
分散良好な微結晶二酸化チタン生成物の調製方法、その生成物、およびその使用
本発明は、分散良好なルチル構造の微結晶二酸化チタン生成物を調製する方法、この方法により調製される生成物、ならびにこの生成物により得られる装飾塗料およびラッカーに関する。この方法においては、まず、二酸化チタン原料を、アルカリ性のpHを有するように塩基で処理し、その後に、8から30g/lの塩酸含有率を有するように、それを酸で処理し、中和、処理、焼成する。特に、この調製方法は、分散性を改善するために、焼成段階前に前処理薬品を使用することを特徴とする。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、分散良好であるルチル構造の微結晶二酸化チタン生成物を調製する方法、その方法により得られる生成物、ならびにその生成物により得られる装飾塗料および木材ラックに関する。
【背景技術】
【0002】
微結晶酸化チタンの特性は、白色顔料として知られている従来の顔料二酸化チタンの特性と異なる。微結晶酸化チタンの結晶サイズ(10から100nm)は、従来の二酸化チタン(160から250nm)の結晶サイズより約5から10倍小さいので、その差は結晶サイズの違いによるものである。結晶サイズが減少するにつれ、可視光の範囲内における二酸化チタンの隠蔽力は消え、顔料は透明になる。他方で、紫外線に対する透過度は減少する。したがって、微結晶二酸化チタンはUV保護剤に適している。その小さい結晶サイズ、および広い比表面積のため、微結晶二酸化チタンは、例えば、化粧品、触媒、セラミック材料の中において、および塗料中の装飾顔料として有用である。
【0003】
微結晶二酸化チタンの調製における1つの問題は、工程の間、所望の結晶および粒子のサイズ分布を作り出し、調節し、保持することであり、所望のサイズ分布には、平均サイズ、および狭い幅のサイズ分布の両方が含まれる。様々な調製方法を評価するときに、二酸化チタン生成物の、純度、ならびに結晶および粒子のサイズ分布の制御は重要な因子である。さらに、調製工程は、エネルギー的に、かつ経済的に有利であり、環境に優しくあるべきである。
【0004】
TiO2は、3種の異なる結晶形で生じ得る。ルチルは、高温で安定な形である。アナターゼは、低温で支配的な形である。低温において、一般に斜方晶の形で発生する鉱物の中にのみ見出されるブルッカイトの形も生じ得る。ルチルは、最も耐久性のある結晶形として知られており、紫外光に対するその透過度はアナターゼの透過度より低い。
【0005】
純粋なルチルより、アナターゼおよびルチルの混合物を調製する方が容易であることは、既知の事実である。例えば、亜鉛等のルチル化薬品と呼ばれるものの存在下でアナターゼの形の二酸化チタンを焼成することにより、アナターゼおよびルチルの混合物をより低い温度でも生成することができる。
【0006】
気相技術または沈殿のいずれかを用い、微結晶二酸化チタンを様々な方法により調製することができる。チタン源には、例えば、イルメナイトから調製される四塩化チタン、チタンアルコキシドまたはチタン水和物が含まれ得る。ルチルの調製について様々な沈殿法に特許が付与されており、それらのいくつかに対する参照が、出願人の以前の特許明細書EP0444798に組み込まれている。
【0007】
特許明細書EP0444798には、微結晶二酸化チタンの調製方法が開示されており、そこでは第1の段階で、二酸化チタン水和物を、アルカリ性のpH値を有するように塩基で処理し、第2の段階で、塩酸の最終的な含有率を8から25g/lの間に調節するように塩酸による処理を続ける。第3の段階では、酸性混合物が塩基により中和され、中和された反応混合物を、例えば焼成により後処理することができる。第2の段階の塩酸含有率を調節することにより、第3の段階の中和を通常より低い4から6のpH値で行うことができ、そのことにより、実施例によれば、25nm等の100nm未満の結晶サイズ、および10から50nm等の適切な結晶サイズ分布を有する有用なルチル結晶が得られ、それらを焼成パラメータにより調節することができる。この方法において、処理段階で必要な薬品量についても、節減を達成できる。しかし、このように得られた二酸化チタンから分散良好な生成物を調製するには、一般に、後処理の前か間に数回の磨砕が必要である。ここでは、余分な処理段階として実質量のエネルギーが消費され、したがって、生産コストが増加する。
【0008】
磨砕性は、最終生成物の品質特性にも実質的に影響する。良好な分散性とともに、小さい結晶サイズの他に、いくつかの用途では、磨砕性が本質的に影響する狭いまたは制御された結晶サイズ分布が要求される。
【0009】
顔料二酸化チタンの調製において、前処理薬品を用いて、二酸化チタンのルチル化を防止または加速し、特に、焼成済の生成物の磨砕性を改善することは周知である。これは、ルチル構造の微結晶二酸化チタンの調製における場合ではなかった。この理由には、次いで焼成に送られる塊はすでにルチルの形である、すなわち、焼成時に結晶形の変化を制御する必要がなかったことが、まず挙げられる。別の理由は、微結晶二酸化チタンの焼成において用いられる温度が、顔料二酸化チタンの調製においてより低いことであった。より低い焼成温度のため、従来は焼成時に用いられる、主に硫酸塩系の前処理薬品塩は、顔料二酸化チタンを焼成する場合と同様に、微結晶二酸化チタンの焼成時には分解せず、そのことにより、これらの塩は微結晶二酸化チタンの焼成から得られる生成物中に残存する。焼成微結晶二酸化チタン生成物中の最終生成物であるこれらの硫酸塩残留物は、その分散性を著しく妨げることが知られている。
【0010】
特許明細書CA962142には、イルメナイトから調製されたチタン水和物の塊を、水酸化ナトリウムで処理してチタン水和物ケーキを形成する、低結晶性二酸化チタンの調製方法が開示されている。初めに、pHを、2.8から3.0の間に塩酸で調整し、処理の後半の段階で、酸と二酸化チタンとの比率を0.26の値に調節する。処理の最後に、スラリーを、アンモニアで6.5のpH値に中和する。この方法において、酸処理の間に、K2OおよびP2O5を、焼成薬品として添加する。その後で、酸化チタンを濾過および洗浄し、その後に、50から150nm、実施例によれば125nmの粒径をもつ二酸化チタンが得られるまで、この濾物を500℃〜800℃で焼成する。しかし、十分に低結晶性の微結晶酸化チタンは、この方法よっては得られず、結晶サイズは100nm超のままである。この方法に係る前処理化学組成物を用いる場合には、約640℃の温度まで溶融物の形成は始まらず、完全な溶融は約800℃で達成される。
【0011】
特許明細書WO0112555には、塩化チタンの水溶液から超微粒酸化チタンを調製する湿式製錬法が開示されている。この方法においては、化学制御剤が使用され、加水分解される塩化チタン溶液に添加され、乾燥するまで蒸発され、これにより、アモルファス二酸化チタンの薄膜が形成される。この化学制御剤は、結晶形および粒径を制御するために添加される。適切な化学制御剤は、有機化合物およびそれらの塩、ナトリウム、カリウム、アルミニウム、スズおよび亜鉛の塩化物、炭酸塩、フッ化物塩、硫酸塩およびリン酸塩等の無機化合物、リン酸、およびそれらの混合物、ポリアクリレート、グリコール、シロキサン、およびそれらの混合物であることがわかった。しかし、実施例には、リン酸を用いることにより得られる結果のみが記載されている。目的は、アナターゼもしくはルチルの形、またはそれらの混合物のいずれかへの所望の方向性で、焼成および薬品により、蒸発後に二酸化チタンの結晶形を調節することである。焼成後に形成される二酸化チタンの薄膜は磨砕されて、基本的な粒子(約30から50nm)を放出し、適切な特定の面積(BET33から43m2/g)を有する微結晶酸化チタンを与える。結晶サイズおよび磨砕性等の物理的特性、ならびに結晶構造の変換を改良するために、上述の化学制御剤を、焼成前に乾燥アモルファス酸化チタンに添加することもできる。この方法には、その沸点より上で、本質的に結晶成長が起きる温度未満で、溶液を完全に蒸発させることが含まれる。この方法では、大量の溶媒を用い、蒸発させなければならず、必然的にエネルギー使用量が多くなり、操業コストが高くなることを意味する。この方法に関して、分散性は論じられておらず、分散性と磨砕性との間の依存性も有していない。
【先行技術文献】
【特許文献】
【0012】
【特許文献1】EP0444798
【特許文献2】CA962142
【特許文献3】WO0112555
【特許文献4】FI62130
【特許文献5】FI57124
【特許文献6】FI20040186
【発明の概要】
【発明が解決しようとする課題】
【0013】
本発明の目的は、上述の方法に関して開示されている不利を除去することである。本発明の別の目的は、例えば自動車塗料および木材ラック組成物において用いるのに適している、磨砕容易で分散良好なルチル構造の微結晶二酸化チタンを提供することである。
【0014】
本発明の別の目的は、上記の有利な特性を有する二酸化チタン生成物を調製する新規な方法を開示することであり、そこでは、容易に入手可能で有利な原料、および単純な処理段階も用いられる。
【0015】
本発明のさらなる目的は、薬品の使用量、およびエネルギーの必要量の点から、できるだけ経済的に上記の二酸化チタン生成物を調製することであり、取扱い容易な処理薬品および処理設備も用いられる。
【課題を解決するための手段】
【0016】
上述の本発明の目的は、独立請求項1に記載の方法、ならびに請求項13および14に記載の生成物により達成される。本発明は、装飾塗料および木材ラック等の上記の二酸化チタン生成物を含有するさらなる生成物も開示する。
【0017】
前記方法で、ルチル構造の微結晶二酸化チタンを焼成する前にリン酸二水素カリウム等の前処理薬品を用い、その光安定性に認め得るいかなる低下もなく、このように得られた最終生成物の分散性および透明度に改善が観測されたことは驚きに値する。
【図面の簡単な説明】
【0018】
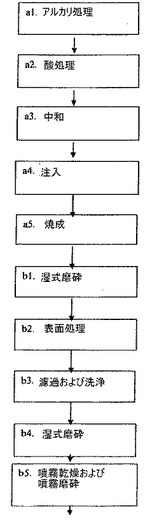
【図1】本発明に係る方法を実施するための実施形態に係る工程流れ図を示す。
【図2】本発明に係る方法により調製される二酸化チタン生成物の分散性を示す。
【図3】本発明に係る方法により調製される二酸化チタン生成物の透明度を示す。
【図4】本発明に係る方法により調製される二酸化チタン生成物の分散性の、磨砕時間に対する依存性を示す。
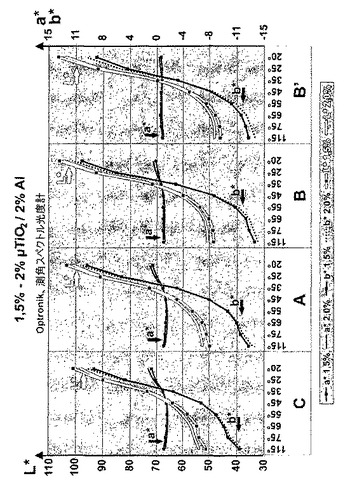
【図5】本発明に係る方法により調製される二酸化チタン生成物を含有する装飾塗料の角度依存反射特性を示す。
【図6】本発明に係るこの方法により調製される生成物の粒径分布を示す。
【図7】本発明に係る方法により調製される生成物から測定した電子顕微鏡的画像である。
【発明を実施するための形態】
【0019】
本発明に係る方法は、以下に詳細に記載する、その下位段階をもつ段階Aと、好ましくはその下位段階をもつ段階Bとを少なくとも含む。
【0020】
既知の方法により、チタン含有物質を、段階Aで処理するのに適切な形の二酸化チタン原料にする。このチタン含有物質には、段階Aに適しているチタン含有二酸化チタン原料を調製することができる、イルメナイト、その濃縮物、またはいくつかの他の純粋でない二酸化チタンの粗原料が含まれ得る。二酸化チタン原料は、好ましくはイルメナイトから作製される。二酸化チタン原料には、沈殿または再結晶が可能な、任意の商業的工程のチタン化合物も含まれ得る。
【0021】
