPSを、10ng/mlにて使用した(Sigma)。
マウスCD40リガンドトランスフェクト細胞株(CD40−L L 細胞)を、DC成熟の刺激因子として使用した。
【0053】
(臍帯血CD34+ HPCからのDCの生成) 臍帯血サンプルを、全期間の送達の後に得た。CD34+抗原を有する細胞を、記載のように(Cauxら,1996,J.Exp.Med.184:695−706;Cauxら,1990,Blood.75:2292−2298)、抗CD34+モノクローナル抗体(Immu−133.3,Immunotech Marseille,France)、ヤギ抗マウスIgG被覆マイクロビーズ(Miltenyi Biotec GmBH,Bergish Gladbach,Germany)およびMinimacs分離カラム(Miltenyi Biotec)を使用して、ポジティブ選択を介して単球画分から単離した。全ての実験において、単離した細胞は、80%〜99% CD34+であった。精製の後、CD34+細胞を、10% DMSO中で低温保存した。
【0054】
SCF、GM−CSFおよびTNFαの存在下で、記載のように(Cauxら,1996,J.Exp.Med.184:695−706)、エンドトキシンを含まない培地(10%(v/v)非働化ウシ胎仔血清(FBS)(Life Techniques,France,Irvine,UK)、10mM Hepes、2mM L−グルタミン、5×10−5M D−メルカプトエタノール、100μg/ml ゲンタマイシン(Schering−Plough,Levallois,France)を補充したRPMI
1640(Gibco,Grand Island,NY)からなる)(完全培地とも
いわれる)中で確立した。融解した後、CD34+細胞を、25〜75cm2培養容器(
Linbro,ICN Biomedicals,Acron,OH)中で2×104細胞/mlに増殖させるために播種した。最適条件を、これらの培養物を、5日目および10日目に新たなGM−CSFおよびTNFαを含む培地を使用して分割することにより維持した(細胞濃度:1〜3×105細胞/ml)。CD1a+DCは、12日目には、70〜90%の細胞の間である。
【0055】
(FACSソーティングによるCD86発現に従う未成熟DCおよび成熟DCの単離)
GM−CSFおよびTNFαの存在下での培養の12日後に、細胞を収集し、FITC結合体化OKT6(CD1a)(Ortho Diagnosis System,Raritan,NJ)およびPE結合体化IT2.2(CD86)(Pharmingen, San Diego,CA)で標識した。細胞を、CD1aおよびCD86発現に従って、未成熟CD1a+CD86−DC集団、および成熟CD1a+CD86+DC集団に、FACStarplus(登録商標)(レーザー設定:出力250mW、励起波長488nm、Becton−Dickinson,Sunnyvale,CA)を使用して分離した。染色およびソーティングの手順全てを、細胞凝集を避けるために、0.5mM
EDTAの存在下で行った。ソートした集団の再分析により、純度>98%であることが示された。
(末梢血単球からのDCの生成) 単球を、PBMCの調製、続いて52% Percoll勾配の後に、免疫磁性除去(Dynabeads,Dynal Oslo,Norway)により精製した。除去を、抗CD3モノクローナル抗体(OKT3)、抗CD19モノクローナル抗体(4G2)、抗CD8モノクローナル抗体(OKT8)、抗CD56モノクローナル抗体(NKH1,Coulter Corporation,Hialeah,FL)および抗CD16モノクローナル抗体(ION16,Immunotech)を使用して行った。単球由来樹状細胞を、GM−CSFおよびIL−4の存在下で精製単球を6〜7日間培養することにより生成した(Sallustoら,1994,J.Exp.Med.179:1109−1118)。
【0056】
(インビトロで生成したDCの成熟の誘導) CD34+HPCを、GM−CSF+TNFαの存在下で6日目まで培養し、GM−CSF単独の存在下で6日目〜12日目まで、それらの未成熟を保つために培養した。CD34+HPC由来の未成熟DCまたは単球由来DCを、TNFα(2.5ng/ml)またはLPS(10ng/ml)またはCD40LトランスフェクトL細胞(5DCについて1つのL細胞)の存在下で、3時間〜72時間の間、記載のように(Cauxら,1994,J.Exp.Med.180:1263−1272)、活性化した。
【0057】
(末梢血または扁桃からのCD11c+DCの精製) CD11c+DCを、末梢血または扁桃から以前に記載のように調製した(Grouardら,1996,Nature
384:364−367)。簡潔には、扁桃除去を受けた子供から得た扁桃を、細かく刻み、コラゲナーゼIVおよびDNase I(Sigma)を使用して消化した。収集した細胞を、SRBC(BioMerieux,Lyon,France)を含有するFicoll−Hypaqueを介して500rpmにて15分間、次いで、2000rpmにて30分間遠心分離した。末梢血単球(PBMC)を、Ficoll−Hypaqueにより単離した。CD3+T細胞(OKT3)、CD19+B細胞(4G7)、およびCD14+単球(MOP9)を、磁性ビーズ(抗マウスIg被覆Dynabeads,Dynal)により、得られた低密度細胞から取り出した。第2の除去を、抗NKH1、抗グリコホリンA(Immunotech)および抗CD20(1F54)を使用して行った。残りの細胞を、以下のmAbを用いて染色した:抗CD1a FITC(OKT6);抗CD14 FITC、抗CD57 FITC、抗CD16 FITC、抗CD7 FITC、抗CD20 FITC、抗CD3 FITC(Becton Dickinson,Mountain View,CA);抗CD4 PE−Cy5(Immunotech)ならびに抗CD11c PE(Becton Dickinson)。CD4+CD11c+系列−DCを、FACStarPlus(登録商標)を使用した細胞ソーティングにより単離した(レーザー設定:出力250mW、励起波長488nm)。除去、染色およびソーティングの手順全てを、0.5mM EDTAの存在下で行った。ソートした集団の再分析により、純度>97%であることが示された。
【0058】
(走化性アッセイ) 細胞移動を、走化性マイクロチャンバ技術(48ウェル Boyden microchamber,Neuroprobe,Pleasanton,CA)(Baconら,1988,Br.J.Pharmacol.95:966−974)を使用して評価した。簡潔には、ヒト組換えMIP−3αおよびMIP−3β、MIP−1αおよびRANTESを、RPMI 1640培地中に1ng/ml〜1000ng/mlの範囲の濃度に希釈し、走化性チャンバの下方のウェルに添加した。50μlのRPMI 1640培地中の105細胞/ウェル(またはCD11c+DCについては5×104細胞/ウェル)を、下方のウェルを分離する標準的な5μm孔のポリビニルピロリドンを含まないポリカーボネートフィルター(Neuroprobe)を備えるチャンバの上方のウェルにアプライした。このチャンバを、5% CO2の加湿空気中、37℃にて1時間インキュベートした。次いで、フィルターの下面に移動した細胞を、Field’s AおよびField’s B(BDH,Dorcet,England)を使用して染色し、2つのランダムに選択した低倍率視野(倍率×20)において、イメージアナライザー(ソフトウェア:Vision ExplorerおよびETC 3000,Graphtek,Mirmande,France)を使用して数えた。各アッセイを二連で行い、結果を、2視野あたりの移動細胞の平均±SDとしてあらわした。
【0059】
(総RNAの抽出およびcDNAの合成) 細胞を、上記のように調製し、総RNAを、製造業者により言及されたように、チオシアン酸グアニジウム法により抽出した(RNAgents total RNA isolation system,Promega)。DNAse I(RQ1 RNAse非含有DNAse,Promega)処理の後に、RNAを、分光光度計により定量し、質を、ホルムアルデヒド変性条件下での電気泳動により評価した。第1鎖cDNAを、RNAse非含有条件下で抽出した総RNAから合成した。この反応を、製造業者により記載されるように、5μgの総RNA、25ng/μlのオリゴdT12−18プライマー(Pharmacia,Orsay,France)およびSuperscriptキット(SuperScript II RNase H−Reverse Transcripase,Gibco BRL)を使用して行った。全てのサンプルに関して、cDNAの合成を制御し、β−アクチンプライマーを使用して21サイクルのRT−PCRにより較正した。
【0060】
(RT−PCR分析) 半定量的PCRを、最終容積100μlの反応混合物(その1×緩衝液とともに2.5U AmpliTaq酵素(5U/μl,Perkin Elmer,Paris,France)、0.2mMの各dNTP(Perkin Elmer,Paris,France)、5% DMSO、および1μMの各正方向および逆方向プライマーを含む)中でPerkin Elmer 9600サーマルサイクラーにおいて行った。CCR6(登録番号Z79784)およびCCR7(登録番号L08176)プライマーを、ケモカインレセプター間の最低の相同性の領域内に指定した。以下:
【0061】
【化1】

を、RT−PCRおよび配列決定に使用した。両方のケモカインレセプターのために、反応混合物を、以下の条件を使用する30サイクルおよび35サイクルのPCRに供した:94℃で1分、61.5℃で2分、および72℃で3分。PCR産物を、0.5μg/mlの臭化エチジウムを含む1.2%アガロースゲル上で可視化した。推定サイズ(CCR
6については1,021bpおよびCCR7については1,067bp)に移動した反応
産物をゲル精製し、色素ターミネーター技術を使用して、ABI 373A配列決定機(Applied Biosystems,Foster City,CA.)で配列決定確認のためにpCRII TAクローニングベクター(Invitrogen,Leek,The Netherlands)にサブクローニングした。2つの他のオリゴヌクレオチド:
【0062】
【化2】
を、1.2%アガロースゲル上で分離したPCR産物とのハイブリダイゼーションのため
のプローブとして使用し、Hybond N+膜(Amersham,Les Ulis,France)にブロットした。
【0063】
(カルシウム蛍光測定) 細胞内Ca2+濃度を、蛍光プローブIndo−1を使用して、Grynkiewiczら(J.Biol.Chem.,1985,260:3440−3450)により報告される技術に従って、測定した。簡潔には、細胞をPBS中で洗浄し、完全RPMI 1640培地中に107細胞/mlに再懸濁した(上記を参照のこと)。次いで、細胞を、遮光して、3μg/mlのIndo−1 AM(Molecular Probes)とともに室温にて45分間インキュベートした。インキュベーションの後、細胞を洗浄し、HBSS/1% FCS中で107細胞/mlに再懸濁した。細胞内Ca2+濃度の測定の前に、細胞を、予め39℃に温めたHBSS/10mM Hepes/1.6mM CaCl2中に10倍に希釈した。サンプルを、連続して攪拌しながら330nmで励起し、Indo−1蛍光を、810光電子倍増管検出システム(ソフトウェア:Felix,Photon Technology International,Monmouth Junction,NJ)にて、405nm(Ca2+と錯体化した色素)および485nm(Ca2+非含有培地)での時間の関数として測定した。結果を、2つの発光波長で得られた値の比率として表す。
【0064】
(インサイチュハイブリダイゼーション) インサイチュハイブリダイゼーションを、記載されたように行った(Peuchmaurら,1990,Am.J.Pathol.136:383−390)。2対のプライマーを、RT−PCRによりMIP−3α遺伝子(登録番号D86955)およびMIP−3β遺伝子(登録番号U77180)のオープンリーディングフレームの大部分を増幅するために使用した。
【0065】
【化3】
を、62℃のアニーリング温度で上記のように使用した。次いで、PCR産物を、適合したプロモーターとともにセンスプローブおよびアンチセンスプローブの生成のために、pCRII TAクローニングベクター(Invitrogen,Leek,The Netherlands)にクローニングした。MIP−3αおよびMIP−3βの35S標識したセンスプローブおよびアンチセンスプローブを、それぞれ、367bpフラグメントおよび435bpフラグメントの転写物を複製する(run off)ことにより得た。6μmのヒト扁桃の切片を、アセトンおよび4% パラホルムアルデヒド、続いて0.1M トリエタノールアミン/0.25%無水酢酸中で固定した。この切片を、一晩ハイ
ブリダイズし、RNAse Aで処理し、24日間曝した。展開させた後、切片を、ヘマトキシリンで染色した。
【0066】
(実施例1:CD34+由来DCの発生の間のMIP−3αおよびMIP−3βに対する差示的応答性)
DC移動(traffic)の調節を理解するために、成熟の異なる段階におけるDCの種々のケモカインに対する応答を研究した。DCを、GM−CSFおよびTNFαの存在下で培養したCD34+HPCから生成し、Boydenマイクロチャンバにおいて、ケモカインに応答して移動するそれらの能力について、異なる培養日数で試験した。MIP−3αおよびMIP−3βは、CD34+由来DCをMIP−1αまたはRANTESより2〜3倍に増加した。しかし、MIP−3αおよびMIP−3βにより誘引されたDCを、培養の異なる時点で回収した。MIP−3αに対する応答は、既に、4日目に検出され、5〜6日目に最大であり、10日目まで続いた。13〜14日目には、MIP−3αに対する応答は、通常、失われた。対照的に、MIP−3βに対応する応答は、10日目より前に検出され得ず、13日目にピークに達し得、15日目を超えて持続し得た。より早い時点では、培養中の細胞の大部分が、なおDC前駆体(CD1a−CD86−)であった場合、MIP−3αに対する応答は、1〜10ng/ml(実験に依存して)の濃度にて検出され得た。対照的に、4日後、ほとんど全ての細胞が未成熟DC(CD1a+CD86−)であった場合、300ng/ml以上が、細胞を誘引するために必要であり、このことは、成熟の間に細胞の連続的な脱感作を示唆する。比較的高濃度のMIP−3β(300ng/ml)もまた、成熟DC(CD1a+CD86+)を増大させるために必要であった。チェッカーボード分析により、MIP−3αおよびMIP−3βが走化性を誘導するが、DCのケモキネシスを誘導しないことが確立された。
【0067】
成熟の段階と、MIP−3αおよびMIP−3βに対する応答との間の関係を確認するために、CD34+由来DCを、CD86発現に従って、未成熟DC(CD1a+CD86−)および成熟DC(CD1a+CD86+)へと培養の10日目にFACSによりソートした。CD1a+CD86−は、MIP−3αに専ら応答したが、CD1a+CD86+は、主にMIP−3βに応答した。これらの観察により、MIP−3αおよびMIP−3βにより増加した細胞が、実際にDC(CD1a+)であったこともまた確認された。DC成熟とケモカイン応答性との間の相関は、DCの未成熟性が、6日目から12日目にTNFαを除去することにより維持され、それらの成熟性が、TNFα、LPSまたはCD40Lの添加により同期化された場合、さらに例示される。MIP−3αに対する応答は、TNFα、LPSおよびCD40Lを使用した48時間の成熟の際に強く減少された。同時に、MIP−3βに対する応答は、3つのシグナルすべてにより誘導され、CD40LおよびLPSは、TNFαより強力であった。動態学的実験において、MIP−3αに対する応答は、CD40活性化のわずか24時間後に50〜70%減少し、72時間目には、完全に失われた。MIP−3βに対する応答は、CD40活性化の24時間後に既に最大になり、比較的高濃度(48時間で100〜300ng/ml)のケモカインを要した。
【0068】
合わせて考えると、これらの結果により、未成熟CD34+由来DCがMIP−3αに応答し、一方で成熟DCがMIP−3βに応答することが確立される。
【0069】
(実施例2:MIP−3αおよびMIP−3βに対する応答は、CD34+由来DCに対するそれぞれのレセプターCCR6およびCCR7の発現と並行する)
MIP−3αおよびMIP−3βの応答性の調節の機構を明らかにするために、それらそれぞれのレセプターCCR6 mRNA(Powerら,1997,J.Exp.Med.186:825−835;Greavesら,1997,J.Exp.Med.186:837−844;Babaら,1997,J.Biol.Chem.272:14893−14898;Liaoら,1997,Biochem.Biophys.Res.Commun.236:212−217)およびCCR7 mRNA(Yoshidaら,199,J.Biol.Chem.272:13803−13809)の発現を、半定量的RT−PCRにより研究した。CD34+ HPCからのDC発生の間、CCR6 mRNAを、6日目に最初に検出し、10日目までに増大し、その後、減少したが、14日目には、わずかに検出可能であった。対照的に、CCR7 mRNAは、10日目に現れ、14日目まで確実に増大した。さらに、CD40L依存性成熟は、CCR6 mRNAの連続的な下方調節を誘導し(これは、72時間後にほとんど検出可能であった)、24時間程度の早さでCCR7 mRNAの上方調節を誘導した。同様な結果は、LPSまたはTNFαのいずれかによる誘導性DC成熟の後に得られた。活性化後のCCR7 mRNAの上方調節を、cDNAライブラリーのサザンブロット分析により確認した。
【0070】
移動アッセイならびにCCR6およびCCR7の発現の調節と調和して、MIP−3αは、休止/未成熟DCにおいて専らCa2+フラックスを誘導し、MIP−3βは、成熟
DCにおいてのみCa2+フラックスを誘導した。最大のCa2+フラックスは、30ng/mlのMIP−3αおよび30ng/mlのMIP−3βを用いて、それぞれ、未成熟DCおよび成熟DCに対して観察された。
【0071】
これらの結果は、MIP−3αおよびMIP−3βに対する応答性の変化が、CCR6
mRNAおよびCCR7 mRNAの発現の調節に関連することを示し、CCR6およびCCR7が、それぞれ、MIP−3αおよびMIP−3βについてのDC上で発現される主要な機能的レセプターであることを示唆する。
【0072】
(実施例3:MIP−3βに対する応答はまた、単球由来DCの成熟の際に誘導される)
GM−CSF+IL−4の存在下で単球を6日間培養することにより生成した単球由来DCは、代表的には、未成熟DCである(CD1a+、CD14−、CD80low、CD86low、CD83−)(Cellaら,1997,Current Opin.Immunol.9:10−16;Sallustoら,1994,J.Exp.Med.179:1109−1118)。それらは、MIP−1αおよびRANTESに応答して移動したが、MIP−3αに対しても、MIP−3βに対しても応答せず、移動しなかった。単球由来DCのMIP−3αに対する応答の欠如は、それらの細胞上でのCCR6発現の非存在に従う(Powerら,1997,J.Exp.Med.186:825−835;Greavesら,1997,J.Exp.Med.186:837−844)。TNFα、LPS、またはCD40Lにより誘導される成熟に際して、MIP−1αおよびRANTESに対する応答は失われたが、MIP−3βに対する応答が誘導された。CD34+由来DCを使用する場合と同様に、MIP−3βに対する応答は、TNFα、LPSまたはCD40Lにより誘導された成熟に際して観察されたCCR7 mRNA発現の上方調節と相関した。繰り返すと、TNFRまたはCD40シグナル伝達の後にCCR7の上方調節は、初期の時点(3h)で生じた。さらに、移動およびケモカインレセプター発現のデータは、Ca2+フラックス結果と一致した。
【0073】
これらの結果により、成熟の際に、DCが種々のケモカインに対するそれらの応答性を失うが、1つのケモカインMIP−3βには感受性になるという概念は、単球由来DCに及ぶ。
【0074】
