
創傷治癒のための電界紡糸絹材料システム
本発明は、絹フィブロイン/ポリエチレンオキシドブレンド材料を調製するプロセス、および、創傷治癒などの生物医学的用途に適したその得られる材料に関する。特に、制御された蒸発、拘束乾燥技法、および/またはアルコール処理、および/またはPEO抽出で処理された、絹:PEOブレンド比が2:1〜4:1の電界紡糸した絹フィブロイン/PEOマットは、創傷被覆材に対する有用性をもつ生体材料系に関する、適した物理的なおよび生体機能特性、例えば繊維構造、トポグラフィー、吸収、水蒸気透過速度、酸素透過性、および生分解性などを実証する。

【発明の詳細な説明】
【技術分野】
【0001】
関連出願の相互参照
本出願は、2009年7月14日出願の米国特許仮出願第61/225,335号の優先権の恩典を主張するものであり、その内容はその全文が参照により本明細書に組み入れられる。
【0002】
政府支援
本発明は、米国国立衛生研究所(National Institutes of Health)(組織工学資源センター(Tissue Engineering Resource Center))により与えられた助成金番号P41 EB002520の下での財政的支援によってなされたものである。米国政府は本発明において一定の権利を有する。
【0003】
発明の分野
本発明は、創傷治癒などの生物医学的用途に適した、絹/ポリエチレンオキシドブレンド材料を調製するためのプロセス、およびその得られる材料に関する。
【背景技術】
【0004】
発明の背景
創傷治癒、または創傷修復は、真皮および上皮組織を再生する身体の自然のプロセスである。創傷治癒のプロセスは複雑で脆弱である。これらの中でも、全層熱傷の治療は、医学において最も取り組みがいのある仕事の一つであり続けている。大きな割合の体表面積(BSA)にわたって全層の損傷を受けている患者は、多くの場合焼痂から合併症を起こし、それは全身の細菌感染、血液量減少、低体温、低灌流、および、横紋筋融解および溶血に起因するヘモグロビン尿症を引き起こす可能性がある。現在、全層熱傷は、一般に自家皮膚移植により最小限の瘢痕形成で治癒する。しかし、自家皮膚移植には制限がある:20%を上回るBSAに全層熱傷を負っている患者は、死体由来の一時的に引き伸ばしたメッシュ状同種移植片か、またはブタ異種移植片およびコラーゲンでコーティングされた半透性合成膜などの人工の真皮再生鋳型に制限される。患者に免疫学的に適合しないこととともに、これらの代用品は、広く不規則なコラーゲンバンドの急性分布によって治癒を誘発し、不均等な格子様の表面および過度の過形成性肥大性瘢痕をもたらす。
【0005】
様々な合成および天然ポリマーを用いて創傷被覆材料(wound dressing materials)を開発することができ、それは例えばポリ(グリコール酸)(PGA)、ポリ(L-乳酸)(PLA)などの加水分解に不安定な合成脂肪族ポリエステルまたはキトサンなどの天然由来ポリマーである。しかし、これらのポリマーは、特定の創傷環境にさらされた場合に、副反応または性能の低下を被る可能性がある。例えば、PGAまたはPLAポリマーの加水分解副生成物の酸度は、全層創傷治癒カスケードを阻害する可能性がある;酸性の創傷環境に浸漬した場合、キトサンはアミン基のプロトン化に起因して可溶性となり、それは機械的完全性の早期喪失をもたらし得る。
【0006】
それ故に、改良された生分解性、生体適合性を有し、天然の皮膚の創傷治癒特性を保有するだけでなく、改良された物理的および機械的特性、ならびに効果的な創傷被覆材に適した満足できる柔軟性も有する、新規な種類の生体材料が必要とされている。
【発明の概要】
【0007】
本発明は、絹ブレンドマットの製造のためのプロセスを提供する。本プロセスは、ポリエチレンオキシド(PEO)と絹フィブロイン水溶液をブレンドする段階;ブレンドした溶液を電界紡糸し、それにより絹タンパク質/PEOブレンドマットを形成する段階;および電界紡糸した絹マットを拘束乾燥する段階を含む。結晶化皿の技法(a crystallization dish technique)を拘束乾燥段階に用いてよい。本プロセスは、乾燥段階の前または後にアルコールおよび/または水溶液中で電子紡糸絹マットを処理する段階をさらに含むことができる。アルコールは、メタノール、エタノール、イソプロピルアルコール(2-プロパノール)またはn-ブタノールであってよい。本プロセスは、PEOを絹マットから抽出する段階をさらに含むことができる。PEOは、水中に浸出することにより絹マットから抽出することができる。その上、本プロセスは、少なくとも一つの活性物質、例えば治療剤または生物材料などを絹マットに埋め込む段階をさらに含むことができる。
【0008】
本発明はまた、ポリエチレンオキシド(PEO)と絹フィブロイン水溶液をブレンドする段階;ブレンドした溶液を電界紡糸し、それにより絹タンパク質/PEOブレンドマットを形成する段階;および電界紡糸した絹マットを拘束乾燥する段階、を含むプロセスにより調製した絹材料も提供する。
【0009】
本発明の一部の態様は、ポリエチレンオキシド(PEO)と、少なくとも一つの活性物質を含む絹フィブロイン水溶液をブレンドする段階;ブレンドした溶液を電界紡糸し、それにより活性物質を封入する絹タンパク質/PEOブレンドマットを形成する段階;および電界紡糸した絹マットを拘束乾燥する段階を含むプロセスにより調製した、創傷治癒を促進するために創傷を手当てするための、少なくとも一つの活性物質を埋め込むかまたは封入する絹材料に関する。あるいは、活性物質は、PEOとブレンドした後に絹フィブロインに添加されるか、または電界紡糸した絹材料に添加されてよい、例えば、電界紡糸した絹/PEOマットを活性物質でコーティングしてよい。
【0010】
本発明はまた、少なくとも絹フィブロインタンパク質を含む電界紡糸絹マットに関し、ここで、絹マット中の絹フィブロインタンパク質の含量は、約50重量%〜約90重量%の範囲に及び、絹マットの厚さは約20〜80μmである。
【0011】
本発明はまた、絹フィブロインタンパク質およびポリエチレンオキシド(PEO)を含む電界紡糸絹マットに関する。電界紡糸絹マットは、2:1〜4:1の絹フィブロインタンパク質/PEOブレンド比を有するか、または絹の割合は約75%w/w〜90%(w/w)であり;かつ、絹マットの厚さは約20〜約80μmである。
【0012】
一態様では、電子紡糸絹マットは、約20〜30μmほどの細さである。
【0013】
一態様では、電子紡糸絹マットは、平均して約0.1〜約1μmの細孔のど径(pore throat size)表面積をもつ相互接続した細孔を有する。
【0014】
本発明のプロセスにより調製される電界紡糸絹マットは、生物医学的用途、特に創傷被覆材に適した、良好な構造特性、形態特性、生体機能特性および生体適合特性を提示する。例えば、得られる本発明の絹マットは、14日未満で約86%を超える重量を分解する;本発明の絹マットの平衡含水率は、約82%より大きく;絹マットの酸素透過速度は、約15460cm3/m2/日より大きく;かつ、絹マットの水蒸気透過速度は、約1934g/m2/日より大きい。
【0015】
本発明の一部の態様は、絹フィブロインタンパク質および任意で少なくとも一つの活性物質を含む、少なくとも一つの拘束乾燥した電界紡糸絹マットに創傷を接触させることを含む、創傷治癒を促進する方法にも関する。電子紡糸絹マットの絹フィブロイン含量は、約50重量%〜約90重量%の範囲に及び;かつ、絹マットの厚さは、約20〜約80μmである。
【0016】
本発明の一部の態様は、絹フィブロインタンパク質、PEO、および任意で少なくとも一つの活性物質を含む、少なくとも一つの拘束乾燥した電界紡糸絹マットに創傷を接触させることを含む、創傷治癒を促進する方法にも関する。電子紡糸絹マットは、約2:1〜約4:1の絹フィブロイン/PEOブレンド比を有し(または電界紡糸絹マット中の絹フィブロインの割合は、約75%w/w〜90%w/wであるか、または電界紡糸絹マット中のPEOの割合は、約10%w/w〜約25%w/wである);かつ、絹マットの厚さは、約20〜約80μmである。
【図面の簡単な説明】
【0017】
【図1】図1Aは、絹/PEO比が4:1、3:1、2:1、3:2、7:6、および1:1の6種類の、それぞれ、各材料群に対して86.5%、82.8%、76%、70.6%、65.1%および61.5%w/wの絹フィブロインタンパク質率に相当する電界紡糸絹マットを示す図である。直径10cmの絹マットを、メタノールで処理し、水に浸漬した。図1Bは、dH2Oに浸漬した直径10cmのS87〜S57絹マットを示す図である。各々のマットは、柔軟な軟質の絹のようなテクスチャーをもつ均一な立体構造を提示した。白色の斑点および襞は、材料中の気泡および折り目を反映する。
【図2】直径3.5cmの風乾した(または拘束乾燥していない)絹サンプルを示す図である。これらの画像は、絹濃度の低下に対して漸進的な材料の変形を反映する。
【図3】直径13cmの拘束乾燥した絹マット(3A)および直径12.5cmの拘束乾燥したS87〜S57絹マット(3B)を示す図である。これらの画像は、絹濃度の低下に対して増加する材料の剪断および変形を明らかにする。
【図4】風乾および拘束乾燥技法後の、小型および大型材料群サンプルについての絹表面積の変化の割合を表すグラフである。表面積は、円形形状の査定によって決定した。線形曲線分析を、全ての材料群で平均表面積減少率に関して行った(SD±%、n=3)。乾燥技法間での表面積減少は、4:1と3:1の材料群で有意に異なった(P=0.001)。傾き(m)およびR2値は、風乾したサンプルの絹濃度の低下に伴う表面積の線形の漸進的な減少を示す。対照的に、拘束乾燥した材料群についての表面積減少は、4:1および3:1マットについての2%から、3:2、7:6および1:1材料群についての60%超まで30倍増大した。
【図5】絹材料群と、(1)電界紡糸マットのサイズ、および(2)風乾および引き伸ばし乾燥技法後に基づく膜の厚さとの間の関係を表すグラフである。材料の厚さは、Ono Sokki EG-225F Digital Indicatorを利用して決定した。線形曲線分析を、全ての材料群で平均材料厚さに関して行った(SD±%、n=6)。全ての10cm風乾材料群は、16.5風乾群および12.5拘束乾燥群よりも有意に厚かった(P=0.001)。絹濃度に対して、傾き(m)およびR2値は、10cm風乾サンプルについて線形の厚さ減少を示す。16.5cm風乾群および12.5拘束乾燥群の厚さの傾きは、絹濃度に対して僅かな相違を伴って水平の痕跡に近づいた。
【図6】6.5倍の倍率の、メタノール処理の前の絹材料群のFE SEM画像を示す図である。PEO濃度の増加とともに、S87P13〜S76P24絹マットの上の繊維ビーディングは減少した。均一なよく分散したS67P33〜S61P39繊維の、不規則な形状の混合繊維への移行が、密集したS57P43構造に示される。
【図7】絹/PEO比がそれぞれ4:1、3:1および2:1の、メタノール処理した絹マットのSEM画像を示す図である。図7Aは、1.5倍の倍率の、3つ全てのマットの広範な図を表す。3:1および2:1マットの画像中の等高線は、方向性の繊維伸張および整列を反映する。図7Bは、1.5倍の倍率での3つ全ての絹マットのクローズアップ写真を示す。3:1および2:1の画像中の矢印は、繊維の凝集および整列を強調する。図7Cは、12倍の倍率の3:1および2:1絹マットを表す。3:1画像中の円は、整列した繊維間の相分散の証拠を露呈する。矢印は、2:1画像中でピンと張った伸張された繊維の詳細な輪郭を際立たせる。図7Dは、2:1絹マットの50倍画像において丸く囲まれた領域が、混合繊維を明らかにすることを示す。図7Eは、3つ全ての絹モデルについての2.5倍の倍率の断面図を表す。画像は繊維密度と絹濃度の逆相関を反映する。
【図8】50×50μmの領域一面の、4:1、3:1、および2:1拘束乾燥マット(8A)またはS87〜76マット(8B)についての細孔のど径分布の柱状グラフを示す図である。減少する絹濃度に対して、細孔の数は、のど径の低下による細孔の集積の増加で139から226に増大した。
【図9】結晶化皿乾燥法を利用する漸進的なポリマー鎖および繊維の立体構造を説明する概念図である。上部は、分散した未整列の二次構造の、親水性環境から、疎水性相互作用により駆動される整列したタンパク質凝集体への移行。繊維が延伸されるときに、β-シートは主に鎖間形成によって集合し、半径応力の方向に伸張する。図の下の繊維形成は、絹濃度および繊維整列および伸張の間の逆相関を反映する。
【図10】メタノール処理後の絹材料群についての50×50μm 3D AFM画像および粗さ値を実証する図である。これらの画像は、Ultrasharp NSC16/AIBSプローブを非接触モードで用いて得た(共振周波数:170kHz、バネ定数:45N/m)。
【図11】さまざまな環境条件下での4:1、3:1および2:1絹材料系についての酸素および水蒸気透過性能を表す図である。線形曲線の当てはめ分析を、全ての材料群での平均OTRおよびWVTR測定値に実行した(SD±%、n=3)。傾き(m)およびR2値は、全ての材料群でOTRおよびWVTR性能の線形の減少を示す。OTR R2値は、材料群でのOTR測定値の最低限の相違を開示するが、各々の群の中の標準偏差は、それぞれ、±14.6、11.2、および16.9パーセントの範囲に及んだ。
【図12】1、3、6、10、および14日の時点に関する絹材料群のインビトロ酵素生分解分析を表すグラフである。三層の25±5mg円形3.5cmサンプルを、pH7.4の1mg/mLプロテアーゼXIVのPBS溶液6mL中、37℃でインキュベートした。対照サンプルを、酵素を含まないPBSに浸漬した。酵素および対照溶液は、毎日補充した。図12Aは、全ての材料群で平均生分解材料損失について行った線形の当てはめ分析を説明する(SD±%、n=3)。図12Bは、各々の絹材料群の全てのサンプルに関する質量損失率の対数変換の散布図表示を表す図である。変換により、全ての絹材料系について3日の時点の直前に分解転移点が示される。図12Cは、各々の絹群の生分解率分析を全ての時点、0日〜3日および3日〜14日の時点について説明する図である。データは、2つの異なる分解傾向を示す。3日までは、全ての絹群は、-10〜-12の間の加速した酵素分解率を有する(単位=経時的質量損失)。3日後、酵素分解の傾きは、全ての絹材料について-3〜-5に著しく低下する。
【図13】図13Aは、非拘束および拘束乾燥S87〜S57絹マットについて絹表面積の変化の割合を表すグラフである。乾燥技法間の表面積の減少は、S87およびS82絹マットについて有意に異なった(P=0.001)(バー=標準偏差、n=3)。図13Bは、S87〜S57サンプルについて非拘束および拘束乾燥技法間の材料厚さの関係を表すグラフである。S87〜S61非拘束乾燥絹マットは、拘束乾燥群よりも有意に厚かった(P=0.001)(SD±%、n=6)。
【図14】それぞれ、拘束乾燥S87、S82、およびS76絹マットのFE-SEM顕微鏡写真を示す図である。図14Aおよび14Bは、1.5倍の倍率で見たS87〜S76マットを示す。画像は、進展する繊維の伸張、凝集および整列を開示する。図14Cは、整列したS82繊維と明確な伸張されたS76繊維との間の相分散をあらわにする12倍の倍率の画像である。図14Dは、混合され絡まったS76繊維の構造を示す50倍の倍率の画像を表す。図14Eは、2.5倍の倍率のS87〜S76マットの断面図を示す。画像は、繊維密度と絹濃度との間の逆相間を反映する。
【図15】図15Aは、全てのS87〜S57非拘束乾燥絹マットの明確な表面の不規則性を表す、S87絹マットの三次元AFM画像である。図15Bは、非拘束乾燥S87〜S57絹マットについて最低高さ分布(valley height distribution)の痕跡と同様のz面ピークを開示する柱状グラフを示すグラフである。
【図16】1、3、6、10、および14日の時点に関してS87〜S57絹マットのインビトロ酵素生分解分析を表すグラフである。三層の25±5mg 3.5cmサンプルを、pH7.4の1mg・mL-1プロテアーゼXIVのPBS溶液6mL中、37℃でインキュベートした。対照サンプルを、酵素を含まないPBSに浸漬した。酵素および対照溶液は、毎日補充した。図16Aは、各々の時点に関するS87〜S57生分解材料群の線形当てはめ表示平均率(the averaged percent linear fit representation)を説明する図である。(SD±%、n=3)。図16Bは、全ての時点に関して全ての生分解サンプルの対数変換を表す図である。変換により、全てのS87〜S75材料系について3日の時点の直前に分解転移点が示される。図16Cは、各々の絹群について、全ての時点、開始から3日、および3日から14日の生分解率分析を説明する図である。データは、2つの異なる分解傾向を示す。3日までは、全ての絹群は、-10〜-12の間の加速した酵素分解率を有する(単位=経時的質量損失)。3日後、酵素分解率は、全ての絹材料について-3〜-5に著しく低下する。
【発明を実施するための形態】
【0018】
発明の詳細な説明
本発明が、本明細書に記載される特定の方法論、プロトコール、および試薬などに制限されないこと、およびそのようなものが変動しうることは当然理解される。本明細書において使用される用語法は、特定の態様を説明する目的だけのものであって、特許請求の範囲によってのみ定義される本発明の範囲を制限するものではない。
【0019】
本明細書および特許請求の範囲において、単数形には、文脈上明らかに示されている場合を除き複数への言及が含まれ、逆の場合も同様である。運用例以外では、または別に指定されていない限り、本明細書において使用される構成成分の量または反応条件を表す全ての数値は、全ての場合において用語「約」によって変更されると理解されるべきである。
【0020】
特定される全ての特許およびその他の刊行物は、例えば、本発明に関連して使用されうるかかる刊行物に記載される方法論を説明および開示する目的で、参照により本明細書に明示的に組み入れられる。これらの刊行物は、単にその開示が本出願の出願日よりも前であるために提供される。この件について、先行発明という理由でまたは任意のその他の理由で、発明者らがかかる開示に先行する権利のないことを承認すると解釈されるべきではない。これらの文書の内容に関する日付または表現に関する全ての記述は、出願者らに利用可能な情報に基づくものであり、これらの文書の日付または内容の正確さに関していかなる承認も成すものでない。
【0021】
別に定義されない限り、本明細書において使用される全ての技術用語および科学用語は、本発明が関係する当業者に一般に理解される意味と同じ意味を有する。あらゆる公知の方法、装置、および材料を本発明の実践または実験に用いてよいが、この件について、方法、装置、および材料は、本明細書に記載されている。
【0022】
本発明は、創傷治癒などの生物医学的用途に適した、絹/ポリエチレンオキシドブレンド材料を調製するプロセス、およびその得られる材料に関する。特に、絹フィブロイン/PEOブレンド比が2:1〜4:1であり、制御蒸発および拘束乾燥技法によって乾燥させた、電界紡糸した絹フィブロイン/PEOマットは、創傷被覆材に有用性をもつ生体材料系に関する、適した物理的特性および生体機能特性、例えば繊維構造、トポグラフィー、多孔性、吸収性、水蒸気透過速度、酸素透過、および生分解性などを実証した。
【0023】
全層熱傷の治療は、医学において最も取り組みがいのある仕事の一つであり続けている。毎年、米国において、3000人が死亡しており、100万人を超える患者が、I度の上皮損傷からIII度の全層皮膚創傷の範囲に及ぶ、熱、放射線、化学物質および電気源から受けた熱傷を治療している。Beers et al.,MERCK MANUAL DIAGNOSIS&THER.(Merck&Co.,Inc.,Boston,MA,2006)。大きな割合のBSAにわたって全層損傷を受けた患者は、多くの場合焼痂から合併症を起こし、それは全身の細菌感染、血液量減少、低体温、低灌流、ならびに、横紋筋融解および溶血に起因するヘモグロビン尿症を誘発する。Ratner et al.,BIOMATS.Sci.INTRO.MATS.MED.(Acad.Press,NY,2004);Beers et al.,2006;Malafaya et al.,59 Adv.Drug Deliv.Rev.207-33(2007)。即時の処置を行わないと、全層熱傷は、血液量減少性ショック、免疫抑制、および、細菌性敗血症を引き起こし得、全身性炎症反応症状群(SIRS)、臓器不全、および死をもたらし得る。
【0024】
現在、全層熱傷は、一般に自家皮膚移植により最小限の瘢痕形成で治癒する。皮膚は、優れた曲げ強度をもつ耐久性のある生体材料複合材料であり、有害な細菌に対する物理的障壁を提供し、バランスのとれた静的血流および血栓性の創傷治癒カスケードを促進する重要な血液-表面の界面をもたらす。皮膚の優れた吸着、ガスおよび水蒸気の透過性により、浮腫および脱水症を抑制すると同時にタンパク質性浸出液のドレナージが可能となり、従って体温調節、細胞浸潤および軟組織再生を促進する。Kim et al.,341 Int.J.Pharm.35-43(2007);Lee et al.,11 J.Mater.Sci.-Mater.Med.817-23(2000)。永久的分層自家皮膚移植片を直ちに適用すると、72時間後に新血管新生を開始し、多くの場合、関節拘縮、虚血、瘢痕または全身毒性の合併症なく完全な真皮再構成をもたらす。Ratner et al.,2004;Beers et al.,2006。
【0025】
しかし、自家皮膚移植には制限がある。20%BSAを上回る全層熱傷を負っている患者は、一般に死体由来の一時的に引き伸ばしたメッシュ状同種移植片か、またはブタ異種移植片およびコラーゲンでコーティングされた半透性合成膜などの人工の真皮再生鋳型で処理される。患者に免疫学的に適合しないこととともに、これらの代用品は、多くの場合、広く不規則なコラーゲンバンドの急性分布によって治癒を誘発し、不均等な格子様の表面および過度の過形成性肥大性瘢痕をもたらす。Queen et al.,8 Biomats.367-71(1987);Ratner et al.,2004;Beers et al.,2006。そのために、天然の皮膚の創傷治癒特性を保有するだけでなく、十分に生分解性である、効果的な創傷被覆材を開発することが必要である。
【0026】
良好な生分解性、生体適合性および機械的特性を有するさまざまな合成および天然ポリマーを用いて創傷被覆材料を開発することができる。加水分解に不安定な合成脂肪族ポリエステル、例えばポリ(グリコール酸)(PGA)、ポリ(L-乳酸)(PLA)、およびポリ(乳酸-コ-グリコール酸)(PLGA)は、手術による移植、骨セメント、吸収性縫合、およびミクロスフェア制御放出系を含む、多くの医学的適用に用いられる。Quynh et al.,43 Eur.Polym.J.1779-85(2007);Wu&Wu 91 Polym.Degrad.Stab.2198-204(2006)。多孔性の電界紡糸PLAおよびPGA繊維は現在創傷被覆材料として研究されているが、これらのポリマーの加水分解副生成物の酸度は、全層創傷治癒カスケードを抑制しうる。Quynh et al.,2007;Wu&Wu,2006。グルコサミンおよびN-アセチルグルコサミンから構成され、ヘパリンに似た二糖の天然由来ポリマーキトサンは、血栓性カスケードを促進し、多形核(PMN)および単核細胞の創傷部位への細胞移動を刺激することにより創傷治癒を加速させることが見出された。Kim et al.,341 Int.J.Pharm.35-43(2007);Malafaya et al.,2007。しかし、酸性の創傷環境に浸漬すると、キトサンはアミン基のプロトン化に起因して可溶性となり、それは機械的完全性の早期喪失をもたらし得る。それ故に、柔軟性をもつフィルム、ゲルまたはスポンジを形成するためのその他の合成ポリマーの共重合は、全層創傷被覆材に必要である。Kim et al.,2007;Malafaya et al.,2007。
【0027】
本発明は、フィルム、繊維、ゲル、および多孔性のスポンジを含む、広い範囲の材料形態にわたって明確な生物学的特性を有する天然のフィブロイン絹を提供する。Vepari&Kaplan 32 Prog.Polym.Sci.991-1007(2007)。カイコおよびクモにより製造された絹フィブロインは、グリシンおよびアラニンから主に成る生体高分子に基づくタンパク質である。Vepari&Kaplan,2007;Zhou et al.12 Nucleic Acids Res.2413-19(2000);Tanaka et al.,29 Insect Biochem.MoI.Biol.269-76(1999)。構造的に、絹フィブロイン生体高分子はアミノ酸の繰り返し配列を含有し、それは結晶化する重鎖、および低結晶性の軽鎖を形成する。フィブロインの両親媒性領域の相互作用は、その他の二次構造とともに有意な含量の結晶性β-シート(およそ55%)を生じて、この特有の生体高分子の機械的および生体機能特性を作り出す。Vepari&Kaplan,2007;Wang et al.,39 Macromol.1102-07(2006);Wang et al.,37 Macromol.6856-64(2004);Jin et al.,15 Adv.Funct.Mater.1241-47(2005);Hu et al.,39 Macromol.6161-70(2006)。しっかりと詰め込まれた結晶性β-シートは水を排除するが、構築したタンパク質中の低結晶性ドメインは水素結合によって組織化されたままであり、含水量の変化に対応し得る。Wong et al.,82 Appl.Phys.A-Mater.293-303(2006)。広範な細胞および組織研究が、とりわけ骨、軟骨、靭帯および血管工学を含む絹タンパク質生体材料を用いて行われ、このタンパク質系の生体適合性かつ有効な組織再生の特色を実証した。Vepari&Kaplan 2007。創傷被覆材に関して、絹フィルムは、従来のブタに基づく創傷被覆材よりも速く、低い炎症応答で、ラットにおいて全層皮膚創傷を治癒することが示された。Sugihara et al.,225 Exp.Biol.Med.58-64(2000)。
【0028】
本発明の態様は、絹フィブロイン/ポリエチレンオキシド(PEO)のブレンドの二流体電界紡糸技法を利用して調製した創傷被覆材に対する潜在的有用性をもつ絹マトリックスを提供する。Wang et al.,39 Macromol.1102-07(2006)。電界紡糸法は、豊富な種類の機能性材料からナノファイバーの膜を作製するための単純で多用途かつ有用な技法である。Doshi&Reneker 35 J.Electro.151-60(1995);Reneker&Chun 7 Nanotech.216-23(1996);Fridrikh et al.,90 Phys.Rev.Let.144502-06(2003)。かなりの数の天然および合成材料が創傷被覆材を形成するために電界紡糸されているが、生体適合性、機械的特性、および総合的な機能性性能に関して課題が残っている。本発明では、絹フィブロインを標的プラットフォームに継続的に紡糸することにより、絹/PEO比濃度および用いた紡糸ドープの容積に相対的な厚さをもつ、層状の繊維シートで構築される大型の融合性(confluent)絹マットが製造される。絹マットはメタノールに浸漬され、β-シート結晶化に関連する物理的架橋を引き起こし、水安定化材料の形成を誘導する。異なる絹/PEOブレンドの特有の繊維多孔性および表面粗さを活用することで(Jin et al.,3 Biomacromol.1233-39(2002);Wang et al.,37 Macromol.6856-64(2004))、さまざまな絹/PEOブレンド比をもつ絹材料系が調製され、創傷治癒の必要性に照らして物理的なおよび生体機能特性について評価された。
【0029】
従って、本発明は、絹ブレンドマットの製造のためのプロセスを提供する。本プロセスは、ポリエチレンオキシド(PEO)と絹フィブロイン水溶液をブレンドする段階;ブレンドした溶液を電界紡糸し、それにより絹タンパク質/PEOブレンドマットを形成する段階;および電界紡糸絹マットを拘束乾燥する段階を含む。所望の口径をもつ結晶化皿またはポリスチレン容器を拘束乾燥段階に用いることができる。
【0030】
電界紡糸法は、当技術分野において公知のあらゆる手段により行うことができる(例えば、米国特許第6,110,590号参照)。例えば、内径1.0〜2.0mmの先端をもつ鋼毛管を、調節可能な電気絶縁スタンドの上に取り付ける。毛管は、一般に高い電位で維持され、平行なプレート配置で取り付けられる。毛管は、絹/生体適合性ポリマー溶液を満たしたシリンジに接続することができる。一定の体積流量は、通常、滴が漏れることなく管の先端に溶液を保持するように設定したシリンジポンプを用いて維持される。表1に示されるように、電位(10〜12kV)、溶液流速(0.014〜0.032mL/分)、および毛管の先端と収集スクリーンの間の作動距離(20〜22.5cm)を、安定した噴出が得られるように調節する。乾燥または湿潤繊維は、毛管の先端と収集スクリーンの間の距離を変えることにより収集される。
【0031】
(表1)各絹/PEOブレンドについての電界紡糸パラメータ。8重量%絹、5%PEO、および6%PEO溶液の粘度は、それぞれ約24、約4464、および約7520センチポイズ(mPa・s)であった。各ブレンドに対して4.5mL、8mL、および10.4mLの3種類のバッチ溶液を作成してそれぞれ直径10、13、および16.5cmの絹マットを作出した。
【0032】
絹繊維を収集するのに適した収集プレートまたは収集スクリーンは、金網またはポリマー網であってよい。あるいは、収集スクリーンはアルミニウム箔(直径10〜16.5cm)である。アルミニウム箔をTeflon流体でコーティングして絹繊維をより簡単に剥離するようにすることができる。繊維溶液が電場を移動するときにそれを収集するその他の手段を当業者は容易に選択することができる。下により詳細に記載されるように、毛管の先端とアルミニウム箔の対極との間の電位差は、約10〜12kVに徐々に増加する可能性があるが、当業者であれば電位を調節して適した噴出流を実現することができるはずである。
【0033】
次に、電界紡糸マットを拘束乾燥する。本発明のプロセスは、乾燥段階の前または後に電界紡糸絹マットをアルコール/水溶液中で処理してβ-シート形成および結晶化を誘導する段階をさらに含むことができる。アルコールは、メタノール、エタノール、イソプロピルアルコール(2-プロパノール)またはn-ブタノールであってよい。さらに、PEOを絹マットから抽出することができる。絹マットからのPEOの抽出は、電界紡糸した絹ブレンドマットを水(例えば、dH2O)中で一定期間、例えば1〜3日にわたって浸出させることにより行うことができる。
【0034】
本明細書において、用語「フィブロイン」には、カイコフィブロインおよび昆虫もしくはクモ絹タンパク質が含まれる。Lucas et al.,13 Adv.Protein Chem.107-242(1958)。例えば、フィブロインは、溶解したカイコ絹またはクモ絹を含有する溶液から得られる。カイコ絹タンパク質は、例えば、カイコ(Bombyx mori)から得られ、クモ絹は、アメリカジョロウグモ(Nephil clavipes)から得られる。しかし、使用することのできる絹は、数多くあり、それには、クモ絹(例えば、アメリカジョロウグモから得たもの)、トランスジェニック絹、遺伝子操作された絹、例えば細菌、酵母、哺乳類細胞、トランスジェニック動物、またはトランスジェニック植物由来の絹(例えば、国際公開公報第97/08315号;米国特許第5,245,012号参照)、ならびにそれらの変異体が含まれる。
【0035】
絹フィブロイン水溶液は、当技術分野において公知の技法を用いてカイコ繭から調製することができる。絹フィブロイン溶液を調製するために適したプロセスは、例えば、米国特許出願第11/247,358号;国際公開公報第2005/012606号;および国際公開公報第2008/127401号に開示されている。一態様では、カイコ(B.mori)繭を約30分間水溶液中で沸騰させる。この水溶液は、0.02M炭酸ナトリウムであってよい。繭を水ですすいでこれらのリシンタンパク質を抽出し、抽出した絹を塩類水溶液に溶解する。この目的に有用な塩としては、臭化リチウム、チオシアン酸リチウム、硝酸カルシウムまたは絹を可溶化する能力のあるその他の化学物質が挙げられるが、それに限定されるわけではない。例えば、抽出した絹を、60℃の約9〜12M LiBr溶液に4時間溶解し、20%(w/v)溶液を得てもよい。塩は結果的に透析を用いて除去される。このプロセスの間に形成されうる少量の絹凝集体、通常繭に存在する環境汚染物質由来のものを除去するために、この溶液を遠心してよい。絹フィブロイン水溶液の終濃度は、およそ8%(w/v)であってよい。より高い濃度の絹フィブロイン溶液を得るために、より低い濃度の絹フィブロイン溶液を吸湿性ポリマー、例えば、PEG、ポリエチレンオキシド、アミロースまたはセリシンに対して透析してよい。例えば、8%絹フィブロイン溶液は、10%(w/v)PEG(10,000g/mol)溶液に対して透析することができる。透析は、10〜30%の間の絹水溶液終濃度をもたらすのに十分な時間なされる。大抵の場合、2〜12時間の透析が十分である。
【0036】
絹フィブロイン溶液は、本明細書に記載されるように、1以上の生体適合性ポリマー、例えば、ポリエチレンオキシド、ポリエチレングリコール、コラーゲン、フィブロネクチン、ケラチン、ポリアスパラギン酸、ポリライシン、アルギン酸塩、キトサン、キチン、ヒアルロン酸、および同類のものなど;または1以上の活性物質、例えば細胞、酵素、タンパク質、核酸、抗体および同類のもの、と混合することができる。例えば、国際公開公報第2004/062697号および国際公開公報第2005/012606号を参照されたい。また、絹フィブロインは、絹タンパク質の物理的特性および官能性を変えるために、例えばジアゾニウムまたはカルボジイミドカップリング反応、アビジン-ビオチン(biodin)相互作用、または遺伝子組み換えおよび同類のものによって、溶液中の活性物質によって化学的に修飾することができる。例えば、PCT/US09/64673号;米国特許出願第61/227,254号;同第61/224,618号;同第12/192,588号を参照されたい。
【0037】
広範囲の水溶液中の絹フィブロインおよびPEO濃度が、絹材料を電界紡糸するためのブレンド溶液を調製するために適している。例えば、溶液中の絹フィブロインの濃度は、ブレンディングの前に約30重量%未満であってよく;溶液中のPEOの濃度は、PEO溶液の溶解度および粘度に応じて、ブレンディングの前に約1%〜約15重量%の範囲に及んでよい。例えば、約5重量%〜15重量%の絹フィブロイン濃度を有する水溶液および約3重量%〜10重量%のPEO濃度を有するPEO溶液を、ブレンディングに使用してよい。一態様では、8重量%の絹フィブロイン溶液および5重量%のPEO溶液がブレンディングに使用される。別の態様では、8重量%の絹フィブロイン溶液および6重量%のPEO溶液がブレンディングに使用される。絹フィブロイン溶液およびPEO溶液の初期濃度、ならびに絹フィブロインタンパク質とPEOの間の初期ブレンディング比は両方ともに、電界紡糸の間に安定した流体の噴出を生じるために望ましい粘弾特性および表面張力特性に依存しうる。Jin et al.,3 Biomacromol.s 1233-39(2002)。絹フィブロイン溶液およびPEO溶液の初期濃度ならびに絹フィブロインタンパク質とPEOの間の初期ブレンディング比は、最終の絹ブレンドマット中の絹フィブロインおよび/またはPEOの所望の重量百分率にも依存しうる。
【0038】
一態様では、本発明の絹生体材料は、少なくとも一つの治療剤を含有してよい。これらの材料を形成するため、マトリックスを形成するよりも前に絹フィブロインまたは絹フィブロイン/PEO溶液を治療剤と混合するか、それを形成した後に材料に添加する。本発明の生体材料と併せて使用することのできる豊富な種類のさまざまな治療剤は膨大である。
