
共有結合グラフトを介した生物活性分子を含む高度に多孔質のポリマー材料

本発明は、共有結合的にグラフトされた生物活性種を含む高度に多孔質のポリマー材料に関する。また、本発明は、生物活性分子種にグラフトすることが可能な機能性モノマーを含む高度に多孔質の材料の調製方法であって、(a)液相と連続相とを含むとともにモノマーを含有するエマルジョン組成物を調製する工程と、(b)エマルジョンを硬化する工程と、(c)任意に、水/液相を除去する工程とを含む方法に関する。本発明はさらに、生物活性種を、かかる高度に多孔質のポリマー材料にグラフトするための方法であって、(i)高度に多孔質の材料を、好適な溶媒中の生物活性種の溶液に曝す工程と、(ii)任意に、活性化剤を加える工程と、(iii)任意に、加熱する工程と、(iv)多孔質の材料を溶媒ですすいで、グラフトしていない種を除去する工程とを含む方法に関する。共有結合的にグラフトされた生物活性種を含む高度に多孔質のポリマー材料は、例えば、不均一触媒として、バイオセンサー、クロマトグラフィー、生物医学装置およびインプラントに使用することが可能である。
【発明の詳細な説明】
【発明の詳細な説明】
【0001】
本発明は、多孔質の材料に結合された生物活性分子種を含む高度に多孔質の材料に関する。また、本発明は、生物活性分子を共有結合的にグラフトすることが可能な高度に多孔質の材料を製造する方法、および多孔質の材料の材料に前記生物活性分子をグラフトするための方法に関する。さらに、本発明は、共有結合グラフトを介した生物活性分子を含む、かかる高度に多孔質の材料の、不均一触媒、バイオセンサー、クロマトグラフィー、生物医学装置およびインプラントへの用途に関する。さらに、本発明は、本発明による共有結合グラフトを介した生物活性分子を含む高度に多孔質の材料をベースとする任意の生物学的および生化学的に活性な装置に関する。
【0002】
酵素などの生物活性分子種はこれまでに、疎水性同士の相互作用によって、疎水性の多孔質ポリマー材料に固定化されている[E.リュッケンシュタイン(Ruckenstein)およびX.ワン(Wang)、Biotech.およびBioeng.、第42巻、821頁(1993年)]。この物理吸着は共有結合ではなく、生物活性分子種(酵素)がその活性の一部を保持する一方、物理吸着の性質は、生物活性分子種がポリマー担体から離脱され(溶脱され(leached))得るため、系の活性が後の再利用では低下するようなものである。このことは、ノボザイム(Novozyme)435などの、酵素が共有結合でない物理吸着プロセスを介してポリマービーズに固定化された市販の系においても見られる。
【0003】
また、生物活性分子種は、ポリマー、例えばアガロースの誘導体に共有結合的に固定化されている[R.G.フロスト(Frost)ら、Biochimica et Biophysica Acta、670、163頁(1981年)]。これは、生物学的または生化学的活性の保持をもたらすことができる。しかし、これらの系は、非孔質または高粘性のポリマーゲルであり、生物触媒反応手順において目的とする反応物質である、すなわち固定化された生物活性分子種と相互作用する化合物の拡散が著しく妨げられる。
【0004】
したがって、生物活性分子種が固体担体表面から溶脱されず、よって生物学的または生化学的活性の低下につながる安定性の問題を生じないように、例えばタンパク質および酵素などの生物活性分子種を固体担体に固定化するための有効な方法が依然として求められている。さらに、固定化ひいては後の生物学的および生化学的作用のために利用できる表面積をできるだけ大きくするため、固体担体材料は機械的完全性を維持しながらできるだけ多孔質であることも望ましい。さらに、高い多孔性は、生物活性分子種が相互作用するか、反応するか、または反応を起こすことを目的とする化合物の液体流に固定化された酵素が曝される用途に有益であろう。
【0005】
意外なことに、生物活性分子種を、共有結合グラフトを介して高度に多孔質のポリマー担体に固定化することによって、固定化された生物活性分子種の優れた活性および安定性が得られることが分かった。このようにして、生物活性分子種は分子の共有結合を介して高度に多孔質のポリマー担体に結合され、よってグラフトされるといわれている。このようにいったんグラフトされると、生物活性分子種はもはや、ある種の分解反応なしに担体から離脱することはなく、よってその生物学的活性を保持する。
【0006】
また、本発明は、前記生物活性分子種をグラフトすることが可能な機能性モノマーを含む高度に多孔質の材料の調製方法であって、
a.液相と連続相とを含むとともにモノマーを含有するエマルジョン組成物を調製する工程と、
b.エマルジョンを硬化する工程と、
c.任意に、水/液相を除去する工程と
を含む方法に関する。
【0007】
上記モノマーとは別に、エマルジョン組成物は、架橋モノマー、機能性モノマー、重合開始剤、界面活性剤および水も含有し得る。
【0008】
エマルジョンの硬化は、例えば、熱的にまたは光化学的に行うことができる。
【0009】
水/液相の除去は、有利なことには、例えば、蒸発、凍結乾燥、吸引ろ過によって行うことができる。
【0010】
本発明の他の実施形態は、生物活性種を共有結合的にグラフトすることが可能な高度に多孔質のポリマー材料の調製方法であって、
a.以下のもの:
A)機能性モノマーを5〜95重量%
B)架橋モノマーを5〜80重量%
C)重合開始剤を0〜10重量%
D)界面活性剤を0〜20重量%
E)機能性モノマーまたは架橋モノマー以外のモノマーを0〜90重量%
(ここで、重量%は、A、B、C、DおよびEの総重量に対するものである)
およびF、液相を構成する液体または液体組成物を74〜93体積%(ここで、体積%は、A、B、C、DおよびEを含む連続相ならびに液相の全体積に対するものである)
を含む組成物から液相と連続相とを含むエマルジョンを調製する工程と、
b.エマルジョンを硬化する工程と、
c.任意に、水/液相を除去する工程と
を含む方法に関する。
【0011】
本発明の文脈内では、高度に多孔質のポリマー材料という用語は、全空隙体積に対して74%を超える多孔率を有する任意のポリマー材料を指す。特に、かかる材料は、高分散相エマルジョン(HIPE)の重合によって調製可能であり、いったん重合されたものはポリHIPEとして当該技術分野で公知である(D.バービー(Barby)およびZ.ハック(Haq)、Eur.Pat.Appl.60138、1982年)。上記のプロセスから得られるこれらの高度に多孔質の材料は、モノリシック材料であり、すなわち、プロセスの結果、一体型の材料が得られる。これに対し、生物活性種がグラフトされた公知のポリマー材料は通常、ビーズ状または粒状である。
【0012】
本発明の第1の方法は、種々のモノマーを含む好適なエマルジョン組成物を調製する工程と、モノマー相を後に硬化または架橋する工程とを含む。
【0013】
ポリHIPEは、高分散相エマルジョン(HIPE)の重合から作製される。HIPEは、液相が全体積の74体積%超を占めるエマルジョンである(K.J.リサント(Lissant)編、Emulsions and Emulsion Technology Part 1、マーセル・デッカー(Marcel Dekker)、ニューヨーク(New York)、1974年、第1章)。HIPEの場合には、連続相は、重合され得るとともに自身の典型的なセル構造をポリHIPEに与え得るモノマーを含有する。ポリマーの収縮は、エマルジョンの液滴構造のために、巨視的レベルでは起こり得ない。その結果、収縮は液滴間の連続相内で起き、セル壁に相互連通した窓が現われ、それによって、ポリHIPEは、液体および気体媒体に対して完全に透過性になり、よってそれらのモノリシック形状においてフロースルー用途に利用可能になる。
【0014】
2種類のポリHIPEがあり、最も一般的なものは、逆エマルジョン(「油中水」型エマルジョンと呼ばれることが多い)から作製されるものであり、他方は、通常のエマルジョン(「水中油」型エマルジョン)から作製される。
【0015】
逆エマルジョンから作製されるポリHIPEの連続相は、モノマー、好ましくは疎水性モノマー、最も好ましくは液相と不混和性のモノマーを含有する相である。本発明の実施例に記載されるスチレンおよびアクリレートベースのポリHIPEは、この種類のポリHIPEに属する。架橋剤と呼ばれる、2つ以上の重合性部分を有する少なくとも1種のモノマーを使用する必要がある。液相と混和性のモノマーを利用可能であるが、それらの液相への溶解が部分的であるために完全には重合され得ない。連続相は、エマルジョンの安定性を向上させるために少なくとも1種の界面活性剤、好ましくは非イオン性界面活性剤を含有する。連続相は、粗度を増して連続気泡構造中の空隙をさらに生成することによってポリHIPEの表面積を増大させるのに用いる際に、少なくとも1つの非重合性種、好ましくは液相と混和しない化学物質、最も好ましくはポロゲンと呼ばれることが多い疎水性溶媒を含有することができる(P.ヘイニー(Hainey)、I.M.ハックスマン(Huxman)、B.ロワット(Rowatt)、D.C.シェリントン(Sherrington)およびL.テトレー(Tetley)、Macromolecules、1991年、24、117;A.バルベッタ(Barbetta)およびN.R.キャメロン(Cameron)、Macromolecules、2004年、37、3202)。
【0016】
逆エマルジョンから作製されるポリHIPEの液相は、疎水性の液体媒体、好ましくは疎水性の溶媒、最も好ましくは水である。それは、連続相との混和性を抑えることによってエマルジョンを安定化させる目的の塩または化学物質、光開始剤、または両方の混合物を含有することができるが、これらはまた、連続相ならびに両方の相中に含まれ得る。最後に、それは、連続相との界面で部分的に重合しやすい少なくとも1種のモノマー、好ましくは連続相中にも存在するモノマーを含有することができる。
【0017】
通常のエマルジョンから作製されるポリHIPEの連続相は、モノマー、好ましくは親水性モノマー、最も好ましくは液相と混和しないモノマーを含有する相である。一例は、アリールエーテルスルホン部分によって終端されたマクロモノマーである。架橋剤と呼ばれる、2つ以上の重合性部分を有する少なくとも1種のモノマーを使用する必要がある。液相と混和性のモノマーを利用可能であるが、それらの液相への溶解が部分的であるために完全には重合されないであろう。連続相は、エマルジョンの安定性を向上させるために少なくとも1種の界面活性剤、好ましくはイオン性界面活性剤を含有する。連続相は、粗度を増して連続気泡構造中の空隙をさらに生成することによってポリHIPEの表面積を増大させるのに用いる際に、少なくとも1つの非重合性種、好ましくは液相と混和しない化学物質、最も好ましくはポロゲンと呼ばれることが多い、水などの親水性溶媒を含有することができる。
【0018】
通常のエマルジョンから作製されるポリHIPEの液相は、疎水性の液体媒体、好ましくは疎水性の溶媒である。例は、石油エーテル、ヘキサン、および超臨界二酸化炭素である。それは、連続相との混和性を抑えることによってエマルジョンを安定化させる目的の化学物質を含有することができる。それはラジカル開始剤または光開始剤などの少なくとも1種の開始剤、または両方の混合物を含有することができるが、これらはまた、連続相ならびに両方の相中に含まれ得る。最後に、それは、連続相との界面で部分的に重合しやすい少なくとも1種のモノマー、好ましくは連続相中にも存在するモノマーを含有することができる。
【0019】
HIPEは、それらの液相の高い体積比(74%超)(乾燥下でモノリスが壊れない限り液相を除去した後に少なくとも74%多孔率のポリマーをもたらす)によって定義される。多孔率99%のポリHIPEの複数の例がある(J.エスクエナ(Esquena)、G.S.R.R.サンカー(Sanker)およびC.ソランス(Solans)、Langmuir、2003年、19、2983)。かかる材料は、セルを相互連通させる窓のためにモノリシック形態で液体媒体および気体に対して非常に透過性である。
【0020】
基本的に、反応時にポリマー材料を形成するいかなる分子も本発明の文脈の範囲内でモノマーとして用いることができる。高分散相エマルジョンの連続相に可溶のモノマーを選択することのみが重要である。有機相が連続相である油中水型のHIPEについては、かかるモノマーは、好ましくは、有機相に十分に可溶であり、水相に不溶であるべきである。水中油型のHIPEについては、逆のことが当てはまる。
【0021】
架橋モノマーは、重合の際に2つ以上のポリマー鎖間に架橋を形成することによって、架橋された網目構造の形成をもたらすような官能基を有するモノマーであるべきである。これらの架橋モノマーの選択は、上述したモノマーの場合のように、連続相への可溶性に基づくべきである。
【0022】
本発明の文脈内では、機能性モノマーは、重合に関与し得る少なくとも1つの化学部分と、第2の段階で生物活性分子種と反応し、よって生物活性分子種をポリマー材料へグラフトすることができる少なくとも1つの他の化学部分とを含む。本発明の一実施形態では、グラフトできる化学部分は、中間段階で他の分子と反応可能であり、次いで生物活性分子種とグラフトすることが可能である。別の実施形態では、架橋モノマーおよび機能性モノマーの両方のモノマーが用いられる。したがって、かかるモノマーは、少なくとも2つ、好ましくは3つの重合性基および1つの化学部分を含み、生物活性種と反応可能である。かかるモノマーは、上記の成分A、B、C、DおよびEの総重量に対して5〜95重量%の量で用いることができる。
【0023】
これは、一般構造式1a)P−G(式中、Pは重合に関与する化学部分を表し、Gは直接または間接的に生物活性分子種をグラフトするために後に用いられる化学部分である)によって、あるいは式1c)P−X−G(式中、PおよびGは上記の通りであり、Xは親水性または疎水性であり得る任意のスペーサー基である)として表すことができる。例としては、アルキル−、パーフルオロアルキル、エチレングリコール、または他のオリゴ−エーテルがある。
【0024】
好ましい実施形態では、機能性モノマーは活性エステル基、最も好ましくは式2のn−ヒドロキシスクシンイミドをベースとする活性化エステル基を含有する。
【化1】

【0025】
生物活性分子種をグラフトするのに使用可能な官能基の他の例としては、マレイミド、チオール、イソチオシアネート、ヨードアセトアミド、2−ピリジル誘導体、アジド、オキシム、エポキシド、イソシアネートおよびアルデヒドが挙げられるがこれらに限定されない。
【0026】
また、これらの機能性モノマーは、上記のモノマーおよび架橋モノマーの場合のように、ある程度、モノマー相へのそれらの可溶性に基づいて選択されるべきである。
【0027】
式3に概略的に示されるように、n−ヒドロキシスクシンイミドをベースとするモノマーのP基とG基との間にスペーサー基を導入することが可能である。
【化2】
【0028】
このようにして、例えば、限定されないが、3個またはそれ以上のメチレン基を有するアルキル鎖などからなるスペーサー基(x)を選択することによって、疎水性を調節することができる。さらに、例えば、限定されないが、様々な長さnを有するエチレンオキシド単位(CH2CH2O)nなどの、本質的に親水性であるスペーサー基(X)を選択することによって、親水性を調節することができる。
