
共役系高分子、該共役系高分子を用いた有機薄膜太陽電池
【課題】本発明は、成膜性に優れると共に、長波長領域における光吸収特性に優れ、フラーレン類との間で高い開放電圧値を実現することができる共役系高分子、および該共役系高分子を用いた光電変換層を備える有機薄膜太陽電池を提供することを目的とする。
【解決手段】一般式(1)で表される繰り返し単位と、一般式(2)で表される繰り返し単位とを有する共役系高分子。

(一般式(1)中、Rは、水素原子または一価の有機基を表す。
一般式(2)中、Xは、一般式(1)で表される繰り返し単位とは異なる二価の共役系連結基を表す。)
【解決手段】一般式(1)で表される繰り返し単位と、一般式(2)で表される繰り返し単位とを有する共役系高分子。
(一般式(1)中、Rは、水素原子または一価の有機基を表す。
一般式(2)中、Xは、一般式(1)で表される繰り返し単位とは異なる二価の共役系連結基を表す。)
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、共役系高分子、および該共役系高分子を用いた有機薄膜太陽電池に関する。より具体的には、本発明は、イミド環を有するチオフェン基と二価の共役構造を有する連結基とを含む共役系高分子と、該共役系高分子を用いて得られる有機薄膜太陽電池に関する。
【背景技術】
【0002】
有機薄膜太陽電池は、p型(電子ドナー性)の有機半導体材料とn型(電子アクセプター性)の有機半導体材料とを組み合わせた簡便な素子構造を有する。この電池は、色素増感型太陽電池に代表される湿式太陽電池とは異なり、電解液を用いないため加工性に優れ、望みの形状に成形可能である。また、該電池は、シリコン太陽電池や色素増感型太陽電池よりもはるかに軽量化できるため、様々な設置場所に適用することができる。さらに、高純度シリコンや貴金属の使用といった資源的制約もないため、市場シェアの拡大と共に大幅に低コスト化できると見積もられている。
【0003】
有機薄膜太陽電池の課題の一つとして、エネルギー変換効率(光電変換効率)の向上が挙げられる。この光電変換効率の向上のためには、開放電圧値の改善が重要である。有機薄膜太陽電池の開放電圧値は、電子供与性材料および電子受容性材料の組み合わせに大きく依存し、用いる材料の最適化にブレークスルーが必要である。
【0004】
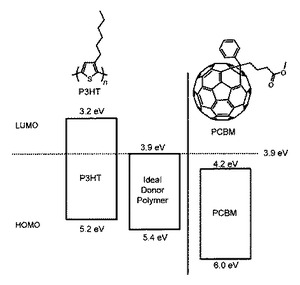
現在、世界中で進められている一連の有機薄膜太陽電池研究において、n型半導体材料としてはフラーレン類(例えば、PCBMなどのフラーレン誘導体)が最適であるとされている。PCBMのHOMO(最高占有分子軌道)は6.0eV、LUMO(最低非占有分子軌道)は4.2eVであることが知られている(非特許文献1)。
【0005】
一方、現在最も一般的に用いられているp型半導体材料としては、ポリチオフェン誘導体の共役高分子化合物(例えば、P3HT:ポリ3−ヘキシルチオフェン−2,5−ジイル)が挙げられる。このP3HTのHOMO(最高占有分子軌道)は3.2eV、LUMO(最低非占有分子軌道)は5.2eVであることが知られている。
【先行技術文献】
【非特許文献】
【0006】
【非特許文献1】Barry C. Thompson他一名、Angewandte Chemie International Edition, 2008, Vol.47, pp.58-77.
【発明の概要】
【発明が解決しようとする課題】
【0007】
一方、非特許文献1に示されるように、P3HTは、n型半導体材料であるフラーレン類に対して、必ずしも理想的なHOMO準位およびLUMO準位を示していなかった(図17 参照)。実際、P3HTとPCBMとを含む光電変換層を備える有機薄膜太陽電池においては、その開放電圧値は実用上の観点からは必ずしも満足できるものではなく、さらなる改良が必要であった。
開放電圧値を改良する方法として、p型半導体材料のHOMO準位を下げる手段が考えられるが、この場合、p型半導体材料のバンドギャップ(LUMO準位とHOMO準位の差)が広がり、長波長領域の光を吸収することができなくなる。つまり、可視光領域の長波長側における光の吸収効率が減少し、入射した光を有効利用できなくなり、結果としてエネルギー変換効率が上がらないという欠点がある。
このように開放電圧値と長波長領域の光吸収とはトレード・オフの関係にあることが多く、両者をより高いレベルで両立させることは困難とされていた。
【0008】
本発明は、上記実情に鑑みて、成膜性に優れると共に、長波長領域における光吸収特性に優れ、フラーレン類との間で高い開放電圧値を実現することができる共役系高分子、および該共役系高分子を用いた光電変換層を備える有機薄膜太陽電池を提供することを目的とする。
【課題を解決するための手段】
【0009】
本発明者らは、鋭意検討を行った結果、所定のイミド環を有するチオフェン構造とπ共役系の連結基とを有する共役系高分子を使用することにより、上記課題を解決できることを見出した。
つまり、本発明者らは、上記課題が下記の構成により解決されることを見出した。
【0010】
<1> 後述する一般式(1)で表される繰り返し単位と、後述する一般式(2)で表される繰り返し単位とを有する共役系高分子。
<2> 一般式(1)中のXが、二価の芳香族炭化水素基、二価の芳香族複素環基、−(CH=CH)−、−(C≡C)−、またはそれらを組み合わせた基である、<1>に記載の共役系高分子。
<3> 第1電極層と、前記第1電極層と対向する電極である第2電極層とを備え、
前記第1電極層と前記第2電極層との間に、少なくとも<1>または<2>に記載の共役系高分子およびフラーレン類を含む光電変換層を備える有機薄膜太陽電池。
【発明の効果】
【0011】
本発明によれば、成膜性に優れると共に、長波長領域における光吸収特性に優れ、フラーレン類との間で高い開放電圧値を実現することができる共役系高分子、および該共役系高分子を用いた光電変換層を備える有機薄膜太陽電池を提供することができる。
【図面の簡単な説明】
【0012】
【図1】本発明の有機薄膜太陽電池の第1の実施形態の模式的断面図である。
【図2】本発明の有機薄膜太陽電池の第2の実施形態の模式的断面図である。
【図3】本発明の有機薄膜太陽電池の第3の実施形態の模式的断面図である。
【図4】本発明の有機薄膜太陽電池の第4の実施形態の模式的断面図である。
【図5】図5AはPDTの1H NMR測定結果を、図5BはIR測定結果を、図5CはUV測定および蛍光測定結果を示す。
【図6】図6AはPOTの1H NMR測定結果を、図6BはIR測定結果を、図6CはUV測定および蛍光測定結果を示す。
【図7】図7AはPHOTの1H NMR測定結果を、図7BはIR測定結果を、図7CはUV測定結果を示す。
【図8】図8AはPHHTの1H NMR測定結果を、図8BはIR測定結果を、図8CはUV測定および蛍光測定結果を示す。
【図9】図9AはPPHTの1H NMR測定結果を、図9BはIR測定結果を、図9CはUV測定および蛍光測定結果を示す。
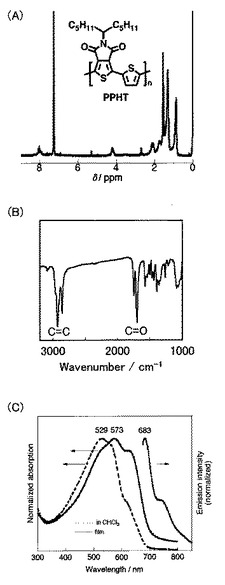
【図10】図10はPTPFの1H NMR測定結果を示す。
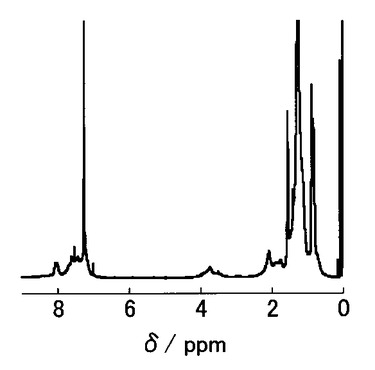
【図11】図11はPTPBのUV測定および蛍光測定結果を示す。
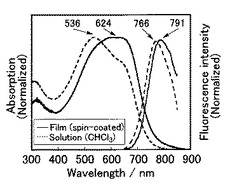
【図12】PDTとP3HTのクロロホルム溶液の吸収スペクトル図である。
【図13】本発明の共役系高分子のエネルギー準位図である。
【図14】図14Aは、実施例で使用された有機薄膜太陽電池の上面図であり、図14Bは模式的断面図である。
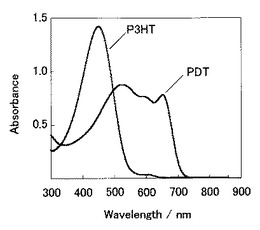
【図15】PDT:PCBM膜とP3HT:PCBM膜の吸収スペクトル図である。
【図16】実施例3の有機薄膜太陽電池の光電流アクションスペクトル図である。
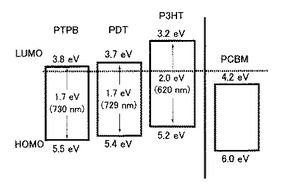
【図17】非特許文献1に記載のP3HTとPCMBのエネルギー準位図である。
【発明を実施するための形態】
【0013】
以下に、本発明に係る一般式(1)で表される繰り返し単位と一般式(2)で表される繰り返し単位とを有する共役系高分子、および、該共役系高分子を用いる光電変換層を備える有機薄膜太陽電池について詳述する。
まず、共役系高分子の構成およびその製造方法について詳述する。
【0014】
<共役系高分子>
本発明の共役系高分子は、以下の一般式(1)で表される繰り返し単位と、一般式(2)で表される繰り返し単位とを主鎖中に有する高分子である。
一般式(1)で表される繰り返し単位中のイミド環部分が電子吸引性の機能を有しており、該部分を共役系高分子に含有させることにより、フラーレン類に対してより好ましい電子状態が実現されたと推測される。通常、チオフェン骨格中の2つのβ位に電子吸引性基を導入した化合物を用いて重合をおこなうと、得られる高分子の分子量が伸びず所望の成膜性が得られない場合が多いが、本発明の共役系高分子は実用的な観点からも満足できる成膜性を有している。
なお、公知文献に記載される一般式(1)で表される繰り返し単位のみから得られるポリマーでは、分子量が伸びず、有機薄膜太陽電池へ応用できる程度の十分な成膜性を有していない。また、そのエネルギー準位もLUMO準位が約6evと計算され、フラーレン類と共に有機薄膜太陽電池に応用するには不適切なエネルギー準位といえる。
【0015】
【化1】
【0016】
(一般式(1)で表される繰り返し単位)
一般式(1)中、Rは水素原子または一価の有機基を表す。
一価の有機基としては特に限定されないが、例えば、アルキル基、アルケニル基、アルキニル基、アリール基、複素環基、アルコキシ基、アリールオキシ基、またはこれらを組み合わせた基などが挙げられる。
これら例示の基は置換基を有していてもよい。この置換基の種類は、本発明の効果を損なわない限り特に制限されない。例えば、ハロゲン原子、ニトロ基、シアノ基、スルホン酸基、カルボニルオキシ基、カルボキシル基、アミノ基、アルキル基、アルケニル基、アルキニル基、アリール基、複素環基、アルコキシ基、アリールオキシ基、カルボン酸エステル、スルホン酸エステル基、アミド基などが挙げられる。
また、該有機基は、本発明の効果を損なわない範囲において、−O−、−S−、−SO2−、−CO−、−NH−などの二価の連結基部位を含んでいてもよい。
【0017】
上記有機基としては、成膜性により優れる点から、アルキル基、アルケニル基、アルキニル基などの脂肪族炭化水素基が好ましく、アルキル基がより好ましい。
脂肪族炭化水素基としては、直鎖状、分岐状、環状のいずれであってもよく、炭素数6〜30が好ましく、炭素数8〜20がより好ましい。
なお、アルキル基の具体例としては、ヘキシル基、ヘプチル基、オクチル基、ノニル基、デシル基、ウンデシル基、ドデシル基などが挙げられる。
【0018】
なお、共役系高分子中に複数の一般式(1)で表される繰り返し単位が含まれる場合、Rは各繰り返し単位中で同一であっても異なっていてもよい。
【0019】
(一般式(2)で表される繰り返し単位)
一般式(2)中、Xは、一般式(1)で表される繰り返し単位構造(連結基)とは異なる二価の共役系連結基を表す。共役系連結基とは、π共役構造を有する連結基を意味する。高分子中にこのようなπ共役構造を有する連結基が存在することによって、高分子全体にπ共役平面が広がり、p型半導体材料としての特性がより向上する。
なお、共役系高分子中に複数の一般式(2)で表される繰り返し単位が含まれる場合、複数のXは同一であっても異なっていてもよい。
【0020】
Xは、一般式(1)で表される繰り返し単位以外の共役構造からなる二価の連結基であれば特に限定されない。なかでも、合成が簡便であり、成膜性など本発明の効果がより優れる点で、二価の芳香族炭化水素基、二価の芳香族複素環基、−(CH=CH)−、−(C≡C)−、または、これらから選択された2種以上の基を組み合わせてできる連結基であることが好ましい。なかでも、本発明の効果がより優れる点より、二価の芳香族炭化水素基、二価の芳香族複素環基、およびそれらを組み合わせた基から選ばれる芳香族連結基が好ましく挙げられる。
【0021】
芳香族炭化水素基の炭素数は特に限定されないが、合成上の観点から、炭素数5〜40が好ましく、炭素数6〜20がより好ましく、アリーレン基などが挙げられる。
芳香族炭化水素基としては、例えば、ベンゼン環基、ナフタレン環基、アントラセン環基、テトラリン環基、フェナントレン環基、フルオレン環基、アズレン環基、ピレン環基、クリセン環基、トリフェニレン環基、ペリレン環基、ペンタレン環基、インダセン環基、フェナレン環基、フルオランテン環基、アセフェナントリレン環基、アセアントリレン環基、ナフタセン環基、プレイアデン環基、ペンタフェン環基、ペンタセン環基、テトラフェニレン環基、ヘキサフェン環基、ヘキサセン環基などが挙げられる。なかでも、フルオレン環基、ベンゼン環基、ナフタレン環基が好ましい。なお、上記基中の結合可能な部分のいずれかに、結合が形成される。
【0022】
芳香族複素環基の炭素数は特に限定されないが、合成上の観点から、炭素数5〜40が好ましく、炭素数6〜20がより好ましく、含窒素又は含硫黄芳香族複素環などが挙げられる。
芳香族複素環基としては、例えば、ピロール環基、インドール環基、イソインドール環基、カルバゾール環基、フラン環基、クマロン環基、イソベンゾフラン環基、チオフェン環基、ベンゾチオフェン環基、ジベンゾチオフェン環基、ピラゾール環基、インダゾール環基、イミダゾール環基、ベンゾイミダゾール環基、オキサゾール環基、ベンゾオキサゾール環基、ベンゾオキサゾリン環基、イソオキサゾール環基、ベンゾイソオキサゾール環基、チアゾール環基、ベンゾチアゾール環基、ベンゾチアゾリン環基、イソチアゾール環基、ベンゾイソチアゾール環基、トリアゾール環基、ベンゾトリアゾール環基、オキサジアゾール環基、チアジアゾール環基、ベンゾオキサジアゾール環基、ベンゾチアジアゾール環基、テトラゾール環基、プリン環基、ピリジン環基、キノリン環基、イソキノリン環基、アクリジン環基、フェナントリジン環基、ベンゾキノリン環基、ベンゾイソキノリン環基、ナフチリジン環基、フェナントロリン環基、ピリダジン環基、ピリミジン環基、ピラジン環基、フタラジン環基、キノキサリン環基、キナゾリン環基、シンノリン環基、フェナジン環基、ペリミジン環基、トリアジン環基、テトラジン環基、プテリジン環基、ベンゾオキサジン環基、フェノキサジン環基、ベンゾチアジン環基、フェノチアジン環基、オキサジアジン環基、チアジアジン環基、ベンゾジオキソール環基、ベンゾジオキサン環基、ピラン環基、クロメン環基、キサンテン環基、クロマン環基、イソクロマン環基などが挙げられる。なかでも、チオフェン環基、ベンゾチアジアゾール環基などが好ましい。なお、上記基中の結合可能な部分のいずれかに、結合が形成される。
【0023】
Xで表される共役系連結基は置換基を有してもよく、置換基としては、例えば、上述したRで表される有機基が有していてもよい置換基などが挙げられる。
【0024】
共役系連結基は、上述した連結基を2種以上組み合わせてできる連結基であってもよく、上述した連結基を2または3つ組み合わせた基がより好ましい。例えば、−(芳香族炭化水素基)−(芳香族炭化水素基)−、−(芳香族炭化水素基)−(芳香族炭化水素基)−(芳香族炭化水素基)−などの芳香族炭化水素基が2以上連結した基、−(芳香族複素環基)−(芳香族複素環基)−、−(芳香族複素環基)−(芳香族複素環基)−(芳香族複素環基)−などの芳香族複素環基が2以上連結した基や、−(芳香族炭化水素基)−(芳香族複素環基)−、−(芳香族炭化水素基)−(CH=CH)−、−(芳香族複素環基)−(CH=CH)−、−(芳香族複素環基)−(芳香族炭化水素基)−(芳香族複素環基)−など異種の連結基が連結した基などが挙げられる。
【0025】
共役系高分子中における一般式(1)で表される繰り返し単位と一般式(2)で表される繰り返し単位との含有割合(モル比)は、発明の効果を損なわない限り特に制限されない。
なかでも、共役系高分子の光吸収特性、および、有機薄膜太陽電池における開放電圧値がより大きい点から、両繰り返し単位の含有割合(一般式(1)で表される繰り返し単位/一般式(2)で表される繰り返し単位)(モル比)は、0.2〜5が好ましく、0.5〜2がより好ましい。
【0026】
本発明の共役系高分子中において、一般式(1)で表される繰り返し単位の含有量は、本発明の効果を損なわない限り特に制限されない。なかでも、本発明の効果がより優れる点で、該繰り返し単位の含有量は、全繰り返し単位(100モル%)に対して、10〜90モル%が好ましく、30〜70モル%がより好ましい。
また、本発明の共役系高分子中において、一般式(2)で表される繰り返し単位の含有量は、本発明の効果を損なわない限り特に制限されない。なかでも、本発明の効果がより優れる点で、該繰り返し単位の含有量は、全繰り返し単位(100モル%)に対して、10〜90モル%が好ましく、30〜70モル%がより好ましい。
【0027】
本発明の共役系高分子中において、一般式(1)で表される繰り返し単位と、一般式(2)で表される繰り返し単位とが主成分であることが好ましい。
より具体的には、両繰り返し単位の合計量は、全繰り返し単位(100モル%)に対して、80モル%以上が好ましく、90モル%以上がより好ましい。上限としては、100モル%である。
【0028】
本発明の共役系高分子中において、一般式(1)で表される繰り返し単位と、一般式(2)で表される繰り返し単位との結合様式は特に制限されず、ランダム共重合体、ブロック共重合体、交互共重合体などが挙げられる。
【0029】
(共役系高分子の好適実施態様)
本発明の共役系高分子の好適実施態様としては、一般式(1)で表される繰り返し単位と、一般式(2)で表される繰り返し単位とが交互に結合した構造を有する共役系高分子(交互共重合体型共役系高分子)が好ましい。該共役系高分子であれば、一般式(1)で表される立体的にかさ高い各チオフェン骨格部が一般式(2)で表される繰り返し単位を介して離れて存在するため、高分子の平面性がより高くなり、膜中での高分子間のパッキングが向上することが予想される。
【0030】
本発明の共役系高分子中において、一般式(1)で表される繰り返し単位と一般式(2)で表される繰り返し単位とが交互に結合した構造の含有量は、本発明の効果を損なわない限り特に制限されない。
なかでも、共役系高分子の光吸収特性、および、有機薄膜太陽電池における開放電圧値がより大きい点から、該構造が実質的に主成分として含まれていることが好ましい。より具体的には、該構造の含有量は、全繰り返し単位(100モル%)に対して、80モル%以上が好ましく、90モル%以上がより好ましい。上限としては、100モル%である。
【0031】
なお、本発明の共役系高分子が上記交互共重合型の場合、本発明の効果を損なわない範囲で、上述した一般式(1)で表される繰り返し単位と、一般式(2)で表される繰り返し単位とが交互に結合した構造以外の共役構造を有する繰り返し単位を有していてもよい。
例えば、一般式(2)で表される繰り返し単位が連結して得られる構造などを一部に含んでいてもよい。
【0032】
本発明の共役系高分子の数平均分子量(Mn)は特に制限されないが、溶剤への溶解性や成膜性により優れる点から、2000〜150000が好ましく、2500〜100000がより好ましい。