【0039】
通例、本発明の薬学的組成物によって投与されうる治療剤としては:抗生物質および抗ウイルス剤などの抗感染薬;化学療法剤(例えば、抗癌剤);拒絶反応抑制剤;鎮痛薬および鎮痛薬合剤;抗炎症剤;ステロイドなどのホルモン類;細胞接着メディエーター、例えば細胞接着に影響を及ぼすことが公知の「RGD」インテグリン結合配列のペプチド含有種、生物活性リガンド、および特定の種類の細胞または組織内殖を強化または排除する物質、例えば骨形態形成タンパク質(例えば、BMP1〜7)、骨形成様タンパク質(例えば、GFD-5、GFD-7、およびGFD-8)、上皮成長因子(EGF)、線維芽細胞成長因子(例えば、FGF1〜9)、血小板由来成長因子(PDGF)、インスリン様成長因子(IGF-IおよびIGF-II)、トランスフォーミング成長因子(例えば、TGF-βI〜III)、TGF-、YIGSRペプチド、グリコサミノグリカン(GAG)、ヒアルロン酸(HA)、インテグリン、セレクチンおよびカドヘリンなど;血管内皮成長因子(VEGF);ならびにその他の天然由来もしくは遺伝子操作されたタンパク質、多糖、糖タンパク質、またはリポタンパク質が挙げられるが、それに限定されるわけではない。成長因子は当技術分野において公知であり、例えば、Rosen&Thies,CELLULAR&MOL.BASIS BONE FORMATION&REPAIR(R.G.Landes Co.,2004)を参照されたい。
【0040】
活性物質は、絹材料に埋め込まれることの可能なあらゆる材料を表し得る。例えば、活性物質は、治療剤、または生物材料、例えば、細胞(幹細胞を含む)、タンパク質、ペプチド、核酸(例えば、DNA、RNA、siRNA)、核酸類似体、ヌクレオチド、オリゴヌクレオチド、ペプチド核酸(PNA)、アプタマー、抗体またはその断片もしくは部分(例えば、パラトープまたは相補性決定領域)、抗原またはエピトープ、ホルモン、ホルモンアンタゴニスト、成長因子または組換え成長因子およびその断片および変異体、細胞接着メディエーター(RGDなど)、サイトカイン、酵素、小分子、薬物、色素、アミノ酸、ビタミン、抗酸化剤、抗生物質または抗菌性化合物、抗炎症剤、抗真菌薬、ウイルス、抗ウイルス薬、毒素、プロドラッグ、化学療法剤、またはそれらの組合せなどであってよい。例えば、PCT/US09/44117号;米国特許出願第61/224,618号を参照されたい)。また、活性物質は、上記の物質のいずれかの組合せであってもよい。封入された生成物は多数の生物医学的目的に使用することができるので、治療剤または生物材料、あるいはそれらの組合せを封入することが望ましい。
【0041】
一部の態様では、活性物質はまた、真菌、植物、動物、細菌、またはウイルス(バクテリオファージを含む)などの生物であってもよい。さらに、活性物質には、神経伝達物質、ホルモン、細胞間シグナル伝達物質、薬学活性物質、毒剤、農薬、化学的毒素、生物学的毒素、微生物、ならびに動物細胞、例えばニューロン、肝細胞、および免疫系細胞が含まれてよい。また、活性物質には、治療用化合物、例えば薬理学的材料、ビタミン、鎮静薬、催眠薬、プロスタグランジンおよび放射性医薬品などが含まれてよい。
【0042】
本明細書での使用に適した例示的な細胞としては、前駆細胞または幹細胞、平滑筋細胞、骨格筋細胞、心筋細胞、上皮細胞、内皮細胞、尿路上皮細胞、線維芽細胞、筋芽細胞、口腔細胞(oscular cell)、軟骨細胞、軟骨芽細胞、骨芽細胞、破骨細胞、ケラチン生成細胞、腎尿細管細胞、腎基底膜細胞、外皮細胞、骨髄細胞、肝細胞、胆管細胞、膵島細胞、甲状腺細胞、上皮小体細胞、副腎細胞、視床下部細胞、下垂体細胞、卵巣細胞、精巣細胞、唾液腺細胞、脂肪細胞、および前駆細胞を挙げることができるが、それに限定されるわけではない。また、活性物質は、上記に列挙した細胞のいずれかの組合せであってもよい。国際公開公報第2008/106485号;PCT/US2009/059547号;国際公開公報第2007/103442号も参照されたい。
【0043】
絹フィブロインに組み込むことのできる例示的な抗体としては、アブシキマブ、アダリムマブ、アレムツズマブ、バシリキシマブ、ベバシズマブ、セツキシマブ、セルトリズマブ・ペゴル、ダクリズマブ、エクリズマブ、エファリズマブ、ゲムツズマブ、イブリツモマブ・ティウキセタン、インフリキシマブ、ムロモナブ-CD3、ナタリズマブ、オファツムマブ、オマリズマブ、パリビズマブ、パニツムマブ、ラニビズマブ、リツキシマブ、トシツモマブ、トラスツズマブ、アルツモマブペンテテート(altumomab pentetate)、アルシツモマブ、アトリズマブ、ベクツモマブ、ベリムマブ、ベシレソマブ、ビシロマブ、カナキヌマブ、カプロマブペンデチド、カツマキソマブ、デノスマブ、エドレコロマブ、エフングマブ(efungumab)、エルツマキソマブ(ertumaxomab)、エタラシズマブ、ファノレソマブ(fanolesomab)、フォントリズマブ、ゲムツズマブオゾガマイシン、ゴリムマブ、イゴボマブ(igovomab)、イミシロマブ(imciromab)、ラベツズマブ、メポリズマブ、モタビズマブ、ニモツズマブ(nimotuzumab)、ノフェツモマブメルペンタン(nofetumomab merpentan)、オレゴボマブ、ペムツモマブ(pemtumomab)、ペルツズマブ、ロベリズマブ(rovelizumab)、ルプリズマブ(ruplizumab)、スレソマブ(sulesomab)、タカツズマブテトラキセタン、テフィバズマブ、トシリズマブ、ウステキヌマブ、ビジリズマブ、ボツムマブ(votumumab)、ザルツムバブ、およびザノリムマブが挙げられるが、それに限定されるわけではない。また、活性物質は、上記に列挙した抗体のいずれかの組合せであってもよい。
【0044】
例示的な抗生剤としては、アクチノマイシン;アミノグリコシド(例えば、ネオマイシン、ゲンタマイシン、トブラマイシン);β-ラクタマーゼ阻害剤(例えば、クラブラン酸、スルバクタム);グリコペプチド(例えば、バンコマイシン、テイコプラニン、ポリミキシン);アンサマイシン;バシトラシン;カルバセフェム;カルバペネム;セファロスポリン(例えば、セファゾリン、セファクロル、セフジトレン、セフトビプロール、セフロキシム、セフォタキシム、セフェピム、セファドロキシル、セフォキシチン、セフプロジル、セフジニル);グラミシジン;イソニアジド;リネゾリド;マクロライド(例えば、エリスロマイシン、クラリスロマイシン、アジスロマイシン);ムピロシン;ペニシリン(例えば、アモキシシリン、アンピシリン、クロキサシリン、ジクロキサシリン、フルクロキサシリン、オキサシリン、ピペラシリン);オキソリン酸;ポリペプチド(例えば、バシトラシン、ポリミキシンB);キノロン(例えば、シプロフロキサシン、ナリジクス酸、エノキサシン、ガチフロキサシン、レバキン、オフロキサシン、など);スルホンアミド(例えば、スルファサラジン、トリメトプリム、トリメトプリム-スルファメトキサゾール(コ-トリモキサゾール)、スルファジアジン);テトラサイクリン(例えば、ドキシシリン(doxycyline)、ミノサイクリン、テトラサイクリンなど);アズトレオナムなどのモノバクタム;クロラムフェニコール;リンコマイシン;クリンダマイシン;エタンブトール;ムピロシン;メトロニダゾール;ペフロキサシン;ピラジンアミド;チアンフェニコール;リファンピシン;チアンフェニコール;ダプソン;クロファジミン;キヌプリスチン;メトロニダゾール;リネゾリド;イソニアジド;ピラシル(piracil);ノボビオシン;トリメトプリム;ホスホマイシン;フシジン酸;またはその他の局所用抗生物質が挙げられが、それに限定されるわけではない。任意で、抗生剤は、抗菌ペプチド、例えばデフェンシン、マゲイニンおよびナイシンなど;または溶菌性バクテリオファージであってもよい。また、抗生剤は、上記に列挙した抗生剤のいずれかの組合せであってもよい。PCT/US2010/026190も参照されたい。
【0045】
本明細書での使用に適した例示的な酵素としては、ペルオキシダーゼ、リパーゼ、アミロース、有機リン酸デヒドロゲナーゼ、リガーゼ、制限エンドヌクレアーゼ、リボヌクレアーゼ、DNAポリメラーゼ、グルコースオキシダーゼ、ラッカーゼ、および同類のものが挙げられるが、それに限定されるわけではない。また、成分間の相互作用を用いて、例えば、アビジンとビオチンとの間の特定の相互作用によって絹フィブロインを官能化してもよい。また、活性物質は、上記に列挙した酵素のいずれかの組合せであってもよい。米国特許出願第61/226,801号を参照されたい。
【0046】
治療剤または生物材料を絹フィブロインの中に導入する場合、当技術分野において公知のその他の材料を該作用物質に添加してもよい。例として、該作用物質の成長(生物材料に関して)を促進する、それが絹マットから放出された後に該作用物質の機能性を促進する、またはそれが絹に埋め込まれている期間の間、該作用物質が残存するかまたはその有効性を保持する能力を増加させる材料を添加することが望ましいであろう。細胞増殖を促進するとして公知の材料としては、細胞増殖培地、例えば、ダルベッコ改変イーグル培地(DMEM)、ウシ胎児血清(FBS)、非必須アミノ酸および抗生物質、ならびに成長因子および形態形成因子、例えば線維芽細胞成長因子(FGF)、トランスフォーミング成長因子(TGF)、血管内皮成長因子(VEGF)、上皮成長因子(EGF)、インスリン様成長因子(IGF-I)、骨形態形成成長因子(BMP)、神経成長因子、および関連タンパク質などを使用してよい。成長因子は、当技術分野において公知である。例えば、Rosen&Thies,CELLULAR&MOLECULAR BASIS BONE FORMATION&REPAIR(R.G.Landes Co.,Austin,TX,1995)を参照されたい。絹マットによる送達のためのさらなる選択肢としては、DNA、siRNA、アンチセンス、プラスミド、リポソームおよび遺伝物質の送達のための関連系;細胞のシグナル伝達カスケードを活性化するペプチドおよびタンパク質;ミネラル化または細胞由来の関連事象を促進するペプチドおよびタンパク質;絹マット-組織界面を改善する接着ペプチドおよびタンパク質;抗菌ペプチド;ならびにタンパク質および関連化合物が挙げられる。
【0047】
また、さらなる生体適合性材料を、絹フィブロインマットにブレンドすることもでき、それは例えばポリエチレングリコール(PCT/US09/64673号参照)、コラーゲン、フィブロネクチン、ケラチン、ポリアスパラギン酸、ポリリジン、アルギン酸塩、キトサン、キチン、ヒアルロン酸、ペクチン、ポリカプロラクトン、ポリ乳酸、ポリグリコール酸、ポリヒドロキシアルカノエート、デキストラン、ポリ無水物、グリセロール(PCT/US2009/060135号参照)、およびその他の生体適合性ポリマーなどである。国際公開公報第2004/0000915号を参照されたい。あるいは、絹をヒドロキシアパタイト粒子と混合してもよい。PCT/US08/82487号を参照されたい。本明細書において述べたように、絹フィブロインは組換え起源であってよく、それは繊維状タンパク質ドメインおよびミネラル化ドメインを含む融合ポリペプチドの封入などの絹のさらなる修飾をもたらし、有機-無機複合材料を形成するために使用される。これらの有機-無機複合材料は、使用する繊維状タンパク質融合ドメインのサイズに応じて、ナノスケールからマクロスケールまで構築することができる。国際公開公報第2006/076711号を参照されたい。また、米国特許出願第12/192,588号も参照されたい。
【0048】
活性物質または生物材料を埋め込んだ絹フィブロインは、細胞および/または活性物質の長期保存および安定化に適しうる。細胞および/または活性物質は、絹マットに組み込まれた場合に、室温にて(すなわち22℃〜25℃)および体温(37℃)で少なくとも30日間安定している(すなわち少なくとも50%の残効性を維持する)ことができる。それ故に、温度感受性活性物質、例えば一部の抗生物質などは、冷凍することなく絹マット中に保存することができる。重要なことには、温度感受性生物活性物質は、絹マット中で身体に(例えば、注射により)送達されて、これまでに想像されたよりも長い期間活性を維持することができる。例えば、PCT/US2010/026190号を参照されたい。
【0049】
活性物質(例えば、治療剤)または生物材料を埋め込んだ絹フィブロインは、生物送達装置に適している。絹フィブロインを生物送達装置として使用するための技法は、例えば、米国特許出願第10/541,182号;同第11/628,930号;同第11/664,234号;同第11/407,373号;PCT/US07/020789号;PCT/US08/55072号;PCT/US09/44117号に見出すことができる。本発明の一部の態様は、医療用移植、組織修復における潜在的有用性のため、かつ医療用具のコーティングのための、治療剤または生物材料を埋め込んだ絹フィブロインの薬物送達システムとしての有用性に関する。
【0050】
絹マット構造は、生物送達媒体が制御放出を有することを可能にする。制御放出は、投薬量を経時的に制御放出速度で投与することを可能にする。場合によっては、治療剤または生物材料の送達は、治療を必要とする部位に連続して、例えば、数週にわたって行われる。経時的な、例えば、数日もしくは数週にわたるかまたはそれよりも長い制御放出は、好ましい治療を得るための治療剤または生物材料の連続送達を可能にする。制御送達媒体は、治療剤または生物材料を体液および組織中で、例えば、プロテアーゼによるインビボ分解から保護するので有利である。例えば、PCT/US09/44117号を参照されたい。
【0051】
生物活性物質の絹マットからの制御放出は、経時的に、例えば、約12時間または24時間以上の間;1ヶ月または2ヶ月または5ヶ月以上起こるように設計することができる。放出の期間は、例えば、約12時間〜24時間、または約12時間〜1週間の期間にわたって起こるように選択することができる。別の態様では、放出は、例えば約1ヶ月以上2ヶ月以内で起こりうる。制御放出期間は処置条件に基づいて選択されてよい。例えば、一貫した放出および高い局所投薬量が望ましい場合には特定の放出プロフィールがより効果的でありうる。
【0052】
あるいは、治療剤を絹材料の上に薬学的に許容される担体で覆うこともあり得る。マトリックスを溶解しないどんな薬学的担体を使用してもよい。治療剤は液体、微粉化された固体、または任意のその他の適切な物理的形態として存在してよい。一般に、だが任意で、マトリックスには、1以上の添加剤、例えば希釈剤、担体、賦形剤、安定剤または同類のものなどが含まれる。
【0053】
治療剤の量は、用いられる特定の薬物および治療される医学的状態によって決まる。例えば、薬物の量は、材料の約0.001%〜約70%、または約0.001%〜約50%、または約0.001%〜約20重量%に相当しうる。体液と接触すると、薬物は放出される。
【0054】
組織工学足場に適した絹材料は、作製後にさらに修飾することができる。例えば、所望の細胞集団に対する受容体または化学誘引物質(chemoattractors)の役割を果たす生物活性物質で足場をコーティングすることができる。コーティングは、吸収または化学結合によって適用することができる。
【0055】
本発明の一部の態様は、ポリエチレンオキシド(PEO)と、少なくとも一つの活性物質を含む絹フィブロイン水溶液をブレンドすること;ブレンドした溶液を電界紡糸し、それにより活性物質を封入する絹タンパク質/PEOブレンドマットを形成すること;および電界紡糸した絹マットを拘束乾燥することにより、創傷治癒を促進するための創傷被覆材としての、少なくとも一つの活性物質を埋め込むかまたは封入する絹材料に関する。あるいは、活性物質は、PEOとブレンドした後に絹フィブロインに添加されるか、または電界紡糸した絹材料に添加されてよい、例えば、電界紡糸した絹/PEOマットは、活性物質でコーティングされてよい。
【0056】
本発明の絹材料は、生理活性分子を局所送達する能力があり、次世代の生体材料となりうる。例えば、ナノスケールの絹繊維でできた、EGFを含有する電界紡糸絹マットは、創傷治癒プロセスの促進のために使用されている。絹マットの中に組み込まれたEGFは、時間依存性にゆっくり放出されることができ得る(例えば、170時間で25%のEGF放出)。本発明の絹材料は、ヒト皮膚と同じ構造を示し、インビボで見出される同じ分子および細胞機構を用いて治癒する能力のある、三次元の負傷したヒト皮膚等価モデルにおいて特徴付けることができる。この生体機能性絹マットを負傷したヒト皮膚等価モデルの上に包帯材として載せると、絹マットは、上皮舌状部(epidermal tongue)による創傷閉鎖の時間を90%まで減少させることにより治癒を助ける。Schneider et al.,Acta Biomater.,(2009)。
【0057】
本発明の一部の態様は、絹フィブロインタンパク質およびPEOを含む電界紡糸絹マットに関する。一態様では、電界紡糸絹マットの絹フィブロインタンパク質/PEOブレンド比は、約2:1〜約4:1である。絹/PEO重量比および次式:
に基づく。
【0058】
絹マットのw/wでの絹の割合は、約75%w/w〜90%w/wの範囲に及びうる。電界紡糸絹マットの厚さは約20μm〜約80μmの範囲である。
【0059】
PEO濃度、または絹/PEOブレンド比は、絹繊維の表面積および電界紡糸プロセスの間のバルク形態に直接影響を及ぼす。Jin et al.,3 Biomacromol.1233-39(2002);Wang et al.,37 Macromol.6856-64(2004)。PEO濃度が増加するにつれて、繊維中で生じたフィブロインミセルおよび小球構造のサイズは低下する。その上、ひとたび素早く動く帯電した流体の噴出の中に入ると、これらの小球構造は整列し、100,000倍まで伸張する。Wang et al.,2006;Kowalewski et al.,53 Bulletin Polish Acad.ScL,Tech.Sci.385-94(2005);Reneker&Yarin,49 Polymer 2387-425(2008)。本発明は、絹/PEOブレンド比が、繊維厚さ、密度、配向、相分散、多孔性およびマット厚さを含む、得られる絹マットの特性に重要な役割を果たすことを実証する。その結果として、増加したPEO濃度で形成された繊維は、幾何学的形状、表面積、ならびに、4:1から1:1までの絹/PEOブレンドマットに観察された漸進的な視覚およびテクスチャーの変化に相関するバルク体積が低下した。
【0060】
一態様では、4:1、3:1、2:1、3:2、7:6、および1:1の絹/PEOブレンド比で調製した6つの絹/PEOブレンド材料系を、融合性の直径16.5cmおよび10cmのマットに電界紡糸した。各々のサンプルの物理的特性は、水飽和状態と乾燥状態の両方で評価した。水に浸すと、6つのマトリックスは、不透明に半透明な灰白色の外観を提示する、均一な立体構造を有し、絹のようなテクスチャーをもち柔軟であったが、図1Aに示されるように、取扱期間が延びると絹濃度に対する(respective of)引張強さの悪化を提示した。絹のようなテクスチャーとは、水分子が非晶質ポリマーマトリックスの全体にわたって継続的に可塑化するフィブロインの動的吸湿性を説明するために言及される。アミノ、ヒドロキシル、またはカルボキシル酸末端基との水素結合を形成することによるか、または、親水性ドメインの至る所に自由に分散することにより;この流動性の環境は、これらの飽和材料系の軟質の絹のようなテクスチャーをもたらす運動エネルギーの最小化に起因して、継続的に移行している。Hu et al.,39 Macromol.6161-70(2006);Agarwal et al.,63 J.Appl.Polym.Sci.401-10(1997);van der Heijden et al.,378 Thermochim.Acta 27-34(2001);Wong et al.,2006。周囲温度で24時間の乾燥期間の後、物理的な特徴は、6つの材料系で漸進的に変化した。図2および3に示されるように、絹濃度の低下(86.5%、82.8%、76%、70.6%、65.1%および61.5%)に関連して、マットは、付着曲げ強度(cohesive flex strength)をもつ雪のように白い柔軟なウエハーのようなテクスチャーから、半透明褐色で超薄の柔軟性の低いフィルムのような材料に変わった。
【0061】
本発明において、乾燥法は、絹/PEOブレンドマットの物理的および機械的特性、例えば電界紡糸絹/PEOブレンドマットの厚さなどにも影響を及ぼす。例えば、ポリスチレンペトリ皿を使用する風乾法を用いることができる。あるいは、拘束乾燥の方法を用いてよい。例えば、電界紡糸絹マットを乾燥させるために結晶化皿技法を用いてよい。
【0062】
本発明の電界紡糸絹マットの厚さは、約20μm〜約80μmである。拘束乾燥法を用いる場合、電界紡糸絹マットの厚さは、平均約20μm〜30μmでありうる。
【0063】
例えば、絹/PEOブレンド比が4:1、3:1、2:1、3:2、7:6、および1:1の電界紡糸絹マットの直径3.5cmのサンプルを、直径10cmのマットから打ち抜き、ポリスチレンペトリ皿法を用いて風乾させた。得られる絹マットを図2Aに示す。飽和したいくつかの直径3.5cmのサンプルは、正味の力(net force)の表面-表面疎水性平衡を実現し、層状の絹シートの分離および変位で親水性挙動を提示するために、取り扱いが困難で、多くの場合半分に折り重なる。水乾燥段階の全体にわたって、極性水分子がこの不織の多孔性生体材料の大きい表面積から蒸発するにつれて、界面での親水性の疎水性ドメインへの転移により表面エネルギーは最小となった。この動的表面構造の再編成は、重鎖の再整列およびβ-シートの結晶化を明示する。Vepari&Kaplan,2007;Jin et al.,200);Hu et al.,39 Macromol.6161-70(2006)。ねじれた襞のあるβ-シート形成の特徴を示して、マトリックスはいずれも完全に平らな配向で乾燥せず、4:1および3:1マトリックスだけが元の円形の形状を維持していた。その上、フィブロインの重鎖の結晶性ドメイン間に交互に配置された非晶質ドメインの末端に位置するプロリン残基がある。Zhou et al.,2000。プロリンは、脱水によって非常に収縮し、従って繊維の収縮する能力を増加させることが示された。Liu et al.,9 Biomacromol.116-21(2008)。図4に示されるように、絹濃度の低下とともに、これらの要素は乾燥したサンプルにおける飽和状態から乾燥状態へのそれらの表面積の51.0±0.0%〜87.5±9.9%の間の減少の一因となる(n=3)。図5に示されるように、サンプルの各々の乾燥したセットの厚さ測定値は、それぞれ、81.7±7.5から77.5±10.5に、66.7±5.1に、53.3±8.1に、46.7±5.1に、および30.0±6.4μmに漸進的に下降した(n=6)。
【0064】
別の態様では、絹/PEOブレンド比が4:1、3:1、2:1、3:2、7:6、および1:1の直径12.5cmの電界紡糸絹マットを、結晶化皿技法を用いて乾燥させた。得られる絹マットを図3Aに示す。水に浸すと、これらのより大型のサンプルは展開がより容易であった。親水性の力に関して、層状のシート分離は、シートの変位なく各々のサンプルの内部領域でのみ観察された。これは、これらのサンプルがより大型のマットから打ち抜かれておらず、従ってサンプルの端部の架橋された結晶化領域を保持しているためでありうる。乾燥段階の間、飽和したサンプルが縁からサンプルの中心に向かって均一に乾燥するとき、マットの各々のセットは皿の口の全体にわたって漸進的に収縮する。4:1および3:1サンプルは、結晶化皿の縁に付着して完全に乾燥し、完全に平坦で柔軟な白色の膜のような材料をもたらした。3:2、7:6、および1:1サンプルが乾燥するとき、結晶化する延伸力が繊維の降伏点伸びを越えて材料に応力を加え、皿の縁での材料の剪断による構造的破壊をもたらし、サンプルの内部領域に伝わった。この乾燥法では、絹濃度の低下は、材料の構造的完全性および曲げ強度に影響を及ぼした。2:1サンプルは皿の縁から切り取られたが、材料変形の痕跡はほとんどなく、特性は4:1および3:1サンプルに類似していた。図4に示されるように、4:1および3:1マトリックスは、元の表面積の98%を保持したが、2:1マットは、11.8%±2.7%を失った。3:2、7:6および1:1サンプルは、それぞれ、68.8%±9.1%、65.9%±4.3%および63.9%±6.5%収縮した(n=3)。図5に示されるように、マットの各々の乾燥したセットの平均厚さは、それぞれ、31.2±1.8、28.7±1.2、24.3±2.3、25.3±2.3、20.0±1.4および26.0±0.9μmであった。
【0065】
本発明において、電界紡糸絹/PEOブレンドマットの結果として得られる繊維は、マット構造全体にわたって実質的に均一な直径分布を有する。図6AのSEM画像は、電界紡糸プロセスの直後、かつメタノール飽和およびPEO浸出処理の前の全ての6つの絹/PEO材料群の画像である。全体に、これらの電界紡糸した絹/PEO繊維は、直径が200nm〜500nmの範囲に及ぶ。Huang et al,2001に報告されるように、繊維ビーズ形成は、PEO濃度の低下とともに次第に顕著となった。Wang et al.,2006;Zhou et al.,2000;Huang et al.,12 J.Biomat.Sci.Polym.Ed.979-93(2001)。具体的には、4:1、3:1、および2:1(フィブロイン:PEO)サンプルは、各々、繊維内のランダムな位置にちょうど幅1μmを上回るものから700nm間での範囲に及ぶビーズセグメントを有した。ビーズは、3:2および7:6サンプルセットで最小化し、明確に定義された微細な円形状の繊維が構造の全体にわたって秩序化された外観を与えた。1:1サンプルセットは特有の外観を有し、繊維は不規則で非円形の形状であって、不均一で高密度のマット構造に移行した。この移行が、装置の接地ステージに集まった場合の、液-液/液-固相分散による繊維の収束に起因しうることは妥当である。1:1の個々の繊維は、300〜500nmの範囲に及び、一方、混合繊維は700〜900nmの間で測定された。
【0066】
「拘束乾燥技法」または「拘束乾燥する」とは、本明細書において、絹材料が延伸力を受けている間に乾燥するように、絹材料を拘束している間に乾燥させるプロセスをさす。例えば、拘束力は、結晶化皿の口の上に付着しながら絹材料が乾燥するときに起こる合成収縮力に起因しうる。記載されるように、これらの飽和した絹材料は、最初に結晶化皿の口の上に掛けられてそれに付着する。水分子が蒸発するにつれて、タンパク質の表面基質の疎水性ドメインおよびバルク領域の全体が、不織のキャスト(non-woven cast)の組織間隙(interstitial space)および材料のバルク領域の中から自由体積の喪失を開始する。自由体積の喪失は、材料を収縮させ、結晶化皿の縁に向かって放射状に延伸させる。結晶化皿の縁に付着しているので、材料は自由体積の継続的な喪失によって束縛されることとなり、繊維は半径応力の方向に整列し、伸張されるようになる。絹体積に応じて、材料繊維が降伏点伸びを越えて収縮すれば、材料の剪断が材料/結晶化皿縁表面界面で起こる。拘束乾燥法に反して、ペトリ皿の中の風乾サンプルは、乾燥してねじれた不規則な立体構造となるまで継続的に収縮する。
【0067】
一態様では、拘束乾燥法は、制御された蒸発によって行われる。この方法は、電界紡糸絹/PEOブレンドマットを水浴から取り出し、マットを、皿の3分の1まで水を入れた結晶化皿の上に掛け、マットを含有する皿を20%〜50%の相対湿度のデシケーターに入れて一晩乾燥させることを含む。結晶化皿乾燥法を利用する漸進的なポリマー鎖および繊維の立体構造を説明する概念図を図9に示す。上部は、分散した未整列の二次構造の、親水性環境から、疎水性結合により駆動される整列したタンパク質凝集体への移行である。繊維が引き伸ばされ始めるとき、水なましされた(water-annealed)β-シートが鎖間形成対鎖内形成によって集合する。図の下部の繊維形成は、絹濃度および繊維整列および伸張の間の逆相関を反映する。
【0068】
一態様では、図7に示されるように、絹/PEOブレンドマットのSEM画像は、拘束乾燥法、例えば結晶化皿法などを用いて調製された。調製したマットの絹/PEOブレンド比は、それぞれ4:1、3:1および2:1である。表面のトポグラフィーは、各々のモデルの全体にわたる繊維の高密度でランダムな分布を反映する。評価は、この乾燥技法によって起こる繊維収縮、伸張、および再整列の痕跡の増加を示す。図7Bを参照すると、4:1マットの繊維は、目立った繊維収縮または整列がなく緩んでねじれた外観を有する。対照的に、3:1および2:1マットの繊維は伸張して整列し付着して、ウェブのような微小なテクスチャー(web-like micro texture)を形成するようになる。図7Cの繊維形成に焦点を合わせると、3:1マットの伸張繊維は、ピンと張ったウェブ構造を形成し、末端が液化性の外観になっている整列した繊維間の相分散の痕跡を示している。2:1フィブロイン:PEOサンプルのウェブ構造は、ロープのような配置を形成する、明確に定義された、伸張され整列された繊維の絡み合った網状構造からなる。
【0069】
拘束乾燥技法により明らかにされる繊維の整列および伸張は、絹フィブロインの水なまし特性によるものでありうる。Jin et al.,15 Adv.Funct.Mater.1241-47(2005);Agarwal et al.,63 J.Appl.Polym.Sci.401-10(1997);Lawrence et al.,43 J.Mat.Sci.6967-85(2008);Wong et al.,2006。ポリマーバルク領域内部で可塑剤の役割を果たすことにより、水分子は、低結合エネルギーのポリマー鎖間で分子間の動きを伝え、ポリマーの流動性および再整列を促進する。水分子が蒸発するときに、ポリマー鎖は、延伸されて、結晶化皿の縁の周囲から発生する半径応力の方向を向く。延伸されたポリマー鎖の整列とともに、非晶質軽鎖の末端で折り畳まれている、両側性の鎖間層構造の段階的拡大および結晶化した鎖内のねじれた立体構造の減少を促進するプロリンは減少する。Zhou et al.,2000;Liu et al.,9 Biomacromol.116-21(2008)。支配的な鎖間疎水性相互作用は、バルク領域から自由体積を圧縮し、結晶性二次構造整列および非晶質絹I状態の結晶性絹II状態への移行に影響を及ぼす。Jin et al.,15 Adv.Funct.Mater.1241-47(2005);Agarwal et al.,1997;Lawrence et al.,43 J.Mat.Sci.6967-85(2008);Wong et al.,82 Appl.Phys.A-Mater.293-203(2006)。水分子の継続的な排出によって、バルク体積は低下し、繊維が結晶化皿の口の上で伸張し始めるまで収縮する。図9に示されるように、これらの構成要素の組合せが、繊維軸に沿って、高い強度材料特性をもつ伸張され整列された繊維によって、マットを形成する。降伏点伸びを越えて、重および軽(H,L)鎖の両方の非晶質二次構造の内部および二次構造に沿って剪断変形が起こる。
【0070】
整列した図7Cの3:1繊維と図7Dの2:1繊維の間の巨視的相分散の外観も、水の可塑化特性に起因しうる。水およびカイコ絹フィブロインフィルム系の示差走査熱量測定により、脱水した絹フィブロインのガラス転移温度(Tg)が、20〜23重量%の吸水量で178℃から40℃より下まで低下したことが明らかとなった。Agarwal et al.,1997。結晶性立体構造は生体材料の全体にわたる希釈剤の吸収に影響を及ぼすが、各々の絹マット群の平衡含水率(EWC)は、80重量%よりも大きく(表4)、相当な親水性相互作用を示し;Tgおよび妥当な相分散の低下を予見する。Hu et al.,39 Macromol.6161-70(2006)。その上、絹ポリマー鎖の全体にわたって交互に配置された疎水性ドメインおよび親水性ドメインは、材料が表面-液体から表面-ガス、表面-表面界面へ移行するときに、動的移動性表面基質をもたらす。Ratner et al.,BIOMATS.Sci.:INTRO.MATS.MED.(Acad.Press,NY,2004;Allcock,INTRO.MATS.CHEM.(Wiley&Sons,Hoboken,NJ,2008)。具体的には、希釈剤が蒸発するときに、親水性のドメインは、表面で疎水性セグメントに置き換わる。上記の相分散を考慮に入れると、この現象が界面の繊維間に起こる場合、非晶質二次構造が散在するようになり、混合繊維となる。また、繊維間の線形の二次構造が整列するようになって熱力学的に安定した結晶性β-ストランド(stands)を形成しうることも考えられる。Lawrence et al.,43 J.Mat.Sci.6967-85(2008);Fink,3 Folding&Design R9-R23(1998)。
【0071】
本発明の絹マットは、平均して約0.1〜約1μmの細孔のど径表面積をもつ相互接続した細孔を有しうる。図7Eの断面図より、数種類の異なる絹/PEOブレンド比をもつ絹マットのさまざまな特色が明らかとなる。これらの画像は、絹濃度の低下とともに増加した繊維密度を示す;そして、マトリックスにわたる繊維凝集も、異なる絹/PEOブレンド比をもつ絹マットに関して異なっている。4:1ブレンドの繊維は、多数の大きな空間の隙間をもつ水平シートに凝集した。3:1および2:1ブレンドの繊維は、繊維束の増加を実証し、絹容積の低下に対する繊維の収縮および空間の間隙の漸進的低下を反映した。これらの所見は、絹濃度に対するマット厚さの低下に一致する。図7Bの3枚の画像は、この生体材料の全体にわたって相互に連絡する多孔性を表す。細孔は、各々のマトリックスに対して、それぞれ平均して294、201、から103nm2ののど径表面積を有した。図9に示される下降する細孔径分布は、断面図に見られる繊維密度の増加に一致し、ビーズ領域の数ならびに4:1、3:1、および2:1マトリックスの繊維直径の低下に関して繊維の集合体に関連する。
【0072】
材料の表面粗さは、細胞の配向、接着、増殖、および遊走を制御する、正味の力の生体力学的平衡を促進する、応力/剪断(sheer)のない平面による細胞接触誘導に影響を及ぼす。拘束乾燥処理を受けた本発明の絹/PEOブレンドマットは、風乾法を受けた絹マットよりも表面粗さが小さい。図10のAFM画像は、ポリスチレン皿風乾法で乾燥させた後のさまざまな絹/PEOブレンド比の本発明の絹マットの、三次元の形態学的イメージおよびサンプルの二乗平均平方根の粗さ値を示す。局地的なドメイン粗さ分析は、画像のXおよびY平面の全体にわたる粗さの変動により特徴付けられる。全体として、これらの絹材料系は、粗さ値が500nmから1.4μmまでの明確な表面不規則性を示すクラス3の表面トポグラフィー(class three surface topographies)を実証した(n=3)。4:1および1:1サンプルは、それぞれ1.17±0.00および0.78±0.01μmを測定する、ナノサイズの不規則性をもつ相対的に均一な粗さを有した。3:1、2:1、および3:2サンプルの粗さの標準偏差は0.1〜0.17μmの間で、それぞれ0.65±0.10、0.88±0.17、および0.76±0.16μmの範囲に及んだ。7:6マットが、局地的な粗さが平均1.01±0.43μmの最も大きい偏差を有した。AFM粗さ評価も結晶化皿技法で拘束乾燥した絹/PEOブレンドマットに行った。4:1、3:1、および2:1サンプルは、16×16μmの面積に関して、それぞれ0.66、0.36および0.25μmの粗さ値を有する。サンプルの面積サイズは低下し、サンプルサイズは制限されるが(n=1)、引き伸ばし乾燥したマットの粗さは、風乾マットよりも少なくとも44%平坦である。拘束乾燥したサンプルは、絹濃度に対して粗さが直線的に低下するように見えるが、風乾サンプルについては顕著な傾向はない。この所見は、風乾サンプルのねじれた不規則さと比較して、拘束乾燥したサンプルの繊維の伸張特性に一致する。
【0073】
