
内在性貯蔵タンパク質の標的化抑制を介した、植物種子中で異種ポリペプチドの蓄積を亢進させる方法
本発明は、限られた資源、例えばアミノ酸、リボゾーム結合部位および翻訳修飾および後翻訳修飾に関与する酵素の集合体など、に関して異種タンパク質と競合する内在性タンパク質の標的化抑制を通じて、分子的農業のための、植物種子における異種タンパク質の蓄積レベルを増加させるための改良された方法に関する。アンチセンス“阻害”または二本鎖RNA-誘導RNA干渉、または後転写遺伝子サイレンシング(PTGS)のいずれかを使用して、ホルデイン遺伝子の転写活性化に関して得られた、オオムギにおける転写因子の発現を抑制するための方法を提供する。
【発明の詳細な説明】
【発明の詳細な説明】
【0001】
発明の属する分野
本発明は、植物分子生物学の分野におけるものであり、そして具体的には、単子葉植物の胚乳において種子貯蔵タンパク質の内在性発現に由来する競合を抑制することにより、植物種子において異種タンパク質の発現を増加する方法に関する。
【0002】
背景
多数の植物が、発生途中の種子の胚乳において、様々な種類の貯蔵タンパク質、代謝酵素、内在性キチナーゼ、タンパク質合成阻害剤、プロテアーゼ阻害剤、アミラーゼ、レクチンおよびペルオキシダーゼなどの様々な機能を発揮する、タンパク質の大規模な蓄積物を発現しそして貯蔵する。胚乳中の貯蔵タンパク質は、多数の単子葉穀物種において一般的であるものの様な15%より多い種子乾燥重量の総量に達する場合がある。従って、特定の発生期において、種子胚乳中の細胞装置は、主としてこれらの特定のタンパク質の産生および貯蔵にかかりきりとなる。さらに、ほとんどの貯蔵タンパク質遺伝子は、多コピー存在することが見いだされ、そのことは、数日〜数週間のスパンでこれらのタンパク質が急速に蓄積することの重要性を反映している。
【0003】
穀物種子の発生中胚乳でのタンパク質蓄積に関する固有の生物学的能力は、多数の穀物植物、特に単子葉植物、が異種組換えタンパク質(例えば、医薬産業用の高価値のポリペプチド)の大規模生産のための実際的でそして効率的なビヒクルとなる可能性を有していることを示す;製造方法はしばしば、分子的農業(molecular farming)とも呼ばれた。さらに、種子中に異種ポリペプチドを蓄積することは、下流の処理コストを低減させる。というのも、これらの種子を、異種ポリペプチドの品質に影響を与えることなく、数年間にわたり保存することができるからである。そのようなタンパク質の発現は、好ましくは、種子-特異的プロモータまたは胚乳-特異的プロモータの調節下にておこなう。
【0004】
適切なプロモータの選択および細胞内(sub-cellular)分布などの一般的な分子生物学的戦略は、分子的農業のために使用される特定の植物における異種タンパク質発現レベルを改善する際に有用である。しかし、発現レベルは発現すべきタンパク質の性質、その複雑性、およびその機能により重大な影響を受けるものでもある。しかしながら、異種タンパク質の低発現レベル(低%の全可溶性タンパク質または%TSP)は、種子中で発現される場合であっても、格別の関心事である。新規のバイオテクノロジー的アプローチが、発現レベルをさらに向上させるために必要とされる。発生途中の種子において好ましくは貯蔵タンパク質および休眠-依存的生残のための一般的な調製物の蓄積にかかりきりであるその内在性の役割のために、細胞機構が既にプログラムされているため特に、このことはかなりの挑戦である。単子葉植物の胚乳または種子における目的の異種遺伝子の発現を駆動するために最も頻繁に使用されるプロモータは、多くの場合、貯蔵タンパク質遺伝子由来のプロモータ、例えばコメ由来の1.3 kbの胚乳-特異的プロモータGluB-1(Patel et al. 2000;GenBank Accession No. X54314)、またはオオムギ由来の0.45 kb D-ホルデイン(hordein)プロモータ(Sorensen et al. 1996; GenBank Accession No. X84368)などである。これらのプロモータは、目的の異種タンパク質の発現を駆動するために強力であるが、それらの活性は、最も豊富な貯蔵タンパク質を駆動する内在性プロモータの活性と、時間的そして空間的に同時に生じることから、アミノ酸、リボゾーム結合部位、および翻訳や後翻訳修飾に関与する酵素の集合体などの限定的な供給源に対する競合を生じる。この競合は、目的の異種ポリペプチドの発現レベルに対して悪い影響を与える。
【0005】
内在性遺伝子の発現(供給源と積極的に競合する多数の内在性貯蔵タンパク質遺伝子など)を時間的そして空間的に、目的の異種タンパク質の発現により抑制する方法を手にすることが非常に望ましいことである。従って、貯蔵タンパク質の抑制には、抑制の実際的な方法が提供されれば、様々な産業的ニーズが存在する可能性がある。このことは、特に内在性遺伝子が複数遺伝子ファミリーの構成分子である場合に、分子的農業においてかなり重要な挑戦となりうるものである。例えば、オオムギにおいて、胚乳における資源のための主要なsinkは、全胚乳タンパク質の50%もの割合を占める可能性があるB-ホルデイン貯蔵タンパク質である。(Shewry 1993: in Barley; Chemistry and Technology)。
【0006】
用語“sink”、“sink遺伝子”および“sinkタンパク質”は、refer 本明細書中でto細胞中で活発に発現されそして実質的な量の利用可能な資源を取り込む遺伝子および遺伝子生成物のことをいい、すなわち、sinkはその資源の実質的な部分の細胞を“枯渇させ(drain)”、そして従ってその他の遺伝子および遺伝子生成物の発現のための利用可能な資源を制限する。
【0007】
アンチセンス技術は、植物細胞中の特定の内在性遺伝子生成物の発現を減少させるための有効な手段として発生したものである(例えば、US特許No. 5,759,829;Orvar et al. 1997;Coles et al. 1999を参照)。しかしながら、この技術は、遺伝子ファミリーの構成分子である遺伝子の発現を直接的に抑制するためには、実際的なアプローチではなく、このことはしばしば、半数体ゲノムあたり少なくとも20であるB-ホルデイン遺伝子などの種子貯蔵タンパク質に当てはまる(Shewry et al. 1985);この技術は、遺伝子ファミリーに属さない遺伝子の発現を操作するために適している。
【0008】
内在性遺伝子発現を減少するための別の方法は、後転写遺伝子サイレンシング(PTGS)のための二本鎖RNA(dsRNA)の利用である。このRNA-誘導性遺伝子サイレンシングまたはRNA-干渉は、特定の内在性遺伝子のセンスRNA鎖およびその相補的アンチセンスRNA鎖の両方を組み合わせて1本のdsRNAにしたものであり、最初はCaenorhabditis elegansにおいて示された(Fire et al. 1998)。Hamiltonら(1999)は、短いヌクレオチドRNA(21〜23ヌクレオチド長)がdsRNAの分解生成物であり、それが植物中のPTGSにおいて内在性標的mRNAの切断を制御することを示した。植物における二本鎖遺伝子サイレンシングのより強力なアプローチは、Wesleyら(2001)により記載された方法であり、ここで遺伝子サイレンシング遺伝子は、転写後に“ヘアピン-ループ”RNA(hpRNA)を形成する様にして、構築される。hpRNA遺伝子サイレンシングの一形式は、ヘアピン-ループ中のスペーサーがイントロンであるイントロン-スプライシングhpRNA(ihpRNA)である。ihpRNAは、植物において非常に強力なPTGSの能力を有する(Waterhouse et al 2001およびSmith et al 2000も参照)。特別なベクター中にてそのようなPTGS構築物を作成する方法は、US特許出願2003-A-0049835に記載されている。
【0009】
分子的農業において、そして特にオオムギなどの単子葉植物において、異種遺伝子の発現を改善する新規な方法に関する要求が存在する。分子的農業において目的の医薬タンパク質の発現を増加させることを目的として、目的の異種遺伝子と内在性“sink”遺伝子とのあいだでの発現の競合を減少させる能力が、当該技術分野においては完成されていない。従来技術は、例えばhpRNA-誘導PTGSを使用して、標的化遺伝子サイレンシングが実際にどのようにして、分子的農業における目的の異種遺伝子の発現レベルを増加することができるのかについても示していなかった。
【0010】
本発明の概要と目的
本発明の第一の目的は、分子的農業における生産ビヒクルとして使用されるトランスジェニック種子における目的の異種ポリペプチドの蓄積レベルを増大させるための方法および核酸構築物を提供するものとして概説できる。最初のアプローチは、単子葉植物における胚乳中で組換え的に産生された目的の異種ポリペプチドの発現の利益となるように、上述した胚乳-特異的貯蔵タンパク質をコードする内在性の所望しないmRNAのタンパク質翻訳とのあいだでの資源に関する競合を制限することである。
【0011】
遺伝子ファミリーの構成分子である遺伝子の発現を抑制するための以前の方法は、個々の構成分子または遺伝子ファミリー構成分子のすべてに特徴的な遺伝子ファミリー構成分子のコード配列中の相同領域を標的とすることである。本発明の一つの目的は、上述した遺伝子ファミリーの複数の構成分子の発現の制御を、協調的な様式で編成する単一-遺伝子転写制御因子の発現を抑制することにより、遺伝子ファミリーの発現を減弱することによって、これらの以前に記載された方法の欠点を防止することである。
【0012】
本発明の別の目的は、転写制御因子のhpRNA-誘導PTGSを使用して、単子葉植物の胚乳における主要な貯蔵タンパク質の発現を抑制すること、そしてそれによりavoid using当該技術分野においてより従来型のいわゆるアンチセンス技術、または同じ目的のためのいわゆる同時抑制、を使用することを回避することである。
【0013】
目的の異種タンパク質をコードするトランスジーンを駆動する特定のプロモータの転写制御因子の発現レベルに影響を与えることなく、主要な貯蔵タンパク質遺伝子(例えばオオムギにおけるホルデイン)の転写制御因子のhpRNA-誘導PTGSが達成された場合、翻訳のための限られた資源についての競合は、目的の異種タンパク質をコードするmRNAの利益となるように減少され、特定の目的の異種タンパク質の蓄積の増加を引き起こす。
【0014】
本発明の目的は、主要な貯蔵タンパク質遺伝子の転写制御因子の発現を抑制し、そしてその結果、植物中で産生される異種タンパク質をコードする目的のトランスジーンの発現を駆動する特定のプロモータの活性に影響を与えることなく、前記貯蔵タンパク質遺伝子の発現レベルを減少させる方法を提供することである。
【0015】
本発明の第一の側面は、植物種子中の目的の異種ポリペプチドの発現および蓄積を亢進する方法を提供し、ここで前記方法は:
(a)種子貯蔵タンパク質をコードする1またはそれ以上の内在性遺伝子の転写を制御する、異なる(1または複数の)転写制御因子(TF)のキメラ結合物を含む、1またはそれ以上のTFまたはその(1または複数の)部分をコードするDNA配列に対して機能可能に連結された種子-特異的プロモータについてのDNA配列を用いて植物細胞を形質転換し、ここで前記TF DNA配列の転写された鎖が前記植物細胞における1またはそれ以上の前記種子貯蔵タンパク質の発現を抑制し、遅延させ、またはそうでなければ減少させることができ;そして
(b)(a)における前記転写制御因子により認識されるcis-作用性要素を有さない種子-特異的プロモータを選択し;そして
(c)目的の異種ポリペプチドをコードするDNA配列と機能可能に連結した(b)に記載のプロモータについてのDNA配列を用いて、(a)に記載したものと同一または別の植物細胞を形質転換し;
(d)前記(1または複数の)形質転換植物宿主細胞由来の植物を再生させ、そして1またはそれ以上のTFまたはその部分をコードする前記(1または複数の)DNA配列が転写される条件下で前記植物を生長させ、それにより前記内在性mRNAの発現を減少させ、そして前記種子貯蔵タンパク質の発現を減少させ、そして目的の前記異種ポリペプチドの発現および蓄積を亢進させる;
ことを含む。
【0016】
有用な態様において、工程(a)の(1または複数の)DNA配列および工程(c)のDNA配列を、同一の植物細胞中に導入する。そのような態様において、配列は、1つのDNA配列に機能可能に連結していてもよい。
【0017】
しかしながら、その他の興味深い態様において、工程(a)の(1または複数の)DNA配列を第一の植物宿主細胞のゲノム中に導入し、そして工程(c)の前記DNA配列を第二の植物宿主細胞のゲノム中に導入する。その後、第一のトランスジェニック植物を前記第一の植物宿主細胞から再生し、そして第二のトランスジェニック植物を前記第二の植物宿主細胞から作製し、そしてトランスジェニック植物の後代個体群は、前記第一のトランスジェニック植物と第二のトランスジェニック植物との有性交配から作製され、後代個体群植物は、前記1またはそれ以上のTFをコードする(1または複数の)DNA配列および目的の異種タンパク質をコードするDNA配列の両方を含む細胞を有し、植物が前記異種タンパク質を発現しそして蓄積することができるようにする。
【0018】
好ましい態様においては、抑制された種子貯蔵タンパク質が、オオムギのホルデイン、例えば、B-ホルデインおよびC-ホルデインの一方または両方である。
目的の異種ポリペプチドをコードするDNA配列は、本発明による植物細胞中に蓄積することができる、原核細胞のタンパク質または真核細胞のタンパク質をコードするDNA配列であってもよい。本発明の産生のために選択することができるタンパク質の例は、コラーゲン、コラゲナーゼ、ホメオボックスポリペプチド、モノクローナル抗体、分泌抗体、一本鎖抗体、マンノース結合レクチン、ペプシン、キモトリプシン、トリプシン、カゼイン、ヒト成長ホルモン、ヒト血清アルブミン、ヒトインスリン、ラクトフェリン、リゾチーム、セルラーゼ、ペクチナーゼ、ヘミセルラーゼ、フィターゼ、ヒドロラーゼ、ペルオキシダーゼ、フィブリノーゲン、第IX因子、第XIII因子、トロンビン,タンパク質C、キシラナーゼ、イソアミラーゼ、グルコアミラーゼ、アミラーゼ、リゾチーム、β-グルカナーゼ、グルコセレブロシダーゼ、カゼイン、ラクターゼ、ウレアーゼ、グルコースイソメラーゼ、インベルターゼ、ストレプトアビジン、エステラーゼ、アルカリホスファターゼ、プロテアーゼ阻害剤、プロテアーゼ、ペプシン、キモトリプシン、トリプシン、パパイン、キナーゼ,ホスファターゼ、デオキシリボヌクレアーゼ、リボヌクレアーゼ、ホスホリパーゼ、リパーゼ、ラッカーゼ、クモの糸タンパク質、凍結防止タンパク質、抗菌性ペプチドまたはデフェンシン、成長因子、およびサイトカインである。
【0019】
いくつかの態様において、前記DNA配列は、例えば、炭水化物結合モジュール(CBM)などの、好熱性生物由来の所望のタンパク質をコードする。適切なCBMの例は、Thermotoga maritima由来のCBM9-2である。CBM9-2ゲノムDNA配列は、GenBank Accession No. Z46264として利用可能であり、そしてそれはCBMのファミリーIXに属する。同様に、前記CBMまたはその他の適切なCBMを含む融合タンパク質は、DNA配列によりコードされ、そして本発明に従って、種子中で過剰発現させることができる。そのようなCBMおよびCBM融合タンパク質を精製するための方法は、本件出願と同時に出願した出願人の同時係属中の国際特許出願“A non-denaturing process to purify recombinant protein from plants”および“A process for proteolytic cleavage and purification of recombinant proteins”中にさらに詳細に記載されており、その内容全体を参考文献として本明細書中に援用する。
【0020】
特定の態様において、前記DNA配列は、ホメオボックスB4(HoxB4)タンパク質をコードするヒトHoxB4遺伝子を含む。前記ヒトHoxB4タンパク質は、好ましくは、SEQ ID NO: 1に示す配列を有するか、またはSEQ ID NO: 1と実質的な配列同一性を有し、その結果、発現タンパク質が好ましくは未処置HoxB4タンパク質のすべての機能的特徴を有する。実質的な配列同一性は、本明細書中の文脈において、少なくとも50%以上の配列同一性、より好ましくは少なくとも60%の配列同一性、少なくとも70%の配列同一性、例えば少なくとも80%の配列同一性、そして好ましくは少なくとも90%の配列同一性、例えば少なくとも95%の配列同一性または99%の配列同一性、を示す。
【0021】
本明細書中で示されるように、1またはそれ以上のTFまたはその部分をコードする前記DNA配列は、好ましくは、1またはそれ以上の前記種子貯蔵タンパク質の発現を、抑制し、遅延させ、またはそうでなければ減少させることができる“ヘアピン”RNAを形成する能力を有する。前記DNA配列は、完全なTF配列を含んでもよいが、しかしながら、ある場合には、TF配列の部分のみが適切な位置に存在しさえすれば抑制が機能する可能性もある。このように、これらの態様において、1またはそれ以上のTF配列の十分に長い部分が、抑制が作用するために必要とされる。ある場合には、長さ20ヌクレオチド程度のTF配列の部分で十分であり、しかしながら好ましくは、約20〜200、例えば長さ20〜100の範囲または長さ50〜100ヌクレオチドの範囲、を含む、少なくとも20〜500ヌクレオチドの範囲の部分が使用される。
【0022】
特定の有用な態様において、1またはそれ以上のTFをコードするDNA配列は、本明細書中に定義する様に、キメラDNA配列であり、TFまたはその部分をコードする2またはそれ以上のDNA配列の領域を含む。別の有用な態様において、キメラDNA配列において1またはそれ以上のTFをコードするDNA配列は、“ヘアピン”RNA中にループを形成することができるイントロン配列を含むこともできる。
【0023】