(実施例4:末梢血CD11c+DCは、成熟後にMIP−3βに応答して移動する)末梢血(または扁桃)から単離された未成熟CD11c+DCに対するMIP−3αおよびMIP−3βの走化性活性もまた研究した。新たに単離したDCは、MIP−3αに応答して移動しなかったが、MIP−3βに応答して移動し、これらの細胞におけるCCR6の非存在およびCCR7 mRNA発現との相関が観察された。しかし、GM−CSFとの一晩の培養の後に生じることが公知の成熟は、MIP−3βに対するCD11c+DCの応答に向けられるが、MIP−3αに対する応答には向けられなかった。再び、MIP−3βに対する応答は、CCR7 mRNA発現の誘導と相関した。
【0075】
従って、たとえ血液から新たに単離された未成熟CD11c+DCが、MIP−3αに応答できないとしても、これらの結果は、MIP−3βに対する応答性に依存した成熟がまた、エキソビボで単離されたDCに適用されることを示す。
【0076】
(実施例5:インビボでMIP−3αは、炎症を生じた上皮において発現され、MIP−3βは、扁桃のT細胞富化領域内で発現される)
実施例4において報告された知見の物理的関連性を、MIP−3αの分析および炎症を生じた扁桃の切片でのインサイチュハイブリダイゼーションによるMIP−3β mRNA発現を通じて取り組んだ。MIP−3αのmRNAは、炎症を生じた上皮陰窩において高レベルで検出されたが、T細胞富化領域においても、B細胞濾胞においても検出されなかった。実際に、MIP−3α発現は、上皮陰窩を覆う細胞に制限されていた。対照的に、MIP−3β mRNAの発現は、T細胞富化領域に制限されていた。最も強いシグナルは、散在した細胞において存在し、分布は、IDCの分布と重なっていた。副皮質(paracortical)領域の外側では、シグナルは、B細胞濾胞においても、上皮陰窩においても検出されなかった。連続切片は、MIP−3αが豊富に発現される上皮陰窩内のMIP−3β発現の明らかな不在を示した。MIP−3αおよびMIP−3βのセンスプローブは、バックグラウンドハイブリダイゼーションを生成しなかった。
【0077】
従って、MIP−3α発現は、未成熟DCが補充されるべき抗原進入の部位において炎症を生じた上皮に制限される。対照的に、MIP−3βは、副皮質領域において検出されるのみであり、ここで成熟IDCは、ホーミングし、第1のT細胞応答を生成する。
【0078】
(実施例6:マウスモデルにおけるインビボでのケモカインMIP−3α投与)
MIP−3αは、本発明者らがインビトロでマウス未成熟樹状細胞についての走化性因子であることを示し、ケモカインMIP−3αがインビボで未成熟DCを誘引し、インビボで腫瘍に対する抗原特異的免疫応答を調節する能力を研究した。腫瘍関連抗原が同じ時間に送達される場合、より多くのDCが抗原を捕捉するために利用可能である。従って、この抗原に対する抗原特異的応答が増加されるはずである。
【0079】
ケモカインはプラスミドベクター(pcDNA3,InVitrogen)を介してインビボで送達され、このベクターは、CMVプロモーター(PMIP−3α)の制御下でマウスMIP−3αをコードするcDNAを含む。使用した抗原は、E.coliから単離したβ−ガラクトシダーゼであった。この抗原を、同じプラスミドベクターpcDNA3(pLaczといわれる)を介してインビボで送達した。腫瘍は、β-ガラクトシダー
ゼをコードする遺伝子で安定にトランスフェクトされた同系BALB/cマウスにおけるC26結腸癌であった。従って、この系において、β−ガラクトシダーゼが、腫瘍関連抗原を規定する。
【0080】
6匹の6週齡の雌性マウスの群を、空のpcDNA3プラスミド(ネガティブコントロール)、抗原単独をコードするプラスミドpLacz、またはpLaczおよびPMIP−3αの混合物のいずれかを注射した。注射(50μgの総プラスミド)を、4週間の間毎週、後足の踵に行った。その後、マウスにβ−ガラクトシダーゼを発現するC26腫瘍細胞株を皮下注射した。代表的には、全てのマウスは、10日後に、皮下に腫瘍を発生させた。これらのマウスの群における腫瘍の出現をモニターした。腫瘍の出現は、pLaczおよびpLacz+PMIP−αの注射の後に遅延した(図1)。このことは、腫瘍関連抗原をコードするプラスミドでの免疫が、腫瘍移植に対する保護効果を有することを示す。この遅延は、pLaczを用いるより、pLacz+PMIP−3αを用いるほうが大きかった。このことは、ケモカインMIP−3αが抗原をとともに送達された場合、腫瘍関連抗原特異的免疫応答を増大させることを示唆する。
【0081】
優れた抗腫瘍応答は、強いT細胞媒介性抗原特異的細胞傷害性(CTL活性)と関連すると考えられる。従って、同じ群のマウスにおけるCTL活性を、腫瘍接種して30日後に分析した。脾細胞を取り出し、インターロイキン−2の存在下で、照射した同系DCおよびβ−ガラクトシダーゼ由来の免疫優性なCTLペプチドを使用して5日間刺激した。次いで、β−ガラクトシダーゼをコードする遺伝子を安定にトランスフェクトした細胞株(P13.1)を溶解するそれらの能力を、β−ガラクトシダーゼを発現しない親細胞株P815を溶解するそれらの能力と並行して、測定した(図2)。これを、異なる比のエフェクター(脾細胞)対標的(P13.1またはP815)を使用して行った。この結果は、腫瘍チャレンジの前にpLacz+P−MIP−3αを注射したマウスは、pLaczまたはPCDNA3単独でのみ注射したマウスより、腫瘍関連抗原β−ガラクトシダーゼに対して大きなCTL活性を有する。
【0082】
(実施例7:インビボマウスモデルにおけるケモカインhMCP−4投与)
本発明者らは、hMCP−4局所的注射が、用量依存性様式でマウスにおいてインビボでの樹状細胞の補充を促進し得ることを示した(図4)。
【0083】
6〜10週齡の雌性BALB/cマウスを、Charles River(Iffa−Credo,L’Arbresle,France)から購入し、標準的な条件下で本発明者らの施設にて維持した。動物およびそれらの世話に関する手順は、EEC(欧州経済共同体)会議の指示(86/609,OJL 358,1,1987年12月12日)に合わせて行った。組換えヒトMCP−4タンパク質(>97%純度(図3))を、Peprotechから得、PBS(Gibco−BRL)中に再懸濁した。3匹のマウスの群
に、PBS単独またはPBS中の種々の量のヒトMCP−4を、50μ容量で右後足踵に皮内注射した。マウスを2〜20時間後に屠殺し、注射部位の皮膚および注射部位から排出する膝窩リンパ節を取り出した。皮膚における局所的細胞補充を、標準的な技術に従って、特異的モノクローナル抗体を使用して、免疫組織化学により試験した。細胞懸濁液を、RPMI 1640および10%ウシ胎仔血清(FCS)(Gibco−BRL)中にリンパ節から調製した。細胞を計数し、標準的な技術に従って、PBSおよび2% FCS中で、ビオチン−CD11c抗体およびFITC−CD11b抗体(Becton Dickinson)、続いて、PE−ストレプトアビジン(Dako)を含有するPBSおよび2% FCS中で染色した。CD11bおよびCD11cの発現(これは、マウス樹状細胞の集団を規定する)を、Facscanフローサイトメーター(Becton Dickinson)で、CellQuestソフトウェアを使用して、分析した。この分析および計数から、各リンパ節におけるCD11b+CD11c+の数を決定した。これらの実験は、以下を示す:(A)hMCP−4の局所的注射が、短期間(2時間)の後に、注射部位におけるCD11bを発現する細胞の補充を誘導し得ること。これらの細胞は、マウス血液樹状細胞または樹状細胞前駆体(例えば、単球)であり得る。なぜなら、両方とも、CD11bを発現し得るからである。マウスにおいて、循環する血液樹状細胞は、技術的な制限に起因して、同定されなかった。しかし、ヒトにおいては、血液樹状細胞が単離され得、それらは、hMCP−4に対する走化性アッセイに応答する(図3)。(B)排出リンパ節において、抗原特異的免疫応答が開始される場合、hMCP−4は、CD11bおよびCD11cの同時発現により同定される樹状細胞の補充を誘導するが、長期間(20時間)の後のみである。この遅延は、排出リンパ節に移動するために、皮膚において最初に補充される樹状細胞またはそれらの前駆体に必要な成熟および移動時間に対応する可能性が最も高い。
【0084】
(実施例8:ヒト血液由来の樹状細胞のhMCP−4に対する応答)
hMCP−4はまた、ヒト樹状細胞(血液から単離された樹状細胞を含む)に対して活性である(図5)。
【0085】
パネルA:ヒト循環血液CD11c+DCを磁性ビーズ除去により富化し、種々のケモカインに応答するトランスウェル(5μm孔サイズ)移動アッセイにおいて研究した。この移動を、2時間後に三重染色:系列マーカーFITC、HLA−DR tricolorおよびCD11c PEにより明らかにし、Facsにより染色した。各ケモカインを広範囲の濃度(1〜1000ng/ml)に対して試験し、最適応答のみを示す。結果を、移動指数として表し、3〜10の独立した実験から得た平均値を示す。血液CD11c+は主にMCP−4ならびにMCP1、MCP2およびMCP3に対して応答する(示さず)。SDF−1は選択性を欠き、CD11c+DCに対して強く活性である唯一の他のケモカインである。
【0086】
パネルB:異なるヒトDCおよびDC前駆体の集団(血液CD11c+DC、単球、単球由来DC、CD1a+ランゲルハンス細胞前駆体およびCD14+間質DC前駆体を含む)を、MCP−1およびMCP−4に応答するトランスウェル(5μm孔サイズ)移動アッセイにおいて研究した。全ての集団は、CD1a+ランゲルハンス細胞前駆体を除いて、MCP−4に応答する。さらに、単球由来DCは、CCR2とは異なるレセプターを介して、MCP−4に応答するが、MCP−1には応答しない。
【0087】
重要なことには、MCP−4は、ヒトDCに対して活性である。特に、他のケモカインと比較すると、MCP−4は、循環血液CD11c+DCの移動を誘導する最も強力なケモカインである。MCP−1、およびMCP−2およびMCP−3は、血液DCに対して類似の活性を示す。MCPは、これらの細胞上で発現されるCCR2を介して血液DCを補充するようである。さらに、MCP−4は、CCR2を発現しないCD1a+ランゲルハンス細胞前駆体を除いて、全てのDCまたはDC前駆体の集団(血液CD11c+DC、単球、単球由来DC、CD14+間質型DC前駆体)に対して活性である。最後に、MCP−4(MCP−1ではない)は、おそらく、CCR2とは異なるレセプターを介して、単球由来DCの移動を誘導する。
【0088】
(実施例9:インビボマウスモデルにおけるhMCP−4およびβ−ガラクトシダーゼの投与)
さらに、hMCP−4を、プラスミドDNAワクチン接種により誘導される抗原特異的免疫応答のアジュバントとして使用し得る。さらに、hMCP−4がプラスミドDNAワクチン接種のアジュバントとして使用される場合、これは、プラスミドDNAによりコードされた抗原を発現する腫瘍でその後チャレンジしたマウスの防御を増大し得る。
【0089】
6〜8週齡の7匹の雌性BALB/cマウス(Iffa−Credo,L’Arbresle,France)の群に、PBS単独またはPBS中の100ngのヒトMCP−4を、50μlの容量で、右後足踵の皮下に注射した。3時間後、マウスの同じ部位に、50μl容量のPBSの下で、コントロールpcDNA3プラスミド(InVitrogen)50μgまたはCMVプロモーター下のβ−ガラクトシダーゼをコードするpcDNA3プラスミド(pLacz,InVitrogen)50μgを注射した。この免疫プロトコルを、1週間間隔で4回繰り返した。
【0090】
最初の免疫の1日前、および最後の免疫の1週間後に血清を回収した。血清中のβ−ガラクトシダーゼ特異的免疫グロブリンのレベルを、以前に記載のように(Mendozaら,1997,J.Immunol.159:5777−5781)、特異的ELISAアッセイを使用して測定した。
【0091】
図6に見られるように、MCP−4注射は、β−ガラクトシダーゼDNA免疫(右後足
踵に100ng hMCP−4注射して3時間後の50μg DNA注射)の後に抗原特
異的体液性応答を増大する。図6は、4回の免疫の後に測定した抗β−ガラクトシダーゼ抗体を示す[PBS+pLaczと比較した場合のhMCP−4+pLaczの有意性:ステューデント検定]。
【0092】
最後に免疫して1週間後、マウスの群に、100μl容量のRPMI−1640の下で、β−ガラクトシダーゼを発現する5×104のC26−BAG結腸癌細胞(親切にも、Mario Colombo,Instituto Nazionale Tumori,Milan,Italyから譲り受けた)を右脇腹に皮下注射してチャレンジした。腫瘍の発生を、触知により1週間3回評価した。
【0093】
図7に見られるように、MCP−4注射は、マウスを、β−ガラクトシダーゼを発現するC26結腸癌細胞株でチャレンジした場合に、β−ガラクトシダーゼDNA免疫(右後
足踵に100ngのhMCP−4を注射して3時間後の50μgのDNA注射、腫瘍チャレンジする前に4回の免疫)により誘導された抗腫瘍効果を増大する[PBS+pLac
zと比較した場合のhMCP−4+pLaczの有意性:p<0.05、logrank
MCP−4対抗(opp):異なる部位に注射したhMCP−4]。
【0094】
従って、実施例7〜9は、ケモカインhMCP−4が、免疫応答、特に抗腫瘍応答のアジュバントとして使用され得ることを示す。MCP−4投与により媒介された増強された免疫応答を、血清中の抗原特異的免疫グロブリンレベルの増強として測定した。従って、MCP−4投与に対するB細胞応答の増強が明らかに存在する。さらに、切り替えにT細胞媒介補助を要する免疫グロブリンのサブクラス(例えば、IgG2a)が増大するので、T細胞媒介性応答もまた増大するようである。
【0095】
(実施例10:ヒト樹状細胞のh6Ckineケモカインに対する応答)
この実施例において、本発明者らは、ヒト6Ckine(h6Ckine)が、インビトロでのヒトの樹状細胞の公知のサブセット全てについての走化性因子であることを示した。特に、h6Ckineは、GM−CSF、IL−3およびCD40Lとの3時間という短時間のインキュベーション後のヒト血液樹状細胞に対して活性である(図8)。
【0096】
異なるヒトDC集団(CD1a+ランゲルハンス細胞、CD14+間質DC、単球由来DC、循環血液CD11c+DC、単球、および循環血液CD11c−プラズマ細胞様DCを含む)を、成熟の前および後に、ヒト6Ckineに対して応答する移動アッセイにおいて研究した。CD34−由来DC前駆体を、GM−CSF+TNFおよびSCFの存在下で6日間培養した後に、CD1a発現およびCD14発現に従ってFacsソーティングにより単離した。細胞を、GM−CSF単独(未成熟)中で12日目まで、またはGM−CSF+CD40−L(成熟)中で最後の2日間培養した。単球由来DCを、単球をGM−CSF+IL−4の存在下で5日間培養することにより生成し、CD40−Lを使用して、最後の2日間、活性化させた(成熟)かまたは活性化させなかった(未成熟)。ヒト循環血液CD11c+DCおよびCD11c−プラズマ細胞様DCを、磁性ビーズの除去により富化し、三重染色を使用して、系統マーカーFITCネガティブ、HLA−DR tricolorポジティブ、ならびにCD11c PEポジティブおよびCD11c PEネガティブにfacsソートした。CD11c+DCおよびCD11c−プラズマ細胞様DCを、GM−CSF+IL−3の存在下で、CD40Lとともに(成熟)またはCD40Lなしで(未成熟)、3時間培養した。
【0097】
移動アッセイを、5μm孔サイズまたは8μm孔サイズのTranswell(6.5mm直径,COSTAR,Cambridge,MA)を使用して、1〜3時間の間行い、facs分析により明らかにした。全ての集団が、CD40−L活性化後にのみ6Ckineに応答した。
【0098】
(実施例11:腫瘍モデルにおけるケモカインm6Ckine遺伝子移入)
この実施例において、本発明者らは、以下を示した:
m6Ckineを発現するように操作されたC26結腸癌腫瘍細胞は、あまり腫瘍形成性ではなく、この効果は、インビボでのCD8+細胞およびナチュラルキラー細胞活性に依存する(図9);
m6Ckineを発現するC26腫瘍は、親腫瘍と比較して、樹状細胞およびCD8+T細胞により有意に浸潤される(図10);および
m6Ckineを発現するように操作されたC26結腸癌腫瘍細胞は、親C26腫瘍よりあまり抗原性ではない(図11)。
【0099】
6〜10週齡の雌性BALB/c(H−2d)マウスを、Charles River(Iffa−Credo,L’Arbresle,France)から購入し、標準的な条件下で維持した。動物およびそれらの世話に関する手順は、EEC(欧州経済共同体)会議の指示(86/609,OJL 358,1,1987年12月12日)に合わせて行った。全ての腫瘍細胞培養を、10% FCS(Gibco−BRL)、1mM hepes(Gibco−BRL)、Gentallin(Schering−Plough,Union,NJ)、2×10−5M β−2メルカプトエタノール(Sigma,St Louis,MO)を補充したDMEM(Gibco−BRL, Life Technologies,Paisley Park,Scotland)中で行った。全ての細胞培養を、37℃にて5% CO2の加湿インキュベーター中で行った。マウス6Ckine/SLCをコードするcDNA(m6Ckine/SLC)を、CMVプロモーターを含むpcDNA3ベクター(InVitrogen,Carlsbad,CA)にクローニングした。C26結腸癌腫瘍細胞(親切にも、Mario P.Colombo,Instituto Nazionale per lo Studio e la Cura dei Tumori, Milano,Italyから譲り受けた)を、製造業者の指示に従って、Fugene試薬(Roche Molecular Diagnostics,Mannheim,Germany)を使用して、この構築物を用いてトランスフェクトした。m6Ckine/SLC mRNA(C26−6CK)を発現する単一のC26クローンを、800μg/mlでのネオマイシン(Sigma)選択の後に得た。C26腫瘍細胞またはC26−6CK腫瘍細胞を、100μlのDMEM中で右脇腹にS.C.注射し、腫瘍増殖を、1週間に3回触知することによりモニターした。抗体除去のために、腫瘍接種の1日前に、200μlのPBS中の0.5mgの抗CD8(クローン2.43)精製抗体、ラットコントロール(GL113)精製抗体または200μlのウサギ抗アシアロGM1血清(Wako Pure Chemicals,Osaka,Japan)を、i.p.注射し、次いで、0.2mgの抗体または100μlの抗アシアロGM1血清を3日後に、および実験過程の間に1週間に1回注射した。図10は、皮下C26−6CK細胞注射が、親腫瘍細胞と比較して、logrank分析により有意に遅延した腫瘍取り込み(intake)を生じる(p<0.01)ことを示す(A
およびB:C26+コントロール 対 C26−6CK+コントロール)。インビボで特
定の抗体を用いたCD8+細胞(A)またはナチュラルキラー細胞活性(B)の除去は、C26−6CK腫瘍細胞の遅延した腫瘍形成性を部分的に戻す。このことは、CD8+細胞およびNK細胞は、腫瘍増殖の遅延において役割を果たすことを示す。
【0100】
腫瘍を、約1cmのサイズに達したときに外科手術により取り出した。腫瘍塊を、小さな断片に細かく刻み、コラゲナーゼA(Roche Molecular Biochemicals)溶液中で37℃にて30分間攪拌しながらインキュベートした。次いで、懸濁液を、数回DMEM中で洗浄した。細胞懸濁液の染色を、PBSおよび5% FCS中で行った。FITC標識特異的抗体、ビオチン標識特異的抗体またはPE標識特異的抗体とのインキュベーションの前に、Fcレセプターを、FcブロックTM CD16/CD32抗体(PharMingen,San Diego,CA)を使用してブロックした。この研究において使用した種々の抗体(全て、PharMingen製)は、CD8β(53−5.8)、CD11c(HL3)、抗MHCクラスII I−Ad/I−Ed(269)、CD3(145−2C11)であった。ビオチン化抗体を、PEストレプトアビジン(Becton Dickinson)を使用して明らかにした。表現型パラメーターを、FacScan(Becton Dickinson,Mountain View,CA)で獲得し、CellQuestソフトウェア(Becton Dickinson)を使用して分析した。図10において、C26野生型腫瘍またはm6Ckineを発現するC26−6CK腫瘍を、腫瘍懸濁液全体のフローサイトメトリー分析によりCD8 T細胞およびCD11c+MHCクラスII+樹状細胞(DC)の浸潤について分析した(n=7)。データは、C26腫瘍と比較して、両方のC26−6CK腫瘍中の白血球サブセットの有意な補充を示す(ステューデントのt検定)。これらの結果は、腫瘍へのm6Ckine遺伝子移入は、免疫応答(抗腫瘍応答を含む)を開始するに必須の細胞である樹状細胞、および適応性免疫応答のエフェクター細胞であるCD8 T細胞の補充の両方を促進することを示唆する。