本発明のプロセスにより調製した電界紡糸絹/PEOブレンドマットは、生体材料用途、例えば創傷被覆材などに適した、良好な構造特性、形態特性、生体機能特性および生体適合特性を示す。例えば、得られる本発明の絹マットは、14日未満で約86%(重量)超を分解する;本発明の絹マットの平衡含水率は約82%を上回る;絹マットの酸素透過速度は、約15460cm3/m2/日よりも大きい;かつ、絹マットの水蒸気透過速度は、約1934g/m2/日よりも大きい。
【0074】
本発明の態様は、時間放出生物療法によって上皮化を促進するための、酵素生分解を備える絹材料を提供する。さまざまなブレンド比をもつ電界紡糸絹/PEOブレンドマットの酵素生分解を、14日以上にわたって評価した。図12Aに示されるように、インビトロ生分解性は、全ての時点で全ての材料群について線形の分解を示し、それぞれ、各々の群に対して1日後に22.6%±3.4%の分解、および14日後に74.0%±8.8%までの材料損失をもたらした。データは、6日までは、全てのブレンドについての分解率は48.2±4.6%で比較的近かったという推論を許容する。10日後、4:1(78%)から1:1(51%)の範囲に及ぶ絹/PEOブレンド比をもつさまざまな材料群について27%の重量減少差が記録された。14日後、酵素分解は85.6±3.8%(4:1)から62.5±5.2%(1:1)に至る範囲に及んだ。形態学的に、目視検査によって、全ての材料系が、最初の6日で表面浸食により主に分解された。10日の時点の後、4:1および3:1サンプルが、マットのほころび、断片化、および微粒子破片への崩壊をもたらす、繊維の切断分解の増加を実証した。4:1および3:1サンプルの生分解挙動は、これらのブレンドの増加した繊維サイズ、マット多孔性および繊維密度特性の低下により、マットの内部繊維構造に酵素が接近することに起因し得る。
【0075】
線形回帰分析は、Minitab(登録商標)15.2.30を用いて各々の絹/PEOブレンド比について全てのサンプルで行った。図12Bの散布図を参照することにより、各々の材料群に関して実行した対数変換は、3日の分解時点の直前の全ての材料群について明確な転移点を明らかにした。次に、各々の材料群について全ての時点、開始から3日の時点および3日〜14日の時点で回帰分析を行った。図12Cに示されるように、全ての材料群につての分解の傾きは3日後に実質的に変化した。開始から3日の時点まで、酵素分解速度は、平均-11.68±0.91傾き単位であった;3日以後14日まで、分解の速度は-3.41±1.12まで横ばいとなった。初期分解段階の間、非晶質領域は、内部結晶領域と比較して加速した速度で分解する。この仮説は、酵素分解の開始時および3日後にこれらの絹材料の赤外吸光度スペクトルにフーリエセルフデコンボリューション(FSD)を用いることにより検証することができる。Hu et al.,2006。
【0076】
正常なヒト皮膚は約21日で再生する。本発明は、時間放出生物療法での上皮化を促進するために、例えば、プロテアーゼが絹材料を断片に分解する時間を比較することにより、マトリックスの設計を所望の分解速度に一致させることを可能にする。結果は、14日後に、4:1絹/PEOブレンドマットの86%が溶解し、それは、リゾチームに曝露した場合に14日後に82%分解したキトサン/ポロキサマー包帯材、およびPBS中で14日後に20%分解するPLGA/PLLA(90/10)と比べても遜色がない。また、酵素レベルは変動する可能性があるため、インビボでの分解速度にも対処し、絹生体材料が、材料形式、位置、および関連する変数によって、インビボで数週間から数年のうちに分解し得ることが示された。Wang et al.,2008。
【0077】
本発明のプロセスにより製造された絹材料は、多様な医学的用途、例えば、血管創傷修復装置、止血包帯材、全層熱傷被覆材、パッチおよびグルー、縫合糸、薬物送達を含む、創傷閉鎖系などにおいて、かつ、組織工学用途、例えば、足場材料、靭帯補綴装置などにおいて、かつ、ヒト身体への長期もしくは生分解性の移植のための製品において、使用することができる。例示的な組織工学足場は、電界紡糸繊維の不織網状構造である。
【0078】
その上、これらの生体材料は、限定されるものではないが、脊椎板、頭蓋組織、硬膜、神経組織、肝臓、膵臓、腎臓、膀胱、脾臓、心筋、骨格筋、腱、靭帯、および乳房組織を含む、これらの特有の足場から利益を受けうる、器官修復置換または再生計画に使用することができる。
【0079】
一態様では、本発明は、材料を包帯に加工し、創傷部位の浮腫およびO2/CO2ガス透過を管理する能力、ならびに同時刻に合わせた(time synchronized)抗生物質、免疫学的、および組織再生生物治療を投与する能力を含む全層熱傷被覆材において有用な特性をもつ絹材料を提供する。本発明において、拘束乾燥技法を用いて、絹濃度が繊維厚さ、密度、配向、相分散、多孔性およびマット厚さを含む特性に大きな役割を果たしたことが見出された。例えば、全層創傷被覆材において、4:1〜2:1の絹/PEOブレンド比をもつ電界紡糸絹マットが使用され、それは最小限の表面積減少で柔軟な膜様材料を提示する有用な物理的特性を保有し、グラム陰性桿菌およびグラム陽性球菌敗血症を惹起する病原性微生物に対して不透過性の障壁をもたらす0.3μm2よりも小さい細孔のど表面積サイズを示す。
【0080】
材料の吸収および平衡含水率(EWC)特性は、細菌の栄養床を提供し得る創傷浸出液の蓄積を制御する際に役割を果たす。一態様では、4:1〜1:1の範囲の絹/PEOブレンド比をもつ絹材料についての全体的な吸収性およびEWC性能は、各群内で比較的接近し、それぞれ400%〜700%および82%〜86%の範囲に及んだ。絹材料群での繊維密度の違いを考慮すると、異なるブレンド比をもつ各々の材料群はそれでも類似する膨潤特性を提示した。これらのモデルと表2中のその他の創傷被覆材候補を比較すると、本発明の絹マットはスポンジのような天然キトサンに基づく包帯材と同様によく機能した。キトサン/ポロキサマー包帯材候補は良好な吸収性およびEWC特性を有したが、本発明の全天然のFDA認可絹材料系は、これらのその他の系と比較して生体適合性および顕著な機械的堅牢性を提供する。
【0081】
(表2)キトサン誘導体に基づく創傷被覆材と本発明の絹材料系との間の平均吸水率および平衡含水率の比較
*Gibran et al., 70 J. Surg. Res. 1-6 (1997);**Quynh et al., 2007;***Wu & Wu, 2006
【0082】
体温恒常性を維持するために、正常な皮膚は、1日あたり204g/m2の速度で体液を透過させる。Lamke et al.,3 Burns 159-65(1977)。また、全層芽創の蒸発水損失は、1日当たり5,138g/m2であり、過剰な脱水を防ぐと同時に適切な水分レベルを許容するために理想的な全層創傷被覆材は、1日当たり2,000〜2,500g/m2の水蒸気透過速度(WVTR)有するべきであることも報告されている。Queen et al.,1987。一態様では、本発明において飽和および乾燥した絹材料は、37℃および50%RHで1,977±35および1,469±81g/m2/日のWVTRを有し、それは、表3に詳細に示されるように、相対温度および湿度で1,180〜2,830g/m2/日の範囲に及ぶキトサン包帯材と遜色がない。本発明の電界紡糸絹マットの厚さ(30〜80μm)を、スポンジのような二層および不斉キトサン包帯材(60〜800μm)に対して考慮すると、多層の絹包帯材を目的に合わせて作り、上記の所望のWVTR基準を実現することが妥当である。その上、図7に示されるように、1日当たり25,000から7,800cm3/m2までの酸素透過速度は、SEM写真において示される各々の材料群の繊維サイズ、多孔性および繊維密度に起因し得る。この生体材料の疎水性の性質は、これらのマットを、創傷浸出液のドレナージ、浮腫および脱水のバランスをとる際に最適なガスおよび水蒸気の透過性能に理想的な厚さに作る能力を促進する。
【0083】
(表3)キトサン誘導体に基づく創傷被覆材と本発明の絹材料系との間の平均水蒸気および酸素透過速度の比較
*Gibran et al., 70 J. Surg. Res. 1-6 (1997);**Quynh et al., 2007;***Hu et al., 2006
【0084】
絹マトリックスの全体にわたって相互連絡する細孔の網状構造は、水の不織構造組織間隙への吸収に有用な材料系であることがわかる。改質した電界紡糸絹繊維(絹/PEOブレンド比は4:1)は、68%までの多孔性を有する。Wang et al.,37 Macromol.6856-64(2004)。増加した表面積は、重鎖および軽鎖の両方の親水性領域内部に存在するヒドロキシル基、カルボキシル基、およびアミノ基に結合している極性水分子によりエネルギーが最小化されるときに、生体高分子のバルク領域への水の吸収を促進する。膨潤は、混和性の希釈剤分子が、ポリマー鎖間を流動して自由体積を生成するときに起こる。絹/PEOブレンディング比が4:1、3:1、2:1、3:2、7:6、および1:1の風乾した2.8cmの電界紡糸絹マットを、直径10cmのマットから打ち抜いた。表4を参照すると、吸収は、461%〜613%の範囲に及び、全ての絹材料群の平均は551%±54%であった。その上、各々の絹モデルについての平均乾燥重量は、22.5mg(絹/PEOブレンド比は4:1)から13.6mg(絹/PEOブレンド比は1:1)まで直線的に低下したが、平衡含水率は、全ての材料系について84%±1%で比較的一定のままであった。データは、吸水が各々の絹濃度の繊維直径、密度、多孔性および二次構造集合特性とは無関係であることを示唆する。
【0085】
(表4)脱イオン水に24時間浸漬した電界紡糸絹/PEOブレンドマットについての平均吸水および平衡含水率測定値(±値=SD、n=6)
【0086】
酸素/二酸化炭素ガス交換を促進する包帯材は、創傷の酸度を低下させ、嫌気性細菌感染を抑制し、従って創傷治癒を促進する環境を形成する。Mi et al.,22 Biomats.165-73(2001);Mi et al.,59 J.Biomed.Mat.Res.438-49(2002)。表5に提示されるように、水和条件下(37℃および80%RH)で評価したサンプルの平均酸素透過速度(OTR)は、4:1サンプルについて25,000cm3/m2/日から、1:1サンプルについて7,800cm3/m2/日に至る平均OTRを提示した。これらのOTRの低下は、それぞれの絹材料群の減少する繊維サイズ、細孔のど径、および増加した繊維密度に起因し得る。既に述べたように、4:1、3:1、および2:1モデルは、緩く分布した繊維密度をもつ多孔性足場を形成するビーズ領域を含むミクロン以上の(micron plus)繊維直径を有した。3:2および7:6繊維は、200nm〜500nmの間に及ぶより小さい直径を有し、繊維密度の増加およびマット多孔性の低下を示した。その上、相の分散した1:1繊維は、多孔性が低下して結晶質-非晶質ガラス障壁を生成したフィブロインのシートを形成した。対照的に、37℃および50% RHで試験した各々の材料群についての全ての飽和および風乾サンプルは、1回の15分間隔の試験の終了よりも前に100,000cm3/m2/日の分析器閾値を上回った。予測されるように、これらの結果は、乾燥またはほぼ乾燥状態ではこれらの多孔質材料は、飽和不織織物の組織間隙およびバルク領域に存在する水分子を通じて酸素が拡散される場合よりも大きい酸素透過速度を有することを反映する。図11の線形曲線分析は、4:1から2:1までの絹ブレンドマトリックスの9,600cm3/m2/日のOTR低下を開示する。
【0087】
(表5)飽和した電界紡糸絹/PEOブレンドマットの平均酸素透過速度を、Illinois Instruments 8001 Oxygen Permeation Analyzerを用いて37℃にて80% RHで測定した。酸素透過性(PO2)および酸素透過係数(P'O2)値は、ASTM 3985-05に基づいて計算した。(±値=SD、n=3)
酸素ガス透過速度:O2GTR:cm3/(m2・d);単位厚さ当たりの酸素透過:cm3/(m2・d)/単位厚さ
【0088】
全層創傷被覆材の水蒸気透過性は、創傷部位での体液蒸発の制御において重要な役割を果たす。過剰な水蒸気透過特性を提示する創傷被覆材は、血液量減少、低体温、および高血圧症を引き起こし得る。Peppas,HYDROGEL MED.&PHARM.II&III(CRC Press,Boca Raton,FL,1987);Beers et al.,2006。水蒸気透過速度(WVTR)は、24時間にわたって、引き伸ばして乾燥した25cm2の4:1、3:1、および2:1絹/PEOマットについて計算した。3:2、7:6、および1:1マットについてのWVTRを確かめる努力は、乾燥段階中の材料変形のために不成功に終わった。拘束乾燥した材料は、水和状態と乾燥状態の両方で評価した。表6に示されるように、飽和および乾燥した4:1、3:1、および2:1材料群についてのWVTRは、平均して、飽和に関して1,977±35g/m2/日、乾燥に関して1469±81g/m2/日であった。飽和したサンプルが乾燥したサンプルよりも優れていたが、これは液体-ガス-膜-ガス界面と比べて直接の液体-膜-ガス界面に起因する可能性が最も高い。水和したマットの吸湿性は、熱力学的反応および蒸発の加速を促進する生体材料表面-ガス界面に水分子を都合よく存在させることを可能にした。図11に示されるように、WVTRは、3つ全ての絹濃度で比較的同じであり、162g/m2/日および82g/m2/日のごく僅かな下降する変化は、繊維サイズ、マット密度、多孔性および二次構造特性に起因するものであった。
【0089】
(表6)飽和および乾燥した4:1、3:1および2:1拘束乾燥絹/PEOマトリックスについての平均水透過速度を、24時間後に、22.8±0.6℃および50%±2%RHで、ウォーターカップ法(water cup method)に続いてASTM D 1653に従ってPerm Cupを用いて測定した。(±値=SD、n=3)
【0090】
本発明の一部の態様はまた、絹フィブロインタンパク質、ポリエチレンオキシド(PEO)、および少なくとも一つの活性物質を含む、少なくとも一つの電界紡糸絹マットと創傷を接触させることを含む、創傷治癒を促進する方法にも関する。電子紡糸絹マットの絹フィブロインタンパク質/PEOブレンド比は、約2:1〜約4:1であり(または絹の割合が約75%w/w〜90%w/wであり);絹マットの厚さは、約20〜約80μmである。
【0091】
本発明は、効果的な創傷被覆材などの生物医学的用途に適した、電界紡糸した絹/PEO材料を提供する。電界紡糸した絹/PEOマトリックスの物理的特性および生体機能特性を評価して、創傷被覆材に関連する構造的形態学的および生体適合性の特徴を査定した。例えば吸収、水蒸気透過性、酸素透過性、および生分解性生体機能特性などの特性は、創傷被覆材用途に有用である。絹/ポリエチレンオキシド(PEO)含量の変化を用いて、形態および構造に関して異なるマトリックスを作成した。二流体絹/PEO電界紡糸技法を適用して、大型の融合性絹マットを製造し、表面のテクスチャーおよびバルク特性を数量化した(繊維構造、トポグラフィー、吸収、水WVTR、酸素透過性、および生分解性を含む)。水和した状態では、全ての材料群が、創傷治癒に適した吸収性およびVTRを提示した。酸素透過速度(OTR)は、創傷部位に適した酸素/二酸化炭素ガス交換の特色を示唆した。インビトロ酵素生分解は、1日で初期重量の23%±3%の分解および14日で74±9%までの分解を確認した。かかる創傷マット系の保存および分布に関連する材料特性に対応するために、複数の乾燥法を検討した。制御蒸発および拘束乾燥技法を用いると、絹濃度が、繊維伸張、整列、密度、多孔性および相分散に影響を及ぼす各々のマトリックスの特性の決定要因であった。絹/PEOブレンド比が2:1〜4:1の、制御蒸発および拘束乾燥技法で処理した電界紡糸絹/PEOマットは、創傷被覆材に対する潜在的有用性をもつ生体材料系に関する特に適した特性を実証した。これらの絹材料系は、抗生物質送達、マクロファージ応答、線維芽細胞/ケラチン生成細胞およびサイトカインの影響、および関連する生物学的問題に有用でありうる。
【0092】
本発明の特定の態様を、限定されない例において説明する。
【0093】
本発明は、以下の番号の付いた項目のいずれか一つに定義される通りでありうる。
1.以下の段階を含む、絹マットを製造するための方法:
ポリエチレンオキシド(PEO)と絹フィブロイン水溶液をブレンドする段階;
前記ブレンドした溶液を電界紡糸する段階であって、それにより絹タンパク質/PEOブレンドマットを形成する、段階;および
前記電界紡糸した絹マットを拘束乾燥する段階。
2.電界紡糸した絹マットをアルコールで処理する段階をさらに含む、項目1記載の方法。
3.PEOを絹マットから抽出する段階をさらに含む、項目1または2記載の方法。
4.少なくとも一つの活性物質を絹マットに埋め込む段階をさらに含む、項目1〜3のいずれか一項記載の方法。
5.活性物質が、細胞、タンパク質、ペプチド、核酸、核酸類似体、ヌクレオチドまたはオリゴヌクレオチド、ペプチド核酸、アプタマー、抗体またはその断片もしくは部分、抗原またはエピトープ、ホルモン、ホルモンアンタゴニスト、成長因子または組換え成長因子およびその断片および変異体、細胞接着メディエーター、サイトカイン、酵素、抗生物質または抗菌性化合物、ウイルス、毒素、プロドラッグ、化学療法剤、小分子、薬物、ならびにそれらの組合せからなる群より選択される治療剤または生物材料である、項目4記載の方法。
6.活性物質が、前駆細胞または幹細胞、平滑筋細胞、骨格筋細胞、心筋細胞、上皮細胞、内皮細胞、尿路上皮細胞、線維芽細胞、筋芽細胞、口腔細胞、軟骨細胞、軟骨芽細胞、骨芽細胞、破骨細胞、ケラチン生成細胞、腎尿細管細胞、腎基底膜細胞、外皮細胞、骨髄細胞、肝細胞、胆管細胞、膵島細胞、甲状腺細胞、上皮小体細胞、副腎細胞、視床下部細胞、下垂体細胞、卵巣細胞、精巣細胞、唾液腺細胞、脂肪細胞、前駆細胞、およびそれらの組合せからなる群より選択される細胞である、項目5記載の方法。
7.活性物質が、細胞増殖培地をさらに含む、項目6記載の方法。
8.活性物質が、抗生物質である、項目6記載の方法。
9.ポリエチレンオキシド(PEO)と絹フィブロイン水溶液をブレンドする段階;
ブレンドした溶液を電界紡糸する段階であって、それにより絹タンパク質/PEOブレンドマットを形成する、段階;および
電界紡糸した絹マットを拘束乾燥する段階
を含む方法から調製された絹材料。
10.ポリエチレンオキシド(PEO)を、少なくとも一つの活性物質を含む絹フィブロイン水溶液とブレンドする段階;
ブレンドした溶液を電界紡糸する段階であって、それにより該活性物質を封入する絹タンパク質/PEOブレンドマットを形成する、段階;および
該活性物質を封入する電界紡糸した絹マットを拘束乾燥する段階
を含む方法から調製された、創傷に巻いて創傷治癒を促進するための少なくとも一つの活性物質を封入する絹材料。
11.活性物質が、細胞、タンパク質、ペプチド、核酸、核酸類似体、ヌクレオチドまたはオリゴヌクレオチド、ペプチド核酸、アプタマー、抗体またはその断片もしくは部分、抗原またはエピトープ、ホルモン、ホルモンアンタゴニスト、成長因子または組換え成長因子およびその断片および変異体、細胞接着メディエーター、サイトカイン、酵素、抗生物質または抗菌性化合物、ウイルス、毒素、プロドラッグ、化学療法剤、小分子、薬物、ならびにそれらの組合せからなる群より選択される治療剤または生物材料である、項目10記載の絹材料。
12.活性物質が、前駆細胞または幹細胞、平滑筋細胞、骨格筋細胞、心筋細胞、上皮細胞、内皮細胞、尿路上皮細胞、線維芽細胞、筋芽細胞、口腔細胞、軟骨細胞、軟骨芽細胞、骨芽細胞、破骨細胞、ケラチン生成細胞、腎尿細管細胞、腎基底膜細胞、外皮細胞、骨髄細胞、肝細胞、胆管細胞、膵島細胞、甲状腺細胞、上皮小体細胞、副腎細胞、視床下部細胞、下垂体細胞、卵巣細胞、精巣細胞、唾液腺細胞、脂肪細胞、前駆細胞、およびそれらの組合せからなる群より選択される細胞である、項目11記載の絹材料。
13.活性物質が、細胞増殖培地をさらに含む、項目12記載の絹材料。
14.活性物質が、抗生物質である、項目12記載の絹材料。
15.電界紡糸絹マットが、アルコールでさらに処理される、項目9〜14のいずれか一項記載の絹材料。
16.PEOが、電界紡糸絹マットから抽出される、項目9〜15のいずれか一項記載の絹材料。
17.電界紡糸絹マットの厚さが約20μm〜約80μmである、約50重量%〜約100重量%の範囲に及ぶ絹フィブロインタンパク質を含む電界紡糸した絹材料。
18.電界紡糸絹マット中の絹フィブロインタンパク質の含量が、約75重量%〜約90重量%の範囲に及ぶ、項目17記載の電界紡糸絹材料。
19.電界紡糸絹マット中のPEOの含量が、約0重量%〜約50重量%の範囲に及ぶ、電界紡糸絹マット中にポリエチレンオキシド(PEO)のブレンドをさらに含む、項目17または18記載の電界紡糸絹材料。
20.電界紡糸絹マット中のPEOの含量が、約10重量%〜約25重量%の範囲に及ぶ、項目19記載の電界紡糸絹材料。
21.少なくとも一つの活性物質をさらに含む、項目17〜20のいずれか一項記載の絹材料。
22.活性物質が、細胞、タンパク質、ペプチド、核酸、核酸類似体、ヌクレオチドまたはオリゴヌクレオチド、ペプチド核酸、アプタマー、抗体またはその断片もしくは部分、抗原またはエピトープ、ホルモン、ホルモンアンタゴニスト、成長因子または組換え成長因子およびその断片および変異体、細胞接着メディエーター、サイトカイン、酵素、抗生物質または抗菌性化合物、ウイルス、毒素、プロドラッグ、化学療法剤、小分子、薬物、およびそれらの組合せからなる群より選択される治療剤または生物材料である、項目21記載の絹材料。
23.絹マットの厚さが約20〜30μmである、項目17〜22のいずれか一項記載の絹材料。
24.絹マットが、平均して約0.1〜約0.3μmの細孔のど径表面積をもつ、相互接続した細孔を有する、項目17〜23のいずれか一項記載の絹材料。
25.得られる絹マットの吸水量が約460%よりも大きい、項目9〜24のいずれか一項記載の絹材料。
26.得られる絹マットの平衡含水率が約82%よりも大きい、項目9〜25のいずれか一項記載の絹材料。
27.得られる絹マットの酸素透過速度が約15460cm3/m2/日よりも大きい、項目9〜26のいずれか一項記載の絹材料。
28.得られる絹マットの水蒸気透過速度が約1934g/m2/日よりも大きい、項目9〜27のいずれか一項記載の絹材料。
29.絹フィブロインタンパク質、および任意で少なくとも一つの活性物質を含む、少なくとも一つの電界紡糸絹マットに創傷を接触させる段階を含む、創傷治癒を促進する方法であって;
前記絹フィブロインタンパク質が約50重量%〜約90重量%の範囲に及び、
前記絹マットの厚さが約20μm〜約80μmであり;
前記絹マットが、約460%よりも大きい吸水量か、または約82%よりも大きい平衡含水率を有し;かつ
得られる絹マットの酸素透過速度が約15460cm3/m2/日よりも大きい、前記方法。
30.絹フィブロインタンパク質が、約75重量%〜約90重量%の範囲に及ぶ、項目29記載の方法。
31.絹フィブロインタンパク質、ポリエチレンオキシド(PEO)、および任意で少なくとも一つの活性物質を含む、少なくとも一つの電界紡糸絹マットに創傷を接触させる段階を含む、創傷治癒を促進する方法であって;
絹/PEOブレンド比が、約4:1〜約2:1であり;
前記絹マットの厚さが約20μm〜約80μmであり;
前記絹マットが、約460%よりも大きい吸水量か、または約82%よりも大きい平衡含水率を有し;かつ
得られる絹マットの酸素透過速度が約15460cm3/m2/日よりも大きい、前記方法。
32.絹マットの水蒸気透過速度が、約1934g/m2/日よりも大きい、項目29〜31のいずれか一項記載の方法。
33.活性物質が、細胞、タンパク質、ペプチド、核酸、核酸類似体、ヌクレオチドまたはオリゴヌクレオチド、ペプチド核酸、アプタマー、抗体またはその断片もしくは部分、抗原またはエピトープ、ホルモン、ホルモンアンタゴニスト、成長因子または組換え成長因子およびその断片および変異体、細胞接着メディエーター、サイトカイン、酵素、抗生物質または抗菌性化合物、ウイルス、毒素、プロドラッグ、化学療法剤、小分子、薬物、およびそれらの組合せからなる群より選択される治療剤または生物材料である、項目29〜32のいずれか一項記載の方法。
【実施例】
【0094】
実施例1
材料
カイコ絹の繭(Tajima Shoji Co.,Yokohama,Japan)を調製して8重量%の絹溶液を生成した。Wang et al.,2006.4:1、3:1、2:1、3:2、7:6、および1:1w/w絹:PEO(920,000g/mol)比溶液を用いて6つの絹材料を調製した。電界紡糸の間に安定した流体の噴出を生じるための粘弾特性および表面張力特性に必要な最低限の7.2%絹/PEOポリマー濃度を維持するために、4:1、3:1、および2:1ブレンドは、5%のPEOを含有し、一方3:2、7:6、および1:1ブレンドは6%のPEOを含有した。Jin et al.,3 Biomacromol.s 1233-39(2002)。
【0095】
この研究のために構築した電界紡糸装置は、高電圧電源(Gamma High Voltage Research ES-30P,Ormond Beach,FL)、10〜60mlシリンジポンプ(Braintree Scientific 8000,Braintree,MA)、電位ステージおよび接地ステージ、1.5mmポリエチレン管および16ゲージ5.08cm鋼毛管を用いて、既に公開された手順(Wang et al.,2002)に従った。例えば、国際公開公報第2004/0000915号;国際公開公報第2004/062697号を参照されたい。
【0096】
絹/PEO溶液を、ポリエチレン管を通じてシリンジポンプから、電位プレートに挿入した12kV DC荷電鋼毛管(steal capillary tube)にポンプで送った。同文献;Reneker&Yarin,49 Polymer 2387-25(2008);Jin et al.,2002。電界紡糸繊維は、電位プレートよりもおよそ17cm低く設置され、毛管の先端の垂直落下線を越えておよそ2cm〜3cmに位置する、接地ステージで収集した。これらの重量比に基づくと、方程式は次の通りである。
【0097】
各々のモデルについての絹の割合w/wは、それぞれ、86.5%、82.8%、76%、70.6%、65.1%および61.5%に等しかった。溶液粘度は、#5スピンドルを69°Fで用いるBrookfield HATD粘度計(Brookfield Engineering Laboratories,Inc.,Stoughton,MA)によって、それぞれ、128、152、240、424、768、および1120mPa-Sに等しいと決定された。直径16.5cmおよび10cmのマットを、6つの材料群の各々について室温にて(RT)60%よりも低い相対湿度で電界紡糸して、電界紡糸プロセスによるマット厚さの違いを評価した。
【0098】
乾燥法
乾燥技法を用いて、絹電界紡糸材料マットの物理的特性を評価した。風乾法では、直径3.5cm、2.8cmおよび2.2cmのサンプルを、水に浸漬した直径10cmのマットから打ち抜いた。秤量紙(VWR,West Chester,PA)の間でプレスした後、サンプルを、ほぼ乾燥するまでポリスチレンペトリ皿の側壁に垂直に置いた。その後、粘着を防ぐためにサンプルを定期的に再配置し、RTにて24時間乾燥させた。
【0099】
拘束乾燥法では、大型の直径16.5cmのサンプルを水浴から取り出し、3分の1まで脱イオン水を入れた125×65mm結晶化皿の口の上に掛け、RTにて20%〜50%の間のRHのデシケーターの中に入れ、一晩乾燥させる。絹/PEOマットは、口の円周全体に沿った縁にマットが接触し、軽く付着するように掛けた。乾燥段階の間、飽和したサンプルが縁からサンプルの中心に向かって均一に乾燥するとき、マットの各々のセットは皿の口の全体にわたって漸進的に収縮する。4:1および3:1サンプルは、結晶化皿の縁に付着して完全に乾燥し、引き伸ばされた、完全に平坦で柔軟な白色の膜のような材料をもたらした。サンプル重量(Mettler Toledo AB54-S/FAC,Columbus,OH)および厚さ(Ono Sokki EG-225F Digital Indicator,Addison,IL;等半径点AA821;力25g)の測定値を、各々の絹系について記録した。
【0100】
実施例2 材料の特徴付け
繊維厚さおよび表面トポグラフィーは、JEOL JSM 740-1F FE-SEM(Tokyo,Japan)を1.5倍、6.5倍および12倍の倍率で用いて特徴付けた(加速電圧:1kV、作動距離:13.6mm)。断面画像は、2.5倍、5倍、10倍、および50倍の倍率を用いて撮影した(加速電圧:5kV、作動距離:6mm)。断面のサンプルを、2×5mmの断片に切断し、液体窒素中で急速冷凍し、ピンセットを用いて半分に割った。サンプルを、断面を上にしてカーボンテープの上に取り付けた。全てのサンプルを、Denten Vacuum Desk IV(Moorestown,NJ)を次の設定で用いて100Å Auでコーティングした:真空:80〜90mtorr、スパッタリング設定点:20〜30%、堆積時間:2分。サンプルの表面形態、粗さ、および三次元の特徴を、PSIA XE-150 AFM(Santa Clara,CA)によって、Ultrasharp NSC16/AIBSプローブを非接触モードで用いて得た(共振周波数:170kHz、バネ定数:45N/m)。XEIデータ分析ソフトウェア(Park Solutions,Santa Clara,CA)を、表面粗さの特徴付けに用いた。5つの繊維層の最小深さ(約1μm)まで伸びる細孔によって定義される材料多孔性を、50×50μmの面積に分布バケツアルゴリズム(distribution bucket algorithm)を適用して統計的に評価した。細孔のど径および細孔表面積を、細孔径直径が0.15、0.30、0.45、0.60、0.75、0.90、1.05および1.25μmの範囲に及ぶ、円形領域に関して幾何学的に推定した。
【0101】
実施例3 水および酸素透過性
吸収
6つの直径2.8cmの試験サンプルを、ポリスチレン皿法で乾燥させ、無菌の6ウェルの組織培養処理したポリスチレンプレートに入れ、脱イオン水に24時間浸漬して膨潤平衡に達した。次に、サンプルを取り出し、Kimwipe(登録商標)組織の上に最小限の安定したペンダントドロップがサンプルの端部に維持されるまで優しく塗りつけた。次に、飽和したサンプルを秤量し、吸水および平衡含水率(EWC)を次の方程式により算出した。
式中、WwおよびWdは、それぞれ、湿潤および乾燥サンプルの重量である。Kim et al.,341 Int.J.Pharm.35-43(2007)。吸収(%)およびEWC(%)の結果を表4に示す。
【0102】
酸素透過速度
酸素透過速度(OTR)を、Illinois 8001 Oxygen Permeation Analyzer(Illinois Instruments,Johnsburg,Illinois;ASTM 3985-05)を用いて測定した。円形の5cm2脱イオン水飽和サンプルを、15分の試験間隔の間中、37℃および80% RHの水和した環境ならびに37℃および50% RHのより乾燥した条件下で試験した。試験は、3つの連続した1%以内の酸素透過速度を記録して上首尾に終わった。酸素透過性は、ASTM 3985-05に従って1日当たりのcm3/m2で記録した。Apiezon Type T Grease(Manchester,UK)を用いてサンプルを5cm2マスクの間に密封した。単位厚さ当たりの酸素ガス透過速度および酸素透過速度の結果を表5に示す。
【0103】
水蒸気透過速度(WVTR)
Perm Cup(Gardner Co.,Pompano Beach,FL)を用いてASTM D 1653ウォーターカップ法Bに従ってWVTRを測定した。飽和および乾燥した25cm2直径サンプルを、上端の6mmまで満たされたカップの開口部に密閉し、73±1°F(22.8±0.6℃)および50±2%RHに維持した、温度と湿度を制御された環境に24時間置いた。装入されたカップ構成を、開始時および24時間後に0.1mgの精度で秤量した。温度および相対湿度は6時間ごとに確認し、水蒸気透過を1日当たりのg/m2でカップの重量減少により計算した。
【0104】
生分解
生分解性に用いたプロテアーゼは、タンパク質構造の複数の位置で絹フィブロインを区別せずに切断することが示された。Horan et al.26 Biomats.3385-93(2005);Li et al.,24 Biomats.357-65(2003)。6つの材料群からの三層の円形の3.5cmサンプルを、25±5mgの重さになるように手入れし(manicured)、3回20分の70%エタノール浴で滅菌し、PBSですすぎ、その後にpH7.4のPBS中1mg/mLプロテアーゼXIV(EC 3.4.24.31,5.6 U mg-1,Streptomyces griseus,Sigma,St.Louis,MO)の溶液6mL中37℃でインキュベートした。Jin et al.,15 Adv.Funct.Mater.1241-47(2005);Horan et al.,2005。対照サンプルを、酵素を含まないPBSに浸漬した。酵素および対照溶液は毎日補充した。サンプルを脱イオン水中で1時間すすいでから1、3、6、10、および14日後に生分解性を測定した。小型のへら、および4mLシリンジにに取り付けられた25ゲージ毛管を用いて、サンプルを培養ウェルプレートから指定された事前に秤量した秤量皿に移した。サンプルを無菌フード下RTにて24時間乾燥させた後、秤量して経時的重量減少率を決定した。線形回帰分析は、Minitab(登録商標)15.1.30(Minitab Inc.,State College,PA)を用いて行った。
【0105】
実施例4
材料
カイコ絹の繭(Tajima Shoji Co.,Yokohama,Japan)を調製して8重量%の絹溶液を生成した。Wang et al.,2006。4:1、3:1、2:1、3:2、7:6、および1:1w/w絹:PEO(900,000g/mol)比溶液を用いて6つの絹材料を調製した。電界紡糸の間に安定した流体の噴出を生じるための粘弾特性および表面張力特性に必要な最低限の7.2%絹/PEOポリマー濃度を維持するために、4:1、3:1、および2:1ブレンドは、5%のPEOを含有し、一方3:2、7:6、および1:1ブレンドは6%のPEOを含有した。
【0106】
溶液粘度を、#5スピンドルを69°Fで用いるBrookfield HATD粘度計(Brookfield Engineering Laboratories,Inc.,Stoughton,MA)によって、それぞれ、128、152、240、424、768、および1120mPa・S-1に等しいと決定した。
【0107】
この研究のために構築した電界紡糸装置は、高電圧電源(Gamma High Voltage Research ES-30P,Ormond Beach,Fl.)、60mlシリンジポンプ(Braintree Scientific 8000,Braintree,MA)、電位ステージおよび接地ステージ、1.5mmポリエチレン管および16ゲージ5.08cm鋼毛管を用いて、既に公開された手順に従った。
【0108】
絹/PEO溶液を、ポリエチレン管を通じてシリンジポンプから、電位プレートに挿入した12kV DC荷電鋼毛管にポンプで送った。電界紡糸繊維を、電位プレートよりもおよそ21cm低く設置され、毛管の先端の垂直落下線を越えておよそ2.5cmに位置する、アルミニウム箔で覆った接地ステージで収集した。
【0109】
上に挙げたた各々のブレンド比の絹/PEOの10および4.5mLバッチ溶液を用いて、それぞれ16.5および10cm絹マットを作成した。溶液は、室温にて(RT、20〜22℃)60%よりも低い相対湿度(RH)で電界紡糸した。絹/PEOマットを、90%MeOH溶液に20分間浸漬してβ-シート形成および結晶化を誘導した。72時間の3回の1L dH2O浴での浸出でPEOを抽出した。
