
凍結乾燥製剤
【課題】再溶解性(reconstitution)を損なうことなく、保存安定性に優れた化合物Aを含有する凍結乾燥製剤を提供する。
【解決手段】(4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸またはその塩、またはそれらの溶媒和物を、0.6%以上の塩化ナトリウムと10〜50%の親水性溶媒を含む水溶液に溶解し、該溶液を凍結乾燥することにより調製された、上記化合物またはその塩、またはそれらの溶媒和物と塩化ナトリウムを含有する、実質的に炭酸塩を含まない凍結乾燥製剤。
【解決手段】(4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸またはその塩、またはそれらの溶媒和物を、0.6%以上の塩化ナトリウムと10〜50%の親水性溶媒を含む水溶液に溶解し、該溶液を凍結乾燥することにより調製された、上記化合物またはその塩、またはそれらの溶媒和物と塩化ナトリウムを含有する、実質的に炭酸塩を含まない凍結乾燥製剤。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、保存安定性に優れたカルバペネム系抗生物質である(4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸を含有する凍結乾燥製剤と、その製造方法に関するものである。
【背景技術】
【0002】
カルバペネム系抗生物質は、一般的に、注射や点滴により非経口的に静脈内投与される。このような投与形態に適した注射用製剤の形態として、一般的には、結晶性粉末を注射用バイアルに充填する粉末充填製剤や、凍結乾燥により原薬結晶の水溶液を容器内で乾燥した凍結乾燥製剤が臨床に提供されている。粉末充填製剤や凍結乾燥製剤は、いずれも用時に注射用水など所定の溶媒をバイアル内に添加して溶液剤を調製する製剤であり、不安定な溶液状態での長期保存を回避できるという利点がある。
【0003】
一方で、カルバペネム系抗生物質は、一般的に、結晶状態に比べて非晶質状態では不安定といった化学的特性を有している(特許文献1)。
【0004】
そのため、一般的には、結晶原薬を充填する粉末充填製剤は、結晶性が低い或いは非晶質状態となりやすい凍結乾燥製剤に比べて保存安定性に優れている。しかし、その反面、医薬品の製造工程において異物が混入しやすく、再溶解性(reconstitution)が凍結乾燥製剤に比べて劣る場合があり、技術的ハードルが高い。
【0005】
下記式(I):
【0006】
【化1】

【0007】
によって示される (4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸(以下、化合物Aと略す)は、カルバペネム系抗生物質として特許文献2で開示されている。
【0008】
炭酸水素ナトリウム等の炭酸イオンを生成する添加剤は、カルバペネム骨格を有する化合物に対して溶解補助効果を示すことが知られている(非特許文献1)。
特許文献3では、カルバペネム化合物を炭酸系水溶液或いは炭酸−エタノール水溶液に溶かし、この液をそのまま凍結乾燥する方法を開示し、溶解補助剤として塩化ナトリウムを使用できると記載している。
特許文献4では、塩化ナトリウムを安定化剤としたカルバペネム凍結乾燥製剤を開示している。
特許文献5では、カプリル酸ナトリウムの添加を開示している。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】米国特許第5,424,306号明細書
【特許文献2】国際公開第02/38564号パンフレット
【特許文献3】特開平11−255653
【特許文献4】特開昭61−172878
【特許文献5】国際公開第2007/142212号パンフレット
【非特許文献】
【0010】
【非特許文献1】“Meropenem Exists in Equilibrium with a Carbon Dioxide Adduct in Bicarbonate Solution” Journal of Pharmaceutical Sciences Vol.87, No.5, May 1998 p663-666
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明の目的は、再溶解性(reconstitution)を損なうことなく、保存安定性に優れた化合物Aを含有する凍結乾燥製剤を提供することにある。
【課題を解決するための手段】
【0012】
化合物Aの注射用の粉末充填製剤は、結晶原薬を充填するため保存安定性に優れているが、医薬品としての品質を保証するためには異物混入の防止策など技術的ハードルが高いため、凍結乾燥製剤について検討した。
【0013】
しかし、汎用の方法で得られた化合物Aの凍結乾燥製剤は、化合物Aの原薬結晶に比べて水或いは生理食塩水への再溶解性(reconstitution)がよく、臨床現場で投与液の調製が行いやすい特徴があったが、非晶質であり保存安定性が悪く、製剤としては不適であった。
また、化合物Aの水への溶解度は約7mg/mLと低いため、臨床上有用な含量の凍結乾燥製剤、例えば0.5gを含有する凍結乾燥製剤を商業的に提供するためには、70mL以上バイアルに充填して凍結乾燥を行わなければならず、大規模な設備が必要であり困難であった。そのため、商業的に臨床上有用な含量の凍結乾燥製剤を提供するためには、化合物Aの高濃度水溶液の調製方法と、それを凍結乾燥することによる凍結乾燥製剤の保存安定性の改良が必要であった。
【0014】
一方、炭酸水素ナトリウム等の炭酸イオンを生成する添加剤は、カルバペネム骨格を有する化合物に対して溶解補助効果を示すことが知られている(非特許文献1)。そのため数多くのカルバペネム骨格を有する医薬品において、溶解補助剤として使用されている。
【0015】
特許文献3では、カルバペネム化合物を炭酸系水溶液或いは炭酸−エタノール水溶液に溶かし、この液をそのまま凍結乾燥する方法を開示し、溶解補助剤として塩化ナトリウムを使用できると記載されている。
【0016】
化合物Aもカルバペネム骨格を有することから、炭酸系水溶液中で高い溶解度を示す。しかし、化合物Aの炭酸系水溶液は高濃度では溶液状態での安定性が悪く、特許文献3の記載の方法では、炭酸系水溶液を凍結乾燥するまでの間に化合物Aの分解が進行した。また、特許文献3の記載の方法のように、炭酸系水溶液をそのまま凍結乾燥して作製した化合物Aの凍結乾燥製剤は、保存安定性が悪かった。
