
創傷を治癒するため制御された酸化窒素放出を提供する超高分子ポリマー複合体
一般式(1)(式中、R1はアルコキシ基で無置換又は置換されているアルカンであり;R2は低級アルカンであり;R3及びR4は任意に置換されていてもよい長鎖アルカンであり;Wは水素結合受容官能基含有実体であり;Yはカルボン酸エステル又はアミドであり;Zは連結基であり;T1、T2、T3及びT4は末端基であり;m1、m2、m3、n1及びn2は少なくとも25から選択される整数であり;かつPは約1×103〜1×107の分子量を有し、Qは約1×103〜1×107の分子量を有する)の生体接着性超高分子複合体。この複合体は、局所送達システムにおける従来技術の化合物より長い期間にわたって制御された酸化窒素放出を与える。複合体を含む新規組成物、複合体の調製方法、レイヤーバイレイヤーアセンブリコーティング、電気紡糸及び超音波噴霧などの装置、複合体を含む皮膚被覆物並びに複合体の医療用途を開示する。
【発明の詳細な説明】
【技術分野】
【0001】
(関連出願)
この出願は、発明の名称が「創傷を治癒するため制御された酸化窒素放出を提供する超高分子複合体」である2007年8月27日提出のカナダ特許出願第2,599,082号の優先権を主張する。その内容を参照によって本明細書に引用したものとする。
この発明は、超高分子(supramacromolecular)酸化窒素放出ポリマー複合体;前記複合体を含む組成物及び含浸物品及び被覆物品;前記複合体の製造方法;並びに創傷、特に糖尿病に起因する立方状潰瘍形成を治癒する処置における前記複合体、組成物及び物品の使用方法に関する。
【背景技術】
【0002】
酸化窒素(NO)がとらえどころのない内皮由来弛緩因子と同一であるという発見以来[1]、NOの多くのさらに難解な生物学的役割が同定かつ解明されている[2〜6]。これらの知見が、創傷治癒、心血管疾患、呼吸器疾患、癌療法、神経系再構築、並びに新機能性医療デバイスにおける外因性NOの潜在的応用のさらなる探索を促した。これに関しては、局所適用したNOガスを用いた創傷感染の治療で実証された成功で明らかなように、NOの局所送達は臨床用途で利益を得る大きな可能性がある[7]。しかし、この小さい気体分子の短い半減期及びその内在する不安的性が、その医薬品剤形及び薬物送達システムに組み入れるための大きな課題を提示した。血管内皮細胞によって内因的に合成されたNOは5秒以下という非常に短い生物学的半減期を有すると報告されている[8、9]。NOはヘモグロビンによって急速に捕捉されるので、組織内のその作用部位は、それが発生されるところに局在化されるだろう。細胞及び組織内におけるNOの化学的な不安定性は、NO2-及びNO3-の両方へのNOの急速な酸化に起因している。
NOの周知起源である有機ニトラート及び亜硝酸ナトリウムのほかに、広範に研究されているNO前駆物質の2つの他ファミリーがある。1つはジアゼニウムジオラートから成り、他方はS-ニトロソチオールから成る。ジアゼニウムジオラートはNONOアート(NONOate)としても知られる構造R1R2NN(O)=NOR3の化合物を包含する。この分類のNOドナーに基づくNO放出物質の開発に多大な努力が払われている[10、11]。これらは、物理的ブレンド又はポリマー骨格若しくは側鎖への共有結合のどちらかによるジアゼニウムジオラートの異なるポリマーマトリックスへの組込みを包含する。ジアゼニウムジオラートに関する関連従来技術について以下に述べる。
【0003】
WO 2005/011575、WO 2005/07008、及びWO 2006/058318において、Smithは、ジアゼニウムジオラート成分がイオン結合又は共有結合のどちらかでポリマーマトリックスに結合している、イオン交換樹脂又はポリエチレンイミン(PEI)繊維の多積層物のどちらかに基づくMO放出デバイスを開示した。活性化又は創傷への適用時に、該NO誘導ポリマーが水、水素カチオン又はアスコルビン酸などのアクチベーターと接触すると、局所的なNO放出が生じうる。しかし、このようなシステムからのNO放出の持続時間は短く、典型的にイオン交換樹脂システムからはたった0.5〜3時間しか持続せず、繊維の多積層物デバイスからは多くても1〜2日持続するだけである。
Meyerhoff及び共同研究者らは、US 6,841,166及びUS 2006/0008529において、酸性アクチベーター及び可塑剤と共に、ジアゼニウムジオラート誘導体化ヒュームドシリカ、分散したジアゼニウムジオラート又は共有結合したジアゼニウムジオラートなどの分離したNOドナーを含有する疎水性ポリマー(例えばシリコーンゴム、ポリ(塩化ビニル)、ポリウレタン等)に基づく抗血栓性血液接触デバイス用のNO放出ポリマー材料を開示した。活性化中、水がゆっくり疎水性ポリマーマトリックス中に浸透し、数日まで水性環境にNOを持続的に放出することとなる。これらのシステムは、心血管の再狭窄及び血液循環障害の治療のためのNO送達用の埋め込み型グラフト、カテーテル又は生物医学的デバイス上コーティングとしても試験されている[12〜15]。生体適合性の懸念に加え、これらの極端に疎水性の材料は、その不十分な吸水性及び創傷部位における不十分な生体接着のため、創傷治癒適用には適さない。
さらに、この分類のNONOアートドナーのin vivo適用における1つの主な制限は、米国特許第6,841,166号で明らかにされているように、浸出しうるジアゼニウムジオラート及びその分解生成物、特にニトロソアミンの毒性の可能性である。上記従来技術のアプローチ及び米国特許第6,703,046号は、疎水性ポリマーを利用して、このような浸出を最小限にしていた。しかし、親水性の酸性添加剤及び可塑剤を含有するこれらのポリマーからはまだ浸出が起こりうる。さらに、1つの確立したジアゼニウムジオラートプロドラッグ、V-PYRRO/NOは、既知の最も強力な実験上の肝癌誘発物質の1つであるN-ニトロソピロリジンを形成する可能性がある[16]。さらに、水性媒体中に放出されたジアミンベース及びポリエチレンイミンベースジアゼニウムジオラートは、測定可能レベルの、既知分類の発癌物質であるニトロソアミンを形成することが分かっている[12]。従って、特に創傷治癒のためのジアゼニウムジオラートのin vivo適用は限界があると思われる。
【0004】
別の主分類のNOドナーは、一般構造R-SNOを有する化合物であるS-ニトロソチオールである。NOの重要な内因性及び外因性起源として、RSNOは広く生体内に分布しており、哺乳類の体内で酸化窒素を貯蔵、輸送及び放出することが分かっている[17]。さらに、生理液内で水によって活性化するとNOを発生するその能力は、全身に負担をかける必要なく、特異的組織だけを標的にするNOの局所送達に特に有利である。種々の内因性RSNOのうち、S-ニトロソグルタチオン(GSNO)は、グルタチオンと亜硝酸ナトリウムの間の室温での自発的反応によるその合成の容易さ及び固体として単離できることのため、非常に注目を集めている[18]。しかし、これらの小分子RSNOの安定性は、S-NO結合が熱分解にも光分解にも不安定であり、かつNOの自発的放出及びその急速な不活性化につながる溶血開裂しやすいことから不十分であり、そのため創傷治癒といった実際の適用に対するその適合性を制限している。
de Oliveira及び共同研究者らは、経皮NO送達のため、ポリ(ビニルアルコール)、ポリ(ビニルピロリドン)、又はPluoronic F127ヒドロゲル等の水溶性ポリマーに基づいたフィルム及びゲル中にS-ニトロソグルタチオン(GSNO)及び/又はS-ニトロソ-N-アセチル-システイン(SNAC)を物理的に組み入れた[19〜22]。彼らの動物結果は、ラットの皮膚創傷回復の初期段階中のGSNO含有ヒドロゲルの反復適用が創傷閉鎖及び再上皮化を加速することを示す[23]。しかし、反復適用を避けるための患者のコンプライアンスの観点から、長期のNO放出がさらに望ましいだろう。
Katsumi及び共同研究者らは、亜硝酸塩をウシ血清アルブミン(BSA)上のシステイン残渣に共有結合させることによって、BSA及びポリ(エチレングリコール)(PEG)結合BSAに基づいた超高分子担体S-ニトロソチオールを合成した[24、25]。同様に、Westらは、米国特許第7,052,711号において、ポリ(エチレングリコール)ヒドロゲル内に固定化されたS-ニトロソシステイン(CysNO)が、in vitro細胞培養における血小板接着及び平滑筋細胞増殖を減少させることを実証した。しかし、これらの報告された親水性システムは、S-NO結合が熱分解にも光分解にも不安定であり、かつNOの自発的放出及びその急速な不活性化につながる溶血開裂しやすいことから、望ましい安定性を欠く。結果として、従来技術の化合物からの酸化窒素放出持続時間は、如何なる長期間をも維持され得ず、一般的に、多くて数時間である。
WO2006/095193に記載されているように適用時にin situ又はWO2008/031182に記載されているように創傷へのその適用前にin vitroでポリマー中でGSNOを物理的に混合して混合物を形成する方法[21〜24]及びNO前駆物質をアクチベーターと混合してGSNOを発生させる従来技術の方法は、一旦GSNOが形成又は放出されると、それはまだ熱、水分及び光による分解に感受性なので、GSNOの短い半減期という問題に対処していない。実際に、ほとんどのこれらの従来技術のアプローチでは、NO又はGSNOの放出は、一般的に非常に速く、せいぜい数時間しか持続しないので、繰返し適用する必要がある。
【0005】

