
共役ジエンの製造方法
【課題】n−ブテン等のモノオレフィンの接触酸化脱水素反応によりブタジエン等の共役ジエンを製造する方法において、反応中にコークのような炭素分が触媒上に蓄積するのを防止し、コーキングの進行を緩和して、安定的に運転が継続できる工業的に有利なブタジエンの製造方法を提供する。
【解決手段】炭素原子数4以上のモノオレフィンを含む原料ガスと分子状酸素含有ガスとを混合した混合ガスを、触媒を有する反応器に供給し、酸化脱水素反応により対応する共役ジエンを生成し、該共役ジエンを含む生成ガスを得る共役ジエンの製造方法において、下記式(1)で示される該反応器中の触媒の破砕率が0.0重量%以上40.0重量%以下であることを特徴とする共役ジエンの製造方法。
破砕率(重量%)={(反応器中の破砕触媒の重量)/(反応器中の全触媒重量)}×100 …(1)
【解決手段】炭素原子数4以上のモノオレフィンを含む原料ガスと分子状酸素含有ガスとを混合した混合ガスを、触媒を有する反応器に供給し、酸化脱水素反応により対応する共役ジエンを生成し、該共役ジエンを含む生成ガスを得る共役ジエンの製造方法において、下記式(1)で示される該反応器中の触媒の破砕率が0.0重量%以上40.0重量%以下であることを特徴とする共役ジエンの製造方法。
破砕率(重量%)={(反応器中の破砕触媒の重量)/(反応器中の全触媒重量)}×100 …(1)
【発明の詳細な説明】
【技術分野】
【0001】
本発明は共役ジエンの製造方法に係り、特にn−ブテン等の炭素原子数4以上のモノオレフィンの接触酸化脱水素反応でブタジエン等の共役ジエンを製造する方法に関する。
【背景技術】
【0002】
n−ブテン等のモノオレフィンを触媒の存在下に酸化脱水素反応させてブタジエン等の共役ジエンを製造する方法は、従来公知である。
この反応は例えば以下の反応式に従って進行し、水が副生する。
C4H8+1/2O2→C4H6+H2O
n−ブテンの接触酸化脱水素反応によるブタジエンの製造は、工業的にはナフサ分解で副生するC4留分(C4炭化水素混合物。以下、「BB」と称す場合がある。)からのブタジエンの抽出分離プロセスにおいて、抽出蒸留塔でブタジエンを分離して得られた、1−ブテンの他、2−ブテン、ブタン等を含む混合物(以下、この混合物を「BBSS]と称す場合がある。)中に含まれるブテンからブタジエンを製造する方法が提案されている。
【0003】
具体的な方法としては、炭素原子数4以上のモノオレフィンを含む原料ガスと酸素ガスとの酸化脱水素反応により生成した共役ジエンを含む生成ガスを得て、その生成ガスを冷却する工程、冷却された生成ガスを脱水する工程、及び脱水された生成ガスを溶媒と接触させて共役ジエンを回収する工程を有する方法(特開2010−90083号公報)や、アセチレン系炭化水素濃度が0.016vol%以下に抑制された生成ガスを得ることで、アセチレン類の分離のための抽出蒸留の負荷を軽減する方法(特開2010−90082号公報)などがある。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特開2010−90083号公報
【特許文献2】特開2010−90082号公報
【発明の概要】
【発明が解決しようとする課題】
【0005】
上記特許文献1〜2には、触媒上に炭素物質が析出するコーキングの問題については一切記載されていないが、本発明者等の検討によれば、触媒の存在下、酸化脱水素反応により、ブテンからブタジエンを製造するに際に、原料ガス(例えば、BB又はBBSS)中に含まれるイソブテンなどの分岐型モノオレフィンや直鎖型n−ブテンが酸化脱水素反応で高沸点の炭素成分を副生し、触媒上でのコーキングが進行し、触媒活性が低下する、更にはコーキングの進行に伴い、反応器の差圧が上昇し、長期の運転に支障をきたす問題があることが判明した。触媒上にコーキングが進行すると運転を止めて、デコーキングなどにより高沸点の炭素成分を除去しなければならない。この運転停止操作ならびにデコーキングはブタジエン製造における生産性の低下原因となる。
【0006】
本発明は上記課題に鑑みてなされたものであって、n−ブテン等のモノオレフィンの接触酸化脱水素反応によりブタジエン等の共役ジエンを製造する方法において、反応中にコークのような炭素分が触媒上に蓄積するのを防止し、コーキングの進行を緩和して、安定的に運転が継続できる工業的に有利なブタジエンの製造方法を提供することを目的とする。
【課題を解決するための手段】
【0007】
本発明者らは、上記課題を解決すべく鋭意検討した結果、炭素原子数4以上のモノオレフィンを含む原料ガスと分子状酸素含有ガスとを混合した混合ガスを反応器に供給し、触媒の存在下、酸化脱水素反応により生成された対応する共役ジエンを含むガスを得る共役ジエンの製造方法において、反応器内の触媒が何らかの原因で割れ、更には粉砕が起こり、それらが反応器内で局所的に凝集すると高沸点の炭素成分を副生する反応が起きやすくなるという考えの下、反応器内に存在する全触媒量に対する割れた触媒や粉砕した触媒の量(重量比)をある特定の範囲で反応を行うことで、プラントの安定運転を阻害するコーキング速度を低減でき、且つ、より安全に運転ができ、更に高い収率で安定的にブタジエンの製造を行うことができることを見出した。
【0008】
本発明はこのような知見に基いて達成されたものであり、以下[1]〜[6]を要旨とする。
[1]炭素原子数4以上のモノオレフィンを含む原料ガスと分子状酸素含有ガスとを混合した混合ガスを、触媒を有する反応器に供給し、酸化脱水素反応により対応する共役ジエンを生成し、該共役ジエンを含む生成ガスを得る共役ジエンの製造方法において、下記式(1)で示される該反応器中の触媒の破砕率が0.0重量%以上40.0重量%以下であることを特徴とする共役ジエンの製造方法。
【0009】
破砕率(重量%)={(反応器中の破砕触媒の重量)/(反応器中の全触媒重量)}×100 …(1)
[2]前記反応器が前記触媒と反応性の無い固形物を更に有することを特徴とする[1]に記載の共役ジエンの製造方法。
[3]前記反応器内の全触媒量に対する前記混合ガスの流量の比(体積比)が500〜5000h-1であることを特徴とする[1]又は[2]に記載の共役ジエンの製造方法。
【0010】
[4]前記触媒が、少なくともモリブデン及びビスマスを含むことを特徴とする[1]〜[3]のいずれか1項に記載の共役ジエンの製造方法。
[5]前記触媒が、下記一般式(2)で表される複合酸化物触媒であることを特徴とする[1]〜[4]のいずれか1項に記載の共役ジエンの製造方法。
MoaBibCocNidFeeXfYgZhSiiOj (2)
(式中、Xはマグネシウム(Mg)、カルシウム(Ca)、亜鉛(Zn)、セリウム(Ce)及びサマリウム(Sm)からなる群から選ばれる少なくとも1種の元素であり、Yはナトリウム(Na)、カリウム(K)、ルビジウム(Rb)、セシウム(Cs)及びタリウム(Tl)からなる群から選ばれる少なくとも1種の元素であり、Zはホウ素(B)、リン(P)、砒素(As)及びタングステン(W)からなる群から選ばれる少なくとも1種の元素である。また、a〜jはそれぞれの元素の原子比を表し、a=12のとき、b=0.1〜7、c=0〜10、d=0〜10(但しc+d=1〜10)、e=0.05〜3、f=0〜2、g=0.04〜2、h=0〜3、i=0〜48の範囲にあり、またjは他の元素の酸化状態を満足させる数値である。)
[6]前記原料ガスが、エチレンの2量化により得られる1−ブテン、シス−2−ブテン、トランス−2−ブテン若しくはこれらの混合物を含有するガス、n−ブタンの脱水素若しくは酸化脱水素反応により生成するブテン留分、又は重油留分を流動接触分解する際に得られる炭素原子数が4の炭化水素を多く含むガスであることを特徴とする[1]〜[5]のいずれか1項に記載の共役ジエンの製造方法。
【発明の効果】
【0011】
本発明によれば、反応器内の触媒形状を保持することで、継続的にプラントの安定運転が可能となる。
【図面の簡単な説明】
【0012】
【図1】本発明の共役ジエンの製造方法の実施の形態を示す系統図である。
【発明を実施するための形態】
【0013】
以下に本発明の共役ジエンの製造方法の実施の形態を詳細に説明するが、以下に記載する説明は、本発明の実施態様の一例(代表例)であり、本発明はこれらの内容に限定されない。
本発明では、炭素原子数4以上のモノオレフィンを含む原料ガスと分子状酸素含有ガスとを触媒層を有する反応器に供給し、酸化脱水素反応により対応する共役ジエンを製造する。
【0014】
<炭素原子数4以上のモノオレフィンを含む原料ガス>
本発明の原料ガスは炭素原子数4以上のモノオレフィンを含むが、炭素原子数4以上のモノオレフィンとしては、ブテン(1−ブテン及び/又は2−ブテン等のn−ブテン、イソブテン)、ペンテン、メチルブテン、ジメチルブテン等の炭素原子数4以上、好ましくは炭素原子数4〜6のモノオレフィンが挙げられ、接触酸化脱水素反応による対応する共役ジエンの製造に有効に適用することができる。この中でも、n−ブテン(1−ブテン及び/又は2−ブテン等のn−ブテン)からのブタジエンの製造に最も好適に用いられる。又、炭素原子数4以上のモノオレフィンを含む原料ガスとしては、単離した炭素原子数4以上のモノオレフィンそのものを使用する必要はなく、必要に応じて任意の混合物の形で用いることができる。例えばブタジエンを得ようとする場合には高純度のn−ブテン(1−ブテン及び/又は2−ブテン)を原料ガスとすることもできるが、前述のナフサ分解で副生するC4留分(BB)からブタジエン及びi−ブテン(イソブテン)を分離して得られるn−ブテン(1−ブテン及び/又は2−ブテン)を主成分とする留分(BBSS)やn−ブタンの脱水素又は酸化脱水素反応により生成するブテン留分を使用することもできる。また、エチレンの2量化により得られる高純度の1−ブテン、シス−2−ブテン、トランス−2−ブテン又はこれらの混合物を含有するガスを原料ガスとして使用しても差し支えない。尚、このエチレンはエタン脱水素、エタノール脱水、又はナフサ分解などの方法で得られるエチレンを使用することができる。更に、石油精製プラントなどで原油を蒸留した際に得られる重油留分を、流動層状態で粉末状の固体触媒を使って分解し、低沸点の炭化水素に変換する流動接触分解 (Fluid Catalytic Cracking)から得られる炭素原子
数4の炭化水素類を多く含むガス(以下、FCC−C4と略記することがある)をそのまま原料ガスとする、又は、FCC−C4からリンや砒素などの不純物を除去したものを原料ガスとして使用しても差し支えない。なお、ここでいう、主成分とは、原料ガスに対して、通常40体積%以上、好ましくは60体積%以上、より好ましくは75体積%以上、特に好ましくは99体積%以上を示す。
【0015】
また、本発明の原料ガス中には、本発明の効果を阻害しない範囲で、任意の不純物を含んでいても良い。n−ブテン(1−ブテン及び2−ブテン)からブタジエンを製造する場合、含んでいても良い不純物として、具体的には、イソブテンなどの分岐型モノオレフィン;プロパン、n−ブタン、i−ブタン、ペンタンなどの飽和炭化水素;プロピレン、ペンテンなどのオレフィン;1,2−ブタジエンなどのジエン;メチルアセチレン、ビニルアセチレン、エチルアセチレンなどのアセチレン類等が挙げられる。この不純物の量は、通常40%以下、好ましくは20%以下、より好ましくは10%以下、特に好ましくは1%以下である。この量が多すぎると、主原料である1−ブテンや2−ブテンの濃度が下がって反応が遅くなったり、目的生成物であるブタジエンの収率が低下する傾向にある。また、本発明では、原料ガス中の炭素原子数4以上の直鎖型モノオレフィンの濃度は、特に限定されないが、通常は、70.00〜99.99vol%である。
【0016】
<酸化脱水素反応触媒>
次に、本発明で好適に用いられる酸化脱水素反応触媒について説明する。本発明で用いる酸化脱水素触媒は、少なくともモリブデン、ビスマス及びコバルトを含有する複合酸化物触媒であることが好ましい。そして、この中でも、下記一般式(2)で表される複合酸化物触媒であることがより好ましい。
【0017】
MoaBibCocNidFeeXfYgZhSiiOj (2)
なお、式中、Xはマグネシウム(Mg)、カルシウム(Ca)、亜鉛(Zn)、セリウム(Ce)及びサマリウム(Sm)からなる群から選ばれる少なくとも1種の元素である。Yはナトリウム(Na)、カリウム(K)、ルビジウム(Rb)、セシウム(Cs)及びタリウム(Tl)からなる群から選ばれる少なくとも1種の元素である。Zはホウ素(B)、リン(P)、砒素(As)及びタングステン(W)からなる群から選ばれる少なくとも1種の元素である。
【0018】
さらに、a〜jはそれぞれの元素の原子比を表し、a=12のとき、b=0.1〜7、c=0〜10、d=0〜10(但しc+d=1〜10)、e=0.05〜3、f=0〜2、g=0.04〜2、h=0〜3、i=0〜48の範囲にあり、またjは他の元素の酸化状態を満足させる数値である。
また、この複合酸化物触媒は、この複合酸化物触媒を構成する各成分元素の供給源化合物を水系内で一体化して加熱する工程を経て製造する方法がよい。例えば、前記各成分元素の供給源化合物の全部を水系内で一体化して加熱してもよい。
【0019】
その中でも、モリブデン化合物、鉄化合物、ニッケル化合物及びコバルト化合物よりなる群から選ばれる少なくとも1種と、必要に応じてシリカとを含む原料化合物の水溶液若しくは水分散液、又はこれを乾燥して得た乾燥物を加熱処理して触媒前駆体を製造する前工程と、この触媒前駆体、モリブデン化合物及びビスマス化合物を水性溶媒とともに一体化し、乾燥、焼成する後工程とを有する方法で製造するのが好ましい。この方法を用いると、得られた複合酸化物触媒は、高い触媒活性を発揮するので、高収率でブタジエン等の共役ジエンを製造することができ、アルデヒド類含有量の少ない反応生成ガスを得ることができる。なお、水性溶媒とは、水、又はメタノール、エタノール等の水と相溶性を有する有機溶媒、又はこれらの混合物をいう。
【0020】
次に、本発明に好適な複合酸化物触媒の製造方法について説明する。
まず、この複合酸化物触媒の製造方法においては、前記前工程で用いられるモリブデンが、モリブデンの全原子比(a)の内の一部の原子比(a1)相当のモリブデンであり、前記後工程で用いられるモリブデンが、モリブデンの全原子比(a)からa1を差し引いた残りの原子比(a2)相当のモリブデンであることが好ましい。そして、前記a1が0.5<a1/(c+d+e)<3を満足する値であることが好ましく、さらに、前記a2が0<a2/b<8を満足する値であることが好ましい。
【0021】
前記成分元素の供給源化合物としては、成分元素の酸化物、硝酸塩、炭酸塩、アンモニウム塩、水酸化物、カルボン酸塩、カルボン酸アンモニウム塩、ハロゲン化アンモニウム塩、水素酸、アセチルアセトナート、アルコキシド等が挙げられ、その具体例としては、下記のようなものが挙げられる。
Moの供給源化合物としては、パラモリブデン酸アンモニウム、三酸化モリブデン、モリブデン酸、リンモリブデン酸アンモニウム、リンモリブデン酸等が挙げられる。
【0022】
Feの供給源化合物としては、硝酸第二鉄、硫酸第二鉄、塩化第二鉄、酢酸第二鉄等が挙げられる。
Coの供給源化合物としては、硝酸コバルト、硫酸コバルト、塩化コバルト、炭酸コバルト、酢酸コバルト等が挙げられる。
Niの供給源化合物としては、硝酸ニッケル、硫酸ニッケル、塩化ニッケル、炭酸ニッケル、酢酸ニッケル等が挙げられる。
【0023】
Siの供給源化合物としては、シリカ、粒状シリカ、コロイダルシリカ、ヒュームドシリカ等が挙げられる。
Biの供給源化合物としては、塩化ビスマス、硝酸ビスマス、酸化ビスマス、次炭酸ビスマス等が挙げられる。また、X成分(Mg,Ca,Zn,Ce,Smの1種又は2種以上)やY成分(Na,K,Rb,Cs,Tlの1種又は2種以上)を固溶させた、BiとX成分やY成分との複合炭酸塩化合物として供給することもできる。
【0024】
例えば、Y成分としてNaを用いた場合、BiとNaとの複合炭酸塩化合物は、炭酸ナトリウム又は重炭酸ナトリウムの水溶液等に、硝酸ビスマス等の水溶性ビスマス化合物の水溶液を滴下混合し、得られた沈殿を水洗、乾燥することによって製造することができる。
また、BiとX成分との複合炭酸塩化合物は、炭酸アンモニウム又は重炭酸アンモニウムの水溶液等に、硝酸ビスマス及びX成分の硝酸塩等の水溶性化合物からなる水溶液を滴下混合し、得られた沈殿を水洗、乾燥することによって製造することができる。
【0025】
前記炭酸アンモニウム又は重炭酸アンモニウムの代わりに、炭酸ナトリウム又は重炭酸ナトリウムを用いると、Bi、Na及びX成分との複合炭酸塩化合物を製造することができる。
その他の成分元素の供給源化合物としては、下記のものが挙げられる。
Kの供給源化合物としては、硝酸カリウム、硫酸カリウム、塩化カリウム、炭酸カリウム、酢酸カリウム等を挙げることができる。
【0026】
Rbの供給源化合物としては、硝酸ルビジウム、硫酸ルビジウム、塩化ルビジウム、炭酸ルビジウム、酢酸ルビジウム等を挙げることができる。
Csの供給源化合物としては、硝酸セシウム、硫酸セシウム、塩化セシウム、炭酸セシウム、酢酸セシウム等を挙げることができる。
