
免疫機能分子の活性を調節する方法
【課題】抗体、蛋白質またはペプチドなどの免疫機能分子の活性を調節する方法、免疫機能分子活性促進剤、及び免疫機能活性が促進された免疫機能分子を提供する。
【解決手段】CHO/dhFr−細胞及びCHO/DG44細胞、マウスミエローマNS0細胞、マウスミエローマSP2/0−Ag14細胞で生産したヒトIgG1サブクラスの各種ヒト化抗体のin vitroの活性評価を行った結果、ラットミエローマYB2/0細胞で生産したヒト化抗体のADCC活性が他の細胞で生産したヒト化抗体に比べ著しく高いこと、更にカニクイザルを用いたin vivoの活性評価を行った結果、YB2/0細胞で生産したヒト化抗体が最も高い効果を示すことを見出し、ADCC活性の高い抗体のヒトの臨床応用での有用性を示した。更に各種動物細胞で生産したヒト化抗体のADCC活性を高める機能を有する糖鎖を特定した。
【解決手段】CHO/dhFr−細胞及びCHO/DG44細胞、マウスミエローマNS0細胞、マウスミエローマSP2/0−Ag14細胞で生産したヒトIgG1サブクラスの各種ヒト化抗体のin vitroの活性評価を行った結果、ラットミエローマYB2/0細胞で生産したヒト化抗体のADCC活性が他の細胞で生産したヒト化抗体に比べ著しく高いこと、更にカニクイザルを用いたin vivoの活性評価を行った結果、YB2/0細胞で生産したヒト化抗体が最も高い効果を示すことを見出し、ADCC活性の高い抗体のヒトの臨床応用での有用性を示した。更に各種動物細胞で生産したヒト化抗体のADCC活性を高める機能を有する糖鎖を特定した。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、抗体、蛋白質またはペプチドなどの免疫機能分子の活性を調節する方法、免疫機能分子活性促進剤、および免疫機能活性が促進された免疫機能分子に関する。
【背景技術】
【0002】
抗体は、高い結合活性、結合特異性及び血中での高い安定性を有することから、ヒトの各種疾患の診断、予防及び治療への応用が試みられてきた[モノクローナル・アンティボディズ:プリンシプルズ・アンド・アプリケーションズ(Monoclonal Antibodies:Principles and Applications),Wiley−Liss,Inc.,Capter 2.1(1995)]。しかし、マウス抗体のようなヒト以外の動物の抗体はヒトに投与すると、異物として認識されることにより、ヒト体内にマウス抗体に対するヒト抗体(Human Anti Mouse Antibody:以下、HAMAと表記する)が誘導され、HAMAは投与したマウス抗体と反応して副作用を引き起こしたり[ジャーナル・オブ・クリニカル・オンコロジー(J.Clin.Oncol.),2,881(1984);ブラッド(Blood),65,1349(1985);ジャーナル・オブ・ザ・ナショナル・キャンサー・インスティテュート(J.Natl.Cancer Inst.),80,932(1988);プロシーディングス・オブ・ザ・ナショナル・アカデミー・オブ・サイエンス(Proc.Natl.Acad.Sci.U.S.A),82,1242(1985)]、投与したマウス抗体の血中からの消失を速め[ジャーナル・オブ・ニュークレアー・メディシン(J.Nucl,Med.),26,1011(1985);ブラッド(Blood),65,1349(1985);ジャーナル・オブ・ザ・ナショナル・キャンサー・インスティテュート(J.Natl.Cancer Inst.),80,937(1988)]、マウス抗体の診断、予防及び治療の効果を減じてしまうことが知られている[ザ・ジャーナル・オブ・イムノロジー(J.Immunol.),135,1530(1985);キャンサー・リサーチ(Cancer Res.),46,6489(1986)]。
【0003】
これらの問題点を解決するため、遺伝子組換え技術を利用してヒト以外の動物の抗体をヒト型キメラ抗体或いはヒト型相補性決定領域(以下、CDRと表記する)移植抗体の様なヒト化抗体を作製することが試みられている。ヒト型キメラ抗体とは、抗体可変領域(以下、V領域と表記する)がヒト以外の動物の抗体で、定常領域(以下、C領域と表記する)がヒト抗体である抗体であり[プロシーディングス・オブ・ザ・ナショナル・アカデミー・オブ・サイエンス(Proc.Natl.Acad.Sci.U.S.A),81,6851(1984)]、ヒトに投与した場合、重篤な副作用は認められず、かつマウス抗体に比べ血中半減期が約6倍伸びたことが報告されている[プロシーディングス・オブ・ザ・ナショナル・アカデミー・オブ・サイエンス(Proc.Natl.Acad.Sci.U.S.A),86,4220(1989)]。ヒト型CDR移植抗体とは、ヒト抗体のCDRをヒト以外の動物の抗体のCDRと置換した抗体であり[ネイチャー(Nature),321,522(1986)]、サルを用いた実験でマウス抗体に比べ免疫原性が低下し、血中半減期が4〜5倍伸びたことが報告されている[ザ・ジャーナル・オブ・イムノロジー(J.Immunol.),147,1352(1991)]。これらの報告は、ヒトの各種疾患の診断、予防及び治療に応用する抗体としては完全なヒト抗体でなくてもヒト化抗体で十分な効果が期待できることを示しているが、実際に、抗腫瘍抗体に関しては、例えば抗CD20ヒト型キメラ抗体であるリツキサン[Rituxan;アイデック(IDEC)社]や抗HER2/neuヒト型CDR移植抗体であるハーセプチン[Herceptin;ジェネンテック(Genentech)社]の臨床試験が行われ、それぞれBリンパ腫、乳癌において安全性及びある程度の治療効果が認められている[ジャーナル・オブ・クリニカル・オンコロジー(J.Clin.Oncol.),16,2825(1998)、ジャーナル・オブ・ザ・ナショナル・キャンサー・インスティテュート(J.National Cancer Institute),90,882(1998)]。また、既に抗GPIIb/IIIaヒト型キメラ抗体の断片(Fab’)であるレオプロ[ReoPro;セントコア(Centocor)社]は経皮経管冠動脈血管形成術後の合併症予防薬として欧米で販売されている。現在、これら以外にも多数のヒト化抗体の臨床試験が行われているが、ヒト化抗体の多くは遺伝子組換え技術を用いて作製され、適当な動物細胞を用いて製造されている。
【0004】
哺乳類の抗体には、IgM、IgD、IgG、IgA、IgEの5種類のクラスが存在することが明らかとなっているが、ヒトの各種疾患の診断、予防及び治療には血中半減期が長く、各種エフェクター機能を有する等の機能特性からヒトIgGクラスの抗体が主として利用されている[モノクローナル・アンティボディズ:プリンシプルズ・アンド・アプリケーションズ(Monoclonal Antibodies:Principles and Applications),Wiley−Liss,Inc.,Capter 1(1995)]。ヒトIgGクラスの抗体は、更にIgG1、IgG2、IgG3、IgG4の4種類のサブクラスに分類されている。IgGクラスの抗体のエフェクター機能である抗体依存性細胞障害活性(以下、ADCC活性と表記する)や補体依存性細胞障害活性(以下、CDC活性と表記する)については、これまでに多数の研究が行われ、ヒトIgGクラスでは、IgG1サブクラスの抗体が最も高いADCC活性、CDC活性を有していることが報告されている[ケミカル・イムノロジー(Chemical Immunology),65,88(1997)]。以上の観点から、上記のリツキサン、ハーセプチンを始めとして、その効果発現に高いエフェクター機能を必要とする抗腫瘍ヒト化抗体の殆どはヒトIgG1サブクラスの抗体である。
【0005】
ヒトIgG1サブクラスの抗体のADCC活性及びCDC活性の発現には、抗体Fc領域と、キラー細胞、ナチュラルキラー細胞、活性化されたマクロファージ等のエフェクター細胞表面上に存在する抗体レセプター(以下、FcγRと表記する)及び各種補体成分との結合が必要であり、その結合については、抗体のヒンジ領域及びC領域の第2番目のドメイン(以下、Cγ2ドメインと表記する)内のいくつかのアミノ酸残基の重要性[ヨーロピアン・ジャーナル・オブ・イムノロジー(Eur.J.Immunol.),23,1098(1993)、イムノロジー(Immunology),86,319(1995)、ケミカル・イムノロジー(Chemical Immunology),65,88(1997)]の他、Cγ2ドメインに結合している糖鎖の重要性[ケミカル・イムノロジー(Chemical Immunology),65,88(1997)]が示唆されている。糖鎖に関しては、ボイド(Boyd)らは、チャイニーズハムスター卵巣細胞(CHO細胞)或いはマウスミエローマNS0細胞で生産したヒト型CDR移植抗体CAMPATH−1H(ヒトIgG1サブクラス)を各種糖分解酵素で処理し、糖鎖のADCC活性、CDC活性に対する影響を検討した結果、非還元末端のシアル酸の除去は、両活性に影響を与えないが、更にガラクトース残基を除去することでCDC活性のみが影響を受け、約50%程度活性が低下すること、糖鎖の完全な除去は、両活性を消失させることを報告した[モレキュラー・イムノロジー(Molecular Immunol.),32,1311(1995)]。また、ライフリー(Lifely)らは、CHO細胞、NS0細胞或いはラットミエローマY0細胞で生産したヒト型CDR移植抗体CAMPATH−1H(ヒトIgG1サブクラス)の糖鎖の分析及びADCC活性を測定した結果、Y0細胞由来のCAMPATH−1Hが最も高いADCC活性を示し、その活性にはバイセクティングに位置するN−アセチル−グルコサミンが重要であることを示唆した[グリコバイオロジー(Glycobiology),5,813(1995):WO99/54342]。これらの報告は、ヒトIgG1サブクラスの抗体のエフェクター機能に糖鎖の構造が極めて重要な役割を果たしており、糖鎖の構造を変えることでより高いエフェクター機能を有する抗体を作製することが可能であることを示している。しかし、実際には糖鎖の構造は多様かつ複雑であり、エフェクター機能に真に重要な構造は未だ特定できていない。
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明の目的は、各種動物細胞で生産したヒトIgG1サブクラスの抗体の糖鎖を分析してADCC活性を高める糖鎖を特定し、免疫機能分子の活性を調節する方法を提供することにある。そのような抗体は、ADCC活性が増強し、抗腫瘍抗体のみならずその他の抗体、さらには蛋白質またはペプチドの場合でも、ヒトの各種疾患に対する治療効果の増大が期待される。特に抗腫瘍抗体の臨床応用では、現状では抗体単独の抗腫瘍効果は不充分な場合が多く、化学療法との併用療法が行われているが[サイエンス(Science),280,1197,1998]、ADCC活性の増強による抗体単独のより強い抗腫瘍効果が認められれば、化学療法に対する依存度が低くなり、副作用の低減も期待できる。
【課題を解決するための手段】
【0007】
本発明者らは、2種類のチャイニーズハムスター卵巣細胞であるCHO/dhFr−細胞(ATCC CRL9096)及びCHO/DG44細胞[ソマティック・セル・アンド・モレキュラー・ジェネティクス(Somatic Cell and Molecular Genetics),12,555(1986)]、マウスミエローマNS0細胞[RCB0213、バイオ/テクノロジー(BIO/TECHNOLOGY),10,169(1992)]、マウスミエローマSP2/0−Ag14細胞(以下、SP2/0細胞と表記する;ATCC CRL1581)、ラットミエローマYB2/3HL.P2.G11.16Ag.20細胞(以下、YB2/0細胞と表記する;ATCC CRL1662)で生産したヒトIgG1サブクラスの各種ヒト化抗体のin vitroの活性評価を行った結果、ラットミエローマYB2/0細胞で生産したヒト化抗体のADCC活性が他の細胞で生産したヒト化抗体に比べ著しく高いことを見出した。更にカニクイザルを用いたin vivoの活性評価を行った結果、YB2/0細胞で生産したヒト化抗体が最も高い効果を示すことを見出し、ADCC活性の高い抗体のヒトの臨床応用での有用性を示した。更に各種動物細胞で生産したヒト化抗体の糖鎖の構造を詳細に分析、比較し、ADCC活性を高める機能を有する糖鎖を特定し、本発明を完成させるに至った。
【0008】
本発明は、以下の(1)〜(27)に関する。
(1)N−グリコシド結合コンプレックス型糖鎖が結合した抗体を含有する抗体組成物であって、該抗体が還元末端のN−アセチルグルコサミンにフコースが結合しないN−グリコシド結合コンプレックス型糖鎖を有する抗体である抗体組成物。
(2)糖鎖が、
【化1】

を含む糖鎖を含有する上記(1)に記載の抗体組成物。
(3)糖鎖が、α1,6−フコシルトランスフェラーゼ活性の低い細胞が合成する糖鎖を含有する上記(1)または(2)に記載の抗体組成物。
(4)α1,6−フコシルトランスフェラーゼ活性の低い細胞を用いることを特徴とする上記(1)または(2)に記載の抗体組成物の製造方法。
(5)抗体が、腫瘍関連抗原を認識する抗体である上記(1)〜(3)のいずれか1項に記載の抗体組成物。
(6)腫瘍関連抗原がガングリオシドGD3である、上記(5)に記載の抗体組成物。
(7)抗体が、7−9−51(FERM BP−6691)より生産される抗体である上記(5)または(6)に記載の抗体組成物。
(8)抗体が、アレルギーまたは炎症に関連する抗原を認識する抗体である上記(1)〜(3)のいずれか1項に記載の抗体組成物。
(9)アレルギーまたは炎症に関連する抗原が、ヒトインターロイキン5レセプターα鎖である上記(8)に記載の抗体組成物。
(10)抗体が、No.3(FERM BP−6690)より生産される抗体である上記(8)または(9)に記載の抗体組成物。
(11)抗体が、循環器疾患に関連する抗原を認識する抗体である上記(1)〜(3)のいずれか1項に記載の抗体組成物。
(12)抗体が、自己免疫疾患に関連する抗原を認識する抗体である上記(1)〜(3)のいずれか1項に記載の抗体組成物。
(13)抗体が、ウィルスまたは細菌感染に関連する抗原を認識する抗体である上記(1)〜(3)のいずれか1項に記載の抗体組成物。
(14)上記(5)〜(7)のいずれか1項に記載の抗体組成物を有効成分として含有する、癌の診断薬。
(15)上記(5)〜(7)のいずれか1項に記載の抗体組成物を有効成分として含有する、癌の治療薬。
(16)上記(5)〜(7)のいずれか1項に記載の抗体組成物を有効成分として含有する、癌の予防薬。
(17)上記(8)〜(10)のいずれか1項に記載の抗体組成物を有効成分として含有する、アレルギーまたは炎症の診断薬。
(18)上記(8)〜(10)のいずれか1項に記載の抗体組成物を有効成分として含有する、アレルギーまたは炎症の治療薬。
(19)上記(8)〜(10)のいずれか1項に記載の抗体組成物を有効成分として含有する、アレルギーまたは炎症の予防薬。
(20)上記(11)に記載の抗体組成物を有効成分として含有する、循環器疾患の診断薬。
(21)上記(11)に記載の抗体組成物を有効成分として含有する、循環器疾患の治療薬。
(22)上記(11)に記載の抗体組成物を有効成分として含有する、循環器疾患の予防薬。
(23)上記(12)に記載の抗体組成物を有効成分として含有する、自己免疫疾患の診断薬。
(24)上記(12)に記載の抗体組成物を有効成分として含有する、自己免疫疾患の治療薬。
(25)上記(12)に記載の抗体組成物を有効成分として含有する、自己免疫疾患の予防薬。
(26)上記(13)に記載の抗体組成物を有効成分として含有する、ウィルスまたは細菌感染症の診断薬。
(27)上記(13)に記載の抗体組成物を有効成分として含有する、ウィルスまたは細菌感染症の治療薬。
(28)上記(13)に記載の抗体組成物を有効成分として含有する、ウィルスまたは細菌感染症の予防薬。
【図面の簡単な説明】
【0009】
【図1】第1図 精製した5種類の抗GD3キメラ抗体のSDS−PAGE(4〜15%グラジエントゲルを使用)の電気泳動パターンを示した図である。上図が非還元条件、下図が還元条件でそれぞれ電気泳動を行った図である。レーン1が高分子量マーカー、2がYB2/0−GD3キメラ抗体、3がCHO/DG44−GD3キメラ抗体、4がSP2/0−GD3キメラ抗体、5がNS0−GD3キメラ抗体(302)、6がNS0−GD3キメラ抗体(GIT)、7が低分子量マーカーの泳動パターンをそれぞれ示す。
【図2】第2図 精製した5種類の抗GD3キメラ抗体のGD3との結合活性を抗体濃度を変化させて測定した図である。縦軸はGD3との結合活性、横軸は抗体濃度をそれぞれ示す。○がYB2/0−GD3キメラ抗体、●がCHO/DG44−GD3キメラ抗体、□がSP2/0−GD3キメラ抗体、■がNS0−GD3キメラ抗体(302)、△がNS0−GD3キメラ抗体(GIT)の活性をそれぞれ示す。
【図3】第3図 精製した5種類の抗GD3キメラ抗体のヒトメラノーマ細胞株G−361に対するADCC活性を示した図である。縦軸に細胞障害活性、横軸に抗体濃度をそれぞれ示す。○がYB2/0−GD3キメラ抗体、●がCHO/DG44−GD3キメラ抗体、□がSP2/0−GD3キメラ抗体、■がNS0−GD3キメラ抗体(302)、△がNS0−GD3キメラ抗体(GIT)の活性をそれぞれ示す。
【図4】第4図 精製した3種類の抗hIL−5RαCDR移植抗体のSDS−PAGE(4〜15%グラジエントゲルを使用)の電気泳動パターンを示した図である。上図が非還元条件、下図が還元条件でそれぞれ電気泳動を行った図である。レーン1が高分子量マーカー、2がYB2/0−hIL−5RCDR抗体、3がCHO/d−hIL−5RCDR抗体、4がNS0−hIL−5RCDR抗体、5が低分子量マーカーの泳動パターンをそれぞれ示す。
【図5】第5図 精製した3種類の抗hIL−5RαCDR移植抗体のhIL−5Rαとの結合活性を抗体濃度を変化させて測定した図である。縦軸はhIL−5Rαとの結合活性、横軸は抗体濃度をそれぞれ示す。○がYB2/0−hIL−5RCDR抗体、●がCHO/d−hIL−5RCDR抗体、□がNS0−hIL−5RCDR抗体の活性をそれぞれ示す。
【図6】第6図 精製した3種類の抗hIL−5RαCDR移植抗体のhIL−5R発現マウスT細胞株CTLL−2(h5R)に対するADCC活性を示した図である。縦軸に細胞障害活性、横軸に抗体濃度をそれぞれ示す。○がYB2/0−hIL−5RCDR抗体、●がCHO/d−hIL−5RCDR抗体、□がNS0−hIL−5RCDR抗体の活性をそれぞれ示す。
【図7】第7図 精製した3種類の抗hIL−5RαCDR移植抗体のカニクイザルのhIL−5誘発好酸球増加モデルに対する抑制作用を示した図である。縦軸に末梢血中好酸球数、横軸に日数(抗体及びhIL−5の投与開始日を0日とした)をそれぞれ示す。101、102が抗体非投与群、301、302、303がYB2/0−hIL−5RCDR抗体投与群、401、402、403がCHO/d−hIL−5RCDR抗体投与群、501、502、503がNS0−hIL−5RCDR抗体投与群の結果をそれぞれ示す。
【図8】第8図 YB2/0が生産した精製抗hIL−5RαCDR移植抗体(上側)およびNS0が生産した精製抗hIL−5RαCDR移植抗体(下側)のPA化糖鎖の逆相HPLC溶離の溶離図(左図)とそのPA化糖鎖をα−L−フコシダーゼ処理した後に逆相HPLCで分析して得た溶離図(右図)を示したものである。縦軸に相対蛍光強度、横軸に溶出時間をそれぞれ示す。
【図9】第9図 CHO/d細胞が生産した精製抗hIL−5RαCDR移植抗体からPA化糖鎖を調製し、逆相HPLCで分析して得た溶離図を示したものである。縦軸に相対蛍光強度、横軸に溶出時間をそれぞれ示す。
【図10】第10図 非吸着画分、吸着画分の一部のGD3との結合活性を、抗体濃度を変化させて測定した図である。縦軸はGD3との結合活性、横軸は抗体濃度をそれぞれ示す。●が非吸着画分、○が吸着画分の一部をそれぞれ示す。下図は非吸着画分、吸着画分の一部のヒトメラノーマ細胞株G−361に対するADCC活性を示した図である。縦軸に細胞障害活性、横軸に抗体濃度をそれぞれ示す。●が非吸着画分、○が吸着画分の一部をそれぞれ示す。
【図11】第11図 非吸着画分、吸着画分の一部から調製したPA化糖鎖を逆相HPLCで分析して得た溶離図を示したものである。左図に非吸着画分の溶離図、右図に吸着画分の一部の溶離図をそれぞれ示す。縦軸に相対蛍光強度、横軸に溶出時間をそれぞれ示す。
【図12】第12図 ラットFUT8配列をスタンダード、内部コントロールに用いた場合の各宿主細胞株におけるFUT8転写産物の量を示す。■がCHO細胞株、□がYB2/0細胞株を宿主細胞として用いた場合をそれぞれ示す。
【発明を実施するための形態】
【0010】
糖鎖は、免疫機能分子の結合様式により、アスパラギンと結合する糖鎖(N−グリコシド結合糖鎖という)ならびに、セリン、スレオニンなどと結合する糖鎖(0−グリコシド結合糖鎖という)の2種類に大別される。
【0011】
本発明にかかるN−グリコシド結合糖鎖は、様々な構造を有しているが[生物化学実験法23−糖蛋白質糖鎖研究法(学会出版センター)高橋禮子編(1989年)]、いずれの場合も下記に示す基本となる共通のコア構造を有する。
【0012】
【化2】
【0013】
上記の構造において、アスパラギンと結合するの糖鎖の末端を還元末端、反対側の非還元末端という。還元末端のN−アセチルグルコサミンへのフコースの結合としては、α1,3結合、α1,6結合などがあげられる。
【0014】
N−グリコシド結合糖鎖には、コア構造の非還元末端にマンノースのみが結合するハイマンノース型、コア構造の非還元末端側にガラクトース−N−アセチルグルコサミン(以下、Gal−GlcNAcと称す。)の枝を並行して1ないしは複数本有し、更にGal−GlcNAcの非還元末端側にシアル酸、バイセクティングのN−アセチルグルコサミンなどの構造を有するコンプレックス型、コア構造の非還元末端側にハイマンノース型とコンプレックス型の両方の枝を持つハイブリッド型などがあげられる。
【0015】
上記のいずれの型において、還元末端側のN−アセチルグルコサミンにフコースが結合する糖鎖もあるが、本発明の糖鎖は、上述の糖鎖のみならず、N−アセチルグルコサミンにフコースが結合しない糖鎖であればいかなるものも包含する。
【0016】
免疫機能分子とは、元来生体内に存在し、種々の免疫反応に関与する分子であり、具体的には、抗体、タンパク質、ペプチドなどを包含する。
【0017】
抗体とは、外来抗原刺激の結果、免疫反応によって生体内に産生される蛋白質で、抗原と特異的に結合する活性を有するものをいう。抗体としては動物に抗原を免疫し、免疫動物の脾臓細胞より作製したハイブリドーマ細胞が分泌する抗体のほか、遺伝子組換え技術により作製された抗体、すなわち、抗体遺伝子を挿入した抗体発現ベクターを、宿主細胞へ導入することにより取得された抗体などがあげられる。具体的には、ハイブリドーマが生産する抗体、ヒト化抗体、ヒト抗体などをあげることができる。
【0018】
ハイブリドーマとは、ヒト以外の哺乳動物に抗原を免疫して取得されたB細胞と、マウス等に由来するミエローマ細胞とを細胞融合させて得られる、所望の抗原特異性を有したモノクローナル抗体を産生する細胞を意味する。
【0019】
ヒト化抗体としては、ヒト型キメラ抗体、ヒト型相同性決定領域(complementarity determining region:以下、CDRと略記する)移植抗体などがあげられる。
【0020】
ヒト型キメラ抗体は、ヒト以外の動物の抗体重鎖可変領域(以下、重鎖はH鎖として、可変領域はV領域としてHVまたはVHとも称す)および抗体軽鎖可変領域(以下、軽鎖はL鎖としてLVまたはVLとも称す)とヒト抗体の重鎖定常領域(以下、定常領域はC領域としてCHとも称す)およびヒト抗体の軽鎖定常領域(以下、CLとも称す)とからなる抗体を意味する。ヒト以外の動物としては、マウス、ラット、ハムスター、ラビット等、ハイブリドーマを作製することが可能であれば、いかなるものも用いることができる。
【0021】
ヒト型キメラ抗体は、モノクローナル抗体を生産するハイブリドーマより、VHおよびVLをコードするcDNAを取得し、ヒト抗体CHおよびヒト抗体CLをコードする遺伝子を有する宿主細胞用発現ベクターにそれぞれ挿入してヒト型キメラ抗体発現ベクターを構築し、宿主細胞へ導入することにより発現させ、製造することができる。
【0022】
ヒト型キメラ抗体のCHとしては、ヒトイムノグロブリン(以下、hIgと表記する)に属すればいかなるものでもよいが、hIgGクラスのものが好適であり、更にhIgGクラスに属するhIgG1、hIgG2、hIgG3、hIgG4といったサブクラスのいずれも用いることができる。また、ヒト型キメラ抗体のCLとしては、hIgに属すればいかなるものでもよく、κクラスあるいはλクラスのものを用いることができる。
【0023】
ヒト型CDR移植抗体は、ヒト以外の動物の抗体のVHおよびVLのCDRのアミノ酸配列をヒト抗体のVHおよびVLの適切な位置に移植した抗体を意味する。
【0024】
ヒト型CDR移植抗体は、ヒト以外の動物の抗体のVHおよびVLのCDR配列を任意のヒト抗体のVHおよびVLのCDR配列に移植したV領域をコードするcDNAを構築し、ヒト抗体のCHおよびヒト抗体のCLをコードする遺伝子を有する宿主細胞用発現ベクターにそれぞれ挿入してヒト型CDR移植抗体発現ベクターを構築し、該発現ベクターを宿主細胞へ導入することによりヒト型CDR移植抗体を発現させ、製造することができる。
【0025】
ヒト型CDR移植抗体のCHとしては、hIgに属すればいかなるものでもよいが、hIgGクラスのものが好適であり、更にhIgGクラスに属するhIgG1、hIgG2、hIgG3、hIgG4といったサブクラスのいずれも用いることができる。また、ヒト型CDR移植抗体のCLとしては、hIgに属すればいかなるものでもよく、κクラスあるいはλクラスのものを用いることができる。
【0026】
ヒト抗体は、元来、ヒト体内に天然に存在する抗体を意味するが、最近の遺伝子工学的、細胞工学的、発生工学的な技術の進歩により作製されたヒト抗体ファージライブラリーならびにヒト抗体産生トランスジェニック動物あるいはヒト抗体産生トランスジェニック植物から得られる抗体等も含まれる。
【0027】
ヒト体内に存在する抗体は、例えば、ヒト末梢血リンパ球を単離し、EBウイルス等を感染させ不死化、クローニングすることにより、該抗体を産生するリンパ球を培養でき、培養物中より該抗体を精製することができる。
【0028】
ヒト抗体ファージライブラリーは、ヒトB細胞から調製した抗体遺伝子をファージ遺伝子に挿入することによりFab、一本鎖抗体等の抗体断片をファージ表面に発現させたライブラリーである。該ライブラリーより、抗原を固定化した基質に対する結合活性を指標として所望の抗原結合活性を有する抗体断片を発現しているファージを回収することができる。該抗体断片は、更に遺伝子工学的手法により、2本の完全なH鎖および2本の完全なL鎖からなるヒト抗体分子へも変換することができる。
【0029】
ヒト抗体産生トランスジェニック非ヒト動物は、ヒト抗体遺伝子が細胞内に組込まれた動物を意味する。具体的には、マウスES細胞へヒト抗体遺伝子を導入し、該ES細胞を他のマウスの初期胚へ移植後、発生させることによりヒト抗体産生トランスジェニック動物を作製することができる。ヒト抗体産生トランスジェニック動物からのヒト抗体の作製方法は、通常のヒト以外の哺乳動物で行われているハイブリドーマ作製方法によりヒト抗体産生ハイブリドーマを得、培養することで培養物中にヒト抗体を産生蓄積させることができる。
【0030】
本発明の抗体の活性とは、ADCC活性を意味する。
ADCC活性とは、腫瘍細胞等に結合した抗体が、抗体Fc領域とキラー細胞、ナチュラルキラー細胞、活性化されたマクロファージ等のエフェクター細胞表面上に存在するFcレセプターの結合を介してエフェクター細胞を活性化し、腫瘍細胞等を障害する活性を意味する[モノクローナル・アンティボディズ:プリンシプルズ・アンド・アプリケーションズ(Monoclonal Antibodies:Principles and Applications),Wiley−Liss,Inc.,Capter 2.1(1995)]。
【0031】
蛋白質およびペプチドとしては、種々の免疫反応を活性化できるものであればいかなるものでも用いることができる。例えば、インターフェロン分子であるインターロイキン−2(IL−2)[サイエンス(Science),193,1007(1976)]、インターロイキン−12(IL−12)[ジャーナル・オブ・ロイコサイト・バイオロジー(J.Leuc.Biol.),55,280(1994)]、コロニー刺激因子である顆粒球コロニー刺激因子(G−CSF)[ジャーナル・オブ・バイオロジカル・ケミストリー(J.Biol.Chem.),258,9017(1983)]、マクロファージコロニー刺激因子(M−CSF)[ジャーナル・オブ・エクスペリメンタル・メディシン(J.Exp.Med.),173,269(1992)]、顆粒球−マクロファージコロニー刺激因子(GM−CSF)[ジャーナル・オブ・バイオロジカル・ケミストリー(J.Biol.Chem.),252,1998(1977)]、増殖因子であるエリスロポイエチン(EPO)[ジャーナル・オブ・バイオロジカル・ケミストリー(J.Biol.Chem.),252,5558(1977)]、トロンボポイエチン(TPO)[ネイチャー(Nature),369,533(1994)]などがあげられる。
【0032】
本発明の蛋白質およびペプチドの活性とは、該糖鎖を有する蛋白質およびペプチドを生体内に投与した際の、リンパ球(T細胞、B細胞等)、マクロファージ等をはじめとする各種免疫担当細胞の活性、または種々の免疫応答反応を意味する。
【0033】
本発明の蛋白質およびペプチドの活性の促進とは、IL−2やIL−12によるNK細胞、T細胞の活性化、EPOによる赤血球産生の促進作用等がさらに増強されることを意味する。
【0034】
1.免疫機能分子の糖鎖の分析方法
(1)中性糖・アミノ糖組成分析
IgG糖鎖は、上記で示したように、ガラクトース、マンノース、フコースなどの中性糖、N−アセチルグルコサミンなどのアミノ糖、シアル酸などの酸性糖から構成されている。
【0035】
抗体の糖鎖の組成分析は、トリフルオロ酢酸等で、糖鎖の酸加水分解を行うことにより、中性糖またはアミノ糖を遊離し、その組成比を分析することができる。
具体的な方法として、Dionex社製糖組成分析装置(BioLC)を用いる方法が挙げられる。BioLCはHPAEC−PAD(high performance anion−exchange chromatography−pulsed amperometric detection)法[ジャーナル・オブ・リキッド・クロマトグラフィー(J.Liq.Chromatogr.),6,1577(1983)]によって糖組成を分析する装置である。
【0036】
また、2−アミノピリジンによる蛍光標識化法でも組成比を分析することができる。具体的には、公知の方法[アグリカルチュラル・アンド・バイオロジカル・ケミストリー(Agruc.Biol.Chem.),55(1),283−284(1991)]に従って酸加水分解した試料を2−アミノピリジル化で蛍光ラベル化し、HPLC分析して組成比を算出することができる。
【0037】
(2)糖鎖構造解析
抗体の糖鎖の構造解析は、2次元糖鎖マップ法[アナリティカル・バイオケミストリー(Anal.Biochem.),171,73(1988)、生物化学実験法23−糖蛋白質糖鎖研究法(学会出版センター)高橋禮子編(1989年)]により行うことができる。2次元糖鎖マップ法は、例えば、X軸には逆相クロマトグラフィー糖鎖の保持時間または溶出位置を、Y軸には順相クロマトグラフィーによる糖鎖の保持時間または溶出位置を、それぞれプロットし、既知糖鎖のそれらの結果と比較することにより、糖鎖構造を推定する方法である。
【0038】
具体的には、抗体をヒドラジン分解して、抗体から糖鎖を遊離し、2−アミノピリジン(以下、PAと略記する)による糖鎖の蛍光標識[ジャーナル・オブ・バイオケミストリー(J.Biochem.),95,197(1984)]を行った後、ゲルろ過により糖鎖を過剰のPA化試薬などと分離し、逆相クロマトグラフィーを行う。次いで、分取した糖鎖の各ピークについて順相クロマトグラフィーを行う。これらの結果をもとに、2次元糖鎖マップ上にプロットし、糖鎖スタンダード(TaKaRa社製)、文献[アナリティカル・バイオケミストリー(Anal.Biochem.),171,73(1988)]とのスポットの比較より糖鎖構造を推定することができる。
【0039】
さらに各糖鎖のMALDI−TOF−MSなどの質量分析を行い、2次元糖鎖マップ法により推定される構造を確認することができる。
【0040】
2.免疫機能分子の活性を調節する方法
本発明の免疫機能分子の活性を調節する方法について、イムノグロブリンG(以下、IgGと略記する)を例として以下に述べる。
【0041】
IgGに結合するN−グルコシド結合糖鎖は、主として次式で示す構造(バイアンテナリーと記す)のバイアンテナリー複合型糖鎖である。
【0042】
【化3】
【0043】
また、N−グルコシド結合糖鎖の非還元末端のGalに、更に酸性糖であるシアル酸が付加する場合もあり、N−グルコシド結合糖鎖に、バイセクティングのN−アセチルグルコサミンが付加する場合もある。
【0044】
IgGは、Fc領域内の1カ所に、N−グルコシド結合糖鎖が結合している。IgGは2本のH鎖から構成されているので、抗体1分子にはFc部分が2カ所存在する。したがって、糖鎖結合部位も2カ所存在する。
【0045】
IgGの活性は、上記2カ所の糖鎖結合部位への、N−アセチルグルコサミンにフコースが結合しないN−グルコシド結合糖鎖の付加する個数により活性が変化する。すなわち、糖鎖結合部位の少なくとも1カ所に、N−アセチルグルコサミンにフコースが結合しないN−グルコシド結合糖鎖が付加されるときには、免疫機能分子の活性が高まる。2カ所の糖鎖結合部位ともN−アセチルグルコサミンにフコースが結合しないN−グルコシド結合糖鎖が付加される抗体をF0構造を有する抗体(F0抗体)、1カ所の糖鎖結合部位にN−アセチルグルコサミンにフコースが結合しないN−グルコシド結合糖鎖が付加される抗体をF1構造を有する抗体(F1抗体)、糖鎖結合部位のどちらにもN−アセチルグルコサミンにフコースが結合したN−グルコシド結合糖鎖が付加する場合をF2構造を有する抗体(F2抗体)とすると、IgGの活性の高さは、F0抗体>F1抗体>F2抗体となる。
【0046】
製造された抗体は、必ずしも単一の糖鎖構造を有しておらず、かつフコースの有無に着目した場合でもF0抗体、F1抗体およびF2抗体が混在して存在している。製造された抗体のADCC活性を調節するためには、上述した免疫機能分子の糖鎖の解析方法により抗体に結合している糖鎖を解析し、解析結果を指標としてADCC活性を調節することができる。
【0047】
製造された抗体のADCC活性を促進するためには、F1抗体およびF0抗体の存在比率を高くすればよい。具体的には、F1抗体およびF0抗体を精製してもよいし、またN−アセチルグルコサミンにフコースが結合しないN−グルコシド結合糖鎖が免疫機能分子に付加するように、宿主細胞での発現を調節してもよい。
【0048】
製造された抗体のADCC活性を抑制するためには、F2抗体の存在比率を高くすればよい。具体的には、F2抗体を精製してもよいし、またN−アセチルグルコサミンにフコースが結合したN−グルコシド結合糖鎖が免疫機能分子に付加するように、宿主細胞での発現を調節してもよい。
【0049】
上述のように、F0抗体、F1抗体およびF2抗体を任意に存在比率を調節することにより、所望の活性の強弱を調節することができる。
【0050】
3.免疫機能分子の製造方法
N−アセチルグルコサミンにフコースが結合しないN−グルコシド結合糖鎖を有する免疫機能分子、またはN−アセチルグルコサミンにフコースが結合するN−グルコシド結合糖鎖を有する免疫機能分子の製造方法について、以下に述べる。