【0101】
いくつかの実験において、動物から腫瘍を取り出し、OCT化合物(Miles laboratory,Elkhart,IN)中に包埋して液体窒素中で急速冷凍し、免疫組織化学手順の前に−80℃で保存した。スライドガラスに適用した5μmの低温保持切片を、アセトン中で固定し、室温にて10分間、1% H2O2とともにインキュベートした。次いで、スライドを、ビオチンブロックTM試薬およびアビジンブロックTM試薬(ともに、Vector,Burlingame,CA)とともにインキュベートした。全てのインキュベーションを、PBS(Gibco−BRL)中の3回の2分間洗浄の後に行った。次いで、スライドを、二次抗体(Dako,Glostrup,Denmark)の同じ種に由来する血清の1/10希釈物とともに30分間プレインキュベートした。次いで、スライドを、5μg/mlの精製CD105(クローンMJ7/18,PharMingen,San Diego,CA)、ビオチン化二次抗体(Vectorのウ
サギ抗ラット)、ストレプトアビジン−アルカリ(VectorのABCキット)と連続
してインキュベートした。酵素反応物を、対応するVector基質を使用して発色させた。脈管形成アッセイを、インビボで発生中の腫瘍細胞を含むMatrigel(Becton Dickinson,Bedford,MA)ペレットのヘモグロビン含有量を決定することにより行った。BALB/cマウスを、2×105のC26細胞またはC26−6CK細胞と混合した0.5mlのMatrigelを、腹部正中線にs.c.注射した。9日後、Matrigelペレットを取り出し、周りの結合組織を解剖して除去し、ペレットを、90分間4℃にてMatriSperse溶液 v/v(Becton Dickinson)中で溶かした。ヘモグロビン含有量を、Drabkin法(Sigmaの試薬)により決定した。図11Aは、C26−6CK腫瘍は、親C26腫瘍よりあまり血管を形成しないことを示す。図11Bは、Matrigelアッセイにおいて、C26−6CK腫瘍細胞がC26細胞よりあまり脈管を形成しないことを示す。全体的に、これらの結果は、m6Ckineケモカインの腫瘍への遺伝子移入が、腫瘍血管に対して血管静止的(angiostatic)効果を有することを示す。
【0102】
これらの結果は、ケモカイン6Ckineが、遺伝子移入を介して癌の処置に使用され得ることを示す。好ましい実施形態は、以下からなるが、これらに制限されない:m6Ckineもしくはh6Ckineまたはm6Ckineもしくはh6Ckineの画分をコードするDNAまたはウイルスベクター(例えば、アデノウイルス)(標的化部分(ペプチドまたは抗体)を伴うかまたは伴わない)。
【0103】
(実施例12:インビボでのケモカイン6Ckineの腫瘍への局所的送達)
この実施例において、本発明者らは、組換えヒトまたは組換えマウス6Ckineタンパク質の既に存在するC26腫瘍への注射が、腫瘍保有マウスの生存を増すことを示した。h6Ckineの注射は、腫瘍増殖を遅延させる(図12)。
【0104】
6〜10週齡の雌性BALB/c(H−2d)マウスを、Charles River(Iffa−Credo,L’Arbresle,France)から購入し、標準的な条件下で維持した。動物およびそれらの世話に関する手順は、EEC(欧州経済共同体)会議の指示(86/609,OJL 358,1,1987年12月12日)に合わせて行った。全ての腫瘍細胞培養を、10% FCS(Gibco−BRL)、1mM hepes(Gibco−BRL)、Gentallin(Schering−Plough,Union,NJ)、2×10−5M β−2メルカプトエタノール(Sigma,St Louis,MO)を補充したDMEM(Gibco−BRL,Life Technologies,Paisley Park,Scotland)中で行った。全ての細胞培養を、37℃にて5% CO2の加湿インキュベーター中で行った。C26細胞を、Mario P.Colombo(Milano, Italy)から譲り受けた。
【0105】
C26腫瘍細胞を、100μlのDMEMにて右脇腹にs.c.注射し、腫瘍増殖を、1週間に3回触知することによりモニターした。いくつかの実験において、腫瘍容積を、カリパスを使用してモニターし、以下のように予測した:腫瘍容積=小さな直径2×大きな直径×0.4。組換えケモカインを用いた処置に関しては、マウスに、50μlのPBS下で10ngの>97%純度の組換えヒトまたはマウス6Ckine/SLC(R&D
Systems,Minneapolis,MN)を腫瘍内注射した。図1は、h6Ckineまたはm6Ckineを使用して注射したマウスが、PBSビヒクル単独と比較した場合に、生存の改善を示すことを示す(A)。h6Ckineの注射はまた、腫瘍の増殖を低減させた(B)。これらのデータは、組換え6Ckineケモカインの腫瘍内送達が抗腫瘍効果を有することを示す。
【0106】
(実施例13:rAd/6Ckineの転移性腫瘍の緩和)
雌性マウス(BALB/c ByJ;Jackson Laboratories)に、0.2ml(培地)の容量中の3×1015 4T1−p53乳腺腫瘍細胞(同系)を使用して、動物の左脇腹に皮下経路にて注射した。腫瘍が50〜100mm3のサイズに成長したときに、動物に、VPBS中の100μlのCMCB(1e10 PN/注射)を腫瘍内注射した。マウスに1週間あたり3回(月曜日、水曜日、金曜日)2週間にわたり注射した。腫瘍を、カリパスを使用して1週間に3回測定(長さ、幅、深さ)し、腫瘍容積を、以下の式に従って計算した:V=4/3r3
ここで、r=(W(mm)+L(mm)+D(mm))/6
動物を、腫瘍が1000mm3を超えた場合に屠殺した。
【0107】
各群から3匹のマウスを屠殺し、腫瘍が50mm3に達したときに(代表的には、10日目)開始して、腫瘍および肺を、以下に記載した生化学的分析のために組織処理のために切除し、肉眼および組織化学的手段により転移の存在を評価した。
【0108】
図13に示されるように、6Ckineは、免疫を増大し、脈管形成を抑制することにより、確立された腫瘍において腫瘍増殖を阻害し、自発的な転移を阻害する。
【0109】
本発明の多くの改変およびバリエーションが本発明の趣旨および範囲から逸脱することなく行われ得、当業者にも明らかである。本明細書中に記載される特定の実施形態は、例示により提供されるに過ぎず、添付の特許請求の範囲によってのみ、このような特許請求の範囲に与えられる等価物の全範囲とともに限定されるべきである。
【図面の簡単な説明】
【0110】
【図1】図1は、MIP−3αおよび腫瘍関連抗原を含むプラスミドを用いた免疫が、腫瘍移植に対する防御効果を有することを示す。
【図2】図2は、ケモカインMIP−3αの投与に伴うより大きなCTL活性を示す。
【図3】図3は、ケモカインhMCP−4のヌクレオチド配列および部分的アミノ酸配列を示す。
【図4】図4は、hMCP−4注射が、マウスにおいて用量依存性様式でインビボで樹状細胞の補充を促進することを示す。
【図5】図5は、hMCP−4がヒト血液における樹状細胞の補充において活性であることを示す。
【図6】図6は、MCP−4注射が、β−ガラクトシダーゼDNA免疫の後に抗原特異的体液性応答を増大させることを示す。
【図7】図7は、マウスがβ−ガラクトシダーゼを発現するC26結腸癌細胞株でチャレンジされたときに、MCP−4が、β−ガラクトシダーゼDNA免疫により誘導される抗腫瘍効果を増大させることを示す。
【図8】図8は、h6Ckineがヒト血液における樹状細胞の補充において活性であることを示す。
【図9】図9は、m6Ckineを発現するように操作されたC26結腸癌腫瘍細胞が、あまり腫瘍形成性ではなく、この効果が、インビボにおいてCD8+細胞およびナチュラルキラー細胞活性に依存することを示す。
【図10】図10は、m6Ckineを発現するC26腫瘍が、親腫瘍と比較した場合に、樹状細胞およびCD8+T細胞により有意に浸潤されることを示す。
【図11】図11は、m6Ckineを発現するように操作されたC26結腸癌腫瘍細胞が、親C26腫瘍よりも抗原性でないことを示す。
【図12】図12は、h6Ckineの注射が、インビボにおいてマウスの腫瘍増殖を遅延させることを示す。
【図13】図13は、6Ckineが、インビボにおいて確立された腫瘍の腫瘍増殖および自発的な転移を阻害することを示す。
【技術分野】
【0001】
本出願は、欧州特許出願第0 974 357 A1(1998年7月16日に出願および2000年1月26日公開)の利益を主張する。
【0002】
(発明の分野)
本発明は、癌を含む疾患状態の処置におけるヒトケモカインの使用に関連する。投与されたケモカインは、すべての抗原提示の樹状細胞または樹状細胞の特定のサブセットのいずれかの移動を方向付ける。1つの実施形態において、樹状細胞を活性化するように設計された疾患特異的抗原および/または部分は、ケモカインとともに投与される。
【背景技術】
【0003】
(発明の背景)
樹状細胞(DC)は、抗原の取り込みおよびT細胞へのそれらの提示に従事している。従って、DCは、抗原特異的免疫応答において重要な役割を果たす。
【0004】
DCは、種々のリンパ組織および非リンパ組織において身体の至る所に広範囲に分布した形態学的に類似な細胞型の多様な集団により表わされる(Cauxら、1995、Immunology Today 16:2;Steinman、1991、Ann.Rev.Immunol.9:271−296)。これらの細胞には、脾臓のリンパDC、およびリンパ節、表皮のランゲルハンス細胞、ならびに血液循環内の明瞭でない細胞が挙げられる。DCは、それらの形態学、表面のMHCクラスII発現の高レベルならびにT細胞の結合および同時刺激を媒介するいくつかのアクセサリー分子(B7−1[CD80]およびB7−2[CD86])(Inabaら、1990、Intern.Rev.Immunol.6:197−206;Frendenthalら、1990、Proc.Natl.Acad.Sci.USA 87:7698)、ならびにT細胞、B細胞、単球、およびナチュラルキラー細胞上で発現された特定の他の表面マーカーの非存在に基づいたグループとして集団的に分類される。
【0005】
DCは、骨髄由来であり、血流から組織の至るところまで前駆体として移動し、ここでDCはこの表皮におけるランゲルハンス細胞のような常在の細胞となる。
【0006】
末梢において、病原体侵入後、新鮮なランゲルハンス細胞のような未成熟DCは、抗原を捕獲しプロセシングする炎症部位(Kaplanら、1992、J.Exp.Med.175:1717−1728;McWilliamら、1994、J.Exp.Med.179:1331−1336)に補充される(Inabaら、1986.J.Exp.Med.164:605−613;Streileinら、1989、J.Immunol.143:3925−3933;Romaniら、1989、J.Exp.Med.169:1169−1178;Pureら、1990.J.Exp.Med.172:1459−1469;Schulerら、1985、J.Exp.Med.161:526−546)。
【0007】
次いで、抗原が負荷されたDCは、リンパ管を経由して末梢組織から、リンパ節のT細胞が豊富な領域まで移動し、ここで成熟DCは、相互連結細胞(IDC)と呼ばれる(Austynら、1988、J.Exp.Med.167:646−651;Kupiec−Weglinskiら、1988、J.Exp.Med.167:632−645;Larsenら、1990、J.Exp.Med.172:1483−1494;Fossum、S.1988、Scand.J.Immunol.27:97−105;Macatoniaら、1987、J.Exp.Med.166:1654−1667;Kripkeら、1990.、J.Immunol.145:2833−2838)。この部位で、これらは、未処置のT細胞に対してプロセシングされた抗原を提示し、そして抗原特異的な初期のT細胞応答を発生する(Liuら、1993、J.Exp.Med.177:1299−1307;Sornasseら、1992、J.Exp.Med.175:15−21;Heuflerら、1988、J.Exp.Med.167:700−705)。
【0008】
末梢組織からリンパ器官までのこれらの移動の間、DCは、表現型および機能において劇的な変化を含む成熟過程を経る(Larsenら、1990、J.Exp.Med.172:1483−1494;Streileinら、1990、Immunol.Rev.117:159−184;De Smedtら、1996、J.Exp.Med.184:1413−1424)。特に、可溶性タンパク質を効率的に捕獲およびプロセシングし特異的な記憶およびエフェクターT細胞を活性化するのに有効な、新鮮なランゲルハンス細胞のような未成熟のDCと対照的に、リンパ器官のIDCのような成熟DCは、未処置のT細胞の初回刺激において顕著に効果的であるが抗原の捕獲およびプロセシングにおいては乏しい(Inabaら、1986.J.Exp.Med.164:605−613;Streileinら、1989、J.Immunol.143:3925−3933;Romaniら、1989、J.Exp.Med.169:1169−1178;Pureら、1990、J.Exp.Med.172:1459−1469;Sallustoら、1995、J.Exp.Med.182:389−400;Cellaら、1997、Current Opin.Immunol.9:10−16)。
【0009】
DCの移動(traffic)パターンを制御するシグナルは、複雑であり、十分には理解されていない。
【0010】
TNFαおよびLPSにより提供されたシグナルが、組織から排出リンパ器官まで常在のDCの移動をインビボにおいて誘導することは公知である(De Smedtら、1996、J.Exp.Med.184:1413−1424;MacPhersonら、1995、J.Immunol.154:1317−1322;Roakeら、1995、J.Exp.Med.181:2237−2247;Cumberbatchら、1992、Immunology.75:257−263;Cumberbatchら、1995、Immunology.84:31−35)。
【0011】
ケモカインは、白血球の移動および活性化を調節する低分子量タンパク質である(Oppenheim、1993、Adv.Exp.Med.Biol.351:183−186;Schallら、1994、Curr.Opin.Immunol.6:865−873;Rollins、1997、Blood 90:909−928;Baggioliniら、1994、Adv.Imunol.55:97−179)。これらは、活性化された白血球自身、ならびに炎症刺激に際しての内皮細胞および上皮細胞を含む間質細胞により分泌される(Oppenheim、1993、Adv.Exp.Med.Biol.351:183−186;Schallら、1994、Curr.Opin.Immunol.6:865−873;Rollins、1997、Blood 90:909−928;Baggioliniら、1994、Adv.Immunol.55:97−179)。ケモカインに対する応答は、7つの膜貫通Gタンパク質共役レセプターにより媒介される(Rollins、1997、Blood 90:909−928;Premackら、1996、Nat.Med.2:1174−1178;Murphy、P.M.1994、Ann.Rev.Immunol.12:593−633)。いくつかのケモカイン(例えば、単球走化性タンパク質(MCP)−3、MCP−4、マクロファージ炎症タンパク質(MIP)−1α、MIP−1β、RANTES(発現および分泌された正常なT細胞の活性化に際する制御)、SDF−1、Teck(胸腺発現ケモカイン)およびMDC(マクロファージ由来ケモカイン))は、インビトロにおいてDCを誘引することが報告されている(Sozzaniら、1995、J.Immunol.155:3292−3295;Sozzaniら、1997、J.Immunol.159:1993−2000;Xuら、1996、J.Leukoc.Biol.60:365−371;MacPhersonら、1995、J.Immunol.154:1317−1322;Roakeら、1995、J.Exp.Med.181:2237−2247)。
【0012】
最近では、研究者らは、癌の処置においてDCの活性化を利用することを試みている。動物のモデルにおいて、わずか2×105の抗原パルスDCは、未処置のマウスに注入した場合、免疫を誘導する(Inabaら、1990、Intern.Rev.Immunol.6:197−206)。Flamandら(Eur.J.Immunol、1994、24:605−610)は、B細胞リンパ腫からのイディオタイプ抗原を用いてマウスDCをパルスし、未処置のマウスにそれらを注射した。この処置は、次の腫瘍攻撃からレシピエントマウスを効果的に保護し、そして免疫を持続する状態を確立した。抗原単独の注入、または抗原を用いてパルスされたB細胞は、効果がなく、このことは、抗腫瘍応答を担うDCの独特な特徴であることを示唆する。DCは抗腫瘍免疫を誘導することが可能なだけでなく、DCが、このプロセスの発生に絶対的に必須であると仮定されている(Ostrand−Rosenberg、1994、Current Opinion in Immunol.6:722−727;Grabbeら、1995、Immunol.Today 16:117−120;Huangら、1994、Science 264:961−965)。Huangおよび共同研究者ら(Huangら、1994、Science 264:961−965)は、抗腫瘍免疫を産生することが公知であるB7−1トランスフェクト腫瘍を用いてマウスに接種した。彼らは、MHC適合性APCを有するマウスだけが腫瘍チャレンジを拒絶することが可能であることを実証した。ヒトにおける研究では、DCについての類似の役割を実証している。ペプチド特異的CTLはペプチドパルスDCを使用して、精製CD8+T細胞から容易に誘導されることが報告されているが、ペプチドパルス単球を使用した場合は誘発されていない(Mehta−Damaniら、1994、J.Immunology153:996−1003)。
【0013】
樹状細胞の原発性腫瘍病変への組織学的浸潤が、膀胱癌、肺癌、食道癌、胃癌および鼻咽頭癌の患者における非常に長期化した患者の生存および転移性疾患の発生の減少と関連していることは、臨床的に非常に興味がある。対照的に、比較的乏しい臨床的予後は、DCを伴う散在性の浸潤を示す病変を有する患者で観察され、転移性の病変は、頻繁に、DC浸潤が欠けている(Becker,1993,In Vivo 7:187;Zeidら,1993,Pathology 25:338;Furihatonら,1992,61:409;Tsujitaniら,1990,Cancer 66:2012;Gianniら,1991,Pathol.Res.Pract.187:496;Murphyら,1993,J.Inv.Dermatol.100:3358)。最近、進行したB細胞リンパ腫の患者を、患者自身の腫瘍イディオタイプでパルスしたDCで処置した(Hsuら,1996,Nature Medicine 2(1):52)。このことにより、患者のB細胞リンパ腫において測定可能な減少が生じた。PSM抗原でパルスしたDCを用いた前立腺癌の処置は、Murphyらにより報告されている(The Prostate 1996 29:371)。
【0014】