【0110】
重量比方程式
に基づいて、各々のモデルの絹含量は、それぞれ86.5重量%、82.8重量%、76.2重量%、66.7重量%、60.9重量%、および57.1重量%であった。未処理の水溶性絹/PEOマットを、S87P13、S83P17、S76P24、S67P33、S61P39、およびS57P43(S87P13〜SP57P43)と名付けた。S87、S83、S76、S67、S61、およびS57(S87〜S13)と名付けられたマットは、メタノール処理しPEO抽出した絹マットを表す。
【0111】
乾燥法
非拘束および拘束乾燥技法を用いて、S87〜S57絹材料群の物理的特性を評価した。
【0112】
非拘束乾燥法では、飽和直径3.5cmのサンプルを10cmの電界紡糸キャストから打ち抜き、秤量紙(VWR,West Chester,PA)の間で手でプレスした後、ほぼ乾燥するまで表面対空気界面を最大化するためにポリスチレンペトリ皿の側壁に垂直に置いた。サンプルは、粘着を防ぐためにサンプルを定期的に再配置し、RTにて24時間乾燥させた。
【0113】
拘束乾燥技法を適用し、絹材料を、結晶化皿の口の上に掛けて付着している間に延伸力を受けながら乾燥させた。水浴から取り出し、直径16.5cmのS87〜S57サンプルを、3分の1をdH2Oで満たした125×65mm2結晶化皿の上に掛け、周囲条件下で一晩乾燥させた。サンプル重量(Mettler Toledo AB54-S/FAC,Columbus,OH)および厚さ(Ono Sokki EG225F Digital Indicator,Addison,IL;等半径点AA821;力25g)の測定値を、各々の絹系について記録した。
【0114】
材料の特徴付け
繊維形態、表面トポグラフィー、および断面の特性を、電界放射型走査電子顕微鏡(FE-SEM,JEOL JSM 740-1F,Tokyo,Japan)により1.5〜50倍の倍率で特徴付けた。繊維形態を、S87P13〜S57P43および拘束乾燥したS87〜S76サンプルセットについて評価した。表面トポグラフィーおよび断面特性を、拘束乾燥したS87〜S76サンプルについて査定した。断面のサンプルを、2×5mm2の断片に切断し、液体窒素中で急速冷凍し、ピンセットを用いて半分に割った。サンプルを、断面を上にしてカーボンテープの上に取り付けた。全てのサンプルを、Denton Vacuum Desk IV(Moorestown,NJ)を次の設定で用いて100Å Auでコーティングした:真空:80〜90mtorr、スパッタリング設定点:20〜30%、堆積時間:2分。
【0115】
S87〜S57サンプルの表面形態は、PSIA XE-150原子間力顕微鏡(AFM Santa Clara,CA)によって、Ultrasharp NSC16/AIBSプローブを非接触モードで用いて測定した(共振周波数:170kHz、バネ定数:45N・m-1)。表面粗さおよび三次元(3D)の特色を、XEI定量分析(Park Solutions Inc.,Santa Clara,CA)によりレンダリングした(rendered)。材料の多孔性は、5つの繊維層の最小深さ(約1μm)まで伸びる細孔により定義した。細孔のど径頻度分布は、50×50μm2の面積にわたって測定した。
【0116】
吸収
非拘束乾燥した、直径2.9cmのS87〜S57サンプルを事前に秤量し、次にdH2Oに24時間浸漬して膨潤平衡に達した。次に、サンプルを、Kimwipe(登録商標)組織の上に最小限の水がサンプル表面に観察されるまで優しく塗りつけた。次に、飽和したサンプルを秤量し、吸水および平衡含水率(EWC)を次の方程式により算出した。
式中、WwおよびWdは、それぞれ、湿潤および乾燥サンプルの重量である。
【0117】
酸素透過速度
酸素透過速度(OTR)を、Illinois 8001 Oxygen Permeation Analyzer(Illinois Instruments,Johnsburg,Illinois;ASTM 3985-05)を用いて測定した。OTRを、37℃および80% RHで15分間隔で試験した、円形の5cm2の水和したS87〜S57サンプルについて測定した。非拘束乾燥S87〜S57マットのOTRは、37℃および50%RHで測定した。酸素透過性は、ASTM 3985-05に従ってcm3・m-2・d-1で記録し、試験は3つの連続した1%以内のOTRを記録して上首尾に終わった。Apiezon Type T Grease(Manchester,UK)を用いてサンプルを5cm2マスクの間に密封した。
【0118】
水蒸気透過速度
水蒸気透過速度(WVTR)は、Perm Cup(Gardner Company,Pompano Beach Florida)を用いてASTM D 1653ウォーターカップ法Bに従って測定した。水和および拘束乾燥した円形の25cm2 S87〜S76サンプルで、上端の4mmまでdH2Oを満たしたPerm Cupの口を覆って密閉した。事前に秤量した集合体を、73±1°F(22.8±0.6℃)および50±2% RHに維持された環境に置き、24時間後に0.1mgの精度で再び秤量した。温度および相対湿度は6時間ごとに確認し、水蒸気透過を集合体の重量減少によりg・m-2・d-1で計算した。
【0119】
生分解
生分解性に用いたプロテアーゼは、タンパク質構造の複数の位置で絹フィブロインを区別せずに切断することが示された。三層の円形3.5cm非拘束乾燥S87〜S57サンプルを、25±5mgの重さになるように切断し、3回20分の70%エタノールへの浸漬によって滅菌し、リン酸緩衝生理食塩水(PBS,pH=7.4,Invitrogen 291,Carlsbad,CA.)ですすぎ、その後に1mg/mLプロテアーゼXIV(EC 3.4.24.31,5.6 U mg-1,Streptomyces griseus,Sigma,MO)の6mL PBS溶液中37℃でインキュベートした。対照サンプルを、酵素を含まないPBSに浸漬した。酵素および対照溶液は、毎日補充した。サンプルをdH2O中で1時間すすいでから1、3、6、10、および14日後に生分解性を測定した。先細りした端の平らなマイクロスパチュラ、および4mLシリンジにに取り付けられた25ゲージ毛管を用いて、サンプルを培養ウェルプレートから指定された事前に秤量した秤量皿に移した。サンプルを無菌フード下RTにて24時間乾燥させた後、秤量して経時的重量減少率を決定した。線形回帰分析は、Minitab(登録商標)15.1.30(Minitab Inc.,State College,Pennsylvania)を用いて行った。
【0120】
初期評価
絹/PEO電界紡糸プロセスを用いて、6つのS87P13〜S57P43材料系を全て直径16.5cmおよび10cmのマットに電界紡糸した。各々のS87〜S57サンプルの物理的特性を、水和状態および乾燥状態で評価した。dH2Oに浸漬されると、6つのS87〜S57材料群は全て、不透明に半透明の外観をもつ均一な立体構造を有し、絹のようなテクスチャーをもち柔軟であったが、取扱期間が延びると絹濃度の低下に対して材料の剪断の増加を提示した(図1B)。絹のようなテクスチャーとは、水分子が非晶質バルクマトリックスの全体にわたって吸収し可塑化するフィブロインの動的吸湿性を説明するために言及される。アミノ、ヒドロキシル、またはカルボキシル酸末端基との水素結合を形成することによるか、または、親水性ドメインの至る所に自由に分散することにより、高移動性水分子は、これらの飽和材料系の軟質の絹のようなテクスチャーを生成する運動エネルギーの最小化によって継続的に移行している。
【0121】
乾燥法1
水和した3.5cm S87〜S57サンプルは、正味の力の表面-表面疎水性平衡を実現し、層状の絹シートの分離および変位をもつ親水性の傾向を示すために、半分に折り重なることがある。しかし、24時間の乾燥時間の後、物理的な特徴はS87〜S57材料系で漸進的に変化した。絹濃度の低下に関連して、マットは、雪のように白い柔軟なウエハーのような構造から、制限された付着曲げ強度をもつ半透明褐色で超薄のフィルムのような材料に変わった(図2B)。
【0122】
タンパク質ポリマーのねじれた襞のあるβ-シート形成の特徴を示して、マトリックスは完全に平坦な配向で乾燥せず、S87およびS82群だけが元の円形の形状を保持した。その上、重鎖結晶領域間に交互に配置された非晶質ドメインの末端に位置するプロリンがある。プロリンは、脱水すると収縮し、繊維の縮小する能力を増加させることが示されている。これらの要素は、51.0±0.0%〜87.5±9.9%の間のそれらの表面積を失う、非拘束乾燥サンプルのねじれた、不規則な立体構造の一因となる(図13)。
【0123】
非拘束乾燥S87〜S57材料群についての厚さ測定値は、それぞれ、81.7±7.5μmから77.5±10.5μm、66.7±5.1μm、53.3±8.1μm、46.7±5.1μm、および30.0±6.4μmに直線的に下降した(図13)。PEO濃度は、電界紡糸プロセスの間の繊維の表面積およびバルク形態に直接影響を及ぼしうる。Wang et al.,2006。PEO濃度が増加するにつれて、絹繊維を形成するフィブロインミセルおよび小球構造のサイズは低下する。その上、素早く動く(whipping)帯電した流体噴出の内部の縦応力が、これらの小球構造を整列させ、10000倍まで伸張させる。Wang et al.,2006;Kowalewski et al.,2005;Reneker&Yarin,2008。その結果として、増加したPEO濃度で形成された絹繊維はバルク体積が低下し、それは非拘束乾燥S87〜S57材料群に観察される漸進的物理的な変化と相互に関連する。
【0124】
乾燥法2
水和した12.5cm S87〜S57サンプルは展開がより容易であった。層状のシート分離は、横変位なしに各々のマットの内部領域においてのみ観察された。恐らくこれは、これらのサンプルが、より大型のサンプルから打ち抜かれたものでなく、従ってサンプルのパラメータに結晶化領域を保持しているという事実に起因しうる。図3Bの拘束乾燥したマットを参照すると、S87およびS82サンプルは、結晶化皿に付着して乾燥し、元の表面積の98%を保持し、平坦で柔軟な白色の膜をもたらす。S67、S61、およびS57群は同様には起こらなかった。これらのサンプルが乾燥するときに、延伸力が繊維の降伏点伸びを越えて材料に応力を加え、構造的破壊および60%の表面積減少がもたらされた。材料の剪断は、一般に皿の縁で開始し、絹マットの内部領域に伝わった。S76サンプルは皿の縁から剪断されたが、たった12%しか表面積減少はなく、物理的特性はS87およびS82マトリックスに類似していた(図13)。
【0125】
拘束乾燥期間の間、表面基質で、かつタンパク質バルク領域の全体にわたって蒸発する水分子は、メチル基の収束および疎水性鎖相互作用を開始した。このカスケードは、自由体積喪失およびフィブロイン収縮を実現した。結晶化皿の縁に付着しているので、材料は拘束された状態になり、繊維を半径応力の方向に延伸および伸張させる。材料が各々のサンプル群で均質であり、12.5cmの拘束乾燥したS76およびS67サンプルの平均厚さがごく僅かであった(図13)ことを考えると、各々の材料群の剪断点は各々の繊維のバルク体積に依存したと思われた。
【0126】
材料の特徴付け
未処理のS87P13〜S57P43マットのFE-SEM顕微鏡写真を、図6Bに示す。全体的に、これらの電界紡糸絹/PEO繊維は、直径が200nm〜500nmの範囲に及び、構造全体にわたって均一に分布し、繊維ビーズ形成は、PEO濃度の低下とともに次第に顕著であった。Wang et al.,2006;Zhou et al.,2000;Huang et al.,2001。S87P13、S82P18、およびS76P24繊維は、直径が1μmをやや上回るものから700nmまでの範囲に及ぶ、ランダムに分散したビーズセグメントを有した。ビーズは、S67P33およびS61P39画像で最小化され、明確な直径200nm〜500nmの繊維の均一な分布を開示する。S57P43サンプルは、不均一で高密度のマット構造に移行する、不規則なおよび非円形の形状の繊維から明らかな、特有の形態を有した。高密度のマットの外観は、装置の接地ステージに集まるときに固化していない繊維の相分散に起因しうる。S57P43繊維の直径は、300〜500nmの範囲に及んだが、一方、混合繊維は700nm〜900nmの間で測定された。
【0127】
図14のFE-SEM画像は、拘束乾燥したS87〜S76材料群を示す。図14Aの表面トポグラフィーは、各々のモデルの全体にわたる繊維の高密度でランダムな分布を反映するが、より精密な評価は、拘束乾燥技法と一緒に起こる繊維の伸張および再整列の痕跡の増加を明らかにする。図14BのS87繊維は、限られた繊維伸張または整列を伴う緩んでねじれた外観を有する。対照的に、S82およびS76繊維は、伸張され整列されて、ウェブのような微小なテクスチャーを形成するようになる。図14CのS82マットは、ピンと張ったウェブ構造を形成し、末端が透明な共連続形態になっている隣接する繊維間の相分散の痕跡を示している。S76サンプルのウェブ構造は、ロープのような配置を形成する、明確な、伸張され整列された繊維の絡み合った網状構造により定義される。
【0128】
拘束乾燥技法により明示される繊維の整列および伸張は、絹フィブロインブロック共重合体設計で示される両親媒性特性に起因し得る。親水性領域の内部で可塑剤として働くので、水分子は、二次構造の移動および再整列を促進する、低凝集エネルギーの非晶質鎖間で分子間の動きを伝える。述べたように、蒸発は、疎水性鎖相互作用および自由体積喪失を作動させ、繊維を半径応力の方向に収縮および延伸させる。二次構造が伸張されるにつれて、両側性の鎖間層構造の整列を促進し、結晶化した鎖内のねじれた立体構造を制限する、非晶質軽鎖の末端のプロリンの折り畳みは制限されるようになる。支配的な鎖間疎水性相互作用も、非晶質絹Iから結晶性絹IIへの結晶性二次構造の移行に影響を及ぼす。これらの作用の組合せにより、機械的安定性および繊維軸に沿った曲げ強度を備えるマットが形成される(図8B)。降伏点伸びを越えると、重鎖と軽鎖の両方の非晶質二次構造の中で、かつそれに沿って、剪断変形が起こる。
【0129】
図14C中の整列したS82繊維と図14D中のS76繊維との間の肉眼的な相分散の外観は、水の可塑化特性に起因し得る。水和したカイコ絹フィルムの示差走査熱量測定は、20〜23重量%の吸水で脱水した絹フィブロインのガラス転移温度(Tg)が178℃から40℃まで低下したことを明らかにした。結晶性立体構造はバルク領域の全体にわたって吸収に影響を及ぼすが、各々の水和した群のEWCは、80重量%よりも大きかった(表7)、これは相当な親水性相互作用を示し、Tgの低下および妥当な相分散を予測する。その上、表面/液体、表面/気体、表面/表面の熱力学的転移の間に、親水性分子および疎水性分子が逆転するときに界面エネルギーが最小となる場合に、移動性材料の表面基質が提示された。延伸乾燥期間の間の相分散と親水性/疎水性表面交換の両方を考慮に入れると、非晶質二次構造は、混合繊維立体構造を作り出す界面の繊維の間に散在するようになる。また、繊維間の線形二次構造が、整列して、熱力学的に安定した結晶性βストランドを形成することも考えられる。
【0130】
(表7)dH2Oに24時間浸漬した直径2.9cmの非拘束乾燥S87〜S57サンプルについての平均吸収およびEWC測定値(±SD、n=6)
【0131】
図14Eの拘束乾燥したS87〜S76材料群の断面図は、低下する絹濃度に対して増加する繊維密度および凝集を開示する。S87繊維は、多数の大きな空間の隙間を有する水平のシートに凝集した。S82およびS76マットは、繊維束の増加および空間の隙間の漸進的減少を実証した。これらの所見は、絹体積に対するマット厚さの減少に一致する。図14BのS87〜S76顕微鏡写真は、これらの立体構造の全体にわたって相互に連絡する多孔性を示す。細孔のど径表面積は、それぞれ平均294nm2、201nm2、および103nm2である。下降する細孔径分布は、断面図に提示される繊維密度の増加に一致し、また、各々のS87、S82、およびS76材料群のビーディング(beading)および繊維直径に関する繊維の集合にも関連する(図9)。
【0132】
材料の表面粗さは、細胞の配向、接着、増殖、および遊走を制御する、正味の力の生体力学的平衡を促進する、応力/剪断のない平面による細胞接触誘導に影響を及ぼす。図15のS87三次元AFM画像に代表されるように、6つのS87〜S57非拘束乾燥サンプルは全て、二乗平均平方根粗さ値が500nmから1.4μmまでの範囲に及ぶ、明確な表面不規則性を提示するクラス3の表面トポグラフィーを実証した(n=3)。S87、S82、S76、S67、およびS57群は、それぞれ1.17±0.00μm、0.65±0.10μm、0.88±0.17μm、0.76±0.16μm、および0.78±0.01μmを測定する、ナノサイズの不規則性をもつ相対的に均一な粗さを有した。S61マットが、局地的な粗さが平均1.01±0.43μmの最も大きい偏差を有した。拘束乾燥したS87、S82、およびS76サンプルについての16×16μm2の面積に関するAFM粗さ値は、それぞれ0.66μm、0.36μm、および0.25μmとなった。低下した面積サイズおよび制限されたサンプルサイズを有しているが(n=1)、拘束乾燥した材料の粗さ値は、非拘束乾燥サンプルよりも少なくとも44%平坦である。拘束乾燥したサンプルは、絹濃度に対して粗さが直線的に低下するが、非拘束乾燥サンプルについては顕著な傾向はない。この所見は、非拘束乾燥サンプルのねじれた不規則さと比較して、拘束乾燥の繊維の伸張特性に一致する。
【0133】
吸収
創傷被覆材の吸収およびEWC特性は、細菌の栄養床を作り出す創傷浸出液の蓄積を制御する際に寄与する。表7を参照すると、吸収は、461%〜613%の範囲に及び、全ての絹材料群は平均して551±54%であった。非拘束乾燥したS87〜S57サンプルの平均乾燥重量は、22.5mgから13.6mgまで直線的に低下したが、EWCは84±1%で比較的一定のままであった。データは、吸水が各々の絹濃度の繊維直径、密度、および二次構造集合特性とは無関係であることを示唆する。同等な吸収およびEWC率は、主にこれらのマトリックスの全体にわたって相互に連絡する細孔の網状構造、および電界紡糸した絹繊維の多孔性に起因すると思われる。Wang et al.,2004。水分子がこれらの不織構造の格子間領域の中に、かつ、生体高分子のバルク領域への中に可塑化するときに、比例する量の膨潤が全ての材料群で起こる。
【0134】
酸素透過速度
酸素/二酸化炭素ガス交換を促進する包帯材は、創傷の酸度を低下させ、嫌気性細菌感染を抑制し、従って創傷治癒を促進する環境を作り出すと思われる。水和条件下(80% RH)で評価したS87〜S57サンプルは、それぞれ、25000cm-3・m-2・d-1から7800cm-3・m-2・d-1までの平均OTRを提示した(表8)。絹濃度の低下に対して(Respective to)、OTRの線形の減少は、各々の絹材料群の減少するマット厚さ、繊維サイズ、細孔のど径、および繊維密度の増加に起因した。S87、S82、およびS76材料群についてのOTR線形回帰分析は、96%の予測二乗相関係数分散および4800cm cm-3・m-2・d-1の低下するOTR率を有した。対照的に、乾燥条件(50%RH)下で試験した水和したS87〜S57サンプルは、1回の試験間隔の終了よりも前に100000cm-3・m-2・d-1の分析器閾値を上回った。これらの結果は、乾燥またはほぼ乾燥状態ではこれらの多孔質材料は、水和した不織織物の組織間隙およびバルク領域に存在する水分子を通じて酸素が拡散される場合よりも大きい酸素透過速度を有することを反映する。
【0135】
(表8)8001 OPA(Illinois Instruments,ASTM 3985-05)およびPerm Cup(Gardner Company,ASTM D1653)で測定したS87〜S57材料群についての平均O2ガス透過速度(GTR)、WVTRおよび厚さ(±SD、n=3)
a)拘束乾燥したS87〜S76絹材料;b)水和したS87〜S76絹材料。
【0136】
水蒸気透過速度
全層創傷被覆材の水蒸気透過性は、体液の蒸発を制御し、創傷部位の感染を抑制する際に役割を果たす。S67、S61、およびS57マトリックスについてのWVTRを確かめる努力は、乾燥段階中の材料変形のために不成功に終わった。水和し拘束乾燥したS87、S82、およびS76材料群についてのWVTRは、それぞれ、平均1977±35g・m-2・d-1および1469±81g・m-2・d-1であった(表8)。液体/ガス/膜/ガス界面と比べて直接の液体/膜/ガス界面の界面エネルギー最小化の増加に起因して、飽和したサンプルが乾燥したサンプルよりも優れていた。これは、蒸発の加速を促進する生体材料表面/ガス界面に水分子が都合よく存在することを可能にする、これらの材料系の親水性の特性に起因しうる。水和し乾燥したS87〜S76材料群についてのWVTR回帰分析は、97%および99%の二乗相関係数分散での線形当てはめ、および81g・m-2・d-1および41g・m-2・d-1の下降するWVTRを予測した。これらのごく僅かなWVTRの差は、材料の厚さ、繊維サイズ、繊維密度、および多孔性に起因しうる。
【0137】
生分解性
時間放出生物療法での上皮化を促進するための、これらの絹材料の酵素生分解を評価した。インビトロでの生分解性研究は、全てのS87〜S57材料群について1日後に平均22.6±3.4%の分解および14日後に74.0±8.8%までの材料損失の、線形の分解傾向を明らかにした(図16A)。データは、6日までは、全てのブレンドについての分解率は48.2±4.6%で比較的近かったことを示唆する。対照的に、10日後に、S87(78%)とS57(51%)サンプルの間で27%の重量減少差が記録された。14日後、酵素分解は、S87〜S57サンプルに関して、それぞれ85.6±3.8から62.5±5.2%に至る範囲に及んだ。目視検査によって、全ての材料系が最初の6日で表面浸食により形態学的に分解された。10日後、S87およびS82マトリックスは、材料のほころび、断片化、および微粒子破片への崩壊をもたらす、繊維の切断の増加を実証した。S87およびS82サンプルの形態的分解は、これらのマトリックスの増加した繊維サイズ、マット多孔性、および繊維密度特性の低下により、内部マット構造に酵素が接近することに起因した。
【0138】
図16Bの散布図を参照すると、各々の材料群に関して実行した対数変換は、3日の分解時点の直前に全ての材料群につての明確な転移点を明らかにした。次に、回帰分析を各々の材料群について全ての時点、0日から3日まで、および3日〜14日の間の時点に関して行った。図16Cに示されるように、全ての材料群についての分解の傾きは、3日後に実質的に変わった。0日から3日まで、酵素分解速度は平均-11.7±0.9であった。3日後から14日まで、分解の速度は-3.4±1.1で安定した。初期の加速した分解速度は、潜在性の結晶領域と比較して表面基質の非晶質領域の分解に起因しうる。
【0139】
全層熱傷被覆材のための材料は、病原性微生物に対する不透過性の障壁をもたらし、創傷部位の浮腫および脱水症を管理し、および同時刻に合わせた抗生物質、免疫学的、および組織再生生物治療を支援する能力を含む、多くの有用な特性を提示することができる。さまざまな電界紡糸した絹/PEOが設計され、これらのPEO抽出絹材料系の立体構造特性および生体機能特性が、全層創傷被覆材としての有用性について研究された。拘束乾燥技法を用いることにより、絹濃度が、材料の厚さ、繊維密度、繊維配向、相分散、および多孔性を含む材料構造特性において役割を果たすことが見出された。この乾燥技法によって、S87、S82、およびS76の絹割合の材料群が、最小限の表面積減少で平坦な柔軟な膜のような立体構造に変化した、それは持続可能な有効期間をもつ流通可能な創傷被覆材に理想的である。
【0140】
安定した恒常状態を維持するために、正常な皮膚は体液を204g・m-2・d-1の速度で透過させる。全層肉芽創の蒸発水損失は5138g・m-2・d-1である。2000〜2500g・m-2・d-1の水透過速度を有する全層創傷被覆材が、適切な水分レベルを許容すると同時に過剰な脱水を防ぐことが決定された。表9の吸収、EWCおよび水蒸気透過特性を参照すると、拘束乾燥したS87〜S76材料は、提案されたスポンジのような天然キトサン創傷被覆材に対して比較的機能した。キトサン/ポロキサマー包帯材候補は、強い吸収およびEWC特性を提示したが、これらの試験は37℃のPBS(pH=7.4)で実施された。その上、S87〜S76材料系と、不斉および二層キトサン材料との間の酸素透過性の相違は、試験条件に起因するものであった。キトサン誘導体のOTRは、0%RHの乾燥条件下で試験されたが、dH2O飽和絹材料は、エキソバサーティングな(exovasating)創傷環境を模倣するために80%RHの水和環境下で評価された。
【0141】
(表9)キトサン誘導体に基づく創傷被覆材とS87〜S76材料系との間の平均吸収、EWC、WVTR、O2 GTR、および厚さの比較
a)酸素透過分析は35℃および0%RHで行った;
b)吸収およびEWCはPBS(pH=7.4)で測定した。
【0142】
正常なヒト皮膚は約21日で再生する。時間放出生物療法を送達する多層創傷被覆材を用いることによって全層創傷の上皮化を促進するために、これらの絹材料の酵素分解時間を評価した。結果は、14日後、S87〜S76マトリックスは80%分解したことを明らかにし、これは同じ時間の間に82%分解したリゾチームに曝露したキトサン/ポロキサマー包帯材と遜色がなかった。対照的に、PLGA/PLLA(90/10)コ-ブロックポリマー系はPBS中14日後に20%の分解率しかなかった。酵素レベルは著しく変動するので、インビボでの分解率も考慮されるべきである。絹生体材料は、材料形式、位置、および関連する変数によって、インビボで数週から数年で分解し得ることが示された。
【図1A】
【図1B】
【図2A】
【図2B】
【図3A】
【図3B】
【技術分野】
【0001】
関連出願の相互参照
本出願は、2009年7月14日出願の米国特許仮出願第61/225,335号の優先権の恩典を主張するものであり、その内容はその全文が参照により本明細書に組み入れられる。
【0002】
政府支援
本発明は、米国国立衛生研究所(National Institutes of Health)(組織工学資源センター(Tissue Engineering Resource Center))により与えられた助成金番号P41 EB002520の下での財政的支援によってなされたものである。米国政府は本発明において一定の権利を有する。
【0003】
発明の分野
本発明は、創傷治癒などの生物医学的用途に適した、絹/ポリエチレンオキシドブレンド材料を調製するためのプロセス、およびその得られる材料に関する。
【背景技術】
【0004】
発明の背景
創傷治癒、または創傷修復は、真皮および上皮組織を再生する身体の自然のプロセスである。創傷治癒のプロセスは複雑で脆弱である。これらの中でも、全層熱傷の治療は、医学において最も取り組みがいのある仕事の一つであり続けている。大きな割合の体表面積(BSA)にわたって全層の損傷を受けている患者は、多くの場合焼痂から合併症を起こし、それは全身の細菌感染、血液量減少、低体温、低灌流、および、横紋筋融解および溶血に起因するヘモグロビン尿症を引き起こす可能性がある。現在、全層熱傷は、一般に自家皮膚移植により最小限の瘢痕形成で治癒する。しかし、自家皮膚移植には制限がある:20%を上回るBSAに全層熱傷を負っている患者は、死体由来の一時的に引き伸ばしたメッシュ状同種移植片か、またはブタ異種移植片およびコラーゲンでコーティングされた半透性合成膜などの人工の真皮再生鋳型に制限される。患者に免疫学的に適合しないこととともに、これらの代用品は、広く不規則なコラーゲンバンドの急性分布によって治癒を誘発し、不均等な格子様の表面および過度の過形成性肥大性瘢痕をもたらす。
【0005】
様々な合成および天然ポリマーを用いて創傷被覆材料(wound dressing materials)を開発することができ、それは例えばポリ(グリコール酸)(PGA)、ポリ(L-乳酸)(PLA)などの加水分解に不安定な合成脂肪族ポリエステルまたはキトサンなどの天然由来ポリマーである。しかし、これらのポリマーは、特定の創傷環境にさらされた場合に、副反応または性能の低下を被る可能性がある。例えば、PGAまたはPLAポリマーの加水分解副生成物の酸度は、全層創傷治癒カスケードを阻害する可能性がある;酸性の創傷環境に浸漬した場合、キトサンはアミン基のプロトン化に起因して可溶性となり、それは機械的完全性の早期喪失をもたらし得る。
【0006】
それ故に、改良された生分解性、生体適合性を有し、天然の皮膚の創傷治癒特性を保有するだけでなく、改良された物理的および機械的特性、ならびに効果的な創傷被覆材に適した満足できる柔軟性も有する、新規な種類の生体材料が必要とされている。
【発明の概要】
【0007】
本発明は、絹ブレンドマットの製造のためのプロセスを提供する。本プロセスは、ポリエチレンオキシド(PEO)と絹フィブロイン水溶液をブレンドする段階;ブレンドした溶液を電界紡糸し、それにより絹タンパク質/PEOブレンドマットを形成する段階;および電界紡糸した絹マットを拘束乾燥する段階を含む。結晶化皿の技法(a crystallization dish technique)を拘束乾燥段階に用いてよい。本プロセスは、乾燥段階の前または後にアルコールおよび/または水溶液中で電子紡糸絹マットを処理する段階をさらに含むことができる。アルコールは、メタノール、エタノール、イソプロピルアルコール(2-プロパノール)またはn-ブタノールであってよい。本プロセスは、PEOを絹マットから抽出する段階をさらに含むことができる。PEOは、水中に浸出することにより絹マットから抽出することができる。その上、本プロセスは、少なくとも一つの活性物質、例えば治療剤または生物材料などを絹マットに埋め込む段階をさらに含むことができる。
【0008】
本発明はまた、ポリエチレンオキシド(PEO)と絹フィブロイン水溶液をブレンドする段階;ブレンドした溶液を電界紡糸し、それにより絹タンパク質/PEOブレンドマットを形成する段階;および電界紡糸した絹マットを拘束乾燥する段階、を含むプロセスにより調製した絹材料も提供する。
【0009】
本発明の一部の態様は、ポリエチレンオキシド(PEO)と、少なくとも一つの活性物質を含む絹フィブロイン水溶液をブレンドする段階;ブレンドした溶液を電界紡糸し、それにより活性物質を封入する絹タンパク質/PEOブレンドマットを形成する段階;および電界紡糸した絹マットを拘束乾燥する段階を含むプロセスにより調製した、創傷治癒を促進するために創傷を手当てするための、少なくとも一つの活性物質を埋め込むかまたは封入する絹材料に関する。あるいは、活性物質は、PEOとブレンドした後に絹フィブロインに添加されるか、または電界紡糸した絹材料に添加されてよい、例えば、電界紡糸した絹/PEOマットを活性物質でコーティングしてよい。
【0010】
本発明はまた、少なくとも絹フィブロインタンパク質を含む電界紡糸絹マットに関し、ここで、絹マット中の絹フィブロインタンパク質の含量は、約50重量%〜約90重量%の範囲に及び、絹マットの厚さは約20〜80μmである。
【0011】
本発明はまた、絹フィブロインタンパク質およびポリエチレンオキシド(PEO)を含む電界紡糸絹マットに関する。電界紡糸絹マットは、2:1〜4:1の絹フィブロインタンパク質/PEOブレンド比を有するか、または絹の割合は約75%w/w〜90%(w/w)であり;かつ、絹マットの厚さは約20〜約80μmである。
【0012】
一態様では、電子紡糸絹マットは、約20〜30μmほどの細さである。
【0013】
一態様では、電子紡糸絹マットは、平均して約0.1〜約1μmの細孔のど径(pore throat size)表面積をもつ相互接続した細孔を有する。
【0014】
本発明のプロセスにより調製される電界紡糸絹マットは、生物医学的用途、特に創傷被覆材に適した、良好な構造特性、形態特性、生体機能特性および生体適合特性を提示する。例えば、得られる本発明の絹マットは、14日未満で約86%を超える重量を分解する;本発明の絹マットの平衡含水率は、約82%より大きく;絹マットの酸素透過速度は、約15460cm3/m2/日より大きく;かつ、絹マットの水蒸気透過速度は、約1934g/m2/日より大きい。
【0015】
本発明の一部の態様は、絹フィブロインタンパク質および任意で少なくとも一つの活性物質を含む、少なくとも一つの拘束乾燥した電界紡糸絹マットに創傷を接触させることを含む、創傷治癒を促進する方法にも関する。電子紡糸絹マットの絹フィブロイン含量は、約50重量%〜約90重量%の範囲に及び;かつ、絹マットの厚さは、約20〜約80μmである。
【0016】
本発明の一部の態様は、絹フィブロインタンパク質、PEO、および任意で少なくとも一つの活性物質を含む、少なくとも一つの拘束乾燥した電界紡糸絹マットに創傷を接触させることを含む、創傷治癒を促進する方法にも関する。電子紡糸絹マットは、約2:1〜約4:1の絹フィブロイン/PEOブレンド比を有し(または電界紡糸絹マット中の絹フィブロインの割合は、約75%w/w〜90%w/wであるか、または電界紡糸絹マット中のPEOの割合は、約10%w/w〜約25%w/wである);かつ、絹マットの厚さは、約20〜約80μmである。
【図面の簡単な説明】
【0017】
【図1】図1Aは、絹/PEO比が4:1、3:1、2:1、3:2、7:6、および1:1の6種類の、それぞれ、各材料群に対して86.5%、82.8%、76%、70.6%、65.1%および61.5%w/wの絹フィブロインタンパク質率に相当する電界紡糸絹マットを示す図である。直径10cmの絹マットを、メタノールで処理し、水に浸漬した。図1Bは、dH2Oに浸漬した直径10cmのS87〜S57絹マットを示す図である。各々のマットは、柔軟な軟質の絹のようなテクスチャーをもつ均一な立体構造を提示した。白色の斑点および襞は、材料中の気泡および折り目を反映する。
【図2】直径3.5cmの風乾した(または拘束乾燥していない)絹サンプルを示す図である。これらの画像は、絹濃度の低下に対して漸進的な材料の変形を反映する。
【図3】直径13cmの拘束乾燥した絹マット(3A)および直径12.5cmの拘束乾燥したS87〜S57絹マット(3B)を示す図である。これらの画像は、絹濃度の低下に対して増加する材料の剪断および変形を明らかにする。
【図4】風乾および拘束乾燥技法後の、小型および大型材料群サンプルについての絹表面積の変化の割合を表すグラフである。表面積は、円形形状の査定によって決定した。線形曲線分析を、全ての材料群で平均表面積減少率に関して行った(SD±%、n=3)。乾燥技法間での表面積減少は、4:1と3:1の材料群で有意に異なった(P=0.001)。傾き(m)およびR2値は、風乾したサンプルの絹濃度の低下に伴う表面積の線形の漸進的な減少を示す。対照的に、拘束乾燥した材料群についての表面積減少は、4:1および3:1マットについての2%から、3:2、7:6および1:1材料群についての60%超まで30倍増大した。
【図5】絹材料群と、(1)電界紡糸マットのサイズ、および(2)風乾および引き伸ばし乾燥技法後に基づく膜の厚さとの間の関係を表すグラフである。材料の厚さは、Ono Sokki EG-225F Digital Indicatorを利用して決定した。線形曲線分析を、全ての材料群で平均材料厚さに関して行った(SD±%、n=6)。全ての10cm風乾材料群は、16.5風乾群および12.5拘束乾燥群よりも有意に厚かった(P=0.001)。絹濃度に対して、傾き(m)およびR2値は、10cm風乾サンプルについて線形の厚さ減少を示す。16.5cm風乾群および12.5拘束乾燥群の厚さの傾きは、絹濃度に対して僅かな相違を伴って水平の痕跡に近づいた。
【図6】6.5倍の倍率の、メタノール処理の前の絹材料群のFE SEM画像を示す図である。PEO濃度の増加とともに、S87P13〜S76P24絹マットの上の繊維ビーディングは減少した。均一なよく分散したS67P33〜S61P39繊維の、不規則な形状の混合繊維への移行が、密集したS57P43構造に示される。
