
出芽酵母形質転換体
【課題】分裂酵母(S.pombe)を用いる方法に代わる生産性に優れた新たなグルクロン酸抱合体の製造方法とこの製造方法に用いる新たな手段を提供する。
【解決手段】UDP-グルコース脱水素酵素をコードする遺伝子及びUDP-グルクロン酸転移酵素をコードする遺伝子を発現可能に挿入して形質転換した出芽酵母。シトクロムP450遺伝子をコードする遺伝子をさらに発現可能に挿入して形質転換した出芽酵母。形質転換出芽酵母をグルコース及び被抱合物質の存在下で培養して、前記被抱合物質のグルクロン酸抱合体を生成させることを含む、グルクロン酸抱合体の製造方法。
【解決手段】UDP-グルコース脱水素酵素をコードする遺伝子及びUDP-グルクロン酸転移酵素をコードする遺伝子を発現可能に挿入して形質転換した出芽酵母。シトクロムP450遺伝子をコードする遺伝子をさらに発現可能に挿入して形質転換した出芽酵母。形質転換出芽酵母をグルコース及び被抱合物質の存在下で培養して、前記被抱合物質のグルクロン酸抱合体を生成させることを含む、グルクロン酸抱合体の製造方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明はグルクロン酸抱合体の製造方法、並びにそれに用いる出芽酵母発現ベクター及び形質転換体に関する。より詳しくは、出芽酵母発現ベクターは、UDP-グルクロン酸転移酵素及び/又はUDP-グルコース脱水素酵素を導入した出芽酵母発現ベクターであり、形質転換体は、前記出芽酵母発現ベクターで形質転換された出芽酵母である。
【背景技術】
【0002】
医薬品開発において、ヒト体内における医薬品代謝物の解析は重要である。グルクロン酸抱合体は医薬品の解毒代謝物として排泄されるが、その一部は反応性代謝物となり毒性を示す可能性が指摘されている。例えば、グルクロン酸抱合体の内、エステル型抱合であるアシル抱合体は、反応性代謝物となり薬物性肝障害の原因となる可能性がある(非特許文献1)。
【0003】
そこで、抱合体自身の安全性評価が必要であるが、有機合成法による部位特異的抱合化はきわめて困難である。そこで、酵素や微生物等を用いて効率良く目的とする抱合体を製造する方法が切望されている。現在、動物肝臓由来ミクロソーム画分を用いた抱合体調製が実用化されているが、生産性や適用範囲が十分とは言えない。また、昆虫細胞発現系を用いたヒトグルクロン酸転移酵素がすでに市販されているが、酵素源として抱合体調製に用いるにはコスト面からも現実的ではない。
【0004】
本発明者らは、これまでに出芽酵母を用いたグルクロン酸転移酵素の発現系を構築し、抱合体調製の酵素源として用いる、医薬品代謝物である抱合体の酵素的合成を提案してきた(非特許文献2)。
【0005】
最近、Draganら(非特許文献7、特許文献1)は分裂酵母(Schizosaccharomyces pombe(S.pombe))を用いたUGTを発現する分裂酵母菌体からグルクロン酸抱合体を産生する系を構築した。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】WO2010/031875
【非特許文献】
【0007】
【非特許文献1】Bailey MJ, Dickinson RG. 1333963959742_0.Pubmed_RVDocSum Chem Biol Interact. 145, 117-37. (2003)
【非特許文献2】S. Ikushiro, M. Sahara, Y. Emi, Y. Yabusaki, T. Iyanogi: Functional co-expression of xenobiotic metabolizing enzymes, rat cytochrome P450 1A1 and UDP-glucuronosylransferase 1A6. Biochimica et Biophysica Acta1672 (2004) 86-92
【非特許文献3】Mackenzie, P. I , Walter Bock, K. ,Burchell, B. ,Guillemette, C. , Ikushiro, S. I. ,Iyanagi, T. ,Miners, J.O. ,Owens, I.S. and Nebert, D.W.: Nomenclature Update for the Mammalian UDP Glycosyltransferase (UGT) Gene Superfamily. Pharmacogenetics and Genomics, 10, 677-685 (2005)
【非特許文献4】生城真一、衣斐義一、井柳堯: UDP-グルクロン酸転移酵素:薬物代謝における最近の進歩, 肝臓, 42 , pp.297-301(2001)
【非特許文献5】T. Sakaki, M. Akiyoshi-Shibata, Y. Yabusaki, H. Ohkawa, Organellatargeted expression of rat liver cytochrome P450c27 in yeast: genetically engineered alteration of mitochondrial P450 into a microsomal form creates a novel functional electron transport chain, J. Biol.Chem. 26716497-16502. (1992)
【非特許文献6】Ikushiro. S., Emi, Y., Kato, Y., Yamada, S. and Sakaki, T.: Monospecific antipeptide antibodies against human hepatic UDP- glucuronosyltransferase 1A subfamily ( UGT1A) isoforms. Drug Metabolism and Pharmacokinetics, 21, 70-75 (2006)
【非特許文献7】Dragan CA, Buchheit D, Bischoff D, Ebner T, Bureik M .:Glucuronide production by whole-cell biotransformation using genetically engineered fission yeast S. pombe. Drug Metab Dispos. 38 509-515. (2010)
【非特許文献8】Jo Wixon :Featured Organism: Schizosaccharomyces pombe, the fission yeast. Comp Funct Genom, 3: 194-204 (2002)
【非特許文献9】Esben H. Hansen,Birger Lindberg Moller, Gertrud R. Kock,Camilla M. Bu¨nner,Charlotte Kristensen,Ole R. Jensen,Finn T. Okkels,Carl E. Olsen,Mohammed S. Motawia,and Jorgen Hansen1: De Novo Biosynthesis of Vanillin in Fission Yeast(Schizosaccharomyces pombe) and Baker’s Yeast(Saccharomyces cerevisiae) APPLIED AND ENVIRONMENTAL MICROBIOLOGY, 75,2765-2774 (2009)
【非特許文献10】Tony K.L. Kiang, Mary H.H. Ensom, Thomas K.H. Chang: UDP-glucuronosyltansferases and clinical drug-drug interactions. Pharmacology & Therapeutics 106 97-132 (2005)
【発明の概要】
【発明が解決しようとする課題】
【0008】
しかしながら、酵母内で発現させて得られるヒト由来UDP-グルクロン酸転移酵素(UGT)を酵素源として試験管内における酵素変換にて抱合体を調製する方法は、生産性や適用範囲が十分とは言えなかった。特に糖供与体として用いるUDP-グルクロン酸は高価であり、抱合体調製に際してコスト低減が可能な方法の出現が待たれている。
【0009】
さらに、非特許文献7に記載の分裂酵母(S.pombe)を用いた系では総じてその生産性は低く、依然として実用性に乏しかった。
【0010】
そこで本発明の目的は、上記方法に代わる生産性に優れた新たなグルクロン酸抱合体の製造方法とこの製造方法に用いる新たな手段を提供することにある。
【課題を解決するための手段】
【0011】
そこで、本発明者らは、出芽酵母(Saccharomyces cerevisiae(S.cerevisiae))を宿主として用い、かつUGTを発現する出芽酵母の代謝系を改変して、糖供与体であるUDP-グルクロン酸産生を可能にすることを企画した。しかし、出芽酵母は、抱合体産生に必要とされるUDP-グルクロン酸の生産能力を欠失しているため、菌体内での抱合体への直接変換は不可能であった。そこで出芽酵母にUDP-グルクロン酸の生産能力を付与するために、他生物由来のUDP-グルコース脱水素酵素遺伝子を導入した。その結果、出芽酵母内においてUDP-グルクロン酸の産生が見られた。さらに、このUDPGDH導入出芽酵母においてUGT分子種を同時に発現させることで、出芽酵母菌体に添加した基質の抱合体への直接変換が高い収率で得られた。このようにして、本発明者は出芽酵母(Saccharomyces cerevisiae(S.cerevisiae))を宿主として用いることにより出芽酵母菌体内での抱合体産生能を著しく向上させた抱合体変換系の構築に成功した。
【0012】
出芽酵母(S.cerevisiae)と分裂酵母(S.pombe)は、酵母に分類されてはいるが、3〜4億年前に分岐したと考えられ、両者の生物種としての違いは、分裂酵母と動物の違いに比べられるほど大きいと言われている(非特許文献8)。両者は、ゲノムサイズ(出芽酵母12Mb、分裂酵母14Mb)や遺伝子数(出芽酵母7000程度、分裂酵母5000程度)は似通っているが、染色体数(一倍体として、出芽酵母17、分裂酵母3)や増殖方法(出芽酵母は出芽、分裂酵母は分裂)に著しい違いが認められ、個々の遺伝子の相同性も低い。さらに、哺乳動物由来の異種タンパク質の発現についても、状況は大きく異なっており、分裂酵母(S.pombe)で高発現したからといって出芽酵母(S.cerevisiae)で高発現するとは限らない(非特許文献9)。このような状況であるので、非特許文献7に記載のように、分裂酵母(S.pombe)でUDPGDHとUGTの同時発現に成功したからといって、それをそのまま出芽酵母(S.cerevisiae)に適用しても、UDPGDHとUGTを同時発現できることは限らない。
【0013】
本発明は以下の通りである。
[1]
UDP-グルコース脱水素酵素をコードする遺伝子及びUDP-グルクロン酸転移酵素をコードする遺伝子を発現可能に挿入して形質転換した出芽酵母。
[2]
シトクロムP450遺伝子をコードする遺伝子をさらに発現可能に挿入して形質転換した、[1]に記載の出芽酵母。
[3]
以下の(A)〜(G)のいずれかから選ばれる形質転換出芽酵母。
(A)UDP-グルコース脱水素酵素発現ベクター及びUDP-グルクロン酸転移酵素発現ベクターで形質転換した出芽酵母、
(B)UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクターで形質転換した出芽酵母、
(C)UDP-グルコース脱水素酵素発現ベクター、UDP-グルクロン酸転移酵素発現ベクター及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母、
(D)UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクター、及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母、
(E)UDP-グルクロン酸転移酵素発現ベクター、及びUDP-グルコース脱水素酵素及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母、
(F)UDP-グルクロン酸転移酵素及びシトクロムP450遺伝子発現ベクター、及びUDP-グルコース脱水素酵素発現ベクターで形質転換した出芽酵母、並びに
(G)UDP-グルコース脱水素酵素、UDP-グルクロン酸転移酵素及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母
[4]
UDP-グルコース脱水素酵素発現ベクターが、出芽酵母発現ベクターにUDP-グルコース脱水素酵素遺伝子を発現可能に挿入したものであり、
UDP-グルクロン酸転移酵素発現ベクターが、出芽酵母発現ベクターにUDP-グルクロン酸転移酵素遺伝子を発現可能に挿入したものであり、
UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクターが、出芽酵母発現ベクターにUDP-グルクロン酸転移酵素遺伝子及びUDP-グルコース脱水素酵素含を発現可能に挿入したものである、[3]に記載の形質転換出芽酵母。
[5]
出芽酵母発現ベクターが自律複製型ベクターまたは染色体組み込み型ベクターである、[4]に記載の形質転換出芽酵母。
[6]
UDP-グルコース脱水素酵素遺伝子が動物または植物由来の遺伝子である、[1]、[2]、[4]〜[5]のいずれかに記載の形質転換出芽酵母。
[7]
UDP-グルコース脱水素酵素遺伝子がシロイヌナズナ由来遺伝子またはラット由来遺伝子である、[1]、[2]、[4]〜[5]のいずれかに記載の形質転換出芽酵母。
[8]
UDP-グルクロン酸転移酵素遺伝子が哺乳動物由来の遺伝子である、[1]、[2]、[4]〜[7]のいずれかに記載の形質転換出芽酵母。
[9]
UDP-グルクロン酸転移酵素遺伝子がヒト由来の遺伝子である、[1]、[2]、[4]〜[7]のいずれかに記載の形質転換出芽酵母。
[10]
シトクロムP450遺伝子発現ベクターが、出芽酵母発現ベクターにシトクロムP450遺伝子を発現可能に挿入したものである、[3]〜[9]のいずれかに記載の形質転換出芽酵母。
[11]
前記UDP-グルクロン酸転移酵素発現ベクターは、UDP-グルクロン酸転移酵素遺伝子が、発現量がUGT1A7の50%以下である低発現量グルクロン酸転移酵素遺伝子である場合、
前記低発現量グルクロン酸転移酵素遺伝子は、シグナル配列遺伝子を、発現量がUGT1A7の80%以上である高発現グルクロン酸転移酵素のシグナル配列遺伝子と置換したものであるか、または
シグナル配列遺伝子を(A)以下に示すアミノ酸配列(a)〜(c)のいずれかをコードする遺伝子、(B)アミノ酸配列(a)〜(c)のアミノ酸の1〜5個が置換若しくは欠失したアミノ酸配列のいずれかをコードする遺伝子、または(C)1〜5個のアミノ酸がアミノ酸配列(a)〜(c)のアミノ酸に付加されたアミノ酸配列のいずれかをコードする遺伝子と置換したものであり、但し、(B)の置換若しくは欠失したアミノ酸配列および(C)の付加されたアミノ酸配列をコードする遺伝子は、シグナル配列遺伝子とした場合にグルクロン酸転移酵素の発現量が野生型の80%以上である、[1]〜[10]のいずれかに記載の形質転換出芽酵母。
(a) MARAGWTGLLPLYVCLLLTCGFAKAG(配列番号1)
(b) MACLLRSFQRISAGVFFLALWGMVVG(配列番号2)
(c) MAPRRVDQPRSFMCVSTADLWLCEAG(配列番号3)
[12]
前記低発現量グルクロン酸転移酵素は、UGT1A1、UGT1A4、UGT1A8またはUGT1A9である[11]に記載の形質転換出芽酵母。
[13]
前記高発現量グルクロン酸転移酵素は、UGT1A7、UGT1A6またはUGT1A10である[11]または[12]に記載の形質転換出芽酵母。
[14]
被抱合物質のグルクロン酸抱合体生成に用いるための、[1]〜[13]のいずれか1項に記載の出芽酵母。
[15]
[1]〜[13]のいずれかに記載の形質転換出芽酵母をグルコース及び被抱合物質の存在下で培養して、前記被抱合物質のグルクロン酸抱合体を生成させることを含む、グルクロン酸抱合体の製造方法。
[16]
被抱合物質がアルコール性水酸基を含む医薬品及び医薬品の候補物質、フェノール性水酸基を複数含むポリフェノール化合物、カルボン酸を含む非ステイロイド性抗炎症薬及びその候補物質、並びに1〜4級のいずれか少なくとも1つのアミンを含む化合物から成る群から選ばれる少なくとも1種である[15]に記載の製造方法。
[17]
被抱合物質が、P450の代謝により抱合化を受ける官能基(主に水酸基)を生成する物質である[15]に記載の製造方法。
[18]
被抱合物質が、メトキシ基またはエトキシ基を含む医薬品及び医薬品の候補物質、メチレンジオキシフェニル基をもつセサミン化合物、並びに水酸基を有しないジアゼピン系の医薬品及び医薬品の候補物質から成る群から選ばれる少なくとも1種である[17]に記載の製造方法。
[19]
出芽酵母発現ベクターにUDP-グルコース脱水素酵素遺伝子を発現可能に挿入したものである、UDP-グルコース脱水素酵素発現ベクター。
[20]
出芽酵母発現ベクターにUDP-グルクロン酸転移酵素遺伝子を発現可能に挿入したものである、UDP-グルクロン酸転移酵素発現ベクター。
[21]
酵母発現ベクターにUDP-グルクロン酸転移酵素遺伝子及びUDP-グルコース脱水素酵素含を発現可能に挿入したものである、UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクター。
[22]
酵母発現ベクターが自律複製型ベクターまたは染色体組み込み型ベクターである、[19]〜[21]のいずれかに記載のベクター。
[23]
UDP-グルコース脱水素酵素遺伝子が動物または植物由来の遺伝子である、[15]、[19]、[21]〜[22]のいずれかに記載のベクター。
[24]
UDP-グルコース脱水素酵素遺伝子がシロイヌナズナ由来遺伝子またはラット由来遺伝子である、[19]、[21]〜[22] のいずれかに記載のベクター。
[25]
UDP-グルクロン酸転移酵素遺伝子が哺乳動物由来の遺伝子である、[20]〜[24]のいずれかに記載のベクター。
[26]
UDP-グルクロン酸転移酵素遺伝子がヒト由来の遺伝子である、[20]〜[24]のいずれかに記載のベクター。
【発明の効果】
【0014】
本発明によれば、UDPGDH及びUGT発現出芽酵母菌体を用いて抱合体を調製することができる。本発明の遺伝子改変出芽酵母菌体を用いた抱合体調製法によれば、従来の有機合成法や試験管内での酵素法や分裂酵母を用いた抱合体調製法に比べて、著しく効率的かつ安価に目的の代謝物を調製することができる。例えば、実施例に示すように、UDPGDH及びUGT1A6同時発現出芽酵母菌体を用いた最適条件下において、7位水酸化クマリンの抱合体を1L反応溶液あたり200mgの産生効率で製造することが可能である。
【0015】
さらに、UGT分子種は、種々のものの遺伝子情報が知られていることから、本発明のUDPGDH及びUGT発現出芽酵母菌体を多様なUGT分子種について調製することができる。その結果、本発明のUDPGDH及びUGT発現出芽酵母菌体は、さまざまな医薬品に対して適用でき、実用性もきわめて高い。
【0016】
さらに本発明の方法では、宿主酵母としてS.cerevisiaeを用い、さらには、いくつかの工夫を加えることで、S. pombeを宿主とする非特許文献7に記載の方法に比べて以下のような利点がある。
(1)S.pombeではUGT1A6の活性がきわめて低く、発現量が低いと推測されるが、S.cerevisiaeにおける発現量は高く、活性はきわめて高い(図5、100%変換を達成)。実際に4メチルウンベリフェロンを基質として分裂酵母のデータと比較したところ複数の分子種において数十倍高い産生能力を示した。(表6参照)
(2)S.cerevisiaeが有するABCトランスポーターの一種が菌体外への分泌に関わっていると推測されるが(図6)、これは予想外の結果であり、S.pombeには見られないS.cerevisiae特有の現象と考えられる。
(3)S.pombeではUGT1A3、1A4、2B7の発現がうまく行ってないが、S. cerevisiaeにおいてはこれらについても高発現を達成することができる。また、1A4の発現については、シグナル配列を改変することで高発現に成功している。
【0017】
このように、S.pombeを宿主としたUGT発現系の最大の欠点は発現量の低さと分子種の違いによる発現量の違いと考えられる。
【0018】
一方で、本発明では、S.cerevisiaeを宿主として用い、必要によりN末端のシグナル配列を変換することにより、いずれのUGT分子種も高発現させることに成功し、S.pombeの発現系の最大の欠点をクリアーすることに成功した。動植物由来のUDPGDHとの同時発現に成功し、菌体を用いたグルクロン酸抱合体の高効率菌体外分泌生産に成功した。
【図面の簡単な説明】
【0019】
【図1】UDP-グルクロン酸転移酵素遺伝子とUDP-グルコース脱水素酵素を同時に含む自律複製型酵母発現ベクターの構築スキームを示す。
【図2】酵母内発現UDP-グルコース脱水素酵素における酵素活性測定結果を示す。
【図3】酵母内発現UDP-グルクロン酸転移酵素遺伝子及びUDP-グルコース脱水素酵素タンパクの発現をウエスタンブロット法により確認した結果を示す。
【図4】UDP-グルコース脱水素酵素遺伝子導入酵母菌体におけるUDP-グルクロン酸の産生結果を示す。
【図5】種々のヒトUDP-グルクロン酸転移酵素との同時発現酵母における抱合体生成結果を示す。
【図6】UGT1A6同時発現株における抱合体産生の時間変化を示す。
【図7】UGT1A6同時発現株における抱合体産生のグルコース濃度の影響を示す。
【図8】同時発現株の抱合体産生における発現ベクターの組み合わせの影響を示す。
【図9】酵母静止菌体を用いたケルセチン抱合体の製造結果を示す。
【図10】酵母静止菌体を用いたマイコフェノール酸抱合体の製造結果を示す。
【図11】酵母静止菌体を用いたメフェナム酸のアシル抱合体の製造結果を示す。
【図12】酵母静止菌体を用いてタモキシフェン抱合体(Nグルクロン酸抱合体)の製造結果を示す。
【図13】シトクロムP450を含む同時発現株の静止菌体を用いた7ヒドロキシクマリン抱合体の製造結果を示す。
【図14】出芽酵母PGK1由来プロモーター及びターミネーター領域を有するヒトシトクロムP450発現プラスミドの構築スキームを示す。
【図15】ヒトシトクロムP450(CYP2D6)とUDP-グルクロン酸転移酵素(UGT1A8)を同時発現させた酵母株静止菌体を用いた7エトキシクマリン代謝物の製造結果を示す。
【図16】組換え酵母菌体を用いたグルクロン酸抱合体の調製の模式説明図を示す。
【発明を実施するための形態】
【0020】
[形質転換した出芽酵母]
本発明は、形質転換した出芽酵母に関し、具体的には、UDP-グルコース脱水素酵素をコードする遺伝子及びUDP-グルクロン酸転移酵素をコードする遺伝子を発現可能に挿入して形質転換した出芽酵母である。本発明の形質転換した出芽酵母は、UDP-グルコース脱水素酵素をコードする遺伝子及びUDP-グルクロン酸転移酵素をコードする遺伝子を発現可能に挿入して形質転換した出芽酵母にさらに、シトクロムP450遺伝子をコードする遺伝子をさらに発現可能に挿入して形質転換した出芽酵母であることもできる。UDP-グルコース脱水素酵素をコードする遺伝子、UDP-グルクロン酸転移酵素をコードする遺伝子、及びシトクロムP450遺伝子をコードする遺伝子は、出芽酵母でこれら遺伝子を出芽酵母発現ベクターに組み込まれた形で、出芽酵母に挿入されて、出芽酵母を形質転換したものであることができ、あるいは、これらの遺伝子を形質転換すべき出芽酵母の染色体に、例えば、相同組換などの公知の手法により、発現可能な状態で挿入したものであることもできる。
【0021】
前記遺伝子を出芽酵母発現ベクターに組み込んだ形で、出芽酵母に挿入して、出芽酵母を形質転換したものの例としては、以下の(A)〜(G)のいずれかから選ばれる形質転換出芽酵母を挙げることができる。
(A)UDP-グルコース脱水素酵素発現ベクター及びUDP-グルクロン酸転移酵素発現ベクターで形質転換した出芽酵母、
(B)UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクターで形質転換した出芽酵母、
(C)UDP-グルコース脱水素酵素発現ベクター、UDP-グルクロン酸転移酵素発現ベクター及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母、
(D)UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクター、及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母、
(E)UDP-グルクロン酸転移酵素発現ベクター、及びUDP-グルコース脱水素酵素及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母、
(F)UDP-グルクロン酸転移酵素及びシトクロムP450遺伝子発現ベクター、及びUDP-グルコース脱水素酵素発現ベクターで形質転換した出芽酵母、並びに
(G)UDP-グルコース脱水素酵素、UDP-グルクロン酸転移酵素及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母
【0022】
例えば、上記UDP-グルコース脱水素酵素発現ベクターは、出芽酵母発現ベクターにUDP-グルコース脱水素酵素遺伝子を発現可能に挿入したものである。UDP-グルクロン酸転移酵素発現ベクターは、出芽酵母発現ベクターにUDP-グルクロン酸転移酵素遺伝子を発現可能に挿入したものである。UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクターは、出芽酵母発現ベクターにUDP-グルクロン酸転移酵素遺伝子及びUDP-グルコース脱水素酵素含を発現可能に挿入したものである。以下、各ベクターについて説明する。
【0023】
[酵素等の発現ベクター]
本発明では、(1)UDP-グルコース脱水素酵素発現ベクター、(2)UDP-グルクロン酸転移酵素発現ベクター、(3)UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクター、(4)シトクロムP450遺伝子発現ベクター、(5)UDP-グルクロン酸転移酵素及びシトクロムP450遺伝子発現ベクター、(6)UDP-グルコース脱水素酵素及びシトクロムP450遺伝子発現ベクター、及び(7)UDP-グルクロン酸転移酵素、UDP-グルコース脱水素酵素及びシトクロムP450遺伝子発現ベクターを用いる。1つのベクターに複数の遺伝子を有する場合、遺伝子配置(順序)に制限はない。以下、(1)、(2)及び(4)の順に個別に説明する。(3)、(5)、(6)、(7)の1つのベクターに複数の遺伝子を有するベクターについては、(1)、(2)及び(4)の説明に基づいて適宜提供できる。
【0024】
<UDP-グルコース脱水素酵素発現ベクター>
UDP-グルコース脱水素酵素発現ベクターは、酵母発現ベクターにUDP-グルコース脱水素酵素(UDPGDH)遺伝子を挿入したものである。
【0025】
酵素遺伝子を挿入する発現ベクターは、宿主である出芽酵母において複製保持され、上記酵素遺伝子を発現し得る自律複製型ベクターであっても、出芽酵母の染色体に組み込まれる、染色体組み込み型プラスミドベクターであってもよい。自律複製型ベクターおよび染色体組み込み型プラスミドベクターは、宿主である出芽酵母において保持されかつ機能するものであれば制限なく利用できる。自律複製型ベクターとしては、例えば、実施例で用いたpGYR、さらには酵母由来のプラスミドYEp352GAP、YEp51、pSH19などを挙げることができる。染色体組み込み型プラスミドベクターとしては、実施例で用いたpAURなどを挙げることができる。
【0026】
上記酵素遺伝子および、必要により、シグナル配列遺伝子を上記発現に適したベクター中のプロモーターの下流に連結して発現ベクターを得ることができる。プロモーターとしては、ENO1プロモーター、GAL10プロモーター、GAPDHプロモーター、ADHプロモーターなどが挙げられる。
【0027】