【0051】
抗体、ペプチドまたは蛋白質に所望の糖鎖を結合させる為には、目的とする抗体、ペプチドまたは蛋白質をコードする遺伝子を宿主細胞に導入し、該細胞を用いて生産させることができる。また、動物個体または植物個体に目的とする抗体、ペプチドまたは蛋白質をコードする遺伝子を導入し、該個体を用いて生産させることもできる。
【0052】
N−アセチルグルコサミンにフコースが結合しないN−グルコシド結合糖鎖を有する免疫機能分子を製造するための宿主細胞または動物個体あるいは植物個体としては、例えば抗体で、抗体のFc領域に結合するN−アセチルグルコサミンにフコースを付加させる酵素活性の低いまたは酵素活性を有しない細胞または個体であればいかなるものでもよい。
抗体のFc領域に結合するN−アセチルグルコサミンにフコースを付加させる酵素活性の低いまたは酵素活性を有しない細胞としては、ラットミエローマ細胞であるYB2/3HL.P2.G11.16Ag.20細胞(ATCC CRL 1662;以下、YB2/0細胞と略記する)などがあげられる。
【0053】
また、宿主細胞または動物個体あるいは植物個体のα1,6結合に関与する酵素の遺伝子を欠損させたり、該遺伝子への変異を与えて酵素活性を下げるか欠失させたりすることにより、α1,6結合に関与する酵素活性の少ない、または有しない細胞または個体を作製して宿主細胞または動物個体あるいは植物個体として用いることもできる。α1,6結合に関与する酵素としては、フコシルトランスフェラーゼ、好ましくはα1,6−フコシルトランスフェラーゼ(以下、FUT8と称す)があげられる。
【0054】
N−アセチルグルコサミンにフコースが結合するN−グルコシド結合糖鎖を有する免疫機能分子を製造するための宿主細胞または動物個体あるいは植物個体としては、例えば抗体で、抗体のFc領域に結合するN−アセチルグルコサミンにフコースを付加させる酵素活性の高い細胞または個体であればいかなるものでもよい。
【0055】
また、宿主細胞または動物個体あるいは植物個体のα1,6結合に関与する酵素の遺伝子を導入したり、該遺伝子への変異を与えて酵素活性をあげることにより、α1,6結合に関与する酵素活性の高い細胞または個体を作製して宿主細胞または動物個体あるいは植物個体として用いることもできる。α1,6結合に関与する酵素としては、フコシルトランスフェラーゼ、好ましくはFUT8があげられる。
【0056】
宿主細胞としては、細菌、酵母、動物細胞、昆虫細胞、植物細胞等、目的とする遺伝子を発現できるものであればいずれも用いることができる。
【0057】
細菌の宿主細胞としては、エシェリヒア属、セラチア属、バチルス属、ブレビバクテリウム属、コリネバクテリウム属、ミクロバクテリウム属、シュードモナス属等に属する微生物、例えば、Escherichiacoli XL1−Blue、Escherichia coli XL2−Blue、Escherichiacoli DH1、Escherichia coli MC1000、Escherichia coliKY3276、Escherichia coli W1485、Escherichia coli JM109、Escherichiacoli HB101、Escherichia
coli No.49、Escherichia coliW3110、Escherichia coli NY49、Escherichia coli GI698、Escherichiacoli TB1、Serratia ficaria Serratiafonticola、Serratialiquefaciens、Serratia marcescens、Bacillus subtilis、Bacillusamyloliquefacines、Brevibacterium ammoniagenes、BrevibacteriumimmariophilumATCC14068、Brevibacterium saccharolyticum ATCC14066、Brevibacteriumflavum ATCC14067、Brevibacterium lactofermentum ATCC13869、Corynebacteriumglutamicum ATCC13032、Corynebacterium glutamicum ATCC13869、Corynebacteriumacetoacidophilum ATCC13870、Microbacterium ammoniaphilum ATCC15354、Pseudomonasputida、Pseudomonas sp.D−0110等をあげることができる。
【0058】
酵母の宿主細胞としては、Saccharomyces属、Schizosaccharomyces属、Kluyveromyces属、Trichosporon属、Schwanniomyces属、Pichia属、Candida属等に属する微生物、例えば、Saccharomycescerevisiae、Schizosaccharomyces pombe、Kluyvermyces lactis、Trichosporonpullulans、Schwanniomyces alluvius、Candidautilis等をあげることができる。
【0059】
動物細胞の宿主細胞としては、マウスミエローマ細胞であるNS0細胞、SP2/0細胞、チャイニーズハムスター卵巣細胞CHO/dhfr−細胞、CHO/DG44細胞、ラットミエローマYB2/0細胞、IR983F細胞、サルの細胞であるCOS細胞、ヒトミエローマ細胞であるナマルバ(Namalwa)細胞などがあげられる。好ましくは、チャイニーズハムスター卵巣細胞であるCHO/DG44細胞等があげられる。
【0060】
昆虫細胞の宿主細胞としては、Spodoptera frugiperdaの卵巣細胞であるSf9、Sf21〔Baculovirus Expression Vectors,A Laboratory Manual,W.H.Freeman and Company,New York(1992)〕、Trichoplusianiの卵巣細胞であるHigh 5(Invitrogen社製)等を用いることができる。
【0061】
植物細胞の宿主細胞としては、タバコ、ジャガイモ、トマト、ニンジン、ダイズ、アブラナ、アルファルファ、イネ、コムギ、オオムギ等の植物細胞等をあげることができる。
以上のようにして得られる本発明の形質転換体を培地に培養し、培養物中に免疫機能分子を生成蓄積させ、該培養物から採取することにより、免疫機能分子を製造することができる。
さらに、遺伝子が導入された動物個体または植物個体を造成し、これらの個体を用いて免疫機能分子を製造することもできる。
【0062】
N−アセチルグルコサミンにフコースが結合しないN−グルコシド結合糖鎖を有する免疫機能分子を製造するための動物個体または植物個体としては、例えば抗体で、抗体のFc領域に結合するN−アセチルグルコサミンにフコースを付加させる酵素活性の低いまたは酵素活性を有しない動物個体または植物個体であればいかなるものでもよい。
【0063】
また、動物個体または植物個体のα1,6結合に関与する酵素の遺伝子を欠損させたり、該遺伝子への変異を与えて酵素活性を下げるか欠失させたりすることにより、α1,6結合に関与する酵素活性の少ない、または有しないノックアウト非ヒト動物またはノックアウト植物を作製して用いてもよい。α1,6結合に関与する酵素としては、フコシルトランスフェラーゼ、好ましくはFUT8があげられる。
【0064】
N−アセチルグルコサミンにフコースが結合するN−グルコシド結合糖鎖を有する免疫機能分子を製造するための動物個体または植物個体としては、例えば抗体で、抗体のFc領域に結合するN−アセチルグルコサミンにフコースを付加させる酵素活性の高い動物個体または植物個体であればいかなるものでもよい。
【0065】
また、動物個体または植物個体のα1,6結合に関与する酵素の遺伝子を導入したり、該遺伝子への変異を与えて酵素活性を上げることにより、α1,6結合に関与する酵素活性の高いトランスジェニック非ヒト動物またはトランスジェニック植物を作製して用いてもよい。α1,6結合に関与する酵素としては、フコシルトランスフェラーゼ、好ましくはFUT8があげられる。
トランスジェニック非ヒト動物は、受精卵へ所望の遺伝子を直接インジェクションすることより取得することができる[Proc.Natl.Acad.Sci.USA,77,7380(1980)]。
【0066】
トランスジェニック非ヒト動物としては、マウス、ラット、ウサギ、ニワトリ、ヤギ、ウシなどがあげられる。
【0067】
また、胚性幹細胞へ所望の遺伝子を導入し、集合キメラ法や注入キメラ法の手法で動物を作製することにより、所望の遺伝子のトランスジェニック非ヒト動物またはノックアウト非ヒト動物を作製することができる[Manipulating the Mouse Embryo A Laboratory Manual,Second Edition,Cold Spring Harbor Laboratory Press(1994);Gene targeting,A practical Approach,IRL Press at Oxford university Press(1993);バイオマテリアルシリーズ8ジーンターゲッティング,ES細胞を用いた変異マウスの作製,羊土社(1995)]。
【0068】
胚性幹細胞としては、マウス[Nature,292,154(1981)]、ラット、ニワトリ、ブタ、サル、ヤギ、ウシなどがあげられる。
【0069】
さらに、所望の遺伝子を導入した核を、脱核した卵へ移植するというクローン技術を用いても、トランスジェニック非ヒト動物またはノックアウト非ヒト動物を作製することができる[Science,280,1256,1998;Science,278,824(1997)]。
上述の方法により作製された動物個体に、免疫機能分子をコードするDNAを導入し、該免疫機能分子を動物個体中に生成・蓄積させ、該動物個体中より該免疫機能分子を採取することにより、該免疫機能分子を製造することができる。該動物中の生成・蓄積場所としては、例えば、該動物のミルク(特開昭63−309192)、卵等をあげることができる。
【0070】
トランスジェニック植物の作製方法としては、文献[Biol.Chem.,380,825(1999)]などがあげられ、ノックアウト植物の作製方法としては、文献[Plant Journal,11,1195(1997)]があげられる。
【0071】
植物個体を用いて免疫機能分子を製造する方法としては、例えば免疫機能分子をコードするDNAを導入したトランスジェニック植物を公知の方法[組織培養,20(1994)、組織培養,21(1995)、Trends in Biotechnology,15,45(1997)]に準じて栽培し、該免疫機能分子を該植物中に生成・蓄積させ、該植物中より該免疫機能分子を採取することにより、該免疫機能分子を製造することができる。
【0072】
また、フコシルトランスフェラーゼ、好ましくはFUT8のトランスジェニック非ヒト動物またはノックアウト非ヒト動物と同系他系統である所望の免疫機能分子のトランスジェニック動物とを交配させることにより、N−アセチルグルコサミンにフコースが結合しないN−グルコシド結合糖鎖を有する免疫機能分子またはN−アセチルグルコサミンにフコースが結合するN−グルコシド結合糖鎖を有する免疫機能分子を製造することができる、遺伝子改変動物を取得することができる。交配の方法としては、自然交配、体外受精などの方法があげられる。
【0073】
また、単離した該酵素等の遺伝子群を酵母、大腸菌等へ導入し、該糖鎖の大量生産[ネイチャー・バイオテクノロジー(Nature Biotechnology),16,847(1998)]をすることもできる。また、生産された該酵素を、抗体、ペプチドまたは蛋白質への該糖鎖による修飾または生産に利用することもできる。
【0074】
さらに、本発明の免疫機能分子の活性を促進する糖鎖をペプチドで代替することもできる[ジャーナル・オブ・イムノロジー(J.Immunol.),160,293(1998)]。該ペプチドは、上述の糖鎖の使用方法において有用性があり、また免疫機能分子との融合も容易であるので簡便性の点においても優れている。
【0075】
以下に、免疫機能活性の促進された免疫機能分子の製造方法について述べる。ここでは一例として、ヒト化抗体の製造方法について記すが、他の免疫機能分子も上述の方法および当該方法に準じて作製することができる。
【0076】
4.ヒト化抗体の製造方法
(1)ヒト化抗体発現用ベクターの構築
ヒト化抗体発現用ベクターとは、ヒト抗体の重鎖(以下、H鎖と表記する)及び軽鎖(以下、L鎖と表記する)C領域をコードする遺伝子が組み込まれた動物細胞用発現ベクターであり、動物細胞用発現ベクターにヒト抗体のH鎖及びL鎖C領域をコードする遺伝子をそれぞれクローニングすることにより構築することができる。
【0077】
ヒト抗体のC領域としては、任意のヒト抗体のH鎖及びL鎖C領域であることができ、例えば、ヒト抗体のH鎖のIgG1サブクラスのC領域(以下、hCγ1と表記する)及びヒト抗体のL鎖のκクラスのC領域(以下、hCκと表記する)等があげられる。
【0078】
ヒト抗体のH鎖及びL鎖C領域をコードする遺伝子としてはエキソンとイントロンから成る染色体DNAを用いることができ、また、cDNAを用いることもできる。
【0079】
動物細胞用発現ベクターとしては、ヒト抗体のC領域をコードする遺伝子を組込み発現できるものであればいかなるものでも用いることができる。例えば、pAGE107[サイトテクノロジー(Cytotechnology),3,133(1990)]、pAGE103[ジャーナル・オブ・バイオケミストリー(J.Biochem.),101,1307(1987)]、pHSG274[ジーン(Gene),27,223(1984)]、pKCR[プロシーディングス・オブ・ザ・ナショナル・アカデミー・オブ・サイエンス(Proc.Natl.Acad.Sci.
U.S.A.),78,1527(1981)]、pSG1 β d2−4[サイトテクノロジー(Cytotechnology),4,173(1990)]等があげられる。動物細胞用発現ベクターに用いるプロモーターとエンハンサーとしては、SV40の初期プロモーターとエンハンサー[ジャーナル・オブ・バイオケミストリー(J.Biochem.),101,1307(1987)]、モロニーマウス白血病ウイルスのLTR[バイオケミカル・アンド・バイオフィジカル・リサーチ・コミュニケーションズ(Biochem.Biophys.Res.Commun.),149,960(1987)]、免疫グロブリンH鎖のプロモーター[セル(Cell),41,479(1985)]とエンハンサー[セル(Cell),33,717(1983)]等があげられる。
【0080】
ヒト化抗体発現用ベクターは、抗体H鎖及びL鎖が別々のベクター上に存在するタイプあるいは同一のベクター上に存在するタイプ(以下、タンデム型と表記する)のどちらでも用いることができるが、ヒト化抗体発現ベクターの構築の容易さ、動物細胞への導入の容易さ、動物細胞内での抗体H鎖及びL鎖の発現量のバランスが均衡する等の点からタンデム型のヒト化抗体発現用ベクターの方が好ましい[ジャーナル・オブ・イムノロジカル・メソッズ(J.Immunol.Methods),167,271(1994)]。
【0081】
構築したヒト化抗体発現用ベクターは、ヒト型キメラ抗体及びヒト型CDR移植抗体の動物細胞での発現に使用できる。
【0082】
(2)ヒト以外の動物の抗体のV領域をコードするcDNAの取得
ヒト以外の動物の抗体、例えば、マウス抗体のH鎖及びL鎖V領域をコードするcDNAは以下のようにして取得することができる。
【0083】
目的のマウス抗体を産生するハイブリドーマ細胞よりmRNAを抽出し、cDNAを合成する。合成したcDNAをファージ或いはプラスミド等のベクターにクローニングしてcDNAライブラリーを作製する。該ライブラリーより、既存のマウス抗体のC領域部分或いはV領域部分をプローブとして用い、H鎖V領域をコードするcDNAを有する組換えファージ或いは組換えプラスミド及びL鎖V領域をコードするcDNAを有する組換えファージ或いは組換えプラスミドをそれぞれ単離する。組換えファージ或いは組換えプラスミド上の目的のマウス抗体のH鎖及びL鎖V領域の全塩基配列を決定し、塩基配列よりH鎖及びL鎖V領域の全アミノ酸配列を推定する。
【0084】
ヒト以外の動物としては、マウス、ラット、ハムスター、ラビット等、ハイブリドーマ細胞を作製することが可能であれば、いかなるものも用いることができる。
【0085】
ハイブリドーマ細胞から全RNAを調製する方法としては、チオシアン酸グアニジン−トリフルオロ酢酸セシウム法[メソッズ・イン・エンザイモロジー(Methods in Enzymol.),154,3(1987)]、また全RNAからmRNAを調製する方法としては、オリゴ(dT)固定化セルロースカラム法[モレキュラー・クローニング:ア・ラボラトリー・マニュアル(Molecular Cloning: A Laboratory Manual),Cold Spring Harbor Lab.Press New York,1989]等があげられる。また、ハイブリドーマ細胞からmRNAを調製するキットとしては、Fast Track mRNA Isolation Kit(Invitrogen社製))、Quick Prep mRNA Purification Kit(Pharmacia社製)等があげられる。
【0086】
cDNAの合成及びcDNAライブラリー作製法としては、常法[モレキュラー・クローニング:ア・ラボラトリー・マニュアル(Molecular Cloning:A Laboratory Manual),Cold Spring Harbor Lab.Press New York,1989;カレント・プロトコールズ・イン・モレキュラー・バイオロジー(Current Protocols in Molecular Biology),Supplement 1−34]、或いは市販のキット、例えば、Super ScriptTMPlasmid System for cDNA Synthesis and Plasmid Cloning(GIBCO BRL社製)やZAP−cDNA Synthesis Kit(Stratagene社製)を用いる方法などがあげられる。
【0087】
cDNAライブラリーの作製の際、ハイブリドーマ細胞から抽出したmRNAを鋳型として合成したcDNAを組み込むベクターは、該cDNAを組み込めるベクターであればいかなるものでも用いることができる。例えば、ZAP Express[ストラテジーズ(Strategies),5,58(1992)]、pBluescript II SK(+)[ヌクレイック・アシッズ・リサーチ(Nucleic Acids Research),17,9494(1989)]、λzapII(Stratagene社製)、λgt10、λgt11[DNAクローニング:ア・プラクティカル・アプローチ(DNA Cloning: A Practical Approach),I,49(1985)]、Lambda BlueMid(Clontech社製)、λExCell、pT7T3 18U(Pharmacia社製)、pcD2[モレキュラー・アンド・セルラー・バイオロジー(Mol.Cell.Biol.),3,280(1983)]及びpUC18[シーン(Gene)33,103(1985)]等が用いられる。
【0088】
ファージ或いはプラスミドベクターにより構築されるcDNAライブラリーを導入する大腸菌としては該cDNAライブラリーを導入、発現及び維持できるものであればいかなるものでも用いることができる。例えば、XL1−Blue MRF’[ストラテジーズ(Strategies),5,81(1992)]、C600[ジェネティックス(Genetics),39,440(1954)]、Y1088、Y1090[サイエンス(Science),222,778(1983)]、NM522[ジャーナル・オブ・モレキュラー・バイオロジー(J.Mol.Biol.),166,1(1983)]、K802[ジャーナル・オブ・モレキュラー・バイオロジー(J.Mol.Biol.),16,118(1966)]及びJM105[ジーン(Gene),38,275(1985)]等が用いられる。
【0089】
cDNAライブラリーからのヒト以外の動物の抗体のH鎖及びL鎖V領域をコードするcDNAクローンの選択法としては、アイソトープ或いは蛍光標識したプローブを用いたコロニー・ハイブリダイゼーション法或いはプラーク・ハイブリダイゼーション法[モレキュラー・クローニング:ア・ラボラトリー・マニュアル(Molecular Cloning:A Laboratory Manual),Cold Spring Harbor Lab.Press NewYork,1989]により選択することができる。また、プライマーを調製し、mRNAから合成したcDNA或いはcDNAライブラリーを鋳型として、Polymerase Chain Reaction[以下、PCR法と表記する;モレキュラー・クローニング:ア・ラボラトリー・マニュアル(Molecular Cloning:A Laboratory Manual),Cold Spring Harbor Lab.Press New York,1989;カレント・プロトコールズ・イン・モレキュラー・バイオロジー(Current Protocols in
Molecular Biology),Supplement 1−34]によりH鎖及びL鎖V領域をコードするcDNAを調製することもできる。
【0090】
上記方法により選択されたcDNAを、適当な制限酵素などで切断後、pBluescriptSK(−)(Stratagene社製)等のプラスミドにクローニングし、通常用いられる塩基配列解析方法、例えば、サンガー(Sanger)らのジデオキシ法[プロシーディングス・オブ・ザ・ナショナル・アカデミー・オブ・サイエンス(Proc.Natl.Acad.Sci.,U.S.A.),74,5463(1977)]等の反応を行い、塩基配列自動分析装置、例えば、A.L.F.DNAシークエンサー(Pharmacia社製)等を用いて解析することで該cDNAの塩基配列を決定することができる。
【0091】
決定した塩基配列からH鎖及びL鎖V領域の全アミノ酸配列を推定し、既知の抗体のH鎖及びL鎖V領域の全アミノ酸配列[シーケンシズ・オブ・プロテインズ・オブ・イムノロジカル・インタレスト(Sequences of Proteins of Immunological Interest),US Dept.Health and Human Services,1991]と比較することにより、取得したcDNAが分泌シグナル配列を含む抗体のH鎖及びL鎖V領域の完全なアミノ酸配列をコードしているかを確認することができる。
【0092】
(3)ヒト以外の動物の抗体のV領域のアミノ酸配列の解析
分泌シグナル配列を含む抗体のH鎖及びL鎖V領域の完全なアミノ酸配列に関しては、既知の抗体のH鎖及びL鎖V領域の全アミノ酸配列[シーケンシズ・オブ・プロテインズ・オブ・イムノロジカル・インタレスト(Sequences of Proteins of Immunological Interest),US Dept.Health and Human Services,1991]と比較することにより、分泌シグナル配列の長さ及びN末端アミノ酸配列を推定でき、更にはそれらが属するサブグループを知ることができる。また、H鎖及びL鎖V領域の各CDRのアミノ酸配列についても、既知の抗体のH鎖及びL鎖V領域のアミノ酸配列[シーケンシズ・オブ・プロテインズ・オブ・イムノロジカル・インタレスト(Sequences of Proteins of Immunological Interest),US Dept.Health and Human Services,1991]と比較することによって見出すことができる。
【0093】
(4)ヒト型キメラ抗体発現ベクターの構築
本項4の(1)に記載のヒト化抗体発現用ベクターのヒト抗体のH鎖及びL鎖C領域をコードする遺伝子の上流に、ヒト以外の動物の抗体のH鎖及びL鎖V領域をコードするcDNAをクローニングし、ヒト型キメラ抗体発現ベクターを構築することができる。例えば、ヒト以外の動物の抗体のH鎖及びL鎖V領域をコードするcDNAを、ヒト以外の動物の抗体H鎖及びL鎖V領域の3’末端側の塩基配列とヒト抗体のH鎖及びL鎖C領域の5’末端側の塩基配列とから成り、かつ適当な制限酵素の認識配列を両端に有する合成DNAとそれぞれ連結し、それぞれを本項4の(1)に記載のヒト化抗体発現用ベクターのヒト抗体のH鎖及びL鎖C領域をコードする遺伝子の上流にそれらが適切な形で発現するようにクローニングし、ヒト型キメラ抗体発現ベクターを構築することができる。
【0094】
(5)ヒト型CDR移植抗体のV領域をコードするcDNAの構築
ヒト型CDR移植抗体のH鎖及びL鎖V領域をコードするcDNAは、以下のようにして構築することができる。まず、目的のヒト以外の動物の抗体のH鎖及びL鎖V領域のCDRを移植するヒト抗体のH鎖及びL鎖V領域のフレームワーク(以下、FRと表記する)のアミノ酸配列を選択する。ヒト抗体のH鎖及びL鎖V領域のFRのアミノ酸配列としては、ヒト抗体由来のものであれば、いかなるものでも用いることができる。例えば、Protein Data Bank等のデータベースに登録されているヒト抗体のH鎖及びL鎖V領域のFRのアミノ酸配列、ヒト抗体のH鎖及びL鎖のV領域のFRの各サブグループの共通アミノ酸配列[シーケンシズ・オブ・プロテインズ・オブ・イムノロジカル・インタレスト(Sequences of Proteins of Immunological Interest),US Dept.Health and Human Services,1991]等があげられるが、その中でも、十分な活性を有するヒト型CDR移植抗体を作製するためには、目的のヒト以外の動物の抗体のH鎖及びL鎖V領域のFRのアミノ酸配列とできるだけ高い相同性(少なくとも60%以上)を有するアミノ酸配列を選択することが望ましい。
【0095】
次に、選択したヒト抗体のH鎖及びL鎖V領域のFRのアミノ酸配列に目的のヒト以外の動物の抗体のH鎖及びL鎖V領域のCDRのアミノ酸配列を移植し、ヒト型CDR移植抗体のH鎖及びL鎖V領域のアミノ酸配列を設計する。設計したアミノ酸配列を抗体の遺伝子の塩基配列に見られるコドンの使用頻度[シーケンシズ・オブ・プロテインズ・オブ・イムノロジカル・インタレスト(Sequences of Proteins of Immunological Interest),US Dept.Health and Human Services,1991]を考慮してDNA配列に変換し、ヒト型CDR移植抗体のH鎖及びL鎖V領域のアミノ酸配列をコードするDNA配列を設計する。設計したDNA配列に基づき、100塩基前後の長さから成る数本の合成DNAを合成し、それらを用いてPCR法を行う。この場合、PCRでの反応効率及び合成可能なDNAの長さから、H鎖、L鎖とも6本の合成DNAを設計することが好ましい。
【0096】
また、両端に位置する合成DNAの5’末端に適当な制限酵素の認識配列を導入することで、本項4の(1)で構築したヒト化抗体発現用ベクターに容易にクローニングすることができる。PCR後、増幅産物をpBluescript SK(−)(Stratagene社製)等のプラスミドにクローニングし、本項4の(2)に記載の方法により、塩基配列を決定し、所望のヒト型CDR移植抗体のH鎖及びL鎖V領域のアミノ酸配列をコードするDNA配列を有するプラスミドを取得する。
【0097】
(6)ヒト型CDR移植抗体のV領域のアミノ酸配列の改変
ヒト型CDR移植抗体は、目的のヒト以外の動物の抗体のH鎖及びL鎖V領域のCDRのみをヒト抗体のH鎖及びL鎖V領域のFRに移植しただけでは、その抗原結合活性は元のヒト以外の動物の抗体に比べて低下してしまうことが知られている[バイオ/テクノロジー(BIO/TECHNOLOGY),9,266(1991)]。この原因としては、元のヒト以外の動物の抗体のH鎖及びL鎖V領域では、CDRのみならず、FRのいくつかのアミノ酸残基が直接的或いは間接的に抗原結合活性に関与しており、それらアミノ酸残基がCDRの移植に伴い、ヒト抗体のH鎖及びL鎖V領域のFRの異なるアミノ酸残基へと変化してしまうことが考えられている。この問題を解決するため、ヒト型CDR移植抗体では、ヒト抗体のH鎖及びL鎖V領域のFRのアミノ酸配列の中で、直接抗原との結合に関与しているアミノ酸残基やCDRのアミノ酸残基と相互作用したり、抗体の立体構造を維持し、間接的に抗原との結合に関与しているアミノ酸残基を同定し、それらを元のヒト以外の動物の抗体に見出されるアミノ酸残基に改変し、低下した抗原結合活性を上昇させることが行われている[バイオ/テクノロジー(BIO/TECHNOLOGY),9,266(1991)]。
【0098】
ヒト型CDR移植抗体の作製においては、それら抗原結合活性に関わるFRのアミノ酸残基を如何に効率よく同定するかが、最も重要な点であり、そのためにX線結晶解析[ジャーナル・オブ・モレキュラー・バイオロジー(J.Mol.Biol.),112,535(1977)]或いはコンピューターモデリング[プロテイン・エンジニアリング(Protein Engineering),7,1501(1994)]等による抗体の立体構造の構築及び解析が行われている。これら抗体の立体構造の情報は、ヒト型CDR移植抗体の作製に多くの有益な情報をもたらして来たが、その一方、あらゆる抗体に適応可能なヒト型CDR移植抗体の作製法は未だ確立されておらず、現状ではそれぞれの抗体について数種の改変体を作製し、それぞれの抗原結合活性との相関を検討する等の種々の試行錯誤が必要である。
【0099】
ヒト抗体のH鎖及びL鎖V領域のFRのアミノ酸残基の改変は、改変用合成DNAを用いて本項4の(5)に記載のPCR法を行うことにより、達成できる。PCR後の増幅産物について本項4の(2)に記載の方法により、塩基配列を決定し、目的の改変が施されたことを確認する。
【0100】
(7)ヒト型CDR移植抗体発現ベクターの構築
本項4の(1)に記載のヒト化抗体発現用ベクターのヒト抗体のH鎖及びL鎖C領域をコードする遺伝子の上流に、本項4の(5)及び(6)で構築したヒト型CDR移植抗体のH鎖及びL鎖V領域をコードするcDNAをクローニングし、ヒト型CDR移植抗体発現ベクターを構築することができる。例えば、本項4の(5)及び(6)でヒト型CDR移植抗体のH鎖及びL鎖V領域を構築する際に用いる合成DNAのうち、両端に位置する合成DNAの5’末端に適当な制限酵素の認識配列を導入することで、本項4の(1)に記載のヒト化抗体発現用ベクターのヒト抗体のH鎖及びL鎖C領域をコードする遺伝子の上流にそれらが適切な形で発現するようにクローニングし、ヒト型CDR移植抗体発現ベクターを構築することができる。
【0101】
(8)ヒト化抗体の安定的生産
本項4の(4)及び(7)に記載のヒト化抗体発現ベクターを適当な動物細胞に導入することによりヒト化抗体を安定に生産する形質転換株を得ることができる。
【0102】
動物細胞への発現ベクターの導入法としては、エレクトロポレーション法[特開平2−257891、サイトテクノロジー(Cytotechnology)3,133(1990)]等があげられる。
【0103】
ヒト化抗体発現ベクターを導入する動物細胞としては、ヒト化抗体を生産させることができる動物細胞であれば、いかなる細胞でも用いることができるが、好ましくは生産される抗体のFc領域に付加するN−アセチルグルコサミンにフコースを付加させる酵素活性の低いまたは酵素活性を有しない細胞があげられる。
【0104】
抗体のFc領域に付加するN−アセチルグルコサミンにフコースを付加させる酵素活性の低いまたは酵素活性を有しない細胞とは、α1,6結合に関与する酵素が少ない、またはない細胞であり、具体的にはフコシルトランスフェラーゼ、好ましくはFUT8活性が少ない、またはない細胞があげられる。
【0105】
抗体のFc領域に付加するN−アセチルグルコサミンにフコースを付加させる酵素活性の低い、または酵素活性を有しない細胞としては、ラットミエローマ細胞であるYB2/0細胞などがあげられるが、α1,6結合に関与する酵素の遺伝子を欠損させたり、該遺伝子への変異を与えて酵素活性を下げるか欠失させた細胞を抗体生産細胞として用いることもできる。
【0106】
具体的には、マウスミエローマ細胞であるNS0細胞、SP2/0細胞、チャイニーズハムスター卵巣細胞CHO/dhfr−細胞、CHO/DG44細胞、ラットミエローマYB2/0細胞、IR983F細胞、ヒトミエローマ細胞であるナマルバ細胞などがあげられる。好ましくは、チャイニーズハムスター卵巣細胞であるCHO/DG44細胞等があげられる。
【0107】
発現ベクターの導入後、ヒト化抗体を安定に生産する形質転換株は、特開平2−257891に開示されている方法に従い、G418 sulfate(以下、G418と表記する;SIGMA社製)等の薬剤を含む動物細胞培養用培地により選択できる。動物細胞培養用培地としては、RPMI1640培地(日水製薬社製)、GIT培地(日本製薬社製)、EX−CELL302培地(JRH社製)、IMDM培地(GIBCO BRL社製)、Hybridoma−SFM培地(GIBCO BRL社製)、またはこれら培地に牛胎児血清(以下、FBSと表記する)等の各種添加物を添加した培地等を用いることができる。得られた形質転換株を培地中で培養することで培養上清中にヒト化抗体を生産蓄積させることができる。培養上清中のヒト化抗体の生産量及び抗原結合活性は酵素免疫抗体法[以下、ELISA法と表記する;アンティボディズ:ア・ラボラトリー・マニュアル(Antibodies:A Laboratory Manual),Cold Spring Harbor Laboratory,Chapter 14,1998、モノクローナル・アンティボディズ:プリンシプルズ・アンド・プラクティス(Monoclonal Antibodies:Principles and Practice),Academic Press Limited,1996]等により測定できる。また、形質転換株は、特開平2−257891に開示されている方法に従い、DHFR遺伝子増幅系等を利用してヒト化抗体の生産量を上昇させることができる。
【0108】
ヒト化抗体は、形質転換株の培養上清よりプロテインAカラムを用いて精製することができる[アンティボディズ:ア・ラボラトリー・マニュアル(Antibodies:A
Laboratory Manual),Cold Spring Harbor Laboratory,Chapter 8,1988、モノクローナル・アンティボディズ:プリンシプルズ・アンド・プラクティス(Monoclonal Antibodies:Principles and Practice),Academic Press Limited,1996]。また、その他に通常、蛋白質の精製で用いられる精製方法を使用することができる。例えば、ゲル濾過、イオン交換クロマトグラフィー及び限外濾過等を組み合わせて行い、精製することができる。精製したヒト化抗体のH鎖、L鎖或いは抗体分子全体の分子量は、ポリアクリルアミドゲル電気泳動[以下、SDS−PAGEと表記する;ネイチャー(Nature),227,680(1970)]やウエスタンブロッティング法[アンティボディズ:ア・ラボラトリー・マニュアル(Antibodies:A Laboratory Manual),Cold Spring Harbor
Laboratory,Chapter 12,1988、モノクローナル・アンティボディズ:プリンシプルズ・アンド・プラクティス(Monoclonal Antibodies:Principles and Practice),Academic Press Limited,1996]等で測定することができる。
【0109】