【図7】絹/PEO比がそれぞれ4:1、3:1および2:1の、メタノール処理した絹マットのSEM画像を示す図である。図7Aは、1.5倍の倍率の、3つ全てのマットの広範な図を表す。3:1および2:1マットの画像中の等高線は、方向性の繊維伸張および整列を反映する。図7Bは、1.5倍の倍率での3つ全ての絹マットのクローズアップ写真を示す。3:1および2:1の画像中の矢印は、繊維の凝集および整列を強調する。図7Cは、12倍の倍率の3:1および2:1絹マットを表す。3:1画像中の円は、整列した繊維間の相分散の証拠を露呈する。矢印は、2:1画像中でピンと張った伸張された繊維の詳細な輪郭を際立たせる。図7Dは、2:1絹マットの50倍画像において丸く囲まれた領域が、混合繊維を明らかにすることを示す。図7Eは、3つ全ての絹モデルについての2.5倍の倍率の断面図を表す。画像は繊維密度と絹濃度の逆相関を反映する。
【図8】50×50μmの領域一面の、4:1、3:1、および2:1拘束乾燥マット(8A)またはS87〜76マット(8B)についての細孔のど径分布の柱状グラフを示す図である。減少する絹濃度に対して、細孔の数は、のど径の低下による細孔の集積の増加で139から226に増大した。
【図9】結晶化皿乾燥法を利用する漸進的なポリマー鎖および繊維の立体構造を説明する概念図である。上部は、分散した未整列の二次構造の、親水性環境から、疎水性相互作用により駆動される整列したタンパク質凝集体への移行。繊維が延伸されるときに、β-シートは主に鎖間形成によって集合し、半径応力の方向に伸張する。図の下の繊維形成は、絹濃度および繊維整列および伸張の間の逆相関を反映する。
【図10】メタノール処理後の絹材料群についての50×50μm 3D AFM画像および粗さ値を実証する図である。これらの画像は、Ultrasharp NSC16/AIBSプローブを非接触モードで用いて得た(共振周波数:170kHz、バネ定数:45N/m)。
【図11】さまざまな環境条件下での4:1、3:1および2:1絹材料系についての酸素および水蒸気透過性能を表す図である。線形曲線の当てはめ分析を、全ての材料群での平均OTRおよびWVTR測定値に実行した(SD±%、n=3)。傾き(m)およびR2値は、全ての材料群でOTRおよびWVTR性能の線形の減少を示す。OTR R2値は、材料群でのOTR測定値の最低限の相違を開示するが、各々の群の中の標準偏差は、それぞれ、±14.6、11.2、および16.9パーセントの範囲に及んだ。
【図12】1、3、6、10、および14日の時点に関する絹材料群のインビトロ酵素生分解分析を表すグラフである。三層の25±5mg円形3.5cmサンプルを、pH7.4の1mg/mLプロテアーゼXIVのPBS溶液6mL中、37℃でインキュベートした。対照サンプルを、酵素を含まないPBSに浸漬した。酵素および対照溶液は、毎日補充した。図12Aは、全ての材料群で平均生分解材料損失について行った線形の当てはめ分析を説明する(SD±%、n=3)。図12Bは、各々の絹材料群の全てのサンプルに関する質量損失率の対数変換の散布図表示を表す図である。変換により、全ての絹材料系について3日の時点の直前に分解転移点が示される。図12Cは、各々の絹群の生分解率分析を全ての時点、0日〜3日および3日〜14日の時点について説明する図である。データは、2つの異なる分解傾向を示す。3日までは、全ての絹群は、-10〜-12の間の加速した酵素分解率を有する(単位=経時的質量損失)。3日後、酵素分解の傾きは、全ての絹材料について-3〜-5に著しく低下する。
【図13】図13Aは、非拘束および拘束乾燥S87〜S57絹マットについて絹表面積の変化の割合を表すグラフである。乾燥技法間の表面積の減少は、S87およびS82絹マットについて有意に異なった(P=0.001)(バー=標準偏差、n=3)。図13Bは、S87〜S57サンプルについて非拘束および拘束乾燥技法間の材料厚さの関係を表すグラフである。S87〜S61非拘束乾燥絹マットは、拘束乾燥群よりも有意に厚かった(P=0.001)(SD±%、n=6)。
【図14】それぞれ、拘束乾燥S87、S82、およびS76絹マットのFE-SEM顕微鏡写真を示す図である。図14Aおよび14Bは、1.5倍の倍率で見たS87〜S76マットを示す。画像は、進展する繊維の伸張、凝集および整列を開示する。図14Cは、整列したS82繊維と明確な伸張されたS76繊維との間の相分散をあらわにする12倍の倍率の画像である。図14Dは、混合され絡まったS76繊維の構造を示す50倍の倍率の画像を表す。図14Eは、2.5倍の倍率のS87〜S76マットの断面図を示す。画像は、繊維密度と絹濃度との間の逆相間を反映する。
【図15】図15Aは、全てのS87〜S57非拘束乾燥絹マットの明確な表面の不規則性を表す、S87絹マットの三次元AFM画像である。図15Bは、非拘束乾燥S87〜S57絹マットについて最低高さ分布(valley height distribution)の痕跡と同様のz面ピークを開示する柱状グラフを示すグラフである。
【図16】1、3、6、10、および14日の時点に関してS87〜S57絹マットのインビトロ酵素生分解分析を表すグラフである。三層の25±5mg 3.5cmサンプルを、pH7.4の1mg・mL-1プロテアーゼXIVのPBS溶液6mL中、37℃でインキュベートした。対照サンプルを、酵素を含まないPBSに浸漬した。酵素および対照溶液は、毎日補充した。図16Aは、各々の時点に関するS87〜S57生分解材料群の線形当てはめ表示平均率(the averaged percent linear fit representation)を説明する図である。(SD±%、n=3)。図16Bは、全ての時点に関して全ての生分解サンプルの対数変換を表す図である。変換により、全てのS87〜S75材料系について3日の時点の直前に分解転移点が示される。図16Cは、各々の絹群について、全ての時点、開始から3日、および3日から14日の生分解率分析を説明する図である。データは、2つの異なる分解傾向を示す。3日までは、全ての絹群は、-10〜-12の間の加速した酵素分解率を有する(単位=経時的質量損失)。3日後、酵素分解率は、全ての絹材料について-3〜-5に著しく低下する。
【発明を実施するための形態】
【0018】
発明の詳細な説明
本発明が、本明細書に記載される特定の方法論、プロトコール、および試薬などに制限されないこと、およびそのようなものが変動しうることは当然理解される。本明細書において使用される用語法は、特定の態様を説明する目的だけのものであって、特許請求の範囲によってのみ定義される本発明の範囲を制限するものではない。
【0019】
本明細書および特許請求の範囲において、単数形には、文脈上明らかに示されている場合を除き複数への言及が含まれ、逆の場合も同様である。運用例以外では、または別に指定されていない限り、本明細書において使用される構成成分の量または反応条件を表す全ての数値は、全ての場合において用語「約」によって変更されると理解されるべきである。
【0020】
特定される全ての特許およびその他の刊行物は、例えば、本発明に関連して使用されうるかかる刊行物に記載される方法論を説明および開示する目的で、参照により本明細書に明示的に組み入れられる。これらの刊行物は、単にその開示が本出願の出願日よりも前であるために提供される。この件について、先行発明という理由でまたは任意のその他の理由で、発明者らがかかる開示に先行する権利のないことを承認すると解釈されるべきではない。これらの文書の内容に関する日付または表現に関する全ての記述は、出願者らに利用可能な情報に基づくものであり、これらの文書の日付または内容の正確さに関していかなる承認も成すものでない。
【0021】
別に定義されない限り、本明細書において使用される全ての技術用語および科学用語は、本発明が関係する当業者に一般に理解される意味と同じ意味を有する。あらゆる公知の方法、装置、および材料を本発明の実践または実験に用いてよいが、この件について、方法、装置、および材料は、本明細書に記載されている。
【0022】
本発明は、創傷治癒などの生物医学的用途に適した、絹/ポリエチレンオキシドブレンド材料を調製するプロセス、およびその得られる材料に関する。特に、絹フィブロイン/PEOブレンド比が2:1〜4:1であり、制御蒸発および拘束乾燥技法によって乾燥させた、電界紡糸した絹フィブロイン/PEOマットは、創傷被覆材に有用性をもつ生体材料系に関する、適した物理的特性および生体機能特性、例えば繊維構造、トポグラフィー、多孔性、吸収性、水蒸気透過速度、酸素透過、および生分解性などを実証した。
【0023】
全層熱傷の治療は、医学において最も取り組みがいのある仕事の一つであり続けている。毎年、米国において、3000人が死亡しており、100万人を超える患者が、I度の上皮損傷からIII度の全層皮膚創傷の範囲に及ぶ、熱、放射線、化学物質および電気源から受けた熱傷を治療している。Beers et al.,MERCK MANUAL DIAGNOSIS&THER.(Merck&Co.,Inc.,Boston,MA,2006)。大きな割合のBSAにわたって全層損傷を受けた患者は、多くの場合焼痂から合併症を起こし、それは全身の細菌感染、血液量減少、低体温、低灌流、ならびに、横紋筋融解および溶血に起因するヘモグロビン尿症を誘発する。Ratner et al.,BIOMATS.Sci.INTRO.MATS.MED.(Acad.Press,NY,2004);Beers et al.,2006;Malafaya et al.,59 Adv.Drug Deliv.Rev.207-33(2007)。即時の処置を行わないと、全層熱傷は、血液量減少性ショック、免疫抑制、および、細菌性敗血症を引き起こし得、全身性炎症反応症状群(SIRS)、臓器不全、および死をもたらし得る。
【0024】
現在、全層熱傷は、一般に自家皮膚移植により最小限の瘢痕形成で治癒する。皮膚は、優れた曲げ強度をもつ耐久性のある生体材料複合材料であり、有害な細菌に対する物理的障壁を提供し、バランスのとれた静的血流および血栓性の創傷治癒カスケードを促進する重要な血液-表面の界面をもたらす。皮膚の優れた吸着、ガスおよび水蒸気の透過性により、浮腫および脱水症を抑制すると同時にタンパク質性浸出液のドレナージが可能となり、従って体温調節、細胞浸潤および軟組織再生を促進する。Kim et al.,341 Int.J.Pharm.35-43(2007);Lee et al.,11 J.Mater.Sci.-Mater.Med.817-23(2000)。永久的分層自家皮膚移植片を直ちに適用すると、72時間後に新血管新生を開始し、多くの場合、関節拘縮、虚血、瘢痕または全身毒性の合併症なく完全な真皮再構成をもたらす。Ratner et al.,2004;Beers et al.,2006。
【0025】
しかし、自家皮膚移植には制限がある。20%BSAを上回る全層熱傷を負っている患者は、一般に死体由来の一時的に引き伸ばしたメッシュ状同種移植片か、またはブタ異種移植片およびコラーゲンでコーティングされた半透性合成膜などの人工の真皮再生鋳型で処理される。患者に免疫学的に適合しないこととともに、これらの代用品は、多くの場合、広く不規則なコラーゲンバンドの急性分布によって治癒を誘発し、不均等な格子様の表面および過度の過形成性肥大性瘢痕をもたらす。Queen et al.,8 Biomats.367-71(1987);Ratner et al.,2004;Beers et al.,2006。そのために、天然の皮膚の創傷治癒特性を保有するだけでなく、十分に生分解性である、効果的な創傷被覆材を開発することが必要である。
【0026】
良好な生分解性、生体適合性および機械的特性を有するさまざまな合成および天然ポリマーを用いて創傷被覆材料を開発することができる。加水分解に不安定な合成脂肪族ポリエステル、例えばポリ(グリコール酸)(PGA)、ポリ(L-乳酸)(PLA)、およびポリ(乳酸-コ-グリコール酸)(PLGA)は、手術による移植、骨セメント、吸収性縫合、およびミクロスフェア制御放出系を含む、多くの医学的適用に用いられる。Quynh et al.,43 Eur.Polym.J.1779-85(2007);Wu&Wu 91 Polym.Degrad.Stab.2198-204(2006)。多孔性の電界紡糸PLAおよびPGA繊維は現在創傷被覆材料として研究されているが、これらのポリマーの加水分解副生成物の酸度は、全層創傷治癒カスケードを抑制しうる。Quynh et al.,2007;Wu&Wu,2006。グルコサミンおよびN-アセチルグルコサミンから構成され、ヘパリンに似た二糖の天然由来ポリマーキトサンは、血栓性カスケードを促進し、多形核(PMN)および単核細胞の創傷部位への細胞移動を刺激することにより創傷治癒を加速させることが見出された。Kim et al.,341 Int.J.Pharm.35-43(2007);Malafaya et al.,2007。しかし、酸性の創傷環境に浸漬すると、キトサンはアミン基のプロトン化に起因して可溶性となり、それは機械的完全性の早期喪失をもたらし得る。それ故に、柔軟性をもつフィルム、ゲルまたはスポンジを形成するためのその他の合成ポリマーの共重合は、全層創傷被覆材に必要である。Kim et al.,2007;Malafaya et al.,2007。
【0027】
本発明は、フィルム、繊維、ゲル、および多孔性のスポンジを含む、広い範囲の材料形態にわたって明確な生物学的特性を有する天然のフィブロイン絹を提供する。Vepari&Kaplan 32 Prog.Polym.Sci.991-1007(2007)。カイコおよびクモにより製造された絹フィブロインは、グリシンおよびアラニンから主に成る生体高分子に基づくタンパク質である。Vepari&Kaplan,2007;Zhou et al.12 Nucleic Acids Res.2413-19(2000);Tanaka et al.,29 Insect Biochem.MoI.Biol.269-76(1999)。構造的に、絹フィブロイン生体高分子はアミノ酸の繰り返し配列を含有し、それは結晶化する重鎖、および低結晶性の軽鎖を形成する。フィブロインの両親媒性領域の相互作用は、その他の二次構造とともに有意な含量の結晶性β-シート(およそ55%)を生じて、この特有の生体高分子の機械的および生体機能特性を作り出す。Vepari&Kaplan,2007;Wang et al.,39 Macromol.1102-07(2006);Wang et al.,37 Macromol.6856-64(2004);Jin et al.,15 Adv.Funct.Mater.1241-47(2005);Hu et al.,39 Macromol.6161-70(2006)。しっかりと詰め込まれた結晶性β-シートは水を排除するが、構築したタンパク質中の低結晶性ドメインは水素結合によって組織化されたままであり、含水量の変化に対応し得る。Wong et al.,82 Appl.Phys.A-Mater.293-303(2006)。広範な細胞および組織研究が、とりわけ骨、軟骨、靭帯および血管工学を含む絹タンパク質生体材料を用いて行われ、このタンパク質系の生体適合性かつ有効な組織再生の特色を実証した。Vepari&Kaplan 2007。創傷被覆材に関して、絹フィルムは、従来のブタに基づく創傷被覆材よりも速く、低い炎症応答で、ラットにおいて全層皮膚創傷を治癒することが示された。Sugihara et al.,225 Exp.Biol.Med.58-64(2000)。
【0028】
本発明の態様は、絹フィブロイン/ポリエチレンオキシド(PEO)のブレンドの二流体電界紡糸技法を利用して調製した創傷被覆材に対する潜在的有用性をもつ絹マトリックスを提供する。Wang et al.,39 Macromol.1102-07(2006)。電界紡糸法は、豊富な種類の機能性材料からナノファイバーの膜を作製するための単純で多用途かつ有用な技法である。Doshi&Reneker 35 J.Electro.151-60(1995);Reneker&Chun 7 Nanotech.216-23(1996);Fridrikh et al.,90 Phys.Rev.Let.144502-06(2003)。かなりの数の天然および合成材料が創傷被覆材を形成するために電界紡糸されているが、生体適合性、機械的特性、および総合的な機能性性能に関して課題が残っている。本発明では、絹フィブロインを標的プラットフォームに継続的に紡糸することにより、絹/PEO比濃度および用いた紡糸ドープの容積に相対的な厚さをもつ、層状の繊維シートで構築される大型の融合性(confluent)絹マットが製造される。絹マットはメタノールに浸漬され、β-シート結晶化に関連する物理的架橋を引き起こし、水安定化材料の形成を誘導する。異なる絹/PEOブレンドの特有の繊維多孔性および表面粗さを活用することで(Jin et al.,3 Biomacromol.1233-39(2002);Wang et al.,37 Macromol.6856-64(2004))、さまざまな絹/PEOブレンド比をもつ絹材料系が調製され、創傷治癒の必要性に照らして物理的なおよび生体機能特性について評価された。
【0029】
従って、本発明は、絹ブレンドマットの製造のためのプロセスを提供する。本プロセスは、ポリエチレンオキシド(PEO)と絹フィブロイン水溶液をブレンドする段階;ブレンドした溶液を電界紡糸し、それにより絹タンパク質/PEOブレンドマットを形成する段階;および電界紡糸絹マットを拘束乾燥する段階を含む。所望の口径をもつ結晶化皿またはポリスチレン容器を拘束乾燥段階に用いることができる。
【0030】
電界紡糸法は、当技術分野において公知のあらゆる手段により行うことができる(例えば、米国特許第6,110,590号参照)。例えば、内径1.0〜2.0mmの先端をもつ鋼毛管を、調節可能な電気絶縁スタンドの上に取り付ける。毛管は、一般に高い電位で維持され、平行なプレート配置で取り付けられる。毛管は、絹/生体適合性ポリマー溶液を満たしたシリンジに接続することができる。一定の体積流量は、通常、滴が漏れることなく管の先端に溶液を保持するように設定したシリンジポンプを用いて維持される。表1に示されるように、電位(10〜12kV)、溶液流速(0.014〜0.032mL/分)、および毛管の先端と収集スクリーンの間の作動距離(20〜22.5cm)を、安定した噴出が得られるように調節する。乾燥または湿潤繊維は、毛管の先端と収集スクリーンの間の距離を変えることにより収集される。
【0031】
(表1)各絹/PEOブレンドについての電界紡糸パラメータ。8重量%絹、5%PEO、および6%PEO溶液の粘度は、それぞれ約24、約4464、および約7520センチポイズ(mPa・s)であった。各ブレンドに対して4.5mL、8mL、および10.4mLの3種類のバッチ溶液を作成してそれぞれ直径10、13、および16.5cmの絹マットを作出した。
【0032】
絹繊維を収集するのに適した収集プレートまたは収集スクリーンは、金網またはポリマー網であってよい。あるいは、収集スクリーンはアルミニウム箔(直径10〜16.5cm)である。アルミニウム箔をTeflon流体でコーティングして絹繊維をより簡単に剥離するようにすることができる。繊維溶液が電場を移動するときにそれを収集するその他の手段を当業者は容易に選択することができる。下により詳細に記載されるように、毛管の先端とアルミニウム箔の対極との間の電位差は、約10〜12kVに徐々に増加する可能性があるが、当業者であれば電位を調節して適した噴出流を実現することができるはずである。
【0033】
次に、電界紡糸マットを拘束乾燥する。本発明のプロセスは、乾燥段階の前または後に電界紡糸絹マットをアルコール/水溶液中で処理してβ-シート形成および結晶化を誘導する段階をさらに含むことができる。アルコールは、メタノール、エタノール、イソプロピルアルコール(2-プロパノール)またはn-ブタノールであってよい。さらに、PEOを絹マットから抽出することができる。絹マットからのPEOの抽出は、電界紡糸した絹ブレンドマットを水(例えば、dH2O)中で一定期間、例えば1〜3日にわたって浸出させることにより行うことができる。
【0034】
本明細書において、用語「フィブロイン」には、カイコフィブロインおよび昆虫もしくはクモ絹タンパク質が含まれる。Lucas et al.,13 Adv.Protein Chem.107-242(1958)。例えば、フィブロインは、溶解したカイコ絹またはクモ絹を含有する溶液から得られる。カイコ絹タンパク質は、例えば、カイコ(Bombyx mori)から得られ、クモ絹は、アメリカジョロウグモ(Nephil clavipes)から得られる。しかし、使用することのできる絹は、数多くあり、それには、クモ絹(例えば、アメリカジョロウグモから得たもの)、トランスジェニック絹、遺伝子操作された絹、例えば細菌、酵母、哺乳類細胞、トランスジェニック動物、またはトランスジェニック植物由来の絹(例えば、国際公開公報第97/08315号;米国特許第5,245,012号参照)、ならびにそれらの変異体が含まれる。
【0035】
絹フィブロイン水溶液は、当技術分野において公知の技法を用いてカイコ繭から調製することができる。絹フィブロイン溶液を調製するために適したプロセスは、例えば、米国特許出願第11/247,358号;国際公開公報第2005/012606号;および国際公開公報第2008/127401号に開示されている。一態様では、カイコ(B.mori)繭を約30分間水溶液中で沸騰させる。この水溶液は、0.02M炭酸ナトリウムであってよい。繭を水ですすいでこれらのリシンタンパク質を抽出し、抽出した絹を塩類水溶液に溶解する。この目的に有用な塩としては、臭化リチウム、チオシアン酸リチウム、硝酸カルシウムまたは絹を可溶化する能力のあるその他の化学物質が挙げられるが、それに限定されるわけではない。例えば、抽出した絹を、60℃の約9〜12M LiBr溶液に4時間溶解し、20%(w/v)溶液を得てもよい。塩は結果的に透析を用いて除去される。このプロセスの間に形成されうる少量の絹凝集体、通常繭に存在する環境汚染物質由来のものを除去するために、この溶液を遠心してよい。絹フィブロイン水溶液の終濃度は、およそ8%(w/v)であってよい。より高い濃度の絹フィブロイン溶液を得るために、より低い濃度の絹フィブロイン溶液を吸湿性ポリマー、例えば、PEG、ポリエチレンオキシド、アミロースまたはセリシンに対して透析してよい。例えば、8%絹フィブロイン溶液は、10%(w/v)PEG(10,000g/mol)溶液に対して透析することができる。透析は、10〜30%の間の絹水溶液終濃度をもたらすのに十分な時間なされる。大抵の場合、2〜12時間の透析が十分である。
【0036】
絹フィブロイン溶液は、本明細書に記載されるように、1以上の生体適合性ポリマー、例えば、ポリエチレンオキシド、ポリエチレングリコール、コラーゲン、フィブロネクチン、ケラチン、ポリアスパラギン酸、ポリライシン、アルギン酸塩、キトサン、キチン、ヒアルロン酸、および同類のものなど;または1以上の活性物質、例えば細胞、酵素、タンパク質、核酸、抗体および同類のもの、と混合することができる。例えば、国際公開公報第2004/062697号および国際公開公報第2005/012606号を参照されたい。また、絹フィブロインは、絹タンパク質の物理的特性および官能性を変えるために、例えばジアゾニウムまたはカルボジイミドカップリング反応、アビジン-ビオチン(biodin)相互作用、または遺伝子組み換えおよび同類のものによって、溶液中の活性物質によって化学的に修飾することができる。例えば、PCT/US09/64673号;米国特許出願第61/227,254号;同第61/224,618号;同第12/192,588号を参照されたい。
【0037】
広範囲の水溶液中の絹フィブロインおよびPEO濃度が、絹材料を電界紡糸するためのブレンド溶液を調製するために適している。例えば、溶液中の絹フィブロインの濃度は、ブレンディングの前に約30重量%未満であってよく;溶液中のPEOの濃度は、PEO溶液の溶解度および粘度に応じて、ブレンディングの前に約1%〜約15重量%の範囲に及んでよい。例えば、約5重量%〜15重量%の絹フィブロイン濃度を有する水溶液および約3重量%〜10重量%のPEO濃度を有するPEO溶液を、ブレンディングに使用してよい。一態様では、8重量%の絹フィブロイン溶液および5重量%のPEO溶液がブレンディングに使用される。別の態様では、8重量%の絹フィブロイン溶液および6重量%のPEO溶液がブレンディングに使用される。絹フィブロイン溶液およびPEO溶液の初期濃度、ならびに絹フィブロインタンパク質とPEOの間の初期ブレンディング比は両方ともに、電界紡糸の間に安定した流体の噴出を生じるために望ましい粘弾特性および表面張力特性に依存しうる。Jin et al.,3 Biomacromol.s 1233-39(2002)。絹フィブロイン溶液およびPEO溶液の初期濃度ならびに絹フィブロインタンパク質とPEOの間の初期ブレンディング比は、最終の絹ブレンドマット中の絹フィブロインおよび/またはPEOの所望の重量百分率にも依存しうる。
【0038】
一態様では、本発明の絹生体材料は、少なくとも一つの治療剤を含有してよい。これらの材料を形成するため、マトリックスを形成するよりも前に絹フィブロインまたは絹フィブロイン/PEO溶液を治療剤と混合するか、それを形成した後に材料に添加する。本発明の生体材料と併せて使用することのできる豊富な種類のさまざまな治療剤は膨大である。
【0039】
通例、本発明の薬学的組成物によって投与されうる治療剤としては:抗生物質および抗ウイルス剤などの抗感染薬;化学療法剤(例えば、抗癌剤);拒絶反応抑制剤;鎮痛薬および鎮痛薬合剤;抗炎症剤;ステロイドなどのホルモン類;細胞接着メディエーター、例えば細胞接着に影響を及ぼすことが公知の「RGD」インテグリン結合配列のペプチド含有種、生物活性リガンド、および特定の種類の細胞または組織内殖を強化または排除する物質、例えば骨形態形成タンパク質(例えば、BMP1〜7)、骨形成様タンパク質(例えば、GFD-5、GFD-7、およびGFD-8)、上皮成長因子(EGF)、線維芽細胞成長因子(例えば、FGF1〜9)、血小板由来成長因子(PDGF)、インスリン様成長因子(IGF-IおよびIGF-II)、トランスフォーミング成長因子(例えば、TGF-βI〜III)、TGF-、YIGSRペプチド、グリコサミノグリカン(GAG)、ヒアルロン酸(HA)、インテグリン、セレクチンおよびカドヘリンなど;血管内皮成長因子(VEGF);ならびにその他の天然由来もしくは遺伝子操作されたタンパク質、多糖、糖タンパク質、またはリポタンパク質が挙げられるが、それに限定されるわけではない。成長因子は当技術分野において公知であり、例えば、Rosen&Thies,CELLULAR&MOL.BASIS BONE FORMATION&REPAIR(R.G.Landes Co.,2004)を参照されたい。
【0040】
活性物質は、絹材料に埋め込まれることの可能なあらゆる材料を表し得る。例えば、活性物質は、治療剤、または生物材料、例えば、細胞(幹細胞を含む)、タンパク質、ペプチド、核酸(例えば、DNA、RNA、siRNA)、核酸類似体、ヌクレオチド、オリゴヌクレオチド、ペプチド核酸(PNA)、アプタマー、抗体またはその断片もしくは部分(例えば、パラトープまたは相補性決定領域)、抗原またはエピトープ、ホルモン、ホルモンアンタゴニスト、成長因子または組換え成長因子およびその断片および変異体、細胞接着メディエーター(RGDなど)、サイトカイン、酵素、小分子、薬物、色素、アミノ酸、ビタミン、抗酸化剤、抗生物質または抗菌性化合物、抗炎症剤、抗真菌薬、ウイルス、抗ウイルス薬、毒素、プロドラッグ、化学療法剤、またはそれらの組合せなどであってよい。例えば、PCT/US09/44117号;米国特許出願第61/224,618号を参照されたい)。また、活性物質は、上記の物質のいずれかの組合せであってもよい。封入された生成物は多数の生物医学的目的に使用することができるので、治療剤または生物材料、あるいはそれらの組合せを封入することが望ましい。
【0041】
一部の態様では、活性物質はまた、真菌、植物、動物、細菌、またはウイルス(バクテリオファージを含む)などの生物であってもよい。さらに、活性物質には、神経伝達物質、ホルモン、細胞間シグナル伝達物質、薬学活性物質、毒剤、農薬、化学的毒素、生物学的毒素、微生物、ならびに動物細胞、例えばニューロン、肝細胞、および免疫系細胞が含まれてよい。また、活性物質には、治療用化合物、例えば薬理学的材料、ビタミン、鎮静薬、催眠薬、プロスタグランジンおよび放射性医薬品などが含まれてよい。
【0042】
本明細書での使用に適した例示的な細胞としては、前駆細胞または幹細胞、平滑筋細胞、骨格筋細胞、心筋細胞、上皮細胞、内皮細胞、尿路上皮細胞、線維芽細胞、筋芽細胞、口腔細胞(oscular cell)、軟骨細胞、軟骨芽細胞、骨芽細胞、破骨細胞、ケラチン生成細胞、腎尿細管細胞、腎基底膜細胞、外皮細胞、骨髄細胞、肝細胞、胆管細胞、膵島細胞、甲状腺細胞、上皮小体細胞、副腎細胞、視床下部細胞、下垂体細胞、卵巣細胞、精巣細胞、唾液腺細胞、脂肪細胞、および前駆細胞を挙げることができるが、それに限定されるわけではない。また、活性物質は、上記に列挙した細胞のいずれかの組合せであってもよい。国際公開公報第2008/106485号;PCT/US2009/059547号;国際公開公報第2007/103442号も参照されたい。
【0043】
絹フィブロインに組み込むことのできる例示的な抗体としては、アブシキマブ、アダリムマブ、アレムツズマブ、バシリキシマブ、ベバシズマブ、セツキシマブ、セルトリズマブ・ペゴル、ダクリズマブ、エクリズマブ、エファリズマブ、ゲムツズマブ、イブリツモマブ・ティウキセタン、インフリキシマブ、ムロモナブ-CD3、ナタリズマブ、オファツムマブ、オマリズマブ、パリビズマブ、パニツムマブ、ラニビズマブ、リツキシマブ、トシツモマブ、トラスツズマブ、アルツモマブペンテテート(altumomab pentetate)、アルシツモマブ、アトリズマブ、ベクツモマブ、ベリムマブ、ベシレソマブ、ビシロマブ、カナキヌマブ、カプロマブペンデチド、カツマキソマブ、デノスマブ、エドレコロマブ、エフングマブ(efungumab)、エルツマキソマブ(ertumaxomab)、エタラシズマブ、ファノレソマブ(fanolesomab)、フォントリズマブ、ゲムツズマブオゾガマイシン、ゴリムマブ、イゴボマブ(igovomab)、イミシロマブ(imciromab)、ラベツズマブ、メポリズマブ、モタビズマブ、ニモツズマブ(nimotuzumab)、ノフェツモマブメルペンタン(nofetumomab merpentan)、オレゴボマブ、ペムツモマブ(pemtumomab)、ペルツズマブ、ロベリズマブ(rovelizumab)、ルプリズマブ(ruplizumab)、スレソマブ(sulesomab)、タカツズマブテトラキセタン、テフィバズマブ、トシリズマブ、ウステキヌマブ、ビジリズマブ、ボツムマブ(votumumab)、ザルツムバブ、およびザノリムマブが挙げられるが、それに限定されるわけではない。また、活性物質は、上記に列挙した抗体のいずれかの組合せであってもよい。
【0044】
例示的な抗生剤としては、アクチノマイシン;アミノグリコシド(例えば、ネオマイシン、ゲンタマイシン、トブラマイシン);β-ラクタマーゼ阻害剤(例えば、クラブラン酸、スルバクタム);グリコペプチド(例えば、バンコマイシン、テイコプラニン、ポリミキシン);アンサマイシン;バシトラシン;カルバセフェム;カルバペネム;セファロスポリン(例えば、セファゾリン、セファクロル、セフジトレン、セフトビプロール、セフロキシム、セフォタキシム、セフェピム、セファドロキシル、セフォキシチン、セフプロジル、セフジニル);グラミシジン;イソニアジド;リネゾリド;マクロライド(例えば、エリスロマイシン、クラリスロマイシン、アジスロマイシン);ムピロシン;ペニシリン(例えば、アモキシシリン、アンピシリン、クロキサシリン、ジクロキサシリン、フルクロキサシリン、オキサシリン、ピペラシリン);オキソリン酸;ポリペプチド(例えば、バシトラシン、ポリミキシンB);キノロン(例えば、シプロフロキサシン、ナリジクス酸、エノキサシン、ガチフロキサシン、レバキン、オフロキサシン、など);スルホンアミド(例えば、スルファサラジン、トリメトプリム、トリメトプリム-スルファメトキサゾール(コ-トリモキサゾール)、スルファジアジン);テトラサイクリン(例えば、ドキシシリン(doxycyline)、ミノサイクリン、テトラサイクリンなど);アズトレオナムなどのモノバクタム;クロラムフェニコール;リンコマイシン;クリンダマイシン;エタンブトール;ムピロシン;メトロニダゾール;ペフロキサシン;ピラジンアミド;チアンフェニコール;リファンピシン;チアンフェニコール;ダプソン;クロファジミン;キヌプリスチン;メトロニダゾール;リネゾリド;イソニアジド;ピラシル(piracil);ノボビオシン;トリメトプリム;ホスホマイシン;フシジン酸;またはその他の局所用抗生物質が挙げられが、それに限定されるわけではない。任意で、抗生剤は、抗菌ペプチド、例えばデフェンシン、マゲイニンおよびナイシンなど;または溶菌性バクテリオファージであってもよい。また、抗生剤は、上記に列挙した抗生剤のいずれかの組合せであってもよい。PCT/US2010/026190も参照されたい。
【0045】
本明細書での使用に適した例示的な酵素としては、ペルオキシダーゼ、リパーゼ、アミロース、有機リン酸デヒドロゲナーゼ、リガーゼ、制限エンドヌクレアーゼ、リボヌクレアーゼ、DNAポリメラーゼ、グルコースオキシダーゼ、ラッカーゼ、および同類のものが挙げられるが、それに限定されるわけではない。また、成分間の相互作用を用いて、例えば、アビジンとビオチンとの間の特定の相互作用によって絹フィブロインを官能化してもよい。また、活性物質は、上記に列挙した酵素のいずれかの組合せであってもよい。米国特許出願第61/226,801号を参照されたい。
【0046】
治療剤または生物材料を絹フィブロインの中に導入する場合、当技術分野において公知のその他の材料を該作用物質に添加してもよい。例として、該作用物質の成長(生物材料に関して)を促進する、それが絹マットから放出された後に該作用物質の機能性を促進する、またはそれが絹に埋め込まれている期間の間、該作用物質が残存するかまたはその有効性を保持する能力を増加させる材料を添加することが望ましいであろう。細胞増殖を促進するとして公知の材料としては、細胞増殖培地、例えば、ダルベッコ改変イーグル培地(DMEM)、ウシ胎児血清(FBS)、非必須アミノ酸および抗生物質、ならびに成長因子および形態形成因子、例えば線維芽細胞成長因子(FGF)、トランスフォーミング成長因子(TGF)、血管内皮成長因子(VEGF)、上皮成長因子(EGF)、インスリン様成長因子(IGF-I)、骨形態形成成長因子(BMP)、神経成長因子、および関連タンパク質などを使用してよい。成長因子は、当技術分野において公知である。例えば、Rosen&Thies,CELLULAR&MOLECULAR BASIS BONE FORMATION&REPAIR(R.G.Landes Co.,Austin,TX,1995)を参照されたい。絹マットによる送達のためのさらなる選択肢としては、DNA、siRNA、アンチセンス、プラスミド、リポソームおよび遺伝物質の送達のための関連系;細胞のシグナル伝達カスケードを活性化するペプチドおよびタンパク質;ミネラル化または細胞由来の関連事象を促進するペプチドおよびタンパク質;絹マット-組織界面を改善する接着ペプチドおよびタンパク質;抗菌ペプチド;ならびにタンパク質および関連化合物が挙げられる。
【0047】