Tlの供給源化合物としては、硝酸第一タリウム、塩化第一タリウム、炭酸タリウム、酢酸第一タリウム等を挙げることができる。
【0027】
Bの供給源化合物としては、ホウ砂、ホウ酸アンモニウム、ホウ酸等を挙げることができる。
Pの供給源化合物としては、リンモリブデン酸アンモニウム、リン酸アンモニウム、リン酸、五酸化リン等を挙げることができる。
Asの供給源化合物としては、ジアルセノ十八モリブデン酸アンモニウム、ジアルセノ十八タングステン酸アンモニウム等を挙げることができる。
【0028】
Wの供給源化合物としては、パラタングステン酸アンモニウム、三酸化タングステン、タングステン酸、リンタングステン酸等を挙げることができる。
Mgの供給源化合物としては、硝酸マグネシウム、硫酸マグネシウム、塩化マグネシウム、炭酸マグネシウム、酢酸マグネシウム等が挙げられる。
Caの供給源化合物としては、硝酸カルシウム、硫酸カルシウム、塩化カルシウム、炭酸カルシウム、酢酸カルシウム等が挙げられる。
【0029】
Znの供給源化合物としては、硝酸亜鉛、硫酸亜鉛、塩化亜鉛、炭酸亜鉛、酢酸亜鉛等が挙げられる。
Ceの供給源化合物としては、硝酸セリウム、硫酸セリウム、塩化セリウム、炭酸セリウム、酢酸セリウム等が挙げられる。
Smの供給源化合物としては、硝酸サマリウム、硫酸サマリウム、塩化サマリウム、炭酸サマリウム、酢酸サマリウム等が挙げられる。
【0030】
前工程において用いる原料化合物の水溶液又は水分散液は、触媒成分として少なくともモリブデン(全原子比aの内のa1相当)、鉄、ニッケル又はコバルトの少なくとも一方を含む水溶液、水スラリー又はケーキである。なお、この水溶液、水スラリー又はケーキは必要に応じてシリカを含んでいても良い。
この原料化合物の水溶液又は水分散液の調製は、供給源化合物の水性系での一体化により行われる。ここで各成分元素の供給源化合物の水性系での一体化とは、各成分元素の供給源化合物の水溶液あるいは水分散液を一括に、あるいは段階的に混合及び/又は熟成処理を行うことをいう。即ち、(イ)前記の各供給源化合物を一括して混合する方法、(ロ)前記の各供給源化合物を一括して混合し、そして熟成処理する方法、(ハ)前記の各供給源化合物を段階的に混合する方法、(ニ)前記の各供給源化合物を段階的に混合・熟成処理を繰り返す方法、及び(イ)〜(ニ)を組み合わせる方法のいずれもが、各成分元素の供給源化合物の水性系での一体化という概念に含まれる。ここで、熟成とは、工業原料もしくは半製品を、一定時間、一定温度等の特定条件のもとに処理して、必要とする物理性、化学性の取得、上昇あるいは所定反応の進行等を図る操作をいい、一定時間とは、通常10分〜24時間の範囲であり、一定温度とは通常室温〜水溶液又は水分散液の沸点範囲をいう。
【0031】
前記の一体化の具体的な方法としては、例えば、触媒成分から選ばれた酸性塩を混合して得られた溶液と、触媒成分から選ばれた塩基性塩を混合して得られた溶液とを混合する方法等が挙げられ、具体例としてモリブデン化合物の水溶液に、鉄化合物とニッケル化合物及び/又はコバルト化合物との混合物を加温下添加し混合する方法等が挙げられる。なお、必要に応じてシリカの添加、混合もこの前工程で行うのが好ましい。
【0032】
このようにして得られた原料化合物の水溶液又は水分散液を60〜90℃に加温し、熟成する。
この熟成とは、前記触媒前駆体用スラリーを所定温度で所定時間、撹拌することをいう。この熟成により、スラリーの粘度が上昇し、スラリー中の固体成分の沈降を緩和し、とりわけ次の乾燥工程での成分の不均一化を抑制するのに有効となり、得られる最終製品である複合酸化物触媒の原料転化率や選択率等の触媒活性がより良好となる。
【0033】
前記熟成における温度は、60〜90℃が好ましく、70〜85℃がより好ましい。熟成温度が60℃未満では、熟成の効果が十分ではなく、良好な活性を得られない場合がある。一方、90℃を超えると、熟成時間中の水の蒸発が多く、工業的な実施には不利である。更に100℃を超えると、溶解槽に耐圧容器が必要となり、また、ハンドリングも複雑になり、経済性及び操作性の面で著しく不利となる。
【0034】
前記熟成にかける時間は、2〜12時間がよく、3〜8時間が好ましい。熟成時間が2時間未満では、触媒の活性及び選択性が十分に発現しない場合がある。一方、12時間を超えても熟成効果が増大することはなく、工業的な実施には不利である。
前記撹拌方法としては、任意の方法を採用することができ、例えば、撹拌翼を有する撹拌機による方法や、ポンプによる外部循環による方法等が挙げられる。
【0035】
熟成されたスラリーは、そのままで、又は乾燥した後、加熱処理を行う。乾燥する場合の乾燥方法及び得られる乾燥物の状態については特に限定はなく、例えば、通常のスプレードライヤー、スラリードライヤー、ドラムドライヤー等を用いて粉体状の乾燥物を得てもよいし、また、通常の箱型乾燥器、トンネル型焼成炉を用いてブロック状又はフレーク状の乾燥物を得てもよい。
【0036】
前記の原料塩水溶液又はこれを乾燥して得た顆粒あるいはケーキ状のものは、空気中で200〜400℃、好ましくは250〜350℃の温度域で短時間の熱処理を行う。その際の炉の形式及びその方法については特に限定はなく、例えば、通常の箱型加熱炉、トンネル型加熱炉等を用いて乾燥物を固定した状態で加熱してもよいし、また、ロータリーキルン等を用いて乾燥物を流動させながら加熱してもよい。
【0037】
加熱処理後に得られた触媒前駆体の灼熱減量は、0.5〜5重量%であることが好ましく、1〜3重量%であるのがより好ましい。灼熱減量をこの範囲とすることで、原料転化率や選択率が高い触媒を得ることができる。なお、灼熱減量は、次式により与えられる値である。
灼熱減量(%)=[(W0−W1)/W0]×100
・W0:触媒前駆体を150℃で3時間乾燥して付着水分を除いたものの重量(g)
・W1:付着水分を除いた前記触媒前駆体を更に500℃で2時間熱処理した後の重量(g)
前記の後工程では、前記の前工程において得られる触媒前駆体とモリブデン化合物(全原子比aからa1相当を差し引いた残りのa2相当)とビスマス化合物の一体化を、水性溶媒下で行う。この際、アンモニア水を添加するのが好ましい。X、Y、Z成分の添加もこの後工程で行うのが好ましい。また、この発明のビスマス供給源化合物は、水に難溶性ないし不溶性のビスマスである。この化合物は、粉末の形態で使用することが好ましい。触媒製造原料としてのこれら化合物は粉末より大きな粒子のものであってもよいが、その熱拡散を行わせるべき加熱工程を考えれば小さい粒子である方が好ましい。従って、原料としてのこれらの化合物がこのように粒子の小さいものでなかった場合は、加熱工程前に粉砕を行うべきである。
【0038】
次に、得られたスラリーを充分に撹拌した後、乾燥する。このようにして得られた乾燥品を、押出し成型、打錠成型、あるいは担持成型等の方法により任意の形状に賦形する。次に、このものを、好ましくは450〜650℃の温度条件にて1〜16時間程度の最終熱処理に付す。以上のようにして、高活性で、かつ目的とする酸化生成物を高い収率で与える複合酸化物触媒が得られる。
【0039】
<分子状酸素含有ガス>
本発明の分子状酸素含有ガスとは、通常、分子状酸素が10体積%以上、好ましくは、15体積%以上、更に好ましくは20体積%以上含まれるガスのことであり、具体的に好ましくは空気である。なお、分子状酸素含有ガスを工業的に用意するのに必要なコストが増加するという観点から、分子状酸素の含有量の上限としては、通常50体積%以下であり、好ましくは、30体積%以下、更に好ましくは25体積%以下である。本発明の分子状酸素含有ガスに含まれうる酸素以外の成分としては、具体的には、窒素、アルゴン、ネオン、ヘリウム、CO、CO2、水等が挙げられる。これら酸素以外の成分の量は、窒素の場合、通常90体積%以下、好ましくは85体積%以下、より好ましくは80体積%以下である。窒素以外の成分の場合、通常10体積%以下、好ましくは1体積%以下である。この量が多すぎると、反応に必要な酸素を供給するのが難しくなる傾向にある。
【0040】
<ガス供給>
本発明では、反応器に原料ガスを供給するにあたり、原料ガスと分子状酸素含有ガスとを混合し、その混合されたガス(以下、「混合ガス」呼ぶことがある)を反応器に供給する必要がある。なお、本発明の混合ガス中の、原料ガスの割合としては、通常、4.2vol%以上であり、好ましくは7.6vol%以上、更に好ましくは8.0vol%以上である。この値が大きくなるほど、反応器のサイズを小さくでき、建設費および運転に要するコストが低減する傾向にある。また、一方、上限は、20.0vol%以下であり、
好ましくは、17.0vol%以下、更に好ましくは、15.0vol%以下である。この値が小さくなるほど、原料ガス中の触媒上へのコーキングの起因物質も低減するため、触媒のコーキングが発生しにくく好ましい。
【0041】
<窒素ガス、水(水蒸気)>
また、混合ガスと共に、窒素ガス、及び水(水蒸気)を反応器に供給してもよい。窒素ガスは、混合ガスが爆鳴気を形成しないように可燃性ガスと酸素の濃度を調整するという理由から、水(水蒸気)は窒素ガスと同様に可燃性ガスと酸素の濃度を調整するという理由と触媒のコーキングを抑制するという理由から、混合ガスに、水(水蒸気)と窒素ガスとを更に混合し反応器に供給するのが好ましい。
【0042】
反応器に水蒸気を供給する場合、前記原料ガスの供給量に対して0.5〜5.0の比率で導入することが好ましい。この比率が大きくなるほど、廃水量が増加する傾向にあり、小さくなるほど、目的生成物であるブタジエンの収率が低下する傾向にある。そのため、水蒸気を前記原料ガスの供給量に対して、好ましくは、0.8〜4.5であり、更に好ましくは、1.0〜4.0である。
【0043】
反応器に窒素ガスを供給する場合、前記原料ガスの供給量に対して0.5〜8.0の比率(体積比)で導入することが好ましい。この比率が大きくなるほど、後工程の生成ガスを圧縮する工程の負荷が上がる傾向にあり、小さくなるほど、反応器に供給する水蒸気の使用量が増加する傾向にある。そのため、窒素ガスを前記原料ガスの供給量に対して、好ましくは、1.0〜6.0、更に好ましくは、2.0〜5.0の比率(体積比)で供給する。
【0044】
原料ガスと分子状酸素含有ガスの混合ガス、及び必要により供給される窒素ガス、及び水(水蒸気)を供給する方法は特に限定されず、別々の配管で供給してもよいが、爆鳴気の形成を確実に回避するために、混合ガスを得る前に、予め窒素ガスを原料ガス、もしくは分子状酸素含有ガスに供給しておき、その状態で、原料ガスと分子状酸素含有ガスとを混合して混合ガスを得、該混合ガスを供給することが好ましい。
【0045】
以下に、混合ガスの代表的な組成を示す。
[混合ガス組成]
・n−ブテン:C4留分合計に対して50〜100vol%
・C4留分合計:5〜15vol%
・O2:C4留分合計に対して40〜120vol/vol%
・N2:C4留分合計に対して500〜1000vol/vol%
・H2O:C4留分合計に対して90〜900vol/vol%
反応器に供給する原料ガスは、分子状酸素含有ガスと混合されると、酸素と可燃性ガスの混合物となることから、爆発範囲に入らないように各々のガス(原料ガス、空気、及び必要に応じて窒素ガスと水(水蒸気))を供給する配管に設置された流量計にて流量を監視しながら、反応器入り口の組成制御を行い、例えば、n−ブテン(1−ブテン及び/又は2−ブテン)からブタジエンを製造する場合、後述の混合ガス組成の範囲に調整される。なお、ここでいう爆発範囲とは、酸素と可燃性ガスの混合ガスが何らかの着火源の存在下で着火するような組成を持つ範囲のことである。可燃性ガスの濃度がある値より低いと着火源が存在しても着火しないことが知られており、この濃度を爆発下限界という。また可燃性ガスの濃度がある値より高いとやはり着火源が存在しても着火しないことが知られており、この濃度を爆発上限界という。各々の値は酸素濃度に依存しており、一般に酸素濃度が低いほど両者の値が近づき、酸素濃度がある値になったとき両者が一致する。このときの酸素濃度を限界酸素濃度と言い、酸素濃度がこれより低ければ可燃性ガスの濃度によらず混合ガスは着火しない。
【0046】
本発明の反応を開始するときは、最初に反応器に供給する分子状酸素含有ガス、窒素、水蒸気の量を調整して反応器入り口の酸素濃度が限界酸素濃度以下になるようにしてから可燃性ガス(主に原料ガス)の供給を開始し、次いで可燃性ガス濃度が爆発上限界よりも濃くなるように可燃性ガス(主に原料ガス)と空気などの分子状酸素含有ガスの供給量を増やしていくのが良い。可燃性ガス(主に原料ガス)と分子状酸素含有ガスの供給量を増やしていくときに窒素および/または水蒸気の供給量を減らして混合ガスの供給量が一定となるようにしても良い。こうすることで、配管および反応器における混合ガスの滞留時間を一定に保ち、圧力の変動を抑えることが出来る。
【0047】
なお、爆発範囲外であっても、ある温度、圧力条件下で、ある時間保持されると発火する場合がある。このときの保持時間を発火遅れ時間という。反応器周りを設計するときは原料配管や生成ガス配管の滞留時間が発火遅れ時間以下になるように設計する必要がある。発火遅れ時間は温度や圧力、組成に依存するので一概には言えないが、混合ガス配管の滞留時間は1000秒以下、生成ガス配管の滞留時間は10秒以下もしくは生成ガスを10秒以内に350℃以下に冷却することが望ましい。
【0048】
また、本発明の混合ガス中の原料ガスの割合について、下限値としては、通常、4.2vol%以上であり、好ましくは7.6vol%以上、更に好ましくは9.3vol%以上である。この下限値が大きくなるほど、反応器のサイズを小さくでき、建設費および運転に要するコストが低減する傾向にある。また、一方、上限値は、通常、20.0vol%以下であり、好ましくは、17.0vol%以下、更に好ましくは、15.0vol%以下である。この上限値が小さくなるほど、原料ガス中の触媒上へのコーキングの起因物質も低減するため、触媒のコーキングが発生しにくくなる。
【0049】
<反応器>
本発明の酸化脱水素反応に用いられる反応器は特に限定されないが、具体的には、管型反応器、槽型反応器、又は流動床反応器が挙げられ、好ましくは、固定床反応器、より好ましくは固定床の多管式反応器やプレート式反応器であり、最も好ましくは固定床の多管式反応器である。これらの反応器は一般に工業的に用いられているものであり特に制限はない。
【0050】
また、反応器が固定床反応器の場合、反応器には、上述の酸化脱水素反応触媒を有する触媒層が存在する。その触媒層は、触媒のみからなる層から構成されていても、触媒と該触媒と反応性の無い固形物とを含む層のみから構成されていても、触媒と該触媒と反応性の無い固形物とを含む層と触媒のみからなる層の複数の層から構成されていてもよいが、触媒層が、触媒と該触媒と反応性の無い固形物とを含む層を含むことで、反応時の発熱による触媒層の急激な温度上昇を抑制できるので、触媒層に反応性の無い固形物を有することが好ましい。尚、複数の層を有する場合、複数の層は反応器の入口から反応器の生成ガス出口の方向に向かって層状に形成される。触媒層が触媒と該触媒と反応性の無い固形物とを含む層を含む場合、下記式(3)で示される触媒希釈率が10体積%以上であることが好ましく、より好ましくは、15体積%以上、更に好ましくは、20体積%以上である。この下限値が大きくなるほど、触媒層中でのホットスポットの発生を抑えることができ、触媒上への炭素分の蓄積を抑制する効果が高くなる。触媒層の希釈率の上限は特に限定されないが、通常、99vol%以下であり、好ましくは90vol%以下、更に好ましくは、80vol%以下である。この上限値が小さくなるほど、反応器の大きさを小さくすることができ、建設費や運転コストを抑えることができる。
【0051】
希釈率(体積%)=[(触媒と反応性の無い固形物の体積)/(触媒の体積+触媒と反
応性の無い固形物の体積)]×100 ・・・(3)
尚、上述の通り、反応器内に設けられる触媒層は、単層でも2層以上でもよいが、好ましくは、2〜5層である。触媒層の数が多くなるほど、触媒充填作業が煩雑になる傾向にあり、触媒層の数が少なくなるほど、容易という傾向にある。また、反応器内に触媒層を2層以上設ける場合は、各触媒層の希釈率は、反応条件や反応温度によって適宜決めることができるが、希釈率が異なる触媒層を設けることが好ましい。
【0052】
本発明に用いられる反応性の無い固形物は、共役ジエン生成反応条件下で安定であり、炭素原子数4以上のモノオレフィン等の原料物質、及び共役ジエン等の生成物と反応性がない材質のものであれば特に限定されず、一般的に、イナートボールとも呼ばれることがある。具体的には、アルミナ、ジルコニア等のセラミック材等が挙げられる。また、その形状は、特に限定されず、球状、円柱状、リング状、不定形のいずれでもよい。また、その大きさは、本発明で使用する触媒と同等の大きさであればよい。その粒径は、通常、2〜10mm程度である。 触媒層の充填長は、充填される触媒の活性(反応性の無い固形物で希釈される場合は、希釈された触媒としての活性)、反応器の大きさ、反応原料ガス温度、反応温度及び反応条件が決まれば、物質収支及び熱収支計算によって求めることができる。
【0053】