二酸化チタン原料は、好ましくは、硫酸化工程によりイルメナイトから得られる固体の二酸化チタン水和物沈殿物である。それは、より好ましくは、特許EP0444798に記載されている方法により、イルメナイト、その濃縮物、または別の純粋でない二酸化チタン粗原料から作製される。
【0022】
本発明に係る方法により、60nm未満、好ましくは50nm未満の結晶サイズをもつ、分散良好なルチル構造の微結晶二酸化チタン生成物が、段階Aの以下の下位段階A1からA5により、上述の既知の二酸化チタン原料から調製される。
【0023】
下位段階A1において、特許EP0444798に本質的に従って、二酸化チタン原料を、アルカリ性のpH値を有するように、高温で、塩基、好ましくは水酸化ナトリウム水溶液で処理する。二酸化チタン原料を水中で水簸し、塩基をこのスラリーに添加する。塩基濃度を、好ましくはH2O1リットルあたりNaOH約300から350gに相当するようにする。高温は、好ましくは、60℃以上である。
【0024】
本発明に係る好ましい実施形態において、二酸化チタン水和物等の二酸化チタン原料は、硫酸化工程の中間生成物であり、塩酸に可溶であるチタン酸ナトリウムへの塩基処理は、好ましくは、約95℃以上、好ましくは2時間行われる。この処理を、間接的な蒸気で加熱され、撹拌されている反応炉中で行うことができる。塩基で処理された生成物のpHは、好ましくは11超である。
【0025】
塩基処理後に得られるチタン塊を、好ましくは熱水で、好ましくは60℃未満の水で洗浄し、濾過する。目的は、硫酸イオンを含まない塊を洗浄することであり、それによる洗浄結果および残留物含有率を、既知の方法での塩化バリウム試験により調査することができる。
【0026】
下位段階A2において、段階A1で得られたナトリウム含有チタン塊を、水中で再水簸する。この硫酸塩不含のナトリウム含有濾過ケーキを、好ましくは140から200g TiO2/l、より好ましくは約180g TiO2/lの濃度に、蒸留水中で水簸する。
【0027】
実施形態によれば、スラリーの温度を、40から50℃に、好ましくは約1℃/分の速度で上昇させる。
【0028】
そのpHを低下させるために、酸、好ましくは塩酸を、スラリーに変化し塩基で処理された二酸化チタン原料、好ましくはチタン酸ナトリウムに添加する。
【0029】
最終的な酸濃度を、8から35g HCl/リットル、好ましくは29から31g HCl/リットルに調節する。HCl含有率を、滴定により再調査し、必要な場合には補正することができる。この後に、このように生成したスラリーの温度を、88から95℃、好ましくは約90℃にゆっくりと上昇させる。温度上昇の速度は好ましくは約1℃/分であり、加熱中はスラリーを撹拌する。処理を約2時間続けて、ルチル結晶を形成する。塩酸濃度および温度等の使用パラメータの値は、このように得られるルチル結晶の形成に影響を与えることが観測されている。
【0030】
酸処理後に得られた生成物のルチル含有率は、99.0%超、好ましくは99.5%超である。
【0031】
下位段階A3において、特許EP0444798に本質的に従い、段階A2から得られた上述の沈殿物を中和し、わずかに酸性のpH値のスラリーにする。中和においては、pHを4以上の値だが、6未満の値、好ましくは4.4から5.0の値に上昇させる。中和を、適切なアルカリ性中和薬品、好ましくは水酸化ナトリウムまたは炭酸ナトリウムにより行うことができる。中和後に得られた二酸化チタン塊を、濾過および洗浄する。中和を注意深く行い6以下、または好ましくは5以下のpH値にすれば、濾過および洗浄は極めてうまくいき、そのことにより、さらなる処理を妨げる本質的にすべてのナトリウムおよび塩化物のイオンを、この塊から除去することができる。
【0032】
下位段階A4において、95.0%超、好ましくは99.5%超がルチルの形である、段階A3から得られたルチル構造の二酸化チタンスラリーに、適切な前処理薬品を注入する。前処理薬品を、撹拌しつつ二酸化チタン塊に添加する。前処理薬品は、好ましくは、微細に磨砕された固体の乾燥粉末として添加される。添加後に、塊を、少なくとも1時間、好ましくは4時間以超、それを炉に供給し始める前に撹拌する。
【0033】
本発明における前処理薬品は、焼成を制御しつつ使用されて、結晶成長と結晶構造の転移温度とに影響を与える薬品を指し、それは当技術分野において一般的な意味の用語である。本発明に関して、それは、好ましくは、TiO2焼成中に用いられる前処理薬品を指す。
【0034】
前処理薬品は、結晶成長を制御し、必要な溶融物の低温での形成もする形の物質である。前処理薬品は、好ましくは、リン酸二水素カリウム、KH2PO4、または焼成の前もしくは間に反応し、リン酸二水素カリウム、もしくはリン酸二水素カリウムの類似化合物になるカリウムおよびリンの化合物の混合物である。その場合において、例えば、硫酸カリウム、K2SO4およびリン酸アンモニウムMAPの混合物を考慮することができる。例えばリン酸は、リン源としても機能することができる。前処理薬品は、最も好ましくは、リン酸二水素カリウム、KH2PO4である。前処理薬品として様々なカリウムおよびリンの化合物を用いると、それらは結晶成長を制御する適切な溶融物を焼成中に形成し、それらを、混合されるべき塊の中に一緒または別々のいずれかで供給できることが観測されている。
【0035】
リン酸二水素カリウムとして表される必要な前処理薬品の量は、0.2から1重量%、好ましくは0.5から0.7重量%、最も好ましくは約0.6重量%である。含有率が0.2重量%未満である場合、十分な効果はもはや得られない。それぞれに必要な薬品量は、焼成温度、焼成時間、および所望の結晶サイズにも依存し、したがって、当技術分野の専門家により決定され得る。含有率が本質的に1重量%を超える場合、同様の焼成条件下で結晶が異常成長し始める。さらに、用いる前処理薬品が多すぎると、例えばカリウムを洗浄するのがより困難となり、処理段階が複雑になり、操業コストが増大する。
【0036】
前処理薬品に必要な特性は、焼成温度で溶融物を形成する能力である。前処理薬品は、好ましくは明らかに焼成温度未満で溶融物を形成し、そのことにより、溶融物の粘度は本発明に係る操業温度で可能な限り低くまで低下する。前処理薬品の溶融温度は、好ましくは730℃未満、より好ましくは670℃未満、最も好ましくは253℃等の350℃未満である。異なる化合物の混合物を用いた場合、完全な溶融は求められないが、溶融物量が十分であれば、明らかに液相線内である、すなわち部分的に溶融していれば十分である。
【0037】
リン酸二水素カリウムを用いることにより、顔料二酸化チタンの調製が有利になる。さらに、リン酸二水素カリウムは、約253℃の非常に低い温度ですでに結晶を形成するのに有利な溶融物を形成し、結晶表面の原子の移動を可能にし、低温ですでに焼成処理を促進し、そのことにより、より小さい結晶サイズをもつ最終生成物が得られる。薬品のリンが結晶表面に残存し、その部分により、水相中における結晶の分散性が改善される。その場合において、磨砕性も改善される。この薬品は、このように得られる生成物のpH値も本質的には変化させない。前処理薬品の使用は、特に焼成後の生成物の磨砕性を改善する効果を示すことが観測された。
【0038】
前処理後に得られたスラリー様生成物を、下位段階A5において焼成する。焼成は、好ましくは、350から800℃の温度で、より好ましくは560から770℃の温度で、最も好ましくは650から750℃の温度で行われる。焼成温度の選択は、例えば、供給材料の組成、使用する水含有率、滞留時間、および焼成ガスにより影響を受ける。様々な要因の組合せにより条件が与えられ、焼成炉の取り出し物の結晶サイズおよび結晶サイズ分布が所望のようになる。本発明に係る塩不含のルチル構造二酸化チタン塊の焼成パラメータを用いて、簡単かつ正確に微結晶二酸化チタンの結晶サイズおよび結晶サイズ分布を調節することができる。焼成中に、結晶を凝集させ、その後に所望の粒径に磨砕する。焼成後の本発明に係る二酸化チタンの結晶サイズは、好ましくは約10nm超、さらに60nm未満、好ましくは50nm未満、最も好ましくは約30nmである。焼成後の生成物のルチル含有率は、99.0%超、好ましくは99.5%超である。焼成された顔料の結晶は、本質的に楕円形である。
【0039】
焼成時間は、0.5から5時間、好ましくは1から3時間である。沈殿段階も焼成段階も、加圧条件でなく常圧で行われる。
【0040】
好ましい実施形態において、前処理薬品としてリン酸二水素カリウムを用いた場合、水中で水簸したときに、従来の未注入焼成された微結晶ルチル構造の二酸化チタンよりはるかに磨砕が容易である焼成取り出し物が得られる。
【0041】
一般に、所望の粒径を達成するために、焼成後に何度も、例えば約5から6回生成物を磨砕し、最終的に、例えばコーティング後に噴霧磨砕する必要があった。その場合において、エネルギー消費が多く、磨砕したにもかかわらず、所望の結晶サイズ、および狭い結晶サイズ分布を達成されなかった。現在では、これらの不利を、本発明に係る方法により回避することができる。さらに、焼成取り出し物の処理後洗浄において、最終生成物に不利になる、前処理薬品の一部であるカリウムを、最終生成物から洗い去ることができる。
【0042】
段階Bにおいて、段階A(A1からA5)から得られたルチル構造の微結晶酸化チタンをさらに処理し、当技術分野において既知の任意の方法により二酸化チタン最終生成物にすることができる。さらなる処理は、好ましくは、特許明細書EP0444798に記載の方法により主に行われる。
【0043】
本発明に係る好ましい実施形態によれば、段階Bには、明細書EO0444798に開示されている以下のさらなる処理段階が含まれ、特別な湿式磨砕段階B4をそれに加えている。すなわち、
B1 焼成から得られた塊を湿式磨砕し、
B2 湿式磨砕から得られた塊を表面処理し、
B3 表面処理した塊を濾過および洗浄し、
B4 段階B3から得られた塊を再度湿式磨砕し、
B5 段階B4から得られた塊を噴霧乾燥および噴霧磨砕して、最終的な二酸化チタン生成物を調製する。
【0044】
処理段階B1において、段階A(A5)から得られた焼成二酸化チタンを蒸留水中で水簸して濃厚スラリーを形成する。水簸は、好ましくは、分散剤、より好ましくはモノイソプロパノールアミンMIPAにより行われる。スラリーの濃度は、500から600g TiO2/l、好ましくは約550TiO2/lである。焼成から得られる塊を、例えば研磨機で既知の方法により湿式磨砕する。磨砕後に得られた生成物のU/V*100値は、好ましくは850超である。
【0045】
処理段階B2において、段階B1から得られた、磨砕された二酸化チタン塊を、その使用目的に従った任意の既知の方法により、例えばアルミニウム、シリカおよび/またはジルコニウム化合物によりコーティングする。使用した方法には、例えば、特許明細書FI62130に開示されている処理方法が含まれ得る。
【0046】
処理段階B3において、コーティング処理B2の後に、生成物を濾過し水で洗浄する。無機コーティングは、微結晶二酸化チタンを激しく凝集させる。
【0047】
処理段階B4において、段階B3から得られた、コーティングされた二酸化チタン塊を、水中で再水簸し、スラリーを湿式磨砕する。磨砕の開始前に、または湿式磨砕段階がない場合には、メチロールプロパン、TMP、メチロールエタンまたはケイ素(FI57124参照)等の、乾燥設備の有機添加剤を、供給スラリーを乾燥設備に供給する前にそれに添加する。使用されるコーティング剤は、好ましくはTMPである。使用されるTMPの量は、2から8重量%、好ましくは2から6重量%、最も好ましくは5重量%である。磨砕の目的は、TMPを乾燥設備への供給物にできるだけ均一に混合させることである。磨砕が、低い濃度、好ましくは130から270g/lで、多い供給量で行われることにより、TiO21トンあたりの磨砕効率が低いままとなる。使用される磨砕効率は、使用される設備および能力に依存し、そのことは当業者には明らかである。
