
前立腺癌の処置

本明細書には、アンドロゲン受容体媒介性の疾患又は疾病を処置するための化合物、そのような化合物を作る方法、そのような化合物を含む医薬組成物及び薬剤、およびそのような化合物を使用する方法が記載される。本発明は、前立腺癌の処置のための治療及び治療レジメンを提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本出願は、2009年8月7日に出願の米国仮特許出願第61/232,257号、及び2009年11月13日に出願の米国仮特許出願第61/261,265号の利益を主張するものであり、これらは各々、その全体における引用により本明細書に組み込まれる。
【0002】
<引用による組み込み>
この明細書で言及されるすべての公報、特許、及び特許出願は、個々の公報、特許、又は特許出願が、それぞれ、明確且つ個々に引用によって組み込まれると示されるのと同じ程度まで、引用によって本明細書に組み込まれる。
【背景技術】
【0003】
前立腺癌は、男性において最もよく見られる癌である。大多数の前立腺癌死亡は、従来のアンドロゲン遮断療法に反応性のない転移性疾患の進行が原因である。アンドロゲン遮断療法は、1940年代以降、前立腺癌を有する被験体における標準治療であった。アンドロゲン遮断にもかかわらず、ほとんどの被験体は、最終的に疾患の進行を経験する。長年、該疾患のこの後の相は、「ホルモン不応性前立腺癌」又は「アンドロゲン非依存性前立腺癌」と呼ばれた。それ以降、数年間のアンドロゲン遮断療法の後に出現する前立腺癌は、アンドロゲンに依存したままであることが明らかになった。生存した前立腺癌細胞は、(副腎から発現した)低レベルの循環するアンドロゲンを移入し、これらの低レベルのテストステロンにかなり感応的になり、及び実際に、前立腺癌細胞自体内のテストステロンを合成する能力を得た。前立腺癌のこの段階は、現在、「去勢抵抗性前立腺癌」又はCRPCと称される。
【発明の概要】
【課題を解決するための手段】
【0004】
幾つかの実施形態において、本発明は、化合物(1):
【0005】
【化1】

【0006】
又はその薬学的に許容可能な塩、N-オキシド、活性代謝物質、プロドラッグ、又は溶媒和物を含む医薬組成物を熟考し、
式中、化合物(1)は、約:a. 13.1°及び14.1°;b. 17.2°、18.5°及び19.1°、及び随意に約16.2°、及び29.6°;c. 12.5°、14.8°、及び25.5°;又はd. 14.4°、16.1°、19.1°及び19.3°の角度 2-シータ(2θ)において表される、特性ピークを有する粉末X線回折パターンによって特徴付けられた、微粉化結晶形態である。
【0007】
幾つかの実施形態において、本発明は、被験体における前立腺癌の処置を提供する方法を熟考し、該方法は、化合物(1):
【0008】
【化2】
【0009】
又はその薬学的に許容可能な塩、N-オキシド、活性代謝物質、プロドラッグ、又は溶媒和物を含む医薬組成物を、それを必要とする又は望んでいる被験体に投与する工程を含み、
式中、化合物(1)は、約:a. 13.1°及び14.1°;b. 17.2°、18.5°及び19.1°、及び随意に約16.2°、及び29.6°;c. 12.5°、14.8°、及び25.5°;又はd. 14.4°、16.1°、19.1°及び19.3°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられた、微粉化結晶形態である。
【図面の簡単な説明】
【0010】
【図1】図1は、化合物(1)フォームIの代表的な粉末X線回折図(XRPD)である。
【図2】図2は、フォームIIを含む化合物(1)のいくつかの形態の代表的な粉末X線回折図(XRPD)である。
【図3】図3は、標準条件下及び40℃で1週間後の、化合物(1)フォームIIIの、代表的な粉末X線回折図(XRPD)である。
【図4】図4は、上昇する温度とともに、パターン5が他のパターンに変換することを示す、化合物(1)パターン5の代表的な粉末X線回折図(XRPD)である。
【図5】図5は、加熱によってフォーム1に戻ることを示す、化合物(1)パターン2の代表的な粉末X線回折図(XRPD)である。
【図6】図6は、様々な温度での非晶質の化合物(1)の代表的な粉末X線回折図(XRPD)である。
【図7】図7は、化合物(1)フォームIの代表的なサーモグラムである。
【図8】図8は、アンドロゲン受容体に結合する(化合物5と記載される)化合物(1)を示す表である。
【図9】図9は、アンドロゲン受容体タンパク質のレベルを示すウエスタンブロットである。
【図10】図10は、腫瘍異種移植片のサイズを示す線グラフである。
【図11】図11は、本明細書に記載される臨床研究の様々な段階の過程を図表で示す。
【図12】図12は、研究の被験体のための手順のスケジュールを表にする。
【図13】図13は、臨床試験の被験体における時間に対する化合物(1)の血清中濃度を示す。コホート群は、図の上部にわたってリストされる。
【発明を実施するための形態】
【0011】
定義
有害事象:本明細書に使用されるような用語「有害事象」は、その技術分野で理解される意味を有し、医薬品を投与された、被験体又は臨床試験の被験体における任意の好ましくない医学的出来事を指す。有害事象は、必ずしも、投与される処置と因果関係がある必要はない。
【0012】
副作用:本明細書に使用されるような用語「有害反応」は、その技術分野で理解される意味を有し、任意の投与量に関連する医薬品に対する任意の有害で意図されない反応を指す。
【0013】
併用療法:用語「併用療法」は、本明細書に使用されるように、2つ以上の異なる医薬品が、レジメンを重複させる際に投与される、これらの状況を指し、その結果、被験体は両方の薬剤に同時に曝される。
【0014】
投薬レジメン:用語「投薬レジメン」は、本明細書に使用されるように、期間によって分けられて個々に投与される一連の単位用量(典型的には1以上)を指す。特定の医薬品のための推奨される一連の投薬(すなわち、量、タイミング、投与経路など)は、その投薬レジメンを構成する。
【0015】
開始:本明細書に使用されるように、投薬レジメンに適用される時の用語「開始」は、以前に医薬品を受けたことのない被験体への、医薬品の最初の投与を指すように使用され得る。代わりに又はさらに、用語「開始」は、被験体の治療の間の医薬品の特定の単位用量の投与を指すように使用され得る。
【0016】
医薬品:本明細書に使用されるように、句「医薬品」は、被験体に投与される時に、治療効果を有している及び/又は所望の生物学的及び/又は薬理学的な効果を誘発する、任意の薬剤を指す。
【0017】
薬学的に許容可能なエステル:本明細書に使用されるように、用語「薬学的に許容可能なエステル」は、インビボで加水分解し、ヒトの身体において容易に分解し、親化合物又はその塩を残すエステルを含む、エステルを指す。
【0018】
重篤有害事象:用語「重篤有害事象」は、本明細書に使用されるように、その技術分野で理解される意味を有し、任意の投与量で、例えば、死に至る、生命を脅かす、被験体の入院(又は既存の入院の延長)を必要とする、(正常な生命機能を実行する被験体の能力の相当な破壊として定義される)持続性の又は重大な障害又は不能という結果になるなどの、任意の好ましくない医学的出来事を指す。幾つかの実施形態において、重篤有害事象は、その用語が、例えば、21 CFR § 310.305(b)において定義されるように、米国食品医薬品局 (the United States Food and Drug Administration)によって使用されるように、「重篤有害薬物経験」であり、ここで、重篤有害事象が、以下の結果のいずれかにつながる、任意の投与量で生じる有害薬物経験であると述べている:死亡、生命を脅かす有害薬物経験、被験体の入院又は既存の入院の延長、持続性の又は重大な障害/不能,又は先天異常/出産時欠損。死に至り得ない、生命を脅かし得ない、入院を必要とし得ない重要な医学的事象は、適切な医学的判断に基づいて、それらが被験体を危険にさらし得る、及びこの定義においてリストされる結果の1つを防ぐために医学的又は外科的な介入を必要とし得る時に、有害薬物経験と考えられ得る。そのような医学的事象の例は、緊急処置室又は家で集中治療を必要とするアレルギー性気管支痙攣、被験体の入院という結果にはならない血液疾患又は痙攣、または薬物依存又は薬物乱用の進行を含む。
【0019】
感受性:用語「〜に感受性がある(susceptible to)」は、一般集団において観察されるより、特定の疾患又は障害、又はその症状を進行させる、(典型的に、遺伝性素因、環境要因、個人歴、又はそれらの組み合わせに基づいた)高いリスクを有する個体に言及するために本明細書において使用される。
【0020】
治療上有効な量:用語、医薬品又は薬剤の組み合わせの「治療上有効な量」は、任意の治療に適用可能な合理的なベネフィット・リスク比で、処置される被験体に対する治療効果を与える薬剤(複数可)の量を指すように意図される。治療効果は、客観的であり得るか(すなわち、幾つかの試験又はマーカーによって測定可能である)、又は主観的であり得る(すなわち、被験体は効果の指標を与える、又は効果を感じる)。治療上有効な量は、通常、複数の単位用量を含み得る投薬レジメンにおいて投与される。任意の特定の医薬品に関して、治療上有効な量(及び/又は有効な投薬レジメン内の適切な単位用量)は、例えば、投与の経路、他の医薬品との組み合わせに依存して、変化し得る。また、任意の特定の被験体のための具体的な治療上有効な量(及び/又は単位用量)は、処置されている障害及びその障害の重症度、用いられる具体的な医薬品の活性、用いられる具体的な組成物、被験体の年齢、体重、健康状態、性別及び食事、投与の時間、投与の経路、用いられる具体的な医薬品の排泄又は代謝の速度、処置の期間、及び医学的技術分野において周知であるような因子を含む、様々な因子に依存し得る。
【0021】
処置:本明細書に使用されるように、用語「処置(treatment)」は(「処置する(treat)」又は「処置すること(treating)」も)、特定の疾患、障害、及び/又は疾病の1以上の症状又は特徴を、部分的に又は完全に緩和する、寛解する、和らげる、阻害する、その発症を遅らせる、重症度を低下させる、及び/又は発生率を減らす、医薬品の任意の投与を指す。そのような処置は、関連する疾患、障害、及び/又は疾病の徴候を示さない被験体、及び/又は疾患、障害、及び/又は疾病の初期の徴候のみを示す被験体の処置であり得る。代わりに又はさらに、そのような処置は、関連する疾患、障害及び/又は疾病の1以上の確立された徴候を示す被験体の処置であり得る。
【0022】
単位用量:用語「単位用量(unit dose)」又は「投与量(dose)」は、本明細書に使用されるように、典型的に投薬レジメンとの関連で、医薬品の別々の投与を指す。標準化学用語の定義は、Carey and Sundberg "ADVANCED ORGANIC CHEMISTRY 4TH ED." Vols. A (2000) and B (2001), Plenum Press, New Yorkを含む、参考資料において見受けられ得、その全体における引用によって本明細書に組み込まれる。特に指示がない限り、当該技術分野の技術内で、質量分析、NMR、HPCL、タンパク質化学、生化学、組換え型DNA技術及び薬理学の従来の方法が用いられる。
【0023】
例示的な生物学的活性
<アンドロゲン受容体(AR)>
アンドロゲンは、標的組織の細胞内で、特定の受容体、アンドロゲン受容体(AR)に結合する。ARは、身体の多数の組織において発現され、それを介してテストステロン(T)及びジヒドロテストステロン(DHT)などの内因性のアンドロゲンリガンドの生理学的効果に加え、病態生理学的効果も現れる受容体である。構造上、ARは、3つの主な機能ドメイン:リガンド結合ドメイン(LBD)、DNA結合ドメイン、及びアミノ末端ドメインからなる。ARに結合し、内因性のARリガンドの効果を模倣する化合物は、ARアゴニストと呼ばれ、一方で内因性のARリガンドの効果を阻害する化合物は、ARアンタゴニストと称される。受容体へのアンドロゲンの結合は、受容体を活性化し、標的遺伝子に隣接しているDNA結合部位へそれを結合させる。そこから、それは、遺伝子の発現を規制するために、コアクチベータータンパク質及び塩基性の転写因子と相互に作用する。したがって、その受容体を介して、アンドロゲンは細胞における遺伝子発現の変化を引き起こす。これらの変化は、最終的に、標的組織の生理機能において目に見える、細胞の代謝的産生(output)、分化又は増殖という結果を引き起こす。前立腺において、アンドロゲンは、アンドロゲン感受性組織の細胞質内に存在するARに結合することにより、前立腺組織及び前立腺癌細胞の成長を刺激する。
【0024】
選択的にARを調節する化合物は、限定されないが、前立腺癌、良性前立腺肥大症、女性型多毛症、脱毛症、神経性食欲不振、乳癌、ざ瘡、骨疾患、造血状態(hematopoietic conditions)、神経筋疾患、リウマチ疾患などの筋骨格系の疾病、癌、AIDS、悪液質を含む、様々な疾患、疾病、及び癌の処置又は予防において、男性避妊法において用いられるホルモン補充療法(HRT)にとって、男性の性能強化にとって、男性の生殖状態にとって、及び一次又は二次の男性性腺機能低下症にとって臨床的に重要である。
【0025】
<去勢抵抗性前立腺癌>
内因性のホルモン(例えば、テストステロン)の作用を遮断する薬剤(抗アンドロゲン)は、前立腺癌の処置に高度に有効であり、日常的に使用される(アンドロゲン除去療法)。最初は腫瘍成長を抑えるに有効であるが、これらのアンドロゲン除去療法は、結局、ほとんどすべての被験体において失敗し、「去勢抵抗性前立腺癌」(「CRPC」)につながる。すべてではないが、ほとんどの前立腺癌細胞は、最初に、アンドロゲン除去療法に反応する。しかしながら、時間とともに、前立腺癌細胞の生存する個体群は、アンドロゲン除去療法によって生じた選択圧に反応したが、今はそれに対して反応がない(refractory)ために出現する。原発性癌が、利用可能な療法に反応がないだけではなく、癌細胞はまた、原発腫瘍から離れ、血流を移動し得、疾患が離れた部位(特に骨)に広がる。他の効果の中で、これは著しい疼痛及び更なる骨の脆弱性を引き起こす。
【0026】
CRPC細胞は、利用可能なままである細胞内のアンドロゲンに対する反応を強めるための、少なくとも3つの異なる経路を増幅させることによる、低レベルの循環するアンドロゲンによって特徴付けられた環境において生存することが予期される。これらは:(1)ARコピー数を増加させ、その結果、内科的去勢療法によって誘発された低レベルの循環するアンドロゲンに対する、細胞の感受性を増加させる、ARの発現のアップレギュレーション、(2)アンドロゲン遮断療法の後に細胞に残るアンドロゲンの移入(importation)に関係する酵素の発現の増加、(3)CRPC細胞がその細胞自体のアンドロゲンを合成することを可能にする、ステロイド産生を規制する遺伝子の発現の増加を含む。ステロイド産生経路における重要な酵素は、チトクロームC17α-ヒドロキシラーゼ/C17,20-リアーゼ(CYP17)であり、該酵素は、副腎、精巣、及び前立腺におけるアンドロゲン産生を制御する。
【0027】
本明細書には、特定の実施形態において、限定されないが、前立腺癌、良性前立腺肥大症、女性型多毛症、脱毛症、神経性食欲不振、乳癌、ざ瘡、骨疾患、造血状態、神経筋疾患、リウマチ疾患などの筋骨格系の疾病、癌、AIDS、悪液質を含む、アンドロゲン受容体媒介性の疾患又は疾病を処置するための、男性避妊法において用いられるホルモン補充療法(HRT)のための、男性の性能強化のための、男性の生殖状態のための、及び一次又は二次の男性性腺機能低下症のための化合物、そのような化合物を作る方法、そのような化合物を含む医薬組成物及び薬剤、およびそのような化合物を使用する方法が記載される。幾つかの実施形態において、アンドロゲン受容体媒介性の疾患又は疾病は、前立腺癌である。幾つかの実施形態において、前立腺癌は、去勢抵抗性前立腺癌である。
【0028】
幾つかの実施形態において、本発明は、アンドロゲン生合成を減少させる、アンドロゲン受容体シグナル伝達を減少させる、及びアンドロゲン受容体感受性を減少させる、化合物、そのような化合物を含む医薬組成物、及び薬剤、及びそのような化合物を使用する方法を提供する。
【0029】
1つの態様において、化合物、そのような化合物を含む医薬組成物及び薬剤、およびそのような化合物を使用する方法は、アンドロゲン生合成を減少させる。幾つかの実施形態において、本明細書に開示される化合物は、アンドロゲン産生を制御する酵素の活性を阻害する。特定の実施形態において、本明細書に開示される化合物は、チトクロームC17α-ヒドロキシラーゼ/C17,20-リアーゼ(CYP17)の活性を阻害する。
【0030】
1つの態様において、化合物、そのような化合物を含む医薬組成物及び薬剤、およびそのような化合物を使用する方法は、アンドロゲン受容体シグナル伝達を減少させる。幾つかの実施形態において、本明細書に開示される化合物は、ARに結合し、テストステロン結合の競合的インヒビターである。
【0031】
1つの態様において、化合物、そのような化合物を含む医薬組成物及び薬剤、およびそのような化合物を使用する方法は、アンドロゲン受容体感受性を減少させる。幾つかの実施形態において、本明細書に開示される化合物は、細胞内のARタンパク質の含量を減少させ、低レベルのアンドロゲン増殖シグナルによって持続される細胞の能力を低減する。
【0032】
典型的な化合物
化合物(1)としても記載される、式(1)の化合物、その薬学的に許容可能な塩、薬学的に許容可能なN-オキシド、薬学的に活性な代謝物質、薬学的に許容可能なプロドラッグ、薬学的に許容可能な多形体及び薬学的に許容可能な溶媒和物は、ステロイドホルモン核内受容体の活性を調節し、それゆえ、アンドロゲン受容体媒介性の疾患又は疾病を処置するのに有用である。
【0033】
【化3】
【0034】
化合物の典型的な合成
式(1)の化合物(化合物(1)又は3-β-ヒドロキシ-17-(1H ベンズイミダゾール-1-イル)アンドロスタ-5,16-ジエンとしても記載される)は、本明細書に記載される方法と組み合わせて、当業者に公知の標準の合成技術を使用して、又は当該技術分野に公知の方法を使用して合成され得る。当業者が理解するであろうように、本明細書に提示される溶媒、温度及び反応条件は、当業者の慣習及び知識によって変えられ得る。
【0035】
化合物(1)の合成に使用される出発物質は、Aldrich Chemical Co. (Milwaukee, Wis.)、Sigma Chemical Co. (St. Louis, Mo.)などの、商業的供給源から得られ得るか、又は出発物質は合成され得る。本明細書に記載される化合物、及び異なる置換基を有する他の関連する化合物は、記載されるように、例えば、March, ADVANCED ORGANIC CHEMISTRY 4th Ed., (Wiley 1992)、Carey and Sundberg, ADVANCED ORGANIC CHEMISTRY 4th Ed., Vols. A and B (Plenum 2000, 2001)、及びGreen and Wuts, PROTECTIVE GROUPS IN ORGANIC SYNTHESIS 3rd Ed., (Wiley 1999)におけるような、当業者に公知の技術及び物質を使用して合成され得る(これら全ては、それらの全体において引用によって組み込まれる)。本明細書に開示される化合物の調製のための一般的な方法は、当該技術分野において公知の反応に由来し、該反応は、本明細書に提供されるように、式において見られる様々な成分を導入するために、当業者によって理解されるように、適切な試薬及び条件を使用することによって変更され得る。
【0036】
化合物(1)は、遊離塩基の形態の化合物を、限定されないが、塩化水素酸、臭化水素酸、硫酸、硝酸、リン酸、メタリン酸などの無機酸、および酢酸、プロピオン酸、ヘキサン酸、シクロペンタンプロピオン酸、グリコール酸、ピルビン酸、乳酸、マロン酸、コハク酸、リンゴ酸、マレイン酸、フマル酸、p-トルエンスルホン酸、酒石酸、トリフルオロ酢酸、クエン酸、安息香酸、3-(4-ヒドロキシベンゾイル)安息香酸、桂皮酸、マンデル酸、アリールスルホン酸、メタンスルホン酸、エタンスルホン酸、1,2-エタンジスルホン酸、2-ヒドロキシエタンスルホン酸、ベンゼンスルホン酸、2-ナフタリンスルホン酸、4-メチルビシクロ-[2.2.2]オクタ-2-エン-1-カルボン酸、グルコペプトン酸、4,4'-メチレンビス-(3-ヒドロキシ-2-エン-1-カルボン酸)、3-フェニルプロピオン酸、トリメチル酢酸、三級ブチル酢酸、ラウリル硫酸、グルコン酸、グルタミン酸、ヒドロキシナフトエ酸、サリチル酸、ステアリン酸、及びムコン酸などの有機酸を含む、薬学的に許容可能な無機酸又は有機酸と反応させることにより、(薬学的に許容可能な塩のタイプである)薬学的に許容可能な酸付加塩として調製され得る。
【0037】
化合物(1)は、プロドラッグとして調製され得る。プロドラッグは、一般的に薬物前駆体であり、この薬物前駆体は、被験体への投与及びその後の吸収に続いて、代謝経路による変換などの、幾つかの過程を介して、活性な、又はより活性な種へと変換される。プロドラッグの中には、活性を和らげ、及び/又は薬物に溶解度又は幾つかの他の特性を与える、プロドラッグ上に存在する化学基を有するものもある。一旦、化学基がプロドラッグから開裂及び/又は変更されると、活性薬物が生じる。幾つかの状況において、プロドラッグは親薬物よりも投与しやすいことがあるため、しばしば有用である。プロドラッグは、例えば、経口投与によって生物学的に利用可能である得る一方で、親薬物はそうではない。プロドラッグはまた、親薬物以上に医薬組成物において改善された溶解度を有し得る。プロドラッグの限定されない例は、式(I)の誘導体であり、これは親水性エステル(「プロドラッグ」)として投与され、改善された水溶性に対して有益である胃腸管における吸収を促進するが、その後、代謝的にカルボン酸及び活性な実体、化合物(1)に加水分解される。プロドラッグの更なる例は、化合物(1)の水酸基に結合した短鎖ペプチドであり、ここで、ペプチドは代謝され、化合物(1)を提供する。
【0038】
プロドラッグは、部位特異的な組織への薬物輸送を増強させる修飾因子として使用するための、可逆的な薬物誘導体として考案され得る。現在までのプロドラッグの考案は、水が主な溶媒である領域を標的とするための、治療用化合物の有効な水溶性を高めるためのものであった。例えば、Fedorak et al., Am. J. Physiol., 269:G210-218 (1995); McLoed et al., Gastroenterol, 106:405-413 (1994)、Hochhaus et al., Biomed. Chrom., 6:283-286 (1992)、J. Larsen and H. Bundgaard, Int. J. Pharmaceutics, 37, 87 (1987)、J. Larsen et al., Int. J. Pharmaceutics, 47, 103 (1988)、Sinkula et al., J. Pharm. Sci., 64:181-210 (1975)、T. Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems, Vol. 14 of the A.C.S. Symposium Series、及びEdward B. Roche, Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press, 1987を参照し、それら全体において、すべてが本明細書に組み込まれる。
【0039】
更に、化合物(1)のプロドラッグ誘導体は、 当業者に公知の方法によって調製され得る(例えば、更なる詳細については、Saulnier et al., (1994), Bioorganic and Medicinal Chemistry Letters, Vol. 4, p. 1985を参照)。プロドラッグがインビボで代謝され、本明細書に述べられるような誘導体を生成する、本明細書に記載される化合物のプロドラッグの形態は、請求の範囲内に含まれる。実際、本明細書に記載される化合物の幾つかは、別の誘導体又は活性化合物に対するプロドラッグであり得る。
【0040】
化合物(1)の芳香環部分の部位は、様々な代謝反応を起こしやすく、それ故、芳香環構造上の適切な置換基、例えば、ハロゲンを取り込むことにより、この代謝経路を減少、最小化又は除去することが可能である。
【0041】
化合物(1)を製造する様々な方法が熟考され、以下の記載は限定しない例として提供される。幾つかの実施形態において、以下の化学反応の1以上は、不活性雰囲気、例えば、窒素、アルゴンにおいて行われる。幾つかの実施形態において、反応の温度が監視される。幾つかの実施形態において、反応は、HPLC又はTLCによって監視される。幾つかの実施形態において、反応のpHが監視される。幾つかの実施形態において、反応の温度が制御される。幾つかの実施形態において、産物の純度はHPLCによって測定される。幾つかの実施形態において、実験は、小規模、中規模、大規模、分析的規模、又は製造規模で実行される。幾つかの実施形態において、産物は、シリカゲル及びセライトの1以上を含むパッドを介するろ過によって明確にされる。
【0042】
幾つかの実施形態において、合成は、大規模で行われる。幾つかの実施形態において、大規模は、約1から約10kgまでの規模を含む。幾つかの実施形態において、合成は、製造規模で行われる。幾つかの実施形態において、製造規模は、約10kgより大きい規模を含む。幾つかの実施形態において、製造規模は、約10から約1,000kgまでの規模を含む。幾つかの実施形態において、製造規模は、約10から約100kgまでの規模を含む。幾つかの実施形態において、製造規模は、約10から約50kgまでの規模を含む。幾つかの実施形態において、製造規模は、約33.4kgの規模を含む。
【0043】
幾つかの実施形態において、製造規模の合成を計画するか又は行うために使用される情報を集めるために、実験は小規模で行われる。幾つかの実施形態において、より小規模で得られた結果は、製造規模で再生可能であると予期される。幾つかの実施形態において、より小規模で得られた結果は、製造規模で再生可能であると予期されない。幾つかの実施形態において、製造規模で得られた収量は、より小規模で得られた収量より多い。幾つかの実施形態において、製造規模で得られた収量は、より小規模で得られた収量より少ない。
【0044】
【化4】
【0045】
1つの実施形態において、溶媒中の式iの化合物の溶液は調製される。その後、式iiの化合物は、溶液に接触され、結果として生じる混合物は、式iiiの化合物を提供するのに十分な期間、塩基の存在下で加熱される。幾つかの実施形態において、期間は、約1時間、約2時間、約4時間、約8時間、約12時間、又は約24時間である。幾つかの実施形態において、時間は、約1時間から約24時間までである。幾つかの実施形態において、塩基は、炭酸リチウム、炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム、リン酸ナトリウム、又はリン酸カリウムを含む。幾つかの実施形態において、溶媒は、DMFを含む。幾つかの実施形態において、温度は、約50℃、約70℃、約100℃、約150℃、又は還流条件を維持するのに有効な温度である。幾つかの実施形態において、温度は、約50℃から約200℃までである。式iiiの化合物は、反応混合物から分離され、当業者に公知の任意の方法によって精製され得る。このような方法は、限定されないが、水性混合物を反応混合物へ注ぐ工程を含み、それによって、固形物として化合物iiiを沈殿させる。式iiiの分離した化合物は、当業者に公知の任意の方法によって随意に精製され得る。このような方法は、限定されないが、水による粉砕を含む。
【0046】
【化5】
【0047】
1つの実施形態において、溶媒中の式iiiの化合物の溶液は調製され、溶液は、式ivの化合物を提供するのに十分な期間、触媒と接触される。幾つかの実施形態において、期間は、約1時間、約2時間、約4時間、約8時間、約12時間、又は約24時間である。幾つかの実施形態において、時間は、約1時間から約24時間までである。幾つかの実施形態において、触媒は、炭素上のパラジウム、炭素上のプラチナ、遷移金属塩又は遷移金属錯体を含む。幾つかの実施形態において、溶媒は、N-メチルピロリドンである。幾つかの実施形態において、温度は、約50℃、約70℃、約100℃、約150℃、約190℃、約200℃、又は還流条件を維持するのに有効な温度である。幾つかの実施形態において、温度は、約50℃から約250℃までである。式ivの化合物は、反応混合物から分離され、当業者に公知の任意の方法によって精製され得る。このような方法は、限定されないが、インラインろ過を含む。式ivの分離した化合物は、当業者に公知の任意の方法によって随意に精製さ得る。
【0048】
【化6】
【0049】
1つの実施形態において、溶媒中の式ivの化合物の溶液は調製され、溶液は、式vの化合物(すなわち、化合物(1))を提供するのに十分な期間、塩基と接触される。幾つかの実施形態において、期間は、約1時間、約2時間、約4時間、約8時間、約12時間、又は約24時間である。幾つかの実施形態において、時間は、約1時間から約24時間までである。幾つかの実施形態において、塩基は、水酸化リチウム、水酸化ナトリウム、水酸化カリウム、ナトリウムメトキシド、カリウムメトキシド、ナトリウムエトキシド、カリウムエトキシド、炭酸リチウム、炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム、リン酸ナトリウム、又はリン酸カリウムを含む。幾つかの実施形態において、溶媒は、水、メタノール、エタノール、2-プロパノール、t-ブタノール、又はそれらの混合物を含む。幾つかの実施形態において、溶媒は、メタノールを含み、塩基はナトリウムメトキシドを含む。幾つかの実施形態において、温度は、約35℃、約50℃、約70℃、約100℃、又は還流条件を維持するのに有効な温度である。幾つかの実施形態において、温度は、約25℃から約100℃までである。式vの化合物は、反応混合物から分離され、当業者に公知の任意の方法によって精製され得る。このような方法は、限定されないが、抽出を含む。式vの分離した化合物は、当業者に公知の方法によって随意に精製され得る。このような方法は、限定されないが、粉砕を含む。
【0050】
化合物(1)の典型的な形態
薬学的に許容可能な塩に対する言及は、その溶媒付加形態又は結晶形態、特に溶媒和物又は多形体を含むことを理解されたい。溶媒和物は、定比又は不定比量の溶媒のいずれかを含み、水、エタノールなどの薬学的に許容可能な溶媒での結晶化の過程の間に形成され得る。水和物は、溶媒が水である時に形成され、又はアルコラートは、溶媒がアルコールの時に形成される。化合物(1)の溶媒和物は、本明細書に記載される過程の間に、都合よく調製又は形成され得る。ほんの一例ではあるが、化合物(1)の水和物は、限定されないが、ジオキサン、テトラヒドロフラン、又はメタノールなどを含む、有機溶媒を用いて、水性/有機溶媒混合物からの再結晶によって都合よく調製され得る。さらに、本明細書に提供される化合物は、溶媒和形態と同様、非溶媒和形態でも存在し得る。一般的に、溶媒和形態は、本明細書に提供される化合物及び方法の目的のため、非溶媒和形態と同等であると考慮される。
【0051】
化合物(1)は、多形体としても知られる結晶形態を含む。多形体は、化合物の同じ元素組成の異なる結晶充填配置を有する。多形体は通常、異なるX線回折パターン、赤外線スペクトル、融点、密度、硬度、結晶形、光学及び電気学上の特性、安定性、及び溶解度を有する。再結晶溶媒、結晶化の速度、及び保管温度等の様々な要因は、支配的な単結晶形態を引き起こし得る。
【0052】
1つの実施形態において、化合物(1)は、フォームIとして示される結晶形態であり、約13.0°、14.6°、16.3°、17.6°、及び19.0°±0.2°2θでの粉末X線反射によって特徴付けられる。幾つかの実施形態において、フォームIは、約11.8°、20.2°、22.9°、及び25.4°±0.2°2θでの更なるピーク粉末X線反射によって特徴付けられる。図1は、化合物(1)形態Iの代表的な粉末X線回折図(XRPD)である。幾つかの実施形態において、フォームIは、一例として、溶媒中の非晶質の化合物(1)を溶解し、溶媒を蒸発させることから得られる。幾つかの実施形態において、非晶質の化合物(1)は、溶媒中に懸濁され、8時間のサイクルで50°/室温で代替的にインキュベートされる。結果として生じる懸濁液中の固形物は、ろ過され分析される。溶媒は、限定されないが、ヘプタン、ジオキサン、tert-ブチルメチルエーテル、酢酸ブチル、酢酸イソプロピル、プロパノール、テトラヒドロフラン、ジクロロメタン、メタノール、ニトロメタン、及び水を含む。
【0053】
1つの態様において、化合物(1)は、フォームIIとして示される結晶形態であり、約13.1°及び14.1°±0.2°2θでの粉末X線反射によって特徴付けられる。図2は、化合物(1)フォームIIの代表的な粉末X線回折図(XRPD)を含む。
【0054】
1つの態様において、化合物(1)は、フォームIIIとして示される結晶形態であり、約17.2°、18.5°、及び19.1°±0.2°2θでの粉末X線反射によって特徴付けられる。幾つかの実施形態において、フォームIIIは、約16.2°及び29.6°±0.2°2θでの更なるピーク粉末X線反射によって特徴付けられる。図3は、化合物(1)フォームIIIの代表的な粉末X線回折図(XRPD)である。
【0055】
1つの態様において、化合物(1)は、パターン5として示される結晶性水和物であり、約12.5°、14.8°、及び25.5°±0.2°2θでの粉末X線反射によって特徴付けられる。幾つかの実施形態において、パターン5は、約40%から約90%までの間の相対湿度の水分を有している。図4は、化合物(1)パターン5の代表的な粉末X線回折図(XRPD)である。
【0056】
1つの態様において、化合物(1)は、パターン2として示される結晶性クメン溶媒和物であり、約14.4°、16.1°、19.1°及び19.3°±0.2°2θでの粉末X線反射によって特徴付けられる。幾つかの実施形態において、パターン2は、約20.4°、23.6°及び28.7°±0.2°2θでの更なるピーク粉末X線反射によって特徴付けられる。幾つかの実施形態において、パターン2は、約20%から約40%までの間のクメン溶媒含有量を有している。他の実施形態において、パターン2は、約25%のクメン溶媒含有量を有している。図5は、化合物(1)パターン2の代表的な粉末X線回折図(XRPD)である。
【0057】
幾つかの実施形態において、化合物(1)は、実質的に、1つの結晶形態又はパターンである。本明細書に使用されるような「実質的に」は、80%より高いことを指す。幾つかの実施形態において、化合物(1)は、2つの形態又はパターンの混合物である。幾つかの実施形態において、化合物(1)は、2つ以上の形態又はパターンの混合物である。2つの形態又はパターンの混合物を有する例において、混合比は、約1:100から100:1までであり得る。他の実施形態において、化合物(1)は、非晶質の形態をさらに含む。特定の実施形態において、化合物(1)は、実質的に非晶質である。
【0058】
典型的な医薬組成物/製剤
本明細書に使用されるような、医薬組成物は、担体、安定化剤、希釈剤、分散剤、懸濁化剤、増粘剤、及び/又は賦形剤などの他の化学成分との、化合物(1)の混合物を指す。医薬組成物は、有機体への化合物の投与を促進する。化合物(1)を含む医薬組成物は、限定されないが、以下のものを含む、当該技術分野に公知の、任意の従来の形態及び経路による医薬組成物として治療上有効な量で投与され得る:静脈内、経口、直腸、エアロゾル、非経口、眼、肺、経皮、膣、耳、鼻、及び局所の投与。
【0059】
当業者は、全身よりもむしろ局所に、例えば、器官への直接的な化合物の注入を介して、しばしばデポー製剤又は持続放出性製剤において化合物を投与し得る。さらに、当業者は、標的薬物送達システムにおいて、例えば、臓器特異性抗体でコーティングされたリポソームにおいて、化合物(1)を含む医薬組成物を投与し得る。リポソームは、臓器の標的とされ、臓器によって選択的に取り込まれる。さらに、化合物(1)を含む医薬組成物は、急速放出製剤の形態、徐放製剤の形態、又は中間放出製剤の形態で提供され得る。
【0060】
経口投与に関して、化合物(1)は、活性化合物を、当該技術分野に周知の薬学的に許容可能な担体又は賦形剤と組み合わせることによって、容易に調剤され得る。このような担体は、本明細書に記載される化合物が、処置される被験体による経口摂取のための、錠剤、粉末剤、丸剤、ドラゼー、カプセル剤、液剤、ゲル剤、シロップ剤、エリキシル剤、スラリー剤、懸濁液などのように調剤されることを可能にする。
【0061】
経口使用のための医薬調製物は、必要に応じて、錠剤又はドラゼーコアを得るために、適切な助剤を加えた後に、1以上の固体の賦形剤を、本明細書に記載される化合物の1以上と混合させ、結果として生じる混合物を随意に粉砕し、顆粒の混合物を処理することによって得られる。ドラゼーコアは、適切なコーティングによって提供される。この目的のために、濃縮された砂糖溶液が使用され得、これは、アラビアゴム、タルク、ポリビニルピロリドン、カルボポールゲル、ポリエチレングリコール、及び/又は二酸化チタン、ラッカー溶液、及び適切な有機溶媒又は溶媒混合液を随意に含有し得る。色素又はピグメントは、識別のために、又は活性化合物の用量の異なる組み合わせを特徴付けるために、錠剤又はドラゼーのコーティングに加えられ得る。
【0062】
経口に使用され得る医薬製剤は、ゼラチンで作られた押し込み型カプセル剤に加え、グリセロール又はソルビトールなどの、ゼラチン及び可塑剤で作られた軟らかい、密閉されたカプセル剤を含む。幾つかの実施形態において、カプセル剤は、製薬ゼラチン、ウシゼラチン、及び植物ゼラチンの1以上を含む硬ゼラチンカプセルを含む。幾つかの例において、ゼラチンは、アルカリ処理される。押し込み型カプセル剤は、ラクトースなどの充填剤、デンプンなどの結合剤、及び/又はタルク又はステアリン酸マグネシウムなどの潤滑剤、及び随意に安定剤との混合で、活性成分を含み得る。軟カプセル剤において、活性化合物は、脂肪油、液動パラフィン、又は液体ポリエチレングリコールなどの適切な液体において溶解又は懸濁され得る。さらに、安定化剤が加えられ得る。経口投与のためのすべての製剤は、そのような投与に適した用量であるべきである。
【0063】
口腔内又は舌下投与のために、組成物は、従来の方法で調剤された、錠剤、ロゼンジ、又はゲルの形態をとり得る。非経口注入は、ボーラス注入又は持続注入に関係する。化合物(1)の医薬組成物は、油性又は水溶性のビヒクルにおける滅菌した懸濁液、溶液又はエマルションとして、非経口注入に適した形態であり得、懸濁化剤、安定化剤及び/又は分散剤などの製剤化剤を含み得る。非経口投与のための医薬製剤は、水溶性の形態で活性化合物の水溶液を含む。さらに、活性化合物の懸濁液は、好適な油性の注射懸濁液として調製され得る。適切な親油性溶媒又はビヒクルは、胡麻油などの脂肪油、オレイン酸エチル又はトリグリセリドなどの合成脂肪酸エステル、またはリポソームを含む。水溶性の注射懸濁液は、例えば、ナトリウムカルボキシメチルセルロース、ソルビトール、又はデキストランなどの、懸濁液の粘性を増加させる物質を含み得る。随意に、懸濁液はまた、化合物の溶解度を増加させる適切な安定化剤または薬剤を含み得、高濃縮溶液の調製を可能にする。あるいは、活性成分は、使用前には、好適なビヒクル、例えば、滅菌したピロゲンを含まない水で構成するための粉末形態であり得る。
【0064】
化合物(1)は、局所的に投与され得、溶液、懸濁液、ローション剤、ゲル、ペースト剤、薬用スティック、バーム、クリーム、軟膏剤などの様々な局所的に投与可能な組成物に調剤され得る。このような医薬組成物は、可溶化剤、安定化剤、等張化促進剤、緩衝液及び防腐剤を含有し得る。
【0065】
式(1)の構造を有する化合物の経皮投与に適した製剤は、経皮送達装置及び経皮送達パッチを用い得、脂溶性のエマルション又は緩衝水溶液であり得、ポリマー又は接着剤中に溶解及び/又は分散され得る。このようなパッチは、連続的、パルス状、又はオンデマンドの医薬品の送達のために構築され得る。またさらに、化合物(1)の経皮送達は、イオン泳動的なパッチなどの手段によって達成され得る。さらに、経皮的なパッチは、化合物(1)の制御された送達を提供し得る。律速膜を使用することによって、またはポリマーマトリックス又はゲル内で化合物を捕捉することによって、吸収の速度は遅れ得る。逆に、吸収促進剤は、吸収を増やすために使用され得る。吸収促進剤又は担体は、皮膚の通過を補助する吸収性の薬学的に許容可能な溶媒を含み得る。例えば、経皮装置は、裏打ち材(backing member)、随意に担体を備える化合物を含有し、随意に、長時間にわたって制御された速度及び予め定められた速度で化合物を宿主の皮膚に送達するための律速バリアを含有するリザーバー、及び装置を皮膚に固定するための手段を含む包帯(bandage)の形態である。
【0066】
吸入による投与に関して、化合物(1)は、エアロゾル、噴霧又は粉末としての形態であり得る。式(1)の医薬組成物は、適切な推進剤、例えば、ジクロロジフルオロメタン、トリクロロフルオロメタン、ジクロロテトラフルオロエタン、二酸化炭素又は他の適切なガスを使用して、加圧包装又は噴霧器からエアロゾルスプレーを示す形態で都合よく送達され得る。加圧エアロゾルの場合、投与ユニットは、測定された量を送達する弁を提供することによって決定され得る。ほんの一例として、吸入器又は注入器において使用するためのゼラチンなどのカプセル及び薬包は、化合物の粉末混合物及びラクトース又はスターチなどの適切な粉末を含んで調剤され得る。
【0067】
化合物(1)は、ココアバター又は他のグリセリドなどの従来の坐剤基剤を含有する、浣腸剤、直腸ゲル、直腸泡(rectal foams)、直腸エアロゾル、坐剤、ゼリー状の坐剤、又は停留浣腸剤などの直腸組成物に加え、ポリビニルピロリドン、PEGなどの合成ポリマーにおいて調剤され得る。組成物の坐剤形態において、限定されないが、脂肪酸グリセリドの混合物などの低融点ワックスは、随意にココアバターと組み合わされて、最初に融解される。
【0068】
本明細書に提供される処置又は使用の方法を実行する際に、本明細書に提供される治療上有効な量の化合物(1)は、医薬組成物において、処置されるべき疾患又は疾病を有する哺乳動物に投与される。幾つかの実施形態において、哺乳動物は、ヒトである。治療上有効な量は、疾患の重症度、被検体の年齢及び相対的な健康、使用される化合物の効力及び他の要因に依存して、幅広く異なり得る。化合物は、単独で又は混合物の成分として1以上の治療薬と組み合わせて使用され得る。
【0069】
医薬組成物は、活性化合物を医薬的に使用され得る製剤へと処理するのを促進する賦形剤及び助剤を含む、1以上の生理学的に許容可能な担体を使用する従来の方法で調剤され得る。適切な製剤は、選択される投与の経路に依存する。周知の技術、担体、及び賦形剤はどれも、適切なものとして、及び当該技術分野において理解されるものとして使用され得る。式(1)の化合物を含む医薬組成物は、ほんの一例ではあるが、従来の混合、溶解、造粒、ドラゼー製造、粉砕、乳化、封入、包括、又は圧迫の過程の手段によるなど、従来の方法で製造され得る。
【0070】
医薬組成物は、少なくとも1つの薬学的に許容可能な担体、希釈剤又は賦形剤、及び遊離塩基形態、又は薬学的に許容可能な塩形態の活性成分として本明細書に記載される式(1)の化合物を含み得る。さらに、本明細書に記載される方法及び医薬組成物は、N-オキシドの使用、(多形体としても知られる)結晶形態に加え、同じタイプの活性を有するこれらの化合物の活性代謝物質も含む。
【0071】
本明細書に記載される化合物を含む組成物の調製のための方法は、固形物、半固形物又は液体を形成するために、1以上の不活性な、薬学的に許容可能な賦形剤又は担体を用いて、化合物を調剤する工程を含む。固形組成物は、限定されないが、粉末、錠剤、分散性顆粒、カプセル剤、カシェ剤、及び坐剤を含む。液体組成物は、本明細書に開示されるような、化合物を溶解する溶液、化合物を含むエマルション、又は化合物を含むリポソーム、ミセル、又はナノ粒子を含む溶液を含む。半固形組成物は、限定されないが、ゲル、懸濁液及びクリームを含む。組成物は、液体溶液又は懸濁液中にあり得るか、使用前に液体中で溶液又は懸濁液に適した固形形態であり得るか、またはエマルションとしてあり得る。これらの組成物はまた、湿潤剤又は乳化剤、pH緩衝剤などの微量の非毒性補助物質を含む。
【0072】
医薬組成物のタイプの要約は、例えば、Remington: The Science and Practice of Pharmacy, Nineteenth Ed (Easton, Pa.: Mack Publishing Company, 1995)、Hoover, John E., Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pennsylvania 1975、Liberman, H.A. and Lachman, L., Eds., Pharmaceutical Dosage Forms, Marcel Decker, New York, N.Y., 1980、及びPharmaceutical Dosage Forms and Drug Delivery Systems, Seventh Ed. (Lippincott Williams & Wilkins 1999)において見出され得、それら各々は、その全体において、引用によって本明細書に組み込まれる。
【0073】
投与の典型的な方法及び処置方法
化合物(1)は、ステロイドホルモン核内受容体の活性が疾患の病因及び/又は症状の一因となる、疾患又は疾病の処置のための薬剤の調製に使用され得る。さらに、本明細書に記載されるあらゆる疾患又は疾病を、そのような処置を必要とする被験体において処置するための方法は、式(1)の少なくとも1つの化合物、又はその薬学的に許容可能な塩、薬学的に許容可能なN-オキシド、薬学的に活性な代謝物質、薬学的に許容可能なプロドラッグ、又は薬学的に許容可能な溶媒和物を含む医薬組成物を、治療上有効な量で前記被験体に投与する工程を含む。
【0074】
本明細書に記載される化合物を含有する組成物は、予防的及び/又は治療的な処置のために投与され得る。治療的な適用において、組成物は、疾患又は疾病の症状を治療する又は少なくとも部分的に抑える、又は症状自体を治療する、治癒する、改善する、又は寛解するのに十分な量で、疾患又は疾病に既に苦しむ被検体に投与される。この用途に有効な量は、疾患又は疾病の重症度及び経過、以前の治療、被検体の健康状態、体重、薬物への反応、及び処置に当たる医師の判断に依存する。
【0075】
一旦、被験体の疾病が改善すると、必要ならば維持量が投与される。続いて、投与の量又は頻度、またはその両方は、症状に応じて、改善された疾患又は疾病が保持されるレベルまで減らされ得る。しかしながら、被験体は、症状が再発すると、長期的に間欠的処置を必要とし得る。