インターロイキン4(IL−4)もしくは腫瘍壊死因子α(TNFα)のいずれかと組み合わせた顆粒球−マクロファージコロニー刺激因子(GM−CSF)に応答して、循環単球またはCD34造血性前駆体から多数のDCをインビトロ増殖させる技術が、最近出てきた(Sallustoら,1994,J.Exp.Med.179:1109−1118;Romaniら,1994,J.Exp.Med.180:83−93:Caux
ら,1992,Nature 360:258)。GM−CSFとIL−4の組み合わせは、末梢血単球を強力なDCへと分化させるように誘導する(KiertscherおよびRoth,1996,J.Leukocyte Biol.59:208−281)。これら2つのサイトカインの組み合わせを用いると、インビトロで末梢血から100倍のDCの収量の増加が達成される。
【0015】
マウスにおいて、種々の群により、腫瘍抗原を負荷したインビトロで生成されたDCは、腫瘍の発生を予防し、より重要なことには、確立された腫瘍の退行を誘導することが示された。黒色腫の患者を、MAGE−1腫瘍抗原由来のペプチドでパルスしたGM−CSF活性化APCを用いて処置する臨床試験を行った(Mehta−Damaniら,1994,J.Immunology 153:996−1003)。免疫する前は、2名の患者に由来する腫瘍浸潤リンパ球は、CD4+が優性であり、それぞれ、特異的腫瘍反応性を欠いていた。対照的に、同じ患者に由来する腫瘍浸潤リンパ球を免疫した後は、CD8+が優性であり、MAGE−1特異的抗腫瘍細胞傷害性が示された。従って、これらの研究から、DCが、免疫応答を刺激する独特かつ強い能力を有することが明らかである。
【発明の開示】
【発明が解決しようとする課題】
【0016】
従って、樹状細胞治療は、疾患(特に、癌)の処置に対する非常に確実な(promising)アプローチを示す。抗原提示樹状細胞を増殖かつ活性化させるだけでなく、治療的に有用かつ予防的に有用であるように、DCの移動を促進するために使用され得る、改善された材料および方法が継続して必要である。
【課題を解決するための手段】
【0017】
(発明の要旨)
本発明は、抗原提示樹状細胞の移動もしくは活性化を促進または阻害することにより疾患状態を処置するための材料および方法を提供することによって、前述の必要性を満たす。現在では、ケモカインは、有用な治療的薬剤であることが発見されている。本発明に従って処置され得る疾患状態としては、寄生生物感染、細菌感染、ウイルス感染、真菌感染、癌、自己免疫疾患、移植片拒絶、およびアレルギーが挙げられる。
【0018】
本発明は、疾患状態を処置する方法を提供し、この方法は、疾患状態の処置が必要な個体に、未成熟樹状細胞の抗原送達部位への移動を増大させるに十分な量のケモカインを投与する工程を包含する。本発明の1つの局面において、ケモカイン(例えば、MCP−1、MCP−2、MCP−3、MCP−4、MIP−1α、MIP−3α、RANTES、SDF−1、Teck、DC tactin−β、6Ckine、MDC、MIP−5またはこれらの組み合わせ)が投与される。本発明の好ましい方法において、疾患関連抗原(例えば、腫瘍関連抗原)は、ケモカインとともに投与される。
【0019】
本発明の別の局面は、疾患状態を処置する方法を提供し、この方法は、疾患状態を処置する必要のある個体に、未成熟樹状細胞の抗原送達部位への移動を減少させるに十分な量のケモカインを投与する工程を包含する。
【0020】
本発明のなお別の局面において、サイトカイン(特に、GM−CSFおよびIL−4)は、ケモカインと組み合わせて、ケモカインの前またはケモカインと同時のいずれかで投与される。GM−CSFおよびIL−4の投与は、前駆体からのDCの生成を刺激し、それにより、抗原を捕捉し、プロセシングするに利用可能なDCの数を増大させる。
【0021】
本発明のなお別の局面は、活性化薬剤(例えば、TNF−α、IFN−α、RANK−LまたはRANKのアゴニスト、およびtoll様レセプターファミリーの分子のアンタゴニスト)は、組織から、排出リンパを通ってリンパ器官へ向かうDCの移動を駆動する成熟シグナルを提供するために投与される。
【0022】
本発明はまた、哺乳動物における免疫応答を増強する方法を提供し、この方法は、ケモカインMCP−4またはMCP−4の生物学的に活性なフラグメントを哺乳動物に投与する工程を包含する。ヒトMCP−4(hMCP−4)は、ヒト血液の樹状細胞に対して活性であり、樹状細胞および樹状細胞前駆体を血液から補充する。好ましい局面において、このケモカインは、組換えケモカインである。最も好ましくは、このケモカインは、例えば、組換えケモカインと抗原の融合タンパク質の形態で、抗原とともに投与される。このような抗原は、腫瘍関連抗原、細菌抗原、ウイルス抗原または真菌抗原であり得る。
【0023】
さらに、本発明は、哺乳動物における免疫応答を増強する方法を提供し、この方法は、ケモカイン6Ckineまたは6Ckineの生物学的に活性なフラグメントを哺乳動物に投与する工程を包含する。ヒト6Ckineは、ヒト血液樹状細胞に対して活性であり、樹状細胞および樹状細胞前駆体を血液から補充する。樹状細胞の補充により、ケモカイン6Ckineは、抗腫瘍薬剤として働き、具体的には、腫瘍血管に対する新脈管形成効果を発揮することが示される。好ましい局面において、このケモカインは、組換えケモカインである。最も好ましくは、このケモカインは、例えば、組換えケモカインと抗原の融合タンパク質の形態で、抗原とともに投与される。このような抗原は、腫瘍関連抗原、細菌抗原、ウイルス抗原または真菌抗原であり得る。
【0024】
本発明のなお別の局面において、サイトカイン(特に、GM−CSFおよびIL−4)は、ケモカインと組み合わせて(ケモカインの前またはケモカインと同時のいずれか)投与される。
【0025】
最後の局面において、本発明は、MCP−4またはMCP−4の生物学的に活性な部分および抗原、ならびに6Ckineまたは6Ckineの生物学的に活性な部分および抗原を含む融合タンパク質を提供する。これらの融合タンパク質は、プラスミド、ウイルスベクターの形態で、または組換えベクターの形態で哺乳動物に投与され得る。
本発明は以下を提供する。
(1)疾患状態の処置のための医薬品の製造における、樹状細胞の移動を方向付け得るケモカインの使用。
(2)項目1に記載の使用であって、前記ケモカインが、MCP−1、MCP−2、MCP−3、MCP−4、MIP−1α、MIP−1β、MIP−3α、RANTES、SDF−1、Teck、DCtactin−β、6Ckine/SLC、LEC、MDC、およびMIP−5からなる群より選択される、使用。
(3)項目1に記載の使用であって、前記ケモカインが、樹状細胞の抗原送達部位への移動を方向付け得る、使用。
(4)項目1に記載の使用であって、前記ケモカインが、樹状細胞のリンパ器官への移動を方向付け得る、使用。
(5)項目1に記載の使用であって、前記疾患状態が、細菌感染、ウイルス感染、真菌感染、寄生生物感染または癌である、使用。
(6)項目1に記載の使用であって、前記疾患状態が、自己免疫疾患、組織拒絶、またはアレルギーである、使用。
(7)項目5に記載の使用であって、前記疾患状態が、黒色腫、乳癌、すい臓癌、結腸癌、肺癌、神経膠腫、肝細胞癌、子宮内膜癌、胃癌、腸癌、腎臓癌、前立腺癌、甲状腺癌、卵巣癌、精巣癌、肝臓癌、頭部および頚部の癌、結腸直腸癌、食道癌、胃癌、眼の癌、膀胱癌、神経膠芽細胞種、および転移性癌腫からなる群より選択される癌である、使用。
(8)項目3に記載の使用であって、前記樹状細胞が、未成熟樹状細胞である、使用。
(9)項目8に記載の使用であって、前記ケモカインが、MCP−1、MCP−2、MCP−3、MCP−4、MIP−1B、MDC、MIP−3α、MIP−1α、RANTESおよびMIP−5からなる群より選択される、使用。
(10)項目4に記載の使用であって、前記ケモカインが、MIP−3βである、使用。
(11)項目3に記載の使用であって、少なくとも1つの疾患関連抗原の使用をさらに包含する、使用。
(12)項目11に記載の使用であって、前記抗原が、腫瘍関連抗原である、使用。
(13)項目11に記載の使用であって、前記抗原が、細菌抗原、ウイルス抗原、または真菌抗原である、使用。
(14)項目12に記載の使用であって、前記腫瘍関連抗原が、Melan−A、チロシナーゼ、p97、β−HCG、GalNAc、MAGE−1、MAGE−2、MAGE−3、MAGE−4、MAGE−12、MART−1、MUC1、MUC2、MUC3、MUC4、MUC18、CEA、DDC、黒色腫抗原gp75、Hker8、高分子量黒色腫抗原、K19、Tyr1およびTyr2、pMel 17遺伝子ファミリーのメンバー、c−Met、PSA、PSM、α−フェトプロテイン、甲状腺ペルオキシダーゼ、gp100、NY−ESO−1、テロメラーゼならびにp53からなる群より選択される、使用。
(15)項目14に記載の使用であって、前記癌が前立腺癌であり、かつ前記腫瘍関連抗原が、PSAおよび/またはPSMである、使用。
(16)項目14に記載の使用であって、前記疾患状態が黒色腫であり、かつ前記腫瘍関連抗原がMelan−A、gp100またはチロシナーゼである、使用。
(17)項目1に記載の使用であって、活性化薬剤の使用をさらに包含する、使用。
(18)項目15に記載の使用であって、前記活性化薬剤が、TNFα、RP−105、抗CD40抗体および非メチル化CpGモチーフを含む核酸、またはtoll様レセプターのリガンドから選択される、使用。
(19)項目1に記載の使用であって、ケモカインとともに、GM−CSFおよびIL−4の組み合わせの使用をさらに包含する、使用。
(20)項目1に記載の使用であって、前記ケモカインが、皮内投与、筋肉内投与、皮下投与、局所投与、またはベクターの形態で投与される、使用。
(21)哺乳動物における免疫応答を増強する方法であって、該方法は、ケモカインMCP−4またはケモカインMCP−4の生物学的に活性な画分を該哺乳動物に投与する工程を包含する、方法。
(22)項目21に記載の方法であって、前記ケモカインが、組換えケモカインである、方法。
(23)項目21に記載の方法であって、前記ケモカインがヒトケモカインである、方法。
(24)項目21に記載の方法であって、該方法は、前記ケモカインの徐放を可能にする物質を送達部位に投与する工程をさらに包含する、方法。
(25)項目21に記載の方法であって、該方法は、前記ケモカインとともに抗原を投与する工程をさらに包含する、方法。
(26)項目25に記載の方法であって、MCP−4および抗原を含む融合タンパク質が、前記哺乳動物に投与される、方法。
(27)項目25に記載の方法であって、前記抗原が、腫瘍関連抗原である、方法。
(28)項目26に記載の方法であって、前記抗原が、腫瘍関連抗原である、方法。
(29)項目25に記載の方法であって、前記抗原が、細菌抗原、ウイルス抗原、または真菌抗原である、方法。
(30)項目26に記載の方法であって、前記抗原が、細菌抗原、ウイルス抗原または真菌抗原である、方法。
(31)項目25に記載の方法であって、前記腫瘍関連抗原が、Melan−A、チロシナーゼ、p97、β−HCG、GalNAc、MAGE−1、MAGE−2、MAGE−3、MAGE−4、MAGE−12、MART−1、MUC1、MUC2、MUC3、MUC4、MUC18、CEA、DDC、黒色腫抗原gp75、Hker8、高分子量黒色腫抗原
、K19、Tyr1およびTyr2、pMel 17遺伝子ファミリーのメンバー、c−Met、PSA、PSM、α−フェトプロテイン、甲状腺ペルオキシダーゼ、gp100、p53ならびにテロメラーゼからなる群より選択される、方法。
(32)項目26に記載の方法であって、前記腫瘍関連抗原が、Melan−A、チロシナーゼ、p97、β−HCG、GalNAc、MAGE−1、MAGE−2、MAGE−3、MAGE−4、MAGE−12、MART−1、MUC1、MUC2、MUC3、MUC4、MUC18、CEA、DDC、黒色腫抗原gp75、Hker8、高分子量黒色腫抗原、K19、Tyr1およびTyr2、pMel 17遺伝子ファミリーのメンバー、c−Met、PSA、PSM、α−フェトプロテイン、甲状腺ペルオキシダーゼ、gp100、p53ならびにテロメラーゼからなる群より選択される、方法。
(33)項目25に記載の方法であって、GM−CSFおよびIL−4の組み合わせを投与する工程をさらに包含する、方法。
(34)項目26に記載の方法であって、GM−CSFおよびIL−4の組み合わせを投与する工程をさらに包含する、方法。
(35)項目21に記載の方法であって、前記ケモカインとともに活性化薬剤を投与する工程をさらに包含する、方法。
(36)項目21に記載の方法であって、前記ケモカインが、皮内投与、筋肉内投与、皮下投与、局所投与、またはベクターの形態で投与される、方法。
(37)MCP−4および抗原を含む、融合タンパク質。
(38)項目37に記載の融合タンパク質であって、前記抗原が腫瘍関連抗原である、融合タンパク質。
(39)項目38に記載の融合タンパク質であって、前記腫瘍関連抗原が、Melan−A、チロシナーゼ、p97、β−HCG、GalNAc、MAGE−1、MAGE−2、MAGE−3、MAGE−4、MAGE−12、MART−1、MUC1、MUC2、MUC3、MUC4、MUC18、CEA、DDC、黒色腫抗原gp75、Hker8、高分子量黒色腫抗原、K19、Tyr1およびTyr2、pMel 17遺伝子ファミリーのメンバー、c−Met、PSA、PSM、α−フェトプロテイン、甲状腺ペルオキシダーゼ、gp100、p53ならびにテロメラーゼからなる群より選択される、融合タンパク質。
(40)項目38に記載の融合タンパク質であって、前記抗原が、細菌抗原、ウイルス抗原または真菌抗原である、融合タンパク質。
(41)項目37に記載の融合タンパク質を含む、プラスミド。
(42)項目39に記載のプラスミドであって、樹状細胞に特に適したプロモーター配列をさらに含む、プラスミド。
(43)項目37に記載の融合タンパク質を含む、ウイルスベクター。
(44)哺乳動物における免疫応答を増強する方法であって、該方法は、ケモカイン6Ckineまたはケモカイン6Ckineの生物学的に活性な画分を該哺乳動物に投与する工程を包含する、方法。
(45)項目44に記載の方法であって、前記ケモカインが、組換えケモカインである、方法。
(46)項目44に記載の方法であって、前記ケモカインがヒトケモカインである、方法。
(47)項目44に記載の方法であって、該方法は、前記ケモカインの徐放を可能にする物質を送達部位に投与する工程をさらに包含する、方法。
(48)項目44に記載の方法であって、該方法は、前記ケモカインとともに抗原を投与する工程をさらに包含する、方法。
(49)項目48に記載の方法であって、6Ckineおよび抗原を含む融合タンパク質が、前記哺乳動物に投与される、方法。
(50)項目48に記載の方法であって、前記抗原が、腫瘍関連抗原である、方法。
(51)項目49に記載の方法であって、前記抗原が、腫瘍関連抗原である、方法。
(52)項目48に記載の方法であって、前記抗原が、細菌抗原、ウイルス抗原、または真菌抗原である、方法。
(53)項目49に記載の方法であって、前記抗原が、細菌抗原、ウイルス抗原、または真菌抗原である、方法。
(54)項目48に記載の方法であって、前記腫瘍関連抗原が、Melan−A、チロシナーゼ、p97、β−HCG、GalNAc、MAGE−1、MAGE−2、MAGE−3、MAGE−4、MAGE−12、MART−1、MUC1、MUC2、MUC3、MUC4、MUC18、CEA、DDC、黒色腫抗原gp75、Hker8、高分子量黒色腫抗原、K19、Tyr1およびTyr2、pMel 17遺伝子ファミリーのメンバー、c−Met、PSA、PSM、α−フェトプロテイン、甲状腺ペルオキシダーゼ、gp100、p53ならびにテロメラーゼならびにC26結腸癌からなる群より選択される、方法。
(55)項目49に記載の方法であって、前記腫瘍関連抗原が、Melan−A、チロシナーゼ、p97、β−HCG、GalNAc、MAGE−1、MAGE−2、MAGE−3、MAGE−4、MAGE−12、MART−1、MUC1、MUC2、MUC3、MUC4、MUC18、CEA、DDC、黒色腫抗原gp75、Hker8、高分子量黒色腫抗原、K19、Tyr1およびTyr2、pMel 17遺伝子ファミリーのメンバー、c−Met、PSA、PSM、α−フェトプロテイン、甲状腺ペルオキシダーゼ、gp100、p53ならびにテロメラーゼならびにC26結腸癌からなる群より選択される、方法。
(56)項目48に記載の方法であって、GM−CSFおよびIL−4の組み合わせを投与する工程をさらに包含する、方法。
(57)項目49に記載の方法であって、GM−CSFおよびIL−4の組み合わせを投与する工程をさらに包含する、方法。
(58)項目44に記載の方法であって、前記ケモカインとともに活性化薬剤を投与する工程をさらに包含する、方法。
(59)項目44に記載の方法であって、前記ケモカインが、皮内投与、筋肉内投与、皮下投与
、局所投与、またはベクターの形態で投与される、方法。
(60)6Ckineおよび抗原を含む、融合タンパク質。
(61)項目60に記載の融合タンパク質であって、前記抗原が腫瘍関連抗原である、融合タンパク質。
(62)項目61に記載の融合タンパク質であって、前記腫瘍関連抗原が、Melan−A、チロシナーゼ、p97、β−HCG、GalNAc、MAGE−1、MAGE−2、MAGE−3、MAGE−4、MAGE−12、MART−1、MUC1、MUC2、MUC3、MUC4、MUC18、CEA、DDC、黒色腫抗原gp75、Hker8、高分子量黒色腫抗原、K19、Tyr1およびTyr2、pMel 17遺伝子ファミリーのメンバー、c−Met、PSA、PSM、α−フェトプロテイン、甲状腺ペルオキシダーゼ、gp100、p53ならびにテロメラーゼからなる群より選択される、融合タンパク質。
(63)項目61に記載の融合タンパク質であって、前記抗原が、細菌抗原、ウイルス抗原または真菌抗原である、融合タンパク質。
(64)項目60に記載の融合タンパク質を含む、プラスミド。
(65)項目64に記載のプラスミドであって、樹状細胞に特に適したプロモーター配列をさらに含む、プラスミド。
(66)項目60に記載の融合タンパク質を含む、ウイルスベクター。
【0026】
(発明の詳細な説明)
本明細書中で引用されるすべての参考文献は、その全体が、参考として援用される。
【0027】
インビボでのDC移動を誘導するシグナルと、それらのケモカインに対する応答との間の関係は、これまで公知ではなかった。本発明者らは、DCにより発現されるケモカインレセプターのパターンは、それらの成熟段階に従って変化し、ケモカインがDCサブセットの移動を駆動するために使用され得、それにより、免疫応答の開始を制御し得ることを発見した。ケモカインは、本発明に従って、未成熟DCサブセットを抗原送達部位に選択的に誘引するアジュバントとして使用され得る。自己免疫疾患、組織拒絶またはアレルギーの状況において、本発明は、例えば、CCR6アゴニストおよびアンタゴニスト、CCR7アゴニストおよびアンタゴニスト、ならびにCCR2アゴニストおよびアンタゴニストの発生を介して、DCの移動を妨害することにより、DC機能をブロックする方法を提供する。
【0028】
免疫系に抗原を提示するDCのサブセットに依存して、応答は劇的に変化する。DCは、寛容を誘導し得る。胸腺の髄質において見出されたDCは、自己選択的胸腺細胞を発生させるネガティブ選択において役割を果たし得る(Brockerら,1997,J.Exp.Med.185(3):541−550)。DCはまた、自己反応性末梢T細胞に寛容させる(Kurtsら,1997,J.Exp.Med.186(2):239−245;Adlerら,1998,J.Exp.Med.187(10):1555−1564)。マウスDCの特定のサブセット(おそらく、リンパ起源の)は、免疫寛容を誘導することが提唱された(Ardavin,1993,Nature 362(6422):761−763)。さらに、リンパDCに対する候補ヒト対応物(DC−2)(Grouardら,1997,J.Exp.Med.185(6):1101−1111)がIL−12を分泌できないという最近の説明は、この亜集団による提示の後に、免疫応答がTH−2型に向かって偏向され得ることを示唆する。
【0029】
目的が免疫応答を減少させることである場合、DCに寛容させること(自己免疫、アレルギー)が増大されるか、または応答の質がDC−2を特異的に増大させることにより改変される(TH2より大きなTH1、すなわち、アレルギーにおいて)。