また、本発明では、固定床反応器内に、上記の触媒層以外に、反応性が無い固形物のみで形成される層を有していても良い。通常、反応器の原料ガスの入口一番近い箇所と生成ガス出口側に最も近い箇所の両方、若しくはどちらか一方に、反応性が無い固形物のみで形成される層を設けるのがよい。これら層の大きさは、触媒層の大きさや反応器の大きさ、及び反応条件に応じて、適宜決めることができる。
【0054】
本発明では、触媒層中の固体触媒を、下記式(1)で示される破砕率(重量%)を0.0重量%以上40.0重量%以下とする必要がある。
破砕率(重量%)={(反応器中の破砕触媒の重量)/(反応器中の全触媒重量)}×100 …(1)
上記式において破砕触媒とは、反応器内で使用される触媒が製造された当初の形状、又は反応器に充填する際の形状に対して、それらの形状と同等の形状ではないものを指す。具体的には、反応器内で触媒が割れたり、或いは粉化したりする際に発生する触媒である。通常、破砕触媒は、その大きさとしては、触媒の製造時、又は反応器に充填する際の大きさに比べて、小さいものが多い。破砕触媒の特定は、酸化脱水素反応停止後に反応器を開放して、反応器内の内容物を抜き出した後、それらを目視にて選別する、又は篩にかけることで特定できる。反応器内の内容物を反応器から抜き出す際は、内容物が破壊されないように抜き出すことが好ましい。反応器から抜き出した内容物を篩にかける際は、篩の目開きは、使用する固体触媒の大きさによって異なるが、目開き4.5mm〜4.9mmの篩を使用することで、割れた触媒や粉化した触媒を分離回収することが可能である。なお、反応器内に反応性の無い固形物が存在する場合は、破砕触媒を特定する前に、予め除いておくことが好ましい。
【0055】
このようにして特定された破砕触媒の重量を測定して、反応器中の破砕触媒の重量を得ることができる。また、反応器中の全触媒重量は、反応器に反応に使用する触媒を充填する前に、予め測定しておけばよい。
反応器が固定床の多管式反応器の場合、即ち、反応器内に同じ種類の触媒と重量が同じ触媒が充填され、且つ同じ長さと管径を持つ反応管が複数存在する際は、全ての反応管について、破砕触媒の特定を行い、破砕触媒の重量を測定してもよいが、複数の反応管の中から少なくとも1本以上の反応管を選定して、破砕触媒の特定とその重量の測定を行い、それら反応管で得られた破砕触媒の重量の平均値を、反応器中の破砕触媒の重量として破砕率を求めてもよい。なお、この際、反応器中の全触媒重量は、上記で選定された反応管の触媒の全重量となる。
【0056】
本発明では、触媒層中の固体触媒を、上記式(1)で示される破砕率(重量%)を0.0重量%以上40.0重量%以下とする必要があるが、好ましくは、1.0重量%以上30.0重量%以下である。更に好ましくは、5.0重量%以上25.0重量%以下である。この値が小さくなるほど、触媒の充填に時間を要し、また製造時の触媒の強度を高くする必要傾向がある。
【0057】
<反応条件>
本発明の酸化脱水素反応は発熱反応であり、反応により温度が上昇するが、本発明では、通常、反応温度は250〜450℃、好ましくは、280〜400℃の範囲に調整される。この温度が大きくなるほど、触媒活性が急激に低下しやすい傾向にあり、小さくなるほど、目的生成物である共役ジエンの収率が低下する傾向にある。反応温度は、熱媒体(例えば、ジベンジルトルエンや亜硝酸塩など)を使用して制御することができる。なお、ここでいう反応温度は熱媒体の温度のことのことである。
【0058】
また、本発明における反応器内温度は、特に限定されないが、通常、250〜450℃、好ましくは、280〜400℃、更に好ましくは、320〜395℃である。触媒層の温度が450℃を超えると、反応を継続するに従って、急激に触媒活性が低下する恐れがある傾向にあり、一方、触媒層の温度が250℃を下回ると、目的生成物である共役ジエンの収率が低下する傾向にある。反応器内温度は、反応条件によって決定されるが、触媒層の希釈率や混合ガスの流量等で制御することができる。なお、ここでいう反応器内温度とは、反応器出口での生成ガスの温度、又は触媒層を有する反応器の場合は、その触媒層の温度のことである。
【0059】
本発明の反応器内の圧力は、特に限定されないが、下限は、通常、0MPaG以上、好ましくは、0.001MPaG以上、更に好ましくは、0.01MPaG以上である。この値が大きくなるほど、反応器に反応ガスを多量に供給できるというメリットがある。一方、上限は、0.5MPaG以下であり、好ましくは0.3MPaG以下、更に好ましくは、0.1MPaGである。この値が小さくなるほど、爆発範囲が狭くなる傾向にある。
【0060】
本発明における反応器の滞留時間は、特に限定されないが、下限は、好ましくは、0.72秒以上、更に好ましくは0.90秒以上である。この値が大きくなるほど、原料ガス中のモノオレフィンの転化率が高くなるというメリットがある。一方、上限は、好ましくは2.80秒以下、更に好ましくは、2.10秒以下である。この値が小さくなるほど、反応器が小さくなる傾向にある。
【0061】
また、本発明では、反応器内の触媒量に対する混合ガスの流量の比(以下、SVと略記することがある)は、特に限定されないが、好ましくは、500〜5000h−1であり、更に好ましくは、1300〜4500h−1であり、特に好ましくは、1700〜4000h−1ある。この値が大きくなるほど、固形物の析出が抑制される傾向にあり、小さくなるほど、固形物が析出しやすい傾向にある。
【0062】
反応器の入口と出口との流量差としては、原料ガスの反応器入口での流量、及び生成ガスの反応器出口での流量に依存するが、通常、入口流量に対する出口の流量の比率が100〜110vol%、好ましくは、102〜107vol%、更に好ましくは103〜105vol%である。n−ブテン(1−ブテン及び2−ブテン)からブタジエンを製造する場合、出口流量が増えるのはブテンが酸化脱水素されてブタジエンと水が生成する反応や副反応でCOやCO2が生成する反応において化学量論的に分子数が増えるためである。出口流量の増加が少ないと反応が進行していないので好ましくなく、出口流量が増えすぎると副反応でCOやCO2が増加しているため好ましくない。
【0063】
かくして、原料ガス中のモノオレフィンの酸化脱水素反応により、該モノオレフィンに対応する共役ジエンが生成することとなり、該共役ジエンを含有する生成ガスを取得する。生成ガス中に含まれる原料ガス中のモノオレフィンに対応する共役ジエンの濃度は、原料ガス中に含まれるモノオレフィンの濃度に依存するが、通常1〜15vol%、好ましくは、5〜13vol%、更に好ましくは9〜11vol%である。共役ジエンの濃度が大きいほど、回収コストが低いというメリットがあり、小さいほど次工程で圧縮したときに重合などの副反応が起き難いというメリットがある。また、生成ガス中には未反応のモノオレフィンも含まれていてもよく、その濃度は、通常0〜7vol%、好ましくは、0〜4vol%、更に好ましくは0〜2vol%である。なお、本発明では、生成ガス中に含まれる高沸点副生物は、使用する原料ガス中に含まれる不純物の種類によって異なるが、常圧下での沸点が200〜500℃のものを言う。n−ブテン(1−ブテン及び2−ブテン)からブタジエンを製造する場合、具体的に、フタル酸、アントラキノン、フルオレノン等である。これらの量は、特に限定されないが、通常、反応ガス中に0.05〜0.10vol%である。
【0064】
<後工程>
本発明の共役ジエンの製造方法においては、共役ジエンを含有する生成ガスから共役ジエンを分離するために、更に、冷却工程、脱水工程、溶媒吸収工程、分離工程、精製工程等を有していてもよい。なお、反応器から得られる生成ガスは、脱水工程で、圧縮ガス、脱水ガスとなる。しかし、これらのガスは、水以外の含有割合は同一であり、また、含まれる水のほとんどは液状なので、各ガスの気体部分の成分割合は、同一と考えてよい。このため、以下において、生成ガス、圧縮ガス、及び脱水ガスについて、単に「生成ガス」と称する場合がある。
【0065】
(冷却工程)
本発明では、共役ジエンを含む生成ガスを冷却する冷却工程を有していてもよい。冷却工程については、反応器出口から抜き出される生成ガスを冷却できる工程であれば、特に限定されないが、好適には、冷却溶媒と生成ガスとを直接接触させて冷却させる方法が用いられる。冷却溶媒としては、特に限定されないが、好ましくは水やアルカリ水溶液であり、最も好ましくは水である。
【0066】
また、生成ガスの冷却温度は、反応器出口から抜き出される生成ガス温度や冷却溶媒の種類などによって異なるが、通常、5〜100℃、好ましくは、10〜50℃、更に好ましくは、15〜40℃に冷却される。冷却される温度が高くなるほど、建設費と運転に要するコストを下げられる傾向にあり、低くなるほど、生成ガスを圧縮する工程の負荷を下げられる傾向にある。冷却塔内の圧力は、特に限定されないが、通常は、0.03MPaGである。
尚、冷却塔を使って冷却する場合、生成ガス中に高沸点副生物が多く含まれていると、高沸点副生物同士の重合や、工程内での高沸点副生物に起因する固形析出物の堆積が起きやすくなる。また、冷却塔で使用される冷却溶媒は、循環使用されることが多いため、共役ジエンの製造を連続的に継続すると、固形析出物での閉塞が起きることがある。
【0067】
そのため、可能な限り、反応器出口から抜き出される生成ガス中の高沸点副生物を冷却工程に持ち込ませないように、予めフィルターなどで除去するすることが好ましい。
(脱水工程)
また、本発明では、反応器から排出される生成ガスに含まれる水分を除去する脱水工程を有していても良い。脱水工程を設けることにより、後段のプロセスにおける各工程における水分による機器腐食や、後述する溶媒吸収工程や溶媒分離工程で使用する溶媒への不純物の蓄積を防止することができるため、好ましい。
【0068】
本発明の脱水工程については、生成ガスに含まれる水分を除去できる工程であれば、特に限定されない。脱水工程は反応器の後段の工程であれば、どこで行ってもよいが、上述の冷却工程の後に脱水工程を行うことが好ましい。通常、反応器から排出される生成ガス中に含まれる水分量は、原料ガスの種類や分子状酸素含有ガスの量、更には、原料ガスと共に混合される水蒸気等により異なるが、通常は、4〜35vol%、好ましくは10〜30vol%である(これが水を使用した冷却工程を経過した場合には、100volppm〜2.0vol%まで水分濃度が低減されている)。また、露点として、0〜100℃、好ましくは、10〜80℃である。
【0069】
生成ガスから水分を除去する手段としては、特に限定されないが、酸化カルシウム、塩化カルシウム、モレキュラーシーブ等の乾燥剤(水分吸着剤)を利用することができる。この中でも、再生の容易さ、取り扱いの容易さという観点から、モレキュラーシーブ等の乾燥剤(水分吸着剤)が好ましく利用される。
脱水工程にモレキュラーシーブ等の乾燥剤を利用する場合は、水以外にも生成ガス中に含まれる高沸点副生物が吸着除去される。ここで除去される高沸点副生物は、アントラキノン、フルオレノン、フタル酸などのことである。
【0070】
脱水工程を経て得られる生成ガス中の水分含有量は、通常は10〜10000volppm、好ましくは、20〜1000volppmであり、露点としては、−60〜80℃、好ましくは、−50〜20℃である。この生成ガス中の水分含有量が多くなるほど、溶媒吸収塔や溶媒分離塔のリボイラーの汚れが増加する傾向にあり、一方で、少なくなると、脱水工程で使用する用役コストが増加する傾向にある。
【0071】
(溶媒吸収工程)
本発明では、生成ガスを吸収溶媒と接触させてオレフィンや共役ジエンなどの炭化水素を吸収溶媒に吸収させ共役ジエンを含む溶媒を得る溶媒吸収工程を有することが好ましい。好ましい理由としては、共役ジエンの分離に要するエネルギーコストの低減という観点から、生成ガスを溶媒に吸収させて共役ジエンの回収することが好ましい。溶媒吸収工程については、反応後の工程であれば、特に限定されないが、上述の脱水工程の後に設けることが好ましい。
【0072】
溶媒吸収工程で生成ガスを溶媒に吸収させる具体的な方法としては、例えば吸収塔を用いる方法が好ましい。吸収塔の種類としては、充填塔、濡れ壁塔、噴霧塔、サイクロンスクラバー、気泡塔、気泡攪拌槽、段塔(泡鐘塔、多孔板塔)、泡沫分離塔などが使用可能である。好ましくは、噴霧塔、泡鐘塔、多孔板塔である。
吸収塔を用いる場合、通常は、吸収溶媒と生成ガスとを向流接触させることで、生成ガス中の共役ジエンと未反応の炭素原子数4以上のモノオレフィン並びに炭素原子数3以下の炭化水素化合物が溶媒に吸収される。炭素原子数3以下の炭化水素化合物としては、例えば、メタン、アセチレン、エチレン、エタン、メチルアセチレン、プロピレン、プロパン、又はアレンなどが挙げられる。
【0073】
溶媒吸収工程において、吸収塔を用いて生成ガスを回収する場合、吸収塔内の圧力は、特に限定されないが、通常、0.1〜2.0MPaG,好ましくは、0.2〜1.5MPaG、更に好ましくは0.25〜1.0MPaGである。この圧力が大きいほど、吸収効率が良くなるというメリットがあり、小さいほど吸収塔へのガス導入時の昇圧に要するエネルギーを削減でき、さらに液中の溶存酸素量を低減できるというメリットがある。
【0074】
また、吸収塔10内の温度は、特に限定されないが、通常0〜50℃、好ましくは、10〜40℃、更に好ましくは20〜30℃である。この温度が高いほど、酸素や窒素など
が溶媒に吸収されにくいというメリットがあり、低いほど共役ジエンなどの炭化水素の吸収効率が良くなるというメリットがある。
本発明の溶媒吸収工程で使用する有機溶媒としては、特に限定されないが、C6〜C10の飽和炭化水素やC6〜C8の芳香族炭化水素、アミド化合物などが用いられる。具体的には、例えばジメチルホルムアミド(DMF)、トルエン、キシレン、N−メチル−2−ピロリドン(NMP)等を用いることができる。これらの中でも、好ましくは、無機ガスを溶解しにくいことからC6〜C8の芳香族炭化水素が好ましく、特にトルエンが好ましい。
【0075】
吸収溶媒の使用量には特に制限はないが、回収工程に供給される目的生成物の流量に対して、通常、1〜100重量倍、好ましくは、2〜50重量倍である。吸収溶媒の使用量が多くなるほど、不経済となる傾向にあり、少なくなるほど、共役ジエンの回収効率が低下する傾向にある。
溶媒吸収工程で得られる共役ジエンを含む溶媒中には、主として目的生成物である共役ジエンが含まれており、その共役ジエンの溶媒吸収液中の濃度としては、通常は1〜20重量%であり、好ましくは3〜10重量%である。この溶媒中の共役ジエンの濃度が高いほど、共役ジエンの重合あるいは揮発による消失分が多くなる傾向にあり、低いほど、同じ生産量での溶媒の循環必要量が増加する為に、運転に要するエネルギーコストが大きくなる傾向にある。
【0076】
また、得られる共役ジエンを含む溶媒に、若干量の窒素、酸素も吸収されているため、溶媒に溶存する窒素や酸素をガス化して除去する脱気工程を有していても良い。脱気工程では、溶媒吸収液中に溶存する窒素や酸素をガス化して除去できる工程であれば、特に限定されない。
(分離工程)
このようにして得られた共役ジエンを含む溶媒から粗共役ジエンの分離を行う分離工程を有していてもよく、この工程により粗共役ジエンを得ることができる。分離工程としては、共役ジエンの溶媒吸収液から粗共役ジエンを分離できる工程であれば、特に限定されないが、通常、蒸留分離により粗共役ジエンを分離することができる。具体的には、例えば、リボイラーとコンデンサーにより共役ジエンの蒸留分離が行われ、塔頂付近より共役ジエン留分が抜き出される。分離された吸収溶媒は塔底から抜き出され、前段工程に溶媒を使用する回収工程を有する場合は、その回収工程で吸収溶媒として循環使用される。溶媒は循環使用するうち不純物が蓄積する場合があり、一部を抜き出して蒸留やデカンテーション、沈降、吸着剤やイオン交換樹脂などとの接触処理などの公知の精製方法により不純物を除去することが望ましい。
【0077】
分離工程で使用する蒸留塔の蒸留時の圧力は任意に設定することができるが、通常は、塔頂圧力を0.05〜2.0MPaGとすることが好ましい。より好ましくは塔頂圧力が0.1〜1.0MPaGであり、特に好ましくは0.15〜0.8MPaGの範囲である。この塔頂圧力が低すぎると、留出した共役ジエンを低温で凝縮するために多大なコストが必要となり、また高すぎると蒸留塔の塔底部の温度が高くなり、蒸気コストの増大となってしまう。
【0078】
塔底温度は通常50〜200℃であり、好ましくは80〜180℃、より好ましくは100〜160℃である。塔底温度が低すぎると共役ジエンを塔頂から留出させるのが困難となる。また温度が高すぎると、溶媒も塔頂から留出してしまう。還流比は1〜10で差し支えなく、好ましくは2〜4である。
蒸留塔としては充填塔、棚段塔のいずれもが使用できるが、多段蒸留が好ましい。共役ジエンと溶媒を分離するには、蒸留塔理論段を5段以上、特に10段〜20段とするのが好ましい。50段を越える蒸留塔は、蒸留塔建設の経済性、運転難易度、及び安全管理の
ためには好ましくない。また段数が小さすぎると分離が困難となる。
【0079】
(精製工程)
