
処理溶液、前処理方法、情報取得方法および検出方法
【課題】大きさが10〜数10μm径の細胞の一つ一つに含まれるタンパク質の種類を高感度で同定する方法を提供すること。また、これを達成するために必要となる検体の処理液を提供すること。
【解決手段】(1)病変組織の切片を、金粒子と消化酵素を含む水溶液で処理し、注目するタンパク質を限定分解分解する工程、(2)TOF−SIMSを用い、断片化したペプチドの二次元分布を測定する工程、(3)プロテオーム解析結果と数値解析的手法を用いることにより、病変組織の切片における注目タンパク質の二次元分布を可視化する工程、の三工程により上記の課題を解決する。
【解決手段】(1)病変組織の切片を、金粒子と消化酵素を含む水溶液で処理し、注目するタンパク質を限定分解分解する工程、(2)TOF−SIMSを用い、断片化したペプチドの二次元分布を測定する工程、(3)プロテオーム解析結果と数値解析的手法を用いることにより、病変組織の切片における注目タンパク質の二次元分布を可視化する工程、の三工程により上記の課題を解決する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、検体中の対象物を分析するための前処理に用いる処理溶液、該前処理方法、検体中の対象物に関する情報を取得する方法および、特有な対象物の有無を検出する方法に関する。特に、生体組織または細胞中に含まれるタンパク質を分析するための前処理に用いる処理溶液、該前処理方法および、生体組織または細胞中に含まれるタンパク質に関する情報を取得する方法、該タンパク質の有無を検出する方法に関する。
【背景技術】
【0002】
近年のゲノム解析の進展により、生体内に存在する遺伝子産物であるタンパク質の解析の重要性が急速にクローズアップされてきている。
【0003】
従来から、タンパク質の発現及び機能解析の重要性が指摘されており、その解析手法の開発が進められている。これらの手法は、
(1)二次元電気泳動や高速液体クロマトグラフ(HPLC)による分離精製と、
(2)放射線分析、光学的分析、質量分析等の検出系
の組み合わせを基本としている。
【0004】
タンパク質解析技術の基盤はプロテオーム解析と呼ばれるもので、これは遺伝子から作り出され実際に生体内で働いているタンパク質を解析し、細胞の機能や疾患の原因を究明することを目的としている。代表的な解析手法としては、
(1)対象とする生体組織や細胞からのタンパク質の抽出、
(2)二次元電気泳動によるタンパク質の分離、
(3)MALDI法(マトリクス支援レーザー脱離−飛行時間型質量分析法:MALDI−TOFMS)などの質量分析によるタンパク質またはその断片の分析、
(4)ゲノムプロジェクトなどのデータベースを利用したタンパク質の同定、
などが挙げられる。
【0005】
この他に、
(1)対象とする生体組織や細胞からのタンパク質の抽出、
(2)抽出したタンパク質の消化(または変性)、
(3)液体クロマトグラフ(LC)とイオントラップ型質量分析計(Ion−trap MS)を組み合わせたシステムを用いた上記消化(または変性)タンパク質の分析、
(4)データベースの構築およびタンパク質の同定、
などもある(例えば非特許文献1)。
【0006】
これらのプロテオーム解析により、癌を例に挙げれば、再発や転移に関わるタンパク質が明らかになりつつあるなど、既に成果が出始めている。
【0007】
一方、プロテインチップや生体組織切片におけるタンパク質の二次元分布の可視化を目的とした、飛行時間型二次イオン質量分析法(以下、TOF−SIMS法と略す。)をベースとする情報取得手法及び装置を本願発明者らが提案した(特許文献1)。この手法は、インクジェット法などを用い、イオン化促進物質かつ/または消化酵素を上記のプロテインチップや生体組織切片に付与し、タンパク質の種類に関する情報(消化酵素により限定分解されたペプチドの情報を含む)を、その位置情報を保持したままTOF−SIMS法により可視化するというものである。
【0008】
また、上記を発展させた手法や装置も本願発明者らが提案した。すなわち、インクジェット法で使用する水溶液のpHを工夫したもの、細胞内代謝物質を検出するために工夫したもの、電気泳動法や薄層クロマトグラフィーなどの分離精製手法を併用しタンパク質の識別能を高めたものなどを提案した。
【0009】
TOF−SIMS法において検出感度を向上させる方法として金属粒子を使う方法が提案されている。例えば、A.MarcusとN.Winogradは、金や銀のナノ粒子をサブモノレイヤー程度、サンプル表面に成膜(deposition)することでTOF−SIMSの検出感度が向上することを報告している(非特許文献2)。また、Y.P.Kimらは、金ナノ粒子上に配置したペプチド分子の(TOF−SIMS法における)イオン化効率が、金ナノ粒子の効果により向上することを報告している(非特許文献3)。
【0010】
また、個々の細胞を対象としたSIMS分析の例としては、S.G.Ostrowskiらの報告(非特許文献4)などが挙げられる。
【0011】
さらに特許第3207706号には、金属コロイドを含む水溶液を液滴化し、インクジェット方式により基板上へ付与する方法が開示されている。
【非特許文献1】磯辺俊明、高橋信弘編「実験医学別冊 プロテオーム解析」、羊土社、2000年
【非特許文献2】A.Marcus and N.Winograd,Anal.Chem.,78,141−148(2006)
【非特許文献3】Y.P.Kim et al.,Anal.Chem.,78,1913−1920(2006)
【非特許文献4】S.G.Ostrowski et al.,“Single−Cell Level Mass Spectrometric Imaging”,Dekker Encyclopedia of Nanoscience and Nanotechnology,pages 1−11(2006)
【特許文献1】特許第3658397号明細書
【特許文献2】特許第3207706号明細書
【発明の開示】
【発明が解決しようとする課題】
【0012】
しかしながら、従来のプロテオーム解析方法は特定の生体組織や体液、血液などを対象としたもので、大きさが10〜数10μm径の細胞を直接対象としたものではなかった。癌細胞など特定の病変細胞の中に含まれるタンパク質、あるいは癌細胞に隣接する細胞中に含まれるタンパク質、さらにはその両者を同定できれば診断デバイスや創薬(薬剤候補スクリーニング)デバイスの開発に寄与できることになる。また、初期の癌細胞や癌組織について、転移や再発に関わるタンパク質の有無を直接調べることも原理的には可能になり、新たな予後診断方法にもなり得る。
【0013】
また、本願発明者らが提案した情報取得方法は、細胞レベルでのタンパク質に関する情報(消化酵素により限定分解されたペプチドの情報を含む)を取得できるが、検出感度が十分でない場合があった。
【0014】
S.G.Ostrowskiらの方法は、SIMS法における空間分解能を高めることで、質量情報を用いた細胞レベルのイメージングを可能としたが、質量/電荷(m/z)比の上限が500程度という制限があった。すなわち、本願発明者らが提案した特許文献2に記載されている、消化分解ペプチドから分解前のタンパク質を決定するのに必要な「m/z比が500〜5000程度の特徴的なフラグメントイオン」を検出するには、その方法は十分でなかった。
【0015】
A.MarcusとN.Winogradの方法や、Y.P.Kimらの方法は、金ナノ粒子を共存させる(成膜させる、または被検出分子の下部に配置する)ことが、TOF−SIMS法の検出感度を向上させることを開示している。しかしながらこれらの方法は、消化分解させたペプチドを対象としたものではないことから、生体組織や細胞中の特定タンパク質の分析に利用することは難しい(均一に消化分解する方法が開示されていない)。
【0016】
また、特許文献2には、金属コロイドを含む水溶液を液滴化し、インクジェット方式により基板上へ付与する方法が開示されているが、電子デバイスの配線形成を目的としたものであり、消化酵素の付与に関する記載がない。すなわち、この方法も、生体組織や細胞中の特定タンパク質を分析するための前処理法としてそのまま利用することは難しい。
【課題を解決するための手段】
【0017】
本発明の処理溶液は、検体中の対象物を分析するための前処理に用いる処理溶液であって、少なくとも金属粒子と消化酵素を含み、かつ該金属粒子が常温常圧下で分散された状態にあることを特徴とする。ここでいう常温常圧とは、摂氏25度±5度、101325±1000Paを意味する。
【0018】
前記の金属粒子は金粒子であることが好ましく、またその直径は1nm〜100nmの範囲にあることが好ましい。本発明の処理溶液は、TOF−SIMS法で分析する被検体の前処理に好適に用いられる。したがって、TOF−SIMS法で感度が向上する微粒子であれば金以外の金属を使用することもできる。また、本発明の処理溶液はインクジェット法を用い、該処理溶液を液滴として所定の位置に付与することを特徴とする。したがって、上記金属粒子は、インクジェット装置の吐出口で目詰まりを起こさないことが必要で、該金属粒子の大きさの上限は100nm程度である。また通常は水溶液として使用するため、該金属粒子は水溶液に分散された状態であることが必要である。なお、該金属粒子の大きさの下限は1nm程度が好ましい。