UDP-グルコース脱水素酵素遺伝子は、動物または植物由来の遺伝子であることができる。動物または植物由来のUDP-グルコース脱水素酵素遺伝子は、公知の遺伝子(例えば、 ヒト遺伝子:AF061016.1,マウス遺伝子:AF061017.1 など )から適宜選択することができ、実施例では、シロイヌナズナ由来の遺伝子及びラット由来の遺伝子を用いたが、いずれかのUDP-グルコース脱水素酵素遺伝子を導入した。出芽酵母発現ベクターで形質転換した出芽酵母では、UDP-グルコース脱水素酵素の活性が得られた。従って、遺伝子の由来に関わらず、公知のUDP-グルコース脱水素酵素遺伝子を用いることができる。尚、シロイヌナズナ由来の遺伝子は、例えば、市販のcDNAライブラリーである、PCR ready First Strand cDNA (Biochain社)から入手することができる。また、ラット由来の遺伝子も、例えば、市販のラット肝臓cDNAライブラリーであるPCR ready First Strand cDNA (Biochain 社)から入手することができる。これら以外にも、市販のcDNAライブラリーからUDP-グルコース脱水素酵素遺伝子を適宜入手することもできる。
【0028】
<UDP-グルクロン酸転移酵素発現ベクター>
UDP-グルクロン酸転移酵素発現ベクターは、出芽酵母発現ベクターにUDP-グルクロン酸転移酵素(UGT)遺伝子を挿入したものである。出芽酵母発現ベクターは上記UDP-グルコース脱水素酵素発現ベクターで説明したものと同様のものを用いることができる。
【0029】
UDP-グルクロン酸転移酵素遺伝子は、公知の遺伝子から適宜選択することができる。UDP-グルクロン酸転移酵素遺伝子は、哺乳動物由来の遺伝子であることができ、具体的にはヒト由来の遺伝子であることができる。UDP-グルクロン酸転移酵素(UGT)は、約530アミノ酸残基からなる小胞体膜タンパク質であり、肝臓や小腸において複数の分子種が存在する。これら分子種の特徴としては、アミノ末端側半分(約290アミノ酸残基)は抱合化基質を認識するドメインを構成し、分子種間で高い相同性を持つカルボキシル末端側半分(約240アミノ酸残基)は共通の基質であるUDP-グルクロン酸が結合するドメインとして機能している(非特許文献3、4、10)。
【0030】
UDP-グルクロン酸転移酵素遺伝子は、これまでの研究により多くの分子種がクローニングされ、その塩基配列が明らかにされた(非特許文献3)。本発明ではこれら公知の遺伝子を適宜利用できる。代表的なグルクロン酸転移酵素は、例えば、ヒトまたはブタ由来の酵素である。それら転移酵素の遺伝子配列およびシグナル配列の遺伝子配列は、ヒト由来の酵素はGenBankに登録されており、ブタ由来の酵素はPEDE(Database of full-length cDNA clones and ESTs in pigs)(http://pede.dna.affrc.go.jp)に登録されており、配列情報は容易に入手可能である。UGT遺伝子の代表例を以下に示す。
【0031】
【表1】

【0032】
出芽酵母を用いたグルクロン酸転移酵素の発現系においては、分子種によっては発現量の低いものが存在する。出芽酵母内での発現量の低かった分子種である、例えば、UGT1A1遺伝子の発現量を上昇させることができれば、医薬品抱合代謝物を調製できる範囲が広がり、出芽酵母発現グルクロン酸転移酵素による抱合体調製法の実用的価値は飛躍的に高まる。本発明では、出芽酵母を用いたグルクロン酸転移酵素の発現系において、出芽酵母内での発現量の低い分子種については、シグナル配列遺伝子を、高発現グルクロン酸転移酵素のシグナル配列遺伝子と置換することが好ましい。
【0033】
前記発現量の低い分子種のグルクロン酸転移酵素遺伝子は、例えば、発現量がUGT1A7の50%以下である低発現量グルクロン酸転移酵素遺伝子であることができる。前記低発現量グルクロン酸転移酵素は、例えば、UGT1A1、UGT1A4、UGT1A8またはUGT1A9であることができる。但し、これらに限定される意図ではない。さらに、前記低発現量グルクロン酸転移酵素遺伝子は、シグナル配列遺伝子を、例えば、発現量がUGT1A7の80%以上である高発現グルクロン酸転移酵素のシグナル配列遺伝子と置換することができる。高発現量グルクロン酸転移酵素は、例えば、UGT1A7、UGT1A6またはUGT1A10であることができる。但し、これらに限定される意図ではない。このようなシグナル配列の置換により、出芽酵母形質転換体によるグルクロン酸転移酵素の発現量がシグナル配列遺伝子未置換の形質転換体に比べて強化される。
【0034】
前記低発現量グルクロン酸転移酵素遺伝子は、シグナル配列遺伝子を(A)以下に示すアミノ酸配列(a)〜(c)のいずれかをコードする遺伝子、(B)アミノ酸配列(a)〜(c)のアミノ酸の1〜5個が置換若しくは欠失したアミノ酸配列のいずれかをコードする遺伝子、または(C)1〜5個のアミノ酸がアミノ酸配列(a)〜(c)のアミノ酸に付加されたアミノ酸配列のいずれかをコードする遺伝子と置換したものであることもできる。但し、(B)の置換若しくは欠失したアミノ酸配列および(C)の付加されたアミノ酸配列をコードする遺伝子は、シグナル配列遺伝子とした場合にグルクロン酸転移酵素の発現量が野生型の80%以上である。
(a) MARAGWTGLLPLYVCLLLTCGFAKAG(配列番号1)
(b) MACLLRSFQRISAGVFFLALWGMVVG(配列番号2)
(c) MAPRRVDQPRSFMCVSTADLWLCEAG(配列番号3)
【0035】
低発現量グルクロン酸転移酵素遺伝子のシグナル配列遺伝子を、(B)アミノ酸配列(a)〜(c)のアミノ酸の1〜5個が置換若しくは欠失したアミノ酸配列のいずれかをコードする遺伝子であって、この遺伝子は、グルクロン酸転移酵素遺伝子のシグナル配列遺伝子として用いた場合に、グルクロン酸転移酵素の発現量がUGT1A7の80%以上である遺伝子と置換する。アミノ酸配列(a)のアミノ酸の1〜5個が置換若しくは欠失したアミノ酸配列をコードする遺伝子をシグナル配列遺伝子として用いた場合に、UGT1A7の発現量が野生型のUGT1A7の80%以上、好ましく90%以上、より好ましくは100%以上である遺伝子とする。アミノ酸配列(b)のアミノ酸の1〜5個が置換若しくは欠失したアミノ酸配列をコードする遺伝子をシグナル配列遺伝子として用いた場合に、UGT1A6の発現量が野生型のUGT1A6の80%以上、好ましく90%以上、より好ましくは100%以上である遺伝子とする。アミノ酸配列(c)のアミノ酸の1〜5個が置換若しくは欠失したアミノ酸配列をコードする遺伝子をシグナル配列遺伝子として用いた場合に、UGT1A6の発現量が野生型のUGT1A10の80%以上、好ましく90%以上、より好ましくは100%以上である遺伝子とする。
【0036】
さらに、前記低発現量グルクロン酸転移酵素遺伝子のシグナル配列遺伝子を、(C)1〜5個のアミノ酸がアミノ酸配列(a)〜(c)のアミノ酸に付加されたアミノ酸配列のいずれかをコードする遺伝子と置換したものであり、この遺伝子は、グルクロン酸転移酵素遺伝子のシグナル配列遺伝子として用いた場合に、グルクロン酸転移酵素の発現量がUGT1A7の80%以上である遺伝子と置換する。アミノ酸配列(a)に1〜5個のアミノ酸が付加されたアミノ酸配列をコードする遺伝子をシグナル配列遺伝子として用いた場合に、UGT1A7の発現量が野生型のUGT1A7の80%以上、好ましく90%以上、より好ましくは100%以上である遺伝子とする。アミノ酸配列(b) に1〜5個のアミノ酸が付加されたアミノ酸配列をコードする遺伝子をシグナル配列遺伝子として用いた場合に、UGT1A6の発現量が野生型のUGT1A6の80%以上、好ましく90%以上、より好ましくは100%以上である遺伝子とする。アミノ酸配列(c)に1〜5個のアミノ酸が付加されたアミノ酸配列をコードする遺伝子をシグナル配列遺伝子として用いた場合に、UGT1A6の発現量が野生型のUGT1A10の80%以上、好ましく90%以上、より好ましくは100%以上である遺伝子とする。
【0037】
尚、野生型のグルクロン酸転移酵素の発現量との対比は、ミクロソーム画分におけるグルクロン酸転移酵素の発現を、例えば、ヒトグルクロン酸転移酵素ファミリー1の共通配列認識ペプチド抗体(非特許文献6)を用いたウエスタンブロット法による解析によって行うことができる。
【0038】
低発現量グルクロン酸転移酵素遺伝子のシグナル配列遺伝子を、上記アミノ酸配列をコードする遺伝子と置換することで、出芽酵母形質転換体によるグルクロン酸転移酵素の発現量がシグナル配列遺伝子未置換の形質転換体に比べて強化される。
【0039】
<UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクター>
UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクターは、1つの出芽酵母発現ベクターにUDP-グルクロン酸転移酵素遺伝子及びUDP-グルコース脱水素酵素遺伝子の両方を挿入したものであり、このベクターで形質転換した出芽酵母では、UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素の活性が同時に得られる。出芽酵母発現ベクターは上記UDP-グルコース脱水素酵素発現ベクターで説明したものと同様のものを用いることができる。また、UDP-グルクロン酸転移酵素遺伝子及びUDP-グルコース脱水素酵素遺伝子は、上記で説明したものと同様のものをそれぞれ利用できる。
【0040】
UDP-グルクロン酸転移酵素遺伝子及びUDP-グルコース脱水素酵素遺伝子のベクター中での挿入の順番は特に制限はなく、どちらが上流にあっても各酵素遺伝子の発現は支障なく行われる。
【0041】
<シトクロムP450遺伝子発現ベクター>
シトクロムP450遺伝子発現ベクターは、非特許文献2に記載されており、この文献の記載に基づいて調製することができる。出芽酵母発現ベクターは上記UDP-グルコース脱水素酵素発現ベクターで説明したものと同様のものを用いることができる。
【0042】
シトクロムP450遺伝子発現ベクターは、小胞体膜酵素であるシトクロムP450の発現で実績のあるpGYRを用いた。発現調節領域に醤油酵母(Zygosaccharomyces rouxii)グリセロアルデヒド3リン酸脱水素酵素遺伝子由来のプロモーター及びターミネーター領域を有し定常的なタンパク発現を可能にしている。さらに出芽酵母由来P450還元酵素遺伝子を含むことによりNADPHからの電子供給を増強することで酵母内でのP450によるモノオキシゲナーゼ反応を促進することができる。
【0043】
<発現ベクターの調製>
酵素遺伝子及び必要により、シトクロムP450遺伝子をそれぞれ機能的に連結し、これらを適切なベクターに挿入する方法は、当業者に通常知られる方法、たとえば、文献Molecular Cloning (1989) (Cold Spring Harbor Lab.)に記載の方法である。組換えベクターにおける挿入位置は、組換えベクターの複製に関与していない領域であればどこでも良く、通常はベクター内のマルチクローニングサイトが利用される。
【0044】
[形質転換体]
本発明の形質転換体は、上記本発明のいずれか1つまたは2つ以上のベクターで形質転換した出芽酵母からなる。宿主として用いる出芽酵母はサッカロマイセス・セレビシエ(Saccharomyces cerevisiae)であり、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)に属する菌株であれば特に制限はない。例えば、Saccharomyces cerevisiae AH22株、NA87-11A株、SHY3株等を用いることができる。
【0045】
S.cerevisiae AH22株はミトコンドリアDNAを欠損した呼吸欠損株であり、小胞体膜に結合するシトクロムP450の発現には最適の宿主であることが知られている。(参考文献 T. Sakaki, M. Akiyoshi-Shibata, Y. Yabusaki, H. Ohkawa, Organella targeted expression of rat liver cytochrome P450c27 in yeast: genetically engineered alteration of mitochondrial P450 into a microsomal form creates a novel functional electron transport chain, J. Biol.Chem. 267 16497-16502.(1992))AH22株は、ミトコンドリアDNAが欠損しているため機能を有するミトコンドリアが形成されず、その代わりに小胞体膜が発達することでP450やUGTが存在する場が増えるため、これらタンパク発現に有利であると考えられる。UDP-グルクロン酸転移酵素(UGT)もP450と同様に小胞体膜に結合するタンパク質であることから、UGTの発現においてもAH22株は適した宿主であると考えられ、実施例では、AH22株を宿主として用いた。このような理由から、シトクロムP450とUGTの同時発現系においても、AH22株は好ましい宿主と考えられる。但し、本発明は、宿主がAH22株の場合に限定されるものではない。
【0046】
本発明の形質転換体は、例えば、
(A)UDP-グルコース脱水素酵素発現ベクター及びUDP-グルクロン酸転移酵素発現ベクターで形質転換した出芽酵母、
(B)UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクターで形質転換した出芽酵母、
(C)UDP-グルコース脱水素酵素発現ベクター、UDP-グルクロン酸転移酵素発現ベクター及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母、
(D)UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクター、及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母、
(E)UDP-グルクロン酸転移酵素発現ベクター、及びUDP-グルコース脱水素酵素及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母、
(F)UDP-グルクロン酸転移酵素及びシトクロムP450遺伝子発現ベクター、及びUDP-グルコース脱水素酵素発現ベクターで形質転換した出芽酵母、並びに
(G)UDP-グルコース脱水素酵素、UDP-グルクロン酸転移酵素及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母
を挙げることができる。
【0047】
形質転換体の調製方法にも特に制限はなく、酵母宿主への組み換えベクターの導入方法としては、例えば、エレクトロポレーション法、スフェロプラスト法、酢酸リチウム法等を挙げることができる。
【0048】
調製された形質転換体は、適宜常法により培養、増殖させた後に、後述するグルクロン酸抱合体の製造方法に使用することができる。増殖させた形質転換体は、導入した遺伝子の種類や宿主の種類により異なるが、一般には対数増殖期にある菌体を用いることが適当であるが、対数増殖期前の菌体や対数増殖期後の菌体を用いることが好ましい場合もある。
【0049】
[グルクロン酸抱合体の製造方法]
本発明のグルクロン酸抱合体の製造方法は、前記本発明の出芽酵母形質転換体をグルコース及び被抱合物質の存在下で培養して、前記被抱合物質のグルクロン酸抱合体を生成させることを含むものである。
【0050】
図16に、本発明の組換え酵母菌体(形質転換体)を用いたグルクロン酸抱合体の調製の模式説明図を示す。
【0051】
一般に、生体内外の異物(脂溶性化合物(疎水性基質)(RH))は、抱合化反応を受けて水溶性の抱合体(Glucuronide)となり、体外へ排泄される。特にグルクロン酸抱合は、脂溶性化合物中の水酸基、アミノ基、カルボキシル基あるいはチオール基などの官能基にグルクロン酸が付加されることによって水溶性の極性代謝物に変換される。極性代謝物(グルクロン酸抱合体)は、多剤耐性薬剤輸送タンパク質(ABCC2)により、生体外に排泄が促進される。従って、本発明のグルクロン酸抱合体の製造方法において用いられる被抱合物質は、上記水酸基、アミノ基、カルボキシル基あるいはチオール基などの官能基を有する化合物であり、これらの化合物は、医薬品または医薬品の候補物質であることができる。被抱合物質が上記水酸基、アミノ基、カルボキシル基あるいはチオール基などの官能基を有する場合には、菌体内のUDP-グルクロン酸転移酵素(UGT, UDP-glucuronic acid transferase)が触媒するグルクロン酸転移反応により、UDP-グルクロン酸(UDP-GlcUA)を糖の供与体として、グルクロン酸抱合体に変換される。
【0052】
一方、被抱合物質が上記水酸基、アミノ基、カルボキシル基あるいはチオール基などの官能基を有さない場合には、被抱合物質である生体内外の脂溶性化合物(疎水性基質)(RH,脂溶性基質)は、シトクロムP450(CYP)により酸化されて水酸化物(ROH)となった後に、菌体内のUDP-グルクロン酸転移酵素(UGT, UDP-glucuronic acid transferase)が触媒するグルクロン酸転移反応により、UDP-グルクロン酸(UDP-GlcUA)を糖の供与体として、グルクロン酸抱合体に変換される。UDP-グルクロン酸転移酵素(UGT)が関与するこの系は、生体外への異物排泄を促進させる役割をもつ異物代謝系のひとつである。
【0053】
一方、UDP-グルクロン酸(UDP-GlcUA)は、グルコースから、UDP-グルコース(UDP-glucose)を経て、UDP-グルコース脱水素酵素(UDPGDH, UDP-glucose dehydrogenase)の作用により生成される。グルコースは、酵母内に取り込まれるとエネルギー代謝系である解糖におけるグルコキナーゼ、ホスホムターゼ及びUTPグルコース1リン酸ウリジルトランスフェラーゼの作用によりUDP-グルコース(UDP-glucose)に変換される。UDP-グルコース(UDP-glucose)は、UDP-グルコース脱水素酵素(UDPGDH )の作用により、NAD+を補酵素として、6位が酸化されてUDP-グルクロン酸(UDP-GlcUA)に変換される。
【0054】
上記のように、原料として水酸基、アミノ基、カルボキシル基あるいはチオール基などの官能基を有する被抱合物質を用いる方法(第1の態様)においては、本発明の形質転換体として、上記(A)、(B)または(E)の形質転換体を用いることで、前記被抱合物質のグルクロン酸抱合体を生成させることができる。
【0055】
水酸基、アミノ基、カルボキシル基あるいはチオール基などの官能基を有する被抱合物質は、特に制限はなく、例えば、アルコール性水酸基を含む医薬品または医薬品の候補物質、フェノール性水酸基を多数含むポリフェノール化合物、カルボン酸を含む非ステイロイド性抗炎症薬またはその候補物質、1〜4級のいずれか少なくとも1つのアミンを含む化合物等を挙げることができる。これら被抱合物質の例は、例えば、非特許文献10の100〜102頁のTable 1に列記されており、非特許文献10の100〜102頁のTable 1を含む全記載はここに参照として援用される。
【0056】
また、被抱合物質が上記水酸基、アミノ基、カルボキシル基あるいはチオール基などの官能基を有さない場合には、例えば、有機合成法を用いた水酸基の導入やシトクロムP450を用いた酵素法による選択的な水酸化によって、事前に被抱合物質に官能基を導入した後に、上記第一の態様の方法に供することができる。あるいは、被抱合物質が上記水酸基、アミノ基、カルボキシル基あるいはチオール基などの官能基を有さない場合には、本発明の形質転換体として、シトクロムP450遺伝子を導入した前記(C)、(D)、(E)、(F)または(G)の形質転換体を用い、かつこの形質転換体をグルコース、及び被抱合物質の存在下で培養して、前記被抱合物質のグルクロン酸抱合体を生成させることもできる(第2の態様)。本発明において、P450とUGT同時発現系による代謝物質は、P450の代謝により抱合化を受ける官能基(主に水酸基)が生成されるものであれば特に制限はなく、例えば、メトキシ基あるいはエトキシ基を含む医薬品または医薬品の候補物質、メチレンジオキシフェニル基をもつセサミン化合物、水酸基を有しないジアゼピン系の医薬品または医薬品の候補物質等を挙げることができる。第2の態様においては、シトクロムP450を含む同時発現株を用いるため、抱合化基質の前駆体(被抱合物質)から抱合体を調製することができる。即ち、被抱合物質を前駆体化(水酸化物への変換)をすること無しに、抱合体を調製することができる。
【0057】
本発明の製造方法において用いられる被抱合物質は、上記のように特に制限はない。これは、上述のように本発明においては、UDP-グルクロン酸転移酵素(UGT)の基質となる被抱合物質の種類に応じて、形質転換体に含まれるUDP-グルクロン酸転移酵素(UGT)遺伝子の種類を適宜選択することができるからである。例えば、実施例に示す例では、7位水酸化クマリンの抱合体(フェノール性水酸基)を調製する場合、用いるUGTは、UGT1A6であることが適当であり、ケルセチンの4’位及び3’位水酸基に対する抱合体(フェノール性水酸基)を調製する場合、用いるUGTは、UGT1A1あるいはUGT1A8であることが適当であり、マイコフェノール酸の7位水酸基に対する抱合体(アルコール性水酸基)を調製する場合、用いるUGTは、UGT1A9であることが適当である。
【0058】
上記本発明の形質転換体の培養は、グルコース及び被抱合物質の存在下、公知の酵母の培養方法を適宜用いて実施できる。酵母宿主が上記AH22株の場合、L-ヒスチジンとL-ロイシン以外の全ての必須アミノ酸を生合成することができる。従って、培地には、これら2つのアミノ酸を添加して培養するか、あるいは発現ベクターとして、いずれか一方のアミノ酸合成遺伝子を持つベクターを用いることで、この発現ベクターを有する酵母宿主を選択的に培養することもできる。
【0059】
培養の条件は、酵母形質転換体の生育に適した条件において適宜に設定することができる。形質転換体の培養に用いる栄養培地としては、炭素源、窒素源、無機物、および必要に応じ使用菌株の必要とする微量栄養素を程よく含有するものであれば、天然培地、合成培地のいずれでもよい。あるいは酵母形質転換体の生育に必要な栄養源の少なくとも一部を欠いた条件で培養することで、グルクロン酸抱合体の生成、蓄積が良好に得られる場合もある。被抱合物質の種類によっては後者の条件が好ましい場合もある。
【0060】
栄養培地の炭素源としては、酵母形質転換体が資化しうる物であればよく、例えば、グルコース、マルトース、フラクトース、マンノース、トレハロース、スクロース、マンニトール、ソルビトール、デンプン、デキストリン、糖蜜などの糖質、またはクエン酸、コハク酸などの有機酸、またはグリセリンなどの脂肪酸も使用することができる。
【0061】
栄養培地の窒素源としては、各種有機および無機の窒素化合物、さらに培地は各種の無機塩を含むことができる。たとえば、コーンスティープリカー、大豆粕、あるいは各種ペプトン類等の有機窒素源、そして塩化アンモニウム、硫酸アンモニウム、尿素、硝酸アンモニウム、硝酸ナトリウム、リン酸アンモニウム等の無機窒素源などの化合物が使用可能である。また、グルタミン酸などのアミノ酸および尿素などの有機窒素源が炭素源にもなることはいうまでもない。さらに、ペプトン、ポリペプトン、バクトペプトン、肉エキス、魚肉エキス、酵母エキス、コーンスティープリカー、大豆粉、大豆粕、乾燥酵母、カザミノ酸、ソリュブルベジタブルプロテイン等の窒素含有天然物も窒素源として使用できる。
【0062】
栄養培地の無機物としては、たとえば、カルシウム塩、マグネシウム塩、カリウム塩、ナトリウム塩、リン酸塩、マンガン塩、亜鉛塩、鉄塩、銅塩、モリブデン塩、コバルト塩などが適宜用いられる。具体的には、リン酸二水素カリウム、リン酸水素二カリウム、硫酸マグネシウム、硫酸第一鉄、硫酸マンガン、硫酸亜鉛、塩化ナトリウム、塩化カリウム、塩化カルシウム等が用いられる。さらに、必要に応じて、アミノ酸ならびにビオチンおよびチアミンなどの微量栄養素ビタミンなども適宜用いられる。
【0063】
培養法としては液体培養法がよく、回分培養、流加培養、連続培養または灌流培養のいずれを用いてもよいが、工業的には通気攪拌培養法が好ましい。培養温度とpHは、使用する形質転換体の増殖に最も適した条件を選べばよい。培養時間は微生物が増殖し始める時間以上の時間であればよく、好ましくは8〜120時間であり、さらに好ましくは組換えタンパク質遺伝子の遺伝子産物が最大に生成する時間までである。たとえば、形質転換体が出芽酵母の場合の培養は、通常、温度20〜40℃、好ましくは25〜35℃、pH2〜9、好ましくは5〜8、培養日数0.5〜7日間から選ばれる条件で振盪または通気攪拌して行われる。酵母の増殖を確認する方法は特に制限はないが、たとえば、培養物を採取して顕微鏡で観察してもよいし、吸光度で観察してもよい。また、培養液の溶存酸素濃度には特に制限はないが、通常は、0.5〜20ppmが好ましい。そのために、通気量を調節したり、撹拌したり、通気に酸素を追加したりすればよい。
【0064】
形質転換体の使用量は、例えば、0.5〜5%(w/v、乾燥重量/培養液体積)の範囲であることができる。さらに、グルコース及び被抱合物質の培地への添加量は、培地における浸透圧及び細胞に対する毒性を考慮して適宜決定され、例えば、グルコースは、4〜20%(w/v)の範囲であり、被抱合物質は、0.5〜25mM の範囲であることが適当である。
【0065】
形質転換体を培養することで培養物中にグルクロン酸抱合体が蓄積する。培養後、酵母形質転換体の培養液からグルクロン酸抱合体を回収する。グルクロン酸抱合体は、形質転換体外に蓄積されることが多いが、一部は形質転換体内にも蓄積される。形質転換体外に蓄積されたグルクロン酸抱合体は、例えば、溶媒抽出により回収することができる。溶媒抽出による回収は、例えば、通常知られる手段によって形質転換体を培養液から除いた後に、得られる培養上清に対して行うことができる。形質転換体は再利用も可能である。
【0066】
また、形質転換体内に蓄積したグルクロン酸抱合体は、例えば、有機溶剤やザイモリアーゼのような酵素によって形質転換体の細胞壁を溶解する方法、および、超音波破砕法、フレンチプレス法、ガラスビーズ破砕法、ダイノミル破砕法等の細胞破砕法で得られた形質転換体の細胞破砕物および/または培養物を遠心分離法、ろ過法等の操作によって形質転換体と培養上清に分離する。このようにして得られた培養上清から、上記と同様にグルクロン酸抱合体を回収することができる。
【実施例】
【0067】
以下本発明を実施例によりさらに詳細に説明する。但し、本発明は実施例に限定される意図ではない。
【0068】
実施例1
1. シロイヌナズナ及びラット由来UDP-グルコース脱水素酵素発現プラスミドの構築
シロイヌナズナ及びラット由来UDP-グルコース脱水素酵素は、cDNAライブラリーを鋳型として、両端に相補的な配列を有するプライマーを用いてPCR法によって増幅し構築した。酵母発現ベクターとしては自律複製型プラスミドであるpGYR(文献番号4)あるいは染色体組み込み型プラスミドであるpAUR101(TaKaRa)を用いた。
【0069】
1-1. シロイヌナズナ及びラット由来UDP-グルコース脱水素酵素遺伝子の作成
1-1-1. シロイヌナズナ由来UDP-グルコース脱水素酵素遺伝子の作成
シロイヌナズナのcDNAライブラリーを用いてPCR法によって遺伝子のクローニングを行った。cDNAライブラリーはPCR ready First Strand cDNA (Biochain 社)を用いた。
【0070】
DNAポリメラーゼとしてはKOD-plus-(TOYOBO)を用いた。反応液組成は製造業者の指示に従い、以下に示すプライマーと反応条件でPCRを行った。
フォワード配列:配列番号4
リバース配列:配列番号5
PCR条件
変性 94℃ 2分
5サイクル 94℃ 15秒、37℃ 30秒、68℃1分45秒
30サイクル 94℃ 15秒、55℃ 30秒、68℃1分45秒
最終伸長 68℃10分
【0071】
PCR産物を1%アガロースゲルで電気泳動した結果、目的サイズ(約1.5kb)に特異的な増幅がみられた。(以下、1%アガロースゲルでの電気泳動は、単に電気泳動と略称する)。PCR増幅溶液を電気泳動して、DNA断片をゲルから分離した後、TAクローニングキット(Target cloneTM-plus-,TOYOBO社)を用いてpTAベクターに挿入した。このクローニングされた遺伝子のシークエンスを行い遺伝子配列を解析したところ、配列番号6に示した塩基配列(GenBank Acc.No. AY056200)であることを確認した。