前記共役ジエンの分離工程で粗共役ジエンが得られるが、この粗共役ジエンを蒸留精製等により、更に精製された高純度の共役ジエンとする精製工程を有していてもよい。ここで使用する蒸留塔の蒸留時の圧力は任意に設定することができるが、通常は、塔頂圧力を0.05〜0.4MPaGとすることが好ましい。より好ましくは塔頂圧力が0.1〜0.3MPaGであり、特に好ましくは0.15〜0.2MPaGの範囲である。この塔頂圧力が低すぎると、留出した共役ジエンを低温で凝縮するために多大なコストが必要となり、また高すぎると蒸留塔の塔底部の温度が高くなり、蒸気コストの増大となってしまう。
【0080】
塔底温度は通常30℃〜100℃であり、好ましくは40℃〜80℃、より好ましくは50℃〜60℃である。塔底温度が低すぎると共役ジエンを塔頂から留出させるのが困難となる。また温度が高すぎると、塔頂で凝縮させる量が増えてコストが増大してしまう。また、還流比は1〜10で差し支えなく、好ましくは2〜4である。
蒸留塔としては充填塔、棚段塔のいずれもが使用できるが、多段蒸留が好ましい。共役ジエンとフランなどの不純物を分離するには、蒸留塔理論段を5段以上、特に10段〜20段とするのが好ましい。50段を越える蒸留塔は、蒸留塔建設の経済性、運転難易度、及び安全管理のためには好ましくない。また段数が小さすぎると分離が困難となる。このようにして得られる精製された共役ジエンは、純度が99.0〜99.9%の共役ジエンである。
【0081】
[プロセスの実施形態]
以下に、図面を参照して、本発明の共役ジエンの製造方法に関するプロセスの実施形態について、ブタジエンを製造する例を挙げて説明する。
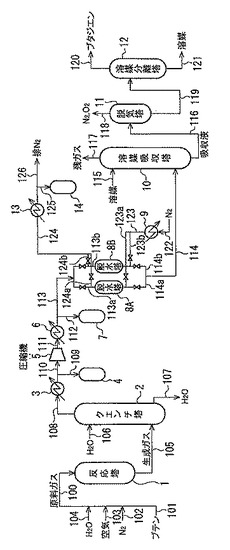
図1は本発明プロセスの実施の態様の一つである。
図1において、1は反応器、2はクエンチ塔、3,6,13は冷却器(熱交換器)、4,7,14はドレンポット、8A,8Bは脱水塔、9は加熱器(熱交換器)、10は溶媒吸収塔、11は脱気塔、12は溶媒分離塔を示し、符号100〜126は配管を示す。原料となるn−ブテン或いは前述のBBSS等のn−ブテンを含む混合物を、気化器(図示せず)でガス化して、配管101より導入すると共に、配管102、103、104より、窒素ガス、空気(分子状酸素含有ガス)、及び水(水蒸気)をそれぞれ導入し、これらの混合ガスを予熱器(図示せず)で150〜400℃程度に加熱した後、配管100より触
媒が充填された多管式の反応器1(酸化脱水素反応器)に供給する。反応器1からの反応
生成ガスは、配管105よりクエンチ塔2に送給され、20〜99℃程度に冷却される。クエンチ塔2には、配管106より冷却水が導入され、生成ガスと向流接触する。そして、この向流接触で生成ガスを冷却した水は、配管107より排出される。なお、この冷却排水は、熱交換器(図示せず)で冷却されて再度クエンチ塔2において循環使用される。クエンチ塔2で冷却された生成ガスは、塔頂から留出され、次いで配管108より冷却器3を経て室温に冷却される。冷却により発生した凝縮水は配管109よりドレンポット4に分離される。水分離後のガスは更に配管110を経て圧縮機5で0.1〜0.5MPa程度に昇圧され、昇圧ガスは配管111を経て冷却器6で再度10〜30℃程度に冷却される。冷却により発生した凝縮水は配管112よりドレンポット7に分離される。水分離後の圧縮ガスは、モレキュラーシーブ等の乾燥剤が充填された脱水塔8A,8Bに導入され脱水処理される。脱水塔8A,8Bは圧縮ガスの脱水と乾燥剤の加熱乾燥による再生とが交互に行われる。即ち、圧縮ガスは、まず、配管113,113aを経て脱水塔8Aに導入されて脱水処理され、配管114a,114を経て溶媒吸収塔10に送給される。この間に、脱水塔8Bには、配管122、加熱器9、配管123,123a,123bを経て150〜250℃程度に加熱された窒素ガスが導入され、乾燥剤の加熱による水分の脱
着が行われる。脱着した水分を含む窒素ガスは、配管124a,124b、124を経て冷却器13で室温まで冷却され、凝縮水が配管125よりドレンポット14に分離された後、配管126より排出される。
【0082】
脱水塔8Aの乾燥剤が飽和に達したら、ガス流路を切り換え、脱水塔8Bで圧縮ガスの脱水処理を行い、脱水塔8A内の乾燥剤の再生を行う。
脱水工程における脱水塔内の乾燥剤の再生時間は、特に限定されないが、通常6〜48時間、好ましくは、12〜36時間、更に好ましくは18〜30時間である。
脱水塔8A,8Bからの脱水ガスは、必要に応じて冷却器(図示せず)で10〜30℃程度に冷却された後、溶媒吸収塔10に送給され、配管115からの溶媒(吸収溶媒)と向流接触される。これにより、脱水ガス中の共役ジエンや未反応の原料ガスが吸収溶媒に吸収される。吸収溶媒に吸収されなかった成分(offガス)は、溶媒吸収塔10の塔頂より配管117を経て排出され燃焼廃棄される。このとき、吸収溶媒として、トルエンのような比較的沸点の低い溶媒を用いると経済的に無視できない量の溶媒が配管117を経て揮散することがある。このような場合はより沸点の高い溶媒を用いて沸点の低い溶媒を回収する工程を配管117の先に設けてもよい。この溶媒吸収塔10で、ブタジエンや未反応の原料ガスを吸収溶媒に吸収した溶媒吸収液は、溶媒吸収塔10の塔底より抜き出され、配管116より脱気塔11に送給される。溶媒吸収塔10で得られるブタジエンの溶媒吸収液には、若干量の窒素、酸素も吸収されているため、次いでこの溶媒吸収液を脱気塔11に供給して加熱することにより、液中に溶存する窒素や酸素をガス化して除去する。この際、ブタジエンや原料ガス、溶媒の中には、その一部がガス化することがあるため、この脱気塔11の塔頂に設けたコンデンサ(図示せず)でこれを液化して溶媒吸収液中に回収する。凝縮しなかった原料ガス、ブタジエン等は窒素、酸素の混合ガスとして配管118より抜き出され、共役ジエンの回収率を高めるために圧縮機5の入口側へ循環され再度処理が行われる。一方、溶媒吸収液を脱気した脱気処理液は配管119より溶媒分離塔12へ送給される。
【0083】
溶媒分離塔12では、リボイラーとコンデンサーにより共役ジエンの蒸留分離が行われ、塔頂より配管120を経て粗ブタジエン留分が抜き出される。分離された吸収溶媒は塔底より配管121を経て抜き出され、溶媒吸収塔10の吸収溶媒として循環使用される。
【実施例】
【0084】
以下に実施例を挙げて本発明をより具体的に説明する。
[製造例1:複合酸化物触媒の調製]
パラモリブデン酸アンモニウム54gを純水250mlに70℃に加温して溶解させた。次に、硝酸第二鉄7.18g、硝酸コバルト31.8g及び硝酸ニッケル31.8gを純水60mlに70℃に加温して溶解させた。これらの溶液を、充分に攪拌しながら徐々に混合した。
【0085】
次に、シリカ64gを加えて、充分に攪拌した。このスラリーを75℃に加温し、5時間熟成した。その後、このスラリーを加熱乾燥した後、空気雰囲気で300℃、1時間の熱処理に付した。
得られた触媒前駆体の粒状固体(灼熱減量:1.4重量%)を粉砕し、パラモリブデン酸アンモニウム40.1gを純水150mlにアンモニア水10mlを加え溶解した溶液に分散した。次に、純水40mlにホウ砂0.85g及び硝酸カリウム0.36gを25℃の加温下に溶解させて、上記スラリーを加えた。
【0086】
次に、Naを0.45%固溶した次炭酸ビスマス58.1gを加えて、攪拌混合した。このスラリーを130℃、12時間加熱乾燥した後、得られた粒状固体を、小型成型機にて径5mm、高さ4mmの錠剤に打錠成型し、次に500℃、4時間の焼成を行って、触
媒を得た。仕込み原料から計算される触媒は、次の原子比を有する複合酸化物であった。
Mo:Bi:Co:Ni:Fe:Na:B:K:Si=12:5:2.5:2.5:0.4:0.35:0.2:0.08:24
なお、触媒調製の際のモリブデンの原子比a1とa2は、それぞれ6.9と5.1であった。
【0087】
[実施例1]
内径23.0mm、高さ500mmのステンレス製反応管に、予めイナートボール(1
粒あたりの大きさ:約0.065mm3)を27ml充填(充填層長:201mm)しておき、そのイナートボールの充填層の上に、製造例1で製造された複合酸化物触媒8.6mlとイナートボール(1粒あたりの大きさ:約0.065mm3)2.9mlとを混合
して充填し、第1の触媒層を形成した(以下、「触媒層1」とする)。この触媒層1の充填層長は32mmで、触媒希釈率は25%であった。次に、触媒層1の上に、製造例1で製造された複合酸化物触媒2.9mlとイナートボール(1粒あたりの大きさ:約0.0
65mm3)8.6mlと混合して充填し、第2の触媒層を形成した(以下、「触媒層2
」とする)。この触媒層2の充填層長は31mmで、触媒希釈率は75%であった。尚、反応器へ充填した触媒量合計は11.5ml(充填した触媒の全重量:7.51g)であった。
【0088】
これらの反応管には外径2.0mmの挿入管を設置し、挿入管の中に熱電対を設置して反応器内温度を測定した。なお、熱媒体は電気炉を使用した。
そして、予め窒素を1.8L/hr、空気を11.8L/hr、及び水蒸気を5.5L/hrで予熱器に供給しておき、その後、表1に示す組成の原料ガスを2.8L/hr供給し、予熱器内で混合して混合ガスとして350℃に昇温した(反応器導入ガス組成=窒素:8.1vol%、空気:53.9vol%、水蒸気:25.0vol%、原料ガス:13.0vol%)。原料ガスに含まれる代表的な成分の組成(mol%)を表1に示す。この際、触媒量11.5mlに対して、合計ガス流量は21.9ml/hであり、上記式(3)で算出したSVは1900h−1であった。
【0089】
その混合ガスを上記の反応器に供給し、酸化脱水素反応を行った。反応器内温度は、平均で367℃で、圧力はゲージ圧で2kPaであった。反応器出口から得られた生成ガスをガスクロマトグラフィー(GL Sciences社製 型番GC4000)で分析したところ、n−ブテン転化率(1−ブテン、シス−2−ブテン、トランス−2−ブテンの合計での転化率)は87.7mol%、ブタジエン選択率は90.7mol%であった。酸化脱水素反応を200時間行なった後、反応管から触媒を全量取り出し、破砕触媒を目視で特定したところ、その重量は、0.07gであり、上記式(1)による破砕率は1.0重量%であった。その後、抜き出した全ての触媒に付着した炭素の量を測定した(測定装置:LECO社製 炭素硫黄分析装置 型番CS600)ところ、3.5wt%であった。結
果を表−1に示す。
【0090】
[比較例1]
実施例1において、予め製造例1で製造された複合酸化物触媒9.8mlを、全てその粒径が半分以下になるように粉砕し(触媒重量:6.57g)、イナートボール(1粒あ
たりの大きさ:約0.065mm3)3.2mlとを混合して充填されたものを触媒層1とし、触媒層1の上に、予め製造例1で製造された複合酸化物触媒3.2mlを、全てその粒径が半分以下になるように粉砕し(触媒重量:2.14g)とイナートボール(1粒
あたりの大きさ:約0.065mm3)7.7mlと混合して充填されたものを触媒層2として(充填した触媒の全重量:8.71g)、窒素と水蒸気の供給量を、それぞれ3.9L/hr、6.2L/hrに変更し、合計ガス流量を24.7L/hr と変更した以外は、同様の方法で、酸化脱水素反応を行った。
【0091】
反応器出口から得られた生成ガスをガスクロマトグラフィー(GL Sciences社製 型番GC4000)で分析したところ、n−ブテン転化率(1−ブテン、シス−2−ブテン、トランス−2−ブテンの合計での転化率)は90.4mol%、ブタジエン選択率は85.8mol%であった。酸化脱水素反応を200時間行なった後、反応管から触媒を全量取り出した。破砕率は100重量%であった。触媒に付着した炭素の量を測定した(測定装置:LECO社製 炭素硫黄分析装置 型番CS600)。測定した粉砕触媒の炭
素濃度は、10.8wt%であった。結果を表−1に示す。
【0092】
[実施例2]
比較例1において、予め製造例1で製造された複合酸化物触媒2.6mlを、全てその粒径が半分以下になるように粉砕し(触媒重量:1.74g)、この粉砕した触媒と、予め製造例1で製造された複合酸化物触媒7.2ml(触媒重量:4.82g)と、イナートボール(1粒あたりの大きさ:約0.065mm3)3.3mlとを混合して充填されたものを触媒層1とし、触媒層1の上に、予め製造例1で製造された複合酸化物触媒3.3ml(触媒重量:2.21g)とイナートボール(1粒あたりの大きさ:約0.065mm3)9.8mlと混合して充填されたものを触媒層2とした(充填した触媒の全重量:8.77g)以外は、比較例1と同様の方法で、酸化脱水素反応を行った。
【0093】
反応器出口から得られた生成ガスをガスクロマトグラフィー(GL Sciences社製 型番GC4000)で分析したところ、n−ブテン転化率(1−ブテン、シス−2−ブテン、トランス−2−ブテンの合計での転化率)は87.4mol%、ブタジエン選択率は84.6mol%であった。酸化脱水素反応を200時間行った後、反応管から触媒を全量取り出した。破砕率は20重量%であった。触媒に付着した炭素の量を測定した(測定装置:LECO社製 炭素硫黄分析装置 型番CS600)。測定した粉砕触媒の炭素濃
度は、3.9wt%であった。結果を表−1に示す。
【0094】
[比較例2]
実施例2において、予め製造例1で製造された複合酸化物触媒6.5mlを、全てその粒径が半分以下になるように粉砕し(触媒重量:4.36g)、この粉砕した触媒と、予め製造例1で製造された複合酸化物触媒3.3ml(触媒重量:2.21g)と、イナートボール(1粒あたりの大きさ:約0.065mm3)3.3mlとを混合して充填されたものを触媒層1とし、触媒層1の上に、予め製造例1で製造された複合酸化物触媒3.3ml(触媒重量:2.21g)とイナートボール(1粒あたりの大きさ:約0.065mm3)9.8mlと混合して充填されたものを触媒層2とした(充填した触媒の全重量:8.78g)以外は、実施例2と同様の方法で、酸化脱水素反応を行った。
【0095】
反応器出口から得られた生成ガスをガスクロマトグラフィー(GL Sciences社製 型番GC4000)で分析したところ、n−ブテン転化率(1−ブテン、シス−2−ブテン、トランス−2−ブテンの合計での転化率)は88.6mol%、ブタジエン選択率は84.4mol%であった。酸化脱水素反応を200時間行った後、反応管から触媒を全量取り出した。破砕率は50重量%であった。触媒に付着した炭素の量を測定した(測定装置:LECO社製 炭素硫黄分析装置 型番CS600)。測定した粉砕触媒の炭素濃
度は、12.8wt%であった。結果を表−1に示す。
【0096】
【表1】

【0097】
実施例1、2と比較例1、2を対比すると、実施例1、2の抜き出し触媒炭素組成が比較例1、2の抜き出し触媒炭素組成よりも小さいことから、破砕率が0.0重量%以上40.0重量%以下であれば、n−ブテンからブタジエンを製造する際に、反応器中の触媒にコークなどの炭素分が付着するのを低減できていることがわかる。
【符号の説明】
【0098】
1 反応器
2 クエンチ塔
3,6,13 冷却器
4,7,14 ドレンポット
8A,8B 脱水器
9 加熱器
10 溶媒吸収塔
11 脱気塔
12 溶媒分離塔
101〜121 配管
【技術分野】
【0001】
本発明は共役ジエンの製造方法に係り、特にn−ブテン等の炭素原子数4以上のモノオレフィンの接触酸化脱水素反応でブタジエン等の共役ジエンを製造する方法に関する。
【背景技術】
【0002】
n−ブテン等のモノオレフィンを触媒の存在下に酸化脱水素反応させてブタジエン等の共役ジエンを製造する方法は、従来公知である。
この反応は例えば以下の反応式に従って進行し、水が副生する。
C4H8+1/2O2→C4H6+H2O
n−ブテンの接触酸化脱水素反応によるブタジエンの製造は、工業的にはナフサ分解で副生するC4留分(C4炭化水素混合物。以下、「BB」と称す場合がある。)からのブタジエンの抽出分離プロセスにおいて、抽出蒸留塔でブタジエンを分離して得られた、1−ブテンの他、2−ブテン、ブタン等を含む混合物(以下、この混合物を「BBSS]と称す場合がある。)中に含まれるブテンからブタジエンを製造する方法が提案されている。
【0003】
具体的な方法としては、炭素原子数4以上のモノオレフィンを含む原料ガスと酸素ガスとの酸化脱水素反応により生成した共役ジエンを含む生成ガスを得て、その生成ガスを冷却する工程、冷却された生成ガスを脱水する工程、及び脱水された生成ガスを溶媒と接触させて共役ジエンを回収する工程を有する方法(特開2010−90083号公報)や、アセチレン系炭化水素濃度が0.016vol%以下に抑制された生成ガスを得ることで、アセチレン類の分離のための抽出蒸留の負荷を軽減する方法(特開2010−90082号公報)などがある。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特開2010−90083号公報
【特許文献2】特開2010−90082号公報
【発明の概要】
【発明が解決しようとする課題】
【0005】