【0019】
また、本発明の処理溶液は、前記の金属粒子と消化酵素とが化学結合(配位結合を含む)していることが好ましい。これは、インクジェット法を用いて該処理溶液を検体に付与するに際し、金属粒子が水溶液に分散された状態を長く保つためである。金属粒子として金を例に挙げれば、金と消化酵素の結合(配位結合を含む)に、消化酵素中のチオール基(メルカプト基)やアミノ基などを利用するのが一般的である。
【0020】
さらに、本発明の処理溶液は、前記消化酵素が人工消化酵素であることを特徴とする。ここでいう人工消化酵素とは、人工的に合成した酵素であって、タンパク質を消化分解する消化部位と上記金属粒子との結合部位との、少なくとも二つの部位を持つ酵素を指す。タンパク質を消化分解する消化部位とは、ペプシン、トリプシン、キモトリプシンなどの消化活性部位と基本的には同一である。
【0021】
また、前記検体中の対象物として、検体中のタンパク質を挙げることができる。
【0022】
本発明の前処理方法は、上記の処理溶液をインクジェット装置に充填し、該装置を用い、該処理溶液を液滴として所定の位置に付与することを特徴とする。インクジェット装置としては、ピエゾ方式、バブルジェット方式のいずれの装置を用いることができる。
【0023】
本発明の情報取得方法は、検体中の対象物に関する情報を取得する方法であって、前記の前処理方法を用いて検体を処理する工程と、処理後の検体に含まれる対象物の質量に関する情報(消化酵素で分解されたペプチド断片の質量に関する情報を含む)をTOF−SIMS法により取得する工程とを備えることを特徴とする。TOF−SIMSで用いる一次イオン種としては、イオン化効率、質量分解能等の観点から、ガリウムイオン、セシウムイオン、金イオン、ビスマスイオンなどの単原子イオンや、Au3イオン、Bi3イオン、C60イオンなどのクラスターイオンなどが用いられる。
【0024】
本発明の情報取得方法は、前記の検体が病変組織の切片であることを特徴とし、また前記の対象物がタンパク質であることを特徴とする。
【0025】
さらに本発明は、検体中における、疾病に関わる特有な対象物の有無を検出する方法であって、上記の情報取得方法を利用して検体中における疾病に関わる特有な対象物の有無を検出することを特徴とする。
【発明の効果】
【0026】
本発明によれば、大きさが10〜数10μm径の細胞の一つ一つに含まれるタンパク質の種類を高感度で同定する方法を提供できる。
【発明を実施するための最良の形態】
【0027】
本発明は上記の特徴を持つものであるが以下に最良の実施形態について述べる。
【0028】
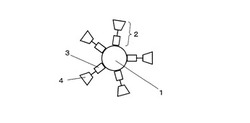
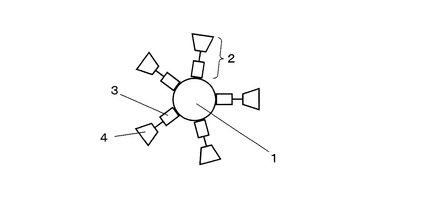
本発明の処理溶液は、直径が1nm〜100nmの範囲にある金粒子と消化酵素を含む水溶液であって、これらが常温常圧下で分散された状態にあることが好ましい。特に、前記金粒子と消化酵素が化学結合(配位結合を含む)していることが好ましい。最良の形態としては、前記消化酵素として、チオール基(メルカプト基)またはアミノ基を有する(金粒子との)結合部位と、ペプシン、トリプシン、キモトリプシンなどの消化活性部位とを有する人工消化酵素を用いることである。この第一の理由は、水溶液中での分散安定性が向上するためである。また第二の理由は、後述のTOF−SIMS分析工程において検出感度が向上するためである。すなわち、二次イオンの生成効率を高める作用のある金粒子の近傍で効率よく消化反応を進行させ、結果として、金粒子の近傍に消化分解ペプチドを集めることができる。図1に金粒子と結合した人工消化酵素の模式図を示す。図1において、1は金粒子、2は人工消化酵素、3は人工消化酵素2内の金粒子との結合部位、4は人工消化酵素2内の消化活性部位、である。
【0029】
本発明において、検体として病変組織の切片を挙げることができ、また対象物としては検体中のタンパク質を挙げることができる。そのほか、検体としては分子量が10000以下のペプチドなどを挙げることができる。
【0030】
本発明の情報取得方法の基本工程は、(1)病変組織の切片を、金粒子と消化酵素を含む水溶液で処理し、注目するタンパク質を限定分解する工程、(2)TOF−SIMSを用い、断片化したペプチドの二次元分布を測定する工程、(3)プロテオーム解析結果と数値解析的手法を用いることにより、病変組織の切片における注目タンパク質の二次元分布を可視化する工程、の三つからなる。
【0031】
上記(1)の工程では、検体に消化酵素等を均一に付与させるためにインクジェット法を用いることが好ましい。
【0032】
上記(2)の工程において、TOF−SIMSの代表的測定条件を以下に示す。
【0033】
一次イオン種:Au3イオン、Bi3イオン、C60イオン等のクラスターイオン
一次イオンビーム径:約1μm
一次イオンフルーエンス(ドーズ量):1×1014 ions/cm2
測定領域:100μm×100μm
測定質量範囲:1〜5000(m/z)
測定時間:約10分。
【0034】
一次イオンとしてクラスターイオンを用いるのは、単原子イオンに比べクラスターイオンの方が質量数の大きい二次イオンの検出に有利であるからである(例えば、本発明者らの特許文献1)。一次イオンビーム径を約1μmとするのは、直径20μm前後の個々の細胞を識別するために必要だからである。一次イオンフルーエンスを上記の値とするのは、一定以上の二次イオン感度(積算信号強度)を得るために必要だからである。測定質量範囲を上記の値とするのは主測定対象が消化分解ペプチドであるからである(例えば、特開2006−010658号公報参照)。測定時間を約10分程度と設定するのは、長時間の測定が定量的評価に悪影響を与える場合があるからである(例えば、H.Hashimoto et al.,Appl.Surf.Sci.,231−232,385−391(2004))。
【0035】
上記(3)の工程における数値解析的手法とは、因子分析などの統計的手法を指す。
【0036】
本発明は、病変組織の切片において、疾病に関わる特有なタンパク質の有無を検出する方法でもある。
【実施例】
【0037】
以下に、実施例を挙げて、本発明をより具体的に説明する。以下に示す具体例は、本発明にかかる最良の実施形態の一例ではあるが、本発明はかかる具体的形態に限定されるものではない。
【0038】
本開示によりトリプシン組換えタンパク質の取得とその方法を示す。以下の実施例では、ウシ膵臓由来のトリプシノーゲン(トリプシン前駆体)の大腸菌発現ベクターを設計・作製し、不溶性顆粒画分として大腸菌に発現させて巻き戻しと精製工程の後、自己触媒により酵素活性を有する組換えトリプシンタンパク質を取得する。また、以下に記す組換えDNA法はSambrookら著書のモレキュラークローニング(実験マニュアル第二版Cold Spring Harbor Laboratory,New York(1989))に基づいて行い、市販のキットを用いる場合は、特記しない限り当業者が推奨プロトコールに準じて行うことができる。また、発現ベクターの構築にはNovagen社のpETBlue−1System(Cat.No.70673−3)に付属のベクターpETBlue−1を用いてコンストラクトを作製する。宿主大腸菌には、サブクローニング用としてJM109株(Novagen社)を、たんぱく質生産用としてRosetta (DE3) pLacI株(Novagen社)を用いる。
【0039】
また、実施例に用いる化学薬品は特記しない限り分析級またはそれ以上であり、シグマ社及びナカライテスク社より購入できる。オリゴDNAはシグマジェノシス社、制限酵素および修飾酵素はタカラバイオ社より購入できる。
【0040】
(実施例1) ウシ膵臓由来のトリプシノーゲン遺伝子発現ベクターの構築
以下、ウシ膵臓由来のトリプシノーゲン組換え遺伝子は、トリプシノーゲン遺伝子コーディング領域(NCBI GenBankアクセッション番号;X54703)の3'末端に、アフィニティー精製用His6配列と金担体に固定するためのCys残基をコードするスペーサー能を有するDNAタグ配列を設けたDNA配列(図2)をさす。
【0041】
(1)上記ウシ膵臓由来のトリプシノーゲン組換え遺伝子の合成
プライマーセット1(配列番号1〜4)、プライマーセット2(配列番号5〜10)と、Pyrobest DNAポリメラーゼ(タカラバイオ社)を用い当業者の推奨する配合で調合し、温度サイクル98℃×1min.→(98℃、10sec.→55℃、30sec.→72℃、30sec.)×40cycles→4℃でオーバーラップPCRを行う。それぞれ目的サイズ約300bpと450bpの断片の増幅を電気泳動とEtBr染色により確認する。
配列番号1(5’→3’)
atgttcccctcggacgacgatgacaagatcgtcgggggctacacctgcgcagagaatt
配列番号2
ggacaccacccactggtcattgatgagggagcccccgcagaagtggtagccagcattcagggacacctggtaagggacggaattctctgcgcaggtgtag
配列番号3
atgaccagtgggtggtgtccgcggctcactgctaccagtaccacatccaggtgaggctgggagaatacaacattgatgtcttggagggtggtgagcagtt