【0048】
コーティング後の湿式磨砕処理により、二酸化チタン最終生成物の分散性および透明度がさらに改善される。湿式磨砕は、二酸化チタン最終生成物の光活性の増加も観測されなかったことは、この生成物の用途には重要である。以前は、コーティングが破壊され、有益な特性が失われる、例えば光安定性が弱まると、後処理の後にもはや効率的な磨砕を用いることができないと考えられていた。しかし、それは事実ではなく、代わりに、本発明に係るコーティングおよび噴霧乾燥の間に湿式磨砕段階を加えることにより、より良好な分散性を、光安定性を失うことなく達成した。
【0049】
処理段階B5において、段階B4からの、コーティングされ湿式磨砕された二酸化チタン塊を、噴霧乾燥および噴霧磨砕して、当技術分野において既知の方法で最終的な二酸化チタン最終生成物を得る。
【0050】
本発明は、請求項13に係る、新規なルチル構造の微結晶二酸化チタン生成物も開示する。生成物の結晶サイズ(d50)は、60nm未満、好ましくは50nm未満であり、結晶サイズは、より好ましくは20から40nm、最も好ましくは約30nmであり、粒径d50は0.150μm未満であり、生成物は良好に分散可能であり、U/V*100 p ratは500以上である。このように得られた生成物の比表面積BETは55m2/g以下であり、その光安定性は好ましくは10未満である。本発明に係る二酸化チタン生成物は、図3B’からわかる可視光の良好な透過度も特徴とする。図3に、本発明に係る二酸化チタン生成物の粒子を含有するラッカー膜の透過曲線を示す。本発明に係る生成物を含有するラッカー膜の透過曲線の透過度は、少なくとも、図3B’に示される曲線の透過度ほどに良好すなわち浸透性がある。一般に、浸透性すなわち透過度は、波長490nmで得られるT=60%の値により説明される。
【0051】
好ましい実施形態によれば、生成物の結晶サイズは、20から40nm、より好ましくは約30nmである。結晶サイズは、使用目的に応じてこの方法の中で変化させることができる。
【0052】
本発明に係る生成物の良好な分散性は、濁度測定により測定される特性を指す。測定において、水相中で終夜分散させた試料の吸収の変化は、当技術分野における既知の方法で分光光度法により波長の関数により観測される。測定曲線から、粒径分布の広さまたは狭さを記述するために使用可能なU/V*100 p ratを算出することができる。U/V*100値が高いほど、粒径分布が狭い、すなわち、生成物が良好に磨砕されているほど、生成物の分散性はさらに良好である。本発明に関して、「良好な分散性」の用語は、300以上、好ましくは500以上のU/V*100 p rat値を有する生成物について得られる特性を指す。
【0053】
本発明に係る生成物のU/V*100 p rat値は、好ましくは550超である。このことは、曲線の急峻さのために、生成物が非常に良好なUV保護も提供することを示している。
【0054】
分散性は様々な因子に依存するが、この場合においては、粒径は最も本質的なものであることがわかった。粒径が減少している場合は、粒子が凝集する自然な傾向のために分散性が悪化する。
【0055】
本発明に係る二酸化チタン生成物の比表面積、BETは、結晶サイズ分布d50が30±5nmである場合で、≦55m2/g、好ましくは≦50m2/gである。最も好ましくは、平均結晶サイズd50が30±3nmである場合で、BET≦45m2/gである。
【0056】
本発明に係る生成物は、意外にも制御が容易である小さな平均粒径と、狭い粒径分布とを有する。この分布は、好ましくは、焼成中にリン酸二水素カリウム等の前処理薬品を用いることにより達成される。さらに、特別な湿式磨砕段階は、より好ましくは噴霧乾燥および噴霧磨砕の前の調製において用いられる。本発明に係る生成物は、最も好ましくは請求項1に記載の方法により調製される。本発明に係る方法を用い、特許明細書EP0444798に言及されている方法と比べて明確に良好な分散性の微結晶生成物を調製することができる。
【0057】
さらに、本発明に係る二酸化チタン生成物は、非常に光安定性である。一般に、その光安定性は、特許明細書FI20040186に記載されているように、アセトンの形成速度により説明されるが、別に、分解される気体としてプロパノールの代わりにアルデヒドとトルエンとの混合物を用いると、光安定性はアセトンの形成速度でなくCO2の形成速度により説明される。本発明に係る二酸化チタン生成物は、10ppm/h未満、好ましくは5ppm/h未満の、アセトンの形成速度を特徴とする。
【0058】
本発明に係る新規な二酸化チタン生成物は、好ましくは、上述の方法により調製される。本発明に係る方法により調製される生成物は、磨砕が容易な焼成取り出し物を特徴とし、それにより、最終生成物の最終的な粒径分布を、明確により少ない磨砕回数により制御することができる。特別な湿式磨砕段階B4を加えることにより、磨砕性をより容易になり、粒径分布がより狭くなることがさらに観測された。必要な磨砕回数を、好ましくは半減することができる。
【0059】
本発明に係る生成物に与えられる、粒径および粒径分布の最適な制御は、かなりより良好な分散可能生成物を示唆し、生成物の特定の用途に必要な金属光沢の特性、フリップフロップ効果をさらに強める。
【0060】
好ましい実施形態において、本発明に係る新規な二酸化チタン生成物を、光沢のある自動車塗料等の装飾塗料組成物中に用いて、可視光波長および反射効率の非常に強い角度依存性を提供することができる。この場合において、20°b*が8以上で、明るい金色のフリップ効果、115°b*が−11未満で、濃い青色のフロップ効果をもたらすために用いることができる塗料組成物が提供される。
【0061】
本発明は、上記に係る二酸化チタン生成物を含有する装飾塗料を開示し、塗料にフリップフロップ効果を与える。この塗料は、好ましくは、20°b*が8以上でフリップ効果、115°b*が−11未満で濃い青色のフロップ効果を提供するために用いられる。
【0062】
本発明は、本発明に係る二酸化チタン生成物をUV保護として用いることができる木材ラック組成物をさらに開示する。
【0063】
本発明を、以下の実施例により例証する。個別に言及がなければ、原料および材料は、市販の製品であるか、それらは既知の技術により製造され得る。
【実施例1】
【0064】
図1は、二酸化チタンの調製において用いられる処理段階を本質的に示す。硫酸塩法による二酸化チタンの調製は、イルメナイト濃縮物を硫酸と反応させることにより開始される。このように得られた固体の反応ケーキを、水および廃酸により溶解する。不純物を除去し、硫酸鉄を晶出させる。チタン含有溶液を濃縮し、二酸化チタン水和物を加水分解により沈殿させる。この沈殿物塊を数段階で洗浄して、塩を除去する。
【0065】
TiO2として表される、この洗浄済沈殿物塊約10トンを取り、水で水簸して、目標350g/lで300から400g/lの濃度を得る。このように得られたスラリーを、NaOH溶液としてNaOH約15トンを60℃で添加することにより高アルカリ性(pH>11)とし、その濃度は700g/lである。スラリーの温度を95℃に上げ、それをこの温度で2時間撹拌する。処理中に、水酸化チタン塊をアルカリ液と反応させ、固体のチタン酸ナトリウムを形成し、そこから、塩化バリウムで沈殿させたときに濾液中に硫化物が見られなくなるまで熱水でスラリーを洗浄することにより、硫酸イオンをさらに除去する(図1、段階A1)。
【0066】
二酸化チタンとして表されるスラリーの濃度が約180g/lになるように、硫化物不含のナトリウム含有濾過ケーキを水中で水簸する。このスラリーを、1℃/分の速度で40から45℃の温度に加熱する。このスラリーの酸含有率を、30%塩酸溶液を添加することにより30g HCl/lに設定して、ルチル構造の結晶を作り出す。必要であれば、HCl含有率を滴定により調節し補正する。スラリーの温度を、1℃/分の速度で、一定した撹拌をしつつゆっくりと90℃に上げる。この温度で、スラリーを撹拌しつつ120分間処理する(図1、段階A2)。
【0067】
最終的に、スラリーを、pHが4.7から4.8に定まるように、炭酸ナトリウムまたは水酸化ナトリウムで中和する。中和済スラリーを、濾過し、蒸留水約4リットルで洗浄する。このように、乾燥した濾過ケーキの含有率は約30%である(図1、段階A3)。
【0068】
TiO2として表される洗浄ケーキ10tを取り、水で水簸して、150から250g/lの濃度を得る、すなわち、スラリー約40から50m3を得る。水簸は撹拌機を備えた容器中で行われ、そこに粉末の市販のKH2PO4約60kgを前処理薬品として添加する。スラリーを、炉へ供給し始める前に、約4時間混合させる(図1、段階A4)。
【0069】
試料を、炉の供給口から取り、乾燥し、X線回折図を取り、微結晶二酸化チタンの99.0%超がルチルの結晶形を有することが見出される。
【0070】
二酸化チタン濾過ケーキを、720℃の温度の連続回転リールオーブン中で焼成する。滞留時間は、平均で約1時間である。焼成されたルチル構造の生成物の電子顕微鏡画像を測定すると、平均結晶サイズは約30nmであり、分布幅は10から50nmである。焼成生成物のナトリウム含有率は0.1%未満であり、塩化物含有率は0.05%未満である(図1、段階A5)。このように作り出された生成物の電子顕微鏡画像を図7に示す(160000倍)。
【0071】
塩不含の二酸化チタンを、分散剤により濃厚スラリーとして蒸留水中で水簸する。湿式磨砕を研磨機で行う。一般に、生成物は3回湿式磨砕される(図1、段階B1)。
【0072】
互いに別々に磨砕した結晶を、使用目的に従って、アルミニウム、ケイ素および/またはジルコニウムの化合物で後処理する。ここで、試料を酸化ジルコニウム(1.0%)および酸化アルミニウム(4.0%)でコーティングする(図1、段階B2)。方法として、例えば特許明細書FI62130に表されるもの等の、二酸化チタン顔料の処理のための他の既知の方法を用いることもできる。
【0073】
TMP5%を、洗浄され濾過されたコーティング済塊試料に添加し、その後に、乾燥噴霧前にそれをもう一度湿式磨砕する(図1、段階B3およびB4)。
【0074】
噴霧乾燥された微結晶TiO2を噴霧磨砕し、約30nmの結晶サイズを有する微細な粉末にする(図1、段階B5)。粒径は、Master Malvernサイズ測定装置により測定され、約0.1μmで明確により大きい。
【0075】
噴霧乾燥後に、このように得られた最終生成物の紫外可視スペクトルを、水性スラリーから測定する。それは焼成段階後の研磨段階での生成物のスペクトルとほとんど同じ水準であることが観測される。
【実施例2】
【0076】
試料の磨砕性、分散性および粒径分布への調製方法の効果を調査する。
【0077】
磨砕性を、分光光度計により測定される濁度曲線と、Malvern Mastersizer 200により測定した粒径分布の両方に基づいて評価する。
【0078】
