
制がん剤
【課題】リポ酸誘導体を有効成分とする制がん剤を提供する。
【解決手段】次の一般式(1)

(式中、R1はOH基、O−アルキル基、N−置換アミノ基、アミノ酸残基またはペプタイド残基を示す。nは0または1の整数であり、nが0のとき隣接するS原子は互いに結合して環を形成し、nが1のときR2及びR3は、同一または異なって、水素原子またはスルホ基を示す。但し、R1はOH基であるとき、nは0ではない。)で表わされる化合物、またはその薬理学的に許容される塩を有効成分とする制がん剤。
【解決手段】次の一般式(1)
(式中、R1はOH基、O−アルキル基、N−置換アミノ基、アミノ酸残基またはペプタイド残基を示す。nは0または1の整数であり、nが0のとき隣接するS原子は互いに結合して環を形成し、nが1のときR2及びR3は、同一または異なって、水素原子またはスルホ基を示す。但し、R1はOH基であるとき、nは0ではない。)で表わされる化合物、またはその薬理学的に許容される塩を有効成分とする制がん剤。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、αリポ酸誘導体を有効成分とする制がん剤に関する。
【背景技術】
【0002】
がんは何らかのきっかけで生じるDNAの突然変異による制御不能な細胞分裂によって生じ、条件を問わず転移と増殖を繰り返し、宿主を死に至らしめる。多くの制がん剤(抗がん剤)は細胞分裂をターゲットとして、その分裂を短時間に効果的に阻害することを標的にしている。
【0003】
従来の制がん剤は、大きくアルキル化剤、代謝拮抗剤、抗がん性抗生物質剤、植物アルカロイド剤、ホルモン療法剤、白金錯体剤およびアスパラギナーゼ剤などに分類される。
【0004】
このような制がん剤は、通常、細胞毒性を有し、またある種の薬剤はアポトーシス(細胞自殺)を引き起こすため、がん細胞のみならず、正常な細胞にも障害を与えてしまう。特に効果が強いとされる制がん剤は、制がん効果と副作用が隣合わせであるため、そのバランスを考えながら使うことが非常に重要になっている。
【0005】
ところで、αリポ酸(チオクト酸)はミトコンドリア中に存在する補酵素で、生体内では一部還元されてジヒドロリポ酸になる。これは酸化型のグルタチオンやビタミンCを還元型に再生させる作用が知られている。しかし、空気中では非常に不安定であり、酸化されて一部αリポ酸に戻ることが知られている。
【0006】
かかるαリポ酸またはリポ酸誘導体の還元体は、金属でキレート化することで安定化させることができる。かかるαリポ酸またはリポ酸誘導体の金属キレート化物、具体的には6,8−ジメルカプトオクタン酸金属キレート化合物またはその誘導体には、抗酸化作用、フリーラジカル抑制作用、チロシナーゼ阻害作用、エラスターゼ阻害作用、メラニン産生抑制作用、頭皮脱毛治癒作用、抗がん作用があることが知られている(特許文献1〜3参照)。
【0007】
αリポ酸の誘導体としては、αリポ酸にグリシン、メチオニン、グルタミン酸、またはバリンなどのアミノ酸残基が結合したαリポイルアミノ酸(特許文献4〜5)、αリポイルアミノエチルスルホン酸のイミダゾール塩(特許文献6)、リポイルエステル、及びジヒドロリポ酸及びジヒドロリポアミドの金属誘導体が知られている。
【0008】
またリポ酸のS原子(2価)を酸化してスルホン酸(6価)に導いたリポ酸のスルホン酸誘導体も知られている。
【0009】
αリポ酸は有機溶媒には溶けるが、水には殆んど溶けない難溶性の化合物である。水溶液のpHをアルカリ性にすることで溶解するものの安定性が悪くなるという問題がある。これを解決する方法として、αリポ酸またはαリポイルアミノ酸に亜硫酸塩やメタ重亜硫酸塩等の亜硫酸塩類を加える方法が提案されている(特許文献7及び8)。かかる方法によればpH7以下の水溶液にも溶解し、経時的にも安定である。その構造は、αリポ酸のSS結合が開環し、生じた少なくとも一方のS原子に亜硫酸が付加したもの(S−スルホ−リポ酸またはS−ジスルホ−リポ酸)であり、上記リポ酸のスルホン酸誘導体とは構造及び性状を異にするものである。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】国際公開第2002/076935号
【特許文献2】国際公開第2004/024139号
【特許文献3】特表2002−528446号公報
【特許文献4】特公昭42−1286号公報(対応米国特許第3238224号)
【特許文献5】特開2003−286168号公報
【特許文献6】特開2000−169371号公報
【特許文献7】特開2005−2096号公報
【特許文献8】特開2003−48833号公報
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明は、リポ酸誘導体を有効成分とする新規な制がん剤を提供することを目的とする。
【課題を解決するための手段】
【0012】
本発明者らは、上記目的を達成すべく鋭意研究を重ねていたところ、下記一般式(1)で示されるαリポ酸の誘導体、特にジヒドロリポ酸(ジメルカプトオクタン酸)またはその誘導体の2つのSH基のどちらか少なくとも一方の水素原子がスルホン酸又はその塩に置換された化合物、並びにαリポイルアミノ酸が、がん細胞に対して高い親和性を有しており、有意な制がん作用があることを見出した。
【0013】
本願発明はかかる知見に基づくものであって、下記の実施形態を包含するものである。
(I)次の一般式(1)
【0014】
【化1】
(式中、R1はOH基、O−アルキル基、N−置換アミノ基、アミノ酸残基またはペプタイド残基を示す。nは0または1の整数であり、nが0のとき隣接するS原子は互いに結合して環を形成し、nが1のときR2及びR3は、同一または異なって、水素原子またはスルホ基を示す。但し、R1はOH基であるとき、nは0ではない。)
で表わされる化合物、またはその薬理学的に許容される塩を有効成分とする制がん剤。
【0015】
(I-1)R1がアミノ酸残基であり、nが0で隣接するS原子が互いに結合して環を形成してなるものである(I)記載の制がん剤。
(I-1-1)一般式(1)で表わされる化合物が、N−α−リポイル−α−アミノ酸、N−α−リポイル−β−アミノ酸、N−α−リポイル−γ−アミノ酸、N−α−リポイル−δ−アミノ酸、N−α−リポイル−ω−アミノ酸、及びN−α−リポイル特殊アミノ酸からなる群から選ばれるものである、(I-1)記載の制がん剤。
(I-1-2)N−α−リポイル−α−アミノ酸が、N−α−リポイルグリシン、N−α−リポイルアラニン、N−α−リポイルスレオニン、N−α−リポイルセリン、N−α−リポイルアスパラギン酸、N−α−リポイルグルタミン酸、N−α−リポイルフェニルアラニン、N−α−リポイルバリン、N−α−リポイルメチオニン、N−α−リポイルノルロイシン、N−α−リポイルシステイン、N−α−リポイルハイドロキシプロリン、N−α−リポイルヒスチジン、N−α−リポイル−5−ハイドロキシトリプトファン、N−α−リポイルペニシラミン、及びN−α−リポイルリジンからなる群から選ばれるものである、(I-1-1)記載の制がん剤。
(I-1-3)N−α−リポイル−ω−アミノ酸及びN−α−リポイル特殊アミノ酸が、N−α−リポイル−3−アミノプロピオン酸、N−α−リポイル−4−アミノ酪酸、N−α−リポイル−6−アミノヘキサン酸、N−α−リポイル−4−トランスアミノメチル−1−シクロヘキサンカルボン酸、N−α−リポイル−2−アミノエタンスルホン酸、N−α−リポイルスルファニル酸、及びN−α−リポイルアントラニル酸からなる群から選ばれるものである、(I-1-1)記載の制がん剤。
【0016】
(I-2)R1がOH基であり、nが1で、R2及びR3のいずれか一方が水素原子で他方がスルホ基であるか、またはR2及びR3の両方がスルホ基である(I)記載の制がん剤。
(I-3)R1がO−アルキル基であり、nが1で、R2及びR3のいずれか一方が水素原子で他方がスルホ基であるか、またはR2及びR3の両方がスルホ基である(I)記載の制がん剤。
(I-4)R1がN−置換アミノ基であり、nが1で、R2及びR3のいずれか一方が水素原子で他方がスルホ基であるか、またはR2及びR3の両方がスルホ基である(I)記載の制がん剤。
【0017】
(I-5)R1がアミノ酸残基であり、nが1で、R2及びR3のいずれか一方が水素原子で他方がスルホ基であるか、またはR2及びR3の両方がスルホ基である(I)記載の制がん剤。
(I-5-1)一般式(1)で表わされる化合物が、S−スルホ−N−リポイル−α−アミノ酸、S−ジスルホ−N−リポイル−α−アミノ酸、S−スルホ−N−リポイル−ω−アミノ酸、S−ジスルホ−N−リポイル−ω−アミノ酸、S−スルホ−N−リポイル特殊アミノ酸、及びS−ジスルホ−N−リポイル特殊アミノ酸からなる群から選ばれるものである、(I-5)記載の制がん剤。
(I-5-2)S−スルホまたはS−ジスルホ−N−リポイル−α−アミノ酸が、S−スルホまたはS−ジスルホ−N−リポイルグリシン、S−スルホまたはS−ジスルホ−N−リポイルアラニン、S−スルホまたはS−ジスルホ−N−リポイルスレオニン、S−スルホまたはS−ジスルホ−N−リポイルセリン、S−スルホまたはS−ジスルホ−N−リポイルアスパラギン酸、S−スルホまたはS−ジスルホ−N−リポイルグルタミン酸、S−スルホまたはS−ジスルホ−N−リポイルフェニルアラニン、S−スルホまたはS−ジスルホ−N−リポイルバリン、S−スルホまたはS−ジスルホ−N−リポイルメチオニン、S−スルホまたはS−ジスルホ−N−リポイルノルロイシン、S−スルホまたはS−ジスルホ−N−リポイルシステイン、S−スルホまたはS−ジスルホ−N−リポイルハイドロキシプロリン、S−スルホまたはS−ジスルホ−N−リポイルヒスチジン、S−スルホまたはS−ジスルホ−N−リポイル−5−ハイドロキシトリプトファン、S−スルホまたはS−ジスルホ−N−リポイルペニシラミン、及びS−スルホまたはS−ジスルホ−N−リポイルリジンからなる群から選ばれるものである、(I-5-1)記載の制がん剤。
(I-5-3)S−スルホまたはS−ジスルホ−N−リポイル−ω−アミノ酸、及びS−スルホまたはS−ジスルホ−N−リポイル特殊アミノ酸が、S−スルホまたはS−ジスルホ−N−リポイル−3−アミノプロピオン酸、S−スルホまたはS−ジスルホ−N−リポイル−4−アミノ酪酸、S−スルホまたはS−ジスルホ−N−リポイル−6−アミノヘキサン酸、S−スルホまたはS−ジスルホ−N−リポイル−4−トランスアミノメチル−1−シクロヘキサンカルボン酸、S−スルホまたはS−ジスルホ−N−リポイル−2−アミノエタンスルホン酸、S−スルホまたはS−ジスルホ−N−リポイルスルファニル酸、及びS−スルホまたはS−ジスルホ−N−リポイルアントラニル酸からなる群から選ばれるものである、(I-5-1)記載の制がん剤。