配列番号4
agagtttgatcagcaggatgtcattgtccagagtccagctgctgtacttggggtggcggatgatcttggacgcatcgatgaactgctcaccaccctccaa
配列番号5
catcctgctgatcaaactctccacgcctgcggtcatcaatgcccgggtgtccaccttgctgctgcccagtgcctgtgcttccgcaggcacagagtgcctc
配列番号6
ctcagcagcggggccaccaggcattgcagcaggtccgggtagttgacgccactgctcagggtgttgccccagccggagatgaggcactctgtgcctgcgg
配列番号7
ctggtggccccgctgctgagccacgccgactgtgaagcctcataccctggacagatcactaacaacatgatctgcgctggcttcctggaaggaggcaagg
配列番号8
acagccgtagccccaggacacaatgccctggagctgtccgttgcaagccacagggccgccagagtcaccctggcaggaatccttgcctccttccaggaag
配列番号9
tgtcctggggctacggctgtgcccagaagggcaagcctggggtctacaccaaggtctgcaactacgtggactggattcaggagaccatcgccgccaacag
配列番号10
ggcttagcaatggtggtgatgatggtggctagcgctgttggcggcgatggtctc
次に、目的サイズの断片を市販のゲル精製キット(Wizard SV Gel and PCR Clean−Up system(Cat No #A9281 Promega社))により精製する。次に精製した上記二つの断片を鋳型にし、プライマーセット3(配列番号11および12)を用いて再度同条件でオーバーラップPCRを行い、最終目的サイズ約730bpの断片が増幅していることを確認し、同様に精製する。
配列番号11
atgttcccctcggacgacgatga
配列番号12
ggcttagcaatggtggtgatgat
(2)大腸菌発現ベクターへのサブクローニング
pETBlue−1発現ベクターをEcoRV酵素で消化した後、BAPにより脱リン酸化処理(37℃、1時間)を行い、Wizard SV Gel and PCR Clean−Up system(Cat No #A9281 Promega社)により脱リン酸化されたDNA断片を精製する。次に、市販のDNAライゲーションキット(Ligation High Code No.LGK−101(TOYOBO社))を用いて、(1)で合成したDNA断片と脱リン酸化ベクターを業者推奨の方法にて調合し連結させる。
【0042】
そして、ライゲーション溶液をJM109株コンピテントセル(Novagen社)にヒートショック法により形質転換した後に、Luria−Bertani medium(LB)+Amp.(100μg/m)+IPTG+X−gal寒天プレートに撒き、37℃にて一晩静置する。
【0043】
次に、プレート中から青白選択を行い任意のコロニーをLB/Amp液体培地3mlに植菌し、37℃にて一晩振盪培養を行う。その後、市販のMiniPrepキット(Plus Minipreps DNA Purification System(Promega社))を使用して、プラスミドDNAを回収する。
【0044】
得られたプラスミドの塩基配列を決定し、目的の断片が正しい方向に挿入され且つ配列が正しいことを確認することができ、結果としてウシ膵臓由来のトリプシノーゲン組換え遺伝子発現ベクターが得られる。
【0045】
(実施例2) トリプシノーゲン組換えタンパク質の大腸菌による発現、巻き戻し実験
(1)形質転換
上記実施例1で取得した発現ベクターを、タンパク質生産用のRosetta(DE3)pLacI株コンピテントセル溶液に形質転換する。形質転換は、ヒートショックを氷中→42℃、90sec.→氷中の条件でおこなう。ヒートショックにより形質転換した上記Rosetta(DE3)pLacI株コンピテントセル溶液にTerrific broth(TB;1%(W/V)yeast extract,2%(W/V)peptone,0.1M リン酸バッファー(pH7.5),1%グリセロール)培地500μLを加え、一時間37℃にて振盪培養を行った。その後、6000r.p.m.で5分間遠心を行い、培養上清650μLを廃棄し、残った培養上清と沈殿となった細胞画分を攪拌し、TB/Amp.100μg/ml/chloramphenicol 34μg/ml寒天プレートに撒き、一晩37℃にて培養する。
【0046】
(2)予備培養
プレート上のコロニーを無作為に選択し、50mlのTB/Amp.200μg/ml/chloramphenicol 34μg/ml培地にて37℃でOD600nm値が0.2〜0.6になるまで振盪培養を行う。次に1,000×g、5分間遠心して上清を捨て、菌を集菌し、新しい培地(上記)で懸濁した後2mlとり、TB/Amp.400μg/ml/chloramphenicol 34μg/ml培地500mlに植菌し37℃でOD600nm値が0.2〜0.6になるまで振盪培養を行う。
【0047】
(3)本培養
上記予備培養溶液を更に培養を30℃にて継続する。D600nm値が1.0を越えた時点で、終濃度が1mMとなるようにIPTGを加え、更に30℃にて終夜培養を行う。
【0048】
(4)精製
目的のポリペプチド鎖を不溶性顆粒画分から以下の工程により精製する。
【0049】
(i)不溶性顆粒の回収
上記3)で得られた培養液を6,000r.p.m.で30分間遠心し、沈殿を菌体画分として得る。得られた菌体を100μg/mlリゾチーム入りトリス溶液(20mM Tris−HCl(pH8.0)/500mM NaCl)15mlに懸濁し、37℃で30分反応後、氷中保管する。得られた懸濁液をフレンチプレスにて破砕し、菌破砕液を得る。10unitのDNaseI酵素を加え、37℃で15分間反応させる。その後、最終2%になるようにTriton X−100を加え不溶性顆粒画分を2回洗浄する。
【0050】
次に、菌破砕液を12,000r.p.m.で15分間遠心を行い、上清を除き、沈殿を不溶性顆粒画分として得る。
【0051】
(ii)不溶性顆粒画分の可溶化
上記(i)で得られた不溶性画分を6M 塩酸グアニジン/トリス溶液(20mMTris−HCl(pH8.0)/500mM NaCl)10mlを加え、超音波で不溶画分を分散させた後室温で一晩浸漬する。次に、12,000r.p.m.で10分間遠心し、上清を可溶化溶液として得る。
【0052】
(iii)金属キレートカラム
金属キレートカラム担体としてHis−Bind(Novagen社製)を用いる。カラム調整やサンプル負荷、及び洗浄工程は、前記業者の推奨方法に準拠し、室温(20℃)にて行う。目的であるHisタグ融合のポリペプチドの溶出は60mMイミダゾール/トリス溶液にて行う。溶出液のSDS−PAGE(アクリルアミド15%)の結果、単一バンドであり、精製されていることを確認する。
【0053】
(iv)透析
上記溶出液に対して、外液を6M塩酸グアニンジン/トリス溶液として4℃にて透析を行い、溶出液中のイミダゾールの除去を行い、上記ポリペプチド鎖溶液を得る。
【0054】
(v)巻き戻し
上記と同様にして、ポリペプチド鎖溶液を以下の工程により、脱塩酸グアニンジンを透析(4℃)にて行いながらタンパク質の巻き戻しを行う。
【0055】
(a)6M塩酸グアニジン/トリス溶液を用い、ポリペプチド鎖のモル吸光係数とΔO.D.(280nm−320nm)値から濃度7.5μMのサンプル(希釈後体積10ml)を調製する。次にβ−メルカプトエタノール(還元剤)を終濃度375μM(タンパク濃度50倍)になるよう添加し、室温、暗所で4時間還元を行う。このサンプル溶液を透析バック(MWCO;8,000)に入れ、透析用サンプルとする
(b)透析外液を6M塩酸グアニンジン/トリス溶液として、透析サンプルを浸漬し、緩やかに攪拌しながら6時間透析する
(c)外液の塩酸グアニジン濃度を3M、2Mと段階的に下げる。それぞれの外液濃度において、6時間透析する
(d)酸化型グルタチオン(GSSG)を終濃度375μM、L−Argを終濃度0.4Mとなるようにトリス溶液に加え、上記(c)の2Mの透析外液を加え、塩酸グアニジン濃度を1Mとし、NaOHでpH8.0(4℃)に調整した溶液にて、12時間緩やかに攪拌しながら透析する
(e)上記(d)と同様の作業にて塩酸グアニジン濃度0.5MのL−Arg含有トリス溶液を調整し、更に12時間透析する
(f)最後にトリス溶液にて12時間透析する
(g)透析終了後、10,000r.p.m.で約20分間遠心分離し凝集体と上清とを分離し、可溶性の巻き戻しトリプシノーゲン組換えタンパク質を得る。
【0056】
(実施例3) トリプシノーゲン組換えタンパク質の活性化と活性組換えトリプシンタンパク質の精製取得
(1)トリプシノーゲン組換えタンパク質の活性化
得られた巻き戻しトリプシノーゲン組換えタンパク質を1時間、室温に放置した後、4℃で12時間静置することで自己触媒能による組換えトリプシンタンパク質を得る。
