
内側側頭葉てんかん(TLE)の処置及び可能性を防止/減少させる方法
本発明は、被験者が内側側頭葉てんかん(TLE)を誘発することが既知の発作を患った後に、NKCC1抑制剤を被験者に投与することにより、被験者における内側側頭葉てんかん(TLE)の発症を防止し、又は被験者における側頭葉てんかん(TLE)の重症度を軽減する処置方法を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
(関連出願)
2009年7月24日出願の表題「内側側頭葉てんかん(TLE)の処置及び可能性を防止/減少させる方法」の特許文献1、及び2009年11月12日出願の同一表題の特許文献2の優先の恩恵を主張し、それらは参照により個々にその全体がここに採り入れられる。
(連邦助成による研究)
本発明はNIH/NRSAによる助成金番号RR15636,FF015614,AA014127及びAA016880の政府の助成により完成された。従って米国政府は本発明に対し権利を有する。
(技術分野)
本発明は、内側側頭葉てんかん(TLE)の処置及び防止に関する新規の方法を提供する。
【背景技術】
【0002】
全ての参照に対する引用文献は実施例の後に記載されている。
内側側頭葉てんかん(TLE)は成人において最も一般的なタイプのてんかんであり(Engel,1989)、しばしば薬物処置に対し耐性を示し、最終的には発作を抑えるために神経手術の選択肢しか残されない。TLEは脳損傷及びてんかん重積(SE)を含む種々の発作の後に発症する。突然の発作の後に、特徴的な無発作の「潜伏期」がヒトでは典型的に2−3カ月、例外的に数年続く(Annegers氏他、1980;Weiss氏他,1986)。リチウム−ピロカルピン誘導てんかん重積(SE)後数週間後に発症するラットのTLEは、ヒトTLEの臨床及び神経病理学的特徴を再現し、そしてその病気の非常に有益な動物モデルを提供する(Ormandy氏他,1989;Turski氏他,1991;Cavalheiro氏,1995;Dube氏他,200;Andre氏他,2007)。当初の原因が異なっても、TLEの行動性及び組織病理学的特徴は全ての病因で驚くほど類似している。このことが多くの研究者に初期の原因の下流に主な共通する経路、恐らくは発作の識別特性である激しい同期活性があると仮定するように誘導した(Du氏他,1995;Wu氏及びSchwarcz氏,1998;Schwarcz氏他,2000)。通常この同期活性は嗅内皮質(EC)を含む海馬及び海馬傍回皮質に見られる(Schwartzkroin氏及びKnowles氏,1984;Bartolomei氏他,2004)。長い間、特定の内側側頭葉萎縮が発作病巣に対して同側性であることが示されて来た(Bartolomei氏他,2005)。外科的に切除された標本の検査により、嗅内皮質(EC)で細胞消失とアストログリオーシスが確認された(Yilmazer−Hanke氏他,2000)。
【0003】
神経生理学において、てんかん重積(SE)の後、どこの場所において重要な初期変化が起こるのかが不明確である;しかしながら、海馬アンモン角領域に対する海馬傍回の神経保護実験は、てんかん発生の初期段階において海馬傍回皮質の重要な役割を示唆した(Andre氏他,2007)。さらに我々は以前の研究において、深部嗅内皮質(EC)が、第2層及び第3層と著しく対照的に、異常に高い網状組織の興奮を発現することを観察した(Gloveli氏他,1999;Egorov氏他,2003)。このことは、深部嗅内皮質(EC)が、てんかん重積(SE)にトリガーされた過剰興奮性の発現に対して特に疑わしいことを示唆する。
【0004】
自発的再発性発作(SRS)を防止する重要な機構は、興奮性ニューロンのCl−依存シナプス抑制である。この抑制の有効性は細胞外Cl−濃度に対する細胞内Cl−の増加と共に低下する。細胞内Cl−濃度はK+Cl−共輸送体、特にKCC2により、K+外向き勾配により駆動されて減少し、Na+K+2Cl−共輸送体、特にNKCC1により、Na+内向き勾配に駆動されて増加する。従って周辺及び中心ニューロンに対しては、両方のCl−輸送体がシナプス抑制の効率性に対して重要な役割を果たしていることが確認されている(Aickin氏他,1984;Misgeld氏他,1986)。
【0005】
特許文献3は種々の疾病、特に、塩化ナトリウムカリウム共輸送体媒介疾病、及びガンマアミノ酪酸(GABA)の損なわれた抑制により激化する脳内の興奮毒性に伴う疾病、の処置に利尿剤を使用することを記載している。
【0006】
TLEを防止又は処置する努力に拘わらず、上記のTLE関連発作を防止又は寛解させる処置方法に対する要求は依然として存在している。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】米国暫定特許出願US61/271,714
【特許文献2】米国暫定特許出願US61/281,095
【特許文献3】米国特許出願 20070043034
【発明の概要】
【0008】
出願人は、被験者がTLEを誘発することが既知の発作を患った後にNKCC1抑制剤を被験者に投与することにより、被験者に内側側頭葉てんかん(TLE)が発症するのを防止する、或いはTLEの重症度を低減する処置法を発見した。
【0009】
従って本発明のある側面では、被験者が、てんかん重積(SE)、小発作性てんかん、欠神、筋クローヌス性発作、クローヌス性(間代性)発作、緊張性発作、緊張性―クローヌス性(間代性)発作、無緊張性発作、後天性失語、てんかんを伴う後天性失語症(ランドウ・クレフナー症候群)、後天性てんかん性失語症、大脳皮質異形成−焦点性てんかん(CDFE)、新生児てんかん、海馬硬化症(HS)、海馬萎縮症、大脳萎縮症、小脳萎縮症、複雑熱性けいれん(CFC)を含む熱性発作、外傷性脳障害、脳卒中、脳腫瘍の診断又は発見後、などの発作を患った後に、被験者にNKCC1抑制剤が投与される。
【0010】
ある側面では、NKCC1抑制剤が、被験者のてんかん重積(SE)発症後の潜伏期に被験者に投与される。
また他の側面では、NKCC1抑制剤が、被験者が原因が未知の最初の発作を患った後に被験者に投与される。
【0011】
さらに他の側面では、NKCC1抑制剤が、被験者がTLE誘発発作(一般的に、SE発作を含む)を患った後約8週間から約24週間以内(或いは約12週間から約16週間以内)に被験者に投与される、又は約1週間から約4週間以内、又は約2−3週間から4週間以内に被験者に投与される。ブメタニド及びGABAA作動性伝達賦活薬との組合せによる、TLE又はTLE発症の効果的処置のためのSE又は他のTLE誘発発作後の時間は、SE又は発作の継続期間と重症度、及び種、特にヒトに依存し、それよりずっと長い。
【0012】
本発明の他の側面では、NKCC1抑制剤とガンマアミノ酪酸(GABA)調節組成物が、被験者がTLEを誘発することが既知の発作を患った後又は原因が未知の発作の最初の発作後に、前記被験者に同時投与される。
【0013】
本発明の好適な実施形態では、NKCC1抑制剤が,トラセミド、フロセミド、アゾセミド、ブメタニド、ピレタニド、トリパミド、エトゾリン(etozoline)及びその代謝体オゾリノン(Ozolinone)、シクレタニン、およびそれらの医薬的に許容可能な派生物、それらの塩及びエステルからなるグループから選択されるループ利尿薬である。
【0014】
本発明の他の側面では、1つ以上のNKCC1抑制剤、及び選択肢として1つ以上のガンマアミノ酪酸(GABA)調節組成物が、被験者がTLEを誘発することが既知の反復性発作を患った後に、慢性的に被験者に投与される。
【0015】
本発明のさらに他の側面では、NKCC1抑制剤が第2の処置薬と組合わされ、そして被験者がTLEを誘発することが既知の発作を患った後に、被験者に同時投与され、
ここにおいて第2の処置薬は、ガンマアミノ酪酸(GABA)調節組成物(例えば、GABAA受容体依存信号伝達の調節剤)、抗けいれん薬、イオンチャネル不活性化剤、抗利尿剤、又はそれらの組合せである。
【0016】
発明者らは、TLEを誘発することが既知の発作に続く期間(例えば、てんかん重積(SE)後の潜伏期)において、嗅内皮質(EC)第5層内の増加する比率のニューロンが単一のシナプス刺激に多シナプス性バースト脱分極/てんかん型活性で応答することを発見した。この変化はCl−内向き輸送体NKCC1の発現量の増大及び同時発生のCl−外向き輸送体KCC2の発現量の減少に明白に起因する、第5層ニューロンにおけるIPSP逆転電位の進行性脱分極シフトと平行して起こり、両方の変化は細胞内Cl−蓄積を助ける。潜伏期におけるCl−取り込みの抑制は、よりネガティブなGABA作動性逆転電位を回復し、そして多シナプス性バーストを消滅させた。Cl−輸送体における変化は、深部嗅内皮質に高度に特異的であった。変化はこの期間、第1−3層、嗅周囲皮質、海馬台、CA1,CA2又は歯状回では起こらない。いかなる理論にも縛られないことを希望するが、我々は、Cl−のホメオスタシスの変化が深部嗅内皮質において過剰興奮性を促進し、そこでのてんかん放電につながり、それは結果的に下流の皮質領域に影響を与えることを提案する。
【0017】
本発明の処置方法は従って、潜伏期及び初期慢性型てんかん期における異常性細胞活性及び発作形成の根底をなす特定の基礎的病理を標的とする。TLEを誘発することが既知の発作の後、又は未知の原因の初期発作の後、のNKCC1の慢性的抑制は、TLEの予防的防止、又は発現の減弱に有用であることを証明する。
本発明のこれら及び他の側面は、発明の詳細においてさらに記述される。
【図面の簡単な説明】
【0018】
【図1】実施例1の実験により画定される、てんかん重積(SE)後の嗅内皮質(EC)第5層ニューロンの増大した多シナプス性バースト興奮を示す図である。
【図2】「材料及び方法」の項、及び実施例2で説明される、てんかん重積(SE)後2週間後と3週間後のGABAA誘発シナプス後電位(PSP)逆転電位のポジティブ変移を示す図である。
【図3】実施例3で画定される、NKCC1のmRNA、Cl−内側方向輸送体が、てんかん重積(SE)後嗅内皮質(EC)第5層内において進行的に増大することを示す図である。
【図4】実施例4の実験において、NKCC1タンパク質も増加したことを示す図である。
【図5】実施例4の実験において測定されるように、SE後、嗅内皮質(EC)第5層内ニューロンがNKCC1タンパク質の発現を増加させたことを示す図である。
【図6】実施例5の実験において測定されたように、KCC2に対するmRNA,ニューロンのCl−の外向き輸送体は、嗅内皮質(EC)第5層内においてSE後進行的に減少することを示す図である。
【図7】実施例6の実験において測定されたように、SE後KCC2タンパク質も減少することを示す図である。
【図8】実施例7の実験において測定されたように、NKCC1のmRNAは他の皮質領域では増加しないことを示す図である。
【図9】実施例7の実験において測定されたように、KCC2のmRNAは深層嗅内皮質(EC)でのみ減少することを示す図である。
【図10】実施例8の実験において測定されたように、NKCC1抑制剤ブメタニドは部分的にE(PSP)を回復し、そして多シナプス性興奮を抑制することを示す図である。
【図11】実施例9の実験において測定されたように、ピクロトキシンは多シナプス性バーストに軽度の影響をあたえることを示す図である。
【図12】リチウム−ピロカルピンSE後12−17週間後における、ブメタニドによる発作の抑制(0.2mg/Kg体重、腹腔内)を示す(n=5ラット)図である。5体のリチウム−ピロカルピンSEラットが連続するブメタニド又は媒体注射(生理食塩水内の5%エタノール、腹腔内、下部に日付を記載)を受け、そしてその後8時間以内に発現する発作がビデオ録画により分析された。1体の同一のラットは、ブメタニドがCl及びIPSP逆転電位を概して基準化した場合でも、2度目及び3度目のブメタニド投与後に発作を発現したが、それは恐らく自発的発作を可能にするこのラット内の2次的な下流の変化によるものである。
【図13】NKCC1タンパク質が正常な成人レベルに向かって回復することを示す図である。プロットは、深層内側嗅内皮質(EC)のNKCC1タンパク質の免疫蛍光信号を、抗体を持たない対照ラット(「免疫グロブリンG(IgG)無し」と表示)、抗体を持つ対照ラット(「対照」と表示)、SE後2週間後及び3週間後(「SE 週間後」と表示)のラットにおいて示す。SE後17週間でNKCC1信号は、ブメタニドが完全に自発的再発性発作(SRS)を防止するラット(「ブメット(Bumet)ポジティブ」と表示)では、SE後3週間後のラットのレベルの50%未満まで回復し、そしてブメタニドが自発的再発性発作(SRS)を防止する効果が無くなったラット(「ブメット(Bumet)ネガティブ」と表示)では95%超に回復した。
【図14】KCC2タンパク質が正常な成体レベルに向かって回復することを示す図である。プロットは、深層内側嗅内皮質(EC)のKCC2タンパク質の免疫蛍光信号を対照ラット(「対照」と表示)、SE後2週間後及び3週間後(「SE後 週間」と表示)のラット、及びSE後17週間後のラットにおいて示す。SE後17週間でKCC2信号は、正常レベルに向かって回復した(ブメタニドが完全に自発的再発性発作(SRS)を防止するラットは「ブメット(Bumet)ポジティブ」と表示、そして、ブメタニドが自発的再発性発作(SRS)を防止する効果が無くなったラットは「ブメット(Bumet)ネガティブ」と表示)。
【図15】海馬の錐体細胞CA1層又はCA3層では、NKCC1のmRNAは変化がないことを示す図である。
【図16】海馬の錐体細胞CA1層又はCA3層では、KCC2のmRNAは変化がないことを示す図である。
【図17】嗅内皮質(EC)第2層、第3層、又は第5層においても、SE後3週間後にニューロンの損失がないことを示す図である。
【図18】ブメタニド処置が有る場合と無い場合の発作発現を示す図である。
【図19】慢性ブメタニド処置が、処置期間中および処置終了後少なくとも6週間、自発的発作の発現を抑制することを示す図である。
【図20】ブメタニド処置終了後6週間後も誘発発作の発現を抑制することを示す図である。
【図21】ブメタニド処置がラットの挙動を回復させることを示す図である。
【図22】内側側頭葉てんかん(TLE)発症の我々の原理、及び本発明による防止を示す図である。
【発明を実施するための形態】
【0019】
以下の用語が本発明の記述に使用される。ここで特別に定義されない言葉は、本発明の記述に使用される言葉と一貫性を持つ、当業者に使用される共通の意味を有する。
特に特定されない限り、「1つの」、および「少なくとも1つ」は交換可能に使用され、1つ又はそれ以上を意味する。
用語「効果的」は、本発明において意図された効果を生むための本発明の薬剤の量を記述するために使用される。
用語「ガンマアミノ酪酸(GABA)調節組成物」は、限定されないが、バルビツール酸化合物(例えばフェノバルビタール)、ベンゾジアゼピン系化合物(例えば、ジアゼパム)、又はGABAの放出を増加させ又はGABAの再摂取を抑制する薬剤、例えば、ガバペンチン、プレガバリン、4アミノ酪酸(GABA),4−アミノ−3−(4−クロロフェニル)ブタン酸(バクロフェン)、4−アミノ−3−フェニルブタン酸、4−アミノ−3−ヒドロキシブタン酸、4−アミノ−3−(4−クロロフェニル)−3−ヒドロキシフェニルブタン酸、4−アミノ−3−(チエン−2−イル)ブタン酸、4−アミノ−3−(5−クロロチエン−2−イル)ブタン酸、4−アミノ−3−(5−ブロモチエン−2−イル)ブタン酸、4−アミノ−3−(5−メチルチエンー2−イル)ブタン酸、4−アミノ−3−(2−イミダゾリル)ブタン酸、4−グアニジノー3−(4−クロロフェニル)ブタン酸、(3−アミノプロピル)亜ホスホン酸、(4−アミノブチ−2−イル)亜ホスホン酸、酪酸ナトリウム、(3−アミノ−2−メチルプロピル)亜ホスホン酸、(3−アミノブチル)亜ホスホン酸、(3−アミノ−2−(4−クロロフェニル)プロピル)亜ホスホン酸、(3−アミノ−2−(4−クロロフェニル)−2−ヒドロキシプロピル)亜ホスホン酸、(3−アミノ−2−(4−フルオロフェニル)プロピル)亜ホスホン酸、(3−アミノ−2−フェニルプロピル)亜ホスホン酸、(3−アミノ−2−ヒドロキシプロピル)亜ホスホン酸、(E)−(3−アミノプロペン−1−イル)亜ホスホン酸、(3−アミノ−2−シクロヘキシルプロピル)亜ホスホン酸、(3−アミノ−2−ベンジルプロピル)亜ホスホン酸、[3−アミノ−2−(4−メチルフェニル)プロピル]亜ホスホン酸、[3−アミノ−2−(4−トリフルオロメチルフェニル)プロピル]亜ホスホン酸、[3−アミノ−2−(4−メトキシフェニル)プロピル]亜ホスホン酸、[3−アミノ−2−(4−クロロフェニル)−2−ヒドロキシプロピル]亜ホスホン酸、(3−アミノプロピル)メチルホスフィン酸、(3−アミノ−2−ヒドロキシプロピル)メチルホスフィン酸、(3−アミノプロピル)(ジフルオロメチル)ホスフィン酸、(4−アミノブチ−2−イル)メチルホスフィン酸、(3−アミノ−1−ヒドロキシプロピル)メチルホスフィン酸、(3−アミノ−2−ヒドロキシプロピル)(ジフルオロメチル)ホスフィン酸、(E)−(3−アミノプロペン−1−イル)メチルホスフィン酸、(3−アミノ−2−オキソ−プロピル)メチルホスフィン酸、(3−アミノプロピル)ヒドロキシメチルホスフィン酸、(5−アミノペンチ−3−イル)メチルホスフィン酸、(4−アミノ−1,1,1−トリフルオロブチ−2−イル)メチルホスフィン酸、(3−アミノ−2−(4−クロロフェニル)プロピル)スルフィン酸、及び、3−アミノプロピルスルフィン酸、を含む。
【0020】
ガンマアミノ酪酸(GABA)調節組成物として有用な本発明の他の組成物は:
拮抗薬:ガボクサドール、イソグバシン、イソニペコチン酸、ムシモール(ベニテングタケ)、プロガビド、SL75102、チオムシモール(Thiomuscimol)。
GABAA受容体ポジティブアロステリック調節剤:
バルビツール酸化合物;
アロバルビタール、アルフェナール(Alphenal)、アモバルビタール、アプロバルビタール、バルベキサクロン、バルビタール、バルビツール酸ベンジルブチル、ブラロバルビタール(Brallobarbital),ブロフェバルビタール(Brophebarbital)、ブコローム、ブタバービタル、ブタルビタール、ブトバルビタール、ブタリロナール、クロチルバルビタール(Crotylbarbital),シクロバルビタール、シクロパール(Cyclopal),エナリルプロピマール(Enallylpropymal)、エサロバルビタール(Ethallobarbital),フェバルバメート、ヘプタバルビタール、ヘキセタール、ヘキソバルビタール、メホバルビタール、メタルビタール、メトヘキシタール、メチルフェノバルビタール、ナルコバルビタール、ニールバルビタール(Nealbarbital),ペントバルビタール、フェノバルビタール、フェタアルビタール(Phetharbital)、プリミドン、プラジトン(Praziton)、プロバルビタール、プロパリロナール(Propallylonal)、プロキシバルバール、プロキシバルビタール、レポサール、セクブタバルビタール、セコバルビタール、シグモダール(Sigmodal)、スピロバルビタール(Spirobarbital)、タルブタール、チアルバルビタール、チアミラール、チオバルビタール、チオブタバルビタール、チオペンタール、バロフェイン(Valofane)、ビンバルビタール(Vinbarbital),ビニルビタール。
【0021】
ベンゾジアゼピン系薬;
アジナゾラム、アルプラゾラム、アルフェンダザム(Arfendazam),アビザホン(Avizafone),ベンタゼパム,ブレタゼニル,ブロマゼパム,ブロチゾラム,カマゼパム,クロルジアゼポキシド,シクロチゾラム(Ciclotizolam),シノラゼパム,クリマゾラム(Climazolam),クロバザム,クロナゼパム,クロラゼペート,クロチアゼパム,クロキサゾラム,シプラゼパム(Cyprazepam),デロラゼパム,ジアゼパム,ドキセファゼパム,エルファゼパム(Elfazepam)、エスタゾラム,エチルカルフルゼペート(Ethyl carfluzepate),エチルジラゼペート(Ethyl Dirazepate),ロフラゼプ酸エチル,エチゾラム,フレタゼパム(Fletazepam),フルジアゼパム,フルニトラゼパム,フルラゼパム,フルタゾラム,フルテマゼパム(Flutemazepam),フルトプラゼパム、ホサゼパム(Fosazepam)、ギダゼパム(Gidazepam)、ギリソパム(Girisopam), ハラゼパム, ハロキサゾラム, イクラゼパム(Iclazepam),イミダゼニル(Imidazenil),ケタゾラム,ロフェンダザム(Lofendazam),ロピラゼパム(Lopirazepam),ロピラゼパム(Lopirazepam),ロプラゾラム,ロラゼパム,ロルメタゼパム,メクロナゼパム(Meclonazepam),メダゼパム,メニトラゼパム(Menitrazepam),メタクラゼパム,メキサゾラム,ミダゾラム,ネリソパム(Nerisopam),ニメタゼパム,ニトラゼパム,ニトラゼペート(Nitrazepate),ノルダゼパム,オキサゼパム,オキサゾラム,フェナゼパム、ピナゼパム、ピボザゼパム(Pivoxazepam),プラゼパム,プレマゼパム,プロフラゼパム(Proflazepam),QH−II−66,クアゼパム,レクラゼパム(Reclazepam),リルマザホン,リパゼパム,Ro48−6791,SH−053−R−CH3−2’F,スラゼパム(Sulazepam),テマゼパム,テトラゼパム,トフィソパム,トリアゾラム,トリフルバザム(Triflubazam),ウルダゼパム(Uldazepam)、ザピゾラム(Zapizolam),ゾラゼパム,ゾメバザム(Zomebazam)。
【0022】
カルバミン酸化合物;
カリスバメート、カリソプロドール、エミルカメート、フェルバメート、メブタメート、メプロバメート、メトカルバモール、フェンプロバメート、プロシメート(Procymate)、チバメート。
抗神経活性ステロイド;
アセブロコール(Acebrochol)、アロプレグナノロン、アルファドロン、アルファキサロン、ゲドカルニル、ガナクソロン(Ganaxolone)、ヒドロキシジオン、ミナキソロン、Org20599、THDOC。
非ベンゾジアゼピン系薬;
アベカーニル、アジピプロン(Adipiplon)、アルピデム、CGS−20625、CGS−9896、CL−218,872、ELB−139、エスゾピクロン、エチホキシン、インジプロン(Indiplon)、L−838,417、ネコピデム(Necopidem)、NS−2664、NS−2710、オシナプロン、パゴクロン(Pagoclone)、パナジプロン(Panadiplon)、パジナクロン(Pazinaclone)、ピペクアリン(Pipequaline)、ROD−188、RWJ−51204、サリピデム(Saripidem)、SB−205,384、SL−651,498、スプロクロン(Suproclone)、スリクロン、SX−3228、TP−003、TPA−023,TP−13,トラカゾレート、U−89843A,U−90042、Y−23684、ザレプロン、ゾルピデム、ゾピクロン。
フェノール;
フォスプロポフォール、プロポフォール、チモール。
ピペリジンジオン;
グルテチミド、メチプリロン、ピペリジオン、ピリチルジオン。
キナゾリノン;
アフロクアロン、クロロカロン(Cloroqualone)、ジプロカロン(Diproqualone)、エタカロン(Etaqualone)、メブロカロン(Mebroqualone)、メクロカロン、メタカロン、メチルメタカロン。
その他;
クロルメザノン、クロメチアゾール、エタゾレート、エタノール(アルコール)、エトミデート、キャバラクトン(Kavalactone)(カバカバ)、ロレクレゾール、プロガビド、プロパニジド、ROD−188、タツナミソウ、スチリペントール、ワレレン酸(Valerenic Acid)(カノコソウ)。
【0023】
Na(+)−K(+)−2Cl(−)の共輸送体(ブメタニド又はエタクリン酸)の抑制剤。Am J Physiol Renal Physiol。2009年2月号;296(2):F446−57
セレンスレオニンキナーゼWNK4は、Na(+)−K(+)−2Cl(−)の共輸送体(NKCC1)及びCl(−)/塩基交換輸送体SLC26A6(CFEX)の両方に対し強い薬効のある抑制剤であることが証明されている(Proc Natl Acad Sci USA.2004年2月17;101(7):2064−9.Epub 2004年2月9日)。
mRNAからのNKCC1タンパク質の発現は再発現期間の間、嗅内皮質を標的とする核酸のアンチセンス鎖により抑制される。
炎症は典型的にNKCC特にNKCC1の増加につながる。炎症の典型的なサインはてんかん誘発性皮質に現れ、そして炎症媒介物質は従ってNKCC1再発現に寄与する可能性がある。従って、遷延性のインターフェロン−ガンマ暴露はイオン輸送及びNKCC1発現を他の組織において減少させ(Am J Physiol Gastrointest Liver Physiol. 2004年1月;286(1):G157-65.Epub 2003 9月4)、従って側頭葉てんかん(TLE)誘発発作の後に、嗅内皮質(EC)におけるNKCC1再発現を抑制するのに有用である可能性がある。
【0024】
GABAB受容体遮断薬は、GABAB受容体アンタゴニストの活性化による放出の負のフィードバックを遮断することにより、GABA放出を増大させるのに有用でありうる:
主要サイト:ファクロフェン、サクロフェン、SCH−50911。
GABA異化反応の遮断もまた有用でありうる:GABA−T抑制薬、例えば、3−ヒドラジノプロピオン酸、アミノオキシ酢酸、ガバクリン、ガバペンチン、イソニアジド、フェネルジン、フェニルエチリデンヒドラジン、プレガバリン、バルノクタミド、バルプロ酸、バルプロミド、ビガバトリン。
【0025】
NKCC1抑制薬の利尿効果の抑制は水及びナトリウムのホメオタシスに対する副作用を最小化するのに有用であり得る。1)プロベネシドでの前処置はブメタニド又は他のNKCC1抑制薬により発症したナトリウム利尿及び高レニン血症の両方を減少させる。プロベネシドのNKCC抑制媒介ナトリウム利尿に対するこのような拮抗作用は明らかにナトリウム排出に対する直接作用に起因せず、おそらくブメタニドの腎尿細管分泌に対する抑制作用の2次性である。インドメタシンはブメタニド処置の間、尿量およびナトリウム排出の増加を鈍化させ、そして、ブメタニド−誘発の血漿レニン活性の増加を抑制する。3)バソプレシン、アルギプレシン又は抗利尿ホルモンとして知られるアルギニンバソプレシン(AVP)及びその変異体、例えば、リジンバソプレシン(LVP)又はリプレシンは、腎臓内の遠位尿細管及び集合尿細管における水の再吸収を増大させることにより、腎臓内のNKCCの抑制に起因する水利尿の増加を鈍化させる。
【0026】
抗けいれん薬は、神経性発作活性の蓄積と拡散を抑制するためにてんかん発作の処置に使用される、イオンチャネル不活性剤を含む広範な薬剤グループである。それらは、てんかん様活性及び興奮性を増大させる下流の変化をさらに抑制するのに有用でありうる。このことは慢性期間において疾病が進行する間、てんかん様活性のNKCC1活性への依存性が低下する場合に、特に重要である。カルバマゼピン及びフェニトインは共に現在での第1選択肢の抗けいれん薬である。これらの薬は共にナトリウムチャネルの非活性化状態を安定させ、ニューロンを発火がより起り難い電位にし、そして他の作用も持ちうる。
【0027】
ガバペンチンの作用の明確なメカニズムは未知であるが、その抗けいれん性作用は電圧ゲートのN型カルシウムイオンチャネルを含んでいると考えられている。バルプロ酸もまた電圧ゲートのナトリウムチャネル及びT型カルシウムチャネルを遮断する。このメカニズムがバルプロ酸を広範囲の抗けいれん剤にする。他の臨床的に使用される抗けいれん薬はレベチラセタム、ラモトリギン、プリミドン、及びフェルバメートであり、それらはカルバマゼピン及びフェニトインが効かなかった場合とくに有益である。有用でありうる他の抗けいれん薬は、臭化物(臭化カリウム)、カルバメート(フェルバメート)、カルボキサミド(カルバマゼピン、オクスカルバゼピン、エスリカルバゼピンアセテート)、脂肪酸(バルプロエート−バルプロン酸、バルプロン酸ナトリウム、ジバルプロエクスナトリウム)、ビガバトリン、プロガビド、チアガビン、果糖誘導体(トピラマート)、GABA誘導体(ガバペンチン、プレガパリン)、ヒダントイン(エトトイン、フェニトイン、メフェニトイン、ホスフェニトイン)、オキサゾリジンジオン(パラメタジオン、トリメタジオン、エタジオン)、プロピオネート(ベクラミド)、ピリミジンジオン(プリミドン)、ピロリジン(ブリバラセタム、レベチラセタム、セレトラセタム(seletracetam))、コハク酸イミド(エトスクシミド、フェンスクシミド、メスクシミド)、スルホンアミド(アセタゾラミド、スルチアム、メタゾラミド、ゾニサミド)、トリアジン(ラモトリギン)、尿素(フェネトライド、フェナセミド)、バルプロイルアミド(valproyamides;バルプロエートのアミド誘導体),アミド(バルプロミド、バルノクタミド)である。
【0028】
「TLEを誘発することが既知の発作」には、限定されないが、てんかん重積(SE)、小発作性てんかん、欠神、筋クローヌス性発作、クローヌス性(間代性)発作、緊張性発作、強直間代発作、無緊張性発作、後天性失語、てんかんを伴う後天性失語症(ランドウ・クレフナー症候群)、後天性てんかん性失語症、大脳皮質異形成−焦点性てんかん(CDFE)、新生児てんかん、海馬硬化症(HS)、海馬萎縮症、大脳萎縮症、小脳萎縮症、複雑熱性けいれん(CFC)を含む熱性発作、外傷性脳障害、脳卒中、脳腫瘍、感染症、脊髄膜炎、血管形成異常を含む。
【0029】
「NKCC1抑制剤」は限定されないが、ループ利尿薬(即ち、ヘンレループの厚い上行脚の中のNa+/K+/2Cl−担体を抑制し、それによりナトリウム、カリウム及び塩素イオンの再吸収を抑制する利尿薬)を含む。有用なループ利尿薬は、トラセミド、フロセミド、アゾセミド、ブメタニド、ピレタニド、トリパミド、エトゾリン(etozoline)及びその代謝体、オゾリノン(ozolinone)、シクレタニン、およびそれらの医薬的に許容可能な派生物、例えばそれらの塩及びエステルである。)他のNKCC1抑制剤はGABA−T抑制薬を含む:3−ヒドラジノプロピオン酸、アミノオキシ酢酸、ガバクリン、ガバペンチン、イソニアジド、フェネルジン、フェニルエチリデンヒドラジン、プレガバリン、バルノクタミド、バルプロ酸、バルプロミド、ビガバトリン。
【0030】
本明細書で使用される「患者」又は「被験者」とは、本発明に基づく組成物によるてんかん処置を含む処置が行われる動物、一般的には哺乳類、好適にはヒトを示す。これらのヒト患者のような特定の動物に特異的な感染症、容態、または疾病の状態の処置において、「患者」とはその特定の動物を示す。
本明細書で使用されているように、「処置」とは症状の進行の防止、速度の低減、遅延、抑止又は留保することを意味する。
ここで使用される「処置上効果的な量」とは、TLEを患い、又は発症するリスクのある患者の処置に十分なNKCC1抑制剤(選択肢として、1つ以上のGABA調節組成物と組み合わせて)の量を意味する。例えば、効果的な量はTLE又は関連症候の発症又は進行を遅延させ、速度を遅くさせ、又は防止するのに十分な量を意味する。
【0031】
本明細書で使用される「抑制的効果的濃度」又は「抑制的効果的量」とは、実質的に又は有意にTLE又は関連する症候を処置または防止する本発明に基づく組成物の濃度又は量を意味する。
本明細書で使用される「防止的効果的濃度」とは、TLE又は関連する症候の発症を防止し可能性を低減するのに予防的に効果的な本発明に基づく組成物の濃度又は量を意味する。「抑制的効果的濃度」及び「防止的効果的濃度」は一般的に「効果的な量」の範疇に含まれる。
【0032】
「被験者におけるTLEの重症度を減ずる」とは、ある有益な程度までTLEに付随する多くの症候のいくつかを抑制又は寛解することを意味する。例示の目的だけのために言えば、本発明の処置法は側頭葉の小さな領域に影響を及ぼし、そして意識には影響を与えない単純部分発作(SPS)の悪影響を寛解又は低減する。これらは初期に感覚を引き起こす発作である。これらの感覚は記憶に関し、例えば、既視感(精通感)、未視感(未精通感)、特定の単一又は1組の記憶、又は記憶喪失である。感覚は実際には存在しない音、又は旋律のような聴覚性、或いは味のような味覚性、又は匂いのような嗅覚性でありうる。感覚はまた視覚的でありえ又は皮膚の感覚又は内臓の感覚でありうる。後者の感覚は体中を移動するようである。不快性又は多幸性感覚、恐怖、怒り、及び他の感情もまた単純部分発作(SPS)の間起こり得る。多くの場合、TLEのSPSを患う人は、感覚を記述するのが困難である。SPSは多くの場合「発作の前兆」と呼ばれ、更に激しい発作の前触れと考えられることもある。このような感覚は、本発明の方法を適用することにより軽減する。
【0033】
他の事例では、複雑部分発作(CPS)の重症度が本発明の方法を適用することにより軽減する。このような発作は意識をある程度障害し、通常はSPSと共に発症し、その後側頭葉の大部分に拡がる。徴候は無動凝視、手及び口の自動的運動、他人への応答不能、異常発言、又は異常行動を含む。
更に他の事例では、本発明の方法は続発性全汎強直間代発作(SGTCS)に付随する徴候の重症性を軽減する。これらは初期にはSPS又はCPSで発症し、その後、腕、体幹、及び脚が曲がった位置又は伸ばした位置で硬化する。この後、脚と体幹の突然の粗いけいれんが起こる。
【0034】
