
内皮前駆細胞およびその使用方法
【課題】インビトロ細胞培養物を提供すること。
【解決手段】上記インビトロ細胞培養物は、ヒトの末梢血に由来する細胞を含み、該培養物中の該細胞は、a.Tie−2+またはVEGFR−2+であり;b.CD144−およびCD45+であり;c.培養物中で培養することができ;ならびにd.インビボで増殖し、内皮細胞および平滑筋細胞に分化することができる。一局面において、上記培養物中の上記細胞は、少なくとも2週間生存できる。他の局面において、上記培養物中の上記細胞は、少なくとも4週間生存できる。他の局面において、上記培養物が付着培養物である。
【解決手段】上記インビトロ細胞培養物は、ヒトの末梢血に由来する細胞を含み、該培養物中の該細胞は、a.Tie−2+またはVEGFR−2+であり;b.CD144−およびCD45+であり;c.培養物中で培養することができ;ならびにd.インビボで増殖し、内皮細胞および平滑筋細胞に分化することができる。一局面において、上記培養物中の上記細胞は、少なくとも2週間生存できる。他の局面において、上記培養物中の上記細胞は、少なくとも4週間生存できる。他の局面において、上記培養物が付着培養物である。
【発明の詳細な説明】
【技術分野】
【0001】
(発明の分野)
本発明は前駆細胞に関する。
【背景技術】
【0002】
(発明の背景)
血管形成は、既存の脈管構造からの新しい血管の発生(脈管新生)のプロセスであり、一方、脈管形成は、インサイチュで分化する内皮前駆細胞からの血管の形成をいう。最近まで、血管形成は成体での脈管新生のみを意味すると考えられ、脈管形成は胚発生に限定されると考えられてきた。しかし、循環している内皮前駆細胞(EPC)の存在により、成体においても出生後脈管形成が生じることの証拠がもたらされた。虚血性損傷からの組織の再生のための治療用ツールとしてのEPCの可能性が示唆されている。
【発明の概要】
【発明が解決しようとする課題】
【0003】
したがって、内皮細胞に分化することができる前駆細胞の供給源が必要とされている。
【課題を解決するための手段】
【0004】
(発明の要旨)
本発明は、内皮前駆細胞の発見に基づく。前駆細胞は多能性である。前駆細胞は、例えば、インビボまたはインビトロで、内皮細胞または平滑筋細胞に分化することができる。したがって、本発明は、内皮前駆細胞の培養物、例えば、インビトロ培養物を特徴とする。培養物は付着培養物である。あるいは、培養物中の細胞は浮遊状態である。細胞は、造血組織、例えば、血液(例えば、末梢血または骨髄)に由来する。組織は、哺乳動物、例えば、ヒト、霊長類、マウス、ラット、イヌ、ネコ、ウシ、ウマ、ブタに由来する。細胞は、VEGFR−2またはTie−2とCD45に免疫反応性であり、CD144とは免疫反応しない。状況によっては、VEGFR+細胞はCD14とも免疫反応する。細胞はインビトロで増殖させられる。細胞は、2倍、3倍、4倍、5倍、6倍、7倍、8倍、9倍、10倍、11倍、12倍、15倍、20倍、25倍またはそれ以上に増殖することができ、内皮細胞または平滑筋細胞に分化する能力を維持することができる。
【0005】
本発明はさらに、VEGFR−2+またはTie−2+、CD45+、およびCD144−前駆細胞、あるいは、VEGFR−2+CD14+、CD45+、およびCD144−前駆細胞を提供し、そしてこれらの細胞を被験体に移植することによって、被験体に前駆細胞または前駆細胞の子孫を移植する方法を特徴とする。移植によっては、臨床的有益性、例えば、特定の血管障害の1つ以上の症状の緩和がもたらされる。血管障害は、当該分野で公知の方法を使用して医師によって診断される。
【0006】
損傷した血管は、VEGFR−2+またはTie−2+、CD45+、およびCD144−前駆細胞を含む組成物、あるいは、VEGFR−2+CD14+、CD45+、およびCD144−前駆細胞を含む組成物を被験体に投与することによって再増殖させられる。これらの組成物は、血管(例えば、静脈または動脈)に、あるいは動脈床に直接投与される。あるいは、これらの組成物は全身的に投与される。
【0007】
血管移植片は、血管移植片をVEGFR−2+またはTie−2、CD45+、およびCD144−前駆細胞を含む組成物、あるいは、VEGFR−2+CD14+、CD45+、およびCD144−前駆細胞を含む組成物と接触させることによって内皮細胞化(endothelized)させられる。血管移植片は静脈である。あるいは、血管移植片は動脈である。移植片は人工血管移植片、同種移植片、または同系移植片である。移植片は、インビトロ、インビボ、またはエキソビボで接触させられる。血管移植片は、被験体への移植の前に接触させられる。あるいは、移植片は、被験体への移植後に組成物と接触させられる。
【0008】
血管を再増殖させるか、または血管移植片を内皮細胞化させるによって、血管または移植片が、前駆細胞で処置されていない血管または移植片と比較して、血管または移植片が多量の内皮細胞を有していることが意味される。例えば、処置された血管または移植片は、処置されていない血管または移植片と比較して大きな血管径と中膜厚を有する。
【0009】
VEGFR−2+またはTie−2+、CD45+、およびCD144−前駆細胞は、インビボで、内皮細胞または平滑筋細胞に分化する能力を維持する。一方、VEGFR−2+CD14+、CD45+、およびCD144−前駆細胞は、インビボで、内皮細胞に分化する能力を維持する。被験体は、例えば、哺乳動物であり、例えば、ヒト、霊長類、マウス、ラット、イヌ、ネコ、ウシ、ウマ、ブタである。被験体は、血管障害または血管組織の損傷に罹患している。例えば、被験体は、末梢動脈障害、動脈瘤、腎(腎臓)動脈障害、レイノー現象、バージャー病、末梢静脈障害、静脈瘤、静脈内血塊に罹患している。移植および/または再増殖によって、臨床的有益性、例えば、特定の血管障害の1つ以上の症状の緩和がもたらされる。血管障害は、当該分野で公知の方法を使用して医師によって診断される。
【0010】
他の場所で明確に定義されていない限りは、本明細書中で使用される全ての技術用語および科学用語は、本発明が属する分野の当業者によって一般的に理解されている意味と同じ意味を有する。本明細書中に記載される方法および材料と同様または同等の方法および材料を本発明の実施または試験において使用することができるが、適切な方法および材料は以下に記載される。本明細書中に言及される全ての刊行物、特許出願、特許、および他の参考文献は、それらの全体が参考として援用される。矛盾する場合は、定義を含む本明細書が統制する。さらに、材料、方法、および実施例は例示にすぎず、限定するようには意図されない。
【0011】
本発明の他の特徴および利点は、以下の詳細な記載および特許請求の範囲から明らかである。
したがって、本発明は、以下の項目を提供する:
(項目1)
ヒトの末梢血に由来する細胞を含む、インビトロ細胞培養物であって、該培養物中の該細胞は、
a.Tie−2+またはVEGFR−2+であり;
b.CD144−およびCD45+であり;
c.培養物中で培養することができ;ならびに
d.インビボで増殖し、内皮細胞および平滑筋細胞に分化することができる、
インビトロ細胞培養物。
(項目2)
上記培養物中の上記細胞が、少なくとも2週間生存できる、項目1に記載の培養物。
(項目3)
上記培養物中の上記細胞が、少なくとも4週間生存できる、項目1に記載の培養物。
(項目4)
上記培養物が付着培養物である、項目1に記載の培養物。
(項目5)
インビボで内皮細胞または平滑筋細胞に分化するヒト前駆細胞の集団を生成する方法であって、ヒト末梢血由来の細胞の集団からCD144−、CD45+、およびTie−2+またはVEGFR−2+である細胞を選択する工程を包含する、方法。
(項目6)
多能性前駆細胞を被験体に移植する方法であって、
a.多能性のヒトCD144−、CD45+、およびTie−2+またはVEGFR−2+前駆細胞を含むインビトロ細胞培養物を提供する工程であって、該細胞はインビボで内皮細胞または平滑筋細胞に分化する多能性を維持している、工程;および
b.上記細胞を上記被験体に移植する工程、
を包含する、方法。
(項目7)
上記被験体が血管障害に罹患している、項目6に記載の方法。
(項目8)
被験体において損傷した血管を再増殖させる方法であって、多能性のヒトCD144−、CD45+、およびTie−2+またはVEGFR−2+前駆細胞の集団を含む組成物を被験体に投与する工程を包含し、ここで、該細胞はインビボで内皮細胞または平滑筋細胞に分化する多能性を維持している、方法。
(項目9)
上記組成物が血管に直接投与される、項目8に記載の方法。
(項目10)
上記組成物が動脈床に直接投与される、項目8に記載の方法。
(項目11)
上記血管が静脈または動脈である、項目8に記載の方法。
(項目12)
上記被験体が血管障害に罹患している、項目8に記載の方法。
(項目13)
血管移植片を内皮細胞化させる方法であって、該血管移植片を、多能性のヒトCD144−、CD45+、およびTie−2+またはVEGFR−2+前駆細胞の集団を含む組成物と接触させる工程を包含し、ここで、該細胞はインビボで内皮細胞または平滑筋細胞に分化する多能性を維持している、方法。
(項目14)
上記移植片が人工血管移植片である、項目13に記載の方法。
(項目15)
上記移植片が同種移植片である、項目13に記載の方法。
(項目16)
上記移植片が同系移植片である、項目13に記載の方法。
(項目17)
上記血管移植片がインビトロ、インビボ、またはエキソビボで接触させられる、項目13に記載の方法。
(項目18)
上記血管移植片が被験体への移植の前に上記組成物と接触させられる、項目13に記載の方法。
(項目19)
上記血管移植片が被験体への移植の後に上記組成物と接触させられる、項目13に記載の方法。
(項目20)
上記血管移植片が静脈または動脈である、項目13に記載の方法。
(項目21)
ヒトの末梢血由来の細胞を含むインビトロ細胞培養物であって、該培養物中の該細胞は、
a.VEGFR−2+;CD14+、CD144−、およびCD45+であり;
b.培養物中で培養することができ;ならびに
c.インビボで増殖し、内皮細胞に分化することができる、
インビトロ細胞培養物。
(項目22)
上記培養物中の上記細胞が、少なくとも2週間生存できる、項目21に記載の培養物。
(項目23)
上記培養物中の上記細胞が、少なくとも4週間生存できる、項目21に記載の培養物。
(項目24)
上記培養物が付着培養物である、項目21に記載の培養物。
(項目25)
インビボで内皮細胞に分化するヒト前駆細胞の集団を生成する方法であって、ヒト末梢血由来の細胞の集団からCD144−、CD45+、CD14+、およびVEGFR−2+である細胞を選択する工程を包含する、方法。
(項目26)
前駆細胞を被験体に移植する方法であって、
a.ヒトCD144−、CD45+、CD14+、およびVEGFR−2+前駆細胞を含むインビトロ細胞培養物を提供する工程であって、該細胞はインビボで内皮細胞に分化する能力を維持している、工程;および
b.該細胞を該被験体に移植する工程、
を包含する、方法。
(項目27)
上記被験体が血管障害に罹患している、項目26に記載の方法。
(項目28)
被験体の損傷した血管を再増殖させる方法であって、CD144−、CD45+、CD14+、およびVEGFR−2+前駆細胞の集団を含む組成物を被験体に投与する工程を包含し、ここで、該細胞はインビボで内皮細胞に分化する能力を維持している、方法。
(項目29)
上記組成物が血管に直接投与される、項目28に記載の方法。
(項目30)
上記組成物が動脈床に直接投与される、項目28に記載の方法。
(項目31)
上記血管が静脈または動脈である、項目28に記載の方法。
(項目32)
上記被験体が血管障害に罹患している、項目28に記載の方法。
(項目33)
血管移植片を内皮細胞化させる方法であって、上記血管移植片を、多能性のヒトCD144−、CD45+、CD14+、およびVEGFR−2+前駆細胞の集団を含む組成物と接触させる工程を包含し、ここで、該細胞はインビボで内皮細胞に分化する能力を維持している、方法。
(項目34)
上記移植片が人工血管移植片である、項目33に記載の方法。
(項目35)
上記移植片が同種移植片である、項目33に記載の方法。
(項目36)
上記移植片が同系移植片である、項目33に記載の方法。
(項目37)
上記血管移植片がインビトロ、インビボ、またはエキソビボで接触させられる、項目33に記載の方法。
(項目38)
上記血管移植片が被験体への移植の前に上記組成物と接触させられる、項目33に記載の方法。
(項目39)
上記血管移植片が被験体への移植の後に上記組成物と接触させられる、項目33に記載の方法。
(項目40)
上記血管移植片が静脈または動脈である、項目33に記載の方法。
【図面の簡単な説明】
【0012】
【図1】図1A〜Cは、抗VEGFR1抗体、抗VEGFR2抗体、およびTie−2抗体で染色した際に、VEGFR−1+、VEGFR−2+、およびTie−2+細胞がそれらのそれぞれの表面マーカーについてポジティブであったことを示す一連の蛍光顕微鏡写真(40倍)である。図1D〜Iは、新たに選別したVEGFR−1+細胞、VEGFR−2+細胞、およびTie−2+細胞の集団に、単核の単球(M)、マクロファージ(M)、リンパ球(L)および多核白血球(PMN)を含む細胞の混合物が含まれていることを示す一連の電子顕微鏡写真である。G〜Iは、VEGFR−2+細胞およびTie−2+細胞が内皮細胞に分化したことを示すTEM顕微鏡写真であり、(G)バイベル・パラーデ小体(丸い、矢印)および内皮細胞に典型的な堅い結合(矢印)の存在、ならびに、(H、I)開口(矢印)を示している。
【図2A】図2Aは、VEGFR−2+およびTie−2+であるが、VEGFR−1+細胞が、VEGFおよびang−1に向かって移動すること、およびこれらの細胞の3つの集団の全てが、マトリゲル細胞浸潤アッセイにおいて浸潤能を示したことを示している、棒グラフである。結果は、±SEMとして示されている。
【図2B】図2Bは、VEGFR−2+およびTie−2+であるが、VEGFR−1+細胞が、VEGFおよびang−1に向かって移動すること、およびこれらの細胞の3つの集団の全てが、マトリゲル細胞浸潤アッセイにおいて浸潤能を示したことを示している、棒グラフである。結果は、±SEMとして示されている。
【図2C】図2Cは、VEGFR−2+およびTie−2+であるが、VEGFR−1+細胞が、VEGFおよびang−1に向かって移動すること、およびこれらの細胞の3つの集団の全てが、マトリゲル細胞浸潤アッセイにおいて浸潤能を示したことを示している、棒グラフである。結果は、±SEMとして示されている。
【図2D】図2Dは、VEGFR−2+およびTie−2+であるが、VEGFR−1+細胞が、マトリゲル中で毛細管様の細管を形成したことを示している写真である。HUVECがコントロール細胞として使用された。
【図2E】図2Eは、VEGFR−2+およびTie−2+であるが、VEGFR−1+細胞が、マトリゲル中で毛細管様の細管を形成したことを示している写真である。HUVECがコントロール細胞として使用された。
【図2F】図2Fは、VEGFR−2+およびTie−2+であるが、VEGFR−1+細胞が、マトリゲル中で毛細管様の細管を形成したことを示している写真である。HUVECがコントロール細胞として使用された。
【図2G】図2Gは、VEGFR−2+およびTie−2+であるが、VEGFR−1+細胞が、マトリゲル中で毛細管様の細管を形成したことを示している写真である。HUVECがコントロール細胞として使用された。
【図3】図3は一連の写真である。AおよびB、ヒト核抗体によって、ヒトにおいては細胞が染色された(黒)が、コントロールであるマウスの動脈においては染色されなかった。バルーンで損傷させたマウスの大腿動脈に由来する切片の免疫組織化学染色は、4週目では、播種された動脈(C)VEGFR−2+細胞(矢印)の、2週目では、Tie−2+細胞(矢印)の、管腔表面での移植されたヒトの局在化を示している。もとの倍率は60倍である。E、正常なヒトの動脈、F、ヒトTie−2+細胞が移植された損傷させられたマウスの動脈、およびG、抗ヒト核抗体での、擬似移植されたマウスの動脈の免疫蛍光染色(矢印)。
【図4】図4は、移植されたヒト細胞のEC表現形を、ヒト核抗体と、A、VEGFR−2+細胞についてはフォン・ヴィレブランド因子に対する抗体(二重ポジティブ細胞は黄色に染色される、矢印)、そしてB、Tie−2+細胞についてはVE−カドヘリンに対する抗体を用いて、二重染色によって確認したことを示す一連の写真である。α−アクチンに対する抗体で標識された場合は、培地中の細胞(緑色のフィラメント状の染色パターン)はポジティブに染色され、このことは、平滑筋細胞の存在を示している。もとの倍率は60倍である。
【図5A】図5A〜Fは、7日目の組織の分析によってインビボでVEGFR−2+を移植されたヒト細胞およびTie−2+を移植されたヒト細胞への多数のBrdUの取り込みが明らかになったことを示す一連の写真である。AおよびBは、擬似移植されたマウスの動脈についての代表的な連続する切片であり、そしてC〜Fは、抗BrdU(茶色の細胞)とヒト核抗体(黒)で染色された移植されたマウスの動脈の代表的な連続する切片であり、それぞれ、移植されたVEGFR−2+細胞とTie−2細胞中での増殖を示している。
【図5B】図5A〜Fは、7日目の組織の分析によってインビボでVEGFR−2+を移植されたヒト細胞およびTie−2+を移植されたヒト細胞への多数のBrdUの取り込みが明らかになったことを示す一連の写真である。AおよびBは、擬似移植されたマウスの動脈についての代表的な連続する切片であり、そしてC〜Fは、抗BrdU(茶色の細胞)とヒト核抗体(黒)で染色された移植されたマウスの動脈の代表的な連続する切片であり、それぞれ、移植されたVEGFR−2+細胞とTie−2細胞中での増殖を示している。
【図5C】図5A〜Fは、7日目の組織の分析によってインビボでVEGFR−2+を移植されたヒト細胞およびTie−2+を移植されたヒト細胞への多数のBrdUの取り込みが明らかになったことを示す一連の写真である。AおよびBは、擬似移植されたマウスの動脈についての代表的な連続する切片であり、そしてC〜Fは、抗BrdU(茶色の細胞)とヒト核抗体(黒)で染色された移植されたマウスの動脈の代表的な連続する切片であり、それぞれ、移植されたVEGFR−2+細胞とTie−2細胞中での増殖を示している。
【図5D】図5A〜Fは、7日目の組織の分析によってインビボでVEGFR−2+を移植されたヒト細胞およびTie−2+を移植されたヒト細胞への多数のBrdUの取り込みが明らかになったことを示す一連の写真である。AおよびBは、擬似移植されたマウスの動脈についての代表的な連続する切片であり、そしてC〜Fは、抗BrdU(茶色の細胞)とヒト核抗体(黒)で染色された移植されたマウスの動脈の代表的な連続する切片であり、それぞれ、移植されたVEGFR−2+細胞とTie−2細胞中での増殖を示している。
【図5E】図5A〜Fは、7日目の組織の分析によってインビボでVEGFR−2+を移植されたヒト細胞およびTie−2+を移植されたヒト細胞への多数のBrdUの取り込みが明らかになったことを示す一連の写真である。AおよびBは、擬似移植されたマウスの動脈についての代表的な連続する切片であり、そしてC〜Fは、抗BrdU(茶色の細胞)とヒト核抗体(黒)で染色された移植されたマウスの動脈の代表的な連続する切片であり、それぞれ、移植されたVEGFR−2+細胞とTie−2細胞中での増殖を示している。
【図5F】図5A〜Fは、7日目の組織の分析によってインビボでVEGFR−2+を移植されたヒト細胞およびTie−2+を移植されたヒト細胞への多数のBrdUの取り込みが明らかになったことを示す一連の写真である。AおよびBは、擬似移植されたマウスの動脈についての代表的な連続する切片であり、そしてC〜Fは、抗BrdU(茶色の細胞)とヒト核抗体(黒)で染色された移植されたマウスの動脈の代表的な連続する切片であり、それぞれ、移植されたVEGFR−2+細胞とTie−2細胞中での増殖を示している。
【図5G】図5Gは、マウスの大腿動脈の中でのバルーン損傷手順によって、媒体だけとともにインキュベートされた損傷した血管においては病変の動脈セグメントの膨張が生じたが、ヒト細胞が移植されたマウスの大腿動脈においては病変の動脈断片の膨張は生じなかったことを示す棒グラフである。断面の中膜面積の減少は、損傷した動脈において観察されたが、VEGFR−2細胞およびTie−2細胞が移植された動脈においては観察されなかった。
【図5H】図5Hは、マウスの大腿動脈の中でのバルーン損傷手順によって、媒体だけとともにインキュベートされた損傷した血管においては病変の動脈セグメントの膨張が生じたが、ヒト細胞が移植されたマウスの大腿動脈においては病変の動脈断片の膨張は生じなかったことを示す棒グラフである。断面の中膜面積の減少は、損傷した動脈において観察されたが、VEGFR−2細胞およびTie−2細胞が移植された動脈においては観察されなかった。
【図6A】図6は、CD14+/VEGFR−2+およびCD14+/VEGFR−2−細胞の特徴を示す一連のグラフおよび写真である。A、末梢血単核細胞からのCD14+/VEGFR−2+およびCD14+/VEGFR−2−細胞のフローサイトメトリーによる選別により、Tリンパ球は激減した。
【図6B】図6は、CD14+/VEGFR−2+およびCD14+/VEGFR−2−細胞の特徴を示す一連のグラフおよび写真である。B、CD14+/VEGFR−2−細胞は内皮の一酸化窒素合成酵素(e−NOS)は発現しなかった。
【図6C】図6は、CD14+/VEGFR−2+およびCD14+/VEGFR−2−細胞の特徴を示す一連のグラフおよび写真である。C、CD14+/VEGFR−2+細胞は、e−NOS(黒)、フォン・ヴィレブランド因子(赤)、およびトロンボモジュリン(赤)のようないくつかの内皮特異的マーカーを発現したが、VE−カドヘリンは発現しなかった。細胞をヘマトキシリンで対比染色した。
【図6D】図6は、CD14+/VEGFR−2+およびCD14+/VEGFR−2−細胞の特徴を示す一連のグラフおよび写真である。D、CD14+/VEGFR−2+細胞由来の溶解物のVEGFR−2に対する抗体でのウェスタンブロッティングによっては、およそ200kDaの予想されたバンドが生じた(レーン1および2)が、CD14+/VEGFR−2−細胞(レーン3)とコントロールである二次抗体(レーン4)はバンドを生じなかった。
【図6E】図6は、CD14+/VEGFR−2+およびCD14+/VEGFR−2−細胞の特徴を示す一連のグラフおよび写真である。E、CD14+/VEGFR−2+細胞においては、e−NOSのmRNAの強い発現が、そしてCD14+/VEGFR−2−細胞においては弱い発現が検出された。
【図7】図7は、CD14+/VEGFR−2+細胞とCD14+/VEGFR−2−細胞のインビトロでの形態学的特性決定を示す一連の写真である。A、TEM顕微鏡写真は、CD14+/VEGFR−2+細胞について、内皮細胞に典型的な堅い結合の存在(矢印、棒;2μm)、B、バイベル・パラーデ小体の存在(矢印、棒;0.5μm)、およびC、基底膜の存在(M、棒;1μm)、D、培養物中のCD14+/VEGFR−2−細胞はコロニーを形成しないが、CD14+/VEGFR−2+細胞は最初に丸い細胞からなるコロニーを形成し、その1週間後、1つの細長い細胞に成長することが明らかになったことを示している。1週間の培養後には、CD14+/VEGFR−2+細胞だけがVE−カドヘリンを発現していた。
【図8A】図8AおよびBは、CD14+/VEGFR−2+細胞とCD14+/VEGFR−2−細胞のインビトロでの移動と細管形成能力を示す散布図である。A、50ng/mlの濃度の血管内皮成長因子(VEGF)と300ng/mlの単球走化性タンパク質−1(MCP−1)を含まない条件と、それらを含む条件下での移動アッセイにおいては、CD14+/VEGFR−2−細胞と比較して、明らかに多数のCD14+/VEGFR−2+細胞がVEGFおよびMCP−1に移動した。
【図8B】図8AおよびBは、CD14+/VEGFR−2+細胞とCD14+/VEGFR−2−細胞のインビトロでの移動と細管形成能力を示す散布図である。B、CD14+/VEGF−2+細胞の集団は、CD14+/VEGFR−2−集団と比較して、血管由来成長因子、VEGF、顆粒球コロニー刺激因子(G−CSF)、および顆粒球−マクロファージコロニー刺激因子(GM−CSF)をより高いレベルで生成した。
【図8C】図8Cは、CD14+/VEGFR−2+細胞はマトリゲルの中で毛細管様細管を形成したが、CD14+/VEGF−2−細胞は形成しなかったことを示している一連の写真である。ヒトの大動脈細胞(HAEC)がコントロール細胞として使用された。