また、共役系高分子の分子量分布(Mw/Mn)は特に制限されないが、溶剤への溶解性や成膜性により優れる点から、1〜8が好ましく、1〜5がより好ましい。
【0033】
本発明の共役系高分子の合成方法は特に制限されず、公知の方法を組み合わせて合成することができる。例えば、上述したイミド環を有するチオフェン化合物と、Xで表される構造を有する化合物とをカップリング反応(例えば、Stilleカップリング反応)を介して重合させる方法が挙げられる。
具体的には、Nielsen, C. B.; Bjornholm, T. Org. Lett. 2004, 6, 3381などの文献を参照することにより合成することができる。
【0034】
<有機薄膜太陽電池、およびその製造方法>
本発明の有機薄膜太陽電池は、第1電極層と、少なくとも上述した共役系高分子およびフラーレン類を含有する光電変換層と、第2電極層とを備える。
以下に、本発明の有機薄膜太陽電池の形態、および、その製造方法について詳述する。
【0035】
<有機薄膜太陽電池(第1の実施形態)>
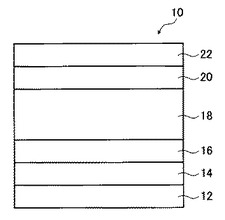
図1は、本発明の有機薄膜太陽電池の第1の実施形態の模式的断面図である。
同図に示す有機薄膜太陽電池10は、透明基板12、透明電極層14、正孔輸送層16、光電変換層18、電子輸送層20、および対向電極層22をこの順で積層した積層構造を有する。該太陽電池は、光電変換層18がp型有機半導体材料とn型有機半導体材料とが混合して存在する混合層である、いわゆるバルクヘテロ接合型有機薄膜太陽電池に該当する。各層の厚みは、該図によっては制限されない。
以下に、各層について詳述する。
【0036】
<透明基板>
透明基板12は、後述する電極層、光電変換層などを支持し、補強するものであれば、任意の材料を使用することができる。なお、該基板は、後述する透明電極層14が十分な強度有していれば、本発明の有機薄膜太陽電池に含まれていなくてもよい。
透明基板12としては、例えば、ガラスや、ポリイミド、PET、PEN、PES、テフロン(登録商標)等の耐熱性の高分子フィルムや、セラミックなどが挙げられる。透明基板12は、高い透明性を有するものが好ましく、例えばガラスが挙げられる。なお、透明基板12の表面はフラットであってもよいし、凹凸を有していてもよい。
透明基板12の厚さは任意の厚さとすることができ、好ましくは0.05mm〜3mmである。
【0037】
<透明電極層(第1電極層)>
透明電極層14は、後述する光電変換層18の一面側に設けられる第1電極層である。本実施形態では、透明電極層14は、正孔輸送層16を介して設けられているが、光電変換層18に接して設けられていてもよい。透明電極層14は、光電変換層18に対してオーミック接触の形成が可能であり、かつ照射光を透過させるものであれば任意の材料を使用することができる。
例えば、ITO、SnO2、ZnO、In2O3などの透明導電材料、または、フッ素ドープ酸化錫(SnO2:F)、アンチモンドープ酸化錫(SnO2:Sb)、錫ドープ酸化インジウム(In2O3:Sn)、Alドープ酸化亜鉛(ZnO:Al)、Gaドープ酸化亜鉛(ZnO:Ga)などの、上記透明導電材料に不純物がドープされたもので構成される。
透明電極層14は、上記材料からなる単独層で構成されていてもよく、複数の層を積層した積層体で構成されていてもよい。透明電極層14の層厚は、電極としての機能を果たすものであれば特に限定されるものではないが、通常は3nm〜10μmである。なお、透明電極層14は、表面がフラットなものでもよく、表面に凹凸を有しているものでもよい。
【0038】
<正孔輸送層>
正孔輸送層16は、透明電極層14と後述する光電変換層18との間に設置され、正孔の取り出し効率の向上に寄与する。また、正孔輸送層16は、光電変換層18から透明電極層14への電子の漏れを抑制する電子ブロッキング効果や、透明電極層14の凹凸を平滑にするバッファー層としても機能する。なお、この正孔輸送層16は、必要に応じて設置され、本発明の有機薄膜太陽電池に含まれていなくてもよい。
正孔輸送層16としては、ポリ−(3,4−エチレンジオキシチオフェン)/ポリエチレンスルフォン酸ナトリウム(通称PEDOT−PSS)など正孔輸送性のある物質であって、最高占有分子軌道(HOMO)のエネルギーレベルが真空準位から比較して透明電極層14よりも低いレベルであれば、特に制限はない。材料としては、例えば、ドープされたポリアニリン、ポリフェニレンビニレン、ポリピロール、ポリパラフェニレン、ポリアセチレン、トリフェニルジアミン(TPD)等の導電性有機化合物などが挙げられる。
正孔輸送層16の層厚については特に制限はないが、5〜500nm程度が好ましい。
【0039】
<光電変換層>
本実施形態の光電変換層18は、1層中にp型有機半導体材料(アクセプター)およびn型半導体材料(ドナー)が混在するバルクヘテロ型の光電変換層(バルクヘテロ型混合層)のことである。本実施形態においてはp型有機半導体材料として上述した一般式(1)で表される繰り返し単位および一般式(2)で表される繰り返し単位を有する共役系高分子が用いられ、n型有機半導体材料としてフラーレン類が主に用いられる。
以下に、使用される材料に関して詳述する。
【0040】
p型有機半導体材料としては、上述した共役系高分子が使用されるが、それ以外の公知の他の材料を併用してもよい。
公知のp型有機半導体材料としては、N,N’−ビス(3−トリル)−N,N’−ジフェニルベンジジン(mTPD)、N,N’−ジナフチル−N,N’−ジフェニルベンジジン(NPD)、4,4’,4’’−トリス(フェニル−3−トリルアミノ)トリフェニルアミン(MTDATA)などのアミン化合物;フタロシアニン(Pc)、銅フタロシアニン(CuPc)、亜鉛フタロシアニン(ZnPc)、チタニルフタロシアニン(TiOPc)などのフタロシアニン類;オクタエチルポルフィリン(OEP)、白金オクタエチルポルフィリン(PtOEP)、亜鉛テトラフェニルポルフィリン(ZnTPP)などのポルフィリン類;ポリヘキシルチオフェン(P3HT)、メトキシエチルヘキシロキシフェニレンビニレン(MEHPPV)などの主鎖型共役高分子類やポリビニルカルバゾールなどの側鎖型高分子類などの高分子化合物などが挙げられる。
【0041】
n型有機半導体材料としては、フラーレン類が用いられる。フラーレン類とは、フラーレン、その誘導体(フラーレン誘導体)、および、その金属内包物等を含む概念である。
フラーレンとしては、種々の立体構造を有するカーボンクラスター、例えば、C60、C70、C74、C76、C78、C82、C84、C720、C860などのフラーレンが挙げられる。フラーレンの形態は、例えば、サッカーボール状、バッキーボール状などが挙げられる。
【0042】
フラーレン誘導体とは、フラーレン骨格に置換基を導入して表面修飾した化合物を意味する。修飾方法は、特に限定されず、例えば、フラーレンの反応性に富む炭素5員環部を化学的に修飾できる。置換基の種類は、特に限定されず、例えば、アルキル基(炭素数1〜10のメチル基、t−ブチル基などのアルキル基等)、アリール基(フェニル基等)、アラルキル基(ベンジル基等)、ジオキソラン単位、ハロゲン原子、または、酸素原子等が例示できる。また、液晶ポリマー、色素類、ポリエチレンオキシド等の導入により修飾されていてもよい。フラーレンの修飾により、溶媒、高分子への可溶化や親和性の改善、フラーレンの配列または配向を可能にする。
【0043】
金属を内包したフラーレンとしては、種々の金属、例えば、周期表第1A族元素(K、Na、Rb等)、周期表第2A族元素、ランタノイド族元素(La等)等の金属がドープされたフラーレンが例示できる。
【0044】
n型有機半導体材料としては、上記の中でも、[6,6]−フェニル−C61ブチルカルボン酸メチルエステル(PCBM)が好ましく挙げられる。
【0045】
なお、本発明の効果を損なわない限り、ペリレン誘導体、多環キノン、キナクリドンなどの他の公知のn型有機半導体材料を併用してもよい。
【0046】
p型有機半導体材料とn型有機半導体材料の混合割合は特に制限されないが、得られる有機薄膜太陽電池の性能がより優れる点から、両者の質量比(p型有機半導体材料/n型有機半導体材料)は、0.3〜1.8が好ましく、0.5〜1.5がより好ましい。
【0047】
光電変換層18の厚みは特に制限されないが、得られる有機薄膜太陽電池の性能がより優れる点から、100〜400nmが好ましく、150〜300nmがより好ましい。
【0048】
<電子輸送層>
電子輸送層20は、光電変換層18と対向電極層22との間に配置され、電子の取り出し効率の向上に寄与する。なお、この電子輸送層20は、必要に応じて設置され、本発明の有機薄膜太陽電池に含まれていなくてもよい。
電子輸送層20としては、例えば、フッ化リチウム等のアルカリ金属、アルカリ土類金属のハロゲン化物、酸化物等を用いることができる。また、酸化チタンなどの無機半導体を用いることもできる。
電子輸送層20の層厚については特に制限はないが、5〜100nm程度が好ましい。
【0049】
<対向電極層(第2電極層)>
対向電極層22は、光電変換層18の他面側に設けられる第2電極層である。他面側とは、光電変換層18に対し、透明電極層14の反対側をいう。本実施形態では、対向電極層22は、電子輸送層20を介して設けられているが、光電変換層18に接して設けられていてもよい。
対向電極層22を構成する材料は、光電変換層18の半導体からの電荷を有効に収集できる仕事関数を有することが望ましい。透明電極層14が、正孔を収集する場合は、対向電極層22としては電子を収集しやすいアルミニウム、マグネシウム、カルシウムなどが用いられる。
対向電極層22の層厚は、発生した光電荷を十分に外部回路へ伝達できる程度のシート抵抗を得ることができる範囲であれば、特に限定されない。対向電極層22の層厚については特に制限はないが、1〜100nmが好ましい。
【0050】
<有機薄膜太陽電池の製造方法>
本発明の有機薄膜太陽電池の各層の形成方法は特に制限されず、真空蒸着、スパッタリング、プラズマ、イオンプレーティングなどの乾式成膜法や、スピンコーティング、ディップコート、キャスティング、ロールコート、フローコーティング、インクジェットなどの湿式成膜法を採用することができる。
より詳細には、まず、透明基板12を設置し、透明基板12上に透明電極層14を形成する。基板の形状および寸法に制限はなく、任意に設置することができる。透明電極層14は任意の方法により形成することができ、例えば、真空蒸着、スパッタなどのドライプロセス、ゾルゲル法などの湿式プロセスの方法が挙げられる。
次に、透明電極層14の表面上に正孔輸送層16を形成する。正孔輸送層16は、使用する材料を溶媒に溶かして、該溶液を使用してスピンコート、ディップコート、キャストコートなどの任意の方法を用いて形成することができる。
次に、上述した共役系高分子とn型有機半導体材料とを含む溶液を用いて、湿式成膜法により透明電極層14の表面上に光電変換層18を成膜する。
さらに、光電変換層18の表面上に、電子輸送層20を形成する。電子輸送層20は、真空蒸着などのドライプロセス、ゾルゲル法などの湿式プロセスなどの方法を用いて形成できる。
最後に、電子輸送層20の表面上に、対向電極層22を形成する。対向電極層22は、真空蒸着、レーザー転写法などの任意の方法で形成できる。
【0051】
乾式成膜法を採用する場合、抵抗加熱法を用いて材料を加熱蒸発させることが好ましい。また、混合層を形成する場合には、例えば、複数の蒸発源からの同時蒸着による成膜方法が好ましい。成膜時には、基板温度を一定に制御することが好ましい。
湿式成膜法を適用する場合、材料を適切な溶媒に溶解または分散させて溶液を調製してから薄膜を形成する。かかる溶媒としては任意の溶媒を使用でき、例えば、ジクロロメタン、ジクロロエタン、クロロホルム、四塩化炭素、テトラクロロエタン、トリクロロエタン、クロロベンゼン、ジクロロベンゼン、クロロトルエンなどのハロゲン系炭化水素系溶媒;ジブチルエーテル、テトラヒドロフラン、ジオキサン、アニソールなどのエーテル系溶媒;メタノール、エタノール、プロパノール、ブタノール、ペンタノール、ヘキサノール、シクロヘキサノール、メチルセロソルブ、エチルセロソルブ、エチレングリコールなどのアルコール系溶媒;ベンゼン、トルエン、キシレン、エチルベンゼン、ヘキサン、オクタン、デカン、テトラリンなどの炭化水素系溶媒;酢酸エチル、酢酸ブチル、酢酸アミルなどのエステル系溶媒などが挙げられる。また、これらの溶媒は、単独で使用しても複数混合して用いてもよい。
また、溶液中の材料濃度は、使用される材料に応じて適宜最適な濃度が選択される。
【0052】
本発明においては、有機薄膜太陽電池のいずれの層においても、成膜性向上、膜のピンホール防止などのため、適切な樹脂や添加剤を含有させてもよい。使用可能な樹脂としては、ポリスチレン、ポリカーボネート、ポリアリレート、ポリエステル、ポリアミド、ポリウレタン、ポリスルフォン、ポリメチルメタクリレート、ポリメチルアクリレート、セルロースなどの絶縁性樹脂およびそれらの共重合体;ポリ−N−ビニルカルバゾール、ポリシランなどの光導電性樹脂;ポリチオフェン、ポリピロールなどの導電性樹脂などが挙げられる。
また、添加剤としては、酸化防止剤、紫外線吸収剤、可塑剤などが挙げられる。
【0053】
<有機薄膜太陽電池(第2の実施形態)>
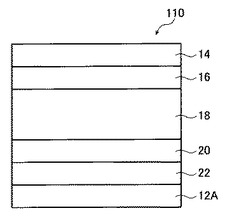
図2は、本発明の有機薄膜太陽電池の第2の実施形態を示す概略図である。
図2に示す有機薄膜太陽電池110は、基板12A上に、対向電極層22、光電変換層18および透明電極層14が順次積層されて構成されている。この構成は、基板12Aからの積層の順を図1に示したのとは逆にした構成である。対向電極層22は基板12A上に設けられており、透明電極層14は有機薄膜太陽電池110の最表面に設けられている。
本実施形態においては、基板12Aは必ずしも透明である必要はなく、金属、セラミックス、プラスチックなどの様々な材料の基板を用いることができる。
本実施形態における各層の成分および塗布液の調製方法は、第一の実施形態と同様とすることができる。本実施形態の有機薄膜太陽電池の製造方法は、基板12A上に、対向電極層22、電子輸送層20、光電変換層18、正孔輸送層16、透明電極層14の順に積層する点において、第一の実施形態と異なる。かかる構成にすることにより、図1に示す構成と比較して、多くの場合に金属薄膜が用いられ、真空蒸着で形成される対抗電極層22を、有機膜形成前に形成できるので、真空蒸着による有機膜への損傷を抑制する事が可能という効果が得られる。
【0054】
<有機薄膜太陽電池(第3の実施形態)>
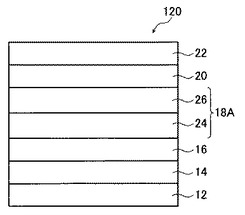
図3は、本発明の有機薄膜太陽電池の第3の実施形態を示す概略図である。
図3に示す有機薄膜太陽電池120は、透明基板12、透明電極層14、正孔輸送層16、光電変換層18A、電子輸送層20、および対向電極層22をこの順で積層した積層構造を有し、光電変換層18Aがp型有機半導体材料からなるp層24とn型有機半導体材料からなるn層26との二層構造(バイレイヤー構造)である。
本実施形態における各層の成分および塗布液の調製方法は、第一の実施形態と同様とすることができる。なお、光電変換層18Aを作製する方法としては、まず、p型有機半導体材料を含む溶液を用いて湿式成膜法によりp層24を作製した後、n型有機半導体材料を含む溶液を用いて湿式成膜法によりn層26を作製する方法が挙げられる。
【0055】
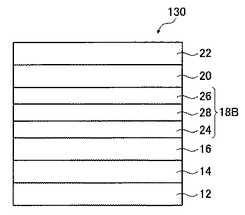
<有機薄膜太陽電池(第4の実施形態)>
図4は、本発明の有機薄膜太陽電池の第4の実施形態を示す概略図である。
図4に示す有機薄膜太陽電池130は、透明基板12、透明電極層14、正孔輸送層16、光電変換層18B、電子輸送層20、および対向電極層22をこの順で積層した積層構造を有し、光電変換層18Bがp型有機半導体材料からなるp層24と、p型有機半導体材料とn型有機半導体材料との混合層28と、n型有機半導体材料からなるn層26との三層構造である。
本実施形態における各層の成分および塗布液の調製方法は、第一の実施形態と同様とすることができる。なお、光電変換層18Bを作製する方法としては、まず、p型有機半導体材料を含む溶液を用いて湿式成膜法によりp層24を作製した後、p型有機半導体材料とn型有機半導体材料とを含む溶液を用いて湿式成膜法により混合層28を作製し、さらにn型有機半導体材料を含む溶液を用いて湿式成膜法によりn層26を作製する方法が挙げられる。
【0056】
本発明で得られる有機薄膜太陽電池材料を用いた有機薄膜太陽電池は、太陽電池モジュール、太陽光発電パネル、時計、携帯情報端末、パーソナルコンピューターなどの装置に有効に利用される。
【実施例】
【0057】
以下、実施例により本発明を詳細に説明するが、本発明はこれらに限定されるものではない。
【0058】
(測定条件等)
GPC測定は、SHIMADZU Prominenceを使用し、カラム温度25℃で、溶出溶媒としてクロロホルムを使用し、数平均分子量は標準ポリスチレンを用いて作製した検量線を用いて計算した。
核磁気共鳴(1H NMR)スペクトルは、JEOL JNM-EX400を用いて測定した。
赤外吸収(IR)スペクトルは、JASCO FT/IR-470 Plusを用いて測定した。
サイクリックボルタンメトリーは、測定装置としてALS 630aを使用し、作用電極として後述する共役系高分子がコートされたITO電極、対電極としてPt線、参照電極としてAg/AgNO3電極を用いて測定した。この測定時の掃引速度は10〜100mV/sec、走査電位領域は−2.0V〜1.6Vであった。還元電位および酸化電位は、後述する共役系高分子のフィルムを、支持電解質としてテトラブチルアンモニウムヘキサフルオロフォスファート0.1mol/Lを溶解させたアセトニトリル溶液中に浸漬して測定した。
【0059】
<合成例1>
N−ドデシルイミド縮環チオフェン(化合物5)を既報(Nielsen, C. B.; Bjornholm, T. Org. Lett. 2004, 6, 3381)の手法に従って合成した(Scheme 1)。
【0060】
【化2】
【0061】
(2,5−ジヨード(N−ドデシルジオキソピロロチオフェン))(化合物6)
上記化合物5(0.838g、2.70mmol)を、濃硫酸(4.2ml)とトリフルオロ酢酸(14ml)の混合液に溶解させた。次に、反応液にN−ヨードコハク酸イミド(NIS)(2.44g、10.8mmol)を二回に分けて加え、反応液を室温で24時間攪拌した。得られた紫色の溶液を氷水(150ml)に加えて、ジクロロメタンで抽出を行った。得られた有機層を10%チオ硫酸ナトリウム水溶液で洗浄し、無水硫酸マグネシウムで乾燥した後、溶媒をエバポレーターで除去して、茶色の粗生成物を得た。シリカゲルと、溶離液としてジクロロメタン/ヘキサン溶液(1:1)とを使用したカラムクロマトグラフィーにより、粗生成物を精製し、さらにエタノールで再結晶することにより、化合物6を得た(0.868g、1.51mmol、収率56%)。化合物6の1H NMR測定の結果は、以下の通りであった。
1H NMR (300 Mhz, CDCl3):δ 3.59 (dd, 2H), 1.62 (quintet, 2H), 1.25 (m, 18H), 0.88 (t, 3H).