【0029】
本発明の他の実施形態では、モノマー、架橋モノマーおよび機能性モノマーは、ビニル不飽和を含み、好ましくはスチレンモノマー、より好ましくはメタクリルモノマー、最も好ましくはアクリルモノマーである。
【0030】
本発明にしたがって用いられる開始剤は、水溶性または有機溶解性であってもよく、全体的にいずれかの相に加えられ、複数の相の間で分割されてもよく、エマルジョン形成の前、その間、またはその後に加えられてもよい。
【0031】
1種または複数種の開始剤または開始剤部分が組み合わせて用いられる場合、これらは、必要に応じて、一緒にまたは別個に加えられてもよい。開始剤は、光開始剤および/または熱開始剤および/またはレドックス開始剤であり得る。
【0032】
開始剤は、モノマーを重合させるのに有効な量で存在するべきである。典型的には、開始剤は、全連続相を基準にして、約0.005〜約20重量%、好ましくは約0.1〜約15重量%、最も好ましくは約0.1〜約10重量%の量で存在し得る。
【0033】
本発明による方法において有用な開始剤は、例えば、光開始剤または熱開始剤であり得る。
【0034】
光開始剤としては、以下の例、すなわち、アセトフェノン、アニソイン、アントラキノン、アントラキノン−2−スルホン酸、ナトリウム塩、トリカルボニルクロム、ベンジル、ベンゾイン、ベンゾインエチルエーテル、ベンゾインイソブチルエーテル、ベンゾインメチルエーテル、ベンゾフェノン、ベンゾフェノン/1−ヒドロキシシクロヘキシルフェニルケトンの50/50ブレンド、3,3’,4,4’−ベンゾフェノンテトラカルボン酸二無水物、1,4−ベンゾイルビフェニル、2−ベンジル−2−(ジメチルアミノ)−4’−モルホリノブチロフェノン、4,4’−ビス(ジエチルアミノ)ベンゾフェノン、4,4’−ビス(ジメチルアミノ)ベンゾフェノン、3−カンファーキノン、2−クロロチオキサンテン−9−オン、(クメン)シクロペンタジエニル鉄(II)、ヘキサフルオロホスフェート、ジベンゾスベレノン、ジエトキシアセトフェノン、4,4’−ジヒドロキシベンゾフェノン、2,2−ジメトキシ−2−フェニルアセトフェノン、4−(ジメチルアミノ)ベンゾフェノン、4,4’−ジメチルベンジル、2,5−ジメチルベンゾフェノン、3,4−ジメチルベンゾフェノン、ジフェニル(2,4,6−トリメチルベンゾイル)ホスフィンオキシド/2−ヒドロキシ−2−メチルプロピオフェノンの50/50ブレンド、4’−エトキシアセトフェノン、2−エチルアントラキノン、フェロセン、3’−ヒドロキシアセトフェノン、4’−ヒドロキシアセトフェノン、3−ヒドロキシベンゾフェノン、4−ヒドロキシベンゾフェノン、1−ヒドロキシシクロヘキシルフェニルケトン、2−ヒドロキシ−2−メチルプロピオフェノン、2−メチルベンゾフェノン、3−メチルベンゾフェノン、メチルベンゾイルホルメート、2−メチル−4’−(メチルチオ)−2−モルホリノプロピオフェノン、フェナントレンキノン、4’−フェノキシアセトフェノン、チオキサンテン−9−オン、トリアリールスルホニウムヘキサフルオロアンチモネート塩、トリアリールスルホニウムヘキサフルオロホスフェート塩が挙げられるがこれらに限定されない。
【0035】
熱開始剤としては、以下の例、すなわち、tert−アミルペルオキシベンゾエート、4,4−アゾビス(4−シアノ吉草酸)、1,1’−アゾビス(シクロヘキサンカルボニトリル)、2,2’−アゾビスイソブチロニトリル(AIBN)、ベンゾイルペルオキシド、2,2−ビス(tert−ブチルペルオキシ)ブタン、1,1−ビス(tert−ブチルペルオキシ)シクロヘキサン、2,5−ビス(tert−ブチルペルオキシ)−2,5−ジメチルヘキサン、2,5−ビス(tert−ブチルペルオキシ)−2,5−ジメチル−3−ヘキシン、ビス(1−(tert−ブチルペルオキシ)1−メチルエチル)ベンゼン、1,1−ビス(1−(tert−ブチルペルオキシ)−3,3,5−トリメチルシクロヘキサン、tert−ブチルヒドロペルオキシド、tert−ブチルペルアセテート、tert−ブチルペルオキシド、tert−ブチルペルオキシベンゾエート、tert−ブチルペルオキシイソプロピルカルボネート、クメンヒドロペルオキシド、シクロヘキサノンペルオキシド、ジクミルペルオキシド、ラウロイルペルオキシド、2,4−ペンタンジオンペルオキシド、過酢酸が挙げられるがこれらに限定されない。
【0036】
開始剤は、単独で、あるいは、他の開始剤、還元剤、および/または触媒と組み合わせて用いることができる。レドックス重合系に有用な還元剤および触媒は周知であり、所定の開始剤のための特定の還元剤または触媒の選択は、当業者のレベルの範囲内である。
【0037】
レドックス系に有用な還元剤の例としては、第一鉄、亜硫酸水素塩、チオ硫酸塩、および種々の還元糖およびアミンが挙げられる。アスコルビン酸、ヒドロ亜硫酸ナトリウムおよび/またはN,N,N’,N’−テトラメチレンジアミンを還元剤として用いることが好都合である。
【0038】
用いられる還元剤または触媒は、典型的には、重合の開始が望まれるとき、すなわち、一般にエマルジョンが形成された後に導入される。開始剤は、それが水溶性か油溶性かに応じて、水相または油相に加えることができる。水溶性および油溶性の開始剤の組合せを用いることもできる。
【0039】
任意に、内部水相は、安定したエマルジョンを形成する際に界面活性剤を助けるための水溶性電解質を含み得る。水溶性電解質としては、無機塩(1価、2価、3価の無機塩またはそれらの混合物)、例えば、アルカリ金属塩、アルカリ土類金属塩、および、ハロゲン化物、硫酸塩、炭酸塩、リン酸塩などの重金属塩、ならびにそれらの混合物が挙げられる。かかる電解質としては、例えば、塩化ナトリウム、硫酸ナトリウム、塩化カリウム、硫酸カリウム、塩化リチウム、塩化マグネシウム、塩化カルシウム、硫酸マグネシウム、塩化アルミニウムおよびそれらの混合物が挙げられる。ハロゲン化物などの、1価のアニオンを含む1価または2価の塩が好ましい。
【0040】
本発明による他の実施形態は、第1の方法にしたがって調製された高度に多孔質のポリマー担体への生物活性分子種の共有結合グラフトであり、共有結合グラフトは、
a.高度に多孔質の材料を、好適な溶剤中の生物活性分子種の溶液に曝す工程と、
b.任意に、活性化剤を加える工程と、
c.任意に、加熱する工程と、
d.多孔質の材料を溶剤ですすいで、グラフトされていない種を除去する工程と
を含む。
【0041】
あるいは、生物学的物質のグラフトは、モノマーの重合とともに起こり得る。かかる手順のための前提条件は、重合プロセスが生物学的物質の活性に実質的に影響しない条件が適用され、生物学的物質を含むことが、エマルジョンまたは重合プロセスの安定性に実質的に影響しないことである。
【0042】
上記の工程b)に任意に用いられる活性化剤は、例えば触媒または開始剤などの、多孔質の材料と生物活性種との間の反応を促進する化合物である。
【0043】
本発明の文脈内では、生物活性分子種は、いったん高度に多孔質のポリマー担体にグラフトされると、生物系と相互作用することができ、生物系と反応するかまたは当業者に公知のような生化学的機序を介して生物学的種または化学種の反応を引き起こす、任意の生物学的、生体由来(bio−derived)または生体模倣(bio−mimetic)の分子種を指す。
【0044】
かかる生物活性分子種としては、核酸、ヌクレオチド、オリゴ糖、ペプチド、ペプチド核酸および糖タンパク質、プロテオグリカン、抗体、脂質あるいは上記の任意のものの模倣体が挙げられるがこれらに限定されない。
【0045】
本発明の好ましい実施形態では、生物活性分子種はタンパク質または酵素であり、ここで、当該技術分野で公知の酵素は、生体触媒タンパク質と呼ばれる。
【0046】
本発明の他の好ましい実施形態では、生物活性分子種は、タンパク質の混合物、酵素とタンパク質との混合物、最も好ましくは酵素の混合物などの異なる種の混合物であってもよい。本発明にしたがって2種以上の酵素を固定化する際に、酵素カスケード合成として当該技術分野で公知のもので、多段階の生体触媒反応を行うことが可能である。
【0047】
第2の方法の工程i)およびiii)で用いられる溶剤は、生物活性分子種の安定溶液を形成可能な任意の溶媒系であり得る。溶剤は水または有機溶媒、より好ましくは水性緩衝溶液あるいは有機溶媒と水性緩衝液との混合物であり得る。工程i)およびiii)で用いられる溶剤は、同じであっても、または異なる溶剤を工程iii)で用いてもよい。
【0048】
また、本発明は、不均一触媒への用途のための、共有結合グラフトを介した生物活性分子を含む高度に多孔質のポリマー材料の使用に関する。さらに、本発明は、10回、より好ましくは50回、最も好ましくは100回の反応およびすすぎサイクルの後に、生体触媒活性が元の活性の90%以上を保持する、不均一触媒へのかかる用途に関する。
【0049】
さらに、本発明は、本発明による、バイオセンサー、クロマトグラフィー、任意の生物医学装置およびインプラントならびに生物学的または生化学的に活性な装置への、共有結合グラフトを介した生物活性分子を含む高度に多孔質のポリマー材料の使用に関する。
【0050】
また、本発明は、分析目的のため、したがって、ある化学物質の存在または不存在を、化学物質の存在または不存在と定性的または定量的に相関する検知可能な信号に変換するための、共有結合グラフトを介した生物活性分子を含む高度に多孔質のポリマー材料の使用に関する。
【0051】
[実施例]
特に記載がなければ、全ての化学物質は、入手したままの状態で用いられる。
【0052】
用いた紫外線硬化系は、I600M D−バルブが装備されたフュージョン(Fusion)DRSE−I20QNL照射器であった。全紫外線照度(A+B+C)を、1.0J/cm2(ベルト速度:20フィート/分)に設定した。
【0053】
走査型電子顕微鏡は、フィリップスXL30CPであった。試料を全て、導電性を高めるために金で被覆し、カーボンペーストでアルミニウムスタブ上に装着し、電子ビームを、倍率に応じて5〜20kVに設定した。
【0054】
蛍光光学顕微鏡は、ライカCC−12カメラが結合されたライカMZFLIIIであった。青色フィルタを用いた(480±50nm)。ポリHIPE試料を、黒色背景のガラススライド上に付着させた。
【0055】
紫外線可視分光光度計は、フローセルとともに用いるための蠕動ポンプを含むヒタチ(Hitachi)U−2000であった。400nmにおける吸光度を監視し、10秒ごとに値を取った。
【0056】
[比較例1]
比較例1は、高分散相エマルジョンから熱重合された高度に多孔質の材料を作製するためのバッチ処理の生成物である。
【0057】
スチレン(4.5ml、アルドリッチ)、ジビニルベンゼン(0.5ml、アルドリッチ)、およびソルビタンモノ−(Z)−9−オクタデセノエートであるSPAN80(1.0ml、アルドリッチ)を50mlの口径の広いプラスチックボトルに入れ、矩形のPTFEパドルが装着されるとともにオーバーヘッド攪拌器モータに接続された鋼製の攪拌ロッドで、300rpmで攪拌した。窒素流束をボトルにわたって保持した。過硫酸カリウム(0.22g、アルドリッチ)および塩化カルシウム(0.50g、無水、アルドリッチ)を含有する脱イオン化した脱気水(45ml)を、絶えず攪拌しながら滴下して加え(約1ml/分)、HIPEを形成した。水相を加える際に、形成しているHIPEの表面のすぐ下での攪拌を保つようにボトルを下げて、水のポケットが形成されないようにする。全ての水相が加えられたら、攪拌をさらに10分間続けて、できるだけ均一なエマルジョンを生成する。次いで、ボトルを窒素でフラッシュしたオーブンに入れ、60℃で48時間加熱した。ボトルを切断し、ポリマーの管状の片をソックスレー(Soxhlet)装置の中に入れ、水(200ml)で24時間、次いでアセトン(200ml)で24時間洗浄した。次いで、モノリスをオーブン中で50℃で24時間、軽い減圧下で乾燥させた。
【0058】
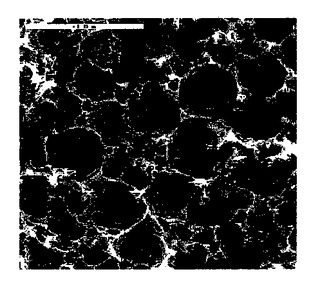
ポリマーは硬質で壊れやすいが、これは純粋なスチレン系ポリHIPEの典型である。水置換によって測定された比較例1の密度は、連続相/液相の比1:9で予測して約0.09g/cm3であった。窒素吸着によって測定されブルナウアー−エメット−テラー(Brunauer−Emmett−Teller)モデル面積を適用する典型的な表面積は、約4m2/gであった。図1は、ポリHIPEを特徴付ける連続気泡構造を示す走査型電子顕微鏡写真である。
【0059】
[比較例2〜5]
比較例2〜5は、種々の比率の主モノマーを含む高分散相エマルジョンから光重合された高度に多孔質の材料を作製するためのバッチ処理から生成される。重量パーセントは、連続相の総重量を指す(比較例2〜5では5.00g)。
【0060】
比較例2では、2−エチルヘキシルアクリレート(アルドリッチ製、表1を参照のこと)、イソボルニルアクリレート(アルドリッチ製、表1を参照のこと)、トリメチロールプロパントリアクリレート(アルドリッチ製、表1を参照のこと)、ソルビタンモノ−(Z)−9−オクタデセノエートであるSPAN80(アルドリッチ製、表1を参照のこと)およびダロキュア4265、すなわちダロキュアTPO(ジフェニル(2,4,6−トリメチルベンゾイル)−ホスフィンオキシド)とダロキュア1173(2−ヒドロキシ−2−メチル−1−フェニル−1−プロパノン)との50/50ブレンド(チバガイギー製、表1を参照のこと)を50mlの口径の広いプラスチックボトルに入れ、矩形のPTFEパドルが装着されるとともにオーバーヘッド攪拌器モータに接続された鋼製の攪拌ロッドで、300rpmで攪拌した。窒素流束をボトルにわたって保持した。脱イオン化した脱気水(表1を参照のこと)を、絶えず攪拌しながら滴下して加え(約1ml/分)、HIPEを形成した。水相を加える際に、形成しているHIPEの表面のすぐ下での攪拌を保つようにボトルを下げて、水のポケットが形成されないようにする。全ての水相が加えられたら、攪拌をさらに10分間続けて、できるだけ均一なエマルジョンを生成する。2時間以内に重合されない場合、HIPEを使用前に再び10分間攪拌して、均一な液滴を確保する必要があった。
【0061】
方形のPTFEフレームを用いて、ガラスプレート上に金型(金型サイズ:5cm辺、厚さ5mm)を形成した。HIPEを中に注ぎ、第2のガラスプレートを用いて金型を閉じた。コンベヤ速度20フィート/分で、焦点を合わせて、100%出力で、I600M D−バルブが装備されたフュージョンDRSE−I20QNL照射器の下に、金型の各辺を交互に3回ずつ通した。光重合されたHIPEを、かみそりの刃を用いて金型から除去した。硬化された湿潤試料を、600mlビーカー中の1:1(vol/vol)のアセトン/水混合物100ml中に浸した。60℃で1時間、ゆっくりと磁気攪拌にかけた。次いで、溶液を別の新鮮な100mlと取り替え、再び60℃で1時間攪拌した。このプロセスを6回繰り返した。最後の洗浄の際には、1:3のアセトン/水混合物(vol/vol)を用いた。次いで、湿潤ポリHIPEを−80℃の冷凍庫内で完全に凍結するまで凍結させ、凍結乾燥機内に24時間入れて、サイズの収縮が5%未満の乾燥ポリHIPEを得た。凍結乾燥を用いずに乾燥した試料は、40〜50%の収縮を示した。いかなる場合も、有機緩衝液混合物を用いて再び試料を湿らせれば収縮を完全に元に戻すことができる。ある有機溶媒が水性緩衝液または水と混合されていない限り、乾燥ポリHIPEの水の吸い上げは非常に遅い。
