
制御された相分離により調製される小球状粒子の作製方法、使用および組成物
本発明は、単一液相状態の溶液を提供することによって、活性薬剤の小球状粒子を調製する方法に関する。この単一液相は、活性薬剤、相分離促進剤、および第1溶剤を含む。この溶液に相変化が制御速度で誘導されて、活性薬剤の液−固相分離を引き起こし、固相および液相を生成する。固相は、活性薬剤の固体小球状粒子を含む。液相は、相分離促進剤および溶剤を含む。小球状粒子は実質的に球状であり、約0.01μm〜約200μmのサイズを有する。

【発明の詳細な説明】
【技術分野】
【0001】
(関連出願の相互参照)
本願は、2003年7月18日に出願された米国仮特許出願番号第60/488,712号に対する優先権を主張し、その全体が、本明細書中に参考として援用され、本明細書の一部をなす。
【0002】
(連邦政府によって後援された研究または開発)
該当なし。
【背景技術】
【0003】
(発明の背景)
(技術分野)
本発明は、活性薬剤の小球状粒子の製造方法、使用方法、および組成に関する。この製造方法に従って、溶解した相分離促進剤(PSEA)を含む水性または水混和性溶剤中に活性薬剤を溶解して、単一液相状態の溶液を生成する。次いで、この溶液は、活性薬剤が固相を構成し、かつPSEAおよび溶剤が液相を構成する、液−固相分離に供される。この液−固相分離は、溶液の温度をこの系の相転移温度より下に変化させるなどの、多数の方法で誘導され得る。この方法は、治療剤を必要とする被験体に送達され得る、治療剤の小球状粒子を生成するのに最適である。この方法はまた、高分子の固体小球状粒子、特に熱不安定性の高分子(例えばタンパク質)を生成するのにも最適である。
【0004】
(背景技術)
いくつかの技術が、バイオポリマーナノ粒子およびミクロ粒子の製造のためにこれまで用いられてきた。従来技術としては、粒子形成のための噴霧乾燥およびミリングが挙げられ、5μm以下のサイズの粒子を生成するのに用いられ得る。
【0005】
米国特許番号第5,654,010号および米国特許番号第5,667,808号には、亜鉛と錯化して非晶質錯体を作製し、次いで、この非晶質錯体を超音波ノズルを通して微粉化し、そして液体窒素中に噴霧してこれらの小滴を凍結させることによる、組換えヒト成長ホルモン(hGH)の固体形態の生成が記載される。次いで、液体窒素を−80℃の温度で蒸発させ、結果として生じる物質を凍結乾燥させる。
【0006】
ミクロ粒子、ミクロスフェア、およびマイクロカプセルは、直径1ミリメートル未満、より好ましくは直径100ミクロン未満、そして最も好ましくは直径10ミクロン未満の固体または半固体粒子であり、タンパク質、合成ポリマー、多糖類およびこれらの組み合わせを含む種々の材料から形成され得る。ミクロスフェアは、多くの種々の用途、一次的分離、診断薬、および薬物送達に用いられている。
【0007】
分離技術において用いられる最も周知のミクロスフェアの例は、合成または天然起源のポリマー(例えば、ポリアクリルアミド、ヒドロキシアパタイトもしくはアガロース)から形成されるミクロスフェアである。制御された薬物送達エリアでは、分子は、小球状粒子中に取り込まれるかまたは小球状粒子内にカプセル化されるか、あるいはその後の放出のためにモノリシックマトリックス中に取り込まれることが多い。相分離、溶剤蒸発、コアセルベーション、乳化、および噴霧乾燥を包含する、多数の異なる技術を常法として用いて、これらのミクロスフェアを合成ポリマー、天然ポリマー、タンパク質および多糖類から作製する。一般に、ポリマーはこれらのミクロスフェアの支持構造を形成し、目的の薬物はこのポリマー構造の中に取り込まれる。
【0008】
標的薬物をカプセル化するために脂質を用いて調製された粒子が、現在利用可能である。リポソームは、単一または複数のリン脂質二重層および/またはコレステロール二重層からなる球状粒子である。リポソームは100ナノメートル以上のサイズであり、種々の水溶性薬物または脂溶性薬物を輸送し得る。例えば、複数の水性区画を取り囲む二分子膜中に配置されて粒子を形成する脂質は、Sinil Kimによって米国特許番号第5,422,120号に記載されるように、その後の送達のために水溶性の薬物をカプセル化するのに用いられ得る。
【0009】
球状ビーズは、長年、生化学者のためのツールとして市販されてきた。例えば、ビーズに結合体化した抗体は、特定のリガンドに結合特異性を有する比較的大きな粒子を作製する。抗体は、細胞活性化のために細胞表面上のレセプターに結合するのに常法として用いられ、免疫親和性精製のために固相に結合されて抗体被覆粒子を形成し、そしてこれらの粒子に結合体化した組織特異的抗体または腫瘍特異的抗体を用いて所望の部位に薬剤を標的化し、経時的に緩徐に放出される治療剤を送達するのに用いられ得る。
【発明の開示】
【発明が解決しようとする課題】
【0010】
粒子(特に薬物送達、分離および診断領域における使用に適合させ得る粒子)を作製するための新たな方法の開発の必要性が、継続的に存在している。有用性の観点から最も望ましい粒子は、以下の特性を有する小球状粒子である:狭いサイズ分布、実質的に球状、実質的に活性薬剤のみで構成、活性薬剤の生化学的完全性の保持および生物活性の保持。これらの粒子は、コーティングによるかまたはマイクロカプセル化によって、粒子をさらに安定化させる適切な固体を生じるはずである。さらに、これらの小球状粒子の成形加工方法は、以下の望ましい特性を有する:単純な成形加工、本質的に水性のプロセス、高収率、およびその後のふるい分けが不要。
【課題を解決するための手段】
【0011】
(発明の概要)
本発明は、活性薬剤の小球状粒子の製造方法および使用方法に関する。この方法に従って、活性薬剤は、溶解した相分離促進剤を含む溶剤中に溶解されて、単一液相状態の溶液を形成する。この溶剤は、好ましくは水性または水混和性溶剤である。次いで、この溶液は、活性薬剤が固相を構成し、かつPSEAおよび溶剤が液相を構成する、液−固相分離に供される。この液−固相分離は、例えば、溶液の温度をこの溶液の相転移温度より下に変化させるなどの、多数の方法で誘導され得る。
【0012】
本発明の好ましい実施形態において、溶液を液−固相分離に供する方法は、溶液中の活性薬剤の相転移温度より下にこの溶液を冷却することによるものである。その温度は、この溶液の凝固点を超えるかまたは凝固点未満であり得る。凝固点が相転移温度を超える溶液について、この溶液は、ポリエチレングリコールまたはプロピレングリコールのような凝固点降下剤を含み得、この溶液の凝固点を降下させて、溶液を凍結させることなく溶液中の相分離を生じることが可能である。
【0013】
この溶液が相変化の工程(この工程において、活性薬剤は凝固して小球状粒子の懸濁液を不連続相として形成し、一方、相分離促進剤は連続相に溶解したままである)に供されると、本発明の相分離促進剤は、溶液中の活性薬剤の液−固相分離を促進するかまたは誘導する。すなわち、相を分離する促進剤は相の変化を受けないが、活性薬剤は相変化を受ける。
【0014】
本発明において粒子を生成する方法はまた、粒子の液−固相分離を制御するさらなる工程も包含し、生成された粒子のサイズおよび形状を制御し得る。相分離を制御する方法は、溶液中のイオン強度、pH、相分離促進剤の濃度、活性薬剤の濃度の制御、または溶液の温度の変化速度の制御を包含し、これらの制御は相分離の前であるか、あるいはこれらの任意またはいくつかの変化が相分離を誘導する。
【0015】
本発明の好ましい実施形態において、小球状粒子は、粒子形成後に連続相中のPSEAから分離される。さらに別の好ましい実施形態において、この分離方法は、これらの粒子を含む溶液を液体媒体で洗浄することによるものであり、その場合、活性薬剤は液体媒体において可溶性ではなく、一方で相分離促進剤は液体媒体において可溶性である。液体洗浄媒体は、この液体媒体中での活性薬剤の溶解度を低下させる薬剤を含み得る。この液体洗浄媒体はまた、1種以上の賦形剤も含み得る。この賦形剤は、小球状粒子または活性薬剤のための安定剤あるいはキャリア剤として機能し得る。この賦形剤はまた、粒子からの活性薬剤の徐放または生物組織への活性薬剤の改質された浸透のようなさらなる特性を、活性薬剤もしくは粒子にもたらし得る。
【0016】
別の好ましい実施形態において、これらの小粒子はPSEAを含まないが、これらの小粒子はその後のプロセス工程のためにPSEA相の存在下で回収され、その後にこのPSEA相から分離され得る。
【0017】
別の好ましい実施形態において、この溶液は、水性または水混和性溶剤を含む水溶液である。
【0018】
本発明の活性薬剤は、好ましくは薬学的に活性な物質であり、治療剤、診断用薬、化粧品、栄養剤、または農薬であり得る。本発明の好ましい実施形態において、この活性薬剤は、例えばタンパク質、ポリペプチド、炭水化物、ポリヌクレオチド、または核酸のような高分子である。さらに別の好ましい実施形態において、この活性薬剤を含む粒子は、適切な経路(例えば、非経口注射、局所、経口、直腸、経鼻、肺、膣、頬側、舌下、経皮、経粘膜、眼球、眼球内または耳)による、薬剤を必要とする被験体へのインビボ送達に適している。
【発明を実施するための最良の形態】
【0019】
(発明の詳細な説明)
本発明は、多数の異なる形態で実施形態の影響を受けやすい。本発明の好ましい実施形態は、本開示が本発明の原理の例証とみなされるべきであるという了解の下で開示され、かつ、本発明の広範な態様を、例示した実施形態に限定することを意図するものではない。
【0020】
本発明は、活性薬剤の小球状粒子の製造方法および使用方法および組成に関する。この製造方法に従い、溶解した相分離促進剤を含む溶剤中に活性薬剤を溶解して、単一連続液相である溶液を生成する。この溶剤は、好ましくは水性または水混和性溶剤である。次いで、この溶液は、例えば溶液の温度を活性薬剤の相転移温度より下に下げることによって相変化を受け、それによって、この活性薬剤は液−固相分離を受けて不連続相を構成する小球状粒子の懸濁液を生成し、一方、相分離促進剤は連続相に残る。
【0021】
(相)
(連続相)
活性薬剤の小球状粒子を調製する本発明の方法は、活性薬剤および第1溶剤中に溶解された相分離促進剤を単一液相状態で含む溶液を生成する工程から始まる。この溶液は、有機溶剤または混和性の有機溶剤の混合物を含む有機系であってもよい。この溶液はまた、水性媒体または水混和性有機溶剤または水混和性有機溶剤の混合物またはこれらの組合せを含む水性溶液であってもよい。この水性媒体は、水、正常生理食塩水、緩衝液、緩衝生理食塩水などであり得る。適切な水混和性有機溶剤としては、N−メチル−2−ピロリジノン(N−メチル−2−ピロリドン)、2−ピロリジノン(2−ピロリドン)、1,3−ジメチル−2−イミダゾリジノン(DMI)、ジメチルスルホキシド、ジメチルアセトアミド、酢酸、乳酸、アセトン、メチルエチルケトン、アセトニトリル、メタノール、エタノール、イソプロパノール、3−ペンタノール、n−プロパノール、ベンジルアルコール、グリセロール、テトラヒドロフラン(THF)、ポリエチレングリコール(PEG)、PEG−4、PEG−8、PEG−9、PEG−12、PEG−14、PEG−16、PEG−120、PEG−75、PEG−150、ポリエチレングリコールエステル、PEG−4ジラウレート、PEG−20ジラウレート、PEG−6イソステアレート、PEG−8 パルミトステアレート、PEG−150 パルミトステアレート、ポリエチレングリコールソルビタン、PEG−20ソルビタンイソステアレート、ポリエチレングリコールモノアルキルエーテル、PEG−3ジメチルエーテル、PEG−4ジメチルエーテル、ポリプロピレングリコール(PPG)、ポリプロピレンアルギレート、PPG−10ブタンジオール、PPG−10メチルグルコースエーテル、PPG−20メチルグルコースエーテル、PPG−15ステアリルエーテル、プロピレングリコールジカプリレート/ジカプレート、プロピレングリコールラウレート、およびグリコフロール(テトラヒドロフルフリルアルコールポリエチレングリコールエーテル)、プロパン、ブタン、ペンタン、ヘキサン、ヘプタン、オクタン、ノナン、デカンを含むアルカン、またはこれらの組み合わせが挙げられるが、これらに限定されない。
【0022】
この単一連続相は、第1溶剤において可溶性であるかまたは第1溶剤と混和性の相分離促進剤の溶液をまず生成することによって調製され得る。続いて、この溶液に活性薬剤を添加する。この活性薬剤はこの溶液に直接添加してもよく、またはこの活性薬剤をまず第2溶剤に溶解し、次いでこの溶液に合わせてもよい。この第2溶剤は、第1溶剤と同じ溶剤であってもよく、または上記リストから選択され、かつこの溶液と混和性の別の溶剤であってもよい。この薬剤は、周囲温度以下でこの溶液に添加されることが好ましく、これは、特に熱不安定分子(例えば、ある種のタンパク質)にとって重要である。「周囲温度」は、約20℃から約40℃の室温前後の温度を意味する。しかし、この系はまた、加熱によって薬剤の活性に顕著な低下が引き起こされない限り、系中の活性薬剤の溶解度を高めるために加熱され得る。
【0023】
(相分離促進剤)
溶液が、相分離の工程(この工程において、活性薬剤は固体または半固体となって不連続相として小球状粒子の懸濁液を生成し、一方、相分離促進剤は連続相に溶解したままである)に供されると、本発明の相分離促進剤(PSEA)は、この溶液からの活性薬剤の液−固相分離を促進するかまたは誘導する。この溶液が相分離条件に付されると、相分離促進剤は活性薬剤の溶解度を低下させる。適切な相分離促進剤としては、ポリマーまたはこの溶液に可溶性もしくは混和性のポリマーの混合物が挙げられるが、これらに限定されない。適切なポリマーとしては、例えば直鎖ポリマーまたは分枝鎖ポリマーが挙げられる。これらのポリマーは、水溶性、半水溶性、水混和性、または不溶性であり得る。
【0024】
本発明の好ましい形態において、この相分離促進剤は、水溶性または水混和性である。用いられ得るポリマーのタイプとしては、炭水化物系ポリマー、ポリ脂肪族アルコール、ポリ(ビニル)ポリマー、ポリアクリル酸、ポリ有機酸、ポリアミノ酸、コポリマーおよびブロックコポリマー(例えば、プルロニックF127またはプルロニックF68のようなポロクサマー)、tert−ポリマー、ポリエーテル、天然に存在するポリマー、ポリイミド、界面活性剤、ポリエステル、分枝ポリマーおよび環状ポリマー、ならびにポリアルデヒドが挙げられる。
【0025】
好ましいポリマーは、活性薬剤粒子の意図された投与経路のための医薬品添加物として受容可能なポリマーである。好ましいポリマーは、種々の分子量のポリエチレングリコール(PEG)(例えば、PEG200、PEG300、PEG3350、PEG8000、PEG10000、PEG20000など)およびポロクサマー(例えば、プルロニックF127またはプルロニックF68)のような薬学的に受容可能な添加物である。さらに別の好ましいポリマーは、ポリビニルピロリドン(PVP)である。さらに別の好ましいポリマーは、ヒドロキシエチルデンプンである。他の両親媒性ポリマーはまた、単独かまたは組み合わせて用いられ得る。この相分離促進剤はまた、プロピレングリコールおよびエタノールの混合物のような非ポリマーであり得る。
【0026】
(液−固相分離)
この溶液中の活性薬剤の液−固相分離は、当該分野で公知の任意の方法(例えば、温度の変化、圧力の変化、pHの変化、溶液のイオン強度の変化、活性薬剤の濃度の変化、相分離促進剤の濃度の変化、溶液の重量モル浸透圧濃度の変化、これらの組合せなど)によって誘導され得る。
【0027】
本発明の好ましい実施形態において、この相変化は、温度を溶液中の活性薬剤の相転移温度より下に下げることによる、温度誘発性相変化である。
【0028】
図1は、溶剤、PSEAおよび活性薬剤を含む溶液についての二次元状態図10である。この図は溶液の温度に対する活性薬剤濃度をプロットしている。PSEAの濃度は一定に維持される。
【0029】
この図は、飽和曲線12;過飽和曲線14;これらの間に準安定エリア16;該飽和曲線12の下に第1エリア18(この系は、全成分が液相中に存在する、均質な単一液相状態である);および該過飽和曲線14の上に第2エリア20(この系は、活性薬剤の固相ならびにPSEAおよび溶剤の液相を有する二相系である)を有する。この状態図は、この系の温度ならびに純液相中、液−固相中およびこれらの二相間の転移の周囲条件における成分の相対濃度を決定するのに役立つ。
【0030】
本明細書中に開示されるように、活性薬剤の小球状粒子の調製は、不飽和溶液(点A’)から冷却することにより、この溶液が、存在し得る任意の固相と平衡状態にある点Aにおいて飽和に達する工程に主に関与する。さらに冷却すると、この溶液は、所定の温度で、平衡溶解度に相当するよりも多くの活性薬剤を含む状態に達し、従って、この溶液は過飽和状態になる。この固相の自然形成は、点Bに到達するまで生じない。この点Bは、準安定ゾーンの境界上の点である。この準安定ゾーン幅は、最大達成可能過冷却ΔTmax=T2−T1または過飽和ΔCmax=C*2−C*1のいずれかで表され得る。これらの2つの式は、熱力学的当量である:
【0031】
【化1】
経路A’−A−Bは、準安定溶液を調製する多重熱方法を表す。等温プロセスにおいて、起点はA’’である。一定温度で濃度を増大させることにより、点Aで再び飽和に達成する。点Cへの濃度の等温増大(例えば、溶剤蒸発によるかまたは活性薬剤の播種/添加による)により、この溶液を、再び準安定限界に達するまで準安定領域中へ移動させる。準安定限界を超えると、この溶液は不安定になり、固相の自然形成が直ちに起こる。
【0032】
等温的に得られた値(ΔCmax)T=C*3−C*2は、多重熱的に得られたΔTmax=T3−T2の換算値とは異なり得る。準安定ゾーンの境界に接近すると、固体粒子形成に必要な時間は、準安定限界に達するまで減少する。
【0033】
多重熱プロセスにおいて、冷却速度は制御速度で行われ、粒子のサイズおよび形状が制御される。制御速度とは、約0.2℃/分から約50℃/分、そしてさらに好ましくは、0.2℃/分から30℃/分を意味する。変化速度は、一定速度または線形速度、非線形速度、断続的速度またはプログラム速度(複数の相サイクルを有する)であり得る。
【0034】
これらの粒子は、下記のように、溶液中のPSEAから分離され得、そして洗浄によって精製され得る。
【0035】
本発明は、活性薬剤の濃度、PSEAの濃度、温度またはこれらの任意の組み合わせを調整して、PSEAおよび溶剤は相変化を受けずに液体として残るが、活性薬剤は液体状態から固体状態に変わる、相変化を引き起こすことを企図する。pH、イオン強度、重量モル浸透圧濃度などを変化させて、相変化を増強、促進、制御または抑制することもまた企図される。凝固点が比較的高いか、または凝固点が相転移温度を超える溶液について、これらの溶液は、プロピレングリコール、スクロース、エチレングリコール、アルコール(例えば、エタノール、メタノール)のような凝固点降下剤または凝固点降下剤の水性混合物を含み得、この系の凝固点を降下させて、この系を凍結させることなく系中の相変化を可能にする。このプロセスはまた、温度を系の凝固点より下に低下させるために実行され得る。本明細書中に記載されるプロセスは、熱不安定性の分子(例えば、タンパク質)に特に適する。
【0036】
(任意の賦形剤)
本発明の粒子は、1以上の賦形剤を含む。この賦形剤は、粒子または活性薬剤またはキャリア剤の安定性の増大、粒子からの活性薬剤の徐放、あるいは生物組織への活性薬剤の改質された浸透のようなさらなる特性を、活性薬剤または粒子にもたらし得る。
【0037】
適切な賦形剤としては、炭水化物(例えば、トレハロース、スクロース、マンニトール)、カチオン(例えば、Zn2+、Mg2+、Ca2+)、アニオン(例えば、SO42−)、アミノ酸(例えば、グリシン)、脂質、リン脂質、脂肪酸、界面活性剤、トリグリセリド、胆汁酸またはそれらの塩(例えば、コール酸またはその塩、例えばコール酸ナトリウム;デオキシコール酸またはその塩)、脂肪酸エステル、およびポリマーであってそれらのPSEAとしての機能よりも低いレベルで存在するものが挙げられるが、これらに限定されない。賦形剤を用いる場合、この賦形剤は、溶液の状態図に有意な影響を及ぼしてはならない。
【0038】
(粒子の分離および洗浄)
本発明の好ましい実施形態において、小球状粒子は、溶液中の相分離促進剤からこれらを分離することによって回収される。さらに別の好ましい実施形態において、分離方法は、活性薬剤は液体媒体において可溶性ではなく、一方相分離促進剤は液体媒体において可溶性である液体媒体で、小球状粒子を含む溶液を洗浄することによるものである。いくつかの洗浄方法は、ダイアフィルトレーションまたは遠心分離によるものであり得る。液体媒体は、水性媒体または有機溶剤であり得る。低い水性溶解度を有する活性薬剤について、液体媒体は、水性媒体または活性薬剤の水性溶解度を低下させる薬剤(例えば2価カチオン)を含む水性媒体であり得る。高い水性溶解度を有する活性薬剤(例えば多くのタンパク質)について、有機溶剤または硫酸アンモニウムのようなタンパク質沈殿剤を含む水性溶剤が用いられ得る。
【0039】
液体媒体としての使用に適した有機溶剤の例としては、連続相に適するように上記に特定された有機溶剤が挙げられ、そしてより好ましくは、塩化メチレン、クロロホルム、アセトニトリル、酢酸エチル、メタノール、エタノール、ペンタンなどが挙げられる。
【0040】
任意のこれらの溶剤の混合物を用いることも企図される。1つの好ましいブレンドは、塩化メチレンまたは塩化メチレンとアセトンの1:1混合物である。液体媒体は、例えば、凍結乾燥、蒸発、または乾燥によって容易に除去されるために、低い沸点を有することが好ましい。
【0041】
液体媒体はまた、液体二酸化炭素またはそのほぼ超臨界点の流体のような超臨界流体でもあり得る。超臨界流体は、相分離促進剤(特にいくつかのポリマー)に適した溶剤であり得るが、タンパク質粒子に適した非溶剤である。超臨界流体は、それら単独でかまたは補助溶剤と共に用いられ得る。以下の超臨界流体が用いられ得る:液体C02、エタン、またはキセノン。潜在的補助溶剤は、アセトニトリル、ジクロロメタン、エタノール、メタノール、水、または2−プロパノールであり得る。
【0042】
本明細書中に記載されるPSEAから小球状粒子を分離するのに用いられる液体媒体は、この液体媒体中における活性薬剤の溶解度を低下させる薬剤を含み得る。これらの粒子は、粒子の収率を最大にするために、液体媒体中において最小の溶解度を示すことが最も望ましい。いくつかのタンパク質、例えばインスリンおよびヒト成長ホルモンについて、溶解度の低下は、このタンパク質への2価カチオン(Zn2+など)の添加によって達成され得る。複合体を形成するのに用いられ得る他のイオンとしては、Ca2+、Cu2+、Fe2+、Fe3+などが挙げられるが、これらに限定されない。
【0043】
インスリン−Zn複合体または成長ホルモン−Zn複合体の溶解度は十分に低いので、水溶液中の複合体のダイアフィルトレーションが可能になる。
【0044】
この液体媒体は1以上の賦形剤も含み得、これらの賦形剤は、上記のように、粒子の安定性および/または活性薬剤もしくはキャリア剤の安定性の増大、粒子からの活性薬剤の徐放、あるいは生物組織への活性薬剤の改質された浸透のようなさらなる特性を、活性薬剤または粒子にもたらし得る。
【0045】
本発明の別の形態において、小球状粒子は、PSEAを含む溶液から分離されない。
【0046】
(水系プロセス)
別の好ましい実施形態において、本系の成形加工プロセスは、水性溶剤または水混和性溶剤を含む水性系の成形加工プロセスである。適切な水混和性溶剤の例としては、連続相について上記に特定したものが挙げられるが、これらに限定されない。水性プロセスを用いる利点の1つは、この溶液が緩衝化され得かつ賦形剤を含み得ることであり、これらの賦形剤は生化学的安定化をもたらして活性薬剤(例えばタンパク質)を保護する。
【0047】
(活性薬剤)
本発明の活性薬剤は、好ましくは薬学的に活性な物質であり、この物質は、治療剤、診断用薬、化粧品、栄養剤、または農薬であり得る。
【0048】
この治療剤は生物製剤であり得、タンパク質、ポリペプチド、炭水化物、ポリヌクレオチド、および核酸が挙げられるがこれらに限定されない。このタンパク質は抗体であり得、ポリクローナルであってもモノクローナルであってもよい。この治療剤は低分子量分子であり得る。さらに、これらの治療剤は、例えば以下の種々の公知の医薬品から選択され得るが、これらに限定されない:鎮痛薬、麻酔薬、興奮薬、アドレナリン作動薬、抗アドレナリン作動薬、抗アドレナリン剤、アドレノコルチコイド、アドレナリン様作動、抗コリン剤、抗コリンエステラーゼ、抗痙攣薬、アルキル化剤、アルカロイド、アロステリック阻害剤、アナボリックステロイド、食欲抑制物、制酸薬、下痢止め薬、解毒薬、葉酸代謝拮抗薬、解熱薬、抗リウマチ薬、精神治療薬、神経遮断薬、抗炎症薬、駆虫薬、抗不整脈薬、抗生物質、抗凝血薬、抗鬱薬、抗糖尿病薬、抗てんかん薬、抗真菌剤、抗ヒスタミン薬、血圧降下薬、ムスカリン性受容体拮抗薬、抗ミコバクテリア薬、抗マラリア薬、防腐薬、抗悪性腫瘍薬、抗原虫薬、免疫抑制薬、免疫賦活薬、抗甲状腺薬、抗ウイルス薬、抗不安鎮静薬、収斂薬、βアドレナリン作動性レセプター遮断薬、造影剤、コルチコステロイド、鎮咳剤、診断用薬、画像診断用薬、利尿薬、ドーパミン作動薬、止血薬、血液作用薬、ヘモグロビン修飾因子、ホルモン、睡眠薬、免疫、抗高脂血症薬および他の脂質調節薬、ムスカリニック、筋弛緩薬、副交感神経作動薬、副甲状腺ホルモン、カルシトニン、プロスタグランジン、放射性医薬品、鎮静薬、性ホルモン、抗アレルギー薬、刺激薬、交感神経模倣薬、甲状腺薬、血管拡張薬、ワクチン、ビタミン、およびキサンチン。抗悪性腫瘍薬、または抗癌薬としては、パクリタキセルおよび誘導体化合物、ならびにアルカロイド、代謝拮抗物質、酵素阻害剤、アルキル化剤および抗生物質からなる群より選択される他の抗悪性腫瘍薬が挙げられるが、これらに限定されない。
【0049】
美容薬は、美容活性を有し得る任意の活性成分である。これらの活性成分の例としては、とりわけ、皮膚軟化薬、湿潤剤、フリーラジカル抑制剤、抗炎症薬、ビタミン、脱色剤、抗アクネ薬、抗脂漏性薬、角質溶解薬、やせ薬、皮膚着色剤および日焼け止め薬、そして特にリノール酸、レチノール、レチノイン酸、アスコルビン酸アルキルエステル、多価不飽和脂肪酸、ニコチンエステル、ニコチン酸トコフェロール、米、ダイズまたはシアバターノキの不鹸化物、セラミド、グリコール酸のようなヒドロキシ酸、セレン誘導体、抗酸化剤、βカロチン、γオリザノールおよびグリセリン酸ステアリルが挙げられ得る。これらの化粧品は市販されており、かつ/または当該分野で公知の技術によって調製され得る。
【0050】
本発明の実施における使用が企図される栄養剤としては、例えば、タンパク質、炭水化物、水溶性ビタミン(例えば、ビタミンC、B−複合ビタミン、など)、脂溶性ビタミン(例えば、ビタミンA、D、E、K、など)、およびハーブエキスが挙げられるが、これらに限定されない。これらの栄養剤は市販されており、かつ/または当該分野で公知の技術によって調製され得る。
【0051】
農薬との用語は、除草剤、殺虫剤、ダニ駆除剤、殺線虫剤、外部奇生生物撲滅薬および殺菌剤を包含すると理解される。本発明における農薬が属し得る化合物クラスとしては、例えば、尿素、トリアジン、トリアゾール、カルバメート、リン酸エステル、ジニトロアニリン、モルホリン、アシルアラニン、ピレスロイド、ベンジル酸エステル、ジフェニルエーテルおよび多環式ハロゲン化炭化水素が挙げられる。これらの各クラスの農薬の具体例は、Pesticide Manual、第9版、British Crop Protection Councilに記載される。これらの農薬は市販されており、かつ/または当該分野で公知の技術によって調製され得る。
【0052】
本発明の好ましい実施形態において、活性薬剤は、高分子、例えばタンパク質、ポリペプチド、炭水化物、ポリヌクレオチド、ウイルス、または核酸である。核酸としては、DNA、オリゴヌクレオチド、アンチセンスオリゴヌクレオチド、アプタマー(aptimer)、RNA、およびSiRNAが挙げられる。この高分子は天然であっても合成であってもよい。このタンパク質は抗体であり得、モノクローナルであってもポリクローナルであってもよい。このタンパク質はまた、天然の起源から単離されたかまたは合成法もしくは組替え法によって生成された任意の公知の治療タンパク質であってもよい。治療タンパク質の例としては、血液凝固カスケードのタンパク質(例えば、第VII因子、第VIII因子、第IX因子など)、サブチリシン、オボアルブミン、α−1−アンチトリプシン(AAT)、DNase、スーパーオキシドジスムターゼ(SOD)、リゾチーム、リボヌクレアーゼ、ヒアルロニダーゼ、コラゲナーゼ、成長ホルモン、エリスロポエチン、インスリン様成長因子またはそれらのアナログ、インターフェロン、グラチラマー、顆粒球マクロファージコロニー刺激因子、顆粒球コロニー刺激因子、抗体、ペグ化タンパク質、グリコシル化または過剰グリコシル化タンパク質、デスモプレシン、LHRHアゴニスト、例えば:ロイプロリド、ゴセレリン、ナフシリン、ブセレリン;LHRHアンタゴニスト、バソプレッシン、シクロスポリン、カルシトニン、副甲状腺ホルモン、副甲状腺ホルモンペプチドおよびインスリンが挙げられるが、これらに限定されない。好ましい治療タンパク質は、インスリン、α−1アンチトリプシン、LHRHアゴニストおよび成長ホルモンである。
【0053】
低分子量治療分子の例としては、ステロイド、βアゴニスト、抗菌剤、抗真菌剤、タキサン(有糸分裂阻害剤および微小管阻害剤)、アミノ酸、脂肪族化合物、芳香族化合物、および尿素化合物が挙げられるが、これらに限定されない。
【0054】
好ましい実施形態において、この活性薬剤は、肺疾患の処置のための治療剤である。このような薬剤の例としては、ステロイド、βアゴニスト、抗真菌剤、抗菌剤化合物、呼吸器作用薬、抗喘息薬、非ステロイド性抗炎症剤(NSAIDS)、α−1アンチトリプシン、および嚢胞性線維症を処置するための薬剤が挙げられるが、これらに限定されない。ステロイドの例としては、ベクロメタゾン(ジプロピオン酸ベクロメタゾンを含む)、フルチカゾン(プロピオン酸フルチカゾンを含む)、ブデソニド、エストラジオール、フルドロコルチゾン、フルオシノニド、トリアムシノロン(トリアムシノロンアセトニドを含む)、およびフルニソリドが挙げられるが、これらに限定されない。βアゴニストの例としては、キシナホ酸サルメテロール、フマル酸フォルモテロール、レボアルブテロール、バンブテロール、およびツロブテロールが挙げられるが、これらに限定されない。
【0055】
抗真菌薬の例としては、イトラコナゾール、フルコナゾール、およびアムホテリシンBが挙げられるが、これらに限定されない。
【0056】
診断用薬としては、X線造影剤および造影剤が挙げられる。X線造影剤の例としては、ジアトラゾ酸(diatrazoic acid)のエチルエステル(EEDA)としても公知のWIN−8883(エチル3,5−ジアセトアミド−2,4,6−トリヨードベンゾエート)、WIN67722、すなわち、(6−エトキシ−6−オキソヘキシル−3,5−ビス(アセトアミド)−2,4,6−トリヨードベンゾエート;エチル−2−(3,5−ビス(アセトアミド)−2,4,6−トリヨードベンゾイルオキシ)ブチラート(WIN16318);ジアトリゾキシ酢酸エチル(ethyl diatrizoxyacetate)(WIN12901);2−(3,5−ビス(アセトアミド)−2,4,6−トリヨードベンゾイルオキシ)プロピオン酸エチル(WIN16923);N−エチル2−(3,5−ビス(アセトアミド)−2,4,6−トリヨードベンゾイルオキシアセトアミド(WIN65312);イソプロピル2−(3,5−ビス(アセトアミド)−2,4,6−トリヨードベンゾイルオキシ)アセトアミド(WIN12855); 2−(3,5−ビス(アセトアミド)−2,4,6−トリヨードベンゾイルオキシマロン酸ジエチル(WIN67721);2−(3,5−ビス(アセトアミド)−2,4,6−トリヨードベンゾイルオキシ)フェニル酢酸エチル(WIN67585);[[3,5−ビス(アセチルアミノ)−2,4,5−トリヨードベンゾイル]オキシ]ビス(l−メチル)プロパン二酸エステル(WIN68165);および3,5−ビス(アセチルアミノ)−2,4,6−トリヨード−4−(エチル−3−エトキシ−2−ブテノエート)安息香酸エステル(WIN68209)が挙げられる。好ましい造影剤としては、生理的条件下で比較的急速に分解され、従って炎症反応に関連する任意の粒子を最小化することが期待される造影剤が挙げられる。分解は、酵素加水分解、生理的pHでのカルボン酸の可溶化、または他のメカニズムによって生じ得る。従って、低溶解性のヨウ化カルボン酸、例えばヨージパミド、ジアトリゾ酸、およびメトリゾ酸が、加水分解的に不安定なヨウ化種、例えばWIN67721、WIN12901、WIN68165、およびWIN68209などと並んで、好ましくあり得る。
【0057】
活性薬剤の多数の組合せが、例えば、ステロイドとβアゴニストの組合せ(例えば、プロピオン酸フルチカゾンとサルメテロール、ブデソニドとフォルメテロール、など)を包含することが望ましくあり得る。
【0058】
炭水化物の例は、デキストラン、ヘタスターチ、シクロデキストリン、アルギン酸塩、キトサン、コンドロイチン、ヘパリンなどである。
【0059】
(小球状粒子)
本発明の粒子および小球状粒子は、動的光散乱法(例えば、光相関分光法、レーザー回折、低角レーザー光散乱(LALLS)、中角レーザー光散乱(MALLS))によって、光暗化法(light obscuration method)(例えば、コールター(Coulter)分析法)によって、あるいはレオロジーまたは(光学もしくは電子)顕微鏡法のような他の方法によって測定されるように、好ましくは約0.01μm〜約200μm、より好ましくは0.1μm〜10μm、さらにより好ましくは約0.5μm〜約5μm、そして最も好ましくは約0.5μm〜約3μmの平均幾何学粒子径を有する。肺送達のための粒子は、飛行時間計測(例えば、エアロゾル化器)またはアンダーセンカスケードインパクター(Andersen Cascade Impactor)計測によって決定される空気力学的粒子サイズを有する。
【0060】
これらの小球状粒子は実質的に球状である。「実質的に球状」とは、粒子断面の最短垂直軸の長さに対する最長垂直軸の長さの比が約1.5以下であることを意味する。実質的に球状は、対称中心線を必要としない。さらに、これらの粒子は、表面テクスチャー加工、例えば粒子全体のサイズと比較して小規模の線または圧痕もしくは隆起などを有し得るが、依然として実質的に球状である。より好ましくは、この粒子の最長軸と最短軸の間の長さの比は、約1.33以下である。より好ましくは、この粒子の最長軸と最短軸の間の長さの比は、約1.25以下である。面接触は、実質的に球状であるミクロスフェアにおいて最小化され、貯蔵の際の粒子の望ましくない凝集を最小化する。多くの結晶または薄片は、大きな面接触領域を可能にし得る平面を有し、ここでイオン性相互作用または非イオン性相互作用によって凝集が生じ得る。球は、はるかに小さな領域にわたる接触を可能にする。
【0061】
これらの粒子はまた、実質的に同じ粒子サイズを有することが好ましい。相対的に大きな粒子および相対的に小さな粒子の両方が存在する広いサイズ分布を有する粒子により、より小さな粒子がより大きな粒子間のギャップを埋めることが可能になり、それによって新しい接触面を生じる。広いサイズ分布が、結合性の凝集のための多くの接触機会を与えることによって、より大きな球を生じ得る。本発明は、狭いサイズ分布を有する球状粒子を作製し、それによって接触凝集のための機会が最小限になる。「狭いサイズ分布」とは、好ましい粒子サイズ分布が、第10百分位数の小球状粒子の体積粒径(volume diameter)に対する第90百分位数の小球状粒子の体積粒径の比が5以下である比を有することを意味する。より好ましくは、この粒子サイズ分布は、第10百分位数の小球状粒子の体積粒径に対する第90百分位数の小球状粒子の体積粒径の比が3以下である比を有する。より好ましくは、この粒子サイズ分布は、第10百分位数の小球状粒子の体積粒径に対する第90百分位数の小球状粒子の体積粒径の比が2以下である。
【0062】
幾何学的標準偏差(GSD)もまた、狭いサイズ分布を示すのに用いられ得る。GSD計算値は、有効カットオフ直径(ECD)を累積で15.9%および84.1%のパーセンテージ未満と決定するのに用いた。GSDは、15.9%未満のECDに対する84.17%未満のECDの比の平方根に等しい。GSDが2.5未満、より好ましくは1.8未満である場合、このGSDは狭いサイズ分布を有する。
【0063】
本発明の好ましい形態において、小球状粒子中の活性薬剤は半晶質または非晶質である。
【0064】
代表的には、本発明のプロセスによって作製される小球状粒子は実質的に無孔であり、0.5g/cm3を超える密度、より好ましくは0.75g/cm3を超える密度、そして最も好ましくは約0.85g/cm3を超える密度を有する。密度について好ましい範囲は約0.5〜約2g/cm3であり、そしてより好ましくは約0.75〜約1.75g/cm3であり、そしてさらにより好ましくは約0.85g/cm3〜約1.5g/cm3である。
【0065】
本発明の粒子は、活性薬剤の高い含有量を示し得る。多くの他の粒子調製方法に必要とされる有意量の増量剤または類似の賦形剤の必要はない。例えば、インスリン小球状粒子は、95重量%と同等かまたは95重量%を超える粒子からなる。しかし、増量剤または賦形剤は、これらの粒子中に含まれ得る。好ましくは、この活性薬剤は、粒子の約0.1重量%〜95重量%を超えて、より好ましくは約30重量%〜約100重量%、さらにより好ましくは約50重量%〜約100重量%、なおより好ましくは約75重量%〜約100重量%、そして最も好ましくは90重量%を超えて存在する。本明細書中に範囲を記載する場合、その中に任意の範囲または範囲の組合せを包含することを意味する。
【0066】
本発明のさらなる態様は、これらの小球状粒子が、賦形剤の封入体を含むかまたは含まずに活性薬剤の生化学的完全性および生化学的活性を保持することである。
【0067】
(粒子のインビボ送達)
本発明における活性薬剤を含む粒子は、適切な経路(例えば、注射可能な、局所、経口、直腸、経鼻、肺、膣、頬側、舌下、経皮、経粘膜、眼球、眼球内または耳)による、薬剤を必要とする被験体へのインビボ送達に適している。これらの粒子は、安定な液体懸濁液として送達され得るか、または錠剤、キャプレッツ、カプセルなどのような固形の剤形として処方され得る。好ましい送達経路は、静脈内、筋肉内、皮下、腹腔内、髄腔内、硬膜外、動脈内、関節内などを包む、注射可能な経路である。別の好ましい送達経路は、肺吸入である。この送達経路において、これらの粒子は、肺深部に、上気道中に、またはこの気道中のどこにでも沈積し得る。これらの粒子は、乾燥粉末吸入器によって乾燥粉末として送達され得るか、または定量噴霧式吸入器もしくは噴霧器によって送達され得る。
【0068】
全身的に機能することを目的とする薬物(例えばインスリン)は、血流中への吸収に利用可能な非常に大きな表面積がある肺胞中に沈積されるのが望ましい。肺内の特定の領域に薬物沈積を標的化する場合、粒子の空気力学的直径は、これらの粒子の基礎物理特性(例えば、形状、密度、および粒子サイズ)を操作することによって、最適範囲に調整され得る。
【0069】
吸入薬物粒子の受容可能な呼吸可能画分は、粒子組成物中に組み込まれるか、薬物粒子との混合物として組み込まれて、処方物に賦形剤を添加することによってしばしば達成される。例えば、微粉化薬物粒子(約5μm)の沈積の改善は、トレハロース、ラクトースまたはマルトデキストリンなどの不活性キャリア粒子のより大きな(30〜90μm)粒子とのブレンディングによってもたらされる。より大きな賦形剤粒子は、粉末流動性を改善し、これは薬力学的効果の改善と相関関係にある。さらなる改善において、これらの賦形剤は小球状粒子中に直接組み込まれて、タンパク質薬物の安定性を強力に高めると同時に、エアロゾル性能に影響を及ぼす。一般に、賦形剤は、以前に吸入がFDAに認可されたもの(例えばラクトース)、または肺にとって内因性の有機分子(例えばアルブミンおよびDL−α−ホスファチジルコリンジパルミトイル(DPPC))が選択される。他の賦形剤、例えば乳酸・グリコール酸共重合体(PLGA)は、望ましい物理的および化学的特性で粒子を操作するのに用いられてきた。しかしながら、FDAに認可された賦形剤を用いる吸入経験の多くは、気管気管支領域中に望ましく沈積する大きな空気力学的粒子サイズを有し、かつ認めうるほどには肺深部に達しない喘息薬を用いてきた。肺深部に送達される吸入タンパク質またはペプチド治療法について、賦形剤が肺胞領域に送達される場合、免疫応答に起因し得るかまたは賦形剤によって引き起こされ得る炎症および刺激のような望ましくない長期副作用が生じ得ることが懸念される。
【0070】
肺深部吸入治療法の潜在的な有害副作用を最小化するために、送達されるべき薬物で実質的に構成される、吸入用の粒子を調製することが有利であり得る。このストラテジーは、賦形剤への肺胞の曝露を最小化し、各々の投与毎に肺胞表面上に沈積される粒子の総質量用量を低減させ、おそらく吸入治療の慢性的使用の間に刺激を最小化する。本質的に完全に治療タンパク質またはペプチドからなる、肺深部沈積に適した空気力学的性質を有する小球状粒子は、肺の肺胞膜に対する慢性的治療投薬の影響に関する単独研究に特に有用であり得る。次いで、吸入による小球状粒子の形態でのタンパク質またはペプチドの全身送達の影響が、関連する賦形剤によって複雑な要素が導入されることなく研究され得る。
【0071】
吸入によって肺深部に粒子を送達する必要条件は、これらの粒子が、0.5〜10マイクロメーターの小さな平均空気力学的直径および狭いサイズ分布を有することである。本発明はまた、異なる粒子サイズ範囲を有する粒子の種々のバッチを一緒に混合することも企図する。本発明のプロセスにより、上記特性を有する小球状粒子の成形加工が可能になる。
【0072】
0.5〜3ミクロンの空気力学的直径を有する粒子を形成するための2つの主要なアプローチがある。第1のアプローチは、比較的大きいが極めて多孔性の(または有孔の)ミクロ粒子を生成することである。空気力学的直径(D空気力学的)と幾何学的直径(D幾何学的)の間の関係は、D空気力学的が、極めて低い質量密度(約0.1g/cm3)を有する粒子の密度の平方根をD幾何学的に乗じたものに等しいので、比較的大きな幾何学的直径(5〜10ミクロン)を有しながら、小さな空気力学的直径(0.5〜3ミクロン)を示し得る。
【0073】
代替のアプローチは、比較的低い多孔率を有する粒子を生成することであり、本発明の場合は、これらの粒子は上に示した範囲内の密度を有し、そしてさらに一般にはこの密度はほぼ1g/cm3である。従って、このような非多孔性の高密度粒子の空気力学的直径は、ほぼそれらの幾何学的直径である。
【0074】
上述の粒子形成のための本方法は、賦形剤を用いるかまたは用いない粒子形成を提供する。
【0075】
添加物なしでのタンパク質単独からのタンパク質小球状粒子の成形加工は、より大きな薬物有効負荷量、安全性の増大および必要吸入数の減少についての選択肢を提供するので、肺送達における使用に優れた利点をもたらす。
【0076】
(前調製小球状粒子のマイクロカプセル化)
本発明の小球状粒子または他の方法で調製された小粒子(ミクロ粒子、ミクロスフェア、ナノスフェア、ナノ粒子などを含む)は、さらに壁形成材料のマトリックス内にカプセル化されて、マイクロカプセル化粒子を形成し得る。マイクロカプセル化は、当該分野で公知の任意のプロセスによって達成され得る。好ましい実施形態において、本発明の小球状粒子または任意の他の小粒子のマイクロカプセル化は、下記のように乳化/溶剤抽出プロセスによって達成される。このマトリックスは、活性薬剤に徐放性を与え得、結果として所望の治療用途によって数分から数時間、数日または数週間持続する放出速度をもたらす。これらのマイクロカプセル化粒子はまた、前調製小球状粒子の遅延放出処方物も生成し得る。好ましい実施形態において、これらの前調製小球状粒子は高分子の粒子である。別の好ましい実施形態において、この高分子はタンパク質またはポリペプチドである。
【0077】
この乳化/溶剤抽出プロセスにおいて、乳化は、2つの不混和相である連続相および不連続相(分散相としてもまた公知である)を混合してエマルジョンを形成することによって生じる。好ましい実施形態において、連続相は水性相(または水相)であり、かつ不連続相は有機相(または油相)であって、水中油(O/W)エマルジョンを形成する。この不連続相は、油中固体(S/O)相を形成する微細懸濁液または微細分散液のいずれかとして存在する固体粒子の分散系をさらに含み得る。この有機相は、好ましくは水不混和性または部分的に水混和性の有機溶剤である。この水性相に対する有機相の重量比は、約1:99〜約99:1であり、より好ましくは約1:99〜約40:60であり、そして最も好ましくは約2:98〜約1:3であるか、あるいはその中の任意の範囲または範囲の組合せである。好ましい実施形態において、この水性相に対する有機相の比は約1:3である。本発明は、油相が連続相を形成しかつ水相が不連続相を形成する、逆エマルジョンすなわち油中水エマルジョン(W/O)の利用をさらに企図する。本発明は、例えば油中水中油エマルジョン(O/W/O)または水中油中水エマルジョン(W/O/W)のような3つ以上の相を有するエマルジョンの利用をさらに企図する。