【0017】
エタノールについては、化合物Aは全く溶解せず、更に、2−プロパノール或いはt−ブタノールについても化合物Aは全く溶解しなかった。
【0018】
一方、特許文献3には、塩化ナトリウムを溶解補助剤として使用できると記載されているが、化合物Aに対しての溶解補助効果は認められなかった。更に、特許文献4には、塩化ナトリウムを安定化剤としたカルバペネム凍結乾燥製剤が開示されているが、特許文献4に記載の方法で作製した化合物Aの凍結乾燥性剤は非晶質であり、保存安定性が悪かった。
【0019】
また、特許文献5には、化合物Aの水への溶解度の向上のために、カプリル酸ナトリウムの添加を開示しているが、この方法を用いて化合物Aの高濃度水溶液を調製して凍結乾燥した製剤でも、保存安定性が悪かった。
【0020】
化合物Aを汎用の方法を用いて作製した凍結乾燥製剤は、結晶性粉末を注射用バイアルに充填した粉末充填製剤に比べて再溶解性(reconstitution)は優れていたが、保存安定性が悪く、医薬品として開発することはできなかった。しかし、本発明者は上記の課題を解決すべく鋭意努力した結果、化合物Aが、塩化ナトリウム水溶液と親水性溶媒の混液に対して高い溶解度を示すことを見出し、更にこの混液を用いて高濃度に調製した化合物A溶液を汎用の方法により凍結乾燥して得られた製剤は、再溶解性(reconstitution)を損なうことなく、非晶質であるにも関わらず優れた保存安定性を示すことを見出し、本発明を完成した。
【0021】
すなわち本発明は;
【0022】
〔1〕 (4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸またはその塩、またはそれらの溶媒和物を、0.6%以上の塩化ナトリウムと10〜50%の親水性溶媒を含む水溶液に溶解し、該溶液を凍結乾燥することにより調製された、 (4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸またはその塩、またはそれらの溶媒和物と塩化ナトリウムを含有する、実質的に炭酸塩を含まない凍結乾燥製剤、
〔2〕 親水性溶媒が、t−ブタノール、エタノール、2−プロパノールまたはその混合溶媒である、〔1〕記載の凍結乾燥製剤、
〔3〕 親水性溶媒が、t−ブタノールである、〔1〕または〔2〕記載の凍結乾燥製剤、
〔4〕 該溶液のpHを5.0〜7.0に調整した後に凍結乾燥する、〔1〕〜〔3〕のいずれかに記載の凍結乾燥製剤、
〔5〕 該溶液中の(4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸またはその塩、またはそれらの溶媒和物の濃度が20mg/mL以上である、〔1〕〜〔4〕のいずれかに記載の凍結乾燥製剤、
【0023】
〔6〕 凍結乾燥製剤の製造方法であって、(4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸またはその塩、またはそれらの溶媒和物を、0.6%以上の塩化ナトリウムと10〜50%の親水性溶媒を含む水溶液に溶解し、実質的に炭酸塩を含まない該溶液を凍結乾燥する方法、
〔7〕 親水性溶媒が、t−ブタノール、エタノール、2−プロパノールまたはその混合溶媒である、〔6〕記載の方法、
〔8〕 親水性溶媒が、t−ブタノールである、〔6〕または〔7〕記載の方法、
〔9〕 該溶液のpHを5.0〜7.0に調整した後に凍結乾燥する、〔6〕〜〔8〕のいずれかに記載の方法、または
〔10〕 該溶液中の(4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸またはその塩、またはそれらの溶媒和物の濃度が20mg/mL以上である、〔6〕〜〔9〕のいずれかに記載の方法に関する。
【発明の効果】
【0024】
本発明によれば、再溶解性(reconstitution)を損なうことなく、保存安定性に優れた化合物Aを含有する凍結乾燥製剤を提供することができる。
【図面の簡単な説明】
【0025】
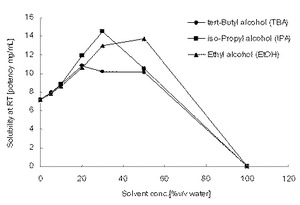
【図1】試験例5に示した、水に添加した親水性溶媒の割合(容量%)と化合物Aの室温における溶解度(mg/mL)を示すグラフである。
【0026】
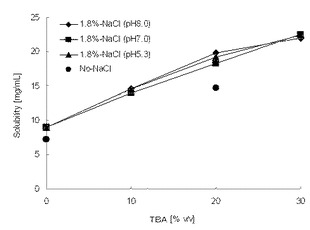
【図2】試験例6に示した、1.8%の塩化ナトリウム水溶液に対して添加したt−ブチルアルコールの添加量(容量%)と化合物Aの室温における溶解度(mg/mL)を示すグラフである。
【0027】
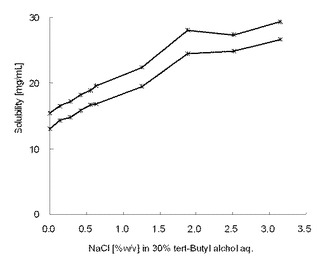
【図3】試験例7に示した、30%のt−ブチルアルコール水溶液に含まれる塩化ナトリウムの配合量(重量%)と、化合物Aの室温における溶解度(mg/mL)を示すグラフである。
【0028】
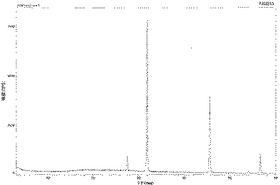
【図4】実施例12の化合物Aの凍結乾燥製剤粉末のX線回折チャートである。
【発明を実施するための形態】
【0029】
本発明の凍結乾燥製剤で有効成分として含有する、(4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸は、フリー体であってももしくはその薬理学的に許容される塩またはそれらの薬理学的に許容される溶媒和物であってもよい。
薬理学上許容される塩としては、カルボキシル基に関してはナトリウム、カリウムなどのアルカリ金属との塩やカルシウム、マグネシウムなどのアルカリ土類金属との塩、およびアンモニウム塩などの無機塩基との塩や、トリエチルアンモニウム塩、ピリジニウム塩、ジイソプロピルアンモニウム塩などの有機塩基との塩や、上記式の(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル基が4級アンモニウム塩となったものとの塩が挙げられる。分子内の塩基性基の塩としては例えば塩酸、硫酸、リン酸などの無機酸との塩、ギ酸、酢酸、シュウ酸、メタンスルホン酸、ベンゼンスルホン酸などの有機酸との塩などが挙げられる。その他、水が付加した水和物や、アセトン、テトラヒドロフランなどの有機溶媒が付加した溶媒和物が挙げられる。
【0030】