上記特許文献1〜2には、触媒上に炭素物質が析出するコーキングの問題については一切記載されていないが、本発明者等の検討によれば、触媒の存在下、酸化脱水素反応により、ブテンからブタジエンを製造するに際に、原料ガス(例えば、BB又はBBSS)中に含まれるイソブテンなどの分岐型モノオレフィンや直鎖型n−ブテンが酸化脱水素反応で高沸点の炭素成分を副生し、触媒上でのコーキングが進行し、触媒活性が低下する、更にはコーキングの進行に伴い、反応器の差圧が上昇し、長期の運転に支障をきたす問題があることが判明した。触媒上にコーキングが進行すると運転を止めて、デコーキングなどにより高沸点の炭素成分を除去しなければならない。この運転停止操作ならびにデコーキングはブタジエン製造における生産性の低下原因となる。
【0006】
本発明は上記課題に鑑みてなされたものであって、n−ブテン等のモノオレフィンの接触酸化脱水素反応によりブタジエン等の共役ジエンを製造する方法において、反応中にコークのような炭素分が触媒上に蓄積するのを防止し、コーキングの進行を緩和して、安定的に運転が継続できる工業的に有利なブタジエンの製造方法を提供することを目的とする。
【課題を解決するための手段】
【0007】
本発明者らは、上記課題を解決すべく鋭意検討した結果、炭素原子数4以上のモノオレフィンを含む原料ガスと分子状酸素含有ガスとを混合した混合ガスを反応器に供給し、触媒の存在下、酸化脱水素反応により生成された対応する共役ジエンを含むガスを得る共役ジエンの製造方法において、反応器内の触媒が何らかの原因で割れ、更には粉砕が起こり、それらが反応器内で局所的に凝集すると高沸点の炭素成分を副生する反応が起きやすくなるという考えの下、反応器内に存在する全触媒量に対する割れた触媒や粉砕した触媒の量(重量比)をある特定の範囲で反応を行うことで、プラントの安定運転を阻害するコーキング速度を低減でき、且つ、より安全に運転ができ、更に高い収率で安定的にブタジエンの製造を行うことができることを見出した。
【0008】
本発明はこのような知見に基いて達成されたものであり、以下[1]〜[6]を要旨とする。
[1]炭素原子数4以上のモノオレフィンを含む原料ガスと分子状酸素含有ガスとを混合した混合ガスを、触媒を有する反応器に供給し、酸化脱水素反応により対応する共役ジエンを生成し、該共役ジエンを含む生成ガスを得る共役ジエンの製造方法において、下記式(1)で示される該反応器中の触媒の破砕率が0.0重量%以上40.0重量%以下であることを特徴とする共役ジエンの製造方法。
【0009】
破砕率(重量%)={(反応器中の破砕触媒の重量)/(反応器中の全触媒重量)}×100 …(1)
[2]前記反応器が前記触媒と反応性の無い固形物を更に有することを特徴とする[1]に記載の共役ジエンの製造方法。
[3]前記反応器内の全触媒量に対する前記混合ガスの流量の比(体積比)が500〜5000h-1であることを特徴とする[1]又は[2]に記載の共役ジエンの製造方法。
【0010】
[4]前記触媒が、少なくともモリブデン及びビスマスを含むことを特徴とする[1]〜[3]のいずれか1項に記載の共役ジエンの製造方法。
[5]前記触媒が、下記一般式(2)で表される複合酸化物触媒であることを特徴とする[1]〜[4]のいずれか1項に記載の共役ジエンの製造方法。
MoaBibCocNidFeeXfYgZhSiiOj (2)
(式中、Xはマグネシウム(Mg)、カルシウム(Ca)、亜鉛(Zn)、セリウム(Ce)及びサマリウム(Sm)からなる群から選ばれる少なくとも1種の元素であり、Yはナトリウム(Na)、カリウム(K)、ルビジウム(Rb)、セシウム(Cs)及びタリウム(Tl)からなる群から選ばれる少なくとも1種の元素であり、Zはホウ素(B)、リン(P)、砒素(As)及びタングステン(W)からなる群から選ばれる少なくとも1種の元素である。また、a〜jはそれぞれの元素の原子比を表し、a=12のとき、b=0.1〜7、c=0〜10、d=0〜10(但しc+d=1〜10)、e=0.05〜3、f=0〜2、g=0.04〜2、h=0〜3、i=0〜48の範囲にあり、またjは他の元素の酸化状態を満足させる数値である。)
[6]前記原料ガスが、エチレンの2量化により得られる1−ブテン、シス−2−ブテン、トランス−2−ブテン若しくはこれらの混合物を含有するガス、n−ブタンの脱水素若しくは酸化脱水素反応により生成するブテン留分、又は重油留分を流動接触分解する際に得られる炭素原子数が4の炭化水素を多く含むガスであることを特徴とする[1]〜[5]のいずれか1項に記載の共役ジエンの製造方法。
【発明の効果】
【0011】
本発明によれば、反応器内の触媒形状を保持することで、継続的にプラントの安定運転が可能となる。
【図面の簡単な説明】
【0012】
【図1】本発明の共役ジエンの製造方法の実施の形態を示す系統図である。
【発明を実施するための形態】
【0013】
以下に本発明の共役ジエンの製造方法の実施の形態を詳細に説明するが、以下に記載する説明は、本発明の実施態様の一例(代表例)であり、本発明はこれらの内容に限定されない。
本発明では、炭素原子数4以上のモノオレフィンを含む原料ガスと分子状酸素含有ガスとを触媒層を有する反応器に供給し、酸化脱水素反応により対応する共役ジエンを製造する。
【0014】
<炭素原子数4以上のモノオレフィンを含む原料ガス>
本発明の原料ガスは炭素原子数4以上のモノオレフィンを含むが、炭素原子数4以上のモノオレフィンとしては、ブテン(1−ブテン及び/又は2−ブテン等のn−ブテン、イソブテン)、ペンテン、メチルブテン、ジメチルブテン等の炭素原子数4以上、好ましくは炭素原子数4〜6のモノオレフィンが挙げられ、接触酸化脱水素反応による対応する共役ジエンの製造に有効に適用することができる。この中でも、n−ブテン(1−ブテン及び/又は2−ブテン等のn−ブテン)からのブタジエンの製造に最も好適に用いられる。又、炭素原子数4以上のモノオレフィンを含む原料ガスとしては、単離した炭素原子数4以上のモノオレフィンそのものを使用する必要はなく、必要に応じて任意の混合物の形で用いることができる。例えばブタジエンを得ようとする場合には高純度のn−ブテン(1−ブテン及び/又は2−ブテン)を原料ガスとすることもできるが、前述のナフサ分解で副生するC4留分(BB)からブタジエン及びi−ブテン(イソブテン)を分離して得られるn−ブテン(1−ブテン及び/又は2−ブテン)を主成分とする留分(BBSS)やn−ブタンの脱水素又は酸化脱水素反応により生成するブテン留分を使用することもできる。また、エチレンの2量化により得られる高純度の1−ブテン、シス−2−ブテン、トランス−2−ブテン又はこれらの混合物を含有するガスを原料ガスとして使用しても差し支えない。尚、このエチレンはエタン脱水素、エタノール脱水、又はナフサ分解などの方法で得られるエチレンを使用することができる。更に、石油精製プラントなどで原油を蒸留した際に得られる重油留分を、流動層状態で粉末状の固体触媒を使って分解し、低沸点の炭化水素に変換する流動接触分解 (Fluid Catalytic Cracking)から得られる炭素原子
数4の炭化水素類を多く含むガス(以下、FCC−C4と略記することがある)をそのまま原料ガスとする、又は、FCC−C4からリンや砒素などの不純物を除去したものを原料ガスとして使用しても差し支えない。なお、ここでいう、主成分とは、原料ガスに対して、通常40体積%以上、好ましくは60体積%以上、より好ましくは75体積%以上、特に好ましくは99体積%以上を示す。
【0015】
また、本発明の原料ガス中には、本発明の効果を阻害しない範囲で、任意の不純物を含んでいても良い。n−ブテン(1−ブテン及び2−ブテン)からブタジエンを製造する場合、含んでいても良い不純物として、具体的には、イソブテンなどの分岐型モノオレフィン;プロパン、n−ブタン、i−ブタン、ペンタンなどの飽和炭化水素;プロピレン、ペンテンなどのオレフィン;1,2−ブタジエンなどのジエン;メチルアセチレン、ビニルアセチレン、エチルアセチレンなどのアセチレン類等が挙げられる。この不純物の量は、通常40%以下、好ましくは20%以下、より好ましくは10%以下、特に好ましくは1%以下である。この量が多すぎると、主原料である1−ブテンや2−ブテンの濃度が下がって反応が遅くなったり、目的生成物であるブタジエンの収率が低下する傾向にある。また、本発明では、原料ガス中の炭素原子数4以上の直鎖型モノオレフィンの濃度は、特に限定されないが、通常は、70.00〜99.99vol%である。
【0016】
<酸化脱水素反応触媒>
次に、本発明で好適に用いられる酸化脱水素反応触媒について説明する。本発明で用いる酸化脱水素触媒は、少なくともモリブデン、ビスマス及びコバルトを含有する複合酸化物触媒であることが好ましい。そして、この中でも、下記一般式(2)で表される複合酸化物触媒であることがより好ましい。
【0017】
MoaBibCocNidFeeXfYgZhSiiOj (2)
なお、式中、Xはマグネシウム(Mg)、カルシウム(Ca)、亜鉛(Zn)、セリウム(Ce)及びサマリウム(Sm)からなる群から選ばれる少なくとも1種の元素である。Yはナトリウム(Na)、カリウム(K)、ルビジウム(Rb)、セシウム(Cs)及びタリウム(Tl)からなる群から選ばれる少なくとも1種の元素である。Zはホウ素(B)、リン(P)、砒素(As)及びタングステン(W)からなる群から選ばれる少なくとも1種の元素である。
【0018】
さらに、a〜jはそれぞれの元素の原子比を表し、a=12のとき、b=0.1〜7、c=0〜10、d=0〜10(但しc+d=1〜10)、e=0.05〜3、f=0〜2、g=0.04〜2、h=0〜3、i=0〜48の範囲にあり、またjは他の元素の酸化状態を満足させる数値である。
また、この複合酸化物触媒は、この複合酸化物触媒を構成する各成分元素の供給源化合物を水系内で一体化して加熱する工程を経て製造する方法がよい。例えば、前記各成分元素の供給源化合物の全部を水系内で一体化して加熱してもよい。
【0019】
その中でも、モリブデン化合物、鉄化合物、ニッケル化合物及びコバルト化合物よりなる群から選ばれる少なくとも1種と、必要に応じてシリカとを含む原料化合物の水溶液若しくは水分散液、又はこれを乾燥して得た乾燥物を加熱処理して触媒前駆体を製造する前工程と、この触媒前駆体、モリブデン化合物及びビスマス化合物を水性溶媒とともに一体化し、乾燥、焼成する後工程とを有する方法で製造するのが好ましい。この方法を用いると、得られた複合酸化物触媒は、高い触媒活性を発揮するので、高収率でブタジエン等の共役ジエンを製造することができ、アルデヒド類含有量の少ない反応生成ガスを得ることができる。なお、水性溶媒とは、水、又はメタノール、エタノール等の水と相溶性を有する有機溶媒、又はこれらの混合物をいう。
【0020】
次に、本発明に好適な複合酸化物触媒の製造方法について説明する。
まず、この複合酸化物触媒の製造方法においては、前記前工程で用いられるモリブデンが、モリブデンの全原子比(a)の内の一部の原子比(a1)相当のモリブデンであり、前記後工程で用いられるモリブデンが、モリブデンの全原子比(a)からa1を差し引いた残りの原子比(a2)相当のモリブデンであることが好ましい。そして、前記a1が0.5<a1/(c+d+e)<3を満足する値であることが好ましく、さらに、前記a2が0<a2/b<8を満足する値であることが好ましい。
【0021】
前記成分元素の供給源化合物としては、成分元素の酸化物、硝酸塩、炭酸塩、アンモニウム塩、水酸化物、カルボン酸塩、カルボン酸アンモニウム塩、ハロゲン化アンモニウム塩、水素酸、アセチルアセトナート、アルコキシド等が挙げられ、その具体例としては、下記のようなものが挙げられる。
Moの供給源化合物としては、パラモリブデン酸アンモニウム、三酸化モリブデン、モリブデン酸、リンモリブデン酸アンモニウム、リンモリブデン酸等が挙げられる。
【0022】
Feの供給源化合物としては、硝酸第二鉄、硫酸第二鉄、塩化第二鉄、酢酸第二鉄等が挙げられる。
Coの供給源化合物としては、硝酸コバルト、硫酸コバルト、塩化コバルト、炭酸コバルト、酢酸コバルト等が挙げられる。
Niの供給源化合物としては、硝酸ニッケル、硫酸ニッケル、塩化ニッケル、炭酸ニッケル、酢酸ニッケル等が挙げられる。
【0023】
Siの供給源化合物としては、シリカ、粒状シリカ、コロイダルシリカ、ヒュームドシリカ等が挙げられる。
Biの供給源化合物としては、塩化ビスマス、硝酸ビスマス、酸化ビスマス、次炭酸ビスマス等が挙げられる。また、X成分(Mg,Ca,Zn,Ce,Smの1種又は2種以上)やY成分(Na,K,Rb,Cs,Tlの1種又は2種以上)を固溶させた、BiとX成分やY成分との複合炭酸塩化合物として供給することもできる。
【0024】
例えば、Y成分としてNaを用いた場合、BiとNaとの複合炭酸塩化合物は、炭酸ナトリウム又は重炭酸ナトリウムの水溶液等に、硝酸ビスマス等の水溶性ビスマス化合物の水溶液を滴下混合し、得られた沈殿を水洗、乾燥することによって製造することができる。
また、BiとX成分との複合炭酸塩化合物は、炭酸アンモニウム又は重炭酸アンモニウムの水溶液等に、硝酸ビスマス及びX成分の硝酸塩等の水溶性化合物からなる水溶液を滴下混合し、得られた沈殿を水洗、乾燥することによって製造することができる。
【0025】
前記炭酸アンモニウム又は重炭酸アンモニウムの代わりに、炭酸ナトリウム又は重炭酸ナトリウムを用いると、Bi、Na及びX成分との複合炭酸塩化合物を製造することができる。
その他の成分元素の供給源化合物としては、下記のものが挙げられる。
Kの供給源化合物としては、硝酸カリウム、硫酸カリウム、塩化カリウム、炭酸カリウム、酢酸カリウム等を挙げることができる。
【0026】
Rbの供給源化合物としては、硝酸ルビジウム、硫酸ルビジウム、塩化ルビジウム、炭酸ルビジウム、酢酸ルビジウム等を挙げることができる。
Csの供給源化合物としては、硝酸セシウム、硫酸セシウム、塩化セシウム、炭酸セシウム、酢酸セシウム等を挙げることができる。
Tlの供給源化合物としては、硝酸第一タリウム、塩化第一タリウム、炭酸タリウム、酢酸第一タリウム等を挙げることができる。
【0027】
Bの供給源化合物としては、ホウ砂、ホウ酸アンモニウム、ホウ酸等を挙げることができる。
Pの供給源化合物としては、リンモリブデン酸アンモニウム、リン酸アンモニウム、リン酸、五酸化リン等を挙げることができる。
Asの供給源化合物としては、ジアルセノ十八モリブデン酸アンモニウム、ジアルセノ十八タングステン酸アンモニウム等を挙げることができる。
【0028】
Wの供給源化合物としては、パラタングステン酸アンモニウム、三酸化タングステン、タングステン酸、リンタングステン酸等を挙げることができる。
Mgの供給源化合物としては、硝酸マグネシウム、硫酸マグネシウム、塩化マグネシウム、炭酸マグネシウム、酢酸マグネシウム等が挙げられる。
Caの供給源化合物としては、硝酸カルシウム、硫酸カルシウム、塩化カルシウム、炭酸カルシウム、酢酸カルシウム等が挙げられる。
【0029】
Znの供給源化合物としては、硝酸亜鉛、硫酸亜鉛、塩化亜鉛、炭酸亜鉛、酢酸亜鉛等が挙げられる。
Ceの供給源化合物としては、硝酸セリウム、硫酸セリウム、塩化セリウム、炭酸セリウム、酢酸セリウム等が挙げられる。
Smの供給源化合物としては、硝酸サマリウム、硫酸サマリウム、塩化サマリウム、炭酸サマリウム、酢酸サマリウム等が挙げられる。
【0030】
前工程において用いる原料化合物の水溶液又は水分散液は、触媒成分として少なくともモリブデン(全原子比aの内のa1相当)、鉄、ニッケル又はコバルトの少なくとも一方を含む水溶液、水スラリー又はケーキである。なお、この水溶液、水スラリー又はケーキは必要に応じてシリカを含んでいても良い。
この原料化合物の水溶液又は水分散液の調製は、供給源化合物の水性系での一体化により行われる。ここで各成分元素の供給源化合物の水性系での一体化とは、各成分元素の供給源化合物の水溶液あるいは水分散液を一括に、あるいは段階的に混合及び/又は熟成処理を行うことをいう。即ち、(イ)前記の各供給源化合物を一括して混合する方法、(ロ)前記の各供給源化合物を一括して混合し、そして熟成処理する方法、(ハ)前記の各供給源化合物を段階的に混合する方法、(ニ)前記の各供給源化合物を段階的に混合・熟成処理を繰り返す方法、及び(イ)〜(ニ)を組み合わせる方法のいずれもが、各成分元素の供給源化合物の水性系での一体化という概念に含まれる。ここで、熟成とは、工業原料もしくは半製品を、一定時間、一定温度等の特定条件のもとに処理して、必要とする物理性、化学性の取得、上昇あるいは所定反応の進行等を図る操作をいい、一定時間とは、通常10分〜24時間の範囲であり、一定温度とは通常室温〜水溶液又は水分散液の沸点範囲をいう。
【0031】
前記の一体化の具体的な方法としては、例えば、触媒成分から選ばれた酸性塩を混合して得られた溶液と、触媒成分から選ばれた塩基性塩を混合して得られた溶液とを混合する方法等が挙げられ、具体例としてモリブデン化合物の水溶液に、鉄化合物とニッケル化合物及び/又はコバルト化合物との混合物を加温下添加し混合する方法等が挙げられる。なお、必要に応じてシリカの添加、混合もこの前工程で行うのが好ましい。
【0032】
このようにして得られた原料化合物の水溶液又は水分散液を60〜90℃に加温し、熟成する。
この熟成とは、前記触媒前駆体用スラリーを所定温度で所定時間、撹拌することをいう。この熟成により、スラリーの粘度が上昇し、スラリー中の固体成分の沈降を緩和し、とりわけ次の乾燥工程での成分の不均一化を抑制するのに有効となり、得られる最終製品である複合酸化物触媒の原料転化率や選択率等の触媒活性がより良好となる。