また、さらなる生体適合性材料を、絹フィブロインマットにブレンドすることもでき、それは例えばポリエチレングリコール(PCT/US09/64673号参照)、コラーゲン、フィブロネクチン、ケラチン、ポリアスパラギン酸、ポリリジン、アルギン酸塩、キトサン、キチン、ヒアルロン酸、ペクチン、ポリカプロラクトン、ポリ乳酸、ポリグリコール酸、ポリヒドロキシアルカノエート、デキストラン、ポリ無水物、グリセロール(PCT/US2009/060135号参照)、およびその他の生体適合性ポリマーなどである。国際公開公報第2004/0000915号を参照されたい。あるいは、絹をヒドロキシアパタイト粒子と混合してもよい。PCT/US08/82487号を参照されたい。本明細書において述べたように、絹フィブロインは組換え起源であってよく、それは繊維状タンパク質ドメインおよびミネラル化ドメインを含む融合ポリペプチドの封入などの絹のさらなる修飾をもたらし、有機-無機複合材料を形成するために使用される。これらの有機-無機複合材料は、使用する繊維状タンパク質融合ドメインのサイズに応じて、ナノスケールからマクロスケールまで構築することができる。国際公開公報第2006/076711号を参照されたい。また、米国特許出願第12/192,588号も参照されたい。
【0048】
活性物質または生物材料を埋め込んだ絹フィブロインは、細胞および/または活性物質の長期保存および安定化に適しうる。細胞および/または活性物質は、絹マットに組み込まれた場合に、室温にて(すなわち22℃〜25℃)および体温(37℃)で少なくとも30日間安定している(すなわち少なくとも50%の残効性を維持する)ことができる。それ故に、温度感受性活性物質、例えば一部の抗生物質などは、冷凍することなく絹マット中に保存することができる。重要なことには、温度感受性生物活性物質は、絹マット中で身体に(例えば、注射により)送達されて、これまでに想像されたよりも長い期間活性を維持することができる。例えば、PCT/US2010/026190号を参照されたい。
【0049】
活性物質(例えば、治療剤)または生物材料を埋め込んだ絹フィブロインは、生物送達装置に適している。絹フィブロインを生物送達装置として使用するための技法は、例えば、米国特許出願第10/541,182号;同第11/628,930号;同第11/664,234号;同第11/407,373号;PCT/US07/020789号;PCT/US08/55072号;PCT/US09/44117号に見出すことができる。本発明の一部の態様は、医療用移植、組織修復における潜在的有用性のため、かつ医療用具のコーティングのための、治療剤または生物材料を埋め込んだ絹フィブロインの薬物送達システムとしての有用性に関する。
【0050】
絹マット構造は、生物送達媒体が制御放出を有することを可能にする。制御放出は、投薬量を経時的に制御放出速度で投与することを可能にする。場合によっては、治療剤または生物材料の送達は、治療を必要とする部位に連続して、例えば、数週にわたって行われる。経時的な、例えば、数日もしくは数週にわたるかまたはそれよりも長い制御放出は、好ましい治療を得るための治療剤または生物材料の連続送達を可能にする。制御送達媒体は、治療剤または生物材料を体液および組織中で、例えば、プロテアーゼによるインビボ分解から保護するので有利である。例えば、PCT/US09/44117号を参照されたい。
【0051】
生物活性物質の絹マットからの制御放出は、経時的に、例えば、約12時間または24時間以上の間;1ヶ月または2ヶ月または5ヶ月以上起こるように設計することができる。放出の期間は、例えば、約12時間〜24時間、または約12時間〜1週間の期間にわたって起こるように選択することができる。別の態様では、放出は、例えば約1ヶ月以上2ヶ月以内で起こりうる。制御放出期間は処置条件に基づいて選択されてよい。例えば、一貫した放出および高い局所投薬量が望ましい場合には特定の放出プロフィールがより効果的でありうる。
【0052】
あるいは、治療剤を絹材料の上に薬学的に許容される担体で覆うこともあり得る。マトリックスを溶解しないどんな薬学的担体を使用してもよい。治療剤は液体、微粉化された固体、または任意のその他の適切な物理的形態として存在してよい。一般に、だが任意で、マトリックスには、1以上の添加剤、例えば希釈剤、担体、賦形剤、安定剤または同類のものなどが含まれる。
【0053】
治療剤の量は、用いられる特定の薬物および治療される医学的状態によって決まる。例えば、薬物の量は、材料の約0.001%〜約70%、または約0.001%〜約50%、または約0.001%〜約20重量%に相当しうる。体液と接触すると、薬物は放出される。
【0054】
組織工学足場に適した絹材料は、作製後にさらに修飾することができる。例えば、所望の細胞集団に対する受容体または化学誘引物質(chemoattractors)の役割を果たす生物活性物質で足場をコーティングすることができる。コーティングは、吸収または化学結合によって適用することができる。
【0055】
本発明の一部の態様は、ポリエチレンオキシド(PEO)と、少なくとも一つの活性物質を含む絹フィブロイン水溶液をブレンドすること;ブレンドした溶液を電界紡糸し、それにより活性物質を封入する絹タンパク質/PEOブレンドマットを形成すること;および電界紡糸した絹マットを拘束乾燥することにより、創傷治癒を促進するための創傷被覆材としての、少なくとも一つの活性物質を埋め込むかまたは封入する絹材料に関する。あるいは、活性物質は、PEOとブレンドした後に絹フィブロインに添加されるか、または電界紡糸した絹材料に添加されてよい、例えば、電界紡糸した絹/PEOマットは、活性物質でコーティングされてよい。
【0056】
本発明の絹材料は、生理活性分子を局所送達する能力があり、次世代の生体材料となりうる。例えば、ナノスケールの絹繊維でできた、EGFを含有する電界紡糸絹マットは、創傷治癒プロセスの促進のために使用されている。絹マットの中に組み込まれたEGFは、時間依存性にゆっくり放出されることができ得る(例えば、170時間で25%のEGF放出)。本発明の絹材料は、ヒト皮膚と同じ構造を示し、インビボで見出される同じ分子および細胞機構を用いて治癒する能力のある、三次元の負傷したヒト皮膚等価モデルにおいて特徴付けることができる。この生体機能性絹マットを負傷したヒト皮膚等価モデルの上に包帯材として載せると、絹マットは、上皮舌状部(epidermal tongue)による創傷閉鎖の時間を90%まで減少させることにより治癒を助ける。Schneider et al.,Acta Biomater.,(2009)。
【0057】
本発明の一部の態様は、絹フィブロインタンパク質およびPEOを含む電界紡糸絹マットに関する。一態様では、電界紡糸絹マットの絹フィブロインタンパク質/PEOブレンド比は、約2:1〜約4:1である。絹/PEO重量比および次式:
に基づく。
【0058】
絹マットのw/wでの絹の割合は、約75%w/w〜90%w/wの範囲に及びうる。電界紡糸絹マットの厚さは約20μm〜約80μmの範囲である。
【0059】
PEO濃度、または絹/PEOブレンド比は、絹繊維の表面積および電界紡糸プロセスの間のバルク形態に直接影響を及ぼす。Jin et al.,3 Biomacromol.1233-39(2002);Wang et al.,37 Macromol.6856-64(2004)。PEO濃度が増加するにつれて、繊維中で生じたフィブロインミセルおよび小球構造のサイズは低下する。その上、ひとたび素早く動く帯電した流体の噴出の中に入ると、これらの小球構造は整列し、100,000倍まで伸張する。Wang et al.,2006;Kowalewski et al.,53 Bulletin Polish Acad.ScL,Tech.Sci.385-94(2005);Reneker&Yarin,49 Polymer 2387-425(2008)。本発明は、絹/PEOブレンド比が、繊維厚さ、密度、配向、相分散、多孔性およびマット厚さを含む、得られる絹マットの特性に重要な役割を果たすことを実証する。その結果として、増加したPEO濃度で形成された繊維は、幾何学的形状、表面積、ならびに、4:1から1:1までの絹/PEOブレンドマットに観察された漸進的な視覚およびテクスチャーの変化に相関するバルク体積が低下した。
【0060】
一態様では、4:1、3:1、2:1、3:2、7:6、および1:1の絹/PEOブレンド比で調製した6つの絹/PEOブレンド材料系を、融合性の直径16.5cmおよび10cmのマットに電界紡糸した。各々のサンプルの物理的特性は、水飽和状態と乾燥状態の両方で評価した。水に浸すと、6つのマトリックスは、不透明に半透明な灰白色の外観を提示する、均一な立体構造を有し、絹のようなテクスチャーをもち柔軟であったが、図1Aに示されるように、取扱期間が延びると絹濃度に対する(respective of)引張強さの悪化を提示した。絹のようなテクスチャーとは、水分子が非晶質ポリマーマトリックスの全体にわたって継続的に可塑化するフィブロインの動的吸湿性を説明するために言及される。アミノ、ヒドロキシル、またはカルボキシル酸末端基との水素結合を形成することによるか、または、親水性ドメインの至る所に自由に分散することにより;この流動性の環境は、これらの飽和材料系の軟質の絹のようなテクスチャーをもたらす運動エネルギーの最小化に起因して、継続的に移行している。Hu et al.,39 Macromol.6161-70(2006);Agarwal et al.,63 J.Appl.Polym.Sci.401-10(1997);van der Heijden et al.,378 Thermochim.Acta 27-34(2001);Wong et al.,2006。周囲温度で24時間の乾燥期間の後、物理的な特徴は、6つの材料系で漸進的に変化した。図2および3に示されるように、絹濃度の低下(86.5%、82.8%、76%、70.6%、65.1%および61.5%)に関連して、マットは、付着曲げ強度(cohesive flex strength)をもつ雪のように白い柔軟なウエハーのようなテクスチャーから、半透明褐色で超薄の柔軟性の低いフィルムのような材料に変わった。
【0061】
本発明において、乾燥法は、絹/PEOブレンドマットの物理的および機械的特性、例えば電界紡糸絹/PEOブレンドマットの厚さなどにも影響を及ぼす。例えば、ポリスチレンペトリ皿を使用する風乾法を用いることができる。あるいは、拘束乾燥の方法を用いてよい。例えば、電界紡糸絹マットを乾燥させるために結晶化皿技法を用いてよい。
【0062】
本発明の電界紡糸絹マットの厚さは、約20μm〜約80μmである。拘束乾燥法を用いる場合、電界紡糸絹マットの厚さは、平均約20μm〜30μmでありうる。
【0063】
例えば、絹/PEOブレンド比が4:1、3:1、2:1、3:2、7:6、および1:1の電界紡糸絹マットの直径3.5cmのサンプルを、直径10cmのマットから打ち抜き、ポリスチレンペトリ皿法を用いて風乾させた。得られる絹マットを図2Aに示す。飽和したいくつかの直径3.5cmのサンプルは、正味の力(net force)の表面-表面疎水性平衡を実現し、層状の絹シートの分離および変位で親水性挙動を提示するために、取り扱いが困難で、多くの場合半分に折り重なる。水乾燥段階の全体にわたって、極性水分子がこの不織の多孔性生体材料の大きい表面積から蒸発するにつれて、界面での親水性の疎水性ドメインへの転移により表面エネルギーは最小となった。この動的表面構造の再編成は、重鎖の再整列およびβ-シートの結晶化を明示する。Vepari&Kaplan,2007;Jin et al.,200);Hu et al.,39 Macromol.6161-70(2006)。ねじれた襞のあるβ-シート形成の特徴を示して、マトリックスはいずれも完全に平らな配向で乾燥せず、4:1および3:1マトリックスだけが元の円形の形状を維持していた。その上、フィブロインの重鎖の結晶性ドメイン間に交互に配置された非晶質ドメインの末端に位置するプロリン残基がある。Zhou et al.,2000。プロリンは、脱水によって非常に収縮し、従って繊維の収縮する能力を増加させることが示された。Liu et al.,9 Biomacromol.116-21(2008)。図4に示されるように、絹濃度の低下とともに、これらの要素は乾燥したサンプルにおける飽和状態から乾燥状態へのそれらの表面積の51.0±0.0%〜87.5±9.9%の間の減少の一因となる(n=3)。図5に示されるように、サンプルの各々の乾燥したセットの厚さ測定値は、それぞれ、81.7±7.5から77.5±10.5に、66.7±5.1に、53.3±8.1に、46.7±5.1に、および30.0±6.4μmに漸進的に下降した(n=6)。
【0064】
別の態様では、絹/PEOブレンド比が4:1、3:1、2:1、3:2、7:6、および1:1の直径12.5cmの電界紡糸絹マットを、結晶化皿技法を用いて乾燥させた。得られる絹マットを図3Aに示す。水に浸すと、これらのより大型のサンプルは展開がより容易であった。親水性の力に関して、層状のシート分離は、シートの変位なく各々のサンプルの内部領域でのみ観察された。これは、これらのサンプルがより大型のマットから打ち抜かれておらず、従ってサンプルの端部の架橋された結晶化領域を保持しているためでありうる。乾燥段階の間、飽和したサンプルが縁からサンプルの中心に向かって均一に乾燥するとき、マットの各々のセットは皿の口の全体にわたって漸進的に収縮する。4:1および3:1サンプルは、結晶化皿の縁に付着して完全に乾燥し、完全に平坦で柔軟な白色の膜のような材料をもたらした。3:2、7:6、および1:1サンプルが乾燥するとき、結晶化する延伸力が繊維の降伏点伸びを越えて材料に応力を加え、皿の縁での材料の剪断による構造的破壊をもたらし、サンプルの内部領域に伝わった。この乾燥法では、絹濃度の低下は、材料の構造的完全性および曲げ強度に影響を及ぼした。2:1サンプルは皿の縁から切り取られたが、材料変形の痕跡はほとんどなく、特性は4:1および3:1サンプルに類似していた。図4に示されるように、4:1および3:1マトリックスは、元の表面積の98%を保持したが、2:1マットは、11.8%±2.7%を失った。3:2、7:6および1:1サンプルは、それぞれ、68.8%±9.1%、65.9%±4.3%および63.9%±6.5%収縮した(n=3)。図5に示されるように、マットの各々の乾燥したセットの平均厚さは、それぞれ、31.2±1.8、28.7±1.2、24.3±2.3、25.3±2.3、20.0±1.4および26.0±0.9μmであった。
【0065】
本発明において、電界紡糸絹/PEOブレンドマットの結果として得られる繊維は、マット構造全体にわたって実質的に均一な直径分布を有する。図6AのSEM画像は、電界紡糸プロセスの直後、かつメタノール飽和およびPEO浸出処理の前の全ての6つの絹/PEO材料群の画像である。全体に、これらの電界紡糸した絹/PEO繊維は、直径が200nm〜500nmの範囲に及ぶ。Huang et al,2001に報告されるように、繊維ビーズ形成は、PEO濃度の低下とともに次第に顕著となった。Wang et al.,2006;Zhou et al.,2000;Huang et al.,12 J.Biomat.Sci.Polym.Ed.979-93(2001)。具体的には、4:1、3:1、および2:1(フィブロイン:PEO)サンプルは、各々、繊維内のランダムな位置にちょうど幅1μmを上回るものから700nm間での範囲に及ぶビーズセグメントを有した。ビーズは、3:2および7:6サンプルセットで最小化し、明確に定義された微細な円形状の繊維が構造の全体にわたって秩序化された外観を与えた。1:1サンプルセットは特有の外観を有し、繊維は不規則で非円形の形状であって、不均一で高密度のマット構造に移行した。この移行が、装置の接地ステージに集まった場合の、液-液/液-固相分散による繊維の収束に起因しうることは妥当である。1:1の個々の繊維は、300〜500nmの範囲に及び、一方、混合繊維は700〜900nmの間で測定された。
【0066】
「拘束乾燥技法」または「拘束乾燥する」とは、本明細書において、絹材料が延伸力を受けている間に乾燥するように、絹材料を拘束している間に乾燥させるプロセスをさす。例えば、拘束力は、結晶化皿の口の上に付着しながら絹材料が乾燥するときに起こる合成収縮力に起因しうる。記載されるように、これらの飽和した絹材料は、最初に結晶化皿の口の上に掛けられてそれに付着する。水分子が蒸発するにつれて、タンパク質の表面基質の疎水性ドメインおよびバルク領域の全体が、不織のキャスト(non-woven cast)の組織間隙(interstitial space)および材料のバルク領域の中から自由体積の喪失を開始する。自由体積の喪失は、材料を収縮させ、結晶化皿の縁に向かって放射状に延伸させる。結晶化皿の縁に付着しているので、材料は自由体積の継続的な喪失によって束縛されることとなり、繊維は半径応力の方向に整列し、伸張されるようになる。絹体積に応じて、材料繊維が降伏点伸びを越えて収縮すれば、材料の剪断が材料/結晶化皿縁表面界面で起こる。拘束乾燥法に反して、ペトリ皿の中の風乾サンプルは、乾燥してねじれた不規則な立体構造となるまで継続的に収縮する。
【0067】
一態様では、拘束乾燥法は、制御された蒸発によって行われる。この方法は、電界紡糸絹/PEOブレンドマットを水浴から取り出し、マットを、皿の3分の1まで水を入れた結晶化皿の上に掛け、マットを含有する皿を20%〜50%の相対湿度のデシケーターに入れて一晩乾燥させることを含む。結晶化皿乾燥法を利用する漸進的なポリマー鎖および繊維の立体構造を説明する概念図を図9に示す。上部は、分散した未整列の二次構造の、親水性環境から、疎水性結合により駆動される整列したタンパク質凝集体への移行である。繊維が引き伸ばされ始めるとき、水なましされた(water-annealed)β-シートが鎖間形成対鎖内形成によって集合する。図の下部の繊維形成は、絹濃度および繊維整列および伸張の間の逆相関を反映する。
【0068】
一態様では、図7に示されるように、絹/PEOブレンドマットのSEM画像は、拘束乾燥法、例えば結晶化皿法などを用いて調製された。調製したマットの絹/PEOブレンド比は、それぞれ4:1、3:1および2:1である。表面のトポグラフィーは、各々のモデルの全体にわたる繊維の高密度でランダムな分布を反映する。評価は、この乾燥技法によって起こる繊維収縮、伸張、および再整列の痕跡の増加を示す。図7Bを参照すると、4:1マットの繊維は、目立った繊維収縮または整列がなく緩んでねじれた外観を有する。対照的に、3:1および2:1マットの繊維は伸張して整列し付着して、ウェブのような微小なテクスチャー(web-like micro texture)を形成するようになる。図7Cの繊維形成に焦点を合わせると、3:1マットの伸張繊維は、ピンと張ったウェブ構造を形成し、末端が液化性の外観になっている整列した繊維間の相分散の痕跡を示している。2:1フィブロイン:PEOサンプルのウェブ構造は、ロープのような配置を形成する、明確に定義された、伸張され整列された繊維の絡み合った網状構造からなる。
【0069】
拘束乾燥技法により明らかにされる繊維の整列および伸張は、絹フィブロインの水なまし特性によるものでありうる。Jin et al.,15 Adv.Funct.Mater.1241-47(2005);Agarwal et al.,63 J.Appl.Polym.Sci.401-10(1997);Lawrence et al.,43 J.Mat.Sci.6967-85(2008);Wong et al.,2006。ポリマーバルク領域内部で可塑剤の役割を果たすことにより、水分子は、低結合エネルギーのポリマー鎖間で分子間の動きを伝え、ポリマーの流動性および再整列を促進する。水分子が蒸発するときに、ポリマー鎖は、延伸されて、結晶化皿の縁の周囲から発生する半径応力の方向を向く。延伸されたポリマー鎖の整列とともに、非晶質軽鎖の末端で折り畳まれている、両側性の鎖間層構造の段階的拡大および結晶化した鎖内のねじれた立体構造の減少を促進するプロリンは減少する。Zhou et al.,2000;Liu et al.,9 Biomacromol.116-21(2008)。支配的な鎖間疎水性相互作用は、バルク領域から自由体積を圧縮し、結晶性二次構造整列および非晶質絹I状態の結晶性絹II状態への移行に影響を及ぼす。Jin et al.,15 Adv.Funct.Mater.1241-47(2005);Agarwal et al.,1997;Lawrence et al.,43 J.Mat.Sci.6967-85(2008);Wong et al.,82 Appl.Phys.A-Mater.293-203(2006)。水分子の継続的な排出によって、バルク体積は低下し、繊維が結晶化皿の口の上で伸張し始めるまで収縮する。図9に示されるように、これらの構成要素の組合せが、繊維軸に沿って、高い強度材料特性をもつ伸張され整列された繊維によって、マットを形成する。降伏点伸びを越えて、重および軽(H,L)鎖の両方の非晶質二次構造の内部および二次構造に沿って剪断変形が起こる。
【0070】
整列した図7Cの3:1繊維と図7Dの2:1繊維の間の巨視的相分散の外観も、水の可塑化特性に起因しうる。水およびカイコ絹フィブロインフィルム系の示差走査熱量測定により、脱水した絹フィブロインのガラス転移温度(Tg)が、20〜23重量%の吸水量で178℃から40℃より下まで低下したことが明らかとなった。Agarwal et al.,1997。結晶性立体構造は生体材料の全体にわたる希釈剤の吸収に影響を及ぼすが、各々の絹マット群の平衡含水率(EWC)は、80重量%よりも大きく(表4)、相当な親水性相互作用を示し;Tgおよび妥当な相分散の低下を予見する。Hu et al.,39 Macromol.6161-70(2006)。その上、絹ポリマー鎖の全体にわたって交互に配置された疎水性ドメインおよび親水性ドメインは、材料が表面-液体から表面-ガス、表面-表面界面へ移行するときに、動的移動性表面基質をもたらす。Ratner et al.,BIOMATS.Sci.:INTRO.MATS.MED.(Acad.Press,NY,2004;Allcock,INTRO.MATS.CHEM.(Wiley&Sons,Hoboken,NJ,2008)。具体的には、希釈剤が蒸発するときに、親水性のドメインは、表面で疎水性セグメントに置き換わる。上記の相分散を考慮に入れると、この現象が界面の繊維間に起こる場合、非晶質二次構造が散在するようになり、混合繊維となる。また、繊維間の線形の二次構造が整列するようになって熱力学的に安定した結晶性β-ストランド(stands)を形成しうることも考えられる。Lawrence et al.,43 J.Mat.Sci.6967-85(2008);Fink,3 Folding&Design R9-R23(1998)。
【0071】
本発明の絹マットは、平均して約0.1〜約1μmの細孔のど径表面積をもつ相互接続した細孔を有しうる。図7Eの断面図より、数種類の異なる絹/PEOブレンド比をもつ絹マットのさまざまな特色が明らかとなる。これらの画像は、絹濃度の低下とともに増加した繊維密度を示す;そして、マトリックスにわたる繊維凝集も、異なる絹/PEOブレンド比をもつ絹マットに関して異なっている。4:1ブレンドの繊維は、多数の大きな空間の隙間をもつ水平シートに凝集した。3:1および2:1ブレンドの繊維は、繊維束の増加を実証し、絹容積の低下に対する繊維の収縮および空間の間隙の漸進的低下を反映した。これらの所見は、絹濃度に対するマット厚さの低下に一致する。図7Bの3枚の画像は、この生体材料の全体にわたって相互に連絡する多孔性を表す。細孔は、各々のマトリックスに対して、それぞれ平均して294、201、から103nm2ののど径表面積を有した。図9に示される下降する細孔径分布は、断面図に見られる繊維密度の増加に一致し、ビーズ領域の数ならびに4:1、3:1、および2:1マトリックスの繊維直径の低下に関して繊維の集合体に関連する。
【0072】
材料の表面粗さは、細胞の配向、接着、増殖、および遊走を制御する、正味の力の生体力学的平衡を促進する、応力/剪断(sheer)のない平面による細胞接触誘導に影響を及ぼす。拘束乾燥処理を受けた本発明の絹/PEOブレンドマットは、風乾法を受けた絹マットよりも表面粗さが小さい。図10のAFM画像は、ポリスチレン皿風乾法で乾燥させた後のさまざまな絹/PEOブレンド比の本発明の絹マットの、三次元の形態学的イメージおよびサンプルの二乗平均平方根の粗さ値を示す。局地的なドメイン粗さ分析は、画像のXおよびY平面の全体にわたる粗さの変動により特徴付けられる。全体として、これらの絹材料系は、粗さ値が500nmから1.4μmまでの明確な表面不規則性を示すクラス3の表面トポグラフィー(class three surface topographies)を実証した(n=3)。4:1および1:1サンプルは、それぞれ1.17±0.00および0.78±0.01μmを測定する、ナノサイズの不規則性をもつ相対的に均一な粗さを有した。3:1、2:1、および3:2サンプルの粗さの標準偏差は0.1〜0.17μmの間で、それぞれ0.65±0.10、0.88±0.17、および0.76±0.16μmの範囲に及んだ。7:6マットが、局地的な粗さが平均1.01±0.43μmの最も大きい偏差を有した。AFM粗さ評価も結晶化皿技法で拘束乾燥した絹/PEOブレンドマットに行った。4:1、3:1、および2:1サンプルは、16×16μmの面積に関して、それぞれ0.66、0.36および0.25μmの粗さ値を有する。サンプルの面積サイズは低下し、サンプルサイズは制限されるが(n=1)、引き伸ばし乾燥したマットの粗さは、風乾マットよりも少なくとも44%平坦である。拘束乾燥したサンプルは、絹濃度に対して粗さが直線的に低下するように見えるが、風乾サンプルについては顕著な傾向はない。この所見は、風乾サンプルのねじれた不規則さと比較して、拘束乾燥したサンプルの繊維の伸張特性に一致する。
【0073】
本発明のプロセスにより調製した電界紡糸絹/PEOブレンドマットは、生体材料用途、例えば創傷被覆材などに適した、良好な構造特性、形態特性、生体機能特性および生体適合特性を示す。例えば、得られる本発明の絹マットは、14日未満で約86%(重量)超を分解する;本発明の絹マットの平衡含水率は約82%を上回る;絹マットの酸素透過速度は、約15460cm3/m2/日よりも大きい;かつ、絹マットの水蒸気透過速度は、約1934g/m2/日よりも大きい。
【0074】
本発明の態様は、時間放出生物療法によって上皮化を促進するための、酵素生分解を備える絹材料を提供する。さまざまなブレンド比をもつ電界紡糸絹/PEOブレンドマットの酵素生分解を、14日以上にわたって評価した。図12Aに示されるように、インビトロ生分解性は、全ての時点で全ての材料群について線形の分解を示し、それぞれ、各々の群に対して1日後に22.6%±3.4%の分解、および14日後に74.0%±8.8%までの材料損失をもたらした。データは、6日までは、全てのブレンドについての分解率は48.2±4.6%で比較的近かったという推論を許容する。10日後、4:1(78%)から1:1(51%)の範囲に及ぶ絹/PEOブレンド比をもつさまざまな材料群について27%の重量減少差が記録された。14日後、酵素分解は85.6±3.8%(4:1)から62.5±5.2%(1:1)に至る範囲に及んだ。形態学的に、目視検査によって、全ての材料系が、最初の6日で表面浸食により主に分解された。10日の時点の後、4:1および3:1サンプルが、マットのほころび、断片化、および微粒子破片への崩壊をもたらす、繊維の切断分解の増加を実証した。4:1および3:1サンプルの生分解挙動は、これらのブレンドの増加した繊維サイズ、マット多孔性および繊維密度特性の低下により、マットの内部繊維構造に酵素が接近することに起因し得る。
【0075】
線形回帰分析は、Minitab(登録商標)15.2.30を用いて各々の絹/PEOブレンド比について全てのサンプルで行った。図12Bの散布図を参照することにより、各々の材料群に関して実行した対数変換は、3日の分解時点の直前の全ての材料群について明確な転移点を明らかにした。次に、各々の材料群について全ての時点、開始から3日の時点および3日〜14日の時点で回帰分析を行った。図12Cに示されるように、全ての材料群につての分解の傾きは3日後に実質的に変化した。開始から3日の時点まで、酵素分解速度は、平均-11.68±0.91傾き単位であった;3日以後14日まで、分解の速度は-3.41±1.12まで横ばいとなった。初期分解段階の間、非晶質領域は、内部結晶領域と比較して加速した速度で分解する。この仮説は、酵素分解の開始時および3日後にこれらの絹材料の赤外吸光度スペクトルにフーリエセルフデコンボリューション(FSD)を用いることにより検証することができる。Hu et al.,2006。
【0076】
正常なヒト皮膚は約21日で再生する。本発明は、時間放出生物療法での上皮化を促進するために、例えば、プロテアーゼが絹材料を断片に分解する時間を比較することにより、マトリックスの設計を所望の分解速度に一致させることを可能にする。結果は、14日後に、4:1絹/PEOブレンドマットの86%が溶解し、それは、リゾチームに曝露した場合に14日後に82%分解したキトサン/ポロキサマー包帯材、およびPBS中で14日後に20%分解するPLGA/PLLA(90/10)と比べても遜色がない。また、酵素レベルは変動する可能性があるため、インビボでの分解速度にも対処し、絹生体材料が、材料形式、位置、および関連する変数によって、インビボで数週間から数年のうちに分解し得ることが示された。Wang et al.,2008。
【0077】
本発明のプロセスにより製造された絹材料は、多様な医学的用途、例えば、血管創傷修復装置、止血包帯材、全層熱傷被覆材、パッチおよびグルー、縫合糸、薬物送達を含む、創傷閉鎖系などにおいて、かつ、組織工学用途、例えば、足場材料、靭帯補綴装置などにおいて、かつ、ヒト身体への長期もしくは生分解性の移植のための製品において、使用することができる。例示的な組織工学足場は、電界紡糸繊維の不織網状構造である。
【0078】
その上、これらの生体材料は、限定されるものではないが、脊椎板、頭蓋組織、硬膜、神経組織、肝臓、膵臓、腎臓、膀胱、脾臓、心筋、骨格筋、腱、靭帯、および乳房組織を含む、これらの特有の足場から利益を受けうる、器官修復置換または再生計画に使用することができる。
【0079】
一態様では、本発明は、材料を包帯に加工し、創傷部位の浮腫およびO2/CO2ガス透過を管理する能力、ならびに同時刻に合わせた(time synchronized)抗生物質、免疫学的、および組織再生生物治療を投与する能力を含む全層熱傷被覆材において有用な特性をもつ絹材料を提供する。本発明において、拘束乾燥技法を用いて、絹濃度が繊維厚さ、密度、配向、相分散、多孔性およびマット厚さを含む特性に大きな役割を果たしたことが見出された。例えば、全層創傷被覆材において、4:1〜2:1の絹/PEOブレンド比をもつ電界紡糸絹マットが使用され、それは最小限の表面積減少で柔軟な膜様材料を提示する有用な物理的特性を保有し、グラム陰性桿菌およびグラム陽性球菌敗血症を惹起する病原性微生物に対して不透過性の障壁をもたらす0.3μm2よりも小さい細孔のど表面積サイズを示す。
【0080】
材料の吸収および平衡含水率(EWC)特性は、細菌の栄養床を提供し得る創傷浸出液の蓄積を制御する際に役割を果たす。一態様では、4:1〜1:1の範囲の絹/PEOブレンド比をもつ絹材料についての全体的な吸収性およびEWC性能は、各群内で比較的接近し、それぞれ400%〜700%および82%〜86%の範囲に及んだ。絹材料群での繊維密度の違いを考慮すると、異なるブレンド比をもつ各々の材料群はそれでも類似する膨潤特性を提示した。これらのモデルと表2中のその他の創傷被覆材候補を比較すると、本発明の絹マットはスポンジのような天然キトサンに基づく包帯材と同様によく機能した。キトサン/ポロキサマー包帯材候補は良好な吸収性およびEWC特性を有したが、本発明の全天然のFDA認可絹材料系は、これらのその他の系と比較して生体適合性および顕著な機械的堅牢性を提供する。
【0081】
(表2)キトサン誘導体に基づく創傷被覆材と本発明の絹材料系との間の平均吸水率および平衡含水率の比較
*Gibran et al., 70 J. Surg. Res. 1-6 (1997);**Quynh et al., 2007;***Wu & Wu, 2006
【0082】
体温恒常性を維持するために、正常な皮膚は、1日あたり204g/m2の速度で体液を透過させる。Lamke et al.,3 Burns 159-65(1977)。また、全層芽創の蒸発水損失は、1日当たり5,138g/m2であり、過剰な脱水を防ぐと同時に適切な水分レベルを許容するために理想的な全層創傷被覆材は、1日当たり2,000〜2,500g/m2の水蒸気透過速度(WVTR)有するべきであることも報告されている。Queen et al.,1987。一態様では、本発明において飽和および乾燥した絹材料は、37℃および50%RHで1,977±35および1,469±81g/m2/日のWVTRを有し、それは、表3に詳細に示されるように、相対温度および湿度で1,180〜2,830g/m2/日の範囲に及ぶキトサン包帯材と遜色がない。本発明の電界紡糸絹マットの厚さ(30〜80μm)を、スポンジのような二層および不斉キトサン包帯材(60〜800μm)に対して考慮すると、多層の絹包帯材を目的に合わせて作り、上記の所望のWVTR基準を実現することが妥当である。その上、図7に示されるように、1日当たり25,000から7,800cm3/m2までの酸素透過速度は、SEM写真において示される各々の材料群の繊維サイズ、多孔性および繊維密度に起因し得る。この生体材料の疎水性の性質は、これらのマットを、創傷浸出液のドレナージ、浮腫および脱水のバランスをとる際に最適なガスおよび水蒸気の透過性能に理想的な厚さに作る能力を促進する。
【0083】
(表3)キトサン誘導体に基づく創傷被覆材と本発明の絹材料系との間の平均水蒸気および酸素透過速度の比較
*Gibran et al., 70 J. Surg. Res. 1-6 (1997);**Quynh et al., 2007;***Hu et al., 2006
【0084】
絹マトリックスの全体にわたって相互連絡する細孔の網状構造は、水の不織構造組織間隙への吸収に有用な材料系であることがわかる。改質した電界紡糸絹繊維(絹/PEOブレンド比は4:1)は、68%までの多孔性を有する。Wang et al.,37 Macromol.6856-64(2004)。増加した表面積は、重鎖および軽鎖の両方の親水性領域内部に存在するヒドロキシル基、カルボキシル基、およびアミノ基に結合している極性水分子によりエネルギーが最小化されるときに、生体高分子のバルク領域への水の吸収を促進する。膨潤は、混和性の希釈剤分子が、ポリマー鎖間を流動して自由体積を生成するときに起こる。絹/PEOブレンディング比が4:1、3:1、2:1、3:2、7:6、および1:1の風乾した2.8cmの電界紡糸絹マットを、直径10cmのマットから打ち抜いた。表4を参照すると、吸収は、461%〜613%の範囲に及び、全ての絹材料群の平均は551%±54%であった。その上、各々の絹モデルについての平均乾燥重量は、22.5mg(絹/PEOブレンド比は4:1)から13.6mg(絹/PEOブレンド比は1:1)まで直線的に低下したが、平衡含水率は、全ての材料系について84%±1%で比較的一定のままであった。データは、吸水が各々の絹濃度の繊維直径、密度、多孔性および二次構造集合特性とは無関係であることを示唆する。
【0085】
(表4)脱イオン水に24時間浸漬した電界紡糸絹/PEOブレンドマットについての平均吸水および平衡含水率測定値(±値=SD、n=6)
【0086】
酸素/二酸化炭素ガス交換を促進する包帯材は、創傷の酸度を低下させ、嫌気性細菌感染を抑制し、従って創傷治癒を促進する環境を形成する。Mi et al.,22 Biomats.165-73(2001);Mi et al.,59 J.Biomed.Mat.Res.438-49(2002)。表5に提示されるように、水和条件下(37℃および80%RH)で評価したサンプルの平均酸素透過速度(OTR)は、4:1サンプルについて25,000cm3/m2/日から、1:1サンプルについて7,800cm3/m2/日に至る平均OTRを提示した。これらのOTRの低下は、それぞれの絹材料群の減少する繊維サイズ、細孔のど径、および増加した繊維密度に起因し得る。既に述べたように、4:1、3:1、および2:1モデルは、緩く分布した繊維密度をもつ多孔性足場を形成するビーズ領域を含むミクロン以上の(micron plus)繊維直径を有した。3:2および7:6繊維は、200nm〜500nmの間に及ぶより小さい直径を有し、繊維密度の増加およびマット多孔性の低下を示した。その上、相の分散した1:1繊維は、多孔性が低下して結晶質-非晶質ガラス障壁を生成したフィブロインのシートを形成した。対照的に、37℃および50% RHで試験した各々の材料群についての全ての飽和および風乾サンプルは、1回の15分間隔の試験の終了よりも前に100,000cm3/m2/日の分析器閾値を上回った。予測されるように、これらの結果は、乾燥またはほぼ乾燥状態ではこれらの多孔質材料は、飽和不織織物の組織間隙およびバルク領域に存在する水分子を通じて酸素が拡散される場合よりも大きい酸素透過速度を有することを反映する。図11の線形曲線分析は、4:1から2:1までの絹ブレンドマトリックスの9,600cm3/m2/日のOTR低下を開示する。