本発明に使用する塩化ナトリウムは、溶解補助剤として一般的に知られているが、化合物Aに対して溶解補助効果はなく、また、親水性溶媒であるt−ブタノール、エタノール、2−プロパノールも化合物Aを全く溶解しない。単独では溶解補助効果を有さない塩化ナトリウムと親水性溶媒を組み合わせることにより高濃度の化合物A溶液を調製できること、更にこの高濃度の化合物A溶液を特段の冷却凍結プロセスを経ることなく汎用の方法により凍結乾燥することにより、汎用の凍結乾燥製剤の特徴である優れた再溶解性(reconstitution)を損なうことなく、保存安定性に優れた凍結乾燥製剤が提供できることは、驚きに値する。
特に、カルバペネム系化合物は非晶質で安定性が悪いと報告されているが、本発明で得られる化合物Aの凍結乾燥製剤は、非晶質にも関わらず保存安定性に優れていることは、まさに驚きに値する。
【0031】
本発明において、化合物Aを溶解する溶液に含まれる塩化ナトリウムの濃度は、0.6%w/v以上が好ましく、0.6%w/v〜3%w/vがより好ましく、1%w/v〜2%w/vが特に好ましく、さらには、1.3%w/v〜1.9%w/vが特段に好ましい。
実質的に炭酸塩を含まない時の炭酸塩の濃度としては、例えば1%以下が挙げられ、好ましくは0.1%以下が挙げられる。
【0032】
本発明において、化合物Aを溶解する溶液に使用する親水性溶媒は特に限定されないが、例えば、t−ブタノール、エタノール、2−プロパノール、またはこれらの混合溶媒が挙げられる。このうち、t−ブタノールは、凝固点約26℃、沸点約80℃と凍結乾燥に適した親水性溶媒として知られており、t−ブタノールを用いる方法は、本発明の好ましい態様である。
【0033】
本発明において、化合物Aを溶解する溶液に含まれる親水性溶媒の濃度は、10〜50%v/vが好ましく、20〜50%v/vがより好ましく、20〜40%v/vが特に好ましく、さらには30%v/vが特段に好ましい。
【0034】
本発明において、凍結乾燥する化合物Aの溶液のpHは5.0〜7.0が好ましく、5.5〜6.5がより好ましく、5.5〜6.0が特に好ましい。pHの調整には、生理学的および製剤学的に許容される緩衝成分や、酸、塩基の使用が可能で、緩衝成分としては、例えば、2−モルホリノエタンスルホン酸およびその水和物、クエン酸塩、リン酸塩、酢酸塩が使用可能で、酸としては、例えば、塩酸、メタンスルホン酸、硫酸、酢酸、硝酸、リン酸が使用可能で、塩基としては、例えば、水酸化ナトリウム、水酸化カリウムが使用可能である。
【0035】
以下に、実施例を挙げて本発明をさらに詳しく説明するが、本発明はこれらに限定されるものではない。
【0036】
なお、以下の実施例、比較例および試験例において、t−ブチルアルコールは和光純薬株式会社製のものを、それ以外の試薬或いは溶媒は、ナカライテスク株式会社製のものを、それぞれ使用した。
【実施例1】
【0037】
試験例1
10mMの燐酸ナトリウム緩衝液に1mg/mLとなるように化合物A無水物結晶を溶解し、塩酸或いは水酸化ナトリウム水溶液を用いて、表1に示したpH初期値に調整した。これらの液を5℃及び室温で24時間保存し、pHを測定すると共に、高速液体クロマトグラム法により化合物A残存率を測定した。ここで述べる化合物A残存率とは、保存前の化合物Aの量を100としたときの比率である。結果を表1に示す。
化合物Aは溶液状態ではpHが7を超えると、急速に分解が進行することが明らかであり、pH5〜7が安定域であると考えられた。
【0038】
【表1】
【0039】
試験例2
表2に示した異なるpHを示す水溶液を調製し、これらに過剰量の化合物A無水物結晶を添加後、5分間超音波処理を施し、25℃で1時間静置した。その後、液中に溶解していない過剰量の化合物A粉末が残存していることを確認後、各液を孔径0.45μmのフィルターに通液してろ液を回収し、pHを測定すると共に、高速液体クロマトグラム法によりろ液中の化合物A濃度を測定し、各水溶液に対する化合物Aの溶解度を求めた。結果を表2に示す。いずれのpH域でも20mg/mLを超える高い溶解度は示さなかった。
【0040】
【表2】
【0041】
試験例3
表3に示した組成の水溶液に、過剰量の化合物A無水物結晶を添加した後、25℃或いは40℃でそれぞれ5分間超音波処理を行った。超音波処理後、液中に溶解していない過剰量の化合物A粉末が残存していることを確認後、各液を孔径0.45μmのフィルターに通液してろ液を回収し、pHを測定すると共に、試験例2と同様の方法によりろ液中の化合物A濃度を測定し、各水溶液に対する化合物Aの溶解度を求めた。結果を表3に示す。
劇的に溶解度を向上させる添加剤は認められなかった。
【0042】
【表3】
【0043】
試験例4
表4に示す処方分量となるように化合物A無水物結晶、炭酸ナトリウム、炭酸水素ナトリウム、カプリル酸ナトリウムを注射用水に溶解し、水酸化ナトリウムと塩酸を用いてpHを7.5に調整した。この液をメンブランフィルターでろ過し、ガラス製バイアルに0.5mLずつ充填した後、凍結乾燥機を行い、化合物Aの凍結乾燥製剤を得た。得られた凍結乾燥製剤を50℃で1週間、3週間、4週間保存し、試験例1と同様の方法により化合物A残存率を測定した。結果を表5に示す。
いずれの比較例においても臨床への適用が可能な保存安定性を有した凍結乾燥製剤を得ることはできなかった。
【0044】
【表4】
【0045】
【表5】
【0046】
試験例5
表6に示す処方分量となるように、化合物A無水物結晶、リン酸水素ナトリウム、塩化ナトリウム、マンニトール、スクロースを注射用水に溶解し、水酸化ナトリウムと塩酸を用いてpHを約6.0に調整した。この液をメンブランフィルターでろ過し、ガラス製バイアルに1mLずつ充填した後、凍結乾燥機に仕込み、表7に示した凍結乾燥条件にて真空乾燥を行い、化合物Aの凍結乾燥製剤を得た。得られた凍結乾燥製剤を60℃で3週間保存し、性状を観察すると共に、試験例1と同様の方法により化合物A残存率を測定した。結果を表8に示す。
いずれの比較例においても臨床への適用が可能な保存安定性を有した凍結乾燥製剤を得ることはできなかった。
【0047】
【表6】
【0048】
【表7】
【0049】
【表8】
【0050】
試験例5
親水性溶媒であるt−ブタノール、エタノール、2−プロパノールを、それぞれ0%、5%、10%、20%、30%、50%、100%(v/v)の割合で含む水溶液に、過剰量の化合物A無水物結晶を添加し、25℃で5分間超音波処理後、25℃で30分間静置した。その後、液中に溶解していない過剰量の化合物A粉末が残存していることを確認し、各液を孔径0.45μmのフィルターに通液してろ液を回収し、試験例2と同様の方法によりろ液中の化合物A濃度を測定し、各水溶液に対する化合物Aの溶解度を求めた。結果を図1に示す。