【0062】
(2,5−ビス(トリブチルスタニル)チオフェン)(化合物7)
2,5−ビス(トリブチルスタニル)チオフェン(化合物7)を既報の手法(Hou, J.; Tan, Z.; Yan, Y.; He, Y.; Yang, C.; Li, Y. J. Am. Chem. Soc. 2006, 128, 4911)にしたがって合成した(Scheme 2)。
【0063】
【化3】
【0064】
(ポリ[(N−ドデシルジオキソピロロチオフェン)−alt−(チオフェン)])(PDT)
さらに、アルゴン置換した乾燥シュレンク管に、化合物6(30mg、0.052mmol)、化合物7(38mg、0.057mmol)、ジクロロビス(トリフェニルホスフィン)パラジウム(II)(3.7g、0.0053mmol)、および蒸留トルエン(1ml)を加えた(Scheme 3)。その後、反応混合液を120℃で72時間攪拌した。攪拌終了後、冷ました反応混合液をメタノールに加えて、生じた黒色沈殿をメンブレンフィルター(ADVANTEC)により回収した。この生成物をメタノールおよびヘキサンで洗浄した後、分取クロマトグラフィー(LC−908 JAi)を用いて精製し、黒色のPDT(8)を得た(5.6mg、収率27%)。得られたポリマーの1H NMR測定の結果を図5Aに、IR測定の結果を図5Bに、UV測定および蛍光測定の結果を図5Cに示す。なお、サイクリックボルタンメトリー測定により、酸化オンセット(EOX,ONSET、onset of the oxidation potential)は0.81Vで、還元オンセット(ERED,ONSET、onset of the reduction potential)は−1.11Vであった。
なお、PDT(8)の数平均分子量(Mn)は87,000で、分子量分布(PDI)は4.1であった。
【0065】
【化4】
【0066】
<合成例2>
(4−オクタデシルカルバモイルチオフェン−3−カルボン酸)(化合物9)
Scheme 4に示すように、合成例1で合成したチオフェン−3,4−カルボン酸無水物(化合物3)(468mg、3.04mmol)とn−オクタデシルアミン(873mg、3.24mmol)とを蒸留トルエン(50m)に加え、反応液を24時間還流した。冷ました反応液から、ろ過により粗生成物を回収した。また、5%塩酸水溶液でろ液を洗浄して粗生成物を沈殿させ、クロロホルムで抽出を行った後、溶媒を除去することにより、ろ液からも粗生成物を回収した。次に、回収した粗生成物をトルエンで再結晶することにより、白色の結晶である化合物9を得た(1.21g、2.86mmol、収率94%)。化合物9の1H NMR測定の結果は、以下の通りであった。
1H NMR (300 MHz, CDCl3):δ 8.40 (d, 1H), 7.73 (d, 1H), 6.46 (broad s, 1H), 3.41 (t, 2H), 1.58 (quintet, 2H), 1.46 - 0.76 (m, 33H).
【0067】
(オクタデシルジオキソピロロチオフェン)(化合物10)
塩化チオニル(100ml)に化合物9(1.21g、2.86mmol)を加えて、反応液を3時間還流した。反応液を濃縮して薄い茶色のオイルを得た後、さらに乾燥して薄い黄色の結晶を得た。次に、結晶をヘキサン中で再結晶することにより、白い結晶として化合物10を得た(903mg、2.23mmol、収率78%)。化合物10の1H NMR測定の結果は、以下の通りであった。
1H NMR (300 MHz, CDCl3):δ 7.80 (s, 2H), 3.60 (t, 2H), 1.63 (quintet, 2H), 1.54 - 0.86 (m, 33H).
【0068】
(1,3−ジブロモオクタデシルジオキソピロロチオフェン)(化合物11)
得られた化合物10(887mg、2.19mmol)を、濃硫酸(3.4ml)とトリフルオロ酢酸(11.3ml)の混合液に溶解させた。次に、反応液にN−ブロモコハク酸イミド(NBS)(1.56g、4.67mmol)を二回に分けて加え、反応液を55℃で24時間攪拌した。得られた茶色の溶液を水(120ml)で希釈して、ジクロロメタン(150ml)で抽出を行った。得られた有機層を無水硫酸マグネシウムで乾燥した後、溶媒をエバポレーターで除去して、オレンジ色の粗生成物を得た。シリカゲルと、溶離液としてクロロホルムとを使用したカラムクロマトグラフィーにより、粗生成物を精製し、さらにエタノールで再結晶することにより、白い結晶として化合物11を得た(716mg、1.27mmol、収率58%)。化合物11の1H NMR測定の結果は、以下の通りであった。
1H NMR (400 MHz, CDCl3):δ 3.58 (t, 2H), 1.62 (quintet, 2H), 1.53 - 086 (m, 33H).
【0069】
【化5】
【0070】
(ポリ[(N−オクタデシルジオキソピロロチオフェン)−alt−(チオフェン)])(POT)
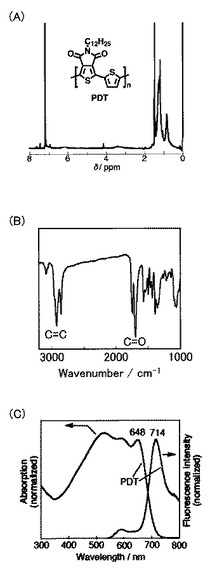
上記で合成した化合物11(200mg、0.355mmol)を、アルゴン雰囲気下、クロロホルム(5ml)に溶解させた(Scheme 5)。次に、化合物7(243mg、0.367mmol)とジクロロビス(トリフェニルホスフィン)パラジウム(II)(12.5mg,0.018mmol)とを反応液に加えて、65℃で96時間攪拌した。冷ました反応液を500mlのメタノールに加えて、ろ過により沈殿物を回収した。その後、メタノールおよびヘキサンで沈殿物を洗浄して、粗生成物POTを黒色固体として得た(160mg、収率93%)。溶離液としてクロロホルムを用いるクロマトグラフィーにより、粗生成物を精製した後、クロロホルム/メタノールにより再沈殿して、POT(12)を得た(131mg、収率76%)。得られたポリマーの1H NMR測定の結果を図6Aに、IR測定の結果を図6Bに、UV測定および蛍光測定の結果を図6Cに示す。なお、サイクリックボルタンメトリー測定により、酸化オンセット(EOX,ONSET、onset of the oxidation potential)は0.57Vで、還元オンセット(ERED,ONSET、onset of the reduction potential)は−1.31Vであった。
なお、POT(12)の数平均分子量(Mn)は39,000であり、分子量分布(PDI)は1.5であった。
【0071】
【化6】
【0072】
<合成例3>
(4−(2−ヘプチルオクチル)カルバモイルチオフェン−3−カルボン酸)(化合物13)
Scheme 6に示すように、合成例1で合成した化合物3(1.00g、6.49mmol)と2−ヘプチルオクチルアミン(1.57g、6.92mmol)とを蒸留トルエン(100ml)に加え、反応液を24時間還流した。冷ました反応液をろ過して、粗生成物を回収した。また、5%塩酸水溶液でろ液を洗浄して粗生成物を沈殿させ、クロロホルムで抽出を行った後、溶媒を除去することにより、ろ液からも粗生成物を回収した。さらに、回収した粗生成物を真空中で乾燥させた後、薄い黄色の液体として化合物13を得た(2.05g、5.39mmol、収率83%)。化合物13の1H NMR測定の結果は、以下の通りであった。
1H NMR (300 MHz, CDCl3):δ8.49 (d, 1H), 7.97 (d, 1H), 6.70 (broad s, 1H), 4.11 (m, 1H), 1.57 (m, 4H), 1.26 (m, 20H), 0.87 (t, 6H).
【0073】
(2−ヘプチルオクチルジオキソピロロチオフェン)(化合物14)
塩化チオニル(150ml)に化合物13(1.51g、3.96mmol)を加えて、反応液を4時間還流した。反応液を濃縮して薄い茶色の液体を得た後、さらに乾燥して薄い黄色の結晶を得た。さらに、真空中で乾燥させた後、薄い茶色の液体として化合物14を得た(1.14g、3.13mmol、収率79%)。化合物14の1H NMR測定の結果は、以下の通りであった。
1H NMR (300 MHz, CDCl3):δ 7.78 (s, 2H), 4.11 (m, 1H), 1.85 (m, 4H), 1.23 (m, 20H), 0.85 (t, 6H).
【0074】
(1,3−ジブロモ(2−ヘプチルオクチル)ジオキソピロロチオフェン)(化合物15)
得られた化合物14(1,00g、2.75mmol)を、濃硫酸(4.3ml)とトリフルオロ酢酸(14.2ml)の混合液に溶解させた。次に、反応液にN−ブロモコハク酸イミド(NBS)(1.96g、5.86mmol)を二つに分けて加え、反応液を室温で24時間攪拌した。得られた茶色の溶液を水(150ml)で希釈して、ジクロロメタン(200ml)で抽出を行った。得られた有機層を無水硫酸マグネシウムで乾燥した後、溶媒をエバポレーターで除去して、オレンジ色の粗生成物を得た。シリカゲルと、溶離液としてクロロホルムとを使用したカラムクロマトグラフィーにより、粗生成物を精製して、茶色の液体として化合物15を得た(816mg、1.56mmol、収率57%)。化合物15の1H NMR測定の結果は、以下の通りであった。
1H NMR (400 MHz, CDCl3):δ 4.14 (m, 1H), 1.83 (m, 4H), 1.23 (m, 20H), 0.86 (t, 6H).
【0075】
【化7】
【0076】
(ポリ[(N−(2−ヘプチルオクチル)ジオキソピロロチオフェン)−alt−(チオフェン)])(PHOT)
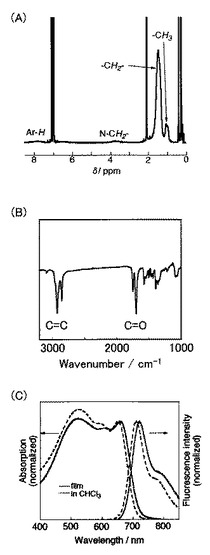
上記で合成した化合物15(50mg、0.096mmol)を、アルゴン雰囲気下、クロロホルム(1ml)に溶解させた(Scheme 7)。次に、化合物7(67mg、0.10mmol)とジクロロビス(トリフェニルホスフィン)パラジウム(II)(3.4mg、0.005mmol)とを反応液に加えて、65℃で48時間攪拌した。冷ました反応液を500mlのメタノールに加えて、ろ過により沈殿物を回収した。その後、メタノールおよびヘキサンで沈殿物を洗浄して、PHOT(16)を黒色固体として得た(37mg、収率86%)。得られたポリマーの1H NMR測定の結果を図7Aに、IR測定の結果を図7Bに、UV測定の結果を図7Cに示す。
なお、PHOT(16)の数平均分子量(Mn)は3900であり、分子量分布は1.3であった。
【0077】
【化8】
【0078】
<合成例4>
(4−(2−ヘキシルヘプチル)カルバモイルチオフェン−3−カルボン酸)(化合物17)
Scheme 8に示すように、合成例1で合成した化合物3(1.64g、10.6mmol)および2−ヘキシルヘプチルアミン(2.25g、11.3mmol)を蒸留トルエンに加えて、反応液を24時間還流した。冷ました反応液をろ過して、粗生成物を回収した。また、5%塩酸水溶液でろ液を洗浄して粗生成物を沈殿させ、クロロホルムで抽出を行った後、溶媒を除去することにより、ろ液からも粗生成物を回収した。さらに、回収した粗生成物を真空中で乾燥させた後、薄い黄色の液体として化合物17を得た(3.52g、収率94%)。化合物17の1H NMR測定の結果は、以下の通りであった。
1H NMR (400 MHz, CDCl3):δ8.46 (d, 1H), 7.87 (d, 1H), 6.42 (broad s, 1H), 4.11 (m, 1H), 1.56 (m, 4H), 1.28 (m, 16H), 0.87 (t, 6H).
【0079】
(2−ヘキシルヘプチルジオキソピロロチオフェン)(化合物18)
塩化チオニル(200ml)に化合物17(3.52g、9.96mmol)を加えて、反応液を8時間還流した。反応液を濃縮して薄い茶色の液体を得た後、さらに乾燥して薄い黄色の結晶を得た。さらに、真空中で乾燥させた後、薄い黄色の固体として化合物18を得た(2.82g、8.41mmol、収率85%)。化合物18の1H NMR測定の結果は、以下の通りであった。
1H NMR (400 MHz, CDCl3):δ 7.84 (s, 2H), 4.11 (m, 1H), 1.60 (m, 4H), 1.28 (m, 16H), 0.86 (t, 6H).