【0072】
本遺伝子を発現ベクターにサブクローニングするために、部位特異的な変異導入によりアミノ酸配列を変えずに内部HindIIIサイトを欠失させた。変異導入は変異体作成キットQuick ChangeTMを用いた。変異導入をおこなったクローンの塩基配列を解析し、目的以外の箇所に変異導入されていないことを確認した。
【0073】
1-1-2. ラット由来UDP-グルコース脱水素酵素遺伝子の作成
ラット肝臓cDNAライブラリーを用いてPCR法によって遺伝子のクローニングを行った。cDNAライブラリーはPCR ready First Strand cDNA (Biochain 社)を用いた。
【0074】
DNAポリメラーゼとしてはKOD-plus-(TOYOBO)を用いた。反応液組成は製造業者の指示に従い、以下に示すプライマーと反応条件でPCRを行った。
フォワード配列:配列番号7
リバース配列:配列番号8
PCR条件
変性 94℃ 2分
5サイクル 94℃ 15秒、37℃ 30秒、68℃1分45秒
30サイクル 94℃ 15秒、55℃ 30秒、68℃1分45秒
最終伸長 68℃10分
【0075】
PCR産物を1%アガロースゲルで電気泳動した結果、目的サイズ(約1.5kb)に特異的な増幅がみられた。(以下、1%アガロースゲルでの電気泳動は、単に電気泳動と略称する)。PCR増幅溶液を電気泳動して、DNA断片をゲルから分離した後、TAクローニングキット(Target cloneTM-plus-,TOYOBO社)を用いてpTAベクターに挿入した。このクローニングされた遺伝子のシークエンスを行い遺伝子配列を解析したところ、配列番号9に示した塩基配列(GenBank Acc.No. O70199)であることを確認した。
【0076】
本遺伝子を発現ベクターにサブクローニングするために、部位特異的な変異導入によりアミノ酸配列を変えずに内部HindIIIサイトを欠失させた。変異導入は変異体作成キットQuick ChangeTMを用いた。変異導入をおこなったクローンの塩基配列を解析し、目的以外の箇所に変異導入されていないことを確認した。
【0077】
1-2. UDP-グルコース脱水素酵素発現ベクターの構築
1-2-1. 自律複製型酵母発現ベクターの構築
1-1-1及び1-1-2で作成したUDP-グルコース脱水素酵素断片を含むプラスミド約1μgを37℃で4時間、HindIII処理し、電気泳動後、遺伝子断片をゲルから切り出した。切り出したゲルからWizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてインサート断片を調製した。
【0078】
酵母発現ベクターとして用いるpGYRを37℃で4時間HindIII処理し、電気泳動後、目的サイズ(約11kb)のバンドを切り出した。その後、Wizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてベクター断片を調製した。
【0079】
インサート断片とpGYR-HindIIベクター断片の濃度を電気泳動で確認後、それらのモル比が3:1〜10:1程度になるように混合し、等量のDNA Ligation Kit Ver. 2 (TaKaRa)のsolution Iを加え、16℃、1時間反応を行った。その後、大腸菌JM109株にヒートショック法を用いてトランスフォーメーションを行い、LBプレート(アンピシリン50μg/ml含有)に展開した。得られた大腸菌コロニーのうち数個を選択し、それらを鋳型としてコロニーPCRを行った。DNAポリメラーゼは、Ex Taq(登録商標)DNAポリメラーゼを用いた。プライマーはYGAP-Pプライマー(5’-aatgacaccgtgtggtgatcttcaagg-3’)(配列番号10)と上記リバースプライマー(配列番号5あるいは8)を用いた。以下の条件でPCRを行った。
【0080】
PCR条件
変性 98℃5分
30サイクル 94℃ 30秒、50℃ 30秒、72℃ 2分30秒
最終伸長 72℃4分
【0081】
PCR後、電気泳動を行い、予想サイズ(約3kb)に特異的な増幅がみられるクローンを数個得た。得られた大腸菌コロニーを5mlのLB培地(アンピシリン100μg/ml含有)に植菌し、37℃、200rpmで16時間振盪培養を行った。その後アルカリSDS法を用いてプラスミドを抽出した。そして、その一部を37℃で1時間、HindII処理し、電気泳動をしてインサートの導入を再度確認し、2種のUDP-グルコース脱水素酵素酵素をそれぞれ含む酵母発現ベクター(シロイヌナズナ由来:pGYR/At.UDPGDH、ラット由来:pGYR/ratUDPGDH)を得た。
【0082】
1-2-2. 染色体組み込み型酵母発現ベクターの構築
1-2-1で作成したUDP-グルコース脱水素酵素断片を含む発現プラスミド(pGYR/At.UDPGDH) 約1μgを37℃で4時間、NotI処理し、電気泳動後、酵母発現プロモーター及びターミネーター領域を含む遺伝子断片(約3kb)をゲルから切り出した。切り出したゲルからWizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてインサート断片を調製した。
【0083】
自律複製型酵母発現ベクターpAUR101(TaKaRa)に上記で得られたNotI断片を挿入するために、部位特異的な変異導入によりマルチクローニングサイトにNotI制限酵素配列(GCGGCCGC)をマルチクローニングサイト領域に導入した。変異導入は変異体作成キットQuick ChangeTMを用いた。変異導入をおこなったクローンの塩基配列を解析し、目的以外の箇所に変異導入されていないことを確認した(pAUR-N)。酵母発現ベクターとして用いるpAUR-Nを37℃で4時間NotI処理し、電気泳動後、目的サイズ(約7kb)のバンドを切り出した。その後、Wizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてベクター断片を調製した。
【0084】
インサート断片とpAUR-NotIベクター断片の濃度を電気泳動で確認後、それらのモル比が3:1〜10:1程度になるように混合し、等量のDNA Ligation Kit Ver. 2 (TaKaRa)のsolution Iを加え、16℃、1時間反応を行った。その後、大腸菌JM109株にヒートショック法を用いてトランスフォーメーションを行い、LBプレート(アンピシリン50μg/ml含有)に展開した。得られた大腸菌コロニーを5mlのLB培地(アンピシリン100μg/ml含有)に植菌し、37℃、200rpmで16時間振盪培養を行った。その後アルカリSDS法を用いてプラスミドを抽出した。そして、その一部を37℃で1時間、NotI処理し、電気泳動をしてインサートの導入を再度確認し、UDP-グルコース脱水素酵素を含む酵母発現ベクター(pAUR/At.UDPGDH)を得た。
【0085】
1-2-3.UDP-グルクロン酸転移酵素遺伝子とUDP-グルコース脱水素酵素を同時に含む自律複製型酵母発現ベクターの構築
1-2-2で作成したpAUR-Nを用いてシロイヌナズナ由来UDP-グルコース脱水素酵素及びUDP-グルクロン酸転移酵素を同時に含む酵母発現ベクターを構築し(図1)た。まず、最初にIn FusionTM Advantage PCRクローニングキット(TaKaRa)を用いて、1-2-2で作成したUDP-グルコース脱水素酵素遺伝子を含むNotI断片におけるプロモーター領域上流のNotIサイトを欠失させた断片と相補的な配列を有するpAUR-Nベクターを融合させてUDP-グルコース脱水素酵素遺伝子を含む染色体組み込み型酵母発現ベクター (pAUR-At.UDPGDH)を作成した。
【0086】
DNAポリメラーゼとしてはPrime STAR Max Premix (TaKaRa)を用いた。反応液組成は製造業者の指示に従い、以下に示すプライマーと反応条件でPCRを行った。
インサートDNAのPCR反応
テンプレート:pGYR/At.UDPGDH
フォワード配列:配列番号11
リバース配列:配列番号12
PCR条件
変性 98℃ 10秒
30サイクル 98℃ 10秒、55℃ 5秒、72℃ 16秒
最終伸長 72℃10分
ベクターDNAのPCR反応
テンプレート:pAUR-N NotI digest
フォワード配列:配列番号13
リバース配列:配列番号14
PCR条件
変性 98℃ 10秒
30サイクル 98℃ 10秒、55℃ 5秒、72℃ 33秒
最終伸長 72℃10分
【0087】
UDP-グルクロン酸転移酵素断片を含む発現プラスミドpGYR/UGT約1μgを37℃で4時間、NotI処理し、電気泳動後、酵母発現プロモーター及びターミネーター領域を含む遺伝子断片(約3.2kb)をゲルから切り出した。切り出したゲルからWizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてインサート断片を調製した。
【0088】
先に作成したpAUR-At.UDPGDHを37℃で4時間NotI処理し、電気泳動後、目的サイズ(約10kb)のバンドを切り出した。その後、Wizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてベクター断片を調製した。
【0089】
インサート断片とpAUR-NotIベクター断片の濃度を電気泳動で確認後、それらのモル比が3:1〜10:1程度になるように混合し、等量のDNA Ligation Kit Ver. 2 (TaKaRa)のsolution Iを加え、16℃、1時間反応を行った。その後、大腸菌JM109株にヒートショック法を用いてトランスフォーメーションを行い、LBプレート(アンピシリン50μg/ml含有)に展開した。得られた大腸菌コロニーを5mlのLB培地(アンピシリン100μg/ml含有)に植菌し、37℃、200rpmで16時間振盪培養を行った。その後アルカリSDS法を用いてプラスミドを抽出した。そして、その一部を37℃で1時間、NotI処理し、電気泳動をしてインサートの導入を再度確認し、UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素を含む酵母発現ベクター(pAUR-At.UDPGDH/UGT)を得た。
【0090】
【表2】
【0091】
【表3】
【0092】
1-2-4. 遺伝子改変グルクロン酸転移酵素発現プラスミドの構築
本発現系で用いるUDP-グルクロン酸転移酵素分子種のうちUGT1A1及びUGT1A9に関しては、発現量を増強するために次に述べるようにN末端シグナル配列の改変(高発現分子種であるUGT1A7に由来するN末端シグナル配列への置換)を行った。
【0093】
ヒトグルクロン酸転移酵素におけるシグナル配列領域の塩基配列を表4に示す(配列番号15〜17)。シグナル配列置換したヒトグルクロン酸転移酵素は、ヒトグルクロン酸転移酵素遺伝子(UGT1A1及びUGT1A9)を含むpUC119クローニングベクターを鋳型として、置換配列を有するプライマー(表5 配列番号18〜20)を用いてPCR法によって増幅し構築した。
【0094】
【表4】
【0095】
【表5】
【0096】
1-2-4-1. N末端置換グルクロン酸転移酵素遺伝子の作成
N末端置換グルクロン酸転移酵素を、ヒトグルクロン酸転移酵素遺伝子(UGT1A1,及びUGT1A9)を含むpUC119クローニングベクターを鋳型としてPCRにより増幅した。DNAポリメラーゼとしてはKOD-plus-(TOYOBO)を用いた。反応液組成は製造業者の指示に従い、以下に示すプライマーと反応条件でPCRを行った。
【0097】
プライマー
フォワード配列:配列番号15あるいは16
リバース配列:配列番号20
【0098】
PCR条件
変性 94℃ 2分
5サイクル 94℃ 15秒、37℃ 30秒、68℃1分45秒
30サイクル 94℃ 15秒、55℃ 30秒、68℃1分45秒
最終伸長 68℃4分
【0099】
PCR産物を1%アガロースゲルで電気泳動した結果、目的サイズ(約1.6kb)に特異的な増幅がみられた。(以下、1%アガロースゲルでの電気泳動は、単に電気泳動と略称する)。
PCR断片を37℃で1時間、HindIII処理し、電気泳動後、グルクロン酸転移酵素遺伝子断片をゲルから切り出した。切り出したゲルからWizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてインサート断片を調製した。
【0100】
サブクローニングベクターとして用いるpUC119を37℃で4時間HindIII処理し、電気泳動後、目的サイズ(約2.8kb)のバンドを切り出した。その後、Wizard(登録商標)SV Gel and PCR Clean-Up System(Promega)を用いてベクター断片を調製した。
【0101】
インサート(グルクロン酸転移酵素遺伝子断片)とベクター(pUC119)を混合し、等量のDNA Ligation Kit Ver. 2 (TaKaRa)のsolution Iを加え、16℃、1時間反応を行った。その後、大腸菌JM109株にヒートショック法を用いてトランスフォーメーションを行い、LBプレート(アンピシリン50μg/ml含有)に展開した。
【0102】
得られた大腸菌コロニーのうち数個を選択し、それらを鋳型としてコロニーPCRを行った。DNAポリメラーゼとして、Ex Taq(登録商標)DNAポリメラーゼ(TaKaRa)を用い、M13-M4プライマー、M13-Rvプライマーを用いて以下の条件でPCRを行った。
【0103】
PCR条件
変性 98℃ 5分
30サイクル 94℃ 30秒、50℃ 30秒、72℃ 2分
最終伸長 72℃ 4分
PCR後、電気泳動を行い、予想サイズ(約1.6kb)に特異的な増幅が見られるクローンを数個得た。
【0104】
得られたクローンのインサート配列にPCRによる変異が入っていないことを確認するために配列決定を行った。まず、得られた大腸菌コロニーを5mlのLB培地(アンピシリン100μg/ml含有)に植菌し、37℃、200rpmで16時間振盪培養を行った。その後、Wizard(登録商標)Plus SV Minipreps DNA Purification System (Promega)を用いてプラスミドを抽出し、プラスミド濃度を260nmの吸光度を用いて測定し、テンプレートDNAとした。サイクルシークエンス反応は、BigDye(登録商標)Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems)を用いて行った。配列決定を行った結果、N末端置換グルクロン酸転移酵素のインサート(UGT1A1及びUGT1A9)は、UGT1A7のN末端シグナル配列をもち、それぞれ分子種に特有の正確な配列であることが確認できた。
【0105】
1-2-4-2. 酵母におけるN末端置換グルクロン酸転移酵素の発現ベクターの構築
1-2-4-1で作成したN末端置換グルクロン酸転移酵素断片を含むプラスミド約1μgを37℃で4時間、HindIII処理し、電気泳動後、グルクロン酸転移酵素遺伝子断片をゲルから切り出した。切り出したゲルからWizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてインサート断片を調製した。
【0106】
酵母発現ベクターとして用いるpGYRを37℃で4時間HindIII処理し、電気泳動後、目的サイズ(約11kb)のバンドを切り出した。その後、Wizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてベクター断片を調製した。
【0107】
インサート断片とpGYR-HindIIベクター断片の濃度を電気泳動で確認後、それらのモル比が3:1〜10:1程度になるように混合し、等量のDNA Ligation Kit Ver. 2 (TaKaRa)のsolution Iを加え、16℃、1時間反応を行った。その後、大腸菌JM109株にヒートショック法を用いてトランスフォーメーションを行い、LBプレート(アンピシリン50μg/ml含有)に展開した。得られた大腸菌コロニーのうち数個を選択し、それらを鋳型としてコロニーPCRを行った。DNAポリメラーゼは、Ex Taq(登録商標) DNAポリメラーゼを用いた。プライマーはYGAP-Pプライマー(5’-aatgacaccgtgtggtgatcttcaagg-3’)(配列番号10)と上記リバースプライマー(配列番号20)を用いた。以下の条件でPCRを行った。
【0108】
PCR条件
変性 98℃5分
30サイクル 94℃ 30秒、50℃ 30秒、72℃ 2分30秒
最終伸長 72℃4分
PCR後、電気泳動を行い、予想サイズ(約3kb)に特異的な増幅がみられるクローンを数個得た。
【0109】
得られた大腸菌コロニーを5mlのLB培地(アンピシリン100μg/ml含有)に植菌し、37℃、200rpmで16時間振盪培養を行った。その後アルカリSDS法を用いてプラスミドを抽出した。そして、その一部を37℃で1時間、HindII処理し、電気泳動をしてインサートの導入を再度確認し、2種のN末端シグナル配列置換グルクロン酸転移酵素(UGT1A1及びUGT1A9)を含む酵母発現ベクター(pGYR/UGT1A1及びpGYRUGT1A9)を得た。
【0110】
実施例2
2. UDP-グルコース脱水素酵素及びUDP-グルクロン酸転移酵素発を同時に発現する出芽酵母形質転換体の作成
2-1. 出芽酵母への発現用プラスミドの形質転換
発現用には、出芽酵母(Saccharomyces cerevisiae)AH22株を用いた。AH22株は、L-ヒスチジンとL-ロイシン以外は全ての必須アミノ酸を生合成することが出来る。pGYRはL-ロイシン合成遺伝子(LEU2)を持つため、培地にL-ヒスチジンを添加するとプラスミドを持った酵母のみが増殖できる。また、pAURは抗生物質であるAureobasdin A耐性遺伝子を有しており、Aureobasidin A存在下で形質転換体を選択することが可能である。発現用コンストラクトを酵母AH22株に塩化リチウム法により形質転換した。表2に作成した同時発現株における発現ベクターの組み合わせを示す。以下に形質転換の詳細なプロトコールを示す。
【0111】
<材料>
YPD培地:1%酵母抽出物、2%ポリペプトン、2%グルコース
SDプレート:2%グルコース、0.67%N-base w/o アミノ酸、1.5%寒天、20μg/ml L-ヒスチジン、0.2M LiCl: 10ml(フィルター滅菌)、1M LiCl: 10ml(フィルター滅菌)
70%(w/v) PEG 4000: 10ml(溶解後、10mlにメスアップ)
【0112】
<方法>
30℃で培養したS.cerevisiae AH22株1.0x107細胞を使用し、卓上遠心機で13,000rpm、4分遠心を行い、上清を除いた。ペレットを0.2M LiClで洗浄した(少しボルテックスにかけた)。卓上遠心機で13,000rpm、4分遠心を行い、上清を完全に取り除いた。ペレットを1M LiCl 20μlにピペッティングにより懸濁した。DNA(プラスミド)溶液10μl(0.5〜1μgのプラスミドを含有)を加えた。70%(w/v) PEG 4000を30μl加え、ピペッティングによりよく混合した。40℃で30分間インキュベートした。滅菌水140μlを加えてpGYRベクターの場合にはSDプレート、またはpAURベクターの場合には0.5μg/ml Aureobasidin A を含むYPDプレートに展開し、30℃にてインキュベートした。プレート上で30℃、3日間培養し、3〜5mm程度の大きさのコロニーを数十個得た。また、pGYR及びpAURの同時形質転換体の場合は、0.5μg/ml Aureobasidin A を含むSDプレートに展開し、30℃にてインキュベートした。プレート上で30℃、5日間培養し、3〜5mm程度の大きさのコロニーを数個得た。
【0113】
2-2. UDP-グルコース脱水素酵素及びUDP-グルクロン酸転移酵素タンパク質の酵母内での発現の確認
実施例2-1で得られた形質転換体について、それぞれの菌体内でタンパク質が機能的に発現されているかどうかを、ウエスタンブロット法及びUDP-グルコース脱水素酵素活性測定によって確認した。
【0114】
形質転換体を選択培地で48時間30℃で培養し、得られた酵母菌体をZymolyase処理にした後に、音波破砕を行い遠心操作を用いて細胞質画分を分離した。UDP-グルコース脱水素酵素活性は基質として5mM UDP-グルコースを、補助因子として0.5mMNAD+を用い、50mM Tris-HCl(pH8.6)という条件で37℃、1時間反応を行い、遠心分離により上清を回収した。C18カラムを用いたHPLCによって、20mM酢酸トリエチルアミン(pH7.0)溶離液を流速1ml/minで流し、検出波長260nmでUDP-グルクロン酸を検出した。その結果、UDP-グルコース脱水素酵素遺伝子を挿入した発現ベクター(pGYR/At.UDPGDH) を含む形質転換体菌体抽出液のみでUDP-グルクロン酸合成活性が検出された(図2)。
【0115】
図2は、酵母内発現UDP-グルコース脱水素酵素タンパクによるUDP-グルクロン酸生成を、逆相HPLCを用いて分析した結果である。A:AH22:pAUR(コントロール)、B:AH22:pAUR/At.UDPGDH。シロイヌナズナ由来UDP-グルコース脱水素酵素遺伝子を挿入した発現ベクター形質転換体抽出溶液においてのみ溶出時間11分付近にUDP-グルクロン酸の生成が見られた。
【0116】
また、形質転換体株より得られた酵母菌体をZymolyase処理にした後に、ヒトグルクロン酸転移酵素ファミリー1の共通配列認識ペプチド抗体(文献番号5)及びシロイヌナズナ由来UDP-グルコース脱水素酵素のC末端ペプチド配列(KPLDQWLKDMPALA)認識抗体を用いたウエスタンブロット法により解析した(図3)。酵母内発現UDP-グルクロン酸転移酵素遺伝子及びUDP-グルコース脱水素酵素タンパクの発現を確かめるために、表2の形質転換体1〜4の抽出液タンパクのウエスタンブロット解析を行った。パネル(A):抗UGT1A共通抗体、パネル(B):抗シロイヌナズナUDPGDH抗体。パネル(B)コントロール(AH22:pGYR/UGT1A6)には検出されないタンパクのバンドが1〜3のレーンで見られ、シロイナズナ由来UDP-グルコース脱水素酵素タンパクの発現が確認された。両遺伝子を挿入した発現ベクターを含む形質転換体菌体抽出液のみでそれぞれのタンパク発現が確かめられた。
【0117】
2-3. 酵母菌体におけるUDP-グルクロン酸の産生
実施例2-1で得られたUDP−グルコース脱水素酵素が発現している形質転換体について、酵母菌体内におけるUDP-グルクロン酸の生成を確認した。
【0118】
ラットUDP−グルコース脱水素酵素が発現している形質転換体を選択培地で24時間30℃で培養し、得られた酵母菌体に2.5倍容のクロロホルム:メタノール=3:1を添加し激しく攪拌した後、上層の水相の代謝物を高速液体クロマトグラフィーにより分析した。UDP-グルクロン酸のHPLCによる分析条件は以下のとおりである。カラム:Wako社製 WakoPack Navi C-30-5 (内径3.0mm × 長さ150mm)、検出波長:260nm、流速:0.35ml/min、カラム温度: 37℃、溶出条件: 20mM酢酸トリエチルアミン(pH7.0)
【0119】
図4は、酵母内におけるUDP-グルクロン酸生成を逆相HPLCを用いて分析した結果である。図中のAはUDP-グルクロン酸、UDP-グルコースの標準物質の溶出パターンを示し、Bはコントロール酵母(AH22:pGYR)、CはラットUDP−グルコース脱水素酵素を遺伝子導入した形質転換体酵母の抽出画分を示す。Bでは糖ヌクレオチドとしてはUDP-グルコースしか検出されないのに対して、CではUDP-グルクロン酸のピークが検出された。既知濃度の標準化合物との比較によりUDP−グルコース脱水素酵素が導入された酵母菌体内には、4〜5mMの濃度でUDP-グルクロン酸が生成されていることが確認された。
【0120】
実施例3
3. 酵母形質転換体を用いたグルクロン酸抱合体の製造
3-1. 酵母静止菌体を用いた7ヒドロキシクマリン抱合体の製造
UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素タンパクの同時発現した酵母静止菌体におけるグルクロン酸抱合体の生成を確かめるために、形質転換体を選択培地で48時間30℃で培養し、得られた酵母菌体を1−8%グルコースを含む適当な緩衝溶液に懸濁し、抱合化基質として0.1−1mM 7ヒドロキシクマリンを添加した。30℃の条件下で24時間振盪培養をおこない、培養液に2倍容のクロロホルム:メタノール=3:1を添加し、激しく攪拌した後、上層の水相と下層の有機相を回収し、有機相は減圧乾固し、アセトニトリルに溶解した。それぞれの代謝物を超高速液体クロマトグラフィーにより分析した。条件は以下のとおりである。カラム:nacalai tesque社製 2.5C18-MS-II (内径2.0mm × 長さ100mm)、検出波長:320nm、流速:0.5ml/min、カラム温度:45 ℃、溶出条件:水/アセトニトリル系(0.1%TFAを含む)、10%アセトニトリル(4分間)、10-70 %アセトニトリル直線濃度勾配(6分間)の後、70-10%アセトニトリル(2分間)、10%アセトニトリル(3分間)
【0121】
3-1-1. 種々のヒトUDP-グルクロン酸転移酵素との同時発現酵母における抱合体生成
シロイヌナズナ由来UDP-グルコース脱水素酵素を含むヒトUDP-グルクロン酸転移酵素との同時発現酵母株 (AH22: pAUR/At.UDPGDH + pGYR/hUGT1A1, 1A6,1A7,1A8,1A9)における7ヒドロキシクマリン抱合体の生成を確かめた。
【0122】
図5に示すように、いずれのヒトUDP-グルクロン酸転移酵素分子種(UGT1A1,1A6,1A7,1A8,1A9) においてもUDP-グルコース脱水素酵素との同時発現により、酵母菌体に直接添加した7ヒドロキシクマリンのグルクロン酸抱合体の生成が見られた。したがって、UDP-グルクロン酸転移酵素とUDP-グルコース脱水素酵素を同時発現させた酵母菌体内において、UDP-グルコース脱水素酵素によってUDP-グルクロン酸が供給され、抱合化基質へのグルクロン酸転移反応がUDP-グルクロン酸転移酵素によって触媒されることが確かめられた。また、UDP-グルクロン酸転移酵素のいずれの分子種に関しても触媒反応がみられたことから、同時発現させるUDP-グルクロン酸転移酵素分子種を選択することにより多様な抱合体調製が可能である。
【0123】
3-1-2. UGT1A6同時発現株における抱合体産生の時間変化
実施例3-1-1でもっとも変換効率の高かった同時発現酵母株 (AH22: pAUR/At.UDPGDH+pGYR/hUGT1A6) における7ヒドロキシクマリン抱合体の生成の時間依存性を調べた(図6)。
【0124】
図6に示されるように、加えた基質(0.5mM 7OHC:△)は反応時間に依存して減少し、抱合体(7GC:○)は直線的に増加してゆき、24時間経過後にはほぼ100%抱合体へ変換されることが示された。
【0125】
3-1-3. UGT1A6同時発現株における抱合体産生のグルコース濃度の影響
実施例3-1-1でもっとも変換効率の高かった同時発現酵母株 (AH22: pAUR/At.UDPGDH+pGYR/hUGT1A6) における7ヒドロキシクマリン抱合体の生成におけるグルコース濃度を調べた(図7)。
【0126】
図7に示されるように、添加したグルコース濃度に依存して抱合体の産生量は増加し、8%の濃度でほぼ100%変換を示した。さらに、生成した抱合体の培地と菌体への分布を調べると、90%以上の抱合体が培地中に分泌されていることがわかった。
【0127】
3-1-4.同時発現株における発現ベクターの組み合わせの影響
同時発現系による抱合体の産生能力における発現ベクターの組み合わせの影響を調べるために、表2に示した同時発現株(1〜4)を選択培地で24時間30℃で培養し、得られた酵母菌体を8%グルコースを含む適当な緩衝溶液に懸濁し、抱合化基質として1mM 7ヒドロキシクマリンを添加した。30℃の条件下で24時間振盪培養をおこない抱合体の培地中の濃度を測定した(図8)。
【0128】
図8に示されるように形質転換体1〜3の比較から発現ベクターの組み合わせに依存せず、抱合体産生能力を示すことが明らかとなった。また、形質転換体3及び4の比較から、酵母に導入されたUDP-グルコース脱水素酵素は生物種の由来に関係なくUDP-グルクロン酸の供給に関与し、抱合体生成に効率よく機能することが示された。
【0129】
3-2. 酵母静止菌体を用いたケルセチン抱合体の製造
