
加速器質量分析による検体の定量化
本発明は、試験サンプル内の、AMS同位体で標識化した検体の量を測定する方法に用いる較正方法を提供する。前記較正方法は、
(i)複数の較正サンプルを提供するために、前記検体にも標識化していない対照検体にも汚染されていない複数のサンプルを、既知量の前記対照検体および、量Cの検体へ接触させるステップにおいて、前記較正サンプルが、既知量でかつ量Cとは異なる量の対照検体含むことを特徴とするステップと、
(ii)複数のサンプルへそれぞれ加えた検体の量Cを、AMSにより測定するステップと、
(iii)複数の精製サンプルを提供するために、複数のサンプル内で、検体および対照検体を、他の化学種から分離するステップと、
(iv)前記精製サンプル内の検体の量Aを、AMSにより測定するステップと、
(v)前記精製サンプル内の対照検体の量Bを、AMSにより測定するステップとを
含むことを特徴とする。
(i)複数の較正サンプルを提供するために、前記検体にも標識化していない対照検体にも汚染されていない複数のサンプルを、既知量の前記対照検体および、量Cの検体へ接触させるステップにおいて、前記較正サンプルが、既知量でかつ量Cとは異なる量の対照検体含むことを特徴とするステップと、
(ii)複数のサンプルへそれぞれ加えた検体の量Cを、AMSにより測定するステップと、
(iii)複数の精製サンプルを提供するために、複数のサンプル内で、検体および対照検体を、他の化学種から分離するステップと、
(iv)前記精製サンプル内の検体の量Aを、AMSにより測定するステップと、
(v)前記精製サンプル内の対照検体の量Bを、AMSにより測定するステップとを
含むことを特徴とする。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、対象のサンプル中の、特定の同位体標識検体の量を決定する正確性を改善する方法に関する。特に、本発明は、加速器質量分析(AMS)を用いた検体の定量化時の内部標準化を可能とする。
【背景技術】
【0002】
代表的には液体クロマトグラフィ質量分析(LC−MS)を用いて行う生物分析は、通常、臨床研究(フェーズI、II、およびIII)中、および動物サンプルによる毒物研究中にもしばしば行われる。
【0003】
LC−MSにおいて、従来のHPLC手法と、質量分析器のイオン源内を通過したHPLCからの溶離剤を用いて検体を分離する。ここで、複数あるイオン技術(例えば電気スプレーもしくは大気圧イオン化(API)など)のうちの1つにより検体をイオン化する。イオン化エネルギーによっては、検体の分子分裂が生じることがある。正もしくは負のどちらかに帯電したイオンは、その荷電のためにイオン源から引き出される。イオンの質量(m)を荷電量(z)で割った値に従って、これらを磁場中もしくは四重極磁場中で分離する。多くの場合、特に小さな分子の場合には、質量スペクトルの結果により検体の分子量を解明できるようにzは1である。使用するイオン化の種類によっては、H+を加えることでしばしば正イオンをつくり、そのため分子量を表すイオン(すなわち分子イオン)は[M+1]+と表記する。その逆である、H+の抽出による負イオンの形成もまた起こり、分子イオンは[M―1]― と表記する。
【0004】
ガス衝突セルにより隔離された、連続する2つの質量分析器を含む装置がある。これらはタンデム質量分析器として知られる。質量分析器が四重極であるとき、タンデム質量分析器は三連四重極質量分析器として知られる。イオンは第1質量分析器から衝突セルに流入して(親イオン)分裂し、第2質料分析器によりその断片(娘イオン)を検出する。これは、LC―MS/MSとして知られる。
【0005】
質量スペクトルは、イオンの絶対分子量を表す。例えば、2−ヒドロキシ―2ピロール―キノリンの平均分子量は、炭素、水素、窒素、および酸素の同位体を考慮すると、210.24である。しかし、質量分析器は個別の同位体の含有量をそれぞれ検出する。2−ヒドロキシ―2ピロール―キノリンでは、炭素同位体の含有量が支配的であり、従って[M+1]+、m/z211イオンは12Cに基づく。m/z212のイオンは13Cに基づき、m/z213イオンは14Cに基づく。14Cの天然存在度は極めて小さく、代表的には7.4×1011個の炭素原子中に、ただ1個の14C原子が含まれる。
【0006】
生物分析は、一般に極めて大規模に行われ、上述の議論にもかかわらず、代表的には放射性標識化薬物の検出および定量化を含まない。
【0007】
生物分析において、代表的には、例えば親薬物(例えば人体、動物、もしくは他の生物系に投与する分子など)および、しばしば多くの特定代謝物のような、標的分子のためにサンプルを分析する。簡単のため、本明細書において「標的分子」といえば、分析対象であり、例えば未知の薬剤候補もしくは推定薬剤候補もしくはその代謝物といった、対象の分子を指すと理解されたい。臨床試験に参加した多くの被験者からの薬物動態データを作るために、生物分析の結果を使用する。
【0008】
LC−MSを用いる生物分析において、(例えば血漿溶媒抽出により)生体サンプルを抽出し、その生成抽出物をLC−MSにかけて標的分子を検出し定量化する。しかし、このような定量化は以下の理由で複雑である。
1.例えば血漿からの標的分子の抽出は完全でなく、未知の量が抽出されずに残留する。さらに、サンプルによりその量は異なり、抽出は濃度に依存する。
2.(MS分析に先立つ)HPLCによる分離中に損失が生じる。言い換えれば、HPLCのカラム回復は100%ではない。1と同様に、前記カラム回復はサンプルにより異なり、濃度に依存する。
3.質量分析器は、検出の前に合成物をイオン化する。イオン化の過程は極めて合成物依存が高く、イオン化効率は時間とともに変化しうる。従って、基準を参照せずに質量分析器により定量化することができない。
【0009】
これらの難点を補うために、標的分子の定量化では、一般に較正が必要である。そのために、代表的には極めて高純度な以下の2種類の物質が必要である。すなわち(1)参照基準としての標的分子自体と、(2)内部標準として使用する、標的分子と極めて似た化学構造を有する合成物とである。クロマトグラフィの保持時間が重水素化していない標的分子に極めて近いが、質量分析のステップ中に分子量が大きいことによりLC−MS内で区別できるため、理想的な内部標準としては、一般的に重水素化した標的分子が考えられる。しかし、重水素化標準は常に入手できるわけではない。重水素化内部標準が入手できない場合、標的分子とは構造が異なる合成物(すなわち類似体)を使用できるが、可能な限り標的分子と構造が似ているべきである。
【0010】
較正のため、一般的に対照基質(例えば血漿)を、対象の標的分子にかつて接触したことのないドナー(例えば人間)から取り出す。一連のそのような制御血漿サンプルそれぞれに、異なる濃度の参照基準標的分子を加える(例えば、8種のサンプルは、10,20,50,100,500,1000,1500および2000pg/mLの参照標的分子を、他の例としては、100,150,200,250,500,1000,1500および2000pg/mLの参照標的分子を含む)。追加的に、既知で等量の内部標準を、各制御血漿サンプルへ加える(例えば500pg/mLの内部標準)。
【0011】
そうして準備した各サンプルを、適切なクロマトグラフィの方法(例えばLC−MS)で分析し、標的分子および内部標準の量を求める。例えば標的分子の濃度(制御血漿サンプルに初めに加えた量から既知)をx軸上にプロットし、分離した標的分子の量と内部標準の量(これらの量はLCMSにより求める)の比をy軸上にプロットした、較正グラフもしくは較正ラインを作成するために、得られるデータを使用する。ここで、そのようなプロットは必ずしも線形ではないことに注意されたい。
【0012】
次に、臨床試験もしくは他の試験により得た実際のサンプル内に存在する標的分子の、未知の濃度を定量化するために、各サンプルへ、正確に既知の量の内部標準を加える。LC−MSにより、標的分子および内部標準の量を求めるために、そのサンプルを抽出し、分析する。対応する標的分子濃度を導くために、検体:内部標準の比と、較正グラフ/ライン上で対応する標的分子濃度とを比較するために、これらの量の比を使用できる。
【0013】
サンプルの準備中などに標的分子の損失が生じたら、較正の比率性により許される内部標準の比例損失により修正する。同様に、LCMS技術により損失もしくは他の失敗もしくは標的分子の検出不足が生じたら、これらの損失を対応する内部標準の損失に反映するべきである。従って、実際には較正ラインによって、質量分析器の方法、理論的損失および反応が特徴づけられる。
【0014】
上記の通常の生物分析に追加して、代表的には14Cで標識化する、放射性標識化薬物を用いた関連実験もまた行える。そのような実験は、一般的に、臨床試験に関連する多数のサンプルにより行われることはないが、薬物の代謝作用や薬物動態を調べるために設計される明確な実験である傾向がある。そのような研究において、一般的に薬物濃度を定量化するためにLC−MSは使用しない。代わりに、代表的には標的分子は、放射性追跡子技術を用いて、放射能レベルを求めることで定量化する。この理由は基本的には、このような研究における標的分子(もしくはおそらく特定の代謝物)は、一般的に既知であり、容易に分離することができるためである。従って、識別は難しくないため、定量化のみが必要である。しかし、親薬物のような特定の標的分子を放射性追跡子技術により定量化する場合、放射性標識化されていない標的分子と14Cで標識化した標的分子をともに入手できるとしても、適切な14Cで標識化した内部標準は入手できないだろう。このため、放射性追跡子を用いた標的分子の定量化は、伝統的に、分析の絶対的な方法により行われている。このとき、HPLC溶出液内の放射能の量の計測が必要で、その結果は直接、初めの基質内の標的分子濃度と等しいと見なされる。
【0015】
放射能追跡子の使用は、放射能の測定が直接、存在する標的分子の量に関係する点で有利である。LC−MSと異なり、考慮すべきイオン化効率の要因はない。それにもかかわらず、サンプル抽出に関連する不正確性およびクロマトグラフィカラムで生じうる損失については、この場合にもあてはまる(すなわち上述の項目1および2)。このため、計測する放射能を、初めの基質内の標的分子濃度に直接的に等しいと見なすことは、はっきり言えば妥当な仮定であるとは言えない。
【0016】
さらに、液体シンチレーション(LSC)のような放射性追跡子技術は、本質的に感度が鈍いことに悩まされる。例えば、14CのLSCによる測定において、初めにサンプルを液体シンチレーションの混合物内に溶解する。崩壊事象中に14Cから放出されるβ線のエネルギーは、液体シンチレーション溶液を活性化する。エネルギーの基底状態に戻る際に、シンチレーション溶液は光量子を放出し、その光量子を光電子増倍管により検出する。従って、シンチレーション溶液が放出する光量子の数は、放射性崩壊の発生数に比例する。しかし、14Cの半減期は5760年であるため、一度に崩壊するサンプル内の14C原子は比較的少ない。実際、平均して1壊変毎分(dpm)を生じるために、10億個以上の14C原子が必要である。
【0017】
放射性追跡子技術を使用する代案として、サンプル中に存在する標的分子の量を直接定量化するために、AMSもまた使用可能である。
【0018】
AMSは特定の同位体の量を測定する技術である。AMSは、1970年代に考古学の炭素年代測定のため発明され、1990年の薬学研究(Garner,R.C.(2000)、「薬学研究における加速器質量分析および開発―同位体測定の新たな超高感度分析方法」、Curr Drug Metab. 1(2)205−213)に初めて使用された。
【0019】
LSCとは異なり、AMSは、検出器に到達するために1000個の原子のみを必要とする極めて高感度な分析方法である(Lappin, G.& Garner,R.C.,「加速器質量分析を用いた、放射性標識化薬物およびその代謝物の超高感度検出」、分析的分離のハンドブック、I.Wilson編、2003、Elsevier:Amsterdam.p.331−349)。AMSは同位体比の技術であるので、分析する物質において珍しい同位体を濃縮しなければならない。他の同位体も使用可能であるが、生物医学研究において、共通して14Cをこの同位体とする(その場合、12C:14Cの同位体比を測定できる)。
【0020】
AMSは生物医学および他の研究に多くの用途を有するが、現在の議論に関連するのは、所与の基質内に極めて低濃度で存在する合成物の測定に使用可能であることである。AMSの最も重要な利点の1つは、比較的短い分析時間で、被験者に与える必要のある放射線量が、規制認可を必要とする規定の放射能レベルを下回るほどに低い放射能レベルの、検出と定量化が可能である点である。代表的には、分析する合成物は製剤原料、もしくはその代謝物であり、その基質は生体サンプル、すなわち被験者もしくは被験動物から得られた例えば血漿といった生体サンプルである。しかし、その分析する合成物は任意の基質中の任意の合成物とすることができる(例えば土壌中の環境汚染物質)。
【0021】
上記のように、基質中における標的分子濃度の共通の測定方法はLC−MSである。生物医学研究において、代表的にはLC−MSは、mLサンプルあたり100pgの合成物を測定できる。執筆時(2007)にはLC−MSはより高感度を達成している(例えば10pg/mL)。しかし、AMSは通常、当業者には容易に達成可能な、通常のある最適化方法により、1pg/mLの感度を達成でき、その感度はフェムトグラムもしくはアトグラム(10−15−10−18g)の範囲まで達することができる。
【0022】
従って、AMSは、より高感度な分析が必要とされる生物医学研究で、分析ツールとして使用されている。分析する合成物は、代表的には14CであるAMS同位体の濃縮した量を含み、合成物の濃度を測定するために、AMSはその同位体比を測定する。LC−MSにおいて合成物は同位体濃縮を必要としないが、その感度はAMSで得られるレベルには達しない。
【0023】
代表的には、AMS分析において、まず分析する合成物を、HPLCによる分離前に見つかる基質中から抽出する。一連の留分としてHPLC溶離剤を収集し、対象の合成物に対応するものを、AMSによる分析のために等分する。執筆時(2007)には、特許文献1(国際公開第95/04369号)に詳細が示されているにもかかわらず、効率的に分離ステップを(例えばHPLCによって)AMSへ統合する、通常のインタフェースが存在すると信じられてはいない。
【先行技術文献】
【特許文献】
【0024】
【特許文献1】国際公開第95/04369号
【発明の概要】
【発明が解決しようとする課題】
【0025】
この方法は、抽出過程および分離過程において分析上の損失がないことを仮定している。抽出という語は、分析する合成物を、それが見つかる基質中から初期精製する1つもしくはそれ以上のステップを意味する(例えば分析する合成物および他の合成物を、血中で見つかるタンパク性材料から分離するステップ)。分離とは、さらなる1つもしくはそれ以上の精製ステップを指し、代表的には抽出ステップ後に存在する、基質中の全ての残留成分から、分析する合成物を分離することで精製することを指す。しかし、その特性によっては、多量の結合を示し、そのために抽出中およびHPLC分析中に損失が生じる合成物がある。従って、従来のAMS分析の結果は、真の値よりもかなり低い結果となる。このことは、AMSによって極めて少量の合成物を分析することが必要なときに、例えば抽出中および精製中の結合効果および実験的損失により、特に問題となる。結果として、AMSに内在する、AMSの同位体標識化標的分子の少量検出の能力は、少量の材料を定量化するとき誤差が増すと考えられる。