【0076】
特定の例において、本明細書に記載される治療上有効な量の少なくとも1つの化合物(又はその薬学的に許容可能な塩、薬学的に許容可能なN-オキシド、薬学的に活性な代謝物質、薬学的に許容可能なプロドラッグ、及び薬学的に許容可能な溶媒和物)を、別の治療薬と組み合わせて投与するのが適切であり得る。ほんの一例ではあるが、本明細書の化合物の1つを受け取ることで被験体が受ける副作用の1つが炎症である場合、抗炎症剤を、最初の治療薬と組み合わせて投与するのが適切であり得る。又は、ほんの一例として、本明細書に記載される化合物の1つの治療的有用性は、アジュバントの投与によって高められ得る(すなわち、アジュバント自体は、最小の治療的有用性を有し得るだけであるが、別の治療薬と組み合わせることで、被験体に対する全体的な治療効果が高められる)。又は、ほんの一例として、被験体が受けるその効果は、本明細書に記載される化合物の1つを、治療効果をまた有する別の治療薬(これはまた治療レジメンを含む)とともに投与することで増幅され得る。いかなる場合においても、処置されている疾患又は疾病にかかわらず、被験体が受ける全体的な効果は、2つの治療薬を単に加えたものであり得るか、又は被験体は相乗効果を受け得る。本明細書に記載される化合物が他の治療と併用して投与される場合、同時投与された化合物の投与量は、当然のことながら、用いられる同時投与の薬物の種類、用いられる具体的な薬物、処置されている疾患又は疾病などに依存して変化する。さらに、1以上の生理活性物質とともに同時投与される時に、本明細書に提供される化合物は、生理活性物質(複数可)と同時に投与され得るか、又は連続して投与され得るかのどちらかである。連続して投与される場合、主治医は、生理活性物質(複数可)と組み合わせてタンパク質を投与する、適切な順序を決定する。
【0077】
いかなる場合においても、複数の治療薬(その1つは本明細書に記載される化合物の1つである)は、任意の順番で又は同時に投与され得る。もし同時であれば、複数の治療薬は、単一の、統一された形態で、又は複数回の形態で(ほんの一例として、単一の丸薬又は2つの別の丸薬のどちらかとして)提供され得る。治療薬の1つが複数回投与で与えられ得るか、又は両方が複数回の投与として与えられ得る。もし同時でなければ、複数回投与の間の期間は0週間以上4週間未満で変化し得る。さらに、組み合わせの方法、組成物及び製剤は、2つのみの薬剤の使用に限定されない。複数の治療上の組み合わせが想定される。
【0078】
さらに、化合物(1)はまた、被験体に更なる又は相乗的な効果を提供し得る手順と組み合わせて使用され得る。ほんの一例ではあるが、被験体は、本明細書に記載される方法において、治療的及び/又は予防的な効果を得ると予期され、ここで、式(I)の医薬組成物及び/又は他の治療法との組み合わせは、その個体が特定の疾患又は疾病と相互に関連することが知られている突然変異遺伝子のキャリアであるかを決定するための遺伝子検査と組み合わせられる。
【0079】
化合物(1)及び併用療法は、疾患又は疾病の発生の前、発生の間又は発生の後に投与され得、化合物を含有する組成物を投与するタイミングは変わり得る。したがって、例えば、疾患又は疾病の発生を防止するために、化合物は、予防薬として使用され得、疾病又は疾患への傾向のある被験体に、連続的に投与され得る。化合物及び組成物は、症状の発症の間に又は発症の後、できるだけすぐに被検体に投与され得る。化合物の投与は、症状の発症後の最初の48時間以内に、好ましくは症状の発症後の最初の48時間以内に、より好ましくは症状の発症後の最初の6時間以内に、及び最も好ましくは症状の発症後の3時間以内に開始され得る。最初の投与は、例えば、静脈注射、ボーラス注入、5分間から約5時間にわたる点滴、錠剤、カプセル、経皮パッチ、口腔送達など、又はそれらの組み合わせなどの任意の実用的な経路を介され得る。化合物は、好ましくは、疾患又は疾病の発症が見つかった又は疑われた後に、実行可能となってすぐに投与され、疾患の処置に必要な期間は、例えば、約1ヶ月から約3ヶ月である。処置の期間は、各被検体ごとに異なり得、その期間は公知の基準を用いて決定され得る。例えば、化合物又は化合物を含む製剤は、少なくとも2週間、好ましくは約1ヶ月から約3年間、及び幾つかの実施形態において約1ヶ月から約10年間投与され得る。他の実施形態において、化合物は、90日から2年間まで一日に一回投与される。
【0080】
本明細書に記載される医薬組成物は、正確な投与量での単回投与に適した単位剤形であり得る。単位剤形において、製剤は、1以上の化合物の適量を含有する単位用量に分割される。単位用量は、製剤の別々の量を含有するパッケージの形態であり得る。制限しない例は、包装された錠剤又はカプセル剤、及びバイアル又はアンプル中の粉末である。水溶性懸濁液組成物は、単回投与の再密閉が不可能な容器に入れられ得る。あるいは、複数回投与の密閉可能な容器が使用され得、その場合、組成物中に防腐剤を含むことが典型的である。ほんの一例として、非経口注射のための製剤は、単位剤形で提供され得、これは、限定されないが、アンプルを含み、又は複数回投与の容器においては、防腐剤が加えられる。
【0081】
本明細書中に記載される化合物(1)に適した1日の投与量は、体重あたり約0.03から60mg/kgまでである。限定されないが、ヒトを含む大型哺乳動物における示される日常的な投与量は、約1mgから約4000mgの範囲であり、限定されないが、1日に4回までを含む、1回以上の投与で、又は遅延の形態(retard form)で都合良く投与される。経口投与のための適切な単位剤形は、約1mgから約4000mgまでの活性成分を含む。幾つかの実施形態において、式(1)の化合物の単一用量は、約50mgから約2,000mgまでの範囲内にある。幾つかの実施形態において、式(1)の化合物の単一用量は、約90mg、約200mg、約250mg、約325mg、約650mg、約975mg、約1300mg、約1625mg、又は約1950mgである。幾つかの実施形態において、約90mg、約325mg、約650mg、約975mg、約1300mg、約1625mg、又は約1950mgの式(1)の化合物の投与は、複数回投与として与えられる。
【0082】
幾つかの実施形態において、式(a)の化合物の単一用量は、90から2500mgまでの間であり、化合物は、90日から2年の間で被験体に投与される。
【0083】
このような投与量は、多くの変数、限定されない使用される化合物の活性、処置される疾患又は疾病、投与の形式、個々の被験体の要件、処置されている疾患又は疾病の重症度、及び医師の判断によって変化し得る。
【0084】
薬物動態学的及び薬理学的な測定
薬物動態学的及び薬理学的なデータは、当該技術分野に公知の技術によって得られ得る。ヒトの被験体の薬物代謝の薬物動態学的及び薬理学的なパラメーターにおける固有の変化により、特定の組成物を表す適切な薬物動態学的及び薬理学的な特性成分は変化し得る。典型的には、薬物動態学的及び薬理学的な特性は、被験体の群の平均パラメーターの測定に基づく。被験体の群は、代表的な平均を測定するのに適した被験体の任意の合理的な数、例えば、5の被験体、10の被験体、16の被験体、20の被験体、25の被験体、30の被験体、35の被験体、又はそれ以上を含む。その平均は、測定される各パラメーターのすべての被験体の測定の平均を算出することによって決定される。
【0085】
薬物動態学的パラメーターは、本発明の組成物を表すのに適切な任意のパラメーターであり得る。例えば、Cmaxは、約500ng/ml以上、約550ng/ml以上、約600ng/ml以上、約700ng/ml以上、約800ng/ml以上、約880ng/ml以上、約900ng/ml以上、約100ng/ml以上、約1250ng/ml以上、約1500ng/ml以上、約1700ng/ml以上、又は化合物(1)の薬物動態学的特性を表すのに適切な任意の他のCmaxであり得る。幾つかの実施形態において、活性代謝物質は、薬物の被験体への投与の後にインビボで形成され、Cmaxは、約500pg/ml以上、約550pg/ml以上、約600pg/ml以上、約700pg/ml以上、約800pg/ml以上、約880pg/ml以上、約900pg/ml以上、約1000pg/ml以上、約1250pg/ml以上、約1500pg/ml以上、約1700pg/ml以上、又は被験体への化合物(1)の投与後にインビボで形成される化合物の薬物動態学的特性を表すのに適切な任意の他のCmaxであり得る。
【0086】
Tmaxは、例えば、約0.5時間以下、約1.0時間以下、約1.5時間以下、約2.0時間以下、約2.5時間以下、約3.0時間以下、又は化合物(1)の薬物動態学的特性を表すのに適切な任意の他のTmaxであり得る。
【0087】
AUC(0-inf)は、例えば、約590ng・hr/mL以上、約1500ng・hr/mL以上、約2000ng・hr/mL以上、約3000ng.times.hr/ml以上、約3500ng・hr/mL以上、約4000ng・hr/mL以上、約5000ng・hr/mL以上、約6000ng・hr/mL以上、約7000ng・hr/mL以上、約8000ng・hr/mL以上、約9000ng・hr/mL以上、又は化合物(1)の薬物動態学的特性を表すのに適切な任意の他のAUC(0-inf)であり得る。幾つかの実施形態において、活性代謝物質は、化合物(1)の被験体への投与の後にインビボで形成され、AUC(0-inf)は、例えば、約590pg・hr/mL以上、約1500pg・hr/mL以上、約2000pg・hr/mL以上、約3000pg・hr/mL以上、約3500pg・hr/mL以上、約4000pg・hr/mL以上、約5000pg・hr/mL以上、約6000pg・hr/mL以上、約7000pg・hr/mL以上、約8000pg・hr/mL以上、約9000pg・hr/mL以上、又は被験体への化合物(1)の投与後にインビボで形成される化合物の薬物動態学的特性を表すのに適切な任意の他のAUC(0-inf)であり得る。
【0088】
投与の約1時間後の化合物(1)の血漿濃度は、例えば、約140ng/ml以上、約425ng/ml以上、約550ng/ml以上、約640ng/ml以上、約720ng/ml以上、約750ng/ml以上、約800ng/ml
以上、約900ng/ml以上、約1000ng/ml以上、約1200ng/ml以上、又は化合物(1)の任意の他の血漿濃度であり得る。
【0089】
薬理学的パラメーターは、本発明の組成物を表すのに適切な任意のパラメーターであり得る。例えば、薬理学的特性は、ほんの一例として、少なくとも約2時間、少なくとも約4時間、少なくとも約8時間、少なくとも約12時間又は少なくとも約24時間、ARタンパク質又は内因性のアンドロゲンの減少を示し得る。薬理学的特性は、ほんの一例として、少なくとも約2時間、少なくとも約4時間、少なくとも約8時間、少なくとも約12時間又は少なくとも約24時間、CYP17を含む、アンドロゲンを合成する酵素の阻害を示し得る。薬理学的特性は、ほんの一例として、少なくとも約2時間、少なくとも約4時間、少なくとも約8時間、少なくとも約12時間又は少なくとも約24時間、アンドロゲンシグナル伝達の低下を示し得る。
【0090】
治療を提供する典型的な方法
本発明は、ヒトにおける前立腺癌の処置のための治療上の方策を提供する。
【0091】
幾つかの実施形態において、本発明は、前立腺癌の処置における化合物1の使用のための調製及びレジメンを提供する。幾つかの実施形態において、前立腺癌は、去勢抵抗性前立腺癌である。幾つかの実施形態において、前立腺癌は、化学療法未治療の前立腺癌である。
【0092】
幾つかの実施形態において、本発明は、化合物1の経口投与を含む治療上のレジメンを提供する。
【0093】
幾つかの実施形態において、本発明は、化合物1の複数回用量の投与を含む治療上のレジメンを提供する。幾つかの実施形態において、異なる投与は、時間において別々に間隔を置かれる。幾つかの実施形態において、すべての投与は、同じ量の化合物1を含む。幾つかの実施形態において、異なる投与は、異なる量の化合物1を含む。幾つかの実施形態において、時間で分離される異なる投与は、同じ時間によって互いから分離され、幾つかの実施形態において、時間で分離される異なる投与は、異なる時間によって互いから分離される。幾つかの実施形態において、本発明は、規則的な時間間隔(複数可)によって分離された複数の用量の投与を含む投薬レジメンを提供し、その後、休息期間があり、随意にその後、規則的な時間間隔(複数可)によって分離された第2の複数の用量を提供する。
【0094】
幾つかの実施形態において、少なくとも2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109,110、111、112、113、114、115、116、117、118、119、120、121、122、123、124、125、126、127、128、129、130、131、132、133、134、135、136、137、138、139、140、141、142、143、144、145、146、147、148、149、150、151, 152, 153, 154, 155, 156, 157, 158, 159, 160, 161, 162, 163, 164, 165, 166, 167, 168又はそれ以上の用量の化合物1が投与される。幾つかの実施形態において、少なくとも7、14、21、28、35、42、49、56、63、70、77、84、91、98、105、112、119、126、133、140、147、154、161、168又はそれ以上の用量の化合物1が投与される。
【0095】
幾つかの実施形態において、本発明は、化合物(1):
【0096】
【化7】
【0097】
を含む医薬組成物を、微粉化結晶性粉末として熟考する。
【0098】
幾つかの実施形態において、本発明は、化合物(1):
【0099】
【化8】
【0100】
を含む医薬組成物を熟考し、式中、化合物(1)は、約:a. 13.0°、14.6°、16.3°、17.6°、及び19.0°、及び随意に約11.8°、20.2°、22.9°、及び25.4°;b. 13.1°及び14.1°;c. 17.2°、18.5°、及び19.1°、及び随意に16.2°、及び29.6°;d. 12.5°、14.8°、及び25.5°;又はe. 14.4°、16.1°、19.1°、及び19.3°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられた、結晶形態である。
【0101】
幾つかの実施形態において、本発明は、化合物(1):
【0102】
【化9】
【0103】
を含む医薬組成物を熟考し、式中、化合物(1)は、約:a. 13.1°及び14.1°;b. 17.2°、18.5°、及び19.1°、及び随意に約16.2°、及び29.6°;c. 12.5°、14.8°、及び25.5°;又はd. 14.4°、16.1°、19.1°及び19.3°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられた、微粉化結晶形態、又はその薬学的に許容可能な塩、N-オキシド、活性代謝物質、プロドラッグ、又は溶媒和物である。
【0104】
幾つかの実施形態において、本発明は、化合物(1):
【0105】
【化10】
【0106】
を含む医薬組成物を熟考し、式中、化合物(1)は、約:a. 13.1°及び14.1°;b. 17.2°、18.5°、及び19.1°、及び随意に約16.2°、及び29.6°;c. 12.5°、14.8°、及び25.5°;又はd. 14.4°、16.1°、19.1°及び19.3°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられた、微粉化結晶形態である。
【0107】
幾つかの実施形態において、化合物(1)は、被験体への投与の後に、アンドロゲン受容体媒介性の疾患又は疾病を処置するのに有効な量で存在する。
【0108】
幾つかの実施形態において、アンドロゲン受容体媒介性の疾患又は疾病は、前立腺癌、良性前立腺肥大症、多毛症、脱毛症、神経性食欲不振、乳癌、及び男性の性腺機能亢進から成る群から選択される。
【0109】
幾つかの実施形態において、アンドロゲン受容体媒介性の疾患又は疾病は、前立腺癌である。
【0110】
幾つかの実施形態において、前立腺癌は、去勢抵抗性前立腺癌である。
【0111】
幾つかの実施形態において、化合物(1)は、被験体への投与の後に、アンドロゲン生合成を阻害し、アンドロゲン受容体シグナル伝達を阻害し、及びアンドロゲン受容体感受性を低下させるのに有効な量で存在する。
【0112】
幾つかの実施形態において、化合物は、アンドロゲン受容体シグナル伝達を阻害するか、又はアンドロゲン受容体感受性を低下させる。
【0113】
幾つかの実施形態において、アンドロゲン生合成阻害は、チトクロームC17α-ヒドロキシラーゼ/C17,20-リアーゼ(CYP17)の活性を阻害することを含む。
【0114】
幾つかの実施形態において、アンドロゲン受容体シグナル伝達の阻害は、テストステロン結合の競合的阻害を含む。
【0115】
幾つかの実施形態において、アンドロゲン受容体感受性の低下は、細胞内のアンドロゲン受容体タンパク質の含量の減少、及び低レベルのアンドロゲンの増殖シグナルによって持続される、細胞の低下した能力を含む。
【0116】
幾つかの実施形態において、組成物は、被験体に、非経口的に、静脈内に、筋肉内に、皮内に、皮下に、腹腔内に、経口的に、口腔に、舌下に、粘膜に、直腸に、経皮的に、皮膚に、眼に、又は吸入によって投与するために調剤される。
【0117】
幾つかの実施形態において、組成物は、錠剤、カプセル剤、クリーム、ローション剤、油剤、軟膏、ゲル、ペースト、粉末、懸濁液、エマルション、又は溶液として被験体に投与するために調剤される。
【0118】
幾つかの実施形態において、組成物は、カプセル剤として被験体に投与するために調剤される。
【0119】
前述の請求項のいずれかの医薬組成物は、錠剤として被験体に投与するために調剤される。
【0120】
幾つかの実施形態において、カプセル剤は、粉末として化合物(1)を含む。
【0121】
幾つかの実施形態において、粉末は微粉化される。
【0122】
幾つかの実施形態において、組成物は、約50mgから約500mgまでの化合物(1)を含む。
【0123】
幾つかの実施形態において、組成物は、約90mgの化合物(1)を含む。
【0124】
幾つかの実施形態において、組成物は、約325mgの化合物(1)を含む。
【0125】
幾つかの実施形態において、組成物は、被験体に、1日当たり、1、2、3、4、5、6、7、8、9、又は10回投与するために調剤される。
【0126】
幾つかの実施形態において、組成物は、前立腺癌の処置に対して被験体に投与されるために調剤される。
【0127】
幾つかの実施形態において、組成物は、去勢抵抗性前立腺癌の処置に対して被験体に投与されるために調剤される。
【0128】
幾つかの実施形態において、組成物は、1以上の薬学的に許容可能な賦形剤を更に含む。
【0129】
幾つかの実施形態において、薬学的に許容可能な賦形剤は、充填剤、崩壊剤、潤滑剤、界面活性剤、流動促進剤、結合剤、砂糖、スターチ、ニス、又はワックスを含む。
【0130】
幾つかの実施形態において、化合物(1)は、薬学的に許容可能な塩、N-オキシド、活性代謝物質、プロドラッグ、結晶多形体、又は溶媒和物である。
【0131】
幾つかの実施形態において、溶媒和物は、クメン溶媒和物又は水和物を含む。
【0132】
幾つかの実施形態において、結晶多形体は、化合物(1)のフォームI、フォームII、又はフォームIIIを含む。
【0133】
幾つかの実施形態において、本発明は、化合物(1)
【0134】
【化11】
【0135】
の結晶形態を熟考し、(フォームI)約13.0°、14.6°、16.3°、17.6°、及び19.0°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられる。
【0136】
幾つかの実施形態において、結晶形態は、約11.8°、20.2°、22.9°、及び25.4°の角度 2-シータにおいて表される、特性ピークによって更に特徴付けられる。
【0137】
幾つかの実施形態において、本発明は、化合物(1)
【0138】
【化12】
【0139】
の結晶形態を熟考し、(フォームII)約13.1°及び14.1°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられる。
【0140】
幾つかの実施形態において、本発明は、化合物(1)
【0141】
【化13】
【0142】
の結晶形態を熟考し、(フォームIII)約17.2°、18.5°、及び19.1°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられる。
【0143】
幾つかの実施形態において、結晶形態は、約16.2°、及び29.6°の角度 2-シータにおいて表される、特性ピークによって更に特徴付けられる。
【0144】
幾つかの実施形態において、本発明は、化合物(1)
【0145】
【化14】
【0146】
の結晶形態を熟考し、(パターン5)約12.5°、14.8°、及び25.5°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられる。
【0147】
幾つかの実施形態において、本発明は、化合物(1)
【0148】
【化15】
【0149】
の結晶形態を熟考し、(パターン2)約14.4°、16.1°、19.1°、及び19.3°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられる。
【0150】
幾つかの実施形態において、本発明は、ジメチルホルムアミド、炭酸カリウム、式:
【0151】
【化16】
【0152】
の化合物、又はそのアナログ、及び式:
【0153】
【化17】
【0154】
の化合物を接触させ、式:
【0155】
【化18】
【0156】
の化合物を作る工程を含む方法を熟考する。
【0157】
幾つかの実施形態において、該方法は、式:
【0158】
【化19】
【0159】
の化合物を、N-メチルピロリドン中の木炭上の10%のパラジウムと接触させ、式:
【0160】
【化20】
【0161】
の化合物を生成する工程を更に含む。
【0162】
幾つかの実施形態において、該方法は、式:
【0163】
【化21】
【0164】
の化合物を、メタノールナトリウムメトキシドと接触させ、式(I):
【0165】
【化22】
【0166】
の化合物を生成する工程を更に含む。
【0167】
幾つかの実施形態において、該方法は、大規模又は製造規模で行われる。幾つかの実施形態において、大規模は、約1から約10kgまでの規模である。幾つかの実施形態において、製造規模は、約10kgより大きい規模である。幾つかの実施形態において、製造規模は、約10から約1,000kgまでの規模である。幾つかの実施形態において、製造規模は、約10から約100kgまでの規模である。幾つかの実施形態において、製造規模は、約10から約50kgまでの規模である。幾つかの実施形態において、製造規模は、約33.4kgの規模である。
【0168】
幾つかの実施形態において、本発明は、被験体において前立腺癌に対する処置を提供する方法を熟考し、該方法は、治療上有効な量の化合物(1):
【0169】
【化23】
【0170】
又はその薬学的に許容可能な塩、N-オキシド、活性代謝物質、プロドラッグ、又は溶媒和物を含む医薬組成物を被験体に投与する工程を含み、
式中、化合物(1)は、約:a. 13.1°及び14.1°;b. 17.2°、18.5°、及び19.1°、及び随意に約16.2°、及び29.6°;c. 12.5°、14.8°、及び25.5°;又はd. 14.4°、16.1°、19.1°及び19.3°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられた、微粉化結晶形態である。
【0171】
幾つかの実施形態において、本発明は、被験体において前立腺癌に対する処置を提供する方法を熟考し、該方法は、治療上有効な量の化合物(1):
【0172】
【化24】
【0173】
を含む医薬組成物を被験体に投与する工程を含み、
式中、化合物(1)は、約:a. 13.0°、14.6°、16.3°、17.6°、及び19.0°、及び随意に約11.8°、20.2°、22.9°、及び25.4°;b. 13.1°及び14.1°;c. 17.2°、18.5°、及び19.1°、及び随意に16.2°、及び29.6°;d. 12.5°、14.8°、及び25.5°;又はe. 14.4°、16.1°、19.1°、及び19.3°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられた、結晶形態である。
【0174】
幾つかの実施形態において、本発明は、被験体において前立腺癌に対する処置を提供する方法を熟考し、該方法は、治療上有効な量の化合物(1):
【0175】
【化25】
【0176】
を含む医薬組成物を被験体に投与する工程を含み、
式中、化合物(1)は、約:a. 13.1°及び14.1°;b. 17.2°、18.5°、及び19.1°、及び随意に約16.2°、及び29.6°;c. 12.5°、14.8°、及び25.5°;又はd. 14.4°、16.1°、19.1°及び19.3°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられた、微粉化結晶形態である。
【0177】
幾つかの実施形態において、本発明は、被験体において前立腺癌の処置のための薬剤を調剤する際の化合物(1)の使用を熟考し、該薬剤は、治療上有効な量の化合物(1):
【0178】
【化26】
【0179】
又はその薬学的に許容可能な塩、N-オキシド、活性代謝物質、プロドラッグ、又は溶媒和物を含み、
式中、化合物(1)は、約:a. 13.1°及び14.1°;b. 17.2°、18.5°、及び19.1°、及び随意に約16.2°、及び29.6°;c. 12.5°、14.8°、及び25.5°;又はd. 14.4°、16.1°、19.1°及び19.3°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられた、微粉化結晶形態である。
【0180】
幾つかの実施形態において、本発明は、被験体において前立腺癌の処置のための薬剤を調剤する際の化合物(1)の使用を熟考し、該薬剤は、治療上有効な量の化合物(1):
【0181】
【化27】
【0182】
を含み、
式中、化合物(1)は、約:a. 13.0°、14.6°、16.3°、17.6°、及び19.0°、及び随意に約11.8°、20.2°、22.9°、及び25.4°;b. 13.1°及び14.1°;c. 17.2°、18.5°、及び19.1°、及び随意に16.2°、及び29.6°;d. 12.5°、14.8°、及び25.5°;又はe. 14.4°、16.1°、19.1°、及び19.3°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられた、結晶形態である。
【0183】
幾つかの実施形態において、本発明は、被験体において前立腺癌の処置のための薬剤を調剤する際の化合物(1)の使用を熟考し、該薬剤は、治療上有効な量の化合物(1):
【0184】
【化28】
【0185】
を含み、
式中、化合物(1)は、約:a. 13.1°及び14.1°;b. 17.2°、18.5°、及び19.1°、及び随意に約16.2°、及び29.6°;c. 12.5°、14.8°、及び25.5°;又はd. 14.4°、16.1°、19.1°及び19.3°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられた、微粉化結晶形態である。
【実施例】
【0186】
以下の実施例は、化合物(1)を生成するための、及び化合物(1)の有効性及び安全性を試験するための例示的方法を提供する。これらの実施例は、例示目的のみに提供され、本明細書に提供される請求項の範囲を制限しない。本明細書に開示され請求される方法のすべては、本開示の観点から、不当な実験なしで作られ実行され得る。
【0187】
請求項の概念、精神及び範囲から逸脱することなく、本明細書に記載される方法に、及び方法の工程において又は工程の順序において変更が適用され得ることは当業者に明らかである。当業者に明らかなすべてのこのような同様の代替及び改変は、添付された請求項の精神、範囲及び概念内にあると考えられる。
【0188】
<実施例1:式(1)の化合物の合成>
<実施例1A:3-β-アセトキシ-17-(1H-ベンズイミダゾール-1-イル)-16-ホルミル-アンドロスタ-5,16-ジエンの合成>
【0189】
【化29】
【0190】
33.4kgの3-β-アセトキシ-17-クロロ-16-ホルミルアンドロスタ-5,16-ジエンを、ジメチルホルムアミド(DMF)中でベンズイミダゾール及び炭酸カリウムと混合させ、残りの出発物質の量によって決定されるように、反応が終了するまで加熱した。反応が終了した後、反応混合物を冷却し、冷却した水と混合し、反応物をクエンチした。固形物をクエンチした反応混合物から分離し、DMFと水の混合物、水、希薄した水性の塩化水素酸、水、希薄した水性の炭酸水素ナトリウム、及び水で連続して洗浄した。中間物質、3-β-アセトキシ-17-(1H-ベンズイミダゾール-1-イル)-16-ホルミルアンドロスタ-5,16-ジエンを、続いて乾燥した。
【0191】
<実施例1B:3-β-アセトキシ-17-(1H-ベンズイミダゾール-1-イル)アンドロスタ-5,16-ジエンの合成及び精製>
【0192】
【化30】
【0193】
3-β-アセトキシ-17-(1H-ベンズイミダゾール-1-イル)-16-ホルミルアンドロスタ-5,16-ジエンを、N-メチルピロリドン(NMP)中で炭素上の約10%のパラジウム(Pd/C)と混合させ、反応混合物中の3-β-アセトキシ-17-(1H-ベンズイミダゾール-1-イル)-16-ホルミルアンドロスタ-5,16-ジエン/3-β-アセトキシ-17-(1H-ベンズイミダゾール-1-イル)アンドロスタ-5,16-ジエンの比率によって決定されるように、反応が終了するまで加熱した。反応が終了した後、反応混合物を冷却した。硫酸マグネシウムを加え、結果として生じる混合物をろ過した。水を濾液に加え、結果として生じる混合物を撹拌した。固体の、粗製の3-β-アセトキシ-17-(1H-ベンズイミダゾール-1-イル)アンドロスタ-5,16-ジエンを、水/NMP混合物から分離し、水とメタノールの混合物で洗浄し、乾燥し、包装した。
【0194】
粗製の3-β-アセトキシ-17-(1H-ベンズイミダゾール-1-イル)アンドロスタ-5,16-ジエンを、酢酸エチル中に溶解し、浄化した。この混合物の容量を、真空蒸留によって低減した。結果として生じる混合物を、冷却し、固形物を分離し、冷たい酢酸エチルで洗浄し、真空下で乾燥した。幾つかの実施形態において、サンプルを、不純物レベルを測定するためにインプロセスの試験にさらした。不純物レベルが許容可能でなかったとき、再結晶過程(recrystallization process)を繰り返した。
【0195】
<実施例1C:3-β-ヒドロキシ-17-(1H-ベンズイミダゾール-1-イル)アンドロスタ-5,16-ジエンの合成及び精製>
【0196】
【化31】
【0197】
3-β-アセトキシ-17-(1H-ベンズイミダゾール-1-イル)アンドロスタ-5,16-ジエンを、メタノール中でナトリウムメトキシドと混合させ、残りの3-β-アセトキシ-17-(1H-ベンズイミダゾール-1-イル)アンドロスタ-5,16-ジエンの量によって決定されるように、反応が終了するまで加熱した。反応が終了した後、反応混合物を冷却し、水と混合し、反応物をクエンチした。結果として生じるスラリーを撹拌し、更に冷却した。固体の、粗製の3-β-ヒドロキシ-17-(1H-ベンズイミダゾール-1-イル)アンドロスタ-5,16-ジエンを、クエンチした反応混合物から分離し、メタノールと水の混合物で洗浄し、その後、洗液が中性になるまで水で洗浄し、乾燥し、及び包装した。
【0198】
粗製の3-β-ヒドロキシ-17-(1H-ベンズイミダゾール-1-イル)アンドロスタ-5,16-ジエンを、メタノールと酢酸エチルの混合物中に溶解し、浄化した。生成物を、溶媒交換単独によって、メタノール/酢酸エチル溶液から酢酸エチルに移した。結果として生じる混合物を冷却し、固形物を分離し、冷たい酢酸エチルで洗浄し、真空下で乾燥した。幾つかの実施形態において、サンプルを、不純物レベルを測定するためにインプロセスの試験にさらした。不純物レベルが許容可能でなかったとき、再結晶過程を繰り返した。
【0199】
<実施例2:化合物(1)の結晶多形相>
<実施例2A:機器及び方法論>
X線粉末回折(XRPD):Cu Kα放射光(40kV, 40mA)、自動XYZステージ、自動サンプル配置のためのレーザービデオ顕微鏡、及びHiStar 2次元面積検出器を使用して、X線粉末回折パターンを、Bruker AXS C2 GADDS回折計上に収集した。X線光学は、0.3mmのピンホールコリメータとつながれた単一のGoebel 多層膜鏡から成った。
【0200】
ビーム発散、すなわちサンプル上のX線ビームの有効径は、約4mmであった。θ-θの連続的な走査方式を、3〜30°2θの有効な2θ範囲を与える20cmのサンプル-検出器距離で利用した。典型的に、サンプルを、120秒間X線ビームに曝した。データ収集に使用したソフトウェアは、WNT 4.1.16用のGADDSであり、データを、Diffrac Plus EVA v 9.0.0.2又はv 13.0.0.2を使用して分析し、提示した。周囲条件下で実行したサンプルを、粉砕なしで受け取られるように、粉末を使用して、平面プレート試料として調製した。非周囲条件下で実行したサンプルを、熱伝導性の化合物とともにシリコンウェーファー上に乗せた。
【0201】
核磁気共鳴(1H-NMR):NMRスペクトルを、自動サンプラーを備える、Bruker 400MHz機器上に収集し、DRX400コンソールによって制御した。自動化した実験を、標準のBruker 荷重実験(standard Bruker loaded experiments)を使用してTopspin v 1.3(パッチレベル8)で実行する、ICON-NMR v4.0.4(build 1)を使用して得た。非ルーチン分光法(non-routinespectroscopy)に関して、データを、Topspin単独の使用を介して得た。他に明記のない限り、サンプルを、d6-DMSO中で調製した。オフライン分析を、ACD SpecManager v 12.00(build 29094)を使用して実行した。
【0202】
示差走査熱量測定法(DSC):DSCデータを、50位置の自動サンプラーを備えた、TA Instruments Q2000上で収集した。該機器を、公認のインジウムを使用して、エネルギー及び温度の較正のために較正した。ピンで穴をあけたアルミニウムパンにおいて、典型的に0.5〜3mgの各サンプルを、25℃から250℃までの10℃.min-1で加熱した。50mL/分での窒素パージを、サンプル上で維持した。
【0203】
熱重量分析(TGA):TGAデータを、16位置の自動サンプラーを備えた、TA Instruments Q500 TGA上で収集した。該機器を、公認のアルメルを使用して、温度較正した。典型的に5〜30mgの各サンプルを、風袋に入れた白金るつぼ及びアルミニウムDSCパンに充填し、周囲温度から300℃までの10℃/分で加熱した。60mL/分での窒素パージを、サンプル上で維持した。
【0204】
重量蒸気収着(GVS):等温収着曲線を、SMS DVS Intrinsicの水分収着分析を使用して獲得し、SMS Analysis Suiteソフトウェアによって制御した。サンプル温度を、機器の制御によって25℃で維持した。湿度を、200mL/分の合計流量で、乾燥した及び湿った窒素の流れを混合することにより制御した。相対湿度を、サンプルの近くにある、較正したRotronic探査(1.0〜100%RHの動作範囲)によって測定した。重量変化、%RHに応じたサンプルの(質量緩和)を、微量天秤(正確度±0.005mg)によって常に監視した。5〜20mgのサンプルを、周囲条件下で、風袋に入れたメッシュステンレス鋼バスケットに置いた。40%RH及び25℃(典型的な室内条件)で、サンプルに負荷を与え、及びサンプルを負荷軽減した。水分収着曲線を、以下のように実行した:
【0205】
【表1】
【0206】
HPLCによる化学的純度測定:ダイオードアレイ検出器を備える、Agilent HP1100シリーズの装置上で、及び以下に詳述される2つの方法の1つを使用するChemStationソフトウェア vB.02.01-SR1を使用して、純分分析を行った。
【0207】
【表2】
【0208】
偏光顕微鏡法(PLM):サンプルを、画像キャプチャー用のデジタルビデオカメラを備えたLeica LM/DM 偏光顕微鏡上で研究した。小量の各サンプルを、スライドガラス上に置き、液浸油に浸け(mounted in)、ガラススリップで覆い、その個々の粒子状物質をできるだけうまく分離した。サンプルを、適切な倍率及び部分偏光で観察し、λの類似色彩フィルターにつなげた。
【0209】
<実施例2B:非晶質の化合物(1)の調製>
化合物(1)を、tert-ブタノール中に溶解した。サンプルを凍結乾燥過程にさらし、その中で、サンプルをろ過し、冷凍し、凍結乾燥した。固形物を収集し、XRPD、1H-NMR、調節したDSC、HPLC純度、及び光学顕微鏡法によって特徴付けた。非晶質物質の調製を、再現性をチェックするために繰り返した。図6は、凍結乾燥によって得られた固形物のXRPDである。
【0210】
<実施例2C:化合物(1)フォームIの調製及び分析>
化合物(1)を、メタノール中に溶解した。溶液中のメタノールを蒸発させ、結果として生じる固形物を、収集し、XRPD、1H-NMR、DSC、HPLC純度、及び光学顕微鏡法によって特徴付けた。図1は、フォームIのXRPDである。TGAによるフォームIの熱分析を、10℃/分の加熱速度で実行した。TGAサーモグラムにおいて体重損失は観察されず(図7)、これは、物質が非溶媒和の結晶形態であったことを示した。サンプルの分解は、230℃までの温度で始まった。DSC分析(図7)は、化合物(1)(フォームI)の融点を示す196℃での急速な吸熱(91.7J/g)を示した。
【0211】
<実施例2D:化合物(1)パターン2の調製及び分析>
フォームI化合物(1)を、周囲条件でクメン中でスラリーにした。混合物を、50℃/室温のヒート・クールサイクル(heat-cool cycles)(8時間のサイクル)で、70時間成熟させた。固形物をろ過し、XRPDによって分析した。固形物を、真空オーブンにおいて約35℃で24時間乾燥した後、別のXRPD分析を得た。固形物(パターン2)の1H-NMR分析は、0.2eqまでのクメン含量を示した。
【0212】
<実施例2E:化合物(1)パターン5の調製及び分析>
非晶質の化合物(1)を、周囲条件で水中でスラリーにした。混合物を、50℃/室温のヒート・クールサイクル(8時間のサイクル)で、70時間成熟させた。固形物をろ過し、XRPDによって分析した。固形物を、真空オーブンにおいて約35℃で24時間乾燥した後、別のXRPD分析を得た。固形物の1H-NMR分析は、残留溶媒を示さなかった。GVS実験は、約50%の相対湿度での水和物(パターン5)の存在を示した。
【0213】
<実施例2F:化合物(1)フォームIIの調製及び分析>
パターン5化合物(1)を、40%の相対湿度を下回るくらいにまでGVSによって脱水した。
XRPD分析を、非溶媒和の結晶上で行い、フォームIIを示した。
【0214】
<実施例2G:化合物(1)フォームIIIの調製及び分析>
温度のエンドポイントで、約5分間10℃/分で、約200℃まで水和物のサンプルを加熱することにより、パターン5化合物(1)を融解した。その後、サンプルを冷却した。サンプルの化学的な完全性(integrity)が温度によって影響を受けなかったことを確認するために、1H-NMR分析を行った。XRPD分析を、非溶媒和の結晶上で行い、フォームIIIを示した。
【0215】
<実施例3:医薬組成物>
<実施例3A:経口組成物>
経口送達のための医薬組成物を調製するために、式(1)の化合物を、約0.20g/mLのバルク密度及び約0.31g/mLのタップ密度を有するように微粉化した。90mgの微粉化した化合物を、経口投与に適したサイズ「3」のカプセル剤にパック充填した。
【0216】
<実施例3B:経口組成物>
経口送達のための医薬組成物を調製するために、式(1)の化合物を、約0.20g/mLのバルク密度及び約0.31ng/mLのタップ密度を有するように微粉化した。325mgの微粉化した化合物を、経口投与に適したサイズ「00」のカプセル剤にパック充填した。
【0217】
<実施例3C:経口組成物>
経口送達のための医薬組成物を調製するために、90mgの式(1)の化合物を、200mgのラクトース及び1%のステアリン酸マグネシウムと混合する。混合物を混ぜ合わせ、経口投与に適するように、錠剤へと直接圧縮する。
【0218】
<実施例3D:非経口組成物>
注入による投与に適した非経口医薬組成物を調製するために、式(1)の化合物の100mgの水溶性塩を、DMSO中に溶解し、その後、0.9%の無菌食塩水、10mLと混合する。混合物を、注入による投与に適した投与ユニット剤形に組み込む。
【0219】
<実施例4:インビトロの薬理学的試験>
<実施例4A:アンドロゲン受容体結合アッセイ>
アンドロゲン受容体の競合的結合を、変異したAR(384nMのIC50)を発現する、アンドロゲン感受性ヒト前立腺癌細胞株(LNCaP)細胞において、及び野生型AR(845nMのIC50)を発現する細胞において、ラベルが付けられたR1881(アンドロゲンアゴニスト)を使用して測定した。図8は、(化合物5と記載される)化合物(1)が、用量依存性の方法で両方のタイプのARに結合するために、ラベルが付けられたR1881と有効に競合したことを実証する。
【0220】
<実施例4B:リアーゼ活性の阻害>
トランスフェクトされたE. coliによって発現される完全なCYP17を分離し、酵素源として精製した。基質として17α-ヒドロキシプレグネノロンを放射標識した。CYP17活性を、基質のC-21側鎖の開裂中に形成された、トリチウム化した酢酸の量によって測定した。図8は、(化合物5と記載される)化合物(1)が、CYP17に対して300.0nMのIC50値を示したことを実証する。
【0221】
<実施例4C:前立腺癌細胞株のテストステロン誘発性の増殖の阻害>
ヒトの前立腺癌細胞株(LNCaP及びLAPC-4)を、培養において成長させ、1nMのジヒドロテストステロン(DHT)によって刺激した。DHTのこの濃度は、前立腺癌細胞の増殖を刺激した。化合物(1)の付加は、Casodex(登録商標)に似た方法で、テストステロン誘発性の増殖の用量依存性の阻害を誘発し、これを、この実験において陽性対照として使用した。
【0222】
<実施例4D:前立腺癌細胞株におけるアンドロゲン受容体(AR)タンパク質の分解>
培養細胞におけるすべてのタンパク質合成を阻害するために、シクロヘキシミドを、ヒトの前立腺癌細胞株(LNCaP)に加えた。抽出タンパク質が、ARタンパク質に向けられたモノクローナル抗体により調べられた時、シクロヘキシミド処置は、単独で、時間依存性の方法によってARレベルを低下させた。図9において、これらの培養物への化合物(1)の付加によって、結果的に、培養における時間とともに、ARタンパク質の減少率が著しくより大きくなった。
【0223】
<実施例5:インビボの薬理学的試験>
<実施例5A:重度障害型免疫不全(SCID)のマウスにおけるヒト前立腺癌の異種移植片の増殖の阻害>
LAPC4前立腺癌細胞腫瘍の異種移植片を、SCIDマウスに移植した。癌を有するマウスは、体重(BW)当たり50mg/kgの化合物(1)の皮下(SC)投与を毎日2回受けた。腫瘍サイズを毎週測定し、ビヒクル、Casodex(登録商標)又は去勢のみを受けた対照マウスと比較した。図10は、対照と比較して、去勢が最終的な腫瘍容積の著しい減少につながったことを示す。化合物(1)で処置されたマウスは、去勢と比較して、同等の又はより顕著な腫瘍成長の減少を示した。
【0224】
<実施例6:化学療法未治療の去勢抵抗性前立腺癌に苦しむヒトを処置するための化合物1の使用>
【0225】
【表3−1】
【0226】
【表3−2】
【0227】
【表3−3】
【0228】
【表3−4】
【0229】
【表3−5】
【0230】
用語の略語及び定義のリスト
表1にリストされる略語は、この試験プロトコルにおいて使用される。
【0231】
【表4−1】
【0232】
【表4−2】
【0233】
導入
1.1 去勢抵抗性前立腺癌の処置に対する化合物1の使用のための科学的及び臨床的根拠
1.1.1 去勢抵抗性前立腺癌
前立腺癌は、男性において最もよく見られる癌である。大多数の前立腺癌死亡は、従来のアンドロゲン遮断療法に反応性のない転移性疾患の進行が原因である。アンドロゲン遮断療法は、1940年代以降、前立腺癌を有する被験体における標準治療であった。アンドロゲン遮断にもかかわらず、ほとんどの被験体は、最終的に、疾患の進行を経験する(Mohler et al., 2004; Scher et al., 2004)。長年、該疾患のこの後の相は、「ホルモン不応性前立腺癌」又は「アンドロゲン非依存性前立腺癌」と呼ばれた。それ以降、数年間のアンドロゲン遮断療法の後に出現する前立腺癌は、アンドロゲンに依存したままであることが明らかになった。生存した前立腺癌細胞は、(副腎から発現した)低レベルの循環するアンドロゲンを移入し、これらの低レベルのテストステロンにかなり感応的になり、及び実際に、前立腺癌細胞自体内のテストステロンを合成する能力を得た。前立腺癌のこの段階は、現在、「去勢抵抗性前立腺癌」又はCRPCと称される。
【0234】
1.1.2 前立腺癌におけるアンドロゲン及びアンドロゲン受容体の役割の概要
1.1.2.1 前立腺癌におけるアンドロゲン及びアンドロゲン受容体の役割
アンドロゲンは、正常な前立腺の成長、及び前立腺癌において重要な役割を果たす。ほぼすべての初期の前立腺癌がアンドロゲン依存性であるため、アンドロゲン除去は、最初に、前立腺癌を有する被験体における反応をもたらす(Canil et al., 2005; Catalona, 1994; Chodak, 2004; Mohler et al., 2004; Pienta and Bradley, 2006)。アンドロゲン除去療法[ゴナドトロピン放出ホルモン(GnRH)アゴニスト±アンドロゲン受容体(AR)アンタゴニストによる、外科又は内科的去勢のいずれか]は、ほとんどの前立腺癌の被験体のための標準治療である。臨床転帰は、アンドロゲン除去療法の性質及びタイミングに関係なく、類似している。初期反応があり、その後、生化学的な、放射線学的な、及び最終的に臨床的な進行において終了する安定性の期間が続く(Bubley and Balk, 1996; Scher et al., 2004; Scher et al., 2008)。前立腺癌細胞によって確立された1つの重要な生存メカニズムが、低レベルの循環する及び/又は細胞内のアンドロゲンに対するその感受性の劇的な増加であることが明らかになった。これは、アンドロゲン除去によって誘発される。低レベルのアンドロゲンに対する感受性の増加は、ARタンパク質の増加した発現によって与えられる(Gregory et al., 2001; Taplin and Ho, 2001; Gelman, 2002; Litvinov et al., 2003; Lee and Chang 2003; Heinlin and Chang, 2004; Chen et al., 2004)。本発明によると、前立腺癌の処置のための有効な治療は、完全なアンドロゲン遮断を含むべきである。