以上、動物細胞を宿主とした抗体の製造方法を示したが、上記3にあるように、細菌、酵母、昆虫細胞、植物細胞または動物個体あるいは植物個体においても製造することができる。
【0110】
(9)ヒト化抗体の活性評価
精製したヒト化抗体の抗原との結合活性、抗原陽性培養細胞株に対する結合活性はELISA法及び蛍光抗体法[キャンサー・イムノロジー・イムノセラピー(Cancer Immunol.Immunother.),36,373(1993)]等により測定できる。抗原陽性培養細胞株に対する細胞障害活性は、CDC活性、ADCC活性等を測定することにより、評価することができる[キャンサー・イムノロジー・イムノセラピー(Cancer Immunol Immunother.),36,373(1993)]。更にヒト化抗体のヒトでの安全性、治療効果は、カニクイザル等のヒトに比較的近い動物種の適当なモデルを用いて評価することができる。
【0111】
5.免疫機能分子の使用方法
上記4記載のヒト化抗体の例にあるように、高いADCC活性を有する抗体は、癌、アレルギー、循環器疾患、またはウィルスあるいは細菌感染をはじめとする各種疾患の予防および治療において有用である。
癌、すなわち悪性腫瘍は癌細胞が増殖する。通常の抗癌剤は癌細胞の増殖を抑制することを特徴とする。しかし、高いADCC活性を有する抗体は、殺細胞効果により癌細胞を障害することにより癌を治療することができるので、通常の抗癌剤よりも治療薬として有効である。
【0112】
アレルギー反応は、免疫細胞によるメディエータ分子の放出により惹起されるため、高いADCC活性を有する抗体を用いて免疫細胞を除去することにより、アレルギー反応を抑えることができる。
【0113】
循環器疾患としては、動脈硬化などがあげられる。動脈硬化は、現在バルーンカテーテルによる治療を行うが、治療後の再狭窄での動脈細胞の増殖を高いADCC活性を有する抗体を用いて抑えることより、循環器疾患を予防および治療することができる。
【0114】
ウィルスまたは細菌に感染細胞を、高いADCC活性を有する抗体を用いてウィルスまたは細菌に感染細胞の増殖を抑えることにより、ウィルスまたは細菌感染をはじめとする各種疾患の予防および治療することができる。
【0115】
また、ADCC活性が抑制された抗体は、自己免疫疾患の予防および治療において有用である。また、ADCC活性が抑制された抗体は、自己免疫疾患において亢進された免疫反応を押さえるという観点から、自己免疫疾患の予防および治療において有用である。
【0116】
本発明の抗体を含有する医薬は、治療薬として単独で投与することも可能ではあるが、通常は薬理学的に許容される一つあるいはそれ以上の担体と一緒に混合し、製剤学の技術分野においてよく知られる任意の方法により製造した医薬製剤として提供するのが望ましい。
【0117】
投与経路は、治療に際して最も効果的なものを使用するのが望ましく、経口投与、または口腔内、気道内、直腸内、皮下、筋肉内および静脈内等の非経口投与をあげることができ、抗体製剤の場合、望ましくは静脈内投与をあげることができる。
【0118】
投与形態としては、噴霧剤、カプセル剤、錠剤、顆粒剤、シロップ剤、乳剤、座剤、注射剤、軟膏、テープ剤等があげられる。
【0119】
経口投与に適当な製剤としては、乳剤、シロップ剤、カプセル剤、錠剤、散剤、顆粒剤等があげられる。
【0120】
乳剤およびシロップ剤のような液体調製物は、水、ショ糖、ソルビトール、果糖等の糖類、ポリエチレングリコール、プロピレングリコール等のグリコール類、ごま油、オリーブ油、大豆油等の油類、p−ヒドロキシ安息香酸エステル類等の防腐剤、ストロベリーフレーバー、ペパーミント等のフレーバー類等を添加剤として用いて製造できる。
【0121】
カプセル剤、錠剤、散剤、顆粒剤等は、乳糖、ブドウ糖、ショ糖、マンニトール等の賦形剤、デンプン、アルギン酸ナトリウム等の崩壊剤、ステアリン酸マグネシウム、タルク等の滑沢剤、ポリビニルアルコール、ヒドロキシプロピルセルロース、ゼラチン等の結合剤、脂肪酸エステル等の界面活性剤、グリセリン等の可塑剤等を添加剤として用いて製造できる。
【0122】
非経口投与に適当な製剤としては、注射剤、座剤、噴霧剤等があげられる。
注射剤は、塩溶液、ブドウ糖溶液、あるいは両者の混合物からなる担体等を用いて調製される。または、ヒト化抗体を常法に従って凍結乾燥し、これに塩化ナトリウムを加えることによって粉末注射剤を調製することもできる。
【0123】
座剤はカカオ脂、水素化脂肪またはカルボン酸等の担体を用いて調製される。
また、噴霧剤は該化合物そのもの、ないしは受容者の口腔および気道粘膜を刺激せず、かつ該化合物を微細な粒子として分散させ吸収を容易にさせる担体等を用いて調製される。
【0124】
担体として具体的には乳糖、グリセリン等が例示される。該化合物および用いる担体の性質により、エアロゾル、ドライパウダー等の製剤が可能である。また、これらの非経口剤においても経口剤で添加剤として例示した成分を添加することもできる。
【0125】
投与量または投与回数は、目的とする治療効果、投与方法、治療期間、年齢、体重等により異なるが、通常成人1日当たり10μg/kg〜20mg/kgである。
【0126】
また、抗体の各種腫瘍細胞に対する抗腫瘍効果を検討する方法は、インビトロ実験としては、CDC活性測定法、ADCC活性測定法等があげられ、インビボ実験としては、マウス等の実験動物での腫瘍系を用いた抗腫瘍実験等があげられる。
【0127】
CDC活性、ADCC活性、抗腫瘍実験は、文献[キャンサー・イムノロジー・イムノセラピー(Cancer Immunology Immunotherapy),36,373(1993);キャンサー・リサーチ(Cancer Research),54,1511(1994)]等記載の方法に従って行うことができる。
【0128】
6.免疫機能分子の活性を促進または抑制させる方法
上述の方法によりフコースが存在しない糖鎖が結合された抗体、ペプチドまたは蛋白質を製造することにより免疫機能分子の活性を促進させることができる。
【0129】
活性が促進された免疫機能分子を生体内に投与することにより、生体内では、ADCC活性を担うエフェクター細胞であるキラー細胞、ナチュラルキラー細胞、活性化マクロファージ等の細胞をはじめとする各種免疫細胞が活性化され、種々の免疫反応を調節することが可能となる。
また、上述の方法によりフコースが存在する糖鎖を結合された抗体、ペプチドまたは蛋白質を製造することにより免疫機能分子の活性を抑制させることができる。
【0130】
活性が抑制された免疫機能分子を生体内に投与することにより、生体内では、ADCC活性を担う各種免疫細胞の活性が弱まり、種々の免疫反応を調節することが可能となる。
【実施例】
【0131】
以下に、本発明の実施例を示すが、これにより本発明の範囲が限定されるものではない。
【実施例1】
【0132】
抗ガングリオシドGD3ヒト型キメラ抗体の作製
1.抗ガングリオシドGD3ヒト型キメラ抗体のタンデム型発現ベクターpChiLHGM4の構築
抗ガングリオシドGD3ヒト型キメラ抗体(以下、抗GD3キメラ抗体と表記する)のL鎖の発現ベクターpChi641LGM4[ジャーナル・オブ・イムノロジカル・メソッズ(J.Immunol.Methods),167,271(1994)]を制限酵素MluI(宝酒造社製)とSalI(宝酒造社製)で切断して得られるL鎖cDNAを含む約4.03kbの断片と動物細胞用発現ベクターpAGE107[サイトテクノロジー(Cytotechnology),3,133(1990)]を制限酵素MluI(宝酒造社製)とSalI(宝酒造社製)で切断して得られるG418耐性遺伝子及びスプライシングシグナルを含む約3.40kbの断片をDNA Ligation Kit(宝酒造社製)を用いて連結、大腸菌HB101株[モレキュラー・クローニング:ア・ラボラトリー・マニュアル(Molecular Cloning:A Laboratory Manual),Cold Spring Harbor Lab.Press New York,1989]を形質転換してプラスミドpChi641LGM40を構築した。
次に、上記で構築したプラスミドpChi641LGM40を制限酵素ClaI(宝酒造社製)で切断後、DNA Blunting Kit(宝酒造社製)を用いて平滑末端化し、更にMluI(宝酒造社製)で切断して得られるL鎖cDNAを含む約5.68kbの断片と抗GD3キメラ抗体のH鎖の発現ベクターpChi641HGM4[ジャーナル・オブ・イムノロジカル・メソッズ(J.Immunol.Methods),167,271(1994)]を制限酵素XhoI(宝酒造社製)で切断後、DNA Blunting Kit(宝酒造社製)を用いて平滑末端化し、更にMluI(宝酒造社製)で切断して得られるH鎖cDNAを含む約8.40kbの断片をDNA Ligation Kit(宝酒造社製)を用いて連結、大腸菌HB101株[モレキュラー・クローニング:ア・ラボラトリー・マニュアル(Molecular Cloning:A Laboratory Manual),Cold Spring Harbor Lab.Press New York,1989]を形質転換して抗GD3キメラ抗体のタンデム型発現ベクターpChi641LHGM4を構築した。
【0133】
2.抗GD3キメラ抗体の安定生産細胞の作製
上記実施例1の1項で構築した抗GD3キメラ抗体のタンデム型発現ベクターpChi641LHGM4を用いて抗GD3キメラ抗体の安定生産細胞を以下のようにして作製した。
(1)ラットミエローマYB2/0細胞を用いた生産細胞の作製
抗GD3キメラ抗体発現ベクターpChi641LHGM4の5μgを4×106細胞のラットミエローマYB2/0細胞へエレクトロポレーション法[サイトテクノロジー(Cytotechnology),3,133(1990)]により導入後、40mlのRPMI1640−FBS(10)[FBS(GIBCO BRL社製)を10%含むRPMI1640培地]に懸濁し、96ウェル培養用プレート(住友ベークライト社製)に200μl/ウェルずつ分注した。5%CO2インキュベーター内で37℃、24時間培養した後、G418を0.5mg/mlになるように添加して1〜2週間培養した。G418耐性を示す形質転換株のコロニーが出現し、増殖の認められたウェルより培養上清を回収し、上清中の抗GD3キメラ抗体の抗原結合活性を実施例1の3項に示すELISA法により測定した。
培養上清中に抗GD3キメラ抗体の生産が認められたウェルの形質転換株については、DHFR遺伝子増幅系を利用して抗体生産量を増加させる目的で、G418を0.5mg/ml、DHFRの阻害剤であるメソトレキセート(以下、MTXと表記する;SIGMA社製)を50nM含むRPMI1640−FBS(10)培地に1〜2×105細胞/mlになるように懸濁し、24ウェルプレート(Greiner社製)に2mlずつ分注した。5%CO2インキュベーター内で37℃で1〜2週間培養して、50nM MTX耐性を示す形質転換株を誘導した。形質転換株の増殖が認められたウェルの培養上清中の抗GD3キメラ抗体の抗原結合活性を実施例1の3項に示すELISA法により測定した。培養上清中に抗GD3キメラ抗体の生産が認められたウェルの形質転換株については、上記と同様の方法により、MTX濃度を100nM、200nMと順次上昇させ、最終的にG418を0.5mg/ml、MTXを200nMの濃度で含むRPMI1640−FBS(10)培地で増殖可能かつ、抗GD3キメラ抗体を高生産する形質転換株を得た。
得られた形質転換株については、2回の限界希釈法による単一細胞化(クローン化)を行った。
このようにして得られた抗GD3キメラ抗体を生産する形質転換細胞クローン7−9−51は平成11年4月5日付で工業技術院生命工学工業技術研究所(日本国茨城県つくば市東1丁目1番3号)にFERM BP−6691として寄託されている。
(2)CHO/DG44細胞を用いた生産細胞の作製
抗GD3キメラ抗体発現ベクターpChi641LHGM4の4μgを1.6×106細胞のCHO/DG44細胞へエレクトロポレーション法[サイトテクノロジー(Cytotechnology),3,133(1990)]により導入後、10mlのIMDM−FBS(10)[FBSを10%、HT supplement(GIBCO BRL社製)を1倍濃度で含むIMDM培地]に懸濁し、96ウェル培養用プレート(岩城硝子社製)に200μl/ウェルずつ分注した。5%CO2インキュベーター内で37℃、24時間培養した後、G418を0.5mg/mlになるように添加して1〜2週間培養した。G418耐性を示す形質転換株のコロニーが出現し、増殖の認められたウェルより培養上清を回収し、上清中の抗GD3キメラ抗体の抗原結合活性を実施例1の3項に示すELISA法により測定した。
培養上清中に抗GD3キメラ抗体の生産が認められたウェルの形質転換株については、DHFR遺伝子増幅系を利用して抗体生産量を増加させる目的で、G418を0.5mg/ml、MTXを10nM含むIMDM−dFBS(10)培地[透析牛胎児血清(以下、dFBSと表記する;GIBCO BRL社製)を10%含むIMDM培地]に1〜2×105細胞/mlになるように懸濁し、24ウェルプレート(岩城硝子社製)に0.5mlずつ分注した。5%CO2インキュベーター内で37℃で1〜2週間培養して、10nM MTX耐性を示す形質転換株を誘導した。増殖が認められたウェルの形質転換株については、上記と同様の方法により、MTX濃度を100nMに上昇させ、最終的にG418を0.5mg/ml、MTXを100nMの濃度で含むIMDM−dFBS(10)培地で増殖可能かつ、抗GD3キメラ抗体を高生産する形質転換株を得た。得られた形質転換株については、2回の限界希釈法による単一細胞化(クローン化)を行った。
(3)マウスミエローマNS0細胞を用いた生産細胞の作製
抗GD3キメラ抗体発現ベクターpChi641LHGM4の5μgを4×106細胞のマウスミエローマNS0細胞へエレクトロポレーション法[サイトテクノロジー(Cytotechnology),3,133,1990]により導入後、40mlのEX−CELL302−FBS(10)[FBSを10%、L−グルタミン(以下、L−Glnと表記する;GIBCO BRL社製)を2mM含むEX−CELL302培地]に懸濁し、96ウェル培養用プレート(住友ベークライト社製)に200μl/ウェルずつ分注した。5%CO2インキュベーター内で37℃、24時間培養した後、G418を0.5mg/mlになるように添加して1〜2週間培養した。G418耐性を示す形質転換株のコロニーが出現し、増殖の認められたウェルより培養上清を回収し、上清中の抗GD3キメラ抗体の抗原結合活性を実施例1の3項に示すELISA法により測定した。
培養上清中に抗GD3キメラ抗体の生産が認められたウェルの形質転換株については、DHFR遺伝子増幅系を利用して抗体生産量を増加させる目的で、G418を0.5mg/ml、MTXを50nM含むEX−CELL302−dFBS(10)培地(dFBSを10%、L−Glnを2mM含むEX−CELL302培地)に1〜2×105細胞/mlになるように懸濁し、24ウェルプレート(Greiner社製)に2mlずつ分注した。5%CO2インキュベーター内で37℃で1〜2週間培養して、50nM MTX耐性を示す形質転換株を誘導した。形質転換株の増殖が認められたウェルの培養上清中の抗GD3キメラ抗体の抗原結合活性を実施例1の3項に示すELISA法により測定した。培養上清中に抗GD3キメラ抗体の生産が認められたウェルの形質転換株については、上記と同様の方法により、MTX濃度を100nM、200nMと順次上昇させ、最終的にG418を0.5mg/ml、MTXを200nMの濃度で含むEX−CELL302−dFBS(10)培地で増殖可能かつ、抗GD3キメラ抗体を高生産する形質転換株を得た。得られた形質転換株については、2回の限界希釈法による単一細胞化(クローン化)を行った。
【0134】
3.抗体のGD3に対する結合活性の測定(ELISA法)
抗体のGD3に対する結合活性は以下のようにして測定した。
4nmolのGD3を10μgのジパルミトイルフォスファチジルコリン(SIGMA社製)と5μgのコレステロール(SIGMA社製)とを含む2mlのエタノール溶液に溶解した。該溶液の20μl(40pmol/ウェルとなる)を96ウェルのELISA用のプレート(Greiner社製)の各ウェルにそれぞれ分注し、風乾後、1%牛血清アルブミン(以下、BSAと表記する;SIGMA社製)を含むPBS(以下、1%BSA−PBSと表記する)を100μl/ウェルで加え、室温で1時間反応させて残存する活性基をブロックした。1%BSA−PBSを捨て、形質転換株の培養上清或いは精製したヒト型キメラ抗体の各種希釈溶液を50μl/ウェルで加え、室温で1時間反応させた。反応後、各ウェルを0.05%Tween20(和光純薬社製)を含むPBS(以下、Tween−PBSと表記する)で洗浄後、1%BSA−PBSで3000倍に希釈したペルオキシダーゼ標識ヤギ抗ヒトIgG(H&L)抗体溶液(American Qualex社製)を二次抗体溶液として、50μl/ウェルで加え、室温で1時間反応させた。反応後、Tween−PBSで洗浄後、ABTS基質液[2,2’−アジノ−ビス(3−エチルベンゾチアゾリン−6−スルホン酸)アンモニウムの0.55gを1Lの0.1Mクエン酸緩衝液(pH4.2)に溶解し、使用直前に過酸化水素を1μl/mlで添加した溶液)を50μl/ウェルで加えて発色させ、415nmの吸光度(以下、OD415と表記する)を測定した。
【0135】
4.抗GD3キメラ抗体の精製
(1)YB2/0細胞由来の生産細胞の培養及び抗体の精製
上記実施例1の2項(1)で得られた抗GD3キメラ抗体を生産する形質転換細胞クローン をBSAを0.2%、MTXを200nM、トリヨードチロニン(以下、T3と表記する;SIGMA社製)を100nMの濃度で含むHybriddoma−SFM培地に3×105細胞/mlとなるように懸濁し、2.0Lスピナーボトル(岩城硝子社製)を用いて50rpmの速度で攪拌培養した。37℃の恒温室内で10日間培養後、培養上清を回収した。培養上清よりProsep−A(Bioprocessing社製)カラムを用いて、添付の説明書に従い、抗GD3キメラ抗体を精製した。精製した抗GD3キメラ抗体は、YB2/0−GD3キメラ抗体と名付けた。
(2)CHO/DG44細胞由来の生産細胞の培養及び抗体の精製
上記実施例1の2項(2)で得られた抗GD3キメラ抗体を生産する形質転換細胞クローンをL−Glnを3mM、脂肪酸濃縮液(以下、CDLCと表記する;GIBCO BRL社製)を0.5%、プルロニックF68(以下、PF68と表記する;GIBCO BRL社製)を0.3%の濃度で含むEX−CELL302培地に1×106細胞/mlとなるように懸濁し、175mm2フラスコ(Greiner社製)に50mlずつ分注した。5%CO2インキュベーター内で37℃で4日間培養後、培養上清を回収した。培養上清よりProsep−A(Bioprocessing社製)カラムを用いて、添付の説明書に従い、抗GD3キメラ抗体を精製した。精製した抗GD3キメラ抗体は、CHO/DG44−GD3キメラ抗体と名付けた。
(3)NS0細胞由来の生産細胞の培養及び抗体の精製
上記実施例1の2項(3)で得られた抗GD3キメラ抗体を生産する形質転換細胞クローンをL−Glnを2mM、G418を0.5mg/ml、MTXを200nM、FBSを1%の濃度で含むEX−CELL302培地に1×106細胞/mlとなるように懸濁し、175mm2フラスコ(Greiner社製)に200mlずつ分注した。5%CO2インキュベーター内で37℃で4日間培養後、培養上清を回収した。培養上清よりProsep−A(Bioprocessing社製)カラムを用いて、添付の説明書に従い、抗GD3キメラ抗体を精製した。精製した抗GD3キメラ抗体は、NS0−GD3キメラ抗体(302)と名付けた。また、該形質転換細胞クローンをG418を0.5mg/ml、MTXを200nMの濃度で含むGIT培地に3×105細胞/mlとなるように懸濁し、175mm2フラスコ(Greiner社製)に200mlずつ分注した。5%CO2インキュベーター内で37℃で10日間培養後、培養上清を回収した。培養上清よりProsep−A(Bioprocessing社製)カラムを用いて、添付の説明書に従い、抗GD3キメラ抗体を精製した。精製した抗GD3キメラ抗体は、NS0−GD3キメラ抗体(GIT)と名付けた。
(4)SP2/0細胞由来の生産細胞の培養及び抗体の精製
特開平5−304989に記載の抗GD3キメラ抗体を生産する形質転換細胞クローンをG418を0.5mg/ml、MTXを200nMの濃度で含むGIT培地に3×105細胞/mlとなるように懸濁し、175mm2フラスコ(Greiner社製)に200mlずつ分注した。5%CO2インキュベーター内で37℃で8日間培養後、培養上清を回収した。培養上清よりProsep−A(Bioprocessing社製)カラムを用いて、添付の説明書に従い、抗GD3キメラ抗体を精製した。精製した抗GD3キメラ抗体は、SP2/0−GD3キメラ抗体と名付けた。
【0136】
5.精製した抗GD3キメラ抗体の解析
上記実施例1の4項で得られた各種動物細胞で生産、精製した5種類の抗GD3キメラ抗体の各4μgを公知の方法[ネイチャー(Nature),227,680,1970]に従ってSDS−PAGEし、分子量及び製精度を解析した。その結果を第1図に示した。第1図に示したように、精製した各抗GD3キメラ抗体は、いずれも非還元条件下では分子量が約150キロダルトン(以下、Kdと表記する)の単一のバンドが、還元条件下では約50Kdと約25Kdの2本のバンドが認められた。これらの分子量は、抗体のH鎖及びL鎖のcDNAの塩基配列から推定される分子量(H鎖:約49Kd、L鎖:約23Kd、分子全体:約144Kd)とほぼ一致し、更に、IgG型の抗体は、非還元条件下では分子量は約150Kdであり、還元条件下では分子内のジスルフィド結合(以下、S−S結合と表記する)が切断され、約50Kdの分子量を持つH鎖と約25Kdの分子量を持つL鎖に分解されるという報告[アンティボディズ:ア・ラボラトリー・マニュアル(Antibodies:A Laboratory Manual),Cold Spring Harbor Laboratory,Chapter14,1988、モノクローナル・アンティボディズ:プリンシプルズ・アンド・プラクティス(Monoclonal Antibodies:principles and Practice),Academic Press Limited,1996]と一致し、各抗GD3キメラ抗体が正しい構造の抗体分子として発現され、かつ精製されたことが確認された。
【実施例2】
【0137】
抗GD3キメラ抗体の活性評価
1.抗GD3キメラ抗体のGD3に対する結合活性(ELISA法)
上記実施例1の4項で得られた5種類の精製抗GD3キメラ抗体のGD3(雪印乳業社製)に対する結合活性を実施例1の3項に示すELISA法により測定した。第2図は、添加する抗GD3キメラ抗体の濃度を変化させて結合活性を検討した結果である。第2図に示したように、5種類の抗GD3キメラ抗体は、ほぼ同等のGD3に対する結合活性を示した。この結果は抗体の抗原結合活性は、抗体を生産する動物細胞やその培養方法に関わらず、一定であることを示している。また、NS0−GD3キメラ抗体(302)とNS0−GD3キメラ抗体(GIT)の比較から抗原結合活性は、培養に用いる培地にも依らず、一定であることが示唆された。
【0138】
2.抗GD3キメラ抗体のin vitro細胞障害活性(ADCC活性)
上記実施例1の4項で得られた5種類の精製抗GD3キメラ抗体のin vitro細胞障害活性を評価するため、以下に示す方法に従い、ADCC活性を測定した。
(1)標的細胞溶液の調製 RPMI1640−FBS(10)培地で培養したヒトメラノーマ培養細胞株G−361(ATCC CRL1424)の1×106細胞を調製し、放射性物質であるNa251CrO4を3.7MBq当量加えて37℃で1時間反応させ、細胞を放射標識した。反応後、RPMI1640−FBS(10)培地で懸濁及び遠心分離操作により3回洗浄し、培地に再懸濁し、4℃で30分間氷中に放置して放射性物質を自然解離させた。遠心分離後、RPMI1640−FBS(10)培地を5ml加え、2×105細胞/mlに調製し、標的細胞溶液とした。
(2)エフェクター細胞溶液の調製 健常人静脈血50mlを採取し、ヘパリンナトリウム(武田薬品社製)0.5mlを加え穏やかに混ぜた。これをLymphoprep(Nycomed Pharma AS社製)を用いて使用説明書に従い、遠心分離して単核球層を分離した。RPMI1640−FBS(10)培地で3回遠心分離して洗浄後、培地を用いて2×106細胞/mlの濃度で再懸濁し、エフェクター細胞溶液とした。
(3)ADCC活性の測定 96ウェルU字底プレート(Falcon社製)の各ウェルに上記(1)で調製した標的細胞溶液の50μl(1×104細胞/ウェル)を分注した。次いで(2)で調製したエフェクター細胞溶液を100μl(2×105細胞/ウェル、エフェクター細胞と標的細胞の比は20:1となる)添加した。更に、各種抗GD3キメラ抗体を各最終濃度0.0025〜2.5μg/mlとなるように加え、37℃で4時間反応させた。反応後、プレートを遠心分離し、上清の51Cr量をγ−カウンターにて測定した。自然解離51Cr量は、エフェクター細胞溶液、抗体溶液の代わりに培地のみを用いて上記と同様の操作を行い、上清の51Cr量を測定することにより求めた。全解離51Cr量は、抗体溶液の代わりに培地のみを、エフェクター細胞溶液の代わりに1規定塩酸を添加し、上記と同様の操作を行い、上清の51Cr量を測定することにより求めた。ADCC活性は下式により求めた。
【0139】
【数1】
【0140】
その結果を第3図に示した。第3図に示したように、5種類の抗GD3キメラ抗体のうち、YB2/0−GD3キメラ抗体が最も高いADCC活性を示し、次いでSP2/0−GD3キメラ抗体、NS0−GD3キメラ抗体、CHO−GD3キメラ抗体の順に高いADCC活性を示した。培養に用いた培地の異なるNS0−GD3キメラ抗体(302)とNS0−GD3キメラ抗体(GIT)では、それらのADCC活性に差は認められなかった。以上の結果は、抗体のADCC活性は、生産に用いる動物細胞によって大きく異なることを示している。その機構としては、抗原結合活性が同等であったことから、抗体のFc領域の構造の差に起因していることが推定された。
【実施例3】
【0141】
抗ヒトインターロイキン5レセプターα鎖ヒト型CDR移植抗体の作製
1.抗ヒトインターロイキン5レセプターα鎖ヒト型CDR移植抗体の安定生産細胞の作製
(1)ラットミエローマYB2/0細胞を用いた生産細胞の作製
WO97/10354に記載の抗ヒトインターロイキン5レセプターα鎖ヒト型CDR移植抗体(以下、抗hIL−5RαCDR移植抗体と表記する)の発現ベクターpKANTEX1259HV3LV0を用いて抗hIL−5RαCDR移植抗体の安定生産細胞を以下のようにして作製した。
抗hIL−5RαCDR移植抗体発現ベクターpKANTEX1259HV3LV0の5μgを4×106細胞のラットミエローマYB2/0細胞へエレクトロポレーション法[サイトテクノロジー(Cytotechnology),3,133,1990]により導入後、40mlのRPMI1640−FBS(10)に懸濁し、96ウェル培養用プレート(住友ベークライト社製)に200μl/ウェルずつ分注した。5%CO2インキュベーター内で37℃、24時間培養した後、G418を0.5mg/mlになるように添加して1〜2週間培養した。G418耐性を示す形質転換株のコロニーが出現し、増殖の認められたウェルより培養上清を回収し、上清中の抗hIL−5RαCDR移植抗体の抗原結合活性を実施例3の2項に示すELISA法により測定した。
培養上清中に抗hIL−5RαCDR移植抗体の生産が認められたウェルの形質転換株については、DHFR遺伝子増幅系を利用して抗体生産量を増加させる目的で、G418を0.5mg/ml、MTXを50nM含むRPMI1640−FBS(10)培地に1〜2×105細胞/mlになるように懸濁し、24ウェルプレート(Greiner社製)に2mlずつ分注した。5%CO2インキュベーター内で37℃で1〜2週間培養して、50nM MTX耐性を示す形質転換株を誘導した。形質転換株の増殖が認められたウェルの培養上清中の抗hIL−5RαCDR移植抗体の抗原結合活性を実施例3の2項に示すELISA法により測定した。培養上清中に抗hIL−5RαCDR移植抗体の生産が認められたウェルの形質転換株については、上記と同様の方法により、MTX濃度を100nM、200nMと順次上昇させ、最終的にG418を0.5mg/ml、MTXを200nMの濃度で含むRPMI1640−FBS(10)培地で増殖可能かつ、抗hIL−5RαCDR移植抗体を高生産する形質転換株を得た。得られた形質転換株については、2回の限界希釈法による単一細胞化(クローン化)を行った。このようにして得られた抗hIL−5RαCDR移植抗体を生産する形質転換細胞クローンNo.3は平成11年4月5日付で工業技術院生命工学工業技術研究所(日本国茨城県つくば市東1丁目1番3号)にFERM BP−6690として寄託されている。
(2)CHO/dhfr−細胞を用いた生産細胞の作製
WO97/10354に記載の抗hIL−5RαCDR移植抗体発現ベクターpKANTEX1259HV3LV0の4μgを1.6×106細胞のCHO/dhfr−細胞へエレクトロポレーション法[サイトテクノロジー(Cytotechnology),3,133(1990)]により導入後、10mlのIMDM−FBS(10)に懸濁し、96ウェル培養用プレート(岩城硝子社製)に200μl/ウェルずつ分注した。5%CO2インキュベーター内で37℃、24時間培養した後、G418を0.5mg/mlになるように添加して1〜2週間培養した。G418耐性を示す形質転換株のコロニーが出現し、増殖の認められたウェルより培養上清を回収し、上清中の抗hIL−5RαCDR移植抗体の抗原結合活性を実施例3の2項に示すELISA法により測定した。
培養上清中に抗hIL−5RαCDR移植抗体の生産が認められたウェルの形質転換株については、DHFR遺伝子増幅系を利用して抗体生産量を増加させる目的で、G418を0.5mg/ml、MTXを10nM含むIMDM−dFBS(10)培地に1〜2×105細胞/mlになるように懸濁し、24ウェルプレート(岩城硝子社製)に0.5mlずつ分注した。5%CO2インキュベーター内で37℃で1〜2週間培養して、10nM MTX耐性を示す形質転換株を誘導した。増殖が認められたウェルの形質転換株については、上記と同様の方法により、MTX濃度を100nM、500nMに上昇させ、最終的にG418を0.5mg/ml、MTXを500nMの濃度で含むIMDM−dFBS(10)培地で増殖可能かつ、抗hIL−5RαCDR移植抗体を高生産する形質転換株を得た。得られた形質転換株については、2回の限界希釈法による単一細胞化(クローン化)を行った。
(3)マウスミエローマNS0細胞を用いた生産細胞の作製
ヤラントン(Yarranton)らの方法[バイオ/テクノロジー(BIO/TECHNOLOGY),10,169(1992)]に従い、WO97/10354に記載の抗hIL−5RαCDR移植抗体発現ベクターpKANTEX1259HV3LV0上の抗体H鎖及びL鎖cDNAを用いて抗hIL−5RαCDR移植抗体発現ベクターを作製し、NS0細胞を形質転換し、抗hIL−5RαCDR移植抗体を高生産する形質転換株を得た。得られた形質転換株については、2回の限界希釈法による単一細胞化(クローン化)を行った。
【0142】
2.抗体のhIL−5Rαに対する結合活性の測定(ELISA法)
抗体のhIL−5Rαに対する結合活性は以下のようにして測定した。
WO97/10354に記載の抗hIL−5Rαマウス抗体KM1257をPBSで10μg/mlの濃度に希釈した溶液の50μlを96ウェルのELISA用のプレート(Greiner社製)の各ウェルにそれぞれ分注し、4℃で20時間反応させた。反応後、1%BSA−PBSを100μl/ウェルで加え、室温で1時間反応させて残存する活性基をブロックした。1%BSA−PBSを捨て、WO97/10354に記載の可溶性hIL−5Rαを1%BSA−PBSで0.5μg/mlの濃度に希釈した溶液を50μl/ウェルで加え、4℃で20時間反応させた。反応後、各ウェルをTween−PBSで洗浄後、形質転換株の培養上清或いは精製したヒト型CDR移植抗体の各種希釈溶液を50μl/ウェルで加え、室温で2時間反応させた。反応後、各ウェルをTween−PBSで洗浄後、1%BSA−PBSで3000倍に希釈したペルオキシダーゼ標識ヤギ抗ヒトIgG(H&L)抗体溶液(American Qualex社製)を二次抗体溶液として、50μl/ウェルで加え、室温で1時間反応させた。反応後、Tween−PBSで洗浄後、ABTS基質液[2,2’−アジノ−ビス(3−エチルベンゾチアゾリン−6−スルホン酸)アンモニウムの0.55gを1Lの0.1Mクエン酸緩衝液(pH4.2)に溶解し、使用直前に過酸化水素を1μl/mlで添加した溶液]を50μl/ウェルで加えて発色させ、OD415を測定した。
【0143】
3.抗hIL−5RαCDR移植抗体の精製
(1)YB2/0細胞由来の生産細胞の培養及び抗体の精製
上記実施例3の1項(1)で得られた抗hIL−5RαCDR移植抗体を生産する形質転換細胞クローンをG418を0.5mg/ml、MTXを200nMの濃度で含むGIT培地に3×105細胞/mlとなるように懸濁し、175mm2フラスコ(Greiner社製)に200mlずつ分注した。5%CO2インキュベーター内で37℃で8日間培養後、培養上清を回収した。培養上清よりイオン交換クロマトグラフィー及びゲル濾過法を用いて抗hIL−5RαCDR移植抗体を精製した。精製した抗hIL−5RαCDR移植抗体は、YB2/0−hIL−5RCDR抗体と名付けた。
(2)CHO/dhfr−細胞由来の生産細胞の培養及び抗体の精製
上記実施例3の1項(2)で得られた抗hIL−5RαCDR移植抗体を生産する形質転換細胞クローンをL−Glnを3mM、CDLCを0.5%、PF68を0.3%の濃度で含むEX−CELL302培地に3×105細胞/mlとなるように懸濁し、4.0Lスピナーボトル(岩城硝子社製)を用いて100rpmの速度で攪拌培養した。37℃の恒温室内で10日間培養後、培養上清を回収した。培養上清よりイオン交換クロマトグラフィー及びゲル濾過法を用いて抗hIL−5RαCDR移植抗体を精製した。精製した抗hIL−5RαCDR移植抗体は、CHO/d−hIL−5RCDR抗体と名付けた。
(3)NS0細胞由来の生産細胞の培養及び抗体の精製
上記実施例3の1項(3)で得られた抗hIL−5RαCDR移植抗体を生産する形質転換細胞クローンをヤラントン(Yarranton)らの方法[バイオ/テクノロジー(BIO/TECHNOLOGY),10,169(1992)]に従い、培養後、培養上清を回収した。培養上清よりイオン交換クロマトグラフィー及びゲル濾過法を用いて抗hIL−5RαCDR移植抗体を精製した。精製した抗hIL−5RαCDR移植抗体は、NS0−hIL−5RCDR抗体と名付けた。
【0144】
4.精製した抗hIL−5RαCDR移植抗体の解析