【0078】
好ましい実施形態において、この乳化/溶剤抽出プロセスを用いるマイクロカプセル化のプロセスは、前調製小球状粒子を以前に記載した方法によって調製する工程および壁形成材料を含む有機相を調製する工程で始まる。これらの前調製小球状粒子を壁形成材料の有機相中に分散させて、油相中に前調製小球状粒子の分散系を含む油中固体(S/O)相を形成する。好ましい実施形態において、この分散は、小球状粒子と有機相の混合物をホモジナイズすることによって達成される。水性媒体は連続相を形成する。この場合、S/O相と水性相を乳化することによって形成されるエマルジョン系は、水中油中固体(S/O/W)エマルジョン系である。
【0079】
壁形成材料とは、マトリックスの構造体を個々にまたは組み合わせて形成可能な材料をいう。生分解性の壁形成材料は、特に注射用用途に好ましい。このような材料の例としては、ポリラクチド/ポリグリコリドポリマー(PLGA)のファミリー、ポリエチレングリコール結合体化PLGA(PLGA−PEG)、およびトリグリセリドが挙げられるが、これらに限定されない。PLGAまたはPLGA−PEGが用いられる実施形態において、PLGAは好ましくは100:0〜0:100、より好ましくは約90:10〜約15:85、そして最も好ましくは約50:50のポリラクチド対ポリグリコリド比を有する。一般に、ポリマー中のポリグリコリド対ポリラクチド比が高いほど、マイクロカプセル化粒子はより親水性になり、結果としてより速い水和およびより速い分解をもたらす。種々の分子量のPLGAもまた用いられ得る。一般に、ポリマー中のポリグリコリドとポリラクチドの比率が同じであれば、PLGAの分子量が高いほど活性薬剤の放出が遅くなり、マイクロカプセル化粒子のサイズ分布が広くなる。
【0080】
水中油(O/W)または水中油中固体(S/O/W)エマルジョンの有機相(油相)中の有機溶剤は、水不混和性であるかまたは部分的に水不混和性であり得る。用語「水不混和性溶剤」は、1:1の比率(O/W)で水溶液と混合した場合に界面メニカスを形成する溶剤を意味する。適切な水不混和性溶剤としては、炭素数5以上の置換または非置換の直鎖、分枝または環状アルカン、炭素数5以上の置換または非置換の直鎖、分枝または環状アルケン、炭素数5以上の置換または非置換の直鎖、分枝または環状アルキン;炭化水素を完全にまたは部分的にハロゲン化した芳香族炭化水素、エーテル、エステル、ケトン、モノ−、ジ−またはトリ−グリセリド、天然油、アルコール、アルデヒド、酸、アミン、直鎖または環状シリコーン、ヘキサメチルジシロキサン、あるいはこれらの溶剤の任意の組合せが挙げられるが、これらに限定されない。ハロゲン化溶剤としては、四塩化炭素、塩化メチレン、クロロホルム、テトラクロロエチレン、トリクロロエチレン、トリクロロエタン、ヒドロフルオロカーボン、塩素化ベンゼン(モノ、ジ、トリ)、トリクロロフルオロメタンが挙げられるが、これらに限定されない。特に適切な溶剤は、塩化メチレン、クロロホルム、ジエチルエーテル、トルエン、キシレンおよび酢酸エチルである。「部分的に水混和性の溶剤」は、ある濃度では水不混和性であり、別のより低い濃度では水混和性である溶剤を意味する。これらの溶剤は、水混和性が制限された溶剤であり、かつ自発的エマルジョン形成が可能である。部分的に水混和性の溶剤の例は、テトラヒドロフラン(THF)、プロピレンカーボネート、ベンジルアルコール、および酢酸エチルである。
【0081】
界面活性化合物は、例えば有機相の湿潤性を増大させるために添加され得る。この界面活性化合物は、乳化プロセス前に水性相、有機相、水性媒体と有機溶液の両方に添加され得るか、または乳化プロセス後にエマルジョンに添加され得る。界面活性化合物の使用により、カプセル化されていないかまたは部分的にカプセル化された小球状粒子の数を減少させ得、結果として放出の間の活性薬剤のイニシャルバーストの低減をもたらす。この界面活性化合物は、化合物の溶解度に応じて、有機相、または水性相、または有機相と水性相の両方に添加され得る。
【0082】
用語「界面活性化合物」は、例えば陰イオン界面活性剤、陽イオン界面活性剤、双性イオン界面活性剤、非イオン性界面活性剤または生物学的界面活性分子のような化合物を意味する。この界面活性化合物は、水性相または有機相またはエマルジョンのいずれの場合にもその約0.01重量%未満〜約30重量%、より好ましくは約0.01重量%未満〜約10重量%、またはその中の任意の範囲もしくは範囲の組合せで存在すべきである。
【0083】
適切な陰イオン界面活性剤としては、以下:ラウリン酸カリウム、ラウリル硫酸ナトリウム、ドデシル硫酸ナトリウム、ポリオキシエチレンアルキル硫酸塩、アルギン酸ナトリウム、スルホコハク酸ジオクチルナトリウム、ホスファチジルコリン、ホスファチジルグリセロール、ホスファチジルイノシン、ホスファチジルセリン、ホスファチジン酸およびそれらの塩、グリセリルエステル、カルボキシメチルセルロースナトリウム、コール酸および他の胆汁酸(例えば、コール酸、デオキシコール酸、グリココール酸、タウロコール酸、グリコデオキシコール酸)およびそれらの塩(例えば、デオキシコール酸ナトリウムなど)が挙げられるが、これらに限定されない。
【0084】
適切な陽イオン界面活性剤としては、第4級アンモニウム化合物、例えば、塩化ベンザルコニウム、臭化セチルトリメチルアミン、塩化ラウリルジメチルベンジルアンモニウム、アシルカルニチンハイドロクロライド、またはアルキルピリジニウムハライドが挙げられるが、これらに限定されない。陰イオン界面活性剤として、リン脂質が用いられ得る。適切なリン脂質としては、例えばホスファチジルコリン、ホスファチジルエタノールアミン、ホスファチジルセリン、ホスファチジルイノシトール、ホスファチジルグリセロール、ホスファチジン酸、リゾリン脂質、卵または大豆リン脂質あるいはこれらの組合せが挙げられる。このリン脂質は、加塩されても脱塩されてもよく、水素化されても部分的に水素化されてもよく、あるいは天然、半合成または合成であってもよい。
【0085】
適切な非イオン性界面活性剤としては、以下:ポリオキシエチレン脂肪アルコールエーテル(MacrogolおよびBrij)、ポリオキシエチレンソルビタン脂肪酸エステル(ポリソルベート)、ポリオキシエチレン脂肪酸エステル(Myrj)、ソルビタンエステル(Span)、グリセロールモノステアレート、ポリエチレングリコール、ポリプロピレングリコール、セチルアルコール、セトステアリルアルコール、ステアリルアルコール、アリールアルキルポリエーテルアルコール、ポリオキシエチレン−ポリオキシプロピレンコポリマー(ポロクサマー)、ポラキサミン(polaxamine)、ポリビニルアルコール、ポリビニルピロリドン、および多糖類(例えばヒドロキシエチルデンプン(HES)、メチルセルロース、ヒドロキシセルロース、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、および非晶質セルロースのような、デンプンおよびデンプン誘導体を含む)が挙げられる。本発明の好ましい形態において、非イオン性界面活性剤はポリオキシエチレンおよびポリオキシプロピレンコポリマーであり、好ましくはプロピレングリコールおよびエチレングリコールのブロックコポリマーである。このようなポリマーは、商品名POLOXAMERのもとで販売され、また時々PLURONIC(登録商標)とも呼ばれ、そしてSpectrum Chemical and Rugerを含むいくつかの業者によって販売される。ポリオキシエチレンの中で、短いアルキル鎖を有する脂肪酸エステルが挙げられる。このような界面活性剤の一例は、BASF Aktiengesellschaft製のポリエチレン−660−水酸化ステロイドである、SOLUTOL(登録商標)HS15である。
【0086】
界面活性生物学的分子としては、アルブミン、カゼイン、ヘパリン、ヒルジン、ヘタスターチまたは他の適切な生体適合性薬剤のような分子が挙げられる。
【0087】
本発明の好ましい形態において、水性相は界面活性化合物としてタンパク質を含む。好ましいタンパク質はアルブミンである。このタンパク質はまた、賦形剤としても機能し得る。タンパク質が界面活性化合物ではない実施形態において、他の賦形剤が、乳化プロセス前または後のいずれかに添加されて、エマルジョン中に含まれ得る。適切な賦形剤としては、糖類、二糖類、および糖アルコールが挙げられるが、これらに限定されない。好ましい二糖類はスクロースであり、好ましい糖アルコールはマンニトールである。
【0088】
さらに、チャネリング剤、例えばポリエチレングリコール(PEG)を用いることによって最終産物の水浸透率を増大させ得、その結果、水和率を改変することによって、マトリックスからの活性薬剤の初期放出動力学ならびにマトリックスの分解速度および分解依存性放出動力学の改変をもたらす。カプセル化の間のチャネリング剤としてPEGを用いることは、PEGを相分離促進剤として用いる小球状粒子の成形加工の間の洗浄プロセスの部分をなくすという点で有利であり得る。さらに、緩衝液を用いて連続相のpHを変動させることにより、粒子表面と有機相の間の湿潤プロセスを有意に増進させ得、従って結果として、マイクロカプセル化粒子のマトリックスからのカプセル化治療剤のイニシャルバーストを有意に減少させる。連続相の性質は、例えば、NaClのような塩を添加してその塩分濃度を増加させることによっても改変され、2つの相の混和性を低下させ得る。
【0089】
小球状粒子を有機相(油相)中に分散した後、次いで水性媒体(水相)の連続相を、例えば均質化または超音波処理によってこの有機相の不連続相と勢いよく混合し、初期マイクロカプセル化粒子の乳化小滴を含むエマルジョンを形成する。この連続水性相は、乳化小滴からの有機溶剤の急速な抽出を最小限にするために、有機相中に用いた有機溶剤で飽和させた後に、水性相と有機相を混合し得る。この乳化プロセスは、この混合物がその流動性を維持し得る任意の温度で実行され得る。エマルジョン安定性は、有機相中または水性相中の、あるいは界面活性化合物が乳化プロセス後にエマルジョンに添加される場合はエマルジョン中の、界面活性化合物の濃度の関数である。これは、エマルジョン系(胚性マイクロカプセル化粒子)の小滴サイズならびにマイクロカプセル化粒子のサイズおよびサイズ分布を決定する要因の1つである。マイクロカプセル化粒子のサイズ分布に影響を及ぼす他の要因は、連続相の粘度、不連続相の粘度、乳化の間の剪断力、界面活性化合物の種類および濃度、ならびに油/水比である。
【0090】
乳化後、次いでこのエマルジョンを硬化媒体中に移す。この硬化媒体は、胚性マイクロカプセル化粒子から不連続相中に溶剤を抽出し、結果として、乳化小滴の近傍内に、前調製小球状粒子の周囲に固体ポリマーマトリックスを有する固体マイクロカプセル化粒子の形成をもたらす。O/W系またはS/O/W系の実施形態において、硬化媒体は水性媒体であり、界面活性化合物、または増粘剤、または他の賦形剤を含み得る。これらのマイクロカプセル化粒子は、好ましくは球状であり、約0.6〜約300μmの粒子サイズを有し、そしてより好ましくは約0.8〜約60μmの粒子サイズを有する。さらに、これらのマイクロカプセル化粒子は、好ましくは狭い粒子サイズ分布を有する。不連続相の抽出時間を短縮するために、硬化媒体に熱または減圧が適用され得る。例えば蒸発(煮沸効果)による不連続相の急速な除去は、マトリックスの連続性の破壊を生じるので、これらの初期マイクロカプセル化粒子からの不連続相の抽出速度は、最終の固体マイクロカプセル化粒子中の多孔度にとって重要な因子である。
【0091】
好ましい実施形態において、この乳化プロセスは、バッチプロセスの代わりに連続様式で実行される。図41は、連続乳化反応器のデザインを示す。
【0092】
別の好ましい実施形態において、活性薬剤の小球状粒子をカプセル化する、硬化壁形成ポリマーマトリックスは、遠心分離および/または濾過(ダイアフィルトレーションを含む)によってさらに回収され、水で洗浄される。残りの液相は、凍結乾燥または蒸発のようなプロセスによってさらに除去され得る。
【0093】
(A.インスリン小球状粒子)
(実施例1:インスリン小球状粒子の調製の一般法)
16.67%のPEG3350を含む、pH5.65に緩衝化した溶液(0.033Mの酢酸ナトリウム緩衝液)を調製した。亜鉛結晶性インスリンの濃縮スラリーを、この溶液に攪拌しながら添加した。最終溶液中のインスリン濃度は0.83mg/mLであった。この溶液を約85〜90℃に加熱した。インスリン結晶をこの温度範囲で5分間以内に完全に溶解した。この溶液の温度が制御速度で低下する場合、インスリン小球状粒子は約60℃で形成を開始した。収率は、PEGの濃度が増大するにつれて増大した。このプロセスにより、平均値1.4μmを有する種々のサイズ分布の小球状粒子が生成される。
【0094】
形成されたインスリン小球状粒子を、これらの小球状粒子が溶解しない条件下でダイアフィルトレーションによってミクロスフェアを洗浄することにより、PEGから分離した。これらのインスリン小球状粒子を、Zn2+を含む水溶液を用いて懸濁液から洗い出した。このZn2+イオンは、インスリンの溶解度を低下させて、収率を低下させかつ小球状粒子凝集を引き起こすインスリンの溶解を妨げる。
【0095】
(実施例2:インスリン小球状粒子を作製するための無攪拌バッチプロセス)
20.2mgの亜鉛結晶性インスリンを1mLの脱イオン水中に室温で懸濁した。50マイクロリットルの0.5N HClをこのインスリンに添加した。1mLの脱イオン水を添加して、l0mg/mLの亜鉛結晶性インスリン溶液を生成した。12.5gのポリエチレングリコール3350(Sigma)と12.5gのポリビニルピロリドン(Sigma)を、50mLの100mM酢酸ナトリウム緩衝液(pH5.7)に溶解した。このポリマー溶液の容量を、酢酸ナトリウム緩衝液で100mLに調整した。エッペンドルフチューブ中の800マイクロリットルのポリマー溶液に、400マイクロリットルの10mg/mLインスリン溶液を添加した。このインスリン/ポリマー溶液は、混合すると濁りを生じた。ポリマー溶液の代わりに水を用いてコントロールを調製した。これらのエッペンドルフチューブを、水浴中で90℃にて30分間、混合または攪拌せずに加熱し、次いで取り出して氷上に10分間置いた。このインスリン/ポリマー溶液は、90℃水浴から取り出した際には透明であったが、冷めるにつれて濁り始めた。ポリマーを含まないコントロールは、実験を通して透明なままであった。粒子を、遠心分離によってこのインスリン/ポリマーチューブから回収し、続いて2回洗浄してポリマーを除去した。最後の水中の懸濁液を凍結乾燥して、乾燥粉末を得た。インスリン/ポリマーチューブからの凍結乾燥粒子のSEM分析により、直径約1マイクロメーターの小球状粒子の一様分布を示した。これらの粒子のコールター光散乱粒子サイズ分析により、1.413マイクロメーターの平均粒子サイズ、0.941〜1.88マイクロメーターの95%信頼限界、および0.241マイクロメーターの標準偏差を有する狭いサイズ分布を示した。ポリマーまたは洗浄工程を含まないが、別な方法で同様に処理および凍結乾燥されたインスリンコントロールは、SEM下で薄片のみ(粒子なし)を示し、外観上はタンパク質の凍結乾燥後に典型的に得られる薄片と類似していた。
【0096】
(実施例3:インスリン小球状粒子作製のためのプロセスを通しての連続フロー)
36.5mgのインスリンを量り取り、3mLの脱イオン水中に懸濁した。30μLの1N HClを添加してインスリンを溶解させた。この溶液の最終容量を、脱イオン水で3.65mLに調整した。次いで、7.3mLのPEG/PVP溶液(100mM NaOAc緩衝液中の25%PEG/PVP(pH5.6))をこのインスリン溶液に添加して、最終全容量が10.95mLのインスリン溶液にした。次いで、この溶液をボルテックスして、インスリンとPEG/PVPの均質な懸濁液を得た。
【0097】
このインスリン懸濁液を、テフロン(登録商標)チュービング(TFE内径1/32ゲージ可撓性チュービング)を介して、0.4mL/分の速度で作動中のBioRad蠕動ポンプに連結した。このポンプからのチュービングを、90℃に維持した水浴中に沈め、その後に氷中に埋めたコレクションチューブ中に挿入した。このインスリン溶液の温度が水浴中での約90℃から氷中の収集管中で約4℃に低下すると、インスリン小球状粒子が形成された。図7は、このプロセスの概略図である。このプロセスのための全実行時間は、10.95mL容量について35分間であった。小球状粒子を収集した後、この収集管を3000rpmで20分間、Beckman J6B遠心機中で遠心分離した。2回目の水洗を完了し、これらの小球状粒子ペレットを2600rpmで15分間遠心分離した。最後の水洗は1500rpmで15分間遠心分離した。アリコートを粒子サイズ分析のために取り出した。これらの小球状粒子を−80℃で凍結し、そして2日間凍結乾燥した。
【0098】
この粒子サイズを、容量で1.397μm、表面積で1.119μm、そしてBeckman Coulter LS230粒子カウンターによる決定のとおり、数で0.691μmと決定した。走査型電子顕微鏡写真により、サイズが均質であり凝集形成していないインスリン小球状粒子が示された(図8)。
【0099】
インスリン溶液を短期間90℃に曝露する連続貫流プロセスの使用により、小球状粒子の生成が可能になった。この方法により、高速液体クロマトグラフィー(HPLC)によって決定されるとおり90%タンパク質である最終組成物を生成した(図9)。HPLC分析によっても、溶解インスリン小球状粒子が約4.74分の溶離時間を有することを示し、これはインスリン標準または天然インスリン出発物質の溶離時間と有意には異ならず、小球状粒子へ成形加工後のインスリンの生化学的完全性の保存を示した。
【0100】
(実施例4:インスリン小球状粒子を作製するための熱交換バッチプロセス)
ヒト亜鉛結晶性インスリンを超音波処理で必要最低限量の脱イオン水中に懸濁して、完全分散を確実にした。このインスリン懸濁液を、0.1M酢酸ナトリウム緩衝液中における最終溶質濃度が0.83%亜鉛結晶性インスリン、18.5%ポリエチレングリコール3350、0.7%塩化ナトリウムとなるように、77℃に予熱した撹拌緩衝化ポリマー溶液(25℃でpH5.65)に添加した。初期に混濁した混合物は、結晶インスリンが溶解するにつれて3分以内に透明になった。清澄後直ちに、この溶液を、熱交換器(カラム内径:25mm、長さ:600mm;Ace Glass Incorporated,Vineland,NJ)として用いられるガラス製の水ジャケット付きクロマトグラフィーカラムに移した。このガラス製カラムを垂直に配置し、そしてこの熱交換機流体はカラムの下端で水ジャケットに入り、上端から出た。この系の熱交換特性を実証するために、熱電対(タイプJ、Cole Parmer)をカラムの上端および下端でインスリン処方物液体の中心に配置し、冷却温度プロフィールを予備試運転の間に得た。これらの熱電対を、外部表面変化が生じないように、この実験のために実施された6回のバッチの間に取り除いた。
【0101】
この熱交換器を65℃に予熱し、インスリン緩衝化ポリマー溶液を、この溶液温度が65℃未満に下がらず、かつ気泡がこの溶液中に導入されない方法で移した。この透明な溶液を熱交換器中で4分間65℃に平衡化させた後、熱交換流体を65℃供給から15℃供給に切り換えた。この熱交換器中のインスリン処方物を20分間にわたって15℃に平衡化させた。温度が60℃を経て55℃に下がるにつれてインスリン小球状粒子が形成され、その結果、均一な安定したクリーム状の白色懸濁液を生じた。
【0102】
これらのインスリン小球状粒子を、5倍容量の0.16%酢酸ナトリウム−0.026%塩化亜鉛緩衝液(pH7.0)に対するダイアフィルトレーション(A/G Technologies、750,000 MWCO限外濾過カートリッジ)によってポリエチレングリコールから分離し、続いて原体積の5分の1に濃縮した。このインスリン小球状粒子懸濁液を、5倍容量の脱イオン水に対するダイアフィルトレーションによってさらに洗浄し、続いて凍結乾燥して水を除去した。ダイアフィルトレーションの間の小球状粒子の凝集(膜表面上のこれらの粒子の分極パッキング)および凍結乾燥の間の小球状粒子の凝集(凍結前のこれらの小球状粒子の沈殿)が起こらないように気を付けた。これらの乾燥小球状粒子は易流動性であり、解凝集またはふるい分けの必要のない、使用準備の整った状態であった。
【0103】
(インスリンの小球状粒子)
上記のプロセスにより、均一なサイズの球状粒子が、賦形剤を添加することなく亜鉛結晶性インスリンから生成する。このプロセスによって調製された小球状粒子は、飛行時間計測(エアロサイザー(商標))およびアンダーセンカスケードインパクター計測によって決定されるように、単純な広く用いられている乾燥粉末吸入器(Cyclohaler(商標))から送達される場合に肺深部送達を示す高度呼吸可能画分を有し、優れた空気力学的性質を有する。インスリンをモデルタンパク質として用いることにより、本発明者らはまた、確立されたU.S.P.方法を用いてこのタンパク質の化学的完全性に対するこのプロセスの影響も検査し得る。
【0104】
乾燥粉末インスリン小球状粒子を、偏光顕微鏡法(Leica EPISTAR(登録商標),Buffalo,NY)によって、および走査型電子顕微鏡(AMRAY 1000,Bedford,MA)を用いてイメージ化した。粒子サイズ分析を、エアロサイザー(登録商標)モデル3292粒子サイジングシステム(この機器に粉末を導入するために、モデル3230 Aero−Disperser(登録商標)乾燥粉末分散器(TSI Incorporated,St.Paul,MN)を備える)を用いて行った。個々の粒子サイズを、エアロサイザーの結果を電子顕微鏡写真と比較することによって確認した。
【0105】
このプロセスの前後のインスリンの化学的完全性を、ヒトインスリンについてのUSPモノグラフ(USP26)に従いHPLCによって決定した。インスリンおよび高分子量タンパク質含量を、276nmでのUV検出を用いる定組成SEC HPLC法を用いて測定した。インスリン、A−21デサミドインスリンおよび他のインスリン関連物質を測定するために、USP勾配逆相HPLC法を用いてサンプルを分析した。インスリン含量を、214nmでのUV検出を用いて測定する。高分子量タンパク質、デサミドインスリン、および他のインスリン関連物質をアッセイして、このプロセスによって引き起こされる任意の化学分解を定量した。
【0106】
インスリン小球状粒子の空気力学的性質を、エアロサイザー(登録商標)機器を用いて検査した。インスリン乾燥粉末についてのサイズ分布測定を、低い剪断力、媒体供給量、および正常な解凝集でAeroDisperser付属装置を用いて行った。この機器のソフトウェアは、飛行時間計測データをサイズに変換し、それを対数的に間隔をあけた範囲に配置する。各サイズビン中で検出された粒子の数、ならびに各サイズビン中で検出された粒子の全体積を、統計分析に用いた。体積分布は数分布よりも大粒子を際立たせ、従って、体積分布は大粒子のみならず非分散粒子の凝集塊の検出時にもより感度が高い。
【0107】
アンダーセンカスケードインパクターアセンブリは、1つのプレセパレータ、9つのステージ、8つの収集プレート、および1つのバックアップフィルターで構成されていた。これらのステージを、−1、−0、1、2、3、4、5、6、およびFと番号付けする。ステージ−1は、オリフィスステージのみである。ステージFは、ステージ6のための収集プレートおよびバックアップフィルターを備える。ステンレス鋼収集プレートを、粒子の「はね返り」を防ぐために食品用シリコーンの薄層でコーティングした。サンプラーによる60LPMのサンプル流空気流速を分析に用いた。正確に秤量した約10mgのサンプルサイズを、4秒でCyclohalerからエアロゾルとして送達された粉末とともに、各デンプンカプセル(Vendor)中に秤量した。各プレート上に沈積したインスリン粉末の量を、ヒトインスリンについてのUSP26アッセイに従い214nmでの逆相HPLC検出によって決定した。
【0108】
空気動力学的中央粒子径(MMAD)を、有効カットオフ直径(ECD)に対する質量パーセント未満の累積性のプロビットフィットを用いるSigma Plotソフトウェアによって算出した。放射線量(ED)を、カスケードインパクター中に沈積したインスリンの全実測質量として決定した。これを、Cyclohalerカプセル中に充填されたインスリン小球状粒子の質量のパーセンテージとして表す。
【0109】
これらの結果により、相変化処方に関連して以下のプロセスパラメータの慎重な制御を実現し得ることを示す:1)約2μmの直径を有する主に球状の粒子;2)狭いサイズ分布;3)およびバッチ間再現性の空気力学的性質;および4)残留水分を除いて95%を超える活性薬物(ヒトインスリン)からなる小球状粒子。本発明者らは、亜鉛結晶性インスリンの溶解度が、溶液温度、pH、ポリマー濃度、およびイオン強度によって制御され得ることを決定した。本発明者らはまた、相変化期間中の冷却速度を制御することが、狭いサイズ範囲内に主に球状の粒子を形成することを可能にする重要なパラメータであることも見出した。
【0110】
図2は、本実施例に相当するプロセスのための冷却温度プロフィールである。このプロフィールを、垂直に配置した水ジャケット付きクロマトグラフィーカラムを用いて測定し、そしてこの熱交換流体は、カラムの下端で水ジャケットに入り上端から出た。2つの熱電対を、カラムの中に、この溶液に接触させて配置した。1つの熱電対をこのカラムの上端に配置し、2番目の熱電対をこのカラムの下端に配置する。これらの温度曲線は時間−温度プロットを別個の領域に分け、事前の最適化実験により、最適な温度変化速度よりも高いかまたは低い温度変化速度で相変化を誘導すると粒子サイズが広範囲になりかつ非球形状となる傾向があることを決定した。60℃を超える温度では、インスリンは緩衝化ポリマー溶液中に可溶性のままである(領域A;図2)。約8.6℃/分〜26.5℃/分の速度で温度が低下する場合、均一化されたサイズの球状粒子の最適形成が支持される(領域B;図2)。25.6℃/分よりも速い冷却速度がこの処方物に適用される場合、容易に凝集するインスリンの極めて微細な(0.5ミクロン未満の)非球状粒子を生成する傾向がある(領域C;図2)。8.6℃/分よりも遅い冷却速度により、非球状かつ非晶質の綿状沈殿とともに、より広いサイズ分布のインスリン小球状粒子を生成する傾向がある(領域D;図2)。
【0111】
熱交換器内部のインスリン緩衝化ポリマー溶液の温度が図2の領域Bの範囲内に入ると相変化が生じ、結果として乳白色の安定なインスリン小球状粒子の懸濁液を生じる。相分離を示すミクロスフェア形成は、温度が60℃を下回ると生じ始め、温度が40℃に達すると完了するようである。この処方物を、PEGポリマーを除去するためにダイアフィルトレーションによって洗浄する前に15℃まで冷却しても、懸濁液中にさらなる変化は観察されなかった。
【0112】
出発ヒト亜鉛結晶性インスリン原料のSEMが、約5〜40μmの粒子サイズを含む非均質サイズおよび結晶形状を示すのに対して、本実施例からのバッチの1つを撮影したSEM写真は、球状かつ均一なサイズのインスリン小球状粒子を示す(図3b)。SEMで示した粒子の形状およびサイズは、本実施例のために調製した他の5回のバッチの代表である。
【0113】
ダイアフィルトレーション洗浄による緩衝化ポリマーからの分離および脱イオン水懸濁液からの凍結乾燥の後、これらの乾燥粉末インスリン小球状粒子は、比較的易流動性であり、かつ秤量および取り扱いが容易であった。インスリン小球状粒子の含水率は、出発亜鉛結晶性インスリン原料について12%であったのと比較して、2.1〜4.4%の水分の範囲内で変動した。HPLCによるインスリン小球状粒子の化学分析は、高分子量化合物の増加が全くなく、このプロセスによるインスリンの化学分解は極めて少ないことを示した(図4)。ダイマー(%)、A21デサミドインスリン(%)、後期溶出ピーク(%)、および他の化合物(%)が(出発インスリン原料を超えて)増大したにもかかわらず、6回のバッチ全てについての結果はUSP限界の範囲内であった。インスリン効力の保持は、出発原料について28.7IU/mgであったのと比較して、28.3〜29.9IU/mgであった。このプロセスに用いられたポリマー(ポリエチレングリコール)の残留レベルは0.13%未満から検出不可能までであり、このことから、このポリマーはインスリン小球状粒子の重要な成分ではないことが示された。
【0114】
(インスリン小球状粒子についての空気力学的性質のバッチ間再現性)
エアロサイザーおよびアンダーセンカスケードインパクターデータによって実証されるように、生成された6回の別個のインスリン小球状粒子のバッチの間に、空気力学的性質について優れた再現性があった。6回のバッチ全てについて、エアロサイザーデータにより、99.5%を超えるこれらの粒子が0.63〜3.4μmのサイズ範囲内にあるとともに、最低限60%の小球状粒子が1.6〜2.5μmの狭いサイズ範囲内にあることを示した(図5)。統計学的に、このデータにより、生成したインスリン小球状粒子バッチの少なくとも99%がこれらの粒子の少なくとも96.52%を0.63〜3.4μmのサイズ範囲(目標直径2μmの−68.5%〜70%)内に有することは、95%信頼し得ることを示す。
【0115】
このアンダーセンカスケードインパクターデータは、Cyclohalerから送達される用量の平均17.6%がこれらの機器の開口部およびプレセパレータ/スロート中に沈積したことを除いては、エアロサイザーデータとよく一致した(図6)。このデータは、エアロサイザーの粉末分散効率がCyclohalerデバイスのものよりも大きいことを示す。しかしながら、これらの6回のバッチについての平均放射線量はCyclohalerに基づいて71.4%であったとともに、72.8%の放射線量がこのインパクターのステージ3上に沈積した。肺深部送達のための呼吸可能画分は、ECDを含む画分が1.1ミクロンと3.3ミクロンの間であると見積もられる場合、平均60.1%の吸入インスリン小球状粒子が肺深部送達およびその後の全身吸収に利用可能である。このプロセスについての優れた再現性を表1に示し、ここで、6回の別個のバッチについてMMAD平均およびGSD平均についての標準偏差値は極めて低い。このことは、このプロセス変量が厳格な制御下にあり、結果として空気力学的性質についてバッチ間均一性をもたらすことを示す。
【0116】
【表1】
表1は、インスリン小球状粒子の空気力学的性質を示す。結果(平均+/−SD)を、アンダーセンカスケードインパクターによる別個のインスリン小球状粒子バッチ(N=6)の分析に基づいて算出した。このプロセスについての非常に良好な再現性が、MMADおよびGSDについての極めて低い標準偏差によって実証される。
【0117】
この冷却プロセスによって生成したインスリン小球状粒子は、表1の空気力学的データによって証明されたように、凝集する傾向をほとんど示さなかった。
【0118】
(実施例5:インスリン小球状粒子を作製するための攪拌容器プロセス)
2880mLの緩衝化ポリマー溶液(0.1M酢酸ナトリウム緩衝液(2℃でpH5.65)中の18.5%ポリエチレングリコール3350、0.7%塩化ナトリウム)を、ガラス製の3リットルの水ジャケット付き攪拌容器に添加し、75℃まで予熱した。2.4グラムのヒト亜鉛結晶性インスリンをこの80mLの緩衝化ポリマー溶液中に超音波処理で懸濁して、完全分散を確実にした。このインスリン懸濁液を、攪拌して予熱した緩衝化ポリマー溶液に添加し、さらに5分間攪拌した。この混合物はこの間に透明になり、これは亜鉛結晶性インスリンが溶解したことを示していた。10℃に設定した冷却装置からの水を、このインスリンポリマー溶液が15〜20℃に低下するまで容器のジャケットを通してポンピングした。結果として生じた懸濁液を、5倍容量の0.16%酢酸ナトリウム−0.026%塩化亜鉛緩衝液(pH7.0)、続いて5倍容量の脱イオン水に対してダイアフィルトレーションし、続いて凍結乾燥して水を除去した。凍結乾燥粉末のSEM分析は、TSIエアロサイザー飛行時間計測分析により1.433マイクロメーターの平均空気力学的直径を有する均一な小球状粒子を示した。アンダーセンカスケードインパクター分析により、ステージ3からフィルターまでの上に沈積した放射線量が73%、MMADが2.2、およびGSDが1.6(全ての指標は、この粉末の優れた空気力学的性質を示す)を結果として得た。
【0119】
(実施例6:小球状粒子生成処方物のイオン強度を調整することによるインスリン分解生成物の形成の低下)
インスリンはまた、長期間または酸性環境でなければ、より低い初期温度(例えば、75℃)でこの溶液中に溶解され得るが、この溶液にNaClを添加することによって著しい凝集を生じる。
【0120】
インスリン小球状粒子の成形加工プロセスの改善を、以下の技術を用いて達成した。亜鉛結晶性インスリンの濃縮スラリーを、約85〜90℃に予熱した0.1M酢酸ナトリウム(pH5.65)中のポリエチレングリコールの16.7%溶液に(攪拌しながら)(室温にて)添加した。このインスリン結晶を、この温度範囲で5分間以内に完全に溶解した。この溶液の温度が低下するにつれて、インスリン小球状粒子が生成した。
【0121】
化学反応によるA21デサミドインスリンおよびインスリンダイマーの顕著な生成が、高温によって85〜90℃の初期温度で生じた。しかしながら、これは75℃では長時間を要した。長時間により、結果として顕著なインスリン分解も生じた。インスリンを酸性環境中に前溶解することにより、大きなパーセンテージのインスリンがA21デサミドインスリン分解生成物への望ましくない変換も生じた。
【0122】
実験中、化学的手段によってインスリンダイマーの形成を低下させるために、塩化ナトリウムを緩衝化ポリマー反応混合物に添加した。添加した塩化ナトリウムは、デサミドまたはダイマーインスリン分解生成物の形成を有意には低下させなかったが、塩化ナトリウムの添加はオリゴマー(高分子量インスリン生成物)の形成を大幅に低下させた(表2)。
【0123】
【表2】
さらに、Zn結晶性インスリンは、NaClを含まないコントロールよりもNaClの存在下でずっと速く溶解した。このことから、塩化ナトリウムの添加によりインスリンの溶解率が改善されることが示唆され、亜鉛インスリン結晶の初期溶解に用いられる温度の低下が可能になった。処方物への0.7%NaClの添加により、亜鉛結晶性インスリン原料を、以前にNaClを添加しなければ必要であった87℃よりも有意に低い温度である75℃で5分以内に溶解することが可能になることを実証した実験において、この仮説を確認した。75℃にて、NaClの非存在下では、インスリンは13分後に完全には溶解しなかった。
【0124】
一連の実験から、塩化ナトリウム濃度の増大(2.5mg/ml、5.0mg/ml、10.0mg/ml、および20.0mg/ml)により、インスリン結晶が溶解する温度がさらに低下し、そしてまた小球状粒子が形成を開始する温度も低下することを実証した(図l0a〜d)。さらに、この処方物中のNaCl濃度が増大すると、より高濃度のZn結晶性インスリンが急速に溶解することを決定した。従って、所定の温度でのインスリンの溶解度は、初期連続相の塩化ナトリウムレベルを調整することによって慎重に制御可能であることを確認した。これにより、分解生成物の形成があまり誘導されない温度でこのプロセスを誘導することが可能になる。
【0125】
塩化ナトリウムが、インスリンを溶解させる温度の低下を可能にする固有の化学的性質を有するか否かを決定するために、等モル濃度の塩化アンモニウムおよび硫酸ナトリウムを、塩化ナトリウムを含むコントロールと比較した。NH4ClおよびNa2S04の両方とも同じように亜鉛結晶性インスリン原料を溶解するのに必要な温度を低下させた。イオン強度が高いほど、溶液温度が低下するにつれて小球状粒子を形成する能力に影響を及ぼすことなく、ミクロスフェア生成処方物中のインスリンの溶解度を増大させるようである。
【0126】
(実施例7:インスリン小球状粒子の収率およびインスリン濃度およびサイズに対するPEG濃度の研究)
ポリエチレングリコール(3350)滴定データにより、PEG−3350の増大によっても小球状粒子の収率が増大することを示す。しかしながら、PEG濃度が高すぎると粒子の球形状が崩れ、収率のわずかな改善を無効にする。
【0127】
インスリン濃度データは、PEGと相反する傾向を示し、インスリン濃度が増大すると結果として小球状粒子の収率が低下する。
【0128】
本発明者らは、インスリン濃度が高いほど、より大きな直径の小球状粒子を生成するという一般的傾向を見出す。この実験において、濃度が高いほど、結果として非球状粒子と小球状粒子の混合も生じた。
【0129】
(実施例8:イヌを用いたインスリン小球状粒子研究)
この実験研究の目的は、ビーグル犬の肺におけるエアロゾル化インスリン粉末の沈積についての定量化および視覚化実験を実施することであった。本明細書中に開示される方法に従って99mTc標識インスリン粒子を作製した。エアロゾル化インスリンの肺沈積を、ガンマシンチグラフィーを用いて評価した。
【0130】
5匹のビーグル犬を本研究に用い、各動物に99mTc放射標識インスリン粒子エアロゾルを投与した。イヌの識別番号は、101、102、103、104、および105であった。
【0131】
エアロゾル投与の前に、これらの動物を、麻酔用の注入ラインを通してプロポフォールで麻酔し、エアロゾル送達のために各動物に気管内チューブを設置した。
【0132】
各イヌを、放射標識エアロゾルの吸入のために「スパングラーボックス」チャンバー中に入れた。放射標識エアロゾル投与の直後に、ガンマカメラコンピュータ画像を前側ならびに後側胸部領域について取得した。
【0133】
2つのインビトロカスケードインパクターコレクションを、1つは最初の動物(101)のエアロゾル投与前に、そしてまた最後の動物(105)の曝露後に評価して、99mTc放射標識インスリン粉末の安定性を確証した。
【0134】
これらの結果を図11に図示した。カスケードインパクターコレクションはいずれの場合も単峰形の分布を示した。
【0135】
図12は、全ての動物についてのP/I比計算の結果を示す。このP/I比は、肺の末梢部、すなわち肺深部に沈積する99mTcインスリン粉末の比率の尺度である。代表的なP/I比は、おそらく約0.7である。0.7を超えるP/I比は、中心肺と比較して末梢肺または気管支領域に顕著な沈積を示す。
【0136】
図13のシンチグラフィー画像は、呼吸器系内のインスリン沈積位置を示し、P/Iデータ(図12)と一致する。イヌ101についてのシンチグラフィー画像は、本研究において5匹全てのイヌを代表する。
【0137】
イヌ101についてのシンチグラフィー画像は、末梢肺に明白な沈積の増加を伴う気管または気管支の沈積をほとんど示さない。肺の外側の放射能は、エアロゾル化粉末の肺深部沈積からの99mTcの急速な吸収に起因する。
【0138】
これらのP/I比および画像データは、99mTc放射標識インスリンが主に肺深部に沈積したことを示す。末梢肺中に沈積した放射標識インスリンの量により、これらの粒子の凝集が低レベルであることを示した。
【0139】
(実施例9:インスリン小球状粒子からポリマーを除去するための亜鉛を含む緩衝液に対するダイアフィルトレーション)
PSEA溶液中のインスリン小球状粒子の成形加工に続いて、この懸濁液から全てのPSEAを除去した後に凍結乾燥することが望ましかった。残留PSEAがほんの数パーセントであっても、結合剤として作用し、小球状粒子の非破砕性凝集塊を形成し得る。この凝集は、DPIデバイスから送達される粉末の放射線量および空気力学的性質に悪影響を及ぼした。さらに、PSEAの反復投与への肺組織の曝露は、毒物学的問題を生じ得た。
【0140】
3つの技術を、凍結乾燥の前にPSEAから小球状粒子を分離するために検討した。濾過は、少量の粒子を収集するのに用いられ得た。しかしながら、小球状粒子がより多量であれば、濾過媒体の細孔を急速にブロックし、数ミリグラムを超える粒子の洗浄および回復が実行不能になる。
【0141】
これらの粒子を回収するために遠心分離(その後に、洗浄溶剤中の再懸濁および再遠心分離を含む数回の洗浄サイクルが続く)を首尾よく用いて、PSEAを除去した。これらのインスリン小球状粒子が容易に溶解されず、かつPSEAが溶液中に残ったので、脱イオン水を洗浄溶剤として用いた。遠心分離の1つの不都合は、小球状粒子が、これらの粒子をスピンダウンさせるのに必要な高いg力によってペレット中に詰まったことであった。連続的な洗浄ごとに、これらのペレットを分散した粒子に再懸濁することがますます困難になった。インスリン粒子の凝集は、しばしば遠心分離プロセスの望ましくない副作用であった。
【0142】
中空繊維カートリッジを用いるダイアフィルトレーションを、インスリン小球状粒子を洗浄するために遠心分離に代わる手段として用いた。ダイアフィルトレーション装置の従来のセットアップにおいて、緩衝化PSEA/インスリン粒子懸濁液を密閉容器中に入れ、そしてこの懸濁液を十分な背圧で繊維を通って再循環させ、濾液が中空繊維膜を通過するようにした。再循環速度および背圧を最適化して、膜の細孔の閉塞(分極)を防止した。懸濁液から除去された濾液の体積を、洗浄溶剤を攪拌密閉容器中に吸い上げることによって、連続的に補充した。ダイアフィルトレーションプロセスの間に懸濁液中のPSEAの濃度は徐々に低下し、そして約1時間にわたって懸濁液の原体積を洗浄溶剤で5〜7回交換した後には、このインスリン小球状粒子懸濁液は本質的にPSEAを含まなかった。
【0143】
ダイアフィルトレーションプロセスはポリマーの除去時に極めて有効であり、かつ商業量への拡大の影響を非常に受けやすかったが、インスリン小球状粒子は、洗浄溶剤として本来用いられた脱イオン水中にゆっくりと溶解した。実験により、インスリンは濾液中に徐々に失われ、そして懸濁液の原体積の20倍に相当する脱イオン水が交換された後にインスリン粒子が完全に溶解することを決定した。これらのインスリン小球状粒子は、脱イオン水中にやや溶けにくいことが分かったが、高性能のダイアフィルトレーションプロセスにより、懸濁液から可溶性インスリン(およびおそらく亜鉛イオン)が持続的に除去された。したがって、所定容量の脱イオン水中の不溶性インスリン濃度と可溶性インスリン濃度の間の平衡はダイアフィルトレーションでは生じず、インスリンの溶解に恵まれた条件となった。
【0144】
表3は、可能性のある洗浄媒体として評価された種々の溶液を示す。10mgの乾燥インスリン小球状粒子を1mLの各溶液中に懸濁し、室温で48時間徐々に混合した。可溶性インスリンのパーセンテージを24時間および48時間に測定した。このインスリンは、24時間足らずで可溶性インスリンの全重量のほんの1%未満で平衡に達し、脱イオン水中にやや溶けにくいことが見出された。しかしながら、先述のとおり、高性能のダイアフィルトレーションによって可溶性インスリン(および亜鉛)を連続的に除去するので、この平衡は決して達成されず、インスリン小球状粒子は溶解し続ける。従って、理想的な洗浄溶液中のインスリン溶解度は、水中のインスリン溶解度よりも低い。インスリンは、その等電点付近で可溶性が最も低いので、2種類のモル濃度のpH5.65の酢酸緩衝液を試験した。このインスリンの溶解度は、緩衝液のモル濃度に左右され、低モル濃度で水に相当することが見出された。エタノールはインスリンの溶解度を大いに低下させたが、ほぼ無水濃度でだけは低下させなかった。このインスリン溶解度は、水溶液と混合したエタノールをダイアフィルトレーションの初期段階においてPSEA/インスリン小球状粒子懸濁液中に用いた場合、実際に増大する。
【0145】
【表3】
注射用の市販亜鉛結晶性インスリン懸濁液中に用いられる緩衝液もまた、溶液中に亜鉛を含む。これらの溶液の2つをインスリン小球状粒子を用いて試験し、脱イオン水と比較してインスリン溶解度を大いに低下させることを見出した。文献によると、亜鉛結晶性インスリンは、各インスリン六量体に結合した2〜4個のZnイオンを有するべきである。インスリン小球状粒子を作製するための原料として用いられる種々の亜鉛結晶性インスリン調製物について、六量体当たりの亜鉛イオンは1.93〜2.46に及ぶ。これは、所定重量の原料亜鉛結晶性インスリン当たり0.36〜0.46%亜鉛に相当した。インスリン小球状粒子の形成および脱イオン水に対するダイアフィルトレーションの後、58〜74%の亜鉛がプロセシングの間に失われた。インスリン粒子からの亜鉛の損失により、インスリンの溶解度の増大およびダイアフィルトレーション中の損失を生じた。
【0146】
0.16%酢酸ナトリウム−0.027%ZnCl2(pH7.0)に対してインスリン小球状粒子をダイアフィルトレーションすることにより、濾液中のインスリン損失を事実上排除した。しかしながら驚くべきことに、これらのインスリン小球状粒子の亜鉛含量は、出発亜鉛結晶性インスリン原料について測定した0.46%を十分に上回る、ほぼ2%まで増加した。亜鉛を含む緩衝液に対するダイアフィルトレーションの別の予想外の結果は、Cyclohaler DPIデバイス(脱イオン水に対してダイアフィルトレーションされた68% 対 亜鉛緩衝液ダイアフィルトレーション後の84〜90%)に基づいて観察された放射線量の劇的な改善およびアンダーセンカスケードインパクターのスロート中に沈積されたインスリン粒子の量の減少であった。亜鉛緩衝液ダイアフィルトレーションは、インスリン小球状粒子乾燥粉末の分散性を改善し、そして粒子の凝集を低下させ、結果としてインパクターのより低いステージ上で、より低いMMADおよびより高い沈積をもたらした。このことは、亜鉛緩衝液ダイアフィルトレーションおよびインスリン小球状粒子中のより高い亜鉛濃度により、肺深部に沈積した線量の割合(%)を改善し得ることを示唆した。