【0062】
比較例2〜5では、2−エチルヘキシルアクリレートおよびイソボルニルアクリレートの量を表1に記載のように変更し、イソボルニルアクリレートの量を増加した結果として、軟質で弾性(比較例2)ないし硬質で壊れやすい(比較例5)の範囲のポリHIPEを得た。
【0063】
これらの例で調製され水置換によって測定される乾燥ポリHIPEの典型的な密度は、連続相/液相の比1:9で予測して約0.10g/cm3であった。上記の面積として測定される典型的な表面積は、約1.9m2/gであった。
【0064】
【表1】
【0065】
[実施例1〜9]
実施例1〜9は、機能性モノマーN−アクリロキシスクシンイミド(NASI)を含む高度に多孔質のポリマーであり、よって生物活性種を共有結合的にグラフトすることが可能である。
【0066】
実施例4では、2−エチルヘキシルアクリレート(アルドリッチ製、表2を参照のこと)、イソボルニルアクリレート(アルドリッチ製、表2を参照のこと)、トリメチロールプロパントリアクリレート(0.50g、アルドリッチ製)、界面活性剤SPAN80(0.65g、アルドリッチ製)およびダロキュア4265(0.35g、チバガイギー製の光開始剤)を50mlの口径の広いプラスチックボトルに入れ、矩形のPTFEパドルが装着されるとともにオーバーヘッド攪拌器モータに接続された鋼製の攪拌ロッドで、300rpmで攪拌した。N−アクリロキシスクシンイミド(アクロス(Acros)製、表2を参照のこと)を3回に分けて加え、次の分が加えられる前に完全に溶解されるように時間を与える。窒素流束をボトルにわたって保持した。N−アクリロキシスクシンイミド(アクロス製、表2を参照のこと)を含有する脱イオン化した脱気水(45g)を、絶えず攪拌しながら滴下して加え(約1ml/分)、HIPEを形成した。水相を加える際に、形成しているHIPEの表面のすぐ下での攪拌を保つようにボトルを下げて、水のポケット(すなわち、水相の2つ以上の液滴が形成されている場合の、反応混合物が不均一である領域)が形成されないようにする。全ての水相が加えられたら、攪拌をさらに10分間続けて、できるだけ均一なエマルジョンを生成する。2時間以内に重合されない場合、HIPEを使用前に再び10分間攪拌して、最適な液滴径を確保する必要があった。
【0067】
この配合物の硬化を、比較例2〜5に示されているように行い、さらに得られた高度に多孔質の機能性ポリマーを洗浄および乾燥した。密度および表面積も比較例2〜5と同様であった。
【0068】
実施例1〜3および5〜9のポリHIPEを同じように作製した。実施例1〜5のそれぞれのための出発材料の量を表2に示す。表2に示されていない量は、実施例4に記載のものと同じである。
【0069】
実施例1〜4を、連続相に10%w/wのイソボルニルアクリレートを含有するエマルジョンから作製した。ここで、重量パーセントは、連続相を構成している材料(すなわち、EHA、IBOA、NASI(液滴相のNASIを除く)SPAN80およびダロキュア)の総重量に対するものである。それらは、連続相内、液相内、またはその両方に導入された様々な量のN−アクリロキシスクシンイミドを含有する。
【0070】
実施例5〜8を、連続相に30%w/wのイソボルニルアクリレートを含有するエマルジョンから作製した。それらは、連続相内、液相内、またはその両方に導入された様々な量のN−アクリロキシスクシンイミドを含有する。
【0071】
実施例9を、連続相に40%w/wのイソボルニルアクリレートを含有するエマルジョンから作製した。N−アクリロキシスクシンイミドは、液相内にのみ導入され得るが;そうでなければエマルジョンを安定化させることができないであろう。表2は、実施例1〜9を調製するための作製されたエマルジョンの違いをまとめている。
【0072】
【表2】
【0073】
図3の実施例の走査型電子顕微鏡写真は、N−アクリロキシスクシンイミドの組込みの効果を示す。N−アクリロキシスクシンイミドは、液相への部分的な可溶性のために、液相に導入された際(図3、実施例7)には、連続気泡構造の規則性が部分的に乱してセルのサイズ分布を広げ、連続相内にのみ導入された際(図3、実施例6)には、より薄いセル壁を生じた。
【0074】
[ブラッド−フォードアッセイを用いた、溶液のタンパク質濃度の判定]
ブラッド−フォードアッセイタンパク質測定試験に適合する、バイオ−ラッド製のタンパク質アッセイ試薬を用いて、純粋な水性緩衝液または30%v/vまでのエタノールを含有する水性緩衝液中のタンパク質または酵素の濃度を判定した。
【0075】
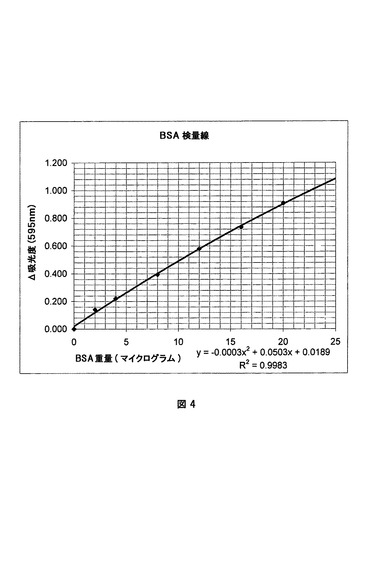
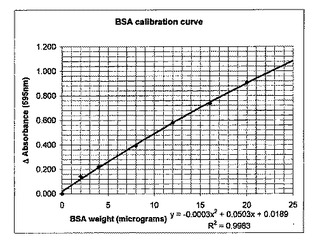
タンパク質溶液を用いて、水で系列希釈を行い、0〜25μg/mlの範囲の濃度を有する0.8mlの試料を調製した。次いで、純粋なタンパク質アッセイ試薬を各希釈試料に加えた。試薬は、G−250クマシーブルー、すなわち、タンパク質の塩基性および芳香族残基と速やかに反応して、明るい青色の錯体を形成する色素を含有する。10分間攪拌した後、試料を紫外線可視分光光度計内に入れ、595nmにおける吸光度を測定した(タンパク質を含有しない試料でゼロ吸光度を設定した)。外部検量線を、0〜20μg/mlの範囲のウシ血清アルブミン(BSA)を含む試料を用いて測定した。この曲線(図4)を用いて試料における吸光度およびタンパク質の量の相関をとった。
【0076】
[緑色蛍光タンパク質の緩衝液交換のためのプロトコル]
この実施例では、スクシンイミドエステル(実施例1〜9)を含有するポリHIPEを共有結合的に固定化するための組み換え緑色蛍光タンパク質(rAce−GFP)を調製するのに用いられるプロセスを記載する。本プロセスは、ほとんどの場合、緩衝液を交換するとともにrAce−GFPが送達された添加剤を除去するためのタンパク質の透析を含んだ。
【0077】
エヴロゲン(Evrogen)製の組み換えAce−緑色蛍光タンパク質を用いた。1つのバイアルのAce−GFP(1mg/mlにおいて0.10ml)を、5種のポリHIPE試料(20μg/試料)のために用いた。1つのバイアルを、ミリポア・マイクロコン(Millipore Microcon)YM−10遠心分離装置(膜の排除分子量:10000)でリン酸緩衝液(66mM、pH8.0)に対して透析した。リン酸緩衝液(0.5ml)を6回加えてから8000Gで20分間遠心分離した後、エタノール(30%v/v)を含有するリン酸緩衝液(66mM、pH8.0)を用いてタンパク質濃縮物を最終的に2.0mlにした。
【0078】
[実施例10]
実施例10では、N−アクリロキシスクシンイミド(実施例1〜9)の仕込み量の様々なポリHIPEに緑色蛍光タンパク質を共有結合的にグラフトするためのプロセスを記載する。比較例2〜4のポリHIPEを陰性対照として用いた。固定化プロセスは各ポリHIPEについて同じであり、すなわち、5mmのポリHIPEキューブを切断し、秤量し、2.0mlのエッペンドルフバイアルに入れた。バイアルを透析したrAce−GFP(0.40ml)で満たし、ローラー攪拌器で4時間攪拌させた。次いで、バイアルの内容物を、直径5.5cmのペーパーフィルタに注ぎ、フィルタ装置に減圧をかけ、エタノール(30%v/v)を含有するリン酸緩衝液(66mM、pH7.0)を、ポリHIPE片に滴下して加えた。吸引は、溶媒をポリマーに勢いよく通すことによって速やかな洗浄を可能にした。20mlの緩衝液を各片のために用い、洗浄されたGFPグラフトされたポリHIPEキューブを、エタノール(30%v/v)を含有するリン酸緩衝液(66mM、pH7.0)の中に入れた。溶出容量が20mlになった後の洗浄液に、ブラッド−フォード試験を用いてrAce−GFPを検出することはできなかった。表3は、どのポリHIPEを固定化のために用いたかを示す。
【0079】
【表3】
【0080】
rAce−GFPに曝されその後洗浄された湿潤ポリHIPEキューブを、黒色背景の顕微鏡ガラススライド上に置き、ライカMZFLIII蛍光顕微鏡で青色ランプ(発光:480±50nm)の下で調べた。ライカCC−12デジタルカメラで写真を撮影した。比較例2、および実施例1、2、4の、タンパク質の塩基性表面残基(おそらく主にリジン)と、N−アクリロキシスクシンイミドの活性化されたエステル官能基との間の反応によって生じると考えられるポリHIPEの写真は、図5に見られ、ポリHIPEの蛍光と機能性モノマー(NASI)との間の関係を明確に示している。比較例2は、機能性モノマーを含まず、蛍光を示さず、このことは、非常にわずかなrAce−GFPがこの非機能性ポリHIPEに物理的に吸着されるかまたは全く吸着されないことを示唆している。
【0081】
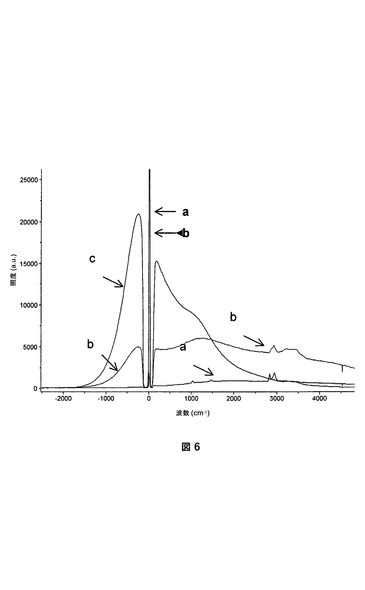
タンパク質の塩基性表面残基(例えばリジン)と、等価な仕込み量のN−アクリロキシスクシンイミドの活性化されたエステル官能基との間の反応によって生じると考えられるポリHIPEにおける固定化プロセスは、ポリHIPE中のイソボルニルアクリレートの量が増加した際に、おそらく、ほとんどのタンパク質が届かないNASIのスクシンイミドエステルを作製するバルキーなイソボルニル基の立体障害効果のために効率性が悪くなる。非機能性ポリHIPE(比較例2)およびrAce−GFPに曝されその後洗浄されたN−アクリロキシスクシンイミド含有ポリHIPE(実施例2)のキューブを、リン酸緩衝液(66mM、pH7.0)およびエタノール(30%v/v)を含有するペトリ(Petri)皿に入れた。各キューブのラマンスペクトルを、524〜532nmのラマンレーザーで取り、rAce−GFPを含まない溶液のラマンスペクトルと比較した。蛍光は、ラマン効果に対して強く競合する効果であるため、蛍光材料に対して、ラマン分光法を用いることは一般に不可能である。意外なことに、rAce−GFP吸着がレーザーの波長とそれほどかけ離れていないため、ラマンレーザーを用いて、図6のポリHIPEキューブの内部の蛍光を判定した。比較例2のキューブは蛍光を示さず(黒色のスペクトル)、このことは、アクリレートをベースとするポリHIPE自体が非蛍光性であるとともにrAce−GFPを共有結合的または物理的に固定化できないことを証明している。実施例2のキューブ(赤色の曲線)は、ほぼレーザーの波長(約505nm)を中心とするとともに溶液中のrAce−GFPの蛍光ピーク(青色スペクトル)に対応する蛍光スペクトルを示した。
【0082】
N−アクリロキシスクシンイミドなどの機能性モノマーを含有するアクリレートをベースとするポリHIPEなどの高度に多孔質のポリマーが、タンパク質、この場合rAce−GFPを共有結合的にグラフトすることが可能であることが実証された。また、実施例2のキューブの厚さにわたって様々なポイントで共焦点レーザーを集束させることによって、蛍光が一定であり、よって固定化がキューブの容積全体で均一であったことが示された。
【0083】
[固定化のためのCAL−Bの調製(透析および緩衝液交換)]
このセクションでは、スクシンイミドエステルを含有するポリHIPEにおける共有結合的な固定化のためにカンジダ・アンタークチカ・リパーゼ(Candida Antarctica Lipase)B(CAL−B)を調製するのに用いられるプロセスを記載する。本プロセスは、ほとんどの場合、緩衝液を交換するとともにCAL−Bが送達された添加剤を除去するためのタンパク質の透析を含んだ。リン酸緩衝液(66mM、pH8.0)とともに記載されるこのプロセスが、用いられる酵素に適した任意の水性緩衝液および任意のpHに適用可能であることは強調されるべきである。
【0084】
純粋なCAL−B源として、ノボザイムN525Lを用いた。N525Lを知られていない緩衝液(pH7.0)内でグリコール(50%v/v)で送達した。2つのミリポア・セントリコン・プラス−20遠心分離装置(膜の排除分子量:20000)を用いて緩衝液を交換し、グリセロールを除去した。各チューブにN525L(8mL)およびリン酸緩衝液(9mL、66mM、pH8.0)を仕込み、次いで、20分間2000Gで8回遠心分離し、各運転の後リン酸緩衝液を用いて容積が最終的に17mLになるようにした。次いで、両方のチューブからCAL−B濃縮物を採取し、リン酸緩衝液(66mM、pH8.0)中に分散し、最終容積が10.5mLになるようにした。ブラッド−フォードタンパク質測定を行い、最終溶液中のCAL−B濃度を判定した。
【0085】
[実施例11]
実施例11では、N−アクリロキシスクシンイミド(実施例4の)を含有する光重合HIPEにおけるカンジダ・アンタークチカ・リパーゼB(CAL−B)の固定化のための一般的なプロセスを記載する。リン酸緩衝液(66mM、pH8.0)とともに記載されるこのプロセスが、用いられる酵素に適した任意の水性緩衝液および任意のpHに適用可能であることは強調されるべきである。この場合、エタノール(20%v/v)を含有するリン酸緩衝液(66mM、pH7.0)が、保管および酵素活性試験のための緩衝液として選択されるが、担持される酵素がどのプロセスを取るかに応じて他の緩衝液または溶媒を用いることが可能である。
【0086】
実施例4の1片のポリHIPEを切断して秤量した(通常100mg)。CAL−B(1ml、前のセクションの透析されたN525L)、リン酸緩衝液(3ml、66mM、pH7.0)およびエタノール(1ml)を含有する10ml透明ガラス試料ボトルにそれを入れた。試料ボトルをローラー攪拌器で室温で4時間振とうした。次いで、試料ボトルの内容物を直径5.5cmのペーパーフィルタに注ぎ、フィルタ装置に減圧をかけ、エタノール(20%v/v)を含有するリン酸緩衝液(66mM、pH7.0)の混合物を、ポリHIPE片に滴下して加えた。吸引効果は、溶媒をポリマーに勢いよく通すことによる速やかな洗浄を可能にした。約50mlを用いてポリHIPEを洗浄し、エタノール(20%v/v)を含有するリン酸緩衝液(66mM、pH7.0)中に入れた。洗浄分画をブラッド−フォード試験にかけて、固定化されていないCAL−Bの量およびポリHIPE中で固定化されたCAL−Bの量を判定した。さらに、この固定化の共有結合的な性質を前提として、固定化された酵素またはポリマーマトリックスを分解させた条件を用いずに、ポリマーから固定化されたCAL−Bを除去できなかったことに留意されたい。
【0087】
[実施例12]
実施例12では、パラ−ニトロフェニルエステル基質の酵素加水分解に基づく、ノボザイムN525L由来のカンジダ・アンタークチカ・リパーゼB(CAL−B)における活性試験を記載する。
【0088】
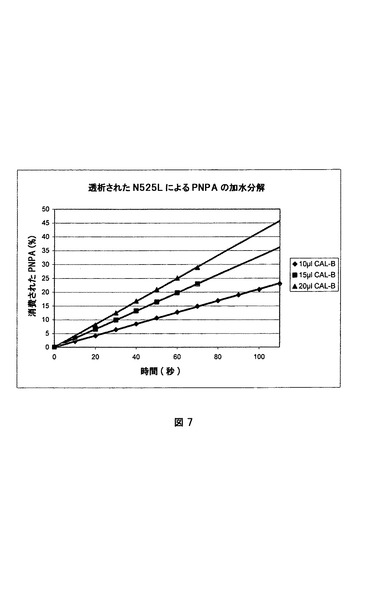