【0080】
(1,3−ジヨード(2−ヘキシルヘプチル)ジオキソピロロチオフェン)(化合物19)
得られた化合物18(3.15g、9.39mmol)を、濃硫酸(15ml)とトリフルオロ酢酸(50ml)の混合液に溶解させた。次に、反応液にN−ヨードコハク酸イミド(NIS)(4.23g、18.7mmol)を二回に分けて加え、反応液を室温で12時間攪拌した。得られた茶色の溶液を水で希釈して、ジクロロメタンで抽出を行った。得られた有機層を無水硫酸マグネシウムで乾燥した後、溶媒をエバポレーターで除去して、オレンジ色の粗生成物を得た。シリカゲルと、溶離液としてクロロホルムとを使用したカラムクロマトグラフィーにより、粗生成物を精製して、白色の結晶として化合物19を得た(3.09g、5.26mmol、収率58%)。化合物19の1H NMR測定の結果は、以下の通りであった。
1H NMR (400 MHz, CDCl3):δ 4.09 (m, 1H), 1.83 (m, 4H), 1.24 (m, 12H), 0.86 (t, 6H).
【0081】
【化9】
【0082】
(ポリ[(N−(2−ヘキシルヘプチル)ジオキソピロロチオフェン)−alt−(チオフェン)])(PHHT)
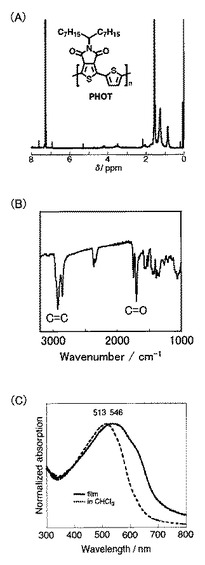
上記で合成した化合物19(58mg、0.10mmol)を、アルゴン雰囲気下、ジメチルホルムアミド(1ml)に溶解させた(Scheme 9)。次に、化合物7(66mg、0.10mmol)とテトラキス(トリフェニルホスフィン)パラジウム(0)(3mg,0.005mmol)とを反応液に加えて、110℃で48時間攪拌した。冷ました反応液を500mlのメタノールに加えて、ろ過により沈殿物を回収した。その後、メタノールおよびヘキサンで沈殿物を洗浄して、PHHT(20)を黒色固体として得た(27mg、収率64%)。得られたポリマーの1H NMR測定の結果を図8Aに、IR測定の結果を図8Bに、UV測定および蛍光測定の結果を図8Cに示す。なお、サイクリックボルタンメトリー測定により、還元オンセット(ERED,ONSET、onset of the reduction potential)は−1.18Vであった。
なお、PHHT(20)の数平均分子量(Mn)は4300であり、分子量分布(PDI)は1.1であった。
【0083】
【化10】
【0084】
<合成例5>
(4−(2−ペンチルヘキシル)カルバモイルチオフェン−3−カルボン酸)(化合物21)
Scheme 10に示すように、合成例1で合成した化合物3(1.54g、11.3mmol)および2−ペンチルヘキシルアミン(1.83g、10.7mmol)を蒸留トルエン(100ml)に加えて、反応液を24時間還流した。冷ました反応液をろ過して、粗生成物を回収した。また、5%塩酸水溶液でろ液を洗浄して粗生成物を沈殿させ、クロロホルムで抽出を行った後、溶媒を除去することにより、ろ液からも粗生成物を回収した。さらに、回収した粗生成物を真空中で乾燥させた後、化合物21を得た(3.60g、11.1mmol、収率98%)。化合物21の1H NMR測定の結果は、以下の通りであった。
1H NMR (400 MHz, CDCl3):δ8.43 (d, 1H), 8.07 (d, 1H), 6.95 (broad s, 1H), 4.11 (m, 1H), 1.56 (m, 4H), 1.30 (m, 12H), 0.87 (t, 6H).
【0085】
(2−ペンチルヘキシルジオキソピロロチオフェン)(化合物22)
塩化チオニル(200ml)に化合物21(3.22g、9.90mmol)を加えて、反応液を8時間還流した。反応液を濃縮して薄い茶色の液体を得た後、さらに乾燥して薄い黄色の結晶を得た。さらに、真空中で乾燥させた後、薄い黄色の固体として化合物22を得た(2.80g、9.11mmol、収率92%)。化合物22の1H NMR測定の結果は、以下の通りであった。
1H NMR (400 MHz, CDCl3):δ 7.84 (s, 2H), 4.11 (m, 1H), 1.60 (m, 4H), 1.28 (m, 16H), 0.86 (t, 6H).
【0086】
(1,3−ジヨード(2−ペンチルヘキシル)ジオキソピロロチオフェン)(化合物23)
得られた化合物22(2.80g、9.11mmol)を、濃硫酸(14ml)とトリフルオロ酢酸(47ml)の混合液に溶解させた。次に、反応液にN−ヨードコハク酸イミド(NIS)(4.10g、18.2mmol)を二回に分けて加え、反応液を室温で12時間攪拌した。得られた茶色の溶液を水で希釈して、ジクロロメタンで抽出を行った。得られた有機層を無水硫酸マグネシウムで乾燥した後、溶媒をエバポレーターで除去して、オレンジ色の粗生成物を得た。シリカゲルと、溶離液としてクロロホルムとを使用したカラムクロマトグラフィーにより、粗生成物を精製して、白色の結晶として化合物23を得た(3.11g、5.56mmol、収率61%)。化合物23の1H NMR測定の結果は、以下の通りであった。
1H NMR (400 MHz, CDCl3):δ 4.09 (m, 1H), 1.83 (m, 4H), 1.24 (m, 12H), 0.86 (t, 6H).
【0087】
【化11】
【0088】
(ポリ[(N−(2−ペンチルヘキシル)ジオキソピロロチオフェン)−alt−(チオフェン)])(PPHT)
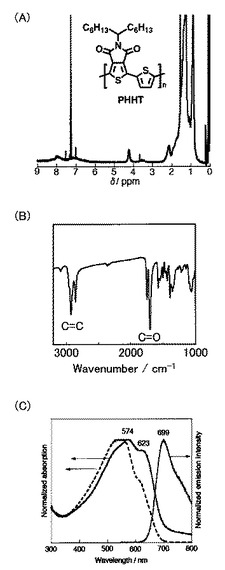
上記で合成した化合物23(56mg、0.10mmol)を、アルゴン雰囲気下、ジメチルホルムアミド(1ml)に溶解させた(Scheme 11)。次に、化合物7(66mg、0.10mmol)とテトラキス(トリフェニルホスフィン)パラジウム(0)(3mg,0.005mmol)とを反応液に加えて、110℃で48時間攪拌した。冷ました反応液を500mlのメタノールに加えて、ろ過により沈殿物を回収した。その後、メタノールおよびヘキサンで沈殿物を洗浄して、PPHT(24)を黒色固体として得た(28mg、収率71%)。得られたポリマーの1H NMR測定の結果を図9Aに、IR測定の結果を図9Bに、UV測定および蛍光測定の結果を図9Cに示す。なお、サイクリックボルタンメトリー測定により、還元オンセット(ERED,ONSET、onset of the reduction potential)は−1.20Vであった。
なお、PPHT(24)の数平均分子量(Mn)は2700であり、分子量分布(PDI)は1.3であった。
【0089】
【化12】
【0090】
<合成例6>
(2,5−ジチエノ(N−ペンタデシルジオキソピロロチオフェン))(化合物27)
Scheme 12に示すように、窒素雰囲気下、2,5−ジブロモ(N−ペンタデシルジオキソピロロチオフェン)(547mg、1.05mmol)(化合物25)、2−(トリブチルスタニル)チオフェン(783mg、2.10mmol)(化合物26)、およびテトラキス(トリフェニルホスフィン)パラジウム(0)(121mg)をジメチルホルムアミド(20ml)に溶解させ、反応液を110℃にて24時間攪拌した。冷ました反応液をメタノール中に加え、黄色の沈殿物を回収した。シリカゲルと、溶離液としてクロロホルム/ヘキサン(1:1)溶液を使用したカラムクロマトグラフィーにより、沈殿物を精製して、黄色の固体として化合物27を得た(274mg、0.51mmol、収率50%)。化合物27の1H NMR測定の結果は、以下の通りであった。
1H NMR (400 MHz, CDCl3):δ 8.02 (d, 2H), 7.44 (d, 2H), 7.13 (t, 2H), 3.66 (dd, 2H), 1.68 (m, 2H), 1.24 (m, 24H), 0.88 (t, 3H).
【0091】
(2,5−ビス(5−ブロモチオフェン−2−イル)(N−ペンタデシルジオキソピロロチオフェン))(化合物28)
化合物27(270mg、0.51mmol)をクロロホルム(4ml)と酢酸(4ml)との混合液に溶解させた。次に、反応液にN−ヨードコハク酸イミド(NIS)(191mg、1.07mmol)を加えて、室温で12時間攪拌した。得られた溶液を氷水(100ml)中に加え、クロロホルムで抽出を行った。得られた有機層を無水硫酸マグネシウムで乾燥し、溶媒をエバポレーターで除去して、黄色の粗生成物を得た。シリカゲルと、溶離液としてクロロホルム/ヘキサン(1:1)溶液とを使用したカラムクロマトグラフィーにより、粗生成物を精製して、黄色の結晶として化合物28を得た(224mg、0.33mmol、収率64%)。化合物28の1H NMR測定の結果は、以下の通りであった。
1H NMR (400 MHz, CDCl3):δ 7.65 (d, 2H), 7.07 (d, 2H), 3.65 (dd, 2H), 1.67 (m, 2H), 1.24 (m, 24H), 0.88 (t, 3H).
【0092】
【化13】
【0093】
化合物30を既報の方法(Ranger, M.; Rondeau, D.; Leclerc, M. Macromolecules 1997, 30, 7686.)により合成した。
【0094】
Scheme 13に示すように、化合物28(36mg、0.05mmol)、化合物30(34mg、0.05mmol)、およびテトラキス(トリフェニルホスフィン)パラジウム(0)(3mg、0.05eq)をトルエン(0.57ml)に溶解させた。さらに、炭酸ナトリウム水溶液(0.12ml)と、Aliquat 336(2Mの炭酸ナトリウム溶液1ml中にAliquat 336を17mg)とを反応液に加えて、100℃にて60時間攪拌した。冷ました溶液をメタノール中に加えて、1M塩酸水溶液で中和した。赤い沈殿物を回収し、メタノールおよびヘキサンで洗浄して、赤い固体としてPTPF(31)を得た。得られたポリマーの1H NMR測定の結果を図10に示す。
なお、PTPF(31)の数平均分子量(Mn)は13,000であり、分子量分布(PDI)は1.9であった。
【0095】
【化14】
【0096】
<合成例7>
(2,5−ビス(5−トリブチルスタニルチオフェン−2−イル)(N−ペンタデシルジオキソピロロチオフェン))(化合物29)
Scheme 14に示すように、窒素雰囲気下、化合物27(181mg、0.34mmol)をヘキサン(5ml)中に溶解させた。次に、ブチルリチウム(0.54ml、0.86mmol、ヘキサン中1.6mol/l)を反応液に滴下して、反応液を1時間還流した。反応液を−78℃まで冷やし、トリブチルスズクロリド(57mg、0.21mmol)を加えた後、さらに室温で12時間攪拌した。攪拌終了後、反応液を水(20ml)に加えて、有機層を分離し、水層をヘキサン(20ml)で抽出を行った。集めた有機層を無水硫酸マグネシウムで乾燥した後、溶媒を除去して、化合物29を得た。化合物29の1H NMR測定の結果は、以下の通りであった。
1H NMR (400 MHz, CDCl3):δ 7.65 (d, 2H), 7.07 (d, 2H), 3.65 (dd, 2H), 1.68 -0.88 (m, 85H).
【0097】
【化15】
【0098】
Scheme 15に示すように、化合物29(54mg、0.048mmol)、化合物32(14mg、0.047mmol)、およびジクロロビス(トリフェニルホスフィン)パラジウム(II)(2mg、0.05eq)をクロロホルム(1ml)に溶解させ、反応液を65℃で72時間加熱した。冷ました反応液をメタノール中に加え、沈殿物を回収した。その後、メタノールおよびヘキサンで沈殿物を洗浄して、黒色固体としてPTPB(33)を得た(26mg、0.039mmol、収率83%)。得られたポリマーのUV測定および蛍光測定の結果を図11に示す。なお、サイクリックボルタンメトリー測定により、酸化オンセット(EOX,ONSET、onset of the oxidation potential)は0.57Vで、還元オンセット(ERED,ONSET、onset of the reduction potential)は−1.31Vであった。
なお、PTPB(33)の数平均分子量(Mn)は9,800であり、分子量分布(PDI)は1.4であった。
【0099】
【化16】
【0100】
<合成例8>
N−オクチルイミド縮環チオフェン(化合物34)を、既報の手法(Nielsen, C. B.; Bjornholm, T. Org. Lett. 2004, 6, 3381)に従って合成した。
【0101】
Scheme 16に示すように、アルゴン雰囲気下、化合物34(42mg、0.10mmol)、化合物7(73mg、0.11mmol)、ジクロロビス(トリフェニルホスフィン)パラジウム(II)(0.1eq)、および蒸留トルエン(1ml)をシュレンク管に入れ、反応液を110℃で96時間攪拌した。冷ました反応液をメタノール中に加えて、メンブレンフィルター(ADVANTEC)にて黒色沈殿物を回収した。その後、メタノールおよびヘキサンで沈殿物を洗浄し、エバポレーターで溶媒を除去して黒色のP8T(35)を得た(18mg、収率38%)。
【0102】
【化17】
【0103】
<吸収スペクトル測定>
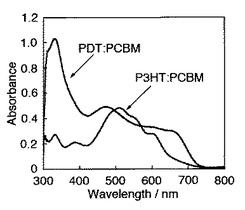
上記で合成したPDTと、公知の材料であるP3HT(ポリ3−ヘキシルチオフェン−2,5−ジイル(レジオレギュラー))(Aldrich社製、Poly(3-hexylthiophene-2,5-diyl)、重量平均分子量60000)とをそれぞれクロロホルムに溶解させ、同じ濃度(27mg/L)の溶液を調整し、吸収スペクトル測定を実施した(図12)。
図12から、PDTのほうが、公知材料のP3HTよりも長波長領域における吸収特性に優れることが分かった。該図からも分かるように、550〜750nmの範囲において、PDTのほうが優れた光吸収特性を示した。例えば、波長650nmにおける吸収係数としては、PDTは29L g-1 cm-1であり、P3HTでは0.30L g-1cm-1であった。また、波長550nmにおける吸収係数としては、PDTは26L g-1 cm-1であり、P3HTでは3.1L g-1cm-1であった。
【0104】
<HOMO−LUMOエネルギー準位>
図13は、本発明の共役系高分子、公知材料のP3HT、およびPCBMの最高被占分子軌道(highest occupied molecular orbit, HOMO)および最低空分子軌道(lowest unoccupied molecular orbit, LUMO)のエネルギー準位図である。
該図より分かるように、公知材料のP3HTに比べて、本発明の共役系高分子(PDTおよびPTPB)のHOMOおよびLUMOはより低くなっていた。つまり、本発明の共役系高分子は、該図および図17から分かるように従来公知の材料と比較して、フラーレン類(PCBM)に対してより適切なエネルギー準位を有する材料であった。
なお、本発明の共役系高分子の該HOMOおよびLUMO準位は、上述したサイクリックボルタンメトリー測定、UV測定などの結果から換算した値である。また、P3HT、PCBMのエネルギー準位は、非特許文献1に記載の値を参照した。
【0105】
<有機薄膜太陽電池の作製と評価>
PDTとPCBMとを光電変換層材料に用い、文献既報(Umeyama, T.; Takamatsu, T.; Tezuka, N.; Matano, Y.; Araki, Y.; Wada, T.; Yoshikawa, O.; Sagawa, T.; Yoshikawa, S.; Imahori, H. J. Phys. Chem. C 2009, 113, 10798-10806.)の手法により、有機薄膜太陽電池の作製と評価を行った。
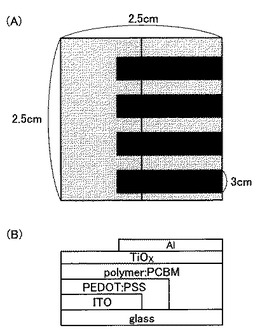
より詳細には、まず、ITO(インジウム錫酸化物)(厚み:300nm)を塗布したガラス基板(Geomatec社製:表面抵抗は5Ω/□)を用意した。次に、ガラス基板をアセトンおよびエタノール中で連続して超音波洗浄した。基板をブロー乾燥して、UVオゾン処理した後、ポリ(3,4−エチレンジオキシチオフェン)/ポリスチレンスルホン酸水溶液(PEDOT:PSS、BaytronP)を基板上に4000rpmでスピンコートし、ホットプレート上で200℃、10分間加熱乾燥した。さらに、窒素雰囲気下、上記で合成したPDTとPCBMとのクロロホルム溶液をPEDOT:PSS層上に1000rpmでスピンコートして、PDT:PCBM層(厚み:170nm)を作製した。その後、チタンテトライソプロポキシド(和光純薬工業)のエタノール溶液をPDT:PCBM層上に2000rpmでスピンコートし、30分間放置して、チタンテトライソプロポキシドの加水分解を進行させ、TiOx層(厚み:5nm)を形成させた。最後に、真空下(5×10‐3Pa)にて、熱蒸着によりアルミニウム層(NilacoCorp製)(厚み:100nm)を堆積させ、ITO/PEDOT:PSS/共役系高分子:PCBM/TiOx/Alの構成をとる有機薄膜太陽電池を得た(図14)
AM1.5,擬似太陽光を100mW/m2の光強度にて照射して、I−V特性を測定し、開放電圧値(VOC)、短絡電流密度(JSC)、曲線因子(FF)、変換効率(η)を求めた。結果を表1に示す。
なお、表1中、PDTとPCBMの濃度(g/L)は、上述したクロロホルム溶液中の濃度を意味する。
【0106】
【表1】
【0107】
表1からわかるように、本発明の共役系高分子を用いた有機薄膜太陽電池においては、0.70V以上と高い開放電圧を実現できた。
一方、実施例1中のPDTの代わりにP3HT(Aldrich社製、Poly(3-hexylthiophene-2,5-diyl)、重量平均分子量60000)を用いて上記有機薄膜太陽電池を作製した場合、その開放電圧は0.6V程度であり、本発明のPDTを使用したほうがより高い開放電圧値を示すことが確認された。
【0108】
また、図15は、PDT:PCBM膜とP3HT:PCBM膜の光吸収スペクトル図である。PDT:PCBM膜は、PDT/PCBM(1mLクロロホルム中、10mg/10mg)をガラス基板上に750rpmにてスピンコートして得られた膜である。また、P3HT:PCBM膜は、P3HT/PCBM(1mLクロロベンゼン中、15mg/7.5mg)をガラス基板上に750rpmにてスピンコートして、150℃で6分間アニーリング処理して得られた膜である。なお、P3HT(Poly(3-hexylthiophene-2,5-diyl))としては、重量平均分子量60000(Aldrich社製)のものを用いた。
該図からわかるように、本発明の共役系高分子を用いた光電変換層であるPDT:PCBM膜においては、P3HTを使用した場合に比べて、可視光の長波長領域における吸収特性に優れることが分かった。
【0109】
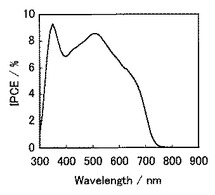
また、図16は、実施例3の有機薄膜太陽電池の光電流アクションスペクトル図である。光電流アクションスペクトルは、入射単色光当たりの光電変換効率(IPCE;Incident Photon-to-Current Efficiency)を測定した結果である。該図からも分かるように、スペクトルの吸収端が長波長領域まで伸びており、幅広い波長の光を有効に利用して高効率の光電変換を実現できる可能性があることを示唆している。
【符号の説明】
【0110】
10、110、120、130 有機薄膜太陽電池
12 透明基板
12A 基板
14 透明電極層
16 正孔輸送層
18、18A、18B 光電変換層
20 電子輸送層
22 対向電極層
24 p層
26 n層
28 混合層
【技術分野】
【0001】
本発明は、共役系高分子、および該共役系高分子を用いた有機薄膜太陽電池に関する。より具体的には、本発明は、イミド環を有するチオフェン基と二価の共役構造を有する連結基とを含む共役系高分子と、該共役系高分子を用いて得られる有機薄膜太陽電池に関する。
【背景技術】
【0002】
有機薄膜太陽電池は、p型(電子ドナー性)の有機半導体材料とn型(電子アクセプター性)の有機半導体材料とを組み合わせた簡便な素子構造を有する。この電池は、色素増感型太陽電池に代表される湿式太陽電池とは異なり、電解液を用いないため加工性に優れ、望みの形状に成形可能である。また、該電池は、シリコン太陽電池や色素増感型太陽電池よりもはるかに軽量化できるため、様々な設置場所に適用することができる。さらに、高純度シリコンや貴金属の使用といった資源的制約もないため、市場シェアの拡大と共に大幅に低コスト化できると見積もられている。
【0003】
有機薄膜太陽電池の課題の一つとして、エネルギー変換効率(光電変換効率)の向上が挙げられる。この光電変換効率の向上のためには、開放電圧値の改善が重要である。有機薄膜太陽電池の開放電圧値は、電子供与性材料および電子受容性材料の組み合わせに大きく依存し、用いる材料の最適化にブレークスルーが必要である。
【0004】
現在、世界中で進められている一連の有機薄膜太陽電池研究において、n型半導体材料としてはフラーレン類(例えば、PCBMなどのフラーレン誘導体)が最適であるとされている。PCBMのHOMO(最高占有分子軌道)は6.0eV、LUMO(最低非占有分子軌道)は4.2eVであることが知られている(非特許文献1)。
【0005】
一方、現在最も一般的に用いられているp型半導体材料としては、ポリチオフェン誘導体の共役高分子化合物(例えば、P3HT:ポリ3−ヘキシルチオフェン−2,5−ジイル)が挙げられる。このP3HTのHOMO(最高占有分子軌道)は3.2eV、LUMO(最低非占有分子軌道)は5.2eVであることが知られている。
【先行技術文献】
【非特許文献】
【0006】
【非特許文献1】Barry C. Thompson他一名、Angewandte Chemie International Edition, 2008, Vol.47, pp.58-77.