【0057】
(2)クロマトグラフィーによる精製
上記(1)で得られた5μM可溶性の巻き戻しトリプシン組換えタンパク質溶液をスーパーデックス200カラム(GEヘルスケアサイエンス社)(バッファー;20mMのTris−HCl(pH8.0)、500mMのNaCl、流速;0.5ml/分)にてゲルろ過し、目的サイズの単ピークのフラクションを得る。
【0058】
(3)トリプシン活性測定
N−Benzoyl−DL‐arginine‐p−nitroanilide hydrochloride(BAPA)を基質とするErlangerらの方法(BAPA法)に準じる。25℃で5分間、基質と上記フラクションのサブフラクションとを反応させ、BAPA分解物であるパラニトロアニリンの405nm吸光度を測定し、トリプシン活性の最も高いフラクションを取得する。結果として、活性を有する組換えトリプシンタンパク質を得る。このタンパク質を後の評価に用いる。
【0059】
(実施例4) 組換えトリプシンタンパク質と金粒子を含む水溶液の調製と、インクジェットプリンタを用いた検体への該水溶液の付与
平均粒径が40nm程度の金粒子を含む溶液(分散剤:クエン酸)を出発材料とし、常法に従い、上記組換えトリプシンタンパク質と金粒子が結合した、本願発明の処理溶液である水溶液を調製する。この水溶液が十分分散された状態にあるか否かは、例えば、二日間程度常温で放置した後に沈殿が生じるか否かなどにより判断することができる。この水溶液を、本発明者らの特許第3658397号明細書に記載されているバブルジェットプリンタに充填する。より具体的には、バブルジェットプリンタBJF−850(キヤノン(株)製)用のプリンターヘッドBC−50(キヤノン(株)製)を、数100μlの溶液を吐出可能とするべく改造したものを使用し、このヘッドのタンク部に、前記水溶液を数100μl注入する。その後、シリコン基板上に配置した検体を前記プリンタに挿入し、検体表面の全面に前記水溶液を液滴として付与する。その際、検体表層のタンパク質を均一に消化するために、最終的に付与される水溶液量が二次元的に均一になるように工夫する必要がある(例えば、液滴を付与する地点を少しずつ移動させながら行う)。なお、液滴量は通常1滴あたり1〜8plとし、ある一定の地点における付与回数(重ねうちの回数)は上記組換えトリプシンタンパク質の濃度に応じて調整する。該組換えトリプシンタンパク質の濃度は1〜100μM程度とする。また、検体に前記水溶液を付与した後は、消化反応を進行させるために、常温、高湿度下で一定時間保持することが必要である。
【0060】
なお、上記バブルジェットプリンタで使用する水溶液は、吐出の安定化などを目的のため、グリセリン、尿素、チオジグリコール、アセチレンアルコール(商品名:アセチレノールEH;川研ファインケミカル社)の中から選ばれる一種または複数を含んでもよい。
【0061】
(実施例5) ヒト血清アルブミン(HSA)のトリプシン消化物のTOF−SIMS分析
ヒト血清アルブミン(HAS、アミノ酸の配列番号13)をシリコン基板などにスピンコートし、これを実施例4に示す方法により消化分解する。
配列番号13
mkwvtfisllflfssaysrgvfrrdahksevahrfkdlgeenfkalvliafaqylqqcpfedhvklvnevtefaktcvadesaencdkslhtlfgdklctatlretygemadccakqepernecflqhkddnpnlprlvrpevdvmctafhdneetflkkylyeiarrhpyfyapellffakrykaafteccqaadkaacllpkldelrdegkassakqrlkcaslqkfgerafkawavarlsqrfpkaefaevsklvtdltkvhtecchgdllecaddadlayicenqdsissklkeccekpllekshciaevendempadlpslaadfveskdvcknyaeakdvflgmflyeyarrhpdysvvlllrlaktyettlekccaaadphecyakvfdefkplveepqnlikqncelfeqlgeykfqnallvrytkkvpqvstptlvevsrnlgkvgskcckhpeakrmpcaedylsvvlnqlcvlhek tpvsdrvtkccteslvnrrpcfsalevdetyvpkefnaetftfhadictlsekerqikkqtalvelvkhkpkatkeqlkavmddfaafvekcckaddketcfaeegkklvaasqaalgl
上記の消化分解処理により、基本的には上記配列13中のkとrの右側が選択的に切断され、多数のペプチド断片が生成する。この断片ペプチドの内、例えば、dahk,sevahr,necflqhk,hpyfyapellffak,caslqk,aefaevsk,lvtdltk,adlayicenqdsissk,dvflgmflyeyar,hpdysvvlllr,vpqvstptlvevsrvgsk,cckhpeak,tpvsdr,ccteslvnr,erqik,avmddfaafvek,lvaasqaalglなどの特徴的なペプチドを、それらの水素付加イオンまたは金付加イオンなどの形でTOF−SIMS法により検出する。これらの分子量は概ね500〜5000m/zの範囲にある。上記のように、特徴的な断片ペプチドを複数検出することで、消化分解前のタンパク質がHSAであることを同定することができる。なお、検体表層に金粒子が共存するため二次イオンの検出感度が向上し、上記同定精度が向上する。
【0062】
(実施例6) ヒト癌組織切片のトリプシン消化物のTOF−SIMS分析
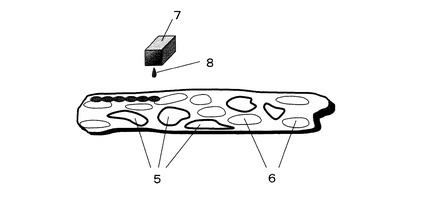
ヒトの癌組織から、厚さ0.5〜5μm程度、大きさ1〜10mm2程度の切片を切り出し、これを基板上に配置し、次に実施例4に示す方法により消化分解する。図3にその模式図を示す。図3において、5は癌組織切片中の異常細胞、6は癌組織切片中の正常細胞、7はインクジェットプリンタヘッド、8は液滴を示す。
【0063】
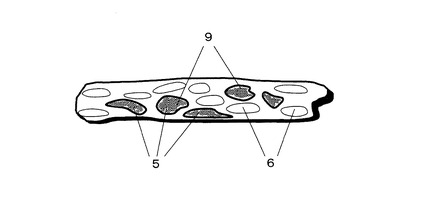
消化分解処理を行った後、実施例5と同様に消化分解ペプチドをTOF−SIMS法により検出する。その後、例えば、再発や転移に関わるタンパク質を分解ペプチドのパターンから同定し、これらの信号強度の二次元分布を再表示させる。この模式図を図4に示す。図4において、9は注目するタンパク質の存在個所(再発や転移に関わるタンパク質の存在個所)である。上記のように、本発明の方法を用いることで、注目するタンパク質の存在個所を細胞レベルで調べることが可能になる。なお、検体表層に金粒子が共存するため二次イオンの検出感度が向上し、結果として上記の注目するタンパク質の検出感度が向上する。
【図面の簡単な説明】
【0064】
【図1】金粒子と結合した人工消化酵素の模式図である。
【図2】金担体に固定するためのCys残基をコードするスペーサー能を有するDNAタグ配列を設けたDNA配列である。
【図3】ヒトの癌組織切片に対し、金粒子と消化酵素を含む水溶液をインクジェット法により付与する工程を示す模式図である。
【図4】注目するタンパク質の存在個所(再発や転移に関わるタンパク質の存在個所)を、断片ペプチドの検出パターンから同定し、これを二次元表示した例である。
【符号の説明】
【0065】
1 金粒子
2 人工消化酵素
3 人工消化酵素2内の金粒子との結合部位
4 人工消化酵素2内の消化活性部位
5 癌組織切片中の異常細胞
6 癌組織切片中の正常細胞
7 インクジェットプリンタヘッド
8 液滴
9 注目するタンパク質の存在個所(再発や転移に関わるタンパク質の存在個所)
【技術分野】
【0001】
本発明は、検体中の対象物を分析するための前処理に用いる処理溶液、該前処理方法、検体中の対象物に関する情報を取得する方法および、特有な対象物の有無を検出する方法に関する。特に、生体組織または細胞中に含まれるタンパク質を分析するための前処理に用いる処理溶液、該前処理方法および、生体組織または細胞中に含まれるタンパク質に関する情報を取得する方法、該タンパク質の有無を検出する方法に関する。
【背景技術】
【0002】
近年のゲノム解析の進展により、生体内に存在する遺伝子産物であるタンパク質の解析の重要性が急速にクローズアップされてきている。
【0003】
従来から、タンパク質の発現及び機能解析の重要性が指摘されており、その解析手法の開発が進められている。これらの手法は、
(1)二次元電気泳動や高速液体クロマトグラフ(HPLC)による分離精製と、
(2)放射線分析、光学的分析、質量分析等の検出系
の組み合わせを基本としている。
【0004】
タンパク質解析技術の基盤はプロテオーム解析と呼ばれるもので、これは遺伝子から作り出され実際に生体内で働いているタンパク質を解析し、細胞の機能や疾患の原因を究明することを目的としている。代表的な解析手法としては、
(1)対象とする生体組織や細胞からのタンパク質の抽出、
(2)二次元電気泳動によるタンパク質の分離、
(3)MALDI法(マトリクス支援レーザー脱離−飛行時間型質量分析法:MALDI−TOFMS)などの質量分析によるタンパク質またはその断片の分析、
(4)ゲノムプロジェクトなどのデータベースを利用したタンパク質の同定、