【0018】
(I-6)R1がペプタイド残基であり、nが1で、R2及びR3のいずれか一方が水素原子で他方がスルホ基であるか、またはR2及びR3の両方がスルホ基である(I)記載の制がん剤。
(I-6-1)一般式(1)で表わされる化合物が、S−スルホ−N−リポイルジペプタイド、及びS−ジスルホ−N−リポイルジペプタイドからなる群から選ばれるものである、(I-6)記載の制がん剤。
(I-6-2)S−スルホまたはS−ジスルホ−N−リポイルジペプタイドが、S−スルホまたはS−ジスルホ−N−リポイルアスパラチイルグリシン、及びS−スルホまたはS−ジスルホ−N−リポイルスレオニルグリシンからなる群から選ばれるものである、(I-6-1)記載の制がん剤。
【0019】
(II)次の一般式(2)
【0020】
【化2】
(式中、R1はOH基、O−アルキル基、N−置換アミノ基、アミノ酸残基またはペプタイド残基を示す。)
で表わされる化合物またはその薬理学的に許容される塩と、亜硫酸塩類またはその水和物とを含有することを特徴とする制がん剤。
【0021】
(II-1)亜硫酸塩類が、亜硫酸塩、重亜硫酸塩、及びメタ重亜硫酸塩からなる群から選択されるいずれかである、(II)に記載する制がん剤。
(II-1-1)亜硫酸塩類の塩が、亜硫酸、重亜硫酸、及びメタ重亜硫酸からなる群から選択される少なくとも一種の酸のアルカリ金属塩である、(II)に記載する制がん剤。
(II-2)一般式(2)で表わされる化合物が、N−α−リポイル−α−アミノ酸、N−α−リポイル−β−アミノ酸、N−α−リポイル−γ−アミノ酸、N−α−リポイル−δ−アミノ酸、N−α−リポイル−ω−アミノ酸、及びN−α−リポイル特殊アミノ酸からなる群から選ばれるものである、(II)または(II-1)に記載する制がん剤。
(II-2-1)N−α−リポイル−α−アミノ酸が、N−α−リポイルグリシン、N−α−リポイルアラニン、N−α−リポイルスレオニン、N−α−リポイルセリン、N−α−リポイルアスパラギン酸、N−α−リポイルグルタミン酸、N−α−リポイルフェニルアラニン、N−α−リポイルバリン、N−α−リポイルメチオニン、N−α−リポイルノルロイシン、N−α−リポイルシステイン、N−α−リポイルハイドロキシプロリン、N−α−リポイルヒスチジン、N−α−リポイル−5−ハイドロキシトリプトファン、N−α−リポイルペニシラミン、及びN−α−リポイルリジンからなる群から選ばれるものである、(II-2)記載の制がん剤。
(II-2-2)N−α−リポイル−ω−アミノ酸及びN−α−リポイル特殊アミノ酸が、N−α−リポイル−3−アミノプロピオン酸、N−α−リポイル−4−アミノ酪酸、N−α−リポイル−6−アミノヘキサン酸、N−α−リポイル−4−トランスアミノメチル−1−シクロヘキサンカルボン酸、N−α−リポイル−2−アミノエタンスルホン酸、N−α−リポイルスルファニル酸、及びN−α−リポイルアントラニル酸からなる群から選ばれるものである、(II-2)記載の制がん剤。
【0022】
(II-3)一般式(1)で表わされる化合物が、N−リポイルジペプタイドからなる群から選ばれるものである、(II)または(II-1)記載の制がん剤。
(II-3-1)N−リポイルジペプタイドが、N−リポイルアスパラチイルグリシン及びN−リポイルスレオニルグリシンからなる群から選ばれるものである、(II-3)記載の制がん剤。
【発明の効果】
【0023】
本発明が対象とするαリポ酸誘導体(1)およびその薬理学的に許容される塩は、がん細胞の増殖を有意に抑制する作用(制がん作用)を有している。このため、これらの化合物は、かかる作用に基づいてがん治療剤の有効成分として有用であり、制がん治療に優れた治療効果を発揮する。
【図面の簡単な説明】
【0024】
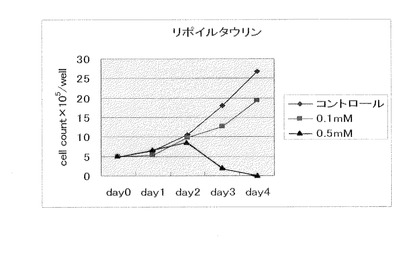
【図1】N−α−リポイルアミノエタンスルホン酸ナトリウム(製造例1)(0.1mM、0.5mM)の大腸癌細胞に対する増殖抑制効果を示す。図中、コントロールはN−α−リポイルアミノエタンスルホン酸ナトリウムを配合しない場合の大腸癌細胞の増殖能を示す(以下、同じ)。
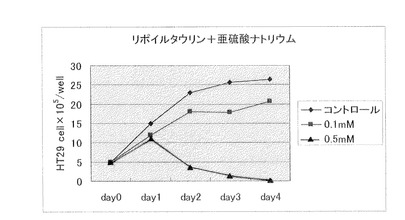
【図2】N−α−リポイルアミノエタンスルホン酸ナトリウム(製造例1)(0.1mM、0.5mM)と亜硫酸ナトリウム(0.1mM、0.5mM)を併用した場合の、大腸癌細胞に対する増殖抑制効果を示す。
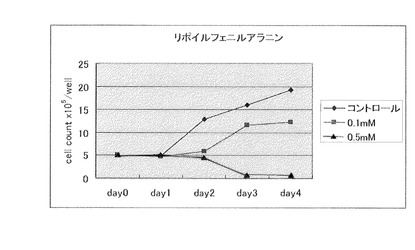
【図3】N−リポイルフェニルアラニン(製造例4)(0.1mM、0.5mM)の大腸癌細胞に対する増殖抑制効果を示す。
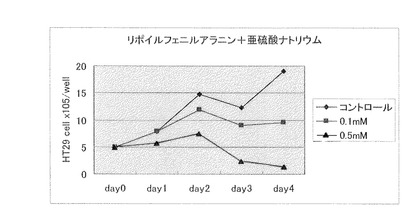
【図4】N−α−リポイルフェニルアラニン(製造例4)(0.1mM、0.5mM)と亜硫酸ナトリウム(0.1mM、0.5mM)を併用した場合の、大腸癌細胞に対する増殖抑制効果を示す。
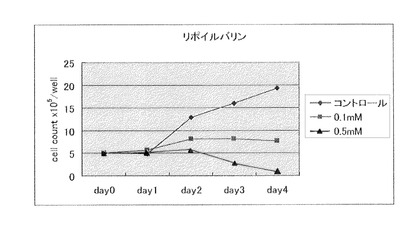
【図5】N−リポイルバリン(製造例5)(0.1mM、0.5mM)の大腸癌細胞に対する増殖抑制効果を示す。
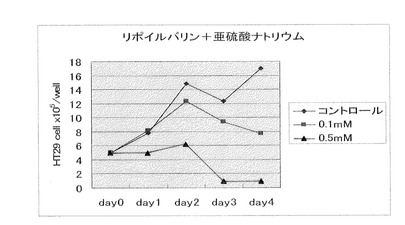
【図6】N−α−リポイルバリン(製造例5)(0.1mM、0.5mM)と亜硫酸ナトリウム(0.1mM、0.5mM)を併用した場合の、大腸癌細胞に対する増殖抑制効果を示す。
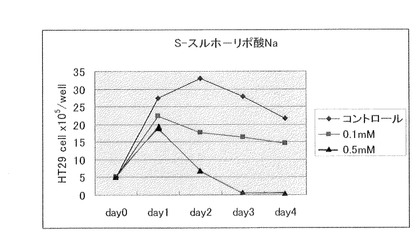
【図7】S−スルホ−リポ酸ナトリウム(製造例13)(0.1mM、0.5mM)の大腸癌細胞に対する増殖抑制効果を示す。
【発明を実施するための形態】
【0025】
本発明の制がん剤は、下記一般式(I)で示されるαリポ酸誘導体またはその薬理学的に許容される塩を有効成分とするものである。
【0026】
【化3】
(式中、R1はOH基、O−アルキル基、N−置換アミノ基、アミノ酸残基またはペプタイド残基を示す。nは0または1の整数であり、nが0のとき隣接するS原子は互いに結合して環を形成し、nが1のときR2及びR3は、同一または異なって、水素原子またはスルホ基を示す。但し、R1はOH基であるとき、nは0ではない。)。
【0027】
上記式中、R1で示すO−アルキル基としては、炭素数1〜6の直鎖状または分岐状の低級アルキル基を挙げることができる。具体的には、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、ペンチル基、イソペンチル基、ネオペンチル基、tert−ペンチル基、ヘキシル基、2−メチルペンチル基、3−メチルペンチル基、4−メチルペンチル基、2,2−ジメチルブチル基、3,3−ジメチルブチル基、2−エチルブチル基などが挙げられる。好ましくはメチル基およびエチル基である。
【0028】
上記式中、R1で示すN−置換アミノ基としては、第一級アミノ基の1または2の水素原子または第二級アミノ基の1の水素原子が、炭素数1〜5の脂肪族炭化水素基;炭素数1〜5の脂肪族アルコール残基;またはピリジン環、ピリミジン環もしくはインドール環などの窒素原子を含むヘテロ環で置換された基を挙げることができる。なお、ここで脂肪族炭化水素基、脂肪族アルコール残基、及び窒素原子を含むヘテロ環は、それぞれ置換基を有するものであってもよく、かかる置換基としては水酸基;ハロゲン原子(塩素原子、フッ素原子、ヨウ素元素等);炭素数1〜6のO−低級アルキル基;炭素数1〜6のアルキル基;並びに修飾されていてもよいピリジン環、ピリミジン環もしくはインドール環などの窒素原子を含むヘテロ環基等を、特に制限なく挙げることができる。
【0029】
炭素数1〜5の脂肪族炭化水素基としては、炭素数1〜5の直鎖状または分岐状の低級アルキル基、炭素数2〜5の直鎖状または分岐状の低級アルケニル基、及び炭素数2〜5の直鎖状または分岐状の低級アルキニル基を挙げることができる。好ましくは低級アルキル基である。かかる低級アルキル基としては、前述するように、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、ペンチル基、イソペンチル基、ネオペンチル基、tert−ペンチル基などが挙げられる。好ましくはメチル基およびエチル基である。
【0030】
また炭素数2〜5の低級アルケニル基としてはビニル基、プロペニル基、ブテニル基およびペンテニル基などを、炭素数2〜5の低級アルキニル基としてはエチニル基、プロピニル基、ブチニル基、ペンチニル基等が挙げられる。
【0031】
炭素数1〜5の脂肪族アルコール残基としては、具体的にはメタノール、エタノール、プロパノール、イソプロパノール、ブタノール、およびイソブタノール等の低級アルコールが挙げられる。
【0032】
R1がN−置換アミノ基である本発明のαリポ酸誘導体(1)として、具体的には、S−スルホまたはS−ジスルホ−N−リポイル−2−アミノエタノール(R1=−NH2の1つの水素原子がエタノール残基で置換された基、n=1、R2及びR3のいずれか1方が水素原子で他方がスルホ基であるか、または両方がスルホ基である)、S−スルホまたはS−ジスルホ−N−リポイル−2−イソプロピルアミン(R=−NH2の1つの水素原子がイソプロピル基で置換された基、n=1、R2及びR3のいずれか1方が水素原子で他方がスルホ基であるか、または両方がスルホ基である)、S−スルホまたはS−ジスルホ−N−リポイル−2−アミノピリジン(R=−NH2の1つの水素原子が2−ピリジル基で置換された基、n=1、R2及びR3のいずれか1方が水素原子で他方がスルホ基であるか、または両方がスルホ基である)、S−スルホまたはS−ジスルホ−N−リポイル−5−O−メチルセロトニン(R=5−O−メチルセロトニンの側鎖のNH2基の水素原子の1つが置換された基、n=1、R2及びR3のいずれか1方が水素原子で他方がスルホ基であるか、または両方がスルホ基である)などを挙げることができる。
【0033】