【0030】
本発明において使用するためのケモカインは、DCの制限されたサブセット、特に、未成熟DCに対して活性な、身体の天然のタンパク質である。これらのケモカインのいくつか(MIP−3α、Teck、MDCおよびMCP−4、ならびに6Ckineが挙げられるが、これらに限定されない)が、本発明者らにより同定された。
【0031】
本発明を実施する際に使用されるケモカインは、天然産物に対して同一のアミノ酸配列を有する組換えタンパク質、または天然産物に由来し得るが、その本来の走化性特性を保持しつつその薬物動態特性を変化させる改変を含む組換えタンパク質であり得る。ケモカインの送達の様式は、注射(皮内、筋肉内および皮下が挙げられる)または局所的(例えば、軟膏またはパッチ)によってであり得る。
【0032】
ケモカインはまた、ベクター(例えば、ウイルスベクター)(例えば、アデノウイルス、ポックスウイルス、レトロウイルス、レンチウイルス)、または操作されたプラスミドDNAによって核酸配列として送達され得る。
【0033】
本明細書中で使用される用語「ケモカイン」は、走化性因子を含み得る。走化性因子は、低分子化合物(これは、未成熟DCにより発現されるケモカインレセプターの選択的アゴニストである)であり得る。CCR6(ケモカインMIP−3αの天然のレセプター)は、このようなレセプターの例である。
【0034】
本発明の特に好ましい実施形態において、ケモカインは、疾患関連抗原とともに投与される。この抗原は、任意の分子部分であり得、これに対して、免疫応答の増大または低下が調べられる。これは、ヒトまたは動物において疾患を引き起こすことが公知の生物(例えば、細菌、ウイルス、寄生生物(例えば、Leishmania)、および真菌)に由来する抗原を含む。これはまた、腫瘍により発現される抗原(腫瘍関連抗原)および植物抗原(アレルゲン)を含む。
【0035】
本発明において使用するための腫瘍関連抗原としては、以下が挙げられるが、これらに限定されない:Melan−A、チロシナーゼ、p97、β−HCG、GalNAc、MAGE−1、MAGE−2、MAGE−3、MAGE−4、MAGE−12、MART−1、MUC1、MUC2、MUC3、MUC4、MUC18、CEA、DDC、黒色腫抗原gp75、HKer 8、高分子量黒色腫抗原、K19、Tyr1およびTyr2、pMel 17遺伝子ファミリーのメンバー、c−Met、PSA、PSM、αフェトプロテイン、甲状腺ペルオキシダーゼ、gp100、NY−ESO−1、テロメラーゼ、ならびにp53。この列挙は、本発明の実施において使用され得る抗原の型を網羅することを意図するのではなく、単に例示するに過ぎない。
【0036】
抗原の異なる組み合わせが使用され得、これは、異なる人種グループ、性別、地理的分布、および疾患の病期に従って最適な機能を示す。本発明の1つの実施形態において、少なくとも2つ以上の異なる抗原が、ケモカインの投与とともに投与される。
【0037】
抗原は、ケモカインと同じ部位および同じ時間に、または48時間を越えない遅延の後に送達または投与され得る。同時投与または組み合わせた投与は、本明細書中で使用される場合、ケモカインおよび抗原が、被験体に、一般的な処置スケジュールの過程の間に、(a)時間内に同時または(b)異なる時間のいずれかに投与される。後者の場合において、2つの化合物は、意図された効果を達成するために十分に近い時間内に投与される。抗原は、タンパク質、または1または数個のペプチドの形態、あるいは送達ベクターに含まれる核酸配列の形態にあり得る。
【0038】
原発性癌および転移制癌が、ともに、本発明に従って処置され得る。処置され得る癌の型としては、以下が挙げられるが、これらに限定されない:黒色腫、乳癌、膵臓癌、結腸癌、肺癌、神経膠腫、肝細胞癌、子宮内膜癌、胃癌、腸癌、腎臓癌、前立腺癌、甲状腺癌、卵巣癌、精巣癌、肝臓癌、頭部および頚部の癌、結腸直腸癌、食道癌、胃癌、眼の癌、膀胱癌、神経膠芽細胞種、および転移性癌。用語「癌(腫)」とは、呼吸器系癌、胃腸管系癌、尿生殖器系癌、前立腺癌、内分泌系癌、および黒色腫を含む、上皮組織または内分泌組織の悪性腫瘍をいう。転移性は、本明細書中でこの用語が使用される場合、原発性腫瘍(局所的リンパ節を含む)から離れた部位への腫瘍の拡がりとして規定される。
【0039】
DCの成熟を達成、誘導または刺激するように設計された部分は、有利に投与され得る。このような薬剤は、組織からリンパ節への移動を促進する成熟シグナルを提供する。この部分は、例えば、TNF−αもしくはRP−105のような身体の天然産物、またはDC上の特異的構造を認識するアゴニスト抗体(例えば、抗CD−40抗体)、または別の物質であり得る。活性化物質は、非メチル化CpGモチーフ、またはDCを刺激することが公知のtoll様レセプターのアゴニストを含む核酸の配列であり得る。ケモカインおよび/または抗原がプラスミドベクターにより送達される本発明の実施形態において、これらの核酸配列は、ベクターの一部であり得る。
【0040】
GM−CSFおよびIL−4は、有利には、ケモカインおよび/または抗原と組み合わせて投与され得る。GM−CSFおよびIL−4の投与組み合わせは、前駆体からDCの生成を刺激する。GM−CSFおよびIL−4は、循環未成熟DCの数を増大させる目的で投与され得る。これは、ケモカインの引き続く注射により、局所的に補充され得る。このプロトコルは、GM−CSFおよびIL−4を使用して、少なくとも5〜7日間の全身的な前処置を含む。別の方法は、GM−CSFおよびIL−4の局所的投与により、同じ部位に送達される抗原を拾い上げ得る、DC前駆体(単球)の未成熟DCへの局所的分化に都合がよい。
【0041】
一般に、ケモカインおよび/または抗原および/または活性化薬剤および/またはサイトカインは、薬学的キャリア中に有効量のケモカインおよび/または抗原および/または活性化薬剤および/またはサイトカインを含む薬学的組成物として投与される。これらの試薬は、生理学的に刺激のない安定剤および賦形剤とともに、例えば、従来の薬学的に受容可能なキャリアまたは賦形剤(例えば、免疫原性アジュバント)中に、さらなる活性成分または不活性成分を用いた治療的用途のために組み合わせられ得る。薬学的キャリアは、患者に本発明の組成物を送達するために適切な任意の適合性の、非毒性物質であり得る。
【0042】
有効な治療に必要な試薬の量は、多くの異なる要因(投与の手段、標的部位、患者の身体的状態、および投与されている他の医薬品が挙げられる)に依存する。従って、処置投薬量は、安全性および有効性を最適化するために力価測定されるべきである。特定の癌を処置するための有効用量を試験する動物は、ヒト投薬量のさらなる予測指標を提供する。種々の考慮事項が、例えば、Gilmanら(編)(1990)Goodman and
Gilman’s:The Pharmacological Bases of Therapeutics,第8版,Pergamon Press;およびRemington’s Pharmaceutical Sciences,第17版(1990),Mack Publishing Co.,Easton,PAに記載される。投与のための方法は、例えば、静脈内投与、腹腔内投与、または筋肉内投与、経皮的拡散などについて、そこでおよび以下に議論される。薬学的に受容可能なキャリアとしては、水、生理食塩水、緩衝液、および例えば、the Merck Index,Merck&Co.,Rahway,New Jerseyに記載される他の化合物が挙げられる。徐放処方物、または徐放装置は、連続的な投与のために使用され得る。
【0043】
ケモカインおよび/または抗原および/または活性化薬剤についての投薬量範囲は、通常は、適切なキャリアとともに、1mM濃度未満、代表的には、約10μM濃度未満、通常は、約100nM未満、好ましくは、約10pM(ピコモル濃度)未満、および最も好ましくは、約1fM(フェムトモル濃度)未満の量にあると予測される。一般に、処置は、化合物の最適な用量未満のより少量の投薬量を用いて開始される。その後、投薬量は、その環境下での最適な効果が達成されるまで、少量ずつ増大される。特定の状況のための適切な用量および投薬レジメンの決定は、当該分野の技術範囲内である。
【0044】
本願発明の実施におけるGM−CSFおよびIL−4の好ましい生物学的に活性な用量は、循環CD14+/CD13+前駆体細胞の数;DC前駆体および成熟DCの表面上での抗原提示分子の発現;T細胞への抗原提示活性;および/または成熟DC機能と一致した抗原依存性T細胞応答の刺激の最大の増加を誘導する投薬組み合わせである。本発明の実施において、皮下投与するために使用されるIL−4の量は、代表的には、約0.05〜約8.0μg/kg/日、好ましくは、0.25〜6.0μg/kg/日、最も好ましくは、0.50〜4.0μg/kg/日の範囲にある。皮下投与に使用されるGM−CSFの量は、代表的には、約0.25μg/kg/日〜約10.0μg/kg/日、好ましくは、約1.0〜8.0μg/kg/日、最も好ましくは、2.5〜5.0μg/kg/日の範囲にある。特定の患者のための有効量は、上記のパラメーターの1以上における有意な変化を測定することにより確立される。
【0045】
ケモカインMCP−4またはMCP−4の生物学的に活性なフラグメントの投与が、用量依存性様式においてインビボでマウスの樹状細胞の補充を促進し、血液から単離されたヒト樹状細胞に対しても活性であることが見出された。生物学的に活性なフラグメントは、測定可能な免疫応答を刺激するに十分なMCP−4分子の部分を意味する。この応答は、血清中の免疫グロブリンレベルの抗原特異的刺激の増強(代表的には、B細胞応答として公知)として測定され得る。さらに、MCP−4の生物学的に活性なフラグメントは、免疫グロブリンの特定のクラス(例えば、T細胞における増大を要するIgG2a)の生成を刺激する。さらに、MCP−4の生物学的に活性なフラグメントは、抗原特異的抗腫瘍応答を増強する。増強した応答は、抗原を発現する腫瘍を使用したチャレンジの後のより遅延した腫瘍増殖またはより遅延した腫瘍指標により測定され得る。増強した免疫応答はまた、リンパ球の規定された集団(血液、脾臓、リンパ節、腫瘍)の抗原特異的細胞傷害性応答の分析により測定され得る。当然のことながら、CCR2アゴニスト(例えば、薬物発見スクリーニングにより見出される)である低分子はまた、抗原特異的抗腫瘍応答を増強することが認識される。根本的理由は、全てのMCP(1〜4)が天然のCCR2アゴニストであり、引き続き人工的な低分子アゴニストが同じ効果を有し得ることである。多くの現在の治療剤は、有機化学合成により得られる低分子である。
【0046】
好ましい実施形態は、組換えhMCP−4タンパク質単独または送達部位における徐放を可能にする物質(デポー);hMCP−4またはhMCP−4の画分および抗原からなる融合タンパク質(9アミノ酸より大きなペプチドまたはタンパク質); hMCP−4またはhMCP−4の画分(抗原を伴うかまたは伴わない)をコードするDNAまたはウイルスベクター(9アミノ酸より大きなペプチドまたはタンパク質)、あるいは送達ベクターに含まれる核酸配列と組み合わせられた組換えhMCP−4タンパク質からなるが、これらに限定されない。
【0047】
ヒトMCP−4(hMCP−4)は、ケモカインのCCファミリーに属する。その配列は、1996年に最初に公開された(Uguccioniら,1996,Monocyte Chemotactic Protein 4(MCP−4),A Novel Structural and Functional Analogue of MCP−3およびEotaxin,J.Exp.Med.183:2379−2394)。ヒトMCP−4は、75アミノ酸残基からなる8.6kDaのペプチドである(図3)。これはまた、CK−β−10、SCY−A13およびNCC−1(Swiss−Prot登録番号Q99616)として公知であり、新たなケモカイン命名においてCCL13と再度名づけられた(Zlotnikら,2000,Chemokines:A New Classification System and Their Role In Immunity,Immunity,12:121−127)。
【0048】
6Ckineは、ケモカインのCCファミリーに属する(Hedrickら,1997,J.Immunol.159:1589−1593)。CK−β−9、exodus−2およびSLC(ヒトタンパク質についてはSwiss−Prot登録番号000585
)として公知であり、CCL21と再度名づけた。ヒト6Ckine(h6Ckine)は、ケモカインCCR7に結合する一方、マウス6Ckine(m6Ckine)は、CCR7およびCXCR3レセプターに結合するが、親和性は低い(Jenhら,1999,J.Immunol.162:3765−3769)。マウス6Ckineは、マウスにおける腫瘍に注射された場合、抗腫瘍効果を有することが示されている(Sharmaら,2000,J.Immunol.164:4558−4563)。
【0049】
6Ckine様MIP−3βおよびMCP−4は、成熟DCの移動を誘導する。興味深いことに、6CkineならびにMIP−3βは、成熟の後に全てのヒトDC集団(CD1a+ランゲルハンス細胞、CD14+間質DC、単球由来DC、循環血液CD11c+DC、単球、および循環血液CD11c−プラズマ細胞様DCを含む)の移動を誘導し得る。6Ckineに対する応答は、いくつかのDCアクチベーター(CD40−L、TNF−α、およびLPSを含む)により誘導される成熟の後に観察される。MIP−3βの場合において見られるように、CCR7は、6Ckineを介してDC活性化の間に上方調節され、おそらく6Ckineに対する応答を説明する。
【0050】
従って、ケモカインh6Ckineは、癌処置において使用され得ることが提唱されている。好ましい実施形態は、以下からなるが、これらに限定されない:組換えh6Ckineタンパク質単独または送達部位におけるその徐放を可能にする物質(腫瘍部位におけるデポー)と組み合わせた組換えh6Ckineタンパク質;融合タンパク質またはh6Ckineもしくはh6Ckineの画分および腫瘍への構築物の送達を可能にする標的化部分(例えば、抗体または抗体のフラグメント、タンパク質リガンド、10アミノ酸よ
り大きなペプチド)からなる、化学的連結により作製された構築物;h6Ckineもし
くはh6Ckineの画分(上記の標的化部分を有するかまたは有さない)をコードするDNAもしくはウイルスベクター(例えば、アデノウイルス)。
【実施例】
【0051】
本発明は、以下の非限定的な実施例により例示され得る。以下の実施例は、以下の材料および方法を参照することによって容易に理解され得る。
【0052】
(造血因子、試薬および細胞株) 組換えGM−CSF(比活性:2.106U/mg
,Schering−Plough Research Institute,Kenilworth,NJ)を、飽和濃度の100ng/mlにて使用した。組換えヒトTNFα(比活性:2×107U/mg,Genzyme,Boston,MA)を、最適濃度の2.5ng/mにて使用した。組換えヒトSCF(比活性:4×105U/mg,R&D Abington,UK)を、25ng/mlの最適濃度にて使用した。組換えヒトIL−4(比活性:2.107U/mg,Schering−Plough Research Institute,Kenilworth,NJ)を、50U/mlの飽和濃度にて使用した。組換えヒトケモカインMIP−1α(比活性:2×105U/mg,9×1012U/M)、RANTES(比活性:1×104U/mg,8×1010U/M)、MIP−3α(比活性:4×105U/mg,3×1012U/M)およびMIP−3β(比活性:1×104U/mg,9×1010U/M)を、R&D(Abington,UK)から得た。LPSを、10ng/mlにて使用した(Sigma)。
マウスCD40リガンドトランスフェクト細胞株(CD40−L L 細胞)を、DC成熟の刺激因子として使用した。
【0053】
(臍帯血CD34+ HPCからのDCの生成) 臍帯血サンプルを、全期間の送達の後に得た。CD34+抗原を有する細胞を、記載のように(Cauxら,1996,J.Exp.Med.184:695−706;Cauxら,1990,Blood.75:2292−2298)、抗CD34+モノクローナル抗体(Immu−133.3,Immunotech Marseille,France)、ヤギ抗マウスIgG被覆マイクロビーズ(Miltenyi Biotec GmBH,Bergish Gladbach,Germany)およびMinimacs分離カラム(Miltenyi Biotec)を使用して、ポジティブ選択を介して単球画分から単離した。全ての実験において、単離した細胞は、80%〜99% CD34+であった。精製の後、CD34+細胞を、10% DMSO中で低温保存した。
【0054】
SCF、GM−CSFおよびTNFαの存在下で、記載のように(Cauxら,1996,J.Exp.Med.184:695−706)、エンドトキシンを含まない培地(10%(v/v)非働化ウシ胎仔血清(FBS)(Life Techniques,France,Irvine,UK)、10mM Hepes、2mM L−グルタミン、5×10−5M D−メルカプトエタノール、100μg/ml ゲンタマイシン(Schering−Plough,Levallois,France)を補充したRPMI
1640(Gibco,Grand Island,NY)からなる)(完全培地とも
いわれる)中で確立した。融解した後、CD34+細胞を、25〜75cm2培養容器(
Linbro,ICN Biomedicals,Acron,OH)中で2×104細胞/mlに増殖させるために播種した。最適条件を、これらの培養物を、5日目および10日目に新たなGM−CSFおよびTNFαを含む培地を使用して分割することにより維持した(細胞濃度:1〜3×105細胞/ml)。CD1a+DCは、12日目には、70〜90%の細胞の間である。
【0055】
(FACSソーティングによるCD86発現に従う未成熟DCおよび成熟DCの単離)
GM−CSFおよびTNFαの存在下での培養の12日後に、細胞を収集し、FITC結合体化OKT6(CD1a)(Ortho Diagnosis System,Raritan,NJ)およびPE結合体化IT2.2(CD86)(Pharmingen, San Diego,CA)で標識した。細胞を、CD1aおよびCD86発現に従って、未成熟CD1a+CD86−DC集団、および成熟CD1a+CD86+DC集団に、FACStarplus(登録商標)(レーザー設定:出力250mW、励起波長488nm、Becton−Dickinson,Sunnyvale,CA)を使用して分離した。染色およびソーティングの手順全てを、細胞凝集を避けるために、0.5mM
EDTAの存在下で行った。ソートした集団の再分析により、純度>98%であることが示された。
(末梢血単球からのDCの生成) 単球を、PBMCの調製、続いて52% Percoll勾配の後に、免疫磁性除去(Dynabeads,Dynal Oslo,Norway)により精製した。除去を、抗CD3モノクローナル抗体(OKT3)、抗CD19モノクローナル抗体(4G2)、抗CD8モノクローナル抗体(OKT8)、抗CD56モノクローナル抗体(NKH1,Coulter Corporation,Hialeah,FL)および抗CD16モノクローナル抗体(ION16,Immunotech)を使用して行った。単球由来樹状細胞を、GM−CSFおよびIL−4の存在下で精製単球を6〜7日間培養することにより生成した(Sallustoら,1994,J.Exp.Med.179:1109−1118)。
【0056】
(インビトロで生成したDCの成熟の誘導) CD34+HPCを、GM−CSF+TNFαの存在下で6日目まで培養し、GM−CSF単独の存在下で6日目〜12日目まで、それらの未成熟を保つために培養した。CD34+HPC由来の未成熟DCまたは単球由来DCを、TNFα(2.5ng/ml)またはLPS(10ng/ml)またはCD40LトランスフェクトL細胞(5DCについて1つのL細胞)の存在下で、3時間〜72時間の間、記載のように(Cauxら,1994,J.Exp.Med.180:1263−1272)、活性化した。
【0057】
(末梢血または扁桃からのCD11c+DCの精製) CD11c+DCを、末梢血または扁桃から以前に記載のように調製した(Grouardら,1996,Nature