【0147】
MDI適用で用いる賦形剤を添加せずに噴霧剤HFA−134a中に懸濁した場合、亜鉛緩衝液で洗浄したインスリン小球状粒子の不可逆的凝集は明白には存在しなかった。これらのインスリン粒子は1分足らずで懸濁液から凝集したが、使用直前に振盪すると容易に再懸濁された。使用直前にMDI容器を振盪する工程は、通常、任意のMDI生成物を使用するために提供された使用説明書の一部である。実際に、これらの粒子はMDI加圧容器の底で高密度充填層に定着しないので、MDI容器の底に定着する遊離した凝集粒子はインスリン粒子の長期凝集を(球形状に起因する最小限の接触に加えて)事実上阻害し得る。従って、インスリン小粒子の亜鉛緩衝液ダイアフィルトレーションによって付与された性質により、インスリンおよび他の亜鉛結合化合物のためのMDI調製物の長期有効期間および分散性を改善し得る。
【0148】
インスリン小球状粒子はXRPD分析によって非晶質であることが見出されたので、亜鉛結合は、インスリンモノマーの亜鉛イオン配位で会合せずに六量体を形成した。従って、イオンの非特異的結合および得られた潜在的利点が、亜鉛以外のイオンの結合にまで及び得た。亜鉛と結合しない種々のタンパク質は、ダイアフィルトレーションプロセスにおいて溶解度を低下させる他のイオンに結合し、類似の有益な効果を付与し得た。
【0149】
これらの小球状粒子を10mg/mLの濃度でヒドロフルオロアルカン(HFA)134a噴霧剤中に懸濁した。HFA134a中に貯蔵後のインスリンの化学安定性を、時間0および1ヶ月目に評価した。図28に示されるデータは、モノマーインスリン、インスリンダイマー、インスリンオリゴマー、インスリン主要ピークおよびA21−デサミドインスリンに関してインスリンミクロスフェアの保存を示す。
【0150】
以下の研究において、実施例4の方法に従って調製したインスリン小球状粒子を、アンダーセンカスケードインパクター法を用いて3つの異なる吸入デバイスでの性能について比較した。Cyclohalerデバイスは市販の乾燥粉末吸入器(DPI)であり、Disphalerは別の乾燥粉末吸入器であり、そして定量噴霧式吸入器(MDI)は、ミクロスフェアが本実施例に記載のようにHFA134a中に懸濁されて100マイクロリットルかまたは他のサイズの絞り弁を通って推進されるデバイスである。図29における結果は、アンダーセンカスケードインパクターデバイスのステージに嵌入する小球状粒子がステージ3および4上に沈積することを明らかに示す。このことは、吸入器として用いられるデバイスにかかわらず、極めて再現性のある小球状粒子の性能を示す。DPIデバイスとMDIデバイスの間の唯一の重大な相違は、MDIを用いてアンダーセンカスケードインパクターのスロート部分中に沈積した小球状粒子の量が有意に大きいことである。このMDIデバイスがアンダーセンインパクターのスロートに対して小球状粒子を推進させる高速度は、DPIデバイスと比較して沈積したインスリンミクロスフェアの割合が高いことを説明する。流出速度を減じたかまたは改変したMDIデバイスを用いてスロート中に沈積する小球状粒子の数を減少させ得ることは、当業者によって当然のこととみなされ得る。さらなる手段は、MDIの末端部でのスペーサーデバイスの使用であり得る。
【0151】
インスリン小球状粒子(ロット番号YQ010302)を、本実施例に記載の方法に従って凍結乾燥インスリン出発物質から作製した。インスリン小球状粒子についての1年間の貯蔵安定性を、25℃および37℃で凍結乾燥インスリン出発物質と比較した。インスリン安定性を、全関連インスリン化合物、インスリンダイマーおよびオリゴマーならびにA21−デサミドインスリンを試験することによって比較した。
【0152】
図30〜35は、1年間にわたって、インスリン小球状粒子は同一条件下で貯蔵したインスリン出発物質と比較して有意に少ない量のインスリンダイマーおよびオリゴマー、A21−デサミドインスリンならびに全関連インスリン化合物を示したことを示す。これにより、インスリンのミクロスフェア形態は出発物質よりも化学変化に対して有意に安定性が高いことを示す。
【0153】
インスリン小球状粒子を製造後0時間および10ヶ月後にアンダーセンカスケードインパクター研究において試験した。Cyclohaler DPIデバイスを用いて長期貯蔵後の空気力学的安定性を決定した。図36は、空気力学的性能が10ヶ月貯蔵後に著しく不変のままであることを示す。
【0154】
ラマン分光研究を行って加工前のインスリンサンプルと本実施例で調製した小球状粒子中のインスリンとの間の構造的相違を明らかにした。小球状粒子中のインスリンは、これらの元の加工前インスリンサンプルよりも実質的により高いβ−シート含量を有し、その結果、より低いα−ヘリックス含量を有することを示した。これらの研究結果は、小球状粒子中における凝集ミクロフィブリル構造物の形成と一致する。しかしながら、水性媒体中に溶解すると、このスペクトルは、加工前のミクロスフェアまたはインスリンのいずれかから生じる本質的に同一のタンパク質構造物を顕示し、ミクロスフェア中の任意の構造変化が溶解に際して十分に可逆的であることを示す。
【0155】
以下の2つのバッチのインスリンを、ラマン分光法を用いて試験した:A)加工前のインスリンUSP(Intergen、カタログ番号4502−10、ロット番号;XDH1350110)およびB)小球状粒子中のインスリン(JKPL072502−2NB32:P.64)。これらの粉末状サンプルまたはインスリン溶液(0.01MのHCl中に約15mg/mL)を標準ガラス毛細管中に詰め、ラマン分析のために12℃に温度調整した。代表的には、2〜15μLのアリコートは、レーザー照明に曝露されたサンプル毛細管の部分を満たすのに十分であった。スペクトルをアルゴンレーザー(Coherent Innova 70−4 Argon Ion Laser,Coherent Inc.,Santa Clara,CA)を用いて514.5nmで励起させ、光子含有検出器(モデルR928P,Hamamatsu,Middlesex,NJ)を用いて走査型二重分光計(Ramalog V/VI,Spex Industries,Edison,NJ)に記録した。1.0cm−1間隔でのデータを、1.5秒の組み込み時間および8cm−1のスペクトルスリット幅で収集した。サンプルを繰り返しスキャンし、そして個々のスキャンを平均する前に表示しそして検討した。代表的には、各サンプルの少なくとも4回のスキャンを収集した。分光計をインデンおよび四塩化炭素を用いて較正した。スペクトルを、SpectraCalcおよびGRAMS/AIバージョン7ソフトウェア(Thermo Galactic,Salem,NH)を用いてデジタル微分法によって比較した。このスペクトルを、溶剤(もしあれば)およびバックグラウンドの寄与について補正した。これらの溶液のスペクトルを、同一条件下で0.01MのHClスペクトルを取得することによって補正し、傾斜したバックグラウンド上に位置する一連の5つの重複したガウス関数−ローレンツ関数に適合させた[S.−D.Yeo,P.G.Debenedetti,S.Y.Patro,T.M.Przybycien,J.Pharm.Sci.,1994,83,1651〜1656]。この適合を1500〜1800cm−1領域で行った。
【0156】
ラマンスペクトルを、粉末状インスリンサンプルおよびそれらの各溶液の両方について得た(図10i)。加工前のサンプルのスペクトルは、市販のインスリンサンプルの前述のスペクトルに非常によく一致する[S.−D.Yeo,P.G.Debenedetti,S.Y.Patro,T.M.Przybycien,J.Pharm.Sci.,1994,83,1651〜1656;J.L.Lippert,D.Tyminski,P.J.Desmueles,J.Amer.Chem.Soc.,1976,98,7075〜7080]。この小球状粒子サンプルは、タンパク質の二次構造において有意な摂動を示す、アミドIモードにおける明白な(約+10〜+15cm−1)シフトを示した。しかしながら、特に、市販の粉末および小球状粒子のスペクトルは、これらのサンプルが水性媒体中に溶解した場合に事実上同一であり、加工の際の二次構造の変化が完全に可逆的であったことを示していた。
【0157】
二次構造パラメータを、平滑化、蛍光および芳香族バックグラウンドの減算、およびアミドIバンドデコンヴォルーションを含むコンピュータアルゴリズムを用いて推定した。指数関数的に減衰する蛍光を、本質的に他に記載されるように減算した[S.−D.Yeo,P.G.Debenedetti,S.Y.Patro,T.M.Przybycien,J.Pharm.Sci.,1994,83,1651〜1656]。推定した構造パラメータを表4に集結する。
【0158】
【表4】
(実施例10:等温法によるヒトインスリンの小球状粒子の調製)
ヒトインスリンUSP(Intergen)をNaClおよびPEG(分子量3350、スペクトルロット番号RP0741)溶液中に分散させ、結果として最終インスリン濃度0.86mg/mL、ならびに0.7重量%のNaCl濃度および8.3重量%のPEG濃度となった。微量の氷酢酸および1MのNaOH溶液の添加によってpHを5.65に調整した。T1=77℃まで加熱した後、結果としてインスリン濃度Ceqの透明なタンパク質溶液を得た。次いで、これらの溶液を所定の速度で温度T2=37℃まで冷却した。T2で、タンパク質沈殿を認めた。これらの沈殿を遠心分離(13,000×g、3分間)によって再び温度37℃で除去し、この生じた上清中のインスリン濃度(C*)をビシンコニンタンパク質アッセイによって0.45mg/mLと決定した。このようにして調製した、37℃に保持されているインスリン溶液を、溶液Aと称する。
【0159】
溶液Bを、0.7重量%NaCl/8.3重量%PEG(HClの添加によってpHを約2.1にした)中にヒトインスリンを溶解させることによって調製し、結果として2mg/mLのインスリン濃度となった。この溶液を7時間攪拌しながら37℃でインキュベートし、続いて2分間超音波処理した。生じた溶液Bのアリコートを溶液Aに添加し、結果として1mg/mLの全インスリン濃度となった。生じた混合物を37℃で一晩激しい攪拌下に保持し、結果としてインスリン沈殿を生じ、これらの沈殿をメンブランフィルター(有効ポア直径、0.22μm)を用いて液体から穏やかに除去した。次いで生じたタンパク質ミクロ粒子を液体窒素中で急速凍結させて、凍結乾燥させた。
【0160】
(B.α−1−アンチトリプシン(AAT)の小球状粒子)
本発明はまた、肺送達に特に適したAATの小球状粒子を調製するのにも用いられ得る。
【0161】
(実施例11:AAT小球状粒子(10〜300mgスケール)のジャケット付きカラムバッチ調製)
16%PEG3350および0.02%プルロニックF−68を含むl0mM酢酸アンモニウムでpH6.0に緩衝化した溶液をジャケット付きビーカー中でマグネティック攪拌棒を用いて混合し、かつ30℃に加熱した。このビーカー温度を、循環水浴を用いて制御した。組換えAAT(rAAT)の濃縮溶液をこの溶液に攪拌しながら添加し、そしてpHを6.0に調整した。最終溶液中のrAAT濃度は2mg/mlであった。rAATは、この溶液組成物中にこの温度で完全に可溶性であった。この容器の全内容物をジャケット付きカラムに移し、そして25〜30℃に加熱した。このカラムのための循環水浴をセットして−5℃まで下降させた。このカラムおよび内容物を約1℃/分で約4℃の温度まで冷却した。rAAT小球状粒子はこの冷却工程の間に形成された。ミクロスフェア懸濁液をガラス結晶皿中で凍結させ、そして凍結乾燥して水および緩衝液を除去した。
【0162】
凍結乾燥後のタンパク質小球状粒子からPEGを抽出するために、PEG/タンパク質ケークを塩化メチレン(MeCl2)で洗浄した。利用される別の洗浄媒体は、塩化メチレン:アセトン1:1または塩化メチレン:ペンタン1:1であった。この洗浄工程は、原体積洗浄を合計3回繰り返した。最終ペレットを小容量のアセトンまたはペンタン中に再懸濁し、窒素ガスへの直接曝露かまたはロータリーエバポレーションのいずれかによって乾燥させた。
【0163】
(実施例12:AAT小球状粒子(200〜2000mgスケール)のジャケット付き容器バッチ調製)
このタイプの調製を、ジャケット付きカラムと同じ処方組成物を用いて行ったが、より大きな容量に適応可能であり、スケールアップにより適していた。このスケールで、処方物をジャケット付き容器(通常500〜1000ml)中でA形のパドル式インペラーを用いて75rpmで混合し、そして30℃に加熱した。この容器温度を循環水浴を用いて制御した。同じ容器中に溶液を保持して、水浴源を30℃浴から2℃浴に切り換えた。この容器および内容物を約1℃/分で4℃の温度まで冷却した。rAAT小球状粒子はこの冷却工程の間に形成された。この温度を、熱電対を用いてモニタリングし、この懸濁液が4℃に達すると、ほぼこの温度をさらに30分間保持した。この保持工程の後、この小球状粒子懸濁液をダイアフィルトレーションによって約4℃で濃縮し、ポリマーおよび容量の約75%を除去した。残りの小球状粒子懸濁液を予冷した凍結乾燥トレー中で薄層として凍結し、そして凍結乾燥して水および残りの緩衝液を除去した。
【0164】
これらのタンパク質小球状粒子を、有機溶剤で遠心分離することによって(実施例10に記載されるように)または超臨界流体(SCF)抽出によって、残った乾燥ポリマーから分離した。SCF抽出のために、この乾燥物質を高圧抽出チャンバー中に移し、このチャンバーをCO2で2500psi(室温で)まで加圧した。一旦作業圧に達すると、エタノールを、70:30のC02:エタノール混合物として吸込流体流に導入した。この超臨界流体は、小球状粒子を残してポリマーを溶解した。このプロセスの終わりに、このシステムをエタノールで洗い流し、ゆっくりと除圧した。
【0165】
(実施例13:プロセス収率−rAATの小球状粒子への変換%)
小球状粒子を実施例10および11に記載のように作製した。冷却プロセスが完了した後、この懸濁液の小アリコートを取り出し、そして0.2μmのシリンジフィルターを通して濾過して固体小球状粒子を除去した。溶液中に残ったrAATである、濾液の吸光度を、UV分光光度計を用いて280nmで決定した。次いで、rAAT濃度を検量線から算出した。変換%を以下のように算出した:
(出発rAAT濃度−濾液rAAT濃度)/出発rAAT濃度*100%=変換%
【0166】
【化2】
上の表に示すように、プロセススケールにかかわらず高いパーセンテージのAATタンパク質を小球状粒子に変換した。
【0167】
(実施例14:種々のプロセススケールでのAAT粒子の粒子サイズ分布)
(エアロサイザーデータ)
最終的なAAT乾燥粉末小球状粒子のサンプルを、飛行時間計測によって粒子サイズを測定する、TSIエアロサイザー3225で分析した。これらの測定値から種々の比の体積粒径を算出して、AAT小球状粒子の粒子サイズ分布を実証し、かつ本発明の方法以外の方法で作製した粒子と比較するのに用いた。
【0168】
【化3】
(アンダーセンデータ)
5〜l0mgのサンプルをゲルカプセル中に秤量し、そしてCyclohaler乾燥粉末吸入器を用いて流速60リットル/分(LPM)でアンダーセンカスケードインパクター中に投与した。小球状粒子を全てのインパクターステージから収集し、0.2Mのトリス塩酸緩衝液中にpH8.0で溶解し、そして逆相HPLCを用いて定量した。このデータを分析し、そして幾何学的標準偏差(GSD)を米国薬局方(USP)に記載のように算出した。このデータは狭いサイズ分布を実証した。
【0169】
【化4】
前掲の全ての分布パラメータは、本発明の成形加工方法によってもたらされる優れた粒子サイズ分布を実証した。
【0170】
(実施例15:AAT生物活性の保持)
比活性を決定するために、rAAT小球状粒子を0.2Mのトリス塩酸(室温でpH8.0)中に溶解した。生じた溶液を、C末端にp−ニトロアニリド基を含む合成ペプチドを加水分解する、ブタ膵エラスターゼ(PPE)の能力を阻害する、rAATの能力を測定するアッセイによって分析した。次いで、rAAT小球状粒子の同じ溶液を、タンパク質濃度についてビシンコニン酸(BCA)アッセイを用いてアッセイした。コントロールrAAT出発物質溶液もまた両方のアッセイで分析した。活性アッセイを進行させて1mg/mlのタンパク質/サンプルの濃度に基づいて活性を決定したので、BCAによって決定されるような実際のタンパク質濃度に基づいて活性値を補正し、以下の比活性値を得た:
【0171】
【化5】
rAATによるブタ膵エラスターゼの阻害
【0172】
【化6】
従って、この比活性は、小球状粒子へのAATの成形加工後の生物活性の保持を実証した。
【0173】
(実施例16:AATの構造完全性の保持)
制御相分離(CPS)技術の重要な識別ポイントの1つは、粒子形成の間に水性系を利用し、かつ他のストレス誘導性の条件(例えば、温度の上昇、剪断など)を回避する、温和な条件下での粒子の形成である。粒子工学の分野において、主要な関心事は、成形加工中のタンパク質の安定性および貯蔵安定性である。タンパク質の主要な分解経路、例えば酸化、脱アミドおよび特に凝集は、免疫原性を含むタンパク質処方の副作用を引き起こすと考えられる。従って、調節の関係上、最終粒子処方物中の分解生成物は極めて低レベルであることが必要である。HPLC、例えばCDおよびDSCのような物理的化学的特性決定を利用して、形成の間にタンパク質修飾が起こったか否かを決定した。
【0174】
円偏光二色性(CD)は、摂動を受けたタンパク質の構造変化の評価、または親タンパク質に対する改変タンパク質の構造比較に通常用いられる。このCD法は、タンパク質の折り畳み、ならびにタンパク質の二次構造および三次構造を評価する。
【0175】
二次構造は、「遠紫外線」スペクトル領域(190〜250nm)におけるCD分光法によって決定され得る。これらの波長では、発色団は、通常の折り畳み環境中にある場合、ペプチド結合である。α−ヘリックス、β−シート、およびランダムコイル構造はそれぞれ特徴的な形状およびサイズのCDスペクトルを生じる。従って、任意のタンパク質中に存在する各二次構造型の概算比率を、その遠紫外線CDスペクトルを分析することによって、各構造型についてのこのような参照スペクトルの分数倍数の合計として決定し得る。
【0176】
「近紫外線」スペクトル領域(250〜350nm)におけるタンパク質のCDスペクトルは、ある態様の三次構造に高感度であり得る。これらの波長では、発色団は芳香族アミノ酸およびジスルフィド結合であり、これらの発色団が生成するCDシグナルはタンパク質の全般的な三次構造に高感度である。250〜270nmの領域のシグナルはフェニルアラニン残基に起因し、270〜290nmのシグナルはチロシンに起因し、そして280〜300nmのシグナルはトリプトファンに起因する。ジスルフィド結合は、近紫外線スペクトル全体にわたって幅広の弱いシグナルを生じる。
【0177】
rAATストック溶液の遠紫外線CDスペクトルおよびリン酸緩衝液(pH7.4、T=25℃、タンパク質濃度0.05mg/mL)中の小球状粒子から放出されたAATを図13に示す。各スペクトルは10回のスキャンの平均を表す。
【0178】
遠紫外線CDスペクトルは識別不能であり、このことは、AATの小球状粒子への成形加工が引き続く放出の際に出発AAT物質の構造と同一の構造を有するAAT分子を結果として生じたことを実証している。
【0179】
(RP−HPLC)
小球状粒子を0.2Mのトリス塩酸中にpH8.0で溶解し、逆相HPLCによって分析した。出発rAATタンパク質のコントロール溶液と比較した場合、クロマトグラムの外観上に明らかな相違はない。
【0180】
HPLCシステム:
HPLCカラム−Pheomenex Jupiter、5ミクロン、C4、300A、250×4.6mm
Waters Alliance 2965ポンプ/オートサンプルラー
波長−280nm
注入容量−75μl
濃度勾配:
移動相1:0.1%TFA/水
移動相2:0.085%TFA/90%(c/v)アセトニトリル/水
実行時間−60分
流速−1.0ml/分
DSC
DSCダイアグラムを作成した。図15〜25bを参照のこと。
【0181】
(実施例17:AAT出発物質の貯蔵安定性に対するAAT小球状粒子の貯蔵安定性)
小球状粒子を、室温および4℃で1週間、1ヶ月間、2ヶ月間、3ヶ月間、6ヶ月間、および12ヶ月間貯蔵後の生物活性の保持について(実施例15に記載のアッセイを用いて)分析した。(図14bおよび14c)。このバルク材料は、rAAT出発溶液を透析し、次いで凍結乾燥したものである。各々の時点および貯蔵条件について、各々二連でアッセイした二連のサンプルが存在した。
【0182】
(C.ヒト成長ホルモン(hGH)の小球状粒子)
本発明はまたhGHの小球状粒子を調製するのにも用いられ得る。
【0183】
(実施例18:hGHの小球状粒子の試験管バッチ調製(20〜50mgスケール))
18%PEG3350を含むpH5.6で緩衝化した溶液(50mM酢酸アンモニウム/50mM重炭酸アンモニウム)(この溶液中におけるhGHの最終濃度は1mg/mlである)を50mlコニカルチューブ中で混合し、固定水浴中で58℃に加熱した。hGHをこれらの条件下でこの溶液中に溶解した。次いで、このチューブを水浴から取り出し、この溶液が10℃に達するまで氷浴中で冷却した。冷却速度を4〜6℃/分に維持した。hGHタンパク質小球状粒子はこの冷却工程の間に形成した。小球状粒子は、溶液の温度が約40℃に達すると形成し始めた。粒子形成後、hGHタンパク質小球状粒子を以下に記載の2つの方法のうち1つによってPEGから分離した。
【0184】
有機溶剤洗浄には、冷却工程および粒子形成の後に小球状粒子懸濁液を液体窒素で瞬間凍結し、そして凍結乾燥して水および緩衝液を除去することが必要であった。凍結乾燥後にPEGからタンパク質小球状粒子を分離するために、PEG/タンパク質ケークを塩化メチレン(MeCl2)中に懸濁した。PEGはMeCl2において可溶性であり、一方タンパク質小球状粒子は不溶性である。この懸濁液を室温で5分間混合した。hGH小球状粒子の密度はMeCl2の密度(d=1.335g/ml)に近いので、この液体密度を低下させて遠心分離を促進するために第2溶剤が必要であった。MeCl2と混和性のアセトンを、MeCl2の容量と等しい容量で添加した。次いで、この小球状粒子懸濁液を室温にて5分間3300rpmで遠心分離した。上清を捨て、ペレットをMeCl2中に再懸濁し、そして再び室温で5分間混合した。この洗浄手順を合計5回の洗浄の間繰り返した。最終洗浄の後、このペレットを小容量のMeCl2中に再懸濁し、ロータリーエバポレーションによって乾燥し、hGH小球状粒子の最終粉末を取り出した。
【0185】
亜鉛緩衝液洗浄には、冷却工程および粒子形成の後に、小球状粒子懸濁液を4℃にて10分間4000rpmで遠心分離して小球状粒子をPEGから分離することが必要であった。上清を除去し、このペレットを、除去した上清の容量と等しい容量の、50mMの酢酸亜鉛を含む冷緩衝液中に再懸濁した。Zn2+イオンはhGHの溶解度を低下させ、洗浄の間の溶解を阻止した。この洗浄緩衝液を氷上に保持した。次いで、この懸濁液を直ちに4℃にて5分間3000rpmで遠心分離した。この上清を除去し、そして亜鉛緩衝液洗浄を合計3回繰り返した。3回の亜鉛緩衝液洗浄に続いて、ペレットを水中で2回洗浄し、そして4℃にて5分間3000rpmで遠心分離して過剰の亜鉛を除去した。最後の水洗に続いて、ペレットを小容量の水に再懸濁し、そして液体窒素を用いて瞬間凍結した。次いで、凍結ペレットを凍結乾燥して水を除去し、hGH小球状粒子の最終粉末を取り出した。
【0186】
(実施例19:hGHの小球状粒子のジャケット付き容器バッチ調製(100mgスケール))
このタイプの調製を、実施例18と類似の処方組成物を用いて行ったが、より大きな容量に適応可能であり、スケールアップにより適している。
【0187】
18%PEG3350および0.02%プルロニックF−68を含む、pH6.1で緩衝化した溶液(80mM酢酸アンモニウム/l0mM重炭酸アンモニウム)をオーバーヘッドインペラーを用いてジャケット付きビーカー中で混合し、58℃に加熱した。この混合物温度を、循環水浴を用いて制御した。hGHの高濃度溶液を、攪拌しながらこの溶液に添加した。この溶液中のhGHの最終濃度は1mg/mlであった。hGHは、この溶液組成物においてこの温度で完全に可溶性であった。次いで、この容器および内容物を8℃/分の速度で約10℃の温度まで冷却した。hGH小球状粒子はこの冷却工程の間に形成された。これらの小球状粒子は約40℃で形成し始め、そしてこのプロセスを、懸濁液をさらに冷却しながら継続した。冷却工程の後、小球状粒子を実施例20aに記載の2つの方法のうち1つによってPEGから分離した。
【0188】
(実施例20:hGHの完全性の保持)
小球状粒子中のhGHのタンパク質完全性を、このプロセスの以下の段階で評価した:粒子形成後、PEG抽出後、および溶剤除去後または乾燥後。小球状粒子への成形加工後のhGHの化学的完全性の測定を、凝集および分解生成物を定量化するためにHPLCアッセイ(サイズ排除クロマトグラフィー(SEC)、逆相(RP))を用いて決定した。結果から、小球状粒子形成プロセスの間に凝集塊または他の関連物質の有意な蓄積はなかったことが証明された。
【0189】
a.有機溶剤洗浄
サイズ排除によるhGH凝集:出発物質を超える凝集の増大
【0190】
【化7】
逆相によるhGH関連物質:出発物質を超える分解の増大
【0191】
【化8】
b.亜鉛緩衝液洗浄
サイズ排除によるhGH凝集:出発物質を超える凝集の増大
【0192】
【化9】
逆相によるhGH関連物質:出発物質を超える分解の増大
【0193】
【化10】
(実施例21:hGHの小球状粒子の粒子サイズ分布)
小球状粒子の粒子サイズ分布の特性決定を、TSIエアロサイザーを用いる空気力学的飛行時間計測(図26)によって、および走査型電子顕微鏡(図27)によって決定した。
【0194】
(実施例22:hGH小球状粒子の溶解速度論)
2つの異なる抽出手順を受けたhGH小球状粒子の溶解速度論を比較した。
【0195】
有機溶剤で洗浄したhGH小球状粒子を、hGH出発物質と同様に水性媒体中に直ちに溶解した。
【0196】
hGH小球状粒子を亜鉛緩衝液で洗浄した場合、溶解度が低下した(図28)。hGH小球状粒子の溶解を、10mMトリス、154mMのNaCl、0.05%のBrij35(pH7.5)中に37℃で行った。タンパク質のより完全な放出が、インビトロで他の媒体中で達成された。溶解速度論は、全hGHの約30%が最初の15分に放出し、そして約50%が最初の24時間に放出したことを実証した。このタンパク質放出は1ヶ月で完了した。小球状粒子溶解が二相様式で進行したという事実は、結果としてインビボで一部の遅延放出をもたらし得る。
【0197】
(D.リゾチーム小球状粒子)
(実施例23.リゾチームの小球状粒子の調製)
以下の溶液:1.6mg/mlのリゾチーム、13.2%のPEG3350、55mMの酢酸アンモニウム(pH9.5)53mMの硫酸アンモニウム、263mMの塩化ナトリウム、26mMの塩化カルシウム。
【0198】
このPEG緩衝液を40℃に加熱した(pH9.55)。生じた懸濁液を液体窒素中で瞬間凍結し、そして多岐管凍結乾燥器で凍結乾燥した。小球状粒子が形成された。
【0199】
(E.DNase小球状粒子)
(実施例24.DNaseの小球状粒子の調製)
処方例:以下の溶液:0.18mg/mlのDNase(1mg/mlストックから)、18.2%のPEG3350(25%ストックから)、9mMの酢酸アンモニウム(pH5.15)(1Mストックから)。
【0200】
この懸濁液を−80℃フリーザー中で冷却し、凍結するとすぐに、多岐管凍結乾燥器で凍結乾燥させ、続いてMeCl2/アセトンで遠心分離によって洗浄した。
【0201】
試験済みの初発濃度は0.1mg/mlのDNaseおよび20%のPEG3350であった。しかし、37℃から0℃まで冷却を試みた後に沈殿が得られない場合は、新たな量のDNaseを添加して上記濃度に至った。この溶液を−80℃フリーザー中で冷却し、凍結するとすぐに、多岐管凍結乾燥器で凍結乾燥させた。MeCl2/アセトンで遠心分離によって洗浄した。試験済みの初発濃度は0.1mg/mlのDNaseおよび20%のPEG3350であった。しかし、37℃から0℃まで冷却を試みた後に沈殿が得られない場合は、新たな量のDNaseを添加して上記濃度に至った。この溶液を−80℃フリーザー中で冷却し、凍結するとすぐに、多岐管凍結乾燥器で凍結乾燥させた。MeCl2/アセトンで遠心分離によって洗浄した(図37、38)。
【0202】
活性(Sigmaから購入したDNA−メチルグリーンを用いるDNase−Iについてのアッセイ)
出発物質についての理論活性を775Ku/mgタンパク質としてリストに記載する。このストック溶液を0.145mg/mlのタンパク質と決定した。この濃度を最終濃度が0.0199mg/mlとなるように5mlに希釈した。この活性は775Ku/mg*0.0199mg/ml=15.46Ku/mlであるはずである。
【0203】
【化11】
Ku/ml=−0.0004×40×1/−0.0011=14.55Ku/ml
理論との比較:
小球状粒子/理論*100%=活性%
14.55Ku/ml/15.46Ku/ml*100%=94.1%
(F.スーパーオキシドジスムターゼ小球状粒子)
(実施例25.スーパーオキシドジスムターゼの小球状粒子の調製)
0.68mg/mlのSOD(5mg/mlストックから)、24.15%のPEG3350(31.25%ストックから)、9.1mMの酢酸アンモニウム(1Mストックから)、最終pH=4.99(水酸化アンモニウムおよび酢酸で調整)の溶液。この溶液を40℃から0℃まで50分間にわたって(約0.8℃/分)冷却し、そして約25℃で沈殿が開始した。この懸濁液を液体窒素中で瞬間凍結し、そして多岐管凍結乾燥器で凍結乾燥し、続いてMeCl2/アセトンで遠心分離によって洗浄した(図39、40)。
【0204】
40℃から0℃まで50分間にわたって(約0.8℃/分)冷却した。約25℃で沈殿が開始した。液体窒素中で瞬間凍結し、そして多岐管凍結乾燥器で凍結乾燥した。MeCl2/アセトンで遠心分離によって洗浄した。小球状粒子が形成され、そして大部分のアセトンが保持された。
【0205】
(G.サブチリシン小球状粒子)
(実施例26:非ポリマー相分離促進剤を用いるサブチリシン小球状粒子)
初期系の連続相は非ポリマー相分離促進剤を含み、冷却中にタンパク質の相分離を誘導し得る。サブチリシン小球状粒子は、ポリマーを全く用いることなくプロピレングリコールとエタノールの混合物を用いて本発明に従って形成され得る。この系において、プロピレングリコールは凝固点降下剤としての機能を果たし、そしてエタノールは相分離促進剤としての機能を果たす。プロピレングリコールはまた、小球状粒子の球形状の形成にも役立つ。
【0206】
35%プロピレングリコール−10%ホルメート−0.02%CaCl2中の20mg/mLサブチリシン溶液を調製した。次いでこの35%プロピレングリコール−サブチリシン溶液を、混合しながら67%エタノールに付した。この溶液は室温で透明のままであった。しかしながら、1時間−20℃に冷却すると、粒子の懸濁液が形成される。遠心分離してこれらの粒子を収集し、そして90%エタノールで洗浄した後、懸濁液流体として無水エタノールを用いてコールター粒子サイズ分析を行った。これらの粒子は、2.2ミクロンの平均直径を有する離散粒子と一致するコールター結果をもたらし、そしてこれらの粒子の95%が0.46ミクロンと3.94ミクロンの間であった。光学顕微鏡評価は、実質的に球状の粒子を示すことによりこれらの結果を確認した。粒子のSEM分析はこれらのコールター結果を確認した。
【0207】
(小球状粒子の形成後のサブチリシン酵素活性の保持)
溶液中のサブチリシンのサブチリシン小球状粒子への変換後の酵素活性の保持を、比色アッセイによって確認した。小球状粒子についての理論上の全活性単位を、冷却の前にエタノール−サブチリシン−プロピレングリコール溶液中でアッセイされたサブチリシンの全単位から、(サブチリシン粒子の分離後に)上清中に見出された全単位を減算することによって算出した。サブチリシン小球状粒子について求めた実際の全単位を理論単位で割ってパーセンテージで表したものは、粒子形成後のサブチリシン活性の保持を表す。この計算によって、107%の理論サブチリシン活性がサブチリシン小球状粒子の形成後に保持された。
【0208】
(H.炭水化物小球状粒子)
(実施例27:炭水化物小球状粒子の形成)
本発明は、炭水化物小球状粒子の調製に適用され得る。相分離は、この系の冷却中にPEG相とデキストラン相の間に誘導され得る。種々の分子量(例えば、5K、40K、144K、および500K)のデキストランが用いられ得る。30%PEG300中の5mg/mlデキストラン40Kの混合物を35℃で平衡化し、次いでこの混合物を0℃に冷却し、そして凍結乾燥した。この混合物を塩化メチレン:アセトン(1:1)で洗浄および遠心分離することによって凍結乾燥粒子を収集した。図49を見ても分かるように、小球状粒子が形成された。他の炭水化物、例えばデンプン、ヒドロキシエチルデンプン、トレハロース、ラクトース、マンニトール、ソルビトール、ハイロース(hylose)、硫酸デキストランなどを、このプロセスを用いて小球状粒子中に処方され得る。
【0209】
(I.前調製小球状粒子のマイクロカプセル化)
(実施例28.PLGA−カプセル化前調製インスリン小球状粒子の調製)
a)20%(w/v)ポリマー溶液(8ml)を塩化メチレン中の1600mgのポリラクチド・グリコリド共重合体(PLGA、MW35k)を溶解することによって調製した。この溶液に、100mgのインスリン小球状粒子(INSms)を添加し、そしてこの媒体をローター/ステーターホモジナイザーを用いて11krpmで激しく混合することによって均質の懸濁液を得た。0.02%メチルセルロース水溶液からなる連続相(24ml)を塩化メチレンで飽和した。この連続相を同じホモジナイザーを用いて11krpmで混合し、そして前記懸濁液を徐々にこの媒体に注入して有機相の初期マイクロカプセル化粒子を生成した。このエマルジョンは1:3のO/W比を有する。乳化を5分間継続した。次に、このエマルジョンを、150mlの脱イオン(DI)水からなる硬化媒体中に、この媒体を400rpmで攪拌しながら直ちに移した。この有機溶剤を−0.7バールの減圧下で1時間にわたって抽出した。硬化したマイクロカプセル化粒子を濾過によって収集し、水で洗浄した。これらの洗浄したマイクロカプセル化粒子を凍結乾燥して過剰な水を除去した。結果として生じるマイクロカプセル化粒子は、約30μmの平均粒子サイズを有するとともに、これらの粒子集団の大部分が90μm未満であり、そして5.7%(w/w)インスリンを含んだ。
【0210】
b)塩化メチレン中に1200mgの50:50ポリラクチド−グリコリド共重合体(PLGA、MW35k)を溶解することによって、30%(w/v)ポリマー溶液(4ml)を調製した。次に上記ポリマー溶液中の100mgのINSmsの懸濁液を、ホモジナイザーを用いて調製した。この懸濁液を用いて、実施例28に記載されるように12mlの0.02%メチルセルロース水溶液中のO/Wエマルジョンを生成した(W/O比=1:3)。実施例28と同じ手順に従って、最終マイクロカプセル化粒子を調製する。形成されたマイクロカプセル化粒子は25μmの平均粒子サイズを有し、0.8〜60μmの範囲にわたった。これらのマイクロカプセル化粒子のインスリン含量は8.8%(w/w)であった。
【0211】
あるいは、ポリマーの10%(w/v)溶液を用いて上記と同じ条件下でマイクロカプセル化プロセスを行った。このプロセスにより、約12μmの平均粒子サイズを有するマイクロカプセル化粒子であってこれらの粒子の大部分が50μm未満であり、かつインスリンの充填が21.1%(w/w)である、マイクロカプセル化粒子がもたらされた。
【0212】
インビトロ放出のための方法:
マイクロカプセル化粒子からのインスリンのインビトロ放出(IVR)は、37℃でインキュベートした3mg当量のカプセル化インスリンを含むガラスバイアル中に10mlの放出緩衝液(10mMトリス、0.05%Brij35、0.9%NaCl、pH7.4)を添加することによって達成される。指定の時間間隔でこのIVR媒体の400μLを微量遠心管中に移し、そして2分間13krpmで遠心する。上清の最上層300μLを取り出し、分析されるまで−80℃で保存する。この取得した容量を300μLの新たな媒体で置き換え、これを用いて残りの上清(100μL)とともにパレットを再構成した。この懸濁液をもとの対応するインビトロ放出媒体へ移す。
【0213】
(実施例29.PLGA/PLA合金マトリックス系中の前調製インスリン小球状粒子のマイクロカプセル化のための手順)
PLGA/PLA合金の30%(w/v)溶液を塩化メチレン(4ml)中で調製した。この合金は、それぞれ40、54および6%(0.48、0.68および0.07g)で、50:50PLGA(MW35k)、D,L−ポリ乳酸(PLA、MW19k)およびポリL−PLA(PLLA、MW180k)からなった。実施例28bと同じ手順に従って、最終マイクロカプセル粒子を調製した。これらのマイクロカプセル化粒子の例は、粒子サイズが0.8〜120μmの範囲にわたり、平均して40μmであって、粒子集団の大部分が90μmよりも小さかった。
【0214】
(実施例30.連続および不連続相の両方においてPEGを用いた、PLGAマトリックス系における前調製インスリン小球状粒子のマイクロカプセル化のための手順)
10%の50:50PLGA(0.4g)および25%ポリエチレングリコール(PEG、MW8k)の4mlの溶液を塩化メチレン中で調製した。ローター/ステーターホモジナイザーを用いて、100mgのINSmsをこの溶液中に11krpmで懸濁した。この連続相は、塩化メチレンで飽和させた0.02%(w/v)メチルセルロースおよび25%PEG(MW8k)の水溶液(12ml)からなった。この連続相を同じホモジナイザーを用いて11krpmで混合し、そして上記懸濁液をこの媒体に徐々に注入して有機相の初期マイクロカプセル化粒子を生成した。このエマルジョンは1:3のO/W比を有する。乳化を5分間継続した。次に、このエマルジョンを、150mlの脱イオン(DI)水からなる硬化媒体中に、この媒体を400rpmで攪拌しながら直ちに移した。この有機溶剤を−0.7バールの減圧下で1時間にわたって抽出した。硬化したマイクロカプセル化粒子を濾過によって収集し、水で洗浄した。これらの洗浄したマイクロカプセル化粒子を凍結乾燥して過剰な水を除去した。本実施例のマイクロカプセル化粒子は、30μmの平均粒子サイズを有し、2〜90μmの範囲にわたるとともに、この集団の大部分が70μmよりも小さかった。これらのミクロスフェアのインスリン含量は16.0%(w/w)であった。
【0215】
(実施例31.リン酸緩衝液を用いる種々の連続相pHでのPLGAマトリックス系における前調製インスリン小球状粒子のマイクロカプセル化のための手順)
20%の50:50 35kD PLGA(0.8g)の4mlの溶液を塩化メチレン中で調製した。ローター/ステーターホモジナイザーを用いて、100mgのINSmsをこの溶液中に11krpmで懸濁した。連続相は、0.1%(w/v)メチルセルロースおよび50mMリン酸緩衝液(pH2.5、5.4および7.8)の水溶液からなった。連続装備(図41A)を用いてマイクロカプセル化を行った。この連続相を11krpmで混合し、そして乳化チャンバー中に12ml/分で送り込んだ。分散相をこのチャンバー中に2.7ml/分で注入して初期マイクロカプセル化粒子を生成した。生成したエマルジョンをこのチャンバーから取り出し、硬化浴中に連続様式で移した。この硬化媒体を400rpmで攪拌した。この有機溶剤を−0.4バールの減圧下で1時間にわたって抽出した。硬化したマイクロカプセル化粒子を濾過によって収集し、水で洗浄した。これらの洗浄したマイクロカプセル化粒子を凍結乾燥して過剰な水を除去した。
【0216】
pH2.5、5.4および7.8で調製した、結果として生じるマイクロカプセル化粒子のインスリン含量を、それぞれ12.5、11.5および10.9と見積もった。これらのマイクロカプセル化粒子のサイズ分布分析の結果を表5に要約する。
【0217】
【表5】
(インビトロ放出のための方法)
マイクロカプセル化粒子からのインスリンのインビトロ放出は、37℃でインキュベートした3mg当量のカプセル化インスリンを含むガラスバイアル中に10mlの放出緩衝液(10mMトリス、0.05%Brij35、0.9%NaCl、pH7.4)を添加することによって達成された。指定の時間間隔でこのIVR媒体の400μLを微量遠心管中に移し、そして2分間13krpmで遠心した。上清の最上層300μLを取り出し、分析されるまで−80℃で保存した。この取得した容量を300μLの新たな媒体で置き換え、これを用いて残りの上清(100μL)とともにパレットを再構成した。この懸濁液をもとの対応するインビトロ放出媒体へ移した。
【0218】
上記調製物のインビトロ放出(IVR)の結果は図44に示し、これらの処方物からのインスリンの放出動力学に対する連続相のpHの有意な影響を示す。
【0219】
(実施例32.PLLAまたはPLLA/PEGマトリックス系における前調製ヒト血清アルブミン(HSA)小球状粒子のマイクロカプセル化のための手順)
25%(w/v、500mg)PEG(MW3kまたは8k)の2mlの溶液を塩化メチレン中で調製した。このPEG溶液または2mlの塩化メチレンを用いて50mgの前調製ヒト血清アルブミン小球状粒子(HSAms)の懸濁液を、ローター/ステーターホモジナイザーを11krpmで用いて生成した。この懸濁液に塩化メチレン中の4%PLLA(80mg、MW180k)2mlを添加し、そしてこの媒体を11〜27krpmでホモジナイズして有機相を生成した。この連続相は、塩化メチレンで飽和させた12mlの0.02%メチルセルロース水溶液からなった。この連続相を1lkrpmで激しく攪拌することによって乳化を惹起し、続いて有機相を徐々に注入した。この媒体を5分間乳化し、次いでこのエマルジョンを400rpmで混合している150mlのDI水中に移した。全ての上記手順を4℃で行った。次いでこの硬化媒体を室温に移し、そしてこの有機溶剤を−0.7バールの減圧下で1時間にわたって抽出した。硬化したマイクロカプセル化粒子を濾過によって収集し、水で洗浄した。洗浄したマイクロカプセル化粒子を凍結乾燥して過剰な水を除去した。上記処方物からのHASのIVRに対するPEGのチャンネル効果を図42に示す。
【0220】
(インビトロ放出のための方法)
カプセル化したマイクロカプセル化粒子からのHASのインビトロ放出(IVR)は、37℃でインキュベートした2.5mg当量のカプセル化HSAを含む15mlポリプロピレン遠心管中に15mlの放出緩衝液(20mMのHEPES、0.01%Tween−80、0.1MのNaCl、1mMのCaCl2、pH7.4)を添加することによって達成される。サンプリング手順は実施例31に記載した。
【0221】
(実施例33.PLGA−カプセル化前調製ロイプロリド/硫酸デキストラン小球状粒子の調製)
塩化メチレン中に1200mgの50:50ポリラクチド−グリコリド共重合体(PLGA、MW35k)を溶解することによって、30%(w/v)ポリマー溶液(4ml)を調製した。次に50mgのロイプロリドを含む65.9mgの前調製ロイプロリド(leuprilide)/硫酸デキストラン小球状粒子(LDS)を、該ポリマー溶液中にホモジナイザーを用いて懸濁した。この懸濁液を用いて、実施例28に記載されるように12mlの0.02%メチルセルロース水溶液中のO/Wエマルジョンを生成した(W/O比=1:3)。実施例28bと同じ手順に従って、最終マイクロカプセル化粒子を調製した。
【0222】
これらのマイクロカプセル化粒子は20μmの平均粒子サイズを有し、それらの大部分が50μm未満であった。これらのマイクロカプセル化粒子からのロイプロリドのIVRの結果を図43に図示する。
【0223】
(インビトロ放出のための方法)
マイクロカプセル化粒子からのロイプロリドのインビトロ放出(IVR)は、37℃でインキュベートした2.5mg当量のカプセル化ロイプロリドを含む15mlポリプロピレン遠心管中に15mlの放出緩衝液(10mMのNa−リン酸緩衝液、0.01%Tween−80、0.9%NaCl、0.04%NaN3、pH7.4)を添加することによって達成される。サンプリング手順は実施例28に記載した。
【0224】
(実施例34.PLGA−カプセル化前調製組換えヒト成長ホルモン小球状粒子の調製)
塩化メチレン中に0.4gのPLGA−PEGを溶解することによって、10%(w/v)ポリマー溶液(4ml)を調製した。次に100mgの前調製組換えヒト成長ホルモン小球状粒子(hGHms)を、上記ポリマー溶液中にホモジナイザーを用いて懸濁した。この連続相は、0.1%(w/v)メチルセルロースおよび50mMリン酸緩衝液(pH7.0)の水溶液からなった。マイクロカプセル化を実施例31に記載のように連続装備(図41A)を用いて行った。これらのマイクロカプセル化粒子の平均粒子サイズは25μmであり、1〜60μmの範囲にわたった。高分子マトリックスからのhGHのIVRプロフィールを図45に示す。
【0225】