【発明の概要】
【発明が解決しようとする課題】
【0006】
従って、当技術分野では、創傷治癒適用のためNOの制御可能かつ持続的な放出を提供する安定したNO送達システムを達成することが要望されている。
【課題を解決するための手段】
【0007】
従って、一態様では、本発明は、物理的に架橋したポリマーネットワーク内で安定化した固定化RSNOを含む超高分子複合体に基づいた新分類のNO送達システムに関する。このシステムでは、担体ポリマーに共有結合したRSNO前駆物質が、第2のポリマーとの分子間複合体形成によって、好ましくは水素結合相互作用を介して安定化される。結果として生じる超高分子複合体は、連続的かつ長期にわたる、貯蔵安定性が改良されたNO放出を提供することができる。ここで、用語「超高分子」を用いて、的確な3D構造化非共有結合高分子を含む分子集合を表す[26]。
さらなる態様では、この発明は、(1)異なるNO装填レベルのRSNO;(2)側鎖内に、RSNO上のアミン基と反応することができる酸無水物官能基を有するポリマーA;及び(3) ポリマーAと強力な水素結合を形成することができるプロトン受容基を骨格内又は側鎖内に含むポリマーB;の付加体を含む医薬組成物をも提供する。
さらなる態様では、本発明は、前記NO放出複合体の製造方法;及び前記複合体の使用方法にも関する。
本発明は、強力な高分子間相互作用によるレイヤーバイレイヤーアセンブリ(layer-by-layer assembly)によって前記NO放出複合体をコーティングにする方法をさらに提供する。
なおさらなる態様では、この発明は、粉末、微粒子、繊維及びフィルムなどの多様な形態の該NO放出超高分子複合体の調製方法を提供する。特に、この新規酸化窒素放出ポリマー複合体を創傷治療用の手当用品及び包帯に組み入れることができ、慢性の治癒不十分な創傷の治療に適した持続性かつ制御された様式で治療量の酸化窒素の放出をもたらす。
この発明は、有効にNO装填する能力及び有意に改良された安定性を示す新規NO前駆物質としての広範なGSNO誘導RSNOの利用にも関する。
さらに、この発明は、慢性創傷の治療方法をも提供する。本NO放出超高分子複合体は、糖尿病動物モデルにおいて加速された創傷治癒を示した。
なおさらに、この発明は、局所組織及び医療デバイス上のコーティングに直接適用できる制御された持続性様式で治療レベルのNOを発生させるための新規プラットフォームを提示する。
ほとんどの小分子NOドナーは水性媒体中で化学的に不安定である。例えば、図1から、室内条件下で酸性及び中性媒体中では、GSNOからNOが連続的に解離することが分かる。本発明は、RSNOを高分子担体に付着させて、ポリマーNO前駆物質又はプロドラッグを形成することによって、RSNOの半減期を延長するための有用な方法を提供する。予想外に、前記ポリマーNOプロドラッグを別のポリマーとの分子間水素結合相互作用によって物理的に架橋することによって、結果として生じたる超高分子複合体が、さらに改良されたRSNO貯蔵安定性で連続的かつ長期にわたるNO発生を提供できることが分かった。さらに、水和すると、本発明の超高分子NO放出複合体は、生体接着性にもなり、そのためNOの局所的制御送達を容易にする。
NO発生超高分子を得るため、全ての上記反応が非常に迅速に起こり、かつ関与する全ての有機溶媒を容易に除去できることが望ましい。
【0008】
本発明の目的は、創傷の治癒において酸化窒素の永続的放出を生じさせるための簡単な安定した生体適合性手段を提供する酸化窒素担体を提供することである。
前記酸化窒素担体の製造方法を提供することがさらなる目的である。
固体剤形は、貯蔵及び輸送中に水性剤形より高い安定性を示すので、粉末、フィルム、繊維、ミクロスフェア又はコーティング等のいくつかの物理的形態の形で前記酸化窒素担体を提供することがさらなる目的である。本システムは、多くの点で従来技術のポリマー及びゲルシステムより優れる。
本発明は、疎水性生体接着性ポリマーポリ酸無水物(引き続き第2のポリマーとの分子間水素結合を形成できる)に共有結合した酸化窒素ドナーの生成物を含む生体接着性超高部分子複合体を提供する。
従って、一態様では、本発明は、下記一般式の生体接着性超高分子複合体を提供する。
【0009】
【化1】
【0010】
式中、R1は無置換又はアルコキシ基で置換されているアルカンであり;R2は低級アルカンであり;R3及びR4は任意に置換されていてもよい長鎖アルカンである。構造の多様性に関して、式の全ての3つのR基を広い範囲にわたって変えて分離できる物質を生成することができ;Wは水素結合受容官能基含有実体(entitiy)であり;Yはカルボン酸エステル又はアミドであり;Zは連結基であり;T1、T2、T3及びT4は末端基であり;m1、m2、m3、n1及びn2は少なくとも25から選択される整数であり;かつPは約1×103〜1×107の分子量を有し、Qは約1×103〜1×107の分子量を有する。
【0011】
超高分子複合体は、好ましくは、R1がマレイン酸コポリマーであり、さらに好ましくは前記マレイン酸コポリマーが、ポリ(メチルビニルエーテル-co-マレイン酸)、ポリ(ビニルピロリドン-co-ジメチルマレイン酸)、ポリ(エチレン-co-マレイン酸)、ポリ(イソブチレン-co-マレイン酸)、ポリ(スチレン-co-マレイン酸)、ポリ(エチレン-co-酢酸エチル-co-マレイン酸)、ポリ(マレイン酸-co-オクタデセン)、ポリエチレン-グラフト-マレイン酸、ポリプロピレン-グラフト-マレイン酸、及びポリイソプレン-グラフト-マレイン酸から成る群より選択される。
さらなる態様では、本発明は、下記一般式の生体接着性超高分子複合体を提供する。
【0012】
【化2】
【0013】
式中、R1は、アルキルビニルエーテル(C1-C5)、エチレン、プロピレン、イソブチレン、ブタジエン、1-オクタデセン、スチレン、マレイン酸、又は無水マレイン酸単位であり;W1及びW2は、ビニルピロリドン、エチレンオキシド又はプロピレンオキシド、酢酸ビニル、アルコキシル置換グルコピラノース、グルコサミン、及びアセチルグルコサミンから成る群より選択される水素結合受容官能基含有実体であり;R2はH、脂肪酸エステル、又は脂肪アルコールであり;Xはカルボン酸エステル又はアミド結合であり;RSNOは、システイン、γ-Glu-Cys、α-Glu-Cys、グルタチオン、ホモグルタチオン、グルタチオンエチルエステル、ヒドロキシメチル-グルタチオン、γ-Glu-Cys-Glu、α-Glu-Cys-Gly、α-Glu-Cys-β-Ala、α-Glu-Cys-Ser、α-Glu-Cys-Glu、-SH及び-NH2及び/又は-OH官能基を含有する他のグルタチオン類似体、或いは下記ペプチドの1つ:(γ-Glu-Cys)q、(γ-Glu-Cys)q-Gly、(γ-Glu-Cys)q-β-Ala、(γ-Glu-Cys)q-Ser、(γ-Glu-Cys)q-Glu、(α-Glu-Cys)q、(α-Glu-Cys)q-Gly、(α-Glu-Cys)q-β-Ala、(α-Glu-Cys)q-Ser、及び(α-Glu-Cys)q-Glu(ここで、q=2〜11)のS-ニトロソチオールであり;T1及びT2は末端基であり;m、n及びpは25より大きい整数である。
【0014】
超高分子複合体は、好ましくは、T1-[-R1-CH(COOH)-CH(X-RSNO)-]m-T2が、RSNOと無水マレイン酸ポリマー又はコポリマーの反応付加体であり、前記無水マレイン酸ポリマー又はコポリマーが、ポリ(メチルビニルエーテル-alt-無水マレイン酸)、ポリ(マレイン酸-co-無水マレイン酸)、ポリ(無水マレイン酸)、ポリ(ビニルピロリドン-co-ジメチル無水マレイン酸)、ポリ(酢酸ビニル-co-無水マレイン酸)、ポリ(エチレン-alt-無水マレイン酸)、ポリ(イソブチレン-alt-無水マレイン酸)、ポリ(スチレン-alt-無水マレイン酸)、ポリ(エチレン-co-酢酸エチル-co-無水マレイン酸)、及びポリ(無水マレイン酸-alt-1-オクタデセン)から成る群より選択される。
本発明では、無水マレイン酸含有ポリマーを利用して、RSNO、好ましくはGSNOを、ペンダント酸無水物基とGSNOの一級アミン基との相互作用によって固体化する。一般的に温和な条件下における無水マレイン酸含有ポリマーの反応性のため該ポリマーは生理活性剤の固定化に適している[27〜29]。例えばUS2001/0046476では、殺菌剤、フレーバー剤及びエッセンシャルオイルをポリ(メチルビニルエーテル-alt-無水マレイン酸)(PVMMA)及びその誘導体に共有結合させて徐放性オーラルケア組成物を提供している。
NOの効率的な局所送達を果たすため、本NO放出超高分子複合体の創傷部位における滞留時間を効率的に長くするその顕著な生体接着特性に鑑み、PVMMAをNO担体として利用することが非常に有利であろう。PVMMAの疎水性及びその表面浸食特性は、長期にわたるNO放出の達成を容易にするだろう。実際に、PVMMAとその変性誘導体は、歯科用接着剤、化粧品及び薬物送達システムで多くの適用を見出した[30〜31、米国特許第6,355,706号、US2007/196459、WO 2006/015093、WO 2001/087276]。
酸化窒素ドナーRSNOは、好ましくは、システイン、γ-Glu-Cys、α-Glu-Cys、グルタチオン(GSH)、グルタチオンエチルエステル、ホモグルタチオン、ヒドロキシメチル-グルタチオン、γ-Glu-Cys-Glu、α-Glu-Cys-Gly、α-Glu-Cys-β-Ala、α-Glu-Cys-Ser、α-Glu-Cys-Glu、-SH及び-NH2及び/又は-OH官能基を含有する他のグルタチオン類似体、或いは下記ペプチドの1つ:(γ-Glu-Cys)n、(γ-Glu-Cys)n-Gly(フィトケラチンとしても知られる)、(γ-Glu-Cys)n-β-Ala、(γ-Glu-Cys)n-Ser、(γ-Glu-Cys)n-Glu、(α-Glu-Cys)n、(α-Glu-Cys)n-Gly、(α-Glu-Cys)n-β-Ala、(α-Glu-Cys)n-Ser、及び(α-Glu-Cys)n-Glu(ここで、n=2〜11)のS-ニトロソチオールから成る群より選択される。
T3-[R2W.]n1-[R3]n2-T4及び[W1]n-[W2]n-R2水素結合受容ポリマーは、好ましくは、ポリ(ビニルピロリドン)、ポリエチレングリコール、ポリ(エチレンオキシド)、ポリ(ビニルピロリドン-co-酢酸ビニル)、ポリエチレンオキシド-ポリプロピレンオキシドブロックコポリマー(プルロニック又はポロキサマー)、ポリエチレングリコール脂肪アルコールエステル、ポリエチレングリコール脂肪酸エステル、エチルセルロース、及びキトサンから成る群より選択され、さらに好ましくはポリ(ビニルピロリドン)である。
好ましくは、Y.Z.SNOは、アミノ-S-ニトロソグルタチオン又はアミド-フィトケラチンである。
【0015】
さらなる態様では、本発明は、生体接着性超高分子酸化窒素発生可能ポリマー複合体の製造方法であって、以下の工程
i. アミノ連結基を有するS-ニトロソ化合物を生体接着性疎水性ポリ酸無水物化合物と共有結合させて酸化窒素ドナーポリマー担体を形成する工程;及び
ii. 前記担体を親水性分子間水素結合受容ポリマーと混合して前記超高分子酸化窒素発生可能複合体を生成する工程
を含む方法を提供する。
好ましい酸化窒素ドナーRSNOは、システイン、γ-Glu-Cys、α-Glu-Cys、グルタチオン(GSH)、ホモグルタチオン、ヒドロキシメチル-グルタチオン、γ-Glu-Cys-Glu、α-Glu-Cys-Gly、α-Glu-Cys-β-Ala、α-Glu-Cys-Ser、α-Glu-Cys-Glu、-SH及び-NH2及び/又は-OH官能基を含有する他のグルタチオン類似体、或いは下記ペプチドの1つ:(γ-Glu-Cys)n、(γ-Glu-Cys)n-Gly(フィトケラチンとしても知られる)、(γ-Glu-Cys)n-β-Ala、(γ-Glu-Cys)n-Ser、(γ-Glu-Cys)n-Glu、(α-Glu-Cys)n、(α-Glu-Cys)n-Gly、(α-Glu-Cys)n-β-Ala、(α-Glu-Cys)n-Ser、及び(α-Glu-Cys)n-Glu(ここで、n=2〜11)のS-ニトロソチオールから成る群より選択される。最も好ましくは、S-ニトロソチオール化合物はGSNO又はフィトケラチンである。
好ましいポリ酸無水物化合物は、分子量(Mw)が約5,000〜2,000,000の範囲の無水マレイン酸ポリマー又はコポリマーであり、前記無水マレイン酸ポリマー又はコポリマーは、例えば、好ましくはポリ(メチルビニルエーテル-alt-無水マレイン酸)、ポリ(マレイン酸-co-無水マレイン酸)、ポリ(無水マレイン酸)、ポリ(ビニルピロリドン-co-ジメチル無水マレイン酸)、ポリ(酢酸ビニル-co-無水マレイン酸)、ポリ(エチレン-alt-無水マレイン酸)、ポリ(イソブチレン-alt-無水マレイン酸)、ポリ(スチレン-alt-無水マレイン酸)、ポリ(エチレン-co-アクリル酸エチル-co-無水マレイン酸)、及びポリ(無水マレイン酸-alt-1-オクタデセン)から成る群より選択される。最も好ましくは、ポリ酸無水物化合物はポリ(メチルビニルエーテル-alt-無水マレイン酸)である。
水素結合受容ポリマーは、好ましくは、ポリ(ビニルピロリドン)、ポリエチレングリコール、ポリ(エチレンオキシド)、ポリ(ビニルピロリドン-co-酢酸ビニル)、ポリエチレンオキシド-ポリプロピレンオキシドブロックコポリマー(プルロニック又はポロキサマー)、ポリエチレングリコール脂肪アルコールエステル、ポリエチレングリコール脂肪酸エステル、エチルセルロース、及びキトサンから成る、分子量(Mw)が約5,000〜7,000,000の群から選択され、最も好ましくは前記水素結合受容ポリマーがポリ(ビニルピロリドン)である、請求項17に記載の方法である。
結果として生じる超高分子酸化窒素発生可能ポリマー複合体は、好ましくはポリ酸無水物化合物と水素結合受容ポリマーを1:9〜9:1、さらに好ましくは2:5〜5:2、最も好ましくは1:2〜2:1の相対質量比で含む。
結果として生じる超高分子酸化窒素発生可能ポリマー複合体中の酸化窒素ドナーRSNOの総装填は、好ましくは1〜50wt%、さらに好ましくは1〜30%、最も好ましくは5〜20%の範囲である。
【0016】
本発明は、さらなる態様では、上述した方法で製造された生体接着性超高分子酸化窒素発生可能複合体を提供する。
なおさらなる態様では、本発明は、上述したように、有効な創傷治癒量の前記超高分子複合体と、生理学的に許容しうる担体とを含む医薬組成物を提供する。
なおさらなる態様では、本発明は、上述したように、前記超高分子複合体を加工してコーティングにするためのレイヤーバイレイヤーアセンブリ(layer-by-layer assembly)方法を提供する。
なおさらなる態様では、本発明は、上述したように、前記超高分子複合体を紡糸繊維として製造するための電気紡糸(electrospinning)方法を提供する。
なおさらなる態様では、本発明は、上述したように、前記超高分子複合体をミクロスフェアとして製造するための超音波噴霧方法を提供する。
それによって、本発明は、上述したように、粉末、マイクロカプセル、紡糸繊維、又は基材、例えば、カテーテル若しくはステント表面上のコーティングという物理的形態で超高分子複合体を提供する。
従って、本発明は、粉末形態で、創傷手当用品として役立ち、また、経皮パッチ、包帯、縫合糸などの中に組み入れることができる新規の酸化窒素放出ポリマー複合体を提供する。それは、該ポリマー複合体を、多層方法によって結束させる前に、医療デバイス上の血液接触面に適用することによって、コーティングの形態を取ることもできる。この超高分子複合体は、持続性かつ制御可能様式で治療量の酸化窒素を生成して、それを患部組織、例えば慢性の治癒が不十分な創傷内の当該組織に送達する。
【0017】
従って、さらなる態様では、本発明は、上述した超高分子複合体に基づいて、装填手順中に不織マットを製造するための電気紡糸装置を使用すること又は紡糸プロセス中にコーティング基材を使用することに関する。
従って、さらなる態様では、本発明は、上述した超高分子複合体に基づいた均一サイズのミクロスフェアを製造するための超音波噴霧技術の利用に関する。結果として生じたミクロスフェアをさらにカプセルに組み入れるか又は噴霧プロセス中に基材上にコーティングすることができる。
従って、さらなる態様では、本発明は、皮膚に適用するための皮膚被覆物を提供し、該被覆物は、上述したように、有効な創傷治癒量の超高分子複合体を組み入れている。皮膚被覆物が包帯又は創傷手当用品であってよい。
【0018】
さらなる態様では、本発明は、皮膚の創傷又は感染症の治癒を促進する方法であって、有効な創傷又は感染症治癒量の、上述した生体接着性超高分子複合体又はその医薬的に許容しうる組成物を前記創傷に適用する工程を含む方法を提供する。
なおさらなる態様では、本発明は、上述したように、生体接着性超高分子複合体又はその医薬的に許容しうる組成物の、皮膚の創傷又は感染症の治癒を促進するための使用を提供する。
【0019】
このように、本発明は、3つの必須の主要素、すなわち、(1)疎水性、生体適合性、生体内分解性であり、かつ酸無水物官能基を含むポリマー担体、例えば、ポリ(メチルビニルエーテル-alt-無水マレイン酸)[PVMMA]等、(2)酸化窒素ドナー、例えば温和な条件下で上記ポリマー担体の高分子骨格又は側鎖上の酸無水物基に共有結合することができるS-ニトロソグルタチオン(GSNO)又は他のS-ニトロソ化グルタチオン誘導体、及び(3) 前記第1のポリマー担体と強力な物理的分子間複合体を形成する第2のポリマー、例えば、ポリ(ビニルピロリドン)[PVP]等を含む。
従って、本発明の分野は、創傷及び感染症を治療するため、さらに詳細には、酸化窒素を長期にわたって局所放出して創傷及び感染症を治療するためのデバイス及び方法に関する。本発明の複合体を粉末にして、創傷治癒を促進するための包帯又は創傷手当用品に組み入れることができる。さらに、本発明の複合体を、肺性高血圧を下げるための吸入製剤の成分として配備させるか又は循環障害の治療に適用することができる。
少なくとも約7日間にわたる生体接着性超高分子複合体からの長期の酸化窒素放出は、創傷及び感染症の有効な治療を提供する。理論によって拘束されるものではないが、我々は、この効力は、生体接着性疎水性ポリマー、例えばPVMMAのカルボン酸基を介して水素結合している水素結合受容官能基、例えばPVPの存在のため、ジスルフィド結合の形成及びPVMMA疎水性マトリックス内に埋まっている立体障害RSNOからの酸化窒素の放出の速度を遅くするためであると考える。
【図面の簡単な説明】
【0020】
以下の図面は、本明細書の一部を形成し、かつ本発明の必須態様をさらに実証するために含まれる。本発明をより良く理解してもらうため、例示としてのみ、以下の図面を参照して特定の好ましい実施形態を説明する。
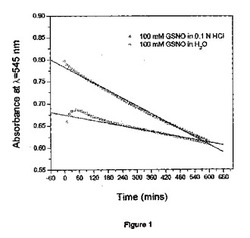
【図1】22℃における酸性及び中性媒体中のGSNOからのNO分解速度論の時間経過を示す。
【図2】GSNO、GSNO-PVMMAのUV-Visスペクトル、UV範囲(a)及び可視範囲(b)内の最大吸収(λ=336nm及びλ=545nmにおけるS-NO結合の特徴的吸光度に相当し、それぞれσ-σ*及びπ-π*電子遷移に割り当てられる。(1)アセトンに溶解した純粋PVMMA;(2)水性媒体中のGSNO;(3)水性媒体中のGSNO結合PVMMA)を示す。
【図3A】グルタチオンエチルエステル(GSHEE)のS-ニトロソ反応のUV-Visスペクトルの特徴づけを示す。
【図3B】グルタチオンエチルエステル(GSHEE)のS-ニトロソ反応のUV-Visスペクトルの特徴づけを示す。
【図4】水性媒体中の(a)純粋PC5(フィトケラチン);(b)PC5と亜硝酸ナトリウムの間のS-ニトロソ化;(c)S-ニトロソPC5結合PVMMA/PVP複合体のUV-Visスペクトルを示す。
【図5】それぞれ22℃におけるGSNO、S-ニトロソ-GSHEE及びS-ニトロソ-PC5からの0.1N HCl中のNO分解速度論を示す。
【図6】純粋GSNO、純粋PVMMA及びGSNO結合PVMMAフィルムのFTIRスペクトルを示す。
【図7】純粋PVMMA、PVP及びPVMMA/PVP複合体のFTIRスペクトルを示す。
【図8】リン酸緩衝食塩水中20mgのGSNO-PVMMA結合体からの37℃(a)及び22℃(b)におけるNOのin vitro放出挙動、並びに16.6wt%のGSNOを含み、PVMMA/PVPの質量比が1:1のGSNO-PVMMA/PVP超高分子複合体からの37℃(c)及び22℃(d)におけるNOのin vitro放出挙動を示す。
【図9】リン酸緩衝食塩水中25℃にて、15.04wt%のGSNOを含み、異なるPVMMA/PVP質量比の20mgのGSNO-PVMMA/PVP超高分子複合体からのNOのin vitro放出挙動を示す。
【図10】リン酸緩衝食塩水中37℃及び25℃にて、15.04wt%のGSNOを含み、PVMMA/PVPの質量比が1:1の20mgのGSNO-PVMMA/PVP超高分子複合体からのNOのin vitro放出挙動を示す。
【図11】リン酸緩衝食塩水中、15.04wt%のGSNOを含み、PVMMA/PVPの質量比が1:1で、異なるPVMMA分子量(a)及びPVP分子量(b)の20mgのGSNO-PVMMA/PVP超高分子複合体からのNOのin vitro放出挙動を示す。
【図12】リン酸緩衝食塩水中室温にて、7.52wt%(231系列)及び15.04wt%(232系列)のGSNOを含み、PVMMA/PVPの質量比が1:1の20mgのGSNO-PVMMA/PVP超高分子複合体からのNOのin vitro放出挙動を示す。
【図13】S-ニトロソPC5-PVMMA/PVP超高分子複合体粉末からのNOのin vitro持続性放出挙動を示す。
【図14】室内条件下で6カ月貯蔵前後のGSNO-PVMMA/PVP超高分子複合体粉末からのNOのin vitro放出挙動を比較する。
【図15】UV照射前後のGSNO-PVMMA/PVP超高分子複合体からのNOのin vitro放出挙動を比較する。
【図16】(a1,a2)12wt%のGSNO-結合純粋PVMMA;(b1,b2)純粋EC;(c1,c2)GSNO-結合PVMMA/EC(ポリマー組成1:1)から電気紡糸されたナノ繊維のそれぞれ低倍率及び高倍率の代表的SEM写真を示す。
【図17】12%GSNO装填の乾燥した1:1のPVMMA/EC電気紡糸ファブリックの、浸漬前(a)PBSに37℃で1時間浸漬後(b);1日浸漬後(c)及び3日浸漬後(d)のSEM写真である。
【図18】紡いだままの(a)純粋PVMMA、(b)GSNO結合PVMMA/EC、(c)GSNOなしのPVMMA/EC及び(d)純粋ECナノコンポジットファブリックのFTIRスペクトルである。
【図19】紡いだままの繊維質マット((a)純粋PVMMA、(b)純粋EC;(c)GSNOなしのPVMMA/EC(1:1)及び(d)12wt%GSNO結合PVMMA/EC(1:1))の機械的性質を示す。
【図20】0.1M PBS(pH7.4)中、(A)室温及び(B)37℃において、12%のGSNOを組み入れたPVMMA/EC電気紡糸マット((a)純粋PVMMA、(b)2:1のPVMMA/EC;(c)1:1のPVMMA/EC;(d)1:2のPVMMA/EC;(e)純粋EC)からのNOのin vitro放出速度に及ぼす組成の効果を示す。
【図21】超音波噴霧法で調製したGSNO-PVMMA(AN139)/ECミクロスフェアの顕微鏡写真を示す。
【図22】超音波噴霧法で調製したGSNO-PVMMA(AN169)/ECミクロスフェアの顕微鏡写真を示す。
【図23】コントロール群と試験群との間の創傷閉鎖速度を比較する(*p<0.05)。
【図24】コントロール(C3)とGSNO治療(T9)動物の創傷前及び創傷後4、10、16日後の創傷の写真を示す。
【発明を実施するための形態】
【0021】
〔好ましい実施形態の詳細な説明〕
以下の実施例を参照することによって本発明はより容易に理解されるだろう。なお、実施例は本発明の特定の態様及び実施形態をさらに説明する目的のためだけに含まれるものであり、いかなる場合にも本発明を制限するつもりはない。
〔RSNO-PVMMA/PVP複合体粉末の調製〕
(材料)
以下の実験において、還元型グルタチオン(GSH)、還元型グルタチオンエチルエステル(GSHEE)、亜硝酸ナトリウム(NaNO2)、スルファニルアミド(SULF)及びN-(1-ナフチル)エチレンジアミン二塩酸塩)(NEDD)はSigma-Aldrich Chemical Co. Oakville, CA)から得た。全てのフィトケラチンはAnaSpec Inc.(San Jose, CA, US)から購入した。
全てのポリマーはISP(New Jersey, USA)及びDow Chemical Company(Midland, MI)から得た。分析試薬グレードの他の化学薬品及び溶媒はSigma Aldrichから得、特に断らない限り、入手したまま使用した。Milli-Qグレード(Millipore, SA, France)脱イオン水を全ての溶液及び緩衝液に使用した。以下の実施例で用いる全てのPVMMA及びPVPは、特に断らない限り、PVMMA AN-169及びPVP K-90である。
【実施例1】
【0022】
(RSNOの合成)
下記反応式に従うチオールのニトロソ化によってRSNOの合成を行う。
【0023】
【数1】
【0024】
この反応は、少なくとも合成の観点からは非常に迅速、効率的、かつ定量的である。しかし、この反応は、不安定な生成物(その純粋状態で)を生じさせることが多い。下記反応式に示すように、ジスルフィド架橋形成をもたらすRSNOのホモリシスが、その熱的不安定性の原因である主な機構である。
【0025】
【数2】
【0026】
この反応についての詳細な情報を以下の実施例で述べる。
A. GSNOを調製するための実験手順
光への露出から保護した酸性媒体中で還元型グルタチオン(GSH)と等モルの亜硝酸塩を反応させることによって、GSNOを容易に調製した。
【0027】
【化3】
スキーム1
【0028】
要するに、5mlの0.2N HCl中のグルタチオン(GSH)(154mg,0.5mmol)の撹拌氷冷溶液にNaNO2の一部(35mg,0.5mmol)を加えた。この反応は80%を超える高収率でGSNOを与える。最終赤色溶液をアルミニウム箔で光から保護すると、該溶液は暗所では安定であり、合成後に精製せずに直接使用することができる。
B. GSHEEのS-ニトロソ反応
同様にグルタチオンエチルエステル(GSHEE)のS-ニトロソ化(スキーム2)を行った。要するに、2mlの0.2N HCl中のGSHEE(67mg,0.2mmol)の撹拌氷冷溶液にNaNO2の一部(14mg,0.2mmol)を加えた。結果として生じた赤色溶液をアルミニウム箔で光から保護したバイアルに貯蔵した。
【0029】
【化4】
スキーム2
【0030】
C. フィトケラチン5のS-ニトロソ反応
各PC5分子中の5個のチオール基を考慮に入れて、PC5対NaNO2のモル比が1:5であることを除き、同様にフィトケラチン5(PC5)(スキーム3)のS-ニトロソ化を達成した。
【0031】
【化5】
スキーム3
【0032】
手短に言えば、まず3mgのPC5(3.2325μmol)を氷浴内で100μlの0.2N HClに溶かしてからこの溶液に即座に100μlの新たに調製したNaNO2溶液(11.152mg/ml)を加えた。結果として生じたピンク色溶液を、アルミニウム箔で光から保護したバイアルに貯蔵した。
D. ホモ-フィトケラチン2のS-ニトロソ反応
