
制御放出ヒドロコドン処方
【課題】痛みの管理の効力および品質を実質的に改善する生物利用性ヒドロコロン(塩酸モルホロン)処方を提供する。
【解決手段】固形経口制御放出の経口剤形のヒドロコドンであって、鎮痛効果を示す有効量のヒドロコドンまたは医薬上許容しうるその塩、およびヒト患者に対して一日二回投与に適当な剤形にさせるために十分な量の制御放出材料を包含し、0.55から0.85のC12/Cmax比を有する処方。そして少なくとも12時間治療的効果を有する処方。
【解決手段】固形経口制御放出の経口剤形のヒドロコドンであって、鎮痛効果を示す有効量のヒドロコドンまたは医薬上許容しうるその塩、およびヒト患者に対して一日二回投与に適当な剤形にさせるために十分な量の制御放出材料を包含し、0.55から0.85のC12/Cmax比を有する処方。そして少なくとも12時間治療的効果を有する処方。
【発明の詳細な説明】
【技術分野】
【0001】
痛み、特に慢性の痛みの薬物療法による困難性のために、オピオイド鎮痛薬は、制御放
出処方として投与されるべき理想的な薬剤である。本発明は、痛みの治療で使用するため
の固形の制御放出経口剤形に関する。
【背景技術】
【0002】
即時放出剤形の投与の後に通常に得られるより投与後の長期間の薬理学的作用を提供す
るのは、全ての制御(遅延)放出処方の目的である。このような長期間の応答は、対応す
る短期作用の即時放出製品では達成されない多くの治療利益をもたらす。したがって、治
療は、例えば、中程度から重篤な痛み(例えば、術後患者、癌患者など)までについて、
または起きているときに偏頭痛を経験する患者のものについて、並びに睡眠が必須である
衰弱した患者について、患者を治療するときに、患者の睡眠を中断せずに継続されうる。
【0003】
従来の迅速作用薬剤療法が、薬剤の有効に安定な状態の血漿濃度を維持するために、頻
繁な間隔で注意深く投与されない限り、化合物の、そして代謝不活性化を通しての迅速な
吸収、全身排出のため、活性薬剤の血漿中濃度における頂点および谷が起り、それにより
患者の維持療法における特別の問題を生じる。長期作用の薬剤製品のさらに一般的な利点
は、患者の健忘を通して抜かされた用量の回避から生じる改善された患者服薬遵守である
。
【0004】
ヒトおよび動物に対する経口投与の後の組成物に含まれる薬理学上活性な物質の制御放
出を生じる組成物を製造することは、製薬業界で知られている。このような遅延放出組成
物は、それが、栄養管のある種の部分に到達するまで、医薬品単位の吸収を遅らせるため
に使用される。消化管における医薬品のこのような制御放出は、さらに、従来の迅速放出
剤形が投与される場合に起るより長い期間、血流での上記医薬品の望ましい濃度をさらに
維持する。
【0005】
担体からの活性化合物の制御放出を提供する組成物の製造および使用の先行技術の教示
は、消化管の生理学的流動体への活性物質の放出で基本的に考慮される。しかし、それは
、胃腸液における活性物質の単なる存在は、それ自身で、生物利用性を確保しないことが
一般に認識される。
【0006】
吸収されるために、活性薬剤物質は、溶液中になければならない。単位剤形からの活性
物質の所定の比率について要求される時間は、標準化条件下で行われた試験方法によって
、特化された時間基本に対する、単位剤形から放出される活性薬剤物質の量の比率として
測定される。胃腸管の生理学上の流動体は、溶解時間を決定するための媒体である。技術
の現状は、多くの満足のいく試験手段が、製薬上の組成物について溶解時間を測定するこ
とを認識し、そしてこれらの試験手段は、世界的な公式解説で記述されている。
【0007】
それの担体からの薬剤物質の溶解に影響する多くの多様な因子があるが、特定の組成物
からの薬理学上の活性物質について測定された溶解時間は、比較的一定で、そして再現性
がある。溶解時間に影響しうる様々の因子の中でも、溶解溶媒媒質、溶液のpH、特定の
溶媒媒質での物質の溶解性、および溶媒溶質中に溶解した材料の飽和濃度の駆動力に表さ
れる薬剤物質の表面領域である。したがって、成分が、組織部位を越えた吸収を通して溶
解媒体から除去されるときに、活性薬剤物質の溶解濃度は、それの安定な状態で動的に改
質される。生理学的条件下で、溶解した材料の飽和レベルは、安定な状態の吸収を生じる
溶媒媒体での相対的均質性および一定の溶解濃度を維持するために剤形から逆に補給され
る。
【0008】
駆動力の方向が、膜のいずれかの側での活性物質の濃度、すなわち、胃腸の流動体で溶
解される量と、血液中に存在する量との間の差異であるので、胃腸管の組織吸収部位にお
ける輸送は、膜の両面でドナンの浸透平衡力によって影響される。血中濃度は、希釈、循
環変化、組織保存、代謝変換および全身排出により一定に変化するので、活性材料の流れ
は、胃腸管から血流に向けられる。
【0009】
種々の技術は、制御放出剤形を製造するために使用された。活性医薬品の遅延放出が、
製造のコーティングの選択的破壊を通して、または薬剤の放出に影響を及ぼす特別のマト
リックスを用いたコンパウンド化を通して起される特別に被覆されたペレット、錠剤およ
びカプセル剤は、当業界で知られている。ある種の制御放出処方は、投与後の所定の期間
に活性化合物の単位用量の関連した連続放出を提供する。
【0010】
本文献で報告される制御放出オピオイド処方の特定の例としては、例えば、共に、本発
明の譲受人に譲渡され、そして参照してここに組込まれる米国特許第4,990,341
号および第4,844,909号(ゴールデイら)に開示されるものが挙げられ、米国薬
局方パドルまたはバスケット法によって測定される場合、37℃で、900ml水性緩衝
液(pH1.6と7.2との間)で、100rmpでの剤形のインビトロでの溶解速度と
して、12.5〜42.5%(重量で)の塩酸モルホロンが1時間後に放出され、25〜
55%(重量で)が2時間後に放出され、45〜75%(重量で)の塩酸モルホロンが4時
間後に放出され、そして55〜85%(重量で)の塩酸モルホロンが6時間後に放出され
、インビトロ放出速度は、pH1.6〜7.2のpHから独立しており、そしてインビボ
で得られた塩酸モルホロンの頂点の血漿中濃度が、剤形の投与の2〜4時間の間で起るよ
うに選択される。少なくとも12時間の痛み解放が、これらの塩酸モルホロン処方で得ら
れる。
【0011】
中程度の痛みのために使用されうる他のオピオイド鎮痛薬剤の制御放出投与量処方を提
供することが、非常の望ましいと考えられる。さらに、痛み療法の必要な患者における最
も有効な痛みの管理を提供する薬理学的特性を示すこのような制御放出処方を提供するこ
とが、非常に望ましいと考えられる。
【発明の開示】
【発明が解決しようとする課題】
【0012】
中程度の痛みを経験するヒト患者での痛みの管理の効力および品質を実質的に改善する
ことが本発明の目的である。
痛みの管理の効力および品質を実質的に改善する生物利用性ヒドロコドン(塩酸モルホ
ロン;hydrocodon)処方を提供することが本発明の目的である。
【課題を解決するための手段】
【0013】
即時放出ヒドロコドン処方に比較した場合に実質的に増大した効果の期間を提供するが
、しかし痛覚脱失症の早期発生を提供する生物利用性ヒドロコドン処方を提供することが
、本発明のさらに別の目的である。
治療効果の早期発生を提供し、そして投与間隔の間に最大限の濃度に上昇した後、相対
的に平坦な血清血漿プロファイルを提供し、すなわちオピオイドの血漿中濃度が、0.5
5から0.85までのC12/Cmax比を提供することを意味し、そして患者に対して有効
な痛み解放を提供する一日二回投与に適する経口で投与できる制御放出オピオイド処方を
提供することが、本発明の別の目的である。代替の実施形態では、剤形は、0.65から
0.75までのC12/Cmax比を提供する。
【0014】
本発明の上記及び他の目的は本発明によって達成される。特定の実施形態では、鎮痛薬
で有効な量のヒドロコドンまたは医薬上許容しうるその塩および一日二回投与に適した剤
形にするのに十分量の制御放出材料を包含する固形の経口制御放出剤形であって、ヒト患
者または患者の集団に対する単回投与後に、約2から約8時間(Tmax)まででインビト
ロでのヒドロコドンの頂点の血漿中濃度に達する時間を提供し、そして最大濃度を得た後
は、0.55から0.85までのC12/Cmax比を提供する。
特定の好ましい実施形態では、制御放出剤形は、37℃での55分間の700mlの模
擬胃液(SGF)中で、その後37℃での900mlの模擬小腸液(SIF)に交換する
100rpmでの米国薬局方バスケット法によって測定されるときに、1時間で、剤形か
らのヒドロコドンまたはその塩の18%から約42.5重量%までのインビトロ放出を提
供する。
【0015】
特定の好ましい実施形態では、37℃での1.2および7.5のpHで、900ml水
性緩衝液中で100rpmでの米国薬局方バスケット法によって測定されるときに、ヒド
ロコドン剤形のインビトロでの溶解速度として、約25から約65重量%までのヒドロコ
ドンまたはその塩2時間後放出され、約45から約85重量%までのヒドロコドンまたは
その塩が4時間後放出され、そして約60重量%より大きいヒドロコドンまたはその塩が8
時間後放出される。インビトロ放出速度は、本発明の好ましい実施形態では、所望される
とおりpH−独立またはpH−従属のいずれかでありうるが、ヒドロコドンの放出は、p
H−独立性である。
特定の好ましい実施形態では、剤形が、投与の12時間後に、少なくとも5または6n
g/mlのヒドロコドン血漿濃度を提供し、そして投与の約2から約8時間後までに、少
なくとも約8ng/mlの血漿濃度を提供する治療的に有効な量のヒドロコドンを包含す
る制御放出剤形の提供がある。
【0016】
本発明の別の好ましい実施形態では、等量用量の即時放出ヒドロコドン対照処方(例え
ば、ロルタボ(登録商標;Lortab)の約50%未満のCmaxであるヒドロコドンの
Cmaxを提供し、そして12時間投与間隔の間、有効な無痛覚を提供するヒドロコドンの
一日二回経口制御放出剤形が提供される。
本発明の他の好ましい実施形態では、等量用量の即時放出ヒドロコドン対照処方、例え
ば、ロルタボ(登録商標)の80%Cmaxに達する時間の約90%から約150%まで、
好ましくは約90%から約110%までである80%Cmaxに達する時間を提供するヒド
ロコドンの一日二回制御放出剤形の提供がある。好ましくは、制御放出剤形についての8
0%Cmaxに達する時間は、約0.5から約1.5時間まで、最も好ましくは約0.8か
ら約1.2時間までである。他の実施形態では、制御放出剤形についてのヒドロコドンの
80%Cmaxに達する時間は、約0.75から約2.0時間まで、最も好ましくは約0.
9から約1.5時間までである。
【0017】
本発明の他の好ましい実施形態では、等量用量の即時放出ヒドロコドン対照処方の90
%Cmaxに達する時間の約150%から約400%まで、好ましくは約150%から約2
50%までである90%Cmaxに達する時間を提供するヒドロコドンの一日二回制御放出
剤形の提供がある。好ましくは、制御放出剤形についての90%Cmaxに達する時間は、
約1.5から約2.5時間まで、最も好ましくは約1.8から約2.2時間までである。
他の実施形態では、制御放出剤形についてのヒドロコドンの90%Cmaxに達する時間は
、約1.5から約4.0時間まで、最も好ましくは約1.8から約2.5時間までである
。
【0018】
本発明の他の好ましい実施形態では、剤形が、約0.5から10時間まで、好ましくは
約1から約9時間まで、または約4から約8時間までのCmaxの80%以内に血漿中濃度
を維持するヒドロコドンの一日二回制御放出剤形が提供される。
本発明の他の好ましい実施形態では、剤形が、約1から6.5時間まで、好ましくは約
2から約5時間まで、または約2から約6.5時間までのCmaxの90%以内に血漿血漿
中濃度を維持するヒドロコドンの一日二回制御放出剤形の提供がある。
【0019】
本発明の他の好ましい実施形態では、約1.5mg/時間から約5mg/時間までの投
与からTmaxまでの平均インビボ吸収速度を提供し、15mgヒドロコドンビタートレー
トを含む剤形の経口投与に基づいて約0.5mg/時間未満であるTmaxから投与間隔の
終点までの平均吸収の速度を提供するヒドロコドンの一日二回制御放出剤形が提供される
。好ましくは、剤形は、約2mg/時間から約4mg/時間までの投与からTmaxまでの
平均インビボ吸収速度を提供し、そして15mgヒドロコドンビタートレートを含む剤形
の経口投与に基づいて、約0.08mg/時間から約0.4mg/時間までであるTmax
から12時間投与間隔の終点の平均インビボ吸収速度を提供する。
本発明の他の好ましい実施形態では、同じ期間の放出速度の約55%から約85%まで
である、剤形の経口投与のTmaxから約12時間までの期間、吸収速度を提供する一日二
回制御放出ヒドロコドン剤形が提供される。
【0020】
本発明の上記実施形態、並びに他の実施形態は、好ましくは、即時放出ヒドロコドン対
照処方の同等用量によって提供されるTmaxより3から4倍遅い時点でTmaxに達する時間
を提供する。好ましくは、持続的放出処方によって供されるTmaxは、経口投与の後約2
から約8時間、約3から約7時間まで、または約4から約6時間までで起る。
【0021】
本発明は、さらに、即時放出ヒドロコドン対照製品の同等用量によって提供されるCma
xの約50%未満、好ましくは約40%未満であるヒドロコドンのCmaxを提供するヒドロ
コドン処方に向けられる。
例えば、ヒドロコドンが、米国特許第4,861,598号および第4,970,07
5号で開示されるとおりデリバリー・システムで処方されるときに、即時放出ヒドロコド
ン対照製品のCmaxの百分率としてデリバリー・システムによって提供されるヒドロコド
ンのCmaxは、同じデリバリー・システムで処方されたオキシコドンについて同じ計算よ
り相当に低かったことが驚くべきことに知見された。この現象は、制御放出オキシコドン
およびヒドロコドン処方が、同様のインビトロ溶解パラメータを示した事実にもかかわら
ず明かである。
【0022】
本発明は、デリバリー・システム米国特許第4,861,598号および第4,970
,075号を用いて配合される場合に、即時放出ヒドロコドン対照製品のCmaxの百分率
としてデリバリー・システムのCmaxは、好ましい実施形態で約50%未満、そして40
%未満であるのに対して、オキシコドンは、50%より大きな計算を示す。
【発明を実施するための最良の形態】
【0023】
「ヒドロコドン」は、医薬上許容しうる塩およびヒドロコドンの複合体と同様に、ヒド
ロコドン遊離塩基を示す場合、本発明の目的のために定義される。
語句「米国薬局方パドルまたはバスケット法」は、例えば、参照してここに組込まれる
米国薬局方XXII(1990年)で記述されるパドルおよびバスケット法である。
本発明の目的のため、用語「pH−依存性」は、環境的pHによって変化する特徴(例
えば、溶解)を示すと定義される。
本発明の目的のため、用語「pH−独立性」は、pHによって実質的に影響されない特
徴(例えば、溶解)を示すと定義される。
本発明の目的のため、用語「生物利用性」は、薬剤(例えば、ヒドロコドン)が、単位
剤形から吸収される範囲として定義される。
本発明の目的のため、用語「制御放出」は、血中(例えば、血漿)濃度が、治療範囲内
に維持されるが、しかし約12時間またはそれより長くの時間の期間かけて毒性濃度を低
下させるような速度での薬剤(例えば、ヒドロコドン)の放出として定義される。
用語「Cmax」は、投与間隔の間に得られる最大血漿濃度を示す。
用語「Tmax」は、最大血漿濃度に達する時間を示す。
用語「T1/2(abs)」は、血漿に移行されるべきオピオイドの吸収可能な用量の2分の1
に必要な時間の量を示す。
【0024】
用語「安定な状態」は、所定の薬剤についての血漿濃度が、達成され、そして所定の薬
剤について最少有効治療濃度で、またはそれより上であり、そして最少毒性血漿濃度より
下である濃度での薬剤の連続用量で維持されることを意味する。オピオイド鎮痛薬につい
て、最少有効治療濃度は、所定の患者で達成される痛み解放の量によって部分的に測定さ
れる。痛みの測定は、非常に主観的であり、そして多くの個人的変動は、患者の中で起り
うることは、医療業界での習熟したものによって理解される。
用語「維持療法」および「慢性療法」は、患者が、上に定義されるとおり安定な状態ま
でオピオイド鎮痛薬で滴定された後に患者に対して投与された薬剤療法として、本発明の
目的のために定義される。
ヒドロコドンのようなオピオイドの濃度に関して、用語「最少有効鎮痛薬濃度」または
「MEAC」は、量を特定するのが極めて困難である。しかし、一般に、それより下で痛
覚脱失(analgesia)が供給されない、血漿ヒドロコドンの最小有効鎮痛薬濃度がある。
例えば、血漿中ヒドロコドン濃度と痛覚脱失の間の間接的関係があるときに、高く、そし
て延長された血漿中濃度は、一般に、優れた痛み解放に関連する。頂点の血漿中ヒドロコ
ドン濃度の時間と頂点の薬剤効果の時間との間に、遅滞時間またはヒステリシス(hyster
esis)がある。これは、一般にオピオイド鎮痛薬を用いた痛みの治療について有効である
。
【0025】
用語「平均共鳴時間」(MRT)は、薬剤分子が体内で留まる平均時間として定義され
る。吸収、分布および放出の関数であるこの計算は、活性成分を含有する剤形に部分的に
依存性がある。
本発明の目的のために、さらに特定されない限り、用語「患者」は、検討(または請求
項)が、個々の患者または対象の薬物動態的パラメータに向けられることを意味する。
用語「患者の集団」は、検討(または請求項)が、少なくとも2名の患者または対象の
平均薬物動態的パラメータに向けられる。
用語「突破痛み」では、患者は、患者が、ヒドロモルホンを含む一般に有効量の本発明
の持続放出の固形経口剤形を投与されているという事実の代わりに経験する痛みを意味す
る。
用語「救援」は、突破痛みを経験している患者に投与される鎮痛薬に該当する。
用語「有効な痛み管理」は、医師による鎮痛療法に対するヒト患者の応答(経験された
痛み対副作用)の客観的評価、並びにこのような治療を受ける患者による治療的治療の主
観的評価を意味する。当業者は、有効な鎮痛薬が、個々の患者の多様性を含めた多くの因
子によって変化することを理解する。
【0026】
本発明の目的のため、用語「即時放出ヒドロコドン対照処方」は、ユーシービー・ファ
ルマ、インク.から商用的に入手可能な登録商標ロルタブの等価量のヒドロコドン部分、
または即時放出のヒドロコドンまたはその塩を提供する医薬製品である。
本発明の目的のために、ここに開示された制御放出処方および即時放出制御処方は、用
量に比例する。このような処方では、薬理学的パラメータ(例えば、AUCおよびCmax
)は、1つの投与強度から別のものまで線状で増加する。したがって、特別の用量の薬理
学的パラメータは、同じ処方の異なる用量のパラメータから推論される。
本発明の目的のために、特に特定されない限り、ここに開示される薬理学的パラメータ
は、個別の患者に対する単回用量のヒドロコドン処方の投与に基づいている。患者集団に
基づいた薬理学的パラメータは、「平均」データとして特定される。
【0027】
用語「第一の投与」は、個々の患者または患者集団に対する治療の開始時での本発明の
単回用量を意味する。
本発明の制御放出の経口固形剤形は、驚くべきことに、オピオイドを倹約しうる。本発
明の制御放出の経口固形剤形は、鎮静薬効能における差異なしに、従来の即時放出製品に
対する比較で、実質的に低い一日投与量で投与されうることは可能である。匹敵する一日
投与量で、大きな効能は、従来の即時放出製品に対する比較で、本発明の制御放出の経口
固形剤形の使用で生じうる。
【0028】
ここに付随する図面は、本発明の実施形態を示し、そして請求項によって包含されると
おり本発明の範囲を制限すると意味されない。
【0029】
本発明の上記実施形態は、当業者に知られる広く多様な制御放出処方によって提供され
うる。例えば、適切な制御放出剤形は、参照してここに組込まれる米国特許第4,861
,598号および第4,970,075号で開示される。
本発明の特定の実施形態で、即時放出形態における有効量のオピオイドが、処方に含ま
れる。オピオイドの即時放出形態は、Tmaxが、例えば約2から約5時間まで、または約
2から約4時間までの時間に短縮されるように血液中のオピオイド(例えば、血漿)の最
大濃度までの時間を短縮するのに有効な量含まれる。単位用量でのこのような有効量の即
時放出オピオイドを含めることによって、患者における痛みの相対的に高いレベルの経験
が明らかに減少されることが知見された。このような実施形態で、即時放出形態での有効
量のオピオイドは、本発明の基質(substrates)に被覆されうる。例えば、その処方から
得た延長放出オピオイドは、制御放出コーティングによる場合、即時放出層は、制御放出
コーティングの上に上塗される。他方、即時放出層は、オピオイドが、制御放出マトリッ
クスに組込まれる基質の表面に被覆されうる。有効単位用量のオピオイド(例えば、ペレ
ット、球体、ビーズおよび同等物を含めた微粒子システム)を含む複数の持続的放出基質
が、硬質ゼラチンカプセルに組込まれるときに、オピオイド用量の即時放出部分は、カプ
セル内の粉末または顆粒として十分量の即時放出オピオイドの封入を介してゼラチンカプ
セルに組込まれうる。代わりに、ゼラチンカプセルそれ自身は、オピオイドの即時放出層
で被覆されうる。当業者は、即時放出オピオイド部分を単位用量に組込むさらに他の代替
手段を認識する。このような代替法は、付随の請求項に包含されると考えられる。
本発明のオピオイド剤形の1つの利点は、治療的濃度が、一般に、むかつき、吐気また
は嗜眠状態のような共存する副作用の強度および/または程度における明らかな増加なし
に実質的に達成され、そしてそれは、しばしば、オピオイドの高い血中濃度に関連するこ
とである。本発明の剤形の使用は、薬剤添加の危険が減少されることに至る証拠もある。
【0030】
活性剤
本発明の制御放出の経口剤形は、好ましくは、約0.5mgから約1250mgまでの
ヒドロコドンまたは等価量の医薬上許容しうるその塩を含む。より好ましい実施形態では
、剤形は、約5mgから約60mgまで、例えば15mgを含みうる。ヒドロコドンの適
切な医薬上許容しうる塩としては、ヒドロコドン・ビタートレート(bitartrate)、ヒド
ロコドン・ビタートレート・ヒドレート、塩酸ヒドロコドン、ヒドロコドン・p−トルエ
ンスルホネート、リン酸ヒドロコドン、ヒドロコドン・チオセミカルバゾン、ヒドロコド
ン・スルフェート、ヒドロコドン・トリフルオロアセテート、ヒドロコドン・ヘミペンタ
ヒドレート、ヒドロコドン・ペンタフルオロプロピオネート、ヒドロコドン・p−ニトロ
フェニルヒドラゾン、ヒドロコドン・o−メチルオキシム、ヒドロコドン・セミカルバゾ
ン、ヒドロコドン・ヒドロブロミド、ヒドロコドン・ムケート、ヒドロコドン・オレート
、ヒドロコドン・ホスフェート二塩基物、ヒドロコドン・ホスフェート一塩基物、ヒドロ
コドン無機塩、ヒドロコドン有機塩、ヒドロコドン・アセテート・トリヒドレート、ヒド
ロコドン・ビス(ヘプタフオロブチレート)、ヒドロコドン・ビス(メチルカルバメート
)、ヒドロコドン・ビス(ペンタフルオロプロピオネート)、ヒドロコドン・ビス(ピリ
ジンカルボキシレート)、ヒドロコドン・ビス(トリフルオロアセテート)、ヒドロコド
ン・クロロヒドレート、およびヒドロコドン・スルフェート・ペンタヒドレートが挙げら
れる。好ましくは、ヒドロコドンは、ビタートレート塩として存在する。
【0031】
本発明の剤形は、さらに、本発明のヒドロコドン鎮静薬と相乗的に作用しうるか、また
はし得ない1つまたはそれ以上の別の薬剤を含みうる。このような別の薬剤の例としては
、イブプロフェン、ジクロフェナック、ナプロキセン、ベノキサプロフェン、フルビプロ
フェン、フェノプロフェン、フルブフェン、ケトプロフェン、インドプロフェン、ピロプ
ロフェン、カルプロフェン、オキサプロジン、プラモプロフェン、ムロプロフェン、トリ
オキサプロフェン、スプロフェン、アミノプロフェン、トリアプロフェン酸、フルプロフ
ェン、ブクロキシン酸、インドメタシン、スリンダック、トルメチン、ゾメピラック、チ
オピナック、ジドメタシン、アセメタシン、フェンチアザック、クリダナック、オキシピ
ナック、メフェナミン酸、メクロフェナミン酸、フルフェナミン酸、ニフルミン酸、トル
フェナミン酸、ジフルリザール、フルフェニザール、ピロキシカム、スドキシカム、また
はイソキシカムおよび同等物を含めた非ステロイド抗炎症性剤が挙げられる。このような
非ステロイド抗炎症剤は、セレコキシブ(SC−58635)のようなシクロオキシゲナ
ーゼ阻害剤、DUP−697、フルスリド(CGP−28238)、メロキシカム、6−
メトキシ−2−ナフチル酢酸(6−MNA)、ビオクス(Vioxx)(MK−966)
、ナブメトン(6−MNAについてのプロドラッグ)、ニメスリド、NS−398、SC
−5766、SC−58215、およびアマンタジン(1−アミノアダマンチン)として
のT−614、およびメマンチン(3,5ジメチルアミノアダマントン)、それらの混合
物および医薬上許容しうるその塩も挙げられる。
他の追加の薬剤としては、デキトロルファンのような非毒性NMDAレセプターアンタ
ゴニスト、デキストロメトルファン、3−(1−ナフタレニル)−5−(ホスホノメチル
)−L−フェニルアラニン、3−(1−ナフタレニル)−5−(ホスホノメチル)−DL
−フェニルアラミン、1−(3,5−ジメチルフェニル)ナフタレン、および2−(3,
5−ジメチルフェニル)ナフタレン、2SR,4RS−4−(((1H−テトラゾル−5
−イル)メチル)オキシ)ピペリジン−2−カルボン酸;2SR,4RS−4−((((
1H−テトラゾル−5−イル)メチル)オキシ)メチル)ピペリジン−2−カルボン酸;
E及びZ 2SR−4−(O−1H−テトラゾル−5−イル)メチル)ケトキシイミノ)ピペリ
ジン−2−カルボン酸;2SR,4RS−4−((1H−テトラゾル−5−イル)チオ)
ピペリジン−2−カルボン酸;2SR,4RS−4−((1H−テトラゾル−5−イル)
チオ)ピペリジン−2−カルボン酸;2SR,4RS−4−(5−メルカプト−1H−テ
トラゾル−1−イル)ピペリジン−2−カルボン酸;2SR,4RS−4−(5−メルカ
プト−2H−テトラゾル−2−イル)ピペリジン−2−カルボン酸;2SR,4RS−4
−(5−メルカプト−1H−テトラゾル−1−イル)ピペリジン−2−カルボン酸;2S
R,4RS−4−(5−メルカプト−2H−テトラゾル−2−イル)ピペリジン−2−カ
ルボン酸;2SR,4RS−4−(5−メルカプト−1H−テトラゾル−1−イル)ピペ
リジン−2−カルボン酸;2SR,4RS−4−(((1H−テトラゾル−5−イル)チ
オ)メチル)ピペリジン−2−カルボン酸;2SR,4RS−4−((5−メルカプト−
1H−テトラゾル−1−イル)メチル)ピペリジン−2−カルボン酸;または2SR,4
RS−4−((5−メルカプト−2H−テトラゾル−2−イル)メチル)ピペリジン−2
−カルボン酸、それらの混合物および医薬上許容しうるその塩が挙げられる。
【0032】