などが挙げられる。
【0005】
この他に、
(1)対象とする生体組織や細胞からのタンパク質の抽出、
(2)抽出したタンパク質の消化(または変性)、
(3)液体クロマトグラフ(LC)とイオントラップ型質量分析計(Ion−trap MS)を組み合わせたシステムを用いた上記消化(または変性)タンパク質の分析、
(4)データベースの構築およびタンパク質の同定、
などもある(例えば非特許文献1)。
【0006】
これらのプロテオーム解析により、癌を例に挙げれば、再発や転移に関わるタンパク質が明らかになりつつあるなど、既に成果が出始めている。
【0007】
一方、プロテインチップや生体組織切片におけるタンパク質の二次元分布の可視化を目的とした、飛行時間型二次イオン質量分析法(以下、TOF−SIMS法と略す。)をベースとする情報取得手法及び装置を本願発明者らが提案した(特許文献1)。この手法は、インクジェット法などを用い、イオン化促進物質かつ/または消化酵素を上記のプロテインチップや生体組織切片に付与し、タンパク質の種類に関する情報(消化酵素により限定分解されたペプチドの情報を含む)を、その位置情報を保持したままTOF−SIMS法により可視化するというものである。
【0008】
また、上記を発展させた手法や装置も本願発明者らが提案した。すなわち、インクジェット法で使用する水溶液のpHを工夫したもの、細胞内代謝物質を検出するために工夫したもの、電気泳動法や薄層クロマトグラフィーなどの分離精製手法を併用しタンパク質の識別能を高めたものなどを提案した。
【0009】
TOF−SIMS法において検出感度を向上させる方法として金属粒子を使う方法が提案されている。例えば、A.MarcusとN.Winogradは、金や銀のナノ粒子をサブモノレイヤー程度、サンプル表面に成膜(deposition)することでTOF−SIMSの検出感度が向上することを報告している(非特許文献2)。また、Y.P.Kimらは、金ナノ粒子上に配置したペプチド分子の(TOF−SIMS法における)イオン化効率が、金ナノ粒子の効果により向上することを報告している(非特許文献3)。
【0010】
また、個々の細胞を対象としたSIMS分析の例としては、S.G.Ostrowskiらの報告(非特許文献4)などが挙げられる。
【0011】
さらに特許第3207706号には、金属コロイドを含む水溶液を液滴化し、インクジェット方式により基板上へ付与する方法が開示されている。
【非特許文献1】磯辺俊明、高橋信弘編「実験医学別冊 プロテオーム解析」、羊土社、2000年
【非特許文献2】A.Marcus and N.Winograd,Anal.Chem.,78,141−148(2006)
【非特許文献3】Y.P.Kim et al.,Anal.Chem.,78,1913−1920(2006)
【非特許文献4】S.G.Ostrowski et al.,“Single−Cell Level Mass Spectrometric Imaging”,Dekker Encyclopedia of Nanoscience and Nanotechnology,pages 1−11(2006)
【特許文献1】特許第3658397号明細書
【特許文献2】特許第3207706号明細書
【発明の開示】
【発明が解決しようとする課題】
【0012】
しかしながら、従来のプロテオーム解析方法は特定の生体組織や体液、血液などを対象としたもので、大きさが10〜数10μm径の細胞を直接対象としたものではなかった。癌細胞など特定の病変細胞の中に含まれるタンパク質、あるいは癌細胞に隣接する細胞中に含まれるタンパク質、さらにはその両者を同定できれば診断デバイスや創薬(薬剤候補スクリーニング)デバイスの開発に寄与できることになる。また、初期の癌細胞や癌組織について、転移や再発に関わるタンパク質の有無を直接調べることも原理的には可能になり、新たな予後診断方法にもなり得る。
【0013】
また、本願発明者らが提案した情報取得方法は、細胞レベルでのタンパク質に関する情報(消化酵素により限定分解されたペプチドの情報を含む)を取得できるが、検出感度が十分でない場合があった。
【0014】
S.G.Ostrowskiらの方法は、SIMS法における空間分解能を高めることで、質量情報を用いた細胞レベルのイメージングを可能としたが、質量/電荷(m/z)比の上限が500程度という制限があった。すなわち、本願発明者らが提案した特許文献2に記載されている、消化分解ペプチドから分解前のタンパク質を決定するのに必要な「m/z比が500〜5000程度の特徴的なフラグメントイオン」を検出するには、その方法は十分でなかった。
【0015】
A.MarcusとN.Winogradの方法や、Y.P.Kimらの方法は、金ナノ粒子を共存させる(成膜させる、または被検出分子の下部に配置する)ことが、TOF−SIMS法の検出感度を向上させることを開示している。しかしながらこれらの方法は、消化分解させたペプチドを対象としたものではないことから、生体組織や細胞中の特定タンパク質の分析に利用することは難しい(均一に消化分解する方法が開示されていない)。
【0016】
また、特許文献2には、金属コロイドを含む水溶液を液滴化し、インクジェット方式により基板上へ付与する方法が開示されているが、電子デバイスの配線形成を目的としたものであり、消化酵素の付与に関する記載がない。すなわち、この方法も、生体組織や細胞中の特定タンパク質を分析するための前処理法としてそのまま利用することは難しい。
【課題を解決するための手段】
【0017】
本発明の処理溶液は、検体中の対象物を分析するための前処理に用いる処理溶液であって、少なくとも金属粒子と消化酵素を含み、かつ該金属粒子が常温常圧下で分散された状態にあることを特徴とする。ここでいう常温常圧とは、摂氏25度±5度、101325±1000Paを意味する。
【0018】
前記の金属粒子は金粒子であることが好ましく、またその直径は1nm〜100nmの範囲にあることが好ましい。本発明の処理溶液は、TOF−SIMS法で分析する被検体の前処理に好適に用いられる。したがって、TOF−SIMS法で感度が向上する微粒子であれば金以外の金属を使用することもできる。また、本発明の処理溶液はインクジェット法を用い、該処理溶液を液滴として所定の位置に付与することを特徴とする。したがって、上記金属粒子は、インクジェット装置の吐出口で目詰まりを起こさないことが必要で、該金属粒子の大きさの上限は100nm程度である。また通常は水溶液として使用するため、該金属粒子は水溶液に分散された状態であることが必要である。なお、該金属粒子の大きさの下限は1nm程度が好ましい。
【0019】
また、本発明の処理溶液は、前記の金属粒子と消化酵素とが化学結合(配位結合を含む)していることが好ましい。これは、インクジェット法を用いて該処理溶液を検体に付与するに際し、金属粒子が水溶液に分散された状態を長く保つためである。金属粒子として金を例に挙げれば、金と消化酵素の結合(配位結合を含む)に、消化酵素中のチオール基(メルカプト基)やアミノ基などを利用するのが一般的である。
【0020】
さらに、本発明の処理溶液は、前記消化酵素が人工消化酵素であることを特徴とする。ここでいう人工消化酵素とは、人工的に合成した酵素であって、タンパク質を消化分解する消化部位と上記金属粒子との結合部位との、少なくとも二つの部位を持つ酵素を指す。タンパク質を消化分解する消化部位とは、ペプシン、トリプシン、キモトリプシンなどの消化活性部位と基本的には同一である。
【0021】
また、前記検体中の対象物として、検体中のタンパク質を挙げることができる。
【0022】
本発明の前処理方法は、上記の処理溶液をインクジェット装置に充填し、該装置を用い、該処理溶液を液滴として所定の位置に付与することを特徴とする。インクジェット装置としては、ピエゾ方式、バブルジェット方式のいずれの装置を用いることができる。
【0023】
本発明の情報取得方法は、検体中の対象物に関する情報を取得する方法であって、前記の前処理方法を用いて検体を処理する工程と、処理後の検体に含まれる対象物の質量に関する情報(消化酵素で分解されたペプチド断片の質量に関する情報を含む)をTOF−SIMS法により取得する工程とを備えることを特徴とする。TOF−SIMSで用いる一次イオン種としては、イオン化効率、質量分解能等の観点から、ガリウムイオン、セシウムイオン、金イオン、ビスマスイオンなどの単原子イオンや、Au3イオン、Bi3イオン、C60イオンなどのクラスターイオンなどが用いられる。
【0024】
本発明の情報取得方法は、前記の検体が病変組織の切片であることを特徴とし、また前記の対象物がタンパク質であることを特徴とする。
【0025】
さらに本発明は、検体中における、疾病に関わる特有な対象物の有無を検出する方法であって、上記の情報取得方法を利用して検体中における疾病に関わる特有な対象物の有無を検出することを特徴とする。
【発明の効果】
【0026】
本発明によれば、大きさが10〜数10μm径の細胞の一つ一つに含まれるタンパク質の種類を高感度で同定する方法を提供できる。
【発明を実施するための最良の形態】
【0027】
本発明は上記の特徴を持つものであるが以下に最良の実施形態について述べる。
【0028】