好ましい態様は、TFまたはその部分をコードするDNA配列を利用し、これはbZIPタンパク質の群、より好ましくは、オオムギBLZ1タンパク質およびBLZ2タンパク質(Onate et al 1999を参照)から選択されたbZIPタンパク質、由来のTFまたはその部分をコードする領域を含む。前記DNA配列は、抑制に影響を与えるために十分な長さのそのようなタンパク質をコードする配列の部分、または前記タンパク質をコードする配列の部分の組み合わせを含んでもよい。
【0024】
前記DNA配列は好ましくは、SEQ ID NO: 2、SEQ ID NO: 3またはSEQ ID NO: 4に記載された配列、または前記配列によりコードされたいずれかのアミノ酸配列と実質的に同一な配列を有するタンパク質をコードする配列、または本発明の方法による抑制を引き起こすために十分な長さを有するSEQ ID NO: 2、またはSEQ ID NO: 4の部分を有する配列を含む。上述したように、前記配列のいずれかの組み合わせ、例えば上述の配列の1またはそれ以上の部分の組み合わせ、が有用である可能性があり、そのような組み合わせの例は、BLZ1およびBLZ2の部分のキメラであるSEQ ID NO: 5として示されるものがある。
【0025】
本発明の別の目的は、hpRNA-誘導PTGS技術を使用して、主要な貯蔵タンパク質遺伝子の転写制御因子の発現を抑制する方法を提供することである。そのため、特定の態様において、本発明の方法は、hpRNA-誘導PTGS技術を、内在性貯蔵タンパク質の発現を抑制するために使用し、そして従って、目的の異種ポリペプチドをコードするmRNAの翻訳のための、資源(例えば、アミノ酸、リボゾーム結合部位および翻訳修飾および後翻訳修飾に関与する酵素の集合物)の利用可能性を増大する。このように、好ましい態様において、1またはそれ以上のTFまたはその部分、またはそれらの組み合わせ、好ましくは上述したbZIPタンパク質の群に由来するもの、をコードする前記DNA配列は、前記植物細胞における“ヘアピン”RNA(hpRNA)を発現することができる。
【0026】
前記DNA配列は好ましくは、SEQ ID NO: 2、SEQ ID NO: 4、SEQ ID NO: 6、またはSEQ ID NO: 7、SEQ ID NO: 8、およびいずれかの部分またはそれらの組み合わせ(本明細書中に記載した方法による抑制、すなわち、特に好ましくはオオムギ中の胚乳などの種子および/または胚乳におけるもの、を引き起こすために十分な長さのもの)から選択される配列を含む。
【0027】
従って、本発明の目的は、オオムギ胚乳におけるB-ホルデインおよびC-ホルデインレベルを減少させるための、分子生物学における方法およびツールを提供することである。より具体的には、本発明の目的は、hpRNAを形成することができるBLZ1遺伝子およびBLZ2遺伝子の領域を含み、そして従って前記胚乳遺伝子の発現を減少させることができる遺伝子構築物を提供することである。
【0028】
より具体的には、本発明は、オオムギ胚乳においてPTGSを可能にするhpRNAを形成する転写物をコードする、BLZ1遺伝子およびBLZ2遺伝子の領域の遺伝子構築物を提供する。
上述した好ましい態様において、胚乳貯蔵タンパク質レベルが減少したトランスジェニック植物を作製する方法は:(1)結実能力のある植物への再生が可能な単子葉植物細胞、特にオオムギを提供すること;(2)5'から3'方向に読む発現カセット、種子-特異的プロモータ、特に胚乳-特異的プロモータ、内在性種子貯蔵タンパク質を制御する転写制御因子をコードする核酸配列、抑制される(1または複数の)貯蔵タンパク質遺伝子を制御する転写制御因子により認識されるcis-作用性要素を有さない別の種子-特異的プロモータ、好ましくは胚乳-特異的プロモータ、目的の異種ポリペプチドをコードする核酸配列、そして3'非翻訳領域を含む核酸構築物により、植物細胞を形質転換すること;(3)前記植物細胞を再生させて種子貯蔵タンパク質の発現を制御する転写制御因子のレベルを減少させ、そして内在性貯蔵タンパク質レベルを減少させた第一のトランスジェニック植物を提供させること;を含む。
【0029】
単子葉植物種子において目的の異種ポリペプチドを高レベルに発現するためのDNA構築物もまた、本発明により包含される。そのようなDNA構築物には:所定の単子葉植物種子において機能しそして目的の異種ポリペプチドをコードする配列に対して機能可能に連結するプロモータ;そして所定の単子葉植物種子中で機能するプロモータに対して機能可能に連結した、オオムギ胚乳中でPTGSを可能にするhpRNAを形成することができる転写物をコードするBLZ1遺伝子とBLZ2遺伝子の領域のキメラ遺伝子構築物;が含まれる。
【0030】
本発明の別の側面において、本明細書中に記載される方法により得ることができる、特に上述したようなものなどのトランスジェニック植物が提供される。前記植物は、上述した所望の特徴を有する、すなわち、組換えにより産生される所望のタンパク質の発現および蓄積を亢進するため、選択された主要な資源“sink”タンパク質の発現を抑制する。本発明の好ましい植物には、例えば、種子中での異種タンパク質の発現および蓄積のために本発明に従って使用することができる、変種Hordeum vulgarisのオオムギ植物が含まれる。本発明のオオムギ植物は、好ましくは、そのゲノム中に、本明細書中で記載される好ましいTF、例えばbZIPタンパク質の群由来のTFまたはその部分をコードする配列を有する。
【0031】
発明の詳細な説明
本明細書の以下において、本発明を詳細に記載する。
用語“タンパク質”は、本明細書中において、“ポリペプチド”および“ペプチド”と、互換的に使用される。
【0032】
本明細書中において使用される用語“異種”は、用語“非天然”または“外来性”または“外因性”と互換的に使用することができ、そして通常は組換えDNA技術が関与する遺伝子操作に供されていない宿主生物においては見いだされない、タンパク質および/またはin-vitro修飾DNA配列に対して利用される。本明細書中において使用される用語“目的の異種ポリペプチド”または“目的のポリペプチド”は、本発明の方法または組成物を使用して宿主生物中で発現させるために意図されたいずれかのポリペプチドのことをいう。限定的ではない例として、薬理学的ポリペプチド(例えば、医薬用途のための)または産業的ポリペプチド(例えば、酵素)を、本発明に従って作製することができる。
【0033】
用語“コード配列”は、特定のアミノ酸配列をコードするヌクレオチド配列のことをいう。
“プロモータ”は、機能可能に連結された核酸の転写を指向する核酸調節配列または転写制御因子結合部位の一連のものとして定義される。プロモータは、コード配列または機能性RNAの発現を調節する核酸配列のことをいう。用語“機能可能に連結”は、プロモータ(核酸発現調節配列、または一連の転写因子結合部位)と第二の核酸配列とのあいだの機能的な連結のことをいい、ここでプロモータは、第二の配列に対応する核酸の転写を指向する。
【0034】
“転写制御因子”または“転写因子”は、遺伝子の上流、イントロンの内部、または停止コドンの下流に位置する短いDNA配列であるcis-作用性要素(またはcis-作用性モチーフ)に結合することができる、trans-作用性制御タンパク質のことをいう。
【0035】
用語“内在性遺伝子”は、生物のゲノム中の天然の位置における天然の遺伝子のことを言う。
用語“キメラ組み合わせ”または“キメラ遺伝子”または“ハイブリッド遺伝子”は、内在性遺伝子ではない遺伝子を形成し、天然には通常は同時に見いだされない制御配列および/またはコード配列を含む、いずれかの2またはそれ以上の連結したDNA配列のことをいう。用語“遺伝子構築物”または“DNA構築物”または“核酸構築物”は、いずれかのDNA配列または一般的な分子生物学的ストラテジーにより組み立てられるDNA配列の組み合わせのことをいう。
【0036】
本明細書中で使用する場合、用語“発現”は、結果としてDNA配列のRNAポリメラーゼ-触媒転写を引き起こす一本鎖または二本鎖形状のいずれかにおける、センス(mRNA)、アンチセンスRNAまたはそれらのその他のRNAポリマーの転写を含む、遺伝子生成物の生合成のことをいう。発現は、ポリペプチド中の前記遺伝子由来のmRNAの翻訳のことをいう場合もある。
【0037】
用語“形質転換”は、結果として遺伝子的に安定な遺伝形質をもたらす、宿主生物のゲノム中の核酸分子の転移のことをいう。形質転換された核酸断片を含有する宿主生物は、“トランスジェニック”生物と呼ばれる。用語“トランスジェニック”は、本発明の植物宿主細胞が、ゲノム中に安定に組み込まれた少なくとも1つの外来性核酸分子、好ましくは2つの外来性核酸分子を含有することを意味する。植物形質転換方法の例には、アグロバクテリウム-媒介性形質転換(De Blaere et al. 1987)および粒子衝撃形質転換技術、または“遺伝子銃”形質転換技術(Klein et al. (1987); U. S. Pat. No. 4,945, 050)が含まれる。
【0038】
用語“アンチセンス阻害”または“アンチセンス”または“アンチセンス抑制”は、内在性転写生成物またはmRNAに対して十分に相補的なアンチセンス鎖のことをいい、それにより内在性転写生成物の翻訳および発現を阻害しまたは減少させることをいう。用語“標的化抑制”は、組換えDNA技術を使用して、いくつかの所定の生体における抑制のために選択された内在性遺伝子の発現を阻害しそして抑制することができるRNA転写物をコードすることができるDNA配列を設計しそして利用することをいう。用語“抑制性トランスジーン”は、内在性遺伝子の発現を阻害しそして抑制することができるRNA転写物をコードするいずれかの異種DNA配列のことをいう。用語“同時抑制”は、形質転換により細胞中に導入され、そしてその同一の細胞中で内在性遺伝子として同一の又は同様の配列を有する遺伝子が、内在性遺伝子と導入遺伝子の両方を抑制する場合のことをいう。
【0039】
本明細書中で使用される“bZIP”タンパク質は、DNA結合および二量体化に関与し、そして5'ACGT'3コアを含有するDNA配列に共通して結合するいわゆる塩基性ヘリックス/ロイシンジッパードメインを含有する制御タンパク質のことをいう。
【0040】
本明細書中で使用される用語“ホルデイン”は、オオムギにおいて、具体的には胚乳中で合成される貯蔵タンパク質のことをいい、そして4つの群:β-ホルデイン(ベータホルデイン、B-ホルデインとも呼ぶ)、D-ホルデイン、C-ホルデインおよびγ-ホルデインに分類される。
【0041】
本明細書中で使用される用語“分子的農業”は、植物を使用してさらにプロセッシングするためのタンパク質などの貴重な生物学的化合物を作製するプロセスのことをいう。
遺伝子操作していてもよい単子葉植物を、本発明において使用することができる。好ましくは、植物は単子葉植物であり、より好ましくは、オオムギ、トウモロコシ、コムギ、オートムギ、およびコメから選択される。オオムギは、オオムギを本発明の好ましい候補物とする多数の所望される特徴を有し、オオムギの種Hordeum vulgarisisが特に好ましい。遺伝子的に形質転換されていてもよい植物は、コード領域用のDNA配列またはhpRNAを形成することができるRNA転写物をコードするDNA配列を含む異種DNA配列を、導入し、発現させ、安定的に維持させ、そして子孫の以降の世代に遺伝させることができる植物である。例えば、ビアラホスまたはバスタなどを含む除草剤、または選択可能マーカーとしてのハイグロマイシンなどの殺虫剤を使用する遺伝子操作および形質転換方法を使用して、オオムギ植物を作製した。
【0042】
適切な宿主植物を選択した後、プロモータが、目的の異種遺伝子の発現を駆動するために選択される。植物細胞において活性な多数のプロモータが、この文献中に記載された。本発明のDNA構築物中で利用されるプロモータは、目的の異種ポリペプチドの蓄積が所望される組織中、例えば単子葉植物の種子の胚乳中で、強力な活性を有することが好ましい。選択されるプロモータは、本発明の方法により抑制されるように標的となる転写制御因子、例えば目的の異種遺伝子の発現と資源について競合する内在性貯蔵タンパク質遺伝子の発現を制御する転写制御因子、により制御されないことがさらに好ましい。そのようなプロモータは、様々な植物遺伝子材料からまたは植物ウィルスから得ることができる。以下に記載したように、選択された特定のプロモータは、単子葉植物種子における、より好ましくはオオムギにおける、そして最も好ましくは種子の胚乳組織における、異種タンパク質の発現に適していることが好ましい。本発明の目的のために有用なそのような適したプロモータのクローニングおよび解析は、実施例2に記載される。
【0043】
目的の異種タンパク質の蓄積に関して選択された標的組織中のタンパク質プロファイル発現パターンが解析され、そして最も豊富な貯蔵タンパク質が本発明による標的化抑制のために同定されることがさらに好ましい。解析は、刊行文献から採取したデータなどの従来技術に依存することができる。このことはまた、マイクロアレイアプローチを使用して、異種タンパク質の発現を駆動するために選択されたプロモータの同時的発現プロファイルと重複する時点で前記組織から抽出したmRNAを使用して、またはノザンブロット解析、RT-PCRおよび/またはウェスタンブロッティングを使用して、数千の遺伝子の発現パターンを同時にモニターすることを含んでいてもよい。集められた情報に基づいて、時間的そして空間的に目的の異種遺伝子の発現と競合する、高い発現を伴う候補遺伝子を、本発明による標的化抑制のために選択する。
【0044】
本発明の組換えプラスミドを、目的のDNA配列を、細菌宿主中で複製可能な適切なプラスミド中にライゲーション(挿入)することにより得ることができる。目的の異種タンパク質をコードする遺伝子などのDNA配列、または内在性遺伝子の標的化抑制のために設計されたDNA配列を、プロモータに加えて、この目的のため、所望される場合には、付加的エンハンサーDNA配列、足場付着領域(scaffold-attachment regions)、イントロン、ポリ(A)付加シグナル、目的のリボソーム結合配列や選択マーカー遺伝子(例えば、ハイグロマイシン耐性遺伝子、アンピシリン耐性遺伝子、ビアラホス耐性遺伝子など)を含有していてもよいプラスミド中に、機能するように組み込むことが好ましい。
【実施例】
【0045】
本発明をよりよく規定するため、そして本発明を実施する際に当業者をガイドするため、以下の実施例を提供する。それ以外の記載をしていない限り、用語は、当業者により従来から使用されている様に理解されるべきものである。
【0046】
方法
RNA抽出およびノザンブロット解析
全RNAを、製造者の指示にしたがってTRIzoI(Gibco BRL)を使用して単離し、そしてレーンあたり10μgの全RNAを電気泳動に供し、そして毛細管作用によりHybond Nメンブレン(Amersham)に転写した。Stratolinker(Stratagene)を使用してRNAをメンブレンに架橋し、そしてフィルターを高ストリンジェントのハイブリダイゼーションによりハイブリダイズさせ、そして記載されたように洗浄する(Davis et al., 1994)。プローブは、製造者の指示にしたがってランダムプライミング(T7 QuickPrime KitTM;Pharmacia)により32Pで放射性標識する。EtBr-染色したブロットのネガデジタル画像を、ロード対照として使用する。
【0047】
オオムギ形質転換
a)遺伝子形質転換用の植物材料
Hordeum vulgarecv Golden Promieの種子を、75%軽ミズゴケピート(light sphagnum peat)と25%軽石(中粒子サイズ)の混合物上に蒔き、そして植物を、日中18℃(16時間)および夜間12℃(8時間)で、そして70%相対湿度、250μモル/m2 sの日中冷白色蛍光での連続光のもと、必要に応じて灌水して、生育させた。これらの条件下で、植物は、約55〜95日間、または未成熟の種子が形質転換用の材料として準備されるまで、すなわち開花後約8〜14日後まで、栄養生長させた。種子は、3%次亜塩素酸ナトリウム中で40分間、回転震盪器で殺菌消毒し、そして無菌水を5回変えることによりすすいだ。
【0048】
b)植物形質転換用の細菌系統および調製物
vir領域を有するTi-プラスミドをトランスで保持するバイナリベクターを保有するAgrobacterium tumefaciensを使用して、目的の異種タンパク質の発現を制御するDNA構築物を有するT-DNA領域をオオムギ中に導入する。
【0049】
E. coli XL-BlueおよびAgrobacterium tumefaciens細菌の両方の形質転換は、Maniatisら(Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N. Y. (1982))により記載されるエレクトロポレーションにより行う。植物の形質転換用の調製物のため、Agrobacterium培養物のシングルコロニーを、5 mlのAgro培地(Tryptone 5 g/L、酵母エキス2.5 g/L、マンニトール5 g/L、グルタミン酸1 g/L、KH2PO4 250 mg/L、MgSO5-7H20 100 mg/L、ビオチン1μg/L、pH 7.0、25μg/mlリファンピシン、50μg/mlスペクチオマイシン)中に接種し、そして27℃にて24〜40時間増殖させる。無菌エッペンドルフチューブ中で、200μlの培養物を200μlの30%グリセロール水溶液(事前に滅菌したもの)に対して添加し、そして培養物をよくボルテックスにかけ、そしてベンチに2時間静置した後、-80℃で保存する。各形質転換につき、1本のチューブを-80℃冷凍庫から取り出し、解凍し、そして約200μlのAgrobacterium細菌ストックを抗生物質を含まない5 mlのAgro培地に対して添加する。その後、培養物を27℃にて17〜20時間生育させた後、植物材料の接種のために使用する(以下を参照)。
【0050】
c)Agrobacterium-誘導形質転換のためのオオムギ外植体の調製
第1日に、約10個のオオムギ頭頂部を摘み、開花後約8〜14日後に、芒(awn)および種子を取り出し、1.5 mm〜2 mmのサイズの胚を選択した。最初の植物材料は、健康で成熟していることが必要であるが、水に浸されていることは必要とされず、そして種子は緑色で病気やカビの兆候がないものであるべきである。種子を50 mlのファルコンチューブ(高々半量)に静置し、そして70%エタノールですすぎ、そしてその後エタノールを捨てた。次いで、20%漂白溶液(White King)を添加し、20分間混合した。