水に対する溶解度は約7mg/mL程度であり、100%の親水性溶媒に対しては、いずれの親水性溶媒においても化合物Aは全く溶けなかった。しかし、水と親水性溶媒の混液では、水単独に比べて高い溶解度を示し、親水性溶媒が20〜50%含むときに最も高い溶解度を示した。
【0051】
試験例6
1.8%の塩化ナトリウム水溶液に、t−ブタノールを0%(v/v)、10%、20%、30%相当添加し、試験例2と同様の方法により、化合物Aの溶解度を求めた。結果を図2に示す。
試験例5における親水性溶媒と水の混液系に比べて、塩化ナトリウムとt−ブタノールの存在下において非常に高い溶解度を示した。特にt−ブタノールの添加量に依存して、化合物Aの溶解度は高くなった。
【0052】
試験例7
0%〜4.5%の塩化ナトリウム水溶液に30%v/vとなるようにt−ブタノールを添加し、0%〜3.15%の塩化ナトリウムを含む30%のt−ブタノール水溶液を調製し、試験例2と同様の方法により、化合物Aの溶解度を求めた。結果を図3に示す。
塩化ナトリウム濃度に比例して、化合物Aの溶解度は高くなった。
【0053】
試験例8
化合物A無水物結晶及び注射用水を用いて、表9に示す処方分量の液を調製し、−7℃、1℃、5℃、25℃、40℃で保存した。保存開始から約24時間後まで、経時的に化合物Aの残存率を試験例1の条件で求め、保存前の化合物Aの量との比率で表した。横軸に保存時間、縦軸に化合物Aの残存率をプロットし、各プロットから近似直線を引き、その傾きを化合物Aの分解速度定数として算出した。結果を表10に示す。
高濃度の化合物Aを溶解できる炭酸水素ナトリウムを配合した水溶液中では、1℃以下に冷却しても化合物Aの分解は進行した。
【0054】
【表9】
【0055】
【表10】
【0056】
実施例1〜9
表11及び表12に示した処方分量にて、それぞれ化合物Aの薬液を調製した。この液をメンブランフィルターでろ過し、ガラス製バイアルに1mLずつ充填し、凍結乾燥機に仕込んだ後、表7に示した条件に従って真空乾燥を行い、実施例1〜9の化合物Aの凍結乾燥製剤を得た。なお、pH調整には水酸化ナトリウム或いは塩酸を用いた。
【0057】
【表11】
【0058】
【表12】
【0059】
実施例11
化合物Aアセトン溶媒和物結晶(WO2009/075323を参照)を20mg/mLとなるように、1.3%の塩化ナトリウムを含む30%t−ブタノール水溶液に溶解し、水酸化ナトリウムと塩酸を用いてpHを約6.0に調整した後、実施例1と同じ方法により、化合物Aの凍結乾燥製剤を得た。
【0060】
実施例12
化合物A無水物結晶を20mg/mLとなるように、1.3%の塩化ナトリウムを含む30%t−ブチルアルコール水溶液に溶解し、水酸化ナトリウムと塩酸を用いてpHを約6.0に調整した後、表13に示した条件に従って真空乾燥を行い、化合物Aの凍結乾燥製剤を得た。
【0061】
【表13】
【0062】
試験例9
実施例1〜12の製剤を60℃で2週間、3週間保存し、化合物A残存率を試験例1と同様に求めると共に、性状と再溶解性についても評価した。
表14に示したように、いずれの実施例の製剤においても比較例1〜9に比べて明らかに保存後の化合物A残存率が向上し、保存安定性の向上が認められた。
また、表15に示したように、性状の変化は、いずれの実施例の製剤においても認められなかった。
さらに、各実施例の製剤に、化合物A濃度が5〜10mg/mLとなるように注射用水を加えて再溶解性を評価したところ、いずれの実施例の製剤においても、速やかに溶解し、無色澄明な液を容易に調製することができた。
【0063】
【表14】
【0064】
【表15】
【0065】
【表16】
【0066】
試験例10
実施例12の化合物Aの凍結乾燥製剤の粉末についてX線回折測定を実施した。図4に示したように、配合している塩化ナトリウムの結晶回折ピークしか認められず、化合物Aに由来する結晶回折ピークは認められなかったことから、凍結乾燥製剤中の化合物Aは非晶質状態で存在していると考えられた。
【産業上の利用可能性】
【0067】
本発明によれば、再溶解性(reconstitution)を損なうことなく、保存安定性に優れた(4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸を含有する凍結乾燥製剤を提供することができる。
【技術分野】
【0001】
本発明は、保存安定性に優れたカルバペネム系抗生物質である(4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸を含有する凍結乾燥製剤と、その製造方法に関するものである。
【背景技術】
【0002】
カルバペネム系抗生物質は、一般的に、注射や点滴により非経口的に静脈内投与される。このような投与形態に適した注射用製剤の形態として、一般的には、結晶性粉末を注射用バイアルに充填する粉末充填製剤や、凍結乾燥により原薬結晶の水溶液を容器内で乾燥した凍結乾燥製剤が臨床に提供されている。粉末充填製剤や凍結乾燥製剤は、いずれも用時に注射用水など所定の溶媒をバイアル内に添加して溶液剤を調製する製剤であり、不安定な溶液状態での長期保存を回避できるという利点がある。
【0003】
一方で、カルバペネム系抗生物質は、一般的に、結晶状態に比べて非晶質状態では不安定といった化学的特性を有している(特許文献1)。
【0004】
そのため、一般的には、結晶原薬を充填する粉末充填製剤は、結晶性が低い或いは非晶質状態となりやすい凍結乾燥製剤に比べて保存安定性に優れている。しかし、その反面、医薬品の製造工程において異物が混入しやすく、再溶解性(reconstitution)が凍結乾燥製剤に比べて劣る場合があり、技術的ハードルが高い。
【0005】
下記式(I):
【0006】
【化1】
【0007】
によって示される (4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸(以下、化合物Aと略す)は、カルバペネム系抗生物質として特許文献2で開示されている。
【0008】
炭酸水素ナトリウム等の炭酸イオンを生成する添加剤は、カルバペネム骨格を有する化合物に対して溶解補助効果を示すことが知られている(非特許文献1)。
特許文献3では、カルバペネム化合物を炭酸系水溶液或いは炭酸−エタノール水溶液に溶かし、この液をそのまま凍結乾燥する方法を開示し、溶解補助剤として塩化ナトリウムを使用できると記載している。