【0087】
(表5)飽和した電界紡糸絹/PEOブレンドマットの平均酸素透過速度を、Illinois Instruments 8001 Oxygen Permeation Analyzerを用いて37℃にて80% RHで測定した。酸素透過性(PO2)および酸素透過係数(P'O2)値は、ASTM 3985-05に基づいて計算した。(±値=SD、n=3)
酸素ガス透過速度:O2GTR:cm3/(m2・d);単位厚さ当たりの酸素透過:cm3/(m2・d)/単位厚さ
【0088】
全層創傷被覆材の水蒸気透過性は、創傷部位での体液蒸発の制御において重要な役割を果たす。過剰な水蒸気透過特性を提示する創傷被覆材は、血液量減少、低体温、および高血圧症を引き起こし得る。Peppas,HYDROGEL MED.&PHARM.II&III(CRC Press,Boca Raton,FL,1987);Beers et al.,2006。水蒸気透過速度(WVTR)は、24時間にわたって、引き伸ばして乾燥した25cm2の4:1、3:1、および2:1絹/PEOマットについて計算した。3:2、7:6、および1:1マットについてのWVTRを確かめる努力は、乾燥段階中の材料変形のために不成功に終わった。拘束乾燥した材料は、水和状態と乾燥状態の両方で評価した。表6に示されるように、飽和および乾燥した4:1、3:1、および2:1材料群についてのWVTRは、平均して、飽和に関して1,977±35g/m2/日、乾燥に関して1469±81g/m2/日であった。飽和したサンプルが乾燥したサンプルよりも優れていたが、これは液体-ガス-膜-ガス界面と比べて直接の液体-膜-ガス界面に起因する可能性が最も高い。水和したマットの吸湿性は、熱力学的反応および蒸発の加速を促進する生体材料表面-ガス界面に水分子を都合よく存在させることを可能にした。図11に示されるように、WVTRは、3つ全ての絹濃度で比較的同じであり、162g/m2/日および82g/m2/日のごく僅かな下降する変化は、繊維サイズ、マット密度、多孔性および二次構造特性に起因するものであった。
【0089】
(表6)飽和および乾燥した4:1、3:1および2:1拘束乾燥絹/PEOマトリックスについての平均水透過速度を、24時間後に、22.8±0.6℃および50%±2%RHで、ウォーターカップ法(water cup method)に続いてASTM D 1653に従ってPerm Cupを用いて測定した。(±値=SD、n=3)
【0090】
本発明の一部の態様はまた、絹フィブロインタンパク質、ポリエチレンオキシド(PEO)、および少なくとも一つの活性物質を含む、少なくとも一つの電界紡糸絹マットと創傷を接触させることを含む、創傷治癒を促進する方法にも関する。電子紡糸絹マットの絹フィブロインタンパク質/PEOブレンド比は、約2:1〜約4:1であり(または絹の割合が約75%w/w〜90%w/wであり);絹マットの厚さは、約20〜約80μmである。
【0091】
本発明は、効果的な創傷被覆材などの生物医学的用途に適した、電界紡糸した絹/PEO材料を提供する。電界紡糸した絹/PEOマトリックスの物理的特性および生体機能特性を評価して、創傷被覆材に関連する構造的形態学的および生体適合性の特徴を査定した。例えば吸収、水蒸気透過性、酸素透過性、および生分解性生体機能特性などの特性は、創傷被覆材用途に有用である。絹/ポリエチレンオキシド(PEO)含量の変化を用いて、形態および構造に関して異なるマトリックスを作成した。二流体絹/PEO電界紡糸技法を適用して、大型の融合性絹マットを製造し、表面のテクスチャーおよびバルク特性を数量化した(繊維構造、トポグラフィー、吸収、水WVTR、酸素透過性、および生分解性を含む)。水和した状態では、全ての材料群が、創傷治癒に適した吸収性およびVTRを提示した。酸素透過速度(OTR)は、創傷部位に適した酸素/二酸化炭素ガス交換の特色を示唆した。インビトロ酵素生分解は、1日で初期重量の23%±3%の分解および14日で74±9%までの分解を確認した。かかる創傷マット系の保存および分布に関連する材料特性に対応するために、複数の乾燥法を検討した。制御蒸発および拘束乾燥技法を用いると、絹濃度が、繊維伸張、整列、密度、多孔性および相分散に影響を及ぼす各々のマトリックスの特性の決定要因であった。絹/PEOブレンド比が2:1〜4:1の、制御蒸発および拘束乾燥技法で処理した電界紡糸絹/PEOマットは、創傷被覆材に対する潜在的有用性をもつ生体材料系に関する特に適した特性を実証した。これらの絹材料系は、抗生物質送達、マクロファージ応答、線維芽細胞/ケラチン生成細胞およびサイトカインの影響、および関連する生物学的問題に有用でありうる。
【0092】
本発明の特定の態様を、限定されない例において説明する。
【0093】
本発明は、以下の番号の付いた項目のいずれか一つに定義される通りでありうる。
1.以下の段階を含む、絹マットを製造するための方法:
ポリエチレンオキシド(PEO)と絹フィブロイン水溶液をブレンドする段階;
前記ブレンドした溶液を電界紡糸する段階であって、それにより絹タンパク質/PEOブレンドマットを形成する、段階;および
前記電界紡糸した絹マットを拘束乾燥する段階。
2.電界紡糸した絹マットをアルコールで処理する段階をさらに含む、項目1記載の方法。
3.PEOを絹マットから抽出する段階をさらに含む、項目1または2記載の方法。
4.少なくとも一つの活性物質を絹マットに埋め込む段階をさらに含む、項目1〜3のいずれか一項記載の方法。
5.活性物質が、細胞、タンパク質、ペプチド、核酸、核酸類似体、ヌクレオチドまたはオリゴヌクレオチド、ペプチド核酸、アプタマー、抗体またはその断片もしくは部分、抗原またはエピトープ、ホルモン、ホルモンアンタゴニスト、成長因子または組換え成長因子およびその断片および変異体、細胞接着メディエーター、サイトカイン、酵素、抗生物質または抗菌性化合物、ウイルス、毒素、プロドラッグ、化学療法剤、小分子、薬物、ならびにそれらの組合せからなる群より選択される治療剤または生物材料である、項目4記載の方法。
6.活性物質が、前駆細胞または幹細胞、平滑筋細胞、骨格筋細胞、心筋細胞、上皮細胞、内皮細胞、尿路上皮細胞、線維芽細胞、筋芽細胞、口腔細胞、軟骨細胞、軟骨芽細胞、骨芽細胞、破骨細胞、ケラチン生成細胞、腎尿細管細胞、腎基底膜細胞、外皮細胞、骨髄細胞、肝細胞、胆管細胞、膵島細胞、甲状腺細胞、上皮小体細胞、副腎細胞、視床下部細胞、下垂体細胞、卵巣細胞、精巣細胞、唾液腺細胞、脂肪細胞、前駆細胞、およびそれらの組合せからなる群より選択される細胞である、項目5記載の方法。
7.活性物質が、細胞増殖培地をさらに含む、項目6記載の方法。
8.活性物質が、抗生物質である、項目6記載の方法。
9.ポリエチレンオキシド(PEO)と絹フィブロイン水溶液をブレンドする段階;
ブレンドした溶液を電界紡糸する段階であって、それにより絹タンパク質/PEOブレンドマットを形成する、段階;および
電界紡糸した絹マットを拘束乾燥する段階
を含む方法から調製された絹材料。
10.ポリエチレンオキシド(PEO)を、少なくとも一つの活性物質を含む絹フィブロイン水溶液とブレンドする段階;
ブレンドした溶液を電界紡糸する段階であって、それにより該活性物質を封入する絹タンパク質/PEOブレンドマットを形成する、段階;および
該活性物質を封入する電界紡糸した絹マットを拘束乾燥する段階
を含む方法から調製された、創傷に巻いて創傷治癒を促進するための少なくとも一つの活性物質を封入する絹材料。
11.活性物質が、細胞、タンパク質、ペプチド、核酸、核酸類似体、ヌクレオチドまたはオリゴヌクレオチド、ペプチド核酸、アプタマー、抗体またはその断片もしくは部分、抗原またはエピトープ、ホルモン、ホルモンアンタゴニスト、成長因子または組換え成長因子およびその断片および変異体、細胞接着メディエーター、サイトカイン、酵素、抗生物質または抗菌性化合物、ウイルス、毒素、プロドラッグ、化学療法剤、小分子、薬物、ならびにそれらの組合せからなる群より選択される治療剤または生物材料である、項目10記載の絹材料。
12.活性物質が、前駆細胞または幹細胞、平滑筋細胞、骨格筋細胞、心筋細胞、上皮細胞、内皮細胞、尿路上皮細胞、線維芽細胞、筋芽細胞、口腔細胞、軟骨細胞、軟骨芽細胞、骨芽細胞、破骨細胞、ケラチン生成細胞、腎尿細管細胞、腎基底膜細胞、外皮細胞、骨髄細胞、肝細胞、胆管細胞、膵島細胞、甲状腺細胞、上皮小体細胞、副腎細胞、視床下部細胞、下垂体細胞、卵巣細胞、精巣細胞、唾液腺細胞、脂肪細胞、前駆細胞、およびそれらの組合せからなる群より選択される細胞である、項目11記載の絹材料。
13.活性物質が、細胞増殖培地をさらに含む、項目12記載の絹材料。
14.活性物質が、抗生物質である、項目12記載の絹材料。
15.電界紡糸絹マットが、アルコールでさらに処理される、項目9〜14のいずれか一項記載の絹材料。
16.PEOが、電界紡糸絹マットから抽出される、項目9〜15のいずれか一項記載の絹材料。
17.電界紡糸絹マットの厚さが約20μm〜約80μmである、約50重量%〜約100重量%の範囲に及ぶ絹フィブロインタンパク質を含む電界紡糸した絹材料。
18.電界紡糸絹マット中の絹フィブロインタンパク質の含量が、約75重量%〜約90重量%の範囲に及ぶ、項目17記載の電界紡糸絹材料。
19.電界紡糸絹マット中のPEOの含量が、約0重量%〜約50重量%の範囲に及ぶ、電界紡糸絹マット中にポリエチレンオキシド(PEO)のブレンドをさらに含む、項目17または18記載の電界紡糸絹材料。
20.電界紡糸絹マット中のPEOの含量が、約10重量%〜約25重量%の範囲に及ぶ、項目19記載の電界紡糸絹材料。
21.少なくとも一つの活性物質をさらに含む、項目17〜20のいずれか一項記載の絹材料。
22.活性物質が、細胞、タンパク質、ペプチド、核酸、核酸類似体、ヌクレオチドまたはオリゴヌクレオチド、ペプチド核酸、アプタマー、抗体またはその断片もしくは部分、抗原またはエピトープ、ホルモン、ホルモンアンタゴニスト、成長因子または組換え成長因子およびその断片および変異体、細胞接着メディエーター、サイトカイン、酵素、抗生物質または抗菌性化合物、ウイルス、毒素、プロドラッグ、化学療法剤、小分子、薬物、およびそれらの組合せからなる群より選択される治療剤または生物材料である、項目21記載の絹材料。
23.絹マットの厚さが約20〜30μmである、項目17〜22のいずれか一項記載の絹材料。
24.絹マットが、平均して約0.1〜約0.3μmの細孔のど径表面積をもつ、相互接続した細孔を有する、項目17〜23のいずれか一項記載の絹材料。
25.得られる絹マットの吸水量が約460%よりも大きい、項目9〜24のいずれか一項記載の絹材料。
26.得られる絹マットの平衡含水率が約82%よりも大きい、項目9〜25のいずれか一項記載の絹材料。
27.得られる絹マットの酸素透過速度が約15460cm3/m2/日よりも大きい、項目9〜26のいずれか一項記載の絹材料。
28.得られる絹マットの水蒸気透過速度が約1934g/m2/日よりも大きい、項目9〜27のいずれか一項記載の絹材料。
29.絹フィブロインタンパク質、および任意で少なくとも一つの活性物質を含む、少なくとも一つの電界紡糸絹マットに創傷を接触させる段階を含む、創傷治癒を促進する方法であって;
前記絹フィブロインタンパク質が約50重量%〜約90重量%の範囲に及び、
前記絹マットの厚さが約20μm〜約80μmであり;
前記絹マットが、約460%よりも大きい吸水量か、または約82%よりも大きい平衡含水率を有し;かつ
得られる絹マットの酸素透過速度が約15460cm3/m2/日よりも大きい、前記方法。
30.絹フィブロインタンパク質が、約75重量%〜約90重量%の範囲に及ぶ、項目29記載の方法。
31.絹フィブロインタンパク質、ポリエチレンオキシド(PEO)、および任意で少なくとも一つの活性物質を含む、少なくとも一つの電界紡糸絹マットに創傷を接触させる段階を含む、創傷治癒を促進する方法であって;
絹/PEOブレンド比が、約4:1〜約2:1であり;
前記絹マットの厚さが約20μm〜約80μmであり;
前記絹マットが、約460%よりも大きい吸水量か、または約82%よりも大きい平衡含水率を有し;かつ
得られる絹マットの酸素透過速度が約15460cm3/m2/日よりも大きい、前記方法。
32.絹マットの水蒸気透過速度が、約1934g/m2/日よりも大きい、項目29〜31のいずれか一項記載の方法。
33.活性物質が、細胞、タンパク質、ペプチド、核酸、核酸類似体、ヌクレオチドまたはオリゴヌクレオチド、ペプチド核酸、アプタマー、抗体またはその断片もしくは部分、抗原またはエピトープ、ホルモン、ホルモンアンタゴニスト、成長因子または組換え成長因子およびその断片および変異体、細胞接着メディエーター、サイトカイン、酵素、抗生物質または抗菌性化合物、ウイルス、毒素、プロドラッグ、化学療法剤、小分子、薬物、およびそれらの組合せからなる群より選択される治療剤または生物材料である、項目29〜32のいずれか一項記載の方法。
【実施例】
【0094】
実施例1
材料
カイコ絹の繭(Tajima Shoji Co.,Yokohama,Japan)を調製して8重量%の絹溶液を生成した。Wang et al.,2006.4:1、3:1、2:1、3:2、7:6、および1:1w/w絹:PEO(920,000g/mol)比溶液を用いて6つの絹材料を調製した。電界紡糸の間に安定した流体の噴出を生じるための粘弾特性および表面張力特性に必要な最低限の7.2%絹/PEOポリマー濃度を維持するために、4:1、3:1、および2:1ブレンドは、5%のPEOを含有し、一方3:2、7:6、および1:1ブレンドは6%のPEOを含有した。Jin et al.,3 Biomacromol.s 1233-39(2002)。
【0095】
この研究のために構築した電界紡糸装置は、高電圧電源(Gamma High Voltage Research ES-30P,Ormond Beach,FL)、10〜60mlシリンジポンプ(Braintree Scientific 8000,Braintree,MA)、電位ステージおよび接地ステージ、1.5mmポリエチレン管および16ゲージ5.08cm鋼毛管を用いて、既に公開された手順(Wang et al.,2002)に従った。例えば、国際公開公報第2004/0000915号;国際公開公報第2004/062697号を参照されたい。
【0096】
絹/PEO溶液を、ポリエチレン管を通じてシリンジポンプから、電位プレートに挿入した12kV DC荷電鋼毛管(steal capillary tube)にポンプで送った。同文献;Reneker&Yarin,49 Polymer 2387-25(2008);Jin et al.,2002。電界紡糸繊維は、電位プレートよりもおよそ17cm低く設置され、毛管の先端の垂直落下線を越えておよそ2cm〜3cmに位置する、接地ステージで収集した。これらの重量比に基づくと、方程式は次の通りである。
【0097】
各々のモデルについての絹の割合w/wは、それぞれ、86.5%、82.8%、76%、70.6%、65.1%および61.5%に等しかった。溶液粘度は、#5スピンドルを69°Fで用いるBrookfield HATD粘度計(Brookfield Engineering Laboratories,Inc.,Stoughton,MA)によって、それぞれ、128、152、240、424、768、および1120mPa-Sに等しいと決定された。直径16.5cmおよび10cmのマットを、6つの材料群の各々について室温にて(RT)60%よりも低い相対湿度で電界紡糸して、電界紡糸プロセスによるマット厚さの違いを評価した。
【0098】
乾燥法
乾燥技法を用いて、絹電界紡糸材料マットの物理的特性を評価した。風乾法では、直径3.5cm、2.8cmおよび2.2cmのサンプルを、水に浸漬した直径10cmのマットから打ち抜いた。秤量紙(VWR,West Chester,PA)の間でプレスした後、サンプルを、ほぼ乾燥するまでポリスチレンペトリ皿の側壁に垂直に置いた。その後、粘着を防ぐためにサンプルを定期的に再配置し、RTにて24時間乾燥させた。
【0099】
拘束乾燥法では、大型の直径16.5cmのサンプルを水浴から取り出し、3分の1まで脱イオン水を入れた125×65mm結晶化皿の口の上に掛け、RTにて20%〜50%の間のRHのデシケーターの中に入れ、一晩乾燥させる。絹/PEOマットは、口の円周全体に沿った縁にマットが接触し、軽く付着するように掛けた。乾燥段階の間、飽和したサンプルが縁からサンプルの中心に向かって均一に乾燥するとき、マットの各々のセットは皿の口の全体にわたって漸進的に収縮する。4:1および3:1サンプルは、結晶化皿の縁に付着して完全に乾燥し、引き伸ばされた、完全に平坦で柔軟な白色の膜のような材料をもたらした。サンプル重量(Mettler Toledo AB54-S/FAC,Columbus,OH)および厚さ(Ono Sokki EG-225F Digital Indicator,Addison,IL;等半径点AA821;力25g)の測定値を、各々の絹系について記録した。
【0100】
実施例2 材料の特徴付け
繊維厚さおよび表面トポグラフィーは、JEOL JSM 740-1F FE-SEM(Tokyo,Japan)を1.5倍、6.5倍および12倍の倍率で用いて特徴付けた(加速電圧:1kV、作動距離:13.6mm)。断面画像は、2.5倍、5倍、10倍、および50倍の倍率を用いて撮影した(加速電圧:5kV、作動距離:6mm)。断面のサンプルを、2×5mmの断片に切断し、液体窒素中で急速冷凍し、ピンセットを用いて半分に割った。サンプルを、断面を上にしてカーボンテープの上に取り付けた。全てのサンプルを、Denten Vacuum Desk IV(Moorestown,NJ)を次の設定で用いて100Å Auでコーティングした:真空:80〜90mtorr、スパッタリング設定点:20〜30%、堆積時間:2分。サンプルの表面形態、粗さ、および三次元の特徴を、PSIA XE-150 AFM(Santa Clara,CA)によって、Ultrasharp NSC16/AIBSプローブを非接触モードで用いて得た(共振周波数:170kHz、バネ定数:45N/m)。XEIデータ分析ソフトウェア(Park Solutions,Santa Clara,CA)を、表面粗さの特徴付けに用いた。5つの繊維層の最小深さ(約1μm)まで伸びる細孔によって定義される材料多孔性を、50×50μmの面積に分布バケツアルゴリズム(distribution bucket algorithm)を適用して統計的に評価した。細孔のど径および細孔表面積を、細孔径直径が0.15、0.30、0.45、0.60、0.75、0.90、1.05および1.25μmの範囲に及ぶ、円形領域に関して幾何学的に推定した。
【0101】
実施例3 水および酸素透過性
吸収
6つの直径2.8cmの試験サンプルを、ポリスチレン皿法で乾燥させ、無菌の6ウェルの組織培養処理したポリスチレンプレートに入れ、脱イオン水に24時間浸漬して膨潤平衡に達した。次に、サンプルを取り出し、Kimwipe(登録商標)組織の上に最小限の安定したペンダントドロップがサンプルの端部に維持されるまで優しく塗りつけた。次に、飽和したサンプルを秤量し、吸水および平衡含水率(EWC)を次の方程式により算出した。
式中、WwおよびWdは、それぞれ、湿潤および乾燥サンプルの重量である。Kim et al.,341 Int.J.Pharm.35-43(2007)。吸収(%)およびEWC(%)の結果を表4に示す。
【0102】
酸素透過速度
酸素透過速度(OTR)を、Illinois 8001 Oxygen Permeation Analyzer(Illinois Instruments,Johnsburg,Illinois;ASTM 3985-05)を用いて測定した。円形の5cm2脱イオン水飽和サンプルを、15分の試験間隔の間中、37℃および80% RHの水和した環境ならびに37℃および50% RHのより乾燥した条件下で試験した。試験は、3つの連続した1%以内の酸素透過速度を記録して上首尾に終わった。酸素透過性は、ASTM 3985-05に従って1日当たりのcm3/m2で記録した。Apiezon Type T Grease(Manchester,UK)を用いてサンプルを5cm2マスクの間に密封した。単位厚さ当たりの酸素ガス透過速度および酸素透過速度の結果を表5に示す。
【0103】
水蒸気透過速度(WVTR)
Perm Cup(Gardner Co.,Pompano Beach,FL)を用いてASTM D 1653ウォーターカップ法Bに従ってWVTRを測定した。飽和および乾燥した25cm2直径サンプルを、上端の6mmまで満たされたカップの開口部に密閉し、73±1°F(22.8±0.6℃)および50±2%RHに維持した、温度と湿度を制御された環境に24時間置いた。装入されたカップ構成を、開始時および24時間後に0.1mgの精度で秤量した。温度および相対湿度は6時間ごとに確認し、水蒸気透過を1日当たりのg/m2でカップの重量減少により計算した。
【0104】
生分解
生分解性に用いたプロテアーゼは、タンパク質構造の複数の位置で絹フィブロインを区別せずに切断することが示された。Horan et al.26 Biomats.3385-93(2005);Li et al.,24 Biomats.357-65(2003)。6つの材料群からの三層の円形の3.5cmサンプルを、25±5mgの重さになるように手入れし(manicured)、3回20分の70%エタノール浴で滅菌し、PBSですすぎ、その後にpH7.4のPBS中1mg/mLプロテアーゼXIV(EC 3.4.24.31,5.6 U mg-1,Streptomyces griseus,Sigma,St.Louis,MO)の溶液6mL中37℃でインキュベートした。Jin et al.,15 Adv.Funct.Mater.1241-47(2005);Horan et al.,2005。対照サンプルを、酵素を含まないPBSに浸漬した。酵素および対照溶液は毎日補充した。サンプルを脱イオン水中で1時間すすいでから1、3、6、10、および14日後に生分解性を測定した。小型のへら、および4mLシリンジにに取り付けられた25ゲージ毛管を用いて、サンプルを培養ウェルプレートから指定された事前に秤量した秤量皿に移した。サンプルを無菌フード下RTにて24時間乾燥させた後、秤量して経時的重量減少率を決定した。線形回帰分析は、Minitab(登録商標)15.1.30(Minitab Inc.,State College,PA)を用いて行った。
【0105】
実施例4
材料
カイコ絹の繭(Tajima Shoji Co.,Yokohama,Japan)を調製して8重量%の絹溶液を生成した。Wang et al.,2006。4:1、3:1、2:1、3:2、7:6、および1:1w/w絹:PEO(900,000g/mol)比溶液を用いて6つの絹材料を調製した。電界紡糸の間に安定した流体の噴出を生じるための粘弾特性および表面張力特性に必要な最低限の7.2%絹/PEOポリマー濃度を維持するために、4:1、3:1、および2:1ブレンドは、5%のPEOを含有し、一方3:2、7:6、および1:1ブレンドは6%のPEOを含有した。
【0106】
溶液粘度を、#5スピンドルを69°Fで用いるBrookfield HATD粘度計(Brookfield Engineering Laboratories,Inc.,Stoughton,MA)によって、それぞれ、128、152、240、424、768、および1120mPa・S-1に等しいと決定した。
【0107】
この研究のために構築した電界紡糸装置は、高電圧電源(Gamma High Voltage Research ES-30P,Ormond Beach,Fl.)、60mlシリンジポンプ(Braintree Scientific 8000,Braintree,MA)、電位ステージおよび接地ステージ、1.5mmポリエチレン管および16ゲージ5.08cm鋼毛管を用いて、既に公開された手順に従った。
【0108】
絹/PEO溶液を、ポリエチレン管を通じてシリンジポンプから、電位プレートに挿入した12kV DC荷電鋼毛管にポンプで送った。電界紡糸繊維を、電位プレートよりもおよそ21cm低く設置され、毛管の先端の垂直落下線を越えておよそ2.5cmに位置する、アルミニウム箔で覆った接地ステージで収集した。
【0109】
上に挙げたた各々のブレンド比の絹/PEOの10および4.5mLバッチ溶液を用いて、それぞれ16.5および10cm絹マットを作成した。溶液は、室温にて(RT、20〜22℃)60%よりも低い相対湿度(RH)で電界紡糸した。絹/PEOマットを、90%MeOH溶液に20分間浸漬してβ-シート形成および結晶化を誘導した。72時間の3回の1L dH2O浴での浸出でPEOを抽出した。
【0110】
重量比方程式
に基づいて、各々のモデルの絹含量は、それぞれ86.5重量%、82.8重量%、76.2重量%、66.7重量%、60.9重量%、および57.1重量%であった。未処理の水溶性絹/PEOマットを、S87P13、S83P17、S76P24、S67P33、S61P39、およびS57P43(S87P13〜SP57P43)と名付けた。S87、S83、S76、S67、S61、およびS57(S87〜S13)と名付けられたマットは、メタノール処理しPEO抽出した絹マットを表す。
【0111】
乾燥法
非拘束および拘束乾燥技法を用いて、S87〜S57絹材料群の物理的特性を評価した。
【0112】
非拘束乾燥法では、飽和直径3.5cmのサンプルを10cmの電界紡糸キャストから打ち抜き、秤量紙(VWR,West Chester,PA)の間で手でプレスした後、ほぼ乾燥するまで表面対空気界面を最大化するためにポリスチレンペトリ皿の側壁に垂直に置いた。サンプルは、粘着を防ぐためにサンプルを定期的に再配置し、RTにて24時間乾燥させた。
【0113】
拘束乾燥技法を適用し、絹材料を、結晶化皿の口の上に掛けて付着している間に延伸力を受けながら乾燥させた。水浴から取り出し、直径16.5cmのS87〜S57サンプルを、3分の1をdH2Oで満たした125×65mm2結晶化皿の上に掛け、周囲条件下で一晩乾燥させた。サンプル重量(Mettler Toledo AB54-S/FAC,Columbus,OH)および厚さ(Ono Sokki EG225F Digital Indicator,Addison,IL;等半径点AA821;力25g)の測定値を、各々の絹系について記録した。
【0114】
材料の特徴付け
繊維形態、表面トポグラフィー、および断面の特性を、電界放射型走査電子顕微鏡(FE-SEM,JEOL JSM 740-1F,Tokyo,Japan)により1.5〜50倍の倍率で特徴付けた。繊維形態を、S87P13〜S57P43および拘束乾燥したS87〜S76サンプルセットについて評価した。表面トポグラフィーおよび断面特性を、拘束乾燥したS87〜S76サンプルについて査定した。断面のサンプルを、2×5mm2の断片に切断し、液体窒素中で急速冷凍し、ピンセットを用いて半分に割った。サンプルを、断面を上にしてカーボンテープの上に取り付けた。全てのサンプルを、Denton Vacuum Desk IV(Moorestown,NJ)を次の設定で用いて100Å Auでコーティングした:真空:80〜90mtorr、スパッタリング設定点:20〜30%、堆積時間:2分。
【0115】
S87〜S57サンプルの表面形態は、PSIA XE-150原子間力顕微鏡(AFM Santa Clara,CA)によって、Ultrasharp NSC16/AIBSプローブを非接触モードで用いて測定した(共振周波数:170kHz、バネ定数:45N・m-1)。表面粗さおよび三次元(3D)の特色を、XEI定量分析(Park Solutions Inc.,Santa Clara,CA)によりレンダリングした(rendered)。材料の多孔性は、5つの繊維層の最小深さ(約1μm)まで伸びる細孔により定義した。細孔のど径頻度分布は、50×50μm2の面積にわたって測定した。
【0116】
吸収
非拘束乾燥した、直径2.9cmのS87〜S57サンプルを事前に秤量し、次にdH2Oに24時間浸漬して膨潤平衡に達した。次に、サンプルを、Kimwipe(登録商標)組織の上に最小限の水がサンプル表面に観察されるまで優しく塗りつけた。次に、飽和したサンプルを秤量し、吸水および平衡含水率(EWC)を次の方程式により算出した。
式中、WwおよびWdは、それぞれ、湿潤および乾燥サンプルの重量である。
【0117】
酸素透過速度
酸素透過速度(OTR)を、Illinois 8001 Oxygen Permeation Analyzer(Illinois Instruments,Johnsburg,Illinois;ASTM 3985-05)を用いて測定した。OTRを、37℃および80% RHで15分間隔で試験した、円形の5cm2の水和したS87〜S57サンプルについて測定した。非拘束乾燥S87〜S57マットのOTRは、37℃および50%RHで測定した。酸素透過性は、ASTM 3985-05に従ってcm3・m-2・d-1で記録し、試験は3つの連続した1%以内のOTRを記録して上首尾に終わった。Apiezon Type T Grease(Manchester,UK)を用いてサンプルを5cm2マスクの間に密封した。
【0118】
水蒸気透過速度
水蒸気透過速度(WVTR)は、Perm Cup(Gardner Company,Pompano Beach Florida)を用いてASTM D 1653ウォーターカップ法Bに従って測定した。水和および拘束乾燥した円形の25cm2 S87〜S76サンプルで、上端の4mmまでdH2Oを満たしたPerm Cupの口を覆って密閉した。事前に秤量した集合体を、73±1°F(22.8±0.6℃)および50±2% RHに維持された環境に置き、24時間後に0.1mgの精度で再び秤量した。温度および相対湿度は6時間ごとに確認し、水蒸気透過を集合体の重量減少によりg・m-2・d-1で計算した。
【0119】
生分解
生分解性に用いたプロテアーゼは、タンパク質構造の複数の位置で絹フィブロインを区別せずに切断することが示された。三層の円形3.5cm非拘束乾燥S87〜S57サンプルを、25±5mgの重さになるように切断し、3回20分の70%エタノールへの浸漬によって滅菌し、リン酸緩衝生理食塩水(PBS,pH=7.4,Invitrogen 291,Carlsbad,CA.)ですすぎ、その後に1mg/mLプロテアーゼXIV(EC 3.4.24.31,5.6 U mg-1,Streptomyces griseus,Sigma,MO)の6mL PBS溶液中37℃でインキュベートした。対照サンプルを、酵素を含まないPBSに浸漬した。酵素および対照溶液は、毎日補充した。サンプルをdH2O中で1時間すすいでから1、3、6、10、および14日後に生分解性を測定した。先細りした端の平らなマイクロスパチュラ、および4mLシリンジにに取り付けられた25ゲージ毛管を用いて、サンプルを培養ウェルプレートから指定された事前に秤量した秤量皿に移した。サンプルを無菌フード下RTにて24時間乾燥させた後、秤量して経時的重量減少率を決定した。線形回帰分析は、Minitab(登録商標)15.1.30(Minitab Inc.,State College,Pennsylvania)を用いて行った。
【0120】
初期評価
絹/PEO電界紡糸プロセスを用いて、6つのS87P13〜S57P43材料系を全て直径16.5cmおよび10cmのマットに電界紡糸した。各々のS87〜S57サンプルの物理的特性を、水和状態および乾燥状態で評価した。dH2Oに浸漬されると、6つのS87〜S57材料群は全て、不透明に半透明の外観をもつ均一な立体構造を有し、絹のようなテクスチャーをもち柔軟であったが、取扱期間が延びると絹濃度の低下に対して材料の剪断の増加を提示した(図1B)。絹のようなテクスチャーとは、水分子が非晶質バルクマトリックスの全体にわたって吸収し可塑化するフィブロインの動的吸湿性を説明するために言及される。アミノ、ヒドロキシル、またはカルボキシル酸末端基との水素結合を形成することによるか、または、親水性ドメインの至る所に自由に分散することにより、高移動性水分子は、これらの飽和材料系の軟質の絹のようなテクスチャーを生成する運動エネルギーの最小化によって継続的に移行している。
【0121】
乾燥法1
水和した3.5cm S87〜S57サンプルは、正味の力の表面-表面疎水性平衡を実現し、層状の絹シートの分離および変位をもつ親水性の傾向を示すために、半分に折り重なることがある。しかし、24時間の乾燥時間の後、物理的な特徴はS87〜S57材料系で漸進的に変化した。絹濃度の低下に関連して、マットは、雪のように白い柔軟なウエハーのような構造から、制限された付着曲げ強度をもつ半透明褐色で超薄のフィルムのような材料に変わった(図2B)。
【0122】
タンパク質ポリマーのねじれた襞のあるβ-シート形成の特徴を示して、マトリックスは完全に平坦な配向で乾燥せず、S87およびS82群だけが元の円形の形状を保持した。その上、重鎖結晶領域間に交互に配置された非晶質ドメインの末端に位置するプロリンがある。プロリンは、脱水すると収縮し、繊維の縮小する能力を増加させることが示されている。これらの要素は、51.0±0.0%〜87.5±9.9%の間のそれらの表面積を失う、非拘束乾燥サンプルのねじれた、不規則な立体構造の一因となる(図13)。
【0123】
非拘束乾燥S87〜S57材料群についての厚さ測定値は、それぞれ、81.7±7.5μmから77.5±10.5μm、66.7±5.1μm、53.3±8.1μm、46.7±5.1μm、および30.0±6.4μmに直線的に下降した(図13)。PEO濃度は、電界紡糸プロセスの間の繊維の表面積およびバルク形態に直接影響を及ぼしうる。Wang et al.,2006。PEO濃度が増加するにつれて、絹繊維を形成するフィブロインミセルおよび小球構造のサイズは低下する。その上、素早く動く(whipping)帯電した流体噴出の内部の縦応力が、これらの小球構造を整列させ、10000倍まで伸張させる。Wang et al.,2006;Kowalewski et al.,2005;Reneker&Yarin,2008。その結果として、増加したPEO濃度で形成された絹繊維はバルク体積が低下し、それは非拘束乾燥S87〜S57材料群に観察される漸進的物理的な変化と相互に関連する。
【0124】
乾燥法2
水和した12.5cm S87〜S57サンプルは展開がより容易であった。層状のシート分離は、横変位なしに各々のマットの内部領域においてのみ観察された。恐らくこれは、これらのサンプルが、より大型のサンプルから打ち抜かれたものでなく、従ってサンプルのパラメータに結晶化領域を保持しているという事実に起因しうる。図3Bの拘束乾燥したマットを参照すると、S87およびS82サンプルは、結晶化皿に付着して乾燥し、元の表面積の98%を保持し、平坦で柔軟な白色の膜をもたらす。S67、S61、およびS57群は同様には起こらなかった。これらのサンプルが乾燥するときに、延伸力が繊維の降伏点伸びを越えて材料に応力を加え、構造的破壊および60%の表面積減少がもたらされた。材料の剪断は、一般に皿の縁で開始し、絹マットの内部領域に伝わった。S76サンプルは皿の縁から剪断されたが、たった12%しか表面積減少はなく、物理的特性はS87およびS82マトリックスに類似していた(図13)。
【0125】