シロイヌナズナ由来UDP-グルコース脱水素酵素を含むヒトUDP-グルクロン酸転移酵素との同時発現酵母株 (pAUR/At.UDPGDH+pGYR/hUGT1A1)におけるケルセチン抱合体の生成を確かめた。形質転換体を選択培地で48時間30℃で培養し、得られた酵母菌体を8%グルコースを含む0.1Mリン酸緩衝溶液(pH7.4)に懸濁し、抱合化基質として0.2mM ケルセチンを添加した。30℃の条件下で24時間振盪培養をおこない、培養液に2倍容のクロロホルム:メタノール=3:1を添加し、激しく攪拌した後、上層の水相と下層の有機相を回収し、有機相は減圧乾固し、アセトニトリルに溶解した。ケルセチン抱合体のHPLCによる分析条件は以下のとおりである。カラム:Wako社製 WakoPack Navi C-30-5(内径3.0mm×長さ150mm)、検出波長:370nm、流速:0.4ml/min、カラム温度:37℃、溶出条件:水/アセトニトリル系(0.5% リン酸を含む)、18 %アセトニトリル(10分間)、18-55%アセトニトリル直線濃度勾配(10分間)の後、55%アセトニトリル(5 分間)
【0130】
結果を図9に示す。シロイヌナズナ由来UDP-グルコース脱水素酵素を含むヒトUDP-グルクロン酸転移酵素との同時発現酵母株 (pAUR/At.UDPGDH+pGYR/hUGT1A1) におけるケルセチン抱合体の生成を確かめた。A:AH22:pGYR/UGT1A1、B:AH22/pGYRUGT1A1+pAUR/At.UDPGDH、C:AH22:pAUR/At.UDPGDH
【0131】
ケルセチンは分子内に抱合化をうける水酸基を4箇所有し、複数の抱合体が産生される。図9に示すように同時発現株においてのみB環の3’位及び4’位水酸基に特異的なケルセチン抱合体の産生が見られ、特に3’位抱合体が優勢に産生された。このようにUDP-グルコース脱水素酵素と同時発現させるUDP-グルクロン酸転移酵素分子種を選択することで、複数の水酸基を有する化合物に対して特異的な抱合体を製造することが可能である。
【0132】
3-3. 酵母静止菌体を用いたマイコフェノール酸抱合体の製造
シロイヌナズナ由来UDP-グルコース脱水素酵素を含むヒトUDP-グルクロン酸転移酵素との同時発現酵母株 (pAUR/At.UDPGDH+pGYR/hUGT1A9)におけるマイコフェノール抱合体の生成を確かめた。形質転換体を選択培地で48時間30℃で培養し、得られた酵母菌体を8%グルコース溶液に懸濁し、抱合化基質として0.2mM マイコフェノール酸を添加した。30℃の条件下で24時間振盪培養をおこない、培養液に2倍容のクロロホルム:メタノール=3:1を添加し、激しく攪拌した後、上層の水相と下層の有機相を回収し、有機相は減圧乾固し、アセトニトリルに溶解した。代謝物を超高速液体クロマトグラフィーにより分析した。条件は以下のとおりである。カラム:nacalai tesque社製 2.5C18-MS-II(内径2.0mm×長さ100mm)、検出波長:250nm、流速:0.5ml/min、カラム温度:45℃、溶出条件:水/アセトニトリル系(0.1%TFAを含む)、20-40 %アセトニトリル直線濃度勾配(7分間)の後、40%アセトニトリル(2分間)、40-20%アセトニトリル(2分間)、20%アセトニトリル(2分間)
【0133】
シロイヌナズナ由来UDP-グルコース脱水素酵素を含むヒトUDP-グルクロン酸転移酵素との同時発現酵母株 (pAUR/At.UDPGDH+pGYR/hUGT1A9) におけるマイコフェノール酸抱合体の生成を確かめた。結果を図10に示す。
A:標準物質(MPA:マイコフェノール酸、G1:O-glucuronide, G2: Acyl glucuronide),
B:AH22/pGYRUGT1A9+pAUR/At.UDPGDH,
C:AH22:pAUR/At.UDPGDH(コントロール)
【0134】
マイコフェノール酸は、分子内にフェノール性水酸基とカルボキシル基を持ち2種の抱合体が産生される。図10に示すようにUGT1A9との同時発現株によってのみフェノール性水酸基に特異的なマイコフェノール酸抱合体(o-glucuronide) の産生が見られた。このようにUDP-グルコース脱水素酵素と同時発現させるUDP-グルクロン酸転移酵素分子種を選択することで、異なる抱合化官能基を有する化合物に対して特異的な抱合体を製造することが可能である。
【0135】
3-4. 酵母静止菌体を用いたアシルグルクロン酸抱合体の製造
グルクロン酸転移酵素を発現した酵母静止菌体におけるアシルグルクロン酸抱合体の生成を確かめるために、形質転換体を選択培地で24時間30℃で培養し、得られた酵母菌体を8%グルコースを含む適当な緩衝溶液に懸濁し、抱合化基質として1mM メフェナム酸を添加した。30℃の条件下で24時間シントウ培養をおこない、培養液に2.5倍容のクロロホルム:メタノール=3:1を添加し、激しく攪拌した後、上層の水相の代謝物を超高速液体クロマトグラフィーにより分析した。条件は以下のとおりである。カラム:Nacalai Tesque社製 2.5C18-MS-II (内径2.0mm × 長さ100mm)、検出波長:320nm、流速:0.5ml/min、カラム温度:45℃、溶出条件:水/アセトニトリル系(0.1%TFAを含む)、5-40%アセトニトリル直線濃度勾配(5分間)、40-100%アセトニトリル直線濃度勾配(3分間)の後、100%アセトニトリル(1分間)、100-5%アセトニトリル直線濃度勾配、5%アセトニトリル(3分間)
【0136】
図11はヒトグルクロン酸転移酵素分子種を発現した酵母静止菌体を用いてメフェナム酸のアシル抱合体の生産量を乾燥菌体重量あたりの日生産量を示したものである。UGT1A1,1A7,1A8,1A9及び1A10がアシル抱合体への変換能を示し、特に1A8,1A9,1A10が高い抱合能をもつことが示された。
【0137】
3-5. 酵母静止菌体を用いたNグルクロン酸抱合体の製造
グルクロン酸転移酵素を発現した酵母静止菌体におけるNグルクロン酸抱合体の生成を確かめるために、形質転換体を選択培地で24時間30℃で培養し、得られた酵母菌体を8%グルコースを含む適当な緩衝溶液に懸濁し、抱合化基質として0.5mM タモキシフェンを添加した。30℃の条件下で24時間シントウ培養をおこない、培養液に2.5倍容のクロロホルム:メタノール=3:1を添加し、激しく攪拌した後、上層の水相の代謝物を高速液体クロマトグラフィーにより分析した。
タモキシフェン抱合体のHPLCによる分析条件は以下のとおりである。カラム:Wako社製 WakoPack Navi C-30-5 (内径3.0mm × 長さ150mm)、検出波長:254nm、流速:0.5ml/min、カラム温度: 37℃、溶出条件:100mM酢酸アンモニウム(pH5.0)/アセトニトリル系、25 %アセトニトリル(5分間)、25-75%アセトニトリル直線濃度勾配(25分間)の後、75%アセトニトリル(10分間)、75-25%アセトニトリル直線濃度勾配(5分間)の後、25%アセトニトリル(5分間)
【0138】
抗がん剤として知られるタモキシフェンは分子内に抱合化を受ける窒素原子が存在する化合物である。図12はヒトグルクロン酸転移酵素分子種(UGT1A4及び1A9)を発現した酵母静止菌体を用いてタモキシフェン抱合体(Nグルクロン酸抱合体)生成を分析したHPLCの溶出パターンを示したものである。Nグルクロン酸抱合能を有するUGT分子種であるUGT1A4を発現した酵母静止菌体をもちいた場合についてのみ抱合体(溶出時間11.8分)の産生が見られた。
【0139】
実施例4
出芽酵母と分裂酵母におけるグルクロン酸抱合体産生能の比較
出芽酵母(Saccharomyces cerevisiae)におけるグルクロン酸抱合体産生能を分裂酵母(Schizosaccharomyces pombe)の系と比較するために、表5に示す形質転換出芽酵母(表2に示す形質転換体No.4、UGT分子種: UGT1A1(N末端シグナル配列置換グルクロン酸転移酵素(UGT1A1)を含む酵母発現ベクター(pGYR/UGT1A1)を用いた)またはUGT1A6)を選択培地で、30℃で24時間培養し、得られた酵母菌体を、8%グルコースを含む緩衝溶液に懸濁し、抱合化基質として0.5mM 4メチルウンベリフェロンを添加した。30℃の条件下で24時間振盪培養を行い、培養液に2倍容のクロロホルム:メタノール=3:1を添加し、激しく攪拌した後、上層の水相と下層の有機相を回収し、有機相は減圧乾固し、アセトニトリルに溶解した。それぞれの代謝物を超高速液体クロマトグラフィーにより分析した。条件は以下のとおりである。
【0140】
カラム:nacalai tesque社製 2.5C18-MS-II (内径2.0mm×長さ100mm)、検出波長:320nm、流速:0.5ml/min、カラム温度:45℃、溶出条件:水/アセトニトリル系(0.1%TFAを含む)、10%アセトニトリル(4分間)、10-70%アセトニトリル直線濃度勾配(6分間)の後、70-10%アセトニトリル(2分間)、10%アセトニトリル(3分間)
【0141】
表6には出芽酵母及び分裂酵母(文献値)における4メチルウンベリフェロングルクロン酸抱合体産生能比較を示した。出芽酵母の抱合体産生能力に関して分裂酵母を用いた場合と比べて、1日あたり抱合体濃度はUGT1A1に関しては10倍、UGT1A6では50倍以上高い値を示した。また、酵母乾燥重量あたりの抱合体生成量も20〜100倍以上高い値を示した。このように複数のUGT分子種に関して分裂酵母を宿主として用いた場合よりも、出芽酵母におけるグルクロン酸抱合体産生系が有利なことが示された。
【0142】
【表6】
【0143】
実施例5
5-1. シトクロムP450を含む同時発現株の静止菌体を用いた7ヒドロキシクマリン抱合体の製造
生体で生じる抱合体の多くは、その前段階としてシトクロムP450によるモノオキシゲナーゼ反応を受け、抱合化をうける官能基としてはたらく水酸基の導入を受ける。酵母においてシトクロムP450及びUDP-グルクロン酸転移酵素の同時発現株より調製されたミクロソーム画分を用いて試験管内の反応により、7エトキシクマリンを基質として、シトクロムP450による脱エチル反応による7ヒドロキシクマリン生成、引き続いてUDP-グルクロン酸転移酵素による抱合化が観測された(文献番号6)。このP450とUDP-グルクロン酸転移酵素の連続的な変換反応を酵母菌体内で可能にするために、pAUR-At.UDPGDH/UGT1A6及びpGYR/ratCYP1A1 の発現ベクターを同時に形質転換した酵母株(表2,形質転換体5)を作成した。形質転換体を選択培地で48時間30℃で培養し、得られた酵母菌体を8%グルコースを含む溶液に懸濁し、抱合化基質として1mM 7エトキシクマリンを添加した。30℃の条件下で72時間振盪培養をおこない、培養液に生成される抱合体生成の時間変化を調べた(図13)。
【0144】
図13にみられるように、添加した7エトキシクマリンはP450により脱エチル化をうけ、7ヒドロキシクマリン(●)に変換された後、UGTによって抱合化を受けて抱合体(○)に変換された。抱合体の生成においては時間的なラグは見られず酵母内にて生成された中間体である7ヒドロキシクマリンが抱合化反応の基質として供給され効率的に抱合体に変換されているものと思われる。したがって、シトクロムP450を含む同時発現株は抱合化基質の前駆体から効率よく抱合体を調製する系として有用である。
【0145】
5-2. ヒト由来シトクロムP450を含む同時発現株の静止菌体を用いた7-エトキシクマリン由来抱合体の製造
実施例3で示したように抱合体の多くはシトクロムP450の代謝に引き続きグルクロン酸抱合をうけて産生される。そこでとくにヒト代謝物を製造するためにヒト由来シトクロムP450を含む同時発現酵母株の構築をおこない7エトキシクマリンの代謝解析を行なった。
【0146】
5-2-1 ヒト由来シトクロムP450発現ベクター(pPYR)の構築
ヒト由来シトクロムP450を含む同時発現株を構築するために、出芽酵母グリセロールリン酸キナーゼ由来のプロモーター及びターミネーター領域をクローニングして前述のpPYRの発現調節領域を置換した自律複製型酵母発現ベクター(pPYR)を作成した(図14)。
【0147】
出芽酵母AH22株より常法にてゲノムDNAを調製し鋳型DNAとした。以下の示す配列を有するプライマーDNAを用いてPCR法によってグリセロールリン酸キナーゼ(PGK1)由来のプロモーター及びターミネーター領域のクローニングを行った。
【0148】
DNAポリメラーゼとしてはKOD-plus-(TOYOBO)を用いた。反応液組成は製造業者の指示に従い、以下に示すプライマーと反応条件でPCRを行った。
PGK1由来のプロモーター領域
フォワード配列 : 配列番号 21
リバース配列 : 配列番号 22
PGK1由来のターミネーター領域
フォワード配列 : 配列番号 23
リバース配列 : 配列番号 24
【0149】
【0150】
PCR条件
変性 94℃ 2分
5サイクル 94℃ 15秒、42℃ 30秒、68℃1分45秒
30サイクル 94℃ 15秒、50℃ 30秒、68℃1分45秒
最終伸長 68℃10分
【0151】
PCR産物を1%アガロースゲルで電気泳動した結果、目的サイズ(グリセロールリン酸キナーゼ由来のプロモーター領域:約1.5kb及びグリセロールリン酸キナーゼ由来のターミネーター領域:0.3kb)に特異的な増幅がみられた。(以下、1%アガロースゲルでの電気泳動は、単に電気泳動と略称する)。PCR増幅溶液を電気泳動して、DNA断片をゲルから分離した後、TAクローニングキット(Target cloneTM-plus-,TOYOBO社)を用いてpTAベクターに挿入した。このクローニングされた遺伝子のシークエンスを行い遺伝子配列を解析したところ、配列番号25(Genebank Acc.No. BK006937, TPA: Saccharomyces cerevisiae S288c chromosome III, complete sequence; 136264〜137743)及び26(Genebank Acc.No. BK006937, TPA: Saccharomyces cerevisiae S288c chromosome III, complete sequence; 138995〜139270)に示した塩基配列であることを確認した。
【0152】
【0153】
PGK1プロモーターあるいはターミネーター領域を含むプラスミド約1μgを37℃で4時間、NotI及びHindIII処理し、電気泳動後、遺伝子断片をゲルから切り出した。切り出したゲルからWizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてインサート断片を調製した。
【0154】
サブクローニングベクターとして用いるpGEM-T-Easy Vector (Promega)を37℃で4時間NotI処理し、電気泳動後、目的サイズ(約3kb)のバンドを切り出した。その後、Wizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてベクター断片を調製した。
【0155】
2種のインサート断片とpGEM-NotIベクター断片の濃度を電気泳動で確認後、それらのモル比が3:1〜10:1程度になるように混合し、等量のDNA Ligation Kit Ver. 2 (TaKaRa)のsolution Iを加え、16℃、1時間反応を行った。その後、大腸菌JM109株にヒートショック法を用いてトランスフォーメーションを行い、LBプレート(アンピシリン50μg/ml含有)に展開した。得られた大腸菌コロニーのうち数個を選択し、それらを鋳型としてコロニーPCRを行った。DNAポリメラーゼは、Ex Taq(登録商標) DNAポリメラーゼを用いた。インサート確認のためのコロニーPCRには、プライマーとして配列番号21と24を用いた。以下の条件でPCRを行った。
【0156】
PCR条件
変性 98℃5分
30サイクル 94℃ 30秒、50℃ 30秒、72℃ 2分30秒
最終伸長 72℃4分
【0157】
PCR後、電気泳動を行い、予想サイズ(約1.8kb)に特異的な増幅がみられるクローンを数個得た。得られた大腸菌コロニーを5mlのLB培地(アンピシリン100μg/ml含有)に植菌し、37℃、200rpmで16時間振盪培養を行った。その後アルカリSDS法を用いてプラスミドを抽出した。そして、その一部を37℃で1時間、NotI及びHindIII処理し、電気泳動をおこない予想されるインサートの導入を再度確認し、発現分子種挿入部位にHindIIIをもつPGK1由来プロモーター及びターミネーター領域を得た(pGEM/PGK-1-P/T)。
【0158】
PGK1プロモーター及びターミネーター領域を含むプラスミド約1μgを37℃で4時間、NotI処理し、電気泳動後、遺伝子断片をゲルから切り出した。切り出したゲルからWizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてインサート断片を調製した。
【0159】
pGYRを37℃で4時間NotI処理し、電気泳動後、目的サイズ(約9kb)のバンドを切り出した。その後、Wizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてベクター断片を調製した。
【0160】
インサート断片とpGYR-NotIベクター断片の濃度を電気泳動で確認後、それらのモル比が3:1〜10:1程度になるように混合し、等量のDNA Ligation Kit Ver. 2(TaKaRa)のsolution Iを加え、16℃、1時間反応を行った。その後、大腸菌JM109株にヒートショック法を用いてトランスフォーメーションを行い、LBプレート(アンピシリン50μg/ml含有)に展開した。得られた大腸菌コロニーのうち数個を選択し、それらを鋳型としてコロニーPCRを行った。DNAポリメラーゼは、Ex Taq(登録商標) DNAポリメラーゼを用いた。インサート確認のためのコロニーPCRには、プライマーとして配列番号21と24を用いた。以下の条件でPCRを行った。
【0161】
PCR条件
変性 98℃5分
30サイクル 94℃ 30秒、50℃ 30秒、72℃ 2分30秒
最終伸長 72℃4分
【0162】
PCR後、電気泳動を行い、予想サイズ(約1.8kb)に特異的な増幅がみられるクローンを数個得た。得られた大腸菌コロニーを5mlのLB培地(アンピシリン100μg/ml含有)に植菌し、37℃、200rpmで16時間振盪培養を行った。その後アルカリSDS法を用いてプラスミドを抽出した。そして、その一部を37℃で1時間、NotI及びHindIII処理した後、電気泳動をおこない予想されるインサートの導入を再度確認し、PGK1由来プロモーター及びターミネーター領域を有する自律複製型酵母発現ベクター(pPYR)を得た。
【0163】
ヒトシトクロムP450断片を含むプラスミド約1μgを37℃で4時間、HindIII処理し、電気泳動後、遺伝子断片をゲルから切り出した。切り出したゲルからWizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてインサート断片を調製した。
【0164】
pPYRを37℃で4時間HindIII処理し、電気泳動後、目的サイズ(約11kb)のバンドを切り出した。その後、Wizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてベクター断片を調製した。
【0165】
ヒトチトクロムP450遺伝子を含むインサート断片とpPYR-NotIベクター断片の濃度を電気泳動で確認後、それらのモル比が3:1〜10:1程度になるように混合し、等量のDNA Ligation Kit Ver. 2 (TaKaRa)のsolution Iを加え、16℃、1時間反応を行った。その後、大腸菌JM109株にヒートショック法を用いてトランスフォーメーションを行い、LBプレート(アンピシリン50μg/ml含有)に展開した。 得られた大腸菌コロニーのうち数個を選択し、それらを鋳型としてコロニーPCRを行った。DNAポリメラーゼは、Ex Taq(登録商標) DNAポリメラーゼを用いた。インサート確認のためのコロニーPCRには、プライマーとして配列番号21と挿入するシトクロムP450内部リバース配列を用いた。以下の条件でPCRを行った。
【0166】
PCR条件
変性 98℃5分
30サイクル 94℃ 30秒、50℃ 30秒、72℃ 2分30秒
最終伸長 72℃4分
【0167】
PCR後、電気泳動を行い、予想サイズに特異的な増幅がみられるクローンを数個得た。得られた大腸菌コロニーを5mlのLB培地(アンピシリン100μg/ml含有)に植菌し、37℃、200rpmで16時間振盪培養を行った。その後アルカリSDS法を用いてプラスミドを抽出した。そして、その一部を37℃で1時間、NotI及びHindIII処理した後、電気泳動をおこない予想されるインサートの導入を再度確認し、ヒトシトクロムP450を含む自律複製型酵母発現ベクター(pPYR/CYP)を得た。
【0168】
5-2-2 ヒト由来シトクロムP450を含む同時発現株の静止菌体を用いた7-エトキシクマリン由来抱合体の製造
ヒトシトクロムP450とUDP-グルクロン酸転移酵素によるヒト特異的な連続的な変換反応を酵母菌体内で可能にするために、pAUR-rat.UGDH/UGT及びpPYR/humanCYPの発現ベクターを同時に形質転換した酵母株を作成した。
形質転換体を選択培地で24時間30℃で培養し、得られた酵母菌体を8%グルコースを含む溶液に懸濁し、抱合化基質として1mM 7エトキシクマリンを添加した。30℃の条件下で24時間シントウ培養をおこない、培養液に生成される代謝物を調べた(図15、表7)。
【0169】
【表7】
【0170】
図15にはヒトシトクロムP450(CYP2D6)とUDP-グルクロン酸転移酵素(UGT1A8)を同時発現させた酵母株静止菌体における7エトキシクマリン代謝物のHPLC分析パターンを示す。添加した7エトキシクマリンはヒトシトクロムP450により脱エチル化あるいは3位水酸化をうけ、7ヒドロキシクマリン(7OHC)あるいは3ヒドロキシ-7エトキシクマリン(3OH−7EC)に変換された後、UGT1A8によってそれぞれ抱合化を受けて抱合体(7GCあるいは3G−7EC)に変換された。
また、表7にはヒトシトクロムP450とUDP-グルクロン酸転移酵素の異なる分子種の組み合わせにより構築した同時発現酵母株を用いた7エトキシクマリンの抱合体製造結果を示す。いずれの組み合わせにおいてもP450の代謝物である7OHCあるいは3OH−7ECの抱合体の生成が見られた。
【産業上の利用可能性】
【0171】
医薬品開発におけるヒト体内における医薬品代謝物の解析に関する分野に有用である。
【技術分野】
【0001】
本発明はグルクロン酸抱合体の製造方法、並びにそれに用いる出芽酵母発現ベクター及び形質転換体に関する。より詳しくは、出芽酵母発現ベクターは、UDP-グルクロン酸転移酵素及び/又はUDP-グルコース脱水素酵素を導入した出芽酵母発現ベクターであり、形質転換体は、前記出芽酵母発現ベクターで形質転換された出芽酵母である。
【背景技術】
【0002】
医薬品開発において、ヒト体内における医薬品代謝物の解析は重要である。グルクロン酸抱合体は医薬品の解毒代謝物として排泄されるが、その一部は反応性代謝物となり毒性を示す可能性が指摘されている。例えば、グルクロン酸抱合体の内、エステル型抱合であるアシル抱合体は、反応性代謝物となり薬物性肝障害の原因となる可能性がある(非特許文献1)。
【0003】
そこで、抱合体自身の安全性評価が必要であるが、有機合成法による部位特異的抱合化はきわめて困難である。そこで、酵素や微生物等を用いて効率良く目的とする抱合体を製造する方法が切望されている。現在、動物肝臓由来ミクロソーム画分を用いた抱合体調製が実用化されているが、生産性や適用範囲が十分とは言えない。また、昆虫細胞発現系を用いたヒトグルクロン酸転移酵素がすでに市販されているが、酵素源として抱合体調製に用いるにはコスト面からも現実的ではない。
【0004】
本発明者らは、これまでに出芽酵母を用いたグルクロン酸転移酵素の発現系を構築し、抱合体調製の酵素源として用いる、医薬品代謝物である抱合体の酵素的合成を提案してきた(非特許文献2)。
【0005】
最近、Draganら(非特許文献7、特許文献1)は分裂酵母(Schizosaccharomyces pombe(S.pombe))を用いたUGTを発現する分裂酵母菌体からグルクロン酸抱合体を産生する系を構築した。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】WO2010/031875
【非特許文献】
【0007】
【非特許文献1】Bailey MJ, Dickinson RG. 1333963959742_0.Pubmed_RVDocSum Chem Biol Interact. 145, 117-37. (2003)
【非特許文献2】S. Ikushiro, M. Sahara, Y. Emi, Y. Yabusaki, T. Iyanogi: Functional co-expression of xenobiotic metabolizing enzymes, rat cytochrome P450 1A1 and UDP-glucuronosylransferase 1A6. Biochimica et Biophysica Acta1672 (2004) 86-92
【非特許文献3】Mackenzie, P. I , Walter Bock, K. ,Burchell, B. ,Guillemette, C. , Ikushiro, S. I. ,Iyanagi, T. ,Miners, J.O. ,Owens, I.S. and Nebert, D.W.: Nomenclature Update for the Mammalian UDP Glycosyltransferase (UGT) Gene Superfamily. Pharmacogenetics and Genomics, 10, 677-685 (2005)
【非特許文献4】生城真一、衣斐義一、井柳堯: UDP-グルクロン酸転移酵素:薬物代謝における最近の進歩, 肝臓, 42 , pp.297-301(2001)
【非特許文献5】T. Sakaki, M. Akiyoshi-Shibata, Y. Yabusaki, H. Ohkawa, Organellatargeted expression of rat liver cytochrome P450c27 in yeast: genetically engineered alteration of mitochondrial P450 into a microsomal form creates a novel functional electron transport chain, J. Biol.Chem. 26716497-16502. (1992)
【非特許文献6】Ikushiro. S., Emi, Y., Kato, Y., Yamada, S. and Sakaki, T.: Monospecific antipeptide antibodies against human hepatic UDP- glucuronosyltransferase 1A subfamily ( UGT1A) isoforms. Drug Metabolism and Pharmacokinetics, 21, 70-75 (2006)
【非特許文献7】Dragan CA, Buchheit D, Bischoff D, Ebner T, Bureik M .:Glucuronide production by whole-cell biotransformation using genetically engineered fission yeast S. pombe. Drug Metab Dispos. 38 509-515. (2010)
【非特許文献8】Jo Wixon :Featured Organism: Schizosaccharomyces pombe, the fission yeast. Comp Funct Genom, 3: 194-204 (2002)
【非特許文献9】Esben H. Hansen,Birger Lindberg Moller, Gertrud R. Kock,Camilla M. Bu¨nner,Charlotte Kristensen,Ole R. Jensen,Finn T. Okkels,Carl E. Olsen,Mohammed S. Motawia,and Jorgen Hansen1: De Novo Biosynthesis of Vanillin in Fission Yeast(Schizosaccharomyces pombe) and Baker’s Yeast(Saccharomyces cerevisiae) APPLIED AND ENVIRONMENTAL MICROBIOLOGY, 75,2765-2774 (2009)