【0235】
1.1.2.2 アンドロゲン作用のメカニズム及び摂動(Perturbation)
アンドロゲンは、アンドロゲン感受性組織の細胞質内に存在する受容体に結合することにより、前立腺組織及び前立腺癌細胞の成長を刺激する。受容体は、熱ショックタンパク質90などの担体タンパク質に結合される(Pratt, 1993)。一旦結合されると、ホルモン受容体複合体は、細胞核へ移行し、遺伝子転写に影響を与える。ARは、男性の二次性徴の発達及び調整において役割を果たす核内受容体スーパーファミリーのリガンド誘致性のホルモン受容体であり:これらは、精子形成の誘発、および骨及び筋肉量、およびアンドロゲン感受性組織の維持を含む(例えば、前立腺;Heinlein and Chang, 2004; Gelman, 2002)。
内因性のホルモン(例えば、テストステロン)の作用を遮断する薬剤(抗アンドロゲン)は、前立腺癌の処置に高度に有効であり、日常的に使用される(アンドロゲン除去療法)。第1非ステロイド性の抗アンドロゲン、フルタミドは、1989年に前立腺癌療法のために認可された(Shahinian et al., 2006)。続いて、他の非ステロイド性のリガンドが開発され、その主な理由は、化合物のこの種類が、ARのみと反応するからである。最近、アンドロゲン作用を阻害する、より特異的な、非ステロイド性のARモジュレーターが開発され、市場に出されている[例えば、Casodex(登録商標)(ビカルタミド); Bohl et al., 2005]。
【0236】
1.1.2.3 アンドロゲン依存性前立腺癌のための利用可能な治療
他の治療は、精巣からの内因性のテストステロンの合成および分泌を減少させるために開発された。アンドロゲンの循環するレベルの低下は、Lupron(登録商標)(ロイプロリド)などの、GnRH受容体アゴニストの投与を介して達成され、それは、1997年に現在のデポー剤のバージョン(depot version)に対する食品医薬品局(FDA)の認可を得た。継続的に与えられた時、Lupron(登録商標)は、脳下垂体前葉から黄体形成ホルモン(LH)の合成及び放出を減少させる。正常な状況下において、LHは、全身循環において精巣に移動し、そこで、テストステロンの合成及び放出を刺激する。GnRHによって誘発された脳下垂体のLHのダウンレギュレーションの結果として、テストステロンの循環するレベルは低下する(Catalona, 1994)。テストステロンの循環するレベルの低下により、ほとんどのアンドロゲン依存性前立腺癌細胞は、細胞分裂を止め、死滅する。
【0237】
GnRH受容体アゴニスト及び抗アンドロゲンの組み合わせは、精巣からのアンドロゲンの放出を減少させるためだけでなく、前立腺などのアンドロゲン感受性組織において、相当する受容体に結合する、残りの低レベルのアンドロゲンの能力を遮断するためにも使用される。
【0238】
1.1.2.4 去勢抵抗性前立腺癌;増加したアンドロゲン受容体及びアンドロゲン
最初は腫瘍成長を抑えるのに有効であるが、これらのアンドロゲン除去療法は、結局、ほとんどすべての被験体において失敗し、CRPCにつながる(Mohler et al., 2004)。前述したように、すべてではないが、ほとんどの前立腺癌細胞は、最初に、アンドロゲン除去療法に反応する。しかしながら、時間とともに、前立腺癌細胞の新しい個体群は、アンドロゲン除去療法によって生じた選択圧に反応したが、それに対して反応しなくなると出現する。原発性癌が、利用可能な療法に反応がないだけではなく、癌細胞はまた、原発腫瘍から離れ、血流を移動し得、疾患が離れた部位(特に骨)に広がる。他の効果の中で、これは、著しい疼痛及び極端な骨脆弱性を引き起こす(Stoch et al., 2001; Greenspan et al., 2005; Ye et al., 2007)。GnRH療法は、循環するテストステロンの濃度において、90〜95%より大きな減少を引き起こし得、前立腺細胞内のステロイドホルモンの濃度は、50%だけ減少する。これは、アンドロゲン感受性腫瘍の刺激を継続するために、十分な量のアンドロゲンを未だ提供する(Mizokami et al., 2004; Mohler et al., 2004; Titus et al., 2005; Page et al., 2006; Mostaghel et al., 2007)。
【0239】
1.1.3 去勢抵抗性前立腺癌におけるアンドロゲン受容体の増加
以前に、治療によって誘発されるような、テストステロンの去勢レベルの存在下における前立腺癌細胞の増殖が、ホルモン非依存性前立腺癌細胞型の出現によるものであると仮定された。しかしながら、ここでは、CRPC細胞が、細胞内のAR密度を増加させることにより、アンドロゲン除去療法によって誘発された低レベルの循環する睾丸アンドロゲンを補うと考えられる。最初の治療から生存し、去勢抵抗性になった前立腺腫瘍組織において、ARの数が増加されることが最近示された(Chen et al., 2004; Litvinov et al., 2003; Scher et al., 2004; Heinlein and Chang, 2004)。
【0240】
このように、前立腺癌細胞は、かなり低いレベルの循環するテストステロンに反応することができ、循環するアンドロゲンの量が低減したにもかかわらず、十分なアンドロゲンの援助を受け続ける。
【0241】
1.1.4 腫瘍内アンドロゲンにおける評価:副腎及び腫瘍内の源(Intratumoral Source)
GnRHアンタゴニスト及びアンドロゲンアンタゴニストで処置された男性における、低いがまだ測定可能な量の循環するアンドロゲンの1つの源は、副腎である(Mizokami et al., 2004; Labrie et al., 2005)。副腎性アンドロゲン産生は、睾丸のテストステロン産生を低減するほとんどの治療による影響を受けない。前立腺癌細胞が、循環する副腎性アンドロゲンを効率的に移入し、処理するその能力を向上させることにより、細胞内の副腎性アンドロゲン含量を増加させることを、いくつかの研究が示した(Bubley and Balk 1996; Mizokami et al., 2004; Labrie et al., 2005; Stanbrough et al., 2006)。
【0242】
副腎性アンドロゲン生合成の適度の阻害は、CRPCの処置に多少有効であると示された抗真菌剤である、ケトコナゾールの投与によって達成された。しかしながら、ケトコナゾールは、前立腺癌の処置における使用のために認可されていない。本発明は、精巣及び副腎の組織においてアンドロゲン合成を阻害する、強力で特異的な化合物が、前立腺癌のこの段階の処置により有効であり得るという認識を包含する。
【0243】
副腎性アンドロゲンの移入に加えて、アンドロゲン除去療法への長期曝露の後に生存する前立腺癌細胞はまた、アンドロゲンの前立腺内の合成に必要な、アップレギュレートされたレベルの酵素を有する(Nakamura et al., 2005; Stigliano et al., 2007; Holzbeierlein et al., 2004)。イントラクラインの仮説(intracrine hypothesis)は、中断されないアンドロゲンの援助を保証するために、CRPC細胞によって利用された生存メカニズムとして示唆された(Loberg 2005; Mostaghel et al., 2007)。この概念は、前立腺癌を研究する科学者および臨床医のコミュニティー内の有意な援助を獲得した。結果として、この概念は、すべてのステロイド産生組織(精巣、副腎及び、CRPCの場合には、前立腺癌細胞自体)においてアンドロゲン合成を阻害する、治療用化合物の増加する需要を刺激した。
【0244】
利用可能なままである細胞内のアンドロゲンに対する反応を強めるための、3つの異なる経路を増幅させることによる、低レベルの循環するアンドロゲンによって特徴付けられた環境において、CRPC細胞は生存する。これらは:
・ARコピー数を増加させ、その結果、内科的去勢療法によって誘発された低レベルの循環するアンドロゲンに対する、細胞の感受性を増加させる、ARの発現のアップレギュレーション、
・アンドロゲン遮断療法の後の循環において残る、副腎性アンドロゲンの移入及び処理に関係する酵素の発現の増加、
・ステロイド産生を規制する遺伝子の発現の増加を含み、それによって、CRPC細胞がその細胞自体のアンドロゲンを合成することが可能となる。
【0245】
CRPCを有する被験体に生じたこれらの変化に取り組むために、新しい治療が必要とされる。
【0246】
1.1.4.1 去勢抵抗性前立腺癌の処置としての化合物1
・CRPC細胞の生存メカニズムの新たな理解を考えると、新しい治療が必要とされる。化合物1は、以下に示されるような必要な特性をすべて示す:
・アンドロゲン生合成を減少させる:化合物1は、副腎、精巣、及び前立腺におけるアンドロゲン産生を制御する酵素である、チトクロームC17α-ヒドロキシラーゼ/C17,20-リアーゼ(CYP17)の活性を阻害する。
・ARシグナル伝達を減少させる:化合物1はARに結合し、テストステロン結合の競合的インヒビターである。
・アンドロゲン感受性を減少させる:化合物1は、前立腺癌細胞内のARタンパク質の含量を減少させ、低レベルのアンドロゲン増殖シグナルによって持続されるべき細胞の能力を低下させる。
【0247】
本発明は、前立腺癌、及び特にCRPCの処置における化合物1の使用のための治療方法及び組成物を提供する。
【0248】
1.2 リスク・ベネフィット評価
これは、化合物1のヒト初回投与試験であり、ヒトにおける潜在性の毒性効果は、まだ完全には知られていない。化合物1が、CRPCの処置における新しい潜在的に活性な薬剤であることと一致する、生物学的な特性を有していることを、非臨床データの評価は示す。データは提案されたMOAを支持し、細胞に基づいたモデル及び動物モデルにおいて最小の毒性を示す。見られるただ1つのシグナル、肝毒性は、抗アンドロゲンによって潜在的に誘発され得る既知の有害反応である(Thole et al., 2004)。更なる既知のクラスの影響、明らかなミネラルコルチコイド過剰症(AME)は、非臨床試験において見られなかった。この試験を、有害な非臨床的結果、理論的なMOA、及び既知のクラスの影響問題の考察とともに考案した。試験は、潜在的な臨床的利点を評価し、概念実証を提供する一方で、被験体に対するリスクを最小化する。
【0249】
本発明は、生物学的活性に関する安全性および潜在性の両方を考慮する投薬スキームを特に提供する。1つの実施形態において、650mgの初回量を利用する。この投与量は、安全シグナルが見られた、イヌにおける投与量よりも18倍少なく、ステロイド産生組織において発現されたリアーゼ酵素の阻害、ARへのアンドロゲンステロイドの結合の阻害、及びアンドロゲン反応性組織におけるARタンパク質の不安定化において、潜在的に生物学上有効であると予期される。さらに、140、280、及び560mgのヒトの皮下投与に相当するレベルで、重症複合免疫不全症(SCID)のマウスモデルにおけるアンドロゲン依存性のヒト前立腺腫瘍のサイズにおいて、劇的な減少が示された。
【0250】
治験計画
本実施例は、化学療法未治療のCRPCの処置に関する化合物1の、相1及び2、非盲検、用量増加、用量比較の試験を記載する。試験の個体群は、確認された前立腺の腺癌、及び最も最近のPSAレベルが≧5ng/mLである、少なくとも1週間離されて、少なくとも2つの連続した機会において上昇する、前立腺特異性抗原(PSA)レベルとして定義される、アンドロゲン除去療法にもかかわらず進行する疾患を有する、18歳以上の男性を含む。
【0251】
この試験は2つの段階に分けられる:相1(用量増加)、その後の相2(選択された用量比較);試験の相1及び/又は相2の段階の終了の後に、資格のある被験体に対する随意の延長の相がある。
【0252】
最初の処置訪問の28日間以内にスクリーニングが行われる。被験体は、夕食とともに、臨床試験薬(化合物1)を12週間毎日1回とる。試験の訪問は、2週ごとに行われ、被験体は、12週間の投薬期間の終わりに、試験終結の訪問のために戻る。その後、資格のある被験体は、継続的な治療のために延長のアームに入ることを許可され得る。処置は、疾患の進行、離脱、被験体の容認できない毒性が出るまで、又は研究員の判断で継続し得る。
【0253】
個々の被験体に関して、試験の相1又は2の段階の最大期間は、[スクリーニングに28日間まで及び89日間までの処置(相1又は2)を含む]117日間までである。処置ウィンドウ計算(treatment window calculation)は、試験終結の訪問(+5日)のための最大ウィンドウを含む。更なる延長投薬は、資格のある被験体に提供され得る。
【0254】
これらの相の各詳細を、下記に記載し、図11において図表で提示する。すべての相1のデータのデータの検討を、相2に入る被験体のための投与量を決定するために行う。相2において最後の被験体が12週目に達した後に、すべての試験データのデータの検討を、今後の試験(及び継続される延長)のための投与量を決定するために行う。
【0255】
1.3 相1の考案及び計画:記載
相1の段階の目的は、許容可能な安全性プロフィールを提供する化合物1の投与量(複数可)を設定することである。許容可能な安全性プロフィールを、≦35%の正確なDLT比率での投与量として定義する。
これは、3つの投与量群を、1の投与量当たり6まで(3以上)の被験体の標的とともに登録する、用量増加スキームである。調査されるべき標的とされる投与量群は、650mgの化合物1;1300mgの化合物1;及び1950mgの化合物1である。
【0256】
内部モニタリング委員会(IMC)によって完了される検討に続いて、これらの投与量群は、より高い又はより低い投与量群、又は中間の投与量までの用量増加又は用量減少によって修正され得る。これらの修正は、調査されている投与量の範囲内にのみ生じ得る。
【0257】
【表5】
【0258】
被験体の群を、650mgの化合物1から開始して、増加する投与量で処置する。次に続く投与量群は、登録(新しい被験体)のために空けられ得、最初の3の被験体が、より低い投与量群に集まった後の、増加のための基準を提供する。≦35%の正確なDLT比率は、許容可能であると考えられる。用量設定規則はこれに基づき、実験の相1の段階の目標は、許容可能な正確なDLT比率を有する最も高い用量を識別することである。DLTは、任意の臨床試験薬に関連するグレード3又はより高い有害事象(AE)として定義され、臨床試験薬と、ひょっとしたら、おそらく、又は明確に関連があると考えられる。
【0259】
臨床試験薬を少なくとも1回用量受け取らない、相1の段階において登録された被験体を、適切な投与量群における更なる被験体と交換する。同様に、登録された被験体が資格がないと発見されるとき、そのような被験体を、適切な投与量群における更なる被験体と交換する。
【0260】
1.3.1 最初の3の被験体の後の増加に対する基準
連続的な投薬のリアルタイムデータが電子症例報告書(eCRF)によって収集される、最小の4週間に一旦すべての3の被験体が達すると、各投与量群における最初の3の被験体のDLTのための検討を行い、検討は、すべての利用可能なデータを含む。データは、この検討に含まれるべきと確認される源(source)である必要はない。DLTは、任意の臨床試験薬に関連するグレード3又はより高いAEとして定義され、臨床試験薬と、ひょっとしたら、おそらく、又は明確に関連があると考えられる。
【0261】
最初の3の被験体のデータの検討の後、もし:
・0/3の被験体がDLTを経験すると、次の投与量群までの増加が生じ得る。
・1/3の被験体がDLTを経験すると、3の更なる被験体は、次の投与量群までの増加前のこの投与量で登録されるべきである。
・2/3又は3/3の被験体がDLTを経験すると、投薬はこの投与量群で停止され、相1の投与量減少が生じる。
【0262】
DLTの検討期間内では、臨床試験薬にひょっとしたら、おそらく、又は明確に関連があると考えられる、任意のグレード2の肝毒性に関連する事象(HTRE)は、最大50%まで次の投与量群に対する増加を制限し得る。
【0263】
1.3.2 最大限の6の被験体への拡張が必要とされるときの増加の基準
3の更なる被験体を増加の前に投与量群に加えなければならないとき、一旦更なる3の被験体が最小の4週の連続的な投薬に達すると、すべての6の被験体におけるDLTのための検討が行われる。
【0264】
拡張の後、もし:
・2/6の被験体がDLTを経験すると、次の投与量群までの増加が生じ得る。
・3/6の被験体がDLTを経験すると、投薬はこの投与量群で停止し、相1の投与量減少が生じる。
【0265】
1.3.3 相1の投与量減少
減少が必要なとき、被験体は、1)次のより低い投与量群まで、又は2)実現可能であれば、DLTが生じた投与量と次のより低い投与量群の間の新しい中間の投与量まで減少される。例えば、3以上の被験体が1300mgの群においてDLTを有すると、1300mgと650mgの間の投与量が被験体に再び与えられ得る。開始する投与量群(650mg)からの減少が必要であると、3の被験体の更なる群は、325mgで登録され得る。
【0266】
減少が生じると、減少が生じてから投与量へ戻る増加が、両方の投与量で登録された被験体の検討に続いて、考慮され得る。
【0267】
1.3.4 相1のデータの検討
相1のデータの検討は、相2の開始前に完了する。安全性及び生物学的シグナルを支持する12週間までのデータを、相2のための投与量を選択するために利用する。相1において検討された及び許容可能なDLT特性を有すると測定された投与量のみ、又は許容可能なDLT特性を有するものより低い新しい投与量は、相2の試験のために考慮される。
【0268】
3.1.5 投与量遅延/投与量修正
被験体を毒性に関して監視し、投与量は、個々の被験体耐性に従って調節され得る。
【0269】
1の投与量レベルによる用量減少は、遭遇する毒性のタイプ及び重症度によって必要とされ得る。最小用量は一日325mgである。用量減少ガイダンスは表3に提供される。
【0270】
最小量の処置に関連する効果を有する4週間の処置を終えた被験体は、表2に記載されるように、1プロトコル当たりの被験体の割り当てられたレベルで投薬を継続し得る。
【0271】
【表6】
【0272】
注:AMEの徴候を示す被験体は、推奨されるAME処置に反応する限り、妨害なしで研究処置を継続し得る。被験体がAMEを有すると疑われると、1のラベル当たりの、エプレレノンでの処置、及び/又は電解質補充が推奨される。医学的に実現可能なとき、AMEに関する任意の他の処置は、トーカイメディカルアドバイザー(又は指名された人)及びメディカルモニターと前もって話し合われるべきである。
【0273】
現在の投与量が325mgであり、更なる用量減少が必要であると、毒性ガイドラインが示すと、被験体は研究から外される。>4週間の投与量中断を必要とする被験体は、研究から外されることが考慮される。
【0274】
1.4 相2の考案及び計画:記載
相2の目的は、生物学的なシグナルを評価し、もたらされる各投与量の許容可能な安全性プロフィールを確認することである。正確なDLT比率が35%を超過することを示唆する、合理的に説得力のある証拠が存在するならば、特定の投与量への登録が停止されるように、停止規則は実施される。
【0275】
新しい20の被験体を、相1から進められた各アームに登録する。2のアームが進められると、被験体は2のアームに無作為化される(最大で40の被験体)。このサンプルのサイズの論理的根拠に関しては、この概要の統計的方法のセクションを参照する。
【0276】
相1に関して、DLTは、任意の臨床試験薬に関連するグレード3又はより高いAEとして定義され、臨床試験薬と、ひょっとしたら、おそらく、又は明確に関連があると考えられる。
【0277】
相1に関して、臨床試験薬を少なくとも1回用量受け取る被験体のみが相2にあると考えられる。臨床試験薬を少なくとも1回用量受け取らない被験体の比率、及び臨床試験薬が投与されなかった理由を記録する。これは非常に少ない頻度で生じ、その理由は通常管理上のものである。
【0278】
1.4.1 相2の投与量選択
2までの投与量を、相2における研究のために選択する。相1のデータの検討を、投与量(複数可)を測定するための基礎として利用する。投与量(複数可)の選択を、相1からの安全性と生物学的作用のデータのIMC検討に続いて決定する。
【0279】
単一用量の相2における診査につながる可能なシナリオは、以下を含む:
・観察されたDLT比率が相1における投与量で35%の閾値を越えると、これが生じたことよりも低い投与量群が単独で診査され得る。
・相1におけるすべての投与量が許容可能なDLT特性を示すと、相1における最大投与量、1950mgは、単独で診査され得る。
【0280】
相2における2の投与量の診査につながる可能なシナリオは、以下を含む:
・観察されたDLT比率が相1において35%の閾値を超過すると、これが生じたことよりも低い投与量群、及びより低い投与量が選択され得る。より低い投与量は、次のより低い投与量、又は35%のDLT比率が超過されたことよりも低い投与量と次のより低い投与量の間の中間の投与量であり得る。
・相1におけるすべての投与量が、<35%のDLT比率を示すと、相1における最大投与量、1950mg、及び第2投与量が選択され得る。第2投与量は、1300mgの投与量又は1950mgと1300mgの間の中間の投与量であり得る。
・相1において診査される1より多い投与量が、<35%のDLT比率を示すと、高用量/低用量のシナリオが考慮され得る。
【0281】
1.4.2 相2の停止規則
相2のデータを、DLTのために継続的に検討する。データは、この検討に含まれるべきと確認される源である必要はない。
【0282】
関連するDLTの発生率が35%を超過すると、その後、IMCはデータを検討し、投与量に対する修正を行う必要があるかを決定する。
【0283】
1.4.3 相2の投与量修正
投与量が相2において停止されると、相2の停止規則により、被験体の投薬は、データの検討に続いて以下のように修正される。
・2の投与量が相2において実行されると、20の被験体が登録されるまで、停止されなかったアームにおいて登録は継続する。停止されたアームが2の投与量群におけるよりも高いと、IMCは前進するためにどの投与量が使用されるべきかを推奨するように要求される。停止された試験のアームにおける被験体は、継続的な投与量で処置され、12週のフォローアップスケジュールを完了する。
・1の投与量が相2において実行されると、続く被験体は、1)相1において許容可能なDLT特性を有すると見られる、次のより低い投与量群において、又は、2)実現可能であると、相1において許容可能なDLT特性を有すると見られる、DLTが生じた投与量と次のより低い投与量の間の新しい中間の投与量まで投薬される(又はそれらに換えられる)べきである。20の被験体がより低い投与量で登録されるまで、投薬のこの減少は起こる。停止規則がこのより低い投与量で適用されるとき、これ以上の用量減少は行われない。
【0284】
1.5 延長の考案及び計画:記載
延長される投薬は、試験の相1又は相2の段階における12週が終了した後に、被験体に提案され得る。最初の資格を、許容可能なリスク・ベネフィット比率、及び研究における10週目にわたる生化学的な進行の徴候の欠如に基づいて、12週目の訪問で決定する。継続される資格を、継続される許容可能なリスク・ベネフィット比率、及び生化学的な進行の徴候の欠如に基づいて、延長の全体にわたって評価する。
【0285】
許容可能なリスク・ベネフィット比率を、メディカルモニター及び/又はメディカルアドバイザーによる確認とともに、主要な研究員(PI)によって決定する。
【0286】
生化学的な進行を、前立腺癌ワーキンググループ2(Prostate Cancer Working Group 2)(Scher et al., 2008)により、a)PSA増加≧25%、及び(基準線からの低下が生じるならば)最下点を超える≧2ng/mL、又は、b)PSA増加≧25%、及び(基準線からの低下が生じないならば)基準線からの≧2ng/mLとして定義する。
【0287】
延長のための投与量は以下のとおりである:
・相1の間:割り当てられた相1の投与量で延長が生じる。
・相2の間:相2において診査される投与量での延長である。相2において使用されない投与量で最初に投薬される相1の被験体に、相2の投与量を再び与える。
・相2の終了後:相1及び2からのデータの検討に続いて、投与量(複数可)を決定する。
【0288】
延長の間に、基準線から12週までのPSA、又はPSA最下点(最初に生ずるどちらか)における50%又はそれ以上の減少を有する被験体のパーセントによって示されるような、[AE、身体検査、生命徴候、12誘導心電図(ECG)、臨床検査室評価のIMCによる定期的な検討による]安全性、処置コンプライアンス、及び有効性、コンピューター断層撮影(CT)/磁気共鳴画像法(MRI)及び骨スキャンの変化、固形腫瘍における反応評価基準(RECIST; v. 1.1)による進行、特殊実験室の変化、及び進行までの時間(TTP)、無進行生存(PFS)、及び全存率(OS)に関して、被験体の試験が行われる。
【0289】
処置は、疾患の進行、被験体の離脱、容認できない毒性が出るまで、又は研究員の判断で継続し得る。
【0290】
3.1.5 内部モニタリング委員会
外部データの安全性を監視する委員会は、この試験のために設立されない。研究員、メディカルモニター、及びメディカルアドバイザーを含む、形式的に認可を得た組織内のIMCが設立される。このプロトコルの目的のために、組織内のIMCは、重篤有害事象(SAE)において収集された簡略なデータを検討し、各投与量群における最後の被験体が登録した後、及び相1において新しい投与量レベルを始める前の各4週の期間の終わりに、及び相2において必要とされるような、臨床的なデータベースを最小限に検討する。IMCは、プロトコル内で提供されるガイダンスに従うが、自らの判断で、報告されるDLT、更に拡張される投与量群、又は投薬の計画を再評価する権利を保持する。会議の結果は、必要に応じてプロジェクトファイルに文書化され、行動がとられる。試験における被験体の管理に関する直接の意味合いを有する結果は、予期されない及び薬物に関連するSAEと関係する時間枠におけるすべてのPIに伝えられる。
【0291】
1.6 エンドポイント
1.6.1 相1のエンドポイント
1.6.1.1 一次エンドポイント
・AEの発生率
・以下の更なる安全パラメーターの基準線からの変化:臨床検査室評価、身体検査、生命徴候、及び12誘導ECG。
【0292】
1.6.1.2 二次エンドポイント
・基準線から12週までのPSA、又はPSA最下点(最初に生ずるどちらか)における50%又はそれ以上の減少を有する被験体のパーセント。
・CT/MRI及び骨スキャンにおける基準線からの変化。
・RECIST基準による進行。
・更なる特殊実験室における基準線からの変化。
・腫瘍内のARタンパク質、テストステロン、及びジヒドロテストステロン(DHT)(生検サブセット中)の基準線からの変化。
【0293】
1.6.2 相2のエンドポイント
1.6.2.1 一次エンドポイント
・基準線から12週までのPSA、又はPSA最下点(最初に生ずるどちらか)における50%又はそれ以上の減少を有する被験体のパーセント。
【0294】
1.6.2.2 二次エンドポイント
・AEの発生率
・以下の更なる安全パラメーターの基準線からの変化:臨床検査室評価、身体検査、生命徴候、及び12誘導ECG。
・CT/MRI及び骨スキャンにおける基準線からの変化。
・RECIST基準による進行。
・更なる特殊実験室における基準線からの変化。
・腫瘍内のARタンパク質、テストステロン、及びDHT(生検サブセット中)の基準線からの変化。
【0295】
1.6.3 延長に特有のエンドポイント
延長のエンドポイントは相2に関し、更なる以下の二次エンドポイントを有する:進行までの時間(TTP)、無進行生存(PF)、及び全存率(OS)。図12(表5)は、研究の相において使用される手順のスケジュールを記載する。以下の注釈は、図12に相当する。
ET:期限前契約解除;ICF:インフォームドコンセント用紙;ECG:心電図;CT:コンピューター断層撮影;MRI:磁気共鳴画像法;PCF:前立腺癌財団;PSA:前立腺特異性抗原;CBC:完全血球計算;PSA:前立腺特異性抗原;AME:明らかなミネラルコルチコイド過剰症;PK:薬物動態学。
1:被験体は、相1及び2の間に毎週、CBC及び化学検査が行われることが必要とされる。臨床現場での実験室においてこれを行うほど、十分近くない被験体に関して、地方の実験室が、これら検査のない週に関して利用され得る
2:これが期限前契約解除の訪問であると、研究員は、試験終結の訪問のために記載される安全性評価を行なうために、あらゆる努力をしなければならない。
3:1日目は、(10:00と14:00の間の食事とともに摂られる)臨床試験薬の最初の投与である。
4:12誘導ECGは、投与の0時間(投与前(predose))及び4時間(±15分)後に行われる(PKサンプル前)。
5:基準線に関して、使用されるCT/MRI及び骨スキャンの結果は[試験の包含の目的のために繰り返されているスキャンを回避するために]、1日目から4週間以内の日付でなければならず、記録される4週のウィンドウ以外の次に空けられた投与(open dose)に対する包含のためのスクリーニングのCT/MRI又は骨スキャンの使用に関して、トーカイメディカルアドバイザー(又は指名された人)と相談して、メディカルモニターによって例外が認められ得る;繰り返すために、相1又は2の終結の訪問又はPSA最下点(最初に生ずるどちらか)でスキャンが行われるべきである。延長投薬の間に、CT/MRI及び骨スキャンが12週ごとに行われる。もし基準線で使用される同じ様式が、試験においてすべての後の評価のために継続的に利用されるならば、CTスキャンの代わりのMRIの使用は許容可能である。万一、予期しない事象が生じ、処置する医師の判断において、任意の他のスキャンの形態を有するのが被験体の利益であるならば、これは、メディカルモニター及びメディカルアドバイザーと話し合われるべきである。
6:アンドロゲン受容体タンパク質、テストステロン、及びジヒドロテストステロンの評価のための生検は、ICFを作成する別々の機関の下で被験体のサブセット(PCFの許可)において提案される。相1又は2の終結の訪問又はPSA最下点(最初に生ずるどちらか)で生検が繰り返されるべきである。
7:被験体がAMEを有すると疑われると、1のラベル当たりの、エプレレノンでの処置、及び/又は電解質補充が推奨される。医学的に実現可能なとき、AMEに対する任意の他の処置は、トーカイの医薬情報提供者及びメディカルモニターと前もって話し合われるべきである。
8:安全性実験室評価が、1日目の7日より前に記録されると、それらの評価は、投薬前に繰り返され、確認されるべきである。
9:訪問が1週間ないときのみの安全性検査(検尿はない)。
10:投与前に行われる試験
11:11-デオキシコルチゾール、コルチコステロン、コルチゾール、及びテストステロンだけが実行される。以下の試験は実行されない:プレグネノロン、17α-ヒドロキシプロゲステロン、デヒドロエピアンドロステロン、デオキシコルチコステロン、及びアンドロステンジオン。
12:すべての相1及び2の被験体は、1日目に0時間(投与前)及び投与(10:00と14:00の間となる投薬)の4時間(±15分)後にPK抽出(draws)を受ける。残りの訪問で:単一のPKサンプルは、(被験体の日記から計算されるように)最後の投与からの時間の記録によって抽出される。
13:延長へと継続する場合。
14:報告はスクリーニングで始まる。
【0296】
【表7】
【0297】
CBC:完全血球計算;EDTA:エチレンジアミン四酢酸;K2EDTA:エチレンジアミン四酢酸ジカリウム。
1: 中央検査室で実行される。
2: 薬物動態学的サンプルを、提供されるK2EDTA管に収集し、すぐに氷上に置き、血漿分離のために遠心分離にかける。サンプルを、血漿分離の8時間以内に冷凍する。
3: 前立腺癌財団(PCF)の許可により完了した生検を受ける被験体のサブセットにおいてのみ行う。
【0298】
2. 被験体の選択及び離脱
試験個体群は、CRPCを有する被験体から成る。この試験のための資格を有するために、被験体は、書面による同意を提供でき、すべての包含基準を満たし、排除基準は1つも満たさない。
【0299】
2.1 被験体の包含基準
被験体は、試験のための資格を有するために、登録前に以下の包含基準のすべてを満たさなければならない:
1. 投薬スケジュールを厳守すること、すべての試験の訪問及び承認を報告すること、使用すること、及び健康及び研究試験の情報を発表することへの同意を提供する、インフォームドコンセント用紙(ICF)への署名。
2. 18歳以上の男性。
3. 前立腺の組織学的に又は細胞学的に確認された腺癌(神経内分泌分化又は小細胞の組織学的検査を除く)。
4. 最も最近のPSAレベルが≧5ng/mLである、少なくとも1週間離されて、少なくとも2つの連続した機会において上昇する、PSAレベルとして定義される、アンドロゲン除去療法にもかかわらず疾患が進行していること。
5. GnRHアナログ又は精巣摘除を伴う進行中の生殖腺のアンドロゲン遮断療法。精巣摘除を伴わなかった被験体は、有効なGnRHアナログ治療を維持されなければならない。
6. 米国東海岸癌臨床試験グループ(ECOG)の活動指標0又は1。
7. >12週の寿命。
8. 複数のカプセル剤を呑み込むことができる。
【0300】
2.2 被験体の排除基準
登録前に以下の排除基準のいずれかを満たす被験体は、試験に参加する資格を有さない:
1. 登録の<4週間前に実験的治療を伴う別の臨床試験への参加。
2. 下記の1つ以上を有する転移性の被験体:1) 肝障害;2) 積極的な疼痛管理を必要とする転移の確認された放射線学的証拠に関係する骨痛
3. 以下の薬物治療:1) MDV3100、アビラテロン、又はTAK-700での以前の処置;2) ケトコナゾールでの以前の処置;3) 化学療法での以前の処置;4) 登録の≦4週間前に終了する以前の放射線療法;5) 登録の≦4週間前にPSAレベルを低下させると知られる他の治療での処置[Megace(登録商標)(酢酸メゲストロール)、Proscar(登録商標)(フィナステリド)、Propecia(登録商標)(フィナステリド)、Avodart(登録商標)(デュタステライド)、Eulexin(登録商標)(フルタミド)、Casodex(登録商標)(ビカルタミド)、Nilandron(登録商標)(ニルタミド)、Aldactone(登録商標)(スピロノラクトン)、Cytadren(登録商標)(アミノグルテチミド)、エストロゲン、PSAレベルを低下させると知られる任意のハーブ製品(例えば、ノコギリヤシなど)、又は(チーフメディカルオフィサー(Chief Medical Officer)及びトーカイメディカルアドバイザーの判断での)任意の全身性のコルチコステロイドの任意の投与を含む]6) 登録の≦4週間前の心室性不整脈に対する抗不整脈治療での処置;7) 登録の≦4週間前のCoumadin(登録商標)(ワルファリンナトリウム)治療での処置;8) 登録の≦1週間前のLasix(登録商標)(フロセミド)又はZaroxolyn(登録商標)(メトラゾン)での処置;9) 登録の≦1週間前のスタチンでの処置[注:Zetia(商標)(エゼチミベ)は除外されない];又は、10) 登録の≦4週間前の三環系抗鬱薬(TCA)での処置。
4. 以下の検査所見:1) テストステロン>50ng/dL;2) 血清クレアチニン>1.5x 正常上限(ULN);3) ビリルビン>1.5x ULN(注:ジルベール症候群による高められたビリルビンを有する被験体は除外されない);4) アスパラギン酸アミノトランスフェラーゼ(AST)及びアラニンアミノトランスフェラーゼ(ALT)>1.5x ULN;5) ヘモグロビン≦9.0g/dL;6) 絶対好中球数(ANC)≦1.5x 109/L;7) 血小板≦100x 109/L;8) 血清K+<3.5mmol/L。
5. 以下の病状:1) ニューヨーク心臓協会クラスIII又はIVのうっ血性心不全;2) 心筋梗塞(登録の6か月前以内);3) 活発な狭心症;4) B型肝炎又はC型肝炎の病歴;5) 公知のヒト免疫不全ウィルス(HIV)感染症;6) 高血圧(少なくとも2回の機会で測定される、収縮期血圧>150mmHg、又は>95mmHgの拡張期血圧として定義される);7) 副腎機能不全又は高アルドステロン症の病歴;8) AMEの病歴又はブラックリコリス過敏性の病歴;9) 胃腸;10) 化合物1の吸収を妨害し得る障害又は胃バイパス手術;11) 全身療法を必要とする重大な活動性感染又は制御されない非悪性の医学的疾患;12) 処置された非黒色腫皮膚癌以外の二次性悪性腫瘍の(過去5年間の)あらゆる病歴;13) 活動性又は抑制されない自己免疫性疾患;又は、14)活動性の胆道障害。
6. 研究員の意見において、試験の手順に従う被験体の能力を妨害し得るあらゆる物理的又は精神的疾病、又は社会的状況。
7. 妊娠の可能性がある女性との性的接触に関係する場合に、避妊の適切な方法を使用しようとしない男性。
【0301】
2.3 被験体の離脱基準
被験体は、罰則なしでいつでも、及び今後の医療を失うことなく任意の理由で試験から離脱し得る。
【0302】
被験体は、以下の状況の下で離脱されなければならない:
・被験体は同意を撤回する。
【0303】
さらに、被験体は、以下の理由で離脱することが必要とされ得る:
・有害事象(複数可)(AE)
・臨床的、放射線学的、及び腫瘍マーカー(PSA)の進行によって評価されるような疾患の進行
・研究員の見解において、被験体が、プロトコルの必要条件を満たすことができるとこれ以上考えられない場合
・資格基準の違反
・プロトコルに明記される治療計画からの逸脱(例えば、臨床試験薬の正しくない投与、試験訪問を行わない)。
【0304】
すべての場合において、離脱の主な理由は、eCRFに記録されなければならない。被験体が任意の理由で早期に離脱すると(12週目の訪問前)、研究員は、試験終結の訪問(12週目の訪問)のために記載される安全性評価を行なうために、あらゆる努力をしなければならない。
【0305】
2.4 試験の早期終結
もし試験が継続するときに被験体に対して起こり得る危険を示唆する条件又は事象に、研究員又はメディカルモニターが気付くと、関係者間の適切な相談の後に試験が終結し得る。
【0306】
終結を保証し得る条件は、限定されないが、試験に登録される被験体に対する予期されない、重大な、又は許容できないリスクの発見、又は許容可能な割合で被験体を登録できないことを含む。
【0307】
3. 被験体の処置
3.1 臨床試験薬の記載
相1に対する臨床試験薬は、3つの投与量群:(1) 650mgの化合物1(2つの325mgのカプセル剤);(2) 1300mgの化合物1(4つの325mgのカプセル剤);(3) 1950mgの化合物1(6つの325mgのカプセル剤)での化合物1から成る。更なる中間の投与量が診査され得る。
【0308】
相1において得られた結果に基づいて、相2に対する投与量(複数可)は決定されるが、相1において診査された投与量の1つ又は2つを含むと予期され得る。
【0309】
相1及び2において得られた結果に基づいて、延長に対する投与量は決定される。
【0310】
投薬の最初の日(1日目)に、被験体は(10:00と14:00の間の)食事とともに臨床試験薬を経口的に摂ることが必要とされる。被験体は、この訪問で、及び(8日目;訪問3に開始する)2週ごとの訪問で、臨床試験薬を供給され、2日目からの(17:00と21:00の間の)夕食とともに臨床試験薬を経口的に毎日1回摂るように指示される。被験体は、相1及び2において12週間、臨床試験薬を服用する。臨床試験薬に耐性があり、進行の徴候を示さない被験体は、試験の延長のアームにおいて継続される投薬に資格を有し得る。延長のアームにおける投薬の持続時間は、少なくとも12週間である。
【0311】
臨床試験薬は、登録時、及び最初の12週の間の2週ごと(訪問の2、4、6、8、10及び12週目)に調剤され、その後、(延長へと継続する場合)試験終結の訪問で開始して、4週ごとに調剤される。
【0312】
3.2 併用薬
被験体は、試験へのエントリー時に未承認の併用処置を受けるべきではないし、化合物1以外のCRPCに対する処置も試験の間に与えられるべきではない。精巣摘除を有さなかった被験体は、有効なGnRHアナログ治療を維持されなければならない。
【0313】
任意の併用処置の投与がPIによって必要であると考えられると、それは、eCRF及び被験体の医療記録において報告されなければならない。
【0314】
試験のエントリーによって、又は試験の間の任意の時間に被験体によって得られたすべての更なる処置は、併用薬と見なされ、eCRFに文書化されなければならない。以下の情報は、各併用薬のeCRFに記録されなければならない:属名、投与経路、開始日、停止日、投与量、及び指標。併用薬の投与量又はレジメンのあらゆる変化は、eCRFに記録されなければならない。
【0315】
スクリーニング訪問では、被験体は、過去30日間でどの薬剤を服用したかを尋ねられる。各々の続く試験の訪問では、被験体は、現在どの併用薬を服用しているかを尋ねられる。禁止される併用薬は以下を含む:Megace(登録商標)(酢酸メゲストロール)、Proscar(登録商標)(フィナステリド)、Propecia(登録商標)(フィナステリド)、Avodart(登録商標)(デュタステライド)、Eulexin(登録商標)(フルタミド)、Casodex(登録商標)(ビカルタミド)、Nilandron(登録商標)(ニルタミド)、Aldactone(登録商標)(スピロノラクトン)、Cytadren(登録商標)(アミノグルテチミド)、エストロゲン、PSAレベルを低下させると知られる任意のハーブ製品(例えば、ノコギリヤシ及びPC-SPESなど)、又は任意の全身性のコルチコステロイド、Coumadin(登録商標)(ワルファリンナトリウム)、Lasix(登録商標)(フロセミド)及び他のカリウム消耗性利尿薬、Zaroxolyn(登録商標)(メトラゾン)、スタチン[注:Zetia(商標)(エゼチミベ)は除外されない]、心室性不整脈に対する抗不整脈薬、及びTCA。
【0316】
非麻薬性及び麻薬性の鎮痛薬を含む、すべての他の処方及び非処方の薬剤は、必要に応じて使用され得る。すべての併用薬の使用は、各研究の訪問で口頭で評価され、eCRFに記録されるものとする。
【0317】
注:全身性のコルチコステロイドに関して:中程度の慢性閉塞性肺疾患(COPD)、制御された喘息、又はステロイドを必要とする他の慢性症状を有する被験体が、登録基準を満たすが、研究の間(例えば、COPD増悪の間など)に短時間でパルス化した中用量ステロイドを必要とすると、これは、試験に対して責任を負うメディカルモニターによる認可により許容可能になり得る。
【0318】
3.3 処置コンプライアンス
被験体は、各々の続く訪問で、任意の未使用の供給を返すように指示される。各訪問では、被験体は、処置コンプライアンスに関して質問され、返された丸剤は、被験体のコンプライアンス及び薬物責任を確認するために数えられ、文書化される。
【0319】
日記カードは、スクリーニング訪問で、及びその後2週ごとに分配される(延長の相の間は4週ごと);完成したカードは、試験を通して収集される。被験体は、日記カードに投薬の時間及び毎日の服用される丸剤の数を記録し、薬物責任及び処置コンプライアンスのモニタリングを可能にする。
【0320】
投薬コンプライアンスが80〜120%の間で維持されないと、被験体は試験から離脱され得る。被験体は、任意の服用し忘れた投与量のスケジュールを組み直さないように指示される。
【0321】
3.4 無作為化及び盲検化
無作為化は、相2における投与量群への割り当てに使用される。臨床試験薬の盲検化はない;割り当ては、被験体及び研究員に公知である。相2における被験体の無作為化に関して(1以上の投与量群が診査されると)、研究員は双方向ウェブ応答システム(IWRS)を使用する。IWRSは、あらかじめ定義された無作為化リストに基づいて、投与量群に被験体を割り当てる。更なる詳細は、試験マスターファイル(TMF)において見られ得る。
【0322】
4. 臨床試験薬の材料及び管理
4.1 臨床試験薬
4.1.1 定量的組成
化合物1は、17-ヘテロアリールで置換された半合成のステロイドである。A、B、及びC環は、デヒドロエピアンドロステロン(DHEA)及びプレグネノロンの環と構造上類似している。化合物1のベンズイミダゾールの機能性は、sp2炭素(Δ-16,17 オレフィン)を介してD環に加えられる。
【0323】
【化32】
【0324】
4.1.2 投与量選択
この試験のための投与量の安全な、及び潜在的に治療効果のある範囲を予想するために、以下の情報が考慮された:CYP17阻害;受容体の結合データ;標的組織におけるARタンパク質の濃度;腫瘍組織における蓄積;及び動物の安全性、有効性、薬物動態学的(PK)、及び毒素動態学的データ。
【0325】
非臨床データのすべての評価は、化合物1がCRPCを処置するのに必要とされるものと一致する生物学的特性を有していることを示す。例えば、この薬物は、前立腺癌の生化学的な、細胞に基づいた、及び動物のモデルにおいて、生物学的活性を有する。インビトロの安全性試験は、化合物1が遺伝毒性を有しておらず、多くの薬物を代謝すると知られるシトクロームP450システムの活性を妨害せず、及び高いミクロモル濃度でのみオフターゲットの受容体と相互に作用することを示した。化合物は、経口的に生物学的利用が可能であり、ラットに慢性的に投与される時に毒性反応を誘発しない。イヌにおける300mg/kg未満の投与量で、化合物1は毒性を有さないと示された。投与量がmg/kgの体重ベースで投与された時、薬物の同様の最大血中濃度は、ラット及びイヌのモデルにおいて達成された。
【0326】
イヌ(150mg/kg)における無有害作用量(NOAEL)で、化合物1への曝露[最大濃度(Cmax)及び曲線下面積(AUC)]は計算され、類似した曝露指標が、ヒトにおける開始投与量(70kgの男性に対して9mg/kg)で予想された。AUCに基づくと、ヒトの開始投与量は、イヌにおけるNOAELの投与量より16倍低くなると予想される。
【0327】