【0051】
ラミナエアフロー中で、漂白溶液を捨て、種子を滅菌水ですすぎ(約5〜8回すすぐ)、そしてチューブを4℃にて一晩静置した。第2日に、種子を顕微鏡プラットフォーム上の滅菌ペトリ皿中に静置した。胚の位置を特定し、種子の末端部を切断し、そして両側を切断した種子を作製した。その後、種子をピンセットを用いてしっかりと固定し、そして胚が飛び出すように種子の中央部に圧をかけた。
【0052】
胚をピンセットを用いて固定し、そしてメス刃を胚盤と胚軸とのあいだの溝に挿入し、そして胚軸をゆっくりと切断した。胚軸を取り除いた胚を、再生培地上に切断面を上にして静置し、ペトリ皿の中央に、約25個の胚を静置させた。
【0053】
d)すべてのバイナリベクターは、37℃にて100μg/mlのスペクチオマイシンを含有するE. coli XI-Blue LB培養培地中で増殖させ、そして引き続いてベクターを、QIAGEN(商標)Plasmid Midi Kitを用いて一晩増殖させた100 ml培養物から精製した。精製したバイナリベクターを、1μl(1μg)のベクターを0.1 cmのギャップを有する滅菌キュベット(BioRad)中に静置し、ベクターを40μlのエレクトロコンピテント細胞とともに洗浄し、そしてエレクトロポレーション用に2.5 kVの電圧、および21μFの電気容量に設定することにより、Agrobacterium tumefaciens中にエレクトロポレーションを用いて導入した。細胞を100μg/mlのスペクチノマイシンおよび20μg/mlのリファンピシンを含有するYEP選択プレート上に広げ、28 ℃にて2日間増殖させた。A. tumefaciens形質転換体由来のプラスミド制限酵素消化物を解析を行い、バイナリベクターの完全性を確認した。
【0054】
e)オオムギ外植体のAgrobacterium感染
約20μlのAgrobacterium培養物を各胚上にピペットでのせ、すべての胚が溶液と確実に接触できるようにする。胚を裏返しにし(切断面を下向き)、そして再生培地上をプレートの外まで引っ張っていき、過剰なAgrobacteriumを除去した。その後、胚を均等な間隔で新鮮な再生培地プレートに移し(切断面を上)(プレートあたり25個)、そして24℃にて暗キャビネット中に静置した。
【0055】
f)Agrobacterium 感染後のオオムギ組織培養における再生および器官形成
3日後、胚を選択マーカー(たとえばビアラフォスまたはハイグロマイシン)を含む新鮮な再生培地に移し、そして4〜6週間そこにそのままにし、2週間ごとにサブカルチャーした。苗条を再生するため、カルスを苗条誘導培地(SIM)に移し、そして生残したカルスおよび再生し始めた苗条を、小さな苗が形成されるまで、2週間ごとに新鮮なSIMへ移した。その後、苗を根誘導培地(RIM)に移し、そして生残した植物をさらにスクリーニングするために土に鉢植えした。
【0056】
DNA抽出、PCRおよびクローニング
PCR増幅のためのゲノムDNAを、NucleoSpinPlant Kit(商標)(Clonetech)を製造者の指示にしたがって用いて約200 mgの若葉組織から抽出した。PCR反応を加熱蓋を有するペルティエ型サーマルサイクラー(MJ Research, PTC-200)中で行った。すべてのプライマーは、完全自動化DNA合成装置(Perkin-Elmer)を用いて化学的に合成した。すべてのDNA断片は、UV光のもと、1%EtBr-染色アガロースゲル上でサイズ画分し、そしてQIAquick(商標)Gel Extration Kit(Qiagen)を用いて精製した。制限酵素は、New England Biolabs Inc(Beverly, Mass.)およびFermentasから購入し、そして製造者の指示にしたがって使用した。クローニングおよびその他のDNA操作は、Maniatisら(Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N. Y. (1982))に記載されるような標準的な手順にしたがって行ったが、すべてのライゲーション反応では、以下の条件を使用した:バッファー〔50 mM Tris-HCl(pH 7.6)、5 mM MgCl2、1 mM DTT、0.5 mM ATP、2.5%ポリエチレングリコール-8000〕中T4 DNAリガーゼ(2.5 U)を16℃にて16時間。
【0057】
コンピテントEscherichia coli XL-Blueを、DNA操作のための形質転換実験において、そしてプラスミドクローンの構築のため、レシピエントとして使用した。すべてのプラスミドDNA midi prepは、Qiagen(商標)Plasmid Midi Kit(Qiagen)を使用して調製し、そしてDNA断片の配列決定は、自動化DNA配列決定装置(ABI, Pharmacia)を使用して行った。
【0058】
実施例1 Hordeum vulgarisの胚乳中の“sink”タンパク質の解析
Hordeum vulgaris種子変種Skeglaにおけるタンパク質発現解析を、開花49-日後(dpa)に回収した種子から抽出したタンパク質のSDS-PAGEを使用して行い、胚乳の全タンパク質レベルに対して主要な寄与を行う胚乳発現タンパク質を同定し、そしてしたがって、標的化抑制のための可能性のある候補物を同定することができる。5つのオオムギ胚軸の種子全体を液体窒素中で手動で細かく磨砕し、その後適切な抽出バッファーを添加した。2種類の抽出バッファーを、水性抽出に加えて、量産プルに使用した。最初に、1%2-メルカプトエタノール、10 mM Tris-HCl pH 8.0および1%ポリビニルピロリジン(MW 360.000)の存在下にて、エタノール抽出(70%EtOH)を伴う還元条件下で全タンパク質の抽出を行った。次に、170 mM NaCl、1%2-メルカプトエタノール、10 mM Tris-HCl pH 8.0および1%ポリビニルピロリジン(MW 360.000)の存在下にて、還元条件下で、全タンパク質の抽出を行った。
【0059】
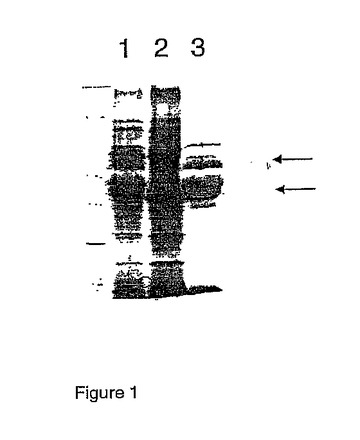
液体窒素中でサンプルを磨砕した後、5 mlの抽出バッファーを抽出バイアルに添加し、その後3分間連続的に磨砕した。抽出物を4℃、4000 rpmにて10分間、遠心分離にかけて清澄化させ、その後分子量5 kDaのカットオフを有するUltrafree-4濃縮装置(UFV4BCC00- Millipore Corp. Bedford, MA, USA)中で、10倍遠心分離濃縮を行った。清澄化させ濃縮した抽出物の100μlのサンプルを、100μlの2×サンプルバッファーに添加し、混合物を加熱水浴中で5分間静置した。冷却後、10μlのサンプルを12%ポリアクリルアミドゲルにかけ、SDS-PAGEにより分離した。SDS-PAGEの後、ゲルをクマシーブルーR250染色により染色し、脱染色してタンパク質バンドを可視化した。結果から、H. vulgare変種Skeglaにおける発生途中の胚乳において最も豊富なタンパク質は、B-ホルデインおよびC-ホルデインであることが示された(図1)。
【0060】
この結果から、BホルデインおよびCホルデインをコードする遺伝子を制御する転写制御因子を抑制することが、資源に関する競合を非常に減少させやすく、そして本発明にしたがって発現された異種ポリペプチドの蓄積を増加させることが示される。
【0061】
実施例2 異種タンパク質のための発現ビヒクルとして使用される、Hordeum vulgareのB-ホルデインおよびC-ホルデイン標的化抑制のためのプロモータの選択
B-ホルデイン(GenBank Accession No. X53690)およびC-ホルデイン(GenBank Accession No. M36941)の遺伝子プロモータは、それら両方において同定された100%同一なcis-作用性要素を含有し、それらはそれらの転写制御に関与する(Muller and Knudsen 1993;Vicente-Carbajosaet al. 1992)。これらのcis-要素は、翻訳開始コドンの約300 bp上流に位置するいわゆる胚乳ボックス(EM)中に存在する。EMは、転写因子結合のための2種の特有のcis-作用性モチーフ、プロラミンボックス(PB)5'-TGTAAAG-3'、そしてGCN4-様モチーフ(GLM)5'-(G/A)TGA(G/C)TCAT-3'を保有する。
【0062】
GLMは、2種の転写因子、BLZ1(GenBank Accession No. X80068)およびBLZ2(GenBank Accession No. X80068)により認識される。結合に際して、特定の遺伝子の転写が活性化される(Onate et al. 1999を参照)。これらの遺伝子は両方とも、1コピーの遺伝子であり、そしてしたがって、本発明により記載される標的化抑制のための良好な候補である。Vicente-Carbajosaら(1998)は、安定的に形質転換した植物に対する作用、または貯蔵タンパク質の蓄積に対する作用を調べることなく、一過性発現系におけるそれらのアンチセンス抑制を試験した。さらに、同一組織における異種タンパク質の蓄積に対するそのような抑制の作用は調べられなかった。本発明による標的化抑制を操作する際の重要な基準は、目的の異種タンパク質をコードするトランスジーンを駆動するプロモータが、貯蔵タンパク質の発現を抑制する原因となる特定の転写制御因子により認識されるcis-作用性モチーフを含有してはいけない、ということである。B-ホルデインおよびC-ホルデインがBLZ1およびBLZ2により認識されるGLMを含有するため、B-ホルデインプロモータおよびC-ホルデインプロモータを使用して、所望の異種タンパク質をコードする異種トランスジーンの発現を駆動することができない。別の重要な基準は、抑制トランスジーンそれ自体を駆動するプロモータが、組織特異的プロモータ、例えば胚乳特異的プロモータ、により駆動され、それによりそれらのプロモータが必要とされるその他の組織における転写制御因子の発現には影響が及ばない、ということである。このことは、多数の転写因子(例えばオオムギにおけるBLZ1など)が1より多い組織中で発現されていることから、重要である。さらに別の基準は、本発明により抑制されるべき転写制御因子により認識されるcis-作用性要素を有さない抑制トランスジーンを駆動するためのプロモータを使用することにより、抑制トランスジーンの“自己抑制(self-suppression)”を回避することである。
【0063】
これらの基準に基づいて、B-ホルデインおよびC-ホルデインの標的化抑制について候補プロモータを選択した。
Hordeum vulgare cv Skegla由来のD-ホルデインプロモータの単離、クローニングおよび解析
プライマーを、-435〜-16由来の隣接するプロモータ領域を同定するために、GenBank配列id. X84368に記述されたD-ホルデイン遺伝子の翻訳開始部位に基づいて、センスプライマーとして5'末端にEcoRI制限酵素部位を導入された5' GGAATTCCEcoRICTTCGAGTGCCCGCCGATTTGCCAGCAATGG(SEQ ID NO: 9)を設計し、そしてアンチセンスプライマーとして5'末端にNotI制限酵素部位を導入された5'ATAAGAATGCGGCCGCNotIAATGAATTGATCTCTAGTTTTGTGG(SEQ ID NO: 10)を設計し、そして合成した。プライマーの5'末端にこれらの制限酵素部位を導入した目的は、増幅断片のクローニングを補助するためであった。Hordeum vulgare cv. Skegla由来のゲノムDNAを鋳型として使用して420 bpの断片を増幅するために使用する反応溶液の組成は、以下の通りであった:
【0064】
【表1】

【0065】
9μlの滅菌水およびTaq DNAポリメラーゼ以外の上述の反応溶液をよく混合し、そして溶液を94℃にて3分間加熱し、その後、80℃まで冷却して30秒間保持し、そしてTaq DNAポリメラーゼおよび9μlの滅菌水を添加した。PCRの最初のサイクルを、以下の様にして行った:反応物を、94℃にて30秒間加熱し、アニーリングのために57℃まで冷却して30秒間保持し、そして次いで伸長反応のため72℃で45秒間加熱した。その後の30サイクルは、以下の様にして行った:94℃にて30秒間の加熱変性、62℃にて30秒間のアニーリング、そして72℃にて45秒間の伸長。30サイクルが終了した後、反応物を72℃にて4分間加熱した。420 bpの増幅断片(図2a)を、EcoRI/NotIで消化し、次いでベクターpKOH122のEcoRI/NotI部位にライゲーションして、組換えプラスミドpDH104を得た。420 bpのDNA挿入物のヌクレオチド配列を、その後決定した(SEQ ID NO: 11)。その配列によれば、D-ホルデインプロモータはGLMを含有せず、そして従って、BLZ1またはBLZ2のいずれかの標的化抑制を介したB-ホルデインおよびC-ホルデインの抑制(以下の検討の項を参照)は、D-ホルデインプロモータの活性に影響を与えず、そして従って、目的の異種タンパク質の発現に影響を与えなかった。さらに、D-ホルデインプロモータは、もっぱら胚乳-特異的なものであるため、このプロモータはBLZ1およびBLZ2の発現を抑制するために設計された抑制トランスジーンを駆動するために有用である。
【0066】
プロモータを使用して本発明により抑制されるべき転写制御因子により認識されるcis-作用性要素を有さない抑制トランスジーンを駆動することにより、抑制トランスジーンの“自己抑制”を回避することが重要であるため、GLMを欠損するD-ホルデインプロモータを使用してBLZ1およびBLZ2を抑制する抑制遺伝子を駆動することは、そのような“自己抑制”を引き起こさないだろう。
【0067】
実施例3 D-ホルデインコード領域のクローニングと、mRNAレベルでのD-ホルデイン遺伝子の内在性発現の解析
センスプライマー、5'GGAATTCCEcoRIATGGCTAAGCGGCTGGTCCTC(SEQ ID NO: 12)およびアンチセンスプライマー、5'GGAATTCCEcoRITTGCAATTGGATAGGTCTCTTG(SEQ ID NO: 13)を設計して、GenBank 配列id. X84368に記載されるD-ホルデイン遺伝子のコード領域由来御727 bpの断片を増幅した。Hordeum vulgare cv. Skegla由来のゲノムDNAを鋳型として使用して、727 bp断片を増幅するために使用される反応溶液の組成は、以下の通りである:
【0068】
【表2】
【0069】
9μlの滅菌水およびTaq DNAポリメラーゼ以外の上述の反応溶液をよく混合し、そして溶液を94℃にて3分間加熱し、その後80℃まで冷却して30秒間保持し、そしてTaq DNAポリメラーゼおよび9μlの滅菌水を添加した。PCRの最初のサイクルを、以下の様にして行った:反応物を、94℃にて30秒間加熱し、アニーリングのために60℃まで冷却して30秒間保持し、そして次いで伸長反応のため72℃で45秒間加熱した。その後の30サイクルは、以下の様にして行った:94℃にて30秒間の加熱変性、65℃にて30秒間のアニーリング、そして72℃にて45秒間の伸長。30サイクルが終了した後、反応物を72℃にて4分間加熱した。727 bpの増幅断片(図2b)を、EcoRIで消化し、次いでベクターpKOH122のEcoRI部位にライゲーションして、組換えプラスミドpDH136を得た。DNA挿入物のヌクレオチド配列を、決定した(SEQ ID NO: 14)。
【0070】
胚乳40 DAP、胚40 DAP、葉、葉軸、および根におけるD-ホルデイン遺伝子のmRNAの定常状態レベルは、ノーザンブロット解析を使用して適切に解析する:Hordeum vulgare cv Skeglaの様々な組織または植物の部分由来の200〜250 mgの植物材料から全RNAを単離し、1.2%アガロース-ホルムアルデヒドゲル上で分離し、そして製造者の指示に従ってZeta-Probeメンブレン(Millipore, Bedford, Mass.)上にブロットした。ブロッティング、ハイブリダイゼーション、および洗浄は、記載されたように行った(Davis et al., 1994)。ハイブリダイゼーションは、D-ホルデイン遺伝子の32P放射標識しランダムプライミングした747bp断片(SEQ ID NO: 14)をプローブとして使用して行う。
【0071】
実施例4 植物形質転換ベクターpbDH101中へのオオムギの胚乳-コドン最適化HoxB4のクローニング
オオムギにおけるD-ホルデインプロモータの制御下での発現に関するコドン最適化HoxB4は、記載されたように(本願出願人の同時係属出願“Methods for high level expression of polypeptides in plants using codon optimization”を参照)、GeneBank ID NM_024015に記載される基本的配列情報およびD-ホルデインプロモータの制御下での胚乳におけるコドン最適化発現に関するコドン利用情報を使用して、GeneArt GmbH(Germany)により合成された。791 bpの断片(SEQ ID NO: 20)は、クローニング用のNcoI部位(CCATGG)に隣接し、そしてリンカー-エンテロキナーゼ切断部位DDDDKPTPTPTをコードする27ヌクレオチド配列を3'末端に含む。HoxB4断片は、pKOH122のNcoI/NcoI部位にライゲーションされ、pDH170が作製される。その後、断片をNcoIにより放出させ、そしてpDH152のNcoI部位にライゲーションさせ、pDH152中のEK-L断片を置換してpDH156を得る。次いで、pDH104由来のD-ホルデインプロモータ断片をpDH156のEcoRI/NotI部位中にライゲーションしてpDH157を得る。D-プロモータ/SP/HoxB4-E-L/CBM/kd/pinII断片をSse83871/SfIにより放出させ、そしてpKOH250のSse83871/SfI部位にライゲーションして植物形質転換ベクターpbDH101を作製する(図3)。