「同時投与」という言葉は2つ以上の活性化合物を、TLE又はそれに関連する徴候を処置又は防止するのに有効な量だけ投与することを意味する。同時投与という言葉は、好適には2つ以上の活性化合物を患者に同時に投与することを意味するが、その化合物が正確に同時刻に投与される必要はなく、効果的な濃度が血液、血清、血漿又は肺組織内で同時に発見されるように化合物の量が患者又は被験者に投与されればよい。
「慢性的に投与される」又は「慢性投与」とは、一連の処置の中で、NKCC1抑制剤及び、選択肢として、GABA調節組成物を、被験者の状態又は被験者から提示される徴候により変化するある期間内に、1回より多い回数投与することを意味する。
【0035】
本発明の方法は、NKCC1抑制剤とGABA調節組成物が担体材料で組合わされる剤形を使用してもよい。剤形と投与経路は処置される被験者及びTLE又は関連徴候の状態により異なる。同じ観点から、本願の方法は、0.25ミリグラム−1グラム、より好適には約1ミリグラム−約750ミリグラム、更に好適には、約10ミリグラム−約500から600ミリグラムの間のNKCC1抑制剤とGABA調節組成物を含む剤形の投与を必要とする。
特定の患者に対する特定の投薬及び投与計画は種々の要素により異なることを理解しなければならない。それら要素は、使用された特定の化合物の活性、年齢、体重、全体的健康、性別、食事制限、投薬の時間、排泄速度、薬の組合せ、及び処置担当医の判断、及び処置中の特定の疾病又は症状の重症度を含む。
【0036】
活性化合物の投与は、連続性(静脈内持続点滴)から日に数回の経口投与(例えば、1日4回(Q.I.D.))までの範囲が有り、また、投与の他の経路の中で、経口、外用、非経口、筋肉内、静脈内、皮下、経皮(これには浸透促進剤を含んでもよい)、バッカル、及び座薬を含んでよい。腸溶性経口錠も化合物の経口投与経路からの生物学的利用能を高めるため使用可能である。最も効果的な剤様は、選択された特定の薬剤の薬物動態および患者の疾病の重症度に依存する。経口剤様は投与の容易性及び予期される患者の応諾から特に好適である。
本発明はさらに以下の実験的セクションで記述されるが、それは説明目的のためだけであり、限定するものではない。
【0037】
(実験の部)
(材料及び方法)
(TLE誘発)
全てのプロセスはニューメキシコ大学健康科学センターに所在の動物介護及び使用委員会により承認され、また全米健康局の実験動物のケア及び使用に関するガイドに従っている。本実験のプロトコルはアンドレ氏他による刊行物のプロトコル(Andre氏他、2007)を僅かに変更したものである。発作誘発の24時間前に生後2−3カ月の雄のウィスター(Wistar)ラットが皮下注射で等浸透圧のNaCl生理食塩水に溶解した3mmol/kgの塩化リチウムを投与された。
その後ピロカルピンが皮下注射(等浸透圧のNaCl内25mg/Kg)で投与された。一度大脳辺縁系のけいれんが始まると、てんかん重積(SE)の発症を確認するため皮下針EEG電極が配置される。てんかん重積は連続高振幅脳波(EEG)スパイキングの発生として定義される(Ormandy氏他、1989)。このような針は25ゲージの皮下注射針(Becton Dickinson)から、ハブを切り取り、そして60度曲げて形成され、それにより針は定位置に留まる。1本の針が頭皮に皮下挿入により頭頂皮質に挿入され、そして対照電極として機能する他の1本の針が肩甲骨に挿入された。グラスモデル8脳波計(アストロ−メド(Astro−Med)社、ウエストワーウィック、イリノイ州)を使用した脳波(EEG)記録は、連続高振幅スパイキングをてんかん重積(SE)発症後1時間に亘って示し、それは頭蓋内に埋め込まれた電極での従前の研究で記録された(Peterson氏他、2005)正常な脳波に対し明確なコントラストを示した。その後記録は全体的に停止された。硫酸アトロピン(10mg/Kg皮下注射)及びジアゼパム(4mg/Kg筋肉内注射)が生存率を上げ、不安を軽減させ、そして筋肉弛緩を誘発するため投与された。ジアゼパムの投与は、発作を停止出来なかったが、1時間以内に筋肉弛緩を誘発し、ラットは脚を使わず腹側面を下にして横になった。アトロピンも発作を停止出来なかった。てんかん重積(SE)発症後12時間後に脳波が再び5−10分間記録され、全てのラットは断続的又は全体に形成される、即ち脳波スパイキングのバーストを有した。この時点でスパイキングのバーストや連続するスパイキングの列のような全体的な脳波スパイキングを示したこれらのラット(ケースの約20%)は、追加の2mg/Kgのジアゼパム筋肉内注射を投与された。てんかん重積(SE)の発症後12時間後にリンゲル溶液内の5%ブドウ糖が皮下注射で投与された。
【0038】
このプロトコルはてんかん重積(SE)の発症後1時間後にジアゼパムを投与した点で、2時間後に投与したアンドレ(Andre氏他、2007)のプロトコルとは異なる。更に我々は末梢性ピロカルピンの影響(Du氏他、1995)を軽減するため、アトロピンを投与した。従って我々のプロトコルはよりマイルドな発作を動物に提供すると信ずる。
【0039】
アンドレ(Andre)氏他のプロトコルは、信頼性を持って成体ラットの100%にてんかんを誘発したと記載されている(Dube氏他,2000b,Andre氏他,2007)。これは、僅かに高いピロカルピン投与量とリチウムを使用したこの研究所のそれより早期のプロトコルからも真実である(Dube氏他,2000b,Andre氏他,2007)。我々のラボでアンドレ(Andre氏他、2007)のプロトコルを使用した研究では、全てのラットが28.6±2.43(標準誤差)日(n=14;24時間/1日の連続ビデオ録画、S.Peterson氏、非公開結果)の潜伏期の後、ランク3,4又は5の自発性発作を発症した。1体のラットだけが15日目に発作を起こした。21日までには4体のラットが発作を起こした。
これらの潜伏期データを正規分布に適合させることにより、平均約25%のラットがピロカルピン/2時間後ジアゼパム処置の3週間以内に最初の発作を起こすことが予期された。
しかし屠殺する前の観察及び取り扱い中にはラットに発作は見られなかった。
我々のマイルドなプロトコルのてんかんの転帰がてんかん重積(SE)後8−16週間、1ラット当り合計36時間までのビデオ録画をしながらテストされた。そして、この期間に13体全てのラットに自発的発作が観察された。てんかん重積(SE)のラットの新しい2グループ(合計12体)によって、我々は潜伏期期間が12.7±1.7(平均±標準誤差)週間であると量定した。全体で203体のラットが使用された、リチウム対照ラット56体、てんかん重積(SE)後2週間のラット42体、SE後3週間のラット47体、ビデオ録画用SE後ラット58体。
【0040】
(スライス準備)
全ての実験は、側頭葉新皮質、嗅内皮質、海馬台及び腹側海馬を含む鋭い形状の水平大脳スライス上で行われた。対照又は発作後2又は3週間のラットは深くケタミンとキシラジン(それぞれ85と15mg/ml)麻酔をかけられそして断頭された。大脳は迅速に移動され酸素負荷された(95%酸素、5%炭酸ガス)氷冷の3 KCL、1.25 NaH2PO4、6 MgSO4、26 NaHCO3、0.2 CaCl2、10 グルコース、220 ショ糖及び0.43 ケタミンを含む(mMで)切断溶液中に置いた。スライス(350ミクロン)は氷冷の切断溶液内でビブラトーム(Dosaka DTK−1000)を使用して薄切りにされ,そこからリカバリー用に95%酸素/5%炭酸ガスで34°Cで平衡化された人工脳脊髄液(ACSF、mMで:126 NaCl,3 KCl,1.25 NaHPO4,1 MgSO4,26 NaHCO3,2 CaCl2及びグルコース)内に移された。1時間後ACSFが取り替えられ、そして記録に使用されるまで室温で保管された。
【0041】
(電気生理)
個々のスライスは固定台顕微鏡(Zeiss Axioskop社、ジュネーブ、ドイツ)に搭載された記録用チャンバに移され、そして暖められ(34°C)、酸素負荷された人工脳脊髄液(ACSF)で2ml/分で灌流された。細胞内記録は、嗅内皮質(EC)第5層ニューロンから、低めの電極抵抗に対しては1M CsSO4又は0.5M酢酸カリウム/0.5M塩化カリウム、又は改良空間クランプ用には2M CsSO4/1M塩化カリウムを充填された、ホウケイ酸ガラスの鋭い微小電極(80−150MΩ)を使用してなされた。Cl−輸送遮断剤がない場合は、全ての細胞内液はGABAA作動性シナプス後電位(PSP)逆転電位に対して同一の結果を示し、逆転電位は従って蓄積された(Misgeld氏他、1986参照)。ブメタニドによるCl−輸送遮断の間、0.5M酢酸カリウム/0.5M塩化カリウムを充填された微小電極がEIPSPを脱分極方向に約20mVだけ変移させるのが確認された。従ってGABAA作動性シナプス後電位(PSP)逆転電位に対するブメタニドの影響に関する全ての最終的な記録/データは、1M K2SO4又は2M CsSO4/1M塩化カリウムで充填された微小電極で得られた。
【0042】
ニューロンは、皮質の厚さの1/4まで、ナノステッパー微小位置決め装置(Scientific Precision Instruments社、オッペンハイム、ドイツ)を使用して、内側嗅内皮質内のアンギュラーバンドル(Angular bundle)のすぐ上に刺入された。記録はAxoclamp2Bアンプを使用して記録され、Axon Instruments Digidata 1322A(Molecular Devices社、ユニオンシティ、カリフォルニア)を使用してデジタル化し記録された。マイナス68mVまでの静止膜電位(RMP)が負のニューロンが、マイナス85mVまでの過分極パルスを使用して、錐体ニューロンを示唆するIhの存在をテストされた(Egorov氏他、2002;Egorov氏他、2003)。この基準では記録されたニューロンの75%は錐体であり、残りは多極ニューロンと推定された。Hamam氏他の研究では鋭い電極を第5層に刺入されたニューロンの85%が錐体ニューロンに典型的な顕著な長い先端樹状突起を示した(Hamam氏他、2000)。我々の研究と対照的にこの研究は非錐体ニューロンにもIhの存在を報告した(Hamam氏他、2000)。同心性2極刺激電極(直径200ミクロン、HFC Inc.社製、メイン州、米国)が嗅内皮質(EC)の第5層に、記録用電極から約150−250ミクロンの距離に配置された。刺激(70−100μ秒、100−800nA)は刺激分離ユニット(A.M.P.I、エルサレム、イスラエル)に接続されたマスター8プログラム可能パルス発生機により制御された。興奮性シナプス後電位(EPSP)及び抑制性シナプス後電位(IPSP)に対する全入力−出力関係が記録され、そして70−90%の最大応答を与える刺激強度が使用された。そこでは計測された刺激強度が最大応答の5−10%を誘発するまで減少した。
【0043】
図2AはGABAA受容体−媒介シナプス後電位の逆転電位を測定するのに使用されるプロトコルを示す。単シナプス性のシナプス後電位(PSP)がグルタミン酸受容体遮断剤CNQX(10μM)及びAPV(50μM)の存在下で誘発された。刺激強度は最大誘起シナプス後電位(PSP)振幅の約80%となるように調整された。電場刺激に先だって、膜電位(Vm)は正又は負の直流(DC)パルス(500ms、400pA以上)で変化された。膜電位が安定に達した後、シナプス後電位(PSP)が誘起された(図中*印)。安定膜電位(図2B、○印)とシナプス後電位(PSP)のピーク電圧(図2B、×印)の両方の電流−電圧関係がプロットされ、そして線形回帰がシグマプロットを使用して計算され、直接、膜電気伝導度の値を与えた。直線的な膜動作の可能性領域内で電流−電圧関係を測定することにより、遅延整流の電圧依存活性化又は他の整流電流に起因する接線コンダクタンスの誤りを避けることができる。逆転電位は回帰直線の交点により決定された。典型的なシナプス後電位(PSP)トレースが対照と3週目の実験動物で示される。CNQX、DL−2−アミノ−5−ホスホノペンタン酸、ピクロトキシン、ブメタニド、ピロカルピン及びアトロピンはシグマ(Sigma)社(セントルイス、ミズーリ州)より入手した。ジアゼパムはHospira Inc.(レイクフォーレスト、イリノイ州)から入手した。1000倍に濃縮された原液はフリーザーに保管され、ACSFで使用する前に希釈された。原液に関しては、CNQXとピクロトキシンはジメチルスルホキシド(DMSO)に溶解され、ブメタニドはエタノールに溶解された。ジメチルスルホキシド(DMSO)(1:1000)もエタノール(1:1000)もシナプス後電位(PSP)、電流−電圧関係、又はバースト応答に影響を与えなかった。
【0044】
(免疫ブロッティング)
免疫ブロッティング研究のため、内側嗅内皮質(EC)の第5−6層が350ミクロンの厚さの切片から切り裂かれ、ドライアイス上でスナップ冷凍され、そしてマイナス80℃で処理まで保管された。膜断片の準備のため、組織は250mMスクロース、10mMトリス−HCl、10mM HEPES、1mM EDTA、及びプロテアーゼ抑制剤(pH7.2)を含む緩衝液内で均質化され、その後、前記のように(Payne氏他、1996)分画遠心分離された。精製された膜断片(50μg)はその後、ドデシル硫酸ナトリウム(SDS)−ポリアクリルアミドゲル電気泳動(7%)及び抗KCC2(1:1000;ニューロマブ(NeuroMab)クローン N1/12、UCデービス)と抗アクチン(1:500;シグマ(Sigma))をそれぞれ使用した免疫ブロッティング用に処理された。
抗原−抗体複合体が、増大した化学発光(Amersham社、アーリントンハイツ、イリノイ州からのECL−プラス)から検知され、X線フィルムはNIH ImageJソフトウェアを使用して定量化された。
【0045】
(蛍光遺伝子プローブ法(FISH))
FISH用に大脳スライスを準備する場合、1つの大脳の半球がドライアイス/エタノールスラリー内で平衡化されたイソペンタンのビーカー内で急速冷凍され、そしてマイナス80℃で保存された。水平脳部位(16ミクロン)がクリオスタットを使用して準備され、スライド(Superfrost Plus,VWR)上に配置され,空気乾燥され、処理までマイナス80℃で冷凍保存された。
【0046】
NKCC1(クローンID4824556、アクセス番号BC033003)及びKCC2(クローンID6838880、アクセス番号BC054808)プラスミドはオープンバイオシステム(Open Biosystem)社(ハンツビル、アラバマ州)から入手した。NKCC1cDNAシークエンスは、Sall/XhoI及びBamHI制限酵素部位間のpBluescriptRベクターに挿入された。KCC2cDNAシークエンスは、EcoRI及びNotI制限酵素部位間のpYX−Ascベクターに挿入された。プラスミドはその後NKCC1及びKCC2シークエンスを確認するため配列決定された(DNAリサーチサービス、ニューメキシコ大学、健康科学センター)。EcoRI及びNotIは共に異なる長さのアンチセンス鎖を生成するのに使用され、KpnIはセンス鎖を生成するのに使用された(ニューイングランド バイオラボ社、Ipswich,マサチューセッツ州)。KCC2のアンチセンス鎖はAscIを使用し、センス鎖はPacIを使用して生成された。制限消化反応は37℃で2時間インキュベートされた。
【0047】
NKCC1a,b及びKCC2a,bに対する単一標識化FISHが前述(Guzowski氏他、1999)のように実行された。概要は、ジゴキシゲニン標識化KCC2及びNKCC1全長アンチセンスリボプローブが、直線化されたcDNAから商業的に入手可能な転写キット(Maxiscript,アンビオン、オースチン、テキサス州)を使用して生成され、そして事前混合されたRNAジゴキシゲニンがT3重合酵素(ロシェ分子生化学、パルアルト、カリフォルニア州)を使用してヌクレオチドを標識化した。NKCC1及びKCC2のジゴキシゲニン標識化リボプローブのハイブリダイゼーションは共に56℃で一晩かけて異なるスライド(1ng/μl)上で行われた。その後、抗ジゴキシゲニン セイヨウワサビ過酸化酵素複合体(HRP;1:200;ロシェ分子生化学、パルアルト、カリフォルニア州)が4℃で一晩インキュベートされた。TSA−シアニン−3(Cy3)(1:50;パーキンエルマー、Waltham, マサチューセッツ州)がHRP複合体を検知するのに使用された。核は、4’,6’−ジアミジノ−2−フェニルインドール(DAPI)(1:500;Invitrogen、Carlsbad,カリフォルニア州)によって対比染色された。画像は、スピンニングディスク共焦点システム(スピンニングディスク共焦点画像化装置CARV、Atto生科学、ロックビル、メリーランド州)及びCoolSNAP−Hq CCDカメラ(Roper科学、ツーソン、アリゾナ州)を備えるニコンTE2000U落射蛍光顕微鏡(20倍対物)により獲得された。画像はMetaMorphソストウェア(ユニバーサル画像)を使用して更に分析され、NIH ImageJソフトウェアを使用して定量化された。信号は背景(画像内の明確にネガティブな領域をカバーする注目画像領域(ROI)で得られた)に対して補正され、核染色に対する比率が計算された。ここにおいてこれらのデータは対照ラットに関して正規化されていた。
【0048】
(免疫組織化学的検査)
スライスは100mMリン酸緩衝食塩水内の4%パラホルムアルデヒドで一晩固定化され、PBS内の15%及び30%ショ糖で凍結保護され、その後最適切断温度(OCT)化合物(サクラファインテック、トーランス、カリフォルニア州)内で凍結された。
免疫組織化学的検査が室温で24時間かけて300ミクロン浮遊部位上で、抗KCC2(ニューロマブ(NeuroMab)クローン N1/12、UCデービス))又はNKCC1抗体(ラビットポリクローナル、Chemicon,Temecula,カリフォルニア州;結果はカリフォルニア大学リバーサイド校のC.Lytle博士から提供されたマウスNKCCモノクローナル抗体MAbT4で得られた結果と合致した)で実施された。スライスはガラススライド上に搭載され、画像がBiorad二光子顕微鏡とスライス表面下75ミクロンに配置された40倍対物レンズにより撮像された。ニューロトレース蛍光染色(モレキュラープローブ社、Eugene,オレゴン州)がクリトスタットを使用して調製された60ミクロン区画内のニューロンを対抗染色するのに使用され、そして画像はCCDカメラと40倍対物レンズを備える落射蛍光顕微鏡で撮像された。NKCC1抗体での免疫染色のため、組織区画は遮断前に1%ドデシル硫酸ナトリウム(SDS)及び8%2−メルカプトエタノールで5分間前処理された(Sung氏他、2000)。この前処理を伴わない多くの試みにおいて、MAbT4抗体で明確なNKCC信号を獲得するのに失敗した。全ての区画はPBS−T(0.2%トリトン−X−100を含むリン酸緩衝食塩水(PBS))内の10%正常ヤギ血清で遮断され、一晩4℃でそれぞれの1次抗体(1:200)と共にインキュベートされた。PBS−Tで広範に洗浄した後、組織区画はCy3接合ヤギ抗マウス又は抗ウサギIgG(1:200、ジャクソン免疫研究所、ウェストグローブ、ペンシルバニア州)内でインキュベートされた。対照は1次抗体の無い同一の処理を受け、そして常にネガティブであった。区画は広範にPBS−T内で洗浄され、観察のためカバーグラスを付けられた。細胞核の対抗染色にはヘキスト33342DNA染色(10ミクロン;Invitrogen,Carlsbad,カリフォルニア州)が使用された。
【0049】
(統計)
平均±標準誤差が別途指定されない限り示される。データのグループ間の統計的比較はシグマプロット(SigmaPlot)ソフトウェアで、必要に応じ独立又は対応のある両側スチューデントのt−検定が帰無仮説を検証するため実行された。0.05未満の確率値Pは統計的に有意であると見做された。
【0050】
(実施例1)
(てんかん重積(SE)後の嗅内皮質(EC)第5層ニューロンにおける多シナプス性バースト応答の発生)
対照ラットからの切片において、嗅内皮質(EC)第5層に対する単一のシナプス前性の刺激が静止膜電位−70mVにおいて振幅20mVまでの単シナプス性の急速な興奮性シナプス後電位(EPSP)を第5層ニューロンに誘発し(図1A)、その後、膜保持電位により振幅が異なる急速な抑制性シナプス後電位(IPSP)を誘発する。対照的に、てんかん重積(SE)後3週間後の記録では、刺激強度を対照調製より大きく減少させた場合でも単一シナプス性刺激に応答して多シナプス性てんかん活性を示した。(図1B)多シナプス性バースト応答は、より高い脱分極電位への活動電位の発火の場合を除いて、DC注入による−95mVから−60mVまでの範囲のシナプス後性膜電位の変化によっては殆ど変化を示さなかった。示された実施例では、弱い刺激での多シナプス性脱分極の僅かな遅れが、初期興奮性シナプス後電位(EPSP)を明白に顕在化させ;そして振幅で僅か1−2mVとして見える。(挿入図、細胞は負のDCにより発火を防ぐため過分極されている。これらの弱い刺激は、高信頼度で多シナプス性バースト応答を、2週目でテストされた10.8%のニューロン(n=37)で,そして3週目でテストされた61.8%のニューロン(n=34)で誘発した(図1C)。この量定のため、多シナプス性バーストは、−75mV<RMP<−68mVから誘発され、半振幅で150ms以上継続するシナプス後性応答として定義された。対照切片では、このような応答は42の記録されたニューロンの内で1つにおいてのみ観察された。
【0051】
図1およびその図に反映されているデータを獲得した実験のさらなる詳細は以下の通りである。
(図1:てんかん重積(SE)後の嗅内皮質(EC)第5層ニューロンの増大した多シナプス性バースト興奮)
A:無処置ラットからの嗅内皮質(EC)第5層ニューロンの単一シナプス前性刺激(*,500nA,70μS)に対するシナプス後性応答。
B:てんかん重積(SE)3週間後の第5層ニューロンのはるかに小さい刺激(*,100nA,70μS、図10及び11も参照)に対する多シナプス性バースト応答。初期に誘起されたシナプス後電位(ボックス内に示す)は、拡大挿入図(30msX5mV)に示される。この記録に対しては、細胞は発火を防ぐため負のDCにより過分極されている。
C:単一の刺激に多シナプス性脱分極で応答する嗅内皮質(EC)第5層ニューロンの比率を示す棒グラフである。単一の刺激は対照ラットから(n=42)の嗅内皮質(EC)第5層細胞の内の僅か2.4%でしか多シナプス性応答を誘起しないが、てんかん重積(SE)後2週間後(n=37)及び3週間後(n=34)のラットではそれぞれ10.8%と61.8%の細胞が多シナプス性応答を誘起した。
【0052】
(実施例2)
(てんかん重積(SE)後、GABAA作動性のシナプス後電位(PSP)が脱分極化する)
長期に渡って確立されたTLEを有する海馬台ニューロン亜集団におけるEIPSP及びCl−輸送体システムの重要な変化の現存する観察(Cohen氏他、2002;de Guzman氏他,2006;Palma氏他,2006;Huberfeld氏他,2007;Munoz氏他,2007;Sen氏他、2007)に依存して、我々は、バースト活性を促進する可能性のある、極めて初期の対象段階におけるGABA作動性伝達の継続的変化を探した。図2Aは、CNQX(10μM)とAPV(50μM)によるグルタミン酸塩受容体の遮断の間の単シナプス性GABAA作動性シナプス後電位(PSP)を示し、そのPSPは電場刺激により嗅内皮質(EC)第5層に誘発され、一方細胞は異なる安定膜電位に保持された(前記(材料及び方法)参照)。PSP逆転電位(E(PSP))は、安定膜電位(図2Bの○)及びPSPのピーク電圧(図2Bの×)の両方の電流−電圧関係をプロットし、線形回帰を計算することにより決定された。膜電位とPSP線形回帰との交点が逆転電位である(図2B、矢印)。対照ラットとSE後2週目と3週目のラットからの代表的ニューロンの電流−電圧プロットは、PSP逆転電位(E(PSP))の進行性変移を示し、それらはそれぞれ、対照の−68.3±2.1mVから、SE後2週目の−52.6±1.8mV、SE後3週目の−34.8±1.4mVであった(図2B1−B3)。データがヒストグラムで示された場合、SE後2週目と3週目の両方で、処置ラットからのスライス内に2つの細胞クラスが観察された(図2C、n=84)。1つのクラスでは平均PSP逆転電位(E(PSP))は対照と類似していた、即ち静止膜電位(RMP)に対し5mV未満ポジティブであった(2週目と3週目に分析した細胞全体のそれぞれ23.5%及び19.2%)。もう1つのクラスは強い変移を示した(2週目と3週目に分析した細胞全体のそれぞれ76.5%及び80.8%)。両方の細胞クラスは同一のラットからの複数のスライス内だけでなく、単一のスライス内にも観察された。SE後3週間で約25%と推定された(前記(方法)参照)屠殺前自発発作は、これらのラットからのニューロンのPSP逆転電位(E(PSP))をさらに変移させ、これによりさらに強く変位したPSP逆転電位(E(PSP))を有する亜集団を確立する可能性が有った。ヒストグラムは、このような顕著なニューロン亜集団のサインを全く示さず、そのことは、自発的発作が全く起らなかったか、あるいはそれがPSP逆転電位(E(PSP))の更なる変移を引き起こさなかったかを示唆した。
【0053】
集団データの棒グラフ(図2D)は、変化が起こらなかった集団の部分を除外した、PSP逆転電位の平均値の時間経過変移をまとめたものである(E(PSP)=静止膜電位(RMP)−71.9±1.46mVに対し3.6±0.5mVポジティブ、n=24;SE後2週目E(PSP)=静止膜電位(RMP)−70.9±2.8mVに対し18.0±3.6mVポジティブ、t56=8.3、P=0.006、n=26;SE後3週目E(PSP)=静止膜電位(RMP)−72.9±2.8mVに対し45.1±6.2mVポジティブ、t48=9.81、P=0.0008、n=21)。3週目の測定点において、変移集団及び非変移集団のそれぞれ81%と75%が錐体ニューロンであり、それぞれ19%と25%が多極性ニューロンであった。これらの分布は、嗅内皮質(EC)第5層のこれら2つの細胞タイプの全体分布から有意に差異がなかった(Egorov氏他、2002;Egorov氏他、2003)。3週目のポジティブな変移がPSP逆転電位(E(PSP))をスパイク開始のレベルにごく近い値に持ってゆくことは注目すべきである。従って、てんかん重積(SE)後のGABA放出は反復性発火を効果的に制限しない可能性が有り、逆にニューロンを興奮させる可能性がある。
【0054】
対照ニューロン内の僅かに脱分極した静止膜電位に対する抑制性シナプス後電位(IPSP)は、明らかにCl−の外向き輸送だけが発現する状態に対して不可解に見えるかもしれない(以下参照)。Cl−の逆転電位は静止膜電位にたいしネガティブ(又はCl−の外向き輸送がCl−の流入の後にのみ活性になった場合、静止膜電位と等しい)でなければならない。しかし、GABAAチャネルは炭酸水素イオンをある程度、陰イオンの約1/6、伝導し、GABA−PSP逆転電位をECl−とEHCO3−との間に置く(Bormann氏他,1987)。細胞内エネルギー代謝は2酸化炭素を生成し、それは炭酸脱水酵素により触媒されて水と逆反応し、炭酸水素イオンを生成する。このプロセスは強い外向きの傾斜をHCO3−に与え、逆転電位を−10mVにまでポジティブ側にする(Ben−Ari氏他,2007)。このことは正常な動物ではシナプス後電位(PSP)逆転電位をECl−よりポジティブな値に変移させる(Staley氏他,1995)。また,Cl−の内側輸送の限定的出現は、僅かに脱分極したシナプス後電位(PSP)逆転電位を生成可能である(以下参照)。
【0055】
逆転電位の変移に加えて、膜接線コンダクタンス、およびコンダクタンスが、シナプス後電位(PSP)がてんかん重積(SE)後に進行的に減少する間、増加する(図2B1−B3参照)。静止コンダクタンスは、対照での5.9±0.7nSから、てんかん重積(SE)後2週間及び3週間でそれぞれ4.8±0.7(t12=3.66、P=0.032)と3.6±0.4nS(t13=3.79、P=0.026)に減少した。これは恐らく漏出イオンチャネルの劣化による(Bernard氏他,2004;Borter氏他,2006)。同様に、GABA活性化ピークコンダクタンスは、対照での8.5±0.69nSから、てんかん重積(SE)後2週間及び3週間でそれぞれ2.1±0.43(t12=13.12、P=0.0018)と0.24±0.07nS(t13=6.16、P=0.0005)に減少した。この減少は、100Hzで3度繰り返された、入出力関係の最大値に近い刺激強度により誘発された、大きなGABA誘発シナプス後電位(PSP)により確認され、それは、3度目の刺激後、3グループ全てに大きな非撹乱PSPを与えた。
【0056】
図2、及びその図に反映されたデータを与えた実験のさらなる詳細は以下の通り。
(図2.てんかん重積(SE)後2週間後と3週間後のGABAA誘発シナプス後電位(PSP)逆転電位のポジティブ変移)
図2A:対照(上側の軌跡)とてんかん重積(SE)後3週間後のラット(下側の軌跡)からの嗅内皮質(EC)第5層内のニューロンの、鋭い微小電極により記録された、5つの異なる膜電位において誘起された、単一シナプス性GABAA誘発シナプス後電位(PSP)逆転電位。膜電位は微小電極からの電流の印加(500m秒パルス)により変化した。*(アスタリスク)はシナプス前性刺激の時点を示す。記録は、AMPA/カイニン酸グルタミン酸受容体拮抗薬CNQX(10μM)及びNメチルDアスパラギン酸(NMDA)受容体拮抗薬APV(50μM)の存在下で行なわれた。
図2B1−2B3:刺激前性膜電位(図内の○印)及び刺激後15m秒後に測定されたGABA誘発シナプス後電位(PSP)の絶対値(図中×印)と電極電流との電流―電圧関係。データは図2Aのプロトコルを使用して獲得され、線形回帰で適合された。膜電位とPSPプロットとの交叉はPSP逆転電位(EPSP,矢印)で起こる。
図2C:測定3時点で2mV未満のGABAA誘発シナプス後電位(PSP)逆転電位を示す嗅内皮質(EC)第5層内のニューロンの数のヒストグラム。包絡線(点線の曲線)は5つの明白な集団のガウス適合である。対照ラットからのニューロンでは、PSP逆転電位(EPSP)ガウス適合は中央値約−69mVを有した。てんかん重積(SE)後2週間及び3週間後のラットからのニューロンは、非変移と有意の変移の2つのクラスに分かれた。2週間目には77%のニューロンが約−54mV、3週間目には81%のニューロンが約−28mVの変移した平均値を示した。非変移ニューロンは集団のそれぞれ23%と19%を占めた。それらのPSP逆転電位(EPSP)分布は対照グループから識別可能であった。対照、SE後2週間後、3週間後の記録された細胞数Nはそれぞれ、24,34及び26であった。
図2D:SE後2週間後(n=26)、3週間後(n=21)の変移集団の静止膜電位(RMP)に対するGABA誘発シナプス後電位(PSP)逆転電位(EPSP)の平均値±標準誤差は、対照と比較して逆転電位の進行性脱分極変移を示す(n=24,SE後2週間後(**)p0.01未満、SE後3週間後(***)p0.001未満)。
【0057】
(実施例3)
(てんかん重積(SE)後NKCC1mRNAの増加)
てんかん重積(SE)後強く脱分極するGABA誘発シナプス後電位(PSP)は、活性Cl−内側方向輸送を必要とする。しかし成体ニューロンには殆どNKCC1の発現がないことが知られている(Delpire,2000;Wang氏他,2002)。我々は、嗅内皮質(EC)第5層内の成人ニューロンで、NKCC1 mRNA発現が、初期の発症段階を再現して、てんかん重積(SE)後の潜伏期に再確立されるか否かを検証した(山田氏他、2004;Ben−Ari氏他,2007)。我々は、従前の研究と適合して、対照ラットの嗅内皮質(EC)第5層内においてNKCC1 mRNA蛍光発光が非常に低いか存在しないことを発見した。対照的に、リチウム−ピロカルピン誘発てんかん重積(SE)後、2週間後、特に3週間後では、明瞭なNKCC1染色を確認した(図3B)。てんかん重積(SE)後、2週間後では、NKCC1 mRNA蛍光発光が対照の237±33.4%に増大し、3週間後では452±33.5%に増大した(図3B、E、n=100注目画像領域(ROI)、前記(方法)参照、各グループ4ラット、それぞれt38=2.15、P=0.037及びt49=3.54、P=0.0009)。これらのパーセントは、対照における発現が低いことを前提にすると、幾分任意であるが、発現の非常に大きな増加を示している。DAPIによる核の対抗染色は、分析期間中には細胞数に変化を与えなかった(対照、SE後2週間後及び3週間後のそれぞれにおいて、12.2±2.7,12.8±3.4及び12.5±3.1細胞核数/500平方ミクロン、それぞれt68=−0.49,P=0.62及びt68=−2.38,P=0.24、n=100注目画像領域(ROI)、各グループ4ラット;図3A2、B2)。DAPI及びNKCC1 mRNAの画像の重なりは標識化mRNAの初期の核周囲の位置を示す(図3B2)。NKCC1ポジティブ細胞と場内の全細胞との比較分析では、対照組織では細胞の5.1±2.1%の標識化に対して、SE後2週間後及び3週間後のそれぞれにおいては28.5±6.7%及び56.7±4.3%を示した(図3D、n=100注目画像領域(ROI)、各グループ4ラット、それぞれt38=3.27,P=0.042及びt49=2.71,P=0.0096)。
【0058】
図3、及びその図に反映されたデータを与えた実験のさらなる詳細は以下の通り。
(図3.てんかん重積(SE)後嗅内皮質(EC)第5層内において進行的に増大する、NKCC1のmRNA、Cl−内側方向輸送)