【0026】
本発明は、1つもしくはそれ以上の上述した従来技術の問題点を改善することを意図するものである。
【課題を解決するための手段】
【0027】
AMSによるサンプル分析の準備として、結合および実験的損失により生じる不正確性や誤差に対処するための手順を、我々は開発した。その手順では、AMS分析用の合成物を、例えば14CのようなAMS同位体で濃縮することを利用する。
【0028】
本発明は、AMSによる、AMS同位体標識化検体(本明細書内では「検体」)の定量化時の内部標準化の方法に関する。従って、検体は、定量化が望まれ、AMS同位体で標識化した全ての対象となる合成物となる、AMS同位体標識化合成物である。上記の生物分析で使用するものと類似の標準化手順でありながら、AMS同位体により標識化していない検体(本明細書内では「対照検体」)と構造的に区別できる合成物を内部標準として使用するという、根本的な相違点を有する内部標準化手順を、本発明は使用する。
【0029】
従って、1つの態様から見ると、本発明は、試験サンプル内のAMS同位体で標識化した検体量の測定に用いる較正方法を提供するものであり、前記較正方法は、
(i)複数の較正サンプルを提供するために、前記検体および標識化しない対照検体のどちらにも汚染されていない複数のサンプルを、既知量の前記対照検体および量Cの検体へ接触するステップにおいて、前記較正サンプルがそれぞれCとは異なる既知量の対照検体を含むことを特徴とするステップと、
(ii)AMSにより、複数のサンプルそれぞれに加えた検体の量Cを測定するステップと、
(iii)複数の精製サンプルを提供するために、検体および対照検体を、複数のサンプル内の他の化学種から分離するステップと、
(iv)AMSにより、前記精製サンプル内の検体の量Aを測定するステップと、
(v)AMSにより、前記精製サンプル内の対照検体の量Bを測定するステップ
とを含むことを特徴とする。
【0030】
前記精製サンプル内の対照検体の量Bを測定するとき、ここでは対照検体のみを測定するだろう。代案として、測定技術が対照検体と検体を識別できないとき、この測定は対照検体と検体にともに可能である。以下でより詳細に説明する後者の場合、このような測定は、存在量が十分に検体よりも多いならば、対照検体のみに対すると考えられる。
【0031】
本発明の第1の態様による方法から得るデータは、その方法を用いる前にAMSにより求める前記サンプル内に存在する検体の絶対量Cを、比率A:Bに関連付けるために使用できる。従って、検体量は未知であるが存在する対照検体量は既知の試験サンプルに対して、上記の方法で実行する条件と同様の条件下でこの比率を求めるとき、本発明の第1の態様による方法から得るデータを参照して、試験サンプル内に存在する検体量を求めることができる。
【0032】
この態様から見ると、本発明は、試験サンプル内のAMS同位体で標識化した検体量の測定に用いる方法を提供するものであり、前記方法は、
(i)前記試験サンプルを、既知量の標識化しない対照検体に接触させるステップと、
(ii)複数の精製試験サンプルを提供するために、もし存在すれば検体と対照検体を、試験サンプル内の他の化学種から分離するステップと、
(iii)AMSにより、前記精製試験サンプル内の検体の量Aを測定するステップと、
(iv)AMSにより、前記精製試験サンプル内の対照検体の量Bを測定するステップ
とを含むことを特徴とする。
【0033】
本発明の第1の態様の実行と同様に、精製試験サンプル内の対照検体の量Bを測定するとき、ここでは対照検体のみを測定するだろう。代案として、測定技術が対照検体と検体を識別できないとき、この測定は対照検体と検体にともに可能である。以下でより詳細に説明する後者の場合、このような測定は、存在量が十分に検体よりも多いならば、対照検体のみに対すると考えられる。
【0034】
精製試験サンプルのA:Bの比率と、本発明の第1の態様の実行により、A:Bの比率を既知の値Cによって補正して得るデータとを比較することで、本発明の第2の態様の方法で分析する試験サンプル内の検体の未知量を決定できる。
【図面の簡単な説明】
【0035】
【図1】本発明の較正方法の実行により得るデータから、Cに対するB:Aの比率をプロットして得る較正曲線である。
【図2】本発明の較正方法の実行により得るデータから、Cに対するB:Aの比率をプロットして得る、さらなる較正曲線である。
【発明を実施するための形態】
【0036】
本発明は、上述したような生物分析とは異なる。例えば、AMSで同位体標識化した検体を含むことが本発明において要求される。第2に、生物分析において内部標準は、できる限り構造的に密接に検体と関連する必要がある。本明細書で説明する方法において、内部標準は、標識化した検体に対して標識化しない対照物である。この方法は、検体が、AMS同位体の存在により内部標準と区別可能であるために可能である。2つの分析方法を用いて、すなわちAMSにより検体の量を測定し、対照検体の量を異なる分析方法で測定する。
【0037】
以下の議論では、本発明の方法を14Cで標識化した検体の測定での使用、特に例えば臨床試験で得る生体サンプル分析における使用に頻繁に言及するが、本発明はそれに限定されるものではないと理解されたい。
【0038】
サンプルが臨床サンプルである場合、臨床試験中、検体の投与後に一定時間が必要となるだろう。さらに、その臨床サンプルは、所望ならばあらかじめ放射化したサンプルを含むこともできる。
【0039】
また当然のことながら、本明細書において、本発明の方法による、効果的な分離を行うときのHPLC使用を重視する。しかし、本発明はそれに限定されるものではないと理解されたい。精製試験サンプルの生成物のAMS分析が、精製試験サンプル内の検体量の正確な定量化を可能とするように、他の標識化した合成物から対象の検体を分離するために使用する場合には、あらゆる分離方法(HPLCもしくは他による)が、本発明の方法の実行によって使用できる。例えば特許文献1に記載されたような、AMS機器との直接的な接触を可能とする分離方法を含みうる。しかし代表的には、分離ステップおよびAMS測定ステップは接触していない。
【0040】
本明細書において以下の定義を使用する。
【0041】
未知の薬剤候補の臨床試験において、例えば検体は、そのような臨床試験により試験する親薬物、もしくは親薬物の代謝物とすることができる。一般的に検体は、14Cで標識化した合成物であり、本明細書の議論はそのような14Cで標識化した合成物に注目する。しかし、少量の他のAMS同位体を検出する能力があれば、検体はAMS同位体で標識化した任意の合成物とすることができる。
【0042】
全ての原子には同位体があり、その中にはAMS分析に適したものがある。例えば、129Iで標識化した生物製剤はAMS検出に有用であるが、131Iで標識化した生物製剤は高活性であり、おそらく安全性の問題から人間に対して使用が制限される。同様に、14Cで標識化した生物製剤はAMS検出に有用である一方で、13Cで標識化した生物製剤が、NMR検出のような他の多くの非放射性技術において広く使用されるが、その生物製剤はAMS分析では使用できない。特に不適切な同位体、とりわけ窒素は、負イオンを形成できない。
【0043】
AMS同位体は、AMS分析に敏感な任意の同位体とすることができる。好適にはAMS同位体は、例えば同位体存在度が1×10−3から1×10−15%の範囲内、特に1×10−5から1×10−15%といった、極めて低い天然存在度である。例えば14Cではおよそ1.4×10−10%であるように、AMS同位体のそのような自然バックグラウンドの低さに、AMSの検出感度は依存する。(13Cの自然バックグラウンドは、それと比較して大きい1.1%である。)好適には、例えば前述のように数週間を超え、特に30日以上、もしくは60日以上で、扱いやすいように上限は数千年の範囲内にある長い半減期を、AMS同位体が有するか、もしくは天然存在度が低い場合には安定同位体である。
【0044】
水素、ベリリウム、炭素、アルミニウム、リン、塩素、カルシウム、マグネシウム、鉄、セレニウム、ヨウ素、バリウム、およびウランやプルトニウムといったランタニドおよびアクチニドのAMS同位体の中から、AMS同位体を選択でき、特に、3H、7Be、10Be、14C、17O、18O、26Mg、26Al、32Si、35S、36Cl、41Ca、55Fe、60Fe、53Mn、79Se、59Ni、および129Iを含む一群から選択できる。より一般に、同位体は1つもしくはそれ以上の3Hおよび14Cから選択する。最も一般的には、同位体は14Cである。
【0045】
本明細書では、AMS同位体として最も一般的に使用する14Cの使用を重要視するが、この理由の一部は、当然のことながら薬物の大多数が有機物であるためである。しかし、前記のAMS同位体を含む他の同位体も使用されており、また使用できる。
【0046】
本発明の方法で用いる内部標準(「対照検体」)は、検体と識別可能な合成物であるが、検体中に存在するAMS同位体は含まない。
【0047】
対照基質とは、例えば、検体もしくは対照検体で汚染されていないサンプルのような、本発明の第1の態様により使用するサンプルを指す。従って、臨床試験の場合には、対照基質は商業的に得られる血漿、臨床前サンプル、もしくは臨床試験に参加していない被験者からのサンプルとすることができる。一般的には、対照基質は血液から得られる血清、もしくは血漿とする。しかし、尿、便、もしくは細胞組織といった他の基質を分析することも必要であることを理解されたい。
【0048】
「対照基質」とは逆に、本明細書において「基質」は、検体の量を求めるために分析の対象となる試験サンプルを指すために用いる。そのため、基質は、対照基質と同じ物質群から選択できる。
【0049】
「較正抽出溶媒」は、既知の濃度の14Cを含有する(もしくは他のAMSで標識化した)検体とともに、対照基質抽出をHPLCにより分析するときに、特定の量を達成する濃度の対照検体を含む溶媒を指す。代表的には、較正抽出溶媒はアセトニトリルであるが、当業者に知られる例えばメタノールなどの他の溶媒も使用できる。較正抽出溶媒は一般的に、本発明の第1の態様の方法による分離ステップの一部で使用する。
【0050】
「標準抽出溶媒」は、本発明の第2の態様の方法による分離ステップの一部で一般的に使用する。較正抽出溶媒と同様に、標準抽出溶媒は代表的にはアセトニトリルであるが、当業者に知られるような、例えばメタノールなどの他の溶媒、および例えば固相抽出などの他の方法も使用できる。代表的には、HPLC内に抽出物を噴射するときに特定の量を達成するための濃度で、標準抽出溶媒は対照検体を含む。較正抽出溶媒とは逆に、標準抽出溶媒は検体を含まない。
【0051】
本発明により検体から分析できる試験サンプルは、任意の発生源に由来しうる。代表的には、これらのサンプルは、人間か、もしくは特定の人間内の動物に由来する。例えば、人間へのマイクロドージング、もしくは特定の人間における絶対的バイオアベイラビリティ研究から起こりうる。
【0052】
しばしば人間フェーズ0臨床試験と呼ばれる、人間へのマイクロドージングは、比較的新しいコンセプトで、AMSの超高感度に依存する。人間へのマイクロドージングにより、薬物を0.5μg程度投与した後に、詳細な人間の代謝作用研究の実施が可能となる。しかしより代表的には、100μgの薬物を投与する(マイクロドーズは、EMEAおよびFDAの両方により、決して100μgを超えない100回の予測される薬理学的投与量として定義されている)。マイクロドージングにおいて、早急にADME(吸収、分布、代謝、排せつ)情報およびPK(薬物動態)情報を得るために、1種類もしくはそれ以上の薬剤候補は追跡量で人間に取り込まれる。次に、マイクロドーズしたどの薬物がさらに取り込まれる適切なPKパラメータを有しているのか選択するためのデシジョンツリーの一部で、この情報を使用する。これらのADME研究をスクリーニングする低量投与の目的は、不適切な代謝作用のために、発達経路の下流に滴下される薬物がより少量となることを保証するための試行である(例えば初回通過、半減期が短すぎること、バイオアベイラビリティが悪い等)。3種類のうち1種類程度の薬物が、PK、薬物動態、もしくは毒物問題のために薬物発達のフェーズ1の段階で滴下される。人間へのマイクロドージングの目的は、フェーズ1での消耗を減らすことである。
【0053】
マイクロドージング方法において、1μgから上限100μgまでの投薬量で、未知の薬剤候補を人間の被験者へ投与することができる。血液、尿もしくは便のサンプルを何度も採取し、ADMEおよびPKのデータを求めるために、AMSにより、生成サンプルの14Cもしくは他のAMS同位体含有量を分析する。
【0054】
従来技術(例えば、G.Lappin,M Rowland and RC Garner, Expert Opin. 「薬物代謝中毒」2006、2(3):419−427を参照)で知られる絶対バイオアビリティは、合成物の静脈内投与(代表的には薬剤候補)を含む。そのような研究は一般的に、溶解度、分析上の感度、およびIV毒物学のデータが通常研究により裏付けられる必要がある事実に関する問題のため、実行することが難しい。しかし、これらの問題には、AMS同位体の標識化合成物のマイクロドーズを用いて対処できる。
【0055】
マイクロドージングおよび絶対バイオアビリティ研究において、対照検体と同時に、対象に検体を投与する。対照検体は通常、標識化していない未知の薬剤候補である。代表的には、混合は「軽い標識化」のみである。軽い標識化とは、放射能の量が極めて少ないことを指し、代表的には、被験者に対して200nCiのみである(既に体内に存在する14Cによる放射能のおよそ2倍の量)。
【0056】
ここで、検体および対照検体という用語は、AMS同位体標識化合成物と、標識化していない対応する(対照の)合成物とを区別するために使用することに注意されたい。実際には、検体は一般的に対照検体と共にみられ、従って同時に投与される。
【0057】
(例えば)マイクロドーズが投与される対象(例えば被験者)の体内で移送されるとき、100μgの投薬(すなわち検体および対照検体を含む)は、およそ500pg/mLの最大濃度まで達する。代表的には、そのサンプルに内部標準として加える対照検体の量は、5μg/mLである。これは、内部標準の量が、対象への投与により上昇する、試験サンプル内に存在した初期の対照検体量の少なくとも10000倍であることを意味する。このことから、その試験サンプルに加えた内部標準を、精製試験サンプル内で測定する対照検体の唯一の発生源であるとみなすことは理にかなう。代表的には、内部標準として存在する対照検体の量は、与えられた試験サンプル内に存在する全ての対照検体の、少なくとも98wt%であり、より代表的には99wt%かもしくはそれ以上であり、より代表的には99.9wt%かもしくはそれ以上であり、より代表的には99.95wt%かもしくはそれ以上である。
【0058】
本発明は、2種類の構造的に識別可能な化学物質(検体および内部標準)の区別、ならびに2種類の定量化の方法に依存する。定量化方法の一方は、検体に対するAMSであり、他方は、対照検体に対して、質量分析を含むクロマトグラフィ検出において(一般的に)有用な、例えば紫外線吸収、蛍光検出もしくは他のあらゆる検出方法である。
【0059】