【0072】
実施例5 D-ホルデインプロモータの制御下でのBLZ1およびBLZ2のヘアピンRNA-誘導標的化抑制による、植物形質転換ベクターpbDH104の構築
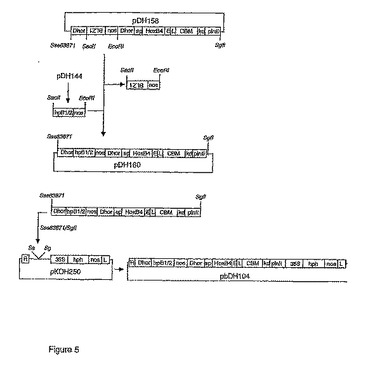
BLZ1およびBLZ2のヘアピンRNA-誘導標的化抑制のための遺伝子が、265 bp nos-末端断片(SEQ ID NO: 8)が伴う592 bpの抑制断片として、GeneArt GmbH(Germany)により合成され、そしてpUC19のHindIII/EcoRI部位にクローニングして、pDH144を得た。断片は、BLZI-遺伝子(SEQ ID NO: 2)およびBLZ2-遺伝子(SEQ ID NO: 4)由来のヌクレオチド配列のキメラ組成であり、これはセンス部分とアンチセンス部分とを形成し、そして88 bpのヌクレオチド配列はBLZ1遺伝子由来のイントロン4を示し(GenBank配列id. X80068)、転写後にRNAヘアピンループを形成する様に設計された(図4)。857 bp断片であるhpB1/2-nosをSacII/EcoRIにより放出させ、そしてpDH158中でアンチセンスBLZ1-nos断片と置換して、pDH160を得る。pDH160中の完全な挿入物を、Sse83871/SgfI消化により放出させ、そしてpKOH250のSse83871/SgfI部位にライゲーションして、ベクターの左側境界(L)と右側境界(R)のあいだのDNA配列を植物ゲノム中に安定的に組み込むことができる、バイナリ植物形質転換ベクターpbDH104を作製した(図5)。
【0073】
実施例6 pbDH101およびpDH104を用いたオオムギの遺伝子形質転換
開花後約8〜14日のHordeum vulgaris cv Golden Promiseの未成熟種子を採取し、そして4℃にて暗所で一晩保存した。冷所でインキュベートした未成熟種子を、70%EtOH中で1分間処理し、次いで0.6%次亜塩素酸塩ナトリウム(natrium hypochloride)中で10分間処理し、その後滅菌条件下でラミナフロー中の解剖顕微鏡のもとで、滅菌ペトリ皿上に静置した状態で滅菌蒸留水で完全に洗浄した(5〜8回)。胚の位置を特定し、種子の末端部を切断し、そして両側を切断した種子を作製した。種子をピンセットで固定し、そして種子の中央部を圧迫して胚を押し出す。胚をピンセットでしっかりと固定し、そしてメス刃を胚盤と胚軸とのあいだの溝に挿入し、そして胚軸をゆっくりと切断した。胚盤をカルス誘導培地上に静置し、切断面を上にし、そして植物形質転換ベクターを保持するそのままの濃度のAgrobacterium tumefaciens25μl〜40μlを1〜5分間接種した。接種の後、胚盤をディッシュの外に引き出し、細菌の負荷を低減し、同時培養フェーズのあいだの過剰増殖を低下させる。感染した胚盤を新しいカルス誘導培地プレートに移し、そしてプレートを暗所、24℃にて3日間インキュベートする。3日後、胚盤をAgrobacteriumを殺すための100μg/mlのチメンチン(timentin)および形質転換細胞を選択するための50μg/mlのハイグロマイシンを含む新しいカルス誘導培地に移し、暗所、24℃にて4週間インキュベートし、2週間後にサブカルチャーする。次いで、カルスを2.5 mg/L BAP、50μg/mlチメンチンおよび25μg/mlハイグロマイシンを含む苗条誘導培地に移し、そして強光下にて4〜10週間インキュベートする。個々の再生しつつある苗を、発根培地(50μg/mlチメンチンおよび25μg/mlのハイグロマイシンを含み、ホルモン類は含まない)に移す。発根している約5〜7 cmの苗条にまで発生させた後、トランスジェニック植物を土壌に移し、そこで完全光(full light)下にて生育させた。
【0074】
実施例9 BLZ1およびBLZ2の標的化抑制の後のHoxB4-CBMタンパク質の蓄積
pbDH10l(hoxB4-cbmのみ)またはpbDH104(hoxB4-cbmにより発現されるBLZ1/2 hpRNA)のいずれかにより形質転換されたトランスジェニック植物におけるB-ホルデインおよびC-ホルデインの蓄積を測定するため、Smith(in The Protein Protocols Handbook ed., Walker, 2nd ed., Humana Press, 2002)により記載されるように、Colloidal Coomassie Brilliant Blue G法を使用することができる。次いで、種子抽出物由来の可溶性タンパク質をSDS-PAGEにより標準的タンパク質と共に分離し、その後、ゲルを染色溶液(3.5%(w/v)過塩素酸中、0.04%(w/v)Brilliant Blue G)を用いて1時間染色し、その後ゲルを蒸留水中で4時間脱染色する。それぞれのB-ホルデインおよびC-ホルデインバンドの強度を、スキャニングデンシトメトリーを用いて解析し、そして相対バンド強度をpbDH101またはpbDH104により形質転換したトランスジェニック植物中で比較する。これにより、BLZ1/2 hpRNAにより引き起こされたB-ホルデインおよびC-ホルデインの抑制を推定する。pbDH101またはpbDH104により形質転換された植物におけるHoxB4-CBMの同一性および発現レベルを、二つに複製したサンプルを含有する同一のゲルの断片をウェスタンブロッティングすることにより、確認しそして測定することができる。バンドをゲル断片からニトロセルロースメンブレン上に、BioradのMini transblot Cellを使用してエレクトロブロットすることができるが、ゲル断片の輪郭をフィルター上にマークした後、ブロッティングする。ゲル/メンブレンサンドイッチのブロッティングおよび取り外しの後、ニトロセルロースをCBMに対するポリクローナル抗血清を用いてハイブリダイズし、そして複数回の洗浄工程(2×5分、2×15分、1×5分)の後、西洋ワサビペルオキシダーゼ(HRP)を抱合させた二次抗体を用いて45分間インキュベートした。上述した後に続く洗浄工程の後、そして基質5-ブロモ-4-クロロ-3-インドリルホスフェートおよびニトロブルー塩化テトラゾリウムをメンブレンに添加した際、HRP呈色反応はバンドをHoxB4-CBMとして陽性に同定し、そしてBLZ1/2 hpRNA発現のhoxB4-CBMレベルに対する作用を推定する。
【0075】
本発明の好ましい態様のみが特に示されるが、上述した実施例において記載される本発明の多数の修飾およびバリエーションが、本発明の範囲からはずれることなく当業者には生じることが予想される。
【0076】
【表3】
【図面の簡単な説明】
【0077】
【図1】図1は、水溶性抽出物(レーン1)、塩抽出物(レーン2)そしてEtOH抽出物(レーン3)を使用した、Hordeum vulgaris cv. Skegla(開花後49日)由来の全タンパク質のドデシル硫酸ナトリウム-ポリアクリルアミド電気泳動である。矢印は、B-ホルデイン(下)およびC-ホルデイン(上)を示す。
【図2】図2は、a)プライマーSEQ ID NO: 9およびSEQ ID NO: 10(実施例2を参照)およびb)プライマーSEQ ID NO: 12およびSEQ ID NO: 13(実施例3)を使用した、PCR増幅生成物のアガロースゲル解析である。生成物を、1.0%アガロースゲル上で分離し、エチジウムブロマイドにより染色し、そしてUVの下で写真撮影した。左側のサイズマーカーは、EcoRI/HindIII切断ラムダである。矢印は、増幅生成物を示す。
【図3】図3は、D-ホルデインプロモータの制御下でオオムギ胚乳組織中で発現させるためのコドン-最適化HoxB4-CBMキメラ遺伝子を用いて、オオムギを形質転換するための、バイナリ植物形質転換ベクターpbDH101のクローニングの概略説明を示す。略語:D-hor、D-ホルデインプロモータ;CBM、Thermatoga maritima由来の炭水化物結合ドメインをコードするコドン-最適化遺伝子;HoxB4、コドン-最適化ホメオボックスB4遺伝子(SEQ ID NO: 20);L、リンカー(PTPTPT;Pはプロリン、Tはトレオニン);kd、KDEL配列;pinII、ポテトプロテナーゼ阻害剤II遺伝子終止シグナル;CaMV 35S、カリフラワーモザイクウィルス35Sプロモータ;hph、E. coli由来のハイグロマイシンホスホトランスフェラーゼ(Genebankaccession # K01193);Nos-ter、ノパリン合成酵素終止シグナル;R、右側境界;L、左側境界。
【図4】図4は、D-ホルデインプロモータの制御下で、オオムギにおけるBLZ1およびBLZ2のヘアピンRNA-誘導標的化抑制のために構築された、遺伝子の概略説明である。センス配向およびアンチセンス配向の114 bpのBLZ1/BLZ2はそれぞれ、ステム部分を形成することができる。一方、88 bpのイントロン4がヘアピンループ中のループを形成する。
【図5】図5は、D-ホルデインプロモータの制御下でオオムギ胚乳組織中のBLZ1およびBLZ2のhpRNA-誘導標的化抑制を用いて、オオムギを形質転換するための、バイナリ植物形質転換ベクターpbDH104のクローニングを概略的に説明したものである。略語:D-hor、D-ホルデインプロモータ;hpBl/2、BLZ1およびBLZ2由来のヌクレオチド配列(SEQ ID NO: 8)からなる620 bpのサプレッサー断片;CBM、Thermatoga maritima由来の炭水化物結合ドメインをコードするコドン-最適化遺伝子;HoxB4、コドン-最適化ホメオボックスB4遺伝子(SEQ ID NO: 20);L、リンカー(PTPTPT;Pはプロリン、Tはトレオニン);pinII、ポテトプロテナーゼ阻害剤II遺伝子終止シグナル;CaMV 35S、カリフラワーモザイクウィルス35Sプロモータ;hph、E. coli由来のハイグロマイシンホスホトランスフェラーゼ(Genebank accession #K01193);Nos-ter、ノパリン合成酵素終止シグナル;R、右側境界;L、左側境界。
【発明の詳細な説明】
【0001】
発明の属する分野
本発明は、植物分子生物学の分野におけるものであり、そして具体的には、単子葉植物の胚乳において種子貯蔵タンパク質の内在性発現に由来する競合を抑制することにより、植物種子において異種タンパク質の発現を増加する方法に関する。
【0002】
背景
多数の植物が、発生途中の種子の胚乳において、様々な種類の貯蔵タンパク質、代謝酵素、内在性キチナーゼ、タンパク質合成阻害剤、プロテアーゼ阻害剤、アミラーゼ、レクチンおよびペルオキシダーゼなどの様々な機能を発揮する、タンパク質の大規模な蓄積物を発現しそして貯蔵する。胚乳中の貯蔵タンパク質は、多数の単子葉穀物種において一般的であるものの様な15%より多い種子乾燥重量の総量に達する場合がある。従って、特定の発生期において、種子胚乳中の細胞装置は、主としてこれらの特定のタンパク質の産生および貯蔵にかかりきりとなる。さらに、ほとんどの貯蔵タンパク質遺伝子は、多コピー存在することが見いだされ、そのことは、数日〜数週間のスパンでこれらのタンパク質が急速に蓄積することの重要性を反映している。
【0003】
穀物種子の発生中胚乳でのタンパク質蓄積に関する固有の生物学的能力は、多数の穀物植物、特に単子葉植物、が異種組換えタンパク質(例えば、医薬産業用の高価値のポリペプチド)の大規模生産のための実際的でそして効率的なビヒクルとなる可能性を有していることを示す;製造方法はしばしば、分子的農業(molecular farming)とも呼ばれた。さらに、種子中に異種ポリペプチドを蓄積することは、下流の処理コストを低減させる。というのも、これらの種子を、異種ポリペプチドの品質に影響を与えることなく、数年間にわたり保存することができるからである。そのようなタンパク質の発現は、好ましくは、種子-特異的プロモータまたは胚乳-特異的プロモータの調節下にておこなう。
【0004】
適切なプロモータの選択および細胞内(sub-cellular)分布などの一般的な分子生物学的戦略は、分子的農業のために使用される特定の植物における異種タンパク質発現レベルを改善する際に有用である。しかし、発現レベルは発現すべきタンパク質の性質、その複雑性、およびその機能により重大な影響を受けるものでもある。しかしながら、異種タンパク質の低発現レベル(低%の全可溶性タンパク質または%TSP)は、種子中で発現される場合であっても、格別の関心事である。新規のバイオテクノロジー的アプローチが、発現レベルをさらに向上させるために必要とされる。発生途中の種子において好ましくは貯蔵タンパク質および休眠-依存的生残のための一般的な調製物の蓄積にかかりきりであるその内在性の役割のために、細胞機構が既にプログラムされているため特に、このことはかなりの挑戦である。単子葉植物の胚乳または種子における目的の異種遺伝子の発現を駆動するために最も頻繁に使用されるプロモータは、多くの場合、貯蔵タンパク質遺伝子由来のプロモータ、例えばコメ由来の1.3 kbの胚乳-特異的プロモータGluB-1(Patel et al. 2000;GenBank Accession No. X54314)、またはオオムギ由来の0.45 kb D-ホルデイン(hordein)プロモータ(Sorensen et al. 1996; GenBank Accession No. X84368)などである。これらのプロモータは、目的の異種タンパク質の発現を駆動するために強力であるが、それらの活性は、最も豊富な貯蔵タンパク質を駆動する内在性プロモータの活性と、時間的そして空間的に同時に生じることから、アミノ酸、リボゾーム結合部位、および翻訳や後翻訳修飾に関与する酵素の集合体などの限定的な供給源に対する競合を生じる。この競合は、目的の異種ポリペプチドの発現レベルに対して悪い影響を与える。
【0005】
内在性遺伝子の発現(供給源と積極的に競合する多数の内在性貯蔵タンパク質遺伝子など)を時間的そして空間的に、目的の異種タンパク質の発現により抑制する方法を手にすることが非常に望ましいことである。従って、貯蔵タンパク質の抑制には、抑制の実際的な方法が提供されれば、様々な産業的ニーズが存在する可能性がある。このことは、特に内在性遺伝子が複数遺伝子ファミリーの構成分子である場合に、分子的農業においてかなり重要な挑戦となりうるものである。例えば、オオムギにおいて、胚乳における資源のための主要なsinkは、全胚乳タンパク質の50%もの割合を占める可能性があるB-ホルデイン貯蔵タンパク質である。(Shewry 1993: in Barley; Chemistry and Technology)。
【0006】
用語“sink”、“sink遺伝子”および“sinkタンパク質”は、refer 本明細書中でto細胞中で活発に発現されそして実質的な量の利用可能な資源を取り込む遺伝子および遺伝子生成物のことをいい、すなわち、sinkはその資源の実質的な部分の細胞を“枯渇させ(drain)”、そして従ってその他の遺伝子および遺伝子生成物の発現のための利用可能な資源を制限する。
【0007】
アンチセンス技術は、植物細胞中の特定の内在性遺伝子生成物の発現を減少させるための有効な手段として発生したものである(例えば、US特許No. 5,759,829;Orvar et al. 1997;Coles et al. 1999を参照)。しかしながら、この技術は、遺伝子ファミリーの構成分子である遺伝子の発現を直接的に抑制するためには、実際的なアプローチではなく、このことはしばしば、半数体ゲノムあたり少なくとも20であるB-ホルデイン遺伝子などの種子貯蔵タンパク質に当てはまる(Shewry et al. 1985);この技術は、遺伝子ファミリーに属さない遺伝子の発現を操作するために適している。
【0008】
内在性遺伝子発現を減少するための別の方法は、後転写遺伝子サイレンシング(PTGS)のための二本鎖RNA(dsRNA)の利用である。このRNA-誘導性遺伝子サイレンシングまたはRNA-干渉は、特定の内在性遺伝子のセンスRNA鎖およびその相補的アンチセンスRNA鎖の両方を組み合わせて1本のdsRNAにしたものであり、最初はCaenorhabditis elegansにおいて示された(Fire et al. 1998)。Hamiltonら(1999)は、短いヌクレオチドRNA(21〜23ヌクレオチド長)がdsRNAの分解生成物であり、それが植物中のPTGSにおいて内在性標的mRNAの切断を制御することを示した。植物における二本鎖遺伝子サイレンシングのより強力なアプローチは、Wesleyら(2001)により記載された方法であり、ここで遺伝子サイレンシング遺伝子は、転写後に“ヘアピン-ループ”RNA(hpRNA)を形成する様にして、構築される。hpRNA遺伝子サイレンシングの一形式は、ヘアピン-ループ中のスペーサーがイントロンであるイントロン-スプライシングhpRNA(ihpRNA)である。ihpRNAは、植物において非常に強力なPTGSの能力を有する(Waterhouse et al 2001およびSmith et al 2000も参照)。特別なベクター中にてそのようなPTGS構築物を作成する方法は、US特許出願2003-A-0049835に記載されている。
【0009】
分子的農業において、そして特にオオムギなどの単子葉植物において、異種遺伝子の発現を改善する新規な方法に関する要求が存在する。分子的農業において目的の医薬タンパク質の発現を増加させることを目的として、目的の異種遺伝子と内在性“sink”遺伝子とのあいだでの発現の競合を減少させる能力が、当該技術分野においては完成されていない。従来技術は、例えばhpRNA-誘導PTGSを使用して、標的化遺伝子サイレンシングが実際にどのようにして、分子的農業における目的の異種遺伝子の発現レベルを増加することができるのかについても示していなかった。
【0010】
本発明の概要と目的
本発明の第一の目的は、分子的農業における生産ビヒクルとして使用されるトランスジェニック種子における目的の異種ポリペプチドの蓄積レベルを増大させるための方法および核酸構築物を提供するものとして概説できる。最初のアプローチは、単子葉植物における胚乳中で組換え的に産生された目的の異種ポリペプチドの発現の利益となるように、上述した胚乳-特異的貯蔵タンパク質をコードする内在性の所望しないmRNAのタンパク質翻訳とのあいだでの資源に関する競合を制限することである。