酸で処理した沈殿物塊が濾過および洗浄されるように、試料を実施例1に従って調製し、その後、リン酸二水素カリウム0.6gをこの塊に添加する。この塊を回転リールオーブン中で730℃で約1時間焼成する。焼成後に、オーブン取り出し物を研磨機中で4回湿式磨砕する。この粒子を、ジルコニウムおよびアルミニウムの酸化物(ZrO2はTiO2の1.0%、Al2O3はTiO2の4.0%)でコーティングし、濾過し水で洗浄する。TMP5%をこの塊に添加し、その後に、試料を2つの部分に分割する。試料Aは、噴霧乾燥および噴霧磨砕の前に特別な湿式磨砕を受ける。試料Bは、特別な湿式磨砕をされずに、乾燥および磨砕される。参照試料Cを、特許EP0444798、実施例1に記載の方法で調製し、粒径30nmにする(EP0444798、実施例9)。焼成生成物Cは、試料Bと同様に後処理段階を受ける。
【0079】
噴霧磨砕された最終生成物試料A、BおよびCを、分析で純粋な0.1体積%の1−アミノ−2−2プロパノール(MIPA)を添加したイオン交換水中に分散する。350から1100nmの範囲内の濁度を、1ナノメートルの間隔で、分光光度計により希釈水性分散液から測定する。
【0080】
表1において、顔料の微細さの程度を4つの数量により測定している。すなわち、
1)吸光度(最大)、最大吸収波長[nm]、
2)U/V*100、波長450nmの吸収で割った最大吸収波長の吸収*100、
3)吸光度(最大)、最大吸収波長での吸収、および
4)吸光度(450nm)、波長450での吸収
である。
【0081】
【表1】
【0082】
図2に、生成物AおよびBの紫外可視スペクトルを、生成物Cの紫外可視スペクトルと比較して示す。参照の生成物に対して、本発明に従って調製される微結晶顔料AまたはBは、明確に良好に磨砕および分散されている。このように、生成物を前処理薬品を用い調製すると、参照生成物Cと比べて分散性が改善する(生成物B)。前処理薬品の他に、噴霧乾燥前に特別な湿式磨砕段階を用いると、分散性は、550以上のU/V*100の値にさらに改善される(生成物A)。
【実施例3】
【0083】
粒径分布を、Malvern Mastersize2000により、実施例2に係る生成物CおよびAから測定する。この方法において、試料を水簸して懸濁液を形成し、連続循環中の懸濁液を、レーザー光線を当てた流通キュベットに導く。試料の粒子は、単色のレーザー光線を、それらのサイズに応じて様々な散乱角で散乱させる。様々な角度で散乱される放射の強度を測定する。試料の粒径分布を、MieまたはFraunhoferの理論に従って、散乱強度から算出する。測定は、装置の取扱説明に従って行われる。
【0084】
図6および表2に粒径分布を示す。参照の生成物Cの試料の粒径分布は、明確に2つの山を有し、d50の値は明確により高い。
【0085】
【表2】
【実施例4】
【0086】
実施例2の生成物A、BおよびCの光活性を、既知の気相の方法により測定する。
【0087】
この方法において、2−プロパノールを閉鎖室内に注入する。ペトリ皿上の粉末試料にXeランプにより照射する。2−プロパノールは、まずアセトンに、最終的には二酸化炭素に分解する。二酸化炭素の形成は、本質的に光触媒活性の結果である。より大きい数は、より高い光活性を表す。
【0088】
このように得られた光活性の結果を表3に示す。
【0089】
【表3】
【0090】
表3における結果に基づくと、この特別な湿式磨砕段階が、生成物Aの光触媒活性を増大させないことを確証することができ、このことは、その用途のために重要である。
【実施例5】
【0091】
実施例1に記載の方法により調製した生成物Gを、できるだけ類似した化学組成を有する市販の他の生成物AからFと比較した。調査した特性は、
1.既知の方法によるX線回折(XRD)により求められるルチル結晶のサイズ(nm)、
2.既知の方法により求められる比表面積、BET(m2/g)、
3.濁度曲線により求められる、分散性を記述する数量U/V*100
4.アセトンの形成速度(ppm/h)として測定される光安定性
であった。
【0092】
表4に示す測定結果に基づいて、本発明に係る生成物Gの光安定性が、他のルチル構造の二酸化チタン生成物と比べて優れていることを観測することができる。同時に、生成物Gは、極めて良好な分散性を提供する。参照の試料に対する結晶サイズ分布の狭さは、より小さいBET値として表れている一方、より狭い粒径分布は、より高いU/V*100値として表れている。
【0093】
【表4】
【実施例6】
【0094】
ラッカー膜A’、B’およびC’(参照の膜)を、実施例2に従って作製した二酸化チタン生成物AおよびB、ならびに参照の生成物Cからさらに作製する。TiO2水性分散液をベース組成物と合わせ、約5分間撹拌(Skandex BA−EL−T)することにより、二酸化チタンを2.0重量%水希釈性アクリルワニスベースと混合する。ラッカー組成物の粘度を、25〜30秒/DIN−Bencher 4mmに調節する。
【0095】
図3に、ラッカー膜の透過スペクトルを示す。膜B’の透過度は、膜C’と比べて可視スペクトルの全領域内で明確に良好である。膜A’についての透過度は、膜B’およびC’と比べて、スペクトルの範囲にわたってさらに改善する。
【0096】
様々な波長についての参照値を表5に示す。0−膜は、二酸化チタンがない基本的なラッカー組成物である。
【0097】
【表5】
【実施例7】
【0098】
実施例2に記載の方法により作製した二酸化チタンA、BおよびCならびにB’(B’は、その焼成温度が670℃である点で生成物Bと異なる)を、金属装飾の塗料に適した1.5および2.0重量%TiO2/2重量%Alの、車の下塗り塗料組成物と混合する。
【0099】
図5に、参照の試料C、ならびに本発明に係る試料A、BおよびB’を含有する装飾塗料試料から、CIELAB色記述モデルに従って、測角スペクトル光度計(Optronic GmbH)により様々な角度で測定した反射を示す。このように調製した装飾塗料により、非常に強い角度依存性が、本発明に係る二酸化チタンを含有するコーティングによりもたらされる。明るい金色のフリップ効果20°b*は8以上であり、濃い青色のフロップ効果115°b*は−11未満である。
【0100】
さらに、このようにもたらされる効果の強度が高いことを述べておくべきである。入手可能な既知の塗料組成物と比べて、本発明に係る二酸化チタン生成物を含有する塗料組成物によりもたらされる効果について、同じ効果を得るために塗料組成物において必要な対応する二酸化チタン量はわずか約25%である。
【技術分野】
【0001】
本発明は、分散良好であるルチル構造の微結晶二酸化チタン生成物を調製する方法、その方法により得られる生成物、ならびにその生成物により得られる装飾塗料および木材ラックに関する。
【背景技術】
【0002】
微結晶酸化チタンの特性は、白色顔料として知られている従来の顔料二酸化チタンの特性と異なる。微結晶酸化チタンの結晶サイズ(10から100nm)は、従来の二酸化チタン(160から250nm)の結晶サイズより約5から10倍小さいので、その差は結晶サイズの違いによるものである。結晶サイズが減少するにつれ、可視光の範囲内における二酸化チタンの隠蔽力は消え、顔料は透明になる。他方で、紫外線に対する透過度は減少する。したがって、微結晶二酸化チタンはUV保護剤に適している。その小さい結晶サイズ、および広い比表面積のため、微結晶二酸化チタンは、例えば、化粧品、触媒、セラミック材料の中において、および塗料中の装飾顔料として有用である。
【0003】
微結晶二酸化チタンの調製における1つの問題は、工程の間、所望の結晶および粒子のサイズ分布を作り出し、調節し、保持することであり、所望のサイズ分布には、平均サイズ、および狭い幅のサイズ分布の両方が含まれる。様々な調製方法を評価するときに、二酸化チタン生成物の、純度、ならびに結晶および粒子のサイズ分布の制御は重要な因子である。さらに、調製工程は、エネルギー的に、かつ経済的に有利であり、環境に優しくあるべきである。
【0004】
TiO2は、3種の異なる結晶形で生じ得る。ルチルは、高温で安定な形である。アナターゼは、低温で支配的な形である。低温において、一般に斜方晶の形で発生する鉱物の中にのみ見出されるブルッカイトの形も生じ得る。ルチルは、最も耐久性のある結晶形として知られており、紫外光に対するその透過度はアナターゼの透過度より低い。
【0005】
純粋なルチルより、アナターゼおよびルチルの混合物を調製する方が容易であることは、既知の事実である。例えば、亜鉛等のルチル化薬品と呼ばれるものの存在下でアナターゼの形の二酸化チタンを焼成することにより、アナターゼおよびルチルの混合物をより低い温度でも生成することができる。
【0006】
気相技術または沈殿のいずれかを用い、微結晶二酸化チタンを様々な方法により調製することができる。チタン源には、例えば、イルメナイトから調製される四塩化チタン、チタンアルコキシドまたはチタン水和物が含まれ得る。ルチルの調製について様々な沈殿法に特許が付与されており、それらのいくつかに対する参照が、出願人の以前の特許明細書EP0444798に組み込まれている。
【0007】
特許明細書EP0444798には、微結晶二酸化チタンの調製方法が開示されており、そこでは第1の段階で、二酸化チタン水和物を、アルカリ性のpH値を有するように塩基で処理し、第2の段階で、塩酸の最終的な含有率を8から25g/lの間に調節するように塩酸による処理を続ける。第3の段階では、酸性混合物が塩基により中和され、中和された反応混合物を、例えば焼成により後処理することができる。第2の段階の塩酸含有率を調節することにより、第3の段階の中和を通常より低い4から6のpH値で行うことができ、そのことにより、実施例によれば、25nm等の100nm未満の結晶サイズ、および10から50nm等の適切な結晶サイズ分布を有する有用なルチル結晶が得られ、それらを焼成パラメータにより調節することができる。この方法において、処理段階で必要な薬品量についても、節減を達成できる。しかし、このように得られた二酸化チタンから分散良好な生成物を調製するには、一般に、後処理の前か間に数回の磨砕が必要である。ここでは、余分な処理段階として実質量のエネルギーが消費され、したがって、生産コストが増加する。
【0008】
磨砕性は、最終生成物の品質特性にも実質的に影響する。良好な分散性とともに、小さい結晶サイズの他に、いくつかの用途では、磨砕性が本質的に影響する狭いまたは制御された結晶サイズ分布が要求される。
【0009】
顔料二酸化チタンの調製において、前処理薬品を用いて、二酸化チタンのルチル化を防止または加速し、特に、焼成済の生成物の磨砕性を改善することは周知である。これは、ルチル構造の微結晶二酸化チタンの調製における場合ではなかった。この理由には、次いで焼成に送られる塊はすでにルチルの形である、すなわち、焼成時に結晶形の変化を制御する必要がなかったことが、まず挙げられる。別の理由は、微結晶二酸化チタンの焼成において用いられる温度が、顔料二酸化チタンの調製においてより低いことであった。より低い焼成温度のため、従来は焼成時に用いられる、主に硫酸塩系の前処理薬品塩は、顔料二酸化チタンを焼成する場合と同様に、微結晶二酸化チタンの焼成時には分解せず、そのことにより、これらの塩は微結晶二酸化チタンの焼成から得られる生成物中に残存する。焼成微結晶二酸化チタン生成物中の最終生成物であるこれらの硫酸塩残留物は、その分散性を著しく妨げることが知られている。
【0010】