図3A、B:蛍光遺伝子プローブ法(FISH)は、ピロカルピン誘発発作の3週間後にNKCC1のmRNAが対照(赤い信号、A1,B1)に比較して発現量の増大を示した。DAPI核染色は細胞数に変化の無いことを示した(青の信号、図3A2、図3B2)。下方パネルA3、B3は図3A1,2及び図3B1,2で概観された領域の拡大統合図であり、NKCC1のmRNAの核周辺配置を示す。
図3C:てんかん重積(SE)後のNKCC1のmRNAの増加を示す棒グラフである。データは、所定の注目画像領域(ROI)内の全NKCC1信号に対する核染色の蛍光の比率を生成することにより定量化された。これらの比率はその後、対照ラットからの値に対して正規化された(平均値±標準誤差、n=100注目画像領域(ROI)、各グループ4ラット、SE後2週間後(*)p0.01未満、SE後3週間後(***)p0.001未満)。
図3D:DAPI染色により決定される、核の数に対する%として表現される、NKCC1のmRNA−ポジティブの細胞数の平均値(平均値±標準誤差、n=約300注目画像領域(ROI)、各グループ4ラット、SE後2週間後(*)p0.05未満、SE後3週間後(**)p0.01未満)。
【0059】
(実施例4)
(NKCC1タンパク質の増加)
潜伏期間中に起こったNKCC1のタンパク質レベルの変化を分析するため、蛍光免疫組織化学的検査を実施した。これらのデータはケミコン(Chemicon)社NKCC1抗体によって得られ、そして図4に示すように、SE後2−3週間において、NKCC1タンパク質の進行的増加を示した。一方対照ラットからのスライスでは、特定の信号はほとんど得られなかった(図4A−4C)。同様の結果がMabT4 NKCC抗体で得られた。NKCC1画像の上に染色核画像(ヘキスト33342)を重ね合わせることにより、特にSE後3週間後において、NKCC1の優先的な核周囲の、そして細胞体性の位置が明らかにされ、ニューロン性タンパク質合成の有意の増加を示唆した(図4B3、4C3及び拡大された図4B4,4C4)。繰り返すが、ヘキストで染色された核の数は3つのラットグループの間で有意の差異を示さなかった(対照85.3±5.3に対し、発作後2週間後は97.7±3.4、発作後3週間後は86.0±4.9、4ラット、それぞれt20=−1.33,P=0.20及びt18=−2.35,P=0.81、n=100注目画像領域(ROI)、各グループ4ラット)。図4Dは細胞体からのNKCC1の蛍光強度の平均値は、発作後2週間後及び3週間後に対照の背景蛍光の約350%及び860%であったことを示す(n=100注目画像領域(ROI)、各グループ4ラット、それぞれt64=5.32,P=0.0006及びt51=4.56,P=0.0008)。神経線維網ではNKCC1の平均蛍光強度は、発作後2週間後及び3週間後に対照の背景蛍光のそれぞれ123±21%及び222±45%であった、その結果は樹状突起内においてNKCC1タンパク質が遅延して増加することを示唆した(n=100注目画像領域(ROI)、各グループ4ラット、それぞれt32=4.25,P=0.025及びt36=5.31,P=0.008)。個々の細胞に関しては、SE後2週間後と3週間後で、それぞれ細胞体の31.2±6.9%及び56.3±11.2%が明確にNKCC1に対しポジティブであったが、対照では明確なものは無かった(図4E、n=100注目画像領域(ROI)、各グループ4ラット、それぞれt42=11.24,P=0.0009及びt37=7.12,P=0.0007)。
【0060】
図5ではニューロン特異ニューロトレース(Neuro Trace)対抗染色が使用され、発作後3週間後には77.1±8.1%のニューロンがNKCC1ポジティブになったことを示した(対照組織:3.0±1.3%、各グループn=3ラット、それぞれt21=3.78,P=0.008及びt33=4.23,P=0.001)。NKCC1ポジティブのニューロンの比率がNKCC1ポジティブの細胞の比率より高いことが、NKCC1がニューロンにおいて増大していることを示した。ニューロトレース(Neuro Trace)ポジティブな細胞数は実験期間中安定し、(対照の35.4±7.2に対し、発作後2週間後は37.1±6.4及び3週間後は36.6±6.7、各グループ3ラット、それぞれt12=−0.51,P=0.6及びt16=−0.30,P=0.8)、それは安定的なH33342細胞数がニューロン損出とグリア細胞の増大との相殺の結果ではないことを示唆した。NKCC1発現及びポジティブニューロン比率の漸増は、GABAA誘発シナプス後電位(PSP)逆転電位の同時発生的進行性脱分極変移を説明するのによく適合した。
【0061】
図4,5、及びそれらの図に反映されたデータを与えた実験のさらなる詳細は以下の通り。
(図4.NKCC1タンパク質も増加した。)
図4A、4B、4C:蛍光免疫組織化学的検査は、嗅内皮質(EC)第5層内においてSE後2−3週間に、NKCC1タンパク質の発現の進行的増加を示した(赤色信号、A1,B1,C1)。ヘキスト(Hoechst)33342核対抗染色は安定的細胞数を示した(A2,B2,C2,緑色信号)。上記パネルセットの統合図(A3,B3,C3)は発現の拡散的増加を示す。これらパネルのボックスに囲まれた領域は、NKCC1の核周辺及び細胞体系の位置をより明示するため、拡大されて下の図に示される(A4,B4,C4)。
図4D:SE後のNKCC1タンパク質の増加を示す棒グラフである。データは、所定の注目画像領域(ROI)におけるNKCC1信号合計に対する核染色の蛍光合計の比率を形成して定量化された。その後これらの比率は対照ラットからの値で正規化された(平均値±標準誤差、3ラット、SE2週間後(**)ではpは0.01未満、SE3週間後(***)ではpは0.001未満)。
図4E:核数の%で表示されるNKCC1ポジティブ細胞の平均数(H33342、平均値±標準誤差、3ラット、SE2週間後及び3週間後(***)ではpは0.01未満)。
【0062】
(図5.SE後、嗅内皮質(EC)第5層内ニューロンがNKCC1タンパク質の発現を増加)
図5A、B:蛍光免疫組織化学的検査は、嗅内皮質(EC)第5層内においてSE後3週間に、NKCC1タンパク質の強い発現を示した(赤色信号、A1,B1)。ニューロン特異ニューロトレース(Neuro Trace)対抗染色は安定的細胞数を示した(A2,B2,緑色信号)。統合図はNKCC1の核周辺及び細胞体系の位置を示す(A3,B3)。ここでは厚さ60ミクロンの組織ブロック中のニューロンが示される。
図5C:ニューロトレース(Neuro Trace)に対する%で示されるNKCC1ポジティブニューロンの平均値であり、嗅内皮質(EC)第5層内においてピロカルピン誘発SE後2週間後及び3週間後に進行的に増加している(平均値±標準誤差、3ラット、SE3週間後(***)ではpは0.01未満)。
【0063】
(KCC2 mRNAの進行的減少)
示されたNKCC1の増加は活性Cl−の摂取を示唆する。ニューロンのKCl共輸送体KCC2による正常型のCl−の放出(Ben−Ari氏他、2007)は、このCl−の摂取と対抗し、そして大なり小なりCl−の摂取を妨げる。もしKCC2出現が維持されれば、それはCl−の摂取の効率を限定し、そして代謝エネルギーを浪費する。これが事実であることを検証するため、KCC2 mRNAの発現を研究した。
【0064】
上記のように、我々は嗅内皮質(EC)第5層内においてKCC2 mRNAの発現レベルを蛍光遺伝子プローブ法により評価した。図6はKCC2 mRNAが対照動物(A1)において多量に発現し、一方SE3週間後(B1)には減少することを示す。核のDAPIによる染色は3時点の間に有意の差異を示さなかった。(11.9±2.9,1±2.33.8及び12.1±3.2核数/500μm2、それぞれt24=−0.36,P=0.17及びt24=−0.41,P=0.24、図6A2、6B2)。統合蛍光画像(図6A3、6B3)は、KCC2 mRNAが核内及び核周辺領域内に配置されることを示した。
【0065】
対照に対し正規化されたKCC2 mRNAの蛍光は、SE後2週間後及び3週間後においてKCC2 mRNA発現がそれぞれ対照の81±8.3及び44±7.8%に進行的に減少することを示した(図6C、nは約100注目画像領域(ROI)、(方法)参照、各グループ4ラット、それぞれt62=1.95,P=0.008及びt62=2.34,P=0.0095)。同様に、核数に対する嗅内皮質(EC)第5層内のKCC2 mRNAを発現する(図6A3、6B3に示すように核の周りの少なくとも2つの赤色の点)細胞数の比率は、対照動物の嗅内皮質(EC)第5層内における全細胞数の63.9±8.5%から、SE2週間後及び3週間後にそれぞれ47.3±6.7及び36.3±4.3%に減少した(図6D、平均値±標準誤差、nは約300細胞、各グループ4ラット、それぞれt34=3.14,P=0.045及びt28=4.21,P=0.007)。
【0066】
図6、及びそれらの図に反映されたデータを与えた実験のさらなる詳細は以下の通り。
(図6.KCC2に対するmRNA,ニューロンのCl−の外向き輸送体は、嗅内皮質(EC)第5層内においてSE後進行的に減少する、A、B)
蛍光遺伝子プローブ法で検知されたKCC2 mRNAの発現は、対照と比較してピロカルピン誘発SE後3週間後においてKCC2 mRNAの減少を示した(A1、B1、赤信号)。DAPI核染色は細胞数が変わらないことを示唆した(A2,B2,青色信号)。図A1,A2,及びB1、B2で囲った領域の拡大統合図は、KCC2 mRNAの核周囲の配置を示す(A3、B3)。
図6C:SE後のKCC2 mRNAの減少を示す棒グラフである。データは、所定の注目画像領域(ROI)におけるKCC2信号合計に対する核染色の蛍光合計の比率を形成して定量化された。その後これらの比率は対照ラットからの値で正規化された(平均値±標準誤差、3ラット、SE2週間後(**)ではpは0.01未満、SE3週間後(***)ではpは0.001未満)。
図6D:核数の%で表示されるKCC2ポジティブ細胞の平均数(DAPI、平均値±標準誤差、nは約300細胞、各グループ4ラット、SE2週間後(*)及びSE3週間後(**)では、pはそれぞれ0.05未満及び0.01未満)。
【0067】
我々はまた、対照ラットとSE後ラットからの嗅内皮質(EC)第5層内におけるKCC2タンパク質レベルを、ウェスタンブロット法と単一細胞分析を使用して調査した。図7A,7Bに示すように結果は、KCC2タンパク質レベルが、SE2週間後及び3週間後にそれぞれ対照レベルの78.8±7.5及び38.1±4.3%へと、有意な減少を示した(平均値±標準誤差、n=5ラット、それぞれt12=2.17,P=0.01及びt18=4.6,P=0.0006)。蛍光免疫組織化学的検査を使用して、KCC2タンパク質の分布及び処置後のその消失について研究した(図7C)。対照ラットでは、KCC2の免疫活性は、散在する神経線維網信号の上の密集した細胞体系の蛍光として検知された(図7C、一番上のパネル、及び挿入図)。SE後2週間及び3週間後では免疫蛍光はニューロンの細胞体内で漸減し(図7C、7D)、嗅内皮質(EC)第5層内ニューロンの細胞体系領域に弱い離散した蛍光が残った(図7C、真中及び一番下のパネル、及び挿入図)。KCC2に対する細胞体性の蛍光は、SE2週間後及び3週間後にそれぞれ対照レベルの66±3.5及び34±1.9%に減少した(図7D、平均値±標準誤差、nは4ラットに対して約100注目画像領域(ROI)、それぞれt34=7.85,P=0.007及びt41=6.39,P=0.00006)。
【0068】
神経線維網において、KCC2信号減衰はさらに遅延する。ここではKCC2信号は2週間及び3週間後にそれぞれ96±4.1%及び60±4.4%に減少した。神経細胞体と神経線維網の体積を概略1:1と推定すると、KCC2免疫蛍光の合計の変化はウェスタンブロットのデータとよく一致する。ニューロンが死滅せず、かつグリアに置き換わることを証明するため、ニューロン特異性の蛍光ニューロトレース染色が対抗染色に使用された。これら染色はニューロンの安定的な数(対照が157.6±6.5に対して、発作後3週間後で154.6±21.3、3ラット、t26=2.34,P=0.44)を示し、そしてKCC2信号がニューロンに高度に特異的(KCC2ポジティブ細胞の95%超がニューロトレースポジティブ)であることを示した。興味深いことに、ピロカルピン多量投与モデルでは、嗅内皮質(EC)からのてんかん様活性の海馬への拡散に対するこの構造のゲートキーパー機能を損なう、下流歯状回におけるKCC2の早期、機能的減少の証拠もある(Pathak氏他、2007)。このゲートキーパー機能の損傷と同時に、生体内てんかん様脳波(EEG)活性の証拠もある(El−Hassar氏他、2007)。
【0069】
図7、及びその図に反映されたデータを与えた実験のさらなる詳細は以下の通り。
(図7.SE後KCC2タンパク質も減少する)
図7A:ウェスタンブロットKCC2バンド(約150kDa)は、ピロカブリン誘発SE後2週間及び3週間後のラットの嗅内皮質(EC)第5層内KCC2が対照と比較して有意に漸減することを示す。βアクチン(約40kDa)は負荷制御として機能した。
図7B:図7Aのブロットからの、対照の%で表示される平均光学密度(平均値±標準誤差、n=5、SE2週間後(**)ではpは0.01未満、SE3週間後(***)ではpは0.001未満)。これらのデータはmRNA及び抑制性シナプス後電位(IPSP)逆転電位データと非常によく一致する。
図7C:免疫組織化学的検査はピロカブリン誘発SE後2週間及び3週間後のKCC2タンパク質の漸減を示し、それは細胞体で最も顕著であった。各パネルの白抜きのボックスは、白矢印で識別される細胞の拡大図である。
図7D:SE後のKCC2免疫組織化学的検査の平均信号で、対照に対して正規化されている(平均値±標準誤差、nは4ラットに対して約100注目画像領域(ROI)、SE2週間後(**)ではpは0.01未満、SE3週間後(***)ではpは0.001未満)。
【0070】
(実施例7)
(NKCC1及びKCC2における初期の変化は嗅内皮質(EC)第5層に特異である)
てんかん重積(SE)中に発生する高振幅脳波(EEG)スパイキングは、海馬及び海馬台を含む他の皮質層の嗅内皮質(EC)第5層で見られるNKCC1及びKCC2出現における進行性変化をトリガーするのに十分でありうる。従って我々は、分析を海馬台、歯状回、嗅周囲皮質だけでなく、嗅内皮質(EC)の浅葉も含むように拡げた。 図8A,Bは拡大率の低い概観で、SE後3週間の嗅内皮質(EC)深層のNKCC1 mRNA発現を示し、一方、浅葉、海馬台、歯状回、嗅周囲皮質では発現しなかった。
これらの領域からの蛍光の平均値のプロット(図8C、倍率のより大きい画像から、対照に対し正規化)は、NKCC1 mRNAの深層内側嗅内皮質(EC)にこの高度に特異的な出現を示し、それよりずっと弱い程度に深層外側嗅内皮質(EC)に出現し、一方他のテスト領域ではネガティブであった(図8C、コラム3−10)。
【0071】
同様の特異性が関連するKCC2出現の研究において観察された。図9A,Bの概観は、SE後3週間において、KCC2 mRNAが特異的に深層嗅内皮質(EC)、特に内側部分で発現するのがわかる。平均蛍光の定量的分析では、KCC2 mRNAの深層内側嗅内皮質(mEC)における進行的減少が56±7.8%(SE後3週間、t62=2.34,P=0.0095)深層外側嗅内皮質において14±4.5%(SE後3週間、t62=2.41,P=0.039)であり、一方海馬台、歯状回、嗅周囲皮質及び嗅内皮質(EC)浅葉では有意の変化を示さなかった(図9C)。
【0072】
拡張的実験のさらなる重要な結果は、「SE発症2時間超後のジアゼパム投与」プロトコル(Du氏他,1995;小林氏他、2003)に対抗してここで使用されるプロトコル「SE発症1時間後ピロカプリンの低量投与−ジアゼパム」後の初期のニューロンの有意の減少が第3層に存在しないことである。第1に嗅内皮質(EC)第3層においてKCC2 mRNAの有意の変化の不存在は、我々のラットでは第3層の錐体ニューロンがSE後3週間後も死滅していないことを示唆する。第2に、ニューロンの生存が、第3層の安定的ニューロン数を示した(対照、SE後2週間後及び3週間後においてそれぞれ、ニューロン数114±14.2,112±18.1及び116±15、それぞれt10=−0.18,P=0.86及びt10=−0.77,P=0.46、3ラット)ニューロトレース(Neuro Trace)染色で確認された。
【0073】
図8,9、及びそれらの図に反映されたデータを与えた実験のさらなる詳細は以下の通り。
(図8.NKCC1のmRNAは他の皮質領域では増加しない)
図8A,B:内側嗅内皮質(mEC)、外側嗅内皮質(lEC)、アンギュラーバンドル(Angular bundle)(ab),海馬台(Sub)、歯状回(DG)及び嗅周囲皮質(PRC)に亘る脳領域を示すNKCC1 mRNAに対する蛍光遺伝子プローブ法(FISH)研究:
A、対照スライス;B、SE後3週間後に採取されたスライス。左のパネルは、DAPI染色された領域を示し、それは構成要素領域のアウトラインが重ねて表示されている(A1,B1)。中央のパネルはNKCC1信号を示す。対照では殆ど見られないか皆無であるが、SE後3週間後では信号は中央の嗅内皮質(EC)第5−6層に明白に存在するが、他の場所では存在しない(A2、B2)。これらの画像は右パネルで統合される(A3,B3,高出力画像は図3参照)。
図8C:SE後にNKCC1 mRNAが、深層嗅内皮質(EC)、(内側嗅内皮質(mEC)第5層、それぞれt54=2.15,P=0.037及びt54=3.54,P=0.0009)及び#2とラベル付けされた外側嗅内皮質第5層(それぞれt54=1.02,P=0.12及びt54=1.28,P=0.047)においてのみ増加するが、内側嗅内皮質(mEC)第3層(#3、それぞれt54=0.93,P=0.24及びt49=0.42,P=0.34)、外側嗅内皮質(lEC)(#4、それぞれt54=0.27,P=0.86及びt54=0.24,P=0.094)内側嗅内皮質(mEC)第1−2層(#5、それぞれt54=−1.25,P=0.32及びt54=0.34,P=0.23)、外側嗅内皮質(lEC)第1−2層(#6、それぞれt54=1.24,P=0.078及びt54=0.52,P=0.27)、海馬台(#7、それぞれt54=1.54,P=0.49及びt54=1.67,P=0.063)、歯状回(#8、それぞれt54=0.45,P=0.098及びt54=−1.35,P=0.26)、深層嗅周囲皮質(#9、それぞれt54=0.53,P=0.47及びt54=2.01,P=0.48)及び嗅周囲皮質(PRC)第1−3層(#10、それぞれt54=−1.36,P=0.81及びt54=0.28,P=0.81;データは高倍率画像より抽出)では、有意の変化が起こらなかったことを示す。
【0074】
(図9.KCC2のmRNAは深層嗅内皮質(EC)でのみ減少する)
図9A、B:内側嗅内皮質(mEC)、外側嗅内皮質(lEC)、アンギュラーバンドル(Angular bundle)(ab),海馬台(Sub)、歯状回(DG)及び嗅周囲皮質(PRC)に亘る脳領域を示すKCC2 mRNAに対する蛍光遺伝子プローブ法(FISH)研究:
A、対照スライス;B、SE後3週間後に採取されたスライス。左のパネルは、KCC2染色された領域を示し、それは構成要素領域のアウトラインが重ねて表示されている(A1,A2)。対照では全体に発現が見られるが、SE後3週間後では嗅内皮質(EC)第5層でのみ信号が消滅した。中央のパネルはDAPI染色を示す(A2、B2)。これらの画像は右パネルで統合される(A3,B3,高出力画像は図6参照)。
図9C:SE後にKCC2 mRNAが、深層嗅内皮質(EC)、(内側嗅内皮質(mEC)第5層、それぞれt62=1.95,P=0.082及びt62=2.34,P=0.0095)及び#2とラベル付けされた外側嗅内皮質第5層(それぞれt62=1.86,P=0.093及びt62=2.41,P=0.039)においてのみ増加するが、内側嗅内皮質(mEC)第3層(#3、それぞれt62=0.34,P=0.32及びt62=−1.22,P=0.51)、外側嗅内皮質(lEC)(#4、それぞれt62=−0.25,P=0.17及びt62=0.24,P=0.36)内側嗅内皮質(mEC)第1−2層(#5、それぞれt62=1.06,P=0.85及びt62=0.43,P=0.62)、外側嗅内皮質(lEC)第1−2層(#6、それぞれt62=−0.45,P=0.68及びt62=0.15,P=0.39)、海馬台(#7、それぞれt62=−1.01,P=0.098及びt62=1.84,P=0.065)、歯状回(#8、それぞれt62=1.05,P=0.77及びt62=−2.03,P=0.49)、深層嗅周囲皮質(#9、それぞれt62=1.33,P=0.28及びt62=−1.52,P=0.53)及び嗅周囲皮質(PRC)第1−3層(#10、それぞれt62=1.12,P=0.085及びt62=0.36,P=0.14;データは高倍率画像より抽出)では、有意の変化が起こらなかったことを示す。
【0075】
(実施例8)
(Cl−輸送遮断抑制バースト活性)
抑制性シナプス後電位(EIPSP)の変移及び発作後の多シナプス性バースト応答の増加に対するNKCC1増加の機能的役割を調査するため、我々はCl−輸送体遮断を通して作用し、NKCC対KCC輸送体の遮断により効果のある利尿剤ブメタニドを採用した(Russel,2000;Sung氏他,2000;Hannaert氏他,2002;Beck氏他,2003)。ここで絶対的特異性の欠如は1)示されたKCC2の強い減少、2)Cl−内向き輸送のみがEClをポジティブに静止膜電位(RMP)に変移させるという事実、により補償される。残存KCC2活性の遮断は、NKCC1の遮断の間、EClの強い過分極変移を阻止しない。図10AはSE後3週間後の膜電圧に対するシナプス後電位(PSP)振幅のプロットであり、E(PSP)の脱分極変移がブメタニド(10μM)での20分灌流の後、消滅したことを示す。これは、Cl−の膜を通過する受動的再分布に対応する。図10BはSE後3週間後の嗅内皮質(EC)第5層の正常記録状態における静止膜電位(RMP)(約−72mV)に対するシナプス後電位(PSP)逆転電位の平均値が+40MV超であり、ブメタニド存在下では+10mVに低下した(E(PSP)絶対平均電圧は、対照状態で−29.7±2.3mV、ブメタニド存在下で−60.9±4.1mVであり(t24=2.26,P=0.00009)、ブメタニドの洗い流し中は−34.23.4mV、n=5,t24=1.52,P=0.0057対ブメタニド)。
【0076】
シナプス後電位E(PSP)の脱分極に対して2次性の脱抑制又は興奮が、SE後の潜伏期における多シナプス性バースト応答の発現に決定的な場合、ブメタニドによる逆転電位の蓄積もまた、多シナプス性バースト応答を遮断することになる。図10Cは、確かに、ブメタニドでの灌流(10μM)は単一ショック刺激に対する多シナプス性応答を効果的に抑制した。図10Cの上段パネルに示すような強い多シナプス性バースト応答を記録した後、ブメタニドでの灌流が始まり、20分後までには、バースト放電は強く減衰した(中段の軌跡)。30分間のブメタニドの洗い出しにより、強い多シナプス性応答が回復した(下段の軌跡)。平均すると、ブメタニドは、ピロカルピン発作後3週間後のラットからの細胞の多シナプス性応答を持つニューロンの数を、64.8%(合計14ニューロンの内9ニューロン)から11.6%(2/14)に減じた。30分間のブメタニドの洗い出しは応答ニューロンの数を35.1%(5/14、図10D)に回復した。半最大振幅における応答期間はブメタニドで対照応答(t13=13.8,P=0.0000000004)の36±9%に減少し、そして洗い出し中に75±11%(t13=−5.07,P=0.0002対ブメタニド)に回復した。
【0077】
図10、及びその図に反映されたデータを与えた実験のさらなる詳細は以下の通り。
(図10.NKCC1抑制剤ブメタニドは部分的にE(PSP)を回復し、そして多シナプス性興奮を抑制する)
図10A:SE後3週間後のラットからの嗅内皮質(EC)第5層ニューロンの膜電位に対するシナプス後電位(PSP)振幅をブメタニドへの20分暴露前(上段パネル、白丸)と後(下段パネル、×印)でプロットしたものであり、PSP逆転電位の過分極変移を示す。データは図2Aのプロトコルから得られた。E(PSP)は−29.7mVから−60.9mVへ変移した。
測定はCNQX(10μM)及びAPV(50μM)による興奮性シナプス後電位(EPSP)遮断中に行われた。
図10B:集団データで、ポジティブ変移したGABAA−PSP逆転電位が、ブメタニドにより強く、かつ可逆的に再分極した(平均値±標準誤差、n=3、ブメタニドへの暴露(***)pは0.001未満)ことを示す。
図10C:最上段の記録は、SE後3週間後に観察された単一刺激(100nA)に対する多シナプス性応答を示し、それがブメタニドにより遮断されるが(中段の記録)、30分間の薬物の洗い出しにより回復する(下段の記録)ことを示す。
図10D:ブメタニドは、単一刺激に応答して多シナプス性脱分極を発現するSE後3週間後の嗅内皮質(EC)第5層ニューロンの数を、64.8%から11.6%(n=14)に減少させることを示す集団データである。この数値は30分間のブメタニド洗い出しにより35.1%に回復した。
【0078】
(実施例9)
(GABAA誘発興奮はバーストに軽度の影響をあたえる)
シナプス後電位(PSP)逆転電位の脱分極変移は、第5層ニューロンの脱抑制及び直接的GABA誘発興奮の両方に繋がる。我々はピクロトキシンでGABAA誘発興奮を遮断してバースト応答における直接的興奮の役割を調べた。ブメタニドと異なり、ピクロトキシン(100μM)はSE3週間後の多シナプス性バースト放電に対し僅かな効果しか示さなかった(図11A1,A2;ピクロトキシンでは、自発的バーストも観察した;自発的バースト後5秒より長く記録された誘発バーストのみ分析した;これらのバーストは明白に従前の自発的バーストに影響されない)。研究された細胞集団において、ピクロトキシンは、SE3週間後に多シナプス性バースト応答におけるスパイクの数を9.8±0.8から7.9±0.7へ減少させた(図11C、n=8,t24=2.26,P=0.000089)。このような同一細胞からの10個の応答の平均をとると、図11B1,B2に示すように、ピクロトキシンが平均バースト応答において活動電位振幅を10.5±3.4%減少させ、それは刺激後の活動電位のタイミングの可変性が増大したことを示唆する(n=8,p=0.094)ことが判った。このように、GABA誘発興奮の存在は、明白にネットワークの発火のタイミング精度を増大させ、それは、GABA誘発伝達がニューロン間興奮の下流であるため遅延するので、恐らく直接的興奮よりは全体的な興奮性の増大に起因している。
ピクロトキシン単独での僅かな効果と対照的に、CNQX(10μM)及びAPV(50μM)のACSF/ピクロトキシン生理食塩水への追加は、ほぼ完全にシナプス応答を遮断し(95.9±0.26%、t7=−10.05,P=0.00002)、それはバーストが主として多シナプス性グルタミン酸作動性伝達により媒介されることを示した(図11A3,B3;図2Aで示すように、ピクロトキシンが無いCNQX及びAPV内で、単一シナプス性PSPが誘起された)。ピクロトキシンでは統計的有意性に達しないが、バースト期間の短縮傾向が存在した(図11D)。
)。
【0079】
図11、及びその図に反映されたデータを与えた実験のさらなる詳細は以下の通り。
(図11.ピクロトキシンは多シナプス性バーストに軽度の影響をあたえる)
図11A:ピクロトキシン(100μM)によるGABAA受容体の遮断が多シナプス性バースト応答における活動電位発火を、小さいが有意な量だけ減少させた(図A1,A2;刺激100nA、70μ秒が*印時点で印加された)。その後のCNQX(10μM)及びAPV(50μM)によるイオンチャネル型グルタミン酸受容体の遮断は、全てのシナプス後性応答を殆ど除去した(図11A3)。
図11B:同一の細胞からの10個のバーストの平均がピクロトキシンでのバースト期間の短縮化を示した。これら平均におけるスパイク振幅の減少は、ピクロトキシンでのバーストにおいて発火パターンの精度が低下することを示唆する。
図11C:ピクロトキシンが、多シナプス性バースト応答における平均スパイク数を、1応答当り9.8±0.8から7.9±0.7へ減少させる(平均値±標準誤差、n=8細胞、5ラット、ピクロトキシン付加(*)pは0.05未満)ことを示す数量データである。
図11D:ピクロトキシンでは多シナプス性バースト期間の10.5%短縮傾向が見られた(平均値±標準誤差、n=8、p=0.094)。
【0080】
図12−14、及びそれらの図に反映されたデータを与えた実験のさらなる詳細は以下の通り。
図12は、リチウム−ピロカルピンSE後12−17週間後における、ブメタニドによる発作の抑制(0.2mg/Kg体重、腹腔内)を示す(n=5ラット)。5体のリチウム−ピロカルピンSEラットが連続するブメタニド又は媒体注射(生理食塩水内の5%エタノール、腹腔内、下部に日付を記載)を受け、そしてその後8時間以内に発現する発作がビデオ録画により分析された。1体の同一のラットは、ブメタニドがCl及びIPSP逆転電位を概して基準化した場合でも、2度目及び3度目のブメタニド投与後に発作を発現したが、それは恐らく自発的発作を可能にするこのラット内の2次的な下流の変化によるものである。
【0081】
図13は、NKCC1タンパク質が正常な成体レベルに向かって回復することを示す。プロットは、深層内側嗅内皮質(EC)のNKCC1タンパク質の免疫蛍光信号を、抗体を持たない対照ラット(「免疫グロブリンG(IgG)無し」と表示)、抗体を持つ対照ラット(「対照」と表示)、SE後2週間後及び3週間後(「SE 週間後」と表示)のラットにおいて示す。SE後17週間でNKCC1信号は、ブメタニドが完全に自発的再発性発作(SRS)を防止するラット(「ブメット(Bumet)ポジティブ」と表示)では、SE後3週間後のラットのレベルの50%未満まで回復し、そしてブメタニドが自発的再発性発作(SRS)を防止する効果が無くなったラット(「ブメット(Bumet)ネガティブ」と表示)では95%超に回復した。
【0082】
図14は、KCC2タンパク質が正常な成人レベルに向かって回復することを示す。プロットは、深層内側嗅内皮質(EC)のKCC2タンパク質の免疫蛍光信号を対照ラット(「対照」と表示)、SE後2週間後及び3週間後(「SE後 週間」と表示)のラット、及びSE後17週間後のラットにおいて示す。SE後17週間でKCC2信号は、正常レベルに向かって回復した(ブメタニドが完全に自発的再発性発作(SRS)を防止するラットは「ブメット(Bumet)ポジティブ」と表示、そして、ブメタニドが自発的再発性発作(SRS)を防止する効果が無くなったラットは「ブメット(Bumet)ネガティブ」と表示)。
【0083】
ブメタニド又は媒体が投与された後の発作活性が図12で定量化された、ラットの新規のNKCC1データが図13に示され、図13は従前のNKCC1データを比較のために含む。抗体を持たない対照の測定(「免疫グロブリンG(IgG)無し」と表示)はさらに、対照ラットにおけるNKCC1タンパク質レベルがゼロに近く非常に低いことを示す。SE後17週間でNKCC1タンパク質レベルは、ブメタニド投与が完全に自発的再発性発作(SRS)を防止したラット(「ブメット(Bumet)ポジティブ」と表示)において正常レベルに向かって有意に回復した。2度目及び3度目のブメタニド投与(約4週間離れて09年6月8日と09年7月1日に)後に発作を発現した1体のラットでは、NKCC1はほぼ完全に基準化された。
【0084】
図14はKCC2タンパク質レベルも、SE後17週間後に正常な成体レベルに回復したことを示す。これらのデータは、NKCC1とKCC2のCl−輸送体の発現は共に長い時間の中で基準化され、従ってCl−分布の基準化およびCl−依存性IPSP逆転電位に関するシナプス性抑制につながるという、我々の仮説を確認した。
【0085】
図15は、海馬の錐体細胞CA1層又はCA3層では、NKCC1のmRNAはSE2週間後及び3週間後に有意の変化がない(「SE後2週間」、「SE後3週間」と表示)ことを示す。棒グラフは、海馬の錐体細胞CA1層又はCA3層では、NKCC1のmRNAはSE2週間後及び3週間後に変化しないことを示す。NKCC1のmRNAの蛍光遺伝子プローブ法(FISH)データは、図8に使用されたものと同一の組織の高倍率画像から抽出された。
【0086】
図16は、海馬の錐体細胞CA1層又はCA3層では、KCC2のmRNAは変化がないことを示す。棒グラフは、海馬の錐体細胞CA1層又はCA3層では、NKCC1のmRNAはSE2週間後及び3週間後(「SE後2週間」、「SE後3週間」と表示)に変化しないことを示す。NKCC1のmRNAの蛍光遺伝子プローブ法(FISH)データは、図9に使用されたものと同一の組織の高倍率画像から抽出された。
【0087】
図17:ニューロン−特異染色「ニューロトレース(NeuroTrace)」で染色されたニューロンの数に、嗅内皮質(EC)第2層(「EC第2層」と表示)、又は第3層、又は第5層においても、SE後3週間後に有意の変化が無いことを示す(ニューロンの数/100ミクロンx100ミクロン領域)。
【0088】
図18は慢性ブメタニド処置が慢性期において強く発作の発現を抑制することを示す。SE後の週単位時間経過に対する発作発現/8時間ビデオ録画のプロットであり、無処置ラット(黒塗りの棒、水消費 平均±標準誤差=水29±6.3ml/日)及び飲料水によりブメタニド(bumet.)を処置されたラット(平均±標準誤差=0.65±0.13mgブメタニド/日;水32±6.4ml/日;点線及び斜線)。4体のラットではブメタニドは完全に発作を防止した(中空の棒)が、他の2体のラットでは、発作の発現及びSE後時間経過に伴う発作の進行は共に減少した(斜線の棒)。
