
全身性エリテマトーデス(SLE)を治療するためのヒト化抗IL−10抗体
炎症促進性サイトカインレベルの許容できない上昇を伴うことなしに被験体に投与することができることを特徴とする、インターロイキン−10(IL−10)に結合可能なヒト化抗体、キメラ抗体又はこれらの断片を提供する。更に、前記抗体又はその断片の使用を含む治療方法を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、インターロイキン10(IL−10)及びIL−10に特異的な剤に関する。特に、本発明は、ヒト化IL−10抗体及びその使用に関する。更に、本発明は、全身性エリテマトーデス(SLE)を治療する方法に関する。
【背景技術】
【0002】
全身性エリテマトーデス(SLE)は、自己免疫疾患であると考えられており、この疾患においては、Bリンパ球の異常な高活性及び免疫グロブリンガンマ(IgG)自己抗体の異常な大量産生が重要な役割を果たしている。この病理過程では、Igでコーティングされた細胞が隔離及び破壊され、補体タンパク質が定着及び分割され、且つケモタキシン、血管作動性ペプチド、及び破壊酵素が組織に放出される(非特許文献1)。
【0003】
SLEは、多様な徴候を特徴とする。この疾患の過程においては、合計95%の患者が筋骨格系疾患を訴え、80%が皮膚病変、85%が血液疾患、60%が神経障害、60%が心肺疾患、30%〜50%が腎臓疾患、40%が胃腸疾患、15%が血栓症、及び15%が眼疾患を示す。また、患者の大部分(95%)が、疲労、倦怠感、発熱、食欲不振、及び体重減少等の全身性症状に苦しみ、これら症状は、殆ど常に発現している。大部分の患者では、寛解と突然再発する疾患期間とが交互に生じる。永続的に寛解する(治療をしなくても症状が出ない)ことは非常に稀である。50年以上前、SLEと診断された患者の大部分は、5年間未満しか生存できなかった。現在は、10年生存率が90%を超えており、これは、早期診断、対症療法的な抗炎症及び免疫抑制治療によるものである。最も一般的な死因は、免疫抑制の結果としての感染症である(非特許文献1)。
【0004】
SLEの治療には、通常、抗マラリア薬、抗炎症薬、及び免疫抑制薬が用いられている。症状の制御が困難になった場合には、非ステロイド性抗炎症薬にコルチコステロイドを加える。更に、多くの臓器に関連する活動性SLEは、シクロホスファミドを用いる積極的治療を必要とする。
【0005】
現在まで、SLEを治癒させる及び/又は長期間に亘って患者の生活の質を向上させるために利用可能な原因療法は存在していない。しかし、近年、抗体技術が進歩し、また、この自己免疫疾患の原因である因子が更に同定されたことにより、治療の選択肢としてモノクローナル抗体を使用する可能性が開かれつつある。特に、SLEを治療するための好ましいアプローチは、ポリクローナル自己抗体を大量に過剰産生させる病的免疫反応と相互作用するか又は補正する特異的な治療である。SLEの病因は、主にB細胞の調節不全に関連しているので、B細胞を標的とすることができるモノクローナル抗体が特に注目されている。非特許文献2に記載の通り、潜在的なB細胞表面抗原は、CD19、CD20、CD21、及びCD22を標的とする。更に、IL−10、IL−1ra、IL−12(非特許文献3)及びIL−6(非特許文献4)は、免疫反応の調節において重要なサイトカインであり、特に、SLE患者における突然の再発中に上昇する。二本鎖DNA(dsDNA)に対する自己抗体及びIL−10の血漿濃度は、SLE患者における疾患の活動性を反映していることが多い。IL−10濃度の上昇は、SLE患者における疾患の活動性と相関している(非特許文献5)。しかし、IL−10は、免疫系に対して多面的な作用を有するサイトカインであり、また、炎症促進反応の低減に関与していることが知られている。
【0006】
モノクローナル抗体を用いた臨床試験がSLE患者で実施されている。具体的には、幾つかの臨床試験では、非ホジキンリンパ腫を治療するために用いられるキメラマウス抗CD20モノクローナル抗体である抗体リタキシマブが用いられた。非特許文献2に記載の通り、これら臨床試験の結果は、SLE患者においてこの抗体が高い活性を有していることを示したので、オファツムマブ、IMMU−106、及びGA−101等のCD20を標的とする幾つかの新規抗体が開発された。SLEにおけるモノクローナル抗体の活性を報告している更なる臨床試験では、抗CD22抗体であるエプラツズマブ、抗TNFα抗体であるインフリキシマブ、抗IL−10抗体であるB−N10(非特許文献6)、抗CD40L抗体であるIDEC131及びBG9588、BLYS阻害剤であるベリムマブ、抗IL−6受容体抗体であるトクリムマブ、並びに抗C5抗体であるエクリズマブが用いられた。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Hahn BH.Systemic Lupus Erythematosus.In:Kasper DL,Braunwald E,Fauci AS,Hauser SL,Longo DL,Jameson,JL,editors.In:Harrison’s Principles of Internal Medicine(16th edition).New York(US):McGraw−Hill;2005.pp.1960−1967
【非特許文献2】Robak and Robak(Current Drug Targets,2009,No.10,pages26−37)
【非特許文献3】Capper et al.,Clin.Exp.Immunol.2004 Nov;138(2):348−56
【非特許文献4】Chun et al.,J.Clin.Immunol.2007 Sep;27(5):461−6
【非特許文献5】Park et al.,Clin.Exp.Rheumatol.1998 May−Jun;16(3):283−8
【非特許文献6】Llorente et al.,Arthritis Rheum.2000 Aug;43(8):1790−800
【発明の概要】
【発明が解決しようとする課題】
【0008】
本発明の目的は、更なる剤、特に、この領域において有用性を有する抗体を提供することにある。
【課題を解決するための手段】
【0009】
したがって、本発明は、炎症促進性サイトカインレベルの許容できない上昇を伴うことなしに被験体に投与することができる、インターロイキン10(IL−10)に結合可能なヒト化抗体、キメラ抗体又はこれらの断片を提供する。
【0010】
IL−10は、抗炎症性サイトカインの代表的な物質であるので、ブロック時には、サイトカイン量が著しく増加すると考えられる。マウスIL−10抗体であるB−N10の場合には、無刺激の細胞培養物(健常個体のインビボの状態を反映する)において、IL−6やTNFαなどの炎症促進性サイトカインの上昇が観察できる。しかしながら、本発明者らは驚くべきことに、本発明の抗体をインビトロで細胞に適用した場合及びこれをインビボで投与した場合に、サイトカイン放出量が遥かに低いことを見出した。サイトカイン放出量が低いことは、結果として、本発明の抗体が投与対象である個体に許容されるという点で有利である。
【図面の簡単な説明】
【0011】
単なる一例として、以下の図面を参照して本発明を例証する。
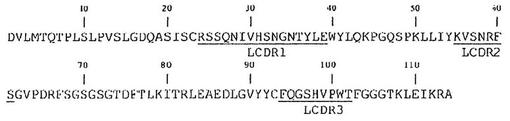
【図1A】図1Aは、マウスB−N10抗体の軽鎖可変領域のアミノ酸配列(配列番号2)を示す。超可変相補性決定領域(CDR)に下線を引く(LCDR1は、配列番号4であり、LCDR2は、配列番号5であり;LCDR3は、配列番号6である)。
【図1B】図1Bは、マウスB−N10抗体の重鎖可変領域のアミノ酸配列(配列番号3)を示す。超可変相補性決定領域(CDR)に下線を引く(HCDR1は、配列番号7であり、HCDR2は、配列番号8であり;HCDR3は、配列番号9である)。
【図2A】図2Aは、マウスB−N10抗体の軽鎖可変領域をコードするヌクレオチド配列(配列番号10)を示す。
【図2B】図2Bは、マウスB−N10抗体の重鎖可変領域をコードするヌクレオチド配列(配列番号11)を示す。
【図3A】図3Aは、マウスB−N10の軽鎖可変領域(配列番号12)、A17(配列番号14)、JK1(配列番号15)、及びマウスB−N10抗体のヒト化中に生じた可変領域hVL1〜hVL12(配列番号18〜29)のアミノ酸配列を示す。
【図3B】図3Bは、マウスB−N10の重鎖可変領域(配列番号13)、3−66+04(配列番号16)、JH4(配列番号17)、及びマウスB−N10抗体のヒト化中に生じた可変領域可変領域hVH1〜hVH12(配列番号30〜41)のアミノ酸配列を示す。
【図3C】図3Cは、マウスB−N10抗体のヒト化中に生じた可変領域hVH13〜hVH29(配列番号42〜58)のアミノ酸配列を示す。
【図4】図4は、hIL−10抗原ELISAを用いてキメラcB−N10抗体と比較した、ヒト化抗体変異体の抗原結合性を提供する。
【図5】図5は、精製抗体製剤を用いてキメラB−N10抗体と比較した、3つのヒト化変異体hVH20/hVL7、hVH20/hVL8、及びhVH26/hVL7の結合性の測定結果を提供する。
【図6A】図6Aは、B−N10とインキュベーションした後の健常ボランティア由来全血培養物におけるTNFαの放出レベルを、BT−063の場合と比較して示す(50μg/mL)。
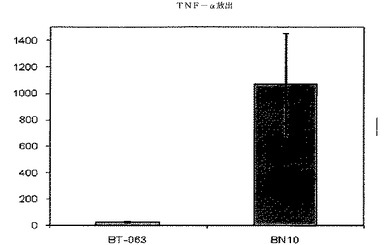
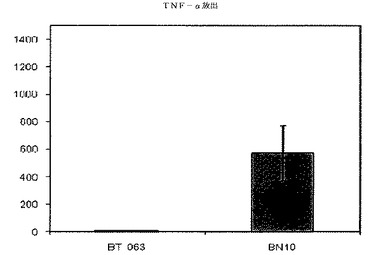
【図6B】図6Bは、B−N10とインキュベーションした後のSLE患者由来全血培養物におけるTNFαの放出レベルを、BT−063の場合と比較して示す(50μg/mL)。
【図7A】図7Aは、B−N10とインキュベーションした後の健常ボランティア由来全血培養物におけるIL−6の放出レベルを、BT−063の場合と比較して示す(50μg/mL)。
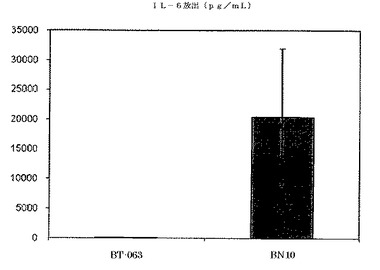
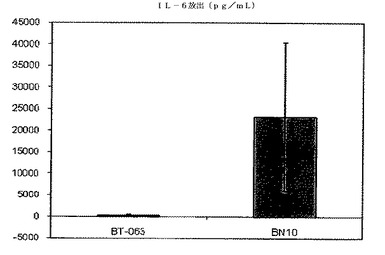
【図7B】図7Bは、B−N10とインキュベーションした後のSLE患者由来全血培養物におけるIL−6の放出レベルを、BT−063の場合と比較して示す(50μg/mL)。
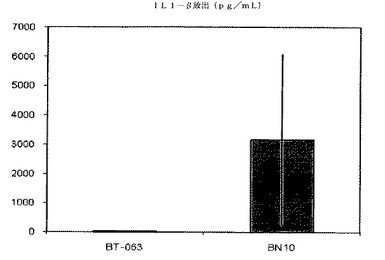
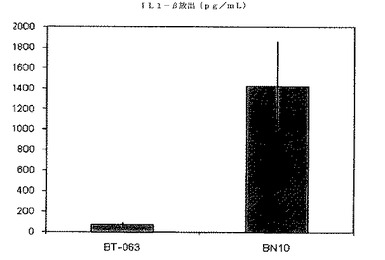
【図8A】図8Aは、B−N10とインキュベーションした後の健常ボランティア由来全血培養物におけるIL−1βの放出レベルを、BT−063の場合と比較して示す(50μg/mL)。
【図8B】図8Bは、B−N10とインキュベーションした後のSLE患者由来全血培養物におけるIL−1βの放出レベルを、BT−063の場合と比較して示す(50μg/mL)。
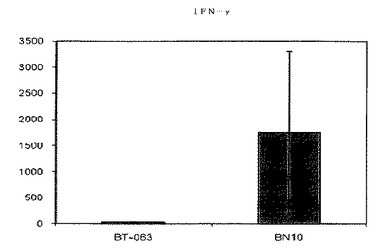
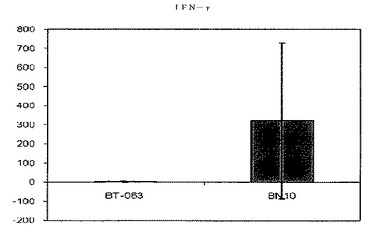
【図9A】図9Aは、B−N10とインキュベーションした後の健常ボランティア由来全血培養物におけるIFN−γの放出レベルを、BT−063の場合と比較して示す(50μg/mL)。
【図9B】図9Bは、B−N10とインキュベーションした後のSLE患者由来全血培養物におけるIFN−γの放出レベルを、BT−063の場合と比較して示す(50μg/mL)。
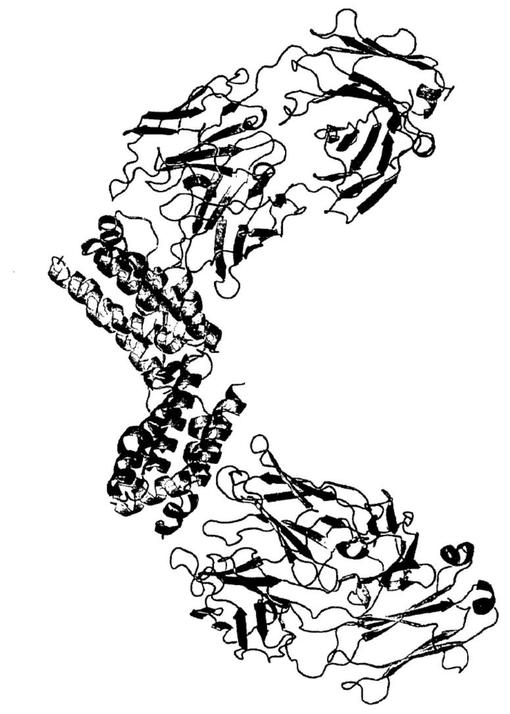
【図10】図10は、IL−10に結合しているBT−063のFab断片の全体構造を示す。IL−10及びFab断片をリボン図で示す。
【図11】図11は、IL−10受容体が結合する部位と同じIL−10の部位に結合するBT−063のFab断片を示す。IL−10、IL−10R1、及びFab断片をリボン図で示す。
【図12】図12は、健常ボランティアにBT−063を投与した後の、投薬量に対するサイトカインの血漿濃度の平均cmaxのグラフを示す。
【図13】図13は、健常ボランティアにBT−063をインビボ投与した後の、経時的なIL−10平均血漿濃度のグラフを示す。
【発明を実施するための形態】
【0012】
本発明は、インターロイキン−10(IL−10)に結合可能であるヒト化抗体、キメラ抗体又はこれらの断片、及び高レベル又は高活性のIL−10により媒介される病状の治療におけるこの抗体又はその断片の使用に関する。
【0013】
ヒトIL−10は、分子量37kDaのホモダイマーである。各モノマーは、160アミノ酸からなり、分子量は18.5kDaである。IL−10のダイマーは、IL−10R受容体α(IL−R又はIL−10R1)と相互作用し、次いで、IL−10受容体β(IL−10Rβ又はIL−10R2)を複合体に動員する。これら受容体は、様々な細胞、特に、単球、マクロファージ、Tリンパ球、及びBリンパ球等の大部分の造血細胞を含む免疫細胞(Asadullah et al.,Pharmacol.Rev.2003 Jun;55(2):241−69)で発現するが、表皮細胞又はケラチン産生細胞等の非造血細胞でも発現する。IL−10受容体αとIL−10との結合及びIL−10受容体βの動員により、Jak1及びTyk2チロシンキナーゼを介するシグナル伝達が導かれ、次いで、STATファミリーの転写因子が活性化される。ヘルパーT細胞、調節性T細胞、単球、マクロファージ、B細胞、好酸球、マスト細胞、ケラチン産生細胞、樹状細胞、更には癌細胞等、様々なIL−10の細胞源が公知である。B細胞におけるIL−10の機能は、アポトーシスの防止、増殖の強化、クラススイッチ事象、及びプラズマ細胞への分化(Asadullah et al.,Pharmacol.Rev.2003 Jun;55(2):241−69)などである。
【0014】
本発明は、炎症促進性サイトカインレベルの許容できない上昇を伴うことなしに被験体に投与することができる、インターロイキン10(IL−10)に結合可能なヒト化抗体、キメラ抗体又はこれらの断片を提供する。
【0015】
治療用抗体の投与は、被験体に望ましくない副作用を引き起こす、炎症促進性サイトカインレベルの許容できない上昇を引き起こし得ることが知られている。特に、この許容できない上昇は、皮膚の発赤を引き起こすことがあると共に、体温の上昇などの発熱又はインフルエンザ様症状をもたらすことがある。したがって、本発明の好ましい態様においては、前記抗体又は抗体断片は、2℃を超える体温の上昇を伴うことなしに被験体に投与することができるものである。
【0016】
更に、前記抗体又はその断片は、投与後の被験体の血漿中における炎症促進性サイトカイン量の実質的な上昇を引き起こさない。これらのサイトカインの許容できないレベルは、通常、サイトカインの正常値上限(ULN)よりも数倍高い。該ULNは、被験体コホートで測定されるサイトカインの平均レベルに2×標準偏差を加えた値として定義される。
【0017】
したがって、本発明の好ましい態様においては、前記抗体又はその断片は、炎症促進性サイトカインレベルが、正常値上限(ULN)の500%を超えて上昇することなしに、より好ましくは300%を超えて上昇することなしに被験体に投与することができる。換言すれば、前記抗体又はその断片は、炎症促進性サイトカインレベルを、当該炎症促進性サイトカインのULNの500%未満、より好ましくは300%未満しか上昇させない。前記炎症促進性サイトカインは、TNF−α、IFN−γ及びIL−1βのうちの少なくとも1つ、好ましくはこれらの全てであることが好ましい。或いは、前記炎症促進性サイトカインは、IL−6とIL−8のいずれでもない。
【0018】
更に、本発明の抗体又はその断片は、抗炎症反応を引き起こす、IL−1受容体アンタゴニスト(IL−1ra)を被験体において誘導することができることが特に好ましい。
【0019】
本発明の好ましい態様においては、前記ヒト化抗体又はその断片は、PBMC、より具体的には免疫細胞から、これらの細胞にインビトロで接触させたときに放出される炎症促進性サイトカイン、特にTNF−α、IL−1β、IL−6、のレベルに500%を超える上昇を引き起こさない。
【0020】
本発明者らは、驚くべきことに、本発明のヒト化抗体又はその断片が引き起こす炎症促進性サイトカインレベルのインビトロアッセイにおける上昇が、マウスB−N10抗体よりも小さいことを見出した。
【0021】
炎症促進性サイトカインの放出レベルは、単離した免疫細胞又はヒト全血培養物を用いるインビトロ研究によって測定することができ、例えば、以下の実施例5に記載の研究によって測定することができる。具体的には、前記測定方法は、(a)前記抗体又はその断片と共に細胞培養物をインキュベートする工程、及び(b)少なくとも1つの炎症促進性サイトカインのレベルを測定する工程を含む。
【0022】
ヒト全血培養物は、健常人又はSLEを罹患している患者などの疾患患者から採取することができる。前記末梢血単核細胞(PBMC)は、免疫細胞であってもよく、特にマクロファージ、単球、樹状細胞、ヘルパーT細胞、及びB細胞から選択することができる。
【0023】
前記少なくとも1つの炎症促進性サイトカインは、インターロイキン−1β(IL−1β)、IL−1α、IL−6、腫瘍壊死因子α(TNF−α)、ヘルパーTサイトカイン(インターフェロンγ、IFN−γ、IL−4)、マクロファージサイトカイン(IL−12)、ケモカイン(IL−8、MCP−1)のそれぞれから選択することができる。これらのサイトカインの放出レベルは、当該技術分野で一般に知られている方法を用い細胞培養物上清中で測定することができる。
【0024】
より具体的には、該インビトロ法は、以下の工程を含むことができる:
a)50μg/mLの抗体又は断片をヒト全血培養物と接触させる工程;
b)前記抗体又は断片を前記全血培養物と共に37℃で48時間インキュベートする工程;及び
c)前記培養物中の1以上の炎症促進性サイトカインの量を測定する工程。
【0025】
特に、前記方法は、前記抗体と接触させないヒト全血培養物と共に行う。このコントロールと比較して、本発明の抗体又は抗体断片は、サイトカインレベルを500%未満、より好ましくは300%未満しか上昇させないことが好ましい。この方法におけるサイトカインは、IL−6とIL−8のいずれでもないことが好ましい。
【0026】