【0011】
遺伝子ファミリーの構成分子である遺伝子の発現を抑制するための以前の方法は、個々の構成分子または遺伝子ファミリー構成分子のすべてに特徴的な遺伝子ファミリー構成分子のコード配列中の相同領域を標的とすることである。本発明の一つの目的は、上述した遺伝子ファミリーの複数の構成分子の発現の制御を、協調的な様式で編成する単一-遺伝子転写制御因子の発現を抑制することにより、遺伝子ファミリーの発現を減弱することによって、これらの以前に記載された方法の欠点を防止することである。
【0012】
本発明の別の目的は、転写制御因子のhpRNA-誘導PTGSを使用して、単子葉植物の胚乳における主要な貯蔵タンパク質の発現を抑制すること、そしてそれによりavoid using当該技術分野においてより従来型のいわゆるアンチセンス技術、または同じ目的のためのいわゆる同時抑制、を使用することを回避することである。
【0013】
目的の異種タンパク質をコードするトランスジーンを駆動する特定のプロモータの転写制御因子の発現レベルに影響を与えることなく、主要な貯蔵タンパク質遺伝子(例えばオオムギにおけるホルデイン)の転写制御因子のhpRNA-誘導PTGSが達成された場合、翻訳のための限られた資源についての競合は、目的の異種タンパク質をコードするmRNAの利益となるように減少され、特定の目的の異種タンパク質の蓄積の増加を引き起こす。
【0014】
本発明の目的は、主要な貯蔵タンパク質遺伝子の転写制御因子の発現を抑制し、そしてその結果、植物中で産生される異種タンパク質をコードする目的のトランスジーンの発現を駆動する特定のプロモータの活性に影響を与えることなく、前記貯蔵タンパク質遺伝子の発現レベルを減少させる方法を提供することである。
【0015】
本発明の第一の側面は、植物種子中の目的の異種ポリペプチドの発現および蓄積を亢進する方法を提供し、ここで前記方法は:
(a)種子貯蔵タンパク質をコードする1またはそれ以上の内在性遺伝子の転写を制御する、異なる(1または複数の)転写制御因子(TF)のキメラ結合物を含む、1またはそれ以上のTFまたはその(1または複数の)部分をコードするDNA配列に対して機能可能に連結された種子-特異的プロモータについてのDNA配列を用いて植物細胞を形質転換し、ここで前記TF DNA配列の転写された鎖が前記植物細胞における1またはそれ以上の前記種子貯蔵タンパク質の発現を抑制し、遅延させ、またはそうでなければ減少させることができ;そして
(b)(a)における前記転写制御因子により認識されるcis-作用性要素を有さない種子-特異的プロモータを選択し;そして
(c)目的の異種ポリペプチドをコードするDNA配列と機能可能に連結した(b)に記載のプロモータについてのDNA配列を用いて、(a)に記載したものと同一または別の植物細胞を形質転換し;
(d)前記(1または複数の)形質転換植物宿主細胞由来の植物を再生させ、そして1またはそれ以上のTFまたはその部分をコードする前記(1または複数の)DNA配列が転写される条件下で前記植物を生長させ、それにより前記内在性mRNAの発現を減少させ、そして前記種子貯蔵タンパク質の発現を減少させ、そして目的の前記異種ポリペプチドの発現および蓄積を亢進させる;
ことを含む。
【0016】
有用な態様において、工程(a)の(1または複数の)DNA配列および工程(c)のDNA配列を、同一の植物細胞中に導入する。そのような態様において、配列は、1つのDNA配列に機能可能に連結していてもよい。
【0017】
しかしながら、その他の興味深い態様において、工程(a)の(1または複数の)DNA配列を第一の植物宿主細胞のゲノム中に導入し、そして工程(c)の前記DNA配列を第二の植物宿主細胞のゲノム中に導入する。その後、第一のトランスジェニック植物を前記第一の植物宿主細胞から再生し、そして第二のトランスジェニック植物を前記第二の植物宿主細胞から作製し、そしてトランスジェニック植物の後代個体群は、前記第一のトランスジェニック植物と第二のトランスジェニック植物との有性交配から作製され、後代個体群植物は、前記1またはそれ以上のTFをコードする(1または複数の)DNA配列および目的の異種タンパク質をコードするDNA配列の両方を含む細胞を有し、植物が前記異種タンパク質を発現しそして蓄積することができるようにする。
【0018】
好ましい態様においては、抑制された種子貯蔵タンパク質が、オオムギのホルデイン、例えば、B-ホルデインおよびC-ホルデインの一方または両方である。
目的の異種ポリペプチドをコードするDNA配列は、本発明による植物細胞中に蓄積することができる、原核細胞のタンパク質または真核細胞のタンパク質をコードするDNA配列であってもよい。本発明の産生のために選択することができるタンパク質の例は、コラーゲン、コラゲナーゼ、ホメオボックスポリペプチド、モノクローナル抗体、分泌抗体、一本鎖抗体、マンノース結合レクチン、ペプシン、キモトリプシン、トリプシン、カゼイン、ヒト成長ホルモン、ヒト血清アルブミン、ヒトインスリン、ラクトフェリン、リゾチーム、セルラーゼ、ペクチナーゼ、ヘミセルラーゼ、フィターゼ、ヒドロラーゼ、ペルオキシダーゼ、フィブリノーゲン、第IX因子、第XIII因子、トロンビン,タンパク質C、キシラナーゼ、イソアミラーゼ、グルコアミラーゼ、アミラーゼ、リゾチーム、β-グルカナーゼ、グルコセレブロシダーゼ、カゼイン、ラクターゼ、ウレアーゼ、グルコースイソメラーゼ、インベルターゼ、ストレプトアビジン、エステラーゼ、アルカリホスファターゼ、プロテアーゼ阻害剤、プロテアーゼ、ペプシン、キモトリプシン、トリプシン、パパイン、キナーゼ,ホスファターゼ、デオキシリボヌクレアーゼ、リボヌクレアーゼ、ホスホリパーゼ、リパーゼ、ラッカーゼ、クモの糸タンパク質、凍結防止タンパク質、抗菌性ペプチドまたはデフェンシン、成長因子、およびサイトカインである。
【0019】
いくつかの態様において、前記DNA配列は、例えば、炭水化物結合モジュール(CBM)などの、好熱性生物由来の所望のタンパク質をコードする。適切なCBMの例は、Thermotoga maritima由来のCBM9-2である。CBM9-2ゲノムDNA配列は、GenBank Accession No. Z46264として利用可能であり、そしてそれはCBMのファミリーIXに属する。同様に、前記CBMまたはその他の適切なCBMを含む融合タンパク質は、DNA配列によりコードされ、そして本発明に従って、種子中で過剰発現させることができる。そのようなCBMおよびCBM融合タンパク質を精製するための方法は、本件出願と同時に出願した出願人の同時係属中の国際特許出願“A non-denaturing process to purify recombinant protein from plants”および“A process for proteolytic cleavage and purification of recombinant proteins”中にさらに詳細に記載されており、その内容全体を参考文献として本明細書中に援用する。
【0020】
特定の態様において、前記DNA配列は、ホメオボックスB4(HoxB4)タンパク質をコードするヒトHoxB4遺伝子を含む。前記ヒトHoxB4タンパク質は、好ましくは、SEQ ID NO: 1に示す配列を有するか、またはSEQ ID NO: 1と実質的な配列同一性を有し、その結果、発現タンパク質が好ましくは未処置HoxB4タンパク質のすべての機能的特徴を有する。実質的な配列同一性は、本明細書中の文脈において、少なくとも50%以上の配列同一性、より好ましくは少なくとも60%の配列同一性、少なくとも70%の配列同一性、例えば少なくとも80%の配列同一性、そして好ましくは少なくとも90%の配列同一性、例えば少なくとも95%の配列同一性または99%の配列同一性、を示す。
【0021】
本明細書中で示されるように、1またはそれ以上のTFまたはその部分をコードする前記DNA配列は、好ましくは、1またはそれ以上の前記種子貯蔵タンパク質の発現を、抑制し、遅延させ、またはそうでなければ減少させることができる“ヘアピン”RNAを形成する能力を有する。前記DNA配列は、完全なTF配列を含んでもよいが、しかしながら、ある場合には、TF配列の部分のみが適切な位置に存在しさえすれば抑制が機能する可能性もある。このように、これらの態様において、1またはそれ以上のTF配列の十分に長い部分が、抑制が作用するために必要とされる。ある場合には、長さ20ヌクレオチド程度のTF配列の部分で十分であり、しかしながら好ましくは、約20〜200、例えば長さ20〜100の範囲または長さ50〜100ヌクレオチドの範囲、を含む、少なくとも20〜500ヌクレオチドの範囲の部分が使用される。
【0022】
特定の有用な態様において、1またはそれ以上のTFをコードするDNA配列は、本明細書中に定義する様に、キメラDNA配列であり、TFまたはその部分をコードする2またはそれ以上のDNA配列の領域を含む。別の有用な態様において、キメラDNA配列において1またはそれ以上のTFをコードするDNA配列は、“ヘアピン”RNA中にループを形成することができるイントロン配列を含むこともできる。
【0023】
好ましい態様は、TFまたはその部分をコードするDNA配列を利用し、これはbZIPタンパク質の群、より好ましくは、オオムギBLZ1タンパク質およびBLZ2タンパク質(Onate et al 1999を参照)から選択されたbZIPタンパク質、由来のTFまたはその部分をコードする領域を含む。前記DNA配列は、抑制に影響を与えるために十分な長さのそのようなタンパク質をコードする配列の部分、または前記タンパク質をコードする配列の部分の組み合わせを含んでもよい。
【0024】
前記DNA配列は好ましくは、SEQ ID NO: 2、SEQ ID NO: 3またはSEQ ID NO: 4に記載された配列、または前記配列によりコードされたいずれかのアミノ酸配列と実質的に同一な配列を有するタンパク質をコードする配列、または本発明の方法による抑制を引き起こすために十分な長さを有するSEQ ID NO: 2、またはSEQ ID NO: 4の部分を有する配列を含む。上述したように、前記配列のいずれかの組み合わせ、例えば上述の配列の1またはそれ以上の部分の組み合わせ、が有用である可能性があり、そのような組み合わせの例は、BLZ1およびBLZ2の部分のキメラであるSEQ ID NO: 5として示されるものがある。
【0025】
本発明の別の目的は、hpRNA-誘導PTGS技術を使用して、主要な貯蔵タンパク質遺伝子の転写制御因子の発現を抑制する方法を提供することである。そのため、特定の態様において、本発明の方法は、hpRNA-誘導PTGS技術を、内在性貯蔵タンパク質の発現を抑制するために使用し、そして従って、目的の異種ポリペプチドをコードするmRNAの翻訳のための、資源(例えば、アミノ酸、リボゾーム結合部位および翻訳修飾および後翻訳修飾に関与する酵素の集合物)の利用可能性を増大する。このように、好ましい態様において、1またはそれ以上のTFまたはその部分、またはそれらの組み合わせ、好ましくは上述したbZIPタンパク質の群に由来するもの、をコードする前記DNA配列は、前記植物細胞における“ヘアピン”RNA(hpRNA)を発現することができる。
【0026】
前記DNA配列は好ましくは、SEQ ID NO: 2、SEQ ID NO: 4、SEQ ID NO: 6、またはSEQ ID NO: 7、SEQ ID NO: 8、およびいずれかの部分またはそれらの組み合わせ(本明細書中に記載した方法による抑制、すなわち、特に好ましくはオオムギ中の胚乳などの種子および/または胚乳におけるもの、を引き起こすために十分な長さのもの)から選択される配列を含む。
【0027】
従って、本発明の目的は、オオムギ胚乳におけるB-ホルデインおよびC-ホルデインレベルを減少させるための、分子生物学における方法およびツールを提供することである。より具体的には、本発明の目的は、hpRNAを形成することができるBLZ1遺伝子およびBLZ2遺伝子の領域を含み、そして従って前記胚乳遺伝子の発現を減少させることができる遺伝子構築物を提供することである。
【0028】
より具体的には、本発明は、オオムギ胚乳においてPTGSを可能にするhpRNAを形成する転写物をコードする、BLZ1遺伝子およびBLZ2遺伝子の領域の遺伝子構築物を提供する。
上述した好ましい態様において、胚乳貯蔵タンパク質レベルが減少したトランスジェニック植物を作製する方法は:(1)結実能力のある植物への再生が可能な単子葉植物細胞、特にオオムギを提供すること;(2)5'から3'方向に読む発現カセット、種子-特異的プロモータ、特に胚乳-特異的プロモータ、内在性種子貯蔵タンパク質を制御する転写制御因子をコードする核酸配列、抑制される(1または複数の)貯蔵タンパク質遺伝子を制御する転写制御因子により認識されるcis-作用性要素を有さない別の種子-特異的プロモータ、好ましくは胚乳-特異的プロモータ、目的の異種ポリペプチドをコードする核酸配列、そして3'非翻訳領域を含む核酸構築物により、植物細胞を形質転換すること;(3)前記植物細胞を再生させて種子貯蔵タンパク質の発現を制御する転写制御因子のレベルを減少させ、そして内在性貯蔵タンパク質レベルを減少させた第一のトランスジェニック植物を提供させること;を含む。
【0029】
単子葉植物種子において目的の異種ポリペプチドを高レベルに発現するためのDNA構築物もまた、本発明により包含される。そのようなDNA構築物には:所定の単子葉植物種子において機能しそして目的の異種ポリペプチドをコードする配列に対して機能可能に連結するプロモータ;そして所定の単子葉植物種子中で機能するプロモータに対して機能可能に連結した、オオムギ胚乳中でPTGSを可能にするhpRNAを形成することができる転写物をコードするBLZ1遺伝子とBLZ2遺伝子の領域のキメラ遺伝子構築物;が含まれる。
【0030】
本発明の別の側面において、本明細書中に記載される方法により得ることができる、特に上述したようなものなどのトランスジェニック植物が提供される。前記植物は、上述した所望の特徴を有する、すなわち、組換えにより産生される所望のタンパク質の発現および蓄積を亢進するため、選択された主要な資源“sink”タンパク質の発現を抑制する。本発明の好ましい植物には、例えば、種子中での異種タンパク質の発現および蓄積のために本発明に従って使用することができる、変種Hordeum vulgarisのオオムギ植物が含まれる。本発明のオオムギ植物は、好ましくは、そのゲノム中に、本明細書中で記載される好ましいTF、例えばbZIPタンパク質の群由来のTFまたはその部分をコードする配列を有する。
【0031】
発明の詳細な説明
本明細書の以下において、本発明を詳細に記載する。
用語“タンパク質”は、本明細書中において、“ポリペプチド”および“ペプチド”と、互換的に使用される。
【0032】
本明細書中において使用される用語“異種”は、用語“非天然”または“外来性”または“外因性”と互換的に使用することができ、そして通常は組換えDNA技術が関与する遺伝子操作に供されていない宿主生物においては見いだされない、タンパク質および/またはin-vitro修飾DNA配列に対して利用される。本明細書中において使用される用語“目的の異種ポリペプチド”または“目的のポリペプチド”は、本発明の方法または組成物を使用して宿主生物中で発現させるために意図されたいずれかのポリペプチドのことをいう。限定的ではない例として、薬理学的ポリペプチド(例えば、医薬用途のための)または産業的ポリペプチド(例えば、酵素)を、本発明に従って作製することができる。
【0033】
用語“コード配列”は、特定のアミノ酸配列をコードするヌクレオチド配列のことをいう。
“プロモータ”は、機能可能に連結された核酸の転写を指向する核酸調節配列または転写制御因子結合部位の一連のものとして定義される。プロモータは、コード配列または機能性RNAの発現を調節する核酸配列のことをいう。用語“機能可能に連結”は、プロモータ(核酸発現調節配列、または一連の転写因子結合部位)と第二の核酸配列とのあいだの機能的な連結のことをいい、ここでプロモータは、第二の配列に対応する核酸の転写を指向する。
【0034】
“転写制御因子”または“転写因子”は、遺伝子の上流、イントロンの内部、または停止コドンの下流に位置する短いDNA配列であるcis-作用性要素(またはcis-作用性モチーフ)に結合することができる、trans-作用性制御タンパク質のことをいう。
【0035】
用語“内在性遺伝子”は、生物のゲノム中の天然の位置における天然の遺伝子のことを言う。
用語“キメラ組み合わせ”または“キメラ遺伝子”または“ハイブリッド遺伝子”は、内在性遺伝子ではない遺伝子を形成し、天然には通常は同時に見いだされない制御配列および/またはコード配列を含む、いずれかの2またはそれ以上の連結したDNA配列のことをいう。用語“遺伝子構築物”または“DNA構築物”または“核酸構築物”は、いずれかのDNA配列または一般的な分子生物学的ストラテジーにより組み立てられるDNA配列の組み合わせのことをいう。
【0036】
本明細書中で使用する場合、用語“発現”は、結果としてDNA配列のRNAポリメラーゼ-触媒転写を引き起こす一本鎖または二本鎖形状のいずれかにおける、センス(mRNA)、アンチセンスRNAまたはそれらのその他のRNAポリマーの転写を含む、遺伝子生成物の生合成のことをいう。発現は、ポリペプチド中の前記遺伝子由来のmRNAの翻訳のことをいう場合もある。
【0037】
用語“形質転換”は、結果として遺伝子的に安定な遺伝形質をもたらす、宿主生物のゲノム中の核酸分子の転移のことをいう。形質転換された核酸断片を含有する宿主生物は、“トランスジェニック”生物と呼ばれる。用語“トランスジェニック”は、本発明の植物宿主細胞が、ゲノム中に安定に組み込まれた少なくとも1つの外来性核酸分子、好ましくは2つの外来性核酸分子を含有することを意味する。植物形質転換方法の例には、アグロバクテリウム-媒介性形質転換(De Blaere et al. 1987)および粒子衝撃形質転換技術、または“遺伝子銃”形質転換技術(Klein et al. (1987); U. S. Pat. No. 4,945, 050)が含まれる。
【0038】