(インビトロ放出のための方法)
マイクロカプセル化粒子からのhGHのIVRは、実施例28に記載のように達成される。
【0226】
(実施例35.マイクロカプセル化前調製インスリン小球状粒子の完全性の決定)
カプセル化前調製インスリン小球状粒子の完全性に対するマイクロカプセル化プロセスの影響を評価するために、前調製INSmsを含む高分子マイクロカプセル化粒子を、二相二重抽出法を用いて分解した。秤量したカプセル化INSmsのサンプルを塩化メチレン中に懸濁し、そして穏やかに混合して高分子マトリックスを溶解させた。タンパク質を抽出するために0.01NのHClを添加し、これらの2つの相を混合してエマルジョンを生成した。次いで、これらの2つの相を分離し、水性相を除去して同じ溶液を補給し、そしてこの抽出プロセスを繰り返した。抽出したインスリンの完全性をサイズ排除クロマトグラフィー(SEC)によって決定した。この方法は、抽出した媒体中のINSのモノマー、ダイマーおよび高分子量(HMW)種の増大を確認する。適切なコントロールを用いて、INSの完全性に対する分解プロセスの影響を確認した。これらの結果は、INS完全性に対するこのプロセスの有意な影響を示さなかった。
【0227】
カプセル化INSmsは、もとの(カプセル化されていない)INSms中のモノマー含量が99.13%であるのと比較して、マイクロカプセル化プロセスの条件および内容に応じて97.5〜98.94%のタンパク質モノマーを含んだ。カプセル化INSms中のダイマー種の含量は、もとのINSms中の0.85%と比較して、1.04%〜1.99%の範囲にわたった。カプセル化INSmsのHMW含量は、もとのINSms中の0.02%に対して、0.02%〜0.06%の範囲にわたった。これらの結果を表6に要約する。高分子マトリックスの影響を図46および47に示す。
【0228】
【表6】
(実施例36.マイクロカプセル化前調製小球状粒子からのインビボ放出インスリン)
前調製インスリン小球状粒子のマイクロカプセル化粒子からのインスリンのインビボ放出をSprague Dawley(SD)ラットにおいて研究した。これらの動物に、初回皮下用量の1IU/kgのカプセル化されていないかまたはカプセル化された前調製インスリン小球状粒子を投与した。ELISAを用いて、収集したサンプル中の組換えヒトインスリン(rhINS)血清レベルを決定した。これらの結果を図48に示す。
【0229】
特定の実施形態を図示しかつ記載してきたが、多数の改変が本発明の精神を逸脱することなく想起され、そして保護の範囲は添付クレームの範囲によってのみ限定される。
【図面の簡単な説明】
【0230】
【図1】図1は、温度に対して活性薬剤濃度をプロットした二次元状態図である。
【図2】図2は、冷却温度プロフィールである。
【図3】図3aは、インスリン出発物質の走査型電子顕微鏡写真(SEM)である。図3bは、インスリンの小球状粒子(実施例4)のSEMである。
【図4】図4は、小球状粒子中に調製された場合のインスリンの化学安定性の総合的維持を示すHPLC分析である。
【図5】図5aおよび5bは、バッチ間再現性を示す概略図である。
【図6】図6は、バッチ間再現性を示す概略図である。
【図7】図7は、実施例3においてインスリン小球状粒子を作製するための連続貫流プロセスの概略図である。
【図8】図8は、実施例3において連続貫流プロセスによって生成されたインスリン小球状粒子の走査型電子顕微鏡写真(10Kvおよび倍率6260×)である。
【図9】図9は、実施例3において連続貫流プロセスによって調製された溶解したインスリン小球状粒子のHPLCクロマトグラフである。
【図10−1】図10aは、インスリン溶解度に対する塩化ナトリウムの影響を示す。図10bは、インスリン溶解度に対する塩化ナトリウムの影響を示す。
【図10−2】図10cは、インスリン溶解度に対する塩化ナトリウムの影響を示す。図10dは、インスリン溶解度に対する塩化ナトリウムの影響を示す。
【図10−3】図10eは、インスリン溶解度に対する異なる塩の影響を示す。図10fは、インスリン溶解度に対する異なる塩の影響を示す。図10gは、インスリン溶解度に対する異なる塩の影響を示す。
【図10−4】図10hは、インスリン溶解度に対する異なる塩の影響を示す。図10iは、原料インスリン、小球状粒子から放出されたインスリンおよび小球状粒子中のインスリンのラマンスペクトルである。
【図11】図11は、実施例10の放射標識インスリンについてのアンダーセンカスケードインパクターの結果である。
【図12】図12は、実施例8についてのP/I比の棒グラフである。
【図13】図13は、実施例8からの肺のシンチグラフィー画像である
【図14−1】図14aは、α−1−アンチトリプシン(AAT)についての円偏光二色性(CD)プロットである。図14bは、実施例17における室温での貯蔵時間に対する活性のプロットである。
【図14−2】図14cは、実施例17における4℃での貯蔵時間に対する活性のプロットである。
【図15】図15は、DSCプロットである。
【図16】図15は、DSCプロットである。
【図17】図15は、DSCプロットである。
【図18】図15は、DSCプロットである。
【図19】図15は、DSCプロットである。
【図20】図15は、DSCプロットである。
【図21】図15は、DSCプロットである。
【図22】図15は、DSCプロットである。
【図23】図15は、DSCプロットである。
【図24】図15は、DSCプロットである。
【図25】図15は、DSCプロットである。
【図26】図26は、TSI社エアロサイザー(Aerosizer)の粒子サイズデータのプロットである。
【図27】図27は、ヒト成長ホルモン(hGH)小球状粒子のSEMである。
【図28】図28は、HFA−134aにおけるインスリン安定性データを示すチャートである。
【図29】図29は、3つの吸入デバイスを用いるインスリンの空気力学的性能を比較するチャートである。
【図30】図30は、25℃で貯蔵したインスリン出発物質と比較した、インスリン小球状粒子の安定性データのチャートである。
【図31】図31は、37℃で貯蔵したインスリン出発物質と比較した、インスリン小球状粒子の安定性データのチャートである。
【図32】図32は、25℃で貯蔵したインスリン出発物質と比較した、インスリン小球状粒子の安定性データのチャートである。
【図33】図33は、37℃で貯蔵したインスリン出発物質と比較した、インスリン小球状粒子の安定性データのチャートである。
【図34】図34は、25℃で貯蔵したインスリン出発物質と比較した、インスリン小球状粒子の安定性データのチャートである。
【図35】図35は、37℃で貯蔵したインスリン出発物質と比較した、インスリン小球状粒子の安定性データのチャートである。
【図36】図36は、Cyclohaler DPIを用いるインスリン空気力学的安定性の棒グラフである。
【図37】図37は、DNase小球状粒子の光学顕微鏡写真である。
【図38】図38は、DNaseの酵素活性のチャートである。
【図39】図39は、SOD小球状粒子の光学顕微鏡写真である。
【図40】図40は、SOD小球状粒子についての酵素学的データのチャートである。
【図41】図41A〜Bは、連続乳化反応器の概略図であり、ここで、図41Aは、乳化前に連続相または分散相に界面活性化合物が添加される場合の連続乳化反応器の概略図であり、そして図41Bは、乳化後に界面活性化合物が添加される場合の連続乳化反応器の概略図である。
【図42】図42は、PLLAカプセル化HSA粒子のIVRプロフィールに対するPEGの影響を図示する(実施例32)。
【図43】図43は、PLGAカプセル化LDS小球状粒子のIVRプロフィールを図示する(実施例33)。
【図44】図44は、PLGAカプセル化インスリン小球状粒子のIVRプロフィールに対する連続相のpHの影響を図示する(実施例31)。
【図45】図45は、PLGAカプセル化hGH小球状粒子のIVRプロフィールを図示する(実施例34)。
【図46】図46は、カプセル化INSms中のINSダイマーの形成に対するマイクロカプセル化変数(連続相およびマトリックス材料のpH)の影響を図示する(実施例35)。
【図47】図47は、カプセル化INSms中のHMW種の形成に対するマイクロカプセル化変数(連続相およびマトリックス材料のpH)の影響を図示する(実施例35)。
【図48】図48は、ラットにおける、カプセル化されていない前調製インスリン小球状粒子およびカプセル化された前調製インスリン小球状粒子からの組換えヒトインスリンのインビボ放出を図示する(実施例36)。
【図49】図49は、実施例27の粒子のSEMである。
【技術分野】
【0001】
(関連出願の相互参照)
本願は、2003年7月18日に出願された米国仮特許出願番号第60/488,712号に対する優先権を主張し、その全体が、本明細書中に参考として援用され、本明細書の一部をなす。
【0002】
(連邦政府によって後援された研究または開発)
該当なし。
【背景技術】
【0003】
(発明の背景)
(技術分野)
本発明は、活性薬剤の小球状粒子の製造方法、使用方法、および組成に関する。この製造方法に従って、溶解した相分離促進剤(PSEA)を含む水性または水混和性溶剤中に活性薬剤を溶解して、単一液相状態の溶液を生成する。次いで、この溶液は、活性薬剤が固相を構成し、かつPSEAおよび溶剤が液相を構成する、液−固相分離に供される。この液−固相分離は、溶液の温度をこの系の相転移温度より下に変化させるなどの、多数の方法で誘導され得る。この方法は、治療剤を必要とする被験体に送達され得る、治療剤の小球状粒子を生成するのに最適である。この方法はまた、高分子の固体小球状粒子、特に熱不安定性の高分子(例えばタンパク質)を生成するのにも最適である。
【0004】
(背景技術)
いくつかの技術が、バイオポリマーナノ粒子およびミクロ粒子の製造のためにこれまで用いられてきた。従来技術としては、粒子形成のための噴霧乾燥およびミリングが挙げられ、5μm以下のサイズの粒子を生成するのに用いられ得る。
【0005】
米国特許番号第5,654,010号および米国特許番号第5,667,808号には、亜鉛と錯化して非晶質錯体を作製し、次いで、この非晶質錯体を超音波ノズルを通して微粉化し、そして液体窒素中に噴霧してこれらの小滴を凍結させることによる、組換えヒト成長ホルモン(hGH)の固体形態の生成が記載される。次いで、液体窒素を−80℃の温度で蒸発させ、結果として生じる物質を凍結乾燥させる。
【0006】
ミクロ粒子、ミクロスフェア、およびマイクロカプセルは、直径1ミリメートル未満、より好ましくは直径100ミクロン未満、そして最も好ましくは直径10ミクロン未満の固体または半固体粒子であり、タンパク質、合成ポリマー、多糖類およびこれらの組み合わせを含む種々の材料から形成され得る。ミクロスフェアは、多くの種々の用途、一次的分離、診断薬、および薬物送達に用いられている。
【0007】
分離技術において用いられる最も周知のミクロスフェアの例は、合成または天然起源のポリマー(例えば、ポリアクリルアミド、ヒドロキシアパタイトもしくはアガロース)から形成されるミクロスフェアである。制御された薬物送達エリアでは、分子は、小球状粒子中に取り込まれるかまたは小球状粒子内にカプセル化されるか、あるいはその後の放出のためにモノリシックマトリックス中に取り込まれることが多い。相分離、溶剤蒸発、コアセルベーション、乳化、および噴霧乾燥を包含する、多数の異なる技術を常法として用いて、これらのミクロスフェアを合成ポリマー、天然ポリマー、タンパク質および多糖類から作製する。一般に、ポリマーはこれらのミクロスフェアの支持構造を形成し、目的の薬物はこのポリマー構造の中に取り込まれる。
【0008】
標的薬物をカプセル化するために脂質を用いて調製された粒子が、現在利用可能である。リポソームは、単一または複数のリン脂質二重層および/またはコレステロール二重層からなる球状粒子である。リポソームは100ナノメートル以上のサイズであり、種々の水溶性薬物または脂溶性薬物を輸送し得る。例えば、複数の水性区画を取り囲む二分子膜中に配置されて粒子を形成する脂質は、Sinil Kimによって米国特許番号第5,422,120号に記載されるように、その後の送達のために水溶性の薬物をカプセル化するのに用いられ得る。
【0009】
球状ビーズは、長年、生化学者のためのツールとして市販されてきた。例えば、ビーズに結合体化した抗体は、特定のリガンドに結合特異性を有する比較的大きな粒子を作製する。抗体は、細胞活性化のために細胞表面上のレセプターに結合するのに常法として用いられ、免疫親和性精製のために固相に結合されて抗体被覆粒子を形成し、そしてこれらの粒子に結合体化した組織特異的抗体または腫瘍特異的抗体を用いて所望の部位に薬剤を標的化し、経時的に緩徐に放出される治療剤を送達するのに用いられ得る。
【発明の開示】
【発明が解決しようとする課題】
【0010】
粒子(特に薬物送達、分離および診断領域における使用に適合させ得る粒子)を作製するための新たな方法の開発の必要性が、継続的に存在している。有用性の観点から最も望ましい粒子は、以下の特性を有する小球状粒子である:狭いサイズ分布、実質的に球状、実質的に活性薬剤のみで構成、活性薬剤の生化学的完全性の保持および生物活性の保持。これらの粒子は、コーティングによるかまたはマイクロカプセル化によって、粒子をさらに安定化させる適切な固体を生じるはずである。さらに、これらの小球状粒子の成形加工方法は、以下の望ましい特性を有する:単純な成形加工、本質的に水性のプロセス、高収率、およびその後のふるい分けが不要。
【課題を解決するための手段】
【0011】
(発明の概要)
本発明は、活性薬剤の小球状粒子の製造方法および使用方法に関する。この方法に従って、活性薬剤は、溶解した相分離促進剤を含む溶剤中に溶解されて、単一液相状態の溶液を形成する。この溶剤は、好ましくは水性または水混和性溶剤である。次いで、この溶液は、活性薬剤が固相を構成し、かつPSEAおよび溶剤が液相を構成する、液−固相分離に供される。この液−固相分離は、例えば、溶液の温度をこの溶液の相転移温度より下に変化させるなどの、多数の方法で誘導され得る。
【0012】
本発明の好ましい実施形態において、溶液を液−固相分離に供する方法は、溶液中の活性薬剤の相転移温度より下にこの溶液を冷却することによるものである。その温度は、この溶液の凝固点を超えるかまたは凝固点未満であり得る。凝固点が相転移温度を超える溶液について、この溶液は、ポリエチレングリコールまたはプロピレングリコールのような凝固点降下剤を含み得、この溶液の凝固点を降下させて、溶液を凍結させることなく溶液中の相分離を生じることが可能である。
【0013】
この溶液が相変化の工程(この工程において、活性薬剤は凝固して小球状粒子の懸濁液を不連続相として形成し、一方、相分離促進剤は連続相に溶解したままである)に供されると、本発明の相分離促進剤は、溶液中の活性薬剤の液−固相分離を促進するかまたは誘導する。すなわち、相を分離する促進剤は相の変化を受けないが、活性薬剤は相変化を受ける。
【0014】
本発明において粒子を生成する方法はまた、粒子の液−固相分離を制御するさらなる工程も包含し、生成された粒子のサイズおよび形状を制御し得る。相分離を制御する方法は、溶液中のイオン強度、pH、相分離促進剤の濃度、活性薬剤の濃度の制御、または溶液の温度の変化速度の制御を包含し、これらの制御は相分離の前であるか、あるいはこれらの任意またはいくつかの変化が相分離を誘導する。
【0015】
本発明の好ましい実施形態において、小球状粒子は、粒子形成後に連続相中のPSEAから分離される。さらに別の好ましい実施形態において、この分離方法は、これらの粒子を含む溶液を液体媒体で洗浄することによるものであり、その場合、活性薬剤は液体媒体において可溶性ではなく、一方で相分離促進剤は液体媒体において可溶性である。液体洗浄媒体は、この液体媒体中での活性薬剤の溶解度を低下させる薬剤を含み得る。この液体洗浄媒体はまた、1種以上の賦形剤も含み得る。この賦形剤は、小球状粒子または活性薬剤のための安定剤あるいはキャリア剤として機能し得る。この賦形剤はまた、粒子からの活性薬剤の徐放または生物組織への活性薬剤の改質された浸透のようなさらなる特性を、活性薬剤もしくは粒子にもたらし得る。
【0016】
別の好ましい実施形態において、これらの小粒子はPSEAを含まないが、これらの小粒子はその後のプロセス工程のためにPSEA相の存在下で回収され、その後にこのPSEA相から分離され得る。
【0017】
別の好ましい実施形態において、この溶液は、水性または水混和性溶剤を含む水溶液である。
【0018】
本発明の活性薬剤は、好ましくは薬学的に活性な物質であり、治療剤、診断用薬、化粧品、栄養剤、または農薬であり得る。本発明の好ましい実施形態において、この活性薬剤は、例えばタンパク質、ポリペプチド、炭水化物、ポリヌクレオチド、または核酸のような高分子である。さらに別の好ましい実施形態において、この活性薬剤を含む粒子は、適切な経路(例えば、非経口注射、局所、経口、直腸、経鼻、肺、膣、頬側、舌下、経皮、経粘膜、眼球、眼球内または耳)による、薬剤を必要とする被験体へのインビボ送達に適している。
【発明を実施するための最良の形態】
【0019】
(発明の詳細な説明)
本発明は、多数の異なる形態で実施形態の影響を受けやすい。本発明の好ましい実施形態は、本開示が本発明の原理の例証とみなされるべきであるという了解の下で開示され、かつ、本発明の広範な態様を、例示した実施形態に限定することを意図するものではない。
【0020】
本発明は、活性薬剤の小球状粒子の製造方法および使用方法および組成に関する。この製造方法に従い、溶解した相分離促進剤を含む溶剤中に活性薬剤を溶解して、単一連続液相である溶液を生成する。この溶剤は、好ましくは水性または水混和性溶剤である。次いで、この溶液は、例えば溶液の温度を活性薬剤の相転移温度より下に下げることによって相変化を受け、それによって、この活性薬剤は液−固相分離を受けて不連続相を構成する小球状粒子の懸濁液を生成し、一方、相分離促進剤は連続相に残る。
【0021】
(相)
(連続相)
活性薬剤の小球状粒子を調製する本発明の方法は、活性薬剤および第1溶剤中に溶解された相分離促進剤を単一液相状態で含む溶液を生成する工程から始まる。この溶液は、有機溶剤または混和性の有機溶剤の混合物を含む有機系であってもよい。この溶液はまた、水性媒体または水混和性有機溶剤または水混和性有機溶剤の混合物またはこれらの組合せを含む水性溶液であってもよい。この水性媒体は、水、正常生理食塩水、緩衝液、緩衝生理食塩水などであり得る。適切な水混和性有機溶剤としては、N−メチル−2−ピロリジノン(N−メチル−2−ピロリドン)、2−ピロリジノン(2−ピロリドン)、1,3−ジメチル−2−イミダゾリジノン(DMI)、ジメチルスルホキシド、ジメチルアセトアミド、酢酸、乳酸、アセトン、メチルエチルケトン、アセトニトリル、メタノール、エタノール、イソプロパノール、3−ペンタノール、n−プロパノール、ベンジルアルコール、グリセロール、テトラヒドロフラン(THF)、ポリエチレングリコール(PEG)、PEG−4、PEG−8、PEG−9、PEG−12、PEG−14、PEG−16、PEG−120、PEG−75、PEG−150、ポリエチレングリコールエステル、PEG−4ジラウレート、PEG−20ジラウレート、PEG−6イソステアレート、PEG−8 パルミトステアレート、PEG−150 パルミトステアレート、ポリエチレングリコールソルビタン、PEG−20ソルビタンイソステアレート、ポリエチレングリコールモノアルキルエーテル、PEG−3ジメチルエーテル、PEG−4ジメチルエーテル、ポリプロピレングリコール(PPG)、ポリプロピレンアルギレート、PPG−10ブタンジオール、PPG−10メチルグルコースエーテル、PPG−20メチルグルコースエーテル、PPG−15ステアリルエーテル、プロピレングリコールジカプリレート/ジカプレート、プロピレングリコールラウレート、およびグリコフロール(テトラヒドロフルフリルアルコールポリエチレングリコールエーテル)、プロパン、ブタン、ペンタン、ヘキサン、ヘプタン、オクタン、ノナン、デカンを含むアルカン、またはこれらの組み合わせが挙げられるが、これらに限定されない。
【0022】
この単一連続相は、第1溶剤において可溶性であるかまたは第1溶剤と混和性の相分離促進剤の溶液をまず生成することによって調製され得る。続いて、この溶液に活性薬剤を添加する。この活性薬剤はこの溶液に直接添加してもよく、またはこの活性薬剤をまず第2溶剤に溶解し、次いでこの溶液に合わせてもよい。この第2溶剤は、第1溶剤と同じ溶剤であってもよく、または上記リストから選択され、かつこの溶液と混和性の別の溶剤であってもよい。この薬剤は、周囲温度以下でこの溶液に添加されることが好ましく、これは、特に熱不安定分子(例えば、ある種のタンパク質)にとって重要である。「周囲温度」は、約20℃から約40℃の室温前後の温度を意味する。しかし、この系はまた、加熱によって薬剤の活性に顕著な低下が引き起こされない限り、系中の活性薬剤の溶解度を高めるために加熱され得る。
【0023】
(相分離促進剤)
溶液が、相分離の工程(この工程において、活性薬剤は固体または半固体となって不連続相として小球状粒子の懸濁液を生成し、一方、相分離促進剤は連続相に溶解したままである)に供されると、本発明の相分離促進剤(PSEA)は、この溶液からの活性薬剤の液−固相分離を促進するかまたは誘導する。この溶液が相分離条件に付されると、相分離促進剤は活性薬剤の溶解度を低下させる。適切な相分離促進剤としては、ポリマーまたはこの溶液に可溶性もしくは混和性のポリマーの混合物が挙げられるが、これらに限定されない。適切なポリマーとしては、例えば直鎖ポリマーまたは分枝鎖ポリマーが挙げられる。これらのポリマーは、水溶性、半水溶性、水混和性、または不溶性であり得る。
【0024】
本発明の好ましい形態において、この相分離促進剤は、水溶性または水混和性である。用いられ得るポリマーのタイプとしては、炭水化物系ポリマー、ポリ脂肪族アルコール、ポリ(ビニル)ポリマー、ポリアクリル酸、ポリ有機酸、ポリアミノ酸、コポリマーおよびブロックコポリマー(例えば、プルロニックF127またはプルロニックF68のようなポロクサマー)、tert−ポリマー、ポリエーテル、天然に存在するポリマー、ポリイミド、界面活性剤、ポリエステル、分枝ポリマーおよび環状ポリマー、ならびにポリアルデヒドが挙げられる。
【0025】
好ましいポリマーは、活性薬剤粒子の意図された投与経路のための医薬品添加物として受容可能なポリマーである。好ましいポリマーは、種々の分子量のポリエチレングリコール(PEG)(例えば、PEG200、PEG300、PEG3350、PEG8000、PEG10000、PEG20000など)およびポロクサマー(例えば、プルロニックF127またはプルロニックF68)のような薬学的に受容可能な添加物である。さらに別の好ましいポリマーは、ポリビニルピロリドン(PVP)である。さらに別の好ましいポリマーは、ヒドロキシエチルデンプンである。他の両親媒性ポリマーはまた、単独かまたは組み合わせて用いられ得る。この相分離促進剤はまた、プロピレングリコールおよびエタノールの混合物のような非ポリマーであり得る。
【0026】
(液−固相分離)
この溶液中の活性薬剤の液−固相分離は、当該分野で公知の任意の方法(例えば、温度の変化、圧力の変化、pHの変化、溶液のイオン強度の変化、活性薬剤の濃度の変化、相分離促進剤の濃度の変化、溶液の重量モル浸透圧濃度の変化、これらの組合せなど)によって誘導され得る。
【0027】
本発明の好ましい実施形態において、この相変化は、温度を溶液中の活性薬剤の相転移温度より下に下げることによる、温度誘発性相変化である。
【0028】
図1は、溶剤、PSEAおよび活性薬剤を含む溶液についての二次元状態図10である。この図は溶液の温度に対する活性薬剤濃度をプロットしている。PSEAの濃度は一定に維持される。
【0029】
この図は、飽和曲線12;過飽和曲線14;これらの間に準安定エリア16;該飽和曲線12の下に第1エリア18(この系は、全成分が液相中に存在する、均質な単一液相状態である);および該過飽和曲線14の上に第2エリア20(この系は、活性薬剤の固相ならびにPSEAおよび溶剤の液相を有する二相系である)を有する。この状態図は、この系の温度ならびに純液相中、液−固相中およびこれらの二相間の転移の周囲条件における成分の相対濃度を決定するのに役立つ。
【0030】
本明細書中に開示されるように、活性薬剤の小球状粒子の調製は、不飽和溶液(点A’)から冷却することにより、この溶液が、存在し得る任意の固相と平衡状態にある点Aにおいて飽和に達する工程に主に関与する。さらに冷却すると、この溶液は、所定の温度で、平衡溶解度に相当するよりも多くの活性薬剤を含む状態に達し、従って、この溶液は過飽和状態になる。この固相の自然形成は、点Bに到達するまで生じない。この点Bは、準安定ゾーンの境界上の点である。この準安定ゾーン幅は、最大達成可能過冷却ΔTmax=T2−T1または過飽和ΔCmax=C*2−C*1のいずれかで表され得る。これらの2つの式は、熱力学的当量である:
【0031】
【化1】
経路A’−A−Bは、準安定溶液を調製する多重熱方法を表す。等温プロセスにおいて、起点はA’’である。一定温度で濃度を増大させることにより、点Aで再び飽和に達成する。点Cへの濃度の等温増大(例えば、溶剤蒸発によるかまたは活性薬剤の播種/添加による)により、この溶液を、再び準安定限界に達するまで準安定領域中へ移動させる。準安定限界を超えると、この溶液は不安定になり、固相の自然形成が直ちに起こる。
【0032】
等温的に得られた値(ΔCmax)T=C*3−C*2は、多重熱的に得られたΔTmax=T3−T2の換算値とは異なり得る。準安定ゾーンの境界に接近すると、固体粒子形成に必要な時間は、準安定限界に達するまで減少する。
【0033】
多重熱プロセスにおいて、冷却速度は制御速度で行われ、粒子のサイズおよび形状が制御される。制御速度とは、約0.2℃/分から約50℃/分、そしてさらに好ましくは、0.2℃/分から30℃/分を意味する。変化速度は、一定速度または線形速度、非線形速度、断続的速度またはプログラム速度(複数の相サイクルを有する)であり得る。
【0034】
これらの粒子は、下記のように、溶液中のPSEAから分離され得、そして洗浄によって精製され得る。
【0035】
本発明は、活性薬剤の濃度、PSEAの濃度、温度またはこれらの任意の組み合わせを調整して、PSEAおよび溶剤は相変化を受けずに液体として残るが、活性薬剤は液体状態から固体状態に変わる、相変化を引き起こすことを企図する。pH、イオン強度、重量モル浸透圧濃度などを変化させて、相変化を増強、促進、制御または抑制することもまた企図される。凝固点が比較的高いか、または凝固点が相転移温度を超える溶液について、これらの溶液は、プロピレングリコール、スクロース、エチレングリコール、アルコール(例えば、エタノール、メタノール)のような凝固点降下剤または凝固点降下剤の水性混合物を含み得、この系の凝固点を降下させて、この系を凍結させることなく系中の相変化を可能にする。このプロセスはまた、温度を系の凝固点より下に低下させるために実行され得る。本明細書中に記載されるプロセスは、熱不安定性の分子(例えば、タンパク質)に特に適する。
【0036】
(任意の賦形剤)
本発明の粒子は、1以上の賦形剤を含む。この賦形剤は、粒子または活性薬剤またはキャリア剤の安定性の増大、粒子からの活性薬剤の徐放、あるいは生物組織への活性薬剤の改質された浸透のようなさらなる特性を、活性薬剤または粒子にもたらし得る。
【0037】
適切な賦形剤としては、炭水化物(例えば、トレハロース、スクロース、マンニトール)、カチオン(例えば、Zn2+、Mg2+、Ca2+)、アニオン(例えば、SO42−)、アミノ酸(例えば、グリシン)、脂質、リン脂質、脂肪酸、界面活性剤、トリグリセリド、胆汁酸またはそれらの塩(例えば、コール酸またはその塩、例えばコール酸ナトリウム;デオキシコール酸またはその塩)、脂肪酸エステル、およびポリマーであってそれらのPSEAとしての機能よりも低いレベルで存在するものが挙げられるが、これらに限定されない。賦形剤を用いる場合、この賦形剤は、溶液の状態図に有意な影響を及ぼしてはならない。
【0038】
(粒子の分離および洗浄)
本発明の好ましい実施形態において、小球状粒子は、溶液中の相分離促進剤からこれらを分離することによって回収される。さらに別の好ましい実施形態において、分離方法は、活性薬剤は液体媒体において可溶性ではなく、一方相分離促進剤は液体媒体において可溶性である液体媒体で、小球状粒子を含む溶液を洗浄することによるものである。いくつかの洗浄方法は、ダイアフィルトレーションまたは遠心分離によるものであり得る。液体媒体は、水性媒体または有機溶剤であり得る。低い水性溶解度を有する活性薬剤について、液体媒体は、水性媒体または活性薬剤の水性溶解度を低下させる薬剤(例えば2価カチオン)を含む水性媒体であり得る。高い水性溶解度を有する活性薬剤(例えば多くのタンパク質)について、有機溶剤または硫酸アンモニウムのようなタンパク質沈殿剤を含む水性溶剤が用いられ得る。
【0039】
液体媒体としての使用に適した有機溶剤の例としては、連続相に適するように上記に特定された有機溶剤が挙げられ、そしてより好ましくは、塩化メチレン、クロロホルム、アセトニトリル、酢酸エチル、メタノール、エタノール、ペンタンなどが挙げられる。
【0040】
任意のこれらの溶剤の混合物を用いることも企図される。1つの好ましいブレンドは、塩化メチレンまたは塩化メチレンとアセトンの1:1混合物である。液体媒体は、例えば、凍結乾燥、蒸発、または乾燥によって容易に除去されるために、低い沸点を有することが好ましい。
【0041】
液体媒体はまた、液体二酸化炭素またはそのほぼ超臨界点の流体のような超臨界流体でもあり得る。超臨界流体は、相分離促進剤(特にいくつかのポリマー)に適した溶剤であり得るが、タンパク質粒子に適した非溶剤である。超臨界流体は、それら単独でかまたは補助溶剤と共に用いられ得る。以下の超臨界流体が用いられ得る:液体C02、エタン、またはキセノン。潜在的補助溶剤は、アセトニトリル、ジクロロメタン、エタノール、メタノール、水、または2−プロパノールであり得る。
【0042】
本明細書中に記載されるPSEAから小球状粒子を分離するのに用いられる液体媒体は、この液体媒体中における活性薬剤の溶解度を低下させる薬剤を含み得る。これらの粒子は、粒子の収率を最大にするために、液体媒体中において最小の溶解度を示すことが最も望ましい。いくつかのタンパク質、例えばインスリンおよびヒト成長ホルモンについて、溶解度の低下は、このタンパク質への2価カチオン(Zn2+など)の添加によって達成され得る。複合体を形成するのに用いられ得る他のイオンとしては、Ca2+、Cu2+、Fe2+、Fe3+などが挙げられるが、これらに限定されない。
【0043】
インスリン−Zn複合体または成長ホルモン−Zn複合体の溶解度は十分に低いので、水溶液中の複合体のダイアフィルトレーションが可能になる。
【0044】
この液体媒体は1以上の賦形剤も含み得、これらの賦形剤は、上記のように、粒子の安定性および/または活性薬剤もしくはキャリア剤の安定性の増大、粒子からの活性薬剤の徐放、あるいは生物組織への活性薬剤の改質された浸透のようなさらなる特性を、活性薬剤または粒子にもたらし得る。
【0045】
本発明の別の形態において、小球状粒子は、PSEAを含む溶液から分離されない。
【0046】
(水系プロセス)
別の好ましい実施形態において、本系の成形加工プロセスは、水性溶剤または水混和性溶剤を含む水性系の成形加工プロセスである。適切な水混和性溶剤の例としては、連続相について上記に特定したものが挙げられるが、これらに限定されない。水性プロセスを用いる利点の1つは、この溶液が緩衝化され得かつ賦形剤を含み得ることであり、これらの賦形剤は生化学的安定化をもたらして活性薬剤(例えばタンパク質)を保護する。
【0047】
(活性薬剤)
本発明の活性薬剤は、好ましくは薬学的に活性な物質であり、この物質は、治療剤、診断用薬、化粧品、栄養剤、または農薬であり得る。
【0048】
この治療剤は生物製剤であり得、タンパク質、ポリペプチド、炭水化物、ポリヌクレオチド、および核酸が挙げられるがこれらに限定されない。このタンパク質は抗体であり得、ポリクローナルであってもモノクローナルであってもよい。この治療剤は低分子量分子であり得る。さらに、これらの治療剤は、例えば以下の種々の公知の医薬品から選択され得るが、これらに限定されない:鎮痛薬、麻酔薬、興奮薬、アドレナリン作動薬、抗アドレナリン作動薬、抗アドレナリン剤、アドレノコルチコイド、アドレナリン様作動、抗コリン剤、抗コリンエステラーゼ、抗痙攣薬、アルキル化剤、アルカロイド、アロステリック阻害剤、アナボリックステロイド、食欲抑制物、制酸薬、下痢止め薬、解毒薬、葉酸代謝拮抗薬、解熱薬、抗リウマチ薬、精神治療薬、神経遮断薬、抗炎症薬、駆虫薬、抗不整脈薬、抗生物質、抗凝血薬、抗鬱薬、抗糖尿病薬、抗てんかん薬、抗真菌剤、抗ヒスタミン薬、血圧降下薬、ムスカリン性受容体拮抗薬、抗ミコバクテリア薬、抗マラリア薬、防腐薬、抗悪性腫瘍薬、抗原虫薬、免疫抑制薬、免疫賦活薬、抗甲状腺薬、抗ウイルス薬、抗不安鎮静薬、収斂薬、βアドレナリン作動性レセプター遮断薬、造影剤、コルチコステロイド、鎮咳剤、診断用薬、画像診断用薬、利尿薬、ドーパミン作動薬、止血薬、血液作用薬、ヘモグロビン修飾因子、ホルモン、睡眠薬、免疫、抗高脂血症薬および他の脂質調節薬、ムスカリニック、筋弛緩薬、副交感神経作動薬、副甲状腺ホルモン、カルシトニン、プロスタグランジン、放射性医薬品、鎮静薬、性ホルモン、抗アレルギー薬、刺激薬、交感神経模倣薬、甲状腺薬、血管拡張薬、ワクチン、ビタミン、およびキサンチン。抗悪性腫瘍薬、または抗癌薬としては、パクリタキセルおよび誘導体化合物、ならびにアルカロイド、代謝拮抗物質、酵素阻害剤、アルキル化剤および抗生物質からなる群より選択される他の抗悪性腫瘍薬が挙げられるが、これらに限定されない。
【0049】
美容薬は、美容活性を有し得る任意の活性成分である。これらの活性成分の例としては、とりわけ、皮膚軟化薬、湿潤剤、フリーラジカル抑制剤、抗炎症薬、ビタミン、脱色剤、抗アクネ薬、抗脂漏性薬、角質溶解薬、やせ薬、皮膚着色剤および日焼け止め薬、そして特にリノール酸、レチノール、レチノイン酸、アスコルビン酸アルキルエステル、多価不飽和脂肪酸、ニコチンエステル、ニコチン酸トコフェロール、米、ダイズまたはシアバターノキの不鹸化物、セラミド、グリコール酸のようなヒドロキシ酸、セレン誘導体、抗酸化剤、βカロチン、γオリザノールおよびグリセリン酸ステアリルが挙げられ得る。これらの化粧品は市販されており、かつ/または当該分野で公知の技術によって調製され得る。
【0050】
本発明の実施における使用が企図される栄養剤としては、例えば、タンパク質、炭水化物、水溶性ビタミン(例えば、ビタミンC、B−複合ビタミン、など)、脂溶性ビタミン(例えば、ビタミンA、D、E、K、など)、およびハーブエキスが挙げられるが、これらに限定されない。これらの栄養剤は市販されており、かつ/または当該分野で公知の技術によって調製され得る。
【0051】
農薬との用語は、除草剤、殺虫剤、ダニ駆除剤、殺線虫剤、外部奇生生物撲滅薬および殺菌剤を包含すると理解される。本発明における農薬が属し得る化合物クラスとしては、例えば、尿素、トリアジン、トリアゾール、カルバメート、リン酸エステル、ジニトロアニリン、モルホリン、アシルアラニン、ピレスロイド、ベンジル酸エステル、ジフェニルエーテルおよび多環式ハロゲン化炭化水素が挙げられる。これらの各クラスの農薬の具体例は、Pesticide Manual、第9版、British Crop Protection Councilに記載される。これらの農薬は市販されており、かつ/または当該分野で公知の技術によって調製され得る。
【0052】
本発明の好ましい実施形態において、活性薬剤は、高分子、例えばタンパク質、ポリペプチド、炭水化物、ポリヌクレオチド、ウイルス、または核酸である。核酸としては、DNA、オリゴヌクレオチド、アンチセンスオリゴヌクレオチド、アプタマー(aptimer)、RNA、およびSiRNAが挙げられる。この高分子は天然であっても合成であってもよい。このタンパク質は抗体であり得、モノクローナルであってもポリクローナルであってもよい。このタンパク質はまた、天然の起源から単離されたかまたは合成法もしくは組替え法によって生成された任意の公知の治療タンパク質であってもよい。治療タンパク質の例としては、血液凝固カスケードのタンパク質(例えば、第VII因子、第VIII因子、第IX因子など)、サブチリシン、オボアルブミン、α−1−アンチトリプシン(AAT)、DNase、スーパーオキシドジスムターゼ(SOD)、リゾチーム、リボヌクレアーゼ、ヒアルロニダーゼ、コラゲナーゼ、成長ホルモン、エリスロポエチン、インスリン様成長因子またはそれらのアナログ、インターフェロン、グラチラマー、顆粒球マクロファージコロニー刺激因子、顆粒球コロニー刺激因子、抗体、ペグ化タンパク質、グリコシル化または過剰グリコシル化タンパク質、デスモプレシン、LHRHアゴニスト、例えば:ロイプロリド、ゴセレリン、ナフシリン、ブセレリン;LHRHアンタゴニスト、バソプレッシン、シクロスポリン、カルシトニン、副甲状腺ホルモン、副甲状腺ホルモンペプチドおよびインスリンが挙げられるが、これらに限定されない。好ましい治療タンパク質は、インスリン、α−1アンチトリプシン、LHRHアゴニストおよび成長ホルモンである。
【0053】
低分子量治療分子の例としては、ステロイド、βアゴニスト、抗菌剤、抗真菌剤、タキサン(有糸分裂阻害剤および微小管阻害剤)、アミノ酸、脂肪族化合物、芳香族化合物、および尿素化合物が挙げられるが、これらに限定されない。
【0054】
好ましい実施形態において、この活性薬剤は、肺疾患の処置のための治療剤である。このような薬剤の例としては、ステロイド、βアゴニスト、抗真菌剤、抗菌剤化合物、呼吸器作用薬、抗喘息薬、非ステロイド性抗炎症剤(NSAIDS)、α−1アンチトリプシン、および嚢胞性線維症を処置するための薬剤が挙げられるが、これらに限定されない。ステロイドの例としては、ベクロメタゾン(ジプロピオン酸ベクロメタゾンを含む)、フルチカゾン(プロピオン酸フルチカゾンを含む)、ブデソニド、エストラジオール、フルドロコルチゾン、フルオシノニド、トリアムシノロン(トリアムシノロンアセトニドを含む)、およびフルニソリドが挙げられるが、これらに限定されない。βアゴニストの例としては、キシナホ酸サルメテロール、フマル酸フォルモテロール、レボアルブテロール、バンブテロール、およびツロブテロールが挙げられるが、これらに限定されない。
【0055】
抗真菌薬の例としては、イトラコナゾール、フルコナゾール、およびアムホテリシンBが挙げられるが、これらに限定されない。
【0056】
診断用薬としては、X線造影剤および造影剤が挙げられる。X線造影剤の例としては、ジアトラゾ酸(diatrazoic acid)のエチルエステル(EEDA)としても公知のWIN−8883(エチル3,5−ジアセトアミド−2,4,6−トリヨードベンゾエート)、WIN67722、すなわち、(6−エトキシ−6−オキソヘキシル−3,5−ビス(アセトアミド)−2,4,6−トリヨードベンゾエート;エチル−2−(3,5−ビス(アセトアミド)−2,4,6−トリヨードベンゾイルオキシ)ブチラート(WIN16318);ジアトリゾキシ酢酸エチル(ethyl diatrizoxyacetate)(WIN12901);2−(3,5−ビス(アセトアミド)−2,4,6−トリヨードベンゾイルオキシ)プロピオン酸エチル(WIN16923);N−エチル2−(3,5−ビス(アセトアミド)−2,4,6−トリヨードベンゾイルオキシアセトアミド(WIN65312);イソプロピル2−(3,5−ビス(アセトアミド)−2,4,6−トリヨードベンゾイルオキシ)アセトアミド(WIN12855); 2−(3,5−ビス(アセトアミド)−2,4,6−トリヨードベンゾイルオキシマロン酸ジエチル(WIN67721);2−(3,5−ビス(アセトアミド)−2,4,6−トリヨードベンゾイルオキシ)フェニル酢酸エチル(WIN67585);[[3,5−ビス(アセチルアミノ)−2,4,5−トリヨードベンゾイル]オキシ]ビス(l−メチル)プロパン二酸エステル(WIN68165);および3,5−ビス(アセチルアミノ)−2,4,6−トリヨード−4−(エチル−3−エトキシ−2−ブテノエート)安息香酸エステル(WIN68209)が挙げられる。好ましい造影剤としては、生理的条件下で比較的急速に分解され、従って炎症反応に関連する任意の粒子を最小化することが期待される造影剤が挙げられる。分解は、酵素加水分解、生理的pHでのカルボン酸の可溶化、または他のメカニズムによって生じ得る。従って、低溶解性のヨウ化カルボン酸、例えばヨージパミド、ジアトリゾ酸、およびメトリゾ酸が、加水分解的に不安定なヨウ化種、例えばWIN67721、WIN12901、WIN68165、およびWIN68209などと並んで、好ましくあり得る。
【0057】
活性薬剤の多数の組合せが、例えば、ステロイドとβアゴニストの組合せ(例えば、プロピオン酸フルチカゾンとサルメテロール、ブデソニドとフォルメテロール、など)を包含することが望ましくあり得る。
【0058】
炭水化物の例は、デキストラン、ヘタスターチ、シクロデキストリン、アルギン酸塩、キトサン、コンドロイチン、ヘパリンなどである。
【0059】
(小球状粒子)
本発明の粒子および小球状粒子は、動的光散乱法(例えば、光相関分光法、レーザー回折、低角レーザー光散乱(LALLS)、中角レーザー光散乱(MALLS))によって、光暗化法(light obscuration method)(例えば、コールター(Coulter)分析法)によって、あるいはレオロジーまたは(光学もしくは電子)顕微鏡法のような他の方法によって測定されるように、好ましくは約0.01μm〜約200μm、より好ましくは0.1μm〜10μm、さらにより好ましくは約0.5μm〜約5μm、そして最も好ましくは約0.5μm〜約3μmの平均幾何学粒子径を有する。肺送達のための粒子は、飛行時間計測(例えば、エアロゾル化器)またはアンダーセンカスケードインパクター(Andersen Cascade Impactor)計測によって決定される空気力学的粒子サイズを有する。
【0060】
これらの小球状粒子は実質的に球状である。「実質的に球状」とは、粒子断面の最短垂直軸の長さに対する最長垂直軸の長さの比が約1.5以下であることを意味する。実質的に球状は、対称中心線を必要としない。さらに、これらの粒子は、表面テクスチャー加工、例えば粒子全体のサイズと比較して小規模の線または圧痕もしくは隆起などを有し得るが、依然として実質的に球状である。より好ましくは、この粒子の最長軸と最短軸の間の長さの比は、約1.33以下である。より好ましくは、この粒子の最長軸と最短軸の間の長さの比は、約1.25以下である。面接触は、実質的に球状であるミクロスフェアにおいて最小化され、貯蔵の際の粒子の望ましくない凝集を最小化する。多くの結晶または薄片は、大きな面接触領域を可能にし得る平面を有し、ここでイオン性相互作用または非イオン性相互作用によって凝集が生じ得る。球は、はるかに小さな領域にわたる接触を可能にする。
【0061】
これらの粒子はまた、実質的に同じ粒子サイズを有することが好ましい。相対的に大きな粒子および相対的に小さな粒子の両方が存在する広いサイズ分布を有する粒子により、より小さな粒子がより大きな粒子間のギャップを埋めることが可能になり、それによって新しい接触面を生じる。広いサイズ分布が、結合性の凝集のための多くの接触機会を与えることによって、より大きな球を生じ得る。本発明は、狭いサイズ分布を有する球状粒子を作製し、それによって接触凝集のための機会が最小限になる。「狭いサイズ分布」とは、好ましい粒子サイズ分布が、第10百分位数の小球状粒子の体積粒径(volume diameter)に対する第90百分位数の小球状粒子の体積粒径の比が5以下である比を有することを意味する。より好ましくは、この粒子サイズ分布は、第10百分位数の小球状粒子の体積粒径に対する第90百分位数の小球状粒子の体積粒径の比が3以下である比を有する。より好ましくは、この粒子サイズ分布は、第10百分位数の小球状粒子の体積粒径に対する第90百分位数の小球状粒子の体積粒径の比が2以下である。
【0062】
幾何学的標準偏差(GSD)もまた、狭いサイズ分布を示すのに用いられ得る。GSD計算値は、有効カットオフ直径(ECD)を累積で15.9%および84.1%のパーセンテージ未満と決定するのに用いた。GSDは、15.9%未満のECDに対する84.17%未満のECDの比の平方根に等しい。GSDが2.5未満、より好ましくは1.8未満である場合、このGSDは狭いサイズ分布を有する。
【0063】
本発明の好ましい形態において、小球状粒子中の活性薬剤は半晶質または非晶質である。
【0064】
代表的には、本発明のプロセスによって作製される小球状粒子は実質的に無孔であり、0.5g/cm3を超える密度、より好ましくは0.75g/cm3を超える密度、そして最も好ましくは約0.85g/cm3を超える密度を有する。密度について好ましい範囲は約0.5〜約2g/cm3であり、そしてより好ましくは約0.75〜約1.75g/cm3であり、そしてさらにより好ましくは約0.85g/cm3〜約1.5g/cm3である。
【0065】