パラ−ニトロフェニルアセテート(PNPA)を基質として用いて、CAL−Bの加水分解活性を評価した。エタノール(20%v/v)を含有するリン酸緩衝液(1.9ml、66mM、pH7.0)を、2mlの紫外線可視石英セルに入れた。次いで、PNPA(0.1ml、4×10−3mmol、絶対エタノール中の7.25mg/ml溶液)を加え、ヒタチU−2000紫外線可視分光光度計中の400nmにおける吸光度の増加を2分間続け、緩衝液中のバックグラウンドPNPA化学的加水分解速度の測定をした。次いで、水中で希釈されたCAL−B(0〜0.10mlの様々な容積)を加え、パラ−ニトロフェニルの解離よる400nmにおける吸光度の増加を、線形性からの逸脱が観察されるまで続けた。計算のために、吸光度の増加曲線の傾斜から活性を推測し、化学的加水分解活性を全活性から差し引いて、単独の酵素加水分解活性を定量した。図7は、様々な量の透析され希釈されたノボザイムN525Lの活性曲線を示す。
【0089】
[実施例13]
実施例13では、再現可能な条件(担体、流量、時間)下で、様々な多孔質の担体(ポリHIPE上に担持されたCAL−B、ビーズ上に担持されたCAL−B)の活性を判定するために用いられる設備を記載する。この設備を用いて、実施例11に記載されるように行われた固定化実験から得られる様々な担持されたCAL−Bの活性を比較する。
【0090】
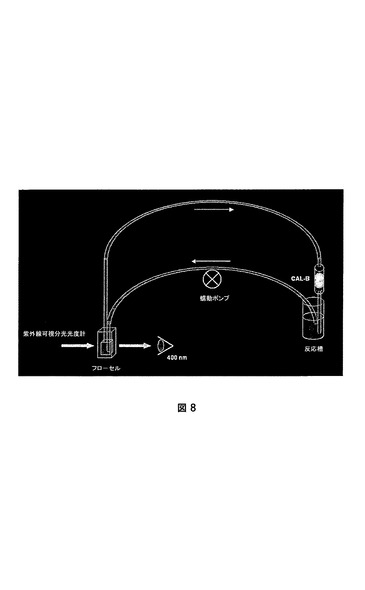
シリコーンゴムチューブ(内径1.5mm)を用いて容器(10mlガラス試料ボトル)の上のミニカラム(長さ20mm、内径5mm)に接続された紫外線可視石英フローセル(内部容量:1ml)を用いて閉ループを構築した。蠕動ポンプをフローセルのすぐ前のチューブに装着して、ループ中に急流(30ml/分)を生成した。様々な担持されたCAL−Bをミニカラムガラスフィルタの上部に充填し、液体流れが担体を通るようにした。400nmにおける吸光度0を規定するために、エタノール(20%v/v)を含有するリン酸緩衝液(9.50ml、66mM、pH7.0)をループに通して再循環させた。次いでPNPA(0.50ml、20×10−3mmol、絶対エタノール中の7.25mg/ml溶液)を反応槽に加え、パラ−ニトロフェノール化学的加水分解のための400nmにおける吸光度の増加を、それが線形になった時点から2分間続けた。次いで、担持されたCAL−Bをミニカラムに加え、400nmにおける吸光度の増加を、それが線形である限り監視した(典型的には1〜5分間)。
【0091】
充填された担持酵素をすすぎ、経時的なおよび連続使用を経たそれらの安定性を評価するために再利用することが可能であった。計算のために、吸光度の増加曲線の傾斜から活性を推測し、化学的加水分解活性を全活性から差し引いて、単独の酵素加水分解活性を定量した。
【0092】
[実施例14]
実施例14は、同じ酵素、カンジダ・アンタークチカ・リパーゼB(CAL−B)(ノボザイムN525L由来)を含有するいくつかの担体間の安定性の比較の実施例である。
【0093】
参照例として、ノボザイムN435を用いた。それは、ポリアクリル酸樹脂のビーズに物理的に吸着されたCAL−Bからなる。CHN分析によって判定されたCAL−Bの仕込み量は約8%w/w(80mg CAL−B/g ビーズ)であり、N435の表面積は105m2/gであった。
【0094】
第2の参照例として、比較例1で作製されるものと同様のスチレン系熱重合HIPEを選択した。これらのスチレン系ポリHIPEは、疎水性相互作用(非共有結合的固定化)を介して酵素を物理的に吸着することができることが知られているためである。共有結合的固定化のために実施例11に記載のようにCAL−BをこのポリHIPEに物理的に吸着させた。酵素の固定化の後の洗浄溶液におけるタンパク質測定によって判定されるCAL−Bの仕込み量は約0.75%w/w(7.5mg CAL−B/g ポリHIPE)であった。この担体を粉末として用いた。
【0095】
陰性対照として、グラフトされたCAL−Bを含まない実施例4のポリHIPEを用いて、このポリマーが単独で加水分解に何ら影響を及ぼさないことを確認した。他の陰性対照は、固定化のための溶媒としてMES緩衝液(100mM、pH6.0)を用いた以外は、実施例11と同様のプロセスを用いてCAL−Bを物理的に吸着することを試みた実施例1のポリHIPEであった。CAL−Bの吸着は検出できなかった。これらのポリHIPEを5mgのモノリスとして用いた。
【0096】
最後に、固定化のための溶媒としてMES緩衝液(100mM、pH6.0)を用いた以外は、実施例11と同様のプロセスを用いてCAL−Bを共有結合的に固定化するために実施例4のポリHIPEを選択した。酵素の固定化の後の洗浄溶液におけるタンパク質測定によって判定されるCAL−Bの仕込み量は約0.80%w/w(8.0mg CAL−B/gポリHIPE)であった。これらのポリHIPEを5mgのモノリスとして用いた。
【0097】
これらの担体の各々を、実施例13に記載のように様々な量で試験した。
【0098】
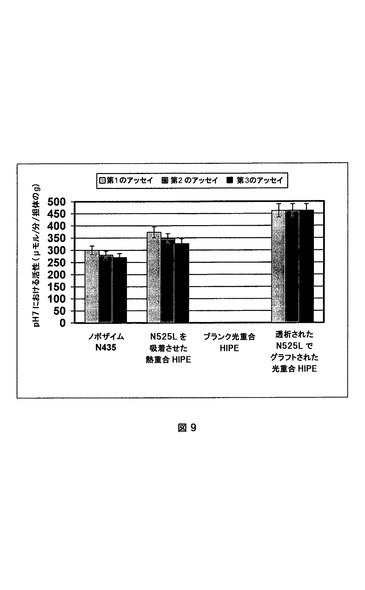
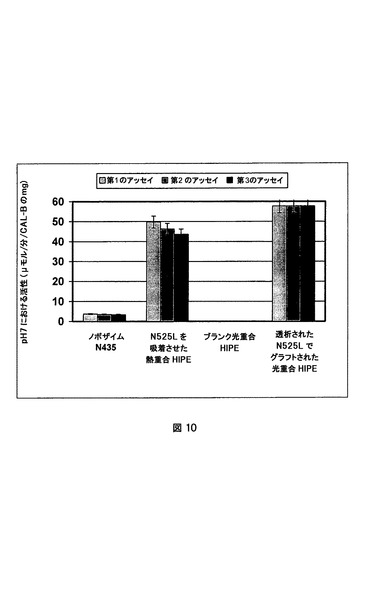
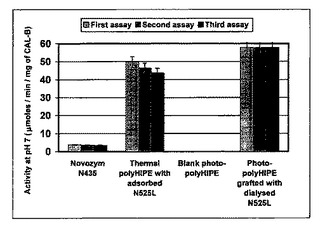
結果を図9および10にまとめた。これらの図は、図9の担体のグラムおよび図10の固定化されたCAL−Bのミリグラムによって正規化された、各担体についての、パラ−ニトロフェニルアセテートをパラ−ニトロフェノールと酢酸とに加水分解するための酵素活性を示す。
【0099】
これらの結果から以下のような3つの明白な結論があった。
a)市販のCAL−BノボザイムN435を、吸着されたCAL−Bを含有するスチレン系熱重合HIPEおよび共有結合的に固定化されたCAL−Bを含有する光重合HIPEの両方によって得られる活性に匹敵する、担体のグラム当たりの全活性を有していた。これらのスチレン系熱重合HIPEおよび光重合HIPEは、溶液中のCAL−B(約50μモル/分/mg CAL−B ノボザイムN525L由来)に匹敵するCAL−Bのミリグラム当たりの活性を示したが、ノボザイムN435は、活性が10分の1より低い。それは、N435中の吸着されたCAL−Bのほとんどが基質に届かないか、または溶液中のCAL−Bほど活性でないかのいずれかを意味する。これは、本発明による方法が低量の酵素を用いるという意味で効率的であることを示す。
b)脂肪族アクリレートベース配合物のために、非機能性光重合HIPEにおけるCAL−Bの物理的吸着はない。さらに、これらのポリHIPEにはエステルの加水分解に対して効果がない。
c)時間の経過にわたるおよび次の再利用にわたる共有結合的にグラフトされたCAL−Bの安定性は、物理的にのみ吸着された場合の担体と比較して非常に良好であった。(実験再現性の限界内の)活性の減少を、パラ−ニトロフェニルアセテートの加水分解のための同じ担体を10回用いることによって検出することができた。実施例4のポリHIPEにおける担持されたCAL−Bのエタノール(20%v/v)を含有するリン酸緩衝液(66mM、pH7.0)を3ヶ月保管した後、活性の減少を検出できなかった。
【0100】
[実施例15〜18]
これらの実施例は、界面活性剤の種類および添加剤を変えながら、EHA:IBOA:TMPTA:NASIの一定のモル比(11.65:65.82:8.19:14.34)に基づいている。HIPEに安定化効果をもたらすために、当業者には予測されるように、CaCl2(実施例15)の添加が見られた。他のところ(国際公開第97/45479号パンフレット)に報告され、実施例18で用いられる混合した界面活性剤系が、粘性のゲルのような稠密度および優れた熱安定性を有するHIPEをもたらす。
【0101】
ハイパーマー(Hypermer)B246の使用は、予想外にも、実施例16および17の両方で84%の仕込み効率をもたらすポリHIPEのNASIの保持率を高めることが分かった。これは、NASIが液相にも加えられる実施例4(62%)、および30%〜59%の範囲の仕込み効率が観察される他の全ての実施例に勝る。
【0102】
実施例15〜18を実施例1〜9と同じように作製した。実施例18を、60℃で窒素雰囲気下において16時間にわたって熱的に重合させた。
【0103】
【表4】
【0104】
[実施例19]
[DERAのカップリング]
大腸菌D−2−デオキシリボース−5−ホスフェートアルドラーゼ(DERA)無細胞抽出液(大腸菌で過剰発現したもの)(50ml)を、22℃およびpH6.60で6時間にわたって、1片のN−アクリロキシスクシンアミド−コ−ポリマー、ポリHIPE(1g)に連続的に通すことによって固定化した。次にポリマーをトリエタノールアミン緩衝液(200ml、50mM、pH7.25)で洗浄して、固定化された酵素、DERAを含有する灰白色のポリマーモノリスを残した。
i.固定化前の溶液中の総タンパク質濃度:23.6mg/ml
ii.n−ヒドロキシスクシンアミド機能性ポリマーを通して流した6時間後の総タンパク質濃度:21.3mg/ml
iii.前述したように、ブラッドフォードアッセイによってタンパク質濃度を判定した。
iv.結果として得られる、ポリHIPEにおけるDERAの仕込み量:115mg/g
【0105】
[実施例20]
[s−HNLのカップリング]
パラゴムノキ(Hevea brasiliensis)s−ヒドロキシニトリルリアーゼ(s−HNL)無細胞抽出液(ピヒア・パストリス(Pichia pastoris)で過剰発現したもの)(50ml)を、22℃およびpH5.75で6時間にわたって、1片のN−アクリロキシスクシンアミド−コ−ポリマー、ポリHIPE(1g)に連続的に通すことによって固定化した。次にポリマーをMES緩衝液(100ml、50mM、pH5.80)で洗浄して、固定化された酵素、s−HNLを含有する灰白色のポリマーモノリスを残した。
i.固定化前の溶液中の総タンパク質濃度:49.9mg/ml
ii.n−ヒドロキシスクシンアミド機能性ポリマーを通して流した6時間後:27.0mg/ml
iii.前述したように、ブラッドフォードアッセイによってタンパク質濃度を判定した。
iv.結果として得られる、ポリHIPEにおけるs−HNLの仕込み量:150mg/g
【0106】
[実施例21]
[DERA共重合]
大腸菌D−2−デオキシリボース−5−ホスフェートアルドラーゼ(DERA)無細胞抽出液(大腸菌で過剰発現したもの)(45ml、タンパク質含量が1mg/ml)を共重合によってポリHIPE内に固定化した。
【0107】
水またはCaCl2および/またはNASIの水溶液の代わりに有機相にDERA無細胞抽出液を加えたことによる以外は、実施例1〜12のようにポリHIPEを形成した。次いで、得られたポリHIPE片をリン酸カリウム緩衝液(5×100ml、50mM、pH7.00)で洗浄して、固定化された酵素、DERAを含有する灰白色のポリマーモノリスを残した。
【図面の簡単な説明】
【0108】
【図1】比較例1の走査型電子顕微鏡写真。
【図2】比較例の走査型電子顕微鏡写真(A.比較例2、B.比較例3、C.比較例4、D.比較例5)。
【図3】スクシンイミドエステルを含有するポリHIPEの走査型電子顕微鏡写真(A.実施例4、B.実施例6、C.実施例7、D.実施例8)。
【図4】タンパク質測定試験のための検量線。
【図5】蛍光ポリHIPEの図(左から順に比較例2、および実施例1、2および4からのポリHIPE)。曝露時間:500ms。倍率:1.6倍。
【図6】いずれもrAce−GFPに曝された、比較例2のキューブのラマンスペクトル(a、黒色)および実施例2のキューブのラマンスペクトル(b、赤色)、ならびに溶液中のrAce−GFPのラマンスペクトル(c、青色)の重畳。照度は正規化されていない。
【図7】ノボザイムN525L(CAL−B水溶液)によるパラ−ニトロフェニルアセテートの加水分解の活性曲線。
【図8】多孔質の担体におけるCAL−B活性の定量のためのフローセル設備。
【図9】異なる担体におけるCAL−B(N525L)によるパラ−ニトロフェニルアセテートの加水分解。活性は、担体のグラムによって正規化されている。
【図10】異なる担体におけるCAL−B(N525L)によるパラ−ニトロフェニルアセテートの加水分解。活性は、担体に最初に存在するCAL−Bのミリグラムによって正規化されている。
【発明の詳細な説明】
【0001】
本発明は、多孔質の材料に結合された生物活性分子種を含む高度に多孔質の材料に関する。また、本発明は、生物活性分子を共有結合的にグラフトすることが可能な高度に多孔質の材料を製造する方法、および多孔質の材料の材料に前記生物活性分子をグラフトするための方法に関する。さらに、本発明は、共有結合グラフトを介した生物活性分子を含む、かかる高度に多孔質の材料の、不均一触媒、バイオセンサー、クロマトグラフィー、生物医学装置およびインプラントへの用途に関する。さらに、本発明は、本発明による共有結合グラフトを介した生物活性分子を含む高度に多孔質の材料をベースとする任意の生物学的および生化学的に活性な装置に関する。
【0002】
酵素などの生物活性分子種はこれまでに、疎水性同士の相互作用によって、疎水性の多孔質ポリマー材料に固定化されている[E.リュッケンシュタイン(Ruckenstein)およびX.ワン(Wang)、Biotech.およびBioeng.、第42巻、821頁(1993年)]。この物理吸着は共有結合ではなく、生物活性分子種(酵素)がその活性の一部を保持する一方、物理吸着の性質は、生物活性分子種がポリマー担体から離脱され(溶脱され(leached))得るため、系の活性が後の再利用では低下するようなものである。このことは、ノボザイム(Novozyme)435などの、酵素が共有結合でない物理吸着プロセスを介してポリマービーズに固定化された市販の系においても見られる。
【0003】
また、生物活性分子種は、ポリマー、例えばアガロースの誘導体に共有結合的に固定化されている[R.G.フロスト(Frost)ら、Biochimica et Biophysica Acta、670、163頁(1981年)]。これは、生物学的または生化学的活性の保持をもたらすことができる。しかし、これらの系は、非孔質または高粘性のポリマーゲルであり、生物触媒反応手順において目的とする反応物質である、すなわち固定化された生物活性分子種と相互作用する化合物の拡散が著しく妨げられる。
【0004】
したがって、生物活性分子種が固体担体表面から溶脱されず、よって生物学的または生化学的活性の低下につながる安定性の問題を生じないように、例えばタンパク質および酵素などの生物活性分子種を固体担体に固定化するための有効な方法が依然として求められている。さらに、固定化ひいては後の生物学的および生化学的作用のために利用できる表面積をできるだけ大きくするため、固体担体材料は機械的完全性を維持しながらできるだけ多孔質であることも望ましい。さらに、高い多孔性は、生物活性分子種が相互作用するか、反応するか、または反応を起こすことを目的とする化合物の液体流に固定化された酵素が曝される用途に有益であろう。
【0005】
意外なことに、生物活性分子種を、共有結合グラフトを介して高度に多孔質のポリマー担体に固定化することによって、固定化された生物活性分子種の優れた活性および安定性が得られることが分かった。このようにして、生物活性分子種は分子の共有結合を介して高度に多孔質のポリマー担体に結合され、よってグラフトされるといわれている。このようにいったんグラフトされると、生物活性分子種はもはや、ある種の分解反応なしに担体から離脱することはなく、よってその生物学的活性を保持する。
【0006】
また、本発明は、前記生物活性分子種をグラフトすることが可能な機能性モノマーを含む高度に多孔質の材料の調製方法であって、