特許明細書CA962142には、イルメナイトから調製されたチタン水和物の塊を、水酸化ナトリウムで処理してチタン水和物ケーキを形成する、低結晶性二酸化チタンの調製方法が開示されている。初めに、pHを、2.8から3.0の間に塩酸で調整し、処理の後半の段階で、酸と二酸化チタンとの比率を0.26の値に調節する。処理の最後に、スラリーを、アンモニアで6.5のpH値に中和する。この方法において、酸処理の間に、K2OおよびP2O5を、焼成薬品として添加する。その後で、酸化チタンを濾過および洗浄し、その後に、50から150nm、実施例によれば125nmの粒径をもつ二酸化チタンが得られるまで、この濾物を500℃〜800℃で焼成する。しかし、十分に低結晶性の微結晶酸化チタンは、この方法よっては得られず、結晶サイズは100nm超のままである。この方法に係る前処理化学組成物を用いる場合には、約640℃の温度まで溶融物の形成は始まらず、完全な溶融は約800℃で達成される。
【0011】
特許明細書WO0112555には、塩化チタンの水溶液から超微粒酸化チタンを調製する湿式製錬法が開示されている。この方法においては、化学制御剤が使用され、加水分解される塩化チタン溶液に添加され、乾燥するまで蒸発され、これにより、アモルファス二酸化チタンの薄膜が形成される。この化学制御剤は、結晶形および粒径を制御するために添加される。適切な化学制御剤は、有機化合物およびそれらの塩、ナトリウム、カリウム、アルミニウム、スズおよび亜鉛の塩化物、炭酸塩、フッ化物塩、硫酸塩およびリン酸塩等の無機化合物、リン酸、およびそれらの混合物、ポリアクリレート、グリコール、シロキサン、およびそれらの混合物であることがわかった。しかし、実施例には、リン酸を用いることにより得られる結果のみが記載されている。目的は、アナターゼもしくはルチルの形、またはそれらの混合物のいずれかへの所望の方向性で、焼成および薬品により、蒸発後に二酸化チタンの結晶形を調節することである。焼成後に形成される二酸化チタンの薄膜は磨砕されて、基本的な粒子(約30から50nm)を放出し、適切な特定の面積(BET33から43m2/g)を有する微結晶酸化チタンを与える。結晶サイズおよび磨砕性等の物理的特性、ならびに結晶構造の変換を改良するために、上述の化学制御剤を、焼成前に乾燥アモルファス酸化チタンに添加することもできる。この方法には、その沸点より上で、本質的に結晶成長が起きる温度未満で、溶液を完全に蒸発させることが含まれる。この方法では、大量の溶媒を用い、蒸発させなければならず、必然的にエネルギー使用量が多くなり、操業コストが高くなることを意味する。この方法に関して、分散性は論じられておらず、分散性と磨砕性との間の依存性も有していない。
【先行技術文献】
【特許文献】
【0012】
【特許文献1】EP0444798
【特許文献2】CA962142
【特許文献3】WO0112555
【特許文献4】FI62130
【特許文献5】FI57124
【特許文献6】FI20040186
【発明の概要】
【発明が解決しようとする課題】
【0013】
本発明の目的は、上述の方法に関して開示されている不利を除去することである。本発明の別の目的は、例えば自動車塗料および木材ラック組成物において用いるのに適している、磨砕容易で分散良好なルチル構造の微結晶二酸化チタンを提供することである。
【0014】
本発明の別の目的は、上記の有利な特性を有する二酸化チタン生成物を調製する新規な方法を開示することであり、そこでは、容易に入手可能で有利な原料、および単純な処理段階も用いられる。
【0015】
本発明のさらなる目的は、薬品の使用量、およびエネルギーの必要量の点から、できるだけ経済的に上記の二酸化チタン生成物を調製することであり、取扱い容易な処理薬品および処理設備も用いられる。
【課題を解決するための手段】
【0016】
上述の本発明の目的は、独立請求項1に記載の方法、ならびに請求項13および14に記載の生成物により達成される。本発明は、装飾塗料および木材ラック等の上記の二酸化チタン生成物を含有するさらなる生成物も開示する。
【0017】
前記方法で、ルチル構造の微結晶二酸化チタンを焼成する前にリン酸二水素カリウム等の前処理薬品を用い、その光安定性に認め得るいかなる低下もなく、このように得られた最終生成物の分散性および透明度に改善が観測されたことは驚きに値する。
【図面の簡単な説明】
【0018】
【図1】本発明に係る方法を実施するための実施形態に係る工程流れ図を示す。
【図2】本発明に係る方法により調製される二酸化チタン生成物の分散性を示す。
【図3】本発明に係る方法により調製される二酸化チタン生成物の透明度を示す。
【図4】本発明に係る方法により調製される二酸化チタン生成物の分散性の、磨砕時間に対する依存性を示す。
【図5】本発明に係る方法により調製される二酸化チタン生成物を含有する装飾塗料の角度依存反射特性を示す。
【図6】本発明に係るこの方法により調製される生成物の粒径分布を示す。
【図7】本発明に係る方法により調製される生成物から測定した電子顕微鏡的画像である。
【発明を実施するための形態】
【0019】
本発明に係る方法は、以下に詳細に記載する、その下位段階をもつ段階Aと、好ましくはその下位段階をもつ段階Bとを少なくとも含む。
【0020】
既知の方法により、チタン含有物質を、段階Aで処理するのに適切な形の二酸化チタン原料にする。このチタン含有物質には、段階Aに適しているチタン含有二酸化チタン原料を調製することができる、イルメナイト、その濃縮物、またはいくつかの他の純粋でない二酸化チタンの粗原料が含まれ得る。二酸化チタン原料は、好ましくはイルメナイトから作製される。二酸化チタン原料には、沈殿または再結晶が可能な、任意の商業的工程のチタン化合物も含まれ得る。
【0021】
二酸化チタン原料は、好ましくは、硫酸化工程によりイルメナイトから得られる固体の二酸化チタン水和物沈殿物である。それは、より好ましくは、特許EP0444798に記載されている方法により、イルメナイト、その濃縮物、または別の純粋でない二酸化チタン粗原料から作製される。
【0022】
本発明に係る方法により、60nm未満、好ましくは50nm未満の結晶サイズをもつ、分散良好なルチル構造の微結晶二酸化チタン生成物が、段階Aの以下の下位段階A1からA5により、上述の既知の二酸化チタン原料から調製される。
【0023】
下位段階A1において、特許EP0444798に本質的に従って、二酸化チタン原料を、アルカリ性のpH値を有するように、高温で、塩基、好ましくは水酸化ナトリウム水溶液で処理する。二酸化チタン原料を水中で水簸し、塩基をこのスラリーに添加する。塩基濃度を、好ましくはH2O1リットルあたりNaOH約300から350gに相当するようにする。高温は、好ましくは、60℃以上である。
【0024】
本発明に係る好ましい実施形態において、二酸化チタン水和物等の二酸化チタン原料は、硫酸化工程の中間生成物であり、塩酸に可溶であるチタン酸ナトリウムへの塩基処理は、好ましくは、約95℃以上、好ましくは2時間行われる。この処理を、間接的な蒸気で加熱され、撹拌されている反応炉中で行うことができる。塩基で処理された生成物のpHは、好ましくは11超である。
【0025】
塩基処理後に得られるチタン塊を、好ましくは熱水で、好ましくは60℃未満の水で洗浄し、濾過する。目的は、硫酸イオンを含まない塊を洗浄することであり、それによる洗浄結果および残留物含有率を、既知の方法での塩化バリウム試験により調査することができる。
【0026】
下位段階A2において、段階A1で得られたナトリウム含有チタン塊を、水中で再水簸する。この硫酸塩不含のナトリウム含有濾過ケーキを、好ましくは140から200g TiO2/l、より好ましくは約180g TiO2/lの濃度に、蒸留水中で水簸する。
【0027】
実施形態によれば、スラリーの温度を、40から50℃に、好ましくは約1℃/分の速度で上昇させる。
【0028】
そのpHを低下させるために、酸、好ましくは塩酸を、スラリーに変化し塩基で処理された二酸化チタン原料、好ましくはチタン酸ナトリウムに添加する。
【0029】
最終的な酸濃度を、8から35g HCl/リットル、好ましくは29から31g HCl/リットルに調節する。HCl含有率を、滴定により再調査し、必要な場合には補正することができる。この後に、このように生成したスラリーの温度を、88から95℃、好ましくは約90℃にゆっくりと上昇させる。温度上昇の速度は好ましくは約1℃/分であり、加熱中はスラリーを撹拌する。処理を約2時間続けて、ルチル結晶を形成する。塩酸濃度および温度等の使用パラメータの値は、このように得られるルチル結晶の形成に影響を与えることが観測されている。
【0030】
酸処理後に得られた生成物のルチル含有率は、99.0%超、好ましくは99.5%超である。
【0031】
下位段階A3において、特許EP0444798に本質的に従い、段階A2から得られた上述の沈殿物を中和し、わずかに酸性のpH値のスラリーにする。中和においては、pHを4以上の値だが、6未満の値、好ましくは4.4から5.0の値に上昇させる。中和を、適切なアルカリ性中和薬品、好ましくは水酸化ナトリウムまたは炭酸ナトリウムにより行うことができる。中和後に得られた二酸化チタン塊を、濾過および洗浄する。中和を注意深く行い6以下、または好ましくは5以下のpH値にすれば、濾過および洗浄は極めてうまくいき、そのことにより、さらなる処理を妨げる本質的にすべてのナトリウムおよび塩化物のイオンを、この塊から除去することができる。
【0032】
下位段階A4において、95.0%超、好ましくは99.5%超がルチルの形である、段階A3から得られたルチル構造の二酸化チタンスラリーに、適切な前処理薬品を注入する。前処理薬品を、撹拌しつつ二酸化チタン塊に添加する。前処理薬品は、好ましくは、微細に磨砕された固体の乾燥粉末として添加される。添加後に、塊を、少なくとも1時間、好ましくは4時間以超、それを炉に供給し始める前に撹拌する。
【0033】
本発明における前処理薬品は、焼成を制御しつつ使用されて、結晶成長と結晶構造の転移温度とに影響を与える薬品を指し、それは当技術分野において一般的な意味の用語である。本発明に関して、それは、好ましくは、TiO2焼成中に用いられる前処理薬品を指す。
【0034】
前処理薬品は、結晶成長を制御し、必要な溶融物の低温での形成もする形の物質である。前処理薬品は、好ましくは、リン酸二水素カリウム、KH2PO4、または焼成の前もしくは間に反応し、リン酸二水素カリウム、もしくはリン酸二水素カリウムの類似化合物になるカリウムおよびリンの化合物の混合物である。その場合において、例えば、硫酸カリウム、K2SO4およびリン酸アンモニウムMAPの混合物を考慮することができる。例えばリン酸は、リン源としても機能することができる。前処理薬品は、最も好ましくは、リン酸二水素カリウム、KH2PO4である。前処理薬品として様々なカリウムおよびリンの化合物を用いると、それらは結晶成長を制御する適切な溶融物を焼成中に形成し、それらを、混合されるべき塊の中に一緒または別々のいずれかで供給できることが観測されている。
【0035】
リン酸二水素カリウムとして表される必要な前処理薬品の量は、0.2から1重量%、好ましくは0.5から0.7重量%、最も好ましくは約0.6重量%である。含有率が0.2重量%未満である場合、十分な効果はもはや得られない。それぞれに必要な薬品量は、焼成温度、焼成時間、および所望の結晶サイズにも依存し、したがって、当技術分野の専門家により決定され得る。含有率が本質的に1重量%を超える場合、同様の焼成条件下で結晶が異常成長し始める。さらに、用いる前処理薬品が多すぎると、例えばカリウムを洗浄するのがより困難となり、処理段階が複雑になり、操業コストが増大する。
【0036】
前処理薬品に必要な特性は、焼成温度で溶融物を形成する能力である。前処理薬品は、好ましくは明らかに焼成温度未満で溶融物を形成し、そのことにより、溶融物の粘度は本発明に係る操業温度で可能な限り低くまで低下する。前処理薬品の溶融温度は、好ましくは730℃未満、より好ましくは670℃未満、最も好ましくは253℃等の350℃未満である。異なる化合物の混合物を用いた場合、完全な溶融は求められないが、溶融物量が十分であれば、明らかに液相線内である、すなわち部分的に溶融していれば十分である。