本発明の粒子は、活性薬剤の高い含有量を示し得る。多くの他の粒子調製方法に必要とされる有意量の増量剤または類似の賦形剤の必要はない。例えば、インスリン小球状粒子は、95重量%と同等かまたは95重量%を超える粒子からなる。しかし、増量剤または賦形剤は、これらの粒子中に含まれ得る。好ましくは、この活性薬剤は、粒子の約0.1重量%〜95重量%を超えて、より好ましくは約30重量%〜約100重量%、さらにより好ましくは約50重量%〜約100重量%、なおより好ましくは約75重量%〜約100重量%、そして最も好ましくは90重量%を超えて存在する。本明細書中に範囲を記載する場合、その中に任意の範囲または範囲の組合せを包含することを意味する。
【0066】
本発明のさらなる態様は、これらの小球状粒子が、賦形剤の封入体を含むかまたは含まずに活性薬剤の生化学的完全性および生化学的活性を保持することである。
【0067】
(粒子のインビボ送達)
本発明における活性薬剤を含む粒子は、適切な経路(例えば、注射可能な、局所、経口、直腸、経鼻、肺、膣、頬側、舌下、経皮、経粘膜、眼球、眼球内または耳)による、薬剤を必要とする被験体へのインビボ送達に適している。これらの粒子は、安定な液体懸濁液として送達され得るか、または錠剤、キャプレッツ、カプセルなどのような固形の剤形として処方され得る。好ましい送達経路は、静脈内、筋肉内、皮下、腹腔内、髄腔内、硬膜外、動脈内、関節内などを包む、注射可能な経路である。別の好ましい送達経路は、肺吸入である。この送達経路において、これらの粒子は、肺深部に、上気道中に、またはこの気道中のどこにでも沈積し得る。これらの粒子は、乾燥粉末吸入器によって乾燥粉末として送達され得るか、または定量噴霧式吸入器もしくは噴霧器によって送達され得る。
【0068】
全身的に機能することを目的とする薬物(例えばインスリン)は、血流中への吸収に利用可能な非常に大きな表面積がある肺胞中に沈積されるのが望ましい。肺内の特定の領域に薬物沈積を標的化する場合、粒子の空気力学的直径は、これらの粒子の基礎物理特性(例えば、形状、密度、および粒子サイズ)を操作することによって、最適範囲に調整され得る。
【0069】
吸入薬物粒子の受容可能な呼吸可能画分は、粒子組成物中に組み込まれるか、薬物粒子との混合物として組み込まれて、処方物に賦形剤を添加することによってしばしば達成される。例えば、微粉化薬物粒子(約5μm)の沈積の改善は、トレハロース、ラクトースまたはマルトデキストリンなどの不活性キャリア粒子のより大きな(30〜90μm)粒子とのブレンディングによってもたらされる。より大きな賦形剤粒子は、粉末流動性を改善し、これは薬力学的効果の改善と相関関係にある。さらなる改善において、これらの賦形剤は小球状粒子中に直接組み込まれて、タンパク質薬物の安定性を強力に高めると同時に、エアロゾル性能に影響を及ぼす。一般に、賦形剤は、以前に吸入がFDAに認可されたもの(例えばラクトース)、または肺にとって内因性の有機分子(例えばアルブミンおよびDL−α−ホスファチジルコリンジパルミトイル(DPPC))が選択される。他の賦形剤、例えば乳酸・グリコール酸共重合体(PLGA)は、望ましい物理的および化学的特性で粒子を操作するのに用いられてきた。しかしながら、FDAに認可された賦形剤を用いる吸入経験の多くは、気管気管支領域中に望ましく沈積する大きな空気力学的粒子サイズを有し、かつ認めうるほどには肺深部に達しない喘息薬を用いてきた。肺深部に送達される吸入タンパク質またはペプチド治療法について、賦形剤が肺胞領域に送達される場合、免疫応答に起因し得るかまたは賦形剤によって引き起こされ得る炎症および刺激のような望ましくない長期副作用が生じ得ることが懸念される。
【0070】
肺深部吸入治療法の潜在的な有害副作用を最小化するために、送達されるべき薬物で実質的に構成される、吸入用の粒子を調製することが有利であり得る。このストラテジーは、賦形剤への肺胞の曝露を最小化し、各々の投与毎に肺胞表面上に沈積される粒子の総質量用量を低減させ、おそらく吸入治療の慢性的使用の間に刺激を最小化する。本質的に完全に治療タンパク質またはペプチドからなる、肺深部沈積に適した空気力学的性質を有する小球状粒子は、肺の肺胞膜に対する慢性的治療投薬の影響に関する単独研究に特に有用であり得る。次いで、吸入による小球状粒子の形態でのタンパク質またはペプチドの全身送達の影響が、関連する賦形剤によって複雑な要素が導入されることなく研究され得る。
【0071】
吸入によって肺深部に粒子を送達する必要条件は、これらの粒子が、0.5〜10マイクロメーターの小さな平均空気力学的直径および狭いサイズ分布を有することである。本発明はまた、異なる粒子サイズ範囲を有する粒子の種々のバッチを一緒に混合することも企図する。本発明のプロセスにより、上記特性を有する小球状粒子の成形加工が可能になる。
【0072】
0.5〜3ミクロンの空気力学的直径を有する粒子を形成するための2つの主要なアプローチがある。第1のアプローチは、比較的大きいが極めて多孔性の(または有孔の)ミクロ粒子を生成することである。空気力学的直径(D空気力学的)と幾何学的直径(D幾何学的)の間の関係は、D空気力学的が、極めて低い質量密度(約0.1g/cm3)を有する粒子の密度の平方根をD幾何学的に乗じたものに等しいので、比較的大きな幾何学的直径(5〜10ミクロン)を有しながら、小さな空気力学的直径(0.5〜3ミクロン)を示し得る。
【0073】
代替のアプローチは、比較的低い多孔率を有する粒子を生成することであり、本発明の場合は、これらの粒子は上に示した範囲内の密度を有し、そしてさらに一般にはこの密度はほぼ1g/cm3である。従って、このような非多孔性の高密度粒子の空気力学的直径は、ほぼそれらの幾何学的直径である。
【0074】
上述の粒子形成のための本方法は、賦形剤を用いるかまたは用いない粒子形成を提供する。
【0075】
添加物なしでのタンパク質単独からのタンパク質小球状粒子の成形加工は、より大きな薬物有効負荷量、安全性の増大および必要吸入数の減少についての選択肢を提供するので、肺送達における使用に優れた利点をもたらす。
【0076】
(前調製小球状粒子のマイクロカプセル化)
本発明の小球状粒子または他の方法で調製された小粒子(ミクロ粒子、ミクロスフェア、ナノスフェア、ナノ粒子などを含む)は、さらに壁形成材料のマトリックス内にカプセル化されて、マイクロカプセル化粒子を形成し得る。マイクロカプセル化は、当該分野で公知の任意のプロセスによって達成され得る。好ましい実施形態において、本発明の小球状粒子または任意の他の小粒子のマイクロカプセル化は、下記のように乳化/溶剤抽出プロセスによって達成される。このマトリックスは、活性薬剤に徐放性を与え得、結果として所望の治療用途によって数分から数時間、数日または数週間持続する放出速度をもたらす。これらのマイクロカプセル化粒子はまた、前調製小球状粒子の遅延放出処方物も生成し得る。好ましい実施形態において、これらの前調製小球状粒子は高分子の粒子である。別の好ましい実施形態において、この高分子はタンパク質またはポリペプチドである。
【0077】
この乳化/溶剤抽出プロセスにおいて、乳化は、2つの不混和相である連続相および不連続相(分散相としてもまた公知である)を混合してエマルジョンを形成することによって生じる。好ましい実施形態において、連続相は水性相(または水相)であり、かつ不連続相は有機相(または油相)であって、水中油(O/W)エマルジョンを形成する。この不連続相は、油中固体(S/O)相を形成する微細懸濁液または微細分散液のいずれかとして存在する固体粒子の分散系をさらに含み得る。この有機相は、好ましくは水不混和性または部分的に水混和性の有機溶剤である。この水性相に対する有機相の重量比は、約1:99〜約99:1であり、より好ましくは約1:99〜約40:60であり、そして最も好ましくは約2:98〜約1:3であるか、あるいはその中の任意の範囲または範囲の組合せである。好ましい実施形態において、この水性相に対する有機相の比は約1:3である。本発明は、油相が連続相を形成しかつ水相が不連続相を形成する、逆エマルジョンすなわち油中水エマルジョン(W/O)の利用をさらに企図する。本発明は、例えば油中水中油エマルジョン(O/W/O)または水中油中水エマルジョン(W/O/W)のような3つ以上の相を有するエマルジョンの利用をさらに企図する。
【0078】
好ましい実施形態において、この乳化/溶剤抽出プロセスを用いるマイクロカプセル化のプロセスは、前調製小球状粒子を以前に記載した方法によって調製する工程および壁形成材料を含む有機相を調製する工程で始まる。これらの前調製小球状粒子を壁形成材料の有機相中に分散させて、油相中に前調製小球状粒子の分散系を含む油中固体(S/O)相を形成する。好ましい実施形態において、この分散は、小球状粒子と有機相の混合物をホモジナイズすることによって達成される。水性媒体は連続相を形成する。この場合、S/O相と水性相を乳化することによって形成されるエマルジョン系は、水中油中固体(S/O/W)エマルジョン系である。
【0079】
壁形成材料とは、マトリックスの構造体を個々にまたは組み合わせて形成可能な材料をいう。生分解性の壁形成材料は、特に注射用用途に好ましい。このような材料の例としては、ポリラクチド/ポリグリコリドポリマー(PLGA)のファミリー、ポリエチレングリコール結合体化PLGA(PLGA−PEG)、およびトリグリセリドが挙げられるが、これらに限定されない。PLGAまたはPLGA−PEGが用いられる実施形態において、PLGAは好ましくは100:0〜0:100、より好ましくは約90:10〜約15:85、そして最も好ましくは約50:50のポリラクチド対ポリグリコリド比を有する。一般に、ポリマー中のポリグリコリド対ポリラクチド比が高いほど、マイクロカプセル化粒子はより親水性になり、結果としてより速い水和およびより速い分解をもたらす。種々の分子量のPLGAもまた用いられ得る。一般に、ポリマー中のポリグリコリドとポリラクチドの比率が同じであれば、PLGAの分子量が高いほど活性薬剤の放出が遅くなり、マイクロカプセル化粒子のサイズ分布が広くなる。
【0080】
水中油(O/W)または水中油中固体(S/O/W)エマルジョンの有機相(油相)中の有機溶剤は、水不混和性であるかまたは部分的に水不混和性であり得る。用語「水不混和性溶剤」は、1:1の比率(O/W)で水溶液と混合した場合に界面メニカスを形成する溶剤を意味する。適切な水不混和性溶剤としては、炭素数5以上の置換または非置換の直鎖、分枝または環状アルカン、炭素数5以上の置換または非置換の直鎖、分枝または環状アルケン、炭素数5以上の置換または非置換の直鎖、分枝または環状アルキン;炭化水素を完全にまたは部分的にハロゲン化した芳香族炭化水素、エーテル、エステル、ケトン、モノ−、ジ−またはトリ−グリセリド、天然油、アルコール、アルデヒド、酸、アミン、直鎖または環状シリコーン、ヘキサメチルジシロキサン、あるいはこれらの溶剤の任意の組合せが挙げられるが、これらに限定されない。ハロゲン化溶剤としては、四塩化炭素、塩化メチレン、クロロホルム、テトラクロロエチレン、トリクロロエチレン、トリクロロエタン、ヒドロフルオロカーボン、塩素化ベンゼン(モノ、ジ、トリ)、トリクロロフルオロメタンが挙げられるが、これらに限定されない。特に適切な溶剤は、塩化メチレン、クロロホルム、ジエチルエーテル、トルエン、キシレンおよび酢酸エチルである。「部分的に水混和性の溶剤」は、ある濃度では水不混和性であり、別のより低い濃度では水混和性である溶剤を意味する。これらの溶剤は、水混和性が制限された溶剤であり、かつ自発的エマルジョン形成が可能である。部分的に水混和性の溶剤の例は、テトラヒドロフラン(THF)、プロピレンカーボネート、ベンジルアルコール、および酢酸エチルである。
【0081】
界面活性化合物は、例えば有機相の湿潤性を増大させるために添加され得る。この界面活性化合物は、乳化プロセス前に水性相、有機相、水性媒体と有機溶液の両方に添加され得るか、または乳化プロセス後にエマルジョンに添加され得る。界面活性化合物の使用により、カプセル化されていないかまたは部分的にカプセル化された小球状粒子の数を減少させ得、結果として放出の間の活性薬剤のイニシャルバーストの低減をもたらす。この界面活性化合物は、化合物の溶解度に応じて、有機相、または水性相、または有機相と水性相の両方に添加され得る。
【0082】
用語「界面活性化合物」は、例えば陰イオン界面活性剤、陽イオン界面活性剤、双性イオン界面活性剤、非イオン性界面活性剤または生物学的界面活性分子のような化合物を意味する。この界面活性化合物は、水性相または有機相またはエマルジョンのいずれの場合にもその約0.01重量%未満〜約30重量%、より好ましくは約0.01重量%未満〜約10重量%、またはその中の任意の範囲もしくは範囲の組合せで存在すべきである。
【0083】
適切な陰イオン界面活性剤としては、以下:ラウリン酸カリウム、ラウリル硫酸ナトリウム、ドデシル硫酸ナトリウム、ポリオキシエチレンアルキル硫酸塩、アルギン酸ナトリウム、スルホコハク酸ジオクチルナトリウム、ホスファチジルコリン、ホスファチジルグリセロール、ホスファチジルイノシン、ホスファチジルセリン、ホスファチジン酸およびそれらの塩、グリセリルエステル、カルボキシメチルセルロースナトリウム、コール酸および他の胆汁酸(例えば、コール酸、デオキシコール酸、グリココール酸、タウロコール酸、グリコデオキシコール酸)およびそれらの塩(例えば、デオキシコール酸ナトリウムなど)が挙げられるが、これらに限定されない。
【0084】
適切な陽イオン界面活性剤としては、第4級アンモニウム化合物、例えば、塩化ベンザルコニウム、臭化セチルトリメチルアミン、塩化ラウリルジメチルベンジルアンモニウム、アシルカルニチンハイドロクロライド、またはアルキルピリジニウムハライドが挙げられるが、これらに限定されない。陰イオン界面活性剤として、リン脂質が用いられ得る。適切なリン脂質としては、例えばホスファチジルコリン、ホスファチジルエタノールアミン、ホスファチジルセリン、ホスファチジルイノシトール、ホスファチジルグリセロール、ホスファチジン酸、リゾリン脂質、卵または大豆リン脂質あるいはこれらの組合せが挙げられる。このリン脂質は、加塩されても脱塩されてもよく、水素化されても部分的に水素化されてもよく、あるいは天然、半合成または合成であってもよい。
【0085】
適切な非イオン性界面活性剤としては、以下:ポリオキシエチレン脂肪アルコールエーテル(MacrogolおよびBrij)、ポリオキシエチレンソルビタン脂肪酸エステル(ポリソルベート)、ポリオキシエチレン脂肪酸エステル(Myrj)、ソルビタンエステル(Span)、グリセロールモノステアレート、ポリエチレングリコール、ポリプロピレングリコール、セチルアルコール、セトステアリルアルコール、ステアリルアルコール、アリールアルキルポリエーテルアルコール、ポリオキシエチレン−ポリオキシプロピレンコポリマー(ポロクサマー)、ポラキサミン(polaxamine)、ポリビニルアルコール、ポリビニルピロリドン、および多糖類(例えばヒドロキシエチルデンプン(HES)、メチルセルロース、ヒドロキシセルロース、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、および非晶質セルロースのような、デンプンおよびデンプン誘導体を含む)が挙げられる。本発明の好ましい形態において、非イオン性界面活性剤はポリオキシエチレンおよびポリオキシプロピレンコポリマーであり、好ましくはプロピレングリコールおよびエチレングリコールのブロックコポリマーである。このようなポリマーは、商品名POLOXAMERのもとで販売され、また時々PLURONIC(登録商標)とも呼ばれ、そしてSpectrum Chemical and Rugerを含むいくつかの業者によって販売される。ポリオキシエチレンの中で、短いアルキル鎖を有する脂肪酸エステルが挙げられる。このような界面活性剤の一例は、BASF Aktiengesellschaft製のポリエチレン−660−水酸化ステロイドである、SOLUTOL(登録商標)HS15である。
【0086】
界面活性生物学的分子としては、アルブミン、カゼイン、ヘパリン、ヒルジン、ヘタスターチまたは他の適切な生体適合性薬剤のような分子が挙げられる。
【0087】
本発明の好ましい形態において、水性相は界面活性化合物としてタンパク質を含む。好ましいタンパク質はアルブミンである。このタンパク質はまた、賦形剤としても機能し得る。タンパク質が界面活性化合物ではない実施形態において、他の賦形剤が、乳化プロセス前または後のいずれかに添加されて、エマルジョン中に含まれ得る。適切な賦形剤としては、糖類、二糖類、および糖アルコールが挙げられるが、これらに限定されない。好ましい二糖類はスクロースであり、好ましい糖アルコールはマンニトールである。
【0088】
さらに、チャネリング剤、例えばポリエチレングリコール(PEG)を用いることによって最終産物の水浸透率を増大させ得、その結果、水和率を改変することによって、マトリックスからの活性薬剤の初期放出動力学ならびにマトリックスの分解速度および分解依存性放出動力学の改変をもたらす。カプセル化の間のチャネリング剤としてPEGを用いることは、PEGを相分離促進剤として用いる小球状粒子の成形加工の間の洗浄プロセスの部分をなくすという点で有利であり得る。さらに、緩衝液を用いて連続相のpHを変動させることにより、粒子表面と有機相の間の湿潤プロセスを有意に増進させ得、従って結果として、マイクロカプセル化粒子のマトリックスからのカプセル化治療剤のイニシャルバーストを有意に減少させる。連続相の性質は、例えば、NaClのような塩を添加してその塩分濃度を増加させることによっても改変され、2つの相の混和性を低下させ得る。
【0089】
小球状粒子を有機相(油相)中に分散した後、次いで水性媒体(水相)の連続相を、例えば均質化または超音波処理によってこの有機相の不連続相と勢いよく混合し、初期マイクロカプセル化粒子の乳化小滴を含むエマルジョンを形成する。この連続水性相は、乳化小滴からの有機溶剤の急速な抽出を最小限にするために、有機相中に用いた有機溶剤で飽和させた後に、水性相と有機相を混合し得る。この乳化プロセスは、この混合物がその流動性を維持し得る任意の温度で実行され得る。エマルジョン安定性は、有機相中または水性相中の、あるいは界面活性化合物が乳化プロセス後にエマルジョンに添加される場合はエマルジョン中の、界面活性化合物の濃度の関数である。これは、エマルジョン系(胚性マイクロカプセル化粒子)の小滴サイズならびにマイクロカプセル化粒子のサイズおよびサイズ分布を決定する要因の1つである。マイクロカプセル化粒子のサイズ分布に影響を及ぼす他の要因は、連続相の粘度、不連続相の粘度、乳化の間の剪断力、界面活性化合物の種類および濃度、ならびに油/水比である。
【0090】
乳化後、次いでこのエマルジョンを硬化媒体中に移す。この硬化媒体は、胚性マイクロカプセル化粒子から不連続相中に溶剤を抽出し、結果として、乳化小滴の近傍内に、前調製小球状粒子の周囲に固体ポリマーマトリックスを有する固体マイクロカプセル化粒子の形成をもたらす。O/W系またはS/O/W系の実施形態において、硬化媒体は水性媒体であり、界面活性化合物、または増粘剤、または他の賦形剤を含み得る。これらのマイクロカプセル化粒子は、好ましくは球状であり、約0.6〜約300μmの粒子サイズを有し、そしてより好ましくは約0.8〜約60μmの粒子サイズを有する。さらに、これらのマイクロカプセル化粒子は、好ましくは狭い粒子サイズ分布を有する。不連続相の抽出時間を短縮するために、硬化媒体に熱または減圧が適用され得る。例えば蒸発(煮沸効果)による不連続相の急速な除去は、マトリックスの連続性の破壊を生じるので、これらの初期マイクロカプセル化粒子からの不連続相の抽出速度は、最終の固体マイクロカプセル化粒子中の多孔度にとって重要な因子である。
【0091】
好ましい実施形態において、この乳化プロセスは、バッチプロセスの代わりに連続様式で実行される。図41は、連続乳化反応器のデザインを示す。
【0092】
別の好ましい実施形態において、活性薬剤の小球状粒子をカプセル化する、硬化壁形成ポリマーマトリックスは、遠心分離および/または濾過(ダイアフィルトレーションを含む)によってさらに回収され、水で洗浄される。残りの液相は、凍結乾燥または蒸発のようなプロセスによってさらに除去され得る。
【0093】
(A.インスリン小球状粒子)
(実施例1:インスリン小球状粒子の調製の一般法)
16.67%のPEG3350を含む、pH5.65に緩衝化した溶液(0.033Mの酢酸ナトリウム緩衝液)を調製した。亜鉛結晶性インスリンの濃縮スラリーを、この溶液に攪拌しながら添加した。最終溶液中のインスリン濃度は0.83mg/mLであった。この溶液を約85〜90℃に加熱した。インスリン結晶をこの温度範囲で5分間以内に完全に溶解した。この溶液の温度が制御速度で低下する場合、インスリン小球状粒子は約60℃で形成を開始した。収率は、PEGの濃度が増大するにつれて増大した。このプロセスにより、平均値1.4μmを有する種々のサイズ分布の小球状粒子が生成される。
【0094】
形成されたインスリン小球状粒子を、これらの小球状粒子が溶解しない条件下でダイアフィルトレーションによってミクロスフェアを洗浄することにより、PEGから分離した。これらのインスリン小球状粒子を、Zn2+を含む水溶液を用いて懸濁液から洗い出した。このZn2+イオンは、インスリンの溶解度を低下させて、収率を低下させかつ小球状粒子凝集を引き起こすインスリンの溶解を妨げる。
【0095】
(実施例2:インスリン小球状粒子を作製するための無攪拌バッチプロセス)
20.2mgの亜鉛結晶性インスリンを1mLの脱イオン水中に室温で懸濁した。50マイクロリットルの0.5N HClをこのインスリンに添加した。1mLの脱イオン水を添加して、l0mg/mLの亜鉛結晶性インスリン溶液を生成した。12.5gのポリエチレングリコール3350(Sigma)と12.5gのポリビニルピロリドン(Sigma)を、50mLの100mM酢酸ナトリウム緩衝液(pH5.7)に溶解した。このポリマー溶液の容量を、酢酸ナトリウム緩衝液で100mLに調整した。エッペンドルフチューブ中の800マイクロリットルのポリマー溶液に、400マイクロリットルの10mg/mLインスリン溶液を添加した。このインスリン/ポリマー溶液は、混合すると濁りを生じた。ポリマー溶液の代わりに水を用いてコントロールを調製した。これらのエッペンドルフチューブを、水浴中で90℃にて30分間、混合または攪拌せずに加熱し、次いで取り出して氷上に10分間置いた。このインスリン/ポリマー溶液は、90℃水浴から取り出した際には透明であったが、冷めるにつれて濁り始めた。ポリマーを含まないコントロールは、実験を通して透明なままであった。粒子を、遠心分離によってこのインスリン/ポリマーチューブから回収し、続いて2回洗浄してポリマーを除去した。最後の水中の懸濁液を凍結乾燥して、乾燥粉末を得た。インスリン/ポリマーチューブからの凍結乾燥粒子のSEM分析により、直径約1マイクロメーターの小球状粒子の一様分布を示した。これらの粒子のコールター光散乱粒子サイズ分析により、1.413マイクロメーターの平均粒子サイズ、0.941〜1.88マイクロメーターの95%信頼限界、および0.241マイクロメーターの標準偏差を有する狭いサイズ分布を示した。ポリマーまたは洗浄工程を含まないが、別な方法で同様に処理および凍結乾燥されたインスリンコントロールは、SEM下で薄片のみ(粒子なし)を示し、外観上はタンパク質の凍結乾燥後に典型的に得られる薄片と類似していた。
【0096】
(実施例3:インスリン小球状粒子作製のためのプロセスを通しての連続フロー)
36.5mgのインスリンを量り取り、3mLの脱イオン水中に懸濁した。30μLの1N HClを添加してインスリンを溶解させた。この溶液の最終容量を、脱イオン水で3.65mLに調整した。次いで、7.3mLのPEG/PVP溶液(100mM NaOAc緩衝液中の25%PEG/PVP(pH5.6))をこのインスリン溶液に添加して、最終全容量が10.95mLのインスリン溶液にした。次いで、この溶液をボルテックスして、インスリンとPEG/PVPの均質な懸濁液を得た。
【0097】
このインスリン懸濁液を、テフロン(登録商標)チュービング(TFE内径1/32ゲージ可撓性チュービング)を介して、0.4mL/分の速度で作動中のBioRad蠕動ポンプに連結した。このポンプからのチュービングを、90℃に維持した水浴中に沈め、その後に氷中に埋めたコレクションチューブ中に挿入した。このインスリン溶液の温度が水浴中での約90℃から氷中の収集管中で約4℃に低下すると、インスリン小球状粒子が形成された。図7は、このプロセスの概略図である。このプロセスのための全実行時間は、10.95mL容量について35分間であった。小球状粒子を収集した後、この収集管を3000rpmで20分間、Beckman J6B遠心機中で遠心分離した。2回目の水洗を完了し、これらの小球状粒子ペレットを2600rpmで15分間遠心分離した。最後の水洗は1500rpmで15分間遠心分離した。アリコートを粒子サイズ分析のために取り出した。これらの小球状粒子を−80℃で凍結し、そして2日間凍結乾燥した。
【0098】
この粒子サイズを、容量で1.397μm、表面積で1.119μm、そしてBeckman Coulter LS230粒子カウンターによる決定のとおり、数で0.691μmと決定した。走査型電子顕微鏡写真により、サイズが均質であり凝集形成していないインスリン小球状粒子が示された(図8)。
【0099】
インスリン溶液を短期間90℃に曝露する連続貫流プロセスの使用により、小球状粒子の生成が可能になった。この方法により、高速液体クロマトグラフィー(HPLC)によって決定されるとおり90%タンパク質である最終組成物を生成した(図9)。HPLC分析によっても、溶解インスリン小球状粒子が約4.74分の溶離時間を有することを示し、これはインスリン標準または天然インスリン出発物質の溶離時間と有意には異ならず、小球状粒子へ成形加工後のインスリンの生化学的完全性の保存を示した。
【0100】
(実施例4:インスリン小球状粒子を作製するための熱交換バッチプロセス)
ヒト亜鉛結晶性インスリンを超音波処理で必要最低限量の脱イオン水中に懸濁して、完全分散を確実にした。このインスリン懸濁液を、0.1M酢酸ナトリウム緩衝液中における最終溶質濃度が0.83%亜鉛結晶性インスリン、18.5%ポリエチレングリコール3350、0.7%塩化ナトリウムとなるように、77℃に予熱した撹拌緩衝化ポリマー溶液(25℃でpH5.65)に添加した。初期に混濁した混合物は、結晶インスリンが溶解するにつれて3分以内に透明になった。清澄後直ちに、この溶液を、熱交換器(カラム内径:25mm、長さ:600mm;Ace Glass Incorporated,Vineland,NJ)として用いられるガラス製の水ジャケット付きクロマトグラフィーカラムに移した。このガラス製カラムを垂直に配置し、そしてこの熱交換機流体はカラムの下端で水ジャケットに入り、上端から出た。この系の熱交換特性を実証するために、熱電対(タイプJ、Cole Parmer)をカラムの上端および下端でインスリン処方物液体の中心に配置し、冷却温度プロフィールを予備試運転の間に得た。これらの熱電対を、外部表面変化が生じないように、この実験のために実施された6回のバッチの間に取り除いた。
【0101】
この熱交換器を65℃に予熱し、インスリン緩衝化ポリマー溶液を、この溶液温度が65℃未満に下がらず、かつ気泡がこの溶液中に導入されない方法で移した。この透明な溶液を熱交換器中で4分間65℃に平衡化させた後、熱交換流体を65℃供給から15℃供給に切り換えた。この熱交換器中のインスリン処方物を20分間にわたって15℃に平衡化させた。温度が60℃を経て55℃に下がるにつれてインスリン小球状粒子が形成され、その結果、均一な安定したクリーム状の白色懸濁液を生じた。
【0102】
これらのインスリン小球状粒子を、5倍容量の0.16%酢酸ナトリウム−0.026%塩化亜鉛緩衝液(pH7.0)に対するダイアフィルトレーション(A/G Technologies、750,000 MWCO限外濾過カートリッジ)によってポリエチレングリコールから分離し、続いて原体積の5分の1に濃縮した。このインスリン小球状粒子懸濁液を、5倍容量の脱イオン水に対するダイアフィルトレーションによってさらに洗浄し、続いて凍結乾燥して水を除去した。ダイアフィルトレーションの間の小球状粒子の凝集(膜表面上のこれらの粒子の分極パッキング)および凍結乾燥の間の小球状粒子の凝集(凍結前のこれらの小球状粒子の沈殿)が起こらないように気を付けた。これらの乾燥小球状粒子は易流動性であり、解凝集またはふるい分けの必要のない、使用準備の整った状態であった。
【0103】
(インスリンの小球状粒子)
上記のプロセスにより、均一なサイズの球状粒子が、賦形剤を添加することなく亜鉛結晶性インスリンから生成する。このプロセスによって調製された小球状粒子は、飛行時間計測(エアロサイザー(商標))およびアンダーセンカスケードインパクター計測によって決定されるように、単純な広く用いられている乾燥粉末吸入器(Cyclohaler(商標))から送達される場合に肺深部送達を示す高度呼吸可能画分を有し、優れた空気力学的性質を有する。インスリンをモデルタンパク質として用いることにより、本発明者らはまた、確立されたU.S.P.方法を用いてこのタンパク質の化学的完全性に対するこのプロセスの影響も検査し得る。
【0104】
乾燥粉末インスリン小球状粒子を、偏光顕微鏡法(Leica EPISTAR(登録商標),Buffalo,NY)によって、および走査型電子顕微鏡(AMRAY 1000,Bedford,MA)を用いてイメージ化した。粒子サイズ分析を、エアロサイザー(登録商標)モデル3292粒子サイジングシステム(この機器に粉末を導入するために、モデル3230 Aero−Disperser(登録商標)乾燥粉末分散器(TSI Incorporated,St.Paul,MN)を備える)を用いて行った。個々の粒子サイズを、エアロサイザーの結果を電子顕微鏡写真と比較することによって確認した。
【0105】
このプロセスの前後のインスリンの化学的完全性を、ヒトインスリンについてのUSPモノグラフ(USP26)に従いHPLCによって決定した。インスリンおよび高分子量タンパク質含量を、276nmでのUV検出を用いる定組成SEC HPLC法を用いて測定した。インスリン、A−21デサミドインスリンおよび他のインスリン関連物質を測定するために、USP勾配逆相HPLC法を用いてサンプルを分析した。インスリン含量を、214nmでのUV検出を用いて測定する。高分子量タンパク質、デサミドインスリン、および他のインスリン関連物質をアッセイして、このプロセスによって引き起こされる任意の化学分解を定量した。
【0106】
インスリン小球状粒子の空気力学的性質を、エアロサイザー(登録商標)機器を用いて検査した。インスリン乾燥粉末についてのサイズ分布測定を、低い剪断力、媒体供給量、および正常な解凝集でAeroDisperser付属装置を用いて行った。この機器のソフトウェアは、飛行時間計測データをサイズに変換し、それを対数的に間隔をあけた範囲に配置する。各サイズビン中で検出された粒子の数、ならびに各サイズビン中で検出された粒子の全体積を、統計分析に用いた。体積分布は数分布よりも大粒子を際立たせ、従って、体積分布は大粒子のみならず非分散粒子の凝集塊の検出時にもより感度が高い。
【0107】
アンダーセンカスケードインパクターアセンブリは、1つのプレセパレータ、9つのステージ、8つの収集プレート、および1つのバックアップフィルターで構成されていた。これらのステージを、−1、−0、1、2、3、4、5、6、およびFと番号付けする。ステージ−1は、オリフィスステージのみである。ステージFは、ステージ6のための収集プレートおよびバックアップフィルターを備える。ステンレス鋼収集プレートを、粒子の「はね返り」を防ぐために食品用シリコーンの薄層でコーティングした。サンプラーによる60LPMのサンプル流空気流速を分析に用いた。正確に秤量した約10mgのサンプルサイズを、4秒でCyclohalerからエアロゾルとして送達された粉末とともに、各デンプンカプセル(Vendor)中に秤量した。各プレート上に沈積したインスリン粉末の量を、ヒトインスリンについてのUSP26アッセイに従い214nmでの逆相HPLC検出によって決定した。
【0108】
空気動力学的中央粒子径(MMAD)を、有効カットオフ直径(ECD)に対する質量パーセント未満の累積性のプロビットフィットを用いるSigma Plotソフトウェアによって算出した。放射線量(ED)を、カスケードインパクター中に沈積したインスリンの全実測質量として決定した。これを、Cyclohalerカプセル中に充填されたインスリン小球状粒子の質量のパーセンテージとして表す。
【0109】
これらの結果により、相変化処方に関連して以下のプロセスパラメータの慎重な制御を実現し得ることを示す:1)約2μmの直径を有する主に球状の粒子;2)狭いサイズ分布;3)およびバッチ間再現性の空気力学的性質;および4)残留水分を除いて95%を超える活性薬物(ヒトインスリン)からなる小球状粒子。本発明者らは、亜鉛結晶性インスリンの溶解度が、溶液温度、pH、ポリマー濃度、およびイオン強度によって制御され得ることを決定した。本発明者らはまた、相変化期間中の冷却速度を制御することが、狭いサイズ範囲内に主に球状の粒子を形成することを可能にする重要なパラメータであることも見出した。
【0110】
図2は、本実施例に相当するプロセスのための冷却温度プロフィールである。このプロフィールを、垂直に配置した水ジャケット付きクロマトグラフィーカラムを用いて測定し、そしてこの熱交換流体は、カラムの下端で水ジャケットに入り上端から出た。2つの熱電対を、カラムの中に、この溶液に接触させて配置した。1つの熱電対をこのカラムの上端に配置し、2番目の熱電対をこのカラムの下端に配置する。これらの温度曲線は時間−温度プロットを別個の領域に分け、事前の最適化実験により、最適な温度変化速度よりも高いかまたは低い温度変化速度で相変化を誘導すると粒子サイズが広範囲になりかつ非球形状となる傾向があることを決定した。60℃を超える温度では、インスリンは緩衝化ポリマー溶液中に可溶性のままである(領域A;図2)。約8.6℃/分〜26.5℃/分の速度で温度が低下する場合、均一化されたサイズの球状粒子の最適形成が支持される(領域B;図2)。25.6℃/分よりも速い冷却速度がこの処方物に適用される場合、容易に凝集するインスリンの極めて微細な(0.5ミクロン未満の)非球状粒子を生成する傾向がある(領域C;図2)。8.6℃/分よりも遅い冷却速度により、非球状かつ非晶質の綿状沈殿とともに、より広いサイズ分布のインスリン小球状粒子を生成する傾向がある(領域D;図2)。
【0111】
熱交換器内部のインスリン緩衝化ポリマー溶液の温度が図2の領域Bの範囲内に入ると相変化が生じ、結果として乳白色の安定なインスリン小球状粒子の懸濁液を生じる。相分離を示すミクロスフェア形成は、温度が60℃を下回ると生じ始め、温度が40℃に達すると完了するようである。この処方物を、PEGポリマーを除去するためにダイアフィルトレーションによって洗浄する前に15℃まで冷却しても、懸濁液中にさらなる変化は観察されなかった。
【0112】
出発ヒト亜鉛結晶性インスリン原料のSEMが、約5〜40μmの粒子サイズを含む非均質サイズおよび結晶形状を示すのに対して、本実施例からのバッチの1つを撮影したSEM写真は、球状かつ均一なサイズのインスリン小球状粒子を示す(図3b)。SEMで示した粒子の形状およびサイズは、本実施例のために調製した他の5回のバッチの代表である。
【0113】
ダイアフィルトレーション洗浄による緩衝化ポリマーからの分離および脱イオン水懸濁液からの凍結乾燥の後、これらの乾燥粉末インスリン小球状粒子は、比較的易流動性であり、かつ秤量および取り扱いが容易であった。インスリン小球状粒子の含水率は、出発亜鉛結晶性インスリン原料について12%であったのと比較して、2.1〜4.4%の水分の範囲内で変動した。HPLCによるインスリン小球状粒子の化学分析は、高分子量化合物の増加が全くなく、このプロセスによるインスリンの化学分解は極めて少ないことを示した(図4)。ダイマー(%)、A21デサミドインスリン(%)、後期溶出ピーク(%)、および他の化合物(%)が(出発インスリン原料を超えて)増大したにもかかわらず、6回のバッチ全てについての結果はUSP限界の範囲内であった。インスリン効力の保持は、出発原料について28.7IU/mgであったのと比較して、28.3〜29.9IU/mgであった。このプロセスに用いられたポリマー(ポリエチレングリコール)の残留レベルは0.13%未満から検出不可能までであり、このことから、このポリマーはインスリン小球状粒子の重要な成分ではないことが示された。
【0114】
(インスリン小球状粒子についての空気力学的性質のバッチ間再現性)
エアロサイザーおよびアンダーセンカスケードインパクターデータによって実証されるように、生成された6回の別個のインスリン小球状粒子のバッチの間に、空気力学的性質について優れた再現性があった。6回のバッチ全てについて、エアロサイザーデータにより、99.5%を超えるこれらの粒子が0.63〜3.4μmのサイズ範囲内にあるとともに、最低限60%の小球状粒子が1.6〜2.5μmの狭いサイズ範囲内にあることを示した(図5)。統計学的に、このデータにより、生成したインスリン小球状粒子バッチの少なくとも99%がこれらの粒子の少なくとも96.52%を0.63〜3.4μmのサイズ範囲(目標直径2μmの−68.5%〜70%)内に有することは、95%信頼し得ることを示す。
【0115】
このアンダーセンカスケードインパクターデータは、Cyclohalerから送達される用量の平均17.6%がこれらの機器の開口部およびプレセパレータ/スロート中に沈積したことを除いては、エアロサイザーデータとよく一致した(図6)。このデータは、エアロサイザーの粉末分散効率がCyclohalerデバイスのものよりも大きいことを示す。しかしながら、これらの6回のバッチについての平均放射線量はCyclohalerに基づいて71.4%であったとともに、72.8%の放射線量がこのインパクターのステージ3上に沈積した。肺深部送達のための呼吸可能画分は、ECDを含む画分が1.1ミクロンと3.3ミクロンの間であると見積もられる場合、平均60.1%の吸入インスリン小球状粒子が肺深部送達およびその後の全身吸収に利用可能である。このプロセスについての優れた再現性を表1に示し、ここで、6回の別個のバッチについてMMAD平均およびGSD平均についての標準偏差値は極めて低い。このことは、このプロセス変量が厳格な制御下にあり、結果として空気力学的性質についてバッチ間均一性をもたらすことを示す。
【0116】
【表1】
表1は、インスリン小球状粒子の空気力学的性質を示す。結果(平均+/−SD)を、アンダーセンカスケードインパクターによる別個のインスリン小球状粒子バッチ(N=6)の分析に基づいて算出した。このプロセスについての非常に良好な再現性が、MMADおよびGSDについての極めて低い標準偏差によって実証される。
【0117】
この冷却プロセスによって生成したインスリン小球状粒子は、表1の空気力学的データによって証明されたように、凝集する傾向をほとんど示さなかった。
【0118】
(実施例5:インスリン小球状粒子を作製するための攪拌容器プロセス)
2880mLの緩衝化ポリマー溶液(0.1M酢酸ナトリウム緩衝液(2℃でpH5.65)中の18.5%ポリエチレングリコール3350、0.7%塩化ナトリウム)を、ガラス製の3リットルの水ジャケット付き攪拌容器に添加し、75℃まで予熱した。2.4グラムのヒト亜鉛結晶性インスリンをこの80mLの緩衝化ポリマー溶液中に超音波処理で懸濁して、完全分散を確実にした。このインスリン懸濁液を、攪拌して予熱した緩衝化ポリマー溶液に添加し、さらに5分間攪拌した。この混合物はこの間に透明になり、これは亜鉛結晶性インスリンが溶解したことを示していた。10℃に設定した冷却装置からの水を、このインスリンポリマー溶液が15〜20℃に低下するまで容器のジャケットを通してポンピングした。結果として生じた懸濁液を、5倍容量の0.16%酢酸ナトリウム−0.026%塩化亜鉛緩衝液(pH7.0)、続いて5倍容量の脱イオン水に対してダイアフィルトレーションし、続いて凍結乾燥して水を除去した。凍結乾燥粉末のSEM分析は、TSIエアロサイザー飛行時間計測分析により1.433マイクロメーターの平均空気力学的直径を有する均一な小球状粒子を示した。アンダーセンカスケードインパクター分析により、ステージ3からフィルターまでの上に沈積した放射線量が73%、MMADが2.2、およびGSDが1.6(全ての指標は、この粉末の優れた空気力学的性質を示す)を結果として得た。
【0119】
(実施例6:小球状粒子生成処方物のイオン強度を調整することによるインスリン分解生成物の形成の低下)
インスリンはまた、長期間または酸性環境でなければ、より低い初期温度(例えば、75℃)でこの溶液中に溶解され得るが、この溶液にNaClを添加することによって著しい凝集を生じる。
【0120】
インスリン小球状粒子の成形加工プロセスの改善を、以下の技術を用いて達成した。亜鉛結晶性インスリンの濃縮スラリーを、約85〜90℃に予熱した0.1M酢酸ナトリウム(pH5.65)中のポリエチレングリコールの16.7%溶液に(攪拌しながら)(室温にて)添加した。このインスリン結晶を、この温度範囲で5分間以内に完全に溶解した。この溶液の温度が低下するにつれて、インスリン小球状粒子が生成した。
【0121】
化学反応によるA21デサミドインスリンおよびインスリンダイマーの顕著な生成が、高温によって85〜90℃の初期温度で生じた。しかしながら、これは75℃では長時間を要した。長時間により、結果として顕著なインスリン分解も生じた。インスリンを酸性環境中に前溶解することにより、大きなパーセンテージのインスリンがA21デサミドインスリン分解生成物への望ましくない変換も生じた。
【0122】
実験中、化学的手段によってインスリンダイマーの形成を低下させるために、塩化ナトリウムを緩衝化ポリマー反応混合物に添加した。添加した塩化ナトリウムは、デサミドまたはダイマーインスリン分解生成物の形成を有意には低下させなかったが、塩化ナトリウムの添加はオリゴマー(高分子量インスリン生成物)の形成を大幅に低下させた(表2)。
【0123】
【表2】
さらに、Zn結晶性インスリンは、NaClを含まないコントロールよりもNaClの存在下でずっと速く溶解した。このことから、塩化ナトリウムの添加によりインスリンの溶解率が改善されることが示唆され、亜鉛インスリン結晶の初期溶解に用いられる温度の低下が可能になった。処方物への0.7%NaClの添加により、亜鉛結晶性インスリン原料を、以前にNaClを添加しなければ必要であった87℃よりも有意に低い温度である75℃で5分以内に溶解することが可能になることを実証した実験において、この仮説を確認した。75℃にて、NaClの非存在下では、インスリンは13分後に完全には溶解しなかった。
【0124】
一連の実験から、塩化ナトリウム濃度の増大(2.5mg/ml、5.0mg/ml、10.0mg/ml、および20.0mg/ml)により、インスリン結晶が溶解する温度がさらに低下し、そしてまた小球状粒子が形成を開始する温度も低下することを実証した(図l0a〜d)。さらに、この処方物中のNaCl濃度が増大すると、より高濃度のZn結晶性インスリンが急速に溶解することを決定した。従って、所定の温度でのインスリンの溶解度は、初期連続相の塩化ナトリウムレベルを調整することによって慎重に制御可能であることを確認した。これにより、分解生成物の形成があまり誘導されない温度でこのプロセスを誘導することが可能になる。
【0125】
塩化ナトリウムが、インスリンを溶解させる温度の低下を可能にする固有の化学的性質を有するか否かを決定するために、等モル濃度の塩化アンモニウムおよび硫酸ナトリウムを、塩化ナトリウムを含むコントロールと比較した。NH4ClおよびNa2S04の両方とも同じように亜鉛結晶性インスリン原料を溶解するのに必要な温度を低下させた。イオン強度が高いほど、溶液温度が低下するにつれて小球状粒子を形成する能力に影響を及ぼすことなく、ミクロスフェア生成処方物中のインスリンの溶解度を増大させるようである。
【0126】
(実施例7:インスリン小球状粒子の収率およびインスリン濃度およびサイズに対するPEG濃度の研究)
ポリエチレングリコール(3350)滴定データにより、PEG−3350の増大によっても小球状粒子の収率が増大することを示す。しかしながら、PEG濃度が高すぎると粒子の球形状が崩れ、収率のわずかな改善を無効にする。
【0127】
インスリン濃度データは、PEGと相反する傾向を示し、インスリン濃度が増大すると結果として小球状粒子の収率が低下する。
【0128】
本発明者らは、インスリン濃度が高いほど、より大きな直径の小球状粒子を生成するという一般的傾向を見出す。この実験において、濃度が高いほど、結果として非球状粒子と小球状粒子の混合も生じた。
【0129】
(実施例8:イヌを用いたインスリン小球状粒子研究)
この実験研究の目的は、ビーグル犬の肺におけるエアロゾル化インスリン粉末の沈積についての定量化および視覚化実験を実施することであった。本明細書中に開示される方法に従って99mTc標識インスリン粒子を作製した。エアロゾル化インスリンの肺沈積を、ガンマシンチグラフィーを用いて評価した。
【0130】
5匹のビーグル犬を本研究に用い、各動物に99mTc放射標識インスリン粒子エアロゾルを投与した。イヌの識別番号は、101、102、103、104、および105であった。
【0131】
エアロゾル投与の前に、これらの動物を、麻酔用の注入ラインを通してプロポフォールで麻酔し、エアロゾル送達のために各動物に気管内チューブを設置した。
【0132】