【発明の概要】
【発明が解決しようとする課題】
【0007】
一方、非特許文献1に示されるように、P3HTは、n型半導体材料であるフラーレン類に対して、必ずしも理想的なHOMO準位およびLUMO準位を示していなかった(図17 参照)。実際、P3HTとPCBMとを含む光電変換層を備える有機薄膜太陽電池においては、その開放電圧値は実用上の観点からは必ずしも満足できるものではなく、さらなる改良が必要であった。
開放電圧値を改良する方法として、p型半導体材料のHOMO準位を下げる手段が考えられるが、この場合、p型半導体材料のバンドギャップ(LUMO準位とHOMO準位の差)が広がり、長波長領域の光を吸収することができなくなる。つまり、可視光領域の長波長側における光の吸収効率が減少し、入射した光を有効利用できなくなり、結果としてエネルギー変換効率が上がらないという欠点がある。
このように開放電圧値と長波長領域の光吸収とはトレード・オフの関係にあることが多く、両者をより高いレベルで両立させることは困難とされていた。
【0008】
本発明は、上記実情に鑑みて、成膜性に優れると共に、長波長領域における光吸収特性に優れ、フラーレン類との間で高い開放電圧値を実現することができる共役系高分子、および該共役系高分子を用いた光電変換層を備える有機薄膜太陽電池を提供することを目的とする。
【課題を解決するための手段】
【0009】
本発明者らは、鋭意検討を行った結果、所定のイミド環を有するチオフェン構造とπ共役系の連結基とを有する共役系高分子を使用することにより、上記課題を解決できることを見出した。
つまり、本発明者らは、上記課題が下記の構成により解決されることを見出した。
【0010】
<1> 後述する一般式(1)で表される繰り返し単位と、後述する一般式(2)で表される繰り返し単位とを有する共役系高分子。
<2> 一般式(1)中のXが、二価の芳香族炭化水素基、二価の芳香族複素環基、−(CH=CH)−、−(C≡C)−、またはそれらを組み合わせた基である、<1>に記載の共役系高分子。
<3> 第1電極層と、前記第1電極層と対向する電極である第2電極層とを備え、
前記第1電極層と前記第2電極層との間に、少なくとも<1>または<2>に記載の共役系高分子およびフラーレン類を含む光電変換層を備える有機薄膜太陽電池。
【発明の効果】
【0011】
本発明によれば、成膜性に優れると共に、長波長領域における光吸収特性に優れ、フラーレン類との間で高い開放電圧値を実現することができる共役系高分子、および該共役系高分子を用いた光電変換層を備える有機薄膜太陽電池を提供することができる。
【図面の簡単な説明】
【0012】
【図1】本発明の有機薄膜太陽電池の第1の実施形態の模式的断面図である。
【図2】本発明の有機薄膜太陽電池の第2の実施形態の模式的断面図である。
【図3】本発明の有機薄膜太陽電池の第3の実施形態の模式的断面図である。
【図4】本発明の有機薄膜太陽電池の第4の実施形態の模式的断面図である。
【図5】図5AはPDTの1H NMR測定結果を、図5BはIR測定結果を、図5CはUV測定および蛍光測定結果を示す。
【図6】図6AはPOTの1H NMR測定結果を、図6BはIR測定結果を、図6CはUV測定および蛍光測定結果を示す。
【図7】図7AはPHOTの1H NMR測定結果を、図7BはIR測定結果を、図7CはUV測定結果を示す。
【図8】図8AはPHHTの1H NMR測定結果を、図8BはIR測定結果を、図8CはUV測定および蛍光測定結果を示す。
【図9】図9AはPPHTの1H NMR測定結果を、図9BはIR測定結果を、図9CはUV測定および蛍光測定結果を示す。
【図10】図10はPTPFの1H NMR測定結果を示す。
【図11】図11はPTPBのUV測定および蛍光測定結果を示す。
【図12】PDTとP3HTのクロロホルム溶液の吸収スペクトル図である。
【図13】本発明の共役系高分子のエネルギー準位図である。
【図14】図14Aは、実施例で使用された有機薄膜太陽電池の上面図であり、図14Bは模式的断面図である。
【図15】PDT:PCBM膜とP3HT:PCBM膜の吸収スペクトル図である。
【図16】実施例3の有機薄膜太陽電池の光電流アクションスペクトル図である。
【図17】非特許文献1に記載のP3HTとPCMBのエネルギー準位図である。
【発明を実施するための形態】
【0013】
以下に、本発明に係る一般式(1)で表される繰り返し単位と一般式(2)で表される繰り返し単位とを有する共役系高分子、および、該共役系高分子を用いる光電変換層を備える有機薄膜太陽電池について詳述する。
まず、共役系高分子の構成およびその製造方法について詳述する。
【0014】
<共役系高分子>
本発明の共役系高分子は、以下の一般式(1)で表される繰り返し単位と、一般式(2)で表される繰り返し単位とを主鎖中に有する高分子である。
一般式(1)で表される繰り返し単位中のイミド環部分が電子吸引性の機能を有しており、該部分を共役系高分子に含有させることにより、フラーレン類に対してより好ましい電子状態が実現されたと推測される。通常、チオフェン骨格中の2つのβ位に電子吸引性基を導入した化合物を用いて重合をおこなうと、得られる高分子の分子量が伸びず所望の成膜性が得られない場合が多いが、本発明の共役系高分子は実用的な観点からも満足できる成膜性を有している。
なお、公知文献に記載される一般式(1)で表される繰り返し単位のみから得られるポリマーでは、分子量が伸びず、有機薄膜太陽電池へ応用できる程度の十分な成膜性を有していない。また、そのエネルギー準位もLUMO準位が約6evと計算され、フラーレン類と共に有機薄膜太陽電池に応用するには不適切なエネルギー準位といえる。
【0015】
【化1】
【0016】
(一般式(1)で表される繰り返し単位)
一般式(1)中、Rは水素原子または一価の有機基を表す。
一価の有機基としては特に限定されないが、例えば、アルキル基、アルケニル基、アルキニル基、アリール基、複素環基、アルコキシ基、アリールオキシ基、またはこれらを組み合わせた基などが挙げられる。
これら例示の基は置換基を有していてもよい。この置換基の種類は、本発明の効果を損なわない限り特に制限されない。例えば、ハロゲン原子、ニトロ基、シアノ基、スルホン酸基、カルボニルオキシ基、カルボキシル基、アミノ基、アルキル基、アルケニル基、アルキニル基、アリール基、複素環基、アルコキシ基、アリールオキシ基、カルボン酸エステル、スルホン酸エステル基、アミド基などが挙げられる。
また、該有機基は、本発明の効果を損なわない範囲において、−O−、−S−、−SO2−、−CO−、−NH−などの二価の連結基部位を含んでいてもよい。
【0017】
上記有機基としては、成膜性により優れる点から、アルキル基、アルケニル基、アルキニル基などの脂肪族炭化水素基が好ましく、アルキル基がより好ましい。
脂肪族炭化水素基としては、直鎖状、分岐状、環状のいずれであってもよく、炭素数6〜30が好ましく、炭素数8〜20がより好ましい。
なお、アルキル基の具体例としては、ヘキシル基、ヘプチル基、オクチル基、ノニル基、デシル基、ウンデシル基、ドデシル基などが挙げられる。
【0018】
なお、共役系高分子中に複数の一般式(1)で表される繰り返し単位が含まれる場合、Rは各繰り返し単位中で同一であっても異なっていてもよい。
【0019】
(一般式(2)で表される繰り返し単位)
一般式(2)中、Xは、一般式(1)で表される繰り返し単位構造(連結基)とは異なる二価の共役系連結基を表す。共役系連結基とは、π共役構造を有する連結基を意味する。高分子中にこのようなπ共役構造を有する連結基が存在することによって、高分子全体にπ共役平面が広がり、p型半導体材料としての特性がより向上する。
なお、共役系高分子中に複数の一般式(2)で表される繰り返し単位が含まれる場合、複数のXは同一であっても異なっていてもよい。
【0020】
Xは、一般式(1)で表される繰り返し単位以外の共役構造からなる二価の連結基であれば特に限定されない。なかでも、合成が簡便であり、成膜性など本発明の効果がより優れる点で、二価の芳香族炭化水素基、二価の芳香族複素環基、−(CH=CH)−、−(C≡C)−、または、これらから選択された2種以上の基を組み合わせてできる連結基であることが好ましい。なかでも、本発明の効果がより優れる点より、二価の芳香族炭化水素基、二価の芳香族複素環基、およびそれらを組み合わせた基から選ばれる芳香族連結基が好ましく挙げられる。
【0021】
芳香族炭化水素基の炭素数は特に限定されないが、合成上の観点から、炭素数5〜40が好ましく、炭素数6〜20がより好ましく、アリーレン基などが挙げられる。
芳香族炭化水素基としては、例えば、ベンゼン環基、ナフタレン環基、アントラセン環基、テトラリン環基、フェナントレン環基、フルオレン環基、アズレン環基、ピレン環基、クリセン環基、トリフェニレン環基、ペリレン環基、ペンタレン環基、インダセン環基、フェナレン環基、フルオランテン環基、アセフェナントリレン環基、アセアントリレン環基、ナフタセン環基、プレイアデン環基、ペンタフェン環基、ペンタセン環基、テトラフェニレン環基、ヘキサフェン環基、ヘキサセン環基などが挙げられる。なかでも、フルオレン環基、ベンゼン環基、ナフタレン環基が好ましい。なお、上記基中の結合可能な部分のいずれかに、結合が形成される。
【0022】
芳香族複素環基の炭素数は特に限定されないが、合成上の観点から、炭素数5〜40が好ましく、炭素数6〜20がより好ましく、含窒素又は含硫黄芳香族複素環などが挙げられる。
芳香族複素環基としては、例えば、ピロール環基、インドール環基、イソインドール環基、カルバゾール環基、フラン環基、クマロン環基、イソベンゾフラン環基、チオフェン環基、ベンゾチオフェン環基、ジベンゾチオフェン環基、ピラゾール環基、インダゾール環基、イミダゾール環基、ベンゾイミダゾール環基、オキサゾール環基、ベンゾオキサゾール環基、ベンゾオキサゾリン環基、イソオキサゾール環基、ベンゾイソオキサゾール環基、チアゾール環基、ベンゾチアゾール環基、ベンゾチアゾリン環基、イソチアゾール環基、ベンゾイソチアゾール環基、トリアゾール環基、ベンゾトリアゾール環基、オキサジアゾール環基、チアジアゾール環基、ベンゾオキサジアゾール環基、ベンゾチアジアゾール環基、テトラゾール環基、プリン環基、ピリジン環基、キノリン環基、イソキノリン環基、アクリジン環基、フェナントリジン環基、ベンゾキノリン環基、ベンゾイソキノリン環基、ナフチリジン環基、フェナントロリン環基、ピリダジン環基、ピリミジン環基、ピラジン環基、フタラジン環基、キノキサリン環基、キナゾリン環基、シンノリン環基、フェナジン環基、ペリミジン環基、トリアジン環基、テトラジン環基、プテリジン環基、ベンゾオキサジン環基、フェノキサジン環基、ベンゾチアジン環基、フェノチアジン環基、オキサジアジン環基、チアジアジン環基、ベンゾジオキソール環基、ベンゾジオキサン環基、ピラン環基、クロメン環基、キサンテン環基、クロマン環基、イソクロマン環基などが挙げられる。なかでも、チオフェン環基、ベンゾチアジアゾール環基などが好ましい。なお、上記基中の結合可能な部分のいずれかに、結合が形成される。
【0023】
Xで表される共役系連結基は置換基を有してもよく、置換基としては、例えば、上述したRで表される有機基が有していてもよい置換基などが挙げられる。
【0024】
共役系連結基は、上述した連結基を2種以上組み合わせてできる連結基であってもよく、上述した連結基を2または3つ組み合わせた基がより好ましい。例えば、−(芳香族炭化水素基)−(芳香族炭化水素基)−、−(芳香族炭化水素基)−(芳香族炭化水素基)−(芳香族炭化水素基)−などの芳香族炭化水素基が2以上連結した基、−(芳香族複素環基)−(芳香族複素環基)−、−(芳香族複素環基)−(芳香族複素環基)−(芳香族複素環基)−などの芳香族複素環基が2以上連結した基や、−(芳香族炭化水素基)−(芳香族複素環基)−、−(芳香族炭化水素基)−(CH=CH)−、−(芳香族複素環基)−(CH=CH)−、−(芳香族複素環基)−(芳香族炭化水素基)−(芳香族複素環基)−など異種の連結基が連結した基などが挙げられる。
【0025】
共役系高分子中における一般式(1)で表される繰り返し単位と一般式(2)で表される繰り返し単位との含有割合(モル比)は、発明の効果を損なわない限り特に制限されない。
なかでも、共役系高分子の光吸収特性、および、有機薄膜太陽電池における開放電圧値がより大きい点から、両繰り返し単位の含有割合(一般式(1)で表される繰り返し単位/一般式(2)で表される繰り返し単位)(モル比)は、0.2〜5が好ましく、0.5〜2がより好ましい。
【0026】
本発明の共役系高分子中において、一般式(1)で表される繰り返し単位の含有量は、本発明の効果を損なわない限り特に制限されない。なかでも、本発明の効果がより優れる点で、該繰り返し単位の含有量は、全繰り返し単位(100モル%)に対して、10〜90モル%が好ましく、30〜70モル%がより好ましい。
また、本発明の共役系高分子中において、一般式(2)で表される繰り返し単位の含有量は、本発明の効果を損なわない限り特に制限されない。なかでも、本発明の効果がより優れる点で、該繰り返し単位の含有量は、全繰り返し単位(100モル%)に対して、10〜90モル%が好ましく、30〜70モル%がより好ましい。
【0027】
本発明の共役系高分子中において、一般式(1)で表される繰り返し単位と、一般式(2)で表される繰り返し単位とが主成分であることが好ましい。
より具体的には、両繰り返し単位の合計量は、全繰り返し単位(100モル%)に対して、80モル%以上が好ましく、90モル%以上がより好ましい。上限としては、100モル%である。
【0028】
本発明の共役系高分子中において、一般式(1)で表される繰り返し単位と、一般式(2)で表される繰り返し単位との結合様式は特に制限されず、ランダム共重合体、ブロック共重合体、交互共重合体などが挙げられる。
【0029】
(共役系高分子の好適実施態様)
本発明の共役系高分子の好適実施態様としては、一般式(1)で表される繰り返し単位と、一般式(2)で表される繰り返し単位とが交互に結合した構造を有する共役系高分子(交互共重合体型共役系高分子)が好ましい。該共役系高分子であれば、一般式(1)で表される立体的にかさ高い各チオフェン骨格部が一般式(2)で表される繰り返し単位を介して離れて存在するため、高分子の平面性がより高くなり、膜中での高分子間のパッキングが向上することが予想される。
【0030】
本発明の共役系高分子中において、一般式(1)で表される繰り返し単位と一般式(2)で表される繰り返し単位とが交互に結合した構造の含有量は、本発明の効果を損なわない限り特に制限されない。
なかでも、共役系高分子の光吸収特性、および、有機薄膜太陽電池における開放電圧値がより大きい点から、該構造が実質的に主成分として含まれていることが好ましい。より具体的には、該構造の含有量は、全繰り返し単位(100モル%)に対して、80モル%以上が好ましく、90モル%以上がより好ましい。上限としては、100モル%である。
【0031】
なお、本発明の共役系高分子が上記交互共重合型の場合、本発明の効果を損なわない範囲で、上述した一般式(1)で表される繰り返し単位と、一般式(2)で表される繰り返し単位とが交互に結合した構造以外の共役構造を有する繰り返し単位を有していてもよい。
例えば、一般式(2)で表される繰り返し単位が連結して得られる構造などを一部に含んでいてもよい。
【0032】
本発明の共役系高分子の数平均分子量(Mn)は特に制限されないが、溶剤への溶解性や成膜性により優れる点から、2000〜150000が好ましく、2500〜100000がより好ましい。
また、共役系高分子の分子量分布(Mw/Mn)は特に制限されないが、溶剤への溶解性や成膜性により優れる点から、1〜8が好ましく、1〜5がより好ましい。
【0033】
本発明の共役系高分子の合成方法は特に制限されず、公知の方法を組み合わせて合成することができる。例えば、上述したイミド環を有するチオフェン化合物と、Xで表される構造を有する化合物とをカップリング反応(例えば、Stilleカップリング反応)を介して重合させる方法が挙げられる。
具体的には、Nielsen, C. B.; Bjornholm, T. Org. Lett. 2004, 6, 3381などの文献を参照することにより合成することができる。
【0034】
<有機薄膜太陽電池、およびその製造方法>
本発明の有機薄膜太陽電池は、第1電極層と、少なくとも上述した共役系高分子およびフラーレン類を含有する光電変換層と、第2電極層とを備える。
以下に、本発明の有機薄膜太陽電池の形態、および、その製造方法について詳述する。
【0035】
<有機薄膜太陽電池(第1の実施形態)>
図1は、本発明の有機薄膜太陽電池の第1の実施形態の模式的断面図である。
同図に示す有機薄膜太陽電池10は、透明基板12、透明電極層14、正孔輸送層16、光電変換層18、電子輸送層20、および対向電極層22をこの順で積層した積層構造を有する。該太陽電池は、光電変換層18がp型有機半導体材料とn型有機半導体材料とが混合して存在する混合層である、いわゆるバルクヘテロ接合型有機薄膜太陽電池に該当する。各層の厚みは、該図によっては制限されない。
以下に、各層について詳述する。
【0036】
<透明基板>