上記実施例3の3項で得られた各種動物細胞で生産、精製した3種類の抗hIL−5RαCDR移植抗体の各4μgを公知の方法[ネイチャー(Nature),227,680(1970)]に従ってSDS−PAGEし、分子量及び製精度を解析した。その結果を第4図に示した。第4図に示したように、精製した各抗hIL−5RαCDR移植抗体は、いずれも非還元条件下では分子量が約150Kdの単一のバンドが、還元条件下では約50Kdと約25Kdの2本のバンドが認められた。これらの分子量は、抗体のH鎖及びL鎖のcDNAの塩基配列から推定される分子量(H鎖:約49Kd、L鎖:約23Kd、分子全体:約144Kd)とほぼ一致し、更に、IgG型の抗体は、非還元条件下では分子量は約150Kdであり、還元条件下では分子内のジスルフィド結合(以下、S−S結合と表記する)が切断され、約50Kdの分子量を持つH鎖と約25Kdの分子量を持つL鎖に分解されるという報告[アンティボディズ:ア・ラボラトリー・マニュアル(Antibodies:A Laboratory Manual),Cold Spring Harbor Laboratory,Chapter 14,1988、モノクローナル・アンティボディズ:プリンシプルズ・アンド・プラクティス(Monoclonal Antibodies:Principles and Practice),Academic Press Limited,1996]と一致し、各抗hIL−5RαCDR移植抗体が正しい構造の抗体分子として発現され、かつ、精製されたことが確認された。
【実施例4】
【0145】
抗hIL−5RαCDR移植抗体の活性評価
1.抗hIL−5RαCDR移植抗体のhIL−5Rαに対する結合活性(ELISA法)
上記実施例3の3項で得られた3種類の精製抗hIL−5RαCDR移植抗体のhIL−5Rαに対する結合活性を実施例3の2項に示すELISA法により測定した。第5図は、添加する抗hIL−5RαCDR移植抗体の濃度を変化させて結合活性を検討した結果である。第5図に示したように、3種類の抗hIL−5RαCDR移植抗体は、ほぼ同等のhIL−5Rαに対する結合活性を示した。この結果は実施例2の1項の結果と同様に、抗体の抗原結合活性は、抗体を生産する動物細胞やその培養方法に関わらず、一定であることを示している。
【0146】
2.抗hIL−5RαCDR移植抗体のin vitro細胞障害活性(ADCC活性)
上記実施例3の3項で得られた3種類の精製抗hIL−5RαCDR移植抗体のin vitro細胞障害活性を評価するため、以下に示す方法に従い、ADCC活性を測定した。
(1)標的細胞溶液の調製
WO97/10354に記載のhIL−5Rα鎖及びβ鎖を発現しているマウスT細胞株CTLL−2(h5R)をRPMI1640−FBS(10)培地で培養し、1×106細胞/0.5mlとなるように調製し、放射性物質であるNa251CrO4を3.7MBq当量加えて37℃で1.5時間反応させ、細胞を放射標識した。反応後、RPMI1640−FBS(10)培地で懸濁及び遠心分離操作により3回洗浄し、培地に再懸濁し、4℃で30分間氷中に放置して放射性物質を自然解離させた。遠心分離後、RPMI1640−FBS(10)培地を5ml加え、2×105細胞/mlに調製し、標的細胞溶液とした。
(2)エフェクター細胞溶液の調製
健常人静脈血50mlを採取し、ヘパリンナトリウム(武田薬品社製)0.5mlを加え穏やかに混ぜた。これをPolymorphprep(Nycomed Pharma AS社製)を用いて使用説明書に従い、遠心分離して単核球層を分離した。RPMI1640−FBS(10)培地で3回遠心分離して洗浄後、培地を用いて9×106細胞/mlの濃度で再懸濁し、エフェクター細胞溶液とした。
(3)ADCC活性の測定
96ウェルU字底プレート(Falcon社製)の各ウェルに上記(1)で調製した標的細胞溶液の50μl(1×104細胞/ウェル)を分注した。次いで(2)で調製したエフェクター細胞溶液を100μl(9×105細胞/ウェル、エフェクター細胞と標的細胞の比は90:1となる)添加した。更に、各種抗hIL−5RαCDR移植抗体を各最終濃度0.001〜0.1μg/mlとなるように加え、37℃で4時間反応させた。
反応後、プレートを遠心分離し、上清の51Cr量をγ−カウンターにて測定した。自然解離51Cr量は、エフェクター細胞溶液、抗体溶液の代わりに培地のみを用いて上記と同様の操作を行い、上清の51Cr量を測定することにより求めた。全解離51Cr量は、抗体溶液の代わりに培地のみを、エフェクター細胞溶液の代わりに1規定塩酸を添加し、上記と同様の操作を行い、上清の51Cr量を測定することにより求めた。
ADCC活性は下式により求めた。
【0147】
【数2】
【0148】
その結果を第6図に示した。第6図に示したように、3種類の抗hIL−5RαCDR移植抗体のうち、YB2/0−hIL−5RCDR抗体が最も高いADCC活性を示し、次いでCHO/d−hIL−5RCDR抗体、NS0−hIL−5RCDR抗体の順に高いADCC活性を示した。以上の結果は実施例2の2項の結果と同ように、抗体のADCC活性は、生産に用いる動物細胞によって大きく異なることを示している。更に、2種類のヒト化抗体のいずれの場合もYB2/0細胞で生産した抗体が最も高いADCC活性を示したことから、YB2/0細胞を用いることにより、ADCC活性の高い抗体を製造できることが明らかとなった。
【0149】
3.抗hIL−5RαCDR移植抗体のin vivoにおける活性評価
上記実施例3の3項で得られた3種類の精製抗hIL−5RαCDR移植抗体のin vivoにおける活性を評価するため、以下に示す方法に従い、カニクイザルのhIL−5誘発好酸球増加モデルに対する抑制作用を検討した。
カニクイザルに初日よりhIL−5(調製方法はWO97/10354に記載)を1μg/kgで1日1回、計14回背部皮下より投与した。各種抗hIL−5RαCDR移植抗体を0日のhIL−5の投与1時間前に0.3mg/kgで静脈内に単回投与した。抗体非投与群をコントロールとして用いた。抗体投与群は各群3頭(No.301、No.302、No.303、No.401、No.402、No.403、No.501、No.502、No.503)、抗体非投与群は2頭(No.101、No.102)のカニクイザルを用いた。投与開始の7日前より投与後42日目まで経時的に約1mlの血液を伏在静脈または大腿静脈より採取し、1μlの末梢血中の好酸球数を測定した。その結果を第7図に示した。第7図に示したように、YB2/0−hIL−5RCDR抗体を投与した群では、血中好酸球の増加が完全に抑制された。一方、CHO/d−hIL−5RCDR抗体の投与群では、1頭で完全な抑制作用が認められたものの、2頭ではその抑制作用は不充分であった。NS0−hIL−5RCDR抗体の投与群では、完全な抑制作用は認められず、その効果は不充分であった。以上の結果は、抗体のin vivo活性は、生産に用いる動物細胞によって大きく異なることを示している。更に、抗hIL−5RαCDR移植抗体ではそのin vivo活性の高さは、実施例4の2項で述べたADCC活性の高さと正の相関が認められたことから、その活性発現には、ADCC活性の高さが極めて重要であることが示唆された。
以上の結果から、ADCC活性の高い抗体は、ヒトの各種疾患の臨床においても有用であることが期待される。
【実施例5】
【0150】
ADCC活性を高める糖鎖の解析
1.2−アミノピリジン標識糖鎖(PA化糖鎖)の調製
本発明のヒト化抗体を塩酸による酸加水分解にてシアル酸を除去した。塩酸を完全に除去した後、ヒドラジン分解により糖鎖を蛋白質から切断した[メソッド・オブ・エンザイモロジー(Method of Enzymology),83,263,1982]。ヒドラジンを除去した後、酢酸アンモニウム水溶液と無水酢酸加えてN−アセチル化を行った。凍結乾燥後、2−アミノピリジンによる蛍光標識を行った[ジャーナル・オブ・バイオケミストリー(J.Biochem.),95,197(1984)]。蛍光標識した糖鎖(PA化糖鎖)を、Surperdex Peptide HR 10/30カラム(Pharmacia社製)を用いて不純物と分離した。糖鎖画分を遠心濃縮機にて乾固させ、精製PA化糖鎖とした。
【0151】
2.精製抗hIL−5RαCDR移植抗体のPA化糖鎖の逆相HPLC分析
上記実施例5の1項で調製した各種抗hIL−5RCDR抗体のPA化糖鎖を用いて、CLC−ODSカラム(Shimadzu社製)による逆相HPLC分析を行った。過剰量のα−L−フコシダーゼ(ウシ腎由来、SIGMA社製)をPA化糖鎖に添加して消化を行い(37℃、15時間)、逆相HPLCで分析した(第8図)。アスパラギン結合糖鎖は30分間から80分間の範囲に溶出することをTaKaRa社製PA化糖鎖スタンダードを用いて確認した。α−L−フコシダーゼ消化によって、逆相HPLCの溶出位置が移動する糖鎖(48分間から78分間に溶出される糖鎖)の全体に占める割合を計算した。結果を第1表に示す。
【0152】
【表1】
【0153】
YB2/0細胞で生産させた抗hIL−5RCDR移植抗体は約47%、NS0細胞で生産させた抗hIL−5RCDR移植抗体は約73%がα1−6フコースをもつ糖鎖であった。よって、YB2/0細胞で生産した抗体は、NS0細胞で生産した抗体と比較してα1−6フコースを持たない糖鎖が多かった。
【0154】
3.精製抗hIL−5RαCDR移植抗体の単糖組成分析
トリフルオロ酢酸による酸加水分解により、YB2/0細胞、NS0細胞およびCHO/d細胞で生産した抗hIL−5RαCDR移植抗体の糖鎖を単糖に分解し、BioLC(Dionex社製)を用いて単糖組成分析を行った。
N−グリコシド結合糖鎖のうち、コンプレックス型では、1本の糖鎖におけるマンノース数は3であるため、マンノースを3として計算した場合の各単糖の相対比を第2表に示す。
【0155】
【表2】
【0156】
フコースの相対比は、YB2/0<CHO/d<NS0であり、本結果でもYB2/0細胞で生産した抗体の糖鎖はフコース含量が最も低かった。
【実施例6】
【0157】
CHO/dhfr−細胞生産抗体の糖鎖解析
CHO/dhfr−細胞で生産した精製抗hIl−5RαCDR移植抗体からPA化糖鎖を調製し、CLC−ODSカラム(島津社製)を用いて逆相HPLC分析を行った(第9図)。第9図において、溶出時間35〜45分間がフコースを持たない糖鎖、45〜60分間がフコースを持つ糖鎖であった。CHO/dhfr−細胞で生産した抗hIl−5RαCDR移植抗体は、マウスミエローマNS0細胞で生産させた抗体と同様に、ラットミエローマYB2/0細胞で生産させた抗体よりもフコースを持たない糖鎖の含量が少なかった。
【実施例7】
【0158】
高ADCC活性抗体の分離
フコースを持つ糖鎖に結合するレクチンカラムを用いて、ラットミエローマYB2/0細胞で生産させた抗hIl−5RαCDR移植抗体の分離を行った。HPLCは島津社製LC−6Aを用い、流速は1ml/分、カラム温度は室温で行った。50mMトリス−硫酸緩衝液(pH7.3)で平衡化し、精製された抗hIL−5RαCDR移植抗体を注入後、0.2Mα−メチルマンノシド(ナカライテスク社製)の直線濃度勾配(60分間)にて溶出した。抗hIl−5RαCDR移植抗体を非吸着画分と吸着画分とに分離した。
非吸着画分、吸着画分の一部をとり、hIL−5Rαに対する結合活性を測定すると、同様の結合活性を示した(第10図 上図)。ADCC活性を測定すると、非吸着画分の方が吸着画分の一部よりも高いADCC活性を示した(第10図 下図)。さらに、非吸着画分、吸着画分の一部からPA化糖鎖を調製し、CLC−ODSカラム(島津社製)を用いて逆相HPLC分析を行った(第11図)。非吸着画分は主としてフコースのない糖鎖をもつ抗体であり、吸着画分の一部は主としてフコースがある糖鎖もつ抗体であった。
【実施例8】
【0159】
宿主細胞株におけるα1,6−フコシルトランスフェラーゼ(FUT8)遺伝子の転写物の定量
(1)各種細胞株由来一本鎖cDNAの調製
チャイニーズハムスター卵巣由来CHO/DG44細胞を、10%FBS(Life Technologies社)および1倍濃度のHT supplement(Life Technologies社)を添加したIMDM培地(Life Technologies社)に懸濁し、2×105cells/mlの密度で接着細胞培養用T75フラスコ(Greiner社)に播種した。またラットミエローマ由来YB2/0細胞を、10%FBS(Life Technlogies社)、4mMグルタミン(Life Technologies社)を添加したRPMI1640培地(Life Technologies社)に懸濁し、2×105cells/mlの密度で浮遊細胞培養用T75フラスコ(Greiner社)に播種した。これらを37℃の5%CO2インキュベーター内で培養し、培養1日目、2日目、3日目、4日目および5日目に各宿主細胞1×107cellsを回収し、RNAeasy(QIAGEN社製)により全RNAを抽出した。
全RNAを45μlの滅菌水に溶解し、RQ1 Rnase−Free DNase(Promega社)0.5U/μl、付属の10×DNase buffer 5μl、RNasin Ribonuclease inhibitor(Promega社)0.5μlをそれぞれに添加して、37℃で30分間反応させた。反応後、RNAeasy(QIAGEN社)により全RNAを再精製し、50μlの滅菌水に溶解した。
得られた各々の全RNA3μgについて、オリゴ(dT)をプライマーとしてSUPERSCRIPTTM Preamplification System for First Strand cDNA Synthesis(Life Technologies社)により、20μlの系で逆転写反応を行い、cDNAを合成した。各宿主細胞由来FUT8およびβ−アクチンのクローニングには逆転写反応後の溶液の1倍濃度液を、競合的PCRによる各遺伝子転写量の定量には逆転写反応後の溶液を水で50倍希釈したものを用い、各々使用するまで−80℃で保管した。
【0160】
(2)チャイニーズハムスターFUT8およびラットFUT8の各cDNA部分断片の取得
チャイニーズハムスターFUT8およびラットFUT8の各cDNA部分断片の取得は、以下のように行った。まず、ヒトFUT8のcDNA[Journal of Biochemistry,121,626(1997)]およびブタFUT8のcDNA[Journal of Biological Chemistry,271,27810(1996)]に共通の塩基配列に対して特異的なプライマー(配列番号1および配列番号2に示す)を設計した。
次にDNAポリメラーゼExTaq(宝酒造社)を用いて、(1)で調製した培養2日目のCHO細胞由来cDNAおよびYB2/0細胞由来cDNAを各々1μlを含む25μlの反応液[ExTaq buffer(宝酒造社)、0.2mM dNTPs、0.5μM上記特異的プライマー(配列番号1および配列番号2)]を調製し、ポリメラーゼ連鎖反応(PCR)を行った。PCRは、94℃で1分間の加熱の後、94℃で30秒間、55℃で30秒間、72℃で2分間からなる反応を1サイクルとして30サイクルの後、さらに72℃で10分間加熱する条件で行った。PCRにより得た各特異的増幅断片979bpをTOPO TA cloning Kit(Invitrogen社)を用いてプラスミドpCR2.1へ連結し、チャイニーズハムスターFUT8およびラットFUT8の各cDNA部分断片を含むプラスミド(CHFT8−pCR2.1およびYBFT8−pCR2.1)を取得した。
取得した各cDNAの塩基配列について、DNAシークエンサー377(Parkin
Elmer社)およびBigDye Terminator Cycle Sequencing FS Ready Reaction Kit(Parkin Elmer社)を用いて決定し、取得したcDNAがチャイニーズハムスターFUT8およびラットFUT8(配列番号3および4に示す)のオープンリーディングフレーム(ORF)部分配列をコードすることを確認した。
【0161】
(3)チャイニーズハムスターβ−アクチンおよびラットβ−アクチンcDNAの取得 β−アクチン遺伝子は各細胞において恒常的に転写されており、その転写量は細胞間で同程度と考えられているため、各細胞由来cDNA合成反応の効率の目安としては、β−アクチン遺伝子の転写量を定量する。
チャイニーズハムスターβ−アクチンおよびラットβ−アクチンの取得は、以下の方法で行った。まず、チャイニーズハムスターβ−アクチンゲノム配列[GenBank,U20114]およびラットβ−アクチンゲノム配列[Nucleic Acid Research,11,1759(1983)]より、翻訳開始コドンを含む共通配列に特異的なフォワードプライマー(配列番号5に示す)および翻訳終止コドンを含む各配列特異的なリバースプライマー(配列番号6および配列番号7に示す)を設計した。
次にDNAポリメラーゼKOD(東洋紡社)を用いて、(1)で調製した培養2日目のCHO細胞由来cDNAおよびYB2/0細胞由来cDNA 1μlを含む25μlの反応液[KOD buffer #1(東洋紡社)、0.2mM dNTPs、1mM MgCl2、0.4μM 上記遺伝子特異的プライマ−(配列番号5および配列番号6、または配列番号5および配列番号7)、5% DMSO]を調製し、ポリメラーゼ連鎖反応(PCR)を行った。PCRは、94℃で4分間の加熱の後、98℃で15秒間、65℃で2秒間、74℃で30秒間からなる反応を1サイクルとして、25サイクル行った。PCRにより得た各特異的増幅断片1128bpの5’末端をMEGALABEL(宝酒造社)によりリン酸化した後、pBluescriptII KS(+)(Strategene社)に、制限酵素EcoRVにより切断して得られる断片(2.9Kb)をLigation High(東洋紡社)で連結し、チャイニーズハムスターβ−アクチンおよびラットβ−アクチンの各cDNAのORF全長を含むプラスミド(CHAc−pBS、YBAc−pBS)を得た。
取得した各cDNAの塩基配列を、DNAシークエンサー377(Parkin Elmer社製)およびBigDye Terminator Cycle Sequencing FS Ready Reaction Kit(Parkin Elmer社製)を用いて決定し、各々チャイニーズハムスターβ−アクチンおよびラットβ−アクチンの各cDNAのORF全長配列をコードすることを確認した。
【0162】
(4)スタンダードおよび内部配列のコントロールの調製
生産細胞内のFUT8遺伝子からのmRNA転写量を測定するために、まず検量線を作成した。
検量線に用いるFUT8のスタンダードとしては、(2)で得たチャイニーズハムスターFUT8およびラットFUT8の各cDNA部分断片をpCR2.1に組み込んだプラスミドであるCHFT8−pCR2.1およびYBFT8−pCR2.1を制限酵素EcoRIで切断して得られたDNAを直鎖化して用いた。
FUT8定量の内部コントロールとしては、CHFT8−pCR2.1およびYBFT8−pCR2.1のうち、チャイニーズハムスターFUT8およびラットFUT8の内部塩基配列のScaI−HindIII間203bpを欠失させることにより得られたCHFT8d−pCR2.1およびYBFT8d−pCR2.1を、制限酵素EcoRIで切断して得られたDNAを直鎖化して用いた。
生産細胞内のβ−アクチン遺伝子からのmRNA転写量のスタンダードとしては、(3)で得たチャイニーズハムスターβ−アクチンおよびラットβ−アクチンのそれぞれのcDNAのORF全長をpBluescriptII KS(+)に組み込んだプラスミドであるCHAc−pBSおよびYBAc−pBSを、前者はHindIIIおよびPstIで、後者はHindIIIおよびKpnIで、各々切断して得られたDNAを直鎖化して用いた。
β−アクチン定量の内部コントロールとしては、CHAc−pBSおよびYBAc−pBSのうち、チャイニーズハムスターβ−アクチンおよびラットβ−アクチンの内部塩基配列のDraIII−DraIII間180bpを欠失させることにより得られたCHAcd−pBSおよびYBAcd−pBSを、前者はHindIIIおよびPstIで、後者はHindIIIおよびKpnIで、各々切断して得られたDNAを直鎖化して用いた。
【0163】
(5)競合的RT−PCRによる転写量の定量
FUT8の転写産物の定量は以下のように行った。まず(2)で得たチャイニーズハムスターFUT8およびラットFUT8のORF部分配列の内部配列に対し、共通配列特異的なプライマーセット(配列番号8および9に示す)を設計した。
次に、(1)で得られた各宿主細胞株由来のcDNA溶液の50倍希釈液 5μlおよび内部コントロール用プラスミド5μl(10fg)を含む総体積20μlの反応液[ExTaq buffer(宝酒造社)、0.2mM dNTPs、0.5μM 上記遺伝子特異的プライマー(配列番号8および配列番号9)、5%DMSO]で、DNAポリメラーゼExTaq(宝酒造社)を用いてPCRを行った。PCRは、94℃で3分間の加熱の後、94℃で1分間、60℃で1分間、72℃で1分間からなる反応を1サイクルとして32サイクル行った。
β−アクチンの転写産物の定量は、以下のように行った。(3)で得たチャイニーズハムスターβ−アクチンおよびラットβ−アクチンORF全長の内部配列に対し、各遺伝子特異的なプライマーセット(前者を配列番号10および配列番号11に、後者を配列番号12および配列番号13に示す)をそれぞれ設計した。
次に、(1)で得られた各宿主細胞株由来のcDNA溶液の50倍希釈液 5μlおよび内部コントロール用プラスミド5μl(1pg)を含む総体積20μlの反応液[ExTaq buffer(宝酒造社)、0.2mM dNTPs、0.5μM 上記遺伝子特異的プライマー(配列番号10および配列番号11、または配列番号12および配列番号13)、5% DMSO]で、DNAポリメラーゼExTaq(宝酒造社)を用いてPCRを行った。PCRは、94℃で3分間の加熱の後、94℃で30秒間、65℃で1分間、72℃で2分間からなる反応を1サイクルとして17サイクル行った。
【0164】
【表3】
【0165】
第3表に記載のプライマーセットを用いて定量的PCRを行った。その結果、各遺伝子転写産物および各スタンダードからは、第3表のターゲット欄に示したサイズのDNA断片を、各内部コントロールからは、第3表のコンペティター欄に示したサイズのDNA断片を増幅させることができた。
PCR後の溶液のうち、7μlを1.75%アガロースゲル電気泳動に供した後、ゲルをSYBR Green I Nucleic Acid Gel Stain(Molecular Probes社)で染色した。増幅された各DNA断片の発光強度をフルオロイメージャー(FluorImager SI;Molecular Dynamics社)で算出することにより、増幅されたDNA断片の量を測定した。
一方、細胞由来cDNAに代えて(4)で調製したスタンダードプラスミド量を0.1fg、1fg、5fg、10fg、50fg、100fgおよび500fgとしてPCRをそれぞれ行い、増幅産物量を測定した。そして、その測定値とスタンダードプラスミド量をプロットして検量線を作成した。
上述の検量線を用いて、各細胞由来全cDNAを鋳型とした場合の増幅産物の量より各細胞中の目的遺伝子cDNA量を算出し、これを各細胞におけるmRNA転写量とした。
ラットFUT8配列をスタンダード、内部コントロールに用いた場合の各宿主細胞株におけるFUT8転写産物の量を第12図に示した。培養期間を通じてCHO細胞株はYB2/0細胞株の10倍以上の転写量を示した。この傾向は、チャイニーズハムスターFUT8配列をスタンダード、内部コントロールに用いた場合にも認められた。
また、第4表にβ−actin転写産物の量との相対値としてFUT8転写量を示した。
【0166】
【表4】
【0167】
YB2/0細胞株のFUT8転写量がβ−actinの0.1%前後であるのに対し、CHO細胞株は0.5〜2%であった。
以上の結果より、YB2/0細胞株のPUT8転写産物量はCHO細胞株のそれよりも有意に少ないことが示された。
【産業上の利用可能性】
【0168】
本発明は、抗体、蛋白質またはペプチドなどの免疫機能分子の有する活性を調節する糖鎖、および糖鎖を有する抗体、蛋白質またはペプチドに関する。本発明は更に、該糖鎖および糖鎖を有する抗体、蛋白質またはペプチドの製造法、ならびにそれらを有効成分として含有する診断薬、予防薬および治療薬に関する。
【技術分野】
【0001】
本発明は、抗体、蛋白質またはペプチドなどの免疫機能分子の活性を調節する方法、免疫機能分子活性促進剤、および免疫機能活性が促進された免疫機能分子に関する。
【背景技術】
【0002】
抗体は、高い結合活性、結合特異性及び血中での高い安定性を有することから、ヒトの各種疾患の診断、予防及び治療への応用が試みられてきた[モノクローナル・アンティボディズ:プリンシプルズ・アンド・アプリケーションズ(Monoclonal Antibodies:Principles and Applications),Wiley−Liss,Inc.,Capter 2.1(1995)]。しかし、マウス抗体のようなヒト以外の動物の抗体はヒトに投与すると、異物として認識されることにより、ヒト体内にマウス抗体に対するヒト抗体(Human Anti Mouse Antibody:以下、HAMAと表記する)が誘導され、HAMAは投与したマウス抗体と反応して副作用を引き起こしたり[ジャーナル・オブ・クリニカル・オンコロジー(J.Clin.Oncol.),2,881(1984);ブラッド(Blood),65,1349(1985);ジャーナル・オブ・ザ・ナショナル・キャンサー・インスティテュート(J.Natl.Cancer Inst.),80,932(1988);プロシーディングス・オブ・ザ・ナショナル・アカデミー・オブ・サイエンス(Proc.Natl.Acad.Sci.U.S.A),82,1242(1985)]、投与したマウス抗体の血中からの消失を速め[ジャーナル・オブ・ニュークレアー・メディシン(J.Nucl,Med.),26,1011(1985);ブラッド(Blood),65,1349(1985);ジャーナル・オブ・ザ・ナショナル・キャンサー・インスティテュート(J.Natl.Cancer Inst.),80,937(1988)]、マウス抗体の診断、予防及び治療の効果を減じてしまうことが知られている[ザ・ジャーナル・オブ・イムノロジー(J.Immunol.),135,1530(1985);キャンサー・リサーチ(Cancer Res.),46,6489(1986)]。
【0003】
これらの問題点を解決するため、遺伝子組換え技術を利用してヒト以外の動物の抗体をヒト型キメラ抗体或いはヒト型相補性決定領域(以下、CDRと表記する)移植抗体の様なヒト化抗体を作製することが試みられている。ヒト型キメラ抗体とは、抗体可変領域(以下、V領域と表記する)がヒト以外の動物の抗体で、定常領域(以下、C領域と表記する)がヒト抗体である抗体であり[プロシーディングス・オブ・ザ・ナショナル・アカデミー・オブ・サイエンス(Proc.Natl.Acad.Sci.U.S.A),81,6851(1984)]、ヒトに投与した場合、重篤な副作用は認められず、かつマウス抗体に比べ血中半減期が約6倍伸びたことが報告されている[プロシーディングス・オブ・ザ・ナショナル・アカデミー・オブ・サイエンス(Proc.Natl.Acad.Sci.U.S.A),86,4220(1989)]。ヒト型CDR移植抗体とは、ヒト抗体のCDRをヒト以外の動物の抗体のCDRと置換した抗体であり[ネイチャー(Nature),321,522(1986)]、サルを用いた実験でマウス抗体に比べ免疫原性が低下し、血中半減期が4〜5倍伸びたことが報告されている[ザ・ジャーナル・オブ・イムノロジー(J.Immunol.),147,1352(1991)]。これらの報告は、ヒトの各種疾患の診断、予防及び治療に応用する抗体としては完全なヒト抗体でなくてもヒト化抗体で十分な効果が期待できることを示しているが、実際に、抗腫瘍抗体に関しては、例えば抗CD20ヒト型キメラ抗体であるリツキサン[Rituxan;アイデック(IDEC)社]や抗HER2/neuヒト型CDR移植抗体であるハーセプチン[Herceptin;ジェネンテック(Genentech)社]の臨床試験が行われ、それぞれBリンパ腫、乳癌において安全性及びある程度の治療効果が認められている[ジャーナル・オブ・クリニカル・オンコロジー(J.Clin.Oncol.),16,2825(1998)、ジャーナル・オブ・ザ・ナショナル・キャンサー・インスティテュート(J.National Cancer Institute),90,882(1998)]。また、既に抗GPIIb/IIIaヒト型キメラ抗体の断片(Fab’)であるレオプロ[ReoPro;セントコア(Centocor)社]は経皮経管冠動脈血管形成術後の合併症予防薬として欧米で販売されている。現在、これら以外にも多数のヒト化抗体の臨床試験が行われているが、ヒト化抗体の多くは遺伝子組換え技術を用いて作製され、適当な動物細胞を用いて製造されている。
【0004】
哺乳類の抗体には、IgM、IgD、IgG、IgA、IgEの5種類のクラスが存在することが明らかとなっているが、ヒトの各種疾患の診断、予防及び治療には血中半減期が長く、各種エフェクター機能を有する等の機能特性からヒトIgGクラスの抗体が主として利用されている[モノクローナル・アンティボディズ:プリンシプルズ・アンド・アプリケーションズ(Monoclonal Antibodies:Principles and Applications),Wiley−Liss,Inc.,Capter 1(1995)]。ヒトIgGクラスの抗体は、更にIgG1、IgG2、IgG3、IgG4の4種類のサブクラスに分類されている。IgGクラスの抗体のエフェクター機能である抗体依存性細胞障害活性(以下、ADCC活性と表記する)や補体依存性細胞障害活性(以下、CDC活性と表記する)については、これまでに多数の研究が行われ、ヒトIgGクラスでは、IgG1サブクラスの抗体が最も高いADCC活性、CDC活性を有していることが報告されている[ケミカル・イムノロジー(Chemical Immunology),65,88(1997)]。以上の観点から、上記のリツキサン、ハーセプチンを始めとして、その効果発現に高いエフェクター機能を必要とする抗腫瘍ヒト化抗体の殆どはヒトIgG1サブクラスの抗体である。
【0005】
ヒトIgG1サブクラスの抗体のADCC活性及びCDC活性の発現には、抗体Fc領域と、キラー細胞、ナチュラルキラー細胞、活性化されたマクロファージ等のエフェクター細胞表面上に存在する抗体レセプター(以下、FcγRと表記する)及び各種補体成分との結合が必要であり、その結合については、抗体のヒンジ領域及びC領域の第2番目のドメイン(以下、Cγ2ドメインと表記する)内のいくつかのアミノ酸残基の重要性[ヨーロピアン・ジャーナル・オブ・イムノロジー(Eur.J.Immunol.),23,1098(1993)、イムノロジー(Immunology),86,319(1995)、ケミカル・イムノロジー(Chemical Immunology),65,88(1997)]の他、Cγ2ドメインに結合している糖鎖の重要性[ケミカル・イムノロジー(Chemical Immunology),65,88(1997)]が示唆されている。糖鎖に関しては、ボイド(Boyd)らは、チャイニーズハムスター卵巣細胞(CHO細胞)或いはマウスミエローマNS0細胞で生産したヒト型CDR移植抗体CAMPATH−1H(ヒトIgG1サブクラス)を各種糖分解酵素で処理し、糖鎖のADCC活性、CDC活性に対する影響を検討した結果、非還元末端のシアル酸の除去は、両活性に影響を与えないが、更にガラクトース残基を除去することでCDC活性のみが影響を受け、約50%程度活性が低下すること、糖鎖の完全な除去は、両活性を消失させることを報告した[モレキュラー・イムノロジー(Molecular Immunol.),32,1311(1995)]。また、ライフリー(Lifely)らは、CHO細胞、NS0細胞或いはラットミエローマY0細胞で生産したヒト型CDR移植抗体CAMPATH−1H(ヒトIgG1サブクラス)の糖鎖の分析及びADCC活性を測定した結果、Y0細胞由来のCAMPATH−1Hが最も高いADCC活性を示し、その活性にはバイセクティングに位置するN−アセチル−グルコサミンが重要であることを示唆した[グリコバイオロジー(Glycobiology),5,813(1995):WO99/54342]。これらの報告は、ヒトIgG1サブクラスの抗体のエフェクター機能に糖鎖の構造が極めて重要な役割を果たしており、糖鎖の構造を変えることでより高いエフェクター機能を有する抗体を作製することが可能であることを示している。しかし、実際には糖鎖の構造は多様かつ複雑であり、エフェクター機能に真に重要な構造は未だ特定できていない。
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明の目的は、各種動物細胞で生産したヒトIgG1サブクラスの抗体の糖鎖を分析してADCC活性を高める糖鎖を特定し、免疫機能分子の活性を調節する方法を提供することにある。そのような抗体は、ADCC活性が増強し、抗腫瘍抗体のみならずその他の抗体、さらには蛋白質またはペプチドの場合でも、ヒトの各種疾患に対する治療効果の増大が期待される。特に抗腫瘍抗体の臨床応用では、現状では抗体単独の抗腫瘍効果は不充分な場合が多く、化学療法との併用療法が行われているが[サイエンス(Science),280,1197,1998]、ADCC活性の増強による抗体単独のより強い抗腫瘍効果が認められれば、化学療法に対する依存度が低くなり、副作用の低減も期待できる。
【課題を解決するための手段】
【0007】
本発明者らは、2種類のチャイニーズハムスター卵巣細胞であるCHO/dhFr−細胞(ATCC CRL9096)及びCHO/DG44細胞[ソマティック・セル・アンド・モレキュラー・ジェネティクス(Somatic Cell and Molecular Genetics),12,555(1986)]、マウスミエローマNS0細胞[RCB0213、バイオ/テクノロジー(BIO/TECHNOLOGY),10,169(1992)]、マウスミエローマSP2/0−Ag14細胞(以下、SP2/0細胞と表記する;ATCC CRL1581)、ラットミエローマYB2/3HL.P2.G11.16Ag.20細胞(以下、YB2/0細胞と表記する;ATCC CRL1662)で生産したヒトIgG1サブクラスの各種ヒト化抗体のin vitroの活性評価を行った結果、ラットミエローマYB2/0細胞で生産したヒト化抗体のADCC活性が他の細胞で生産したヒト化抗体に比べ著しく高いことを見出した。更にカニクイザルを用いたin vivoの活性評価を行った結果、YB2/0細胞で生産したヒト化抗体が最も高い効果を示すことを見出し、ADCC活性の高い抗体のヒトの臨床応用での有用性を示した。更に各種動物細胞で生産したヒト化抗体の糖鎖の構造を詳細に分析、比較し、ADCC活性を高める機能を有する糖鎖を特定し、本発明を完成させるに至った。
【0008】
本発明は、以下の(1)〜(27)に関する。
(1)N−グリコシド結合コンプレックス型糖鎖が結合した抗体を含有する抗体組成物であって、該抗体が還元末端のN−アセチルグルコサミンにフコースが結合しないN−グリコシド結合コンプレックス型糖鎖を有する抗体である抗体組成物。
(2)糖鎖が、
【化1】
を含む糖鎖を含有する上記(1)に記載の抗体組成物。
(3)糖鎖が、α1,6−フコシルトランスフェラーゼ活性の低い細胞が合成する糖鎖を含有する上記(1)または(2)に記載の抗体組成物。
(4)α1,6−フコシルトランスフェラーゼ活性の低い細胞を用いることを特徴とする上記(1)または(2)に記載の抗体組成物の製造方法。
(5)抗体が、腫瘍関連抗原を認識する抗体である上記(1)〜(3)のいずれか1項に記載の抗体組成物。
(6)腫瘍関連抗原がガングリオシドGD3である、上記(5)に記載の抗体組成物。
(7)抗体が、7−9−51(FERM BP−6691)より生産される抗体である上記(5)または(6)に記載の抗体組成物。
(8)抗体が、アレルギーまたは炎症に関連する抗原を認識する抗体である上記(1)〜(3)のいずれか1項に記載の抗体組成物。
(9)アレルギーまたは炎症に関連する抗原が、ヒトインターロイキン5レセプターα鎖である上記(8)に記載の抗体組成物。
(10)抗体が、No.3(FERM BP−6690)より生産される抗体である上記(8)または(9)に記載の抗体組成物。
(11)抗体が、循環器疾患に関連する抗原を認識する抗体である上記(1)〜(3)のいずれか1項に記載の抗体組成物。