上記式中、R1で示すアミノ酸残基の「アミノ酸」としては、同一分子内にカルボキシル基とアミノ基を有する、α−アミノ酸;β−アミノ酸、γ−アミノ酸、δ−アミノ酸、及びε−アミノ酸(本明細書では、β−アミノ酸〜ε−アミノ酸を総称して「ω−アミノ酸」という);アミノメチルシクロヘキサンカルボン酸、アントラニル酸およびアントラニル酸エチル;及び同一分子内にスルホン酸基とアミノ基を有するアミノエタンスルホン酸(タウリン)やp−アミノベンゼンスルホン酸(スルファニル酸)などのアミノ酸(以上のアミノ酸を総称して「特殊アミノ酸」という)を挙げることができる。
【0034】
α−アミノ酸としては、たとえばグリシン、アラニン、バリン、ロイシン、イソロイシン、ノルロイシン、セリン、スレオニン、チロシン、システイン、メチオニン、アスパラギン酸、アスパラギン、グルタミン酸、グルタミン、アルギニン、リジン、ヒスチジン、フェニルアラニン、トリプトファン及びペニシラミンなどが挙げられる。好ましくはグリシン、アラニン、スレオニン、セリン、アスパラギン酸、グルタミン酸、フェニルアラニン、バリン、メチオニン、システイン、リジン、ヒスチジン、及びペニシラミンである。なお、これらのα−アミノ酸は、置換基として水酸基を有するものであってもよい。水酸基を有するα−アミノ酸としては、制限されないが、例えばハイドロキシプロリンやハイドロキシトリプトファン等を挙げることができる。
【0035】
β−アミノ酸としてはβ−アラニンが挙げられる。γ−アミノ酸としてはγ−アミノ−n−酪酸(GABA)やカルニチンが挙げられる。δ−アミノ酸としては5−アミノレブリン酸や5−アミノ吉草酸が、ε−アミノ酸としては6−アミノヘキサン酸が挙げられる(前述するように、これらのアミノ酸を総称して「ω−アミノ酸」という)。
【0036】
これらのアミノ酸のうち、好ましくはメチオニン、ヒスチジン、リジン、フェニルアラニン、バリン等のα−アミノ酸;γ−アミノ−n−酪酸や6−アミノヘキサン酸等のω−アミノ酸;及びアントラニル酸やアミノエタンスルホン酸等の特殊アミノ酸であり、より好ましくはフェニルアラニン、バリン、アミノエタンスルホン酸である。
【0037】
上記式中、R1で示すペプタイド残基の「ペプタイド」としては、好ましくは同種または異種の2個のアミノ酸(前記と同義)が、互いに一方のカルボキシル基と他方のアミノ酸のアミノ基とが酸アミド結合したジペプチドを挙げることができる。かかるジペプチドとして、具体的にはアスパラチイルグリシンやスレオニイルグリシンなどを挙げることができる。
【0038】
本発明のαリポ酸誘導体(1)の薬理学的に許容される塩としては、ナトリウム塩やカリウム塩などのアルカリ金属塩、およびカルシウム塩やマグネシウム塩などのアルカリ土類金属塩を挙げることができる。なお、これら以外の塩であっても薬理学的に許容できる塩であればいずれのものであっても本発明の目的のため適宜に用いることができる。また、本発明が対象とするαリポ酸誘導体(1)には、その水和物も含まれる。
【0039】
本発明の制がん剤の有効成分として使用するαリポ酸誘導体(1)のうち、nが0であり、隣接する硫黄原子が互いに結合して環を形成してなるαルポ酸誘導体(1)は、制限されないが、例えば、R1がアミノ酸残基である化合物(1a)は下記に説明する混合酸無水物法(MA法)により、αリポ酸を原料として合成することができる。
【0040】
【化4】
(式中、R1aは、アミノ酸残基を意味する。)
【0041】
具体的にはαリポ酸を有機溶媒(たとえばクロロホルム、テトラヒドロフラン、アセトニトリルなど)に溶かし、これに3級アミン(トリエチルアミン、トリブチルアミンやN−メチルモルホリン(NMM)など)の存在下−15℃〜−5℃でハロゲン化炭酸エステル(クロル炭酸エチル、クロル炭酸ブチルなど)、イソブチルオキシカルボニルクロリド、塩化ジエチルアセチルまたは塩化トリメチルアセチルなどの混合酸無水物化試薬を反応させてαリポ酸の混合酸無水物とする。反応時間は1〜2分から数10分程度である。さらにアミノ酸を塩基(水酸化ナトリウム、水酸化カリウムやトリエチルアミン、トリブチルアミンなどの3級アミン)存在下でアルコール、水またはそれらの混液などの溶媒に溶かしたものを加えて反応させた後、適当な溶媒、たとえば水またはアルコールから再結晶させると、所望のαリポイルアミノ酸(1a)を得ることができる。
【0042】
また本発明の制がん剤の有効成分として使用するαリポ酸誘導体(1)のうち、nが1であり、R2及びR3が、同一または異なって水素原子またはスルホ基である化合物(1c)及び(1d)は、制限されないが、下記に説明する方法により、αリポ酸を原料として合成することができる。
【0043】
【化5】
(式中、R2及びR3は前記と同義である。R1bは、前記のうち、OH基以外の基を意味する。)
【0044】
具体的には、本発明が対象とするαリポ酸誘導体(1c)及び(1d)は、αリポ酸、またはこれを所望によりアルキルエステル、アミノ酸もしくはペプタイド(上記式中、「R1b−OH」として示す)と反応させ、上記式(1b)で示すαリポ酸誘導体としたものに、亜硫酸塩類を付加させて製造することが出来る。
【0045】
ここで亜硫酸塩類としては、亜硫酸ナトリウムや亜硫酸カリウムなどの亜硫酸のアルカリ金属塩等の亜硫酸塩;亜硫酸水素ナトリウムや亜硫酸水素カリウムなどの亜硫酸水素化物のアルカリ金属塩等の重亜硫酸塩;およびメタ重亜硫酸塩を挙げることができる。好ましくは亜硫酸ナトリウムや亜硫酸カリウムなどの亜硫酸塩である。
【0046】
αリポ酸をアルキルエステルまたはアミド体とする反応は、混合酸無水物法(MA法)により実施することが出来る。すなわち、αリポ酸を有機溶媒(例えば、クロロホルム、THF,アセトニトリルなど)に溶かし、これに3級アミン(トリエチルアミン、トチブチルアミン、N−メチルモルホリンなど)の存在下、−15〜−5℃で、ハロゲン化炭酸エステル(例えば、クロル炭酸エチル、クロル炭酸ブチルなど)を加えて5〜10分後に、さらに、所望のアルコール、アミン、アミノ酸、ペプタイドを必要に応じた塩基(水酸化ナトリウム、トリエチルアミンやトリブチルアミンなどの3級アミンなど)の存在下、水またはアルコールまたはそれらの混液などの溶媒に溶かしたものを一挙に加えて反応させる。次に反応温度を徐々に室温に戻して反応を終結させ、反応液を処理して粗結晶を得た後、適当な溶媒、例えば、水またはアルコールから再結晶させて、αリポ酸のアルキルエステル、または対応したアミド体を得ることが出来る。
【0047】
本発明が対象とするαリポ酸誘導体(1c)及び(1d)は、αリポ酸または上記方法で調製されるαリポ酸誘導体(1b)を水に溶かすか、またはサスペンドしておき、これに亜硫酸塩類を徐々に加えて攪拌することによって調製することができる。亜硫酸塩類の配合割合は、特に制限されないが、S−スルホ体を調製する場合は、αリポ酸またはαリポ酸誘導体1モルに対して亜硫酸塩類が等モル量になるように、S−ジスルホ体を調製する場合は、αリポ酸またはαリポ酸誘導体1モルに対して亜硫酸塩類が2倍モル量になるように調整することが好ましい。
【0048】
斯くして得られる反応物(反応液)はこのまま本発明の制がん剤の有効成分として使用することも出来るが、通常はこの水溶液を減圧下、40℃以下で濃縮した後、エタノールを加えて、上記式(1d)で示すS−スルホまたはS−ジスルホ−N−リポ酸またはその塩、または上記式(1c)で示すS−スルホまたはS−ジスルホ−N−リポ酸誘導体にそれぞれ対応した固形物を得ることが出来る。
【0049】
本発明のαリポ誘導体(1)は構造上、ジヒドロリポ酸誘導体でその薬理学的に許容できる塩は、還元作用並びにラジカル抑制作用も強く、安定性もよく、組成からしても安全性の高い化合物で問題ない優れた化合物である。
【0050】
上記αリポ酸誘導体(1)またはその薬理学上許容される塩は、制がん剤、並びにがん転移抑制剤の有効成分として有効である。
【0051】
従って、上記αリポ酸誘導体(1)またはその薬理学上許容される塩を有効成分とする医薬品は、制がん剤(がん転移抑制剤を包含する)として、がん細胞の増殖を抑制し、がんの治療、及びがんの進展やがんの転移を抑制するために有効に使用することができる。なお、αリポ酸誘導体(1)またはその薬理学上許容される塩は、1種単独で使用してもよいが、目的と必要に応じて、2種以上を適宜組み合わせて用いることもできる。
【0052】
本発明の制がん剤は、経口的にあるいは非経口的に用いることができ、投与形態に応じた製剤形態に調製することができる。製剤の形態としては、例えば、錠剤、顆粒剤、散剤、カプセル剤等の固形製剤、または注射剤等の液剤などいずれの形にも公知の方法により調製することができる。これらの製剤には通常用いられる賦形剤、結合剤、増粘剤、分散剤、再吸収促進剤、緩衝材、界面活性剤、溶解補助剤、保存剤、乳化剤、等張化剤、安定化剤やpH調整化剤等の各種添加剤を適宜使用しても良い。
【0053】
本発明の制がん剤の投与量は、使用する本化合物の種類、患者の体重や年齢、対象とする疾患の種類やその状態および投与方法などによっても異なるが、例えば注射剤の場合、成人1日1回、約10mg〜100mg、内服剤の場合は、成人1日数回、10mg〜1000mg程度投与するのがよい。
【実施例】
【0054】
次に製造例、実験例および実施例を挙げて本発明を説明するが、本発明はこれらに何ら限定されるものではない。
【0055】
製造例1 N−α−リポイルアミノエタンスルホン酸ナトリウム(N−α−リポイルタウリンナトリウム)
αリポ酸6.2gをクロロホルム60mlに溶かし、トリエチルアミン3.2gを加えて−5℃に冷却した。これにクロル炭酸エチル3.3gを徐々に滴下し、滴下終了15分後に、これにアミノエタンスルホン酸4.5gおよび水酸化ナトリウム1.5gをメタノール60mlに溶かしたものを一挙に加えて、その温度(−5℃)で15分間、さらに室温に戻して1時間攪拌した。次に、これに水酸化ナトリウム1.5gをメタノール50mlに溶かした溶液を加えて、減圧下で溶媒を1/3程度になるまで濃縮した。次いで、これにエタノール60mlを加えて析出した結晶を濾取した。これを水−メタノールを用いて再結晶させて、掲題化合物の白色結晶5.8gを得た。融点235〜237℃。
元素分析: C10H17NO4S3Na・H2Oとして理論値 C:34.08、 H:5.43、N:3.97、実測値 C:34.23、H:5.54、N:3.80。
【0056】
製造例2 N−α−リポイルアミノエタンスルホン酸カリウム(N−α−リポイルタウリンカリウム)