透明基板12は、後述する電極層、光電変換層などを支持し、補強するものであれば、任意の材料を使用することができる。なお、該基板は、後述する透明電極層14が十分な強度有していれば、本発明の有機薄膜太陽電池に含まれていなくてもよい。
透明基板12としては、例えば、ガラスや、ポリイミド、PET、PEN、PES、テフロン(登録商標)等の耐熱性の高分子フィルムや、セラミックなどが挙げられる。透明基板12は、高い透明性を有するものが好ましく、例えばガラスが挙げられる。なお、透明基板12の表面はフラットであってもよいし、凹凸を有していてもよい。
透明基板12の厚さは任意の厚さとすることができ、好ましくは0.05mm〜3mmである。
【0037】
<透明電極層(第1電極層)>
透明電極層14は、後述する光電変換層18の一面側に設けられる第1電極層である。本実施形態では、透明電極層14は、正孔輸送層16を介して設けられているが、光電変換層18に接して設けられていてもよい。透明電極層14は、光電変換層18に対してオーミック接触の形成が可能であり、かつ照射光を透過させるものであれば任意の材料を使用することができる。
例えば、ITO、SnO2、ZnO、In2O3などの透明導電材料、または、フッ素ドープ酸化錫(SnO2:F)、アンチモンドープ酸化錫(SnO2:Sb)、錫ドープ酸化インジウム(In2O3:Sn)、Alドープ酸化亜鉛(ZnO:Al)、Gaドープ酸化亜鉛(ZnO:Ga)などの、上記透明導電材料に不純物がドープされたもので構成される。
透明電極層14は、上記材料からなる単独層で構成されていてもよく、複数の層を積層した積層体で構成されていてもよい。透明電極層14の層厚は、電極としての機能を果たすものであれば特に限定されるものではないが、通常は3nm〜10μmである。なお、透明電極層14は、表面がフラットなものでもよく、表面に凹凸を有しているものでもよい。
【0038】
<正孔輸送層>
正孔輸送層16は、透明電極層14と後述する光電変換層18との間に設置され、正孔の取り出し効率の向上に寄与する。また、正孔輸送層16は、光電変換層18から透明電極層14への電子の漏れを抑制する電子ブロッキング効果や、透明電極層14の凹凸を平滑にするバッファー層としても機能する。なお、この正孔輸送層16は、必要に応じて設置され、本発明の有機薄膜太陽電池に含まれていなくてもよい。
正孔輸送層16としては、ポリ−(3,4−エチレンジオキシチオフェン)/ポリエチレンスルフォン酸ナトリウム(通称PEDOT−PSS)など正孔輸送性のある物質であって、最高占有分子軌道(HOMO)のエネルギーレベルが真空準位から比較して透明電極層14よりも低いレベルであれば、特に制限はない。材料としては、例えば、ドープされたポリアニリン、ポリフェニレンビニレン、ポリピロール、ポリパラフェニレン、ポリアセチレン、トリフェニルジアミン(TPD)等の導電性有機化合物などが挙げられる。
正孔輸送層16の層厚については特に制限はないが、5〜500nm程度が好ましい。
【0039】
<光電変換層>
本実施形態の光電変換層18は、1層中にp型有機半導体材料(アクセプター)およびn型半導体材料(ドナー)が混在するバルクヘテロ型の光電変換層(バルクヘテロ型混合層)のことである。本実施形態においてはp型有機半導体材料として上述した一般式(1)で表される繰り返し単位および一般式(2)で表される繰り返し単位を有する共役系高分子が用いられ、n型有機半導体材料としてフラーレン類が主に用いられる。
以下に、使用される材料に関して詳述する。
【0040】
p型有機半導体材料としては、上述した共役系高分子が使用されるが、それ以外の公知の他の材料を併用してもよい。
公知のp型有機半導体材料としては、N,N’−ビス(3−トリル)−N,N’−ジフェニルベンジジン(mTPD)、N,N’−ジナフチル−N,N’−ジフェニルベンジジン(NPD)、4,4’,4’’−トリス(フェニル−3−トリルアミノ)トリフェニルアミン(MTDATA)などのアミン化合物;フタロシアニン(Pc)、銅フタロシアニン(CuPc)、亜鉛フタロシアニン(ZnPc)、チタニルフタロシアニン(TiOPc)などのフタロシアニン類;オクタエチルポルフィリン(OEP)、白金オクタエチルポルフィリン(PtOEP)、亜鉛テトラフェニルポルフィリン(ZnTPP)などのポルフィリン類;ポリヘキシルチオフェン(P3HT)、メトキシエチルヘキシロキシフェニレンビニレン(MEHPPV)などの主鎖型共役高分子類やポリビニルカルバゾールなどの側鎖型高分子類などの高分子化合物などが挙げられる。
【0041】
n型有機半導体材料としては、フラーレン類が用いられる。フラーレン類とは、フラーレン、その誘導体(フラーレン誘導体)、および、その金属内包物等を含む概念である。
フラーレンとしては、種々の立体構造を有するカーボンクラスター、例えば、C60、C70、C74、C76、C78、C82、C84、C720、C860などのフラーレンが挙げられる。フラーレンの形態は、例えば、サッカーボール状、バッキーボール状などが挙げられる。
【0042】
フラーレン誘導体とは、フラーレン骨格に置換基を導入して表面修飾した化合物を意味する。修飾方法は、特に限定されず、例えば、フラーレンの反応性に富む炭素5員環部を化学的に修飾できる。置換基の種類は、特に限定されず、例えば、アルキル基(炭素数1〜10のメチル基、t−ブチル基などのアルキル基等)、アリール基(フェニル基等)、アラルキル基(ベンジル基等)、ジオキソラン単位、ハロゲン原子、または、酸素原子等が例示できる。また、液晶ポリマー、色素類、ポリエチレンオキシド等の導入により修飾されていてもよい。フラーレンの修飾により、溶媒、高分子への可溶化や親和性の改善、フラーレンの配列または配向を可能にする。
【0043】
金属を内包したフラーレンとしては、種々の金属、例えば、周期表第1A族元素(K、Na、Rb等)、周期表第2A族元素、ランタノイド族元素(La等)等の金属がドープされたフラーレンが例示できる。
【0044】
n型有機半導体材料としては、上記の中でも、[6,6]−フェニル−C61ブチルカルボン酸メチルエステル(PCBM)が好ましく挙げられる。
【0045】
なお、本発明の効果を損なわない限り、ペリレン誘導体、多環キノン、キナクリドンなどの他の公知のn型有機半導体材料を併用してもよい。
【0046】
p型有機半導体材料とn型有機半導体材料の混合割合は特に制限されないが、得られる有機薄膜太陽電池の性能がより優れる点から、両者の質量比(p型有機半導体材料/n型有機半導体材料)は、0.3〜1.8が好ましく、0.5〜1.5がより好ましい。
【0047】
光電変換層18の厚みは特に制限されないが、得られる有機薄膜太陽電池の性能がより優れる点から、100〜400nmが好ましく、150〜300nmがより好ましい。
【0048】
<電子輸送層>
電子輸送層20は、光電変換層18と対向電極層22との間に配置され、電子の取り出し効率の向上に寄与する。なお、この電子輸送層20は、必要に応じて設置され、本発明の有機薄膜太陽電池に含まれていなくてもよい。
電子輸送層20としては、例えば、フッ化リチウム等のアルカリ金属、アルカリ土類金属のハロゲン化物、酸化物等を用いることができる。また、酸化チタンなどの無機半導体を用いることもできる。
電子輸送層20の層厚については特に制限はないが、5〜100nm程度が好ましい。
【0049】
<対向電極層(第2電極層)>
対向電極層22は、光電変換層18の他面側に設けられる第2電極層である。他面側とは、光電変換層18に対し、透明電極層14の反対側をいう。本実施形態では、対向電極層22は、電子輸送層20を介して設けられているが、光電変換層18に接して設けられていてもよい。
対向電極層22を構成する材料は、光電変換層18の半導体からの電荷を有効に収集できる仕事関数を有することが望ましい。透明電極層14が、正孔を収集する場合は、対向電極層22としては電子を収集しやすいアルミニウム、マグネシウム、カルシウムなどが用いられる。
対向電極層22の層厚は、発生した光電荷を十分に外部回路へ伝達できる程度のシート抵抗を得ることができる範囲であれば、特に限定されない。対向電極層22の層厚については特に制限はないが、1〜100nmが好ましい。
【0050】
<有機薄膜太陽電池の製造方法>
本発明の有機薄膜太陽電池の各層の形成方法は特に制限されず、真空蒸着、スパッタリング、プラズマ、イオンプレーティングなどの乾式成膜法や、スピンコーティング、ディップコート、キャスティング、ロールコート、フローコーティング、インクジェットなどの湿式成膜法を採用することができる。
より詳細には、まず、透明基板12を設置し、透明基板12上に透明電極層14を形成する。基板の形状および寸法に制限はなく、任意に設置することができる。透明電極層14は任意の方法により形成することができ、例えば、真空蒸着、スパッタなどのドライプロセス、ゾルゲル法などの湿式プロセスの方法が挙げられる。
次に、透明電極層14の表面上に正孔輸送層16を形成する。正孔輸送層16は、使用する材料を溶媒に溶かして、該溶液を使用してスピンコート、ディップコート、キャストコートなどの任意の方法を用いて形成することができる。
次に、上述した共役系高分子とn型有機半導体材料とを含む溶液を用いて、湿式成膜法により透明電極層14の表面上に光電変換層18を成膜する。
さらに、光電変換層18の表面上に、電子輸送層20を形成する。電子輸送層20は、真空蒸着などのドライプロセス、ゾルゲル法などの湿式プロセスなどの方法を用いて形成できる。
最後に、電子輸送層20の表面上に、対向電極層22を形成する。対向電極層22は、真空蒸着、レーザー転写法などの任意の方法で形成できる。
【0051】
乾式成膜法を採用する場合、抵抗加熱法を用いて材料を加熱蒸発させることが好ましい。また、混合層を形成する場合には、例えば、複数の蒸発源からの同時蒸着による成膜方法が好ましい。成膜時には、基板温度を一定に制御することが好ましい。
湿式成膜法を適用する場合、材料を適切な溶媒に溶解または分散させて溶液を調製してから薄膜を形成する。かかる溶媒としては任意の溶媒を使用でき、例えば、ジクロロメタン、ジクロロエタン、クロロホルム、四塩化炭素、テトラクロロエタン、トリクロロエタン、クロロベンゼン、ジクロロベンゼン、クロロトルエンなどのハロゲン系炭化水素系溶媒;ジブチルエーテル、テトラヒドロフラン、ジオキサン、アニソールなどのエーテル系溶媒;メタノール、エタノール、プロパノール、ブタノール、ペンタノール、ヘキサノール、シクロヘキサノール、メチルセロソルブ、エチルセロソルブ、エチレングリコールなどのアルコール系溶媒;ベンゼン、トルエン、キシレン、エチルベンゼン、ヘキサン、オクタン、デカン、テトラリンなどの炭化水素系溶媒;酢酸エチル、酢酸ブチル、酢酸アミルなどのエステル系溶媒などが挙げられる。また、これらの溶媒は、単独で使用しても複数混合して用いてもよい。
また、溶液中の材料濃度は、使用される材料に応じて適宜最適な濃度が選択される。
【0052】
本発明においては、有機薄膜太陽電池のいずれの層においても、成膜性向上、膜のピンホール防止などのため、適切な樹脂や添加剤を含有させてもよい。使用可能な樹脂としては、ポリスチレン、ポリカーボネート、ポリアリレート、ポリエステル、ポリアミド、ポリウレタン、ポリスルフォン、ポリメチルメタクリレート、ポリメチルアクリレート、セルロースなどの絶縁性樹脂およびそれらの共重合体;ポリ−N−ビニルカルバゾール、ポリシランなどの光導電性樹脂;ポリチオフェン、ポリピロールなどの導電性樹脂などが挙げられる。
また、添加剤としては、酸化防止剤、紫外線吸収剤、可塑剤などが挙げられる。
【0053】
<有機薄膜太陽電池(第2の実施形態)>
図2は、本発明の有機薄膜太陽電池の第2の実施形態を示す概略図である。
図2に示す有機薄膜太陽電池110は、基板12A上に、対向電極層22、光電変換層18および透明電極層14が順次積層されて構成されている。この構成は、基板12Aからの積層の順を図1に示したのとは逆にした構成である。対向電極層22は基板12A上に設けられており、透明電極層14は有機薄膜太陽電池110の最表面に設けられている。
本実施形態においては、基板12Aは必ずしも透明である必要はなく、金属、セラミックス、プラスチックなどの様々な材料の基板を用いることができる。
本実施形態における各層の成分および塗布液の調製方法は、第一の実施形態と同様とすることができる。本実施形態の有機薄膜太陽電池の製造方法は、基板12A上に、対向電極層22、電子輸送層20、光電変換層18、正孔輸送層16、透明電極層14の順に積層する点において、第一の実施形態と異なる。かかる構成にすることにより、図1に示す構成と比較して、多くの場合に金属薄膜が用いられ、真空蒸着で形成される対抗電極層22を、有機膜形成前に形成できるので、真空蒸着による有機膜への損傷を抑制する事が可能という効果が得られる。
【0054】
<有機薄膜太陽電池(第3の実施形態)>
図3は、本発明の有機薄膜太陽電池の第3の実施形態を示す概略図である。
図3に示す有機薄膜太陽電池120は、透明基板12、透明電極層14、正孔輸送層16、光電変換層18A、電子輸送層20、および対向電極層22をこの順で積層した積層構造を有し、光電変換層18Aがp型有機半導体材料からなるp層24とn型有機半導体材料からなるn層26との二層構造(バイレイヤー構造)である。
本実施形態における各層の成分および塗布液の調製方法は、第一の実施形態と同様とすることができる。なお、光電変換層18Aを作製する方法としては、まず、p型有機半導体材料を含む溶液を用いて湿式成膜法によりp層24を作製した後、n型有機半導体材料を含む溶液を用いて湿式成膜法によりn層26を作製する方法が挙げられる。
【0055】
<有機薄膜太陽電池(第4の実施形態)>
図4は、本発明の有機薄膜太陽電池の第4の実施形態を示す概略図である。
図4に示す有機薄膜太陽電池130は、透明基板12、透明電極層14、正孔輸送層16、光電変換層18B、電子輸送層20、および対向電極層22をこの順で積層した積層構造を有し、光電変換層18Bがp型有機半導体材料からなるp層24と、p型有機半導体材料とn型有機半導体材料との混合層28と、n型有機半導体材料からなるn層26との三層構造である。
本実施形態における各層の成分および塗布液の調製方法は、第一の実施形態と同様とすることができる。なお、光電変換層18Bを作製する方法としては、まず、p型有機半導体材料を含む溶液を用いて湿式成膜法によりp層24を作製した後、p型有機半導体材料とn型有機半導体材料とを含む溶液を用いて湿式成膜法により混合層28を作製し、さらにn型有機半導体材料を含む溶液を用いて湿式成膜法によりn層26を作製する方法が挙げられる。
【0056】
本発明で得られる有機薄膜太陽電池材料を用いた有機薄膜太陽電池は、太陽電池モジュール、太陽光発電パネル、時計、携帯情報端末、パーソナルコンピューターなどの装置に有効に利用される。
【実施例】
【0057】
以下、実施例により本発明を詳細に説明するが、本発明はこれらに限定されるものではない。
【0058】
(測定条件等)
GPC測定は、SHIMADZU Prominenceを使用し、カラム温度25℃で、溶出溶媒としてクロロホルムを使用し、数平均分子量は標準ポリスチレンを用いて作製した検量線を用いて計算した。
核磁気共鳴(1H NMR)スペクトルは、JEOL JNM-EX400を用いて測定した。
赤外吸収(IR)スペクトルは、JASCO FT/IR-470 Plusを用いて測定した。
サイクリックボルタンメトリーは、測定装置としてALS 630aを使用し、作用電極として後述する共役系高分子がコートされたITO電極、対電極としてPt線、参照電極としてAg/AgNO3電極を用いて測定した。この測定時の掃引速度は10〜100mV/sec、走査電位領域は−2.0V〜1.6Vであった。還元電位および酸化電位は、後述する共役系高分子のフィルムを、支持電解質としてテトラブチルアンモニウムヘキサフルオロフォスファート0.1mol/Lを溶解させたアセトニトリル溶液中に浸漬して測定した。
【0059】
<合成例1>
N−ドデシルイミド縮環チオフェン(化合物5)を既報(Nielsen, C. B.; Bjornholm, T. Org. Lett. 2004, 6, 3381)の手法に従って合成した(Scheme 1)。
【0060】
【化2】
【0061】
(2,5−ジヨード(N−ドデシルジオキソピロロチオフェン))(化合物6)
上記化合物5(0.838g、2.70mmol)を、濃硫酸(4.2ml)とトリフルオロ酢酸(14ml)の混合液に溶解させた。次に、反応液にN−ヨードコハク酸イミド(NIS)(2.44g、10.8mmol)を二回に分けて加え、反応液を室温で24時間攪拌した。得られた紫色の溶液を氷水(150ml)に加えて、ジクロロメタンで抽出を行った。得られた有機層を10%チオ硫酸ナトリウム水溶液で洗浄し、無水硫酸マグネシウムで乾燥した後、溶媒をエバポレーターで除去して、茶色の粗生成物を得た。シリカゲルと、溶離液としてジクロロメタン/ヘキサン溶液(1:1)とを使用したカラムクロマトグラフィーにより、粗生成物を精製し、さらにエタノールで再結晶することにより、化合物6を得た(0.868g、1.51mmol、収率56%)。化合物6の1H NMR測定の結果は、以下の通りであった。
1H NMR (300 Mhz, CDCl3):δ 3.59 (dd, 2H), 1.62 (quintet, 2H), 1.25 (m, 18H), 0.88 (t, 3H).