本発明の剤形に含まれうる他の適切な追加の薬剤としては、アセトアミノフェン、神経
−活性ステロイド(参照してここに組込まれる、1998年2月20に提出された米国特
許出願番号第09/026,520号に開示されるもののような)および他の非オピオイ
ド鎮静薬(analgesics)が挙げられる。
【0033】
例えば、第二(非オピオイド)薬剤が、処方に含まれる場合、このような薬剤は、制御
放出形態中、または即時放出形態中に含まれ得る。追加の薬剤は、オピオイドと一緒に制
御放出マトリックスに組込まれうる;制御放出コーティングに組込まれる;分離された制
御放出層または即時放出層として組込まれる;または本発明の基質を有するゼラチンカプ
セル中の粉末、顆粒などとして組込まれうる。
本発明のある種の好ましい実施形態では、即時放出形態中の有効量のヒドロコドンは、
投与されるべき制御放出単位用量ヒドロコドン処方に含まれる。即時放出形態のヒドロコ
ドンは、血液(例えば、血漿)中のヒドロコドンのCmaxに達する時間を短縮するのに有
効である量で含まれる。このような実施形態では、即時放出形態中の有効量のヒドロコド
ンは、本発明の基質上に被覆されうる。例えば、処方からの遅延放出ヒドロコドンが、制
御放出コーティングによる場合、即時放出層は、制御放出コーティングの上に上塗りされ
る。他方、即時放出層は、ヒドロコドンが制御放出マトリックスに組込まれる基質の表面
に被覆されうる。有効な単位用量のヒドロコドンを含む複数の持続的放出基質(例えば、
ペレット、球体、ビーズおよび同等物を含む微粒子システム)が、硬質カプセルに組込ま
れる場合、オピオイドの即時放出部分は、カプセル内で粉末またはカルシウム粒として十
分量の即時放出ヒドロコドンの封入を介して、ゼラチンカプセルに組込まれうる。代わり
に、ゼラチンカプセルそれ自身は、ヒドロコドンの即時放出層で被覆されうる。当業者は
、即時放出ヒドロモルホン部分を、単位用量に組込むさらに他の代替手段を認識する。こ
のような代替法は、付随の請求項に包含すると考えられる。単位用量中のこのような有効
量の即時放出ヒドロコドンを含むことによって、患者における相対的に高いレベルの痛み
の経験は、明らかに減少される。
【0034】
剤形
制御放出剤形は、都合により、ヒドロコドンと一緒にマトリックスに組込まれるか、ま
たは薬剤を包含する基質(語句「基質」は、ビーズ、ペレット、スフェロイド、錠剤、錠
剤コアなどを包含する)の上に持続的放出コーティングとして塗布される制御放出材料を
含みうる。制御放出材料は、所望される場合、疎水性または親水性でありうる。本発明に
よる経口剤形は、例えば、顆粒、球状体、ペレット(以降、「マルチパーチパーティキュ
レート(multiparticulates)」として集約的に引用される)として提供されうる。単位
時間あたり所望の用量のオピオイドを提供するために有効であるマルチパーティキュレー
ト(微粒子)の量は、カプセルに入れられうるか、または例えば錠剤に圧縮された任意の
他の適切な経口固形形態に組込まれうる。他方、本発明による経口投与剤形は、制御放出
コーティングで被覆された錠剤コアとして、または薬剤のマトリックス、制御放出材料、
および都合により他の医薬上所望の成分(例えば、希釈剤、バインダ、着色剤、潤滑剤な
ど)を含む錠剤として製造されうる。
【0035】
制御放出マトリックス処方
本発明の特定の好ましい実施形態では、制御放出処方は、上に説明されるとおり制御放
出材料を含むマトリックス(例えば、マトリックス錠剤)を介して達成される。制御放出
マトリックスを含む剤形は、pH依存性またはpH独立性手段で、好まれる範囲内で、そ
してオピオイドを放出するオピオイドのインビトロ溶解速度を提供する。制御放出マトリ
ックスで封入に適した材料は、マトリックスを形成するために使用される方法に依存する
。経口剤形は、少なくとも1種の親水性または疎水性制御放出材料の1%および8%(重
量で)の間で含有しうる。
【0036】
本発明による制御放出マトリックスに含まれうる適切な制御放出材料の制限なしのリス
トは、親水性および/または疎水性材料、例えば、ゴム、セルロースエーテル、アクリル
酸樹脂、タンパク質由来の材料、蝋(waxes)、シェラック、および水素化ひまし油、水
素化植物性油のような油が挙げられる。しかし、オピオイドの制御放出を与える能力のあ
る任意の医薬上許容しうる疎水性または親水性の制御放出材料は、本発明によって使用さ
れうる。好ましい制御放出高分子としては、エチルセルロースのようなアルキルセルロー
ス、アクリル酸またはメタクリル酸重合体および共重合体、およびセルロースエーテル、
特にヒドロキシアルキルセルロース(特にヒドロキシプロピルメチルセルロース)および
カルボキシアルキルセルロースが挙げられる。好ましいアクリル酸およびメタクリル酸重
合体および共重合体としては、メチルメタクリレート、メチル・メタクリレート共重合体
、エトキシエチル・メタクリレート、シアノエチル・メタクリレート、アミノアルキル・
メタクリレート共重合体、ポリ(アクリル酸)、ポリ(メタクリル酸)、メタクリル酸ア
ルキルアミン共重合体、ポリ(メチル・メタクリレート)、ポリ(メタクリル酸)(無水
物)、ポリメタクリレート、ポリアクリルアミド、ポリ(メタクリル酸無水物)、および
グリシジル・メタクリレート共重合体が挙げられる。特定の好ましい実施形態は、本発明
のマトリックス中の前述の制御放出材料のいずれかの混合物を利用する。
【0037】
マトリックスは、バインダとしても含有しうる。このような実施形態では、バインダは
、制御放出マトリックスからのヒドロコドンの制御放出に寄与することが好ましい。
好ましい疎水性バインダ材料は、多かれ少なかれ顕著な親水性および/または疎水性傾
向を示すが、水不溶性である。好ましくは、本発明に有用な疎水性バインダ材料は、約3
0から約200℃まで、好ましくは約45から約90℃までの融点を示す。疎水性材料が
、炭化水素であるときに、炭化水素は、好ましくは、25°および90℃の間の融点を示
す。長鎖(C8−C50)ヒドロコドン材料の中でも、脂肪酸(脂肪族)アルコールが好ま
しい。経口剤形は、少なくとも1種の消化可能な長鎖炭化水素の80%(重量で)までを
含有しうる。
好ましくは、経口剤形は、少なくとも1種のポリアルキレングリコールの80%(重量
で)までを含有する。特に、疎水性バインダ材料は、天然または合成ワックス、脂肪酸ア
ルコール(ラウリル、ミリスチル、ステアリル、セチルまたは好ましくはセトステアリル
アルコールのような)、脂肪酸を包含でき、そしてそれに限定されないが、脂肪酸エステ
ル、脂肪酸グリセリド(モノ−、ジ−、およびトリ−グリセリド)、水素化脂肪、炭化水
素、正常なワックス、ステリン酸、ステアリルアルコール、および炭化水素骨格を有する
疎水性および親水性材料が挙げられる。適切なワックスとしては、例えば、蜜蝋、糖ワッ
クス、ヒマシワックスおよびカルナバ蝋が挙げられる。本発明の目的のために、ワックス
様物質は、室温で正常に固形であり、そして約30から約110℃までの融解点を示す任
意の材料として定義される。
【0038】
本発明により使用されうる好ましい疎水性バインダ材料としては、消化のよい長鎖(C
8−C50、特にC12−C40)、脂肪酸、脂肪酸アルコール、脂肪酸のグリセリルエステル
、鉱物油および植物性油、天然および合成ワックスおよびポリアルキレングリコールのよ
うな置換または未置換炭化水素が挙げられる。25℃および90℃の間の融点を示す炭化
水素が、好ましい。長鎖炭化水素バインダ材料の、脂肪酸(脂肪族)アルコールは、特定
の実施形態で好まれる。経口剤形は、80%(重量で)までの少なくとも1種の消化のよ
い長鎖ヒドロコドンを含有しうる。
特定の好ましい実施形態では、2種またはそれ以上の疎水性バインダ材料は、マトリッ
クス処方に含まれる。追加の疎水性バインダ材料が含まれる場合、それは、天然および合
成ワックス、脂肪酸、脂肪酸アルコール、および同じものの混合物から選択されるのが好
ましい。例としては、蜜蝋、カルナバ蝋、ステアリン酸およびステアリルアルコールが挙
げられる。このリストは、限定されることを意味しない。
【0039】
1つの特定の適切な制御放出マトリックスは、少なくとも1種の水溶性ヒドロキシアル
キルセルロース、少なくとも1つのC12−C36、好ましくはC14−C22、脂肪族アルコー
ルおよび都合により少なくとも1つのポリアルキレングリコールを包含する。ヒドロキシ
アルキルセルロースは、好ましくは、ヒドロキシプロピルセルロース、ヒドリキシプロピ
ルメチルセルロース、および特にヒドロキシエチルセルロースのようなヒドロキシ(C1
からC6まで)アルキルセルロースである。本発明の経口剤形における少なくとも1種の
ヒドロキシアルキルセルロースの量は、中でも、必要とされる正確な速度のオピオイド放
出によって決定される。脂肪族アルコールは、例えば、ラウリルアルコール、ミリスチル
アルコールまたはステアリルアルコールでありうる。しかし、本発明の経口剤形の特に好
ましい実施形態で、少なくとも1種の脂肪族アルコールは、セチルアルコールまたはセト
ステアリルアルコールである。本発明の経口剤形における脂肪族アルコールの量は、上記
のとおり、必要とされる正確な速度のオピオイド放出によって決定される。それは、少な
くとも1種のポリアルキレングリコールが、経口剤形で存在、または不在であるかどうか
にもよる。少なくとも1種のポリアルキレングリコールの不在で、経口剤形は、好ましく
は、20%〜50%(重量で)の脂肪族アルコールを含む。ポリアルキレングリコールは
、経口剤形で存在するとき、それにより脂肪族アルコールおよびポリアルキレングリコー
ルの合わせた重量は、好ましくは、総投与量の20%および50%(重量で)の間で構築
する。
【0040】
1つの好ましい実施形態では、例えば、少なくとも1種のヒドロキシアルキルセルロー
スまたはアクリル酸樹脂対少なくとも1種の脂肪族アルコール/ポリアルキレングリコー
ルの比は、相当の範囲まで、処方からのオピオイドの放出速度を決定する。1:2および
1:4の間のヒドロキシアルキルセルロース対脂肪族アルコール/ホリアルキレングリコ
ールの比が好ましく、そして1:3および1:4の間の比は、特に好ましい。
【0041】
ポリアルキレングリコールは、例えば、ポリプロピレングリコールまたは好ましくはポ
リエチレングリコールでありうる。少なくとも1種のポリアルキレングリコールの数平均
分子量は、好ましくは、1,000および15,000の間、特に1,500および12
,000の間である。
別の適切な制御放出マトリックスは、アルキルセルロース(特に、エチルセルロース)
、C12からC36までの脂肪族アルコールおよび都合によりポリアルキレングリコールを包
含する。
【0042】
上記成分に加えて、制御放出マトリックスは、適切な量の他の材料、例えば希釈剤、潤
滑剤、バインダ、造粒助剤、着色剤、風味剤および製薬業界で従来のものであるグライダ
ント(glidants)をも含有しうる。
本発明による固形の制御放出の経口剤形の製造を促進するために、本発明の別の態様で
は、オピオイドまたはその塩を、制御放出マトリックスに組込む本発明による固形の制御
放出の経口剤形を製造する方法が提供される。マトリックス中の組込みは、例えば、
(a)ヒドロコドンと一緒に、上に説明されるとおりの少なくとも1種の疎水性および
/または親水性材料(例えば、水溶性ヒドロキシアルキルセルロース)を含む顆粒を形成
すること、
(b)顆粒を含む少なくとも1種の疎水性および/または親水性材料を、少なくとも1
種のC12−C36の脂肪族アルコールと混合すること、および
(c)都合により、顆粒を圧縮および成形すること
によってもたらされる。
顆粒は、製薬処方の当業者によく知られる手段のいずれかによって形成されうる。例え
ば、1つの好ましい方法では、顆粒は、水を用いてヒドロキシアルキルセルロース/オピ
オイドを湿式造粒することによって形成されうる。この方法の特に好ましい実施形態では
、湿式造粒段階の間に添加される水の量は、好ましくは、オピオイドの乾燥重量の1.5
および5倍の間、特に1.75および3.5倍の間である。
【0043】
本発明のマトリックスは、溶融ペリティゼーション技術を介しても製造されうる。この
ような環境で、細かく分割された形態でのオピオイドを、バインダ(粒子形態でも)およ
び他の任意の不活性成分と合わせ、そしてその後、混合物を、例えば高せん断混合装置で
の混合物を機械的作業して、ペレット(顆粒、球状体)を形成することによってペレット
化する。その後、ペレット(顆粒、球状体)は、必要とされるサイズのペレットを得るた
めに篩にかけられる。バインダ材料は、好ましくは、粒子形態にあり、そして約40℃よ
り上の融点を示す。適切なバインダ物質としては、例えば、水素化ヒマシ油、水素化植物
性油、他の水素化脂肪、脂肪酸アルコール、脂肪酸エステル、脂肪酸グリセリドおよび同
等物が挙げられる。
制御放出材料は、例えば、溶融造粒または溶融押出技術によっても製造されうる。一般
に、溶融造粒技術は、正常に固形の疎水性バインダ材料、例えばワックスを融解させ、そ
してそこに粉末薬剤を組込むことに関与する。制御放出剤形を得るために、疎水性制御放
出材料、例えばエチルセルロースまたは水不溶性アクリル酸重合体を、溶融ワックス疎水
性バインダ材料に組込むことは必要でありうる。溶融造粒技術を介して製造される制御放
出処方の例は、本発明の譲受人に譲渡され、そしてそれの全体で参照して組込まれた米国
特許第4,861,598号で見られる。
【0044】
別の疎水性バインダ材料は、上記1種またはそれ以上の水不溶性ワックス様物質より疎
水性が低い1種またはそれ以上のワックス様熱可塑性物質と混合された1種またはそれ以
上の水不溶性ワックス様熱可塑性物質を包含しうる。制御放出を達成するために、処方中
の個々のワックス様物質は、当初の放出相の間じゅう胃腸流動体で実質的に非分解性で、
そして不溶性であるべきである。有用な水不溶性ワックス様バインダ物質は、約1:50
00(w/w)より低い水不溶性を示すものでありうる。
上記成分に加えて、制御放出マトリックスは、適切な量の他の材料、例えば、希釈剤、
潤滑剤、バインダ、造粒助剤、着色剤、風味剤、および所望の場合、約50重量%の粒子
までの量で、製薬業界で従来のものであるグライダントをも含有しうる。これらの追加の
材料の量は、所望の処方に所望の効果を提供するのに十分である。
経口剤形を処方するために使用されうる医薬上許容しうる担体および賦形剤の特別の例
は、ここに参照して組込まれるHandbook of Pharmaceutical
Excipients,American Pharmaceutical Asso
siation(1986)で記述される。
【0045】
本発明による適切な溶融押出マトリックスの製造は、例えば、制御放出材料、そして好
ましくはバインダ材料と一緒に、オピオイド鎮静薬を混錬して、均質な混合物を得る段階
を包含しうる。その後、均質な混合物は、少なくとも混合物を、同じものを押出すために
十分に軟化するのに十分な温度まで加熱される。その後、生じた均質混合物は、例えば、
二重スクリュー押出装置を用いて押出して、ストランドを形成される。押出物を、好まし
くは、冷却し、そして当業界で知られる任意の手段によってマルチパーティキュレートに
切断される。ストランドを冷却し、そしてマルチパーティキュレートに切断する。その後
、マルチパーティキュレートを単位用量に分割する。押出物は、好ましくは、約0.1か
ら約5mmまでの直径を有し、そして約8から約24時間の期間に、制御放出の治療的な
活性剤を提供する。
【0046】
本発明の溶融押出された処方を製造する任意の方法は、押出装置に、疎水性制御放出材
料、治療的な活性剤、および任意のバインダ材料を直接メーターで測定すること;均質な
混合物を加熱すること;均質な混合物を押出して、それによりストランドを形成すること
;均質な混合物を含むストランドを冷却すること;ストランドを、約0.1mmから約1
2mmまでのサイズを示す粒子に切断すること;および上記粒子を単位用量に分割するこ
とを含む。本発明のこの態様で、相対的に継続的に製造手段が認識される。
ここで上に記述されるもののような可塑剤は、溶融押出マトリックスに含まれうる。可
塑剤は、好ましくは、約0.1から約30重量%までのマトリックスとして含まれうる。
他の製薬上の賦形剤、例えばタルク、単または多糖類、着色剤、風味剤、潤滑剤および同
等物は、所望のとおり本発明の制御放出マトリックスで含まれうる。含まれる量は、達成
されるべき所望の特徴による。
【0047】
押出装置または出口の直径は、押出ストランドの厚みを変化させるために調節されうる
。さらに、押出装置の出口部分は、丸型である必要はない;それは、長方形、矩形などで
ありうる。存在するストランドを、熱いワイヤー状カッター、断裁機などを用いて粒子に
変えさせる。溶融押出マルチパーティキュレートシステムは、押出装置の出口オリフィス
によって、例えば、顆粒、球状体またはペレットの形態にありうる。本発明の目的のため
に、語句「溶融押出マルチパーティキュレート(類)」および「溶融押出マルチパーティ
キュレートシステム(類)」および「溶融押出粒子」は、好ましくは、類似のサイズおよ
び/または形状の範囲内にあり、そして1種またはそれ以上の活性剤および1種またはそ
れ以上の賦形剤を含有し、好ましくはここに記述されるとおり疎水性制御放出を含む複数
の単位に該当すべきである。好ましくは、溶融押出マルチパーティキュレートは、長さ約
0.1から約12mmまでの範囲のものであり、そして約0.1から約5mmまでの直径
を示す。さらに、溶融押出マルチパーティキュレートは、単にビーズ、種子、ペレットな
どの手段によるようなこのサイズ範囲内の任意の幾何学的形状でありうることと理解すべ
きである。代わりに、押出物は、単に、所望の長さに切断され、そして球形化段階の必要
なしに、治療上活性剤の単位用量に分割されうる。
【0048】
1つの好ましい実施形態では、カプセル内に有効量の溶融押出マルチパーティキュレー
トを含む経口剤形が、製造される。例えば、複数の溶融押出マルチパーティキュレートは
、胃液によって摂取および接触されるときに、有効な制御放出用量を提供するのに十分な
量でゼラチンカプセルに注がれうる。
別の好ましい実施形態では、適切な量のマルチパーティキュレート押出物を、標準技術
を用いた従来の打錠装置を用いて、経口錠剤に圧縮する。錠剤(圧縮および注型された)
、カプセル(硬質および軟質ゼラチン)および丸剤を製造するための技術および組成物は
、ここに参照して組込まれるRemington's
Pharmaceutical Science,(アーサー・オソル、編集者)、1
553−1593(1980)にも記述される。
さらに別の好ましい実施形態では、押出物は、上にさらに詳細に記述され、そして参照
してここに組込まれる米国特許第4,957,681号(クリメッシュら)で説明される
とおり錠剤に成形されうる。
都合により、制御放出のマトリックスマルチパーティキュレートシステムまたは錠剤を
、被覆しうるか、またはゼラチン、カプセルを、上に記述される制御放出コーティングの
ような制御放出コーティングでさらに被覆しうる。上塗は、例えば使用される特定のオピ
オイド鎮静薬の物理的特性、および所望の放出速度により大きくなりうるが、このような
コーティングは、好ましくは、十分量の疎水性および/または親水性の制御放出材料を含
んで、約2から約25パーセントまでの重量増レベルを得る。
【0049】
本発明の剤形は、さらに、1つまたはそれ以上のオピオイド鎮静薬を含む溶融押出マル
チパーティキュレートの組合せを包含しうる。さらに、剤形は、即座治療効果についての
多量の即時放出の治療的に有効な剤をも包含しうる。即時放出の治療的な活性剤は、例え
ば、ゼラチンカプセル内の別個のペレットとして組込まれうるか、または、例えば、ビー
ズまたは溶融押出マルチパーティキュレートの表面に被覆されうる。本発明の単位剤形は
、例えば、制御放出ビーズおよびマトリックスマルチパーティキュレートの組合せをも含
んで、所望の効果を達成しうる。
本発明の制御放出処方は、摂取され、そして胃液に、そしてその後腸液にさらされると
きに、治療的に有効な剤をゆっくりと放出することが好ましい。本発明の溶融押出処方の
制御放出プロフィルは、例えば、制御放出材料の量を変化させることにより、他のマトリ
ックス構成物、疎水性材料に比べて可塑剤の量を変化させることにより、追加の成分また
は賦形剤の封入により、製造の方法を改変されるなどにより変えられうる。
【0050】
本発明の他の実施形態では、溶融押出処方は、その後押出物に添加される治療的に有効
な量の封入なしに製造される。このような処方は、典型的には、押出マトリックス材料と
一緒に混錬された治療上有効な剤を有し、そしてその後、その混合物は、遅延放出処方を
提供するために錠剤化される。このような処方は、例えば、処方に含まれる治療上活性剤
が、疎水性材料および/または遅延反応剤材料を軟化させるために必要とされる温度に感
受性があるときに、有益でありうる。
本発明により使用するのに適した典型的な溶融押出製造システムは、可変速度および一
定トルク制御を示す適切な押出装置駆動モータ、開始停止制御および電流計を含む。さら
に、製造システムは、押出装置の長さを通して、温度センサー、冷却手段および温度イン
ジケーターを含む温度制御コンソールを含む。さらに、製造システムは、その出口で開口
部またはダイを有するシリンダーまたはバレル内に含まれる2つの計測計回転かみ合いス
クリューから構成される二重スクリュー押出装置のような押出装置を含む。供給材料は、
供給ホッパーを通って入り、そしてスクリューによるバレルを通して移動され、そしてダ
イを通して、冷却に対処する連続的可動ベルトによるようにその後運搬されるストランド
に向けられ、そして押出ロープをマルチパーティキュレートシステムにさせるためにペレ
ット化装置または他の適切なデバイスに向けられる。ペレット化装置は、ローラー、固定
ナイフ、回転カッターおよび同等物から構成されうる。適切な装置およびシステムは、ニ
ュージャージー州サザン・ハッケンサックのシー・ダブリュー・ブラベンダー・インスト
ルメンツ、インク.のようなディストリビューターから利用できる。他の適切な装置は、
当業者に明らかである。
【0051】
本発明の別の態様は、押出製品に含まれる空気の量を制御する方法で、上に説明される
とおり溶融押出マルチパーティキュレートの製造に関する。押出物に含まれる量を制御す
ることにより、例えば、マルチパーティキュレート押出物からの治療上活性剤の放出速度
は、明らかに変えられうることが、驚くべきことに分かった。特定の実施形態では、押出
製品のpH依存性は、同様に変えられうることが、驚くべきことに分かった。
したがって、本発明の別の態様で、溶融押出製品は、工程の押出層を通して空気を実質
的に押出す手段で製造される。これは、例えば、真空付属物を有するレイズトリッツの押
出装置を使用することによって達成されうる。レイズトリッツの押出装置を用いた本発明
によって製造される押出マルチパーティキュレートが、様々の物理特性を示す溶融押出製
品を提供することが、驚くべきことに分かった。特に、押出物は、例えば、SEM(走査
電子顕微鏡写真)を提供する走査電子顕微鏡を用いて拡大されるときに、実質的に非多孔
性である。従来の考えに対比して、このような実質的に非多孔性処方が、真空なしに製造
された同じ処方に比べて、治療上有効な剤の早い放出を提供することが分かった。真空下
で押出装置を用いて製造されたマルチパーティキュレートのSEMは、非常に平滑である
ように見え、そしてマルチパーティキュレートは、真空なしに製造されたマルチパーティ
キュレートのものよりいっそう強い傾向にある。少なくとも特定の処方で、真空下の押出
の使用は、真空なしに製造されたそれの対抗の処方よりいっそうpH依存性である押出さ
れたマルチパーティキュレート製品を提供することが観察された。
【0052】
マトリックス・ビーズを製造する方法
本発明による制御放出の剤形は、マトリックスビーズ処方としても製造されうる。マト
リックス・ビーズは、球体化剤およびヒドロコドンを含む。
ヒドロコドンは、重量で約0.01から約99重量%までのマトリックスビーズを包含
することが好ましい。ヒドロコドンは、約0.1から約50重量%までのマトリックスビ
ーズとして包含されることが好ましい。
本発明のマトリックス・ビーズ処方を製造するために使用されうる球体化剤(spheroni
sing agents)は、任意の当業界で知られる球体化剤を含む。セルロース誘導体が好まれ
、そして微小結晶性セルロースが、特に好ましい。適切な微小結晶性セルロースは、例え
ば、アビセルPH101(商標、エフ・エム・シー・コーポレーション)として販売され
ている材料である。球体化剤は、重量で約1から約99%のマトリックス・ビーズとして
含まれることが好ましい。
【0053】
活性成分および球状化剤に加えて、スフェロイドは、バインダをも含みうる。低粘度水
溶性高分子のような適切なバインダは、製薬業界での当業者によく知られている。しかし
、ヒドロキシプロピルセルロースのような水溶性ヒドロキシ低級アルキルセルロースが好
ましい。
オピオイド鎮痛薬および球状化剤に加えて、本発明のマトリックス・ビーズ処方は、上
述されるもののような制御放出材料を含みうる。マトリックス・ビーズ処方中の封入のた
めの好ましい制御放出材料は、アクリル酸およびメタクリル酸重合体または共重合体およ
びエチルセルロースを含む。処方に存在する場合、制御放出材料は、重量で約1から約8
0%のマトリックス・ビーズの量で含まれる。制御放出材料は、好ましくは、ビーズから
のオピオイド鎮痛薬の制御放出を提供するのに有効な量で、マトリックス・ビーズ処方に
含まれる。
【0054】
バインダ、希釈剤、および同等物のような製薬的加工助剤は、マトリックス・ビーズ処
方に含まれ得る。処方に含まれるこれらの剤の量は、処方により示されるべき所望の効果
で変化する。
マトリックス・ビーズは、ここで上に記述されるもののような制御放出材料を含めた制
御放出コーティングで上塗りされうる。制御放出のコーティングは、約5から約30%ま
での重量増まで塗布される。使用されるべき制御放出コーティングの量は、マトリックス
・ビーズおよび化合物の組成、および/またはオピオイド鎮痛薬(すなわち、ヒドロコド
ン)の物理特性のような多様な因子によって変化する。
マトリックス・ビーズは、一般に、例えば湿式造粒により、オピオイド鎮痛薬と一緒に
球状化剤を造粒することによって製造される。その後、顆粒は、マトリックス・ビーズを
生成するために球状化される。その後、マトリックス・ビーズは、都合により、ここで上
に記述されるもののような方法によって、制御放出コーティングで上塗りされる。
【0055】
マトリックス・ビーズを製造する別の方法は、(a)少なくとも1種の水溶性ヒドロキ
シアルキルセルロースおよびオピオイドまたはオピオイド塩を含む顆粒を成形し、(b)
顆粒を含むヒドロキシアルキルセルロースを、少なくとも1種のC12−C36脂肪族アルコ
ールと混合し、そして(c)都合により、顆粒を圧縮および成形することによる。好まし
くは、顆粒は、水を用いてヒドロキシアルキルセルロース/オピオイドを湿式造粒するこ
とによって成形される。本発明の特に好ましい実施形態では、湿式造粒段階の間に添加さ
れる水の量は、オピオイドの乾燥重量の1.5および5倍の間、特に1.75および3.