上記のように、本発明は、特に低濃度の検体測定によく適している。AMS分析で代表的な状況では、サンプル内の検体のレベルは極めて小さい(例えば10−18から10−9g)。そのような少量の物質を扱うのは特に難しい。例示のように、それ以上はなく、表面(例えばサンプル管の内部)はわずかな活性結合部位を有し、上限100fg(10−13g)の物質を吸収できる。LC−MSによる代表的な検出限界(LOD)はおよそ100pgであり、無視できる量の結合による損失(すなわち0.1%)の1000倍も高い。しかし、より低濃度では、結合による損失量の比率は高い。濃度が1pg/mLの場合、100fgの結合は検体の10%の損失となり大きい。しかし、本発明によると、試験サンプル内に存在する検体を、代表的には14Cにより標識化し、対照検体をこの試験サンプルへ内部標準として加える。有利なことに、これによって検体と対照検体の濃度の合計値を、例えば1fg/mLレベルから200pg/mLレベルへと(100万倍も高い)上昇させる効果があり、従って極めて少量のサンプルの分析に関連する問題を、改善もしくは解決できる。
【0060】
本発明の較正方法を実行する前に、代表的にはいくつかの予備調査を行う。予備調査に含まれるものに、AMSによって、全ての対照検体のサンプルが、検体中に存在するAMS同位体を持たないこと、すなわち全てのサンプルが最大で通常のバックグラウンドレベルの同位体を含むことの確認がある。
【0061】
同様に、検体と対照検体を、検体と対照検体の定量化において障害となりうる他の合成物(例えば推定代謝物)と分離するための適切な分離手順の開発は、本発明の範囲ではなく、本明細書において議論しない。しかし、適切な分離手順の開発は、当業者の技術内にある。既に述べたように、本発明による方法を用いた分離様式を、電気泳動法、もしくは高性能液体クロマトグラフィ(HPLC)、ガスクロマトグラフィ(GC)、もしくは薄層クロマトグラフィ(TLC)などのクロマトグラフィ法とすることができる。代表的にはHPLCを用いる。
【0062】
同様に、あらゆる所与の溶媒に対する検体の溶解度および安定性の測定は、本発明の範囲ではない。当然のことながら、検体によって溶解度および安定性は変化し、当業者は測定できるであろう。この点から、サンプルから対照検体および検体を抽出するために使用する溶媒を、ある程度、本発明により定量化を所望する検体の溶解度および安定性により決定する。
【0063】
本発明により使用する分離手順において、検体の存在を検出するために、紫外線吸収を使用するとき、□maxを初めに測定する。一般的にHPLCを用いる場合、対照検体に対して、紫外線吸収が代表的で便利な検出方法である。しかし、当然のことながら、当業者は他の検出方法を使用できる。
【0064】
HPLCで対照検体を検出するために紫外線応答を用いる場合、HPLC−紫外線応答の再現性は、代表的には3回もしくはそれ以上の噴射で、そのうち好適には少なくとも1回は別の日に実行される噴射により確かめる。これにもかかわらず、再現性を確かめる正確な方法は、対象の検体の特性に依存する。同様に、全てのクロマトグラフィのキャリーオーバーの程度を、同様の方法で確かめる。
【0065】
さらに、本発明の実行の前に、分離系用の適切なHPLC分析の開発において、一般的に当業者は、HPLCカラム上に噴射された対照検体のどれだけの量が、ピーク形状(例えばカラムの過負荷による)に不利に影響することなく、ノイズ比に対して適切な信号を与えるのか、また対照検体のどれだけの量が使用する濃度に溶解するのか、測定するだろう。この量は、本明細書において「標準量」と呼び、紫外線検出による定量化に使用するHPLCカラム上に噴射した対照検体の量を指す。
【0066】
標準量の測定後、例えばxμlの標準対照検体をyμlの基質内に混合し、その後でHPLC上にzμl噴射することで、噴射する対照検体の標準量が結果として出るといったように、対照検体の標準溶解濃度を計算できる。本明細書において、このことを「標準溶解」と呼ぶ。
【0067】
しばしば、所与の基質から検体を抽出するおおよその効率を定量化するため、初期検査を行うことが有効である。これを行うために、基質からのおおよその抽出効率が計算できるような濃度まで、対照基質サンプルを検体とともに混合する。これは、例えば液体シンチレーション計測(LSC)を用いて、便利に達成できる。
【0068】
例えば、3000dpmの14Cで標識化した検体を、1.5mlの対照基質内に混合でき、LSC分析のために200μlのアリコートを3回、すなわちいずれの抽出ステップも干渉しないように採取する。次に、別の200μlを3回、200μlの適切な溶媒(例えばアセトニトリル)内に抽出し、乾燥し、再構成することができ、その200μlのアリコートをLSCで分析できる。回復が100%ならば、LSC分析用の200μlのアリコートは、それぞれ400dpm含み、その計測時間(計測は95.5%の信頼時間間隔を仮定すると)は25分である。(計測時間はT=1/dpm(200/2)2より計算できる)(計測は95.5%の信頼区間内にあると仮定する)
【0069】
前記のように、これらの予備段階は本発明の方法の一部ではなく、前述のおよその抽出効率の測定の観点から、抽出効率は100%近くを達成する必要がないことは、当然のことながら重要である。実際、本発明は、必ずしもそうではないという認識に起因する。本明細書における予備段階の目的は、本明細書の方法が有効であるように、十分な量の検体を、実際に対照基質から抽出できることを保証することである。
【0070】
本発明の較正方法の実行は、A:Bの比率を、検体の存在量の絶対値に関連付けるために使用できるデータを提供する。例えば、前述の生物分析と類似の、Cに対するA:Bの較正ラインを引くために、そのデータを使用できる。(それぞれの、また全ての本明細書におけるA:Bという呼称は、B:Aの代わりに使用でき、重要な点はただ比率が得られるということである)
【0071】
そのようなデータを得るために、例えば臨床試験では、対照基質(例えば血漿)を、検体もしくは対照検体により汚染されていない物質源から採取する。汚染されていない、というのは、検体および対照検体がともに存在しないか、もしくは検出できないレベルで存在することを意味する。臨床試験において、分析する薬物もしくは代謝物に触れていない被験者から、対照基質を得ることで、このことを好都合に達成できる。
【0072】
そのような複数の対照基質サンプルぞれぞれに対して、異なる濃度の検体を加える。さらに、正確に既知で等量の内部標準(例えば対照検体)を、各対照基質サンプルへ加える。検体と対照検体を加えた後、準備した各検体を精製する。これは一般的に、従来技術で知られる初期抽出ステップを含む。
【0073】
検体と対照検体を含む複数の対照基質サンプルを準備するとき、検体を基質内に加えることが可能で、その各対照基質サンプルに加えた検体の量Cを、個別の対照基質サンプルのAMS分析により、測定することが可能である。しかし、AMS同位体が14Cである場合、基質(例えば血漿)がバックグラウンド14Cを含むため、この方法には問題が生じうる。例えば、血液はおよそ12%w/vの炭素を、血漿はおよそ4%の炭素を、尿はおよそ1%の炭素を含む。比較的高い濃度で炭素が存在することは、検出限界(LOD)に影響する「ノイズ」とみなしうる、内生の14Cが存在することを意味する。
【0074】
実際、他の分析技術と同様に、AMSのLODは信号とノイズの比率により決まる。ここで信号は、14Cを濃縮した検体からの14Cの量に依存する。14C:12Cの比率は、大気中に含まれるこれらの同位体の比率と等しく、そのため全ての生物で一定である。従って、内生の14Cは、サンプル内の全炭素量に比例して増加する。従って、便サンプルのLODは、便が血清よりも内生の炭素含有量が高いため、血清サンプルよりも高い。
【0075】
従来技術で知られているように、検体からの14Cの量を低減することなく、サンプル内の内生の炭素量を少なくする方法がある。例えば、例えばアセトニトリルのような水混和性の有機溶媒により、炭素リッチのたんぱく質は沈殿し、続いて遠心分離により除去できる。事前のAMS溶媒抽出ステップも同様に、便における内生の14Cの量を低減できる。
【0076】
生体サンプル内に内生の14Cが存在するために、対照基質を直接混合するとき、例えばおよそ0.06dpm/mLまで低下した場合のみ14C濃度を確認することができる、という結果となる(Lappin, G.& Garner, R.C.以下)。反対に、HPLCにおいて、ほぼ14Cのバックグラウンドが存在しないため、そのレベルはこの場合よりも低くできる。
【0077】
この根本的な問題に対処するために、好適には、14Cで標識化した検体および対照検体を含む複数の対照基質サンプルを準備するときに、検体を(および一般的には、例えば、対照検体を)較正抽出溶媒内に加え(混合し)、較正抽出溶媒はほぼ不揮発性の14Cのバックグラウンドを含むため、量Cを知るために、較正抽出溶媒内に存在する検体をAMSにより分析する。(AMSサンプルを準備するとき、アセトニトリルを除去する。)このように、分析したい試験サンプルへ、より関連づけて(より低い)濃度に至るまで較正曲線を作成することが可能である。
【0078】
このように、それぞれが随意的に(しかし通常)、同量の対照検体(通常は、標準量をHPLC上に噴射できるような濃度で)と異なる14C検体の濃度とを含む、一連の較正抽出溶媒を準備する。複数の同じ抽出溶媒を使用でき、この方法の正確性が改善される。代表的には、5回の較正抽出溶媒を使用する(代表的には、0.005から2dpmをHPLCカラム上へ噴射する)が、その正確な回数および使用濃度は研究の特性に依存する。
【0079】
較正抽出溶媒の(もしくは適切に検体および対照検体を混合した対照基質の)アリコートを、一般的には少なくとも二重に、AMS分析で採取する。その結果は、対照基質サンプルに加えた、14Cの、もしくは他のAMS同位体の正確な濃度を規定する。
【0080】
対照検体の貯蔵液もまた準備し、そこから較正抽出溶媒を作成する。以下で説明する標準抽出溶媒を作るために、代表的には同じ貯蔵液を使用する。このように、試験サンプルの分析に使用したものと同じ濃度の内部標準を使用して、較正曲線を作成する。
【0081】
較正抽出溶媒を使用して、代表的には二重に、対照基質を抽出する。次に、(適切ならば、乾燥および再構成の後で)サンプル内の他の要素から、最終抽出物を分離する。この操作は、代表的にはHPLCにより達成し、HPLCからの溶出液は一連の留分として収集する。検体(および構造的に識別可能な内部標準)の保持時間に対応するその留分を貯蔵し、14C、もしくは他のAMS同位体の、AMSに含まれる単体サンプルとして分析する。また、選択したサンプルの個別の留分を、留分層が検体を代表することを保証するために分析する。
【0082】
別個に、対照検体の量を測定するために、代表的には紫外線検出などの、サンプル定量化の他の方法を使用する。
【0083】
較正の方法により得るデータ、すなわち複数の精製サンプルそれぞれに対する、1セットのC,AおよびBの値は、A:Bの比率とCとの間に関係性を構築できる。Cは、各試験サンプルに加えた検体の量に対応する。Aは、精製試験サンプル内に存在する検体の量に対応する(そして、Cの100%は、その抽出(および精製)効率100%である)。Bは、精製試験サンプル内に存在する対照検体の量に対応する。
【0084】
当然のことながら、本発明の較正方法において、AMS同位体に関して得られるデータは、AMS測定は対象の検体のみであることを仮定する。しかしこのことは、その標準がAMS標識化した化学種をその標準以外に含む場合には、必ずしもあてはまらず、そのため、少なくとも量Cの測定の妨げとなる。しかし実際には、本発明の較正方法に使用できる複数の検体を他の方法で測定できるため、そのような可能性は問題とはならない。代表的には、検体の供給者により、その純度が確かめられる。
【0085】
一般的に、通常は検体と対照検体を相互に分離することはなく、ならびに/もしくは対照検体の定量化に使用する方法はそれらを識別することができないため、対照検体の定量化は、検体および対照検体をともに含むサンプルに対して実行する。このことは、例えば50倍かもしくはそれ以上、一般的には100倍かそれ以上、対照検体の量が検体の量よりも、はるかに多いことを仮定している。当然のことながら、例えば、対照検体の総量と精製サンプル内に存在する検体の、簡単に標識化した人間マイクロドーズ内に存在する検体は極めて少量であるため、多くの生体サンプル中に存在する検体は、実際には極めて少量である。
【0086】
しかし、対照検体を定量化するために用いる方法により、対照検体と検体とを識別することができることも考えられる。そのような方法の例として、精密質量分析がある。
【0087】
Cに対するA:Bの比率をプロットすることで、較正ラインを得ることができる。従って、対照検体が内部標準としてどう機能するのか理解できるだろう。任意の摂動(例えばその損失および/もしくは未検出)は、検体それ自体が引き起こす、損失および/もしくは未検出に比例する。この2つの比率を用いるため、これらの損失は相殺される。
【0088】
所与の検体/対照検体の組に対して較正ラインを一度作ると、次に試験(例えば生体)サンプルを分析でき、存在する(もし存在するならば)検体量が測定できる。
【0089】
一般的に、標準抽出溶媒を準備する。これは、既知濃度の対照検体を含む抽出溶媒であり(代表的にはアセトニトリル系)、その標準量を抽出後にHPLCへ噴射できる。上述のように、好適には、標準抽出溶媒は、較正抽出溶媒と同じ濃度の対照検体を含む。このことは一般的に、標準抽出溶媒および較正抽出溶媒をともに作るために、同じ貯蔵液を使用することで達成する。このように、試験サンプルの分析に使用するものと同じ濃度の内部標準を用いて、較正曲線を作る。
【0090】
従って、試験サンプルに対して、既知の正確な量の対照検体を加える。これは対照検体として機能し、また有利なことに、上述の理由により、サンプル内の検体および対照検体の濃度の合計を増加させる。さらに代表的には、存在する検体の推定量をはるかに超えた量であって、代表的には本発明の較正方法において各サンプルに加えた対照検体と同じ量の、対照検体を加える。試験サンプルに用いた検体および対照検体の総量が、(a)抽出および精製中の損失を十分に抑制するため、また(b)検体および対照検体の合計と同量を、較正方法におけるサンプルおよび試験サンプルに使用するときには、試験サンプルについて得られたデータを摂動する濃度効果は存在しないために、濃度依存効果が抑制される点で、このことは有利である。
【0091】
抽出後、抽出した試験サンプルを例えばHPLCにより精製する。検体および対照検体を表す分率を、本発明の較正方法により得られる値と全く類似の方法で、(検体のための)AMS、および(対照検体のための)分離技術により分析する。
【0092】
精製試験サンプルにおいて得られた比率A:Bを算出することで、未処理の試験サンプル内に存在する初期濃度Cを、標準曲線から外挿することができる。
【0093】
量Bを測定するときに、紫外線応答を確認するため、HPLCの移動相に溶解した標準対照検体の少量のアリコートを、HPLC−紫外線により繰り返し(代表的には1日に2回)分析する。代案として、任意の簡便なHPLC較正は、効果的な、例えば製造者の特許もしくは推奨の方法になりうる。AMSは長期にわたり一貫する(そして実際に、データは同位体比率に基づくため、装置間で一貫する)ことが知られている。しかし、紫外線応答がドリフトするとき、対照検体濃度の計算において、A:Bの比率がエラーを生じる方へ変わってしまう。代表的には、20%以下かもしくは同等のCVは容認できる。