サイトカインは、Multiplex Bead Immunoassays(96ウェルフィルタープレートで行う固相タンパク質アッセイ)を用いて培養物中で検出することができる。該方法は、特定のサイトカインに対する特定の抗体が結合していると共に所定の分光特性を示すビーズを用いる。これにより、ビーズを特異的に同定することができ、これらを既知の結合抗体に帰することができる。ビーズに結合したサイトカインは、検出抗体を該サイトカインに更に結合させることにより定量することができる。これは、GM−CSF、IL−1β、IL−2、IL−4、IL−5、IL−6、IL−8、IL−10、IFN−γ、及びTNF−αなどの最大10種のヒトサイトカインを同時に検出することを可能にする。湿ったウェルにサンプル、標準物質、及びビーズ溶液がピペットで注入されたフィルタープレートを前記目的のために用いる。インキュベーション後、液体を真空ポンプで吸引除去し、残ったビーズを洗浄し、ビオチン化検出抗体の混合物を添加する。液体を吸引除去し且つウェルに残っているビーズを洗浄することにより、結合していない検出抗体を除去し、次いでストレプトアビジンコンジュゲートR−フィコエリスリン(ストレプトアビジン−RPE)を添加する。これは、インキュベーション中にビオチン化検出抗体を認識し、再度洗浄した後に、形成された免疫複合体をBio−Plex200Systemで分析することができる。この免疫学的アッセイは、Bio−Plex200装置を用いて分析する。この装置は、2つのレーザーシステムを用いており、これらのうちの一方は、ビーズの分光特性に基づいてビーズを同定し、他方は、第2抗体を用いる検出により結合したサイトカインの量を測定する。結合量の定量は、既知量のサイトカインを含む標準物質を並行して用いることで達成される。
【0027】
更に、本発明は、インターロイキン10(IL−10)に結合可能なヒト化抗体、キメラ抗体又はこれらの断片であって、被験体に投与されたときにIL−10の血漿中のレベルを上昇させることができるヒト化抗体、キメラ抗体又はこれらの断片を提供する。
【0028】
以下の実施例8に示すように、本発明の抗体は、炎症促進性サイトカインレベルの許容できない上昇を伴うことなしに投与することができる一方で、投与により、血漿サンプル中に検出可能なIL−10の量が用量依存的に上昇する。中和抗体を適用したときにIL−10レベルが上昇するという知見は、予想外である。通常、サイトカイン中和剤を投与すると遊離サイトカインレベルが低下することが予想される(Strandら、Nature Reviews Drug Discovery、2007、Vol.6、pp.75−92)。何ら理論に拘束されることを望むものではないが、抗体がIL−10に結合することにより、IL−10のIL−10受容体への結合を阻害し、より多くのIL−10をB細胞に産生させるネガティブフィードバックループを誘導すると考えられる。にもかかわらず、このアップレギュレーションが本発明の抗体の治療有用性を阻害することはない。これは、実施例8にも示されるように、該抗体は、全てのIL−10を中和するのに十分高いレベルでも安全に投与されるからである。
【0029】
本願において、用語「キメラ抗体」は、重鎖及び/又は軽鎖の一部が、特定の種に由来するか、或いは特定の抗体クラス又はサブクラスに属する抗体の配列と同一であるが、一方、抗体の残りの部分は、異なる種、抗体クラス、又は抗体サブクラスに由来する抗体の配列と同一である抗体を指す。キメラ抗体のCDRはある起源に由来するが、抗体の残りの部分は異なる起源に由来することが特に好ましい。具体的には、本発明において、キメラ抗体は、非ヒト抗体の抗原結合配列/可変ドメインがヒト抗体のフレームワーク領域にグラフトされているヒト化抗体であってよい。
【0030】
本願において、用語「断片」は、望ましい生物活性を保持している抗体の断片又は誘導体を指す。断片は、一般的に、抗体の抗原結合領域、具体的には、Fab、Fab’、F(ab)’2、Fv、及びscFv断片、並びに多価抗体誘導体、具体的には、二重特異性抗体(diabody)又はタンデム二重特異性抗体を含む。断片は、少なくとも25アミノ酸が好ましく、50アミノ酸がより好ましく、200アミノ酸〜500アミノ酸が更により好ましい。或いは、断片は、30KDa〜150kDaのサイズを有する断片として定義することもできる。更に、抗体断片は、2以上のペプチド/ポリペプチド鎖を含んでいてもよい。例えば、各長さが200アミノ酸〜300アミノ酸である2本の鎖を含むFab断片、又は各長さが400アミノ酸〜500アミノ酸である2本の鎖を含むTandAbs(登録商標)(四価二重特異的抗体フォーマット)である。
【0031】
前記抗体又はその断片は、本明細書に記載されるマウスB−N10抗体又はヒト化BT−063(変異体hVH26/hVL7)抗体に由来することが、本発明の好ましい特徴である。具体的には、かかる抗体又はその断片は、B−N10/BT−063可変軽鎖のCDR1、CDR2、及びCDR3のアミノ酸配列と少なくとも80%同一である、及び/又はB−N10/BT−063可変重鎖のCDR1、CDR2、及びCDR3のアミノ酸配列と少なくとも80%同一であるアミノ酸配列を含むCDRを含む。マウスCDRのアミノ酸配列を図1に示す。BT−063のCDRのアミノ酸配列を実施例6に示す。前記配列は、B−N10/BT−063抗体のCDRの配列と少なくとも90%又は少なくとも95%同一であることがより好ましい。本明細書中の実施例6に記載するX線結晶構造解析は、CDR内のどの残基がIL−10との結合に重要であるかを示す。
【0032】
或いは、本発明の抗体又は断片は、B−N10/BT−063抗体に由来するものではあるが、抗体又はその断片の親和性及び/又は特異性を実質的に変化させることのない変異を配列中に任意で含む、B−N10/BT−063可変軽鎖のCDR1、CDR2、及びCDR3のアミノ酸配列、及び/又はB−N10/BT−063可変重鎖のCDR1、CDR2、及びCDR3のアミノ酸配列を含んでいてもよい。具体的には、マウスB−N10抗体又はBT−063(変異体hVH26/hVL7)抗体のCDRを含む抗体又は断片と比べて、前記配列中の変異は、IL−10に対する抗体又は断片の親和性又は特異性を低下させない。
【0033】
特定の実施形態において、本発明は、B−N10/BT−063の可変軽鎖及び/又は可変重鎖のCDR1、CDR2、及びCDR3のアミノ酸配列を含むヒト化抗体、キメラ抗体又はこれらの断片を提供する。より好ましくは、本発明は、図1に示すマウス抗体B−N10の可変ドメインのアミノ酸配列を含むヒト化抗体、キメラ抗体又はこれらの断片を提供する。最も好ましくは、前記抗体又は断片は、BT−063の可変ドメインのアミノ酸配列(配列番号69及び70)のうちのいずれか一方又は両方を含む。
【0034】
一般的に、本発明の抗体は、ヒト定常領域(Fc)を更に含む。これは、IgM、IgG、IgD、IgA、及びIgEを含む免疫グロブリンの任意のクラス、並びにIgG1、IgG2、IgG3、及びIgG4を含む任意のアイソタイプに由来する定常ドメインの中から選択することができる。好ましい定常領域は、IgG、特にIgG1の定常ドメインの中から選択される。
【0035】
他の生成物
本発明は、更に、上記抗体又は抗体断片をコードする核酸配列を提供する。前記核酸配列は、DNAであってもRNAであってもよいが、DNAが好ましい。前記配列は、発現カセット又はベクター内で用いることができ、本明細書に開示される抗体及びその断片の産生において特に用いられる。
【0036】
本発明は、更に、これらポリヌクレオチド、発現カセット又はベクターで形質転換されたホスト細胞を提供する。好適なホスト細胞は、原核細胞であっても真核細胞であってもよい。
【0037】
或いは、ホスト細胞は、本発明の抗体を産生する細胞と骨髄腫細胞とを融合させることにより得られるハイブリドーマであってもよい。
【0038】
上記ホスト細胞は、抗体又はその断片を産生するための方法で利用することができる。具体的には、かかる方法は、抗体又はその断片を発現させる条件下で好適な培養培地中にてホスト細胞を培養する工程と、培養培地から前記抗体又はその断片を分離する工程とを含んでいてもよい。この種の方法は、当技術分野において公知であり、報告されている。
【0039】
医療的用途
本明細書に記載される抗体又はその断片は、高レベル又は高活性のIL−10により媒介される疾患又は病状の治療において有用性を有する。結果として、被験体における病状を治療又は予防するための方法であって、前記病状が高レベル又は高活性のIL−10により媒介され、治療上有効な量の本明細書に記載される抗体又はその断片を投与することを含む方法を提供する。
【0040】
具体的には、高レベル又は高活性のIL−10により媒介される病状は、SLEである。したがって、また、本発明は、SLEの治療において使用するための本明細書に記載される抗体又はその断片を提供する。
【0041】
更なる例は、血小板減少性紫斑病、ループス腎炎、HIV、hCMV、及びC型肝炎である。別の例は、免疫反応の増殖又は抑制を直接支持することによりIL−10に依存している腫瘍細胞を治療することである。
【0042】
本発明の更なる実施形態は、薬学的に許容し得る担体又は希釈剤と共に、上記抗体又はその断片を含む医薬組成物である。1つの実施形態では、前記組成物は、抗体又はその断片がカップリングしているリポソームを含む。
【0043】
かかる組成物は、患者に対して、非経口的に、静脈内に、又は皮下に投与することができる。SLEの治療において、前記抗体又はその断片は、静脈内又は皮下に投与されることが好ましい。
【0044】
前記抗体及びその断片は、炎症促進性サイトカインレベルを大幅に上昇させることがなく、患者にとって許容できない「サイトカインストーム」を防ぐためのコルチコステロイドの併用投与の必要がないので、疾患の治療に特に有用である。
【0045】
したがって、特定の態様においては、本発明は、被験体における、高レベル又は高活性のIL−10により媒介される病状を治療又は予防する方法であって、治療上有効な量の本明細書に記載の抗体又はその断片を前記被験体に投与する工程を含み、前記被験体は、コルチコステロイドによる治療を同時にも別々にも受けていない(即ち、患者は、コルチコステロイドを併用していない)を提供する。この好ましい態様の実施形態においては、高レベル又は高活性のIL−10により媒介される病状は、SLEである。
【0046】
多くの患者において、SLEの治療にコルチコステロイドが併用される。しかしながら、本発明のこの態様は、コルチコステロイドの投与が望ましくない患者の治療において特に有用である。
【0047】
非医療的用途
更に、本明細書に記載される抗体又はその断片と標識とを含む、標識されているヒト化抗体、キメラ抗体又はこれらの断片を提供する。前記標識は、サンプル中の抗体の存在を検出するために当技術分野において公知である任意の好適な種類であってよい。具体的には、前記標識は、蛍光標識であってよい。
【0048】
標識されている抗体又は標識されていない抗体、具体的には、標識されている抗体は、サンプル中に存在するIL−10の存在を検出するためのインビトロ方法において特定の有用性を有する。前記方法は、標識されていない抗体若しくは標識されている抗体又はこれらの断片とサンプルとを接触させる工程と、前記サンプルを洗浄して、前記サンプルに結合していない抗体及びその断片(即ち、非結合抗体又は抗体断片)を除去する工程と、例えば標識を介して、前記サンプル中の前記抗体(又は断片)の存在を検出する工程とを含む。
【0049】
或いは、サンプル中のIL−10を中和するためのインビトロ方法に、標識されていない抗体又は断片を使用してもよい。かかる方法は、抗体又はその断片をIL−10に結合させるために、前記抗体又は断片とサンプルとを接触させる工程を含む。
【実施例】
【0050】
次に、以下の特定の実施形態に関連して本発明を更に説明する。
【0051】
実施例1−マウス抗IL10抗体B−N10の特性評価
1.1 B−N10抗体の可変ドメインをコードするDNAの単離
マウスBN−10の可変配列を同定するために、細胞ペレットを用いた。−80℃で保存しておいたサンプル(3×B−N10、3代継代、1×107細胞)の細胞からmRNAを単離し、cDNAを合成した後、B−N10の可変配列をPCRにより増幅させ、次いで、クローニングした。
【0052】
合計14クローンの配列を決定し(SEQ Laboratories,Goettingen)、可変軽鎖及び可変重鎖について分析した。B−N10の可変配列を明確に決定した。N−末端のプライマー領域でのみ差が生じた(表1を参照されたい)。可変重鎖については、プライマー領域における配列変異QVQLKQ(配列番号59)が9クローンで生じていたが、他の変異は1クローン又は2クローンでしか生じていなかった。サブクローニングするためにこの変異体を選択した。可変軽鎖については、2つの変異体が同比率で存在していた。マウス生殖細胞系配列と比較したところ、3つの突然変異しか有しないcr1配列が、同定されたVL配列と高い相同性を示した。これは、DVLMTQ(配列番号60)配列が正確な配列である可能性が非常に高いことを意味する。配列DIVMTQ(配列番号61)は、典型的な別のクラスの生殖細胞系配列であるので、除外した。
【表1】

【0053】
可変軽鎖VL及び可変重鎖VHのタンパク質配列を、それぞれ図1A及び1Bに示す。超可変相補性決定領域(CDR)に下線を引く。対応するDNA配列を、それぞれ図2A及び2Bに示す。
【0054】
実施例2−キメラB−N10抗体の作製
組換え抗体を発現させるために、実施例1で同定された抗体の重鎖及び軽鎖の可変配列をベクター系にクローニングした。第1の工程として、前記配列をBSリーダーにクローニングし、N末端に分泌シグナルを付加し、C末端にスプライスドナー配列を付加した。第2の工程として、それぞれ定常ヒトカッパ鎖及び定常ヒトガンマ−1鎖を含有する発現ベクターにこれら配列をクローニングした。軽鎖用ベクター及び重鎖用ベクターを調製し、次いで、リン酸カルシウム沈殿又はリポフェクションによってCOS−7細胞に一過的にコトランスフェクトした。2日間後、細胞培養上清を回収した。COS−7細胞中でキメラB−N10を発現させ、上清中の抗体の力価を検出した(サンドイッチELISA)後、ヒトインターロイキン−10(R&D Systems、カタログ番号217−IL/CF、ロット番号ET114021、−20℃で保存)に対する結合能をELISAで試験した。
【0055】
サンドイッチELISAの場合、マウス抗ヒトカッパ鎖抗体(Becton Dickinson)をキャッチャー抗体としてプレート表面に結合させ、次いで、細胞培養上清と共にインキュベートし、PODと複合体化しているウサギ抗ヒトIgG(H+L)抗体(Dianova)を用いてキメラ抗体の存在を検出した。規定の濃度(0.125μg/mL〜12μg/mL)のキメラ対照抗体をポジティブコントロールとして用いた。
【0056】
抗原ELISAの場合、0.5μg/mL〜5μg/mLの濃度でプレート表面にヒトIL−10を結合させた。細胞培養上清(未希釈及び1:5希釈)と共にインキュベートした後、PODと複合体化しているウサギ抗ヒトIgG(H+L)抗体(Dianova)を用いてキメラB−N10の結合を検出した。マウスB−N10をポジティブコントロールとして用いた。0.5μg/mL〜5μg/mLの濃度の抗体を用い、PODと複合体化しているウサギ抗マウスIgG/IgM抗体(Dako)で結合を検出した。
【0057】
ELISAの結果については、実施例3で論じる。
【0058】
実施例3−抗IL−10抗体のヒト化
げっ歯類抗体のヒトにおける免疫原性を低下させようとする最初の試みは、げっ歯類抗体の定常ドメインをヒト抗体の定常ドメインに置き換えることによりキメラ抗体を作製することであった。依然として可変ドメイン内のげっ歯類のフレームワーク領域が免疫反応を誘導する場合があったので、より進歩したCDRのグラフト方法、即ち、抗原結合配列(相補性決定領域、CDR)を完全にヒト抗体フレームワークに転移させる(ヒト化)方法が開発された。通常、マウスドナー抗体に最も類似するヒトアクセプターフレームワークを選択して、ヒト化プロセス中にオリジナル抗原の特異性及び親和性が回復される可能性を高める。ヒト抗体生殖細胞系配列、発現抗体のコンセンサス配列、CDRループ構造の分析、及び抗体/抗原複合体のX線構造を用いる異なるアプローチを用いてもよく、これらと組み合わせて前記プロセスを改善してもよい。通常、幾つかのヒト化抗体変異体は、この方法で作製され、後に、互いに異なる及びオリジナル抗体と異なる可能性のある生物学的作用に関して分析される。最後に、抗体の望ましい機能に従って、好適なヒト定常領域を選択することができる。
【0059】
3.1 B−N10のマウス可変配列とヒト配列との配列比較、並びにヒト化VL(hVL)及びVH(hVH)の配列のセットの設計
マウス抗IL−10抗体であるB−N10を選択した(Llorente et al.,Eur.Cytokine Netw.1993 Nov−Dec;4(6):421−7;及びLlorente et al.,Arthritis Rheum.2000 Aug;43(8):1790−80)。ヒト化抗体を得るための方法は、相補性決定領域(CDR)をヒトアクセプター領域に融合させるCDRグラフト手順に基づく。
【0060】
以下の3つのデータセットの複合分析に基づいて、ヒトアクセプターフレームワークを選択した:
1. 体細胞突然変異のリスクを最小化するための、マウス配列とヒト生殖細胞系配列との相同性、
2. 異常なアミノ酸残基を同定するための、マウス配列とヒトコンセンサス配列との比較、及び
3. 重要な構造フレームワークアミノ酸残基についての情報を得るための、CDR配列の基準となる構造クラスの同定。
【0061】
B−N10のマウス可変軽鎖は、ヒト生殖細胞系可変セグメント2−30*01(A17(配列番号14))及び接合セグメントJK1(配列番号15)に対して最も高い相同性を示す。B−N10に対して最も高い相同性を有するヒトコンセンサス配列は、HuKIIである。可変軽鎖の相補性決定領域(CDR)は、L1の場合クラス4に、L2及びL3の場合クラス1に分類することができた。重要なアミノ酸残基を同定した。
【0062】
クラス4−1−1の基準構造を有するマウスCDRとヒト生殖細胞系VL遺伝子との配列比較により、2−30*01と最も高い相同性を有する(一致しないアミノ酸数が最も少ない)ことが明らかになった。