【0037】
リン酸二水素カリウムを用いることにより、顔料二酸化チタンの調製が有利になる。さらに、リン酸二水素カリウムは、約253℃の非常に低い温度ですでに結晶を形成するのに有利な溶融物を形成し、結晶表面の原子の移動を可能にし、低温ですでに焼成処理を促進し、そのことにより、より小さい結晶サイズをもつ最終生成物が得られる。薬品のリンが結晶表面に残存し、その部分により、水相中における結晶の分散性が改善される。その場合において、磨砕性も改善される。この薬品は、このように得られる生成物のpH値も本質的には変化させない。前処理薬品の使用は、特に焼成後の生成物の磨砕性を改善する効果を示すことが観測された。
【0038】
前処理後に得られたスラリー様生成物を、下位段階A5において焼成する。焼成は、好ましくは、350から800℃の温度で、より好ましくは560から770℃の温度で、最も好ましくは650から750℃の温度で行われる。焼成温度の選択は、例えば、供給材料の組成、使用する水含有率、滞留時間、および焼成ガスにより影響を受ける。様々な要因の組合せにより条件が与えられ、焼成炉の取り出し物の結晶サイズおよび結晶サイズ分布が所望のようになる。本発明に係る塩不含のルチル構造二酸化チタン塊の焼成パラメータを用いて、簡単かつ正確に微結晶二酸化チタンの結晶サイズおよび結晶サイズ分布を調節することができる。焼成中に、結晶を凝集させ、その後に所望の粒径に磨砕する。焼成後の本発明に係る二酸化チタンの結晶サイズは、好ましくは約10nm超、さらに60nm未満、好ましくは50nm未満、最も好ましくは約30nmである。焼成後の生成物のルチル含有率は、99.0%超、好ましくは99.5%超である。焼成された顔料の結晶は、本質的に楕円形である。
【0039】
焼成時間は、0.5から5時間、好ましくは1から3時間である。沈殿段階も焼成段階も、加圧条件でなく常圧で行われる。
【0040】
好ましい実施形態において、前処理薬品としてリン酸二水素カリウムを用いた場合、水中で水簸したときに、従来の未注入焼成された微結晶ルチル構造の二酸化チタンよりはるかに磨砕が容易である焼成取り出し物が得られる。
【0041】
一般に、所望の粒径を達成するために、焼成後に何度も、例えば約5から6回生成物を磨砕し、最終的に、例えばコーティング後に噴霧磨砕する必要があった。その場合において、エネルギー消費が多く、磨砕したにもかかわらず、所望の結晶サイズ、および狭い結晶サイズ分布を達成されなかった。現在では、これらの不利を、本発明に係る方法により回避することができる。さらに、焼成取り出し物の処理後洗浄において、最終生成物に不利になる、前処理薬品の一部であるカリウムを、最終生成物から洗い去ることができる。
【0042】
段階Bにおいて、段階A(A1からA5)から得られたルチル構造の微結晶酸化チタンをさらに処理し、当技術分野において既知の任意の方法により二酸化チタン最終生成物にすることができる。さらなる処理は、好ましくは、特許明細書EP0444798に記載の方法により主に行われる。
【0043】
本発明に係る好ましい実施形態によれば、段階Bには、明細書EO0444798に開示されている以下のさらなる処理段階が含まれ、特別な湿式磨砕段階B4をそれに加えている。すなわち、
B1 焼成から得られた塊を湿式磨砕し、
B2 湿式磨砕から得られた塊を表面処理し、
B3 表面処理した塊を濾過および洗浄し、
B4 段階B3から得られた塊を再度湿式磨砕し、
B5 段階B4から得られた塊を噴霧乾燥および噴霧磨砕して、最終的な二酸化チタン生成物を調製する。
【0044】
処理段階B1において、段階A(A5)から得られた焼成二酸化チタンを蒸留水中で水簸して濃厚スラリーを形成する。水簸は、好ましくは、分散剤、より好ましくはモノイソプロパノールアミンMIPAにより行われる。スラリーの濃度は、500から600g TiO2/l、好ましくは約550TiO2/lである。焼成から得られる塊を、例えば研磨機で既知の方法により湿式磨砕する。磨砕後に得られた生成物のU/V*100値は、好ましくは850超である。
【0045】
処理段階B2において、段階B1から得られた、磨砕された二酸化チタン塊を、その使用目的に従った任意の既知の方法により、例えばアルミニウム、シリカおよび/またはジルコニウム化合物によりコーティングする。使用した方法には、例えば、特許明細書FI62130に開示されている処理方法が含まれ得る。
【0046】
処理段階B3において、コーティング処理B2の後に、生成物を濾過し水で洗浄する。無機コーティングは、微結晶二酸化チタンを激しく凝集させる。
【0047】
処理段階B4において、段階B3から得られた、コーティングされた二酸化チタン塊を、水中で再水簸し、スラリーを湿式磨砕する。磨砕の開始前に、または湿式磨砕段階がない場合には、メチロールプロパン、TMP、メチロールエタンまたはケイ素(FI57124参照)等の、乾燥設備の有機添加剤を、供給スラリーを乾燥設備に供給する前にそれに添加する。使用されるコーティング剤は、好ましくはTMPである。使用されるTMPの量は、2から8重量%、好ましくは2から6重量%、最も好ましくは5重量%である。磨砕の目的は、TMPを乾燥設備への供給物にできるだけ均一に混合させることである。磨砕が、低い濃度、好ましくは130から270g/lで、多い供給量で行われることにより、TiO21トンあたりの磨砕効率が低いままとなる。使用される磨砕効率は、使用される設備および能力に依存し、そのことは当業者には明らかである。
【0048】
コーティング後の湿式磨砕処理により、二酸化チタン最終生成物の分散性および透明度がさらに改善される。湿式磨砕は、二酸化チタン最終生成物の光活性の増加も観測されなかったことは、この生成物の用途には重要である。以前は、コーティングが破壊され、有益な特性が失われる、例えば光安定性が弱まると、後処理の後にもはや効率的な磨砕を用いることができないと考えられていた。しかし、それは事実ではなく、代わりに、本発明に係るコーティングおよび噴霧乾燥の間に湿式磨砕段階を加えることにより、より良好な分散性を、光安定性を失うことなく達成した。
【0049】
処理段階B5において、段階B4からの、コーティングされ湿式磨砕された二酸化チタン塊を、噴霧乾燥および噴霧磨砕して、当技術分野において既知の方法で最終的な二酸化チタン最終生成物を得る。
【0050】
本発明は、請求項13に係る、新規なルチル構造の微結晶二酸化チタン生成物も開示する。生成物の結晶サイズ(d50)は、60nm未満、好ましくは50nm未満であり、結晶サイズは、より好ましくは20から40nm、最も好ましくは約30nmであり、粒径d50は0.150μm未満であり、生成物は良好に分散可能であり、U/V*100 p ratは500以上である。このように得られた生成物の比表面積BETは55m2/g以下であり、その光安定性は好ましくは10未満である。本発明に係る二酸化チタン生成物は、図3B’からわかる可視光の良好な透過度も特徴とする。図3に、本発明に係る二酸化チタン生成物の粒子を含有するラッカー膜の透過曲線を示す。本発明に係る生成物を含有するラッカー膜の透過曲線の透過度は、少なくとも、図3B’に示される曲線の透過度ほどに良好すなわち浸透性がある。一般に、浸透性すなわち透過度は、波長490nmで得られるT=60%の値により説明される。
【0051】
好ましい実施形態によれば、生成物の結晶サイズは、20から40nm、より好ましくは約30nmである。結晶サイズは、使用目的に応じてこの方法の中で変化させることができる。
【0052】
本発明に係る生成物の良好な分散性は、濁度測定により測定される特性を指す。測定において、水相中で終夜分散させた試料の吸収の変化は、当技術分野における既知の方法で分光光度法により波長の関数により観測される。測定曲線から、粒径分布の広さまたは狭さを記述するために使用可能なU/V*100 p ratを算出することができる。U/V*100値が高いほど、粒径分布が狭い、すなわち、生成物が良好に磨砕されているほど、生成物の分散性はさらに良好である。本発明に関して、「良好な分散性」の用語は、300以上、好ましくは500以上のU/V*100 p rat値を有する生成物について得られる特性を指す。
【0053】
本発明に係る生成物のU/V*100 p rat値は、好ましくは550超である。このことは、曲線の急峻さのために、生成物が非常に良好なUV保護も提供することを示している。
【0054】
分散性は様々な因子に依存するが、この場合においては、粒径は最も本質的なものであることがわかった。粒径が減少している場合は、粒子が凝集する自然な傾向のために分散性が悪化する。
【0055】
本発明に係る二酸化チタン生成物の比表面積、BETは、結晶サイズ分布d50が30±5nmである場合で、≦55m2/g、好ましくは≦50m2/gである。最も好ましくは、平均結晶サイズd50が30±3nmである場合で、BET≦45m2/gである。
【0056】
本発明に係る生成物は、意外にも制御が容易である小さな平均粒径と、狭い粒径分布とを有する。この分布は、好ましくは、焼成中にリン酸二水素カリウム等の前処理薬品を用いることにより達成される。さらに、特別な湿式磨砕段階は、より好ましくは噴霧乾燥および噴霧磨砕の前の調製において用いられる。本発明に係る生成物は、最も好ましくは請求項1に記載の方法により調製される。本発明に係る方法を用い、特許明細書EP0444798に言及されている方法と比べて明確に良好な分散性の微結晶生成物を調製することができる。
【0057】
さらに、本発明に係る二酸化チタン生成物は、非常に光安定性である。一般に、その光安定性は、特許明細書FI20040186に記載されているように、アセトンの形成速度により説明されるが、別に、分解される気体としてプロパノールの代わりにアルデヒドとトルエンとの混合物を用いると、光安定性はアセトンの形成速度でなくCO2の形成速度により説明される。本発明に係る二酸化チタン生成物は、10ppm/h未満、好ましくは5ppm/h未満の、アセトンの形成速度を特徴とする。
【0058】
本発明に係る新規な二酸化チタン生成物は、好ましくは、上述の方法により調製される。本発明に係る方法により調製される生成物は、磨砕が容易な焼成取り出し物を特徴とし、それにより、最終生成物の最終的な粒径分布を、明確により少ない磨砕回数により制御することができる。特別な湿式磨砕段階B4を加えることにより、磨砕性をより容易になり、粒径分布がより狭くなることがさらに観測された。必要な磨砕回数を、好ましくは半減することができる。
【0059】
本発明に係る生成物に与えられる、粒径および粒径分布の最適な制御は、かなりより良好な分散可能生成物を示唆し、生成物の特定の用途に必要な金属光沢の特性、フリップフロップ効果をさらに強める。
【0060】
好ましい実施形態において、本発明に係る新規な二酸化チタン生成物を、光沢のある自動車塗料等の装飾塗料組成物中に用いて、可視光波長および反射効率の非常に強い角度依存性を提供することができる。この場合において、20°b*が8以上で、明るい金色のフリップ効果、115°b*が−11未満で、濃い青色のフロップ効果をもたらすために用いることができる塗料組成物が提供される。
【0061】
本発明は、上記に係る二酸化チタン生成物を含有する装飾塗料を開示し、塗料にフリップフロップ効果を与える。この塗料は、好ましくは、20°b*が8以上でフリップ効果、115°b*が−11未満で濃い青色のフロップ効果を提供するために用いられる。