【0062】
(2,5−ビス(トリブチルスタニル)チオフェン)(化合物7)
2,5−ビス(トリブチルスタニル)チオフェン(化合物7)を既報の手法(Hou, J.; Tan, Z.; Yan, Y.; He, Y.; Yang, C.; Li, Y. J. Am. Chem. Soc. 2006, 128, 4911)にしたがって合成した(Scheme 2)。
【0063】
【化3】
【0064】
(ポリ[(N−ドデシルジオキソピロロチオフェン)−alt−(チオフェン)])(PDT)
さらに、アルゴン置換した乾燥シュレンク管に、化合物6(30mg、0.052mmol)、化合物7(38mg、0.057mmol)、ジクロロビス(トリフェニルホスフィン)パラジウム(II)(3.7g、0.0053mmol)、および蒸留トルエン(1ml)を加えた(Scheme 3)。その後、反応混合液を120℃で72時間攪拌した。攪拌終了後、冷ました反応混合液をメタノールに加えて、生じた黒色沈殿をメンブレンフィルター(ADVANTEC)により回収した。この生成物をメタノールおよびヘキサンで洗浄した後、分取クロマトグラフィー(LC−908 JAi)を用いて精製し、黒色のPDT(8)を得た(5.6mg、収率27%)。得られたポリマーの1H NMR測定の結果を図5Aに、IR測定の結果を図5Bに、UV測定および蛍光測定の結果を図5Cに示す。なお、サイクリックボルタンメトリー測定により、酸化オンセット(EOX,ONSET、onset of the oxidation potential)は0.81Vで、還元オンセット(ERED,ONSET、onset of the reduction potential)は−1.11Vであった。
なお、PDT(8)の数平均分子量(Mn)は87,000で、分子量分布(PDI)は4.1であった。
【0065】
【化4】
【0066】
<合成例2>
(4−オクタデシルカルバモイルチオフェン−3−カルボン酸)(化合物9)
Scheme 4に示すように、合成例1で合成したチオフェン−3,4−カルボン酸無水物(化合物3)(468mg、3.04mmol)とn−オクタデシルアミン(873mg、3.24mmol)とを蒸留トルエン(50m)に加え、反応液を24時間還流した。冷ました反応液から、ろ過により粗生成物を回収した。また、5%塩酸水溶液でろ液を洗浄して粗生成物を沈殿させ、クロロホルムで抽出を行った後、溶媒を除去することにより、ろ液からも粗生成物を回収した。次に、回収した粗生成物をトルエンで再結晶することにより、白色の結晶である化合物9を得た(1.21g、2.86mmol、収率94%)。化合物9の1H NMR測定の結果は、以下の通りであった。
1H NMR (300 MHz, CDCl3):δ 8.40 (d, 1H), 7.73 (d, 1H), 6.46 (broad s, 1H), 3.41 (t, 2H), 1.58 (quintet, 2H), 1.46 - 0.76 (m, 33H).
【0067】
(オクタデシルジオキソピロロチオフェン)(化合物10)
塩化チオニル(100ml)に化合物9(1.21g、2.86mmol)を加えて、反応液を3時間還流した。反応液を濃縮して薄い茶色のオイルを得た後、さらに乾燥して薄い黄色の結晶を得た。次に、結晶をヘキサン中で再結晶することにより、白い結晶として化合物10を得た(903mg、2.23mmol、収率78%)。化合物10の1H NMR測定の結果は、以下の通りであった。
1H NMR (300 MHz, CDCl3):δ 7.80 (s, 2H), 3.60 (t, 2H), 1.63 (quintet, 2H), 1.54 - 0.86 (m, 33H).
【0068】
(1,3−ジブロモオクタデシルジオキソピロロチオフェン)(化合物11)
得られた化合物10(887mg、2.19mmol)を、濃硫酸(3.4ml)とトリフルオロ酢酸(11.3ml)の混合液に溶解させた。次に、反応液にN−ブロモコハク酸イミド(NBS)(1.56g、4.67mmol)を二回に分けて加え、反応液を55℃で24時間攪拌した。得られた茶色の溶液を水(120ml)で希釈して、ジクロロメタン(150ml)で抽出を行った。得られた有機層を無水硫酸マグネシウムで乾燥した後、溶媒をエバポレーターで除去して、オレンジ色の粗生成物を得た。シリカゲルと、溶離液としてクロロホルムとを使用したカラムクロマトグラフィーにより、粗生成物を精製し、さらにエタノールで再結晶することにより、白い結晶として化合物11を得た(716mg、1.27mmol、収率58%)。化合物11の1H NMR測定の結果は、以下の通りであった。
1H NMR (400 MHz, CDCl3):δ 3.58 (t, 2H), 1.62 (quintet, 2H), 1.53 - 086 (m, 33H).
【0069】
【化5】
【0070】
(ポリ[(N−オクタデシルジオキソピロロチオフェン)−alt−(チオフェン)])(POT)
上記で合成した化合物11(200mg、0.355mmol)を、アルゴン雰囲気下、クロロホルム(5ml)に溶解させた(Scheme 5)。次に、化合物7(243mg、0.367mmol)とジクロロビス(トリフェニルホスフィン)パラジウム(II)(12.5mg,0.018mmol)とを反応液に加えて、65℃で96時間攪拌した。冷ました反応液を500mlのメタノールに加えて、ろ過により沈殿物を回収した。その後、メタノールおよびヘキサンで沈殿物を洗浄して、粗生成物POTを黒色固体として得た(160mg、収率93%)。溶離液としてクロロホルムを用いるクロマトグラフィーにより、粗生成物を精製した後、クロロホルム/メタノールにより再沈殿して、POT(12)を得た(131mg、収率76%)。得られたポリマーの1H NMR測定の結果を図6Aに、IR測定の結果を図6Bに、UV測定および蛍光測定の結果を図6Cに示す。なお、サイクリックボルタンメトリー測定により、酸化オンセット(EOX,ONSET、onset of the oxidation potential)は0.57Vで、還元オンセット(ERED,ONSET、onset of the reduction potential)は−1.31Vであった。
なお、POT(12)の数平均分子量(Mn)は39,000であり、分子量分布(PDI)は1.5であった。
【0071】
【化6】
【0072】
<合成例3>
(4−(2−ヘプチルオクチル)カルバモイルチオフェン−3−カルボン酸)(化合物13)
Scheme 6に示すように、合成例1で合成した化合物3(1.00g、6.49mmol)と2−ヘプチルオクチルアミン(1.57g、6.92mmol)とを蒸留トルエン(100ml)に加え、反応液を24時間還流した。冷ました反応液をろ過して、粗生成物を回収した。また、5%塩酸水溶液でろ液を洗浄して粗生成物を沈殿させ、クロロホルムで抽出を行った後、溶媒を除去することにより、ろ液からも粗生成物を回収した。さらに、回収した粗生成物を真空中で乾燥させた後、薄い黄色の液体として化合物13を得た(2.05g、5.39mmol、収率83%)。化合物13の1H NMR測定の結果は、以下の通りであった。
1H NMR (300 MHz, CDCl3):δ8.49 (d, 1H), 7.97 (d, 1H), 6.70 (broad s, 1H), 4.11 (m, 1H), 1.57 (m, 4H), 1.26 (m, 20H), 0.87 (t, 6H).
【0073】
(2−ヘプチルオクチルジオキソピロロチオフェン)(化合物14)
塩化チオニル(150ml)に化合物13(1.51g、3.96mmol)を加えて、反応液を4時間還流した。反応液を濃縮して薄い茶色の液体を得た後、さらに乾燥して薄い黄色の結晶を得た。さらに、真空中で乾燥させた後、薄い茶色の液体として化合物14を得た(1.14g、3.13mmol、収率79%)。化合物14の1H NMR測定の結果は、以下の通りであった。
1H NMR (300 MHz, CDCl3):δ 7.78 (s, 2H), 4.11 (m, 1H), 1.85 (m, 4H), 1.23 (m, 20H), 0.85 (t, 6H).
【0074】
(1,3−ジブロモ(2−ヘプチルオクチル)ジオキソピロロチオフェン)(化合物15)
得られた化合物14(1,00g、2.75mmol)を、濃硫酸(4.3ml)とトリフルオロ酢酸(14.2ml)の混合液に溶解させた。次に、反応液にN−ブロモコハク酸イミド(NBS)(1.96g、5.86mmol)を二つに分けて加え、反応液を室温で24時間攪拌した。得られた茶色の溶液を水(150ml)で希釈して、ジクロロメタン(200ml)で抽出を行った。得られた有機層を無水硫酸マグネシウムで乾燥した後、溶媒をエバポレーターで除去して、オレンジ色の粗生成物を得た。シリカゲルと、溶離液としてクロロホルムとを使用したカラムクロマトグラフィーにより、粗生成物を精製して、茶色の液体として化合物15を得た(816mg、1.56mmol、収率57%)。化合物15の1H NMR測定の結果は、以下の通りであった。
1H NMR (400 MHz, CDCl3):δ 4.14 (m, 1H), 1.83 (m, 4H), 1.23 (m, 20H), 0.86 (t, 6H).
【0075】
【化7】
【0076】
(ポリ[(N−(2−ヘプチルオクチル)ジオキソピロロチオフェン)−alt−(チオフェン)])(PHOT)
上記で合成した化合物15(50mg、0.096mmol)を、アルゴン雰囲気下、クロロホルム(1ml)に溶解させた(Scheme 7)。次に、化合物7(67mg、0.10mmol)とジクロロビス(トリフェニルホスフィン)パラジウム(II)(3.4mg、0.005mmol)とを反応液に加えて、65℃で48時間攪拌した。冷ました反応液を500mlのメタノールに加えて、ろ過により沈殿物を回収した。その後、メタノールおよびヘキサンで沈殿物を洗浄して、PHOT(16)を黒色固体として得た(37mg、収率86%)。得られたポリマーの1H NMR測定の結果を図7Aに、IR測定の結果を図7Bに、UV測定の結果を図7Cに示す。
なお、PHOT(16)の数平均分子量(Mn)は3900であり、分子量分布は1.3であった。
【0077】
【化8】
【0078】
<合成例4>
(4−(2−ヘキシルヘプチル)カルバモイルチオフェン−3−カルボン酸)(化合物17)
Scheme 8に示すように、合成例1で合成した化合物3(1.64g、10.6mmol)および2−ヘキシルヘプチルアミン(2.25g、11.3mmol)を蒸留トルエンに加えて、反応液を24時間還流した。冷ました反応液をろ過して、粗生成物を回収した。また、5%塩酸水溶液でろ液を洗浄して粗生成物を沈殿させ、クロロホルムで抽出を行った後、溶媒を除去することにより、ろ液からも粗生成物を回収した。さらに、回収した粗生成物を真空中で乾燥させた後、薄い黄色の液体として化合物17を得た(3.52g、収率94%)。化合物17の1H NMR測定の結果は、以下の通りであった。
1H NMR (400 MHz, CDCl3):δ8.46 (d, 1H), 7.87 (d, 1H), 6.42 (broad s, 1H), 4.11 (m, 1H), 1.56 (m, 4H), 1.28 (m, 16H), 0.87 (t, 6H).
【0079】
(2−ヘキシルヘプチルジオキソピロロチオフェン)(化合物18)
塩化チオニル(200ml)に化合物17(3.52g、9.96mmol)を加えて、反応液を8時間還流した。反応液を濃縮して薄い茶色の液体を得た後、さらに乾燥して薄い黄色の結晶を得た。さらに、真空中で乾燥させた後、薄い黄色の固体として化合物18を得た(2.82g、8.41mmol、収率85%)。化合物18の1H NMR測定の結果は、以下の通りであった。
1H NMR (400 MHz, CDCl3):δ 7.84 (s, 2H), 4.11 (m, 1H), 1.60 (m, 4H), 1.28 (m, 16H), 0.86 (t, 6H).
【0080】
(1,3−ジヨード(2−ヘキシルヘプチル)ジオキソピロロチオフェン)(化合物19)
得られた化合物18(3.15g、9.39mmol)を、濃硫酸(15ml)とトリフルオロ酢酸(50ml)の混合液に溶解させた。次に、反応液にN−ヨードコハク酸イミド(NIS)(4.23g、18.7mmol)を二回に分けて加え、反応液を室温で12時間攪拌した。得られた茶色の溶液を水で希釈して、ジクロロメタンで抽出を行った。得られた有機層を無水硫酸マグネシウムで乾燥した後、溶媒をエバポレーターで除去して、オレンジ色の粗生成物を得た。シリカゲルと、溶離液としてクロロホルムとを使用したカラムクロマトグラフィーにより、粗生成物を精製して、白色の結晶として化合物19を得た(3.09g、5.26mmol、収率58%)。化合物19の1H NMR測定の結果は、以下の通りであった。
1H NMR (400 MHz, CDCl3):δ 4.09 (m, 1H), 1.83 (m, 4H), 1.24 (m, 12H), 0.86 (t, 6H).
【0081】
【化9】
【0082】
(ポリ[(N−(2−ヘキシルヘプチル)ジオキソピロロチオフェン)−alt−(チオフェン)])(PHHT)
上記で合成した化合物19(58mg、0.10mmol)を、アルゴン雰囲気下、ジメチルホルムアミド(1ml)に溶解させた(Scheme 9)。次に、化合物7(66mg、0.10mmol)とテトラキス(トリフェニルホスフィン)パラジウム(0)(3mg,0.005mmol)とを反応液に加えて、110℃で48時間攪拌した。冷ました反応液を500mlのメタノールに加えて、ろ過により沈殿物を回収した。その後、メタノールおよびヘキサンで沈殿物を洗浄して、PHHT(20)を黒色固体として得た(27mg、収率64%)。得られたポリマーの1H NMR測定の結果を図8Aに、IR測定の結果を図8Bに、UV測定および蛍光測定の結果を図8Cに示す。なお、サイクリックボルタンメトリー測定により、還元オンセット(ERED,ONSET、onset of the reduction potential)は−1.18Vであった。
なお、PHHT(20)の数平均分子量(Mn)は4300であり、分子量分布(PDI)は1.1であった。
【0083】
【化10】
【0084】
<合成例5>
(4−(2−ペンチルヘキシル)カルバモイルチオフェン−3−カルボン酸)(化合物21)
Scheme 10に示すように、合成例1で合成した化合物3(1.54g、11.3mmol)および2−ペンチルヘキシルアミン(1.83g、10.7mmol)を蒸留トルエン(100ml)に加えて、反応液を24時間還流した。冷ました反応液をろ過して、粗生成物を回収した。また、5%塩酸水溶液でろ液を洗浄して粗生成物を沈殿させ、クロロホルムで抽出を行った後、溶媒を除去することにより、ろ液からも粗生成物を回収した。さらに、回収した粗生成物を真空中で乾燥させた後、化合物21を得た(3.60g、11.1mmol、収率98%)。化合物21の1H NMR測定の結果は、以下の通りであった。
1H NMR (400 MHz, CDCl3):δ8.43 (d, 1H), 8.07 (d, 1H), 6.95 (broad s, 1H), 4.11 (m, 1H), 1.56 (m, 4H), 1.30 (m, 12H), 0.87 (t, 6H).