【図9】図9は、CD14+/VEGFR−2+細胞とCD14+/VEGFR−2−細胞のインビボでの生着能力を示す一連の写真である。抗緑色蛍光タンパク質(GFP)抗体での、バルーン損傷させたマウスの大腿動脈由来の切片の免疫組織化学染色により、A、擬似移植されたGFPポジティブである領域の局在化はないこと、および、B、播種された動脈の管腔表面のCD14+/VEGFR−2−が移植された細胞が示された。C、切片はコントロールである二次抗体での染色を示している。D、2週間目の有意な数のGFP−ポジティブである(赤)CD14+/VEGFR−2+細胞(矢印)、そしてEおよびF、4週間目の有意な数のGFP−ポジティブである(赤)CD14+/VEGFR−2+細胞が、これらの細胞が移植された動物において見られた。もとの倍率は60倍であった。
【図10】図10は、移植後のCD14+/VEGFR−2+細胞とCD14+/VEGFR−2−細胞の検出のための免疫蛍光染色を示す一連の写真である。A、正常なヒトの動脈中にはヒト核抗体で染色された細胞(赤)があったが、B、コントロールの損傷させた擬似移植したマウスの動脈にはなかった。C、バルーン損傷させたマウスの大腿動脈は、4週目には、播種された大動脈(赤色の細胞、矢印)の管腔表面で移植されたヒトCD14+/VEGFR−2+細胞の局在化を示した。D、しかし、4週間目には、移植されたCD14+/VEGFR−2−細胞の局在化は観察されなかった。E、移植されたヒト細胞の内皮細胞表現形が、ヒト核抗体とVE−カドヘリンに対する抗体での二重染色によって確認された(二重ポジティブである細胞は黄色く染色された、矢印)。もとの倍率は60倍であった。
【図11】図11は、CD14+/VEGFR−2+細胞とCD14+/VEGFR−2−細胞のインビボでの生着能力および機能的能力を示すウェスタンブロットの写真である。マウスの動脈の中でのヒトの内皮特異的遺伝子の転写。ヒトCD31とe−NOSは、ヒトCD14+/VEGFR−2+細胞が移植されたバルーン損傷させられたマウスの大腿動脈の中で検出されたが、擬似移植されたマウス、またはCD14+/VEGFR−2−細胞が移植されたマウスにおいては検出されなかった。ABLがハウスキーピング遺伝子として使用された。
【発明を実施するための形態】
【0013】
(詳細な説明)
本発明は、ヒトの末梢血の中の明確な前駆細胞の集団の予想外の発見に基づく。これは、インビトロで培養することができ、前駆細胞の表現形を維持することができる。さらに、これらの細胞をマトリゲル上でインビトロで培養すると、これらは、細管様構造を形成することができる。これらの細胞は、動脈が損傷している動物に移植されると、内皮細胞および/または平滑筋細胞への機能的な分化を示した。
【0014】
血管障害には、世界中で顕著な罹患率および死亡率のある、広い範囲の急性の状態と慢性の状態のものが含まれる。内皮細胞の移植は、虚血によって損傷した組織の処置においては、非常にすばらしい治療能力を有している。血管障害には、循環系に影響を及ぼす任意の状態が含まれる。これは、動脈、静脈、およびリンパ管の疾患から、循環に影響を及ぼす血液疾患にまでの範囲に及ぶ。血管障害の例としては、末梢動脈障害、動脈瘤、腎(腎臓)動脈障害、レイノー現象、バージャー病、末梢静脈障害、静脈瘤、静脈内血塊が挙げられる。
【0015】
冠動脈疾患は、少なくとも2つのグループにカテゴリー分類することができる。急性の冠動脈疾患としては心筋梗塞が挙げられ、慢性の冠動脈疾患としては慢性冠動脈虚血、動脈硬化症、うっ血性心不全、狭心症、アテローム性動脈硬化症、および心筋肥大が挙げられる。他の冠動脈疾患としては、脳卒中、心筋梗塞、拡張型心筋症、再狭窄、冠状動脈疾患、心不全、不整脈、狭心症、または高血圧が挙げられる。
【0016】
急性の冠動脈疾患によって、心臓への血液の供給の突然の遮断が生じ、これによって心臓組織には酸素や栄養分が届かなくなり、結果として心臓組織の損傷および死が生じる。対照的に、慢性の冠動脈疾患は、時間とともに心臓組織への酸素および血液の供給が段階的に減少し、これによって心臓組織の進行性の損傷が生じ、最終的には死に至ることを特徴とする。内皮前駆細胞のインビトロおよびインビボでの増殖および分化をうまく行うことは、内皮細胞の移植に、および遺伝子治療用のビヒクルに有用である。
【0017】
本発明によって、インビボで内皮細胞および/または平滑筋細胞に分化することができる、末梢血由来の多能性内皮前駆細胞を生成するための方法が提供される。
【0018】
(内皮前駆細胞)
本発明によって、内皮前駆細胞(本明細書中ではEPCと呼ばれる)が提供される。EPC細胞は、本発明の方法を使用して増殖するように誘導することができる未分化の細胞である、EPCは自己維持(self−maintenance)することができ、その結果、どの細胞分裂によっても、少なくとも1つの娘細胞がまたEPC細胞となる。EPC細胞は100倍、250倍、500倍、1000倍、2000倍、3000倍、4000倍、5000倍、またはそれ以上に増殖させることができる。
【0019】
EPCの表現形は、これらの細胞が約束された造血細胞のマーカーであるCD45を発現することを明らかに示している。さらに、EPCは、VEGFR−2および/またはTei−2と免疫反応性であり、カドヘリン5(CD144)とは非免疫反応性である。状況によっては、EPCはCD14と免疫反応する。CD14とは免疫反応しないEPCは、インビボでは、内皮細胞または平滑筋細胞に分化することができる。別に、CD14と免疫反応する細胞は、内皮細胞だけに分化することができる。EPCは多能性前駆細胞である。多能性前駆細胞によって、細胞が1つ以上のタイプの細胞に分化することができることが意味される。例えば、細胞は、内皮細胞または平滑筋細胞に分化することができる。
【0020】
血管内皮成長因子(VEGF)は、特異的チロシンキナーゼ受容体を介して作用する。特異的チロシンキナーゼ受容体には、VFGFR−1(flt−1)およびVEGFR−2(flk−1/KDR)およびVEGFR−3/Flt−4が含まれ、これらは、胚血管形成および造血に不可欠なシグナルを伝達する。VEGFは3つの受容体全てに結合するが、最大の生物学的機能はVEGFR−2によって媒介され、VEGFR−1の役割は現在は不明である。VEGFR3/Flt4によるシグナル伝達は、リンパ腺の内皮細胞の発達に重要であることが知られており、VEGFR3によるシグナル伝達は、内皮細胞にリンパ球内皮細胞様表現形を付与し得る。VEGFRは、血管の増殖の刺激、血管緊張低下、血管透過性の誘導、内皮細胞の移動、増殖、および生存に不可欠なプロセスについてのシグナルをリレーする。内皮細胞は全ての異なるVEGF−Rを発現する。胚発生の間には、1つの前駆細胞である、血管芽細胞が造血系と脈管系の両方を生じ得ることが報告されている。
【0021】
Tie−2は内皮特異的受容体チロシンキナーゼであり、アンギオポエチン1の受容体である。これはI型膜タンパク質であり、活発に増殖している血管の内皮において主に発現され、最も早く発見された哺乳動物の内皮細胞系統のマーカーであり得る。Tie−2はおそらく、内皮細胞の増殖と分化の調節に関係しており、血管の形成の間に内皮細胞の特別な方向付けを指示し得る。
【0022】
CD14抗原は、リポ多糖(LPS)とLPS結合タンパク質(LBP)の複合体についての高親和性受容体である。CD14抗原は、CD14、TLR4、およびMD−2から構成される機能性のヘテロマーであるLPS受容体複合体の一部である。CD14は、末梢血の中のほとんどのヒトの単球およびマクロファージ上、他の体液および種々の組織、例えば、リンパ節および脾臓の上で強く発現される。CD14は、ヒト好中球および骨髄樹状細胞の亜集団上では、おそらく、弱く発現される。
【0023】
CD45抗原はチロシンホスファターゼであり、これは白血球共通抗原(LCA)としても知られている。CD45は、赤血球系細胞、血小板、およびそれらの前駆細胞を除く、造血系を起源とする全てのヒト細胞上に存在する。CD45分子にはT細胞とB細胞の活性化が必要であり、これは、細胞の活性化状態によって少なくとも5つのイソ型で発現される。
【0024】
CD144はまた、カドヘリン5またはVE−カドヘリンとも呼ばれ、細胞接着分子のカドへリンファミリーに属する140kDaのタンパク質である。CD144抗原は内皮細胞に特異的であり、そして、内皮組織内の接合部位の細胞間の裂隙部位に存在する。カドヘリン5分子は、脈管内皮の透過特性において役割を果たすことができる。
【0025】
血液は、任意の動物、例えば、魚類、爬虫類、鳥類、両生類、および哺乳動物、例えば、好ましくは、齧歯類、および例えば、マウスおよびヒトから得ることができる。
【0026】
VEGFR−1+、VEGFR−2+、およびTie−2+細胞は、それぞれ、血液中の単核細胞の全集団のおよそ3.0±0.2%、0.8±0.5%、2.0±0.3%を占める。CD14+/VEGFR−2+細胞は、単球の全集団のおよそ2.0±0.5%を占め、そして血液中の単核細胞の0.08±0.04%を占める。
【0027】
EPCは、長期培養物中でインビトロで維持することができる。EPCは、2回、3回、4回、5回、6回、7回、8回、9回、10回、11回、12回、またはそれ以上、培養物中で継代培養することができる。
【0028】
(内皮前駆細胞の単離)
本発明は、内皮前駆細胞または平滑筋前駆細胞の集団を単離する方法に関する。細胞の集団は、ポジティブ選択の手段によって、またはいずれかの順序でのポジティブ選択とネガティブ選択の両方の混合によって単離される。前駆細胞の集団が精製される。精製されたEPCの集団には、それから細胞が単離された細胞の粗集団よりも明らかに多い割合のEPCの集団が含まれる。
【0029】
例えば、精製手順によって、集団全体に対してEPCは、少なくとも5倍の増加、好ましくは少なくとも10倍の増加、より好ましくは少なくとも15倍の増加、最も好ましくは少なくとも20倍の増加が、そして状況によっては、少なくとも25倍の増加が導かれるはずである。精製されたEPCの集団には、少なくとも15%、好ましくは少なくとも20%、より好ましくは少なくとも25%、最も好ましくは少なくとも35%、そして最適には少なくとも50%のEPCが含まれるはずである。
【0030】
本明細書中に記載される方法によっては、75%まで、好ましくは80%まで、より好ましくは85%まで、最も好ましくは90%まで、そして最適には95%までを幹細胞が占める混合物を導くことができる。このような方法は、99%、99.90%、そしてさらには100%をEPCが占める混合物を生じることができる。したがって、本発明の精製された集団には、上記のような自然界に存在している集団よりも有意に高いレベルのEPCが含まれる。
【0031】
精製されたEPCの集団は、EPCに特徴的な抗原を発現する幹細胞の集団を含む細胞の粗混合物を、抗原の細胞外部分に特異的に結合する分子と接触させることによって単離される。このような技術は、ポジティブ選択として知られている。EPCのこれらの分子への結合によって、抗原を発現しない混入している細胞とEPCを十分に識別することができ、混入している細胞から幹細胞を単離することができる。抗原はVEGFRであることが好ましく、より好ましくはVEGFR−2である。
【0032】
混入している細胞から前駆細胞を分離するために使用される分子は、EPCに特徴的な抗原に特異的に結合する任意の分子であり得る。分子は、例えば、モノクローナル抗体、モノクローナル抗体の断片、または受容体である抗原の場合には、受容体のリガンドであり得る。例えば、VEGF受容体(例えば、FLK−1)の場合には、リガンドはVEGFである。
【0033】
本発明の特有の単離された細胞は、それらのCD45+状態と、血管内皮成長因子受容体(VEGFR)(例えば、VEGFR−2)の保有によって他の細胞から分離される。細胞は、Civin,米国特許第4,714,680号、同第4,965,204号、同第5,035,994号、および同第5,130,144号、Tsukamotoら、米国特許第5,750,397号、およびLokenら米国特許第5,137,809号(これらの全ては、それらの全体が参考として本明細書中に援用される)に記載されている技術のような、細胞を分離するための従来技術によって単離することができる。したがって、例えば、CD45特異的モノクローナル抗体またはVEGFR特異的抗体を、ニトロセルロース、アガロースビーズ、ポリスチレンビーズ、中空線維膜、磁気ビーズ、およびプラスチック製のペトリ皿のような固体支持体上に固定することができる。その後、細胞集団全体が固体支持体の上を通過させられるか、またはビーズに添加される。
【0034】
結合分子に結合した細胞は、残っている細胞懸濁液から固体支持体を物理的に分離することによって、細胞懸濁液から取り出される。例えば、結合していない細胞は、幹細胞を固体支持体に結合させるために十分な時間の後、生理学的緩衝液で溶出させることも、また洗浄することもできる。
【0035】
結合した細胞は、固相および結合分子の性質に主に依存して、任意の適切な方法によって固相から分離される。例えば、結合した細胞は激しい攪拌によってプラスチック製のペトリ皿から溶出させられ得る。あるいは、結合した細胞は、酵素による「ニッキング」または固相と抗体との間の酵素に敏感な「スペーサー」配列の消化によって溶出させることができる。アガロースビーズに結合させられた適切なスペーサー配列は、例えば、Pharmaciaから市販されている。
【0036】
溶出された富化させられた細胞の画分は、その後、遠心分離によって緩衝液で洗浄され、従来技術にしたがって後で使用されるまで低温で生存している状態で保存される。細胞はまた、例えば、レシピエントに静脈内注射されることによって、すぐに使用される場合もある。
【0037】
固相支持体に付着したままである細胞は、使用される抗体によって認識されるマーカーを含む細胞である。したがって、抗CD45抗体が使用されると、その後、得られる集団はCD45+細胞について大幅に富化させられる。使用される抗体がVFGFRである場合には、得られる集団はVFGFR+細胞が大幅に富化させられる。この集団は、その後、他のマーカーに対する抗体がそれに付着している固相を使用する工程を繰り返し行うことによって、他のマーカーについて富化させることができる。
【0038】
CD45+VEGFR+細胞を選別するための別の方法は、フローサイトメトリーによる方法であり、Becton−Dickinsonによって製品名FACScanまたはFACSCaliburとして製造されている選別装置のような蛍光活性化細胞選別装置(FACS)による方法が最も好ましい。この技術によって、その上に抗CD45抗体を有している細胞が、そのような色素に結合させられた抗CD45抗体によって特定の蛍光色素でタグ化される。同様に、細胞のVEGFRマーカーは、他の色素に結合させられた抗VEGFR抗体によって別の蛍光色素でタグ化される。染色された細胞が機器の上に置かれると、細胞の流れがアルゴンレーザー光線によって方向付けられ、蛍光色素が励起されて光を発する。この発光は、光学フィルターのセットによって蛍光色素の発光波長に特異的な光電子倍増管(PMT)によって検出される。PMTによって検出されたシグナルは、その自身のチャネルにおいて増幅され、種々の異なる形態(例えば、柱状グラフ、点表示、または曲線表示)でコンピューターによって表示される。したがって、1つの波長で発光する蛍光細胞は、特異的な蛍光色素標識された試薬と反応する分子であり、一方、非蛍光細胞または異なる波長で発光する蛍光細胞はこの分子を発現しないが、他の波長で蛍光を発する蛍光色素標識された試薬と反応する分子を発現し得る。フローサイトメーターはまた、細胞によって発現される蛍光の量(蛍光強度)を表示する点において半定量的でもある。これは、比較の意味では、細胞によって発現される分子の数に関係する。
【0039】
フローサイトメーターはまた、非蛍光パラメーター、例えば、細胞容量、または、それがレーザー光線を通過する際に細胞によって散乱させられた光を測定するために、取り付けられる。細胞容量は、通常、直接測定される。光散乱PMTによって、前角(前方散乱;FSC)または直角(側方散乱;SSC)のいずれかで細胞によって散乱させられた光が検出される。FSCは、通常は大きさの指標であり、一方、SSCは、細胞の複雑さの指標であるが、いずれのパラメーターも他の要因による影響を受け得る。
【0040】
好ましくは、フローサイトメーターには、1つ以上のPMT発光検出器が取り付けられる。さらなるPMTによって、他の発光波長を検出することができ、1つ以上の蛍光色素を同時に、個々の別々のチャネルにおいてそれぞれ検出することができる。コンピューターによりそれぞれのチャネル、または個々のパラメーターと別のパラメーターとの相関関係を分析することができる。通常、FACS機器と共に使用される蛍光色素としては、フルオレセインイソチオシアネート(FITC)(これは、525nmに発光ピークを有している)(緑)、R−フィコエリスリン(PE)(これは、575nmに発光ピークを有している)(赤−オレンジ)、ヨウ化プロピジウム(PI)(これは、620nmに発光ピークを有している)(赤)、7−アミノアクチノマイシンD(7−AAD)(これは660nmに発光ピークを有している)(赤)、R−フィコエリスリンCy5(RPE−Cy5)(これは、670nmに発光ピークを有している)(赤)、およびアロフィコシアニン(APC)(これは、655〜750nmに発光ピークを有している)(深紅)が挙げられる。
【0041】
これらおよび他のタイプのFACS機器は、異なる特性の細胞を異なる容器に屈折させることによって、種々の画分を物理的に分離するさらなる能力を有し得る。
【0042】
出発物質、例えば、骨髄、末梢血、または臍帯血のCD45+VEGFR+集団を単離するための任意の他の方法もまた、本発明にしたがって使用することができる。本発明の種々の亜集団(例えば、CD14+、Tie2+、CD144−)を同様の様式で単離することができる。
【0043】
粗細胞集団が上記のように精製される前またはその後のいずれかに、前駆細胞の集団が当該分野で公知の方法によってさらに濃縮される場合がある。例えば、前駆細胞は、EPCに特徴的な1つ以上の抗原についてのポジティブ選択によって富化させることができる。このような抗原としては、例えば、CD14またはTie−2が挙げられる。
【0044】
上記の方法の特に好ましいバリエーションにおいては、血液がドナーの循環している末梢血から直接採血される。血液は、固相に結合させた結合分子(例えば、EPCを捕捉するための抗体VEGFR−2)を含むカラム全体に連続的に浸透させられる。前駆細胞が除去された血液は、アフェレーシス療法のような当該分野で公知の方法によって、すぐにドナーの循環系に戻される。血液は、十分な数の前駆細胞がカラムに結合するまでこの方法で処理される。その後、幹細胞が当該分野で公知の方法によってカラムから単離される。この方法により、数少ない末梢血前駆細胞をきわめて多量の血液から回収することができ、ドナーの費用がかからず、骨髄の採取の痛みもなく、そしてそれに伴う麻酔、無痛覚症、輸血、および感染のリスクもない。
【0045】
(培養条件)
EPCは、本明細書中に記載される方法を使用して培養され、増殖させられる。細胞は、密度勾配遠心分離によって末梢血単核細胞(PBMC)を単離することによって末梢血から得られる。
【0046】
細胞懸濁液は、細胞を維持することができる任意の容器、特に、培養フラスコ、培養プレート、またはローラーボトル、そして具体的には、25cm2の培養フラスコのような小さい培養フラスコに播種される。懸濁液の中で培養された細胞は、およそ5×104から2×105細胞/ml(例えば、1×105細胞/ml)で再懸濁される。固定された担体の上にプレートされる細胞は、およそ2〜3×10310細胞/cm2でプレートされる。状況に応じて、培養プレートは、コラーゲンのようなマトリックスタンパク質でコーティングされる。細胞は、細胞の代謝に必要な補助物質(例えば、グルタミンおよび他のアミノ酸、ビタミン、ミネラル、およびトランスフェリンのようなタンパク質など)を含む細胞増殖をサポートすることができる任意の公知の培養培地(HEM、DMEM、RPMI、F−12などを含む)に入れることができる。培養培地にはまた、酵母、細菌、および真菌の混入を防ぐための抗生物質(例えば、ペニシリン、ストレプトマイシン、ゲンタマイシンなど)を含めることもできる。培養培地には、ウシ、ウマ、ニワトリなどに由来する血清が含まれる場合がある。
【0047】
培養のための条件は、生理学的条件に近づけるべきである。培養培地のpHは、生理学的pH(例えば、pH6〜8の間、約pH7から7.8の間、またはpH7.4)に近づけるべきである。生理学的温度は、約30℃から40℃の間の範囲である。EPCは、約32℃から約38℃の間の温度(例えば、約35℃から約37℃)で培養される。
【0048】
状況によっては、培養培地には、少なくとも1種の増殖を誘導する(「細胞分裂促進」増殖因子が補充される。「増殖因子」は、EPCに対して成長効果、増殖を誘導する効果、分化を誘導する効果、または栄養効果を有している、タンパク質、ペプチド、または他の分子である。「増殖を誘導する成長因子」は、EPCを増殖させる栄養素であり、これには、細胞に対して栄養効果または増殖誘導効果を発揮するために細胞の表面上の受容体に結合する任意の分子が含まれる。増殖を誘導する成長因子としては、EGF、アンフィレグリン、酸性線維芽細胞増殖因子(aFGFまたはFGF−1)、塩基性線維芽細胞増殖因子(bFGFまたはFGF−2)、形質転換増殖因子α(TGFα)、VEGFおよびそれらの組み合わせが挙げられる。増殖因子は、通常、約1fg/mlから1mg/mlの範囲の濃度で培養培地に添加される。約1から100ng/mlの間の濃度で、通常は十分である。試料滴定アッセイは、特定の増殖因子の最適濃度を決定するために容易に行うことができる。
【0049】
成長因子および栄養素の生物学的効果は、一般的には、細胞表面受容体に対する結合によって媒介される。多数のこれらの因子の受容体が同定されており、特異的受容体についての抗体および分子プローブを利用することができる。EPCは、分化の全ての段階で増殖因子受容体の存在について分析することができる。多くの場合には、特定の受容体の同定によって、外来の増殖因子または栄養素の添加を伴う特異的な発達経路に沿って細胞をさらに分化させることにおいて使用するためのストラテジーについての指針が提供される。
【0050】
一般的には、インビトロでは約3〜10日後に、EPCの培養培地が、培地を吸引し、培養フラスコに新しい培地を添加することによって再び補給される。状況によっては、吸引された培地が回収され、濾過され、続けてEPCを継代培養するための馴化培地として使用される。例えば、10%、20%、30%、40%、またはそれ以上の馴化培地が使用される。
【0051】
EPC細胞培養物は、再度増殖を開始するように容易に継代培養することができる。例えば、インビトロでは3〜7日後に、培養フラスコが十分に揺らされ、その後、EPCは50mlの遠心分離管に移され、低速で遠心分離される。培地は吸引され、EPCは少量の培養培地中に再懸濁される。その後、細胞は数えられ、所望される密度で再度プレートされて、増殖が再開させられる。この手順を毎週繰り返すことができ、その結果、それぞれの継代においては、生存している細胞の数は対数的に増加する。この手順は、所望される数のEPCが得られるまで続けて行うことができる。
【0052】
EPCおよびEPCの子孫は、それらが必要になるまで、任意の当該分野で公知の方法によって低温保存することができる(例えば、米国特許第5,071,741号、PCT国際公開第93/14191号、同第95/07611号、同第96/27287号、同第96/29862号、および同第98/14058号、Karlssonら、65 Biophysical J.2524−2536(1993)を参照のこと)。EPCは、特定の低温保存剤を含む等張溶液、好ましくは、細胞培養培地の中に懸濁させることができる。このような低温保存剤としては、ジメチルスルフォキシド(DMSO)、グリセロールなどが挙げられる。これらの低温保存剤は、5〜15%(例えば、8〜10%)の濃度で使用される。細胞は、−10℃から−150℃(例えば、−20℃から−100℃、または−70℃から−80℃)の温度に徐々に凍結させられる。
【0053】
(内皮前駆細胞の分化)
培養条件に応じて、EPCは内皮細胞または平滑筋細胞へと分化させることができる。
【0054】
EPCは、分化を誘導する成長因子を含む培養培地中の固定された担体上で、内皮細胞または平滑筋細胞EPCへと分化させることができる。EPCの分化はまた、増殖を誘導する生物学的事象のカスケードを活性化させる当該分野で公知の任意の方法によっても誘導することができ、これにはイノシトール三リン酸および細胞内Ca2+の遊離、ジアシルグリセロールの遊離、ならびに、プロテインキナーゼCおよび他の細胞キナーゼの活性化などが含まれる。ホルボールエステル、分化を誘導する成長因子、および他の化学的シグナルでの処理によって分化を誘導することができる。EPCの増殖のための増殖を誘導する成長因子(上記を参照のこと)の代わりに、分化を誘導する成長因子を、EPCの分化に影響を与えるために培養培地に添加することができる。他の分化を誘導する成長因子としては、血小板由来成長因子(PDGF)、チロトロピン放出ホルモン(TRH)、トランスフォーミング成長因子β(TGFβ)、インシュリン様成長因子(IGF−1)などが挙げられる。
【0055】