【0033】
前記熟成における温度は、60〜90℃が好ましく、70〜85℃がより好ましい。熟成温度が60℃未満では、熟成の効果が十分ではなく、良好な活性を得られない場合がある。一方、90℃を超えると、熟成時間中の水の蒸発が多く、工業的な実施には不利である。更に100℃を超えると、溶解槽に耐圧容器が必要となり、また、ハンドリングも複雑になり、経済性及び操作性の面で著しく不利となる。
【0034】
前記熟成にかける時間は、2〜12時間がよく、3〜8時間が好ましい。熟成時間が2時間未満では、触媒の活性及び選択性が十分に発現しない場合がある。一方、12時間を超えても熟成効果が増大することはなく、工業的な実施には不利である。
前記撹拌方法としては、任意の方法を採用することができ、例えば、撹拌翼を有する撹拌機による方法や、ポンプによる外部循環による方法等が挙げられる。
【0035】
熟成されたスラリーは、そのままで、又は乾燥した後、加熱処理を行う。乾燥する場合の乾燥方法及び得られる乾燥物の状態については特に限定はなく、例えば、通常のスプレードライヤー、スラリードライヤー、ドラムドライヤー等を用いて粉体状の乾燥物を得てもよいし、また、通常の箱型乾燥器、トンネル型焼成炉を用いてブロック状又はフレーク状の乾燥物を得てもよい。
【0036】
前記の原料塩水溶液又はこれを乾燥して得た顆粒あるいはケーキ状のものは、空気中で200〜400℃、好ましくは250〜350℃の温度域で短時間の熱処理を行う。その際の炉の形式及びその方法については特に限定はなく、例えば、通常の箱型加熱炉、トンネル型加熱炉等を用いて乾燥物を固定した状態で加熱してもよいし、また、ロータリーキルン等を用いて乾燥物を流動させながら加熱してもよい。
【0037】
加熱処理後に得られた触媒前駆体の灼熱減量は、0.5〜5重量%であることが好ましく、1〜3重量%であるのがより好ましい。灼熱減量をこの範囲とすることで、原料転化率や選択率が高い触媒を得ることができる。なお、灼熱減量は、次式により与えられる値である。
灼熱減量(%)=[(W0−W1)/W0]×100
・W0:触媒前駆体を150℃で3時間乾燥して付着水分を除いたものの重量(g)
・W1:付着水分を除いた前記触媒前駆体を更に500℃で2時間熱処理した後の重量(g)
前記の後工程では、前記の前工程において得られる触媒前駆体とモリブデン化合物(全原子比aからa1相当を差し引いた残りのa2相当)とビスマス化合物の一体化を、水性溶媒下で行う。この際、アンモニア水を添加するのが好ましい。X、Y、Z成分の添加もこの後工程で行うのが好ましい。また、この発明のビスマス供給源化合物は、水に難溶性ないし不溶性のビスマスである。この化合物は、粉末の形態で使用することが好ましい。触媒製造原料としてのこれら化合物は粉末より大きな粒子のものであってもよいが、その熱拡散を行わせるべき加熱工程を考えれば小さい粒子である方が好ましい。従って、原料としてのこれらの化合物がこのように粒子の小さいものでなかった場合は、加熱工程前に粉砕を行うべきである。
【0038】
次に、得られたスラリーを充分に撹拌した後、乾燥する。このようにして得られた乾燥品を、押出し成型、打錠成型、あるいは担持成型等の方法により任意の形状に賦形する。次に、このものを、好ましくは450〜650℃の温度条件にて1〜16時間程度の最終熱処理に付す。以上のようにして、高活性で、かつ目的とする酸化生成物を高い収率で与える複合酸化物触媒が得られる。
【0039】
<分子状酸素含有ガス>
本発明の分子状酸素含有ガスとは、通常、分子状酸素が10体積%以上、好ましくは、15体積%以上、更に好ましくは20体積%以上含まれるガスのことであり、具体的に好ましくは空気である。なお、分子状酸素含有ガスを工業的に用意するのに必要なコストが増加するという観点から、分子状酸素の含有量の上限としては、通常50体積%以下であり、好ましくは、30体積%以下、更に好ましくは25体積%以下である。本発明の分子状酸素含有ガスに含まれうる酸素以外の成分としては、具体的には、窒素、アルゴン、ネオン、ヘリウム、CO、CO2、水等が挙げられる。これら酸素以外の成分の量は、窒素の場合、通常90体積%以下、好ましくは85体積%以下、より好ましくは80体積%以下である。窒素以外の成分の場合、通常10体積%以下、好ましくは1体積%以下である。この量が多すぎると、反応に必要な酸素を供給するのが難しくなる傾向にある。
【0040】
<ガス供給>
本発明では、反応器に原料ガスを供給するにあたり、原料ガスと分子状酸素含有ガスとを混合し、その混合されたガス(以下、「混合ガス」呼ぶことがある)を反応器に供給する必要がある。なお、本発明の混合ガス中の、原料ガスの割合としては、通常、4.2vol%以上であり、好ましくは7.6vol%以上、更に好ましくは8.0vol%以上である。この値が大きくなるほど、反応器のサイズを小さくでき、建設費および運転に要するコストが低減する傾向にある。また、一方、上限は、20.0vol%以下であり、
好ましくは、17.0vol%以下、更に好ましくは、15.0vol%以下である。この値が小さくなるほど、原料ガス中の触媒上へのコーキングの起因物質も低減するため、触媒のコーキングが発生しにくく好ましい。
【0041】
<窒素ガス、水(水蒸気)>
また、混合ガスと共に、窒素ガス、及び水(水蒸気)を反応器に供給してもよい。窒素ガスは、混合ガスが爆鳴気を形成しないように可燃性ガスと酸素の濃度を調整するという理由から、水(水蒸気)は窒素ガスと同様に可燃性ガスと酸素の濃度を調整するという理由と触媒のコーキングを抑制するという理由から、混合ガスに、水(水蒸気)と窒素ガスとを更に混合し反応器に供給するのが好ましい。
【0042】
反応器に水蒸気を供給する場合、前記原料ガスの供給量に対して0.5〜5.0の比率で導入することが好ましい。この比率が大きくなるほど、廃水量が増加する傾向にあり、小さくなるほど、目的生成物であるブタジエンの収率が低下する傾向にある。そのため、水蒸気を前記原料ガスの供給量に対して、好ましくは、0.8〜4.5であり、更に好ましくは、1.0〜4.0である。
【0043】
反応器に窒素ガスを供給する場合、前記原料ガスの供給量に対して0.5〜8.0の比率(体積比)で導入することが好ましい。この比率が大きくなるほど、後工程の生成ガスを圧縮する工程の負荷が上がる傾向にあり、小さくなるほど、反応器に供給する水蒸気の使用量が増加する傾向にある。そのため、窒素ガスを前記原料ガスの供給量に対して、好ましくは、1.0〜6.0、更に好ましくは、2.0〜5.0の比率(体積比)で供給する。
【0044】
原料ガスと分子状酸素含有ガスの混合ガス、及び必要により供給される窒素ガス、及び水(水蒸気)を供給する方法は特に限定されず、別々の配管で供給してもよいが、爆鳴気の形成を確実に回避するために、混合ガスを得る前に、予め窒素ガスを原料ガス、もしくは分子状酸素含有ガスに供給しておき、その状態で、原料ガスと分子状酸素含有ガスとを混合して混合ガスを得、該混合ガスを供給することが好ましい。
【0045】
以下に、混合ガスの代表的な組成を示す。
[混合ガス組成]
・n−ブテン:C4留分合計に対して50〜100vol%
・C4留分合計:5〜15vol%
・O2:C4留分合計に対して40〜120vol/vol%
・N2:C4留分合計に対して500〜1000vol/vol%
・H2O:C4留分合計に対して90〜900vol/vol%
反応器に供給する原料ガスは、分子状酸素含有ガスと混合されると、酸素と可燃性ガスの混合物となることから、爆発範囲に入らないように各々のガス(原料ガス、空気、及び必要に応じて窒素ガスと水(水蒸気))を供給する配管に設置された流量計にて流量を監視しながら、反応器入り口の組成制御を行い、例えば、n−ブテン(1−ブテン及び/又は2−ブテン)からブタジエンを製造する場合、後述の混合ガス組成の範囲に調整される。なお、ここでいう爆発範囲とは、酸素と可燃性ガスの混合ガスが何らかの着火源の存在下で着火するような組成を持つ範囲のことである。可燃性ガスの濃度がある値より低いと着火源が存在しても着火しないことが知られており、この濃度を爆発下限界という。また可燃性ガスの濃度がある値より高いとやはり着火源が存在しても着火しないことが知られており、この濃度を爆発上限界という。各々の値は酸素濃度に依存しており、一般に酸素濃度が低いほど両者の値が近づき、酸素濃度がある値になったとき両者が一致する。このときの酸素濃度を限界酸素濃度と言い、酸素濃度がこれより低ければ可燃性ガスの濃度によらず混合ガスは着火しない。
【0046】
本発明の反応を開始するときは、最初に反応器に供給する分子状酸素含有ガス、窒素、水蒸気の量を調整して反応器入り口の酸素濃度が限界酸素濃度以下になるようにしてから可燃性ガス(主に原料ガス)の供給を開始し、次いで可燃性ガス濃度が爆発上限界よりも濃くなるように可燃性ガス(主に原料ガス)と空気などの分子状酸素含有ガスの供給量を増やしていくのが良い。可燃性ガス(主に原料ガス)と分子状酸素含有ガスの供給量を増やしていくときに窒素および/または水蒸気の供給量を減らして混合ガスの供給量が一定となるようにしても良い。こうすることで、配管および反応器における混合ガスの滞留時間を一定に保ち、圧力の変動を抑えることが出来る。
【0047】
なお、爆発範囲外であっても、ある温度、圧力条件下で、ある時間保持されると発火する場合がある。このときの保持時間を発火遅れ時間という。反応器周りを設計するときは原料配管や生成ガス配管の滞留時間が発火遅れ時間以下になるように設計する必要がある。発火遅れ時間は温度や圧力、組成に依存するので一概には言えないが、混合ガス配管の滞留時間は1000秒以下、生成ガス配管の滞留時間は10秒以下もしくは生成ガスを10秒以内に350℃以下に冷却することが望ましい。
【0048】
また、本発明の混合ガス中の原料ガスの割合について、下限値としては、通常、4.2vol%以上であり、好ましくは7.6vol%以上、更に好ましくは9.3vol%以上である。この下限値が大きくなるほど、反応器のサイズを小さくでき、建設費および運転に要するコストが低減する傾向にある。また、一方、上限値は、通常、20.0vol%以下であり、好ましくは、17.0vol%以下、更に好ましくは、15.0vol%以下である。この上限値が小さくなるほど、原料ガス中の触媒上へのコーキングの起因物質も低減するため、触媒のコーキングが発生しにくくなる。
【0049】
<反応器>
本発明の酸化脱水素反応に用いられる反応器は特に限定されないが、具体的には、管型反応器、槽型反応器、又は流動床反応器が挙げられ、好ましくは、固定床反応器、より好ましくは固定床の多管式反応器やプレート式反応器であり、最も好ましくは固定床の多管式反応器である。これらの反応器は一般に工業的に用いられているものであり特に制限はない。
【0050】
また、反応器が固定床反応器の場合、反応器には、上述の酸化脱水素反応触媒を有する触媒層が存在する。その触媒層は、触媒のみからなる層から構成されていても、触媒と該触媒と反応性の無い固形物とを含む層のみから構成されていても、触媒と該触媒と反応性の無い固形物とを含む層と触媒のみからなる層の複数の層から構成されていてもよいが、触媒層が、触媒と該触媒と反応性の無い固形物とを含む層を含むことで、反応時の発熱による触媒層の急激な温度上昇を抑制できるので、触媒層に反応性の無い固形物を有することが好ましい。尚、複数の層を有する場合、複数の層は反応器の入口から反応器の生成ガス出口の方向に向かって層状に形成される。触媒層が触媒と該触媒と反応性の無い固形物とを含む層を含む場合、下記式(3)で示される触媒希釈率が10体積%以上であることが好ましく、より好ましくは、15体積%以上、更に好ましくは、20体積%以上である。この下限値が大きくなるほど、触媒層中でのホットスポットの発生を抑えることができ、触媒上への炭素分の蓄積を抑制する効果が高くなる。触媒層の希釈率の上限は特に限定されないが、通常、99vol%以下であり、好ましくは90vol%以下、更に好ましくは、80vol%以下である。この上限値が小さくなるほど、反応器の大きさを小さくすることができ、建設費や運転コストを抑えることができる。
【0051】
希釈率(体積%)=[(触媒と反応性の無い固形物の体積)/(触媒の体積+触媒と反
応性の無い固形物の体積)]×100 ・・・(3)
尚、上述の通り、反応器内に設けられる触媒層は、単層でも2層以上でもよいが、好ましくは、2〜5層である。触媒層の数が多くなるほど、触媒充填作業が煩雑になる傾向にあり、触媒層の数が少なくなるほど、容易という傾向にある。また、反応器内に触媒層を2層以上設ける場合は、各触媒層の希釈率は、反応条件や反応温度によって適宜決めることができるが、希釈率が異なる触媒層を設けることが好ましい。
【0052】
本発明に用いられる反応性の無い固形物は、共役ジエン生成反応条件下で安定であり、炭素原子数4以上のモノオレフィン等の原料物質、及び共役ジエン等の生成物と反応性がない材質のものであれば特に限定されず、一般的に、イナートボールとも呼ばれることがある。具体的には、アルミナ、ジルコニア等のセラミック材等が挙げられる。また、その形状は、特に限定されず、球状、円柱状、リング状、不定形のいずれでもよい。また、その大きさは、本発明で使用する触媒と同等の大きさであればよい。その粒径は、通常、2〜10mm程度である。 触媒層の充填長は、充填される触媒の活性(反応性の無い固形物で希釈される場合は、希釈された触媒としての活性)、反応器の大きさ、反応原料ガス温度、反応温度及び反応条件が決まれば、物質収支及び熱収支計算によって求めることができる。
【0053】
また、本発明では、固定床反応器内に、上記の触媒層以外に、反応性が無い固形物のみで形成される層を有していても良い。通常、反応器の原料ガスの入口一番近い箇所と生成ガス出口側に最も近い箇所の両方、若しくはどちらか一方に、反応性が無い固形物のみで形成される層を設けるのがよい。これら層の大きさは、触媒層の大きさや反応器の大きさ、及び反応条件に応じて、適宜決めることができる。
【0054】
本発明では、触媒層中の固体触媒を、下記式(1)で示される破砕率(重量%)を0.0重量%以上40.0重量%以下とする必要がある。
破砕率(重量%)={(反応器中の破砕触媒の重量)/(反応器中の全触媒重量)}×100 …(1)
上記式において破砕触媒とは、反応器内で使用される触媒が製造された当初の形状、又は反応器に充填する際の形状に対して、それらの形状と同等の形状ではないものを指す。具体的には、反応器内で触媒が割れたり、或いは粉化したりする際に発生する触媒である。通常、破砕触媒は、その大きさとしては、触媒の製造時、又は反応器に充填する際の大きさに比べて、小さいものが多い。破砕触媒の特定は、酸化脱水素反応停止後に反応器を開放して、反応器内の内容物を抜き出した後、それらを目視にて選別する、又は篩にかけることで特定できる。反応器内の内容物を反応器から抜き出す際は、内容物が破壊されないように抜き出すことが好ましい。反応器から抜き出した内容物を篩にかける際は、篩の目開きは、使用する固体触媒の大きさによって異なるが、目開き4.5mm〜4.9mmの篩を使用することで、割れた触媒や粉化した触媒を分離回収することが可能である。なお、反応器内に反応性の無い固形物が存在する場合は、破砕触媒を特定する前に、予め除いておくことが好ましい。
【0055】
このようにして特定された破砕触媒の重量を測定して、反応器中の破砕触媒の重量を得ることができる。また、反応器中の全触媒重量は、反応器に反応に使用する触媒を充填する前に、予め測定しておけばよい。
反応器が固定床の多管式反応器の場合、即ち、反応器内に同じ種類の触媒と重量が同じ触媒が充填され、且つ同じ長さと管径を持つ反応管が複数存在する際は、全ての反応管について、破砕触媒の特定を行い、破砕触媒の重量を測定してもよいが、複数の反応管の中から少なくとも1本以上の反応管を選定して、破砕触媒の特定とその重量の測定を行い、それら反応管で得られた破砕触媒の重量の平均値を、反応器中の破砕触媒の重量として破砕率を求めてもよい。なお、この際、反応器中の全触媒重量は、上記で選定された反応管の触媒の全重量となる。
【0056】
本発明では、触媒層中の固体触媒を、上記式(1)で示される破砕率(重量%)を0.0重量%以上40.0重量%以下とする必要があるが、好ましくは、1.0重量%以上30.0重量%以下である。更に好ましくは、5.0重量%以上25.0重量%以下である。この値が小さくなるほど、触媒の充填に時間を要し、また製造時の触媒の強度を高くする必要傾向がある。
【0057】
<反応条件>
本発明の酸化脱水素反応は発熱反応であり、反応により温度が上昇するが、本発明では、通常、反応温度は250〜450℃、好ましくは、280〜400℃の範囲に調整される。この温度が大きくなるほど、触媒活性が急激に低下しやすい傾向にあり、小さくなるほど、目的生成物である共役ジエンの収率が低下する傾向にある。反応温度は、熱媒体(例えば、ジベンジルトルエンや亜硝酸塩など)を使用して制御することができる。なお、ここでいう反応温度は熱媒体の温度のことのことである。
【0058】