【0094】
当然のことながら、本発明の方法を実行するとき、および実際には本発明の較正方法により複数のサンプルを準備するとき、異なる濃度の対照検体を使用できる。主に、より多くのデータ操作を含むため、異なる濃度の対照検体を用いることはより不便であり、特定の条件下では信頼性の劣る結果を生じるかもしれないが、同じ濃度(もしくは量)の対照検体を使用する必要はない。例えば、較正曲線を作るときにxμgの対照検体を使用する場合、精製した較正サンプルに対して行う定量化ステップ(v)の間、「B」の値を与えると考えられる。2xμgの対照検体を試験サンプルへ加えるとき、対応する定量化ステップ(iv)の間、「2B」の値を与えると考えられる。次にそのような試験サンプルに対して、較正グラフから試験サンプル内の初期検体濃度を読み取ることを可能とするために、A:Bの比率は半分となるべきである。しかし、そのような試験サンプルから得られるA:Bの比率を関連付けるために必要なことは、ただこの比率を2倍することである。
【0095】
しかし当然のことながら、この方法の条件を(異なる量の内部標準を用いることで)変えることで、較正曲線が試験サンプルに対して妥当であるという仮定は、必ずしも全ての場合で正しくない。実際には、「B」の値は正確に「2B」とはならず、そのために較正曲線を参照して算出する検体濃度は、絶対的に真の値ではない。それにもかかわらず、好適ではないが、較正データを作るときに用いることのできる試験サンプル分析を行うときに、同量の内部標準を使用する必要はない。初期の試験サンプル内に存在した検体の算出量は、おそらく特定の場合に、より少量の内部標準化を受けている可能性があり、より正確ではない。
【0096】
本発明の較正方法により、複数のサンプルをそれぞれ準備するとき、同量の対照検体を使用する必要がないという点で、同様のコメントを必要な変更を加えて適用する。従って、xμgの対照検体が1つのサンプル内に存在し、2xμgの対照検体が他のサンプル内に存在するとき、「B」および「2B」の値がステップ(v)により得られる。そのような場合、xμgの対照検体にもとづいてA:B(もしくはB:A)の比率を準備するために、「2B」の値を2で割ることができる。
【0097】
前述の議論は、より一般的な点を示す。すなわち、本明細書内で前述した、本発明の第1および第2の態様は、本発明の異なる態様に関連する。第1の態様は、試験サンプル内の検体量を測定するための、次の方法で使用するデータを提供する。
【0098】
本発明の第2の態様による好適な実施例において、分離ステップおよび接触ステップの全ての態様は、本発明の較正方法による対応する分離ステップと、同じ条件で実行する。このことは、本発明の第1の態様を実行するときに行う対応するステップと、これらの条件が実用的に近く類似であることを意味する。同様に、好適には、本発明の較正方法による対応の測定ステップと同じ条件で、測定ステップを実行する。このように、本発明の第2の態様により実行するものと同じ条件、もしくは実用的な類似の条件において、本発明の第1の態様の実行により得られるデータを得る。従って、本発明の第1の態様で得られるデータによって、本発明の第2の態様の実行により得られるデータから検体を定量化することが可能となる。実用的な類似の条件において両データを得るため、後者のデータを、前者の正確性をもとに修正できる。例えば、本発明の第2の態様による分離ステップの一部としての抽出ステップが、乾燥ステップを含むようなとき、乾燥ステップ中に生じうる検体の損失を確認するために、好適には、乾燥の前後で例えばLSC分析を行うために、アリコートを採取する。
【0099】
試験サンプルを分析するときに、本発明の第1の態様の方法で得られるデータを適用する最も直接的な方法は、いわゆるpMC法である。代案の方法に、いわゆるdpm法がある。AMS同位体が14Cである方法に関して、両者を以下に説明する。
【0100】
全ての生物は、大気中の天然存在度と同じ14Cを含む。「100%現在」もしくは100pMCとして任意に参照、もしくは定義する14Cのレベルは、1.18×1012個の炭素原子の中に1個の14C原子があること、もしくは1ミリグラムの炭素中に97.6アトモルの14C原子があることに対応する。必要ならばデータを標準化するために、正確に既知のpMC値のAMS標準を、装置による確認により入手することが可能である。2種類の最も広範に使用する14C標準は、米国のNational Institute of Standards Technology(NIST)の標準的なシュウ酸と、1960年代に収穫されAustralian National University(ANU)により認証された糖料作物である。NISTのシュウ酸のpMCは95、ANU砂糖のpMCは150.61である(後者は、原子爆弾からの放射性降下物が依然として比較的高い時期に収穫されたため、100を超える)。
【0101】
この「pMC」法により、直接的にデータを処理し計算することが可能となる。しかし、この方法を用いるとき、好適には、dpm法を用いるときよりも、より厳しい標準化方法をとる。例えば、好適には、本発明の2つの態様において、同じ実験的手順を実行する。
【0102】
基質抽出手順において変更を行うことは通常できず、そのため一般的には難しくない。しかし、サンプルを一度HPLC上に噴射したら、全てのサンプルを全く同様に扱うべきである。特に、
1.検体のHPLCピークにまたがって、同数の留分をためるべきである。
2.留分プールを形成するために、各留分から同量の体積を採取するべきである。
3.AMS分析のため、留分プールを同量の体積に等分するべきである。
4.同量の炭素キャリヤ(例えば液体パラフィン(LP))を加えるべきでる。
【0103】
項目4の観点から、また従来技術で知られるように、同位体の量が実質無視できるだけの量の物質(キャリヤ物質)を含み、AMSにより分析したいサンプルよりもその物質が十分に多量であることが、AMSサンプルの準備において通常である。代表的には、存在するキャリヤ物質の重量は、分析したい精製サンプル(蒸発により除去される精製溶媒を除く)の重量の、100から1000000倍、代表的には100から10000倍大きい。これは2つの点で有利である。
第1に、従来のAMS分析において、そうでなければ取り扱うことが難しい定量サンプル(例えばマイクログラム程度の量)の分析が可能となる。
第2に、本発明において、過剰な対照検体がサンプルおよび試験サンプル内に含まれるため、当業者に知られるように、これらの量は、AMS機器は同位体比率の算出にもとづくため、AMS機器により得られる値に影響する。
【0104】
そのため正確に検体の量を算出するために、検体を標識化するAMS同位体以外の、同位体の量を知る必要がある。実際には、使用するキャリヤ物質の正確な重量測定により達成する。キャリヤ物質の量は、サンプルよりも極めて過剰であるため(以下で議論する)、AMS同位体と検体の定量化のために使用する比率を得るための、非AMS同位体源のみをサンプルが構成することを、仮定することが妥当である。
【0105】
キャリヤ物質をキャリヤと呼び、測定下で希少同位体を無視できる量しか含んでいない任意の物質とすることができる。
【0106】
上記の条件1から4が満たされるとき、標準曲線を準備するときに、例えば、留分プールのアリコートの分析により得られるpMC値は、紫外線ピーク領域(A:Bの比率で)で割ることができ、x軸上のC値に対してy軸上にプロットすることができる。試験サンプルに対して、検体ピークのpMC値は、紫外線ピーク領域で割ることができ、次に、試験サンプル内の検体濃度を確立するためy軸からx軸へ修正できる。
【0107】
x軸は1mLサンプルあたりのdpmか、もしくは1mLサンプル辺りの重量とすることができる。例えば、投与する14C薬物の比活性度から、dpm/mLで割ることで、pgもしくはng/mLを算出できる。較正ラインを作るために使用する14C検体の比活性度は、高くなるだろう(例えばおよそ2GBqもしくは50mCi/mmol)。しかし、標準曲線から臨床サンプルのpgもしくはng値を算出するために、投与した薬物の比活性度を用いる必要がある。例えば、100μgおよび200nCiのマイクロドーズを投与したとき、比活性度は2nCi/μgである。
【0108】
pMC法を使用することで、pMC値をdpm値に変換する必要がなくなり、次に行う通常の算出を含まない。このため、これは好適な方法であるが、分析にpMC法が不可能である場合に、以下で説明するdpm法を使用できることがわかる。
【0109】
おそらくこの状況では、同位体比率に基づきAMSが1グラムの炭素あたりのdpm値を与え、生体サンプル1グラムあたりのdpmのような絶対値を与えないことを、理解することは有用である。これが、AMSとLSCの結果の根本的な違いである。AMSは、放射性崩壊の事象数ではなく、14C原子の数を測定する。同位体比率をdpm値に変換することは、単に生物医学研究者になじみの単位を与えるためである。サンプル値のdpm/gを算出するために、サンプル中の炭素比率を知る必要があり、CHN分析により容易に求めることができる。
【0110】
この「dpm」法では分析手順において変更が可能であるが、より多くのデータ抽出が必要である。例えば、HPLCカラムが劣化するにつれピーク形状は広がり、当初の想定で標準曲線を作るために使用するよりも多くの、ピークをまたがる留分をプールする必要が生じる。従ってpMC法が好適ではあるが、例えば以下すぐに例示するHPLC精製の実用性からは、dpm法の使用を意味するだろう。
【0111】
上述のように、基質抽出方法において変更を行うことは通常できず、そのため、おそらくは難しくない。しかし、一度サンプルをHLPC上に噴射すると、
(a)検体のHPLCピークをまたいだ異なる数の留分をプールして標準曲線上で使用した留分と比較すること、
(b)AMS分析用のプールの投薬計画およびサンプルサイズを調整すること
が必要となる。
【0112】
条件(a)(b)が必要なとき、pMCデータをdpmデータに変換するべきである。この過程で、留分体積およびアリコート体積を算出に使用し、分析手順の相違を考慮に入れている。
【実施例】
【0113】
以下の例は、本発明の説明のためであり、限定するものではない。
【0114】
例1:較正曲線の作成
6個の1mLの対照血漿(分析する薬物に接触したことのない提供者からの人間血漿)サンプルを、個別のチューブに採取した。表1に示す標的濃度を達成するために、各チューブ内にいくらかの14C薬物を加えた。
【0115】
各チューブからアリコートを採取し、以下に説明するように、AMSを用いて直接分析した。表1に示すように達成した、各チューブの実際の濃度を、AMSの結果から測定した。
【0116】
【表1】

【0117】
次に、6個の血漿サンプル(それぞれ200μLのアリコート)を、標識化していない薬物を40μg/mL含む、正確な量の有機溶媒を用いて抽出した。その抽出は、窒素流上での乾燥により行い、200μLの溶媒内で再構成した。
【0118】
再構成した各サンプルのアリコート(50μL)を、HPLC上に噴射し、そのHPLC溶出液を、96wellプレート内に一連の留分として収集した。HPLCには、紫外線吸収検出機を備えた。薬物に対応するHPLCピークのピーク領域を、紫外線吸収により測定して記録した。
【0119】
分析する薬物を含む96wellプレート内の留分を特定するために、紫外線ピークの保持時間を使用した。この留分はAMSにより14Cを分析した。
【0120】
上記の分析により、以下の情報が得られた。
1)AMSにより直接測定した、dpm/mLでの、血漿サンプル内の実際の薬物濃度(全ての抽出もしくはHPLCに先立つ)
2)各サンプル内の、標識化していない薬物の濃度(血漿サンプルの抽出に、正確に等量が用いられる)
3)抽出およびHPLC分析に続く、各分析における標識化していない薬物に対応する紫外線ピーク領域(標識化していない薬物の量はμg程度の存在量であることに注意されたい。14C薬物もまた存在するが、pg程度の量であるため、紫外線ピーク領域に重大な寄与をしない)
4)AMS分析からの、HPLC留分用のdpm/mL値
【0121】
計算
まず、この結果は本発明により算出せず、従来の方法を参照する。従来の方法は単純に、HPLC画分(留分)内に存在する14C薬物の濃度をとり(以下の「試験サンプル測定」と題した章で説明する)、初めの血漿サンプルのdpm/mL濃度の推定値を算出する。この従来の方法は、多くの機会に文献で報告されている(RC Garnerら、薬物代謝排せつ,30(7),823−30.(2002);N Sarapaら,臨床薬理学,45(10),1198−205,(2005);JS Vogel,バイオテクノロジー,Suppl,25−9(2005);G Lappin ら,臨床薬理学,80(3),203−215(2006))。
【0122】
HPLC留分のAMSによる分析結果を表2に示す。
【0123】
【表2】
*抽出した200μLのうちの50μLを、HPLC上に噴射した。そのため、血漿内の濃度(dpm/mLと表記している)は、HPLC留分で測定される量の20倍である。
【0124】
分析が100%の効率(すなわち損失なし)で行われるならば、表2に記載の濃度は、初期血漿サンプルの分析結果(すなわち、表1で得た濃度)と同じはずである。表1と表2のデータの比較を、表3に示す。
【0125】
【表3】
【0126】
表3の結果は、チューブ1−5において、分析中におよそ58%にのぼる重大な損失が生じることを示す。しかし、表3に示す結果は、この分析において分析損失が重大で、従来のサンプル定量化方法はひどく間違っていることを、明確に示している。
【0127】
以下に、本発明により、分析結果が内部標準(標識化していない薬物)に関連することを仮定して算出し直した結果を示す。標識化していない薬物のピーク領域の結果を、表4に示す。
【0128】
【表4】
【0129】
表5は、留分中のdpmを紫外線ピーク領域(表4より)で割った値である。次にこれらの比率をグラフのy軸にプロットし、血漿中の実際の薬物濃度(表1で達成した濃度(dpm/mL))をx軸にプロットする。
【0130】
【表5】
【0131】
例2:試験サンプル測定
6.9498dpm/mLの薬物を含むように、血漿サンプルを準備した。40μg/mLの標識化していない薬物を含む抽出溶媒を用いて(200μLの)サンプルを抽出した。乾燥および再構成のため抽出物を200μLの溶媒内に採取し、50μLをHPLC上に噴射した。薬物のピーク領域を、紫外線吸収により記録した。一連の留分として溶出液を収集し、薬物に対応する留分の14CをAMSにより分析した。その分析を2回行った。このサンプルの分析結果を表6に示す。
【0132】
【表6】
*抽出した200μLのうちの50μLを、HPLC上に噴射した。そのため、血漿内の濃度は、HPLC留分で測定される量の20倍である。
【0133】
直接の方法(すなわち血漿濃度を、HPLC留分のAMSデータから直接算出し、較正曲線を用いない)による試験サンプルの分析により、4.306dpm/mLの平均値を得た。しかし、真の値は6.9498dpm/mLであった。従って、HPLCおよびAMS分析の結果は61%低かった。本発明を用いたサンプル分析により、(AMSにより測定した)留分中のdpm比率は、紫外線ピーク領域に対して0.000026567であった。較正曲線のy軸にこの値をとり(図1)、x軸の濃度により修正することで、7.04dpm/mLの平均濃度が得られ、真の値と1.3%のみ離れている。
【0134】