【0089】
図19:SE後2週間後に開始され20週間継続する慢性ブメタニド処置が、処置期間中および処置終了後少なくとも6週間、発作の発現を強く抑制する。棒グラフは、SE後第11−21週における8時間ビデオ録画セッション中のラット当りの平均発作発現回数を示し(一番左の棒のペア)、無処置ラット(黒)又はSE後2週間目から20週間に亘って慢性的に飲料水経由でブメタニド処置されたラット(斜線)である。その後ブメタニド処置を終了し、その後3週間後と6週間後の発作発現回数を示す、無処置ラットは約3回の発作/8時間録画のレベルに到達した(黒の棒、ラット当り3回の録画セッション)が、一方事前にブメタニド処置されたラットでは発作の発現はずっと少なかった(斜線の棒)、即ち、SE後時間適合対照SEラットが発現する発作回数の15−22%であった(各グループn=6ラット)。
【0090】
図20は、SE後1週間後に開始する慢性ブメタニド処置が、ブメタニド処置終了後少なくとも6週間のオープンフィールド暴露に誘発される発作の発現を強く抑制することを示す。棒グラフは、SE発作後27週後のピロカプリンSEラットをオープンフィールド(1.4x1.4m)の中央に置き、その後1分後から始まる15分間に発現する発作の回数を示す。ラットは無処置(黒の棒)、又はSE後第2週目から21週間に亘り慢性的に飲料水経由でブメタニド処置された(斜線の棒)である。このテストでは、ブメタニド処置終了後6週間後でも処置済みラットは、SE後時間適合対照SEラットが発現する発作回数の15%未満の発作発現回数を示した(各グループn=6ラット)。
【0091】
図21はSE後16日目から12週間に亘る慢性ブメタニド処置がオープンフィールドの暗箱に入るラットの挙動を回復させることを示す。ラットは箱を含むオープンフィールド(1.4x1.4m)の中央に置かれ、箱は内部が暗く、0.2(長さ)x0.3(幅)x0.15(高さ)mの寸法であり、0.3mの幅側が開口しており、オープンフィールドの中央と囲み壁の間の中間点に置かれる。挙動は1分間ビデオ録画される(3回の試技/ラット、各グループ6ラット)。対照ラットは殆ど箱に入り、箱内に留まった(試技の78%)。対照的に、SE後のラット(SEは後14−15週)は殆ど箱を無視して入らなかった(試技の15%、7ラット)。同じSEグループからのブメタニド処置ラットは、対照ラットと類似の挙動を示し、殆ど箱に入った(試技の67%、8ラット)。
【0092】
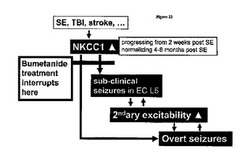
図22は内側側頭葉てんかん(TLE)発症の我々の原理、及び我々の新規の予防的及び抗てんかん慢性ブメタニド処置を示す。TLE誘発の発作、例えば、てんかん重積(SE)、外傷性脳障害(TBI)、脳卒中他は、潜伏期(SE後2及び3週間)に進行するNKCC1の病理学的再発現を誘発する。GABAA受容体依存シナプス性抑制の進行性消失は嗅内皮質(EC)第5層のてんかん様活性/無症候性発作をもたらす。このてんかん様活性は嗅内皮質(EC)及び下流脳領域において下流可塑性変化をトリガーし、それは易興奮性を増大させる2次的変化につながる。易興奮性を増大させる変化の進行に伴って、顕性発作の可能性も増大する。顕性発作もまた下流可塑性変化をトリガーし、それは更に大脳易興奮性を増大させ、発作発現の進行につながる(図18及び19に示す)。最終的に発作はNKCC1活性に依存性でなくなり、そしてNKCC1が基準化した時でも継続する(図12、13に示す)。ブメタニド処置は低い細胞内Cl−レベル及びより効率的な抑制(図10A、B)を回復し、潜伏期においててんかん様活性(図10C、D)及び慢性期の発作(図12,18,19,20)を抑制する。無症候性及び顕性発作のブメタニド処置は、下流病理学的変化を防止し、処置後少なくとも6週間継続する(図19、20)発作出現率及びテストされた挙動(図21)の基準化につながる。
【0093】
(実験結果の議論)
発作中のてんかん様ニューロン活性は多くの大脳領域で観察されるが、しかしそのてんかん重積(SE)状態に後続する発生源は明確ではない。我々は深層嗅内皮質(EC)におけるてんかん様活性の初期発現を検証するため、内側側頭葉てんかん(TLE)のリチウム−低投与量ピロカプリンラットモデルを使用した。我々はSE後3週間の潜在期間内に増加する比率の嗅内皮質(EC)第5層のニューロンが単一シナプス刺激に多シナプス性バースト脱分極で応答することを示した。この変化は並行して第5層ニューロンのIPSP逆転電位の進行性脱分極変移を伴い、これは明らかにCl−内側輸送体NKCC1の増加及び同時にCl−外側輸送体KCC2の減少に起因し、この両方の変化は、細胞内Cl−蓄積に役立っている。潜伏期におけるCl−摂取の抑制はよりネガティブなGABA作動性逆転電位を回復し、そして多シナプス性バーストを終了させた。Cl−輸送体の変化は深層嗅内皮質(EC)に強く特異的であった。それらはこの時期に第1−3層、嗅周囲皮質、海馬台又は歯状回では発現しなかった。我々はCl−ホメオスタシスの変化が深層嗅内皮質における過剰興奮性を促進し、それがそこでのてんかん様放電に繋がり、それは結果的に下流皮質領域に影響を及ぼすと提案する。
【0094】
従って、我々はSE後の潜伏期に深層嗅内皮質における大幅に増加した興奮性の発現を示した。第5層のニューロンは通常は閾値以下のシナプス性刺激に対し多シナプス性バースト放電で応答を始める。さらに、我々はこの潜伏期にCl−輸送体NKCC1(内向き)およびKCC2(外向き)の変化した発現に起因するGABAA受容体逆転電位の大きな脱分極変移が起こることを示した。このてんかん発生の初期段階では、これらの輸送体発現の変化は深層嗅内皮質においてのみ発現し、隣接する海馬領域及び皮質領域では発現しなかった。Cl−輸送活性の変化と増加する多シナプス性バーストとの間の直接的な関係は、ブメタニドによるNKCC1の遮断がIPSP逆転電位を大きく回復し、多シナプス性バースト放電を大幅に減少させた、という発見よって支持された。我々の結果は、嗅内皮質第5層が内側側頭葉てんかん(TLE)の発症において重要な部位であることを意味している。
【0095】
従って、上記の実験結果は、SE後の潜伏期において、行動性発作の発現前に、深層内側嗅内皮質においてニューロ興奮性の強い増加、即ち弱いシナプス刺激に対し多シナプス性バースト放電で応答する嗅内皮質第5層ニューロンの比率の進行性増加があることを示唆している。従って嗅内皮質における自然活性パターンは、ますます自発的発作をトリガーする可能性が高くなり、自発的発作は定義により潜伏期を終了させる。我々の発見は、SE後、内側側頭葉てんかん(TLE)は初期において嗅内皮質深層に発現することを示唆している。我々の知る限りでは、潜伏期に他の脳領域においてシナプス刺激に応答するてんかん活性を示す他の研究は無かった。この領域の特異性は、嗅内皮質深層に対する脱抑制に繋がるNKCC1及びKCC2発現の変化の特異性により実証された。内側側頭葉てんかん(TLE)のピロカプリン高投与モデルでCA1に対し潜伏期発作間活性が生体内で示されたが(El−Hassar氏他,2007)、検査した幾つかの他の皮質領域及び海馬領域は変化を示さなかった。我々のレポートした第5層ニューロンの易興奮性の変化が、嗅内皮質第3層の錐体ニューロンの明確に認識可能な消失がなく発現した、ことは非常に重要である。嗅内皮質第3層の錐体ニューロンの高度の消失は後期LTEの症状に典型的であることが示されて来たし(Du氏他,1993;Du氏他,1995)、そしてそれは遷延性SEの24時間以内に発現可能であった(Du氏他,1995)が、明確に認識可能な消失が発現しないため、これらの発見は本論とは無関係である。
【0096】
ジアゼパムがSE発症後2時間後でなく1時間後に投与され、かつアトロピンが投与される、我々のSE発現プロトコルはアンドレ氏他(Andre氏他,2007)のプロトコルよりも幾分弱く見えるかもしれない。ジアゼパムもアトロピンもいずれかの時点で即座にSEを停止させないが、この変更がラットの回復を促進し、より良い生存率の脳スライスを生成したことを我々は発見した。良かれ悪しかれ我々はまた、弱い初期の発作はより徐々に進行する細胞又は神経網の変化を生成し、従って分析が容易であるかも知れないと理由づけた。ビデオモニタリングで検査した場合、我々のプロトコルは、18週までの潜伏期を持つ全てのラットにおいて実証可能な発作活性を生成した(平均=12.7週、材料と方法の項参照)。重要なことに、これらの潜伏期間は非常によく患者に対し報告された潜伏期間と適合した(Annegers氏他,1980)。これは、潜伏期が平均して1−4週間しか継続せず、殆どは2週間未満である公開されたげっ歯類TLEモデル(Dube氏他,2000a;Arzimanoglou氏他,2002;Curia氏他,2008;Li氏他,2008)に対する重要な改良である。はるかに短い潜伏期は、恐らく追加又は全体的な発作を促進する他のメカニズムを活性化させるより激しい発作を示唆するが、それは殆どの場合明らかに患者に関係が無い。このようにLi氏他(Li氏他,2008)はマウスに対するピロカプリン高投与と組み合わせた異常なリチウム侵襲を使用して(確立されたTLEモデルではない)、海馬領域CA1に本願で研究される期間まで継続するNKCC1の急性再発現を示した。この発見は、我々のラットにおいてはCA1ではNKCC1の変化が無いことと対照的である。我々の示した嗅内皮質第5層における細胞性変化は、より強いリチウム−ピロカプリンSE及び他のプロトコルによっても殆どの場合トリガーされる。
【0097】
嗅内皮質第5層における易興奮性の変化は、嗅内皮質第5層のニューロンの約80%で発現する、GABA作動性PSP逆転電位における強いそして進行性の脱分極変移、と同時性を有し、そして我々はそれにより補助されるものと提案する。この比率はCl−内向き輸送体NKCC1の増加と非常によく適合する。殆どの嗅内皮質第5層のニューロンで同時に発現するCl−外向き輸送体KCC2の減少は、NKCC1によるネットのCl−蓄積を促進するが、それ自身では静止膜電位に対するEClポジティブをもたらさない。NKCC1及びKCC2の進行性変化は、SE後3週間後の強く変位したE(PSP)と非常によく適合した、ただし後者の量定は、PSP振幅の外挿及び傾斜の差異の減少に起因して精度が劣り、従って測定可変性を追加した。
【0098】
GABA作動性PSP逆転電位の弱い変移はTLE疾病進行のずっと後の期、即ち、後期慢性てんかんにおいて海馬台ニューロンで観察され、それは明らかにNKCC1発現のずっと弱い変移に起因した(Cohen氏他,2002;de Guzman氏他,2006;Palma氏他,2006;Huberfeld氏他,2007;Munoz氏他,2007;Sen氏他,2007)。領域、疾病進行期、発作間及び発作活性のレベル、及び発現レベルの差異は、NKCC1とKCC2の変移した発現レベルを開始しそして更に制御する信号ネットワークにおける、潜伏期と後期慢性TLEにおける基本的差異を示唆する。
【0099】
最近になって、大規模な発作が、海馬において急性の45日以上に渡るNKCC1の部分回復性再発現、及び発作後2週間以内の自発性再発性発作(SRS)の発現に繋がることが示された(Kang氏他,2002;Li氏他,2008)。しかしこれらのマウスにおいて、1)NKCC1がニューロンにより再発現したか、グリア細胞により再発現したか、2)GABAA−PSP逆転電位が変位したか、3)潜伏期間中にてんかん様活性があったか、4)発作がNKCC1活性に依存するか、が示されておらず、従って不明確である。対照的に、これらの研究(Li氏他)に基づく最も最近の研究は、高投与量の注入によってもブメタニド処置はSRSの抑制に完全に失敗したことを示した(Brandt氏他,2010)。Brandt氏他はこの結果の原因について議論している:「最近の研究の成体ラットTLEモデルにおいて、ブメタニドを使用した予防的処置において有意な抗てんかん活性が見られなかったことは、下記を含む幾つかの原因が考えられる:(1)ブメタニドの全身性投与が脳に到達しそしてこの薬剤が脳で十分な高濃度を保つことができないことに伴う、薬物動態的問題点(速い排出、脳浸透が乏しい)、(2)NKCC1の変化がピロカプリン誘発てんかん発作に決定的には関与していない、又は、(3)このモデルにおいて予防的処置に使用される時間窓、即ち、SE後2週間後が適切でなかった。」。我々のデータにおける低投与量ブメタニドでの発作抑制は、薬物動態的問題点がBradt氏他の発作処置の失敗の理由となりえないことを示している(Brandt氏他,2010)。我々のデータは、SE後最初の2週間以内ではNKCC1の変化はてんかん発作に決定的には関与していないことを示唆している。従って時間窓も間違っている。さらに、回復NKCC1レベルは潜伏期の最後の発作発現の開始の理由にはなりえない(Li氏他,2008)。このマウスモデルは患者に見られた(上記参照)潜伏期より遥かに短い潜伏期を示す(Li氏他,2008)のみならず、示されたCA1におけるNKCC1発現は、我々のデータと矛盾する(Kang氏他、2002;Li氏他,2008)。これら事実の両方は、使用されたSEモデルが現実的なTLEモデルではないことを示唆している(上記参照)。本発明の新規性は、1)我々の改良ラットTLEモデルによる潜伏期の現実的適合、2)潜伏期ニューロンにおけるNKCC1の進行性再出現とIPSP逆転電位の変移の両方、3)潜伏期及び慢性期(SE後3−18週)におけるブメタニドによるてんかん様活性及び発作の効果的抑制、そして、4)遷延性ブメタニド処置終了後少なくとも6週間を超えること、に反映されている。
【0100】
嗅内皮質第5層ニューロンにおけるGABA−PSPの通常の逆転電位、即ちほぼVrestは2つの別個の慢性てんかん中のSE−TLEモデル(Fountain氏他,1998;de Guzman氏他,2006)で発見され,我々の報告する非常に大きなGABAAE(PSP)は、一度慢性てんかんが確立されると大部分回復することを支持した。他の説明も可能である:第1に、記録は外側嗅内皮質から得られ、そこではNKCC1およびKCC2に小さな変化しか見られないことを我々は発見した。更に、異なるTLEモデルが使用された。de Guzman氏他(2006)は、TLEのピロカルピン高投与量ラットモデルを研究し、それは,リチウム−ピロカルピンラットモデルとは基礎となるSE中活性化される信号伝達経路が有意に異なり(Ormandy氏他,1989)、はるかに短い潜伏期(2週間未満)、及び第3層ニューロン消失の差異(上記)において異なることが示された。後者2者の差異はより激しい発作を示唆する(上記)。これはE(PSP)変移、及び慢性てんかんから回復するか否かにおいて、差異を生成可能である。我々はこれら重要な問題について将来研究する予定である。
【0101】
GABA作動性PSPの強い脱分極変移が、深層嗅内皮質の易興奮性の観察された変化の主たる原因であるという推論は、Cl−内向き輸送体NKCC1の遮断剤ブメタニドの効果により支持された。ブメタニドはより過分極されたPSP逆転電位を回復し、そして刺激に対するてんかん様バースト応答をほぼ消滅させた。これらの発見は、SE後のこの病期におけるてんかん様バースト応答の発現に対し、強く脱分極したE(PSP)の必要性を意味するが、しかし、他の同時性変化、例えば、第5層ニューロンの観察された入力コンダクタンスの減少が追加的に必要となる可能性を除外しない。ブメタニドの効果はさらに、GABAAR遺伝子の減少、又は抑制性シナプス及び介在ニューロンの消失に起因すると思われる、観察された入力コンダクタンスの減少にも拘わらず、SE後3週間後に抑制が依然非常に効果的でありうることを意味する(Bernard氏他,2004;Gorter氏他,2006;Kumar氏とBuckmaster氏,2006)。対照ニューロンではGABA−PSPは既にわずかに脱分極しており、そして、SE後3週間後、ブメタニドはそれを完全にはそのレベルに回復させなかった。従って、抑制は「過分極している」のではなく、常に「短絡している」と言える。しかし、AP発火のトリガーを興奮電流により防止するための最も重要な要素は、発火閾値に近い膜電位での抑止性電流の振幅であるように思われることを忘れないようにする必要がある。この電流振幅は、発火閾値(約45mV)に近いE(PSP)の変移と共にCl−駆動力に従って徐々に変化する。その結果SE3週間後かつブメタニドの存在下で(E(PSP)は約−60mV)−45mVでの駆動力は、対照ラットの約26mVに対し約15mVであり、従って約42%減少する。結果としてこの臨界膜電位領域でのCl−電流は、駆動力の減少及びGABA−PSPコンダクタンスの減少の両方によって減衰されるが、ブメタニドの存在下では、てんかん様活性の抑制に依然として全く効果的である。このことは、全体的にCl−勾配を回復することが生体内で処置的利益をもたらすという希望を与える。てんかん発生、及びNKCC1活性の抑制に対する処置的及び予防的干渉に関する我々の原理は、図22に要約されている。そこに示された、潜伏期に始まるブメタニド処置中及び以降の発作の非常に効果的な抑制は、原理の実証を提供する。
さらに、初期のそして高度に特異的な病理的NKCC1活性の基準化、及びこの処置による変化した挙動の基準化は、ブメタニドでの提案された抗てんかん処置が、少なくとも幾つかの現在の抗てんかん剤による知性と脳の能力に対する副作用を回避する、という重要な追加的利益を有することを示唆する。
【0102】
我々は、我々の発見がTLEの初期の発現において作用する主要な要素を開示するものと信じ、また、疾患につながる変化の起点及び進行中の深層嗅内皮質の重要性を強調する。
【0103】
(略語)
ACSF、人工脳脊髄液;CA、アンモン角;DAPI、4’,6’−ジアミジノ−2−フェニルインドール;EC,嗅内皮質;E(PSP)、PSP逆転電位;FISH,蛍光遺伝子プローブ法;IPSP,抑制性シナプス後電位;PBS,リン酸緩衝食塩水;PBS−T,0.2%のトリトン−X−100を含むPBS;PSP, シナプス後電位;RMP,静止膜電位;SE、てんかん重積;TLE、内側側頭葉てんかん。
【0104】
(参照)
【表1】

【技術分野】
【0001】
(関連出願)
2009年7月24日出願の表題「内側側頭葉てんかん(TLE)の処置及び可能性を防止/減少させる方法」の特許文献1、及び2009年11月12日出願の同一表題の特許文献2の優先の恩恵を主張し、それらは参照により個々にその全体がここに採り入れられる。
(連邦助成による研究)
本発明はNIH/NRSAによる助成金番号RR15636,FF015614,AA014127及びAA016880の政府の助成により完成された。従って米国政府は本発明に対し権利を有する。
(技術分野)
本発明は、内側側頭葉てんかん(TLE)の処置及び防止に関する新規の方法を提供する。
【背景技術】
【0002】
全ての参照に対する引用文献は実施例の後に記載されている。
内側側頭葉てんかん(TLE)は成人において最も一般的なタイプのてんかんであり(Engel,1989)、しばしば薬物処置に対し耐性を示し、最終的には発作を抑えるために神経手術の選択肢しか残されない。TLEは脳損傷及びてんかん重積(SE)を含む種々の発作の後に発症する。突然の発作の後に、特徴的な無発作の「潜伏期」がヒトでは典型的に2−3カ月、例外的に数年続く(Annegers氏他、1980;Weiss氏他,1986)。リチウム−ピロカルピン誘導てんかん重積(SE)後数週間後に発症するラットのTLEは、ヒトTLEの臨床及び神経病理学的特徴を再現し、そしてその病気の非常に有益な動物モデルを提供する(Ormandy氏他,1989;Turski氏他,1991;Cavalheiro氏,1995;Dube氏他,200;Andre氏他,2007)。当初の原因が異なっても、TLEの行動性及び組織病理学的特徴は全ての病因で驚くほど類似している。このことが多くの研究者に初期の原因の下流に主な共通する経路、恐らくは発作の識別特性である激しい同期活性があると仮定するように誘導した(Du氏他,1995;Wu氏及びSchwarcz氏,1998;Schwarcz氏他,2000)。通常この同期活性は嗅内皮質(EC)を含む海馬及び海馬傍回皮質に見られる(Schwartzkroin氏及びKnowles氏,1984;Bartolomei氏他,2004)。長い間、特定の内側側頭葉萎縮が発作病巣に対して同側性であることが示されて来た(Bartolomei氏他,2005)。外科的に切除された標本の検査により、嗅内皮質(EC)で細胞消失とアストログリオーシスが確認された(Yilmazer−Hanke氏他,2000)。
【0003】
神経生理学において、てんかん重積(SE)の後、どこの場所において重要な初期変化が起こるのかが不明確である;しかしながら、海馬アンモン角領域に対する海馬傍回の神経保護実験は、てんかん発生の初期段階において海馬傍回皮質の重要な役割を示唆した(Andre氏他,2007)。さらに我々は以前の研究において、深部嗅内皮質(EC)が、第2層及び第3層と著しく対照的に、異常に高い網状組織の興奮を発現することを観察した(Gloveli氏他,1999;Egorov氏他,2003)。このことは、深部嗅内皮質(EC)が、てんかん重積(SE)にトリガーされた過剰興奮性の発現に対して特に疑わしいことを示唆する。
【0004】
自発的再発性発作(SRS)を防止する重要な機構は、興奮性ニューロンのCl−依存シナプス抑制である。この抑制の有効性は細胞外Cl−濃度に対する細胞内Cl−の増加と共に低下する。細胞内Cl−濃度はK+Cl−共輸送体、特にKCC2により、K+外向き勾配により駆動されて減少し、Na+K+2Cl−共輸送体、特にNKCC1により、Na+内向き勾配に駆動されて増加する。従って周辺及び中心ニューロンに対しては、両方のCl−輸送体がシナプス抑制の効率性に対して重要な役割を果たしていることが確認されている(Aickin氏他,1984;Misgeld氏他,1986)。
【0005】
特許文献3は種々の疾病、特に、塩化ナトリウムカリウム共輸送体媒介疾病、及びガンマアミノ酪酸(GABA)の損なわれた抑制により激化する脳内の興奮毒性に伴う疾病、の処置に利尿剤を使用することを記載している。
【0006】
TLEを防止又は処置する努力に拘わらず、上記のTLE関連発作を防止又は寛解させる処置方法に対する要求は依然として存在している。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】米国暫定特許出願US61/271,714
【特許文献2】米国暫定特許出願US61/281,095
【特許文献3】米国特許出願 20070043034
【発明の概要】
【0008】
出願人は、被験者がTLEを誘発することが既知の発作を患った後にNKCC1抑制剤を被験者に投与することにより、被験者に内側側頭葉てんかん(TLE)が発症するのを防止する、或いはTLEの重症度を低減する処置法を発見した。
【0009】
従って本発明のある側面では、被験者が、てんかん重積(SE)、小発作性てんかん、欠神、筋クローヌス性発作、クローヌス性(間代性)発作、緊張性発作、緊張性―クローヌス性(間代性)発作、無緊張性発作、後天性失語、てんかんを伴う後天性失語症(ランドウ・クレフナー症候群)、後天性てんかん性失語症、大脳皮質異形成−焦点性てんかん(CDFE)、新生児てんかん、海馬硬化症(HS)、海馬萎縮症、大脳萎縮症、小脳萎縮症、複雑熱性けいれん(CFC)を含む熱性発作、外傷性脳障害、脳卒中、脳腫瘍の診断又は発見後、などの発作を患った後に、被験者にNKCC1抑制剤が投与される。
【0010】
ある側面では、NKCC1抑制剤が、被験者のてんかん重積(SE)発症後の潜伏期に被験者に投与される。
また他の側面では、NKCC1抑制剤が、被験者が原因が未知の最初の発作を患った後に被験者に投与される。
【0011】
さらに他の側面では、NKCC1抑制剤が、被験者がTLE誘発発作(一般的に、SE発作を含む)を患った後約8週間から約24週間以内(或いは約12週間から約16週間以内)に被験者に投与される、又は約1週間から約4週間以内、又は約2−3週間から4週間以内に被験者に投与される。ブメタニド及びGABAA作動性伝達賦活薬との組合せによる、TLE又はTLE発症の効果的処置のためのSE又は他のTLE誘発発作後の時間は、SE又は発作の継続期間と重症度、及び種、特にヒトに依存し、それよりずっと長い。
【0012】
本発明の他の側面では、NKCC1抑制剤とガンマアミノ酪酸(GABA)調節組成物が、被験者がTLEを誘発することが既知の発作を患った後又は原因が未知の発作の最初の発作後に、前記被験者に同時投与される。
【0013】
本発明の好適な実施形態では、NKCC1抑制剤が,トラセミド、フロセミド、アゾセミド、ブメタニド、ピレタニド、トリパミド、エトゾリン(etozoline)及びその代謝体オゾリノン(Ozolinone)、シクレタニン、およびそれらの医薬的に許容可能な派生物、それらの塩及びエステルからなるグループから選択されるループ利尿薬である。
【0014】
本発明の他の側面では、1つ以上のNKCC1抑制剤、及び選択肢として1つ以上のガンマアミノ酪酸(GABA)調節組成物が、被験者がTLEを誘発することが既知の反復性発作を患った後に、慢性的に被験者に投与される。
【0015】
本発明のさらに他の側面では、NKCC1抑制剤が第2の処置薬と組合わされ、そして被験者がTLEを誘発することが既知の発作を患った後に、被験者に同時投与され、
ここにおいて第2の処置薬は、ガンマアミノ酪酸(GABA)調節組成物(例えば、GABAA受容体依存信号伝達の調節剤)、抗けいれん薬、イオンチャネル不活性化剤、抗利尿剤、又はそれらの組合せである。
【0016】
発明者らは、TLEを誘発することが既知の発作に続く期間(例えば、てんかん重積(SE)後の潜伏期)において、嗅内皮質(EC)第5層内の増加する比率のニューロンが単一のシナプス刺激に多シナプス性バースト脱分極/てんかん型活性で応答することを発見した。この変化はCl−内向き輸送体NKCC1の発現量の増大及び同時発生のCl−外向き輸送体KCC2の発現量の減少に明白に起因する、第5層ニューロンにおけるIPSP逆転電位の進行性脱分極シフトと平行して起こり、両方の変化は細胞内Cl−蓄積を助ける。潜伏期におけるCl−取り込みの抑制は、よりネガティブなGABA作動性逆転電位を回復し、そして多シナプス性バーストを消滅させた。Cl−輸送体における変化は、深部嗅内皮質に高度に特異的であった。変化はこの期間、第1−3層、嗅周囲皮質、海馬台、CA1,CA2又は歯状回では起こらない。いかなる理論にも縛られないことを希望するが、我々は、Cl−のホメオスタシスの変化が深部嗅内皮質において過剰興奮性を促進し、そこでのてんかん放電につながり、それは結果的に下流の皮質領域に影響を与えることを提案する。
【0017】
本発明の処置方法は従って、潜伏期及び初期慢性型てんかん期における異常性細胞活性及び発作形成の根底をなす特定の基礎的病理を標的とする。TLEを誘発することが既知の発作の後、又は未知の原因の初期発作の後、のNKCC1の慢性的抑制は、TLEの予防的防止、又は発現の減弱に有用であることを証明する。
本発明のこれら及び他の側面は、発明の詳細においてさらに記述される。
【図面の簡単な説明】
【0018】
【図1】実施例1の実験により画定される、てんかん重積(SE)後の嗅内皮質(EC)第5層ニューロンの増大した多シナプス性バースト興奮を示す図である。
【図2】「材料及び方法」の項、及び実施例2で説明される、てんかん重積(SE)後2週間後と3週間後のGABAA誘発シナプス後電位(PSP)逆転電位のポジティブ変移を示す図である。
【図3】実施例3で画定される、NKCC1のmRNA、Cl−内側方向輸送体が、てんかん重積(SE)後嗅内皮質(EC)第5層内において進行的に増大することを示す図である。
【図4】実施例4の実験において、NKCC1タンパク質も増加したことを示す図である。
【図5】実施例4の実験において測定されるように、SE後、嗅内皮質(EC)第5層内ニューロンがNKCC1タンパク質の発現を増加させたことを示す図である。
【図6】実施例5の実験において測定されたように、KCC2に対するmRNA,ニューロンのCl−の外向き輸送体は、嗅内皮質(EC)第5層内においてSE後進行的に減少することを示す図である。
【図7】実施例6の実験において測定されたように、SE後KCC2タンパク質も減少することを示す図である。
【図8】実施例7の実験において測定されたように、NKCC1のmRNAは他の皮質領域では増加しないことを示す図である。
【図9】実施例7の実験において測定されたように、KCC2のmRNAは深層嗅内皮質(EC)でのみ減少することを示す図である。
【図10】実施例8の実験において測定されたように、NKCC1抑制剤ブメタニドは部分的にE(PSP)を回復し、そして多シナプス性興奮を抑制することを示す図である。
【図11】実施例9の実験において測定されたように、ピクロトキシンは多シナプス性バーストに軽度の影響をあたえることを示す図である。
【図12】リチウム−ピロカルピンSE後12−17週間後における、ブメタニドによる発作の抑制(0.2mg/Kg体重、腹腔内)を示す(n=5ラット)図である。5体のリチウム−ピロカルピンSEラットが連続するブメタニド又は媒体注射(生理食塩水内の5%エタノール、腹腔内、下部に日付を記載)を受け、そしてその後8時間以内に発現する発作がビデオ録画により分析された。1体の同一のラットは、ブメタニドがCl及びIPSP逆転電位を概して基準化した場合でも、2度目及び3度目のブメタニド投与後に発作を発現したが、それは恐らく自発的発作を可能にするこのラット内の2次的な下流の変化によるものである。
【図13】NKCC1タンパク質が正常な成人レベルに向かって回復することを示す図である。プロットは、深層内側嗅内皮質(EC)のNKCC1タンパク質の免疫蛍光信号を、抗体を持たない対照ラット(「免疫グロブリンG(IgG)無し」と表示)、抗体を持つ対照ラット(「対照」と表示)、SE後2週間後及び3週間後(「SE 週間後」と表示)のラットにおいて示す。SE後17週間でNKCC1信号は、ブメタニドが完全に自発的再発性発作(SRS)を防止するラット(「ブメット(Bumet)ポジティブ」と表示)では、SE後3週間後のラットのレベルの50%未満まで回復し、そしてブメタニドが自発的再発性発作(SRS)を防止する効果が無くなったラット(「ブメット(Bumet)ネガティブ」と表示)では95%超に回復した。
【図14】KCC2タンパク質が正常な成体レベルに向かって回復することを示す図である。プロットは、深層内側嗅内皮質(EC)のKCC2タンパク質の免疫蛍光信号を対照ラット(「対照」と表示)、SE後2週間後及び3週間後(「SE後 週間」と表示)のラット、及びSE後17週間後のラットにおいて示す。SE後17週間でKCC2信号は、正常レベルに向かって回復した(ブメタニドが完全に自発的再発性発作(SRS)を防止するラットは「ブメット(Bumet)ポジティブ」と表示、そして、ブメタニドが自発的再発性発作(SRS)を防止する効果が無くなったラットは「ブメット(Bumet)ネガティブ」と表示)。
【図15】海馬の錐体細胞CA1層又はCA3層では、NKCC1のmRNAは変化がないことを示す図である。
【図16】海馬の錐体細胞CA1層又はCA3層では、KCC2のmRNAは変化がないことを示す図である。
【図17】嗅内皮質(EC)第2層、第3層、又は第5層においても、SE後3週間後にニューロンの損失がないことを示す図である。
【図18】ブメタニド処置が有る場合と無い場合の発作発現を示す図である。
【図19】慢性ブメタニド処置が、処置期間中および処置終了後少なくとも6週間、自発的発作の発現を抑制することを示す図である。
【図20】ブメタニド処置終了後6週間後も誘発発作の発現を抑制することを示す図である。
【図21】ブメタニド処置がラットの挙動を回復させることを示す図である。
【図22】内側側頭葉てんかん(TLE)発症の我々の原理、及び本発明による防止を示す図である。
【発明を実施するための形態】
【0019】
以下の用語が本発明の記述に使用される。ここで特別に定義されない言葉は、本発明の記述に使用される言葉と一貫性を持つ、当業者に使用される共通の意味を有する。
特に特定されない限り、「1つの」、および「少なくとも1つ」は交換可能に使用され、1つ又はそれ以上を意味する。
用語「効果的」は、本発明において意図された効果を生むための本発明の薬剤の量を記述するために使用される。