製造例1のN−α−リポイルアミノエタンスルホン酸ナトリウムの製造方法において、アミノエタンスルホン酸4.5gと混合する水酸化ナトリウム1.5gに代えて水酸化カリウム4.0gを用いる以外は、製造例1と同じ方法を実施して結晶を濾取した。これを水−メタノールを用いて再結晶させて、掲題化合物の白色結晶6.5gを得た。融点240〜242℃。
【0057】
製造例3 N−α−リポイルアミノエタンスルホン酸のカルシウムまたはマグネシウム塩
製造例1で得られたN−α−リポイルアミノエタンスルホン酸ナトリウムを水に溶かし、スルホン酸樹脂で脱塩して遊離酸とした。これを炭酸カルシウムまたは塩基性炭酸マグネシウムで、それぞれ中和させて、N−α−リポイルアミノエタンスルホン酸の水溶性のカルシウム塩またはマグネシウム塩を得た。それぞれ融点300℃以上。
【0058】
製造例4 N−α−リポイルフェニルアラニン
原料としてαリポ酸6.2gに代えてαリポ酸4.2g、およびアミノエタンスルホン酸4.5gに代えてL−フェニルアラニン3.5gを用いる以外は、製造例1と同様に反応させた。得られた反応液を減圧下で40℃以下で溶媒を留去濃縮した後、塩酸で酸性として酢酸エチルで抽出し、水洗した後、酢酸エチルを留去した。残渣結晶をエタノール−水を用いて再結晶させて、掲題化合物の淡黄色結晶4.6gを得た。融点154℃〜156℃。
【0059】
製造例5 N−α−リポイルバリン
原料としてαリポ酸4.2g、およびL−フェニルアラニン3.5g に代えてL−バリン2.4gを用いる以外は、製造例4と同様に反応処理して、掲題化合物の淡黄色板状4.1gを得た。
【0060】
製造例6 N−α−リポイルグリシンナトリウム
原料としてαリポ酸6.2gに代えてαリポ酸4.2g、およびアミノエタンスルホン酸4.5gに代えてグリシン1.9gを用いる以外は、製造例1と同様に反応処理して、掲題化合物4.5gを得た。融点218℃〜220℃。
【0061】
製造例7 N−α−リポイルアスパラギン酸ナトリウム
原料としてαリポ酸6.2gに代えてαリポ酸4.2g、およびアミノエタンスルホン酸4.5gに代えてL−アスパラギン酸2.9gを用いる以外は、製造例1と同様に反応処理して、掲題化合物5.1gを得た。融点300℃以上。
【0062】
製造例8 N−α−リポイルメチオニン
原料としてαリポ酸4.2g、およびL−フェニルアラニン3.5g に代えてL−メチオニン3.5gを用いる以外は、製造例4と同様に処理して、掲題化合物の淡黄色結晶4.0gを得た。融点108℃〜109℃。
【0063】
製造例9 N−α−リポイルシステインナトリウム
原料としてαリポ酸4.2g、およびアミノエタンスルホン酸4.5g に代えてL−システイン2.6gを用いる以外は、製造例1と同様に処理して、掲題化合物の白色結晶4.5gを得た。融点150℃付近から徐々に分解。
【0064】
製造例10 N−α−リポイル−6−アミノヘキサン酸ナトリウム
原料としてαリポ酸6.2gに代えてαリポ酸4.2g、およびアミノエタンスルホン酸4.5g に代えて6−アミノヘキサン酸3.0gを用いる以外は、製造例1と同様の方法で反応させて結晶を濾取した。これをエタノールを用いて再結晶させて、標題化合物の黄色がかった白色結晶5.5gを得た。融点200〜202℃(分解)。
【0065】
製造例11 N−α−リポイルアントラニル酸ナトリウム
原料としてαリポ酸6.2gに代えてαリポ酸4.2g、およびアミノエタンスルホン酸4.5g に代えてアントラニル酸2.9gを用いる以外は、製造例1と同様に処理して、掲題化合物の白色結晶3.5gを得た。融点300℃以上。
【0066】
製造例12 N−α−リポイル−4−アミノ酪酸
原料としてαリポ酸4.2g、およびL−フェニルアラニン3.5g に代えて4−アミノ酪酸2.3gを用いる以外は、製造例4と同様に処理して、掲題化合物の淡黄色結晶4.3gを得た。融点235℃付近から分解。
【0067】
製造例13 S−スルホ−リポ酸ナトリウム
αリポ酸2.1g(0.01 モル)を水50mlに懸濁したものに、亜硫酸ナトリウム1.3g(0.01モル)を加えて撹拌しながら溶解した。これを減圧下、40℃以下で濃縮した後、エタノールを加え、白色固形物2.3gを濾取した。これを水−エタノールを用いて再結晶させて、掲題化合物の白色結晶を得た。
【0068】
製造例14 S−スルホ−N−リポイルフェニルアラニンナトリウム
製造例13において、αリポ酸2.1gの代わりに製造例4で調製したN−リポイルフェニルアラニン3.5gを用いて、同様に反応処理した。具体的には、N−リポイルフェニルアラニン3.5g(0.01 モル)を水50mlに懸濁したものに、亜硫酸ナトリウム1.3g(0.01モル)を加えて撹拌しながら溶解した。これを減圧下、40℃以下で濃縮した後、エタノールを加え、白色固形物2.3gを濾取した。これを水−エタノールを用いて再結晶させて、掲題化合物の白色結晶を得た。
【0069】
製造例15 S−スルホ−N−リポイルバリンナトリウム溶液
製造例5で調製したN−リポイルバリン305mgを水100mlに懸濁しておき、これに無水亜硫酸ナトリウム150mgを加えて溶解して濾過し、掲題化合物の水溶液を調製した。
【0070】
製造例16 S−スルホ−N−リポイルタウリンナトリウム溶液
製造例1で調製したN−リポイルタウリンナトリウム335mgを、水100mlに溶解し、これに無水亜硫酸ナトリウム150mgを加えて溶解して濾過して、掲題化合物の水溶液を取得した。
【0071】
実験例1 癌細胞増殖能に対する抑制効果
上記で製造したリポ酸誘導体のうち、N−α−リポイルアミノエタンスルホン酸ナトリウム(製造例1)、N−α−リポイルフェニルアラニン(製造例4)、N−α−リポイルバリン(製造例5)、及びS−スルホ−リポ酸ナトリウム(製造例13)を被験化合物とし、これらの化合物の癌細胞増殖能に対する抑制効果を評価した。なお、N−α−リポイルアミノエタンスルホン酸ナトリウム(製造例1)、N−α−リポイルフェニルアラニン(製造例4)、及びN−α−リポイルバリン(製造例5)については、それぞれ各化合物を単独で使用した場合と、亜硫酸ナトリウムを併用した場合との両方で実験を行った。
【0072】
(1)被験化合物
1.N−α−リポイルアミノエタンスルホン酸ナトリウム(製造例1)
2.N−α−リポイルアミノエタンスルホン酸ナトリウム+亜硫酸ナトリウム
3.N−リポイルフェニルアラニン(製造例4)
4.N−リポイルフェニルアラニン+亜硫酸ナトリウム
5.N−リポイルバリン(製造例5)
6.N−リポイルバリン+亜硫酸ナトリウム
7.S−スルホ−リポ酸ナトリウム(製造例13)。
【0073】
(2)試験方法
コントロール培地として、RPMI 1640に10%ウシ胎児血清と抗菌剤(100 IU/ml penicillin,0.1mg/ml streptomycin,2.5×10-4mg/ml amphotericin B) を加え調製したものを使用した。また試験培地として、上記コントロール培地に、上記各被験化合物をαリポ酸誘導体の最終濃度が0.1 mMまたは0.5mMに、被験化合物2、4及び6については、培地中の亜硫酸ナトリウムの最終濃度が0.1 mMまたは0.5mMになるように調整した。
【0074】
かかる培地を用いて、大腸癌細胞(HT29)(5.0×105/wellまたは1.0×106well)を5% CO2,37℃のもとで培養した。培養開始後24時間ごとに0.25%トリプシンを用いて細胞を剥離し,トリパンブルー排除法にて,Haemocytometerで生細胞数を測定した。
【0075】
(3)試験結果
大腸癌細胞増殖能に対する被験化合物1〜7の効果(癌細胞増殖抑制効果)を図1〜7にそれぞれ示す。またその総合評価を表1に示す。
【0076】
【表1】
【0077】
図1〜7及び表1の結果から、N-α-リポイルフェニルアラニン、N-α-リポイルバリン、及びN-α-リポイルアミノエタンスルホン酸等のαリポイルアミノ酸、並びにS−スルホ−リポ酸ナトリウムは0.1mM以上の濃度、特に0.5mM以上の濃度で、大腸癌細胞HT29の増殖を顕著に抑制することが確認された。またαリポニルアミノ酸は、亜硫酸塩と併用することで、がん細胞発育の抑制効果が増強される傾向にあることが確認された。
【0078】
これらの実験結果から、本願発明が対象とするαリポ酸誘導体は大腸がんなどのがん細胞の増殖を抑制する作用があり、かかる作用に基づいて制がん剤(抗がん剤)としての治療効果があると考えられる。
【0079】
〔製剤実施例1〕 内服錠
製造例1〜14のいずれかの化合物 30mg
乳糖 80mg
馬鈴薯澱粉 17mg
ポリエチレングリコール6000 3mg
以上の成分を1錠分の材料として常法により成型する。
【0080】
〔製剤実施例2〕 注射剤
製造例1〜14のいずれかの化合物 1.0g
マニトール 4.0g
注射用蒸留水 全量 100mL
以上を常法により混合溶解させ注射剤とする。
【技術分野】
【0001】
本発明は、αリポ酸誘導体を有効成分とする制がん剤に関する。
【背景技術】
【0002】
がんは何らかのきっかけで生じるDNAの突然変異による制御不能な細胞分裂によって生じ、条件を問わず転移と増殖を繰り返し、宿主を死に至らしめる。多くの制がん剤(抗がん剤)は細胞分裂をターゲットとして、その分裂を短時間に効果的に阻害することを標的にしている。
【0003】
従来の制がん剤は、大きくアルキル化剤、代謝拮抗剤、抗がん性抗生物質剤、植物アルカロイド剤、ホルモン療法剤、白金錯体剤およびアスパラギナーゼ剤などに分類される。
【0004】
このような制がん剤は、通常、細胞毒性を有し、またある種の薬剤はアポトーシス(細胞自殺)を引き起こすため、がん細胞のみならず、正常な細胞にも障害を与えてしまう。特に効果が強いとされる制がん剤は、制がん効果と副作用が隣合わせであるため、そのバランスを考えながら使うことが非常に重要になっている。
【0005】
ところで、αリポ酸(チオクト酸)はミトコンドリア中に存在する補酵素で、生体内では一部還元されてジヒドロリポ酸になる。これは酸化型のグルタチオンやビタミンCを還元型に再生させる作用が知られている。しかし、空気中では非常に不安定であり、酸化されて一部αリポ酸に戻ることが知られている。
【0006】
かかるαリポ酸またはリポ酸誘導体の還元体は、金属でキレート化することで安定化させることができる。かかるαリポ酸またはリポ酸誘導体の金属キレート化物、具体的には6,8−ジメルカプトオクタン酸金属キレート化合物またはその誘導体には、抗酸化作用、フリーラジカル抑制作用、チロシナーゼ阻害作用、エラスターゼ阻害作用、メラニン産生抑制作用、頭皮脱毛治癒作用、抗がん作用があることが知られている(特許文献1〜3参照)。