各イヌを、放射標識エアロゾルの吸入のために「スパングラーボックス」チャンバー中に入れた。放射標識エアロゾル投与の直後に、ガンマカメラコンピュータ画像を前側ならびに後側胸部領域について取得した。
【0133】
2つのインビトロカスケードインパクターコレクションを、1つは最初の動物(101)のエアロゾル投与前に、そしてまた最後の動物(105)の曝露後に評価して、99mTc放射標識インスリン粉末の安定性を確証した。
【0134】
これらの結果を図11に図示した。カスケードインパクターコレクションはいずれの場合も単峰形の分布を示した。
【0135】
図12は、全ての動物についてのP/I比計算の結果を示す。このP/I比は、肺の末梢部、すなわち肺深部に沈積する99mTcインスリン粉末の比率の尺度である。代表的なP/I比は、おそらく約0.7である。0.7を超えるP/I比は、中心肺と比較して末梢肺または気管支領域に顕著な沈積を示す。
【0136】
図13のシンチグラフィー画像は、呼吸器系内のインスリン沈積位置を示し、P/Iデータ(図12)と一致する。イヌ101についてのシンチグラフィー画像は、本研究において5匹全てのイヌを代表する。
【0137】
イヌ101についてのシンチグラフィー画像は、末梢肺に明白な沈積の増加を伴う気管または気管支の沈積をほとんど示さない。肺の外側の放射能は、エアロゾル化粉末の肺深部沈積からの99mTcの急速な吸収に起因する。
【0138】
これらのP/I比および画像データは、99mTc放射標識インスリンが主に肺深部に沈積したことを示す。末梢肺中に沈積した放射標識インスリンの量により、これらの粒子の凝集が低レベルであることを示した。
【0139】
(実施例9:インスリン小球状粒子からポリマーを除去するための亜鉛を含む緩衝液に対するダイアフィルトレーション)
PSEA溶液中のインスリン小球状粒子の成形加工に続いて、この懸濁液から全てのPSEAを除去した後に凍結乾燥することが望ましかった。残留PSEAがほんの数パーセントであっても、結合剤として作用し、小球状粒子の非破砕性凝集塊を形成し得る。この凝集は、DPIデバイスから送達される粉末の放射線量および空気力学的性質に悪影響を及ぼした。さらに、PSEAの反復投与への肺組織の曝露は、毒物学的問題を生じ得た。
【0140】
3つの技術を、凍結乾燥の前にPSEAから小球状粒子を分離するために検討した。濾過は、少量の粒子を収集するのに用いられ得た。しかしながら、小球状粒子がより多量であれば、濾過媒体の細孔を急速にブロックし、数ミリグラムを超える粒子の洗浄および回復が実行不能になる。
【0141】
これらの粒子を回収するために遠心分離(その後に、洗浄溶剤中の再懸濁および再遠心分離を含む数回の洗浄サイクルが続く)を首尾よく用いて、PSEAを除去した。これらのインスリン小球状粒子が容易に溶解されず、かつPSEAが溶液中に残ったので、脱イオン水を洗浄溶剤として用いた。遠心分離の1つの不都合は、小球状粒子が、これらの粒子をスピンダウンさせるのに必要な高いg力によってペレット中に詰まったことであった。連続的な洗浄ごとに、これらのペレットを分散した粒子に再懸濁することがますます困難になった。インスリン粒子の凝集は、しばしば遠心分離プロセスの望ましくない副作用であった。
【0142】
中空繊維カートリッジを用いるダイアフィルトレーションを、インスリン小球状粒子を洗浄するために遠心分離に代わる手段として用いた。ダイアフィルトレーション装置の従来のセットアップにおいて、緩衝化PSEA/インスリン粒子懸濁液を密閉容器中に入れ、そしてこの懸濁液を十分な背圧で繊維を通って再循環させ、濾液が中空繊維膜を通過するようにした。再循環速度および背圧を最適化して、膜の細孔の閉塞(分極)を防止した。懸濁液から除去された濾液の体積を、洗浄溶剤を攪拌密閉容器中に吸い上げることによって、連続的に補充した。ダイアフィルトレーションプロセスの間に懸濁液中のPSEAの濃度は徐々に低下し、そして約1時間にわたって懸濁液の原体積を洗浄溶剤で5〜7回交換した後には、このインスリン小球状粒子懸濁液は本質的にPSEAを含まなかった。
【0143】
ダイアフィルトレーションプロセスはポリマーの除去時に極めて有効であり、かつ商業量への拡大の影響を非常に受けやすかったが、インスリン小球状粒子は、洗浄溶剤として本来用いられた脱イオン水中にゆっくりと溶解した。実験により、インスリンは濾液中に徐々に失われ、そして懸濁液の原体積の20倍に相当する脱イオン水が交換された後にインスリン粒子が完全に溶解することを決定した。これらのインスリン小球状粒子は、脱イオン水中にやや溶けにくいことが分かったが、高性能のダイアフィルトレーションプロセスにより、懸濁液から可溶性インスリン(およびおそらく亜鉛イオン)が持続的に除去された。したがって、所定容量の脱イオン水中の不溶性インスリン濃度と可溶性インスリン濃度の間の平衡はダイアフィルトレーションでは生じず、インスリンの溶解に恵まれた条件となった。
【0144】
表3は、可能性のある洗浄媒体として評価された種々の溶液を示す。10mgの乾燥インスリン小球状粒子を1mLの各溶液中に懸濁し、室温で48時間徐々に混合した。可溶性インスリンのパーセンテージを24時間および48時間に測定した。このインスリンは、24時間足らずで可溶性インスリンの全重量のほんの1%未満で平衡に達し、脱イオン水中にやや溶けにくいことが見出された。しかしながら、先述のとおり、高性能のダイアフィルトレーションによって可溶性インスリン(および亜鉛)を連続的に除去するので、この平衡は決して達成されず、インスリン小球状粒子は溶解し続ける。従って、理想的な洗浄溶液中のインスリン溶解度は、水中のインスリン溶解度よりも低い。インスリンは、その等電点付近で可溶性が最も低いので、2種類のモル濃度のpH5.65の酢酸緩衝液を試験した。このインスリンの溶解度は、緩衝液のモル濃度に左右され、低モル濃度で水に相当することが見出された。エタノールはインスリンの溶解度を大いに低下させたが、ほぼ無水濃度でだけは低下させなかった。このインスリン溶解度は、水溶液と混合したエタノールをダイアフィルトレーションの初期段階においてPSEA/インスリン小球状粒子懸濁液中に用いた場合、実際に増大する。
【0145】
【表3】
注射用の市販亜鉛結晶性インスリン懸濁液中に用いられる緩衝液もまた、溶液中に亜鉛を含む。これらの溶液の2つをインスリン小球状粒子を用いて試験し、脱イオン水と比較してインスリン溶解度を大いに低下させることを見出した。文献によると、亜鉛結晶性インスリンは、各インスリン六量体に結合した2〜4個のZnイオンを有するべきである。インスリン小球状粒子を作製するための原料として用いられる種々の亜鉛結晶性インスリン調製物について、六量体当たりの亜鉛イオンは1.93〜2.46に及ぶ。これは、所定重量の原料亜鉛結晶性インスリン当たり0.36〜0.46%亜鉛に相当した。インスリン小球状粒子の形成および脱イオン水に対するダイアフィルトレーションの後、58〜74%の亜鉛がプロセシングの間に失われた。インスリン粒子からの亜鉛の損失により、インスリンの溶解度の増大およびダイアフィルトレーション中の損失を生じた。
【0146】
0.16%酢酸ナトリウム−0.027%ZnCl2(pH7.0)に対してインスリン小球状粒子をダイアフィルトレーションすることにより、濾液中のインスリン損失を事実上排除した。しかしながら驚くべきことに、これらのインスリン小球状粒子の亜鉛含量は、出発亜鉛結晶性インスリン原料について測定した0.46%を十分に上回る、ほぼ2%まで増加した。亜鉛を含む緩衝液に対するダイアフィルトレーションの別の予想外の結果は、Cyclohaler DPIデバイス(脱イオン水に対してダイアフィルトレーションされた68% 対 亜鉛緩衝液ダイアフィルトレーション後の84〜90%)に基づいて観察された放射線量の劇的な改善およびアンダーセンカスケードインパクターのスロート中に沈積されたインスリン粒子の量の減少であった。亜鉛緩衝液ダイアフィルトレーションは、インスリン小球状粒子乾燥粉末の分散性を改善し、そして粒子の凝集を低下させ、結果としてインパクターのより低いステージ上で、より低いMMADおよびより高い沈積をもたらした。このことは、亜鉛緩衝液ダイアフィルトレーションおよびインスリン小球状粒子中のより高い亜鉛濃度により、肺深部に沈積した線量の割合(%)を改善し得ることを示唆した。
【0147】
MDI適用で用いる賦形剤を添加せずに噴霧剤HFA−134a中に懸濁した場合、亜鉛緩衝液で洗浄したインスリン小球状粒子の不可逆的凝集は明白には存在しなかった。これらのインスリン粒子は1分足らずで懸濁液から凝集したが、使用直前に振盪すると容易に再懸濁された。使用直前にMDI容器を振盪する工程は、通常、任意のMDI生成物を使用するために提供された使用説明書の一部である。実際に、これらの粒子はMDI加圧容器の底で高密度充填層に定着しないので、MDI容器の底に定着する遊離した凝集粒子はインスリン粒子の長期凝集を(球形状に起因する最小限の接触に加えて)事実上阻害し得る。従って、インスリン小粒子の亜鉛緩衝液ダイアフィルトレーションによって付与された性質により、インスリンおよび他の亜鉛結合化合物のためのMDI調製物の長期有効期間および分散性を改善し得る。
【0148】
インスリン小球状粒子はXRPD分析によって非晶質であることが見出されたので、亜鉛結合は、インスリンモノマーの亜鉛イオン配位で会合せずに六量体を形成した。従って、イオンの非特異的結合および得られた潜在的利点が、亜鉛以外のイオンの結合にまで及び得た。亜鉛と結合しない種々のタンパク質は、ダイアフィルトレーションプロセスにおいて溶解度を低下させる他のイオンに結合し、類似の有益な効果を付与し得た。
【0149】
これらの小球状粒子を10mg/mLの濃度でヒドロフルオロアルカン(HFA)134a噴霧剤中に懸濁した。HFA134a中に貯蔵後のインスリンの化学安定性を、時間0および1ヶ月目に評価した。図28に示されるデータは、モノマーインスリン、インスリンダイマー、インスリンオリゴマー、インスリン主要ピークおよびA21−デサミドインスリンに関してインスリンミクロスフェアの保存を示す。
【0150】
以下の研究において、実施例4の方法に従って調製したインスリン小球状粒子を、アンダーセンカスケードインパクター法を用いて3つの異なる吸入デバイスでの性能について比較した。Cyclohalerデバイスは市販の乾燥粉末吸入器(DPI)であり、Disphalerは別の乾燥粉末吸入器であり、そして定量噴霧式吸入器(MDI)は、ミクロスフェアが本実施例に記載のようにHFA134a中に懸濁されて100マイクロリットルかまたは他のサイズの絞り弁を通って推進されるデバイスである。図29における結果は、アンダーセンカスケードインパクターデバイスのステージに嵌入する小球状粒子がステージ3および4上に沈積することを明らかに示す。このことは、吸入器として用いられるデバイスにかかわらず、極めて再現性のある小球状粒子の性能を示す。DPIデバイスとMDIデバイスの間の唯一の重大な相違は、MDIを用いてアンダーセンカスケードインパクターのスロート部分中に沈積した小球状粒子の量が有意に大きいことである。このMDIデバイスがアンダーセンインパクターのスロートに対して小球状粒子を推進させる高速度は、DPIデバイスと比較して沈積したインスリンミクロスフェアの割合が高いことを説明する。流出速度を減じたかまたは改変したMDIデバイスを用いてスロート中に沈積する小球状粒子の数を減少させ得ることは、当業者によって当然のこととみなされ得る。さらなる手段は、MDIの末端部でのスペーサーデバイスの使用であり得る。
【0151】
インスリン小球状粒子(ロット番号YQ010302)を、本実施例に記載の方法に従って凍結乾燥インスリン出発物質から作製した。インスリン小球状粒子についての1年間の貯蔵安定性を、25℃および37℃で凍結乾燥インスリン出発物質と比較した。インスリン安定性を、全関連インスリン化合物、インスリンダイマーおよびオリゴマーならびにA21−デサミドインスリンを試験することによって比較した。
【0152】
図30〜35は、1年間にわたって、インスリン小球状粒子は同一条件下で貯蔵したインスリン出発物質と比較して有意に少ない量のインスリンダイマーおよびオリゴマー、A21−デサミドインスリンならびに全関連インスリン化合物を示したことを示す。これにより、インスリンのミクロスフェア形態は出発物質よりも化学変化に対して有意に安定性が高いことを示す。
【0153】
インスリン小球状粒子を製造後0時間および10ヶ月後にアンダーセンカスケードインパクター研究において試験した。Cyclohaler DPIデバイスを用いて長期貯蔵後の空気力学的安定性を決定した。図36は、空気力学的性能が10ヶ月貯蔵後に著しく不変のままであることを示す。
【0154】
ラマン分光研究を行って加工前のインスリンサンプルと本実施例で調製した小球状粒子中のインスリンとの間の構造的相違を明らかにした。小球状粒子中のインスリンは、これらの元の加工前インスリンサンプルよりも実質的により高いβ−シート含量を有し、その結果、より低いα−ヘリックス含量を有することを示した。これらの研究結果は、小球状粒子中における凝集ミクロフィブリル構造物の形成と一致する。しかしながら、水性媒体中に溶解すると、このスペクトルは、加工前のミクロスフェアまたはインスリンのいずれかから生じる本質的に同一のタンパク質構造物を顕示し、ミクロスフェア中の任意の構造変化が溶解に際して十分に可逆的であることを示す。
【0155】
以下の2つのバッチのインスリンを、ラマン分光法を用いて試験した:A)加工前のインスリンUSP(Intergen、カタログ番号4502−10、ロット番号;XDH1350110)およびB)小球状粒子中のインスリン(JKPL072502−2NB32:P.64)。これらの粉末状サンプルまたはインスリン溶液(0.01MのHCl中に約15mg/mL)を標準ガラス毛細管中に詰め、ラマン分析のために12℃に温度調整した。代表的には、2〜15μLのアリコートは、レーザー照明に曝露されたサンプル毛細管の部分を満たすのに十分であった。スペクトルをアルゴンレーザー(Coherent Innova 70−4 Argon Ion Laser,Coherent Inc.,Santa Clara,CA)を用いて514.5nmで励起させ、光子含有検出器(モデルR928P,Hamamatsu,Middlesex,NJ)を用いて走査型二重分光計(Ramalog V/VI,Spex Industries,Edison,NJ)に記録した。1.0cm−1間隔でのデータを、1.5秒の組み込み時間および8cm−1のスペクトルスリット幅で収集した。サンプルを繰り返しスキャンし、そして個々のスキャンを平均する前に表示しそして検討した。代表的には、各サンプルの少なくとも4回のスキャンを収集した。分光計をインデンおよび四塩化炭素を用いて較正した。スペクトルを、SpectraCalcおよびGRAMS/AIバージョン7ソフトウェア(Thermo Galactic,Salem,NH)を用いてデジタル微分法によって比較した。このスペクトルを、溶剤(もしあれば)およびバックグラウンドの寄与について補正した。これらの溶液のスペクトルを、同一条件下で0.01MのHClスペクトルを取得することによって補正し、傾斜したバックグラウンド上に位置する一連の5つの重複したガウス関数−ローレンツ関数に適合させた[S.−D.Yeo,P.G.Debenedetti,S.Y.Patro,T.M.Przybycien,J.Pharm.Sci.,1994,83,1651〜1656]。この適合を1500〜1800cm−1領域で行った。
【0156】
ラマンスペクトルを、粉末状インスリンサンプルおよびそれらの各溶液の両方について得た(図10i)。加工前のサンプルのスペクトルは、市販のインスリンサンプルの前述のスペクトルに非常によく一致する[S.−D.Yeo,P.G.Debenedetti,S.Y.Patro,T.M.Przybycien,J.Pharm.Sci.,1994,83,1651〜1656;J.L.Lippert,D.Tyminski,P.J.Desmueles,J.Amer.Chem.Soc.,1976,98,7075〜7080]。この小球状粒子サンプルは、タンパク質の二次構造において有意な摂動を示す、アミドIモードにおける明白な(約+10〜+15cm−1)シフトを示した。しかしながら、特に、市販の粉末および小球状粒子のスペクトルは、これらのサンプルが水性媒体中に溶解した場合に事実上同一であり、加工の際の二次構造の変化が完全に可逆的であったことを示していた。
【0157】
二次構造パラメータを、平滑化、蛍光および芳香族バックグラウンドの減算、およびアミドIバンドデコンヴォルーションを含むコンピュータアルゴリズムを用いて推定した。指数関数的に減衰する蛍光を、本質的に他に記載されるように減算した[S.−D.Yeo,P.G.Debenedetti,S.Y.Patro,T.M.Przybycien,J.Pharm.Sci.,1994,83,1651〜1656]。推定した構造パラメータを表4に集結する。
【0158】
【表4】
(実施例10:等温法によるヒトインスリンの小球状粒子の調製)
ヒトインスリンUSP(Intergen)をNaClおよびPEG(分子量3350、スペクトルロット番号RP0741)溶液中に分散させ、結果として最終インスリン濃度0.86mg/mL、ならびに0.7重量%のNaCl濃度および8.3重量%のPEG濃度となった。微量の氷酢酸および1MのNaOH溶液の添加によってpHを5.65に調整した。T1=77℃まで加熱した後、結果としてインスリン濃度Ceqの透明なタンパク質溶液を得た。次いで、これらの溶液を所定の速度で温度T2=37℃まで冷却した。T2で、タンパク質沈殿を認めた。これらの沈殿を遠心分離(13,000×g、3分間)によって再び温度37℃で除去し、この生じた上清中のインスリン濃度(C*)をビシンコニンタンパク質アッセイによって0.45mg/mLと決定した。このようにして調製した、37℃に保持されているインスリン溶液を、溶液Aと称する。
【0159】
溶液Bを、0.7重量%NaCl/8.3重量%PEG(HClの添加によってpHを約2.1にした)中にヒトインスリンを溶解させることによって調製し、結果として2mg/mLのインスリン濃度となった。この溶液を7時間攪拌しながら37℃でインキュベートし、続いて2分間超音波処理した。生じた溶液Bのアリコートを溶液Aに添加し、結果として1mg/mLの全インスリン濃度となった。生じた混合物を37℃で一晩激しい攪拌下に保持し、結果としてインスリン沈殿を生じ、これらの沈殿をメンブランフィルター(有効ポア直径、0.22μm)を用いて液体から穏やかに除去した。次いで生じたタンパク質ミクロ粒子を液体窒素中で急速凍結させて、凍結乾燥させた。
【0160】
(B.α−1−アンチトリプシン(AAT)の小球状粒子)
本発明はまた、肺送達に特に適したAATの小球状粒子を調製するのにも用いられ得る。
【0161】
(実施例11:AAT小球状粒子(10〜300mgスケール)のジャケット付きカラムバッチ調製)
16%PEG3350および0.02%プルロニックF−68を含むl0mM酢酸アンモニウムでpH6.0に緩衝化した溶液をジャケット付きビーカー中でマグネティック攪拌棒を用いて混合し、かつ30℃に加熱した。このビーカー温度を、循環水浴を用いて制御した。組換えAAT(rAAT)の濃縮溶液をこの溶液に攪拌しながら添加し、そしてpHを6.0に調整した。最終溶液中のrAAT濃度は2mg/mlであった。rAATは、この溶液組成物中にこの温度で完全に可溶性であった。この容器の全内容物をジャケット付きカラムに移し、そして25〜30℃に加熱した。このカラムのための循環水浴をセットして−5℃まで下降させた。このカラムおよび内容物を約1℃/分で約4℃の温度まで冷却した。rAAT小球状粒子はこの冷却工程の間に形成された。ミクロスフェア懸濁液をガラス結晶皿中で凍結させ、そして凍結乾燥して水および緩衝液を除去した。
【0162】
凍結乾燥後のタンパク質小球状粒子からPEGを抽出するために、PEG/タンパク質ケークを塩化メチレン(MeCl2)で洗浄した。利用される別の洗浄媒体は、塩化メチレン:アセトン1:1または塩化メチレン:ペンタン1:1であった。この洗浄工程は、原体積洗浄を合計3回繰り返した。最終ペレットを小容量のアセトンまたはペンタン中に再懸濁し、窒素ガスへの直接曝露かまたはロータリーエバポレーションのいずれかによって乾燥させた。
【0163】
(実施例12:AAT小球状粒子(200〜2000mgスケール)のジャケット付き容器バッチ調製)
このタイプの調製を、ジャケット付きカラムと同じ処方組成物を用いて行ったが、より大きな容量に適応可能であり、スケールアップにより適していた。このスケールで、処方物をジャケット付き容器(通常500〜1000ml)中でA形のパドル式インペラーを用いて75rpmで混合し、そして30℃に加熱した。この容器温度を循環水浴を用いて制御した。同じ容器中に溶液を保持して、水浴源を30℃浴から2℃浴に切り換えた。この容器および内容物を約1℃/分で4℃の温度まで冷却した。rAAT小球状粒子はこの冷却工程の間に形成された。この温度を、熱電対を用いてモニタリングし、この懸濁液が4℃に達すると、ほぼこの温度をさらに30分間保持した。この保持工程の後、この小球状粒子懸濁液をダイアフィルトレーションによって約4℃で濃縮し、ポリマーおよび容量の約75%を除去した。残りの小球状粒子懸濁液を予冷した凍結乾燥トレー中で薄層として凍結し、そして凍結乾燥して水および残りの緩衝液を除去した。
【0164】
これらのタンパク質小球状粒子を、有機溶剤で遠心分離することによって(実施例10に記載されるように)または超臨界流体(SCF)抽出によって、残った乾燥ポリマーから分離した。SCF抽出のために、この乾燥物質を高圧抽出チャンバー中に移し、このチャンバーをCO2で2500psi(室温で)まで加圧した。一旦作業圧に達すると、エタノールを、70:30のC02:エタノール混合物として吸込流体流に導入した。この超臨界流体は、小球状粒子を残してポリマーを溶解した。このプロセスの終わりに、このシステムをエタノールで洗い流し、ゆっくりと除圧した。
【0165】
(実施例13:プロセス収率−rAATの小球状粒子への変換%)
小球状粒子を実施例10および11に記載のように作製した。冷却プロセスが完了した後、この懸濁液の小アリコートを取り出し、そして0.2μmのシリンジフィルターを通して濾過して固体小球状粒子を除去した。溶液中に残ったrAATである、濾液の吸光度を、UV分光光度計を用いて280nmで決定した。次いで、rAAT濃度を検量線から算出した。変換%を以下のように算出した:
(出発rAAT濃度−濾液rAAT濃度)/出発rAAT濃度*100%=変換%
【0166】
【化2】
上の表に示すように、プロセススケールにかかわらず高いパーセンテージのAATタンパク質を小球状粒子に変換した。
【0167】
(実施例14:種々のプロセススケールでのAAT粒子の粒子サイズ分布)
(エアロサイザーデータ)
最終的なAAT乾燥粉末小球状粒子のサンプルを、飛行時間計測によって粒子サイズを測定する、TSIエアロサイザー3225で分析した。これらの測定値から種々の比の体積粒径を算出して、AAT小球状粒子の粒子サイズ分布を実証し、かつ本発明の方法以外の方法で作製した粒子と比較するのに用いた。
【0168】
【化3】
(アンダーセンデータ)
5〜l0mgのサンプルをゲルカプセル中に秤量し、そしてCyclohaler乾燥粉末吸入器を用いて流速60リットル/分(LPM)でアンダーセンカスケードインパクター中に投与した。小球状粒子を全てのインパクターステージから収集し、0.2Mのトリス塩酸緩衝液中にpH8.0で溶解し、そして逆相HPLCを用いて定量した。このデータを分析し、そして幾何学的標準偏差(GSD)を米国薬局方(USP)に記載のように算出した。このデータは狭いサイズ分布を実証した。
【0169】
【化4】
前掲の全ての分布パラメータは、本発明の成形加工方法によってもたらされる優れた粒子サイズ分布を実証した。
【0170】
(実施例15:AAT生物活性の保持)
比活性を決定するために、rAAT小球状粒子を0.2Mのトリス塩酸(室温でpH8.0)中に溶解した。生じた溶液を、C末端にp−ニトロアニリド基を含む合成ペプチドを加水分解する、ブタ膵エラスターゼ(PPE)の能力を阻害する、rAATの能力を測定するアッセイによって分析した。次いで、rAAT小球状粒子の同じ溶液を、タンパク質濃度についてビシンコニン酸(BCA)アッセイを用いてアッセイした。コントロールrAAT出発物質溶液もまた両方のアッセイで分析した。活性アッセイを進行させて1mg/mlのタンパク質/サンプルの濃度に基づいて活性を決定したので、BCAによって決定されるような実際のタンパク質濃度に基づいて活性値を補正し、以下の比活性値を得た:
【0171】
【化5】
rAATによるブタ膵エラスターゼの阻害
【0172】
【化6】
従って、この比活性は、小球状粒子へのAATの成形加工後の生物活性の保持を実証した。
【0173】
(実施例16:AATの構造完全性の保持)
制御相分離(CPS)技術の重要な識別ポイントの1つは、粒子形成の間に水性系を利用し、かつ他のストレス誘導性の条件(例えば、温度の上昇、剪断など)を回避する、温和な条件下での粒子の形成である。粒子工学の分野において、主要な関心事は、成形加工中のタンパク質の安定性および貯蔵安定性である。タンパク質の主要な分解経路、例えば酸化、脱アミドおよび特に凝集は、免疫原性を含むタンパク質処方の副作用を引き起こすと考えられる。従って、調節の関係上、最終粒子処方物中の分解生成物は極めて低レベルであることが必要である。HPLC、例えばCDおよびDSCのような物理的化学的特性決定を利用して、形成の間にタンパク質修飾が起こったか否かを決定した。
【0174】
円偏光二色性(CD)は、摂動を受けたタンパク質の構造変化の評価、または親タンパク質に対する改変タンパク質の構造比較に通常用いられる。このCD法は、タンパク質の折り畳み、ならびにタンパク質の二次構造および三次構造を評価する。
【0175】
二次構造は、「遠紫外線」スペクトル領域(190〜250nm)におけるCD分光法によって決定され得る。これらの波長では、発色団は、通常の折り畳み環境中にある場合、ペプチド結合である。α−ヘリックス、β−シート、およびランダムコイル構造はそれぞれ特徴的な形状およびサイズのCDスペクトルを生じる。従って、任意のタンパク質中に存在する各二次構造型の概算比率を、その遠紫外線CDスペクトルを分析することによって、各構造型についてのこのような参照スペクトルの分数倍数の合計として決定し得る。
【0176】
「近紫外線」スペクトル領域(250〜350nm)におけるタンパク質のCDスペクトルは、ある態様の三次構造に高感度であり得る。これらの波長では、発色団は芳香族アミノ酸およびジスルフィド結合であり、これらの発色団が生成するCDシグナルはタンパク質の全般的な三次構造に高感度である。250〜270nmの領域のシグナルはフェニルアラニン残基に起因し、270〜290nmのシグナルはチロシンに起因し、そして280〜300nmのシグナルはトリプトファンに起因する。ジスルフィド結合は、近紫外線スペクトル全体にわたって幅広の弱いシグナルを生じる。
【0177】
rAATストック溶液の遠紫外線CDスペクトルおよびリン酸緩衝液(pH7.4、T=25℃、タンパク質濃度0.05mg/mL)中の小球状粒子から放出されたAATを図13に示す。各スペクトルは10回のスキャンの平均を表す。
【0178】
遠紫外線CDスペクトルは識別不能であり、このことは、AATの小球状粒子への成形加工が引き続く放出の際に出発AAT物質の構造と同一の構造を有するAAT分子を結果として生じたことを実証している。
【0179】
(RP−HPLC)
小球状粒子を0.2Mのトリス塩酸中にpH8.0で溶解し、逆相HPLCによって分析した。出発rAATタンパク質のコントロール溶液と比較した場合、クロマトグラムの外観上に明らかな相違はない。
【0180】
HPLCシステム:
HPLCカラム−Pheomenex Jupiter、5ミクロン、C4、300A、250×4.6mm
Waters Alliance 2965ポンプ/オートサンプルラー
波長−280nm
注入容量−75μl
濃度勾配:
移動相1:0.1%TFA/水
移動相2:0.085%TFA/90%(c/v)アセトニトリル/水
実行時間−60分
流速−1.0ml/分
DSC
DSCダイアグラムを作成した。図15〜25bを参照のこと。
【0181】
(実施例17:AAT出発物質の貯蔵安定性に対するAAT小球状粒子の貯蔵安定性)
小球状粒子を、室温および4℃で1週間、1ヶ月間、2ヶ月間、3ヶ月間、6ヶ月間、および12ヶ月間貯蔵後の生物活性の保持について(実施例15に記載のアッセイを用いて)分析した。(図14bおよび14c)。このバルク材料は、rAAT出発溶液を透析し、次いで凍結乾燥したものである。各々の時点および貯蔵条件について、各々二連でアッセイした二連のサンプルが存在した。
【0182】
(C.ヒト成長ホルモン(hGH)の小球状粒子)
本発明はまたhGHの小球状粒子を調製するのにも用いられ得る。
【0183】
(実施例18:hGHの小球状粒子の試験管バッチ調製(20〜50mgスケール))
18%PEG3350を含むpH5.6で緩衝化した溶液(50mM酢酸アンモニウム/50mM重炭酸アンモニウム)(この溶液中におけるhGHの最終濃度は1mg/mlである)を50mlコニカルチューブ中で混合し、固定水浴中で58℃に加熱した。hGHをこれらの条件下でこの溶液中に溶解した。次いで、このチューブを水浴から取り出し、この溶液が10℃に達するまで氷浴中で冷却した。冷却速度を4〜6℃/分に維持した。hGHタンパク質小球状粒子はこの冷却工程の間に形成した。小球状粒子は、溶液の温度が約40℃に達すると形成し始めた。粒子形成後、hGHタンパク質小球状粒子を以下に記載の2つの方法のうち1つによってPEGから分離した。
【0184】
有機溶剤洗浄には、冷却工程および粒子形成の後に小球状粒子懸濁液を液体窒素で瞬間凍結し、そして凍結乾燥して水および緩衝液を除去することが必要であった。凍結乾燥後にPEGからタンパク質小球状粒子を分離するために、PEG/タンパク質ケークを塩化メチレン(MeCl2)中に懸濁した。PEGはMeCl2において可溶性であり、一方タンパク質小球状粒子は不溶性である。この懸濁液を室温で5分間混合した。hGH小球状粒子の密度はMeCl2の密度(d=1.335g/ml)に近いので、この液体密度を低下させて遠心分離を促進するために第2溶剤が必要であった。MeCl2と混和性のアセトンを、MeCl2の容量と等しい容量で添加した。次いで、この小球状粒子懸濁液を室温にて5分間3300rpmで遠心分離した。上清を捨て、ペレットをMeCl2中に再懸濁し、そして再び室温で5分間混合した。この洗浄手順を合計5回の洗浄の間繰り返した。最終洗浄の後、このペレットを小容量のMeCl2中に再懸濁し、ロータリーエバポレーションによって乾燥し、hGH小球状粒子の最終粉末を取り出した。
【0185】
亜鉛緩衝液洗浄には、冷却工程および粒子形成の後に、小球状粒子懸濁液を4℃にて10分間4000rpmで遠心分離して小球状粒子をPEGから分離することが必要であった。上清を除去し、このペレットを、除去した上清の容量と等しい容量の、50mMの酢酸亜鉛を含む冷緩衝液中に再懸濁した。Zn2+イオンはhGHの溶解度を低下させ、洗浄の間の溶解を阻止した。この洗浄緩衝液を氷上に保持した。次いで、この懸濁液を直ちに4℃にて5分間3000rpmで遠心分離した。この上清を除去し、そして亜鉛緩衝液洗浄を合計3回繰り返した。3回の亜鉛緩衝液洗浄に続いて、ペレットを水中で2回洗浄し、そして4℃にて5分間3000rpmで遠心分離して過剰の亜鉛を除去した。最後の水洗に続いて、ペレットを小容量の水に再懸濁し、そして液体窒素を用いて瞬間凍結した。次いで、凍結ペレットを凍結乾燥して水を除去し、hGH小球状粒子の最終粉末を取り出した。
【0186】
(実施例19:hGHの小球状粒子のジャケット付き容器バッチ調製(100mgスケール))
このタイプの調製を、実施例18と類似の処方組成物を用いて行ったが、より大きな容量に適応可能であり、スケールアップにより適している。
【0187】
18%PEG3350および0.02%プルロニックF−68を含む、pH6.1で緩衝化した溶液(80mM酢酸アンモニウム/l0mM重炭酸アンモニウム)をオーバーヘッドインペラーを用いてジャケット付きビーカー中で混合し、58℃に加熱した。この混合物温度を、循環水浴を用いて制御した。hGHの高濃度溶液を、攪拌しながらこの溶液に添加した。この溶液中のhGHの最終濃度は1mg/mlであった。hGHは、この溶液組成物においてこの温度で完全に可溶性であった。次いで、この容器および内容物を8℃/分の速度で約10℃の温度まで冷却した。hGH小球状粒子はこの冷却工程の間に形成された。これらの小球状粒子は約40℃で形成し始め、そしてこのプロセスを、懸濁液をさらに冷却しながら継続した。冷却工程の後、小球状粒子を実施例20aに記載の2つの方法のうち1つによってPEGから分離した。
【0188】
(実施例20:hGHの完全性の保持)
小球状粒子中のhGHのタンパク質完全性を、このプロセスの以下の段階で評価した:粒子形成後、PEG抽出後、および溶剤除去後または乾燥後。小球状粒子への成形加工後のhGHの化学的完全性の測定を、凝集および分解生成物を定量化するためにHPLCアッセイ(サイズ排除クロマトグラフィー(SEC)、逆相(RP))を用いて決定した。結果から、小球状粒子形成プロセスの間に凝集塊または他の関連物質の有意な蓄積はなかったことが証明された。
【0189】
a.有機溶剤洗浄
サイズ排除によるhGH凝集:出発物質を超える凝集の増大
【0190】
【化7】
逆相によるhGH関連物質:出発物質を超える分解の増大
【0191】
【化8】
b.亜鉛緩衝液洗浄
サイズ排除によるhGH凝集:出発物質を超える凝集の増大
【0192】
【化9】
逆相によるhGH関連物質:出発物質を超える分解の増大
【0193】
【化10】
(実施例21:hGHの小球状粒子の粒子サイズ分布)
小球状粒子の粒子サイズ分布の特性決定を、TSIエアロサイザーを用いる空気力学的飛行時間計測(図26)によって、および走査型電子顕微鏡(図27)によって決定した。
【0194】
(実施例22:hGH小球状粒子の溶解速度論)
2つの異なる抽出手順を受けたhGH小球状粒子の溶解速度論を比較した。
【0195】
有機溶剤で洗浄したhGH小球状粒子を、hGH出発物質と同様に水性媒体中に直ちに溶解した。
【0196】
hGH小球状粒子を亜鉛緩衝液で洗浄した場合、溶解度が低下した(図28)。hGH小球状粒子の溶解を、10mMトリス、154mMのNaCl、0.05%のBrij35(pH7.5)中に37℃で行った。タンパク質のより完全な放出が、インビトロで他の媒体中で達成された。溶解速度論は、全hGHの約30%が最初の15分に放出し、そして約50%が最初の24時間に放出したことを実証した。このタンパク質放出は1ヶ月で完了した。小球状粒子溶解が二相様式で進行したという事実は、結果としてインビボで一部の遅延放出をもたらし得る。
【0197】
(D.リゾチーム小球状粒子)
(実施例23.リゾチームの小球状粒子の調製)
以下の溶液:1.6mg/mlのリゾチーム、13.2%のPEG3350、55mMの酢酸アンモニウム(pH9.5)53mMの硫酸アンモニウム、263mMの塩化ナトリウム、26mMの塩化カルシウム。
【0198】
このPEG緩衝液を40℃に加熱した(pH9.55)。生じた懸濁液を液体窒素中で瞬間凍結し、そして多岐管凍結乾燥器で凍結乾燥した。小球状粒子が形成された。
【0199】
(E.DNase小球状粒子)
(実施例24.DNaseの小球状粒子の調製)
処方例:以下の溶液:0.18mg/mlのDNase(1mg/mlストックから)、18.2%のPEG3350(25%ストックから)、9mMの酢酸アンモニウム(pH5.15)(1Mストックから)。
【0200】
この懸濁液を−80℃フリーザー中で冷却し、凍結するとすぐに、多岐管凍結乾燥器で凍結乾燥させ、続いてMeCl2/アセトンで遠心分離によって洗浄した。
【0201】
試験済みの初発濃度は0.1mg/mlのDNaseおよび20%のPEG3350であった。しかし、37℃から0℃まで冷却を試みた後に沈殿が得られない場合は、新たな量のDNaseを添加して上記濃度に至った。この溶液を−80℃フリーザー中で冷却し、凍結するとすぐに、多岐管凍結乾燥器で凍結乾燥させた。MeCl2/アセトンで遠心分離によって洗浄した。試験済みの初発濃度は0.1mg/mlのDNaseおよび20%のPEG3350であった。しかし、37℃から0℃まで冷却を試みた後に沈殿が得られない場合は、新たな量のDNaseを添加して上記濃度に至った。この溶液を−80℃フリーザー中で冷却し、凍結するとすぐに、多岐管凍結乾燥器で凍結乾燥させた。MeCl2/アセトンで遠心分離によって洗浄した(図37、38)。
【0202】
活性(Sigmaから購入したDNA−メチルグリーンを用いるDNase−Iについてのアッセイ)
出発物質についての理論活性を775Ku/mgタンパク質としてリストに記載する。このストック溶液を0.145mg/mlのタンパク質と決定した。この濃度を最終濃度が0.0199mg/mlとなるように5mlに希釈した。この活性は775Ku/mg*0.0199mg/ml=15.46Ku/mlであるはずである。
【0203】
【化11】
Ku/ml=−0.0004×40×1/−0.0011=14.55Ku/ml
理論との比較:
小球状粒子/理論*100%=活性%
14.55Ku/ml/15.46Ku/ml*100%=94.1%
(F.スーパーオキシドジスムターゼ小球状粒子)
(実施例25.スーパーオキシドジスムターゼの小球状粒子の調製)
0.68mg/mlのSOD(5mg/mlストックから)、24.15%のPEG3350(31.25%ストックから)、9.1mMの酢酸アンモニウム(1Mストックから)、最終pH=4.99(水酸化アンモニウムおよび酢酸で調整)の溶液。この溶液を40℃から0℃まで50分間にわたって(約0.8℃/分)冷却し、そして約25℃で沈殿が開始した。この懸濁液を液体窒素中で瞬間凍結し、そして多岐管凍結乾燥器で凍結乾燥し、続いてMeCl2/アセトンで遠心分離によって洗浄した(図39、40)。
【0204】
40℃から0℃まで50分間にわたって(約0.8℃/分)冷却した。約25℃で沈殿が開始した。液体窒素中で瞬間凍結し、そして多岐管凍結乾燥器で凍結乾燥した。MeCl2/アセトンで遠心分離によって洗浄した。小球状粒子が形成され、そして大部分のアセトンが保持された。
【0205】
(G.サブチリシン小球状粒子)
(実施例26:非ポリマー相分離促進剤を用いるサブチリシン小球状粒子)
初期系の連続相は非ポリマー相分離促進剤を含み、冷却中にタンパク質の相分離を誘導し得る。サブチリシン小球状粒子は、ポリマーを全く用いることなくプロピレングリコールとエタノールの混合物を用いて本発明に従って形成され得る。この系において、プロピレングリコールは凝固点降下剤としての機能を果たし、そしてエタノールは相分離促進剤としての機能を果たす。プロピレングリコールはまた、小球状粒子の球形状の形成にも役立つ。
【0206】
35%プロピレングリコール−10%ホルメート−0.02%CaCl2中の20mg/mLサブチリシン溶液を調製した。次いでこの35%プロピレングリコール−サブチリシン溶液を、混合しながら67%エタノールに付した。この溶液は室温で透明のままであった。しかしながら、1時間−20℃に冷却すると、粒子の懸濁液が形成される。遠心分離してこれらの粒子を収集し、そして90%エタノールで洗浄した後、懸濁液流体として無水エタノールを用いてコールター粒子サイズ分析を行った。これらの粒子は、2.2ミクロンの平均直径を有する離散粒子と一致するコールター結果をもたらし、そしてこれらの粒子の95%が0.46ミクロンと3.94ミクロンの間であった。光学顕微鏡評価は、実質的に球状の粒子を示すことによりこれらの結果を確認した。粒子のSEM分析はこれらのコールター結果を確認した。
【0207】
(小球状粒子の形成後のサブチリシン酵素活性の保持)
溶液中のサブチリシンのサブチリシン小球状粒子への変換後の酵素活性の保持を、比色アッセイによって確認した。小球状粒子についての理論上の全活性単位を、冷却の前にエタノール−サブチリシン−プロピレングリコール溶液中でアッセイされたサブチリシンの全単位から、(サブチリシン粒子の分離後に)上清中に見出された全単位を減算することによって算出した。サブチリシン小球状粒子について求めた実際の全単位を理論単位で割ってパーセンテージで表したものは、粒子形成後のサブチリシン活性の保持を表す。この計算によって、107%の理論サブチリシン活性がサブチリシン小球状粒子の形成後に保持された。
【0208】
(H.炭水化物小球状粒子)
(実施例27:炭水化物小球状粒子の形成)
本発明は、炭水化物小球状粒子の調製に適用され得る。相分離は、この系の冷却中にPEG相とデキストラン相の間に誘導され得る。種々の分子量(例えば、5K、40K、144K、および500K)のデキストランが用いられ得る。30%PEG300中の5mg/mlデキストラン40Kの混合物を35℃で平衡化し、次いでこの混合物を0℃に冷却し、そして凍結乾燥した。この混合物を塩化メチレン:アセトン(1:1)で洗浄および遠心分離することによって凍結乾燥粒子を収集した。図49を見ても分かるように、小球状粒子が形成された。他の炭水化物、例えばデンプン、ヒドロキシエチルデンプン、トレハロース、ラクトース、マンニトール、ソルビトール、ハイロース(hylose)、硫酸デキストランなどを、このプロセスを用いて小球状粒子中に処方され得る。
【0209】
(I.前調製小球状粒子のマイクロカプセル化)
(実施例28.PLGA−カプセル化前調製インスリン小球状粒子の調製)
a)20%(w/v)ポリマー溶液(8ml)を塩化メチレン中の1600mgのポリラクチド・グリコリド共重合体(PLGA、MW35k)を溶解することによって調製した。この溶液に、100mgのインスリン小球状粒子(INSms)を添加し、そしてこの媒体をローター/ステーターホモジナイザーを用いて11krpmで激しく混合することによって均質の懸濁液を得た。0.02%メチルセルロース水溶液からなる連続相(24ml)を塩化メチレンで飽和した。この連続相を同じホモジナイザーを用いて11krpmで混合し、そして前記懸濁液を徐々にこの媒体に注入して有機相の初期マイクロカプセル化粒子を生成した。このエマルジョンは1:3のO/W比を有する。乳化を5分間継続した。次に、このエマルジョンを、150mlの脱イオン(DI)水からなる硬化媒体中に、この媒体を400rpmで攪拌しながら直ちに移した。この有機溶剤を−0.7バールの減圧下で1時間にわたって抽出した。硬化したマイクロカプセル化粒子を濾過によって収集し、水で洗浄した。これらの洗浄したマイクロカプセル化粒子を凍結乾燥して過剰な水を除去した。結果として生じるマイクロカプセル化粒子は、約30μmの平均粒子サイズを有するとともに、これらの粒子集団の大部分が90μm未満であり、そして5.7%(w/w)インスリンを含んだ。
【0210】
b)塩化メチレン中に1200mgの50:50ポリラクチド−グリコリド共重合体(PLGA、MW35k)を溶解することによって、30%(w/v)ポリマー溶液(4ml)を調製した。次に上記ポリマー溶液中の100mgのINSmsの懸濁液を、ホモジナイザーを用いて調製した。この懸濁液を用いて、実施例28に記載されるように12mlの0.02%メチルセルロース水溶液中のO/Wエマルジョンを生成した(W/O比=1:3)。実施例28と同じ手順に従って、最終マイクロカプセル化粒子を調製する。形成されたマイクロカプセル化粒子は25μmの平均粒子サイズを有し、0.8〜60μmの範囲にわたった。これらのマイクロカプセル化粒子のインスリン含量は8.8%(w/w)であった。
【0211】
あるいは、ポリマーの10%(w/v)溶液を用いて上記と同じ条件下でマイクロカプセル化プロセスを行った。このプロセスにより、約12μmの平均粒子サイズを有するマイクロカプセル化粒子であってこれらの粒子の大部分が50μm未満であり、かつインスリンの充填が21.1%(w/w)である、マイクロカプセル化粒子がもたらされた。
【0212】
インビトロ放出のための方法:
マイクロカプセル化粒子からのインスリンのインビトロ放出(IVR)は、37℃でインキュベートした3mg当量のカプセル化インスリンを含むガラスバイアル中に10mlの放出緩衝液(10mMトリス、0.05%Brij35、0.9%NaCl、pH7.4)を添加することによって達成される。指定の時間間隔でこのIVR媒体の400μLを微量遠心管中に移し、そして2分間13krpmで遠心する。上清の最上層300μLを取り出し、分析されるまで−80℃で保存する。この取得した容量を300μLの新たな媒体で置き換え、これを用いて残りの上清(100μL)とともにパレットを再構成した。この懸濁液をもとの対応するインビトロ放出媒体へ移す。
【0213】
(実施例29.PLGA/PLA合金マトリックス系中の前調製インスリン小球状粒子のマイクロカプセル化のための手順)
PLGA/PLA合金の30%(w/v)溶液を塩化メチレン(4ml)中で調製した。この合金は、それぞれ40、54および6%(0.48、0.68および0.07g)で、50:50PLGA(MW35k)、D,L−ポリ乳酸(PLA、MW19k)およびポリL−PLA(PLLA、MW180k)からなった。実施例28bと同じ手順に従って、最終マイクロカプセル粒子を調製した。これらのマイクロカプセル化粒子の例は、粒子サイズが0.8〜120μmの範囲にわたり、平均して40μmであって、粒子集団の大部分が90μmよりも小さかった。