用語“アンチセンス阻害”または“アンチセンス”または“アンチセンス抑制”は、内在性転写生成物またはmRNAに対して十分に相補的なアンチセンス鎖のことをいい、それにより内在性転写生成物の翻訳および発現を阻害しまたは減少させることをいう。用語“標的化抑制”は、組換えDNA技術を使用して、いくつかの所定の生体における抑制のために選択された内在性遺伝子の発現を阻害しそして抑制することができるRNA転写物をコードすることができるDNA配列を設計しそして利用することをいう。用語“抑制性トランスジーン”は、内在性遺伝子の発現を阻害しそして抑制することができるRNA転写物をコードするいずれかの異種DNA配列のことをいう。用語“同時抑制”は、形質転換により細胞中に導入され、そしてその同一の細胞中で内在性遺伝子として同一の又は同様の配列を有する遺伝子が、内在性遺伝子と導入遺伝子の両方を抑制する場合のことをいう。
【0039】
本明細書中で使用される“bZIP”タンパク質は、DNA結合および二量体化に関与し、そして5'ACGT'3コアを含有するDNA配列に共通して結合するいわゆる塩基性ヘリックス/ロイシンジッパードメインを含有する制御タンパク質のことをいう。
【0040】
本明細書中で使用される用語“ホルデイン”は、オオムギにおいて、具体的には胚乳中で合成される貯蔵タンパク質のことをいい、そして4つの群:β-ホルデイン(ベータホルデイン、B-ホルデインとも呼ぶ)、D-ホルデイン、C-ホルデインおよびγ-ホルデインに分類される。
【0041】
本明細書中で使用される用語“分子的農業”は、植物を使用してさらにプロセッシングするためのタンパク質などの貴重な生物学的化合物を作製するプロセスのことをいう。
遺伝子操作していてもよい単子葉植物を、本発明において使用することができる。好ましくは、植物は単子葉植物であり、より好ましくは、オオムギ、トウモロコシ、コムギ、オートムギ、およびコメから選択される。オオムギは、オオムギを本発明の好ましい候補物とする多数の所望される特徴を有し、オオムギの種Hordeum vulgarisisが特に好ましい。遺伝子的に形質転換されていてもよい植物は、コード領域用のDNA配列またはhpRNAを形成することができるRNA転写物をコードするDNA配列を含む異種DNA配列を、導入し、発現させ、安定的に維持させ、そして子孫の以降の世代に遺伝させることができる植物である。例えば、ビアラホスまたはバスタなどを含む除草剤、または選択可能マーカーとしてのハイグロマイシンなどの殺虫剤を使用する遺伝子操作および形質転換方法を使用して、オオムギ植物を作製した。
【0042】
適切な宿主植物を選択した後、プロモータが、目的の異種遺伝子の発現を駆動するために選択される。植物細胞において活性な多数のプロモータが、この文献中に記載された。本発明のDNA構築物中で利用されるプロモータは、目的の異種ポリペプチドの蓄積が所望される組織中、例えば単子葉植物の種子の胚乳中で、強力な活性を有することが好ましい。選択されるプロモータは、本発明の方法により抑制されるように標的となる転写制御因子、例えば目的の異種遺伝子の発現と資源について競合する内在性貯蔵タンパク質遺伝子の発現を制御する転写制御因子、により制御されないことがさらに好ましい。そのようなプロモータは、様々な植物遺伝子材料からまたは植物ウィルスから得ることができる。以下に記載したように、選択された特定のプロモータは、単子葉植物種子における、より好ましくはオオムギにおける、そして最も好ましくは種子の胚乳組織における、異種タンパク質の発現に適していることが好ましい。本発明の目的のために有用なそのような適したプロモータのクローニングおよび解析は、実施例2に記載される。
【0043】
目的の異種タンパク質の蓄積に関して選択された標的組織中のタンパク質プロファイル発現パターンが解析され、そして最も豊富な貯蔵タンパク質が本発明による標的化抑制のために同定されることがさらに好ましい。解析は、刊行文献から採取したデータなどの従来技術に依存することができる。このことはまた、マイクロアレイアプローチを使用して、異種タンパク質の発現を駆動するために選択されたプロモータの同時的発現プロファイルと重複する時点で前記組織から抽出したmRNAを使用して、またはノザンブロット解析、RT-PCRおよび/またはウェスタンブロッティングを使用して、数千の遺伝子の発現パターンを同時にモニターすることを含んでいてもよい。集められた情報に基づいて、時間的そして空間的に目的の異種遺伝子の発現と競合する、高い発現を伴う候補遺伝子を、本発明による標的化抑制のために選択する。
【0044】
本発明の組換えプラスミドを、目的のDNA配列を、細菌宿主中で複製可能な適切なプラスミド中にライゲーション(挿入)することにより得ることができる。目的の異種タンパク質をコードする遺伝子などのDNA配列、または内在性遺伝子の標的化抑制のために設計されたDNA配列を、プロモータに加えて、この目的のため、所望される場合には、付加的エンハンサーDNA配列、足場付着領域(scaffold-attachment regions)、イントロン、ポリ(A)付加シグナル、目的のリボソーム結合配列や選択マーカー遺伝子(例えば、ハイグロマイシン耐性遺伝子、アンピシリン耐性遺伝子、ビアラホス耐性遺伝子など)を含有していてもよいプラスミド中に、機能するように組み込むことが好ましい。
【実施例】
【0045】
本発明をよりよく規定するため、そして本発明を実施する際に当業者をガイドするため、以下の実施例を提供する。それ以外の記載をしていない限り、用語は、当業者により従来から使用されている様に理解されるべきものである。
【0046】
方法
RNA抽出およびノザンブロット解析
全RNAを、製造者の指示にしたがってTRIzoI(Gibco BRL)を使用して単離し、そしてレーンあたり10μgの全RNAを電気泳動に供し、そして毛細管作用によりHybond Nメンブレン(Amersham)に転写した。Stratolinker(Stratagene)を使用してRNAをメンブレンに架橋し、そしてフィルターを高ストリンジェントのハイブリダイゼーションによりハイブリダイズさせ、そして記載されたように洗浄する(Davis et al., 1994)。プローブは、製造者の指示にしたがってランダムプライミング(T7 QuickPrime KitTM;Pharmacia)により32Pで放射性標識する。EtBr-染色したブロットのネガデジタル画像を、ロード対照として使用する。
【0047】
オオムギ形質転換
a)遺伝子形質転換用の植物材料
Hordeum vulgarecv Golden Promieの種子を、75%軽ミズゴケピート(light sphagnum peat)と25%軽石(中粒子サイズ)の混合物上に蒔き、そして植物を、日中18℃(16時間)および夜間12℃(8時間)で、そして70%相対湿度、250μモル/m2 sの日中冷白色蛍光での連続光のもと、必要に応じて灌水して、生育させた。これらの条件下で、植物は、約55〜95日間、または未成熟の種子が形質転換用の材料として準備されるまで、すなわち開花後約8〜14日後まで、栄養生長させた。種子は、3%次亜塩素酸ナトリウム中で40分間、回転震盪器で殺菌消毒し、そして無菌水を5回変えることによりすすいだ。
【0048】
b)植物形質転換用の細菌系統および調製物
vir領域を有するTi-プラスミドをトランスで保持するバイナリベクターを保有するAgrobacterium tumefaciensを使用して、目的の異種タンパク質の発現を制御するDNA構築物を有するT-DNA領域をオオムギ中に導入する。
【0049】
E. coli XL-BlueおよびAgrobacterium tumefaciens細菌の両方の形質転換は、Maniatisら(Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N. Y. (1982))により記載されるエレクトロポレーションにより行う。植物の形質転換用の調製物のため、Agrobacterium培養物のシングルコロニーを、5 mlのAgro培地(Tryptone 5 g/L、酵母エキス2.5 g/L、マンニトール5 g/L、グルタミン酸1 g/L、KH2PO4 250 mg/L、MgSO5-7H20 100 mg/L、ビオチン1μg/L、pH 7.0、25μg/mlリファンピシン、50μg/mlスペクチオマイシン)中に接種し、そして27℃にて24〜40時間増殖させる。無菌エッペンドルフチューブ中で、200μlの培養物を200μlの30%グリセロール水溶液(事前に滅菌したもの)に対して添加し、そして培養物をよくボルテックスにかけ、そしてベンチに2時間静置した後、-80℃で保存する。各形質転換につき、1本のチューブを-80℃冷凍庫から取り出し、解凍し、そして約200μlのAgrobacterium細菌ストックを抗生物質を含まない5 mlのAgro培地に対して添加する。その後、培養物を27℃にて17〜20時間生育させた後、植物材料の接種のために使用する(以下を参照)。
【0050】
c)Agrobacterium-誘導形質転換のためのオオムギ外植体の調製
第1日に、約10個のオオムギ頭頂部を摘み、開花後約8〜14日後に、芒(awn)および種子を取り出し、1.5 mm〜2 mmのサイズの胚を選択した。最初の植物材料は、健康で成熟していることが必要であるが、水に浸されていることは必要とされず、そして種子は緑色で病気やカビの兆候がないものであるべきである。種子を50 mlのファルコンチューブ(高々半量)に静置し、そして70%エタノールですすぎ、そしてその後エタノールを捨てた。次いで、20%漂白溶液(White King)を添加し、20分間混合した。
【0051】
ラミナエアフロー中で、漂白溶液を捨て、種子を滅菌水ですすぎ(約5〜8回すすぐ)、そしてチューブを4℃にて一晩静置した。第2日に、種子を顕微鏡プラットフォーム上の滅菌ペトリ皿中に静置した。胚の位置を特定し、種子の末端部を切断し、そして両側を切断した種子を作製した。その後、種子をピンセットを用いてしっかりと固定し、そして胚が飛び出すように種子の中央部に圧をかけた。
【0052】
胚をピンセットを用いて固定し、そしてメス刃を胚盤と胚軸とのあいだの溝に挿入し、そして胚軸をゆっくりと切断した。胚軸を取り除いた胚を、再生培地上に切断面を上にして静置し、ペトリ皿の中央に、約25個の胚を静置させた。
【0053】
d)すべてのバイナリベクターは、37℃にて100μg/mlのスペクチオマイシンを含有するE. coli XI-Blue LB培養培地中で増殖させ、そして引き続いてベクターを、QIAGEN(商標)Plasmid Midi Kitを用いて一晩増殖させた100 ml培養物から精製した。精製したバイナリベクターを、1μl(1μg)のベクターを0.1 cmのギャップを有する滅菌キュベット(BioRad)中に静置し、ベクターを40μlのエレクトロコンピテント細胞とともに洗浄し、そしてエレクトロポレーション用に2.5 kVの電圧、および21μFの電気容量に設定することにより、Agrobacterium tumefaciens中にエレクトロポレーションを用いて導入した。細胞を100μg/mlのスペクチノマイシンおよび20μg/mlのリファンピシンを含有するYEP選択プレート上に広げ、28 ℃にて2日間増殖させた。A. tumefaciens形質転換体由来のプラスミド制限酵素消化物を解析を行い、バイナリベクターの完全性を確認した。
【0054】
e)オオムギ外植体のAgrobacterium感染
約20μlのAgrobacterium培養物を各胚上にピペットでのせ、すべての胚が溶液と確実に接触できるようにする。胚を裏返しにし(切断面を下向き)、そして再生培地上をプレートの外まで引っ張っていき、過剰なAgrobacteriumを除去した。その後、胚を均等な間隔で新鮮な再生培地プレートに移し(切断面を上)(プレートあたり25個)、そして24℃にて暗キャビネット中に静置した。
【0055】
f)Agrobacterium 感染後のオオムギ組織培養における再生および器官形成
3日後、胚を選択マーカー(たとえばビアラフォスまたはハイグロマイシン)を含む新鮮な再生培地に移し、そして4〜6週間そこにそのままにし、2週間ごとにサブカルチャーした。苗条を再生するため、カルスを苗条誘導培地(SIM)に移し、そして生残したカルスおよび再生し始めた苗条を、小さな苗が形成されるまで、2週間ごとに新鮮なSIMへ移した。その後、苗を根誘導培地(RIM)に移し、そして生残した植物をさらにスクリーニングするために土に鉢植えした。
【0056】
DNA抽出、PCRおよびクローニング
PCR増幅のためのゲノムDNAを、NucleoSpinPlant Kit(商標)(Clonetech)を製造者の指示にしたがって用いて約200 mgの若葉組織から抽出した。PCR反応を加熱蓋を有するペルティエ型サーマルサイクラー(MJ Research, PTC-200)中で行った。すべてのプライマーは、完全自動化DNA合成装置(Perkin-Elmer)を用いて化学的に合成した。すべてのDNA断片は、UV光のもと、1%EtBr-染色アガロースゲル上でサイズ画分し、そしてQIAquick(商標)Gel Extration Kit(Qiagen)を用いて精製した。制限酵素は、New England Biolabs Inc(Beverly, Mass.)およびFermentasから購入し、そして製造者の指示にしたがって使用した。クローニングおよびその他のDNA操作は、Maniatisら(Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N. Y. (1982))に記載されるような標準的な手順にしたがって行ったが、すべてのライゲーション反応では、以下の条件を使用した:バッファー〔50 mM Tris-HCl(pH 7.6)、5 mM MgCl2、1 mM DTT、0.5 mM ATP、2.5%ポリエチレングリコール-8000〕中T4 DNAリガーゼ(2.5 U)を16℃にて16時間。
【0057】
コンピテントEscherichia coli XL-Blueを、DNA操作のための形質転換実験において、そしてプラスミドクローンの構築のため、レシピエントとして使用した。すべてのプラスミドDNA midi prepは、Qiagen(商標)Plasmid Midi Kit(Qiagen)を使用して調製し、そしてDNA断片の配列決定は、自動化DNA配列決定装置(ABI, Pharmacia)を使用して行った。
【0058】
実施例1 Hordeum vulgarisの胚乳中の“sink”タンパク質の解析
Hordeum vulgaris種子変種Skeglaにおけるタンパク質発現解析を、開花49-日後(dpa)に回収した種子から抽出したタンパク質のSDS-PAGEを使用して行い、胚乳の全タンパク質レベルに対して主要な寄与を行う胚乳発現タンパク質を同定し、そしてしたがって、標的化抑制のための可能性のある候補物を同定することができる。5つのオオムギ胚軸の種子全体を液体窒素中で手動で細かく磨砕し、その後適切な抽出バッファーを添加した。2種類の抽出バッファーを、水性抽出に加えて、量産プルに使用した。最初に、1%2-メルカプトエタノール、10 mM Tris-HCl pH 8.0および1%ポリビニルピロリジン(MW 360.000)の存在下にて、エタノール抽出(70%EtOH)を伴う還元条件下で全タンパク質の抽出を行った。次に、170 mM NaCl、1%2-メルカプトエタノール、10 mM Tris-HCl pH 8.0および1%ポリビニルピロリジン(MW 360.000)の存在下にて、還元条件下で、全タンパク質の抽出を行った。
【0059】
液体窒素中でサンプルを磨砕した後、5 mlの抽出バッファーを抽出バイアルに添加し、その後3分間連続的に磨砕した。抽出物を4℃、4000 rpmにて10分間、遠心分離にかけて清澄化させ、その後分子量5 kDaのカットオフを有するUltrafree-4濃縮装置(UFV4BCC00- Millipore Corp. Bedford, MA, USA)中で、10倍遠心分離濃縮を行った。清澄化させ濃縮した抽出物の100μlのサンプルを、100μlの2×サンプルバッファーに添加し、混合物を加熱水浴中で5分間静置した。冷却後、10μlのサンプルを12%ポリアクリルアミドゲルにかけ、SDS-PAGEにより分離した。SDS-PAGEの後、ゲルをクマシーブルーR250染色により染色し、脱染色してタンパク質バンドを可視化した。結果から、H. vulgare変種Skeglaにおける発生途中の胚乳において最も豊富なタンパク質は、B-ホルデインおよびC-ホルデインであることが示された(図1)。
【0060】
この結果から、BホルデインおよびCホルデインをコードする遺伝子を制御する転写制御因子を抑制することが、資源に関する競合を非常に減少させやすく、そして本発明にしたがって発現された異種ポリペプチドの蓄積を増加させることが示される。
【0061】
実施例2 異種タンパク質のための発現ビヒクルとして使用される、Hordeum vulgareのB-ホルデインおよびC-ホルデイン標的化抑制のためのプロモータの選択
B-ホルデイン(GenBank Accession No. X53690)およびC-ホルデイン(GenBank Accession No. M36941)の遺伝子プロモータは、それら両方において同定された100%同一なcis-作用性要素を含有し、それらはそれらの転写制御に関与する(Muller and Knudsen 1993;Vicente-Carbajosaet al. 1992)。これらのcis-要素は、翻訳開始コドンの約300 bp上流に位置するいわゆる胚乳ボックス(EM)中に存在する。EMは、転写因子結合のための2種の特有のcis-作用性モチーフ、プロラミンボックス(PB)5'-TGTAAAG-3'、そしてGCN4-様モチーフ(GLM)5'-(G/A)TGA(G/C)TCAT-3'を保有する。
【0062】