(12)抗体が、自己免疫疾患に関連する抗原を認識する抗体である上記(1)〜(3)のいずれか1項に記載の抗体組成物。
(13)抗体が、ウィルスまたは細菌感染に関連する抗原を認識する抗体である上記(1)〜(3)のいずれか1項に記載の抗体組成物。
(14)上記(5)〜(7)のいずれか1項に記載の抗体組成物を有効成分として含有する、癌の診断薬。
(15)上記(5)〜(7)のいずれか1項に記載の抗体組成物を有効成分として含有する、癌の治療薬。
(16)上記(5)〜(7)のいずれか1項に記載の抗体組成物を有効成分として含有する、癌の予防薬。
(17)上記(8)〜(10)のいずれか1項に記載の抗体組成物を有効成分として含有する、アレルギーまたは炎症の診断薬。
(18)上記(8)〜(10)のいずれか1項に記載の抗体組成物を有効成分として含有する、アレルギーまたは炎症の治療薬。
(19)上記(8)〜(10)のいずれか1項に記載の抗体組成物を有効成分として含有する、アレルギーまたは炎症の予防薬。
(20)上記(11)に記載の抗体組成物を有効成分として含有する、循環器疾患の診断薬。
(21)上記(11)に記載の抗体組成物を有効成分として含有する、循環器疾患の治療薬。
(22)上記(11)に記載の抗体組成物を有効成分として含有する、循環器疾患の予防薬。
(23)上記(12)に記載の抗体組成物を有効成分として含有する、自己免疫疾患の診断薬。
(24)上記(12)に記載の抗体組成物を有効成分として含有する、自己免疫疾患の治療薬。
(25)上記(12)に記載の抗体組成物を有効成分として含有する、自己免疫疾患の予防薬。
(26)上記(13)に記載の抗体組成物を有効成分として含有する、ウィルスまたは細菌感染症の診断薬。
(27)上記(13)に記載の抗体組成物を有効成分として含有する、ウィルスまたは細菌感染症の治療薬。
(28)上記(13)に記載の抗体組成物を有効成分として含有する、ウィルスまたは細菌感染症の予防薬。
【図面の簡単な説明】
【0009】
【図1】第1図 精製した5種類の抗GD3キメラ抗体のSDS−PAGE(4〜15%グラジエントゲルを使用)の電気泳動パターンを示した図である。上図が非還元条件、下図が還元条件でそれぞれ電気泳動を行った図である。レーン1が高分子量マーカー、2がYB2/0−GD3キメラ抗体、3がCHO/DG44−GD3キメラ抗体、4がSP2/0−GD3キメラ抗体、5がNS0−GD3キメラ抗体(302)、6がNS0−GD3キメラ抗体(GIT)、7が低分子量マーカーの泳動パターンをそれぞれ示す。
【図2】第2図 精製した5種類の抗GD3キメラ抗体のGD3との結合活性を抗体濃度を変化させて測定した図である。縦軸はGD3との結合活性、横軸は抗体濃度をそれぞれ示す。○がYB2/0−GD3キメラ抗体、●がCHO/DG44−GD3キメラ抗体、□がSP2/0−GD3キメラ抗体、■がNS0−GD3キメラ抗体(302)、△がNS0−GD3キメラ抗体(GIT)の活性をそれぞれ示す。
【図3】第3図 精製した5種類の抗GD3キメラ抗体のヒトメラノーマ細胞株G−361に対するADCC活性を示した図である。縦軸に細胞障害活性、横軸に抗体濃度をそれぞれ示す。○がYB2/0−GD3キメラ抗体、●がCHO/DG44−GD3キメラ抗体、□がSP2/0−GD3キメラ抗体、■がNS0−GD3キメラ抗体(302)、△がNS0−GD3キメラ抗体(GIT)の活性をそれぞれ示す。
【図4】第4図 精製した3種類の抗hIL−5RαCDR移植抗体のSDS−PAGE(4〜15%グラジエントゲルを使用)の電気泳動パターンを示した図である。上図が非還元条件、下図が還元条件でそれぞれ電気泳動を行った図である。レーン1が高分子量マーカー、2がYB2/0−hIL−5RCDR抗体、3がCHO/d−hIL−5RCDR抗体、4がNS0−hIL−5RCDR抗体、5が低分子量マーカーの泳動パターンをそれぞれ示す。
【図5】第5図 精製した3種類の抗hIL−5RαCDR移植抗体のhIL−5Rαとの結合活性を抗体濃度を変化させて測定した図である。縦軸はhIL−5Rαとの結合活性、横軸は抗体濃度をそれぞれ示す。○がYB2/0−hIL−5RCDR抗体、●がCHO/d−hIL−5RCDR抗体、□がNS0−hIL−5RCDR抗体の活性をそれぞれ示す。
【図6】第6図 精製した3種類の抗hIL−5RαCDR移植抗体のhIL−5R発現マウスT細胞株CTLL−2(h5R)に対するADCC活性を示した図である。縦軸に細胞障害活性、横軸に抗体濃度をそれぞれ示す。○がYB2/0−hIL−5RCDR抗体、●がCHO/d−hIL−5RCDR抗体、□がNS0−hIL−5RCDR抗体の活性をそれぞれ示す。
【図7】第7図 精製した3種類の抗hIL−5RαCDR移植抗体のカニクイザルのhIL−5誘発好酸球増加モデルに対する抑制作用を示した図である。縦軸に末梢血中好酸球数、横軸に日数(抗体及びhIL−5の投与開始日を0日とした)をそれぞれ示す。101、102が抗体非投与群、301、302、303がYB2/0−hIL−5RCDR抗体投与群、401、402、403がCHO/d−hIL−5RCDR抗体投与群、501、502、503がNS0−hIL−5RCDR抗体投与群の結果をそれぞれ示す。
【図8】第8図 YB2/0が生産した精製抗hIL−5RαCDR移植抗体(上側)およびNS0が生産した精製抗hIL−5RαCDR移植抗体(下側)のPA化糖鎖の逆相HPLC溶離の溶離図(左図)とそのPA化糖鎖をα−L−フコシダーゼ処理した後に逆相HPLCで分析して得た溶離図(右図)を示したものである。縦軸に相対蛍光強度、横軸に溶出時間をそれぞれ示す。
【図9】第9図 CHO/d細胞が生産した精製抗hIL−5RαCDR移植抗体からPA化糖鎖を調製し、逆相HPLCで分析して得た溶離図を示したものである。縦軸に相対蛍光強度、横軸に溶出時間をそれぞれ示す。
【図10】第10図 非吸着画分、吸着画分の一部のGD3との結合活性を、抗体濃度を変化させて測定した図である。縦軸はGD3との結合活性、横軸は抗体濃度をそれぞれ示す。●が非吸着画分、○が吸着画分の一部をそれぞれ示す。下図は非吸着画分、吸着画分の一部のヒトメラノーマ細胞株G−361に対するADCC活性を示した図である。縦軸に細胞障害活性、横軸に抗体濃度をそれぞれ示す。●が非吸着画分、○が吸着画分の一部をそれぞれ示す。
【図11】第11図 非吸着画分、吸着画分の一部から調製したPA化糖鎖を逆相HPLCで分析して得た溶離図を示したものである。左図に非吸着画分の溶離図、右図に吸着画分の一部の溶離図をそれぞれ示す。縦軸に相対蛍光強度、横軸に溶出時間をそれぞれ示す。
【図12】第12図 ラットFUT8配列をスタンダード、内部コントロールに用いた場合の各宿主細胞株におけるFUT8転写産物の量を示す。■がCHO細胞株、□がYB2/0細胞株を宿主細胞として用いた場合をそれぞれ示す。
【発明を実施するための形態】
【0010】
糖鎖は、免疫機能分子の結合様式により、アスパラギンと結合する糖鎖(N−グリコシド結合糖鎖という)ならびに、セリン、スレオニンなどと結合する糖鎖(0−グリコシド結合糖鎖という)の2種類に大別される。
【0011】
本発明にかかるN−グリコシド結合糖鎖は、様々な構造を有しているが[生物化学実験法23−糖蛋白質糖鎖研究法(学会出版センター)高橋禮子編(1989年)]、いずれの場合も下記に示す基本となる共通のコア構造を有する。
【0012】
【化2】
【0013】
上記の構造において、アスパラギンと結合するの糖鎖の末端を還元末端、反対側の非還元末端という。還元末端のN−アセチルグルコサミンへのフコースの結合としては、α1,3結合、α1,6結合などがあげられる。
【0014】
N−グリコシド結合糖鎖には、コア構造の非還元末端にマンノースのみが結合するハイマンノース型、コア構造の非還元末端側にガラクトース−N−アセチルグルコサミン(以下、Gal−GlcNAcと称す。)の枝を並行して1ないしは複数本有し、更にGal−GlcNAcの非還元末端側にシアル酸、バイセクティングのN−アセチルグルコサミンなどの構造を有するコンプレックス型、コア構造の非還元末端側にハイマンノース型とコンプレックス型の両方の枝を持つハイブリッド型などがあげられる。
【0015】
上記のいずれの型において、還元末端側のN−アセチルグルコサミンにフコースが結合する糖鎖もあるが、本発明の糖鎖は、上述の糖鎖のみならず、N−アセチルグルコサミンにフコースが結合しない糖鎖であればいかなるものも包含する。
【0016】
免疫機能分子とは、元来生体内に存在し、種々の免疫反応に関与する分子であり、具体的には、抗体、タンパク質、ペプチドなどを包含する。
【0017】
抗体とは、外来抗原刺激の結果、免疫反応によって生体内に産生される蛋白質で、抗原と特異的に結合する活性を有するものをいう。抗体としては動物に抗原を免疫し、免疫動物の脾臓細胞より作製したハイブリドーマ細胞が分泌する抗体のほか、遺伝子組換え技術により作製された抗体、すなわち、抗体遺伝子を挿入した抗体発現ベクターを、宿主細胞へ導入することにより取得された抗体などがあげられる。具体的には、ハイブリドーマが生産する抗体、ヒト化抗体、ヒト抗体などをあげることができる。
【0018】
ハイブリドーマとは、ヒト以外の哺乳動物に抗原を免疫して取得されたB細胞と、マウス等に由来するミエローマ細胞とを細胞融合させて得られる、所望の抗原特異性を有したモノクローナル抗体を産生する細胞を意味する。
【0019】
ヒト化抗体としては、ヒト型キメラ抗体、ヒト型相同性決定領域(complementarity determining region:以下、CDRと略記する)移植抗体などがあげられる。
【0020】
ヒト型キメラ抗体は、ヒト以外の動物の抗体重鎖可変領域(以下、重鎖はH鎖として、可変領域はV領域としてHVまたはVHとも称す)および抗体軽鎖可変領域(以下、軽鎖はL鎖としてLVまたはVLとも称す)とヒト抗体の重鎖定常領域(以下、定常領域はC領域としてCHとも称す)およびヒト抗体の軽鎖定常領域(以下、CLとも称す)とからなる抗体を意味する。ヒト以外の動物としては、マウス、ラット、ハムスター、ラビット等、ハイブリドーマを作製することが可能であれば、いかなるものも用いることができる。
【0021】
ヒト型キメラ抗体は、モノクローナル抗体を生産するハイブリドーマより、VHおよびVLをコードするcDNAを取得し、ヒト抗体CHおよびヒト抗体CLをコードする遺伝子を有する宿主細胞用発現ベクターにそれぞれ挿入してヒト型キメラ抗体発現ベクターを構築し、宿主細胞へ導入することにより発現させ、製造することができる。
【0022】
ヒト型キメラ抗体のCHとしては、ヒトイムノグロブリン(以下、hIgと表記する)に属すればいかなるものでもよいが、hIgGクラスのものが好適であり、更にhIgGクラスに属するhIgG1、hIgG2、hIgG3、hIgG4といったサブクラスのいずれも用いることができる。また、ヒト型キメラ抗体のCLとしては、hIgに属すればいかなるものでもよく、κクラスあるいはλクラスのものを用いることができる。
【0023】
ヒト型CDR移植抗体は、ヒト以外の動物の抗体のVHおよびVLのCDRのアミノ酸配列をヒト抗体のVHおよびVLの適切な位置に移植した抗体を意味する。
【0024】
ヒト型CDR移植抗体は、ヒト以外の動物の抗体のVHおよびVLのCDR配列を任意のヒト抗体のVHおよびVLのCDR配列に移植したV領域をコードするcDNAを構築し、ヒト抗体のCHおよびヒト抗体のCLをコードする遺伝子を有する宿主細胞用発現ベクターにそれぞれ挿入してヒト型CDR移植抗体発現ベクターを構築し、該発現ベクターを宿主細胞へ導入することによりヒト型CDR移植抗体を発現させ、製造することができる。
【0025】
ヒト型CDR移植抗体のCHとしては、hIgに属すればいかなるものでもよいが、hIgGクラスのものが好適であり、更にhIgGクラスに属するhIgG1、hIgG2、hIgG3、hIgG4といったサブクラスのいずれも用いることができる。また、ヒト型CDR移植抗体のCLとしては、hIgに属すればいかなるものでもよく、κクラスあるいはλクラスのものを用いることができる。
【0026】
ヒト抗体は、元来、ヒト体内に天然に存在する抗体を意味するが、最近の遺伝子工学的、細胞工学的、発生工学的な技術の進歩により作製されたヒト抗体ファージライブラリーならびにヒト抗体産生トランスジェニック動物あるいはヒト抗体産生トランスジェニック植物から得られる抗体等も含まれる。
【0027】
ヒト体内に存在する抗体は、例えば、ヒト末梢血リンパ球を単離し、EBウイルス等を感染させ不死化、クローニングすることにより、該抗体を産生するリンパ球を培養でき、培養物中より該抗体を精製することができる。
【0028】
ヒト抗体ファージライブラリーは、ヒトB細胞から調製した抗体遺伝子をファージ遺伝子に挿入することによりFab、一本鎖抗体等の抗体断片をファージ表面に発現させたライブラリーである。該ライブラリーより、抗原を固定化した基質に対する結合活性を指標として所望の抗原結合活性を有する抗体断片を発現しているファージを回収することができる。該抗体断片は、更に遺伝子工学的手法により、2本の完全なH鎖および2本の完全なL鎖からなるヒト抗体分子へも変換することができる。
【0029】
ヒト抗体産生トランスジェニック非ヒト動物は、ヒト抗体遺伝子が細胞内に組込まれた動物を意味する。具体的には、マウスES細胞へヒト抗体遺伝子を導入し、該ES細胞を他のマウスの初期胚へ移植後、発生させることによりヒト抗体産生トランスジェニック動物を作製することができる。ヒト抗体産生トランスジェニック動物からのヒト抗体の作製方法は、通常のヒト以外の哺乳動物で行われているハイブリドーマ作製方法によりヒト抗体産生ハイブリドーマを得、培養することで培養物中にヒト抗体を産生蓄積させることができる。
【0030】
本発明の抗体の活性とは、ADCC活性を意味する。
ADCC活性とは、腫瘍細胞等に結合した抗体が、抗体Fc領域とキラー細胞、ナチュラルキラー細胞、活性化されたマクロファージ等のエフェクター細胞表面上に存在するFcレセプターの結合を介してエフェクター細胞を活性化し、腫瘍細胞等を障害する活性を意味する[モノクローナル・アンティボディズ:プリンシプルズ・アンド・アプリケーションズ(Monoclonal Antibodies:Principles and Applications),Wiley−Liss,Inc.,Capter 2.1(1995)]。
【0031】
蛋白質およびペプチドとしては、種々の免疫反応を活性化できるものであればいかなるものでも用いることができる。例えば、インターフェロン分子であるインターロイキン−2(IL−2)[サイエンス(Science),193,1007(1976)]、インターロイキン−12(IL−12)[ジャーナル・オブ・ロイコサイト・バイオロジー(J.Leuc.Biol.),55,280(1994)]、コロニー刺激因子である顆粒球コロニー刺激因子(G−CSF)[ジャーナル・オブ・バイオロジカル・ケミストリー(J.Biol.Chem.),258,9017(1983)]、マクロファージコロニー刺激因子(M−CSF)[ジャーナル・オブ・エクスペリメンタル・メディシン(J.Exp.Med.),173,269(1992)]、顆粒球−マクロファージコロニー刺激因子(GM−CSF)[ジャーナル・オブ・バイオロジカル・ケミストリー(J.Biol.Chem.),252,1998(1977)]、増殖因子であるエリスロポイエチン(EPO)[ジャーナル・オブ・バイオロジカル・ケミストリー(J.Biol.Chem.),252,5558(1977)]、トロンボポイエチン(TPO)[ネイチャー(Nature),369,533(1994)]などがあげられる。
【0032】
本発明の蛋白質およびペプチドの活性とは、該糖鎖を有する蛋白質およびペプチドを生体内に投与した際の、リンパ球(T細胞、B細胞等)、マクロファージ等をはじめとする各種免疫担当細胞の活性、または種々の免疫応答反応を意味する。
【0033】
本発明の蛋白質およびペプチドの活性の促進とは、IL−2やIL−12によるNK細胞、T細胞の活性化、EPOによる赤血球産生の促進作用等がさらに増強されることを意味する。
【0034】
1.免疫機能分子の糖鎖の分析方法
(1)中性糖・アミノ糖組成分析
IgG糖鎖は、上記で示したように、ガラクトース、マンノース、フコースなどの中性糖、N−アセチルグルコサミンなどのアミノ糖、シアル酸などの酸性糖から構成されている。
【0035】
抗体の糖鎖の組成分析は、トリフルオロ酢酸等で、糖鎖の酸加水分解を行うことにより、中性糖またはアミノ糖を遊離し、その組成比を分析することができる。
具体的な方法として、Dionex社製糖組成分析装置(BioLC)を用いる方法が挙げられる。BioLCはHPAEC−PAD(high performance anion−exchange chromatography−pulsed amperometric detection)法[ジャーナル・オブ・リキッド・クロマトグラフィー(J.Liq.Chromatogr.),6,1577(1983)]によって糖組成を分析する装置である。
【0036】
また、2−アミノピリジンによる蛍光標識化法でも組成比を分析することができる。具体的には、公知の方法[アグリカルチュラル・アンド・バイオロジカル・ケミストリー(Agruc.Biol.Chem.),55(1),283−284(1991)]に従って酸加水分解した試料を2−アミノピリジル化で蛍光ラベル化し、HPLC分析して組成比を算出することができる。
【0037】
(2)糖鎖構造解析
抗体の糖鎖の構造解析は、2次元糖鎖マップ法[アナリティカル・バイオケミストリー(Anal.Biochem.),171,73(1988)、生物化学実験法23−糖蛋白質糖鎖研究法(学会出版センター)高橋禮子編(1989年)]により行うことができる。2次元糖鎖マップ法は、例えば、X軸には逆相クロマトグラフィー糖鎖の保持時間または溶出位置を、Y軸には順相クロマトグラフィーによる糖鎖の保持時間または溶出位置を、それぞれプロットし、既知糖鎖のそれらの結果と比較することにより、糖鎖構造を推定する方法である。
【0038】
具体的には、抗体をヒドラジン分解して、抗体から糖鎖を遊離し、2−アミノピリジン(以下、PAと略記する)による糖鎖の蛍光標識[ジャーナル・オブ・バイオケミストリー(J.Biochem.),95,197(1984)]を行った後、ゲルろ過により糖鎖を過剰のPA化試薬などと分離し、逆相クロマトグラフィーを行う。次いで、分取した糖鎖の各ピークについて順相クロマトグラフィーを行う。これらの結果をもとに、2次元糖鎖マップ上にプロットし、糖鎖スタンダード(TaKaRa社製)、文献[アナリティカル・バイオケミストリー(Anal.Biochem.),171,73(1988)]とのスポットの比較より糖鎖構造を推定することができる。
【0039】
さらに各糖鎖のMALDI−TOF−MSなどの質量分析を行い、2次元糖鎖マップ法により推定される構造を確認することができる。
【0040】
2.免疫機能分子の活性を調節する方法
本発明の免疫機能分子の活性を調節する方法について、イムノグロブリンG(以下、IgGと略記する)を例として以下に述べる。
【0041】
IgGに結合するN−グルコシド結合糖鎖は、主として次式で示す構造(バイアンテナリーと記す)のバイアンテナリー複合型糖鎖である。
【0042】
【化3】
【0043】
また、N−グルコシド結合糖鎖の非還元末端のGalに、更に酸性糖であるシアル酸が付加する場合もあり、N−グルコシド結合糖鎖に、バイセクティングのN−アセチルグルコサミンが付加する場合もある。
【0044】
IgGは、Fc領域内の1カ所に、N−グルコシド結合糖鎖が結合している。IgGは2本のH鎖から構成されているので、抗体1分子にはFc部分が2カ所存在する。したがって、糖鎖結合部位も2カ所存在する。
【0045】
IgGの活性は、上記2カ所の糖鎖結合部位への、N−アセチルグルコサミンにフコースが結合しないN−グルコシド結合糖鎖の付加する個数により活性が変化する。すなわち、糖鎖結合部位の少なくとも1カ所に、N−アセチルグルコサミンにフコースが結合しないN−グルコシド結合糖鎖が付加されるときには、免疫機能分子の活性が高まる。2カ所の糖鎖結合部位ともN−アセチルグルコサミンにフコースが結合しないN−グルコシド結合糖鎖が付加される抗体をF0構造を有する抗体(F0抗体)、1カ所の糖鎖結合部位にN−アセチルグルコサミンにフコースが結合しないN−グルコシド結合糖鎖が付加される抗体をF1構造を有する抗体(F1抗体)、糖鎖結合部位のどちらにもN−アセチルグルコサミンにフコースが結合したN−グルコシド結合糖鎖が付加する場合をF2構造を有する抗体(F2抗体)とすると、IgGの活性の高さは、F0抗体>F1抗体>F2抗体となる。
【0046】
製造された抗体は、必ずしも単一の糖鎖構造を有しておらず、かつフコースの有無に着目した場合でもF0抗体、F1抗体およびF2抗体が混在して存在している。製造された抗体のADCC活性を調節するためには、上述した免疫機能分子の糖鎖の解析方法により抗体に結合している糖鎖を解析し、解析結果を指標としてADCC活性を調節することができる。
【0047】
製造された抗体のADCC活性を促進するためには、F1抗体およびF0抗体の存在比率を高くすればよい。具体的には、F1抗体およびF0抗体を精製してもよいし、またN−アセチルグルコサミンにフコースが結合しないN−グルコシド結合糖鎖が免疫機能分子に付加するように、宿主細胞での発現を調節してもよい。
【0048】
製造された抗体のADCC活性を抑制するためには、F2抗体の存在比率を高くすればよい。具体的には、F2抗体を精製してもよいし、またN−アセチルグルコサミンにフコースが結合したN−グルコシド結合糖鎖が免疫機能分子に付加するように、宿主細胞での発現を調節してもよい。
【0049】
上述のように、F0抗体、F1抗体およびF2抗体を任意に存在比率を調節することにより、所望の活性の強弱を調節することができる。
【0050】
3.免疫機能分子の製造方法
N−アセチルグルコサミンにフコースが結合しないN−グルコシド結合糖鎖を有する免疫機能分子、またはN−アセチルグルコサミンにフコースが結合するN−グルコシド結合糖鎖を有する免疫機能分子の製造方法について、以下に述べる。
【0051】
抗体、ペプチドまたは蛋白質に所望の糖鎖を結合させる為には、目的とする抗体、ペプチドまたは蛋白質をコードする遺伝子を宿主細胞に導入し、該細胞を用いて生産させることができる。また、動物個体または植物個体に目的とする抗体、ペプチドまたは蛋白質をコードする遺伝子を導入し、該個体を用いて生産させることもできる。
【0052】
N−アセチルグルコサミンにフコースが結合しないN−グルコシド結合糖鎖を有する免疫機能分子を製造するための宿主細胞または動物個体あるいは植物個体としては、例えば抗体で、抗体のFc領域に結合するN−アセチルグルコサミンにフコースを付加させる酵素活性の低いまたは酵素活性を有しない細胞または個体であればいかなるものでもよい。
抗体のFc領域に結合するN−アセチルグルコサミンにフコースを付加させる酵素活性の低いまたは酵素活性を有しない細胞としては、ラットミエローマ細胞であるYB2/3HL.P2.G11.16Ag.20細胞(ATCC CRL 1662;以下、YB2/0細胞と略記する)などがあげられる。
【0053】
また、宿主細胞または動物個体あるいは植物個体のα1,6結合に関与する酵素の遺伝子を欠損させたり、該遺伝子への変異を与えて酵素活性を下げるか欠失させたりすることにより、α1,6結合に関与する酵素活性の少ない、または有しない細胞または個体を作製して宿主細胞または動物個体あるいは植物個体として用いることもできる。α1,6結合に関与する酵素としては、フコシルトランスフェラーゼ、好ましくはα1,6−フコシルトランスフェラーゼ(以下、FUT8と称す)があげられる。
【0054】
N−アセチルグルコサミンにフコースが結合するN−グルコシド結合糖鎖を有する免疫機能分子を製造するための宿主細胞または動物個体あるいは植物個体としては、例えば抗体で、抗体のFc領域に結合するN−アセチルグルコサミンにフコースを付加させる酵素活性の高い細胞または個体であればいかなるものでもよい。
【0055】
また、宿主細胞または動物個体あるいは植物個体のα1,6結合に関与する酵素の遺伝子を導入したり、該遺伝子への変異を与えて酵素活性をあげることにより、α1,6結合に関与する酵素活性の高い細胞または個体を作製して宿主細胞または動物個体あるいは植物個体として用いることもできる。α1,6結合に関与する酵素としては、フコシルトランスフェラーゼ、好ましくはFUT8があげられる。
【0056】
宿主細胞としては、細菌、酵母、動物細胞、昆虫細胞、植物細胞等、目的とする遺伝子を発現できるものであればいずれも用いることができる。
【0057】
細菌の宿主細胞としては、エシェリヒア属、セラチア属、バチルス属、ブレビバクテリウム属、コリネバクテリウム属、ミクロバクテリウム属、シュードモナス属等に属する微生物、例えば、Escherichiacoli XL1−Blue、Escherichia coli XL2−Blue、Escherichiacoli DH1、Escherichia coli MC1000、Escherichia coliKY3276、Escherichia coli W1485、Escherichia coli JM109、Escherichiacoli HB101、Escherichia
coli No.49、Escherichia coliW3110、Escherichia coli NY49、Escherichia coli GI698、Escherichiacoli TB1、Serratia ficaria Serratiafonticola、Serratialiquefaciens、Serratia marcescens、Bacillus subtilis、Bacillusamyloliquefacines、Brevibacterium ammoniagenes、BrevibacteriumimmariophilumATCC14068、Brevibacterium saccharolyticum ATCC14066、Brevibacteriumflavum ATCC14067、Brevibacterium lactofermentum ATCC13869、Corynebacteriumglutamicum ATCC13032、Corynebacterium glutamicum ATCC13869、Corynebacteriumacetoacidophilum ATCC13870、Microbacterium ammoniaphilum ATCC15354、Pseudomonasputida、Pseudomonas sp.D−0110等をあげることができる。
【0058】
酵母の宿主細胞としては、Saccharomyces属、Schizosaccharomyces属、Kluyveromyces属、Trichosporon属、Schwanniomyces属、Pichia属、Candida属等に属する微生物、例えば、Saccharomycescerevisiae、Schizosaccharomyces pombe、Kluyvermyces lactis、Trichosporonpullulans、Schwanniomyces alluvius、Candidautilis等をあげることができる。
【0059】
動物細胞の宿主細胞としては、マウスミエローマ細胞であるNS0細胞、SP2/0細胞、チャイニーズハムスター卵巣細胞CHO/dhfr−細胞、CHO/DG44細胞、ラットミエローマYB2/0細胞、IR983F細胞、サルの細胞であるCOS細胞、ヒトミエローマ細胞であるナマルバ(Namalwa)細胞などがあげられる。好ましくは、チャイニーズハムスター卵巣細胞であるCHO/DG44細胞等があげられる。
【0060】
昆虫細胞の宿主細胞としては、Spodoptera frugiperdaの卵巣細胞であるSf9、Sf21〔Baculovirus Expression Vectors,A Laboratory Manual,W.H.Freeman and Company,New York(1992)〕、Trichoplusianiの卵巣細胞であるHigh 5(Invitrogen社製)等を用いることができる。
【0061】
植物細胞の宿主細胞としては、タバコ、ジャガイモ、トマト、ニンジン、ダイズ、アブラナ、アルファルファ、イネ、コムギ、オオムギ等の植物細胞等をあげることができる。
以上のようにして得られる本発明の形質転換体を培地に培養し、培養物中に免疫機能分子を生成蓄積させ、該培養物から採取することにより、免疫機能分子を製造することができる。
さらに、遺伝子が導入された動物個体または植物個体を造成し、これらの個体を用いて免疫機能分子を製造することもできる。
【0062】
N−アセチルグルコサミンにフコースが結合しないN−グルコシド結合糖鎖を有する免疫機能分子を製造するための動物個体または植物個体としては、例えば抗体で、抗体のFc領域に結合するN−アセチルグルコサミンにフコースを付加させる酵素活性の低いまたは酵素活性を有しない動物個体または植物個体であればいかなるものでもよい。
【0063】
また、動物個体または植物個体のα1,6結合に関与する酵素の遺伝子を欠損させたり、該遺伝子への変異を与えて酵素活性を下げるか欠失させたりすることにより、α1,6結合に関与する酵素活性の少ない、または有しないノックアウト非ヒト動物またはノックアウト植物を作製して用いてもよい。α1,6結合に関与する酵素としては、フコシルトランスフェラーゼ、好ましくはFUT8があげられる。
【0064】
N−アセチルグルコサミンにフコースが結合するN−グルコシド結合糖鎖を有する免疫機能分子を製造するための動物個体または植物個体としては、例えば抗体で、抗体のFc領域に結合するN−アセチルグルコサミンにフコースを付加させる酵素活性の高い動物個体または植物個体であればいかなるものでもよい。
【0065】
また、動物個体または植物個体のα1,6結合に関与する酵素の遺伝子を導入したり、該遺伝子への変異を与えて酵素活性を上げることにより、α1,6結合に関与する酵素活性の高いトランスジェニック非ヒト動物またはトランスジェニック植物を作製して用いてもよい。α1,6結合に関与する酵素としては、フコシルトランスフェラーゼ、好ましくはFUT8があげられる。
トランスジェニック非ヒト動物は、受精卵へ所望の遺伝子を直接インジェクションすることより取得することができる[Proc.Natl.Acad.Sci.USA,77,7380(1980)]。
【0066】
トランスジェニック非ヒト動物としては、マウス、ラット、ウサギ、ニワトリ、ヤギ、ウシなどがあげられる。
【0067】
また、胚性幹細胞へ所望の遺伝子を導入し、集合キメラ法や注入キメラ法の手法で動物を作製することにより、所望の遺伝子のトランスジェニック非ヒト動物またはノックアウト非ヒト動物を作製することができる[Manipulating the Mouse Embryo A Laboratory Manual,Second Edition,Cold Spring Harbor Laboratory Press(1994);Gene targeting,A practical Approach,IRL Press at Oxford university Press(1993);バイオマテリアルシリーズ8ジーンターゲッティング,ES細胞を用いた変異マウスの作製,羊土社(1995)]。
【0068】
胚性幹細胞としては、マウス[Nature,292,154(1981)]、ラット、ニワトリ、ブタ、サル、ヤギ、ウシなどがあげられる。
【0069】
さらに、所望の遺伝子を導入した核を、脱核した卵へ移植するというクローン技術を用いても、トランスジェニック非ヒト動物またはノックアウト非ヒト動物を作製することができる[Science,280,1256,1998;Science,278,824(1997)]。
上述の方法により作製された動物個体に、免疫機能分子をコードするDNAを導入し、該免疫機能分子を動物個体中に生成・蓄積させ、該動物個体中より該免疫機能分子を採取することにより、該免疫機能分子を製造することができる。該動物中の生成・蓄積場所としては、例えば、該動物のミルク(特開昭63−309192)、卵等をあげることができる。
【0070】
トランスジェニック植物の作製方法としては、文献[Biol.Chem.,380,825(1999)]などがあげられ、ノックアウト植物の作製方法としては、文献[Plant Journal,11,1195(1997)]があげられる。
【0071】
植物個体を用いて免疫機能分子を製造する方法としては、例えば免疫機能分子をコードするDNAを導入したトランスジェニック植物を公知の方法[組織培養,20(1994)、組織培養,21(1995)、Trends in Biotechnology,15,45(1997)]に準じて栽培し、該免疫機能分子を該植物中に生成・蓄積させ、該植物中より該免疫機能分子を採取することにより、該免疫機能分子を製造することができる。
【0072】
また、フコシルトランスフェラーゼ、好ましくはFUT8のトランスジェニック非ヒト動物またはノックアウト非ヒト動物と同系他系統である所望の免疫機能分子のトランスジェニック動物とを交配させることにより、N−アセチルグルコサミンにフコースが結合しないN−グルコシド結合糖鎖を有する免疫機能分子またはN−アセチルグルコサミンにフコースが結合するN−グルコシド結合糖鎖を有する免疫機能分子を製造することができる、遺伝子改変動物を取得することができる。交配の方法としては、自然交配、体外受精などの方法があげられる。
【0073】
また、単離した該酵素等の遺伝子群を酵母、大腸菌等へ導入し、該糖鎖の大量生産[ネイチャー・バイオテクノロジー(Nature Biotechnology),16,847(1998)]をすることもできる。また、生産された該酵素を、抗体、ペプチドまたは蛋白質への該糖鎖による修飾または生産に利用することもできる。
【0074】
さらに、本発明の免疫機能分子の活性を促進する糖鎖をペプチドで代替することもできる[ジャーナル・オブ・イムノロジー(J.Immunol.),160,293(1998)]。該ペプチドは、上述の糖鎖の使用方法において有用性があり、また免疫機能分子との融合も容易であるので簡便性の点においても優れている。
【0075】
以下に、免疫機能活性の促進された免疫機能分子の製造方法について述べる。ここでは一例として、ヒト化抗体の製造方法について記すが、他の免疫機能分子も上述の方法および当該方法に準じて作製することができる。
【0076】
4.ヒト化抗体の製造方法
(1)ヒト化抗体発現用ベクターの構築
ヒト化抗体発現用ベクターとは、ヒト抗体の重鎖(以下、H鎖と表記する)及び軽鎖(以下、L鎖と表記する)C領域をコードする遺伝子が組み込まれた動物細胞用発現ベクターであり、動物細胞用発現ベクターにヒト抗体のH鎖及びL鎖C領域をコードする遺伝子をそれぞれクローニングすることにより構築することができる。
【0077】
ヒト抗体のC領域としては、任意のヒト抗体のH鎖及びL鎖C領域であることができ、例えば、ヒト抗体のH鎖のIgG1サブクラスのC領域(以下、hCγ1と表記する)及びヒト抗体のL鎖のκクラスのC領域(以下、hCκと表記する)等があげられる。
【0078】
ヒト抗体のH鎖及びL鎖C領域をコードする遺伝子としてはエキソンとイントロンから成る染色体DNAを用いることができ、また、cDNAを用いることもできる。
【0079】
動物細胞用発現ベクターとしては、ヒト抗体のC領域をコードする遺伝子を組込み発現できるものであればいかなるものでも用いることができる。例えば、pAGE107[サイトテクノロジー(Cytotechnology),3,133(1990)]、pAGE103[ジャーナル・オブ・バイオケミストリー(J.Biochem.),101,1307(1987)]、pHSG274[ジーン(Gene),27,223(1984)]、pKCR[プロシーディングス・オブ・ザ・ナショナル・アカデミー・オブ・サイエンス(Proc.Natl.Acad.Sci.