分化した内皮細胞または平滑筋細胞は、当該分野で公知の免疫細胞化学的技術を使用して検出される。免疫細胞化学(例えば、二重標識免疫蛍光および免疫ペルオキシダーゼ法)では、内皮細胞または平滑筋細胞の、細胞の特徴または表現形の特性を区別するために、細胞タンパク質を検出する抗体が使用される。内皮細胞についての細胞マーカーとしては、例えば、VE−カドヘリン、CD144、CD141、CD106、またはCD142が挙げられ、一方、平滑筋細胞についての細胞マーカーとしては、Flkが挙げられる。免疫細胞化学はまた、CD31およびe−NOSのような内皮細胞遺伝子の発現を検出することによって内皮細胞を同定するために使用することもできる。
【0056】
インサイチュハイブリダイゼーション組織化学もまた、内皮遺伝子のmRNAに特異的なcDNAまたはRNAプローブを使用して行うことができる。これらの技術は、特異的表現形の同定を促進するために、免疫細胞化学的方法と組み合わせることができる。必要であれば、上記で議論された抗体および分子プローブを、細胞の同定を助けるために、それぞれ、ウェスタンブロットおよびノーザンブロット手順に適応することができる。
【0057】
(内皮前駆細胞の移植)
損傷した血管への新しい細胞の移植には、損傷した血管組織、例えば、静脈、動脈、毛細血管を修復する能力があり、それによって血管機能を回復させることができる。しかし、移植の目的に適している細胞がないことによって、それによってもたらされるこの手順の全ての可能性が妨げられる。「適している」細胞は、以下の基準の1つ以上を満たす細胞である:(1)多数を得ることができる;(2)必要に応じて、遺伝的物質を挿入するためにインビトロで増殖させることができる;(3)永久に生存することができ、移植されると、血管の修復を促進することができる;ならびに(4)免疫原性ではない、好ましくは、患者自身の組織から、または適合するドナーから得られたものである。血管障害の処置におけるEPCの使用は、動物モデルを使用して実証することができる。
【0058】
EPCは、異常な血管または冠不全の兆候を有している任意の動物に投与される。EPCは、宿主に対して異種であるドナー組織から調製することができる。異種移植を成功させるためには、移植された組織に対する免疫応答を軽減させるかまたは排除するいくつかの方法が通常使用される。したがって、EPCレシピエントは、シクロスポリンのような免疫抑制剤の使用によって、または局所投与される免疫抑制剤を使用する局所免疫抑制ストラテジーによってのいずれかで、免疫抑制することができる。局所免疫抑制は、Gruber,54 Transplantation 1−11(1992)に開示されている。米国特許第5,026,365号には、局所免疫抑制に適しているカプセル化方法が開示されている。
【0059】
免疫抑制技術を使用することに代わるものとしては、Smithiesら、317 Nature 230−234(1985)によって教示されている胚性幹細胞における相同組み換えを使用した遺伝子の置き換えまたはノックアウトの方法と、細胞株での遺伝子の置き換えまたはノックアウトにまで拡大された方法(Zhengら、88 Proc.Natl.Acad.Sci.8067−8071(1991))を、主要組織適合複合体(MHC)遺伝子の除去のためにEPCに行うことができる。MHCの発現が欠失しているEPCは、同種の、そしておそらくは、異種である組織適合性バリアをも超えて、レシピエントを免疫抑制する必要なく、富化させられた内皮細胞の集団を移植することができる。ドナー細胞の抗原性を低下させるための組み換え方法の使用についての一般的な概要および基準もまた、Gruber,54 Transplantation 1−11(1992)によって開示されている。表面の修飾により移植片の免疫原性を低下させる例示的なアプローチは、PCT国際公開第92/04033号およびPCT/99/24630に開示されている。あるいは、移植片の免疫原性は、MHC抗原が変化させられているかまたは欠失させられているトランスジェニック動物からEPCを調製することによって低下させることもできる。
【0060】
EPCは、マイクロカプセル化(例えば、参考として本明細書中に援用される米国特許第4,352,883号;同第4,353,888号;および同第5,084,350号を参照のこと)およびマイクロカプセル化(例えば、それぞれが参考として本明細書中に援用される米国特許第5,284,761号、同第5,158,881号、同第4,976,859号、および同第4,968,733号、ならびにPCT国際公開第92/19195号および同第95/05452号を参照のこと)を含む、公知のカプセル化技術によってカプセル化することができ、宿主に因子を送達するために使用することができる。マイクロカプセル化は、米国特許第5,284,761号;同第5,158,881号;同第4,976,859号;同第4,968,733号;同第5,800,828号、およびPCT国際公開第95/05452号に記載されており、これらはそれぞれが参考として本明細書中に援用される。デバイスの中の細胞の数は変動し得る;好ましくは、個々のデバイスには103個〜109個の細胞(例えば、105個〜107個の細胞)が含まれる。複数のマイクロカプセル化デバイスを宿主に埋め込むことができる。
【0061】
レシピエントの組織と同種である組織から調製されたEPCは、レシピエントの組織適合性のタイプと厳密に適合させるために、周知の組織分類の方法によって使用について試験される。
【0062】
EPCは、しばしば、レシピエント自身の血液または骨髄から調製することができる。このような場合には、EPCは切り取られた組織から作成することができ、上記の方法を使用してインビトロで増殖させることができる。細胞数が適切に増加させられると、EPCを回収することができ、必要であれば遺伝的に修飾することができ、そしてレシピエントの血管への直接の注射のために準備することができる。
【0063】
EPCは、血管移植片を形成することができる血管に投与され、その結果、細胞は近隣の血管細胞とともに正常な結合を形成し、移植された内皮細胞または既存の内皮細胞との接触を維持することができる。このように、移植されたEPCは、疾患および加齢が原因で損傷した血管組織を再度構築させる。
【0064】
宿主の血管組織への移植片の機能的な一体化は、種々の機能を回復させることに対する移植片の有効性を試験することによって評価することができる。
【0065】
移植に使用されるEPCをインビトロで増殖させる能力はまた、エキソビボでの遺伝子治療にも有効である。したがって、EPCによって、エキソビボでの遺伝子治療試験においてビヒクルとして使用される内皮細胞を取り出し、増殖させる別の方法が提供される。
【0066】
(内皮前駆細胞の遺伝的修飾)
EPCは形質転換されていない初代細胞であるが、これらは、連続細胞株の特徴を有している。未分化の状態では、EPCは分裂し続け、したがって、遺伝的修飾の標的である。いくつかの実施形態においては、遺伝的に修飾された細胞は、上記の方法のいずれかによって内皮細胞または平滑筋細胞に分化するように誘導される。
【0067】
用語「遺伝的修飾」は、外来DNAの意図的な導入によるEPCの遺伝子型の安定な、または一時的な変更を意味する。DNAは合成のものである場合も、また自然界から導かれたものである場合もあり、そしてこれには、遺伝子、遺伝子の一部、または他の有用なDNA配列が含まれ得る。用語「遺伝的修飾」は、本明細書中で使用される場合は、自然界でのウイルス活性、自然界での遺伝子組み換えなどによって生じるような、自然に生じる変更が含まれるようには意味されない。
【0068】
細胞の任意の有用な遺伝的修飾は、本発明の範囲内である。例えば、EPCは、成長因子などの生物学的活性のある物質を生成するように、またはそれらの生成を増加させるように修飾することができる。1つの実施形態においては、生物学的活性のある物質は、遺伝的差異を調節する転写因子のような転写因子である。別の実施形態においては、生物学的活性のある物質は、非細胞分裂促進増殖因子であり、例えば、v−myc、SV−40ラージT、またはテロメラーゼである。
【0069】
遺伝的修飾は、ウイルスベクター(レトロウイルス、修飾されたヘルペスウイルス、へルペスウイルス、アデノウイルス、アデノ随伴ウイルスなど)での感染、または当該分野で公知の方法を使用するトランスフェクション(リポフェクション、リン酸カルシウムトランスフェクション、DEAE−デキストラン、エレクトロポレーションなど)のいずれかによって行われる(Maniatisら、Molecular Cloning:A Laboratory Manual(Cold Spring Harbor Laboratory,N.Y.,1982)を参照のこと)。例えば、キメラ遺伝子構築物には、ウイルス、例えば、レトロウイルス長末端反復(LTR)、シミアンウイルス40(SV40)、サイトメガロウイルス(CMV);または哺乳動物細胞特異的プロモーター(例えば、チロシンヒドロキシラーゼ(TH、ドーパミン細胞のマーカー)、DBH、フェニルエタノールアミンN−メチルトランスフェラーゼ(PNMT)、ChAT、GFAP、NSE、NFタンパク質(NE−L、NF−M、NF−Hなど)(これらは、所望されるタンパク質をコードする構造遺伝子の発現を指示する))が含まれ得る。さらに、ベクターには、薬物選択マーカー、例えば、E.coliアミノグリコシドホスホトランスフェラーゼ遺伝子を含めることができる。これは、試験遺伝子と同時に感染させられると、タンパク質合成の阻害因子であるゲネチシン(G418)に対する耐性を付与する。
【0070】
EPCは、発現ベクターでのトランスフェクションを使用して遺伝的に修飾することができる。1つのプロトコールにおいては、遺伝子を含むベクターDNAは、0.1×TE(1mMのTris、pH8.0、0.1mMのEDTA)の中に、40μg/mlの濃度になるように稀釈される。22μlのDNAが、使い捨ての滅菌の5mlのプラスチックチューブの中の250μlの2×HBS(280mMのNaCl、10mMのKCl、1.5mMのNa2HPO4、12mMのデキストロース、50mMのHEPES)に添加される。31μlの2MのCaCl2がゆっくりと添加され、混合液は室温で30分間(min)インキュベートされる。この30分間のインキュベーションの間に、細胞は4℃で5分間、800gで遠心分離される。細胞は、20倍容量の氷冷されたPBSに再懸濁させられ、1×107個の細胞のアリコートに分けられる。これは再び遠心分離される。細胞の個々のアリコートは、1mlのDNA−CaCl2懸濁液に再懸濁させられ、室温で20分間インキュベートされる。その後、細胞は増殖培地の中に稀釈され、5%〜7%のCO2の中で37℃で6〜24時間インキュベートされる。細胞は再び遠心分離され、PBS中で洗浄され、そして10mlの増殖培地に48時間戻される。
【0071】
EPCはまた、リン酸カルシウムトランスフェクション技術を使用して遺伝的に修飾される。標準的なリン酸カルシウムトランスフェクションについては、細胞が機械的に分離されて単細胞懸濁液とされ、50%の細胞集密度(50,000〜75,000細胞/cm2)で組織培養物処理皿にプレートされ、一晩付着させられる。1つのプロトコールにおいては、修飾されたリン酸カルシウムトランスフェクション手順が以下のように行われる:滅菌のTE緩衝液(10mMのTris、0.25mMのEDTA、pH7.5)中のDNA(15〜25μg)がTEで440μLに稀釈され、60μLの2MのCaCl2(1MのHEPES緩衝液でpHが5.8にされたもの)がDNA/TE緩衝液に添加される。全部で500μLの2×HeBS(HEPES緩衝生理食塩水;275mMのNaCl、10mMのKCl、1.4mMのNa2HPO4、12mMのデキストロース、40mMのHEPES緩衝液粉末、pH6.92)が、この混合物に一滴ずつ添加される。混合物は、室温で20分間静置される、細胞は、1×HeBSで軽く洗浄され、1mlのリン酸カルシウム沈殿させられたDNA溶液がそれぞれのプレートに添加され、細胞が37℃で20分間インキュベートされる。このインキュベーション後、10mlの培地が細胞に添加され、プレートはインキュベーター(37℃、9.5%のCO2)の中にさらに3〜6時間入れられる。DNAと培地は、インキュベーション時間が終わると吸引によって除去され、細胞が3回洗浄され、その後、インキュベーターに戻される。
【0072】
遺伝的修飾が生物学的活性のある物質の生成のためである場合には、この物質は、所定の血管障害の処置に有用である物質であり得る。EPCは、生物学的活性のある物質、例えば、成長因子、成長因子受容体を発現するように遺伝的に修飾される。例えば、細胞が増殖を誘導する成長因子または分化を誘導する成長因子を分泌するように細胞を遺伝的に修飾することが所望される場合がある。
【0073】
遺伝的に修飾されたEPCは、細胞治療または遺伝子治療のために、遺伝的修飾された細胞によって生成される生物学的活性のある分子が必要であるレシピエントのCNSに移植することができる。移植技術は以下に詳細に記載される。
【0074】
あるいは、遺伝的修飾されたEPCは、移植の前にインビトロで種々の分化プロトコールに供することができる。一旦細胞が分化させられると、これらは再び、所望されるタンパク質の発現についてアッセイされる。所望される表現形を有している細胞を単離することができ、これを、遺伝的修飾された細胞によって発現されるタンパク質または生物学的活性のある分子が必要であるレシピエントに移植することができる。
【0075】
本発明は、以下の実施例によってさらに説明されるが、これらに限定はされない。
【実施例】
【0076】
(実施例1)
(一般的方法)
(末梢血からのVEGFR−1+細胞、VEGFR−2+細胞、およびTie−2+細胞の単離)
VEGFR−1、VEGFR−2、またはTie−2のいずれかを発現する末梢血単核細胞(PBMC)を、ヒトの血液軟膜のFicoll密度勾配遠心分離を使用して単核細胞を単離することによって最初に得た。PBMCをいくつかのチューブに分けて入れ、それぞれが40×106個の細胞を含むようにした。15μlの抗VEGFR−1抗体(4μg/ml)、抗VEGFR−2抗体(20μg/ml)、または抗Tie−2抗体(5μg/ml)(抗体の詳細については表1を参照のこと)をそれぞれのチューブに添加し、室温で30分間インキュベートした。リン酸緩衝生理食塩水(PBS)で1回洗浄した後、細胞を10μlの1:10稀釈した二次ヤギ抗マウスモノクローナル抗体で標識した。細胞を、ストリンジェントなゲートを使用してフローサイトメトリーによって選別して、高度に精製されたこれらの細胞の集団を得た。
【0077】
(末梢血からのCD14+/VEGFR−2+細胞およびCD14+/VEGFR−2−細胞の単離)
末梢血単核細胞(PBMC)を、標準的な手順にしたがって、健常なヒトのボランティアの血液からFicollを用いた密度勾配遠心分離によって単離した。CD14+/VEGFR−2+細胞は、蛍光活性化細胞選別方法によって単離した。染色は以下のように行った:PBMCを、最初に、抗CD3抗体でコーティングされた磁気粒子(DYNAL,ノルウェー)とともにインキュベートして、Tリンパ球を除去した。この手順は製造業者によって記載されているように行った。残りの細胞をいくつかのチューブに分けて入れ、それぞれが20×106個の細胞を含むようにした。15μlの抗VEGFR−2抗体(20μg/ml、Reliatech,ドイツ)をそれぞれのチューブに添加し、室温で30分間インキュベートした。リン酸緩衝生理食塩水(PBS)で1回洗浄した後、細胞を、20μlの1:10稀釈した二次FITC結合ヤギ抗マウスモノクローナル抗体(Serotec,スウェーデン)と20μlのPE結合抗CD14抗体(4μg/ml、BD Biosciences,米国)で標識した。ストリンジェントなゲートを使用した細胞の選別の間に、高度に精製されたCD14+/VEGFR−2+画分、ならびに、CD14+/VEGFR−2−画分を、さらなる分析のために回収した。
【0078】
(ウェスタンブロッティング)
CD14+/VEGFR−2+細胞とCD14+/VEGFR−2−細胞(それぞれの集団について1×106個の細胞)の溶解物をSDS−PAGEによって分離させ、ニトロセルロース膜に移し、そして抗VEGFR−2抗体(1:200)で免疫ブロットした。西洋ワサビペルオキシダーゼに結合させたヤギ抗マウス二次抗体のF(ab’)2断片(1:2000)(Jackson Immunoresearch,米国)を、結合した抗体を同定するためにECLキット(Amersham)とともに使用した。
【0079】
(フローサイトメトリー、表現形分類、および細胞培養)
内皮前駆細胞の表現形分類は、造血細胞および内皮細胞上で発現される特異的マーカーに対する抗体のアレイを使用して、以前に記載された15ように行った(表1)。抗VEGFR−2はReliaTech(ドイツ)から入手し;抗VEGFR−1はR&D(英国)から;抗AcLDLはMolecular Probes(米国)から;抗フォン・ヴィレブランド因子−HLA、抗CD142、および抗HLAクラスIはDAKO(デンマーク)から;抗α−アクチンおよび抗線維芽細胞抗体はSerotec(スウェーデン)から;抗CD144、抗CD141、ヒト核抗体はChemicon(スウェーデン)から、そしてUlex europaeusはSigma(スウェーデン)から入手した。全ての他の抗体は、Becton Dickinson(米国)から購入した。特定のマーカーについては、細胞内染色をリン酸緩衝生理食塩水(PBS)中の5%のサポニンを使用して細胞の透過性を上昇させた後に行った。対応するコントロールのイソ型を、モノクローナル抗体(Mab)の非特異的結合の評価のために使用した。細胞は、Becton Dickinsonフローサイトメーター(FACSorter)上で分析した。選別したVEGFR−1+細胞、VEGFR−2+細胞、およびTie−2細胞を、フィブロネクチンでコーティングした組織培養プレートの上で、5mMのL−グルタミンと100μg/mlのペニシリン/ストレプトマイシン(PEST)を含む内皮選択培地MCDB 131(GIBCO,Gaithersburg,MD,米国)中で培養した。培地には、さらに、内皮細胞成長培地(EGM−2)シングルコート(singlequots)(Clonetics,Bio Whittaker,米国)を補充した。ヒトの臍帯静脈内皮細胞(HUVEC)はCloneticsから購入し、推奨される、EGM−2−MVシングルコートを補充したEGM−2−MV培地中で培養した。HUVECは、表現形分析および毛細血管形成アッセイにおいてコントロールとして使用した。
【0080】
FACSで選別した細胞をサイトスピンを使用してガラススライド上に置いた。免疫細胞化学を酵素染色によって行った。細胞を以下の抗体:抗CD144(VE−カドヘリン)、抗CD141(トロンボモジュリン)(いずれも、Beckton Dickinson,米国による)、抗VEGFR−1(R&D Systems,英国)、および内皮一酸化窒素合成酵素(e−NOS)に対する抗体(全ての抗体を1:50希釈で使用した)で室温で1時間染色し、PBSで3回洗浄し、二次ビオチニル化ヤギ抗マウス抗体(1:500)で染色した。アビジン−ペルオキシダーゼ手順を、Vectastain Elite ABCキット(ImmunKemi,Stockholm,スウェーデン)を使用して、製造業者によって記載されているように行った。ACE(赤色の染色を生じる)またはDABNickel(茶/黒色の染色を生じる)基質キットを発色現像液として使用した。多数の細胞がCD14+/VEGFR−2−集団から得られたので、これらの細胞をさらに、先に記載された16ようにフローサイトメトリーによって、単球および内皮細胞上で発現される特異的マーカーに対する抗体のアレイを使用して特性決定した(表3)。FITC結合アセチル化低密度リポタンパク質(AcLDL)(Molecular Probes,米国)、フォン・ヴィレブランド因子に対する抗体(DAKOPATTS,デンマーク)、α−アクチン、線維芽細胞(Serotec,スウェーデン)、およびUlex europaeus(Sigma,スウェーデン)を使用した。全ての他の抗体は、Becton Dickinson(米国)から購入した。対応するコントロールのイソ型を、モノクローナル抗体(Mab)の非特異的結合の評価のために使用した。細胞をBecton Dickinsonフローサイトメーター(FACSorter)上で分析した。
【0081】
いくつかの実験については、選別したCD14+/VEGFR−2+細胞とCD14+/VEGFR−2−細胞をフィブロネクチン(20μg/ml)をコーティングした組織培養プレート上で、内皮選択培地EndoCultTM(Stem Cell Technologies,カナダ)の中で培養した。これらの培養物を、増殖および形態について観察した。1週間の培養後、細胞をガラス上でサイトスピンし、VE−カドヘリンの発現について染色した。
【0082】
ヒトの大動脈内皮細胞(HAEC)はCloneticsから購入し、推奨される培地の中で培養した。HAECを、表現形の分析と細管形成アッセイにおいてコントロールとして使用した。
【0083】
(透過型電子顕微鏡)
細胞を、24ウェルプレートの中で膜フィルター上で増殖させた。電子顕微鏡分析を、Karolinska University hospital−Huddinge(スウェーデン)で電子顕微鏡についての中心となる設備で行った。CD14+/VEGFR−2+細胞とCD14+/VEGFR−2−細胞を、24ウェルプレート中の膜フィルター上で別々に増殖させた。膜を2%のグルタルアルデヒド中に固定し、蒸留水で軽くリンスし、70%のエタノール中に10分間入れ、そして99.5%のエタノール中に15分間入れ(全て4℃で)、乾燥させた。固定後、膜を切り、0.15Mのカコジル酸ナトリウム、1%の四酸化オスミウム、および3mMのCaCl2(pH7.4)を含む緩衝液中で4℃で1時間固定させた。その後、ウェルを0.15Mのカコジル酸ナトリウム緩衝液中で軽くリンスし、上記のようにエタノールで脱水し、スパー樹脂(Agar Scientific LTD,Essex,英国)に包埋した。切片を、酢酸ウラニル、その後、クエン酸鉛で対比させ、Leo 906(Oberkochen,ドイツ)透過型電子顕微鏡において80kVで試験した。
【0084】
(移動アッセイ)
新しく単離したVEGFR−1+細胞、VEGFR−2+細胞、およびTie−2+細胞(5×104個)を、それぞれの細胞タイプについて、3.0μmの孔径を有している4つのフィブロネクチンでコーティングされたTranswellインサートに添加した。内皮前駆細胞の移動を、5%のウシ胎児血清を含む培地中で、下部チャンバーに血管内皮成長因子(VEGF)を含まない条件、ならびに、0ng/ml、0.5ng/ml、2ng/ml、および10ng/mlの種々の濃度の血管内皮成長因子(VEGF)を含む条件でアッセイした。細胞を22時間移動させた。移動した細胞を回収し、血球計数器において数えた。同様の実験を、アンジオポイエチン−2(Tie−2のリガンド)を、0ng/ml、100ng/ml、200ng/ml、および300ng/mlの濃度で使用して行った。それぞれの実験を3連のウェルで行い、4回繰り返した。
【0085】
新しく単離したCD14+/VEGFR−2+細胞とCD14+/VEGFR−2−細胞(5×104個)を、全て3.0μmの膜孔径を有している4つのフィブロネクチンでコーティングされたTranswellインサートに添加した。細胞の移動を、5%のウシ胎児血清を含む培地中で、下部チャンバーに50ng/mlの血管内皮成長因子(VEGF)または300ng/mlの単球走化性因子−1(MCP−1)を含まない条件、またはそれらを含む条件でアッセイした。細胞を22時間移動させた。移動した細胞を回収し、血球計数器において数えた。それぞれの実験を3連のウェルで行い、4回繰り返した。
【0086】
(CD14+/VEGFR−2+細胞とCD14+/VEGFR−2−細胞の集団による血管由来成長因子の分泌)
成長因子の分泌を評価するために、新しく単離した細胞を、成長因子および血清を含まない培地の中で72時間培養した。全ての培養物から上清を回収し、さらに分析するまで−70℃で保存した。馴化培地を、血管由来成長因子、VEGF、顆粒球コロニー刺激因子(G−CSF)、顆粒球マクロファージコロニー刺激因子(GM−CSF)、線維芽細胞成長因子(FGF)、および間質由来因子−1(SDF−1)についてアッセイした。これらの成長因子は、ELISA(R&D systems)によってアッセイした。
【0087】
(インビトロでの血管形成アッセイ)
毛細管様の細管構造の形成を、原則として先に記載された16ように、マトリゲルでコーティングしたマルチウェルプレートにおいて評価した。新しく単離したVEGFR−1+細胞、VEGFR−2+細胞、およびTie−2+細胞を、マトリゲルでコーティングしたマルチウェルプレート中の増殖培地に30×104個の細胞/mlの密度で播種し、37℃でインキュベートした。細管構造の形成を、Nikon TE3000 microscopeを使用して22時間後に調べた。
【0088】