また、本発明における反応器内温度は、特に限定されないが、通常、250〜450℃、好ましくは、280〜400℃、更に好ましくは、320〜395℃である。触媒層の温度が450℃を超えると、反応を継続するに従って、急激に触媒活性が低下する恐れがある傾向にあり、一方、触媒層の温度が250℃を下回ると、目的生成物である共役ジエンの収率が低下する傾向にある。反応器内温度は、反応条件によって決定されるが、触媒層の希釈率や混合ガスの流量等で制御することができる。なお、ここでいう反応器内温度とは、反応器出口での生成ガスの温度、又は触媒層を有する反応器の場合は、その触媒層の温度のことである。
【0059】
本発明の反応器内の圧力は、特に限定されないが、下限は、通常、0MPaG以上、好ましくは、0.001MPaG以上、更に好ましくは、0.01MPaG以上である。この値が大きくなるほど、反応器に反応ガスを多量に供給できるというメリットがある。一方、上限は、0.5MPaG以下であり、好ましくは0.3MPaG以下、更に好ましくは、0.1MPaGである。この値が小さくなるほど、爆発範囲が狭くなる傾向にある。
【0060】
本発明における反応器の滞留時間は、特に限定されないが、下限は、好ましくは、0.72秒以上、更に好ましくは0.90秒以上である。この値が大きくなるほど、原料ガス中のモノオレフィンの転化率が高くなるというメリットがある。一方、上限は、好ましくは2.80秒以下、更に好ましくは、2.10秒以下である。この値が小さくなるほど、反応器が小さくなる傾向にある。
【0061】
また、本発明では、反応器内の触媒量に対する混合ガスの流量の比(以下、SVと略記することがある)は、特に限定されないが、好ましくは、500〜5000h−1であり、更に好ましくは、1300〜4500h−1であり、特に好ましくは、1700〜4000h−1ある。この値が大きくなるほど、固形物の析出が抑制される傾向にあり、小さくなるほど、固形物が析出しやすい傾向にある。
【0062】
反応器の入口と出口との流量差としては、原料ガスの反応器入口での流量、及び生成ガスの反応器出口での流量に依存するが、通常、入口流量に対する出口の流量の比率が100〜110vol%、好ましくは、102〜107vol%、更に好ましくは103〜105vol%である。n−ブテン(1−ブテン及び2−ブテン)からブタジエンを製造する場合、出口流量が増えるのはブテンが酸化脱水素されてブタジエンと水が生成する反応や副反応でCOやCO2が生成する反応において化学量論的に分子数が増えるためである。出口流量の増加が少ないと反応が進行していないので好ましくなく、出口流量が増えすぎると副反応でCOやCO2が増加しているため好ましくない。
【0063】
かくして、原料ガス中のモノオレフィンの酸化脱水素反応により、該モノオレフィンに対応する共役ジエンが生成することとなり、該共役ジエンを含有する生成ガスを取得する。生成ガス中に含まれる原料ガス中のモノオレフィンに対応する共役ジエンの濃度は、原料ガス中に含まれるモノオレフィンの濃度に依存するが、通常1〜15vol%、好ましくは、5〜13vol%、更に好ましくは9〜11vol%である。共役ジエンの濃度が大きいほど、回収コストが低いというメリットがあり、小さいほど次工程で圧縮したときに重合などの副反応が起き難いというメリットがある。また、生成ガス中には未反応のモノオレフィンも含まれていてもよく、その濃度は、通常0〜7vol%、好ましくは、0〜4vol%、更に好ましくは0〜2vol%である。なお、本発明では、生成ガス中に含まれる高沸点副生物は、使用する原料ガス中に含まれる不純物の種類によって異なるが、常圧下での沸点が200〜500℃のものを言う。n−ブテン(1−ブテン及び2−ブテン)からブタジエンを製造する場合、具体的に、フタル酸、アントラキノン、フルオレノン等である。これらの量は、特に限定されないが、通常、反応ガス中に0.05〜0.10vol%である。
【0064】
<後工程>
本発明の共役ジエンの製造方法においては、共役ジエンを含有する生成ガスから共役ジエンを分離するために、更に、冷却工程、脱水工程、溶媒吸収工程、分離工程、精製工程等を有していてもよい。なお、反応器から得られる生成ガスは、脱水工程で、圧縮ガス、脱水ガスとなる。しかし、これらのガスは、水以外の含有割合は同一であり、また、含まれる水のほとんどは液状なので、各ガスの気体部分の成分割合は、同一と考えてよい。このため、以下において、生成ガス、圧縮ガス、及び脱水ガスについて、単に「生成ガス」と称する場合がある。
【0065】
(冷却工程)
本発明では、共役ジエンを含む生成ガスを冷却する冷却工程を有していてもよい。冷却工程については、反応器出口から抜き出される生成ガスを冷却できる工程であれば、特に限定されないが、好適には、冷却溶媒と生成ガスとを直接接触させて冷却させる方法が用いられる。冷却溶媒としては、特に限定されないが、好ましくは水やアルカリ水溶液であり、最も好ましくは水である。
【0066】
また、生成ガスの冷却温度は、反応器出口から抜き出される生成ガス温度や冷却溶媒の種類などによって異なるが、通常、5〜100℃、好ましくは、10〜50℃、更に好ましくは、15〜40℃に冷却される。冷却される温度が高くなるほど、建設費と運転に要するコストを下げられる傾向にあり、低くなるほど、生成ガスを圧縮する工程の負荷を下げられる傾向にある。冷却塔内の圧力は、特に限定されないが、通常は、0.03MPaGである。
尚、冷却塔を使って冷却する場合、生成ガス中に高沸点副生物が多く含まれていると、高沸点副生物同士の重合や、工程内での高沸点副生物に起因する固形析出物の堆積が起きやすくなる。また、冷却塔で使用される冷却溶媒は、循環使用されることが多いため、共役ジエンの製造を連続的に継続すると、固形析出物での閉塞が起きることがある。
【0067】
そのため、可能な限り、反応器出口から抜き出される生成ガス中の高沸点副生物を冷却工程に持ち込ませないように、予めフィルターなどで除去するすることが好ましい。
(脱水工程)
また、本発明では、反応器から排出される生成ガスに含まれる水分を除去する脱水工程を有していても良い。脱水工程を設けることにより、後段のプロセスにおける各工程における水分による機器腐食や、後述する溶媒吸収工程や溶媒分離工程で使用する溶媒への不純物の蓄積を防止することができるため、好ましい。
【0068】
本発明の脱水工程については、生成ガスに含まれる水分を除去できる工程であれば、特に限定されない。脱水工程は反応器の後段の工程であれば、どこで行ってもよいが、上述の冷却工程の後に脱水工程を行うことが好ましい。通常、反応器から排出される生成ガス中に含まれる水分量は、原料ガスの種類や分子状酸素含有ガスの量、更には、原料ガスと共に混合される水蒸気等により異なるが、通常は、4〜35vol%、好ましくは10〜30vol%である(これが水を使用した冷却工程を経過した場合には、100volppm〜2.0vol%まで水分濃度が低減されている)。また、露点として、0〜100℃、好ましくは、10〜80℃である。
【0069】
生成ガスから水分を除去する手段としては、特に限定されないが、酸化カルシウム、塩化カルシウム、モレキュラーシーブ等の乾燥剤(水分吸着剤)を利用することができる。この中でも、再生の容易さ、取り扱いの容易さという観点から、モレキュラーシーブ等の乾燥剤(水分吸着剤)が好ましく利用される。
脱水工程にモレキュラーシーブ等の乾燥剤を利用する場合は、水以外にも生成ガス中に含まれる高沸点副生物が吸着除去される。ここで除去される高沸点副生物は、アントラキノン、フルオレノン、フタル酸などのことである。
【0070】
脱水工程を経て得られる生成ガス中の水分含有量は、通常は10〜10000volppm、好ましくは、20〜1000volppmであり、露点としては、−60〜80℃、好ましくは、−50〜20℃である。この生成ガス中の水分含有量が多くなるほど、溶媒吸収塔や溶媒分離塔のリボイラーの汚れが増加する傾向にあり、一方で、少なくなると、脱水工程で使用する用役コストが増加する傾向にある。
【0071】
(溶媒吸収工程)
本発明では、生成ガスを吸収溶媒と接触させてオレフィンや共役ジエンなどの炭化水素を吸収溶媒に吸収させ共役ジエンを含む溶媒を得る溶媒吸収工程を有することが好ましい。好ましい理由としては、共役ジエンの分離に要するエネルギーコストの低減という観点から、生成ガスを溶媒に吸収させて共役ジエンの回収することが好ましい。溶媒吸収工程については、反応後の工程であれば、特に限定されないが、上述の脱水工程の後に設けることが好ましい。
【0072】
溶媒吸収工程で生成ガスを溶媒に吸収させる具体的な方法としては、例えば吸収塔を用いる方法が好ましい。吸収塔の種類としては、充填塔、濡れ壁塔、噴霧塔、サイクロンスクラバー、気泡塔、気泡攪拌槽、段塔(泡鐘塔、多孔板塔)、泡沫分離塔などが使用可能である。好ましくは、噴霧塔、泡鐘塔、多孔板塔である。
吸収塔を用いる場合、通常は、吸収溶媒と生成ガスとを向流接触させることで、生成ガス中の共役ジエンと未反応の炭素原子数4以上のモノオレフィン並びに炭素原子数3以下の炭化水素化合物が溶媒に吸収される。炭素原子数3以下の炭化水素化合物としては、例えば、メタン、アセチレン、エチレン、エタン、メチルアセチレン、プロピレン、プロパン、又はアレンなどが挙げられる。
【0073】
溶媒吸収工程において、吸収塔を用いて生成ガスを回収する場合、吸収塔内の圧力は、特に限定されないが、通常、0.1〜2.0MPaG,好ましくは、0.2〜1.5MPaG、更に好ましくは0.25〜1.0MPaGである。この圧力が大きいほど、吸収効率が良くなるというメリットがあり、小さいほど吸収塔へのガス導入時の昇圧に要するエネルギーを削減でき、さらに液中の溶存酸素量を低減できるというメリットがある。
【0074】
また、吸収塔10内の温度は、特に限定されないが、通常0〜50℃、好ましくは、10〜40℃、更に好ましくは20〜30℃である。この温度が高いほど、酸素や窒素など
が溶媒に吸収されにくいというメリットがあり、低いほど共役ジエンなどの炭化水素の吸収効率が良くなるというメリットがある。
本発明の溶媒吸収工程で使用する有機溶媒としては、特に限定されないが、C6〜C10の飽和炭化水素やC6〜C8の芳香族炭化水素、アミド化合物などが用いられる。具体的には、例えばジメチルホルムアミド(DMF)、トルエン、キシレン、N−メチル−2−ピロリドン(NMP)等を用いることができる。これらの中でも、好ましくは、無機ガスを溶解しにくいことからC6〜C8の芳香族炭化水素が好ましく、特にトルエンが好ましい。
【0075】
吸収溶媒の使用量には特に制限はないが、回収工程に供給される目的生成物の流量に対して、通常、1〜100重量倍、好ましくは、2〜50重量倍である。吸収溶媒の使用量が多くなるほど、不経済となる傾向にあり、少なくなるほど、共役ジエンの回収効率が低下する傾向にある。
溶媒吸収工程で得られる共役ジエンを含む溶媒中には、主として目的生成物である共役ジエンが含まれており、その共役ジエンの溶媒吸収液中の濃度としては、通常は1〜20重量%であり、好ましくは3〜10重量%である。この溶媒中の共役ジエンの濃度が高いほど、共役ジエンの重合あるいは揮発による消失分が多くなる傾向にあり、低いほど、同じ生産量での溶媒の循環必要量が増加する為に、運転に要するエネルギーコストが大きくなる傾向にある。
【0076】
また、得られる共役ジエンを含む溶媒に、若干量の窒素、酸素も吸収されているため、溶媒に溶存する窒素や酸素をガス化して除去する脱気工程を有していても良い。脱気工程では、溶媒吸収液中に溶存する窒素や酸素をガス化して除去できる工程であれば、特に限定されない。
(分離工程)
このようにして得られた共役ジエンを含む溶媒から粗共役ジエンの分離を行う分離工程を有していてもよく、この工程により粗共役ジエンを得ることができる。分離工程としては、共役ジエンの溶媒吸収液から粗共役ジエンを分離できる工程であれば、特に限定されないが、通常、蒸留分離により粗共役ジエンを分離することができる。具体的には、例えば、リボイラーとコンデンサーにより共役ジエンの蒸留分離が行われ、塔頂付近より共役ジエン留分が抜き出される。分離された吸収溶媒は塔底から抜き出され、前段工程に溶媒を使用する回収工程を有する場合は、その回収工程で吸収溶媒として循環使用される。溶媒は循環使用するうち不純物が蓄積する場合があり、一部を抜き出して蒸留やデカンテーション、沈降、吸着剤やイオン交換樹脂などとの接触処理などの公知の精製方法により不純物を除去することが望ましい。
【0077】
分離工程で使用する蒸留塔の蒸留時の圧力は任意に設定することができるが、通常は、塔頂圧力を0.05〜2.0MPaGとすることが好ましい。より好ましくは塔頂圧力が0.1〜1.0MPaGであり、特に好ましくは0.15〜0.8MPaGの範囲である。この塔頂圧力が低すぎると、留出した共役ジエンを低温で凝縮するために多大なコストが必要となり、また高すぎると蒸留塔の塔底部の温度が高くなり、蒸気コストの増大となってしまう。
【0078】
塔底温度は通常50〜200℃であり、好ましくは80〜180℃、より好ましくは100〜160℃である。塔底温度が低すぎると共役ジエンを塔頂から留出させるのが困難となる。また温度が高すぎると、溶媒も塔頂から留出してしまう。還流比は1〜10で差し支えなく、好ましくは2〜4である。
蒸留塔としては充填塔、棚段塔のいずれもが使用できるが、多段蒸留が好ましい。共役ジエンと溶媒を分離するには、蒸留塔理論段を5段以上、特に10段〜20段とするのが好ましい。50段を越える蒸留塔は、蒸留塔建設の経済性、運転難易度、及び安全管理の
ためには好ましくない。また段数が小さすぎると分離が困難となる。
【0079】
(精製工程)
前記共役ジエンの分離工程で粗共役ジエンが得られるが、この粗共役ジエンを蒸留精製等により、更に精製された高純度の共役ジエンとする精製工程を有していてもよい。ここで使用する蒸留塔の蒸留時の圧力は任意に設定することができるが、通常は、塔頂圧力を0.05〜0.4MPaGとすることが好ましい。より好ましくは塔頂圧力が0.1〜0.3MPaGであり、特に好ましくは0.15〜0.2MPaGの範囲である。この塔頂圧力が低すぎると、留出した共役ジエンを低温で凝縮するために多大なコストが必要となり、また高すぎると蒸留塔の塔底部の温度が高くなり、蒸気コストの増大となってしまう。
【0080】
塔底温度は通常30℃〜100℃であり、好ましくは40℃〜80℃、より好ましくは50℃〜60℃である。塔底温度が低すぎると共役ジエンを塔頂から留出させるのが困難となる。また温度が高すぎると、塔頂で凝縮させる量が増えてコストが増大してしまう。また、還流比は1〜10で差し支えなく、好ましくは2〜4である。
蒸留塔としては充填塔、棚段塔のいずれもが使用できるが、多段蒸留が好ましい。共役ジエンとフランなどの不純物を分離するには、蒸留塔理論段を5段以上、特に10段〜20段とするのが好ましい。50段を越える蒸留塔は、蒸留塔建設の経済性、運転難易度、及び安全管理のためには好ましくない。また段数が小さすぎると分離が困難となる。このようにして得られる精製された共役ジエンは、純度が99.0〜99.9%の共役ジエンである。
【0081】
[プロセスの実施形態]
以下に、図面を参照して、本発明の共役ジエンの製造方法に関するプロセスの実施形態について、ブタジエンを製造する例を挙げて説明する。
図1は本発明プロセスの実施の態様の一つである。
図1において、1は反応器、2はクエンチ塔、3,6,13は冷却器(熱交換器)、4,7,14はドレンポット、8A,8Bは脱水塔、9は加熱器(熱交換器)、10は溶媒吸収塔、11は脱気塔、12は溶媒分離塔を示し、符号100〜126は配管を示す。原料となるn−ブテン或いは前述のBBSS等のn−ブテンを含む混合物を、気化器(図示せず)でガス化して、配管101より導入すると共に、配管102、103、104より、窒素ガス、空気(分子状酸素含有ガス)、及び水(水蒸気)をそれぞれ導入し、これらの混合ガスを予熱器(図示せず)で150〜400℃程度に加熱した後、配管100より触
媒が充填された多管式の反応器1(酸化脱水素反応器)に供給する。反応器1からの反応
生成ガスは、配管105よりクエンチ塔2に送給され、20〜99℃程度に冷却される。クエンチ塔2には、配管106より冷却水が導入され、生成ガスと向流接触する。そして、この向流接触で生成ガスを冷却した水は、配管107より排出される。なお、この冷却排水は、熱交換器(図示せず)で冷却されて再度クエンチ塔2において循環使用される。クエンチ塔2で冷却された生成ガスは、塔頂から留出され、次いで配管108より冷却器3を経て室温に冷却される。冷却により発生した凝縮水は配管109よりドレンポット4に分離される。水分離後のガスは更に配管110を経て圧縮機5で0.1〜0.5MPa程度に昇圧され、昇圧ガスは配管111を経て冷却器6で再度10〜30℃程度に冷却される。冷却により発生した凝縮水は配管112よりドレンポット7に分離される。水分離後の圧縮ガスは、モレキュラーシーブ等の乾燥剤が充填された脱水塔8A,8Bに導入され脱水処理される。脱水塔8A,8Bは圧縮ガスの脱水と乾燥剤の加熱乾燥による再生とが交互に行われる。即ち、圧縮ガスは、まず、配管113,113aを経て脱水塔8Aに導入されて脱水処理され、配管114a,114を経て溶媒吸収塔10に送給される。この間に、脱水塔8Bには、配管122、加熱器9、配管123,123a,123bを経て150〜250℃程度に加熱された窒素ガスが導入され、乾燥剤の加熱による水分の脱