U.S.A.),78,1527(1981)]、pSG1 β d2−4[サイトテクノロジー(Cytotechnology),4,173(1990)]等があげられる。動物細胞用発現ベクターに用いるプロモーターとエンハンサーとしては、SV40の初期プロモーターとエンハンサー[ジャーナル・オブ・バイオケミストリー(J.Biochem.),101,1307(1987)]、モロニーマウス白血病ウイルスのLTR[バイオケミカル・アンド・バイオフィジカル・リサーチ・コミュニケーションズ(Biochem.Biophys.Res.Commun.),149,960(1987)]、免疫グロブリンH鎖のプロモーター[セル(Cell),41,479(1985)]とエンハンサー[セル(Cell),33,717(1983)]等があげられる。
【0080】
ヒト化抗体発現用ベクターは、抗体H鎖及びL鎖が別々のベクター上に存在するタイプあるいは同一のベクター上に存在するタイプ(以下、タンデム型と表記する)のどちらでも用いることができるが、ヒト化抗体発現ベクターの構築の容易さ、動物細胞への導入の容易さ、動物細胞内での抗体H鎖及びL鎖の発現量のバランスが均衡する等の点からタンデム型のヒト化抗体発現用ベクターの方が好ましい[ジャーナル・オブ・イムノロジカル・メソッズ(J.Immunol.Methods),167,271(1994)]。
【0081】
構築したヒト化抗体発現用ベクターは、ヒト型キメラ抗体及びヒト型CDR移植抗体の動物細胞での発現に使用できる。
【0082】
(2)ヒト以外の動物の抗体のV領域をコードするcDNAの取得
ヒト以外の動物の抗体、例えば、マウス抗体のH鎖及びL鎖V領域をコードするcDNAは以下のようにして取得することができる。
【0083】
目的のマウス抗体を産生するハイブリドーマ細胞よりmRNAを抽出し、cDNAを合成する。合成したcDNAをファージ或いはプラスミド等のベクターにクローニングしてcDNAライブラリーを作製する。該ライブラリーより、既存のマウス抗体のC領域部分或いはV領域部分をプローブとして用い、H鎖V領域をコードするcDNAを有する組換えファージ或いは組換えプラスミド及びL鎖V領域をコードするcDNAを有する組換えファージ或いは組換えプラスミドをそれぞれ単離する。組換えファージ或いは組換えプラスミド上の目的のマウス抗体のH鎖及びL鎖V領域の全塩基配列を決定し、塩基配列よりH鎖及びL鎖V領域の全アミノ酸配列を推定する。
【0084】
ヒト以外の動物としては、マウス、ラット、ハムスター、ラビット等、ハイブリドーマ細胞を作製することが可能であれば、いかなるものも用いることができる。
【0085】
ハイブリドーマ細胞から全RNAを調製する方法としては、チオシアン酸グアニジン−トリフルオロ酢酸セシウム法[メソッズ・イン・エンザイモロジー(Methods in Enzymol.),154,3(1987)]、また全RNAからmRNAを調製する方法としては、オリゴ(dT)固定化セルロースカラム法[モレキュラー・クローニング:ア・ラボラトリー・マニュアル(Molecular Cloning: A Laboratory Manual),Cold Spring Harbor Lab.Press New York,1989]等があげられる。また、ハイブリドーマ細胞からmRNAを調製するキットとしては、Fast Track mRNA Isolation Kit(Invitrogen社製))、Quick Prep mRNA Purification Kit(Pharmacia社製)等があげられる。
【0086】
cDNAの合成及びcDNAライブラリー作製法としては、常法[モレキュラー・クローニング:ア・ラボラトリー・マニュアル(Molecular Cloning:A Laboratory Manual),Cold Spring Harbor Lab.Press New York,1989;カレント・プロトコールズ・イン・モレキュラー・バイオロジー(Current Protocols in Molecular Biology),Supplement 1−34]、或いは市販のキット、例えば、Super ScriptTMPlasmid System for cDNA Synthesis and Plasmid Cloning(GIBCO BRL社製)やZAP−cDNA Synthesis Kit(Stratagene社製)を用いる方法などがあげられる。
【0087】
cDNAライブラリーの作製の際、ハイブリドーマ細胞から抽出したmRNAを鋳型として合成したcDNAを組み込むベクターは、該cDNAを組み込めるベクターであればいかなるものでも用いることができる。例えば、ZAP Express[ストラテジーズ(Strategies),5,58(1992)]、pBluescript II SK(+)[ヌクレイック・アシッズ・リサーチ(Nucleic Acids Research),17,9494(1989)]、λzapII(Stratagene社製)、λgt10、λgt11[DNAクローニング:ア・プラクティカル・アプローチ(DNA Cloning: A Practical Approach),I,49(1985)]、Lambda BlueMid(Clontech社製)、λExCell、pT7T3 18U(Pharmacia社製)、pcD2[モレキュラー・アンド・セルラー・バイオロジー(Mol.Cell.Biol.),3,280(1983)]及びpUC18[シーン(Gene)33,103(1985)]等が用いられる。
【0088】
ファージ或いはプラスミドベクターにより構築されるcDNAライブラリーを導入する大腸菌としては該cDNAライブラリーを導入、発現及び維持できるものであればいかなるものでも用いることができる。例えば、XL1−Blue MRF’[ストラテジーズ(Strategies),5,81(1992)]、C600[ジェネティックス(Genetics),39,440(1954)]、Y1088、Y1090[サイエンス(Science),222,778(1983)]、NM522[ジャーナル・オブ・モレキュラー・バイオロジー(J.Mol.Biol.),166,1(1983)]、K802[ジャーナル・オブ・モレキュラー・バイオロジー(J.Mol.Biol.),16,118(1966)]及びJM105[ジーン(Gene),38,275(1985)]等が用いられる。
【0089】
cDNAライブラリーからのヒト以外の動物の抗体のH鎖及びL鎖V領域をコードするcDNAクローンの選択法としては、アイソトープ或いは蛍光標識したプローブを用いたコロニー・ハイブリダイゼーション法或いはプラーク・ハイブリダイゼーション法[モレキュラー・クローニング:ア・ラボラトリー・マニュアル(Molecular Cloning:A Laboratory Manual),Cold Spring Harbor Lab.Press NewYork,1989]により選択することができる。また、プライマーを調製し、mRNAから合成したcDNA或いはcDNAライブラリーを鋳型として、Polymerase Chain Reaction[以下、PCR法と表記する;モレキュラー・クローニング:ア・ラボラトリー・マニュアル(Molecular Cloning:A Laboratory Manual),Cold Spring Harbor Lab.Press New York,1989;カレント・プロトコールズ・イン・モレキュラー・バイオロジー(Current Protocols in
Molecular Biology),Supplement 1−34]によりH鎖及びL鎖V領域をコードするcDNAを調製することもできる。
【0090】
上記方法により選択されたcDNAを、適当な制限酵素などで切断後、pBluescriptSK(−)(Stratagene社製)等のプラスミドにクローニングし、通常用いられる塩基配列解析方法、例えば、サンガー(Sanger)らのジデオキシ法[プロシーディングス・オブ・ザ・ナショナル・アカデミー・オブ・サイエンス(Proc.Natl.Acad.Sci.,U.S.A.),74,5463(1977)]等の反応を行い、塩基配列自動分析装置、例えば、A.L.F.DNAシークエンサー(Pharmacia社製)等を用いて解析することで該cDNAの塩基配列を決定することができる。
【0091】
決定した塩基配列からH鎖及びL鎖V領域の全アミノ酸配列を推定し、既知の抗体のH鎖及びL鎖V領域の全アミノ酸配列[シーケンシズ・オブ・プロテインズ・オブ・イムノロジカル・インタレスト(Sequences of Proteins of Immunological Interest),US Dept.Health and Human Services,1991]と比較することにより、取得したcDNAが分泌シグナル配列を含む抗体のH鎖及びL鎖V領域の完全なアミノ酸配列をコードしているかを確認することができる。
【0092】
(3)ヒト以外の動物の抗体のV領域のアミノ酸配列の解析
分泌シグナル配列を含む抗体のH鎖及びL鎖V領域の完全なアミノ酸配列に関しては、既知の抗体のH鎖及びL鎖V領域の全アミノ酸配列[シーケンシズ・オブ・プロテインズ・オブ・イムノロジカル・インタレスト(Sequences of Proteins of Immunological Interest),US Dept.Health and Human Services,1991]と比較することにより、分泌シグナル配列の長さ及びN末端アミノ酸配列を推定でき、更にはそれらが属するサブグループを知ることができる。また、H鎖及びL鎖V領域の各CDRのアミノ酸配列についても、既知の抗体のH鎖及びL鎖V領域のアミノ酸配列[シーケンシズ・オブ・プロテインズ・オブ・イムノロジカル・インタレスト(Sequences of Proteins of Immunological Interest),US Dept.Health and Human Services,1991]と比較することによって見出すことができる。
【0093】
(4)ヒト型キメラ抗体発現ベクターの構築
本項4の(1)に記載のヒト化抗体発現用ベクターのヒト抗体のH鎖及びL鎖C領域をコードする遺伝子の上流に、ヒト以外の動物の抗体のH鎖及びL鎖V領域をコードするcDNAをクローニングし、ヒト型キメラ抗体発現ベクターを構築することができる。例えば、ヒト以外の動物の抗体のH鎖及びL鎖V領域をコードするcDNAを、ヒト以外の動物の抗体H鎖及びL鎖V領域の3’末端側の塩基配列とヒト抗体のH鎖及びL鎖C領域の5’末端側の塩基配列とから成り、かつ適当な制限酵素の認識配列を両端に有する合成DNAとそれぞれ連結し、それぞれを本項4の(1)に記載のヒト化抗体発現用ベクターのヒト抗体のH鎖及びL鎖C領域をコードする遺伝子の上流にそれらが適切な形で発現するようにクローニングし、ヒト型キメラ抗体発現ベクターを構築することができる。
【0094】
(5)ヒト型CDR移植抗体のV領域をコードするcDNAの構築
ヒト型CDR移植抗体のH鎖及びL鎖V領域をコードするcDNAは、以下のようにして構築することができる。まず、目的のヒト以外の動物の抗体のH鎖及びL鎖V領域のCDRを移植するヒト抗体のH鎖及びL鎖V領域のフレームワーク(以下、FRと表記する)のアミノ酸配列を選択する。ヒト抗体のH鎖及びL鎖V領域のFRのアミノ酸配列としては、ヒト抗体由来のものであれば、いかなるものでも用いることができる。例えば、Protein Data Bank等のデータベースに登録されているヒト抗体のH鎖及びL鎖V領域のFRのアミノ酸配列、ヒト抗体のH鎖及びL鎖のV領域のFRの各サブグループの共通アミノ酸配列[シーケンシズ・オブ・プロテインズ・オブ・イムノロジカル・インタレスト(Sequences of Proteins of Immunological Interest),US Dept.Health and Human Services,1991]等があげられるが、その中でも、十分な活性を有するヒト型CDR移植抗体を作製するためには、目的のヒト以外の動物の抗体のH鎖及びL鎖V領域のFRのアミノ酸配列とできるだけ高い相同性(少なくとも60%以上)を有するアミノ酸配列を選択することが望ましい。
【0095】
次に、選択したヒト抗体のH鎖及びL鎖V領域のFRのアミノ酸配列に目的のヒト以外の動物の抗体のH鎖及びL鎖V領域のCDRのアミノ酸配列を移植し、ヒト型CDR移植抗体のH鎖及びL鎖V領域のアミノ酸配列を設計する。設計したアミノ酸配列を抗体の遺伝子の塩基配列に見られるコドンの使用頻度[シーケンシズ・オブ・プロテインズ・オブ・イムノロジカル・インタレスト(Sequences of Proteins of Immunological Interest),US Dept.Health and Human Services,1991]を考慮してDNA配列に変換し、ヒト型CDR移植抗体のH鎖及びL鎖V領域のアミノ酸配列をコードするDNA配列を設計する。設計したDNA配列に基づき、100塩基前後の長さから成る数本の合成DNAを合成し、それらを用いてPCR法を行う。この場合、PCRでの反応効率及び合成可能なDNAの長さから、H鎖、L鎖とも6本の合成DNAを設計することが好ましい。
【0096】
また、両端に位置する合成DNAの5’末端に適当な制限酵素の認識配列を導入することで、本項4の(1)で構築したヒト化抗体発現用ベクターに容易にクローニングすることができる。PCR後、増幅産物をpBluescript SK(−)(Stratagene社製)等のプラスミドにクローニングし、本項4の(2)に記載の方法により、塩基配列を決定し、所望のヒト型CDR移植抗体のH鎖及びL鎖V領域のアミノ酸配列をコードするDNA配列を有するプラスミドを取得する。
【0097】
(6)ヒト型CDR移植抗体のV領域のアミノ酸配列の改変
ヒト型CDR移植抗体は、目的のヒト以外の動物の抗体のH鎖及びL鎖V領域のCDRのみをヒト抗体のH鎖及びL鎖V領域のFRに移植しただけでは、その抗原結合活性は元のヒト以外の動物の抗体に比べて低下してしまうことが知られている[バイオ/テクノロジー(BIO/TECHNOLOGY),9,266(1991)]。この原因としては、元のヒト以外の動物の抗体のH鎖及びL鎖V領域では、CDRのみならず、FRのいくつかのアミノ酸残基が直接的或いは間接的に抗原結合活性に関与しており、それらアミノ酸残基がCDRの移植に伴い、ヒト抗体のH鎖及びL鎖V領域のFRの異なるアミノ酸残基へと変化してしまうことが考えられている。この問題を解決するため、ヒト型CDR移植抗体では、ヒト抗体のH鎖及びL鎖V領域のFRのアミノ酸配列の中で、直接抗原との結合に関与しているアミノ酸残基やCDRのアミノ酸残基と相互作用したり、抗体の立体構造を維持し、間接的に抗原との結合に関与しているアミノ酸残基を同定し、それらを元のヒト以外の動物の抗体に見出されるアミノ酸残基に改変し、低下した抗原結合活性を上昇させることが行われている[バイオ/テクノロジー(BIO/TECHNOLOGY),9,266(1991)]。
【0098】
ヒト型CDR移植抗体の作製においては、それら抗原結合活性に関わるFRのアミノ酸残基を如何に効率よく同定するかが、最も重要な点であり、そのためにX線結晶解析[ジャーナル・オブ・モレキュラー・バイオロジー(J.Mol.Biol.),112,535(1977)]或いはコンピューターモデリング[プロテイン・エンジニアリング(Protein Engineering),7,1501(1994)]等による抗体の立体構造の構築及び解析が行われている。これら抗体の立体構造の情報は、ヒト型CDR移植抗体の作製に多くの有益な情報をもたらして来たが、その一方、あらゆる抗体に適応可能なヒト型CDR移植抗体の作製法は未だ確立されておらず、現状ではそれぞれの抗体について数種の改変体を作製し、それぞれの抗原結合活性との相関を検討する等の種々の試行錯誤が必要である。
【0099】
ヒト抗体のH鎖及びL鎖V領域のFRのアミノ酸残基の改変は、改変用合成DNAを用いて本項4の(5)に記載のPCR法を行うことにより、達成できる。PCR後の増幅産物について本項4の(2)に記載の方法により、塩基配列を決定し、目的の改変が施されたことを確認する。
【0100】
(7)ヒト型CDR移植抗体発現ベクターの構築
本項4の(1)に記載のヒト化抗体発現用ベクターのヒト抗体のH鎖及びL鎖C領域をコードする遺伝子の上流に、本項4の(5)及び(6)で構築したヒト型CDR移植抗体のH鎖及びL鎖V領域をコードするcDNAをクローニングし、ヒト型CDR移植抗体発現ベクターを構築することができる。例えば、本項4の(5)及び(6)でヒト型CDR移植抗体のH鎖及びL鎖V領域を構築する際に用いる合成DNAのうち、両端に位置する合成DNAの5’末端に適当な制限酵素の認識配列を導入することで、本項4の(1)に記載のヒト化抗体発現用ベクターのヒト抗体のH鎖及びL鎖C領域をコードする遺伝子の上流にそれらが適切な形で発現するようにクローニングし、ヒト型CDR移植抗体発現ベクターを構築することができる。
【0101】
(8)ヒト化抗体の安定的生産
本項4の(4)及び(7)に記載のヒト化抗体発現ベクターを適当な動物細胞に導入することによりヒト化抗体を安定に生産する形質転換株を得ることができる。
【0102】
動物細胞への発現ベクターの導入法としては、エレクトロポレーション法[特開平2−257891、サイトテクノロジー(Cytotechnology)3,133(1990)]等があげられる。
【0103】
ヒト化抗体発現ベクターを導入する動物細胞としては、ヒト化抗体を生産させることができる動物細胞であれば、いかなる細胞でも用いることができるが、好ましくは生産される抗体のFc領域に付加するN−アセチルグルコサミンにフコースを付加させる酵素活性の低いまたは酵素活性を有しない細胞があげられる。
【0104】
抗体のFc領域に付加するN−アセチルグルコサミンにフコースを付加させる酵素活性の低いまたは酵素活性を有しない細胞とは、α1,6結合に関与する酵素が少ない、またはない細胞であり、具体的にはフコシルトランスフェラーゼ、好ましくはFUT8活性が少ない、またはない細胞があげられる。
【0105】
抗体のFc領域に付加するN−アセチルグルコサミンにフコースを付加させる酵素活性の低い、または酵素活性を有しない細胞としては、ラットミエローマ細胞であるYB2/0細胞などがあげられるが、α1,6結合に関与する酵素の遺伝子を欠損させたり、該遺伝子への変異を与えて酵素活性を下げるか欠失させた細胞を抗体生産細胞として用いることもできる。
【0106】
具体的には、マウスミエローマ細胞であるNS0細胞、SP2/0細胞、チャイニーズハムスター卵巣細胞CHO/dhfr−細胞、CHO/DG44細胞、ラットミエローマYB2/0細胞、IR983F細胞、ヒトミエローマ細胞であるナマルバ細胞などがあげられる。好ましくは、チャイニーズハムスター卵巣細胞であるCHO/DG44細胞等があげられる。
【0107】
発現ベクターの導入後、ヒト化抗体を安定に生産する形質転換株は、特開平2−257891に開示されている方法に従い、G418 sulfate(以下、G418と表記する;SIGMA社製)等の薬剤を含む動物細胞培養用培地により選択できる。動物細胞培養用培地としては、RPMI1640培地(日水製薬社製)、GIT培地(日本製薬社製)、EX−CELL302培地(JRH社製)、IMDM培地(GIBCO BRL社製)、Hybridoma−SFM培地(GIBCO BRL社製)、またはこれら培地に牛胎児血清(以下、FBSと表記する)等の各種添加物を添加した培地等を用いることができる。得られた形質転換株を培地中で培養することで培養上清中にヒト化抗体を生産蓄積させることができる。培養上清中のヒト化抗体の生産量及び抗原結合活性は酵素免疫抗体法[以下、ELISA法と表記する;アンティボディズ:ア・ラボラトリー・マニュアル(Antibodies:A Laboratory Manual),Cold Spring Harbor Laboratory,Chapter 14,1998、モノクローナル・アンティボディズ:プリンシプルズ・アンド・プラクティス(Monoclonal Antibodies:Principles and Practice),Academic Press Limited,1996]等により測定できる。また、形質転換株は、特開平2−257891に開示されている方法に従い、DHFR遺伝子増幅系等を利用してヒト化抗体の生産量を上昇させることができる。
【0108】
ヒト化抗体は、形質転換株の培養上清よりプロテインAカラムを用いて精製することができる[アンティボディズ:ア・ラボラトリー・マニュアル(Antibodies:A
Laboratory Manual),Cold Spring Harbor Laboratory,Chapter 8,1988、モノクローナル・アンティボディズ:プリンシプルズ・アンド・プラクティス(Monoclonal Antibodies:Principles and Practice),Academic Press Limited,1996]。また、その他に通常、蛋白質の精製で用いられる精製方法を使用することができる。例えば、ゲル濾過、イオン交換クロマトグラフィー及び限外濾過等を組み合わせて行い、精製することができる。精製したヒト化抗体のH鎖、L鎖或いは抗体分子全体の分子量は、ポリアクリルアミドゲル電気泳動[以下、SDS−PAGEと表記する;ネイチャー(Nature),227,680(1970)]やウエスタンブロッティング法[アンティボディズ:ア・ラボラトリー・マニュアル(Antibodies:A Laboratory Manual),Cold Spring Harbor
Laboratory,Chapter 12,1988、モノクローナル・アンティボディズ:プリンシプルズ・アンド・プラクティス(Monoclonal Antibodies:Principles and Practice),Academic Press Limited,1996]等で測定することができる。
【0109】
以上、動物細胞を宿主とした抗体の製造方法を示したが、上記3にあるように、細菌、酵母、昆虫細胞、植物細胞または動物個体あるいは植物個体においても製造することができる。
【0110】
(9)ヒト化抗体の活性評価
精製したヒト化抗体の抗原との結合活性、抗原陽性培養細胞株に対する結合活性はELISA法及び蛍光抗体法[キャンサー・イムノロジー・イムノセラピー(Cancer Immunol.Immunother.),36,373(1993)]等により測定できる。抗原陽性培養細胞株に対する細胞障害活性は、CDC活性、ADCC活性等を測定することにより、評価することができる[キャンサー・イムノロジー・イムノセラピー(Cancer Immunol Immunother.),36,373(1993)]。更にヒト化抗体のヒトでの安全性、治療効果は、カニクイザル等のヒトに比較的近い動物種の適当なモデルを用いて評価することができる。
【0111】
5.免疫機能分子の使用方法
上記4記載のヒト化抗体の例にあるように、高いADCC活性を有する抗体は、癌、アレルギー、循環器疾患、またはウィルスあるいは細菌感染をはじめとする各種疾患の予防および治療において有用である。
癌、すなわち悪性腫瘍は癌細胞が増殖する。通常の抗癌剤は癌細胞の増殖を抑制することを特徴とする。しかし、高いADCC活性を有する抗体は、殺細胞効果により癌細胞を障害することにより癌を治療することができるので、通常の抗癌剤よりも治療薬として有効である。
【0112】
アレルギー反応は、免疫細胞によるメディエータ分子の放出により惹起されるため、高いADCC活性を有する抗体を用いて免疫細胞を除去することにより、アレルギー反応を抑えることができる。
【0113】
循環器疾患としては、動脈硬化などがあげられる。動脈硬化は、現在バルーンカテーテルによる治療を行うが、治療後の再狭窄での動脈細胞の増殖を高いADCC活性を有する抗体を用いて抑えることより、循環器疾患を予防および治療することができる。
【0114】
ウィルスまたは細菌に感染細胞を、高いADCC活性を有する抗体を用いてウィルスまたは細菌に感染細胞の増殖を抑えることにより、ウィルスまたは細菌感染をはじめとする各種疾患の予防および治療することができる。
【0115】
また、ADCC活性が抑制された抗体は、自己免疫疾患の予防および治療において有用である。また、ADCC活性が抑制された抗体は、自己免疫疾患において亢進された免疫反応を押さえるという観点から、自己免疫疾患の予防および治療において有用である。
【0116】
本発明の抗体を含有する医薬は、治療薬として単独で投与することも可能ではあるが、通常は薬理学的に許容される一つあるいはそれ以上の担体と一緒に混合し、製剤学の技術分野においてよく知られる任意の方法により製造した医薬製剤として提供するのが望ましい。
【0117】
投与経路は、治療に際して最も効果的なものを使用するのが望ましく、経口投与、または口腔内、気道内、直腸内、皮下、筋肉内および静脈内等の非経口投与をあげることができ、抗体製剤の場合、望ましくは静脈内投与をあげることができる。
【0118】
投与形態としては、噴霧剤、カプセル剤、錠剤、顆粒剤、シロップ剤、乳剤、座剤、注射剤、軟膏、テープ剤等があげられる。
【0119】
経口投与に適当な製剤としては、乳剤、シロップ剤、カプセル剤、錠剤、散剤、顆粒剤等があげられる。
【0120】
乳剤およびシロップ剤のような液体調製物は、水、ショ糖、ソルビトール、果糖等の糖類、ポリエチレングリコール、プロピレングリコール等のグリコール類、ごま油、オリーブ油、大豆油等の油類、p−ヒドロキシ安息香酸エステル類等の防腐剤、ストロベリーフレーバー、ペパーミント等のフレーバー類等を添加剤として用いて製造できる。
【0121】
カプセル剤、錠剤、散剤、顆粒剤等は、乳糖、ブドウ糖、ショ糖、マンニトール等の賦形剤、デンプン、アルギン酸ナトリウム等の崩壊剤、ステアリン酸マグネシウム、タルク等の滑沢剤、ポリビニルアルコール、ヒドロキシプロピルセルロース、ゼラチン等の結合剤、脂肪酸エステル等の界面活性剤、グリセリン等の可塑剤等を添加剤として用いて製造できる。
【0122】
非経口投与に適当な製剤としては、注射剤、座剤、噴霧剤等があげられる。
注射剤は、塩溶液、ブドウ糖溶液、あるいは両者の混合物からなる担体等を用いて調製される。または、ヒト化抗体を常法に従って凍結乾燥し、これに塩化ナトリウムを加えることによって粉末注射剤を調製することもできる。
【0123】
座剤はカカオ脂、水素化脂肪またはカルボン酸等の担体を用いて調製される。
また、噴霧剤は該化合物そのもの、ないしは受容者の口腔および気道粘膜を刺激せず、かつ該化合物を微細な粒子として分散させ吸収を容易にさせる担体等を用いて調製される。
【0124】
担体として具体的には乳糖、グリセリン等が例示される。該化合物および用いる担体の性質により、エアロゾル、ドライパウダー等の製剤が可能である。また、これらの非経口剤においても経口剤で添加剤として例示した成分を添加することもできる。
【0125】
投与量または投与回数は、目的とする治療効果、投与方法、治療期間、年齢、体重等により異なるが、通常成人1日当たり10μg/kg〜20mg/kgである。
【0126】
また、抗体の各種腫瘍細胞に対する抗腫瘍効果を検討する方法は、インビトロ実験としては、CDC活性測定法、ADCC活性測定法等があげられ、インビボ実験としては、マウス等の実験動物での腫瘍系を用いた抗腫瘍実験等があげられる。
【0127】
CDC活性、ADCC活性、抗腫瘍実験は、文献[キャンサー・イムノロジー・イムノセラピー(Cancer Immunology Immunotherapy),36,373(1993);キャンサー・リサーチ(Cancer Research),54,1511(1994)]等記載の方法に従って行うことができる。
【0128】
6.免疫機能分子の活性を促進または抑制させる方法
上述の方法によりフコースが存在しない糖鎖が結合された抗体、ペプチドまたは蛋白質を製造することにより免疫機能分子の活性を促進させることができる。
【0129】
活性が促進された免疫機能分子を生体内に投与することにより、生体内では、ADCC活性を担うエフェクター細胞であるキラー細胞、ナチュラルキラー細胞、活性化マクロファージ等の細胞をはじめとする各種免疫細胞が活性化され、種々の免疫反応を調節することが可能となる。
また、上述の方法によりフコースが存在する糖鎖を結合された抗体、ペプチドまたは蛋白質を製造することにより免疫機能分子の活性を抑制させることができる。
【0130】
活性が抑制された免疫機能分子を生体内に投与することにより、生体内では、ADCC活性を担う各種免疫細胞の活性が弱まり、種々の免疫反応を調節することが可能となる。
【実施例】
【0131】
以下に、本発明の実施例を示すが、これにより本発明の範囲が限定されるものではない。
【実施例1】
【0132】
抗ガングリオシドGD3ヒト型キメラ抗体の作製
1.抗ガングリオシドGD3ヒト型キメラ抗体のタンデム型発現ベクターpChiLHGM4の構築
抗ガングリオシドGD3ヒト型キメラ抗体(以下、抗GD3キメラ抗体と表記する)のL鎖の発現ベクターpChi641LGM4[ジャーナル・オブ・イムノロジカル・メソッズ(J.Immunol.Methods),167,271(1994)]を制限酵素MluI(宝酒造社製)とSalI(宝酒造社製)で切断して得られるL鎖cDNAを含む約4.03kbの断片と動物細胞用発現ベクターpAGE107[サイトテクノロジー(Cytotechnology),3,133(1990)]を制限酵素MluI(宝酒造社製)とSalI(宝酒造社製)で切断して得られるG418耐性遺伝子及びスプライシングシグナルを含む約3.40kbの断片をDNA Ligation Kit(宝酒造社製)を用いて連結、大腸菌HB101株[モレキュラー・クローニング:ア・ラボラトリー・マニュアル(Molecular Cloning:A Laboratory Manual),Cold Spring Harbor Lab.Press New York,1989]を形質転換してプラスミドpChi641LGM40を構築した。