【0007】
αリポ酸の誘導体としては、αリポ酸にグリシン、メチオニン、グルタミン酸、またはバリンなどのアミノ酸残基が結合したαリポイルアミノ酸(特許文献4〜5)、αリポイルアミノエチルスルホン酸のイミダゾール塩(特許文献6)、リポイルエステル、及びジヒドロリポ酸及びジヒドロリポアミドの金属誘導体が知られている。
【0008】
またリポ酸のS原子(2価)を酸化してスルホン酸(6価)に導いたリポ酸のスルホン酸誘導体も知られている。
【0009】
αリポ酸は有機溶媒には溶けるが、水には殆んど溶けない難溶性の化合物である。水溶液のpHをアルカリ性にすることで溶解するものの安定性が悪くなるという問題がある。これを解決する方法として、αリポ酸またはαリポイルアミノ酸に亜硫酸塩やメタ重亜硫酸塩等の亜硫酸塩類を加える方法が提案されている(特許文献7及び8)。かかる方法によればpH7以下の水溶液にも溶解し、経時的にも安定である。その構造は、αリポ酸のSS結合が開環し、生じた少なくとも一方のS原子に亜硫酸が付加したもの(S−スルホ−リポ酸またはS−ジスルホ−リポ酸)であり、上記リポ酸のスルホン酸誘導体とは構造及び性状を異にするものである。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】国際公開第2002/076935号
【特許文献2】国際公開第2004/024139号
【特許文献3】特表2002−528446号公報
【特許文献4】特公昭42−1286号公報(対応米国特許第3238224号)
【特許文献5】特開2003−286168号公報
【特許文献6】特開2000−169371号公報
【特許文献7】特開2005−2096号公報
【特許文献8】特開2003−48833号公報
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明は、リポ酸誘導体を有効成分とする新規な制がん剤を提供することを目的とする。
【課題を解決するための手段】
【0012】
本発明者らは、上記目的を達成すべく鋭意研究を重ねていたところ、下記一般式(1)で示されるαリポ酸の誘導体、特にジヒドロリポ酸(ジメルカプトオクタン酸)またはその誘導体の2つのSH基のどちらか少なくとも一方の水素原子がスルホン酸又はその塩に置換された化合物、並びにαリポイルアミノ酸が、がん細胞に対して高い親和性を有しており、有意な制がん作用があることを見出した。
【0013】
本願発明はかかる知見に基づくものであって、下記の実施形態を包含するものである。
(I)次の一般式(1)
【0014】
【化1】
(式中、R1はOH基、O−アルキル基、N−置換アミノ基、アミノ酸残基またはペプタイド残基を示す。nは0または1の整数であり、nが0のとき隣接するS原子は互いに結合して環を形成し、nが1のときR2及びR3は、同一または異なって、水素原子またはスルホ基を示す。但し、R1はOH基であるとき、nは0ではない。)
で表わされる化合物、またはその薬理学的に許容される塩を有効成分とする制がん剤。
【0015】
(I-1)R1がアミノ酸残基であり、nが0で隣接するS原子が互いに結合して環を形成してなるものである(I)記載の制がん剤。
(I-1-1)一般式(1)で表わされる化合物が、N−α−リポイル−α−アミノ酸、N−α−リポイル−β−アミノ酸、N−α−リポイル−γ−アミノ酸、N−α−リポイル−δ−アミノ酸、N−α−リポイル−ω−アミノ酸、及びN−α−リポイル特殊アミノ酸からなる群から選ばれるものである、(I-1)記載の制がん剤。
(I-1-2)N−α−リポイル−α−アミノ酸が、N−α−リポイルグリシン、N−α−リポイルアラニン、N−α−リポイルスレオニン、N−α−リポイルセリン、N−α−リポイルアスパラギン酸、N−α−リポイルグルタミン酸、N−α−リポイルフェニルアラニン、N−α−リポイルバリン、N−α−リポイルメチオニン、N−α−リポイルノルロイシン、N−α−リポイルシステイン、N−α−リポイルハイドロキシプロリン、N−α−リポイルヒスチジン、N−α−リポイル−5−ハイドロキシトリプトファン、N−α−リポイルペニシラミン、及びN−α−リポイルリジンからなる群から選ばれるものである、(I-1-1)記載の制がん剤。
(I-1-3)N−α−リポイル−ω−アミノ酸及びN−α−リポイル特殊アミノ酸が、N−α−リポイル−3−アミノプロピオン酸、N−α−リポイル−4−アミノ酪酸、N−α−リポイル−6−アミノヘキサン酸、N−α−リポイル−4−トランスアミノメチル−1−シクロヘキサンカルボン酸、N−α−リポイル−2−アミノエタンスルホン酸、N−α−リポイルスルファニル酸、及びN−α−リポイルアントラニル酸からなる群から選ばれるものである、(I-1-1)記載の制がん剤。
【0016】
(I-2)R1がOH基であり、nが1で、R2及びR3のいずれか一方が水素原子で他方がスルホ基であるか、またはR2及びR3の両方がスルホ基である(I)記載の制がん剤。
(I-3)R1がO−アルキル基であり、nが1で、R2及びR3のいずれか一方が水素原子で他方がスルホ基であるか、またはR2及びR3の両方がスルホ基である(I)記載の制がん剤。
(I-4)R1がN−置換アミノ基であり、nが1で、R2及びR3のいずれか一方が水素原子で他方がスルホ基であるか、またはR2及びR3の両方がスルホ基である(I)記載の制がん剤。
【0017】
(I-5)R1がアミノ酸残基であり、nが1で、R2及びR3のいずれか一方が水素原子で他方がスルホ基であるか、またはR2及びR3の両方がスルホ基である(I)記載の制がん剤。
(I-5-1)一般式(1)で表わされる化合物が、S−スルホ−N−リポイル−α−アミノ酸、S−ジスルホ−N−リポイル−α−アミノ酸、S−スルホ−N−リポイル−ω−アミノ酸、S−ジスルホ−N−リポイル−ω−アミノ酸、S−スルホ−N−リポイル特殊アミノ酸、及びS−ジスルホ−N−リポイル特殊アミノ酸からなる群から選ばれるものである、(I-5)記載の制がん剤。
(I-5-2)S−スルホまたはS−ジスルホ−N−リポイル−α−アミノ酸が、S−スルホまたはS−ジスルホ−N−リポイルグリシン、S−スルホまたはS−ジスルホ−N−リポイルアラニン、S−スルホまたはS−ジスルホ−N−リポイルスレオニン、S−スルホまたはS−ジスルホ−N−リポイルセリン、S−スルホまたはS−ジスルホ−N−リポイルアスパラギン酸、S−スルホまたはS−ジスルホ−N−リポイルグルタミン酸、S−スルホまたはS−ジスルホ−N−リポイルフェニルアラニン、S−スルホまたはS−ジスルホ−N−リポイルバリン、S−スルホまたはS−ジスルホ−N−リポイルメチオニン、S−スルホまたはS−ジスルホ−N−リポイルノルロイシン、S−スルホまたはS−ジスルホ−N−リポイルシステイン、S−スルホまたはS−ジスルホ−N−リポイルハイドロキシプロリン、S−スルホまたはS−ジスルホ−N−リポイルヒスチジン、S−スルホまたはS−ジスルホ−N−リポイル−5−ハイドロキシトリプトファン、S−スルホまたはS−ジスルホ−N−リポイルペニシラミン、及びS−スルホまたはS−ジスルホ−N−リポイルリジンからなる群から選ばれるものである、(I-5-1)記載の制がん剤。
(I-5-3)S−スルホまたはS−ジスルホ−N−リポイル−ω−アミノ酸、及びS−スルホまたはS−ジスルホ−N−リポイル特殊アミノ酸が、S−スルホまたはS−ジスルホ−N−リポイル−3−アミノプロピオン酸、S−スルホまたはS−ジスルホ−N−リポイル−4−アミノ酪酸、S−スルホまたはS−ジスルホ−N−リポイル−6−アミノヘキサン酸、S−スルホまたはS−ジスルホ−N−リポイル−4−トランスアミノメチル−1−シクロヘキサンカルボン酸、S−スルホまたはS−ジスルホ−N−リポイル−2−アミノエタンスルホン酸、S−スルホまたはS−ジスルホ−N−リポイルスルファニル酸、及びS−スルホまたはS−ジスルホ−N−リポイルアントラニル酸からなる群から選ばれるものである、(I-5-1)記載の制がん剤。
【0018】
(I-6)R1がペプタイド残基であり、nが1で、R2及びR3のいずれか一方が水素原子で他方がスルホ基であるか、またはR2及びR3の両方がスルホ基である(I)記載の制がん剤。
(I-6-1)一般式(1)で表わされる化合物が、S−スルホ−N−リポイルジペプタイド、及びS−ジスルホ−N−リポイルジペプタイドからなる群から選ばれるものである、(I-6)記載の制がん剤。
(I-6-2)S−スルホまたはS−ジスルホ−N−リポイルジペプタイドが、S−スルホまたはS−ジスルホ−N−リポイルアスパラチイルグリシン、及びS−スルホまたはS−ジスルホ−N−リポイルスレオニルグリシンからなる群から選ばれるものである、(I-6-1)記載の制がん剤。
【0019】
(II)次の一般式(2)
【0020】
【化2】
(式中、R1はOH基、O−アルキル基、N−置換アミノ基、アミノ酸残基またはペプタイド残基を示す。)
で表わされる化合物またはその薬理学的に許容される塩と、亜硫酸塩類またはその水和物とを含有することを特徴とする制がん剤。
【0021】
(II-1)亜硫酸塩類が、亜硫酸塩、重亜硫酸塩、及びメタ重亜硫酸塩からなる群から選択されるいずれかである、(II)に記載する制がん剤。
(II-1-1)亜硫酸塩類の塩が、亜硫酸、重亜硫酸、及びメタ重亜硫酸からなる群から選択される少なくとも一種の酸のアルカリ金属塩である、(II)に記載する制がん剤。
(II-2)一般式(2)で表わされる化合物が、N−α−リポイル−α−アミノ酸、N−α−リポイル−β−アミノ酸、N−α−リポイル−γ−アミノ酸、N−α−リポイル−δ−アミノ酸、N−α−リポイル−ω−アミノ酸、及びN−α−リポイル特殊アミノ酸からなる群から選ばれるものである、(II)または(II-1)に記載する制がん剤。
(II-2-1)N−α−リポイル−α−アミノ酸が、N−α−リポイルグリシン、N−α−リポイルアラニン、N−α−リポイルスレオニン、N−α−リポイルセリン、N−α−リポイルアスパラギン酸、N−α−リポイルグルタミン酸、N−α−リポイルフェニルアラニン、N−α−リポイルバリン、N−α−リポイルメチオニン、N−α−リポイルノルロイシン、N−α−リポイルシステイン、N−α−リポイルハイドロキシプロリン、N−α−リポイルヒスチジン、N−α−リポイル−5−ハイドロキシトリプトファン、N−α−リポイルペニシラミン、及びN−α−リポイルリジンからなる群から選ばれるものである、(II-2)記載の制がん剤。