【非特許文献10】Tony K.L. Kiang, Mary H.H. Ensom, Thomas K.H. Chang: UDP-glucuronosyltansferases and clinical drug-drug interactions. Pharmacology & Therapeutics 106 97-132 (2005)
【発明の概要】
【発明が解決しようとする課題】
【0008】
しかしながら、酵母内で発現させて得られるヒト由来UDP-グルクロン酸転移酵素(UGT)を酵素源として試験管内における酵素変換にて抱合体を調製する方法は、生産性や適用範囲が十分とは言えなかった。特に糖供与体として用いるUDP-グルクロン酸は高価であり、抱合体調製に際してコスト低減が可能な方法の出現が待たれている。
【0009】
さらに、非特許文献7に記載の分裂酵母(S.pombe)を用いた系では総じてその生産性は低く、依然として実用性に乏しかった。
【0010】
そこで本発明の目的は、上記方法に代わる生産性に優れた新たなグルクロン酸抱合体の製造方法とこの製造方法に用いる新たな手段を提供することにある。
【課題を解決するための手段】
【0011】
そこで、本発明者らは、出芽酵母(Saccharomyces cerevisiae(S.cerevisiae))を宿主として用い、かつUGTを発現する出芽酵母の代謝系を改変して、糖供与体であるUDP-グルクロン酸産生を可能にすることを企画した。しかし、出芽酵母は、抱合体産生に必要とされるUDP-グルクロン酸の生産能力を欠失しているため、菌体内での抱合体への直接変換は不可能であった。そこで出芽酵母にUDP-グルクロン酸の生産能力を付与するために、他生物由来のUDP-グルコース脱水素酵素遺伝子を導入した。その結果、出芽酵母内においてUDP-グルクロン酸の産生が見られた。さらに、このUDPGDH導入出芽酵母においてUGT分子種を同時に発現させることで、出芽酵母菌体に添加した基質の抱合体への直接変換が高い収率で得られた。このようにして、本発明者は出芽酵母(Saccharomyces cerevisiae(S.cerevisiae))を宿主として用いることにより出芽酵母菌体内での抱合体産生能を著しく向上させた抱合体変換系の構築に成功した。
【0012】
出芽酵母(S.cerevisiae)と分裂酵母(S.pombe)は、酵母に分類されてはいるが、3〜4億年前に分岐したと考えられ、両者の生物種としての違いは、分裂酵母と動物の違いに比べられるほど大きいと言われている(非特許文献8)。両者は、ゲノムサイズ(出芽酵母12Mb、分裂酵母14Mb)や遺伝子数(出芽酵母7000程度、分裂酵母5000程度)は似通っているが、染色体数(一倍体として、出芽酵母17、分裂酵母3)や増殖方法(出芽酵母は出芽、分裂酵母は分裂)に著しい違いが認められ、個々の遺伝子の相同性も低い。さらに、哺乳動物由来の異種タンパク質の発現についても、状況は大きく異なっており、分裂酵母(S.pombe)で高発現したからといって出芽酵母(S.cerevisiae)で高発現するとは限らない(非特許文献9)。このような状況であるので、非特許文献7に記載のように、分裂酵母(S.pombe)でUDPGDHとUGTの同時発現に成功したからといって、それをそのまま出芽酵母(S.cerevisiae)に適用しても、UDPGDHとUGTを同時発現できることは限らない。
【0013】
本発明は以下の通りである。
[1]
UDP-グルコース脱水素酵素をコードする遺伝子及びUDP-グルクロン酸転移酵素をコードする遺伝子を発現可能に挿入して形質転換した出芽酵母。
[2]
シトクロムP450遺伝子をコードする遺伝子をさらに発現可能に挿入して形質転換した、[1]に記載の出芽酵母。
[3]
以下の(A)〜(G)のいずれかから選ばれる形質転換出芽酵母。
(A)UDP-グルコース脱水素酵素発現ベクター及びUDP-グルクロン酸転移酵素発現ベクターで形質転換した出芽酵母、
(B)UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクターで形質転換した出芽酵母、
(C)UDP-グルコース脱水素酵素発現ベクター、UDP-グルクロン酸転移酵素発現ベクター及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母、
(D)UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクター、及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母、
(E)UDP-グルクロン酸転移酵素発現ベクター、及びUDP-グルコース脱水素酵素及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母、
(F)UDP-グルクロン酸転移酵素及びシトクロムP450遺伝子発現ベクター、及びUDP-グルコース脱水素酵素発現ベクターで形質転換した出芽酵母、並びに
(G)UDP-グルコース脱水素酵素、UDP-グルクロン酸転移酵素及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母
[4]
UDP-グルコース脱水素酵素発現ベクターが、出芽酵母発現ベクターにUDP-グルコース脱水素酵素遺伝子を発現可能に挿入したものであり、
UDP-グルクロン酸転移酵素発現ベクターが、出芽酵母発現ベクターにUDP-グルクロン酸転移酵素遺伝子を発現可能に挿入したものであり、
UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクターが、出芽酵母発現ベクターにUDP-グルクロン酸転移酵素遺伝子及びUDP-グルコース脱水素酵素含を発現可能に挿入したものである、[3]に記載の形質転換出芽酵母。
[5]
出芽酵母発現ベクターが自律複製型ベクターまたは染色体組み込み型ベクターである、[4]に記載の形質転換出芽酵母。
[6]
UDP-グルコース脱水素酵素遺伝子が動物または植物由来の遺伝子である、[1]、[2]、[4]〜[5]のいずれかに記載の形質転換出芽酵母。
[7]
UDP-グルコース脱水素酵素遺伝子がシロイヌナズナ由来遺伝子またはラット由来遺伝子である、[1]、[2]、[4]〜[5]のいずれかに記載の形質転換出芽酵母。
[8]
UDP-グルクロン酸転移酵素遺伝子が哺乳動物由来の遺伝子である、[1]、[2]、[4]〜[7]のいずれかに記載の形質転換出芽酵母。
[9]
UDP-グルクロン酸転移酵素遺伝子がヒト由来の遺伝子である、[1]、[2]、[4]〜[7]のいずれかに記載の形質転換出芽酵母。
[10]
シトクロムP450遺伝子発現ベクターが、出芽酵母発現ベクターにシトクロムP450遺伝子を発現可能に挿入したものである、[3]〜[9]のいずれかに記載の形質転換出芽酵母。
[11]
前記UDP-グルクロン酸転移酵素発現ベクターは、UDP-グルクロン酸転移酵素遺伝子が、発現量がUGT1A7の50%以下である低発現量グルクロン酸転移酵素遺伝子である場合、
前記低発現量グルクロン酸転移酵素遺伝子は、シグナル配列遺伝子を、発現量がUGT1A7の80%以上である高発現グルクロン酸転移酵素のシグナル配列遺伝子と置換したものであるか、または
シグナル配列遺伝子を(A)以下に示すアミノ酸配列(a)〜(c)のいずれかをコードする遺伝子、(B)アミノ酸配列(a)〜(c)のアミノ酸の1〜5個が置換若しくは欠失したアミノ酸配列のいずれかをコードする遺伝子、または(C)1〜5個のアミノ酸がアミノ酸配列(a)〜(c)のアミノ酸に付加されたアミノ酸配列のいずれかをコードする遺伝子と置換したものであり、但し、(B)の置換若しくは欠失したアミノ酸配列および(C)の付加されたアミノ酸配列をコードする遺伝子は、シグナル配列遺伝子とした場合にグルクロン酸転移酵素の発現量が野生型の80%以上である、[1]〜[10]のいずれかに記載の形質転換出芽酵母。
(a) MARAGWTGLLPLYVCLLLTCGFAKAG(配列番号1)
(b) MACLLRSFQRISAGVFFLALWGMVVG(配列番号2)
(c) MAPRRVDQPRSFMCVSTADLWLCEAG(配列番号3)
[12]
前記低発現量グルクロン酸転移酵素は、UGT1A1、UGT1A4、UGT1A8またはUGT1A9である[11]に記載の形質転換出芽酵母。
[13]
前記高発現量グルクロン酸転移酵素は、UGT1A7、UGT1A6またはUGT1A10である[11]または[12]に記載の形質転換出芽酵母。
[14]
被抱合物質のグルクロン酸抱合体生成に用いるための、[1]〜[13]のいずれか1項に記載の出芽酵母。
[15]
[1]〜[13]のいずれかに記載の形質転換出芽酵母をグルコース及び被抱合物質の存在下で培養して、前記被抱合物質のグルクロン酸抱合体を生成させることを含む、グルクロン酸抱合体の製造方法。
[16]
被抱合物質がアルコール性水酸基を含む医薬品及び医薬品の候補物質、フェノール性水酸基を複数含むポリフェノール化合物、カルボン酸を含む非ステイロイド性抗炎症薬及びその候補物質、並びに1〜4級のいずれか少なくとも1つのアミンを含む化合物から成る群から選ばれる少なくとも1種である[15]に記載の製造方法。
[17]
被抱合物質が、P450の代謝により抱合化を受ける官能基(主に水酸基)を生成する物質である[15]に記載の製造方法。
[18]
被抱合物質が、メトキシ基またはエトキシ基を含む医薬品及び医薬品の候補物質、メチレンジオキシフェニル基をもつセサミン化合物、並びに水酸基を有しないジアゼピン系の医薬品及び医薬品の候補物質から成る群から選ばれる少なくとも1種である[17]に記載の製造方法。
[19]
出芽酵母発現ベクターにUDP-グルコース脱水素酵素遺伝子を発現可能に挿入したものである、UDP-グルコース脱水素酵素発現ベクター。
[20]
出芽酵母発現ベクターにUDP-グルクロン酸転移酵素遺伝子を発現可能に挿入したものである、UDP-グルクロン酸転移酵素発現ベクター。
[21]
酵母発現ベクターにUDP-グルクロン酸転移酵素遺伝子及びUDP-グルコース脱水素酵素含を発現可能に挿入したものである、UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクター。
[22]
酵母発現ベクターが自律複製型ベクターまたは染色体組み込み型ベクターである、[19]〜[21]のいずれかに記載のベクター。
[23]
UDP-グルコース脱水素酵素遺伝子が動物または植物由来の遺伝子である、[15]、[19]、[21]〜[22]のいずれかに記載のベクター。
[24]
UDP-グルコース脱水素酵素遺伝子がシロイヌナズナ由来遺伝子またはラット由来遺伝子である、[19]、[21]〜[22] のいずれかに記載のベクター。
[25]
UDP-グルクロン酸転移酵素遺伝子が哺乳動物由来の遺伝子である、[20]〜[24]のいずれかに記載のベクター。
[26]
UDP-グルクロン酸転移酵素遺伝子がヒト由来の遺伝子である、[20]〜[24]のいずれかに記載のベクター。
【発明の効果】
【0014】
本発明によれば、UDPGDH及びUGT発現出芽酵母菌体を用いて抱合体を調製することができる。本発明の遺伝子改変出芽酵母菌体を用いた抱合体調製法によれば、従来の有機合成法や試験管内での酵素法や分裂酵母を用いた抱合体調製法に比べて、著しく効率的かつ安価に目的の代謝物を調製することができる。例えば、実施例に示すように、UDPGDH及びUGT1A6同時発現出芽酵母菌体を用いた最適条件下において、7位水酸化クマリンの抱合体を1L反応溶液あたり200mgの産生効率で製造することが可能である。
【0015】
さらに、UGT分子種は、種々のものの遺伝子情報が知られていることから、本発明のUDPGDH及びUGT発現出芽酵母菌体を多様なUGT分子種について調製することができる。その結果、本発明のUDPGDH及びUGT発現出芽酵母菌体は、さまざまな医薬品に対して適用でき、実用性もきわめて高い。
【0016】
さらに本発明の方法では、宿主酵母としてS.cerevisiaeを用い、さらには、いくつかの工夫を加えることで、S. pombeを宿主とする非特許文献7に記載の方法に比べて以下のような利点がある。
(1)S.pombeではUGT1A6の活性がきわめて低く、発現量が低いと推測されるが、S.cerevisiaeにおける発現量は高く、活性はきわめて高い(図5、100%変換を達成)。実際に4メチルウンベリフェロンを基質として分裂酵母のデータと比較したところ複数の分子種において数十倍高い産生能力を示した。(表6参照)
(2)S.cerevisiaeが有するABCトランスポーターの一種が菌体外への分泌に関わっていると推測されるが(図6)、これは予想外の結果であり、S.pombeには見られないS.cerevisiae特有の現象と考えられる。
(3)S.pombeではUGT1A3、1A4、2B7の発現がうまく行ってないが、S. cerevisiaeにおいてはこれらについても高発現を達成することができる。また、1A4の発現については、シグナル配列を改変することで高発現に成功している。
【0017】
このように、S.pombeを宿主としたUGT発現系の最大の欠点は発現量の低さと分子種の違いによる発現量の違いと考えられる。
【0018】
一方で、本発明では、S.cerevisiaeを宿主として用い、必要によりN末端のシグナル配列を変換することにより、いずれのUGT分子種も高発現させることに成功し、S.pombeの発現系の最大の欠点をクリアーすることに成功した。動植物由来のUDPGDHとの同時発現に成功し、菌体を用いたグルクロン酸抱合体の高効率菌体外分泌生産に成功した。
【図面の簡単な説明】
【0019】
【図1】UDP-グルクロン酸転移酵素遺伝子とUDP-グルコース脱水素酵素を同時に含む自律複製型酵母発現ベクターの構築スキームを示す。
【図2】酵母内発現UDP-グルコース脱水素酵素における酵素活性測定結果を示す。
【図3】酵母内発現UDP-グルクロン酸転移酵素遺伝子及びUDP-グルコース脱水素酵素タンパクの発現をウエスタンブロット法により確認した結果を示す。
【図4】UDP-グルコース脱水素酵素遺伝子導入酵母菌体におけるUDP-グルクロン酸の産生結果を示す。
【図5】種々のヒトUDP-グルクロン酸転移酵素との同時発現酵母における抱合体生成結果を示す。
【図6】UGT1A6同時発現株における抱合体産生の時間変化を示す。
【図7】UGT1A6同時発現株における抱合体産生のグルコース濃度の影響を示す。
【図8】同時発現株の抱合体産生における発現ベクターの組み合わせの影響を示す。
【図9】酵母静止菌体を用いたケルセチン抱合体の製造結果を示す。
【図10】酵母静止菌体を用いたマイコフェノール酸抱合体の製造結果を示す。
【図11】酵母静止菌体を用いたメフェナム酸のアシル抱合体の製造結果を示す。
【図12】酵母静止菌体を用いてタモキシフェン抱合体(Nグルクロン酸抱合体)の製造結果を示す。
【図13】シトクロムP450を含む同時発現株の静止菌体を用いた7ヒドロキシクマリン抱合体の製造結果を示す。
【図14】出芽酵母PGK1由来プロモーター及びターミネーター領域を有するヒトシトクロムP450発現プラスミドの構築スキームを示す。
【図15】ヒトシトクロムP450(CYP2D6)とUDP-グルクロン酸転移酵素(UGT1A8)を同時発現させた酵母株静止菌体を用いた7エトキシクマリン代謝物の製造結果を示す。
【図16】組換え酵母菌体を用いたグルクロン酸抱合体の調製の模式説明図を示す。
【発明を実施するための形態】
【0020】
[形質転換した出芽酵母]
本発明は、形質転換した出芽酵母に関し、具体的には、UDP-グルコース脱水素酵素をコードする遺伝子及びUDP-グルクロン酸転移酵素をコードする遺伝子を発現可能に挿入して形質転換した出芽酵母である。本発明の形質転換した出芽酵母は、UDP-グルコース脱水素酵素をコードする遺伝子及びUDP-グルクロン酸転移酵素をコードする遺伝子を発現可能に挿入して形質転換した出芽酵母にさらに、シトクロムP450遺伝子をコードする遺伝子をさらに発現可能に挿入して形質転換した出芽酵母であることもできる。UDP-グルコース脱水素酵素をコードする遺伝子、UDP-グルクロン酸転移酵素をコードする遺伝子、及びシトクロムP450遺伝子をコードする遺伝子は、出芽酵母でこれら遺伝子を出芽酵母発現ベクターに組み込まれた形で、出芽酵母に挿入されて、出芽酵母を形質転換したものであることができ、あるいは、これらの遺伝子を形質転換すべき出芽酵母の染色体に、例えば、相同組換などの公知の手法により、発現可能な状態で挿入したものであることもできる。
【0021】
前記遺伝子を出芽酵母発現ベクターに組み込んだ形で、出芽酵母に挿入して、出芽酵母を形質転換したものの例としては、以下の(A)〜(G)のいずれかから選ばれる形質転換出芽酵母を挙げることができる。
(A)UDP-グルコース脱水素酵素発現ベクター及びUDP-グルクロン酸転移酵素発現ベクターで形質転換した出芽酵母、
(B)UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクターで形質転換した出芽酵母、
(C)UDP-グルコース脱水素酵素発現ベクター、UDP-グルクロン酸転移酵素発現ベクター及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母、
(D)UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクター、及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母、
(E)UDP-グルクロン酸転移酵素発現ベクター、及びUDP-グルコース脱水素酵素及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母、
(F)UDP-グルクロン酸転移酵素及びシトクロムP450遺伝子発現ベクター、及びUDP-グルコース脱水素酵素発現ベクターで形質転換した出芽酵母、並びに
(G)UDP-グルコース脱水素酵素、UDP-グルクロン酸転移酵素及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母
【0022】
例えば、上記UDP-グルコース脱水素酵素発現ベクターは、出芽酵母発現ベクターにUDP-グルコース脱水素酵素遺伝子を発現可能に挿入したものである。UDP-グルクロン酸転移酵素発現ベクターは、出芽酵母発現ベクターにUDP-グルクロン酸転移酵素遺伝子を発現可能に挿入したものである。UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクターは、出芽酵母発現ベクターにUDP-グルクロン酸転移酵素遺伝子及びUDP-グルコース脱水素酵素含を発現可能に挿入したものである。以下、各ベクターについて説明する。
【0023】
[酵素等の発現ベクター]
本発明では、(1)UDP-グルコース脱水素酵素発現ベクター、(2)UDP-グルクロン酸転移酵素発現ベクター、(3)UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクター、(4)シトクロムP450遺伝子発現ベクター、(5)UDP-グルクロン酸転移酵素及びシトクロムP450遺伝子発現ベクター、(6)UDP-グルコース脱水素酵素及びシトクロムP450遺伝子発現ベクター、及び(7)UDP-グルクロン酸転移酵素、UDP-グルコース脱水素酵素及びシトクロムP450遺伝子発現ベクターを用いる。1つのベクターに複数の遺伝子を有する場合、遺伝子配置(順序)に制限はない。以下、(1)、(2)及び(4)の順に個別に説明する。(3)、(5)、(6)、(7)の1つのベクターに複数の遺伝子を有するベクターについては、(1)、(2)及び(4)の説明に基づいて適宜提供できる。
【0024】
<UDP-グルコース脱水素酵素発現ベクター>
UDP-グルコース脱水素酵素発現ベクターは、酵母発現ベクターにUDP-グルコース脱水素酵素(UDPGDH)遺伝子を挿入したものである。
【0025】
酵素遺伝子を挿入する発現ベクターは、宿主である出芽酵母において複製保持され、上記酵素遺伝子を発現し得る自律複製型ベクターであっても、出芽酵母の染色体に組み込まれる、染色体組み込み型プラスミドベクターであってもよい。自律複製型ベクターおよび染色体組み込み型プラスミドベクターは、宿主である出芽酵母において保持されかつ機能するものであれば制限なく利用できる。自律複製型ベクターとしては、例えば、実施例で用いたpGYR、さらには酵母由来のプラスミドYEp352GAP、YEp51、pSH19などを挙げることができる。染色体組み込み型プラスミドベクターとしては、実施例で用いたpAURなどを挙げることができる。
【0026】
上記酵素遺伝子および、必要により、シグナル配列遺伝子を上記発現に適したベクター中のプロモーターの下流に連結して発現ベクターを得ることができる。プロモーターとしては、ENO1プロモーター、GAL10プロモーター、GAPDHプロモーター、ADHプロモーターなどが挙げられる。
【0027】
UDP-グルコース脱水素酵素遺伝子は、動物または植物由来の遺伝子であることができる。動物または植物由来のUDP-グルコース脱水素酵素遺伝子は、公知の遺伝子(例えば、 ヒト遺伝子:AF061016.1,マウス遺伝子:AF061017.1 など )から適宜選択することができ、実施例では、シロイヌナズナ由来の遺伝子及びラット由来の遺伝子を用いたが、いずれかのUDP-グルコース脱水素酵素遺伝子を導入した。出芽酵母発現ベクターで形質転換した出芽酵母では、UDP-グルコース脱水素酵素の活性が得られた。従って、遺伝子の由来に関わらず、公知のUDP-グルコース脱水素酵素遺伝子を用いることができる。尚、シロイヌナズナ由来の遺伝子は、例えば、市販のcDNAライブラリーである、PCR ready First Strand cDNA (Biochain社)から入手することができる。また、ラット由来の遺伝子も、例えば、市販のラット肝臓cDNAライブラリーであるPCR ready First Strand cDNA (Biochain 社)から入手することができる。これら以外にも、市販のcDNAライブラリーからUDP-グルコース脱水素酵素遺伝子を適宜入手することもできる。
【0028】
<UDP-グルクロン酸転移酵素発現ベクター>
UDP-グルクロン酸転移酵素発現ベクターは、出芽酵母発現ベクターにUDP-グルクロン酸転移酵素(UGT)遺伝子を挿入したものである。出芽酵母発現ベクターは上記UDP-グルコース脱水素酵素発現ベクターで説明したものと同様のものを用いることができる。
【0029】
UDP-グルクロン酸転移酵素遺伝子は、公知の遺伝子から適宜選択することができる。UDP-グルクロン酸転移酵素遺伝子は、哺乳動物由来の遺伝子であることができ、具体的にはヒト由来の遺伝子であることができる。UDP-グルクロン酸転移酵素(UGT)は、約530アミノ酸残基からなる小胞体膜タンパク質であり、肝臓や小腸において複数の分子種が存在する。これら分子種の特徴としては、アミノ末端側半分(約290アミノ酸残基)は抱合化基質を認識するドメインを構成し、分子種間で高い相同性を持つカルボキシル末端側半分(約240アミノ酸残基)は共通の基質であるUDP-グルクロン酸が結合するドメインとして機能している(非特許文献3、4、10)。
【0030】
UDP-グルクロン酸転移酵素遺伝子は、これまでの研究により多くの分子種がクローニングされ、その塩基配列が明らかにされた(非特許文献3)。本発明ではこれら公知の遺伝子を適宜利用できる。代表的なグルクロン酸転移酵素は、例えば、ヒトまたはブタ由来の酵素である。それら転移酵素の遺伝子配列およびシグナル配列の遺伝子配列は、ヒト由来の酵素はGenBankに登録されており、ブタ由来の酵素はPEDE(Database of full-length cDNA clones and ESTs in pigs)(http://pede.dna.affrc.go.jp)に登録されており、配列情報は容易に入手可能である。UGT遺伝子の代表例を以下に示す。
【0031】
【表1】
【0032】
出芽酵母を用いたグルクロン酸転移酵素の発現系においては、分子種によっては発現量の低いものが存在する。出芽酵母内での発現量の低かった分子種である、例えば、UGT1A1遺伝子の発現量を上昇させることができれば、医薬品抱合代謝物を調製できる範囲が広がり、出芽酵母発現グルクロン酸転移酵素による抱合体調製法の実用的価値は飛躍的に高まる。本発明では、出芽酵母を用いたグルクロン酸転移酵素の発現系において、出芽酵母内での発現量の低い分子種については、シグナル配列遺伝子を、高発現グルクロン酸転移酵素のシグナル配列遺伝子と置換することが好ましい。
【0033】
前記発現量の低い分子種のグルクロン酸転移酵素遺伝子は、例えば、発現量がUGT1A7の50%以下である低発現量グルクロン酸転移酵素遺伝子であることができる。前記低発現量グルクロン酸転移酵素は、例えば、UGT1A1、UGT1A4、UGT1A8またはUGT1A9であることができる。但し、これらに限定される意図ではない。さらに、前記低発現量グルクロン酸転移酵素遺伝子は、シグナル配列遺伝子を、例えば、発現量がUGT1A7の80%以上である高発現グルクロン酸転移酵素のシグナル配列遺伝子と置換することができる。高発現量グルクロン酸転移酵素は、例えば、UGT1A7、UGT1A6またはUGT1A10であることができる。但し、これらに限定される意図ではない。このようなシグナル配列の置換により、出芽酵母形質転換体によるグルクロン酸転移酵素の発現量がシグナル配列遺伝子未置換の形質転換体に比べて強化される。
【0034】
前記低発現量グルクロン酸転移酵素遺伝子は、シグナル配列遺伝子を(A)以下に示すアミノ酸配列(a)〜(c)のいずれかをコードする遺伝子、(B)アミノ酸配列(a)〜(c)のアミノ酸の1〜5個が置換若しくは欠失したアミノ酸配列のいずれかをコードする遺伝子、または(C)1〜5個のアミノ酸がアミノ酸配列(a)〜(c)のアミノ酸に付加されたアミノ酸配列のいずれかをコードする遺伝子と置換したものであることもできる。但し、(B)の置換若しくは欠失したアミノ酸配列および(C)の付加されたアミノ酸配列をコードする遺伝子は、シグナル配列遺伝子とした場合にグルクロン酸転移酵素の発現量が野生型の80%以上である。
(a) MARAGWTGLLPLYVCLLLTCGFAKAG(配列番号1)
(b) MACLLRSFQRISAGVFFLALWGMVVG(配列番号2)
(c) MAPRRVDQPRSFMCVSTADLWLCEAG(配列番号3)
【0035】
低発現量グルクロン酸転移酵素遺伝子のシグナル配列遺伝子を、(B)アミノ酸配列(a)〜(c)のアミノ酸の1〜5個が置換若しくは欠失したアミノ酸配列のいずれかをコードする遺伝子であって、この遺伝子は、グルクロン酸転移酵素遺伝子のシグナル配列遺伝子として用いた場合に、グルクロン酸転移酵素の発現量がUGT1A7の80%以上である遺伝子と置換する。アミノ酸配列(a)のアミノ酸の1〜5個が置換若しくは欠失したアミノ酸配列をコードする遺伝子をシグナル配列遺伝子として用いた場合に、UGT1A7の発現量が野生型のUGT1A7の80%以上、好ましく90%以上、より好ましくは100%以上である遺伝子とする。アミノ酸配列(b)のアミノ酸の1〜5個が置換若しくは欠失したアミノ酸配列をコードする遺伝子をシグナル配列遺伝子として用いた場合に、UGT1A6の発現量が野生型のUGT1A6の80%以上、好ましく90%以上、より好ましくは100%以上である遺伝子とする。アミノ酸配列(c)のアミノ酸の1〜5個が置換若しくは欠失したアミノ酸配列をコードする遺伝子をシグナル配列遺伝子として用いた場合に、UGT1A6の発現量が野生型のUGT1A10の80%以上、好ましく90%以上、より好ましくは100%以上である遺伝子とする。
【0036】