5倍の間であるのが好ましい。
【0056】
さらに他の代替の実施形態では、活性成分と一緒の球状化剤は、球状化されて、スフェ
ロイドを形成しうる。微小結晶性セルロースが好ましい。適切な微小結晶性セルロースは
、例えば、アビセルPH101(商標、エフ・エム・シー・コーポレーション)として販
売されている材料である。このような実施形態では、活性成分および球状化剤に加えて、
スフェロイドは、バインダをも含みうる。低粘度水溶性重合体のような適切なバインダは
、製薬業界での当業者によく知られる。しかし、ヒドロキシプロピルセルロースのような
水溶性ヒドロキシ低級アルキルセルロースが、好まれる。さらに(または代わりに)、ス
フェロイドは、メタクリル酸−エチルアクリレート共重合体、またはエチルセルロースの
ような水不溶性重合体、特にアクリル酸重合体、アクリル酸共重合体を含みうる。このよ
うな実施形態では、持続的放出コーティングは、一般に、(a)単独、または脂肪酸アル
コールとの混和物でのいずれかのワックス;または(b)シェラックまたはゼインのよう
な水不溶性材料を含む。
【0057】
制御放出ビーズ処方
1つの特に好ましい実施形態では、経口剤形は、ゼラチンカプセル内に含まれる有効数
の制御放出スフェロイドを包む。
本発明の別の好ましい実施形態では、制御放出剤形は、制御放出材料を含む制御放出コ
ーティングで被覆される活性成分を含むスフェロイドを包む。語句スフェロイドは、製薬
業界で知られており、そして例えば、0.1mmおよび25mmの間、特に0.5mmお
よび2mmの間の直径を示す球状の顆粒を意味する。
スフェロイドは、好ましくは、水性媒体中で、制御速度でのオピオイド(または塩)の
放出を許す制御放出材料で被覆されたフィルムである。フィルムコートは、他の記述され
た特性と組合せて、上に概説されるインビトロ放出速度(例えば、少なくとも約12.5
%が、1時間後に放出される)を達成するために選択される。本発明の制御放出のコーテ
ィング処方は、好ましくは、平滑であり、そして素晴らしく、色素または他のコーティン
グ添加剤を支持する能力のあり、非毒性で、不活性で、そして粘着性なしである、強力な
連続フィルムを生じる。
【0058】
コーティング
本発明の剤形は、都合により、放出の制御のために、または処方の保護のために適する
1つまたはそれ以上のコーティングで被覆されうる。1つの実施形態では、コーティング
は、例えば、胃腸液にさらされる場合に、pH依存性またはpH独立性の放出のいずれか
を許すために供される。pH独立性コーティングが望まれる場合、コーティングは、環境
流動体、例えば胃腸管でのpH変化にもかかわらず、最適な放出を達成するように設計さ
れる。他の好ましい実施形態は、患者に対して少なくとも約12時間、および好ましくは
24時間までの無痛覚を提供する能力がある吸収プロファイルが、提供されるように、胃
腸(GI)管の所望な領域、例えば胃または小腸にオピオイドを放出するpH依存的コー
ティングを含む。胃腸管の1つの所望の領域、例えば胃での用量の一部を放出し、そして
胃腸管の別の領域、例えば小腸でその用量の残りを放出する組成物を処方することも可能
である。
pH依存性コーティングを利用する本発明による処方は、それによって未保護が、腸溶
性コートに上に被覆され、そして胃で放出される反復作用効果をも与え得る一方で、腸溶
性コーティングによって保護される残りは、胃腸管にさらに放出される。本発明によって
使用されうるpH依存性であるコーティングは、例えば、シェラック、セルロースアセテ
ートフタレート(CAP)、ポリビニルアセテートフタレート(PVAP)、ヒドロキシ
プロピルメチルセルロースフタレート、およびメタクリル酸エステル共重合体、ゼインお
よび同等物のような制御放出材料を含む。
【0059】
別の好ましい実施形態では、本発明は、(i)アルキルセルロース;(ii)アクリル
酸重合体;または(iii)その混合物から選択される疎水性制御放出材料で被覆された
オピオイドを包含する固形の制御放出剤形に関する。コーティングは、有機または水性溶
液または分散物の形態で塗布されうる。
特定の好ましい実施形態では、制御放出コーティングは、疎水性制御放出材料の水性分
散物から誘導される。その後、オピオイド(類)(例えば、錠剤コアまたは不活性の製薬
ビーズまたはスフェロイド)を含有する被覆基質は、基質が安定な溶解を提供する終点に
達するまで、硬化される。硬化終点は、硬化直後の剤形の溶解プロファイル(曲線)を、
例えば40℃の温度および75%の相対湿度での少なくとも1ヶ月の促進された保存条件
にさらされた後の剤形の溶解プロファイル(曲線)と比較することによって決定されうる
。これらの処方は、本発明の譲受人に譲渡され、そして参照してここに組込まれる米国特
許第5,273,760号および第5,286,493号で詳細に説明される。本発明に
よって使用されうる制御放出の処方およびコーティングの他の例としては、参照してここ
に組込まれる米国特許第5,324,351号;第5,356,467号、および第5,
472,712号が挙げられる。
【0060】
好ましい実施形態では、制御放出のコーティングは、ここで下に記述されるもののよう
な可塑剤を含む。
特定の実施形態では、制御放出処方を得るために、アルキルセルロースまたはアクリル
酸重合体の十分量の水性分散物と共にオピオイド鎮痛薬を含む基質に上塗りして、約2か
ら約50%、例えば約2から約25%までの重量増を得ることが必要である。上塗りコー
トは、多かれ少なかれ、例えば、治療的な活性剤の物理特性および所望の放出速度、水性
分散物における可塑剤の封入および同じものの組込みの手段による。
【0061】
アルキルセルロース重合体
アルキルセルロースを含めたセルロース性材料および重合体は、本発明による基質、例
えばビーズ、錠剤などを被覆するのによく適合した制御放出材料である。習熟者は、他の
セルロースおよび/またはアルキルセルロース重合体が、本発明による疎水性コーティン
グの全部または一部として、単独で、または任意の組合せで容易に使用されうることを認
識するが、実施例の方法によって簡単に、1つの好ましいアルキルセルロース性重合体は
、エチルセルロースである。
エチルセルロースの1つの商用的に利用できる水性分散物は、アクアコート(登録商標
;Aquacoat)エフ・エム・シー・コープ、米国ペンシルベニア州フィラデルフィ
ア)である。アクアコート(登録商標)は、水混和しない有機溶媒でのエチルセルロース
を溶解し、そしてその後、それを界面活性剤および安定化剤の存在下で水中に乳化させる
ことによって製造される。ミクロンより小さい小滴を生じるための均質化の後、有機溶媒
を、真空で蒸散させて、偽ラテックスを形成する。可塑剤は、製造相の間じゅう、偽ラテ
ックスに組込まれない。したがって、コーティングと同じものを使用する前に、アクアコ
ート(登録商標)を、使用する前に適切な可塑剤と最終的に混合することが必要である。
【0062】
エチルセルロースの別の水性分散物は、シュアレーズ(登録商標;Surelease
)(カラーコン、インク、米国ペンシルベニア州ウエストポイント)として市販で入手で
きる。この製品は、製造工程の間に可塑剤を分散物に組込むことによって製造される。ホ
ットメルトの重合体、可塑剤(ジブチルセバケート)、および安定化剤(オレイン酸)は
、均質な混合物として製造され、そしてそれは、その後、アルカリ性溶液で希釈されて、
基質上に直接塗布されうる水性分散物を得る。
【0063】
アクリル酸重合体
本発明の他の好ましい実施形態では、制御放出コーティングを含む制御放出材料は、製
薬上許容しうるアクリル酸重合体であり、そしてそれに限定されないが、アクリル酸およ
びメタクリル酸共重合体、メチル・メタクリレート共重合体、エトキシエチル・メタクリ
レート、シアノエチル・メタクリレート、ポリ(アクリル酸)、ポリ(メタクリル酸)、
メタクリル酸アルキルアミド共重合体、ポリ(メチル・メタクリレート)、ポリメタクリ
レート、ポリ(メチル・メタクリレート)共重合体、ポリアクリルアミド、アミノアルキ
ル・メタクリレート共重合体、ポリ(メタクリル酸無水物)、およびグリジジル・メタク
リレート共重合体が挙げられる。
特定の好ましい実施形態では、アクリル酸重合体は、1種またはそれ以上のアンモニオ
メタクリレート共重合体から構成される。アンモニオ・メタクリレート共重合体は、当業
界でよく知られており、そして低含有量の四級アンモニウム基を示すアクリル酸およびメ
タクリル酸エステルの十分に重合化した共重合体としてNF XVIIで記述されている
。
【0064】
所望の溶解プロファイルを得るために、様々のモル比の四級アンモニア基のような様々
の物理的特性を示す2つまたはそれ以上のアンモニオメタクリレート共重合体を、天然の
(メト)アクリル酸エステルに組込むことが必要でありうる。
特定のメタクリル酸エステル型重合体は、本発明によって使用されうるpH依存性コー
ティングを製造するために有用である。例えば、ローム・テク、インク.からオイドラギ
ッド(登録商標;Eudragit)として商用的に入手できる、メタクリル酸共重合体
または重合性メタクリレートとしても知られるジエチルアミノエチル・メタクリレートお
よび他の天然のメタクリル酸エステルから合成される共重合体のファミリーがある。幾ら
かの異なる型のオイドラギッド(登録商標)がある。例えば、オイドラギッドEは、酸性
媒体で膨張および溶解するメタクリル酸共重合体の例である。オイドラギッドLは、約p
H<5.7で膨張せず、そして約pH>6で溶解性であるメタクリル酸共重合体の例であ
る。オイドラギッドSは、約pH<6.5で膨張せず、そして約pH>7で溶解性である
メタクリル酸共重合体の例である。オイドラギッドRLおよびオイドラギッドRSは、水
膨張性であり、そしてこれらの重合体によって吸収される水の量は、pH依存性であるが
、しかし、オイドラギッドRLおよびオイドラギッドRSで被覆された剤形は、pH独立
性である。
【0065】
特定の好ましい実施形態では、アクリル酸コーティングは、それぞれ、商標名オイドラ
ギッド(登録商標)RL30DおよびオイドラギッドRS30Dの下でローム・ファルマ
から商用的に入手可能な2つのアクリル酸樹脂ラッカーの混合物を包含する。オイドラギ
ッド(登録商標)RL30Dおよびオイドラギッド(登録商標)RS30Dは、低い含有
量の四級アンモニウム基を有するアクリル酸およびメタクリル酸エステルの共重合体であ
り、アンモニウム基対残りの中性(メト)アクリル酸エスエルのモル比は、オイドラギッ
ド(登録商標)RL30Dで1:20であり、そしてオイドラギッド(登録商標)RS3
0Dで1:40である。平均分子量は、約150,000である。コード認識RL(非常
に透過性)およびRS(低い透過性)は、これらの剤の透過性特性に該当する。オイドラ
ギッド(登録商標)RL/RS混合物は、水に、そして消化性流動体で不溶性である。し
かし、同じものから形成されるコーティングは、膨張性であり、そして水性溶液および消
化性流動体で透過性である。
【0066】
本発明のオイドラギッド(登録商標)RL/RS分散物は、所望の溶解プロファイルを
示す最終的に制御放出処方を得るために、任意の所望の比で一緒に混合されうる。望まれ
る制御放出の処方は、例えば、100%オイドラギッド(登録商標)RL、50%オイド
ラギッド(登録商標)RL、および50%オイドラギッド(登録商標)RSおよび10%
オイドラギッド(登録商標)RL:オイドラギッド(登録商標)90%RSから誘導され
る遅延反応剤コーティングから得られうる。もちろん、当業者は、例えばオイドラギッド
(登録商標)Lのような他のアクリル酸重合体も使用されうることを認識する。
【0067】
可塑剤
コーティングが、疎水性制御放出材料の水性分散物を包含する本発明の実施形態では、
疎水性材料の水性分散物中の有効量の可塑剤の封入は、さらに、制御放出コーティングの
物理的特性を改善する。例えば、エチルセルロースが、比較的高いガラス遷移温度を示し
、そして正常なコーティング条件下で柔軟性フィルムを形成しないので、コーティング材
料と同じものを使用する前に、可塑剤を、制御放出コーティングを含有するエチルセルロ
ースコーティングに組込むことが好ましい。一般に、コーティング溶液に含まれる可塑剤
の量は、フィルム形成剤の濃度、例えば、最もしばしば、約1から約50重量パーセント
までのフィルム形成剤に基づく。しかし、可塑剤の濃度は、特定のコーティング溶液およ
び使用の方法を用いた注意深い実験の後にのみ適切に決定されうる。
他の水不溶性可塑剤(アセチレート化モノグリセリド、フタレートエステル、ヒマシ油
など)が、使用されうる可能性があるが、エチルセルロースについての適切な可塑剤の例
は、ビブチルセバケート、ジエチルフタレート、トリエチルシトレート、トリブチルシト
レート、およびトリアセチンのような水不溶性可塑剤が挙げられる。トリエチルシトレー
トは、本発明のエチルセルロースの水性分散物についての特に好ましい可塑剤である。
【0068】
本発明のアクリル酸重合体についての適切な可塑剤の例としては、それに限定されない
が、トリエチルシトレートNF XVI、トリブチルシトレート、ジブチルフタレート、
および可能性のある1,2−プロピレングリコールのようなクエン酸エステルが挙げられ
る。登録商標オイドラギッドRL/RSラッカー溶液のようなアクリル酸フィルムから形
成されるフィルムの弾性を増強するために適切であると立証された他の可塑剤としては、
ポリエチレングリコール、プロピレングリコール、ジメチルフタレート、ヒマシ油および
トリアセチンが挙げられる。トリエチルシトレートは、本発明のエチルセルロースの水性
分散物についての特に好ましい可塑剤である。
制御放出コーティングに対しての少量のタルクの添加は、加工の間に粘着する水性分散
物の傾向を減少し、そして艶出剤として作用することがさらに分かった。
【0069】
被覆ビーズ処方の製造
疎水性材料の水性分散物は、基質例えば、ヌ・パリエル18/20ビーズのような不活
性の製薬的ビーズを被覆するために使用される場合、その後、複数の結果物で安定化され
た固形制御放出ビーズは、摂取され、そして胃液または溶解媒体のような環境流動体によ
って接触されたときに、有効な制御放出用量を提供するのに十分な量でゼラチンカプセル
に注がれうる。
本発明の安定化された制御放出ビーズ処方は、例えば、摂取され、そして胃液に、そし
てその後小腸液にさらされたときに、オピオイド鎮痛薬をゆっくりと放出する。本発明の
処方の制御放出プロファイルは、例えば、疎水性制御放出材料の水性分散物を用いた上塗
りの量を変化させることによって変えられ得て、そして疎水性制御放出材料に関して可塑
剤の量を変化させることによって、追加の成分または賦形剤の封入によって、製造の方法
を変えることによってなど、可塑剤が、疎水性制御放出材料の水性分散物に添加される手
段を変えうる。最終的製品の溶解プロファイルは、例えば、制御放出コーティングの厚み
を増加または減少させることによって、改質もされうる。
【0070】
治療上活性剤で被覆された基質は、例えば、治療上活性剤を水に溶解させる、そしてそ
の後、ウスター挿入物を用いて、溶液を、ヌ・パリエル18/20ビーズのような基質上
に噴霧することによって製造される。都合により、追加の成分は、オピオイドのビーズへ
の結合を助成し、および/または溶液を着色するなどのためにビーズを被覆する前にも添
加される。例えば、着色剤(例えば、オパドライ(登録商標;Opadry)、カラーコ
ン、インク.から商用的に入手可能な)と共に、またはなしに、ヒドロキシプロピル・メ
チルセルロースなどを含む製品は、溶液に添加され得て、そして溶液は、同じものを基質
上に使用する前に(例えば、約1時間)混合されうる。その後、結果物である被覆基質は
、都合により、バリヤ剤で上塗りされて、疎水性制御放出コーティングから治療上活性剤
を分離しうる。
適切なバリヤ剤の例は、ヒドロキシプロピル・メチルセルロースを包含するものである
。しかし、当業界で知られる任意のフィルム形成剤が使用されうる。バリヤ剤は、最終製
品の溶解速度に影響しないことが好ましい。
【0071】
その後、基質は、疎水性制御放出材料の水性分散物で上塗りされうる。疎水性制御放出
材料の水性分散物は、好ましくは、さらに、トリエチルシトレートのような有効量の可塑
剤を含む。アクアコート(登録商標)またはシュアレーズ(登録商標)のような予備処方
されたエチルセルロースの水性分散物が、使用されうる。登録商標シュアレーズが使用さ
れる場合、可塑剤を別個に添加することは、必要でない。代わりに、オイドラギッド(登
録商標)のようなアクリル酸重合体の予備処方された水性分散物が、使用されうる。
【0072】
本発明のコーティング溶液は、フィルム形成剤に加えて、可塑剤および溶媒システム(
すなわち、水)、優雅さを提供し、そして製品差別化を生じる着色剤を含有することが好
ましい。色は、疎水性材料の水性分散物の代わりに、またはそれに加えて、治療上活性剤
の溶液に添加されうる。例えば、色は、せん断を示す色を、水溶性重合体溶液に添加し、
そしてその後、可塑化されたアクアコート(登録商標)に対して低せん断を用いることに
よって、アルコールまたはプロピレングリコール基本の色分散物、挽かれたアルミニウム
レーキおよび二酸化チタンのような懸濁化剤の使用を介して、アクアコート(登録商標)
に添加されうる。代わりに、色を本発明の処方に供する任意の適切な方法が使用されうる
。アクリル酸重合体の水性分散物が使用されるときに、色を処方に供するための適切な成
分は、二酸化チタンおよび酸化鉄色素のような色素を含む。しかし、色素の組込みは、コ
ーティングの阻止効果を増大しうる。
【0073】
疎水性制御放出材料の可塑化水性分散物は、当業界で知られる任意の適切な噴霧装置を
使用して噴霧することによって、治療上活性剤を含む基質上に塗布されうる。好ましい方
法では、底面から注入される空気ジェットが、コア材料を流動化し、そして乾燥を有効に
する一方で、アクリル酸重合体コーティングが噴霧されるウスター流動床システムが使用
される。前記被覆基質が、水性溶液、例えば胃液にさらされるときに、上記治療上活性剤
の予備測定された制御放出を得るために十分量の疎水性材料の水性分散物が、使用される
ことが好ましく、そして可塑剤などの組込みの手段として、治療上活性剤の物理的特性を
考慮する。疎水性制御放出材料で被覆した後、登録商標オパドライのようなフィルム形成
剤の別の上塗りは、都合により、ビーズに塗布される。この上塗りは、ビーズの凝集を実
質的に減少させるために、ともかく供給される。
【0074】
本発明の制御放出処方からの治療上活性剤の放出は、1種またはそれ以上の放出改質剤
の添加により、またはコーティングを通して1種またはそれ以上の放出経路を提供するこ
とによって、さらに影響され、すなわち、所望の速度に調節されうる。疎水性制御放出材
料対水溶性材料の比は、他の因子の中でも、要求された放出速度および選択された材料の
溶解性特徴によって測定される。
多孔形成剤として機能する放出改質剤は、有機または無機であり得て、そして使用の環
境にあるコーティングから溶解、抽出または浸出されうる材料を含む。多孔形成剤は、ヒ
ドロキシプロピルメチルセルロースのような1種またはそれ以上の親水性材料を包含しう
る。
本発明の制御放出コーティングは、スターチおよびゴムのような浸食促進剤も含みうる
。
本発明の制御放出コーティングは、カルボネート基が、重合鎖に再び生じるカルボン酸
の線状ポリエステルから構成されるポリカーボネートのような使用の環境での微小多孔性
板を作成するのに有用な材料をも含みうる。
【0075】
放出改質剤は、半透過性重合体をも包含しうる。特定の好ましい実施形態では、放出改
質剤は、ヒドロキシプロピルメチルセルロース、ラクトース、金属ステアレート、および
前述のもののいずれかの混合物から選択される。
本発明の制御放出コーティングは、少なくとも1種の放出通路、オリフィスまたは同等
物を包含する出口手段をも含みうる。放出通路は、米国特許第3,845,770号;第
3,916,889号;第4,063,064号;および第4,088,864号に開示
されるもののような方法によって形成され得て、そしてその全ては、参照してここに組込
まれうる。放出通路は、丸型、三角、四角、長円形、不均整などのような任意の形状を示
しうる。
【0076】
約24時間投与のために適する制御放出ビーズ処方を生じる別の方法は、粉末積層を介
することである。本発明の譲受人に譲渡され、そしてそれの全体で参照してここに組込ま
れた米国特許第5,411,745号は、基本的に触知できない水性のラクトースから構
成される加工助剤を利用する粉末積層技術を介して製造される24時間モルフィン処方の
製造を教示する。粉末積層ビーズは、水性バインダ溶液を、不活性ビーズに噴霧して、粘
着性表面を提供し、そして粘着性ビーズ上に、硫酸モルフィンおよび触知できない水性ラ
クトースの均質混合物である粉末を噴霧することにより製造される。その後、ビーズは、