(II-2-2)N−α−リポイル−ω−アミノ酸及びN−α−リポイル特殊アミノ酸が、N−α−リポイル−3−アミノプロピオン酸、N−α−リポイル−4−アミノ酪酸、N−α−リポイル−6−アミノヘキサン酸、N−α−リポイル−4−トランスアミノメチル−1−シクロヘキサンカルボン酸、N−α−リポイル−2−アミノエタンスルホン酸、N−α−リポイルスルファニル酸、及びN−α−リポイルアントラニル酸からなる群から選ばれるものである、(II-2)記載の制がん剤。
【0022】
(II-3)一般式(1)で表わされる化合物が、N−リポイルジペプタイドからなる群から選ばれるものである、(II)または(II-1)記載の制がん剤。
(II-3-1)N−リポイルジペプタイドが、N−リポイルアスパラチイルグリシン及びN−リポイルスレオニルグリシンからなる群から選ばれるものである、(II-3)記載の制がん剤。
【発明の効果】
【0023】
本発明が対象とするαリポ酸誘導体(1)およびその薬理学的に許容される塩は、がん細胞の増殖を有意に抑制する作用(制がん作用)を有している。このため、これらの化合物は、かかる作用に基づいてがん治療剤の有効成分として有用であり、制がん治療に優れた治療効果を発揮する。
【図面の簡単な説明】
【0024】
【図1】N−α−リポイルアミノエタンスルホン酸ナトリウム(製造例1)(0.1mM、0.5mM)の大腸癌細胞に対する増殖抑制効果を示す。図中、コントロールはN−α−リポイルアミノエタンスルホン酸ナトリウムを配合しない場合の大腸癌細胞の増殖能を示す(以下、同じ)。
【図2】N−α−リポイルアミノエタンスルホン酸ナトリウム(製造例1)(0.1mM、0.5mM)と亜硫酸ナトリウム(0.1mM、0.5mM)を併用した場合の、大腸癌細胞に対する増殖抑制効果を示す。
【図3】N−リポイルフェニルアラニン(製造例4)(0.1mM、0.5mM)の大腸癌細胞に対する増殖抑制効果を示す。
【図4】N−α−リポイルフェニルアラニン(製造例4)(0.1mM、0.5mM)と亜硫酸ナトリウム(0.1mM、0.5mM)を併用した場合の、大腸癌細胞に対する増殖抑制効果を示す。
【図5】N−リポイルバリン(製造例5)(0.1mM、0.5mM)の大腸癌細胞に対する増殖抑制効果を示す。
【図6】N−α−リポイルバリン(製造例5)(0.1mM、0.5mM)と亜硫酸ナトリウム(0.1mM、0.5mM)を併用した場合の、大腸癌細胞に対する増殖抑制効果を示す。
【図7】S−スルホ−リポ酸ナトリウム(製造例13)(0.1mM、0.5mM)の大腸癌細胞に対する増殖抑制効果を示す。
【発明を実施するための形態】
【0025】
本発明の制がん剤は、下記一般式(I)で示されるαリポ酸誘導体またはその薬理学的に許容される塩を有効成分とするものである。
【0026】
【化3】
(式中、R1はOH基、O−アルキル基、N−置換アミノ基、アミノ酸残基またはペプタイド残基を示す。nは0または1の整数であり、nが0のとき隣接するS原子は互いに結合して環を形成し、nが1のときR2及びR3は、同一または異なって、水素原子またはスルホ基を示す。但し、R1はOH基であるとき、nは0ではない。)。
【0027】
上記式中、R1で示すO−アルキル基としては、炭素数1〜6の直鎖状または分岐状の低級アルキル基を挙げることができる。具体的には、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、ペンチル基、イソペンチル基、ネオペンチル基、tert−ペンチル基、ヘキシル基、2−メチルペンチル基、3−メチルペンチル基、4−メチルペンチル基、2,2−ジメチルブチル基、3,3−ジメチルブチル基、2−エチルブチル基などが挙げられる。好ましくはメチル基およびエチル基である。
【0028】
上記式中、R1で示すN−置換アミノ基としては、第一級アミノ基の1または2の水素原子または第二級アミノ基の1の水素原子が、炭素数1〜5の脂肪族炭化水素基;炭素数1〜5の脂肪族アルコール残基;またはピリジン環、ピリミジン環もしくはインドール環などの窒素原子を含むヘテロ環で置換された基を挙げることができる。なお、ここで脂肪族炭化水素基、脂肪族アルコール残基、及び窒素原子を含むヘテロ環は、それぞれ置換基を有するものであってもよく、かかる置換基としては水酸基;ハロゲン原子(塩素原子、フッ素原子、ヨウ素元素等);炭素数1〜6のO−低級アルキル基;炭素数1〜6のアルキル基;並びに修飾されていてもよいピリジン環、ピリミジン環もしくはインドール環などの窒素原子を含むヘテロ環基等を、特に制限なく挙げることができる。
【0029】
炭素数1〜5の脂肪族炭化水素基としては、炭素数1〜5の直鎖状または分岐状の低級アルキル基、炭素数2〜5の直鎖状または分岐状の低級アルケニル基、及び炭素数2〜5の直鎖状または分岐状の低級アルキニル基を挙げることができる。好ましくは低級アルキル基である。かかる低級アルキル基としては、前述するように、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、ペンチル基、イソペンチル基、ネオペンチル基、tert−ペンチル基などが挙げられる。好ましくはメチル基およびエチル基である。
【0030】
また炭素数2〜5の低級アルケニル基としてはビニル基、プロペニル基、ブテニル基およびペンテニル基などを、炭素数2〜5の低級アルキニル基としてはエチニル基、プロピニル基、ブチニル基、ペンチニル基等が挙げられる。
【0031】
炭素数1〜5の脂肪族アルコール残基としては、具体的にはメタノール、エタノール、プロパノール、イソプロパノール、ブタノール、およびイソブタノール等の低級アルコールが挙げられる。
【0032】
R1がN−置換アミノ基である本発明のαリポ酸誘導体(1)として、具体的には、S−スルホまたはS−ジスルホ−N−リポイル−2−アミノエタノール(R1=−NH2の1つの水素原子がエタノール残基で置換された基、n=1、R2及びR3のいずれか1方が水素原子で他方がスルホ基であるか、または両方がスルホ基である)、S−スルホまたはS−ジスルホ−N−リポイル−2−イソプロピルアミン(R=−NH2の1つの水素原子がイソプロピル基で置換された基、n=1、R2及びR3のいずれか1方が水素原子で他方がスルホ基であるか、または両方がスルホ基である)、S−スルホまたはS−ジスルホ−N−リポイル−2−アミノピリジン(R=−NH2の1つの水素原子が2−ピリジル基で置換された基、n=1、R2及びR3のいずれか1方が水素原子で他方がスルホ基であるか、または両方がスルホ基である)、S−スルホまたはS−ジスルホ−N−リポイル−5−O−メチルセロトニン(R=5−O−メチルセロトニンの側鎖のNH2基の水素原子の1つが置換された基、n=1、R2及びR3のいずれか1方が水素原子で他方がスルホ基であるか、または両方がスルホ基である)などを挙げることができる。
【0033】
上記式中、R1で示すアミノ酸残基の「アミノ酸」としては、同一分子内にカルボキシル基とアミノ基を有する、α−アミノ酸;β−アミノ酸、γ−アミノ酸、δ−アミノ酸、及びε−アミノ酸(本明細書では、β−アミノ酸〜ε−アミノ酸を総称して「ω−アミノ酸」という);アミノメチルシクロヘキサンカルボン酸、アントラニル酸およびアントラニル酸エチル;及び同一分子内にスルホン酸基とアミノ基を有するアミノエタンスルホン酸(タウリン)やp−アミノベンゼンスルホン酸(スルファニル酸)などのアミノ酸(以上のアミノ酸を総称して「特殊アミノ酸」という)を挙げることができる。
【0034】
α−アミノ酸としては、たとえばグリシン、アラニン、バリン、ロイシン、イソロイシン、ノルロイシン、セリン、スレオニン、チロシン、システイン、メチオニン、アスパラギン酸、アスパラギン、グルタミン酸、グルタミン、アルギニン、リジン、ヒスチジン、フェニルアラニン、トリプトファン及びペニシラミンなどが挙げられる。好ましくはグリシン、アラニン、スレオニン、セリン、アスパラギン酸、グルタミン酸、フェニルアラニン、バリン、メチオニン、システイン、リジン、ヒスチジン、及びペニシラミンである。なお、これらのα−アミノ酸は、置換基として水酸基を有するものであってもよい。水酸基を有するα−アミノ酸としては、制限されないが、例えばハイドロキシプロリンやハイドロキシトリプトファン等を挙げることができる。
【0035】
β−アミノ酸としてはβ−アラニンが挙げられる。γ−アミノ酸としてはγ−アミノ−n−酪酸(GABA)やカルニチンが挙げられる。δ−アミノ酸としては5−アミノレブリン酸や5−アミノ吉草酸が、ε−アミノ酸としては6−アミノヘキサン酸が挙げられる(前述するように、これらのアミノ酸を総称して「ω−アミノ酸」という)。
【0036】
これらのアミノ酸のうち、好ましくはメチオニン、ヒスチジン、リジン、フェニルアラニン、バリン等のα−アミノ酸;γ−アミノ−n−酪酸や6−アミノヘキサン酸等のω−アミノ酸;及びアントラニル酸やアミノエタンスルホン酸等の特殊アミノ酸であり、より好ましくはフェニルアラニン、バリン、アミノエタンスルホン酸である。
【0037】
上記式中、R1で示すペプタイド残基の「ペプタイド」としては、好ましくは同種または異種の2個のアミノ酸(前記と同義)が、互いに一方のカルボキシル基と他方のアミノ酸のアミノ基とが酸アミド結合したジペプチドを挙げることができる。かかるジペプチドとして、具体的にはアスパラチイルグリシンやスレオニイルグリシンなどを挙げることができる。
【0038】
本発明のαリポ酸誘導体(1)の薬理学的に許容される塩としては、ナトリウム塩やカリウム塩などのアルカリ金属塩、およびカルシウム塩やマグネシウム塩などのアルカリ土類金属塩を挙げることができる。なお、これら以外の塩であっても薬理学的に許容できる塩であればいずれのものであっても本発明の目的のため適宜に用いることができる。また、本発明が対象とするαリポ酸誘導体(1)には、その水和物も含まれる。
【0039】
本発明の制がん剤の有効成分として使用するαリポ酸誘導体(1)のうち、nが0であり、隣接する硫黄原子が互いに結合して環を形成してなるαルポ酸誘導体(1)は、制限されないが、例えば、R1がアミノ酸残基である化合物(1a)は下記に説明する混合酸無水物法(MA法)により、αリポ酸を原料として合成することができる。
【0040】
【化4】
(式中、R1aは、アミノ酸残基を意味する。)
【0041】