これらの研究の結果の調整は、650mg/日のヒト初回投与試験の投与量の選択を支持する。この投与量は、安全シグナルが見られた、イヌにおける投与量よりも18倍少なく、ステロイド産生組織において発現されたリアーゼ酵素の阻害、ARへのアンドロゲンステロイドの結合の阻害、及びアンドロゲン反応性組織におけるARタンパク質の不安定化において、潜在的に生物学上有効であると予期される。さらに、140、280、及び560mgのヒトの皮下投与に相当するレベルで、SCIDのマウスモデルにおけるアンドロゲン依存性のヒト前立腺腫瘍のサイズにおいて、劇的な減少が示された。
【0328】
4.2 臨床試験薬の供給
製剤は、微粉化された粉末の入ったカプセル剤である。製剤の組成物は、微粉化された化合物1及びサイズ「00」のCapsugel Coni-Snap(登録商標)カプセルから成る。幾つかの実施形態において、カプセル剤は、325mgの化合物を含む。
【0329】
4.3 臨床試験薬のパッケージング及びラベル付け
製剤は、950ccの白色不透明の高密度ポリエチレン(HDPE)タンパーの明白なホイルで密閉した丸いジャーにおいて、バルク包装され、各臨床現場の薬局に送られる。325mg、サイズ「00」のカプセル剤は、1本のボトル当たり400が包装される。臨床的使用に関して、臨床現場の薬局は、カプセル剤を再包装し、以下の情報によりラベルを付けられた、個々の被験体の使用のためのHDPEボトルへ分配する:被験体の識別;研究使用のみのための化合物1の新薬;製造日及び仕切番号;臨床現場;分類番号。
【0330】
4.4 臨床試験薬の保存
臨床試験薬は、周囲温度(15〜30℃(華氏59〜86度))で保存されるべきであり、標準の制御加熱、換気、及びエアコンディショニング(HVAC)システムを使用して、高熱及び湿気から保護される。
【0331】
4.5 無作為化
2の投与量が相2へもたらされると、1:1の無作為化スケジュールが準備される。
【0332】
4.6 管理
臨床試験薬は経口的に服用され、食事とともに服用されるべきである。臨床試験薬の最初の投与量は、10:00と14:00の間に、臨床現場において被験体によって投与される。最初の投与量(1日目)の受け取りは登録と考えられる。被験体は、その後、上述されるラベルが付けられる要求と一致する薬局からの臨床試験薬を受け取り、17:00と21:00の間で、毎日投与量を服用する。被験体は、任意の服用し忘れた投与量のスケジュールを組み直さないように指示される。臨床試験薬は、登録時、及び最初の12週の間の2週ごと(訪問の2、4、6、8、10及び12週目)に調剤され、その後、(延長へと継続する場合)試験終結の訪問で開始して、4週ごとに調剤される。
【0333】
3.1.5 薬局の責任
臨床試験薬の受け取りによって、棚卸が行われなければならず、指定の職員によって責任の記録(Accountability Log)が記入され、署名されなければならない。指定の職員が、輸送目録に書き留められたすべての品目を含むことを数え、確認することは重要である。所与の輸送における任意の行方不明の、破損された、又は使用できない臨床試験薬は文書化されければならない。更に、PI及び/又は信頼できる現場人員は、輸送の責任がある間に留意された問題を、Pharm-Olamに通知しなければならない。輸送の受け取りの時間に加えて、責任の記録は、薬物が被験体に分配される度に完成されなければならない。
【0334】
8.3.2 臨床試験薬の取り扱い及び処分
研究員は、試験の全体にわたって正確な臨床試験薬の責任の記録を維持する責任を負う。臨床試験薬の各分配は、eCRFに文書化される。現場の薬局によって分配されたすべての未使用の臨床試験薬は、臨床現場に返されなければならず、各臨床現場での標準作業方法に従って適切な方法で破棄されなけばならない。これは、空き瓶と未使用の臨床試験薬を含む瓶の両方の臨床試験薬の瓶を含む。このような供給の破棄は、責任の記録に文書化され、代表者は処分の記録を確認する。
【0335】
5. 有効性の評価
このセクションで詳述されるように、疾患の進行及び生物学的シグナルは、本発明の研究において診査される。
【0336】
5.1 疾患の進行
疾患の進行は以下の評価を実施することにより評価される。
【0337】
5.1.1 臨床的評価
疾患の進行(例えば、疼痛)に関する臨床的評価は、安全性データとして提示される。
【0338】
5.1.2 バイオマーカー
5.1.2.1 前立腺特異性抗原
前立腺特異性抗原は、手順のスケジュールに従って測定される。
【0339】
5.1.3 放射線学的評価
5.1.3.1 固形腫瘍における反応評価基準(RECIST)
固形腫瘍における反応評価基準(RECIST)は、X線、CT、及び磁気共鳴画像法(MRI)を使用して、腫瘍反応を測定するための統一された、容易に適用可能な基準である。該技術は、試験のスポンサーである、国立癌研究所(National Cancer Institute)(NCI)に推薦され、腫瘍標的病変の測定のための形式化された規則を含む。RECIST基準は、任意の国際基準であり、NCI標準ではない。それらは、以前の方法の単純化に基づき[世界保健機構(WHO)、ECOG]、測定可能な疾患(すなわち、少なくとも1つの測定可能な病変の存在)に基づく。RECIST基準は、臨床試験における広範囲の適用のための、単純化された、保存的な、画像データの抽出を提示する。RECIST(Eisenhauer et al., 2009, 本明細書に引用によって組み込まれた)は、この試験に使用される。
【0340】
5.1.3.2 MRI及び骨スキャン
CT/MRI及び骨スキャンは、標準治療の一部として行われると予期され、手順のスケジュールに従って行われる(表5)。もし基準線で使用される同じ様式が、試験においてすべての後の評価のために継続的に利用されるならば、CTスキャンの代わりのMRIの使用は許容可能である。万一、予期しない事象が生じ、処置する医師の判断において、任意の他のスキャンの形態を有するのが被験体の利益であるならば、これは、メディカルモニター及びメディカルアドバイザーと話し合われるべきである。
【0341】
試験包含の目的のために繰り返されているスキャンを回避するために、記録される4週のウィンドウ以外の次に空けられた投与に対する包含のためのスクリーニングスキャン(CT/MRI又は骨)の使用に関して(トーカイメディカルアドバイザー又は指名された人と相談して)、メディカルモニターによって例外が認められ得る。しかしながら、この例外は以下の基準に基づく:被験体は、競合的なスクリーニングにより、以前の相1の投与量における漸増を逃しておかなければならず;すべての他のスクリーニング試験は繰り返されなければならず、被験体が資格を有するままであることを確認しなければならず;臨床試験薬の最初の投与前に、6週以降のスキャンは許容されず;及び、PIの医学的所見において、被験体は放射線学的進行を有することが疑われず、繰り返しスキャンのための臨床的指標がない申し立てが受理されなければならない。
【0342】
この例外の許可は、標準治療を超える増加した頻度でスキャンを繰り返す被験体に対する不必要なリスクを低減する(3〜6か月ごと)。
【0343】
5.1.3.3 進行までの時間(TTP)、無進行生存(PFS)及び全存率(OS)
生存データの獲得のために試験の全体にわたって被験体は追跡調査される。
【0344】
5.2 特殊実験(Special Laboratories)
特殊実験は、手順のスケジュールに従って決定される(表5)。
【0345】
特殊実験は下記を含む:プレグネノロン、17-ヒドロキシプロゲステロン、デオキシコルチコステロン、11-デオキシコルチゾール、コルチコステロン、コルチゾール、DHEA、アンドロステンジオン、及びテストステロン。
【0346】
5.3 腫瘍内のARタンパク質、テストステロン及びジヒドロテストステロン
腫瘍内のパラメーター(ARタンパク質、テストステロン、及びDHT)の分析のための生検は、ICFを作成する別々の機関の下で被験体のサブセットにおいて提案される。相1又は2の試験終結の訪問又はPSA最下点(最初に生ずるどちらか)で生検が繰り返されるべきである。
【0347】
5.4 腫瘍内のパラメーターは、手順のスケジュールに従って決定される。化合物1の濃度
PKサンプルを、手順のスケジュールに従って収集する(表5)。
【0348】
サンプルを、提供されるエチレンジアミン四酢酸ジカリウム(K2EDTA)管に収集し、すぐに氷上に置き、血漿分離のために遠心分離にかける。サンプルを、血漿分離の8時間以内に冷凍する。
【0349】
6. 安全性の評価
6.1 安全パラメーター
6.1.1 研究所のパラメーター
中央検査室:接触リストにおいて識別されるように、研究所の評価は、中央検査室によって行われる。
【0350】
地方の実験室:実験室の評価は、確立された方法によって、各現場の実験室で局所的に行われる。
【0351】
更なる詳細に関しては、臨床検査の改善修正(CLIA)を参照する。
【0352】
安全性の研究所のパラメーターは、研究所のマニュアルに提示される手順のスケジュールに従って決定される。
【0353】
研究所の異常性が、臨床的に有意か、臨床的に有意でないかのいずれかとして、研究員の判断で記録される。
【0354】
6.1.2 生命徴候
以下の生命徴候が、手順のスケジュールに従って評価される:血圧(収縮期及び拡張期;mmHg);心拍度数(拍/分);体温(℃)、経口;呼吸速度(呼吸/分);重量;及び高さ(基準線のみ)。被験体が少なくとも10分間座っているとき、生命徴候が得られる。
【0355】
6.1.3 心電図
12誘導ECGは、手順のスケジュールに従って行われる。被験体が≧3分間背臥位であった後、すべての12誘導ECGが行われる。12誘導ECGモニターは、臨床現場の標準処理手順に従って較正され標準化される。すべての臨床上有意なECG(先に存在する異常性を除く)は、心臓病専門医によって検討される。
【0356】
6.1.4 身体検査
身体検査は、手順のスケジュールに従って行われる。身体検査は、以下の体組織を含む:一般的な様相、HEENT(頭、耳、目、鼻、及び咽喉)、呼吸器、腹、腎(泌尿器科学)、生殖器、筋骨格、神経系、リンパ節、皮膚、及び他のもの。ECOGパフォーマンスステータスが記録される。被験体の疾患がどのように進行しているかを評価し、その疾患が被験体の日常生活能力にどのように影響を与えるかを評価し、および適切な処置及び予後を決定するために、ECOGの規模及び基準は、医者及び研究員によって使用される。
【0357】
【表8】
【0358】
6.1.5 有害事象(AE)
有害事象(AE)は、治験薬を受け取った被験体において生じる任意の好ましくない医学的事象であり、必ずしもこの処置との因果関係を有していない。それ故、AEは、治験薬に関連があろうとなかろうと、治験薬の使用により一時的に関係する、任意の好ましくない及び意図しない徴候(異常な検査所見を含む)、症状、又は疾患であり得る。臨床試験の間に記録されたすべてのAEは、国際医薬用語集(MedDRA)システムに従ってコード化され、器官別大分類に割り当てられる。
【0359】
処置により現れるAE(TEAE)は、臨床試験薬の少なくとも1回用量が投与された後に、始まるか、または重症度及び/又は頻度において悪化するか、または性質が変化するAEとして定義される。試験の間に生じる、併発性疾患を含む、すべてのAEは、eCRFに文書化される。試験へのエントリー前に存在した、併発疾患は、治療期間に悪化しない限り、AEとは考えられない。既往歴はeCRFに記録される。
【0360】
DLTは、任意の臨床試験薬に関連するグレード3又はより高いAEとして定義され、臨床試験薬と、ひょっとしたら、おそらく、又は明確に関連があると考えられる。たとえ重大なものとして考えられなくても、AEは、DLTと考えられ得ることは可能である。
【0361】
6.2 臨床試験薬との関係性
6.2.1 有害事象の評価
各AEは、以下のカテゴリーに関して研究員によって評価される。
【0362】
6.2.1.1 重篤度
重篤AE(SAE)は、任意の投与量で、死に至る、 又は生命を脅かす、任意の好ましくない医学的出来事として定義される。AEとしての分類は、被験体が事象の時に死亡の直接のリスクがあることを必要とし、それは、もし事象がより重篤ならば仮説的に死を引き起こし得ることを意味しない。AEは、以下の1つ以上によって更に特徴付けられる:被験体の入院を必要とする又は延長する;持続的な又は重大な障害又は不能という結果となる;先天異常又は出産時欠損である;すぐに生命を脅かし得ない、または死に至り得ない、または入院の結果になり得ないが、被験体を危険にさらし得るか、または上記の結果のうちの1つを防ぐために介入を必要とし得る、重要な医学的事象(複数可)である。
【0363】
症例が重大であるか、および施設内治験審査委員会(IRB)及び取り締まり機関への事象の促進される報告が適切かどうかを決定する際に、医学的及び科学的な判断が行使される。
【0364】
6.2.1.2 重症度
各AEの重症度は、AE(CTCAE)v4.0のためのNCI共通用語基準を使用して、研究員によって評価され、eCRFに記録されなければならない。
【0365】
AEの重症度は、以下の臨床的記載に従って類別される:
・グレード1:軽度;無症候性又は軽度の徴候;臨床的又は診断上の観察のみ;示されない介入;
・グレード2:中程度;示される、最小限の、局所的な、又は非侵襲性の介入;限定される年齢に応じた手段的日常生活動作(ADL)*;
・グレード3:重度又は医学的に重大であるが、すぐには生命を脅かさない;示される、入院又は入院の延長;不能状態;限定されるセルフケアADL**;
・グレード4:生命を脅かす結果;示される緊急の介入;及び
・グレード5:AEに関連する死。
* 手段的ADLは、食事を準備すること、食料品又は衣類の買い物をすること、電話を使用すること、お金を管理することなどを指す。
** セルフケアADLは,入浴、着衣及び脱衣、自給、トイレの使用、薬剤の服用、及び寝たきりでないことを指す。
【0366】
6.2.1.3 因果関係
研究員は、臨床試験薬とAEの間の因果関係/関係性を評価し、eCRFにその評価を記録する。因果関係は次のように示される:明確に関連する;おそらく関連する;ひょっとしたら関連する;関連しそうにない;又は、関連しない。
【0367】
6.3 有害事象の記録
有害事象報告は、臨床試験薬の最後の投与の後、30日間まで、又は事象までが回復(resolution)/安定するまで、スクリーニングから延長する。試験の終了後に生じる有害事象は、臨床試験薬との因果関係があると研究者が考えると、報告される。
【0368】
すべてのAEは、臨床試験薬との関係性にかかわらず、eCRFに記録される。
【0369】
すべてのAEの報告は、事象の簡単な記載、発生の日及び時間、回復の日及び時間、強度、必要とされる処置、臨床試験薬との関係性、臨床試験薬とともにとられた行動、結果、及び事象が重大なものとして分類されるかを含む。
【0370】
6.3.1 有害事象のフォローアップ
被験体によって経験されたすべてのAEは、疑われる因果関係に関係なく、事象が回復/安定するまで、任意の異常な臨床検査値が基準線に戻る又は研究員とメディカルモニターに許容可能なレベルで安定するまで、観察される変化の満足な説明がなされるまで、または被験体がフォローアップされなくなる(lost)まで、監視される。
【0371】
8.3.2 有害事象の報告
研究員は、事象を発見してから24時間以内に、あらゆるSAEを、Pharm-Olam Pharmacovigilance Unitに報告する。SAEの報告を行うとき、SAE用語、研究員の名前、報告者の名前、報告者と連絡するときの電話番号、およびプロトコルの分類番号及びタイトルが与えられる。最後の投与後の30日以内に生じ、少なくとも臨床試験薬とひょっとしたら関連があると考えられ、それ故、副作用の可能性がある、任意のSAEが報告される。事象の評価に基づいて、規定上の報告を含む更なる行動の必要性に関して決定が下される。
【0372】
7. 訪問による観察
治療期間中の訪問が、予定された訪問の±3日以内に行われる。被験体が訪問を逃すと、その訪問を埋め合わせることはできないが、次の通常の予定された訪問を行う。24時間計(例えば、11:20ではなく23:20)を使用して、すべての時間が記録される。
【0373】
7.1 スクリーニング(訪問1)
スクリーニング訪問が、最初の投与(1日目)から28日間以内に行われる。スクリーニングでは、以下の評価が行われ、測定が記録される:書面によるインフォームドコンセント(署名されたICF)を得る;資格基準を確認する;医学的/腫瘍学的病歴を記録する;人口統計学的詳細;身体検査;生命徴候;12誘導ECG;CT/MRI及び骨スキャン - 使用されるCT/MRI及び骨スキャンの結果は、1日目から4週間以内の日付でなければならない;生検(被験体のサブセット;別々のICFによって表紙をつけられた);実験室の安全性試験(血液学、血清化学検査、検尿) - 安全性実験室評価が1日目の7日より前に記録されると、それらは投薬の前に繰り返され、確認されるべきである;併用薬を記録する。AEは、この訪問から記録される。
【0374】
7.2 登録(訪問2)
登録訪問(1日目、訪問2)では、以下の評価が行われ、測定が記録された:12誘導ECG(投与前);特殊実験室試験;PSA;PKサンプル(投与前);併用薬;AE。
【0375】
臨床試験薬を、10:00と14:00の間に、食事とともに、臨床現場で投与する。12誘導ECGを、投与の4時間後(±15分)に行う(PKサンプル前)。PKサンプルを、投与の4時間後(±15分)に収集する(ECG後)。臨床試験薬を分配し、被験体は臨床試験薬の使用に備える。コンプライアンスの日記を発行し、被験体はその使用に備える。被験体は、2日目から次の訪問まで、夕食(17:00と21:00の間)とともに毎日1回処置を継続することが必要とされる。
【0376】
7.3 1週間はない訪問(訪問3、5、7、9、及び11)
被験体は、8日目(±3日)、訪問2の1週間後、及び実験室試験が1週間ないためにその後2週ごとに臨床現場に戻る。臨床現場から地理的に遠く離れているそれらの被験体のために、地方の実験室が利用され得る。これらの訪問で、実験室の安全性試験(血液学及び血清化学検査のみ)が行われる。
【0377】
7.4 2週ごとの隔週の訪問(訪問4、6、8、及び10)
被験体は、15日目(±3日)、訪問3の1週間後、及びその後2週ごとに臨床現場に戻る。これらの訪問で、以下の評価が行われ、測定が記録される:身体検査;生命徴候;12誘導ECG;実験室の安全性試験(血液学、血清化学検査、検尿);特殊実験室試験;PSA;PKサンプル(投与前);併用薬;AE;コンプライアンスの日記の回収;発行される新しいコンプライアンスの日記;責任手順;及び分配される臨床試験薬。被験体は、2週間後の訪問まで、夕食(17:00と21:00の間)とともに毎日1回処置を継続することが必要とされる。
【0378】
7.5 試験終結の訪問
試験終結の訪問が、85日目(±5日)、最初の投与の12週間後に行われる。以下の詳細及び評価が行われ、測定が記録される:身体検査;生命徴候;12誘導ECG;CT/MRI及び骨スキャン - これは、試験終結の訪問前に行われると、PSA最下点で行われる;生検(被験体のサブセット;別々のICFによって表紙をつけられた) - これは、試験終結の訪問前に行われると、PSA最下点で行われる;実験室の安全性試験(血液学、血清化学検査、検尿);特殊実験室試験;PSA;PKサンプル(投与前);併用薬;AE;コンプライアンスの日記の回収;発行される新しいコンプライアンスの日記(延長へと継続する場合);責任手順;及び分配される臨床試験薬(延長へと継続する場合)。
【0379】
7.6 延長の相
被験体が延長の相へ継続する資格を有すると、以下の評価がなされる、4週ごとの臨床現場に被験体は戻り、測定が記録される:身体検査;生命徴候;CT/MRI及び骨スキャン(12週ごと);実験室の安全性試験(血液学、血清化学検査、検尿);特殊実験室試験;PSA;併用薬;AE;コンプライアンスの日記の回収;発行される新しいコンプライアンスの日記;責任手順;及び分配される臨床試験薬。
【0380】
7.7 期限前契約解除の訪問
試験から初期に中止する被験体は、可能であれば、期限前契約解除の訪問を受けるべきである。被験体が臨床試験薬を服用するのをやめた後、できるだけすぐにこの訪問を行う。被験体が任意の理由で早期に離脱すると(12週目の訪問前)、研究員は、試験終結の訪問(12週目の訪問)のために記載される安全性評価を行なうために、あらゆる努力をする。
【0381】
8. 統計
この試験の相1の段階の1つの目的は、TOK-001が許容可能な安全性プロフィールを提供する投与量(複数可)を設定することである。許容可能な安全性プロフィールは、≦35%の正確なDLT比率での投与量として定義される。相2の段階の目的は、もたらされる各投与量(複数可)のための生物学的なシグナルを評価するのみでなく、許容可能な安全性プロフィールを確認することである。
【0382】
以下の3の投与量群は、相1のために計画される:(1) 650mgの化合物1;(2) 1300mgの化合物1;(3) 1950mgの化合物1。
【0383】
中間の投与量を得る可能性は別にして、各投与量を、許容可能な毒性プロフィールを有する投与量として識別する確率の計算は、本明細書に記載される用量増加及び用量減少の規則に従ってなされ得る。3の投与量の各々に対する3の正確だと想定されるDLT比率は、各々が最大投与量(相1の段階から現れる投与量として定義される)と指定される確率とともに、表6に示される。
【0384】
【表9】
【0385】
650mgの化合物1が「毒性が強すぎる」確率は、上記の各シナリオに対して0.008である。
【0386】
試験の相1の段階の終了で、2までの投与量が、試験の相2の段階に進むために選択される。
【0387】
相2に進められる投与量の安全性が、相1における比較的少数の被験体(6まで且つ3以上)において評価されるため、その投与量での正確なDLT比率が>35%であることを示唆する合理的に説得力のある証拠があると、特定の投与量に対する登録が停止され得るように、停止規則が実施される。あらゆる5番目の被験体がDLTに対して評価可能となる後に、これらの制限が計算される。操作上、以下のいずれかはこのような制限につながり、DLTの数値/被験体の数:4/5(又はより少ない)、6/10(又はより少ない)、7/15(又はより少ない)、又は9/20(又はより少ない)と表される。
【0388】
DLTの正確な確率が20%又は50%であると、過度のDLTにより試験を停止する確率は、それぞれ、およそ0.03及び0.81である(5,000回のシミュレーションから推測される)。投薬がアームにおいて停止されると、相2の投与量の修正が行われる。
【0389】
8.1 一般的な考察及び基準線
他に明記のない限り、すべての統計試験を、5%の有意水準で両側検定を使用して行う。これが相1/2の試験であるため、複数のエンドポイント及び訪問のための多重比較の調整を、最終的な分析では行わない;分析は主として、本来記述的である。基準線は、他に明記のない限り、最初の投与の前に最後の観察として定義される。すべての推論的試験は、両側検定を使用する、処置の直接の比較を含む。試験の変数を、記述統計[連続変数に対する、N、平均、標準偏差(SD)、中央値、最小値、及び最大値、およびカテゴリー変数に対する頻度及びパーセンテージ]によってまとめる。
【0390】
8.2 被験体の処置
試験の各相に入り終了する被験体の数及びパーセンテージが、処置によって階層化されて示される。
【0391】
8.3 分析個体群
分析個体群の選択に対する分析の感受性を評価するために、二次分析もパープロトコル(PP)個体群に基づいて行われるが、主要な有効性分析は、包括解析(Intent-to-treat)(ITT)個体群に基づく。すべての安全性分析は安全性個体群に基づく。
【0392】
投与が相2において停止されると、停止される試験のアームにおける被験体は継続的な投与量で処置され、12週のフォローアップスケジュールを終了するが、継続的な投与量のための有効性分析には含まれない。
【0393】
8.3.1 安全個体群
臨床試験薬を少なくとも1回用量受け取る被験体はすべて、安全個体群に含まれる。
【0394】
8.3.2 包括解析個体群
スクリーニングを終了し、臨床試験薬での処置を予定している被験体はすべて、ITTの個体群に含まれる。
【0395】
8.3.3 パープロトコル個体群
PP個体群は、以下の基準の両方を満たす被験体を含む:1) 臨床試験薬を少なくとも1回用量受け取る;及び 2) あらゆる主要なプロトコル違反を有さない。
【0396】
8.4 プロトコルの逸脱
包含/排除の基準の違反を含むプロトコルからの逸脱は、「主要でない」又は「主要である」として、評価される。プロトコルからの主要な逸脱は、PP個体群からの被験体の排除につながる。
【0397】
8.5 人口統計及びベースライン特性
登録時の年齢などの連続的な人口統計学的パラメーターを、記述統計(N、平均、中央値、SD、最小値、及び最大値)を使用して、安全個体群のためにまとめる。性別などの分類別の人口統計学的パラメーターを、安全個体群の頻度及び割合としてまとめる。
【0398】
8.6 併用薬
併用薬を、WHO医薬品辞書のバージョン12.0又はそれ以上を使用してコード化する。データを、記述統計を使用してまとめる。
【0399】
8.7 処置曝露及び処置コンプライアンス
処置曝露及び処置コンプライアンスを、処置によって階層化して、頻度及びパーセンテージによってまとめる。
【0400】
8.8 安全性分析
安全性分析を、安全個体群を使用して実行する。試験の安全性エンドポイントは以下のものを含む:AEの発生率;生命徴候及び12誘導ECGの変化;身体検査の変化;実験室評価(血清化学検査、血液学、及び検尿)の変化。
【0401】
8.8.1 有害事象
臨床試験の間に記録されたすべてのAEを、MedDRAシステムに従ってコード化し、器官別大分類に割り当てる。TEAEは、治療の開始の後に、重症度及び/又は頻度において最初に生じる又は悪化するAEとして定義される。開始時間が不明の、臨床試験薬の最初の投与の日に開始するあらゆる事象は、処置により現れると想定される。
【0402】
体組織によって任意のAEを経験する被験体の頻度、関連性、及び重症度を、計算及びパーセンテージを使用して各処置群においてまとめる。あらゆる所与のMedDRAの優先使用語に関して、たとえ被験体が複数回の処置にわたって複数回の出現を有したとしても、被験体は、発生に対して1回のみのカウントに貢献する。臨床試験薬との関係性は、(明確に、おそらく、ひょっとしたら)関連する及び関連しない(ありそうにない、関連しない)ものとして分類される。1の被験体に対して複数の記録が存在すると、最大の重症度及び臨床試験薬に対する最も強力な関係性だけが、パーセンテージを計算するために数えられる。重篤AEを、全体的に、および体組織及び優先使用語によってまとめる。
【0403】
8.8.2 実験室評価、心電図、生命徴候、及び身体検査
臨床検査、ECG、及び生命徴候パラメーターにおける基準線からの変化を、処置群及び訪問によって計算する。基準線からの変化に関する記述統計(N、平均、SD、中央値、最小値、及び最大値)を表にする。基準線から基準線後(post-baseline)までの変化の表を、臨床検査及びECGのパラメーター及び身体検査のための処置群及び訪問によって作成する。頻度及びパーセンテージを、変化の表に提示する。研究所評価における臨床的に著しい異常性を、データリストに示す。
【0404】
8.9 有効性分析
ITTの個体群を、すべての有効性分析に使用する。
【0405】
8.9.1 主要な有効性分析
主要な有効性エンドポイントは、基準線から12週までのPSA、又はPSA最下点(最初に生ずるどちらか)における50%又はそれ以上の減少を有する被験体のパーセントである。頻度及びパーセンテージを、各処置群に関して計算する。主要な分析も、PP個体群を使用して繰り返す。
【0406】
8.9.2 二次的な有効性分析
記述統計を、二次的な有効性パラメーターの各々のために表にし、提示する。すべての二次的な有効性分析を、ITT個体群を使用して行う。
【0407】
8.9.2.1 固形腫瘍における反応評価基準(RECIST)による進行。
反応率は、全奏効率(ORR)を含む。ORRは、以下のように定義され:
ORR = (CR + PR)/Nr
ここで、CRは、完全奏効を有する被験体の数であり、PRは、部分奏効を有する被験体の数であり、及びNrは、ITT個体群に含まれる被験体の数である。
【0408】
腫瘍反応評価は、以下のRECIST定義を使用して行われる。
【0409】
【表10】
【0410】
被験体の反応は、表8に概説されるように、処置の開始から疾患の進行/再発まで記録された最良の反応である。
【0411】
【表11】
【0412】
8.9.2.2 CT/MRI及び骨スキャン、更なる特殊実験、及び生検サブセット中のパラメーターにおける基準線からの変化
CT/MRI及び骨スキャン、更なる特殊実験、および生検サブセット中の腫瘍内のパラメーター(腫瘍内のARタンパク質、テストステロン、およびDHT)における基準線からの変化を、処置群及び訪問よって計算する。基準線からの変化に関する記述統計(N、平均、SD、中央値、最小値、及び最大値)を、処置群によって表にする。
【0413】
8.9.2.3 進行までの時間、無進行生存及び全存率
TTP、PFS、及びOSは、延長の相の特異的なエンドポイントである。
【0414】
進行までの時間(TTP)は、臨床経過、放射線学的証拠、及び生化学的マーカー(PSA)の結果に基づいて、臨床試験薬の最初の投与から、より悪化する疾患の最初の文書化されたPI評価までの時間として定義される。
【0415】
無進行生存(PFS)は、被験体が悪化しない疾患(CRPC)を有して生活している試験における処置の間及びその後の時間の長さである。
【0416】
全存率(OS)は、任意の原因により、臨床試験薬の最初の投与から死の最初の文書化までの時間として定義される。この試験の目的で、OSは、臨床試験薬の最初の投与を受けた5年後の生存した被験体のパーセンテージとして報告される。
【0417】
TTP、PFS、及びOSの分析は、ITT個体群を使用する化合物1の投与量群の間で、比較のためのログランク検定を使用する。カプラン・マイヤー推定値は、処置群によって図示される。95%の信頼区間を有する事象までの平均期間(Median time)も、推定できるならば、処置群によって表にされる。
【0418】
打切日で事象を経験しなかった被験体は、中断日をカットオフの日(cut-off date)として使用し、分析から打切りされる。初期に離脱する被験体は、最後の接触日をカットオフの日として使用し、分析から打切りされる。
【0419】
8.9.2.4 診査のエンドポイント
以下の分析は、試験の相2の段階のための可能な診査のエンドポイントである:1) 12週にわたるPSA変化(最初の投与から始まり、12週間の処置をカバーする);及び/又はPSA最下点及びPSA最下点までの時間(最下点は、化合物1の投薬期間中に観察された最低のPSA値である)。
【0420】
記述統計(N、平均、SD、中央値、最小値、及び最大値)を、処置群によって表にする。
【0421】
8.10 化合物1の濃度
化合物1の血漿濃度を、1日目の0時間目(C0hrs)及び4時間目(C4hrs)に測定し、および続く訪問の利用可能な時点について測定する。
【0422】
8.11 相2のサンプルサイズの決定
2の投与量が相1から選択されると、その後、40の被験体は、2の投与量の間で1:1の比率で無作為化される。1の投与量だけがもたらされると、20の被験体は、この投与量を割り当てられ、無作為化は生じない。このセクションは、1アーム当たり20の被験体のサンプルサイズの選択の正当化を記載する。
【0423】
初回量、650mgの化合物1は、忍容性が良好であると思われ、研究の意図は、12〜15の被験体が相1に登録されることである[相2の投与量(複数可)で処置されるいくらかの被験体を含む]。相2に使用される投与量で相1において処置される被験体は、無作為化されるが、一貫した処置及びフォローアップスケジュールを受ける。それ故、試験の相2の段階において生物学的作用の潜在性を検査するときに被験体は含まれ、特定の投与量での少なくとも23の被験体(26まで)が、相2における生物学的シグナルの分析に利用可能であり得ることが予期される。2の投与量が相1から生じると、これらの投与量の1つは、好ましい投与量として識別される。この投与量が有意に強力な生物学的シグナルを有すると考えられると、この投与量は今後の試験に使用される。1アーム当たり20の被験体の選択は、相2における特定の投与量で少なくとも23の被験体(及び26もの被験体)を処置させる予想に基づく。一連の23(23)の被験体は、想定される正確な比率が75%であるときに、45%の固定比率と比較して、統計学的に著しく改善された「奏効率」(PSAの減少)を観察する、92%の力(power)を提供する。
【0424】
生物学的作用の検査に使用されるエンドポイントは、基準線から12週までのPSA、又はPSA最下点(最初に生ずるどちらか)における50%又はそれ以上の減少を有する被験体のパーセンテージである。2の投与量が相1から生じると、そのような被験体の中で最大のパーセンテージを有する投与量は、相2からの好ましい投与量として識別される。この好ましい投与量(又は1のみが続く試験のために相2にもたらされるときの単一用量)の今後(future)は、以下のガイドラインによって管理される:1) 好ましい投与量に対する観察されたPSA反応が30〜45%であると、化合物1は、更なる用量設定作業を恐らく受ける;2) 観察されたPSA反応が45〜75%であると、化合物1は、潜在性の生物学的作用を有すると考えられ、より大規模な相2又は2/3の試験は、全存率及び進行までの時間などの有効性の測定に加えて、PSA反応を確認し、より正確に推測するように考案される;及び3) 観察されたPSA反応が75%を超過すると、この投与量を直接、相3の試験に与えることが強く考慮に入れられる。
【0425】
特定の投与量で23又は26の被験体の間の45%以上のPSA反応を観察する確率は、様々な想定される正確な奏効率のために表7にまとめられる。
【0426】
【表12】
【0427】
さらに、正確なPSA反応率が75%であると、一連の23の被験体は、(0.05の片側の有意水準での)45%の固定比率と比較して、統計学的に有意に改善された奏効率を観察する、92%の力を提供する。n=26の同じパラメーターは、94%の力をもたらす。ベンチマーク奏効率が30%で、想定される正確な奏効率が60%であると、一連の23の被験体はまた、91%の力を提供する。n=26のこれらの想定は、94%をもたらす。
【0428】
具体的なベンチマークに関連する奏効率における統計学的に有意な改善が、続く試験に進むためには必要とされないが、これらの力の計算は、相2におけるサンプルサイズに関して1アーム当たり20の被験体の選択の論理的根拠に加えて、次の試験の方向を決定する助けとなる、必要とされる観察される固定比率のためのいくらかの正当化を与える。2の投与量が相2にもたらされ、アームに対する正確な奏効率が45%及び60%であると、正確なより高い奏効率を有するアームが選択され、1アーム当たり23の被験体でおよそ0.88の確率を有し(シミュレーションから推測される確率);23のサンプルサイズは、相2において各アームに無作為化された20に、相1において各投与量で処置される少なくとも3を加えたものである。
【0429】
<実施例7:化合物(1)での前立腺癌の処置のための臨床研究結果。>
実施例6のプロトコルに従って臨床研究を行った。化合物(1)を、4つのコホートに各々異なる量で投与した。各個々の被験体は、被験体が関係している間同じ量の化合物(1)を受けた。
【0430】
【表13】
【0431】
各群における大多数の被験体が、治療に好意的に反応したことを結果は示す。大量の化合物(1)を受ける、コホート3及び4は、より高い奏効率を生み出した。
【0432】
化合物(1)の血清濃度を、被験体の参加の間、各被験体において追跡した。その濃度を、図13に示す。被験体がすべて同じスケジュールに従ったわけではなく、コホートもすべてが同じスケジュールに従ったわけではなかった。図13において留意されるのは、コホート2及び3の被験体が、それぞれ、コホート1の被験体が受け取った量の2倍以下及び2倍を受け取ったが、コホート2及び3の被験体において見られる化合物(1)の血清濃度は、しばしば、コホート1において見られる濃度の2倍以上であったことである。より高い投与量が、ユニット効果当たり、より大きな有効性を有しており、より生産的且つより経済的な治療が、化合物(1)のより高い投与量により達成可能であることを、これらのデータは示す。
【技術分野】
【0001】
本出願は、2009年8月7日に出願の米国仮特許出願第61/232,257号、及び2009年11月13日に出願の米国仮特許出願第61/261,265号の利益を主張するものであり、これらは各々、その全体における引用により本明細書に組み込まれる。
【0002】
<引用による組み込み>
この明細書で言及されるすべての公報、特許、及び特許出願は、個々の公報、特許、又は特許出願が、それぞれ、明確且つ個々に引用によって組み込まれると示されるのと同じ程度まで、引用によって本明細書に組み込まれる。
【背景技術】
【0003】
前立腺癌は、男性において最もよく見られる癌である。大多数の前立腺癌死亡は、従来のアンドロゲン遮断療法に反応性のない転移性疾患の進行が原因である。アンドロゲン遮断療法は、1940年代以降、前立腺癌を有する被験体における標準治療であった。アンドロゲン遮断にもかかわらず、ほとんどの被験体は、最終的に疾患の進行を経験する。長年、該疾患のこの後の相は、「ホルモン不応性前立腺癌」又は「アンドロゲン非依存性前立腺癌」と呼ばれた。それ以降、数年間のアンドロゲン遮断療法の後に出現する前立腺癌は、アンドロゲンに依存したままであることが明らかになった。生存した前立腺癌細胞は、(副腎から発現した)低レベルの循環するアンドロゲンを移入し、これらの低レベルのテストステロンにかなり感応的になり、及び実際に、前立腺癌細胞自体内のテストステロンを合成する能力を得た。前立腺癌のこの段階は、現在、「去勢抵抗性前立腺癌」又はCRPCと称される。
【発明の概要】
【課題を解決するための手段】
【0004】
幾つかの実施形態において、本発明は、化合物(1):
【0005】
【化1】
【0006】
又はその薬学的に許容可能な塩、N-オキシド、活性代謝物質、プロドラッグ、又は溶媒和物を含む医薬組成物を熟考し、
式中、化合物(1)は、約:a. 13.1°及び14.1°;b. 17.2°、18.5°及び19.1°、及び随意に約16.2°、及び29.6°;c. 12.5°、14.8°、及び25.5°;又はd. 14.4°、16.1°、19.1°及び19.3°の角度 2-シータ(2θ)において表される、特性ピークを有する粉末X線回折パターンによって特徴付けられた、微粉化結晶形態である。
【0007】
幾つかの実施形態において、本発明は、被験体における前立腺癌の処置を提供する方法を熟考し、該方法は、化合物(1):
【0008】
【化2】
【0009】
又はその薬学的に許容可能な塩、N-オキシド、活性代謝物質、プロドラッグ、又は溶媒和物を含む医薬組成物を、それを必要とする又は望んでいる被験体に投与する工程を含み、
式中、化合物(1)は、約:a. 13.1°及び14.1°;b. 17.2°、18.5°及び19.1°、及び随意に約16.2°、及び29.6°;c. 12.5°、14.8°、及び25.5°;又はd. 14.4°、16.1°、19.1°及び19.3°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられた、微粉化結晶形態である。
【図面の簡単な説明】
【0010】
【図1】図1は、化合物(1)フォームIの代表的な粉末X線回折図(XRPD)である。
【図2】図2は、フォームIIを含む化合物(1)のいくつかの形態の代表的な粉末X線回折図(XRPD)である。
【図3】図3は、標準条件下及び40℃で1週間後の、化合物(1)フォームIIIの、代表的な粉末X線回折図(XRPD)である。
【図4】図4は、上昇する温度とともに、パターン5が他のパターンに変換することを示す、化合物(1)パターン5の代表的な粉末X線回折図(XRPD)である。
【図5】図5は、加熱によってフォーム1に戻ることを示す、化合物(1)パターン2の代表的な粉末X線回折図(XRPD)である。
【図6】図6は、様々な温度での非晶質の化合物(1)の代表的な粉末X線回折図(XRPD)である。
【図7】図7は、化合物(1)フォームIの代表的なサーモグラムである。
【図8】図8は、アンドロゲン受容体に結合する(化合物5と記載される)化合物(1)を示す表である。
【図9】図9は、アンドロゲン受容体タンパク質のレベルを示すウエスタンブロットである。
【図10】図10は、腫瘍異種移植片のサイズを示す線グラフである。
【図11】図11は、本明細書に記載される臨床研究の様々な段階の過程を図表で示す。
【図12】図12は、研究の被験体のための手順のスケジュールを表にする。
【図13】図13は、臨床試験の被験体における時間に対する化合物(1)の血清中濃度を示す。コホート群は、図の上部にわたってリストされる。
【発明を実施するための形態】
【0011】
定義
有害事象:本明細書に使用されるような用語「有害事象」は、その技術分野で理解される意味を有し、医薬品を投与された、被験体又は臨床試験の被験体における任意の好ましくない医学的出来事を指す。有害事象は、必ずしも、投与される処置と因果関係がある必要はない。
【0012】
副作用:本明細書に使用されるような用語「有害反応」は、その技術分野で理解される意味を有し、任意の投与量に関連する医薬品に対する任意の有害で意図されない反応を指す。
【0013】
併用療法:用語「併用療法」は、本明細書に使用されるように、2つ以上の異なる医薬品が、レジメンを重複させる際に投与される、これらの状況を指し、その結果、被験体は両方の薬剤に同時に曝される。
【0014】
投薬レジメン:用語「投薬レジメン」は、本明細書に使用されるように、期間によって分けられて個々に投与される一連の単位用量(典型的には1以上)を指す。特定の医薬品のための推奨される一連の投薬(すなわち、量、タイミング、投与経路など)は、その投薬レジメンを構成する。
【0015】
開始:本明細書に使用されるように、投薬レジメンに適用される時の用語「開始」は、以前に医薬品を受けたことのない被験体への、医薬品の最初の投与を指すように使用され得る。代わりに又はさらに、用語「開始」は、被験体の治療の間の医薬品の特定の単位用量の投与を指すように使用され得る。
【0016】
医薬品:本明細書に使用されるように、句「医薬品」は、被験体に投与される時に、治療効果を有している及び/又は所望の生物学的及び/又は薬理学的な効果を誘発する、任意の薬剤を指す。
【0017】
薬学的に許容可能なエステル:本明細書に使用されるように、用語「薬学的に許容可能なエステル」は、インビボで加水分解し、ヒトの身体において容易に分解し、親化合物又はその塩を残すエステルを含む、エステルを指す。
【0018】
重篤有害事象:用語「重篤有害事象」は、本明細書に使用されるように、その技術分野で理解される意味を有し、任意の投与量で、例えば、死に至る、生命を脅かす、被験体の入院(又は既存の入院の延長)を必要とする、(正常な生命機能を実行する被験体の能力の相当な破壊として定義される)持続性の又は重大な障害又は不能という結果になるなどの、任意の好ましくない医学的出来事を指す。幾つかの実施形態において、重篤有害事象は、その用語が、例えば、21 CFR § 310.305(b)において定義されるように、米国食品医薬品局 (the United States Food and Drug Administration)によって使用されるように、「重篤有害薬物経験」であり、ここで、重篤有害事象が、以下の結果のいずれかにつながる、任意の投与量で生じる有害薬物経験であると述べている:死亡、生命を脅かす有害薬物経験、被験体の入院又は既存の入院の延長、持続性の又は重大な障害/不能,又は先天異常/出産時欠損。死に至り得ない、生命を脅かし得ない、入院を必要とし得ない重要な医学的事象は、適切な医学的判断に基づいて、それらが被験体を危険にさらし得る、及びこの定義においてリストされる結果の1つを防ぐために医学的又は外科的な介入を必要とし得る時に、有害薬物経験と考えられ得る。そのような医学的事象の例は、緊急処置室又は家で集中治療を必要とするアレルギー性気管支痙攣、被験体の入院という結果にはならない血液疾患又は痙攣、または薬物依存又は薬物乱用の進行を含む。
【0019】
感受性:用語「〜に感受性がある(susceptible to)」は、一般集団において観察されるより、特定の疾患又は障害、又はその症状を進行させる、(典型的に、遺伝性素因、環境要因、個人歴、又はそれらの組み合わせに基づいた)高いリスクを有する個体に言及するために本明細書において使用される。
【0020】
治療上有効な量:用語、医薬品又は薬剤の組み合わせの「治療上有効な量」は、任意の治療に適用可能な合理的なベネフィット・リスク比で、処置される被験体に対する治療効果を与える薬剤(複数可)の量を指すように意図される。治療効果は、客観的であり得るか(すなわち、幾つかの試験又はマーカーによって測定可能である)、又は主観的であり得る(すなわち、被験体は効果の指標を与える、又は効果を感じる)。治療上有効な量は、通常、複数の単位用量を含み得る投薬レジメンにおいて投与される。任意の特定の医薬品に関して、治療上有効な量(及び/又は有効な投薬レジメン内の適切な単位用量)は、例えば、投与の経路、他の医薬品との組み合わせに依存して、変化し得る。また、任意の特定の被験体のための具体的な治療上有効な量(及び/又は単位用量)は、処置されている障害及びその障害の重症度、用いられる具体的な医薬品の活性、用いられる具体的な組成物、被験体の年齢、体重、健康状態、性別及び食事、投与の時間、投与の経路、用いられる具体的な医薬品の排泄又は代謝の速度、処置の期間、及び医学的技術分野において周知であるような因子を含む、様々な因子に依存し得る。
【0021】
処置:本明細書に使用されるように、用語「処置(treatment)」は(「処置する(treat)」又は「処置すること(treating)」も)、特定の疾患、障害、及び/又は疾病の1以上の症状又は特徴を、部分的に又は完全に緩和する、寛解する、和らげる、阻害する、その発症を遅らせる、重症度を低下させる、及び/又は発生率を減らす、医薬品の任意の投与を指す。そのような処置は、関連する疾患、障害、及び/又は疾病の徴候を示さない被験体、及び/又は疾患、障害、及び/又は疾病の初期の徴候のみを示す被験体の処置であり得る。代わりに又はさらに、そのような処置は、関連する疾患、障害及び/又は疾病の1以上の確立された徴候を示す被験体の処置であり得る。
【0022】
単位用量:用語「単位用量(unit dose)」又は「投与量(dose)」は、本明細書に使用されるように、典型的に投薬レジメンとの関連で、医薬品の別々の投与を指す。標準化学用語の定義は、Carey and Sundberg "ADVANCED ORGANIC CHEMISTRY 4TH ED." Vols. A (2000) and B (2001), Plenum Press, New Yorkを含む、参考資料において見受けられ得、その全体における引用によって本明細書に組み込まれる。特に指示がない限り、当該技術分野の技術内で、質量分析、NMR、HPCL、タンパク質化学、生化学、組換え型DNA技術及び薬理学の従来の方法が用いられる。