【0063】
B−N10のマウス可変重鎖は、ヒト生殖細胞系可変セグメントVH3−33及び接合セグメントJH4(配列番号17)に対して最も高い相同性を示す。B−N10に対して最も高い相同性を有するヒトコンセンサス配列は、HuHIIIである。可変重鎖の相補性決定領域(CDR)は、H1及びH2の場合クラス1に分類することができた。重要なアミノ酸残基を同定した。クラス1−1の基準構造を有するマウスCDRとヒト生殖細胞系VH遺伝子との配列比較により、3−66*04(配列番号16)と最も高い相同性を有する(一致しないアミノ酸数が最も少ない)ことが明らかになった。したがって、生殖細胞系配列VH3−66を考慮した。
【0064】
ヒト化可変軽鎖(12変異体)と可変重鎖(29変異体)との様々な可変配列のセットを設計するために、得られた全てのデータを検討した。
【0065】
3.2 ヒト化hIL−10結合抗体断片の小さなライブラリの構築及び選択
hIL−10に結合する可能性のある抗体断片のライブラリを作製して最適なヒトIL−10結合抗体を得るために、真核細胞のコドン使用頻度を考慮した上で、図3に示す12個のhVL断片及び29個のhVH断片をコードするcDNA配列を作製した。
【0066】
次いで、得られたcDNAをクローニングベクターにクローニングし、SEQ Laboratories(Goettingen,Germany)において配列を決定した。hVL断片をコードする12個の各cDNAとhVH断片をコードする29個のcDNAとを組み合わせて、発現する可能性のある348個の抗体断片を得るという方法でライブラリを構築した。
【0067】
細菌で発現させ、ヒトIL−10(R&D Systems、カタログ番号217−IL/CF)に対して2ラウンドの選択を行った後、hIL−10(同様に選択した)に対する結合についてELISAにより前記抗体断片を分析した。簡潔に述べると、4℃で一晩、PBS中1μg/mLのhIL−10でMaxisorbプレート(Nunc,Germany)をコーティングした。前記プレートをブロッキング及び洗浄した後、抗体断片を産生する細菌の上清を添加した。結合しているヒト化抗体断片を検出するために、PODと複合体化している二次抗体を用いた。
【0068】
優れた結合剤のコード配列を分析し、同定されたhVL断片及びhVH断片の発生について列記した(表2)。
【表2】
【0069】
太字にした配列は、抗体全体について結合特性を分析するために、適切な真核発現ベクターにサブクローニングするために選択した配列である。括弧内に示す配列は、発現ベクターにサブクローニングするために選択したが、欠陥コンストラクトしか得られなかった配列である。
【0070】
3.3 選択したBT−063のヒト化軽鎖及び重鎖の変異体用の発現ベクターの作製
スクリーニングアプローチにより求められた統計値に基づいて、ベクター系にクローニングするためのBT−063のヒト化VL及びヒト化VHの変異体のセットを選択した。第1の工程では、分泌シグナル5’及びスプライスドナー配列3’をコードしている配列とクローニングしたcDNAとを融合させるために、ヒト化VL及びVHの変異体をコードするcDNAを適切なベクターに転移させた。第2の及び最後のサブクローニング工程で、それぞれ、ヒト定常カッパ鎖及びヒト定常ガンマ−1鎖をコードする発現ベクターにこれらcDNAコンストラクトを転移させた。内毒素を含まないQiagen Midi−prepキット(Qiagen,Germany)によって、独立に得られたhVL及びhVHを含有する発現ベクターのプラスミドを調製した。
【0071】
3.4 COS−7細胞における選択されたヒト化BT−063変異体の一過的発現及びhIL−10に対する抗体結合の比較
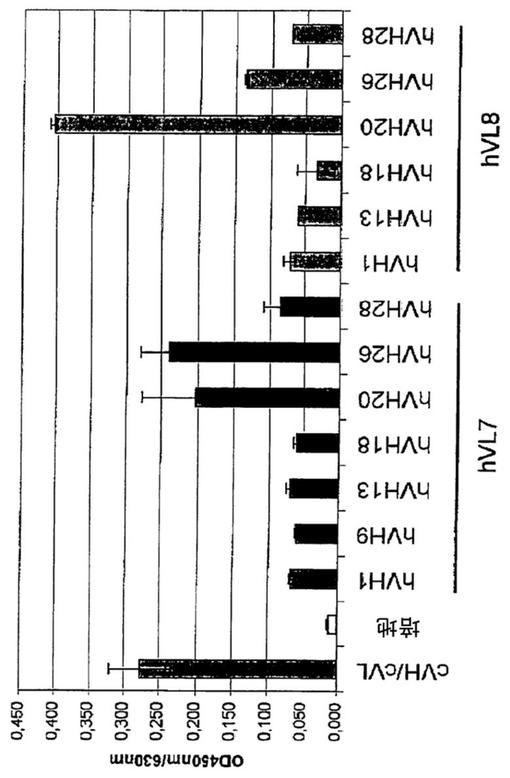
COS−7細胞におけるヒト化抗体変異体を一過的に発現させるために、選択した各ヒト化VL変異体(hVL7及びhVL8)と、選択した各ヒト化VH変異体(hVH1、hVH9、hVH13、hVH18、hVH20、hVH26、hVH28)とを組み合わせて、14個の異なるヒト化抗体を得た。
【0072】
簡潔に述べると、24ウェルフォーマットにおいて10%FCSを含有するDMEM中で、リン酸カルシウム沈殿により、軽鎖及び重鎖をコードする発現ベクターをCOS−7細胞に一過的にコトランスフェクトした。トランスフェクション後、培地を無血清培地CHO−S−SFM II(Invitrogen,Germany)に置換し、トランスフェクションの2日間後〜3日間後にCOS−7細胞の上清を回収した。トランスフェクトされたCOS−7細胞の上清に分泌されたヒト化抗体の抗体力価をサンドイッチELISAにより分析した。求められた抗体濃度に基づいて、全てのサンプルの上清を同じ抗体濃度に調整した。PBS中2μg/mLのhIL−10でコーティングされたMaxisorbプレート(Nunc,Germany)を用いる抗原ELISAにより、全てのサンプルを用いてヒトIL−10に対する結合を分析した。
【0073】
図4に示す通り、分析した変異体は全てhIL−10に結合するが、結合性は異なっている。有意に、抗原ELISAにおいて最も高いシグナルは、キメラB−N10抗体で得られた強度に匹敵するシグナル強度を示すBT−063変異体hVH20/hVL7、hVH26/hVL7、及びhVH20/hVL8で得られた。これら3つの抗体内のシグナル強度の差(hVH20/hVL8のシグナルがより強く、hVH20/hVL7及びhVH26/hVL7のシグナルがより弱い)は、定量サンドイッチELISA(上記を参照されたい)の結果、抗体濃度の差に起因していた可能性がある。調べた他の全ての変異体からは、キメラB−N10抗体と比べてかなり弱いシグナルが得られた。
【0074】
3.5 キメラ及びヒト化抗体の変異体の産生及びアフィニティー精製
選択されたヒト化BT−063変異体(hVH20/hVL7、hVH20/hVL8、hVH26/hVL7)及びキメラcB−N10(実施例2で論じた)をCOS−7細胞で産生させた。
【0075】
10cmの組織プレートを用いて、セクション3.4に記載の通り一過的に発現させた。トランスフェクションの5日間後、各変異体から約0.5Lの無血清上清を回収した。
【0076】
前記無血清上清からプロテインAアフィニティークロマトグラフィーによって抗体を精製した。2MのNaClの存在下で上清をロードした。0.1Mのクエン酸バッファ(pH4.0)により抗体を溶出し、2Mのリン酸バッファ(pH7.2)を含有するチューブに分画した。PBSでバッファ交換し、30kDaでカットオフする膜を用いて遠心分離することにより個々の抗体のプローブを濃縮した。抗原ELISA、非還元条件及び還元条件下におけるSDS−PAGE、並びに260nm及び280nmにおけるUV測定により、精製された物質の品質をチェックした。
【0077】
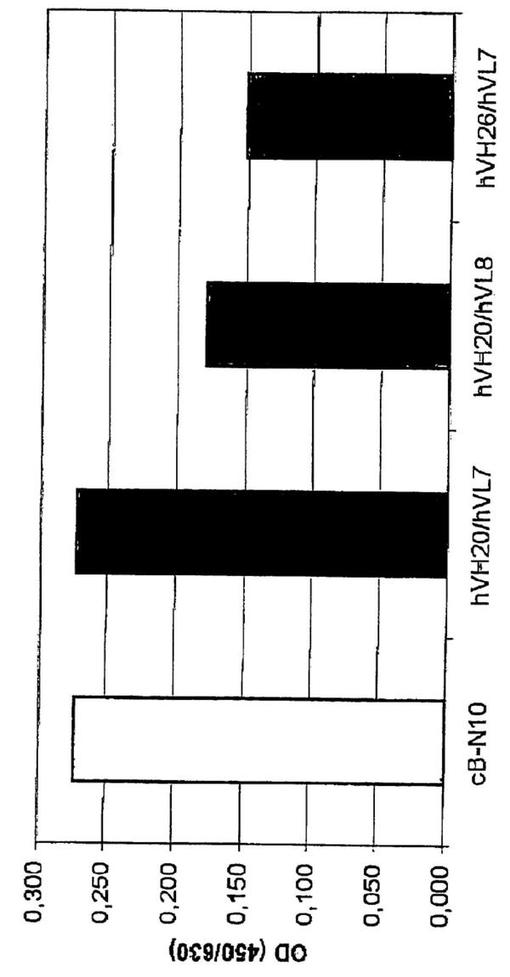
精製したキメラB−N10及びヒト化変異体のhIL−10に対する結合を、実施例2に記載した方法に従ってELISAにより試験した。hIL10をコーティングし、変異体cB−N10、BT−063−1(hVH20/hVL7)、BT−063−2(hVH20/hVL8)、及びBT−063−3(hVH26/hVL7)について抗体の結合を測定した。結果を図5に示す。
【0078】
キメラB−N10及びhVH20/hVL7変異体のシグナル強度は同程度であったが、変異体hVH20/hVL8及びhVH26/hVL7のシグナル強度は僅かに低かった。
【0079】
3.6 BiacoreヒトIL−10による親和性の測定
BIACORE2000(Biacore AB,Uppsala,Sweden)を用いて、hIL−10に対する様々な抗体(マウス、キメラ、3つのヒト化変異体)の結合についての会合速度定数及び解離速度定数を測定するために表面プラズモン共鳴分析を用いた。製造業者の条件に従って、hIL−10をCM−5センサチップに固定化した。5μL/分の流速で20μg/mLのアリコート50μLを添加することによりhIL−10を固定化して、固定化密度320RUにした。50μL/分の流速で0.1Mの炭酸バッファ(pH9.2)及び0.01MのHCL/1MのNaClを各1分間用いることにより2段階サイクルで、固定化hIL−10表面を再生した。20μg/mL〜0.15μg/mLの抗体濃度範囲で少なくとも4回各抗体サンプルを分析した。BIA evaluationバージョン3(1999)ソフトウェアを用いることにより、センサグラムからの計算を実施した。
【0080】
表3に、全てのBiacore測定の結果を要約する。全ての変異体が同程度hIL−10に結合する。しかし、僅かな差が検出可能である。結果として、マウスモノクローナル抗体B−N10、キメラcB−N10、及びヒト化変異体BT−063−1(hVH20/hVL7)は、同程度の親和性で結合するが、2つの他のヒト化変異体BT−063−2(hVH20/hVL8)及びBT−063−3(hVH26/hVL7)は、(マウスB−N10に比べて約3倍)低い親和性を示す。会合速度及び解離速度の僅かな差も検出可能である。
【表3】
【0081】
カニクイザルのIL−10
Biacore T100(Biacore AB,Uppsala,Sweden)を用いた更なる表面プラズモン共鳴実験により、BT−63変異体3(hVH26/hVL7)のカニクイザルIL−10に対する親和性を分析した。
【0082】
BT−063を10mMの酢酸(pH5.5)で5μg/mLに希釈し、アミンカップリング手順を用いて固定化して、最終レベルを約1,000RUにした。10mMのグリシン−HCl(pH1.8)を30秒間注入して、センサチップ表面を再生した。フローセル及びレファレンスセルに異なる濃度のサンプルを注入した。検出器フローセルから得られたシグナルからレファレンスセルから得られたシグナルを減じ、1:1Langmuir結合モデルを用いて、得られた結合プロファイルを評価した。濃度依存性の結合プロファイルが得られ、カニクイザルのIL−10についての平均KDは194pMであると計算された。ポジティブコントロールとして、rhIL−10を分析したところ、KDは4.6nMであった。結果を表4に要約する。
【表4】
【0083】
実施例4−インビトロにおける抗IL−10抗体の活性
BT−063の能力を確認するために、末梢血単核細胞(PBMC)におけるIL−6放出の遮断について調べた。PBMCは、リポ多糖類(LPS)で刺激されるとインターロイキン−6(IL−6)を放出する。インターロイキン−10(IL−10)の生理学的活性は、サイトカイン、例えば、IL−6の分泌阻害である。したがって、LPSで刺激された細胞にIL−10を添加すると、IL−6の分泌が阻害され、細胞培養培地中に存在するIL−6が著しく低下する。しかし、細胞培養物にBT−063を添加するとIL−10と結合するので、IL−10が細胞表面上の受容体に結合することができなくなる。IL−10の阻害作用は補償され、IL−6分泌は回復し、培地中にIL−6が放出される。
【0084】
Ficoll勾配によりヒト血液からPBMCを単離した。単離した細胞を1×106細胞/mLで播種し、IL−6を分泌させるためにLPSで刺激したが、IL−10の添加により阻害された。BT−063の添加によりIL−10の阻害作用が中和されたので、IL−6分泌が回復した。添加したBT−063の目的(レファレンス、又は低品質若しくは高品質の対照サンプル)によって、異なる力価濃度のBT−063を用い、濃度依依存的にIL−6を分泌させた。このIL−6の分布は、細胞培養の上清において検出した。
【表5】
【0085】
表5に示す通り、分泌されたIL−6の量は、BT−063の濃度と直接相関している。BT−063の濃度が高くなるほど、より多くの量のIL−6がPBMCから分泌され、上清中に存在する。ポジティブコントロール(IL−10を含まない、刺激されたPBMCをインキュベートしたもの)と比べて、40μg/mLのBT−063と共に細胞をインキュベートした場合はIL−6分泌が約73%回復するが、一方、0.988μg/mLのBT−063と共にインキュベートしても(用量設定の下限)、培地中で検出可能であったIL−6濃度は僅か17.5%であった。
【0086】
実施例5−ヒト全血培養物におけるサイトカイン合成/レベルに対するB−N10及びBT−063の異なる作用
BT−063(変異体hVH26/hVL7)の免疫薬理学的プロファイルを、ヒト全血培養物において実験的に誘導したサイトカイン合成の薬物依存的モジュレーションにより評価した。この方法を用いて、免疫細胞の活性に対するBT−063の直接的及び間接的影響を求めることができる。BT−063活性は、B−N10の作用と比較した。
【0087】
前記アッセイでは、健常ボランティアとSLE患者の両方に由来する全血培養物を用い、これらをBT−063又はB−N10存在下におけるサイトカイン放出量について分析した。全血培養物中の免疫細胞によるサイトカイン放出は、B−N10抗体又はBT−063抗体それぞれとのインキュベーション後の静止状態で試験した。健常ボランティアに由来する白血球と全身性エリテマトーデス(SLE)の罹患患者に由来する細胞を含む。
【0088】
以下に記載する実験に用いた細胞は、ボランティア又はSLE患者を出血させることで採取し、BT−063又はB−N10それぞれとのインキュベーションは、マイクロ培養プレートで2日間行った。
【0089】
これらの実験で測定した主なメディエータは、Th−1−、Th−2−、又は単球/マクロファージ−活性化などに関連するサイトカイン及びケモカインであり、例えば、インターフェロンγ(IFN−γ)、インターロイキン−1β(IL−1β)、IL−12、IL−4、IL−8、腫瘍壊死因子α(TNF−α)などである。培養物上清中の各測定パラメータの濃度は、マルチパラメトリックなビーズを利用した読み出しシステム(マルチアナライトプロファイル(MAP)試験と呼ばれるLuminex(登録商標)に基づく技術、Rules−Based Medicine、RBM(オースティン、テキサス州、USA))。これらのアッセイは、EDI GmbH(ロイトリンゲン、ドイツ連邦共和国)によって行われた。Luminex(登録商標)技術は、ELISAとフローサイトメトリーの組合せに類似した技術であり、アナライト1個当たり100個のビーズをカウントする。したがって、各濃度は、100個の各測定値の平均蛍光強度から逆算される。
【0090】
50μg/mLの抗体濃度でのみ、サイトカイン放出の誘導が観察された。
【0091】
B−N10又はBT−063とインキュベートした健常ドナー及びSLE患者由来の細胞培養物で測定されたサイトカインレベルの絶対値を次の表6及び7にまとめる。
【表6】
【表7】
【0092】
図6A、6B、7A、7B、8A、8B、9A及び9Bは、健常ボランティアとSLE患者の双方に由来する全血培養物において無刺激条件下で炎症促進性サイトカインの制御が異なることを示す。これらの図は、健常ボランティアとSLE患者由来の全血培養物は、BT−063(50μg/mL)と比較してB−N10とインキュベートした後にTNFα、IL−6、IL−1β、及びIFN−γの放出量がより多いことを示している。IL−6は、炎症促進機能に加えて、B細胞のプラズマ細胞への分化のための因子としても働くことは注目に値する。
【0093】
注目すべきことに、これらの結果から、IL−10及び炎症促進性サイトカインのB−N10によるアップレギュレーションの程度が、BT−063とインキュベートされた健常ドナー及びSLEドナーの双方に由来する培養物の場合よりも高く、より安全なプロファイルを示唆していることが分かる。このことは、ヒト化の手順の結果として予想されるものではない。
【0094】
実施例6−X線結晶構造解析
6.1 ヒトIL−10との複合体におけるBT−063 Fabの結晶化
公開されている構造データに従って幾つかのIL−10コンストラクトを設計し(Zdanov et al.,Structure,Vol.3,1995,pp.591)、標準的な手順により大腸菌で異種発現させるためのベクターにクローニングした。クローニングしたコンストラクトを標準的なプロトコールに従って試験的に発現させたところ、約18kDaの予測範囲でバンドが増加したことにより示される通り、IL−10の高発現を示した。
【0095】
後でタンパク質を精製するために、最適な条件下で発現したIL−10タンパク質を様々な量得た。再折り畳みの後、固定化アフィニティークロマトグラフィー、サイズ排除クロマトグラフィー、及びイオン交換クロマトグラフィーによりタンパク質を精製して、クマシー染色したSDS−PAGEにより判定したところ95%を超える均質性を有するタンパク質が得られた。精製タンパク質の収量は、発現培養物1リットル当たり約0.3mgであり、これは結晶化試験に十分であった。
【0096】
BT−063(変異体hVH26/hVL7)のFab断片を、プロテアーゼパパインを用いてインタクトな抗体から切断し、プロテインAにより精製した。次いで、サイズ排除クロマトグラフィーによりFab断片を更に精製した。
【0097】
精製タンパク質をモル過剰のIL−10と混合することによりIL−10:BT−063 Fab複合体を形成し、サイズ排除クロマトグラフィーにより更に精製した。保持体積は、複合体のサイズと一致していた。次いで、結晶化に好適な濃度にタンパク質を濃縮した。
【0098】
IL−10:BT−063 Fab複合体の結晶を共結晶化法により調製した。
【0099】
6.2 データ収集及び処理
結晶を急速冷凍させ、100Kの温度で測定した。低温条件を用いてSWISS LIGHT SOURCE(SLS,Villigen,Switzerland)でBT−063のFab断片と共にIL−10の共結晶からX線回折データを収集した。
【0100】
結晶は、非対称単位における2つの複合体と共に空間群P6に属する。プログラムXDS及びXSCALEを用いてデータを処理した。データ収集統計値を表8に要約する。
【表8】
【0101】
6.3 構造のモデリング及び構造の精密化
分子置換により、構造を決定及び分析するのに必要な位相情報を得た。IL−10及びFab断片の公開モデルをサーチモデルとして用いた。次いで、ソフトウェアパッケージCCP4及びCOOTを用いて標準的なプロトコールに従って、モデルの構築及び構造の精密化を実施した。最終モデルの正確さを交差検定するための尺度であるフリーR因子を計算するために、構造の精密化手順から測定された反射の4.2%を除外した(表9を参照されたい)。