着が行われる。脱着した水分を含む窒素ガスは、配管124a,124b、124を経て冷却器13で室温まで冷却され、凝縮水が配管125よりドレンポット14に分離された後、配管126より排出される。
【0082】
脱水塔8Aの乾燥剤が飽和に達したら、ガス流路を切り換え、脱水塔8Bで圧縮ガスの脱水処理を行い、脱水塔8A内の乾燥剤の再生を行う。
脱水工程における脱水塔内の乾燥剤の再生時間は、特に限定されないが、通常6〜48時間、好ましくは、12〜36時間、更に好ましくは18〜30時間である。
脱水塔8A,8Bからの脱水ガスは、必要に応じて冷却器(図示せず)で10〜30℃程度に冷却された後、溶媒吸収塔10に送給され、配管115からの溶媒(吸収溶媒)と向流接触される。これにより、脱水ガス中の共役ジエンや未反応の原料ガスが吸収溶媒に吸収される。吸収溶媒に吸収されなかった成分(offガス)は、溶媒吸収塔10の塔頂より配管117を経て排出され燃焼廃棄される。このとき、吸収溶媒として、トルエンのような比較的沸点の低い溶媒を用いると経済的に無視できない量の溶媒が配管117を経て揮散することがある。このような場合はより沸点の高い溶媒を用いて沸点の低い溶媒を回収する工程を配管117の先に設けてもよい。この溶媒吸収塔10で、ブタジエンや未反応の原料ガスを吸収溶媒に吸収した溶媒吸収液は、溶媒吸収塔10の塔底より抜き出され、配管116より脱気塔11に送給される。溶媒吸収塔10で得られるブタジエンの溶媒吸収液には、若干量の窒素、酸素も吸収されているため、次いでこの溶媒吸収液を脱気塔11に供給して加熱することにより、液中に溶存する窒素や酸素をガス化して除去する。この際、ブタジエンや原料ガス、溶媒の中には、その一部がガス化することがあるため、この脱気塔11の塔頂に設けたコンデンサ(図示せず)でこれを液化して溶媒吸収液中に回収する。凝縮しなかった原料ガス、ブタジエン等は窒素、酸素の混合ガスとして配管118より抜き出され、共役ジエンの回収率を高めるために圧縮機5の入口側へ循環され再度処理が行われる。一方、溶媒吸収液を脱気した脱気処理液は配管119より溶媒分離塔12へ送給される。
【0083】
溶媒分離塔12では、リボイラーとコンデンサーにより共役ジエンの蒸留分離が行われ、塔頂より配管120を経て粗ブタジエン留分が抜き出される。分離された吸収溶媒は塔底より配管121を経て抜き出され、溶媒吸収塔10の吸収溶媒として循環使用される。
【実施例】
【0084】
以下に実施例を挙げて本発明をより具体的に説明する。
[製造例1:複合酸化物触媒の調製]
パラモリブデン酸アンモニウム54gを純水250mlに70℃に加温して溶解させた。次に、硝酸第二鉄7.18g、硝酸コバルト31.8g及び硝酸ニッケル31.8gを純水60mlに70℃に加温して溶解させた。これらの溶液を、充分に攪拌しながら徐々に混合した。
【0085】
次に、シリカ64gを加えて、充分に攪拌した。このスラリーを75℃に加温し、5時間熟成した。その後、このスラリーを加熱乾燥した後、空気雰囲気で300℃、1時間の熱処理に付した。
得られた触媒前駆体の粒状固体(灼熱減量:1.4重量%)を粉砕し、パラモリブデン酸アンモニウム40.1gを純水150mlにアンモニア水10mlを加え溶解した溶液に分散した。次に、純水40mlにホウ砂0.85g及び硝酸カリウム0.36gを25℃の加温下に溶解させて、上記スラリーを加えた。
【0086】
次に、Naを0.45%固溶した次炭酸ビスマス58.1gを加えて、攪拌混合した。このスラリーを130℃、12時間加熱乾燥した後、得られた粒状固体を、小型成型機にて径5mm、高さ4mmの錠剤に打錠成型し、次に500℃、4時間の焼成を行って、触
媒を得た。仕込み原料から計算される触媒は、次の原子比を有する複合酸化物であった。
Mo:Bi:Co:Ni:Fe:Na:B:K:Si=12:5:2.5:2.5:0.4:0.35:0.2:0.08:24
なお、触媒調製の際のモリブデンの原子比a1とa2は、それぞれ6.9と5.1であった。
【0087】
[実施例1]
内径23.0mm、高さ500mmのステンレス製反応管に、予めイナートボール(1
粒あたりの大きさ:約0.065mm3)を27ml充填(充填層長:201mm)しておき、そのイナートボールの充填層の上に、製造例1で製造された複合酸化物触媒8.6mlとイナートボール(1粒あたりの大きさ:約0.065mm3)2.9mlとを混合
して充填し、第1の触媒層を形成した(以下、「触媒層1」とする)。この触媒層1の充填層長は32mmで、触媒希釈率は25%であった。次に、触媒層1の上に、製造例1で製造された複合酸化物触媒2.9mlとイナートボール(1粒あたりの大きさ:約0.0
65mm3)8.6mlと混合して充填し、第2の触媒層を形成した(以下、「触媒層2
」とする)。この触媒層2の充填層長は31mmで、触媒希釈率は75%であった。尚、反応器へ充填した触媒量合計は11.5ml(充填した触媒の全重量:7.51g)であった。
【0088】
これらの反応管には外径2.0mmの挿入管を設置し、挿入管の中に熱電対を設置して反応器内温度を測定した。なお、熱媒体は電気炉を使用した。
そして、予め窒素を1.8L/hr、空気を11.8L/hr、及び水蒸気を5.5L/hrで予熱器に供給しておき、その後、表1に示す組成の原料ガスを2.8L/hr供給し、予熱器内で混合して混合ガスとして350℃に昇温した(反応器導入ガス組成=窒素:8.1vol%、空気:53.9vol%、水蒸気:25.0vol%、原料ガス:13.0vol%)。原料ガスに含まれる代表的な成分の組成(mol%)を表1に示す。この際、触媒量11.5mlに対して、合計ガス流量は21.9ml/hであり、上記式(3)で算出したSVは1900h−1であった。
【0089】
その混合ガスを上記の反応器に供給し、酸化脱水素反応を行った。反応器内温度は、平均で367℃で、圧力はゲージ圧で2kPaであった。反応器出口から得られた生成ガスをガスクロマトグラフィー(GL Sciences社製 型番GC4000)で分析したところ、n−ブテン転化率(1−ブテン、シス−2−ブテン、トランス−2−ブテンの合計での転化率)は87.7mol%、ブタジエン選択率は90.7mol%であった。酸化脱水素反応を200時間行なった後、反応管から触媒を全量取り出し、破砕触媒を目視で特定したところ、その重量は、0.07gであり、上記式(1)による破砕率は1.0重量%であった。その後、抜き出した全ての触媒に付着した炭素の量を測定した(測定装置:LECO社製 炭素硫黄分析装置 型番CS600)ところ、3.5wt%であった。結
果を表−1に示す。
【0090】
[比較例1]
実施例1において、予め製造例1で製造された複合酸化物触媒9.8mlを、全てその粒径が半分以下になるように粉砕し(触媒重量:6.57g)、イナートボール(1粒あ
たりの大きさ:約0.065mm3)3.2mlとを混合して充填されたものを触媒層1とし、触媒層1の上に、予め製造例1で製造された複合酸化物触媒3.2mlを、全てその粒径が半分以下になるように粉砕し(触媒重量:2.14g)とイナートボール(1粒
あたりの大きさ:約0.065mm3)7.7mlと混合して充填されたものを触媒層2として(充填した触媒の全重量:8.71g)、窒素と水蒸気の供給量を、それぞれ3.9L/hr、6.2L/hrに変更し、合計ガス流量を24.7L/hr と変更した以外は、同様の方法で、酸化脱水素反応を行った。
【0091】
反応器出口から得られた生成ガスをガスクロマトグラフィー(GL Sciences社製 型番GC4000)で分析したところ、n−ブテン転化率(1−ブテン、シス−2−ブテン、トランス−2−ブテンの合計での転化率)は90.4mol%、ブタジエン選択率は85.8mol%であった。酸化脱水素反応を200時間行なった後、反応管から触媒を全量取り出した。破砕率は100重量%であった。触媒に付着した炭素の量を測定した(測定装置:LECO社製 炭素硫黄分析装置 型番CS600)。測定した粉砕触媒の炭
素濃度は、10.8wt%であった。結果を表−1に示す。
【0092】
[実施例2]
比較例1において、予め製造例1で製造された複合酸化物触媒2.6mlを、全てその粒径が半分以下になるように粉砕し(触媒重量:1.74g)、この粉砕した触媒と、予め製造例1で製造された複合酸化物触媒7.2ml(触媒重量:4.82g)と、イナートボール(1粒あたりの大きさ:約0.065mm3)3.3mlとを混合して充填されたものを触媒層1とし、触媒層1の上に、予め製造例1で製造された複合酸化物触媒3.3ml(触媒重量:2.21g)とイナートボール(1粒あたりの大きさ:約0.065mm3)9.8mlと混合して充填されたものを触媒層2とした(充填した触媒の全重量:8.77g)以外は、比較例1と同様の方法で、酸化脱水素反応を行った。
【0093】
反応器出口から得られた生成ガスをガスクロマトグラフィー(GL Sciences社製 型番GC4000)で分析したところ、n−ブテン転化率(1−ブテン、シス−2−ブテン、トランス−2−ブテンの合計での転化率)は87.4mol%、ブタジエン選択率は84.6mol%であった。酸化脱水素反応を200時間行った後、反応管から触媒を全量取り出した。破砕率は20重量%であった。触媒に付着した炭素の量を測定した(測定装置:LECO社製 炭素硫黄分析装置 型番CS600)。測定した粉砕触媒の炭素濃
度は、3.9wt%であった。結果を表−1に示す。
【0094】
[比較例2]
実施例2において、予め製造例1で製造された複合酸化物触媒6.5mlを、全てその粒径が半分以下になるように粉砕し(触媒重量:4.36g)、この粉砕した触媒と、予め製造例1で製造された複合酸化物触媒3.3ml(触媒重量:2.21g)と、イナートボール(1粒あたりの大きさ:約0.065mm3)3.3mlとを混合して充填されたものを触媒層1とし、触媒層1の上に、予め製造例1で製造された複合酸化物触媒3.3ml(触媒重量:2.21g)とイナートボール(1粒あたりの大きさ:約0.065mm3)9.8mlと混合して充填されたものを触媒層2とした(充填した触媒の全重量:8.78g)以外は、実施例2と同様の方法で、酸化脱水素反応を行った。
【0095】
反応器出口から得られた生成ガスをガスクロマトグラフィー(GL Sciences社製 型番GC4000)で分析したところ、n−ブテン転化率(1−ブテン、シス−2−ブテン、トランス−2−ブテンの合計での転化率)は88.6mol%、ブタジエン選択率は84.4mol%であった。酸化脱水素反応を200時間行った後、反応管から触媒を全量取り出した。破砕率は50重量%であった。触媒に付着した炭素の量を測定した(測定装置:LECO社製 炭素硫黄分析装置 型番CS600)。測定した粉砕触媒の炭素濃
度は、12.8wt%であった。結果を表−1に示す。
【0096】
【表1】
【0097】
実施例1、2と比較例1、2を対比すると、実施例1、2の抜き出し触媒炭素組成が比較例1、2の抜き出し触媒炭素組成よりも小さいことから、破砕率が0.0重量%以上40.0重量%以下であれば、n−ブテンからブタジエンを製造する際に、反応器中の触媒にコークなどの炭素分が付着するのを低減できていることがわかる。
【符号の説明】
【0098】
1 反応器
2 クエンチ塔
3,6,13 冷却器
4,7,14 ドレンポット
8A,8B 脱水器
9 加熱器
10 溶媒吸収塔
11 脱気塔
12 溶媒分離塔
101〜121 配管
【特許請求の範囲】
【請求項1】
炭素原子数4以上のモノオレフィンを含む原料ガスと分子状酸素含有ガスとを混合した混合ガスを、触媒を有する反応器に供給し、酸化脱水素反応により対応する共役ジエンを生成し、該共役ジエンを含む生成ガスを得る共役ジエンの製造方法において、下記式(1)で示される該反応器中の触媒の破砕率が0.0重量%以上40.0重量%以下であることを特徴とする共役ジエンの製造方法。
破砕率(重量%)={(反応器中の破砕触媒の重量)/(反応器中の全触媒重量)}×100 …(1)
【請求項2】
前記反応器が前記触媒と反応性の無い固形物を更に有することを特徴とする請求項1に記載の共役ジエンの製造方法。
【請求項3】
前記反応器内の全触媒量に対する前記混合ガスの流量の比(体積比)が500〜5000h-1であることを特徴とする請求項1又は2に記載の共役ジエンの製造方法。
【請求項4】
前記触媒が、少なくともモリブデン及びビスマスを含むことを特徴とする請求項1〜3のいずれか1項に記載の共役ジエンの製造方法。
【請求項5】
前記触媒が、下記一般式(2)で表される複合酸化物触媒であることを特徴とする請求項1〜4のいずれか1項に記載の共役ジエンの製造方法。
MoaBibCocNidFeeXfYgZhSiiOj (2)
(式中、Xはマグネシウム(Mg)、カルシウム(Ca)、亜鉛(Zn)、セリウム(Ce)及びサマリウム(Sm)からなる群から選ばれる少なくとも1種の元素であり、Yはナトリウム(Na)、カリウム(K)、ルビジウム(Rb)、セシウム(Cs)及びタリウム(Tl)からなる群から選ばれる少なくとも1種の元素であり、Zはホウ素(B)、リン(P)、砒素(As)及びタングステン(W)からなる群から選ばれる少なくとも1種の元素である。また、a〜jはそれぞれの元素の原子比を表し、a=12のとき、b=0.1〜7、c=0〜10、d=0〜10(但しc+d=1〜10)、e=0.05〜3、f=0〜2、g=0.04〜2、h=0〜3、i=0〜48の範囲にあり、またjは他の元素の酸化状態を満足させる数値である。)
【請求項6】
前記原料ガスが、エチレンの2量化により得られる1−ブテン、シス−2−ブテン、トランス−2−ブテン若しくはこれらの混合物を含有するガス、n−ブタンの脱水素若しくは酸化脱水素反応により生成するブテン留分、又は重油留分を流動接触分解する際に得られる炭素原子数が4の炭化水素を多く含むガスであることを特徴とする請求項1〜5のいずれか1項に記載の共役ジエンの製造方法。
【請求項1】
炭素原子数4以上のモノオレフィンを含む原料ガスと分子状酸素含有ガスとを混合した混合ガスを、触媒を有する反応器に供給し、酸化脱水素反応により対応する共役ジエンを生成し、該共役ジエンを含む生成ガスを得る共役ジエンの製造方法において、下記式(1)で示される該反応器中の触媒の破砕率が0.0重量%以上40.0重量%以下であることを特徴とする共役ジエンの製造方法。
破砕率(重量%)={(反応器中の破砕触媒の重量)/(反応器中の全触媒重量)}×100 …(1)
【請求項2】
前記反応器が前記触媒と反応性の無い固形物を更に有することを特徴とする請求項1に記載の共役ジエンの製造方法。
【請求項3】
前記反応器内の全触媒量に対する前記混合ガスの流量の比(体積比)が500〜5000h-1であることを特徴とする請求項1又は2に記載の共役ジエンの製造方法。
【請求項4】
前記触媒が、少なくともモリブデン及びビスマスを含むことを特徴とする請求項1〜3のいずれか1項に記載の共役ジエンの製造方法。
【請求項5】
前記触媒が、下記一般式(2)で表される複合酸化物触媒であることを特徴とする請求項1〜4のいずれか1項に記載の共役ジエンの製造方法。
MoaBibCocNidFeeXfYgZhSiiOj (2)
(式中、Xはマグネシウム(Mg)、カルシウム(Ca)、亜鉛(Zn)、セリウム(Ce)及びサマリウム(Sm)からなる群から選ばれる少なくとも1種の元素であり、Yはナトリウム(Na)、カリウム(K)、ルビジウム(Rb)、セシウム(Cs)及びタリウム(Tl)からなる群から選ばれる少なくとも1種の元素であり、Zはホウ素(B)、リン(P)、砒素(As)及びタングステン(W)からなる群から選ばれる少なくとも1種の元素である。また、a〜jはそれぞれの元素の原子比を表し、a=12のとき、b=0.1〜7、c=0〜10、d=0〜10(但しc+d=1〜10)、e=0.05〜3、f=0〜2、g=0.04〜2、h=0〜3、i=0〜48の範囲にあり、またjは他の元素の酸化状態を満足させる数値である。)
【請求項6】
前記原料ガスが、エチレンの2量化により得られる1−ブテン、シス−2−ブテン、トランス−2−ブテン若しくはこれらの混合物を含有するガス、n−ブタンの脱水素若しくは酸化脱水素反応により生成するブテン留分、又は重油留分を流動接触分解する際に得られる炭素原子数が4の炭化水素を多く含むガスであることを特徴とする請求項1〜5のいずれか1項に記載の共役ジエンの製造方法。
【図1】

【公開番号】特開2012−46509(P2012−46509A)
【公開日】平成24年3月8日(2012.3.8)
【国際特許分類】
【出願番号】特願2011−165936(P2011−165936)
【出願日】平成23年7月28日(2011.7.28)
【出願人】(000005968)三菱化学株式会社 (4,356)
【Fターム(参考)】
【公開日】平成24年3月8日(2012.3.8)
【国際特許分類】
【出願日】平成23年7月28日(2011.7.28)
【出願人】(000005968)三菱化学株式会社 (4,356)
【Fターム(参考)】
[ Back to top ]