乾燥され、そしてここで上に記述されるもののような疎水性材料で被覆して、最終処方が
、環境流動体にさらされるときに、薬剤の所望の放出を得る。その後、適切な量の制御放
出ビーズが、封入されて、約12時間、モルフィンの有効な血漿中濃度を提供する最終剤
形を提供する。
【0077】
以下の実施例は、本発明の種々の態様を示す。それらは、あらゆる手段であればどれで
も、請求項を制限すると解釈されることは意図されない。
【0078】
実施例1
ヒドロコドン持続的放出錠剤を、下の表1で説明される処方で製造された。
【0079】
【表1】

【0080】
以下の手段により:
1.遅延反応剤分散:ライトニンの混合装置を用いてオイドラギッドRS30Dおよびト
リアセチンを混錬する。
2.ステアリルアルコールを溶融させる。
3.流動床造粒装置を用いて、ヒドロコドン・ビタートレート、噴霧乾燥ラクトース、お
よびポビドンにおける噴霧遅延反応剤で分散する。
4.15分間、または一定重量までのステンレス鋼のトレイ上での乾燥バッチ。5.ホー
バートの混合装置を用いて、溶融ステアリルアルコールを、バッチに組込む。
6.30分間、ステンレス鋼のトレイ上で顆粒を乾式ワックス掛けするか、または造粒の
温度は、35℃以下に達する。
7.コミルを通して冷却された顆粒を挽く。
8.ホーバート混合装置を用いて、タルクおよびステアリン酸マグネシウムを顆粒に注入
する。
9.錠剤プレスを使用して、顆粒を錠剤に圧縮する。
【0081】
その後、錠剤は、以下の手段を用いて、溶解(dissolution)について試験した。
1.装置:米国薬局方(USP)の方法I(バスケット)、100rpm。
2.媒体:55分間の700mlのSGF、その後、酵素なしの900mlのSIF
3.サンプル採取時間:1、2、4、8および12時間。
4.解析:高速液体クロマトグラフィー。
溶解パラメータは、下の表2で説明される:
【0082】
【表2】
【0083】
その後、CmaxおよびTmaxが、実施例1について得られ、そして生物利用性研究での即
時放出対照標準は、下の表3に説明されるとおり、即時放出処方(ロルタブ7.5mg×
2)として投与されたヒドロコドン15mgを、健全なヒト対象における上記CR処方と
比較する。
【0084】
【表3】
【0085】
実施例2
ヒドロコドン持続的放出錠剤を、下の表4に説明された処方で製造した。
【0086】
【表4】
【0087】
実施例1の手段による。
その後、溶解パラメータを、実施例1の手段を用いて得た。結果は、下の表5に説明さ
れる。
【0088】
【表5】
【0089】
実施例3
ヒドロコドン持続的放出カプセルを、下の表6に説明された処方で製造した。
【0090】
【表6】
【0091】
以下の手段により:
1.ホーバートの混合装置を用いて、ステアリルアルコール、オイドラギッド(Eudragit
)RLPO、ヒドロコドン・ビタートレート、およびオイドラギッド(Eudragit)RSP
Oを混錬粉砕した。
2.以下の条件下で、粉末供給装置、溶融押出装置(6×1mmダイヘッドを具備した)
、コンベヤー、レーザーマイク、およびペレタイザーを用いて造粒物を押出す。
【0092】
領域1 10℃
領域2 20℃
領域3 120℃
領域4 120℃
領域5 120℃
領域6 120℃
領域7 95℃
領域8 95℃
MGA 120℃
ダイ 117℃
【0093】
粉末供給速度−40g/分;スクリュー速度−185rpm;真空−980ミリバール
押出物の直径が1mmであるような−コンベヤー
ペレットが、長さ1mmまでに切断されるような−ペレタイザー
3.16番メッシュおよび20番メッシュスクリーンを用いてペレットをスクリーニング
する。16番メッシュスクリーンを通過し、そして20番メッシュスクリーンに保有され
る材料を収集する。
4.ペレットでサイズ2番の透明なゼラチンカプセルを充填する。範囲:NLT114m
gおよびNMT126mg。
【0094】
その後、溶解パラメータは、実施例1の手段を用いて得られた。結果は、下の表7で説
明される。
【0095】
【表7】
【0096】
実施例4
オキシコドン持続的放出錠剤を、下の表8に説明された処方で製造した。
【0097】
【表8】
【0098】
以下の手段により:
1.造粒:オキシコドンHCl上にオイドラギッド/トリアセチン分散物を噴霧し、流動
床造粒装置を用いて、乾燥ラクトースおよびポビドンを噴霧する。
2.破砕:顆粒を放出し、そしてミルに通す。
3.ワックスがけ:ステアリルアルコールを溶融させ、そして混合装置を用いて、破砕し
た顆粒に加える。冷却させる。
4.破砕:冷却した顆粒をミルに通す。
5.潤滑:混合装置を用いて、タルクおよびステアリン酸マグネシウムで顆粒を潤滑させ
る。
6.圧縮:錠剤プレスを用いて、顆粒を錠剤に圧縮する。
7.フィルムコーティング:水性フィルムコートを錠剤に塗布する。
その後、錠剤を、以下の手段を用いて、溶解について試験した。
【0099】
1.装置:米国薬局方の方法II(パドル)、150rpm。
2.媒体:第一の時間、700mlのSGF、その後、リン酸緩衝液でpH7.5まで9
00mlになる。
3.サンプル採取時間:1、2、4、8、12、18および24時間。
4.解析:高速液体クロマトグラフィー。
溶解パラメータは、下の表9で説明される:
【0100】
【表9】
【0101】
その後、CmaxおよびTmaxが、実施例4について得られ、そして生物利用性研究での即
時放出対照標準は、下の表10に説明されるとおりである。
【0102】
【表10】
【0103】
実施例5
モルフィン持続的放出錠剤は、下の表11で説明される処方で製造された。
【0104】
【表11】
【0105】
以下の手段により:
1.造粒:硫酸モルフィンに水を添加し、混合装置中で乾燥ラクトースおよびヒドロキシ
エチルセルロースを噴霧し、流動床造粒装置を用いて乾燥させる。
2.スクリーニング:顆粒を放出し、そしてシーブを通過させる。
3.ワックスがけ:セトステアリルアルコールを溶融させ、そして混合装置を用いて、破
砕した顆粒に加える。冷却させる。
4.スクリーニング:冷却した顆粒をシーブに通す。
5.潤滑:混合装置を用いて、タルクおよびステアリン酸マグネシウムで顆粒を潤滑させ
る。
6.圧縮:錠剤プレスを用いて、顆粒を錠剤に圧縮する。
7.フィルムコーティング:水性フィルムコートを錠剤に塗布する。
その後、錠剤を、以下の手段を用いて、溶解について試験した:
【0106】
1.装置:米国薬局方の方法I(バスケット)、50rpm。
2.媒体:37℃、900mlの精製水。
3.サンプル採取時間:1、2、4および6時間。
4.解析:UV検出、285nmおよび305nm、5cmセルを用いた2点法。
【0107】
溶解パラメータは、下の表12で説明される。
【0108】
【表12】
【0109】
その後、CmaxおよびTmaxが、実施例5について得られ、そして生物利用性研究での即
時放出対照標準は、下の表13に説明されるとおりである。
【0110】
【表13】
【0111】
実施例6
実施例1、実施例4および実施例5の薬理学的パラメータを、互いに比較した。実施例
1のヒドロコドンHCl制御放出錠剤の溶解が、実施例4の制御放出オキシコドン錠剤お
よび実施例5の硫酸モルフィン制御放出錠剤の溶解に類似していてさえ、ヒドロコドン処
方についてのIRに対するCRのCmax比は、38%であるのに対して、オキシコドン錠
剤およびモルフィン錠剤は、50%を越えている。比較結果は、下の表14に説明される
。
【0112】
【表14】
【0113】
実施例7
実施例1、絶食中の正常な自発参加者での実施例2、実施例3の制御放出ヒドロコドン
処方と、2つの即時放出ヒドロコドン・ビタートレート7.5mg/アセトアミノフェン
500mg錠剤(IR実施例)の単回用量、4回治療、開放ラベル、薬理学的比較が行わ
れた。これらの処方のついての血漿濃度は、下の表15〜18に説明される。
【0114】
【表15】
【0115】
【表16】
【0116】
【表17】
【0117】
【表18】
【0118】
薬理学的パラメータは、下の表19で説明される。
【0119】
【表19】
【0120】
a AUC(0、最後)およびCmaxについての幾何学的手段およびTmax、W50、T1
/2(abs)およびT1/2(elim)についての計算手段。
b 比および90%Clは、最少の平方手段に基づく。
c 比(%):最少平方手段に基づいて、(試験手段/対照手段)×100。
【0121】
実施例8
ヒドロコドン持続的放出錠剤は、下の表20に説明される処方で製造された。
【0122】
【表20】
【0123】
1 加工の間に蒸散させ、そして最終製品の一部でない。
【0124】
以下の手段により:
1.破砕:ステアリルアルコールのフレークをミルに通す。
2.混錬:適切なブレンダーで、ヒドロコドン・ビタートレート、二塩基性リン酸カルシ
ウム、グリシジルベヘナート、ステアリルアルコールおよび微小結晶性セルロースを混合
する。
3.押出し:上昇温度で、混錬材料を、二重スクリュー押出装置に継続的に供給して、押
出物を軟化および成形する。
4.冷却:押出物をコンベヤーで冷却させる。
5.破砕:冷却した押出物をミルに通して、適切な粒子サイズの顆粒を得る。
6.混錬:破砕された押出物を、ステアリン酸マグネシウムと混錬させる。
7.圧縮:錠剤プレスを用いて、結果物である顆粒を圧縮する。
8.コーティング:精製水中のオパドライを分散させ、そしてそれを錠剤コアに塗布する
ことによって、フィルムコーティング溶液を製造する。
【0125】
その後、錠剤を、以下の手段を用いて、溶解について試験した:
1.装置:米国薬局方の方法I(バスケット)、100rpm。
2.媒体:最初の55分間、700mlSGF(酵素なし)、その後、リン酸緩衝液でp
H7.5まで900mlになる。
3.サンプル採取時間:1、2、4、8および12時間。
4.解析:高速液体クロマトグラフィー。
溶解パラメータは、下の表21で説明される:
【0126】
【表21】
【0127】
実施例9
給食および絶食にある15mgヒドロコドン制御放出錠剤(実施例8)の、15mgヒ
ドロコドン即時放出(2×7.5mg錠剤)の単回用量の3法交差(3 way crossover)
の薬理学的比較研究は、絶食した正常な自主参加者における2つのQ6H用量に付与され
た。
その後、CmaxおよびTmaxが、実施例8について得られ、そして生物利用性研究での即
時放出対照標準は、下の表22および23に説明されるとおりである。
【0128】
【表22】
【0129】
【表23】
【図面の簡単な説明】
【0130】
【図1】実施例1、実施例2、実施例3および等量用量の即時放出ヒドロコドンの平均ヒドロコドン血漿中濃度のグラフ表示である。
【図2】実施例4の手段によって製造された制御放出オキシコドンの様々のサンプル、および実施例5の手段によって製造された制御放出オキシコドンの様々のサンプルに対する、実施例1、実施例2および実施例3の平均ヒドロコドン血漿中濃度のグラフ表示である。
【図3】実施例1、実施例2、実施例3および等量用量の即時放出ヒドロコドンの時間に対して吸収された分画ヒドロコドン含有率のグラフ表示である。
【技術分野】
【0001】
痛み、特に慢性の痛みの薬物療法による困難性のために、オピオイド鎮痛薬は、制御放
出処方として投与されるべき理想的な薬剤である。本発明は、痛みの治療で使用するため
の固形の制御放出経口剤形に関する。
【背景技術】
【0002】
即時放出剤形の投与の後に通常に得られるより投与後の長期間の薬理学的作用を提供す
るのは、全ての制御(遅延)放出処方の目的である。このような長期間の応答は、対応す
る短期作用の即時放出製品では達成されない多くの治療利益をもたらす。したがって、治
療は、例えば、中程度から重篤な痛み(例えば、術後患者、癌患者など)までについて、
または起きているときに偏頭痛を経験する患者のものについて、並びに睡眠が必須である
衰弱した患者について、患者を治療するときに、患者の睡眠を中断せずに継続されうる。
【0003】
従来の迅速作用薬剤療法が、薬剤の有効に安定な状態の血漿濃度を維持するために、頻
繁な間隔で注意深く投与されない限り、化合物の、そして代謝不活性化を通しての迅速な
吸収、全身排出のため、活性薬剤の血漿中濃度における頂点および谷が起り、それにより
患者の維持療法における特別の問題を生じる。長期作用の薬剤製品のさらに一般的な利点
は、患者の健忘を通して抜かされた用量の回避から生じる改善された患者服薬遵守である
。
【0004】
ヒトおよび動物に対する経口投与の後の組成物に含まれる薬理学上活性な物質の制御放
出を生じる組成物を製造することは、製薬業界で知られている。このような遅延放出組成
物は、それが、栄養管のある種の部分に到達するまで、医薬品単位の吸収を遅らせるため
に使用される。消化管における医薬品のこのような制御放出は、さらに、従来の迅速放出
剤形が投与される場合に起るより長い期間、血流での上記医薬品の望ましい濃度をさらに
維持する。
【0005】
担体からの活性化合物の制御放出を提供する組成物の製造および使用の先行技術の教示
は、消化管の生理学的流動体への活性物質の放出で基本的に考慮される。しかし、それは
、胃腸液における活性物質の単なる存在は、それ自身で、生物利用性を確保しないことが
一般に認識される。
【0006】
吸収されるために、活性薬剤物質は、溶液中になければならない。単位剤形からの活性
物質の所定の比率について要求される時間は、標準化条件下で行われた試験方法によって
、特化された時間基本に対する、単位剤形から放出される活性薬剤物質の量の比率として
測定される。胃腸管の生理学上の流動体は、溶解時間を決定するための媒体である。技術
の現状は、多くの満足のいく試験手段が、製薬上の組成物について溶解時間を測定するこ
とを認識し、そしてこれらの試験手段は、世界的な公式解説で記述されている。
【0007】
それの担体からの薬剤物質の溶解に影響する多くの多様な因子があるが、特定の組成物
からの薬理学上の活性物質について測定された溶解時間は、比較的一定で、そして再現性
がある。溶解時間に影響しうる様々の因子の中でも、溶解溶媒媒質、溶液のpH、特定の
溶媒媒質での物質の溶解性、および溶媒溶質中に溶解した材料の飽和濃度の駆動力に表さ
れる薬剤物質の表面領域である。したがって、成分が、組織部位を越えた吸収を通して溶
解媒体から除去されるときに、活性薬剤物質の溶解濃度は、それの安定な状態で動的に改
質される。生理学的条件下で、溶解した材料の飽和レベルは、安定な状態の吸収を生じる
溶媒媒体での相対的均質性および一定の溶解濃度を維持するために剤形から逆に補給され
る。
【0008】
駆動力の方向が、膜のいずれかの側での活性物質の濃度、すなわち、胃腸の流動体で溶
解される量と、血液中に存在する量との間の差異であるので、胃腸管の組織吸収部位にお
ける輸送は、膜の両面でドナンの浸透平衡力によって影響される。血中濃度は、希釈、循
環変化、組織保存、代謝変換および全身排出により一定に変化するので、活性材料の流れ
は、胃腸管から血流に向けられる。
【0009】
種々の技術は、制御放出剤形を製造するために使用された。活性医薬品の遅延放出が、
製造のコーティングの選択的破壊を通して、または薬剤の放出に影響を及ぼす特別のマト
リックスを用いたコンパウンド化を通して起される特別に被覆されたペレット、錠剤およ
びカプセル剤は、当業界で知られている。ある種の制御放出処方は、投与後の所定の期間
に活性化合物の単位用量の関連した連続放出を提供する。
【0010】
本文献で報告される制御放出オピオイド処方の特定の例としては、例えば、共に、本発
明の譲受人に譲渡され、そして参照してここに組込まれる米国特許第4,990,341
号および第4,844,909号(ゴールデイら)に開示されるものが挙げられ、米国薬
局方パドルまたはバスケット法によって測定される場合、37℃で、900ml水性緩衝
液(pH1.6と7.2との間)で、100rmpでの剤形のインビトロでの溶解速度と
して、12.5〜42.5%(重量で)の塩酸モルホロンが1時間後に放出され、25〜
55%(重量で)が2時間後に放出され、45〜75%(重量で)の塩酸モルホロンが4時
間後に放出され、そして55〜85%(重量で)の塩酸モルホロンが6時間後に放出され
、インビトロ放出速度は、pH1.6〜7.2のpHから独立しており、そしてインビボ
で得られた塩酸モルホロンの頂点の血漿中濃度が、剤形の投与の2〜4時間の間で起るよ
うに選択される。少なくとも12時間の痛み解放が、これらの塩酸モルホロン処方で得ら
れる。
【0011】
中程度の痛みのために使用されうる他のオピオイド鎮痛薬剤の制御放出投与量処方を提
供することが、非常の望ましいと考えられる。さらに、痛み療法の必要な患者における最
も有効な痛みの管理を提供する薬理学的特性を示すこのような制御放出処方を提供するこ
とが、非常に望ましいと考えられる。
【発明の開示】
【発明が解決しようとする課題】
【0012】
中程度の痛みを経験するヒト患者での痛みの管理の効力および品質を実質的に改善する
ことが本発明の目的である。
痛みの管理の効力および品質を実質的に改善する生物利用性ヒドロコドン(塩酸モルホ
ロン;hydrocodon)処方を提供することが本発明の目的である。
【課題を解決するための手段】
【0013】
即時放出ヒドロコドン処方に比較した場合に実質的に増大した効果の期間を提供するが
、しかし痛覚脱失症の早期発生を提供する生物利用性ヒドロコドン処方を提供することが
、本発明のさらに別の目的である。
治療効果の早期発生を提供し、そして投与間隔の間に最大限の濃度に上昇した後、相対
的に平坦な血清血漿プロファイルを提供し、すなわちオピオイドの血漿中濃度が、0.5
5から0.85までのC12/Cmax比を提供することを意味し、そして患者に対して有効
な痛み解放を提供する一日二回投与に適する経口で投与できる制御放出オピオイド処方を
提供することが、本発明の別の目的である。代替の実施形態では、剤形は、0.65から
0.75までのC12/Cmax比を提供する。
【0014】
本発明の上記及び他の目的は本発明によって達成される。特定の実施形態では、鎮痛薬
で有効な量のヒドロコドンまたは医薬上許容しうるその塩および一日二回投与に適した剤
形にするのに十分量の制御放出材料を包含する固形の経口制御放出剤形であって、ヒト患
者または患者の集団に対する単回投与後に、約2から約8時間(Tmax)まででインビト
ロでのヒドロコドンの頂点の血漿中濃度に達する時間を提供し、そして最大濃度を得た後
は、0.55から0.85までのC12/Cmax比を提供する。
特定の好ましい実施形態では、制御放出剤形は、37℃での55分間の700mlの模
擬胃液(SGF)中で、その後37℃での900mlの模擬小腸液(SIF)に交換する
100rpmでの米国薬局方バスケット法によって測定されるときに、1時間で、剤形か
らのヒドロコドンまたはその塩の18%から約42.5重量%までのインビトロ放出を提
供する。
【0015】
特定の好ましい実施形態では、37℃での1.2および7.5のpHで、900ml水
性緩衝液中で100rpmでの米国薬局方バスケット法によって測定されるときに、ヒド
ロコドン剤形のインビトロでの溶解速度として、約25から約65重量%までのヒドロコ
ドンまたはその塩2時間後放出され、約45から約85重量%までのヒドロコドンまたは
その塩が4時間後放出され、そして約60重量%より大きいヒドロコドンまたはその塩が8
時間後放出される。インビトロ放出速度は、本発明の好ましい実施形態では、所望される
とおりpH−独立またはpH−従属のいずれかでありうるが、ヒドロコドンの放出は、p
H−独立性である。
特定の好ましい実施形態では、剤形が、投与の12時間後に、少なくとも5または6n
g/mlのヒドロコドン血漿濃度を提供し、そして投与の約2から約8時間後までに、少
なくとも約8ng/mlの血漿濃度を提供する治療的に有効な量のヒドロコドンを包含す
る制御放出剤形の提供がある。
【0016】
本発明の別の好ましい実施形態では、等量用量の即時放出ヒドロコドン対照処方(例え
ば、ロルタボ(登録商標;Lortab)の約50%未満のCmaxであるヒドロコドンの
Cmaxを提供し、そして12時間投与間隔の間、有効な無痛覚を提供するヒドロコドンの
一日二回経口制御放出剤形が提供される。
本発明の他の好ましい実施形態では、等量用量の即時放出ヒドロコドン対照処方、例え
ば、ロルタボ(登録商標)の80%Cmaxに達する時間の約90%から約150%まで、
好ましくは約90%から約110%までである80%Cmaxに達する時間を提供するヒド
ロコドンの一日二回制御放出剤形の提供がある。好ましくは、制御放出剤形についての8
0%Cmaxに達する時間は、約0.5から約1.5時間まで、最も好ましくは約0.8か
ら約1.2時間までである。他の実施形態では、制御放出剤形についてのヒドロコドンの
80%Cmaxに達する時間は、約0.75から約2.0時間まで、最も好ましくは約0.