【0023】
例示的な生物学的活性
<アンドロゲン受容体(AR)>
アンドロゲンは、標的組織の細胞内で、特定の受容体、アンドロゲン受容体(AR)に結合する。ARは、身体の多数の組織において発現され、それを介してテストステロン(T)及びジヒドロテストステロン(DHT)などの内因性のアンドロゲンリガンドの生理学的効果に加え、病態生理学的効果も現れる受容体である。構造上、ARは、3つの主な機能ドメイン:リガンド結合ドメイン(LBD)、DNA結合ドメイン、及びアミノ末端ドメインからなる。ARに結合し、内因性のARリガンドの効果を模倣する化合物は、ARアゴニストと呼ばれ、一方で内因性のARリガンドの効果を阻害する化合物は、ARアンタゴニストと称される。受容体へのアンドロゲンの結合は、受容体を活性化し、標的遺伝子に隣接しているDNA結合部位へそれを結合させる。そこから、それは、遺伝子の発現を規制するために、コアクチベータータンパク質及び塩基性の転写因子と相互に作用する。したがって、その受容体を介して、アンドロゲンは細胞における遺伝子発現の変化を引き起こす。これらの変化は、最終的に、標的組織の生理機能において目に見える、細胞の代謝的産生(output)、分化又は増殖という結果を引き起こす。前立腺において、アンドロゲンは、アンドロゲン感受性組織の細胞質内に存在するARに結合することにより、前立腺組織及び前立腺癌細胞の成長を刺激する。
【0024】
選択的にARを調節する化合物は、限定されないが、前立腺癌、良性前立腺肥大症、女性型多毛症、脱毛症、神経性食欲不振、乳癌、ざ瘡、骨疾患、造血状態(hematopoietic conditions)、神経筋疾患、リウマチ疾患などの筋骨格系の疾病、癌、AIDS、悪液質を含む、様々な疾患、疾病、及び癌の処置又は予防において、男性避妊法において用いられるホルモン補充療法(HRT)にとって、男性の性能強化にとって、男性の生殖状態にとって、及び一次又は二次の男性性腺機能低下症にとって臨床的に重要である。
【0025】
<去勢抵抗性前立腺癌>
内因性のホルモン(例えば、テストステロン)の作用を遮断する薬剤(抗アンドロゲン)は、前立腺癌の処置に高度に有効であり、日常的に使用される(アンドロゲン除去療法)。最初は腫瘍成長を抑えるに有効であるが、これらのアンドロゲン除去療法は、結局、ほとんどすべての被験体において失敗し、「去勢抵抗性前立腺癌」(「CRPC」)につながる。すべてではないが、ほとんどの前立腺癌細胞は、最初に、アンドロゲン除去療法に反応する。しかしながら、時間とともに、前立腺癌細胞の生存する個体群は、アンドロゲン除去療法によって生じた選択圧に反応したが、今はそれに対して反応がない(refractory)ために出現する。原発性癌が、利用可能な療法に反応がないだけではなく、癌細胞はまた、原発腫瘍から離れ、血流を移動し得、疾患が離れた部位(特に骨)に広がる。他の効果の中で、これは著しい疼痛及び更なる骨の脆弱性を引き起こす。
【0026】
CRPC細胞は、利用可能なままである細胞内のアンドロゲンに対する反応を強めるための、少なくとも3つの異なる経路を増幅させることによる、低レベルの循環するアンドロゲンによって特徴付けられた環境において生存することが予期される。これらは:(1)ARコピー数を増加させ、その結果、内科的去勢療法によって誘発された低レベルの循環するアンドロゲンに対する、細胞の感受性を増加させる、ARの発現のアップレギュレーション、(2)アンドロゲン遮断療法の後に細胞に残るアンドロゲンの移入(importation)に関係する酵素の発現の増加、(3)CRPC細胞がその細胞自体のアンドロゲンを合成することを可能にする、ステロイド産生を規制する遺伝子の発現の増加を含む。ステロイド産生経路における重要な酵素は、チトクロームC17α-ヒドロキシラーゼ/C17,20-リアーゼ(CYP17)であり、該酵素は、副腎、精巣、及び前立腺におけるアンドロゲン産生を制御する。
【0027】
本明細書には、特定の実施形態において、限定されないが、前立腺癌、良性前立腺肥大症、女性型多毛症、脱毛症、神経性食欲不振、乳癌、ざ瘡、骨疾患、造血状態、神経筋疾患、リウマチ疾患などの筋骨格系の疾病、癌、AIDS、悪液質を含む、アンドロゲン受容体媒介性の疾患又は疾病を処置するための、男性避妊法において用いられるホルモン補充療法(HRT)のための、男性の性能強化のための、男性の生殖状態のための、及び一次又は二次の男性性腺機能低下症のための化合物、そのような化合物を作る方法、そのような化合物を含む医薬組成物及び薬剤、およびそのような化合物を使用する方法が記載される。幾つかの実施形態において、アンドロゲン受容体媒介性の疾患又は疾病は、前立腺癌である。幾つかの実施形態において、前立腺癌は、去勢抵抗性前立腺癌である。
【0028】
幾つかの実施形態において、本発明は、アンドロゲン生合成を減少させる、アンドロゲン受容体シグナル伝達を減少させる、及びアンドロゲン受容体感受性を減少させる、化合物、そのような化合物を含む医薬組成物、及び薬剤、及びそのような化合物を使用する方法を提供する。
【0029】
1つの態様において、化合物、そのような化合物を含む医薬組成物及び薬剤、およびそのような化合物を使用する方法は、アンドロゲン生合成を減少させる。幾つかの実施形態において、本明細書に開示される化合物は、アンドロゲン産生を制御する酵素の活性を阻害する。特定の実施形態において、本明細書に開示される化合物は、チトクロームC17α-ヒドロキシラーゼ/C17,20-リアーゼ(CYP17)の活性を阻害する。
【0030】
1つの態様において、化合物、そのような化合物を含む医薬組成物及び薬剤、およびそのような化合物を使用する方法は、アンドロゲン受容体シグナル伝達を減少させる。幾つかの実施形態において、本明細書に開示される化合物は、ARに結合し、テストステロン結合の競合的インヒビターである。
【0031】
1つの態様において、化合物、そのような化合物を含む医薬組成物及び薬剤、およびそのような化合物を使用する方法は、アンドロゲン受容体感受性を減少させる。幾つかの実施形態において、本明細書に開示される化合物は、細胞内のARタンパク質の含量を減少させ、低レベルのアンドロゲン増殖シグナルによって持続される細胞の能力を低減する。
【0032】
典型的な化合物
化合物(1)としても記載される、式(1)の化合物、その薬学的に許容可能な塩、薬学的に許容可能なN-オキシド、薬学的に活性な代謝物質、薬学的に許容可能なプロドラッグ、薬学的に許容可能な多形体及び薬学的に許容可能な溶媒和物は、ステロイドホルモン核内受容体の活性を調節し、それゆえ、アンドロゲン受容体媒介性の疾患又は疾病を処置するのに有用である。
【0033】
【化3】
【0034】
化合物の典型的な合成
式(1)の化合物(化合物(1)又は3-β-ヒドロキシ-17-(1H ベンズイミダゾール-1-イル)アンドロスタ-5,16-ジエンとしても記載される)は、本明細書に記載される方法と組み合わせて、当業者に公知の標準の合成技術を使用して、又は当該技術分野に公知の方法を使用して合成され得る。当業者が理解するであろうように、本明細書に提示される溶媒、温度及び反応条件は、当業者の慣習及び知識によって変えられ得る。
【0035】
化合物(1)の合成に使用される出発物質は、Aldrich Chemical Co. (Milwaukee, Wis.)、Sigma Chemical Co. (St. Louis, Mo.)などの、商業的供給源から得られ得るか、又は出発物質は合成され得る。本明細書に記載される化合物、及び異なる置換基を有する他の関連する化合物は、記載されるように、例えば、March, ADVANCED ORGANIC CHEMISTRY 4th Ed., (Wiley 1992)、Carey and Sundberg, ADVANCED ORGANIC CHEMISTRY 4th Ed., Vols. A and B (Plenum 2000, 2001)、及びGreen and Wuts, PROTECTIVE GROUPS IN ORGANIC SYNTHESIS 3rd Ed., (Wiley 1999)におけるような、当業者に公知の技術及び物質を使用して合成され得る(これら全ては、それらの全体において引用によって組み込まれる)。本明細書に開示される化合物の調製のための一般的な方法は、当該技術分野において公知の反応に由来し、該反応は、本明細書に提供されるように、式において見られる様々な成分を導入するために、当業者によって理解されるように、適切な試薬及び条件を使用することによって変更され得る。
【0036】
化合物(1)は、遊離塩基の形態の化合物を、限定されないが、塩化水素酸、臭化水素酸、硫酸、硝酸、リン酸、メタリン酸などの無機酸、および酢酸、プロピオン酸、ヘキサン酸、シクロペンタンプロピオン酸、グリコール酸、ピルビン酸、乳酸、マロン酸、コハク酸、リンゴ酸、マレイン酸、フマル酸、p-トルエンスルホン酸、酒石酸、トリフルオロ酢酸、クエン酸、安息香酸、3-(4-ヒドロキシベンゾイル)安息香酸、桂皮酸、マンデル酸、アリールスルホン酸、メタンスルホン酸、エタンスルホン酸、1,2-エタンジスルホン酸、2-ヒドロキシエタンスルホン酸、ベンゼンスルホン酸、2-ナフタリンスルホン酸、4-メチルビシクロ-[2.2.2]オクタ-2-エン-1-カルボン酸、グルコペプトン酸、4,4'-メチレンビス-(3-ヒドロキシ-2-エン-1-カルボン酸)、3-フェニルプロピオン酸、トリメチル酢酸、三級ブチル酢酸、ラウリル硫酸、グルコン酸、グルタミン酸、ヒドロキシナフトエ酸、サリチル酸、ステアリン酸、及びムコン酸などの有機酸を含む、薬学的に許容可能な無機酸又は有機酸と反応させることにより、(薬学的に許容可能な塩のタイプである)薬学的に許容可能な酸付加塩として調製され得る。
【0037】
化合物(1)は、プロドラッグとして調製され得る。プロドラッグは、一般的に薬物前駆体であり、この薬物前駆体は、被験体への投与及びその後の吸収に続いて、代謝経路による変換などの、幾つかの過程を介して、活性な、又はより活性な種へと変換される。プロドラッグの中には、活性を和らげ、及び/又は薬物に溶解度又は幾つかの他の特性を与える、プロドラッグ上に存在する化学基を有するものもある。一旦、化学基がプロドラッグから開裂及び/又は変更されると、活性薬物が生じる。幾つかの状況において、プロドラッグは親薬物よりも投与しやすいことがあるため、しばしば有用である。プロドラッグは、例えば、経口投与によって生物学的に利用可能である得る一方で、親薬物はそうではない。プロドラッグはまた、親薬物以上に医薬組成物において改善された溶解度を有し得る。プロドラッグの限定されない例は、式(I)の誘導体であり、これは親水性エステル(「プロドラッグ」)として投与され、改善された水溶性に対して有益である胃腸管における吸収を促進するが、その後、代謝的にカルボン酸及び活性な実体、化合物(1)に加水分解される。プロドラッグの更なる例は、化合物(1)の水酸基に結合した短鎖ペプチドであり、ここで、ペプチドは代謝され、化合物(1)を提供する。
【0038】
プロドラッグは、部位特異的な組織への薬物輸送を増強させる修飾因子として使用するための、可逆的な薬物誘導体として考案され得る。現在までのプロドラッグの考案は、水が主な溶媒である領域を標的とするための、治療用化合物の有効な水溶性を高めるためのものであった。例えば、Fedorak et al., Am. J. Physiol., 269:G210-218 (1995); McLoed et al., Gastroenterol, 106:405-413 (1994)、Hochhaus et al., Biomed. Chrom., 6:283-286 (1992)、J. Larsen and H. Bundgaard, Int. J. Pharmaceutics, 37, 87 (1987)、J. Larsen et al., Int. J. Pharmaceutics, 47, 103 (1988)、Sinkula et al., J. Pharm. Sci., 64:181-210 (1975)、T. Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems, Vol. 14 of the A.C.S. Symposium Series、及びEdward B. Roche, Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press, 1987を参照し、それら全体において、すべてが本明細書に組み込まれる。
【0039】
更に、化合物(1)のプロドラッグ誘導体は、 当業者に公知の方法によって調製され得る(例えば、更なる詳細については、Saulnier et al., (1994), Bioorganic and Medicinal Chemistry Letters, Vol. 4, p. 1985を参照)。プロドラッグがインビボで代謝され、本明細書に述べられるような誘導体を生成する、本明細書に記載される化合物のプロドラッグの形態は、請求の範囲内に含まれる。実際、本明細書に記載される化合物の幾つかは、別の誘導体又は活性化合物に対するプロドラッグであり得る。
【0040】
化合物(1)の芳香環部分の部位は、様々な代謝反応を起こしやすく、それ故、芳香環構造上の適切な置換基、例えば、ハロゲンを取り込むことにより、この代謝経路を減少、最小化又は除去することが可能である。
【0041】
化合物(1)を製造する様々な方法が熟考され、以下の記載は限定しない例として提供される。幾つかの実施形態において、以下の化学反応の1以上は、不活性雰囲気、例えば、窒素、アルゴンにおいて行われる。幾つかの実施形態において、反応の温度が監視される。幾つかの実施形態において、反応は、HPLC又はTLCによって監視される。幾つかの実施形態において、反応のpHが監視される。幾つかの実施形態において、反応の温度が制御される。幾つかの実施形態において、産物の純度はHPLCによって測定される。幾つかの実施形態において、実験は、小規模、中規模、大規模、分析的規模、又は製造規模で実行される。幾つかの実施形態において、産物は、シリカゲル及びセライトの1以上を含むパッドを介するろ過によって明確にされる。
【0042】
幾つかの実施形態において、合成は、大規模で行われる。幾つかの実施形態において、大規模は、約1から約10kgまでの規模を含む。幾つかの実施形態において、合成は、製造規模で行われる。幾つかの実施形態において、製造規模は、約10kgより大きい規模を含む。幾つかの実施形態において、製造規模は、約10から約1,000kgまでの規模を含む。幾つかの実施形態において、製造規模は、約10から約100kgまでの規模を含む。幾つかの実施形態において、製造規模は、約10から約50kgまでの規模を含む。幾つかの実施形態において、製造規模は、約33.4kgの規模を含む。
【0043】
幾つかの実施形態において、製造規模の合成を計画するか又は行うために使用される情報を集めるために、実験は小規模で行われる。幾つかの実施形態において、より小規模で得られた結果は、製造規模で再生可能であると予期される。幾つかの実施形態において、より小規模で得られた結果は、製造規模で再生可能であると予期されない。幾つかの実施形態において、製造規模で得られた収量は、より小規模で得られた収量より多い。幾つかの実施形態において、製造規模で得られた収量は、より小規模で得られた収量より少ない。
【0044】
【化4】
【0045】
1つの実施形態において、溶媒中の式iの化合物の溶液は調製される。その後、式iiの化合物は、溶液に接触され、結果として生じる混合物は、式iiiの化合物を提供するのに十分な期間、塩基の存在下で加熱される。幾つかの実施形態において、期間は、約1時間、約2時間、約4時間、約8時間、約12時間、又は約24時間である。幾つかの実施形態において、時間は、約1時間から約24時間までである。幾つかの実施形態において、塩基は、炭酸リチウム、炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム、リン酸ナトリウム、又はリン酸カリウムを含む。幾つかの実施形態において、溶媒は、DMFを含む。幾つかの実施形態において、温度は、約50℃、約70℃、約100℃、約150℃、又は還流条件を維持するのに有効な温度である。幾つかの実施形態において、温度は、約50℃から約200℃までである。式iiiの化合物は、反応混合物から分離され、当業者に公知の任意の方法によって精製され得る。このような方法は、限定されないが、水性混合物を反応混合物へ注ぐ工程を含み、それによって、固形物として化合物iiiを沈殿させる。式iiiの分離した化合物は、当業者に公知の任意の方法によって随意に精製され得る。このような方法は、限定されないが、水による粉砕を含む。
【0046】
【化5】
【0047】
1つの実施形態において、溶媒中の式iiiの化合物の溶液は調製され、溶液は、式ivの化合物を提供するのに十分な期間、触媒と接触される。幾つかの実施形態において、期間は、約1時間、約2時間、約4時間、約8時間、約12時間、又は約24時間である。幾つかの実施形態において、時間は、約1時間から約24時間までである。幾つかの実施形態において、触媒は、炭素上のパラジウム、炭素上のプラチナ、遷移金属塩又は遷移金属錯体を含む。幾つかの実施形態において、溶媒は、N-メチルピロリドンである。幾つかの実施形態において、温度は、約50℃、約70℃、約100℃、約150℃、約190℃、約200℃、又は還流条件を維持するのに有効な温度である。幾つかの実施形態において、温度は、約50℃から約250℃までである。式ivの化合物は、反応混合物から分離され、当業者に公知の任意の方法によって精製され得る。このような方法は、限定されないが、インラインろ過を含む。式ivの分離した化合物は、当業者に公知の任意の方法によって随意に精製さ得る。
【0048】
【化6】
【0049】
1つの実施形態において、溶媒中の式ivの化合物の溶液は調製され、溶液は、式vの化合物(すなわち、化合物(1))を提供するのに十分な期間、塩基と接触される。幾つかの実施形態において、期間は、約1時間、約2時間、約4時間、約8時間、約12時間、又は約24時間である。幾つかの実施形態において、時間は、約1時間から約24時間までである。幾つかの実施形態において、塩基は、水酸化リチウム、水酸化ナトリウム、水酸化カリウム、ナトリウムメトキシド、カリウムメトキシド、ナトリウムエトキシド、カリウムエトキシド、炭酸リチウム、炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム、リン酸ナトリウム、又はリン酸カリウムを含む。幾つかの実施形態において、溶媒は、水、メタノール、エタノール、2-プロパノール、t-ブタノール、又はそれらの混合物を含む。幾つかの実施形態において、溶媒は、メタノールを含み、塩基はナトリウムメトキシドを含む。幾つかの実施形態において、温度は、約35℃、約50℃、約70℃、約100℃、又は還流条件を維持するのに有効な温度である。幾つかの実施形態において、温度は、約25℃から約100℃までである。式vの化合物は、反応混合物から分離され、当業者に公知の任意の方法によって精製され得る。このような方法は、限定されないが、抽出を含む。式vの分離した化合物は、当業者に公知の方法によって随意に精製され得る。このような方法は、限定されないが、粉砕を含む。
【0050】
化合物(1)の典型的な形態
薬学的に許容可能な塩に対する言及は、その溶媒付加形態又は結晶形態、特に溶媒和物又は多形体を含むことを理解されたい。溶媒和物は、定比又は不定比量の溶媒のいずれかを含み、水、エタノールなどの薬学的に許容可能な溶媒での結晶化の過程の間に形成され得る。水和物は、溶媒が水である時に形成され、又はアルコラートは、溶媒がアルコールの時に形成される。化合物(1)の溶媒和物は、本明細書に記載される過程の間に、都合よく調製又は形成され得る。ほんの一例ではあるが、化合物(1)の水和物は、限定されないが、ジオキサン、テトラヒドロフラン、又はメタノールなどを含む、有機溶媒を用いて、水性/有機溶媒混合物からの再結晶によって都合よく調製され得る。さらに、本明細書に提供される化合物は、溶媒和形態と同様、非溶媒和形態でも存在し得る。一般的に、溶媒和形態は、本明細書に提供される化合物及び方法の目的のため、非溶媒和形態と同等であると考慮される。
【0051】
化合物(1)は、多形体としても知られる結晶形態を含む。多形体は、化合物の同じ元素組成の異なる結晶充填配置を有する。多形体は通常、異なるX線回折パターン、赤外線スペクトル、融点、密度、硬度、結晶形、光学及び電気学上の特性、安定性、及び溶解度を有する。再結晶溶媒、結晶化の速度、及び保管温度等の様々な要因は、支配的な単結晶形態を引き起こし得る。
【0052】
1つの実施形態において、化合物(1)は、フォームIとして示される結晶形態であり、約13.0°、14.6°、16.3°、17.6°、及び19.0°±0.2°2θでの粉末X線反射によって特徴付けられる。幾つかの実施形態において、フォームIは、約11.8°、20.2°、22.9°、及び25.4°±0.2°2θでの更なるピーク粉末X線反射によって特徴付けられる。図1は、化合物(1)形態Iの代表的な粉末X線回折図(XRPD)である。幾つかの実施形態において、フォームIは、一例として、溶媒中の非晶質の化合物(1)を溶解し、溶媒を蒸発させることから得られる。幾つかの実施形態において、非晶質の化合物(1)は、溶媒中に懸濁され、8時間のサイクルで50°/室温で代替的にインキュベートされる。結果として生じる懸濁液中の固形物は、ろ過され分析される。溶媒は、限定されないが、ヘプタン、ジオキサン、tert-ブチルメチルエーテル、酢酸ブチル、酢酸イソプロピル、プロパノール、テトラヒドロフラン、ジクロロメタン、メタノール、ニトロメタン、及び水を含む。
【0053】
1つの態様において、化合物(1)は、フォームIIとして示される結晶形態であり、約13.1°及び14.1°±0.2°2θでの粉末X線反射によって特徴付けられる。図2は、化合物(1)フォームIIの代表的な粉末X線回折図(XRPD)を含む。
【0054】
1つの態様において、化合物(1)は、フォームIIIとして示される結晶形態であり、約17.2°、18.5°、及び19.1°±0.2°2θでの粉末X線反射によって特徴付けられる。幾つかの実施形態において、フォームIIIは、約16.2°及び29.6°±0.2°2θでの更なるピーク粉末X線反射によって特徴付けられる。図3は、化合物(1)フォームIIIの代表的な粉末X線回折図(XRPD)である。
【0055】
1つの態様において、化合物(1)は、パターン5として示される結晶性水和物であり、約12.5°、14.8°、及び25.5°±0.2°2θでの粉末X線反射によって特徴付けられる。幾つかの実施形態において、パターン5は、約40%から約90%までの間の相対湿度の水分を有している。図4は、化合物(1)パターン5の代表的な粉末X線回折図(XRPD)である。
【0056】
1つの態様において、化合物(1)は、パターン2として示される結晶性クメン溶媒和物であり、約14.4°、16.1°、19.1°及び19.3°±0.2°2θでの粉末X線反射によって特徴付けられる。幾つかの実施形態において、パターン2は、約20.4°、23.6°及び28.7°±0.2°2θでの更なるピーク粉末X線反射によって特徴付けられる。幾つかの実施形態において、パターン2は、約20%から約40%までの間のクメン溶媒含有量を有している。他の実施形態において、パターン2は、約25%のクメン溶媒含有量を有している。図5は、化合物(1)パターン2の代表的な粉末X線回折図(XRPD)である。
【0057】
幾つかの実施形態において、化合物(1)は、実質的に、1つの結晶形態又はパターンである。本明細書に使用されるような「実質的に」は、80%より高いことを指す。幾つかの実施形態において、化合物(1)は、2つの形態又はパターンの混合物である。幾つかの実施形態において、化合物(1)は、2つ以上の形態又はパターンの混合物である。2つの形態又はパターンの混合物を有する例において、混合比は、約1:100から100:1までであり得る。他の実施形態において、化合物(1)は、非晶質の形態をさらに含む。特定の実施形態において、化合物(1)は、実質的に非晶質である。
【0058】
典型的な医薬組成物/製剤
本明細書に使用されるような、医薬組成物は、担体、安定化剤、希釈剤、分散剤、懸濁化剤、増粘剤、及び/又は賦形剤などの他の化学成分との、化合物(1)の混合物を指す。医薬組成物は、有機体への化合物の投与を促進する。化合物(1)を含む医薬組成物は、限定されないが、以下のものを含む、当該技術分野に公知の、任意の従来の形態及び経路による医薬組成物として治療上有効な量で投与され得る:静脈内、経口、直腸、エアロゾル、非経口、眼、肺、経皮、膣、耳、鼻、及び局所の投与。
【0059】
当業者は、全身よりもむしろ局所に、例えば、器官への直接的な化合物の注入を介して、しばしばデポー製剤又は持続放出性製剤において化合物を投与し得る。さらに、当業者は、標的薬物送達システムにおいて、例えば、臓器特異性抗体でコーティングされたリポソームにおいて、化合物(1)を含む医薬組成物を投与し得る。リポソームは、臓器の標的とされ、臓器によって選択的に取り込まれる。さらに、化合物(1)を含む医薬組成物は、急速放出製剤の形態、徐放製剤の形態、又は中間放出製剤の形態で提供され得る。
【0060】
経口投与に関して、化合物(1)は、活性化合物を、当該技術分野に周知の薬学的に許容可能な担体又は賦形剤と組み合わせることによって、容易に調剤され得る。このような担体は、本明細書に記載される化合物が、処置される被験体による経口摂取のための、錠剤、粉末剤、丸剤、ドラゼー、カプセル剤、液剤、ゲル剤、シロップ剤、エリキシル剤、スラリー剤、懸濁液などのように調剤されることを可能にする。
【0061】
経口使用のための医薬調製物は、必要に応じて、錠剤又はドラゼーコアを得るために、適切な助剤を加えた後に、1以上の固体の賦形剤を、本明細書に記載される化合物の1以上と混合させ、結果として生じる混合物を随意に粉砕し、顆粒の混合物を処理することによって得られる。ドラゼーコアは、適切なコーティングによって提供される。この目的のために、濃縮された砂糖溶液が使用され得、これは、アラビアゴム、タルク、ポリビニルピロリドン、カルボポールゲル、ポリエチレングリコール、及び/又は二酸化チタン、ラッカー溶液、及び適切な有機溶媒又は溶媒混合液を随意に含有し得る。色素又はピグメントは、識別のために、又は活性化合物の用量の異なる組み合わせを特徴付けるために、錠剤又はドラゼーのコーティングに加えられ得る。
【0062】
経口に使用され得る医薬製剤は、ゼラチンで作られた押し込み型カプセル剤に加え、グリセロール又はソルビトールなどの、ゼラチン及び可塑剤で作られた軟らかい、密閉されたカプセル剤を含む。幾つかの実施形態において、カプセル剤は、製薬ゼラチン、ウシゼラチン、及び植物ゼラチンの1以上を含む硬ゼラチンカプセルを含む。幾つかの例において、ゼラチンは、アルカリ処理される。押し込み型カプセル剤は、ラクトースなどの充填剤、デンプンなどの結合剤、及び/又はタルク又はステアリン酸マグネシウムなどの潤滑剤、及び随意に安定剤との混合で、活性成分を含み得る。軟カプセル剤において、活性化合物は、脂肪油、液動パラフィン、又は液体ポリエチレングリコールなどの適切な液体において溶解又は懸濁され得る。さらに、安定化剤が加えられ得る。経口投与のためのすべての製剤は、そのような投与に適した用量であるべきである。
【0063】
口腔内又は舌下投与のために、組成物は、従来の方法で調剤された、錠剤、ロゼンジ、又はゲルの形態をとり得る。非経口注入は、ボーラス注入又は持続注入に関係する。化合物(1)の医薬組成物は、油性又は水溶性のビヒクルにおける滅菌した懸濁液、溶液又はエマルションとして、非経口注入に適した形態であり得、懸濁化剤、安定化剤及び/又は分散剤などの製剤化剤を含み得る。非経口投与のための医薬製剤は、水溶性の形態で活性化合物の水溶液を含む。さらに、活性化合物の懸濁液は、好適な油性の注射懸濁液として調製され得る。適切な親油性溶媒又はビヒクルは、胡麻油などの脂肪油、オレイン酸エチル又はトリグリセリドなどの合成脂肪酸エステル、またはリポソームを含む。水溶性の注射懸濁液は、例えば、ナトリウムカルボキシメチルセルロース、ソルビトール、又はデキストランなどの、懸濁液の粘性を増加させる物質を含み得る。随意に、懸濁液はまた、化合物の溶解度を増加させる適切な安定化剤または薬剤を含み得、高濃縮溶液の調製を可能にする。あるいは、活性成分は、使用前には、好適なビヒクル、例えば、滅菌したピロゲンを含まない水で構成するための粉末形態であり得る。
【0064】
化合物(1)は、局所的に投与され得、溶液、懸濁液、ローション剤、ゲル、ペースト剤、薬用スティック、バーム、クリーム、軟膏剤などの様々な局所的に投与可能な組成物に調剤され得る。このような医薬組成物は、可溶化剤、安定化剤、等張化促進剤、緩衝液及び防腐剤を含有し得る。
【0065】
式(1)の構造を有する化合物の経皮投与に適した製剤は、経皮送達装置及び経皮送達パッチを用い得、脂溶性のエマルション又は緩衝水溶液であり得、ポリマー又は接着剤中に溶解及び/又は分散され得る。このようなパッチは、連続的、パルス状、又はオンデマンドの医薬品の送達のために構築され得る。またさらに、化合物(1)の経皮送達は、イオン泳動的なパッチなどの手段によって達成され得る。さらに、経皮的なパッチは、化合物(1)の制御された送達を提供し得る。律速膜を使用することによって、またはポリマーマトリックス又はゲル内で化合物を捕捉することによって、吸収の速度は遅れ得る。逆に、吸収促進剤は、吸収を増やすために使用され得る。吸収促進剤又は担体は、皮膚の通過を補助する吸収性の薬学的に許容可能な溶媒を含み得る。例えば、経皮装置は、裏打ち材(backing member)、随意に担体を備える化合物を含有し、随意に、長時間にわたって制御された速度及び予め定められた速度で化合物を宿主の皮膚に送達するための律速バリアを含有するリザーバー、及び装置を皮膚に固定するための手段を含む包帯(bandage)の形態である。
【0066】
吸入による投与に関して、化合物(1)は、エアロゾル、噴霧又は粉末としての形態であり得る。式(1)の医薬組成物は、適切な推進剤、例えば、ジクロロジフルオロメタン、トリクロロフルオロメタン、ジクロロテトラフルオロエタン、二酸化炭素又は他の適切なガスを使用して、加圧包装又は噴霧器からエアロゾルスプレーを示す形態で都合よく送達され得る。加圧エアロゾルの場合、投与ユニットは、測定された量を送達する弁を提供することによって決定され得る。ほんの一例として、吸入器又は注入器において使用するためのゼラチンなどのカプセル及び薬包は、化合物の粉末混合物及びラクトース又はスターチなどの適切な粉末を含んで調剤され得る。
【0067】
化合物(1)は、ココアバター又は他のグリセリドなどの従来の坐剤基剤を含有する、浣腸剤、直腸ゲル、直腸泡(rectal foams)、直腸エアロゾル、坐剤、ゼリー状の坐剤、又は停留浣腸剤などの直腸組成物に加え、ポリビニルピロリドン、PEGなどの合成ポリマーにおいて調剤され得る。組成物の坐剤形態において、限定されないが、脂肪酸グリセリドの混合物などの低融点ワックスは、随意にココアバターと組み合わされて、最初に融解される。
【0068】
本明細書に提供される処置又は使用の方法を実行する際に、本明細書に提供される治療上有効な量の化合物(1)は、医薬組成物において、処置されるべき疾患又は疾病を有する哺乳動物に投与される。幾つかの実施形態において、哺乳動物は、ヒトである。治療上有効な量は、疾患の重症度、被検体の年齢及び相対的な健康、使用される化合物の効力及び他の要因に依存して、幅広く異なり得る。化合物は、単独で又は混合物の成分として1以上の治療薬と組み合わせて使用され得る。
【0069】
医薬組成物は、活性化合物を医薬的に使用され得る製剤へと処理するのを促進する賦形剤及び助剤を含む、1以上の生理学的に許容可能な担体を使用する従来の方法で調剤され得る。適切な製剤は、選択される投与の経路に依存する。周知の技術、担体、及び賦形剤はどれも、適切なものとして、及び当該技術分野において理解されるものとして使用され得る。式(1)の化合物を含む医薬組成物は、ほんの一例ではあるが、従来の混合、溶解、造粒、ドラゼー製造、粉砕、乳化、封入、包括、又は圧迫の過程の手段によるなど、従来の方法で製造され得る。
【0070】
医薬組成物は、少なくとも1つの薬学的に許容可能な担体、希釈剤又は賦形剤、及び遊離塩基形態、又は薬学的に許容可能な塩形態の活性成分として本明細書に記載される式(1)の化合物を含み得る。さらに、本明細書に記載される方法及び医薬組成物は、N-オキシドの使用、(多形体としても知られる)結晶形態に加え、同じタイプの活性を有するこれらの化合物の活性代謝物質も含む。
【0071】
本明細書に記載される化合物を含む組成物の調製のための方法は、固形物、半固形物又は液体を形成するために、1以上の不活性な、薬学的に許容可能な賦形剤又は担体を用いて、化合物を調剤する工程を含む。固形組成物は、限定されないが、粉末、錠剤、分散性顆粒、カプセル剤、カシェ剤、及び坐剤を含む。液体組成物は、本明細書に開示されるような、化合物を溶解する溶液、化合物を含むエマルション、又は化合物を含むリポソーム、ミセル、又はナノ粒子を含む溶液を含む。半固形組成物は、限定されないが、ゲル、懸濁液及びクリームを含む。組成物は、液体溶液又は懸濁液中にあり得るか、使用前に液体中で溶液又は懸濁液に適した固形形態であり得るか、またはエマルションとしてあり得る。これらの組成物はまた、湿潤剤又は乳化剤、pH緩衝剤などの微量の非毒性補助物質を含む。
【0072】
医薬組成物のタイプの要約は、例えば、Remington: The Science and Practice of Pharmacy, Nineteenth Ed (Easton, Pa.: Mack Publishing Company, 1995)、Hoover, John E., Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pennsylvania 1975、Liberman, H.A. and Lachman, L., Eds., Pharmaceutical Dosage Forms, Marcel Decker, New York, N.Y., 1980、及びPharmaceutical Dosage Forms and Drug Delivery Systems, Seventh Ed. (Lippincott Williams & Wilkins 1999)において見出され得、それら各々は、その全体において、引用によって本明細書に組み込まれる。
【0073】
投与の典型的な方法及び処置方法
化合物(1)は、ステロイドホルモン核内受容体の活性が疾患の病因及び/又は症状の一因となる、疾患又は疾病の処置のための薬剤の調製に使用され得る。さらに、本明細書に記載されるあらゆる疾患又は疾病を、そのような処置を必要とする被験体において処置するための方法は、式(1)の少なくとも1つの化合物、又はその薬学的に許容可能な塩、薬学的に許容可能なN-オキシド、薬学的に活性な代謝物質、薬学的に許容可能なプロドラッグ、又は薬学的に許容可能な溶媒和物を含む医薬組成物を、治療上有効な量で前記被験体に投与する工程を含む。
【0074】
本明細書に記載される化合物を含有する組成物は、予防的及び/又は治療的な処置のために投与され得る。治療的な適用において、組成物は、疾患又は疾病の症状を治療する又は少なくとも部分的に抑える、又は症状自体を治療する、治癒する、改善する、又は寛解するのに十分な量で、疾患又は疾病に既に苦しむ被検体に投与される。この用途に有効な量は、疾患又は疾病の重症度及び経過、以前の治療、被検体の健康状態、体重、薬物への反応、及び処置に当たる医師の判断に依存する。
【0075】
一旦、被験体の疾病が改善すると、必要ならば維持量が投与される。続いて、投与の量又は頻度、またはその両方は、症状に応じて、改善された疾患又は疾病が保持されるレベルまで減らされ得る。しかしながら、被験体は、症状が再発すると、長期的に間欠的処置を必要とし得る。
【0076】
特定の例において、本明細書に記載される治療上有効な量の少なくとも1つの化合物(又はその薬学的に許容可能な塩、薬学的に許容可能なN-オキシド、薬学的に活性な代謝物質、薬学的に許容可能なプロドラッグ、及び薬学的に許容可能な溶媒和物)を、別の治療薬と組み合わせて投与するのが適切であり得る。ほんの一例ではあるが、本明細書の化合物の1つを受け取ることで被験体が受ける副作用の1つが炎症である場合、抗炎症剤を、最初の治療薬と組み合わせて投与するのが適切であり得る。又は、ほんの一例として、本明細書に記載される化合物の1つの治療的有用性は、アジュバントの投与によって高められ得る(すなわち、アジュバント自体は、最小の治療的有用性を有し得るだけであるが、別の治療薬と組み合わせることで、被験体に対する全体的な治療効果が高められる)。又は、ほんの一例として、被験体が受けるその効果は、本明細書に記載される化合物の1つを、治療効果をまた有する別の治療薬(これはまた治療レジメンを含む)とともに投与することで増幅され得る。いかなる場合においても、処置されている疾患又は疾病にかかわらず、被験体が受ける全体的な効果は、2つの治療薬を単に加えたものであり得るか、又は被験体は相乗効果を受け得る。本明細書に記載される化合物が他の治療と併用して投与される場合、同時投与された化合物の投与量は、当然のことながら、用いられる同時投与の薬物の種類、用いられる具体的な薬物、処置されている疾患又は疾病などに依存して変化する。さらに、1以上の生理活性物質とともに同時投与される時に、本明細書に提供される化合物は、生理活性物質(複数可)と同時に投与され得るか、又は連続して投与され得るかのどちらかである。連続して投与される場合、主治医は、生理活性物質(複数可)と組み合わせてタンパク質を投与する、適切な順序を決定する。
【0077】
いかなる場合においても、複数の治療薬(その1つは本明細書に記載される化合物の1つである)は、任意の順番で又は同時に投与され得る。もし同時であれば、複数の治療薬は、単一の、統一された形態で、又は複数回の形態で(ほんの一例として、単一の丸薬又は2つの別の丸薬のどちらかとして)提供され得る。治療薬の1つが複数回投与で与えられ得るか、又は両方が複数回の投与として与えられ得る。もし同時でなければ、複数回投与の間の期間は0週間以上4週間未満で変化し得る。さらに、組み合わせの方法、組成物及び製剤は、2つのみの薬剤の使用に限定されない。複数の治療上の組み合わせが想定される。
【0078】
さらに、化合物(1)はまた、被験体に更なる又は相乗的な効果を提供し得る手順と組み合わせて使用され得る。ほんの一例ではあるが、被験体は、本明細書に記載される方法において、治療的及び/又は予防的な効果を得ると予期され、ここで、式(I)の医薬組成物及び/又は他の治療法との組み合わせは、その個体が特定の疾患又は疾病と相互に関連することが知られている突然変異遺伝子のキャリアであるかを決定するための遺伝子検査と組み合わせられる。
【0079】
化合物(1)及び併用療法は、疾患又は疾病の発生の前、発生の間又は発生の後に投与され得、化合物を含有する組成物を投与するタイミングは変わり得る。したがって、例えば、疾患又は疾病の発生を防止するために、化合物は、予防薬として使用され得、疾病又は疾患への傾向のある被験体に、連続的に投与され得る。化合物及び組成物は、症状の発症の間に又は発症の後、できるだけすぐに被検体に投与され得る。化合物の投与は、症状の発症後の最初の48時間以内に、好ましくは症状の発症後の最初の48時間以内に、より好ましくは症状の発症後の最初の6時間以内に、及び最も好ましくは症状の発症後の3時間以内に開始され得る。最初の投与は、例えば、静脈注射、ボーラス注入、5分間から約5時間にわたる点滴、錠剤、カプセル、経皮パッチ、口腔送達など、又はそれらの組み合わせなどの任意の実用的な経路を介され得る。化合物は、好ましくは、疾患又は疾病の発症が見つかった又は疑われた後に、実行可能となってすぐに投与され、疾患の処置に必要な期間は、例えば、約1ヶ月から約3ヶ月である。処置の期間は、各被検体ごとに異なり得、その期間は公知の基準を用いて決定され得る。例えば、化合物又は化合物を含む製剤は、少なくとも2週間、好ましくは約1ヶ月から約3年間、及び幾つかの実施形態において約1ヶ月から約10年間投与され得る。他の実施形態において、化合物は、90日から2年間まで一日に一回投与される。
【0080】
本明細書に記載される医薬組成物は、正確な投与量での単回投与に適した単位剤形であり得る。単位剤形において、製剤は、1以上の化合物の適量を含有する単位用量に分割される。単位用量は、製剤の別々の量を含有するパッケージの形態であり得る。制限しない例は、包装された錠剤又はカプセル剤、及びバイアル又はアンプル中の粉末である。水溶性懸濁液組成物は、単回投与の再密閉が不可能な容器に入れられ得る。あるいは、複数回投与の密閉可能な容器が使用され得、その場合、組成物中に防腐剤を含むことが典型的である。ほんの一例として、非経口注射のための製剤は、単位剤形で提供され得、これは、限定されないが、アンプルを含み、又は複数回投与の容器においては、防腐剤が加えられる。
【0081】
本明細書中に記載される化合物(1)に適した1日の投与量は、体重あたり約0.03から60mg/kgまでである。限定されないが、ヒトを含む大型哺乳動物における示される日常的な投与量は、約1mgから約4000mgの範囲であり、限定されないが、1日に4回までを含む、1回以上の投与で、又は遅延の形態(retard form)で都合良く投与される。経口投与のための適切な単位剤形は、約1mgから約4000mgまでの活性成分を含む。幾つかの実施形態において、式(1)の化合物の単一用量は、約50mgから約2,000mgまでの範囲内にある。幾つかの実施形態において、式(1)の化合物の単一用量は、約90mg、約200mg、約250mg、約325mg、約650mg、約975mg、約1300mg、約1625mg、又は約1950mgである。幾つかの実施形態において、約90mg、約325mg、約650mg、約975mg、約1300mg、約1625mg、又は約1950mgの式(1)の化合物の投与は、複数回投与として与えられる。
【0082】
幾つかの実施形態において、式(a)の化合物の単一用量は、90から2500mgまでの間であり、化合物は、90日から2年の間で被験体に投与される。
【0083】
このような投与量は、多くの変数、限定されない使用される化合物の活性、処置される疾患又は疾病、投与の形式、個々の被験体の要件、処置されている疾患又は疾病の重症度、及び医師の判断によって変化し得る。
【0084】
薬物動態学的及び薬理学的な測定
薬物動態学的及び薬理学的なデータは、当該技術分野に公知の技術によって得られ得る。ヒトの被験体の薬物代謝の薬物動態学的及び薬理学的なパラメーターにおける固有の変化により、特定の組成物を表す適切な薬物動態学的及び薬理学的な特性成分は変化し得る。典型的には、薬物動態学的及び薬理学的な特性は、被験体の群の平均パラメーターの測定に基づく。被験体の群は、代表的な平均を測定するのに適した被験体の任意の合理的な数、例えば、5の被験体、10の被験体、16の被験体、20の被験体、25の被験体、30の被験体、35の被験体、又はそれ以上を含む。その平均は、測定される各パラメーターのすべての被験体の測定の平均を算出することによって決定される。
【0085】
薬物動態学的パラメーターは、本発明の組成物を表すのに適切な任意のパラメーターであり得る。例えば、Cmaxは、約500ng/ml以上、約550ng/ml以上、約600ng/ml以上、約700ng/ml以上、約800ng/ml以上、約880ng/ml以上、約900ng/ml以上、約100ng/ml以上、約1250ng/ml以上、約1500ng/ml以上、約1700ng/ml以上、又は化合物(1)の薬物動態学的特性を表すのに適切な任意の他のCmaxであり得る。幾つかの実施形態において、活性代謝物質は、薬物の被験体への投与の後にインビボで形成され、Cmaxは、約500pg/ml以上、約550pg/ml以上、約600pg/ml以上、約700pg/ml以上、約800pg/ml以上、約880pg/ml以上、約900pg/ml以上、約1000pg/ml以上、約1250pg/ml以上、約1500pg/ml以上、約1700pg/ml以上、又は被験体への化合物(1)の投与後にインビボで形成される化合物の薬物動態学的特性を表すのに適切な任意の他のCmaxであり得る。
【0086】
Tmaxは、例えば、約0.5時間以下、約1.0時間以下、約1.5時間以下、約2.0時間以下、約2.5時間以下、約3.0時間以下、又は化合物(1)の薬物動態学的特性を表すのに適切な任意の他のTmaxであり得る。
【0087】
AUC(0-inf)は、例えば、約590ng・hr/mL以上、約1500ng・hr/mL以上、約2000ng・hr/mL以上、約3000ng.times.hr/ml以上、約3500ng・hr/mL以上、約4000ng・hr/mL以上、約5000ng・hr/mL以上、約6000ng・hr/mL以上、約7000ng・hr/mL以上、約8000ng・hr/mL以上、約9000ng・hr/mL以上、又は化合物(1)の薬物動態学的特性を表すのに適切な任意の他のAUC(0-inf)であり得る。幾つかの実施形態において、活性代謝物質は、化合物(1)の被験体への投与の後にインビボで形成され、AUC(0-inf)は、例えば、約590pg・hr/mL以上、約1500pg・hr/mL以上、約2000pg・hr/mL以上、約3000pg・hr/mL以上、約3500pg・hr/mL以上、約4000pg・hr/mL以上、約5000pg・hr/mL以上、約6000pg・hr/mL以上、約7000pg・hr/mL以上、約8000pg・hr/mL以上、約9000pg・hr/mL以上、又は被験体への化合物(1)の投与後にインビボで形成される化合物の薬物動態学的特性を表すのに適切な任意の他のAUC(0-inf)であり得る。
【0088】
投与の約1時間後の化合物(1)の血漿濃度は、例えば、約140ng/ml以上、約425ng/ml以上、約550ng/ml以上、約640ng/ml以上、約720ng/ml以上、約750ng/ml以上、約800ng/ml
以上、約900ng/ml以上、約1000ng/ml以上、約1200ng/ml以上、又は化合物(1)の任意の他の血漿濃度であり得る。
【0089】
薬理学的パラメーターは、本発明の組成物を表すのに適切な任意のパラメーターであり得る。例えば、薬理学的特性は、ほんの一例として、少なくとも約2時間、少なくとも約4時間、少なくとも約8時間、少なくとも約12時間又は少なくとも約24時間、ARタンパク質又は内因性のアンドロゲンの減少を示し得る。薬理学的特性は、ほんの一例として、少なくとも約2時間、少なくとも約4時間、少なくとも約8時間、少なくとも約12時間又は少なくとも約24時間、CYP17を含む、アンドロゲンを合成する酵素の阻害を示し得る。薬理学的特性は、ほんの一例として、少なくとも約2時間、少なくとも約4時間、少なくとも約8時間、少なくとも約12時間又は少なくとも約24時間、アンドロゲンシグナル伝達の低下を示し得る。