本発明の処理溶液は、直径が1nm〜100nmの範囲にある金粒子と消化酵素を含む水溶液であって、これらが常温常圧下で分散された状態にあることが好ましい。特に、前記金粒子と消化酵素が化学結合(配位結合を含む)していることが好ましい。最良の形態としては、前記消化酵素として、チオール基(メルカプト基)またはアミノ基を有する(金粒子との)結合部位と、ペプシン、トリプシン、キモトリプシンなどの消化活性部位とを有する人工消化酵素を用いることである。この第一の理由は、水溶液中での分散安定性が向上するためである。また第二の理由は、後述のTOF−SIMS分析工程において検出感度が向上するためである。すなわち、二次イオンの生成効率を高める作用のある金粒子の近傍で効率よく消化反応を進行させ、結果として、金粒子の近傍に消化分解ペプチドを集めることができる。図1に金粒子と結合した人工消化酵素の模式図を示す。図1において、1は金粒子、2は人工消化酵素、3は人工消化酵素2内の金粒子との結合部位、4は人工消化酵素2内の消化活性部位、である。
【0029】
本発明において、検体として病変組織の切片を挙げることができ、また対象物としては検体中のタンパク質を挙げることができる。そのほか、検体としては分子量が10000以下のペプチドなどを挙げることができる。
【0030】
本発明の情報取得方法の基本工程は、(1)病変組織の切片を、金粒子と消化酵素を含む水溶液で処理し、注目するタンパク質を限定分解する工程、(2)TOF−SIMSを用い、断片化したペプチドの二次元分布を測定する工程、(3)プロテオーム解析結果と数値解析的手法を用いることにより、病変組織の切片における注目タンパク質の二次元分布を可視化する工程、の三つからなる。
【0031】
上記(1)の工程では、検体に消化酵素等を均一に付与させるためにインクジェット法を用いることが好ましい。
【0032】
上記(2)の工程において、TOF−SIMSの代表的測定条件を以下に示す。
【0033】
一次イオン種:Au3イオン、Bi3イオン、C60イオン等のクラスターイオン
一次イオンビーム径:約1μm
一次イオンフルーエンス(ドーズ量):1×1014 ions/cm2
測定領域:100μm×100μm
測定質量範囲:1〜5000(m/z)
測定時間:約10分。
【0034】
一次イオンとしてクラスターイオンを用いるのは、単原子イオンに比べクラスターイオンの方が質量数の大きい二次イオンの検出に有利であるからである(例えば、本発明者らの特許文献1)。一次イオンビーム径を約1μmとするのは、直径20μm前後の個々の細胞を識別するために必要だからである。一次イオンフルーエンスを上記の値とするのは、一定以上の二次イオン感度(積算信号強度)を得るために必要だからである。測定質量範囲を上記の値とするのは主測定対象が消化分解ペプチドであるからである(例えば、特開2006−010658号公報参照)。測定時間を約10分程度と設定するのは、長時間の測定が定量的評価に悪影響を与える場合があるからである(例えば、H.Hashimoto et al.,Appl.Surf.Sci.,231−232,385−391(2004))。
【0035】
上記(3)の工程における数値解析的手法とは、因子分析などの統計的手法を指す。
【0036】
本発明は、病変組織の切片において、疾病に関わる特有なタンパク質の有無を検出する方法でもある。
【実施例】
【0037】
以下に、実施例を挙げて、本発明をより具体的に説明する。以下に示す具体例は、本発明にかかる最良の実施形態の一例ではあるが、本発明はかかる具体的形態に限定されるものではない。
【0038】
本開示によりトリプシン組換えタンパク質の取得とその方法を示す。以下の実施例では、ウシ膵臓由来のトリプシノーゲン(トリプシン前駆体)の大腸菌発現ベクターを設計・作製し、不溶性顆粒画分として大腸菌に発現させて巻き戻しと精製工程の後、自己触媒により酵素活性を有する組換えトリプシンタンパク質を取得する。また、以下に記す組換えDNA法はSambrookら著書のモレキュラークローニング(実験マニュアル第二版Cold Spring Harbor Laboratory,New York(1989))に基づいて行い、市販のキットを用いる場合は、特記しない限り当業者が推奨プロトコールに準じて行うことができる。また、発現ベクターの構築にはNovagen社のpETBlue−1System(Cat.No.70673−3)に付属のベクターpETBlue−1を用いてコンストラクトを作製する。宿主大腸菌には、サブクローニング用としてJM109株(Novagen社)を、たんぱく質生産用としてRosetta (DE3) pLacI株(Novagen社)を用いる。
【0039】
また、実施例に用いる化学薬品は特記しない限り分析級またはそれ以上であり、シグマ社及びナカライテスク社より購入できる。オリゴDNAはシグマジェノシス社、制限酵素および修飾酵素はタカラバイオ社より購入できる。
【0040】
(実施例1) ウシ膵臓由来のトリプシノーゲン遺伝子発現ベクターの構築
以下、ウシ膵臓由来のトリプシノーゲン組換え遺伝子は、トリプシノーゲン遺伝子コーディング領域(NCBI GenBankアクセッション番号;X54703)の3'末端に、アフィニティー精製用His6配列と金担体に固定するためのCys残基をコードするスペーサー能を有するDNAタグ配列を設けたDNA配列(図2)をさす。
【0041】
(1)上記ウシ膵臓由来のトリプシノーゲン組換え遺伝子の合成
プライマーセット1(配列番号1〜4)、プライマーセット2(配列番号5〜10)と、Pyrobest DNAポリメラーゼ(タカラバイオ社)を用い当業者の推奨する配合で調合し、温度サイクル98℃×1min.→(98℃、10sec.→55℃、30sec.→72℃、30sec.)×40cycles→4℃でオーバーラップPCRを行う。それぞれ目的サイズ約300bpと450bpの断片の増幅を電気泳動とEtBr染色により確認する。
配列番号1(5’→3’)
atgttcccctcggacgacgatgacaagatcgtcgggggctacacctgcgcagagaatt
配列番号2
ggacaccacccactggtcattgatgagggagcccccgcagaagtggtagccagcattcagggacacctggtaagggacggaattctctgcgcaggtgtag
配列番号3
atgaccagtgggtggtgtccgcggctcactgctaccagtaccacatccaggtgaggctgggagaatacaacattgatgtcttggagggtggtgagcagtt
配列番号4
agagtttgatcagcaggatgtcattgtccagagtccagctgctgtacttggggtggcggatgatcttggacgcatcgatgaactgctcaccaccctccaa
配列番号5
catcctgctgatcaaactctccacgcctgcggtcatcaatgcccgggtgtccaccttgctgctgcccagtgcctgtgcttccgcaggcacagagtgcctc
配列番号6
ctcagcagcggggccaccaggcattgcagcaggtccgggtagttgacgccactgctcagggtgttgccccagccggagatgaggcactctgtgcctgcgg
配列番号7
ctggtggccccgctgctgagccacgccgactgtgaagcctcataccctggacagatcactaacaacatgatctgcgctggcttcctggaaggaggcaagg
配列番号8
acagccgtagccccaggacacaatgccctggagctgtccgttgcaagccacagggccgccagagtcaccctggcaggaatccttgcctccttccaggaag
配列番号9
tgtcctggggctacggctgtgcccagaagggcaagcctggggtctacaccaaggtctgcaactacgtggactggattcaggagaccatcgccgccaacag
配列番号10
ggcttagcaatggtggtgatgatggtggctagcgctgttggcggcgatggtctc
次に、目的サイズの断片を市販のゲル精製キット(Wizard SV Gel and PCR Clean−Up system(Cat No #A9281 Promega社))により精製する。次に精製した上記二つの断片を鋳型にし、プライマーセット3(配列番号11および12)を用いて再度同条件でオーバーラップPCRを行い、最終目的サイズ約730bpの断片が増幅していることを確認し、同様に精製する。
配列番号11
atgttcccctcggacgacgatga
配列番号12
ggcttagcaatggtggtgatgat
(2)大腸菌発現ベクターへのサブクローニング
pETBlue−1発現ベクターをEcoRV酵素で消化した後、BAPにより脱リン酸化処理(37℃、1時間)を行い、Wizard SV Gel and PCR Clean−Up system(Cat No #A9281 Promega社)により脱リン酸化されたDNA断片を精製する。次に、市販のDNAライゲーションキット(Ligation High Code No.LGK−101(TOYOBO社))を用いて、(1)で合成したDNA断片と脱リン酸化ベクターを業者推奨の方法にて調合し連結させる。
【0042】