a.液相と連続相とを含むとともにモノマーを含有するエマルジョン組成物を調製する工程と、
b.エマルジョンを硬化する工程と、
c.任意に、水/液相を除去する工程と
を含む方法に関する。
【0007】
上記モノマーとは別に、エマルジョン組成物は、架橋モノマー、機能性モノマー、重合開始剤、界面活性剤および水も含有し得る。
【0008】
エマルジョンの硬化は、例えば、熱的にまたは光化学的に行うことができる。
【0009】
水/液相の除去は、有利なことには、例えば、蒸発、凍結乾燥、吸引ろ過によって行うことができる。
【0010】
本発明の他の実施形態は、生物活性種を共有結合的にグラフトすることが可能な高度に多孔質のポリマー材料の調製方法であって、
a.以下のもの:
A)機能性モノマーを5〜95重量%
B)架橋モノマーを5〜80重量%
C)重合開始剤を0〜10重量%
D)界面活性剤を0〜20重量%
E)機能性モノマーまたは架橋モノマー以外のモノマーを0〜90重量%
(ここで、重量%は、A、B、C、DおよびEの総重量に対するものである)
およびF、液相を構成する液体または液体組成物を74〜93体積%(ここで、体積%は、A、B、C、DおよびEを含む連続相ならびに液相の全体積に対するものである)
を含む組成物から液相と連続相とを含むエマルジョンを調製する工程と、
b.エマルジョンを硬化する工程と、
c.任意に、水/液相を除去する工程と
を含む方法に関する。
【0011】
本発明の文脈内では、高度に多孔質のポリマー材料という用語は、全空隙体積に対して74%を超える多孔率を有する任意のポリマー材料を指す。特に、かかる材料は、高分散相エマルジョン(HIPE)の重合によって調製可能であり、いったん重合されたものはポリHIPEとして当該技術分野で公知である(D.バービー(Barby)およびZ.ハック(Haq)、Eur.Pat.Appl.60138、1982年)。上記のプロセスから得られるこれらの高度に多孔質の材料は、モノリシック材料であり、すなわち、プロセスの結果、一体型の材料が得られる。これに対し、生物活性種がグラフトされた公知のポリマー材料は通常、ビーズ状または粒状である。
【0012】
本発明の第1の方法は、種々のモノマーを含む好適なエマルジョン組成物を調製する工程と、モノマー相を後に硬化または架橋する工程とを含む。
【0013】
ポリHIPEは、高分散相エマルジョン(HIPE)の重合から作製される。HIPEは、液相が全体積の74体積%超を占めるエマルジョンである(K.J.リサント(Lissant)編、Emulsions and Emulsion Technology Part 1、マーセル・デッカー(Marcel Dekker)、ニューヨーク(New York)、1974年、第1章)。HIPEの場合には、連続相は、重合され得るとともに自身の典型的なセル構造をポリHIPEに与え得るモノマーを含有する。ポリマーの収縮は、エマルジョンの液滴構造のために、巨視的レベルでは起こり得ない。その結果、収縮は液滴間の連続相内で起き、セル壁に相互連通した窓が現われ、それによって、ポリHIPEは、液体および気体媒体に対して完全に透過性になり、よってそれらのモノリシック形状においてフロースルー用途に利用可能になる。
【0014】
2種類のポリHIPEがあり、最も一般的なものは、逆エマルジョン(「油中水」型エマルジョンと呼ばれることが多い)から作製されるものであり、他方は、通常のエマルジョン(「水中油」型エマルジョン)から作製される。
【0015】
逆エマルジョンから作製されるポリHIPEの連続相は、モノマー、好ましくは疎水性モノマー、最も好ましくは液相と不混和性のモノマーを含有する相である。本発明の実施例に記載されるスチレンおよびアクリレートベースのポリHIPEは、この種類のポリHIPEに属する。架橋剤と呼ばれる、2つ以上の重合性部分を有する少なくとも1種のモノマーを使用する必要がある。液相と混和性のモノマーを利用可能であるが、それらの液相への溶解が部分的であるために完全には重合され得ない。連続相は、エマルジョンの安定性を向上させるために少なくとも1種の界面活性剤、好ましくは非イオン性界面活性剤を含有する。連続相は、粗度を増して連続気泡構造中の空隙をさらに生成することによってポリHIPEの表面積を増大させるのに用いる際に、少なくとも1つの非重合性種、好ましくは液相と混和しない化学物質、最も好ましくはポロゲンと呼ばれることが多い疎水性溶媒を含有することができる(P.ヘイニー(Hainey)、I.M.ハックスマン(Huxman)、B.ロワット(Rowatt)、D.C.シェリントン(Sherrington)およびL.テトレー(Tetley)、Macromolecules、1991年、24、117;A.バルベッタ(Barbetta)およびN.R.キャメロン(Cameron)、Macromolecules、2004年、37、3202)。
【0016】
逆エマルジョンから作製されるポリHIPEの液相は、疎水性の液体媒体、好ましくは疎水性の溶媒、最も好ましくは水である。それは、連続相との混和性を抑えることによってエマルジョンを安定化させる目的の塩または化学物質、光開始剤、または両方の混合物を含有することができるが、これらはまた、連続相ならびに両方の相中に含まれ得る。最後に、それは、連続相との界面で部分的に重合しやすい少なくとも1種のモノマー、好ましくは連続相中にも存在するモノマーを含有することができる。
【0017】
通常のエマルジョンから作製されるポリHIPEの連続相は、モノマー、好ましくは親水性モノマー、最も好ましくは液相と混和しないモノマーを含有する相である。一例は、アリールエーテルスルホン部分によって終端されたマクロモノマーである。架橋剤と呼ばれる、2つ以上の重合性部分を有する少なくとも1種のモノマーを使用する必要がある。液相と混和性のモノマーを利用可能であるが、それらの液相への溶解が部分的であるために完全には重合されないであろう。連続相は、エマルジョンの安定性を向上させるために少なくとも1種の界面活性剤、好ましくはイオン性界面活性剤を含有する。連続相は、粗度を増して連続気泡構造中の空隙をさらに生成することによってポリHIPEの表面積を増大させるのに用いる際に、少なくとも1つの非重合性種、好ましくは液相と混和しない化学物質、最も好ましくはポロゲンと呼ばれることが多い、水などの親水性溶媒を含有することができる。
【0018】
通常のエマルジョンから作製されるポリHIPEの液相は、疎水性の液体媒体、好ましくは疎水性の溶媒である。例は、石油エーテル、ヘキサン、および超臨界二酸化炭素である。それは、連続相との混和性を抑えることによってエマルジョンを安定化させる目的の化学物質を含有することができる。それはラジカル開始剤または光開始剤などの少なくとも1種の開始剤、または両方の混合物を含有することができるが、これらはまた、連続相ならびに両方の相中に含まれ得る。最後に、それは、連続相との界面で部分的に重合しやすい少なくとも1種のモノマー、好ましくは連続相中にも存在するモノマーを含有することができる。
【0019】
HIPEは、それらの液相の高い体積比(74%超)(乾燥下でモノリスが壊れない限り液相を除去した後に少なくとも74%多孔率のポリマーをもたらす)によって定義される。多孔率99%のポリHIPEの複数の例がある(J.エスクエナ(Esquena)、G.S.R.R.サンカー(Sanker)およびC.ソランス(Solans)、Langmuir、2003年、19、2983)。かかる材料は、セルを相互連通させる窓のためにモノリシック形態で液体媒体および気体に対して非常に透過性である。
【0020】
基本的に、反応時にポリマー材料を形成するいかなる分子も本発明の文脈の範囲内でモノマーとして用いることができる。高分散相エマルジョンの連続相に可溶のモノマーを選択することのみが重要である。有機相が連続相である油中水型のHIPEについては、かかるモノマーは、好ましくは、有機相に十分に可溶であり、水相に不溶であるべきである。水中油型のHIPEについては、逆のことが当てはまる。
【0021】
架橋モノマーは、重合の際に2つ以上のポリマー鎖間に架橋を形成することによって、架橋された網目構造の形成をもたらすような官能基を有するモノマーであるべきである。これらの架橋モノマーの選択は、上述したモノマーの場合のように、連続相への可溶性に基づくべきである。
【0022】
本発明の文脈内では、機能性モノマーは、重合に関与し得る少なくとも1つの化学部分と、第2の段階で生物活性分子種と反応し、よって生物活性分子種をポリマー材料へグラフトすることができる少なくとも1つの他の化学部分とを含む。本発明の一実施形態では、グラフトできる化学部分は、中間段階で他の分子と反応可能であり、次いで生物活性分子種とグラフトすることが可能である。別の実施形態では、架橋モノマーおよび機能性モノマーの両方のモノマーが用いられる。したがって、かかるモノマーは、少なくとも2つ、好ましくは3つの重合性基および1つの化学部分を含み、生物活性種と反応可能である。かかるモノマーは、上記の成分A、B、C、DおよびEの総重量に対して5〜95重量%の量で用いることができる。
【0023】
これは、一般構造式1a)P−G(式中、Pは重合に関与する化学部分を表し、Gは直接または間接的に生物活性分子種をグラフトするために後に用いられる化学部分である)によって、あるいは式1c)P−X−G(式中、PおよびGは上記の通りであり、Xは親水性または疎水性であり得る任意のスペーサー基である)として表すことができる。例としては、アルキル−、パーフルオロアルキル、エチレングリコール、または他のオリゴ−エーテルがある。
【0024】
好ましい実施形態では、機能性モノマーは活性エステル基、最も好ましくは式2のn−ヒドロキシスクシンイミドをベースとする活性化エステル基を含有する。
【化1】
【0025】
生物活性分子種をグラフトするのに使用可能な官能基の他の例としては、マレイミド、チオール、イソチオシアネート、ヨードアセトアミド、2−ピリジル誘導体、アジド、オキシム、エポキシド、イソシアネートおよびアルデヒドが挙げられるがこれらに限定されない。
【0026】
また、これらの機能性モノマーは、上記のモノマーおよび架橋モノマーの場合のように、ある程度、モノマー相へのそれらの可溶性に基づいて選択されるべきである。
【0027】
式3に概略的に示されるように、n−ヒドロキシスクシンイミドをベースとするモノマーのP基とG基との間にスペーサー基を導入することが可能である。
【化2】
【0028】
このようにして、例えば、限定されないが、3個またはそれ以上のメチレン基を有するアルキル鎖などからなるスペーサー基(x)を選択することによって、疎水性を調節することができる。さらに、例えば、限定されないが、様々な長さnを有するエチレンオキシド単位(CH2CH2O)nなどの、本質的に親水性であるスペーサー基(X)を選択することによって、親水性を調節することができる。
【0029】
本発明の他の実施形態では、モノマー、架橋モノマーおよび機能性モノマーは、ビニル不飽和を含み、好ましくはスチレンモノマー、より好ましくはメタクリルモノマー、最も好ましくはアクリルモノマーである。
【0030】
本発明にしたがって用いられる開始剤は、水溶性または有機溶解性であってもよく、全体的にいずれかの相に加えられ、複数の相の間で分割されてもよく、エマルジョン形成の前、その間、またはその後に加えられてもよい。
【0031】
1種または複数種の開始剤または開始剤部分が組み合わせて用いられる場合、これらは、必要に応じて、一緒にまたは別個に加えられてもよい。開始剤は、光開始剤および/または熱開始剤および/またはレドックス開始剤であり得る。
【0032】
開始剤は、モノマーを重合させるのに有効な量で存在するべきである。典型的には、開始剤は、全連続相を基準にして、約0.005〜約20重量%、好ましくは約0.1〜約15重量%、最も好ましくは約0.1〜約10重量%の量で存在し得る。
【0033】
本発明による方法において有用な開始剤は、例えば、光開始剤または熱開始剤であり得る。
【0034】
光開始剤としては、以下の例、すなわち、アセトフェノン、アニソイン、アントラキノン、アントラキノン−2−スルホン酸、ナトリウム塩、トリカルボニルクロム、ベンジル、ベンゾイン、ベンゾインエチルエーテル、ベンゾインイソブチルエーテル、ベンゾインメチルエーテル、ベンゾフェノン、ベンゾフェノン/1−ヒドロキシシクロヘキシルフェニルケトンの50/50ブレンド、3,3’,4,4’−ベンゾフェノンテトラカルボン酸二無水物、1,4−ベンゾイルビフェニル、2−ベンジル−2−(ジメチルアミノ)−4’−モルホリノブチロフェノン、4,4’−ビス(ジエチルアミノ)ベンゾフェノン、4,4’−ビス(ジメチルアミノ)ベンゾフェノン、3−カンファーキノン、2−クロロチオキサンテン−9−オン、(クメン)シクロペンタジエニル鉄(II)、ヘキサフルオロホスフェート、ジベンゾスベレノン、ジエトキシアセトフェノン、4,4’−ジヒドロキシベンゾフェノン、2,2−ジメトキシ−2−フェニルアセトフェノン、4−(ジメチルアミノ)ベンゾフェノン、4,4’−ジメチルベンジル、2,5−ジメチルベンゾフェノン、3,4−ジメチルベンゾフェノン、ジフェニル(2,4,6−トリメチルベンゾイル)ホスフィンオキシド/2−ヒドロキシ−2−メチルプロピオフェノンの50/50ブレンド、4’−エトキシアセトフェノン、2−エチルアントラキノン、フェロセン、3’−ヒドロキシアセトフェノン、4’−ヒドロキシアセトフェノン、3−ヒドロキシベンゾフェノン、4−ヒドロキシベンゾフェノン、1−ヒドロキシシクロヘキシルフェニルケトン、2−ヒドロキシ−2−メチルプロピオフェノン、2−メチルベンゾフェノン、3−メチルベンゾフェノン、メチルベンゾイルホルメート、2−メチル−4’−(メチルチオ)−2−モルホリノプロピオフェノン、フェナントレンキノン、4’−フェノキシアセトフェノン、チオキサンテン−9−オン、トリアリールスルホニウムヘキサフルオロアンチモネート塩、トリアリールスルホニウムヘキサフルオロホスフェート塩が挙げられるがこれらに限定されない。
【0035】
熱開始剤としては、以下の例、すなわち、tert−アミルペルオキシベンゾエート、4,4−アゾビス(4−シアノ吉草酸)、1,1’−アゾビス(シクロヘキサンカルボニトリル)、2,2’−アゾビスイソブチロニトリル(AIBN)、ベンゾイルペルオキシド、2,2−ビス(tert−ブチルペルオキシ)ブタン、1,1−ビス(tert−ブチルペルオキシ)シクロヘキサン、2,5−ビス(tert−ブチルペルオキシ)−2,5−ジメチルヘキサン、2,5−ビス(tert−ブチルペルオキシ)−2,5−ジメチル−3−ヘキシン、ビス(1−(tert−ブチルペルオキシ)1−メチルエチル)ベンゼン、1,1−ビス(1−(tert−ブチルペルオキシ)−3,3,5−トリメチルシクロヘキサン、tert−ブチルヒドロペルオキシド、tert−ブチルペルアセテート、tert−ブチルペルオキシド、tert−ブチルペルオキシベンゾエート、tert−ブチルペルオキシイソプロピルカルボネート、クメンヒドロペルオキシド、シクロヘキサノンペルオキシド、ジクミルペルオキシド、ラウロイルペルオキシド、2,4−ペンタンジオンペルオキシド、過酢酸が挙げられるがこれらに限定されない。
【0036】