特許文献4では、塩化ナトリウムを安定化剤としたカルバペネム凍結乾燥製剤を開示している。
特許文献5では、カプリル酸ナトリウムの添加を開示している。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】米国特許第5,424,306号明細書
【特許文献2】国際公開第02/38564号パンフレット
【特許文献3】特開平11−255653
【特許文献4】特開昭61−172878
【特許文献5】国際公開第2007/142212号パンフレット
【非特許文献】
【0010】
【非特許文献1】“Meropenem Exists in Equilibrium with a Carbon Dioxide Adduct in Bicarbonate Solution” Journal of Pharmaceutical Sciences Vol.87, No.5, May 1998 p663-666
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明の目的は、再溶解性(reconstitution)を損なうことなく、保存安定性に優れた化合物Aを含有する凍結乾燥製剤を提供することにある。
【課題を解決するための手段】
【0012】
化合物Aの注射用の粉末充填製剤は、結晶原薬を充填するため保存安定性に優れているが、医薬品としての品質を保証するためには異物混入の防止策など技術的ハードルが高いため、凍結乾燥製剤について検討した。
【0013】
しかし、汎用の方法で得られた化合物Aの凍結乾燥製剤は、化合物Aの原薬結晶に比べて水或いは生理食塩水への再溶解性(reconstitution)がよく、臨床現場で投与液の調製が行いやすい特徴があったが、非晶質であり保存安定性が悪く、製剤としては不適であった。
また、化合物Aの水への溶解度は約7mg/mLと低いため、臨床上有用な含量の凍結乾燥製剤、例えば0.5gを含有する凍結乾燥製剤を商業的に提供するためには、70mL以上バイアルに充填して凍結乾燥を行わなければならず、大規模な設備が必要であり困難であった。そのため、商業的に臨床上有用な含量の凍結乾燥製剤を提供するためには、化合物Aの高濃度水溶液の調製方法と、それを凍結乾燥することによる凍結乾燥製剤の保存安定性の改良が必要であった。
【0014】
一方、炭酸水素ナトリウム等の炭酸イオンを生成する添加剤は、カルバペネム骨格を有する化合物に対して溶解補助効果を示すことが知られている(非特許文献1)。そのため数多くのカルバペネム骨格を有する医薬品において、溶解補助剤として使用されている。
【0015】
特許文献3では、カルバペネム化合物を炭酸系水溶液或いは炭酸−エタノール水溶液に溶かし、この液をそのまま凍結乾燥する方法を開示し、溶解補助剤として塩化ナトリウムを使用できると記載されている。
【0016】
化合物Aもカルバペネム骨格を有することから、炭酸系水溶液中で高い溶解度を示す。しかし、化合物Aの炭酸系水溶液は高濃度では溶液状態での安定性が悪く、特許文献3の記載の方法では、炭酸系水溶液を凍結乾燥するまでの間に化合物Aの分解が進行した。また、特許文献3の記載の方法のように、炭酸系水溶液をそのまま凍結乾燥して作製した化合物Aの凍結乾燥製剤は、保存安定性が悪かった。
【0017】
エタノールについては、化合物Aは全く溶解せず、更に、2−プロパノール或いはt−ブタノールについても化合物Aは全く溶解しなかった。
【0018】
一方、特許文献3には、塩化ナトリウムを溶解補助剤として使用できると記載されているが、化合物Aに対しての溶解補助効果は認められなかった。更に、特許文献4には、塩化ナトリウムを安定化剤としたカルバペネム凍結乾燥製剤が開示されているが、特許文献4に記載の方法で作製した化合物Aの凍結乾燥性剤は非晶質であり、保存安定性が悪かった。
【0019】
また、特許文献5には、化合物Aの水への溶解度の向上のために、カプリル酸ナトリウムの添加を開示しているが、この方法を用いて化合物Aの高濃度水溶液を調製して凍結乾燥した製剤でも、保存安定性が悪かった。
【0020】
化合物Aを汎用の方法を用いて作製した凍結乾燥製剤は、結晶性粉末を注射用バイアルに充填した粉末充填製剤に比べて再溶解性(reconstitution)は優れていたが、保存安定性が悪く、医薬品として開発することはできなかった。しかし、本発明者は上記の課題を解決すべく鋭意努力した結果、化合物Aが、塩化ナトリウム水溶液と親水性溶媒の混液に対して高い溶解度を示すことを見出し、更にこの混液を用いて高濃度に調製した化合物A溶液を汎用の方法により凍結乾燥して得られた製剤は、再溶解性(reconstitution)を損なうことなく、非晶質であるにも関わらず優れた保存安定性を示すことを見出し、本発明を完成した。
【0021】
すなわち本発明は;
【0022】
〔1〕 (4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸またはその塩、またはそれらの溶媒和物を、0.6%以上の塩化ナトリウムと10〜50%の親水性溶媒を含む水溶液に溶解し、該溶液を凍結乾燥することにより調製された、 (4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸またはその塩、またはそれらの溶媒和物と塩化ナトリウムを含有する、実質的に炭酸塩を含まない凍結乾燥製剤、
〔2〕 親水性溶媒が、t−ブタノール、エタノール、2−プロパノールまたはその混合溶媒である、〔1〕記載の凍結乾燥製剤、
〔3〕 親水性溶媒が、t−ブタノールである、〔1〕または〔2〕記載の凍結乾燥製剤、
〔4〕 該溶液のpHを5.0〜7.0に調整した後に凍結乾燥する、〔1〕〜〔3〕のいずれかに記載の凍結乾燥製剤、
〔5〕 該溶液中の(4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸またはその塩、またはそれらの溶媒和物の濃度が20mg/mL以上である、〔1〕〜〔4〕のいずれかに記載の凍結乾燥製剤、
【0023】
〔6〕 凍結乾燥製剤の製造方法であって、(4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸またはその塩、またはそれらの溶媒和物を、0.6%以上の塩化ナトリウムと10〜50%の親水性溶媒を含む水溶液に溶解し、実質的に炭酸塩を含まない該溶液を凍結乾燥する方法、