さらに、前記低発現量グルクロン酸転移酵素遺伝子のシグナル配列遺伝子を、(C)1〜5個のアミノ酸がアミノ酸配列(a)〜(c)のアミノ酸に付加されたアミノ酸配列のいずれかをコードする遺伝子と置換したものであり、この遺伝子は、グルクロン酸転移酵素遺伝子のシグナル配列遺伝子として用いた場合に、グルクロン酸転移酵素の発現量がUGT1A7の80%以上である遺伝子と置換する。アミノ酸配列(a)に1〜5個のアミノ酸が付加されたアミノ酸配列をコードする遺伝子をシグナル配列遺伝子として用いた場合に、UGT1A7の発現量が野生型のUGT1A7の80%以上、好ましく90%以上、より好ましくは100%以上である遺伝子とする。アミノ酸配列(b) に1〜5個のアミノ酸が付加されたアミノ酸配列をコードする遺伝子をシグナル配列遺伝子として用いた場合に、UGT1A6の発現量が野生型のUGT1A6の80%以上、好ましく90%以上、より好ましくは100%以上である遺伝子とする。アミノ酸配列(c)に1〜5個のアミノ酸が付加されたアミノ酸配列をコードする遺伝子をシグナル配列遺伝子として用いた場合に、UGT1A6の発現量が野生型のUGT1A10の80%以上、好ましく90%以上、より好ましくは100%以上である遺伝子とする。
【0037】
尚、野生型のグルクロン酸転移酵素の発現量との対比は、ミクロソーム画分におけるグルクロン酸転移酵素の発現を、例えば、ヒトグルクロン酸転移酵素ファミリー1の共通配列認識ペプチド抗体(非特許文献6)を用いたウエスタンブロット法による解析によって行うことができる。
【0038】
低発現量グルクロン酸転移酵素遺伝子のシグナル配列遺伝子を、上記アミノ酸配列をコードする遺伝子と置換することで、出芽酵母形質転換体によるグルクロン酸転移酵素の発現量がシグナル配列遺伝子未置換の形質転換体に比べて強化される。
【0039】
<UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクター>
UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクターは、1つの出芽酵母発現ベクターにUDP-グルクロン酸転移酵素遺伝子及びUDP-グルコース脱水素酵素遺伝子の両方を挿入したものであり、このベクターで形質転換した出芽酵母では、UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素の活性が同時に得られる。出芽酵母発現ベクターは上記UDP-グルコース脱水素酵素発現ベクターで説明したものと同様のものを用いることができる。また、UDP-グルクロン酸転移酵素遺伝子及びUDP-グルコース脱水素酵素遺伝子は、上記で説明したものと同様のものをそれぞれ利用できる。
【0040】
UDP-グルクロン酸転移酵素遺伝子及びUDP-グルコース脱水素酵素遺伝子のベクター中での挿入の順番は特に制限はなく、どちらが上流にあっても各酵素遺伝子の発現は支障なく行われる。
【0041】
<シトクロムP450遺伝子発現ベクター>
シトクロムP450遺伝子発現ベクターは、非特許文献2に記載されており、この文献の記載に基づいて調製することができる。出芽酵母発現ベクターは上記UDP-グルコース脱水素酵素発現ベクターで説明したものと同様のものを用いることができる。
【0042】
シトクロムP450遺伝子発現ベクターは、小胞体膜酵素であるシトクロムP450の発現で実績のあるpGYRを用いた。発現調節領域に醤油酵母(Zygosaccharomyces rouxii)グリセロアルデヒド3リン酸脱水素酵素遺伝子由来のプロモーター及びターミネーター領域を有し定常的なタンパク発現を可能にしている。さらに出芽酵母由来P450還元酵素遺伝子を含むことによりNADPHからの電子供給を増強することで酵母内でのP450によるモノオキシゲナーゼ反応を促進することができる。
【0043】
<発現ベクターの調製>
酵素遺伝子及び必要により、シトクロムP450遺伝子をそれぞれ機能的に連結し、これらを適切なベクターに挿入する方法は、当業者に通常知られる方法、たとえば、文献Molecular Cloning (1989) (Cold Spring Harbor Lab.)に記載の方法である。組換えベクターにおける挿入位置は、組換えベクターの複製に関与していない領域であればどこでも良く、通常はベクター内のマルチクローニングサイトが利用される。
【0044】
[形質転換体]
本発明の形質転換体は、上記本発明のいずれか1つまたは2つ以上のベクターで形質転換した出芽酵母からなる。宿主として用いる出芽酵母はサッカロマイセス・セレビシエ(Saccharomyces cerevisiae)であり、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)に属する菌株であれば特に制限はない。例えば、Saccharomyces cerevisiae AH22株、NA87-11A株、SHY3株等を用いることができる。
【0045】
S.cerevisiae AH22株はミトコンドリアDNAを欠損した呼吸欠損株であり、小胞体膜に結合するシトクロムP450の発現には最適の宿主であることが知られている。(参考文献 T. Sakaki, M. Akiyoshi-Shibata, Y. Yabusaki, H. Ohkawa, Organella targeted expression of rat liver cytochrome P450c27 in yeast: genetically engineered alteration of mitochondrial P450 into a microsomal form creates a novel functional electron transport chain, J. Biol.Chem. 267 16497-16502.(1992))AH22株は、ミトコンドリアDNAが欠損しているため機能を有するミトコンドリアが形成されず、その代わりに小胞体膜が発達することでP450やUGTが存在する場が増えるため、これらタンパク発現に有利であると考えられる。UDP-グルクロン酸転移酵素(UGT)もP450と同様に小胞体膜に結合するタンパク質であることから、UGTの発現においてもAH22株は適した宿主であると考えられ、実施例では、AH22株を宿主として用いた。このような理由から、シトクロムP450とUGTの同時発現系においても、AH22株は好ましい宿主と考えられる。但し、本発明は、宿主がAH22株の場合に限定されるものではない。
【0046】
本発明の形質転換体は、例えば、
(A)UDP-グルコース脱水素酵素発現ベクター及びUDP-グルクロン酸転移酵素発現ベクターで形質転換した出芽酵母、
(B)UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクターで形質転換した出芽酵母、
(C)UDP-グルコース脱水素酵素発現ベクター、UDP-グルクロン酸転移酵素発現ベクター及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母、
(D)UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクター、及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母、
(E)UDP-グルクロン酸転移酵素発現ベクター、及びUDP-グルコース脱水素酵素及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母、
(F)UDP-グルクロン酸転移酵素及びシトクロムP450遺伝子発現ベクター、及びUDP-グルコース脱水素酵素発現ベクターで形質転換した出芽酵母、並びに
(G)UDP-グルコース脱水素酵素、UDP-グルクロン酸転移酵素及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母
を挙げることができる。
【0047】
形質転換体の調製方法にも特に制限はなく、酵母宿主への組み換えベクターの導入方法としては、例えば、エレクトロポレーション法、スフェロプラスト法、酢酸リチウム法等を挙げることができる。
【0048】
調製された形質転換体は、適宜常法により培養、増殖させた後に、後述するグルクロン酸抱合体の製造方法に使用することができる。増殖させた形質転換体は、導入した遺伝子の種類や宿主の種類により異なるが、一般には対数増殖期にある菌体を用いることが適当であるが、対数増殖期前の菌体や対数増殖期後の菌体を用いることが好ましい場合もある。
【0049】
[グルクロン酸抱合体の製造方法]
本発明のグルクロン酸抱合体の製造方法は、前記本発明の出芽酵母形質転換体をグルコース及び被抱合物質の存在下で培養して、前記被抱合物質のグルクロン酸抱合体を生成させることを含むものである。
【0050】
図16に、本発明の組換え酵母菌体(形質転換体)を用いたグルクロン酸抱合体の調製の模式説明図を示す。
【0051】
一般に、生体内外の異物(脂溶性化合物(疎水性基質)(RH))は、抱合化反応を受けて水溶性の抱合体(Glucuronide)となり、体外へ排泄される。特にグルクロン酸抱合は、脂溶性化合物中の水酸基、アミノ基、カルボキシル基あるいはチオール基などの官能基にグルクロン酸が付加されることによって水溶性の極性代謝物に変換される。極性代謝物(グルクロン酸抱合体)は、多剤耐性薬剤輸送タンパク質(ABCC2)により、生体外に排泄が促進される。従って、本発明のグルクロン酸抱合体の製造方法において用いられる被抱合物質は、上記水酸基、アミノ基、カルボキシル基あるいはチオール基などの官能基を有する化合物であり、これらの化合物は、医薬品または医薬品の候補物質であることができる。被抱合物質が上記水酸基、アミノ基、カルボキシル基あるいはチオール基などの官能基を有する場合には、菌体内のUDP-グルクロン酸転移酵素(UGT, UDP-glucuronic acid transferase)が触媒するグルクロン酸転移反応により、UDP-グルクロン酸(UDP-GlcUA)を糖の供与体として、グルクロン酸抱合体に変換される。
【0052】
一方、被抱合物質が上記水酸基、アミノ基、カルボキシル基あるいはチオール基などの官能基を有さない場合には、被抱合物質である生体内外の脂溶性化合物(疎水性基質)(RH,脂溶性基質)は、シトクロムP450(CYP)により酸化されて水酸化物(ROH)となった後に、菌体内のUDP-グルクロン酸転移酵素(UGT, UDP-glucuronic acid transferase)が触媒するグルクロン酸転移反応により、UDP-グルクロン酸(UDP-GlcUA)を糖の供与体として、グルクロン酸抱合体に変換される。UDP-グルクロン酸転移酵素(UGT)が関与するこの系は、生体外への異物排泄を促進させる役割をもつ異物代謝系のひとつである。
【0053】
一方、UDP-グルクロン酸(UDP-GlcUA)は、グルコースから、UDP-グルコース(UDP-glucose)を経て、UDP-グルコース脱水素酵素(UDPGDH, UDP-glucose dehydrogenase)の作用により生成される。グルコースは、酵母内に取り込まれるとエネルギー代謝系である解糖におけるグルコキナーゼ、ホスホムターゼ及びUTPグルコース1リン酸ウリジルトランスフェラーゼの作用によりUDP-グルコース(UDP-glucose)に変換される。UDP-グルコース(UDP-glucose)は、UDP-グルコース脱水素酵素(UDPGDH )の作用により、NAD+を補酵素として、6位が酸化されてUDP-グルクロン酸(UDP-GlcUA)に変換される。
【0054】
上記のように、原料として水酸基、アミノ基、カルボキシル基あるいはチオール基などの官能基を有する被抱合物質を用いる方法(第1の態様)においては、本発明の形質転換体として、上記(A)、(B)または(E)の形質転換体を用いることで、前記被抱合物質のグルクロン酸抱合体を生成させることができる。
【0055】
水酸基、アミノ基、カルボキシル基あるいはチオール基などの官能基を有する被抱合物質は、特に制限はなく、例えば、アルコール性水酸基を含む医薬品または医薬品の候補物質、フェノール性水酸基を多数含むポリフェノール化合物、カルボン酸を含む非ステイロイド性抗炎症薬またはその候補物質、1〜4級のいずれか少なくとも1つのアミンを含む化合物等を挙げることができる。これら被抱合物質の例は、例えば、非特許文献10の100〜102頁のTable 1に列記されており、非特許文献10の100〜102頁のTable 1を含む全記載はここに参照として援用される。
【0056】
また、被抱合物質が上記水酸基、アミノ基、カルボキシル基あるいはチオール基などの官能基を有さない場合には、例えば、有機合成法を用いた水酸基の導入やシトクロムP450を用いた酵素法による選択的な水酸化によって、事前に被抱合物質に官能基を導入した後に、上記第一の態様の方法に供することができる。あるいは、被抱合物質が上記水酸基、アミノ基、カルボキシル基あるいはチオール基などの官能基を有さない場合には、本発明の形質転換体として、シトクロムP450遺伝子を導入した前記(C)、(D)、(E)、(F)または(G)の形質転換体を用い、かつこの形質転換体をグルコース、及び被抱合物質の存在下で培養して、前記被抱合物質のグルクロン酸抱合体を生成させることもできる(第2の態様)。本発明において、P450とUGT同時発現系による代謝物質は、P450の代謝により抱合化を受ける官能基(主に水酸基)が生成されるものであれば特に制限はなく、例えば、メトキシ基あるいはエトキシ基を含む医薬品または医薬品の候補物質、メチレンジオキシフェニル基をもつセサミン化合物、水酸基を有しないジアゼピン系の医薬品または医薬品の候補物質等を挙げることができる。第2の態様においては、シトクロムP450を含む同時発現株を用いるため、抱合化基質の前駆体(被抱合物質)から抱合体を調製することができる。即ち、被抱合物質を前駆体化(水酸化物への変換)をすること無しに、抱合体を調製することができる。
【0057】
本発明の製造方法において用いられる被抱合物質は、上記のように特に制限はない。これは、上述のように本発明においては、UDP-グルクロン酸転移酵素(UGT)の基質となる被抱合物質の種類に応じて、形質転換体に含まれるUDP-グルクロン酸転移酵素(UGT)遺伝子の種類を適宜選択することができるからである。例えば、実施例に示す例では、7位水酸化クマリンの抱合体(フェノール性水酸基)を調製する場合、用いるUGTは、UGT1A6であることが適当であり、ケルセチンの4’位及び3’位水酸基に対する抱合体(フェノール性水酸基)を調製する場合、用いるUGTは、UGT1A1あるいはUGT1A8であることが適当であり、マイコフェノール酸の7位水酸基に対する抱合体(アルコール性水酸基)を調製する場合、用いるUGTは、UGT1A9であることが適当である。
【0058】
上記本発明の形質転換体の培養は、グルコース及び被抱合物質の存在下、公知の酵母の培養方法を適宜用いて実施できる。酵母宿主が上記AH22株の場合、L-ヒスチジンとL-ロイシン以外の全ての必須アミノ酸を生合成することができる。従って、培地には、これら2つのアミノ酸を添加して培養するか、あるいは発現ベクターとして、いずれか一方のアミノ酸合成遺伝子を持つベクターを用いることで、この発現ベクターを有する酵母宿主を選択的に培養することもできる。
【0059】
培養の条件は、酵母形質転換体の生育に適した条件において適宜に設定することができる。形質転換体の培養に用いる栄養培地としては、炭素源、窒素源、無機物、および必要に応じ使用菌株の必要とする微量栄養素を程よく含有するものであれば、天然培地、合成培地のいずれでもよい。あるいは酵母形質転換体の生育に必要な栄養源の少なくとも一部を欠いた条件で培養することで、グルクロン酸抱合体の生成、蓄積が良好に得られる場合もある。被抱合物質の種類によっては後者の条件が好ましい場合もある。
【0060】
栄養培地の炭素源としては、酵母形質転換体が資化しうる物であればよく、例えば、グルコース、マルトース、フラクトース、マンノース、トレハロース、スクロース、マンニトール、ソルビトール、デンプン、デキストリン、糖蜜などの糖質、またはクエン酸、コハク酸などの有機酸、またはグリセリンなどの脂肪酸も使用することができる。
【0061】
栄養培地の窒素源としては、各種有機および無機の窒素化合物、さらに培地は各種の無機塩を含むことができる。たとえば、コーンスティープリカー、大豆粕、あるいは各種ペプトン類等の有機窒素源、そして塩化アンモニウム、硫酸アンモニウム、尿素、硝酸アンモニウム、硝酸ナトリウム、リン酸アンモニウム等の無機窒素源などの化合物が使用可能である。また、グルタミン酸などのアミノ酸および尿素などの有機窒素源が炭素源にもなることはいうまでもない。さらに、ペプトン、ポリペプトン、バクトペプトン、肉エキス、魚肉エキス、酵母エキス、コーンスティープリカー、大豆粉、大豆粕、乾燥酵母、カザミノ酸、ソリュブルベジタブルプロテイン等の窒素含有天然物も窒素源として使用できる。
【0062】
栄養培地の無機物としては、たとえば、カルシウム塩、マグネシウム塩、カリウム塩、ナトリウム塩、リン酸塩、マンガン塩、亜鉛塩、鉄塩、銅塩、モリブデン塩、コバルト塩などが適宜用いられる。具体的には、リン酸二水素カリウム、リン酸水素二カリウム、硫酸マグネシウム、硫酸第一鉄、硫酸マンガン、硫酸亜鉛、塩化ナトリウム、塩化カリウム、塩化カルシウム等が用いられる。さらに、必要に応じて、アミノ酸ならびにビオチンおよびチアミンなどの微量栄養素ビタミンなども適宜用いられる。
【0063】
培養法としては液体培養法がよく、回分培養、流加培養、連続培養または灌流培養のいずれを用いてもよいが、工業的には通気攪拌培養法が好ましい。培養温度とpHは、使用する形質転換体の増殖に最も適した条件を選べばよい。培養時間は微生物が増殖し始める時間以上の時間であればよく、好ましくは8〜120時間であり、さらに好ましくは組換えタンパク質遺伝子の遺伝子産物が最大に生成する時間までである。たとえば、形質転換体が出芽酵母の場合の培養は、通常、温度20〜40℃、好ましくは25〜35℃、pH2〜9、好ましくは5〜8、培養日数0.5〜7日間から選ばれる条件で振盪または通気攪拌して行われる。酵母の増殖を確認する方法は特に制限はないが、たとえば、培養物を採取して顕微鏡で観察してもよいし、吸光度で観察してもよい。また、培養液の溶存酸素濃度には特に制限はないが、通常は、0.5〜20ppmが好ましい。そのために、通気量を調節したり、撹拌したり、通気に酸素を追加したりすればよい。
【0064】
形質転換体の使用量は、例えば、0.5〜5%(w/v、乾燥重量/培養液体積)の範囲であることができる。さらに、グルコース及び被抱合物質の培地への添加量は、培地における浸透圧及び細胞に対する毒性を考慮して適宜決定され、例えば、グルコースは、4〜20%(w/v)の範囲であり、被抱合物質は、0.5〜25mM の範囲であることが適当である。
【0065】
形質転換体を培養することで培養物中にグルクロン酸抱合体が蓄積する。培養後、酵母形質転換体の培養液からグルクロン酸抱合体を回収する。グルクロン酸抱合体は、形質転換体外に蓄積されることが多いが、一部は形質転換体内にも蓄積される。形質転換体外に蓄積されたグルクロン酸抱合体は、例えば、溶媒抽出により回収することができる。溶媒抽出による回収は、例えば、通常知られる手段によって形質転換体を培養液から除いた後に、得られる培養上清に対して行うことができる。形質転換体は再利用も可能である。
【0066】
また、形質転換体内に蓄積したグルクロン酸抱合体は、例えば、有機溶剤やザイモリアーゼのような酵素によって形質転換体の細胞壁を溶解する方法、および、超音波破砕法、フレンチプレス法、ガラスビーズ破砕法、ダイノミル破砕法等の細胞破砕法で得られた形質転換体の細胞破砕物および/または培養物を遠心分離法、ろ過法等の操作によって形質転換体と培養上清に分離する。このようにして得られた培養上清から、上記と同様にグルクロン酸抱合体を回収することができる。
【実施例】
【0067】
以下本発明を実施例によりさらに詳細に説明する。但し、本発明は実施例に限定される意図ではない。
【0068】
実施例1
1. シロイヌナズナ及びラット由来UDP-グルコース脱水素酵素発現プラスミドの構築
シロイヌナズナ及びラット由来UDP-グルコース脱水素酵素は、cDNAライブラリーを鋳型として、両端に相補的な配列を有するプライマーを用いてPCR法によって増幅し構築した。酵母発現ベクターとしては自律複製型プラスミドであるpGYR(文献番号4)あるいは染色体組み込み型プラスミドであるpAUR101(TaKaRa)を用いた。
【0069】
1-1. シロイヌナズナ及びラット由来UDP-グルコース脱水素酵素遺伝子の作成
1-1-1. シロイヌナズナ由来UDP-グルコース脱水素酵素遺伝子の作成
シロイヌナズナのcDNAライブラリーを用いてPCR法によって遺伝子のクローニングを行った。cDNAライブラリーはPCR ready First Strand cDNA (Biochain 社)を用いた。
【0070】
DNAポリメラーゼとしてはKOD-plus-(TOYOBO)を用いた。反応液組成は製造業者の指示に従い、以下に示すプライマーと反応条件でPCRを行った。
フォワード配列:配列番号4
リバース配列:配列番号5
PCR条件
変性 94℃ 2分
5サイクル 94℃ 15秒、37℃ 30秒、68℃1分45秒
30サイクル 94℃ 15秒、55℃ 30秒、68℃1分45秒
最終伸長 68℃10分
【0071】
PCR産物を1%アガロースゲルで電気泳動した結果、目的サイズ(約1.5kb)に特異的な増幅がみられた。(以下、1%アガロースゲルでの電気泳動は、単に電気泳動と略称する)。PCR増幅溶液を電気泳動して、DNA断片をゲルから分離した後、TAクローニングキット(Target cloneTM-plus-,TOYOBO社)を用いてpTAベクターに挿入した。このクローニングされた遺伝子のシークエンスを行い遺伝子配列を解析したところ、配列番号6に示した塩基配列(GenBank Acc.No. AY056200)であることを確認した。
【0072】
本遺伝子を発現ベクターにサブクローニングするために、部位特異的な変異導入によりアミノ酸配列を変えずに内部HindIIIサイトを欠失させた。変異導入は変異体作成キットQuick ChangeTMを用いた。変異導入をおこなったクローンの塩基配列を解析し、目的以外の箇所に変異導入されていないことを確認した。
【0073】
1-1-2. ラット由来UDP-グルコース脱水素酵素遺伝子の作成
ラット肝臓cDNAライブラリーを用いてPCR法によって遺伝子のクローニングを行った。cDNAライブラリーはPCR ready First Strand cDNA (Biochain 社)を用いた。
【0074】
DNAポリメラーゼとしてはKOD-plus-(TOYOBO)を用いた。反応液組成は製造業者の指示に従い、以下に示すプライマーと反応条件でPCRを行った。
フォワード配列:配列番号7
リバース配列:配列番号8
PCR条件
変性 94℃ 2分
5サイクル 94℃ 15秒、37℃ 30秒、68℃1分45秒
30サイクル 94℃ 15秒、55℃ 30秒、68℃1分45秒
最終伸長 68℃10分
【0075】
PCR産物を1%アガロースゲルで電気泳動した結果、目的サイズ(約1.5kb)に特異的な増幅がみられた。(以下、1%アガロースゲルでの電気泳動は、単に電気泳動と略称する)。PCR増幅溶液を電気泳動して、DNA断片をゲルから分離した後、TAクローニングキット(Target cloneTM-plus-,TOYOBO社)を用いてpTAベクターに挿入した。このクローニングされた遺伝子のシークエンスを行い遺伝子配列を解析したところ、配列番号9に示した塩基配列(GenBank Acc.No. O70199)であることを確認した。
【0076】
本遺伝子を発現ベクターにサブクローニングするために、部位特異的な変異導入によりアミノ酸配列を変えずに内部HindIIIサイトを欠失させた。変異導入は変異体作成キットQuick ChangeTMを用いた。変異導入をおこなったクローンの塩基配列を解析し、目的以外の箇所に変異導入されていないことを確認した。
【0077】
1-2. UDP-グルコース脱水素酵素発現ベクターの構築
1-2-1. 自律複製型酵母発現ベクターの構築
1-1-1及び1-1-2で作成したUDP-グルコース脱水素酵素断片を含むプラスミド約1μgを37℃で4時間、HindIII処理し、電気泳動後、遺伝子断片をゲルから切り出した。切り出したゲルからWizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてインサート断片を調製した。
【0078】
酵母発現ベクターとして用いるpGYRを37℃で4時間HindIII処理し、電気泳動後、目的サイズ(約11kb)のバンドを切り出した。その後、Wizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてベクター断片を調製した。
【0079】
インサート断片とpGYR-HindIIベクター断片の濃度を電気泳動で確認後、それらのモル比が3:1〜10:1程度になるように混合し、等量のDNA Ligation Kit Ver. 2 (TaKaRa)のsolution Iを加え、16℃、1時間反応を行った。その後、大腸菌JM109株にヒートショック法を用いてトランスフォーメーションを行い、LBプレート(アンピシリン50μg/ml含有)に展開した。得られた大腸菌コロニーのうち数個を選択し、それらを鋳型としてコロニーPCRを行った。DNAポリメラーゼは、Ex Taq(登録商標)DNAポリメラーゼを用いた。プライマーはYGAP-Pプライマー(5’-aatgacaccgtgtggtgatcttcaagg-3’)(配列番号10)と上記リバースプライマー(配列番号5あるいは8)を用いた。以下の条件でPCRを行った。
【0080】
PCR条件
変性 98℃5分
30サイクル 94℃ 30秒、50℃ 30秒、72℃ 2分30秒
最終伸長 72℃4分
【0081】
PCR後、電気泳動を行い、予想サイズ(約3kb)に特異的な増幅がみられるクローンを数個得た。得られた大腸菌コロニーを5mlのLB培地(アンピシリン100μg/ml含有)に植菌し、37℃、200rpmで16時間振盪培養を行った。その後アルカリSDS法を用いてプラスミドを抽出した。そして、その一部を37℃で1時間、HindII処理し、電気泳動をしてインサートの導入を再度確認し、2種のUDP-グルコース脱水素酵素酵素をそれぞれ含む酵母発現ベクター(シロイヌナズナ由来:pGYR/At.UDPGDH、ラット由来:pGYR/ratUDPGDH)を得た。
【0082】
1-2-2. 染色体組み込み型酵母発現ベクターの構築
1-2-1で作成したUDP-グルコース脱水素酵素断片を含む発現プラスミド(pGYR/At.UDPGDH) 約1μgを37℃で4時間、NotI処理し、電気泳動後、酵母発現プロモーター及びターミネーター領域を含む遺伝子断片(約3kb)をゲルから切り出した。切り出したゲルからWizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてインサート断片を調製した。
【0083】
自律複製型酵母発現ベクターpAUR101(TaKaRa)に上記で得られたNotI断片を挿入するために、部位特異的な変異導入によりマルチクローニングサイトにNotI制限酵素配列(GCGGCCGC)をマルチクローニングサイト領域に導入した。変異導入は変異体作成キットQuick ChangeTMを用いた。変異導入をおこなったクローンの塩基配列を解析し、目的以外の箇所に変異導入されていないことを確認した(pAUR-N)。酵母発現ベクターとして用いるpAUR-Nを37℃で4時間NotI処理し、電気泳動後、目的サイズ(約7kb)のバンドを切り出した。その後、Wizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてベクター断片を調製した。
【0084】
インサート断片とpAUR-NotIベクター断片の濃度を電気泳動で確認後、それらのモル比が3:1〜10:1程度になるように混合し、等量のDNA Ligation Kit Ver. 2 (TaKaRa)のsolution Iを加え、16℃、1時間反応を行った。その後、大腸菌JM109株にヒートショック法を用いてトランスフォーメーションを行い、LBプレート(アンピシリン50μg/ml含有)に展開した。得られた大腸菌コロニーを5mlのLB培地(アンピシリン100μg/ml含有)に植菌し、37℃、200rpmで16時間振盪培養を行った。その後アルカリSDS法を用いてプラスミドを抽出した。そして、その一部を37℃で1時間、NotI処理し、電気泳動をしてインサートの導入を再度確認し、UDP-グルコース脱水素酵素を含む酵母発現ベクター(pAUR/At.UDPGDH)を得た。
【0085】
1-2-3.UDP-グルクロン酸転移酵素遺伝子とUDP-グルコース脱水素酵素を同時に含む自律複製型酵母発現ベクターの構築