GLMは、2種の転写因子、BLZ1(GenBank Accession No. X80068)およびBLZ2(GenBank Accession No. X80068)により認識される。結合に際して、特定の遺伝子の転写が活性化される(Onate et al. 1999を参照)。これらの遺伝子は両方とも、1コピーの遺伝子であり、そしてしたがって、本発明により記載される標的化抑制のための良好な候補である。Vicente-Carbajosaら(1998)は、安定的に形質転換した植物に対する作用、または貯蔵タンパク質の蓄積に対する作用を調べることなく、一過性発現系におけるそれらのアンチセンス抑制を試験した。さらに、同一組織における異種タンパク質の蓄積に対するそのような抑制の作用は調べられなかった。本発明による標的化抑制を操作する際の重要な基準は、目的の異種タンパク質をコードするトランスジーンを駆動するプロモータが、貯蔵タンパク質の発現を抑制する原因となる特定の転写制御因子により認識されるcis-作用性モチーフを含有してはいけない、ということである。B-ホルデインおよびC-ホルデインがBLZ1およびBLZ2により認識されるGLMを含有するため、B-ホルデインプロモータおよびC-ホルデインプロモータを使用して、所望の異種タンパク質をコードする異種トランスジーンの発現を駆動することができない。別の重要な基準は、抑制トランスジーンそれ自体を駆動するプロモータが、組織特異的プロモータ、例えば胚乳特異的プロモータ、により駆動され、それによりそれらのプロモータが必要とされるその他の組織における転写制御因子の発現には影響が及ばない、ということである。このことは、多数の転写因子(例えばオオムギにおけるBLZ1など)が1より多い組織中で発現されていることから、重要である。さらに別の基準は、本発明により抑制されるべき転写制御因子により認識されるcis-作用性要素を有さない抑制トランスジーンを駆動するためのプロモータを使用することにより、抑制トランスジーンの“自己抑制(self-suppression)”を回避することである。
【0063】
これらの基準に基づいて、B-ホルデインおよびC-ホルデインの標的化抑制について候補プロモータを選択した。
Hordeum vulgare cv Skegla由来のD-ホルデインプロモータの単離、クローニングおよび解析
プライマーを、-435〜-16由来の隣接するプロモータ領域を同定するために、GenBank配列id. X84368に記述されたD-ホルデイン遺伝子の翻訳開始部位に基づいて、センスプライマーとして5'末端にEcoRI制限酵素部位を導入された5' GGAATTCCEcoRICTTCGAGTGCCCGCCGATTTGCCAGCAATGG(SEQ ID NO: 9)を設計し、そしてアンチセンスプライマーとして5'末端にNotI制限酵素部位を導入された5'ATAAGAATGCGGCCGCNotIAATGAATTGATCTCTAGTTTTGTGG(SEQ ID NO: 10)を設計し、そして合成した。プライマーの5'末端にこれらの制限酵素部位を導入した目的は、増幅断片のクローニングを補助するためであった。Hordeum vulgare cv. Skegla由来のゲノムDNAを鋳型として使用して420 bpの断片を増幅するために使用する反応溶液の組成は、以下の通りであった:
【0064】
【表1】
【0065】
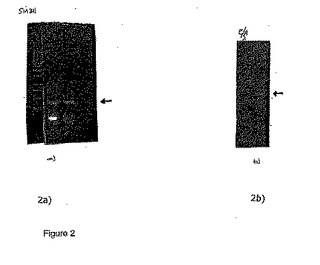
9μlの滅菌水およびTaq DNAポリメラーゼ以外の上述の反応溶液をよく混合し、そして溶液を94℃にて3分間加熱し、その後、80℃まで冷却して30秒間保持し、そしてTaq DNAポリメラーゼおよび9μlの滅菌水を添加した。PCRの最初のサイクルを、以下の様にして行った:反応物を、94℃にて30秒間加熱し、アニーリングのために57℃まで冷却して30秒間保持し、そして次いで伸長反応のため72℃で45秒間加熱した。その後の30サイクルは、以下の様にして行った:94℃にて30秒間の加熱変性、62℃にて30秒間のアニーリング、そして72℃にて45秒間の伸長。30サイクルが終了した後、反応物を72℃にて4分間加熱した。420 bpの増幅断片(図2a)を、EcoRI/NotIで消化し、次いでベクターpKOH122のEcoRI/NotI部位にライゲーションして、組換えプラスミドpDH104を得た。420 bpのDNA挿入物のヌクレオチド配列を、その後決定した(SEQ ID NO: 11)。その配列によれば、D-ホルデインプロモータはGLMを含有せず、そして従って、BLZ1またはBLZ2のいずれかの標的化抑制を介したB-ホルデインおよびC-ホルデインの抑制(以下の検討の項を参照)は、D-ホルデインプロモータの活性に影響を与えず、そして従って、目的の異種タンパク質の発現に影響を与えなかった。さらに、D-ホルデインプロモータは、もっぱら胚乳-特異的なものであるため、このプロモータはBLZ1およびBLZ2の発現を抑制するために設計された抑制トランスジーンを駆動するために有用である。
【0066】
プロモータを使用して本発明により抑制されるべき転写制御因子により認識されるcis-作用性要素を有さない抑制トランスジーンを駆動することにより、抑制トランスジーンの“自己抑制”を回避することが重要であるため、GLMを欠損するD-ホルデインプロモータを使用してBLZ1およびBLZ2を抑制する抑制遺伝子を駆動することは、そのような“自己抑制”を引き起こさないだろう。
【0067】
実施例3 D-ホルデインコード領域のクローニングと、mRNAレベルでのD-ホルデイン遺伝子の内在性発現の解析
センスプライマー、5'GGAATTCCEcoRIATGGCTAAGCGGCTGGTCCTC(SEQ ID NO: 12)およびアンチセンスプライマー、5'GGAATTCCEcoRITTGCAATTGGATAGGTCTCTTG(SEQ ID NO: 13)を設計して、GenBank 配列id. X84368に記載されるD-ホルデイン遺伝子のコード領域由来御727 bpの断片を増幅した。Hordeum vulgare cv. Skegla由来のゲノムDNAを鋳型として使用して、727 bp断片を増幅するために使用される反応溶液の組成は、以下の通りである:
【0068】
【表2】
【0069】
9μlの滅菌水およびTaq DNAポリメラーゼ以外の上述の反応溶液をよく混合し、そして溶液を94℃にて3分間加熱し、その後80℃まで冷却して30秒間保持し、そしてTaq DNAポリメラーゼおよび9μlの滅菌水を添加した。PCRの最初のサイクルを、以下の様にして行った:反応物を、94℃にて30秒間加熱し、アニーリングのために60℃まで冷却して30秒間保持し、そして次いで伸長反応のため72℃で45秒間加熱した。その後の30サイクルは、以下の様にして行った:94℃にて30秒間の加熱変性、65℃にて30秒間のアニーリング、そして72℃にて45秒間の伸長。30サイクルが終了した後、反応物を72℃にて4分間加熱した。727 bpの増幅断片(図2b)を、EcoRIで消化し、次いでベクターpKOH122のEcoRI部位にライゲーションして、組換えプラスミドpDH136を得た。DNA挿入物のヌクレオチド配列を、決定した(SEQ ID NO: 14)。
【0070】
胚乳40 DAP、胚40 DAP、葉、葉軸、および根におけるD-ホルデイン遺伝子のmRNAの定常状態レベルは、ノーザンブロット解析を使用して適切に解析する:Hordeum vulgare cv Skeglaの様々な組織または植物の部分由来の200〜250 mgの植物材料から全RNAを単離し、1.2%アガロース-ホルムアルデヒドゲル上で分離し、そして製造者の指示に従ってZeta-Probeメンブレン(Millipore, Bedford, Mass.)上にブロットした。ブロッティング、ハイブリダイゼーション、および洗浄は、記載されたように行った(Davis et al., 1994)。ハイブリダイゼーションは、D-ホルデイン遺伝子の32P放射標識しランダムプライミングした747bp断片(SEQ ID NO: 14)をプローブとして使用して行う。
【0071】
実施例4 植物形質転換ベクターpbDH101中へのオオムギの胚乳-コドン最適化HoxB4のクローニング
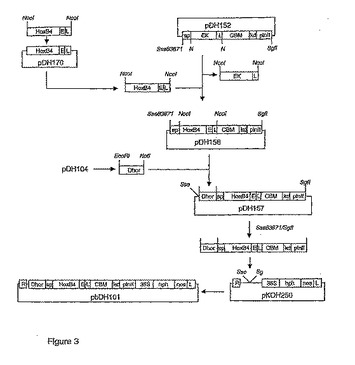
オオムギにおけるD-ホルデインプロモータの制御下での発現に関するコドン最適化HoxB4は、記載されたように(本願出願人の同時係属出願“Methods for high level expression of polypeptides in plants using codon optimization”を参照)、GeneBank ID NM_024015に記載される基本的配列情報およびD-ホルデインプロモータの制御下での胚乳におけるコドン最適化発現に関するコドン利用情報を使用して、GeneArt GmbH(Germany)により合成された。791 bpの断片(SEQ ID NO: 20)は、クローニング用のNcoI部位(CCATGG)に隣接し、そしてリンカー-エンテロキナーゼ切断部位DDDDKPTPTPTをコードする27ヌクレオチド配列を3'末端に含む。HoxB4断片は、pKOH122のNcoI/NcoI部位にライゲーションされ、pDH170が作製される。その後、断片をNcoIにより放出させ、そしてpDH152のNcoI部位にライゲーションさせ、pDH152中のEK-L断片を置換してpDH156を得る。次いで、pDH104由来のD-ホルデインプロモータ断片をpDH156のEcoRI/NotI部位中にライゲーションしてpDH157を得る。D-プロモータ/SP/HoxB4-E-L/CBM/kd/pinII断片をSse83871/SfIにより放出させ、そしてpKOH250のSse83871/SfI部位にライゲーションして植物形質転換ベクターpbDH101を作製する(図3)。
【0072】
実施例5 D-ホルデインプロモータの制御下でのBLZ1およびBLZ2のヘアピンRNA-誘導標的化抑制による、植物形質転換ベクターpbDH104の構築
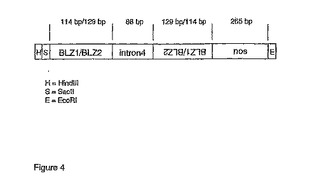
BLZ1およびBLZ2のヘアピンRNA-誘導標的化抑制のための遺伝子が、265 bp nos-末端断片(SEQ ID NO: 8)が伴う592 bpの抑制断片として、GeneArt GmbH(Germany)により合成され、そしてpUC19のHindIII/EcoRI部位にクローニングして、pDH144を得た。断片は、BLZI-遺伝子(SEQ ID NO: 2)およびBLZ2-遺伝子(SEQ ID NO: 4)由来のヌクレオチド配列のキメラ組成であり、これはセンス部分とアンチセンス部分とを形成し、そして88 bpのヌクレオチド配列はBLZ1遺伝子由来のイントロン4を示し(GenBank配列id. X80068)、転写後にRNAヘアピンループを形成する様に設計された(図4)。857 bp断片であるhpB1/2-nosをSacII/EcoRIにより放出させ、そしてpDH158中でアンチセンスBLZ1-nos断片と置換して、pDH160を得る。pDH160中の完全な挿入物を、Sse83871/SgfI消化により放出させ、そしてpKOH250のSse83871/SgfI部位にライゲーションして、ベクターの左側境界(L)と右側境界(R)のあいだのDNA配列を植物ゲノム中に安定的に組み込むことができる、バイナリ植物形質転換ベクターpbDH104を作製した(図5)。
【0073】
実施例6 pbDH101およびpDH104を用いたオオムギの遺伝子形質転換
開花後約8〜14日のHordeum vulgaris cv Golden Promiseの未成熟種子を採取し、そして4℃にて暗所で一晩保存した。冷所でインキュベートした未成熟種子を、70%EtOH中で1分間処理し、次いで0.6%次亜塩素酸塩ナトリウム(natrium hypochloride)中で10分間処理し、その後滅菌条件下でラミナフロー中の解剖顕微鏡のもとで、滅菌ペトリ皿上に静置した状態で滅菌蒸留水で完全に洗浄した(5〜8回)。胚の位置を特定し、種子の末端部を切断し、そして両側を切断した種子を作製した。種子をピンセットで固定し、そして種子の中央部を圧迫して胚を押し出す。胚をピンセットでしっかりと固定し、そしてメス刃を胚盤と胚軸とのあいだの溝に挿入し、そして胚軸をゆっくりと切断した。胚盤をカルス誘導培地上に静置し、切断面を上にし、そして植物形質転換ベクターを保持するそのままの濃度のAgrobacterium tumefaciens25μl〜40μlを1〜5分間接種した。接種の後、胚盤をディッシュの外に引き出し、細菌の負荷を低減し、同時培養フェーズのあいだの過剰増殖を低下させる。感染した胚盤を新しいカルス誘導培地プレートに移し、そしてプレートを暗所、24℃にて3日間インキュベートする。3日後、胚盤をAgrobacteriumを殺すための100μg/mlのチメンチン(timentin)および形質転換細胞を選択するための50μg/mlのハイグロマイシンを含む新しいカルス誘導培地に移し、暗所、24℃にて4週間インキュベートし、2週間後にサブカルチャーする。次いで、カルスを2.5 mg/L BAP、50μg/mlチメンチンおよび25μg/mlハイグロマイシンを含む苗条誘導培地に移し、そして強光下にて4〜10週間インキュベートする。個々の再生しつつある苗を、発根培地(50μg/mlチメンチンおよび25μg/mlのハイグロマイシンを含み、ホルモン類は含まない)に移す。発根している約5〜7 cmの苗条にまで発生させた後、トランスジェニック植物を土壌に移し、そこで完全光(full light)下にて生育させた。
【0074】
実施例9 BLZ1およびBLZ2の標的化抑制の後のHoxB4-CBMタンパク質の蓄積
pbDH10l(hoxB4-cbmのみ)またはpbDH104(hoxB4-cbmにより発現されるBLZ1/2 hpRNA)のいずれかにより形質転換されたトランスジェニック植物におけるB-ホルデインおよびC-ホルデインの蓄積を測定するため、Smith(in The Protein Protocols Handbook ed., Walker, 2nd ed., Humana Press, 2002)により記載されるように、Colloidal Coomassie Brilliant Blue G法を使用することができる。次いで、種子抽出物由来の可溶性タンパク質をSDS-PAGEにより標準的タンパク質と共に分離し、その後、ゲルを染色溶液(3.5%(w/v)過塩素酸中、0.04%(w/v)Brilliant Blue G)を用いて1時間染色し、その後ゲルを蒸留水中で4時間脱染色する。それぞれのB-ホルデインおよびC-ホルデインバンドの強度を、スキャニングデンシトメトリーを用いて解析し、そして相対バンド強度をpbDH101またはpbDH104により形質転換したトランスジェニック植物中で比較する。これにより、BLZ1/2 hpRNAにより引き起こされたB-ホルデインおよびC-ホルデインの抑制を推定する。pbDH101またはpbDH104により形質転換された植物におけるHoxB4-CBMの同一性および発現レベルを、二つに複製したサンプルを含有する同一のゲルの断片をウェスタンブロッティングすることにより、確認しそして測定することができる。バンドをゲル断片からニトロセルロースメンブレン上に、BioradのMini transblot Cellを使用してエレクトロブロットすることができるが、ゲル断片の輪郭をフィルター上にマークした後、ブロッティングする。ゲル/メンブレンサンドイッチのブロッティングおよび取り外しの後、ニトロセルロースをCBMに対するポリクローナル抗血清を用いてハイブリダイズし、そして複数回の洗浄工程(2×5分、2×15分、1×5分)の後、西洋ワサビペルオキシダーゼ(HRP)を抱合させた二次抗体を用いて45分間インキュベートした。上述した後に続く洗浄工程の後、そして基質5-ブロモ-4-クロロ-3-インドリルホスフェートおよびニトロブルー塩化テトラゾリウムをメンブレンに添加した際、HRP呈色反応はバンドをHoxB4-CBMとして陽性に同定し、そしてBLZ1/2 hpRNA発現のhoxB4-CBMレベルに対する作用を推定する。
【0075】
本発明の好ましい態様のみが特に示されるが、上述した実施例において記載される本発明の多数の修飾およびバリエーションが、本発明の範囲からはずれることなく当業者には生じることが予想される。
【0076】
【表3】
【図面の簡単な説明】
【0077】
【図1】図1は、水溶性抽出物(レーン1)、塩抽出物(レーン2)そしてEtOH抽出物(レーン3)を使用した、Hordeum vulgaris cv. Skegla(開花後49日)由来の全タンパク質のドデシル硫酸ナトリウム-ポリアクリルアミド電気泳動である。矢印は、B-ホルデイン(下)およびC-ホルデイン(上)を示す。
【図2】図2は、a)プライマーSEQ ID NO: 9およびSEQ ID NO: 10(実施例2を参照)およびb)プライマーSEQ ID NO: 12およびSEQ ID NO: 13(実施例3)を使用した、PCR増幅生成物のアガロースゲル解析である。生成物を、1.0%アガロースゲル上で分離し、エチジウムブロマイドにより染色し、そしてUVの下で写真撮影した。左側のサイズマーカーは、EcoRI/HindIII切断ラムダである。矢印は、増幅生成物を示す。
【図3】図3は、D-ホルデインプロモータの制御下でオオムギ胚乳組織中で発現させるためのコドン-最適化HoxB4-CBMキメラ遺伝子を用いて、オオムギを形質転換するための、バイナリ植物形質転換ベクターpbDH101のクローニングの概略説明を示す。略語:D-hor、D-ホルデインプロモータ;CBM、Thermatoga maritima由来の炭水化物結合ドメインをコードするコドン-最適化遺伝子;HoxB4、コドン-最適化ホメオボックスB4遺伝子(SEQ ID NO: 20);L、リンカー(PTPTPT;Pはプロリン、Tはトレオニン);kd、KDEL配列;pinII、ポテトプロテナーゼ阻害剤II遺伝子終止シグナル;CaMV 35S、カリフラワーモザイクウィルス35Sプロモータ;hph、E. coli由来のハイグロマイシンホスホトランスフェラーゼ(Genebankaccession # K01193);Nos-ter、ノパリン合成酵素終止シグナル;R、右側境界;L、左側境界。
【図4】図4は、D-ホルデインプロモータの制御下で、オオムギにおけるBLZ1およびBLZ2のヘアピンRNA-誘導標的化抑制のために構築された、遺伝子の概略説明である。センス配向およびアンチセンス配向の114 bpのBLZ1/BLZ2はそれぞれ、ステム部分を形成することができる。一方、88 bpのイントロン4がヘアピンループ中のループを形成する。