9から約1.5時間までである。
【0017】
本発明の他の好ましい実施形態では、等量用量の即時放出ヒドロコドン対照処方の90
%Cmaxに達する時間の約150%から約400%まで、好ましくは約150%から約2
50%までである90%Cmaxに達する時間を提供するヒドロコドンの一日二回制御放出
剤形の提供がある。好ましくは、制御放出剤形についての90%Cmaxに達する時間は、
約1.5から約2.5時間まで、最も好ましくは約1.8から約2.2時間までである。
他の実施形態では、制御放出剤形についてのヒドロコドンの90%Cmaxに達する時間は
、約1.5から約4.0時間まで、最も好ましくは約1.8から約2.5時間までである
。
【0018】
本発明の他の好ましい実施形態では、剤形が、約0.5から10時間まで、好ましくは
約1から約9時間まで、または約4から約8時間までのCmaxの80%以内に血漿中濃度
を維持するヒドロコドンの一日二回制御放出剤形が提供される。
本発明の他の好ましい実施形態では、剤形が、約1から6.5時間まで、好ましくは約
2から約5時間まで、または約2から約6.5時間までのCmaxの90%以内に血漿血漿
中濃度を維持するヒドロコドンの一日二回制御放出剤形の提供がある。
【0019】
本発明の他の好ましい実施形態では、約1.5mg/時間から約5mg/時間までの投
与からTmaxまでの平均インビボ吸収速度を提供し、15mgヒドロコドンビタートレー
トを含む剤形の経口投与に基づいて約0.5mg/時間未満であるTmaxから投与間隔の
終点までの平均吸収の速度を提供するヒドロコドンの一日二回制御放出剤形が提供される
。好ましくは、剤形は、約2mg/時間から約4mg/時間までの投与からTmaxまでの
平均インビボ吸収速度を提供し、そして15mgヒドロコドンビタートレートを含む剤形
の経口投与に基づいて、約0.08mg/時間から約0.4mg/時間までであるTmax
から12時間投与間隔の終点の平均インビボ吸収速度を提供する。
本発明の他の好ましい実施形態では、同じ期間の放出速度の約55%から約85%まで
である、剤形の経口投与のTmaxから約12時間までの期間、吸収速度を提供する一日二
回制御放出ヒドロコドン剤形が提供される。
【0020】
本発明の上記実施形態、並びに他の実施形態は、好ましくは、即時放出ヒドロコドン対
照処方の同等用量によって提供されるTmaxより3から4倍遅い時点でTmaxに達する時間
を提供する。好ましくは、持続的放出処方によって供されるTmaxは、経口投与の後約2
から約8時間、約3から約7時間まで、または約4から約6時間までで起る。
【0021】
本発明は、さらに、即時放出ヒドロコドン対照製品の同等用量によって提供されるCma
xの約50%未満、好ましくは約40%未満であるヒドロコドンのCmaxを提供するヒドロ
コドン処方に向けられる。
例えば、ヒドロコドンが、米国特許第4,861,598号および第4,970,07
5号で開示されるとおりデリバリー・システムで処方されるときに、即時放出ヒドロコド
ン対照製品のCmaxの百分率としてデリバリー・システムによって提供されるヒドロコド
ンのCmaxは、同じデリバリー・システムで処方されたオキシコドンについて同じ計算よ
り相当に低かったことが驚くべきことに知見された。この現象は、制御放出オキシコドン
およびヒドロコドン処方が、同様のインビトロ溶解パラメータを示した事実にもかかわら
ず明かである。
【0022】
本発明は、デリバリー・システム米国特許第4,861,598号および第4,970
,075号を用いて配合される場合に、即時放出ヒドロコドン対照製品のCmaxの百分率
としてデリバリー・システムのCmaxは、好ましい実施形態で約50%未満、そして40
%未満であるのに対して、オキシコドンは、50%より大きな計算を示す。
【発明を実施するための最良の形態】
【0023】
「ヒドロコドン」は、医薬上許容しうる塩およびヒドロコドンの複合体と同様に、ヒド
ロコドン遊離塩基を示す場合、本発明の目的のために定義される。
語句「米国薬局方パドルまたはバスケット法」は、例えば、参照してここに組込まれる
米国薬局方XXII(1990年)で記述されるパドルおよびバスケット法である。
本発明の目的のため、用語「pH−依存性」は、環境的pHによって変化する特徴(例
えば、溶解)を示すと定義される。
本発明の目的のため、用語「pH−独立性」は、pHによって実質的に影響されない特
徴(例えば、溶解)を示すと定義される。
本発明の目的のため、用語「生物利用性」は、薬剤(例えば、ヒドロコドン)が、単位
剤形から吸収される範囲として定義される。
本発明の目的のため、用語「制御放出」は、血中(例えば、血漿)濃度が、治療範囲内
に維持されるが、しかし約12時間またはそれより長くの時間の期間かけて毒性濃度を低
下させるような速度での薬剤(例えば、ヒドロコドン)の放出として定義される。
用語「Cmax」は、投与間隔の間に得られる最大血漿濃度を示す。
用語「Tmax」は、最大血漿濃度に達する時間を示す。
用語「T1/2(abs)」は、血漿に移行されるべきオピオイドの吸収可能な用量の2分の1
に必要な時間の量を示す。
【0024】
用語「安定な状態」は、所定の薬剤についての血漿濃度が、達成され、そして所定の薬
剤について最少有効治療濃度で、またはそれより上であり、そして最少毒性血漿濃度より
下である濃度での薬剤の連続用量で維持されることを意味する。オピオイド鎮痛薬につい
て、最少有効治療濃度は、所定の患者で達成される痛み解放の量によって部分的に測定さ
れる。痛みの測定は、非常に主観的であり、そして多くの個人的変動は、患者の中で起り
うることは、医療業界での習熟したものによって理解される。
用語「維持療法」および「慢性療法」は、患者が、上に定義されるとおり安定な状態ま
でオピオイド鎮痛薬で滴定された後に患者に対して投与された薬剤療法として、本発明の
目的のために定義される。
ヒドロコドンのようなオピオイドの濃度に関して、用語「最少有効鎮痛薬濃度」または
「MEAC」は、量を特定するのが極めて困難である。しかし、一般に、それより下で痛
覚脱失(analgesia)が供給されない、血漿ヒドロコドンの最小有効鎮痛薬濃度がある。
例えば、血漿中ヒドロコドン濃度と痛覚脱失の間の間接的関係があるときに、高く、そし
て延長された血漿中濃度は、一般に、優れた痛み解放に関連する。頂点の血漿中ヒドロコ
ドン濃度の時間と頂点の薬剤効果の時間との間に、遅滞時間またはヒステリシス(hyster
esis)がある。これは、一般にオピオイド鎮痛薬を用いた痛みの治療について有効である
。
【0025】
用語「平均共鳴時間」(MRT)は、薬剤分子が体内で留まる平均時間として定義され
る。吸収、分布および放出の関数であるこの計算は、活性成分を含有する剤形に部分的に
依存性がある。
本発明の目的のために、さらに特定されない限り、用語「患者」は、検討(または請求
項)が、個々の患者または対象の薬物動態的パラメータに向けられることを意味する。
用語「患者の集団」は、検討(または請求項)が、少なくとも2名の患者または対象の
平均薬物動態的パラメータに向けられる。
用語「突破痛み」では、患者は、患者が、ヒドロモルホンを含む一般に有効量の本発明
の持続放出の固形経口剤形を投与されているという事実の代わりに経験する痛みを意味す
る。
用語「救援」は、突破痛みを経験している患者に投与される鎮痛薬に該当する。
用語「有効な痛み管理」は、医師による鎮痛療法に対するヒト患者の応答(経験された
痛み対副作用)の客観的評価、並びにこのような治療を受ける患者による治療的治療の主
観的評価を意味する。当業者は、有効な鎮痛薬が、個々の患者の多様性を含めた多くの因
子によって変化することを理解する。
【0026】
本発明の目的のため、用語「即時放出ヒドロコドン対照処方」は、ユーシービー・ファ
ルマ、インク.から商用的に入手可能な登録商標ロルタブの等価量のヒドロコドン部分、
または即時放出のヒドロコドンまたはその塩を提供する医薬製品である。
本発明の目的のために、ここに開示された制御放出処方および即時放出制御処方は、用
量に比例する。このような処方では、薬理学的パラメータ(例えば、AUCおよびCmax
)は、1つの投与強度から別のものまで線状で増加する。したがって、特別の用量の薬理
学的パラメータは、同じ処方の異なる用量のパラメータから推論される。
本発明の目的のために、特に特定されない限り、ここに開示される薬理学的パラメータ
は、個別の患者に対する単回用量のヒドロコドン処方の投与に基づいている。患者集団に
基づいた薬理学的パラメータは、「平均」データとして特定される。
【0027】
用語「第一の投与」は、個々の患者または患者集団に対する治療の開始時での本発明の
単回用量を意味する。
本発明の制御放出の経口固形剤形は、驚くべきことに、オピオイドを倹約しうる。本発
明の制御放出の経口固形剤形は、鎮静薬効能における差異なしに、従来の即時放出製品に
対する比較で、実質的に低い一日投与量で投与されうることは可能である。匹敵する一日
投与量で、大きな効能は、従来の即時放出製品に対する比較で、本発明の制御放出の経口
固形剤形の使用で生じうる。
【0028】
ここに付随する図面は、本発明の実施形態を示し、そして請求項によって包含されると
おり本発明の範囲を制限すると意味されない。
【0029】
本発明の上記実施形態は、当業者に知られる広く多様な制御放出処方によって提供され
うる。例えば、適切な制御放出剤形は、参照してここに組込まれる米国特許第4,861
,598号および第4,970,075号で開示される。
本発明の特定の実施形態で、即時放出形態における有効量のオピオイドが、処方に含ま
れる。オピオイドの即時放出形態は、Tmaxが、例えば約2から約5時間まで、または約
2から約4時間までの時間に短縮されるように血液中のオピオイド(例えば、血漿)の最
大濃度までの時間を短縮するのに有効な量含まれる。単位用量でのこのような有効量の即
時放出オピオイドを含めることによって、患者における痛みの相対的に高いレベルの経験
が明らかに減少されることが知見された。このような実施形態で、即時放出形態での有効
量のオピオイドは、本発明の基質(substrates)に被覆されうる。例えば、その処方から
得た延長放出オピオイドは、制御放出コーティングによる場合、即時放出層は、制御放出
コーティングの上に上塗される。他方、即時放出層は、オピオイドが、制御放出マトリッ
クスに組込まれる基質の表面に被覆されうる。有効単位用量のオピオイド(例えば、ペレ
ット、球体、ビーズおよび同等物を含めた微粒子システム)を含む複数の持続的放出基質
が、硬質ゼラチンカプセルに組込まれるときに、オピオイド用量の即時放出部分は、カプ
セル内の粉末または顆粒として十分量の即時放出オピオイドの封入を介してゼラチンカプ
セルに組込まれうる。代わりに、ゼラチンカプセルそれ自身は、オピオイドの即時放出層
で被覆されうる。当業者は、即時放出オピオイド部分を単位用量に組込むさらに他の代替
手段を認識する。このような代替法は、付随の請求項に包含されると考えられる。
本発明のオピオイド剤形の1つの利点は、治療的濃度が、一般に、むかつき、吐気また
は嗜眠状態のような共存する副作用の強度および/または程度における明らかな増加なし
に実質的に達成され、そしてそれは、しばしば、オピオイドの高い血中濃度に関連するこ
とである。本発明の剤形の使用は、薬剤添加の危険が減少されることに至る証拠もある。
【0030】
活性剤
本発明の制御放出の経口剤形は、好ましくは、約0.5mgから約1250mgまでの
ヒドロコドンまたは等価量の医薬上許容しうるその塩を含む。より好ましい実施形態では
、剤形は、約5mgから約60mgまで、例えば15mgを含みうる。ヒドロコドンの適
切な医薬上許容しうる塩としては、ヒドロコドン・ビタートレート(bitartrate)、ヒド
ロコドン・ビタートレート・ヒドレート、塩酸ヒドロコドン、ヒドロコドン・p−トルエ
ンスルホネート、リン酸ヒドロコドン、ヒドロコドン・チオセミカルバゾン、ヒドロコド
ン・スルフェート、ヒドロコドン・トリフルオロアセテート、ヒドロコドン・ヘミペンタ
ヒドレート、ヒドロコドン・ペンタフルオロプロピオネート、ヒドロコドン・p−ニトロ
フェニルヒドラゾン、ヒドロコドン・o−メチルオキシム、ヒドロコドン・セミカルバゾ
ン、ヒドロコドン・ヒドロブロミド、ヒドロコドン・ムケート、ヒドロコドン・オレート
、ヒドロコドン・ホスフェート二塩基物、ヒドロコドン・ホスフェート一塩基物、ヒドロ
コドン無機塩、ヒドロコドン有機塩、ヒドロコドン・アセテート・トリヒドレート、ヒド
ロコドン・ビス(ヘプタフオロブチレート)、ヒドロコドン・ビス(メチルカルバメート
)、ヒドロコドン・ビス(ペンタフルオロプロピオネート)、ヒドロコドン・ビス(ピリ
ジンカルボキシレート)、ヒドロコドン・ビス(トリフルオロアセテート)、ヒドロコド
ン・クロロヒドレート、およびヒドロコドン・スルフェート・ペンタヒドレートが挙げら
れる。好ましくは、ヒドロコドンは、ビタートレート塩として存在する。
【0031】
本発明の剤形は、さらに、本発明のヒドロコドン鎮静薬と相乗的に作用しうるか、また
はし得ない1つまたはそれ以上の別の薬剤を含みうる。このような別の薬剤の例としては
、イブプロフェン、ジクロフェナック、ナプロキセン、ベノキサプロフェン、フルビプロ
フェン、フェノプロフェン、フルブフェン、ケトプロフェン、インドプロフェン、ピロプ
ロフェン、カルプロフェン、オキサプロジン、プラモプロフェン、ムロプロフェン、トリ
オキサプロフェン、スプロフェン、アミノプロフェン、トリアプロフェン酸、フルプロフ
ェン、ブクロキシン酸、インドメタシン、スリンダック、トルメチン、ゾメピラック、チ
オピナック、ジドメタシン、アセメタシン、フェンチアザック、クリダナック、オキシピ
ナック、メフェナミン酸、メクロフェナミン酸、フルフェナミン酸、ニフルミン酸、トル
フェナミン酸、ジフルリザール、フルフェニザール、ピロキシカム、スドキシカム、また
はイソキシカムおよび同等物を含めた非ステロイド抗炎症性剤が挙げられる。このような
非ステロイド抗炎症剤は、セレコキシブ(SC−58635)のようなシクロオキシゲナ
ーゼ阻害剤、DUP−697、フルスリド(CGP−28238)、メロキシカム、6−
メトキシ−2−ナフチル酢酸(6−MNA)、ビオクス(Vioxx)(MK−966)
、ナブメトン(6−MNAについてのプロドラッグ)、ニメスリド、NS−398、SC
−5766、SC−58215、およびアマンタジン(1−アミノアダマンチン)として
のT−614、およびメマンチン(3,5ジメチルアミノアダマントン)、それらの混合
物および医薬上許容しうるその塩も挙げられる。
他の追加の薬剤としては、デキトロルファンのような非毒性NMDAレセプターアンタ
ゴニスト、デキストロメトルファン、3−(1−ナフタレニル)−5−(ホスホノメチル
)−L−フェニルアラニン、3−(1−ナフタレニル)−5−(ホスホノメチル)−DL
−フェニルアラミン、1−(3,5−ジメチルフェニル)ナフタレン、および2−(3,
5−ジメチルフェニル)ナフタレン、2SR,4RS−4−(((1H−テトラゾル−5
−イル)メチル)オキシ)ピペリジン−2−カルボン酸;2SR,4RS−4−((((
1H−テトラゾル−5−イル)メチル)オキシ)メチル)ピペリジン−2−カルボン酸;
E及びZ 2SR−4−(O−1H−テトラゾル−5−イル)メチル)ケトキシイミノ)ピペリ
ジン−2−カルボン酸;2SR,4RS−4−((1H−テトラゾル−5−イル)チオ)
ピペリジン−2−カルボン酸;2SR,4RS−4−((1H−テトラゾル−5−イル)
チオ)ピペリジン−2−カルボン酸;2SR,4RS−4−(5−メルカプト−1H−テ
トラゾル−1−イル)ピペリジン−2−カルボン酸;2SR,4RS−4−(5−メルカ
プト−2H−テトラゾル−2−イル)ピペリジン−2−カルボン酸;2SR,4RS−4
−(5−メルカプト−1H−テトラゾル−1−イル)ピペリジン−2−カルボン酸;2S
R,4RS−4−(5−メルカプト−2H−テトラゾル−2−イル)ピペリジン−2−カ
ルボン酸;2SR,4RS−4−(5−メルカプト−1H−テトラゾル−1−イル)ピペ
リジン−2−カルボン酸;2SR,4RS−4−(((1H−テトラゾル−5−イル)チ
オ)メチル)ピペリジン−2−カルボン酸;2SR,4RS−4−((5−メルカプト−
1H−テトラゾル−1−イル)メチル)ピペリジン−2−カルボン酸;または2SR,4
RS−4−((5−メルカプト−2H−テトラゾル−2−イル)メチル)ピペリジン−2
−カルボン酸、それらの混合物および医薬上許容しうるその塩が挙げられる。
【0032】
本発明の剤形に含まれうる他の適切な追加の薬剤としては、アセトアミノフェン、神経
−活性ステロイド(参照してここに組込まれる、1998年2月20に提出された米国特
許出願番号第09/026,520号に開示されるもののような)および他の非オピオイ
ド鎮静薬(analgesics)が挙げられる。
【0033】
例えば、第二(非オピオイド)薬剤が、処方に含まれる場合、このような薬剤は、制御
放出形態中、または即時放出形態中に含まれ得る。追加の薬剤は、オピオイドと一緒に制
御放出マトリックスに組込まれうる;制御放出コーティングに組込まれる;分離された制
御放出層または即時放出層として組込まれる;または本発明の基質を有するゼラチンカプ
セル中の粉末、顆粒などとして組込まれうる。
本発明のある種の好ましい実施形態では、即時放出形態中の有効量のヒドロコドンは、
投与されるべき制御放出単位用量ヒドロコドン処方に含まれる。即時放出形態のヒドロコ
ドンは、血液(例えば、血漿)中のヒドロコドンのCmaxに達する時間を短縮するのに有
効である量で含まれる。このような実施形態では、即時放出形態中の有効量のヒドロコド
ンは、本発明の基質上に被覆されうる。例えば、処方からの遅延放出ヒドロコドンが、制
御放出コーティングによる場合、即時放出層は、制御放出コーティングの上に上塗りされ
る。他方、即時放出層は、ヒドロコドンが制御放出マトリックスに組込まれる基質の表面
に被覆されうる。有効な単位用量のヒドロコドンを含む複数の持続的放出基質(例えば、
ペレット、球体、ビーズおよび同等物を含む微粒子システム)が、硬質カプセルに組込ま
れる場合、オピオイドの即時放出部分は、カプセル内で粉末またはカルシウム粒として十
分量の即時放出ヒドロコドンの封入を介して、ゼラチンカプセルに組込まれうる。代わり
に、ゼラチンカプセルそれ自身は、ヒドロコドンの即時放出層で被覆されうる。当業者は
、即時放出ヒドロモルホン部分を、単位用量に組込むさらに他の代替手段を認識する。こ
のような代替法は、付随の請求項に包含すると考えられる。単位用量中のこのような有効
量の即時放出ヒドロコドンを含むことによって、患者における相対的に高いレベルの痛み
の経験は、明らかに減少される。
【0034】
剤形
制御放出剤形は、都合により、ヒドロコドンと一緒にマトリックスに組込まれるか、ま
たは薬剤を包含する基質(語句「基質」は、ビーズ、ペレット、スフェロイド、錠剤、錠
剤コアなどを包含する)の上に持続的放出コーティングとして塗布される制御放出材料を
含みうる。制御放出材料は、所望される場合、疎水性または親水性でありうる。本発明に
よる経口剤形は、例えば、顆粒、球状体、ペレット(以降、「マルチパーチパーティキュ
レート(multiparticulates)」として集約的に引用される)として提供されうる。単位
時間あたり所望の用量のオピオイドを提供するために有効であるマルチパーティキュレー
ト(微粒子)の量は、カプセルに入れられうるか、または例えば錠剤に圧縮された任意の
他の適切な経口固形形態に組込まれうる。他方、本発明による経口投与剤形は、制御放出
コーティングで被覆された錠剤コアとして、または薬剤のマトリックス、制御放出材料、
および都合により他の医薬上所望の成分(例えば、希釈剤、バインダ、着色剤、潤滑剤な
ど)を含む錠剤として製造されうる。
【0035】
制御放出マトリックス処方
本発明の特定の好ましい実施形態では、制御放出処方は、上に説明されるとおり制御放
出材料を含むマトリックス(例えば、マトリックス錠剤)を介して達成される。制御放出
マトリックスを含む剤形は、pH依存性またはpH独立性手段で、好まれる範囲内で、そ
してオピオイドを放出するオピオイドのインビトロ溶解速度を提供する。制御放出マトリ
ックスで封入に適した材料は、マトリックスを形成するために使用される方法に依存する
。経口剤形は、少なくとも1種の親水性または疎水性制御放出材料の1%および8%(重
量で)の間で含有しうる。
【0036】
本発明による制御放出マトリックスに含まれうる適切な制御放出材料の制限なしのリス
トは、親水性および/または疎水性材料、例えば、ゴム、セルロースエーテル、アクリル
酸樹脂、タンパク質由来の材料、蝋(waxes)、シェラック、および水素化ひまし油、水
素化植物性油のような油が挙げられる。しかし、オピオイドの制御放出を与える能力のあ
る任意の医薬上許容しうる疎水性または親水性の制御放出材料は、本発明によって使用さ
れうる。好ましい制御放出高分子としては、エチルセルロースのようなアルキルセルロー
ス、アクリル酸またはメタクリル酸重合体および共重合体、およびセルロースエーテル、
特にヒドロキシアルキルセルロース(特にヒドロキシプロピルメチルセルロース)および
カルボキシアルキルセルロースが挙げられる。好ましいアクリル酸およびメタクリル酸重
合体および共重合体としては、メチルメタクリレート、メチル・メタクリレート共重合体
、エトキシエチル・メタクリレート、シアノエチル・メタクリレート、アミノアルキル・
メタクリレート共重合体、ポリ(アクリル酸)、ポリ(メタクリル酸)、メタクリル酸ア
ルキルアミン共重合体、ポリ(メチル・メタクリレート)、ポリ(メタクリル酸)(無水
物)、ポリメタクリレート、ポリアクリルアミド、ポリ(メタクリル酸無水物)、および
グリシジル・メタクリレート共重合体が挙げられる。特定の好ましい実施形態は、本発明
のマトリックス中の前述の制御放出材料のいずれかの混合物を利用する。