開始剤は、単独で、あるいは、他の開始剤、還元剤、および/または触媒と組み合わせて用いることができる。レドックス重合系に有用な還元剤および触媒は周知であり、所定の開始剤のための特定の還元剤または触媒の選択は、当業者のレベルの範囲内である。
【0037】
レドックス系に有用な還元剤の例としては、第一鉄、亜硫酸水素塩、チオ硫酸塩、および種々の還元糖およびアミンが挙げられる。アスコルビン酸、ヒドロ亜硫酸ナトリウムおよび/またはN,N,N’,N’−テトラメチレンジアミンを還元剤として用いることが好都合である。
【0038】
用いられる還元剤または触媒は、典型的には、重合の開始が望まれるとき、すなわち、一般にエマルジョンが形成された後に導入される。開始剤は、それが水溶性か油溶性かに応じて、水相または油相に加えることができる。水溶性および油溶性の開始剤の組合せを用いることもできる。
【0039】
任意に、内部水相は、安定したエマルジョンを形成する際に界面活性剤を助けるための水溶性電解質を含み得る。水溶性電解質としては、無機塩(1価、2価、3価の無機塩またはそれらの混合物)、例えば、アルカリ金属塩、アルカリ土類金属塩、および、ハロゲン化物、硫酸塩、炭酸塩、リン酸塩などの重金属塩、ならびにそれらの混合物が挙げられる。かかる電解質としては、例えば、塩化ナトリウム、硫酸ナトリウム、塩化カリウム、硫酸カリウム、塩化リチウム、塩化マグネシウム、塩化カルシウム、硫酸マグネシウム、塩化アルミニウムおよびそれらの混合物が挙げられる。ハロゲン化物などの、1価のアニオンを含む1価または2価の塩が好ましい。
【0040】
本発明による他の実施形態は、第1の方法にしたがって調製された高度に多孔質のポリマー担体への生物活性分子種の共有結合グラフトであり、共有結合グラフトは、
a.高度に多孔質の材料を、好適な溶剤中の生物活性分子種の溶液に曝す工程と、
b.任意に、活性化剤を加える工程と、
c.任意に、加熱する工程と、
d.多孔質の材料を溶剤ですすいで、グラフトされていない種を除去する工程と
を含む。
【0041】
あるいは、生物学的物質のグラフトは、モノマーの重合とともに起こり得る。かかる手順のための前提条件は、重合プロセスが生物学的物質の活性に実質的に影響しない条件が適用され、生物学的物質を含むことが、エマルジョンまたは重合プロセスの安定性に実質的に影響しないことである。
【0042】
上記の工程b)に任意に用いられる活性化剤は、例えば触媒または開始剤などの、多孔質の材料と生物活性種との間の反応を促進する化合物である。
【0043】
本発明の文脈内では、生物活性分子種は、いったん高度に多孔質のポリマー担体にグラフトされると、生物系と相互作用することができ、生物系と反応するかまたは当業者に公知のような生化学的機序を介して生物学的種または化学種の反応を引き起こす、任意の生物学的、生体由来(bio−derived)または生体模倣(bio−mimetic)の分子種を指す。
【0044】
かかる生物活性分子種としては、核酸、ヌクレオチド、オリゴ糖、ペプチド、ペプチド核酸および糖タンパク質、プロテオグリカン、抗体、脂質あるいは上記の任意のものの模倣体が挙げられるがこれらに限定されない。
【0045】
本発明の好ましい実施形態では、生物活性分子種はタンパク質または酵素であり、ここで、当該技術分野で公知の酵素は、生体触媒タンパク質と呼ばれる。
【0046】
本発明の他の好ましい実施形態では、生物活性分子種は、タンパク質の混合物、酵素とタンパク質との混合物、最も好ましくは酵素の混合物などの異なる種の混合物であってもよい。本発明にしたがって2種以上の酵素を固定化する際に、酵素カスケード合成として当該技術分野で公知のもので、多段階の生体触媒反応を行うことが可能である。
【0047】
第2の方法の工程i)およびiii)で用いられる溶剤は、生物活性分子種の安定溶液を形成可能な任意の溶媒系であり得る。溶剤は水または有機溶媒、より好ましくは水性緩衝溶液あるいは有機溶媒と水性緩衝液との混合物であり得る。工程i)およびiii)で用いられる溶剤は、同じであっても、または異なる溶剤を工程iii)で用いてもよい。
【0048】
また、本発明は、不均一触媒への用途のための、共有結合グラフトを介した生物活性分子を含む高度に多孔質のポリマー材料の使用に関する。さらに、本発明は、10回、より好ましくは50回、最も好ましくは100回の反応およびすすぎサイクルの後に、生体触媒活性が元の活性の90%以上を保持する、不均一触媒へのかかる用途に関する。
【0049】
さらに、本発明は、本発明による、バイオセンサー、クロマトグラフィー、任意の生物医学装置およびインプラントならびに生物学的または生化学的に活性な装置への、共有結合グラフトを介した生物活性分子を含む高度に多孔質のポリマー材料の使用に関する。
【0050】
また、本発明は、分析目的のため、したがって、ある化学物質の存在または不存在を、化学物質の存在または不存在と定性的または定量的に相関する検知可能な信号に変換するための、共有結合グラフトを介した生物活性分子を含む高度に多孔質のポリマー材料の使用に関する。
【0051】
[実施例]
特に記載がなければ、全ての化学物質は、入手したままの状態で用いられる。
【0052】
用いた紫外線硬化系は、I600M D−バルブが装備されたフュージョン(Fusion)DRSE−I20QNL照射器であった。全紫外線照度(A+B+C)を、1.0J/cm2(ベルト速度:20フィート/分)に設定した。
【0053】
走査型電子顕微鏡は、フィリップスXL30CPであった。試料を全て、導電性を高めるために金で被覆し、カーボンペーストでアルミニウムスタブ上に装着し、電子ビームを、倍率に応じて5〜20kVに設定した。
【0054】
蛍光光学顕微鏡は、ライカCC−12カメラが結合されたライカMZFLIIIであった。青色フィルタを用いた(480±50nm)。ポリHIPE試料を、黒色背景のガラススライド上に付着させた。
【0055】
紫外線可視分光光度計は、フローセルとともに用いるための蠕動ポンプを含むヒタチ(Hitachi)U−2000であった。400nmにおける吸光度を監視し、10秒ごとに値を取った。
【0056】
[比較例1]
比較例1は、高分散相エマルジョンから熱重合された高度に多孔質の材料を作製するためのバッチ処理の生成物である。
【0057】
スチレン(4.5ml、アルドリッチ)、ジビニルベンゼン(0.5ml、アルドリッチ)、およびソルビタンモノ−(Z)−9−オクタデセノエートであるSPAN80(1.0ml、アルドリッチ)を50mlの口径の広いプラスチックボトルに入れ、矩形のPTFEパドルが装着されるとともにオーバーヘッド攪拌器モータに接続された鋼製の攪拌ロッドで、300rpmで攪拌した。窒素流束をボトルにわたって保持した。過硫酸カリウム(0.22g、アルドリッチ)および塩化カルシウム(0.50g、無水、アルドリッチ)を含有する脱イオン化した脱気水(45ml)を、絶えず攪拌しながら滴下して加え(約1ml/分)、HIPEを形成した。水相を加える際に、形成しているHIPEの表面のすぐ下での攪拌を保つようにボトルを下げて、水のポケットが形成されないようにする。全ての水相が加えられたら、攪拌をさらに10分間続けて、できるだけ均一なエマルジョンを生成する。次いで、ボトルを窒素でフラッシュしたオーブンに入れ、60℃で48時間加熱した。ボトルを切断し、ポリマーの管状の片をソックスレー(Soxhlet)装置の中に入れ、水(200ml)で24時間、次いでアセトン(200ml)で24時間洗浄した。次いで、モノリスをオーブン中で50℃で24時間、軽い減圧下で乾燥させた。
【0058】
ポリマーは硬質で壊れやすいが、これは純粋なスチレン系ポリHIPEの典型である。水置換によって測定された比較例1の密度は、連続相/液相の比1:9で予測して約0.09g/cm3であった。窒素吸着によって測定されブルナウアー−エメット−テラー(Brunauer−Emmett−Teller)モデル面積を適用する典型的な表面積は、約4m2/gであった。図1は、ポリHIPEを特徴付ける連続気泡構造を示す走査型電子顕微鏡写真である。
【0059】
[比較例2〜5]
比較例2〜5は、種々の比率の主モノマーを含む高分散相エマルジョンから光重合された高度に多孔質の材料を作製するためのバッチ処理から生成される。重量パーセントは、連続相の総重量を指す(比較例2〜5では5.00g)。
【0060】
比較例2では、2−エチルヘキシルアクリレート(アルドリッチ製、表1を参照のこと)、イソボルニルアクリレート(アルドリッチ製、表1を参照のこと)、トリメチロールプロパントリアクリレート(アルドリッチ製、表1を参照のこと)、ソルビタンモノ−(Z)−9−オクタデセノエートであるSPAN80(アルドリッチ製、表1を参照のこと)およびダロキュア4265、すなわちダロキュアTPO(ジフェニル(2,4,6−トリメチルベンゾイル)−ホスフィンオキシド)とダロキュア1173(2−ヒドロキシ−2−メチル−1−フェニル−1−プロパノン)との50/50ブレンド(チバガイギー製、表1を参照のこと)を50mlの口径の広いプラスチックボトルに入れ、矩形のPTFEパドルが装着されるとともにオーバーヘッド攪拌器モータに接続された鋼製の攪拌ロッドで、300rpmで攪拌した。窒素流束をボトルにわたって保持した。脱イオン化した脱気水(表1を参照のこと)を、絶えず攪拌しながら滴下して加え(約1ml/分)、HIPEを形成した。水相を加える際に、形成しているHIPEの表面のすぐ下での攪拌を保つようにボトルを下げて、水のポケットが形成されないようにする。全ての水相が加えられたら、攪拌をさらに10分間続けて、できるだけ均一なエマルジョンを生成する。2時間以内に重合されない場合、HIPEを使用前に再び10分間攪拌して、均一な液滴を確保する必要があった。
【0061】
方形のPTFEフレームを用いて、ガラスプレート上に金型(金型サイズ:5cm辺、厚さ5mm)を形成した。HIPEを中に注ぎ、第2のガラスプレートを用いて金型を閉じた。コンベヤ速度20フィート/分で、焦点を合わせて、100%出力で、I600M D−バルブが装備されたフュージョンDRSE−I20QNL照射器の下に、金型の各辺を交互に3回ずつ通した。光重合されたHIPEを、かみそりの刃を用いて金型から除去した。硬化された湿潤試料を、600mlビーカー中の1:1(vol/vol)のアセトン/水混合物100ml中に浸した。60℃で1時間、ゆっくりと磁気攪拌にかけた。次いで、溶液を別の新鮮な100mlと取り替え、再び60℃で1時間攪拌した。このプロセスを6回繰り返した。最後の洗浄の際には、1:3のアセトン/水混合物(vol/vol)を用いた。次いで、湿潤ポリHIPEを−80℃の冷凍庫内で完全に凍結するまで凍結させ、凍結乾燥機内に24時間入れて、サイズの収縮が5%未満の乾燥ポリHIPEを得た。凍結乾燥を用いずに乾燥した試料は、40〜50%の収縮を示した。いかなる場合も、有機緩衝液混合物を用いて再び試料を湿らせれば収縮を完全に元に戻すことができる。ある有機溶媒が水性緩衝液または水と混合されていない限り、乾燥ポリHIPEの水の吸い上げは非常に遅い。
【0062】
比較例2〜5では、2−エチルヘキシルアクリレートおよびイソボルニルアクリレートの量を表1に記載のように変更し、イソボルニルアクリレートの量を増加した結果として、軟質で弾性(比較例2)ないし硬質で壊れやすい(比較例5)の範囲のポリHIPEを得た。
【0063】
これらの例で調製され水置換によって測定される乾燥ポリHIPEの典型的な密度は、連続相/液相の比1:9で予測して約0.10g/cm3であった。上記の面積として測定される典型的な表面積は、約1.9m2/gであった。
【0064】
【表1】
【0065】
[実施例1〜9]
実施例1〜9は、機能性モノマーN−アクリロキシスクシンイミド(NASI)を含む高度に多孔質のポリマーであり、よって生物活性種を共有結合的にグラフトすることが可能である。
【0066】
実施例4では、2−エチルヘキシルアクリレート(アルドリッチ製、表2を参照のこと)、イソボルニルアクリレート(アルドリッチ製、表2を参照のこと)、トリメチロールプロパントリアクリレート(0.50g、アルドリッチ製)、界面活性剤SPAN80(0.65g、アルドリッチ製)およびダロキュア4265(0.35g、チバガイギー製の光開始剤)を50mlの口径の広いプラスチックボトルに入れ、矩形のPTFEパドルが装着されるとともにオーバーヘッド攪拌器モータに接続された鋼製の攪拌ロッドで、300rpmで攪拌した。N−アクリロキシスクシンイミド(アクロス(Acros)製、表2を参照のこと)を3回に分けて加え、次の分が加えられる前に完全に溶解されるように時間を与える。窒素流束をボトルにわたって保持した。N−アクリロキシスクシンイミド(アクロス製、表2を参照のこと)を含有する脱イオン化した脱気水(45g)を、絶えず攪拌しながら滴下して加え(約1ml/分)、HIPEを形成した。水相を加える際に、形成しているHIPEの表面のすぐ下での攪拌を保つようにボトルを下げて、水のポケット(すなわち、水相の2つ以上の液滴が形成されている場合の、反応混合物が不均一である領域)が形成されないようにする。全ての水相が加えられたら、攪拌をさらに10分間続けて、できるだけ均一なエマルジョンを生成する。2時間以内に重合されない場合、HIPEを使用前に再び10分間攪拌して、最適な液滴径を確保する必要があった。
【0067】
この配合物の硬化を、比較例2〜5に示されているように行い、さらに得られた高度に多孔質の機能性ポリマーを洗浄および乾燥した。密度および表面積も比較例2〜5と同様であった。
【0068】
実施例1〜3および5〜9のポリHIPEを同じように作製した。実施例1〜5のそれぞれのための出発材料の量を表2に示す。表2に示されていない量は、実施例4に記載のものと同じである。
【0069】
実施例1〜4を、連続相に10%w/wのイソボルニルアクリレートを含有するエマルジョンから作製した。ここで、重量パーセントは、連続相を構成している材料(すなわち、EHA、IBOA、NASI(液滴相のNASIを除く)SPAN80およびダロキュア)の総重量に対するものである。それらは、連続相内、液相内、またはその両方に導入された様々な量のN−アクリロキシスクシンイミドを含有する。
【0070】
実施例5〜8を、連続相に30%w/wのイソボルニルアクリレートを含有するエマルジョンから作製した。それらは、連続相内、液相内、またはその両方に導入された様々な量のN−アクリロキシスクシンイミドを含有する。
【0071】
実施例9を、連続相に40%w/wのイソボルニルアクリレートを含有するエマルジョンから作製した。N−アクリロキシスクシンイミドは、液相内にのみ導入され得るが;そうでなければエマルジョンを安定化させることができないであろう。表2は、実施例1〜9を調製するための作製されたエマルジョンの違いをまとめている。
【0072】
【表2】
【0073】
図3の実施例の走査型電子顕微鏡写真は、N−アクリロキシスクシンイミドの組込みの効果を示す。N−アクリロキシスクシンイミドは、液相への部分的な可溶性のために、液相に導入された際(図3、実施例7)には、連続気泡構造の規則性が部分的に乱してセルのサイズ分布を広げ、連続相内にのみ導入された際(図3、実施例6)には、より薄いセル壁を生じた。
【0074】
[ブラッド−フォードアッセイを用いた、溶液のタンパク質濃度の判定]
ブラッド−フォードアッセイタンパク質測定試験に適合する、バイオ−ラッド製のタンパク質アッセイ試薬を用いて、純粋な水性緩衝液または30%v/vまでのエタノールを含有する水性緩衝液中のタンパク質または酵素の濃度を判定した。