【0102】
プログラムCHEMSKETCHを用いてナノボディのパラメータ化を実施した。対応するライブラリファイルを作製するためにLIBCHECK(CCP4)を用いた。
【0103】
3.0σで一致するFo−Fcマップのピークに水分子を置くことによりCOOTの「Find waters…」アルゴリズムを用いて水モデルを構築し、次いで、REFMAC5で構造を精密化し、COOTの検証ツールを用いて全ての水をチェックした。疑わしい水のリストの基準は、以下の通りであった:B因子:80超、2Fo−Fcマップ:1.2σ未満、最も近接する接触までの距離:2.3Å未満又は3.5Å超。疑わしい水分子及び活性部位(阻害剤までの距離が10Å未満)におけるものを用手的にチェックした。Fo−Fcマップ(−3.0σに一致)におけるネガティブピークに存在していた側鎖の占有率をゼロに設定し、次いで、次の精密化サイクル後にポジティブピークが生じた場合、0.5に設定した。
【0104】
最終モデルのラマチャンドランプロットは、全ての残基の80.8%が最も好ましい領域に、17.9%が更に許容される領域に、残基の0.7%が寛容に許容される領域にあることを示す。残基Val86(A)、His14(B)、Asp86(B)、Ser131(C)、Val56(D)、及びVal56(F)は、ラマチャンドランプロットの許容されない領域にみられる(表9)。これらは、電子密度マップにより確認される、即ち別の知覚可能な立体構造ではモデル化することができなかった。最終構造及び精密化プロセスの統計値を表9に示す。
【表9】
【0105】
6.4 X線構造解析
BT−063 Fab抗体断片が結合するヒトIL−10の複合体構造を3.48Åの解像度で分析し、Fab抗体断片の詳細な結合様式を明らかにする。
【0106】
得られた電子密度は、Fab断片の配向及び立体構造を含むFab断片の明確な結合様式を示す。空間群P6の結晶は、非対称単位における2つの複合体を含有する。
【0107】
Fabとの複合体におけるIL−10の構造を図7に示す。2つのFab断片は、CDRループでIL−10の各ホモダイマーに結合する。
【0108】
以下のIL−10の残基(分子A及びB)は、3.9Åの最大距離内でCDRループの近傍に見出すことができる:Arg27、Lys34、Gln38、Met39、Asp41、Gln42、Asp44、Leu46、Glu50、Leu53、Glu142、Asp144、Ile145、Asn148、Tyr149、Glu151、及びThr155。
【0109】
CDRループの以下の残基は、3.9Åの最大距離内でIL−10の近傍に見出すことができる:Phe27、Ser28、Ala30、Thr31、Tyr32、Trp52、Arg53、Gly54、Ser56、Asn73、Ser74、Tyr100、Gly101、Tyr103(分子C及びE)、Ser32、Asn33、Asn35、Tyr37、Lys55(分子D及びF)。
【0110】
IL−10:IL−10R1受容体複合体の公開されている構造にIL−10:BT−063の複合体構造を重ねることにより示される通り、BT−063の結合部位は、IL−10の表面上のIL−10受容体の結合部位と一致する(図7)。
【0111】
X線解析によって同定されたヒトIL−10と接触しているBT−063のアミノ酸残基を、以下に示すBT−063抗体可変ドメインの直鎖アミノ酸配列において強調する。
【表10】
CDR領域(Honegger and Plueckthun(2001)J.Mol.Biol.,309,657−670)に下線を引く(軽鎖のCDR1、CDR2、及びCDR3は、それぞれ、配列番号71、72、及び73であり;重鎖のCDR1、CDR2、及びCDR3は、それぞれ、配列番号74、75、及び76である)。IL−10と接触する残基を太字で示す。
【0112】
軽鎖内において、接触残基は、CDR1及びCDR2にはみられるが、CDR3にはみられない。3つ全てのCDRの重鎖残基は抗原結合に関与していると考えられる。FR3の2つの残基(Asn73及びSer74)も抗原結合に寄与している。
【0113】
CDR1の最初のSer28及びAla30は、マウスVHの元になった配列(BN−10)の一部であり、選択されるヒトフレームワーク(3−66*04)中には存在しない。両方の位置は、それほど高い頻度で抗原結合に関与しておらず、ヒト化プロセス中に代替アミノ酸として導入された。
【0114】
残基Asn73及びSer74は、マウスにみられ、ヒト抗体フレームワーク配列でも頻繁にみられるが、通常、抗原結合には関与していない(www.bioc.uzh.ch/antibody;Honegger and Plueckthun,2001)。抗原結合に対するこれらの寄与は、予測されていない。
【0115】
BT−063結合に関与するIL−10アミノ酸残基を以下に示す。また、高親和性IL−10受容体鎖(IL−10R1)及び低親和性受容体鎖(IL−10R2)に対する結合に関与しているIL−10の残基も示す。両方の受容体鎖が、IL−10ホモダイマーの結合に関与しており、シグナル伝達に必要である。配列Aでは、BT−063に接触する残基を太字で示す。配列Bでは、IL−10R1に対する接触残基を太字で示し、IL−1R2に対する接触残基をイタリック体で示し、下線を引き、IL−10R1とIL−10R2により共有されている接触残基を太字、イタリック体、及び下線で示す(Pletnev et al 2005)。
【表11】
【0116】
BT−063は、第1のIL−10モノマーのへリックスAの連結ループ配列(Glu42、Asp44、Leu46)を含む残基(Arg27、Lys34、Gln38、Met39、Asp41)、及びへリックスB(Glu50及びLeu53)のN−末端部分と、第2のIL−10モノマーのへリックスF’の残基(Glu142、Asp144、Ile145、Asn148、Tyr149、Glu151、Thr155)とを含むIL−10の不連続なエピトープに結合することが分かる。
【0117】
実施例7−カニクイザルにおけるインビボ単回用量毒性試験
カニクイザルで、BT−063(変異体hVH26/hVL7)の単回静脈内注射後、安全性薬理学的パラメータを含む単回用量毒性試験を実施した。4匹/性別/群になるように動物を4群に分けた(プラセボ、1mg/kg、7mg/kg、及び50mg/kg)。1日目にBT−063を静脈内注射した。5日間後に動物の半数を剖検し、残りの動物は28日目に屠殺した。
【0118】
この実験における低用量レベルである1mg/kgは、ヒトにおける約300μg/kgの用量と等しく、合計ヒト(体重60kg)用量は18mgになる。少数の研究者で開始した実験では、BT−063の元になった抗体B−N100を同用量21日間毎日投与した(Llorente,2000)。この量の抗体は、薬理学的効果及び臨床的有効性を示した。したがって、この結果を踏まえて低用量レベルを選択した。高用量は、出発用量の倍数(50μg)として選択した。中間用量は、低用量と高用量との幾何平均である。
【0119】
試験中、生理学的パラメータ又は組織病理学的パラメータにおいて毒物学的に有意又は明らかな変化はみられなかった。更に、臨床化学パラメータの変化は、毒物学的有意性が僅かである又は存在しないとみなされた。
【0120】
実施例8−健常ボランティアにおけるインビボ単回用量毒性試験
健常ボランティアにおいて漸増用量の抗体を用いて、BT−063(変異体hVH26/hVL7)の安全性及び忍容性、並びにBT−063の投与の効果をモニターするために実験を行った。23人のボランティアを8用量群に分け、BT−063を単回静脈内投与した。用量群は、以下の通りであった:0.175mg、0.75mg、3.5mg、7mg、15mg、30mg、60mg、及び100mg。100mg用量群のボランティアが2人であったことを除いて、1群当たりのボランティアは3人であった。
【0121】
各用量を0.9%塩化ナトリウム注射液で合計体積20mLに希釈した。2時間に亘って用量を単回連続静脈内注射した。注入後85日間に亘ってボランティアを評価し、この期間中の複数の時点で血液を採取した。
【0122】
サイトカイン
採取した血漿から、BT−63の注入前後のサイトカインIFNγ、IL−1β、IL−2、IL−4、IL−5、IL−6、IL−8、IL−10、及びTNFαの濃度を評価した。
【0123】
表10は、投与前の値とスクリーニングの値とから算出したIL−6、IL−8、及びIL−10の正常値上限を示す。
【表12】
【0124】
サイトカインの測定値から得られた更なる結果は、図12及び13に示す。
【0125】
注入後24時間以内では、IL−6及びIL−8が非用量依存的に一過的に増加したが、3日間後には投与前のレベルに戻った。この作用は、抗体ではなく注入事象に関連すると考えられる。その理由は、用量との相関がなく、また最低用量で既に前記事象が生じたためである。
【0126】
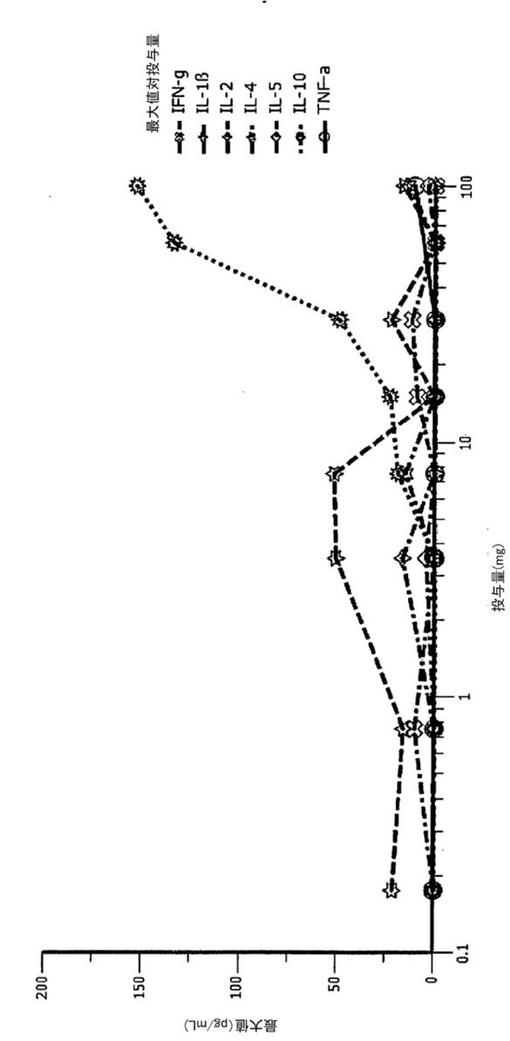
驚くべきことに、IL−10が重要な調節性サイトカインであるにも関わらず、図12に示される通り、IFNγ、IL−1β、IL−2、IL−4、IL−5、及びTNFαの濃度増加は検出されなかった。また、IL−4、IL−5、IFN−γ、IL−2の濃度が増加しないことにより、BT−063の投与がヘルパーT細胞を活性化させないことが確認される。更に、体温も炎症促進性サイトカインの大量放出の指標であるので、処理されたボランティアにおいてこのパラメータも測定した。しかし、投与後、体温の上昇は検出されなかった。
【0127】
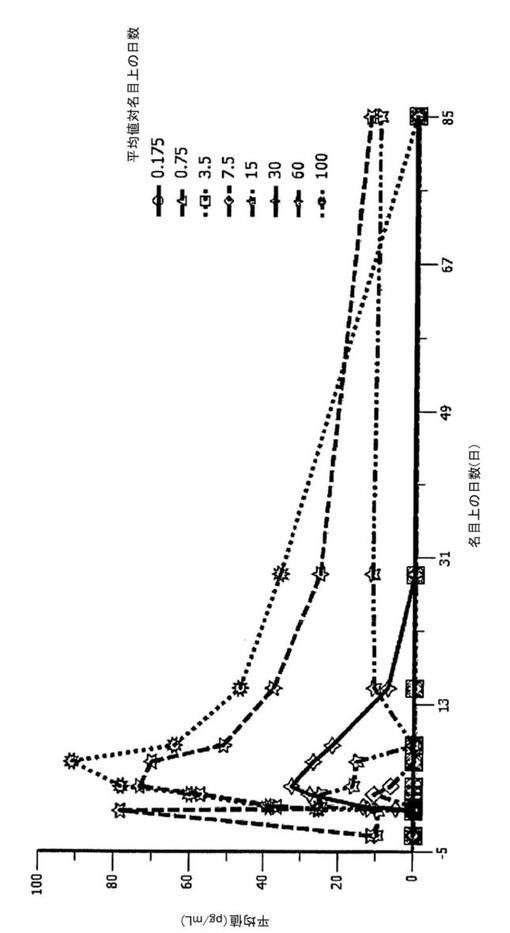
図13に示す通り、IL−10の血漿濃度のみが、BT−063の投与により影響を受け、BT−063の用量が増加するにつれて検出される血漿IL−10も増加する。用いたアッセイは、遊離IL−10及びBT−063に結合しているIL−10の両方を検出することに留意すべきである。増加の理由としては、以下の2つの可能性がある:(1)IL−10の半減期の延長、IL−10のBT−063への結合がIL−10の内部移行を妨げ、同時に、血液からIL−10を排出させなくする(通常、IL−10は速やかにターンオーバーし、分泌されたIL−10の半減期は約2.3時間〜3.5時間である(Huhn et al.,Blood(1996)Jan 15:87(2):699−705);及び/又は(2)ネガティブフィードバックループの誘導−BT−063は、IL−10のその受容体への結合をブロックするので、IL−10が細胞に取り込まれることができなくなり、したがって、B細胞がより多くのIL−10を産生するようになる。BT−063及びマウスIL−10Rノックアウト細胞を用いた全血培養実験から得られたインビトロにおけるデータにより確認されているので(Mahnke et al.、未公開データ)、仮説(2)の可能性の方が高いと考えられる。
【0128】
薬物動態
薬物動態データは、BT−063のCmax、AUC、及び半減期が、予測される理論値の範囲内であることを示した。BT−063の終末半減期は、15日間〜30日間である。30mg以上の用量を投与した後、85日間後も依然としてBT−063を血漿中で検出することが可能である。
【0129】
他のサイトカイン中和抗体(例えば、アダリムマブ、ヒト化抗TNF−α)ではヒト抗ヒト抗体(HAHA)応答がみられるという事実にも関わらず、処理したボランティアにおいてはHAHAがみられなかった。
【0130】
BT−063投与後、IL−10の検出量は増加したが、この実験が、更に多い用量のBT−063の安全性及び許容性を示したことに留意すべきであり、したがって、過剰のIL−10の効果を相殺するために十分な用量のBT−063をSLE患者に安全に投与できる(具体的には、炎症促進性サイトカインレベルの許容できない上昇がない)と結論付けることができる。
【技術分野】
【0001】
本発明は、インターロイキン10(IL−10)及びIL−10に特異的な剤に関する。特に、本発明は、ヒト化IL−10抗体及びその使用に関する。更に、本発明は、全身性エリテマトーデス(SLE)を治療する方法に関する。
【背景技術】
【0002】
全身性エリテマトーデス(SLE)は、自己免疫疾患であると考えられており、この疾患においては、Bリンパ球の異常な高活性及び免疫グロブリンガンマ(IgG)自己抗体の異常な大量産生が重要な役割を果たしている。この病理過程では、Igでコーティングされた細胞が隔離及び破壊され、補体タンパク質が定着及び分割され、且つケモタキシン、血管作動性ペプチド、及び破壊酵素が組織に放出される(非特許文献1)。
【0003】
SLEは、多様な徴候を特徴とする。この疾患の過程においては、合計95%の患者が筋骨格系疾患を訴え、80%が皮膚病変、85%が血液疾患、60%が神経障害、60%が心肺疾患、30%〜50%が腎臓疾患、40%が胃腸疾患、15%が血栓症、及び15%が眼疾患を示す。また、患者の大部分(95%)が、疲労、倦怠感、発熱、食欲不振、及び体重減少等の全身性症状に苦しみ、これら症状は、殆ど常に発現している。大部分の患者では、寛解と突然再発する疾患期間とが交互に生じる。永続的に寛解する(治療をしなくても症状が出ない)ことは非常に稀である。50年以上前、SLEと診断された患者の大部分は、5年間未満しか生存できなかった。現在は、10年生存率が90%を超えており、これは、早期診断、対症療法的な抗炎症及び免疫抑制治療によるものである。最も一般的な死因は、免疫抑制の結果としての感染症である(非特許文献1)。
【0004】
SLEの治療には、通常、抗マラリア薬、抗炎症薬、及び免疫抑制薬が用いられている。症状の制御が困難になった場合には、非ステロイド性抗炎症薬にコルチコステロイドを加える。更に、多くの臓器に関連する活動性SLEは、シクロホスファミドを用いる積極的治療を必要とする。
【0005】
現在まで、SLEを治癒させる及び/又は長期間に亘って患者の生活の質を向上させるために利用可能な原因療法は存在していない。しかし、近年、抗体技術が進歩し、また、この自己免疫疾患の原因である因子が更に同定されたことにより、治療の選択肢としてモノクローナル抗体を使用する可能性が開かれつつある。特に、SLEを治療するための好ましいアプローチは、ポリクローナル自己抗体を大量に過剰産生させる病的免疫反応と相互作用するか又は補正する特異的な治療である。SLEの病因は、主にB細胞の調節不全に関連しているので、B細胞を標的とすることができるモノクローナル抗体が特に注目されている。非特許文献2に記載の通り、潜在的なB細胞表面抗原は、CD19、CD20、CD21、及びCD22を標的とする。更に、IL−10、IL−1ra、IL−12(非特許文献3)及びIL−6(非特許文献4)は、免疫反応の調節において重要なサイトカインであり、特に、SLE患者における突然の再発中に上昇する。二本鎖DNA(dsDNA)に対する自己抗体及びIL−10の血漿濃度は、SLE患者における疾患の活動性を反映していることが多い。IL−10濃度の上昇は、SLE患者における疾患の活動性と相関している(非特許文献5)。しかし、IL−10は、免疫系に対して多面的な作用を有するサイトカインであり、また、炎症促進反応の低減に関与していることが知られている。
【0006】
モノクローナル抗体を用いた臨床試験がSLE患者で実施されている。具体的には、幾つかの臨床試験では、非ホジキンリンパ腫を治療するために用いられるキメラマウス抗CD20モノクローナル抗体である抗体リタキシマブが用いられた。非特許文献2に記載の通り、これら臨床試験の結果は、SLE患者においてこの抗体が高い活性を有していることを示したので、オファツムマブ、IMMU−106、及びGA−101等のCD20を標的とする幾つかの新規抗体が開発された。SLEにおけるモノクローナル抗体の活性を報告している更なる臨床試験では、抗CD22抗体であるエプラツズマブ、抗TNFα抗体であるインフリキシマブ、抗IL−10抗体であるB−N10(非特許文献6)、抗CD40L抗体であるIDEC131及びBG9588、BLYS阻害剤であるベリムマブ、抗IL−6受容体抗体であるトクリムマブ、並びに抗C5抗体であるエクリズマブが用いられた。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Hahn BH.Systemic Lupus Erythematosus.In:Kasper DL,Braunwald E,Fauci AS,Hauser SL,Longo DL,Jameson,JL,editors.In:Harrison’s Principles of Internal Medicine(16th edition).New York(US):McGraw−Hill;2005.pp.1960−1967
【非特許文献2】Robak and Robak(Current Drug Targets,2009,No.10,pages26−37)
【非特許文献3】Capper et al.,Clin.Exp.Immunol.2004 Nov;138(2):348−56
【非特許文献4】Chun et al.,J.Clin.Immunol.2007 Sep;27(5):461−6
【非特許文献5】Park et al.,Clin.Exp.Rheumatol.1998 May−Jun;16(3):283−8
【非特許文献6】Llorente et al.,Arthritis Rheum.2000 Aug;43(8):1790−800
【発明の概要】
【発明が解決しようとする課題】
【0008】
本発明の目的は、更なる剤、特に、この領域において有用性を有する抗体を提供することにある。
【課題を解決するための手段】
【0009】
したがって、本発明は、炎症促進性サイトカインレベルの許容できない上昇を伴うことなしに被験体に投与することができる、インターロイキン10(IL−10)に結合可能なヒト化抗体、キメラ抗体又はこれらの断片を提供する。
【0010】
IL−10は、抗炎症性サイトカインの代表的な物質であるので、ブロック時には、サイトカイン量が著しく増加すると考えられる。マウスIL−10抗体であるB−N10の場合には、無刺激の細胞培養物(健常個体のインビボの状態を反映する)において、IL−6やTNFαなどの炎症促進性サイトカインの上昇が観察できる。しかしながら、本発明者らは驚くべきことに、本発明の抗体をインビトロで細胞に適用した場合及びこれをインビボで投与した場合に、サイトカイン放出量が遥かに低いことを見出した。サイトカイン放出量が低いことは、結果として、本発明の抗体が投与対象である個体に許容されるという点で有利である。