【0037】
マトリックスは、バインダとしても含有しうる。このような実施形態では、バインダは
、制御放出マトリックスからのヒドロコドンの制御放出に寄与することが好ましい。
好ましい疎水性バインダ材料は、多かれ少なかれ顕著な親水性および/または疎水性傾
向を示すが、水不溶性である。好ましくは、本発明に有用な疎水性バインダ材料は、約3
0から約200℃まで、好ましくは約45から約90℃までの融点を示す。疎水性材料が
、炭化水素であるときに、炭化水素は、好ましくは、25°および90℃の間の融点を示
す。長鎖(C8−C50)ヒドロコドン材料の中でも、脂肪酸(脂肪族)アルコールが好ま
しい。経口剤形は、少なくとも1種の消化可能な長鎖炭化水素の80%(重量で)までを
含有しうる。
好ましくは、経口剤形は、少なくとも1種のポリアルキレングリコールの80%(重量
で)までを含有する。特に、疎水性バインダ材料は、天然または合成ワックス、脂肪酸ア
ルコール(ラウリル、ミリスチル、ステアリル、セチルまたは好ましくはセトステアリル
アルコールのような)、脂肪酸を包含でき、そしてそれに限定されないが、脂肪酸エステ
ル、脂肪酸グリセリド(モノ−、ジ−、およびトリ−グリセリド)、水素化脂肪、炭化水
素、正常なワックス、ステリン酸、ステアリルアルコール、および炭化水素骨格を有する
疎水性および親水性材料が挙げられる。適切なワックスとしては、例えば、蜜蝋、糖ワッ
クス、ヒマシワックスおよびカルナバ蝋が挙げられる。本発明の目的のために、ワックス
様物質は、室温で正常に固形であり、そして約30から約110℃までの融解点を示す任
意の材料として定義される。
【0038】
本発明により使用されうる好ましい疎水性バインダ材料としては、消化のよい長鎖(C
8−C50、特にC12−C40)、脂肪酸、脂肪酸アルコール、脂肪酸のグリセリルエステル
、鉱物油および植物性油、天然および合成ワックスおよびポリアルキレングリコールのよ
うな置換または未置換炭化水素が挙げられる。25℃および90℃の間の融点を示す炭化
水素が、好ましい。長鎖炭化水素バインダ材料の、脂肪酸(脂肪族)アルコールは、特定
の実施形態で好まれる。経口剤形は、80%(重量で)までの少なくとも1種の消化のよ
い長鎖ヒドロコドンを含有しうる。
特定の好ましい実施形態では、2種またはそれ以上の疎水性バインダ材料は、マトリッ
クス処方に含まれる。追加の疎水性バインダ材料が含まれる場合、それは、天然および合
成ワックス、脂肪酸、脂肪酸アルコール、および同じものの混合物から選択されるのが好
ましい。例としては、蜜蝋、カルナバ蝋、ステアリン酸およびステアリルアルコールが挙
げられる。このリストは、限定されることを意味しない。
【0039】
1つの特定の適切な制御放出マトリックスは、少なくとも1種の水溶性ヒドロキシアル
キルセルロース、少なくとも1つのC12−C36、好ましくはC14−C22、脂肪族アルコー
ルおよび都合により少なくとも1つのポリアルキレングリコールを包含する。ヒドロキシ
アルキルセルロースは、好ましくは、ヒドロキシプロピルセルロース、ヒドリキシプロピ
ルメチルセルロース、および特にヒドロキシエチルセルロースのようなヒドロキシ(C1
からC6まで)アルキルセルロースである。本発明の経口剤形における少なくとも1種の
ヒドロキシアルキルセルロースの量は、中でも、必要とされる正確な速度のオピオイド放
出によって決定される。脂肪族アルコールは、例えば、ラウリルアルコール、ミリスチル
アルコールまたはステアリルアルコールでありうる。しかし、本発明の経口剤形の特に好
ましい実施形態で、少なくとも1種の脂肪族アルコールは、セチルアルコールまたはセト
ステアリルアルコールである。本発明の経口剤形における脂肪族アルコールの量は、上記
のとおり、必要とされる正確な速度のオピオイド放出によって決定される。それは、少な
くとも1種のポリアルキレングリコールが、経口剤形で存在、または不在であるかどうか
にもよる。少なくとも1種のポリアルキレングリコールの不在で、経口剤形は、好ましく
は、20%〜50%(重量で)の脂肪族アルコールを含む。ポリアルキレングリコールは
、経口剤形で存在するとき、それにより脂肪族アルコールおよびポリアルキレングリコー
ルの合わせた重量は、好ましくは、総投与量の20%および50%(重量で)の間で構築
する。
【0040】
1つの好ましい実施形態では、例えば、少なくとも1種のヒドロキシアルキルセルロー
スまたはアクリル酸樹脂対少なくとも1種の脂肪族アルコール/ポリアルキレングリコー
ルの比は、相当の範囲まで、処方からのオピオイドの放出速度を決定する。1:2および
1:4の間のヒドロキシアルキルセルロース対脂肪族アルコール/ホリアルキレングリコ
ールの比が好ましく、そして1:3および1:4の間の比は、特に好ましい。
【0041】
ポリアルキレングリコールは、例えば、ポリプロピレングリコールまたは好ましくはポ
リエチレングリコールでありうる。少なくとも1種のポリアルキレングリコールの数平均
分子量は、好ましくは、1,000および15,000の間、特に1,500および12
,000の間である。
別の適切な制御放出マトリックスは、アルキルセルロース(特に、エチルセルロース)
、C12からC36までの脂肪族アルコールおよび都合によりポリアルキレングリコールを包
含する。
【0042】
上記成分に加えて、制御放出マトリックスは、適切な量の他の材料、例えば希釈剤、潤
滑剤、バインダ、造粒助剤、着色剤、風味剤および製薬業界で従来のものであるグライダ
ント(glidants)をも含有しうる。
本発明による固形の制御放出の経口剤形の製造を促進するために、本発明の別の態様で
は、オピオイドまたはその塩を、制御放出マトリックスに組込む本発明による固形の制御
放出の経口剤形を製造する方法が提供される。マトリックス中の組込みは、例えば、
(a)ヒドロコドンと一緒に、上に説明されるとおりの少なくとも1種の疎水性および
/または親水性材料(例えば、水溶性ヒドロキシアルキルセルロース)を含む顆粒を形成
すること、
(b)顆粒を含む少なくとも1種の疎水性および/または親水性材料を、少なくとも1
種のC12−C36の脂肪族アルコールと混合すること、および
(c)都合により、顆粒を圧縮および成形すること
によってもたらされる。
顆粒は、製薬処方の当業者によく知られる手段のいずれかによって形成されうる。例え
ば、1つの好ましい方法では、顆粒は、水を用いてヒドロキシアルキルセルロース/オピ
オイドを湿式造粒することによって形成されうる。この方法の特に好ましい実施形態では
、湿式造粒段階の間に添加される水の量は、好ましくは、オピオイドの乾燥重量の1.5
および5倍の間、特に1.75および3.5倍の間である。
【0043】
本発明のマトリックスは、溶融ペリティゼーション技術を介しても製造されうる。この
ような環境で、細かく分割された形態でのオピオイドを、バインダ(粒子形態でも)およ
び他の任意の不活性成分と合わせ、そしてその後、混合物を、例えば高せん断混合装置で
の混合物を機械的作業して、ペレット(顆粒、球状体)を形成することによってペレット
化する。その後、ペレット(顆粒、球状体)は、必要とされるサイズのペレットを得るた
めに篩にかけられる。バインダ材料は、好ましくは、粒子形態にあり、そして約40℃よ
り上の融点を示す。適切なバインダ物質としては、例えば、水素化ヒマシ油、水素化植物
性油、他の水素化脂肪、脂肪酸アルコール、脂肪酸エステル、脂肪酸グリセリドおよび同
等物が挙げられる。
制御放出材料は、例えば、溶融造粒または溶融押出技術によっても製造されうる。一般
に、溶融造粒技術は、正常に固形の疎水性バインダ材料、例えばワックスを融解させ、そ
してそこに粉末薬剤を組込むことに関与する。制御放出剤形を得るために、疎水性制御放
出材料、例えばエチルセルロースまたは水不溶性アクリル酸重合体を、溶融ワックス疎水
性バインダ材料に組込むことは必要でありうる。溶融造粒技術を介して製造される制御放
出処方の例は、本発明の譲受人に譲渡され、そしてそれの全体で参照して組込まれた米国
特許第4,861,598号で見られる。
【0044】
別の疎水性バインダ材料は、上記1種またはそれ以上の水不溶性ワックス様物質より疎
水性が低い1種またはそれ以上のワックス様熱可塑性物質と混合された1種またはそれ以
上の水不溶性ワックス様熱可塑性物質を包含しうる。制御放出を達成するために、処方中
の個々のワックス様物質は、当初の放出相の間じゅう胃腸流動体で実質的に非分解性で、
そして不溶性であるべきである。有用な水不溶性ワックス様バインダ物質は、約1:50
00(w/w)より低い水不溶性を示すものでありうる。
上記成分に加えて、制御放出マトリックスは、適切な量の他の材料、例えば、希釈剤、
潤滑剤、バインダ、造粒助剤、着色剤、風味剤、および所望の場合、約50重量%の粒子
までの量で、製薬業界で従来のものであるグライダントをも含有しうる。これらの追加の
材料の量は、所望の処方に所望の効果を提供するのに十分である。
経口剤形を処方するために使用されうる医薬上許容しうる担体および賦形剤の特別の例
は、ここに参照して組込まれるHandbook of Pharmaceutical
Excipients,American Pharmaceutical Asso
siation(1986)で記述される。
【0045】
本発明による適切な溶融押出マトリックスの製造は、例えば、制御放出材料、そして好
ましくはバインダ材料と一緒に、オピオイド鎮静薬を混錬して、均質な混合物を得る段階
を包含しうる。その後、均質な混合物は、少なくとも混合物を、同じものを押出すために
十分に軟化するのに十分な温度まで加熱される。その後、生じた均質混合物は、例えば、
二重スクリュー押出装置を用いて押出して、ストランドを形成される。押出物を、好まし
くは、冷却し、そして当業界で知られる任意の手段によってマルチパーティキュレートに
切断される。ストランドを冷却し、そしてマルチパーティキュレートに切断する。その後
、マルチパーティキュレートを単位用量に分割する。押出物は、好ましくは、約0.1か
ら約5mmまでの直径を有し、そして約8から約24時間の期間に、制御放出の治療的な
活性剤を提供する。
【0046】
本発明の溶融押出された処方を製造する任意の方法は、押出装置に、疎水性制御放出材
料、治療的な活性剤、および任意のバインダ材料を直接メーターで測定すること;均質な
混合物を加熱すること;均質な混合物を押出して、それによりストランドを形成すること
;均質な混合物を含むストランドを冷却すること;ストランドを、約0.1mmから約1
2mmまでのサイズを示す粒子に切断すること;および上記粒子を単位用量に分割するこ
とを含む。本発明のこの態様で、相対的に継続的に製造手段が認識される。
ここで上に記述されるもののような可塑剤は、溶融押出マトリックスに含まれうる。可
塑剤は、好ましくは、約0.1から約30重量%までのマトリックスとして含まれうる。
他の製薬上の賦形剤、例えばタルク、単または多糖類、着色剤、風味剤、潤滑剤および同
等物は、所望のとおり本発明の制御放出マトリックスで含まれうる。含まれる量は、達成
されるべき所望の特徴による。
【0047】
押出装置または出口の直径は、押出ストランドの厚みを変化させるために調節されうる
。さらに、押出装置の出口部分は、丸型である必要はない;それは、長方形、矩形などで
ありうる。存在するストランドを、熱いワイヤー状カッター、断裁機などを用いて粒子に
変えさせる。溶融押出マルチパーティキュレートシステムは、押出装置の出口オリフィス
によって、例えば、顆粒、球状体またはペレットの形態にありうる。本発明の目的のため
に、語句「溶融押出マルチパーティキュレート(類)」および「溶融押出マルチパーティ
キュレートシステム(類)」および「溶融押出粒子」は、好ましくは、類似のサイズおよ
び/または形状の範囲内にあり、そして1種またはそれ以上の活性剤および1種またはそ
れ以上の賦形剤を含有し、好ましくはここに記述されるとおり疎水性制御放出を含む複数
の単位に該当すべきである。好ましくは、溶融押出マルチパーティキュレートは、長さ約
0.1から約12mmまでの範囲のものであり、そして約0.1から約5mmまでの直径
を示す。さらに、溶融押出マルチパーティキュレートは、単にビーズ、種子、ペレットな
どの手段によるようなこのサイズ範囲内の任意の幾何学的形状でありうることと理解すべ
きである。代わりに、押出物は、単に、所望の長さに切断され、そして球形化段階の必要
なしに、治療上活性剤の単位用量に分割されうる。
【0048】
1つの好ましい実施形態では、カプセル内に有効量の溶融押出マルチパーティキュレー
トを含む経口剤形が、製造される。例えば、複数の溶融押出マルチパーティキュレートは
、胃液によって摂取および接触されるときに、有効な制御放出用量を提供するのに十分な
量でゼラチンカプセルに注がれうる。
別の好ましい実施形態では、適切な量のマルチパーティキュレート押出物を、標準技術
を用いた従来の打錠装置を用いて、経口錠剤に圧縮する。錠剤(圧縮および注型された)
、カプセル(硬質および軟質ゼラチン)および丸剤を製造するための技術および組成物は
、ここに参照して組込まれるRemington's
Pharmaceutical Science,(アーサー・オソル、編集者)、1
553−1593(1980)にも記述される。
さらに別の好ましい実施形態では、押出物は、上にさらに詳細に記述され、そして参照
してここに組込まれる米国特許第4,957,681号(クリメッシュら)で説明される
とおり錠剤に成形されうる。
都合により、制御放出のマトリックスマルチパーティキュレートシステムまたは錠剤を
、被覆しうるか、またはゼラチン、カプセルを、上に記述される制御放出コーティングの
ような制御放出コーティングでさらに被覆しうる。上塗は、例えば使用される特定のオピ
オイド鎮静薬の物理的特性、および所望の放出速度により大きくなりうるが、このような
コーティングは、好ましくは、十分量の疎水性および/または親水性の制御放出材料を含
んで、約2から約25パーセントまでの重量増レベルを得る。
【0049】
本発明の剤形は、さらに、1つまたはそれ以上のオピオイド鎮静薬を含む溶融押出マル
チパーティキュレートの組合せを包含しうる。さらに、剤形は、即座治療効果についての
多量の即時放出の治療的に有効な剤をも包含しうる。即時放出の治療的な活性剤は、例え
ば、ゼラチンカプセル内の別個のペレットとして組込まれうるか、または、例えば、ビー
ズまたは溶融押出マルチパーティキュレートの表面に被覆されうる。本発明の単位剤形は
、例えば、制御放出ビーズおよびマトリックスマルチパーティキュレートの組合せをも含
んで、所望の効果を達成しうる。
本発明の制御放出処方は、摂取され、そして胃液に、そしてその後腸液にさらされると
きに、治療的に有効な剤をゆっくりと放出することが好ましい。本発明の溶融押出処方の
制御放出プロフィルは、例えば、制御放出材料の量を変化させることにより、他のマトリ
ックス構成物、疎水性材料に比べて可塑剤の量を変化させることにより、追加の成分また
は賦形剤の封入により、製造の方法を改変されるなどにより変えられうる。
【0050】
本発明の他の実施形態では、溶融押出処方は、その後押出物に添加される治療的に有効
な量の封入なしに製造される。このような処方は、典型的には、押出マトリックス材料と
一緒に混錬された治療上有効な剤を有し、そしてその後、その混合物は、遅延放出処方を
提供するために錠剤化される。このような処方は、例えば、処方に含まれる治療上活性剤
が、疎水性材料および/または遅延反応剤材料を軟化させるために必要とされる温度に感
受性があるときに、有益でありうる。
本発明により使用するのに適した典型的な溶融押出製造システムは、可変速度および一
定トルク制御を示す適切な押出装置駆動モータ、開始停止制御および電流計を含む。さら
に、製造システムは、押出装置の長さを通して、温度センサー、冷却手段および温度イン
ジケーターを含む温度制御コンソールを含む。さらに、製造システムは、その出口で開口
部またはダイを有するシリンダーまたはバレル内に含まれる2つの計測計回転かみ合いス
クリューから構成される二重スクリュー押出装置のような押出装置を含む。供給材料は、
供給ホッパーを通って入り、そしてスクリューによるバレルを通して移動され、そしてダ
イを通して、冷却に対処する連続的可動ベルトによるようにその後運搬されるストランド
に向けられ、そして押出ロープをマルチパーティキュレートシステムにさせるためにペレ
ット化装置または他の適切なデバイスに向けられる。ペレット化装置は、ローラー、固定
ナイフ、回転カッターおよび同等物から構成されうる。適切な装置およびシステムは、ニ
ュージャージー州サザン・ハッケンサックのシー・ダブリュー・ブラベンダー・インスト
ルメンツ、インク.のようなディストリビューターから利用できる。他の適切な装置は、
当業者に明らかである。
【0051】
本発明の別の態様は、押出製品に含まれる空気の量を制御する方法で、上に説明される
とおり溶融押出マルチパーティキュレートの製造に関する。押出物に含まれる量を制御す
ることにより、例えば、マルチパーティキュレート押出物からの治療上活性剤の放出速度
は、明らかに変えられうることが、驚くべきことに分かった。特定の実施形態では、押出
製品のpH依存性は、同様に変えられうることが、驚くべきことに分かった。
したがって、本発明の別の態様で、溶融押出製品は、工程の押出層を通して空気を実質
的に押出す手段で製造される。これは、例えば、真空付属物を有するレイズトリッツの押
出装置を使用することによって達成されうる。レイズトリッツの押出装置を用いた本発明
によって製造される押出マルチパーティキュレートが、様々の物理特性を示す溶融押出製
品を提供することが、驚くべきことに分かった。特に、押出物は、例えば、SEM(走査
電子顕微鏡写真)を提供する走査電子顕微鏡を用いて拡大されるときに、実質的に非多孔
性である。従来の考えに対比して、このような実質的に非多孔性処方が、真空なしに製造
された同じ処方に比べて、治療上有効な剤の早い放出を提供することが分かった。真空下
で押出装置を用いて製造されたマルチパーティキュレートのSEMは、非常に平滑である
ように見え、そしてマルチパーティキュレートは、真空なしに製造されたマルチパーティ
キュレートのものよりいっそう強い傾向にある。少なくとも特定の処方で、真空下の押出
の使用は、真空なしに製造されたそれの対抗の処方よりいっそうpH依存性である押出さ
れたマルチパーティキュレート製品を提供することが観察された。
【0052】
マトリックス・ビーズを製造する方法
本発明による制御放出の剤形は、マトリックスビーズ処方としても製造されうる。マト
リックス・ビーズは、球体化剤およびヒドロコドンを含む。
ヒドロコドンは、重量で約0.01から約99重量%までのマトリックスビーズを包含
することが好ましい。ヒドロコドンは、約0.1から約50重量%までのマトリックスビ
ーズとして包含されることが好ましい。
本発明のマトリックス・ビーズ処方を製造するために使用されうる球体化剤(spheroni
sing agents)は、任意の当業界で知られる球体化剤を含む。セルロース誘導体が好まれ
、そして微小結晶性セルロースが、特に好ましい。適切な微小結晶性セルロースは、例え
ば、アビセルPH101(商標、エフ・エム・シー・コーポレーション)として販売され
ている材料である。球体化剤は、重量で約1から約99%のマトリックス・ビーズとして
含まれることが好ましい。
【0053】
活性成分および球状化剤に加えて、スフェロイドは、バインダをも含みうる。低粘度水
溶性高分子のような適切なバインダは、製薬業界での当業者によく知られている。しかし
、ヒドロキシプロピルセルロースのような水溶性ヒドロキシ低級アルキルセルロースが好
ましい。
オピオイド鎮痛薬および球状化剤に加えて、本発明のマトリックス・ビーズ処方は、上
述されるもののような制御放出材料を含みうる。マトリックス・ビーズ処方中の封入のた
めの好ましい制御放出材料は、アクリル酸およびメタクリル酸重合体または共重合体およ
びエチルセルロースを含む。処方に存在する場合、制御放出材料は、重量で約1から約8
0%のマトリックス・ビーズの量で含まれる。制御放出材料は、好ましくは、ビーズから
のオピオイド鎮痛薬の制御放出を提供するのに有効な量で、マトリックス・ビーズ処方に
含まれる。
【0054】
バインダ、希釈剤、および同等物のような製薬的加工助剤は、マトリックス・ビーズ処
方に含まれ得る。処方に含まれるこれらの剤の量は、処方により示されるべき所望の効果
で変化する。
マトリックス・ビーズは、ここで上に記述されるもののような制御放出材料を含めた制
御放出コーティングで上塗りされうる。制御放出のコーティングは、約5から約30%ま
での重量増まで塗布される。使用されるべき制御放出コーティングの量は、マトリックス
・ビーズおよび化合物の組成、および/またはオピオイド鎮痛薬(すなわち、ヒドロコド
ン)の物理特性のような多様な因子によって変化する。
マトリックス・ビーズは、一般に、例えば湿式造粒により、オピオイド鎮痛薬と一緒に
球状化剤を造粒することによって製造される。その後、顆粒は、マトリックス・ビーズを
生成するために球状化される。その後、マトリックス・ビーズは、都合により、ここで上
に記述されるもののような方法によって、制御放出コーティングで上塗りされる。
【0055】
マトリックス・ビーズを製造する別の方法は、(a)少なくとも1種の水溶性ヒドロキ
シアルキルセルロースおよびオピオイドまたはオピオイド塩を含む顆粒を成形し、(b)
顆粒を含むヒドロキシアルキルセルロースを、少なくとも1種のC12−C36脂肪族アルコ
ールと混合し、そして(c)都合により、顆粒を圧縮および成形することによる。好まし
くは、顆粒は、水を用いてヒドロキシアルキルセルロース/オピオイドを湿式造粒するこ
とによって成形される。本発明の特に好ましい実施形態では、湿式造粒段階の間に添加さ
れる水の量は、オピオイドの乾燥重量の1.5および5倍の間、特に1.75および3.