【0214】
(実施例30.連続および不連続相の両方においてPEGを用いた、PLGAマトリックス系における前調製インスリン小球状粒子のマイクロカプセル化のための手順)
10%の50:50PLGA(0.4g)および25%ポリエチレングリコール(PEG、MW8k)の4mlの溶液を塩化メチレン中で調製した。ローター/ステーターホモジナイザーを用いて、100mgのINSmsをこの溶液中に11krpmで懸濁した。この連続相は、塩化メチレンで飽和させた0.02%(w/v)メチルセルロースおよび25%PEG(MW8k)の水溶液(12ml)からなった。この連続相を同じホモジナイザーを用いて11krpmで混合し、そして上記懸濁液をこの媒体に徐々に注入して有機相の初期マイクロカプセル化粒子を生成した。このエマルジョンは1:3のO/W比を有する。乳化を5分間継続した。次に、このエマルジョンを、150mlの脱イオン(DI)水からなる硬化媒体中に、この媒体を400rpmで攪拌しながら直ちに移した。この有機溶剤を−0.7バールの減圧下で1時間にわたって抽出した。硬化したマイクロカプセル化粒子を濾過によって収集し、水で洗浄した。これらの洗浄したマイクロカプセル化粒子を凍結乾燥して過剰な水を除去した。本実施例のマイクロカプセル化粒子は、30μmの平均粒子サイズを有し、2〜90μmの範囲にわたるとともに、この集団の大部分が70μmよりも小さかった。これらのミクロスフェアのインスリン含量は16.0%(w/w)であった。
【0215】
(実施例31.リン酸緩衝液を用いる種々の連続相pHでのPLGAマトリックス系における前調製インスリン小球状粒子のマイクロカプセル化のための手順)
20%の50:50 35kD PLGA(0.8g)の4mlの溶液を塩化メチレン中で調製した。ローター/ステーターホモジナイザーを用いて、100mgのINSmsをこの溶液中に11krpmで懸濁した。連続相は、0.1%(w/v)メチルセルロースおよび50mMリン酸緩衝液(pH2.5、5.4および7.8)の水溶液からなった。連続装備(図41A)を用いてマイクロカプセル化を行った。この連続相を11krpmで混合し、そして乳化チャンバー中に12ml/分で送り込んだ。分散相をこのチャンバー中に2.7ml/分で注入して初期マイクロカプセル化粒子を生成した。生成したエマルジョンをこのチャンバーから取り出し、硬化浴中に連続様式で移した。この硬化媒体を400rpmで攪拌した。この有機溶剤を−0.4バールの減圧下で1時間にわたって抽出した。硬化したマイクロカプセル化粒子を濾過によって収集し、水で洗浄した。これらの洗浄したマイクロカプセル化粒子を凍結乾燥して過剰な水を除去した。
【0216】
pH2.5、5.4および7.8で調製した、結果として生じるマイクロカプセル化粒子のインスリン含量を、それぞれ12.5、11.5および10.9と見積もった。これらのマイクロカプセル化粒子のサイズ分布分析の結果を表5に要約する。
【0217】
【表5】
(インビトロ放出のための方法)
マイクロカプセル化粒子からのインスリンのインビトロ放出は、37℃でインキュベートした3mg当量のカプセル化インスリンを含むガラスバイアル中に10mlの放出緩衝液(10mMトリス、0.05%Brij35、0.9%NaCl、pH7.4)を添加することによって達成された。指定の時間間隔でこのIVR媒体の400μLを微量遠心管中に移し、そして2分間13krpmで遠心した。上清の最上層300μLを取り出し、分析されるまで−80℃で保存した。この取得した容量を300μLの新たな媒体で置き換え、これを用いて残りの上清(100μL)とともにパレットを再構成した。この懸濁液をもとの対応するインビトロ放出媒体へ移した。
【0218】
上記調製物のインビトロ放出(IVR)の結果は図44に示し、これらの処方物からのインスリンの放出動力学に対する連続相のpHの有意な影響を示す。
【0219】
(実施例32.PLLAまたはPLLA/PEGマトリックス系における前調製ヒト血清アルブミン(HSA)小球状粒子のマイクロカプセル化のための手順)
25%(w/v、500mg)PEG(MW3kまたは8k)の2mlの溶液を塩化メチレン中で調製した。このPEG溶液または2mlの塩化メチレンを用いて50mgの前調製ヒト血清アルブミン小球状粒子(HSAms)の懸濁液を、ローター/ステーターホモジナイザーを11krpmで用いて生成した。この懸濁液に塩化メチレン中の4%PLLA(80mg、MW180k)2mlを添加し、そしてこの媒体を11〜27krpmでホモジナイズして有機相を生成した。この連続相は、塩化メチレンで飽和させた12mlの0.02%メチルセルロース水溶液からなった。この連続相を1lkrpmで激しく攪拌することによって乳化を惹起し、続いて有機相を徐々に注入した。この媒体を5分間乳化し、次いでこのエマルジョンを400rpmで混合している150mlのDI水中に移した。全ての上記手順を4℃で行った。次いでこの硬化媒体を室温に移し、そしてこの有機溶剤を−0.7バールの減圧下で1時間にわたって抽出した。硬化したマイクロカプセル化粒子を濾過によって収集し、水で洗浄した。洗浄したマイクロカプセル化粒子を凍結乾燥して過剰な水を除去した。上記処方物からのHASのIVRに対するPEGのチャンネル効果を図42に示す。
【0220】
(インビトロ放出のための方法)
カプセル化したマイクロカプセル化粒子からのHASのインビトロ放出(IVR)は、37℃でインキュベートした2.5mg当量のカプセル化HSAを含む15mlポリプロピレン遠心管中に15mlの放出緩衝液(20mMのHEPES、0.01%Tween−80、0.1MのNaCl、1mMのCaCl2、pH7.4)を添加することによって達成される。サンプリング手順は実施例31に記載した。
【0221】
(実施例33.PLGA−カプセル化前調製ロイプロリド/硫酸デキストラン小球状粒子の調製)
塩化メチレン中に1200mgの50:50ポリラクチド−グリコリド共重合体(PLGA、MW35k)を溶解することによって、30%(w/v)ポリマー溶液(4ml)を調製した。次に50mgのロイプロリドを含む65.9mgの前調製ロイプロリド(leuprilide)/硫酸デキストラン小球状粒子(LDS)を、該ポリマー溶液中にホモジナイザーを用いて懸濁した。この懸濁液を用いて、実施例28に記載されるように12mlの0.02%メチルセルロース水溶液中のO/Wエマルジョンを生成した(W/O比=1:3)。実施例28bと同じ手順に従って、最終マイクロカプセル化粒子を調製した。
【0222】
これらのマイクロカプセル化粒子は20μmの平均粒子サイズを有し、それらの大部分が50μm未満であった。これらのマイクロカプセル化粒子からのロイプロリドのIVRの結果を図43に図示する。
【0223】
(インビトロ放出のための方法)
マイクロカプセル化粒子からのロイプロリドのインビトロ放出(IVR)は、37℃でインキュベートした2.5mg当量のカプセル化ロイプロリドを含む15mlポリプロピレン遠心管中に15mlの放出緩衝液(10mMのNa−リン酸緩衝液、0.01%Tween−80、0.9%NaCl、0.04%NaN3、pH7.4)を添加することによって達成される。サンプリング手順は実施例28に記載した。
【0224】
(実施例34.PLGA−カプセル化前調製組換えヒト成長ホルモン小球状粒子の調製)
塩化メチレン中に0.4gのPLGA−PEGを溶解することによって、10%(w/v)ポリマー溶液(4ml)を調製した。次に100mgの前調製組換えヒト成長ホルモン小球状粒子(hGHms)を、上記ポリマー溶液中にホモジナイザーを用いて懸濁した。この連続相は、0.1%(w/v)メチルセルロースおよび50mMリン酸緩衝液(pH7.0)の水溶液からなった。マイクロカプセル化を実施例31に記載のように連続装備(図41A)を用いて行った。これらのマイクロカプセル化粒子の平均粒子サイズは25μmであり、1〜60μmの範囲にわたった。高分子マトリックスからのhGHのIVRプロフィールを図45に示す。
【0225】
(インビトロ放出のための方法)
マイクロカプセル化粒子からのhGHのIVRは、実施例28に記載のように達成される。
【0226】
(実施例35.マイクロカプセル化前調製インスリン小球状粒子の完全性の決定)
カプセル化前調製インスリン小球状粒子の完全性に対するマイクロカプセル化プロセスの影響を評価するために、前調製INSmsを含む高分子マイクロカプセル化粒子を、二相二重抽出法を用いて分解した。秤量したカプセル化INSmsのサンプルを塩化メチレン中に懸濁し、そして穏やかに混合して高分子マトリックスを溶解させた。タンパク質を抽出するために0.01NのHClを添加し、これらの2つの相を混合してエマルジョンを生成した。次いで、これらの2つの相を分離し、水性相を除去して同じ溶液を補給し、そしてこの抽出プロセスを繰り返した。抽出したインスリンの完全性をサイズ排除クロマトグラフィー(SEC)によって決定した。この方法は、抽出した媒体中のINSのモノマー、ダイマーおよび高分子量(HMW)種の増大を確認する。適切なコントロールを用いて、INSの完全性に対する分解プロセスの影響を確認した。これらの結果は、INS完全性に対するこのプロセスの有意な影響を示さなかった。
【0227】
カプセル化INSmsは、もとの(カプセル化されていない)INSms中のモノマー含量が99.13%であるのと比較して、マイクロカプセル化プロセスの条件および内容に応じて97.5〜98.94%のタンパク質モノマーを含んだ。カプセル化INSms中のダイマー種の含量は、もとのINSms中の0.85%と比較して、1.04%〜1.99%の範囲にわたった。カプセル化INSmsのHMW含量は、もとのINSms中の0.02%に対して、0.02%〜0.06%の範囲にわたった。これらの結果を表6に要約する。高分子マトリックスの影響を図46および47に示す。
【0228】
【表6】
(実施例36.マイクロカプセル化前調製小球状粒子からのインビボ放出インスリン)
前調製インスリン小球状粒子のマイクロカプセル化粒子からのインスリンのインビボ放出をSprague Dawley(SD)ラットにおいて研究した。これらの動物に、初回皮下用量の1IU/kgのカプセル化されていないかまたはカプセル化された前調製インスリン小球状粒子を投与した。ELISAを用いて、収集したサンプル中の組換えヒトインスリン(rhINS)血清レベルを決定した。これらの結果を図48に示す。
【0229】
特定の実施形態を図示しかつ記載してきたが、多数の改変が本発明の精神を逸脱することなく想起され、そして保護の範囲は添付クレームの範囲によってのみ限定される。
【図面の簡単な説明】
【0230】
【図1】図1は、温度に対して活性薬剤濃度をプロットした二次元状態図である。
【図2】図2は、冷却温度プロフィールである。
【図3】図3aは、インスリン出発物質の走査型電子顕微鏡写真(SEM)である。図3bは、インスリンの小球状粒子(実施例4)のSEMである。
【図4】図4は、小球状粒子中に調製された場合のインスリンの化学安定性の総合的維持を示すHPLC分析である。
【図5】図5aおよび5bは、バッチ間再現性を示す概略図である。
【図6】図6は、バッチ間再現性を示す概略図である。
【図7】図7は、実施例3においてインスリン小球状粒子を作製するための連続貫流プロセスの概略図である。
【図8】図8は、実施例3において連続貫流プロセスによって生成されたインスリン小球状粒子の走査型電子顕微鏡写真(10Kvおよび倍率6260×)である。
【図9】図9は、実施例3において連続貫流プロセスによって調製された溶解したインスリン小球状粒子のHPLCクロマトグラフである。
【図10−1】図10aは、インスリン溶解度に対する塩化ナトリウムの影響を示す。図10bは、インスリン溶解度に対する塩化ナトリウムの影響を示す。
【図10−2】図10cは、インスリン溶解度に対する塩化ナトリウムの影響を示す。図10dは、インスリン溶解度に対する塩化ナトリウムの影響を示す。
【図10−3】図10eは、インスリン溶解度に対する異なる塩の影響を示す。図10fは、インスリン溶解度に対する異なる塩の影響を示す。図10gは、インスリン溶解度に対する異なる塩の影響を示す。
【図10−4】図10hは、インスリン溶解度に対する異なる塩の影響を示す。図10iは、原料インスリン、小球状粒子から放出されたインスリンおよび小球状粒子中のインスリンのラマンスペクトルである。
【図11】図11は、実施例10の放射標識インスリンについてのアンダーセンカスケードインパクターの結果である。
【図12】図12は、実施例8についてのP/I比の棒グラフである。
【図13】図13は、実施例8からの肺のシンチグラフィー画像である
【図14−1】図14aは、α−1−アンチトリプシン(AAT)についての円偏光二色性(CD)プロットである。図14bは、実施例17における室温での貯蔵時間に対する活性のプロットである。
【図14−2】図14cは、実施例17における4℃での貯蔵時間に対する活性のプロットである。
【図15】図15は、DSCプロットである。
【図16】図15は、DSCプロットである。
【図17】図15は、DSCプロットである。
【図18】図15は、DSCプロットである。
【図19】図15は、DSCプロットである。
【図20】図15は、DSCプロットである。
【図21】図15は、DSCプロットである。
【図22】図15は、DSCプロットである。
【図23】図15は、DSCプロットである。
【図24】図15は、DSCプロットである。
【図25】図15は、DSCプロットである。
【図26】図26は、TSI社エアロサイザー(Aerosizer)の粒子サイズデータのプロットである。
【図27】図27は、ヒト成長ホルモン(hGH)小球状粒子のSEMである。
【図28】図28は、HFA−134aにおけるインスリン安定性データを示すチャートである。
【図29】図29は、3つの吸入デバイスを用いるインスリンの空気力学的性能を比較するチャートである。
【図30】図30は、25℃で貯蔵したインスリン出発物質と比較した、インスリン小球状粒子の安定性データのチャートである。
【図31】図31は、37℃で貯蔵したインスリン出発物質と比較した、インスリン小球状粒子の安定性データのチャートである。
【図32】図32は、25℃で貯蔵したインスリン出発物質と比較した、インスリン小球状粒子の安定性データのチャートである。
【図33】図33は、37℃で貯蔵したインスリン出発物質と比較した、インスリン小球状粒子の安定性データのチャートである。
【図34】図34は、25℃で貯蔵したインスリン出発物質と比較した、インスリン小球状粒子の安定性データのチャートである。
【図35】図35は、37℃で貯蔵したインスリン出発物質と比較した、インスリン小球状粒子の安定性データのチャートである。
【図36】図36は、Cyclohaler DPIを用いるインスリン空気力学的安定性の棒グラフである。
【図37】図37は、DNase小球状粒子の光学顕微鏡写真である。
【図38】図38は、DNaseの酵素活性のチャートである。
【図39】図39は、SOD小球状粒子の光学顕微鏡写真である。
【図40】図40は、SOD小球状粒子についての酵素学的データのチャートである。
【図41】図41A〜Bは、連続乳化反応器の概略図であり、ここで、図41Aは、乳化前に連続相または分散相に界面活性化合物が添加される場合の連続乳化反応器の概略図であり、そして図41Bは、乳化後に界面活性化合物が添加される場合の連続乳化反応器の概略図である。
【図42】図42は、PLLAカプセル化HSA粒子のIVRプロフィールに対するPEGの影響を図示する(実施例32)。
【図43】図43は、PLGAカプセル化LDS小球状粒子のIVRプロフィールを図示する(実施例33)。
【図44】図44は、PLGAカプセル化インスリン小球状粒子のIVRプロフィールに対する連続相のpHの影響を図示する(実施例31)。
【図45】図45は、PLGAカプセル化hGH小球状粒子のIVRプロフィールを図示する(実施例34)。
【図46】図46は、カプセル化INSms中のINSダイマーの形成に対するマイクロカプセル化変数(連続相およびマトリックス材料のpH)の影響を図示する(実施例35)。
【図47】図47は、カプセル化INSms中のHMW種の形成に対するマイクロカプセル化変数(連続相およびマトリックス材料のpH)の影響を図示する(実施例35)。
【図48】図48は、ラットにおける、カプセル化されていない前調製インスリン小球状粒子およびカプセル化された前調製インスリン小球状粒子からの組換えヒトインスリンのインビボ放出を図示する(実施例36)。
【図49】図49は、実施例27の粒子のSEMである。
【特許請求の範囲】
【請求項1】
活性薬剤の小球状粒子を調製するための方法であって、該方法は以下の工程:
活性薬剤、相分離促進剤および第1溶剤を含む単一液相状態の溶液を提供する工程;ならびに
該溶液に制御速度で相変化を誘導して活性薬剤の液−固相分離を引き起こし、固相および液相を形成する工程であって、該固相は該活性薬剤の固体小球状粒子を含み、該液相は該相分離促進剤および該溶剤を含み、該小球状粒子は実質的に球状である、工程、
を包含する、方法。
【請求項2】
前記誘導工程が、以下:
前記溶液の温度を調整する工程;
前記活性薬剤の濃度を調整する工程;
前記相分離促進剤の濃度を調整する工程;
該溶液のイオン強度を調整する工程;
pHを調整する工程;および
該溶液の重量モル浸透圧濃度を調整する工程、
からなる群より選択される工程を包含する、請求項1に記載の方法。
【請求項3】
前記相変化が、前記活性薬剤の濃度の変化によって誘導される、請求項1に記載の方法。
【請求項4】
前記相変化が、前記相分離促進剤の濃度の変化によって誘導される、請求項1に記載の方法。
【請求項5】
前記溶液が、相転移温度、第1温度および第2温度を有し、該溶液を相変化に供する工程は、該第1温度から該第2温度までの該溶液の冷却によるものであり、ここで、該第1温度は該溶液の相転移温度よりも高く、かつ該第2温度は該溶液の相転移温度よりも低い、請求項1に記載の方法。
【請求項6】
前記第2温度が、前記溶液の凝固点よりも高い、請求項5に記載の方法。
【請求項7】
前記第2温度が、前記溶液の凝固点よりも低い、請求項5に記載の方法。
【請求項8】
前記冷却工程が、制御速度で行われる、請求項5に記載の方法。
【請求項9】
前記制御速度が、約0.2℃/分から約50℃/分までを含む、請求項8に記載の方法。
【請求項10】
前記制御速度が、約0.2℃/分から約30℃/分までを含む、請求項8に記載の方法。
【請求項11】
前記溶液を提供する工程が、以下の工程:
前記第1溶剤中に相分離促進剤を溶解して混合物を生成する工程;および
前記活性薬剤を該混合物に添加して溶液を形成する工程、
を包含する、請求項1に記載の方法。
【請求項12】
前記活性薬剤を、前記第1溶剤または該第1溶剤と混和性の第2溶剤中に溶解し、その後に該活性薬剤を前記混合物に添加する工程をさらに包含する、請求項11に記載の方法。
【請求項13】
前記溶液が、該溶液の凝固点を降下させるための凝固点降下剤をさらに含む、請求項6に記載の方法。
【請求項14】
前記凝固点降下剤が、ポリエチレングリコールおよびプロピレングリコールからなる群より選択される、請求項13に記載の方法。
【請求項15】
前記相分離促進剤が、水溶性または水混和性の薬剤である、請求項1に記載の方法。
【請求項16】
前記相分離促進剤が、直線状または分岐状ポリマー、炭水化物系ポリマー、ポリ脂肪族アルコール、ポリ(ビニル)ポリマー、ポリアクリル酸、ポリ有機酸、ポリアミノ酸、コポリマーおよびブロックコポリマー、tert−ポリマー、ポリエーテル、天然に存在するポリマー、ポリイミド、界面活性剤、ポリエステル、分岐状および環状ポリマー、ポリアルデヒド、デンプン、置換デンプン、ポリエチレングリコール、ポリビニルピロリドン、ポロクサマー、エタノール、アセトン、ならびにイソプロパノールからなる群より選択される、請求項1に記載の方法。
【請求項17】
前記PSEAが、ポリエチレングリコール(PEG)である、請求項1に記載の方法。
【請求項18】
前記小球状粒子が、該小球状粒子の安定性を高めるために、該小球状粒子に徐放をもたらすために、または生物組織への該小球状粒子の浸透を高めるために、賦形剤をさらに含む、請求項1に記載の方法。
【請求項19】
前記賦形剤が、炭水化物、カチオン、アニオン、アミノ酸、脂質、脂肪酸、界面活性剤、トリグリセリド、胆汁酸またはそれらの塩、脂肪酸エステル、およびポリマーからなる群より選択される、請求項18に記載の方法。
【請求項20】
前記カチオンがZn2+、Mg2+、およびCa2+からなる群より選択される、請求項19に記載の方法。
【請求項21】
前記胆汁酸がコール酸またはその塩である、請求項19に記載の方法。
【請求項22】
前記小球状粒子を収集する工程をさらに含む、請求項1に記載の方法。
【請求項23】
前記小球状粒子を収集する工程が、前記活性薬剤が前記液体媒体において可溶性ではなく、かつ前記相分離促進剤が該液体媒体において可溶性である温度にて、該粒子を液体媒体で洗浄することによる、請求項22に記載の方法。
【請求項24】
前記洗浄工程が、ダイアフィルトレーションまたは遠心分離によるものである、請求項23に記載の方法。
【請求項25】
前記液体媒体が、水性または有機性である、請求項23に記載の方法。
【請求項26】
前記液体媒体が、超臨界流体または超臨界流体と超臨界流体混和性溶剤との混合物である、請求項23に記載の方法。
【請求項27】
前記有機性液体媒体が、塩化メチレン、クロロホルム、アセトニトレート(acetonitrate)、酢酸エチル、エタノール、およびペンタンからなる群より選択される、請求項25に記載の方法。
【請求項28】
前記液体媒体が、該液体媒体中の活性薬剤の溶解度を低下させる薬剤をさらに含む、請求項23に記載の方法。
【請求項29】
前記液体媒体中の活性薬剤の溶解度を低下させる薬剤が、錯イオンを含む、請求項28に記載の方法。
【請求項30】
前記カチオンが、Zn2+、Ca2+、Fe2+、Mg2+、Mn2+、Na+およびNH4+からなる群より選択される、請求項29に記載の方法。
【請求項31】
前記液体媒体を除去する工程をさらに含む、請求項23に記載の方法。
【請求項32】
前記液体媒体を除去する工程が、凍結乾燥、乾燥または蒸発による、請求項31に記載の方法。
【請求項33】
前記液体媒体が、賦形剤をさらに含む、請求項23に記載の方法。
【請求項34】
前記賦形剤が、前記小球状粒子に徐放をもたらし、生物組織への該小球状粒子の浸透を高めるために、該小球状粒子の安定性を高める、請求項33に記載の方法。
【請求項35】
前記賦形剤が、炭水化物、カチオン、アニオン、アミノ酸、脂質、脂肪酸、界面活性剤、トリグリセリド、胆汁酸またはそれらの塩、脂肪酸エステル、およびポリマーからなる群より選択される、請求項34に記載の方法。
【請求項36】
前記カチオンが、Zn2+、Mg2+、およびCa2+からなる群より選択される、請求項35に記載の方法。
【請求項37】
前記賦形剤がコール酸またはその塩である、請求項35に記載の方法。
【請求項38】
前記ポリマーがポリエチレングリコールである、請求項37に記載の方法。
【請求項39】
前記溶液が、水性または水混和性溶剤を含む、請求項1に記載の方法。
【請求項40】
前記水混和性溶剤が、N−メチル−2−ピロリジノン(N−メチル−2−ピロリドン)、2−ピロリジノン(2−ピロリドン)、1,3−ジメチル−2−イミダゾリジノン(DMI)、ジメチルスルホキシド、ジメチルアセトアミド、酢酸、乳酸、メタノール、エタノール、イソプロパノール、3−ペンタノール、n−プロパノール、ベンジルアルコール、グリセロール、ポリエチレングリコール(PEG)、PEG−4、PEG−8、PEG−9、PEG−12、PEG−14、PEG−16、PEG−120、PEG−75、PEG−150、ポリエチレングリコールエステル、PEG−4ジラウレート、PEG−20ジラウレート、PEG−6イソステアレート、PEG−8パルミトステアレート、PEG−150パルミトステアレート、ポリエチレングリコールソルビタン、PEG−20ソルビタンイソステアレート、ポリエチレングリコールモノアルキルエーテル、PEG−3ジメチルエーテル、PEG−4ジメチルエーテル、ポリプロピレングリコール(PPG)、ポリプロピレンアルギレート、PPG−10ブタンジオール、PPG−10メチルグルコースエーテル、PPG−20メチルグルコースエーテル、PPG−15ステアリルエーテル、プロピレングリコールジカプリレート/ジカプレート、プロピレングリコールラウレート、およびグリコフロール(テトラヒドロフルフリルアルコールポリエチレングリコールエーテル)、またはこれらの組合せからなる群より選択される、請求項39に記載の方法。
【請求項41】
前記活性薬剤が、薬学的に活性な物質である、請求項1に記載の方法。
【請求項42】
前記薬学的に活性な化合物が、治療剤、診断用薬、化粧品、栄養剤、および農薬からなる群より選択される、請求項41に記載の方法。
【請求項43】
前記活性薬剤が高分子である、請求項1に記載の方法。
【請求項44】
前記高分子が、タンパク質、ポリペプチド、炭水化物、ポリヌクレオチド、ウイルスおよび核酸からなる群より選択される、請求項43に記載の方法。
【請求項45】
前記タンパク質が、血液凝固カスケードのタンパク質、VII因子、VIII因子、IX因子、サブチリシン、オボアルブミン、α−1−アンチトリプシン、DNAse、スーパーオキシドジスムターゼ、リゾチーム、リボヌクレアーゼ、ヒアルロニダーゼ、コラゲナーゼ、成長ホルモン、エリスロポエチン、インスリン様成長因子またはそれらのアナログ、インターフェロン、グラチラマー、顆粒球マクロファージコロニー刺激因子、顆粒球コロニー刺激因子、抗体、モノクローナル抗体、ポリクローナル抗体、Fabフラグメント、単鎖抗体、ペグ化タンパク質、グリコシル化または高グリコシル化タンパク質、デスモプレシン、LHRHアゴニスト、例えばロイプロリド、ゴセレリン、ナファレリン、ブセレリン、LHRHアンタゴニスト、バソプレッシン、シクロスポリン、カルシトニン、副甲状腺ホルモン、副甲状腺ホルモンペプチドおよびインスリンからなる群より選択される、請求項44に記載の方法。
【請求項46】
前記粒子が、前記治療剤を必要とする被験体へのインビボ送達に適した、請求項1に記載の方法。
【請求項47】
前記送達方法が、注射可能送達、吸入可能送達、非経口送達、局所送達、経口送達、直腸送達、経鼻送達、肺送達、膣送達、頬側送達、舌下送達、経皮送達、経粘膜送達、耳送達、眼球送達、眼球内送達および眼送達からなる群より選択される、請求項46に記載の方法。
【請求項48】
前記送達方法が、肺送達によるものである、請求項47に記載の方法。
【請求項49】
前記粒子が、前記被験体の肺の中心エリアまたは末梢エリア中の沈積に適した、請求項48に記載の方法。
【請求項50】
前記粒子が、乾燥粉末吸入器、定量噴霧式吸入器、および噴霧器からなる群より選択されるデバイスによって送達される、請求項48に記載の方法。
【請求項51】
前記粒子が、安定な液体懸濁液として送達される、請求項46に記載の方法。
【請求項52】
前記粒子が、実質的に同じ粒子サイズを有する、請求項1に記載の方法。
【請求項53】
前記粒子が、約0.01μmから約200μmまでの平均粒子サイズを有する、請求項1に記載の方法。
【請求項54】
前記粒子が、約0.5μmから約10μmまでの平均粒子サイズを有する、請求項1に記載の方法。
【請求項55】
前記活性薬剤が、前記粒子の約0.1重量%から約100重量%までである、請求項1に記載の方法。
【請求項56】
前記活性薬剤が、前記粒子の約75重量%から約100重量%までである、請求項1に記載の方法。
【請求項57】
前記活性薬剤が、前記粒子の90重量%以上である、請求項1に記載の方法。
【請求項58】
前記小球状粒子が、狭いサイズ分布を有する、請求項1に記載の方法。
【請求項59】
前記狭いサイズ分布が、前記小球状粒子の第90百分位数の体積粒径の第10百分位数の体積粒径に対する比が約5以下であることを含む、請求項58に記載の方法。
【請求項60】
前記小球状粒子が、半晶質または非晶質である、請求項1に記載の方法。
【請求項61】
活性薬剤の小球状粒子を調製する方法であって、該方法は、以下の工程:
該活性薬剤および相分離促進剤を水性または水混和性溶剤中に溶解して、単一連続相の溶液を形成する工程、および
相変化を誘導する工程であって、それによって該活性薬剤が液−固相分離を受けて、該活性薬剤の固体小球状粒子を含む固相および該相分離促進剤の液相を生成する、工程
を包含する、方法。
【請求項62】
前記溶液が、相転移温度、第1温度および第2温度を有し、該溶液を相変化に供する工程は、該溶液を該第1温度から該第2温度まで冷却することによるものであり、ここで、該第1温度は該相転移温度よりも高く、かつ該第2温度は該相転移温度よりも低い、請求項61に記載の方法。
【請求項63】
前記制御速度が、約0.2℃/分から約50℃/分までである、請求項62に記載の方法。
【請求項64】
実質的に球状であり、かつ狭いサイズ分布および約0.5〜約2g/cm3の密度を有する、治療剤の固体球状粒子を含む、インビボ送達のための治療剤の小粒子。
【請求項65】
前記固体小球状粒子が、約0.5〜約1.5g/cm3の密度を有する、請求項64に記載の粒子。
【請求項66】
前記固体小球状粒子が、0.75g/cm3よりも大きい密度を有する、請求項64に記載の粒子。
【請求項67】
前記固体小球状粒子が、0.85g/cm3よりも大きい密度を有する、請求項64に記載の粒子。
【請求項68】
前記固体小球状粒子が、実質的に非多孔性である、請求項64に記載の粒子。
【請求項69】
前記固体小球状粒子が、該固体小球状粒子の安定性を高めるために、該固体小球状粒子の徐放をもたらすために、または生物組織への該固体小球状粒子の浸透を高めるために、賦形剤をさらに含む、請求項64に記載の粒子。
【請求項70】
前記賦形剤が、炭水化物、カチオン、アニオン、アミノ酸、脂質、脂肪酸、界面活性剤、トリグリセリド、胆汁酸またはそれらの塩、脂肪酸エステル、およびポリマーからなる群より選択される、請求項64に記載の粒子。
【請求項71】
前記カチオンが、Zn2+、Mg2+、およびCa2+からなる群より選択される、請求項64に記載の粒子。
【請求項72】
前記小球状粒子は、各々が一定の長さを有する2つの横軸を有し、該2つの軸の長さの比が約1.5以下である、請求項64に記載の粒子。
【請求項73】
前記小球状粒子が、約0.01μmから約200μmまでの平均サイズを有する、請求項64に記載の粒子。
【請求項74】
前記小球状粒子が、約0.1μmから約10μmまでの平均サイズを有する、請求項64に記載の粒子。
【請求項75】
前記小球状粒子が、約0.1μmから約5μmまでの平均サイズを有する、請求項64に記載の粒子。
【請求項76】
前記狭いサイズ分布が、前記小球状粒子の第90百分位数の体積粒径の第10百分位数の体積粒径に対する比が約5以下であることを含む、請求項76に記載の粒子。
【請求項77】
前記小球状粒子が、半晶質または非晶質である、請求項64に記載の粒子。
【請求項1】
活性薬剤の小球状粒子を調製するための方法であって、該方法は以下の工程:
活性薬剤、相分離促進剤および第1溶剤を含む単一液相状態の溶液を提供する工程;ならびに
該溶液に制御速度で相変化を誘導して活性薬剤の液−固相分離を引き起こし、固相および液相を形成する工程であって、該固相は該活性薬剤の固体小球状粒子を含み、該液相は該相分離促進剤および該溶剤を含み、該小球状粒子は実質的に球状である、工程、
を包含する、方法。
【請求項2】
前記誘導工程が、以下:
前記溶液の温度を調整する工程;
前記活性薬剤の濃度を調整する工程;
前記相分離促進剤の濃度を調整する工程;
該溶液のイオン強度を調整する工程;
pHを調整する工程;および
該溶液の重量モル浸透圧濃度を調整する工程、
からなる群より選択される工程を包含する、請求項1に記載の方法。
【請求項3】
前記相変化が、前記活性薬剤の濃度の変化によって誘導される、請求項1に記載の方法。
【請求項4】
前記相変化が、前記相分離促進剤の濃度の変化によって誘導される、請求項1に記載の方法。
【請求項5】
前記溶液が、相転移温度、第1温度および第2温度を有し、該溶液を相変化に供する工程は、該第1温度から該第2温度までの該溶液の冷却によるものであり、ここで、該第1温度は該溶液の相転移温度よりも高く、かつ該第2温度は該溶液の相転移温度よりも低い、請求項1に記載の方法。
【請求項6】
前記第2温度が、前記溶液の凝固点よりも高い、請求項5に記載の方法。
【請求項7】
前記第2温度が、前記溶液の凝固点よりも低い、請求項5に記載の方法。
【請求項8】
前記冷却工程が、制御速度で行われる、請求項5に記載の方法。
【請求項9】
前記制御速度が、約0.2℃/分から約50℃/分までを含む、請求項8に記載の方法。
【請求項10】
前記制御速度が、約0.2℃/分から約30℃/分までを含む、請求項8に記載の方法。
【請求項11】
前記溶液を提供する工程が、以下の工程:
前記第1溶剤中に相分離促進剤を溶解して混合物を生成する工程;および
前記活性薬剤を該混合物に添加して溶液を形成する工程、
を包含する、請求項1に記載の方法。
【請求項12】
前記活性薬剤を、前記第1溶剤または該第1溶剤と混和性の第2溶剤中に溶解し、その後に該活性薬剤を前記混合物に添加する工程をさらに包含する、請求項11に記載の方法。
【請求項13】
前記溶液が、該溶液の凝固点を降下させるための凝固点降下剤をさらに含む、請求項6に記載の方法。
【請求項14】
前記凝固点降下剤が、ポリエチレングリコールおよびプロピレングリコールからなる群より選択される、請求項13に記載の方法。
【請求項15】
前記相分離促進剤が、水溶性または水混和性の薬剤である、請求項1に記載の方法。
【請求項16】
前記相分離促進剤が、直線状または分岐状ポリマー、炭水化物系ポリマー、ポリ脂肪族アルコール、ポリ(ビニル)ポリマー、ポリアクリル酸、ポリ有機酸、ポリアミノ酸、コポリマーおよびブロックコポリマー、tert−ポリマー、ポリエーテル、天然に存在するポリマー、ポリイミド、界面活性剤、ポリエステル、分岐状および環状ポリマー、ポリアルデヒド、デンプン、置換デンプン、ポリエチレングリコール、ポリビニルピロリドン、ポロクサマー、エタノール、アセトン、ならびにイソプロパノールからなる群より選択される、請求項1に記載の方法。
【請求項17】
前記PSEAが、ポリエチレングリコール(PEG)である、請求項1に記載の方法。
【請求項18】
前記小球状粒子が、該小球状粒子の安定性を高めるために、該小球状粒子に徐放をもたらすために、または生物組織への該小球状粒子の浸透を高めるために、賦形剤をさらに含む、請求項1に記載の方法。
【請求項19】
前記賦形剤が、炭水化物、カチオン、アニオン、アミノ酸、脂質、脂肪酸、界面活性剤、トリグリセリド、胆汁酸またはそれらの塩、脂肪酸エステル、およびポリマーからなる群より選択される、請求項18に記載の方法。
【請求項20】
前記カチオンがZn2+、Mg2+、およびCa2+からなる群より選択される、請求項19に記載の方法。
【請求項21】
前記胆汁酸がコール酸またはその塩である、請求項19に記載の方法。
【請求項22】
前記小球状粒子を収集する工程をさらに含む、請求項1に記載の方法。
【請求項23】
前記小球状粒子を収集する工程が、前記活性薬剤が前記液体媒体において可溶性ではなく、かつ前記相分離促進剤が該液体媒体において可溶性である温度にて、該粒子を液体媒体で洗浄することによる、請求項22に記載の方法。
【請求項24】
前記洗浄工程が、ダイアフィルトレーションまたは遠心分離によるものである、請求項23に記載の方法。
【請求項25】
前記液体媒体が、水性または有機性である、請求項23に記載の方法。
【請求項26】
前記液体媒体が、超臨界流体または超臨界流体と超臨界流体混和性溶剤との混合物である、請求項23に記載の方法。
【請求項27】
前記有機性液体媒体が、塩化メチレン、クロロホルム、アセトニトレート(acetonitrate)、酢酸エチル、エタノール、およびペンタンからなる群より選択される、請求項25に記載の方法。
【請求項28】
前記液体媒体が、該液体媒体中の活性薬剤の溶解度を低下させる薬剤をさらに含む、請求項23に記載の方法。
【請求項29】
前記液体媒体中の活性薬剤の溶解度を低下させる薬剤が、錯イオンを含む、請求項28に記載の方法。
【請求項30】
前記カチオンが、Zn2+、Ca2+、Fe2+、Mg2+、Mn2+、Na+およびNH4+からなる群より選択される、請求項29に記載の方法。
【請求項31】
前記液体媒体を除去する工程をさらに含む、請求項23に記載の方法。
【請求項32】
前記液体媒体を除去する工程が、凍結乾燥、乾燥または蒸発による、請求項31に記載の方法。
【請求項33】
前記液体媒体が、賦形剤をさらに含む、請求項23に記載の方法。
【請求項34】
前記賦形剤が、前記小球状粒子に徐放をもたらし、生物組織への該小球状粒子の浸透を高めるために、該小球状粒子の安定性を高める、請求項33に記載の方法。
【請求項35】
前記賦形剤が、炭水化物、カチオン、アニオン、アミノ酸、脂質、脂肪酸、界面活性剤、トリグリセリド、胆汁酸またはそれらの塩、脂肪酸エステル、およびポリマーからなる群より選択される、請求項34に記載の方法。
【請求項36】
前記カチオンが、Zn2+、Mg2+、およびCa2+からなる群より選択される、請求項35に記載の方法。
【請求項37】
前記賦形剤がコール酸またはその塩である、請求項35に記載の方法。
【請求項38】
前記ポリマーがポリエチレングリコールである、請求項37に記載の方法。
【請求項39】
前記溶液が、水性または水混和性溶剤を含む、請求項1に記載の方法。
【請求項40】
前記水混和性溶剤が、N−メチル−2−ピロリジノン(N−メチル−2−ピロリドン)、2−ピロリジノン(2−ピロリドン)、1,3−ジメチル−2−イミダゾリジノン(DMI)、ジメチルスルホキシド、ジメチルアセトアミド、酢酸、乳酸、メタノール、エタノール、イソプロパノール、3−ペンタノール、n−プロパノール、ベンジルアルコール、グリセロール、ポリエチレングリコール(PEG)、PEG−4、PEG−8、PEG−9、PEG−12、PEG−14、PEG−16、PEG−120、PEG−75、PEG−150、ポリエチレングリコールエステル、PEG−4ジラウレート、PEG−20ジラウレート、PEG−6イソステアレート、PEG−8パルミトステアレート、PEG−150パルミトステアレート、ポリエチレングリコールソルビタン、PEG−20ソルビタンイソステアレート、ポリエチレングリコールモノアルキルエーテル、PEG−3ジメチルエーテル、PEG−4ジメチルエーテル、ポリプロピレングリコール(PPG)、ポリプロピレンアルギレート、PPG−10ブタンジオール、PPG−10メチルグルコースエーテル、PPG−20メチルグルコースエーテル、PPG−15ステアリルエーテル、プロピレングリコールジカプリレート/ジカプレート、プロピレングリコールラウレート、およびグリコフロール(テトラヒドロフルフリルアルコールポリエチレングリコールエーテル)、またはこれらの組合せからなる群より選択される、請求項39に記載の方法。
【請求項41】
前記活性薬剤が、薬学的に活性な物質である、請求項1に記載の方法。
【請求項42】
前記薬学的に活性な化合物が、治療剤、診断用薬、化粧品、栄養剤、および農薬からなる群より選択される、請求項41に記載の方法。
【請求項43】
前記活性薬剤が高分子である、請求項1に記載の方法。
【請求項44】
前記高分子が、タンパク質、ポリペプチド、炭水化物、ポリヌクレオチド、ウイルスおよび核酸からなる群より選択される、請求項43に記載の方法。
【請求項45】
前記タンパク質が、血液凝固カスケードのタンパク質、VII因子、VIII因子、IX因子、サブチリシン、オボアルブミン、α−1−アンチトリプシン、DNAse、スーパーオキシドジスムターゼ、リゾチーム、リボヌクレアーゼ、ヒアルロニダーゼ、コラゲナーゼ、成長ホルモン、エリスロポエチン、インスリン様成長因子またはそれらのアナログ、インターフェロン、グラチラマー、顆粒球マクロファージコロニー刺激因子、顆粒球コロニー刺激因子、抗体、モノクローナル抗体、ポリクローナル抗体、Fabフラグメント、単鎖抗体、ペグ化タンパク質、グリコシル化または高グリコシル化タンパク質、デスモプレシン、LHRHアゴニスト、例えばロイプロリド、ゴセレリン、ナファレリン、ブセレリン、LHRHアンタゴニスト、バソプレッシン、シクロスポリン、カルシトニン、副甲状腺ホルモン、副甲状腺ホルモンペプチドおよびインスリンからなる群より選択される、請求項44に記載の方法。
【請求項46】
前記粒子が、前記治療剤を必要とする被験体へのインビボ送達に適した、請求項1に記載の方法。
【請求項47】
前記送達方法が、注射可能送達、吸入可能送達、非経口送達、局所送達、経口送達、直腸送達、経鼻送達、肺送達、膣送達、頬側送達、舌下送達、経皮送達、経粘膜送達、耳送達、眼球送達、眼球内送達および眼送達からなる群より選択される、請求項46に記載の方法。
【請求項48】
前記送達方法が、肺送達によるものである、請求項47に記載の方法。
【請求項49】
前記粒子が、前記被験体の肺の中心エリアまたは末梢エリア中の沈積に適した、請求項48に記載の方法。
【請求項50】
前記粒子が、乾燥粉末吸入器、定量噴霧式吸入器、および噴霧器からなる群より選択されるデバイスによって送達される、請求項48に記載の方法。
【請求項51】
前記粒子が、安定な液体懸濁液として送達される、請求項46に記載の方法。
【請求項52】
前記粒子が、実質的に同じ粒子サイズを有する、請求項1に記載の方法。
【請求項53】
前記粒子が、約0.01μmから約200μmまでの平均粒子サイズを有する、請求項1に記載の方法。
【請求項54】
前記粒子が、約0.5μmから約10μmまでの平均粒子サイズを有する、請求項1に記載の方法。
【請求項55】
前記活性薬剤が、前記粒子の約0.1重量%から約100重量%までである、請求項1に記載の方法。
【請求項56】
前記活性薬剤が、前記粒子の約75重量%から約100重量%までである、請求項1に記載の方法。
【請求項57】
前記活性薬剤が、前記粒子の90重量%以上である、請求項1に記載の方法。
【請求項58】
前記小球状粒子が、狭いサイズ分布を有する、請求項1に記載の方法。
【請求項59】
前記狭いサイズ分布が、前記小球状粒子の第90百分位数の体積粒径の第10百分位数の体積粒径に対する比が約5以下であることを含む、請求項58に記載の方法。
【請求項60】
前記小球状粒子が、半晶質または非晶質である、請求項1に記載の方法。
【請求項61】
活性薬剤の小球状粒子を調製する方法であって、該方法は、以下の工程:
該活性薬剤および相分離促進剤を水性または水混和性溶剤中に溶解して、単一連続相の溶液を形成する工程、および
相変化を誘導する工程であって、それによって該活性薬剤が液−固相分離を受けて、該活性薬剤の固体小球状粒子を含む固相および該相分離促進剤の液相を生成する、工程
を包含する、方法。
【請求項62】
前記溶液が、相転移温度、第1温度および第2温度を有し、該溶液を相変化に供する工程は、該溶液を該第1温度から該第2温度まで冷却することによるものであり、ここで、該第1温度は該相転移温度よりも高く、かつ該第2温度は該相転移温度よりも低い、請求項61に記載の方法。
【請求項63】
前記制御速度が、約0.2℃/分から約50℃/分までである、請求項62に記載の方法。
【請求項64】
実質的に球状であり、かつ狭いサイズ分布および約0.5〜約2g/cm3の密度を有する、治療剤の固体球状粒子を含む、インビボ送達のための治療剤の小粒子。
【請求項65】
前記固体小球状粒子が、約0.5〜約1.5g/cm3の密度を有する、請求項64に記載の粒子。
【請求項66】
前記固体小球状粒子が、0.75g/cm3よりも大きい密度を有する、請求項64に記載の粒子。
【請求項67】
前記固体小球状粒子が、0.85g/cm3よりも大きい密度を有する、請求項64に記載の粒子。
【請求項68】
前記固体小球状粒子が、実質的に非多孔性である、請求項64に記載の粒子。
【請求項69】
前記固体小球状粒子が、該固体小球状粒子の安定性を高めるために、該固体小球状粒子の徐放をもたらすために、または生物組織への該固体小球状粒子の浸透を高めるために、賦形剤をさらに含む、請求項64に記載の粒子。
【請求項70】
前記賦形剤が、炭水化物、カチオン、アニオン、アミノ酸、脂質、脂肪酸、界面活性剤、トリグリセリド、胆汁酸またはそれらの塩、脂肪酸エステル、およびポリマーからなる群より選択される、請求項64に記載の粒子。
【請求項71】
前記カチオンが、Zn2+、Mg2+、およびCa2+からなる群より選択される、請求項64に記載の粒子。
【請求項72】
前記小球状粒子は、各々が一定の長さを有する2つの横軸を有し、該2つの軸の長さの比が約1.5以下である、請求項64に記載の粒子。
【請求項73】
前記小球状粒子が、約0.01μmから約200μmまでの平均サイズを有する、請求項64に記載の粒子。
【請求項74】
前記小球状粒子が、約0.1μmから約10μmまでの平均サイズを有する、請求項64に記載の粒子。
【請求項75】
前記小球状粒子が、約0.1μmから約5μmまでの平均サイズを有する、請求項64に記載の粒子。
【請求項76】
前記狭いサイズ分布が、前記小球状粒子の第90百分位数の体積粒径の第10百分位数の体積粒径に対する比が約5以下であることを含む、請求項76に記載の粒子。
【請求項77】
前記小球状粒子が、半晶質または非晶質である、請求項64に記載の粒子。
【図1】