【0075】
タンパク質溶液を用いて、水で系列希釈を行い、0〜25μg/mlの範囲の濃度を有する0.8mlの試料を調製した。次いで、純粋なタンパク質アッセイ試薬を各希釈試料に加えた。試薬は、G−250クマシーブルー、すなわち、タンパク質の塩基性および芳香族残基と速やかに反応して、明るい青色の錯体を形成する色素を含有する。10分間攪拌した後、試料を紫外線可視分光光度計内に入れ、595nmにおける吸光度を測定した(タンパク質を含有しない試料でゼロ吸光度を設定した)。外部検量線を、0〜20μg/mlの範囲のウシ血清アルブミン(BSA)を含む試料を用いて測定した。この曲線(図4)を用いて試料における吸光度およびタンパク質の量の相関をとった。
【0076】
[緑色蛍光タンパク質の緩衝液交換のためのプロトコル]
この実施例では、スクシンイミドエステル(実施例1〜9)を含有するポリHIPEを共有結合的に固定化するための組み換え緑色蛍光タンパク質(rAce−GFP)を調製するのに用いられるプロセスを記載する。本プロセスは、ほとんどの場合、緩衝液を交換するとともにrAce−GFPが送達された添加剤を除去するためのタンパク質の透析を含んだ。
【0077】
エヴロゲン(Evrogen)製の組み換えAce−緑色蛍光タンパク質を用いた。1つのバイアルのAce−GFP(1mg/mlにおいて0.10ml)を、5種のポリHIPE試料(20μg/試料)のために用いた。1つのバイアルを、ミリポア・マイクロコン(Millipore Microcon)YM−10遠心分離装置(膜の排除分子量:10000)でリン酸緩衝液(66mM、pH8.0)に対して透析した。リン酸緩衝液(0.5ml)を6回加えてから8000Gで20分間遠心分離した後、エタノール(30%v/v)を含有するリン酸緩衝液(66mM、pH8.0)を用いてタンパク質濃縮物を最終的に2.0mlにした。
【0078】
[実施例10]
実施例10では、N−アクリロキシスクシンイミド(実施例1〜9)の仕込み量の様々なポリHIPEに緑色蛍光タンパク質を共有結合的にグラフトするためのプロセスを記載する。比較例2〜4のポリHIPEを陰性対照として用いた。固定化プロセスは各ポリHIPEについて同じであり、すなわち、5mmのポリHIPEキューブを切断し、秤量し、2.0mlのエッペンドルフバイアルに入れた。バイアルを透析したrAce−GFP(0.40ml)で満たし、ローラー攪拌器で4時間攪拌させた。次いで、バイアルの内容物を、直径5.5cmのペーパーフィルタに注ぎ、フィルタ装置に減圧をかけ、エタノール(30%v/v)を含有するリン酸緩衝液(66mM、pH7.0)を、ポリHIPE片に滴下して加えた。吸引は、溶媒をポリマーに勢いよく通すことによって速やかな洗浄を可能にした。20mlの緩衝液を各片のために用い、洗浄されたGFPグラフトされたポリHIPEキューブを、エタノール(30%v/v)を含有するリン酸緩衝液(66mM、pH7.0)の中に入れた。溶出容量が20mlになった後の洗浄液に、ブラッド−フォード試験を用いてrAce−GFPを検出することはできなかった。表3は、どのポリHIPEを固定化のために用いたかを示す。
【0079】
【表3】
【0080】
rAce−GFPに曝されその後洗浄された湿潤ポリHIPEキューブを、黒色背景の顕微鏡ガラススライド上に置き、ライカMZFLIII蛍光顕微鏡で青色ランプ(発光:480±50nm)の下で調べた。ライカCC−12デジタルカメラで写真を撮影した。比較例2、および実施例1、2、4の、タンパク質の塩基性表面残基(おそらく主にリジン)と、N−アクリロキシスクシンイミドの活性化されたエステル官能基との間の反応によって生じると考えられるポリHIPEの写真は、図5に見られ、ポリHIPEの蛍光と機能性モノマー(NASI)との間の関係を明確に示している。比較例2は、機能性モノマーを含まず、蛍光を示さず、このことは、非常にわずかなrAce−GFPがこの非機能性ポリHIPEに物理的に吸着されるかまたは全く吸着されないことを示唆している。
【0081】
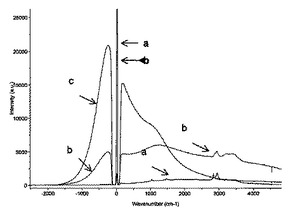
タンパク質の塩基性表面残基(例えばリジン)と、等価な仕込み量のN−アクリロキシスクシンイミドの活性化されたエステル官能基との間の反応によって生じると考えられるポリHIPEにおける固定化プロセスは、ポリHIPE中のイソボルニルアクリレートの量が増加した際に、おそらく、ほとんどのタンパク質が届かないNASIのスクシンイミドエステルを作製するバルキーなイソボルニル基の立体障害効果のために効率性が悪くなる。非機能性ポリHIPE(比較例2)およびrAce−GFPに曝されその後洗浄されたN−アクリロキシスクシンイミド含有ポリHIPE(実施例2)のキューブを、リン酸緩衝液(66mM、pH7.0)およびエタノール(30%v/v)を含有するペトリ(Petri)皿に入れた。各キューブのラマンスペクトルを、524〜532nmのラマンレーザーで取り、rAce−GFPを含まない溶液のラマンスペクトルと比較した。蛍光は、ラマン効果に対して強く競合する効果であるため、蛍光材料に対して、ラマン分光法を用いることは一般に不可能である。意外なことに、rAce−GFP吸着がレーザーの波長とそれほどかけ離れていないため、ラマンレーザーを用いて、図6のポリHIPEキューブの内部の蛍光を判定した。比較例2のキューブは蛍光を示さず(黒色のスペクトル)、このことは、アクリレートをベースとするポリHIPE自体が非蛍光性であるとともにrAce−GFPを共有結合的または物理的に固定化できないことを証明している。実施例2のキューブ(赤色の曲線)は、ほぼレーザーの波長(約505nm)を中心とするとともに溶液中のrAce−GFPの蛍光ピーク(青色スペクトル)に対応する蛍光スペクトルを示した。
【0082】
N−アクリロキシスクシンイミドなどの機能性モノマーを含有するアクリレートをベースとするポリHIPEなどの高度に多孔質のポリマーが、タンパク質、この場合rAce−GFPを共有結合的にグラフトすることが可能であることが実証された。また、実施例2のキューブの厚さにわたって様々なポイントで共焦点レーザーを集束させることによって、蛍光が一定であり、よって固定化がキューブの容積全体で均一であったことが示された。
【0083】
[固定化のためのCAL−Bの調製(透析および緩衝液交換)]
このセクションでは、スクシンイミドエステルを含有するポリHIPEにおける共有結合的な固定化のためにカンジダ・アンタークチカ・リパーゼ(Candida Antarctica Lipase)B(CAL−B)を調製するのに用いられるプロセスを記載する。本プロセスは、ほとんどの場合、緩衝液を交換するとともにCAL−Bが送達された添加剤を除去するためのタンパク質の透析を含んだ。リン酸緩衝液(66mM、pH8.0)とともに記載されるこのプロセスが、用いられる酵素に適した任意の水性緩衝液および任意のpHに適用可能であることは強調されるべきである。
【0084】
純粋なCAL−B源として、ノボザイムN525Lを用いた。N525Lを知られていない緩衝液(pH7.0)内でグリコール(50%v/v)で送達した。2つのミリポア・セントリコン・プラス−20遠心分離装置(膜の排除分子量:20000)を用いて緩衝液を交換し、グリセロールを除去した。各チューブにN525L(8mL)およびリン酸緩衝液(9mL、66mM、pH8.0)を仕込み、次いで、20分間2000Gで8回遠心分離し、各運転の後リン酸緩衝液を用いて容積が最終的に17mLになるようにした。次いで、両方のチューブからCAL−B濃縮物を採取し、リン酸緩衝液(66mM、pH8.0)中に分散し、最終容積が10.5mLになるようにした。ブラッド−フォードタンパク質測定を行い、最終溶液中のCAL−B濃度を判定した。
【0085】
[実施例11]
実施例11では、N−アクリロキシスクシンイミド(実施例4の)を含有する光重合HIPEにおけるカンジダ・アンタークチカ・リパーゼB(CAL−B)の固定化のための一般的なプロセスを記載する。リン酸緩衝液(66mM、pH8.0)とともに記載されるこのプロセスが、用いられる酵素に適した任意の水性緩衝液および任意のpHに適用可能であることは強調されるべきである。この場合、エタノール(20%v/v)を含有するリン酸緩衝液(66mM、pH7.0)が、保管および酵素活性試験のための緩衝液として選択されるが、担持される酵素がどのプロセスを取るかに応じて他の緩衝液または溶媒を用いることが可能である。
【0086】
実施例4の1片のポリHIPEを切断して秤量した(通常100mg)。CAL−B(1ml、前のセクションの透析されたN525L)、リン酸緩衝液(3ml、66mM、pH7.0)およびエタノール(1ml)を含有する10ml透明ガラス試料ボトルにそれを入れた。試料ボトルをローラー攪拌器で室温で4時間振とうした。次いで、試料ボトルの内容物を直径5.5cmのペーパーフィルタに注ぎ、フィルタ装置に減圧をかけ、エタノール(20%v/v)を含有するリン酸緩衝液(66mM、pH7.0)の混合物を、ポリHIPE片に滴下して加えた。吸引効果は、溶媒をポリマーに勢いよく通すことによる速やかな洗浄を可能にした。約50mlを用いてポリHIPEを洗浄し、エタノール(20%v/v)を含有するリン酸緩衝液(66mM、pH7.0)中に入れた。洗浄分画をブラッド−フォード試験にかけて、固定化されていないCAL−Bの量およびポリHIPE中で固定化されたCAL−Bの量を判定した。さらに、この固定化の共有結合的な性質を前提として、固定化された酵素またはポリマーマトリックスを分解させた条件を用いずに、ポリマーから固定化されたCAL−Bを除去できなかったことに留意されたい。
【0087】
[実施例12]
実施例12では、パラ−ニトロフェニルエステル基質の酵素加水分解に基づく、ノボザイムN525L由来のカンジダ・アンタークチカ・リパーゼB(CAL−B)における活性試験を記載する。
【0088】
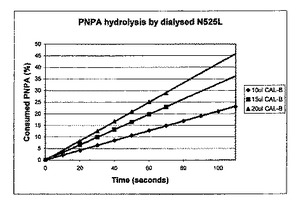
パラ−ニトロフェニルアセテート(PNPA)を基質として用いて、CAL−Bの加水分解活性を評価した。エタノール(20%v/v)を含有するリン酸緩衝液(1.9ml、66mM、pH7.0)を、2mlの紫外線可視石英セルに入れた。次いで、PNPA(0.1ml、4×10−3mmol、絶対エタノール中の7.25mg/ml溶液)を加え、ヒタチU−2000紫外線可視分光光度計中の400nmにおける吸光度の増加を2分間続け、緩衝液中のバックグラウンドPNPA化学的加水分解速度の測定をした。次いで、水中で希釈されたCAL−B(0〜0.10mlの様々な容積)を加え、パラ−ニトロフェニルの解離よる400nmにおける吸光度の増加を、線形性からの逸脱が観察されるまで続けた。計算のために、吸光度の増加曲線の傾斜から活性を推測し、化学的加水分解活性を全活性から差し引いて、単独の酵素加水分解活性を定量した。図7は、様々な量の透析され希釈されたノボザイムN525Lの活性曲線を示す。
【0089】
[実施例13]
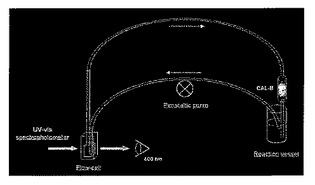
実施例13では、再現可能な条件(担体、流量、時間)下で、様々な多孔質の担体(ポリHIPE上に担持されたCAL−B、ビーズ上に担持されたCAL−B)の活性を判定するために用いられる設備を記載する。この設備を用いて、実施例11に記載されるように行われた固定化実験から得られる様々な担持されたCAL−Bの活性を比較する。
【0090】
シリコーンゴムチューブ(内径1.5mm)を用いて容器(10mlガラス試料ボトル)の上のミニカラム(長さ20mm、内径5mm)に接続された紫外線可視石英フローセル(内部容量:1ml)を用いて閉ループを構築した。蠕動ポンプをフローセルのすぐ前のチューブに装着して、ループ中に急流(30ml/分)を生成した。様々な担持されたCAL−Bをミニカラムガラスフィルタの上部に充填し、液体流れが担体を通るようにした。400nmにおける吸光度0を規定するために、エタノール(20%v/v)を含有するリン酸緩衝液(9.50ml、66mM、pH7.0)をループに通して再循環させた。次いでPNPA(0.50ml、20×10−3mmol、絶対エタノール中の7.25mg/ml溶液)を反応槽に加え、パラ−ニトロフェノール化学的加水分解のための400nmにおける吸光度の増加を、それが線形になった時点から2分間続けた。次いで、担持されたCAL−Bをミニカラムに加え、400nmにおける吸光度の増加を、それが線形である限り監視した(典型的には1〜5分間)。
【0091】
充填された担持酵素をすすぎ、経時的なおよび連続使用を経たそれらの安定性を評価するために再利用することが可能であった。計算のために、吸光度の増加曲線の傾斜から活性を推測し、化学的加水分解活性を全活性から差し引いて、単独の酵素加水分解活性を定量した。
【0092】
[実施例14]
実施例14は、同じ酵素、カンジダ・アンタークチカ・リパーゼB(CAL−B)(ノボザイムN525L由来)を含有するいくつかの担体間の安定性の比較の実施例である。
【0093】
参照例として、ノボザイムN435を用いた。それは、ポリアクリル酸樹脂のビーズに物理的に吸着されたCAL−Bからなる。CHN分析によって判定されたCAL−Bの仕込み量は約8%w/w(80mg CAL−B/g ビーズ)であり、N435の表面積は105m2/gであった。
【0094】
第2の参照例として、比較例1で作製されるものと同様のスチレン系熱重合HIPEを選択した。これらのスチレン系ポリHIPEは、疎水性相互作用(非共有結合的固定化)を介して酵素を物理的に吸着することができることが知られているためである。共有結合的固定化のために実施例11に記載のようにCAL−BをこのポリHIPEに物理的に吸着させた。酵素の固定化の後の洗浄溶液におけるタンパク質測定によって判定されるCAL−Bの仕込み量は約0.75%w/w(7.5mg CAL−B/g ポリHIPE)であった。この担体を粉末として用いた。
【0095】
陰性対照として、グラフトされたCAL−Bを含まない実施例4のポリHIPEを用いて、このポリマーが単独で加水分解に何ら影響を及ぼさないことを確認した。他の陰性対照は、固定化のための溶媒としてMES緩衝液(100mM、pH6.0)を用いた以外は、実施例11と同様のプロセスを用いてCAL−Bを物理的に吸着することを試みた実施例1のポリHIPEであった。CAL−Bの吸着は検出できなかった。これらのポリHIPEを5mgのモノリスとして用いた。
【0096】
最後に、固定化のための溶媒としてMES緩衝液(100mM、pH6.0)を用いた以外は、実施例11と同様のプロセスを用いてCAL−Bを共有結合的に固定化するために実施例4のポリHIPEを選択した。酵素の固定化の後の洗浄溶液におけるタンパク質測定によって判定されるCAL−Bの仕込み量は約0.80%w/w(8.0mg CAL−B/gポリHIPE)であった。これらのポリHIPEを5mgのモノリスとして用いた。
【0097】
これらの担体の各々を、実施例13に記載のように様々な量で試験した。
【0098】
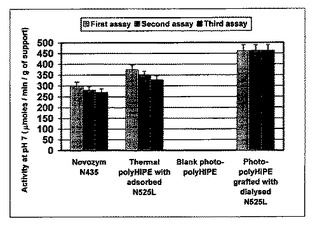