各ホモ-PC2分子中の2個のチオール基を考慮に入れて、ホモ-PC2対NaNO2のモル比が1:2であることを除き、同様にホモ-フィトケラチン2(ホモ-PC2)(スキーム4)のS-ニトロソ化を達成した。
【0033】
【化6】
スキーム4
【0034】
手短に言えば、まず1mgのホモ-PC2(1.8031μmol)を氷浴内で50μlの0.2N HClに溶かしてからこの溶液に即座に50μlの新たに調製したNaNO2溶液(2.4883mg/ml)を加えた。結果として生じたピンク色の溶液を、アルミ箔で光から保護したバイアルに貯蔵した。
【実施例2】
【0035】
(RSNOのPVMMAへの結合)
無水マレイン酸コポリマーの注目すべき特徴は、周知の酸無水物成分と一級アミン基との高い反応性であり、より低度までアルコールと反応する。この反応は、コポリマーの溶解状態でか又は何らかの生理活性分子の界面提示後の表面化学によって遂行される。このようなアシル化反応は一般的に温和な条件下で起こり、本発明の場合、室温で20分以内に自発的に遂行された。
原則として、一級アミン基を含有する全てのRSNOは、スキーム5に従ってPVMMA等の無水マレイン酸コポリマーと反応することができる。該反応は、フリーのカルボン酸基の形成をももたらし、これは、第2のポリマーと分子間高分子複合体を形成するという次の必須工程用のプロトンを与えるのに必要不可欠である。酸無水物基と反応することについては、チオール基がアミン基より反応性が高いので、PVMMAとの結合前にまずRSNOを調製すべきことが非常に重要である。
【0036】
【化7】
スキーム5
【0037】
A. GSNOのPVMMAへの結合
GSNOのPVMMAへの容易な付着は、GSNOは0.1N HClに、PVMMAはアセトンにそれぞれ別々に溶解させなければならず、かつアセトン及び水性HClが偶然それぞれGSNO及びPVMMA用の沈殿剤であるという事実のため、GSNOとPVMMAの不均一反応によって達成された。従って、グラフト反応は溶液中でGSNOとPVMMAの界面で起こる。以下の実施例では、PVMMAの質量に対して50%までGSNO装填を達成できる。
A1. 7.52%装填を伴うGSNOのPVMMAへの結合
まず、500mgのPVMMAを10mlのアセトンに均一に溶かした。次に、実施例1Aに従って得た1mlのGSNO溶液を、氷浴内で撹拌下のPVMMA溶液に滴下した。引き続き、溶液をTeflon皿に注ぎ、ドラフト内に置き;室温下での空気乾燥又は真空乾燥のいずれかでアセトンを除去し、露光から保護した。得られたピンク粉末形態のGSNO-PVMMAを収集してデシケーター内で貯蔵した。さらに、結果として生じた溶液の一部を乾燥させずに次工程のために保管した。
A2. 15.04%装填を伴うGSNOのPVMMAへの結合
まず、500mgのPVMMAを10mlのアセトンに均一に溶かした。次に、実施例1Aに従って得た2mlのGSNO溶液を、氷浴内で撹拌下のPVMMA溶液に滴下した。引き続き、溶液をTeflon皿に注ぎ、ドラフト内に置き、室温下での空気乾燥又は真空乾燥のいずれかでアセトンを除去し、露光から保護した。得られたピンク粉末形態のGSNO-PVMMAを収集してデシケーター内で貯蔵した。さらに、結果として生じた溶液の一部を乾燥させずに次工程のために保管した。
A3. 30%装填を伴うGSNOのPVMMAへの結合
まず、500mgのPVMMAを10mlのアセトンに均一に溶かした。次に、実施例1Aに従って得た4mlのGSNO溶液を、氷浴内で撹拌下のPVMMA溶液に滴下した。引き続き、溶液をTeflon皿に注ぎ、ドラフト内に置き、室温下での空気乾燥又は真空乾燥のいずれかでアセトンを除去し、露光から保護した。得られたピンク粉末形態のGSNO-PVMMAを収集してデシケーター内で貯蔵した。さらに、結果として生じた溶液の一部を乾燥させずに次工程のために保管した。
【0038】
B. S-ニトロソ-GSHEEのPVMMAへの結合
上記と同じ方法で8.1wt%装填を伴うS-ニトロソ-GSHEEのPVMMAへの付着を達成した。要するに、1mlのS-ニトロソ-GSHEE(実施例1Bに従う)を、氷浴内で撹拌下の10mlの5%PVMMAアセトン溶液に滴下した。混合物を10分間反応させてからTeflon皿に注いで暗所で空気乾燥させた。アセトンの急速な揮発のため、結果として生じたピンク色粉末を1時間で収集し、引き続きデシケーター内で貯蔵した。同様に、結果として生じた溶液の一部を乾燥させずに次工程のために保管した。
C. S-ニトロソPC5のPVMMAへの結合
上記と同じ方法で6wt%装填を伴うS-ニトロソ-PC5のPVMMAへの付着を達成した。要するに、まず50mgのPVMMAを5mlのアセトンに溶かしてから200μlのS-ニトロソ-CP5溶液(実施例1Cに従う)を、氷浴内で撹拌下のPVMMA溶液に滴下し、混合物を10分間反応させてから、この合成の次工程で即座に使用した。
D. S-ニトロソ-ホモ-PC2のPVMMAへの結合
上記と同じ方法でS-ニトロソ-PC5のPVMMAへの付着を達成した。まず20mgのPVMMAを2mlのアセトンに溶かしてから100μlのS-ニトロソ-CP5溶液(実施例1Dに従う)を、氷浴内で撹拌下のPVMMA溶液に滴下し、混合物を10分間反応させてから、この合成の次工程で即座に使用した。
【実施例3】
【0039】
(RSNO結合PVMMAとPVPの複合体形成)
RSNO結合PVMMAとPVPの複合体形成は、下記スキーム6に示す共重合体(interpolymer)の水素結合相互作用に基づく。
【0040】
【化8】
スキーム6
【0041】
A. GSNO-PVMMA/PVP複合体の調製
GSNO-PVMMA/PVP複合体を調製するため、まず10:1(体積比)のアセトンとエタノールの混合物中6.36wt%のPVP溶液を調製した。PVPは純粋なアセトンには溶けないので、対応するGSNO-PVMMA溶液の組成に従う溶液調製を容易にするために一定量のエタノールを添加しなければならない。
A1. PVMMAに対して7.52%のGSNO装填を伴うGSNO-PVMMA/PVP複合体の調製
複合体形成前にまず3mlのエタノールをGSNO-PVMMA溶液(実施例2A1に従う10/1のアセトン/0.1N HCl)に添加した。引き続き、氷浴内で激しく撹拌しているGSNO-PVMMA溶液に測定量のPVP溶液を速く注いだ。分子間水素結合によって複合体形成が起こるにつれて、結果として生じた混合物の粘度が、組成(異なる量のPVP溶液を導入することによって、PVMMA/PVP質量比を1:9〜9:1に調整した)によって変化するゲル化度のピンク色のゲル様生成物をもたらす特有の増加を示した。
A2. PVMMAに対して15.04%のGSNOを伴うGSNO-PVMMA/PVP複合体の調製
複合体形成前にまず4mlのエタノールをGSNO-PVMMA溶液(実施例2A2に従う10/2のアセトン/0.1N HCl)に添加した。引き続き、氷浴内で激しく撹拌しているGSNO-PVMMA溶液に測定量のPVP溶液を速く注いだ。分子間水素結合によって複合体形成が起こるにつれて、結果として生じた混合物の粘度が、組成(異なる量のPVP溶液を導入することによって、PVMMA/PVP質量比を1:9〜9:1に調整した)によって変化するゲル化度のピンク色のゲル様生成物をもたらす特有の増加を示した。
A3. PVMMAに対して30%のGSNOを伴うGSNO-PVMMA/PVP複合体の調製
複合体形成前にまず5mlのエタノールをGSNO-PVMMA溶液(実施例2A2に従う10/4のアセトン/0.1N HCl)に添加した。引き続き、氷浴内で激しく撹拌しているGSNO-PVMMA溶液に測定量のPVP溶液を速く注いだ。分子間水素結合によって複合体形成が起こるにつれて、結果として生じた混合物の粘度が、組成(異なる量のPVP溶液を導入することによって、PVMMA/PVP質量比を1:9〜9:1に調整した)によって変化するゲル化度のピンク色のゲル様生成物をもたらす特有の増加を示した。
その後、実施例3のA1、A2、及びA3の結果の全ての半固体生成物をTeflon皿に移してドラフト内で空気乾燥させた。ピンク色ポリマー複合体が完全に凝固した後、そのようにして得られた脆性生成物をドライアイスと混合し、Micro-MillTM内で粉砕して粉末にした。最終ピンク色粉末の異なる大きさのフラクションをMini-Sieve Micro Sieve Setで分離し、使用するまで琥珀色容器内で貯蔵した。
【0042】
B. S-ニトロソ-GSHEE-PVMMA/PVP複合体の調製
S-ニトロソ-GSHEE-PVMMA/PVP複合体を調製するため、まず10:1(体積比)のアセトンとエタノールの混合物中6.36wt%のPVP溶液を調製した。PVPはアセトンに溶けないので、複合体形成前に1mlのエタノールをS-ニトロソ-GSHEE-PVMMAの(10/1のアセトン/0.1N HCl)溶液(実施例2Bに従う)に添加した。引き続き、氷浴内で激しく撹拌しているS-ニトロソ-GSHEE-PVMMA溶液に測定量のPVP溶液を迅速に加え、即座にピンク色のゲル様複合体を生じさせた;異なる量のPVP溶液によって、PVMMA/PVPの質量比を9:1〜1:9に調整した。結果の複合体を空気乾燥させ、ドライアイスと混合し、Micro-MillTM実験室粉砕機で粉砕して粉末にした。Mini-Sieve Micro Sieve Setで最終ピンク色粉末の異なる大きさのフラクションを分離し、使用するまで琥珀色容器内で貯蔵した。
C. S-ニトロソ-PC5-PVMMA/PVP複合体の調製
S-ニトロソ-PC5-PVMMA/PVP複合体を調製するため、まず10:1(体積比)のアセトンとエタノールの混合物中6.36wt%のPVP溶液を調製した。複合体形成前に0.5mlのエタノールを5mlのS-ニトロソ-PC5-PVMMA溶液(実施例2Cに従う)に加えた。引き続き、測定量のPVP溶液をS-ニトロソ-PC5-PVMMA溶液に速く注ぎ、即座にピンク色のゲル様複合体を生じさせた。異なる量のPVP溶液によって、PVMMA/PVPの質量比を9:1〜1:9に調整した。結果の複合体を空気乾燥させ、ドライアイスと混合し、Micro-MillTM実験室粉砕機で粉砕して粉末にした。Mini-Sieve Micro Sieve Setで最終ピンク色粉末の異なる大きさのフラクションを分離し、使用するまで琥珀色容器内で貯蔵した。
D. S-ニトロソ-ホモPC2-PVMMA/PVP複合体の調製
S-ニトロソ-ホモPC2-PVMMA/PVP複合体を作製するため、まず10:1(体積比)のアセトンとエタノールの混合物中6.36wt%のPVP溶液を調製した。複合体形成前に0.2mlのエタノールを2mlのS-ニトロソ-ホモPC2-PVMMA溶液(実施例2Dに従う)に加えた。引き続き、測定量のPVP溶液をS-ニトロソ-ホモPC2-PVMMA溶液に速く注ぎ、即座にピンク色のゲル様複合体を生じさせた。異なる量のPVP溶液によって、PVMMA/PVPの質量比を9:1〜1:9に調整した。結果の複合体を空気乾燥させ、ドライアイスと混合し、Micro-MillTM実験室粉砕機で粉砕して粉末にした。Mini-Sieve Micro Sieve Setで最終ピンク色粉末の異なる大きさのフラクションを分離し、使用するまで琥珀色容器内で貯蔵した。
【実施例4】
【0043】
(RSNO-PVMMA/PVP複合体粉末の特徴づけ)
UV-visスペクトル
それぞれUV及び可視範囲の最大吸収に相当するλ=336nm及びλ=545nmにおけるS-NO結合の特徴的な吸光度の出現によって、RSNO及びRSNO結合PVMMA/PVP複合体の両方のS-NO基の形成を実証することができる。これはσ→σ*及びπ→π*電子遷移に割り当てられる。Cary 50 UV-Vis分光光度計(Varian Inc.)を用いて室温で200〜800nmの範囲のスペクトル変化を記録した。図2、3及び4は、RSNO種としてそれぞれGSNO、GSHEE及びPC5を用いた場合のスペクトル変化を示す。
【実施例5】
【0044】
(水性媒体中のRSNOからのNO分解の速度論)
この発明では、NOドナーとしてまず最初にGSHEE及びフィトケラチンを使用する。NOを運ぶそれらの能力を前記UVスペクトルで実証した。UV-Vis分光光度計を用いて、水性媒体中でのそれらの安定性を探索した。この安定性の研究のため全てのRSNOの溶液を実施例1に従って合成した。10分の時間間隔内の545nmにおける時間依存性吸光度変化から、室温でのこれらの溶液中におけるそれらの分解速度論を得た。
図5は、同様の初期溶液濃度でのGSNO、S-ニトロソ-GSHEE、及びS-ニトロソ-PC5のNO分解速度論のプロファイルを示す。全ての3つの曲線について減衰勾配から良い直線性が得られ、NO分解速度がGSNO>S-ニトロソ-GSHEE>S-ニトロソ-PC5の順序で低減することが分かった。
【実施例6】
【0045】
(FTIRスペクトル)
フーリエ変換赤外分光法(FTIR)及び汎用の減衰全反射(Attenuated Total Reflectance)(ATR)Spectrum-oneTMPerkin-Elmer分光光度計(Perkin Elmer, Connecticut, USA)で記録されたスペクトルによってGSNOのPVMMAへの結合及びPVMMAとPVPの間の水素結合相互作用を特徴づけた。2cm-1の分解能で一区画のサンプルから全てのスペクトルを収集し、3回繰り返した。全てのスペクトルから、いかなるサンプルもないバックグラウンドスペクトルを減じた。スペクトルは4000〜650cm-1で記録された。
図6が示すように、純粋PVMMAフィルムの無水マレイン酸サイクルに典型的な1855及び1773cm-1の2つの肩部がGSNO-結合PVMMAのスペクトルでは完全に消失した。原材料中の微量カルボン酸基から生じる1707cm-1のバンドは、GSNOカップリングプロセス中のPVMMAとエタノールの間のエステル化に起因しうる1724cm-1のカルボニル特有のバンドの存在に置き換えられた。GSNO-結合PVMMAのスペクトルには、結果として生じたアミド基中のC=O基に特有の1642cm-1における別の主な吸収特徴も現れた。これは、PVMMA中の酸無水物とGSNO中の一級アミノ基との間のアシル化反応の存在を意味する。
図7中、1706cm-1で検出された純粋PVMMA中のフリーのCOOH基は、カルボニル基の伸縮振動に基づく可能性がある。複合体形成後、1706から1732cm-1へのこのカルボニル伸縮振動数の観察された上方シフトは、PVPの付加による強力な分子間水素結合相互作用に起因する「フリーの」C=O基の増加を反映している。一方、PVMMA/PVP複合体では、環式イミド基から生じるPVP中の1652cm-1のバンドも1664cm-1にシフトすることが観察される。これらの2つのバンドシフトは、複合体形成が酸のO-HとPVP分子中のイミド酸素との間の水素結合に関与することを支持する強力な証拠である。
【実施例7】
【0046】
(NOのin vitro放出の研究)
20mgのRSNO-PVMMA粉末を10mlの0.1M PBS(pH7.4)に長期間浸漬させることによって、in vitro放出の研究を行った。室温又は37℃で維持したインキュベーター内で15rpmの速度で運転するロータリーシェーカー上に全サンプルを置いた。所定の時間間隔で、2mlのNO-放出媒体をサンプリングして2mlの新鮮なPBSと交換した。
RSNO-PVMMAからのNO放出を標準的なGriessアッセイで定量化した。この比色法は、NOの全ての酸化生成物を定量化することができる。NOは、中性水溶液中で下記反応に従って容易にO2と反応してNO2を生成し、次にNO2-とNO3-を形成することが分かっている。
【0047】
【数3】
【0048】
【数4】
【0049】
要するに、1mlのGriess試薬(NEDD)(0.1%w/v)及び1mlのスルファニルアミド(5%v/vのH3PO4中1%w/v)を室温で当量(1ml)のサンプルとインキュベートした。結果溶液の540nmの波長におけるUV吸光度を決定し、3〜120μmol/LのNaNO2の標準曲線からサンプル溶液中の総[NO2-]を計算し、結果をμmolとして表した。
RSNO-PVMMA/PVP複合体からのNOのin vitro放出挙動をRSNO-PVMMA粉末について上述したのと同様に行った。20mgのRSNO-PVMMA/PVP複合体粉末を10mlの0.1M PBS(pH7.4)に長期間浸した。室温又は37℃で維持したインキュベーター内で15rpmの速度で運転するロータリーシェーカー上に全サンプルを置いた。所定の時間間隔で、2mlのNO-放出溶液をサンプリングして2mlの新鮮なPBSと交換した。NO濃度をGriessアッセイで決定した。
【0050】
【化9】
スキーム7
【0051】
スキーム7にはRSNO-PVMMA複合体のNO放出挙動が示されている。複合体から酸化窒素が徐々に遊離されるにつれて、さらにジスルフィド結合が形成し、RSNOの側鎖間のジスルフィド架橋を生じさせ、さらに複合体のネットワーク構造を強化する。ポリマー構造と鎖パッキングの状態に基づき、成分ポリマーの分子量と濃度比、及び沈殿条件を調整することによって、異なる持続性かつ制御可能な放出速度を得ることができる。
A. GSNO-PVMMA及びGSNO-PVMMA/PVP複合体からのNOのin vitro放出
図8に示すように、PVPとの超高分子複合体を形成しないと、GSNO-PVMMAからのNOの放出は比較的速く、放出時間はたった3日までである。対照的に、PVPとの超高分子複合体の形成によって、水性媒体中におけるその溶解速度が低減するため、酸化窒素放出速度を有意に遅くすることができる。該NO放出の典型的プロファイルは、9日以上まで延長しうる。
B. 異なる組成のGSNO-PVMMA/PVP複合体からのNOのin vitro放出
PVMMA/PVPの種々の質量比(1/0.5、1/1、1/2、1/3)を調査した。図9に示すように、複合体中のPVP含量の増加と共にNO放出速度が上昇するが、1/1比は最も遅いNO放出速度を示し、少なくとも12日持続した。
C. 異なる温度におけるGSNO-PVMMA/PVP複合体からのNOのin vitro放出
図10は、本GSNO-PVMMA/PVP複合体(1/1)からのNO放出で温度が重要な役割を果たすことを実証し;温度が高いほど有意にその放出速度を加速する。
D. PVMMA及びPVPのMwが異なるGSNO-PVMMA/PVP複合体からのNOのin vitro放出
分子量が大きいポリマーほど、複合体形成が促進されるため、ポリマー溶解が遅くなると考えられる。図11は、NO放出挙動に及ぼすポリマー分子量の効果を示す。試験サンプルの対応分子量を表1に列挙する。図11から、PVMMA又はPVPのいずれかの分子量が小さいほどNO放出が速くなることが明らかである。
【0052】
【0053】
E. 異なる粒径のGSNO-PVMMA/PVP複合体からのNOのin vitro放出
3つの異なる平均粒径(それぞれ約0.065、0.125及び0.3mm)のGSNO-PVMMA/PVP複合体のNO放出パターンを図12に示す。粒径がNO放出挙動で重要な役割を果たすことは明白であり、粒径が小さいほどNO放出速度が速くなる。
F. S-ニトロソPC5-PVMMA/PVP複合体からのNOのin vitro放出
図13は、S-ニトロソPC5-PVMMA/PVP複合体の持続性NO放出を実証し、放出期間を少なくとも9日まで延長することができる。この場合もやはり、温度が高いほど放出が速く、かつNO装填を調整することによって放出速度を制御できることが分かる。さらに、NOドナーとしてPC5を選択すると、少なくとも90%のNO装填効率が可能であり、これはGSNOのNO装填効率より高い。
【実施例8】
【0054】
(GSNO-PVMMA/PVP複合体の安定性の研究)
A. 室内条件下の安定性
GSNO-PVMMA/PVP複合体粉末(実施例3A1参照)を6カ月の持続期間、光から保護せずにRT(相対湿度:22%)でバイアル内で貯蔵した。図14から、この安定性期間後にNO放出プロファイルに有意な変化がないことが分かる。これは、室内条件下で貯蔵した場合、本発明のGSNO-PVMMA/PVP複合体が非常に安定していることを示唆している。
B. 照射下の安定性
GSNO-PVMMA/PVP複合体粉末(実施例3A1参照)を24時間UV照射にさらした。図15は、短期のUV照射を受けた後にGSNO-PVMMA/PVP複合体が安定していることを示す。
(電気紡糸装置によるナノ繊維形態のGSNO-PVMMA/EC複合体の調製)
電気紡糸は、直径がマイクロメートル〜ナノメートルの範囲の繊維を含むポリマーの不織、多孔性、及び三次元スキャフォールドを加工するために広く適用されている。この一工程技術は、生体材料の組成、構造及び機械的性質を制御する可能性を提供する。特に、この方法は、薬物分子を軟組織に組み入れることを可能にし、その高い水蒸気透過性、良い機械的強度及び優れた可撓性のため創傷手当用品に理想的に適している。この方法では、薬物装填と最終製剤の調製を一工程で達成することができる。特に、的確な材料の選択と繊維構造設計を通じて、結果として生じる物質はさらなる望ましい特性、例えば生体接着性、弾力性及び薬物放出制御能力に恵まれる可能性がある。本発明では、この方法で、ナノ繊維に基づいたRSNO装填NO送達システムを濃縮溶液から調製することができる。
【実施例9】
【0055】
(電気紡糸用原液の調製)
A. PVMMA/ECブレンドの調製
2gのPVMMA及び1gのエチルセルロース(EC)を別々にN-ジメチルホルムアミド(DMF)とアセトン(体積比=2:3)の混合物15mlに溶かした。2種の溶液を均一にブレンドすることによって、質量比が1:0、2:1、1:1、1:2〜0:1の範囲の一連のPVMMA/ECブレンド溶液を連続的に得た。
B. GSNOのPVMMA/ECブレンドへの結合
脱イオン水とエタノールの混合物(体積比=1:1)1ml中、室温下で約308mgのGSHを69mgのNaNO2と反応させた。その直後に、結果として生じたピンク色のGSNO溶液を、激しく撹拌している上記ポリマー溶液にゆっくり滴下して安定したピンク色エマルションを得、さらに20分撹拌を続けると澄んできた。
【実施例10】
【0056】
(GSNO-PVMMA/ECブレンド溶液の電気紡糸)
ノズルとして先端が平坦のステンレススチールのゲージ20針を備えた5mlの注射器に上記ブレンド溶液を詰めた。典型的手順では、水平マウント内にある注射器ポンプ(KDS 200, KD Scientific, USA)を用いて0.2〜0.8ml/時間の速度でGSNO-PVMMA/ECブレンド溶液を供給した。高圧電源(EL 50PO.8, Glassman High Voltage Inc., USA)を用いてノズルと接地アルミニウムコレクターとの間に高電圧(12〜18kV)を印加した。先端とコレクターの間の距離を12〜16cmに調整した。GSNOの感光性と感熱性を最小限にするため、直接光がなく、かつ20℃で維持されたドラフト内に装備全体を置いて、プロセス中のNO損失を減らした。全ての紡いだままのファブリックを、直接光から保護されたデシケーター内で貯蔵し、次に使用するまで4℃で冷蔵した。
【実施例11】
【0057】
(紡いだままのマットの形態学的特徴づけ)
1kV及び2kVの加速電圧を用いて環境制御型走査電子顕微鏡(environmental scanning electron microscope)(HITACHI S-3400N SEM、日本国)によって、紡いだままのファブリックの形態学的外観及びサイズ分布を調査した。図16は、GSNO-PVMMA(13.33wt%の濃度で)、EC(6.67wt%の濃度で)、及びGSNO-PVMMA/EC複合物から紡いだナノ繊維のSEM画像を示す。調査画像中の挿入図は、対応する繊維サイズ分布を表示する。GSNO-結合PVMMA及びEC超薄繊維の平均径は、それぞれ0.82μm及び0.25μmであり、GSNO-PVMMA/EC複合物のナノ繊維は中間の平均径0.64μmを示す。
PVMMAは、典型的な浸食性(erodable)ポリマーであり、純粋なPVMMAのみに基づいた電気紡糸ナノ繊維は、PBS中37℃でのキャスティングフィルムより速く溶解するであろうことから、創傷手当用品へのその適用の主な限界を呈している。PVMMA/ECナノ繊維にECを添加すると、紡いだままのファブリックの水中における統合性を有意に改善する。図17に示すように、1:1のGSNO-PVMMA/ECナノ繊維製の膜は、37℃で3日の水中浸漬後にその繊維構造を保持した。画像cとdの間には繊維の形態の変化が実質的に存在せず、ECを添加すると、ナノ繊維がその統合性を留める能力を延長できることを示唆しており、創傷手当用品にとって望ましいことである。
【実施例12】
【0058】
(FTIRスペクトル)
スキーム8に示すように、フーリエ変換赤外分光法(FTIR)でGSNO-PVMMA/ECシステムの水素結合相互作用を特徴づけた。汎用の減衰全反射(ATR)Spectrum-oneTMPerkin-Elmer分光光度計(Perkin Elmer, Connecticut, USA)で4000〜650cm-1のスペクトルが記録された。2cm-1の分解能で一区画のサンプルから全スペクトルを収集し、3回繰り返した。全スペクトルから、いかなるサンプルもないバックグラウンドスペクトルを減じた。
【0059】
【化10】
スキーム8
【0060】
図18の純粋PVMMAのFTIRスペクトルから、1707cm-1におけるカルボニル基の伸縮振動の原因である微量カルボキシル基は、ナノ繊維形成プロセス中のわずかな加水分解に起因するか又は原材料に起因しうる。PVMMA/ECフィルムサンプルにおけるカルボニル伸縮振動数の1707から1720cm-1への明白な上方シフトは、ECの添加による強力な分子間水素結合相互作用のための「フリーの」C=O基の増加を反映している。さらに、PVMMA/ECブレンドフィルムでは、酸無水物に典型的な1855及び1773cm-1における吸光度ピークが未だ顕著である。
【実施例13】
【0061】
(紡いだままのファブリックのマイクロ引張り試験)
5kgのロードセルを備えたテクスチャーアナライザー(TA.XTplus, Stable Micro Systems, Haslemere, Surrey, UK)を用いて、GSNO無し及びGSNOを組み入れたPVMMA/EC電子紡糸ファブリックの機械的性質を評価した。引張り試験では、均一厚の電気紡糸繊維マットを30×20mmのサンプル片にカットした。この試験用の2つのクランプ間にサンプルを保持した。測定中、上クランプによって0.5mm/秒の速度で破壊するまでフィルムを引いた。力と伸びは機器によって自動的に記録された。各測定を4回繰り返し、結果を図19に示す。図19から、PVMMA/EC及びGSNO-PVMMA/ECフィルムは、一成分のPVMMA又はECフィルムの機械的性質を有意に超える改善された機械的性質を示すことが明らかである。この改善は、PVMMAとECの間の水素結合相互作用に起因すると考えられる。
【実施例14】
【0062】
(紡いだままのマットからのNOのin vitro放出)
20mgの電気紡糸マット(約2×2cm2)を10mlの0.1M PBSに長時間浸すことによって、NOのin vitro放出の研究を行った。37℃で維持したインキュベーター内のロータリーシェーカー上に全サンプルを置いた。所定時間間隔で、5mlの放出媒体を5mlの新鮮なPBSと交換した。
実施例7で述べたGriessアッセイによって、繊維からのNO放出を定量化した。図20A及び20Bにそれぞれ室温及び37℃について、異なる組成の紡いだままのナノ繊維からの、pH7.4の緩衝液中におけるNO放出の結果を示す。GSNO-PVMMA/EC(1:1)からのNO放出速度は、温度に依存して約35〜40μmol/g(マット)であり、NO放出は、EC含量の増加とともに有意に遅くなることが分かる。複合物フィルムは、1週間にわたる長期放出を示す。