用語「ガンマアミノ酪酸(GABA)調節組成物」は、限定されないが、バルビツール酸化合物(例えばフェノバルビタール)、ベンゾジアゼピン系化合物(例えば、ジアゼパム)、又はGABAの放出を増加させ又はGABAの再摂取を抑制する薬剤、例えば、ガバペンチン、プレガバリン、4アミノ酪酸(GABA),4−アミノ−3−(4−クロロフェニル)ブタン酸(バクロフェン)、4−アミノ−3−フェニルブタン酸、4−アミノ−3−ヒドロキシブタン酸、4−アミノ−3−(4−クロロフェニル)−3−ヒドロキシフェニルブタン酸、4−アミノ−3−(チエン−2−イル)ブタン酸、4−アミノ−3−(5−クロロチエン−2−イル)ブタン酸、4−アミノ−3−(5−ブロモチエン−2−イル)ブタン酸、4−アミノ−3−(5−メチルチエンー2−イル)ブタン酸、4−アミノ−3−(2−イミダゾリル)ブタン酸、4−グアニジノー3−(4−クロロフェニル)ブタン酸、(3−アミノプロピル)亜ホスホン酸、(4−アミノブチ−2−イル)亜ホスホン酸、酪酸ナトリウム、(3−アミノ−2−メチルプロピル)亜ホスホン酸、(3−アミノブチル)亜ホスホン酸、(3−アミノ−2−(4−クロロフェニル)プロピル)亜ホスホン酸、(3−アミノ−2−(4−クロロフェニル)−2−ヒドロキシプロピル)亜ホスホン酸、(3−アミノ−2−(4−フルオロフェニル)プロピル)亜ホスホン酸、(3−アミノ−2−フェニルプロピル)亜ホスホン酸、(3−アミノ−2−ヒドロキシプロピル)亜ホスホン酸、(E)−(3−アミノプロペン−1−イル)亜ホスホン酸、(3−アミノ−2−シクロヘキシルプロピル)亜ホスホン酸、(3−アミノ−2−ベンジルプロピル)亜ホスホン酸、[3−アミノ−2−(4−メチルフェニル)プロピル]亜ホスホン酸、[3−アミノ−2−(4−トリフルオロメチルフェニル)プロピル]亜ホスホン酸、[3−アミノ−2−(4−メトキシフェニル)プロピル]亜ホスホン酸、[3−アミノ−2−(4−クロロフェニル)−2−ヒドロキシプロピル]亜ホスホン酸、(3−アミノプロピル)メチルホスフィン酸、(3−アミノ−2−ヒドロキシプロピル)メチルホスフィン酸、(3−アミノプロピル)(ジフルオロメチル)ホスフィン酸、(4−アミノブチ−2−イル)メチルホスフィン酸、(3−アミノ−1−ヒドロキシプロピル)メチルホスフィン酸、(3−アミノ−2−ヒドロキシプロピル)(ジフルオロメチル)ホスフィン酸、(E)−(3−アミノプロペン−1−イル)メチルホスフィン酸、(3−アミノ−2−オキソ−プロピル)メチルホスフィン酸、(3−アミノプロピル)ヒドロキシメチルホスフィン酸、(5−アミノペンチ−3−イル)メチルホスフィン酸、(4−アミノ−1,1,1−トリフルオロブチ−2−イル)メチルホスフィン酸、(3−アミノ−2−(4−クロロフェニル)プロピル)スルフィン酸、及び、3−アミノプロピルスルフィン酸、を含む。
【0020】
ガンマアミノ酪酸(GABA)調節組成物として有用な本発明の他の組成物は:
拮抗薬:ガボクサドール、イソグバシン、イソニペコチン酸、ムシモール(ベニテングタケ)、プロガビド、SL75102、チオムシモール(Thiomuscimol)。
GABAA受容体ポジティブアロステリック調節剤:
バルビツール酸化合物;
アロバルビタール、アルフェナール(Alphenal)、アモバルビタール、アプロバルビタール、バルベキサクロン、バルビタール、バルビツール酸ベンジルブチル、ブラロバルビタール(Brallobarbital),ブロフェバルビタール(Brophebarbital)、ブコローム、ブタバービタル、ブタルビタール、ブトバルビタール、ブタリロナール、クロチルバルビタール(Crotylbarbital),シクロバルビタール、シクロパール(Cyclopal),エナリルプロピマール(Enallylpropymal)、エサロバルビタール(Ethallobarbital),フェバルバメート、ヘプタバルビタール、ヘキセタール、ヘキソバルビタール、メホバルビタール、メタルビタール、メトヘキシタール、メチルフェノバルビタール、ナルコバルビタール、ニールバルビタール(Nealbarbital),ペントバルビタール、フェノバルビタール、フェタアルビタール(Phetharbital)、プリミドン、プラジトン(Praziton)、プロバルビタール、プロパリロナール(Propallylonal)、プロキシバルバール、プロキシバルビタール、レポサール、セクブタバルビタール、セコバルビタール、シグモダール(Sigmodal)、スピロバルビタール(Spirobarbital)、タルブタール、チアルバルビタール、チアミラール、チオバルビタール、チオブタバルビタール、チオペンタール、バロフェイン(Valofane)、ビンバルビタール(Vinbarbital),ビニルビタール。
【0021】
ベンゾジアゼピン系薬;
アジナゾラム、アルプラゾラム、アルフェンダザム(Arfendazam),アビザホン(Avizafone),ベンタゼパム,ブレタゼニル,ブロマゼパム,ブロチゾラム,カマゼパム,クロルジアゼポキシド,シクロチゾラム(Ciclotizolam),シノラゼパム,クリマゾラム(Climazolam),クロバザム,クロナゼパム,クロラゼペート,クロチアゼパム,クロキサゾラム,シプラゼパム(Cyprazepam),デロラゼパム,ジアゼパム,ドキセファゼパム,エルファゼパム(Elfazepam)、エスタゾラム,エチルカルフルゼペート(Ethyl carfluzepate),エチルジラゼペート(Ethyl Dirazepate),ロフラゼプ酸エチル,エチゾラム,フレタゼパム(Fletazepam),フルジアゼパム,フルニトラゼパム,フルラゼパム,フルタゾラム,フルテマゼパム(Flutemazepam),フルトプラゼパム、ホサゼパム(Fosazepam)、ギダゼパム(Gidazepam)、ギリソパム(Girisopam), ハラゼパム, ハロキサゾラム, イクラゼパム(Iclazepam),イミダゼニル(Imidazenil),ケタゾラム,ロフェンダザム(Lofendazam),ロピラゼパム(Lopirazepam),ロピラゼパム(Lopirazepam),ロプラゾラム,ロラゼパム,ロルメタゼパム,メクロナゼパム(Meclonazepam),メダゼパム,メニトラゼパム(Menitrazepam),メタクラゼパム,メキサゾラム,ミダゾラム,ネリソパム(Nerisopam),ニメタゼパム,ニトラゼパム,ニトラゼペート(Nitrazepate),ノルダゼパム,オキサゼパム,オキサゾラム,フェナゼパム、ピナゼパム、ピボザゼパム(Pivoxazepam),プラゼパム,プレマゼパム,プロフラゼパム(Proflazepam),QH−II−66,クアゼパム,レクラゼパム(Reclazepam),リルマザホン,リパゼパム,Ro48−6791,SH−053−R−CH3−2’F,スラゼパム(Sulazepam),テマゼパム,テトラゼパム,トフィソパム,トリアゾラム,トリフルバザム(Triflubazam),ウルダゼパム(Uldazepam)、ザピゾラム(Zapizolam),ゾラゼパム,ゾメバザム(Zomebazam)。
【0022】
カルバミン酸化合物;
カリスバメート、カリソプロドール、エミルカメート、フェルバメート、メブタメート、メプロバメート、メトカルバモール、フェンプロバメート、プロシメート(Procymate)、チバメート。
抗神経活性ステロイド;
アセブロコール(Acebrochol)、アロプレグナノロン、アルファドロン、アルファキサロン、ゲドカルニル、ガナクソロン(Ganaxolone)、ヒドロキシジオン、ミナキソロン、Org20599、THDOC。
非ベンゾジアゼピン系薬;
アベカーニル、アジピプロン(Adipiplon)、アルピデム、CGS−20625、CGS−9896、CL−218,872、ELB−139、エスゾピクロン、エチホキシン、インジプロン(Indiplon)、L−838,417、ネコピデム(Necopidem)、NS−2664、NS−2710、オシナプロン、パゴクロン(Pagoclone)、パナジプロン(Panadiplon)、パジナクロン(Pazinaclone)、ピペクアリン(Pipequaline)、ROD−188、RWJ−51204、サリピデム(Saripidem)、SB−205,384、SL−651,498、スプロクロン(Suproclone)、スリクロン、SX−3228、TP−003、TPA−023,TP−13,トラカゾレート、U−89843A,U−90042、Y−23684、ザレプロン、ゾルピデム、ゾピクロン。
フェノール;
フォスプロポフォール、プロポフォール、チモール。
ピペリジンジオン;
グルテチミド、メチプリロン、ピペリジオン、ピリチルジオン。
キナゾリノン;
アフロクアロン、クロロカロン(Cloroqualone)、ジプロカロン(Diproqualone)、エタカロン(Etaqualone)、メブロカロン(Mebroqualone)、メクロカロン、メタカロン、メチルメタカロン。
その他;
クロルメザノン、クロメチアゾール、エタゾレート、エタノール(アルコール)、エトミデート、キャバラクトン(Kavalactone)(カバカバ)、ロレクレゾール、プロガビド、プロパニジド、ROD−188、タツナミソウ、スチリペントール、ワレレン酸(Valerenic Acid)(カノコソウ)。
【0023】
Na(+)−K(+)−2Cl(−)の共輸送体(ブメタニド又はエタクリン酸)の抑制剤。Am J Physiol Renal Physiol。2009年2月号;296(2):F446−57
セレンスレオニンキナーゼWNK4は、Na(+)−K(+)−2Cl(−)の共輸送体(NKCC1)及びCl(−)/塩基交換輸送体SLC26A6(CFEX)の両方に対し強い薬効のある抑制剤であることが証明されている(Proc Natl Acad Sci USA.2004年2月17;101(7):2064−9.Epub 2004年2月9日)。
mRNAからのNKCC1タンパク質の発現は再発現期間の間、嗅内皮質を標的とする核酸のアンチセンス鎖により抑制される。
炎症は典型的にNKCC特にNKCC1の増加につながる。炎症の典型的なサインはてんかん誘発性皮質に現れ、そして炎症媒介物質は従ってNKCC1再発現に寄与する可能性がある。従って、遷延性のインターフェロン−ガンマ暴露はイオン輸送及びNKCC1発現を他の組織において減少させ(Am J Physiol Gastrointest Liver Physiol. 2004年1月;286(1):G157-65.Epub 2003 9月4)、従って側頭葉てんかん(TLE)誘発発作の後に、嗅内皮質(EC)におけるNKCC1再発現を抑制するのに有用である可能性がある。
【0024】
GABAB受容体遮断薬は、GABAB受容体アンタゴニストの活性化による放出の負のフィードバックを遮断することにより、GABA放出を増大させるのに有用でありうる:
主要サイト:ファクロフェン、サクロフェン、SCH−50911。
GABA異化反応の遮断もまた有用でありうる:GABA−T抑制薬、例えば、3−ヒドラジノプロピオン酸、アミノオキシ酢酸、ガバクリン、ガバペンチン、イソニアジド、フェネルジン、フェニルエチリデンヒドラジン、プレガバリン、バルノクタミド、バルプロ酸、バルプロミド、ビガバトリン。
【0025】
NKCC1抑制薬の利尿効果の抑制は水及びナトリウムのホメオタシスに対する副作用を最小化するのに有用であり得る。1)プロベネシドでの前処置はブメタニド又は他のNKCC1抑制薬により発症したナトリウム利尿及び高レニン血症の両方を減少させる。プロベネシドのNKCC抑制媒介ナトリウム利尿に対するこのような拮抗作用は明らかにナトリウム排出に対する直接作用に起因せず、おそらくブメタニドの腎尿細管分泌に対する抑制作用の2次性である。インドメタシンはブメタニド処置の間、尿量およびナトリウム排出の増加を鈍化させ、そして、ブメタニド−誘発の血漿レニン活性の増加を抑制する。3)バソプレシン、アルギプレシン又は抗利尿ホルモンとして知られるアルギニンバソプレシン(AVP)及びその変異体、例えば、リジンバソプレシン(LVP)又はリプレシンは、腎臓内の遠位尿細管及び集合尿細管における水の再吸収を増大させることにより、腎臓内のNKCCの抑制に起因する水利尿の増加を鈍化させる。
【0026】
抗けいれん薬は、神経性発作活性の蓄積と拡散を抑制するためにてんかん発作の処置に使用される、イオンチャネル不活性剤を含む広範な薬剤グループである。それらは、てんかん様活性及び興奮性を増大させる下流の変化をさらに抑制するのに有用でありうる。このことは慢性期間において疾病が進行する間、てんかん様活性のNKCC1活性への依存性が低下する場合に、特に重要である。カルバマゼピン及びフェニトインは共に現在での第1選択肢の抗けいれん薬である。これらの薬は共にナトリウムチャネルの非活性化状態を安定させ、ニューロンを発火がより起り難い電位にし、そして他の作用も持ちうる。
【0027】
ガバペンチンの作用の明確なメカニズムは未知であるが、その抗けいれん性作用は電圧ゲートのN型カルシウムイオンチャネルを含んでいると考えられている。バルプロ酸もまた電圧ゲートのナトリウムチャネル及びT型カルシウムチャネルを遮断する。このメカニズムがバルプロ酸を広範囲の抗けいれん剤にする。他の臨床的に使用される抗けいれん薬はレベチラセタム、ラモトリギン、プリミドン、及びフェルバメートであり、それらはカルバマゼピン及びフェニトインが効かなかった場合とくに有益である。有用でありうる他の抗けいれん薬は、臭化物(臭化カリウム)、カルバメート(フェルバメート)、カルボキサミド(カルバマゼピン、オクスカルバゼピン、エスリカルバゼピンアセテート)、脂肪酸(バルプロエート−バルプロン酸、バルプロン酸ナトリウム、ジバルプロエクスナトリウム)、ビガバトリン、プロガビド、チアガビン、果糖誘導体(トピラマート)、GABA誘導体(ガバペンチン、プレガパリン)、ヒダントイン(エトトイン、フェニトイン、メフェニトイン、ホスフェニトイン)、オキサゾリジンジオン(パラメタジオン、トリメタジオン、エタジオン)、プロピオネート(ベクラミド)、ピリミジンジオン(プリミドン)、ピロリジン(ブリバラセタム、レベチラセタム、セレトラセタム(seletracetam))、コハク酸イミド(エトスクシミド、フェンスクシミド、メスクシミド)、スルホンアミド(アセタゾラミド、スルチアム、メタゾラミド、ゾニサミド)、トリアジン(ラモトリギン)、尿素(フェネトライド、フェナセミド)、バルプロイルアミド(valproyamides;バルプロエートのアミド誘導体),アミド(バルプロミド、バルノクタミド)である。
【0028】
「TLEを誘発することが既知の発作」には、限定されないが、てんかん重積(SE)、小発作性てんかん、欠神、筋クローヌス性発作、クローヌス性(間代性)発作、緊張性発作、強直間代発作、無緊張性発作、後天性失語、てんかんを伴う後天性失語症(ランドウ・クレフナー症候群)、後天性てんかん性失語症、大脳皮質異形成−焦点性てんかん(CDFE)、新生児てんかん、海馬硬化症(HS)、海馬萎縮症、大脳萎縮症、小脳萎縮症、複雑熱性けいれん(CFC)を含む熱性発作、外傷性脳障害、脳卒中、脳腫瘍、感染症、脊髄膜炎、血管形成異常を含む。
【0029】
「NKCC1抑制剤」は限定されないが、ループ利尿薬(即ち、ヘンレループの厚い上行脚の中のNa+/K+/2Cl−担体を抑制し、それによりナトリウム、カリウム及び塩素イオンの再吸収を抑制する利尿薬)を含む。有用なループ利尿薬は、トラセミド、フロセミド、アゾセミド、ブメタニド、ピレタニド、トリパミド、エトゾリン(etozoline)及びその代謝体、オゾリノン(ozolinone)、シクレタニン、およびそれらの医薬的に許容可能な派生物、例えばそれらの塩及びエステルである。)他のNKCC1抑制剤はGABA−T抑制薬を含む:3−ヒドラジノプロピオン酸、アミノオキシ酢酸、ガバクリン、ガバペンチン、イソニアジド、フェネルジン、フェニルエチリデンヒドラジン、プレガバリン、バルノクタミド、バルプロ酸、バルプロミド、ビガバトリン。
【0030】
本明細書で使用される「患者」又は「被験者」とは、本発明に基づく組成物によるてんかん処置を含む処置が行われる動物、一般的には哺乳類、好適にはヒトを示す。これらのヒト患者のような特定の動物に特異的な感染症、容態、または疾病の状態の処置において、「患者」とはその特定の動物を示す。
本明細書で使用されているように、「処置」とは症状の進行の防止、速度の低減、遅延、抑止又は留保することを意味する。
ここで使用される「処置上効果的な量」とは、TLEを患い、又は発症するリスクのある患者の処置に十分なNKCC1抑制剤(選択肢として、1つ以上のGABA調節組成物と組み合わせて)の量を意味する。例えば、効果的な量はTLE又は関連症候の発症又は進行を遅延させ、速度を遅くさせ、又は防止するのに十分な量を意味する。
【0031】
本明細書で使用される「抑制的効果的濃度」又は「抑制的効果的量」とは、実質的に又は有意にTLE又は関連する症候を処置または防止する本発明に基づく組成物の濃度又は量を意味する。
本明細書で使用される「防止的効果的濃度」とは、TLE又は関連する症候の発症を防止し可能性を低減するのに予防的に効果的な本発明に基づく組成物の濃度又は量を意味する。「抑制的効果的濃度」及び「防止的効果的濃度」は一般的に「効果的な量」の範疇に含まれる。
【0032】
「被験者におけるTLEの重症度を減ずる」とは、ある有益な程度までTLEに付随する多くの症候のいくつかを抑制又は寛解することを意味する。例示の目的だけのために言えば、本発明の処置法は側頭葉の小さな領域に影響を及ぼし、そして意識には影響を与えない単純部分発作(SPS)の悪影響を寛解又は低減する。これらは初期に感覚を引き起こす発作である。これらの感覚は記憶に関し、例えば、既視感(精通感)、未視感(未精通感)、特定の単一又は1組の記憶、又は記憶喪失である。感覚は実際には存在しない音、又は旋律のような聴覚性、或いは味のような味覚性、又は匂いのような嗅覚性でありうる。感覚はまた視覚的でありえ又は皮膚の感覚又は内臓の感覚でありうる。後者の感覚は体中を移動するようである。不快性又は多幸性感覚、恐怖、怒り、及び他の感情もまた単純部分発作(SPS)の間起こり得る。多くの場合、TLEのSPSを患う人は、感覚を記述するのが困難である。SPSは多くの場合「発作の前兆」と呼ばれ、更に激しい発作の前触れと考えられることもある。このような感覚は、本発明の方法を適用することにより軽減する。
【0033】
他の事例では、複雑部分発作(CPS)の重症度が本発明の方法を適用することにより軽減する。このような発作は意識をある程度障害し、通常はSPSと共に発症し、その後側頭葉の大部分に拡がる。徴候は無動凝視、手及び口の自動的運動、他人への応答不能、異常発言、又は異常行動を含む。
更に他の事例では、本発明の方法は続発性全汎強直間代発作(SGTCS)に付随する徴候の重症性を軽減する。これらは初期にはSPS又はCPSで発症し、その後、腕、体幹、及び脚が曲がった位置又は伸ばした位置で硬化する。この後、脚と体幹の突然の粗いけいれんが起こる。
【0034】
「同時投与」という言葉は2つ以上の活性化合物を、TLE又はそれに関連する徴候を処置又は防止するのに有効な量だけ投与することを意味する。同時投与という言葉は、好適には2つ以上の活性化合物を患者に同時に投与することを意味するが、その化合物が正確に同時刻に投与される必要はなく、効果的な濃度が血液、血清、血漿又は肺組織内で同時に発見されるように化合物の量が患者又は被験者に投与されればよい。
「慢性的に投与される」又は「慢性投与」とは、一連の処置の中で、NKCC1抑制剤及び、選択肢として、GABA調節組成物を、被験者の状態又は被験者から提示される徴候により変化するある期間内に、1回より多い回数投与することを意味する。
【0035】
本発明の方法は、NKCC1抑制剤とGABA調節組成物が担体材料で組合わされる剤形を使用してもよい。剤形と投与経路は処置される被験者及びTLE又は関連徴候の状態により異なる。同じ観点から、本願の方法は、0.25ミリグラム−1グラム、より好適には約1ミリグラム−約750ミリグラム、更に好適には、約10ミリグラム−約500から600ミリグラムの間のNKCC1抑制剤とGABA調節組成物を含む剤形の投与を必要とする。
特定の患者に対する特定の投薬及び投与計画は種々の要素により異なることを理解しなければならない。それら要素は、使用された特定の化合物の活性、年齢、体重、全体的健康、性別、食事制限、投薬の時間、排泄速度、薬の組合せ、及び処置担当医の判断、及び処置中の特定の疾病又は症状の重症度を含む。
【0036】
活性化合物の投与は、連続性(静脈内持続点滴)から日に数回の経口投与(例えば、1日4回(Q.I.D.))までの範囲が有り、また、投与の他の経路の中で、経口、外用、非経口、筋肉内、静脈内、皮下、経皮(これには浸透促進剤を含んでもよい)、バッカル、及び座薬を含んでよい。腸溶性経口錠も化合物の経口投与経路からの生物学的利用能を高めるため使用可能である。最も効果的な剤様は、選択された特定の薬剤の薬物動態および患者の疾病の重症度に依存する。経口剤様は投与の容易性及び予期される患者の応諾から特に好適である。
本発明はさらに以下の実験的セクションで記述されるが、それは説明目的のためだけであり、限定するものではない。
【0037】
(実験の部)
(材料及び方法)
(TLE誘発)
全てのプロセスはニューメキシコ大学健康科学センターに所在の動物介護及び使用委員会により承認され、また全米健康局の実験動物のケア及び使用に関するガイドに従っている。本実験のプロトコルはアンドレ氏他による刊行物のプロトコル(Andre氏他、2007)を僅かに変更したものである。発作誘発の24時間前に生後2−3カ月の雄のウィスター(Wistar)ラットが皮下注射で等浸透圧のNaCl生理食塩水に溶解した3mmol/kgの塩化リチウムを投与された。
その後ピロカルピンが皮下注射(等浸透圧のNaCl内25mg/Kg)で投与された。一度大脳辺縁系のけいれんが始まると、てんかん重積(SE)の発症を確認するため皮下針EEG電極が配置される。てんかん重積は連続高振幅脳波(EEG)スパイキングの発生として定義される(Ormandy氏他、1989)。このような針は25ゲージの皮下注射針(Becton Dickinson)から、ハブを切り取り、そして60度曲げて形成され、それにより針は定位置に留まる。1本の針が頭皮に皮下挿入により頭頂皮質に挿入され、そして対照電極として機能する他の1本の針が肩甲骨に挿入された。グラスモデル8脳波計(アストロ−メド(Astro−Med)社、ウエストワーウィック、イリノイ州)を使用した脳波(EEG)記録は、連続高振幅スパイキングをてんかん重積(SE)発症後1時間に亘って示し、それは頭蓋内に埋め込まれた電極での従前の研究で記録された(Peterson氏他、2005)正常な脳波に対し明確なコントラストを示した。その後記録は全体的に停止された。硫酸アトロピン(10mg/Kg皮下注射)及びジアゼパム(4mg/Kg筋肉内注射)が生存率を上げ、不安を軽減させ、そして筋肉弛緩を誘発するため投与された。ジアゼパムの投与は、発作を停止出来なかったが、1時間以内に筋肉弛緩を誘発し、ラットは脚を使わず腹側面を下にして横になった。アトロピンも発作を停止出来なかった。てんかん重積(SE)発症後12時間後に脳波が再び5−10分間記録され、全てのラットは断続的又は全体に形成される、即ち脳波スパイキングのバーストを有した。この時点でスパイキングのバーストや連続するスパイキングの列のような全体的な脳波スパイキングを示したこれらのラット(ケースの約20%)は、追加の2mg/Kgのジアゼパム筋肉内注射を投与された。てんかん重積(SE)の発症後12時間後にリンゲル溶液内の5%ブドウ糖が皮下注射で投与された。
【0038】
このプロトコルはてんかん重積(SE)の発症後1時間後にジアゼパムを投与した点で、2時間後に投与したアンドレ(Andre氏他、2007)のプロトコルとは異なる。更に我々は末梢性ピロカルピンの影響(Du氏他、1995)を軽減するため、アトロピンを投与した。従って我々のプロトコルはよりマイルドな発作を動物に提供すると信ずる。
【0039】
アンドレ(Andre)氏他のプロトコルは、信頼性を持って成体ラットの100%にてんかんを誘発したと記載されている(Dube氏他,2000b,Andre氏他,2007)。これは、僅かに高いピロカルピン投与量とリチウムを使用したこの研究所のそれより早期のプロトコルからも真実である(Dube氏他,2000b,Andre氏他,2007)。我々のラボでアンドレ(Andre氏他、2007)のプロトコルを使用した研究では、全てのラットが28.6±2.43(標準誤差)日(n=14;24時間/1日の連続ビデオ録画、S.Peterson氏、非公開結果)の潜伏期の後、ランク3,4又は5の自発性発作を発症した。1体のラットだけが15日目に発作を起こした。21日までには4体のラットが発作を起こした。
これらの潜伏期データを正規分布に適合させることにより、平均約25%のラットがピロカルピン/2時間後ジアゼパム処置の3週間以内に最初の発作を起こすことが予期された。
しかし屠殺する前の観察及び取り扱い中にはラットに発作は見られなかった。
我々のマイルドなプロトコルのてんかんの転帰がてんかん重積(SE)後8−16週間、1ラット当り合計36時間までのビデオ録画をしながらテストされた。そして、この期間に13体全てのラットに自発的発作が観察された。てんかん重積(SE)のラットの新しい2グループ(合計12体)によって、我々は潜伏期期間が12.7±1.7(平均±標準誤差)週間であると量定した。全体で203体のラットが使用された、リチウム対照ラット56体、てんかん重積(SE)後2週間のラット42体、SE後3週間のラット47体、ビデオ録画用SE後ラット58体。
【0040】
(スライス準備)
全ての実験は、側頭葉新皮質、嗅内皮質、海馬台及び腹側海馬を含む鋭い形状の水平大脳スライス上で行われた。対照又は発作後2又は3週間のラットは深くケタミンとキシラジン(それぞれ85と15mg/ml)麻酔をかけられそして断頭された。大脳は迅速に移動され酸素負荷された(95%酸素、5%炭酸ガス)氷冷の3 KCL、1.25 NaH2PO4、6 MgSO4、26 NaHCO3、0.2 CaCl2、10 グルコース、220 ショ糖及び0.43 ケタミンを含む(mMで)切断溶液中に置いた。スライス(350ミクロン)は氷冷の切断溶液内でビブラトーム(Dosaka DTK−1000)を使用して薄切りにされ,そこからリカバリー用に95%酸素/5%炭酸ガスで34°Cで平衡化された人工脳脊髄液(ACSF、mMで:126 NaCl,3 KCl,1.25 NaHPO4,1 MgSO4,26 NaHCO3,2 CaCl2及びグルコース)内に移された。1時間後ACSFが取り替えられ、そして記録に使用されるまで室温で保管された。
【0041】
(電気生理)
個々のスライスは固定台顕微鏡(Zeiss Axioskop社、ジュネーブ、ドイツ)に搭載された記録用チャンバに移され、そして暖められ(34°C)、酸素負荷された人工脳脊髄液(ACSF)で2ml/分で灌流された。細胞内記録は、嗅内皮質(EC)第5層ニューロンから、低めの電極抵抗に対しては1M CsSO4又は0.5M酢酸カリウム/0.5M塩化カリウム、又は改良空間クランプ用には2M CsSO4/1M塩化カリウムを充填された、ホウケイ酸ガラスの鋭い微小電極(80−150MΩ)を使用してなされた。Cl−輸送遮断剤がない場合は、全ての細胞内液はGABAA作動性シナプス後電位(PSP)逆転電位に対して同一の結果を示し、逆転電位は従って蓄積された(Misgeld氏他、1986参照)。ブメタニドによるCl−輸送遮断の間、0.5M酢酸カリウム/0.5M塩化カリウムを充填された微小電極がEIPSPを脱分極方向に約20mVだけ変移させるのが確認された。従ってGABAA作動性シナプス後電位(PSP)逆転電位に対するブメタニドの影響に関する全ての最終的な記録/データは、1M K2SO4又は2M CsSO4/1M塩化カリウムで充填された微小電極で得られた。
【0042】
ニューロンは、皮質の厚さの1/4まで、ナノステッパー微小位置決め装置(Scientific Precision Instruments社、オッペンハイム、ドイツ)を使用して、内側嗅内皮質内のアンギュラーバンドル(Angular bundle)のすぐ上に刺入された。記録はAxoclamp2Bアンプを使用して記録され、Axon Instruments Digidata 1322A(Molecular Devices社、ユニオンシティ、カリフォルニア)を使用してデジタル化し記録された。マイナス68mVまでの静止膜電位(RMP)が負のニューロンが、マイナス85mVまでの過分極パルスを使用して、錐体ニューロンを示唆するIhの存在をテストされた(Egorov氏他、2002;Egorov氏他、2003)。この基準では記録されたニューロンの75%は錐体であり、残りは多極ニューロンと推定された。Hamam氏他の研究では鋭い電極を第5層に刺入されたニューロンの85%が錐体ニューロンに典型的な顕著な長い先端樹状突起を示した(Hamam氏他、2000)。我々の研究と対照的にこの研究は非錐体ニューロンにもIhの存在を報告した(Hamam氏他、2000)。同心性2極刺激電極(直径200ミクロン、HFC Inc.社製、メイン州、米国)が嗅内皮質(EC)の第5層に、記録用電極から約150−250ミクロンの距離に配置された。刺激(70−100μ秒、100−800nA)は刺激分離ユニット(A.M.P.I、エルサレム、イスラエル)に接続されたマスター8プログラム可能パルス発生機により制御された。興奮性シナプス後電位(EPSP)及び抑制性シナプス後電位(IPSP)に対する全入力−出力関係が記録され、そして70−90%の最大応答を与える刺激強度が使用された。そこでは計測された刺激強度が最大応答の5−10%を誘発するまで減少した。
【0043】
図2AはGABAA受容体−媒介シナプス後電位の逆転電位を測定するのに使用されるプロトコルを示す。単シナプス性のシナプス後電位(PSP)がグルタミン酸受容体遮断剤CNQX(10μM)及びAPV(50μM)の存在下で誘発された。刺激強度は最大誘起シナプス後電位(PSP)振幅の約80%となるように調整された。電場刺激に先だって、膜電位(Vm)は正又は負の直流(DC)パルス(500ms、400pA以上)で変化された。膜電位が安定に達した後、シナプス後電位(PSP)が誘起された(図中*印)。安定膜電位(図2B、○印)とシナプス後電位(PSP)のピーク電圧(図2B、×印)の両方の電流−電圧関係がプロットされ、そして線形回帰がシグマプロットを使用して計算され、直接、膜電気伝導度の値を与えた。直線的な膜動作の可能性領域内で電流−電圧関係を測定することにより、遅延整流の電圧依存活性化又は他の整流電流に起因する接線コンダクタンスの誤りを避けることができる。逆転電位は回帰直線の交点により決定された。典型的なシナプス後電位(PSP)トレースが対照と3週目の実験動物で示される。CNQX、DL−2−アミノ−5−ホスホノペンタン酸、ピクロトキシン、ブメタニド、ピロカルピン及びアトロピンはシグマ(Sigma)社(セントルイス、ミズーリ州)より入手した。ジアゼパムはHospira Inc.(レイクフォーレスト、イリノイ州)から入手した。1000倍に濃縮された原液はフリーザーに保管され、ACSFで使用する前に希釈された。原液に関しては、CNQXとピクロトキシンはジメチルスルホキシド(DMSO)に溶解され、ブメタニドはエタノールに溶解された。ジメチルスルホキシド(DMSO)(1:1000)もエタノール(1:1000)もシナプス後電位(PSP)、電流−電圧関係、又はバースト応答に影響を与えなかった。
【0044】
(免疫ブロッティング)
免疫ブロッティング研究のため、内側嗅内皮質(EC)の第5−6層が350ミクロンの厚さの切片から切り裂かれ、ドライアイス上でスナップ冷凍され、そしてマイナス80℃で処理まで保管された。膜断片の準備のため、組織は250mMスクロース、10mMトリス−HCl、10mM HEPES、1mM EDTA、及びプロテアーゼ抑制剤(pH7.2)を含む緩衝液内で均質化され、その後、前記のように(Payne氏他、1996)分画遠心分離された。精製された膜断片(50μg)はその後、ドデシル硫酸ナトリウム(SDS)−ポリアクリルアミドゲル電気泳動(7%)及び抗KCC2(1:1000;ニューロマブ(NeuroMab)クローン N1/12、UCデービス)と抗アクチン(1:500;シグマ(Sigma))をそれぞれ使用した免疫ブロッティング用に処理された。
抗原−抗体複合体が、増大した化学発光(Amersham社、アーリントンハイツ、イリノイ州からのECL−プラス)から検知され、X線フィルムはNIH ImageJソフトウェアを使用して定量化された。
【0045】
(蛍光遺伝子プローブ法(FISH))
FISH用に大脳スライスを準備する場合、1つの大脳の半球がドライアイス/エタノールスラリー内で平衡化されたイソペンタンのビーカー内で急速冷凍され、そしてマイナス80℃で保存された。水平脳部位(16ミクロン)がクリオスタットを使用して準備され、スライド(Superfrost Plus,VWR)上に配置され,空気乾燥され、処理までマイナス80℃で冷凍保存された。
【0046】
NKCC1(クローンID4824556、アクセス番号BC033003)及びKCC2(クローンID6838880、アクセス番号BC054808)プラスミドはオープンバイオシステム(Open Biosystem)社(ハンツビル、アラバマ州)から入手した。NKCC1cDNAシークエンスは、Sall/XhoI及びBamHI制限酵素部位間のpBluescriptRベクターに挿入された。KCC2cDNAシークエンスは、EcoRI及びNotI制限酵素部位間のpYX−Ascベクターに挿入された。プラスミドはその後NKCC1及びKCC2シークエンスを確認するため配列決定された(DNAリサーチサービス、ニューメキシコ大学、健康科学センター)。EcoRI及びNotIは共に異なる長さのアンチセンス鎖を生成するのに使用され、KpnIはセンス鎖を生成するのに使用された(ニューイングランド バイオラボ社、Ipswich,マサチューセッツ州)。KCC2のアンチセンス鎖はAscIを使用し、センス鎖はPacIを使用して生成された。制限消化反応は37℃で2時間インキュベートされた。
【0047】