〔7〕 親水性溶媒が、t−ブタノール、エタノール、2−プロパノールまたはその混合溶媒である、〔6〕記載の方法、
〔8〕 親水性溶媒が、t−ブタノールである、〔6〕または〔7〕記載の方法、
〔9〕 該溶液のpHを5.0〜7.0に調整した後に凍結乾燥する、〔6〕〜〔8〕のいずれかに記載の方法、または
〔10〕 該溶液中の(4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸またはその塩、またはそれらの溶媒和物の濃度が20mg/mL以上である、〔6〕〜〔9〕のいずれかに記載の方法に関する。
【発明の効果】
【0024】
本発明によれば、再溶解性(reconstitution)を損なうことなく、保存安定性に優れた化合物Aを含有する凍結乾燥製剤を提供することができる。
【図面の簡単な説明】
【0025】
【図1】試験例5に示した、水に添加した親水性溶媒の割合(容量%)と化合物Aの室温における溶解度(mg/mL)を示すグラフである。
【0026】
【図2】試験例6に示した、1.8%の塩化ナトリウム水溶液に対して添加したt−ブチルアルコールの添加量(容量%)と化合物Aの室温における溶解度(mg/mL)を示すグラフである。
【0027】
【図3】試験例7に示した、30%のt−ブチルアルコール水溶液に含まれる塩化ナトリウムの配合量(重量%)と、化合物Aの室温における溶解度(mg/mL)を示すグラフである。
【0028】
【図4】実施例12の化合物Aの凍結乾燥製剤粉末のX線回折チャートである。
【発明を実施するための形態】
【0029】
本発明の凍結乾燥製剤で有効成分として含有する、(4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸は、フリー体であってももしくはその薬理学的に許容される塩またはそれらの薬理学的に許容される溶媒和物であってもよい。
薬理学上許容される塩としては、カルボキシル基に関してはナトリウム、カリウムなどのアルカリ金属との塩やカルシウム、マグネシウムなどのアルカリ土類金属との塩、およびアンモニウム塩などの無機塩基との塩や、トリエチルアンモニウム塩、ピリジニウム塩、ジイソプロピルアンモニウム塩などの有機塩基との塩や、上記式の(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル基が4級アンモニウム塩となったものとの塩が挙げられる。分子内の塩基性基の塩としては例えば塩酸、硫酸、リン酸などの無機酸との塩、ギ酸、酢酸、シュウ酸、メタンスルホン酸、ベンゼンスルホン酸などの有機酸との塩などが挙げられる。その他、水が付加した水和物や、アセトン、テトラヒドロフランなどの有機溶媒が付加した溶媒和物が挙げられる。
【0030】
本発明に使用する塩化ナトリウムは、溶解補助剤として一般的に知られているが、化合物Aに対して溶解補助効果はなく、また、親水性溶媒であるt−ブタノール、エタノール、2−プロパノールも化合物Aを全く溶解しない。単独では溶解補助効果を有さない塩化ナトリウムと親水性溶媒を組み合わせることにより高濃度の化合物A溶液を調製できること、更にこの高濃度の化合物A溶液を特段の冷却凍結プロセスを経ることなく汎用の方法により凍結乾燥することにより、汎用の凍結乾燥製剤の特徴である優れた再溶解性(reconstitution)を損なうことなく、保存安定性に優れた凍結乾燥製剤が提供できることは、驚きに値する。
特に、カルバペネム系化合物は非晶質で安定性が悪いと報告されているが、本発明で得られる化合物Aの凍結乾燥製剤は、非晶質にも関わらず保存安定性に優れていることは、まさに驚きに値する。
【0031】
本発明において、化合物Aを溶解する溶液に含まれる塩化ナトリウムの濃度は、0.6%w/v以上が好ましく、0.6%w/v〜3%w/vがより好ましく、1%w/v〜2%w/vが特に好ましく、さらには、1.3%w/v〜1.9%w/vが特段に好ましい。
実質的に炭酸塩を含まない時の炭酸塩の濃度としては、例えば1%以下が挙げられ、好ましくは0.1%以下が挙げられる。
【0032】
本発明において、化合物Aを溶解する溶液に使用する親水性溶媒は特に限定されないが、例えば、t−ブタノール、エタノール、2−プロパノール、またはこれらの混合溶媒が挙げられる。このうち、t−ブタノールは、凝固点約26℃、沸点約80℃と凍結乾燥に適した親水性溶媒として知られており、t−ブタノールを用いる方法は、本発明の好ましい態様である。
【0033】
本発明において、化合物Aを溶解する溶液に含まれる親水性溶媒の濃度は、10〜50%v/vが好ましく、20〜50%v/vがより好ましく、20〜40%v/vが特に好ましく、さらには30%v/vが特段に好ましい。
【0034】
本発明において、凍結乾燥する化合物Aの溶液のpHは5.0〜7.0が好ましく、5.5〜6.5がより好ましく、5.5〜6.0が特に好ましい。pHの調整には、生理学的および製剤学的に許容される緩衝成分や、酸、塩基の使用が可能で、緩衝成分としては、例えば、2−モルホリノエタンスルホン酸およびその水和物、クエン酸塩、リン酸塩、酢酸塩が使用可能で、酸としては、例えば、塩酸、メタンスルホン酸、硫酸、酢酸、硝酸、リン酸が使用可能で、塩基としては、例えば、水酸化ナトリウム、水酸化カリウムが使用可能である。
【0035】
以下に、実施例を挙げて本発明をさらに詳しく説明するが、本発明はこれらに限定されるものではない。
【0036】
なお、以下の実施例、比較例および試験例において、t−ブチルアルコールは和光純薬株式会社製のものを、それ以外の試薬或いは溶媒は、ナカライテスク株式会社製のものを、それぞれ使用した。
【実施例1】
【0037】
試験例1
10mMの燐酸ナトリウム緩衝液に1mg/mLとなるように化合物A無水物結晶を溶解し、塩酸或いは水酸化ナトリウム水溶液を用いて、表1に示したpH初期値に調整した。これらの液を5℃及び室温で24時間保存し、pHを測定すると共に、高速液体クロマトグラム法により化合物A残存率を測定した。ここで述べる化合物A残存率とは、保存前の化合物Aの量を100としたときの比率である。結果を表1に示す。
化合物Aは溶液状態ではpHが7を超えると、急速に分解が進行することが明らかであり、pH5〜7が安定域であると考えられた。
【0038】
【表1】
【0039】
試験例2