拘束乾燥期間の間、表面基質で、かつタンパク質バルク領域の全体にわたって蒸発する水分子は、メチル基の収束および疎水性鎖相互作用を開始した。このカスケードは、自由体積喪失およびフィブロイン収縮を実現した。結晶化皿の縁に付着しているので、材料は拘束された状態になり、繊維を半径応力の方向に延伸および伸張させる。材料が各々のサンプル群で均質であり、12.5cmの拘束乾燥したS76およびS67サンプルの平均厚さがごく僅かであった(図13)ことを考えると、各々の材料群の剪断点は各々の繊維のバルク体積に依存したと思われた。
【0126】
材料の特徴付け
未処理のS87P13〜S57P43マットのFE-SEM顕微鏡写真を、図6Bに示す。全体的に、これらの電界紡糸絹/PEO繊維は、直径が200nm〜500nmの範囲に及び、構造全体にわたって均一に分布し、繊維ビーズ形成は、PEO濃度の低下とともに次第に顕著であった。Wang et al.,2006;Zhou et al.,2000;Huang et al.,2001。S87P13、S82P18、およびS76P24繊維は、直径が1μmをやや上回るものから700nmまでの範囲に及ぶ、ランダムに分散したビーズセグメントを有した。ビーズは、S67P33およびS61P39画像で最小化され、明確な直径200nm〜500nmの繊維の均一な分布を開示する。S57P43サンプルは、不均一で高密度のマット構造に移行する、不規則なおよび非円形の形状の繊維から明らかな、特有の形態を有した。高密度のマットの外観は、装置の接地ステージに集まるときに固化していない繊維の相分散に起因しうる。S57P43繊維の直径は、300〜500nmの範囲に及んだが、一方、混合繊維は700nm〜900nmの間で測定された。
【0127】
図14のFE-SEM画像は、拘束乾燥したS87〜S76材料群を示す。図14Aの表面トポグラフィーは、各々のモデルの全体にわたる繊維の高密度でランダムな分布を反映するが、より精密な評価は、拘束乾燥技法と一緒に起こる繊維の伸張および再整列の痕跡の増加を明らかにする。図14BのS87繊維は、限られた繊維伸張または整列を伴う緩んでねじれた外観を有する。対照的に、S82およびS76繊維は、伸張され整列されて、ウェブのような微小なテクスチャーを形成するようになる。図14CのS82マットは、ピンと張ったウェブ構造を形成し、末端が透明な共連続形態になっている隣接する繊維間の相分散の痕跡を示している。S76サンプルのウェブ構造は、ロープのような配置を形成する、明確な、伸張され整列された繊維の絡み合った網状構造により定義される。
【0128】
拘束乾燥技法により明示される繊維の整列および伸張は、絹フィブロインブロック共重合体設計で示される両親媒性特性に起因し得る。親水性領域の内部で可塑剤として働くので、水分子は、二次構造の移動および再整列を促進する、低凝集エネルギーの非晶質鎖間で分子間の動きを伝える。述べたように、蒸発は、疎水性鎖相互作用および自由体積喪失を作動させ、繊維を半径応力の方向に収縮および延伸させる。二次構造が伸張されるにつれて、両側性の鎖間層構造の整列を促進し、結晶化した鎖内のねじれた立体構造を制限する、非晶質軽鎖の末端のプロリンの折り畳みは制限されるようになる。支配的な鎖間疎水性相互作用も、非晶質絹Iから結晶性絹IIへの結晶性二次構造の移行に影響を及ぼす。これらの作用の組合せにより、機械的安定性および繊維軸に沿った曲げ強度を備えるマットが形成される(図8B)。降伏点伸びを越えると、重鎖と軽鎖の両方の非晶質二次構造の中で、かつそれに沿って、剪断変形が起こる。
【0129】
図14C中の整列したS82繊維と図14D中のS76繊維との間の肉眼的な相分散の外観は、水の可塑化特性に起因し得る。水和したカイコ絹フィルムの示差走査熱量測定は、20〜23重量%の吸水で脱水した絹フィブロインのガラス転移温度(Tg)が178℃から40℃まで低下したことを明らかにした。結晶性立体構造はバルク領域の全体にわたって吸収に影響を及ぼすが、各々の水和した群のEWCは、80重量%よりも大きかった(表7)、これは相当な親水性相互作用を示し、Tgの低下および妥当な相分散を予測する。その上、表面/液体、表面/気体、表面/表面の熱力学的転移の間に、親水性分子および疎水性分子が逆転するときに界面エネルギーが最小となる場合に、移動性材料の表面基質が提示された。延伸乾燥期間の間の相分散と親水性/疎水性表面交換の両方を考慮に入れると、非晶質二次構造は、混合繊維立体構造を作り出す界面の繊維の間に散在するようになる。また、繊維間の線形二次構造が、整列して、熱力学的に安定した結晶性βストランドを形成することも考えられる。
【0130】
(表7)dH2Oに24時間浸漬した直径2.9cmの非拘束乾燥S87〜S57サンプルについての平均吸収およびEWC測定値(±SD、n=6)
【0131】
図14Eの拘束乾燥したS87〜S76材料群の断面図は、低下する絹濃度に対して増加する繊維密度および凝集を開示する。S87繊維は、多数の大きな空間の隙間を有する水平のシートに凝集した。S82およびS76マットは、繊維束の増加および空間の隙間の漸進的減少を実証した。これらの所見は、絹体積に対するマット厚さの減少に一致する。図14BのS87〜S76顕微鏡写真は、これらの立体構造の全体にわたって相互に連絡する多孔性を示す。細孔のど径表面積は、それぞれ平均294nm2、201nm2、および103nm2である。下降する細孔径分布は、断面図に提示される繊維密度の増加に一致し、また、各々のS87、S82、およびS76材料群のビーディング(beading)および繊維直径に関する繊維の集合にも関連する(図9)。
【0132】
材料の表面粗さは、細胞の配向、接着、増殖、および遊走を制御する、正味の力の生体力学的平衡を促進する、応力/剪断のない平面による細胞接触誘導に影響を及ぼす。図15のS87三次元AFM画像に代表されるように、6つのS87〜S57非拘束乾燥サンプルは全て、二乗平均平方根粗さ値が500nmから1.4μmまでの範囲に及ぶ、明確な表面不規則性を提示するクラス3の表面トポグラフィーを実証した(n=3)。S87、S82、S76、S67、およびS57群は、それぞれ1.17±0.00μm、0.65±0.10μm、0.88±0.17μm、0.76±0.16μm、および0.78±0.01μmを測定する、ナノサイズの不規則性をもつ相対的に均一な粗さを有した。S61マットが、局地的な粗さが平均1.01±0.43μmの最も大きい偏差を有した。拘束乾燥したS87、S82、およびS76サンプルについての16×16μm2の面積に関するAFM粗さ値は、それぞれ0.66μm、0.36μm、および0.25μmとなった。低下した面積サイズおよび制限されたサンプルサイズを有しているが(n=1)、拘束乾燥した材料の粗さ値は、非拘束乾燥サンプルよりも少なくとも44%平坦である。拘束乾燥したサンプルは、絹濃度に対して粗さが直線的に低下するが、非拘束乾燥サンプルについては顕著な傾向はない。この所見は、非拘束乾燥サンプルのねじれた不規則さと比較して、拘束乾燥の繊維の伸張特性に一致する。
【0133】
吸収
創傷被覆材の吸収およびEWC特性は、細菌の栄養床を作り出す創傷浸出液の蓄積を制御する際に寄与する。表7を参照すると、吸収は、461%〜613%の範囲に及び、全ての絹材料群は平均して551±54%であった。非拘束乾燥したS87〜S57サンプルの平均乾燥重量は、22.5mgから13.6mgまで直線的に低下したが、EWCは84±1%で比較的一定のままであった。データは、吸水が各々の絹濃度の繊維直径、密度、および二次構造集合特性とは無関係であることを示唆する。同等な吸収およびEWC率は、主にこれらのマトリックスの全体にわたって相互に連絡する細孔の網状構造、および電界紡糸した絹繊維の多孔性に起因すると思われる。Wang et al.,2004。水分子がこれらの不織構造の格子間領域の中に、かつ、生体高分子のバルク領域への中に可塑化するときに、比例する量の膨潤が全ての材料群で起こる。
【0134】
酸素透過速度
酸素/二酸化炭素ガス交換を促進する包帯材は、創傷の酸度を低下させ、嫌気性細菌感染を抑制し、従って創傷治癒を促進する環境を作り出すと思われる。水和条件下(80% RH)で評価したS87〜S57サンプルは、それぞれ、25000cm-3・m-2・d-1から7800cm-3・m-2・d-1までの平均OTRを提示した(表8)。絹濃度の低下に対して(Respective to)、OTRの線形の減少は、各々の絹材料群の減少するマット厚さ、繊維サイズ、細孔のど径、および繊維密度の増加に起因した。S87、S82、およびS76材料群についてのOTR線形回帰分析は、96%の予測二乗相関係数分散および4800cm cm-3・m-2・d-1の低下するOTR率を有した。対照的に、乾燥条件(50%RH)下で試験した水和したS87〜S57サンプルは、1回の試験間隔の終了よりも前に100000cm-3・m-2・d-1の分析器閾値を上回った。これらの結果は、乾燥またはほぼ乾燥状態ではこれらの多孔質材料は、水和した不織織物の組織間隙およびバルク領域に存在する水分子を通じて酸素が拡散される場合よりも大きい酸素透過速度を有することを反映する。
【0135】
(表8)8001 OPA(Illinois Instruments,ASTM 3985-05)およびPerm Cup(Gardner Company,ASTM D1653)で測定したS87〜S57材料群についての平均O2ガス透過速度(GTR)、WVTRおよび厚さ(±SD、n=3)
a)拘束乾燥したS87〜S76絹材料;b)水和したS87〜S76絹材料。
【0136】
水蒸気透過速度
全層創傷被覆材の水蒸気透過性は、体液の蒸発を制御し、創傷部位の感染を抑制する際に役割を果たす。S67、S61、およびS57マトリックスについてのWVTRを確かめる努力は、乾燥段階中の材料変形のために不成功に終わった。水和し拘束乾燥したS87、S82、およびS76材料群についてのWVTRは、それぞれ、平均1977±35g・m-2・d-1および1469±81g・m-2・d-1であった(表8)。液体/ガス/膜/ガス界面と比べて直接の液体/膜/ガス界面の界面エネルギー最小化の増加に起因して、飽和したサンプルが乾燥したサンプルよりも優れていた。これは、蒸発の加速を促進する生体材料表面/ガス界面に水分子が都合よく存在することを可能にする、これらの材料系の親水性の特性に起因しうる。水和し乾燥したS87〜S76材料群についてのWVTR回帰分析は、97%および99%の二乗相関係数分散での線形当てはめ、および81g・m-2・d-1および41g・m-2・d-1の下降するWVTRを予測した。これらのごく僅かなWVTRの差は、材料の厚さ、繊維サイズ、繊維密度、および多孔性に起因しうる。
【0137】
生分解性
時間放出生物療法での上皮化を促進するための、これらの絹材料の酵素生分解を評価した。インビトロでの生分解性研究は、全てのS87〜S57材料群について1日後に平均22.6±3.4%の分解および14日後に74.0±8.8%までの材料損失の、線形の分解傾向を明らかにした(図16A)。データは、6日までは、全てのブレンドについての分解率は48.2±4.6%で比較的近かったことを示唆する。対照的に、10日後に、S87(78%)とS57(51%)サンプルの間で27%の重量減少差が記録された。14日後、酵素分解は、S87〜S57サンプルに関して、それぞれ85.6±3.8から62.5±5.2%に至る範囲に及んだ。目視検査によって、全ての材料系が最初の6日で表面浸食により形態学的に分解された。10日後、S87およびS82マトリックスは、材料のほころび、断片化、および微粒子破片への崩壊をもたらす、繊維の切断の増加を実証した。S87およびS82サンプルの形態的分解は、これらのマトリックスの増加した繊維サイズ、マット多孔性、および繊維密度特性の低下により、内部マット構造に酵素が接近することに起因した。
【0138】
図16Bの散布図を参照すると、各々の材料群に関して実行した対数変換は、3日の分解時点の直前に全ての材料群につての明確な転移点を明らかにした。次に、回帰分析を各々の材料群について全ての時点、0日から3日まで、および3日〜14日の間の時点に関して行った。図16Cに示されるように、全ての材料群についての分解の傾きは、3日後に実質的に変わった。0日から3日まで、酵素分解速度は平均-11.7±0.9であった。3日後から14日まで、分解の速度は-3.4±1.1で安定した。初期の加速した分解速度は、潜在性の結晶領域と比較して表面基質の非晶質領域の分解に起因しうる。
【0139】
全層熱傷被覆材のための材料は、病原性微生物に対する不透過性の障壁をもたらし、創傷部位の浮腫および脱水症を管理し、および同時刻に合わせた抗生物質、免疫学的、および組織再生生物治療を支援する能力を含む、多くの有用な特性を提示することができる。さまざまな電界紡糸した絹/PEOが設計され、これらのPEO抽出絹材料系の立体構造特性および生体機能特性が、全層創傷被覆材としての有用性について研究された。拘束乾燥技法を用いることにより、絹濃度が、材料の厚さ、繊維密度、繊維配向、相分散、および多孔性を含む材料構造特性において役割を果たすことが見出された。この乾燥技法によって、S87、S82、およびS76の絹割合の材料群が、最小限の表面積減少で平坦な柔軟な膜のような立体構造に変化した、それは持続可能な有効期間をもつ流通可能な創傷被覆材に理想的である。
【0140】
安定した恒常状態を維持するために、正常な皮膚は体液を204g・m-2・d-1の速度で透過させる。全層肉芽創の蒸発水損失は5138g・m-2・d-1である。2000〜2500g・m-2・d-1の水透過速度を有する全層創傷被覆材が、適切な水分レベルを許容すると同時に過剰な脱水を防ぐことが決定された。表9の吸収、EWCおよび水蒸気透過特性を参照すると、拘束乾燥したS87〜S76材料は、提案されたスポンジのような天然キトサン創傷被覆材に対して比較的機能した。キトサン/ポロキサマー包帯材候補は、強い吸収およびEWC特性を提示したが、これらの試験は37℃のPBS(pH=7.4)で実施された。その上、S87〜S76材料系と、不斉および二層キトサン材料との間の酸素透過性の相違は、試験条件に起因するものであった。キトサン誘導体のOTRは、0%RHの乾燥条件下で試験されたが、dH2O飽和絹材料は、エキソバサーティングな(exovasating)創傷環境を模倣するために80%RHの水和環境下で評価された。
【0141】
(表9)キトサン誘導体に基づく創傷被覆材とS87〜S76材料系との間の平均吸収、EWC、WVTR、O2 GTR、および厚さの比較
a)酸素透過分析は35℃および0%RHで行った;
b)吸収およびEWCはPBS(pH=7.4)で測定した。
【0142】
正常なヒト皮膚は約21日で再生する。時間放出生物療法を送達する多層創傷被覆材を用いることによって全層創傷の上皮化を促進するために、これらの絹材料の酵素分解時間を評価した。結果は、14日後、S87〜S76マトリックスは80%分解したことを明らかにし、これは同じ時間の間に82%分解したリゾチームに曝露したキトサン/ポロキサマー包帯材と遜色がなかった。対照的に、PLGA/PLLA(90/10)コ-ブロックポリマー系はPBS中14日後に20%の分解率しかなかった。酵素レベルは著しく変動するので、インビボでの分解率も考慮されるべきである。絹生体材料は、材料形式、位置、および関連する変数によって、インビボで数週から数年で分解し得ることが示された。
【図1A】
【図1B】
【図2A】
【図2B】
【図3A】
【図3B】
【特許請求の範囲】
【請求項1】
以下の段階を含む、絹マットを製造するための方法:
ポリエチレンオキシド(PEO)と絹フィブロイン水溶液をブレンドする段階;
前記ブレンドした溶液を電界紡糸する段階であって、それにより絹タンパク質/PEOブレンドマットを形成する、段階;および
前記電界紡糸した絹マットを拘束乾燥する段階。
【請求項2】
電界紡糸した絹マットをアルコールで処理する段階をさらに含む、請求項1記載の方法。
【請求項3】
PEOを絹マットから抽出する段階をさらに含む、請求項1または2記載の方法。
【請求項4】
少なくとも一つの活性物質を絹マットに埋め込む段階をさらに含む、請求項1〜3のいずれか一項記載の方法。
【請求項5】
活性物質が、細胞、タンパク質、ペプチド、核酸、核酸類似体、ヌクレオチドまたはオリゴヌクレオチド、ペプチド核酸、アプタマー、抗体またはその断片もしくは部分、抗原またはエピトープ、ホルモン、ホルモンアンタゴニスト、成長因子または組換え成長因子およびその断片および変異体、細胞接着メディエーター、サイトカイン、酵素、抗生物質または抗菌性化合物、ウイルス、毒素、プロドラッグ、化学療法剤、小分子、薬物、ならびにそれらの組合せからなる群より選択される治療剤または生物材料である、請求項4記載の方法。
【請求項6】
活性物質が、前駆細胞または幹細胞、平滑筋細胞、骨格筋細胞、心筋細胞、上皮細胞、内皮細胞、尿路上皮細胞、線維芽細胞、筋芽細胞、口腔細胞、軟骨細胞、軟骨芽細胞、骨芽細胞、破骨細胞、ケラチン生成細胞、腎尿細管細胞、腎基底膜細胞、外皮細胞、骨髄細胞、肝細胞、胆管細胞、膵島細胞、甲状腺細胞、上皮小体細胞、副腎細胞、視床下部細胞、下垂体細胞、卵巣細胞、精巣細胞、唾液腺細胞、脂肪細胞、前駆細胞、およびそれらの組合せからなる群より選択される細胞である、請求項5記載の方法。
【請求項7】
活性物質が、細胞増殖培地をさらに含む、請求項6記載の方法。
【請求項8】
活性物質が、抗生物質である、請求項6記載の方法。
【請求項9】
ポリエチレンオキシド(PEO)と絹フィブロイン水溶液をブレンドする段階;
ブレンドした溶液を電界紡糸する段階であって、それにより絹タンパク質/PEOブレンドマットを形成する、段階;および
電界紡糸した絹マットを拘束乾燥する段階
を含む方法から調製された絹材料。
【請求項10】
ポリエチレンオキシド(PEO)を、少なくとも一つの活性物質を含む絹フィブロイン水溶液とブレンドする段階;
ブレンドした溶液を電界紡糸する段階であって、それにより該活性物質を封入する絹タンパク質/PEOブレンドマットを形成する、段階;および
該活性物質を封入する電界紡糸した絹マットを拘束乾燥する段階
を含む方法から調製された、創傷に巻いて創傷治癒を促進するための少なくとも一つの活性物質を封入する絹材料。
【請求項11】
活性物質が、細胞、タンパク質、ペプチド、核酸、核酸類似体、ヌクレオチドまたはオリゴヌクレオチド、ペプチド核酸、アプタマー、抗体またはその断片もしくは部分、抗原またはエピトープ、ホルモン、ホルモンアンタゴニスト、成長因子または組換え成長因子およびその断片および変異体、細胞接着メディエーター、サイトカイン、酵素、抗生物質または抗菌性化合物、ウイルス、毒素、プロドラッグ、化学療法剤、小分子、薬物、ならびにそれらの組合せからなる群より選択される治療剤または生物材料である、請求項10記載の絹材料。
【請求項12】
活性物質が、前駆細胞または幹細胞、平滑筋細胞、骨格筋細胞、心筋細胞、上皮細胞、内皮細胞、尿路上皮細胞、線維芽細胞、筋芽細胞、口腔細胞、軟骨細胞、軟骨芽細胞、骨芽細胞、破骨細胞、ケラチン生成細胞、腎尿細管細胞、腎基底膜細胞、外皮細胞、骨髄細胞、肝細胞、胆管細胞、膵島細胞、甲状腺細胞、上皮小体細胞、副腎細胞、視床下部細胞、下垂体細胞、卵巣細胞、精巣細胞、唾液腺細胞、脂肪細胞、前駆細胞、およびそれらの組合せからなる群より選択される細胞である、請求項11記載の絹材料。
【請求項13】
活性物質が、細胞増殖培地をさらに含む、請求項12記載の絹材料。
【請求項14】
活性物質が、抗生物質である、請求項12記載の絹材料。
【請求項15】
電界紡糸絹マットが、アルコールでさらに処理される、請求項9〜14のいずれか一項記載の絹材料。
【請求項16】
PEOが、電界紡糸絹マットから抽出される、請求項9〜15のいずれか一項記載の絹材料。
【請求項17】
電界紡糸絹マットの厚さが約20μm〜約80μmである、約50重量%〜約100重量%の範囲に及ぶ絹フィブロインタンパク質を含む電界紡糸した絹材料。
【請求項18】
電界紡糸絹マット中の絹フィブロインタンパク質の含量が、約75重量%〜約90重量%の範囲に及ぶ、請求項17記載の電界紡糸絹材料。
【請求項19】
電界紡糸絹マット中のPEOの含量が、約0重量%〜約50重量%の範囲に及ぶ、電界紡糸絹マット中にポリエチレンオキシド(PEO)のブレンドをさらに含む、請求項17または18記載の電界紡糸絹材料。
【請求項20】
電界紡糸絹マット中のPEOの含量が、約10重量%〜約25重量%の範囲に及ぶ、請求項19記載の電界紡糸絹材料。
【請求項21】
少なくとも一つの活性物質をさらに含む、請求項17〜20のいずれか一項記載の絹材料。
【請求項22】
活性物質が、細胞、タンパク質、ペプチド、核酸、核酸類似体、ヌクレオチドまたはオリゴヌクレオチド、ペプチド核酸、アプタマー、抗体またはその断片もしくは部分、抗原またはエピトープ、ホルモン、ホルモンアンタゴニスト、成長因子または組換え成長因子およびその断片および変異体、細胞接着メディエーター、サイトカイン、酵素、抗生物質または抗菌性化合物、ウイルス、毒素、プロドラッグ、化学療法剤、小分子、薬物、およびそれらの組合せからなる群より選択される治療剤または生物材料である、請求項21記載の絹材料。
【請求項23】
絹マットの厚さが約20〜30μmである、請求項17〜22のいずれか一項記載の絹材料。
【請求項24】
絹マットが、平均して約0.1〜約0.3μmの細孔のど径表面積をもつ、相互接続した細孔を有する、請求項17〜23のいずれか一項記載の絹材料。
【請求項25】
得られる絹マットの吸水量が約460%よりも大きい、請求項9〜24のいずれか一項記載の絹材料。
【請求項26】
得られる絹マットの平衡含水率が約82%よりも大きい、請求項9〜25のいずれか一項記載の絹材料。
【請求項27】
得られる絹マットの酸素透過速度が約15460cm3/m2/日よりも大きい、請求項9〜26のいずれか一項記載の絹材料。
【請求項28】
得られる絹マットの水蒸気透過速度が約1934g/m2/日よりも大きい、請求項9〜27のいずれか一項記載の絹材料。
【請求項29】
絹フィブロインタンパク質、および任意で少なくとも一つの活性物質を含む、少なくとも一つの電界紡糸絹マットに創傷を接触させる段階を含む、創傷治癒を促進する方法であって;
前記絹フィブロインタンパク質が約50重量%〜約90重量%の範囲に及び、
前記絹マットの厚さが約20μm〜約80μmであり;
前記絹マットが、約460%よりも大きい吸水量か、または約82%よりも大きい平衡含水率を有し;かつ
得られる絹マットの酸素透過速度が約15460cm3/m2/日よりも大きい、前記方法。
【請求項30】
絹フィブロインタンパク質が、約75重量%〜約90重量%の範囲に及ぶ、請求項29記載の方法。
【請求項31】
絹フィブロインタンパク質、ポリエチレンオキシド(PEO)、および任意で少なくとも一つの活性物質を含む、少なくとも一つの電界紡糸絹マットに創傷を接触させる段階を含む、創傷治癒を促進する方法であって;
絹/PEOブレンド比が、約4:1〜約2:1であり;
前記絹マットの厚さが約20μm〜約80μmであり;
前記絹マットが、約460%よりも大きい吸水量か、または約82%よりも大きい平衡含水率を有し;かつ
得られる絹マットの酸素透過速度が約15460cm3/m2/日よりも大きい、前記方法。
【請求項32】
絹マットの水蒸気透過速度が、約1934g/m2/日よりも大きい、請求項29〜31のいずれか一項記載の方法。
【請求項33】
活性物質が、細胞、タンパク質、ペプチド、核酸、核酸類似体、ヌクレオチドまたはオリゴヌクレオチド、ペプチド核酸、アプタマー、抗体またはその断片もしくは部分、抗原またはエピトープ、ホルモン、ホルモンアンタゴニスト、成長因子または組換え成長因子およびその断片および変異体、細胞接着メディエーター、サイトカイン、酵素、抗生物質または抗菌性化合物、ウイルス、毒素、プロドラッグ、化学療法剤、小分子、薬物、およびそれらの組合せからなる群より選択される治療剤または生物材料である、請求項29〜32のいずれか一項記載の方法。
【請求項1】
以下の段階を含む、絹マットを製造するための方法:
ポリエチレンオキシド(PEO)と絹フィブロイン水溶液をブレンドする段階;
前記ブレンドした溶液を電界紡糸する段階であって、それにより絹タンパク質/PEOブレンドマットを形成する、段階;および
前記電界紡糸した絹マットを拘束乾燥する段階。
【請求項2】
電界紡糸した絹マットをアルコールで処理する段階をさらに含む、請求項1記載の方法。
【請求項3】
PEOを絹マットから抽出する段階をさらに含む、請求項1または2記載の方法。
【請求項4】
少なくとも一つの活性物質を絹マットに埋め込む段階をさらに含む、請求項1〜3のいずれか一項記載の方法。
【請求項5】
活性物質が、細胞、タンパク質、ペプチド、核酸、核酸類似体、ヌクレオチドまたはオリゴヌクレオチド、ペプチド核酸、アプタマー、抗体またはその断片もしくは部分、抗原またはエピトープ、ホルモン、ホルモンアンタゴニスト、成長因子または組換え成長因子およびその断片および変異体、細胞接着メディエーター、サイトカイン、酵素、抗生物質または抗菌性化合物、ウイルス、毒素、プロドラッグ、化学療法剤、小分子、薬物、ならびにそれらの組合せからなる群より選択される治療剤または生物材料である、請求項4記載の方法。
【請求項6】
活性物質が、前駆細胞または幹細胞、平滑筋細胞、骨格筋細胞、心筋細胞、上皮細胞、内皮細胞、尿路上皮細胞、線維芽細胞、筋芽細胞、口腔細胞、軟骨細胞、軟骨芽細胞、骨芽細胞、破骨細胞、ケラチン生成細胞、腎尿細管細胞、腎基底膜細胞、外皮細胞、骨髄細胞、肝細胞、胆管細胞、膵島細胞、甲状腺細胞、上皮小体細胞、副腎細胞、視床下部細胞、下垂体細胞、卵巣細胞、精巣細胞、唾液腺細胞、脂肪細胞、前駆細胞、およびそれらの組合せからなる群より選択される細胞である、請求項5記載の方法。
【請求項7】
活性物質が、細胞増殖培地をさらに含む、請求項6記載の方法。
【請求項8】
活性物質が、抗生物質である、請求項6記載の方法。
【請求項9】
ポリエチレンオキシド(PEO)と絹フィブロイン水溶液をブレンドする段階;
ブレンドした溶液を電界紡糸する段階であって、それにより絹タンパク質/PEOブレンドマットを形成する、段階;および
電界紡糸した絹マットを拘束乾燥する段階
を含む方法から調製された絹材料。
【請求項10】
ポリエチレンオキシド(PEO)を、少なくとも一つの活性物質を含む絹フィブロイン水溶液とブレンドする段階;
ブレンドした溶液を電界紡糸する段階であって、それにより該活性物質を封入する絹タンパク質/PEOブレンドマットを形成する、段階;および
該活性物質を封入する電界紡糸した絹マットを拘束乾燥する段階
を含む方法から調製された、創傷に巻いて創傷治癒を促進するための少なくとも一つの活性物質を封入する絹材料。
【請求項11】
活性物質が、細胞、タンパク質、ペプチド、核酸、核酸類似体、ヌクレオチドまたはオリゴヌクレオチド、ペプチド核酸、アプタマー、抗体またはその断片もしくは部分、抗原またはエピトープ、ホルモン、ホルモンアンタゴニスト、成長因子または組換え成長因子およびその断片および変異体、細胞接着メディエーター、サイトカイン、酵素、抗生物質または抗菌性化合物、ウイルス、毒素、プロドラッグ、化学療法剤、小分子、薬物、ならびにそれらの組合せからなる群より選択される治療剤または生物材料である、請求項10記載の絹材料。
【請求項12】
活性物質が、前駆細胞または幹細胞、平滑筋細胞、骨格筋細胞、心筋細胞、上皮細胞、内皮細胞、尿路上皮細胞、線維芽細胞、筋芽細胞、口腔細胞、軟骨細胞、軟骨芽細胞、骨芽細胞、破骨細胞、ケラチン生成細胞、腎尿細管細胞、腎基底膜細胞、外皮細胞、骨髄細胞、肝細胞、胆管細胞、膵島細胞、甲状腺細胞、上皮小体細胞、副腎細胞、視床下部細胞、下垂体細胞、卵巣細胞、精巣細胞、唾液腺細胞、脂肪細胞、前駆細胞、およびそれらの組合せからなる群より選択される細胞である、請求項11記載の絹材料。
【請求項13】
活性物質が、細胞増殖培地をさらに含む、請求項12記載の絹材料。
【請求項14】
活性物質が、抗生物質である、請求項12記載の絹材料。
【請求項15】
電界紡糸絹マットが、アルコールでさらに処理される、請求項9〜14のいずれか一項記載の絹材料。
【請求項16】
PEOが、電界紡糸絹マットから抽出される、請求項9〜15のいずれか一項記載の絹材料。
【請求項17】
電界紡糸絹マットの厚さが約20μm〜約80μmである、約50重量%〜約100重量%の範囲に及ぶ絹フィブロインタンパク質を含む電界紡糸した絹材料。
【請求項18】
電界紡糸絹マット中の絹フィブロインタンパク質の含量が、約75重量%〜約90重量%の範囲に及ぶ、請求項17記載の電界紡糸絹材料。
【請求項19】
電界紡糸絹マット中のPEOの含量が、約0重量%〜約50重量%の範囲に及ぶ、電界紡糸絹マット中にポリエチレンオキシド(PEO)のブレンドをさらに含む、請求項17または18記載の電界紡糸絹材料。
【請求項20】
電界紡糸絹マット中のPEOの含量が、約10重量%〜約25重量%の範囲に及ぶ、請求項19記載の電界紡糸絹材料。
【請求項21】
少なくとも一つの活性物質をさらに含む、請求項17〜20のいずれか一項記載の絹材料。
【請求項22】
活性物質が、細胞、タンパク質、ペプチド、核酸、核酸類似体、ヌクレオチドまたはオリゴヌクレオチド、ペプチド核酸、アプタマー、抗体またはその断片もしくは部分、抗原またはエピトープ、ホルモン、ホルモンアンタゴニスト、成長因子または組換え成長因子およびその断片および変異体、細胞接着メディエーター、サイトカイン、酵素、抗生物質または抗菌性化合物、ウイルス、毒素、プロドラッグ、化学療法剤、小分子、薬物、およびそれらの組合せからなる群より選択される治療剤または生物材料である、請求項21記載の絹材料。
【請求項23】
絹マットの厚さが約20〜30μmである、請求項17〜22のいずれか一項記載の絹材料。
【請求項24】
絹マットが、平均して約0.1〜約0.3μmの細孔のど径表面積をもつ、相互接続した細孔を有する、請求項17〜23のいずれか一項記載の絹材料。
【請求項25】
得られる絹マットの吸水量が約460%よりも大きい、請求項9〜24のいずれか一項記載の絹材料。
【請求項26】
得られる絹マットの平衡含水率が約82%よりも大きい、請求項9〜25のいずれか一項記載の絹材料。
【請求項27】
得られる絹マットの酸素透過速度が約15460cm3/m2/日よりも大きい、請求項9〜26のいずれか一項記載の絹材料。
【請求項28】
得られる絹マットの水蒸気透過速度が約1934g/m2/日よりも大きい、請求項9〜27のいずれか一項記載の絹材料。
【請求項29】
絹フィブロインタンパク質、および任意で少なくとも一つの活性物質を含む、少なくとも一つの電界紡糸絹マットに創傷を接触させる段階を含む、創傷治癒を促進する方法であって;
前記絹フィブロインタンパク質が約50重量%〜約90重量%の範囲に及び、
前記絹マットの厚さが約20μm〜約80μmであり;
前記絹マットが、約460%よりも大きい吸水量か、または約82%よりも大きい平衡含水率を有し;かつ
得られる絹マットの酸素透過速度が約15460cm3/m2/日よりも大きい、前記方法。
【請求項30】
絹フィブロインタンパク質が、約75重量%〜約90重量%の範囲に及ぶ、請求項29記載の方法。
【請求項31】
絹フィブロインタンパク質、ポリエチレンオキシド(PEO)、および任意で少なくとも一つの活性物質を含む、少なくとも一つの電界紡糸絹マットに創傷を接触させる段階を含む、創傷治癒を促進する方法であって;
絹/PEOブレンド比が、約4:1〜約2:1であり;
前記絹マットの厚さが約20μm〜約80μmであり;
前記絹マットが、約460%よりも大きい吸水量か、または約82%よりも大きい平衡含水率を有し;かつ
得られる絹マットの酸素透過速度が約15460cm3/m2/日よりも大きい、前記方法。
【請求項32】
絹マットの水蒸気透過速度が、約1934g/m2/日よりも大きい、請求項29〜31のいずれか一項記載の方法。
【請求項33】
活性物質が、細胞、タンパク質、ペプチド、核酸、核酸類似体、ヌクレオチドまたはオリゴヌクレオチド、ペプチド核酸、アプタマー、抗体またはその断片もしくは部分、抗原またはエピトープ、ホルモン、ホルモンアンタゴニスト、成長因子または組換え成長因子およびその断片および変異体、細胞接着メディエーター、サイトカイン、酵素、抗生物質または抗菌性化合物、ウイルス、毒素、プロドラッグ、化学療法剤、小分子、薬物、およびそれらの組合せからなる群より選択される治療剤または生物材料である、請求項29〜32のいずれか一項記載の方法。
【図4】

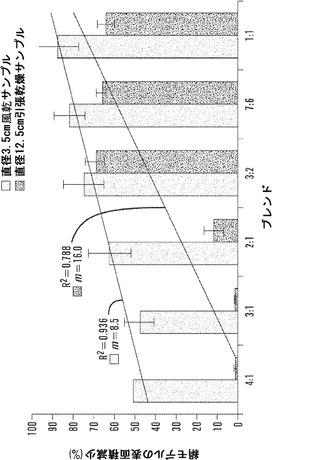
【図5】
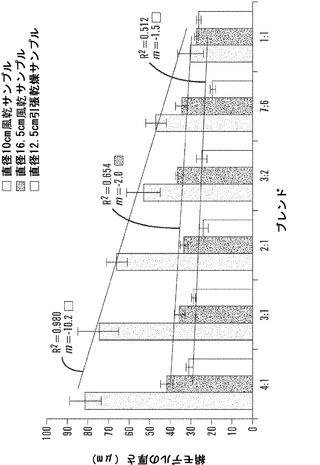
【図6A】
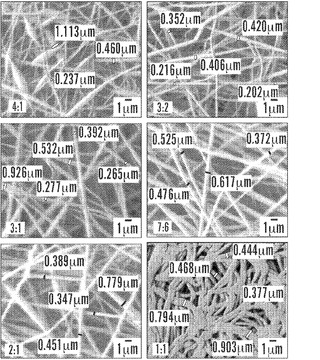
【図6B】

【図7A】

【図7B】

【図7C】
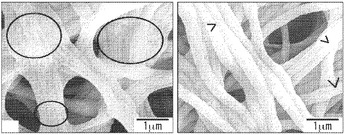
【図7D】
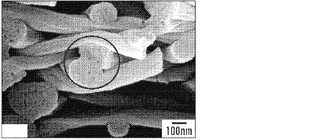
【図7E】

【図8】
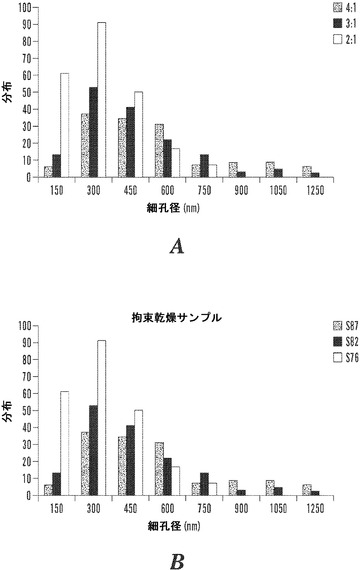
【図9】
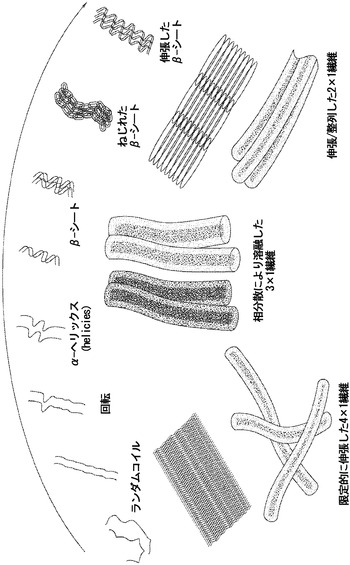
【図10】
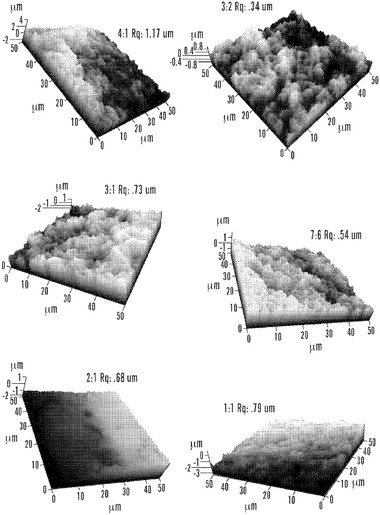
【図11】
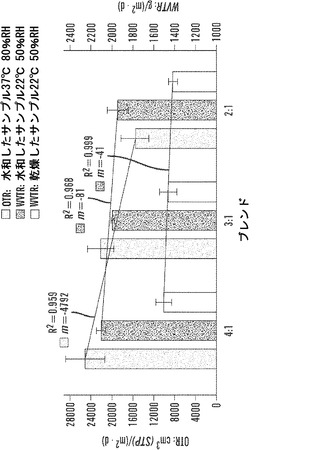
【図12A】
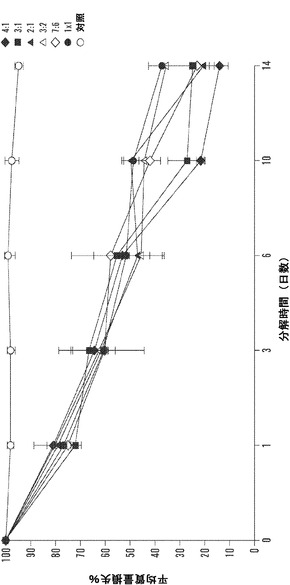
【図12B】
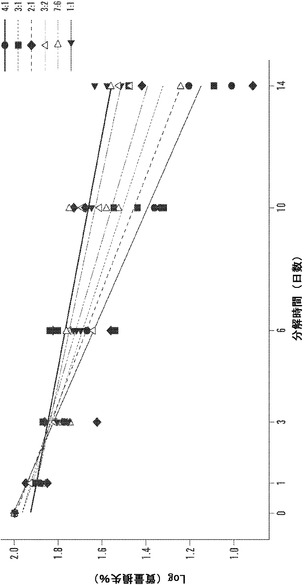
【図12C】
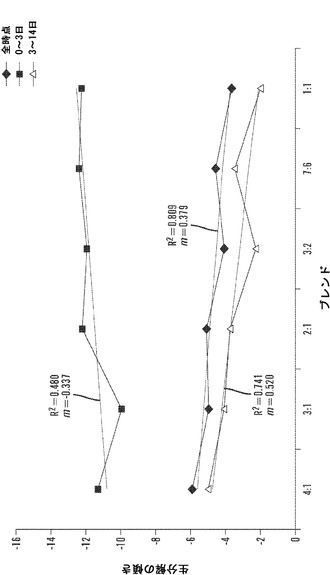
【図13】
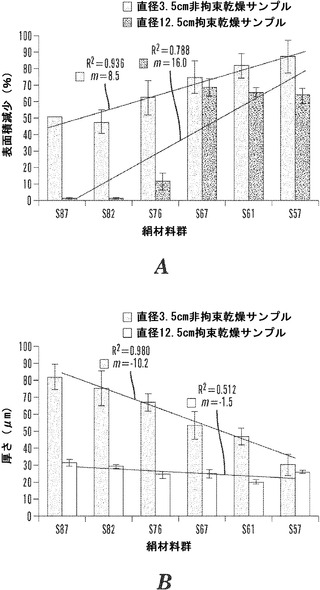
【図14A】

【図14B】

【図14C】

【図14D】

【図14E】

【図15】
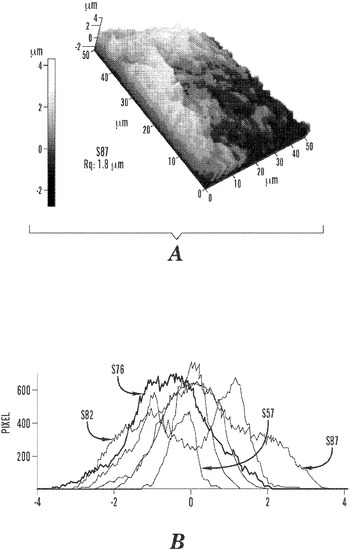
【図16A】
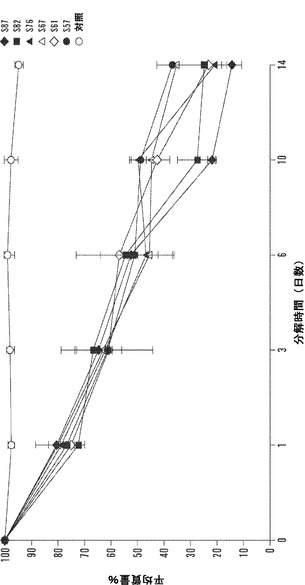
【図16B】
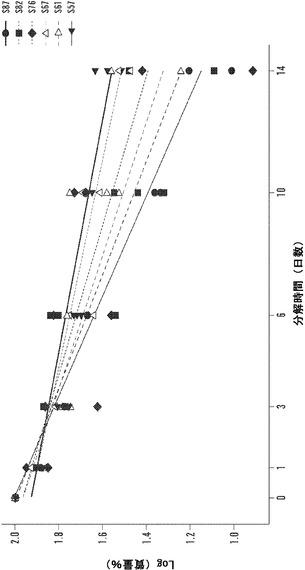
【図16C】
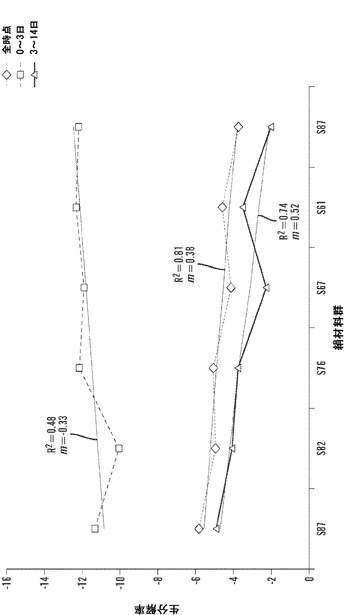
【図5】
【図6A】
【図6B】
【図7A】
【図7B】
【図7C】
【図7D】
【図7E】
【図8】
【図9】
【図10】
【図11】
【図12A】
【図12B】
【図12C】
【図13】
【図14A】
【図14B】
【図14C】
【図14D】
【図14E】
【図15】
【図16A】
【図16B】
【図16C】
【公表番号】特表2012−533354(P2012−533354A)
【公表日】平成24年12月27日(2012.12.27)
【国際特許分類】
【出願番号】特願2012−520744(P2012−520744)
【出願日】平成22年7月14日(2010.7.14)
【国際出願番号】PCT/US2010/041953
【国際公開番号】WO2011/008842
【国際公開日】平成23年1月20日(2011.1.20)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.TEFLON
【出願人】(510300430)タフツ ユニバーシティー/トラスティーズ オブ タフツ カレッジ (12)
【出願人】(512010812)ユニバーシティ オブ マサチューセッツ (1)
【Fターム(参考)】
【公表日】平成24年12月27日(2012.12.27)
【国際特許分類】
【出願日】平成22年7月14日(2010.7.14)
【国際出願番号】PCT/US2010/041953
【国際公開番号】WO2011/008842
【国際公開日】平成23年1月20日(2011.1.20)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.TEFLON
【出願人】(510300430)タフツ ユニバーシティー/トラスティーズ オブ タフツ カレッジ (12)
【出願人】(512010812)ユニバーシティ オブ マサチューセッツ (1)
【Fターム(参考)】
[ Back to top ]