【図面の簡単な説明】
【0011】
単なる一例として、以下の図面を参照して本発明を例証する。
【図1A】図1Aは、マウスB−N10抗体の軽鎖可変領域のアミノ酸配列(配列番号2)を示す。超可変相補性決定領域(CDR)に下線を引く(LCDR1は、配列番号4であり、LCDR2は、配列番号5であり;LCDR3は、配列番号6である)。
【図1B】図1Bは、マウスB−N10抗体の重鎖可変領域のアミノ酸配列(配列番号3)を示す。超可変相補性決定領域(CDR)に下線を引く(HCDR1は、配列番号7であり、HCDR2は、配列番号8であり;HCDR3は、配列番号9である)。
【図2A】図2Aは、マウスB−N10抗体の軽鎖可変領域をコードするヌクレオチド配列(配列番号10)を示す。
【図2B】図2Bは、マウスB−N10抗体の重鎖可変領域をコードするヌクレオチド配列(配列番号11)を示す。
【図3A】図3Aは、マウスB−N10の軽鎖可変領域(配列番号12)、A17(配列番号14)、JK1(配列番号15)、及びマウスB−N10抗体のヒト化中に生じた可変領域hVL1〜hVL12(配列番号18〜29)のアミノ酸配列を示す。
【図3B】図3Bは、マウスB−N10の重鎖可変領域(配列番号13)、3−66+04(配列番号16)、JH4(配列番号17)、及びマウスB−N10抗体のヒト化中に生じた可変領域可変領域hVH1〜hVH12(配列番号30〜41)のアミノ酸配列を示す。
【図3C】図3Cは、マウスB−N10抗体のヒト化中に生じた可変領域hVH13〜hVH29(配列番号42〜58)のアミノ酸配列を示す。
【図4】図4は、hIL−10抗原ELISAを用いてキメラcB−N10抗体と比較した、ヒト化抗体変異体の抗原結合性を提供する。
【図5】図5は、精製抗体製剤を用いてキメラB−N10抗体と比較した、3つのヒト化変異体hVH20/hVL7、hVH20/hVL8、及びhVH26/hVL7の結合性の測定結果を提供する。
【図6A】図6Aは、B−N10とインキュベーションした後の健常ボランティア由来全血培養物におけるTNFαの放出レベルを、BT−063の場合と比較して示す(50μg/mL)。
【図6B】図6Bは、B−N10とインキュベーションした後のSLE患者由来全血培養物におけるTNFαの放出レベルを、BT−063の場合と比較して示す(50μg/mL)。
【図7A】図7Aは、B−N10とインキュベーションした後の健常ボランティア由来全血培養物におけるIL−6の放出レベルを、BT−063の場合と比較して示す(50μg/mL)。
【図7B】図7Bは、B−N10とインキュベーションした後のSLE患者由来全血培養物におけるIL−6の放出レベルを、BT−063の場合と比較して示す(50μg/mL)。
【図8A】図8Aは、B−N10とインキュベーションした後の健常ボランティア由来全血培養物におけるIL−1βの放出レベルを、BT−063の場合と比較して示す(50μg/mL)。
【図8B】図8Bは、B−N10とインキュベーションした後のSLE患者由来全血培養物におけるIL−1βの放出レベルを、BT−063の場合と比較して示す(50μg/mL)。
【図9A】図9Aは、B−N10とインキュベーションした後の健常ボランティア由来全血培養物におけるIFN−γの放出レベルを、BT−063の場合と比較して示す(50μg/mL)。
【図9B】図9Bは、B−N10とインキュベーションした後のSLE患者由来全血培養物におけるIFN−γの放出レベルを、BT−063の場合と比較して示す(50μg/mL)。
【図10】図10は、IL−10に結合しているBT−063のFab断片の全体構造を示す。IL−10及びFab断片をリボン図で示す。
【図11】図11は、IL−10受容体が結合する部位と同じIL−10の部位に結合するBT−063のFab断片を示す。IL−10、IL−10R1、及びFab断片をリボン図で示す。
【図12】図12は、健常ボランティアにBT−063を投与した後の、投薬量に対するサイトカインの血漿濃度の平均cmaxのグラフを示す。
【図13】図13は、健常ボランティアにBT−063をインビボ投与した後の、経時的なIL−10平均血漿濃度のグラフを示す。
【発明を実施するための形態】
【0012】
本発明は、インターロイキン−10(IL−10)に結合可能であるヒト化抗体、キメラ抗体又はこれらの断片、及び高レベル又は高活性のIL−10により媒介される病状の治療におけるこの抗体又はその断片の使用に関する。
【0013】
ヒトIL−10は、分子量37kDaのホモダイマーである。各モノマーは、160アミノ酸からなり、分子量は18.5kDaである。IL−10のダイマーは、IL−10R受容体α(IL−R又はIL−10R1)と相互作用し、次いで、IL−10受容体β(IL−10Rβ又はIL−10R2)を複合体に動員する。これら受容体は、様々な細胞、特に、単球、マクロファージ、Tリンパ球、及びBリンパ球等の大部分の造血細胞を含む免疫細胞(Asadullah et al.,Pharmacol.Rev.2003 Jun;55(2):241−69)で発現するが、表皮細胞又はケラチン産生細胞等の非造血細胞でも発現する。IL−10受容体αとIL−10との結合及びIL−10受容体βの動員により、Jak1及びTyk2チロシンキナーゼを介するシグナル伝達が導かれ、次いで、STATファミリーの転写因子が活性化される。ヘルパーT細胞、調節性T細胞、単球、マクロファージ、B細胞、好酸球、マスト細胞、ケラチン産生細胞、樹状細胞、更には癌細胞等、様々なIL−10の細胞源が公知である。B細胞におけるIL−10の機能は、アポトーシスの防止、増殖の強化、クラススイッチ事象、及びプラズマ細胞への分化(Asadullah et al.,Pharmacol.Rev.2003 Jun;55(2):241−69)などである。
【0014】
本発明は、炎症促進性サイトカインレベルの許容できない上昇を伴うことなしに被験体に投与することができる、インターロイキン10(IL−10)に結合可能なヒト化抗体、キメラ抗体又はこれらの断片を提供する。
【0015】
治療用抗体の投与は、被験体に望ましくない副作用を引き起こす、炎症促進性サイトカインレベルの許容できない上昇を引き起こし得ることが知られている。特に、この許容できない上昇は、皮膚の発赤を引き起こすことがあると共に、体温の上昇などの発熱又はインフルエンザ様症状をもたらすことがある。したがって、本発明の好ましい態様においては、前記抗体又は抗体断片は、2℃を超える体温の上昇を伴うことなしに被験体に投与することができるものである。
【0016】
更に、前記抗体又はその断片は、投与後の被験体の血漿中における炎症促進性サイトカイン量の実質的な上昇を引き起こさない。これらのサイトカインの許容できないレベルは、通常、サイトカインの正常値上限(ULN)よりも数倍高い。該ULNは、被験体コホートで測定されるサイトカインの平均レベルに2×標準偏差を加えた値として定義される。
【0017】
したがって、本発明の好ましい態様においては、前記抗体又はその断片は、炎症促進性サイトカインレベルが、正常値上限(ULN)の500%を超えて上昇することなしに、より好ましくは300%を超えて上昇することなしに被験体に投与することができる。換言すれば、前記抗体又はその断片は、炎症促進性サイトカインレベルを、当該炎症促進性サイトカインのULNの500%未満、より好ましくは300%未満しか上昇させない。前記炎症促進性サイトカインは、TNF−α、IFN−γ及びIL−1βのうちの少なくとも1つ、好ましくはこれらの全てであることが好ましい。或いは、前記炎症促進性サイトカインは、IL−6とIL−8のいずれでもない。
【0018】
更に、本発明の抗体又はその断片は、抗炎症反応を引き起こす、IL−1受容体アンタゴニスト(IL−1ra)を被験体において誘導することができることが特に好ましい。
【0019】
本発明の好ましい態様においては、前記ヒト化抗体又はその断片は、PBMC、より具体的には免疫細胞から、これらの細胞にインビトロで接触させたときに放出される炎症促進性サイトカイン、特にTNF−α、IL−1β、IL−6、のレベルに500%を超える上昇を引き起こさない。
【0020】
本発明者らは、驚くべきことに、本発明のヒト化抗体又はその断片が引き起こす炎症促進性サイトカインレベルのインビトロアッセイにおける上昇が、マウスB−N10抗体よりも小さいことを見出した。
【0021】
炎症促進性サイトカインの放出レベルは、単離した免疫細胞又はヒト全血培養物を用いるインビトロ研究によって測定することができ、例えば、以下の実施例5に記載の研究によって測定することができる。具体的には、前記測定方法は、(a)前記抗体又はその断片と共に細胞培養物をインキュベートする工程、及び(b)少なくとも1つの炎症促進性サイトカインのレベルを測定する工程を含む。
【0022】
ヒト全血培養物は、健常人又はSLEを罹患している患者などの疾患患者から採取することができる。前記末梢血単核細胞(PBMC)は、免疫細胞であってもよく、特にマクロファージ、単球、樹状細胞、ヘルパーT細胞、及びB細胞から選択することができる。
【0023】
前記少なくとも1つの炎症促進性サイトカインは、インターロイキン−1β(IL−1β)、IL−1α、IL−6、腫瘍壊死因子α(TNF−α)、ヘルパーTサイトカイン(インターフェロンγ、IFN−γ、IL−4)、マクロファージサイトカイン(IL−12)、ケモカイン(IL−8、MCP−1)のそれぞれから選択することができる。これらのサイトカインの放出レベルは、当該技術分野で一般に知られている方法を用い細胞培養物上清中で測定することができる。
【0024】
より具体的には、該インビトロ法は、以下の工程を含むことができる:
a)50μg/mLの抗体又は断片をヒト全血培養物と接触させる工程;
b)前記抗体又は断片を前記全血培養物と共に37℃で48時間インキュベートする工程;及び
c)前記培養物中の1以上の炎症促進性サイトカインの量を測定する工程。
【0025】
特に、前記方法は、前記抗体と接触させないヒト全血培養物と共に行う。このコントロールと比較して、本発明の抗体又は抗体断片は、サイトカインレベルを500%未満、より好ましくは300%未満しか上昇させないことが好ましい。この方法におけるサイトカインは、IL−6とIL−8のいずれでもないことが好ましい。
【0026】
サイトカインは、Multiplex Bead Immunoassays(96ウェルフィルタープレートで行う固相タンパク質アッセイ)を用いて培養物中で検出することができる。該方法は、特定のサイトカインに対する特定の抗体が結合していると共に所定の分光特性を示すビーズを用いる。これにより、ビーズを特異的に同定することができ、これらを既知の結合抗体に帰することができる。ビーズに結合したサイトカインは、検出抗体を該サイトカインに更に結合させることにより定量することができる。これは、GM−CSF、IL−1β、IL−2、IL−4、IL−5、IL−6、IL−8、IL−10、IFN−γ、及びTNF−αなどの最大10種のヒトサイトカインを同時に検出することを可能にする。湿ったウェルにサンプル、標準物質、及びビーズ溶液がピペットで注入されたフィルタープレートを前記目的のために用いる。インキュベーション後、液体を真空ポンプで吸引除去し、残ったビーズを洗浄し、ビオチン化検出抗体の混合物を添加する。液体を吸引除去し且つウェルに残っているビーズを洗浄することにより、結合していない検出抗体を除去し、次いでストレプトアビジンコンジュゲートR−フィコエリスリン(ストレプトアビジン−RPE)を添加する。これは、インキュベーション中にビオチン化検出抗体を認識し、再度洗浄した後に、形成された免疫複合体をBio−Plex200Systemで分析することができる。この免疫学的アッセイは、Bio−Plex200装置を用いて分析する。この装置は、2つのレーザーシステムを用いており、これらのうちの一方は、ビーズの分光特性に基づいてビーズを同定し、他方は、第2抗体を用いる検出により結合したサイトカインの量を測定する。結合量の定量は、既知量のサイトカインを含む標準物質を並行して用いることで達成される。
【0027】
更に、本発明は、インターロイキン10(IL−10)に結合可能なヒト化抗体、キメラ抗体又はこれらの断片であって、被験体に投与されたときにIL−10の血漿中のレベルを上昇させることができるヒト化抗体、キメラ抗体又はこれらの断片を提供する。
【0028】
以下の実施例8に示すように、本発明の抗体は、炎症促進性サイトカインレベルの許容できない上昇を伴うことなしに投与することができる一方で、投与により、血漿サンプル中に検出可能なIL−10の量が用量依存的に上昇する。中和抗体を適用したときにIL−10レベルが上昇するという知見は、予想外である。通常、サイトカイン中和剤を投与すると遊離サイトカインレベルが低下することが予想される(Strandら、Nature Reviews Drug Discovery、2007、Vol.6、pp.75−92)。何ら理論に拘束されることを望むものではないが、抗体がIL−10に結合することにより、IL−10のIL−10受容体への結合を阻害し、より多くのIL−10をB細胞に産生させるネガティブフィードバックループを誘導すると考えられる。にもかかわらず、このアップレギュレーションが本発明の抗体の治療有用性を阻害することはない。これは、実施例8にも示されるように、該抗体は、全てのIL−10を中和するのに十分高いレベルでも安全に投与されるからである。
【0029】
本願において、用語「キメラ抗体」は、重鎖及び/又は軽鎖の一部が、特定の種に由来するか、或いは特定の抗体クラス又はサブクラスに属する抗体の配列と同一であるが、一方、抗体の残りの部分は、異なる種、抗体クラス、又は抗体サブクラスに由来する抗体の配列と同一である抗体を指す。キメラ抗体のCDRはある起源に由来するが、抗体の残りの部分は異なる起源に由来することが特に好ましい。具体的には、本発明において、キメラ抗体は、非ヒト抗体の抗原結合配列/可変ドメインがヒト抗体のフレームワーク領域にグラフトされているヒト化抗体であってよい。
【0030】
本願において、用語「断片」は、望ましい生物活性を保持している抗体の断片又は誘導体を指す。断片は、一般的に、抗体の抗原結合領域、具体的には、Fab、Fab’、F(ab)’2、Fv、及びscFv断片、並びに多価抗体誘導体、具体的には、二重特異性抗体(diabody)又はタンデム二重特異性抗体を含む。断片は、少なくとも25アミノ酸が好ましく、50アミノ酸がより好ましく、200アミノ酸〜500アミノ酸が更により好ましい。或いは、断片は、30KDa〜150kDaのサイズを有する断片として定義することもできる。更に、抗体断片は、2以上のペプチド/ポリペプチド鎖を含んでいてもよい。例えば、各長さが200アミノ酸〜300アミノ酸である2本の鎖を含むFab断片、又は各長さが400アミノ酸〜500アミノ酸である2本の鎖を含むTandAbs(登録商標)(四価二重特異的抗体フォーマット)である。
【0031】
前記抗体又はその断片は、本明細書に記載されるマウスB−N10抗体又はヒト化BT−063(変異体hVH26/hVL7)抗体に由来することが、本発明の好ましい特徴である。具体的には、かかる抗体又はその断片は、B−N10/BT−063可変軽鎖のCDR1、CDR2、及びCDR3のアミノ酸配列と少なくとも80%同一である、及び/又はB−N10/BT−063可変重鎖のCDR1、CDR2、及びCDR3のアミノ酸配列と少なくとも80%同一であるアミノ酸配列を含むCDRを含む。マウスCDRのアミノ酸配列を図1に示す。BT−063のCDRのアミノ酸配列を実施例6に示す。前記配列は、B−N10/BT−063抗体のCDRの配列と少なくとも90%又は少なくとも95%同一であることがより好ましい。本明細書中の実施例6に記載するX線結晶構造解析は、CDR内のどの残基がIL−10との結合に重要であるかを示す。
【0032】
或いは、本発明の抗体又は断片は、B−N10/BT−063抗体に由来するものではあるが、抗体又はその断片の親和性及び/又は特異性を実質的に変化させることのない変異を配列中に任意で含む、B−N10/BT−063可変軽鎖のCDR1、CDR2、及びCDR3のアミノ酸配列、及び/又はB−N10/BT−063可変重鎖のCDR1、CDR2、及びCDR3のアミノ酸配列を含んでいてもよい。具体的には、マウスB−N10抗体又はBT−063(変異体hVH26/hVL7)抗体のCDRを含む抗体又は断片と比べて、前記配列中の変異は、IL−10に対する抗体又は断片の親和性又は特異性を低下させない。
【0033】
特定の実施形態において、本発明は、B−N10/BT−063の可変軽鎖及び/又は可変重鎖のCDR1、CDR2、及びCDR3のアミノ酸配列を含むヒト化抗体、キメラ抗体又はこれらの断片を提供する。より好ましくは、本発明は、図1に示すマウス抗体B−N10の可変ドメインのアミノ酸配列を含むヒト化抗体、キメラ抗体又はこれらの断片を提供する。最も好ましくは、前記抗体又は断片は、BT−063の可変ドメインのアミノ酸配列(配列番号69及び70)のうちのいずれか一方又は両方を含む。
【0034】
一般的に、本発明の抗体は、ヒト定常領域(Fc)を更に含む。これは、IgM、IgG、IgD、IgA、及びIgEを含む免疫グロブリンの任意のクラス、並びにIgG1、IgG2、IgG3、及びIgG4を含む任意のアイソタイプに由来する定常ドメインの中から選択することができる。好ましい定常領域は、IgG、特にIgG1の定常ドメインの中から選択される。
【0035】
他の生成物
本発明は、更に、上記抗体又は抗体断片をコードする核酸配列を提供する。前記核酸配列は、DNAであってもRNAであってもよいが、DNAが好ましい。前記配列は、発現カセット又はベクター内で用いることができ、本明細書に開示される抗体及びその断片の産生において特に用いられる。
【0036】
本発明は、更に、これらポリヌクレオチド、発現カセット又はベクターで形質転換されたホスト細胞を提供する。好適なホスト細胞は、原核細胞であっても真核細胞であってもよい。
【0037】
或いは、ホスト細胞は、本発明の抗体を産生する細胞と骨髄腫細胞とを融合させることにより得られるハイブリドーマであってもよい。
【0038】