結果を図9および10にまとめた。これらの図は、図9の担体のグラムおよび図10の固定化されたCAL−Bのミリグラムによって正規化された、各担体についての、パラ−ニトロフェニルアセテートをパラ−ニトロフェノールと酢酸とに加水分解するための酵素活性を示す。
【0099】
これらの結果から以下のような3つの明白な結論があった。
a)市販のCAL−BノボザイムN435を、吸着されたCAL−Bを含有するスチレン系熱重合HIPEおよび共有結合的に固定化されたCAL−Bを含有する光重合HIPEの両方によって得られる活性に匹敵する、担体のグラム当たりの全活性を有していた。これらのスチレン系熱重合HIPEおよび光重合HIPEは、溶液中のCAL−B(約50μモル/分/mg CAL−B ノボザイムN525L由来)に匹敵するCAL−Bのミリグラム当たりの活性を示したが、ノボザイムN435は、活性が10分の1より低い。それは、N435中の吸着されたCAL−Bのほとんどが基質に届かないか、または溶液中のCAL−Bほど活性でないかのいずれかを意味する。これは、本発明による方法が低量の酵素を用いるという意味で効率的であることを示す。
b)脂肪族アクリレートベース配合物のために、非機能性光重合HIPEにおけるCAL−Bの物理的吸着はない。さらに、これらのポリHIPEにはエステルの加水分解に対して効果がない。
c)時間の経過にわたるおよび次の再利用にわたる共有結合的にグラフトされたCAL−Bの安定性は、物理的にのみ吸着された場合の担体と比較して非常に良好であった。(実験再現性の限界内の)活性の減少を、パラ−ニトロフェニルアセテートの加水分解のための同じ担体を10回用いることによって検出することができた。実施例4のポリHIPEにおける担持されたCAL−Bのエタノール(20%v/v)を含有するリン酸緩衝液(66mM、pH7.0)を3ヶ月保管した後、活性の減少を検出できなかった。
【0100】
[実施例15〜18]
これらの実施例は、界面活性剤の種類および添加剤を変えながら、EHA:IBOA:TMPTA:NASIの一定のモル比(11.65:65.82:8.19:14.34)に基づいている。HIPEに安定化効果をもたらすために、当業者には予測されるように、CaCl2(実施例15)の添加が見られた。他のところ(国際公開第97/45479号パンフレット)に報告され、実施例18で用いられる混合した界面活性剤系が、粘性のゲルのような稠密度および優れた熱安定性を有するHIPEをもたらす。
【0101】
ハイパーマー(Hypermer)B246の使用は、予想外にも、実施例16および17の両方で84%の仕込み効率をもたらすポリHIPEのNASIの保持率を高めることが分かった。これは、NASIが液相にも加えられる実施例4(62%)、および30%〜59%の範囲の仕込み効率が観察される他の全ての実施例に勝る。
【0102】
実施例15〜18を実施例1〜9と同じように作製した。実施例18を、60℃で窒素雰囲気下において16時間にわたって熱的に重合させた。
【0103】
【表4】
【0104】
[実施例19]
[DERAのカップリング]
大腸菌D−2−デオキシリボース−5−ホスフェートアルドラーゼ(DERA)無細胞抽出液(大腸菌で過剰発現したもの)(50ml)を、22℃およびpH6.60で6時間にわたって、1片のN−アクリロキシスクシンアミド−コ−ポリマー、ポリHIPE(1g)に連続的に通すことによって固定化した。次にポリマーをトリエタノールアミン緩衝液(200ml、50mM、pH7.25)で洗浄して、固定化された酵素、DERAを含有する灰白色のポリマーモノリスを残した。
i.固定化前の溶液中の総タンパク質濃度:23.6mg/ml
ii.n−ヒドロキシスクシンアミド機能性ポリマーを通して流した6時間後の総タンパク質濃度:21.3mg/ml
iii.前述したように、ブラッドフォードアッセイによってタンパク質濃度を判定した。
iv.結果として得られる、ポリHIPEにおけるDERAの仕込み量:115mg/g
【0105】
[実施例20]
[s−HNLのカップリング]
パラゴムノキ(Hevea brasiliensis)s−ヒドロキシニトリルリアーゼ(s−HNL)無細胞抽出液(ピヒア・パストリス(Pichia pastoris)で過剰発現したもの)(50ml)を、22℃およびpH5.75で6時間にわたって、1片のN−アクリロキシスクシンアミド−コ−ポリマー、ポリHIPE(1g)に連続的に通すことによって固定化した。次にポリマーをMES緩衝液(100ml、50mM、pH5.80)で洗浄して、固定化された酵素、s−HNLを含有する灰白色のポリマーモノリスを残した。
i.固定化前の溶液中の総タンパク質濃度:49.9mg/ml
ii.n−ヒドロキシスクシンアミド機能性ポリマーを通して流した6時間後:27.0mg/ml
iii.前述したように、ブラッドフォードアッセイによってタンパク質濃度を判定した。
iv.結果として得られる、ポリHIPEにおけるs−HNLの仕込み量:150mg/g
【0106】
[実施例21]
[DERA共重合]
大腸菌D−2−デオキシリボース−5−ホスフェートアルドラーゼ(DERA)無細胞抽出液(大腸菌で過剰発現したもの)(45ml、タンパク質含量が1mg/ml)を共重合によってポリHIPE内に固定化した。
【0107】
水またはCaCl2および/またはNASIの水溶液の代わりに有機相にDERA無細胞抽出液を加えたことによる以外は、実施例1〜12のようにポリHIPEを形成した。次いで、得られたポリHIPE片をリン酸カリウム緩衝液(5×100ml、50mM、pH7.00)で洗浄して、固定化された酵素、DERAを含有する灰白色のポリマーモノリスを残した。
【図面の簡単な説明】
【0108】
【図1】比較例1の走査型電子顕微鏡写真。
【図2】比較例の走査型電子顕微鏡写真(A.比較例2、B.比較例3、C.比較例4、D.比較例5)。
【図3】スクシンイミドエステルを含有するポリHIPEの走査型電子顕微鏡写真(A.実施例4、B.実施例6、C.実施例7、D.実施例8)。
【図4】タンパク質測定試験のための検量線。
【図5】蛍光ポリHIPEの図(左から順に比較例2、および実施例1、2および4からのポリHIPE)。曝露時間:500ms。倍率:1.6倍。
【図6】いずれもrAce−GFPに曝された、比較例2のキューブのラマンスペクトル(a、黒色)および実施例2のキューブのラマンスペクトル(b、赤色)、ならびに溶液中のrAce−GFPのラマンスペクトル(c、青色)の重畳。照度は正規化されていない。
【図7】ノボザイムN525L(CAL−B水溶液)によるパラ−ニトロフェニルアセテートの加水分解の活性曲線。
【図8】多孔質の担体におけるCAL−B活性の定量のためのフローセル設備。
【図9】異なる担体におけるCAL−B(N525L)によるパラ−ニトロフェニルアセテートの加水分解。活性は、担体のグラムによって正規化されている。
【図10】異なる担体におけるCAL−B(N525L)によるパラ−ニトロフェニルアセテートの加水分解。活性は、担体に最初に存在するCAL−Bのミリグラムによって正規化されている。
【特許請求の範囲】
【請求項1】
共有結合的にグラフトされた生物活性種を含む高度に多孔質のポリマー材料。
【請求項2】
材料がモノリシックである、請求項1に記載の高度に多孔質の材料。
【請求項3】
前記共有結合的にグラフトされた生物活性種がタンパク質であり、好ましくは酵素であり、より好ましくは2種以上の酵素である、請求項1または2に記載の高度に多孔質のポリマー材料。
【請求項4】
生物活性分子種にグラフトすることが可能な機能性モノマーを含む高度に多孔質の材料の調製方法であって、
a.液相と連続相とを含むとともにモノマーを含有するエマルジョン組成物を調製する工程と、
b.前記エマルジョンを硬化する工程と、
c.任意に、前記水/液相を除去する工程と
を含む方法。
【請求項5】
工程a.の前記エマルジョン組成物が、架橋モノマー、機能性モノマー、重合開始剤、界面活性剤および水からなる群のうちの少なくとも1つをさらに含有する、請求項4に記載の方法。
【請求項6】
生物活性種を共有結合的にグラフトすることが可能な高度に多孔質のポリマー材料の調製方法であって、
a.以下のもの:
A.機能性モノマーを5〜95重量%、
B.架橋モノマーを5〜80重量%、
C.重合開始剤を0〜10重量%、
D.界面活性剤を0〜20重量%、
E.機能性モノマーまたは架橋モノマー以外のモノマーを0〜90重量%
(ここで、重量%は、A、B、C、DおよびEの総重量に対するものである)
およびF、前記液相を構成する液体または液体組成物を74〜93体積%(ここで、体積%は、A、B、C、DおよびEを含む連続相ならびに液相の全体積に対するものである)
を含む組成物から液相と連続相とを含むとエマルジョンを調製する工程と、
b.前記エマルジョンを硬化する工程と、
c.任意に、前記水/液相を除去する工程と
を含む方法。
【請求項7】
前記モノマー、架橋モノマーおよび機能性モノマーがビニル不飽和を含む、請求項4に記載の方法。
【請求項8】
前記モノマーが活性エステル基を含む、請求項4または5に記載の方法。
【請求項9】
請求項1〜3のいずれか一項に記載の高度に多孔質のポリマー材料に生物活性種をグラフトするための方法であって、
a.前記高度に多孔質の材料を、好適な溶剤中の生物活性種の溶液に曝す工程と、
b.任意に、活性化剤を加える工程と、
c.任意に、加熱する工程と、
d.前記多孔質の材料を溶剤ですすいで、グラフトされていない種を除去する工程と
を含む方法。
【請求項10】
前記生物活性種の溶液に用いられる溶剤が、水、より好ましくは水性緩衝液である、請求項9に記載の方法。
【請求項11】
前記生物活性種の溶液に用いられる溶剤が、有機溶媒である、請求項9に記載の方法。
【請求項12】
前記生物活性種の溶液に用いられる溶剤が、水と有機溶媒との混合物、またはより好ましくは水性緩衝液と有機溶媒との混合物である、請求項9に記載の方法。
【請求項13】
不均一触媒としての、請求項1〜3のいずれか一項に記載の生物活性種を含む高度に多孔質のポリマー材料の使用。
【請求項14】
不均一触媒としての、請求項1に記載の生物活性種を含む高度に多孔質の材料の使用であって、10回の反応およびすすぎサイクルの後に、同じ反応条件下で、触媒活性が元の活性の90%超を保持する使用。
【請求項15】
バイオセンサー、クロマトグラフィー、生物医学装置およびインプラントにおける、請求項1〜3のいずれか一項に記載の生物活性種を含む高度に多孔質のポリマー材料の使用。
【請求項16】
生物学的および生化学的に活性な装置、請求項1〜3のいずれか一項に記載の生物活性種を含む高度に多孔質のポリマー材料。
【請求項1】
共有結合的にグラフトされた生物活性種を含む高度に多孔質のポリマー材料。
【請求項2】
材料がモノリシックである、請求項1に記載の高度に多孔質の材料。
【請求項3】
前記共有結合的にグラフトされた生物活性種がタンパク質であり、好ましくは酵素であり、より好ましくは2種以上の酵素である、請求項1または2に記載の高度に多孔質のポリマー材料。
【請求項4】
生物活性分子種にグラフトすることが可能な機能性モノマーを含む高度に多孔質の材料の調製方法であって、
a.液相と連続相とを含むとともにモノマーを含有するエマルジョン組成物を調製する工程と、
b.前記エマルジョンを硬化する工程と、
c.任意に、前記水/液相を除去する工程と
を含む方法。
【請求項5】
工程a.の前記エマルジョン組成物が、架橋モノマー、機能性モノマー、重合開始剤、界面活性剤および水からなる群のうちの少なくとも1つをさらに含有する、請求項4に記載の方法。
【請求項6】
生物活性種を共有結合的にグラフトすることが可能な高度に多孔質のポリマー材料の調製方法であって、
a.以下のもの:
A.機能性モノマーを5〜95重量%、
B.架橋モノマーを5〜80重量%、
C.重合開始剤を0〜10重量%、
D.界面活性剤を0〜20重量%、
E.機能性モノマーまたは架橋モノマー以外のモノマーを0〜90重量%
(ここで、重量%は、A、B、C、DおよびEの総重量に対するものである)
およびF、前記液相を構成する液体または液体組成物を74〜93体積%(ここで、体積%は、A、B、C、DおよびEを含む連続相ならびに液相の全体積に対するものである)
を含む組成物から液相と連続相とを含むとエマルジョンを調製する工程と、
b.前記エマルジョンを硬化する工程と、
c.任意に、前記水/液相を除去する工程と
を含む方法。
【請求項7】
前記モノマー、架橋モノマーおよび機能性モノマーがビニル不飽和を含む、請求項4に記載の方法。
【請求項8】
前記モノマーが活性エステル基を含む、請求項4または5に記載の方法。
【請求項9】
請求項1〜3のいずれか一項に記載の高度に多孔質のポリマー材料に生物活性種をグラフトするための方法であって、
a.前記高度に多孔質の材料を、好適な溶剤中の生物活性種の溶液に曝す工程と、
b.任意に、活性化剤を加える工程と、
c.任意に、加熱する工程と、
d.前記多孔質の材料を溶剤ですすいで、グラフトされていない種を除去する工程と
を含む方法。
【請求項10】
前記生物活性種の溶液に用いられる溶剤が、水、より好ましくは水性緩衝液である、請求項9に記載の方法。
【請求項11】
前記生物活性種の溶液に用いられる溶剤が、有機溶媒である、請求項9に記載の方法。
【請求項12】
前記生物活性種の溶液に用いられる溶剤が、水と有機溶媒との混合物、またはより好ましくは水性緩衝液と有機溶媒との混合物である、請求項9に記載の方法。
【請求項13】
不均一触媒としての、請求項1〜3のいずれか一項に記載の生物活性種を含む高度に多孔質のポリマー材料の使用。
【請求項14】
不均一触媒としての、請求項1に記載の生物活性種を含む高度に多孔質の材料の使用であって、10回の反応およびすすぎサイクルの後に、同じ反応条件下で、触媒活性が元の活性の90%超を保持する使用。
【請求項15】
バイオセンサー、クロマトグラフィー、生物医学装置およびインプラントにおける、請求項1〜3のいずれか一項に記載の生物活性種を含む高度に多孔質のポリマー材料の使用。
【請求項16】
生物学的および生化学的に活性な装置、請求項1〜3のいずれか一項に記載の生物活性種を含む高度に多孔質のポリマー材料。
【図4】

【図6】
【図7】
【図8】
【図9】
【図10】



【図6】
【図7】
【図8】
【図9】
【図10】
【公表番号】特表2008−536986(P2008−536986A)
【公表日】平成20年9月11日(2008.9.11)
【国際特許分類】
【出願番号】特願2008−507009(P2008−507009)
【出願日】平成18年4月21日(2006.4.21)
【国際出願番号】PCT/EP2006/003677
【国際公開番号】WO2006/111399
【国際公開日】平成18年10月26日(2006.10.26)
【出願人】(503220392)ディーエスエム アイピー アセッツ ビー.ブイ. (873)
【Fターム(参考)】
【公表日】平成20年9月11日(2008.9.11)
【国際特許分類】
【出願日】平成18年4月21日(2006.4.21)
【国際出願番号】PCT/EP2006/003677
【国際公開番号】WO2006/111399
【国際公開日】平成18年10月26日(2006.10.26)
【出願人】(503220392)ディーエスエム アイピー アセッツ ビー.ブイ. (873)
【Fターム(参考)】
[ Back to top ]