AMSによるサンプル分析
黒鉛生成の方法は、Vogel, J.S.による「生物医学AMS用の、汚染を生じない黒鉛の迅速生産」Radiocarbon, 34 344−350(1992)に従った。
【0135】
血漿サンプルを、黒鉛化チューブ内で直接等分(60μL)した。HPLC留分(代表的には100μL)を黒鉛化チューブ内で炭素キャリヤ(液体パラフィン、2.5μL)とともに等分した。黒鉛化チューブを、前もって熱した酸化銅のワイヤ(50±10mg)を含むサンプルチューブ内に投入し、Savant AES2010 Speed Vac.を用いて、真空中で完全に乾燥した。黒鉛化する炭素の最終的な量は、各ケースでおよそ2mgであった。
【0136】
工程管理したANU砂糖(AMS装置の較正用のよく知られたAMS標準;5−7mg)および人造黒鉛(2−3mg)を、前もって熱した酸化銅のワイヤを含む個別のサンプルチューブに投入する。2.5μLの液体パラフィン制御もまた、酸化銅を有する個別のサンプルチューブ内に投入した。全ての標準および対照を、上述のように真空中で乾燥した。
【0137】
燃焼(酸化)
乾燥したサンプルおよび酸化銅を含むガラスのサンプルチューブを、より大きなガラスの燃焼チューブへ投入し、カーボライト(Carbolite)加熱機において真空下で熱遮蔽して900℃で2時間熱した。燃焼後、チューブを大気温度までゆっくりと冷却した。サンプルおよび対照基質の酸化により、遮蔽したチューブ内で二酸化炭素が生じた。
【0138】
黒鉛化(減少)
より大きな燃焼チューブの終端部をY字マニホールドに接続した。コバルト粉末を含む(6.5±1.5mg)ホウケイ酸ガラスチューブを、甚句粉末と水酸化チタンの混合物を25:3w/wの比率(120−200mg)で含むより大きなガラスの黒鉛化チューブ内に投入し、その黒鉛化チューブをY字マニホールドのもう一方の端部へ接続した。燃焼チューブを、イソプロパノール/乾燥氷浴内に浸し、黒鉛化チューブを液体窒素浴に浸す。全体の系を真空下で設置する。燃焼チューブの端部を破壊した後、酸化したサンプルから生じる二酸化炭素を、黒鉛化チューブへと低温で移す。一度移したら、黒鉛化チューブを真空化で熱遮蔽し、加熱機内に設置して500℃で4時間熱し、その後さらに550℃で6時間加熱したあと大気温度までゆっくりと冷却した。
【0139】
カソードの黒鉛による梱包
黒鉛化過程が完了したら、カソード内で梱包する準備ができるまで、その黒鉛を遮蔽した黒鉛化チューブ内に残した。カソードを梱包するために、黒鉛化チューブを開き、黒鉛を含むホウケイ酸ガラスチューブは、コバルト触媒上に吸収して除去した。そのコバルト/黒鉛を、慎重にアルミカソード内へ出し、Parr Pellet Press内で100−200psiで圧縮し、カソード内に黒鉛のタブレットを形成した。全ての圧縮後、メタノールで湿したティッシュで拭くことで、圧縮機を洗浄した。次に、サンプルおよび工程管理カソードを、室温での貯蔵のため標識化したプラスチックキャップのチューブ内に設置した。分析が必要な時、これらのカソードを、以下に示す他の機械の標準および対照により、134−位置AMSサンプルホイール内に設置する。
【0140】
【表7】
【0141】
AMS装置を調整するため、もしくは、データの正規化に使用するプールしたANU砂糖カソードを除いて工程の効率を測定するために、上述のカソードを使用した。
【0142】
AMS手順
5MV 15SDH−2 ペレトロン(Pelletron)AMSシステム(National Electrostatics社製)を用いて、AMS分析を行った。黒鉛含有カソードを設置したサンプルホイールを、AMS装置のイオン源内部に挿入した。マルチカソードの陰イオン源(MC−SNICS)は、黒鉛の表面上で加速されるセシウムイオン(Cs+)ビームを生じた。結果の炭素陰イオンビームは、12C-,13C−,および14C−、および16O−のような他のイオンを含んでいた。同重体14N−は不安定で、そのため14Cの測定を阻害しない。
【0143】
炭素イオンビームを前もって加速し、球面静電分析器を通し、次に入射磁石へ向け進ませた。12C−の出力は、代表的には1−100μAである。12C−(150μs)、13C−(600μs)、および14C−(0.1s)を通常68keVで順番に射出し、対応する1サイクルに対して各同位体の測定に1つを結合するように、磁石を設定した。タンデムペレトロン(Pelletron)加速器の中心正極へ向けて、エインゼル(Einzel)レンズを通して炭素イオンビームを加速した。およそ17.5から22.5MeVの粒子エネルギーをもつよう、この一連の分析に用いるターミナル電圧は3.5から4.5MVとした。中心電極では、正に帯電した炭素イオン(12,13,14C+1から+6)を生じるために、炭素原子から電子を奪った。測定のために、このエネルギーにおいて最も豊富となるように、C4+イオンを選択した。これらのイオンを中心電極から遠ざかる方向へ、静電四重極トリプレットおよび分析磁石へ向けて加速した。
【0144】
事後分析磁石を通過してすぐに、12C4+および13C4+イオンを、オフセットファラデーカップ内でイオン電流として計測した。14C4+イオンは、静電四重極ダブレットおよび円筒形静電分析器を通して、高エネルギービーム線上を通過させた。ここから、ガスイオン化検出器へイオンを入れ、それらのイオンを、各イオンのエネルギー損失および全エネルギーを測定する(全部で4個の)アノード上に集めた。一般的に、他の非干渉14C4+イオンは、静電分析器、磁石、スリット、および荷電分離の組み合わせにより、ガスイオン化検出器には入らなかった。ビーム線上でおよそ10−9Torrの真空圧力を、イオン源で10−6Torrの真空圧力を維持した。装置によるイオン転移は30−60%であった。
【0145】
データの処理
AMSデータを、準備したサンプルのdpm/mL値の算出に使用した。AMSの結果はpMCで表現した。100pMCは、
13.56dpm/g C もしくは 0.01356dpm/mg Cに等しいか、
もしくは、98フェムトモルの14C/g C(1フェムトモル=10−15モル)に等しいか、
もしくは、98アトモルの14C/mg C(1アトモル=10−18モル)に等しい。
従って、サンプルの密度を1g/cm3と仮定すると、以下のようになる。
pMC×0.1356 = dpm14C/g C
および (dpm14C/g C)×(サンプル内の%w/v C)=dpm14C/mL
サンプルの[14C]/[12C]比率 =
全[14C](薬物+生体サンプル+キャリヤ1)
全[12C](薬物+生体サンプル+キャリヤ1)
1−適用可能な場合
【0146】
数学的処理
以下の議論は、本明細書内で上述した、本発明と同様のコンセプトの説明であるが、本発明により提供する内部標準化の数学的な表式に関する議論である。上述の議論と同様に、以下の議論では14Cで標識化した合成物により例示するが、当然のことながら、そのコンセプトは他のAMS同位体標識合成物に対して等しく適用できる。
【0147】
薬物の発展の間、例えば特定の研究形態における14C薬物濃度測定において、AMSがますます用いられ、生体サンプル内の全薬物および代謝物(例えば14Cもしくは他のAMS同位体の濃度)の測定のために式が十分に確立されている一方で、検体のクロマトグラフィ分離およびAMSによる分析の結果生じる損失を求める式は導かれていない。そのような式を導けば、本明細書の上述の議論から当然のこととして、正確に検体濃度を測定するために、分析中に生じるあらゆる質量損失を考慮することができる。本発明の恩恵により、分析中のあらゆる質量損失の見積もりに関する本発明の観点による方法において、14C−および他のAMS同位体含有検体のクロマトグラフィ分離およびAMS分析を説明する式を導くことができる。
【0148】
同位体比率の方法として、AMSは検体内に、代表的には12Cおよび14Cのような少なくとも2種の同位体を、薬剤物質として必要とする。同位体比率を測定するために、代表的にはサンプルを黒鉛に変換し、次に分析のためAMS内に設置する(J.S.Vogel,生物医学AMS用の、非汚染な黒鉛の迅速生産、Radiocarbon,34(1992)344−350)。AMSは極めて高感度で、14C薬物の特定の活性度に応じて、フェムトグラムもしくはアトグラムの範囲(10−15から10−18g)の14C薬物濃度の測定が可能である(G.Lappin,R.C.Garner, 加速質量分析器を用いた、放射性標識薬物およびその代謝物の超高感度検出,I.Wilson編,分析用分離のハンドブック,Elsevier,Amsterdam,2003,pp.331−349)。その超高感度のため、本明細書に上述のように、マイクロドージングおよび絶対バイオアビリティ研究のような技術に対して、AMSを使用している。共通に入手可能なHPLCとAMSとの間のインタフェースがないため、HPLCにより血漿抽出を分析し、対象の検体の保持時間に対応するHPLC溶離剤の留分を、12C:14Cの同位体比率を測定する前に「オフライン」で黒鉛化する(I.N.White,K.Brown,Techniques,薬学および毒物学への加速器質量分析の適用,Trends Pharamacol Sci 25(2004)442−7)。
【0149】
VogelおよびLoveは、生体サンプルで測定した同位体比率を薬物濃度へ変換する方法を説明し、式1により表わされる(J.Vogel,A.H.Love,加速器質量分析器による同位体分子標識の定量化,A.L.Burlingame編,Methods in Enzymology,Academic Press,New York,2005)。
K=(RM−RN).Ψ.W/L (式1)
この式において、Kは検体の濃度、RMは同位体比率、RNは生体サンプル中の自然バックグラウンドの同位体比率、Ψはサンプル中の炭素質量分率、Wは検体の分子量、そしてLは検体の比分子活性度である。
【0150】
同位体比率をAMSによりModernという単位で表し、1 Modern=98アトモル14C/mg 炭素 である。同位体比率は炭素の質量に対して表わすので、サンプル中の質量分率(Ψ)は、炭素の質量あたりの濃度を、サンプルの質量あたりの濃度へ変換する式中に含まれる。通常の炭素分析器により、質量分率を測定する。
【0151】
(RM−RN)をRnetとして表わして式1を簡略化することができ、また式2のように、モルの代わりに比活性度を質量(LMass)に対して表わす。
K=RnetΨ / Lmass (式2)
【0152】
式1および式2は一般的に確立されているが、薬物が代謝化されているとき、式の結果は親薬物と代謝物(すなわち質量等価物)の混合物の全14C(これより例として14Cを参照する)しか提供しない。存在する個別の検体の濃度を求めるために、本明細書に前述して説明したように、AMS分析の前に、代表的にはサンプルを抽出し、検体をHPLCのようなクロマトグラフィ技術により分離しなければならない。今までのところ、この過程を記述する式は報告されておらず、本発明の恩恵により、ここに初めて導かれる。
【0153】
検体がHPLCに続く全てのバックグラウンド炭素から分離されたと仮定すると、RN=0である。従って、分離検体の同位体比率からRnetを判別するために、後者をRAと指定する。実験動物もしくは人間へ投与した薬物の比活性度(Lmass)は生化学過程では変化せず、そのためHPLCにより分離した検体の比活性度は、親薬物の比活性度と等しいはずである(すなわちRA=L)。式2に代入すると、K=Ψであり、言い換えれば、HPLC留分内の検体量は、その留分内の検体からの炭素量と等しく、十分に同値である。そのため、分離した検体の濃度は、その同位体比率から求めることができないことがわかった。しかし、上述の説明のように、分離した検体へ14Cではなく12Cを加えて同位体希釈を行うことで、この制約を克服することが可能である。このような同位体希釈物(本明細書に上述したキャリヤ物質)は、かなり古くそのため14C(14Cのt1/2=5760年=106年あたり173半減期)が崩壊して失われた石油化学源からの炭素として入手可能である。この種の代表的な同位体希釈物は、液体パラフィンである。AMS分析における液体パラフィン使用は十分に確立され、液体パラフィンキャリヤ物質は、主に極めて小さなサンプルを扱えるサイズまで多くするために使用するため、「炭素キャリヤ」と呼ばれる(G.Lappin,S.Temple,加速器質量分析器,薬物発達における放射性追跡子,Tayler and Francis CRC Press,Florida,USA,2006)。しかし、HPLCにより分離した留分の場合には、これもまた本明細書で上述したが、炭素キャリヤは、同位体希釈という追加の目的を果たす。
【0154】
クロマトグラフィによりサンプルから精製した検体内の14C:12Cの比率をRAとする(上述のように)。同位体希釈のためにRAに加える12Cの量(すなわち炭素キャリヤ内の12Cの量)をΦとする。同位体希釈後の生成物の14C:12Cの比率をRDとする。式2に代入することで、式3を得る。
K=RDΦ / Lmass (式3)
【0155】
式3は、同位体希釈が正確に既知であることが好適であることを示す。すなわち、あらゆる誤差は、最終結果の誤差へ直接影響する。炭素キャリヤの量を正確に投与し、それにより既知とするが、例えば抽出中やHPLCカラム上での、あらゆる検体の分析損失もまたKの測定に対して誤差を導く。従って、分析回復を表す他のパラメータ(θ)を、式3に導入し、式4を得る。
K=RDΦ/ Lmassθ (式4)
【0156】
一般的な実験的回復を簡単に行うことでθを計測することが可能であり、またその回復が再現可能であることが示されているケースではその計測は容認できる一方で、分析する各サンプルに対して個別に回復を測定することがより望ましい。このことは、本明細書に上述したように、内部標準を使用する定量的HPLC分析で通常用いるものと同様の方法で達成できる。これらの合成物は限定的なアベイラビリティを有し、しばしば製造が比較的高価であるため、一般的に、14Cで標識化した内部標準の使用は実用的ではない。しかし、その代わりに、内部標準として放射性標識化をしない検体(対照検体)を使用し、紫外線吸収のような通常の検出方法により各サンプル内の濃度を測定することが可能である。
【0157】
対照検体をサンプルへ追加しても、代表的には14CであるAMS同位体の存在により識別可能であるため、検体の測定に支障は生じない。対照検体からサンプルへ加えた少量の12Cは、炭素キャリヤ内の12Cと比べて多くない(上述のように、また当業者によく知られるように、代表的にはμg程度の量の対照検体を加え、mg程度の量の炭素キャリヤを用いる)。さらに、一定に等量に正確に既知の量の対照検体を加えるとき(代表的にはそうであるが必ずしも必要ではない)、全ての有効なサンプルは同じ全濃度の検体を含み、そのため濃度に依存する影響は最小限となる。AMS分析において、サンプル中の検体濃度はしばしば極めて小さく、非特異結合による損失が大きくなりうることに留意しておくことは有利である。余剰の対照検体を追加することで、これらの非特異結合の効果を克服し、それにより回復を改善する。
【0158】