5倍の間であるのが好ましい。
【0056】
さらに他の代替の実施形態では、活性成分と一緒の球状化剤は、球状化されて、スフェ
ロイドを形成しうる。微小結晶性セルロースが好ましい。適切な微小結晶性セルロースは
、例えば、アビセルPH101(商標、エフ・エム・シー・コーポレーション)として販
売されている材料である。このような実施形態では、活性成分および球状化剤に加えて、
スフェロイドは、バインダをも含みうる。低粘度水溶性重合体のような適切なバインダは
、製薬業界での当業者によく知られる。しかし、ヒドロキシプロピルセルロースのような
水溶性ヒドロキシ低級アルキルセルロースが、好まれる。さらに(または代わりに)、ス
フェロイドは、メタクリル酸−エチルアクリレート共重合体、またはエチルセルロースの
ような水不溶性重合体、特にアクリル酸重合体、アクリル酸共重合体を含みうる。このよ
うな実施形態では、持続的放出コーティングは、一般に、(a)単独、または脂肪酸アル
コールとの混和物でのいずれかのワックス;または(b)シェラックまたはゼインのよう
な水不溶性材料を含む。
【0057】
制御放出ビーズ処方
1つの特に好ましい実施形態では、経口剤形は、ゼラチンカプセル内に含まれる有効数
の制御放出スフェロイドを包む。
本発明の別の好ましい実施形態では、制御放出剤形は、制御放出材料を含む制御放出コ
ーティングで被覆される活性成分を含むスフェロイドを包む。語句スフェロイドは、製薬
業界で知られており、そして例えば、0.1mmおよび25mmの間、特に0.5mmお
よび2mmの間の直径を示す球状の顆粒を意味する。
スフェロイドは、好ましくは、水性媒体中で、制御速度でのオピオイド(または塩)の
放出を許す制御放出材料で被覆されたフィルムである。フィルムコートは、他の記述され
た特性と組合せて、上に概説されるインビトロ放出速度(例えば、少なくとも約12.5
%が、1時間後に放出される)を達成するために選択される。本発明の制御放出のコーテ
ィング処方は、好ましくは、平滑であり、そして素晴らしく、色素または他のコーティン
グ添加剤を支持する能力のあり、非毒性で、不活性で、そして粘着性なしである、強力な
連続フィルムを生じる。
【0058】
コーティング
本発明の剤形は、都合により、放出の制御のために、または処方の保護のために適する
1つまたはそれ以上のコーティングで被覆されうる。1つの実施形態では、コーティング
は、例えば、胃腸液にさらされる場合に、pH依存性またはpH独立性の放出のいずれか
を許すために供される。pH独立性コーティングが望まれる場合、コーティングは、環境
流動体、例えば胃腸管でのpH変化にもかかわらず、最適な放出を達成するように設計さ
れる。他の好ましい実施形態は、患者に対して少なくとも約12時間、および好ましくは
24時間までの無痛覚を提供する能力がある吸収プロファイルが、提供されるように、胃
腸(GI)管の所望な領域、例えば胃または小腸にオピオイドを放出するpH依存的コー
ティングを含む。胃腸管の1つの所望の領域、例えば胃での用量の一部を放出し、そして
胃腸管の別の領域、例えば小腸でその用量の残りを放出する組成物を処方することも可能
である。
pH依存性コーティングを利用する本発明による処方は、それによって未保護が、腸溶
性コートに上に被覆され、そして胃で放出される反復作用効果をも与え得る一方で、腸溶
性コーティングによって保護される残りは、胃腸管にさらに放出される。本発明によって
使用されうるpH依存性であるコーティングは、例えば、シェラック、セルロースアセテ
ートフタレート(CAP)、ポリビニルアセテートフタレート(PVAP)、ヒドロキシ
プロピルメチルセルロースフタレート、およびメタクリル酸エステル共重合体、ゼインお
よび同等物のような制御放出材料を含む。
【0059】
別の好ましい実施形態では、本発明は、(i)アルキルセルロース;(ii)アクリル
酸重合体;または(iii)その混合物から選択される疎水性制御放出材料で被覆された
オピオイドを包含する固形の制御放出剤形に関する。コーティングは、有機または水性溶
液または分散物の形態で塗布されうる。
特定の好ましい実施形態では、制御放出コーティングは、疎水性制御放出材料の水性分
散物から誘導される。その後、オピオイド(類)(例えば、錠剤コアまたは不活性の製薬
ビーズまたはスフェロイド)を含有する被覆基質は、基質が安定な溶解を提供する終点に
達するまで、硬化される。硬化終点は、硬化直後の剤形の溶解プロファイル(曲線)を、
例えば40℃の温度および75%の相対湿度での少なくとも1ヶ月の促進された保存条件
にさらされた後の剤形の溶解プロファイル(曲線)と比較することによって決定されうる
。これらの処方は、本発明の譲受人に譲渡され、そして参照してここに組込まれる米国特
許第5,273,760号および第5,286,493号で詳細に説明される。本発明に
よって使用されうる制御放出の処方およびコーティングの他の例としては、参照してここ
に組込まれる米国特許第5,324,351号;第5,356,467号、および第5,
472,712号が挙げられる。
【0060】
好ましい実施形態では、制御放出のコーティングは、ここで下に記述されるもののよう
な可塑剤を含む。
特定の実施形態では、制御放出処方を得るために、アルキルセルロースまたはアクリル
酸重合体の十分量の水性分散物と共にオピオイド鎮痛薬を含む基質に上塗りして、約2か
ら約50%、例えば約2から約25%までの重量増を得ることが必要である。上塗りコー
トは、多かれ少なかれ、例えば、治療的な活性剤の物理特性および所望の放出速度、水性
分散物における可塑剤の封入および同じものの組込みの手段による。
【0061】
アルキルセルロース重合体
アルキルセルロースを含めたセルロース性材料および重合体は、本発明による基質、例
えばビーズ、錠剤などを被覆するのによく適合した制御放出材料である。習熟者は、他の
セルロースおよび/またはアルキルセルロース重合体が、本発明による疎水性コーティン
グの全部または一部として、単独で、または任意の組合せで容易に使用されうることを認
識するが、実施例の方法によって簡単に、1つの好ましいアルキルセルロース性重合体は
、エチルセルロースである。
エチルセルロースの1つの商用的に利用できる水性分散物は、アクアコート(登録商標
;Aquacoat)エフ・エム・シー・コープ、米国ペンシルベニア州フィラデルフィ
ア)である。アクアコート(登録商標)は、水混和しない有機溶媒でのエチルセルロース
を溶解し、そしてその後、それを界面活性剤および安定化剤の存在下で水中に乳化させる
ことによって製造される。ミクロンより小さい小滴を生じるための均質化の後、有機溶媒
を、真空で蒸散させて、偽ラテックスを形成する。可塑剤は、製造相の間じゅう、偽ラテ
ックスに組込まれない。したがって、コーティングと同じものを使用する前に、アクアコ
ート(登録商標)を、使用する前に適切な可塑剤と最終的に混合することが必要である。
【0062】
エチルセルロースの別の水性分散物は、シュアレーズ(登録商標;Surelease
)(カラーコン、インク、米国ペンシルベニア州ウエストポイント)として市販で入手で
きる。この製品は、製造工程の間に可塑剤を分散物に組込むことによって製造される。ホ
ットメルトの重合体、可塑剤(ジブチルセバケート)、および安定化剤(オレイン酸)は
、均質な混合物として製造され、そしてそれは、その後、アルカリ性溶液で希釈されて、
基質上に直接塗布されうる水性分散物を得る。
【0063】
アクリル酸重合体
本発明の他の好ましい実施形態では、制御放出コーティングを含む制御放出材料は、製
薬上許容しうるアクリル酸重合体であり、そしてそれに限定されないが、アクリル酸およ
びメタクリル酸共重合体、メチル・メタクリレート共重合体、エトキシエチル・メタクリ
レート、シアノエチル・メタクリレート、ポリ(アクリル酸)、ポリ(メタクリル酸)、
メタクリル酸アルキルアミド共重合体、ポリ(メチル・メタクリレート)、ポリメタクリ
レート、ポリ(メチル・メタクリレート)共重合体、ポリアクリルアミド、アミノアルキ
ル・メタクリレート共重合体、ポリ(メタクリル酸無水物)、およびグリジジル・メタク
リレート共重合体が挙げられる。
特定の好ましい実施形態では、アクリル酸重合体は、1種またはそれ以上のアンモニオ
メタクリレート共重合体から構成される。アンモニオ・メタクリレート共重合体は、当業
界でよく知られており、そして低含有量の四級アンモニウム基を示すアクリル酸およびメ
タクリル酸エステルの十分に重合化した共重合体としてNF XVIIで記述されている
。
【0064】
所望の溶解プロファイルを得るために、様々のモル比の四級アンモニア基のような様々
の物理的特性を示す2つまたはそれ以上のアンモニオメタクリレート共重合体を、天然の
(メト)アクリル酸エステルに組込むことが必要でありうる。
特定のメタクリル酸エステル型重合体は、本発明によって使用されうるpH依存性コー
ティングを製造するために有用である。例えば、ローム・テク、インク.からオイドラギ
ッド(登録商標;Eudragit)として商用的に入手できる、メタクリル酸共重合体
または重合性メタクリレートとしても知られるジエチルアミノエチル・メタクリレートお
よび他の天然のメタクリル酸エステルから合成される共重合体のファミリーがある。幾ら
かの異なる型のオイドラギッド(登録商標)がある。例えば、オイドラギッドEは、酸性
媒体で膨張および溶解するメタクリル酸共重合体の例である。オイドラギッドLは、約p
H<5.7で膨張せず、そして約pH>6で溶解性であるメタクリル酸共重合体の例であ
る。オイドラギッドSは、約pH<6.5で膨張せず、そして約pH>7で溶解性である
メタクリル酸共重合体の例である。オイドラギッドRLおよびオイドラギッドRSは、水
膨張性であり、そしてこれらの重合体によって吸収される水の量は、pH依存性であるが
、しかし、オイドラギッドRLおよびオイドラギッドRSで被覆された剤形は、pH独立
性である。
【0065】
特定の好ましい実施形態では、アクリル酸コーティングは、それぞれ、商標名オイドラ
ギッド(登録商標)RL30DおよびオイドラギッドRS30Dの下でローム・ファルマ
から商用的に入手可能な2つのアクリル酸樹脂ラッカーの混合物を包含する。オイドラギ
ッド(登録商標)RL30Dおよびオイドラギッド(登録商標)RS30Dは、低い含有
量の四級アンモニウム基を有するアクリル酸およびメタクリル酸エステルの共重合体であ
り、アンモニウム基対残りの中性(メト)アクリル酸エスエルのモル比は、オイドラギッ
ド(登録商標)RL30Dで1:20であり、そしてオイドラギッド(登録商標)RS3
0Dで1:40である。平均分子量は、約150,000である。コード認識RL(非常
に透過性)およびRS(低い透過性)は、これらの剤の透過性特性に該当する。オイドラ
ギッド(登録商標)RL/RS混合物は、水に、そして消化性流動体で不溶性である。し
かし、同じものから形成されるコーティングは、膨張性であり、そして水性溶液および消
化性流動体で透過性である。
【0066】
本発明のオイドラギッド(登録商標)RL/RS分散物は、所望の溶解プロファイルを
示す最終的に制御放出処方を得るために、任意の所望の比で一緒に混合されうる。望まれ
る制御放出の処方は、例えば、100%オイドラギッド(登録商標)RL、50%オイド
ラギッド(登録商標)RL、および50%オイドラギッド(登録商標)RSおよび10%
オイドラギッド(登録商標)RL:オイドラギッド(登録商標)90%RSから誘導され
る遅延反応剤コーティングから得られうる。もちろん、当業者は、例えばオイドラギッド
(登録商標)Lのような他のアクリル酸重合体も使用されうることを認識する。
【0067】
可塑剤
コーティングが、疎水性制御放出材料の水性分散物を包含する本発明の実施形態では、
疎水性材料の水性分散物中の有効量の可塑剤の封入は、さらに、制御放出コーティングの
物理的特性を改善する。例えば、エチルセルロースが、比較的高いガラス遷移温度を示し
、そして正常なコーティング条件下で柔軟性フィルムを形成しないので、コーティング材
料と同じものを使用する前に、可塑剤を、制御放出コーティングを含有するエチルセルロ
ースコーティングに組込むことが好ましい。一般に、コーティング溶液に含まれる可塑剤
の量は、フィルム形成剤の濃度、例えば、最もしばしば、約1から約50重量パーセント
までのフィルム形成剤に基づく。しかし、可塑剤の濃度は、特定のコーティング溶液およ
び使用の方法を用いた注意深い実験の後にのみ適切に決定されうる。
他の水不溶性可塑剤(アセチレート化モノグリセリド、フタレートエステル、ヒマシ油
など)が、使用されうる可能性があるが、エチルセルロースについての適切な可塑剤の例
は、ビブチルセバケート、ジエチルフタレート、トリエチルシトレート、トリブチルシト
レート、およびトリアセチンのような水不溶性可塑剤が挙げられる。トリエチルシトレー
トは、本発明のエチルセルロースの水性分散物についての特に好ましい可塑剤である。
【0068】
本発明のアクリル酸重合体についての適切な可塑剤の例としては、それに限定されない
が、トリエチルシトレートNF XVI、トリブチルシトレート、ジブチルフタレート、
および可能性のある1,2−プロピレングリコールのようなクエン酸エステルが挙げられ
る。登録商標オイドラギッドRL/RSラッカー溶液のようなアクリル酸フィルムから形
成されるフィルムの弾性を増強するために適切であると立証された他の可塑剤としては、
ポリエチレングリコール、プロピレングリコール、ジメチルフタレート、ヒマシ油および
トリアセチンが挙げられる。トリエチルシトレートは、本発明のエチルセルロースの水性
分散物についての特に好ましい可塑剤である。
制御放出コーティングに対しての少量のタルクの添加は、加工の間に粘着する水性分散
物の傾向を減少し、そして艶出剤として作用することがさらに分かった。
【0069】
被覆ビーズ処方の製造
疎水性材料の水性分散物は、基質例えば、ヌ・パリエル18/20ビーズのような不活
性の製薬的ビーズを被覆するために使用される場合、その後、複数の結果物で安定化され
た固形制御放出ビーズは、摂取され、そして胃液または溶解媒体のような環境流動体によ
って接触されたときに、有効な制御放出用量を提供するのに十分な量でゼラチンカプセル
に注がれうる。
本発明の安定化された制御放出ビーズ処方は、例えば、摂取され、そして胃液に、そし
てその後小腸液にさらされたときに、オピオイド鎮痛薬をゆっくりと放出する。本発明の
処方の制御放出プロファイルは、例えば、疎水性制御放出材料の水性分散物を用いた上塗
りの量を変化させることによって変えられ得て、そして疎水性制御放出材料に関して可塑
剤の量を変化させることによって、追加の成分または賦形剤の封入によって、製造の方法
を変えることによってなど、可塑剤が、疎水性制御放出材料の水性分散物に添加される手
段を変えうる。最終的製品の溶解プロファイルは、例えば、制御放出コーティングの厚み
を増加または減少させることによって、改質もされうる。
【0070】
治療上活性剤で被覆された基質は、例えば、治療上活性剤を水に溶解させる、そしてそ
の後、ウスター挿入物を用いて、溶液を、ヌ・パリエル18/20ビーズのような基質上
に噴霧することによって製造される。都合により、追加の成分は、オピオイドのビーズへ
の結合を助成し、および/または溶液を着色するなどのためにビーズを被覆する前にも添
加される。例えば、着色剤(例えば、オパドライ(登録商標;Opadry)、カラーコ
ン、インク.から商用的に入手可能な)と共に、またはなしに、ヒドロキシプロピル・メ
チルセルロースなどを含む製品は、溶液に添加され得て、そして溶液は、同じものを基質
上に使用する前に(例えば、約1時間)混合されうる。その後、結果物である被覆基質は
、都合により、バリヤ剤で上塗りされて、疎水性制御放出コーティングから治療上活性剤
を分離しうる。
適切なバリヤ剤の例は、ヒドロキシプロピル・メチルセルロースを包含するものである
。しかし、当業界で知られる任意のフィルム形成剤が使用されうる。バリヤ剤は、最終製
品の溶解速度に影響しないことが好ましい。
【0071】
その後、基質は、疎水性制御放出材料の水性分散物で上塗りされうる。疎水性制御放出
材料の水性分散物は、好ましくは、さらに、トリエチルシトレートのような有効量の可塑
剤を含む。アクアコート(登録商標)またはシュアレーズ(登録商標)のような予備処方
されたエチルセルロースの水性分散物が、使用されうる。登録商標シュアレーズが使用さ
れる場合、可塑剤を別個に添加することは、必要でない。代わりに、オイドラギッド(登
録商標)のようなアクリル酸重合体の予備処方された水性分散物が、使用されうる。
【0072】
本発明のコーティング溶液は、フィルム形成剤に加えて、可塑剤および溶媒システム(
すなわち、水)、優雅さを提供し、そして製品差別化を生じる着色剤を含有することが好
ましい。色は、疎水性材料の水性分散物の代わりに、またはそれに加えて、治療上活性剤
の溶液に添加されうる。例えば、色は、せん断を示す色を、水溶性重合体溶液に添加し、
そしてその後、可塑化されたアクアコート(登録商標)に対して低せん断を用いることに
よって、アルコールまたはプロピレングリコール基本の色分散物、挽かれたアルミニウム
レーキおよび二酸化チタンのような懸濁化剤の使用を介して、アクアコート(登録商標)
に添加されうる。代わりに、色を本発明の処方に供する任意の適切な方法が使用されうる
。アクリル酸重合体の水性分散物が使用されるときに、色を処方に供するための適切な成
分は、二酸化チタンおよび酸化鉄色素のような色素を含む。しかし、色素の組込みは、コ
ーティングの阻止効果を増大しうる。
【0073】
疎水性制御放出材料の可塑化水性分散物は、当業界で知られる任意の適切な噴霧装置を
使用して噴霧することによって、治療上活性剤を含む基質上に塗布されうる。好ましい方
法では、底面から注入される空気ジェットが、コア材料を流動化し、そして乾燥を有効に
する一方で、アクリル酸重合体コーティングが噴霧されるウスター流動床システムが使用
される。前記被覆基質が、水性溶液、例えば胃液にさらされるときに、上記治療上活性剤
の予備測定された制御放出を得るために十分量の疎水性材料の水性分散物が、使用される
ことが好ましく、そして可塑剤などの組込みの手段として、治療上活性剤の物理的特性を
考慮する。疎水性制御放出材料で被覆した後、登録商標オパドライのようなフィルム形成
剤の別の上塗りは、都合により、ビーズに塗布される。この上塗りは、ビーズの凝集を実
質的に減少させるために、ともかく供給される。
【0074】
本発明の制御放出処方からの治療上活性剤の放出は、1種またはそれ以上の放出改質剤
の添加により、またはコーティングを通して1種またはそれ以上の放出経路を提供するこ
とによって、さらに影響され、すなわち、所望の速度に調節されうる。疎水性制御放出材
料対水溶性材料の比は、他の因子の中でも、要求された放出速度および選択された材料の
溶解性特徴によって測定される。
多孔形成剤として機能する放出改質剤は、有機または無機であり得て、そして使用の環
境にあるコーティングから溶解、抽出または浸出されうる材料を含む。多孔形成剤は、ヒ
ドロキシプロピルメチルセルロースのような1種またはそれ以上の親水性材料を包含しう
る。
本発明の制御放出コーティングは、スターチおよびゴムのような浸食促進剤も含みうる
。
本発明の制御放出コーティングは、カルボネート基が、重合鎖に再び生じるカルボン酸
の線状ポリエステルから構成されるポリカーボネートのような使用の環境での微小多孔性
板を作成するのに有用な材料をも含みうる。
【0075】
放出改質剤は、半透過性重合体をも包含しうる。特定の好ましい実施形態では、放出改
質剤は、ヒドロキシプロピルメチルセルロース、ラクトース、金属ステアレート、および
前述のもののいずれかの混合物から選択される。
本発明の制御放出コーティングは、少なくとも1種の放出通路、オリフィスまたは同等
物を包含する出口手段をも含みうる。放出通路は、米国特許第3,845,770号;第
3,916,889号;第4,063,064号;および第4,088,864号に開示
されるもののような方法によって形成され得て、そしてその全ては、参照してここに組込
まれうる。放出通路は、丸型、三角、四角、長円形、不均整などのような任意の形状を示
しうる。
【0076】
約24時間投与のために適する制御放出ビーズ処方を生じる別の方法は、粉末積層を介
することである。本発明の譲受人に譲渡され、そしてそれの全体で参照してここに組込ま
れた米国特許第5,411,745号は、基本的に触知できない水性のラクトースから構
成される加工助剤を利用する粉末積層技術を介して製造される24時間モルフィン処方の
製造を教示する。粉末積層ビーズは、水性バインダ溶液を、不活性ビーズに噴霧して、粘
着性表面を提供し、そして粘着性ビーズ上に、硫酸モルフィンおよび触知できない水性ラ
クトースの均質混合物である粉末を噴霧することにより製造される。その後、ビーズは、
乾燥され、そしてここで上に記述されるもののような疎水性材料で被覆して、最終処方が
、環境流動体にさらされるときに、薬剤の所望の放出を得る。その後、適切な量の制御放
出ビーズが、封入されて、約12時間、モルフィンの有効な血漿中濃度を提供する最終剤
形を提供する。
【0077】
以下の実施例は、本発明の種々の態様を示す。それらは、あらゆる手段であればどれで
も、請求項を制限すると解釈されることは意図されない。
【0078】
実施例1
ヒドロコドン持続的放出錠剤を、下の表1で説明される処方で製造された。
【0079】
【表1】
【0080】
以下の手段により:
1.遅延反応剤分散:ライトニンの混合装置を用いてオイドラギッドRS30Dおよびト
リアセチンを混錬する。
2.ステアリルアルコールを溶融させる。
3.流動床造粒装置を用いて、ヒドロコドン・ビタートレート、噴霧乾燥ラクトース、お
よびポビドンにおける噴霧遅延反応剤で分散する。
4.15分間、または一定重量までのステンレス鋼のトレイ上での乾燥バッチ。5.ホー
バートの混合装置を用いて、溶融ステアリルアルコールを、バッチに組込む。
6.30分間、ステンレス鋼のトレイ上で顆粒を乾式ワックス掛けするか、または造粒の
温度は、35℃以下に達する。
7.コミルを通して冷却された顆粒を挽く。
8.ホーバート混合装置を用いて、タルクおよびステアリン酸マグネシウムを顆粒に注入
する。
9.錠剤プレスを使用して、顆粒を錠剤に圧縮する。
【0081】
その後、錠剤は、以下の手段を用いて、溶解(dissolution)について試験した。
1.装置:米国薬局方(USP)の方法I(バスケット)、100rpm。
2.媒体:55分間の700mlのSGF、その後、酵素なしの900mlのSIF
3.サンプル採取時間:1、2、4、8および12時間。
4.解析:高速液体クロマトグラフィー。
溶解パラメータは、下の表2で説明される:
【0082】
【表2】
【0083】
その後、CmaxおよびTmaxが、実施例1について得られ、そして生物利用性研究での即
時放出対照標準は、下の表3に説明されるとおり、即時放出処方(ロルタブ7.5mg×
2)として投与されたヒドロコドン15mgを、健全なヒト対象における上記CR処方と
比較する。
【0084】
【表3】
【0085】
実施例2
ヒドロコドン持続的放出錠剤を、下の表4に説明された処方で製造した。
【0086】
【表4】
【0087】
実施例1の手段による。
その後、溶解パラメータを、実施例1の手段を用いて得た。結果は、下の表5に説明さ
れる。
【0088】
【表5】
【0089】
実施例3
ヒドロコドン持続的放出カプセルを、下の表6に説明された処方で製造した。
【0090】
【表6】
【0091】
以下の手段により:
1.ホーバートの混合装置を用いて、ステアリルアルコール、オイドラギッド(Eudragit
)RLPO、ヒドロコドン・ビタートレート、およびオイドラギッド(Eudragit)RSP
Oを混錬粉砕した。
2.以下の条件下で、粉末供給装置、溶融押出装置(6×1mmダイヘッドを具備した)
、コンベヤー、レーザーマイク、およびペレタイザーを用いて造粒物を押出す。
【0092】
領域1 10℃
領域2 20℃
領域3 120℃
領域4 120℃
領域5 120℃
領域6 120℃
領域7 95℃
領域8 95℃
MGA 120℃
ダイ 117℃
【0093】
粉末供給速度−40g/分;スクリュー速度−185rpm;真空−980ミリバール
押出物の直径が1mmであるような−コンベヤー
ペレットが、長さ1mmまでに切断されるような−ペレタイザー
3.16番メッシュおよび20番メッシュスクリーンを用いてペレットをスクリーニング
する。16番メッシュスクリーンを通過し、そして20番メッシュスクリーンに保有され
る材料を収集する。
4.ペレットでサイズ2番の透明なゼラチンカプセルを充填する。範囲:NLT114m