1-2-2で作成したpAUR-Nを用いてシロイヌナズナ由来UDP-グルコース脱水素酵素及びUDP-グルクロン酸転移酵素を同時に含む酵母発現ベクターを構築し(図1)た。まず、最初にIn FusionTM Advantage PCRクローニングキット(TaKaRa)を用いて、1-2-2で作成したUDP-グルコース脱水素酵素遺伝子を含むNotI断片におけるプロモーター領域上流のNotIサイトを欠失させた断片と相補的な配列を有するpAUR-Nベクターを融合させてUDP-グルコース脱水素酵素遺伝子を含む染色体組み込み型酵母発現ベクター (pAUR-At.UDPGDH)を作成した。
【0086】
DNAポリメラーゼとしてはPrime STAR Max Premix (TaKaRa)を用いた。反応液組成は製造業者の指示に従い、以下に示すプライマーと反応条件でPCRを行った。
インサートDNAのPCR反応
テンプレート:pGYR/At.UDPGDH
フォワード配列:配列番号11
リバース配列:配列番号12
PCR条件
変性 98℃ 10秒
30サイクル 98℃ 10秒、55℃ 5秒、72℃ 16秒
最終伸長 72℃10分
ベクターDNAのPCR反応
テンプレート:pAUR-N NotI digest
フォワード配列:配列番号13
リバース配列:配列番号14
PCR条件
変性 98℃ 10秒
30サイクル 98℃ 10秒、55℃ 5秒、72℃ 33秒
最終伸長 72℃10分
【0087】
UDP-グルクロン酸転移酵素断片を含む発現プラスミドpGYR/UGT約1μgを37℃で4時間、NotI処理し、電気泳動後、酵母発現プロモーター及びターミネーター領域を含む遺伝子断片(約3.2kb)をゲルから切り出した。切り出したゲルからWizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてインサート断片を調製した。
【0088】
先に作成したpAUR-At.UDPGDHを37℃で4時間NotI処理し、電気泳動後、目的サイズ(約10kb)のバンドを切り出した。その後、Wizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてベクター断片を調製した。
【0089】
インサート断片とpAUR-NotIベクター断片の濃度を電気泳動で確認後、それらのモル比が3:1〜10:1程度になるように混合し、等量のDNA Ligation Kit Ver. 2 (TaKaRa)のsolution Iを加え、16℃、1時間反応を行った。その後、大腸菌JM109株にヒートショック法を用いてトランスフォーメーションを行い、LBプレート(アンピシリン50μg/ml含有)に展開した。得られた大腸菌コロニーを5mlのLB培地(アンピシリン100μg/ml含有)に植菌し、37℃、200rpmで16時間振盪培養を行った。その後アルカリSDS法を用いてプラスミドを抽出した。そして、その一部を37℃で1時間、NotI処理し、電気泳動をしてインサートの導入を再度確認し、UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素を含む酵母発現ベクター(pAUR-At.UDPGDH/UGT)を得た。
【0090】
【表2】
【0091】
【表3】
【0092】
1-2-4. 遺伝子改変グルクロン酸転移酵素発現プラスミドの構築
本発現系で用いるUDP-グルクロン酸転移酵素分子種のうちUGT1A1及びUGT1A9に関しては、発現量を増強するために次に述べるようにN末端シグナル配列の改変(高発現分子種であるUGT1A7に由来するN末端シグナル配列への置換)を行った。
【0093】
ヒトグルクロン酸転移酵素におけるシグナル配列領域の塩基配列を表4に示す(配列番号15〜17)。シグナル配列置換したヒトグルクロン酸転移酵素は、ヒトグルクロン酸転移酵素遺伝子(UGT1A1及びUGT1A9)を含むpUC119クローニングベクターを鋳型として、置換配列を有するプライマー(表5 配列番号18〜20)を用いてPCR法によって増幅し構築した。
【0094】
【表4】
【0095】
【表5】
【0096】
1-2-4-1. N末端置換グルクロン酸転移酵素遺伝子の作成
N末端置換グルクロン酸転移酵素を、ヒトグルクロン酸転移酵素遺伝子(UGT1A1,及びUGT1A9)を含むpUC119クローニングベクターを鋳型としてPCRにより増幅した。DNAポリメラーゼとしてはKOD-plus-(TOYOBO)を用いた。反応液組成は製造業者の指示に従い、以下に示すプライマーと反応条件でPCRを行った。
【0097】
プライマー
フォワード配列:配列番号15あるいは16
リバース配列:配列番号20
【0098】
PCR条件
変性 94℃ 2分
5サイクル 94℃ 15秒、37℃ 30秒、68℃1分45秒
30サイクル 94℃ 15秒、55℃ 30秒、68℃1分45秒
最終伸長 68℃4分
【0099】
PCR産物を1%アガロースゲルで電気泳動した結果、目的サイズ(約1.6kb)に特異的な増幅がみられた。(以下、1%アガロースゲルでの電気泳動は、単に電気泳動と略称する)。
PCR断片を37℃で1時間、HindIII処理し、電気泳動後、グルクロン酸転移酵素遺伝子断片をゲルから切り出した。切り出したゲルからWizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてインサート断片を調製した。
【0100】
サブクローニングベクターとして用いるpUC119を37℃で4時間HindIII処理し、電気泳動後、目的サイズ(約2.8kb)のバンドを切り出した。その後、Wizard(登録商標)SV Gel and PCR Clean-Up System(Promega)を用いてベクター断片を調製した。
【0101】
インサート(グルクロン酸転移酵素遺伝子断片)とベクター(pUC119)を混合し、等量のDNA Ligation Kit Ver. 2 (TaKaRa)のsolution Iを加え、16℃、1時間反応を行った。その後、大腸菌JM109株にヒートショック法を用いてトランスフォーメーションを行い、LBプレート(アンピシリン50μg/ml含有)に展開した。
【0102】
得られた大腸菌コロニーのうち数個を選択し、それらを鋳型としてコロニーPCRを行った。DNAポリメラーゼとして、Ex Taq(登録商標)DNAポリメラーゼ(TaKaRa)を用い、M13-M4プライマー、M13-Rvプライマーを用いて以下の条件でPCRを行った。
【0103】
PCR条件
変性 98℃ 5分
30サイクル 94℃ 30秒、50℃ 30秒、72℃ 2分
最終伸長 72℃ 4分
PCR後、電気泳動を行い、予想サイズ(約1.6kb)に特異的な増幅が見られるクローンを数個得た。
【0104】
得られたクローンのインサート配列にPCRによる変異が入っていないことを確認するために配列決定を行った。まず、得られた大腸菌コロニーを5mlのLB培地(アンピシリン100μg/ml含有)に植菌し、37℃、200rpmで16時間振盪培養を行った。その後、Wizard(登録商標)Plus SV Minipreps DNA Purification System (Promega)を用いてプラスミドを抽出し、プラスミド濃度を260nmの吸光度を用いて測定し、テンプレートDNAとした。サイクルシークエンス反応は、BigDye(登録商標)Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems)を用いて行った。配列決定を行った結果、N末端置換グルクロン酸転移酵素のインサート(UGT1A1及びUGT1A9)は、UGT1A7のN末端シグナル配列をもち、それぞれ分子種に特有の正確な配列であることが確認できた。
【0105】
1-2-4-2. 酵母におけるN末端置換グルクロン酸転移酵素の発現ベクターの構築
1-2-4-1で作成したN末端置換グルクロン酸転移酵素断片を含むプラスミド約1μgを37℃で4時間、HindIII処理し、電気泳動後、グルクロン酸転移酵素遺伝子断片をゲルから切り出した。切り出したゲルからWizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてインサート断片を調製した。
【0106】
酵母発現ベクターとして用いるpGYRを37℃で4時間HindIII処理し、電気泳動後、目的サイズ(約11kb)のバンドを切り出した。その後、Wizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてベクター断片を調製した。
【0107】
インサート断片とpGYR-HindIIベクター断片の濃度を電気泳動で確認後、それらのモル比が3:1〜10:1程度になるように混合し、等量のDNA Ligation Kit Ver. 2 (TaKaRa)のsolution Iを加え、16℃、1時間反応を行った。その後、大腸菌JM109株にヒートショック法を用いてトランスフォーメーションを行い、LBプレート(アンピシリン50μg/ml含有)に展開した。得られた大腸菌コロニーのうち数個を選択し、それらを鋳型としてコロニーPCRを行った。DNAポリメラーゼは、Ex Taq(登録商標) DNAポリメラーゼを用いた。プライマーはYGAP-Pプライマー(5’-aatgacaccgtgtggtgatcttcaagg-3’)(配列番号10)と上記リバースプライマー(配列番号20)を用いた。以下の条件でPCRを行った。
【0108】
PCR条件
変性 98℃5分
30サイクル 94℃ 30秒、50℃ 30秒、72℃ 2分30秒
最終伸長 72℃4分
PCR後、電気泳動を行い、予想サイズ(約3kb)に特異的な増幅がみられるクローンを数個得た。
【0109】
得られた大腸菌コロニーを5mlのLB培地(アンピシリン100μg/ml含有)に植菌し、37℃、200rpmで16時間振盪培養を行った。その後アルカリSDS法を用いてプラスミドを抽出した。そして、その一部を37℃で1時間、HindII処理し、電気泳動をしてインサートの導入を再度確認し、2種のN末端シグナル配列置換グルクロン酸転移酵素(UGT1A1及びUGT1A9)を含む酵母発現ベクター(pGYR/UGT1A1及びpGYRUGT1A9)を得た。
【0110】
実施例2
2. UDP-グルコース脱水素酵素及びUDP-グルクロン酸転移酵素発を同時に発現する出芽酵母形質転換体の作成
2-1. 出芽酵母への発現用プラスミドの形質転換
発現用には、出芽酵母(Saccharomyces cerevisiae)AH22株を用いた。AH22株は、L-ヒスチジンとL-ロイシン以外は全ての必須アミノ酸を生合成することが出来る。pGYRはL-ロイシン合成遺伝子(LEU2)を持つため、培地にL-ヒスチジンを添加するとプラスミドを持った酵母のみが増殖できる。また、pAURは抗生物質であるAureobasdin A耐性遺伝子を有しており、Aureobasidin A存在下で形質転換体を選択することが可能である。発現用コンストラクトを酵母AH22株に塩化リチウム法により形質転換した。表2に作成した同時発現株における発現ベクターの組み合わせを示す。以下に形質転換の詳細なプロトコールを示す。
【0111】
<材料>
YPD培地:1%酵母抽出物、2%ポリペプトン、2%グルコース
SDプレート:2%グルコース、0.67%N-base w/o アミノ酸、1.5%寒天、20μg/ml L-ヒスチジン、0.2M LiCl: 10ml(フィルター滅菌)、1M LiCl: 10ml(フィルター滅菌)
70%(w/v) PEG 4000: 10ml(溶解後、10mlにメスアップ)
【0112】
<方法>
30℃で培養したS.cerevisiae AH22株1.0x107細胞を使用し、卓上遠心機で13,000rpm、4分遠心を行い、上清を除いた。ペレットを0.2M LiClで洗浄した(少しボルテックスにかけた)。卓上遠心機で13,000rpm、4分遠心を行い、上清を完全に取り除いた。ペレットを1M LiCl 20μlにピペッティングにより懸濁した。DNA(プラスミド)溶液10μl(0.5〜1μgのプラスミドを含有)を加えた。70%(w/v) PEG 4000を30μl加え、ピペッティングによりよく混合した。40℃で30分間インキュベートした。滅菌水140μlを加えてpGYRベクターの場合にはSDプレート、またはpAURベクターの場合には0.5μg/ml Aureobasidin A を含むYPDプレートに展開し、30℃にてインキュベートした。プレート上で30℃、3日間培養し、3〜5mm程度の大きさのコロニーを数十個得た。また、pGYR及びpAURの同時形質転換体の場合は、0.5μg/ml Aureobasidin A を含むSDプレートに展開し、30℃にてインキュベートした。プレート上で30℃、5日間培養し、3〜5mm程度の大きさのコロニーを数個得た。
【0113】
2-2. UDP-グルコース脱水素酵素及びUDP-グルクロン酸転移酵素タンパク質の酵母内での発現の確認
実施例2-1で得られた形質転換体について、それぞれの菌体内でタンパク質が機能的に発現されているかどうかを、ウエスタンブロット法及びUDP-グルコース脱水素酵素活性測定によって確認した。
【0114】
形質転換体を選択培地で48時間30℃で培養し、得られた酵母菌体をZymolyase処理にした後に、音波破砕を行い遠心操作を用いて細胞質画分を分離した。UDP-グルコース脱水素酵素活性は基質として5mM UDP-グルコースを、補助因子として0.5mMNAD+を用い、50mM Tris-HCl(pH8.6)という条件で37℃、1時間反応を行い、遠心分離により上清を回収した。C18カラムを用いたHPLCによって、20mM酢酸トリエチルアミン(pH7.0)溶離液を流速1ml/minで流し、検出波長260nmでUDP-グルクロン酸を検出した。その結果、UDP-グルコース脱水素酵素遺伝子を挿入した発現ベクター(pGYR/At.UDPGDH) を含む形質転換体菌体抽出液のみでUDP-グルクロン酸合成活性が検出された(図2)。
【0115】
図2は、酵母内発現UDP-グルコース脱水素酵素タンパクによるUDP-グルクロン酸生成を、逆相HPLCを用いて分析した結果である。A:AH22:pAUR(コントロール)、B:AH22:pAUR/At.UDPGDH。シロイヌナズナ由来UDP-グルコース脱水素酵素遺伝子を挿入した発現ベクター形質転換体抽出溶液においてのみ溶出時間11分付近にUDP-グルクロン酸の生成が見られた。
【0116】
また、形質転換体株より得られた酵母菌体をZymolyase処理にした後に、ヒトグルクロン酸転移酵素ファミリー1の共通配列認識ペプチド抗体(文献番号5)及びシロイヌナズナ由来UDP-グルコース脱水素酵素のC末端ペプチド配列(KPLDQWLKDMPALA)認識抗体を用いたウエスタンブロット法により解析した(図3)。酵母内発現UDP-グルクロン酸転移酵素遺伝子及びUDP-グルコース脱水素酵素タンパクの発現を確かめるために、表2の形質転換体1〜4の抽出液タンパクのウエスタンブロット解析を行った。パネル(A):抗UGT1A共通抗体、パネル(B):抗シロイヌナズナUDPGDH抗体。パネル(B)コントロール(AH22:pGYR/UGT1A6)には検出されないタンパクのバンドが1〜3のレーンで見られ、シロイナズナ由来UDP-グルコース脱水素酵素タンパクの発現が確認された。両遺伝子を挿入した発現ベクターを含む形質転換体菌体抽出液のみでそれぞれのタンパク発現が確かめられた。
【0117】
2-3. 酵母菌体におけるUDP-グルクロン酸の産生
実施例2-1で得られたUDP−グルコース脱水素酵素が発現している形質転換体について、酵母菌体内におけるUDP-グルクロン酸の生成を確認した。
【0118】
ラットUDP−グルコース脱水素酵素が発現している形質転換体を選択培地で24時間30℃で培養し、得られた酵母菌体に2.5倍容のクロロホルム:メタノール=3:1を添加し激しく攪拌した後、上層の水相の代謝物を高速液体クロマトグラフィーにより分析した。UDP-グルクロン酸のHPLCによる分析条件は以下のとおりである。カラム:Wako社製 WakoPack Navi C-30-5 (内径3.0mm × 長さ150mm)、検出波長:260nm、流速:0.35ml/min、カラム温度: 37℃、溶出条件: 20mM酢酸トリエチルアミン(pH7.0)
【0119】
図4は、酵母内におけるUDP-グルクロン酸生成を逆相HPLCを用いて分析した結果である。図中のAはUDP-グルクロン酸、UDP-グルコースの標準物質の溶出パターンを示し、Bはコントロール酵母(AH22:pGYR)、CはラットUDP−グルコース脱水素酵素を遺伝子導入した形質転換体酵母の抽出画分を示す。Bでは糖ヌクレオチドとしてはUDP-グルコースしか検出されないのに対して、CではUDP-グルクロン酸のピークが検出された。既知濃度の標準化合物との比較によりUDP−グルコース脱水素酵素が導入された酵母菌体内には、4〜5mMの濃度でUDP-グルクロン酸が生成されていることが確認された。
【0120】
実施例3
3. 酵母形質転換体を用いたグルクロン酸抱合体の製造
3-1. 酵母静止菌体を用いた7ヒドロキシクマリン抱合体の製造
UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素タンパクの同時発現した酵母静止菌体におけるグルクロン酸抱合体の生成を確かめるために、形質転換体を選択培地で48時間30℃で培養し、得られた酵母菌体を1−8%グルコースを含む適当な緩衝溶液に懸濁し、抱合化基質として0.1−1mM 7ヒドロキシクマリンを添加した。30℃の条件下で24時間振盪培養をおこない、培養液に2倍容のクロロホルム:メタノール=3:1を添加し、激しく攪拌した後、上層の水相と下層の有機相を回収し、有機相は減圧乾固し、アセトニトリルに溶解した。それぞれの代謝物を超高速液体クロマトグラフィーにより分析した。条件は以下のとおりである。カラム:nacalai tesque社製 2.5C18-MS-II (内径2.0mm × 長さ100mm)、検出波長:320nm、流速:0.5ml/min、カラム温度:45 ℃、溶出条件:水/アセトニトリル系(0.1%TFAを含む)、10%アセトニトリル(4分間)、10-70 %アセトニトリル直線濃度勾配(6分間)の後、70-10%アセトニトリル(2分間)、10%アセトニトリル(3分間)
【0121】
3-1-1. 種々のヒトUDP-グルクロン酸転移酵素との同時発現酵母における抱合体生成
シロイヌナズナ由来UDP-グルコース脱水素酵素を含むヒトUDP-グルクロン酸転移酵素との同時発現酵母株 (AH22: pAUR/At.UDPGDH + pGYR/hUGT1A1, 1A6,1A7,1A8,1A9)における7ヒドロキシクマリン抱合体の生成を確かめた。
【0122】
図5に示すように、いずれのヒトUDP-グルクロン酸転移酵素分子種(UGT1A1,1A6,1A7,1A8,1A9) においてもUDP-グルコース脱水素酵素との同時発現により、酵母菌体に直接添加した7ヒドロキシクマリンのグルクロン酸抱合体の生成が見られた。したがって、UDP-グルクロン酸転移酵素とUDP-グルコース脱水素酵素を同時発現させた酵母菌体内において、UDP-グルコース脱水素酵素によってUDP-グルクロン酸が供給され、抱合化基質へのグルクロン酸転移反応がUDP-グルクロン酸転移酵素によって触媒されることが確かめられた。また、UDP-グルクロン酸転移酵素のいずれの分子種に関しても触媒反応がみられたことから、同時発現させるUDP-グルクロン酸転移酵素分子種を選択することにより多様な抱合体調製が可能である。
【0123】
3-1-2. UGT1A6同時発現株における抱合体産生の時間変化
実施例3-1-1でもっとも変換効率の高かった同時発現酵母株 (AH22: pAUR/At.UDPGDH+pGYR/hUGT1A6) における7ヒドロキシクマリン抱合体の生成の時間依存性を調べた(図6)。
【0124】
図6に示されるように、加えた基質(0.5mM 7OHC:△)は反応時間に依存して減少し、抱合体(7GC:○)は直線的に増加してゆき、24時間経過後にはほぼ100%抱合体へ変換されることが示された。
【0125】
3-1-3. UGT1A6同時発現株における抱合体産生のグルコース濃度の影響
実施例3-1-1でもっとも変換効率の高かった同時発現酵母株 (AH22: pAUR/At.UDPGDH+pGYR/hUGT1A6) における7ヒドロキシクマリン抱合体の生成におけるグルコース濃度を調べた(図7)。
【0126】
図7に示されるように、添加したグルコース濃度に依存して抱合体の産生量は増加し、8%の濃度でほぼ100%変換を示した。さらに、生成した抱合体の培地と菌体への分布を調べると、90%以上の抱合体が培地中に分泌されていることがわかった。
【0127】
3-1-4.同時発現株における発現ベクターの組み合わせの影響
同時発現系による抱合体の産生能力における発現ベクターの組み合わせの影響を調べるために、表2に示した同時発現株(1〜4)を選択培地で24時間30℃で培養し、得られた酵母菌体を8%グルコースを含む適当な緩衝溶液に懸濁し、抱合化基質として1mM 7ヒドロキシクマリンを添加した。30℃の条件下で24時間振盪培養をおこない抱合体の培地中の濃度を測定した(図8)。
【0128】
図8に示されるように形質転換体1〜3の比較から発現ベクターの組み合わせに依存せず、抱合体産生能力を示すことが明らかとなった。また、形質転換体3及び4の比較から、酵母に導入されたUDP-グルコース脱水素酵素は生物種の由来に関係なくUDP-グルクロン酸の供給に関与し、抱合体生成に効率よく機能することが示された。
【0129】
3-2. 酵母静止菌体を用いたケルセチン抱合体の製造
シロイヌナズナ由来UDP-グルコース脱水素酵素を含むヒトUDP-グルクロン酸転移酵素との同時発現酵母株 (pAUR/At.UDPGDH+pGYR/hUGT1A1)におけるケルセチン抱合体の生成を確かめた。形質転換体を選択培地で48時間30℃で培養し、得られた酵母菌体を8%グルコースを含む0.1Mリン酸緩衝溶液(pH7.4)に懸濁し、抱合化基質として0.2mM ケルセチンを添加した。30℃の条件下で24時間振盪培養をおこない、培養液に2倍容のクロロホルム:メタノール=3:1を添加し、激しく攪拌した後、上層の水相と下層の有機相を回収し、有機相は減圧乾固し、アセトニトリルに溶解した。ケルセチン抱合体のHPLCによる分析条件は以下のとおりである。カラム:Wako社製 WakoPack Navi C-30-5(内径3.0mm×長さ150mm)、検出波長:370nm、流速:0.4ml/min、カラム温度:37℃、溶出条件:水/アセトニトリル系(0.5% リン酸を含む)、18 %アセトニトリル(10分間)、18-55%アセトニトリル直線濃度勾配(10分間)の後、55%アセトニトリル(5 分間)
【0130】
結果を図9に示す。シロイヌナズナ由来UDP-グルコース脱水素酵素を含むヒトUDP-グルクロン酸転移酵素との同時発現酵母株 (pAUR/At.UDPGDH+pGYR/hUGT1A1) におけるケルセチン抱合体の生成を確かめた。A:AH22:pGYR/UGT1A1、B:AH22/pGYRUGT1A1+pAUR/At.UDPGDH、C:AH22:pAUR/At.UDPGDH
【0131】
ケルセチンは分子内に抱合化をうける水酸基を4箇所有し、複数の抱合体が産生される。図9に示すように同時発現株においてのみB環の3’位及び4’位水酸基に特異的なケルセチン抱合体の産生が見られ、特に3’位抱合体が優勢に産生された。このようにUDP-グルコース脱水素酵素と同時発現させるUDP-グルクロン酸転移酵素分子種を選択することで、複数の水酸基を有する化合物に対して特異的な抱合体を製造することが可能である。
【0132】
3-3. 酵母静止菌体を用いたマイコフェノール酸抱合体の製造
シロイヌナズナ由来UDP-グルコース脱水素酵素を含むヒトUDP-グルクロン酸転移酵素との同時発現酵母株 (pAUR/At.UDPGDH+pGYR/hUGT1A9)におけるマイコフェノール抱合体の生成を確かめた。形質転換体を選択培地で48時間30℃で培養し、得られた酵母菌体を8%グルコース溶液に懸濁し、抱合化基質として0.2mM マイコフェノール酸を添加した。30℃の条件下で24時間振盪培養をおこない、培養液に2倍容のクロロホルム:メタノール=3:1を添加し、激しく攪拌した後、上層の水相と下層の有機相を回収し、有機相は減圧乾固し、アセトニトリルに溶解した。代謝物を超高速液体クロマトグラフィーにより分析した。条件は以下のとおりである。カラム:nacalai tesque社製 2.5C18-MS-II(内径2.0mm×長さ100mm)、検出波長:250nm、流速:0.5ml/min、カラム温度:45℃、溶出条件:水/アセトニトリル系(0.1%TFAを含む)、20-40 %アセトニトリル直線濃度勾配(7分間)の後、40%アセトニトリル(2分間)、40-20%アセトニトリル(2分間)、20%アセトニトリル(2分間)
【0133】
シロイヌナズナ由来UDP-グルコース脱水素酵素を含むヒトUDP-グルクロン酸転移酵素との同時発現酵母株 (pAUR/At.UDPGDH+pGYR/hUGT1A9) におけるマイコフェノール酸抱合体の生成を確かめた。結果を図10に示す。
A:標準物質(MPA:マイコフェノール酸、G1:O-glucuronide, G2: Acyl glucuronide),
B:AH22/pGYRUGT1A9+pAUR/At.UDPGDH,
C:AH22:pAUR/At.UDPGDH(コントロール)
【0134】
マイコフェノール酸は、分子内にフェノール性水酸基とカルボキシル基を持ち2種の抱合体が産生される。図10に示すようにUGT1A9との同時発現株によってのみフェノール性水酸基に特異的なマイコフェノール酸抱合体(o-glucuronide) の産生が見られた。このようにUDP-グルコース脱水素酵素と同時発現させるUDP-グルクロン酸転移酵素分子種を選択することで、異なる抱合化官能基を有する化合物に対して特異的な抱合体を製造することが可能である。
【0135】
3-4. 酵母静止菌体を用いたアシルグルクロン酸抱合体の製造
グルクロン酸転移酵素を発現した酵母静止菌体におけるアシルグルクロン酸抱合体の生成を確かめるために、形質転換体を選択培地で24時間30℃で培養し、得られた酵母菌体を8%グルコースを含む適当な緩衝溶液に懸濁し、抱合化基質として1mM メフェナム酸を添加した。30℃の条件下で24時間シントウ培養をおこない、培養液に2.5倍容のクロロホルム:メタノール=3:1を添加し、激しく攪拌した後、上層の水相の代謝物を超高速液体クロマトグラフィーにより分析した。条件は以下のとおりである。カラム:Nacalai Tesque社製 2.5C18-MS-II (内径2.0mm × 長さ100mm)、検出波長:320nm、流速:0.5ml/min、カラム温度:45℃、溶出条件:水/アセトニトリル系(0.1%TFAを含む)、5-40%アセトニトリル直線濃度勾配(5分間)、40-100%アセトニトリル直線濃度勾配(3分間)の後、100%アセトニトリル(1分間)、100-5%アセトニトリル直線濃度勾配、5%アセトニトリル(3分間)
【0136】
図11はヒトグルクロン酸転移酵素分子種を発現した酵母静止菌体を用いてメフェナム酸のアシル抱合体の生産量を乾燥菌体重量あたりの日生産量を示したものである。UGT1A1,1A7,1A8,1A9及び1A10がアシル抱合体への変換能を示し、特に1A8,1A9,1A10が高い抱合能をもつことが示された。
【0137】
3-5. 酵母静止菌体を用いたNグルクロン酸抱合体の製造
グルクロン酸転移酵素を発現した酵母静止菌体におけるNグルクロン酸抱合体の生成を確かめるために、形質転換体を選択培地で24時間30℃で培養し、得られた酵母菌体を8%グルコースを含む適当な緩衝溶液に懸濁し、抱合化基質として0.5mM タモキシフェンを添加した。30℃の条件下で24時間シントウ培養をおこない、培養液に2.5倍容のクロロホルム:メタノール=3:1を添加し、激しく攪拌した後、上層の水相の代謝物を高速液体クロマトグラフィーにより分析した。
タモキシフェン抱合体のHPLCによる分析条件は以下のとおりである。カラム:Wako社製 WakoPack Navi C-30-5 (内径3.0mm × 長さ150mm)、検出波長:254nm、流速:0.5ml/min、カラム温度: 37℃、溶出条件:100mM酢酸アンモニウム(pH5.0)/アセトニトリル系、25 %アセトニトリル(5分間)、25-75%アセトニトリル直線濃度勾配(25分間)の後、75%アセトニトリル(10分間)、75-25%アセトニトリル直線濃度勾配(5分間)の後、25%アセトニトリル(5分間)
【0138】