上述のように、内部標準として対照検体を使用する手順は、HPLC分析で用いるものと類似の手順に従う。まず標準曲線を作り、それにより一連の血漿サンプルを混合して、内部標準となる既知で一般的には等量の対照検体とともに、14C−もしくは他のAMS同位体標識化薬物の濃度を上昇させる。各混合較正剤内の、14C−もしくは他のAMS同位体標識化薬物の「真の」濃度を、正確に投与した量から求める。さらに、この環境下では、14Cの全濃度は14C薬物の濃度と等しいため、実際の測定値(本明細書内で上述の量「C」)を得るために、較正剤のアリコートを直接AMS(式2)により分析することができる。各血漿サンプルを抽出し、その抽出はHPLC上で行い、検体の保持時間に対応する留分を収集して(代表的には)黒鉛化して、12C:14Cの比率(代表的にはModernで表す)をAMSにより測定する。さらに、対照検体の紫外線応答のピーク(内部標準)を測定する(上述のように、例示として紫外線を使用するが、任意の適切な測定技術を用いることができる)。次に、真の濃度(「C」)をx軸に、HPLC留分のModern値(「B」)をその紫外線応答(「A」)で割った値をy軸にとって、標準曲線を作る。AMSは、14C検出が飽和する点まで線形に応答する。飽和により検出器は損傷してしまうが、飽和することは避けるため、実際にはその応答は常に線形である(L.K.Fifield,加速器質量分析およびその応用,Rep Prog Phys62(1999)1223−1274)。そのため、ラインは線形回帰による較正データに一致する。ここに説明する標準曲線は、装置応答を較正しないため、通常のHPLC較正ラインとは異なることを、理解しておくことは重要である。当業者によく知られるように、例えばAustralian National University砂糖もしくはシュウ酸のような、正確な量の12C:14Cの比率の分離標準を用いて、AMS装置を較正できる(G.Lappin,S.Temple,加速器質量分析,薬物発達における放射性追跡子,Taylor and Francis CRC Press,Florida,USA,2006)。
【0159】
分析下の各サンプルに対して、既知の(通常は上述した較正方法で使用するものと同様の)量の対照検体を内部標準として加える。サンプルを抽出し、HPLC上で実行し、検体の保持時間に対応する留分を収集し、黒鉛化し、Modern値をAMSにより測定する。上述の標準曲線と同じ方法において、紫外線応答もまた測定する。次に、y軸上の紫外線応答で割った留分のModern値は、x軸上の対応値から薬物濃度を決定する。y軸とx軸の間の対応は、曲線の傾きを規定し(線形のとき、その式はy=mx+c)、次に式3に代入して式5を得る。
K= (RA/mU+C)Φ/Lmass (式5)
式中、KはHPLC留分中の検体量、RAは同位体希釈後の検体のModern値、Φは同位体希釈物である12Cの量、Lmassは検体の質量比活性度、mは標準曲線の傾き、Uは紫外線応答(もしくは任意の適切な検出方法の検出応答)、およびCは標準曲線のy切片である。
【0160】
上述のように作る代表的な標準曲線を図2に示す。図2は、それぞれが5つの分離複製からなる14Cで標識化した薬物の7つの異なる濃度に対して、C値に対するB:Aの比率をプロットして得られたさらなる較正曲線である。この特定の標準曲線において、5つの分離複製からなる14C薬物の7つの濃度を使用した。炭素キャリヤとして液体パラフィンを使用した(1.6266mg 12C)。内部標準である放射性標識化していない検体の濃度は、35μg/mLであった。ラインの傾きは0.1359で、cの値は−0.0866であった。実際には、cの値はしばしばわずかで無視できる。図2に示す標準曲線の範囲は、1fgから160pg/HPLC留分であった。
【0161】
4fgから120pg/HPLC留分と等価な範囲を達成するために、14C薬物を混合して正確に希釈した血漿を、HPLCおよびAMSにより抽出した。標準曲線から求めた濃度は、平均で真の値の97.2%(n=25)であった(84.2−118.8%の範囲)。標準曲線を用いず、分析損失を考慮しないとき(すなわち式3)、混合血漿サンプルの濃度は、真の値の52.8−92.3%の範囲で、平均で69.1%(n=25)と求まった。従って、平均分析回復はおよそ72%であった。そのため、全く回復を考慮しないと(すなわち式4のθを1と仮定する)、結果は平均の誤差が−28%を示した。この誤差は、本発明による上述の内部標準方法を用いて、平均で2.8%減少した。
【技術分野】
【0001】
本発明は、対象のサンプル中の、特定の同位体標識検体の量を決定する正確性を改善する方法に関する。特に、本発明は、加速器質量分析(AMS)を用いた検体の定量化時の内部標準化を可能とする。
【背景技術】
【0002】
代表的には液体クロマトグラフィ質量分析(LC−MS)を用いて行う生物分析は、通常、臨床研究(フェーズI、II、およびIII)中、および動物サンプルによる毒物研究中にもしばしば行われる。
【0003】
LC−MSにおいて、従来のHPLC手法と、質量分析器のイオン源内を通過したHPLCからの溶離剤を用いて検体を分離する。ここで、複数あるイオン技術(例えば電気スプレーもしくは大気圧イオン化(API)など)のうちの1つにより検体をイオン化する。イオン化エネルギーによっては、検体の分子分裂が生じることがある。正もしくは負のどちらかに帯電したイオンは、その荷電のためにイオン源から引き出される。イオンの質量(m)を荷電量(z)で割った値に従って、これらを磁場中もしくは四重極磁場中で分離する。多くの場合、特に小さな分子の場合には、質量スペクトルの結果により検体の分子量を解明できるようにzは1である。使用するイオン化の種類によっては、H+を加えることでしばしば正イオンをつくり、そのため分子量を表すイオン(すなわち分子イオン)は[M+1]+と表記する。その逆である、H+の抽出による負イオンの形成もまた起こり、分子イオンは[M―1]― と表記する。
【0004】
ガス衝突セルにより隔離された、連続する2つの質量分析器を含む装置がある。これらはタンデム質量分析器として知られる。質量分析器が四重極であるとき、タンデム質量分析器は三連四重極質量分析器として知られる。イオンは第1質量分析器から衝突セルに流入して(親イオン)分裂し、第2質料分析器によりその断片(娘イオン)を検出する。これは、LC―MS/MSとして知られる。
【0005】
質量スペクトルは、イオンの絶対分子量を表す。例えば、2−ヒドロキシ―2ピロール―キノリンの平均分子量は、炭素、水素、窒素、および酸素の同位体を考慮すると、210.24である。しかし、質量分析器は個別の同位体の含有量をそれぞれ検出する。2−ヒドロキシ―2ピロール―キノリンでは、炭素同位体の含有量が支配的であり、従って[M+1]+、m/z211イオンは12Cに基づく。m/z212のイオンは13Cに基づき、m/z213イオンは14Cに基づく。14Cの天然存在度は極めて小さく、代表的には7.4×1011個の炭素原子中に、ただ1個の14C原子が含まれる。
【0006】
生物分析は、一般に極めて大規模に行われ、上述の議論にもかかわらず、代表的には放射性標識化薬物の検出および定量化を含まない。
【0007】
生物分析において、代表的には、例えば親薬物(例えば人体、動物、もしくは他の生物系に投与する分子など)および、しばしば多くの特定代謝物のような、標的分子のためにサンプルを分析する。簡単のため、本明細書において「標的分子」といえば、分析対象であり、例えば未知の薬剤候補もしくは推定薬剤候補もしくはその代謝物といった、対象の分子を指すと理解されたい。臨床試験に参加した多くの被験者からの薬物動態データを作るために、生物分析の結果を使用する。
【0008】
LC−MSを用いる生物分析において、(例えば血漿溶媒抽出により)生体サンプルを抽出し、その生成抽出物をLC−MSにかけて標的分子を検出し定量化する。しかし、このような定量化は以下の理由で複雑である。
1.例えば血漿からの標的分子の抽出は完全でなく、未知の量が抽出されずに残留する。さらに、サンプルによりその量は異なり、抽出は濃度に依存する。
2.(MS分析に先立つ)HPLCによる分離中に損失が生じる。言い換えれば、HPLCのカラム回復は100%ではない。1と同様に、前記カラム回復はサンプルにより異なり、濃度に依存する。
3.質量分析器は、検出の前に合成物をイオン化する。イオン化の過程は極めて合成物依存が高く、イオン化効率は時間とともに変化しうる。従って、基準を参照せずに質量分析器により定量化することができない。
【0009】
これらの難点を補うために、標的分子の定量化では、一般に較正が必要である。そのために、代表的には極めて高純度な以下の2種類の物質が必要である。すなわち(1)参照基準としての標的分子自体と、(2)内部標準として使用する、標的分子と極めて似た化学構造を有する合成物とである。クロマトグラフィの保持時間が重水素化していない標的分子に極めて近いが、質量分析のステップ中に分子量が大きいことによりLC−MS内で区別できるため、理想的な内部標準としては、一般的に重水素化した標的分子が考えられる。しかし、重水素化標準は常に入手できるわけではない。重水素化内部標準が入手できない場合、標的分子とは構造が異なる合成物(すなわち類似体)を使用できるが、可能な限り標的分子と構造が似ているべきである。
【0010】
較正のため、一般的に対照基質(例えば血漿)を、対象の標的分子にかつて接触したことのないドナー(例えば人間)から取り出す。一連のそのような制御血漿サンプルそれぞれに、異なる濃度の参照基準標的分子を加える(例えば、8種のサンプルは、10,20,50,100,500,1000,1500および2000pg/mLの参照標的分子を、他の例としては、100,150,200,250,500,1000,1500および2000pg/mLの参照標的分子を含む)。追加的に、既知で等量の内部標準を、各制御血漿サンプルへ加える(例えば500pg/mLの内部標準)。
【0011】
そうして準備した各サンプルを、適切なクロマトグラフィの方法(例えばLC−MS)で分析し、標的分子および内部標準の量を求める。例えば標的分子の濃度(制御血漿サンプルに初めに加えた量から既知)をx軸上にプロットし、分離した標的分子の量と内部標準の量(これらの量はLCMSにより求める)の比をy軸上にプロットした、較正グラフもしくは較正ラインを作成するために、得られるデータを使用する。ここで、そのようなプロットは必ずしも線形ではないことに注意されたい。
【0012】
次に、臨床試験もしくは他の試験により得た実際のサンプル内に存在する標的分子の、未知の濃度を定量化するために、各サンプルへ、正確に既知の量の内部標準を加える。LC−MSにより、標的分子および内部標準の量を求めるために、そのサンプルを抽出し、分析する。対応する標的分子濃度を導くために、検体:内部標準の比と、較正グラフ/ライン上で対応する標的分子濃度とを比較するために、これらの量の比を使用できる。
【0013】
サンプルの準備中などに標的分子の損失が生じたら、較正の比率性により許される内部標準の比例損失により修正する。同様に、LCMS技術により損失もしくは他の失敗もしくは標的分子の検出不足が生じたら、これらの損失を対応する内部標準の損失に反映するべきである。従って、実際には較正ラインによって、質量分析器の方法、理論的損失および反応が特徴づけられる。
【0014】
上記の通常の生物分析に追加して、代表的には14Cで標識化する、放射性標識化薬物を用いた関連実験もまた行える。そのような実験は、一般的に、臨床試験に関連する多数のサンプルにより行われることはないが、薬物の代謝作用や薬物動態を調べるために設計される明確な実験である傾向がある。そのような研究において、一般的に薬物濃度を定量化するためにLC−MSは使用しない。代わりに、代表的には標的分子は、放射性追跡子技術を用いて、放射能レベルを求めることで定量化する。この理由は基本的には、このような研究における標的分子(もしくはおそらく特定の代謝物)は、一般的に既知であり、容易に分離することができるためである。従って、識別は難しくないため、定量化のみが必要である。しかし、親薬物のような特定の標的分子を放射性追跡子技術により定量化する場合、放射性標識化されていない標的分子と14Cで標識化した標的分子をともに入手できるとしても、適切な14Cで標識化した内部標準は入手できないだろう。このため、放射性追跡子を用いた標的分子の定量化は、伝統的に、分析の絶対的な方法により行われている。このとき、HPLC溶出液内の放射能の量の計測が必要で、その結果は直接、初めの基質内の標的分子濃度と等しいと見なされる。
【0015】
放射能追跡子の使用は、放射能の測定が直接、存在する標的分子の量に関係する点で有利である。LC−MSと異なり、考慮すべきイオン化効率の要因はない。それにもかかわらず、サンプル抽出に関連する不正確性およびクロマトグラフィカラムで生じうる損失については、この場合にもあてはまる(すなわち上述の項目1および2)。このため、計測する放射能を、初めの基質内の標的分子濃度に直接的に等しいと見なすことは、はっきり言えば妥当な仮定であるとは言えない。
【0016】
さらに、液体シンチレーション(LSC)のような放射性追跡子技術は、本質的に感度が鈍いことに悩まされる。例えば、14CのLSCによる測定において、初めにサンプルを液体シンチレーションの混合物内に溶解する。崩壊事象中に14Cから放出されるβ線のエネルギーは、液体シンチレーション溶液を活性化する。エネルギーの基底状態に戻る際に、シンチレーション溶液は光量子を放出し、その光量子を光電子増倍管により検出する。従って、シンチレーション溶液が放出する光量子の数は、放射性崩壊の発生数に比例する。しかし、14Cの半減期は5760年であるため、一度に崩壊するサンプル内の14C原子は比較的少ない。実際、平均して1壊変毎分(dpm)を生じるために、10億個以上の14C原子が必要である。
【0017】
放射性追跡子技術を使用する代案として、サンプル中に存在する標的分子の量を直接定量化するために、AMSもまた使用可能である。
【0018】
AMSは特定の同位体の量を測定する技術である。AMSは、1970年代に考古学の炭素年代測定のため発明され、1990年の薬学研究(Garner,R.C.(2000)、「薬学研究における加速器質量分析および開発―同位体測定の新たな超高感度分析方法」、Curr Drug Metab. 1(2)205−213)に初めて使用された。
【0019】
LSCとは異なり、AMSは、検出器に到達するために1000個の原子のみを必要とする極めて高感度な分析方法である(Lappin, G.& Garner,R.C.,「加速器質量分析を用いた、放射性標識化薬物およびその代謝物の超高感度検出」、分析的分離のハンドブック、I.Wilson編、2003、Elsevier:Amsterdam.p.331−349)。AMSは同位体比の技術であるので、分析する物質において珍しい同位体を濃縮しなければならない。他の同位体も使用可能であるが、生物医学研究において、共通して14Cをこの同位体とする(その場合、12C:14Cの同位体比を測定できる)。
【0020】
AMSは生物医学および他の研究に多くの用途を有するが、現在の議論に関連するのは、所与の基質内に極めて低濃度で存在する合成物の測定に使用可能であることである。AMSの最も重要な利点の1つは、比較的短い分析時間で、被験者に与える必要のある放射線量が、規制認可を必要とする規定の放射能レベルを下回るほどに低い放射能レベルの、検出と定量化が可能である点である。代表的には、分析する合成物は製剤原料、もしくはその代謝物であり、その基質は生体サンプル、すなわち被験者もしくは被験動物から得られた例えば血漿といった生体サンプルである。しかし、その分析する合成物は任意の基質中の任意の合成物とすることができる(例えば土壌中の環境汚染物質)。
【0021】