表2に示した異なるpHを示す水溶液を調製し、これらに過剰量の化合物A無水物結晶を添加後、5分間超音波処理を施し、25℃で1時間静置した。その後、液中に溶解していない過剰量の化合物A粉末が残存していることを確認後、各液を孔径0.45μmのフィルターに通液してろ液を回収し、pHを測定すると共に、高速液体クロマトグラム法によりろ液中の化合物A濃度を測定し、各水溶液に対する化合物Aの溶解度を求めた。結果を表2に示す。いずれのpH域でも20mg/mLを超える高い溶解度は示さなかった。
【0040】
【表2】
【0041】
試験例3
表3に示した組成の水溶液に、過剰量の化合物A無水物結晶を添加した後、25℃或いは40℃でそれぞれ5分間超音波処理を行った。超音波処理後、液中に溶解していない過剰量の化合物A粉末が残存していることを確認後、各液を孔径0.45μmのフィルターに通液してろ液を回収し、pHを測定すると共に、試験例2と同様の方法によりろ液中の化合物A濃度を測定し、各水溶液に対する化合物Aの溶解度を求めた。結果を表3に示す。
劇的に溶解度を向上させる添加剤は認められなかった。
【0042】
【表3】
【0043】
試験例4
表4に示す処方分量となるように化合物A無水物結晶、炭酸ナトリウム、炭酸水素ナトリウム、カプリル酸ナトリウムを注射用水に溶解し、水酸化ナトリウムと塩酸を用いてpHを7.5に調整した。この液をメンブランフィルターでろ過し、ガラス製バイアルに0.5mLずつ充填した後、凍結乾燥機を行い、化合物Aの凍結乾燥製剤を得た。得られた凍結乾燥製剤を50℃で1週間、3週間、4週間保存し、試験例1と同様の方法により化合物A残存率を測定した。結果を表5に示す。
いずれの比較例においても臨床への適用が可能な保存安定性を有した凍結乾燥製剤を得ることはできなかった。
【0044】
【表4】
【0045】
【表5】
【0046】
試験例5
表6に示す処方分量となるように、化合物A無水物結晶、リン酸水素ナトリウム、塩化ナトリウム、マンニトール、スクロースを注射用水に溶解し、水酸化ナトリウムと塩酸を用いてpHを約6.0に調整した。この液をメンブランフィルターでろ過し、ガラス製バイアルに1mLずつ充填した後、凍結乾燥機に仕込み、表7に示した凍結乾燥条件にて真空乾燥を行い、化合物Aの凍結乾燥製剤を得た。得られた凍結乾燥製剤を60℃で3週間保存し、性状を観察すると共に、試験例1と同様の方法により化合物A残存率を測定した。結果を表8に示す。
いずれの比較例においても臨床への適用が可能な保存安定性を有した凍結乾燥製剤を得ることはできなかった。
【0047】
【表6】
【0048】
【表7】
【0049】
【表8】
【0050】
試験例5
親水性溶媒であるt−ブタノール、エタノール、2−プロパノールを、それぞれ0%、5%、10%、20%、30%、50%、100%(v/v)の割合で含む水溶液に、過剰量の化合物A無水物結晶を添加し、25℃で5分間超音波処理後、25℃で30分間静置した。その後、液中に溶解していない過剰量の化合物A粉末が残存していることを確認し、各液を孔径0.45μmのフィルターに通液してろ液を回収し、試験例2と同様の方法によりろ液中の化合物A濃度を測定し、各水溶液に対する化合物Aの溶解度を求めた。結果を図1に示す。
水に対する溶解度は約7mg/mL程度であり、100%の親水性溶媒に対しては、いずれの親水性溶媒においても化合物Aは全く溶けなかった。しかし、水と親水性溶媒の混液では、水単独に比べて高い溶解度を示し、親水性溶媒が20〜50%含むときに最も高い溶解度を示した。
【0051】
試験例6
1.8%の塩化ナトリウム水溶液に、t−ブタノールを0%(v/v)、10%、20%、30%相当添加し、試験例2と同様の方法により、化合物Aの溶解度を求めた。結果を図2に示す。
試験例5における親水性溶媒と水の混液系に比べて、塩化ナトリウムとt−ブタノールの存在下において非常に高い溶解度を示した。特にt−ブタノールの添加量に依存して、化合物Aの溶解度は高くなった。
【0052】
試験例7
0%〜4.5%の塩化ナトリウム水溶液に30%v/vとなるようにt−ブタノールを添加し、0%〜3.15%の塩化ナトリウムを含む30%のt−ブタノール水溶液を調製し、試験例2と同様の方法により、化合物Aの溶解度を求めた。結果を図3に示す。
塩化ナトリウム濃度に比例して、化合物Aの溶解度は高くなった。
【0053】
試験例8
化合物A無水物結晶及び注射用水を用いて、表9に示す処方分量の液を調製し、−7℃、1℃、5℃、25℃、40℃で保存した。保存開始から約24時間後まで、経時的に化合物Aの残存率を試験例1の条件で求め、保存前の化合物Aの量との比率で表した。横軸に保存時間、縦軸に化合物Aの残存率をプロットし、各プロットから近似直線を引き、その傾きを化合物Aの分解速度定数として算出した。結果を表10に示す。
高濃度の化合物Aを溶解できる炭酸水素ナトリウムを配合した水溶液中では、1℃以下に冷却しても化合物Aの分解は進行した。
【0054】
【表9】
【0055】
【表10】
【0056】
実施例1〜9
表11及び表12に示した処方分量にて、それぞれ化合物Aの薬液を調製した。この液をメンブランフィルターでろ過し、ガラス製バイアルに1mLずつ充填し、凍結乾燥機に仕込んだ後、表7に示した条件に従って真空乾燥を行い、実施例1〜9の化合物Aの凍結乾燥製剤を得た。なお、pH調整には水酸化ナトリウム或いは塩酸を用いた。
【0057】
【表11】
【0058】
【表12】
【0059】
実施例11
化合物Aアセトン溶媒和物結晶(WO2009/075323を参照)を20mg/mLとなるように、1.3%の塩化ナトリウムを含む30%t−ブタノール水溶液に溶解し、水酸化ナトリウムと塩酸を用いてpHを約6.0に調整した後、実施例1と同じ方法により、化合物Aの凍結乾燥製剤を得た。
【0060】
実施例12
化合物A無水物結晶を20mg/mLとなるように、1.3%の塩化ナトリウムを含む30%t−ブチルアルコール水溶液に溶解し、水酸化ナトリウムと塩酸を用いてpHを約6.0に調整した後、表13に示した条件に従って真空乾燥を行い、化合物Aの凍結乾燥製剤を得た。
【0061】
【表13】
【0062】
試験例9
実施例1〜12の製剤を60℃で2週間、3週間保存し、化合物A残存率を試験例1と同様に求めると共に、性状と再溶解性についても評価した。
表14に示したように、いずれの実施例の製剤においても比較例1〜9に比べて明らかに保存後の化合物A残存率が向上し、保存安定性の向上が認められた。
また、表15に示したように、性状の変化は、いずれの実施例の製剤においても認められなかった。