新しく単離したCD14+/VEGFR−2+細胞とCD14+/VEGFR−2−細胞による毛細管様構造の形成を、インビトロでの血管形成の評価のために頻繁に使用されるEngelbreth−Holm−Swamマウス肉腫から抽出した可溶化基底膜調製物中でアッセイした。24ウェルプレートを200μlのマトリゲル(5%のCO2中で37℃で30分間予めゲル状にした)でコーティングし、細胞を、重合させたマトリックス上に、5×104個の細胞/ウェルの密度で播種した。得られた管様構造を、細胞を5%のCO2中で37℃で1週間培養した後、位相差顕微鏡を使用して調べた。HAECをポジティブコントロール細胞として使用した。
【0089】
(インビトロでの細胞侵襲アッセイ)
再構成させた基底膜マトリゲルからの細胞侵襲を、以前に報告された17方法によってアッセイした。移動した細胞を、×100倍の倍率で光学顕微鏡下で5つの異なる領域において数えた。それぞれの実験を3連のウェルで行い、4回繰り返した。
【0090】
(FACSによって選別したCD14+/VEGFR−2+細胞とCD14+/VEGFR−2−細胞のレンチウイルスでの形質導入)
組み換え体であるレンチウイルスベクターを、先に記載された173プラスミド発現システムを使用して生成した。形質導入のために、FACSで選別した細胞を、1mlの培養培地を含む10mlのチューブに移し、0.1のMOIの濃縮されたpHR’EF1−GFPSINビリオンとともに、5%のCO2雰囲気下で一晩37℃でインキュベートした。ウイルスのインキュベーションの後、細胞をPBS中で1回洗浄し、ヌードマウスの露出させた大腿動脈に直接注入した(下記を参照のこと)。GFPの発現を検出するために、同じ形質導入手順にしたがって、形質導入した細胞を6ウェルプレート中で培養し、形質導入の6日後に蛍光顕微鏡によって観察した。
【0091】
(マウス)
右大腿動脈のバルーン損傷を以下のように行った:体重20〜25gのnude C57blackマウス(Taconic M&B,デンマーク)をイソフルランで麻酔し、腹部横切開によって大動脈分岐部のレベルまで右大腿動脈を露出させた。マイクロクランプ(S&T,スイス)を下部大動脈、左腸骨動脈、および右大腿動脈の遠位部分の上に配置した。2F Fogartyバルーンカテーテル(Baxter)を右大腿動脈に挿入し、膨張させ、回転させながら3回前後に動かした。膨張は、カニューレと、1mlのリンガー溶液によって、微小切開部分から自由に流出させて行った。100μlのMCDB培地中の細胞(5×105細胞/動物)(CD14+/VEGFR−2+、n=10;CD14+/VEGFE−2−、n=10)を同じカニューレから染み込ませ、15〜20分間、新しく損傷させた動脈床の中でインキュベートした。一方、コントロール移植マウスには、培養培地だけを投与した(マウス、n=6)。インキュベーション後、結合していない細胞を吸引し、カテーテルを抜き取り、微小切開を11−0ナイロン(S&T,スイス)結節縫合によって縫合した。マイクロクランプを取りはずすことによって血流を回復させた。全ての動物への手順は、機関のガイドライン、スウェーデンのHuddinge University hospitalの実験動物の管理と使用に関する指針(Guide for the Care and Use of Laboratory Animals)に則って行った。動物を、ヒトの細胞の移植の2週間後および4週間後に屠殺し、右大腿動脈を組織病理学的試験のために回収した。血管をO.C.T.に包埋し、液体窒素で凍結させた。
【0092】
インビボでのヒトVEGFR−2+細胞およびTie−2+細胞中でのDNA合成の活性化を明らかにするために、チミジンアナログであるBrdU(Sigma,スウェーデン)を、終了の12時間前および24時間前に、100mg/kg体重で1回i.p.注射として投与した。
【0093】
(免疫組織化学)
マウスの動脈内にヒト細胞を局在化させるために、5μmの凍結切片を抗GFP抗体(1:50)で染色した。免疫ペルオキシダーゼ手順をVectastain Elite
ABCキット(ImmunKemi,Stockholm,スウェーデン)を使用して、製造業者によって記載されているように行った。ACE(赤色の染色を生じる)基質キットを発色現像液として使用し、ヘマトキシリンで対比染色した。さらに、マウス抗ヒト核モノクローナル抗体(Chemicon,CA,米国)を使用して、免疫蛍光18によってマウスの動脈中のヒト細胞を検出した。移植されたヒト細胞の表現形を、ヒト核に対する抗体(1:50)と抗VE−カドヘリン抗体(1:100)での二重染色によって検出した。この後、二次抗マウスサブクラス特異的FITC抗体またはテキサスレッド結合抗体(1:500)で染色した。内皮細胞化を、ヒトACEポジティブ細胞に覆われた表面と全発光表面の割合として計算した。組織形態学(1つのグループについて6匹の動物、6つの切片/大腿動脈)については、動脈の横切片をヘマトキシリン/エオシンで染色し、血管径、中膜の面積、および中膜に対する内膜の面積の比を調べた。
【0094】
(逆転写ポリメラーゼ連鎖反応(RT−PCR))
全RNAを3つのヒト−マウスキメラ大腿動脈(1つはCD14+/VEGFR−2−細胞を投与したマウスであり、1つは擬似移植したマウス)から、移植の4週間後に、Micro−FastTrack RNA単離キット(Invitrogen,Groningen,オランダ)を使用して抽出した。ヒト特異的プライマーを使用して、ヒトCD31およびe−NOSを、ヒト細胞を移植したマウスの動脈の中で検出した。新しく単離したCD14+/VEGFR−2+細胞およびCD14+/VEGFR−2−細胞もまた、e−NOSの発現について試験した。ABLを内因性の参照遺伝子として使用した。プライマーのセットは、CyberGene(Huddinge,スウェーデン)によって商業的に合成された。プライマー配列は、
【0095】
【数1】

PCR反応は先に記載された19ように行った。
【0096】
(統計分析)
適切である場合には、結果を平均±SDとして表した。対応のないt検定を、EPC移植したグループおよび擬似(媒体のみで)処置したグループの間での比較のために使用した。p<0.05を有意と考えた。Mann−Whitney U検定を、グループ間での比較のために使用した。p<0.05を有意と考えた。
【0097】
(実施例2)
(末梢血由来のVEGFR−1+細胞、VEGFR−2+細胞、およびTie−2+細胞は単球/マクロファージ系統に属する)
VEGFR−1+細胞、VEGFR−2+細胞、およびTie−2+細胞は、それぞれ、血液中の単核細胞の集団全体のおよそ3±0.2%、0.8±5%、および2±0.3%を占めることが明らかになった。これらの3つの集団の表現形の分析により、複数のタイプの細胞の混合物が、主に、単球/マクロファージ細胞系統に属することが明らかになった(表1)。新しく単離したVEGFR−1+細胞およびTie−2+細胞は、内皮細胞マーカーであるVE−カドヘリン(CD144)は発現しなかった。しかし、新しく単離したVEGFR−2+細胞の小さい割合がこのマーカーを発現した。さらに、3つの集団の全てはCD45マーカーを発現しており、これは、これらの細胞が造血起源のものであることを示している。VEGFR−1+細胞、VEGFR−2+細胞、およびTie−2+細胞は、それらのそれぞれのマーカーについてポジティブに染色された(図1A〜C)。新しく単離したVEGFR−1+細胞、VEGFR−2+細胞、およびTie−2+細胞の電子顕微鏡による実験により、表現形の分析による発見がさらにサポートされた。3つの集団の全部に、単核単球、マクロファージ、リンパ球、および多形核細胞を含む細胞の混合物が含まれていた(図1D〜F)。
【0098】
【表1−1】
【0099】
【表1−2】
(実施例3)
(培養されたVEGFR−2+細胞とTie−2+細胞は内皮細胞マーカーだけを示す)
フィブロネクチンでコーティングされた培養ウェル上でのこれらの細胞の培養により、付着細胞(80%)と非付着細胞(20%)の2つの集団が示された。1週間後、非付着細胞を廃棄した。これは内皮細胞について記載した条件下ではさらに培養することはできなかった。2週間の培養後、VEGFR−2+培養物およびTie2+培養物中の付着細胞の大部分は内皮細胞マーカーであるCD144、CD141を発現し、活性化されると、CD106とCD142を発現した(表2)。電子顕微鏡を使用したさらなる分析によってこれらの細胞の内皮形態を確認すると、開口、バイベル・パラーデ小体、および内皮細胞に典型的な堅い結合部分の存在が明らかになった(図1G〜I)。一方、VEGFR−1+細胞の小さい画分しか2週間後には付着したままではなく、さらなる分析のために十分な細胞を得ることはできなかった。VEGFR−2+細胞もTie−2+細胞も、培養物中では増殖しなかった。しかし、これらの集団は、>4週間の間、増殖せずに培養物で生存した状態で維持することができた。
【0100】
(表2 2週間の培養後のVEGFR−2+細胞とTie−2+細胞の表現形の特徴)
【0101】
【表2】
(実施例5)
(VEGFR−2+細胞とTie−2+細胞は移動能力と侵襲能力を有しており、細管様構造を形成する)
新しく単離したVEGFR−2+細胞とTie−2細胞は、VEGFとアンジオポイエチン−2に向かって移動したが、VEGFR−1+細胞は移動しなかった(図2AおよびB)。しかし、マトリゲルに添加したVEGFR−1+細胞の60%、VEGFR−2+細胞の70%、そしてTie−2+細胞の65%が、侵襲アッセイにおいて下部チャンバーに移動し、このことは、3つの細胞タイプの全てが侵襲能力を有していることを示している(図2C)。マトリゲルの中では、これらの新しく単離した細胞は、VEGFR−2+細胞とTie−2+細胞だけが細管様構造を形成することができ、VEGFR−1+細胞はそのような能力を有していないことが示された(図2D〜G)。HUVECをコントロールとして使用した。本発明者らのインビトロでの実験では、VEGFR−2+細胞とTie−2+細胞は、表現形の上でも、形態学的にも似ている内皮細胞であることを示していたが、VEGFR−1+細胞は内皮様の特徴を示さなかった。したがって、VEGFR−2+細胞とTie−2+細胞の、バルーン損傷した動脈の再内皮細胞化に関与するインビボでの機能的能力をさらに分析した。
【0102】
(実施例6)
(VEGFR−2+細胞とTie−2+細胞は剥離したマウスの大腿動脈の効率のよい再生に関与しており、インビボで増殖した)
実験用の血管形成術手順を、ヌードマウスの大腿動脈に行った。この介入の直後、VEGFR−2+細胞またはTie−2+細胞を新しく損傷させた動脈床に局所注入した。移植された血管は、血栓の兆候は示さなかった。ヒト核抗体の特異性を、正常なヒトおよびマウスの動脈を使用して最初に決定した。ヒト細胞は黒色に染色された(図3A)。コントロールのマウスの動脈はヒト核抗体では染色されなかった(図3B)。移植の2週間後には、ヒトVEGFR−2+細胞は播種されたバルーン損傷させられた大腿動脈の内腔表面で検出された(DAB−Ni−ポジティブ、図3C)。移植の4週間後には、高度な再生が見られ、およそ75±8%の病変がヒトVEGFR−2+細胞で覆われていた。同様に、Tie−2+細胞が移植後2週間および4週間に検出された(図3D)。Tie−2+細胞を移植した動物は、移植の4週間後には、高度な再生を示し、病変のおよそ70±5%がヒトTie−2+細胞で覆われていた。
【0103】
マウスの動脈内でのヒト細胞の存在を、今一度、免疫蛍光試験において確認した(図3E〜F)。インビボでのEC表現形を、フォン・ヴィレブランド因子に対する抗体とVE−カドヘリン(CD144)に対する抗体での染色によって確認した(図4AおよびB)。それぞれの切片を、ヒト細胞を宿主細胞と区別するために、抗ヒト核抗体で二重染色した(黄色い染色は二重にポジティブである細胞を示している)。興味深いことに、移植されたヒト細胞は、播種された動脈の内腔表面だけではなく、間質中膜においても見ることができた。α−アクチンに対する抗体で標識した場合には、中膜の中の細胞はポジティブに染色され、これは平滑筋細胞の存在を示していた(図4G〜I)。したがって、VEGFR−2+細胞とTie−2+細胞はいずれも、インビボでは、内皮細胞だけではなく、平滑筋細胞にも分化した。VEGFR−2+細胞もTie−2+細胞も、インビトロでは増殖しなかったが(データは示さない)、移植されたVEGFR−2+細胞とTie−2+細胞の増殖についての証拠を得るために、BrdUの取り込み試験を、8匹の動物(VEGFR−2+:n=4、Tie−2+:n=4)において行った。7日目での組織の分析は、移植されたヒト細胞中での広範囲に及ぶBrdUの取り込みを示した。抗BrdU抗体と抗ヒト核抗体の両方でDNAを染色したので、同じ組織切片の二重染色はできなかった。したがって、連続する切片を抗BrdU(茶色)抗体と抗ヒト核抗体(黒色)で染色して、移植された細胞の増殖を示した(図5A〜F)。これらの研究結果は、VEGFR−2+細胞とTie−2+細胞は損傷した動脈を再生させるだけではなく、インビボでは増殖もすることを示している。
【0104】
(実施例7)
(内皮前駆細胞での再内皮細胞化によっては、マウスの大腿動脈のバルーン損傷後に新生内膜の増殖は生じなかった)
損傷した動脈のセグメントの横断面を、移植の2週間後およびよび4週間後に、組織形態学的変化について調べた。マウスの大腿動脈の中でのバルーン損傷手順によって、病変の動脈セグメントの膨張を生じさせ、その結果、血管の直径は顕著に大きくなり、損傷させていないコントロールの動脈についての0.362±0.060mmから、媒体だけと一緒にインキュベートした損傷させた血管については0.601±0.036mmにまでなった。この拡大は、ヒト細胞を移植したマウスの大腿動脈においては見られなかった(図5G)。断片の中膜面積の0.020±0.004から0.010±0.009mm2までの減少が、損傷した動脈において観察された。中膜面積の薄層化は、VEGFR−2細胞およびTie−2細胞を移植した動脈においては見られなかった(図5H)。EPCの播種によって媒介される再内皮細胞化によっては、損傷の2週間後、4週間後、または6週間後には、大腿動脈において新生内膜形成は生じなかった。実際、中膜に対する血管内膜の面積比は、VEGFR−2およびTie−2を移植した血管セグメントにおいては、有意には高くなかった(I/M比:正常な損傷させていない動脈 0.01±0.01、媒体のコントロール 0.02±0.01、VEGFR−2+ 0.02±0.01;Tie−2+ 0.02±0.01,p=ns)。
【0105】
(実施例8)
(CD14+/VEGFR−2+とCD14+/VEGFR−2−の特性決定)
CD14+/VEGFR−2+細胞が単球の集団全体のおよそ2±0.5%(図1A)を占め、血液中のPBMCの0.08±0.04%を占めることが明らかになった。免疫細胞化学分析によっては、新しく単離したCD14+/VEGFR−2−細胞画分はe−NOSを発現しないことが明らかにされた(図6B)。しかし、新しく単離したCD14+/VEGFR−2+細胞は、e−NOS、フォン・ヴィレブランド因子、およびトロンボモジュリンを発現していたが(図6C)、VE−カドヘリンは発現していなかった。CD14+/VEGFR−2+細胞上でのVEGFR−2受容体の発現は、この集団の溶解物が、抗VEGFR−2抗体で免疫ブロットした場合には、およそ200kDaの予想されたバンドを生じ、これはCD14+/VEGFR−2−細胞の集団においては存在しなかったかまたはわずかに発現されていたという事実によってさらにサポートされた(図6D)。さらに、e−NOS mRNAはCD14+/VEGFR−2+細胞中では強く発現されていたが、VEGFR−2ネガティブ画分においては、かすかなe−NOS mRNAのバンドしか見ることができなかった(図6E)。本発明者らは、このかすかなバンドが、CD14+/VEGFR−2−細胞の集団の中に少数のVEGFR−2+細胞が混入しているとの理由から起こり得るとの事実を排除することはできなかった。CD14+/VEGFR−2−集団によって発現される表現形マーカーを表3に示す。いずれの集団もCD45マーカーを発現し、このことは、これらの細胞が造血起源であることを示している。Transwell組織培養インサート上で培養したCD14+/VEGFR−2+細胞の電子顕微鏡による分析によっては、基底膜の形成、バイベル・パラーデ小体の存在、および内皮細胞の特徴である堅い結合が明らかにされた(図7A〜C)。一方、ネガティブな画分はこれらの特徴を示さなかった。
【0106】
CD14+/VEGFR−2+細胞は、フィブロネクチンでコーティングした培養ウェル上で培養した場合には、3日間の培養後にコロニーを形成した。これらのコロニーは、その後数日のうちに、固まった丸い細胞の中心から、一見成長した平たい長く伸びた細胞へと形態を変化させた。一方、CD14+/VEGFR−2集団は、2週間の間、若干丸い細胞として付着したままであったが、クラスターは形成しなかった(図7D)。2つの集団のいずれも、本実験で使用した培養条件下では広範囲の増殖能力は示さなかった。培養したCD14+/VEGFR−2+細胞は、1週間の培養後にVE−カドヘリンを発現していたが(図7D)、VEGFR−2−画分は培養の間のいずれの時点でも、このマーカーを発現することはなかった。
【0107】
【表3】
(実施例9)
(CD14+/VEGFR−2+末梢血細胞とCD14+/VEGFR−2−末梢血細胞のインビトロでの機能的能力)
明らかに多数の新しく単離したCD14+/VEGFR−2+細胞が、CD14+/VEGFR−2−細胞と比較して、VEGFおよびMCP−1に対して移動した(図8A)(それぞれ、p=0.009およびp=0.047)。2つの集団によって生成された血管由来成長因子には有意な相違はなかった。いずれの集団もVEGF、G−CSF、およびGM−CSFを生成したが、SDF−1またはFGFは生成しなかった。
【0108】
しかし、成長因子のレベルは、CD14+/VEGFR−2−集団と比較して、CD14+/VEGFR−2+集団において高かった(図8B)。マトリゲルの中では、3日間培養した細胞は、CD14+/VEGFR−2+細胞だけではなくCD14+/VEGFR−2−細胞も、細管様構造を形成することができた(図8C)。HAECをコントロールとして使用した。
【0109】
したがって、インビトロでのデータは、新しく単離したCD14+/VEGFR−2+細胞が、CD14+/VEGFR−2−細胞と比較して内皮様の特徴を示すことを示している。
【0110】
(実施例10)
(CD14+/VEGFR−2+細胞は内皮細胞へと分化し、剥離されたマウスの大腿動脈を効率よく再生する)
GFPを発現するレンチウイルス粒子に対する2つの細胞集団の暴露によっては、一貫して、>70%の形質導入効率が導かれた。ヒト細胞の動脈切片のスクリーニングは、高い自己蛍光が原因で、GFP+細胞を視覚化することが困難であることを示した。したがって、酵素をベースとする免疫組織化学染色を、抗GFP抗体を使用して行い、GFP+細胞を検出した。
【0111】
この抗体を使用すると、擬似移植したマウスおよびCD14+/VEGFR−2−を移植したマウスにおいては、GFP−ポジティブである領域は見られなかった(図9A〜C)。しかし、GFP−ポジティブ細胞(赤)を有している動脈の内膜表面の範囲が、CD14+/VEGFR−2+細胞を移植したマウスにおいて移植の2週間後にすでに見られた(図9D)。GFP−ポジティブである領域は、播種の4週間後に回収された血管においては明らかに増大していた(図9EおよびF)。さらに、ヒト細胞もまた、免疫蛍光を使用して抗ヒト核抗体によって同定した。ヒト核抗体の特異性を、最初に正常なヒト動脈およびマウス動脈を使用して決定した。ヒト細胞−核は赤色に染色された(図10A)。コントロールマウスの動脈は、ヒト核抗体では染色されなかった(図5B)。移植の4週間後、CD14+/VEGFR−2+ヒト細胞を投与したマウスにおいては高い程度の再生が見られ、病変のおよそ70±8%が、ヒト細胞(赤)で覆われていたが(図10C)、CD14+/VEGFR−2−細胞を移植したマウスにおいては覆われていなかった(図10D)。CD14+/VEGFR−2−細胞を移植した動物は、剥離した動脈、脾臓、肺、または循環においても、移植の2週間後または4週間後には、これらの細胞の存在は示さなかった(データは示さない)。インビボでの内皮細胞の表現形を、VE−カドヘリンに対する抗体での染色(緑)によって確認し、それぞれの切片を、抗ヒト核抗体で二重染色して、ヒト細胞(赤)を検出した。二重染色された細胞は黄色に染色された(図10E)。
【0112】
損傷した動脈切片の横断切片を、組織形態学的変化について移植の2週間後および4週間後に調べた。マウスの大腿動脈でのバルーン損傷手順によって、病変の動脈セグメントの膨張を生じさせ、その結果、血管内径を顕著に大きくし、損傷させていないコントロールの動脈についての0.301±0.041mmから、媒体だけまたはCD14+/VEGFR−2−と一緒にインキュベートした損傷させた血管については、それぞれ、0.601±0.043mmおよび0.631±0.028mmにまでなった(p<0.001)。この拡大は、CD14+/VEGFR−2+細胞を移植したマウスの大腿動脈においては見られなかった(0.404±0.08mm)。横断面の中膜面積の0.028±0.010から0.012±0.007mm2までの減少が、損傷した動脈において観察された。中膜面積の薄層化は、CD14+/VEGFR−2+細胞を移植した動脈においては見られなかった(0.033±0.013)。これらの細胞によって媒介される再内皮細胞化によっては、損傷の2週間後または4週間後には、大腿動脈において新生内膜形成は生じなかった。中膜に対する血管内膜の面積比は、CD14+/VEGFR−2+を移植した血管セグメントにおいては、有意には高くなかった(I/M比:正常な損傷させていない動脈 0.03±0.009、媒体のコントロール 0.02±0.008、CD14+/VEGFR−2− 0.02±0.009;CD14+/VEGFR−2+ 0.04±0.02,p=ns)。
【0113】
(実施例11)
(CD14+/VEGFR−2+細胞を移植したマウスにおけるヒト内皮特異的遺伝子の転写)
移植した細胞の生着を、移植したマウスの中でのヒト遺伝子の発現を決定することによって確認した。ヒト細胞の移植後1ヶ月で屠殺したマウスの大腿動脈を、ヒト内皮特異的遺伝子CD31およびe−NOSに特異的なプライマーを使用して、RT−PCRによって分析した。CD31およびe−NOSプライマーは、それらがそれぞれのマウス遺伝子を増幅しなかったので、ヒトに特異的な種であった(図11A)。これらの結果は、CD14+/VEGFR−2+細胞を投与したマウスに由来する動脈について得られたが、CD14+/VEGFR−2−細胞を移植したマウスにおいては得られなかった。これらのデータは、移植したヒトCD14+/VEGFR−2+細胞が損傷したマウスの動脈に生着したことを示している。
【0114】
(他の実施形態)
特定の実施形態が本明細書中に詳細に開示されているが、これは例示の目的のための例であり、以下に添付される特許請求の範囲の範囲に関する限定とは意図されない。具体的には、種々の置換、変更、および改変を、特許請求の範囲によって定義される本発明の精神および範囲から逸脱することなく本発明に対して行うことができることが、発明者らによって企図される。核酸出発物質、目的のクローン、またはライブラリーのタイプの選択は、当業者にとっては、本明細書中に記載された実施形態の知識にしたがって当業者が日常的に行われている事柄であると考えられる。他の局面、利点、および改変も、以下の特許請求の範囲内であると考えられる。
【技術分野】
【0001】
(発明の分野)
本発明は前駆細胞に関する。
【背景技術】
【0002】
(発明の背景)
血管形成は、既存の脈管構造からの新しい血管の発生(脈管新生)のプロセスであり、一方、脈管形成は、インサイチュで分化する内皮前駆細胞からの血管の形成をいう。最近まで、血管形成は成体での脈管新生のみを意味すると考えられ、脈管形成は胚発生に限定されると考えられてきた。しかし、循環している内皮前駆細胞(EPC)の存在により、成体においても出生後脈管形成が生じることの証拠がもたらされた。虚血性損傷からの組織の再生のための治療用ツールとしてのEPCの可能性が示唆されている。
【発明の概要】
【発明が解決しようとする課題】
【0003】
したがって、内皮細胞に分化することができる前駆細胞の供給源が必要とされている。
【課題を解決するための手段】
【0004】
(発明の要旨)
本発明は、内皮前駆細胞の発見に基づく。前駆細胞は多能性である。前駆細胞は、例えば、インビボまたはインビトロで、内皮細胞または平滑筋細胞に分化することができる。したがって、本発明は、内皮前駆細胞の培養物、例えば、インビトロ培養物を特徴とする。培養物は付着培養物である。あるいは、培養物中の細胞は浮遊状態である。細胞は、造血組織、例えば、血液(例えば、末梢血または骨髄)に由来する。組織は、哺乳動物、例えば、ヒト、霊長類、マウス、ラット、イヌ、ネコ、ウシ、ウマ、ブタに由来する。細胞は、VEGFR−2またはTie−2とCD45に免疫反応性であり、CD144とは免疫反応しない。状況によっては、VEGFR+細胞はCD14とも免疫反応する。細胞はインビトロで増殖させられる。細胞は、2倍、3倍、4倍、5倍、6倍、7倍、8倍、9倍、10倍、11倍、12倍、15倍、20倍、25倍またはそれ以上に増殖することができ、内皮細胞または平滑筋細胞に分化する能力を維持することができる。