【図5】図5は、D-ホルデインプロモータの制御下でオオムギ胚乳組織中のBLZ1およびBLZ2のhpRNA-誘導標的化抑制を用いて、オオムギを形質転換するための、バイナリ植物形質転換ベクターpbDH104のクローニングを概略的に説明したものである。略語:D-hor、D-ホルデインプロモータ;hpBl/2、BLZ1およびBLZ2由来のヌクレオチド配列(SEQ ID NO: 8)からなる620 bpのサプレッサー断片;CBM、Thermatoga maritima由来の炭水化物結合ドメインをコードするコドン-最適化遺伝子;HoxB4、コドン-最適化ホメオボックスB4遺伝子(SEQ ID NO: 20);L、リンカー(PTPTPT;Pはプロリン、Tはトレオニン);pinII、ポテトプロテナーゼ阻害剤II遺伝子終止シグナル;CaMV 35S、カリフラワーモザイクウィルス35Sプロモータ;hph、E. coli由来のハイグロマイシンホスホトランスフェラーゼ(Genebank accession #K01193);Nos-ter、ノパリン合成酵素終止シグナル;R、右側境界;L、左側境界。
【特許請求の範囲】
【請求項1】
植物種子において目的の異種ポリペプチドの発現および蓄積を亢進させる方法であって、前記方法が:
(a)種子貯蔵タンパク質をコードする1またはそれ以上の内在性遺伝子の転写を制御する、異なる(1または複数の)転写制御因子(TF)のキメラ結合物を含む、1またはそれ以上のTFまたはその(1または複数の)部分をコードするDNA配列に対して機能可能に連結された種子-特異的プロモータについてのDNA配列を用いて植物細胞を形質転換し、ここで前記配列の転写された鎖が前記植物細胞における1またはそれ以上の前記種子貯蔵タンパク質の発現を抑制し、遅延させ、またはそうでなければ減少させることができ、そして
(b)(a)における前記転写制御因子により認識されるcis-作用性要素を有さない種子-特異的プロモータを選択し、そして
(c)目的の異種ポリペプチドをコードするDNA配列と機能可能に連結した(b)における前記種子-特異的プロモータについてのDNA配列を用いて、(a)におけるものと同一または別の植物細胞を形質転換し、;
(d)前記(1または複数の)形質転換植物宿主細胞由来の植物を再生させ、そして1またはそれ以上のTFまたはその部分をコードする前記(1または複数の)DNA配列が転写される条件下で前記植物を生長させ、それにより前記内在性mRNAの発現を減少させ、そして前記種子貯蔵タンパク質の発現を減少させ、そして目的の前記異種ポリペプチドの発現および蓄積を亢進させる、
ことを含む、前記方法。
【請求項2】
1またはそれ以上の転写制御因子(TF)またはその部分をコードする前記配列の転写された鎖が、前記植物細胞における1またはそれ以上の前記種子貯蔵タンパク質の発現を抑制し、遅延させ、またはそうでなければ減少させることができる“ヘアピン”RNAを形成することができる、請求項1に記載の方法。
【請求項3】
前記植物宿主細胞が、単子葉植物の群から選択される、請求項1または2に記載の方法。
【請求項4】
前記植物宿主細胞が、オオムギ、トウモロコシ、コムギ、オートムギ、およびコメを含有する単子葉植物の群から選択される、請求項3に記載の方法。
【請求項5】
工程(a)の前記(1または複数の)DNA配列および工程(b)の前記DNA配列が、一つのDNA配列中にて機能可能に連結される、請求項1または2に記載の方法。
【請求項6】
工程(a)の前記(1または複数の)DNA配列および工程(b)の前記DNA配列が、同一の植物細胞中に導入される、請求項1または2に記載の方法。
【請求項7】
工程(a)の前記(1または複数の)DNA配列が第一の植物宿主細胞のゲノム中に導入され、そして工程(b)の前記DNA配列が第二の植物宿主細胞のゲノム中に導入される、請求項1または2に記載の方法。
【請求項8】
第一のトランスジェニック植物が前記第一の植物宿主細胞から再生され、そして第二のトランスジェニック植物が前記第二の植物宿主細胞から再生され、そしてトランスジェニック植物の子孫個体群は前記第一のトランスジェニック植物と第二のトランスジェニック植物との有性交配から作製される、請求項7に記載の方法。
【請求項9】
前記1またはそれ以上の種子貯蔵タンパク質が、オオムギのホルデイン(hordein)である、請求項1〜8のいずれか1項に記載の方法。
【請求項10】
目的の異種ポリペプチドをコードする前記DNA配列が、原核細胞のタンパク質または真核細胞のタンパク質の群由来のタンパク質をコードする、請求項1〜9のいずれか1項に記載の方法。
【請求項11】
目的の異種ポリペプチドをコードする前記DNA配列が、好熱性生物由来のタンパク質をコードする、請求項10に記載の方法。
【請求項12】
前記配列が、炭水化物結合モジュール(CBM)をコードする、請求項10または11に記載の方法。
【請求項13】
原核細胞のタンパク質または真核細胞のタンパク質をコードする前記DNA配列が、コラーゲン、コラゲナーゼ、ホメオボックスポリペプチド、モノクローナル抗体、分泌抗体、一本鎖抗体、マンノース結合レクチン、ペプシン、キモトリプシン、トリプシン、カゼイン、ヒト成長ホルモン、ヒト血清アルブミン、ヒトインスリン、セルラーゼ、ペクチナーゼ、ヘミセルラーゼ、フィターゼ、ヒドロラーゼ、ペルオキシダーゼ、フィブリノーゲン、第IX因子、第XIII因子、トロンビン、タンパク質C、キシラナーゼ、イソアミラーゼ、グルコアミラーゼ、アミラーゼ、リゾチーム、β-グルカナーゼ、グルコセレブロシダーゼ、カゼイン、ラクターゼ、ウレアーゼ、グルコースイソメラーゼ、インベルターゼ、ストレプトアビジン、エステラーゼ、アルカリホスファターゼ、プロテアーゼ阻害剤、ペプシン、キモトリプシン、トリプシン、パパイン、キナーゼ、ホスファターゼ、デオキシリボヌクレアーゼ、リボヌクレアーゼ、ホスホリパーゼ、リパーゼ、ラッカーゼ、クモの糸タンパク質、凍結防止タンパク質、抗菌性ペプチドまたはデフェンシン、成長因子、およびサイトカインをコードするDNA配列からなる群から選択される、請求項10に記載の方法。
【請求項14】
真核細胞のタンパク質をコードする前記DNA配列が、ホメオボックスB4(HoxB4)タンパク質をコードするヒトHoxB4遺伝子である、請求項13に記載の方法。
【請求項15】
前記HoxB4タンパク質が、SEQ ID NO: 1のアミノ酸配列に対して少なくとも70%の配列同一性を有する、請求項14に記載の方法。
【請求項16】
1またはそれ以上のTFをコードする前記DNA配列が、TFまたはその部分をコードする2またはそれ以上のDNA配列の領域からなるキメラDNA配列である、請求項1〜15のいずれか1項に記載の方法。
【請求項17】
TFまたはその部分をコードする前記DNA配列が、bZIPタンパク質の群由来のTFまたはその部分をコードする領域を含む、請求項1〜16のいずれか1項に記載の方法。
【請求項18】
bZIPタンパク質をコードする前記DNA配列が、
a)オオムギBLZ1タンパク質またはその部分をコードするDNA配列;
b)オオムギBLZ2タンパク質またはその部分をコードするDNA配列;
c)a)およびb)の組み合わせ
から選択されるDNA配列を含む、請求項17に記載の方法。
【請求項19】
前記DNA配列が、SEQ IDNO: 2、SEQ ID NO: 3、SEQ ID NO: 4、SEQ ID NO: 5、およびそのいずれかの部分または組み合わせから選択される、請求項18に記載の方法。
【請求項20】
TFをコードする前記DNA配列が、前記植物細胞中でヘアピンRNA(hpRNA)を発現することができる、請求項1に記載の方法。
【請求項21】
TFをコードする前記DNA配列が、bZIPタンパク質の群由来のTFをコードするその領域または部分を含む、請求項20に記載の方法。
【請求項22】
前記bZIPタンパク質が、BLZ1、BLZ2およびBLZ1とBLZ2のキメラ結合物から選択される、請求項21に記載の方法。
【請求項23】
前記DNA配列が、SEQ ID NO: 2、SEQ ID NO: 4、SEQ ID NO: 6、またはSEQ ID NO: 7、SEQ ID NO: 8、およびそれらのいずれかの部分または組み合わせから選択される配列を含む、請求項22に記載の方法。
【請求項24】
請求項1〜23のいずれか1項に記載の方法により得ることができる、トランスジェニック植物。
【請求項25】
植物がオオムギ植物である、請求項24に記載のトランスジェニック植物。
【請求項26】
ゲノム中に:
a)種子貯蔵タンパク質をコードする1またはそれ以上の内在性遺伝子の転写を制御する、異なる(1または複数の)転写制御因子(TF)のキメラ結合物を含む、1またはそれ以上のTFまたはその部分をコードするDNA配列に対して機能可能に連結した種子-特異的プロモータをコードするDNA配列;
b)目的の異種タンパク質をコードするDNA配列に対して機能可能に連結された前記転写制御因子により認識されるcis-作用性要素を有さない種子-特異的プロモータ(ここで前記植物は、その種子中で前記異種タンパク質を発現し、そして対応する非-組換え植物よりも実質的に少ない量の種子貯蔵タンパク質を発現する)
を有する、請求項25に記載のトランスジェニックオオムギ植物。
【請求項1】
植物種子において目的の異種ポリペプチドの発現および蓄積を亢進させる方法であって、前記方法が:
(a)種子貯蔵タンパク質をコードする1またはそれ以上の内在性遺伝子の転写を制御する、異なる(1または複数の)転写制御因子(TF)のキメラ結合物を含む、1またはそれ以上のTFまたはその(1または複数の)部分をコードするDNA配列に対して機能可能に連結された種子-特異的プロモータについてのDNA配列を用いて植物細胞を形質転換し、ここで前記配列の転写された鎖が前記植物細胞における1またはそれ以上の前記種子貯蔵タンパク質の発現を抑制し、遅延させ、またはそうでなければ減少させることができ、そして
(b)(a)における前記転写制御因子により認識されるcis-作用性要素を有さない種子-特異的プロモータを選択し、そして
(c)目的の異種ポリペプチドをコードするDNA配列と機能可能に連結した(b)における前記種子-特異的プロモータについてのDNA配列を用いて、(a)におけるものと同一または別の植物細胞を形質転換し、;
(d)前記(1または複数の)形質転換植物宿主細胞由来の植物を再生させ、そして1またはそれ以上のTFまたはその部分をコードする前記(1または複数の)DNA配列が転写される条件下で前記植物を生長させ、それにより前記内在性mRNAの発現を減少させ、そして前記種子貯蔵タンパク質の発現を減少させ、そして目的の前記異種ポリペプチドの発現および蓄積を亢進させる、
ことを含む、前記方法。
【請求項2】
1またはそれ以上の転写制御因子(TF)またはその部分をコードする前記配列の転写された鎖が、前記植物細胞における1またはそれ以上の前記種子貯蔵タンパク質の発現を抑制し、遅延させ、またはそうでなければ減少させることができる“ヘアピン”RNAを形成することができる、請求項1に記載の方法。
【請求項3】
前記植物宿主細胞が、単子葉植物の群から選択される、請求項1または2に記載の方法。
【請求項4】
前記植物宿主細胞が、オオムギ、トウモロコシ、コムギ、オートムギ、およびコメを含有する単子葉植物の群から選択される、請求項3に記載の方法。
【請求項5】
工程(a)の前記(1または複数の)DNA配列および工程(b)の前記DNA配列が、一つのDNA配列中にて機能可能に連結される、請求項1または2に記載の方法。
【請求項6】
工程(a)の前記(1または複数の)DNA配列および工程(b)の前記DNA配列が、同一の植物細胞中に導入される、請求項1または2に記載の方法。
【請求項7】
工程(a)の前記(1または複数の)DNA配列が第一の植物宿主細胞のゲノム中に導入され、そして工程(b)の前記DNA配列が第二の植物宿主細胞のゲノム中に導入される、請求項1または2に記載の方法。
【請求項8】
第一のトランスジェニック植物が前記第一の植物宿主細胞から再生され、そして第二のトランスジェニック植物が前記第二の植物宿主細胞から再生され、そしてトランスジェニック植物の子孫個体群は前記第一のトランスジェニック植物と第二のトランスジェニック植物との有性交配から作製される、請求項7に記載の方法。
【請求項9】
前記1またはそれ以上の種子貯蔵タンパク質が、オオムギのホルデイン(hordein)である、請求項1〜8のいずれか1項に記載の方法。
【請求項10】
目的の異種ポリペプチドをコードする前記DNA配列が、原核細胞のタンパク質または真核細胞のタンパク質の群由来のタンパク質をコードする、請求項1〜9のいずれか1項に記載の方法。
【請求項11】
目的の異種ポリペプチドをコードする前記DNA配列が、好熱性生物由来のタンパク質をコードする、請求項10に記載の方法。
【請求項12】
前記配列が、炭水化物結合モジュール(CBM)をコードする、請求項10または11に記載の方法。
【請求項13】
原核細胞のタンパク質または真核細胞のタンパク質をコードする前記DNA配列が、コラーゲン、コラゲナーゼ、ホメオボックスポリペプチド、モノクローナル抗体、分泌抗体、一本鎖抗体、マンノース結合レクチン、ペプシン、キモトリプシン、トリプシン、カゼイン、ヒト成長ホルモン、ヒト血清アルブミン、ヒトインスリン、セルラーゼ、ペクチナーゼ、ヘミセルラーゼ、フィターゼ、ヒドロラーゼ、ペルオキシダーゼ、フィブリノーゲン、第IX因子、第XIII因子、トロンビン、タンパク質C、キシラナーゼ、イソアミラーゼ、グルコアミラーゼ、アミラーゼ、リゾチーム、β-グルカナーゼ、グルコセレブロシダーゼ、カゼイン、ラクターゼ、ウレアーゼ、グルコースイソメラーゼ、インベルターゼ、ストレプトアビジン、エステラーゼ、アルカリホスファターゼ、プロテアーゼ阻害剤、ペプシン、キモトリプシン、トリプシン、パパイン、キナーゼ、ホスファターゼ、デオキシリボヌクレアーゼ、リボヌクレアーゼ、ホスホリパーゼ、リパーゼ、ラッカーゼ、クモの糸タンパク質、凍結防止タンパク質、抗菌性ペプチドまたはデフェンシン、成長因子、およびサイトカインをコードするDNA配列からなる群から選択される、請求項10に記載の方法。
【請求項14】
真核細胞のタンパク質をコードする前記DNA配列が、ホメオボックスB4(HoxB4)タンパク質をコードするヒトHoxB4遺伝子である、請求項13に記載の方法。
【請求項15】
前記HoxB4タンパク質が、SEQ ID NO: 1のアミノ酸配列に対して少なくとも70%の配列同一性を有する、請求項14に記載の方法。
【請求項16】
1またはそれ以上のTFをコードする前記DNA配列が、TFまたはその部分をコードする2またはそれ以上のDNA配列の領域からなるキメラDNA配列である、請求項1〜15のいずれか1項に記載の方法。
【請求項17】
TFまたはその部分をコードする前記DNA配列が、bZIPタンパク質の群由来のTFまたはその部分をコードする領域を含む、請求項1〜16のいずれか1項に記載の方法。
【請求項18】
bZIPタンパク質をコードする前記DNA配列が、
a)オオムギBLZ1タンパク質またはその部分をコードするDNA配列;
b)オオムギBLZ2タンパク質またはその部分をコードするDNA配列;
c)a)およびb)の組み合わせ
から選択されるDNA配列を含む、請求項17に記載の方法。
【請求項19】
前記DNA配列が、SEQ IDNO: 2、SEQ ID NO: 3、SEQ ID NO: 4、SEQ ID NO: 5、およびそのいずれかの部分または組み合わせから選択される、請求項18に記載の方法。
【請求項20】
TFをコードする前記DNA配列が、前記植物細胞中でヘアピンRNA(hpRNA)を発現することができる、請求項1に記載の方法。
【請求項21】
TFをコードする前記DNA配列が、bZIPタンパク質の群由来のTFをコードするその領域または部分を含む、請求項20に記載の方法。
【請求項22】
前記bZIPタンパク質が、BLZ1、BLZ2およびBLZ1とBLZ2のキメラ結合物から選択される、請求項21に記載の方法。
【請求項23】
前記DNA配列が、SEQ ID NO: 2、SEQ ID NO: 4、SEQ ID NO: 6、またはSEQ ID NO: 7、SEQ ID NO: 8、およびそれらのいずれかの部分または組み合わせから選択される配列を含む、請求項22に記載の方法。
【請求項24】
請求項1〜23のいずれか1項に記載の方法により得ることができる、トランスジェニック植物。
【請求項25】
植物がオオムギ植物である、請求項24に記載のトランスジェニック植物。
【請求項26】
ゲノム中に:
a)種子貯蔵タンパク質をコードする1またはそれ以上の内在性遺伝子の転写を制御する、異なる(1または複数の)転写制御因子(TF)のキメラ結合物を含む、1またはそれ以上のTFまたはその部分をコードするDNA配列に対して機能可能に連結した種子-特異的プロモータをコードするDNA配列;
b)目的の異種タンパク質をコードするDNA配列に対して機能可能に連結された前記転写制御因子により認識されるcis-作用性要素を有さない種子-特異的プロモータ(ここで前記植物は、その種子中で前記異種タンパク質を発現し、そして対応する非-組換え植物よりも実質的に少ない量の種子貯蔵タンパク質を発現する)
を有する、請求項25に記載のトランスジェニックオオムギ植物。
【図1】

【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公表番号】特表2007−503212(P2007−503212A)
【公表日】平成19年2月22日(2007.2.22)
【国際特許分類】
【出願番号】特願2006−524530(P2006−524530)
【出願日】平成16年8月27日(2004.8.27)
【国際出願番号】PCT/IS2004/000012
【国際公開番号】WO2005/021765
【国際公開日】平成17年3月10日(2005.3.10)
【出願人】(506066847)
【Fターム(参考)】
【公表日】平成19年2月22日(2007.2.22)
【国際特許分類】
【出願日】平成16年8月27日(2004.8.27)
【国際出願番号】PCT/IS2004/000012
【国際公開番号】WO2005/021765
【国際公開日】平成17年3月10日(2005.3.10)
【出願人】(506066847)
【Fターム(参考)】
[ Back to top ]