NKCC1a,b及びKCC2a,bに対する単一標識化FISHが前述(Guzowski氏他、1999)のように実行された。概要は、ジゴキシゲニン標識化KCC2及びNKCC1全長アンチセンスリボプローブが、直線化されたcDNAから商業的に入手可能な転写キット(Maxiscript,アンビオン、オースチン、テキサス州)を使用して生成され、そして事前混合されたRNAジゴキシゲニンがT3重合酵素(ロシェ分子生化学、パルアルト、カリフォルニア州)を使用してヌクレオチドを標識化した。NKCC1及びKCC2のジゴキシゲニン標識化リボプローブのハイブリダイゼーションは共に56℃で一晩かけて異なるスライド(1ng/μl)上で行われた。その後、抗ジゴキシゲニン セイヨウワサビ過酸化酵素複合体(HRP;1:200;ロシェ分子生化学、パルアルト、カリフォルニア州)が4℃で一晩インキュベートされた。TSA−シアニン−3(Cy3)(1:50;パーキンエルマー、Waltham, マサチューセッツ州)がHRP複合体を検知するのに使用された。核は、4’,6’−ジアミジノ−2−フェニルインドール(DAPI)(1:500;Invitrogen、Carlsbad,カリフォルニア州)によって対比染色された。画像は、スピンニングディスク共焦点システム(スピンニングディスク共焦点画像化装置CARV、Atto生科学、ロックビル、メリーランド州)及びCoolSNAP−Hq CCDカメラ(Roper科学、ツーソン、アリゾナ州)を備えるニコンTE2000U落射蛍光顕微鏡(20倍対物)により獲得された。画像はMetaMorphソストウェア(ユニバーサル画像)を使用して更に分析され、NIH ImageJソフトウェアを使用して定量化された。信号は背景(画像内の明確にネガティブな領域をカバーする注目画像領域(ROI)で得られた)に対して補正され、核染色に対する比率が計算された。ここにおいてこれらのデータは対照ラットに関して正規化されていた。
【0048】
(免疫組織化学的検査)
スライスは100mMリン酸緩衝食塩水内の4%パラホルムアルデヒドで一晩固定化され、PBS内の15%及び30%ショ糖で凍結保護され、その後最適切断温度(OCT)化合物(サクラファインテック、トーランス、カリフォルニア州)内で凍結された。
免疫組織化学的検査が室温で24時間かけて300ミクロン浮遊部位上で、抗KCC2(ニューロマブ(NeuroMab)クローン N1/12、UCデービス))又はNKCC1抗体(ラビットポリクローナル、Chemicon,Temecula,カリフォルニア州;結果はカリフォルニア大学リバーサイド校のC.Lytle博士から提供されたマウスNKCCモノクローナル抗体MAbT4で得られた結果と合致した)で実施された。スライスはガラススライド上に搭載され、画像がBiorad二光子顕微鏡とスライス表面下75ミクロンに配置された40倍対物レンズにより撮像された。ニューロトレース蛍光染色(モレキュラープローブ社、Eugene,オレゴン州)がクリトスタットを使用して調製された60ミクロン区画内のニューロンを対抗染色するのに使用され、そして画像はCCDカメラと40倍対物レンズを備える落射蛍光顕微鏡で撮像された。NKCC1抗体での免疫染色のため、組織区画は遮断前に1%ドデシル硫酸ナトリウム(SDS)及び8%2−メルカプトエタノールで5分間前処理された(Sung氏他、2000)。この前処理を伴わない多くの試みにおいて、MAbT4抗体で明確なNKCC信号を獲得するのに失敗した。全ての区画はPBS−T(0.2%トリトン−X−100を含むリン酸緩衝食塩水(PBS))内の10%正常ヤギ血清で遮断され、一晩4℃でそれぞれの1次抗体(1:200)と共にインキュベートされた。PBS−Tで広範に洗浄した後、組織区画はCy3接合ヤギ抗マウス又は抗ウサギIgG(1:200、ジャクソン免疫研究所、ウェストグローブ、ペンシルバニア州)内でインキュベートされた。対照は1次抗体の無い同一の処理を受け、そして常にネガティブであった。区画は広範にPBS−T内で洗浄され、観察のためカバーグラスを付けられた。細胞核の対抗染色にはヘキスト33342DNA染色(10ミクロン;Invitrogen,Carlsbad,カリフォルニア州)が使用された。
【0049】
(統計)
平均±標準誤差が別途指定されない限り示される。データのグループ間の統計的比較はシグマプロット(SigmaPlot)ソフトウェアで、必要に応じ独立又は対応のある両側スチューデントのt−検定が帰無仮説を検証するため実行された。0.05未満の確率値Pは統計的に有意であると見做された。
【0050】
(実施例1)
(てんかん重積(SE)後の嗅内皮質(EC)第5層ニューロンにおける多シナプス性バースト応答の発生)
対照ラットからの切片において、嗅内皮質(EC)第5層に対する単一のシナプス前性の刺激が静止膜電位−70mVにおいて振幅20mVまでの単シナプス性の急速な興奮性シナプス後電位(EPSP)を第5層ニューロンに誘発し(図1A)、その後、膜保持電位により振幅が異なる急速な抑制性シナプス後電位(IPSP)を誘発する。対照的に、てんかん重積(SE)後3週間後の記録では、刺激強度を対照調製より大きく減少させた場合でも単一シナプス性刺激に応答して多シナプス性てんかん活性を示した。(図1B)多シナプス性バースト応答は、より高い脱分極電位への活動電位の発火の場合を除いて、DC注入による−95mVから−60mVまでの範囲のシナプス後性膜電位の変化によっては殆ど変化を示さなかった。示された実施例では、弱い刺激での多シナプス性脱分極の僅かな遅れが、初期興奮性シナプス後電位(EPSP)を明白に顕在化させ;そして振幅で僅か1−2mVとして見える。(挿入図、細胞は負のDCにより発火を防ぐため過分極されている。これらの弱い刺激は、高信頼度で多シナプス性バースト応答を、2週目でテストされた10.8%のニューロン(n=37)で,そして3週目でテストされた61.8%のニューロン(n=34)で誘発した(図1C)。この量定のため、多シナプス性バーストは、−75mV<RMP<−68mVから誘発され、半振幅で150ms以上継続するシナプス後性応答として定義された。対照切片では、このような応答は42の記録されたニューロンの内で1つにおいてのみ観察された。
【0051】
図1およびその図に反映されているデータを獲得した実験のさらなる詳細は以下の通りである。
(図1:てんかん重積(SE)後の嗅内皮質(EC)第5層ニューロンの増大した多シナプス性バースト興奮)
A:無処置ラットからの嗅内皮質(EC)第5層ニューロンの単一シナプス前性刺激(*,500nA,70μS)に対するシナプス後性応答。
B:てんかん重積(SE)3週間後の第5層ニューロンのはるかに小さい刺激(*,100nA,70μS、図10及び11も参照)に対する多シナプス性バースト応答。初期に誘起されたシナプス後電位(ボックス内に示す)は、拡大挿入図(30msX5mV)に示される。この記録に対しては、細胞は発火を防ぐため負のDCにより過分極されている。
C:単一の刺激に多シナプス性脱分極で応答する嗅内皮質(EC)第5層ニューロンの比率を示す棒グラフである。単一の刺激は対照ラットから(n=42)の嗅内皮質(EC)第5層細胞の内の僅か2.4%でしか多シナプス性応答を誘起しないが、てんかん重積(SE)後2週間後(n=37)及び3週間後(n=34)のラットではそれぞれ10.8%と61.8%の細胞が多シナプス性応答を誘起した。
【0052】
(実施例2)
(てんかん重積(SE)後、GABAA作動性のシナプス後電位(PSP)が脱分極化する)
長期に渡って確立されたTLEを有する海馬台ニューロン亜集団におけるEIPSP及びCl−輸送体システムの重要な変化の現存する観察(Cohen氏他、2002;de Guzman氏他,2006;Palma氏他,2006;Huberfeld氏他,2007;Munoz氏他,2007;Sen氏他、2007)に依存して、我々は、バースト活性を促進する可能性のある、極めて初期の対象段階におけるGABA作動性伝達の継続的変化を探した。図2Aは、CNQX(10μM)とAPV(50μM)によるグルタミン酸塩受容体の遮断の間の単シナプス性GABAA作動性シナプス後電位(PSP)を示し、そのPSPは電場刺激により嗅内皮質(EC)第5層に誘発され、一方細胞は異なる安定膜電位に保持された(前記(材料及び方法)参照)。PSP逆転電位(E(PSP))は、安定膜電位(図2Bの○)及びPSPのピーク電圧(図2Bの×)の両方の電流−電圧関係をプロットし、線形回帰を計算することにより決定された。膜電位とPSP線形回帰との交点が逆転電位である(図2B、矢印)。対照ラットとSE後2週目と3週目のラットからの代表的ニューロンの電流−電圧プロットは、PSP逆転電位(E(PSP))の進行性変移を示し、それらはそれぞれ、対照の−68.3±2.1mVから、SE後2週目の−52.6±1.8mV、SE後3週目の−34.8±1.4mVであった(図2B1−B3)。データがヒストグラムで示された場合、SE後2週目と3週目の両方で、処置ラットからのスライス内に2つの細胞クラスが観察された(図2C、n=84)。1つのクラスでは平均PSP逆転電位(E(PSP))は対照と類似していた、即ち静止膜電位(RMP)に対し5mV未満ポジティブであった(2週目と3週目に分析した細胞全体のそれぞれ23.5%及び19.2%)。もう1つのクラスは強い変移を示した(2週目と3週目に分析した細胞全体のそれぞれ76.5%及び80.8%)。両方の細胞クラスは同一のラットからの複数のスライス内だけでなく、単一のスライス内にも観察された。SE後3週間で約25%と推定された(前記(方法)参照)屠殺前自発発作は、これらのラットからのニューロンのPSP逆転電位(E(PSP))をさらに変移させ、これによりさらに強く変位したPSP逆転電位(E(PSP))を有する亜集団を確立する可能性が有った。ヒストグラムは、このような顕著なニューロン亜集団のサインを全く示さず、そのことは、自発的発作が全く起らなかったか、あるいはそれがPSP逆転電位(E(PSP))の更なる変移を引き起こさなかったかを示唆した。
【0053】
集団データの棒グラフ(図2D)は、変化が起こらなかった集団の部分を除外した、PSP逆転電位の平均値の時間経過変移をまとめたものである(E(PSP)=静止膜電位(RMP)−71.9±1.46mVに対し3.6±0.5mVポジティブ、n=24;SE後2週目E(PSP)=静止膜電位(RMP)−70.9±2.8mVに対し18.0±3.6mVポジティブ、t56=8.3、P=0.006、n=26;SE後3週目E(PSP)=静止膜電位(RMP)−72.9±2.8mVに対し45.1±6.2mVポジティブ、t48=9.81、P=0.0008、n=21)。3週目の測定点において、変移集団及び非変移集団のそれぞれ81%と75%が錐体ニューロンであり、それぞれ19%と25%が多極性ニューロンであった。これらの分布は、嗅内皮質(EC)第5層のこれら2つの細胞タイプの全体分布から有意に差異がなかった(Egorov氏他、2002;Egorov氏他、2003)。3週目のポジティブな変移がPSP逆転電位(E(PSP))をスパイク開始のレベルにごく近い値に持ってゆくことは注目すべきである。従って、てんかん重積(SE)後のGABA放出は反復性発火を効果的に制限しない可能性が有り、逆にニューロンを興奮させる可能性がある。
【0054】
対照ニューロン内の僅かに脱分極した静止膜電位に対する抑制性シナプス後電位(IPSP)は、明らかにCl−の外向き輸送だけが発現する状態に対して不可解に見えるかもしれない(以下参照)。Cl−の逆転電位は静止膜電位にたいしネガティブ(又はCl−の外向き輸送がCl−の流入の後にのみ活性になった場合、静止膜電位と等しい)でなければならない。しかし、GABAAチャネルは炭酸水素イオンをある程度、陰イオンの約1/6、伝導し、GABA−PSP逆転電位をECl−とEHCO3−との間に置く(Bormann氏他,1987)。細胞内エネルギー代謝は2酸化炭素を生成し、それは炭酸脱水酵素により触媒されて水と逆反応し、炭酸水素イオンを生成する。このプロセスは強い外向きの傾斜をHCO3−に与え、逆転電位を−10mVにまでポジティブ側にする(Ben−Ari氏他,2007)。このことは正常な動物ではシナプス後電位(PSP)逆転電位をECl−よりポジティブな値に変移させる(Staley氏他,1995)。また,Cl−の内側輸送の限定的出現は、僅かに脱分極したシナプス後電位(PSP)逆転電位を生成可能である(以下参照)。
【0055】
逆転電位の変移に加えて、膜接線コンダクタンス、およびコンダクタンスが、シナプス後電位(PSP)がてんかん重積(SE)後に進行的に減少する間、増加する(図2B1−B3参照)。静止コンダクタンスは、対照での5.9±0.7nSから、てんかん重積(SE)後2週間及び3週間でそれぞれ4.8±0.7(t12=3.66、P=0.032)と3.6±0.4nS(t13=3.79、P=0.026)に減少した。これは恐らく漏出イオンチャネルの劣化による(Bernard氏他,2004;Borter氏他,2006)。同様に、GABA活性化ピークコンダクタンスは、対照での8.5±0.69nSから、てんかん重積(SE)後2週間及び3週間でそれぞれ2.1±0.43(t12=13.12、P=0.0018)と0.24±0.07nS(t13=6.16、P=0.0005)に減少した。この減少は、100Hzで3度繰り返された、入出力関係の最大値に近い刺激強度により誘発された、大きなGABA誘発シナプス後電位(PSP)により確認され、それは、3度目の刺激後、3グループ全てに大きな非撹乱PSPを与えた。
【0056】
図2、及びその図に反映されたデータを与えた実験のさらなる詳細は以下の通り。
(図2.てんかん重積(SE)後2週間後と3週間後のGABAA誘発シナプス後電位(PSP)逆転電位のポジティブ変移)
図2A:対照(上側の軌跡)とてんかん重積(SE)後3週間後のラット(下側の軌跡)からの嗅内皮質(EC)第5層内のニューロンの、鋭い微小電極により記録された、5つの異なる膜電位において誘起された、単一シナプス性GABAA誘発シナプス後電位(PSP)逆転電位。膜電位は微小電極からの電流の印加(500m秒パルス)により変化した。*(アスタリスク)はシナプス前性刺激の時点を示す。記録は、AMPA/カイニン酸グルタミン酸受容体拮抗薬CNQX(10μM)及びNメチルDアスパラギン酸(NMDA)受容体拮抗薬APV(50μM)の存在下で行なわれた。
図2B1−2B3:刺激前性膜電位(図内の○印)及び刺激後15m秒後に測定されたGABA誘発シナプス後電位(PSP)の絶対値(図中×印)と電極電流との電流―電圧関係。データは図2Aのプロトコルを使用して獲得され、線形回帰で適合された。膜電位とPSPプロットとの交叉はPSP逆転電位(EPSP,矢印)で起こる。
図2C:測定3時点で2mV未満のGABAA誘発シナプス後電位(PSP)逆転電位を示す嗅内皮質(EC)第5層内のニューロンの数のヒストグラム。包絡線(点線の曲線)は5つの明白な集団のガウス適合である。対照ラットからのニューロンでは、PSP逆転電位(EPSP)ガウス適合は中央値約−69mVを有した。てんかん重積(SE)後2週間及び3週間後のラットからのニューロンは、非変移と有意の変移の2つのクラスに分かれた。2週間目には77%のニューロンが約−54mV、3週間目には81%のニューロンが約−28mVの変移した平均値を示した。非変移ニューロンは集団のそれぞれ23%と19%を占めた。それらのPSP逆転電位(EPSP)分布は対照グループから識別可能であった。対照、SE後2週間後、3週間後の記録された細胞数Nはそれぞれ、24,34及び26であった。
図2D:SE後2週間後(n=26)、3週間後(n=21)の変移集団の静止膜電位(RMP)に対するGABA誘発シナプス後電位(PSP)逆転電位(EPSP)の平均値±標準誤差は、対照と比較して逆転電位の進行性脱分極変移を示す(n=24,SE後2週間後(**)p0.01未満、SE後3週間後(***)p0.001未満)。
【0057】
(実施例3)
(てんかん重積(SE)後NKCC1mRNAの増加)
てんかん重積(SE)後強く脱分極するGABA誘発シナプス後電位(PSP)は、活性Cl−内側方向輸送を必要とする。しかし成体ニューロンには殆どNKCC1の発現がないことが知られている(Delpire,2000;Wang氏他,2002)。我々は、嗅内皮質(EC)第5層内の成人ニューロンで、NKCC1 mRNA発現が、初期の発症段階を再現して、てんかん重積(SE)後の潜伏期に再確立されるか否かを検証した(山田氏他、2004;Ben−Ari氏他,2007)。我々は、従前の研究と適合して、対照ラットの嗅内皮質(EC)第5層内においてNKCC1 mRNA蛍光発光が非常に低いか存在しないことを発見した。対照的に、リチウム−ピロカルピン誘発てんかん重積(SE)後、2週間後、特に3週間後では、明瞭なNKCC1染色を確認した(図3B)。てんかん重積(SE)後、2週間後では、NKCC1 mRNA蛍光発光が対照の237±33.4%に増大し、3週間後では452±33.5%に増大した(図3B、E、n=100注目画像領域(ROI)、前記(方法)参照、各グループ4ラット、それぞれt38=2.15、P=0.037及びt49=3.54、P=0.0009)。これらのパーセントは、対照における発現が低いことを前提にすると、幾分任意であるが、発現の非常に大きな増加を示している。DAPIによる核の対抗染色は、分析期間中には細胞数に変化を与えなかった(対照、SE後2週間後及び3週間後のそれぞれにおいて、12.2±2.7,12.8±3.4及び12.5±3.1細胞核数/500平方ミクロン、それぞれt68=−0.49,P=0.62及びt68=−2.38,P=0.24、n=100注目画像領域(ROI)、各グループ4ラット;図3A2、B2)。DAPI及びNKCC1 mRNAの画像の重なりは標識化mRNAの初期の核周囲の位置を示す(図3B2)。NKCC1ポジティブ細胞と場内の全細胞との比較分析では、対照組織では細胞の5.1±2.1%の標識化に対して、SE後2週間後及び3週間後のそれぞれにおいては28.5±6.7%及び56.7±4.3%を示した(図3D、n=100注目画像領域(ROI)、各グループ4ラット、それぞれt38=3.27,P=0.042及びt49=2.71,P=0.0096)。
【0058】
図3、及びその図に反映されたデータを与えた実験のさらなる詳細は以下の通り。
(図3.てんかん重積(SE)後嗅内皮質(EC)第5層内において進行的に増大する、NKCC1のmRNA、Cl−内側方向輸送)
図3A、B:蛍光遺伝子プローブ法(FISH)は、ピロカルピン誘発発作の3週間後にNKCC1のmRNAが対照(赤い信号、A1,B1)に比較して発現量の増大を示した。DAPI核染色は細胞数に変化の無いことを示した(青の信号、図3A2、図3B2)。下方パネルA3、B3は図3A1,2及び図3B1,2で概観された領域の拡大統合図であり、NKCC1のmRNAの核周辺配置を示す。
図3C:てんかん重積(SE)後のNKCC1のmRNAの増加を示す棒グラフである。データは、所定の注目画像領域(ROI)内の全NKCC1信号に対する核染色の蛍光の比率を生成することにより定量化された。これらの比率はその後、対照ラットからの値に対して正規化された(平均値±標準誤差、n=100注目画像領域(ROI)、各グループ4ラット、SE後2週間後(*)p0.01未満、SE後3週間後(***)p0.001未満)。
図3D:DAPI染色により決定される、核の数に対する%として表現される、NKCC1のmRNA−ポジティブの細胞数の平均値(平均値±標準誤差、n=約300注目画像領域(ROI)、各グループ4ラット、SE後2週間後(*)p0.05未満、SE後3週間後(**)p0.01未満)。
【0059】
(実施例4)
(NKCC1タンパク質の増加)
潜伏期間中に起こったNKCC1のタンパク質レベルの変化を分析するため、蛍光免疫組織化学的検査を実施した。これらのデータはケミコン(Chemicon)社NKCC1抗体によって得られ、そして図4に示すように、SE後2−3週間において、NKCC1タンパク質の進行的増加を示した。一方対照ラットからのスライスでは、特定の信号はほとんど得られなかった(図4A−4C)。同様の結果がMabT4 NKCC抗体で得られた。NKCC1画像の上に染色核画像(ヘキスト33342)を重ね合わせることにより、特にSE後3週間後において、NKCC1の優先的な核周囲の、そして細胞体性の位置が明らかにされ、ニューロン性タンパク質合成の有意の増加を示唆した(図4B3、4C3及び拡大された図4B4,4C4)。繰り返すが、ヘキストで染色された核の数は3つのラットグループの間で有意の差異を示さなかった(対照85.3±5.3に対し、発作後2週間後は97.7±3.4、発作後3週間後は86.0±4.9、4ラット、それぞれt20=−1.33,P=0.20及びt18=−2.35,P=0.81、n=100注目画像領域(ROI)、各グループ4ラット)。図4Dは細胞体からのNKCC1の蛍光強度の平均値は、発作後2週間後及び3週間後に対照の背景蛍光の約350%及び860%であったことを示す(n=100注目画像領域(ROI)、各グループ4ラット、それぞれt64=5.32,P=0.0006及びt51=4.56,P=0.0008)。神経線維網ではNKCC1の平均蛍光強度は、発作後2週間後及び3週間後に対照の背景蛍光のそれぞれ123±21%及び222±45%であった、その結果は樹状突起内においてNKCC1タンパク質が遅延して増加することを示唆した(n=100注目画像領域(ROI)、各グループ4ラット、それぞれt32=4.25,P=0.025及びt36=5.31,P=0.008)。個々の細胞に関しては、SE後2週間後と3週間後で、それぞれ細胞体の31.2±6.9%及び56.3±11.2%が明確にNKCC1に対しポジティブであったが、対照では明確なものは無かった(図4E、n=100注目画像領域(ROI)、各グループ4ラット、それぞれt42=11.24,P=0.0009及びt37=7.12,P=0.0007)。
【0060】
図5ではニューロン特異ニューロトレース(Neuro Trace)対抗染色が使用され、発作後3週間後には77.1±8.1%のニューロンがNKCC1ポジティブになったことを示した(対照組織:3.0±1.3%、各グループn=3ラット、それぞれt21=3.78,P=0.008及びt33=4.23,P=0.001)。NKCC1ポジティブのニューロンの比率がNKCC1ポジティブの細胞の比率より高いことが、NKCC1がニューロンにおいて増大していることを示した。ニューロトレース(Neuro Trace)ポジティブな細胞数は実験期間中安定し、(対照の35.4±7.2に対し、発作後2週間後は37.1±6.4及び3週間後は36.6±6.7、各グループ3ラット、それぞれt12=−0.51,P=0.6及びt16=−0.30,P=0.8)、それは安定的なH33342細胞数がニューロン損出とグリア細胞の増大との相殺の結果ではないことを示唆した。NKCC1発現及びポジティブニューロン比率の漸増は、GABAA誘発シナプス後電位(PSP)逆転電位の同時発生的進行性脱分極変移を説明するのによく適合した。
【0061】
図4,5、及びそれらの図に反映されたデータを与えた実験のさらなる詳細は以下の通り。
(図4.NKCC1タンパク質も増加した。)
図4A、4B、4C:蛍光免疫組織化学的検査は、嗅内皮質(EC)第5層内においてSE後2−3週間に、NKCC1タンパク質の発現の進行的増加を示した(赤色信号、A1,B1,C1)。ヘキスト(Hoechst)33342核対抗染色は安定的細胞数を示した(A2,B2,C2,緑色信号)。上記パネルセットの統合図(A3,B3,C3)は発現の拡散的増加を示す。これらパネルのボックスに囲まれた領域は、NKCC1の核周辺及び細胞体系の位置をより明示するため、拡大されて下の図に示される(A4,B4,C4)。
図4D:SE後のNKCC1タンパク質の増加を示す棒グラフである。データは、所定の注目画像領域(ROI)におけるNKCC1信号合計に対する核染色の蛍光合計の比率を形成して定量化された。その後これらの比率は対照ラットからの値で正規化された(平均値±標準誤差、3ラット、SE2週間後(**)ではpは0.01未満、SE3週間後(***)ではpは0.001未満)。
図4E:核数の%で表示されるNKCC1ポジティブ細胞の平均数(H33342、平均値±標準誤差、3ラット、SE2週間後及び3週間後(***)ではpは0.01未満)。
【0062】
(図5.SE後、嗅内皮質(EC)第5層内ニューロンがNKCC1タンパク質の発現を増加)
図5A、B:蛍光免疫組織化学的検査は、嗅内皮質(EC)第5層内においてSE後3週間に、NKCC1タンパク質の強い発現を示した(赤色信号、A1,B1)。ニューロン特異ニューロトレース(Neuro Trace)対抗染色は安定的細胞数を示した(A2,B2,緑色信号)。統合図はNKCC1の核周辺及び細胞体系の位置を示す(A3,B3)。ここでは厚さ60ミクロンの組織ブロック中のニューロンが示される。
図5C:ニューロトレース(Neuro Trace)に対する%で示されるNKCC1ポジティブニューロンの平均値であり、嗅内皮質(EC)第5層内においてピロカルピン誘発SE後2週間後及び3週間後に進行的に増加している(平均値±標準誤差、3ラット、SE3週間後(***)ではpは0.01未満)。
【0063】
(KCC2 mRNAの進行的減少)
示されたNKCC1の増加は活性Cl−の摂取を示唆する。ニューロンのKCl共輸送体KCC2による正常型のCl−の放出(Ben−Ari氏他、2007)は、このCl−の摂取と対抗し、そして大なり小なりCl−の摂取を妨げる。もしKCC2出現が維持されれば、それはCl−の摂取の効率を限定し、そして代謝エネルギーを浪費する。これが事実であることを検証するため、KCC2 mRNAの発現を研究した。
【0064】
上記のように、我々は嗅内皮質(EC)第5層内においてKCC2 mRNAの発現レベルを蛍光遺伝子プローブ法により評価した。図6はKCC2 mRNAが対照動物(A1)において多量に発現し、一方SE3週間後(B1)には減少することを示す。核のDAPIによる染色は3時点の間に有意の差異を示さなかった。(11.9±2.9,1±2.33.8及び12.1±3.2核数/500μm2、それぞれt24=−0.36,P=0.17及びt24=−0.41,P=0.24、図6A2、6B2)。統合蛍光画像(図6A3、6B3)は、KCC2 mRNAが核内及び核周辺領域内に配置されることを示した。
【0065】
対照に対し正規化されたKCC2 mRNAの蛍光は、SE後2週間後及び3週間後においてKCC2 mRNA発現がそれぞれ対照の81±8.3及び44±7.8%に進行的に減少することを示した(図6C、nは約100注目画像領域(ROI)、(方法)参照、各グループ4ラット、それぞれt62=1.95,P=0.008及びt62=2.34,P=0.0095)。同様に、核数に対する嗅内皮質(EC)第5層内のKCC2 mRNAを発現する(図6A3、6B3に示すように核の周りの少なくとも2つの赤色の点)細胞数の比率は、対照動物の嗅内皮質(EC)第5層内における全細胞数の63.9±8.5%から、SE2週間後及び3週間後にそれぞれ47.3±6.7及び36.3±4.3%に減少した(図6D、平均値±標準誤差、nは約300細胞、各グループ4ラット、それぞれt34=3.14,P=0.045及びt28=4.21,P=0.007)。
【0066】
図6、及びそれらの図に反映されたデータを与えた実験のさらなる詳細は以下の通り。
(図6.KCC2に対するmRNA,ニューロンのCl−の外向き輸送体は、嗅内皮質(EC)第5層内においてSE後進行的に減少する、A、B)
蛍光遺伝子プローブ法で検知されたKCC2 mRNAの発現は、対照と比較してピロカルピン誘発SE後3週間後においてKCC2 mRNAの減少を示した(A1、B1、赤信号)。DAPI核染色は細胞数が変わらないことを示唆した(A2,B2,青色信号)。図A1,A2,及びB1、B2で囲った領域の拡大統合図は、KCC2 mRNAの核周囲の配置を示す(A3、B3)。
図6C:SE後のKCC2 mRNAの減少を示す棒グラフである。データは、所定の注目画像領域(ROI)におけるKCC2信号合計に対する核染色の蛍光合計の比率を形成して定量化された。その後これらの比率は対照ラットからの値で正規化された(平均値±標準誤差、3ラット、SE2週間後(**)ではpは0.01未満、SE3週間後(***)ではpは0.001未満)。
図6D:核数の%で表示されるKCC2ポジティブ細胞の平均数(DAPI、平均値±標準誤差、nは約300細胞、各グループ4ラット、SE2週間後(*)及びSE3週間後(**)では、pはそれぞれ0.05未満及び0.01未満)。
【0067】
我々はまた、対照ラットとSE後ラットからの嗅内皮質(EC)第5層内におけるKCC2タンパク質レベルを、ウェスタンブロット法と単一細胞分析を使用して調査した。図7A,7Bに示すように結果は、KCC2タンパク質レベルが、SE2週間後及び3週間後にそれぞれ対照レベルの78.8±7.5及び38.1±4.3%へと、有意な減少を示した(平均値±標準誤差、n=5ラット、それぞれt12=2.17,P=0.01及びt18=4.6,P=0.0006)。蛍光免疫組織化学的検査を使用して、KCC2タンパク質の分布及び処置後のその消失について研究した(図7C)。対照ラットでは、KCC2の免疫活性は、散在する神経線維網信号の上の密集した細胞体系の蛍光として検知された(図7C、一番上のパネル、及び挿入図)。SE後2週間及び3週間後では免疫蛍光はニューロンの細胞体内で漸減し(図7C、7D)、嗅内皮質(EC)第5層内ニューロンの細胞体系領域に弱い離散した蛍光が残った(図7C、真中及び一番下のパネル、及び挿入図)。KCC2に対する細胞体性の蛍光は、SE2週間後及び3週間後にそれぞれ対照レベルの66±3.5及び34±1.9%に減少した(図7D、平均値±標準誤差、nは4ラットに対して約100注目画像領域(ROI)、それぞれt34=7.85,P=0.007及びt41=6.39,P=0.00006)。
【0068】
神経線維網において、KCC2信号減衰はさらに遅延する。ここではKCC2信号は2週間及び3週間後にそれぞれ96±4.1%及び60±4.4%に減少した。神経細胞体と神経線維網の体積を概略1:1と推定すると、KCC2免疫蛍光の合計の変化はウェスタンブロットのデータとよく一致する。ニューロンが死滅せず、かつグリアに置き換わることを証明するため、ニューロン特異性の蛍光ニューロトレース染色が対抗染色に使用された。これら染色はニューロンの安定的な数(対照が157.6±6.5に対して、発作後3週間後で154.6±21.3、3ラット、t26=2.34,P=0.44)を示し、そしてKCC2信号がニューロンに高度に特異的(KCC2ポジティブ細胞の95%超がニューロトレースポジティブ)であることを示した。興味深いことに、ピロカルピン多量投与モデルでは、嗅内皮質(EC)からのてんかん様活性の海馬への拡散に対するこの構造のゲートキーパー機能を損なう、下流歯状回におけるKCC2の早期、機能的減少の証拠もある(Pathak氏他、2007)。このゲートキーパー機能の損傷と同時に、生体内てんかん様脳波(EEG)活性の証拠もある(El−Hassar氏他、2007)。
【0069】
図7、及びその図に反映されたデータを与えた実験のさらなる詳細は以下の通り。
(図7.SE後KCC2タンパク質も減少する)
図7A:ウェスタンブロットKCC2バンド(約150kDa)は、ピロカブリン誘発SE後2週間及び3週間後のラットの嗅内皮質(EC)第5層内KCC2が対照と比較して有意に漸減することを示す。βアクチン(約40kDa)は負荷制御として機能した。
図7B:図7Aのブロットからの、対照の%で表示される平均光学密度(平均値±標準誤差、n=5、SE2週間後(**)ではpは0.01未満、SE3週間後(***)ではpは0.001未満)。これらのデータはmRNA及び抑制性シナプス後電位(IPSP)逆転電位データと非常によく一致する。
図7C:免疫組織化学的検査はピロカブリン誘発SE後2週間及び3週間後のKCC2タンパク質の漸減を示し、それは細胞体で最も顕著であった。各パネルの白抜きのボックスは、白矢印で識別される細胞の拡大図である。
図7D:SE後のKCC2免疫組織化学的検査の平均信号で、対照に対して正規化されている(平均値±標準誤差、nは4ラットに対して約100注目画像領域(ROI)、SE2週間後(**)ではpは0.01未満、SE3週間後(***)ではpは0.001未満)。
【0070】
(実施例7)
(NKCC1及びKCC2における初期の変化は嗅内皮質(EC)第5層に特異である)
てんかん重積(SE)中に発生する高振幅脳波(EEG)スパイキングは、海馬及び海馬台を含む他の皮質層の嗅内皮質(EC)第5層で見られるNKCC1及びKCC2出現における進行性変化をトリガーするのに十分でありうる。従って我々は、分析を海馬台、歯状回、嗅周囲皮質だけでなく、嗅内皮質(EC)の浅葉も含むように拡げた。 図8A,Bは拡大率の低い概観で、SE後3週間の嗅内皮質(EC)深層のNKCC1 mRNA発現を示し、一方、浅葉、海馬台、歯状回、嗅周囲皮質では発現しなかった。
これらの領域からの蛍光の平均値のプロット(図8C、倍率のより大きい画像から、対照に対し正規化)は、NKCC1 mRNAの深層内側嗅内皮質(EC)にこの高度に特異的な出現を示し、それよりずっと弱い程度に深層外側嗅内皮質(EC)に出現し、一方他のテスト領域ではネガティブであった(図8C、コラム3−10)。
【0071】
同様の特異性が関連するKCC2出現の研究において観察された。図9A,Bの概観は、SE後3週間において、KCC2 mRNAが特異的に深層嗅内皮質(EC)、特に内側部分で発現するのがわかる。平均蛍光の定量的分析では、KCC2 mRNAの深層内側嗅内皮質(mEC)における進行的減少が56±7.8%(SE後3週間、t62=2.34,P=0.0095)深層外側嗅内皮質において14±4.5%(SE後3週間、t62=2.41,P=0.039)であり、一方海馬台、歯状回、嗅周囲皮質及び嗅内皮質(EC)浅葉では有意の変化を示さなかった(図9C)。
【0072】
拡張的実験のさらなる重要な結果は、「SE発症2時間超後のジアゼパム投与」プロトコル(Du氏他,1995;小林氏他、2003)に対抗してここで使用されるプロトコル「SE発症1時間後ピロカプリンの低量投与−ジアゼパム」後の初期のニューロンの有意の減少が第3層に存在しないことである。第1に嗅内皮質(EC)第3層においてKCC2 mRNAの有意の変化の不存在は、我々のラットでは第3層の錐体ニューロンがSE後3週間後も死滅していないことを示唆する。第2に、ニューロンの生存が、第3層の安定的ニューロン数を示した(対照、SE後2週間後及び3週間後においてそれぞれ、ニューロン数114±14.2,112±18.1及び116±15、それぞれt10=−0.18,P=0.86及びt10=−0.77,P=0.46、3ラット)ニューロトレース(Neuro Trace)染色で確認された。