そして、ライゲーション溶液をJM109株コンピテントセル(Novagen社)にヒートショック法により形質転換した後に、Luria−Bertani medium(LB)+Amp.(100μg/m)+IPTG+X−gal寒天プレートに撒き、37℃にて一晩静置する。
【0043】
次に、プレート中から青白選択を行い任意のコロニーをLB/Amp液体培地3mlに植菌し、37℃にて一晩振盪培養を行う。その後、市販のMiniPrepキット(Plus Minipreps DNA Purification System(Promega社))を使用して、プラスミドDNAを回収する。
【0044】
得られたプラスミドの塩基配列を決定し、目的の断片が正しい方向に挿入され且つ配列が正しいことを確認することができ、結果としてウシ膵臓由来のトリプシノーゲン組換え遺伝子発現ベクターが得られる。
【0045】
(実施例2) トリプシノーゲン組換えタンパク質の大腸菌による発現、巻き戻し実験
(1)形質転換
上記実施例1で取得した発現ベクターを、タンパク質生産用のRosetta(DE3)pLacI株コンピテントセル溶液に形質転換する。形質転換は、ヒートショックを氷中→42℃、90sec.→氷中の条件でおこなう。ヒートショックにより形質転換した上記Rosetta(DE3)pLacI株コンピテントセル溶液にTerrific broth(TB;1%(W/V)yeast extract,2%(W/V)peptone,0.1M リン酸バッファー(pH7.5),1%グリセロール)培地500μLを加え、一時間37℃にて振盪培養を行った。その後、6000r.p.m.で5分間遠心を行い、培養上清650μLを廃棄し、残った培養上清と沈殿となった細胞画分を攪拌し、TB/Amp.100μg/ml/chloramphenicol 34μg/ml寒天プレートに撒き、一晩37℃にて培養する。
【0046】
(2)予備培養
プレート上のコロニーを無作為に選択し、50mlのTB/Amp.200μg/ml/chloramphenicol 34μg/ml培地にて37℃でOD600nm値が0.2〜0.6になるまで振盪培養を行う。次に1,000×g、5分間遠心して上清を捨て、菌を集菌し、新しい培地(上記)で懸濁した後2mlとり、TB/Amp.400μg/ml/chloramphenicol 34μg/ml培地500mlに植菌し37℃でOD600nm値が0.2〜0.6になるまで振盪培養を行う。
【0047】
(3)本培養
上記予備培養溶液を更に培養を30℃にて継続する。D600nm値が1.0を越えた時点で、終濃度が1mMとなるようにIPTGを加え、更に30℃にて終夜培養を行う。
【0048】
(4)精製
目的のポリペプチド鎖を不溶性顆粒画分から以下の工程により精製する。
【0049】
(i)不溶性顆粒の回収
上記3)で得られた培養液を6,000r.p.m.で30分間遠心し、沈殿を菌体画分として得る。得られた菌体を100μg/mlリゾチーム入りトリス溶液(20mM Tris−HCl(pH8.0)/500mM NaCl)15mlに懸濁し、37℃で30分反応後、氷中保管する。得られた懸濁液をフレンチプレスにて破砕し、菌破砕液を得る。10unitのDNaseI酵素を加え、37℃で15分間反応させる。その後、最終2%になるようにTriton X−100を加え不溶性顆粒画分を2回洗浄する。
【0050】
次に、菌破砕液を12,000r.p.m.で15分間遠心を行い、上清を除き、沈殿を不溶性顆粒画分として得る。
【0051】
(ii)不溶性顆粒画分の可溶化
上記(i)で得られた不溶性画分を6M 塩酸グアニジン/トリス溶液(20mMTris−HCl(pH8.0)/500mM NaCl)10mlを加え、超音波で不溶画分を分散させた後室温で一晩浸漬する。次に、12,000r.p.m.で10分間遠心し、上清を可溶化溶液として得る。
【0052】
(iii)金属キレートカラム
金属キレートカラム担体としてHis−Bind(Novagen社製)を用いる。カラム調整やサンプル負荷、及び洗浄工程は、前記業者の推奨方法に準拠し、室温(20℃)にて行う。目的であるHisタグ融合のポリペプチドの溶出は60mMイミダゾール/トリス溶液にて行う。溶出液のSDS−PAGE(アクリルアミド15%)の結果、単一バンドであり、精製されていることを確認する。
【0053】
(iv)透析
上記溶出液に対して、外液を6M塩酸グアニンジン/トリス溶液として4℃にて透析を行い、溶出液中のイミダゾールの除去を行い、上記ポリペプチド鎖溶液を得る。
【0054】
(v)巻き戻し
上記と同様にして、ポリペプチド鎖溶液を以下の工程により、脱塩酸グアニンジンを透析(4℃)にて行いながらタンパク質の巻き戻しを行う。
【0055】
(a)6M塩酸グアニジン/トリス溶液を用い、ポリペプチド鎖のモル吸光係数とΔO.D.(280nm−320nm)値から濃度7.5μMのサンプル(希釈後体積10ml)を調製する。次にβ−メルカプトエタノール(還元剤)を終濃度375μM(タンパク濃度50倍)になるよう添加し、室温、暗所で4時間還元を行う。このサンプル溶液を透析バック(MWCO;8,000)に入れ、透析用サンプルとする
(b)透析外液を6M塩酸グアニンジン/トリス溶液として、透析サンプルを浸漬し、緩やかに攪拌しながら6時間透析する
(c)外液の塩酸グアニジン濃度を3M、2Mと段階的に下げる。それぞれの外液濃度において、6時間透析する
(d)酸化型グルタチオン(GSSG)を終濃度375μM、L−Argを終濃度0.4Mとなるようにトリス溶液に加え、上記(c)の2Mの透析外液を加え、塩酸グアニジン濃度を1Mとし、NaOHでpH8.0(4℃)に調整した溶液にて、12時間緩やかに攪拌しながら透析する
(e)上記(d)と同様の作業にて塩酸グアニジン濃度0.5MのL−Arg含有トリス溶液を調整し、更に12時間透析する
(f)最後にトリス溶液にて12時間透析する
(g)透析終了後、10,000r.p.m.で約20分間遠心分離し凝集体と上清とを分離し、可溶性の巻き戻しトリプシノーゲン組換えタンパク質を得る。
【0056】
(実施例3) トリプシノーゲン組換えタンパク質の活性化と活性組換えトリプシンタンパク質の精製取得
(1)トリプシノーゲン組換えタンパク質の活性化
得られた巻き戻しトリプシノーゲン組換えタンパク質を1時間、室温に放置した後、4℃で12時間静置することで自己触媒能による組換えトリプシンタンパク質を得る。
【0057】
(2)クロマトグラフィーによる精製
上記(1)で得られた5μM可溶性の巻き戻しトリプシン組換えタンパク質溶液をスーパーデックス200カラム(GEヘルスケアサイエンス社)(バッファー;20mMのTris−HCl(pH8.0)、500mMのNaCl、流速;0.5ml/分)にてゲルろ過し、目的サイズの単ピークのフラクションを得る。
【0058】
(3)トリプシン活性測定
N−Benzoyl−DL‐arginine‐p−nitroanilide hydrochloride(BAPA)を基質とするErlangerらの方法(BAPA法)に準じる。25℃で5分間、基質と上記フラクションのサブフラクションとを反応させ、BAPA分解物であるパラニトロアニリンの405nm吸光度を測定し、トリプシン活性の最も高いフラクションを取得する。結果として、活性を有する組換えトリプシンタンパク質を得る。このタンパク質を後の評価に用いる。
【0059】
(実施例4) 組換えトリプシンタンパク質と金粒子を含む水溶液の調製と、インクジェットプリンタを用いた検体への該水溶液の付与
平均粒径が40nm程度の金粒子を含む溶液(分散剤:クエン酸)を出発材料とし、常法に従い、上記組換えトリプシンタンパク質と金粒子が結合した、本願発明の処理溶液である水溶液を調製する。この水溶液が十分分散された状態にあるか否かは、例えば、二日間程度常温で放置した後に沈殿が生じるか否かなどにより判断することができる。この水溶液を、本発明者らの特許第3658397号明細書に記載されているバブルジェットプリンタに充填する。より具体的には、バブルジェットプリンタBJF−850(キヤノン(株)製)用のプリンターヘッドBC−50(キヤノン(株)製)を、数100μlの溶液を吐出可能とするべく改造したものを使用し、このヘッドのタンク部に、前記水溶液を数100μl注入する。その後、シリコン基板上に配置した検体を前記プリンタに挿入し、検体表面の全面に前記水溶液を液滴として付与する。その際、検体表層のタンパク質を均一に消化するために、最終的に付与される水溶液量が二次元的に均一になるように工夫する必要がある(例えば、液滴を付与する地点を少しずつ移動させながら行う)。なお、液滴量は通常1滴あたり1〜8plとし、ある一定の地点における付与回数(重ねうちの回数)は上記組換えトリプシンタンパク質の濃度に応じて調整する。該組換えトリプシンタンパク質の濃度は1〜100μM程度とする。また、検体に前記水溶液を付与した後は、消化反応を進行させるために、常温、高湿度下で一定時間保持することが必要である。
【0060】
なお、上記バブルジェットプリンタで使用する水溶液は、吐出の安定化などを目的のため、グリセリン、尿素、チオジグリコール、アセチレンアルコール(商品名:アセチレノールEH;川研ファインケミカル社)の中から選ばれる一種または複数を含んでもよい。
【0061】
(実施例5) ヒト血清アルブミン(HSA)のトリプシン消化物のTOF−SIMS分析