gおよびNMT126mg。
【0094】
その後、溶解パラメータは、実施例1の手段を用いて得られた。結果は、下の表7で説
明される。
【0095】
【表7】
【0096】
実施例4
オキシコドン持続的放出錠剤を、下の表8に説明された処方で製造した。
【0097】
【表8】
【0098】
以下の手段により:
1.造粒:オキシコドンHCl上にオイドラギッド/トリアセチン分散物を噴霧し、流動
床造粒装置を用いて、乾燥ラクトースおよびポビドンを噴霧する。
2.破砕:顆粒を放出し、そしてミルに通す。
3.ワックスがけ:ステアリルアルコールを溶融させ、そして混合装置を用いて、破砕し
た顆粒に加える。冷却させる。
4.破砕:冷却した顆粒をミルに通す。
5.潤滑:混合装置を用いて、タルクおよびステアリン酸マグネシウムで顆粒を潤滑させ
る。
6.圧縮:錠剤プレスを用いて、顆粒を錠剤に圧縮する。
7.フィルムコーティング:水性フィルムコートを錠剤に塗布する。
その後、錠剤を、以下の手段を用いて、溶解について試験した。
【0099】
1.装置:米国薬局方の方法II(パドル)、150rpm。
2.媒体:第一の時間、700mlのSGF、その後、リン酸緩衝液でpH7.5まで9
00mlになる。
3.サンプル採取時間:1、2、4、8、12、18および24時間。
4.解析:高速液体クロマトグラフィー。
溶解パラメータは、下の表9で説明される:
【0100】
【表9】
【0101】
その後、CmaxおよびTmaxが、実施例4について得られ、そして生物利用性研究での即
時放出対照標準は、下の表10に説明されるとおりである。
【0102】
【表10】
【0103】
実施例5
モルフィン持続的放出錠剤は、下の表11で説明される処方で製造された。
【0104】
【表11】
【0105】
以下の手段により:
1.造粒:硫酸モルフィンに水を添加し、混合装置中で乾燥ラクトースおよびヒドロキシ
エチルセルロースを噴霧し、流動床造粒装置を用いて乾燥させる。
2.スクリーニング:顆粒を放出し、そしてシーブを通過させる。
3.ワックスがけ:セトステアリルアルコールを溶融させ、そして混合装置を用いて、破
砕した顆粒に加える。冷却させる。
4.スクリーニング:冷却した顆粒をシーブに通す。
5.潤滑:混合装置を用いて、タルクおよびステアリン酸マグネシウムで顆粒を潤滑させ
る。
6.圧縮:錠剤プレスを用いて、顆粒を錠剤に圧縮する。
7.フィルムコーティング:水性フィルムコートを錠剤に塗布する。
その後、錠剤を、以下の手段を用いて、溶解について試験した:
【0106】
1.装置:米国薬局方の方法I(バスケット)、50rpm。
2.媒体:37℃、900mlの精製水。
3.サンプル採取時間:1、2、4および6時間。
4.解析:UV検出、285nmおよび305nm、5cmセルを用いた2点法。
【0107】
溶解パラメータは、下の表12で説明される。
【0108】
【表12】
【0109】
その後、CmaxおよびTmaxが、実施例5について得られ、そして生物利用性研究での即
時放出対照標準は、下の表13に説明されるとおりである。
【0110】
【表13】
【0111】
実施例6
実施例1、実施例4および実施例5の薬理学的パラメータを、互いに比較した。実施例
1のヒドロコドンHCl制御放出錠剤の溶解が、実施例4の制御放出オキシコドン錠剤お
よび実施例5の硫酸モルフィン制御放出錠剤の溶解に類似していてさえ、ヒドロコドン処
方についてのIRに対するCRのCmax比は、38%であるのに対して、オキシコドン錠
剤およびモルフィン錠剤は、50%を越えている。比較結果は、下の表14に説明される
。
【0112】
【表14】
【0113】
実施例7
実施例1、絶食中の正常な自発参加者での実施例2、実施例3の制御放出ヒドロコドン
処方と、2つの即時放出ヒドロコドン・ビタートレート7.5mg/アセトアミノフェン
500mg錠剤(IR実施例)の単回用量、4回治療、開放ラベル、薬理学的比較が行わ
れた。これらの処方のついての血漿濃度は、下の表15〜18に説明される。
【0114】
【表15】
【0115】
【表16】
【0116】
【表17】
【0117】
【表18】
【0118】
薬理学的パラメータは、下の表19で説明される。
【0119】
【表19】
【0120】
a AUC(0、最後)およびCmaxについての幾何学的手段およびTmax、W50、T1
/2(abs)およびT1/2(elim)についての計算手段。
b 比および90%Clは、最少の平方手段に基づく。
c 比(%):最少平方手段に基づいて、(試験手段/対照手段)×100。
【0121】
実施例8
ヒドロコドン持続的放出錠剤は、下の表20に説明される処方で製造された。
【0122】
【表20】
【0123】
1 加工の間に蒸散させ、そして最終製品の一部でない。
【0124】
以下の手段により:
1.破砕:ステアリルアルコールのフレークをミルに通す。
2.混錬:適切なブレンダーで、ヒドロコドン・ビタートレート、二塩基性リン酸カルシ
ウム、グリシジルベヘナート、ステアリルアルコールおよび微小結晶性セルロースを混合
する。
3.押出し:上昇温度で、混錬材料を、二重スクリュー押出装置に継続的に供給して、押
出物を軟化および成形する。
4.冷却:押出物をコンベヤーで冷却させる。
5.破砕:冷却した押出物をミルに通して、適切な粒子サイズの顆粒を得る。
6.混錬:破砕された押出物を、ステアリン酸マグネシウムと混錬させる。
7.圧縮:錠剤プレスを用いて、結果物である顆粒を圧縮する。
8.コーティング:精製水中のオパドライを分散させ、そしてそれを錠剤コアに塗布する
ことによって、フィルムコーティング溶液を製造する。
【0125】
その後、錠剤を、以下の手段を用いて、溶解について試験した:
1.装置:米国薬局方の方法I(バスケット)、100rpm。
2.媒体:最初の55分間、700mlSGF(酵素なし)、その後、リン酸緩衝液でp
H7.5まで900mlになる。
3.サンプル採取時間:1、2、4、8および12時間。
4.解析:高速液体クロマトグラフィー。
溶解パラメータは、下の表21で説明される:
【0126】
【表21】
【0127】
実施例9
給食および絶食にある15mgヒドロコドン制御放出錠剤(実施例8)の、15mgヒ
ドロコドン即時放出(2×7.5mg錠剤)の単回用量の3法交差(3 way crossover)
の薬理学的比較研究は、絶食した正常な自主参加者における2つのQ6H用量に付与され
た。
その後、CmaxおよびTmaxが、実施例8について得られ、そして生物利用性研究での即
時放出対照標準は、下の表22および23に説明されるとおりである。
【0128】
【表22】
【0129】
【表23】
【図面の簡単な説明】
【0130】
【図1】実施例1、実施例2、実施例3および等量用量の即時放出ヒドロコドンの平均ヒドロコドン血漿中濃度のグラフ表示である。
【図2】実施例4の手段によって製造された制御放出オキシコドンの様々のサンプル、および実施例5の手段によって製造された制御放出オキシコドンの様々のサンプルに対する、実施例1、実施例2および実施例3の平均ヒドロコドン血漿中濃度のグラフ表示である。
【図3】実施例1、実施例2、実施例3および等量用量の即時放出ヒドロコドンの時間に対して吸収された分画ヒドロコドン含有率のグラフ表示である。
【特許請求の範囲】
【請求項1】
鎮痛効果を示す有効量のヒドロコドンまたは医薬上許容しうるその塩を包含する剤形で
あって、該ヒドロコドンまたは医薬上許容しうるその塩は、a)医薬上許容し得るマトリ
ックスに制御放出材料とともに組込まれるか、b)基質に包含され、またはその上にコー
トされ、さらに制御放出材料で塗布されるか、のいずれかであり、
該剤形は、ヒト患者に対する一日二回投与のために適し、
患者集団に対する第一の投与後に、0.55から0.85までのヒドロコドンの平均C
12/Cmax比を提供し、
少なくとも12時間、治療的効果を提供し、かつ、
37℃での55分間の700mlの模擬胃液(SGF)中で、その後37℃での900
mlの模擬小腸液(SIF)に交換する100rpmでの米国薬局方バスケット法によっ
て測定されるときに4時間で、剤形からのヒドロコドンまたはその塩の51.7重量%か
ら67.4重量%までのインビトロ放出を提供する制御放出経口固形剤形。
【請求項2】
前記ヒドロコドンが、前記医薬上許容し得るマトリックス中に分散されている請求項1
に記載の剤形。
【請求項3】
前記マトリックスが、マルチパーティキュレートの形態である請求項2に記載の剤形。
【請求項4】
前記マルチパーティキュレートが、錠剤に圧縮されている請求項3に記載の剤形。
【請求項5】
前記マルチパーティキュレートが、医薬上許容しうるカプセルに分散されている請求項
3に記載の剤形。
【請求項6】
0.65から0.75までのヒドロコドンの平均C12/Cmax比を提供する請求項1に
記載の剤形。
【請求項7】
37℃での55分間の700mlの模擬胃液(SGF)中で、その後37℃での900
mlの模擬小腸液(SIF)に交換する100rpmでの米国薬局方バスケット法によっ
て測定されるときに1時間で、剤形からのヒドロコドンまたはその塩の23.9重量%か
ら39.7重量%までのインビトロ放出を提供する請求項1に記載の剤形。
【請求項8】
37℃での55分間の700mlの模擬胃液(SGF)中で、その後37℃での900
mlの模擬小腸液(SIF)に交換する100rpmでの米国薬局方バスケット法によっ
て測定されるときに2時間で、剤形からのヒドロコドンまたはその塩の34.7重量%か
ら51.5重量%までのインビトロ放出を提供する請求項1に記載の剤形。
【請求項9】
37℃での55分間の700mlの模擬胃液(SGF)中で、その後37℃での900
mlの模擬小腸液(SIF)に交換する100rpmでの米国薬局方バスケット法によっ
て測定されるときに8時間で、剤形からのヒドロコドンまたはその塩の74.6重量%か
ら86.4重量%までのインビトロ放出を提供する請求項1に記載の剤形。
【請求項10】
前記制御放出材料が、メタクリル酸エステル型重合体を含有する請求項1に記載の剤形
。
【請求項11】
剤形の経口投与の2から8時間後までに、前記患者におけるヒドロコドンのTmaxを提
供する請求項1に記載の剤形。
【請求項12】
剤形の経口投与の3から7時間後までに、前記患者におけるヒドロコドンのTmaxを提
供する請求項1に記載の剤形。
【請求項13】
剤形の経口投与の4から6時間後までに、前記患者におけるヒドロコドンのTmaxを提
供する請求項1に記載の剤形。
【請求項14】
15mgヒドロコドン・ビタートレートを含む剤形の経口投与に基づいて、投与の2か
ら8時間後までに、少なくとも8ng/mlのヒドロコドンの血漿中濃度を提供し、そし
て投与の12時間後に、少なくとも6ng/mlのヒドロコドン血漿血漿濃度を提供する
請求項1に記載の剤形。
【請求項15】
投与の3から7時間後までに、少なくとも8ng/mlのヒドロコドンの血漿血漿濃度
を提供する請求項14に記載の剤形。
【請求項16】
等量用量の即時放出ヒドロコドン照合処方のCmaxの50%未満であるヒドロコドンま
たはその塩のCmaxを提供する請求項1に記載の剤形。
【請求項17】
鎮痛効果を示す有効量のヒドロコドンまたは医薬上許容しうるその塩を包含する剤形で
あって、該ヒドロコドンまたは医薬上許容しうるその塩は、a)医薬上許容し得るマトリ
ックスに制御放出材料とともに組込まれるか、b)基質に包含され、またはその上にコー
トされ、さらに制御放出材料で塗布されるか、のいずれかであり、
該剤形は、ヒト患者に対する一日二回投与のために適し、
患者集団に対する第一の投与後に、同じ期間の放出速度の55%から85%までである
Tmaxから剤形の経口投与の12時間までの期間の吸収速度を提供し、
少なくとも12時間、治療的効果を提供し、かつ、
37℃での55分間の700mlの模擬胃液(SGF)中で、その後37℃での900
mlの模擬小腸液(SIF)に交換する100rpmでの米国薬局方バスケット法によっ
て測定されるときに4時間で、剤形からのヒドロコドンまたはその塩の51.7重量%か
ら67.4重量%までのインビトロ放出を提供する制御放出経口固形剤形。
【請求項18】
鎮痛効果を示す有効な量のヒドロコドンまたは医薬上許容しうるその塩を包含する剤形
であって、該ヒドロコドンまたは医薬上許容しうるその塩は、a)医薬上許容し得るマト
リックスに制御放出材料とともに組込まれるか、b)基質に包含され、またはその上にコ
ートされ、さらに制御放出材料で塗布されるか、のいずれかであり、
該剤形は、ヒト患者に対する一日二回投与のために適し、
患者集団に対する第一の投与後に、2から8時間まででインビボでのヒドロコドンのT
maxを提供し、そして0.55から0.85までのヒドロコドンの平均C12/Cmax比を提
供し、
少なくとも12時間についての治療効果を提供し、かつ、
37℃での55分間の700mlの模擬胃液(SGF)中で、その後37℃での900
mlの模擬小腸液(SIF)に交換する100rpmでの米国薬局方バスケット法によっ
て測定されるときに4時間で、剤形からのヒドロコドンまたはその塩の51.7重量%か
ら67.4重量%までのインビトロ放出を提供する制御放出経口固形剤形。
【請求項19】
鎮痛効果を示す有効な量のヒドロコドンまたは医薬上許容しうるその塩を包含する剤形
であって、該ヒドロコドンまたは医薬上許容しうるその塩は、a)医薬上許容し得るマト
リックスに制御放出材料とともに組込まれるか、b)基質に包含され、またはその上にコ
ートされ、さらに制御放出材料で塗布されるか、のいずれかであり、
該剤形は、ヒト患者に対する一日二回投与のために適し、かつ、
37℃での55分間の700mlの模擬胃液(SGF)中で、その後37℃での900
mlの模擬小腸液(SIF)に交換する100rpmでの米国薬局方バスケット法によっ
て測定されるときに4時間で、剤形からのヒドロコドンまたはその塩の51.7重量%か
ら67.4重量%までのインビトロ放出を提供する制御放出経口固形剤形。
【請求項20】
等量用量の即時放出ヒドロコドン対照処方のCmaxの50%未満であるヒドロコドンま
たはその塩のCmaxを提供する請求項17から19のいずれかに記載の剤形。
【請求項21】
37℃での55分間の700mlの模擬胃液(SGF)中で、その後37℃での900
mlの模擬小腸液(SIF)に交換する100rpmでの米国薬局方バスケット法によっ
て測定されるときに1時間で、剤形からのヒドロコドンまたはその塩の23.9重量%か
ら39.7重量%までのインビトロ放出を提供する請求項17から19のいずれかに記載
の剤形。
【請求項22】
37℃での55分間の700mlの模擬胃液(SGF)中で、その後37℃での900
mlの模擬小腸液(SIF)に交換する100rpmでの米国薬局方バスケット法によっ
て測定されるときに2時間で、剤形からのヒドロコドンまたはその塩の34.7重量%か
ら51.5重量%までのインビトロ放出を提供する請求項17から19のいずれかに記載
の剤形。
【請求項23】
37℃での55分間の700mlの模擬胃液(SGF)中で、その後37℃での900
mlの模擬小腸液(SIF)に交換する100rpmでの米国薬局方バスケット法によっ
て測定されるときに8時間で、剤形からのヒドロコドンまたはその塩の74.6重量%か
ら86.4重量%までのインビトロ放出を提供する請求項17から19のいずれかに記載
の剤形。
【請求項1】
鎮痛効果を示す有効量のヒドロコドンまたは医薬上許容しうるその塩を包含する剤形で
あって、該ヒドロコドンまたは医薬上許容しうるその塩は、a)医薬上許容し得るマトリ
ックスに制御放出材料とともに組込まれるか、b)基質に包含され、またはその上にコー
トされ、さらに制御放出材料で塗布されるか、のいずれかであり、
該剤形は、ヒト患者に対する一日二回投与のために適し、
患者集団に対する第一の投与後に、0.55から0.85までのヒドロコドンの平均C
12/Cmax比を提供し、
少なくとも12時間、治療的効果を提供し、かつ、
37℃での55分間の700mlの模擬胃液(SGF)中で、その後37℃での900
mlの模擬小腸液(SIF)に交換する100rpmでの米国薬局方バスケット法によっ
て測定されるときに4時間で、剤形からのヒドロコドンまたはその塩の51.7重量%か
ら67.4重量%までのインビトロ放出を提供する制御放出経口固形剤形。
【請求項2】
前記ヒドロコドンが、前記医薬上許容し得るマトリックス中に分散されている請求項1
に記載の剤形。
【請求項3】
前記マトリックスが、マルチパーティキュレートの形態である請求項2に記載の剤形。
【請求項4】
前記マルチパーティキュレートが、錠剤に圧縮されている請求項3に記載の剤形。
【請求項5】
前記マルチパーティキュレートが、医薬上許容しうるカプセルに分散されている請求項
3に記載の剤形。
【請求項6】
0.65から0.75までのヒドロコドンの平均C12/Cmax比を提供する請求項1に
記載の剤形。
【請求項7】
37℃での55分間の700mlの模擬胃液(SGF)中で、その後37℃での900
mlの模擬小腸液(SIF)に交換する100rpmでの米国薬局方バスケット法によっ
て測定されるときに1時間で、剤形からのヒドロコドンまたはその塩の23.9重量%か
ら39.7重量%までのインビトロ放出を提供する請求項1に記載の剤形。
【請求項8】
37℃での55分間の700mlの模擬胃液(SGF)中で、その後37℃での900
mlの模擬小腸液(SIF)に交換する100rpmでの米国薬局方バスケット法によっ
て測定されるときに2時間で、剤形からのヒドロコドンまたはその塩の34.7重量%か
ら51.5重量%までのインビトロ放出を提供する請求項1に記載の剤形。
【請求項9】
37℃での55分間の700mlの模擬胃液(SGF)中で、その後37℃での900
mlの模擬小腸液(SIF)に交換する100rpmでの米国薬局方バスケット法によっ
て測定されるときに8時間で、剤形からのヒドロコドンまたはその塩の74.6重量%か
ら86.4重量%までのインビトロ放出を提供する請求項1に記載の剤形。
【請求項10】
前記制御放出材料が、メタクリル酸エステル型重合体を含有する請求項1に記載の剤形
。
【請求項11】
剤形の経口投与の2から8時間後までに、前記患者におけるヒドロコドンのTmaxを提
供する請求項1に記載の剤形。
【請求項12】
剤形の経口投与の3から7時間後までに、前記患者におけるヒドロコドンのTmaxを提
供する請求項1に記載の剤形。
【請求項13】
剤形の経口投与の4から6時間後までに、前記患者におけるヒドロコドンのTmaxを提
供する請求項1に記載の剤形。
【請求項14】
15mgヒドロコドン・ビタートレートを含む剤形の経口投与に基づいて、投与の2か
ら8時間後までに、少なくとも8ng/mlのヒドロコドンの血漿中濃度を提供し、そし
て投与の12時間後に、少なくとも6ng/mlのヒドロコドン血漿血漿濃度を提供する
請求項1に記載の剤形。
【請求項15】
投与の3から7時間後までに、少なくとも8ng/mlのヒドロコドンの血漿血漿濃度
を提供する請求項14に記載の剤形。
【請求項16】
等量用量の即時放出ヒドロコドン照合処方のCmaxの50%未満であるヒドロコドンま
たはその塩のCmaxを提供する請求項1に記載の剤形。
【請求項17】
鎮痛効果を示す有効量のヒドロコドンまたは医薬上許容しうるその塩を包含する剤形で
あって、該ヒドロコドンまたは医薬上許容しうるその塩は、a)医薬上許容し得るマトリ
ックスに制御放出材料とともに組込まれるか、b)基質に包含され、またはその上にコー
トされ、さらに制御放出材料で塗布されるか、のいずれかであり、
該剤形は、ヒト患者に対する一日二回投与のために適し、
患者集団に対する第一の投与後に、同じ期間の放出速度の55%から85%までである
Tmaxから剤形の経口投与の12時間までの期間の吸収速度を提供し、
少なくとも12時間、治療的効果を提供し、かつ、
37℃での55分間の700mlの模擬胃液(SGF)中で、その後37℃での900
mlの模擬小腸液(SIF)に交換する100rpmでの米国薬局方バスケット法によっ
て測定されるときに4時間で、剤形からのヒドロコドンまたはその塩の51.7重量%か
ら67.4重量%までのインビトロ放出を提供する制御放出経口固形剤形。
【請求項18】
鎮痛効果を示す有効な量のヒドロコドンまたは医薬上許容しうるその塩を包含する剤形
であって、該ヒドロコドンまたは医薬上許容しうるその塩は、a)医薬上許容し得るマト
リックスに制御放出材料とともに組込まれるか、b)基質に包含され、またはその上にコ
ートされ、さらに制御放出材料で塗布されるか、のいずれかであり、
該剤形は、ヒト患者に対する一日二回投与のために適し、
患者集団に対する第一の投与後に、2から8時間まででインビボでのヒドロコドンのT
maxを提供し、そして0.55から0.85までのヒドロコドンの平均C12/Cmax比を提
供し、
少なくとも12時間についての治療効果を提供し、かつ、
37℃での55分間の700mlの模擬胃液(SGF)中で、その後37℃での900
mlの模擬小腸液(SIF)に交換する100rpmでの米国薬局方バスケット法によっ
て測定されるときに4時間で、剤形からのヒドロコドンまたはその塩の51.7重量%か
ら67.4重量%までのインビトロ放出を提供する制御放出経口固形剤形。
【請求項19】
鎮痛効果を示す有効な量のヒドロコドンまたは医薬上許容しうるその塩を包含する剤形
であって、該ヒドロコドンまたは医薬上許容しうるその塩は、a)医薬上許容し得るマト
リックスに制御放出材料とともに組込まれるか、b)基質に包含され、またはその上にコ
ートされ、さらに制御放出材料で塗布されるか、のいずれかであり、
該剤形は、ヒト患者に対する一日二回投与のために適し、かつ、
37℃での55分間の700mlの模擬胃液(SGF)中で、その後37℃での900
mlの模擬小腸液(SIF)に交換する100rpmでの米国薬局方バスケット法によっ
て測定されるときに4時間で、剤形からのヒドロコドンまたはその塩の51.7重量%か
ら67.4重量%までのインビトロ放出を提供する制御放出経口固形剤形。
【請求項20】
等量用量の即時放出ヒドロコドン対照処方のCmaxの50%未満であるヒドロコドンま
たはその塩のCmaxを提供する請求項17から19のいずれかに記載の剤形。
【請求項21】
37℃での55分間の700mlの模擬胃液(SGF)中で、その後37℃での900
mlの模擬小腸液(SIF)に交換する100rpmでの米国薬局方バスケット法によっ
て測定されるときに1時間で、剤形からのヒドロコドンまたはその塩の23.9重量%か
ら39.7重量%までのインビトロ放出を提供する請求項17から19のいずれかに記載
の剤形。
【請求項22】
37℃での55分間の700mlの模擬胃液(SGF)中で、その後37℃での900
mlの模擬小腸液(SIF)に交換する100rpmでの米国薬局方バスケット法によっ
て測定されるときに2時間で、剤形からのヒドロコドンまたはその塩の34.7重量%か
ら51.5重量%までのインビトロ放出を提供する請求項17から19のいずれかに記載
の剤形。
【請求項23】
37℃での55分間の700mlの模擬胃液(SGF)中で、その後37℃での900
mlの模擬小腸液(SIF)に交換する100rpmでの米国薬局方バスケット法によっ
て測定されるときに8時間で、剤形からのヒドロコドンまたはその塩の74.6重量%か
ら86.4重量%までのインビトロ放出を提供する請求項17から19のいずれかに記載
の剤形。
【図1】

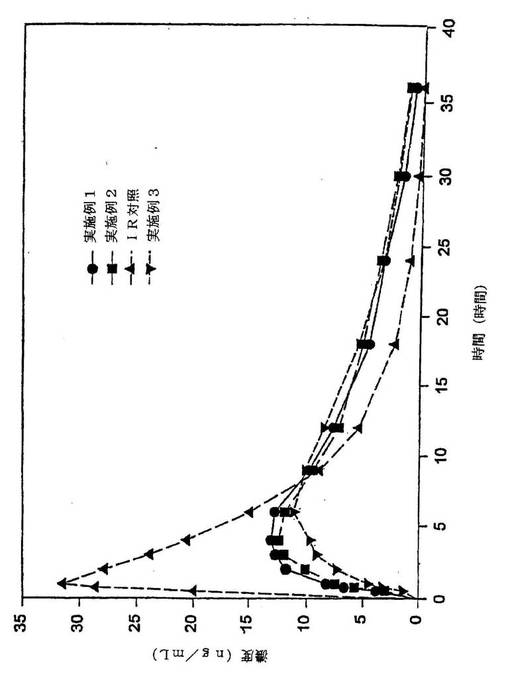
【図2】
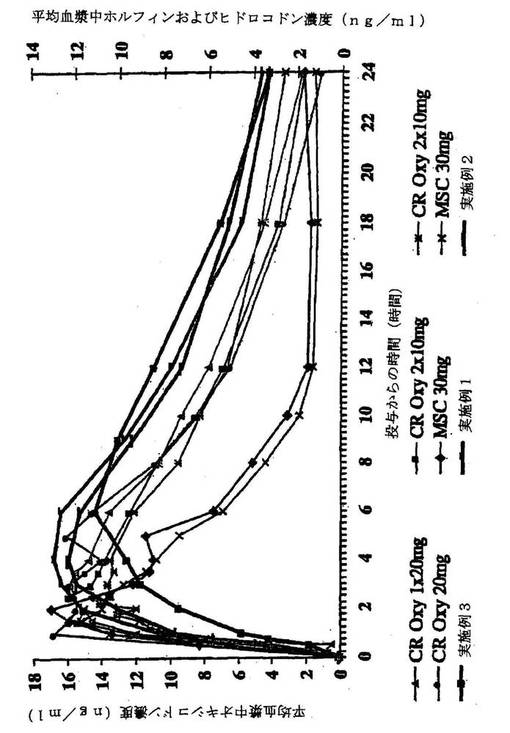
【図3】
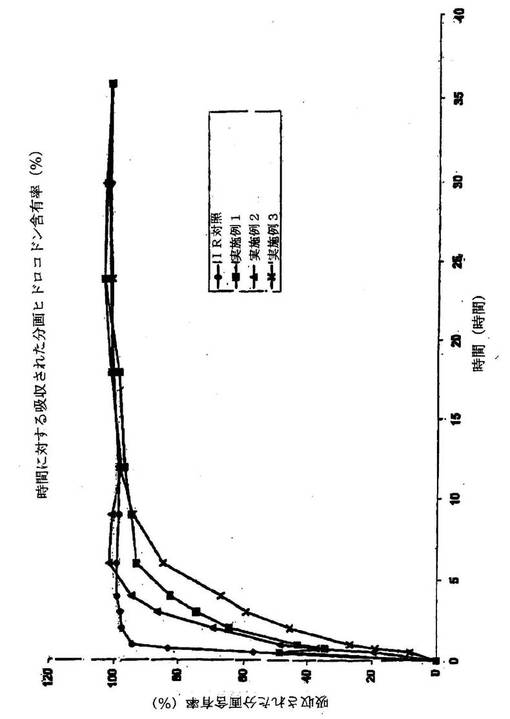
【図2】
【図3】
【公開番号】特開2012−36199(P2012−36199A)
【公開日】平成24年2月23日(2012.2.23)
【国際特許分類】
【出願番号】特願2011−205840(P2011−205840)
【出願日】平成23年9月21日(2011.9.21)
【分割の表示】特願2006−316067(P2006−316067)の分割
【原出願日】平成12年10月30日(2000.10.30)
【出願人】(599108792)ユーロ−セルティーク エス.エイ. (134)
【Fターム(参考)】
【公開日】平成24年2月23日(2012.2.23)
【国際特許分類】
【出願日】平成23年9月21日(2011.9.21)
【分割の表示】特願2006−316067(P2006−316067)の分割
【原出願日】平成12年10月30日(2000.10.30)
【出願人】(599108792)ユーロ−セルティーク エス.エイ. (134)
【Fターム(参考)】
[ Back to top ]