上記ホスト細胞は、抗体又はその断片を産生するための方法で利用することができる。具体的には、かかる方法は、抗体又はその断片を発現させる条件下で好適な培養培地中にてホスト細胞を培養する工程と、培養培地から前記抗体又はその断片を分離する工程とを含んでいてもよい。この種の方法は、当技術分野において公知であり、報告されている。
【0039】
医療的用途
本明細書に記載される抗体又はその断片は、高レベル又は高活性のIL−10により媒介される疾患又は病状の治療において有用性を有する。結果として、被験体における病状を治療又は予防するための方法であって、前記病状が高レベル又は高活性のIL−10により媒介され、治療上有効な量の本明細書に記載される抗体又はその断片を投与することを含む方法を提供する。
【0040】
具体的には、高レベル又は高活性のIL−10により媒介される病状は、SLEである。したがって、また、本発明は、SLEの治療において使用するための本明細書に記載される抗体又はその断片を提供する。
【0041】
更なる例は、血小板減少性紫斑病、ループス腎炎、HIV、hCMV、及びC型肝炎である。別の例は、免疫反応の増殖又は抑制を直接支持することによりIL−10に依存している腫瘍細胞を治療することである。
【0042】
本発明の更なる実施形態は、薬学的に許容し得る担体又は希釈剤と共に、上記抗体又はその断片を含む医薬組成物である。1つの実施形態では、前記組成物は、抗体又はその断片がカップリングしているリポソームを含む。
【0043】
かかる組成物は、患者に対して、非経口的に、静脈内に、又は皮下に投与することができる。SLEの治療において、前記抗体又はその断片は、静脈内又は皮下に投与されることが好ましい。
【0044】
前記抗体及びその断片は、炎症促進性サイトカインレベルを大幅に上昇させることがなく、患者にとって許容できない「サイトカインストーム」を防ぐためのコルチコステロイドの併用投与の必要がないので、疾患の治療に特に有用である。
【0045】
したがって、特定の態様においては、本発明は、被験体における、高レベル又は高活性のIL−10により媒介される病状を治療又は予防する方法であって、治療上有効な量の本明細書に記載の抗体又はその断片を前記被験体に投与する工程を含み、前記被験体は、コルチコステロイドによる治療を同時にも別々にも受けていない(即ち、患者は、コルチコステロイドを併用していない)を提供する。この好ましい態様の実施形態においては、高レベル又は高活性のIL−10により媒介される病状は、SLEである。
【0046】
多くの患者において、SLEの治療にコルチコステロイドが併用される。しかしながら、本発明のこの態様は、コルチコステロイドの投与が望ましくない患者の治療において特に有用である。
【0047】
非医療的用途
更に、本明細書に記載される抗体又はその断片と標識とを含む、標識されているヒト化抗体、キメラ抗体又はこれらの断片を提供する。前記標識は、サンプル中の抗体の存在を検出するために当技術分野において公知である任意の好適な種類であってよい。具体的には、前記標識は、蛍光標識であってよい。
【0048】
標識されている抗体又は標識されていない抗体、具体的には、標識されている抗体は、サンプル中に存在するIL−10の存在を検出するためのインビトロ方法において特定の有用性を有する。前記方法は、標識されていない抗体若しくは標識されている抗体又はこれらの断片とサンプルとを接触させる工程と、前記サンプルを洗浄して、前記サンプルに結合していない抗体及びその断片(即ち、非結合抗体又は抗体断片)を除去する工程と、例えば標識を介して、前記サンプル中の前記抗体(又は断片)の存在を検出する工程とを含む。
【0049】
或いは、サンプル中のIL−10を中和するためのインビトロ方法に、標識されていない抗体又は断片を使用してもよい。かかる方法は、抗体又はその断片をIL−10に結合させるために、前記抗体又は断片とサンプルとを接触させる工程を含む。
【実施例】
【0050】
次に、以下の特定の実施形態に関連して本発明を更に説明する。
【0051】
実施例1−マウス抗IL10抗体B−N10の特性評価
1.1 B−N10抗体の可変ドメインをコードするDNAの単離
マウスBN−10の可変配列を同定するために、細胞ペレットを用いた。−80℃で保存しておいたサンプル(3×B−N10、3代継代、1×107細胞)の細胞からmRNAを単離し、cDNAを合成した後、B−N10の可変配列をPCRにより増幅させ、次いで、クローニングした。
【0052】
合計14クローンの配列を決定し(SEQ Laboratories,Goettingen)、可変軽鎖及び可変重鎖について分析した。B−N10の可変配列を明確に決定した。N−末端のプライマー領域でのみ差が生じた(表1を参照されたい)。可変重鎖については、プライマー領域における配列変異QVQLKQ(配列番号59)が9クローンで生じていたが、他の変異は1クローン又は2クローンでしか生じていなかった。サブクローニングするためにこの変異体を選択した。可変軽鎖については、2つの変異体が同比率で存在していた。マウス生殖細胞系配列と比較したところ、3つの突然変異しか有しないcr1配列が、同定されたVL配列と高い相同性を示した。これは、DVLMTQ(配列番号60)配列が正確な配列である可能性が非常に高いことを意味する。配列DIVMTQ(配列番号61)は、典型的な別のクラスの生殖細胞系配列であるので、除外した。
【表1】
【0053】
可変軽鎖VL及び可変重鎖VHのタンパク質配列を、それぞれ図1A及び1Bに示す。超可変相補性決定領域(CDR)に下線を引く。対応するDNA配列を、それぞれ図2A及び2Bに示す。
【0054】
実施例2−キメラB−N10抗体の作製
組換え抗体を発現させるために、実施例1で同定された抗体の重鎖及び軽鎖の可変配列をベクター系にクローニングした。第1の工程として、前記配列をBSリーダーにクローニングし、N末端に分泌シグナルを付加し、C末端にスプライスドナー配列を付加した。第2の工程として、それぞれ定常ヒトカッパ鎖及び定常ヒトガンマ−1鎖を含有する発現ベクターにこれら配列をクローニングした。軽鎖用ベクター及び重鎖用ベクターを調製し、次いで、リン酸カルシウム沈殿又はリポフェクションによってCOS−7細胞に一過的にコトランスフェクトした。2日間後、細胞培養上清を回収した。COS−7細胞中でキメラB−N10を発現させ、上清中の抗体の力価を検出した(サンドイッチELISA)後、ヒトインターロイキン−10(R&D Systems、カタログ番号217−IL/CF、ロット番号ET114021、−20℃で保存)に対する結合能をELISAで試験した。
【0055】
サンドイッチELISAの場合、マウス抗ヒトカッパ鎖抗体(Becton Dickinson)をキャッチャー抗体としてプレート表面に結合させ、次いで、細胞培養上清と共にインキュベートし、PODと複合体化しているウサギ抗ヒトIgG(H+L)抗体(Dianova)を用いてキメラ抗体の存在を検出した。規定の濃度(0.125μg/mL〜12μg/mL)のキメラ対照抗体をポジティブコントロールとして用いた。
【0056】
抗原ELISAの場合、0.5μg/mL〜5μg/mLの濃度でプレート表面にヒトIL−10を結合させた。細胞培養上清(未希釈及び1:5希釈)と共にインキュベートした後、PODと複合体化しているウサギ抗ヒトIgG(H+L)抗体(Dianova)を用いてキメラB−N10の結合を検出した。マウスB−N10をポジティブコントロールとして用いた。0.5μg/mL〜5μg/mLの濃度の抗体を用い、PODと複合体化しているウサギ抗マウスIgG/IgM抗体(Dako)で結合を検出した。
【0057】
ELISAの結果については、実施例3で論じる。
【0058】
実施例3−抗IL−10抗体のヒト化
げっ歯類抗体のヒトにおける免疫原性を低下させようとする最初の試みは、げっ歯類抗体の定常ドメインをヒト抗体の定常ドメインに置き換えることによりキメラ抗体を作製することであった。依然として可変ドメイン内のげっ歯類のフレームワーク領域が免疫反応を誘導する場合があったので、より進歩したCDRのグラフト方法、即ち、抗原結合配列(相補性決定領域、CDR)を完全にヒト抗体フレームワークに転移させる(ヒト化)方法が開発された。通常、マウスドナー抗体に最も類似するヒトアクセプターフレームワークを選択して、ヒト化プロセス中にオリジナル抗原の特異性及び親和性が回復される可能性を高める。ヒト抗体生殖細胞系配列、発現抗体のコンセンサス配列、CDRループ構造の分析、及び抗体/抗原複合体のX線構造を用いる異なるアプローチを用いてもよく、これらと組み合わせて前記プロセスを改善してもよい。通常、幾つかのヒト化抗体変異体は、この方法で作製され、後に、互いに異なる及びオリジナル抗体と異なる可能性のある生物学的作用に関して分析される。最後に、抗体の望ましい機能に従って、好適なヒト定常領域を選択することができる。
【0059】
3.1 B−N10のマウス可変配列とヒト配列との配列比較、並びにヒト化VL(hVL)及びVH(hVH)の配列のセットの設計
マウス抗IL−10抗体であるB−N10を選択した(Llorente et al.,Eur.Cytokine Netw.1993 Nov−Dec;4(6):421−7;及びLlorente et al.,Arthritis Rheum.2000 Aug;43(8):1790−80)。ヒト化抗体を得るための方法は、相補性決定領域(CDR)をヒトアクセプター領域に融合させるCDRグラフト手順に基づく。
【0060】
以下の3つのデータセットの複合分析に基づいて、ヒトアクセプターフレームワークを選択した:
1. 体細胞突然変異のリスクを最小化するための、マウス配列とヒト生殖細胞系配列との相同性、
2. 異常なアミノ酸残基を同定するための、マウス配列とヒトコンセンサス配列との比較、及び
3. 重要な構造フレームワークアミノ酸残基についての情報を得るための、CDR配列の基準となる構造クラスの同定。
【0061】
B−N10のマウス可変軽鎖は、ヒト生殖細胞系可変セグメント2−30*01(A17(配列番号14))及び接合セグメントJK1(配列番号15)に対して最も高い相同性を示す。B−N10に対して最も高い相同性を有するヒトコンセンサス配列は、HuKIIである。可変軽鎖の相補性決定領域(CDR)は、L1の場合クラス4に、L2及びL3の場合クラス1に分類することができた。重要なアミノ酸残基を同定した。
【0062】
クラス4−1−1の基準構造を有するマウスCDRとヒト生殖細胞系VL遺伝子との配列比較により、2−30*01と最も高い相同性を有する(一致しないアミノ酸数が最も少ない)ことが明らかになった。
【0063】
B−N10のマウス可変重鎖は、ヒト生殖細胞系可変セグメントVH3−33及び接合セグメントJH4(配列番号17)に対して最も高い相同性を示す。B−N10に対して最も高い相同性を有するヒトコンセンサス配列は、HuHIIIである。可変重鎖の相補性決定領域(CDR)は、H1及びH2の場合クラス1に分類することができた。重要なアミノ酸残基を同定した。クラス1−1の基準構造を有するマウスCDRとヒト生殖細胞系VH遺伝子との配列比較により、3−66*04(配列番号16)と最も高い相同性を有する(一致しないアミノ酸数が最も少ない)ことが明らかになった。したがって、生殖細胞系配列VH3−66を考慮した。
【0064】
ヒト化可変軽鎖(12変異体)と可変重鎖(29変異体)との様々な可変配列のセットを設計するために、得られた全てのデータを検討した。
【0065】
3.2 ヒト化hIL−10結合抗体断片の小さなライブラリの構築及び選択
hIL−10に結合する可能性のある抗体断片のライブラリを作製して最適なヒトIL−10結合抗体を得るために、真核細胞のコドン使用頻度を考慮した上で、図3に示す12個のhVL断片及び29個のhVH断片をコードするcDNA配列を作製した。
【0066】
次いで、得られたcDNAをクローニングベクターにクローニングし、SEQ Laboratories(Goettingen,Germany)において配列を決定した。hVL断片をコードする12個の各cDNAとhVH断片をコードする29個のcDNAとを組み合わせて、発現する可能性のある348個の抗体断片を得るという方法でライブラリを構築した。
【0067】
細菌で発現させ、ヒトIL−10(R&D Systems、カタログ番号217−IL/CF)に対して2ラウンドの選択を行った後、hIL−10(同様に選択した)に対する結合についてELISAにより前記抗体断片を分析した。簡潔に述べると、4℃で一晩、PBS中1μg/mLのhIL−10でMaxisorbプレート(Nunc,Germany)をコーティングした。前記プレートをブロッキング及び洗浄した後、抗体断片を産生する細菌の上清を添加した。結合しているヒト化抗体断片を検出するために、PODと複合体化している二次抗体を用いた。
【0068】
優れた結合剤のコード配列を分析し、同定されたhVL断片及びhVH断片の発生について列記した(表2)。
【表2】
【0069】
太字にした配列は、抗体全体について結合特性を分析するために、適切な真核発現ベクターにサブクローニングするために選択した配列である。括弧内に示す配列は、発現ベクターにサブクローニングするために選択したが、欠陥コンストラクトしか得られなかった配列である。
【0070】
3.3 選択したBT−063のヒト化軽鎖及び重鎖の変異体用の発現ベクターの作製
スクリーニングアプローチにより求められた統計値に基づいて、ベクター系にクローニングするためのBT−063のヒト化VL及びヒト化VHの変異体のセットを選択した。第1の工程では、分泌シグナル5’及びスプライスドナー配列3’をコードしている配列とクローニングしたcDNAとを融合させるために、ヒト化VL及びVHの変異体をコードするcDNAを適切なベクターに転移させた。第2の及び最後のサブクローニング工程で、それぞれ、ヒト定常カッパ鎖及びヒト定常ガンマ−1鎖をコードする発現ベクターにこれらcDNAコンストラクトを転移させた。内毒素を含まないQiagen Midi−prepキット(Qiagen,Germany)によって、独立に得られたhVL及びhVHを含有する発現ベクターのプラスミドを調製した。
【0071】
3.4 COS−7細胞における選択されたヒト化BT−063変異体の一過的発現及びhIL−10に対する抗体結合の比較
COS−7細胞におけるヒト化抗体変異体を一過的に発現させるために、選択した各ヒト化VL変異体(hVL7及びhVL8)と、選択した各ヒト化VH変異体(hVH1、hVH9、hVH13、hVH18、hVH20、hVH26、hVH28)とを組み合わせて、14個の異なるヒト化抗体を得た。
【0072】
簡潔に述べると、24ウェルフォーマットにおいて10%FCSを含有するDMEM中で、リン酸カルシウム沈殿により、軽鎖及び重鎖をコードする発現ベクターをCOS−7細胞に一過的にコトランスフェクトした。トランスフェクション後、培地を無血清培地CHO−S−SFM II(Invitrogen,Germany)に置換し、トランスフェクションの2日間後〜3日間後にCOS−7細胞の上清を回収した。トランスフェクトされたCOS−7細胞の上清に分泌されたヒト化抗体の抗体力価をサンドイッチELISAにより分析した。求められた抗体濃度に基づいて、全てのサンプルの上清を同じ抗体濃度に調整した。PBS中2μg/mLのhIL−10でコーティングされたMaxisorbプレート(Nunc,Germany)を用いる抗原ELISAにより、全てのサンプルを用いてヒトIL−10に対する結合を分析した。
【0073】
図4に示す通り、分析した変異体は全てhIL−10に結合するが、結合性は異なっている。有意に、抗原ELISAにおいて最も高いシグナルは、キメラB−N10抗体で得られた強度に匹敵するシグナル強度を示すBT−063変異体hVH20/hVL7、hVH26/hVL7、及びhVH20/hVL8で得られた。これら3つの抗体内のシグナル強度の差(hVH20/hVL8のシグナルがより強く、hVH20/hVL7及びhVH26/hVL7のシグナルがより弱い)は、定量サンドイッチELISA(上記を参照されたい)の結果、抗体濃度の差に起因していた可能性がある。調べた他の全ての変異体からは、キメラB−N10抗体と比べてかなり弱いシグナルが得られた。
【0074】
3.5 キメラ及びヒト化抗体の変異体の産生及びアフィニティー精製
選択されたヒト化BT−063変異体(hVH20/hVL7、hVH20/hVL8、hVH26/hVL7)及びキメラcB−N10(実施例2で論じた)をCOS−7細胞で産生させた。
【0075】
10cmの組織プレートを用いて、セクション3.4に記載の通り一過的に発現させた。トランスフェクションの5日間後、各変異体から約0.5Lの無血清上清を回収した。
【0076】
前記無血清上清からプロテインAアフィニティークロマトグラフィーによって抗体を精製した。2MのNaClの存在下で上清をロードした。0.1Mのクエン酸バッファ(pH4.0)により抗体を溶出し、2Mのリン酸バッファ(pH7.2)を含有するチューブに分画した。PBSでバッファ交換し、30kDaでカットオフする膜を用いて遠心分離することにより個々の抗体のプローブを濃縮した。抗原ELISA、非還元条件及び還元条件下におけるSDS−PAGE、並びに260nm及び280nmにおけるUV測定により、精製された物質の品質をチェックした。
【0077】
精製したキメラB−N10及びヒト化変異体のhIL−10に対する結合を、実施例2に記載した方法に従ってELISAにより試験した。hIL10をコーティングし、変異体cB−N10、BT−063−1(hVH20/hVL7)、BT−063−2(hVH20/hVL8)、及びBT−063−3(hVH26/hVL7)について抗体の結合を測定した。結果を図5に示す。
【0078】
キメラB−N10及びhVH20/hVL7変異体のシグナル強度は同程度であったが、変異体hVH20/hVL8及びhVH26/hVL7のシグナル強度は僅かに低かった。
【0079】
3.6 BiacoreヒトIL−10による親和性の測定