【0090】
治療を提供する典型的な方法
本発明は、ヒトにおける前立腺癌の処置のための治療上の方策を提供する。
【0091】
幾つかの実施形態において、本発明は、前立腺癌の処置における化合物1の使用のための調製及びレジメンを提供する。幾つかの実施形態において、前立腺癌は、去勢抵抗性前立腺癌である。幾つかの実施形態において、前立腺癌は、化学療法未治療の前立腺癌である。
【0092】
幾つかの実施形態において、本発明は、化合物1の経口投与を含む治療上のレジメンを提供する。
【0093】
幾つかの実施形態において、本発明は、化合物1の複数回用量の投与を含む治療上のレジメンを提供する。幾つかの実施形態において、異なる投与は、時間において別々に間隔を置かれる。幾つかの実施形態において、すべての投与は、同じ量の化合物1を含む。幾つかの実施形態において、異なる投与は、異なる量の化合物1を含む。幾つかの実施形態において、時間で分離される異なる投与は、同じ時間によって互いから分離され、幾つかの実施形態において、時間で分離される異なる投与は、異なる時間によって互いから分離される。幾つかの実施形態において、本発明は、規則的な時間間隔(複数可)によって分離された複数の用量の投与を含む投薬レジメンを提供し、その後、休息期間があり、随意にその後、規則的な時間間隔(複数可)によって分離された第2の複数の用量を提供する。
【0094】
幾つかの実施形態において、少なくとも2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109,110、111、112、113、114、115、116、117、118、119、120、121、122、123、124、125、126、127、128、129、130、131、132、133、134、135、136、137、138、139、140、141、142、143、144、145、146、147、148、149、150、151, 152, 153, 154, 155, 156, 157, 158, 159, 160, 161, 162, 163, 164, 165, 166, 167, 168又はそれ以上の用量の化合物1が投与される。幾つかの実施形態において、少なくとも7、14、21、28、35、42、49、56、63、70、77、84、91、98、105、112、119、126、133、140、147、154、161、168又はそれ以上の用量の化合物1が投与される。
【0095】
幾つかの実施形態において、本発明は、化合物(1):
【0096】
【化7】
【0097】
を含む医薬組成物を、微粉化結晶性粉末として熟考する。
【0098】
幾つかの実施形態において、本発明は、化合物(1):
【0099】
【化8】
【0100】
を含む医薬組成物を熟考し、式中、化合物(1)は、約:a. 13.0°、14.6°、16.3°、17.6°、及び19.0°、及び随意に約11.8°、20.2°、22.9°、及び25.4°;b. 13.1°及び14.1°;c. 17.2°、18.5°、及び19.1°、及び随意に16.2°、及び29.6°;d. 12.5°、14.8°、及び25.5°;又はe. 14.4°、16.1°、19.1°、及び19.3°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられた、結晶形態である。
【0101】
幾つかの実施形態において、本発明は、化合物(1):
【0102】
【化9】
【0103】
を含む医薬組成物を熟考し、式中、化合物(1)は、約:a. 13.1°及び14.1°;b. 17.2°、18.5°、及び19.1°、及び随意に約16.2°、及び29.6°;c. 12.5°、14.8°、及び25.5°;又はd. 14.4°、16.1°、19.1°及び19.3°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられた、微粉化結晶形態、又はその薬学的に許容可能な塩、N-オキシド、活性代謝物質、プロドラッグ、又は溶媒和物である。
【0104】
幾つかの実施形態において、本発明は、化合物(1):
【0105】
【化10】
【0106】
を含む医薬組成物を熟考し、式中、化合物(1)は、約:a. 13.1°及び14.1°;b. 17.2°、18.5°、及び19.1°、及び随意に約16.2°、及び29.6°;c. 12.5°、14.8°、及び25.5°;又はd. 14.4°、16.1°、19.1°及び19.3°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられた、微粉化結晶形態である。
【0107】
幾つかの実施形態において、化合物(1)は、被験体への投与の後に、アンドロゲン受容体媒介性の疾患又は疾病を処置するのに有効な量で存在する。
【0108】
幾つかの実施形態において、アンドロゲン受容体媒介性の疾患又は疾病は、前立腺癌、良性前立腺肥大症、多毛症、脱毛症、神経性食欲不振、乳癌、及び男性の性腺機能亢進から成る群から選択される。
【0109】
幾つかの実施形態において、アンドロゲン受容体媒介性の疾患又は疾病は、前立腺癌である。
【0110】
幾つかの実施形態において、前立腺癌は、去勢抵抗性前立腺癌である。
【0111】
幾つかの実施形態において、化合物(1)は、被験体への投与の後に、アンドロゲン生合成を阻害し、アンドロゲン受容体シグナル伝達を阻害し、及びアンドロゲン受容体感受性を低下させるのに有効な量で存在する。
【0112】
幾つかの実施形態において、化合物は、アンドロゲン受容体シグナル伝達を阻害するか、又はアンドロゲン受容体感受性を低下させる。
【0113】
幾つかの実施形態において、アンドロゲン生合成阻害は、チトクロームC17α-ヒドロキシラーゼ/C17,20-リアーゼ(CYP17)の活性を阻害することを含む。
【0114】
幾つかの実施形態において、アンドロゲン受容体シグナル伝達の阻害は、テストステロン結合の競合的阻害を含む。
【0115】
幾つかの実施形態において、アンドロゲン受容体感受性の低下は、細胞内のアンドロゲン受容体タンパク質の含量の減少、及び低レベルのアンドロゲンの増殖シグナルによって持続される、細胞の低下した能力を含む。
【0116】
幾つかの実施形態において、組成物は、被験体に、非経口的に、静脈内に、筋肉内に、皮内に、皮下に、腹腔内に、経口的に、口腔に、舌下に、粘膜に、直腸に、経皮的に、皮膚に、眼に、又は吸入によって投与するために調剤される。
【0117】
幾つかの実施形態において、組成物は、錠剤、カプセル剤、クリーム、ローション剤、油剤、軟膏、ゲル、ペースト、粉末、懸濁液、エマルション、又は溶液として被験体に投与するために調剤される。
【0118】
幾つかの実施形態において、組成物は、カプセル剤として被験体に投与するために調剤される。
【0119】
前述の請求項のいずれかの医薬組成物は、錠剤として被験体に投与するために調剤される。
【0120】
幾つかの実施形態において、カプセル剤は、粉末として化合物(1)を含む。
【0121】
幾つかの実施形態において、粉末は微粉化される。
【0122】
幾つかの実施形態において、組成物は、約50mgから約500mgまでの化合物(1)を含む。
【0123】
幾つかの実施形態において、組成物は、約90mgの化合物(1)を含む。
【0124】
幾つかの実施形態において、組成物は、約325mgの化合物(1)を含む。
【0125】
幾つかの実施形態において、組成物は、被験体に、1日当たり、1、2、3、4、5、6、7、8、9、又は10回投与するために調剤される。
【0126】
幾つかの実施形態において、組成物は、前立腺癌の処置に対して被験体に投与されるために調剤される。
【0127】
幾つかの実施形態において、組成物は、去勢抵抗性前立腺癌の処置に対して被験体に投与されるために調剤される。
【0128】
幾つかの実施形態において、組成物は、1以上の薬学的に許容可能な賦形剤を更に含む。
【0129】
幾つかの実施形態において、薬学的に許容可能な賦形剤は、充填剤、崩壊剤、潤滑剤、界面活性剤、流動促進剤、結合剤、砂糖、スターチ、ニス、又はワックスを含む。
【0130】
幾つかの実施形態において、化合物(1)は、薬学的に許容可能な塩、N-オキシド、活性代謝物質、プロドラッグ、結晶多形体、又は溶媒和物である。
【0131】
幾つかの実施形態において、溶媒和物は、クメン溶媒和物又は水和物を含む。
【0132】
幾つかの実施形態において、結晶多形体は、化合物(1)のフォームI、フォームII、又はフォームIIIを含む。
【0133】
幾つかの実施形態において、本発明は、化合物(1)
【0134】
【化11】
【0135】
の結晶形態を熟考し、(フォームI)約13.0°、14.6°、16.3°、17.6°、及び19.0°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられる。
【0136】
幾つかの実施形態において、結晶形態は、約11.8°、20.2°、22.9°、及び25.4°の角度 2-シータにおいて表される、特性ピークによって更に特徴付けられる。
【0137】
幾つかの実施形態において、本発明は、化合物(1)
【0138】
【化12】
【0139】
の結晶形態を熟考し、(フォームII)約13.1°及び14.1°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられる。
【0140】
幾つかの実施形態において、本発明は、化合物(1)
【0141】
【化13】
【0142】
の結晶形態を熟考し、(フォームIII)約17.2°、18.5°、及び19.1°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられる。
【0143】
幾つかの実施形態において、結晶形態は、約16.2°、及び29.6°の角度 2-シータにおいて表される、特性ピークによって更に特徴付けられる。
【0144】
幾つかの実施形態において、本発明は、化合物(1)
【0145】
【化14】
【0146】
の結晶形態を熟考し、(パターン5)約12.5°、14.8°、及び25.5°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられる。
【0147】
幾つかの実施形態において、本発明は、化合物(1)
【0148】
【化15】
【0149】
の結晶形態を熟考し、(パターン2)約14.4°、16.1°、19.1°、及び19.3°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられる。
【0150】
幾つかの実施形態において、本発明は、ジメチルホルムアミド、炭酸カリウム、式:
【0151】
【化16】
【0152】
の化合物、又はそのアナログ、及び式:
【0153】
【化17】
【0154】
の化合物を接触させ、式:
【0155】
【化18】
【0156】
の化合物を作る工程を含む方法を熟考する。
【0157】
幾つかの実施形態において、該方法は、式:
【0158】
【化19】
【0159】
の化合物を、N-メチルピロリドン中の木炭上の10%のパラジウムと接触させ、式:
【0160】
【化20】
【0161】
の化合物を生成する工程を更に含む。
【0162】
幾つかの実施形態において、該方法は、式:
【0163】
【化21】
【0164】
の化合物を、メタノールナトリウムメトキシドと接触させ、式(I):
【0165】
【化22】
【0166】
の化合物を生成する工程を更に含む。
【0167】
幾つかの実施形態において、該方法は、大規模又は製造規模で行われる。幾つかの実施形態において、大規模は、約1から約10kgまでの規模である。幾つかの実施形態において、製造規模は、約10kgより大きい規模である。幾つかの実施形態において、製造規模は、約10から約1,000kgまでの規模である。幾つかの実施形態において、製造規模は、約10から約100kgまでの規模である。幾つかの実施形態において、製造規模は、約10から約50kgまでの規模である。幾つかの実施形態において、製造規模は、約33.4kgの規模である。
【0168】
幾つかの実施形態において、本発明は、被験体において前立腺癌に対する処置を提供する方法を熟考し、該方法は、治療上有効な量の化合物(1):
【0169】
【化23】
【0170】
又はその薬学的に許容可能な塩、N-オキシド、活性代謝物質、プロドラッグ、又は溶媒和物を含む医薬組成物を被験体に投与する工程を含み、
式中、化合物(1)は、約:a. 13.1°及び14.1°;b. 17.2°、18.5°、及び19.1°、及び随意に約16.2°、及び29.6°;c. 12.5°、14.8°、及び25.5°;又はd. 14.4°、16.1°、19.1°及び19.3°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられた、微粉化結晶形態である。
【0171】
幾つかの実施形態において、本発明は、被験体において前立腺癌に対する処置を提供する方法を熟考し、該方法は、治療上有効な量の化合物(1):
【0172】
【化24】
【0173】
を含む医薬組成物を被験体に投与する工程を含み、
式中、化合物(1)は、約:a. 13.0°、14.6°、16.3°、17.6°、及び19.0°、及び随意に約11.8°、20.2°、22.9°、及び25.4°;b. 13.1°及び14.1°;c. 17.2°、18.5°、及び19.1°、及び随意に16.2°、及び29.6°;d. 12.5°、14.8°、及び25.5°;又はe. 14.4°、16.1°、19.1°、及び19.3°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられた、結晶形態である。
【0174】
幾つかの実施形態において、本発明は、被験体において前立腺癌に対する処置を提供する方法を熟考し、該方法は、治療上有効な量の化合物(1):
【0175】
【化25】
【0176】
を含む医薬組成物を被験体に投与する工程を含み、
式中、化合物(1)は、約:a. 13.1°及び14.1°;b. 17.2°、18.5°、及び19.1°、及び随意に約16.2°、及び29.6°;c. 12.5°、14.8°、及び25.5°;又はd. 14.4°、16.1°、19.1°及び19.3°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられた、微粉化結晶形態である。
【0177】
幾つかの実施形態において、本発明は、被験体において前立腺癌の処置のための薬剤を調剤する際の化合物(1)の使用を熟考し、該薬剤は、治療上有効な量の化合物(1):
【0178】
【化26】
【0179】
又はその薬学的に許容可能な塩、N-オキシド、活性代謝物質、プロドラッグ、又は溶媒和物を含み、
式中、化合物(1)は、約:a. 13.1°及び14.1°;b. 17.2°、18.5°、及び19.1°、及び随意に約16.2°、及び29.6°;c. 12.5°、14.8°、及び25.5°;又はd. 14.4°、16.1°、19.1°及び19.3°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられた、微粉化結晶形態である。
【0180】
幾つかの実施形態において、本発明は、被験体において前立腺癌の処置のための薬剤を調剤する際の化合物(1)の使用を熟考し、該薬剤は、治療上有効な量の化合物(1):
【0181】
【化27】
【0182】
を含み、
式中、化合物(1)は、約:a. 13.0°、14.6°、16.3°、17.6°、及び19.0°、及び随意に約11.8°、20.2°、22.9°、及び25.4°;b. 13.1°及び14.1°;c. 17.2°、18.5°、及び19.1°、及び随意に16.2°、及び29.6°;d. 12.5°、14.8°、及び25.5°;又はe. 14.4°、16.1°、19.1°、及び19.3°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられた、結晶形態である。
【0183】
幾つかの実施形態において、本発明は、被験体において前立腺癌の処置のための薬剤を調剤する際の化合物(1)の使用を熟考し、該薬剤は、治療上有効な量の化合物(1):
【0184】
【化28】
【0185】
を含み、
式中、化合物(1)は、約:a. 13.1°及び14.1°;b. 17.2°、18.5°、及び19.1°、及び随意に約16.2°、及び29.6°;c. 12.5°、14.8°、及び25.5°;又はd. 14.4°、16.1°、19.1°及び19.3°の角度 2-シータにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられた、微粉化結晶形態である。
【実施例】
【0186】
以下の実施例は、化合物(1)を生成するための、及び化合物(1)の有効性及び安全性を試験するための例示的方法を提供する。これらの実施例は、例示目的のみに提供され、本明細書に提供される請求項の範囲を制限しない。本明細書に開示され請求される方法のすべては、本開示の観点から、不当な実験なしで作られ実行され得る。
【0187】
請求項の概念、精神及び範囲から逸脱することなく、本明細書に記載される方法に、及び方法の工程において又は工程の順序において変更が適用され得ることは当業者に明らかである。当業者に明らかなすべてのこのような同様の代替及び改変は、添付された請求項の精神、範囲及び概念内にあると考えられる。
【0188】
<実施例1:式(1)の化合物の合成>
<実施例1A:3-β-アセトキシ-17-(1H-ベンズイミダゾール-1-イル)-16-ホルミル-アンドロスタ-5,16-ジエンの合成>
【0189】
【化29】
【0190】
33.4kgの3-β-アセトキシ-17-クロロ-16-ホルミルアンドロスタ-5,16-ジエンを、ジメチルホルムアミド(DMF)中でベンズイミダゾール及び炭酸カリウムと混合させ、残りの出発物質の量によって決定されるように、反応が終了するまで加熱した。反応が終了した後、反応混合物を冷却し、冷却した水と混合し、反応物をクエンチした。固形物をクエンチした反応混合物から分離し、DMFと水の混合物、水、希薄した水性の塩化水素酸、水、希薄した水性の炭酸水素ナトリウム、及び水で連続して洗浄した。中間物質、3-β-アセトキシ-17-(1H-ベンズイミダゾール-1-イル)-16-ホルミルアンドロスタ-5,16-ジエンを、続いて乾燥した。
【0191】
<実施例1B:3-β-アセトキシ-17-(1H-ベンズイミダゾール-1-イル)アンドロスタ-5,16-ジエンの合成及び精製>
【0192】
【化30】
【0193】
3-β-アセトキシ-17-(1H-ベンズイミダゾール-1-イル)-16-ホルミルアンドロスタ-5,16-ジエンを、N-メチルピロリドン(NMP)中で炭素上の約10%のパラジウム(Pd/C)と混合させ、反応混合物中の3-β-アセトキシ-17-(1H-ベンズイミダゾール-1-イル)-16-ホルミルアンドロスタ-5,16-ジエン/3-β-アセトキシ-17-(1H-ベンズイミダゾール-1-イル)アンドロスタ-5,16-ジエンの比率によって決定されるように、反応が終了するまで加熱した。反応が終了した後、反応混合物を冷却した。硫酸マグネシウムを加え、結果として生じる混合物をろ過した。水を濾液に加え、結果として生じる混合物を撹拌した。固体の、粗製の3-β-アセトキシ-17-(1H-ベンズイミダゾール-1-イル)アンドロスタ-5,16-ジエンを、水/NMP混合物から分離し、水とメタノールの混合物で洗浄し、乾燥し、包装した。
【0194】
粗製の3-β-アセトキシ-17-(1H-ベンズイミダゾール-1-イル)アンドロスタ-5,16-ジエンを、酢酸エチル中に溶解し、浄化した。この混合物の容量を、真空蒸留によって低減した。結果として生じる混合物を、冷却し、固形物を分離し、冷たい酢酸エチルで洗浄し、真空下で乾燥した。幾つかの実施形態において、サンプルを、不純物レベルを測定するためにインプロセスの試験にさらした。不純物レベルが許容可能でなかったとき、再結晶過程(recrystallization process)を繰り返した。
【0195】
<実施例1C:3-β-ヒドロキシ-17-(1H-ベンズイミダゾール-1-イル)アンドロスタ-5,16-ジエンの合成及び精製>
【0196】
【化31】
【0197】
3-β-アセトキシ-17-(1H-ベンズイミダゾール-1-イル)アンドロスタ-5,16-ジエンを、メタノール中でナトリウムメトキシドと混合させ、残りの3-β-アセトキシ-17-(1H-ベンズイミダゾール-1-イル)アンドロスタ-5,16-ジエンの量によって決定されるように、反応が終了するまで加熱した。反応が終了した後、反応混合物を冷却し、水と混合し、反応物をクエンチした。結果として生じるスラリーを撹拌し、更に冷却した。固体の、粗製の3-β-ヒドロキシ-17-(1H-ベンズイミダゾール-1-イル)アンドロスタ-5,16-ジエンを、クエンチした反応混合物から分離し、メタノールと水の混合物で洗浄し、その後、洗液が中性になるまで水で洗浄し、乾燥し、及び包装した。
【0198】
粗製の3-β-ヒドロキシ-17-(1H-ベンズイミダゾール-1-イル)アンドロスタ-5,16-ジエンを、メタノールと酢酸エチルの混合物中に溶解し、浄化した。生成物を、溶媒交換単独によって、メタノール/酢酸エチル溶液から酢酸エチルに移した。結果として生じる混合物を冷却し、固形物を分離し、冷たい酢酸エチルで洗浄し、真空下で乾燥した。幾つかの実施形態において、サンプルを、不純物レベルを測定するためにインプロセスの試験にさらした。不純物レベルが許容可能でなかったとき、再結晶過程を繰り返した。
【0199】
<実施例2:化合物(1)の結晶多形相>
<実施例2A:機器及び方法論>
X線粉末回折(XRPD):Cu Kα放射光(40kV, 40mA)、自動XYZステージ、自動サンプル配置のためのレーザービデオ顕微鏡、及びHiStar 2次元面積検出器を使用して、X線粉末回折パターンを、Bruker AXS C2 GADDS回折計上に収集した。X線光学は、0.3mmのピンホールコリメータとつながれた単一のGoebel 多層膜鏡から成った。
【0200】
ビーム発散、すなわちサンプル上のX線ビームの有効径は、約4mmであった。θ-θの連続的な走査方式を、3〜30°2θの有効な2θ範囲を与える20cmのサンプル-検出器距離で利用した。典型的に、サンプルを、120秒間X線ビームに曝した。データ収集に使用したソフトウェアは、WNT 4.1.16用のGADDSであり、データを、Diffrac Plus EVA v 9.0.0.2又はv 13.0.0.2を使用して分析し、提示した。周囲条件下で実行したサンプルを、粉砕なしで受け取られるように、粉末を使用して、平面プレート試料として調製した。非周囲条件下で実行したサンプルを、熱伝導性の化合物とともにシリコンウェーファー上に乗せた。
【0201】
核磁気共鳴(1H-NMR):NMRスペクトルを、自動サンプラーを備える、Bruker 400MHz機器上に収集し、DRX400コンソールによって制御した。自動化した実験を、標準のBruker 荷重実験(standard Bruker loaded experiments)を使用してTopspin v 1.3(パッチレベル8)で実行する、ICON-NMR v4.0.4(build 1)を使用して得た。非ルーチン分光法(non-routinespectroscopy)に関して、データを、Topspin単独の使用を介して得た。他に明記のない限り、サンプルを、d6-DMSO中で調製した。オフライン分析を、ACD SpecManager v 12.00(build 29094)を使用して実行した。
【0202】
示差走査熱量測定法(DSC):DSCデータを、50位置の自動サンプラーを備えた、TA Instruments Q2000上で収集した。該機器を、公認のインジウムを使用して、エネルギー及び温度の較正のために較正した。ピンで穴をあけたアルミニウムパンにおいて、典型的に0.5〜3mgの各サンプルを、25℃から250℃までの10℃.min-1で加熱した。50mL/分での窒素パージを、サンプル上で維持した。
【0203】
熱重量分析(TGA):TGAデータを、16位置の自動サンプラーを備えた、TA Instruments Q500 TGA上で収集した。該機器を、公認のアルメルを使用して、温度較正した。典型的に5〜30mgの各サンプルを、風袋に入れた白金るつぼ及びアルミニウムDSCパンに充填し、周囲温度から300℃までの10℃/分で加熱した。60mL/分での窒素パージを、サンプル上で維持した。
【0204】
重量蒸気収着(GVS):等温収着曲線を、SMS DVS Intrinsicの水分収着分析を使用して獲得し、SMS Analysis Suiteソフトウェアによって制御した。サンプル温度を、機器の制御によって25℃で維持した。湿度を、200mL/分の合計流量で、乾燥した及び湿った窒素の流れを混合することにより制御した。相対湿度を、サンプルの近くにある、較正したRotronic探査(1.0〜100%RHの動作範囲)によって測定した。重量変化、%RHに応じたサンプルの(質量緩和)を、微量天秤(正確度±0.005mg)によって常に監視した。5〜20mgのサンプルを、周囲条件下で、風袋に入れたメッシュステンレス鋼バスケットに置いた。40%RH及び25℃(典型的な室内条件)で、サンプルに負荷を与え、及びサンプルを負荷軽減した。水分収着曲線を、以下のように実行した:
【0205】
【表1】
【0206】
HPLCによる化学的純度測定:ダイオードアレイ検出器を備える、Agilent HP1100シリーズの装置上で、及び以下に詳述される2つの方法の1つを使用するChemStationソフトウェア vB.02.01-SR1を使用して、純分分析を行った。
【0207】
【表2】
【0208】
偏光顕微鏡法(PLM):サンプルを、画像キャプチャー用のデジタルビデオカメラを備えたLeica LM/DM 偏光顕微鏡上で研究した。小量の各サンプルを、スライドガラス上に置き、液浸油に浸け(mounted in)、ガラススリップで覆い、その個々の粒子状物質をできるだけうまく分離した。サンプルを、適切な倍率及び部分偏光で観察し、λの類似色彩フィルターにつなげた。
【0209】
<実施例2B:非晶質の化合物(1)の調製>
化合物(1)を、tert-ブタノール中に溶解した。サンプルを凍結乾燥過程にさらし、その中で、サンプルをろ過し、冷凍し、凍結乾燥した。固形物を収集し、XRPD、1H-NMR、調節したDSC、HPLC純度、及び光学顕微鏡法によって特徴付けた。非晶質物質の調製を、再現性をチェックするために繰り返した。図6は、凍結乾燥によって得られた固形物のXRPDである。
【0210】
<実施例2C:化合物(1)フォームIの調製及び分析>
化合物(1)を、メタノール中に溶解した。溶液中のメタノールを蒸発させ、結果として生じる固形物を、収集し、XRPD、1H-NMR、DSC、HPLC純度、及び光学顕微鏡法によって特徴付けた。図1は、フォームIのXRPDである。TGAによるフォームIの熱分析を、10℃/分の加熱速度で実行した。TGAサーモグラムにおいて体重損失は観察されず(図7)、これは、物質が非溶媒和の結晶形態であったことを示した。サンプルの分解は、230℃までの温度で始まった。DSC分析(図7)は、化合物(1)(フォームI)の融点を示す196℃での急速な吸熱(91.7J/g)を示した。
【0211】
<実施例2D:化合物(1)パターン2の調製及び分析>
フォームI化合物(1)を、周囲条件でクメン中でスラリーにした。混合物を、50℃/室温のヒート・クールサイクル(heat-cool cycles)(8時間のサイクル)で、70時間成熟させた。固形物をろ過し、XRPDによって分析した。固形物を、真空オーブンにおいて約35℃で24時間乾燥した後、別のXRPD分析を得た。固形物(パターン2)の1H-NMR分析は、0.2eqまでのクメン含量を示した。
【0212】
<実施例2E:化合物(1)パターン5の調製及び分析>
非晶質の化合物(1)を、周囲条件で水中でスラリーにした。混合物を、50℃/室温のヒート・クールサイクル(8時間のサイクル)で、70時間成熟させた。固形物をろ過し、XRPDによって分析した。固形物を、真空オーブンにおいて約35℃で24時間乾燥した後、別のXRPD分析を得た。固形物の1H-NMR分析は、残留溶媒を示さなかった。GVS実験は、約50%の相対湿度での水和物(パターン5)の存在を示した。
【0213】
<実施例2F:化合物(1)フォームIIの調製及び分析>
パターン5化合物(1)を、40%の相対湿度を下回るくらいにまでGVSによって脱水した。
XRPD分析を、非溶媒和の結晶上で行い、フォームIIを示した。
【0214】
<実施例2G:化合物(1)フォームIIIの調製及び分析>
温度のエンドポイントで、約5分間10℃/分で、約200℃まで水和物のサンプルを加熱することにより、パターン5化合物(1)を融解した。その後、サンプルを冷却した。サンプルの化学的な完全性(integrity)が温度によって影響を受けなかったことを確認するために、1H-NMR分析を行った。XRPD分析を、非溶媒和の結晶上で行い、フォームIIIを示した。
【0215】
<実施例3:医薬組成物>
<実施例3A:経口組成物>
経口送達のための医薬組成物を調製するために、式(1)の化合物を、約0.20g/mLのバルク密度及び約0.31g/mLのタップ密度を有するように微粉化した。90mgの微粉化した化合物を、経口投与に適したサイズ「3」のカプセル剤にパック充填した。
【0216】
<実施例3B:経口組成物>
経口送達のための医薬組成物を調製するために、式(1)の化合物を、約0.20g/mLのバルク密度及び約0.31ng/mLのタップ密度を有するように微粉化した。325mgの微粉化した化合物を、経口投与に適したサイズ「00」のカプセル剤にパック充填した。
【0217】
<実施例3C:経口組成物>
経口送達のための医薬組成物を調製するために、90mgの式(1)の化合物を、200mgのラクトース及び1%のステアリン酸マグネシウムと混合する。混合物を混ぜ合わせ、経口投与に適するように、錠剤へと直接圧縮する。
【0218】
<実施例3D:非経口組成物>
注入による投与に適した非経口医薬組成物を調製するために、式(1)の化合物の100mgの水溶性塩を、DMSO中に溶解し、その後、0.9%の無菌食塩水、10mLと混合する。混合物を、注入による投与に適した投与ユニット剤形に組み込む。
【0219】
<実施例4:インビトロの薬理学的試験>
<実施例4A:アンドロゲン受容体結合アッセイ>
アンドロゲン受容体の競合的結合を、変異したAR(384nMのIC50)を発現する、アンドロゲン感受性ヒト前立腺癌細胞株(LNCaP)細胞において、及び野生型AR(845nMのIC50)を発現する細胞において、ラベルが付けられたR1881(アンドロゲンアゴニスト)を使用して測定した。図8は、(化合物5と記載される)化合物(1)が、用量依存性の方法で両方のタイプのARに結合するために、ラベルが付けられたR1881と有効に競合したことを実証する。
【0220】
<実施例4B:リアーゼ活性の阻害>
トランスフェクトされたE. coliによって発現される完全なCYP17を分離し、酵素源として精製した。基質として17α-ヒドロキシプレグネノロンを放射標識した。CYP17活性を、基質のC-21側鎖の開裂中に形成された、トリチウム化した酢酸の量によって測定した。図8は、(化合物5と記載される)化合物(1)が、CYP17に対して300.0nMのIC50値を示したことを実証する。
【0221】
<実施例4C:前立腺癌細胞株のテストステロン誘発性の増殖の阻害>
ヒトの前立腺癌細胞株(LNCaP及びLAPC-4)を、培養において成長させ、1nMのジヒドロテストステロン(DHT)によって刺激した。DHTのこの濃度は、前立腺癌細胞の増殖を刺激した。化合物(1)の付加は、Casodex(登録商標)に似た方法で、テストステロン誘発性の増殖の用量依存性の阻害を誘発し、これを、この実験において陽性対照として使用した。
【0222】
<実施例4D:前立腺癌細胞株におけるアンドロゲン受容体(AR)タンパク質の分解>
培養細胞におけるすべてのタンパク質合成を阻害するために、シクロヘキシミドを、ヒトの前立腺癌細胞株(LNCaP)に加えた。抽出タンパク質が、ARタンパク質に向けられたモノクローナル抗体により調べられた時、シクロヘキシミド処置は、単独で、時間依存性の方法によってARレベルを低下させた。図9において、これらの培養物への化合物(1)の付加によって、結果的に、培養における時間とともに、ARタンパク質の減少率が著しくより大きくなった。
【0223】
<実施例5:インビボの薬理学的試験>
<実施例5A:重度障害型免疫不全(SCID)のマウスにおけるヒト前立腺癌の異種移植片の増殖の阻害>
LAPC4前立腺癌細胞腫瘍の異種移植片を、SCIDマウスに移植した。癌を有するマウスは、体重(BW)当たり50mg/kgの化合物(1)の皮下(SC)投与を毎日2回受けた。腫瘍サイズを毎週測定し、ビヒクル、Casodex(登録商標)又は去勢のみを受けた対照マウスと比較した。図10は、対照と比較して、去勢が最終的な腫瘍容積の著しい減少につながったことを示す。化合物(1)で処置されたマウスは、去勢と比較して、同等の又はより顕著な腫瘍成長の減少を示した。
【0224】
<実施例6:化学療法未治療の去勢抵抗性前立腺癌に苦しむヒトを処置するための化合物1の使用>
【0225】
【表3−1】
【0226】
【表3−2】
【0227】
【表3−3】
【0228】
【表3−4】
【0229】
【表3−5】
【0230】
用語の略語及び定義のリスト
表1にリストされる略語は、この試験プロトコルにおいて使用される。
【0231】
【表4−1】
【0232】
【表4−2】
【0233】
導入
1.1 去勢抵抗性前立腺癌の処置に対する化合物1の使用のための科学的及び臨床的根拠
1.1.1 去勢抵抗性前立腺癌
前立腺癌は、男性において最もよく見られる癌である。大多数の前立腺癌死亡は、従来のアンドロゲン遮断療法に反応性のない転移性疾患の進行が原因である。アンドロゲン遮断療法は、1940年代以降、前立腺癌を有する被験体における標準治療であった。アンドロゲン遮断にもかかわらず、ほとんどの被験体は、最終的に、疾患の進行を経験する(Mohler et al., 2004; Scher et al., 2004)。長年、該疾患のこの後の相は、「ホルモン不応性前立腺癌」又は「アンドロゲン非依存性前立腺癌」と呼ばれた。それ以降、数年間のアンドロゲン遮断療法の後に出現する前立腺癌は、アンドロゲンに依存したままであることが明らかになった。生存した前立腺癌細胞は、(副腎から発現した)低レベルの循環するアンドロゲンを移入し、これらの低レベルのテストステロンにかなり感応的になり、及び実際に、前立腺癌細胞自体内のテストステロンを合成する能力を得た。前立腺癌のこの段階は、現在、「去勢抵抗性前立腺癌」又はCRPCと称される。
【0234】
1.1.2 前立腺癌におけるアンドロゲン及びアンドロゲン受容体の役割の概要
1.1.2.1 前立腺癌におけるアンドロゲン及びアンドロゲン受容体の役割
アンドロゲンは、正常な前立腺の成長、及び前立腺癌において重要な役割を果たす。ほぼすべての初期の前立腺癌がアンドロゲン依存性であるため、アンドロゲン除去は、最初に、前立腺癌を有する被験体における反応をもたらす(Canil et al., 2005; Catalona, 1994; Chodak, 2004; Mohler et al., 2004; Pienta and Bradley, 2006)。アンドロゲン除去療法[ゴナドトロピン放出ホルモン(GnRH)アゴニスト±アンドロゲン受容体(AR)アンタゴニストによる、外科又は内科的去勢のいずれか]は、ほとんどの前立腺癌の被験体のための標準治療である。臨床転帰は、アンドロゲン除去療法の性質及びタイミングに関係なく、類似している。初期反応があり、その後、生化学的な、放射線学的な、及び最終的に臨床的な進行において終了する安定性の期間が続く(Bubley and Balk, 1996; Scher et al., 2004; Scher et al., 2008)。前立腺癌細胞によって確立された1つの重要な生存メカニズムが、低レベルの循環する及び/又は細胞内のアンドロゲンに対するその感受性の劇的な増加であることが明らかになった。これは、アンドロゲン除去によって誘発される。低レベルのアンドロゲンに対する感受性の増加は、ARタンパク質の増加した発現によって与えられる(Gregory et al., 2001; Taplin and Ho, 2001; Gelman, 2002; Litvinov et al., 2003; Lee and Chang 2003; Heinlin and Chang, 2004; Chen et al., 2004)。本発明によると、前立腺癌の処置のための有効な治療は、完全なアンドロゲン遮断を含むべきである。
【0235】
1.1.2.2 アンドロゲン作用のメカニズム及び摂動(Perturbation)
アンドロゲンは、アンドロゲン感受性組織の細胞質内に存在する受容体に結合することにより、前立腺組織及び前立腺癌細胞の成長を刺激する。受容体は、熱ショックタンパク質90などの担体タンパク質に結合される(Pratt, 1993)。一旦結合されると、ホルモン受容体複合体は、細胞核へ移行し、遺伝子転写に影響を与える。ARは、男性の二次性徴の発達及び調整において役割を果たす核内受容体スーパーファミリーのリガンド誘致性のホルモン受容体であり:これらは、精子形成の誘発、および骨及び筋肉量、およびアンドロゲン感受性組織の維持を含む(例えば、前立腺;Heinlein and Chang, 2004; Gelman, 2002)。
内因性のホルモン(例えば、テストステロン)の作用を遮断する薬剤(抗アンドロゲン)は、前立腺癌の処置に高度に有効であり、日常的に使用される(アンドロゲン除去療法)。第1非ステロイド性の抗アンドロゲン、フルタミドは、1989年に前立腺癌療法のために認可された(Shahinian et al., 2006)。続いて、他の非ステロイド性のリガンドが開発され、その主な理由は、化合物のこの種類が、ARのみと反応するからである。最近、アンドロゲン作用を阻害する、より特異的な、非ステロイド性のARモジュレーターが開発され、市場に出されている[例えば、Casodex(登録商標)(ビカルタミド); Bohl et al., 2005]。
【0236】
1.1.2.3 アンドロゲン依存性前立腺癌のための利用可能な治療
他の治療は、精巣からの内因性のテストステロンの合成および分泌を減少させるために開発された。アンドロゲンの循環するレベルの低下は、Lupron(登録商標)(ロイプロリド)などの、GnRH受容体アゴニストの投与を介して達成され、それは、1997年に現在のデポー剤のバージョン(depot version)に対する食品医薬品局(FDA)の認可を得た。継続的に与えられた時、Lupron(登録商標)は、脳下垂体前葉から黄体形成ホルモン(LH)の合成及び放出を減少させる。正常な状況下において、LHは、全身循環において精巣に移動し、そこで、テストステロンの合成及び放出を刺激する。GnRHによって誘発された脳下垂体のLHのダウンレギュレーションの結果として、テストステロンの循環するレベルは低下する(Catalona, 1994)。テストステロンの循環するレベルの低下により、ほとんどのアンドロゲン依存性前立腺癌細胞は、細胞分裂を止め、死滅する。
【0237】
GnRH受容体アゴニスト及び抗アンドロゲンの組み合わせは、精巣からのアンドロゲンの放出を減少させるためだけでなく、前立腺などのアンドロゲン感受性組織において、相当する受容体に結合する、残りの低レベルのアンドロゲンの能力を遮断するためにも使用される。
【0238】
1.1.2.4 去勢抵抗性前立腺癌;増加したアンドロゲン受容体及びアンドロゲン
最初は腫瘍成長を抑えるのに有効であるが、これらのアンドロゲン除去療法は、結局、ほとんどすべての被験体において失敗し、CRPCにつながる(Mohler et al., 2004)。前述したように、すべてではないが、ほとんどの前立腺癌細胞は、最初に、アンドロゲン除去療法に反応する。しかしながら、時間とともに、前立腺癌細胞の新しい個体群は、アンドロゲン除去療法によって生じた選択圧に反応したが、それに対して反応しなくなると出現する。原発性癌が、利用可能な療法に反応がないだけではなく、癌細胞はまた、原発腫瘍から離れ、血流を移動し得、疾患が離れた部位(特に骨)に広がる。他の効果の中で、これは、著しい疼痛及び極端な骨脆弱性を引き起こす(Stoch et al., 2001; Greenspan et al., 2005; Ye et al., 2007)。GnRH療法は、循環するテストステロンの濃度において、90〜95%より大きな減少を引き起こし得、前立腺細胞内のステロイドホルモンの濃度は、50%だけ減少する。これは、アンドロゲン感受性腫瘍の刺激を継続するために、十分な量のアンドロゲンを未だ提供する(Mizokami et al., 2004; Mohler et al., 2004; Titus et al., 2005; Page et al., 2006; Mostaghel et al., 2007)。
【0239】
1.1.3 去勢抵抗性前立腺癌におけるアンドロゲン受容体の増加
以前に、治療によって誘発されるような、テストステロンの去勢レベルの存在下における前立腺癌細胞の増殖が、ホルモン非依存性前立腺癌細胞型の出現によるものであると仮定された。しかしながら、ここでは、CRPC細胞が、細胞内のAR密度を増加させることにより、アンドロゲン除去療法によって誘発された低レベルの循環する睾丸アンドロゲンを補うと考えられる。最初の治療から生存し、去勢抵抗性になった前立腺腫瘍組織において、ARの数が増加されることが最近示された(Chen et al., 2004; Litvinov et al., 2003; Scher et al., 2004; Heinlein and Chang, 2004)。
【0240】
このように、前立腺癌細胞は、かなり低いレベルの循環するテストステロンに反応することができ、循環するアンドロゲンの量が低減したにもかかわらず、十分なアンドロゲンの援助を受け続ける。
【0241】
1.1.4 腫瘍内アンドロゲンにおける評価:副腎及び腫瘍内の源(Intratumoral Source)
GnRHアンタゴニスト及びアンドロゲンアンタゴニストで処置された男性における、低いがまだ測定可能な量の循環するアンドロゲンの1つの源は、副腎である(Mizokami et al., 2004; Labrie et al., 2005)。副腎性アンドロゲン産生は、睾丸のテストステロン産生を低減するほとんどの治療による影響を受けない。前立腺癌細胞が、循環する副腎性アンドロゲンを効率的に移入し、処理するその能力を向上させることにより、細胞内の副腎性アンドロゲン含量を増加させることを、いくつかの研究が示した(Bubley and Balk 1996; Mizokami et al., 2004; Labrie et al., 2005; Stanbrough et al., 2006)。
【0242】
副腎性アンドロゲン生合成の適度の阻害は、CRPCの処置に多少有効であると示された抗真菌剤である、ケトコナゾールの投与によって達成された。しかしながら、ケトコナゾールは、前立腺癌の処置における使用のために認可されていない。本発明は、精巣及び副腎の組織においてアンドロゲン合成を阻害する、強力で特異的な化合物が、前立腺癌のこの段階の処置により有効であり得るという認識を包含する。
【0243】
副腎性アンドロゲンの移入に加えて、アンドロゲン除去療法への長期曝露の後に生存する前立腺癌細胞はまた、アンドロゲンの前立腺内の合成に必要な、アップレギュレートされたレベルの酵素を有する(Nakamura et al., 2005; Stigliano et al., 2007; Holzbeierlein et al., 2004)。イントラクラインの仮説(intracrine hypothesis)は、中断されないアンドロゲンの援助を保証するために、CRPC細胞によって利用された生存メカニズムとして示唆された(Loberg 2005; Mostaghel et al., 2007)。この概念は、前立腺癌を研究する科学者および臨床医のコミュニティー内の有意な援助を獲得した。結果として、この概念は、すべてのステロイド産生組織(精巣、副腎及び、CRPCの場合には、前立腺癌細胞自体)においてアンドロゲン合成を阻害する、治療用化合物の増加する需要を刺激した。
【0244】
利用可能なままである細胞内のアンドロゲンに対する反応を強めるための、3つの異なる経路を増幅させることによる、低レベルの循環するアンドロゲンによって特徴付けられた環境において、CRPC細胞は生存する。これらは:
・ARコピー数を増加させ、その結果、内科的去勢療法によって誘発された低レベルの循環するアンドロゲンに対する、細胞の感受性を増加させる、ARの発現のアップレギュレーション、
・アンドロゲン遮断療法の後の循環において残る、副腎性アンドロゲンの移入及び処理に関係する酵素の発現の増加、
・ステロイド産生を規制する遺伝子の発現の増加を含み、それによって、CRPC細胞がその細胞自体のアンドロゲンを合成することが可能となる。
【0245】
CRPCを有する被験体に生じたこれらの変化に取り組むために、新しい治療が必要とされる。
【0246】
1.1.4.1 去勢抵抗性前立腺癌の処置としての化合物1
・CRPC細胞の生存メカニズムの新たな理解を考えると、新しい治療が必要とされる。化合物1は、以下に示されるような必要な特性をすべて示す:
・アンドロゲン生合成を減少させる:化合物1は、副腎、精巣、及び前立腺におけるアンドロゲン産生を制御する酵素である、チトクロームC17α-ヒドロキシラーゼ/C17,20-リアーゼ(CYP17)の活性を阻害する。
・ARシグナル伝達を減少させる:化合物1はARに結合し、テストステロン結合の競合的インヒビターである。
・アンドロゲン感受性を減少させる:化合物1は、前立腺癌細胞内のARタンパク質の含量を減少させ、低レベルのアンドロゲン増殖シグナルによって持続されるべき細胞の能力を低下させる。
【0247】
本発明は、前立腺癌、及び特にCRPCの処置における化合物1の使用のための治療方法及び組成物を提供する。
【0248】
1.2 リスク・ベネフィット評価
これは、化合物1のヒト初回投与試験であり、ヒトにおける潜在性の毒性効果は、まだ完全には知られていない。化合物1が、CRPCの処置における新しい潜在的に活性な薬剤であることと一致する、生物学的な特性を有していることを、非臨床データの評価は示す。データは提案されたMOAを支持し、細胞に基づいたモデル及び動物モデルにおいて最小の毒性を示す。見られるただ1つのシグナル、肝毒性は、抗アンドロゲンによって潜在的に誘発され得る既知の有害反応である(Thole et al., 2004)。更なる既知のクラスの影響、明らかなミネラルコルチコイド過剰症(AME)は、非臨床試験において見られなかった。この試験を、有害な非臨床的結果、理論的なMOA、及び既知のクラスの影響問題の考察とともに考案した。試験は、潜在的な臨床的利点を評価し、概念実証を提供する一方で、被験体に対するリスクを最小化する。