具体的にはαリポ酸を有機溶媒(たとえばクロロホルム、テトラヒドロフラン、アセトニトリルなど)に溶かし、これに3級アミン(トリエチルアミン、トリブチルアミンやN−メチルモルホリン(NMM)など)の存在下−15℃〜−5℃でハロゲン化炭酸エステル(クロル炭酸エチル、クロル炭酸ブチルなど)、イソブチルオキシカルボニルクロリド、塩化ジエチルアセチルまたは塩化トリメチルアセチルなどの混合酸無水物化試薬を反応させてαリポ酸の混合酸無水物とする。反応時間は1〜2分から数10分程度である。さらにアミノ酸を塩基(水酸化ナトリウム、水酸化カリウムやトリエチルアミン、トリブチルアミンなどの3級アミン)存在下でアルコール、水またはそれらの混液などの溶媒に溶かしたものを加えて反応させた後、適当な溶媒、たとえば水またはアルコールから再結晶させると、所望のαリポイルアミノ酸(1a)を得ることができる。
【0042】
また本発明の制がん剤の有効成分として使用するαリポ酸誘導体(1)のうち、nが1であり、R2及びR3が、同一または異なって水素原子またはスルホ基である化合物(1c)及び(1d)は、制限されないが、下記に説明する方法により、αリポ酸を原料として合成することができる。
【0043】
【化5】
(式中、R2及びR3は前記と同義である。R1bは、前記のうち、OH基以外の基を意味する。)
【0044】
具体的には、本発明が対象とするαリポ酸誘導体(1c)及び(1d)は、αリポ酸、またはこれを所望によりアルキルエステル、アミノ酸もしくはペプタイド(上記式中、「R1b−OH」として示す)と反応させ、上記式(1b)で示すαリポ酸誘導体としたものに、亜硫酸塩類を付加させて製造することが出来る。
【0045】
ここで亜硫酸塩類としては、亜硫酸ナトリウムや亜硫酸カリウムなどの亜硫酸のアルカリ金属塩等の亜硫酸塩;亜硫酸水素ナトリウムや亜硫酸水素カリウムなどの亜硫酸水素化物のアルカリ金属塩等の重亜硫酸塩;およびメタ重亜硫酸塩を挙げることができる。好ましくは亜硫酸ナトリウムや亜硫酸カリウムなどの亜硫酸塩である。
【0046】
αリポ酸をアルキルエステルまたはアミド体とする反応は、混合酸無水物法(MA法)により実施することが出来る。すなわち、αリポ酸を有機溶媒(例えば、クロロホルム、THF,アセトニトリルなど)に溶かし、これに3級アミン(トリエチルアミン、トチブチルアミン、N−メチルモルホリンなど)の存在下、−15〜−5℃で、ハロゲン化炭酸エステル(例えば、クロル炭酸エチル、クロル炭酸ブチルなど)を加えて5〜10分後に、さらに、所望のアルコール、アミン、アミノ酸、ペプタイドを必要に応じた塩基(水酸化ナトリウム、トリエチルアミンやトリブチルアミンなどの3級アミンなど)の存在下、水またはアルコールまたはそれらの混液などの溶媒に溶かしたものを一挙に加えて反応させる。次に反応温度を徐々に室温に戻して反応を終結させ、反応液を処理して粗結晶を得た後、適当な溶媒、例えば、水またはアルコールから再結晶させて、αリポ酸のアルキルエステル、または対応したアミド体を得ることが出来る。
【0047】
本発明が対象とするαリポ酸誘導体(1c)及び(1d)は、αリポ酸または上記方法で調製されるαリポ酸誘導体(1b)を水に溶かすか、またはサスペンドしておき、これに亜硫酸塩類を徐々に加えて攪拌することによって調製することができる。亜硫酸塩類の配合割合は、特に制限されないが、S−スルホ体を調製する場合は、αリポ酸またはαリポ酸誘導体1モルに対して亜硫酸塩類が等モル量になるように、S−ジスルホ体を調製する場合は、αリポ酸またはαリポ酸誘導体1モルに対して亜硫酸塩類が2倍モル量になるように調整することが好ましい。
【0048】
斯くして得られる反応物(反応液)はこのまま本発明の制がん剤の有効成分として使用することも出来るが、通常はこの水溶液を減圧下、40℃以下で濃縮した後、エタノールを加えて、上記式(1d)で示すS−スルホまたはS−ジスルホ−N−リポ酸またはその塩、または上記式(1c)で示すS−スルホまたはS−ジスルホ−N−リポ酸誘導体にそれぞれ対応した固形物を得ることが出来る。
【0049】
本発明のαリポ誘導体(1)は構造上、ジヒドロリポ酸誘導体でその薬理学的に許容できる塩は、還元作用並びにラジカル抑制作用も強く、安定性もよく、組成からしても安全性の高い化合物で問題ない優れた化合物である。
【0050】
上記αリポ酸誘導体(1)またはその薬理学上許容される塩は、制がん剤、並びにがん転移抑制剤の有効成分として有効である。
【0051】
従って、上記αリポ酸誘導体(1)またはその薬理学上許容される塩を有効成分とする医薬品は、制がん剤(がん転移抑制剤を包含する)として、がん細胞の増殖を抑制し、がんの治療、及びがんの進展やがんの転移を抑制するために有効に使用することができる。なお、αリポ酸誘導体(1)またはその薬理学上許容される塩は、1種単独で使用してもよいが、目的と必要に応じて、2種以上を適宜組み合わせて用いることもできる。
【0052】
本発明の制がん剤は、経口的にあるいは非経口的に用いることができ、投与形態に応じた製剤形態に調製することができる。製剤の形態としては、例えば、錠剤、顆粒剤、散剤、カプセル剤等の固形製剤、または注射剤等の液剤などいずれの形にも公知の方法により調製することができる。これらの製剤には通常用いられる賦形剤、結合剤、増粘剤、分散剤、再吸収促進剤、緩衝材、界面活性剤、溶解補助剤、保存剤、乳化剤、等張化剤、安定化剤やpH調整化剤等の各種添加剤を適宜使用しても良い。
【0053】
本発明の制がん剤の投与量は、使用する本化合物の種類、患者の体重や年齢、対象とする疾患の種類やその状態および投与方法などによっても異なるが、例えば注射剤の場合、成人1日1回、約10mg〜100mg、内服剤の場合は、成人1日数回、10mg〜1000mg程度投与するのがよい。
【実施例】
【0054】
次に製造例、実験例および実施例を挙げて本発明を説明するが、本発明はこれらに何ら限定されるものではない。
【0055】
製造例1 N−α−リポイルアミノエタンスルホン酸ナトリウム(N−α−リポイルタウリンナトリウム)
αリポ酸6.2gをクロロホルム60mlに溶かし、トリエチルアミン3.2gを加えて−5℃に冷却した。これにクロル炭酸エチル3.3gを徐々に滴下し、滴下終了15分後に、これにアミノエタンスルホン酸4.5gおよび水酸化ナトリウム1.5gをメタノール60mlに溶かしたものを一挙に加えて、その温度(−5℃)で15分間、さらに室温に戻して1時間攪拌した。次に、これに水酸化ナトリウム1.5gをメタノール50mlに溶かした溶液を加えて、減圧下で溶媒を1/3程度になるまで濃縮した。次いで、これにエタノール60mlを加えて析出した結晶を濾取した。これを水−メタノールを用いて再結晶させて、掲題化合物の白色結晶5.8gを得た。融点235〜237℃。
元素分析: C10H17NO4S3Na・H2Oとして理論値 C:34.08、 H:5.43、N:3.97、実測値 C:34.23、H:5.54、N:3.80。
【0056】
製造例2 N−α−リポイルアミノエタンスルホン酸カリウム(N−α−リポイルタウリンカリウム)
製造例1のN−α−リポイルアミノエタンスルホン酸ナトリウムの製造方法において、アミノエタンスルホン酸4.5gと混合する水酸化ナトリウム1.5gに代えて水酸化カリウム4.0gを用いる以外は、製造例1と同じ方法を実施して結晶を濾取した。これを水−メタノールを用いて再結晶させて、掲題化合物の白色結晶6.5gを得た。融点240〜242℃。
【0057】
製造例3 N−α−リポイルアミノエタンスルホン酸のカルシウムまたはマグネシウム塩
製造例1で得られたN−α−リポイルアミノエタンスルホン酸ナトリウムを水に溶かし、スルホン酸樹脂で脱塩して遊離酸とした。これを炭酸カルシウムまたは塩基性炭酸マグネシウムで、それぞれ中和させて、N−α−リポイルアミノエタンスルホン酸の水溶性のカルシウム塩またはマグネシウム塩を得た。それぞれ融点300℃以上。
【0058】
製造例4 N−α−リポイルフェニルアラニン
原料としてαリポ酸6.2gに代えてαリポ酸4.2g、およびアミノエタンスルホン酸4.5gに代えてL−フェニルアラニン3.5gを用いる以外は、製造例1と同様に反応させた。得られた反応液を減圧下で40℃以下で溶媒を留去濃縮した後、塩酸で酸性として酢酸エチルで抽出し、水洗した後、酢酸エチルを留去した。残渣結晶をエタノール−水を用いて再結晶させて、掲題化合物の淡黄色結晶4.6gを得た。融点154℃〜156℃。
【0059】
製造例5 N−α−リポイルバリン
原料としてαリポ酸4.2g、およびL−フェニルアラニン3.5g に代えてL−バリン2.4gを用いる以外は、製造例4と同様に反応処理して、掲題化合物の淡黄色板状4.1gを得た。
【0060】
製造例6 N−α−リポイルグリシンナトリウム
原料としてαリポ酸6.2gに代えてαリポ酸4.2g、およびアミノエタンスルホン酸4.5gに代えてグリシン1.9gを用いる以外は、製造例1と同様に反応処理して、掲題化合物4.5gを得た。融点218℃〜220℃。
【0061】
製造例7 N−α−リポイルアスパラギン酸ナトリウム
原料としてαリポ酸6.2gに代えてαリポ酸4.2g、およびアミノエタンスルホン酸4.5gに代えてL−アスパラギン酸2.9gを用いる以外は、製造例1と同様に反応処理して、掲題化合物5.1gを得た。融点300℃以上。
【0062】
製造例8 N−α−リポイルメチオニン
原料としてαリポ酸4.2g、およびL−フェニルアラニン3.5g に代えてL−メチオニン3.5gを用いる以外は、製造例4と同様に処理して、掲題化合物の淡黄色結晶4.0gを得た。融点108℃〜109℃。
【0063】
製造例9 N−α−リポイルシステインナトリウム
原料としてαリポ酸4.2g、およびアミノエタンスルホン酸4.5g に代えてL−システイン2.6gを用いる以外は、製造例1と同様に処理して、掲題化合物の白色結晶4.5gを得た。融点150℃付近から徐々に分解。
【0064】
製造例10 N−α−リポイル−6−アミノヘキサン酸ナトリウム
原料としてαリポ酸6.2gに代えてαリポ酸4.2g、およびアミノエタンスルホン酸4.5g に代えて6−アミノヘキサン酸3.0gを用いる以外は、製造例1と同様の方法で反応させて結晶を濾取した。これをエタノールを用いて再結晶させて、標題化合物の黄色がかった白色結晶5.5gを得た。融点200〜202℃(分解)。
【0065】
製造例11 N−α−リポイルアントラニル酸ナトリウム
原料としてαリポ酸6.2gに代えてαリポ酸4.2g、およびアミノエタンスルホン酸4.5g に代えてアントラニル酸2.9gを用いる以外は、製造例1と同様に処理して、掲題化合物の白色結晶3.5gを得た。融点300℃以上。