【0062】
本発明は、本発明に係る二酸化チタン生成物をUV保護として用いることができる木材ラック組成物をさらに開示する。
【0063】
本発明を、以下の実施例により例証する。個別に言及がなければ、原料および材料は、市販の製品であるか、それらは既知の技術により製造され得る。
【実施例1】
【0064】
図1は、二酸化チタンの調製において用いられる処理段階を本質的に示す。硫酸塩法による二酸化チタンの調製は、イルメナイト濃縮物を硫酸と反応させることにより開始される。このように得られた固体の反応ケーキを、水および廃酸により溶解する。不純物を除去し、硫酸鉄を晶出させる。チタン含有溶液を濃縮し、二酸化チタン水和物を加水分解により沈殿させる。この沈殿物塊を数段階で洗浄して、塩を除去する。
【0065】
TiO2として表される、この洗浄済沈殿物塊約10トンを取り、水で水簸して、目標350g/lで300から400g/lの濃度を得る。このように得られたスラリーを、NaOH溶液としてNaOH約15トンを60℃で添加することにより高アルカリ性(pH>11)とし、その濃度は700g/lである。スラリーの温度を95℃に上げ、それをこの温度で2時間撹拌する。処理中に、水酸化チタン塊をアルカリ液と反応させ、固体のチタン酸ナトリウムを形成し、そこから、塩化バリウムで沈殿させたときに濾液中に硫化物が見られなくなるまで熱水でスラリーを洗浄することにより、硫酸イオンをさらに除去する(図1、段階A1)。
【0066】
二酸化チタンとして表されるスラリーの濃度が約180g/lになるように、硫化物不含のナトリウム含有濾過ケーキを水中で水簸する。このスラリーを、1℃/分の速度で40から45℃の温度に加熱する。このスラリーの酸含有率を、30%塩酸溶液を添加することにより30g HCl/lに設定して、ルチル構造の結晶を作り出す。必要であれば、HCl含有率を滴定により調節し補正する。スラリーの温度を、1℃/分の速度で、一定した撹拌をしつつゆっくりと90℃に上げる。この温度で、スラリーを撹拌しつつ120分間処理する(図1、段階A2)。
【0067】
最終的に、スラリーを、pHが4.7から4.8に定まるように、炭酸ナトリウムまたは水酸化ナトリウムで中和する。中和済スラリーを、濾過し、蒸留水約4リットルで洗浄する。このように、乾燥した濾過ケーキの含有率は約30%である(図1、段階A3)。
【0068】
TiO2として表される洗浄ケーキ10tを取り、水で水簸して、150から250g/lの濃度を得る、すなわち、スラリー約40から50m3を得る。水簸は撹拌機を備えた容器中で行われ、そこに粉末の市販のKH2PO4約60kgを前処理薬品として添加する。スラリーを、炉へ供給し始める前に、約4時間混合させる(図1、段階A4)。
【0069】
試料を、炉の供給口から取り、乾燥し、X線回折図を取り、微結晶二酸化チタンの99.0%超がルチルの結晶形を有することが見出される。
【0070】
二酸化チタン濾過ケーキを、720℃の温度の連続回転リールオーブン中で焼成する。滞留時間は、平均で約1時間である。焼成されたルチル構造の生成物の電子顕微鏡画像を測定すると、平均結晶サイズは約30nmであり、分布幅は10から50nmである。焼成生成物のナトリウム含有率は0.1%未満であり、塩化物含有率は0.05%未満である(図1、段階A5)。このように作り出された生成物の電子顕微鏡画像を図7に示す(160000倍)。
【0071】
塩不含の二酸化チタンを、分散剤により濃厚スラリーとして蒸留水中で水簸する。湿式磨砕を研磨機で行う。一般に、生成物は3回湿式磨砕される(図1、段階B1)。
【0072】
互いに別々に磨砕した結晶を、使用目的に従って、アルミニウム、ケイ素および/またはジルコニウムの化合物で後処理する。ここで、試料を酸化ジルコニウム(1.0%)および酸化アルミニウム(4.0%)でコーティングする(図1、段階B2)。方法として、例えば特許明細書FI62130に表されるもの等の、二酸化チタン顔料の処理のための他の既知の方法を用いることもできる。
【0073】
TMP5%を、洗浄され濾過されたコーティング済塊試料に添加し、その後に、乾燥噴霧前にそれをもう一度湿式磨砕する(図1、段階B3およびB4)。
【0074】
噴霧乾燥された微結晶TiO2を噴霧磨砕し、約30nmの結晶サイズを有する微細な粉末にする(図1、段階B5)。粒径は、Master Malvernサイズ測定装置により測定され、約0.1μmで明確により大きい。
【0075】
噴霧乾燥後に、このように得られた最終生成物の紫外可視スペクトルを、水性スラリーから測定する。それは焼成段階後の研磨段階での生成物のスペクトルとほとんど同じ水準であることが観測される。
【実施例2】
【0076】
試料の磨砕性、分散性および粒径分布への調製方法の効果を調査する。
【0077】
磨砕性を、分光光度計により測定される濁度曲線と、Malvern Mastersizer 200により測定した粒径分布の両方に基づいて評価する。
【0078】
酸で処理した沈殿物塊が濾過および洗浄されるように、試料を実施例1に従って調製し、その後、リン酸二水素カリウム0.6gをこの塊に添加する。この塊を回転リールオーブン中で730℃で約1時間焼成する。焼成後に、オーブン取り出し物を研磨機中で4回湿式磨砕する。この粒子を、ジルコニウムおよびアルミニウムの酸化物(ZrO2はTiO2の1.0%、Al2O3はTiO2の4.0%)でコーティングし、濾過し水で洗浄する。TMP5%をこの塊に添加し、その後に、試料を2つの部分に分割する。試料Aは、噴霧乾燥および噴霧磨砕の前に特別な湿式磨砕を受ける。試料Bは、特別な湿式磨砕をされずに、乾燥および磨砕される。参照試料Cを、特許EP0444798、実施例1に記載の方法で調製し、粒径30nmにする(EP0444798、実施例9)。焼成生成物Cは、試料Bと同様に後処理段階を受ける。
【0079】
噴霧磨砕された最終生成物試料A、BおよびCを、分析で純粋な0.1体積%の1−アミノ−2−2プロパノール(MIPA)を添加したイオン交換水中に分散する。350から1100nmの範囲内の濁度を、1ナノメートルの間隔で、分光光度計により希釈水性分散液から測定する。
【0080】
表1において、顔料の微細さの程度を4つの数量により測定している。すなわち、
1)吸光度(最大)、最大吸収波長[nm]、
2)U/V*100、波長450nmの吸収で割った最大吸収波長の吸収*100、
3)吸光度(最大)、最大吸収波長での吸収、および
4)吸光度(450nm)、波長450での吸収
である。
【0081】
【表1】
【0082】
図2に、生成物AおよびBの紫外可視スペクトルを、生成物Cの紫外可視スペクトルと比較して示す。参照の生成物に対して、本発明に従って調製される微結晶顔料AまたはBは、明確に良好に磨砕および分散されている。このように、生成物を前処理薬品を用い調製すると、参照生成物Cと比べて分散性が改善する(生成物B)。前処理薬品の他に、噴霧乾燥前に特別な湿式磨砕段階を用いると、分散性は、550以上のU/V*100の値にさらに改善される(生成物A)。
【実施例3】
【0083】
粒径分布を、Malvern Mastersize2000により、実施例2に係る生成物CおよびAから測定する。この方法において、試料を水簸して懸濁液を形成し、連続循環中の懸濁液を、レーザー光線を当てた流通キュベットに導く。試料の粒子は、単色のレーザー光線を、それらのサイズに応じて様々な散乱角で散乱させる。様々な角度で散乱される放射の強度を測定する。試料の粒径分布を、MieまたはFraunhoferの理論に従って、散乱強度から算出する。測定は、装置の取扱説明に従って行われる。
【0084】
図6および表2に粒径分布を示す。参照の生成物Cの試料の粒径分布は、明確に2つの山を有し、d50の値は明確により高い。
【0085】
【表2】
【実施例4】
【0086】
実施例2の生成物A、BおよびCの光活性を、既知の気相の方法により測定する。
【0087】
この方法において、2−プロパノールを閉鎖室内に注入する。ペトリ皿上の粉末試料にXeランプにより照射する。2−プロパノールは、まずアセトンに、最終的には二酸化炭素に分解する。二酸化炭素の形成は、本質的に光触媒活性の結果である。より大きい数は、より高い光活性を表す。
【0088】
このように得られた光活性の結果を表3に示す。
【0089】
【表3】
【0090】
表3における結果に基づくと、この特別な湿式磨砕段階が、生成物Aの光触媒活性を増大させないことを確証することができ、このことは、その用途のために重要である。
【実施例5】
【0091】
実施例1に記載の方法により調製した生成物Gを、できるだけ類似した化学組成を有する市販の他の生成物AからFと比較した。調査した特性は、
1.既知の方法によるX線回折(XRD)により求められるルチル結晶のサイズ(nm)、
2.既知の方法により求められる比表面積、BET(m2/g)、
3.濁度曲線により求められる、分散性を記述する数量U/V*100
4.アセトンの形成速度(ppm/h)として測定される光安定性
であった。
【0092】
表4に示す測定結果に基づいて、本発明に係る生成物Gの光安定性が、他のルチル構造の二酸化チタン生成物と比べて優れていることを観測することができる。同時に、生成物Gは、極めて良好な分散性を提供する。参照の試料に対する結晶サイズ分布の狭さは、より小さいBET値として表れている一方、より狭い粒径分布は、より高いU/V*100値として表れている。
【0093】
【表4】
【実施例6】
【0094】
ラッカー膜A’、B’およびC’(参照の膜)を、実施例2に従って作製した二酸化チタン生成物AおよびB、ならびに参照の生成物Cからさらに作製する。TiO2水性分散液をベース組成物と合わせ、約5分間撹拌(Skandex BA−EL−T)することにより、二酸化チタンを2.0重量%水希釈性アクリルワニスベースと混合する。ラッカー組成物の粘度を、25〜30秒/DIN−Bencher 4mmに調節する。
【0095】
図3に、ラッカー膜の透過スペクトルを示す。膜B’の透過度は、膜C’と比べて可視スペクトルの全領域内で明確に良好である。膜A’についての透過度は、膜B’およびC’と比べて、スペクトルの範囲にわたってさらに改善する。
【0096】
様々な波長についての参照値を表5に示す。0−膜は、二酸化チタンがない基本的なラッカー組成物である。
【0097】
【表5】
【実施例7】
【0098】
実施例2に記載の方法により作製した二酸化チタンA、BおよびCならびにB’(B’は、その焼成温度が670℃である点で生成物Bと異なる)を、金属装飾の塗料に適した1.5および2.0重量%TiO2/2重量%Alの、車の下塗り塗料組成物と混合する。
【0099】
図5に、参照の試料C、ならびに本発明に係る試料A、BおよびB’を含有する装飾塗料試料から、CIELAB色記述モデルに従って、測角スペクトル光度計(Optronic GmbH)により様々な角度で測定した反射を示す。このように調製した装飾塗料により、非常に強い角度依存性が、本発明に係る二酸化チタンを含有するコーティングによりもたらされる。明るい金色のフリップ効果20°b*は8以上であり、濃い青色のフロップ効果115°b*は−11未満である。
【0100】
さらに、このようにもたらされる効果の強度が高いことを述べておくべきである。入手可能な既知の塗料組成物と比べて、本発明に係る二酸化チタン生成物を含有する塗料組成物によりもたらされる効果について、同じ効果を得るために塗料組成物において必要な対応する二酸化チタン量はわずか約25%である。
【特許請求の範囲】
【請求項1】
A.