384:364−367)。簡潔には、扁桃除去を受けた子供から得た扁桃を、細かく刻み、コラゲナーゼIVおよびDNase I(Sigma)を使用して消化した。収集した細胞を、SRBC(BioMerieux,Lyon,France)を含有するFicoll−Hypaqueを介して500rpmにて15分間、次いで、2000rpmにて30分間遠心分離した。末梢血単球(PBMC)を、Ficoll−Hypaqueにより単離した。CD3+T細胞(OKT3)、CD19+B細胞(4G7)、およびCD14+単球(MOP9)を、磁性ビーズ(抗マウスIg被覆Dynabeads,Dynal)により、得られた低密度細胞から取り出した。第2の除去を、抗NKH1、抗グリコホリンA(Immunotech)および抗CD20(1F54)を使用して行った。残りの細胞を、以下のmAbを用いて染色した:抗CD1a FITC(OKT6);抗CD14 FITC、抗CD57 FITC、抗CD16 FITC、抗CD7 FITC、抗CD20 FITC、抗CD3 FITC(Becton Dickinson,Mountain View,CA);抗CD4 PE−Cy5(Immunotech)ならびに抗CD11c PE(Becton Dickinson)。CD4+CD11c+系列−DCを、FACStarPlus(登録商標)を使用した細胞ソーティングにより単離した(レーザー設定:出力250mW、励起波長488nm)。除去、染色およびソーティングの手順全てを、0.5mM EDTAの存在下で行った。ソートした集団の再分析により、純度>97%であることが示された。
【0058】
(走化性アッセイ) 細胞移動を、走化性マイクロチャンバ技術(48ウェル Boyden microchamber,Neuroprobe,Pleasanton,CA)(Baconら,1988,Br.J.Pharmacol.95:966−974)を使用して評価した。簡潔には、ヒト組換えMIP−3αおよびMIP−3β、MIP−1αおよびRANTESを、RPMI 1640培地中に1ng/ml〜1000ng/mlの範囲の濃度に希釈し、走化性チャンバの下方のウェルに添加した。50μlのRPMI 1640培地中の105細胞/ウェル(またはCD11c+DCについては5×104細胞/ウェル)を、下方のウェルを分離する標準的な5μm孔のポリビニルピロリドンを含まないポリカーボネートフィルター(Neuroprobe)を備えるチャンバの上方のウェルにアプライした。このチャンバを、5% CO2の加湿空気中、37℃にて1時間インキュベートした。次いで、フィルターの下面に移動した細胞を、Field’s AおよびField’s B(BDH,Dorcet,England)を使用して染色し、2つのランダムに選択した低倍率視野(倍率×20)において、イメージアナライザー(ソフトウェア:Vision ExplorerおよびETC 3000,Graphtek,Mirmande,France)を使用して数えた。各アッセイを二連で行い、結果を、2視野あたりの移動細胞の平均±SDとしてあらわした。
【0059】
(総RNAの抽出およびcDNAの合成) 細胞を、上記のように調製し、総RNAを、製造業者により言及されたように、チオシアン酸グアニジウム法により抽出した(RNAgents total RNA isolation system,Promega)。DNAse I(RQ1 RNAse非含有DNAse,Promega)処理の後に、RNAを、分光光度計により定量し、質を、ホルムアルデヒド変性条件下での電気泳動により評価した。第1鎖cDNAを、RNAse非含有条件下で抽出した総RNAから合成した。この反応を、製造業者により記載されるように、5μgの総RNA、25ng/μlのオリゴdT12−18プライマー(Pharmacia,Orsay,France)およびSuperscriptキット(SuperScript II RNase H−Reverse Transcripase,Gibco BRL)を使用して行った。全てのサンプルに関して、cDNAの合成を制御し、β−アクチンプライマーを使用して21サイクルのRT−PCRにより較正した。
【0060】
(RT−PCR分析) 半定量的PCRを、最終容積100μlの反応混合物(その1×緩衝液とともに2.5U AmpliTaq酵素(5U/μl,Perkin Elmer,Paris,France)、0.2mMの各dNTP(Perkin Elmer,Paris,France)、5% DMSO、および1μMの各正方向および逆方向プライマーを含む)中でPerkin Elmer 9600サーマルサイクラーにおいて行った。CCR6(登録番号Z79784)およびCCR7(登録番号L08176)プライマーを、ケモカインレセプター間の最低の相同性の領域内に指定した。以下:
【0061】
【化1】
を、RT−PCRおよび配列決定に使用した。両方のケモカインレセプターのために、反応混合物を、以下の条件を使用する30サイクルおよび35サイクルのPCRに供した:94℃で1分、61.5℃で2分、および72℃で3分。PCR産物を、0.5μg/mlの臭化エチジウムを含む1.2%アガロースゲル上で可視化した。推定サイズ(CCR
6については1,021bpおよびCCR7については1,067bp)に移動した反応
産物をゲル精製し、色素ターミネーター技術を使用して、ABI 373A配列決定機(Applied Biosystems,Foster City,CA.)で配列決定確認のためにpCRII TAクローニングベクター(Invitrogen,Leek,The Netherlands)にサブクローニングした。2つの他のオリゴヌクレオチド:
【0062】
【化2】
を、1.2%アガロースゲル上で分離したPCR産物とのハイブリダイゼーションのため
のプローブとして使用し、Hybond N+膜(Amersham,Les Ulis,France)にブロットした。
【0063】
(カルシウム蛍光測定) 細胞内Ca2+濃度を、蛍光プローブIndo−1を使用して、Grynkiewiczら(J.Biol.Chem.,1985,260:3440−3450)により報告される技術に従って、測定した。簡潔には、細胞をPBS中で洗浄し、完全RPMI 1640培地中に107細胞/mlに再懸濁した(上記を参照のこと)。次いで、細胞を、遮光して、3μg/mlのIndo−1 AM(Molecular Probes)とともに室温にて45分間インキュベートした。インキュベーションの後、細胞を洗浄し、HBSS/1% FCS中で107細胞/mlに再懸濁した。細胞内Ca2+濃度の測定の前に、細胞を、予め39℃に温めたHBSS/10mM Hepes/1.6mM CaCl2中に10倍に希釈した。サンプルを、連続して攪拌しながら330nmで励起し、Indo−1蛍光を、810光電子倍増管検出システム(ソフトウェア:Felix,Photon Technology International,Monmouth Junction,NJ)にて、405nm(Ca2+と錯体化した色素)および485nm(Ca2+非含有培地)での時間の関数として測定した。結果を、2つの発光波長で得られた値の比率として表す。
【0064】
(インサイチュハイブリダイゼーション) インサイチュハイブリダイゼーションを、記載されたように行った(Peuchmaurら,1990,Am.J.Pathol.136:383−390)。2対のプライマーを、RT−PCRによりMIP−3α遺伝子(登録番号D86955)およびMIP−3β遺伝子(登録番号U77180)のオープンリーディングフレームの大部分を増幅するために使用した。
【0065】
【化3】
を、62℃のアニーリング温度で上記のように使用した。次いで、PCR産物を、適合したプロモーターとともにセンスプローブおよびアンチセンスプローブの生成のために、pCRII TAクローニングベクター(Invitrogen,Leek,The Netherlands)にクローニングした。MIP−3αおよびMIP−3βの35S標識したセンスプローブおよびアンチセンスプローブを、それぞれ、367bpフラグメントおよび435bpフラグメントの転写物を複製する(run off)ことにより得た。6μmのヒト扁桃の切片を、アセトンおよび4% パラホルムアルデヒド、続いて0.1M トリエタノールアミン/0.25%無水酢酸中で固定した。この切片を、一晩ハイ
ブリダイズし、RNAse Aで処理し、24日間曝した。展開させた後、切片を、ヘマトキシリンで染色した。
【0066】
(実施例1:CD34+由来DCの発生の間のMIP−3αおよびMIP−3βに対する差示的応答性)
DC移動(traffic)の調節を理解するために、成熟の異なる段階におけるDCの種々のケモカインに対する応答を研究した。DCを、GM−CSFおよびTNFαの存在下で培養したCD34+HPCから生成し、Boydenマイクロチャンバにおいて、ケモカインに応答して移動するそれらの能力について、異なる培養日数で試験した。MIP−3αおよびMIP−3βは、CD34+由来DCをMIP−1αまたはRANTESより2〜3倍に増加した。しかし、MIP−3αおよびMIP−3βにより誘引されたDCを、培養の異なる時点で回収した。MIP−3αに対する応答は、既に、4日目に検出され、5〜6日目に最大であり、10日目まで続いた。13〜14日目には、MIP−3αに対する応答は、通常、失われた。対照的に、MIP−3βに対応する応答は、10日目より前に検出され得ず、13日目にピークに達し得、15日目を超えて持続し得た。より早い時点では、培養中の細胞の大部分が、なおDC前駆体(CD1a−CD86−)であった場合、MIP−3αに対する応答は、1〜10ng/ml(実験に依存して)の濃度にて検出され得た。対照的に、4日後、ほとんど全ての細胞が未成熟DC(CD1a+CD86−)であった場合、300ng/ml以上が、細胞を誘引するために必要であり、このことは、成熟の間に細胞の連続的な脱感作を示唆する。比較的高濃度のMIP−3β(300ng/ml)もまた、成熟DC(CD1a+CD86+)を増大させるために必要であった。チェッカーボード分析により、MIP−3αおよびMIP−3βが走化性を誘導するが、DCのケモキネシスを誘導しないことが確立された。
【0067】
成熟の段階と、MIP−3αおよびMIP−3βに対する応答との間の関係を確認するために、CD34+由来DCを、CD86発現に従って、未成熟DC(CD1a+CD86−)および成熟DC(CD1a+CD86+)へと培養の10日目にFACSによりソートした。CD1a+CD86−は、MIP−3αに専ら応答したが、CD1a+CD86+は、主にMIP−3βに応答した。これらの観察により、MIP−3αおよびMIP−3βにより増加した細胞が、実際にDC(CD1a+)であったこともまた確認された。DC成熟とケモカイン応答性との間の相関は、DCの未成熟性が、6日目から12日目にTNFαを除去することにより維持され、それらの成熟性が、TNFα、LPSまたはCD40Lの添加により同期化された場合、さらに例示される。MIP−3αに対する応答は、TNFα、LPSおよびCD40Lを使用した48時間の成熟の際に強く減少された。同時に、MIP−3βに対する応答は、3つのシグナルすべてにより誘導され、CD40LおよびLPSは、TNFαより強力であった。動態学的実験において、MIP−3αに対する応答は、CD40活性化のわずか24時間後に50〜70%減少し、72時間目には、完全に失われた。MIP−3βに対する応答は、CD40活性化の24時間後に既に最大になり、比較的高濃度(48時間で100〜300ng/ml)のケモカインを要した。
【0068】
合わせて考えると、これらの結果により、未成熟CD34+由来DCがMIP−3αに応答し、一方で成熟DCがMIP−3βに応答することが確立される。
【0069】
(実施例2:MIP−3αおよびMIP−3βに対する応答は、CD34+由来DCに対するそれぞれのレセプターCCR6およびCCR7の発現と並行する)
MIP−3αおよびMIP−3βの応答性の調節の機構を明らかにするために、それらそれぞれのレセプターCCR6 mRNA(Powerら,1997,J.Exp.Med.186:825−835;Greavesら,1997,J.Exp.Med.186:837−844;Babaら,1997,J.Biol.Chem.272:14893−14898;Liaoら,1997,Biochem.Biophys.Res.Commun.236:212−217)およびCCR7 mRNA(Yoshidaら,199,J.Biol.Chem.272:13803−13809)の発現を、半定量的RT−PCRにより研究した。CD34+ HPCからのDC発生の間、CCR6 mRNAを、6日目に最初に検出し、10日目までに増大し、その後、減少したが、14日目には、わずかに検出可能であった。対照的に、CCR7 mRNAは、10日目に現れ、14日目まで確実に増大した。さらに、CD40L依存性成熟は、CCR6 mRNAの連続的な下方調節を誘導し(これは、72時間後にほとんど検出可能であった)、24時間程度の早さでCCR7 mRNAの上方調節を誘導した。同様な結果は、LPSまたはTNFαのいずれかによる誘導性DC成熟の後に得られた。活性化後のCCR7 mRNAの上方調節を、cDNAライブラリーのサザンブロット分析により確認した。
【0070】
移動アッセイならびにCCR6およびCCR7の発現の調節と調和して、MIP−3αは、休止/未成熟DCにおいて専らCa2+フラックスを誘導し、MIP−3βは、成熟
DCにおいてのみCa2+フラックスを誘導した。最大のCa2+フラックスは、30ng/mlのMIP−3αおよび30ng/mlのMIP−3βを用いて、それぞれ、未成熟DCおよび成熟DCに対して観察された。
【0071】
これらの結果は、MIP−3αおよびMIP−3βに対する応答性の変化が、CCR6
mRNAおよびCCR7 mRNAの発現の調節に関連することを示し、CCR6およびCCR7が、それぞれ、MIP−3αおよびMIP−3βについてのDC上で発現される主要な機能的レセプターであることを示唆する。
【0072】
(実施例3:MIP−3βに対する応答はまた、単球由来DCの成熟の際に誘導される)
GM−CSF+IL−4の存在下で単球を6日間培養することにより生成した単球由来DCは、代表的には、未成熟DCである(CD1a+、CD14−、CD80low、CD86low、CD83−)(Cellaら,1997,Current Opin.Immunol.9:10−16;Sallustoら,1994,J.Exp.Med.179:1109−1118)。それらは、MIP−1αおよびRANTESに応答して移動したが、MIP−3αに対しても、MIP−3βに対しても応答せず、移動しなかった。単球由来DCのMIP−3αに対する応答の欠如は、それらの細胞上でのCCR6発現の非存在に従う(Powerら,1997,J.Exp.Med.186:825−835;Greavesら,1997,J.Exp.Med.186:837−844)。TNFα、LPS、またはCD40Lにより誘導される成熟に際して、MIP−1αおよびRANTESに対する応答は失われたが、MIP−3βに対する応答が誘導された。CD34+由来DCを使用する場合と同様に、MIP−3βに対する応答は、TNFα、LPSまたはCD40Lにより誘導された成熟に際して観察されたCCR7 mRNA発現の上方調節と相関した。繰り返すと、TNFRまたはCD40シグナル伝達の後にCCR7の上方調節は、初期の時点(3h)で生じた。さらに、移動およびケモカインレセプター発現のデータは、Ca2+フラックス結果と一致した。
【0073】
これらの結果により、成熟の際に、DCが種々のケモカインに対するそれらの応答性を失うが、1つのケモカインMIP−3βには感受性になるという概念は、単球由来DCに及ぶ。
【0074】
(実施例4:末梢血CD11c+DCは、成熟後にMIP−3βに応答して移動する)末梢血(または扁桃)から単離された未成熟CD11c+DCに対するMIP−3αおよびMIP−3βの走化性活性もまた研究した。新たに単離したDCは、MIP−3αに応答して移動しなかったが、MIP−3βに応答して移動し、これらの細胞におけるCCR6の非存在およびCCR7 mRNA発現との相関が観察された。しかし、GM−CSFとの一晩の培養の後に生じることが公知の成熟は、MIP−3βに対するCD11c+DCの応答に向けられるが、MIP−3αに対する応答には向けられなかった。再び、MIP−3βに対する応答は、CCR7 mRNA発現の誘導と相関した。
【0075】
従って、たとえ血液から新たに単離された未成熟CD11c+DCが、MIP−3αに応答できないとしても、これらの結果は、MIP−3βに対する応答性に依存した成熟がまた、エキソビボで単離されたDCに適用されることを示す。
【0076】
(実施例5:インビボでMIP−3αは、炎症を生じた上皮において発現され、MIP−3βは、扁桃のT細胞富化領域内で発現される)
実施例4において報告された知見の物理的関連性を、MIP−3αの分析および炎症を生じた扁桃の切片でのインサイチュハイブリダイゼーションによるMIP−3β mRNA発現を通じて取り組んだ。MIP−3αのmRNAは、炎症を生じた上皮陰窩において高レベルで検出されたが、T細胞富化領域においても、B細胞濾胞においても検出されなかった。実際に、MIP−3α発現は、上皮陰窩を覆う細胞に制限されていた。対照的に、MIP−3β mRNAの発現は、T細胞富化領域に制限されていた。最も強いシグナルは、散在した細胞において存在し、分布は、IDCの分布と重なっていた。副皮質(paracortical)領域の外側では、シグナルは、B細胞濾胞においても、上皮陰窩においても検出されなかった。連続切片は、MIP−3αが豊富に発現される上皮陰窩内のMIP−3β発現の明らかな不在を示した。MIP−3αおよびMIP−3βのセンスプローブは、バックグラウンドハイブリダイゼーションを生成しなかった。
【0077】
従って、MIP−3α発現は、未成熟DCが補充されるべき抗原進入の部位において炎症を生じた上皮に制限される。対照的に、MIP−3βは、副皮質領域において検出されるのみであり、ここで成熟IDCは、ホーミングし、第1のT細胞応答を生成する。
【0078】
(実施例6:マウスモデルにおけるインビボでのケモカインMIP−3α投与)
MIP−3αは、本発明者らがインビトロでマウス未成熟樹状細胞についての走化性因子であることを示し、ケモカインMIP−3αがインビボで未成熟DCを誘引し、インビボで腫瘍に対する抗原特異的免疫応答を調節する能力を研究した。腫瘍関連抗原が同じ時間に送達される場合、より多くのDCが抗原を捕捉するために利用可能である。従って、この抗原に対する抗原特異的応答が増加されるはずである。
【0079】
ケモカインはプラスミドベクター(pcDNA3,InVitrogen)を介してインビボで送達され、このベクターは、CMVプロモーター(PMIP−3α)の制御下でマウスMIP−3αをコードするcDNAを含む。使用した抗原は、E.coliから単離したβ−ガラクトシダーゼであった。この抗原を、同じプラスミドベクターpcDNA3(pLaczといわれる)を介してインビボで送達した。腫瘍は、β-ガラクトシダー
ゼをコードする遺伝子で安定にトランスフェクトされた同系BALB/cマウスにおけるC26結腸癌であった。従って、この系において、β−ガラクトシダーゼが、腫瘍関連抗原を規定する。
【0080】
6匹の6週齡の雌性マウスの群を、空のpcDNA3プラスミド(ネガティブコントロール)、抗原単独をコードするプラスミドpLacz、またはpLaczおよびPMIP−3αの混合物のいずれかを注射した。注射(50μgの総プラスミド)を、4週間の間毎週、後足の踵に行った。その後、マウスにβ−ガラクトシダーゼを発現するC26腫瘍細胞株を皮下注射した。代表的には、全てのマウスは、10日後に、皮下に腫瘍を発生させた。これらのマウスの群における腫瘍の出現をモニターした。腫瘍の出現は、pLaczおよびpLacz+PMIP−αの注射の後に遅延した(図1)。このことは、腫瘍関連抗原をコードするプラスミドでの免疫が、腫瘍移植に対する保護効果を有することを示す。この遅延は、pLaczを用いるより、pLacz+PMIP−3αを用いるほうが大きかった。このことは、ケモカインMIP−3αが抗原をとともに送達された場合、腫瘍関連抗原特異的免疫応答を増大させることを示唆する。
【0081】