(超音波噴霧装置を用いたRSNO-PVMMA装填ミクロスフェアの調製)
超音波噴霧は、噴霧乾燥、マイクロカプセル化及び基材コーティングに広く適用されている。この一工程法は、精度の高い均質ミクロスフェア及び薄膜コーティングを効率的に製造することができる。単一又は二重供給ノズルから噴霧された液滴は、空中及び収集浴内で凝固しうる。濃縮ポリマー溶液に適用される電気紡糸法と異なり、この方法は特に希釈ポリマー溶液に適している。以下の実施例では、RSNO装填超高分子複合体に基づいたミクロスフェアの、この方法による製造について説明する。
【実施例15】
【0063】
(GSNO-PVMMA(Gantrez(登録商標)AN 139)/P(VP/VAc)(Plasdone(登録商標)S-630)ブレンド溶液の超音波噴霧)
それぞれ分子量が1.0×106及び2.4×104の低分子量のPVMMA(Gantrez(登録商標)AN139)及びポリ(ビニルピロリドン-co-酢酸ビニル)(Plasdone(登録商標)S-630)に基づいた超高分子NO放出複合体をこの実施例のために選択した。下記手順に従い、NOプロドラッグとしてGSNO-PVMMAを含むミクロスフェアを調製した。
1gのP(VP/VAc)を40mlのアセトンに溶かし、1gのPVMMAを10mlのアセトンに溶かし、このPVMMAに1mlのGSNO(実施例1A参照)を結合させ(実施例2A1参照)、結果のGSNO-PVMMA溶液を40mlのアセトンと10mlのエタノールの混合物中で希釈し、引き続きP(VP/VAc)溶液とブレンドした。
超音波発生装置を5.0wの電力で操作しながら、注射器ポンプ(KD-Scientific, Model 200)によって駆動された超音波ノズル(SONO-TEK Corp. 8700-60 MS)の入口を通じて0.5ml/分の流速で最終溶液をポンピングした。噴霧ミストは、ガラスカラム(直径15cm、高さ60cm)を通ってその沈降中に空気乾燥された。
乾燥したミクロスフェアを収集し、顕微鏡下で形態学的に特徴づけた。図21は、収集された粒子(完全に凝固したミクロスフェアに相当する)の大きさと形状を示す。
【実施例16】
【0064】
(GSNO-PVMMA(Gantrez(登録商標)AN169)/エチルセルロース(Ethocel(登録商標)NF100)ブレンド溶液の調製)
それぞれ高分子量のPVMMA(Gantrez(登録商標)AN169)(Mw:1.98×106)及びエチルセルロース(Ethocel(登録商標)NF100)(エトキシ含量、48.8%DS;粘度、80%及び20%のアルコール中5%の溶液について100cP)に基づいた超高分子NO放出複合体をこの実施例のために選択した。NOプロドラッグとしてGSNO-PVMMAを含むミクロスフェアを下記手順に従って調製した。
最初に、500mgのエチルセルロースを40mlのアセトンに均一に溶解させて1.25%の濃度を得た。一方、500mgのPVMMAを10mlのアセトンに溶かし、このPVMMAに1mlのGSNO(実施例1A参照)を結合させ(実施例2A1)、結果のGSNO-PVMMA溶液を40mlのアセトンと10mlのエタノールの混合物中で希釈し、引き続きエチルセルロース溶液とブレンドした。
超音波発生装置を5.0wの電力で操作しながら、注射器ポンプ(KD-Scientific, Model 200)によって駆動された超音波ノズル(SONO-TEK Corp. 8700-60 MS)の入口を通じて0.5ml/分の流速で最終溶液をポンピングした。噴霧ミストは、ガラスカラム(直径15cm、高さ60cm)を通ってその沈降中に空気乾燥された。
乾燥したミクロスフェアを収集し、顕微鏡下で形態学的に特徴づけた。図22は、収集された粒子(完全に凝固したミクロスフェアに相当する)の大きさと形状を示す。
【実施例17】
【0065】
(多層集合によるGSNO-PVMMA/PVP超薄コーティング)
超薄複合体コーティングを下記手順に従って製作した。まず、0.025mMのGSNO-PVMMA溶液を実施例2Aに従って調製し、アセトンとエタノールの混合物(体積比4/1)50mlに1.3gのPVPを溶かして0.02mMのPVP溶液を作製した。その溶液濃度を維持するため、全コーティング手順中、これらの2つの溶液を氷浴内に入れた。
次に、基材(スライドガラス及びPTFEシート)を最初にPVP溶液に10分間さらしてからアセトンとエタノールの溶媒混合物の3つの浴に順次、全部で4分間浸して過剰のPVPポリマーを洗い流した。その直後に、基材をGSNO-PVMMA溶液に10分間浸した後、アセトン溶液の3つの浴に順次、全部で4分間浸した。所望の厚さまで続けうる、このサイクルを20の二重層のため繰り返した。この集合プロセス後、コーティング基材を空気乾燥させて、室内条件下にてデシケーター内で貯蔵した。
【実施例18】
【0066】
(GSNO-PVMMA/PVP複合体粉末のin vivo評価)
A. 材料と方法
糖尿病性潰瘍などの慢性創傷は創傷治癒障害を被ることが多い。最近の証拠は、創傷治癒、特に創傷組織内のNOレベルの低減によって特徴づけられる糖尿病性足部潰瘍の治癒プロセスにおいて、NOが重要な役割を果たしうることを示唆している。NOドナーDETA NONOアート及びL-アルギニンによる外因性NO補充が糖尿病ラットの創傷治癒を促すことが分かった[32、33]。我々の場合、創傷治癒にけるRSNO-PVMMA/PVP超高分子複合体システム(実施例3A1から得た)の利益を評価するため糖尿病ラットモデルを用いた。前記実験を行って、(1)GSNO結合PVMMA/PVP複合体粉末から連続的にNOを発生させられるか、及び(2)このNO含有粉末製剤が糖尿病ラットモデルの創傷治癒を促進できるかを決定した。トロント大学動物管理委員会(The University of Toronto Animal Care Committee)の承認済みの動物プロトコルに従って全ての実験を行った。
B. 急性糖尿病の誘発
15匹のオスのスプラーグドーリーラット(Charles River, Montrealから)を1週間気候順応させ、適宜食物と水を与えた。傷つける7日前、ストレプトゾトシンを腹腔内(IP)注射して(体重1kg当たり60mg、クエン酸緩衝液中0.1mol/L、pH 4.5)、糖尿病を誘発した。14mmol/Lを超える血糖値と頻尿によって糖尿病の証拠を確認した。24時間後に糖尿病状態に達しなかった4匹の動物にストレプトゾトシンを再注射し、そのうちの1匹は血糖値が14mmol/L未満のままだったので、研究から除外した。糖尿病の誘発後、血糖値を1週間に2回モニターして、創傷治癒実験全体を通じて確実に糖尿病状態のままであるようにした。
C. 外科手術手技
外科手術の前日に、動物の体重を量り、2つの群に割り当てた(コントロール群の7匹及び試験群の8匹)。動物にイソフルラン吸入麻酔しながら下記手技を行った。まず、背面を剪毛し、皮膚をポピドンヨード液と70%アルコールで洗浄した。手術直前にラットに鎮痛薬(ケトプロフェン、3mg/kg、S.C.)を与えた。引き続き、8mmの生検パンチを用いて皮膚と皮筋層(panniculus carnosus)を除去して全厚切除創傷(full thickness excisional woun)を生じさせた。創傷部位において、コントロール群には20mgのブランクPVMMA/PVP複合体粉末(NO装填なし)で治療し、試験群には20mgのGSNO-PVMMA/PVP複合体粉末(実施例3A1から)で治療した。全てのポリマー粉末は数滴の無菌食塩水の助けを借りて迅速に創傷組織に付着した。
ポリマー粉末の適用後、チンキBenzodine化合物(Xenex Laboratories, Ferndale, WA)を周辺皮膚に適用して創傷を準閉鎖性ポリウレタン手当用品(TegadermTM, 3M, St. Paul, Minn)で覆った。その後、動物を個々のケージに移して標準的な食餌で維持し、適宜、水を自由に飲めるようにした。
D. 術後の創傷治療
傷つけた後最初の7日間、動物にイソフルラン吸入麻酔しながらTegaderm手当用品を毎日交換し、デジタルカメラを用いて創傷部位の写真を記録した。各写真を用いて較正スケールを記録した。傷害後7日目から、創傷はもはや手当用品によって閉鎖されなかった。
【0067】
表2
HI: > 33.3mmol/L
【0068】
表2は、Ascensia(登録商標)CONTOUR(登録商標)Blood Glucose Meterを用いて測定した動物の血糖値、及び創傷治癒持続期間の動物の体重減少を示す。糖尿病誘発後、有意な体重減少(>20%)及び多尿によって悪化した2匹の糖尿病ラットを外科手術前に安楽死させなければならなかった。
E. 画像解析
Image-Pro Plus 5.0ソフトウェアを用いて各病変の表面積を定量化し、時間の関数としてプロットした。このソフトウェアを用いて、開いた創傷の面積を決定した。結果を時間の関数として初期創傷面積の百分率で表す(図23)。各データ点について、平均及び標準偏差を計算した。
F. 統計解析
n個の観察の平均の平均値±標準誤差として本文及び図面中の全ての値を表した。独立した両側スチューデントt-検定を利用して、実験群間の統計解析を行った。対応のある両側スチューデントt-検定を利用して、右の外科的分裂と左の未傷害の間の統計解析を行った。0.05のαレベルで信頼限界を予め定めた。
G. 創傷治癒の評価
NOは、血管内皮増殖因子発現の誘発及び上方制御に関与し、線維芽細胞及びケラチノサイトの遊走をさらに助長することが分かっている[34、35]。NOの周知の抗菌及び血管拡張作用、特に血管拡張作用は微小血管系内の血流を増やし、ひいては栄養と細胞の両方の損傷部位への送達を容易にすることから、創傷治癒のプロセスでも重要でありうる。
図23の結果は、本発明のNO放出超高分子GSNO-PVMMA/PVP複合体システムが効率的に創傷閉鎖を加速する(p<0.05)ことを実証すると期待できる。コントロール群と試験群の間の創傷閉鎖の傾向には統計的に有意な差異がある。各群の0日目、4日目、10日目及び16日目の全厚創傷の代表的写真を図24に示す。開いた面積及び顆粒化組織についての明白な創傷状態も、傷つけた後4日目、10日目及び16日目では、コントロール群におけるより試験群の方がずっと良い。
【0069】
この開示は、本発明の特定の好ましい実施形態について記載かつ説明したが、本発明は、当該特定の実施形態に制限されるものでないことを理解すべきである。むしろ、本発明は、記載かつ説明した特有の実施形態及び特徴の機能的又は機械的に等価な全ての実施形態を包含する。
【技術分野】
【0001】
(関連出願)
この出願は、発明の名称が「創傷を治癒するため制御された酸化窒素放出を提供する超高分子複合体」である2007年8月27日提出のカナダ特許出願第2,599,082号の優先権を主張する。その内容を参照によって本明細書に引用したものとする。
この発明は、超高分子(supramacromolecular)酸化窒素放出ポリマー複合体;前記複合体を含む組成物及び含浸物品及び被覆物品;前記複合体の製造方法;並びに創傷、特に糖尿病に起因する立方状潰瘍形成を治癒する処置における前記複合体、組成物及び物品の使用方法に関する。
【背景技術】
【0002】
酸化窒素(NO)がとらえどころのない内皮由来弛緩因子と同一であるという発見以来[1]、NOの多くのさらに難解な生物学的役割が同定かつ解明されている[2〜6]。これらの知見が、創傷治癒、心血管疾患、呼吸器疾患、癌療法、神経系再構築、並びに新機能性医療デバイスにおける外因性NOの潜在的応用のさらなる探索を促した。これに関しては、局所適用したNOガスを用いた創傷感染の治療で実証された成功で明らかなように、NOの局所送達は臨床用途で利益を得る大きな可能性がある[7]。しかし、この小さい気体分子の短い半減期及びその内在する不安的性が、その医薬品剤形及び薬物送達システムに組み入れるための大きな課題を提示した。血管内皮細胞によって内因的に合成されたNOは5秒以下という非常に短い生物学的半減期を有すると報告されている[8、9]。NOはヘモグロビンによって急速に捕捉されるので、組織内のその作用部位は、それが発生されるところに局在化されるだろう。細胞及び組織内におけるNOの化学的な不安定性は、NO2-及びNO3-の両方へのNOの急速な酸化に起因している。
NOの周知起源である有機ニトラート及び亜硝酸ナトリウムのほかに、広範に研究されているNO前駆物質の2つの他ファミリーがある。1つはジアゼニウムジオラートから成り、他方はS-ニトロソチオールから成る。ジアゼニウムジオラートはNONOアート(NONOate)としても知られる構造R1R2NN(O)=NOR3の化合物を包含する。この分類のNOドナーに基づくNO放出物質の開発に多大な努力が払われている[10、11]。これらは、物理的ブレンド又はポリマー骨格若しくは側鎖への共有結合のどちらかによるジアゼニウムジオラートの異なるポリマーマトリックスへの組込みを包含する。ジアゼニウムジオラートに関する関連従来技術について以下に述べる。
【0003】
WO 2005/011575、WO 2005/07008、及びWO 2006/058318において、Smithは、ジアゼニウムジオラート成分がイオン結合又は共有結合のどちらかでポリマーマトリックスに結合している、イオン交換樹脂又はポリエチレンイミン(PEI)繊維の多積層物のどちらかに基づくMO放出デバイスを開示した。活性化又は創傷への適用時に、該NO誘導ポリマーが水、水素カチオン又はアスコルビン酸などのアクチベーターと接触すると、局所的なNO放出が生じうる。しかし、このようなシステムからのNO放出の持続時間は短く、典型的にイオン交換樹脂システムからはたった0.5〜3時間しか持続せず、繊維の多積層物デバイスからは多くても1〜2日持続するだけである。
Meyerhoff及び共同研究者らは、US 6,841,166及びUS 2006/0008529において、酸性アクチベーター及び可塑剤と共に、ジアゼニウムジオラート誘導体化ヒュームドシリカ、分散したジアゼニウムジオラート又は共有結合したジアゼニウムジオラートなどの分離したNOドナーを含有する疎水性ポリマー(例えばシリコーンゴム、ポリ(塩化ビニル)、ポリウレタン等)に基づく抗血栓性血液接触デバイス用のNO放出ポリマー材料を開示した。活性化中、水がゆっくり疎水性ポリマーマトリックス中に浸透し、数日まで水性環境にNOを持続的に放出することとなる。これらのシステムは、心血管の再狭窄及び血液循環障害の治療のためのNO送達用の埋め込み型グラフト、カテーテル又は生物医学的デバイス上コーティングとしても試験されている[12〜15]。生体適合性の懸念に加え、これらの極端に疎水性の材料は、その不十分な吸水性及び創傷部位における不十分な生体接着のため、創傷治癒適用には適さない。
さらに、この分類のNONOアートドナーのin vivo適用における1つの主な制限は、米国特許第6,841,166号で明らかにされているように、浸出しうるジアゼニウムジオラート及びその分解生成物、特にニトロソアミンの毒性の可能性である。上記従来技術のアプローチ及び米国特許第6,703,046号は、疎水性ポリマーを利用して、このような浸出を最小限にしていた。しかし、親水性の酸性添加剤及び可塑剤を含有するこれらのポリマーからはまだ浸出が起こりうる。さらに、1つの確立したジアゼニウムジオラートプロドラッグ、V-PYRRO/NOは、既知の最も強力な実験上の肝癌誘発物質の1つであるN-ニトロソピロリジンを形成する可能性がある[16]。さらに、水性媒体中に放出されたジアミンベース及びポリエチレンイミンベースジアゼニウムジオラートは、測定可能レベルの、既知分類の発癌物質であるニトロソアミンを形成することが分かっている[12]。従って、特に創傷治癒のためのジアゼニウムジオラートのin vivo適用は限界があると思われる。
【0004】
別の主分類のNOドナーは、一般構造R-SNOを有する化合物であるS-ニトロソチオールである。NOの重要な内因性及び外因性起源として、RSNOは広く生体内に分布しており、哺乳類の体内で酸化窒素を貯蔵、輸送及び放出することが分かっている[17]。さらに、生理液内で水によって活性化するとNOを発生するその能力は、全身に負担をかける必要なく、特異的組織だけを標的にするNOの局所送達に特に有利である。種々の内因性RSNOのうち、S-ニトロソグルタチオン(GSNO)は、グルタチオンと亜硝酸ナトリウムの間の室温での自発的反応によるその合成の容易さ及び固体として単離できることのため、非常に注目を集めている[18]。しかし、これらの小分子RSNOの安定性は、S-NO結合が熱分解にも光分解にも不安定であり、かつNOの自発的放出及びその急速な不活性化につながる溶血開裂しやすいことから不十分であり、そのため創傷治癒といった実際の適用に対するその適合性を制限している。
de Oliveira及び共同研究者らは、経皮NO送達のため、ポリ(ビニルアルコール)、ポリ(ビニルピロリドン)、又はPluoronic F127ヒドロゲル等の水溶性ポリマーに基づいたフィルム及びゲル中にS-ニトロソグルタチオン(GSNO)及び/又はS-ニトロソ-N-アセチル-システイン(SNAC)を物理的に組み入れた[19〜22]。彼らの動物結果は、ラットの皮膚創傷回復の初期段階中のGSNO含有ヒドロゲルの反復適用が創傷閉鎖及び再上皮化を加速することを示す[23]。しかし、反復適用を避けるための患者のコンプライアンスの観点から、長期のNO放出がさらに望ましいだろう。
Katsumi及び共同研究者らは、亜硝酸塩をウシ血清アルブミン(BSA)上のシステイン残渣に共有結合させることによって、BSA及びポリ(エチレングリコール)(PEG)結合BSAに基づいた超高分子担体S-ニトロソチオールを合成した[24、25]。同様に、Westらは、米国特許第7,052,711号において、ポリ(エチレングリコール)ヒドロゲル内に固定化されたS-ニトロソシステイン(CysNO)が、in vitro細胞培養における血小板接着及び平滑筋細胞増殖を減少させることを実証した。しかし、これらの報告された親水性システムは、S-NO結合が熱分解にも光分解にも不安定であり、かつNOの自発的放出及びその急速な不活性化につながる溶血開裂しやすいことから、望ましい安定性を欠く。結果として、従来技術の化合物からの酸化窒素放出持続時間は、如何なる長期間をも維持され得ず、一般的に、多くて数時間である。
WO2006/095193に記載されているように適用時にin situ又はWO2008/031182に記載されているように創傷へのその適用前にin vitroでポリマー中でGSNOを物理的に混合して混合物を形成する方法[21〜24]及びNO前駆物質をアクチベーターと混合してGSNOを発生させる従来技術の方法は、一旦GSNOが形成又は放出されると、それはまだ熱、水分及び光による分解に感受性なので、GSNOの短い半減期という問題に対処していない。実際に、ほとんどのこれらの従来技術のアプローチでは、NO又はGSNOの放出は、一般的に非常に速く、せいぜい数時間しか持続しないので、繰返し適用する必要がある。
【0005】
【発明の概要】
【発明が解決しようとする課題】
【0006】
従って、当技術分野では、創傷治癒適用のためNOの制御可能かつ持続的な放出を提供する安定したNO送達システムを達成することが要望されている。
【課題を解決するための手段】
【0007】
従って、一態様では、本発明は、物理的に架橋したポリマーネットワーク内で安定化した固定化RSNOを含む超高分子複合体に基づいた新分類のNO送達システムに関する。このシステムでは、担体ポリマーに共有結合したRSNO前駆物質が、第2のポリマーとの分子間複合体形成によって、好ましくは水素結合相互作用を介して安定化される。結果として生じる超高分子複合体は、連続的かつ長期にわたる、貯蔵安定性が改良されたNO放出を提供することができる。ここで、用語「超高分子」を用いて、的確な3D構造化非共有結合高分子を含む分子集合を表す[26]。
さらなる態様では、この発明は、(1)異なるNO装填レベルのRSNO;(2)側鎖内に、RSNO上のアミン基と反応することができる酸無水物官能基を有するポリマーA;及び(3) ポリマーAと強力な水素結合を形成することができるプロトン受容基を骨格内又は側鎖内に含むポリマーB;の付加体を含む医薬組成物をも提供する。
さらなる態様では、本発明は、前記NO放出複合体の製造方法;及び前記複合体の使用方法にも関する。
本発明は、強力な高分子間相互作用によるレイヤーバイレイヤーアセンブリ(layer-by-layer assembly)によって前記NO放出複合体をコーティングにする方法をさらに提供する。
なおさらなる態様では、この発明は、粉末、微粒子、繊維及びフィルムなどの多様な形態の該NO放出超高分子複合体の調製方法を提供する。特に、この新規酸化窒素放出ポリマー複合体を創傷治療用の手当用品及び包帯に組み入れることができ、慢性の治癒不十分な創傷の治療に適した持続性かつ制御された様式で治療量の酸化窒素の放出をもたらす。
この発明は、有効にNO装填する能力及び有意に改良された安定性を示す新規NO前駆物質としての広範なGSNO誘導RSNOの利用にも関する。
さらに、この発明は、慢性創傷の治療方法をも提供する。本NO放出超高分子複合体は、糖尿病動物モデルにおいて加速された創傷治癒を示した。
なおさらに、この発明は、局所組織及び医療デバイス上のコーティングに直接適用できる制御された持続性様式で治療レベルのNOを発生させるための新規プラットフォームを提示する。
ほとんどの小分子NOドナーは水性媒体中で化学的に不安定である。例えば、図1から、室内条件下で酸性及び中性媒体中では、GSNOからNOが連続的に解離することが分かる。本発明は、RSNOを高分子担体に付着させて、ポリマーNO前駆物質又はプロドラッグを形成することによって、RSNOの半減期を延長するための有用な方法を提供する。予想外に、前記ポリマーNOプロドラッグを別のポリマーとの分子間水素結合相互作用によって物理的に架橋することによって、結果として生じたる超高分子複合体が、さらに改良されたRSNO貯蔵安定性で連続的かつ長期にわたるNO発生を提供できることが分かった。さらに、水和すると、本発明の超高分子NO放出複合体は、生体接着性にもなり、そのためNOの局所的制御送達を容易にする。
NO発生超高分子を得るため、全ての上記反応が非常に迅速に起こり、かつ関与する全ての有機溶媒を容易に除去できることが望ましい。
【0008】
本発明の目的は、創傷の治癒において酸化窒素の永続的放出を生じさせるための簡単な安定した生体適合性手段を提供する酸化窒素担体を提供することである。
前記酸化窒素担体の製造方法を提供することがさらなる目的である。
固体剤形は、貯蔵及び輸送中に水性剤形より高い安定性を示すので、粉末、フィルム、繊維、ミクロスフェア又はコーティング等のいくつかの物理的形態の形で前記酸化窒素担体を提供することがさらなる目的である。本システムは、多くの点で従来技術のポリマー及びゲルシステムより優れる。
本発明は、疎水性生体接着性ポリマーポリ酸無水物(引き続き第2のポリマーとの分子間水素結合を形成できる)に共有結合した酸化窒素ドナーの生成物を含む生体接着性超高部分子複合体を提供する。
従って、一態様では、本発明は、下記一般式の生体接着性超高分子複合体を提供する。
【0009】
【化1】
【0010】
式中、R1は無置換又はアルコキシ基で置換されているアルカンであり;R2は低級アルカンであり;R3及びR4は任意に置換されていてもよい長鎖アルカンである。構造の多様性に関して、式の全ての3つのR基を広い範囲にわたって変えて分離できる物質を生成することができ;Wは水素結合受容官能基含有実体(entitiy)であり;Yはカルボン酸エステル又はアミドであり;Zは連結基であり;T1、T2、T3及びT4は末端基であり;m1、m2、m3、n1及びn2は少なくとも25から選択される整数であり;かつPは約1×103〜1×107の分子量を有し、Qは約1×103〜1×107の分子量を有する。
【0011】
超高分子複合体は、好ましくは、R1がマレイン酸コポリマーであり、さらに好ましくは前記マレイン酸コポリマーが、ポリ(メチルビニルエーテル-co-マレイン酸)、ポリ(ビニルピロリドン-co-ジメチルマレイン酸)、ポリ(エチレン-co-マレイン酸)、ポリ(イソブチレン-co-マレイン酸)、ポリ(スチレン-co-マレイン酸)、ポリ(エチレン-co-酢酸エチル-co-マレイン酸)、ポリ(マレイン酸-co-オクタデセン)、ポリエチレン-グラフト-マレイン酸、ポリプロピレン-グラフト-マレイン酸、及びポリイソプレン-グラフト-マレイン酸から成る群より選択される。
さらなる態様では、本発明は、下記一般式の生体接着性超高分子複合体を提供する。
【0012】
【化2】
【0013】
式中、R1は、アルキルビニルエーテル(C1-C5)、エチレン、プロピレン、イソブチレン、ブタジエン、1-オクタデセン、スチレン、マレイン酸、又は無水マレイン酸単位であり;W1及びW2は、ビニルピロリドン、エチレンオキシド又はプロピレンオキシド、酢酸ビニル、アルコキシル置換グルコピラノース、グルコサミン、及びアセチルグルコサミンから成る群より選択される水素結合受容官能基含有実体であり;R2はH、脂肪酸エステル、又は脂肪アルコールであり;Xはカルボン酸エステル又はアミド結合であり;RSNOは、システイン、γ-Glu-Cys、α-Glu-Cys、グルタチオン、ホモグルタチオン、グルタチオンエチルエステル、ヒドロキシメチル-グルタチオン、γ-Glu-Cys-Glu、α-Glu-Cys-Gly、α-Glu-Cys-β-Ala、α-Glu-Cys-Ser、α-Glu-Cys-Glu、-SH及び-NH2及び/又は-OH官能基を含有する他のグルタチオン類似体、或いは下記ペプチドの1つ:(γ-Glu-Cys)q、(γ-Glu-Cys)q-Gly、(γ-Glu-Cys)q-β-Ala、(γ-Glu-Cys)q-Ser、(γ-Glu-Cys)q-Glu、(α-Glu-Cys)q、(α-Glu-Cys)q-Gly、(α-Glu-Cys)q-β-Ala、(α-Glu-Cys)q-Ser、及び(α-Glu-Cys)q-Glu(ここで、q=2〜11)のS-ニトロソチオールであり;T1及びT2は末端基であり;m、n及びpは25より大きい整数である。
【0014】
超高分子複合体は、好ましくは、T1-[-R1-CH(COOH)-CH(X-RSNO)-]m-T2が、RSNOと無水マレイン酸ポリマー又はコポリマーの反応付加体であり、前記無水マレイン酸ポリマー又はコポリマーが、ポリ(メチルビニルエーテル-alt-無水マレイン酸)、ポリ(マレイン酸-co-無水マレイン酸)、ポリ(無水マレイン酸)、ポリ(ビニルピロリドン-co-ジメチル無水マレイン酸)、ポリ(酢酸ビニル-co-無水マレイン酸)、ポリ(エチレン-alt-無水マレイン酸)、ポリ(イソブチレン-alt-無水マレイン酸)、ポリ(スチレン-alt-無水マレイン酸)、ポリ(エチレン-co-酢酸エチル-co-無水マレイン酸)、及びポリ(無水マレイン酸-alt-1-オクタデセン)から成る群より選択される。
本発明では、無水マレイン酸含有ポリマーを利用して、RSNO、好ましくはGSNOを、ペンダント酸無水物基とGSNOの一級アミン基との相互作用によって固体化する。一般的に温和な条件下における無水マレイン酸含有ポリマーの反応性のため該ポリマーは生理活性剤の固定化に適している[27〜29]。例えばUS2001/0046476では、殺菌剤、フレーバー剤及びエッセンシャルオイルをポリ(メチルビニルエーテル-alt-無水マレイン酸)(PVMMA)及びその誘導体に共有結合させて徐放性オーラルケア組成物を提供している。