上記のように、基質中における標的分子濃度の共通の測定方法はLC−MSである。生物医学研究において、代表的にはLC−MSは、mLサンプルあたり100pgの合成物を測定できる。執筆時(2007)にはLC−MSはより高感度を達成している(例えば10pg/mL)。しかし、AMSは通常、当業者には容易に達成可能な、通常のある最適化方法により、1pg/mLの感度を達成でき、その感度はフェムトグラムもしくはアトグラム(10−15−10−18g)の範囲まで達することができる。
【0022】
従って、AMSは、より高感度な分析が必要とされる生物医学研究で、分析ツールとして使用されている。分析する合成物は、代表的には14CであるAMS同位体の濃縮した量を含み、合成物の濃度を測定するために、AMSはその同位体比を測定する。LC−MSにおいて合成物は同位体濃縮を必要としないが、その感度はAMSで得られるレベルには達しない。
【0023】
代表的には、AMS分析において、まず分析する合成物を、HPLCによる分離前に見つかる基質中から抽出する。一連の留分としてHPLC溶離剤を収集し、対象の合成物に対応するものを、AMSによる分析のために等分する。執筆時(2007)には、特許文献1(国際公開第95/04369号)に詳細が示されているにもかかわらず、効率的に分離ステップを(例えばHPLCによって)AMSへ統合する、通常のインタフェースが存在すると信じられてはいない。
【先行技術文献】
【特許文献】
【0024】
【特許文献1】国際公開第95/04369号
【発明の概要】
【発明が解決しようとする課題】
【0025】
この方法は、抽出過程および分離過程において分析上の損失がないことを仮定している。抽出という語は、分析する合成物を、それが見つかる基質中から初期精製する1つもしくはそれ以上のステップを意味する(例えば分析する合成物および他の合成物を、血中で見つかるタンパク性材料から分離するステップ)。分離とは、さらなる1つもしくはそれ以上の精製ステップを指し、代表的には抽出ステップ後に存在する、基質中の全ての残留成分から、分析する合成物を分離することで精製することを指す。しかし、その特性によっては、多量の結合を示し、そのために抽出中およびHPLC分析中に損失が生じる合成物がある。従って、従来のAMS分析の結果は、真の値よりもかなり低い結果となる。このことは、AMSによって極めて少量の合成物を分析することが必要なときに、例えば抽出中および精製中の結合効果および実験的損失により、特に問題となる。結果として、AMSに内在する、AMSの同位体標識化標的分子の少量検出の能力は、少量の材料を定量化するとき誤差が増すと考えられる。
【0026】
本発明は、1つもしくはそれ以上の上述した従来技術の問題点を改善することを意図するものである。
【課題を解決するための手段】
【0027】
AMSによるサンプル分析の準備として、結合および実験的損失により生じる不正確性や誤差に対処するための手順を、我々は開発した。その手順では、AMS分析用の合成物を、例えば14CのようなAMS同位体で濃縮することを利用する。
【0028】
本発明は、AMSによる、AMS同位体標識化検体(本明細書内では「検体」)の定量化時の内部標準化の方法に関する。従って、検体は、定量化が望まれ、AMS同位体で標識化した全ての対象となる合成物となる、AMS同位体標識化合成物である。上記の生物分析で使用するものと類似の標準化手順でありながら、AMS同位体により標識化していない検体(本明細書内では「対照検体」)と構造的に区別できる合成物を内部標準として使用するという、根本的な相違点を有する内部標準化手順を、本発明は使用する。
【0029】
従って、1つの態様から見ると、本発明は、試験サンプル内のAMS同位体で標識化した検体量の測定に用いる較正方法を提供するものであり、前記較正方法は、
(i)複数の較正サンプルを提供するために、前記検体および標識化しない対照検体のどちらにも汚染されていない複数のサンプルを、既知量の前記対照検体および量Cの検体へ接触するステップにおいて、前記較正サンプルがそれぞれCとは異なる既知量の対照検体を含むことを特徴とするステップと、
(ii)AMSにより、複数のサンプルそれぞれに加えた検体の量Cを測定するステップと、
(iii)複数の精製サンプルを提供するために、検体および対照検体を、複数のサンプル内の他の化学種から分離するステップと、
(iv)AMSにより、前記精製サンプル内の検体の量Aを測定するステップと、
(v)AMSにより、前記精製サンプル内の対照検体の量Bを測定するステップ
とを含むことを特徴とする。
【0030】
前記精製サンプル内の対照検体の量Bを測定するとき、ここでは対照検体のみを測定するだろう。代案として、測定技術が対照検体と検体を識別できないとき、この測定は対照検体と検体にともに可能である。以下でより詳細に説明する後者の場合、このような測定は、存在量が十分に検体よりも多いならば、対照検体のみに対すると考えられる。
【0031】
本発明の第1の態様による方法から得るデータは、その方法を用いる前にAMSにより求める前記サンプル内に存在する検体の絶対量Cを、比率A:Bに関連付けるために使用できる。従って、検体量は未知であるが存在する対照検体量は既知の試験サンプルに対して、上記の方法で実行する条件と同様の条件下でこの比率を求めるとき、本発明の第1の態様による方法から得るデータを参照して、試験サンプル内に存在する検体量を求めることができる。
【0032】
この態様から見ると、本発明は、試験サンプル内のAMS同位体で標識化した検体量の測定に用いる方法を提供するものであり、前記方法は、
(i)前記試験サンプルを、既知量の標識化しない対照検体に接触させるステップと、
(ii)複数の精製試験サンプルを提供するために、もし存在すれば検体と対照検体を、試験サンプル内の他の化学種から分離するステップと、
(iii)AMSにより、前記精製試験サンプル内の検体の量Aを測定するステップと、
(iv)AMSにより、前記精製試験サンプル内の対照検体の量Bを測定するステップ
とを含むことを特徴とする。
【0033】
本発明の第1の態様の実行と同様に、精製試験サンプル内の対照検体の量Bを測定するとき、ここでは対照検体のみを測定するだろう。代案として、測定技術が対照検体と検体を識別できないとき、この測定は対照検体と検体にともに可能である。以下でより詳細に説明する後者の場合、このような測定は、存在量が十分に検体よりも多いならば、対照検体のみに対すると考えられる。
【0034】
精製試験サンプルのA:Bの比率と、本発明の第1の態様の実行により、A:Bの比率を既知の値Cによって補正して得るデータとを比較することで、本発明の第2の態様の方法で分析する試験サンプル内の検体の未知量を決定できる。
【図面の簡単な説明】
【0035】
【図1】本発明の較正方法の実行により得るデータから、Cに対するB:Aの比率をプロットして得る較正曲線である。
【図2】本発明の較正方法の実行により得るデータから、Cに対するB:Aの比率をプロットして得る、さらなる較正曲線である。
【発明を実施するための形態】
【0036】
本発明は、上述したような生物分析とは異なる。例えば、AMSで同位体標識化した検体を含むことが本発明において要求される。第2に、生物分析において内部標準は、できる限り構造的に密接に検体と関連する必要がある。本明細書で説明する方法において、内部標準は、標識化した検体に対して標識化しない対照物である。この方法は、検体が、AMS同位体の存在により内部標準と区別可能であるために可能である。2つの分析方法を用いて、すなわちAMSにより検体の量を測定し、対照検体の量を異なる分析方法で測定する。
【0037】
以下の議論では、本発明の方法を14Cで標識化した検体の測定での使用、特に例えば臨床試験で得る生体サンプル分析における使用に頻繁に言及するが、本発明はそれに限定されるものではないと理解されたい。
【0038】
サンプルが臨床サンプルである場合、臨床試験中、検体の投与後に一定時間が必要となるだろう。さらに、その臨床サンプルは、所望ならばあらかじめ放射化したサンプルを含むこともできる。
【0039】
また当然のことながら、本明細書において、本発明の方法による、効果的な分離を行うときのHPLC使用を重視する。しかし、本発明はそれに限定されるものではないと理解されたい。精製試験サンプルの生成物のAMS分析が、精製試験サンプル内の検体量の正確な定量化を可能とするように、他の標識化した合成物から対象の検体を分離するために使用する場合には、あらゆる分離方法(HPLCもしくは他による)が、本発明の方法の実行によって使用できる。例えば特許文献1に記載されたような、AMS機器との直接的な接触を可能とする分離方法を含みうる。しかし代表的には、分離ステップおよびAMS測定ステップは接触していない。
【0040】
本明細書において以下の定義を使用する。
【0041】
未知の薬剤候補の臨床試験において、例えば検体は、そのような臨床試験により試験する親薬物、もしくは親薬物の代謝物とすることができる。一般的に検体は、14Cで標識化した合成物であり、本明細書の議論はそのような14Cで標識化した合成物に注目する。しかし、少量の他のAMS同位体を検出する能力があれば、検体はAMS同位体で標識化した任意の合成物とすることができる。
【0042】
全ての原子には同位体があり、その中にはAMS分析に適したものがある。例えば、129Iで標識化した生物製剤はAMS検出に有用であるが、131Iで標識化した生物製剤は高活性であり、おそらく安全性の問題から人間に対して使用が制限される。同様に、14Cで標識化した生物製剤はAMS検出に有用である一方で、13Cで標識化した生物製剤が、NMR検出のような他の多くの非放射性技術において広く使用されるが、その生物製剤はAMS分析では使用できない。特に不適切な同位体、とりわけ窒素は、負イオンを形成できない。
【0043】
AMS同位体は、AMS分析に敏感な任意の同位体とすることができる。好適にはAMS同位体は、例えば同位体存在度が1×10−3から1×10−15%の範囲内、特に1×10−5から1×10−15%といった、極めて低い天然存在度である。例えば14Cではおよそ1.4×10−10%であるように、AMS同位体のそのような自然バックグラウンドの低さに、AMSの検出感度は依存する。(13Cの自然バックグラウンドは、それと比較して大きい1.1%である。)好適には、例えば前述のように数週間を超え、特に30日以上、もしくは60日以上で、扱いやすいように上限は数千年の範囲内にある長い半減期を、AMS同位体が有するか、もしくは天然存在度が低い場合には安定同位体である。
【0044】
水素、ベリリウム、炭素、アルミニウム、リン、塩素、カルシウム、マグネシウム、鉄、セレニウム、ヨウ素、バリウム、およびウランやプルトニウムといったランタニドおよびアクチニドのAMS同位体の中から、AMS同位体を選択でき、特に、3H、7Be、10Be、14C、17O、18O、26Mg、26Al、32Si、35S、36Cl、41Ca、55Fe、60Fe、53Mn、79Se、59Ni、および129Iを含む一群から選択できる。より一般に、同位体は1つもしくはそれ以上の3Hおよび14Cから選択する。最も一般的には、同位体は14Cである。
【0045】
本明細書では、AMS同位体として最も一般的に使用する14Cの使用を重要視するが、この理由の一部は、当然のことながら薬物の大多数が有機物であるためである。しかし、前記のAMS同位体を含む他の同位体も使用されており、また使用できる。
【0046】
本発明の方法で用いる内部標準(「対照検体」)は、検体と識別可能な合成物であるが、検体中に存在するAMS同位体は含まない。
【0047】
対照基質とは、例えば、検体もしくは対照検体で汚染されていないサンプルのような、本発明の第1の態様により使用するサンプルを指す。従って、臨床試験の場合には、対照基質は商業的に得られる血漿、臨床前サンプル、もしくは臨床試験に参加していない被験者からのサンプルとすることができる。一般的には、対照基質は血液から得られる血清、もしくは血漿とする。しかし、尿、便、もしくは細胞組織といった他の基質を分析することも必要であることを理解されたい。
【0048】
「対照基質」とは逆に、本明細書において「基質」は、検体の量を求めるために分析の対象となる試験サンプルを指すために用いる。そのため、基質は、対照基質と同じ物質群から選択できる。
【0049】
「較正抽出溶媒」は、既知の濃度の14Cを含有する(もしくは他のAMSで標識化した)検体とともに、対照基質抽出をHPLCにより分析するときに、特定の量を達成する濃度の対照検体を含む溶媒を指す。代表的には、較正抽出溶媒はアセトニトリルであるが、当業者に知られる例えばメタノールなどの他の溶媒も使用できる。較正抽出溶媒は一般的に、本発明の第1の態様の方法による分離ステップの一部で使用する。
【0050】
「標準抽出溶媒」は、本発明の第2の態様の方法による分離ステップの一部で一般的に使用する。較正抽出溶媒と同様に、標準抽出溶媒は代表的にはアセトニトリルであるが、当業者に知られるような、例えばメタノールなどの他の溶媒、および例えば固相抽出などの他の方法も使用できる。代表的には、HPLC内に抽出物を噴射するときに特定の量を達成するための濃度で、標準抽出溶媒は対照検体を含む。較正抽出溶媒とは逆に、標準抽出溶媒は検体を含まない。
【0051】
本発明により検体から分析できる試験サンプルは、任意の発生源に由来しうる。代表的には、これらのサンプルは、人間か、もしくは特定の人間内の動物に由来する。例えば、人間へのマイクロドージング、もしくは特定の人間における絶対的バイオアベイラビリティ研究から起こりうる。
【0052】
しばしば人間フェーズ0臨床試験と呼ばれる、人間へのマイクロドージングは、比較的新しいコンセプトで、AMSの超高感度に依存する。人間へのマイクロドージングにより、薬物を0.5μg程度投与した後に、詳細な人間の代謝作用研究の実施が可能となる。しかしより代表的には、100μgの薬物を投与する(マイクロドーズは、EMEAおよびFDAの両方により、決して100μgを超えない100回の予測される薬理学的投与量として定義されている)。マイクロドージングにおいて、早急にADME(吸収、分布、代謝、排せつ)情報およびPK(薬物動態)情報を得るために、1種類もしくはそれ以上の薬剤候補は追跡量で人間に取り込まれる。次に、マイクロドーズしたどの薬物がさらに取り込まれる適切なPKパラメータを有しているのか選択するためのデシジョンツリーの一部で、この情報を使用する。これらのADME研究をスクリーニングする低量投与の目的は、不適切な代謝作用のために、発達経路の下流に滴下される薬物がより少量となることを保証するための試行である(例えば初回通過、半減期が短すぎること、バイオアベイラビリティが悪い等)。3種類のうち1種類程度の薬物が、PK、薬物動態、もしくは毒物問題のために薬物発達のフェーズ1の段階で滴下される。人間へのマイクロドージングの目的は、フェーズ1での消耗を減らすことである。
【0053】
マイクロドージング方法において、1μgから上限100μgまでの投薬量で、未知の薬剤候補を人間の被験者へ投与することができる。血液、尿もしくは便のサンプルを何度も採取し、ADMEおよびPKのデータを求めるために、AMSにより、生成サンプルの14Cもしくは他のAMS同位体含有量を分析する。
【0054】
従来技術(例えば、G.Lappin,M Rowland and RC Garner, Expert Opin. 「薬物代謝中毒」2006、2(3):419−427を参照)で知られる絶対バイオアビリティは、合成物の静脈内投与(代表的には薬剤候補)を含む。そのような研究は一般的に、溶解度、分析上の感度、およびIV毒物学のデータが通常研究により裏付けられる必要がある事実に関する問題のため、実行することが難しい。しかし、これらの問題には、AMS同位体の標識化合成物のマイクロドーズを用いて対処できる。