【0005】
本発明はさらに、VEGFR−2+またはTie−2+、CD45+、およびCD144−前駆細胞、あるいは、VEGFR−2+CD14+、CD45+、およびCD144−前駆細胞を提供し、そしてこれらの細胞を被験体に移植することによって、被験体に前駆細胞または前駆細胞の子孫を移植する方法を特徴とする。移植によっては、臨床的有益性、例えば、特定の血管障害の1つ以上の症状の緩和がもたらされる。血管障害は、当該分野で公知の方法を使用して医師によって診断される。
【0006】
損傷した血管は、VEGFR−2+またはTie−2+、CD45+、およびCD144−前駆細胞を含む組成物、あるいは、VEGFR−2+CD14+、CD45+、およびCD144−前駆細胞を含む組成物を被験体に投与することによって再増殖させられる。これらの組成物は、血管(例えば、静脈または動脈)に、あるいは動脈床に直接投与される。あるいは、これらの組成物は全身的に投与される。
【0007】
血管移植片は、血管移植片をVEGFR−2+またはTie−2、CD45+、およびCD144−前駆細胞を含む組成物、あるいは、VEGFR−2+CD14+、CD45+、およびCD144−前駆細胞を含む組成物と接触させることによって内皮細胞化(endothelized)させられる。血管移植片は静脈である。あるいは、血管移植片は動脈である。移植片は人工血管移植片、同種移植片、または同系移植片である。移植片は、インビトロ、インビボ、またはエキソビボで接触させられる。血管移植片は、被験体への移植の前に接触させられる。あるいは、移植片は、被験体への移植後に組成物と接触させられる。
【0008】
血管を再増殖させるか、または血管移植片を内皮細胞化させるによって、血管または移植片が、前駆細胞で処置されていない血管または移植片と比較して、血管または移植片が多量の内皮細胞を有していることが意味される。例えば、処置された血管または移植片は、処置されていない血管または移植片と比較して大きな血管径と中膜厚を有する。
【0009】
VEGFR−2+またはTie−2+、CD45+、およびCD144−前駆細胞は、インビボで、内皮細胞または平滑筋細胞に分化する能力を維持する。一方、VEGFR−2+CD14+、CD45+、およびCD144−前駆細胞は、インビボで、内皮細胞に分化する能力を維持する。被験体は、例えば、哺乳動物であり、例えば、ヒト、霊長類、マウス、ラット、イヌ、ネコ、ウシ、ウマ、ブタである。被験体は、血管障害または血管組織の損傷に罹患している。例えば、被験体は、末梢動脈障害、動脈瘤、腎(腎臓)動脈障害、レイノー現象、バージャー病、末梢静脈障害、静脈瘤、静脈内血塊に罹患している。移植および/または再増殖によって、臨床的有益性、例えば、特定の血管障害の1つ以上の症状の緩和がもたらされる。血管障害は、当該分野で公知の方法を使用して医師によって診断される。
【0010】
他の場所で明確に定義されていない限りは、本明細書中で使用される全ての技術用語および科学用語は、本発明が属する分野の当業者によって一般的に理解されている意味と同じ意味を有する。本明細書中に記載される方法および材料と同様または同等の方法および材料を本発明の実施または試験において使用することができるが、適切な方法および材料は以下に記載される。本明細書中に言及される全ての刊行物、特許出願、特許、および他の参考文献は、それらの全体が参考として援用される。矛盾する場合は、定義を含む本明細書が統制する。さらに、材料、方法、および実施例は例示にすぎず、限定するようには意図されない。
【0011】
本発明の他の特徴および利点は、以下の詳細な記載および特許請求の範囲から明らかである。
したがって、本発明は、以下の項目を提供する:
(項目1)
ヒトの末梢血に由来する細胞を含む、インビトロ細胞培養物であって、該培養物中の該細胞は、
a.Tie−2+またはVEGFR−2+であり;
b.CD144−およびCD45+であり;
c.培養物中で培養することができ;ならびに
d.インビボで増殖し、内皮細胞および平滑筋細胞に分化することができる、
インビトロ細胞培養物。
(項目2)
上記培養物中の上記細胞が、少なくとも2週間生存できる、項目1に記載の培養物。
(項目3)
上記培養物中の上記細胞が、少なくとも4週間生存できる、項目1に記載の培養物。
(項目4)
上記培養物が付着培養物である、項目1に記載の培養物。
(項目5)
インビボで内皮細胞または平滑筋細胞に分化するヒト前駆細胞の集団を生成する方法であって、ヒト末梢血由来の細胞の集団からCD144−、CD45+、およびTie−2+またはVEGFR−2+である細胞を選択する工程を包含する、方法。
(項目6)
多能性前駆細胞を被験体に移植する方法であって、
a.多能性のヒトCD144−、CD45+、およびTie−2+またはVEGFR−2+前駆細胞を含むインビトロ細胞培養物を提供する工程であって、該細胞はインビボで内皮細胞または平滑筋細胞に分化する多能性を維持している、工程;および
b.上記細胞を上記被験体に移植する工程、
を包含する、方法。
(項目7)
上記被験体が血管障害に罹患している、項目6に記載の方法。
(項目8)
被験体において損傷した血管を再増殖させる方法であって、多能性のヒトCD144−、CD45+、およびTie−2+またはVEGFR−2+前駆細胞の集団を含む組成物を被験体に投与する工程を包含し、ここで、該細胞はインビボで内皮細胞または平滑筋細胞に分化する多能性を維持している、方法。
(項目9)
上記組成物が血管に直接投与される、項目8に記載の方法。
(項目10)
上記組成物が動脈床に直接投与される、項目8に記載の方法。
(項目11)
上記血管が静脈または動脈である、項目8に記載の方法。
(項目12)
上記被験体が血管障害に罹患している、項目8に記載の方法。
(項目13)
血管移植片を内皮細胞化させる方法であって、該血管移植片を、多能性のヒトCD144−、CD45+、およびTie−2+またはVEGFR−2+前駆細胞の集団を含む組成物と接触させる工程を包含し、ここで、該細胞はインビボで内皮細胞または平滑筋細胞に分化する多能性を維持している、方法。
(項目14)
上記移植片が人工血管移植片である、項目13に記載の方法。
(項目15)
上記移植片が同種移植片である、項目13に記載の方法。
(項目16)
上記移植片が同系移植片である、項目13に記載の方法。
(項目17)
上記血管移植片がインビトロ、インビボ、またはエキソビボで接触させられる、項目13に記載の方法。
(項目18)
上記血管移植片が被験体への移植の前に上記組成物と接触させられる、項目13に記載の方法。
(項目19)
上記血管移植片が被験体への移植の後に上記組成物と接触させられる、項目13に記載の方法。
(項目20)
上記血管移植片が静脈または動脈である、項目13に記載の方法。
(項目21)
ヒトの末梢血由来の細胞を含むインビトロ細胞培養物であって、該培養物中の該細胞は、
a.VEGFR−2+;CD14+、CD144−、およびCD45+であり;
b.培養物中で培養することができ;ならびに
c.インビボで増殖し、内皮細胞に分化することができる、
インビトロ細胞培養物。
(項目22)
上記培養物中の上記細胞が、少なくとも2週間生存できる、項目21に記載の培養物。
(項目23)
上記培養物中の上記細胞が、少なくとも4週間生存できる、項目21に記載の培養物。
(項目24)
上記培養物が付着培養物である、項目21に記載の培養物。
(項目25)
インビボで内皮細胞に分化するヒト前駆細胞の集団を生成する方法であって、ヒト末梢血由来の細胞の集団からCD144−、CD45+、CD14+、およびVEGFR−2+である細胞を選択する工程を包含する、方法。
(項目26)
前駆細胞を被験体に移植する方法であって、
a.ヒトCD144−、CD45+、CD14+、およびVEGFR−2+前駆細胞を含むインビトロ細胞培養物を提供する工程であって、該細胞はインビボで内皮細胞に分化する能力を維持している、工程;および
b.該細胞を該被験体に移植する工程、
を包含する、方法。
(項目27)
上記被験体が血管障害に罹患している、項目26に記載の方法。
(項目28)
被験体の損傷した血管を再増殖させる方法であって、CD144−、CD45+、CD14+、およびVEGFR−2+前駆細胞の集団を含む組成物を被験体に投与する工程を包含し、ここで、該細胞はインビボで内皮細胞に分化する能力を維持している、方法。
(項目29)
上記組成物が血管に直接投与される、項目28に記載の方法。
(項目30)
上記組成物が動脈床に直接投与される、項目28に記載の方法。
(項目31)
上記血管が静脈または動脈である、項目28に記載の方法。
(項目32)
上記被験体が血管障害に罹患している、項目28に記載の方法。
(項目33)
血管移植片を内皮細胞化させる方法であって、上記血管移植片を、多能性のヒトCD144−、CD45+、CD14+、およびVEGFR−2+前駆細胞の集団を含む組成物と接触させる工程を包含し、ここで、該細胞はインビボで内皮細胞に分化する能力を維持している、方法。
(項目34)
上記移植片が人工血管移植片である、項目33に記載の方法。
(項目35)
上記移植片が同種移植片である、項目33に記載の方法。
(項目36)
上記移植片が同系移植片である、項目33に記載の方法。
(項目37)
上記血管移植片がインビトロ、インビボ、またはエキソビボで接触させられる、項目33に記載の方法。
(項目38)
上記血管移植片が被験体への移植の前に上記組成物と接触させられる、項目33に記載の方法。
(項目39)
上記血管移植片が被験体への移植の後に上記組成物と接触させられる、項目33に記載の方法。
(項目40)
上記血管移植片が静脈または動脈である、項目33に記載の方法。
【図面の簡単な説明】
【0012】
【図1】図1A〜Cは、抗VEGFR1抗体、抗VEGFR2抗体、およびTie−2抗体で染色した際に、VEGFR−1+、VEGFR−2+、およびTie−2+細胞がそれらのそれぞれの表面マーカーについてポジティブであったことを示す一連の蛍光顕微鏡写真(40倍)である。図1D〜Iは、新たに選別したVEGFR−1+細胞、VEGFR−2+細胞、およびTie−2+細胞の集団に、単核の単球(M)、マクロファージ(M)、リンパ球(L)および多核白血球(PMN)を含む細胞の混合物が含まれていることを示す一連の電子顕微鏡写真である。G〜Iは、VEGFR−2+細胞およびTie−2+細胞が内皮細胞に分化したことを示すTEM顕微鏡写真であり、(G)バイベル・パラーデ小体(丸い、矢印)および内皮細胞に典型的な堅い結合(矢印)の存在、ならびに、(H、I)開口(矢印)を示している。
【図2A】図2Aは、VEGFR−2+およびTie−2+であるが、VEGFR−1+細胞が、VEGFおよびang−1に向かって移動すること、およびこれらの細胞の3つの集団の全てが、マトリゲル細胞浸潤アッセイにおいて浸潤能を示したことを示している、棒グラフである。結果は、±SEMとして示されている。
【図2B】図2Bは、VEGFR−2+およびTie−2+であるが、VEGFR−1+細胞が、VEGFおよびang−1に向かって移動すること、およびこれらの細胞の3つの集団の全てが、マトリゲル細胞浸潤アッセイにおいて浸潤能を示したことを示している、棒グラフである。結果は、±SEMとして示されている。
【図2C】図2Cは、VEGFR−2+およびTie−2+であるが、VEGFR−1+細胞が、VEGFおよびang−1に向かって移動すること、およびこれらの細胞の3つの集団の全てが、マトリゲル細胞浸潤アッセイにおいて浸潤能を示したことを示している、棒グラフである。結果は、±SEMとして示されている。
【図2D】図2Dは、VEGFR−2+およびTie−2+であるが、VEGFR−1+細胞が、マトリゲル中で毛細管様の細管を形成したことを示している写真である。HUVECがコントロール細胞として使用された。
【図2E】図2Eは、VEGFR−2+およびTie−2+であるが、VEGFR−1+細胞が、マトリゲル中で毛細管様の細管を形成したことを示している写真である。HUVECがコントロール細胞として使用された。
【図2F】図2Fは、VEGFR−2+およびTie−2+であるが、VEGFR−1+細胞が、マトリゲル中で毛細管様の細管を形成したことを示している写真である。HUVECがコントロール細胞として使用された。
【図2G】図2Gは、VEGFR−2+およびTie−2+であるが、VEGFR−1+細胞が、マトリゲル中で毛細管様の細管を形成したことを示している写真である。HUVECがコントロール細胞として使用された。
【図3】図3は一連の写真である。AおよびB、ヒト核抗体によって、ヒトにおいては細胞が染色された(黒)が、コントロールであるマウスの動脈においては染色されなかった。バルーンで損傷させたマウスの大腿動脈に由来する切片の免疫組織化学染色は、4週目では、播種された動脈(C)VEGFR−2+細胞(矢印)の、2週目では、Tie−2+細胞(矢印)の、管腔表面での移植されたヒトの局在化を示している。もとの倍率は60倍である。E、正常なヒトの動脈、F、ヒトTie−2+細胞が移植された損傷させられたマウスの動脈、およびG、抗ヒト核抗体での、擬似移植されたマウスの動脈の免疫蛍光染色(矢印)。
【図4】図4は、移植されたヒト細胞のEC表現形を、ヒト核抗体と、A、VEGFR−2+細胞についてはフォン・ヴィレブランド因子に対する抗体(二重ポジティブ細胞は黄色に染色される、矢印)、そしてB、Tie−2+細胞についてはVE−カドヘリンに対する抗体を用いて、二重染色によって確認したことを示す一連の写真である。α−アクチンに対する抗体で標識された場合は、培地中の細胞(緑色のフィラメント状の染色パターン)はポジティブに染色され、このことは、平滑筋細胞の存在を示している。もとの倍率は60倍である。
【図5A】図5A〜Fは、7日目の組織の分析によってインビボでVEGFR−2+を移植されたヒト細胞およびTie−2+を移植されたヒト細胞への多数のBrdUの取り込みが明らかになったことを示す一連の写真である。AおよびBは、擬似移植されたマウスの動脈についての代表的な連続する切片であり、そしてC〜Fは、抗BrdU(茶色の細胞)とヒト核抗体(黒)で染色された移植されたマウスの動脈の代表的な連続する切片であり、それぞれ、移植されたVEGFR−2+細胞とTie−2細胞中での増殖を示している。
【図5B】図5A〜Fは、7日目の組織の分析によってインビボでVEGFR−2+を移植されたヒト細胞およびTie−2+を移植されたヒト細胞への多数のBrdUの取り込みが明らかになったことを示す一連の写真である。AおよびBは、擬似移植されたマウスの動脈についての代表的な連続する切片であり、そしてC〜Fは、抗BrdU(茶色の細胞)とヒト核抗体(黒)で染色された移植されたマウスの動脈の代表的な連続する切片であり、それぞれ、移植されたVEGFR−2+細胞とTie−2細胞中での増殖を示している。
【図5C】図5A〜Fは、7日目の組織の分析によってインビボでVEGFR−2+を移植されたヒト細胞およびTie−2+を移植されたヒト細胞への多数のBrdUの取り込みが明らかになったことを示す一連の写真である。AおよびBは、擬似移植されたマウスの動脈についての代表的な連続する切片であり、そしてC〜Fは、抗BrdU(茶色の細胞)とヒト核抗体(黒)で染色された移植されたマウスの動脈の代表的な連続する切片であり、それぞれ、移植されたVEGFR−2+細胞とTie−2細胞中での増殖を示している。
【図5D】図5A〜Fは、7日目の組織の分析によってインビボでVEGFR−2+を移植されたヒト細胞およびTie−2+を移植されたヒト細胞への多数のBrdUの取り込みが明らかになったことを示す一連の写真である。AおよびBは、擬似移植されたマウスの動脈についての代表的な連続する切片であり、そしてC〜Fは、抗BrdU(茶色の細胞)とヒト核抗体(黒)で染色された移植されたマウスの動脈の代表的な連続する切片であり、それぞれ、移植されたVEGFR−2+細胞とTie−2細胞中での増殖を示している。
【図5E】図5A〜Fは、7日目の組織の分析によってインビボでVEGFR−2+を移植されたヒト細胞およびTie−2+を移植されたヒト細胞への多数のBrdUの取り込みが明らかになったことを示す一連の写真である。AおよびBは、擬似移植されたマウスの動脈についての代表的な連続する切片であり、そしてC〜Fは、抗BrdU(茶色の細胞)とヒト核抗体(黒)で染色された移植されたマウスの動脈の代表的な連続する切片であり、それぞれ、移植されたVEGFR−2+細胞とTie−2細胞中での増殖を示している。
【図5F】図5A〜Fは、7日目の組織の分析によってインビボでVEGFR−2+を移植されたヒト細胞およびTie−2+を移植されたヒト細胞への多数のBrdUの取り込みが明らかになったことを示す一連の写真である。AおよびBは、擬似移植されたマウスの動脈についての代表的な連続する切片であり、そしてC〜Fは、抗BrdU(茶色の細胞)とヒト核抗体(黒)で染色された移植されたマウスの動脈の代表的な連続する切片であり、それぞれ、移植されたVEGFR−2+細胞とTie−2細胞中での増殖を示している。
【図5G】図5Gは、マウスの大腿動脈の中でのバルーン損傷手順によって、媒体だけとともにインキュベートされた損傷した血管においては病変の動脈セグメントの膨張が生じたが、ヒト細胞が移植されたマウスの大腿動脈においては病変の動脈断片の膨張は生じなかったことを示す棒グラフである。断面の中膜面積の減少は、損傷した動脈において観察されたが、VEGFR−2細胞およびTie−2細胞が移植された動脈においては観察されなかった。
【図5H】図5Hは、マウスの大腿動脈の中でのバルーン損傷手順によって、媒体だけとともにインキュベートされた損傷した血管においては病変の動脈セグメントの膨張が生じたが、ヒト細胞が移植されたマウスの大腿動脈においては病変の動脈断片の膨張は生じなかったことを示す棒グラフである。断面の中膜面積の減少は、損傷した動脈において観察されたが、VEGFR−2細胞およびTie−2細胞が移植された動脈においては観察されなかった。
【図6A】図6は、CD14+/VEGFR−2+およびCD14+/VEGFR−2−細胞の特徴を示す一連のグラフおよび写真である。A、末梢血単核細胞からのCD14+/VEGFR−2+およびCD14+/VEGFR−2−細胞のフローサイトメトリーによる選別により、Tリンパ球は激減した。
【図6B】図6は、CD14+/VEGFR−2+およびCD14+/VEGFR−2−細胞の特徴を示す一連のグラフおよび写真である。B、CD14+/VEGFR−2−細胞は内皮の一酸化窒素合成酵素(e−NOS)は発現しなかった。
【図6C】図6は、CD14+/VEGFR−2+およびCD14+/VEGFR−2−細胞の特徴を示す一連のグラフおよび写真である。C、CD14+/VEGFR−2+細胞は、e−NOS(黒)、フォン・ヴィレブランド因子(赤)、およびトロンボモジュリン(赤)のようないくつかの内皮特異的マーカーを発現したが、VE−カドヘリンは発現しなかった。細胞をヘマトキシリンで対比染色した。
【図6D】図6は、CD14+/VEGFR−2+およびCD14+/VEGFR−2−細胞の特徴を示す一連のグラフおよび写真である。D、CD14+/VEGFR−2+細胞由来の溶解物のVEGFR−2に対する抗体でのウェスタンブロッティングによっては、およそ200kDaの予想されたバンドが生じた(レーン1および2)が、CD14+/VEGFR−2−細胞(レーン3)とコントロールである二次抗体(レーン4)はバンドを生じなかった。
【図6E】図6は、CD14+/VEGFR−2+およびCD14+/VEGFR−2−細胞の特徴を示す一連のグラフおよび写真である。E、CD14+/VEGFR−2+細胞においては、e−NOSのmRNAの強い発現が、そしてCD14+/VEGFR−2−細胞においては弱い発現が検出された。
【図7】図7は、CD14+/VEGFR−2+細胞とCD14+/VEGFR−2−細胞のインビトロでの形態学的特性決定を示す一連の写真である。A、TEM顕微鏡写真は、CD14+/VEGFR−2+細胞について、内皮細胞に典型的な堅い結合の存在(矢印、棒;2μm)、B、バイベル・パラーデ小体の存在(矢印、棒;0.5μm)、およびC、基底膜の存在(M、棒;1μm)、D、培養物中のCD14+/VEGFR−2−細胞はコロニーを形成しないが、CD14+/VEGFR−2+細胞は最初に丸い細胞からなるコロニーを形成し、その1週間後、1つの細長い細胞に成長することが明らかになったことを示している。1週間の培養後には、CD14+/VEGFR−2+細胞だけがVE−カドヘリンを発現していた。
【図8A】図8AおよびBは、CD14+/VEGFR−2+細胞とCD14+/VEGFR−2−細胞のインビトロでの移動と細管形成能力を示す散布図である。A、50ng/mlの濃度の血管内皮成長因子(VEGF)と300ng/mlの単球走化性タンパク質−1(MCP−1)を含まない条件と、それらを含む条件下での移動アッセイにおいては、CD14+/VEGFR−2−細胞と比較して、明らかに多数のCD14+/VEGFR−2+細胞がVEGFおよびMCP−1に移動した。
【図8B】図8AおよびBは、CD14+/VEGFR−2+細胞とCD14+/VEGFR−2−細胞のインビトロでの移動と細管形成能力を示す散布図である。B、CD14+/VEGF−2+細胞の集団は、CD14+/VEGFR−2−集団と比較して、血管由来成長因子、VEGF、顆粒球コロニー刺激因子(G−CSF)、および顆粒球−マクロファージコロニー刺激因子(GM−CSF)をより高いレベルで生成した。
【図8C】図8Cは、CD14+/VEGFR−2+細胞はマトリゲルの中で毛細管様細管を形成したが、CD14+/VEGF−2−細胞は形成しなかったことを示している一連の写真である。ヒトの大動脈細胞(HAEC)がコントロール細胞として使用された。
【図9】図9は、CD14+/VEGFR−2+細胞とCD14+/VEGFR−2−細胞のインビボでの生着能力を示す一連の写真である。抗緑色蛍光タンパク質(GFP)抗体での、バルーン損傷させたマウスの大腿動脈由来の切片の免疫組織化学染色により、A、擬似移植されたGFPポジティブである領域の局在化はないこと、および、B、播種された動脈の管腔表面のCD14+/VEGFR−2−が移植された細胞が示された。C、切片はコントロールである二次抗体での染色を示している。D、2週間目の有意な数のGFP−ポジティブである(赤)CD14+/VEGFR−2+細胞(矢印)、そしてEおよびF、4週間目の有意な数のGFP−ポジティブである(赤)CD14+/VEGFR−2+細胞が、これらの細胞が移植された動物において見られた。もとの倍率は60倍であった。
【図10】図10は、移植後のCD14+/VEGFR−2+細胞とCD14+/VEGFR−2−細胞の検出のための免疫蛍光染色を示す一連の写真である。A、正常なヒトの動脈中にはヒト核抗体で染色された細胞(赤)があったが、B、コントロールの損傷させた擬似移植したマウスの動脈にはなかった。C、バルーン損傷させたマウスの大腿動脈は、4週目には、播種された大動脈(赤色の細胞、矢印)の管腔表面で移植されたヒトCD14+/VEGFR−2+細胞の局在化を示した。D、しかし、4週間目には、移植されたCD14+/VEGFR−2−細胞の局在化は観察されなかった。E、移植されたヒト細胞の内皮細胞表現形が、ヒト核抗体とVE−カドヘリンに対する抗体での二重染色によって確認された(二重ポジティブである細胞は黄色く染色された、矢印)。もとの倍率は60倍であった。
【図11】図11は、CD14+/VEGFR−2+細胞とCD14+/VEGFR−2−細胞のインビボでの生着能力および機能的能力を示すウェスタンブロットの写真である。マウスの動脈の中でのヒトの内皮特異的遺伝子の転写。ヒトCD31とe−NOSは、ヒトCD14+/VEGFR−2+細胞が移植されたバルーン損傷させられたマウスの大腿動脈の中で検出されたが、擬似移植されたマウス、またはCD14+/VEGFR−2−細胞が移植されたマウスにおいては検出されなかった。ABLがハウスキーピング遺伝子として使用された。
【発明を実施するための形態】
【0013】
(詳細な説明)
本発明は、ヒトの末梢血の中の明確な前駆細胞の集団の予想外の発見に基づく。これは、インビトロで培養することができ、前駆細胞の表現形を維持することができる。さらに、これらの細胞をマトリゲル上でインビトロで培養すると、これらは、細管様構造を形成することができる。これらの細胞は、動脈が損傷している動物に移植されると、内皮細胞および/または平滑筋細胞への機能的な分化を示した。
【0014】
血管障害には、世界中で顕著な罹患率および死亡率のある、広い範囲の急性の状態と慢性の状態のものが含まれる。内皮細胞の移植は、虚血によって損傷した組織の処置においては、非常にすばらしい治療能力を有している。血管障害には、循環系に影響を及ぼす任意の状態が含まれる。これは、動脈、静脈、およびリンパ管の疾患から、循環に影響を及ぼす血液疾患にまでの範囲に及ぶ。血管障害の例としては、末梢動脈障害、動脈瘤、腎(腎臓)動脈障害、レイノー現象、バージャー病、末梢静脈障害、静脈瘤、静脈内血塊が挙げられる。