NOの効率的な局所送達を果たすため、本NO放出超高分子複合体の創傷部位における滞留時間を効率的に長くするその顕著な生体接着特性に鑑み、PVMMAをNO担体として利用することが非常に有利であろう。PVMMAの疎水性及びその表面浸食特性は、長期にわたるNO放出の達成を容易にするだろう。実際に、PVMMAとその変性誘導体は、歯科用接着剤、化粧品及び薬物送達システムで多くの適用を見出した[30〜31、米国特許第6,355,706号、US2007/196459、WO 2006/015093、WO 2001/087276]。
酸化窒素ドナーRSNOは、好ましくは、システイン、γ-Glu-Cys、α-Glu-Cys、グルタチオン(GSH)、グルタチオンエチルエステル、ホモグルタチオン、ヒドロキシメチル-グルタチオン、γ-Glu-Cys-Glu、α-Glu-Cys-Gly、α-Glu-Cys-β-Ala、α-Glu-Cys-Ser、α-Glu-Cys-Glu、-SH及び-NH2及び/又は-OH官能基を含有する他のグルタチオン類似体、或いは下記ペプチドの1つ:(γ-Glu-Cys)n、(γ-Glu-Cys)n-Gly(フィトケラチンとしても知られる)、(γ-Glu-Cys)n-β-Ala、(γ-Glu-Cys)n-Ser、(γ-Glu-Cys)n-Glu、(α-Glu-Cys)n、(α-Glu-Cys)n-Gly、(α-Glu-Cys)n-β-Ala、(α-Glu-Cys)n-Ser、及び(α-Glu-Cys)n-Glu(ここで、n=2〜11)のS-ニトロソチオールから成る群より選択される。
T3-[R2W.]n1-[R3]n2-T4及び[W1]n-[W2]n-R2水素結合受容ポリマーは、好ましくは、ポリ(ビニルピロリドン)、ポリエチレングリコール、ポリ(エチレンオキシド)、ポリ(ビニルピロリドン-co-酢酸ビニル)、ポリエチレンオキシド-ポリプロピレンオキシドブロックコポリマー(プルロニック又はポロキサマー)、ポリエチレングリコール脂肪アルコールエステル、ポリエチレングリコール脂肪酸エステル、エチルセルロース、及びキトサンから成る群より選択され、さらに好ましくはポリ(ビニルピロリドン)である。
好ましくは、Y.Z.SNOは、アミノ-S-ニトロソグルタチオン又はアミド-フィトケラチンである。
【0015】
さらなる態様では、本発明は、生体接着性超高分子酸化窒素発生可能ポリマー複合体の製造方法であって、以下の工程
i. アミノ連結基を有するS-ニトロソ化合物を生体接着性疎水性ポリ酸無水物化合物と共有結合させて酸化窒素ドナーポリマー担体を形成する工程;及び
ii. 前記担体を親水性分子間水素結合受容ポリマーと混合して前記超高分子酸化窒素発生可能複合体を生成する工程
を含む方法を提供する。
好ましい酸化窒素ドナーRSNOは、システイン、γ-Glu-Cys、α-Glu-Cys、グルタチオン(GSH)、ホモグルタチオン、ヒドロキシメチル-グルタチオン、γ-Glu-Cys-Glu、α-Glu-Cys-Gly、α-Glu-Cys-β-Ala、α-Glu-Cys-Ser、α-Glu-Cys-Glu、-SH及び-NH2及び/又は-OH官能基を含有する他のグルタチオン類似体、或いは下記ペプチドの1つ:(γ-Glu-Cys)n、(γ-Glu-Cys)n-Gly(フィトケラチンとしても知られる)、(γ-Glu-Cys)n-β-Ala、(γ-Glu-Cys)n-Ser、(γ-Glu-Cys)n-Glu、(α-Glu-Cys)n、(α-Glu-Cys)n-Gly、(α-Glu-Cys)n-β-Ala、(α-Glu-Cys)n-Ser、及び(α-Glu-Cys)n-Glu(ここで、n=2〜11)のS-ニトロソチオールから成る群より選択される。最も好ましくは、S-ニトロソチオール化合物はGSNO又はフィトケラチンである。
好ましいポリ酸無水物化合物は、分子量(Mw)が約5,000〜2,000,000の範囲の無水マレイン酸ポリマー又はコポリマーであり、前記無水マレイン酸ポリマー又はコポリマーは、例えば、好ましくはポリ(メチルビニルエーテル-alt-無水マレイン酸)、ポリ(マレイン酸-co-無水マレイン酸)、ポリ(無水マレイン酸)、ポリ(ビニルピロリドン-co-ジメチル無水マレイン酸)、ポリ(酢酸ビニル-co-無水マレイン酸)、ポリ(エチレン-alt-無水マレイン酸)、ポリ(イソブチレン-alt-無水マレイン酸)、ポリ(スチレン-alt-無水マレイン酸)、ポリ(エチレン-co-アクリル酸エチル-co-無水マレイン酸)、及びポリ(無水マレイン酸-alt-1-オクタデセン)から成る群より選択される。最も好ましくは、ポリ酸無水物化合物はポリ(メチルビニルエーテル-alt-無水マレイン酸)である。
水素結合受容ポリマーは、好ましくは、ポリ(ビニルピロリドン)、ポリエチレングリコール、ポリ(エチレンオキシド)、ポリ(ビニルピロリドン-co-酢酸ビニル)、ポリエチレンオキシド-ポリプロピレンオキシドブロックコポリマー(プルロニック又はポロキサマー)、ポリエチレングリコール脂肪アルコールエステル、ポリエチレングリコール脂肪酸エステル、エチルセルロース、及びキトサンから成る、分子量(Mw)が約5,000〜7,000,000の群から選択され、最も好ましくは前記水素結合受容ポリマーがポリ(ビニルピロリドン)である、請求項17に記載の方法である。
結果として生じる超高分子酸化窒素発生可能ポリマー複合体は、好ましくはポリ酸無水物化合物と水素結合受容ポリマーを1:9〜9:1、さらに好ましくは2:5〜5:2、最も好ましくは1:2〜2:1の相対質量比で含む。
結果として生じる超高分子酸化窒素発生可能ポリマー複合体中の酸化窒素ドナーRSNOの総装填は、好ましくは1〜50wt%、さらに好ましくは1〜30%、最も好ましくは5〜20%の範囲である。
【0016】
本発明は、さらなる態様では、上述した方法で製造された生体接着性超高分子酸化窒素発生可能複合体を提供する。
なおさらなる態様では、本発明は、上述したように、有効な創傷治癒量の前記超高分子複合体と、生理学的に許容しうる担体とを含む医薬組成物を提供する。
なおさらなる態様では、本発明は、上述したように、前記超高分子複合体を加工してコーティングにするためのレイヤーバイレイヤーアセンブリ(layer-by-layer assembly)方法を提供する。
なおさらなる態様では、本発明は、上述したように、前記超高分子複合体を紡糸繊維として製造するための電気紡糸(electrospinning)方法を提供する。
なおさらなる態様では、本発明は、上述したように、前記超高分子複合体をミクロスフェアとして製造するための超音波噴霧方法を提供する。
それによって、本発明は、上述したように、粉末、マイクロカプセル、紡糸繊維、又は基材、例えば、カテーテル若しくはステント表面上のコーティングという物理的形態で超高分子複合体を提供する。
従って、本発明は、粉末形態で、創傷手当用品として役立ち、また、経皮パッチ、包帯、縫合糸などの中に組み入れることができる新規の酸化窒素放出ポリマー複合体を提供する。それは、該ポリマー複合体を、多層方法によって結束させる前に、医療デバイス上の血液接触面に適用することによって、コーティングの形態を取ることもできる。この超高分子複合体は、持続性かつ制御可能様式で治療量の酸化窒素を生成して、それを患部組織、例えば慢性の治癒が不十分な創傷内の当該組織に送達する。
【0017】
従って、さらなる態様では、本発明は、上述した超高分子複合体に基づいて、装填手順中に不織マットを製造するための電気紡糸装置を使用すること又は紡糸プロセス中にコーティング基材を使用することに関する。
従って、さらなる態様では、本発明は、上述した超高分子複合体に基づいた均一サイズのミクロスフェアを製造するための超音波噴霧技術の利用に関する。結果として生じたミクロスフェアをさらにカプセルに組み入れるか又は噴霧プロセス中に基材上にコーティングすることができる。
従って、さらなる態様では、本発明は、皮膚に適用するための皮膚被覆物を提供し、該被覆物は、上述したように、有効な創傷治癒量の超高分子複合体を組み入れている。皮膚被覆物が包帯又は創傷手当用品であってよい。
【0018】
さらなる態様では、本発明は、皮膚の創傷又は感染症の治癒を促進する方法であって、有効な創傷又は感染症治癒量の、上述した生体接着性超高分子複合体又はその医薬的に許容しうる組成物を前記創傷に適用する工程を含む方法を提供する。
なおさらなる態様では、本発明は、上述したように、生体接着性超高分子複合体又はその医薬的に許容しうる組成物の、皮膚の創傷又は感染症の治癒を促進するための使用を提供する。
【0019】
このように、本発明は、3つの必須の主要素、すなわち、(1)疎水性、生体適合性、生体内分解性であり、かつ酸無水物官能基を含むポリマー担体、例えば、ポリ(メチルビニルエーテル-alt-無水マレイン酸)[PVMMA]等、(2)酸化窒素ドナー、例えば温和な条件下で上記ポリマー担体の高分子骨格又は側鎖上の酸無水物基に共有結合することができるS-ニトロソグルタチオン(GSNO)又は他のS-ニトロソ化グルタチオン誘導体、及び(3) 前記第1のポリマー担体と強力な物理的分子間複合体を形成する第2のポリマー、例えば、ポリ(ビニルピロリドン)[PVP]等を含む。
従って、本発明の分野は、創傷及び感染症を治療するため、さらに詳細には、酸化窒素を長期にわたって局所放出して創傷及び感染症を治療するためのデバイス及び方法に関する。本発明の複合体を粉末にして、創傷治癒を促進するための包帯又は創傷手当用品に組み入れることができる。さらに、本発明の複合体を、肺性高血圧を下げるための吸入製剤の成分として配備させるか又は循環障害の治療に適用することができる。
少なくとも約7日間にわたる生体接着性超高分子複合体からの長期の酸化窒素放出は、創傷及び感染症の有効な治療を提供する。理論によって拘束されるものではないが、我々は、この効力は、生体接着性疎水性ポリマー、例えばPVMMAのカルボン酸基を介して水素結合している水素結合受容官能基、例えばPVPの存在のため、ジスルフィド結合の形成及びPVMMA疎水性マトリックス内に埋まっている立体障害RSNOからの酸化窒素の放出の速度を遅くするためであると考える。
【図面の簡単な説明】
【0020】
以下の図面は、本明細書の一部を形成し、かつ本発明の必須態様をさらに実証するために含まれる。本発明をより良く理解してもらうため、例示としてのみ、以下の図面を参照して特定の好ましい実施形態を説明する。
【図1】22℃における酸性及び中性媒体中のGSNOからのNO分解速度論の時間経過を示す。
【図2】GSNO、GSNO-PVMMAのUV-Visスペクトル、UV範囲(a)及び可視範囲(b)内の最大吸収(λ=336nm及びλ=545nmにおけるS-NO結合の特徴的吸光度に相当し、それぞれσ-σ*及びπ-π*電子遷移に割り当てられる。(1)アセトンに溶解した純粋PVMMA;(2)水性媒体中のGSNO;(3)水性媒体中のGSNO結合PVMMA)を示す。
【図3A】グルタチオンエチルエステル(GSHEE)のS-ニトロソ反応のUV-Visスペクトルの特徴づけを示す。
【図3B】グルタチオンエチルエステル(GSHEE)のS-ニトロソ反応のUV-Visスペクトルの特徴づけを示す。
【図4】水性媒体中の(a)純粋PC5(フィトケラチン);(b)PC5と亜硝酸ナトリウムの間のS-ニトロソ化;(c)S-ニトロソPC5結合PVMMA/PVP複合体のUV-Visスペクトルを示す。
【図5】それぞれ22℃におけるGSNO、S-ニトロソ-GSHEE及びS-ニトロソ-PC5からの0.1N HCl中のNO分解速度論を示す。
【図6】純粋GSNO、純粋PVMMA及びGSNO結合PVMMAフィルムのFTIRスペクトルを示す。
【図7】純粋PVMMA、PVP及びPVMMA/PVP複合体のFTIRスペクトルを示す。
【図8】リン酸緩衝食塩水中20mgのGSNO-PVMMA結合体からの37℃(a)及び22℃(b)におけるNOのin vitro放出挙動、並びに16.6wt%のGSNOを含み、PVMMA/PVPの質量比が1:1のGSNO-PVMMA/PVP超高分子複合体からの37℃(c)及び22℃(d)におけるNOのin vitro放出挙動を示す。
【図9】リン酸緩衝食塩水中25℃にて、15.04wt%のGSNOを含み、異なるPVMMA/PVP質量比の20mgのGSNO-PVMMA/PVP超高分子複合体からのNOのin vitro放出挙動を示す。
【図10】リン酸緩衝食塩水中37℃及び25℃にて、15.04wt%のGSNOを含み、PVMMA/PVPの質量比が1:1の20mgのGSNO-PVMMA/PVP超高分子複合体からのNOのin vitro放出挙動を示す。
【図11】リン酸緩衝食塩水中、15.04wt%のGSNOを含み、PVMMA/PVPの質量比が1:1で、異なるPVMMA分子量(a)及びPVP分子量(b)の20mgのGSNO-PVMMA/PVP超高分子複合体からのNOのin vitro放出挙動を示す。
【図12】リン酸緩衝食塩水中室温にて、7.52wt%(231系列)及び15.04wt%(232系列)のGSNOを含み、PVMMA/PVPの質量比が1:1の20mgのGSNO-PVMMA/PVP超高分子複合体からのNOのin vitro放出挙動を示す。
【図13】S-ニトロソPC5-PVMMA/PVP超高分子複合体粉末からのNOのin vitro持続性放出挙動を示す。
【図14】室内条件下で6カ月貯蔵前後のGSNO-PVMMA/PVP超高分子複合体粉末からのNOのin vitro放出挙動を比較する。
【図15】UV照射前後のGSNO-PVMMA/PVP超高分子複合体からのNOのin vitro放出挙動を比較する。
【図16】(a1,a2)12wt%のGSNO-結合純粋PVMMA;(b1,b2)純粋EC;(c1,c2)GSNO-結合PVMMA/EC(ポリマー組成1:1)から電気紡糸されたナノ繊維のそれぞれ低倍率及び高倍率の代表的SEM写真を示す。
【図17】12%GSNO装填の乾燥した1:1のPVMMA/EC電気紡糸ファブリックの、浸漬前(a)PBSに37℃で1時間浸漬後(b);1日浸漬後(c)及び3日浸漬後(d)のSEM写真である。
【図18】紡いだままの(a)純粋PVMMA、(b)GSNO結合PVMMA/EC、(c)GSNOなしのPVMMA/EC及び(d)純粋ECナノコンポジットファブリックのFTIRスペクトルである。
【図19】紡いだままの繊維質マット((a)純粋PVMMA、(b)純粋EC;(c)GSNOなしのPVMMA/EC(1:1)及び(d)12wt%GSNO結合PVMMA/EC(1:1))の機械的性質を示す。
【図20】0.1M PBS(pH7.4)中、(A)室温及び(B)37℃において、12%のGSNOを組み入れたPVMMA/EC電気紡糸マット((a)純粋PVMMA、(b)2:1のPVMMA/EC;(c)1:1のPVMMA/EC;(d)1:2のPVMMA/EC;(e)純粋EC)からのNOのin vitro放出速度に及ぼす組成の効果を示す。
【図21】超音波噴霧法で調製したGSNO-PVMMA(AN139)/ECミクロスフェアの顕微鏡写真を示す。
【図22】超音波噴霧法で調製したGSNO-PVMMA(AN169)/ECミクロスフェアの顕微鏡写真を示す。
【図23】コントロール群と試験群との間の創傷閉鎖速度を比較する(*p<0.05)。
【図24】コントロール(C3)とGSNO治療(T9)動物の創傷前及び創傷後4、10、16日後の創傷の写真を示す。
【発明を実施するための形態】
【0021】
〔好ましい実施形態の詳細な説明〕
以下の実施例を参照することによって本発明はより容易に理解されるだろう。なお、実施例は本発明の特定の態様及び実施形態をさらに説明する目的のためだけに含まれるものであり、いかなる場合にも本発明を制限するつもりはない。
〔RSNO-PVMMA/PVP複合体粉末の調製〕
(材料)
以下の実験において、還元型グルタチオン(GSH)、還元型グルタチオンエチルエステル(GSHEE)、亜硝酸ナトリウム(NaNO2)、スルファニルアミド(SULF)及びN-(1-ナフチル)エチレンジアミン二塩酸塩)(NEDD)はSigma-Aldrich Chemical Co. Oakville, CA)から得た。全てのフィトケラチンはAnaSpec Inc.(San Jose, CA, US)から購入した。
全てのポリマーはISP(New Jersey, USA)及びDow Chemical Company(Midland, MI)から得た。分析試薬グレードの他の化学薬品及び溶媒はSigma Aldrichから得、特に断らない限り、入手したまま使用した。Milli-Qグレード(Millipore, SA, France)脱イオン水を全ての溶液及び緩衝液に使用した。以下の実施例で用いる全てのPVMMA及びPVPは、特に断らない限り、PVMMA AN-169及びPVP K-90である。
【実施例1】
【0022】
(RSNOの合成)
下記反応式に従うチオールのニトロソ化によってRSNOの合成を行う。
【0023】
【数1】
【0024】
この反応は、少なくとも合成の観点からは非常に迅速、効率的、かつ定量的である。しかし、この反応は、不安定な生成物(その純粋状態で)を生じさせることが多い。下記反応式に示すように、ジスルフィド架橋形成をもたらすRSNOのホモリシスが、その熱的不安定性の原因である主な機構である。
【0025】
【数2】
【0026】
この反応についての詳細な情報を以下の実施例で述べる。
A. GSNOを調製するための実験手順
光への露出から保護した酸性媒体中で還元型グルタチオン(GSH)と等モルの亜硝酸塩を反応させることによって、GSNOを容易に調製した。
【0027】
【化3】
スキーム1
【0028】
要するに、5mlの0.2N HCl中のグルタチオン(GSH)(154mg,0.5mmol)の撹拌氷冷溶液にNaNO2の一部(35mg,0.5mmol)を加えた。この反応は80%を超える高収率でGSNOを与える。最終赤色溶液をアルミニウム箔で光から保護すると、該溶液は暗所では安定であり、合成後に精製せずに直接使用することができる。
B. GSHEEのS-ニトロソ反応
同様にグルタチオンエチルエステル(GSHEE)のS-ニトロソ化(スキーム2)を行った。要するに、2mlの0.2N HCl中のGSHEE(67mg,0.2mmol)の撹拌氷冷溶液にNaNO2の一部(14mg,0.2mmol)を加えた。結果として生じた赤色溶液をアルミニウム箔で光から保護したバイアルに貯蔵した。
【0029】
【化4】
スキーム2
【0030】
C. フィトケラチン5のS-ニトロソ反応
各PC5分子中の5個のチオール基を考慮に入れて、PC5対NaNO2のモル比が1:5であることを除き、同様にフィトケラチン5(PC5)(スキーム3)のS-ニトロソ化を達成した。
【0031】
【化5】
スキーム3
【0032】
手短に言えば、まず3mgのPC5(3.2325μmol)を氷浴内で100μlの0.2N HClに溶かしてからこの溶液に即座に100μlの新たに調製したNaNO2溶液(11.152mg/ml)を加えた。結果として生じたピンク色溶液を、アルミニウム箔で光から保護したバイアルに貯蔵した。
D. ホモ-フィトケラチン2のS-ニトロソ反応
各ホモ-PC2分子中の2個のチオール基を考慮に入れて、ホモ-PC2対NaNO2のモル比が1:2であることを除き、同様にホモ-フィトケラチン2(ホモ-PC2)(スキーム4)のS-ニトロソ化を達成した。
【0033】
【化6】
スキーム4
【0034】
手短に言えば、まず1mgのホモ-PC2(1.8031μmol)を氷浴内で50μlの0.2N HClに溶かしてからこの溶液に即座に50μlの新たに調製したNaNO2溶液(2.4883mg/ml)を加えた。結果として生じたピンク色の溶液を、アルミ箔で光から保護したバイアルに貯蔵した。
【実施例2】
【0035】
(RSNOのPVMMAへの結合)
無水マレイン酸コポリマーの注目すべき特徴は、周知の酸無水物成分と一級アミン基との高い反応性であり、より低度までアルコールと反応する。この反応は、コポリマーの溶解状態でか又は何らかの生理活性分子の界面提示後の表面化学によって遂行される。このようなアシル化反応は一般的に温和な条件下で起こり、本発明の場合、室温で20分以内に自発的に遂行された。
原則として、一級アミン基を含有する全てのRSNOは、スキーム5に従ってPVMMA等の無水マレイン酸コポリマーと反応することができる。該反応は、フリーのカルボン酸基の形成をももたらし、これは、第2のポリマーと分子間高分子複合体を形成するという次の必須工程用のプロトンを与えるのに必要不可欠である。酸無水物基と反応することについては、チオール基がアミン基より反応性が高いので、PVMMAとの結合前にまずRSNOを調製すべきことが非常に重要である。
【0036】
【化7】
スキーム5
【0037】
A. GSNOのPVMMAへの結合
GSNOのPVMMAへの容易な付着は、GSNOは0.1N HClに、PVMMAはアセトンにそれぞれ別々に溶解させなければならず、かつアセトン及び水性HClが偶然それぞれGSNO及びPVMMA用の沈殿剤であるという事実のため、GSNOとPVMMAの不均一反応によって達成された。従って、グラフト反応は溶液中でGSNOとPVMMAの界面で起こる。以下の実施例では、PVMMAの質量に対して50%までGSNO装填を達成できる。
A1. 7.52%装填を伴うGSNOのPVMMAへの結合
まず、500mgのPVMMAを10mlのアセトンに均一に溶かした。次に、実施例1Aに従って得た1mlのGSNO溶液を、氷浴内で撹拌下のPVMMA溶液に滴下した。引き続き、溶液をTeflon皿に注ぎ、ドラフト内に置き;室温下での空気乾燥又は真空乾燥のいずれかでアセトンを除去し、露光から保護した。得られたピンク粉末形態のGSNO-PVMMAを収集してデシケーター内で貯蔵した。さらに、結果として生じた溶液の一部を乾燥させずに次工程のために保管した。
A2. 15.04%装填を伴うGSNOのPVMMAへの結合
まず、500mgのPVMMAを10mlのアセトンに均一に溶かした。次に、実施例1Aに従って得た2mlのGSNO溶液を、氷浴内で撹拌下のPVMMA溶液に滴下した。引き続き、溶液をTeflon皿に注ぎ、ドラフト内に置き、室温下での空気乾燥又は真空乾燥のいずれかでアセトンを除去し、露光から保護した。得られたピンク粉末形態のGSNO-PVMMAを収集してデシケーター内で貯蔵した。さらに、結果として生じた溶液の一部を乾燥させずに次工程のために保管した。
A3. 30%装填を伴うGSNOのPVMMAへの結合
まず、500mgのPVMMAを10mlのアセトンに均一に溶かした。次に、実施例1Aに従って得た4mlのGSNO溶液を、氷浴内で撹拌下のPVMMA溶液に滴下した。引き続き、溶液をTeflon皿に注ぎ、ドラフト内に置き、室温下での空気乾燥又は真空乾燥のいずれかでアセトンを除去し、露光から保護した。得られたピンク粉末形態のGSNO-PVMMAを収集してデシケーター内で貯蔵した。さらに、結果として生じた溶液の一部を乾燥させずに次工程のために保管した。
【0038】
B. S-ニトロソ-GSHEEのPVMMAへの結合
上記と同じ方法で8.1wt%装填を伴うS-ニトロソ-GSHEEのPVMMAへの付着を達成した。要するに、1mlのS-ニトロソ-GSHEE(実施例1Bに従う)を、氷浴内で撹拌下の10mlの5%PVMMAアセトン溶液に滴下した。混合物を10分間反応させてからTeflon皿に注いで暗所で空気乾燥させた。アセトンの急速な揮発のため、結果として生じたピンク色粉末を1時間で収集し、引き続きデシケーター内で貯蔵した。同様に、結果として生じた溶液の一部を乾燥させずに次工程のために保管した。
C. S-ニトロソPC5のPVMMAへの結合
上記と同じ方法で6wt%装填を伴うS-ニトロソ-PC5のPVMMAへの付着を達成した。要するに、まず50mgのPVMMAを5mlのアセトンに溶かしてから200μlのS-ニトロソ-CP5溶液(実施例1Cに従う)を、氷浴内で撹拌下のPVMMA溶液に滴下し、混合物を10分間反応させてから、この合成の次工程で即座に使用した。
D. S-ニトロソ-ホモ-PC2のPVMMAへの結合
上記と同じ方法でS-ニトロソ-PC5のPVMMAへの付着を達成した。まず20mgのPVMMAを2mlのアセトンに溶かしてから100μlのS-ニトロソ-CP5溶液(実施例1Dに従う)を、氷浴内で撹拌下のPVMMA溶液に滴下し、混合物を10分間反応させてから、この合成の次工程で即座に使用した。
【実施例3】
【0039】
(RSNO結合PVMMAとPVPの複合体形成)
RSNO結合PVMMAとPVPの複合体形成は、下記スキーム6に示す共重合体(interpolymer)の水素結合相互作用に基づく。
【0040】
【化8】
スキーム6
【0041】
A. GSNO-PVMMA/PVP複合体の調製
GSNO-PVMMA/PVP複合体を調製するため、まず10:1(体積比)のアセトンとエタノールの混合物中6.36wt%のPVP溶液を調製した。PVPは純粋なアセトンには溶けないので、対応するGSNO-PVMMA溶液の組成に従う溶液調製を容易にするために一定量のエタノールを添加しなければならない。
A1. PVMMAに対して7.52%のGSNO装填を伴うGSNO-PVMMA/PVP複合体の調製
複合体形成前にまず3mlのエタノールをGSNO-PVMMA溶液(実施例2A1に従う10/1のアセトン/0.1N HCl)に添加した。引き続き、氷浴内で激しく撹拌しているGSNO-PVMMA溶液に測定量のPVP溶液を速く注いだ。分子間水素結合によって複合体形成が起こるにつれて、結果として生じた混合物の粘度が、組成(異なる量のPVP溶液を導入することによって、PVMMA/PVP質量比を1:9〜9:1に調整した)によって変化するゲル化度のピンク色のゲル様生成物をもたらす特有の増加を示した。
A2. PVMMAに対して15.04%のGSNOを伴うGSNO-PVMMA/PVP複合体の調製
複合体形成前にまず4mlのエタノールをGSNO-PVMMA溶液(実施例2A2に従う10/2のアセトン/0.1N HCl)に添加した。引き続き、氷浴内で激しく撹拌しているGSNO-PVMMA溶液に測定量のPVP溶液を速く注いだ。分子間水素結合によって複合体形成が起こるにつれて、結果として生じた混合物の粘度が、組成(異なる量のPVP溶液を導入することによって、PVMMA/PVP質量比を1:9〜9:1に調整した)によって変化するゲル化度のピンク色のゲル様生成物をもたらす特有の増加を示した。
A3. PVMMAに対して30%のGSNOを伴うGSNO-PVMMA/PVP複合体の調製
複合体形成前にまず5mlのエタノールをGSNO-PVMMA溶液(実施例2A2に従う10/4のアセトン/0.1N HCl)に添加した。引き続き、氷浴内で激しく撹拌しているGSNO-PVMMA溶液に測定量のPVP溶液を速く注いだ。分子間水素結合によって複合体形成が起こるにつれて、結果として生じた混合物の粘度が、組成(異なる量のPVP溶液を導入することによって、PVMMA/PVP質量比を1:9〜9:1に調整した)によって変化するゲル化度のピンク色のゲル様生成物をもたらす特有の増加を示した。
その後、実施例3のA1、A2、及びA3の結果の全ての半固体生成物をTeflon皿に移してドラフト内で空気乾燥させた。ピンク色ポリマー複合体が完全に凝固した後、そのようにして得られた脆性生成物をドライアイスと混合し、Micro-MillTM内で粉砕して粉末にした。最終ピンク色粉末の異なる大きさのフラクションをMini-Sieve Micro Sieve Setで分離し、使用するまで琥珀色容器内で貯蔵した。
【0042】
B. S-ニトロソ-GSHEE-PVMMA/PVP複合体の調製