次に、上記で構築したプラスミドpChi641LGM40を制限酵素ClaI(宝酒造社製)で切断後、DNA Blunting Kit(宝酒造社製)を用いて平滑末端化し、更にMluI(宝酒造社製)で切断して得られるL鎖cDNAを含む約5.68kbの断片と抗GD3キメラ抗体のH鎖の発現ベクターpChi641HGM4[ジャーナル・オブ・イムノロジカル・メソッズ(J.Immunol.Methods),167,271(1994)]を制限酵素XhoI(宝酒造社製)で切断後、DNA Blunting Kit(宝酒造社製)を用いて平滑末端化し、更にMluI(宝酒造社製)で切断して得られるH鎖cDNAを含む約8.40kbの断片をDNA Ligation Kit(宝酒造社製)を用いて連結、大腸菌HB101株[モレキュラー・クローニング:ア・ラボラトリー・マニュアル(Molecular Cloning:A Laboratory Manual),Cold Spring Harbor Lab.Press New York,1989]を形質転換して抗GD3キメラ抗体のタンデム型発現ベクターpChi641LHGM4を構築した。
【0133】
2.抗GD3キメラ抗体の安定生産細胞の作製
上記実施例1の1項で構築した抗GD3キメラ抗体のタンデム型発現ベクターpChi641LHGM4を用いて抗GD3キメラ抗体の安定生産細胞を以下のようにして作製した。
(1)ラットミエローマYB2/0細胞を用いた生産細胞の作製
抗GD3キメラ抗体発現ベクターpChi641LHGM4の5μgを4×106細胞のラットミエローマYB2/0細胞へエレクトロポレーション法[サイトテクノロジー(Cytotechnology),3,133(1990)]により導入後、40mlのRPMI1640−FBS(10)[FBS(GIBCO BRL社製)を10%含むRPMI1640培地]に懸濁し、96ウェル培養用プレート(住友ベークライト社製)に200μl/ウェルずつ分注した。5%CO2インキュベーター内で37℃、24時間培養した後、G418を0.5mg/mlになるように添加して1〜2週間培養した。G418耐性を示す形質転換株のコロニーが出現し、増殖の認められたウェルより培養上清を回収し、上清中の抗GD3キメラ抗体の抗原結合活性を実施例1の3項に示すELISA法により測定した。
培養上清中に抗GD3キメラ抗体の生産が認められたウェルの形質転換株については、DHFR遺伝子増幅系を利用して抗体生産量を増加させる目的で、G418を0.5mg/ml、DHFRの阻害剤であるメソトレキセート(以下、MTXと表記する;SIGMA社製)を50nM含むRPMI1640−FBS(10)培地に1〜2×105細胞/mlになるように懸濁し、24ウェルプレート(Greiner社製)に2mlずつ分注した。5%CO2インキュベーター内で37℃で1〜2週間培養して、50nM MTX耐性を示す形質転換株を誘導した。形質転換株の増殖が認められたウェルの培養上清中の抗GD3キメラ抗体の抗原結合活性を実施例1の3項に示すELISA法により測定した。培養上清中に抗GD3キメラ抗体の生産が認められたウェルの形質転換株については、上記と同様の方法により、MTX濃度を100nM、200nMと順次上昇させ、最終的にG418を0.5mg/ml、MTXを200nMの濃度で含むRPMI1640−FBS(10)培地で増殖可能かつ、抗GD3キメラ抗体を高生産する形質転換株を得た。
得られた形質転換株については、2回の限界希釈法による単一細胞化(クローン化)を行った。
このようにして得られた抗GD3キメラ抗体を生産する形質転換細胞クローン7−9−51は平成11年4月5日付で工業技術院生命工学工業技術研究所(日本国茨城県つくば市東1丁目1番3号)にFERM BP−6691として寄託されている。
(2)CHO/DG44細胞を用いた生産細胞の作製
抗GD3キメラ抗体発現ベクターpChi641LHGM4の4μgを1.6×106細胞のCHO/DG44細胞へエレクトロポレーション法[サイトテクノロジー(Cytotechnology),3,133(1990)]により導入後、10mlのIMDM−FBS(10)[FBSを10%、HT supplement(GIBCO BRL社製)を1倍濃度で含むIMDM培地]に懸濁し、96ウェル培養用プレート(岩城硝子社製)に200μl/ウェルずつ分注した。5%CO2インキュベーター内で37℃、24時間培養した後、G418を0.5mg/mlになるように添加して1〜2週間培養した。G418耐性を示す形質転換株のコロニーが出現し、増殖の認められたウェルより培養上清を回収し、上清中の抗GD3キメラ抗体の抗原結合活性を実施例1の3項に示すELISA法により測定した。
培養上清中に抗GD3キメラ抗体の生産が認められたウェルの形質転換株については、DHFR遺伝子増幅系を利用して抗体生産量を増加させる目的で、G418を0.5mg/ml、MTXを10nM含むIMDM−dFBS(10)培地[透析牛胎児血清(以下、dFBSと表記する;GIBCO BRL社製)を10%含むIMDM培地]に1〜2×105細胞/mlになるように懸濁し、24ウェルプレート(岩城硝子社製)に0.5mlずつ分注した。5%CO2インキュベーター内で37℃で1〜2週間培養して、10nM MTX耐性を示す形質転換株を誘導した。増殖が認められたウェルの形質転換株については、上記と同様の方法により、MTX濃度を100nMに上昇させ、最終的にG418を0.5mg/ml、MTXを100nMの濃度で含むIMDM−dFBS(10)培地で増殖可能かつ、抗GD3キメラ抗体を高生産する形質転換株を得た。得られた形質転換株については、2回の限界希釈法による単一細胞化(クローン化)を行った。
(3)マウスミエローマNS0細胞を用いた生産細胞の作製
抗GD3キメラ抗体発現ベクターpChi641LHGM4の5μgを4×106細胞のマウスミエローマNS0細胞へエレクトロポレーション法[サイトテクノロジー(Cytotechnology),3,133,1990]により導入後、40mlのEX−CELL302−FBS(10)[FBSを10%、L−グルタミン(以下、L−Glnと表記する;GIBCO BRL社製)を2mM含むEX−CELL302培地]に懸濁し、96ウェル培養用プレート(住友ベークライト社製)に200μl/ウェルずつ分注した。5%CO2インキュベーター内で37℃、24時間培養した後、G418を0.5mg/mlになるように添加して1〜2週間培養した。G418耐性を示す形質転換株のコロニーが出現し、増殖の認められたウェルより培養上清を回収し、上清中の抗GD3キメラ抗体の抗原結合活性を実施例1の3項に示すELISA法により測定した。
培養上清中に抗GD3キメラ抗体の生産が認められたウェルの形質転換株については、DHFR遺伝子増幅系を利用して抗体生産量を増加させる目的で、G418を0.5mg/ml、MTXを50nM含むEX−CELL302−dFBS(10)培地(dFBSを10%、L−Glnを2mM含むEX−CELL302培地)に1〜2×105細胞/mlになるように懸濁し、24ウェルプレート(Greiner社製)に2mlずつ分注した。5%CO2インキュベーター内で37℃で1〜2週間培養して、50nM MTX耐性を示す形質転換株を誘導した。形質転換株の増殖が認められたウェルの培養上清中の抗GD3キメラ抗体の抗原結合活性を実施例1の3項に示すELISA法により測定した。培養上清中に抗GD3キメラ抗体の生産が認められたウェルの形質転換株については、上記と同様の方法により、MTX濃度を100nM、200nMと順次上昇させ、最終的にG418を0.5mg/ml、MTXを200nMの濃度で含むEX−CELL302−dFBS(10)培地で増殖可能かつ、抗GD3キメラ抗体を高生産する形質転換株を得た。得られた形質転換株については、2回の限界希釈法による単一細胞化(クローン化)を行った。
【0134】
3.抗体のGD3に対する結合活性の測定(ELISA法)
抗体のGD3に対する結合活性は以下のようにして測定した。
4nmolのGD3を10μgのジパルミトイルフォスファチジルコリン(SIGMA社製)と5μgのコレステロール(SIGMA社製)とを含む2mlのエタノール溶液に溶解した。該溶液の20μl(40pmol/ウェルとなる)を96ウェルのELISA用のプレート(Greiner社製)の各ウェルにそれぞれ分注し、風乾後、1%牛血清アルブミン(以下、BSAと表記する;SIGMA社製)を含むPBS(以下、1%BSA−PBSと表記する)を100μl/ウェルで加え、室温で1時間反応させて残存する活性基をブロックした。1%BSA−PBSを捨て、形質転換株の培養上清或いは精製したヒト型キメラ抗体の各種希釈溶液を50μl/ウェルで加え、室温で1時間反応させた。反応後、各ウェルを0.05%Tween20(和光純薬社製)を含むPBS(以下、Tween−PBSと表記する)で洗浄後、1%BSA−PBSで3000倍に希釈したペルオキシダーゼ標識ヤギ抗ヒトIgG(H&L)抗体溶液(American Qualex社製)を二次抗体溶液として、50μl/ウェルで加え、室温で1時間反応させた。反応後、Tween−PBSで洗浄後、ABTS基質液[2,2’−アジノ−ビス(3−エチルベンゾチアゾリン−6−スルホン酸)アンモニウムの0.55gを1Lの0.1Mクエン酸緩衝液(pH4.2)に溶解し、使用直前に過酸化水素を1μl/mlで添加した溶液)を50μl/ウェルで加えて発色させ、415nmの吸光度(以下、OD415と表記する)を測定した。
【0135】
4.抗GD3キメラ抗体の精製
(1)YB2/0細胞由来の生産細胞の培養及び抗体の精製
上記実施例1の2項(1)で得られた抗GD3キメラ抗体を生産する形質転換細胞クローン をBSAを0.2%、MTXを200nM、トリヨードチロニン(以下、T3と表記する;SIGMA社製)を100nMの濃度で含むHybriddoma−SFM培地に3×105細胞/mlとなるように懸濁し、2.0Lスピナーボトル(岩城硝子社製)を用いて50rpmの速度で攪拌培養した。37℃の恒温室内で10日間培養後、培養上清を回収した。培養上清よりProsep−A(Bioprocessing社製)カラムを用いて、添付の説明書に従い、抗GD3キメラ抗体を精製した。精製した抗GD3キメラ抗体は、YB2/0−GD3キメラ抗体と名付けた。
(2)CHO/DG44細胞由来の生産細胞の培養及び抗体の精製
上記実施例1の2項(2)で得られた抗GD3キメラ抗体を生産する形質転換細胞クローンをL−Glnを3mM、脂肪酸濃縮液(以下、CDLCと表記する;GIBCO BRL社製)を0.5%、プルロニックF68(以下、PF68と表記する;GIBCO BRL社製)を0.3%の濃度で含むEX−CELL302培地に1×106細胞/mlとなるように懸濁し、175mm2フラスコ(Greiner社製)に50mlずつ分注した。5%CO2インキュベーター内で37℃で4日間培養後、培養上清を回収した。培養上清よりProsep−A(Bioprocessing社製)カラムを用いて、添付の説明書に従い、抗GD3キメラ抗体を精製した。精製した抗GD3キメラ抗体は、CHO/DG44−GD3キメラ抗体と名付けた。
(3)NS0細胞由来の生産細胞の培養及び抗体の精製
上記実施例1の2項(3)で得られた抗GD3キメラ抗体を生産する形質転換細胞クローンをL−Glnを2mM、G418を0.5mg/ml、MTXを200nM、FBSを1%の濃度で含むEX−CELL302培地に1×106細胞/mlとなるように懸濁し、175mm2フラスコ(Greiner社製)に200mlずつ分注した。5%CO2インキュベーター内で37℃で4日間培養後、培養上清を回収した。培養上清よりProsep−A(Bioprocessing社製)カラムを用いて、添付の説明書に従い、抗GD3キメラ抗体を精製した。精製した抗GD3キメラ抗体は、NS0−GD3キメラ抗体(302)と名付けた。また、該形質転換細胞クローンをG418を0.5mg/ml、MTXを200nMの濃度で含むGIT培地に3×105細胞/mlとなるように懸濁し、175mm2フラスコ(Greiner社製)に200mlずつ分注した。5%CO2インキュベーター内で37℃で10日間培養後、培養上清を回収した。培養上清よりProsep−A(Bioprocessing社製)カラムを用いて、添付の説明書に従い、抗GD3キメラ抗体を精製した。精製した抗GD3キメラ抗体は、NS0−GD3キメラ抗体(GIT)と名付けた。
(4)SP2/0細胞由来の生産細胞の培養及び抗体の精製
特開平5−304989に記載の抗GD3キメラ抗体を生産する形質転換細胞クローンをG418を0.5mg/ml、MTXを200nMの濃度で含むGIT培地に3×105細胞/mlとなるように懸濁し、175mm2フラスコ(Greiner社製)に200mlずつ分注した。5%CO2インキュベーター内で37℃で8日間培養後、培養上清を回収した。培養上清よりProsep−A(Bioprocessing社製)カラムを用いて、添付の説明書に従い、抗GD3キメラ抗体を精製した。精製した抗GD3キメラ抗体は、SP2/0−GD3キメラ抗体と名付けた。
【0136】
5.精製した抗GD3キメラ抗体の解析
上記実施例1の4項で得られた各種動物細胞で生産、精製した5種類の抗GD3キメラ抗体の各4μgを公知の方法[ネイチャー(Nature),227,680,1970]に従ってSDS−PAGEし、分子量及び製精度を解析した。その結果を第1図に示した。第1図に示したように、精製した各抗GD3キメラ抗体は、いずれも非還元条件下では分子量が約150キロダルトン(以下、Kdと表記する)の単一のバンドが、還元条件下では約50Kdと約25Kdの2本のバンドが認められた。これらの分子量は、抗体のH鎖及びL鎖のcDNAの塩基配列から推定される分子量(H鎖:約49Kd、L鎖:約23Kd、分子全体:約144Kd)とほぼ一致し、更に、IgG型の抗体は、非還元条件下では分子量は約150Kdであり、還元条件下では分子内のジスルフィド結合(以下、S−S結合と表記する)が切断され、約50Kdの分子量を持つH鎖と約25Kdの分子量を持つL鎖に分解されるという報告[アンティボディズ:ア・ラボラトリー・マニュアル(Antibodies:A Laboratory Manual),Cold Spring Harbor Laboratory,Chapter14,1988、モノクローナル・アンティボディズ:プリンシプルズ・アンド・プラクティス(Monoclonal Antibodies:principles and Practice),Academic Press Limited,1996]と一致し、各抗GD3キメラ抗体が正しい構造の抗体分子として発現され、かつ精製されたことが確認された。
【実施例2】
【0137】
抗GD3キメラ抗体の活性評価
1.抗GD3キメラ抗体のGD3に対する結合活性(ELISA法)
上記実施例1の4項で得られた5種類の精製抗GD3キメラ抗体のGD3(雪印乳業社製)に対する結合活性を実施例1の3項に示すELISA法により測定した。第2図は、添加する抗GD3キメラ抗体の濃度を変化させて結合活性を検討した結果である。第2図に示したように、5種類の抗GD3キメラ抗体は、ほぼ同等のGD3に対する結合活性を示した。この結果は抗体の抗原結合活性は、抗体を生産する動物細胞やその培養方法に関わらず、一定であることを示している。また、NS0−GD3キメラ抗体(302)とNS0−GD3キメラ抗体(GIT)の比較から抗原結合活性は、培養に用いる培地にも依らず、一定であることが示唆された。
【0138】
2.抗GD3キメラ抗体のin vitro細胞障害活性(ADCC活性)
上記実施例1の4項で得られた5種類の精製抗GD3キメラ抗体のin vitro細胞障害活性を評価するため、以下に示す方法に従い、ADCC活性を測定した。
(1)標的細胞溶液の調製 RPMI1640−FBS(10)培地で培養したヒトメラノーマ培養細胞株G−361(ATCC CRL1424)の1×106細胞を調製し、放射性物質であるNa251CrO4を3.7MBq当量加えて37℃で1時間反応させ、細胞を放射標識した。反応後、RPMI1640−FBS(10)培地で懸濁及び遠心分離操作により3回洗浄し、培地に再懸濁し、4℃で30分間氷中に放置して放射性物質を自然解離させた。遠心分離後、RPMI1640−FBS(10)培地を5ml加え、2×105細胞/mlに調製し、標的細胞溶液とした。
(2)エフェクター細胞溶液の調製 健常人静脈血50mlを採取し、ヘパリンナトリウム(武田薬品社製)0.5mlを加え穏やかに混ぜた。これをLymphoprep(Nycomed Pharma AS社製)を用いて使用説明書に従い、遠心分離して単核球層を分離した。RPMI1640−FBS(10)培地で3回遠心分離して洗浄後、培地を用いて2×106細胞/mlの濃度で再懸濁し、エフェクター細胞溶液とした。
(3)ADCC活性の測定 96ウェルU字底プレート(Falcon社製)の各ウェルに上記(1)で調製した標的細胞溶液の50μl(1×104細胞/ウェル)を分注した。次いで(2)で調製したエフェクター細胞溶液を100μl(2×105細胞/ウェル、エフェクター細胞と標的細胞の比は20:1となる)添加した。更に、各種抗GD3キメラ抗体を各最終濃度0.0025〜2.5μg/mlとなるように加え、37℃で4時間反応させた。反応後、プレートを遠心分離し、上清の51Cr量をγ−カウンターにて測定した。自然解離51Cr量は、エフェクター細胞溶液、抗体溶液の代わりに培地のみを用いて上記と同様の操作を行い、上清の51Cr量を測定することにより求めた。全解離51Cr量は、抗体溶液の代わりに培地のみを、エフェクター細胞溶液の代わりに1規定塩酸を添加し、上記と同様の操作を行い、上清の51Cr量を測定することにより求めた。ADCC活性は下式により求めた。
【0139】
【数1】
【0140】
その結果を第3図に示した。第3図に示したように、5種類の抗GD3キメラ抗体のうち、YB2/0−GD3キメラ抗体が最も高いADCC活性を示し、次いでSP2/0−GD3キメラ抗体、NS0−GD3キメラ抗体、CHO−GD3キメラ抗体の順に高いADCC活性を示した。培養に用いた培地の異なるNS0−GD3キメラ抗体(302)とNS0−GD3キメラ抗体(GIT)では、それらのADCC活性に差は認められなかった。以上の結果は、抗体のADCC活性は、生産に用いる動物細胞によって大きく異なることを示している。その機構としては、抗原結合活性が同等であったことから、抗体のFc領域の構造の差に起因していることが推定された。
【実施例3】
【0141】
抗ヒトインターロイキン5レセプターα鎖ヒト型CDR移植抗体の作製
1.抗ヒトインターロイキン5レセプターα鎖ヒト型CDR移植抗体の安定生産細胞の作製
(1)ラットミエローマYB2/0細胞を用いた生産細胞の作製
WO97/10354に記載の抗ヒトインターロイキン5レセプターα鎖ヒト型CDR移植抗体(以下、抗hIL−5RαCDR移植抗体と表記する)の発現ベクターpKANTEX1259HV3LV0を用いて抗hIL−5RαCDR移植抗体の安定生産細胞を以下のようにして作製した。
抗hIL−5RαCDR移植抗体発現ベクターpKANTEX1259HV3LV0の5μgを4×106細胞のラットミエローマYB2/0細胞へエレクトロポレーション法[サイトテクノロジー(Cytotechnology),3,133,1990]により導入後、40mlのRPMI1640−FBS(10)に懸濁し、96ウェル培養用プレート(住友ベークライト社製)に200μl/ウェルずつ分注した。5%CO2インキュベーター内で37℃、24時間培養した後、G418を0.5mg/mlになるように添加して1〜2週間培養した。G418耐性を示す形質転換株のコロニーが出現し、増殖の認められたウェルより培養上清を回収し、上清中の抗hIL−5RαCDR移植抗体の抗原結合活性を実施例3の2項に示すELISA法により測定した。
培養上清中に抗hIL−5RαCDR移植抗体の生産が認められたウェルの形質転換株については、DHFR遺伝子増幅系を利用して抗体生産量を増加させる目的で、G418を0.5mg/ml、MTXを50nM含むRPMI1640−FBS(10)培地に1〜2×105細胞/mlになるように懸濁し、24ウェルプレート(Greiner社製)に2mlずつ分注した。5%CO2インキュベーター内で37℃で1〜2週間培養して、50nM MTX耐性を示す形質転換株を誘導した。形質転換株の増殖が認められたウェルの培養上清中の抗hIL−5RαCDR移植抗体の抗原結合活性を実施例3の2項に示すELISA法により測定した。培養上清中に抗hIL−5RαCDR移植抗体の生産が認められたウェルの形質転換株については、上記と同様の方法により、MTX濃度を100nM、200nMと順次上昇させ、最終的にG418を0.5mg/ml、MTXを200nMの濃度で含むRPMI1640−FBS(10)培地で増殖可能かつ、抗hIL−5RαCDR移植抗体を高生産する形質転換株を得た。得られた形質転換株については、2回の限界希釈法による単一細胞化(クローン化)を行った。このようにして得られた抗hIL−5RαCDR移植抗体を生産する形質転換細胞クローンNo.3は平成11年4月5日付で工業技術院生命工学工業技術研究所(日本国茨城県つくば市東1丁目1番3号)にFERM BP−6690として寄託されている。
(2)CHO/dhfr−細胞を用いた生産細胞の作製
WO97/10354に記載の抗hIL−5RαCDR移植抗体発現ベクターpKANTEX1259HV3LV0の4μgを1.6×106細胞のCHO/dhfr−細胞へエレクトロポレーション法[サイトテクノロジー(Cytotechnology),3,133(1990)]により導入後、10mlのIMDM−FBS(10)に懸濁し、96ウェル培養用プレート(岩城硝子社製)に200μl/ウェルずつ分注した。5%CO2インキュベーター内で37℃、24時間培養した後、G418を0.5mg/mlになるように添加して1〜2週間培養した。G418耐性を示す形質転換株のコロニーが出現し、増殖の認められたウェルより培養上清を回収し、上清中の抗hIL−5RαCDR移植抗体の抗原結合活性を実施例3の2項に示すELISA法により測定した。
培養上清中に抗hIL−5RαCDR移植抗体の生産が認められたウェルの形質転換株については、DHFR遺伝子増幅系を利用して抗体生産量を増加させる目的で、G418を0.5mg/ml、MTXを10nM含むIMDM−dFBS(10)培地に1〜2×105細胞/mlになるように懸濁し、24ウェルプレート(岩城硝子社製)に0.5mlずつ分注した。5%CO2インキュベーター内で37℃で1〜2週間培養して、10nM MTX耐性を示す形質転換株を誘導した。増殖が認められたウェルの形質転換株については、上記と同様の方法により、MTX濃度を100nM、500nMに上昇させ、最終的にG418を0.5mg/ml、MTXを500nMの濃度で含むIMDM−dFBS(10)培地で増殖可能かつ、抗hIL−5RαCDR移植抗体を高生産する形質転換株を得た。得られた形質転換株については、2回の限界希釈法による単一細胞化(クローン化)を行った。
(3)マウスミエローマNS0細胞を用いた生産細胞の作製
ヤラントン(Yarranton)らの方法[バイオ/テクノロジー(BIO/TECHNOLOGY),10,169(1992)]に従い、WO97/10354に記載の抗hIL−5RαCDR移植抗体発現ベクターpKANTEX1259HV3LV0上の抗体H鎖及びL鎖cDNAを用いて抗hIL−5RαCDR移植抗体発現ベクターを作製し、NS0細胞を形質転換し、抗hIL−5RαCDR移植抗体を高生産する形質転換株を得た。得られた形質転換株については、2回の限界希釈法による単一細胞化(クローン化)を行った。
【0142】
2.抗体のhIL−5Rαに対する結合活性の測定(ELISA法)
抗体のhIL−5Rαに対する結合活性は以下のようにして測定した。
WO97/10354に記載の抗hIL−5Rαマウス抗体KM1257をPBSで10μg/mlの濃度に希釈した溶液の50μlを96ウェルのELISA用のプレート(Greiner社製)の各ウェルにそれぞれ分注し、4℃で20時間反応させた。反応後、1%BSA−PBSを100μl/ウェルで加え、室温で1時間反応させて残存する活性基をブロックした。1%BSA−PBSを捨て、WO97/10354に記載の可溶性hIL−5Rαを1%BSA−PBSで0.5μg/mlの濃度に希釈した溶液を50μl/ウェルで加え、4℃で20時間反応させた。反応後、各ウェルをTween−PBSで洗浄後、形質転換株の培養上清或いは精製したヒト型CDR移植抗体の各種希釈溶液を50μl/ウェルで加え、室温で2時間反応させた。反応後、各ウェルをTween−PBSで洗浄後、1%BSA−PBSで3000倍に希釈したペルオキシダーゼ標識ヤギ抗ヒトIgG(H&L)抗体溶液(American Qualex社製)を二次抗体溶液として、50μl/ウェルで加え、室温で1時間反応させた。反応後、Tween−PBSで洗浄後、ABTS基質液[2,2’−アジノ−ビス(3−エチルベンゾチアゾリン−6−スルホン酸)アンモニウムの0.55gを1Lの0.1Mクエン酸緩衝液(pH4.2)に溶解し、使用直前に過酸化水素を1μl/mlで添加した溶液]を50μl/ウェルで加えて発色させ、OD415を測定した。
【0143】
3.抗hIL−5RαCDR移植抗体の精製
(1)YB2/0細胞由来の生産細胞の培養及び抗体の精製
上記実施例3の1項(1)で得られた抗hIL−5RαCDR移植抗体を生産する形質転換細胞クローンをG418を0.5mg/ml、MTXを200nMの濃度で含むGIT培地に3×105細胞/mlとなるように懸濁し、175mm2フラスコ(Greiner社製)に200mlずつ分注した。5%CO2インキュベーター内で37℃で8日間培養後、培養上清を回収した。培養上清よりイオン交換クロマトグラフィー及びゲル濾過法を用いて抗hIL−5RαCDR移植抗体を精製した。精製した抗hIL−5RαCDR移植抗体は、YB2/0−hIL−5RCDR抗体と名付けた。
(2)CHO/dhfr−細胞由来の生産細胞の培養及び抗体の精製
上記実施例3の1項(2)で得られた抗hIL−5RαCDR移植抗体を生産する形質転換細胞クローンをL−Glnを3mM、CDLCを0.5%、PF68を0.3%の濃度で含むEX−CELL302培地に3×105細胞/mlとなるように懸濁し、4.0Lスピナーボトル(岩城硝子社製)を用いて100rpmの速度で攪拌培養した。37℃の恒温室内で10日間培養後、培養上清を回収した。培養上清よりイオン交換クロマトグラフィー及びゲル濾過法を用いて抗hIL−5RαCDR移植抗体を精製した。精製した抗hIL−5RαCDR移植抗体は、CHO/d−hIL−5RCDR抗体と名付けた。
(3)NS0細胞由来の生産細胞の培養及び抗体の精製
上記実施例3の1項(3)で得られた抗hIL−5RαCDR移植抗体を生産する形質転換細胞クローンをヤラントン(Yarranton)らの方法[バイオ/テクノロジー(BIO/TECHNOLOGY),10,169(1992)]に従い、培養後、培養上清を回収した。培養上清よりイオン交換クロマトグラフィー及びゲル濾過法を用いて抗hIL−5RαCDR移植抗体を精製した。精製した抗hIL−5RαCDR移植抗体は、NS0−hIL−5RCDR抗体と名付けた。
【0144】
4.精製した抗hIL−5RαCDR移植抗体の解析
上記実施例3の3項で得られた各種動物細胞で生産、精製した3種類の抗hIL−5RαCDR移植抗体の各4μgを公知の方法[ネイチャー(Nature),227,680(1970)]に従ってSDS−PAGEし、分子量及び製精度を解析した。その結果を第4図に示した。第4図に示したように、精製した各抗hIL−5RαCDR移植抗体は、いずれも非還元条件下では分子量が約150Kdの単一のバンドが、還元条件下では約50Kdと約25Kdの2本のバンドが認められた。これらの分子量は、抗体のH鎖及びL鎖のcDNAの塩基配列から推定される分子量(H鎖:約49Kd、L鎖:約23Kd、分子全体:約144Kd)とほぼ一致し、更に、IgG型の抗体は、非還元条件下では分子量は約150Kdであり、還元条件下では分子内のジスルフィド結合(以下、S−S結合と表記する)が切断され、約50Kdの分子量を持つH鎖と約25Kdの分子量を持つL鎖に分解されるという報告[アンティボディズ:ア・ラボラトリー・マニュアル(Antibodies:A Laboratory Manual),Cold Spring Harbor Laboratory,Chapter 14,1988、モノクローナル・アンティボディズ:プリンシプルズ・アンド・プラクティス(Monoclonal Antibodies:Principles and Practice),Academic Press Limited,1996]と一致し、各抗hIL−5RαCDR移植抗体が正しい構造の抗体分子として発現され、かつ、精製されたことが確認された。
【実施例4】
【0145】
抗hIL−5RαCDR移植抗体の活性評価
1.抗hIL−5RαCDR移植抗体のhIL−5Rαに対する結合活性(ELISA法)
上記実施例3の3項で得られた3種類の精製抗hIL−5RαCDR移植抗体のhIL−5Rαに対する結合活性を実施例3の2項に示すELISA法により測定した。第5図は、添加する抗hIL−5RαCDR移植抗体の濃度を変化させて結合活性を検討した結果である。第5図に示したように、3種類の抗hIL−5RαCDR移植抗体は、ほぼ同等のhIL−5Rαに対する結合活性を示した。この結果は実施例2の1項の結果と同様に、抗体の抗原結合活性は、抗体を生産する動物細胞やその培養方法に関わらず、一定であることを示している。
【0146】
2.抗hIL−5RαCDR移植抗体のin vitro細胞障害活性(ADCC活性)
上記実施例3の3項で得られた3種類の精製抗hIL−5RαCDR移植抗体のin vitro細胞障害活性を評価するため、以下に示す方法に従い、ADCC活性を測定した。
(1)標的細胞溶液の調製
WO97/10354に記載のhIL−5Rα鎖及びβ鎖を発現しているマウスT細胞株CTLL−2(h5R)をRPMI1640−FBS(10)培地で培養し、1×106細胞/0.5mlとなるように調製し、放射性物質であるNa251CrO4を3.7MBq当量加えて37℃で1.5時間反応させ、細胞を放射標識した。反応後、RPMI1640−FBS(10)培地で懸濁及び遠心分離操作により3回洗浄し、培地に再懸濁し、4℃で30分間氷中に放置して放射性物質を自然解離させた。遠心分離後、RPMI1640−FBS(10)培地を5ml加え、2×105細胞/mlに調製し、標的細胞溶液とした。
(2)エフェクター細胞溶液の調製