【0015】
冠動脈疾患は、少なくとも2つのグループにカテゴリー分類することができる。急性の冠動脈疾患としては心筋梗塞が挙げられ、慢性の冠動脈疾患としては慢性冠動脈虚血、動脈硬化症、うっ血性心不全、狭心症、アテローム性動脈硬化症、および心筋肥大が挙げられる。他の冠動脈疾患としては、脳卒中、心筋梗塞、拡張型心筋症、再狭窄、冠状動脈疾患、心不全、不整脈、狭心症、または高血圧が挙げられる。
【0016】
急性の冠動脈疾患によって、心臓への血液の供給の突然の遮断が生じ、これによって心臓組織には酸素や栄養分が届かなくなり、結果として心臓組織の損傷および死が生じる。対照的に、慢性の冠動脈疾患は、時間とともに心臓組織への酸素および血液の供給が段階的に減少し、これによって心臓組織の進行性の損傷が生じ、最終的には死に至ることを特徴とする。内皮前駆細胞のインビトロおよびインビボでの増殖および分化をうまく行うことは、内皮細胞の移植に、および遺伝子治療用のビヒクルに有用である。
【0017】
本発明によって、インビボで内皮細胞および/または平滑筋細胞に分化することができる、末梢血由来の多能性内皮前駆細胞を生成するための方法が提供される。
【0018】
(内皮前駆細胞)
本発明によって、内皮前駆細胞(本明細書中ではEPCと呼ばれる)が提供される。EPC細胞は、本発明の方法を使用して増殖するように誘導することができる未分化の細胞である、EPCは自己維持(self−maintenance)することができ、その結果、どの細胞分裂によっても、少なくとも1つの娘細胞がまたEPC細胞となる。EPC細胞は100倍、250倍、500倍、1000倍、2000倍、3000倍、4000倍、5000倍、またはそれ以上に増殖させることができる。
【0019】
EPCの表現形は、これらの細胞が約束された造血細胞のマーカーであるCD45を発現することを明らかに示している。さらに、EPCは、VEGFR−2および/またはTei−2と免疫反応性であり、カドヘリン5(CD144)とは非免疫反応性である。状況によっては、EPCはCD14と免疫反応する。CD14とは免疫反応しないEPCは、インビボでは、内皮細胞または平滑筋細胞に分化することができる。別に、CD14と免疫反応する細胞は、内皮細胞だけに分化することができる。EPCは多能性前駆細胞である。多能性前駆細胞によって、細胞が1つ以上のタイプの細胞に分化することができることが意味される。例えば、細胞は、内皮細胞または平滑筋細胞に分化することができる。
【0020】
血管内皮成長因子(VEGF)は、特異的チロシンキナーゼ受容体を介して作用する。特異的チロシンキナーゼ受容体には、VFGFR−1(flt−1)およびVEGFR−2(flk−1/KDR)およびVEGFR−3/Flt−4が含まれ、これらは、胚血管形成および造血に不可欠なシグナルを伝達する。VEGFは3つの受容体全てに結合するが、最大の生物学的機能はVEGFR−2によって媒介され、VEGFR−1の役割は現在は不明である。VEGFR3/Flt4によるシグナル伝達は、リンパ腺の内皮細胞の発達に重要であることが知られており、VEGFR3によるシグナル伝達は、内皮細胞にリンパ球内皮細胞様表現形を付与し得る。VEGFRは、血管の増殖の刺激、血管緊張低下、血管透過性の誘導、内皮細胞の移動、増殖、および生存に不可欠なプロセスについてのシグナルをリレーする。内皮細胞は全ての異なるVEGF−Rを発現する。胚発生の間には、1つの前駆細胞である、血管芽細胞が造血系と脈管系の両方を生じ得ることが報告されている。
【0021】
Tie−2は内皮特異的受容体チロシンキナーゼであり、アンギオポエチン1の受容体である。これはI型膜タンパク質であり、活発に増殖している血管の内皮において主に発現され、最も早く発見された哺乳動物の内皮細胞系統のマーカーであり得る。Tie−2はおそらく、内皮細胞の増殖と分化の調節に関係しており、血管の形成の間に内皮細胞の特別な方向付けを指示し得る。
【0022】
CD14抗原は、リポ多糖(LPS)とLPS結合タンパク質(LBP)の複合体についての高親和性受容体である。CD14抗原は、CD14、TLR4、およびMD−2から構成される機能性のヘテロマーであるLPS受容体複合体の一部である。CD14は、末梢血の中のほとんどのヒトの単球およびマクロファージ上、他の体液および種々の組織、例えば、リンパ節および脾臓の上で強く発現される。CD14は、ヒト好中球および骨髄樹状細胞の亜集団上では、おそらく、弱く発現される。
【0023】
CD45抗原はチロシンホスファターゼであり、これは白血球共通抗原(LCA)としても知られている。CD45は、赤血球系細胞、血小板、およびそれらの前駆細胞を除く、造血系を起源とする全てのヒト細胞上に存在する。CD45分子にはT細胞とB細胞の活性化が必要であり、これは、細胞の活性化状態によって少なくとも5つのイソ型で発現される。
【0024】
CD144はまた、カドヘリン5またはVE−カドヘリンとも呼ばれ、細胞接着分子のカドへリンファミリーに属する140kDaのタンパク質である。CD144抗原は内皮細胞に特異的であり、そして、内皮組織内の接合部位の細胞間の裂隙部位に存在する。カドヘリン5分子は、脈管内皮の透過特性において役割を果たすことができる。
【0025】
血液は、任意の動物、例えば、魚類、爬虫類、鳥類、両生類、および哺乳動物、例えば、好ましくは、齧歯類、および例えば、マウスおよびヒトから得ることができる。
【0026】
VEGFR−1+、VEGFR−2+、およびTie−2+細胞は、それぞれ、血液中の単核細胞の全集団のおよそ3.0±0.2%、0.8±0.5%、2.0±0.3%を占める。CD14+/VEGFR−2+細胞は、単球の全集団のおよそ2.0±0.5%を占め、そして血液中の単核細胞の0.08±0.04%を占める。
【0027】
EPCは、長期培養物中でインビトロで維持することができる。EPCは、2回、3回、4回、5回、6回、7回、8回、9回、10回、11回、12回、またはそれ以上、培養物中で継代培養することができる。
【0028】
(内皮前駆細胞の単離)
本発明は、内皮前駆細胞または平滑筋前駆細胞の集団を単離する方法に関する。細胞の集団は、ポジティブ選択の手段によって、またはいずれかの順序でのポジティブ選択とネガティブ選択の両方の混合によって単離される。前駆細胞の集団が精製される。精製されたEPCの集団には、それから細胞が単離された細胞の粗集団よりも明らかに多い割合のEPCの集団が含まれる。
【0029】
例えば、精製手順によって、集団全体に対してEPCは、少なくとも5倍の増加、好ましくは少なくとも10倍の増加、より好ましくは少なくとも15倍の増加、最も好ましくは少なくとも20倍の増加が、そして状況によっては、少なくとも25倍の増加が導かれるはずである。精製されたEPCの集団には、少なくとも15%、好ましくは少なくとも20%、より好ましくは少なくとも25%、最も好ましくは少なくとも35%、そして最適には少なくとも50%のEPCが含まれるはずである。
【0030】
本明細書中に記載される方法によっては、75%まで、好ましくは80%まで、より好ましくは85%まで、最も好ましくは90%まで、そして最適には95%までを幹細胞が占める混合物を導くことができる。このような方法は、99%、99.90%、そしてさらには100%をEPCが占める混合物を生じることができる。したがって、本発明の精製された集団には、上記のような自然界に存在している集団よりも有意に高いレベルのEPCが含まれる。
【0031】
精製されたEPCの集団は、EPCに特徴的な抗原を発現する幹細胞の集団を含む細胞の粗混合物を、抗原の細胞外部分に特異的に結合する分子と接触させることによって単離される。このような技術は、ポジティブ選択として知られている。EPCのこれらの分子への結合によって、抗原を発現しない混入している細胞とEPCを十分に識別することができ、混入している細胞から幹細胞を単離することができる。抗原はVEGFRであることが好ましく、より好ましくはVEGFR−2である。
【0032】
混入している細胞から前駆細胞を分離するために使用される分子は、EPCに特徴的な抗原に特異的に結合する任意の分子であり得る。分子は、例えば、モノクローナル抗体、モノクローナル抗体の断片、または受容体である抗原の場合には、受容体のリガンドであり得る。例えば、VEGF受容体(例えば、FLK−1)の場合には、リガンドはVEGFである。
【0033】
本発明の特有の単離された細胞は、それらのCD45+状態と、血管内皮成長因子受容体(VEGFR)(例えば、VEGFR−2)の保有によって他の細胞から分離される。細胞は、Civin,米国特許第4,714,680号、同第4,965,204号、同第5,035,994号、および同第5,130,144号、Tsukamotoら、米国特許第5,750,397号、およびLokenら米国特許第5,137,809号(これらの全ては、それらの全体が参考として本明細書中に援用される)に記載されている技術のような、細胞を分離するための従来技術によって単離することができる。したがって、例えば、CD45特異的モノクローナル抗体またはVEGFR特異的抗体を、ニトロセルロース、アガロースビーズ、ポリスチレンビーズ、中空線維膜、磁気ビーズ、およびプラスチック製のペトリ皿のような固体支持体上に固定することができる。その後、細胞集団全体が固体支持体の上を通過させられるか、またはビーズに添加される。
【0034】
結合分子に結合した細胞は、残っている細胞懸濁液から固体支持体を物理的に分離することによって、細胞懸濁液から取り出される。例えば、結合していない細胞は、幹細胞を固体支持体に結合させるために十分な時間の後、生理学的緩衝液で溶出させることも、また洗浄することもできる。
【0035】
結合した細胞は、固相および結合分子の性質に主に依存して、任意の適切な方法によって固相から分離される。例えば、結合した細胞は激しい攪拌によってプラスチック製のペトリ皿から溶出させられ得る。あるいは、結合した細胞は、酵素による「ニッキング」または固相と抗体との間の酵素に敏感な「スペーサー」配列の消化によって溶出させることができる。アガロースビーズに結合させられた適切なスペーサー配列は、例えば、Pharmaciaから市販されている。
【0036】
溶出された富化させられた細胞の画分は、その後、遠心分離によって緩衝液で洗浄され、従来技術にしたがって後で使用されるまで低温で生存している状態で保存される。細胞はまた、例えば、レシピエントに静脈内注射されることによって、すぐに使用される場合もある。
【0037】
固相支持体に付着したままである細胞は、使用される抗体によって認識されるマーカーを含む細胞である。したがって、抗CD45抗体が使用されると、その後、得られる集団はCD45+細胞について大幅に富化させられる。使用される抗体がVFGFRである場合には、得られる集団はVFGFR+細胞が大幅に富化させられる。この集団は、その後、他のマーカーに対する抗体がそれに付着している固相を使用する工程を繰り返し行うことによって、他のマーカーについて富化させることができる。
【0038】
CD45+VEGFR+細胞を選別するための別の方法は、フローサイトメトリーによる方法であり、Becton−Dickinsonによって製品名FACScanまたはFACSCaliburとして製造されている選別装置のような蛍光活性化細胞選別装置(FACS)による方法が最も好ましい。この技術によって、その上に抗CD45抗体を有している細胞が、そのような色素に結合させられた抗CD45抗体によって特定の蛍光色素でタグ化される。同様に、細胞のVEGFRマーカーは、他の色素に結合させられた抗VEGFR抗体によって別の蛍光色素でタグ化される。染色された細胞が機器の上に置かれると、細胞の流れがアルゴンレーザー光線によって方向付けられ、蛍光色素が励起されて光を発する。この発光は、光学フィルターのセットによって蛍光色素の発光波長に特異的な光電子倍増管(PMT)によって検出される。PMTによって検出されたシグナルは、その自身のチャネルにおいて増幅され、種々の異なる形態(例えば、柱状グラフ、点表示、または曲線表示)でコンピューターによって表示される。したがって、1つの波長で発光する蛍光細胞は、特異的な蛍光色素標識された試薬と反応する分子であり、一方、非蛍光細胞または異なる波長で発光する蛍光細胞はこの分子を発現しないが、他の波長で蛍光を発する蛍光色素標識された試薬と反応する分子を発現し得る。フローサイトメーターはまた、細胞によって発現される蛍光の量(蛍光強度)を表示する点において半定量的でもある。これは、比較の意味では、細胞によって発現される分子の数に関係する。
【0039】
フローサイトメーターはまた、非蛍光パラメーター、例えば、細胞容量、または、それがレーザー光線を通過する際に細胞によって散乱させられた光を測定するために、取り付けられる。細胞容量は、通常、直接測定される。光散乱PMTによって、前角(前方散乱;FSC)または直角(側方散乱;SSC)のいずれかで細胞によって散乱させられた光が検出される。FSCは、通常は大きさの指標であり、一方、SSCは、細胞の複雑さの指標であるが、いずれのパラメーターも他の要因による影響を受け得る。
【0040】
好ましくは、フローサイトメーターには、1つ以上のPMT発光検出器が取り付けられる。さらなるPMTによって、他の発光波長を検出することができ、1つ以上の蛍光色素を同時に、個々の別々のチャネルにおいてそれぞれ検出することができる。コンピューターによりそれぞれのチャネル、または個々のパラメーターと別のパラメーターとの相関関係を分析することができる。通常、FACS機器と共に使用される蛍光色素としては、フルオレセインイソチオシアネート(FITC)(これは、525nmに発光ピークを有している)(緑)、R−フィコエリスリン(PE)(これは、575nmに発光ピークを有している)(赤−オレンジ)、ヨウ化プロピジウム(PI)(これは、620nmに発光ピークを有している)(赤)、7−アミノアクチノマイシンD(7−AAD)(これは660nmに発光ピークを有している)(赤)、R−フィコエリスリンCy5(RPE−Cy5)(これは、670nmに発光ピークを有している)(赤)、およびアロフィコシアニン(APC)(これは、655〜750nmに発光ピークを有している)(深紅)が挙げられる。
【0041】
これらおよび他のタイプのFACS機器は、異なる特性の細胞を異なる容器に屈折させることによって、種々の画分を物理的に分離するさらなる能力を有し得る。
【0042】
出発物質、例えば、骨髄、末梢血、または臍帯血のCD45+VEGFR+集団を単離するための任意の他の方法もまた、本発明にしたがって使用することができる。本発明の種々の亜集団(例えば、CD14+、Tie2+、CD144−)を同様の様式で単離することができる。
【0043】
粗細胞集団が上記のように精製される前またはその後のいずれかに、前駆細胞の集団が当該分野で公知の方法によってさらに濃縮される場合がある。例えば、前駆細胞は、EPCに特徴的な1つ以上の抗原についてのポジティブ選択によって富化させることができる。このような抗原としては、例えば、CD14またはTie−2が挙げられる。
【0044】
上記の方法の特に好ましいバリエーションにおいては、血液がドナーの循環している末梢血から直接採血される。血液は、固相に結合させた結合分子(例えば、EPCを捕捉するための抗体VEGFR−2)を含むカラム全体に連続的に浸透させられる。前駆細胞が除去された血液は、アフェレーシス療法のような当該分野で公知の方法によって、すぐにドナーの循環系に戻される。血液は、十分な数の前駆細胞がカラムに結合するまでこの方法で処理される。その後、幹細胞が当該分野で公知の方法によってカラムから単離される。この方法により、数少ない末梢血前駆細胞をきわめて多量の血液から回収することができ、ドナーの費用がかからず、骨髄の採取の痛みもなく、そしてそれに伴う麻酔、無痛覚症、輸血、および感染のリスクもない。
【0045】
(培養条件)
EPCは、本明細書中に記載される方法を使用して培養され、増殖させられる。細胞は、密度勾配遠心分離によって末梢血単核細胞(PBMC)を単離することによって末梢血から得られる。
【0046】
細胞懸濁液は、細胞を維持することができる任意の容器、特に、培養フラスコ、培養プレート、またはローラーボトル、そして具体的には、25cm2の培養フラスコのような小さい培養フラスコに播種される。懸濁液の中で培養された細胞は、およそ5×104から2×105細胞/ml(例えば、1×105細胞/ml)で再懸濁される。固定された担体の上にプレートされる細胞は、およそ2〜3×10310細胞/cm2でプレートされる。状況に応じて、培養プレートは、コラーゲンのようなマトリックスタンパク質でコーティングされる。細胞は、細胞の代謝に必要な補助物質(例えば、グルタミンおよび他のアミノ酸、ビタミン、ミネラル、およびトランスフェリンのようなタンパク質など)を含む細胞増殖をサポートすることができる任意の公知の培養培地(HEM、DMEM、RPMI、F−12などを含む)に入れることができる。培養培地にはまた、酵母、細菌、および真菌の混入を防ぐための抗生物質(例えば、ペニシリン、ストレプトマイシン、ゲンタマイシンなど)を含めることもできる。培養培地には、ウシ、ウマ、ニワトリなどに由来する血清が含まれる場合がある。
【0047】
培養のための条件は、生理学的条件に近づけるべきである。培養培地のpHは、生理学的pH(例えば、pH6〜8の間、約pH7から7.8の間、またはpH7.4)に近づけるべきである。生理学的温度は、約30℃から40℃の間の範囲である。EPCは、約32℃から約38℃の間の温度(例えば、約35℃から約37℃)で培養される。
【0048】
状況によっては、培養培地には、少なくとも1種の増殖を誘導する(「細胞分裂促進」増殖因子が補充される。「増殖因子」は、EPCに対して成長効果、増殖を誘導する効果、分化を誘導する効果、または栄養効果を有している、タンパク質、ペプチド、または他の分子である。「増殖を誘導する成長因子」は、EPCを増殖させる栄養素であり、これには、細胞に対して栄養効果または増殖誘導効果を発揮するために細胞の表面上の受容体に結合する任意の分子が含まれる。増殖を誘導する成長因子としては、EGF、アンフィレグリン、酸性線維芽細胞増殖因子(aFGFまたはFGF−1)、塩基性線維芽細胞増殖因子(bFGFまたはFGF−2)、形質転換増殖因子α(TGFα)、VEGFおよびそれらの組み合わせが挙げられる。増殖因子は、通常、約1fg/mlから1mg/mlの範囲の濃度で培養培地に添加される。約1から100ng/mlの間の濃度で、通常は十分である。試料滴定アッセイは、特定の増殖因子の最適濃度を決定するために容易に行うことができる。
【0049】
成長因子および栄養素の生物学的効果は、一般的には、細胞表面受容体に対する結合によって媒介される。多数のこれらの因子の受容体が同定されており、特異的受容体についての抗体および分子プローブを利用することができる。EPCは、分化の全ての段階で増殖因子受容体の存在について分析することができる。多くの場合には、特定の受容体の同定によって、外来の増殖因子または栄養素の添加を伴う特異的な発達経路に沿って細胞をさらに分化させることにおいて使用するためのストラテジーについての指針が提供される。
【0050】
一般的には、インビトロでは約3〜10日後に、EPCの培養培地が、培地を吸引し、培養フラスコに新しい培地を添加することによって再び補給される。状況によっては、吸引された培地が回収され、濾過され、続けてEPCを継代培養するための馴化培地として使用される。例えば、10%、20%、30%、40%、またはそれ以上の馴化培地が使用される。
【0051】
EPC細胞培養物は、再度増殖を開始するように容易に継代培養することができる。例えば、インビトロでは3〜7日後に、培養フラスコが十分に揺らされ、その後、EPCは50mlの遠心分離管に移され、低速で遠心分離される。培地は吸引され、EPCは少量の培養培地中に再懸濁される。その後、細胞は数えられ、所望される密度で再度プレートされて、増殖が再開させられる。この手順を毎週繰り返すことができ、その結果、それぞれの継代においては、生存している細胞の数は対数的に増加する。この手順は、所望される数のEPCが得られるまで続けて行うことができる。
【0052】
EPCおよびEPCの子孫は、それらが必要になるまで、任意の当該分野で公知の方法によって低温保存することができる(例えば、米国特許第5,071,741号、PCT国際公開第93/14191号、同第95/07611号、同第96/27287号、同第96/29862号、および同第98/14058号、Karlssonら、65 Biophysical J.2524−2536(1993)を参照のこと)。EPCは、特定の低温保存剤を含む等張溶液、好ましくは、細胞培養培地の中に懸濁させることができる。このような低温保存剤としては、ジメチルスルフォキシド(DMSO)、グリセロールなどが挙げられる。これらの低温保存剤は、5〜15%(例えば、8〜10%)の濃度で使用される。細胞は、−10℃から−150℃(例えば、−20℃から−100℃、または−70℃から−80℃)の温度に徐々に凍結させられる。
【0053】
(内皮前駆細胞の分化)
培養条件に応じて、EPCは内皮細胞または平滑筋細胞へと分化させることができる。
【0054】
EPCは、分化を誘導する成長因子を含む培養培地中の固定された担体上で、内皮細胞または平滑筋細胞EPCへと分化させることができる。EPCの分化はまた、増殖を誘導する生物学的事象のカスケードを活性化させる当該分野で公知の任意の方法によっても誘導することができ、これにはイノシトール三リン酸および細胞内Ca2+の遊離、ジアシルグリセロールの遊離、ならびに、プロテインキナーゼCおよび他の細胞キナーゼの活性化などが含まれる。ホルボールエステル、分化を誘導する成長因子、および他の化学的シグナルでの処理によって分化を誘導することができる。EPCの増殖のための増殖を誘導する成長因子(上記を参照のこと)の代わりに、分化を誘導する成長因子を、EPCの分化に影響を与えるために培養培地に添加することができる。他の分化を誘導する成長因子としては、血小板由来成長因子(PDGF)、チロトロピン放出ホルモン(TRH)、トランスフォーミング成長因子β(TGFβ)、インシュリン様成長因子(IGF−1)などが挙げられる。
【0055】
分化した内皮細胞または平滑筋細胞は、当該分野で公知の免疫細胞化学的技術を使用して検出される。免疫細胞化学(例えば、二重標識免疫蛍光および免疫ペルオキシダーゼ法)では、内皮細胞または平滑筋細胞の、細胞の特徴または表現形の特性を区別するために、細胞タンパク質を検出する抗体が使用される。内皮細胞についての細胞マーカーとしては、例えば、VE−カドヘリン、CD144、CD141、CD106、またはCD142が挙げられ、一方、平滑筋細胞についての細胞マーカーとしては、Flkが挙げられる。免疫細胞化学はまた、CD31およびe−NOSのような内皮細胞遺伝子の発現を検出することによって内皮細胞を同定するために使用することもできる。
【0056】
インサイチュハイブリダイゼーション組織化学もまた、内皮遺伝子のmRNAに特異的なcDNAまたはRNAプローブを使用して行うことができる。これらの技術は、特異的表現形の同定を促進するために、免疫細胞化学的方法と組み合わせることができる。必要であれば、上記で議論された抗体および分子プローブを、細胞の同定を助けるために、それぞれ、ウェスタンブロットおよびノーザンブロット手順に適応することができる。
【0057】
(内皮前駆細胞の移植)
損傷した血管への新しい細胞の移植には、損傷した血管組織、例えば、静脈、動脈、毛細血管を修復する能力があり、それによって血管機能を回復させることができる。しかし、移植の目的に適している細胞がないことによって、それによってもたらされるこの手順の全ての可能性が妨げられる。「適している」細胞は、以下の基準の1つ以上を満たす細胞である:(1)多数を得ることができる;(2)必要に応じて、遺伝的物質を挿入するためにインビトロで増殖させることができる;(3)永久に生存することができ、移植されると、血管の修復を促進することができる;ならびに(4)免疫原性ではない、好ましくは、患者自身の組織から、または適合するドナーから得られたものである。血管障害の処置におけるEPCの使用は、動物モデルを使用して実証することができる。
【0058】
EPCは、異常な血管または冠不全の兆候を有している任意の動物に投与される。EPCは、宿主に対して異種であるドナー組織から調製することができる。異種移植を成功させるためには、移植された組織に対する免疫応答を軽減させるかまたは排除するいくつかの方法が通常使用される。したがって、EPCレシピエントは、シクロスポリンのような免疫抑制剤の使用によって、または局所投与される免疫抑制剤を使用する局所免疫抑制ストラテジーによってのいずれかで、免疫抑制することができる。局所免疫抑制は、Gruber,54 Transplantation 1−11(1992)に開示されている。米国特許第5,026,365号には、局所免疫抑制に適しているカプセル化方法が開示されている。