さらに、各実施例の製剤に、化合物A濃度が5〜10mg/mLとなるように注射用水を加えて再溶解性を評価したところ、いずれの実施例の製剤においても、速やかに溶解し、無色澄明な液を容易に調製することができた。
【0063】
【表14】
【0064】
【表15】
【0065】
【表16】
【0066】
試験例10
実施例12の化合物Aの凍結乾燥製剤の粉末についてX線回折測定を実施した。図4に示したように、配合している塩化ナトリウムの結晶回折ピークしか認められず、化合物Aに由来する結晶回折ピークは認められなかったことから、凍結乾燥製剤中の化合物Aは非晶質状態で存在していると考えられた。
【産業上の利用可能性】
【0067】
本発明によれば、再溶解性(reconstitution)を損なうことなく、保存安定性に優れた(4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸を含有する凍結乾燥製剤を提供することができる。
【特許請求の範囲】
【請求項1】
(4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸またはその塩、またはそれらの溶媒和物を、0.6%以上の塩化ナトリウムと10〜50%の親水性溶媒を含む水溶液に溶解し、該溶液を凍結乾燥することにより調製された、 (4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸またはその塩、またはそれらの溶媒和物と塩化ナトリウムを含有する、実質的に炭酸塩を含まない凍結乾燥製剤。
【請求項2】
親水性溶媒が、t−ブタノール、エタノール、2−プロパノールまたはその混合溶媒である、請求項1記載の凍結乾燥製剤。
【請求項3】
親水性溶媒が、t−ブタノールである、請求項1または2記載の凍結乾燥製剤。
【請求項4】
該溶液のpHを5.0〜7.0に調整した後に凍結乾燥する、請求項1〜3のいずれか一項に記載の凍結乾燥製剤。
【請求項5】
該溶液中の(4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸またはその塩、またはそれらの溶媒和物の濃度が20mg/mL以上である、請求項1〜4のいずれか一項に記載の凍結乾燥製剤。
【請求項6】
凍結乾燥製剤の製造方法であって、(4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸またはその塩、またはそれらの溶媒和物を、0.6%以上の塩化ナトリウムと10〜50%の親水性溶媒を含む水溶液に溶解し、実質的に炭酸塩を含まない該溶液を凍結乾燥する方法。
【請求項7】
親水性溶媒が、t−ブタノール、エタノール、2−プロパノールまたはその混合溶媒である、請求項6記載の方法。
【請求項8】
親水性溶媒が、t−ブタノールである、請求項6または7記載の方法。
【請求項9】
該溶液のpHを5.0〜7.0に調整した後に凍結乾燥する、請求項6〜8のいずれか一項に記載の方法。
【請求項10】
該溶液中の(4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸またはその塩、またはそれらの溶媒和物の濃度が20mg/mL以上である、請求項6〜9のいずれか一項に記載の方法。
【請求項1】
(4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸またはその塩、またはそれらの溶媒和物を、0.6%以上の塩化ナトリウムと10〜50%の親水性溶媒を含む水溶液に溶解し、該溶液を凍結乾燥することにより調製された、 (4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸またはその塩、またはそれらの溶媒和物と塩化ナトリウムを含有する、実質的に炭酸塩を含まない凍結乾燥製剤。
【請求項2】
親水性溶媒が、t−ブタノール、エタノール、2−プロパノールまたはその混合溶媒である、請求項1記載の凍結乾燥製剤。
【請求項3】
親水性溶媒が、t−ブタノールである、請求項1または2記載の凍結乾燥製剤。
【請求項4】
該溶液のpHを5.0〜7.0に調整した後に凍結乾燥する、請求項1〜3のいずれか一項に記載の凍結乾燥製剤。
【請求項5】
該溶液中の(4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸またはその塩、またはそれらの溶媒和物の濃度が20mg/mL以上である、請求項1〜4のいずれか一項に記載の凍結乾燥製剤。
【請求項6】
凍結乾燥製剤の製造方法であって、(4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸またはその塩、またはそれらの溶媒和物を、0.6%以上の塩化ナトリウムと10〜50%の親水性溶媒を含む水溶液に溶解し、実質的に炭酸塩を含まない該溶液を凍結乾燥する方法。
【請求項7】
親水性溶媒が、t−ブタノール、エタノール、2−プロパノールまたはその混合溶媒である、請求項6記載の方法。
【請求項8】
親水性溶媒が、t−ブタノールである、請求項6または7記載の方法。
【請求項9】
該溶液のpHを5.0〜7.0に調整した後に凍結乾燥する、請求項6〜8のいずれか一項に記載の方法。
【請求項10】
該溶液中の(4R,5S,6S)−6−[(1R)−1−ヒドロキシエチル]−4−メチル−3−({4−[(5S)−5−メチル−2,5−ジヒドロ−1H−ピロール−3−イル]−1,3−チアゾール−2−イル}スルファニル)−7−オキソ−1−アザビシクロ[3.2.0]ヘプト−2−エン−2−カルボン酸またはその塩、またはそれらの溶媒和物の濃度が20mg/mL以上である、請求項6〜9のいずれか一項に記載の方法。
【図1】

【図2】
【図3】
【図4】
【図2】
【図3】
【図4】
【公開番号】特開2011−6336(P2011−6336A)
【公開日】平成23年1月13日(2011.1.13)
【国際特許分類】
【出願番号】特願2009−149292(P2009−149292)
【出願日】平成21年6月24日(2009.6.24)
【出願人】(000002912)大日本住友製薬株式会社 (332)
【Fターム(参考)】
【公開日】平成23年1月13日(2011.1.13)
【国際特許分類】
【出願日】平成21年6月24日(2009.6.24)
【出願人】(000002912)大日本住友製薬株式会社 (332)
【Fターム(参考)】
[ Back to top ]