ヒト血清アルブミン(HAS、アミノ酸の配列番号13)をシリコン基板などにスピンコートし、これを実施例4に示す方法により消化分解する。
配列番号13
mkwvtfisllflfssaysrgvfrrdahksevahrfkdlgeenfkalvliafaqylqqcpfedhvklvnevtefaktcvadesaencdkslhtlfgdklctatlretygemadccakqepernecflqhkddnpnlprlvrpevdvmctafhdneetflkkylyeiarrhpyfyapellffakrykaafteccqaadkaacllpkldelrdegkassakqrlkcaslqkfgerafkawavarlsqrfpkaefaevsklvtdltkvhtecchgdllecaddadlayicenqdsissklkeccekpllekshciaevendempadlpslaadfveskdvcknyaeakdvflgmflyeyarrhpdysvvlllrlaktyettlekccaaadphecyakvfdefkplveepqnlikqncelfeqlgeykfqnallvrytkkvpqvstptlvevsrnlgkvgskcckhpeakrmpcaedylsvvlnqlcvlhek tpvsdrvtkccteslvnrrpcfsalevdetyvpkefnaetftfhadictlsekerqikkqtalvelvkhkpkatkeqlkavmddfaafvekcckaddketcfaeegkklvaasqaalgl
上記の消化分解処理により、基本的には上記配列13中のkとrの右側が選択的に切断され、多数のペプチド断片が生成する。この断片ペプチドの内、例えば、dahk,sevahr,necflqhk,hpyfyapellffak,caslqk,aefaevsk,lvtdltk,adlayicenqdsissk,dvflgmflyeyar,hpdysvvlllr,vpqvstptlvevsrvgsk,cckhpeak,tpvsdr,ccteslvnr,erqik,avmddfaafvek,lvaasqaalglなどの特徴的なペプチドを、それらの水素付加イオンまたは金付加イオンなどの形でTOF−SIMS法により検出する。これらの分子量は概ね500〜5000m/zの範囲にある。上記のように、特徴的な断片ペプチドを複数検出することで、消化分解前のタンパク質がHSAであることを同定することができる。なお、検体表層に金粒子が共存するため二次イオンの検出感度が向上し、上記同定精度が向上する。
【0062】
(実施例6) ヒト癌組織切片のトリプシン消化物のTOF−SIMS分析
ヒトの癌組織から、厚さ0.5〜5μm程度、大きさ1〜10mm2程度の切片を切り出し、これを基板上に配置し、次に実施例4に示す方法により消化分解する。図3にその模式図を示す。図3において、5は癌組織切片中の異常細胞、6は癌組織切片中の正常細胞、7はインクジェットプリンタヘッド、8は液滴を示す。
【0063】
消化分解処理を行った後、実施例5と同様に消化分解ペプチドをTOF−SIMS法により検出する。その後、例えば、再発や転移に関わるタンパク質を分解ペプチドのパターンから同定し、これらの信号強度の二次元分布を再表示させる。この模式図を図4に示す。図4において、9は注目するタンパク質の存在個所(再発や転移に関わるタンパク質の存在個所)である。上記のように、本発明の方法を用いることで、注目するタンパク質の存在個所を細胞レベルで調べることが可能になる。なお、検体表層に金粒子が共存するため二次イオンの検出感度が向上し、結果として上記の注目するタンパク質の検出感度が向上する。
【図面の簡単な説明】
【0064】
【図1】金粒子と結合した人工消化酵素の模式図である。
【図2】金担体に固定するためのCys残基をコードするスペーサー能を有するDNAタグ配列を設けたDNA配列である。
【図3】ヒトの癌組織切片に対し、金粒子と消化酵素を含む水溶液をインクジェット法により付与する工程を示す模式図である。
【図4】注目するタンパク質の存在個所(再発や転移に関わるタンパク質の存在個所)を、断片ペプチドの検出パターンから同定し、これを二次元表示した例である。
【符号の説明】
【0065】
1 金粒子
2 人工消化酵素
3 人工消化酵素2内の金粒子との結合部位
4 人工消化酵素2内の消化活性部位
5 癌組織切片中の異常細胞
6 癌組織切片中の正常細胞
7 インクジェットプリンタヘッド
8 液滴
9 注目するタンパク質の存在個所(再発や転移に関わるタンパク質の存在個所)
【特許請求の範囲】
【請求項1】
検体中の対象物を分析するための前処理に用いる処理溶液であって、少なくとも金属粒子と消化酵素を含み、かつ該金属粒子が常温常圧下で分散された状態にあることを特徴とする処理溶液。
【請求項2】
前記金属粒子が金粒子であることを特徴とする請求項1に記載の処理溶液。
【請求項3】
前記金属粒子の直径が1nmから100nmの範囲であることを特徴とする請求項1または2に記載の処理溶液。
【請求項4】
前記金属粒子と前記消化酵素とが化学結合していることを特徴とする請求項1から3のいずれかに記載の処理溶液。
【請求項5】
前記消化酵素が人工消化酵素であることを特徴とする請求項1から4のいずれかに記載の処理溶液。
【請求項6】
前記対象物がタンパク質であることを特徴とする請求項1から5のいずれかに記載の処理溶液。
【請求項7】
請求項1から6に記載の処理溶液をインクジェット装置に充填し、該装置を用い、該処理溶液を液滴として所定の位置に付与することを特徴とする前処理方法。
【請求項8】
検体中の対象物に関する情報を取得する方法であって、
請求項7に記載の前処理方法を用いて検体を処理する工程と、
処理後の検体に含まれる対象物の質量に関する情報(消化酵素で分解されたペプチド断片の質量に関する情報を含む)を飛行時間型二次イオン質量分析法により取得する工程とを備えることを特徴とする情報取得方法。
【請求項9】
前記検体が病変組織の切片であることを特徴とする請求項8に記載の情報取得方法。
【請求項10】
前記対象物がタンパク質であることを特徴とする請求項8または9に記載の情報取得方法。
【請求項11】
検体中における疾病に関わる特有な対象物の有無を検出する方法であって、請求項8から10に記載の情報取得方法を利用して検体中における疾病に関わる特有な対象物の有無を検出することを特徴とする検出方法。
【請求項1】
検体中の対象物を分析するための前処理に用いる処理溶液であって、少なくとも金属粒子と消化酵素を含み、かつ該金属粒子が常温常圧下で分散された状態にあることを特徴とする処理溶液。
【請求項2】
前記金属粒子が金粒子であることを特徴とする請求項1に記載の処理溶液。
【請求項3】
前記金属粒子の直径が1nmから100nmの範囲であることを特徴とする請求項1または2に記載の処理溶液。
【請求項4】
前記金属粒子と前記消化酵素とが化学結合していることを特徴とする請求項1から3のいずれかに記載の処理溶液。
【請求項5】
前記消化酵素が人工消化酵素であることを特徴とする請求項1から4のいずれかに記載の処理溶液。
【請求項6】
前記対象物がタンパク質であることを特徴とする請求項1から5のいずれかに記載の処理溶液。
【請求項7】
請求項1から6に記載の処理溶液をインクジェット装置に充填し、該装置を用い、該処理溶液を液滴として所定の位置に付与することを特徴とする前処理方法。
【請求項8】
検体中の対象物に関する情報を取得する方法であって、
請求項7に記載の前処理方法を用いて検体を処理する工程と、
処理後の検体に含まれる対象物の質量に関する情報(消化酵素で分解されたペプチド断片の質量に関する情報を含む)を飛行時間型二次イオン質量分析法により取得する工程とを備えることを特徴とする情報取得方法。
【請求項9】
前記検体が病変組織の切片であることを特徴とする請求項8に記載の情報取得方法。
【請求項10】
前記対象物がタンパク質であることを特徴とする請求項8または9に記載の情報取得方法。
【請求項11】
検体中における疾病に関わる特有な対象物の有無を検出する方法であって、請求項8から10に記載の情報取得方法を利用して検体中における疾病に関わる特有な対象物の有無を検出することを特徴とする検出方法。
【図1】


【図2】

【図3】
【図4】
【図2】
【図3】
【図4】
【公開番号】特開2008−275422(P2008−275422A)
【公開日】平成20年11月13日(2008.11.13)
【国際特許分類】
【出願番号】特願2007−118899(P2007−118899)
【出願日】平成19年4月27日(2007.4.27)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.バブルジェット
【出願人】(000001007)キヤノン株式会社 (59,756)
【Fターム(参考)】
【公開日】平成20年11月13日(2008.11.13)
【国際特許分類】
【出願日】平成19年4月27日(2007.4.27)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.バブルジェット
【出願人】(000001007)キヤノン株式会社 (59,756)
【Fターム(参考)】
[ Back to top ]