【0059】
免疫抑制技術を使用することに代わるものとしては、Smithiesら、317 Nature 230−234(1985)によって教示されている胚性幹細胞における相同組み換えを使用した遺伝子の置き換えまたはノックアウトの方法と、細胞株での遺伝子の置き換えまたはノックアウトにまで拡大された方法(Zhengら、88 Proc.Natl.Acad.Sci.8067−8071(1991))を、主要組織適合複合体(MHC)遺伝子の除去のためにEPCに行うことができる。MHCの発現が欠失しているEPCは、同種の、そしておそらくは、異種である組織適合性バリアをも超えて、レシピエントを免疫抑制する必要なく、富化させられた内皮細胞の集団を移植することができる。ドナー細胞の抗原性を低下させるための組み換え方法の使用についての一般的な概要および基準もまた、Gruber,54 Transplantation 1−11(1992)によって開示されている。表面の修飾により移植片の免疫原性を低下させる例示的なアプローチは、PCT国際公開第92/04033号およびPCT/99/24630に開示されている。あるいは、移植片の免疫原性は、MHC抗原が変化させられているかまたは欠失させられているトランスジェニック動物からEPCを調製することによって低下させることもできる。
【0060】
EPCは、マイクロカプセル化(例えば、参考として本明細書中に援用される米国特許第4,352,883号;同第4,353,888号;および同第5,084,350号を参照のこと)およびマイクロカプセル化(例えば、それぞれが参考として本明細書中に援用される米国特許第5,284,761号、同第5,158,881号、同第4,976,859号、および同第4,968,733号、ならびにPCT国際公開第92/19195号および同第95/05452号を参照のこと)を含む、公知のカプセル化技術によってカプセル化することができ、宿主に因子を送達するために使用することができる。マイクロカプセル化は、米国特許第5,284,761号;同第5,158,881号;同第4,976,859号;同第4,968,733号;同第5,800,828号、およびPCT国際公開第95/05452号に記載されており、これらはそれぞれが参考として本明細書中に援用される。デバイスの中の細胞の数は変動し得る;好ましくは、個々のデバイスには103個〜109個の細胞(例えば、105個〜107個の細胞)が含まれる。複数のマイクロカプセル化デバイスを宿主に埋め込むことができる。
【0061】
レシピエントの組織と同種である組織から調製されたEPCは、レシピエントの組織適合性のタイプと厳密に適合させるために、周知の組織分類の方法によって使用について試験される。
【0062】
EPCは、しばしば、レシピエント自身の血液または骨髄から調製することができる。このような場合には、EPCは切り取られた組織から作成することができ、上記の方法を使用してインビトロで増殖させることができる。細胞数が適切に増加させられると、EPCを回収することができ、必要であれば遺伝的に修飾することができ、そしてレシピエントの血管への直接の注射のために準備することができる。
【0063】
EPCは、血管移植片を形成することができる血管に投与され、その結果、細胞は近隣の血管細胞とともに正常な結合を形成し、移植された内皮細胞または既存の内皮細胞との接触を維持することができる。このように、移植されたEPCは、疾患および加齢が原因で損傷した血管組織を再度構築させる。
【0064】
宿主の血管組織への移植片の機能的な一体化は、種々の機能を回復させることに対する移植片の有効性を試験することによって評価することができる。
【0065】
移植に使用されるEPCをインビトロで増殖させる能力はまた、エキソビボでの遺伝子治療にも有効である。したがって、EPCによって、エキソビボでの遺伝子治療試験においてビヒクルとして使用される内皮細胞を取り出し、増殖させる別の方法が提供される。
【0066】
(内皮前駆細胞の遺伝的修飾)
EPCは形質転換されていない初代細胞であるが、これらは、連続細胞株の特徴を有している。未分化の状態では、EPCは分裂し続け、したがって、遺伝的修飾の標的である。いくつかの実施形態においては、遺伝的に修飾された細胞は、上記の方法のいずれかによって内皮細胞または平滑筋細胞に分化するように誘導される。
【0067】
用語「遺伝的修飾」は、外来DNAの意図的な導入によるEPCの遺伝子型の安定な、または一時的な変更を意味する。DNAは合成のものである場合も、また自然界から導かれたものである場合もあり、そしてこれには、遺伝子、遺伝子の一部、または他の有用なDNA配列が含まれ得る。用語「遺伝的修飾」は、本明細書中で使用される場合は、自然界でのウイルス活性、自然界での遺伝子組み換えなどによって生じるような、自然に生じる変更が含まれるようには意味されない。
【0068】
細胞の任意の有用な遺伝的修飾は、本発明の範囲内である。例えば、EPCは、成長因子などの生物学的活性のある物質を生成するように、またはそれらの生成を増加させるように修飾することができる。1つの実施形態においては、生物学的活性のある物質は、遺伝的差異を調節する転写因子のような転写因子である。別の実施形態においては、生物学的活性のある物質は、非細胞分裂促進増殖因子であり、例えば、v−myc、SV−40ラージT、またはテロメラーゼである。
【0069】
遺伝的修飾は、ウイルスベクター(レトロウイルス、修飾されたヘルペスウイルス、へルペスウイルス、アデノウイルス、アデノ随伴ウイルスなど)での感染、または当該分野で公知の方法を使用するトランスフェクション(リポフェクション、リン酸カルシウムトランスフェクション、DEAE−デキストラン、エレクトロポレーションなど)のいずれかによって行われる(Maniatisら、Molecular Cloning:A Laboratory Manual(Cold Spring Harbor Laboratory,N.Y.,1982)を参照のこと)。例えば、キメラ遺伝子構築物には、ウイルス、例えば、レトロウイルス長末端反復(LTR)、シミアンウイルス40(SV40)、サイトメガロウイルス(CMV);または哺乳動物細胞特異的プロモーター(例えば、チロシンヒドロキシラーゼ(TH、ドーパミン細胞のマーカー)、DBH、フェニルエタノールアミンN−メチルトランスフェラーゼ(PNMT)、ChAT、GFAP、NSE、NFタンパク質(NE−L、NF−M、NF−Hなど)(これらは、所望されるタンパク質をコードする構造遺伝子の発現を指示する))が含まれ得る。さらに、ベクターには、薬物選択マーカー、例えば、E.coliアミノグリコシドホスホトランスフェラーゼ遺伝子を含めることができる。これは、試験遺伝子と同時に感染させられると、タンパク質合成の阻害因子であるゲネチシン(G418)に対する耐性を付与する。
【0070】
EPCは、発現ベクターでのトランスフェクションを使用して遺伝的に修飾することができる。1つのプロトコールにおいては、遺伝子を含むベクターDNAは、0.1×TE(1mMのTris、pH8.0、0.1mMのEDTA)の中に、40μg/mlの濃度になるように稀釈される。22μlのDNAが、使い捨ての滅菌の5mlのプラスチックチューブの中の250μlの2×HBS(280mMのNaCl、10mMのKCl、1.5mMのNa2HPO4、12mMのデキストロース、50mMのHEPES)に添加される。31μlの2MのCaCl2がゆっくりと添加され、混合液は室温で30分間(min)インキュベートされる。この30分間のインキュベーションの間に、細胞は4℃で5分間、800gで遠心分離される。細胞は、20倍容量の氷冷されたPBSに再懸濁させられ、1×107個の細胞のアリコートに分けられる。これは再び遠心分離される。細胞の個々のアリコートは、1mlのDNA−CaCl2懸濁液に再懸濁させられ、室温で20分間インキュベートされる。その後、細胞は増殖培地の中に稀釈され、5%〜7%のCO2の中で37℃で6〜24時間インキュベートされる。細胞は再び遠心分離され、PBS中で洗浄され、そして10mlの増殖培地に48時間戻される。
【0071】
EPCはまた、リン酸カルシウムトランスフェクション技術を使用して遺伝的に修飾される。標準的なリン酸カルシウムトランスフェクションについては、細胞が機械的に分離されて単細胞懸濁液とされ、50%の細胞集密度(50,000〜75,000細胞/cm2)で組織培養物処理皿にプレートされ、一晩付着させられる。1つのプロトコールにおいては、修飾されたリン酸カルシウムトランスフェクション手順が以下のように行われる:滅菌のTE緩衝液(10mMのTris、0.25mMのEDTA、pH7.5)中のDNA(15〜25μg)がTEで440μLに稀釈され、60μLの2MのCaCl2(1MのHEPES緩衝液でpHが5.8にされたもの)がDNA/TE緩衝液に添加される。全部で500μLの2×HeBS(HEPES緩衝生理食塩水;275mMのNaCl、10mMのKCl、1.4mMのNa2HPO4、12mMのデキストロース、40mMのHEPES緩衝液粉末、pH6.92)が、この混合物に一滴ずつ添加される。混合物は、室温で20分間静置される、細胞は、1×HeBSで軽く洗浄され、1mlのリン酸カルシウム沈殿させられたDNA溶液がそれぞれのプレートに添加され、細胞が37℃で20分間インキュベートされる。このインキュベーション後、10mlの培地が細胞に添加され、プレートはインキュベーター(37℃、9.5%のCO2)の中にさらに3〜6時間入れられる。DNAと培地は、インキュベーション時間が終わると吸引によって除去され、細胞が3回洗浄され、その後、インキュベーターに戻される。
【0072】
遺伝的修飾が生物学的活性のある物質の生成のためである場合には、この物質は、所定の血管障害の処置に有用である物質であり得る。EPCは、生物学的活性のある物質、例えば、成長因子、成長因子受容体を発現するように遺伝的に修飾される。例えば、細胞が増殖を誘導する成長因子または分化を誘導する成長因子を分泌するように細胞を遺伝的に修飾することが所望される場合がある。
【0073】
遺伝的に修飾されたEPCは、細胞治療または遺伝子治療のために、遺伝的修飾された細胞によって生成される生物学的活性のある分子が必要であるレシピエントのCNSに移植することができる。移植技術は以下に詳細に記載される。
【0074】
あるいは、遺伝的修飾されたEPCは、移植の前にインビトロで種々の分化プロトコールに供することができる。一旦細胞が分化させられると、これらは再び、所望されるタンパク質の発現についてアッセイされる。所望される表現形を有している細胞を単離することができ、これを、遺伝的修飾された細胞によって発現されるタンパク質または生物学的活性のある分子が必要であるレシピエントに移植することができる。
【0075】
本発明は、以下の実施例によってさらに説明されるが、これらに限定はされない。
【実施例】
【0076】
(実施例1)
(一般的方法)
(末梢血からのVEGFR−1+細胞、VEGFR−2+細胞、およびTie−2+細胞の単離)
VEGFR−1、VEGFR−2、またはTie−2のいずれかを発現する末梢血単核細胞(PBMC)を、ヒトの血液軟膜のFicoll密度勾配遠心分離を使用して単核細胞を単離することによって最初に得た。PBMCをいくつかのチューブに分けて入れ、それぞれが40×106個の細胞を含むようにした。15μlの抗VEGFR−1抗体(4μg/ml)、抗VEGFR−2抗体(20μg/ml)、または抗Tie−2抗体(5μg/ml)(抗体の詳細については表1を参照のこと)をそれぞれのチューブに添加し、室温で30分間インキュベートした。リン酸緩衝生理食塩水(PBS)で1回洗浄した後、細胞を10μlの1:10稀釈した二次ヤギ抗マウスモノクローナル抗体で標識した。細胞を、ストリンジェントなゲートを使用してフローサイトメトリーによって選別して、高度に精製されたこれらの細胞の集団を得た。
【0077】
(末梢血からのCD14+/VEGFR−2+細胞およびCD14+/VEGFR−2−細胞の単離)
末梢血単核細胞(PBMC)を、標準的な手順にしたがって、健常なヒトのボランティアの血液からFicollを用いた密度勾配遠心分離によって単離した。CD14+/VEGFR−2+細胞は、蛍光活性化細胞選別方法によって単離した。染色は以下のように行った:PBMCを、最初に、抗CD3抗体でコーティングされた磁気粒子(DYNAL,ノルウェー)とともにインキュベートして、Tリンパ球を除去した。この手順は製造業者によって記載されているように行った。残りの細胞をいくつかのチューブに分けて入れ、それぞれが20×106個の細胞を含むようにした。15μlの抗VEGFR−2抗体(20μg/ml、Reliatech,ドイツ)をそれぞれのチューブに添加し、室温で30分間インキュベートした。リン酸緩衝生理食塩水(PBS)で1回洗浄した後、細胞を、20μlの1:10稀釈した二次FITC結合ヤギ抗マウスモノクローナル抗体(Serotec,スウェーデン)と20μlのPE結合抗CD14抗体(4μg/ml、BD Biosciences,米国)で標識した。ストリンジェントなゲートを使用した細胞の選別の間に、高度に精製されたCD14+/VEGFR−2+画分、ならびに、CD14+/VEGFR−2−画分を、さらなる分析のために回収した。
【0078】
(ウェスタンブロッティング)
CD14+/VEGFR−2+細胞とCD14+/VEGFR−2−細胞(それぞれの集団について1×106個の細胞)の溶解物をSDS−PAGEによって分離させ、ニトロセルロース膜に移し、そして抗VEGFR−2抗体(1:200)で免疫ブロットした。西洋ワサビペルオキシダーゼに結合させたヤギ抗マウス二次抗体のF(ab’)2断片(1:2000)(Jackson Immunoresearch,米国)を、結合した抗体を同定するためにECLキット(Amersham)とともに使用した。
【0079】
(フローサイトメトリー、表現形分類、および細胞培養)
内皮前駆細胞の表現形分類は、造血細胞および内皮細胞上で発現される特異的マーカーに対する抗体のアレイを使用して、以前に記載された15ように行った(表1)。抗VEGFR−2はReliaTech(ドイツ)から入手し;抗VEGFR−1はR&D(英国)から;抗AcLDLはMolecular Probes(米国)から;抗フォン・ヴィレブランド因子−HLA、抗CD142、および抗HLAクラスIはDAKO(デンマーク)から;抗α−アクチンおよび抗線維芽細胞抗体はSerotec(スウェーデン)から;抗CD144、抗CD141、ヒト核抗体はChemicon(スウェーデン)から、そしてUlex europaeusはSigma(スウェーデン)から入手した。全ての他の抗体は、Becton Dickinson(米国)から購入した。特定のマーカーについては、細胞内染色をリン酸緩衝生理食塩水(PBS)中の5%のサポニンを使用して細胞の透過性を上昇させた後に行った。対応するコントロールのイソ型を、モノクローナル抗体(Mab)の非特異的結合の評価のために使用した。細胞は、Becton Dickinsonフローサイトメーター(FACSorter)上で分析した。選別したVEGFR−1+細胞、VEGFR−2+細胞、およびTie−2細胞を、フィブロネクチンでコーティングした組織培養プレートの上で、5mMのL−グルタミンと100μg/mlのペニシリン/ストレプトマイシン(PEST)を含む内皮選択培地MCDB 131(GIBCO,Gaithersburg,MD,米国)中で培養した。培地には、さらに、内皮細胞成長培地(EGM−2)シングルコート(singlequots)(Clonetics,Bio Whittaker,米国)を補充した。ヒトの臍帯静脈内皮細胞(HUVEC)はCloneticsから購入し、推奨される、EGM−2−MVシングルコートを補充したEGM−2−MV培地中で培養した。HUVECは、表現形分析および毛細血管形成アッセイにおいてコントロールとして使用した。
【0080】
FACSで選別した細胞をサイトスピンを使用してガラススライド上に置いた。免疫細胞化学を酵素染色によって行った。細胞を以下の抗体:抗CD144(VE−カドヘリン)、抗CD141(トロンボモジュリン)(いずれも、Beckton Dickinson,米国による)、抗VEGFR−1(R&D Systems,英国)、および内皮一酸化窒素合成酵素(e−NOS)に対する抗体(全ての抗体を1:50希釈で使用した)で室温で1時間染色し、PBSで3回洗浄し、二次ビオチニル化ヤギ抗マウス抗体(1:500)で染色した。アビジン−ペルオキシダーゼ手順を、Vectastain Elite ABCキット(ImmunKemi,Stockholm,スウェーデン)を使用して、製造業者によって記載されているように行った。ACE(赤色の染色を生じる)またはDABNickel(茶/黒色の染色を生じる)基質キットを発色現像液として使用した。多数の細胞がCD14+/VEGFR−2−集団から得られたので、これらの細胞をさらに、先に記載された16ようにフローサイトメトリーによって、単球および内皮細胞上で発現される特異的マーカーに対する抗体のアレイを使用して特性決定した(表3)。FITC結合アセチル化低密度リポタンパク質(AcLDL)(Molecular Probes,米国)、フォン・ヴィレブランド因子に対する抗体(DAKOPATTS,デンマーク)、α−アクチン、線維芽細胞(Serotec,スウェーデン)、およびUlex europaeus(Sigma,スウェーデン)を使用した。全ての他の抗体は、Becton Dickinson(米国)から購入した。対応するコントロールのイソ型を、モノクローナル抗体(Mab)の非特異的結合の評価のために使用した。細胞をBecton Dickinsonフローサイトメーター(FACSorter)上で分析した。
【0081】
いくつかの実験については、選別したCD14+/VEGFR−2+細胞とCD14+/VEGFR−2−細胞をフィブロネクチン(20μg/ml)をコーティングした組織培養プレート上で、内皮選択培地EndoCultTM(Stem Cell Technologies,カナダ)の中で培養した。これらの培養物を、増殖および形態について観察した。1週間の培養後、細胞をガラス上でサイトスピンし、VE−カドヘリンの発現について染色した。
【0082】
ヒトの大動脈内皮細胞(HAEC)はCloneticsから購入し、推奨される培地の中で培養した。HAECを、表現形の分析と細管形成アッセイにおいてコントロールとして使用した。
【0083】
(透過型電子顕微鏡)
細胞を、24ウェルプレートの中で膜フィルター上で増殖させた。電子顕微鏡分析を、Karolinska University hospital−Huddinge(スウェーデン)で電子顕微鏡についての中心となる設備で行った。CD14+/VEGFR−2+細胞とCD14+/VEGFR−2−細胞を、24ウェルプレート中の膜フィルター上で別々に増殖させた。膜を2%のグルタルアルデヒド中に固定し、蒸留水で軽くリンスし、70%のエタノール中に10分間入れ、そして99.5%のエタノール中に15分間入れ(全て4℃で)、乾燥させた。固定後、膜を切り、0.15Mのカコジル酸ナトリウム、1%の四酸化オスミウム、および3mMのCaCl2(pH7.4)を含む緩衝液中で4℃で1時間固定させた。その後、ウェルを0.15Mのカコジル酸ナトリウム緩衝液中で軽くリンスし、上記のようにエタノールで脱水し、スパー樹脂(Agar Scientific LTD,Essex,英国)に包埋した。切片を、酢酸ウラニル、その後、クエン酸鉛で対比させ、Leo 906(Oberkochen,ドイツ)透過型電子顕微鏡において80kVで試験した。
【0084】
(移動アッセイ)
新しく単離したVEGFR−1+細胞、VEGFR−2+細胞、およびTie−2+細胞(5×104個)を、それぞれの細胞タイプについて、3.0μmの孔径を有している4つのフィブロネクチンでコーティングされたTranswellインサートに添加した。内皮前駆細胞の移動を、5%のウシ胎児血清を含む培地中で、下部チャンバーに血管内皮成長因子(VEGF)を含まない条件、ならびに、0ng/ml、0.5ng/ml、2ng/ml、および10ng/mlの種々の濃度の血管内皮成長因子(VEGF)を含む条件でアッセイした。細胞を22時間移動させた。移動した細胞を回収し、血球計数器において数えた。同様の実験を、アンジオポイエチン−2(Tie−2のリガンド)を、0ng/ml、100ng/ml、200ng/ml、および300ng/mlの濃度で使用して行った。それぞれの実験を3連のウェルで行い、4回繰り返した。
【0085】
新しく単離したCD14+/VEGFR−2+細胞とCD14+/VEGFR−2−細胞(5×104個)を、全て3.0μmの膜孔径を有している4つのフィブロネクチンでコーティングされたTranswellインサートに添加した。細胞の移動を、5%のウシ胎児血清を含む培地中で、下部チャンバーに50ng/mlの血管内皮成長因子(VEGF)または300ng/mlの単球走化性因子−1(MCP−1)を含まない条件、またはそれらを含む条件でアッセイした。細胞を22時間移動させた。移動した細胞を回収し、血球計数器において数えた。それぞれの実験を3連のウェルで行い、4回繰り返した。
【0086】
(CD14+/VEGFR−2+細胞とCD14+/VEGFR−2−細胞の集団による血管由来成長因子の分泌)
成長因子の分泌を評価するために、新しく単離した細胞を、成長因子および血清を含まない培地の中で72時間培養した。全ての培養物から上清を回収し、さらに分析するまで−70℃で保存した。馴化培地を、血管由来成長因子、VEGF、顆粒球コロニー刺激因子(G−CSF)、顆粒球マクロファージコロニー刺激因子(GM−CSF)、線維芽細胞成長因子(FGF)、および間質由来因子−1(SDF−1)についてアッセイした。これらの成長因子は、ELISA(R&D systems)によってアッセイした。
【0087】
(インビトロでの血管形成アッセイ)
毛細管様の細管構造の形成を、原則として先に記載された16ように、マトリゲルでコーティングしたマルチウェルプレートにおいて評価した。新しく単離したVEGFR−1+細胞、VEGFR−2+細胞、およびTie−2+細胞を、マトリゲルでコーティングしたマルチウェルプレート中の増殖培地に30×104個の細胞/mlの密度で播種し、37℃でインキュベートした。細管構造の形成を、Nikon TE3000 microscopeを使用して22時間後に調べた。
【0088】
新しく単離したCD14+/VEGFR−2+細胞とCD14+/VEGFR−2−細胞による毛細管様構造の形成を、インビトロでの血管形成の評価のために頻繁に使用されるEngelbreth−Holm−Swamマウス肉腫から抽出した可溶化基底膜調製物中でアッセイした。24ウェルプレートを200μlのマトリゲル(5%のCO2中で37℃で30分間予めゲル状にした)でコーティングし、細胞を、重合させたマトリックス上に、5×104個の細胞/ウェルの密度で播種した。得られた管様構造を、細胞を5%のCO2中で37℃で1週間培養した後、位相差顕微鏡を使用して調べた。HAECをポジティブコントロール細胞として使用した。
【0089】
(インビトロでの細胞侵襲アッセイ)
再構成させた基底膜マトリゲルからの細胞侵襲を、以前に報告された17方法によってアッセイした。移動した細胞を、×100倍の倍率で光学顕微鏡下で5つの異なる領域において数えた。それぞれの実験を3連のウェルで行い、4回繰り返した。
【0090】
(FACSによって選別したCD14+/VEGFR−2+細胞とCD14+/VEGFR−2−細胞のレンチウイルスでの形質導入)
組み換え体であるレンチウイルスベクターを、先に記載された173プラスミド発現システムを使用して生成した。形質導入のために、FACSで選別した細胞を、1mlの培養培地を含む10mlのチューブに移し、0.1のMOIの濃縮されたpHR’EF1−GFPSINビリオンとともに、5%のCO2雰囲気下で一晩37℃でインキュベートした。ウイルスのインキュベーションの後、細胞をPBS中で1回洗浄し、ヌードマウスの露出させた大腿動脈に直接注入した(下記を参照のこと)。GFPの発現を検出するために、同じ形質導入手順にしたがって、形質導入した細胞を6ウェルプレート中で培養し、形質導入の6日後に蛍光顕微鏡によって観察した。
【0091】
(マウス)