【0073】
図8,9、及びそれらの図に反映されたデータを与えた実験のさらなる詳細は以下の通り。
(図8.NKCC1のmRNAは他の皮質領域では増加しない)
図8A,B:内側嗅内皮質(mEC)、外側嗅内皮質(lEC)、アンギュラーバンドル(Angular bundle)(ab),海馬台(Sub)、歯状回(DG)及び嗅周囲皮質(PRC)に亘る脳領域を示すNKCC1 mRNAに対する蛍光遺伝子プローブ法(FISH)研究:
A、対照スライス;B、SE後3週間後に採取されたスライス。左のパネルは、DAPI染色された領域を示し、それは構成要素領域のアウトラインが重ねて表示されている(A1,B1)。中央のパネルはNKCC1信号を示す。対照では殆ど見られないか皆無であるが、SE後3週間後では信号は中央の嗅内皮質(EC)第5−6層に明白に存在するが、他の場所では存在しない(A2、B2)。これらの画像は右パネルで統合される(A3,B3,高出力画像は図3参照)。
図8C:SE後にNKCC1 mRNAが、深層嗅内皮質(EC)、(内側嗅内皮質(mEC)第5層、それぞれt54=2.15,P=0.037及びt54=3.54,P=0.0009)及び#2とラベル付けされた外側嗅内皮質第5層(それぞれt54=1.02,P=0.12及びt54=1.28,P=0.047)においてのみ増加するが、内側嗅内皮質(mEC)第3層(#3、それぞれt54=0.93,P=0.24及びt49=0.42,P=0.34)、外側嗅内皮質(lEC)(#4、それぞれt54=0.27,P=0.86及びt54=0.24,P=0.094)内側嗅内皮質(mEC)第1−2層(#5、それぞれt54=−1.25,P=0.32及びt54=0.34,P=0.23)、外側嗅内皮質(lEC)第1−2層(#6、それぞれt54=1.24,P=0.078及びt54=0.52,P=0.27)、海馬台(#7、それぞれt54=1.54,P=0.49及びt54=1.67,P=0.063)、歯状回(#8、それぞれt54=0.45,P=0.098及びt54=−1.35,P=0.26)、深層嗅周囲皮質(#9、それぞれt54=0.53,P=0.47及びt54=2.01,P=0.48)及び嗅周囲皮質(PRC)第1−3層(#10、それぞれt54=−1.36,P=0.81及びt54=0.28,P=0.81;データは高倍率画像より抽出)では、有意の変化が起こらなかったことを示す。
【0074】
(図9.KCC2のmRNAは深層嗅内皮質(EC)でのみ減少する)
図9A、B:内側嗅内皮質(mEC)、外側嗅内皮質(lEC)、アンギュラーバンドル(Angular bundle)(ab),海馬台(Sub)、歯状回(DG)及び嗅周囲皮質(PRC)に亘る脳領域を示すKCC2 mRNAに対する蛍光遺伝子プローブ法(FISH)研究:
A、対照スライス;B、SE後3週間後に採取されたスライス。左のパネルは、KCC2染色された領域を示し、それは構成要素領域のアウトラインが重ねて表示されている(A1,A2)。対照では全体に発現が見られるが、SE後3週間後では嗅内皮質(EC)第5層でのみ信号が消滅した。中央のパネルはDAPI染色を示す(A2、B2)。これらの画像は右パネルで統合される(A3,B3,高出力画像は図6参照)。
図9C:SE後にKCC2 mRNAが、深層嗅内皮質(EC)、(内側嗅内皮質(mEC)第5層、それぞれt62=1.95,P=0.082及びt62=2.34,P=0.0095)及び#2とラベル付けされた外側嗅内皮質第5層(それぞれt62=1.86,P=0.093及びt62=2.41,P=0.039)においてのみ増加するが、内側嗅内皮質(mEC)第3層(#3、それぞれt62=0.34,P=0.32及びt62=−1.22,P=0.51)、外側嗅内皮質(lEC)(#4、それぞれt62=−0.25,P=0.17及びt62=0.24,P=0.36)内側嗅内皮質(mEC)第1−2層(#5、それぞれt62=1.06,P=0.85及びt62=0.43,P=0.62)、外側嗅内皮質(lEC)第1−2層(#6、それぞれt62=−0.45,P=0.68及びt62=0.15,P=0.39)、海馬台(#7、それぞれt62=−1.01,P=0.098及びt62=1.84,P=0.065)、歯状回(#8、それぞれt62=1.05,P=0.77及びt62=−2.03,P=0.49)、深層嗅周囲皮質(#9、それぞれt62=1.33,P=0.28及びt62=−1.52,P=0.53)及び嗅周囲皮質(PRC)第1−3層(#10、それぞれt62=1.12,P=0.085及びt62=0.36,P=0.14;データは高倍率画像より抽出)では、有意の変化が起こらなかったことを示す。
【0075】
(実施例8)
(Cl−輸送遮断抑制バースト活性)
抑制性シナプス後電位(EIPSP)の変移及び発作後の多シナプス性バースト応答の増加に対するNKCC1増加の機能的役割を調査するため、我々はCl−輸送体遮断を通して作用し、NKCC対KCC輸送体の遮断により効果のある利尿剤ブメタニドを採用した(Russel,2000;Sung氏他,2000;Hannaert氏他,2002;Beck氏他,2003)。ここで絶対的特異性の欠如は1)示されたKCC2の強い減少、2)Cl−内向き輸送のみがEClをポジティブに静止膜電位(RMP)に変移させるという事実、により補償される。残存KCC2活性の遮断は、NKCC1の遮断の間、EClの強い過分極変移を阻止しない。図10AはSE後3週間後の膜電圧に対するシナプス後電位(PSP)振幅のプロットであり、E(PSP)の脱分極変移がブメタニド(10μM)での20分灌流の後、消滅したことを示す。これは、Cl−の膜を通過する受動的再分布に対応する。図10BはSE後3週間後の嗅内皮質(EC)第5層の正常記録状態における静止膜電位(RMP)(約−72mV)に対するシナプス後電位(PSP)逆転電位の平均値が+40MV超であり、ブメタニド存在下では+10mVに低下した(E(PSP)絶対平均電圧は、対照状態で−29.7±2.3mV、ブメタニド存在下で−60.9±4.1mVであり(t24=2.26,P=0.00009)、ブメタニドの洗い流し中は−34.23.4mV、n=5,t24=1.52,P=0.0057対ブメタニド)。
【0076】
シナプス後電位E(PSP)の脱分極に対して2次性の脱抑制又は興奮が、SE後の潜伏期における多シナプス性バースト応答の発現に決定的な場合、ブメタニドによる逆転電位の蓄積もまた、多シナプス性バースト応答を遮断することになる。図10Cは、確かに、ブメタニドでの灌流(10μM)は単一ショック刺激に対する多シナプス性応答を効果的に抑制した。図10Cの上段パネルに示すような強い多シナプス性バースト応答を記録した後、ブメタニドでの灌流が始まり、20分後までには、バースト放電は強く減衰した(中段の軌跡)。30分間のブメタニドの洗い出しにより、強い多シナプス性応答が回復した(下段の軌跡)。平均すると、ブメタニドは、ピロカルピン発作後3週間後のラットからの細胞の多シナプス性応答を持つニューロンの数を、64.8%(合計14ニューロンの内9ニューロン)から11.6%(2/14)に減じた。30分間のブメタニドの洗い出しは応答ニューロンの数を35.1%(5/14、図10D)に回復した。半最大振幅における応答期間はブメタニドで対照応答(t13=13.8,P=0.0000000004)の36±9%に減少し、そして洗い出し中に75±11%(t13=−5.07,P=0.0002対ブメタニド)に回復した。
【0077】
図10、及びその図に反映されたデータを与えた実験のさらなる詳細は以下の通り。
(図10.NKCC1抑制剤ブメタニドは部分的にE(PSP)を回復し、そして多シナプス性興奮を抑制する)
図10A:SE後3週間後のラットからの嗅内皮質(EC)第5層ニューロンの膜電位に対するシナプス後電位(PSP)振幅をブメタニドへの20分暴露前(上段パネル、白丸)と後(下段パネル、×印)でプロットしたものであり、PSP逆転電位の過分極変移を示す。データは図2Aのプロトコルから得られた。E(PSP)は−29.7mVから−60.9mVへ変移した。
測定はCNQX(10μM)及びAPV(50μM)による興奮性シナプス後電位(EPSP)遮断中に行われた。
図10B:集団データで、ポジティブ変移したGABAA−PSP逆転電位が、ブメタニドにより強く、かつ可逆的に再分極した(平均値±標準誤差、n=3、ブメタニドへの暴露(***)pは0.001未満)ことを示す。
図10C:最上段の記録は、SE後3週間後に観察された単一刺激(100nA)に対する多シナプス性応答を示し、それがブメタニドにより遮断されるが(中段の記録)、30分間の薬物の洗い出しにより回復する(下段の記録)ことを示す。
図10D:ブメタニドは、単一刺激に応答して多シナプス性脱分極を発現するSE後3週間後の嗅内皮質(EC)第5層ニューロンの数を、64.8%から11.6%(n=14)に減少させることを示す集団データである。この数値は30分間のブメタニド洗い出しにより35.1%に回復した。
【0078】
(実施例9)
(GABAA誘発興奮はバーストに軽度の影響をあたえる)
シナプス後電位(PSP)逆転電位の脱分極変移は、第5層ニューロンの脱抑制及び直接的GABA誘発興奮の両方に繋がる。我々はピクロトキシンでGABAA誘発興奮を遮断してバースト応答における直接的興奮の役割を調べた。ブメタニドと異なり、ピクロトキシン(100μM)はSE3週間後の多シナプス性バースト放電に対し僅かな効果しか示さなかった(図11A1,A2;ピクロトキシンでは、自発的バーストも観察した;自発的バースト後5秒より長く記録された誘発バーストのみ分析した;これらのバーストは明白に従前の自発的バーストに影響されない)。研究された細胞集団において、ピクロトキシンは、SE3週間後に多シナプス性バースト応答におけるスパイクの数を9.8±0.8から7.9±0.7へ減少させた(図11C、n=8,t24=2.26,P=0.000089)。このような同一細胞からの10個の応答の平均をとると、図11B1,B2に示すように、ピクロトキシンが平均バースト応答において活動電位振幅を10.5±3.4%減少させ、それは刺激後の活動電位のタイミングの可変性が増大したことを示唆する(n=8,p=0.094)ことが判った。このように、GABA誘発興奮の存在は、明白にネットワークの発火のタイミング精度を増大させ、それは、GABA誘発伝達がニューロン間興奮の下流であるため遅延するので、恐らく直接的興奮よりは全体的な興奮性の増大に起因している。
ピクロトキシン単独での僅かな効果と対照的に、CNQX(10μM)及びAPV(50μM)のACSF/ピクロトキシン生理食塩水への追加は、ほぼ完全にシナプス応答を遮断し(95.9±0.26%、t7=−10.05,P=0.00002)、それはバーストが主として多シナプス性グルタミン酸作動性伝達により媒介されることを示した(図11A3,B3;図2Aで示すように、ピクロトキシンが無いCNQX及びAPV内で、単一シナプス性PSPが誘起された)。ピクロトキシンでは統計的有意性に達しないが、バースト期間の短縮傾向が存在した(図11D)。
)。
【0079】
図11、及びその図に反映されたデータを与えた実験のさらなる詳細は以下の通り。
(図11.ピクロトキシンは多シナプス性バーストに軽度の影響をあたえる)
図11A:ピクロトキシン(100μM)によるGABAA受容体の遮断が多シナプス性バースト応答における活動電位発火を、小さいが有意な量だけ減少させた(図A1,A2;刺激100nA、70μ秒が*印時点で印加された)。その後のCNQX(10μM)及びAPV(50μM)によるイオンチャネル型グルタミン酸受容体の遮断は、全てのシナプス後性応答を殆ど除去した(図11A3)。
図11B:同一の細胞からの10個のバーストの平均がピクロトキシンでのバースト期間の短縮化を示した。これら平均におけるスパイク振幅の減少は、ピクロトキシンでのバーストにおいて発火パターンの精度が低下することを示唆する。
図11C:ピクロトキシンが、多シナプス性バースト応答における平均スパイク数を、1応答当り9.8±0.8から7.9±0.7へ減少させる(平均値±標準誤差、n=8細胞、5ラット、ピクロトキシン付加(*)pは0.05未満)ことを示す数量データである。
図11D:ピクロトキシンでは多シナプス性バースト期間の10.5%短縮傾向が見られた(平均値±標準誤差、n=8、p=0.094)。
【0080】
図12−14、及びそれらの図に反映されたデータを与えた実験のさらなる詳細は以下の通り。
図12は、リチウム−ピロカルピンSE後12−17週間後における、ブメタニドによる発作の抑制(0.2mg/Kg体重、腹腔内)を示す(n=5ラット)。5体のリチウム−ピロカルピンSEラットが連続するブメタニド又は媒体注射(生理食塩水内の5%エタノール、腹腔内、下部に日付を記載)を受け、そしてその後8時間以内に発現する発作がビデオ録画により分析された。1体の同一のラットは、ブメタニドがCl及びIPSP逆転電位を概して基準化した場合でも、2度目及び3度目のブメタニド投与後に発作を発現したが、それは恐らく自発的発作を可能にするこのラット内の2次的な下流の変化によるものである。
【0081】
図13は、NKCC1タンパク質が正常な成体レベルに向かって回復することを示す。プロットは、深層内側嗅内皮質(EC)のNKCC1タンパク質の免疫蛍光信号を、抗体を持たない対照ラット(「免疫グロブリンG(IgG)無し」と表示)、抗体を持つ対照ラット(「対照」と表示)、SE後2週間後及び3週間後(「SE 週間後」と表示)のラットにおいて示す。SE後17週間でNKCC1信号は、ブメタニドが完全に自発的再発性発作(SRS)を防止するラット(「ブメット(Bumet)ポジティブ」と表示)では、SE後3週間後のラットのレベルの50%未満まで回復し、そしてブメタニドが自発的再発性発作(SRS)を防止する効果が無くなったラット(「ブメット(Bumet)ネガティブ」と表示)では95%超に回復した。
【0082】
図14は、KCC2タンパク質が正常な成人レベルに向かって回復することを示す。プロットは、深層内側嗅内皮質(EC)のKCC2タンパク質の免疫蛍光信号を対照ラット(「対照」と表示)、SE後2週間後及び3週間後(「SE後 週間」と表示)のラット、及びSE後17週間後のラットにおいて示す。SE後17週間でKCC2信号は、正常レベルに向かって回復した(ブメタニドが完全に自発的再発性発作(SRS)を防止するラットは「ブメット(Bumet)ポジティブ」と表示、そして、ブメタニドが自発的再発性発作(SRS)を防止する効果が無くなったラットは「ブメット(Bumet)ネガティブ」と表示)。
【0083】
ブメタニド又は媒体が投与された後の発作活性が図12で定量化された、ラットの新規のNKCC1データが図13に示され、図13は従前のNKCC1データを比較のために含む。抗体を持たない対照の測定(「免疫グロブリンG(IgG)無し」と表示)はさらに、対照ラットにおけるNKCC1タンパク質レベルがゼロに近く非常に低いことを示す。SE後17週間でNKCC1タンパク質レベルは、ブメタニド投与が完全に自発的再発性発作(SRS)を防止したラット(「ブメット(Bumet)ポジティブ」と表示)において正常レベルに向かって有意に回復した。2度目及び3度目のブメタニド投与(約4週間離れて09年6月8日と09年7月1日に)後に発作を発現した1体のラットでは、NKCC1はほぼ完全に基準化された。
【0084】
図14はKCC2タンパク質レベルも、SE後17週間後に正常な成体レベルに回復したことを示す。これらのデータは、NKCC1とKCC2のCl−輸送体の発現は共に長い時間の中で基準化され、従ってCl−分布の基準化およびCl−依存性IPSP逆転電位に関するシナプス性抑制につながるという、我々の仮説を確認した。
【0085】
図15は、海馬の錐体細胞CA1層又はCA3層では、NKCC1のmRNAはSE2週間後及び3週間後に有意の変化がない(「SE後2週間」、「SE後3週間」と表示)ことを示す。棒グラフは、海馬の錐体細胞CA1層又はCA3層では、NKCC1のmRNAはSE2週間後及び3週間後に変化しないことを示す。NKCC1のmRNAの蛍光遺伝子プローブ法(FISH)データは、図8に使用されたものと同一の組織の高倍率画像から抽出された。
【0086】
図16は、海馬の錐体細胞CA1層又はCA3層では、KCC2のmRNAは変化がないことを示す。棒グラフは、海馬の錐体細胞CA1層又はCA3層では、NKCC1のmRNAはSE2週間後及び3週間後(「SE後2週間」、「SE後3週間」と表示)に変化しないことを示す。NKCC1のmRNAの蛍光遺伝子プローブ法(FISH)データは、図9に使用されたものと同一の組織の高倍率画像から抽出された。
【0087】
図17:ニューロン−特異染色「ニューロトレース(NeuroTrace)」で染色されたニューロンの数に、嗅内皮質(EC)第2層(「EC第2層」と表示)、又は第3層、又は第5層においても、SE後3週間後に有意の変化が無いことを示す(ニューロンの数/100ミクロンx100ミクロン領域)。
【0088】
図18は慢性ブメタニド処置が慢性期において強く発作の発現を抑制することを示す。SE後の週単位時間経過に対する発作発現/8時間ビデオ録画のプロットであり、無処置ラット(黒塗りの棒、水消費 平均±標準誤差=水29±6.3ml/日)及び飲料水によりブメタニド(bumet.)を処置されたラット(平均±標準誤差=0.65±0.13mgブメタニド/日;水32±6.4ml/日;点線及び斜線)。4体のラットではブメタニドは完全に発作を防止した(中空の棒)が、他の2体のラットでは、発作の発現及びSE後時間経過に伴う発作の進行は共に減少した(斜線の棒)。
【0089】
図19:SE後2週間後に開始され20週間継続する慢性ブメタニド処置が、処置期間中および処置終了後少なくとも6週間、発作の発現を強く抑制する。棒グラフは、SE後第11−21週における8時間ビデオ録画セッション中のラット当りの平均発作発現回数を示し(一番左の棒のペア)、無処置ラット(黒)又はSE後2週間目から20週間に亘って慢性的に飲料水経由でブメタニド処置されたラット(斜線)である。その後ブメタニド処置を終了し、その後3週間後と6週間後の発作発現回数を示す、無処置ラットは約3回の発作/8時間録画のレベルに到達した(黒の棒、ラット当り3回の録画セッション)が、一方事前にブメタニド処置されたラットでは発作の発現はずっと少なかった(斜線の棒)、即ち、SE後時間適合対照SEラットが発現する発作回数の15−22%であった(各グループn=6ラット)。
【0090】
図20は、SE後1週間後に開始する慢性ブメタニド処置が、ブメタニド処置終了後少なくとも6週間のオープンフィールド暴露に誘発される発作の発現を強く抑制することを示す。棒グラフは、SE発作後27週後のピロカプリンSEラットをオープンフィールド(1.4x1.4m)の中央に置き、その後1分後から始まる15分間に発現する発作の回数を示す。ラットは無処置(黒の棒)、又はSE後第2週目から21週間に亘り慢性的に飲料水経由でブメタニド処置された(斜線の棒)である。このテストでは、ブメタニド処置終了後6週間後でも処置済みラットは、SE後時間適合対照SEラットが発現する発作回数の15%未満の発作発現回数を示した(各グループn=6ラット)。
【0091】
図21はSE後16日目から12週間に亘る慢性ブメタニド処置がオープンフィールドの暗箱に入るラットの挙動を回復させることを示す。ラットは箱を含むオープンフィールド(1.4x1.4m)の中央に置かれ、箱は内部が暗く、0.2(長さ)x0.3(幅)x0.15(高さ)mの寸法であり、0.3mの幅側が開口しており、オープンフィールドの中央と囲み壁の間の中間点に置かれる。挙動は1分間ビデオ録画される(3回の試技/ラット、各グループ6ラット)。対照ラットは殆ど箱に入り、箱内に留まった(試技の78%)。対照的に、SE後のラット(SEは後14−15週)は殆ど箱を無視して入らなかった(試技の15%、7ラット)。同じSEグループからのブメタニド処置ラットは、対照ラットと類似の挙動を示し、殆ど箱に入った(試技の67%、8ラット)。
【0092】
図22は内側側頭葉てんかん(TLE)発症の我々の原理、及び我々の新規の予防的及び抗てんかん慢性ブメタニド処置を示す。TLE誘発の発作、例えば、てんかん重積(SE)、外傷性脳障害(TBI)、脳卒中他は、潜伏期(SE後2及び3週間)に進行するNKCC1の病理学的再発現を誘発する。GABAA受容体依存シナプス性抑制の進行性消失は嗅内皮質(EC)第5層のてんかん様活性/無症候性発作をもたらす。このてんかん様活性は嗅内皮質(EC)及び下流脳領域において下流可塑性変化をトリガーし、それは易興奮性を増大させる2次的変化につながる。易興奮性を増大させる変化の進行に伴って、顕性発作の可能性も増大する。顕性発作もまた下流可塑性変化をトリガーし、それは更に大脳易興奮性を増大させ、発作発現の進行につながる(図18及び19に示す)。最終的に発作はNKCC1活性に依存性でなくなり、そしてNKCC1が基準化した時でも継続する(図12、13に示す)。ブメタニド処置は低い細胞内Cl−レベル及びより効率的な抑制(図10A、B)を回復し、潜伏期においててんかん様活性(図10C、D)及び慢性期の発作(図12,18,19,20)を抑制する。無症候性及び顕性発作のブメタニド処置は、下流病理学的変化を防止し、処置後少なくとも6週間継続する(図19、20)発作出現率及びテストされた挙動(図21)の基準化につながる。
【0093】
(実験結果の議論)
発作中のてんかん様ニューロン活性は多くの大脳領域で観察されるが、しかしそのてんかん重積(SE)状態に後続する発生源は明確ではない。我々は深層嗅内皮質(EC)におけるてんかん様活性の初期発現を検証するため、内側側頭葉てんかん(TLE)のリチウム−低投与量ピロカプリンラットモデルを使用した。我々はSE後3週間の潜在期間内に増加する比率の嗅内皮質(EC)第5層のニューロンが単一シナプス刺激に多シナプス性バースト脱分極で応答することを示した。この変化は並行して第5層ニューロンのIPSP逆転電位の進行性脱分極変移を伴い、これは明らかにCl−内側輸送体NKCC1の増加及び同時にCl−外側輸送体KCC2の減少に起因し、この両方の変化は、細胞内Cl−蓄積に役立っている。潜伏期におけるCl−摂取の抑制はよりネガティブなGABA作動性逆転電位を回復し、そして多シナプス性バーストを終了させた。Cl−輸送体の変化は深層嗅内皮質(EC)に強く特異的であった。それらはこの時期に第1−3層、嗅周囲皮質、海馬台又は歯状回では発現しなかった。我々はCl−ホメオスタシスの変化が深層嗅内皮質における過剰興奮性を促進し、それがそこでのてんかん様放電に繋がり、それは結果的に下流皮質領域に影響を及ぼすと提案する。
【0094】
従って、我々はSE後の潜伏期に深層嗅内皮質における大幅に増加した興奮性の発現を示した。第5層のニューロンは通常は閾値以下のシナプス性刺激に対し多シナプス性バースト放電で応答を始める。さらに、我々はこの潜伏期にCl−輸送体NKCC1(内向き)およびKCC2(外向き)の変化した発現に起因するGABAA受容体逆転電位の大きな脱分極変移が起こることを示した。このてんかん発生の初期段階では、これらの輸送体発現の変化は深層嗅内皮質においてのみ発現し、隣接する海馬領域及び皮質領域では発現しなかった。Cl−輸送活性の変化と増加する多シナプス性バーストとの間の直接的な関係は、ブメタニドによるNKCC1の遮断がIPSP逆転電位を大きく回復し、多シナプス性バースト放電を大幅に減少させた、という発見よって支持された。我々の結果は、嗅内皮質第5層が内側側頭葉てんかん(TLE)の発症において重要な部位であることを意味している。
【0095】
従って、上記の実験結果は、SE後の潜伏期において、行動性発作の発現前に、深層内側嗅内皮質においてニューロ興奮性の強い増加、即ち弱いシナプス刺激に対し多シナプス性バースト放電で応答する嗅内皮質第5層ニューロンの比率の進行性増加があることを示唆している。従って嗅内皮質における自然活性パターンは、ますます自発的発作をトリガーする可能性が高くなり、自発的発作は定義により潜伏期を終了させる。我々の発見は、SE後、内側側頭葉てんかん(TLE)は初期において嗅内皮質深層に発現することを示唆している。我々の知る限りでは、潜伏期に他の脳領域においてシナプス刺激に応答するてんかん活性を示す他の研究は無かった。この領域の特異性は、嗅内皮質深層に対する脱抑制に繋がるNKCC1及びKCC2発現の変化の特異性により実証された。内側側頭葉てんかん(TLE)のピロカプリン高投与モデルでCA1に対し潜伏期発作間活性が生体内で示されたが(El−Hassar氏他,2007)、検査した幾つかの他の皮質領域及び海馬領域は変化を示さなかった。我々のレポートした第5層ニューロンの易興奮性の変化が、嗅内皮質第3層の錐体ニューロンの明確に認識可能な消失がなく発現した、ことは非常に重要である。嗅内皮質第3層の錐体ニューロンの高度の消失は後期LTEの症状に典型的であることが示されて来たし(Du氏他,1993;Du氏他,1995)、そしてそれは遷延性SEの24時間以内に発現可能であった(Du氏他,1995)が、明確に認識可能な消失が発現しないため、これらの発見は本論とは無関係である。
【0096】
ジアゼパムがSE発症後2時間後でなく1時間後に投与され、かつアトロピンが投与される、我々のSE発現プロトコルはアンドレ氏他(Andre氏他,2007)のプロトコルよりも幾分弱く見えるかもしれない。ジアゼパムもアトロピンもいずれかの時点で即座にSEを停止させないが、この変更がラットの回復を促進し、より良い生存率の脳スライスを生成したことを我々は発見した。良かれ悪しかれ我々はまた、弱い初期の発作はより徐々に進行する細胞又は神経網の変化を生成し、従って分析が容易であるかも知れないと理由づけた。ビデオモニタリングで検査した場合、我々のプロトコルは、18週までの潜伏期を持つ全てのラットにおいて実証可能な発作活性を生成した(平均=12.7週、材料と方法の項参照)。重要なことに、これらの潜伏期間は非常によく患者に対し報告された潜伏期間と適合した(Annegers氏他,1980)。これは、潜伏期が平均して1−4週間しか継続せず、殆どは2週間未満である公開されたげっ歯類TLEモデル(Dube氏他,2000a;Arzimanoglou氏他,2002;Curia氏他,2008;Li氏他,2008)に対する重要な改良である。はるかに短い潜伏期は、恐らく追加又は全体的な発作を促進する他のメカニズムを活性化させるより激しい発作を示唆するが、それは殆どの場合明らかに患者に関係が無い。このようにLi氏他(Li氏他,2008)はマウスに対するピロカプリン高投与と組み合わせた異常なリチウム侵襲を使用して(確立されたTLEモデルではない)、海馬領域CA1に本願で研究される期間まで継続するNKCC1の急性再発現を示した。この発見は、我々のラットにおいてはCA1ではNKCC1の変化が無いことと対照的である。我々の示した嗅内皮質第5層における細胞性変化は、より強いリチウム−ピロカプリンSE及び他のプロトコルによっても殆どの場合トリガーされる。
【0097】
嗅内皮質第5層における易興奮性の変化は、嗅内皮質第5層のニューロンの約80%で発現する、GABA作動性PSP逆転電位における強いそして進行性の脱分極変移、と同時性を有し、そして我々はそれにより補助されるものと提案する。この比率はCl−内向き輸送体NKCC1の増加と非常によく適合する。殆どの嗅内皮質第5層のニューロンで同時に発現するCl−外向き輸送体KCC2の減少は、NKCC1によるネットのCl−蓄積を促進するが、それ自身では静止膜電位に対するEClポジティブをもたらさない。NKCC1及びKCC2の進行性変化は、SE後3週間後の強く変位したE(PSP)と非常によく適合した、ただし後者の量定は、PSP振幅の外挿及び傾斜の差異の減少に起因して精度が劣り、従って測定可変性を追加した。
【0098】
GABA作動性PSP逆転電位の弱い変移はTLE疾病進行のずっと後の期、即ち、後期慢性てんかんにおいて海馬台ニューロンで観察され、それは明らかにNKCC1発現のずっと弱い変移に起因した(Cohen氏他,2002;de Guzman氏他,2006;Palma氏他,2006;Huberfeld氏他,2007;Munoz氏他,2007;Sen氏他,2007)。領域、疾病進行期、発作間及び発作活性のレベル、及び発現レベルの差異は、NKCC1とKCC2の変移した発現レベルを開始しそして更に制御する信号ネットワークにおける、潜伏期と後期慢性TLEにおける基本的差異を示唆する。
【0099】
最近になって、大規模な発作が、海馬において急性の45日以上に渡るNKCC1の部分回復性再発現、及び発作後2週間以内の自発性再発性発作(SRS)の発現に繋がることが示された(Kang氏他,2002;Li氏他,2008)。しかしこれらのマウスにおいて、1)NKCC1がニューロンにより再発現したか、グリア細胞により再発現したか、2)GABAA−PSP逆転電位が変位したか、3)潜伏期間中にてんかん様活性があったか、4)発作がNKCC1活性に依存するか、が示されておらず、従って不明確である。対照的に、これらの研究(Li氏他)に基づく最も最近の研究は、高投与量の注入によってもブメタニド処置はSRSの抑制に完全に失敗したことを示した(Brandt氏他,2010)。Brandt氏他はこの結果の原因について議論している:「最近の研究の成体ラットTLEモデルにおいて、ブメタニドを使用した予防的処置において有意な抗てんかん活性が見られなかったことは、下記を含む幾つかの原因が考えられる:(1)ブメタニドの全身性投与が脳に到達しそしてこの薬剤が脳で十分な高濃度を保つことができないことに伴う、薬物動態的問題点(速い排出、脳浸透が乏しい)、(2)NKCC1の変化がピロカプリン誘発てんかん発作に決定的には関与していない、又は、(3)このモデルにおいて予防的処置に使用される時間窓、即ち、SE後2週間後が適切でなかった。」。我々のデータにおける低投与量ブメタニドでの発作抑制は、薬物動態的問題点がBradt氏他の発作処置の失敗の理由となりえないことを示している(Brandt氏他,2010)。我々のデータは、SE後最初の2週間以内ではNKCC1の変化はてんかん発作に決定的には関与していないことを示唆している。従って時間窓も間違っている。さらに、回復NKCC1レベルは潜伏期の最後の発作発現の開始の理由にはなりえない(Li氏他,2008)。このマウスモデルは患者に見られた(上記参照)潜伏期より遥かに短い潜伏期を示す(Li氏他,2008)のみならず、示されたCA1におけるNKCC1発現は、我々のデータと矛盾する(Kang氏他、2002;Li氏他,2008)。これら事実の両方は、使用されたSEモデルが現実的なTLEモデルではないことを示唆している(上記参照)。本発明の新規性は、1)我々の改良ラットTLEモデルによる潜伏期の現実的適合、2)潜伏期ニューロンにおけるNKCC1の進行性再出現とIPSP逆転電位の変移の両方、3)潜伏期及び慢性期(SE後3−18週)におけるブメタニドによるてんかん様活性及び発作の効果的抑制、そして、4)遷延性ブメタニド処置終了後少なくとも6週間を超えること、に反映されている。
【0100】
嗅内皮質第5層ニューロンにおけるGABA−PSPの通常の逆転電位、即ちほぼVrestは2つの別個の慢性てんかん中のSE−TLEモデル(Fountain氏他,1998;de Guzman氏他,2006)で発見され,我々の報告する非常に大きなGABAAE(PSP)は、一度慢性てんかんが確立されると大部分回復することを支持した。他の説明も可能である:第1に、記録は外側嗅内皮質から得られ、そこではNKCC1およびKCC2に小さな変化しか見られないことを我々は発見した。更に、異なるTLEモデルが使用された。de Guzman氏他(2006)は、TLEのピロカルピン高投与量ラットモデルを研究し、それは,リチウム−ピロカルピンラットモデルとは基礎となるSE中活性化される信号伝達経路が有意に異なり(Ormandy氏他,1989)、はるかに短い潜伏期(2週間未満)、及び第3層ニューロン消失の差異(上記)において異なることが示された。後者2者の差異はより激しい発作を示唆する(上記)。これはE(PSP)変移、及び慢性てんかんから回復するか否かにおいて、差異を生成可能である。我々はこれら重要な問題について将来研究する予定である。
【0101】
GABA作動性PSPの強い脱分極変移が、深層嗅内皮質の易興奮性の観察された変化の主たる原因であるという推論は、Cl−内向き輸送体NKCC1の遮断剤ブメタニドの効果により支持された。ブメタニドはより過分極されたPSP逆転電位を回復し、そして刺激に対するてんかん様バースト応答をほぼ消滅させた。これらの発見は、SE後のこの病期におけるてんかん様バースト応答の発現に対し、強く脱分極したE(PSP)の必要性を意味するが、しかし、他の同時性変化、例えば、第5層ニューロンの観察された入力コンダクタンスの減少が追加的に必要となる可能性を除外しない。ブメタニドの効果はさらに、GABAAR遺伝子の減少、又は抑制性シナプス及び介在ニューロンの消失に起因すると思われる、観察された入力コンダクタンスの減少にも拘わらず、SE後3週間後に抑制が依然非常に効果的でありうることを意味する(Bernard氏他,2004;Gorter氏他,2006;Kumar氏とBuckmaster氏,2006)。対照ニューロンではGABA−PSPは既にわずかに脱分極しており、そして、SE後3週間後、ブメタニドはそれを完全にはそのレベルに回復させなかった。従って、抑制は「過分極している」のではなく、常に「短絡している」と言える。しかし、AP発火のトリガーを興奮電流により防止するための最も重要な要素は、発火閾値に近い膜電位での抑止性電流の振幅であるように思われることを忘れないようにする必要がある。この電流振幅は、発火閾値(約45mV)に近いE(PSP)の変移と共にCl−駆動力に従って徐々に変化する。その結果SE3週間後かつブメタニドの存在下で(E(PSP)は約−60mV)−45mVでの駆動力は、対照ラットの約26mVに対し約15mVであり、従って約42%減少する。結果としてこの臨界膜電位領域でのCl−電流は、駆動力の減少及びGABA−PSPコンダクタンスの減少の両方によって減衰されるが、ブメタニドの存在下では、てんかん様活性の抑制に依然として全く効果的である。このことは、全体的にCl−勾配を回復することが生体内で処置的利益をもたらすという希望を与える。てんかん発生、及びNKCC1活性の抑制に対する処置的及び予防的干渉に関する我々の原理は、図22に要約されている。そこに示された、潜伏期に始まるブメタニド処置中及び以降の発作の非常に効果的な抑制は、原理の実証を提供する。