A1.二酸化チタン原料を、アルカリ性のpH値を有するように塩基で処理し、
A2.最終的な塩酸含有率が8から35g/lに調節されるように、段階A1から得られた沈殿物を塩酸でさらに処理し、
A3.段階A2から得られた沈殿物を中和して、4.0から6.0のpH値のスラリーにし、
A4.段階A3から得られた中和済スラリーに前処理薬品を注入し、
A5.段階A4から得られた注入済スラリーを焼成する
ように、前記二酸化チタン原料を処理して、それを二酸化チタンに変換する段階と、
B.段階Aから得られた二酸化チタンをさらに処理して、それを二酸化チタン最終生成物に変換する段階と
を含む、60nm未満の結晶サイズを有する、分散良好なルチル構造の微結晶二酸化チタン生成物を、二酸化チタン原料から調製する方法。
【請求項2】
前記前処理薬品が、リン酸二水素カリウム、KH2PO4、または焼成の前もしくは間に反応し、リン酸二水素カリウム、もしくはリン酸二水素カリウムの類似化合物になる、カリウムおよびリンの化合物の混合物であることを特徴とする、請求項1に記載の方法。
【請求項3】
前記前処理薬品を、リン酸二水素カリウムとして算出される0.2から1重量%の量で添加することを特徴とする、請求項1または2に記載の方法。
【請求項4】
前記前処理薬品を、段階A3から得られた混合中の中和済スラリーに、固体粉末として添加することを特徴とする、請求項1乃至3のいずれか1項に記載の方法。
【請求項5】
前記二酸化チタン原料が、固体二酸化チタン水和物であることを特徴とする、請求項1乃至4のいずれか1項に記載の方法。
【請求項6】
前記二酸化チタン水和物を、硫化物法により、イルメナイトから調製することを特徴とする、請求項5に記載の方法。
【請求項7】
前記結晶サイズが50nm未満であることを特徴とする、請求項1に記載の方法。
【請求項8】
段階A2において、段階A1から得られた生成物を、前記スラリーの温度を40〜45℃で上昇させ、その後に、塩酸含有率を段階A2の前記最終的な塩酸含有率に調節することにより水中で水簸することを特徴とする、請求項1乃至7のいずれか1項に記載の方法。
【請求項9】
段階A3において、前記中和を水酸化ナトリウムまたは炭酸ナトリウムにより行い、その後に生成物を濾過および洗浄することを特徴とする、請求項1乃至8のいずれか1項に記載の方法。
【請求項10】
前記さらなる処理段階Bが、以下の逐次的な段階
B1 焼成から得られた塊を湿式磨砕し、
B2 湿式磨砕から得られた塊を表面処理し、
B3 表面処理した塊を濾過および洗浄し、
B4 段階B3から得られた塊を再度湿式磨砕し、
B5 段階B4から得られた塊を噴霧乾燥および噴霧磨砕して、最終的な二酸化チタン生成物を調製する、
を含むことを特徴とする、請求項1乃至9のいずれか1項に記載の方法。
【請求項11】
段階B4において、段階B3から得られた塊を水中で水簸し、有機添加剤を前記スラリーに添加することを特徴とする、請求項10に記載の方法。
【請求項12】
前記有機添加剤がトリメチロールプロパンを含有することを特徴とする、請求項11に記載の方法。
【請求項13】
前記有機添加剤を2から8重量%の量で添加することを特徴とする、請求項10または11に記載の方法。
【請求項14】
a)その濁度曲線から求められるU/V*100p rat値が500以上であり、
b)その比表面積、BETが55m2/g以下であり、
c)その粒径分布d50が0.150μm未満であり、
d)粒子を含有するラッカー膜の透過曲線の透過度が、少なくとも、図3に示す曲線B’の透過度ほどの浸透性である、
60nm未満の結晶サイズを有する分散良好なルチル構造の微結晶二酸化チタン生成物。
【請求項15】
請求項1乃至13のいずれか1項に記載の方法により調製されることを特徴とする、60nm未満の結晶サイズを有する分散良好なルチル構造の微結晶二酸化チタン生成物。
【請求項16】
前記結晶サイズが50nm未満であることを特徴とする、請求項14または15に記載の二酸化チタン生成物。
【請求項17】
アセトンの形成速度として表されるその光安定性が10ppm/h未満であることを特徴とする、請求項14乃至16のいずれか1項に記載の二酸化チタン生成物。
【請求項18】
その結晶サイズが20から40nmであることを特徴とする、請求項14乃至17のいずれか1項に記載の二酸化チタン生成物。
【請求項19】
フリップフロップ効果を塗料に付与するための、請求項14乃至18のいずれか1項に記載の二酸化チタン生成物を含有する装飾塗料。
【請求項20】
前記装飾塗料によりもたらされるフリップフロップ効果において、フリップ効果20°b*が8以上であり、フロップ効果115°b*が−11以下であることを特徴とする、請求項19に記載の装飾塗料。
【請求項21】
UV保護剤として、請求項14乃至18のいずれか1項に記載の二酸化チタン生成物を含有する木材ラッカー組成物。
【請求項1】
A.
A1.二酸化チタン原料を、アルカリ性のpH値を有するように塩基で処理し、
A2.最終的な塩酸含有率が8から35g/lに調節されるように、段階A1から得られた沈殿物を塩酸でさらに処理し、
A3.段階A2から得られた沈殿物を中和して、4.0から6.0のpH値のスラリーにし、
A4.段階A3から得られた中和済スラリーに前処理薬品を注入し、
A5.段階A4から得られた注入済スラリーを焼成する
ように、前記二酸化チタン原料を処理して、それを二酸化チタンに変換する段階と、
B.段階Aから得られた二酸化チタンをさらに処理して、それを二酸化チタン最終生成物に変換する段階と
を含む、60nm未満の結晶サイズを有する、分散良好なルチル構造の微結晶二酸化チタン生成物を、二酸化チタン原料から調製する方法。
【請求項2】
前記前処理薬品が、リン酸二水素カリウム、KH2PO4、または焼成の前もしくは間に反応し、リン酸二水素カリウム、もしくはリン酸二水素カリウムの類似化合物になる、カリウムおよびリンの化合物の混合物であることを特徴とする、請求項1に記載の方法。
【請求項3】
前記前処理薬品を、リン酸二水素カリウムとして算出される0.2から1重量%の量で添加することを特徴とする、請求項1または2に記載の方法。
【請求項4】
前記前処理薬品を、段階A3から得られた混合中の中和済スラリーに、固体粉末として添加することを特徴とする、請求項1乃至3のいずれか1項に記載の方法。
【請求項5】
前記二酸化チタン原料が、固体二酸化チタン水和物であることを特徴とする、請求項1乃至4のいずれか1項に記載の方法。
【請求項6】
前記二酸化チタン水和物を、硫化物法により、イルメナイトから調製することを特徴とする、請求項5に記載の方法。
【請求項7】
前記結晶サイズが50nm未満であることを特徴とする、請求項1に記載の方法。
【請求項8】
段階A2において、段階A1から得られた生成物を、前記スラリーの温度を40〜45℃で上昇させ、その後に、塩酸含有率を段階A2の前記最終的な塩酸含有率に調節することにより水中で水簸することを特徴とする、請求項1乃至7のいずれか1項に記載の方法。
【請求項9】
段階A3において、前記中和を水酸化ナトリウムまたは炭酸ナトリウムにより行い、その後に生成物を濾過および洗浄することを特徴とする、請求項1乃至8のいずれか1項に記載の方法。
【請求項10】
前記さらなる処理段階Bが、以下の逐次的な段階
B1 焼成から得られた塊を湿式磨砕し、
B2 湿式磨砕から得られた塊を表面処理し、
B3 表面処理した塊を濾過および洗浄し、
B4 段階B3から得られた塊を再度湿式磨砕し、
B5 段階B4から得られた塊を噴霧乾燥および噴霧磨砕して、最終的な二酸化チタン生成物を調製する、
を含むことを特徴とする、請求項1乃至9のいずれか1項に記載の方法。
【請求項11】
段階B4において、段階B3から得られた塊を水中で水簸し、有機添加剤を前記スラリーに添加することを特徴とする、請求項10に記載の方法。
【請求項12】
前記有機添加剤がトリメチロールプロパンを含有することを特徴とする、請求項11に記載の方法。
【請求項13】
前記有機添加剤を2から8重量%の量で添加することを特徴とする、請求項10または11に記載の方法。
【請求項14】
a)その濁度曲線から求められるU/V*100p rat値が500以上であり、
b)その比表面積、BETが55m2/g以下であり、
c)その粒径分布d50が0.150μm未満であり、
d)粒子を含有するラッカー膜の透過曲線の透過度が、少なくとも、図3に示す曲線B’の透過度ほどの浸透性である、
60nm未満の結晶サイズを有する分散良好なルチル構造の微結晶二酸化チタン生成物。
【請求項15】
請求項1乃至13のいずれか1項に記載の方法により調製されることを特徴とする、60nm未満の結晶サイズを有する分散良好なルチル構造の微結晶二酸化チタン生成物。
【請求項16】
前記結晶サイズが50nm未満であることを特徴とする、請求項14または15に記載の二酸化チタン生成物。
【請求項17】
アセトンの形成速度として表されるその光安定性が10ppm/h未満であることを特徴とする、請求項14乃至16のいずれか1項に記載の二酸化チタン生成物。
【請求項18】
その結晶サイズが20から40nmであることを特徴とする、請求項14乃至17のいずれか1項に記載の二酸化チタン生成物。
【請求項19】
フリップフロップ効果を塗料に付与するための、請求項14乃至18のいずれか1項に記載の二酸化チタン生成物を含有する装飾塗料。
【請求項20】
前記装飾塗料によりもたらされるフリップフロップ効果において、フリップ効果20°b*が8以上であり、フロップ効果115°b*が−11以下であることを特徴とする、請求項19に記載の装飾塗料。
【請求項21】
UV保護剤として、請求項14乃至18のいずれか1項に記載の二酸化チタン生成物を含有する木材ラッカー組成物。
【図1】

【図2】

【図3】

【図4】

【図5】
【図6】

【図7】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公表番号】特表2010−536689(P2010−536689A)
【公表日】平成22年12月2日(2010.12.2)
【国際特許分類】
【出願番号】特願2010−520605(P2010−520605)
【出願日】平成20年8月15日(2008.8.15)
【国際出願番号】PCT/FI2008/050466
【国際公開番号】WO2009/022061
【国際公開日】平成21年2月19日(2009.2.19)
【出願人】(510041821)
【Fターム(参考)】
【公表日】平成22年12月2日(2010.12.2)
【国際特許分類】
【出願日】平成20年8月15日(2008.8.15)
【国際出願番号】PCT/FI2008/050466
【国際公開番号】WO2009/022061
【国際公開日】平成21年2月19日(2009.2.19)
【出願人】(510041821)
【Fターム(参考)】
[ Back to top ]