右大腿動脈のバルーン損傷を以下のように行った:体重20〜25gのnude C57blackマウス(Taconic M&B,デンマーク)をイソフルランで麻酔し、腹部横切開によって大動脈分岐部のレベルまで右大腿動脈を露出させた。マイクロクランプ(S&T,スイス)を下部大動脈、左腸骨動脈、および右大腿動脈の遠位部分の上に配置した。2F Fogartyバルーンカテーテル(Baxter)を右大腿動脈に挿入し、膨張させ、回転させながら3回前後に動かした。膨張は、カニューレと、1mlのリンガー溶液によって、微小切開部分から自由に流出させて行った。100μlのMCDB培地中の細胞(5×105細胞/動物)(CD14+/VEGFR−2+、n=10;CD14+/VEGFE−2−、n=10)を同じカニューレから染み込ませ、15〜20分間、新しく損傷させた動脈床の中でインキュベートした。一方、コントロール移植マウスには、培養培地だけを投与した(マウス、n=6)。インキュベーション後、結合していない細胞を吸引し、カテーテルを抜き取り、微小切開を11−0ナイロン(S&T,スイス)結節縫合によって縫合した。マイクロクランプを取りはずすことによって血流を回復させた。全ての動物への手順は、機関のガイドライン、スウェーデンのHuddinge University hospitalの実験動物の管理と使用に関する指針(Guide for the Care and Use of Laboratory Animals)に則って行った。動物を、ヒトの細胞の移植の2週間後および4週間後に屠殺し、右大腿動脈を組織病理学的試験のために回収した。血管をO.C.T.に包埋し、液体窒素で凍結させた。
【0092】
インビボでのヒトVEGFR−2+細胞およびTie−2+細胞中でのDNA合成の活性化を明らかにするために、チミジンアナログであるBrdU(Sigma,スウェーデン)を、終了の12時間前および24時間前に、100mg/kg体重で1回i.p.注射として投与した。
【0093】
(免疫組織化学)
マウスの動脈内にヒト細胞を局在化させるために、5μmの凍結切片を抗GFP抗体(1:50)で染色した。免疫ペルオキシダーゼ手順をVectastain Elite
ABCキット(ImmunKemi,Stockholm,スウェーデン)を使用して、製造業者によって記載されているように行った。ACE(赤色の染色を生じる)基質キットを発色現像液として使用し、ヘマトキシリンで対比染色した。さらに、マウス抗ヒト核モノクローナル抗体(Chemicon,CA,米国)を使用して、免疫蛍光18によってマウスの動脈中のヒト細胞を検出した。移植されたヒト細胞の表現形を、ヒト核に対する抗体(1:50)と抗VE−カドヘリン抗体(1:100)での二重染色によって検出した。この後、二次抗マウスサブクラス特異的FITC抗体またはテキサスレッド結合抗体(1:500)で染色した。内皮細胞化を、ヒトACEポジティブ細胞に覆われた表面と全発光表面の割合として計算した。組織形態学(1つのグループについて6匹の動物、6つの切片/大腿動脈)については、動脈の横切片をヘマトキシリン/エオシンで染色し、血管径、中膜の面積、および中膜に対する内膜の面積の比を調べた。
【0094】
(逆転写ポリメラーゼ連鎖反応(RT−PCR))
全RNAを3つのヒト−マウスキメラ大腿動脈(1つはCD14+/VEGFR−2−細胞を投与したマウスであり、1つは擬似移植したマウス)から、移植の4週間後に、Micro−FastTrack RNA単離キット(Invitrogen,Groningen,オランダ)を使用して抽出した。ヒト特異的プライマーを使用して、ヒトCD31およびe−NOSを、ヒト細胞を移植したマウスの動脈の中で検出した。新しく単離したCD14+/VEGFR−2+細胞およびCD14+/VEGFR−2−細胞もまた、e−NOSの発現について試験した。ABLを内因性の参照遺伝子として使用した。プライマーのセットは、CyberGene(Huddinge,スウェーデン)によって商業的に合成された。プライマー配列は、
【0095】
【数1】
PCR反応は先に記載された19ように行った。
【0096】
(統計分析)
適切である場合には、結果を平均±SDとして表した。対応のないt検定を、EPC移植したグループおよび擬似(媒体のみで)処置したグループの間での比較のために使用した。p<0.05を有意と考えた。Mann−Whitney U検定を、グループ間での比較のために使用した。p<0.05を有意と考えた。
【0097】
(実施例2)
(末梢血由来のVEGFR−1+細胞、VEGFR−2+細胞、およびTie−2+細胞は単球/マクロファージ系統に属する)
VEGFR−1+細胞、VEGFR−2+細胞、およびTie−2+細胞は、それぞれ、血液中の単核細胞の集団全体のおよそ3±0.2%、0.8±5%、および2±0.3%を占めることが明らかになった。これらの3つの集団の表現形の分析により、複数のタイプの細胞の混合物が、主に、単球/マクロファージ細胞系統に属することが明らかになった(表1)。新しく単離したVEGFR−1+細胞およびTie−2+細胞は、内皮細胞マーカーであるVE−カドヘリン(CD144)は発現しなかった。しかし、新しく単離したVEGFR−2+細胞の小さい割合がこのマーカーを発現した。さらに、3つの集団の全てはCD45マーカーを発現しており、これは、これらの細胞が造血起源のものであることを示している。VEGFR−1+細胞、VEGFR−2+細胞、およびTie−2+細胞は、それらのそれぞれのマーカーについてポジティブに染色された(図1A〜C)。新しく単離したVEGFR−1+細胞、VEGFR−2+細胞、およびTie−2+細胞の電子顕微鏡による実験により、表現形の分析による発見がさらにサポートされた。3つの集団の全部に、単核単球、マクロファージ、リンパ球、および多形核細胞を含む細胞の混合物が含まれていた(図1D〜F)。
【0098】
【表1−1】
【0099】
【表1−2】
(実施例3)
(培養されたVEGFR−2+細胞とTie−2+細胞は内皮細胞マーカーだけを示す)
フィブロネクチンでコーティングされた培養ウェル上でのこれらの細胞の培養により、付着細胞(80%)と非付着細胞(20%)の2つの集団が示された。1週間後、非付着細胞を廃棄した。これは内皮細胞について記載した条件下ではさらに培養することはできなかった。2週間の培養後、VEGFR−2+培養物およびTie2+培養物中の付着細胞の大部分は内皮細胞マーカーであるCD144、CD141を発現し、活性化されると、CD106とCD142を発現した(表2)。電子顕微鏡を使用したさらなる分析によってこれらの細胞の内皮形態を確認すると、開口、バイベル・パラーデ小体、および内皮細胞に典型的な堅い結合部分の存在が明らかになった(図1G〜I)。一方、VEGFR−1+細胞の小さい画分しか2週間後には付着したままではなく、さらなる分析のために十分な細胞を得ることはできなかった。VEGFR−2+細胞もTie−2+細胞も、培養物中では増殖しなかった。しかし、これらの集団は、>4週間の間、増殖せずに培養物で生存した状態で維持することができた。
【0100】
(表2 2週間の培養後のVEGFR−2+細胞とTie−2+細胞の表現形の特徴)
【0101】
【表2】
(実施例5)
(VEGFR−2+細胞とTie−2+細胞は移動能力と侵襲能力を有しており、細管様構造を形成する)
新しく単離したVEGFR−2+細胞とTie−2細胞は、VEGFとアンジオポイエチン−2に向かって移動したが、VEGFR−1+細胞は移動しなかった(図2AおよびB)。しかし、マトリゲルに添加したVEGFR−1+細胞の60%、VEGFR−2+細胞の70%、そしてTie−2+細胞の65%が、侵襲アッセイにおいて下部チャンバーに移動し、このことは、3つの細胞タイプの全てが侵襲能力を有していることを示している(図2C)。マトリゲルの中では、これらの新しく単離した細胞は、VEGFR−2+細胞とTie−2+細胞だけが細管様構造を形成することができ、VEGFR−1+細胞はそのような能力を有していないことが示された(図2D〜G)。HUVECをコントロールとして使用した。本発明者らのインビトロでの実験では、VEGFR−2+細胞とTie−2+細胞は、表現形の上でも、形態学的にも似ている内皮細胞であることを示していたが、VEGFR−1+細胞は内皮様の特徴を示さなかった。したがって、VEGFR−2+細胞とTie−2+細胞の、バルーン損傷した動脈の再内皮細胞化に関与するインビボでの機能的能力をさらに分析した。
【0102】
(実施例6)
(VEGFR−2+細胞とTie−2+細胞は剥離したマウスの大腿動脈の効率のよい再生に関与しており、インビボで増殖した)
実験用の血管形成術手順を、ヌードマウスの大腿動脈に行った。この介入の直後、VEGFR−2+細胞またはTie−2+細胞を新しく損傷させた動脈床に局所注入した。移植された血管は、血栓の兆候は示さなかった。ヒト核抗体の特異性を、正常なヒトおよびマウスの動脈を使用して最初に決定した。ヒト細胞は黒色に染色された(図3A)。コントロールのマウスの動脈はヒト核抗体では染色されなかった(図3B)。移植の2週間後には、ヒトVEGFR−2+細胞は播種されたバルーン損傷させられた大腿動脈の内腔表面で検出された(DAB−Ni−ポジティブ、図3C)。移植の4週間後には、高度な再生が見られ、およそ75±8%の病変がヒトVEGFR−2+細胞で覆われていた。同様に、Tie−2+細胞が移植後2週間および4週間に検出された(図3D)。Tie−2+細胞を移植した動物は、移植の4週間後には、高度な再生を示し、病変のおよそ70±5%がヒトTie−2+細胞で覆われていた。
【0103】
マウスの動脈内でのヒト細胞の存在を、今一度、免疫蛍光試験において確認した(図3E〜F)。インビボでのEC表現形を、フォン・ヴィレブランド因子に対する抗体とVE−カドヘリン(CD144)に対する抗体での染色によって確認した(図4AおよびB)。それぞれの切片を、ヒト細胞を宿主細胞と区別するために、抗ヒト核抗体で二重染色した(黄色い染色は二重にポジティブである細胞を示している)。興味深いことに、移植されたヒト細胞は、播種された動脈の内腔表面だけではなく、間質中膜においても見ることができた。α−アクチンに対する抗体で標識した場合には、中膜の中の細胞はポジティブに染色され、これは平滑筋細胞の存在を示していた(図4G〜I)。したがって、VEGFR−2+細胞とTie−2+細胞はいずれも、インビボでは、内皮細胞だけではなく、平滑筋細胞にも分化した。VEGFR−2+細胞もTie−2+細胞も、インビトロでは増殖しなかったが(データは示さない)、移植されたVEGFR−2+細胞とTie−2+細胞の増殖についての証拠を得るために、BrdUの取り込み試験を、8匹の動物(VEGFR−2+:n=4、Tie−2+:n=4)において行った。7日目での組織の分析は、移植されたヒト細胞中での広範囲に及ぶBrdUの取り込みを示した。抗BrdU抗体と抗ヒト核抗体の両方でDNAを染色したので、同じ組織切片の二重染色はできなかった。したがって、連続する切片を抗BrdU(茶色)抗体と抗ヒト核抗体(黒色)で染色して、移植された細胞の増殖を示した(図5A〜F)。これらの研究結果は、VEGFR−2+細胞とTie−2+細胞は損傷した動脈を再生させるだけではなく、インビボでは増殖もすることを示している。
【0104】
(実施例7)
(内皮前駆細胞での再内皮細胞化によっては、マウスの大腿動脈のバルーン損傷後に新生内膜の増殖は生じなかった)
損傷した動脈のセグメントの横断面を、移植の2週間後およびよび4週間後に、組織形態学的変化について調べた。マウスの大腿動脈の中でのバルーン損傷手順によって、病変の動脈セグメントの膨張を生じさせ、その結果、血管の直径は顕著に大きくなり、損傷させていないコントロールの動脈についての0.362±0.060mmから、媒体だけと一緒にインキュベートした損傷させた血管については0.601±0.036mmにまでなった。この拡大は、ヒト細胞を移植したマウスの大腿動脈においては見られなかった(図5G)。断片の中膜面積の0.020±0.004から0.010±0.009mm2までの減少が、損傷した動脈において観察された。中膜面積の薄層化は、VEGFR−2細胞およびTie−2細胞を移植した動脈においては見られなかった(図5H)。EPCの播種によって媒介される再内皮細胞化によっては、損傷の2週間後、4週間後、または6週間後には、大腿動脈において新生内膜形成は生じなかった。実際、中膜に対する血管内膜の面積比は、VEGFR−2およびTie−2を移植した血管セグメントにおいては、有意には高くなかった(I/M比:正常な損傷させていない動脈 0.01±0.01、媒体のコントロール 0.02±0.01、VEGFR−2+ 0.02±0.01;Tie−2+ 0.02±0.01,p=ns)。
【0105】
(実施例8)
(CD14+/VEGFR−2+とCD14+/VEGFR−2−の特性決定)
CD14+/VEGFR−2+細胞が単球の集団全体のおよそ2±0.5%(図1A)を占め、血液中のPBMCの0.08±0.04%を占めることが明らかになった。免疫細胞化学分析によっては、新しく単離したCD14+/VEGFR−2−細胞画分はe−NOSを発現しないことが明らかにされた(図6B)。しかし、新しく単離したCD14+/VEGFR−2+細胞は、e−NOS、フォン・ヴィレブランド因子、およびトロンボモジュリンを発現していたが(図6C)、VE−カドヘリンは発現していなかった。CD14+/VEGFR−2+細胞上でのVEGFR−2受容体の発現は、この集団の溶解物が、抗VEGFR−2抗体で免疫ブロットした場合には、およそ200kDaの予想されたバンドを生じ、これはCD14+/VEGFR−2−細胞の集団においては存在しなかったかまたはわずかに発現されていたという事実によってさらにサポートされた(図6D)。さらに、e−NOS mRNAはCD14+/VEGFR−2+細胞中では強く発現されていたが、VEGFR−2ネガティブ画分においては、かすかなe−NOS mRNAのバンドしか見ることができなかった(図6E)。本発明者らは、このかすかなバンドが、CD14+/VEGFR−2−細胞の集団の中に少数のVEGFR−2+細胞が混入しているとの理由から起こり得るとの事実を排除することはできなかった。CD14+/VEGFR−2−集団によって発現される表現形マーカーを表3に示す。いずれの集団もCD45マーカーを発現し、このことは、これらの細胞が造血起源であることを示している。Transwell組織培養インサート上で培養したCD14+/VEGFR−2+細胞の電子顕微鏡による分析によっては、基底膜の形成、バイベル・パラーデ小体の存在、および内皮細胞の特徴である堅い結合が明らかにされた(図7A〜C)。一方、ネガティブな画分はこれらの特徴を示さなかった。
【0106】
CD14+/VEGFR−2+細胞は、フィブロネクチンでコーティングした培養ウェル上で培養した場合には、3日間の培養後にコロニーを形成した。これらのコロニーは、その後数日のうちに、固まった丸い細胞の中心から、一見成長した平たい長く伸びた細胞へと形態を変化させた。一方、CD14+/VEGFR−2集団は、2週間の間、若干丸い細胞として付着したままであったが、クラスターは形成しなかった(図7D)。2つの集団のいずれも、本実験で使用した培養条件下では広範囲の増殖能力は示さなかった。培養したCD14+/VEGFR−2+細胞は、1週間の培養後にVE−カドヘリンを発現していたが(図7D)、VEGFR−2−画分は培養の間のいずれの時点でも、このマーカーを発現することはなかった。
【0107】
【表3】
(実施例9)
(CD14+/VEGFR−2+末梢血細胞とCD14+/VEGFR−2−末梢血細胞のインビトロでの機能的能力)
明らかに多数の新しく単離したCD14+/VEGFR−2+細胞が、CD14+/VEGFR−2−細胞と比較して、VEGFおよびMCP−1に対して移動した(図8A)(それぞれ、p=0.009およびp=0.047)。2つの集団によって生成された血管由来成長因子には有意な相違はなかった。いずれの集団もVEGF、G−CSF、およびGM−CSFを生成したが、SDF−1またはFGFは生成しなかった。
【0108】
しかし、成長因子のレベルは、CD14+/VEGFR−2−集団と比較して、CD14+/VEGFR−2+集団において高かった(図8B)。マトリゲルの中では、3日間培養した細胞は、CD14+/VEGFR−2+細胞だけではなくCD14+/VEGFR−2−細胞も、細管様構造を形成することができた(図8C)。HAECをコントロールとして使用した。
【0109】
したがって、インビトロでのデータは、新しく単離したCD14+/VEGFR−2+細胞が、CD14+/VEGFR−2−細胞と比較して内皮様の特徴を示すことを示している。
【0110】
(実施例10)
(CD14+/VEGFR−2+細胞は内皮細胞へと分化し、剥離されたマウスの大腿動脈を効率よく再生する)
GFPを発現するレンチウイルス粒子に対する2つの細胞集団の暴露によっては、一貫して、>70%の形質導入効率が導かれた。ヒト細胞の動脈切片のスクリーニングは、高い自己蛍光が原因で、GFP+細胞を視覚化することが困難であることを示した。したがって、酵素をベースとする免疫組織化学染色を、抗GFP抗体を使用して行い、GFP+細胞を検出した。
【0111】
この抗体を使用すると、擬似移植したマウスおよびCD14+/VEGFR−2−を移植したマウスにおいては、GFP−ポジティブである領域は見られなかった(図9A〜C)。しかし、GFP−ポジティブ細胞(赤)を有している動脈の内膜表面の範囲が、CD14+/VEGFR−2+細胞を移植したマウスにおいて移植の2週間後にすでに見られた(図9D)。GFP−ポジティブである領域は、播種の4週間後に回収された血管においては明らかに増大していた(図9EおよびF)。さらに、ヒト細胞もまた、免疫蛍光を使用して抗ヒト核抗体によって同定した。ヒト核抗体の特異性を、最初に正常なヒト動脈およびマウス動脈を使用して決定した。ヒト細胞−核は赤色に染色された(図10A)。コントロールマウスの動脈は、ヒト核抗体では染色されなかった(図5B)。移植の4週間後、CD14+/VEGFR−2+ヒト細胞を投与したマウスにおいては高い程度の再生が見られ、病変のおよそ70±8%が、ヒト細胞(赤)で覆われていたが(図10C)、CD14+/VEGFR−2−細胞を移植したマウスにおいては覆われていなかった(図10D)。CD14+/VEGFR−2−細胞を移植した動物は、剥離した動脈、脾臓、肺、または循環においても、移植の2週間後または4週間後には、これらの細胞の存在は示さなかった(データは示さない)。インビボでの内皮細胞の表現形を、VE−カドヘリンに対する抗体での染色(緑)によって確認し、それぞれの切片を、抗ヒト核抗体で二重染色して、ヒト細胞(赤)を検出した。二重染色された細胞は黄色に染色された(図10E)。
【0112】
損傷した動脈切片の横断切片を、組織形態学的変化について移植の2週間後および4週間後に調べた。マウスの大腿動脈でのバルーン損傷手順によって、病変の動脈セグメントの膨張を生じさせ、その結果、血管内径を顕著に大きくし、損傷させていないコントロールの動脈についての0.301±0.041mmから、媒体だけまたはCD14+/VEGFR−2−と一緒にインキュベートした損傷させた血管については、それぞれ、0.601±0.043mmおよび0.631±0.028mmにまでなった(p<0.001)。この拡大は、CD14+/VEGFR−2+細胞を移植したマウスの大腿動脈においては見られなかった(0.404±0.08mm)。横断面の中膜面積の0.028±0.010から0.012±0.007mm2までの減少が、損傷した動脈において観察された。中膜面積の薄層化は、CD14+/VEGFR−2+細胞を移植した動脈においては見られなかった(0.033±0.013)。これらの細胞によって媒介される再内皮細胞化によっては、損傷の2週間後または4週間後には、大腿動脈において新生内膜形成は生じなかった。中膜に対する血管内膜の面積比は、CD14+/VEGFR−2+を移植した血管セグメントにおいては、有意には高くなかった(I/M比:正常な損傷させていない動脈 0.03±0.009、媒体のコントロール 0.02±0.008、CD14+/VEGFR−2− 0.02±0.009;CD14+/VEGFR−2+ 0.04±0.02,p=ns)。
【0113】
(実施例11)
(CD14+/VEGFR−2+細胞を移植したマウスにおけるヒト内皮特異的遺伝子の転写)
移植した細胞の生着を、移植したマウスの中でのヒト遺伝子の発現を決定することによって確認した。ヒト細胞の移植後1ヶ月で屠殺したマウスの大腿動脈を、ヒト内皮特異的遺伝子CD31およびe−NOSに特異的なプライマーを使用して、RT−PCRによって分析した。CD31およびe−NOSプライマーは、それらがそれぞれのマウス遺伝子を増幅しなかったので、ヒトに特異的な種であった(図11A)。これらの結果は、CD14+/VEGFR−2+細胞を投与したマウスに由来する動脈について得られたが、CD14+/VEGFR−2−細胞を移植したマウスにおいては得られなかった。これらのデータは、移植したヒトCD14+/VEGFR−2+細胞が損傷したマウスの動脈に生着したことを示している。
【0114】
(他の実施形態)
特定の実施形態が本明細書中に詳細に開示されているが、これは例示の目的のための例であり、以下に添付される特許請求の範囲の範囲に関する限定とは意図されない。具体的には、種々の置換、変更、および改変を、特許請求の範囲によって定義される本発明の精神および範囲から逸脱することなく本発明に対して行うことができることが、発明者らによって企図される。核酸出発物質、目的のクローン、またはライブラリーのタイプの選択は、当業者にとっては、本明細書中に記載された実施形態の知識にしたがって当業者が日常的に行われている事柄であると考えられる。他の局面、利点、および改変も、以下の特許請求の範囲内であると考えられる。
【特許請求の範囲】
【請求項1】
本明細書に記載の発明。
【請求項1】
本明細書に記載の発明。
【図2A】

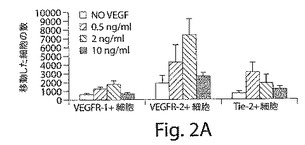
【図2B】
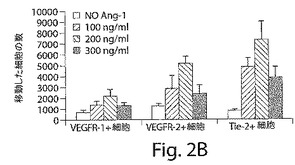
【図2C】

【図5G】
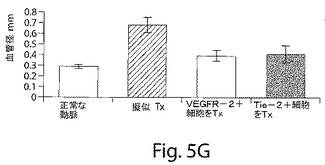
【図5H】
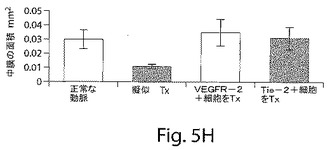
【図6A】
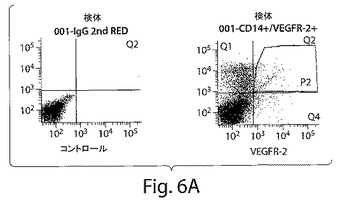
【図6B】

【図6C】
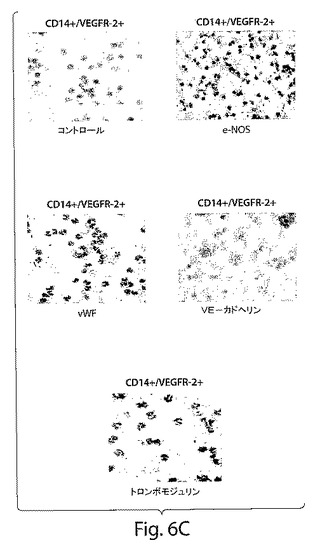
【図6E】
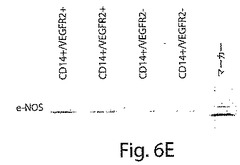
【図7】
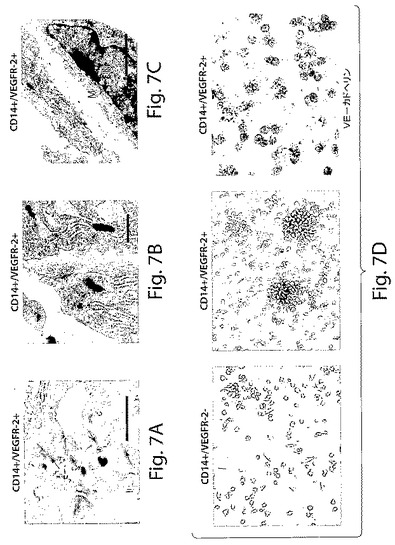
【図8A】
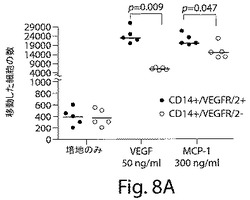
【図11】
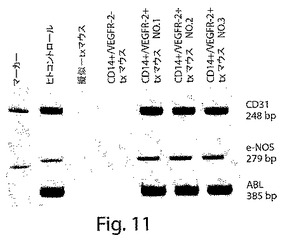
【図1】
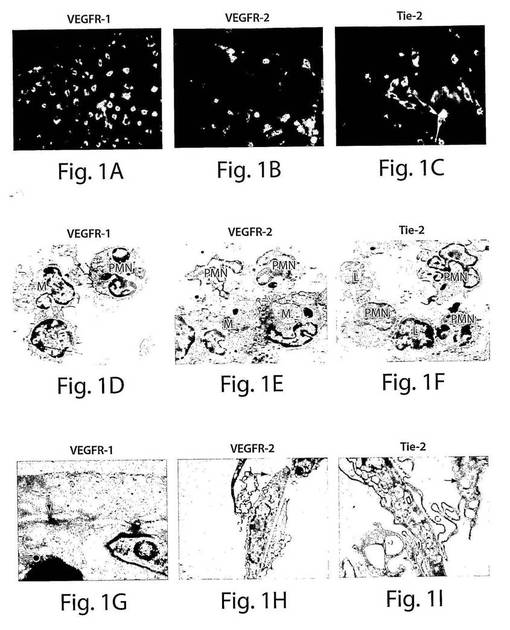
【図2D】

【図2E】

【図2F】
【図2G】
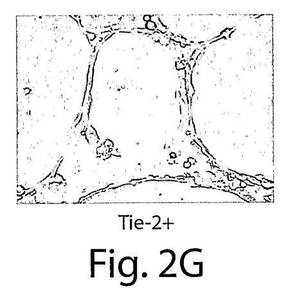
【図3】
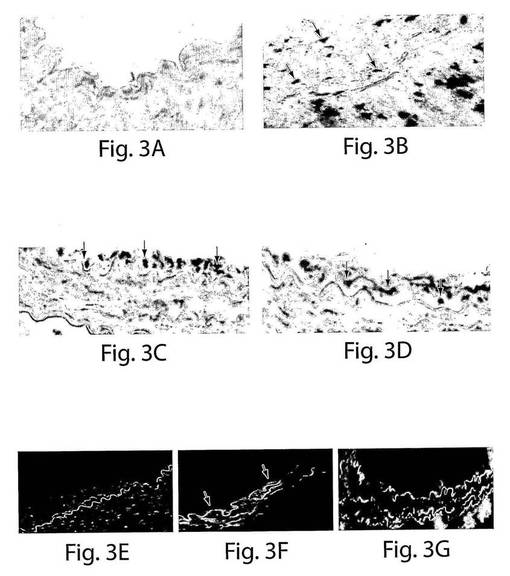
【図4】

【図5A】
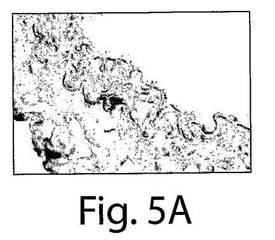
【図5B】
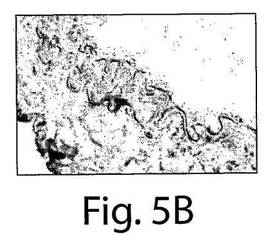
【図5C】
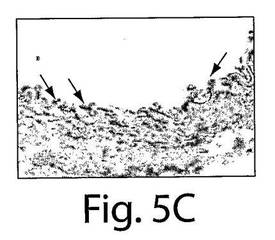
【図5D】
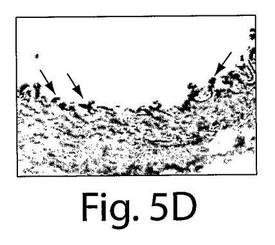
【図5E】

【図5F】
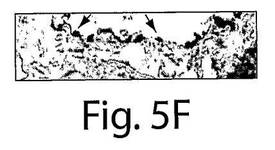
【図6D】
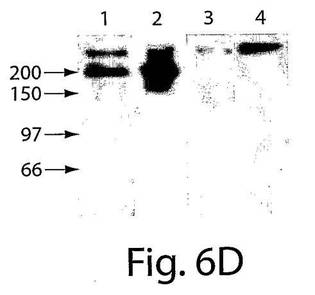
【図8B】
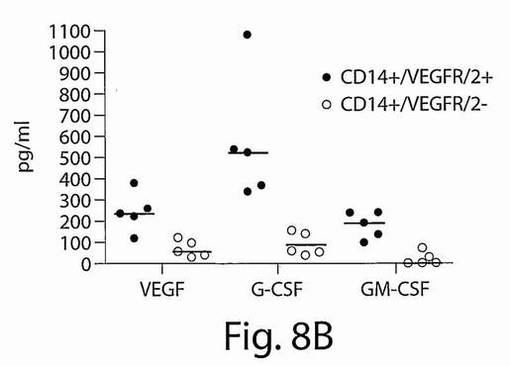
【図8C】
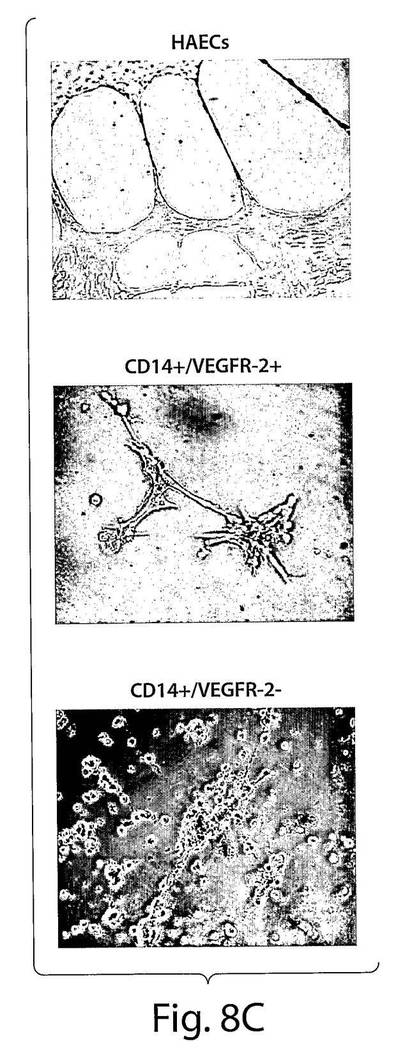
【図9】
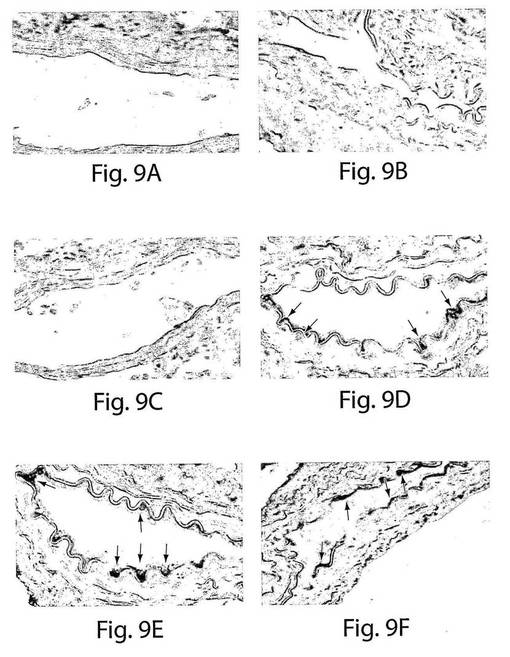
【図10】
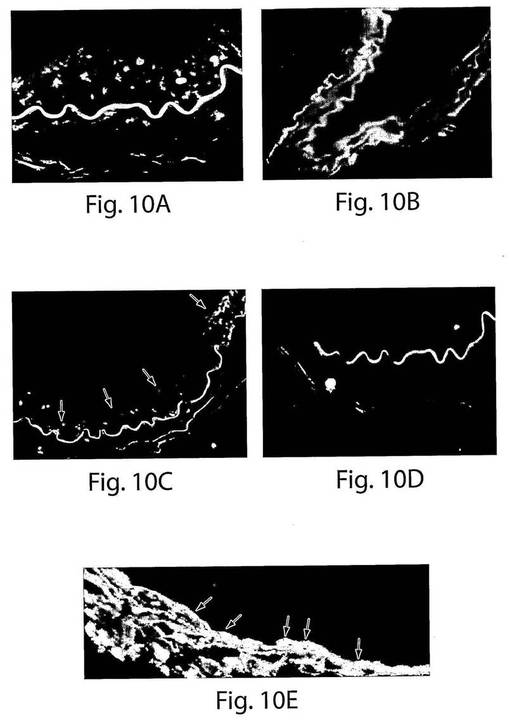
【図2B】
【図2C】
【図5G】
【図5H】
【図6A】
【図6B】
【図6C】
【図6E】
【図7】
【図8A】
【図11】
【図1】
【図2D】
【図2E】
【図2F】
【図2G】
【図3】
【図4】
【図5A】
【図5B】
【図5C】
【図5D】
【図5E】
【図5F】
【図6D】
【図8B】
【図8C】
【図9】
【図10】
【公開番号】特開2012−210227(P2012−210227A)
【公開日】平成24年11月1日(2012.11.1)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−164885(P2012−164885)
【出願日】平成24年7月25日(2012.7.25)
【分割の表示】特願2007−502440(P2007−502440)の分割
【原出願日】平成17年3月9日(2005.3.9)
【出願人】(506304990)アブソルバー, アーベー (4)
【Fターム(参考)】
【公開日】平成24年11月1日(2012.11.1)
【国際特許分類】
【出願番号】特願2012−164885(P2012−164885)
【出願日】平成24年7月25日(2012.7.25)
【分割の表示】特願2007−502440(P2007−502440)の分割
【原出願日】平成17年3月9日(2005.3.9)
【出願人】(506304990)アブソルバー, アーベー (4)
【Fターム(参考)】
[ Back to top ]