【0066】
製造例12 N−α−リポイル−4−アミノ酪酸
原料としてαリポ酸4.2g、およびL−フェニルアラニン3.5g に代えて4−アミノ酪酸2.3gを用いる以外は、製造例4と同様に処理して、掲題化合物の淡黄色結晶4.3gを得た。融点235℃付近から分解。
【0067】
製造例13 S−スルホ−リポ酸ナトリウム
αリポ酸2.1g(0.01 モル)を水50mlに懸濁したものに、亜硫酸ナトリウム1.3g(0.01モル)を加えて撹拌しながら溶解した。これを減圧下、40℃以下で濃縮した後、エタノールを加え、白色固形物2.3gを濾取した。これを水−エタノールを用いて再結晶させて、掲題化合物の白色結晶を得た。
【0068】
製造例14 S−スルホ−N−リポイルフェニルアラニンナトリウム
製造例13において、αリポ酸2.1gの代わりに製造例4で調製したN−リポイルフェニルアラニン3.5gを用いて、同様に反応処理した。具体的には、N−リポイルフェニルアラニン3.5g(0.01 モル)を水50mlに懸濁したものに、亜硫酸ナトリウム1.3g(0.01モル)を加えて撹拌しながら溶解した。これを減圧下、40℃以下で濃縮した後、エタノールを加え、白色固形物2.3gを濾取した。これを水−エタノールを用いて再結晶させて、掲題化合物の白色結晶を得た。
【0069】
製造例15 S−スルホ−N−リポイルバリンナトリウム溶液
製造例5で調製したN−リポイルバリン305mgを水100mlに懸濁しておき、これに無水亜硫酸ナトリウム150mgを加えて溶解して濾過し、掲題化合物の水溶液を調製した。
【0070】
製造例16 S−スルホ−N−リポイルタウリンナトリウム溶液
製造例1で調製したN−リポイルタウリンナトリウム335mgを、水100mlに溶解し、これに無水亜硫酸ナトリウム150mgを加えて溶解して濾過して、掲題化合物の水溶液を取得した。
【0071】
実験例1 癌細胞増殖能に対する抑制効果
上記で製造したリポ酸誘導体のうち、N−α−リポイルアミノエタンスルホン酸ナトリウム(製造例1)、N−α−リポイルフェニルアラニン(製造例4)、N−α−リポイルバリン(製造例5)、及びS−スルホ−リポ酸ナトリウム(製造例13)を被験化合物とし、これらの化合物の癌細胞増殖能に対する抑制効果を評価した。なお、N−α−リポイルアミノエタンスルホン酸ナトリウム(製造例1)、N−α−リポイルフェニルアラニン(製造例4)、及びN−α−リポイルバリン(製造例5)については、それぞれ各化合物を単独で使用した場合と、亜硫酸ナトリウムを併用した場合との両方で実験を行った。
【0072】
(1)被験化合物
1.N−α−リポイルアミノエタンスルホン酸ナトリウム(製造例1)
2.N−α−リポイルアミノエタンスルホン酸ナトリウム+亜硫酸ナトリウム
3.N−リポイルフェニルアラニン(製造例4)
4.N−リポイルフェニルアラニン+亜硫酸ナトリウム
5.N−リポイルバリン(製造例5)
6.N−リポイルバリン+亜硫酸ナトリウム
7.S−スルホ−リポ酸ナトリウム(製造例13)。
【0073】
(2)試験方法
コントロール培地として、RPMI 1640に10%ウシ胎児血清と抗菌剤(100 IU/ml penicillin,0.1mg/ml streptomycin,2.5×10-4mg/ml amphotericin B) を加え調製したものを使用した。また試験培地として、上記コントロール培地に、上記各被験化合物をαリポ酸誘導体の最終濃度が0.1 mMまたは0.5mMに、被験化合物2、4及び6については、培地中の亜硫酸ナトリウムの最終濃度が0.1 mMまたは0.5mMになるように調整した。
【0074】
かかる培地を用いて、大腸癌細胞(HT29)(5.0×105/wellまたは1.0×106well)を5% CO2,37℃のもとで培養した。培養開始後24時間ごとに0.25%トリプシンを用いて細胞を剥離し,トリパンブルー排除法にて,Haemocytometerで生細胞数を測定した。
【0075】
(3)試験結果
大腸癌細胞増殖能に対する被験化合物1〜7の効果(癌細胞増殖抑制効果)を図1〜7にそれぞれ示す。またその総合評価を表1に示す。
【0076】
【表1】
【0077】
図1〜7及び表1の結果から、N-α-リポイルフェニルアラニン、N-α-リポイルバリン、及びN-α-リポイルアミノエタンスルホン酸等のαリポイルアミノ酸、並びにS−スルホ−リポ酸ナトリウムは0.1mM以上の濃度、特に0.5mM以上の濃度で、大腸癌細胞HT29の増殖を顕著に抑制することが確認された。またαリポニルアミノ酸は、亜硫酸塩と併用することで、がん細胞発育の抑制効果が増強される傾向にあることが確認された。
【0078】
これらの実験結果から、本願発明が対象とするαリポ酸誘導体は大腸がんなどのがん細胞の増殖を抑制する作用があり、かかる作用に基づいて制がん剤(抗がん剤)としての治療効果があると考えられる。
【0079】
〔製剤実施例1〕 内服錠
製造例1〜14のいずれかの化合物 30mg
乳糖 80mg
馬鈴薯澱粉 17mg
ポリエチレングリコール6000 3mg
以上の成分を1錠分の材料として常法により成型する。
【0080】
〔製剤実施例2〕 注射剤
製造例1〜14のいずれかの化合物 1.0g
マニトール 4.0g
注射用蒸留水 全量 100mL
以上を常法により混合溶解させ注射剤とする。
【特許請求の範囲】
【請求項1】
次の一般式(1)
【化1】
(式中、R1はOH基、O−アルキル基、N−置換アミノ基、アミノ酸残基またはペプタイド残基を示す。nは0または1の整数であり、nが0のとき隣接するS原子は互いに結合して環を形成し、nが1のときR2及びR3は、同一または異なって、水素原子またはスルホ基を示す。但し、R1はOH基であるとき、nは0ではない。)
で表わされる化合物、またはその薬理学的に許容される塩を有効成分とする制がん剤。
【請求項2】
R1がアミノ酸残基であり、nが0で隣接するS原子が互いに結合して環を形成してなるものである請求項1記載の制がん剤。
【請求項3】
R1がOH基であり、nが1で、R2及びR3のいずれか一方が水素原子で他方がスルホ基であるか、またはR2及びR3の両方がスルホ基である請求項1記載の制がん剤。
【請求項4】
R1がO−アルキル基であり、nが1で、R2及びR3のいずれか一方が水素原子で他方がスルホ基であるか、またはR2及びR3の両方がスルホ基である請求項1記載の制がん剤。
【請求項5】
R1がN−置換アミノ基であり、nが1で、R2及びR3のいずれか一方が水素原子で他方がスルホ基であるか、またはR2及びR3の両方がスルホ基である請求項1記載の制がん剤。
【請求項6】
R1がアミノ酸残基であり、nが1で、R2及びR3のいずれか一方が水素原子で他方がスルホ基であるか、またはR2及びR3の両方がスルホ基である請求項1記載の制がん剤。
【請求項7】
R1がペプタイド残基であり、nが1で、R2及びR3のいずれか一方が水素原子で他方がスルホ基であるか、またはR2及びR3の両方がスルホ基である請求項1記載の制がん剤。
【請求項8】
次の一般式(1)
【化2】
(式中、R1はOH基、O−アルキル基、N−置換アミノ基、アミノ酸残基またはペプタイド残基を示す。)
で表わされる化合物またはその薬理学的に許容される塩と、亜硫酸塩類またはその水和物とを含有することを特徴とする制がん剤。
【請求項9】
亜硫酸塩類が、亜硫酸塩、重亜硫酸塩、及びメタ重亜硫酸塩からなる群から選択されるいずれかである、請求項8に記載する制がん剤。
【請求項1】
次の一般式(1)
【化1】
(式中、R1はOH基、O−アルキル基、N−置換アミノ基、アミノ酸残基またはペプタイド残基を示す。nは0または1の整数であり、nが0のとき隣接するS原子は互いに結合して環を形成し、nが1のときR2及びR3は、同一または異なって、水素原子またはスルホ基を示す。但し、R1はOH基であるとき、nは0ではない。)
で表わされる化合物、またはその薬理学的に許容される塩を有効成分とする制がん剤。
【請求項2】
R1がアミノ酸残基であり、nが0で隣接するS原子が互いに結合して環を形成してなるものである請求項1記載の制がん剤。
【請求項3】
R1がOH基であり、nが1で、R2及びR3のいずれか一方が水素原子で他方がスルホ基であるか、またはR2及びR3の両方がスルホ基である請求項1記載の制がん剤。
【請求項4】
R1がO−アルキル基であり、nが1で、R2及びR3のいずれか一方が水素原子で他方がスルホ基であるか、またはR2及びR3の両方がスルホ基である請求項1記載の制がん剤。
【請求項5】
R1がN−置換アミノ基であり、nが1で、R2及びR3のいずれか一方が水素原子で他方がスルホ基であるか、またはR2及びR3の両方がスルホ基である請求項1記載の制がん剤。
【請求項6】
R1がアミノ酸残基であり、nが1で、R2及びR3のいずれか一方が水素原子で他方がスルホ基であるか、またはR2及びR3の両方がスルホ基である請求項1記載の制がん剤。
【請求項7】
R1がペプタイド残基であり、nが1で、R2及びR3のいずれか一方が水素原子で他方がスルホ基であるか、またはR2及びR3の両方がスルホ基である請求項1記載の制がん剤。
【請求項8】
次の一般式(1)
【化2】
(式中、R1はOH基、O−アルキル基、N−置換アミノ基、アミノ酸残基またはペプタイド残基を示す。)
で表わされる化合物またはその薬理学的に許容される塩と、亜硫酸塩類またはその水和物とを含有することを特徴とする制がん剤。
【請求項9】
亜硫酸塩類が、亜硫酸塩、重亜硫酸塩、及びメタ重亜硫酸塩からなる群から選択されるいずれかである、請求項8に記載する制がん剤。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2011−195516(P2011−195516A)
【公開日】平成23年10月6日(2011.10.6)
【国際特許分類】
【出願番号】特願2010−64850(P2010−64850)
【出願日】平成22年3月19日(2010.3.19)
【出願人】(304028726)国立大学法人 大分大学 (181)
【出願人】(502384060)有限会社オガ リサーチ (14)
【Fターム(参考)】
【公開日】平成23年10月6日(2011.10.6)
【国際特許分類】
【出願日】平成22年3月19日(2010.3.19)
【出願人】(304028726)国立大学法人 大分大学 (181)
【出願人】(502384060)有限会社オガ リサーチ (14)
【Fターム(参考)】
[ Back to top ]