優れた抗腫瘍応答は、強いT細胞媒介性抗原特異的細胞傷害性(CTL活性)と関連すると考えられる。従って、同じ群のマウスにおけるCTL活性を、腫瘍接種して30日後に分析した。脾細胞を取り出し、インターロイキン−2の存在下で、照射した同系DCおよびβ−ガラクトシダーゼ由来の免疫優性なCTLペプチドを使用して5日間刺激した。次いで、β−ガラクトシダーゼをコードする遺伝子を安定にトランスフェクトした細胞株(P13.1)を溶解するそれらの能力を、β−ガラクトシダーゼを発現しない親細胞株P815を溶解するそれらの能力と並行して、測定した(図2)。これを、異なる比のエフェクター(脾細胞)対標的(P13.1またはP815)を使用して行った。この結果は、腫瘍チャレンジの前にpLacz+P−MIP−3αを注射したマウスは、pLaczまたはPCDNA3単独でのみ注射したマウスより、腫瘍関連抗原β−ガラクトシダーゼに対して大きなCTL活性を有する。
【0082】
(実施例7:インビボマウスモデルにおけるケモカインhMCP−4投与)
本発明者らは、hMCP−4局所的注射が、用量依存性様式でマウスにおいてインビボでの樹状細胞の補充を促進し得ることを示した(図4)。
【0083】
6〜10週齡の雌性BALB/cマウスを、Charles River(Iffa−Credo,L’Arbresle,France)から購入し、標準的な条件下で本発明者らの施設にて維持した。動物およびそれらの世話に関する手順は、EEC(欧州経済共同体)会議の指示(86/609,OJL 358,1,1987年12月12日)に合わせて行った。組換えヒトMCP−4タンパク質(>97%純度(図3))を、Peprotechから得、PBS(Gibco−BRL)中に再懸濁した。3匹のマウスの群
に、PBS単独またはPBS中の種々の量のヒトMCP−4を、50μ容量で右後足踵に皮内注射した。マウスを2〜20時間後に屠殺し、注射部位の皮膚および注射部位から排出する膝窩リンパ節を取り出した。皮膚における局所的細胞補充を、標準的な技術に従って、特異的モノクローナル抗体を使用して、免疫組織化学により試験した。細胞懸濁液を、RPMI 1640および10%ウシ胎仔血清(FCS)(Gibco−BRL)中にリンパ節から調製した。細胞を計数し、標準的な技術に従って、PBSおよび2% FCS中で、ビオチン−CD11c抗体およびFITC−CD11b抗体(Becton Dickinson)、続いて、PE−ストレプトアビジン(Dako)を含有するPBSおよび2% FCS中で染色した。CD11bおよびCD11cの発現(これは、マウス樹状細胞の集団を規定する)を、Facscanフローサイトメーター(Becton Dickinson)で、CellQuestソフトウェアを使用して、分析した。この分析および計数から、各リンパ節におけるCD11b+CD11c+の数を決定した。これらの実験は、以下を示す:(A)hMCP−4の局所的注射が、短期間(2時間)の後に、注射部位におけるCD11bを発現する細胞の補充を誘導し得ること。これらの細胞は、マウス血液樹状細胞または樹状細胞前駆体(例えば、単球)であり得る。なぜなら、両方とも、CD11bを発現し得るからである。マウスにおいて、循環する血液樹状細胞は、技術的な制限に起因して、同定されなかった。しかし、ヒトにおいては、血液樹状細胞が単離され得、それらは、hMCP−4に対する走化性アッセイに応答する(図3)。(B)排出リンパ節において、抗原特異的免疫応答が開始される場合、hMCP−4は、CD11bおよびCD11cの同時発現により同定される樹状細胞の補充を誘導するが、長期間(20時間)の後のみである。この遅延は、排出リンパ節に移動するために、皮膚において最初に補充される樹状細胞またはそれらの前駆体に必要な成熟および移動時間に対応する可能性が最も高い。
【0084】
(実施例8:ヒト血液由来の樹状細胞のhMCP−4に対する応答)
hMCP−4はまた、ヒト樹状細胞(血液から単離された樹状細胞を含む)に対して活性である(図5)。
【0085】
パネルA:ヒト循環血液CD11c+DCを磁性ビーズ除去により富化し、種々のケモカインに応答するトランスウェル(5μm孔サイズ)移動アッセイにおいて研究した。この移動を、2時間後に三重染色:系列マーカーFITC、HLA−DR tricolorおよびCD11c PEにより明らかにし、Facsにより染色した。各ケモカインを広範囲の濃度(1〜1000ng/ml)に対して試験し、最適応答のみを示す。結果を、移動指数として表し、3〜10の独立した実験から得た平均値を示す。血液CD11c+は主にMCP−4ならびにMCP1、MCP2およびMCP3に対して応答する(示さず)。SDF−1は選択性を欠き、CD11c+DCに対して強く活性である唯一の他のケモカインである。
【0086】
パネルB:異なるヒトDCおよびDC前駆体の集団(血液CD11c+DC、単球、単球由来DC、CD1a+ランゲルハンス細胞前駆体およびCD14+間質DC前駆体を含む)を、MCP−1およびMCP−4に応答するトランスウェル(5μm孔サイズ)移動アッセイにおいて研究した。全ての集団は、CD1a+ランゲルハンス細胞前駆体を除いて、MCP−4に応答する。さらに、単球由来DCは、CCR2とは異なるレセプターを介して、MCP−4に応答するが、MCP−1には応答しない。
【0087】
重要なことには、MCP−4は、ヒトDCに対して活性である。特に、他のケモカインと比較すると、MCP−4は、循環血液CD11c+DCの移動を誘導する最も強力なケモカインである。MCP−1、およびMCP−2およびMCP−3は、血液DCに対して類似の活性を示す。MCPは、これらの細胞上で発現されるCCR2を介して血液DCを補充するようである。さらに、MCP−4は、CCR2を発現しないCD1a+ランゲルハンス細胞前駆体を除いて、全てのDCまたはDC前駆体の集団(血液CD11c+DC、単球、単球由来DC、CD14+間質型DC前駆体)に対して活性である。最後に、MCP−4(MCP−1ではない)は、おそらく、CCR2とは異なるレセプターを介して、単球由来DCの移動を誘導する。
【0088】
(実施例9:インビボマウスモデルにおけるhMCP−4およびβ−ガラクトシダーゼの投与)
さらに、hMCP−4を、プラスミドDNAワクチン接種により誘導される抗原特異的免疫応答のアジュバントとして使用し得る。さらに、hMCP−4がプラスミドDNAワクチン接種のアジュバントとして使用される場合、これは、プラスミドDNAによりコードされた抗原を発現する腫瘍でその後チャレンジしたマウスの防御を増大し得る。
【0089】
6〜8週齡の7匹の雌性BALB/cマウス(Iffa−Credo,L’Arbresle,France)の群に、PBS単独またはPBS中の100ngのヒトMCP−4を、50μlの容量で、右後足踵の皮下に注射した。3時間後、マウスの同じ部位に、50μl容量のPBSの下で、コントロールpcDNA3プラスミド(InVitrogen)50μgまたはCMVプロモーター下のβ−ガラクトシダーゼをコードするpcDNA3プラスミド(pLacz,InVitrogen)50μgを注射した。この免疫プロトコルを、1週間間隔で4回繰り返した。
【0090】
最初の免疫の1日前、および最後の免疫の1週間後に血清を回収した。血清中のβ−ガラクトシダーゼ特異的免疫グロブリンのレベルを、以前に記載のように(Mendozaら,1997,J.Immunol.159:5777−5781)、特異的ELISAアッセイを使用して測定した。
【0091】
図6に見られるように、MCP−4注射は、β−ガラクトシダーゼDNA免疫(右後足
踵に100ng hMCP−4注射して3時間後の50μg DNA注射)の後に抗原特
異的体液性応答を増大する。図6は、4回の免疫の後に測定した抗β−ガラクトシダーゼ抗体を示す[PBS+pLaczと比較した場合のhMCP−4+pLaczの有意性:ステューデント検定]。
【0092】
最後に免疫して1週間後、マウスの群に、100μl容量のRPMI−1640の下で、β−ガラクトシダーゼを発現する5×104のC26−BAG結腸癌細胞(親切にも、Mario Colombo,Instituto Nazionale Tumori,Milan,Italyから譲り受けた)を右脇腹に皮下注射してチャレンジした。腫瘍の発生を、触知により1週間3回評価した。
【0093】
図7に見られるように、MCP−4注射は、マウスを、β−ガラクトシダーゼを発現するC26結腸癌細胞株でチャレンジした場合に、β−ガラクトシダーゼDNA免疫(右後
足踵に100ngのhMCP−4を注射して3時間後の50μgのDNA注射、腫瘍チャレンジする前に4回の免疫)により誘導された抗腫瘍効果を増大する[PBS+pLac
zと比較した場合のhMCP−4+pLaczの有意性:p<0.05、logrank
MCP−4対抗(opp):異なる部位に注射したhMCP−4]。
【0094】
従って、実施例7〜9は、ケモカインhMCP−4が、免疫応答、特に抗腫瘍応答のアジュバントとして使用され得ることを示す。MCP−4投与により媒介された増強された免疫応答を、血清中の抗原特異的免疫グロブリンレベルの増強として測定した。従って、MCP−4投与に対するB細胞応答の増強が明らかに存在する。さらに、切り替えにT細胞媒介補助を要する免疫グロブリンのサブクラス(例えば、IgG2a)が増大するので、T細胞媒介性応答もまた増大するようである。
【0095】
(実施例10:ヒト樹状細胞のh6Ckineケモカインに対する応答)
この実施例において、本発明者らは、ヒト6Ckine(h6Ckine)が、インビトロでのヒトの樹状細胞の公知のサブセット全てについての走化性因子であることを示した。特に、h6Ckineは、GM−CSF、IL−3およびCD40Lとの3時間という短時間のインキュベーション後のヒト血液樹状細胞に対して活性である(図8)。
【0096】
異なるヒトDC集団(CD1a+ランゲルハンス細胞、CD14+間質DC、単球由来DC、循環血液CD11c+DC、単球、および循環血液CD11c−プラズマ細胞様DCを含む)を、成熟の前および後に、ヒト6Ckineに対して応答する移動アッセイにおいて研究した。CD34−由来DC前駆体を、GM−CSF+TNFおよびSCFの存在下で6日間培養した後に、CD1a発現およびCD14発現に従ってFacsソーティングにより単離した。細胞を、GM−CSF単独(未成熟)中で12日目まで、またはGM−CSF+CD40−L(成熟)中で最後の2日間培養した。単球由来DCを、単球をGM−CSF+IL−4の存在下で5日間培養することにより生成し、CD40−Lを使用して、最後の2日間、活性化させた(成熟)かまたは活性化させなかった(未成熟)。ヒト循環血液CD11c+DCおよびCD11c−プラズマ細胞様DCを、磁性ビーズの除去により富化し、三重染色を使用して、系統マーカーFITCネガティブ、HLA−DR tricolorポジティブ、ならびにCD11c PEポジティブおよびCD11c PEネガティブにfacsソートした。CD11c+DCおよびCD11c−プラズマ細胞様DCを、GM−CSF+IL−3の存在下で、CD40Lとともに(成熟)またはCD40Lなしで(未成熟)、3時間培養した。
【0097】
移動アッセイを、5μm孔サイズまたは8μm孔サイズのTranswell(6.5mm直径,COSTAR,Cambridge,MA)を使用して、1〜3時間の間行い、facs分析により明らかにした。全ての集団が、CD40−L活性化後にのみ6Ckineに応答した。
【0098】
(実施例11:腫瘍モデルにおけるケモカインm6Ckine遺伝子移入)
この実施例において、本発明者らは、以下を示した:
m6Ckineを発現するように操作されたC26結腸癌腫瘍細胞は、あまり腫瘍形成性ではなく、この効果は、インビボでのCD8+細胞およびナチュラルキラー細胞活性に依存する(図9);
m6Ckineを発現するC26腫瘍は、親腫瘍と比較して、樹状細胞およびCD8+T細胞により有意に浸潤される(図10);および
m6Ckineを発現するように操作されたC26結腸癌腫瘍細胞は、親C26腫瘍よりあまり抗原性ではない(図11)。
【0099】
6〜10週齡の雌性BALB/c(H−2d)マウスを、Charles River(Iffa−Credo,L’Arbresle,France)から購入し、標準的な条件下で維持した。動物およびそれらの世話に関する手順は、EEC(欧州経済共同体)会議の指示(86/609,OJL 358,1,1987年12月12日)に合わせて行った。全ての腫瘍細胞培養を、10% FCS(Gibco−BRL)、1mM hepes(Gibco−BRL)、Gentallin(Schering−Plough,Union,NJ)、2×10−5M β−2メルカプトエタノール(Sigma,St Louis,MO)を補充したDMEM(Gibco−BRL, Life Technologies,Paisley Park,Scotland)中で行った。全ての細胞培養を、37℃にて5% CO2の加湿インキュベーター中で行った。マウス6Ckine/SLCをコードするcDNA(m6Ckine/SLC)を、CMVプロモーターを含むpcDNA3ベクター(InVitrogen,Carlsbad,CA)にクローニングした。C26結腸癌腫瘍細胞(親切にも、Mario P.Colombo,Instituto Nazionale per lo Studio e la Cura dei Tumori, Milano,Italyから譲り受けた)を、製造業者の指示に従って、Fugene試薬(Roche Molecular Diagnostics,Mannheim,Germany)を使用して、この構築物を用いてトランスフェクトした。m6Ckine/SLC mRNA(C26−6CK)を発現する単一のC26クローンを、800μg/mlでのネオマイシン(Sigma)選択の後に得た。C26腫瘍細胞またはC26−6CK腫瘍細胞を、100μlのDMEM中で右脇腹にS.C.注射し、腫瘍増殖を、1週間に3回触知することによりモニターした。抗体除去のために、腫瘍接種の1日前に、200μlのPBS中の0.5mgの抗CD8(クローン2.43)精製抗体、ラットコントロール(GL113)精製抗体または200μlのウサギ抗アシアロGM1血清(Wako Pure Chemicals,Osaka,Japan)を、i.p.注射し、次いで、0.2mgの抗体または100μlの抗アシアロGM1血清を3日後に、および実験過程の間に1週間に1回注射した。図10は、皮下C26−6CK細胞注射が、親腫瘍細胞と比較して、logrank分析により有意に遅延した腫瘍取り込み(intake)を生じる(p<0.01)ことを示す(A
およびB:C26+コントロール 対 C26−6CK+コントロール)。インビボで特
定の抗体を用いたCD8+細胞(A)またはナチュラルキラー細胞活性(B)の除去は、C26−6CK腫瘍細胞の遅延した腫瘍形成性を部分的に戻す。このことは、CD8+細胞およびNK細胞は、腫瘍増殖の遅延において役割を果たすことを示す。
【0100】
腫瘍を、約1cmのサイズに達したときに外科手術により取り出した。腫瘍塊を、小さな断片に細かく刻み、コラゲナーゼA(Roche Molecular Biochemicals)溶液中で37℃にて30分間攪拌しながらインキュベートした。次いで、懸濁液を、数回DMEM中で洗浄した。細胞懸濁液の染色を、PBSおよび5% FCS中で行った。FITC標識特異的抗体、ビオチン標識特異的抗体またはPE標識特異的抗体とのインキュベーションの前に、Fcレセプターを、FcブロックTM CD16/CD32抗体(PharMingen,San Diego,CA)を使用してブロックした。この研究において使用した種々の抗体(全て、PharMingen製)は、CD8β(53−5.8)、CD11c(HL3)、抗MHCクラスII I−Ad/I−Ed(269)、CD3(145−2C11)であった。ビオチン化抗体を、PEストレプトアビジン(Becton Dickinson)を使用して明らかにした。表現型パラメーターを、FacScan(Becton Dickinson,Mountain View,CA)で獲得し、CellQuestソフトウェア(Becton Dickinson)を使用して分析した。図10において、C26野生型腫瘍またはm6Ckineを発現するC26−6CK腫瘍を、腫瘍懸濁液全体のフローサイトメトリー分析によりCD8 T細胞およびCD11c+MHCクラスII+樹状細胞(DC)の浸潤について分析した(n=7)。データは、C26腫瘍と比較して、両方のC26−6CK腫瘍中の白血球サブセットの有意な補充を示す(ステューデントのt検定)。これらの結果は、腫瘍へのm6Ckine遺伝子移入は、免疫応答(抗腫瘍応答を含む)を開始するに必須の細胞である樹状細胞、および適応性免疫応答のエフェクター細胞であるCD8 T細胞の補充の両方を促進することを示唆する。
【0101】
いくつかの実験において、動物から腫瘍を取り出し、OCT化合物(Miles laboratory,Elkhart,IN)中に包埋して液体窒素中で急速冷凍し、免疫組織化学手順の前に−80℃で保存した。スライドガラスに適用した5μmの低温保持切片を、アセトン中で固定し、室温にて10分間、1% H2O2とともにインキュベートした。次いで、スライドを、ビオチンブロックTM試薬およびアビジンブロックTM試薬(ともに、Vector,Burlingame,CA)とともにインキュベートした。全てのインキュベーションを、PBS(Gibco−BRL)中の3回の2分間洗浄の後に行った。次いで、スライドを、二次抗体(Dako,Glostrup,Denmark)の同じ種に由来する血清の1/10希釈物とともに30分間プレインキュベートした。次いで、スライドを、5μg/mlの精製CD105(クローンMJ7/18,PharMingen,San Diego,CA)、ビオチン化二次抗体(Vectorのウ
サギ抗ラット)、ストレプトアビジン−アルカリ(VectorのABCキット)と連続
してインキュベートした。酵素反応物を、対応するVector基質を使用して発色させた。脈管形成アッセイを、インビボで発生中の腫瘍細胞を含むMatrigel(Becton Dickinson,Bedford,MA)ペレットのヘモグロビン含有量を決定することにより行った。BALB/cマウスを、2×105のC26細胞またはC26−6CK細胞と混合した0.5mlのMatrigelを、腹部正中線にs.c.注射した。9日後、Matrigelペレットを取り出し、周りの結合組織を解剖して除去し、ペレットを、90分間4℃にてMatriSperse溶液 v/v(Becton Dickinson)中で溶かした。ヘモグロビン含有量を、Drabkin法(Sigmaの試薬)により決定した。図11Aは、C26−6CK腫瘍は、親C26腫瘍よりあまり血管を形成しないことを示す。図11Bは、Matrigelアッセイにおいて、C26−6CK腫瘍細胞がC26細胞よりあまり脈管を形成しないことを示す。全体的に、これらの結果は、m6Ckineケモカインの腫瘍への遺伝子移入が、腫瘍血管に対して血管静止的(angiostatic)効果を有することを示す。
【0102】
これらの結果は、ケモカイン6Ckineが、遺伝子移入を介して癌の処置に使用され得ることを示す。好ましい実施形態は、以下からなるが、これらに制限されない:m6Ckineもしくはh6Ckineまたはm6Ckineもしくはh6Ckineの画分をコードするDNAまたはウイルスベクター(例えば、アデノウイルス)(標的化部分(ペプチドまたは抗体)を伴うかまたは伴わない)。
【0103】
(実施例12:インビボでのケモカイン6Ckineの腫瘍への局所的送達)
この実施例において、本発明者らは、組換えヒトまたは組換えマウス6Ckineタンパク質の既に存在するC26腫瘍への注射が、腫瘍保有マウスの生存を増すことを示した。h6Ckineの注射は、腫瘍増殖を遅延させる(図12)。
【0104】
6〜10週齡の雌性BALB/c(H−2d)マウスを、Charles River(Iffa−Credo,L’Arbresle,France)から購入し、標準的な条件下で維持した。動物およびそれらの世話に関する手順は、EEC(欧州経済共同体)会議の指示(86/609,OJL 358,1,1987年12月12日)に合わせて行った。全ての腫瘍細胞培養を、10% FCS(Gibco−BRL)、1mM hepes(Gibco−BRL)、Gentallin(Schering−Plough,Union,NJ)、2×10−5M β−2メルカプトエタノール(Sigma,St Louis,MO)を補充したDMEM(Gibco−BRL,Life Technologies,Paisley Park,Scotland)中で行った。全ての細胞培養を、37℃にて5% CO2の加湿インキュベーター中で行った。C26細胞を、Mario P.Colombo(Milano, Italy)から譲り受けた。