抗がん剤として知られるタモキシフェンは分子内に抱合化を受ける窒素原子が存在する化合物である。図12はヒトグルクロン酸転移酵素分子種(UGT1A4及び1A9)を発現した酵母静止菌体を用いてタモキシフェン抱合体(Nグルクロン酸抱合体)生成を分析したHPLCの溶出パターンを示したものである。Nグルクロン酸抱合能を有するUGT分子種であるUGT1A4を発現した酵母静止菌体をもちいた場合についてのみ抱合体(溶出時間11.8分)の産生が見られた。
【0139】
実施例4
出芽酵母と分裂酵母におけるグルクロン酸抱合体産生能の比較
出芽酵母(Saccharomyces cerevisiae)におけるグルクロン酸抱合体産生能を分裂酵母(Schizosaccharomyces pombe)の系と比較するために、表5に示す形質転換出芽酵母(表2に示す形質転換体No.4、UGT分子種: UGT1A1(N末端シグナル配列置換グルクロン酸転移酵素(UGT1A1)を含む酵母発現ベクター(pGYR/UGT1A1)を用いた)またはUGT1A6)を選択培地で、30℃で24時間培養し、得られた酵母菌体を、8%グルコースを含む緩衝溶液に懸濁し、抱合化基質として0.5mM 4メチルウンベリフェロンを添加した。30℃の条件下で24時間振盪培養を行い、培養液に2倍容のクロロホルム:メタノール=3:1を添加し、激しく攪拌した後、上層の水相と下層の有機相を回収し、有機相は減圧乾固し、アセトニトリルに溶解した。それぞれの代謝物を超高速液体クロマトグラフィーにより分析した。条件は以下のとおりである。
【0140】
カラム:nacalai tesque社製 2.5C18-MS-II (内径2.0mm×長さ100mm)、検出波長:320nm、流速:0.5ml/min、カラム温度:45℃、溶出条件:水/アセトニトリル系(0.1%TFAを含む)、10%アセトニトリル(4分間)、10-70%アセトニトリル直線濃度勾配(6分間)の後、70-10%アセトニトリル(2分間)、10%アセトニトリル(3分間)
【0141】
表6には出芽酵母及び分裂酵母(文献値)における4メチルウンベリフェロングルクロン酸抱合体産生能比較を示した。出芽酵母の抱合体産生能力に関して分裂酵母を用いた場合と比べて、1日あたり抱合体濃度はUGT1A1に関しては10倍、UGT1A6では50倍以上高い値を示した。また、酵母乾燥重量あたりの抱合体生成量も20〜100倍以上高い値を示した。このように複数のUGT分子種に関して分裂酵母を宿主として用いた場合よりも、出芽酵母におけるグルクロン酸抱合体産生系が有利なことが示された。
【0142】
【表6】
【0143】
実施例5
5-1. シトクロムP450を含む同時発現株の静止菌体を用いた7ヒドロキシクマリン抱合体の製造
生体で生じる抱合体の多くは、その前段階としてシトクロムP450によるモノオキシゲナーゼ反応を受け、抱合化をうける官能基としてはたらく水酸基の導入を受ける。酵母においてシトクロムP450及びUDP-グルクロン酸転移酵素の同時発現株より調製されたミクロソーム画分を用いて試験管内の反応により、7エトキシクマリンを基質として、シトクロムP450による脱エチル反応による7ヒドロキシクマリン生成、引き続いてUDP-グルクロン酸転移酵素による抱合化が観測された(文献番号6)。このP450とUDP-グルクロン酸転移酵素の連続的な変換反応を酵母菌体内で可能にするために、pAUR-At.UDPGDH/UGT1A6及びpGYR/ratCYP1A1 の発現ベクターを同時に形質転換した酵母株(表2,形質転換体5)を作成した。形質転換体を選択培地で48時間30℃で培養し、得られた酵母菌体を8%グルコースを含む溶液に懸濁し、抱合化基質として1mM 7エトキシクマリンを添加した。30℃の条件下で72時間振盪培養をおこない、培養液に生成される抱合体生成の時間変化を調べた(図13)。
【0144】
図13にみられるように、添加した7エトキシクマリンはP450により脱エチル化をうけ、7ヒドロキシクマリン(●)に変換された後、UGTによって抱合化を受けて抱合体(○)に変換された。抱合体の生成においては時間的なラグは見られず酵母内にて生成された中間体である7ヒドロキシクマリンが抱合化反応の基質として供給され効率的に抱合体に変換されているものと思われる。したがって、シトクロムP450を含む同時発現株は抱合化基質の前駆体から効率よく抱合体を調製する系として有用である。
【0145】
5-2. ヒト由来シトクロムP450を含む同時発現株の静止菌体を用いた7-エトキシクマリン由来抱合体の製造
実施例3で示したように抱合体の多くはシトクロムP450の代謝に引き続きグルクロン酸抱合をうけて産生される。そこでとくにヒト代謝物を製造するためにヒト由来シトクロムP450を含む同時発現酵母株の構築をおこない7エトキシクマリンの代謝解析を行なった。
【0146】
5-2-1 ヒト由来シトクロムP450発現ベクター(pPYR)の構築
ヒト由来シトクロムP450を含む同時発現株を構築するために、出芽酵母グリセロールリン酸キナーゼ由来のプロモーター及びターミネーター領域をクローニングして前述のpPYRの発現調節領域を置換した自律複製型酵母発現ベクター(pPYR)を作成した(図14)。
【0147】
出芽酵母AH22株より常法にてゲノムDNAを調製し鋳型DNAとした。以下の示す配列を有するプライマーDNAを用いてPCR法によってグリセロールリン酸キナーゼ(PGK1)由来のプロモーター及びターミネーター領域のクローニングを行った。
【0148】
DNAポリメラーゼとしてはKOD-plus-(TOYOBO)を用いた。反応液組成は製造業者の指示に従い、以下に示すプライマーと反応条件でPCRを行った。
PGK1由来のプロモーター領域
フォワード配列 : 配列番号 21
リバース配列 : 配列番号 22
PGK1由来のターミネーター領域
フォワード配列 : 配列番号 23
リバース配列 : 配列番号 24
【0149】
【0150】
PCR条件
変性 94℃ 2分
5サイクル 94℃ 15秒、42℃ 30秒、68℃1分45秒
30サイクル 94℃ 15秒、50℃ 30秒、68℃1分45秒
最終伸長 68℃10分
【0151】
PCR産物を1%アガロースゲルで電気泳動した結果、目的サイズ(グリセロールリン酸キナーゼ由来のプロモーター領域:約1.5kb及びグリセロールリン酸キナーゼ由来のターミネーター領域:0.3kb)に特異的な増幅がみられた。(以下、1%アガロースゲルでの電気泳動は、単に電気泳動と略称する)。PCR増幅溶液を電気泳動して、DNA断片をゲルから分離した後、TAクローニングキット(Target cloneTM-plus-,TOYOBO社)を用いてpTAベクターに挿入した。このクローニングされた遺伝子のシークエンスを行い遺伝子配列を解析したところ、配列番号25(Genebank Acc.No. BK006937, TPA: Saccharomyces cerevisiae S288c chromosome III, complete sequence; 136264〜137743)及び26(Genebank Acc.No. BK006937, TPA: Saccharomyces cerevisiae S288c chromosome III, complete sequence; 138995〜139270)に示した塩基配列であることを確認した。
【0152】
【0153】
PGK1プロモーターあるいはターミネーター領域を含むプラスミド約1μgを37℃で4時間、NotI及びHindIII処理し、電気泳動後、遺伝子断片をゲルから切り出した。切り出したゲルからWizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてインサート断片を調製した。
【0154】
サブクローニングベクターとして用いるpGEM-T-Easy Vector (Promega)を37℃で4時間NotI処理し、電気泳動後、目的サイズ(約3kb)のバンドを切り出した。その後、Wizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてベクター断片を調製した。
【0155】
2種のインサート断片とpGEM-NotIベクター断片の濃度を電気泳動で確認後、それらのモル比が3:1〜10:1程度になるように混合し、等量のDNA Ligation Kit Ver. 2 (TaKaRa)のsolution Iを加え、16℃、1時間反応を行った。その後、大腸菌JM109株にヒートショック法を用いてトランスフォーメーションを行い、LBプレート(アンピシリン50μg/ml含有)に展開した。得られた大腸菌コロニーのうち数個を選択し、それらを鋳型としてコロニーPCRを行った。DNAポリメラーゼは、Ex Taq(登録商標) DNAポリメラーゼを用いた。インサート確認のためのコロニーPCRには、プライマーとして配列番号21と24を用いた。以下の条件でPCRを行った。
【0156】
PCR条件
変性 98℃5分
30サイクル 94℃ 30秒、50℃ 30秒、72℃ 2分30秒
最終伸長 72℃4分
【0157】
PCR後、電気泳動を行い、予想サイズ(約1.8kb)に特異的な増幅がみられるクローンを数個得た。得られた大腸菌コロニーを5mlのLB培地(アンピシリン100μg/ml含有)に植菌し、37℃、200rpmで16時間振盪培養を行った。その後アルカリSDS法を用いてプラスミドを抽出した。そして、その一部を37℃で1時間、NotI及びHindIII処理し、電気泳動をおこない予想されるインサートの導入を再度確認し、発現分子種挿入部位にHindIIIをもつPGK1由来プロモーター及びターミネーター領域を得た(pGEM/PGK-1-P/T)。
【0158】
PGK1プロモーター及びターミネーター領域を含むプラスミド約1μgを37℃で4時間、NotI処理し、電気泳動後、遺伝子断片をゲルから切り出した。切り出したゲルからWizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてインサート断片を調製した。
【0159】
pGYRを37℃で4時間NotI処理し、電気泳動後、目的サイズ(約9kb)のバンドを切り出した。その後、Wizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてベクター断片を調製した。
【0160】
インサート断片とpGYR-NotIベクター断片の濃度を電気泳動で確認後、それらのモル比が3:1〜10:1程度になるように混合し、等量のDNA Ligation Kit Ver. 2(TaKaRa)のsolution Iを加え、16℃、1時間反応を行った。その後、大腸菌JM109株にヒートショック法を用いてトランスフォーメーションを行い、LBプレート(アンピシリン50μg/ml含有)に展開した。得られた大腸菌コロニーのうち数個を選択し、それらを鋳型としてコロニーPCRを行った。DNAポリメラーゼは、Ex Taq(登録商標) DNAポリメラーゼを用いた。インサート確認のためのコロニーPCRには、プライマーとして配列番号21と24を用いた。以下の条件でPCRを行った。
【0161】
PCR条件
変性 98℃5分
30サイクル 94℃ 30秒、50℃ 30秒、72℃ 2分30秒
最終伸長 72℃4分
【0162】
PCR後、電気泳動を行い、予想サイズ(約1.8kb)に特異的な増幅がみられるクローンを数個得た。得られた大腸菌コロニーを5mlのLB培地(アンピシリン100μg/ml含有)に植菌し、37℃、200rpmで16時間振盪培養を行った。その後アルカリSDS法を用いてプラスミドを抽出した。そして、その一部を37℃で1時間、NotI及びHindIII処理した後、電気泳動をおこない予想されるインサートの導入を再度確認し、PGK1由来プロモーター及びターミネーター領域を有する自律複製型酵母発現ベクター(pPYR)を得た。
【0163】
ヒトシトクロムP450断片を含むプラスミド約1μgを37℃で4時間、HindIII処理し、電気泳動後、遺伝子断片をゲルから切り出した。切り出したゲルからWizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてインサート断片を調製した。
【0164】
pPYRを37℃で4時間HindIII処理し、電気泳動後、目的サイズ(約11kb)のバンドを切り出した。その後、Wizard(登録商標)SV Gel and PCR Clean-Up System (Promega)を用いてベクター断片を調製した。
【0165】
ヒトチトクロムP450遺伝子を含むインサート断片とpPYR-NotIベクター断片の濃度を電気泳動で確認後、それらのモル比が3:1〜10:1程度になるように混合し、等量のDNA Ligation Kit Ver. 2 (TaKaRa)のsolution Iを加え、16℃、1時間反応を行った。その後、大腸菌JM109株にヒートショック法を用いてトランスフォーメーションを行い、LBプレート(アンピシリン50μg/ml含有)に展開した。 得られた大腸菌コロニーのうち数個を選択し、それらを鋳型としてコロニーPCRを行った。DNAポリメラーゼは、Ex Taq(登録商標) DNAポリメラーゼを用いた。インサート確認のためのコロニーPCRには、プライマーとして配列番号21と挿入するシトクロムP450内部リバース配列を用いた。以下の条件でPCRを行った。
【0166】
PCR条件
変性 98℃5分
30サイクル 94℃ 30秒、50℃ 30秒、72℃ 2分30秒
最終伸長 72℃4分
【0167】
PCR後、電気泳動を行い、予想サイズに特異的な増幅がみられるクローンを数個得た。得られた大腸菌コロニーを5mlのLB培地(アンピシリン100μg/ml含有)に植菌し、37℃、200rpmで16時間振盪培養を行った。その後アルカリSDS法を用いてプラスミドを抽出した。そして、その一部を37℃で1時間、NotI及びHindIII処理した後、電気泳動をおこない予想されるインサートの導入を再度確認し、ヒトシトクロムP450を含む自律複製型酵母発現ベクター(pPYR/CYP)を得た。
【0168】
5-2-2 ヒト由来シトクロムP450を含む同時発現株の静止菌体を用いた7-エトキシクマリン由来抱合体の製造
ヒトシトクロムP450とUDP-グルクロン酸転移酵素によるヒト特異的な連続的な変換反応を酵母菌体内で可能にするために、pAUR-rat.UGDH/UGT及びpPYR/humanCYPの発現ベクターを同時に形質転換した酵母株を作成した。
形質転換体を選択培地で24時間30℃で培養し、得られた酵母菌体を8%グルコースを含む溶液に懸濁し、抱合化基質として1mM 7エトキシクマリンを添加した。30℃の条件下で24時間シントウ培養をおこない、培養液に生成される代謝物を調べた(図15、表7)。
【0169】
【表7】
【0170】
図15にはヒトシトクロムP450(CYP2D6)とUDP-グルクロン酸転移酵素(UGT1A8)を同時発現させた酵母株静止菌体における7エトキシクマリン代謝物のHPLC分析パターンを示す。添加した7エトキシクマリンはヒトシトクロムP450により脱エチル化あるいは3位水酸化をうけ、7ヒドロキシクマリン(7OHC)あるいは3ヒドロキシ-7エトキシクマリン(3OH−7EC)に変換された後、UGT1A8によってそれぞれ抱合化を受けて抱合体(7GCあるいは3G−7EC)に変換された。
また、表7にはヒトシトクロムP450とUDP-グルクロン酸転移酵素の異なる分子種の組み合わせにより構築した同時発現酵母株を用いた7エトキシクマリンの抱合体製造結果を示す。いずれの組み合わせにおいてもP450の代謝物である7OHCあるいは3OH−7ECの抱合体の生成が見られた。
【産業上の利用可能性】
【0171】
医薬品開発におけるヒト体内における医薬品代謝物の解析に関する分野に有用である。
【特許請求の範囲】
【請求項1】
UDP-グルコース脱水素酵素をコードする遺伝子及びUDP-グルクロン酸転移酵素をコードする遺伝子を発現可能に挿入して形質転換した出芽酵母。
【請求項2】
シトクロムP450遺伝子をコードする遺伝子をさらに発現可能に挿入して形質転換した、請求項1に記載の出芽酵母。
【請求項3】
以下の(A)〜(G)のいずれかから選ばれる形質転換出芽酵母。
(A)UDP-グルコース脱水素酵素発現ベクター及びUDP-グルクロン酸転移酵素発現ベクターで形質転換した出芽酵母、
(B)UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクターで形質転換した出芽酵母、
(C)UDP-グルコース脱水素酵素発現ベクター、UDP-グルクロン酸転移酵素発現ベクター及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母、
(D)UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクター、及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母、
(E)UDP-グルクロン酸転移酵素発現ベクター、及びUDP-グルコース脱水素酵素及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母、
(F)UDP-グルクロン酸転移酵素及びシトクロムP450遺伝子発現ベクター、及びUDP-グルコース脱水素酵素発現ベクターで形質転換した出芽酵母、並びに
(G)UDP-グルコース脱水素酵素、UDP-グルクロン酸転移酵素及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母
【請求項4】
UDP-グルコース脱水素酵素発現ベクターが、出芽酵母発現ベクターにUDP-グルコース脱水素酵素遺伝子を発現可能に挿入したものであり、
UDP-グルクロン酸転移酵素発現ベクターが、出芽酵母発現ベクターにUDP-グルクロン酸転移酵素遺伝子を発現可能に挿入したものであり、
UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクターが、出芽酵母発現ベクターにUDP-グルクロン酸転移酵素遺伝子及びUDP-グルコース脱水素酵素含を発現可能に挿入したものである、請求項3に記載の形質転換出芽酵母。
【請求項5】
出芽酵母発現ベクターが自律複製型ベクターまたは染色体組み込み型ベクターである、請求項4に記載の形質転換出芽酵母。
【請求項6】
UDP-グルコース脱水素酵素遺伝子が動物または植物由来の遺伝子である、請求項1、2、4〜5のいずれかに記載の形質転換出芽酵母。
【請求項7】
UDP-グルコース脱水素酵素遺伝子がシロイヌナズナ由来遺伝子またはラット由来遺伝子である、請求項1、2、4〜5のいずれかに記載の形質転換出芽酵母。
【請求項8】
UDP-グルクロン酸転移酵素遺伝子が哺乳動物由来の遺伝子である、請求項1、2、4〜7のいずれかに記載の形質転換出芽酵母。
【請求項9】
UDP-グルクロン酸転移酵素遺伝子がヒト由来の遺伝子である、請求項1、2、4〜7のいずれかに記載の形質転換出芽酵母。
【請求項10】
シトクロムP450遺伝子発現ベクターが、出芽酵母発現ベクターにシトクロムP450遺伝子を発現可能に挿入したものである、請求項3〜9のいずれかに記載の形質転換出芽酵母。
【請求項11】
前記UDP-グルクロン酸転移酵素発現ベクターは、UDP-グルクロン酸転移酵素遺伝子が、発現量がUGT1A7の50%以下である低発現量グルクロン酸転移酵素遺伝子である場合、
前記低発現量グルクロン酸転移酵素遺伝子は、シグナル配列遺伝子を、発現量がUGT1A7の80%以上である高発現グルクロン酸転移酵素のシグナル配列遺伝子と置換したものであるか、または
シグナル配列遺伝子を(A)以下に示すアミノ酸配列(a)〜(c)のいずれかをコードする遺伝子、(B)アミノ酸配列(a)〜(c)のアミノ酸の1〜5個が置換若しくは欠失したアミノ酸配列のいずれかをコードする遺伝子、または(C)1〜5個のアミノ酸がアミノ酸配列(a)〜(c)のアミノ酸に付加されたアミノ酸配列のいずれかをコードする遺伝子と置換したものであり、但し、(B)の置換若しくは欠失したアミノ酸配列および(C)の付加されたアミノ酸配列をコードする遺伝子は、シグナル配列遺伝子とした場合にグルクロン酸転移酵素の発現量が野生型の80%以上である、請求項1〜10のいずれかに記載の形質転換出芽酵母。
(a) MARAGWTGLLPLYVCLLLTCGFAKAG(配列番号1)
(b) MACLLRSFQRISAGVFFLALWGMVVG(配列番号2)
(c) MAPRRVDQPRSFMCVSTADLWLCEAG(配列番号3)
【請求項12】
前記低発現量グルクロン酸転移酵素は、UGT1A1、UGT1A4、UGT1A8またはUGT1A9である請求項11に記載の形質転換出芽酵母。
【請求項13】
前記高発現量グルクロン酸転移酵素は、UGT1A7、UGT1A6またはUGT1A10である請求項11または12に記載の形質転換出芽酵母。
【請求項14】
被抱合物質のグルクロン酸抱合体生成に用いるための、請求項1〜13のいずれか1項に記載の出芽酵母。
【請求項1】
UDP-グルコース脱水素酵素をコードする遺伝子及びUDP-グルクロン酸転移酵素をコードする遺伝子を発現可能に挿入して形質転換した出芽酵母。
【請求項2】
シトクロムP450遺伝子をコードする遺伝子をさらに発現可能に挿入して形質転換した、請求項1に記載の出芽酵母。
【請求項3】
以下の(A)〜(G)のいずれかから選ばれる形質転換出芽酵母。
(A)UDP-グルコース脱水素酵素発現ベクター及びUDP-グルクロン酸転移酵素発現ベクターで形質転換した出芽酵母、
(B)UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクターで形質転換した出芽酵母、
(C)UDP-グルコース脱水素酵素発現ベクター、UDP-グルクロン酸転移酵素発現ベクター及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母、
(D)UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクター、及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母、
(E)UDP-グルクロン酸転移酵素発現ベクター、及びUDP-グルコース脱水素酵素及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母、
(F)UDP-グルクロン酸転移酵素及びシトクロムP450遺伝子発現ベクター、及びUDP-グルコース脱水素酵素発現ベクターで形質転換した出芽酵母、並びに
(G)UDP-グルコース脱水素酵素、UDP-グルクロン酸転移酵素及びシトクロムP450遺伝子発現ベクターで形質転換した出芽酵母
【請求項4】
UDP-グルコース脱水素酵素発現ベクターが、出芽酵母発現ベクターにUDP-グルコース脱水素酵素遺伝子を発現可能に挿入したものであり、
UDP-グルクロン酸転移酵素発現ベクターが、出芽酵母発現ベクターにUDP-グルクロン酸転移酵素遺伝子を発現可能に挿入したものであり、
UDP-グルクロン酸転移酵素及びUDP-グルコース脱水素酵素発現ベクターが、出芽酵母発現ベクターにUDP-グルクロン酸転移酵素遺伝子及びUDP-グルコース脱水素酵素含を発現可能に挿入したものである、請求項3に記載の形質転換出芽酵母。
【請求項5】
出芽酵母発現ベクターが自律複製型ベクターまたは染色体組み込み型ベクターである、請求項4に記載の形質転換出芽酵母。
【請求項6】
UDP-グルコース脱水素酵素遺伝子が動物または植物由来の遺伝子である、請求項1、2、4〜5のいずれかに記載の形質転換出芽酵母。
【請求項7】
UDP-グルコース脱水素酵素遺伝子がシロイヌナズナ由来遺伝子またはラット由来遺伝子である、請求項1、2、4〜5のいずれかに記載の形質転換出芽酵母。
【請求項8】
UDP-グルクロン酸転移酵素遺伝子が哺乳動物由来の遺伝子である、請求項1、2、4〜7のいずれかに記載の形質転換出芽酵母。
【請求項9】
UDP-グルクロン酸転移酵素遺伝子がヒト由来の遺伝子である、請求項1、2、4〜7のいずれかに記載の形質転換出芽酵母。
【請求項10】
シトクロムP450遺伝子発現ベクターが、出芽酵母発現ベクターにシトクロムP450遺伝子を発現可能に挿入したものである、請求項3〜9のいずれかに記載の形質転換出芽酵母。
【請求項11】
前記UDP-グルクロン酸転移酵素発現ベクターは、UDP-グルクロン酸転移酵素遺伝子が、発現量がUGT1A7の50%以下である低発現量グルクロン酸転移酵素遺伝子である場合、
前記低発現量グルクロン酸転移酵素遺伝子は、シグナル配列遺伝子を、発現量がUGT1A7の80%以上である高発現グルクロン酸転移酵素のシグナル配列遺伝子と置換したものであるか、または
シグナル配列遺伝子を(A)以下に示すアミノ酸配列(a)〜(c)のいずれかをコードする遺伝子、(B)アミノ酸配列(a)〜(c)のアミノ酸の1〜5個が置換若しくは欠失したアミノ酸配列のいずれかをコードする遺伝子、または(C)1〜5個のアミノ酸がアミノ酸配列(a)〜(c)のアミノ酸に付加されたアミノ酸配列のいずれかをコードする遺伝子と置換したものであり、但し、(B)の置換若しくは欠失したアミノ酸配列および(C)の付加されたアミノ酸配列をコードする遺伝子は、シグナル配列遺伝子とした場合にグルクロン酸転移酵素の発現量が野生型の80%以上である、請求項1〜10のいずれかに記載の形質転換出芽酵母。
(a) MARAGWTGLLPLYVCLLLTCGFAKAG(配列番号1)
(b) MACLLRSFQRISAGVFFLALWGMVVG(配列番号2)
(c) MAPRRVDQPRSFMCVSTADLWLCEAG(配列番号3)
【請求項12】
前記低発現量グルクロン酸転移酵素は、UGT1A1、UGT1A4、UGT1A8またはUGT1A9である請求項11に記載の形質転換出芽酵母。
【請求項13】
前記高発現量グルクロン酸転移酵素は、UGT1A7、UGT1A6またはUGT1A10である請求項11または12に記載の形質転換出芽酵母。
【請求項14】
被抱合物質のグルクロン酸抱合体生成に用いるための、請求項1〜13のいずれか1項に記載の出芽酵母。
【図1】

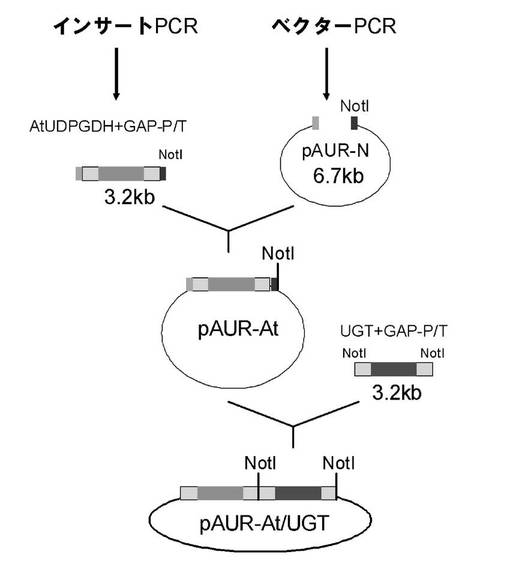
【図2】
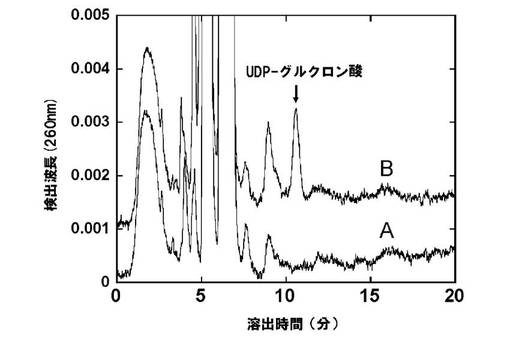
【図3】
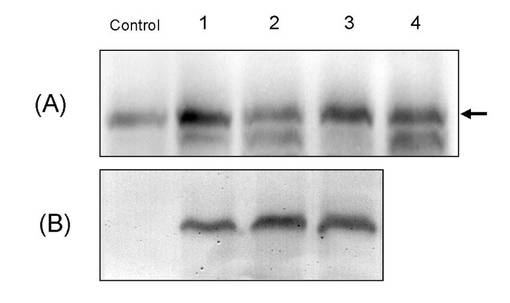
【図4】
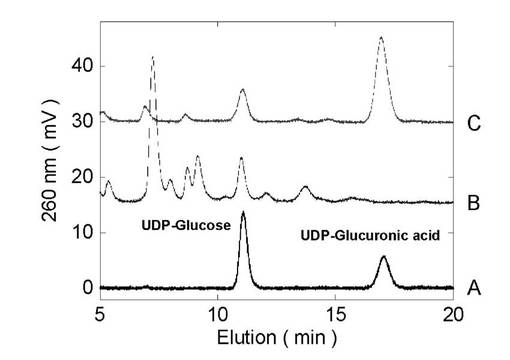
【図5】
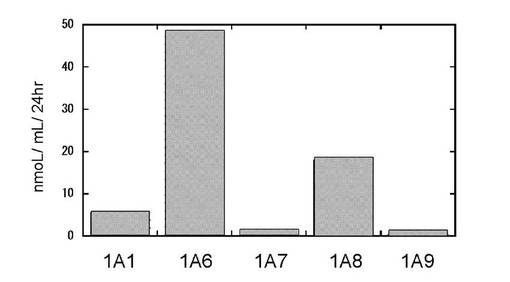
【図6】
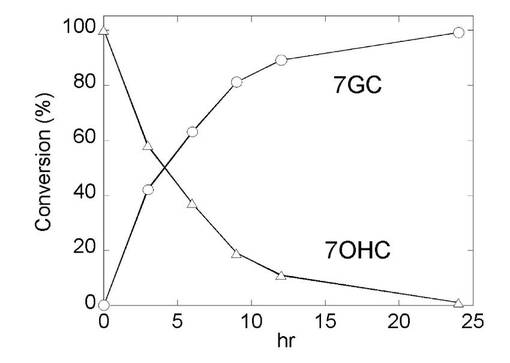
【図7】
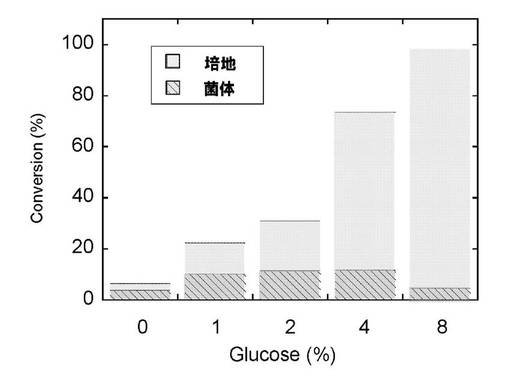
【図8】
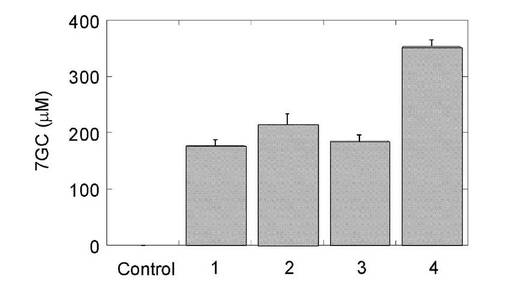
【図9】
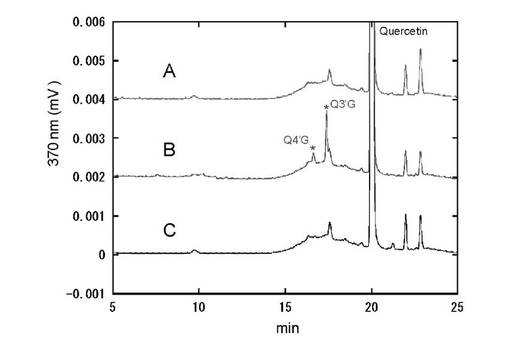
【図10】
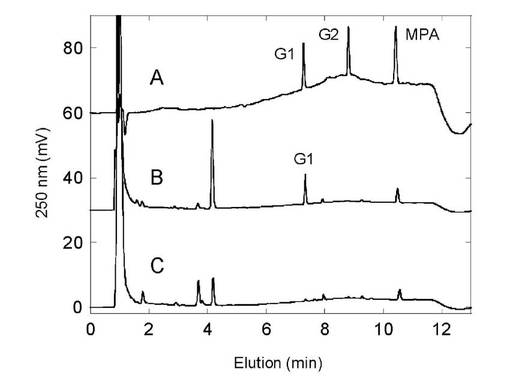
【図11】
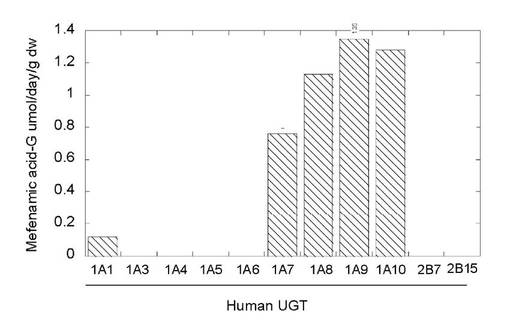
【図12】
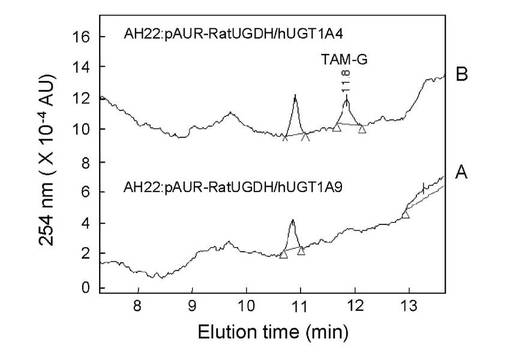
【図13】
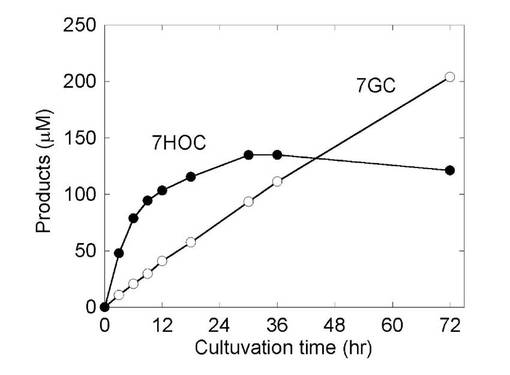
【図14】
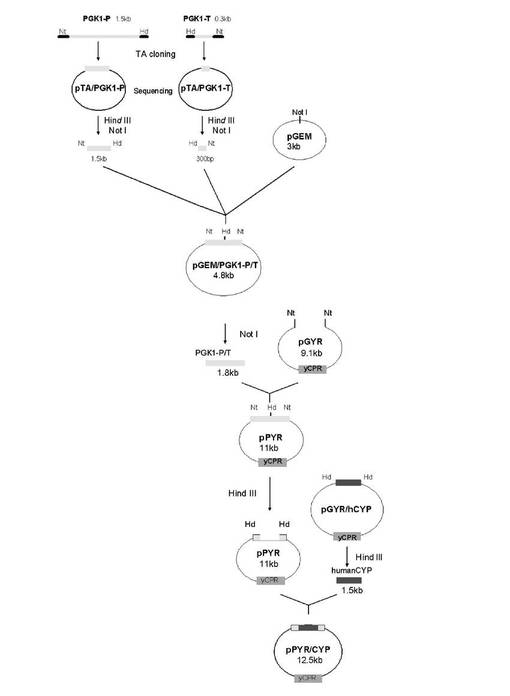
【図15】
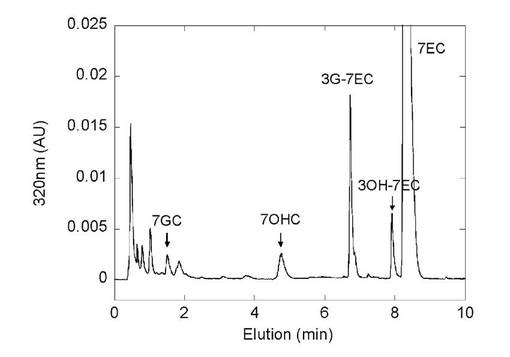
【図16】
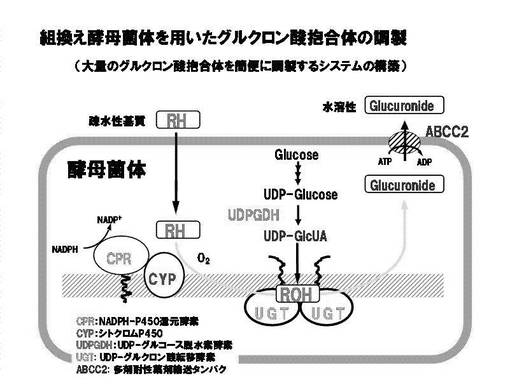
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【公開番号】特開2012−130357(P2012−130357A)
【公開日】平成24年7月12日(2012.7.12)
【国際特許分類】
【出願番号】特願2012−89372(P2012−89372)
【出願日】平成24年4月10日(2012.4.10)
【分割の表示】特願2011−28516(P2011−28516)の分割
【原出願日】平成23年2月14日(2011.2.14)
【出願人】(000236920)富山県 (197)
【Fターム(参考)】
【公開日】平成24年7月12日(2012.7.12)
【国際特許分類】
【出願日】平成24年4月10日(2012.4.10)
【分割の表示】特願2011−28516(P2011−28516)の分割
【原出願日】平成23年2月14日(2011.2.14)
【出願人】(000236920)富山県 (197)
【Fターム(参考)】
[ Back to top ]