S-ニトロソ-GSHEE-PVMMA/PVP複合体を調製するため、まず10:1(体積比)のアセトンとエタノールの混合物中6.36wt%のPVP溶液を調製した。PVPはアセトンに溶けないので、複合体形成前に1mlのエタノールをS-ニトロソ-GSHEE-PVMMAの(10/1のアセトン/0.1N HCl)溶液(実施例2Bに従う)に添加した。引き続き、氷浴内で激しく撹拌しているS-ニトロソ-GSHEE-PVMMA溶液に測定量のPVP溶液を迅速に加え、即座にピンク色のゲル様複合体を生じさせた;異なる量のPVP溶液によって、PVMMA/PVPの質量比を9:1〜1:9に調整した。結果の複合体を空気乾燥させ、ドライアイスと混合し、Micro-MillTM実験室粉砕機で粉砕して粉末にした。Mini-Sieve Micro Sieve Setで最終ピンク色粉末の異なる大きさのフラクションを分離し、使用するまで琥珀色容器内で貯蔵した。
C. S-ニトロソ-PC5-PVMMA/PVP複合体の調製
S-ニトロソ-PC5-PVMMA/PVP複合体を調製するため、まず10:1(体積比)のアセトンとエタノールの混合物中6.36wt%のPVP溶液を調製した。複合体形成前に0.5mlのエタノールを5mlのS-ニトロソ-PC5-PVMMA溶液(実施例2Cに従う)に加えた。引き続き、測定量のPVP溶液をS-ニトロソ-PC5-PVMMA溶液に速く注ぎ、即座にピンク色のゲル様複合体を生じさせた。異なる量のPVP溶液によって、PVMMA/PVPの質量比を9:1〜1:9に調整した。結果の複合体を空気乾燥させ、ドライアイスと混合し、Micro-MillTM実験室粉砕機で粉砕して粉末にした。Mini-Sieve Micro Sieve Setで最終ピンク色粉末の異なる大きさのフラクションを分離し、使用するまで琥珀色容器内で貯蔵した。
D. S-ニトロソ-ホモPC2-PVMMA/PVP複合体の調製
S-ニトロソ-ホモPC2-PVMMA/PVP複合体を作製するため、まず10:1(体積比)のアセトンとエタノールの混合物中6.36wt%のPVP溶液を調製した。複合体形成前に0.2mlのエタノールを2mlのS-ニトロソ-ホモPC2-PVMMA溶液(実施例2Dに従う)に加えた。引き続き、測定量のPVP溶液をS-ニトロソ-ホモPC2-PVMMA溶液に速く注ぎ、即座にピンク色のゲル様複合体を生じさせた。異なる量のPVP溶液によって、PVMMA/PVPの質量比を9:1〜1:9に調整した。結果の複合体を空気乾燥させ、ドライアイスと混合し、Micro-MillTM実験室粉砕機で粉砕して粉末にした。Mini-Sieve Micro Sieve Setで最終ピンク色粉末の異なる大きさのフラクションを分離し、使用するまで琥珀色容器内で貯蔵した。
【実施例4】
【0043】
(RSNO-PVMMA/PVP複合体粉末の特徴づけ)
UV-visスペクトル
それぞれUV及び可視範囲の最大吸収に相当するλ=336nm及びλ=545nmにおけるS-NO結合の特徴的な吸光度の出現によって、RSNO及びRSNO結合PVMMA/PVP複合体の両方のS-NO基の形成を実証することができる。これはσ→σ*及びπ→π*電子遷移に割り当てられる。Cary 50 UV-Vis分光光度計(Varian Inc.)を用いて室温で200〜800nmの範囲のスペクトル変化を記録した。図2、3及び4は、RSNO種としてそれぞれGSNO、GSHEE及びPC5を用いた場合のスペクトル変化を示す。
【実施例5】
【0044】
(水性媒体中のRSNOからのNO分解の速度論)
この発明では、NOドナーとしてまず最初にGSHEE及びフィトケラチンを使用する。NOを運ぶそれらの能力を前記UVスペクトルで実証した。UV-Vis分光光度計を用いて、水性媒体中でのそれらの安定性を探索した。この安定性の研究のため全てのRSNOの溶液を実施例1に従って合成した。10分の時間間隔内の545nmにおける時間依存性吸光度変化から、室温でのこれらの溶液中におけるそれらの分解速度論を得た。
図5は、同様の初期溶液濃度でのGSNO、S-ニトロソ-GSHEE、及びS-ニトロソ-PC5のNO分解速度論のプロファイルを示す。全ての3つの曲線について減衰勾配から良い直線性が得られ、NO分解速度がGSNO>S-ニトロソ-GSHEE>S-ニトロソ-PC5の順序で低減することが分かった。
【実施例6】
【0045】
(FTIRスペクトル)
フーリエ変換赤外分光法(FTIR)及び汎用の減衰全反射(Attenuated Total Reflectance)(ATR)Spectrum-oneTMPerkin-Elmer分光光度計(Perkin Elmer, Connecticut, USA)で記録されたスペクトルによってGSNOのPVMMAへの結合及びPVMMAとPVPの間の水素結合相互作用を特徴づけた。2cm-1の分解能で一区画のサンプルから全てのスペクトルを収集し、3回繰り返した。全てのスペクトルから、いかなるサンプルもないバックグラウンドスペクトルを減じた。スペクトルは4000〜650cm-1で記録された。
図6が示すように、純粋PVMMAフィルムの無水マレイン酸サイクルに典型的な1855及び1773cm-1の2つの肩部がGSNO-結合PVMMAのスペクトルでは完全に消失した。原材料中の微量カルボン酸基から生じる1707cm-1のバンドは、GSNOカップリングプロセス中のPVMMAとエタノールの間のエステル化に起因しうる1724cm-1のカルボニル特有のバンドの存在に置き換えられた。GSNO-結合PVMMAのスペクトルには、結果として生じたアミド基中のC=O基に特有の1642cm-1における別の主な吸収特徴も現れた。これは、PVMMA中の酸無水物とGSNO中の一級アミノ基との間のアシル化反応の存在を意味する。
図7中、1706cm-1で検出された純粋PVMMA中のフリーのCOOH基は、カルボニル基の伸縮振動に基づく可能性がある。複合体形成後、1706から1732cm-1へのこのカルボニル伸縮振動数の観察された上方シフトは、PVPの付加による強力な分子間水素結合相互作用に起因する「フリーの」C=O基の増加を反映している。一方、PVMMA/PVP複合体では、環式イミド基から生じるPVP中の1652cm-1のバンドも1664cm-1にシフトすることが観察される。これらの2つのバンドシフトは、複合体形成が酸のO-HとPVP分子中のイミド酸素との間の水素結合に関与することを支持する強力な証拠である。
【実施例7】
【0046】
(NOのin vitro放出の研究)
20mgのRSNO-PVMMA粉末を10mlの0.1M PBS(pH7.4)に長期間浸漬させることによって、in vitro放出の研究を行った。室温又は37℃で維持したインキュベーター内で15rpmの速度で運転するロータリーシェーカー上に全サンプルを置いた。所定の時間間隔で、2mlのNO-放出媒体をサンプリングして2mlの新鮮なPBSと交換した。
RSNO-PVMMAからのNO放出を標準的なGriessアッセイで定量化した。この比色法は、NOの全ての酸化生成物を定量化することができる。NOは、中性水溶液中で下記反応に従って容易にO2と反応してNO2を生成し、次にNO2-とNO3-を形成することが分かっている。
【0047】
【数3】
【0048】
【数4】
【0049】
要するに、1mlのGriess試薬(NEDD)(0.1%w/v)及び1mlのスルファニルアミド(5%v/vのH3PO4中1%w/v)を室温で当量(1ml)のサンプルとインキュベートした。結果溶液の540nmの波長におけるUV吸光度を決定し、3〜120μmol/LのNaNO2の標準曲線からサンプル溶液中の総[NO2-]を計算し、結果をμmolとして表した。
RSNO-PVMMA/PVP複合体からのNOのin vitro放出挙動をRSNO-PVMMA粉末について上述したのと同様に行った。20mgのRSNO-PVMMA/PVP複合体粉末を10mlの0.1M PBS(pH7.4)に長期間浸した。室温又は37℃で維持したインキュベーター内で15rpmの速度で運転するロータリーシェーカー上に全サンプルを置いた。所定の時間間隔で、2mlのNO-放出溶液をサンプリングして2mlの新鮮なPBSと交換した。NO濃度をGriessアッセイで決定した。
【0050】
【化9】
スキーム7
【0051】
スキーム7にはRSNO-PVMMA複合体のNO放出挙動が示されている。複合体から酸化窒素が徐々に遊離されるにつれて、さらにジスルフィド結合が形成し、RSNOの側鎖間のジスルフィド架橋を生じさせ、さらに複合体のネットワーク構造を強化する。ポリマー構造と鎖パッキングの状態に基づき、成分ポリマーの分子量と濃度比、及び沈殿条件を調整することによって、異なる持続性かつ制御可能な放出速度を得ることができる。
A. GSNO-PVMMA及びGSNO-PVMMA/PVP複合体からのNOのin vitro放出
図8に示すように、PVPとの超高分子複合体を形成しないと、GSNO-PVMMAからのNOの放出は比較的速く、放出時間はたった3日までである。対照的に、PVPとの超高分子複合体の形成によって、水性媒体中におけるその溶解速度が低減するため、酸化窒素放出速度を有意に遅くすることができる。該NO放出の典型的プロファイルは、9日以上まで延長しうる。
B. 異なる組成のGSNO-PVMMA/PVP複合体からのNOのin vitro放出
PVMMA/PVPの種々の質量比(1/0.5、1/1、1/2、1/3)を調査した。図9に示すように、複合体中のPVP含量の増加と共にNO放出速度が上昇するが、1/1比は最も遅いNO放出速度を示し、少なくとも12日持続した。
C. 異なる温度におけるGSNO-PVMMA/PVP複合体からのNOのin vitro放出
図10は、本GSNO-PVMMA/PVP複合体(1/1)からのNO放出で温度が重要な役割を果たすことを実証し;温度が高いほど有意にその放出速度を加速する。
D. PVMMA及びPVPのMwが異なるGSNO-PVMMA/PVP複合体からのNOのin vitro放出
分子量が大きいポリマーほど、複合体形成が促進されるため、ポリマー溶解が遅くなると考えられる。図11は、NO放出挙動に及ぼすポリマー分子量の効果を示す。試験サンプルの対応分子量を表1に列挙する。図11から、PVMMA又はPVPのいずれかの分子量が小さいほどNO放出が速くなることが明らかである。
【0052】
【0053】
E. 異なる粒径のGSNO-PVMMA/PVP複合体からのNOのin vitro放出
3つの異なる平均粒径(それぞれ約0.065、0.125及び0.3mm)のGSNO-PVMMA/PVP複合体のNO放出パターンを図12に示す。粒径がNO放出挙動で重要な役割を果たすことは明白であり、粒径が小さいほどNO放出速度が速くなる。
F. S-ニトロソPC5-PVMMA/PVP複合体からのNOのin vitro放出
図13は、S-ニトロソPC5-PVMMA/PVP複合体の持続性NO放出を実証し、放出期間を少なくとも9日まで延長することができる。この場合もやはり、温度が高いほど放出が速く、かつNO装填を調整することによって放出速度を制御できることが分かる。さらに、NOドナーとしてPC5を選択すると、少なくとも90%のNO装填効率が可能であり、これはGSNOのNO装填効率より高い。
【実施例8】
【0054】
(GSNO-PVMMA/PVP複合体の安定性の研究)
A. 室内条件下の安定性
GSNO-PVMMA/PVP複合体粉末(実施例3A1参照)を6カ月の持続期間、光から保護せずにRT(相対湿度:22%)でバイアル内で貯蔵した。図14から、この安定性期間後にNO放出プロファイルに有意な変化がないことが分かる。これは、室内条件下で貯蔵した場合、本発明のGSNO-PVMMA/PVP複合体が非常に安定していることを示唆している。
B. 照射下の安定性
GSNO-PVMMA/PVP複合体粉末(実施例3A1参照)を24時間UV照射にさらした。図15は、短期のUV照射を受けた後にGSNO-PVMMA/PVP複合体が安定していることを示す。
(電気紡糸装置によるナノ繊維形態のGSNO-PVMMA/EC複合体の調製)
電気紡糸は、直径がマイクロメートル〜ナノメートルの範囲の繊維を含むポリマーの不織、多孔性、及び三次元スキャフォールドを加工するために広く適用されている。この一工程技術は、生体材料の組成、構造及び機械的性質を制御する可能性を提供する。特に、この方法は、薬物分子を軟組織に組み入れることを可能にし、その高い水蒸気透過性、良い機械的強度及び優れた可撓性のため創傷手当用品に理想的に適している。この方法では、薬物装填と最終製剤の調製を一工程で達成することができる。特に、的確な材料の選択と繊維構造設計を通じて、結果として生じる物質はさらなる望ましい特性、例えば生体接着性、弾力性及び薬物放出制御能力に恵まれる可能性がある。本発明では、この方法で、ナノ繊維に基づいたRSNO装填NO送達システムを濃縮溶液から調製することができる。
【実施例9】
【0055】
(電気紡糸用原液の調製)
A. PVMMA/ECブレンドの調製
2gのPVMMA及び1gのエチルセルロース(EC)を別々にN-ジメチルホルムアミド(DMF)とアセトン(体積比=2:3)の混合物15mlに溶かした。2種の溶液を均一にブレンドすることによって、質量比が1:0、2:1、1:1、1:2〜0:1の範囲の一連のPVMMA/ECブレンド溶液を連続的に得た。
B. GSNOのPVMMA/ECブレンドへの結合
脱イオン水とエタノールの混合物(体積比=1:1)1ml中、室温下で約308mgのGSHを69mgのNaNO2と反応させた。その直後に、結果として生じたピンク色のGSNO溶液を、激しく撹拌している上記ポリマー溶液にゆっくり滴下して安定したピンク色エマルションを得、さらに20分撹拌を続けると澄んできた。
【実施例10】
【0056】
(GSNO-PVMMA/ECブレンド溶液の電気紡糸)
ノズルとして先端が平坦のステンレススチールのゲージ20針を備えた5mlの注射器に上記ブレンド溶液を詰めた。典型的手順では、水平マウント内にある注射器ポンプ(KDS 200, KD Scientific, USA)を用いて0.2〜0.8ml/時間の速度でGSNO-PVMMA/ECブレンド溶液を供給した。高圧電源(EL 50PO.8, Glassman High Voltage Inc., USA)を用いてノズルと接地アルミニウムコレクターとの間に高電圧(12〜18kV)を印加した。先端とコレクターの間の距離を12〜16cmに調整した。GSNOの感光性と感熱性を最小限にするため、直接光がなく、かつ20℃で維持されたドラフト内に装備全体を置いて、プロセス中のNO損失を減らした。全ての紡いだままのファブリックを、直接光から保護されたデシケーター内で貯蔵し、次に使用するまで4℃で冷蔵した。
【実施例11】
【0057】
(紡いだままのマットの形態学的特徴づけ)
1kV及び2kVの加速電圧を用いて環境制御型走査電子顕微鏡(environmental scanning electron microscope)(HITACHI S-3400N SEM、日本国)によって、紡いだままのファブリックの形態学的外観及びサイズ分布を調査した。図16は、GSNO-PVMMA(13.33wt%の濃度で)、EC(6.67wt%の濃度で)、及びGSNO-PVMMA/EC複合物から紡いだナノ繊維のSEM画像を示す。調査画像中の挿入図は、対応する繊維サイズ分布を表示する。GSNO-結合PVMMA及びEC超薄繊維の平均径は、それぞれ0.82μm及び0.25μmであり、GSNO-PVMMA/EC複合物のナノ繊維は中間の平均径0.64μmを示す。
PVMMAは、典型的な浸食性(erodable)ポリマーであり、純粋なPVMMAのみに基づいた電気紡糸ナノ繊維は、PBS中37℃でのキャスティングフィルムより速く溶解するであろうことから、創傷手当用品へのその適用の主な限界を呈している。PVMMA/ECナノ繊維にECを添加すると、紡いだままのファブリックの水中における統合性を有意に改善する。図17に示すように、1:1のGSNO-PVMMA/ECナノ繊維製の膜は、37℃で3日の水中浸漬後にその繊維構造を保持した。画像cとdの間には繊維の形態の変化が実質的に存在せず、ECを添加すると、ナノ繊維がその統合性を留める能力を延長できることを示唆しており、創傷手当用品にとって望ましいことである。
【実施例12】
【0058】
(FTIRスペクトル)
スキーム8に示すように、フーリエ変換赤外分光法(FTIR)でGSNO-PVMMA/ECシステムの水素結合相互作用を特徴づけた。汎用の減衰全反射(ATR)Spectrum-oneTMPerkin-Elmer分光光度計(Perkin Elmer, Connecticut, USA)で4000〜650cm-1のスペクトルが記録された。2cm-1の分解能で一区画のサンプルから全スペクトルを収集し、3回繰り返した。全スペクトルから、いかなるサンプルもないバックグラウンドスペクトルを減じた。
【0059】
【化10】
スキーム8
【0060】
図18の純粋PVMMAのFTIRスペクトルから、1707cm-1におけるカルボニル基の伸縮振動の原因である微量カルボキシル基は、ナノ繊維形成プロセス中のわずかな加水分解に起因するか又は原材料に起因しうる。PVMMA/ECフィルムサンプルにおけるカルボニル伸縮振動数の1707から1720cm-1への明白な上方シフトは、ECの添加による強力な分子間水素結合相互作用のための「フリーの」C=O基の増加を反映している。さらに、PVMMA/ECブレンドフィルムでは、酸無水物に典型的な1855及び1773cm-1における吸光度ピークが未だ顕著である。
【実施例13】
【0061】
(紡いだままのファブリックのマイクロ引張り試験)
5kgのロードセルを備えたテクスチャーアナライザー(TA.XTplus, Stable Micro Systems, Haslemere, Surrey, UK)を用いて、GSNO無し及びGSNOを組み入れたPVMMA/EC電子紡糸ファブリックの機械的性質を評価した。引張り試験では、均一厚の電気紡糸繊維マットを30×20mmのサンプル片にカットした。この試験用の2つのクランプ間にサンプルを保持した。測定中、上クランプによって0.5mm/秒の速度で破壊するまでフィルムを引いた。力と伸びは機器によって自動的に記録された。各測定を4回繰り返し、結果を図19に示す。図19から、PVMMA/EC及びGSNO-PVMMA/ECフィルムは、一成分のPVMMA又はECフィルムの機械的性質を有意に超える改善された機械的性質を示すことが明らかである。この改善は、PVMMAとECの間の水素結合相互作用に起因すると考えられる。
【実施例14】
【0062】
(紡いだままのマットからのNOのin vitro放出)
20mgの電気紡糸マット(約2×2cm2)を10mlの0.1M PBSに長時間浸すことによって、NOのin vitro放出の研究を行った。37℃で維持したインキュベーター内のロータリーシェーカー上に全サンプルを置いた。所定時間間隔で、5mlの放出媒体を5mlの新鮮なPBSと交換した。
実施例7で述べたGriessアッセイによって、繊維からのNO放出を定量化した。図20A及び20Bにそれぞれ室温及び37℃について、異なる組成の紡いだままのナノ繊維からの、pH7.4の緩衝液中におけるNO放出の結果を示す。GSNO-PVMMA/EC(1:1)からのNO放出速度は、温度に依存して約35〜40μmol/g(マット)であり、NO放出は、EC含量の増加とともに有意に遅くなることが分かる。複合物フィルムは、1週間にわたる長期放出を示す。
(超音波噴霧装置を用いたRSNO-PVMMA装填ミクロスフェアの調製)
超音波噴霧は、噴霧乾燥、マイクロカプセル化及び基材コーティングに広く適用されている。この一工程法は、精度の高い均質ミクロスフェア及び薄膜コーティングを効率的に製造することができる。単一又は二重供給ノズルから噴霧された液滴は、空中及び収集浴内で凝固しうる。濃縮ポリマー溶液に適用される電気紡糸法と異なり、この方法は特に希釈ポリマー溶液に適している。以下の実施例では、RSNO装填超高分子複合体に基づいたミクロスフェアの、この方法による製造について説明する。
【実施例15】
【0063】
(GSNO-PVMMA(Gantrez(登録商標)AN 139)/P(VP/VAc)(Plasdone(登録商標)S-630)ブレンド溶液の超音波噴霧)
それぞれ分子量が1.0×106及び2.4×104の低分子量のPVMMA(Gantrez(登録商標)AN139)及びポリ(ビニルピロリドン-co-酢酸ビニル)(Plasdone(登録商標)S-630)に基づいた超高分子NO放出複合体をこの実施例のために選択した。下記手順に従い、NOプロドラッグとしてGSNO-PVMMAを含むミクロスフェアを調製した。
1gのP(VP/VAc)を40mlのアセトンに溶かし、1gのPVMMAを10mlのアセトンに溶かし、このPVMMAに1mlのGSNO(実施例1A参照)を結合させ(実施例2A1参照)、結果のGSNO-PVMMA溶液を40mlのアセトンと10mlのエタノールの混合物中で希釈し、引き続きP(VP/VAc)溶液とブレンドした。
超音波発生装置を5.0wの電力で操作しながら、注射器ポンプ(KD-Scientific, Model 200)によって駆動された超音波ノズル(SONO-TEK Corp. 8700-60 MS)の入口を通じて0.5ml/分の流速で最終溶液をポンピングした。噴霧ミストは、ガラスカラム(直径15cm、高さ60cm)を通ってその沈降中に空気乾燥された。
乾燥したミクロスフェアを収集し、顕微鏡下で形態学的に特徴づけた。図21は、収集された粒子(完全に凝固したミクロスフェアに相当する)の大きさと形状を示す。
【実施例16】
【0064】
(GSNO-PVMMA(Gantrez(登録商標)AN169)/エチルセルロース(Ethocel(登録商標)NF100)ブレンド溶液の調製)
それぞれ高分子量のPVMMA(Gantrez(登録商標)AN169)(Mw:1.98×106)及びエチルセルロース(Ethocel(登録商標)NF100)(エトキシ含量、48.8%DS;粘度、80%及び20%のアルコール中5%の溶液について100cP)に基づいた超高分子NO放出複合体をこの実施例のために選択した。NOプロドラッグとしてGSNO-PVMMAを含むミクロスフェアを下記手順に従って調製した。
最初に、500mgのエチルセルロースを40mlのアセトンに均一に溶解させて1.25%の濃度を得た。一方、500mgのPVMMAを10mlのアセトンに溶かし、このPVMMAに1mlのGSNO(実施例1A参照)を結合させ(実施例2A1)、結果のGSNO-PVMMA溶液を40mlのアセトンと10mlのエタノールの混合物中で希釈し、引き続きエチルセルロース溶液とブレンドした。
超音波発生装置を5.0wの電力で操作しながら、注射器ポンプ(KD-Scientific, Model 200)によって駆動された超音波ノズル(SONO-TEK Corp. 8700-60 MS)の入口を通じて0.5ml/分の流速で最終溶液をポンピングした。噴霧ミストは、ガラスカラム(直径15cm、高さ60cm)を通ってその沈降中に空気乾燥された。
乾燥したミクロスフェアを収集し、顕微鏡下で形態学的に特徴づけた。図22は、収集された粒子(完全に凝固したミクロスフェアに相当する)の大きさと形状を示す。
【実施例17】
【0065】
(多層集合によるGSNO-PVMMA/PVP超薄コーティング)
超薄複合体コーティングを下記手順に従って製作した。まず、0.025mMのGSNO-PVMMA溶液を実施例2Aに従って調製し、アセトンとエタノールの混合物(体積比4/1)50mlに1.3gのPVPを溶かして0.02mMのPVP溶液を作製した。その溶液濃度を維持するため、全コーティング手順中、これらの2つの溶液を氷浴内に入れた。
次に、基材(スライドガラス及びPTFEシート)を最初にPVP溶液に10分間さらしてからアセトンとエタノールの溶媒混合物の3つの浴に順次、全部で4分間浸して過剰のPVPポリマーを洗い流した。その直後に、基材をGSNO-PVMMA溶液に10分間浸した後、アセトン溶液の3つの浴に順次、全部で4分間浸した。所望の厚さまで続けうる、このサイクルを20の二重層のため繰り返した。この集合プロセス後、コーティング基材を空気乾燥させて、室内条件下にてデシケーター内で貯蔵した。
【実施例18】
【0066】
(GSNO-PVMMA/PVP複合体粉末のin vivo評価)
A. 材料と方法
糖尿病性潰瘍などの慢性創傷は創傷治癒障害を被ることが多い。最近の証拠は、創傷治癒、特に創傷組織内のNOレベルの低減によって特徴づけられる糖尿病性足部潰瘍の治癒プロセスにおいて、NOが重要な役割を果たしうることを示唆している。NOドナーDETA NONOアート及びL-アルギニンによる外因性NO補充が糖尿病ラットの創傷治癒を促すことが分かった[32、33]。我々の場合、創傷治癒にけるRSNO-PVMMA/PVP超高分子複合体システム(実施例3A1から得た)の利益を評価するため糖尿病ラットモデルを用いた。前記実験を行って、(1)GSNO結合PVMMA/PVP複合体粉末から連続的にNOを発生させられるか、及び(2)このNO含有粉末製剤が糖尿病ラットモデルの創傷治癒を促進できるかを決定した。トロント大学動物管理委員会(The University of Toronto Animal Care Committee)の承認済みの動物プロトコルに従って全ての実験を行った。
B. 急性糖尿病の誘発
15匹のオスのスプラーグドーリーラット(Charles River, Montrealから)を1週間気候順応させ、適宜食物と水を与えた。傷つける7日前、ストレプトゾトシンを腹腔内(IP)注射して(体重1kg当たり60mg、クエン酸緩衝液中0.1mol/L、pH 4.5)、糖尿病を誘発した。14mmol/Lを超える血糖値と頻尿によって糖尿病の証拠を確認した。24時間後に糖尿病状態に達しなかった4匹の動物にストレプトゾトシンを再注射し、そのうちの1匹は血糖値が14mmol/L未満のままだったので、研究から除外した。糖尿病の誘発後、血糖値を1週間に2回モニターして、創傷治癒実験全体を通じて確実に糖尿病状態のままであるようにした。
C. 外科手術手技
外科手術の前日に、動物の体重を量り、2つの群に割り当てた(コントロール群の7匹及び試験群の8匹)。動物にイソフルラン吸入麻酔しながら下記手技を行った。まず、背面を剪毛し、皮膚をポピドンヨード液と70%アルコールで洗浄した。手術直前にラットに鎮痛薬(ケトプロフェン、3mg/kg、S.C.)を与えた。引き続き、8mmの生検パンチを用いて皮膚と皮筋層(panniculus carnosus)を除去して全厚切除創傷(full thickness excisional woun)を生じさせた。創傷部位において、コントロール群には20mgのブランクPVMMA/PVP複合体粉末(NO装填なし)で治療し、試験群には20mgのGSNO-PVMMA/PVP複合体粉末(実施例3A1から)で治療した。全てのポリマー粉末は数滴の無菌食塩水の助けを借りて迅速に創傷組織に付着した。
ポリマー粉末の適用後、チンキBenzodine化合物(Xenex Laboratories, Ferndale, WA)を周辺皮膚に適用して創傷を準閉鎖性ポリウレタン手当用品(TegadermTM, 3M, St. Paul, Minn)で覆った。その後、動物を個々のケージに移して標準的な食餌で維持し、適宜、水を自由に飲めるようにした。
D. 術後の創傷治療
傷つけた後最初の7日間、動物にイソフルラン吸入麻酔しながらTegaderm手当用品を毎日交換し、デジタルカメラを用いて創傷部位の写真を記録した。各写真を用いて較正スケールを記録した。傷害後7日目から、創傷はもはや手当用品によって閉鎖されなかった。
【0067】
表2
HI: > 33.3mmol/L
【0068】