BIACORE2000(Biacore AB,Uppsala,Sweden)を用いて、hIL−10に対する様々な抗体(マウス、キメラ、3つのヒト化変異体)の結合についての会合速度定数及び解離速度定数を測定するために表面プラズモン共鳴分析を用いた。製造業者の条件に従って、hIL−10をCM−5センサチップに固定化した。5μL/分の流速で20μg/mLのアリコート50μLを添加することによりhIL−10を固定化して、固定化密度320RUにした。50μL/分の流速で0.1Mの炭酸バッファ(pH9.2)及び0.01MのHCL/1MのNaClを各1分間用いることにより2段階サイクルで、固定化hIL−10表面を再生した。20μg/mL〜0.15μg/mLの抗体濃度範囲で少なくとも4回各抗体サンプルを分析した。BIA evaluationバージョン3(1999)ソフトウェアを用いることにより、センサグラムからの計算を実施した。
【0080】
表3に、全てのBiacore測定の結果を要約する。全ての変異体が同程度hIL−10に結合する。しかし、僅かな差が検出可能である。結果として、マウスモノクローナル抗体B−N10、キメラcB−N10、及びヒト化変異体BT−063−1(hVH20/hVL7)は、同程度の親和性で結合するが、2つの他のヒト化変異体BT−063−2(hVH20/hVL8)及びBT−063−3(hVH26/hVL7)は、(マウスB−N10に比べて約3倍)低い親和性を示す。会合速度及び解離速度の僅かな差も検出可能である。
【表3】
【0081】
カニクイザルのIL−10
Biacore T100(Biacore AB,Uppsala,Sweden)を用いた更なる表面プラズモン共鳴実験により、BT−63変異体3(hVH26/hVL7)のカニクイザルIL−10に対する親和性を分析した。
【0082】
BT−063を10mMの酢酸(pH5.5)で5μg/mLに希釈し、アミンカップリング手順を用いて固定化して、最終レベルを約1,000RUにした。10mMのグリシン−HCl(pH1.8)を30秒間注入して、センサチップ表面を再生した。フローセル及びレファレンスセルに異なる濃度のサンプルを注入した。検出器フローセルから得られたシグナルからレファレンスセルから得られたシグナルを減じ、1:1Langmuir結合モデルを用いて、得られた結合プロファイルを評価した。濃度依存性の結合プロファイルが得られ、カニクイザルのIL−10についての平均KDは194pMであると計算された。ポジティブコントロールとして、rhIL−10を分析したところ、KDは4.6nMであった。結果を表4に要約する。
【表4】
【0083】
実施例4−インビトロにおける抗IL−10抗体の活性
BT−063の能力を確認するために、末梢血単核細胞(PBMC)におけるIL−6放出の遮断について調べた。PBMCは、リポ多糖類(LPS)で刺激されるとインターロイキン−6(IL−6)を放出する。インターロイキン−10(IL−10)の生理学的活性は、サイトカイン、例えば、IL−6の分泌阻害である。したがって、LPSで刺激された細胞にIL−10を添加すると、IL−6の分泌が阻害され、細胞培養培地中に存在するIL−6が著しく低下する。しかし、細胞培養物にBT−063を添加するとIL−10と結合するので、IL−10が細胞表面上の受容体に結合することができなくなる。IL−10の阻害作用は補償され、IL−6分泌は回復し、培地中にIL−6が放出される。
【0084】
Ficoll勾配によりヒト血液からPBMCを単離した。単離した細胞を1×106細胞/mLで播種し、IL−6を分泌させるためにLPSで刺激したが、IL−10の添加により阻害された。BT−063の添加によりIL−10の阻害作用が中和されたので、IL−6分泌が回復した。添加したBT−063の目的(レファレンス、又は低品質若しくは高品質の対照サンプル)によって、異なる力価濃度のBT−063を用い、濃度依依存的にIL−6を分泌させた。このIL−6の分布は、細胞培養の上清において検出した。
【表5】
【0085】
表5に示す通り、分泌されたIL−6の量は、BT−063の濃度と直接相関している。BT−063の濃度が高くなるほど、より多くの量のIL−6がPBMCから分泌され、上清中に存在する。ポジティブコントロール(IL−10を含まない、刺激されたPBMCをインキュベートしたもの)と比べて、40μg/mLのBT−063と共に細胞をインキュベートした場合はIL−6分泌が約73%回復するが、一方、0.988μg/mLのBT−063と共にインキュベートしても(用量設定の下限)、培地中で検出可能であったIL−6濃度は僅か17.5%であった。
【0086】
実施例5−ヒト全血培養物におけるサイトカイン合成/レベルに対するB−N10及びBT−063の異なる作用
BT−063(変異体hVH26/hVL7)の免疫薬理学的プロファイルを、ヒト全血培養物において実験的に誘導したサイトカイン合成の薬物依存的モジュレーションにより評価した。この方法を用いて、免疫細胞の活性に対するBT−063の直接的及び間接的影響を求めることができる。BT−063活性は、B−N10の作用と比較した。
【0087】
前記アッセイでは、健常ボランティアとSLE患者の両方に由来する全血培養物を用い、これらをBT−063又はB−N10存在下におけるサイトカイン放出量について分析した。全血培養物中の免疫細胞によるサイトカイン放出は、B−N10抗体又はBT−063抗体それぞれとのインキュベーション後の静止状態で試験した。健常ボランティアに由来する白血球と全身性エリテマトーデス(SLE)の罹患患者に由来する細胞を含む。
【0088】
以下に記載する実験に用いた細胞は、ボランティア又はSLE患者を出血させることで採取し、BT−063又はB−N10それぞれとのインキュベーションは、マイクロ培養プレートで2日間行った。
【0089】
これらの実験で測定した主なメディエータは、Th−1−、Th−2−、又は単球/マクロファージ−活性化などに関連するサイトカイン及びケモカインであり、例えば、インターフェロンγ(IFN−γ)、インターロイキン−1β(IL−1β)、IL−12、IL−4、IL−8、腫瘍壊死因子α(TNF−α)などである。培養物上清中の各測定パラメータの濃度は、マルチパラメトリックなビーズを利用した読み出しシステム(マルチアナライトプロファイル(MAP)試験と呼ばれるLuminex(登録商標)に基づく技術、Rules−Based Medicine、RBM(オースティン、テキサス州、USA))。これらのアッセイは、EDI GmbH(ロイトリンゲン、ドイツ連邦共和国)によって行われた。Luminex(登録商標)技術は、ELISAとフローサイトメトリーの組合せに類似した技術であり、アナライト1個当たり100個のビーズをカウントする。したがって、各濃度は、100個の各測定値の平均蛍光強度から逆算される。
【0090】
50μg/mLの抗体濃度でのみ、サイトカイン放出の誘導が観察された。
【0091】
B−N10又はBT−063とインキュベートした健常ドナー及びSLE患者由来の細胞培養物で測定されたサイトカインレベルの絶対値を次の表6及び7にまとめる。
【表6】
【表7】
【0092】
図6A、6B、7A、7B、8A、8B、9A及び9Bは、健常ボランティアとSLE患者の双方に由来する全血培養物において無刺激条件下で炎症促進性サイトカインの制御が異なることを示す。これらの図は、健常ボランティアとSLE患者由来の全血培養物は、BT−063(50μg/mL)と比較してB−N10とインキュベートした後にTNFα、IL−6、IL−1β、及びIFN−γの放出量がより多いことを示している。IL−6は、炎症促進機能に加えて、B細胞のプラズマ細胞への分化のための因子としても働くことは注目に値する。
【0093】
注目すべきことに、これらの結果から、IL−10及び炎症促進性サイトカインのB−N10によるアップレギュレーションの程度が、BT−063とインキュベートされた健常ドナー及びSLEドナーの双方に由来する培養物の場合よりも高く、より安全なプロファイルを示唆していることが分かる。このことは、ヒト化の手順の結果として予想されるものではない。
【0094】
実施例6−X線結晶構造解析
6.1 ヒトIL−10との複合体におけるBT−063 Fabの結晶化
公開されている構造データに従って幾つかのIL−10コンストラクトを設計し(Zdanov et al.,Structure,Vol.3,1995,pp.591)、標準的な手順により大腸菌で異種発現させるためのベクターにクローニングした。クローニングしたコンストラクトを標準的なプロトコールに従って試験的に発現させたところ、約18kDaの予測範囲でバンドが増加したことにより示される通り、IL−10の高発現を示した。
【0095】
後でタンパク質を精製するために、最適な条件下で発現したIL−10タンパク質を様々な量得た。再折り畳みの後、固定化アフィニティークロマトグラフィー、サイズ排除クロマトグラフィー、及びイオン交換クロマトグラフィーによりタンパク質を精製して、クマシー染色したSDS−PAGEにより判定したところ95%を超える均質性を有するタンパク質が得られた。精製タンパク質の収量は、発現培養物1リットル当たり約0.3mgであり、これは結晶化試験に十分であった。
【0096】
BT−063(変異体hVH26/hVL7)のFab断片を、プロテアーゼパパインを用いてインタクトな抗体から切断し、プロテインAにより精製した。次いで、サイズ排除クロマトグラフィーによりFab断片を更に精製した。
【0097】
精製タンパク質をモル過剰のIL−10と混合することによりIL−10:BT−063 Fab複合体を形成し、サイズ排除クロマトグラフィーにより更に精製した。保持体積は、複合体のサイズと一致していた。次いで、結晶化に好適な濃度にタンパク質を濃縮した。
【0098】
IL−10:BT−063 Fab複合体の結晶を共結晶化法により調製した。
【0099】
6.2 データ収集及び処理
結晶を急速冷凍させ、100Kの温度で測定した。低温条件を用いてSWISS LIGHT SOURCE(SLS,Villigen,Switzerland)でBT−063のFab断片と共にIL−10の共結晶からX線回折データを収集した。
【0100】
結晶は、非対称単位における2つの複合体と共に空間群P6に属する。プログラムXDS及びXSCALEを用いてデータを処理した。データ収集統計値を表8に要約する。
【表8】
【0101】
6.3 構造のモデリング及び構造の精密化
分子置換により、構造を決定及び分析するのに必要な位相情報を得た。IL−10及びFab断片の公開モデルをサーチモデルとして用いた。次いで、ソフトウェアパッケージCCP4及びCOOTを用いて標準的なプロトコールに従って、モデルの構築及び構造の精密化を実施した。最終モデルの正確さを交差検定するための尺度であるフリーR因子を計算するために、構造の精密化手順から測定された反射の4.2%を除外した(表9を参照されたい)。
【0102】
プログラムCHEMSKETCHを用いてナノボディのパラメータ化を実施した。対応するライブラリファイルを作製するためにLIBCHECK(CCP4)を用いた。
【0103】
3.0σで一致するFo−Fcマップのピークに水分子を置くことによりCOOTの「Find waters…」アルゴリズムを用いて水モデルを構築し、次いで、REFMAC5で構造を精密化し、COOTの検証ツールを用いて全ての水をチェックした。疑わしい水のリストの基準は、以下の通りであった:B因子:80超、2Fo−Fcマップ:1.2σ未満、最も近接する接触までの距離:2.3Å未満又は3.5Å超。疑わしい水分子及び活性部位(阻害剤までの距離が10Å未満)におけるものを用手的にチェックした。Fo−Fcマップ(−3.0σに一致)におけるネガティブピークに存在していた側鎖の占有率をゼロに設定し、次いで、次の精密化サイクル後にポジティブピークが生じた場合、0.5に設定した。
【0104】
最終モデルのラマチャンドランプロットは、全ての残基の80.8%が最も好ましい領域に、17.9%が更に許容される領域に、残基の0.7%が寛容に許容される領域にあることを示す。残基Val86(A)、His14(B)、Asp86(B)、Ser131(C)、Val56(D)、及びVal56(F)は、ラマチャンドランプロットの許容されない領域にみられる(表9)。これらは、電子密度マップにより確認される、即ち別の知覚可能な立体構造ではモデル化することができなかった。最終構造及び精密化プロセスの統計値を表9に示す。
【表9】
【0105】
6.4 X線構造解析
BT−063 Fab抗体断片が結合するヒトIL−10の複合体構造を3.48Åの解像度で分析し、Fab抗体断片の詳細な結合様式を明らかにする。
【0106】
得られた電子密度は、Fab断片の配向及び立体構造を含むFab断片の明確な結合様式を示す。空間群P6の結晶は、非対称単位における2つの複合体を含有する。
【0107】
Fabとの複合体におけるIL−10の構造を図7に示す。2つのFab断片は、CDRループでIL−10の各ホモダイマーに結合する。
【0108】
以下のIL−10の残基(分子A及びB)は、3.9Åの最大距離内でCDRループの近傍に見出すことができる:Arg27、Lys34、Gln38、Met39、Asp41、Gln42、Asp44、Leu46、Glu50、Leu53、Glu142、Asp144、Ile145、Asn148、Tyr149、Glu151、及びThr155。
【0109】
CDRループの以下の残基は、3.9Åの最大距離内でIL−10の近傍に見出すことができる:Phe27、Ser28、Ala30、Thr31、Tyr32、Trp52、Arg53、Gly54、Ser56、Asn73、Ser74、Tyr100、Gly101、Tyr103(分子C及びE)、Ser32、Asn33、Asn35、Tyr37、Lys55(分子D及びF)。
【0110】
IL−10:IL−10R1受容体複合体の公開されている構造にIL−10:BT−063の複合体構造を重ねることにより示される通り、BT−063の結合部位は、IL−10の表面上のIL−10受容体の結合部位と一致する(図7)。
【0111】
X線解析によって同定されたヒトIL−10と接触しているBT−063のアミノ酸残基を、以下に示すBT−063抗体可変ドメインの直鎖アミノ酸配列において強調する。
【表10】
CDR領域(Honegger and Plueckthun(2001)J.Mol.Biol.,309,657−670)に下線を引く(軽鎖のCDR1、CDR2、及びCDR3は、それぞれ、配列番号71、72、及び73であり;重鎖のCDR1、CDR2、及びCDR3は、それぞれ、配列番号74、75、及び76である)。IL−10と接触する残基を太字で示す。
【0112】
軽鎖内において、接触残基は、CDR1及びCDR2にはみられるが、CDR3にはみられない。3つ全てのCDRの重鎖残基は抗原結合に関与していると考えられる。FR3の2つの残基(Asn73及びSer74)も抗原結合に寄与している。
【0113】
CDR1の最初のSer28及びAla30は、マウスVHの元になった配列(BN−10)の一部であり、選択されるヒトフレームワーク(3−66*04)中には存在しない。両方の位置は、それほど高い頻度で抗原結合に関与しておらず、ヒト化プロセス中に代替アミノ酸として導入された。
【0114】
残基Asn73及びSer74は、マウスにみられ、ヒト抗体フレームワーク配列でも頻繁にみられるが、通常、抗原結合には関与していない(www.bioc.uzh.ch/antibody;Honegger and Plueckthun,2001)。抗原結合に対するこれらの寄与は、予測されていない。
【0115】
BT−063結合に関与するIL−10アミノ酸残基を以下に示す。また、高親和性IL−10受容体鎖(IL−10R1)及び低親和性受容体鎖(IL−10R2)に対する結合に関与しているIL−10の残基も示す。両方の受容体鎖が、IL−10ホモダイマーの結合に関与しており、シグナル伝達に必要である。配列Aでは、BT−063に接触する残基を太字で示す。配列Bでは、IL−10R1に対する接触残基を太字で示し、IL−1R2に対する接触残基をイタリック体で示し、下線を引き、IL−10R1とIL−10R2により共有されている接触残基を太字、イタリック体、及び下線で示す(Pletnev et al 2005)。
【表11】
【0116】
BT−063は、第1のIL−10モノマーのへリックスAの連結ループ配列(Glu42、Asp44、Leu46)を含む残基(Arg27、Lys34、Gln38、Met39、Asp41)、及びへリックスB(Glu50及びLeu53)のN−末端部分と、第2のIL−10モノマーのへリックスF’の残基(Glu142、Asp144、Ile145、Asn148、Tyr149、Glu151、Thr155)とを含むIL−10の不連続なエピトープに結合することが分かる。
【0117】
実施例7−カニクイザルにおけるインビボ単回用量毒性試験
カニクイザルで、BT−063(変異体hVH26/hVL7)の単回静脈内注射後、安全性薬理学的パラメータを含む単回用量毒性試験を実施した。4匹/性別/群になるように動物を4群に分けた(プラセボ、1mg/kg、7mg/kg、及び50mg/kg)。1日目にBT−063を静脈内注射した。5日間後に動物の半数を剖検し、残りの動物は28日目に屠殺した。
【0118】
この実験における低用量レベルである1mg/kgは、ヒトにおける約300μg/kgの用量と等しく、合計ヒト(体重60kg)用量は18mgになる。少数の研究者で開始した実験では、BT−063の元になった抗体B−N100を同用量21日間毎日投与した(Llorente,2000)。この量の抗体は、薬理学的効果及び臨床的有効性を示した。したがって、この結果を踏まえて低用量レベルを選択した。高用量は、出発用量の倍数(50μg)として選択した。中間用量は、低用量と高用量との幾何平均である。
【0119】
試験中、生理学的パラメータ又は組織病理学的パラメータにおいて毒物学的に有意又は明らかな変化はみられなかった。更に、臨床化学パラメータの変化は、毒物学的有意性が僅かである又は存在しないとみなされた。
【0120】
実施例8−健常ボランティアにおけるインビボ単回用量毒性試験
健常ボランティアにおいて漸増用量の抗体を用いて、BT−063(変異体hVH26/hVL7)の安全性及び忍容性、並びにBT−063の投与の効果をモニターするために実験を行った。23人のボランティアを8用量群に分け、BT−063を単回静脈内投与した。用量群は、以下の通りであった:0.175mg、0.75mg、3.5mg、7mg、15mg、30mg、60mg、及び100mg。100mg用量群のボランティアが2人であったことを除いて、1群当たりのボランティアは3人であった。
【0121】