【0105】
C26腫瘍細胞を、100μlのDMEMにて右脇腹にs.c.注射し、腫瘍増殖を、1週間に3回触知することによりモニターした。いくつかの実験において、腫瘍容積を、カリパスを使用してモニターし、以下のように予測した:腫瘍容積=小さな直径2×大きな直径×0.4。組換えケモカインを用いた処置に関しては、マウスに、50μlのPBS下で10ngの>97%純度の組換えヒトまたはマウス6Ckine/SLC(R&D
Systems,Minneapolis,MN)を腫瘍内注射した。図1は、h6Ckineまたはm6Ckineを使用して注射したマウスが、PBSビヒクル単独と比較した場合に、生存の改善を示すことを示す(A)。h6Ckineの注射はまた、腫瘍の増殖を低減させた(B)。これらのデータは、組換え6Ckineケモカインの腫瘍内送達が抗腫瘍効果を有することを示す。
【0106】
(実施例13:rAd/6Ckineの転移性腫瘍の緩和)
雌性マウス(BALB/c ByJ;Jackson Laboratories)に、0.2ml(培地)の容量中の3×1015 4T1−p53乳腺腫瘍細胞(同系)を使用して、動物の左脇腹に皮下経路にて注射した。腫瘍が50〜100mm3のサイズに成長したときに、動物に、VPBS中の100μlのCMCB(1e10 PN/注射)を腫瘍内注射した。マウスに1週間あたり3回(月曜日、水曜日、金曜日)2週間にわたり注射した。腫瘍を、カリパスを使用して1週間に3回測定(長さ、幅、深さ)し、腫瘍容積を、以下の式に従って計算した:V=4/3r3
ここで、r=(W(mm)+L(mm)+D(mm))/6
動物を、腫瘍が1000mm3を超えた場合に屠殺した。
【0107】
各群から3匹のマウスを屠殺し、腫瘍が50mm3に達したときに(代表的には、10日目)開始して、腫瘍および肺を、以下に記載した生化学的分析のために組織処理のために切除し、肉眼および組織化学的手段により転移の存在を評価した。
【0108】
図13に示されるように、6Ckineは、免疫を増大し、脈管形成を抑制することにより、確立された腫瘍において腫瘍増殖を阻害し、自発的な転移を阻害する。
【0109】
本発明の多くの改変およびバリエーションが本発明の趣旨および範囲から逸脱することなく行われ得、当業者にも明らかである。本明細書中に記載される特定の実施形態は、例示により提供されるに過ぎず、添付の特許請求の範囲によってのみ、このような特許請求の範囲に与えられる等価物の全範囲とともに限定されるべきである。
【図面の簡単な説明】
【0110】
【図1】図1は、MIP−3αおよび腫瘍関連抗原を含むプラスミドを用いた免疫が、腫瘍移植に対する防御効果を有することを示す。
【図2】図2は、ケモカインMIP−3αの投与に伴うより大きなCTL活性を示す。
【図3】図3は、ケモカインhMCP−4のヌクレオチド配列および部分的アミノ酸配列を示す。
【図4】図4は、hMCP−4注射が、マウスにおいて用量依存性様式でインビボで樹状細胞の補充を促進することを示す。
【図5】図5は、hMCP−4がヒト血液における樹状細胞の補充において活性であることを示す。
【図6】図6は、MCP−4注射が、β−ガラクトシダーゼDNA免疫の後に抗原特異的体液性応答を増大させることを示す。
【図7】図7は、マウスがβ−ガラクトシダーゼを発現するC26結腸癌細胞株でチャレンジされたときに、MCP−4が、β−ガラクトシダーゼDNA免疫により誘導される抗腫瘍効果を増大させることを示す。
【図8】図8は、h6Ckineがヒト血液における樹状細胞の補充において活性であることを示す。
【図9】図9は、m6Ckineを発現するように操作されたC26結腸癌腫瘍細胞が、あまり腫瘍形成性ではなく、この効果が、インビボにおいてCD8+細胞およびナチュラルキラー細胞活性に依存することを示す。
【図10】図10は、m6Ckineを発現するC26腫瘍が、親腫瘍と比較した場合に、樹状細胞およびCD8+T細胞により有意に浸潤されることを示す。
【図11】図11は、m6Ckineを発現するように操作されたC26結腸癌腫瘍細胞が、親C26腫瘍よりも抗原性でないことを示す。
【図12】図12は、h6Ckineの注射が、インビボにおいてマウスの腫瘍増殖を遅延させることを示す。
【図13】図13は、6Ckineが、インビボにおいて確立された腫瘍の腫瘍増殖および自発的な転移を阻害することを示す。
【特許請求の範囲】
【請求項1】
疾患状態の処置のための医薬品の製造における、樹状細胞の移動を方向付け得るケモカインの使用であって、前記ケモカインが、MCP−1、MCP−2、MCP−3、MCP−4、MIP−1α、MIP−1β、MIP−3α、RANTES、SDF−1、Teck、DCtactin−β、6Ckine/SLC、LEC、MDC、およびMIP−5からなる群より選択される、使用。
【請求項2】
請求項1に記載の使用であって、前記樹状細胞が、未成熟樹状細胞である、使用。
【請求項3】
請求項2に記載の使用であって、前記ケモカインが、MCP−1、MCP−2、MCP−3、MCP−4、MIP−1B、MDC、MIP−3α、MIP−1α、RANTESおよびMIP−5からなる群より選択される、使用。
【請求項4】
請求項1に記載の使用であって、前記ケモカインが、MIP−3βである、使用。
【請求項5】
哺乳動物における免疫応答を増強する方法であって、該方法は、ケモカインMCP−4またはケモカインMCP−4の生物学的に活性な画分を該哺乳動物に投与する工程を包含する、方法。
【請求項6】
請求項5に記載の方法であって、前記ケモカインが、組換えケモカインである、方法。
【請求項7】
請求項5に記載の方法であって、前記ケモカインがヒトケモカインである、方法。
【請求項8】
請求項5に記載の方法であって、該方法は、前記ケモカインの徐放を可能にする物質を送達部位に投与する工程をさらに包含する、方法。
【請求項9】
請求項5に記載の方法であって、該方法は、前記ケモカインとともに抗原を投与する工程をさらに包含する、方法。
【請求項10】
請求項9に記載の方法であって、MCP−4および抗原を含む融合タンパク質が、前記哺乳動物に投与される、方法。
【請求項11】
請求項9に記載の方法であって、前記抗原が、腫瘍関連抗原である、方法。
【請求項12】
請求項10に記載の方法であって、前記抗原が、腫瘍関連抗原である、方法。
【請求項13】
請求項9に記載の方法であって、前記抗原が、細菌抗原、ウイルス抗原、または真菌抗原である、方法。
【請求項14】
請求項10に記載の方法であって、前記抗原が、細菌抗原、ウイルス抗原または真菌抗原である、方法。
【請求項15】
請求項9に記載の方法であって、前記腫瘍関連抗原が、Melan−A、チロシナーゼ、p97、β−HCG、GalNAc、MAGE−1、MAGE−2、MAGE−3、MAGE−4、MAGE−12、MART−1、MUC1、MUC2、MUC3、MUC4、MUC18、CEA、DDC、黒色腫抗原gp75、Hker8、高分子量黒色腫抗原、K19、Tyr1およびTyr2、pMel 17遺伝子ファミリーのメンバー、c−Met、PSA、PSM、α−フェトプロテイン、甲状腺ペルオキシダーゼ、gp100、p53ならびにテロメラーゼからなる群より選択される、方法。
【請求項16】
請求項10に記載の方法であって、前記腫瘍関連抗原が、Melan−A、チロシナーゼ、p97、β−HCG、GalNAc、MAGE−1、MAGE−2、MAGE−3、MAGE−4、MAGE−12、MART−1、MUC1、MUC2、MUC3、MUC4、MUC18、CEA、DDC、黒色腫抗原gp75、Hker8、高分子量黒色腫抗原、K19、Tyr1およびTyr2、pMel 17遺伝子ファミリーのメンバー、c−Met、PSA、PSM、α−フェトプロテイン、甲状腺ペルオキシダーゼ、gp100、p53ならびにテロメラーゼからなる群より選択される、方法。
【請求項17】
請求項9に記載の方法であって、GM−CSFおよびIL−4の組み合わせを投与する工程をさらに包含する、方法。
【請求項18】
請求項10に記載の方法であって、GM−CSFおよびIL−4の組み合わせを投与する工程をさらに包含する、方法。
【請求項19】
請求項5に記載の方法であって、前記ケモカインとともに活性化薬剤を投与する工程をさらに包含する、方法。
【請求項20】
請求項5に記載の方法であって、前記ケモカインが、皮内投与、筋肉内投与、皮下投与、局所投与、またはベクターの形態で投与される、方法。
【請求項21】
MCP−4および抗原を含む、融合タンパク質。
【請求項22】
請求項21に記載の融合タンパク質であって、前記抗原が腫瘍関連抗原である、融合タンパク質。
【請求項23】
請求項22に記載の融合タンパク質であって、前記腫瘍関連抗原が、Melan−A、チロシナーゼ、p97、β−HCG、GalNAc、MAGE−1、MAGE−2、MAGE−3、MAGE−4、MAGE−12、MART−1、MUC1、MUC2、MUC3、MUC4、MUC18、CEA、DDC、黒色腫抗原gp75、Hker8、高分子量黒色腫抗原、K19、Tyr1およびTyr2、pMel 17遺伝子ファミリーのメンバー、c−Met、PSA、PSM、α−フェトプロテイン、甲状腺ペルオキシダーゼ、gp100、p53ならびにテロメラーゼからなる群より選択される、融合タンパク質。
【請求項24】
請求項22に記載の融合タンパク質であって、前記抗原が、細菌抗原、ウイルス抗原または真菌抗原である、融合タンパク質。
【請求項25】
請求項21に記載の融合タンパク質を含む、プラスミド。
【請求項26】
請求項23に記載のプラスミドであって、樹状細胞に特に適したプロモーター配列をさらに含む、プラスミド。
【請求項27】
請求項21に記載の融合タンパク質を含む、ウイルスベクター。
【請求項28】
6Ckineおよび抗原を含む、融合タンパク質。
【請求項29】
請求項28に記載の融合タンパク質であって、前記抗原が腫瘍関連抗原である、融合タンパク質。
【請求項30】
請求項29に記載の融合タンパク質であって、前記腫瘍関連抗原が、Melan−A、チロシナーゼ、p97、β−HCG、GalNAc、MAGE−1、MAGE−2、MAGE−3、MAGE−4、MAGE−12、MART−1、MUC1、MUC2、MUC3、MUC4、MUC18、CEA、DDC、黒色腫抗原gp75、Hker8、高分子量黒色腫抗原、K19、Tyr1およびTyr2、pMel 17遺伝子ファミリーのメンバー、c−Met、PSA、PSM、α−フェトプロテイン、甲状腺ペルオキシダーゼ、gp100、p53ならびにテロメラーゼからなる群より選択される、融合タンパク質。
【請求項31】
請求項29に記載の融合タンパク質であって、前記抗原が、細菌抗原、ウイルス抗原または真菌抗原である、融合タンパク質。
【請求項32】
請求項28に記載の融合タンパク質を含む、プラスミド。
【請求項33】
請求項32に記載のプラスミドであって、樹状細胞に特に適したプロモーター配列をさらに含む、プラスミド。
【請求項34】
請求項28に記載の融合タンパク質を含む、ウイルスベクター。
【請求項35】
明細書に記載のケモカインおよび使用。
【請求項1】
疾患状態の処置のための医薬品の製造における、樹状細胞の移動を方向付け得るケモカインの使用であって、前記ケモカインが、MCP−1、MCP−2、MCP−3、MCP−4、MIP−1α、MIP−1β、MIP−3α、RANTES、SDF−1、Teck、DCtactin−β、6Ckine/SLC、LEC、MDC、およびMIP−5からなる群より選択される、使用。
【請求項2】
請求項1に記載の使用であって、前記樹状細胞が、未成熟樹状細胞である、使用。
【請求項3】
請求項2に記載の使用であって、前記ケモカインが、MCP−1、MCP−2、MCP−3、MCP−4、MIP−1B、MDC、MIP−3α、MIP−1α、RANTESおよびMIP−5からなる群より選択される、使用。
【請求項4】
請求項1に記載の使用であって、前記ケモカインが、MIP−3βである、使用。
【請求項5】
哺乳動物における免疫応答を増強する方法であって、該方法は、ケモカインMCP−4またはケモカインMCP−4の生物学的に活性な画分を該哺乳動物に投与する工程を包含する、方法。
【請求項6】
請求項5に記載の方法であって、前記ケモカインが、組換えケモカインである、方法。
【請求項7】
請求項5に記載の方法であって、前記ケモカインがヒトケモカインである、方法。
【請求項8】
請求項5に記載の方法であって、該方法は、前記ケモカインの徐放を可能にする物質を送達部位に投与する工程をさらに包含する、方法。
【請求項9】
請求項5に記載の方法であって、該方法は、前記ケモカインとともに抗原を投与する工程をさらに包含する、方法。
【請求項10】
請求項9に記載の方法であって、MCP−4および抗原を含む融合タンパク質が、前記哺乳動物に投与される、方法。
【請求項11】
請求項9に記載の方法であって、前記抗原が、腫瘍関連抗原である、方法。
【請求項12】
請求項10に記載の方法であって、前記抗原が、腫瘍関連抗原である、方法。
【請求項13】
請求項9に記載の方法であって、前記抗原が、細菌抗原、ウイルス抗原、または真菌抗原である、方法。
【請求項14】
請求項10に記載の方法であって、前記抗原が、細菌抗原、ウイルス抗原または真菌抗原である、方法。
【請求項15】
請求項9に記載の方法であって、前記腫瘍関連抗原が、Melan−A、チロシナーゼ、p97、β−HCG、GalNAc、MAGE−1、MAGE−2、MAGE−3、MAGE−4、MAGE−12、MART−1、MUC1、MUC2、MUC3、MUC4、MUC18、CEA、DDC、黒色腫抗原gp75、Hker8、高分子量黒色腫抗原、K19、Tyr1およびTyr2、pMel 17遺伝子ファミリーのメンバー、c−Met、PSA、PSM、α−フェトプロテイン、甲状腺ペルオキシダーゼ、gp100、p53ならびにテロメラーゼからなる群より選択される、方法。
【請求項16】
請求項10に記載の方法であって、前記腫瘍関連抗原が、Melan−A、チロシナーゼ、p97、β−HCG、GalNAc、MAGE−1、MAGE−2、MAGE−3、MAGE−4、MAGE−12、MART−1、MUC1、MUC2、MUC3、MUC4、MUC18、CEA、DDC、黒色腫抗原gp75、Hker8、高分子量黒色腫抗原、K19、Tyr1およびTyr2、pMel 17遺伝子ファミリーのメンバー、c−Met、PSA、PSM、α−フェトプロテイン、甲状腺ペルオキシダーゼ、gp100、p53ならびにテロメラーゼからなる群より選択される、方法。
【請求項17】
請求項9に記載の方法であって、GM−CSFおよびIL−4の組み合わせを投与する工程をさらに包含する、方法。
【請求項18】
請求項10に記載の方法であって、GM−CSFおよびIL−4の組み合わせを投与する工程をさらに包含する、方法。
【請求項19】
請求項5に記載の方法であって、前記ケモカインとともに活性化薬剤を投与する工程をさらに包含する、方法。
【請求項20】
請求項5に記載の方法であって、前記ケモカインが、皮内投与、筋肉内投与、皮下投与、局所投与、またはベクターの形態で投与される、方法。
【請求項21】
MCP−4および抗原を含む、融合タンパク質。
【請求項22】
請求項21に記載の融合タンパク質であって、前記抗原が腫瘍関連抗原である、融合タンパク質。
【請求項23】
請求項22に記載の融合タンパク質であって、前記腫瘍関連抗原が、Melan−A、チロシナーゼ、p97、β−HCG、GalNAc、MAGE−1、MAGE−2、MAGE−3、MAGE−4、MAGE−12、MART−1、MUC1、MUC2、MUC3、MUC4、MUC18、CEA、DDC、黒色腫抗原gp75、Hker8、高分子量黒色腫抗原、K19、Tyr1およびTyr2、pMel 17遺伝子ファミリーのメンバー、c−Met、PSA、PSM、α−フェトプロテイン、甲状腺ペルオキシダーゼ、gp100、p53ならびにテロメラーゼからなる群より選択される、融合タンパク質。
【請求項24】
請求項22に記載の融合タンパク質であって、前記抗原が、細菌抗原、ウイルス抗原または真菌抗原である、融合タンパク質。
【請求項25】
請求項21に記載の融合タンパク質を含む、プラスミド。
【請求項26】
請求項23に記載のプラスミドであって、樹状細胞に特に適したプロモーター配列をさらに含む、プラスミド。
【請求項27】
請求項21に記載の融合タンパク質を含む、ウイルスベクター。
【請求項28】
6Ckineおよび抗原を含む、融合タンパク質。
【請求項29】
請求項28に記載の融合タンパク質であって、前記抗原が腫瘍関連抗原である、融合タンパク質。
【請求項30】
請求項29に記載の融合タンパク質であって、前記腫瘍関連抗原が、Melan−A、チロシナーゼ、p97、β−HCG、GalNAc、MAGE−1、MAGE−2、MAGE−3、MAGE−4、MAGE−12、MART−1、MUC1、MUC2、MUC3、MUC4、MUC18、CEA、DDC、黒色腫抗原gp75、Hker8、高分子量黒色腫抗原、K19、Tyr1およびTyr2、pMel 17遺伝子ファミリーのメンバー、c−Met、PSA、PSM、α−フェトプロテイン、甲状腺ペルオキシダーゼ、gp100、p53ならびにテロメラーゼからなる群より選択される、融合タンパク質。
【請求項31】
請求項29に記載の融合タンパク質であって、前記抗原が、細菌抗原、ウイルス抗原または真菌抗原である、融合タンパク質。
【請求項32】
請求項28に記載の融合タンパク質を含む、プラスミド。
【請求項33】
請求項32に記載のプラスミドであって、樹状細胞に特に適したプロモーター配列をさらに含む、プラスミド。
【請求項34】
請求項28に記載の融合タンパク質を含む、ウイルスベクター。
【請求項35】
明細書に記載のケモカインおよび使用。
【図1】

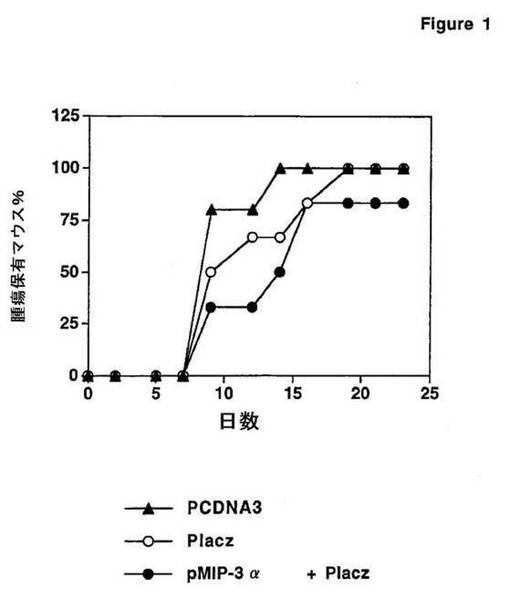
【図2】
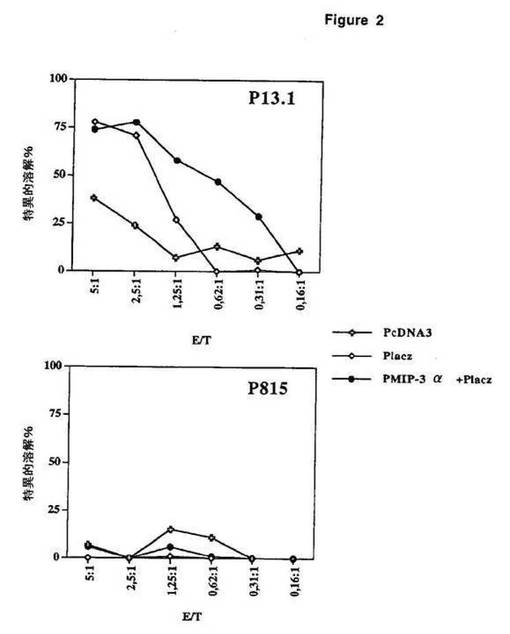
【図3】
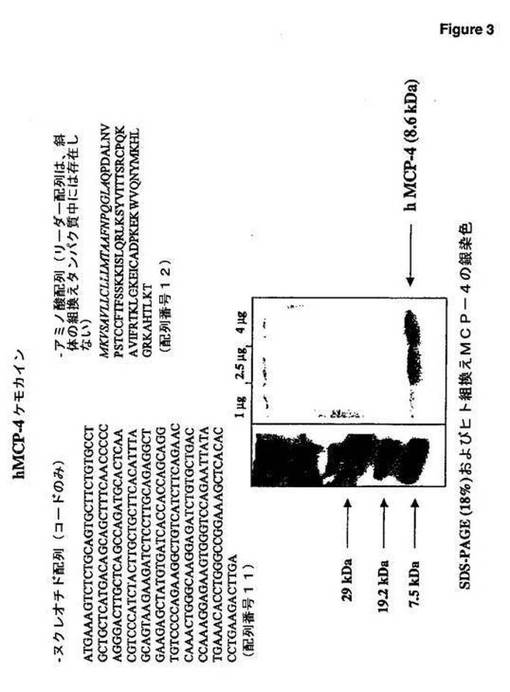
【図4】
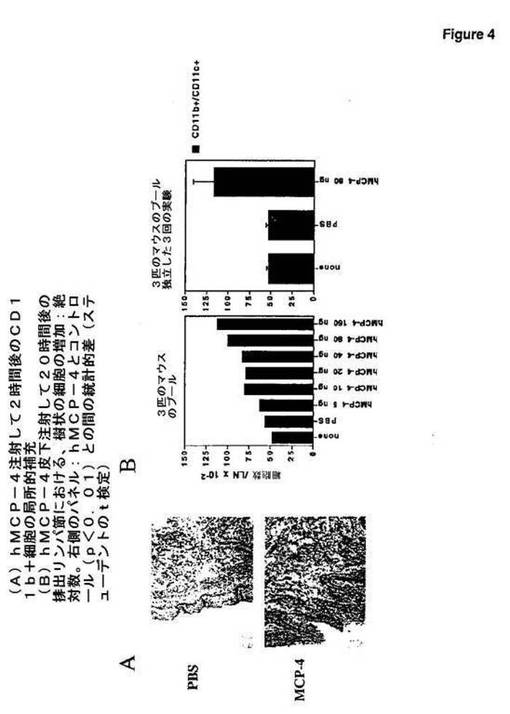
【図5】
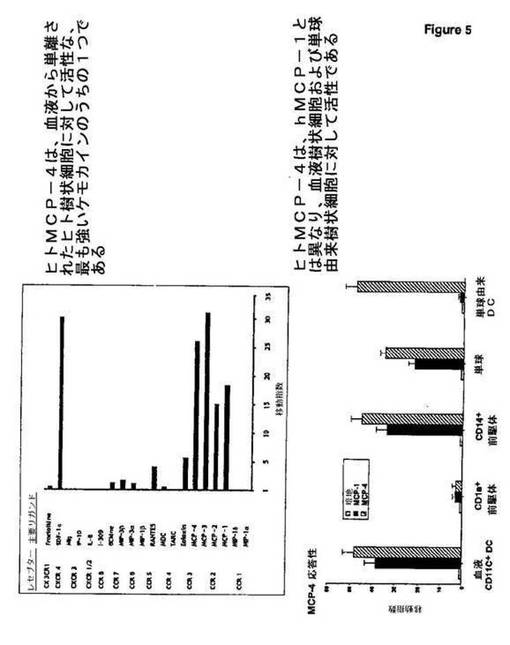
【図6】
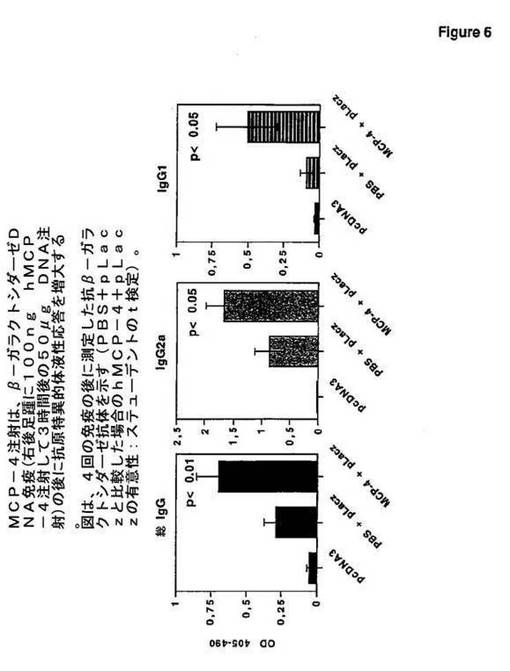
【図7】
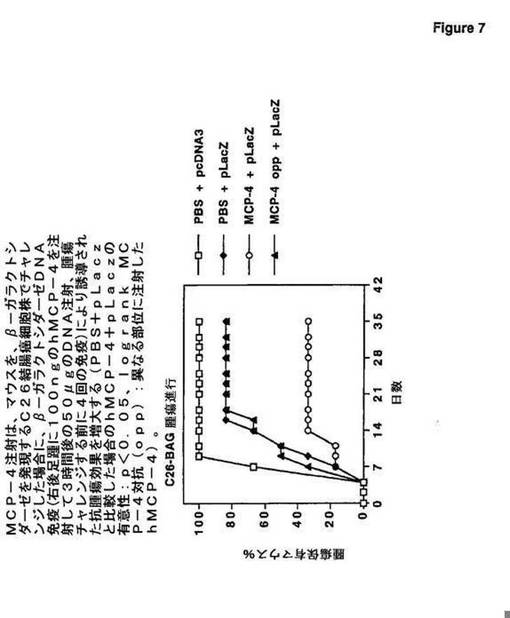
【図8】
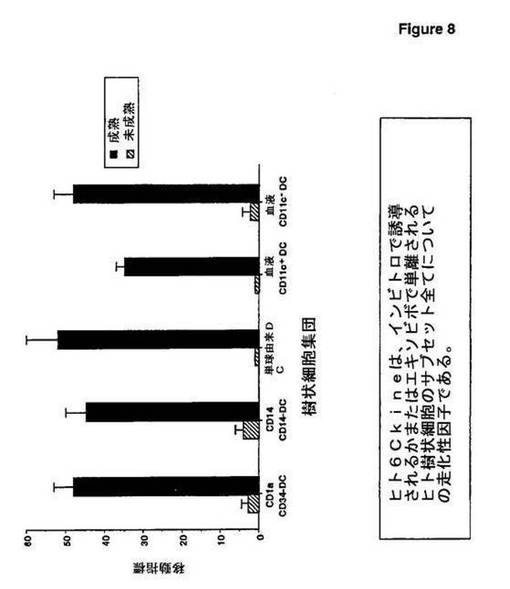
【図9】
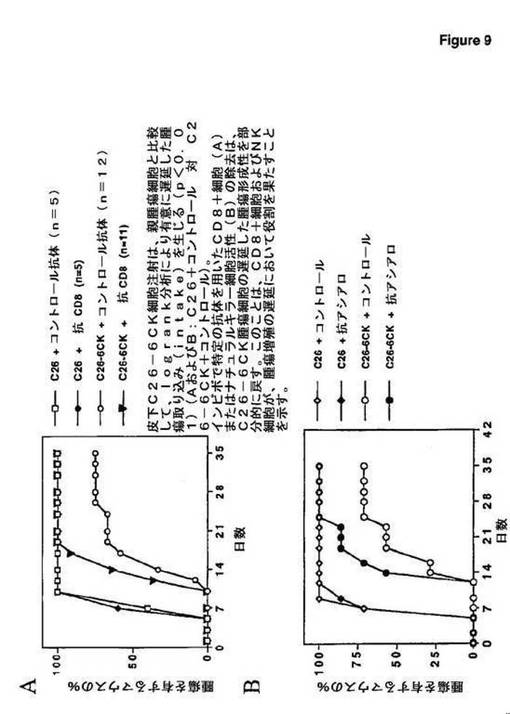
【図10】
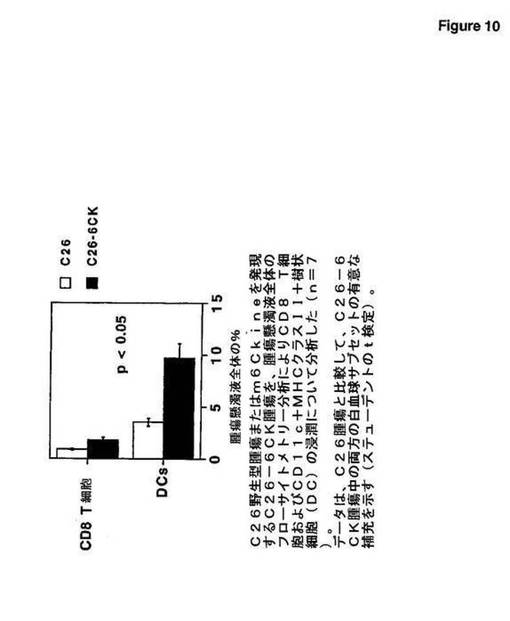
【図11】

【図12】
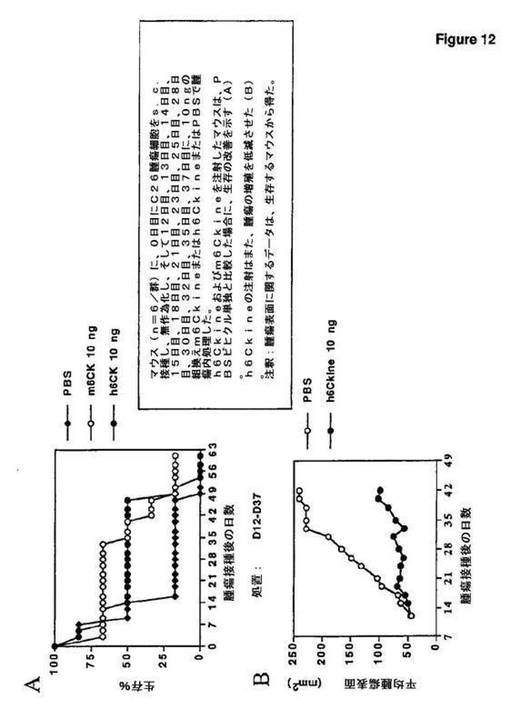
【図13】
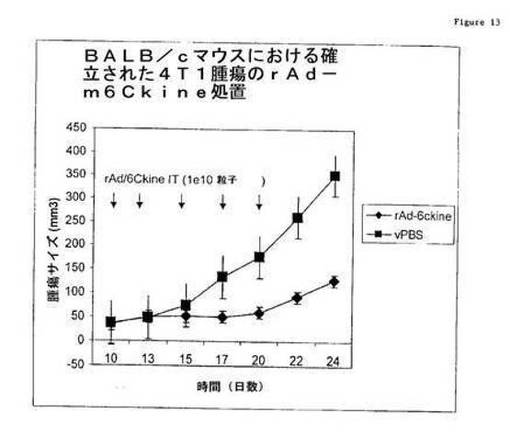
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【公開番号】特開2008−133296(P2008−133296A)
【公開日】平成20年6月12日(2008.6.12)
【国際特許分類】
【出願番号】特願2008−24609(P2008−24609)
【出願日】平成20年2月4日(2008.2.4)
【分割の表示】特願2002−559057(P2002−559057)の分割
【原出願日】平成14年1月22日(2002.1.22)
【出願人】(596129215)シェーリング コーポレイション (785)
【氏名又は名称原語表記】Schering Corporation
【Fターム(参考)】
【公開日】平成20年6月12日(2008.6.12)
【国際特許分類】
【出願日】平成20年2月4日(2008.2.4)
【分割の表示】特願2002−559057(P2002−559057)の分割
【原出願日】平成14年1月22日(2002.1.22)
【出願人】(596129215)シェーリング コーポレイション (785)
【氏名又は名称原語表記】Schering Corporation
【Fターム(参考)】
[ Back to top ]