表2は、Ascensia(登録商標)CONTOUR(登録商標)Blood Glucose Meterを用いて測定した動物の血糖値、及び創傷治癒持続期間の動物の体重減少を示す。糖尿病誘発後、有意な体重減少(>20%)及び多尿によって悪化した2匹の糖尿病ラットを外科手術前に安楽死させなければならなかった。
E. 画像解析
Image-Pro Plus 5.0ソフトウェアを用いて各病変の表面積を定量化し、時間の関数としてプロットした。このソフトウェアを用いて、開いた創傷の面積を決定した。結果を時間の関数として初期創傷面積の百分率で表す(図23)。各データ点について、平均及び標準偏差を計算した。
F. 統計解析
n個の観察の平均の平均値±標準誤差として本文及び図面中の全ての値を表した。独立した両側スチューデントt-検定を利用して、実験群間の統計解析を行った。対応のある両側スチューデントt-検定を利用して、右の外科的分裂と左の未傷害の間の統計解析を行った。0.05のαレベルで信頼限界を予め定めた。
G. 創傷治癒の評価
NOは、血管内皮増殖因子発現の誘発及び上方制御に関与し、線維芽細胞及びケラチノサイトの遊走をさらに助長することが分かっている[34、35]。NOの周知の抗菌及び血管拡張作用、特に血管拡張作用は微小血管系内の血流を増やし、ひいては栄養と細胞の両方の損傷部位への送達を容易にすることから、創傷治癒のプロセスでも重要でありうる。
図23の結果は、本発明のNO放出超高分子GSNO-PVMMA/PVP複合体システムが効率的に創傷閉鎖を加速する(p<0.05)ことを実証すると期待できる。コントロール群と試験群の間の創傷閉鎖の傾向には統計的に有意な差異がある。各群の0日目、4日目、10日目及び16日目の全厚創傷の代表的写真を図24に示す。開いた面積及び顆粒化組織についての明白な創傷状態も、傷つけた後4日目、10日目及び16日目では、コントロール群におけるより試験群の方がずっと良い。
【0069】
この開示は、本発明の特定の好ましい実施形態について記載かつ説明したが、本発明は、当該特定の実施形態に制限されるものでないことを理解すべきである。むしろ、本発明は、記載かつ説明した特有の実施形態及び特徴の機能的又は機械的に等価な全ての実施形態を包含する。
【特許請求の範囲】
【請求項1】
下記一般式:
【化1】
(式中、R1はアルコキシ基で無置換又は置換されているアルカンであり;R2は低級アルカンであり;R3及びR4は任意に置換されていてもよい長鎖アルカンであり;Wは水素結合受容官能基含有実体であり;Yはカルボン酸エステル又はアミドであり;Zは連結基であり;T1、T2、T3及びT4は末端基であり;m1、m2、m3、n1及びn2は少なくとも25から選択される整数であり;かつPは約1×103〜1×107の分子量を有し、Qは約1×103〜1×107の分子量を有する)
の生体接着性超高分子複合体。
【請求項2】
R1がマレイン酸コポリマーである、請求項1に記載の超高分子複合体。
【請求項3】
前記マレイン酸コポリマーが、ポリ(メチルビニルエーテル-co-マレイン酸)ポリ(ビニルピロリドン-co-ジメチルマレイン酸)、ポリ(エチレン-co-マレイン酸)、ポリ(イソブチレン-co-マレイン酸)、ポリ(スチレン-co-マレイン酸)、ポリ(エチレン-co-酢酸エチル-co-マレイン酸)、ポリ(マレイン酸-co-オクタデセン)、ポリエチレン-グラフト-無水マレイン酸、ポリプロピレン-グラフト-マレイン酸、及びポリイソプレン-グラフト-マレイン酸から成る群より選択される、請求項2に記載の超高分子複合体。
【請求項4】
前記T3-[R2W]n1-[R3]n2-T4が、ポリ(ビニルピロリドン)、ポリエチレングリコール、ポリ(エチレンオキシド)、ポリ(ビニルピロリドン-co-酢酸ビニル)、ポリエチレンオキシド-ポリプロピレンオキシドブロックコポリマー(プルロニック又はポロキサマー)、ポリエチレングリコール脂肪アルコールエステル、ポリエチレングリコール脂肪酸エステル、エチルセルロース、及びキトサンから成る群より選択される、請求項1〜3のいずれか1項に記載の超高分子複合体。
【請求項5】
前記T3-[R2W]n1-[R3]n2-T4がポリ(ビニルピロリドン)である、請求項4に記載の超高分子複合体。
【請求項6】
Y-Z-SNOが、アミド-S-ニトロソグルタチオン又はアミド-フィトケラチンである、請求項1〜5のいずれか1項に記載の超高分子複合体。
【請求項7】
下記一般式:
【化2】
(式中、R1はアルキルビニルエーテル(C1-C5)、エチレン、プロピレン、イソブチレン、ブタジエン、1-オクタデセン、スチレン、マレイン酸、又は無水マレイン酸単位であり;W1及びW2は、ビニルピロリドン、エチレンオキシド又はプロピレンオキシド、酢酸ビニル、アルコキシル置換グルコピラノース、グルコサミン、及びアセチルグルコサミンから選択される水素結合受容官能基含有実体であり;R2はH、脂肪酸エステル、又は脂肪アルコールであり;Xはカルボン酸エステル又はアミド結合であり;RSNOは、システイン、γ-Glu-Cys、α-Glu-Cys、グルタチオン、ホモグルタチオン、ヒドロキシメチル-グルタチオン、γ-Glu-Cys-Glu、α-Glu-Cys-Gly、α-Glu-Cys-β-Ala、α-Glu-Cys-Ser、α-Glu-Cys-Glu、-SH及び-NH2及び/又は-OH官能基を含有する他のグルタチオン類似体、或いは下記ペプチドの1つ:(γ-Glu-Cys)q、(γ-Glu-Cys)q-Gly、(γ-Glu-Cys)q-β-Ala、(γ-Glu-Cys)q-Ser、(γ-Glu-Cys)q-Glu、(α-Glu-Cys)q、(α-Glu-Cys)q-Gly、(α-Glu-Cys)q-β-Ala、(α-Glu-Cys)q-Ser、及び(α-Glu-Cys)q-Glu(ここで、q=2〜11)のS-ニトロソチオールであり;T1及びT2は末端基であり;m、n及びpは25より大きい整数である)
の生体接着性超高分子複合体。
【請求項8】
T1-[-R1-CH(COOH)-CH(X-RSNO)-]m-T2が、請求項1に記載のRSNOと無水マレイン酸ポリマー又はコポリマーの反応付加体である、請求項7に記載の超高分子複合体。
【請求項9】
前記無水マレイン酸ポリマー又はコポリマーが、ポリ(メチルビニルエーテル-alt-無水マレイン酸)、ポリ(マレイン酸-co-無水マレイン酸)、ポリ(無水マレイン酸)、ポリ(ビニルピロリドン-co-ジメチル無水マレイン酸)、ポリ(酢酸ビニル-co-無水マレイン酸)、ポリ(エチレン-alt-無水マレイン酸)、ポリ(イソブチレン-alt-無水マレイン酸)、ポリ(スチレン-alt-無水マレイン酸)、ポリ(エチレン-co-酢酸エチル-co-無水マレイン酸)、及びポリ(無水マレイン酸-alt-1-オクタデセン)から成る群より選択される、請求項8に記載の超高分子複合体。
【請求項10】
前記酸化窒素ドナーRSNOが、システイン、γ-Glu-Cys、α-Glu-Cys、グルタチオン、ホモグルタチオン、ヒドロキシメチル-グルタチオン、γ-Glu-Cys-Glu、α-Glu-Cys-Gly、α-Glu-Cys-β-Ala、α-Glu-Cys-Ser、α-Glu-Cys-Glu、-SH及び-NH2及び/又は-OH官能基を含有する他のグルタチオン類似体、或いは下記ペプチドの1つ:(γ-Glu-Cys)q、(γ-Glu-Cys)q-Gly、(γ-Glu-Cys)q-β-Ala、(γ-Glu-Cys)q-Ser、(γ-Glu-Cys)q-Glu、(α-Glu-Cys)q、(α-Glu-Cys)q-Gly、(α-Glu-Cys)q-β-Ala、(α-Glu-Cys)q-Ser、及び(α-Glu-Cys)q-Glu(ここで、q=2〜11)のS-ニトロソチオールから成る群より選択される、請求項8に記載の超高分子複合体。
【請求項11】
前記[W1]n-[W2]n-R2が、ポリ(ビニルピロリドン)、ポリエチレングリコール、ポリ(エチレンオキシド)、ポリ(ビニルピロリドン-co-酢酸ビニル)、ポリエチレンオキシド-ポリプロピレンオキシドブロックコポリマー(プルロニック又はポロキサマー)、ポリエチレングリコール脂肪アルコール、及びポリエチレングリコール脂肪酸エステル、エチルセルロース、及びキトサンから成る群より選択される、請求項7〜10のいずれか1項に記載の超高分子複合体。
【請求項12】
前記[W1]n-[W2]n-R2がポリ(ビニルピロリドン)である、請求項11に記載の超高分子複合体。
【請求項13】
X-RSNOが、アミド-S-ニトロソグルタチオン又はアミド-フィトケラチンである、請求項7〜12のいずれか1項に記載の超高分子複合体。
【請求項14】
生体接着性超高分子酸化窒素発生可能ポリマー複合体の製造方法であって、以下の工程:
(i) アミノ連結基を有するS-ニトロソ化合物を生体接着性疎水性ポリ酸無水物化合物と共有結合させて酸化窒素ドナーポリマー担体を形成する工程;及び
(ii) 前記担体を分子間水素結合受容ポリマーと混合して前記超高分子酸化窒素発生可能複合体を生成する工程、
を含む方法。
【請求項15】
前記S-ニトロソ化合物を有機溶媒中で周囲温度にて前記ポリ酸無水物化合物と混合して、前記ポリ酸無水物化合物の酸無水物基と前記S-ニトロソ化合物のアミノ連結基との間の縮合反応を生じさせて、前記ポリマー担体中にアミド基及びフリーのカルボン酸基を生成する、請求項14に記載の方法。
【請求項16】
前記有機溶媒が、アセトン、ジメチルスルホキシド、N,N-ジメチルホルムアミド及びN-メチルピロリドン及びエタノールから選択される、請求項15に記載の方法。
【請求項17】
前記S-ニトロソ化合物が、S-ニトロソグルタチオン(GSNO)又はアミド-フィトケラチンである、請求項14〜16のいずれか1項に記載の方法。
【請求項18】
前記ポリ酸無水物化合物が無水マレイン酸ポリマー又はコポリマーである、請求項14〜17のいずれか1項に記載の方法。
【請求項19】
前記無水マレイン酸コポリマーが、ポリ(メチルビニルエーテル-co-無水マレイン酸)ポリ(ビニルピロリドン-co-ジメチル無水マレイン酸)、ポリ(エチレン-co-無水マレイン酸)、ポリ(イソブチレン-co-無水マレイン酸)、ポリ(スチレン-co-無水マレイン酸)、ポリ(エチレン-co-アクリル酸エチル-co-無水マレイン酸)、ポリ(無水マレイン酸-co-オクタデセン)、ポリエチレン-グラフト-無水マレイン酸、ポリプロピレン-グラフト-無水マレイン酸、及びポリイソプレン-グラフト-無水マレイン酸から成る群より選択される、請求項18に記載の方法。
【請求項20】
前記無水マレイン酸コポリマーがポリ(メチルビニルエーテル-co-無水マレイン酸)である、請求項19に記載の方法。
【請求項21】
前記水素結合受容ポリマーが、ポリ(ビニルピロリドン)、ポリエチレングリコール、ポリ(エチレンオキシド)、ポリ(ビニルピロリドン-co-酢酸ビニル)、ポリエチレンオキシド-ポリプロピレンオキシドブロックコポリマー(プルロニック又はポロキサマー)、ポリエチレングリコール脂肪アルコールエステル、及びポリエチレングリコール脂肪酸エステル、エチルセルロース、及びキトサンから成る群より選択される、請求項14〜20のいずれか1項に記載の方法。
【請求項22】
前記水素結合受容ポリマーが、ポリ(ビニルピロリドン)である、請求項21に記載の方法。
【請求項23】
請求項14〜22のいずれか1項に記載の方法で製造された、生体接着性超高分子酸化窒素発生可能複合体。
【請求項24】
有効な創傷治癒量の請求項1〜13、又は23のいずれか1項に記載の前記超高分子複合体と、生理学的に許容しうる担体とを含む医薬組成物。
【請求項25】
請求項1〜13、若しくは23のいずれか1項に記載の前記酸化窒素超高分子複合体、又は請求項24に記載のその医薬的に許容しうる組成物を含むレイヤーバイレイヤーアセンブリコーティングの調製方法であって、以下の工程:
(i) アミノ連結基を有するS-ニトロソ化合物を生体接着性疎水性ポリ酸無水物化合物と共有結合させて酸化窒素ドナーポリマー担体を形成する工程;及び
(ii) 前記担体と分子間水素結合受容ポリマーを溶液から交互に堆積させて、強力な水素結合相互作用で連結された高度に互いにかみ合った薄層を形成する工程、
を含む方法。
【請求項26】
請求項1〜13、若しくは23のいずれか1項に記載の前記超高分子複合体、又は請求項24に記載のその医薬的に許容しうる組成物を含むナノ繊維の調製方法であって、以下の工程:
(i) アミノ連結基を有するS-ニトロソ化合物を生体接着性疎水性ポリ酸無水物化合物と共有結合させて酸化窒素ドナーポリマー担体を形成する工程;及び
(ii) 前記担体を分子間水素結合受容ポリマーと混合して前記ポリマーブレンド混合物を生成する工程;
(iii) この濃縮ブレンド系を高電圧場内で電気紡糸装置を用いて紡いで、
(iv) プレートコレクター上に堆積して不織マットを形成できるか又は
(v) ロールコレクター上に堆積してコーティングを形成できる、
連続ナノ繊維を生成する工程、
を含む方法。
【請求項27】
前記水素結合受容ポリマーがエチルセルロースである、請求項26に記載の方法。
【請求項28】
請求項1〜13、若しくは23のいずれか1項に記載の前記超高分子複合体、又は請求項24に記載のその医薬的に許容しうる組成物を含むミクロスフェアの調製方法であって、以下の工程:
(i) アミノ連結基を有するS-ニトロソ化合物を生体接着性疎水性ポリ酸無水物化合物と共有結合させて酸化窒素ドナーポリマー担体を形成する工程;及び
(ii) 前記担体を水素結合受容ポリマーと混合して前記ポリマーブレンド混合物を生成する工程;
(iii) このポリマーブレンド溶液を高電圧場内で超音波噴霧装置を用いて噴霧して、
(iv) 乾燥後に収集できるか又は
(v) ロールデバイス上に堆積してコーティングを形成できる
ミクロスフェアを生成する工程、
を含む方法。
【請求項29】
前記水素結合受容ポリマーがエチルセルロース又はポリ(ビニルピロリドン-co-酢酸ビニル)である、請求項28に記載の方法。
【請求項30】
物理的形態が粉末、紡糸繊維、又は基材表面上のコーティングである、請求項1〜13及び23のいずれか1項に記載の超高分子複合体又は請求項24に記載の組成物。
【請求項31】
前記基材がカテーテル又はステントである、請求項25に記載の超高分子複合体。
【請求項32】
有効な創傷治癒量の請求項1〜13、若しくは23のいずれか1項に記載の超高分子複合体、又は請求項24に記載の組成物を組み入れた、皮膚に適用するための皮膚被覆物。
【請求項33】
包帯又は創傷手当用品の形態の請求項32に記載の皮膚被覆物。
【請求項34】
皮膚の創傷又は感染症の治癒を促進する方法であって、有効な創傷又は感染症治癒量の請求項1〜13、若しくは23のいずれか1項に記載の超高分子複合体、又は請求項24に記載の組成物を前記創傷に適用する工程を含む方法。
【請求項35】
請求項1〜13、若しくは23のいずれか1項に記載の超高分子複合体、又は請求項24に記載のその医薬的に許容しうる組成物の、皮膚の創傷又は感染症の治癒を促進するための使用。
【請求項1】
下記一般式:
【化1】
(式中、R1はアルコキシ基で無置換又は置換されているアルカンであり;R2は低級アルカンであり;R3及びR4は任意に置換されていてもよい長鎖アルカンであり;Wは水素結合受容官能基含有実体であり;Yはカルボン酸エステル又はアミドであり;Zは連結基であり;T1、T2、T3及びT4は末端基であり;m1、m2、m3、n1及びn2は少なくとも25から選択される整数であり;かつPは約1×103〜1×107の分子量を有し、Qは約1×103〜1×107の分子量を有する)
の生体接着性超高分子複合体。
【請求項2】
R1がマレイン酸コポリマーである、請求項1に記載の超高分子複合体。
【請求項3】
前記マレイン酸コポリマーが、ポリ(メチルビニルエーテル-co-マレイン酸)ポリ(ビニルピロリドン-co-ジメチルマレイン酸)、ポリ(エチレン-co-マレイン酸)、ポリ(イソブチレン-co-マレイン酸)、ポリ(スチレン-co-マレイン酸)、ポリ(エチレン-co-酢酸エチル-co-マレイン酸)、ポリ(マレイン酸-co-オクタデセン)、ポリエチレン-グラフト-無水マレイン酸、ポリプロピレン-グラフト-マレイン酸、及びポリイソプレン-グラフト-マレイン酸から成る群より選択される、請求項2に記載の超高分子複合体。
【請求項4】
前記T3-[R2W]n1-[R3]n2-T4が、ポリ(ビニルピロリドン)、ポリエチレングリコール、ポリ(エチレンオキシド)、ポリ(ビニルピロリドン-co-酢酸ビニル)、ポリエチレンオキシド-ポリプロピレンオキシドブロックコポリマー(プルロニック又はポロキサマー)、ポリエチレングリコール脂肪アルコールエステル、ポリエチレングリコール脂肪酸エステル、エチルセルロース、及びキトサンから成る群より選択される、請求項1〜3のいずれか1項に記載の超高分子複合体。
【請求項5】
前記T3-[R2W]n1-[R3]n2-T4がポリ(ビニルピロリドン)である、請求項4に記載の超高分子複合体。
【請求項6】
Y-Z-SNOが、アミド-S-ニトロソグルタチオン又はアミド-フィトケラチンである、請求項1〜5のいずれか1項に記載の超高分子複合体。
【請求項7】
下記一般式:
【化2】
(式中、R1はアルキルビニルエーテル(C1-C5)、エチレン、プロピレン、イソブチレン、ブタジエン、1-オクタデセン、スチレン、マレイン酸、又は無水マレイン酸単位であり;W1及びW2は、ビニルピロリドン、エチレンオキシド又はプロピレンオキシド、酢酸ビニル、アルコキシル置換グルコピラノース、グルコサミン、及びアセチルグルコサミンから選択される水素結合受容官能基含有実体であり;R2はH、脂肪酸エステル、又は脂肪アルコールであり;Xはカルボン酸エステル又はアミド結合であり;RSNOは、システイン、γ-Glu-Cys、α-Glu-Cys、グルタチオン、ホモグルタチオン、ヒドロキシメチル-グルタチオン、γ-Glu-Cys-Glu、α-Glu-Cys-Gly、α-Glu-Cys-β-Ala、α-Glu-Cys-Ser、α-Glu-Cys-Glu、-SH及び-NH2及び/又は-OH官能基を含有する他のグルタチオン類似体、或いは下記ペプチドの1つ:(γ-Glu-Cys)q、(γ-Glu-Cys)q-Gly、(γ-Glu-Cys)q-β-Ala、(γ-Glu-Cys)q-Ser、(γ-Glu-Cys)q-Glu、(α-Glu-Cys)q、(α-Glu-Cys)q-Gly、(α-Glu-Cys)q-β-Ala、(α-Glu-Cys)q-Ser、及び(α-Glu-Cys)q-Glu(ここで、q=2〜11)のS-ニトロソチオールであり;T1及びT2は末端基であり;m、n及びpは25より大きい整数である)
の生体接着性超高分子複合体。
【請求項8】
T1-[-R1-CH(COOH)-CH(X-RSNO)-]m-T2が、請求項1に記載のRSNOと無水マレイン酸ポリマー又はコポリマーの反応付加体である、請求項7に記載の超高分子複合体。
【請求項9】
前記無水マレイン酸ポリマー又はコポリマーが、ポリ(メチルビニルエーテル-alt-無水マレイン酸)、ポリ(マレイン酸-co-無水マレイン酸)、ポリ(無水マレイン酸)、ポリ(ビニルピロリドン-co-ジメチル無水マレイン酸)、ポリ(酢酸ビニル-co-無水マレイン酸)、ポリ(エチレン-alt-無水マレイン酸)、ポリ(イソブチレン-alt-無水マレイン酸)、ポリ(スチレン-alt-無水マレイン酸)、ポリ(エチレン-co-酢酸エチル-co-無水マレイン酸)、及びポリ(無水マレイン酸-alt-1-オクタデセン)から成る群より選択される、請求項8に記載の超高分子複合体。
【請求項10】
前記酸化窒素ドナーRSNOが、システイン、γ-Glu-Cys、α-Glu-Cys、グルタチオン、ホモグルタチオン、ヒドロキシメチル-グルタチオン、γ-Glu-Cys-Glu、α-Glu-Cys-Gly、α-Glu-Cys-β-Ala、α-Glu-Cys-Ser、α-Glu-Cys-Glu、-SH及び-NH2及び/又は-OH官能基を含有する他のグルタチオン類似体、或いは下記ペプチドの1つ:(γ-Glu-Cys)q、(γ-Glu-Cys)q-Gly、(γ-Glu-Cys)q-β-Ala、(γ-Glu-Cys)q-Ser、(γ-Glu-Cys)q-Glu、(α-Glu-Cys)q、(α-Glu-Cys)q-Gly、(α-Glu-Cys)q-β-Ala、(α-Glu-Cys)q-Ser、及び(α-Glu-Cys)q-Glu(ここで、q=2〜11)のS-ニトロソチオールから成る群より選択される、請求項8に記載の超高分子複合体。
【請求項11】
前記[W1]n-[W2]n-R2が、ポリ(ビニルピロリドン)、ポリエチレングリコール、ポリ(エチレンオキシド)、ポリ(ビニルピロリドン-co-酢酸ビニル)、ポリエチレンオキシド-ポリプロピレンオキシドブロックコポリマー(プルロニック又はポロキサマー)、ポリエチレングリコール脂肪アルコール、及びポリエチレングリコール脂肪酸エステル、エチルセルロース、及びキトサンから成る群より選択される、請求項7〜10のいずれか1項に記載の超高分子複合体。
【請求項12】
前記[W1]n-[W2]n-R2がポリ(ビニルピロリドン)である、請求項11に記載の超高分子複合体。
【請求項13】
X-RSNOが、アミド-S-ニトロソグルタチオン又はアミド-フィトケラチンである、請求項7〜12のいずれか1項に記載の超高分子複合体。
【請求項14】
生体接着性超高分子酸化窒素発生可能ポリマー複合体の製造方法であって、以下の工程:
(i) アミノ連結基を有するS-ニトロソ化合物を生体接着性疎水性ポリ酸無水物化合物と共有結合させて酸化窒素ドナーポリマー担体を形成する工程;及び
(ii) 前記担体を分子間水素結合受容ポリマーと混合して前記超高分子酸化窒素発生可能複合体を生成する工程、
を含む方法。
【請求項15】
前記S-ニトロソ化合物を有機溶媒中で周囲温度にて前記ポリ酸無水物化合物と混合して、前記ポリ酸無水物化合物の酸無水物基と前記S-ニトロソ化合物のアミノ連結基との間の縮合反応を生じさせて、前記ポリマー担体中にアミド基及びフリーのカルボン酸基を生成する、請求項14に記載の方法。
【請求項16】
前記有機溶媒が、アセトン、ジメチルスルホキシド、N,N-ジメチルホルムアミド及びN-メチルピロリドン及びエタノールから選択される、請求項15に記載の方法。
【請求項17】
前記S-ニトロソ化合物が、S-ニトロソグルタチオン(GSNO)又はアミド-フィトケラチンである、請求項14〜16のいずれか1項に記載の方法。
【請求項18】
前記ポリ酸無水物化合物が無水マレイン酸ポリマー又はコポリマーである、請求項14〜17のいずれか1項に記載の方法。
【請求項19】
前記無水マレイン酸コポリマーが、ポリ(メチルビニルエーテル-co-無水マレイン酸)ポリ(ビニルピロリドン-co-ジメチル無水マレイン酸)、ポリ(エチレン-co-無水マレイン酸)、ポリ(イソブチレン-co-無水マレイン酸)、ポリ(スチレン-co-無水マレイン酸)、ポリ(エチレン-co-アクリル酸エチル-co-無水マレイン酸)、ポリ(無水マレイン酸-co-オクタデセン)、ポリエチレン-グラフト-無水マレイン酸、ポリプロピレン-グラフト-無水マレイン酸、及びポリイソプレン-グラフト-無水マレイン酸から成る群より選択される、請求項18に記載の方法。
【請求項20】
前記無水マレイン酸コポリマーがポリ(メチルビニルエーテル-co-無水マレイン酸)である、請求項19に記載の方法。
【請求項21】
前記水素結合受容ポリマーが、ポリ(ビニルピロリドン)、ポリエチレングリコール、ポリ(エチレンオキシド)、ポリ(ビニルピロリドン-co-酢酸ビニル)、ポリエチレンオキシド-ポリプロピレンオキシドブロックコポリマー(プルロニック又はポロキサマー)、ポリエチレングリコール脂肪アルコールエステル、及びポリエチレングリコール脂肪酸エステル、エチルセルロース、及びキトサンから成る群より選択される、請求項14〜20のいずれか1項に記載の方法。
【請求項22】
前記水素結合受容ポリマーが、ポリ(ビニルピロリドン)である、請求項21に記載の方法。
【請求項23】
請求項14〜22のいずれか1項に記載の方法で製造された、生体接着性超高分子酸化窒素発生可能複合体。
【請求項24】
有効な創傷治癒量の請求項1〜13、又は23のいずれか1項に記載の前記超高分子複合体と、生理学的に許容しうる担体とを含む医薬組成物。
【請求項25】
請求項1〜13、若しくは23のいずれか1項に記載の前記酸化窒素超高分子複合体、又は請求項24に記載のその医薬的に許容しうる組成物を含むレイヤーバイレイヤーアセンブリコーティングの調製方法であって、以下の工程:
(i) アミノ連結基を有するS-ニトロソ化合物を生体接着性疎水性ポリ酸無水物化合物と共有結合させて酸化窒素ドナーポリマー担体を形成する工程;及び
(ii) 前記担体と分子間水素結合受容ポリマーを溶液から交互に堆積させて、強力な水素結合相互作用で連結された高度に互いにかみ合った薄層を形成する工程、
を含む方法。
【請求項26】
請求項1〜13、若しくは23のいずれか1項に記載の前記超高分子複合体、又は請求項24に記載のその医薬的に許容しうる組成物を含むナノ繊維の調製方法であって、以下の工程:
(i) アミノ連結基を有するS-ニトロソ化合物を生体接着性疎水性ポリ酸無水物化合物と共有結合させて酸化窒素ドナーポリマー担体を形成する工程;及び
(ii) 前記担体を分子間水素結合受容ポリマーと混合して前記ポリマーブレンド混合物を生成する工程;
(iii) この濃縮ブレンド系を高電圧場内で電気紡糸装置を用いて紡いで、
(iv) プレートコレクター上に堆積して不織マットを形成できるか又は
(v) ロールコレクター上に堆積してコーティングを形成できる、
連続ナノ繊維を生成する工程、
を含む方法。
【請求項27】
前記水素結合受容ポリマーがエチルセルロースである、請求項26に記載の方法。
【請求項28】
請求項1〜13、若しくは23のいずれか1項に記載の前記超高分子複合体、又は請求項24に記載のその医薬的に許容しうる組成物を含むミクロスフェアの調製方法であって、以下の工程:
(i) アミノ連結基を有するS-ニトロソ化合物を生体接着性疎水性ポリ酸無水物化合物と共有結合させて酸化窒素ドナーポリマー担体を形成する工程;及び
(ii) 前記担体を水素結合受容ポリマーと混合して前記ポリマーブレンド混合物を生成する工程;
(iii) このポリマーブレンド溶液を高電圧場内で超音波噴霧装置を用いて噴霧して、
(iv) 乾燥後に収集できるか又は
(v) ロールデバイス上に堆積してコーティングを形成できる
ミクロスフェアを生成する工程、
を含む方法。
【請求項29】
前記水素結合受容ポリマーがエチルセルロース又はポリ(ビニルピロリドン-co-酢酸ビニル)である、請求項28に記載の方法。
【請求項30】
物理的形態が粉末、紡糸繊維、又は基材表面上のコーティングである、請求項1〜13及び23のいずれか1項に記載の超高分子複合体又は請求項24に記載の組成物。
【請求項31】
前記基材がカテーテル又はステントである、請求項25に記載の超高分子複合体。
【請求項32】
有効な創傷治癒量の請求項1〜13、若しくは23のいずれか1項に記載の超高分子複合体、又は請求項24に記載の組成物を組み入れた、皮膚に適用するための皮膚被覆物。
【請求項33】
包帯又は創傷手当用品の形態の請求項32に記載の皮膚被覆物。
【請求項34】
皮膚の創傷又は感染症の治癒を促進する方法であって、有効な創傷又は感染症治癒量の請求項1〜13、若しくは23のいずれか1項に記載の超高分子複合体、又は請求項24に記載の組成物を前記創傷に適用する工程を含む方法。
【請求項35】
請求項1〜13、若しくは23のいずれか1項に記載の超高分子複合体、又は請求項24に記載のその医薬的に許容しうる組成物の、皮膚の創傷又は感染症の治癒を促進するための使用。
【図1】