各用量を0.9%塩化ナトリウム注射液で合計体積20mLに希釈した。2時間に亘って用量を単回連続静脈内注射した。注入後85日間に亘ってボランティアを評価し、この期間中の複数の時点で血液を採取した。
【0122】
サイトカイン
採取した血漿から、BT−63の注入前後のサイトカインIFNγ、IL−1β、IL−2、IL−4、IL−5、IL−6、IL−8、IL−10、及びTNFαの濃度を評価した。
【0123】
表10は、投与前の値とスクリーニングの値とから算出したIL−6、IL−8、及びIL−10の正常値上限を示す。
【表12】
【0124】
サイトカインの測定値から得られた更なる結果は、図12及び13に示す。
【0125】
注入後24時間以内では、IL−6及びIL−8が非用量依存的に一過的に増加したが、3日間後には投与前のレベルに戻った。この作用は、抗体ではなく注入事象に関連すると考えられる。その理由は、用量との相関がなく、また最低用量で既に前記事象が生じたためである。
【0126】
驚くべきことに、IL−10が重要な調節性サイトカインであるにも関わらず、図12に示される通り、IFNγ、IL−1β、IL−2、IL−4、IL−5、及びTNFαの濃度増加は検出されなかった。また、IL−4、IL−5、IFN−γ、IL−2の濃度が増加しないことにより、BT−063の投与がヘルパーT細胞を活性化させないことが確認される。更に、体温も炎症促進性サイトカインの大量放出の指標であるので、処理されたボランティアにおいてこのパラメータも測定した。しかし、投与後、体温の上昇は検出されなかった。
【0127】
図13に示す通り、IL−10の血漿濃度のみが、BT−063の投与により影響を受け、BT−063の用量が増加するにつれて検出される血漿IL−10も増加する。用いたアッセイは、遊離IL−10及びBT−063に結合しているIL−10の両方を検出することに留意すべきである。増加の理由としては、以下の2つの可能性がある:(1)IL−10の半減期の延長、IL−10のBT−063への結合がIL−10の内部移行を妨げ、同時に、血液からIL−10を排出させなくする(通常、IL−10は速やかにターンオーバーし、分泌されたIL−10の半減期は約2.3時間〜3.5時間である(Huhn et al.,Blood(1996)Jan 15:87(2):699−705);及び/又は(2)ネガティブフィードバックループの誘導−BT−063は、IL−10のその受容体への結合をブロックするので、IL−10が細胞に取り込まれることができなくなり、したがって、B細胞がより多くのIL−10を産生するようになる。BT−063及びマウスIL−10Rノックアウト細胞を用いた全血培養実験から得られたインビトロにおけるデータにより確認されているので(Mahnke et al.、未公開データ)、仮説(2)の可能性の方が高いと考えられる。
【0128】
薬物動態
薬物動態データは、BT−063のCmax、AUC、及び半減期が、予測される理論値の範囲内であることを示した。BT−063の終末半減期は、15日間〜30日間である。30mg以上の用量を投与した後、85日間後も依然としてBT−063を血漿中で検出することが可能である。
【0129】
他のサイトカイン中和抗体(例えば、アダリムマブ、ヒト化抗TNF−α)ではヒト抗ヒト抗体(HAHA)応答がみられるという事実にも関わらず、処理したボランティアにおいてはHAHAがみられなかった。
【0130】
BT−063投与後、IL−10の検出量は増加したが、この実験が、更に多い用量のBT−063の安全性及び許容性を示したことに留意すべきであり、したがって、過剰のIL−10の効果を相殺するために十分な用量のBT−063をSLE患者に安全に投与できる(具体的には、炎症促進性サイトカインレベルの許容できない上昇がない)と結論付けることができる。
【特許請求の範囲】
【請求項1】
炎症促進性サイトカインレベルの許容できない上昇を伴うことなしに被験体に投与することができることを特徴とする、インターロイキン10(IL−10)に結合可能なヒト化抗体、キメラ抗体又はこれらの断片。
【請求項2】
2℃を超える体温の上昇を伴うことなしに被験体に投与することができる請求項1に記載のヒト化抗体、キメラ抗体又はこれらの断片。
【請求項3】
炎症促進性サイトカインの上昇が、正常値上限(ULN)の500%を超えることなしに被験体に投与することができ、前記炎症促進性サイトカインがTNF−α、IFN−γ及びIL−1βから選択される請求項1又は2に記載のヒト化抗体、キメラ抗体又はこれらの断片。
【請求項4】
炎症促進性サイトカインの上昇が、正常値上限(ULN)の300%を超えることなしに被験体に投与することができる請求項3に記載のヒト化抗体、キメラ抗体又はこれらの断片。
【請求項5】
被験体において、IL−1受容体アンタゴニスト(IL−1ra)を誘導することができる請求項1から4のいずれかに記載のヒト化抗体、キメラ抗体又はこれらの断片。
【請求項6】
末梢血単核細胞(PBMC)にインビトロで接触させたときに、前記細胞から放出される炎症促進性サイトカインのレベルにおいて500%を超える上昇を引き起こさない請求項1に記載のヒト化抗体、キメラ抗体又はこれらの断片。
【請求項7】
炎症促進性サイトカインがTNF−α、IFN−γ及びIL−1βのうちの少なくとも1つである請求項1から6のいずれかに記載のヒト化抗体、キメラ抗体又はこれらの断片。
【請求項8】
マウスB−N10抗体に由来する請求項1から7のいずれかに記載のヒト化抗体、キメラ抗体又はこれらの断片。
【請求項9】
マウス抗体B−N10の可変軽鎖のCDR1、CDR2、及びCDR3のアミノ酸配列と少なくとも80%同一であるアミノ酸配列を含む、
マウス抗体B−N10の可変重鎖のCDR1、CDR2、及びCDR3のアミノ酸配列と少なくとも80%同一であるアミノ酸配列を含む、又は
マウス抗体B−N10の可変軽鎖のCDR1、CDR2、及びCDR3のアミノ酸配列と少なくとも80%同一であるアミノ酸配列を含み、且つマウス抗体B−N10の可変重鎖のCDR1、CDR2、及びCDR3のアミノ酸配列と少なくとも80%同一であるアミノ酸配列を含む請求項1から8のいずれかに記載のヒト化抗体、キメラ抗体又はこれらの断片。
【請求項10】
マウス抗体B−N10の可変軽鎖及び可変重鎖のCDR1、CDR2、及びCDR3のアミノ酸配列を含む請求項9に記載のヒト化抗体、キメラ抗体又はこれらの断片。
【請求項11】
マウス抗体B−N10の可変軽鎖のCDR1、CDR2、及びCDR3のアミノ酸配列、
マウス抗体B−N10の可変重鎖のCDR1、CDR2、及びCDR3のアミノ酸配列、又は
マウス抗体B−N10の可変軽鎖のCDR1、CDR2、及びCDR3のアミノ酸配列とマウス抗体B−N10の可変重鎖のCDR1、CDR2、及びCDR3のアミノ酸配列とを含み、
これら配列中に、親和性及び特異性の少なくともいずれかを実質的に変化させることのない変異が任意で存在する請求項1から7のいずれかに記載のヒト化抗体、キメラ抗体又はこれらの断片。
【請求項12】
マウス抗体B−N10の可変軽鎖のアミノ酸配列、及びマウス抗体B−N10の可変重鎖のアミノ酸配列の少なくともいずれかを含む請求項1から11のいずれかに記載のヒト化抗体、キメラ抗体又はこれらの断片。
【請求項13】
請求項1から12のいずれかに記載の抗体又はその断片をコードすることを特徴とする核酸。
【請求項14】
請求項13に記載の核酸を含むことを特徴とするベクター。
【請求項15】
請求項13に記載の核酸又は請求項14に記載のベクターを含むことを特徴とするホスト細胞。
【請求項16】
請求項1から12のいずれかに記載の抗体又はその断片を産生する方法であって、前記抗体又はその断片を発現させる条件下で培養培地中にて請求項15に記載のホスト細胞を培養し、前記培養培地から前記抗体又はその断片を分離する工程を含むことを特徴とする方法。
【請求項17】
請求項1から12のいずれかに記載の抗体又はその断片を含み、且つ薬学的に許容し得る担体を更に含むことを特徴とする医薬組成物。
【請求項18】
被験体における、高レベル又は高活性のIL−10により媒介される病状を治療又は予防する方法であって、治療上有効な量の請求項1から12のいずれかに記載の抗体又はその断片を前記被験体に投与する工程を含むことを特徴とする方法。
【請求項19】
被験体における、高レベル又は高活性のIL−10により媒介される病状を治療又は予防する方法であって、治療上有効な量の請求項1から12のいずれかに記載の抗体又はその断片を前記被験体に投与する工程を含み、前記被験体がコルチコステロイドによる治療を同時にも別々にも受けていないことを特徴とする方法。
【請求項20】
病状が、全身性エリテマトーデス(SLE)である請求項18又は19に記載の被験体における病状を治療又は予防する方法。
【請求項21】
医療において使用するための請求項1から12のいずれかに記載の抗体又はその断片。
【請求項22】
患者におけるSLEの治療において使用するための請求項1から12のいずれかに記載の抗体又はその断片。
【請求項23】
患者がコルチコステロイドによる治療を同時にも別々にも受けていない請求項22に記載の使用のための抗体又はその断片。
【請求項24】
患者におけるSLEを治療する医薬の製造における請求項1から12のいずれかに記載の抗体又はその断片の使用。
【請求項25】
患者がコルチコステロイドによる治療を同時にも別々にも受けていない請求項24に記載の抗体又はその断片の使用。
【請求項26】
標識と請求項1から12のいずれかに記載の抗体又はその断片とを含むことを特徴とする標識されている抗体又はこれらの断片。
【請求項27】
サンプル中のIL−10を中和するためのインビトロにおける方法であって、前記サンプルと請求項1から12のいずれかに記載の抗体又はその断片とを接触させて、前記抗体又はその断片をIL−10に結合させる工程を含むことを特徴とする方法。
【請求項28】
サンプル中のIL−10の存在を検出するためのインビトロにおける方法であって、前記サンプルと、請求項1又は2に記載の標識されていない抗体若しくはその断片又は請求項26に記載の標識されている抗体若しくはその断片とを接触させる工程と、前記サンプルに結合していない抗体又は抗体断片を除去するために洗浄する工程と、前記サンプル中の前記抗体若しくは前記断片又は前記標識の存在を検出する工程とを含むことを特徴とする方法。
【請求項1】
炎症促進性サイトカインレベルの許容できない上昇を伴うことなしに被験体に投与することができることを特徴とする、インターロイキン10(IL−10)に結合可能なヒト化抗体、キメラ抗体又はこれらの断片。
【請求項2】
2℃を超える体温の上昇を伴うことなしに被験体に投与することができる請求項1に記載のヒト化抗体、キメラ抗体又はこれらの断片。
【請求項3】
炎症促進性サイトカインの上昇が、正常値上限(ULN)の500%を超えることなしに被験体に投与することができ、前記炎症促進性サイトカインがTNF−α、IFN−γ及びIL−1βから選択される請求項1又は2に記載のヒト化抗体、キメラ抗体又はこれらの断片。
【請求項4】
炎症促進性サイトカインの上昇が、正常値上限(ULN)の300%を超えることなしに被験体に投与することができる請求項3に記載のヒト化抗体、キメラ抗体又はこれらの断片。
【請求項5】
被験体において、IL−1受容体アンタゴニスト(IL−1ra)を誘導することができる請求項1から4のいずれかに記載のヒト化抗体、キメラ抗体又はこれらの断片。
【請求項6】
末梢血単核細胞(PBMC)にインビトロで接触させたときに、前記細胞から放出される炎症促進性サイトカインのレベルにおいて500%を超える上昇を引き起こさない請求項1に記載のヒト化抗体、キメラ抗体又はこれらの断片。
【請求項7】
炎症促進性サイトカインがTNF−α、IFN−γ及びIL−1βのうちの少なくとも1つである請求項1から6のいずれかに記載のヒト化抗体、キメラ抗体又はこれらの断片。
【請求項8】
マウスB−N10抗体に由来する請求項1から7のいずれかに記載のヒト化抗体、キメラ抗体又はこれらの断片。
【請求項9】
マウス抗体B−N10の可変軽鎖のCDR1、CDR2、及びCDR3のアミノ酸配列と少なくとも80%同一であるアミノ酸配列を含む、
マウス抗体B−N10の可変重鎖のCDR1、CDR2、及びCDR3のアミノ酸配列と少なくとも80%同一であるアミノ酸配列を含む、又は
マウス抗体B−N10の可変軽鎖のCDR1、CDR2、及びCDR3のアミノ酸配列と少なくとも80%同一であるアミノ酸配列を含み、且つマウス抗体B−N10の可変重鎖のCDR1、CDR2、及びCDR3のアミノ酸配列と少なくとも80%同一であるアミノ酸配列を含む請求項1から8のいずれかに記載のヒト化抗体、キメラ抗体又はこれらの断片。
【請求項10】
マウス抗体B−N10の可変軽鎖及び可変重鎖のCDR1、CDR2、及びCDR3のアミノ酸配列を含む請求項9に記載のヒト化抗体、キメラ抗体又はこれらの断片。
【請求項11】
マウス抗体B−N10の可変軽鎖のCDR1、CDR2、及びCDR3のアミノ酸配列、
マウス抗体B−N10の可変重鎖のCDR1、CDR2、及びCDR3のアミノ酸配列、又は
マウス抗体B−N10の可変軽鎖のCDR1、CDR2、及びCDR3のアミノ酸配列とマウス抗体B−N10の可変重鎖のCDR1、CDR2、及びCDR3のアミノ酸配列とを含み、
これら配列中に、親和性及び特異性の少なくともいずれかを実質的に変化させることのない変異が任意で存在する請求項1から7のいずれかに記載のヒト化抗体、キメラ抗体又はこれらの断片。
【請求項12】
マウス抗体B−N10の可変軽鎖のアミノ酸配列、及びマウス抗体B−N10の可変重鎖のアミノ酸配列の少なくともいずれかを含む請求項1から11のいずれかに記載のヒト化抗体、キメラ抗体又はこれらの断片。
【請求項13】
請求項1から12のいずれかに記載の抗体又はその断片をコードすることを特徴とする核酸。
【請求項14】
請求項13に記載の核酸を含むことを特徴とするベクター。
【請求項15】
請求項13に記載の核酸又は請求項14に記載のベクターを含むことを特徴とするホスト細胞。
【請求項16】
請求項1から12のいずれかに記載の抗体又はその断片を産生する方法であって、前記抗体又はその断片を発現させる条件下で培養培地中にて請求項15に記載のホスト細胞を培養し、前記培養培地から前記抗体又はその断片を分離する工程を含むことを特徴とする方法。
【請求項17】
請求項1から12のいずれかに記載の抗体又はその断片を含み、且つ薬学的に許容し得る担体を更に含むことを特徴とする医薬組成物。
【請求項18】
被験体における、高レベル又は高活性のIL−10により媒介される病状を治療又は予防する方法であって、治療上有効な量の請求項1から12のいずれかに記載の抗体又はその断片を前記被験体に投与する工程を含むことを特徴とする方法。
【請求項19】
被験体における、高レベル又は高活性のIL−10により媒介される病状を治療又は予防する方法であって、治療上有効な量の請求項1から12のいずれかに記載の抗体又はその断片を前記被験体に投与する工程を含み、前記被験体がコルチコステロイドによる治療を同時にも別々にも受けていないことを特徴とする方法。
【請求項20】
病状が、全身性エリテマトーデス(SLE)である請求項18又は19に記載の被験体における病状を治療又は予防する方法。
【請求項21】
医療において使用するための請求項1から12のいずれかに記載の抗体又はその断片。
【請求項22】
患者におけるSLEの治療において使用するための請求項1から12のいずれかに記載の抗体又はその断片。
【請求項23】
患者がコルチコステロイドによる治療を同時にも別々にも受けていない請求項22に記載の使用のための抗体又はその断片。
【請求項24】
患者におけるSLEを治療する医薬の製造における請求項1から12のいずれかに記載の抗体又はその断片の使用。
【請求項25】
患者がコルチコステロイドによる治療を同時にも別々にも受けていない請求項24に記載の抗体又はその断片の使用。
【請求項26】
標識と請求項1から12のいずれかに記載の抗体又はその断片とを含むことを特徴とする標識されている抗体又はこれらの断片。
【請求項27】
サンプル中のIL−10を中和するためのインビトロにおける方法であって、前記サンプルと請求項1から12のいずれかに記載の抗体又はその断片とを接触させて、前記抗体又はその断片をIL−10に結合させる工程を含むことを特徴とする方法。
【請求項28】
サンプル中のIL−10の存在を検出するためのインビトロにおける方法であって、前記サンプルと、請求項1又は2に記載の標識されていない抗体若しくはその断片又は請求項26に記載の標識されている抗体若しくはその断片とを接触させる工程と、前記サンプルに結合していない抗体又は抗体断片を除去するために洗浄する工程と、前記サンプル中の前記抗体若しくは前記断片又は前記標識の存在を検出する工程とを含むことを特徴とする方法。
【図3B】


【図6A】
【図6B】
【図7A】
【図7B】
【図8A】
【図8B】
【図9A】
【図9B】
【図1A】
【図1B】
【図2A】

【図2B】

【図3A】

【図3C】

【図4】
【図5】
【図10】
【図11】

【図12】
【図13】
【図6A】
【図6B】
【図7A】
【図7B】
【図8A】
【図8B】
【図9A】
【図9B】
【図1A】
【図1B】
【図2A】
【図2B】
【図3A】
【図3C】
【図4】
【図5】
【図10】
【図11】
【図12】
【図13】
【公表番号】特表2013−511994(P2013−511994A)
【公表日】平成25年4月11日(2013.4.11)
【国際特許分類】
【出願番号】特願2012−541466(P2012−541466)
【出願日】平成22年11月30日(2010.11.30)
【国際出願番号】PCT/EP2010/068562
【国際公開番号】WO2011/064398
【国際公開日】平成23年6月3日(2011.6.3)
【出願人】(390035378)バイオテスト・アクチエンゲゼルシヤフト (13)
【Fターム(参考)】
【公表日】平成25年4月11日(2013.4.11)
【国際特許分類】
【出願日】平成22年11月30日(2010.11.30)
【国際出願番号】PCT/EP2010/068562
【国際公開番号】WO2011/064398
【国際公開日】平成23年6月3日(2011.6.3)
【出願人】(390035378)バイオテスト・アクチエンゲゼルシヤフト (13)
【Fターム(参考)】
[ Back to top ]