さらに、初期のそして高度に特異的な病理的NKCC1活性の基準化、及びこの処置による変化した挙動の基準化は、ブメタニドでの提案された抗てんかん処置が、少なくとも幾つかの現在の抗てんかん剤による知性と脳の能力に対する副作用を回避する、という重要な追加的利益を有することを示唆する。
【0102】
我々は、我々の発見がTLEの初期の発現において作用する主要な要素を開示するものと信じ、また、疾患につながる変化の起点及び進行中の深層嗅内皮質の重要性を強調する。
【0103】
(略語)
ACSF、人工脳脊髄液;CA、アンモン角;DAPI、4’,6’−ジアミジノ−2−フェニルインドール;EC,嗅内皮質;E(PSP)、PSP逆転電位;FISH,蛍光遺伝子プローブ法;IPSP,抑制性シナプス後電位;PBS,リン酸緩衝食塩水;PBS−T,0.2%のトリトン−X−100を含むPBS;PSP, シナプス後電位;RMP,静止膜電位;SE、てんかん重積;TLE、内側側頭葉てんかん。
【0104】
(参照)
【表1】
【特許請求の範囲】
【請求項1】
被験者における内側側頭葉てんかん(TLE)の発症の可能性を抑制又は減少させる方法であって、被験者がTLEを誘発することが既知の発作を患った後に、治療的に有効な量のNKCC1抑制剤を被験者に投与するステップを有することを特徴とする方法。
【請求項2】
前記TLEを誘発することが既知の発作は、てんかん重積(SE)、小発作性てんかん、欠神、筋クローヌス性発作、クローヌス性(間代性)発作、緊張性発作、強直間代発作、無緊張性発作、後天性失語、てんかんを伴う後天性失語症(ランドウ・クレフナー症候群)、後天性てんかん性失語症、大脳皮質異形成−焦点性てんかん(CDFE)、新生児てんかん、海馬硬化症(HS)、海馬萎縮症、大脳萎縮症、小脳萎縮症、複雑熱性けいれん(CFC)を含む熱性発作、外傷性脳障害、脳卒中、低酸素性虚血性症状、代謝不全、脳腫瘍、からなるグループから選択される、ことを特徴とする請求項1に記載の方法。
【請求項3】
前記NKCC1抑制剤が、被験者のてんかん重積(SE)発症後の潜伏期に被験者に投与される、ことを特徴とする請求項1に記載の方法。
【請求項4】
前記NKCC1抑制剤が、原因が未知の最初の発作を被験者が患った後に被験者に投与される、ことを特徴とする請求項1に記載の方法。
【請求項5】
前記NKCC1抑制剤が、被験者が請求項2に記載の発作の1つを患った後約8週間から約24週間以内に被験者に投与される、ことを特徴とする請求項3に記載の方法。
【請求項6】
前記NKCC1抑制剤が、被験者が請求項2に記載の発作の1つを患った後約1週間から約4週間以内に被験者に投与される、ことを特徴とする請求項3に記載の方法。
【請求項7】
前記NKCC1抑制剤が第2の処置薬と組合わされ、そして前記被験者がTLEを誘発することが既知の発作を患った後に、前記被験者に同時投与され、
ここにおいて前記第2の処置薬は、ガンマアミノ酪酸(GABA)調節組成物、抗けいれん薬、イオンチャネル不活性化剤、抗利尿剤、又はそれらの組合せである、ことを特徴とする請求項1に記載の方法。
【請求項8】
前記NKCC1抑制剤が,トラセミド、フロセミド、アゾセミド、ブメタニド、ピレタニド、トリパミド、エトゾリン(etozoline)及びその代謝体、オゾリノン(Ozolinone)、シクレタニン、およびそれらの医薬的に許容可能な派生物、塩及びエステルからなるグループから選択されるループ利尿薬である、ことを特徴とする請求項1に記載の方法。
【請求項9】
前記NKCC1抑制剤が, 3−ヒドラジノプロピオン酸、アミノオキシ酢酸、ガバクリン、ガバペンチン、イソニアジド、フェネルジン、フェニルエチリデンヒドラジン、プレガバリン、バルノクタミド、バルプロ酸、バルプロミド、ビガバトリン、及びそれらの混合物からなるグループから選択される化合物である、ことを特徴とする請求項1に記載の方法。
【請求項10】
前記ガンマアミノ酪酸(GABA)調節組成物が、バルビツール酸化合物、ベンゾジアゼピン系化合物、ガバペンチン、プレガバリン、4アミノ酪酸(GABA),4−アミノ−3−(4−クロロフェニル)ブタン酸(バクロフェン)、4−アミノ−3−フェニルブタン酸、4−アミノ−3−ヒドロキシブタン酸、4−アミノ−3−(4−クロロフェニル)−3−ヒドロキシフェニルブタン酸、4−アミノ−3−(チエン−2−イル)ブタン酸、4−アミノ−3−(5−クロロチエン−2−イル)ブタン酸、4−アミノ−3−(5−ブロモチエン−2−イル)ブタン酸、4−アミノ−3−(5−メチルチエンー2−イル)ブタン酸、4−アミノ−3−(2−イミダゾリル)ブタン酸、4−グアニジノー3−(4−クロロフェニル)ブタン酸、(3−アミノプロピル)亜ホスホン酸、(4−アミノブチ−2−イル)亜ホスホン酸、酪酸ナトリウム、(3−アミノ−2−メチルプロピル)亜ホスホン酸、(3−アミノブチル)亜ホスホン酸、(3−アミノ−2−(4−クロロフェニル)プロピル)亜ホスホン酸、(3−アミノ−2−(4−クロロフェニル)−2−ヒドロキシプロピル)亜ホスホン酸、(3−アミノ−2−(4−フルオロフェニル)プロピル)亜ホスホン酸、(3−アミノ−2−フェニルプロピル)亜ホスホン酸、(3−アミノ−2−ヒドロキシプロピル)亜ホスホン酸、(E)−(3−アミノプロペン−1−イル)亜ホスホン酸、(3−アミノ−2−シクロヘキシルプロピル)亜ホスホン酸、(3−アミノ−2−ベンジルプロピル)亜ホスホン酸、[3−アミノ−2−(4−メチルフェニル)プロピル]亜ホスホン酸、[3−アミノ−2−(4−トリフルオロメチルフェニル)プロピル]亜ホスホン酸、[3−アミノ−2−(4−メトキシフェニル)プロピル]亜ホスホン酸、[3−アミノ−2−(4−クロロフェニル)−2−ヒドロキシプロピル]亜ホスホン酸、(3−アミノプロピル)メチルホスフィン酸、(3−アミノ−2−ヒドロキシプロピル)メチルホスフィン酸、(3−アミノプロピル)(ジフルオロメチル)ホスフィン酸、(4−アミノブチ−2−イル)メチルホスフィン酸、(3−アミノ−1−ヒドロキシプロピル)メチルホスフィン酸、(3−アミノ−2−ヒドロキシプロピル)(ジフルオロメチル)ホスフィン酸、(E)−(3−アミノプロペン−1−イル)メチルホスフィン酸、(3−アミノ−2−オキソ−プロピル)メチルホスフィン酸、(3−アミノプロピル)ヒドロキシメチルホスフィン酸、(5−アミノペンチ−3−イル)メチルホスフィン酸、(4−アミノ−1,1,1−トリフルオロブチ−2−イル)メチルホスフィン酸、(3−アミノ−2−(4−クロロフェニル)プロピル)スルフィン酸、3−アミノプロピルスルフィン酸、及びここに記載される他の組成物からなるグループから選択される、ことを特徴とする請求項7に記載の方法。
【請求項11】
被験者における内側側頭葉てんかん(TLE)の重症度を抑制又は減少させる方法であって、被験者がTLEを誘発することが既知の発作を患った後に、治療的に有効な量のNKCC1抑制剤を被験者に投与するステップを有することを特徴とする方法。
【請求項12】
前記TLEを誘発することが既知の発作は、てんかん重積(SE)、小発作性てんかん、欠神、筋クローヌス性発作、クローヌス性(間代性)発作、緊張性発作、強直間代発作、無緊張性発作、後天性失語、てんかんを伴う後天性失語症(ランドウ・クレフナー症候群)、後天性てんかん性失語症、大脳皮質異形成−焦点性てんかん(CDFE)、新生児てんかん、海馬硬化症(HS)、海馬萎縮症、大脳萎縮症、小脳萎縮症、複雑熱性けいれん(CFC)を含む熱性発作、外傷性脳障害、脳卒中、低酸素性虚血性症状、代謝不全、脳腫瘍からなるグループから選択される、ことを特徴とする請求項11に記載の方法。
【請求項13】
前記NKCC1抑制剤が、被験者のてんかん重積発症後の潜伏期に被験者に投与される、ことを特徴とする請求項11に記載の方法。
【請求項14】
前記NKCC1抑制剤が、原因が未知の最初の発作を被験者が患った後に被験者に投与される、ことを特徴とする請求項11に記載の方法。
【請求項15】
前記NKCC1抑制剤が、被験者がSE発作を発症後約8週間から約24週間以内に被験者に投与される、ことを特徴とする請求項11に記載の方法。
【請求項16】
前記NKCC1抑制剤が、被験者がSE発作を発症後約2週間から約4週間以内に被験者に投与される、ことを特徴とする請求項11に記載の方法。
【請求項17】
前記被験者がTLEを誘発することが既知の発作を患った後に、前記被験者に前記NKCC1抑制剤と第2の処置薬が同時投与され、
ここにおいて前記第2の処置薬は、ガンマアミノ酪酸(GABA)調節組成物、抗けいれん薬、イオンチャネル不活性化剤、抗利尿剤、又はそれらの組合せである、ことを特徴とする請求項11に記載の方法。
【請求項18】
前記NKCC1抑制剤が, トラセミド、フロセミド、アゾセミド、ブメタニド、ピレタニド、トリパミド、エトゾリン(etozoline)及びその代謝体、オゾリノン(Ozolinone)、シクレタニン、およびそれらの医薬的に許容可能な派生物、塩及びエステルからなるグループから選択されるループ利尿薬である、ことを特徴とする請求項11に記載の方法。
【請求項19】
前記NKCC1抑制剤が, 3−ヒドラジノプロピオン酸、アミノオキシ酢酸、ガバクリン、ガバペンチン、イソニアジド、フェネルジン、フェニルエチリデンヒドラジン、プレガバリン、バルノクタミド、バルプロ酸、バルプロミド、ビガバトリン、及びそれらの混合物からなるグループから選択される化合物である、ことを特徴とする請求項11に記載の方法。
【請求項20】
前記ガンマアミノ酪酸(GABA)調節組成物が、バルビツール酸化合物、ベンゾジアゼピン系化合物、ガバペンチン、プレガバリン、4アミノ酪酸(GABA),4−アミノ−3−(4−クロロフェニル)ブタン酸(バクロフェン)、4−アミノ−3−フェニルブタン酸、4−アミノ−3−ヒドロキシブタン酸、4−アミノ−3−(4−クロロフェニル)−3−ヒドロキシフェニルブタン酸、4−アミノ−3−(チエン−2−イル)ブタン酸、4−アミノ−3−(5−クロロチエン−2−イル)ブタン酸、4−アミノ−3−(5−ブロモチエン−2−イル)ブタン酸、4−アミノ−3−(5−メチルチエンー2−イル)ブタン酸、4−アミノ−3−(2−イミダゾリル)ブタン酸、4−グアニジノー3−(4−クロロフェニル)ブタン酸、(3−アミノプロピル)亜ホスホン酸、(4−アミノブチ−2−イル)亜ホスホン酸、酪酸ナトリウム、(3−アミノ−2−メチルプロピル)亜ホスホン酸、(3−アミノブチル)亜ホスホン酸、(3−アミノ−2−(4−クロロフェニル)プロピル)亜ホスホン酸、(3−アミノ−2−(4−クロロフェニル)−2−ヒドロキシプロピル)亜ホスホン酸、(3−アミノ−2−(4−フルオロフェニル)プロピル)亜ホスホン酸、(3−アミノ−2−フェニルプロピル)亜ホスホン酸、(3−アミノ−2−ヒドロキシプロピル)亜ホスホン酸、(E)−(3−アミノプロペン−1−イル)亜ホスホン酸、(3−アミノ−2−シクロヘキシルプロピル)亜ホスホン酸、(3−アミノ−2−ベンジルプロピル)亜ホスホン酸、[3−アミノ−2−(4−メチルフェニル)プロピル]亜ホスホン酸、[3−アミノ−2−(4−トリフルオロメチルフェニル)プロピル]亜ホスホン酸、[3−アミノ−2−(4−メトキシフェニル)プロピル]亜ホスホン酸、[3−アミノ−2−(4−クロロフェニル)−2−ヒドロキシプロピル]亜ホスホン酸、(3−アミノプロピル)メチルホスフィン酸、(3−アミノ−2−ヒドロキシプロピル)メチルホスフィン酸、(3−アミノプロピル)(ジフルオロメチル)ホスフィン酸、(4−アミノブチ−2−イル)メチルホスフィン酸、(3−アミノ−1−ヒドロキシプロピル)メチルホスフィン酸、(3−アミノ−2−ヒドロキシプロピル)(ジフルオロメチル)ホスフィン酸、(E)−(3−アミノプロペン−1−イル)メチルホスフィン酸、(3−アミノ−2−オキソ−プロピル)メチルホスフィン酸、(3−アミノプロピル)ヒドロキシメチルホスフィン酸、(5−アミノペンチ−3−イル)メチルホスフィン酸、(4−アミノ−1,1,1−トリフルオロブチ−2−イル)メチルホスフィン酸、(3−アミノ−2−(4−クロロフェニル)プロピル)スルフィン酸、3−アミノプロピルスルフィン酸、及びここに記載される他の組成物からなるグループから選択される、ことを特徴とする請求項17に記載の方法。
【請求項21】
前記NKCC1抑制剤が、被験者がTLEを誘発することが既知の反復性発作を患った後に、慢性的に被験者に投与される、ことを特徴とする請求項11に記載の方法。
【請求項22】
前記被験者が反復性てんかん重積(SE)を患う、ことを特徴とする請求項21に記載の方法。
【請求項23】
前記NKCC1抑制剤が、発作後2−4週間以内のNKCC1活性の抑制、又はNKCC1の再発現の抑制を通じて作用する、ことを特徴とする請求項2又は12に記載の方法。
【請求項24】
前記抗利尿剤が末梢作用性抗利尿剤である、ことを特徴とする請求項7又は17に記載の方法。
【請求項25】
前記抗利尿剤が、NKCC1抑制剤の利尿作用を抑制することにより作用することを特徴とする請求項7又は17に記載の方法。
【請求項26】
被験者がTLEを誘発することが既知の発作を患った後に、被験者における内側側頭葉てんかん(TLE)の発症の可能性を抑制又は減少させるための薬剤の生産において、NKCC1抑制剤を使用する方法。
【請求項27】
前記TLEを誘発することが既知の発作は、てんかん重積(SE)、小発作性てんかん、欠神、筋クローヌス性発作、クローヌス性(間代性)発作、緊張性発作、強直間代発作、無緊張性発作、後天性失語、てんかんを伴う後天性失語症(ランドウ・クレフナー症候群)、後天性てんかん性失語症、大脳皮質異形成−焦点性てんかん(CDFE)、新生児てんかん、海馬硬化症(HS)、海馬萎縮症、大脳萎縮症、小脳萎縮症、複雑熱性けいれん(CFC)を含む熱性発作、外傷性脳障害、脳卒中、低酸素性虚血性症状、代謝不全、脳腫瘍、からなるグループから選択される、ことを特徴とする請求項26に記載の方法。
【請求項28】
前記NKCC1抑制剤は、ガンマアミノ酪酸A(GABAA)受容体依存信号伝達の調節剤、抗けいれん薬、イオンチャネル不活性化剤、抗利尿剤、又はそれらの組合せからなるグループから選択される第2の処置剤と組合わされる、ことを特徴とする請求項26又は27に記載の方法。
【請求項29】
被験者がTLEを誘発することが既知の発作を患った後に、被験者における内側側頭葉てんかん(TLE)の重症度を抑制又は減少させるための薬剤の生産において、NKCC1抑制剤を使用する方法。
【請求項30】
前記TLEを誘発することが既知の発作は、てんかん重積(SE)、小発作性てんかん、欠神、筋クローヌス性発作、クローヌス性(間代性)発作、緊張性発作、強直間代発作、無緊張性発作、後天性失語、てんかんを伴う後天性失語症(ランドウ・クレフナー症候群)、後天性てんかん性失語症、大脳皮質異形成−焦点性てんかん(CDFE)、新生児てんかん、海馬硬化症(HS)、海馬萎縮症、大脳萎縮症、小脳萎縮症、複雑熱性けいれん(CFC)を含む熱性発作、外傷性脳障害、脳卒中、低酸素性虚血性症状、代謝不全、脳腫瘍、からなるグループから選択される、ことを特徴とする請求項29に記載の方法。
【請求項31】
前記発作がてんかん重積(SE)である、ことを特徴とする請求項30に記載の方法。
【請求項32】
原因が未知の最初の発作を前記被験者が患う、ことを特徴とする請求項26に記載の方法。
【請求項1】
被験者における内側側頭葉てんかん(TLE)の発症の可能性を抑制又は減少させる方法であって、被験者がTLEを誘発することが既知の発作を患った後に、治療的に有効な量のNKCC1抑制剤を被験者に投与するステップを有することを特徴とする方法。
【請求項2】
前記TLEを誘発することが既知の発作は、てんかん重積(SE)、小発作性てんかん、欠神、筋クローヌス性発作、クローヌス性(間代性)発作、緊張性発作、強直間代発作、無緊張性発作、後天性失語、てんかんを伴う後天性失語症(ランドウ・クレフナー症候群)、後天性てんかん性失語症、大脳皮質異形成−焦点性てんかん(CDFE)、新生児てんかん、海馬硬化症(HS)、海馬萎縮症、大脳萎縮症、小脳萎縮症、複雑熱性けいれん(CFC)を含む熱性発作、外傷性脳障害、脳卒中、低酸素性虚血性症状、代謝不全、脳腫瘍、からなるグループから選択される、ことを特徴とする請求項1に記載の方法。
【請求項3】
前記NKCC1抑制剤が、被験者のてんかん重積(SE)発症後の潜伏期に被験者に投与される、ことを特徴とする請求項1に記載の方法。
【請求項4】
前記NKCC1抑制剤が、原因が未知の最初の発作を被験者が患った後に被験者に投与される、ことを特徴とする請求項1に記載の方法。
【請求項5】
前記NKCC1抑制剤が、被験者が請求項2に記載の発作の1つを患った後約8週間から約24週間以内に被験者に投与される、ことを特徴とする請求項3に記載の方法。
【請求項6】
前記NKCC1抑制剤が、被験者が請求項2に記載の発作の1つを患った後約1週間から約4週間以内に被験者に投与される、ことを特徴とする請求項3に記載の方法。
【請求項7】
前記NKCC1抑制剤が第2の処置薬と組合わされ、そして前記被験者がTLEを誘発することが既知の発作を患った後に、前記被験者に同時投与され、
ここにおいて前記第2の処置薬は、ガンマアミノ酪酸(GABA)調節組成物、抗けいれん薬、イオンチャネル不活性化剤、抗利尿剤、又はそれらの組合せである、ことを特徴とする請求項1に記載の方法。
【請求項8】
前記NKCC1抑制剤が,トラセミド、フロセミド、アゾセミド、ブメタニド、ピレタニド、トリパミド、エトゾリン(etozoline)及びその代謝体、オゾリノン(Ozolinone)、シクレタニン、およびそれらの医薬的に許容可能な派生物、塩及びエステルからなるグループから選択されるループ利尿薬である、ことを特徴とする請求項1に記載の方法。
【請求項9】
前記NKCC1抑制剤が, 3−ヒドラジノプロピオン酸、アミノオキシ酢酸、ガバクリン、ガバペンチン、イソニアジド、フェネルジン、フェニルエチリデンヒドラジン、プレガバリン、バルノクタミド、バルプロ酸、バルプロミド、ビガバトリン、及びそれらの混合物からなるグループから選択される化合物である、ことを特徴とする請求項1に記載の方法。
【請求項10】
前記ガンマアミノ酪酸(GABA)調節組成物が、バルビツール酸化合物、ベンゾジアゼピン系化合物、ガバペンチン、プレガバリン、4アミノ酪酸(GABA),4−アミノ−3−(4−クロロフェニル)ブタン酸(バクロフェン)、4−アミノ−3−フェニルブタン酸、4−アミノ−3−ヒドロキシブタン酸、4−アミノ−3−(4−クロロフェニル)−3−ヒドロキシフェニルブタン酸、4−アミノ−3−(チエン−2−イル)ブタン酸、4−アミノ−3−(5−クロロチエン−2−イル)ブタン酸、4−アミノ−3−(5−ブロモチエン−2−イル)ブタン酸、4−アミノ−3−(5−メチルチエンー2−イル)ブタン酸、4−アミノ−3−(2−イミダゾリル)ブタン酸、4−グアニジノー3−(4−クロロフェニル)ブタン酸、(3−アミノプロピル)亜ホスホン酸、(4−アミノブチ−2−イル)亜ホスホン酸、酪酸ナトリウム、(3−アミノ−2−メチルプロピル)亜ホスホン酸、(3−アミノブチル)亜ホスホン酸、(3−アミノ−2−(4−クロロフェニル)プロピル)亜ホスホン酸、(3−アミノ−2−(4−クロロフェニル)−2−ヒドロキシプロピル)亜ホスホン酸、(3−アミノ−2−(4−フルオロフェニル)プロピル)亜ホスホン酸、(3−アミノ−2−フェニルプロピル)亜ホスホン酸、(3−アミノ−2−ヒドロキシプロピル)亜ホスホン酸、(E)−(3−アミノプロペン−1−イル)亜ホスホン酸、(3−アミノ−2−シクロヘキシルプロピル)亜ホスホン酸、(3−アミノ−2−ベンジルプロピル)亜ホスホン酸、[3−アミノ−2−(4−メチルフェニル)プロピル]亜ホスホン酸、[3−アミノ−2−(4−トリフルオロメチルフェニル)プロピル]亜ホスホン酸、[3−アミノ−2−(4−メトキシフェニル)プロピル]亜ホスホン酸、[3−アミノ−2−(4−クロロフェニル)−2−ヒドロキシプロピル]亜ホスホン酸、(3−アミノプロピル)メチルホスフィン酸、(3−アミノ−2−ヒドロキシプロピル)メチルホスフィン酸、(3−アミノプロピル)(ジフルオロメチル)ホスフィン酸、(4−アミノブチ−2−イル)メチルホスフィン酸、(3−アミノ−1−ヒドロキシプロピル)メチルホスフィン酸、(3−アミノ−2−ヒドロキシプロピル)(ジフルオロメチル)ホスフィン酸、(E)−(3−アミノプロペン−1−イル)メチルホスフィン酸、(3−アミノ−2−オキソ−プロピル)メチルホスフィン酸、(3−アミノプロピル)ヒドロキシメチルホスフィン酸、(5−アミノペンチ−3−イル)メチルホスフィン酸、(4−アミノ−1,1,1−トリフルオロブチ−2−イル)メチルホスフィン酸、(3−アミノ−2−(4−クロロフェニル)プロピル)スルフィン酸、3−アミノプロピルスルフィン酸、及びここに記載される他の組成物からなるグループから選択される、ことを特徴とする請求項7に記載の方法。
【請求項11】
被験者における内側側頭葉てんかん(TLE)の重症度を抑制又は減少させる方法であって、被験者がTLEを誘発することが既知の発作を患った後に、治療的に有効な量のNKCC1抑制剤を被験者に投与するステップを有することを特徴とする方法。
【請求項12】
前記TLEを誘発することが既知の発作は、てんかん重積(SE)、小発作性てんかん、欠神、筋クローヌス性発作、クローヌス性(間代性)発作、緊張性発作、強直間代発作、無緊張性発作、後天性失語、てんかんを伴う後天性失語症(ランドウ・クレフナー症候群)、後天性てんかん性失語症、大脳皮質異形成−焦点性てんかん(CDFE)、新生児てんかん、海馬硬化症(HS)、海馬萎縮症、大脳萎縮症、小脳萎縮症、複雑熱性けいれん(CFC)を含む熱性発作、外傷性脳障害、脳卒中、低酸素性虚血性症状、代謝不全、脳腫瘍からなるグループから選択される、ことを特徴とする請求項11に記載の方法。
【請求項13】
前記NKCC1抑制剤が、被験者のてんかん重積発症後の潜伏期に被験者に投与される、ことを特徴とする請求項11に記載の方法。
【請求項14】
前記NKCC1抑制剤が、原因が未知の最初の発作を被験者が患った後に被験者に投与される、ことを特徴とする請求項11に記載の方法。
【請求項15】
前記NKCC1抑制剤が、被験者がSE発作を発症後約8週間から約24週間以内に被験者に投与される、ことを特徴とする請求項11に記載の方法。
【請求項16】
前記NKCC1抑制剤が、被験者がSE発作を発症後約2週間から約4週間以内に被験者に投与される、ことを特徴とする請求項11に記載の方法。
【請求項17】
前記被験者がTLEを誘発することが既知の発作を患った後に、前記被験者に前記NKCC1抑制剤と第2の処置薬が同時投与され、
ここにおいて前記第2の処置薬は、ガンマアミノ酪酸(GABA)調節組成物、抗けいれん薬、イオンチャネル不活性化剤、抗利尿剤、又はそれらの組合せである、ことを特徴とする請求項11に記載の方法。
【請求項18】
前記NKCC1抑制剤が, トラセミド、フロセミド、アゾセミド、ブメタニド、ピレタニド、トリパミド、エトゾリン(etozoline)及びその代謝体、オゾリノン(Ozolinone)、シクレタニン、およびそれらの医薬的に許容可能な派生物、塩及びエステルからなるグループから選択されるループ利尿薬である、ことを特徴とする請求項11に記載の方法。
【請求項19】
前記NKCC1抑制剤が, 3−ヒドラジノプロピオン酸、アミノオキシ酢酸、ガバクリン、ガバペンチン、イソニアジド、フェネルジン、フェニルエチリデンヒドラジン、プレガバリン、バルノクタミド、バルプロ酸、バルプロミド、ビガバトリン、及びそれらの混合物からなるグループから選択される化合物である、ことを特徴とする請求項11に記載の方法。
【請求項20】
前記ガンマアミノ酪酸(GABA)調節組成物が、バルビツール酸化合物、ベンゾジアゼピン系化合物、ガバペンチン、プレガバリン、4アミノ酪酸(GABA),4−アミノ−3−(4−クロロフェニル)ブタン酸(バクロフェン)、4−アミノ−3−フェニルブタン酸、4−アミノ−3−ヒドロキシブタン酸、4−アミノ−3−(4−クロロフェニル)−3−ヒドロキシフェニルブタン酸、4−アミノ−3−(チエン−2−イル)ブタン酸、4−アミノ−3−(5−クロロチエン−2−イル)ブタン酸、4−アミノ−3−(5−ブロモチエン−2−イル)ブタン酸、4−アミノ−3−(5−メチルチエンー2−イル)ブタン酸、4−アミノ−3−(2−イミダゾリル)ブタン酸、4−グアニジノー3−(4−クロロフェニル)ブタン酸、(3−アミノプロピル)亜ホスホン酸、(4−アミノブチ−2−イル)亜ホスホン酸、酪酸ナトリウム、(3−アミノ−2−メチルプロピル)亜ホスホン酸、(3−アミノブチル)亜ホスホン酸、(3−アミノ−2−(4−クロロフェニル)プロピル)亜ホスホン酸、(3−アミノ−2−(4−クロロフェニル)−2−ヒドロキシプロピル)亜ホスホン酸、(3−アミノ−2−(4−フルオロフェニル)プロピル)亜ホスホン酸、(3−アミノ−2−フェニルプロピル)亜ホスホン酸、(3−アミノ−2−ヒドロキシプロピル)亜ホスホン酸、(E)−(3−アミノプロペン−1−イル)亜ホスホン酸、(3−アミノ−2−シクロヘキシルプロピル)亜ホスホン酸、(3−アミノ−2−ベンジルプロピル)亜ホスホン酸、[3−アミノ−2−(4−メチルフェニル)プロピル]亜ホスホン酸、[3−アミノ−2−(4−トリフルオロメチルフェニル)プロピル]亜ホスホン酸、[3−アミノ−2−(4−メトキシフェニル)プロピル]亜ホスホン酸、[3−アミノ−2−(4−クロロフェニル)−2−ヒドロキシプロピル]亜ホスホン酸、(3−アミノプロピル)メチルホスフィン酸、(3−アミノ−2−ヒドロキシプロピル)メチルホスフィン酸、(3−アミノプロピル)(ジフルオロメチル)ホスフィン酸、(4−アミノブチ−2−イル)メチルホスフィン酸、(3−アミノ−1−ヒドロキシプロピル)メチルホスフィン酸、(3−アミノ−2−ヒドロキシプロピル)(ジフルオロメチル)ホスフィン酸、(E)−(3−アミノプロペン−1−イル)メチルホスフィン酸、(3−アミノ−2−オキソ−プロピル)メチルホスフィン酸、(3−アミノプロピル)ヒドロキシメチルホスフィン酸、(5−アミノペンチ−3−イル)メチルホスフィン酸、(4−アミノ−1,1,1−トリフルオロブチ−2−イル)メチルホスフィン酸、(3−アミノ−2−(4−クロロフェニル)プロピル)スルフィン酸、3−アミノプロピルスルフィン酸、及びここに記載される他の組成物からなるグループから選択される、ことを特徴とする請求項17に記載の方法。
【請求項21】
前記NKCC1抑制剤が、被験者がTLEを誘発することが既知の反復性発作を患った後に、慢性的に被験者に投与される、ことを特徴とする請求項11に記載の方法。
【請求項22】
前記被験者が反復性てんかん重積(SE)を患う、ことを特徴とする請求項21に記載の方法。
【請求項23】
前記NKCC1抑制剤が、発作後2−4週間以内のNKCC1活性の抑制、又はNKCC1の再発現の抑制を通じて作用する、ことを特徴とする請求項2又は12に記載の方法。
【請求項24】
前記抗利尿剤が末梢作用性抗利尿剤である、ことを特徴とする請求項7又は17に記載の方法。
【請求項25】
前記抗利尿剤が、NKCC1抑制剤の利尿作用を抑制することにより作用することを特徴とする請求項7又は17に記載の方法。
【請求項26】
被験者がTLEを誘発することが既知の発作を患った後に、被験者における内側側頭葉てんかん(TLE)の発症の可能性を抑制又は減少させるための薬剤の生産において、NKCC1抑制剤を使用する方法。
【請求項27】
前記TLEを誘発することが既知の発作は、てんかん重積(SE)、小発作性てんかん、欠神、筋クローヌス性発作、クローヌス性(間代性)発作、緊張性発作、強直間代発作、無緊張性発作、後天性失語、てんかんを伴う後天性失語症(ランドウ・クレフナー症候群)、後天性てんかん性失語症、大脳皮質異形成−焦点性てんかん(CDFE)、新生児てんかん、海馬硬化症(HS)、海馬萎縮症、大脳萎縮症、小脳萎縮症、複雑熱性けいれん(CFC)を含む熱性発作、外傷性脳障害、脳卒中、低酸素性虚血性症状、代謝不全、脳腫瘍、からなるグループから選択される、ことを特徴とする請求項26に記載の方法。
【請求項28】
前記NKCC1抑制剤は、ガンマアミノ酪酸A(GABAA)受容体依存信号伝達の調節剤、抗けいれん薬、イオンチャネル不活性化剤、抗利尿剤、又はそれらの組合せからなるグループから選択される第2の処置剤と組合わされる、ことを特徴とする請求項26又は27に記載の方法。
【請求項29】
被験者がTLEを誘発することが既知の発作を患った後に、被験者における内側側頭葉てんかん(TLE)の重症度を抑制又は減少させるための薬剤の生産において、NKCC1抑制剤を使用する方法。
【請求項30】
前記TLEを誘発することが既知の発作は、てんかん重積(SE)、小発作性てんかん、欠神、筋クローヌス性発作、クローヌス性(間代性)発作、緊張性発作、強直間代発作、無緊張性発作、後天性失語、てんかんを伴う後天性失語症(ランドウ・クレフナー症候群)、後天性てんかん性失語症、大脳皮質異形成−焦点性てんかん(CDFE)、新生児てんかん、海馬硬化症(HS)、海馬萎縮症、大脳萎縮症、小脳萎縮症、複雑熱性けいれん(CFC)を含む熱性発作、外傷性脳障害、脳卒中、低酸素性虚血性症状、代謝不全、脳腫瘍、からなるグループから選択される、ことを特徴とする請求項29に記載の方法。
【請求項31】
前記発作がてんかん重積(SE)である、ことを特徴とする請求項30に記載の方法。
【請求項32】
原因が未知の最初の発作を前記被験者が患う、ことを特徴とする請求項26に記載の方法。
【図1】

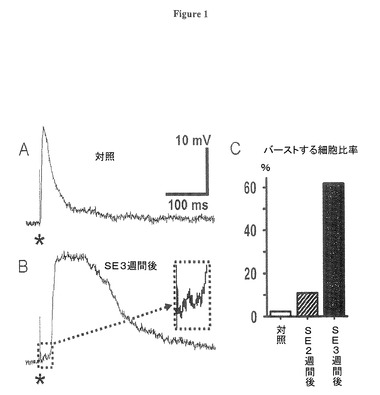
【図2】
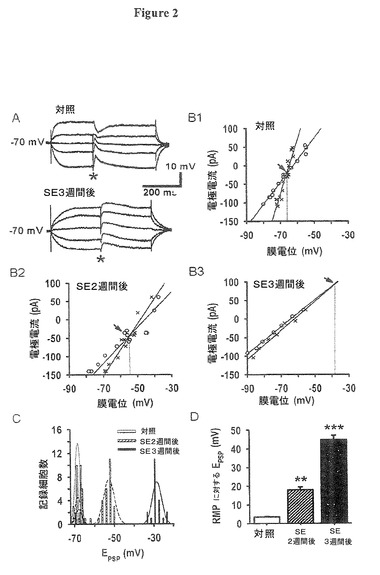
【図3】
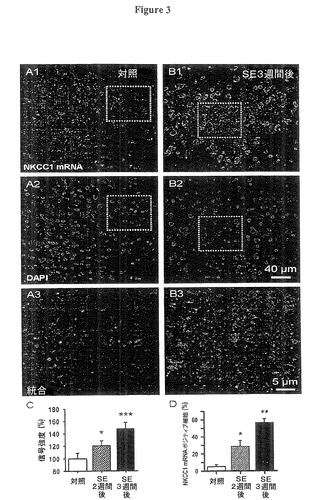
【図4】

【図5】
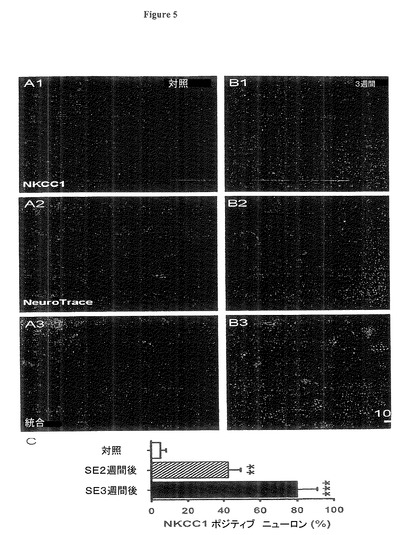
【図6】
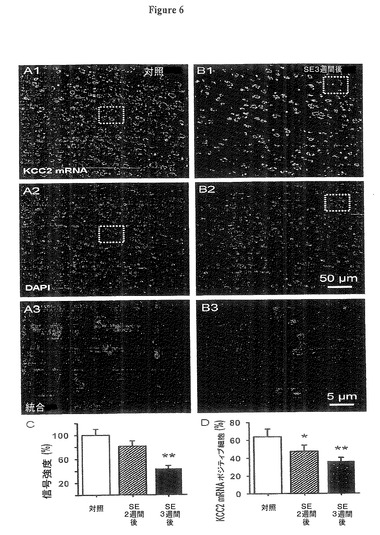
【図7】
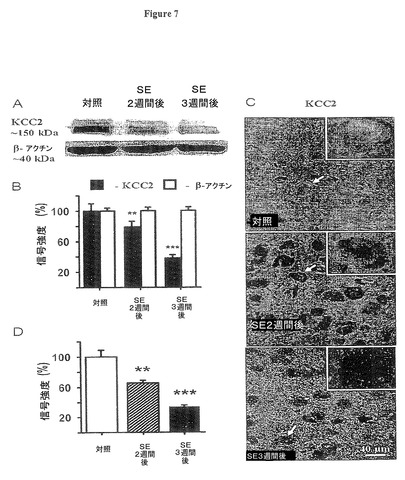
【図8】
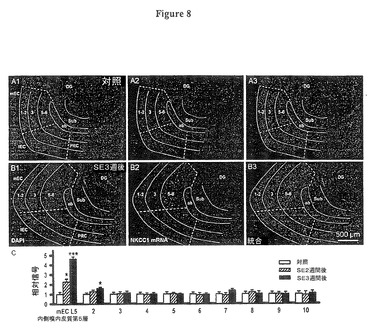
【図9】
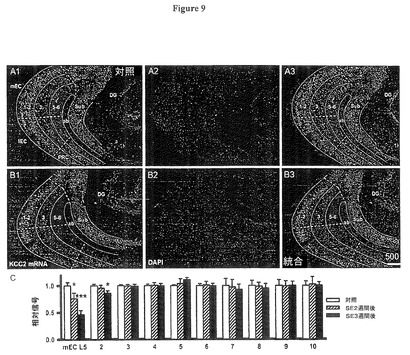
【図10】
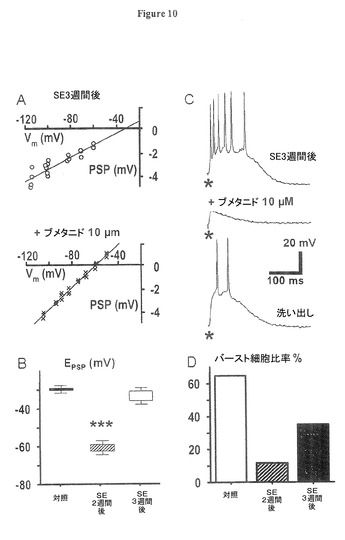
【図11】
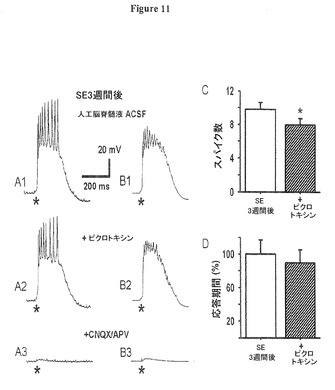
【図12】
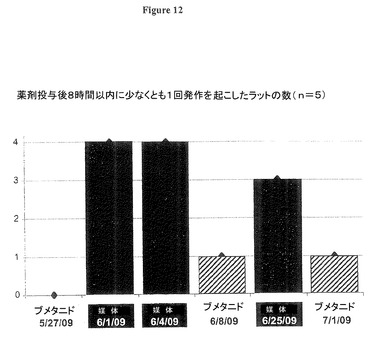
【図13】
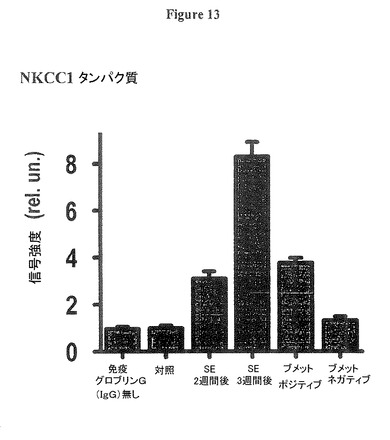
【図14】
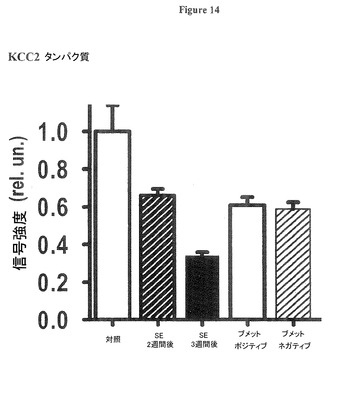
【図15】
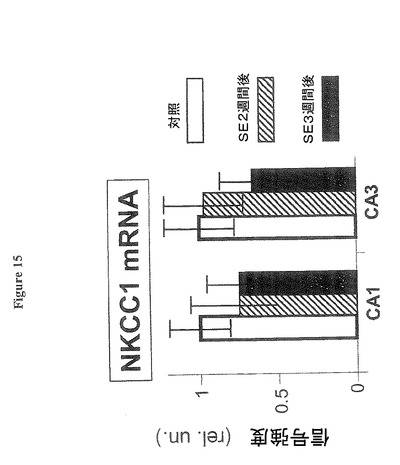
【図16】
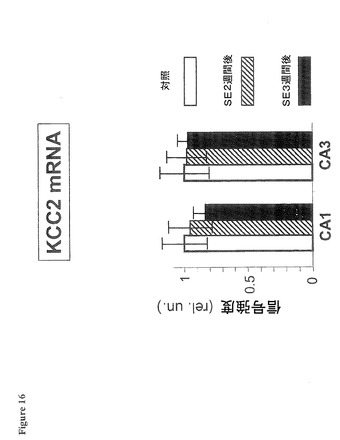
【図17】
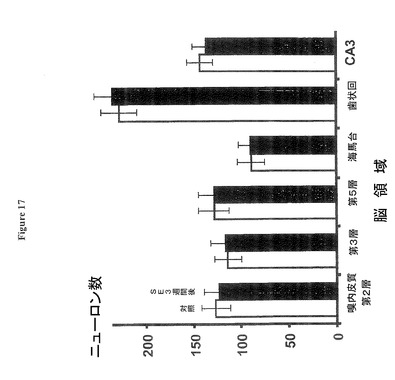
【図18】
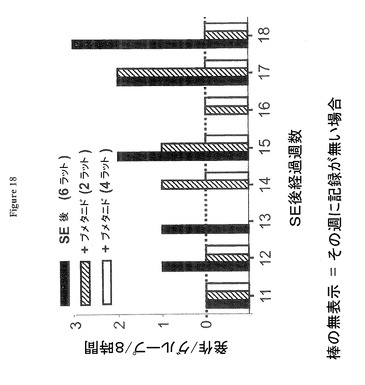
【図19】
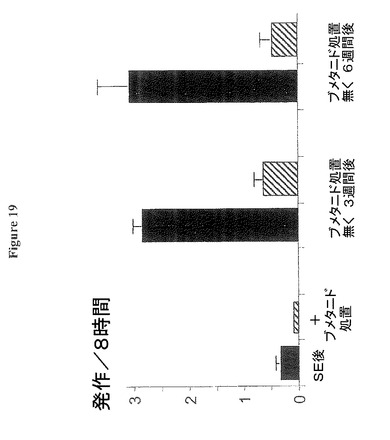
【図20】
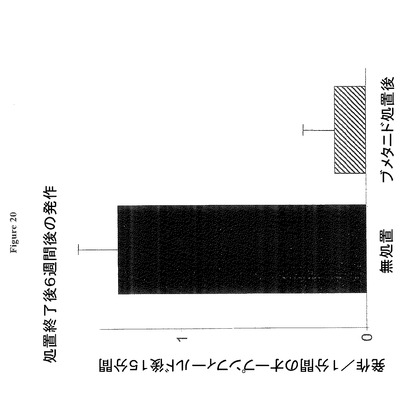
【図21】
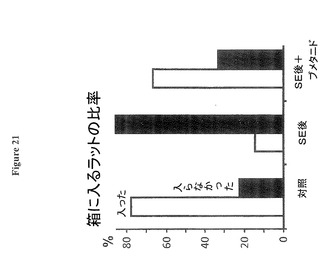
【図22】
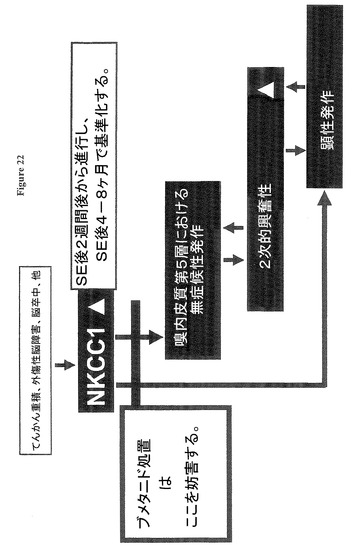
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【公表番号】特表2013−500262(P2013−500262A)
【公表日】平成25年1月7日(2013.1.7)
【国際特許分類】
【出願番号】特願2012−521831(P2012−521831)
【出願日】平成22年7月23日(2010.7.23)
【国際出願番号】PCT/US2010/043063
【国際公開番号】WO2011/011692
【国際公開日】平成23年1月27日(2011.1.27)
【出願人】(508266650)
【Fターム(参考)】
【公表日】平成25年1月7日(2013.1.7)
【国際特許分類】
【出願日】平成22年7月23日(2010.7.23)
【国際出願番号】PCT/US2010/043063
【国際公開番号】WO2011/011692
【国際公開日】平成23年1月27日(2011.1.27)
【出願人】(508266650)
【Fターム(参考)】
[ Back to top ]