【0249】
本発明は、生物学的活性に関する安全性および潜在性の両方を考慮する投薬スキームを特に提供する。1つの実施形態において、650mgの初回量を利用する。この投与量は、安全シグナルが見られた、イヌにおける投与量よりも18倍少なく、ステロイド産生組織において発現されたリアーゼ酵素の阻害、ARへのアンドロゲンステロイドの結合の阻害、及びアンドロゲン反応性組織におけるARタンパク質の不安定化において、潜在的に生物学上有効であると予期される。さらに、140、280、及び560mgのヒトの皮下投与に相当するレベルで、重症複合免疫不全症(SCID)のマウスモデルにおけるアンドロゲン依存性のヒト前立腺腫瘍のサイズにおいて、劇的な減少が示された。
【0250】
治験計画
本実施例は、化学療法未治療のCRPCの処置に関する化合物1の、相1及び2、非盲検、用量増加、用量比較の試験を記載する。試験の個体群は、確認された前立腺の腺癌、及び最も最近のPSAレベルが≧5ng/mLである、少なくとも1週間離されて、少なくとも2つの連続した機会において上昇する、前立腺特異性抗原(PSA)レベルとして定義される、アンドロゲン除去療法にもかかわらず進行する疾患を有する、18歳以上の男性を含む。
【0251】
この試験は2つの段階に分けられる:相1(用量増加)、その後の相2(選択された用量比較);試験の相1及び/又は相2の段階の終了の後に、資格のある被験体に対する随意の延長の相がある。
【0252】
最初の処置訪問の28日間以内にスクリーニングが行われる。被験体は、夕食とともに、臨床試験薬(化合物1)を12週間毎日1回とる。試験の訪問は、2週ごとに行われ、被験体は、12週間の投薬期間の終わりに、試験終結の訪問のために戻る。その後、資格のある被験体は、継続的な治療のために延長のアームに入ることを許可され得る。処置は、疾患の進行、離脱、被験体の容認できない毒性が出るまで、又は研究員の判断で継続し得る。
【0253】
個々の被験体に関して、試験の相1又は2の段階の最大期間は、[スクリーニングに28日間まで及び89日間までの処置(相1又は2)を含む]117日間までである。処置ウィンドウ計算(treatment window calculation)は、試験終結の訪問(+5日)のための最大ウィンドウを含む。更なる延長投薬は、資格のある被験体に提供され得る。
【0254】
これらの相の各詳細を、下記に記載し、図11において図表で提示する。すべての相1のデータのデータの検討を、相2に入る被験体のための投与量を決定するために行う。相2において最後の被験体が12週目に達した後に、すべての試験データのデータの検討を、今後の試験(及び継続される延長)のための投与量を決定するために行う。
【0255】
1.3 相1の考案及び計画:記載
相1の段階の目的は、許容可能な安全性プロフィールを提供する化合物1の投与量(複数可)を設定することである。許容可能な安全性プロフィールを、≦35%の正確なDLT比率での投与量として定義する。
これは、3つの投与量群を、1の投与量当たり6まで(3以上)の被験体の標的とともに登録する、用量増加スキームである。調査されるべき標的とされる投与量群は、650mgの化合物1;1300mgの化合物1;及び1950mgの化合物1である。
【0256】
内部モニタリング委員会(IMC)によって完了される検討に続いて、これらの投与量群は、より高い又はより低い投与量群、又は中間の投与量までの用量増加又は用量減少によって修正され得る。これらの修正は、調査されている投与量の範囲内にのみ生じ得る。
【0257】
【表5】
【0258】
被験体の群を、650mgの化合物1から開始して、増加する投与量で処置する。次に続く投与量群は、登録(新しい被験体)のために空けられ得、最初の3の被験体が、より低い投与量群に集まった後の、増加のための基準を提供する。≦35%の正確なDLT比率は、許容可能であると考えられる。用量設定規則はこれに基づき、実験の相1の段階の目標は、許容可能な正確なDLT比率を有する最も高い用量を識別することである。DLTは、任意の臨床試験薬に関連するグレード3又はより高い有害事象(AE)として定義され、臨床試験薬と、ひょっとしたら、おそらく、又は明確に関連があると考えられる。
【0259】
臨床試験薬を少なくとも1回用量受け取らない、相1の段階において登録された被験体を、適切な投与量群における更なる被験体と交換する。同様に、登録された被験体が資格がないと発見されるとき、そのような被験体を、適切な投与量群における更なる被験体と交換する。
【0260】
1.3.1 最初の3の被験体の後の増加に対する基準
連続的な投薬のリアルタイムデータが電子症例報告書(eCRF)によって収集される、最小の4週間に一旦すべての3の被験体が達すると、各投与量群における最初の3の被験体のDLTのための検討を行い、検討は、すべての利用可能なデータを含む。データは、この検討に含まれるべきと確認される源(source)である必要はない。DLTは、任意の臨床試験薬に関連するグレード3又はより高いAEとして定義され、臨床試験薬と、ひょっとしたら、おそらく、又は明確に関連があると考えられる。
【0261】
最初の3の被験体のデータの検討の後、もし:
・0/3の被験体がDLTを経験すると、次の投与量群までの増加が生じ得る。
・1/3の被験体がDLTを経験すると、3の更なる被験体は、次の投与量群までの増加前のこの投与量で登録されるべきである。
・2/3又は3/3の被験体がDLTを経験すると、投薬はこの投与量群で停止され、相1の投与量減少が生じる。
【0262】
DLTの検討期間内では、臨床試験薬にひょっとしたら、おそらく、又は明確に関連があると考えられる、任意のグレード2の肝毒性に関連する事象(HTRE)は、最大50%まで次の投与量群に対する増加を制限し得る。
【0263】
1.3.2 最大限の6の被験体への拡張が必要とされるときの増加の基準
3の更なる被験体を増加の前に投与量群に加えなければならないとき、一旦更なる3の被験体が最小の4週の連続的な投薬に達すると、すべての6の被験体におけるDLTのための検討が行われる。
【0264】
拡張の後、もし:
・2/6の被験体がDLTを経験すると、次の投与量群までの増加が生じ得る。
・3/6の被験体がDLTを経験すると、投薬はこの投与量群で停止し、相1の投与量減少が生じる。
【0265】
1.3.3 相1の投与量減少
減少が必要なとき、被験体は、1)次のより低い投与量群まで、又は2)実現可能であれば、DLTが生じた投与量と次のより低い投与量群の間の新しい中間の投与量まで減少される。例えば、3以上の被験体が1300mgの群においてDLTを有すると、1300mgと650mgの間の投与量が被験体に再び与えられ得る。開始する投与量群(650mg)からの減少が必要であると、3の被験体の更なる群は、325mgで登録され得る。
【0266】
減少が生じると、減少が生じてから投与量へ戻る増加が、両方の投与量で登録された被験体の検討に続いて、考慮され得る。
【0267】
1.3.4 相1のデータの検討
相1のデータの検討は、相2の開始前に完了する。安全性及び生物学的シグナルを支持する12週間までのデータを、相2のための投与量を選択するために利用する。相1において検討された及び許容可能なDLT特性を有すると測定された投与量のみ、又は許容可能なDLT特性を有するものより低い新しい投与量は、相2の試験のために考慮される。
【0268】
3.1.5 投与量遅延/投与量修正
被験体を毒性に関して監視し、投与量は、個々の被験体耐性に従って調節され得る。
【0269】
1の投与量レベルによる用量減少は、遭遇する毒性のタイプ及び重症度によって必要とされ得る。最小用量は一日325mgである。用量減少ガイダンスは表3に提供される。
【0270】
最小量の処置に関連する効果を有する4週間の処置を終えた被験体は、表2に記載されるように、1プロトコル当たりの被験体の割り当てられたレベルで投薬を継続し得る。
【0271】
【表6】
【0272】
注:AMEの徴候を示す被験体は、推奨されるAME処置に反応する限り、妨害なしで研究処置を継続し得る。被験体がAMEを有すると疑われると、1のラベル当たりの、エプレレノンでの処置、及び/又は電解質補充が推奨される。医学的に実現可能なとき、AMEに関する任意の他の処置は、トーカイメディカルアドバイザー(又は指名された人)及びメディカルモニターと前もって話し合われるべきである。
【0273】
現在の投与量が325mgであり、更なる用量減少が必要であると、毒性ガイドラインが示すと、被験体は研究から外される。>4週間の投与量中断を必要とする被験体は、研究から外されることが考慮される。
【0274】
1.4 相2の考案及び計画:記載
相2の目的は、生物学的なシグナルを評価し、もたらされる各投与量の許容可能な安全性プロフィールを確認することである。正確なDLT比率が35%を超過することを示唆する、合理的に説得力のある証拠が存在するならば、特定の投与量への登録が停止されるように、停止規則は実施される。
【0275】
新しい20の被験体を、相1から進められた各アームに登録する。2のアームが進められると、被験体は2のアームに無作為化される(最大で40の被験体)。このサンプルのサイズの論理的根拠に関しては、この概要の統計的方法のセクションを参照する。
【0276】
相1に関して、DLTは、任意の臨床試験薬に関連するグレード3又はより高いAEとして定義され、臨床試験薬と、ひょっとしたら、おそらく、又は明確に関連があると考えられる。
【0277】
相1に関して、臨床試験薬を少なくとも1回用量受け取る被験体のみが相2にあると考えられる。臨床試験薬を少なくとも1回用量受け取らない被験体の比率、及び臨床試験薬が投与されなかった理由を記録する。これは非常に少ない頻度で生じ、その理由は通常管理上のものである。
【0278】
1.4.1 相2の投与量選択
2までの投与量を、相2における研究のために選択する。相1のデータの検討を、投与量(複数可)を測定するための基礎として利用する。投与量(複数可)の選択を、相1からの安全性と生物学的作用のデータのIMC検討に続いて決定する。
【0279】
単一用量の相2における診査につながる可能なシナリオは、以下を含む:
・観察されたDLT比率が相1における投与量で35%の閾値を越えると、これが生じたことよりも低い投与量群が単独で診査され得る。
・相1におけるすべての投与量が許容可能なDLT特性を示すと、相1における最大投与量、1950mgは、単独で診査され得る。
【0280】
相2における2の投与量の診査につながる可能なシナリオは、以下を含む:
・観察されたDLT比率が相1において35%の閾値を超過すると、これが生じたことよりも低い投与量群、及びより低い投与量が選択され得る。より低い投与量は、次のより低い投与量、又は35%のDLT比率が超過されたことよりも低い投与量と次のより低い投与量の間の中間の投与量であり得る。
・相1におけるすべての投与量が、<35%のDLT比率を示すと、相1における最大投与量、1950mg、及び第2投与量が選択され得る。第2投与量は、1300mgの投与量又は1950mgと1300mgの間の中間の投与量であり得る。
・相1において診査される1より多い投与量が、<35%のDLT比率を示すと、高用量/低用量のシナリオが考慮され得る。
【0281】
1.4.2 相2の停止規則
相2のデータを、DLTのために継続的に検討する。データは、この検討に含まれるべきと確認される源である必要はない。
【0282】
関連するDLTの発生率が35%を超過すると、その後、IMCはデータを検討し、投与量に対する修正を行う必要があるかを決定する。
【0283】
1.4.3 相2の投与量修正
投与量が相2において停止されると、相2の停止規則により、被験体の投薬は、データの検討に続いて以下のように修正される。
・2の投与量が相2において実行されると、20の被験体が登録されるまで、停止されなかったアームにおいて登録は継続する。停止されたアームが2の投与量群におけるよりも高いと、IMCは前進するためにどの投与量が使用されるべきかを推奨するように要求される。停止された試験のアームにおける被験体は、継続的な投与量で処置され、12週のフォローアップスケジュールを完了する。
・1の投与量が相2において実行されると、続く被験体は、1)相1において許容可能なDLT特性を有すると見られる、次のより低い投与量群において、又は、2)実現可能であると、相1において許容可能なDLT特性を有すると見られる、DLTが生じた投与量と次のより低い投与量の間の新しい中間の投与量まで投薬される(又はそれらに換えられる)べきである。20の被験体がより低い投与量で登録されるまで、投薬のこの減少は起こる。停止規則がこのより低い投与量で適用されるとき、これ以上の用量減少は行われない。
【0284】
1.5 延長の考案及び計画:記載
延長される投薬は、試験の相1又は相2の段階における12週が終了した後に、被験体に提案され得る。最初の資格を、許容可能なリスク・ベネフィット比率、及び研究における10週目にわたる生化学的な進行の徴候の欠如に基づいて、12週目の訪問で決定する。継続される資格を、継続される許容可能なリスク・ベネフィット比率、及び生化学的な進行の徴候の欠如に基づいて、延長の全体にわたって評価する。
【0285】
許容可能なリスク・ベネフィット比率を、メディカルモニター及び/又はメディカルアドバイザーによる確認とともに、主要な研究員(PI)によって決定する。
【0286】
生化学的な進行を、前立腺癌ワーキンググループ2(Prostate Cancer Working Group 2)(Scher et al., 2008)により、a)PSA増加≧25%、及び(基準線からの低下が生じるならば)最下点を超える≧2ng/mL、又は、b)PSA増加≧25%、及び(基準線からの低下が生じないならば)基準線からの≧2ng/mLとして定義する。
【0287】
延長のための投与量は以下のとおりである:
・相1の間:割り当てられた相1の投与量で延長が生じる。
・相2の間:相2において診査される投与量での延長である。相2において使用されない投与量で最初に投薬される相1の被験体に、相2の投与量を再び与える。
・相2の終了後:相1及び2からのデータの検討に続いて、投与量(複数可)を決定する。
【0288】
延長の間に、基準線から12週までのPSA、又はPSA最下点(最初に生ずるどちらか)における50%又はそれ以上の減少を有する被験体のパーセントによって示されるような、[AE、身体検査、生命徴候、12誘導心電図(ECG)、臨床検査室評価のIMCによる定期的な検討による]安全性、処置コンプライアンス、及び有効性、コンピューター断層撮影(CT)/磁気共鳴画像法(MRI)及び骨スキャンの変化、固形腫瘍における反応評価基準(RECIST; v. 1.1)による進行、特殊実験室の変化、及び進行までの時間(TTP)、無進行生存(PFS)、及び全存率(OS)に関して、被験体の試験が行われる。
【0289】
処置は、疾患の進行、被験体の離脱、容認できない毒性が出るまで、又は研究員の判断で継続し得る。
【0290】
3.1.5 内部モニタリング委員会
外部データの安全性を監視する委員会は、この試験のために設立されない。研究員、メディカルモニター、及びメディカルアドバイザーを含む、形式的に認可を得た組織内のIMCが設立される。このプロトコルの目的のために、組織内のIMCは、重篤有害事象(SAE)において収集された簡略なデータを検討し、各投与量群における最後の被験体が登録した後、及び相1において新しい投与量レベルを始める前の各4週の期間の終わりに、及び相2において必要とされるような、臨床的なデータベースを最小限に検討する。IMCは、プロトコル内で提供されるガイダンスに従うが、自らの判断で、報告されるDLT、更に拡張される投与量群、又は投薬の計画を再評価する権利を保持する。会議の結果は、必要に応じてプロジェクトファイルに文書化され、行動がとられる。試験における被験体の管理に関する直接の意味合いを有する結果は、予期されない及び薬物に関連するSAEと関係する時間枠におけるすべてのPIに伝えられる。
【0291】
1.6 エンドポイント
1.6.1 相1のエンドポイント
1.6.1.1 一次エンドポイント
・AEの発生率
・以下の更なる安全パラメーターの基準線からの変化:臨床検査室評価、身体検査、生命徴候、及び12誘導ECG。
【0292】
1.6.1.2 二次エンドポイント
・基準線から12週までのPSA、又はPSA最下点(最初に生ずるどちらか)における50%又はそれ以上の減少を有する被験体のパーセント。
・CT/MRI及び骨スキャンにおける基準線からの変化。
・RECIST基準による進行。
・更なる特殊実験室における基準線からの変化。
・腫瘍内のARタンパク質、テストステロン、及びジヒドロテストステロン(DHT)(生検サブセット中)の基準線からの変化。
【0293】
1.6.2 相2のエンドポイント
1.6.2.1 一次エンドポイント
・基準線から12週までのPSA、又はPSA最下点(最初に生ずるどちらか)における50%又はそれ以上の減少を有する被験体のパーセント。
【0294】
1.6.2.2 二次エンドポイント
・AEの発生率
・以下の更なる安全パラメーターの基準線からの変化:臨床検査室評価、身体検査、生命徴候、及び12誘導ECG。
・CT/MRI及び骨スキャンにおける基準線からの変化。
・RECIST基準による進行。
・更なる特殊実験室における基準線からの変化。
・腫瘍内のARタンパク質、テストステロン、及びDHT(生検サブセット中)の基準線からの変化。
【0295】
1.6.3 延長に特有のエンドポイント
延長のエンドポイントは相2に関し、更なる以下の二次エンドポイントを有する:進行までの時間(TTP)、無進行生存(PF)、及び全存率(OS)。図12(表5)は、研究の相において使用される手順のスケジュールを記載する。以下の注釈は、図12に相当する。
ET:期限前契約解除;ICF:インフォームドコンセント用紙;ECG:心電図;CT:コンピューター断層撮影;MRI:磁気共鳴画像法;PCF:前立腺癌財団;PSA:前立腺特異性抗原;CBC:完全血球計算;PSA:前立腺特異性抗原;AME:明らかなミネラルコルチコイド過剰症;PK:薬物動態学。
1:被験体は、相1及び2の間に毎週、CBC及び化学検査が行われることが必要とされる。臨床現場での実験室においてこれを行うほど、十分近くない被験体に関して、地方の実験室が、これら検査のない週に関して利用され得る
2:これが期限前契約解除の訪問であると、研究員は、試験終結の訪問のために記載される安全性評価を行なうために、あらゆる努力をしなければならない。
3:1日目は、(10:00と14:00の間の食事とともに摂られる)臨床試験薬の最初の投与である。
4:12誘導ECGは、投与の0時間(投与前(predose))及び4時間(±15分)後に行われる(PKサンプル前)。
5:基準線に関して、使用されるCT/MRI及び骨スキャンの結果は[試験の包含の目的のために繰り返されているスキャンを回避するために]、1日目から4週間以内の日付でなければならず、記録される4週のウィンドウ以外の次に空けられた投与(open dose)に対する包含のためのスクリーニングのCT/MRI又は骨スキャンの使用に関して、トーカイメディカルアドバイザー(又は指名された人)と相談して、メディカルモニターによって例外が認められ得る;繰り返すために、相1又は2の終結の訪問又はPSA最下点(最初に生ずるどちらか)でスキャンが行われるべきである。延長投薬の間に、CT/MRI及び骨スキャンが12週ごとに行われる。もし基準線で使用される同じ様式が、試験においてすべての後の評価のために継続的に利用されるならば、CTスキャンの代わりのMRIの使用は許容可能である。万一、予期しない事象が生じ、処置する医師の判断において、任意の他のスキャンの形態を有するのが被験体の利益であるならば、これは、メディカルモニター及びメディカルアドバイザーと話し合われるべきである。
6:アンドロゲン受容体タンパク質、テストステロン、及びジヒドロテストステロンの評価のための生検は、ICFを作成する別々の機関の下で被験体のサブセット(PCFの許可)において提案される。相1又は2の終結の訪問又はPSA最下点(最初に生ずるどちらか)で生検が繰り返されるべきである。
7:被験体がAMEを有すると疑われると、1のラベル当たりの、エプレレノンでの処置、及び/又は電解質補充が推奨される。医学的に実現可能なとき、AMEに対する任意の他の処置は、トーカイの医薬情報提供者及びメディカルモニターと前もって話し合われるべきである。
8:安全性実験室評価が、1日目の7日より前に記録されると、それらの評価は、投薬前に繰り返され、確認されるべきである。
9:訪問が1週間ないときのみの安全性検査(検尿はない)。
10:投与前に行われる試験
11:11-デオキシコルチゾール、コルチコステロン、コルチゾール、及びテストステロンだけが実行される。以下の試験は実行されない:プレグネノロン、17α-ヒドロキシプロゲステロン、デヒドロエピアンドロステロン、デオキシコルチコステロン、及びアンドロステンジオン。
12:すべての相1及び2の被験体は、1日目に0時間(投与前)及び投与(10:00と14:00の間となる投薬)の4時間(±15分)後にPK抽出(draws)を受ける。残りの訪問で:単一のPKサンプルは、(被験体の日記から計算されるように)最後の投与からの時間の記録によって抽出される。
13:延長へと継続する場合。
14:報告はスクリーニングで始まる。
【0296】
【表7】
【0297】
CBC:完全血球計算;EDTA:エチレンジアミン四酢酸;K2EDTA:エチレンジアミン四酢酸ジカリウム。
1: 中央検査室で実行される。
2: 薬物動態学的サンプルを、提供されるK2EDTA管に収集し、すぐに氷上に置き、血漿分離のために遠心分離にかける。サンプルを、血漿分離の8時間以内に冷凍する。
3: 前立腺癌財団(PCF)の許可により完了した生検を受ける被験体のサブセットにおいてのみ行う。
【0298】
2. 被験体の選択及び離脱
試験個体群は、CRPCを有する被験体から成る。この試験のための資格を有するために、被験体は、書面による同意を提供でき、すべての包含基準を満たし、排除基準は1つも満たさない。
【0299】
2.1 被験体の包含基準
被験体は、試験のための資格を有するために、登録前に以下の包含基準のすべてを満たさなければならない:
1. 投薬スケジュールを厳守すること、すべての試験の訪問及び承認を報告すること、使用すること、及び健康及び研究試験の情報を発表することへの同意を提供する、インフォームドコンセント用紙(ICF)への署名。
2. 18歳以上の男性。
3. 前立腺の組織学的に又は細胞学的に確認された腺癌(神経内分泌分化又は小細胞の組織学的検査を除く)。
4. 最も最近のPSAレベルが≧5ng/mLである、少なくとも1週間離されて、少なくとも2つの連続した機会において上昇する、PSAレベルとして定義される、アンドロゲン除去療法にもかかわらず疾患が進行していること。
5. GnRHアナログ又は精巣摘除を伴う進行中の生殖腺のアンドロゲン遮断療法。精巣摘除を伴わなかった被験体は、有効なGnRHアナログ治療を維持されなければならない。
6. 米国東海岸癌臨床試験グループ(ECOG)の活動指標0又は1。
7. >12週の寿命。
8. 複数のカプセル剤を呑み込むことができる。
【0300】
2.2 被験体の排除基準
登録前に以下の排除基準のいずれかを満たす被験体は、試験に参加する資格を有さない:
1. 登録の<4週間前に実験的治療を伴う別の臨床試験への参加。
2. 下記の1つ以上を有する転移性の被験体:1) 肝障害;2) 積極的な疼痛管理を必要とする転移の確認された放射線学的証拠に関係する骨痛
3. 以下の薬物治療:1) MDV3100、アビラテロン、又はTAK-700での以前の処置;2) ケトコナゾールでの以前の処置;3) 化学療法での以前の処置;4) 登録の≦4週間前に終了する以前の放射線療法;5) 登録の≦4週間前にPSAレベルを低下させると知られる他の治療での処置[Megace(登録商標)(酢酸メゲストロール)、Proscar(登録商標)(フィナステリド)、Propecia(登録商標)(フィナステリド)、Avodart(登録商標)(デュタステライド)、Eulexin(登録商標)(フルタミド)、Casodex(登録商標)(ビカルタミド)、Nilandron(登録商標)(ニルタミド)、Aldactone(登録商標)(スピロノラクトン)、Cytadren(登録商標)(アミノグルテチミド)、エストロゲン、PSAレベルを低下させると知られる任意のハーブ製品(例えば、ノコギリヤシなど)、又は(チーフメディカルオフィサー(Chief Medical Officer)及びトーカイメディカルアドバイザーの判断での)任意の全身性のコルチコステロイドの任意の投与を含む]6) 登録の≦4週間前の心室性不整脈に対する抗不整脈治療での処置;7) 登録の≦4週間前のCoumadin(登録商標)(ワルファリンナトリウム)治療での処置;8) 登録の≦1週間前のLasix(登録商標)(フロセミド)又はZaroxolyn(登録商標)(メトラゾン)での処置;9) 登録の≦1週間前のスタチンでの処置[注:Zetia(商標)(エゼチミベ)は除外されない];又は、10) 登録の≦4週間前の三環系抗鬱薬(TCA)での処置。
4. 以下の検査所見:1) テストステロン>50ng/dL;2) 血清クレアチニン>1.5x 正常上限(ULN);3) ビリルビン>1.5x ULN(注:ジルベール症候群による高められたビリルビンを有する被験体は除外されない);4) アスパラギン酸アミノトランスフェラーゼ(AST)及びアラニンアミノトランスフェラーゼ(ALT)>1.5x ULN;5) ヘモグロビン≦9.0g/dL;6) 絶対好中球数(ANC)≦1.5x 109/L;7) 血小板≦100x 109/L;8) 血清K+<3.5mmol/L。
5. 以下の病状:1) ニューヨーク心臓協会クラスIII又はIVのうっ血性心不全;2) 心筋梗塞(登録の6か月前以内);3) 活発な狭心症;4) B型肝炎又はC型肝炎の病歴;5) 公知のヒト免疫不全ウィルス(HIV)感染症;6) 高血圧(少なくとも2回の機会で測定される、収縮期血圧>150mmHg、又は>95mmHgの拡張期血圧として定義される);7) 副腎機能不全又は高アルドステロン症の病歴;8) AMEの病歴又はブラックリコリス過敏性の病歴;9) 胃腸;10) 化合物1の吸収を妨害し得る障害又は胃バイパス手術;11) 全身療法を必要とする重大な活動性感染又は制御されない非悪性の医学的疾患;12) 処置された非黒色腫皮膚癌以外の二次性悪性腫瘍の(過去5年間の)あらゆる病歴;13) 活動性又は抑制されない自己免疫性疾患;又は、14)活動性の胆道障害。
6. 研究員の意見において、試験の手順に従う被験体の能力を妨害し得るあらゆる物理的又は精神的疾病、又は社会的状況。
7. 妊娠の可能性がある女性との性的接触に関係する場合に、避妊の適切な方法を使用しようとしない男性。
【0301】
2.3 被験体の離脱基準
被験体は、罰則なしでいつでも、及び今後の医療を失うことなく任意の理由で試験から離脱し得る。
【0302】
被験体は、以下の状況の下で離脱されなければならない:
・被験体は同意を撤回する。
【0303】
さらに、被験体は、以下の理由で離脱することが必要とされ得る:
・有害事象(複数可)(AE)
・臨床的、放射線学的、及び腫瘍マーカー(PSA)の進行によって評価されるような疾患の進行
・研究員の見解において、被験体が、プロトコルの必要条件を満たすことができるとこれ以上考えられない場合
・資格基準の違反
・プロトコルに明記される治療計画からの逸脱(例えば、臨床試験薬の正しくない投与、試験訪問を行わない)。
【0304】
すべての場合において、離脱の主な理由は、eCRFに記録されなければならない。被験体が任意の理由で早期に離脱すると(12週目の訪問前)、研究員は、試験終結の訪問(12週目の訪問)のために記載される安全性評価を行なうために、あらゆる努力をしなければならない。
【0305】
2.4 試験の早期終結
もし試験が継続するときに被験体に対して起こり得る危険を示唆する条件又は事象に、研究員又はメディカルモニターが気付くと、関係者間の適切な相談の後に試験が終結し得る。
【0306】
終結を保証し得る条件は、限定されないが、試験に登録される被験体に対する予期されない、重大な、又は許容できないリスクの発見、又は許容可能な割合で被験体を登録できないことを含む。
【0307】
3. 被験体の処置
3.1 臨床試験薬の記載
相1に対する臨床試験薬は、3つの投与量群:(1) 650mgの化合物1(2つの325mgのカプセル剤);(2) 1300mgの化合物1(4つの325mgのカプセル剤);(3) 1950mgの化合物1(6つの325mgのカプセル剤)での化合物1から成る。更なる中間の投与量が診査され得る。
【0308】
相1において得られた結果に基づいて、相2に対する投与量(複数可)は決定されるが、相1において診査された投与量の1つ又は2つを含むと予期され得る。
【0309】
相1及び2において得られた結果に基づいて、延長に対する投与量は決定される。
【0310】
投薬の最初の日(1日目)に、被験体は(10:00と14:00の間の)食事とともに臨床試験薬を経口的に摂ることが必要とされる。被験体は、この訪問で、及び(8日目;訪問3に開始する)2週ごとの訪問で、臨床試験薬を供給され、2日目からの(17:00と21:00の間の)夕食とともに臨床試験薬を経口的に毎日1回摂るように指示される。被験体は、相1及び2において12週間、臨床試験薬を服用する。臨床試験薬に耐性があり、進行の徴候を示さない被験体は、試験の延長のアームにおいて継続される投薬に資格を有し得る。延長のアームにおける投薬の持続時間は、少なくとも12週間である。
【0311】
臨床試験薬は、登録時、及び最初の12週の間の2週ごと(訪問の2、4、6、8、10及び12週目)に調剤され、その後、(延長へと継続する場合)試験終結の訪問で開始して、4週ごとに調剤される。
【0312】
3.2 併用薬
被験体は、試験へのエントリー時に未承認の併用処置を受けるべきではないし、化合物1以外のCRPCに対する処置も試験の間に与えられるべきではない。精巣摘除を有さなかった被験体は、有効なGnRHアナログ治療を維持されなければならない。
【0313】
任意の併用処置の投与がPIによって必要であると考えられると、それは、eCRF及び被験体の医療記録において報告されなければならない。
【0314】
試験のエントリーによって、又は試験の間の任意の時間に被験体によって得られたすべての更なる処置は、併用薬と見なされ、eCRFに文書化されなければならない。以下の情報は、各併用薬のeCRFに記録されなければならない:属名、投与経路、開始日、停止日、投与量、及び指標。併用薬の投与量又はレジメンのあらゆる変化は、eCRFに記録されなければならない。
【0315】
スクリーニング訪問では、被験体は、過去30日間でどの薬剤を服用したかを尋ねられる。各々の続く試験の訪問では、被験体は、現在どの併用薬を服用しているかを尋ねられる。禁止される併用薬は以下を含む:Megace(登録商標)(酢酸メゲストロール)、Proscar(登録商標)(フィナステリド)、Propecia(登録商標)(フィナステリド)、Avodart(登録商標)(デュタステライド)、Eulexin(登録商標)(フルタミド)、Casodex(登録商標)(ビカルタミド)、Nilandron(登録商標)(ニルタミド)、Aldactone(登録商標)(スピロノラクトン)、Cytadren(登録商標)(アミノグルテチミド)、エストロゲン、PSAレベルを低下させると知られる任意のハーブ製品(例えば、ノコギリヤシ及びPC-SPESなど)、又は任意の全身性のコルチコステロイド、Coumadin(登録商標)(ワルファリンナトリウム)、Lasix(登録商標)(フロセミド)及び他のカリウム消耗性利尿薬、Zaroxolyn(登録商標)(メトラゾン)、スタチン[注:Zetia(商標)(エゼチミベ)は除外されない]、心室性不整脈に対する抗不整脈薬、及びTCA。
【0316】
非麻薬性及び麻薬性の鎮痛薬を含む、すべての他の処方及び非処方の薬剤は、必要に応じて使用され得る。すべての併用薬の使用は、各研究の訪問で口頭で評価され、eCRFに記録されるものとする。
【0317】
注:全身性のコルチコステロイドに関して:中程度の慢性閉塞性肺疾患(COPD)、制御された喘息、又はステロイドを必要とする他の慢性症状を有する被験体が、登録基準を満たすが、研究の間(例えば、COPD増悪の間など)に短時間でパルス化した中用量ステロイドを必要とすると、これは、試験に対して責任を負うメディカルモニターによる認可により許容可能になり得る。
【0318】
3.3 処置コンプライアンス
被験体は、各々の続く訪問で、任意の未使用の供給を返すように指示される。各訪問では、被験体は、処置コンプライアンスに関して質問され、返された丸剤は、被験体のコンプライアンス及び薬物責任を確認するために数えられ、文書化される。
【0319】
日記カードは、スクリーニング訪問で、及びその後2週ごとに分配される(延長の相の間は4週ごと);完成したカードは、試験を通して収集される。被験体は、日記カードに投薬の時間及び毎日の服用される丸剤の数を記録し、薬物責任及び処置コンプライアンスのモニタリングを可能にする。
【0320】
投薬コンプライアンスが80〜120%の間で維持されないと、被験体は試験から離脱され得る。被験体は、任意の服用し忘れた投与量のスケジュールを組み直さないように指示される。
【0321】
3.4 無作為化及び盲検化
無作為化は、相2における投与量群への割り当てに使用される。臨床試験薬の盲検化はない;割り当ては、被験体及び研究員に公知である。相2における被験体の無作為化に関して(1以上の投与量群が診査されると)、研究員は双方向ウェブ応答システム(IWRS)を使用する。IWRSは、あらかじめ定義された無作為化リストに基づいて、投与量群に被験体を割り当てる。更なる詳細は、試験マスターファイル(TMF)において見られ得る。
【0322】
4. 臨床試験薬の材料及び管理
4.1 臨床試験薬
4.1.1 定量的組成
化合物1は、17-ヘテロアリールで置換された半合成のステロイドである。A、B、及びC環は、デヒドロエピアンドロステロン(DHEA)及びプレグネノロンの環と構造上類似している。化合物1のベンズイミダゾールの機能性は、sp2炭素(Δ-16,17 オレフィン)を介してD環に加えられる。
【0323】
【化32】
【0324】
4.1.2 投与量選択
この試験のための投与量の安全な、及び潜在的に治療効果のある範囲を予想するために、以下の情報が考慮された:CYP17阻害;受容体の結合データ;標的組織におけるARタンパク質の濃度;腫瘍組織における蓄積;及び動物の安全性、有効性、薬物動態学的(PK)、及び毒素動態学的データ。
【0325】
非臨床データのすべての評価は、化合物1がCRPCを処置するのに必要とされるものと一致する生物学的特性を有していることを示す。例えば、この薬物は、前立腺癌の生化学的な、細胞に基づいた、及び動物のモデルにおいて、生物学的活性を有する。インビトロの安全性試験は、化合物1が遺伝毒性を有しておらず、多くの薬物を代謝すると知られるシトクロームP450システムの活性を妨害せず、及び高いミクロモル濃度でのみオフターゲットの受容体と相互に作用することを示した。化合物は、経口的に生物学的利用が可能であり、ラットに慢性的に投与される時に毒性反応を誘発しない。イヌにおける300mg/kg未満の投与量で、化合物1は毒性を有さないと示された。投与量がmg/kgの体重ベースで投与された時、薬物の同様の最大血中濃度は、ラット及びイヌのモデルにおいて達成された。
【0326】
イヌ(150mg/kg)における無有害作用量(NOAEL)で、化合物1への曝露[最大濃度(Cmax)及び曲線下面積(AUC)]は計算され、類似した曝露指標が、ヒトにおける開始投与量(70kgの男性に対して9mg/kg)で予想された。AUCに基づくと、ヒトの開始投与量は、イヌにおけるNOAELの投与量より16倍低くなると予想される。
【0327】
これらの研究の結果の調整は、650mg/日のヒト初回投与試験の投与量の選択を支持する。この投与量は、安全シグナルが見られた、イヌにおける投与量よりも18倍少なく、ステロイド産生組織において発現されたリアーゼ酵素の阻害、ARへのアンドロゲンステロイドの結合の阻害、及びアンドロゲン反応性組織におけるARタンパク質の不安定化において、潜在的に生物学上有効であると予期される。さらに、140、280、及び560mgのヒトの皮下投与に相当するレベルで、SCIDのマウスモデルにおけるアンドロゲン依存性のヒト前立腺腫瘍のサイズにおいて、劇的な減少が示された。
【0328】
4.2 臨床試験薬の供給
製剤は、微粉化された粉末の入ったカプセル剤である。製剤の組成物は、微粉化された化合物1及びサイズ「00」のCapsugel Coni-Snap(登録商標)カプセルから成る。幾つかの実施形態において、カプセル剤は、325mgの化合物を含む。
【0329】
4.3 臨床試験薬のパッケージング及びラベル付け
製剤は、950ccの白色不透明の高密度ポリエチレン(HDPE)タンパーの明白なホイルで密閉した丸いジャーにおいて、バルク包装され、各臨床現場の薬局に送られる。325mg、サイズ「00」のカプセル剤は、1本のボトル当たり400が包装される。臨床的使用に関して、臨床現場の薬局は、カプセル剤を再包装し、以下の情報によりラベルを付けられた、個々の被験体の使用のためのHDPEボトルへ分配する:被験体の識別;研究使用のみのための化合物1の新薬;製造日及び仕切番号;臨床現場;分類番号。
【0330】
4.4 臨床試験薬の保存
臨床試験薬は、周囲温度(15〜30℃(華氏59〜86度))で保存されるべきであり、標準の制御加熱、換気、及びエアコンディショニング(HVAC)システムを使用して、高熱及び湿気から保護される。
【0331】
4.5 無作為化
2の投与量が相2へもたらされると、1:1の無作為化スケジュールが準備される。
【0332】
4.6 管理
臨床試験薬は経口的に服用され、食事とともに服用されるべきである。臨床試験薬の最初の投与量は、10:00と14:00の間に、臨床現場において被験体によって投与される。最初の投与量(1日目)の受け取りは登録と考えられる。被験体は、その後、上述されるラベルが付けられる要求と一致する薬局からの臨床試験薬を受け取り、17:00と21:00の間で、毎日投与量を服用する。被験体は、任意の服用し忘れた投与量のスケジュールを組み直さないように指示される。臨床試験薬は、登録時、及び最初の12週の間の2週ごと(訪問の2、4、6、8、10及び12週目)に調剤され、その後、(延長へと継続する場合)試験終結の訪問で開始して、4週ごとに調剤される。
【0333】
3.1.5 薬局の責任
臨床試験薬の受け取りによって、棚卸が行われなければならず、指定の職員によって責任の記録(Accountability Log)が記入され、署名されなければならない。指定の職員が、輸送目録に書き留められたすべての品目を含むことを数え、確認することは重要である。所与の輸送における任意の行方不明の、破損された、又は使用できない臨床試験薬は文書化されければならない。更に、PI及び/又は信頼できる現場人員は、輸送の責任がある間に留意された問題を、Pharm-Olamに通知しなければならない。輸送の受け取りの時間に加えて、責任の記録は、薬物が被験体に分配される度に完成されなければならない。
【0334】
8.3.2 臨床試験薬の取り扱い及び処分
研究員は、試験の全体にわたって正確な臨床試験薬の責任の記録を維持する責任を負う。臨床試験薬の各分配は、eCRFに文書化される。現場の薬局によって分配されたすべての未使用の臨床試験薬は、臨床現場に返されなければならず、各臨床現場での標準作業方法に従って適切な方法で破棄されなけばならない。これは、空き瓶と未使用の臨床試験薬を含む瓶の両方の臨床試験薬の瓶を含む。このような供給の破棄は、責任の記録に文書化され、代表者は処分の記録を確認する。
【0335】
5. 有効性の評価
このセクションで詳述されるように、疾患の進行及び生物学的シグナルは、本発明の研究において診査される。
【0336】
5.1 疾患の進行
疾患の進行は以下の評価を実施することにより評価される。
【0337】
5.1.1 臨床的評価
疾患の進行(例えば、疼痛)に関する臨床的評価は、安全性データとして提示される。
【0338】
5.1.2 バイオマーカー
5.1.2.1 前立腺特異性抗原
前立腺特異性抗原は、手順のスケジュールに従って測定される。
【0339】
5.1.3 放射線学的評価
5.1.3.1 固形腫瘍における反応評価基準(RECIST)
固形腫瘍における反応評価基準(RECIST)は、X線、CT、及び磁気共鳴画像法(MRI)を使用して、腫瘍反応を測定するための統一された、容易に適用可能な基準である。該技術は、試験のスポンサーである、国立癌研究所(National Cancer Institute)(NCI)に推薦され、腫瘍標的病変の測定のための形式化された規則を含む。RECIST基準は、任意の国際基準であり、NCI標準ではない。それらは、以前の方法の単純化に基づき[世界保健機構(WHO)、ECOG]、測定可能な疾患(すなわち、少なくとも1つの測定可能な病変の存在)に基づく。RECIST基準は、臨床試験における広範囲の適用のための、単純化された、保存的な、画像データの抽出を提示する。RECIST(Eisenhauer et al., 2009, 本明細書に引用によって組み込まれた)は、この試験に使用される。
【0340】
5.1.3.2 MRI及び骨スキャン
CT/MRI及び骨スキャンは、標準治療の一部として行われると予期され、手順のスケジュールに従って行われる(表5)。もし基準線で使用される同じ様式が、試験においてすべての後の評価のために継続的に利用されるならば、CTスキャンの代わりのMRIの使用は許容可能である。万一、予期しない事象が生じ、処置する医師の判断において、任意の他のスキャンの形態を有するのが被験体の利益であるならば、これは、メディカルモニター及びメディカルアドバイザーと話し合われるべきである。
【0341】
試験包含の目的のために繰り返されているスキャンを回避するために、記録される4週のウィンドウ以外の次に空けられた投与に対する包含のためのスクリーニングスキャン(CT/MRI又は骨)の使用に関して(トーカイメディカルアドバイザー又は指名された人と相談して)、メディカルモニターによって例外が認められ得る。しかしながら、この例外は以下の基準に基づく:被験体は、競合的なスクリーニングにより、以前の相1の投与量における漸増を逃しておかなければならず;すべての他のスクリーニング試験は繰り返されなければならず、被験体が資格を有するままであることを確認しなければならず;臨床試験薬の最初の投与前に、6週以降のスキャンは許容されず;及び、PIの医学的所見において、被験体は放射線学的進行を有することが疑われず、繰り返しスキャンのための臨床的指標がない申し立てが受理されなければならない。
【0342】
この例外の許可は、標準治療を超える増加した頻度でスキャンを繰り返す被験体に対する不必要なリスクを低減する(3〜6か月ごと)。
【0343】
5.1.3.3 進行までの時間(TTP)、無進行生存(PFS)及び全存率(OS)
生存データの獲得のために試験の全体にわたって被験体は追跡調査される。
【0344】
5.2 特殊実験(Special Laboratories)
特殊実験は、手順のスケジュールに従って決定される(表5)。
【0345】
特殊実験は下記を含む:プレグネノロン、17-ヒドロキシプロゲステロン、デオキシコルチコステロン、11-デオキシコルチゾール、コルチコステロン、コルチゾール、DHEA、アンドロステンジオン、及びテストステロン。
【0346】
5.3 腫瘍内のARタンパク質、テストステロン及びジヒドロテストステロン
腫瘍内のパラメーター(ARタンパク質、テストステロン、及びDHT)の分析のための生検は、ICFを作成する別々の機関の下で被験体のサブセットにおいて提案される。相1又は2の試験終結の訪問又はPSA最下点(最初に生ずるどちらか)で生検が繰り返されるべきである。
【0347】
5.4 腫瘍内のパラメーターは、手順のスケジュールに従って決定される。化合物1の濃度
PKサンプルを、手順のスケジュールに従って収集する(表5)。
【0348】
サンプルを、提供されるエチレンジアミン四酢酸ジカリウム(K2EDTA)管に収集し、すぐに氷上に置き、血漿分離のために遠心分離にかける。サンプルを、血漿分離の8時間以内に冷凍する。
【0349】
6. 安全性の評価
6.1 安全パラメーター
6.1.1 研究所のパラメーター
中央検査室:接触リストにおいて識別されるように、研究所の評価は、中央検査室によって行われる。
【0350】
地方の実験室:実験室の評価は、確立された方法によって、各現場の実験室で局所的に行われる。
【0351】
更なる詳細に関しては、臨床検査の改善修正(CLIA)を参照する。
【0352】
安全性の研究所のパラメーターは、研究所のマニュアルに提示される手順のスケジュールに従って決定される。
【0353】
研究所の異常性が、臨床的に有意か、臨床的に有意でないかのいずれかとして、研究員の判断で記録される。
【0354】
6.1.2 生命徴候
以下の生命徴候が、手順のスケジュールに従って評価される:血圧(収縮期及び拡張期;mmHg);心拍度数(拍/分);体温(℃)、経口;呼吸速度(呼吸/分);重量;及び高さ(基準線のみ)。被験体が少なくとも10分間座っているとき、生命徴候が得られる。
【0355】
6.1.3 心電図
12誘導ECGは、手順のスケジュールに従って行われる。被験体が≧3分間背臥位であった後、すべての12誘導ECGが行われる。12誘導ECGモニターは、臨床現場の標準処理手順に従って較正され標準化される。すべての臨床上有意なECG(先に存在する異常性を除く)は、心臓病専門医によって検討される。
【0356】
6.1.4 身体検査
身体検査は、手順のスケジュールに従って行われる。身体検査は、以下の体組織を含む:一般的な様相、HEENT(頭、耳、目、鼻、及び咽喉)、呼吸器、腹、腎(泌尿器科学)、生殖器、筋骨格、神経系、リンパ節、皮膚、及び他のもの。ECOGパフォーマンスステータスが記録される。被験体の疾患がどのように進行しているかを評価し、その疾患が被験体の日常生活能力にどのように影響を与えるかを評価し、および適切な処置及び予後を決定するために、ECOGの規模及び基準は、医者及び研究員によって使用される。
【0357】
【表8】
【0358】
6.1.5 有害事象(AE)
有害事象(AE)は、治験薬を受け取った被験体において生じる任意の好ましくない医学的事象であり、必ずしもこの処置との因果関係を有していない。それ故、AEは、治験薬に関連があろうとなかろうと、治験薬の使用により一時的に関係する、任意の好ましくない及び意図しない徴候(異常な検査所見を含む)、症状、又は疾患であり得る。臨床試験の間に記録されたすべてのAEは、国際医薬用語集(MedDRA)システムに従ってコード化され、器官別大分類に割り当てられる。
【0359】