【0085】
(2−ペンチルヘキシルジオキソピロロチオフェン)(化合物22)
塩化チオニル(200ml)に化合物21(3.22g、9.90mmol)を加えて、反応液を8時間還流した。反応液を濃縮して薄い茶色の液体を得た後、さらに乾燥して薄い黄色の結晶を得た。さらに、真空中で乾燥させた後、薄い黄色の固体として化合物22を得た(2.80g、9.11mmol、収率92%)。化合物22の1H NMR測定の結果は、以下の通りであった。
1H NMR (400 MHz, CDCl3):δ 7.84 (s, 2H), 4.11 (m, 1H), 1.60 (m, 4H), 1.28 (m, 16H), 0.86 (t, 6H).
【0086】
(1,3−ジヨード(2−ペンチルヘキシル)ジオキソピロロチオフェン)(化合物23)
得られた化合物22(2.80g、9.11mmol)を、濃硫酸(14ml)とトリフルオロ酢酸(47ml)の混合液に溶解させた。次に、反応液にN−ヨードコハク酸イミド(NIS)(4.10g、18.2mmol)を二回に分けて加え、反応液を室温で12時間攪拌した。得られた茶色の溶液を水で希釈して、ジクロロメタンで抽出を行った。得られた有機層を無水硫酸マグネシウムで乾燥した後、溶媒をエバポレーターで除去して、オレンジ色の粗生成物を得た。シリカゲルと、溶離液としてクロロホルムとを使用したカラムクロマトグラフィーにより、粗生成物を精製して、白色の結晶として化合物23を得た(3.11g、5.56mmol、収率61%)。化合物23の1H NMR測定の結果は、以下の通りであった。
1H NMR (400 MHz, CDCl3):δ 4.09 (m, 1H), 1.83 (m, 4H), 1.24 (m, 12H), 0.86 (t, 6H).
【0087】
【化11】
【0088】
(ポリ[(N−(2−ペンチルヘキシル)ジオキソピロロチオフェン)−alt−(チオフェン)])(PPHT)
上記で合成した化合物23(56mg、0.10mmol)を、アルゴン雰囲気下、ジメチルホルムアミド(1ml)に溶解させた(Scheme 11)。次に、化合物7(66mg、0.10mmol)とテトラキス(トリフェニルホスフィン)パラジウム(0)(3mg,0.005mmol)とを反応液に加えて、110℃で48時間攪拌した。冷ました反応液を500mlのメタノールに加えて、ろ過により沈殿物を回収した。その後、メタノールおよびヘキサンで沈殿物を洗浄して、PPHT(24)を黒色固体として得た(28mg、収率71%)。得られたポリマーの1H NMR測定の結果を図9Aに、IR測定の結果を図9Bに、UV測定および蛍光測定の結果を図9Cに示す。なお、サイクリックボルタンメトリー測定により、還元オンセット(ERED,ONSET、onset of the reduction potential)は−1.20Vであった。
なお、PPHT(24)の数平均分子量(Mn)は2700であり、分子量分布(PDI)は1.3であった。
【0089】
【化12】
【0090】
<合成例6>
(2,5−ジチエノ(N−ペンタデシルジオキソピロロチオフェン))(化合物27)
Scheme 12に示すように、窒素雰囲気下、2,5−ジブロモ(N−ペンタデシルジオキソピロロチオフェン)(547mg、1.05mmol)(化合物25)、2−(トリブチルスタニル)チオフェン(783mg、2.10mmol)(化合物26)、およびテトラキス(トリフェニルホスフィン)パラジウム(0)(121mg)をジメチルホルムアミド(20ml)に溶解させ、反応液を110℃にて24時間攪拌した。冷ました反応液をメタノール中に加え、黄色の沈殿物を回収した。シリカゲルと、溶離液としてクロロホルム/ヘキサン(1:1)溶液を使用したカラムクロマトグラフィーにより、沈殿物を精製して、黄色の固体として化合物27を得た(274mg、0.51mmol、収率50%)。化合物27の1H NMR測定の結果は、以下の通りであった。
1H NMR (400 MHz, CDCl3):δ 8.02 (d, 2H), 7.44 (d, 2H), 7.13 (t, 2H), 3.66 (dd, 2H), 1.68 (m, 2H), 1.24 (m, 24H), 0.88 (t, 3H).
【0091】
(2,5−ビス(5−ブロモチオフェン−2−イル)(N−ペンタデシルジオキソピロロチオフェン))(化合物28)
化合物27(270mg、0.51mmol)をクロロホルム(4ml)と酢酸(4ml)との混合液に溶解させた。次に、反応液にN−ヨードコハク酸イミド(NIS)(191mg、1.07mmol)を加えて、室温で12時間攪拌した。得られた溶液を氷水(100ml)中に加え、クロロホルムで抽出を行った。得られた有機層を無水硫酸マグネシウムで乾燥し、溶媒をエバポレーターで除去して、黄色の粗生成物を得た。シリカゲルと、溶離液としてクロロホルム/ヘキサン(1:1)溶液とを使用したカラムクロマトグラフィーにより、粗生成物を精製して、黄色の結晶として化合物28を得た(224mg、0.33mmol、収率64%)。化合物28の1H NMR測定の結果は、以下の通りであった。
1H NMR (400 MHz, CDCl3):δ 7.65 (d, 2H), 7.07 (d, 2H), 3.65 (dd, 2H), 1.67 (m, 2H), 1.24 (m, 24H), 0.88 (t, 3H).
【0092】
【化13】
【0093】
化合物30を既報の方法(Ranger, M.; Rondeau, D.; Leclerc, M. Macromolecules 1997, 30, 7686.)により合成した。
【0094】
Scheme 13に示すように、化合物28(36mg、0.05mmol)、化合物30(34mg、0.05mmol)、およびテトラキス(トリフェニルホスフィン)パラジウム(0)(3mg、0.05eq)をトルエン(0.57ml)に溶解させた。さらに、炭酸ナトリウム水溶液(0.12ml)と、Aliquat 336(2Mの炭酸ナトリウム溶液1ml中にAliquat 336を17mg)とを反応液に加えて、100℃にて60時間攪拌した。冷ました溶液をメタノール中に加えて、1M塩酸水溶液で中和した。赤い沈殿物を回収し、メタノールおよびヘキサンで洗浄して、赤い固体としてPTPF(31)を得た。得られたポリマーの1H NMR測定の結果を図10に示す。
なお、PTPF(31)の数平均分子量(Mn)は13,000であり、分子量分布(PDI)は1.9であった。
【0095】
【化14】
【0096】
<合成例7>
(2,5−ビス(5−トリブチルスタニルチオフェン−2−イル)(N−ペンタデシルジオキソピロロチオフェン))(化合物29)
Scheme 14に示すように、窒素雰囲気下、化合物27(181mg、0.34mmol)をヘキサン(5ml)中に溶解させた。次に、ブチルリチウム(0.54ml、0.86mmol、ヘキサン中1.6mol/l)を反応液に滴下して、反応液を1時間還流した。反応液を−78℃まで冷やし、トリブチルスズクロリド(57mg、0.21mmol)を加えた後、さらに室温で12時間攪拌した。攪拌終了後、反応液を水(20ml)に加えて、有機層を分離し、水層をヘキサン(20ml)で抽出を行った。集めた有機層を無水硫酸マグネシウムで乾燥した後、溶媒を除去して、化合物29を得た。化合物29の1H NMR測定の結果は、以下の通りであった。
1H NMR (400 MHz, CDCl3):δ 7.65 (d, 2H), 7.07 (d, 2H), 3.65 (dd, 2H), 1.68 -0.88 (m, 85H).
【0097】
【化15】
【0098】
Scheme 15に示すように、化合物29(54mg、0.048mmol)、化合物32(14mg、0.047mmol)、およびジクロロビス(トリフェニルホスフィン)パラジウム(II)(2mg、0.05eq)をクロロホルム(1ml)に溶解させ、反応液を65℃で72時間加熱した。冷ました反応液をメタノール中に加え、沈殿物を回収した。その後、メタノールおよびヘキサンで沈殿物を洗浄して、黒色固体としてPTPB(33)を得た(26mg、0.039mmol、収率83%)。得られたポリマーのUV測定および蛍光測定の結果を図11に示す。なお、サイクリックボルタンメトリー測定により、酸化オンセット(EOX,ONSET、onset of the oxidation potential)は0.57Vで、還元オンセット(ERED,ONSET、onset of the reduction potential)は−1.31Vであった。
なお、PTPB(33)の数平均分子量(Mn)は9,800であり、分子量分布(PDI)は1.4であった。
【0099】
【化16】
【0100】
<合成例8>
N−オクチルイミド縮環チオフェン(化合物34)を、既報の手法(Nielsen, C. B.; Bjornholm, T. Org. Lett. 2004, 6, 3381)に従って合成した。
【0101】
Scheme 16に示すように、アルゴン雰囲気下、化合物34(42mg、0.10mmol)、化合物7(73mg、0.11mmol)、ジクロロビス(トリフェニルホスフィン)パラジウム(II)(0.1eq)、および蒸留トルエン(1ml)をシュレンク管に入れ、反応液を110℃で96時間攪拌した。冷ました反応液をメタノール中に加えて、メンブレンフィルター(ADVANTEC)にて黒色沈殿物を回収した。その後、メタノールおよびヘキサンで沈殿物を洗浄し、エバポレーターで溶媒を除去して黒色のP8T(35)を得た(18mg、収率38%)。
【0102】
【化17】
【0103】
<吸収スペクトル測定>
上記で合成したPDTと、公知の材料であるP3HT(ポリ3−ヘキシルチオフェン−2,5−ジイル(レジオレギュラー))(Aldrich社製、Poly(3-hexylthiophene-2,5-diyl)、重量平均分子量60000)とをそれぞれクロロホルムに溶解させ、同じ濃度(27mg/L)の溶液を調整し、吸収スペクトル測定を実施した(図12)。
図12から、PDTのほうが、公知材料のP3HTよりも長波長領域における吸収特性に優れることが分かった。該図からも分かるように、550〜750nmの範囲において、PDTのほうが優れた光吸収特性を示した。例えば、波長650nmにおける吸収係数としては、PDTは29L g-1 cm-1であり、P3HTでは0.30L g-1cm-1であった。また、波長550nmにおける吸収係数としては、PDTは26L g-1 cm-1であり、P3HTでは3.1L g-1cm-1であった。
【0104】
<HOMO−LUMOエネルギー準位>
図13は、本発明の共役系高分子、公知材料のP3HT、およびPCBMの最高被占分子軌道(highest occupied molecular orbit, HOMO)および最低空分子軌道(lowest unoccupied molecular orbit, LUMO)のエネルギー準位図である。
該図より分かるように、公知材料のP3HTに比べて、本発明の共役系高分子(PDTおよびPTPB)のHOMOおよびLUMOはより低くなっていた。つまり、本発明の共役系高分子は、該図および図17から分かるように従来公知の材料と比較して、フラーレン類(PCBM)に対してより適切なエネルギー準位を有する材料であった。
なお、本発明の共役系高分子の該HOMOおよびLUMO準位は、上述したサイクリックボルタンメトリー測定、UV測定などの結果から換算した値である。また、P3HT、PCBMのエネルギー準位は、非特許文献1に記載の値を参照した。
【0105】
<有機薄膜太陽電池の作製と評価>
PDTとPCBMとを光電変換層材料に用い、文献既報(Umeyama, T.; Takamatsu, T.; Tezuka, N.; Matano, Y.; Araki, Y.; Wada, T.; Yoshikawa, O.; Sagawa, T.; Yoshikawa, S.; Imahori, H. J. Phys. Chem. C 2009, 113, 10798-10806.)の手法により、有機薄膜太陽電池の作製と評価を行った。
より詳細には、まず、ITO(インジウム錫酸化物)(厚み:300nm)を塗布したガラス基板(Geomatec社製:表面抵抗は5Ω/□)を用意した。次に、ガラス基板をアセトンおよびエタノール中で連続して超音波洗浄した。基板をブロー乾燥して、UVオゾン処理した後、ポリ(3,4−エチレンジオキシチオフェン)/ポリスチレンスルホン酸水溶液(PEDOT:PSS、BaytronP)を基板上に4000rpmでスピンコートし、ホットプレート上で200℃、10分間加熱乾燥した。さらに、窒素雰囲気下、上記で合成したPDTとPCBMとのクロロホルム溶液をPEDOT:PSS層上に1000rpmでスピンコートして、PDT:PCBM層(厚み:170nm)を作製した。その後、チタンテトライソプロポキシド(和光純薬工業)のエタノール溶液をPDT:PCBM層上に2000rpmでスピンコートし、30分間放置して、チタンテトライソプロポキシドの加水分解を進行させ、TiOx層(厚み:5nm)を形成させた。最後に、真空下(5×10‐3Pa)にて、熱蒸着によりアルミニウム層(NilacoCorp製)(厚み:100nm)を堆積させ、ITO/PEDOT:PSS/共役系高分子:PCBM/TiOx/Alの構成をとる有機薄膜太陽電池を得た(図14)
AM1.5,擬似太陽光を100mW/m2の光強度にて照射して、I−V特性を測定し、開放電圧値(VOC)、短絡電流密度(JSC)、曲線因子(FF)、変換効率(η)を求めた。結果を表1に示す。
なお、表1中、PDTとPCBMの濃度(g/L)は、上述したクロロホルム溶液中の濃度を意味する。
【0106】
【表1】
【0107】
表1からわかるように、本発明の共役系高分子を用いた有機薄膜太陽電池においては、0.70V以上と高い開放電圧を実現できた。
一方、実施例1中のPDTの代わりにP3HT(Aldrich社製、Poly(3-hexylthiophene-2,5-diyl)、重量平均分子量60000)を用いて上記有機薄膜太陽電池を作製した場合、その開放電圧は0.6V程度であり、本発明のPDTを使用したほうがより高い開放電圧値を示すことが確認された。
【0108】
また、図15は、PDT:PCBM膜とP3HT:PCBM膜の光吸収スペクトル図である。PDT:PCBM膜は、PDT/PCBM(1mLクロロホルム中、10mg/10mg)をガラス基板上に750rpmにてスピンコートして得られた膜である。また、P3HT:PCBM膜は、P3HT/PCBM(1mLクロロベンゼン中、15mg/7.5mg)をガラス基板上に750rpmにてスピンコートして、150℃で6分間アニーリング処理して得られた膜である。なお、P3HT(Poly(3-hexylthiophene-2,5-diyl))としては、重量平均分子量60000(Aldrich社製)のものを用いた。
該図からわかるように、本発明の共役系高分子を用いた光電変換層であるPDT:PCBM膜においては、P3HTを使用した場合に比べて、可視光の長波長領域における吸収特性に優れることが分かった。
【0109】
また、図16は、実施例3の有機薄膜太陽電池の光電流アクションスペクトル図である。光電流アクションスペクトルは、入射単色光当たりの光電変換効率(IPCE;Incident Photon-to-Current Efficiency)を測定した結果である。該図からも分かるように、スペクトルの吸収端が長波長領域まで伸びており、幅広い波長の光を有効に利用して高効率の光電変換を実現できる可能性があることを示唆している。
【符号の説明】
【0110】
10、110、120、130 有機薄膜太陽電池
12 透明基板
12A 基板
14 透明電極層
16 正孔輸送層
18、18A、18B 光電変換層
20 電子輸送層
22 対向電極層
24 p層
26 n層
28 混合層
【特許請求の範囲】
【請求項1】
一般式(1)で表される繰り返し単位と、一般式(2)で表される繰り返し単位とを有する共役系高分子。
【化1】
(一般式(1)中、Rは、水素原子または一価の有機基を表す。
一般式(2)中、Xは、一般式(1)で表される繰り返し単位とは異なる二価の共役系連結基を表す。)
【請求項2】
一般式(1)中のXが、二価の芳香族炭化水素基、二価の芳香族複素環基、−(CH=CH)−、−(C≡C)−、またはそれらを組み合わせた基である、請求項1に記載の共役系高分子。
【請求項3】
第1電極層と、前記第1電極層と対向する電極である第2電極層とを備え、
前記第1電極層と前記第2電極層との間に、少なくとも請求項1または2に記載の共役系高分子およびフラーレン類を含む光電変換層を備える有機薄膜太陽電池。
【請求項1】
一般式(1)で表される繰り返し単位と、一般式(2)で表される繰り返し単位とを有する共役系高分子。
【化1】
(一般式(1)中、Rは、水素原子または一価の有機基を表す。
一般式(2)中、Xは、一般式(1)で表される繰り返し単位とは異なる二価の共役系連結基を表す。)
【請求項2】
一般式(1)中のXが、二価の芳香族炭化水素基、二価の芳香族複素環基、−(CH=CH)−、−(C≡C)−、またはそれらを組み合わせた基である、請求項1に記載の共役系高分子。
【請求項3】
第1電極層と、前記第1電極層と対向する電極である第2電極層とを備え、
前記第1電極層と前記第2電極層との間に、少なくとも請求項1または2に記載の共役系高分子およびフラーレン類を含む光電変換層を備える有機薄膜太陽電池。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【公開番号】特開2011−168747(P2011−168747A)
【公開日】平成23年9月1日(2011.9.1)
【国際特許分類】
【出願番号】特願2010−36159(P2010−36159)
【出願日】平成22年2月22日(2010.2.22)
【出願人】(504132272)国立大学法人京都大学 (1,269)
【Fターム(参考)】
【公開日】平成23年9月1日(2011.9.1)
【国際特許分類】
【出願日】平成22年2月22日(2010.2.22)
【出願人】(504132272)国立大学法人京都大学 (1,269)
【Fターム(参考)】
[ Back to top ]