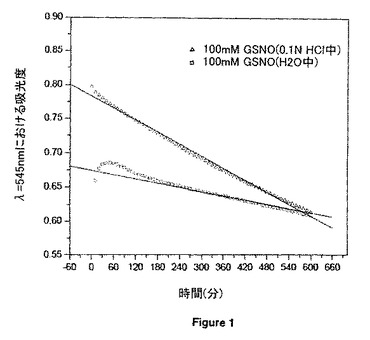
【図2】

【図3】
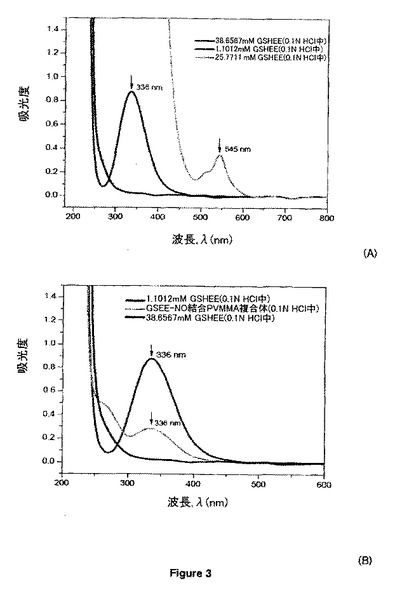
【図4】
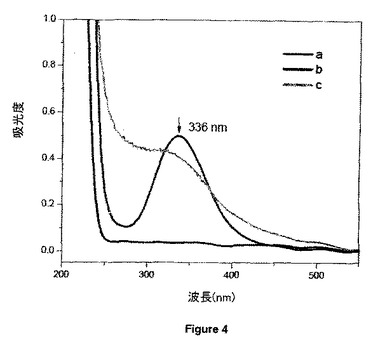
【図5】
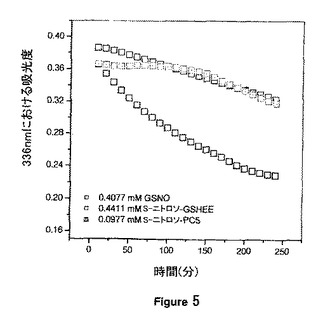
【図6】
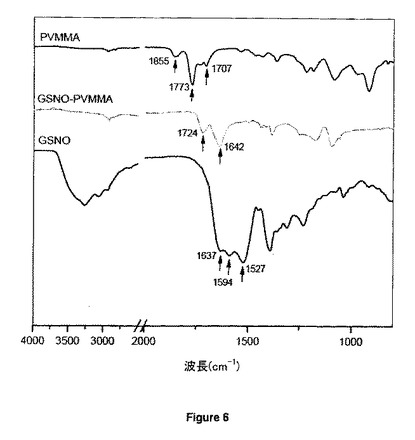
【図7】
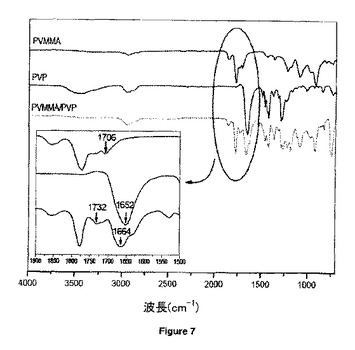
【図8】
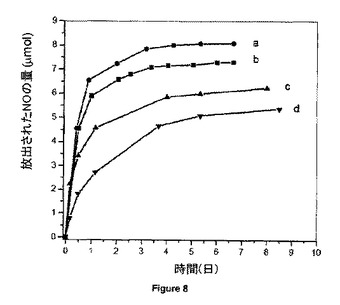
【図9】
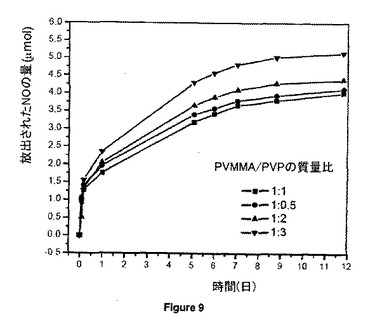
【図10】
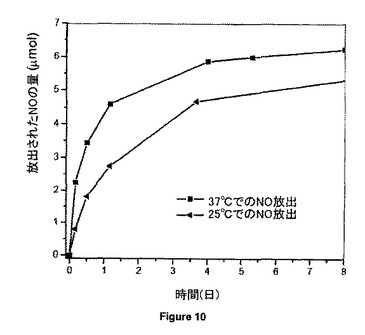
【図11】

【図12】
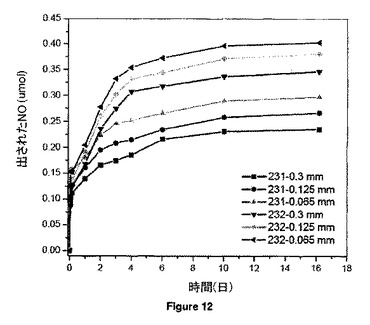
【図13】

【図14】
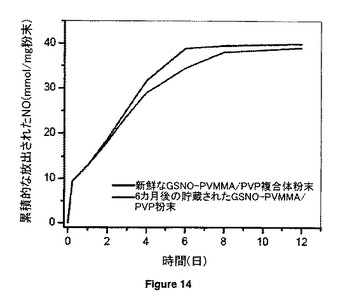
【図15】

【図16a1】

【図16a2】

【図16b1】

【図16b2】

【図16c1】

【図16c2】

【図17】

【図18】

【図19】

【図20】
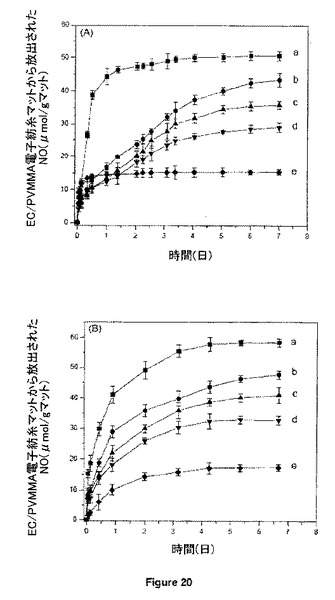
【図21】

【図22】

【図23】

【図24】
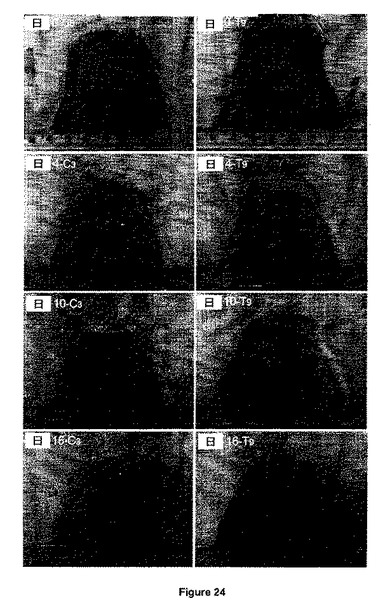
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16a1】
【図16a2】
【図16b1】
【図16b2】
【図16c1】
【図16c2】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【公表番号】特表2010−537008(P2010−537008A)
【公表日】平成22年12月2日(2010.12.2)
【国際特許分類】
【出願番号】特願2010−522143(P2010−522143)
【出願日】平成20年8月20日(2008.8.20)
【国際出願番号】PCT/CA2008/001484
【国際公開番号】WO2009/026680
【国際公開日】平成21年3月5日(2009.3.5)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.TEFLON
【出願人】(510054588)ユニヴァーシティー オブ トロント (1)
【Fターム(参考)】
【公表日】平成22年12月2日(2010.12.2)
【国際特許分類】
【出願日】平成20年8月20日(2008.8.20)
【国際出願番号】PCT/CA2008/001484
【国際公開番号】WO2009/026680
【国際公開日】平成21年3月5日(2009.3.5)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.TEFLON
【出願人】(510054588)ユニヴァーシティー オブ トロント (1)
【Fターム(参考)】
[ Back to top ]