処置により現れるAE(TEAE)は、臨床試験薬の少なくとも1回用量が投与された後に、始まるか、または重症度及び/又は頻度において悪化するか、または性質が変化するAEとして定義される。試験の間に生じる、併発性疾患を含む、すべてのAEは、eCRFに文書化される。試験へのエントリー前に存在した、併発疾患は、治療期間に悪化しない限り、AEとは考えられない。既往歴はeCRFに記録される。
【0360】
DLTは、任意の臨床試験薬に関連するグレード3又はより高いAEとして定義され、臨床試験薬と、ひょっとしたら、おそらく、又は明確に関連があると考えられる。たとえ重大なものとして考えられなくても、AEは、DLTと考えられ得ることは可能である。
【0361】
6.2 臨床試験薬との関係性
6.2.1 有害事象の評価
各AEは、以下のカテゴリーに関して研究員によって評価される。
【0362】
6.2.1.1 重篤度
重篤AE(SAE)は、任意の投与量で、死に至る、 又は生命を脅かす、任意の好ましくない医学的出来事として定義される。AEとしての分類は、被験体が事象の時に死亡の直接のリスクがあることを必要とし、それは、もし事象がより重篤ならば仮説的に死を引き起こし得ることを意味しない。AEは、以下の1つ以上によって更に特徴付けられる:被験体の入院を必要とする又は延長する;持続的な又は重大な障害又は不能という結果となる;先天異常又は出産時欠損である;すぐに生命を脅かし得ない、または死に至り得ない、または入院の結果になり得ないが、被験体を危険にさらし得るか、または上記の結果のうちの1つを防ぐために介入を必要とし得る、重要な医学的事象(複数可)である。
【0363】
症例が重大であるか、および施設内治験審査委員会(IRB)及び取り締まり機関への事象の促進される報告が適切かどうかを決定する際に、医学的及び科学的な判断が行使される。
【0364】
6.2.1.2 重症度
各AEの重症度は、AE(CTCAE)v4.0のためのNCI共通用語基準を使用して、研究員によって評価され、eCRFに記録されなければならない。
【0365】
AEの重症度は、以下の臨床的記載に従って類別される:
・グレード1:軽度;無症候性又は軽度の徴候;臨床的又は診断上の観察のみ;示されない介入;
・グレード2:中程度;示される、最小限の、局所的な、又は非侵襲性の介入;限定される年齢に応じた手段的日常生活動作(ADL)*;
・グレード3:重度又は医学的に重大であるが、すぐには生命を脅かさない;示される、入院又は入院の延長;不能状態;限定されるセルフケアADL**;
・グレード4:生命を脅かす結果;示される緊急の介入;及び
・グレード5:AEに関連する死。
* 手段的ADLは、食事を準備すること、食料品又は衣類の買い物をすること、電話を使用すること、お金を管理することなどを指す。
** セルフケアADLは,入浴、着衣及び脱衣、自給、トイレの使用、薬剤の服用、及び寝たきりでないことを指す。
【0366】
6.2.1.3 因果関係
研究員は、臨床試験薬とAEの間の因果関係/関係性を評価し、eCRFにその評価を記録する。因果関係は次のように示される:明確に関連する;おそらく関連する;ひょっとしたら関連する;関連しそうにない;又は、関連しない。
【0367】
6.3 有害事象の記録
有害事象報告は、臨床試験薬の最後の投与の後、30日間まで、又は事象までが回復(resolution)/安定するまで、スクリーニングから延長する。試験の終了後に生じる有害事象は、臨床試験薬との因果関係があると研究者が考えると、報告される。
【0368】
すべてのAEは、臨床試験薬との関係性にかかわらず、eCRFに記録される。
【0369】
すべてのAEの報告は、事象の簡単な記載、発生の日及び時間、回復の日及び時間、強度、必要とされる処置、臨床試験薬との関係性、臨床試験薬とともにとられた行動、結果、及び事象が重大なものとして分類されるかを含む。
【0370】
6.3.1 有害事象のフォローアップ
被験体によって経験されたすべてのAEは、疑われる因果関係に関係なく、事象が回復/安定するまで、任意の異常な臨床検査値が基準線に戻る又は研究員とメディカルモニターに許容可能なレベルで安定するまで、観察される変化の満足な説明がなされるまで、または被験体がフォローアップされなくなる(lost)まで、監視される。
【0371】
8.3.2 有害事象の報告
研究員は、事象を発見してから24時間以内に、あらゆるSAEを、Pharm-Olam Pharmacovigilance Unitに報告する。SAEの報告を行うとき、SAE用語、研究員の名前、報告者の名前、報告者と連絡するときの電話番号、およびプロトコルの分類番号及びタイトルが与えられる。最後の投与後の30日以内に生じ、少なくとも臨床試験薬とひょっとしたら関連があると考えられ、それ故、副作用の可能性がある、任意のSAEが報告される。事象の評価に基づいて、規定上の報告を含む更なる行動の必要性に関して決定が下される。
【0372】
7. 訪問による観察
治療期間中の訪問が、予定された訪問の±3日以内に行われる。被験体が訪問を逃すと、その訪問を埋め合わせることはできないが、次の通常の予定された訪問を行う。24時間計(例えば、11:20ではなく23:20)を使用して、すべての時間が記録される。
【0373】
7.1 スクリーニング(訪問1)
スクリーニング訪問が、最初の投与(1日目)から28日間以内に行われる。スクリーニングでは、以下の評価が行われ、測定が記録される:書面によるインフォームドコンセント(署名されたICF)を得る;資格基準を確認する;医学的/腫瘍学的病歴を記録する;人口統計学的詳細;身体検査;生命徴候;12誘導ECG;CT/MRI及び骨スキャン - 使用されるCT/MRI及び骨スキャンの結果は、1日目から4週間以内の日付でなければならない;生検(被験体のサブセット;別々のICFによって表紙をつけられた);実験室の安全性試験(血液学、血清化学検査、検尿) - 安全性実験室評価が1日目の7日より前に記録されると、それらは投薬の前に繰り返され、確認されるべきである;併用薬を記録する。AEは、この訪問から記録される。
【0374】
7.2 登録(訪問2)
登録訪問(1日目、訪問2)では、以下の評価が行われ、測定が記録された:12誘導ECG(投与前);特殊実験室試験;PSA;PKサンプル(投与前);併用薬;AE。
【0375】
臨床試験薬を、10:00と14:00の間に、食事とともに、臨床現場で投与する。12誘導ECGを、投与の4時間後(±15分)に行う(PKサンプル前)。PKサンプルを、投与の4時間後(±15分)に収集する(ECG後)。臨床試験薬を分配し、被験体は臨床試験薬の使用に備える。コンプライアンスの日記を発行し、被験体はその使用に備える。被験体は、2日目から次の訪問まで、夕食(17:00と21:00の間)とともに毎日1回処置を継続することが必要とされる。
【0376】
7.3 1週間はない訪問(訪問3、5、7、9、及び11)
被験体は、8日目(±3日)、訪問2の1週間後、及び実験室試験が1週間ないためにその後2週ごとに臨床現場に戻る。臨床現場から地理的に遠く離れているそれらの被験体のために、地方の実験室が利用され得る。これらの訪問で、実験室の安全性試験(血液学及び血清化学検査のみ)が行われる。
【0377】
7.4 2週ごとの隔週の訪問(訪問4、6、8、及び10)
被験体は、15日目(±3日)、訪問3の1週間後、及びその後2週ごとに臨床現場に戻る。これらの訪問で、以下の評価が行われ、測定が記録される:身体検査;生命徴候;12誘導ECG;実験室の安全性試験(血液学、血清化学検査、検尿);特殊実験室試験;PSA;PKサンプル(投与前);併用薬;AE;コンプライアンスの日記の回収;発行される新しいコンプライアンスの日記;責任手順;及び分配される臨床試験薬。被験体は、2週間後の訪問まで、夕食(17:00と21:00の間)とともに毎日1回処置を継続することが必要とされる。
【0378】
7.5 試験終結の訪問
試験終結の訪問が、85日目(±5日)、最初の投与の12週間後に行われる。以下の詳細及び評価が行われ、測定が記録される:身体検査;生命徴候;12誘導ECG;CT/MRI及び骨スキャン - これは、試験終結の訪問前に行われると、PSA最下点で行われる;生検(被験体のサブセット;別々のICFによって表紙をつけられた) - これは、試験終結の訪問前に行われると、PSA最下点で行われる;実験室の安全性試験(血液学、血清化学検査、検尿);特殊実験室試験;PSA;PKサンプル(投与前);併用薬;AE;コンプライアンスの日記の回収;発行される新しいコンプライアンスの日記(延長へと継続する場合);責任手順;及び分配される臨床試験薬(延長へと継続する場合)。
【0379】
7.6 延長の相
被験体が延長の相へ継続する資格を有すると、以下の評価がなされる、4週ごとの臨床現場に被験体は戻り、測定が記録される:身体検査;生命徴候;CT/MRI及び骨スキャン(12週ごと);実験室の安全性試験(血液学、血清化学検査、検尿);特殊実験室試験;PSA;併用薬;AE;コンプライアンスの日記の回収;発行される新しいコンプライアンスの日記;責任手順;及び分配される臨床試験薬。
【0380】
7.7 期限前契約解除の訪問
試験から初期に中止する被験体は、可能であれば、期限前契約解除の訪問を受けるべきである。被験体が臨床試験薬を服用するのをやめた後、できるだけすぐにこの訪問を行う。被験体が任意の理由で早期に離脱すると(12週目の訪問前)、研究員は、試験終結の訪問(12週目の訪問)のために記載される安全性評価を行なうために、あらゆる努力をする。
【0381】
8. 統計
この試験の相1の段階の1つの目的は、TOK-001が許容可能な安全性プロフィールを提供する投与量(複数可)を設定することである。許容可能な安全性プロフィールは、≦35%の正確なDLT比率での投与量として定義される。相2の段階の目的は、もたらされる各投与量(複数可)のための生物学的なシグナルを評価するのみでなく、許容可能な安全性プロフィールを確認することである。
【0382】
以下の3の投与量群は、相1のために計画される:(1) 650mgの化合物1;(2) 1300mgの化合物1;(3) 1950mgの化合物1。
【0383】
中間の投与量を得る可能性は別にして、各投与量を、許容可能な毒性プロフィールを有する投与量として識別する確率の計算は、本明細書に記載される用量増加及び用量減少の規則に従ってなされ得る。3の投与量の各々に対する3の正確だと想定されるDLT比率は、各々が最大投与量(相1の段階から現れる投与量として定義される)と指定される確率とともに、表6に示される。
【0384】
【表9】
【0385】
650mgの化合物1が「毒性が強すぎる」確率は、上記の各シナリオに対して0.008である。
【0386】
試験の相1の段階の終了で、2までの投与量が、試験の相2の段階に進むために選択される。
【0387】
相2に進められる投与量の安全性が、相1における比較的少数の被験体(6まで且つ3以上)において評価されるため、その投与量での正確なDLT比率が>35%であることを示唆する合理的に説得力のある証拠があると、特定の投与量に対する登録が停止され得るように、停止規則が実施される。あらゆる5番目の被験体がDLTに対して評価可能となる後に、これらの制限が計算される。操作上、以下のいずれかはこのような制限につながり、DLTの数値/被験体の数:4/5(又はより少ない)、6/10(又はより少ない)、7/15(又はより少ない)、又は9/20(又はより少ない)と表される。
【0388】
DLTの正確な確率が20%又は50%であると、過度のDLTにより試験を停止する確率は、それぞれ、およそ0.03及び0.81である(5,000回のシミュレーションから推測される)。投薬がアームにおいて停止されると、相2の投与量の修正が行われる。
【0389】
8.1 一般的な考察及び基準線
他に明記のない限り、すべての統計試験を、5%の有意水準で両側検定を使用して行う。これが相1/2の試験であるため、複数のエンドポイント及び訪問のための多重比較の調整を、最終的な分析では行わない;分析は主として、本来記述的である。基準線は、他に明記のない限り、最初の投与の前に最後の観察として定義される。すべての推論的試験は、両側検定を使用する、処置の直接の比較を含む。試験の変数を、記述統計[連続変数に対する、N、平均、標準偏差(SD)、中央値、最小値、及び最大値、およびカテゴリー変数に対する頻度及びパーセンテージ]によってまとめる。
【0390】
8.2 被験体の処置
試験の各相に入り終了する被験体の数及びパーセンテージが、処置によって階層化されて示される。
【0391】
8.3 分析個体群
分析個体群の選択に対する分析の感受性を評価するために、二次分析もパープロトコル(PP)個体群に基づいて行われるが、主要な有効性分析は、包括解析(Intent-to-treat)(ITT)個体群に基づく。すべての安全性分析は安全性個体群に基づく。
【0392】
投与が相2において停止されると、停止される試験のアームにおける被験体は継続的な投与量で処置され、12週のフォローアップスケジュールを終了するが、継続的な投与量のための有効性分析には含まれない。
【0393】
8.3.1 安全個体群
臨床試験薬を少なくとも1回用量受け取る被験体はすべて、安全個体群に含まれる。
【0394】
8.3.2 包括解析個体群
スクリーニングを終了し、臨床試験薬での処置を予定している被験体はすべて、ITTの個体群に含まれる。
【0395】
8.3.3 パープロトコル個体群
PP個体群は、以下の基準の両方を満たす被験体を含む:1) 臨床試験薬を少なくとも1回用量受け取る;及び 2) あらゆる主要なプロトコル違反を有さない。
【0396】
8.4 プロトコルの逸脱
包含/排除の基準の違反を含むプロトコルからの逸脱は、「主要でない」又は「主要である」として、評価される。プロトコルからの主要な逸脱は、PP個体群からの被験体の排除につながる。
【0397】
8.5 人口統計及びベースライン特性
登録時の年齢などの連続的な人口統計学的パラメーターを、記述統計(N、平均、中央値、SD、最小値、及び最大値)を使用して、安全個体群のためにまとめる。性別などの分類別の人口統計学的パラメーターを、安全個体群の頻度及び割合としてまとめる。
【0398】
8.6 併用薬
併用薬を、WHO医薬品辞書のバージョン12.0又はそれ以上を使用してコード化する。データを、記述統計を使用してまとめる。
【0399】
8.7 処置曝露及び処置コンプライアンス
処置曝露及び処置コンプライアンスを、処置によって階層化して、頻度及びパーセンテージによってまとめる。
【0400】
8.8 安全性分析
安全性分析を、安全個体群を使用して実行する。試験の安全性エンドポイントは以下のものを含む:AEの発生率;生命徴候及び12誘導ECGの変化;身体検査の変化;実験室評価(血清化学検査、血液学、及び検尿)の変化。
【0401】
8.8.1 有害事象
臨床試験の間に記録されたすべてのAEを、MedDRAシステムに従ってコード化し、器官別大分類に割り当てる。TEAEは、治療の開始の後に、重症度及び/又は頻度において最初に生じる又は悪化するAEとして定義される。開始時間が不明の、臨床試験薬の最初の投与の日に開始するあらゆる事象は、処置により現れると想定される。
【0402】
体組織によって任意のAEを経験する被験体の頻度、関連性、及び重症度を、計算及びパーセンテージを使用して各処置群においてまとめる。あらゆる所与のMedDRAの優先使用語に関して、たとえ被験体が複数回の処置にわたって複数回の出現を有したとしても、被験体は、発生に対して1回のみのカウントに貢献する。臨床試験薬との関係性は、(明確に、おそらく、ひょっとしたら)関連する及び関連しない(ありそうにない、関連しない)ものとして分類される。1の被験体に対して複数の記録が存在すると、最大の重症度及び臨床試験薬に対する最も強力な関係性だけが、パーセンテージを計算するために数えられる。重篤AEを、全体的に、および体組織及び優先使用語によってまとめる。
【0403】
8.8.2 実験室評価、心電図、生命徴候、及び身体検査
臨床検査、ECG、及び生命徴候パラメーターにおける基準線からの変化を、処置群及び訪問によって計算する。基準線からの変化に関する記述統計(N、平均、SD、中央値、最小値、及び最大値)を表にする。基準線から基準線後(post-baseline)までの変化の表を、臨床検査及びECGのパラメーター及び身体検査のための処置群及び訪問によって作成する。頻度及びパーセンテージを、変化の表に提示する。研究所評価における臨床的に著しい異常性を、データリストに示す。
【0404】
8.9 有効性分析
ITTの個体群を、すべての有効性分析に使用する。
【0405】
8.9.1 主要な有効性分析
主要な有効性エンドポイントは、基準線から12週までのPSA、又はPSA最下点(最初に生ずるどちらか)における50%又はそれ以上の減少を有する被験体のパーセントである。頻度及びパーセンテージを、各処置群に関して計算する。主要な分析も、PP個体群を使用して繰り返す。
【0406】
8.9.2 二次的な有効性分析
記述統計を、二次的な有効性パラメーターの各々のために表にし、提示する。すべての二次的な有効性分析を、ITT個体群を使用して行う。
【0407】
8.9.2.1 固形腫瘍における反応評価基準(RECIST)による進行。
反応率は、全奏効率(ORR)を含む。ORRは、以下のように定義され:
ORR = (CR + PR)/Nr
ここで、CRは、完全奏効を有する被験体の数であり、PRは、部分奏効を有する被験体の数であり、及びNrは、ITT個体群に含まれる被験体の数である。
【0408】
腫瘍反応評価は、以下のRECIST定義を使用して行われる。
【0409】
【表10】
【0410】
被験体の反応は、表8に概説されるように、処置の開始から疾患の進行/再発まで記録された最良の反応である。
【0411】
【表11】
【0412】
8.9.2.2 CT/MRI及び骨スキャン、更なる特殊実験、及び生検サブセット中のパラメーターにおける基準線からの変化
CT/MRI及び骨スキャン、更なる特殊実験、および生検サブセット中の腫瘍内のパラメーター(腫瘍内のARタンパク質、テストステロン、およびDHT)における基準線からの変化を、処置群及び訪問よって計算する。基準線からの変化に関する記述統計(N、平均、SD、中央値、最小値、及び最大値)を、処置群によって表にする。
【0413】
8.9.2.3 進行までの時間、無進行生存及び全存率
TTP、PFS、及びOSは、延長の相の特異的なエンドポイントである。
【0414】
進行までの時間(TTP)は、臨床経過、放射線学的証拠、及び生化学的マーカー(PSA)の結果に基づいて、臨床試験薬の最初の投与から、より悪化する疾患の最初の文書化されたPI評価までの時間として定義される。
【0415】
無進行生存(PFS)は、被験体が悪化しない疾患(CRPC)を有して生活している試験における処置の間及びその後の時間の長さである。
【0416】
全存率(OS)は、任意の原因により、臨床試験薬の最初の投与から死の最初の文書化までの時間として定義される。この試験の目的で、OSは、臨床試験薬の最初の投与を受けた5年後の生存した被験体のパーセンテージとして報告される。
【0417】
TTP、PFS、及びOSの分析は、ITT個体群を使用する化合物1の投与量群の間で、比較のためのログランク検定を使用する。カプラン・マイヤー推定値は、処置群によって図示される。95%の信頼区間を有する事象までの平均期間(Median time)も、推定できるならば、処置群によって表にされる。
【0418】
打切日で事象を経験しなかった被験体は、中断日をカットオフの日(cut-off date)として使用し、分析から打切りされる。初期に離脱する被験体は、最後の接触日をカットオフの日として使用し、分析から打切りされる。
【0419】
8.9.2.4 診査のエンドポイント
以下の分析は、試験の相2の段階のための可能な診査のエンドポイントである:1) 12週にわたるPSA変化(最初の投与から始まり、12週間の処置をカバーする);及び/又はPSA最下点及びPSA最下点までの時間(最下点は、化合物1の投薬期間中に観察された最低のPSA値である)。
【0420】
記述統計(N、平均、SD、中央値、最小値、及び最大値)を、処置群によって表にする。
【0421】
8.10 化合物1の濃度
化合物1の血漿濃度を、1日目の0時間目(C0hrs)及び4時間目(C4hrs)に測定し、および続く訪問の利用可能な時点について測定する。
【0422】
8.11 相2のサンプルサイズの決定
2の投与量が相1から選択されると、その後、40の被験体は、2の投与量の間で1:1の比率で無作為化される。1の投与量だけがもたらされると、20の被験体は、この投与量を割り当てられ、無作為化は生じない。このセクションは、1アーム当たり20の被験体のサンプルサイズの選択の正当化を記載する。
【0423】
初回量、650mgの化合物1は、忍容性が良好であると思われ、研究の意図は、12〜15の被験体が相1に登録されることである[相2の投与量(複数可)で処置されるいくらかの被験体を含む]。相2に使用される投与量で相1において処置される被験体は、無作為化されるが、一貫した処置及びフォローアップスケジュールを受ける。それ故、試験の相2の段階において生物学的作用の潜在性を検査するときに被験体は含まれ、特定の投与量での少なくとも23の被験体(26まで)が、相2における生物学的シグナルの分析に利用可能であり得ることが予期される。2の投与量が相1から生じると、これらの投与量の1つは、好ましい投与量として識別される。この投与量が有意に強力な生物学的シグナルを有すると考えられると、この投与量は今後の試験に使用される。1アーム当たり20の被験体の選択は、相2における特定の投与量で少なくとも23の被験体(及び26もの被験体)を処置させる予想に基づく。一連の23(23)の被験体は、想定される正確な比率が75%であるときに、45%の固定比率と比較して、統計学的に著しく改善された「奏効率」(PSAの減少)を観察する、92%の力(power)を提供する。
【0424】
生物学的作用の検査に使用されるエンドポイントは、基準線から12週までのPSA、又はPSA最下点(最初に生ずるどちらか)における50%又はそれ以上の減少を有する被験体のパーセンテージである。2の投与量が相1から生じると、そのような被験体の中で最大のパーセンテージを有する投与量は、相2からの好ましい投与量として識別される。この好ましい投与量(又は1のみが続く試験のために相2にもたらされるときの単一用量)の今後(future)は、以下のガイドラインによって管理される:1) 好ましい投与量に対する観察されたPSA反応が30〜45%であると、化合物1は、更なる用量設定作業を恐らく受ける;2) 観察されたPSA反応が45〜75%であると、化合物1は、潜在性の生物学的作用を有すると考えられ、より大規模な相2又は2/3の試験は、全存率及び進行までの時間などの有効性の測定に加えて、PSA反応を確認し、より正確に推測するように考案される;及び3) 観察されたPSA反応が75%を超過すると、この投与量を直接、相3の試験に与えることが強く考慮に入れられる。
【0425】
特定の投与量で23又は26の被験体の間の45%以上のPSA反応を観察する確率は、様々な想定される正確な奏効率のために表7にまとめられる。
【0426】
【表12】
【0427】
さらに、正確なPSA反応率が75%であると、一連の23の被験体は、(0.05の片側の有意水準での)45%の固定比率と比較して、統計学的に有意に改善された奏効率を観察する、92%の力を提供する。n=26の同じパラメーターは、94%の力をもたらす。ベンチマーク奏効率が30%で、想定される正確な奏効率が60%であると、一連の23の被験体はまた、91%の力を提供する。n=26のこれらの想定は、94%をもたらす。
【0428】
具体的なベンチマークに関連する奏効率における統計学的に有意な改善が、続く試験に進むためには必要とされないが、これらの力の計算は、相2におけるサンプルサイズに関して1アーム当たり20の被験体の選択の論理的根拠に加えて、次の試験の方向を決定する助けとなる、必要とされる観察される固定比率のためのいくらかの正当化を与える。2の投与量が相2にもたらされ、アームに対する正確な奏効率が45%及び60%であると、正確なより高い奏効率を有するアームが選択され、1アーム当たり23の被験体でおよそ0.88の確率を有し(シミュレーションから推測される確率);23のサンプルサイズは、相2において各アームに無作為化された20に、相1において各投与量で処置される少なくとも3を加えたものである。
【0429】
<実施例7:化合物(1)での前立腺癌の処置のための臨床研究結果。>
実施例6のプロトコルに従って臨床研究を行った。化合物(1)を、4つのコホートに各々異なる量で投与した。各個々の被験体は、被験体が関係している間同じ量の化合物(1)を受けた。
【0430】
【表13】
【0431】
各群における大多数の被験体が、治療に好意的に反応したことを結果は示す。大量の化合物(1)を受ける、コホート3及び4は、より高い奏効率を生み出した。
【0432】
化合物(1)の血清濃度を、被験体の参加の間、各被験体において追跡した。その濃度を、図13に示す。被験体がすべて同じスケジュールに従ったわけではなく、コホートもすべてが同じスケジュールに従ったわけではなかった。図13において留意されるのは、コホート2及び3の被験体が、それぞれ、コホート1の被験体が受け取った量の2倍以下及び2倍を受け取ったが、コホート2及び3の被験体において見られる化合物(1)の血清濃度は、しばしば、コホート1において見られる濃度の2倍以上であったことである。より高い投与量が、ユニット効果当たり、より大きな有効性を有しており、より生産的且つより経済的な治療が、化合物(1)のより高い投与量により達成可能であることを、これらのデータは示す。
【特許請求の範囲】
【請求項1】
化合物(1):
【化1】
又はその薬学的に許容可能な塩、N-オキシド、活性代謝物、プロドラッグ、又は溶媒和物を含む医薬組成物であって、式中、化合物(1)は、約:
a. 13.1°及び14.1°;
b. 17.2°、18.5°、及び19.1°、及び随意に約16.2°、及び29.6°;
c. 12.5°、14.8°、及び25.5°;又は
d. 14.4°、16.1°、19.1°、及び19.3°
の角度 2θにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられた、微粉化結晶形態であることを特徴とする医薬組成物。
【請求項2】
化合物(1)が、被験体への投与後に、アンドロゲン受容体媒介性の疾患又は疾病を処置するのに有効な量で存在することを特徴とする請求項1に記載の医薬組成物。
【請求項3】
前記アンドロゲン受容体媒介性の疾患又は疾病が、前立腺癌、良性前立腺肥大症、多毛症、脱毛症、神経性食欲不振、乳癌、及び男性の性腺機能亢進から成る群から選択されることを特徴とする請求項2に記載の医薬組成物。
【請求項4】
前記アンドロゲン受容体媒介性の疾患又は疾病が、前立腺癌であることを特徴とする請求項3に記載の医薬組成物。
【請求項5】
前記前立腺癌が、去勢抵抗性前立腺癌であることを特徴とする請求項4に記載の医薬組成物。
【請求項6】
化合物(1)が、被験体への投与後に、アンドロゲン生合成を阻害し、アンドロゲン受容体シグナル伝達を阻害し、及びアンドロゲン受容体感受性を低下させるのに有効な量で存在することを特徴とする請求項1に記載の医薬組成物。
【請求項7】
前記化合物が、アンドロゲン受容体シグナル伝達を阻害するか、又はアンドロゲン受容体感受性を減少させることを特徴とする請求項6に記載の医薬組成物。
【請求項8】
前記アンドロゲン生合成阻害が、チトクロームC17α-ヒドロキシラーゼ/C17,20-リアーゼ(CYP17)の活性を阻害することを含むことを特徴とする請求項6に記載の医薬組成物。
【請求項9】
前記アンドロゲン受容体シグナル伝達阻害が、テストステロン結合の競合的阻害を含むことを特徴とする請求項6に記載の医薬組成物。
【請求項10】
前記アンドロゲン受容体感受性の減少が、細胞内のアンドロゲン受容体タンパク質の含量の減少、及び低レベルのアンドロゲンの増殖シグナルによって持続されるべき、細胞の低下した能力を含むことを特徴とする請求項6に記載の医薬組成物。
【請求項11】
前記組成物が、被験体に、非経口的に、静脈内に、筋肉内に、皮内に、皮下に、腹腔内に、経口的に、口腔に、舌下に、粘膜に、直腸に、経皮的に、皮膚に、眼に、又は吸入によって投与するために調剤されることを特徴とする請求項1に記載の医薬組成物。
【請求項12】
前記組成物が、錠剤、カプセル剤、クリーム、ローション剤、油剤、軟膏、ゲル、ペースト、粉末、懸濁液、エマルション、又は溶液として被験体に投与するために調剤されることを特徴とする請求項1に記載の医薬組成物。
【請求項13】
前記組成物が、カプセル剤として被験体に投与するために調剤されることを特徴とする請求項1に記載の医薬組成物。
【請求項14】
前記組成物が、錠剤として被験体に投与するために調剤されることを特徴とする請求項1に記載の医薬組成物。
【請求項15】
前記組成物が、被験体に投与するために調剤され、約25mg/kgから約50mg/kgの間の化合物(1)を含むことを特徴とする請求項1に記載の医薬組成物。
【請求項16】
前記組成物が、約650mgから約3500mgまでの化合物(1)を含むことを特徴とする請求項1に記載の医薬組成物。
【請求項17】
前記組成物が、約1950mgの化合物(1)を含むことを特徴とする請求項1に記載の医薬組成物。
【請求項18】
前記組成物が、約1300mgの化合物(1)を含むことを特徴とする請求項1に記載の医薬組成物。
【請求項19】
前記組成物が、約650mg又は約975mgの化合物(1)を含むことを特徴とする請求項1に記載の医薬組成物。
【請求項20】
前記組成物が、被験体に、1日当たり、1、2、3、4、5、6、7、8、9、又は10回投与するために調剤されることを特徴とする請求項1に記載の医薬組成物。
【請求項21】
前記組成物が、1以上の薬学的に許容可能な賦形剤を更に含むことを特徴とする請求項1に記載の医薬組成物。
【請求項22】
前記薬学的に許容可能な賦形剤が、充填剤、崩壊剤、潤滑剤、界面活性剤、流動促進剤、結合剤、砂糖、スターチ、ニス、又はワックスを含むことを特徴とする請求項21に記載の医薬組成物。
【請求項23】
被験体において前立腺癌に対する処置を提供する方法であって、該方法は、治療上有効な量の化合物(1):
【化2】
又はその薬学的に許容可能な塩、N-オキシド、活性代謝物、プロドラッグ、又は溶媒和物を含む医薬組成物を被験体に投与する工程を含み、式中、化合物(1)は、約:
a. 13.1°及び14.1°;
b. 17.2°、18.5°、及び19.1°、及び随意に約16.2°、及び29.6°;
c. 12.5°、14.8°、及び25.5°;又は
d. 14.4°、16.1°、19.1°、及び19.3°
の角度 2θにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられた、微粉化結晶形態であることを特徴とする方法。
【請求項24】
前記被験体が、ヒトであることを特徴とする請求項23に記載の方法。
【請求項25】
前記前立腺癌が、去勢抵抗性前立腺癌であることを特徴とする請求項23に記載の方法。
【請求項26】
前記前立腺癌が、前立腺腫瘍を含むことを特徴とする請求項23に記載の方法。
【請求項27】
前記組成物が、錠剤又はカプセル剤の形態で被験体に投与されることを特徴とする請求項23に記載の方法。
【請求項28】
前記組成物が、約25mg/kgから約50mg/kgの間の化合物(1)を含むことを特徴とする請求項23に記載の方法。
【請求項29】
前記組成物が、約650mgから約3500mgまでの化合物(1)を含むことを特徴とする請求項23に記載の方法。
【請求項30】
前記組成物が、約1950mgの化合物(1)を含むことを特徴とする請求項23に記載の方法。
【請求項31】
前記組成物が、約1300mgの化合物(1)を含むことを特徴とする請求項23に記載の方法。
【請求項32】
前記組成物が、約650mgから約975mgまでの化合物(1)を含むことを特徴とする請求項23に記載の方法。
【請求項33】
前記組成物が、900mgと1950mgの間の化合物(1)を含むことを特徴とする請求項23に記載の方法。
【請求項34】
前記前立腺腫瘍の容積が、前記組成物の投与後、少なくとも30日間減少することを特徴とする請求項26に記載の方法。
【請求項35】
前記前立腺腫瘍の容積が、前記組成物の投与後、少なくとも60日間減少することを特徴とする請求項26に記載の方法。
【請求項36】
前記前立腺腫瘍の容積が、前記組成物の投与後、少なくとも90日間減少することを特徴とする請求項26に記載の方法。
【請求項37】
患者における前立腺特異性抗原(PSA)レベルが、前記組成物の投与後、少なくとも30日間減少することを特徴とする請求項32に記載の方法。
【請求項38】
患者における前立腺特異性抗原(PSA)レベルが、前記組成物の投与後、少なくとも60日間減少することを特徴とする請求項32に記載の方法。
【請求項39】
患者における前立腺特異性抗原(PSA)レベルが、前記組成物の投与後、少なくとも90日間減少することを特徴とする請求項32に記載の方法。
【請求項40】
患者における前立腺特異性抗原(PSA)レベルが、前記組成物の投与後、30から90日の間減少することを特徴とする請求項32に記載の方法。
【請求項41】
患者における前立腺特異性抗原(PSA)レベルが、前記組成物の投与後、少なくとも30日間減少することを特徴とする請求項31に記載の方法。
【請求項42】
患者における前立腺特異性抗原(PSA)レベルが、前記組成物の投与後、少なくとも60日間減少することを特徴とする請求項31に記載の方法。
【請求項43】
患者における前立腺特異性抗原(PSA)レベルが、前記組成物の投与後、少なくとも90日間減少することを特徴とする請求項31に記載の方法。
【請求項44】
患者における前立腺特異性抗原(PSA)レベルが、前記組成物の投与後、30から90日の間減少することを特徴とする請求項31に記載の方法。
【請求項45】
前記前立腺腫瘍の容積が、前記組成物の投与後、少なくとも30日間減少することを特徴とする請求項33に記載の方法。
【請求項46】
前記前立腺腫瘍の容積が、前記組成物の投与後、少なくとも60日間減少することを特徴とする請求項33に記載の方法。
【請求項47】
前記前立腺腫瘍の容積が、前記組成物の投与後、少なくとも90日間減少することを特徴とする請求項33に記載の方法。
【請求項48】
患者における前立腺特異性抗原(PSA)レベルが、前記組成物の投与後、少なくとも30日間減少することを特徴とする請求項33に記載の方法。
【請求項49】
患者における前立腺特異性抗原(PSA)レベルが、前記組成物の投与後、少なくとも60日間減少することを特徴とする請求項33に記載の方法。
【請求項50】
患者における前立腺特異性抗原(PSA)レベルが、前記組成物の投与後、少なくとも90日間減少することを特徴とする請求項33に記載の方法。
【請求項51】
患者における前立腺特異性抗原(PSA)レベルが、前記組成物の投与後、30から90日の間減少することを特徴とする請求項33に記載の方法。
【請求項52】
患者における前立腺特異性抗原(PSA)レベルが、前記組成物の投与後、30日から2年の間減少することを特徴とする請求項33に記載の方法。
【請求項1】
化合物(1):
【化1】
又はその薬学的に許容可能な塩、N-オキシド、活性代謝物、プロドラッグ、又は溶媒和物を含む医薬組成物であって、式中、化合物(1)は、約:
a. 13.1°及び14.1°;
b. 17.2°、18.5°、及び19.1°、及び随意に約16.2°、及び29.6°;
c. 12.5°、14.8°、及び25.5°;又は
d. 14.4°、16.1°、19.1°、及び19.3°
の角度 2θにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられた、微粉化結晶形態であることを特徴とする医薬組成物。
【請求項2】
化合物(1)が、被験体への投与後に、アンドロゲン受容体媒介性の疾患又は疾病を処置するのに有効な量で存在することを特徴とする請求項1に記載の医薬組成物。
【請求項3】
前記アンドロゲン受容体媒介性の疾患又は疾病が、前立腺癌、良性前立腺肥大症、多毛症、脱毛症、神経性食欲不振、乳癌、及び男性の性腺機能亢進から成る群から選択されることを特徴とする請求項2に記載の医薬組成物。
【請求項4】
前記アンドロゲン受容体媒介性の疾患又は疾病が、前立腺癌であることを特徴とする請求項3に記載の医薬組成物。
【請求項5】
前記前立腺癌が、去勢抵抗性前立腺癌であることを特徴とする請求項4に記載の医薬組成物。
【請求項6】
化合物(1)が、被験体への投与後に、アンドロゲン生合成を阻害し、アンドロゲン受容体シグナル伝達を阻害し、及びアンドロゲン受容体感受性を低下させるのに有効な量で存在することを特徴とする請求項1に記載の医薬組成物。
【請求項7】
前記化合物が、アンドロゲン受容体シグナル伝達を阻害するか、又はアンドロゲン受容体感受性を減少させることを特徴とする請求項6に記載の医薬組成物。
【請求項8】
前記アンドロゲン生合成阻害が、チトクロームC17α-ヒドロキシラーゼ/C17,20-リアーゼ(CYP17)の活性を阻害することを含むことを特徴とする請求項6に記載の医薬組成物。
【請求項9】
前記アンドロゲン受容体シグナル伝達阻害が、テストステロン結合の競合的阻害を含むことを特徴とする請求項6に記載の医薬組成物。
【請求項10】
前記アンドロゲン受容体感受性の減少が、細胞内のアンドロゲン受容体タンパク質の含量の減少、及び低レベルのアンドロゲンの増殖シグナルによって持続されるべき、細胞の低下した能力を含むことを特徴とする請求項6に記載の医薬組成物。
【請求項11】
前記組成物が、被験体に、非経口的に、静脈内に、筋肉内に、皮内に、皮下に、腹腔内に、経口的に、口腔に、舌下に、粘膜に、直腸に、経皮的に、皮膚に、眼に、又は吸入によって投与するために調剤されることを特徴とする請求項1に記載の医薬組成物。
【請求項12】
前記組成物が、錠剤、カプセル剤、クリーム、ローション剤、油剤、軟膏、ゲル、ペースト、粉末、懸濁液、エマルション、又は溶液として被験体に投与するために調剤されることを特徴とする請求項1に記載の医薬組成物。
【請求項13】
前記組成物が、カプセル剤として被験体に投与するために調剤されることを特徴とする請求項1に記載の医薬組成物。
【請求項14】
前記組成物が、錠剤として被験体に投与するために調剤されることを特徴とする請求項1に記載の医薬組成物。
【請求項15】
前記組成物が、被験体に投与するために調剤され、約25mg/kgから約50mg/kgの間の化合物(1)を含むことを特徴とする請求項1に記載の医薬組成物。
【請求項16】
前記組成物が、約650mgから約3500mgまでの化合物(1)を含むことを特徴とする請求項1に記載の医薬組成物。
【請求項17】
前記組成物が、約1950mgの化合物(1)を含むことを特徴とする請求項1に記載の医薬組成物。
【請求項18】
前記組成物が、約1300mgの化合物(1)を含むことを特徴とする請求項1に記載の医薬組成物。
【請求項19】
前記組成物が、約650mg又は約975mgの化合物(1)を含むことを特徴とする請求項1に記載の医薬組成物。
【請求項20】
前記組成物が、被験体に、1日当たり、1、2、3、4、5、6、7、8、9、又は10回投与するために調剤されることを特徴とする請求項1に記載の医薬組成物。
【請求項21】
前記組成物が、1以上の薬学的に許容可能な賦形剤を更に含むことを特徴とする請求項1に記載の医薬組成物。
【請求項22】
前記薬学的に許容可能な賦形剤が、充填剤、崩壊剤、潤滑剤、界面活性剤、流動促進剤、結合剤、砂糖、スターチ、ニス、又はワックスを含むことを特徴とする請求項21に記載の医薬組成物。
【請求項23】
被験体において前立腺癌に対する処置を提供する方法であって、該方法は、治療上有効な量の化合物(1):
【化2】
又はその薬学的に許容可能な塩、N-オキシド、活性代謝物、プロドラッグ、又は溶媒和物を含む医薬組成物を被験体に投与する工程を含み、式中、化合物(1)は、約:
a. 13.1°及び14.1°;
b. 17.2°、18.5°、及び19.1°、及び随意に約16.2°、及び29.6°;
c. 12.5°、14.8°、及び25.5°;又は
d. 14.4°、16.1°、19.1°、及び19.3°
の角度 2θにおいて表される、特性ピークを有する粉末X線回折パターンによって特徴付けられた、微粉化結晶形態であることを特徴とする方法。
【請求項24】
前記被験体が、ヒトであることを特徴とする請求項23に記載の方法。
【請求項25】
前記前立腺癌が、去勢抵抗性前立腺癌であることを特徴とする請求項23に記載の方法。
【請求項26】
前記前立腺癌が、前立腺腫瘍を含むことを特徴とする請求項23に記載の方法。
【請求項27】
前記組成物が、錠剤又はカプセル剤の形態で被験体に投与されることを特徴とする請求項23に記載の方法。
【請求項28】
前記組成物が、約25mg/kgから約50mg/kgの間の化合物(1)を含むことを特徴とする請求項23に記載の方法。
【請求項29】
前記組成物が、約650mgから約3500mgまでの化合物(1)を含むことを特徴とする請求項23に記載の方法。
【請求項30】
前記組成物が、約1950mgの化合物(1)を含むことを特徴とする請求項23に記載の方法。
【請求項31】
前記組成物が、約1300mgの化合物(1)を含むことを特徴とする請求項23に記載の方法。
【請求項32】
前記組成物が、約650mgから約975mgまでの化合物(1)を含むことを特徴とする請求項23に記載の方法。
【請求項33】
前記組成物が、900mgと1950mgの間の化合物(1)を含むことを特徴とする請求項23に記載の方法。
【請求項34】
前記前立腺腫瘍の容積が、前記組成物の投与後、少なくとも30日間減少することを特徴とする請求項26に記載の方法。
【請求項35】
前記前立腺腫瘍の容積が、前記組成物の投与後、少なくとも60日間減少することを特徴とする請求項26に記載の方法。
【請求項36】
前記前立腺腫瘍の容積が、前記組成物の投与後、少なくとも90日間減少することを特徴とする請求項26に記載の方法。
【請求項37】
患者における前立腺特異性抗原(PSA)レベルが、前記組成物の投与後、少なくとも30日間減少することを特徴とする請求項32に記載の方法。
【請求項38】
患者における前立腺特異性抗原(PSA)レベルが、前記組成物の投与後、少なくとも60日間減少することを特徴とする請求項32に記載の方法。
【請求項39】
患者における前立腺特異性抗原(PSA)レベルが、前記組成物の投与後、少なくとも90日間減少することを特徴とする請求項32に記載の方法。
【請求項40】
患者における前立腺特異性抗原(PSA)レベルが、前記組成物の投与後、30から90日の間減少することを特徴とする請求項32に記載の方法。
【請求項41】
患者における前立腺特異性抗原(PSA)レベルが、前記組成物の投与後、少なくとも30日間減少することを特徴とする請求項31に記載の方法。
【請求項42】
患者における前立腺特異性抗原(PSA)レベルが、前記組成物の投与後、少なくとも60日間減少することを特徴とする請求項31に記載の方法。
【請求項43】
患者における前立腺特異性抗原(PSA)レベルが、前記組成物の投与後、少なくとも90日間減少することを特徴とする請求項31に記載の方法。
【請求項44】
患者における前立腺特異性抗原(PSA)レベルが、前記組成物の投与後、30から90日の間減少することを特徴とする請求項31に記載の方法。
【請求項45】
前記前立腺腫瘍の容積が、前記組成物の投与後、少なくとも30日間減少することを特徴とする請求項33に記載の方法。
【請求項46】
前記前立腺腫瘍の容積が、前記組成物の投与後、少なくとも60日間減少することを特徴とする請求項33に記載の方法。
【請求項47】
前記前立腺腫瘍の容積が、前記組成物の投与後、少なくとも90日間減少することを特徴とする請求項33に記載の方法。
【請求項48】
患者における前立腺特異性抗原(PSA)レベルが、前記組成物の投与後、少なくとも30日間減少することを特徴とする請求項33に記載の方法。
【請求項49】
患者における前立腺特異性抗原(PSA)レベルが、前記組成物の投与後、少なくとも60日間減少することを特徴とする請求項33に記載の方法。
【請求項50】
患者における前立腺特異性抗原(PSA)レベルが、前記組成物の投与後、少なくとも90日間減少することを特徴とする請求項33に記載の方法。
【請求項51】
患者における前立腺特異性抗原(PSA)レベルが、前記組成物の投与後、30から90日の間減少することを特徴とする請求項33に記載の方法。
【請求項52】
患者における前立腺特異性抗原(PSA)レベルが、前記組成物の投与後、30日から2年の間減少することを特徴とする請求項33に記載の方法。
【図1】

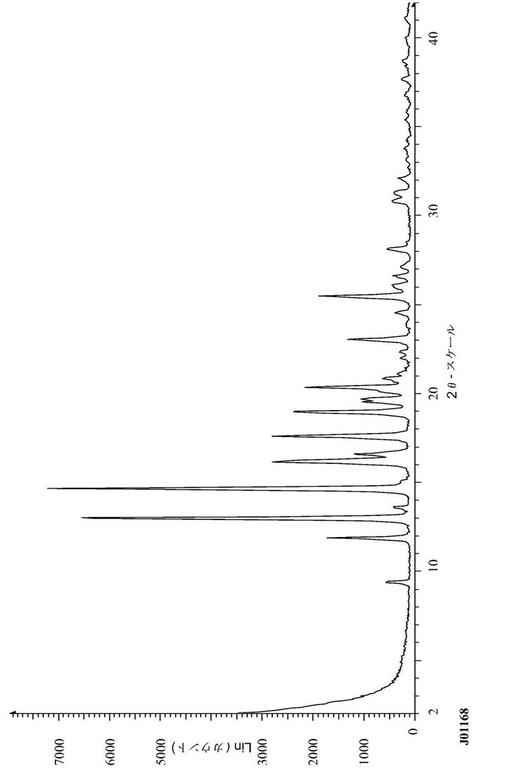
【図2】
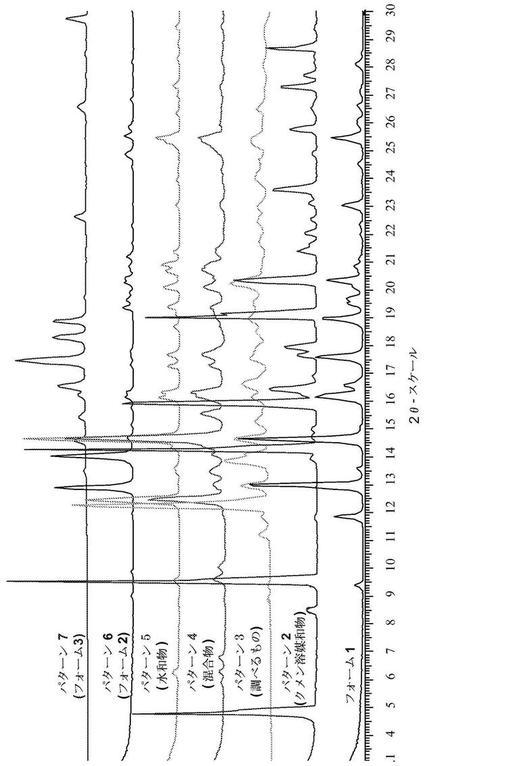
【図3】
【図4】
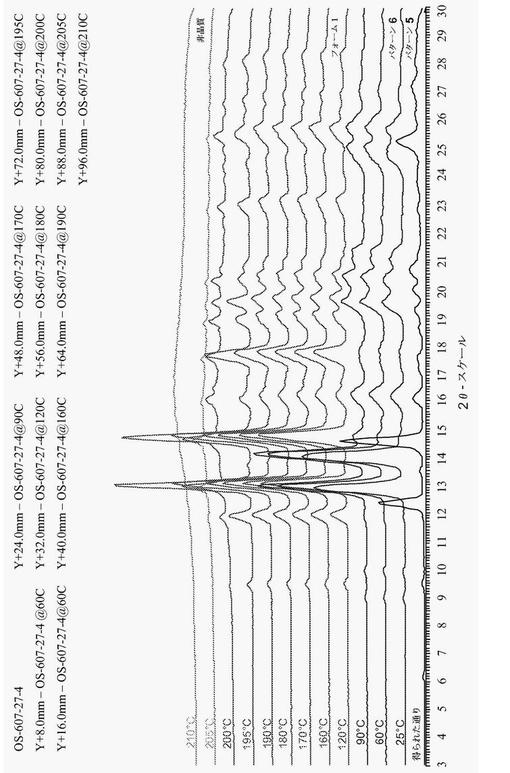
【図5】
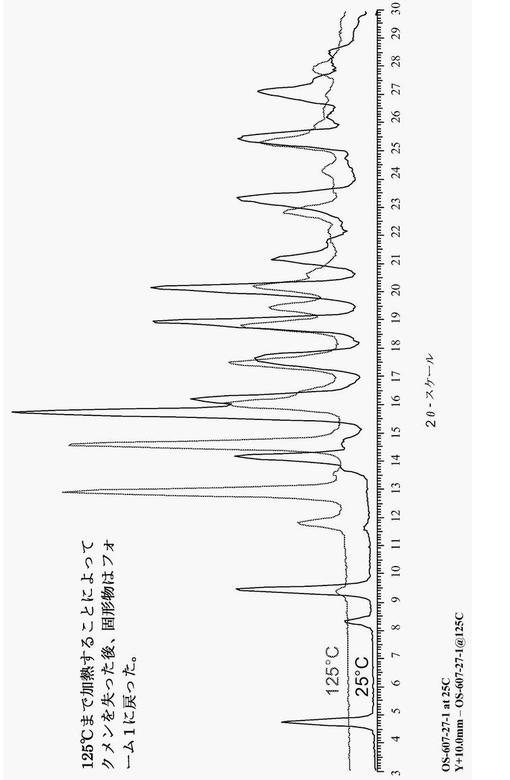
【図6】
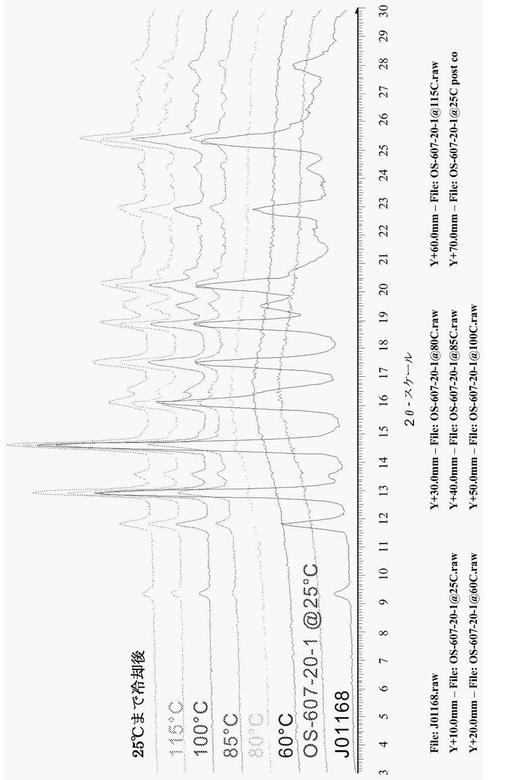
【図7】
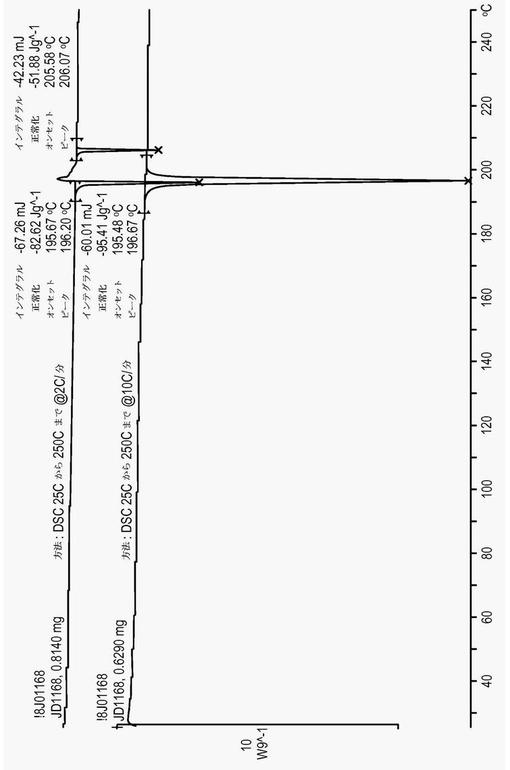
【図8】

【図9】
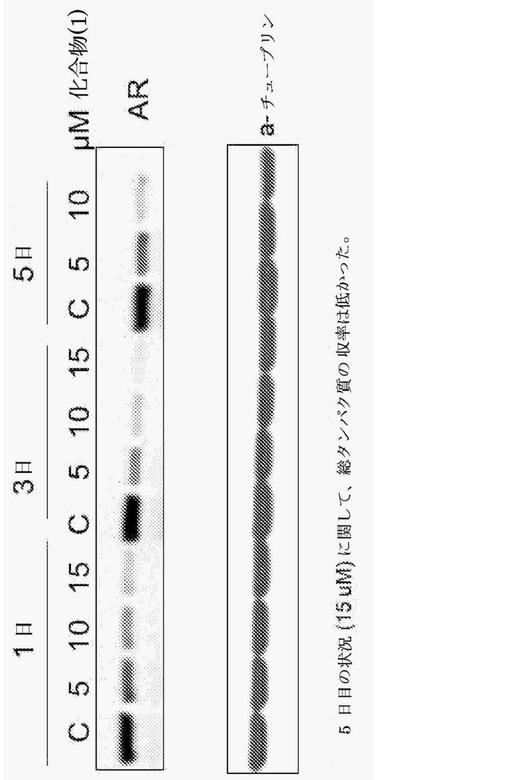
【図10】
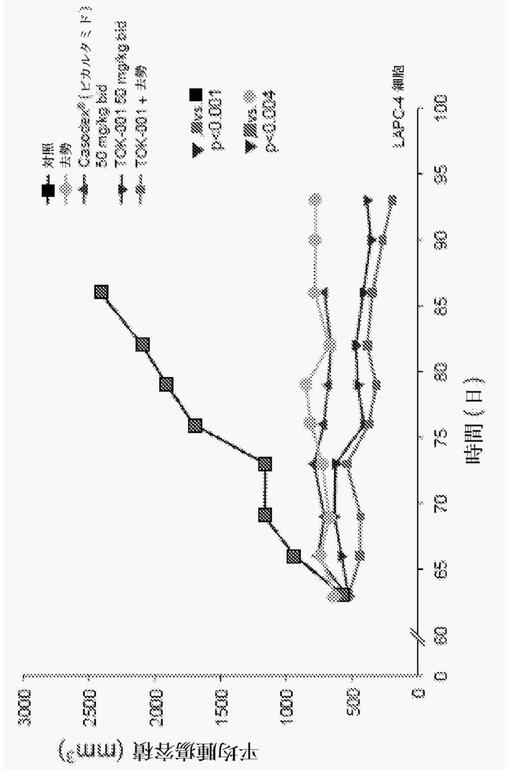
【図11】
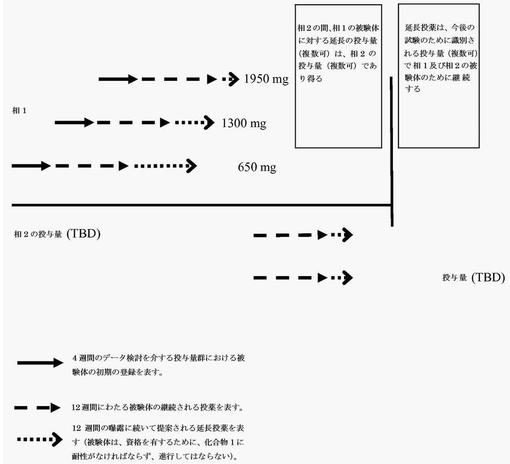
【図12】
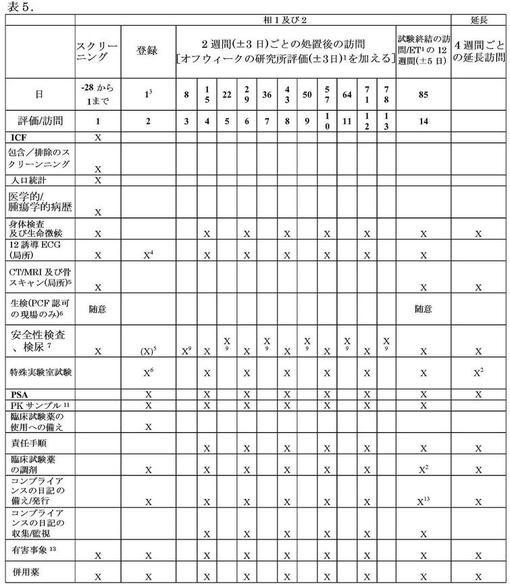
【図13】
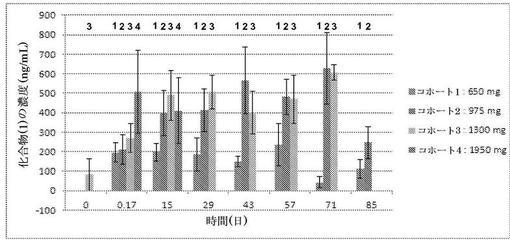
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【公表番号】特表2013−501722(P2013−501722A)
【公表日】平成25年1月17日(2013.1.17)
【国際特許分類】
【出願番号】特願2012−523949(P2012−523949)
【出願日】平成22年8月5日(2010.8.5)
【国際出願番号】PCT/US2010/044570
【国際公開番号】WO2011/017534
【国際公開日】平成23年2月10日(2011.2.10)
【出願人】(511187720)トーカイ ファーマシューティカルズ,インク. (3)
【Fターム(参考)】
【公表日】平成25年1月17日(2013.1.17)
【国際特許分類】
【出願日】平成22年8月5日(2010.8.5)
【国際出願番号】PCT/US2010/044570
【国際公開番号】WO2011/017534
【国際公開日】平成23年2月10日(2011.2.10)
【出願人】(511187720)トーカイ ファーマシューティカルズ,インク. (3)
【Fターム(参考)】
[ Back to top ]