【図2】
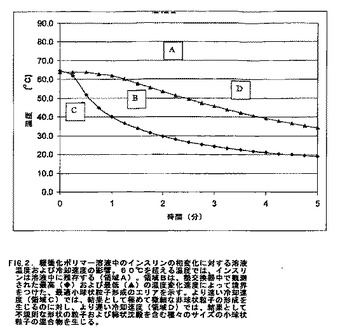
【図3】

【図4】
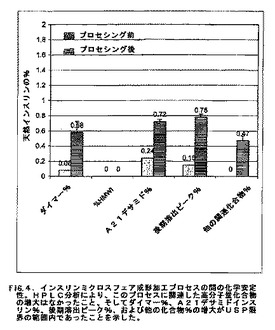
【図5】

【図6】
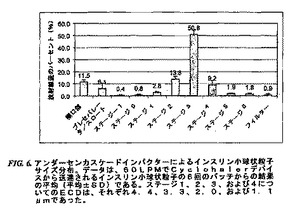
【図7】
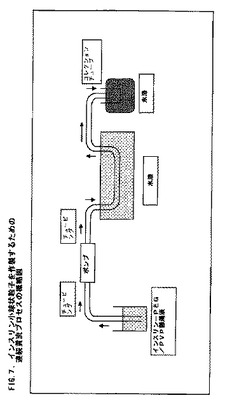
【図8】
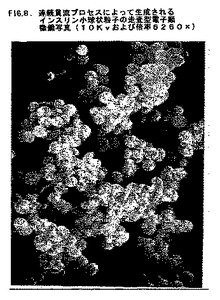
【図9】

【図10−1】

【図10−2】
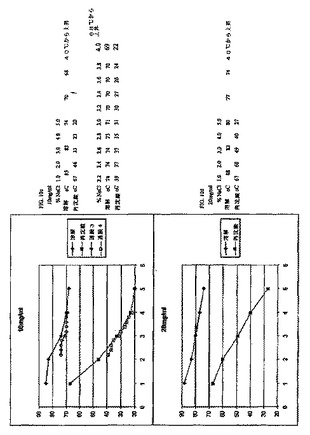
【図10−3】
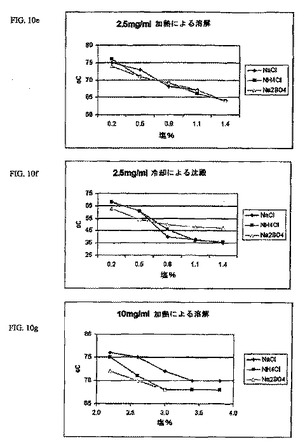
【図10−4】
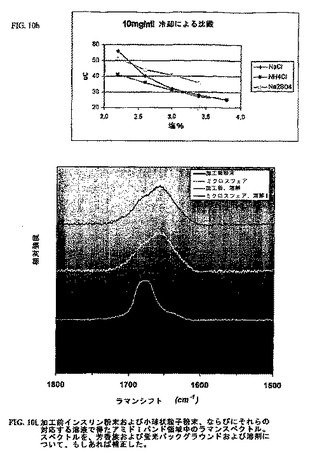
【図11】

【図12】

【図13】

【図14−1】

【図14−2】
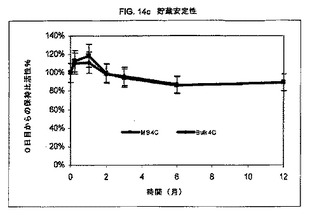
【図15】
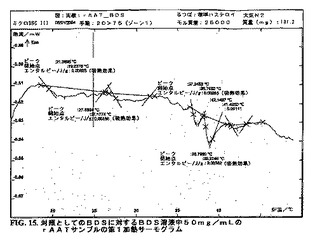
【図16】
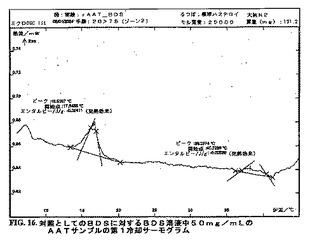
【図17】
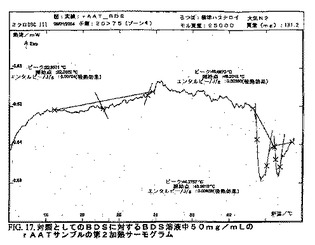
【図18】
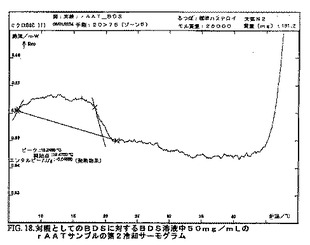
【図19】

【図20】

【図21】
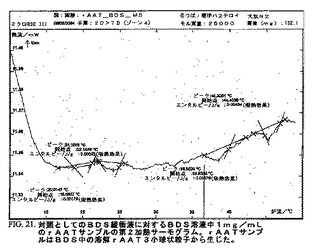
【図22】
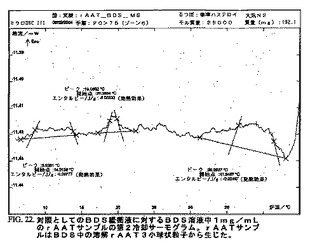
【図23】

【図24】
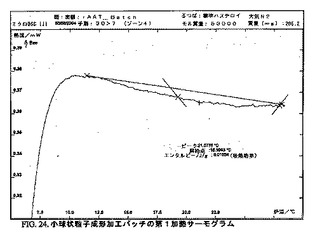
【図25】
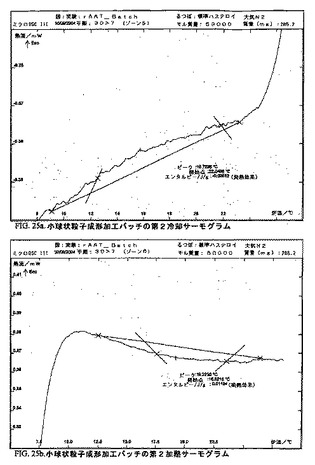
【図26】
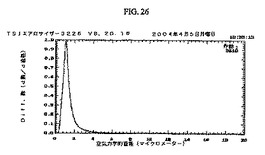
【図27】

【図28】
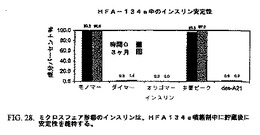
【図29】
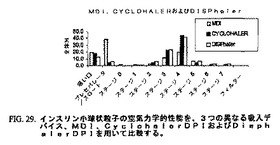
【図30】

【図31】
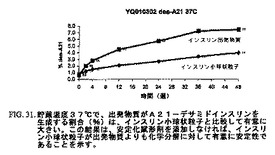
【図32】

【図33】
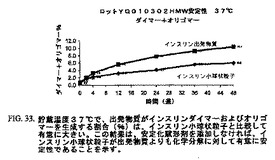
【図34】

【図35】
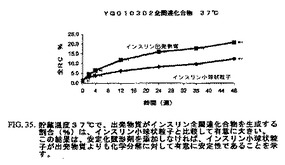
【図36】

【図37】

【図38】
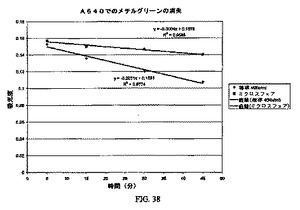
【図39】

【図40】
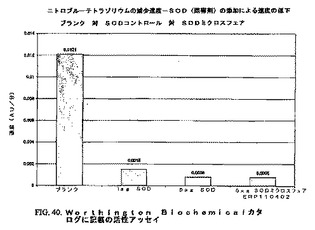
【図41】
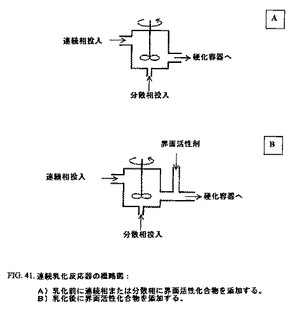
【図42】
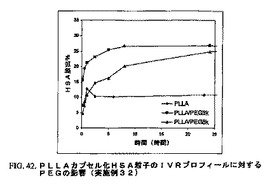
【図43】

【図44】
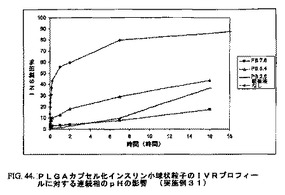
【図45】
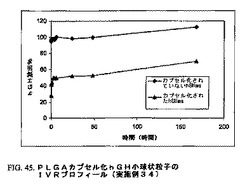
【図46】
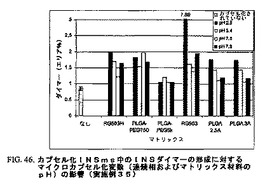
【図47】
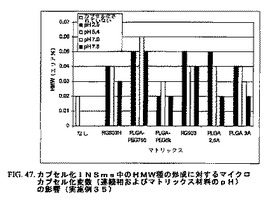
【図48】

【図49】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10−1】
【図10−2】
【図10−3】
【図10−4】
【図11】
【図12】
【図13】
【図14−1】
【図14−2】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図31】
【図32】
【図33】
【図34】
【図35】
【図36】
【図37】
【図38】
【図39】
【図40】
【図41】
【図42】
【図43】
【図44】
【図45】
【図46】
【図47】
【図48】
【図49】
【公表番号】特表2007−531701(P2007−531701A)
【公表日】平成19年11月8日(2007.11.8)
【国際特許分類】
【出願番号】特願2006−520406(P2006−520406)
【出願日】平成16年7月19日(2004.7.19)
【国際出願番号】PCT/US2004/023182
【国際公開番号】WO2005/035088
【国際公開日】平成17年4月21日(2005.4.21)
【出願人】(591013229)バクスター・インターナショナル・インコーポレイテッド (448)
【氏名又は名称原語表記】BAXTER INTERNATIONAL INCORP0RATED
【Fターム(参考)】
【公表日】平成19年11月8日(2007.11.8)
【国際特許分類】
【出願日】平成16年7月19日(2004.7.19)
【国際出願番号】PCT/US2004/023182
【国際公開番号】WO2005/035088
【国際公開日】平成17年4月21日(2005.4.21)
【出願人】(591013229)バクスター・インターナショナル・インコーポレイテッド (448)
【氏名又は名称原語表記】BAXTER INTERNATIONAL INCORP0RATED
【Fターム(参考)】
[ Back to top ]