健常人静脈血50mlを採取し、ヘパリンナトリウム(武田薬品社製)0.5mlを加え穏やかに混ぜた。これをPolymorphprep(Nycomed Pharma AS社製)を用いて使用説明書に従い、遠心分離して単核球層を分離した。RPMI1640−FBS(10)培地で3回遠心分離して洗浄後、培地を用いて9×106細胞/mlの濃度で再懸濁し、エフェクター細胞溶液とした。
(3)ADCC活性の測定
96ウェルU字底プレート(Falcon社製)の各ウェルに上記(1)で調製した標的細胞溶液の50μl(1×104細胞/ウェル)を分注した。次いで(2)で調製したエフェクター細胞溶液を100μl(9×105細胞/ウェル、エフェクター細胞と標的細胞の比は90:1となる)添加した。更に、各種抗hIL−5RαCDR移植抗体を各最終濃度0.001〜0.1μg/mlとなるように加え、37℃で4時間反応させた。
反応後、プレートを遠心分離し、上清の51Cr量をγ−カウンターにて測定した。自然解離51Cr量は、エフェクター細胞溶液、抗体溶液の代わりに培地のみを用いて上記と同様の操作を行い、上清の51Cr量を測定することにより求めた。全解離51Cr量は、抗体溶液の代わりに培地のみを、エフェクター細胞溶液の代わりに1規定塩酸を添加し、上記と同様の操作を行い、上清の51Cr量を測定することにより求めた。
ADCC活性は下式により求めた。
【0147】
【数2】
【0148】
その結果を第6図に示した。第6図に示したように、3種類の抗hIL−5RαCDR移植抗体のうち、YB2/0−hIL−5RCDR抗体が最も高いADCC活性を示し、次いでCHO/d−hIL−5RCDR抗体、NS0−hIL−5RCDR抗体の順に高いADCC活性を示した。以上の結果は実施例2の2項の結果と同ように、抗体のADCC活性は、生産に用いる動物細胞によって大きく異なることを示している。更に、2種類のヒト化抗体のいずれの場合もYB2/0細胞で生産した抗体が最も高いADCC活性を示したことから、YB2/0細胞を用いることにより、ADCC活性の高い抗体を製造できることが明らかとなった。
【0149】
3.抗hIL−5RαCDR移植抗体のin vivoにおける活性評価
上記実施例3の3項で得られた3種類の精製抗hIL−5RαCDR移植抗体のin vivoにおける活性を評価するため、以下に示す方法に従い、カニクイザルのhIL−5誘発好酸球増加モデルに対する抑制作用を検討した。
カニクイザルに初日よりhIL−5(調製方法はWO97/10354に記載)を1μg/kgで1日1回、計14回背部皮下より投与した。各種抗hIL−5RαCDR移植抗体を0日のhIL−5の投与1時間前に0.3mg/kgで静脈内に単回投与した。抗体非投与群をコントロールとして用いた。抗体投与群は各群3頭(No.301、No.302、No.303、No.401、No.402、No.403、No.501、No.502、No.503)、抗体非投与群は2頭(No.101、No.102)のカニクイザルを用いた。投与開始の7日前より投与後42日目まで経時的に約1mlの血液を伏在静脈または大腿静脈より採取し、1μlの末梢血中の好酸球数を測定した。その結果を第7図に示した。第7図に示したように、YB2/0−hIL−5RCDR抗体を投与した群では、血中好酸球の増加が完全に抑制された。一方、CHO/d−hIL−5RCDR抗体の投与群では、1頭で完全な抑制作用が認められたものの、2頭ではその抑制作用は不充分であった。NS0−hIL−5RCDR抗体の投与群では、完全な抑制作用は認められず、その効果は不充分であった。以上の結果は、抗体のin vivo活性は、生産に用いる動物細胞によって大きく異なることを示している。更に、抗hIL−5RαCDR移植抗体ではそのin vivo活性の高さは、実施例4の2項で述べたADCC活性の高さと正の相関が認められたことから、その活性発現には、ADCC活性の高さが極めて重要であることが示唆された。
以上の結果から、ADCC活性の高い抗体は、ヒトの各種疾患の臨床においても有用であることが期待される。
【実施例5】
【0150】
ADCC活性を高める糖鎖の解析
1.2−アミノピリジン標識糖鎖(PA化糖鎖)の調製
本発明のヒト化抗体を塩酸による酸加水分解にてシアル酸を除去した。塩酸を完全に除去した後、ヒドラジン分解により糖鎖を蛋白質から切断した[メソッド・オブ・エンザイモロジー(Method of Enzymology),83,263,1982]。ヒドラジンを除去した後、酢酸アンモニウム水溶液と無水酢酸加えてN−アセチル化を行った。凍結乾燥後、2−アミノピリジンによる蛍光標識を行った[ジャーナル・オブ・バイオケミストリー(J.Biochem.),95,197(1984)]。蛍光標識した糖鎖(PA化糖鎖)を、Surperdex Peptide HR 10/30カラム(Pharmacia社製)を用いて不純物と分離した。糖鎖画分を遠心濃縮機にて乾固させ、精製PA化糖鎖とした。
【0151】
2.精製抗hIL−5RαCDR移植抗体のPA化糖鎖の逆相HPLC分析
上記実施例5の1項で調製した各種抗hIL−5RCDR抗体のPA化糖鎖を用いて、CLC−ODSカラム(Shimadzu社製)による逆相HPLC分析を行った。過剰量のα−L−フコシダーゼ(ウシ腎由来、SIGMA社製)をPA化糖鎖に添加して消化を行い(37℃、15時間)、逆相HPLCで分析した(第8図)。アスパラギン結合糖鎖は30分間から80分間の範囲に溶出することをTaKaRa社製PA化糖鎖スタンダードを用いて確認した。α−L−フコシダーゼ消化によって、逆相HPLCの溶出位置が移動する糖鎖(48分間から78分間に溶出される糖鎖)の全体に占める割合を計算した。結果を第1表に示す。
【0152】
【表1】
【0153】
YB2/0細胞で生産させた抗hIL−5RCDR移植抗体は約47%、NS0細胞で生産させた抗hIL−5RCDR移植抗体は約73%がα1−6フコースをもつ糖鎖であった。よって、YB2/0細胞で生産した抗体は、NS0細胞で生産した抗体と比較してα1−6フコースを持たない糖鎖が多かった。
【0154】
3.精製抗hIL−5RαCDR移植抗体の単糖組成分析
トリフルオロ酢酸による酸加水分解により、YB2/0細胞、NS0細胞およびCHO/d細胞で生産した抗hIL−5RαCDR移植抗体の糖鎖を単糖に分解し、BioLC(Dionex社製)を用いて単糖組成分析を行った。
N−グリコシド結合糖鎖のうち、コンプレックス型では、1本の糖鎖におけるマンノース数は3であるため、マンノースを3として計算した場合の各単糖の相対比を第2表に示す。
【0155】
【表2】
【0156】
フコースの相対比は、YB2/0<CHO/d<NS0であり、本結果でもYB2/0細胞で生産した抗体の糖鎖はフコース含量が最も低かった。
【実施例6】
【0157】
CHO/dhfr−細胞生産抗体の糖鎖解析
CHO/dhfr−細胞で生産した精製抗hIl−5RαCDR移植抗体からPA化糖鎖を調製し、CLC−ODSカラム(島津社製)を用いて逆相HPLC分析を行った(第9図)。第9図において、溶出時間35〜45分間がフコースを持たない糖鎖、45〜60分間がフコースを持つ糖鎖であった。CHO/dhfr−細胞で生産した抗hIl−5RαCDR移植抗体は、マウスミエローマNS0細胞で生産させた抗体と同様に、ラットミエローマYB2/0細胞で生産させた抗体よりもフコースを持たない糖鎖の含量が少なかった。
【実施例7】
【0158】
高ADCC活性抗体の分離
フコースを持つ糖鎖に結合するレクチンカラムを用いて、ラットミエローマYB2/0細胞で生産させた抗hIl−5RαCDR移植抗体の分離を行った。HPLCは島津社製LC−6Aを用い、流速は1ml/分、カラム温度は室温で行った。50mMトリス−硫酸緩衝液(pH7.3)で平衡化し、精製された抗hIL−5RαCDR移植抗体を注入後、0.2Mα−メチルマンノシド(ナカライテスク社製)の直線濃度勾配(60分間)にて溶出した。抗hIl−5RαCDR移植抗体を非吸着画分と吸着画分とに分離した。
非吸着画分、吸着画分の一部をとり、hIL−5Rαに対する結合活性を測定すると、同様の結合活性を示した(第10図 上図)。ADCC活性を測定すると、非吸着画分の方が吸着画分の一部よりも高いADCC活性を示した(第10図 下図)。さらに、非吸着画分、吸着画分の一部からPA化糖鎖を調製し、CLC−ODSカラム(島津社製)を用いて逆相HPLC分析を行った(第11図)。非吸着画分は主としてフコースのない糖鎖をもつ抗体であり、吸着画分の一部は主としてフコースがある糖鎖もつ抗体であった。
【実施例8】
【0159】
宿主細胞株におけるα1,6−フコシルトランスフェラーゼ(FUT8)遺伝子の転写物の定量
(1)各種細胞株由来一本鎖cDNAの調製
チャイニーズハムスター卵巣由来CHO/DG44細胞を、10%FBS(Life Technologies社)および1倍濃度のHT supplement(Life Technologies社)を添加したIMDM培地(Life Technologies社)に懸濁し、2×105cells/mlの密度で接着細胞培養用T75フラスコ(Greiner社)に播種した。またラットミエローマ由来YB2/0細胞を、10%FBS(Life Technlogies社)、4mMグルタミン(Life Technologies社)を添加したRPMI1640培地(Life Technologies社)に懸濁し、2×105cells/mlの密度で浮遊細胞培養用T75フラスコ(Greiner社)に播種した。これらを37℃の5%CO2インキュベーター内で培養し、培養1日目、2日目、3日目、4日目および5日目に各宿主細胞1×107cellsを回収し、RNAeasy(QIAGEN社製)により全RNAを抽出した。
全RNAを45μlの滅菌水に溶解し、RQ1 Rnase−Free DNase(Promega社)0.5U/μl、付属の10×DNase buffer 5μl、RNasin Ribonuclease inhibitor(Promega社)0.5μlをそれぞれに添加して、37℃で30分間反応させた。反応後、RNAeasy(QIAGEN社)により全RNAを再精製し、50μlの滅菌水に溶解した。
得られた各々の全RNA3μgについて、オリゴ(dT)をプライマーとしてSUPERSCRIPTTM Preamplification System for First Strand cDNA Synthesis(Life Technologies社)により、20μlの系で逆転写反応を行い、cDNAを合成した。各宿主細胞由来FUT8およびβ−アクチンのクローニングには逆転写反応後の溶液の1倍濃度液を、競合的PCRによる各遺伝子転写量の定量には逆転写反応後の溶液を水で50倍希釈したものを用い、各々使用するまで−80℃で保管した。
【0160】
(2)チャイニーズハムスターFUT8およびラットFUT8の各cDNA部分断片の取得
チャイニーズハムスターFUT8およびラットFUT8の各cDNA部分断片の取得は、以下のように行った。まず、ヒトFUT8のcDNA[Journal of Biochemistry,121,626(1997)]およびブタFUT8のcDNA[Journal of Biological Chemistry,271,27810(1996)]に共通の塩基配列に対して特異的なプライマー(配列番号1および配列番号2に示す)を設計した。
次にDNAポリメラーゼExTaq(宝酒造社)を用いて、(1)で調製した培養2日目のCHO細胞由来cDNAおよびYB2/0細胞由来cDNAを各々1μlを含む25μlの反応液[ExTaq buffer(宝酒造社)、0.2mM dNTPs、0.5μM上記特異的プライマー(配列番号1および配列番号2)]を調製し、ポリメラーゼ連鎖反応(PCR)を行った。PCRは、94℃で1分間の加熱の後、94℃で30秒間、55℃で30秒間、72℃で2分間からなる反応を1サイクルとして30サイクルの後、さらに72℃で10分間加熱する条件で行った。PCRにより得た各特異的増幅断片979bpをTOPO TA cloning Kit(Invitrogen社)を用いてプラスミドpCR2.1へ連結し、チャイニーズハムスターFUT8およびラットFUT8の各cDNA部分断片を含むプラスミド(CHFT8−pCR2.1およびYBFT8−pCR2.1)を取得した。
取得した各cDNAの塩基配列について、DNAシークエンサー377(Parkin
Elmer社)およびBigDye Terminator Cycle Sequencing FS Ready Reaction Kit(Parkin Elmer社)を用いて決定し、取得したcDNAがチャイニーズハムスターFUT8およびラットFUT8(配列番号3および4に示す)のオープンリーディングフレーム(ORF)部分配列をコードすることを確認した。
【0161】
(3)チャイニーズハムスターβ−アクチンおよびラットβ−アクチンcDNAの取得 β−アクチン遺伝子は各細胞において恒常的に転写されており、その転写量は細胞間で同程度と考えられているため、各細胞由来cDNA合成反応の効率の目安としては、β−アクチン遺伝子の転写量を定量する。
チャイニーズハムスターβ−アクチンおよびラットβ−アクチンの取得は、以下の方法で行った。まず、チャイニーズハムスターβ−アクチンゲノム配列[GenBank,U20114]およびラットβ−アクチンゲノム配列[Nucleic Acid Research,11,1759(1983)]より、翻訳開始コドンを含む共通配列に特異的なフォワードプライマー(配列番号5に示す)および翻訳終止コドンを含む各配列特異的なリバースプライマー(配列番号6および配列番号7に示す)を設計した。
次にDNAポリメラーゼKOD(東洋紡社)を用いて、(1)で調製した培養2日目のCHO細胞由来cDNAおよびYB2/0細胞由来cDNA 1μlを含む25μlの反応液[KOD buffer #1(東洋紡社)、0.2mM dNTPs、1mM MgCl2、0.4μM 上記遺伝子特異的プライマ−(配列番号5および配列番号6、または配列番号5および配列番号7)、5% DMSO]を調製し、ポリメラーゼ連鎖反応(PCR)を行った。PCRは、94℃で4分間の加熱の後、98℃で15秒間、65℃で2秒間、74℃で30秒間からなる反応を1サイクルとして、25サイクル行った。PCRにより得た各特異的増幅断片1128bpの5’末端をMEGALABEL(宝酒造社)によりリン酸化した後、pBluescriptII KS(+)(Strategene社)に、制限酵素EcoRVにより切断して得られる断片(2.9Kb)をLigation High(東洋紡社)で連結し、チャイニーズハムスターβ−アクチンおよびラットβ−アクチンの各cDNAのORF全長を含むプラスミド(CHAc−pBS、YBAc−pBS)を得た。
取得した各cDNAの塩基配列を、DNAシークエンサー377(Parkin Elmer社製)およびBigDye Terminator Cycle Sequencing FS Ready Reaction Kit(Parkin Elmer社製)を用いて決定し、各々チャイニーズハムスターβ−アクチンおよびラットβ−アクチンの各cDNAのORF全長配列をコードすることを確認した。
【0162】
(4)スタンダードおよび内部配列のコントロールの調製
生産細胞内のFUT8遺伝子からのmRNA転写量を測定するために、まず検量線を作成した。
検量線に用いるFUT8のスタンダードとしては、(2)で得たチャイニーズハムスターFUT8およびラットFUT8の各cDNA部分断片をpCR2.1に組み込んだプラスミドであるCHFT8−pCR2.1およびYBFT8−pCR2.1を制限酵素EcoRIで切断して得られたDNAを直鎖化して用いた。
FUT8定量の内部コントロールとしては、CHFT8−pCR2.1およびYBFT8−pCR2.1のうち、チャイニーズハムスターFUT8およびラットFUT8の内部塩基配列のScaI−HindIII間203bpを欠失させることにより得られたCHFT8d−pCR2.1およびYBFT8d−pCR2.1を、制限酵素EcoRIで切断して得られたDNAを直鎖化して用いた。
生産細胞内のβ−アクチン遺伝子からのmRNA転写量のスタンダードとしては、(3)で得たチャイニーズハムスターβ−アクチンおよびラットβ−アクチンのそれぞれのcDNAのORF全長をpBluescriptII KS(+)に組み込んだプラスミドであるCHAc−pBSおよびYBAc−pBSを、前者はHindIIIおよびPstIで、後者はHindIIIおよびKpnIで、各々切断して得られたDNAを直鎖化して用いた。
β−アクチン定量の内部コントロールとしては、CHAc−pBSおよびYBAc−pBSのうち、チャイニーズハムスターβ−アクチンおよびラットβ−アクチンの内部塩基配列のDraIII−DraIII間180bpを欠失させることにより得られたCHAcd−pBSおよびYBAcd−pBSを、前者はHindIIIおよびPstIで、後者はHindIIIおよびKpnIで、各々切断して得られたDNAを直鎖化して用いた。
【0163】
(5)競合的RT−PCRによる転写量の定量
FUT8の転写産物の定量は以下のように行った。まず(2)で得たチャイニーズハムスターFUT8およびラットFUT8のORF部分配列の内部配列に対し、共通配列特異的なプライマーセット(配列番号8および9に示す)を設計した。
次に、(1)で得られた各宿主細胞株由来のcDNA溶液の50倍希釈液 5μlおよび内部コントロール用プラスミド5μl(10fg)を含む総体積20μlの反応液[ExTaq buffer(宝酒造社)、0.2mM dNTPs、0.5μM 上記遺伝子特異的プライマー(配列番号8および配列番号9)、5%DMSO]で、DNAポリメラーゼExTaq(宝酒造社)を用いてPCRを行った。PCRは、94℃で3分間の加熱の後、94℃で1分間、60℃で1分間、72℃で1分間からなる反応を1サイクルとして32サイクル行った。
β−アクチンの転写産物の定量は、以下のように行った。(3)で得たチャイニーズハムスターβ−アクチンおよびラットβ−アクチンORF全長の内部配列に対し、各遺伝子特異的なプライマーセット(前者を配列番号10および配列番号11に、後者を配列番号12および配列番号13に示す)をそれぞれ設計した。
次に、(1)で得られた各宿主細胞株由来のcDNA溶液の50倍希釈液 5μlおよび内部コントロール用プラスミド5μl(1pg)を含む総体積20μlの反応液[ExTaq buffer(宝酒造社)、0.2mM dNTPs、0.5μM 上記遺伝子特異的プライマー(配列番号10および配列番号11、または配列番号12および配列番号13)、5% DMSO]で、DNAポリメラーゼExTaq(宝酒造社)を用いてPCRを行った。PCRは、94℃で3分間の加熱の後、94℃で30秒間、65℃で1分間、72℃で2分間からなる反応を1サイクルとして17サイクル行った。
【0164】
【表3】
【0165】
第3表に記載のプライマーセットを用いて定量的PCRを行った。その結果、各遺伝子転写産物および各スタンダードからは、第3表のターゲット欄に示したサイズのDNA断片を、各内部コントロールからは、第3表のコンペティター欄に示したサイズのDNA断片を増幅させることができた。
PCR後の溶液のうち、7μlを1.75%アガロースゲル電気泳動に供した後、ゲルをSYBR Green I Nucleic Acid Gel Stain(Molecular Probes社)で染色した。増幅された各DNA断片の発光強度をフルオロイメージャー(FluorImager SI;Molecular Dynamics社)で算出することにより、増幅されたDNA断片の量を測定した。
一方、細胞由来cDNAに代えて(4)で調製したスタンダードプラスミド量を0.1fg、1fg、5fg、10fg、50fg、100fgおよび500fgとしてPCRをそれぞれ行い、増幅産物量を測定した。そして、その測定値とスタンダードプラスミド量をプロットして検量線を作成した。
上述の検量線を用いて、各細胞由来全cDNAを鋳型とした場合の増幅産物の量より各細胞中の目的遺伝子cDNA量を算出し、これを各細胞におけるmRNA転写量とした。
ラットFUT8配列をスタンダード、内部コントロールに用いた場合の各宿主細胞株におけるFUT8転写産物の量を第12図に示した。培養期間を通じてCHO細胞株はYB2/0細胞株の10倍以上の転写量を示した。この傾向は、チャイニーズハムスターFUT8配列をスタンダード、内部コントロールに用いた場合にも認められた。
また、第4表にβ−actin転写産物の量との相対値としてFUT8転写量を示した。
【0166】
【表4】
【0167】
YB2/0細胞株のFUT8転写量がβ−actinの0.1%前後であるのに対し、CHO細胞株は0.5〜2%であった。
以上の結果より、YB2/0細胞株のPUT8転写産物量はCHO細胞株のそれよりも有意に少ないことが示された。
【産業上の利用可能性】
【0168】
本発明は、抗体、蛋白質またはペプチドなどの免疫機能分子の有する活性を調節する糖鎖、および糖鎖を有する抗体、蛋白質またはペプチドに関する。本発明は更に、該糖鎖および糖鎖を有する抗体、蛋白質またはペプチドの製造法、ならびにそれらを有効成分として含有する診断薬、予防薬および治療薬に関する。
【特許請求の範囲】
【請求項1】
N−グリコシド結合コンプレックス型糖鎖が結合した抗体を含有する抗体組成物であって、該抗体が還元末端のN−アセチルグルコサミンにフコースが結合しないN−グリコシド結合コンプレックス型糖鎖を有する抗体である抗体組成物。
【請求項2】
糖鎖が、
【化4】
を含む糖鎖を含有する請求項1に記載の抗体組成物。
【請求項3】
糖鎖が、α1,6−フコシルトランスフェラーゼ活性の低い細胞が合成する糖鎖を含有する請求項1または2に記載の抗体組成物。
【請求項4】
α1,6−フコシルトランスフェラーゼ活性の低い細胞を用いることを特徴とする請求項1または2に記載の抗体組成物の製造方法。
【請求項5】
抗体が、腫瘍関連抗原を認識する抗体である請求項1〜3のいずれか1項に記載の抗体組成物。
【請求項6】
腫瘍関連抗原がガングリオシドGD3である、請求項5に記載の抗体組成物。
【請求項7】
抗体が、7−9−51(FERM BP−6691)より生産される抗体である請求項5または6に記載の抗体組成物。
【請求項8】
抗体が、アレルギーまたは炎症に関連する抗原を認識する抗体である請求項1〜3のいずれか1項に記載の抗体組成物。
【請求項9】
アレルギーまたは炎症に関連する抗原が、ヒトインターロイキン5レセプターα鎖である請求項8に記載の抗体組成物。
【請求項10】
抗体が、No.3(FERM BP−6690)より生産される抗体である請求項8または9に記載の抗体組成物。
【請求項11】
抗体が、循環器疾患に関連する抗原を認識する抗体である請求項1〜3のいずれか1項に記載の抗体組成物。
【請求項12】
抗体が、自己免疫疾患に関連する抗原を認識する抗体である請求項1〜3のいずれか1項に記載の抗体組成物。
【請求項13】
抗体が、ウィルスまたは細菌感染に関連する抗原を認識する抗体である請求項1〜3のいずれか1項に記載の抗体組成物。
【請求項14】
請求項5〜7のいずれか1項に記載の抗体組成物を有効成分として含有する、癌の診断薬。
【請求項15】
請求項5〜7のいずれか1項に記載の抗体組成物を有効成分として含有する、癌の治療薬。
【請求項16】
請求項5〜7のいずれか1項に記載の抗体組成物を有効成分として含有する、癌の予防薬。
【請求項17】
請求項8〜10のいずれか1項に記載の抗体組成物を有効成分として含有する、アレルギーまたは炎症の診断薬。
【請求項18】
請求項8〜10のいずれか1項に記載の抗体組成物を有効成分として含有する、アレルギーまたは炎症の治療薬。
【請求項19】
請求項8〜10のいずれか1項に記載の抗体組成物を有効成分として含有する、アレルギーまたは炎症の予防薬。
【請求項20】
請求項11に記載の抗体組成物を有効成分として含有する、循環器疾患の診断薬。
【請求項21】
請求項11に記載の抗体組成物を有効成分として含有する、循環器疾患の治療薬。
【請求項22】
請求項11に記載の抗体組成物を有効成分として含有する、循環器疾患の予防薬。
【請求項23】
請求項12に記載の抗体組成物を有効成分として含有する、自己免疫疾患の診断薬。
【請求項24】
請求項12に記載の抗体組成物を有効成分として含有する、自己免疫疾患の治療薬。
【請求項25】
請求項12に記載の抗体組成物を有効成分として含有する、自己免疫疾患の予防薬。
【請求項26】
請求項13に記載の抗体組成物を有効成分として含有する、ウィルスまたは細菌感染症の診断薬。
【請求項27】
請求項13に記載の抗体組成物を有効成分として含有する、ウィルスまたは細菌感染症の治療薬。
【請求項28】
請求項13に記載の抗体組成物を有効成分として含有する、ウィルスまたは細菌感染症の予防薬。
【請求項1】
N−グリコシド結合コンプレックス型糖鎖が結合した抗体を含有する抗体組成物であって、該抗体が還元末端のN−アセチルグルコサミンにフコースが結合しないN−グリコシド結合コンプレックス型糖鎖を有する抗体である抗体組成物。
【請求項2】
糖鎖が、
【化4】
を含む糖鎖を含有する請求項1に記載の抗体組成物。
【請求項3】
糖鎖が、α1,6−フコシルトランスフェラーゼ活性の低い細胞が合成する糖鎖を含有する請求項1または2に記載の抗体組成物。
【請求項4】
α1,6−フコシルトランスフェラーゼ活性の低い細胞を用いることを特徴とする請求項1または2に記載の抗体組成物の製造方法。
【請求項5】
抗体が、腫瘍関連抗原を認識する抗体である請求項1〜3のいずれか1項に記載の抗体組成物。
【請求項6】
腫瘍関連抗原がガングリオシドGD3である、請求項5に記載の抗体組成物。
【請求項7】
抗体が、7−9−51(FERM BP−6691)より生産される抗体である請求項5または6に記載の抗体組成物。
【請求項8】
抗体が、アレルギーまたは炎症に関連する抗原を認識する抗体である請求項1〜3のいずれか1項に記載の抗体組成物。
【請求項9】
アレルギーまたは炎症に関連する抗原が、ヒトインターロイキン5レセプターα鎖である請求項8に記載の抗体組成物。
【請求項10】
抗体が、No.3(FERM BP−6690)より生産される抗体である請求項8または9に記載の抗体組成物。
【請求項11】
抗体が、循環器疾患に関連する抗原を認識する抗体である請求項1〜3のいずれか1項に記載の抗体組成物。
【請求項12】
抗体が、自己免疫疾患に関連する抗原を認識する抗体である請求項1〜3のいずれか1項に記載の抗体組成物。
【請求項13】
抗体が、ウィルスまたは細菌感染に関連する抗原を認識する抗体である請求項1〜3のいずれか1項に記載の抗体組成物。
【請求項14】
請求項5〜7のいずれか1項に記載の抗体組成物を有効成分として含有する、癌の診断薬。
【請求項15】
請求項5〜7のいずれか1項に記載の抗体組成物を有効成分として含有する、癌の治療薬。
【請求項16】
請求項5〜7のいずれか1項に記載の抗体組成物を有効成分として含有する、癌の予防薬。
【請求項17】
請求項8〜10のいずれか1項に記載の抗体組成物を有効成分として含有する、アレルギーまたは炎症の診断薬。
【請求項18】
請求項8〜10のいずれか1項に記載の抗体組成物を有効成分として含有する、アレルギーまたは炎症の治療薬。
【請求項19】
請求項8〜10のいずれか1項に記載の抗体組成物を有効成分として含有する、アレルギーまたは炎症の予防薬。
【請求項20】
請求項11に記載の抗体組成物を有効成分として含有する、循環器疾患の診断薬。
【請求項21】
請求項11に記載の抗体組成物を有効成分として含有する、循環器疾患の治療薬。
【請求項22】
請求項11に記載の抗体組成物を有効成分として含有する、循環器疾患の予防薬。
【請求項23】
請求項12に記載の抗体組成物を有効成分として含有する、自己免疫疾患の診断薬。
【請求項24】
請求項12に記載の抗体組成物を有効成分として含有する、自己免疫疾患の治療薬。
【請求項25】
請求項12に記載の抗体組成物を有効成分として含有する、自己免疫疾患の予防薬。
【請求項26】
請求項13に記載の抗体組成物を有効成分として含有する、ウィルスまたは細菌感染症の診断薬。
【請求項27】
請求項13に記載の抗体組成物を有効成分として含有する、ウィルスまたは細菌感染症の治療薬。
【請求項28】
請求項13に記載の抗体組成物を有効成分として含有する、ウィルスまたは細菌感染症の予防薬。
【図2】

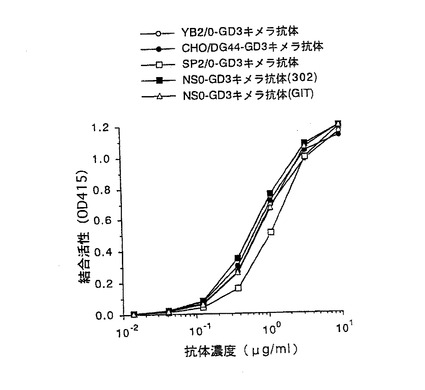
【図3】
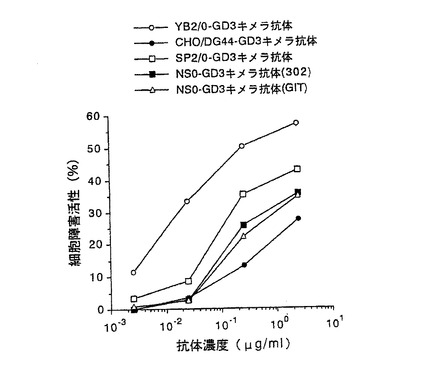
【図5】
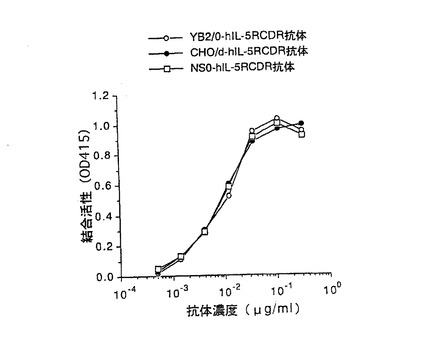
【図6】
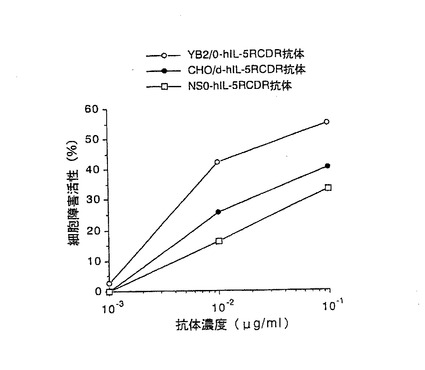
【図7】
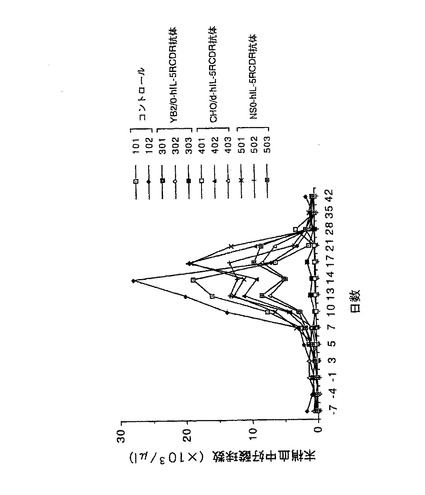
【図8】
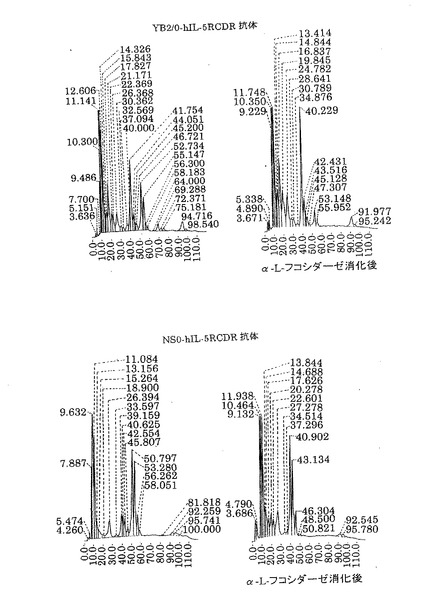
【図9】
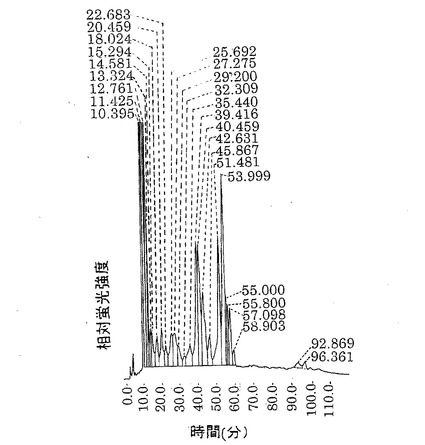
【図10】
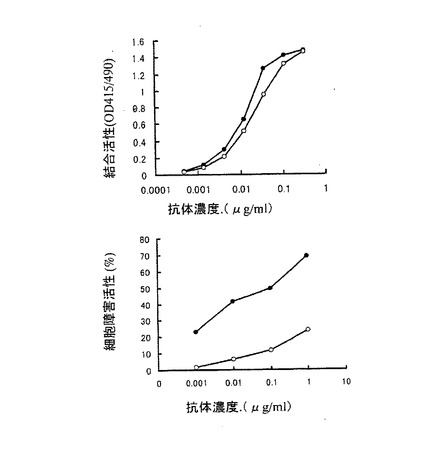
【図11】
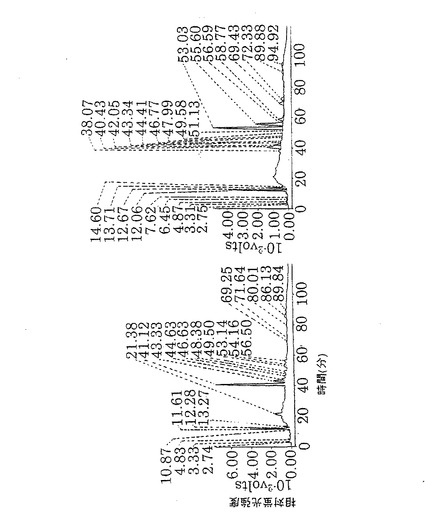
【図12】
【図1】
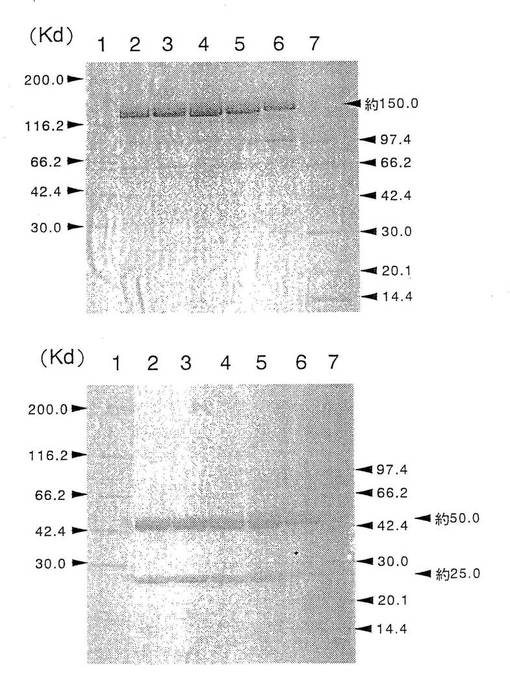
【図4】

【図3】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図1】
【図4】
【公開番号】特開2009−275049(P2009−275049A)
【公開日】平成21年11月26日(2009.11.26)
【国際特許分類】
【出願番号】特願2009−160787(P2009−160787)
【出願日】平成21年7月7日(2009.7.7)
【分割の表示】特願2000−611663(P2000−611663)の分割
【原出願日】平成12年4月7日(2000.4.7)
【出願人】(000001029)協和発酵キリン株式会社 (276)
【Fターム(参考)】
【公開日】平成21年11月26日(2009.11.26)
【国際特許分類】
【出願日】平成21年7月7日(2009.7.7)
【分割の表示】特願2000−611663(P2000−611663)の分割
【原出願日】平成12年4月7日(2000.4.7)
【出願人】(000001029)協和発酵キリン株式会社 (276)
【Fターム(参考)】
[ Back to top ]