【0055】
マイクロドージングおよび絶対バイオアビリティ研究において、対照検体と同時に、対象に検体を投与する。対照検体は通常、標識化していない未知の薬剤候補である。代表的には、混合は「軽い標識化」のみである。軽い標識化とは、放射能の量が極めて少ないことを指し、代表的には、被験者に対して200nCiのみである(既に体内に存在する14Cによる放射能のおよそ2倍の量)。
【0056】
ここで、検体および対照検体という用語は、AMS同位体標識化合成物と、標識化していない対応する(対照の)合成物とを区別するために使用することに注意されたい。実際には、検体は一般的に対照検体と共にみられ、従って同時に投与される。
【0057】
(例えば)マイクロドーズが投与される対象(例えば被験者)の体内で移送されるとき、100μgの投薬(すなわち検体および対照検体を含む)は、およそ500pg/mLの最大濃度まで達する。代表的には、そのサンプルに内部標準として加える対照検体の量は、5μg/mLである。これは、内部標準の量が、対象への投与により上昇する、試験サンプル内に存在した初期の対照検体量の少なくとも10000倍であることを意味する。このことから、その試験サンプルに加えた内部標準を、精製試験サンプル内で測定する対照検体の唯一の発生源であるとみなすことは理にかなう。代表的には、内部標準として存在する対照検体の量は、与えられた試験サンプル内に存在する全ての対照検体の、少なくとも98wt%であり、より代表的には99wt%かもしくはそれ以上であり、より代表的には99.9wt%かもしくはそれ以上であり、より代表的には99.95wt%かもしくはそれ以上である。
【0058】
本発明は、2種類の構造的に識別可能な化学物質(検体および内部標準)の区別、ならびに2種類の定量化の方法に依存する。定量化方法の一方は、検体に対するAMSであり、他方は、対照検体に対して、質量分析を含むクロマトグラフィ検出において(一般的に)有用な、例えば紫外線吸収、蛍光検出もしくは他のあらゆる検出方法である。
【0059】
上記のように、本発明は、特に低濃度の検体測定によく適している。AMS分析で代表的な状況では、サンプル内の検体のレベルは極めて小さい(例えば10−18から10−9g)。そのような少量の物質を扱うのは特に難しい。例示のように、それ以上はなく、表面(例えばサンプル管の内部)はわずかな活性結合部位を有し、上限100fg(10−13g)の物質を吸収できる。LC−MSによる代表的な検出限界(LOD)はおよそ100pgであり、無視できる量の結合による損失(すなわち0.1%)の1000倍も高い。しかし、より低濃度では、結合による損失量の比率は高い。濃度が1pg/mLの場合、100fgの結合は検体の10%の損失となり大きい。しかし、本発明によると、試験サンプル内に存在する検体を、代表的には14Cにより標識化し、対照検体をこの試験サンプルへ内部標準として加える。有利なことに、これによって検体と対照検体の濃度の合計値を、例えば1fg/mLレベルから200pg/mLレベルへと(100万倍も高い)上昇させる効果があり、従って極めて少量のサンプルの分析に関連する問題を、改善もしくは解決できる。
【0060】
本発明の較正方法を実行する前に、代表的にはいくつかの予備調査を行う。予備調査に含まれるものに、AMSによって、全ての対照検体のサンプルが、検体中に存在するAMS同位体を持たないこと、すなわち全てのサンプルが最大で通常のバックグラウンドレベルの同位体を含むことの確認がある。
【0061】
同様に、検体と対照検体を、検体と対照検体の定量化において障害となりうる他の合成物(例えば推定代謝物)と分離するための適切な分離手順の開発は、本発明の範囲ではなく、本明細書において議論しない。しかし、適切な分離手順の開発は、当業者の技術内にある。既に述べたように、本発明による方法を用いた分離様式を、電気泳動法、もしくは高性能液体クロマトグラフィ(HPLC)、ガスクロマトグラフィ(GC)、もしくは薄層クロマトグラフィ(TLC)などのクロマトグラフィ法とすることができる。代表的にはHPLCを用いる。
【0062】
同様に、あらゆる所与の溶媒に対する検体の溶解度および安定性の測定は、本発明の範囲ではない。当然のことながら、検体によって溶解度および安定性は変化し、当業者は測定できるであろう。この点から、サンプルから対照検体および検体を抽出するために使用する溶媒を、ある程度、本発明により定量化を所望する検体の溶解度および安定性により決定する。
【0063】
本発明により使用する分離手順において、検体の存在を検出するために、紫外線吸収を使用するとき、□maxを初めに測定する。一般的にHPLCを用いる場合、対照検体に対して、紫外線吸収が代表的で便利な検出方法である。しかし、当然のことながら、当業者は他の検出方法を使用できる。
【0064】
HPLCで対照検体を検出するために紫外線応答を用いる場合、HPLC−紫外線応答の再現性は、代表的には3回もしくはそれ以上の噴射で、そのうち好適には少なくとも1回は別の日に実行される噴射により確かめる。これにもかかわらず、再現性を確かめる正確な方法は、対象の検体の特性に依存する。同様に、全てのクロマトグラフィのキャリーオーバーの程度を、同様の方法で確かめる。
【0065】
さらに、本発明の実行の前に、分離系用の適切なHPLC分析の開発において、一般的に当業者は、HPLCカラム上に噴射された対照検体のどれだけの量が、ピーク形状(例えばカラムの過負荷による)に不利に影響することなく、ノイズ比に対して適切な信号を与えるのか、また対照検体のどれだけの量が使用する濃度に溶解するのか、測定するだろう。この量は、本明細書において「標準量」と呼び、紫外線検出による定量化に使用するHPLCカラム上に噴射した対照検体の量を指す。
【0066】
標準量の測定後、例えばxμlの標準対照検体をyμlの基質内に混合し、その後でHPLC上にzμl噴射することで、噴射する対照検体の標準量が結果として出るといったように、対照検体の標準溶解濃度を計算できる。本明細書において、このことを「標準溶解」と呼ぶ。
【0067】
しばしば、所与の基質から検体を抽出するおおよその効率を定量化するため、初期検査を行うことが有効である。これを行うために、基質からのおおよその抽出効率が計算できるような濃度まで、対照基質サンプルを検体とともに混合する。これは、例えば液体シンチレーション計測(LSC)を用いて、便利に達成できる。
【0068】
例えば、3000dpmの14Cで標識化した検体を、1.5mlの対照基質内に混合でき、LSC分析のために200μlのアリコートを3回、すなわちいずれの抽出ステップも干渉しないように採取する。次に、別の200μlを3回、200μlの適切な溶媒(例えばアセトニトリル)内に抽出し、乾燥し、再構成することができ、その200μlのアリコートをLSCで分析できる。回復が100%ならば、LSC分析用の200μlのアリコートは、それぞれ400dpm含み、その計測時間(計測は95.5%の信頼時間間隔を仮定すると)は25分である。(計測時間はT=1/dpm(200/2)2より計算できる)(計測は95.5%の信頼区間内にあると仮定する)
【0069】
前記のように、これらの予備段階は本発明の方法の一部ではなく、前述のおよその抽出効率の測定の観点から、抽出効率は100%近くを達成する必要がないことは、当然のことながら重要である。実際、本発明は、必ずしもそうではないという認識に起因する。本明細書における予備段階の目的は、本明細書の方法が有効であるように、十分な量の検体を、実際に対照基質から抽出できることを保証することである。
【0070】
本発明の較正方法の実行は、A:Bの比率を、検体の存在量の絶対値に関連付けるために使用できるデータを提供する。例えば、前述の生物分析と類似の、Cに対するA:Bの較正ラインを引くために、そのデータを使用できる。(それぞれの、また全ての本明細書におけるA:Bという呼称は、B:Aの代わりに使用でき、重要な点はただ比率が得られるということである)
【0071】
そのようなデータを得るために、例えば臨床試験では、対照基質(例えば血漿)を、検体もしくは対照検体により汚染されていない物質源から採取する。汚染されていない、というのは、検体および対照検体がともに存在しないか、もしくは検出できないレベルで存在することを意味する。臨床試験において、分析する薬物もしくは代謝物に触れていない被験者から、対照基質を得ることで、このことを好都合に達成できる。
【0072】
そのような複数の対照基質サンプルぞれぞれに対して、異なる濃度の検体を加える。さらに、正確に既知で等量の内部標準(例えば対照検体)を、各対照基質サンプルへ加える。検体と対照検体を加えた後、準備した各検体を精製する。これは一般的に、従来技術で知られる初期抽出ステップを含む。
【0073】
検体と対照検体を含む複数の対照基質サンプルを準備するとき、検体を基質内に加えることが可能で、その各対照基質サンプルに加えた検体の量Cを、個別の対照基質サンプルのAMS分析により、測定することが可能である。しかし、AMS同位体が14Cである場合、基質(例えば血漿)がバックグラウンド14Cを含むため、この方法には問題が生じうる。例えば、血液はおよそ12%w/vの炭素を、血漿はおよそ4%の炭素を、尿はおよそ1%の炭素を含む。比較的高い濃度で炭素が存在することは、検出限界(LOD)に影響する「ノイズ」とみなしうる、内生の14Cが存在することを意味する。
【0074】
実際、他の分析技術と同様に、AMSのLODは信号とノイズの比率により決まる。ここで信号は、14Cを濃縮した検体からの14Cの量に依存する。14C:12Cの比率は、大気中に含まれるこれらの同位体の比率と等しく、そのため全ての生物で一定である。従って、内生の14Cは、サンプル内の全炭素量に比例して増加する。従って、便サンプルのLODは、便が血清よりも内生の炭素含有量が高いため、血清サンプルよりも高い。
【0075】
従来技術で知られているように、検体からの14Cの量を低減することなく、サンプル内の内生の炭素量を少なくする方法がある。例えば、例えばアセトニトリルのような水混和性の有機溶媒により、炭素リッチのたんぱく質は沈殿し、続いて遠心分離により除去できる。事前のAMS溶媒抽出ステップも同様に、便における内生の14Cの量を低減できる。
【0076】
生体サンプル内に内生の14Cが存在するために、対照基質を直接混合するとき、例えばおよそ0.06dpm/mLまで低下した場合のみ14C濃度を確認することができる、という結果となる(Lappin, G.& Garner, R.C.以下)。反対に、HPLCにおいて、ほぼ14Cのバックグラウンドが存在しないため、そのレベルはこの場合よりも低くできる。
【0077】
この根本的な問題に対処するために、好適には、14Cで標識化した検体および対照検体を含む複数の対照基質サンプルを準備するときに、検体を(および一般的には、例えば、対照検体を)較正抽出溶媒内に加え(混合し)、較正抽出溶媒はほぼ不揮発性の14Cのバックグラウンドを含むため、量Cを知るために、較正抽出溶媒内に存在する検体をAMSにより分析する。(AMSサンプルを準備するとき、アセトニトリルを除去する。)このように、分析したい試験サンプルへ、より関連づけて(より低い)濃度に至るまで較正曲線を作成することが可能である。
【0078】
このように、それぞれが随意的に(しかし通常)、同量の対照検体(通常は、標準量をHPLC上に噴射できるような濃度で)と異なる14C検体の濃度とを含む、一連の較正抽出溶媒を準備する。複数の同じ抽出溶媒を使用でき、この方法の正確性が改善される。代表的には、5回の較正抽出溶媒を使用する(代表的には、0.005から2dpmをHPLCカラム上へ噴射する)が、その正確な回数および使用濃度は研究の特性に依存する。
【0079】
較正抽出溶媒の(もしくは適切に検体および対照検体を混合した対照基質の)アリコートを、一般的には少なくとも二重に、AMS分析で採取する。その結果は、対照基質サンプルに加えた、14Cの、もしくは他のAMS同位体の正確な濃度を規定する。
【0080】
対照検体の貯蔵液もまた準備し、そこから較正抽出溶媒を作成する。以下で説明する標準抽出溶媒を作るために、代表的には同じ貯蔵液を使用する。このように、試験サンプルの分析に使用したものと同じ濃度の内部標準を使用して、較正曲線を作成する。
【0081】
較正抽出溶媒を使用して、代表的には二重に、対照基質を抽出する。次に、(適切ならば、乾燥および再構成の後で)サンプル内の他の要素から、最終抽出物を分離する。この操作は、代表的にはHPLCにより達成し、HPLCからの溶出液は一連の留分として収集する。検体(および構造的に識別可能な内部標準)の保持時間に対応するその留分を貯蔵し、14C、もしくは他のAMS同位体の、AMSに含まれる単体サンプルとして分析する。また、選択したサンプルの個別の留分を、留分層が検体を代表することを保証するために分析する。
【0082】
別個に、対照検体の量を測定するために、代表的には紫外線検出などの、サンプル定量化の他の方法を使用する。
【0083】
較正の方法により得るデータ、すなわち複数の精製サンプルそれぞれに対する、1セットのC,AおよびBの値は、A:Bの比率とCとの間に関係性を構築できる。Cは、各試験サンプルに加えた検体の量に対応する。Aは、精製試験サンプル内に存在する検体の量に対応する(そして、Cの100%は、その抽出(および精製)効率100%である)。Bは、精製試験サンプル内に存在する対照検体の量に対応する。
【0084】
当然のことながら、本発明の較正方法において、AMS同位体に関して得られるデータは、AMS測定は対象の検体のみであることを仮定する。しかしこのことは、その標準がAMS標識化した化学種をその標準以外に含む場合には、必ずしもあてはまらず、そのため、少なくとも量Cの測定の妨げとなる。しかし実際には、本発明の較正方法に使用できる複数の検体を他の方法で測定できるため、そのような可能性は問題とはならない。代表的には、検体の供給者により、その純度が確かめられる。
【0085】
一般的に、通常は検体と対照検体を相互に分離することはなく、ならびに/もしくは対照検体の定量化に使用する方法はそれらを識別することができないため、対照検体の定量化は、検体および対照検体をともに含むサンプルに対して実行する。このことは、例えば50倍かもしくはそれ以上、一般的には100倍かそれ以上、対照検体の量が検体の量よりも、はるかに多いことを仮定している。当然のことながら、例えば、対照検体の総量と精製サンプル内に存在する検体の、簡単に標識化した人間マイクロドーズ内に存在する検体は極めて少量であるため、多くの生体サンプル中に存在する検体は、実際には極めて少量である。
【0086】
しかし、対照検体を定量化するために用いる方法により、対照検体と検体とを識別することができることも考えられる。そのような方法の例として、精密質量分析がある。
【0087】
Cに対するA:Bの比率をプロットすることで、較正ラインを得ることができる。従って、対照検体が内部標準としてどう機能するのか理解できるだろう。任意の摂動(例えばその損失および/もしくは未検出)は、検体それ自体が引き起こす、損失および/もしくは未検出に比例する。この2つの比率を用いるため、これらの損失は相殺される。
【0088】
所与の検体/対照検体の組に対して較正ラインを一度作ると、次に試験(例えば生体)サンプルを分析でき、存在する(もし存在するならば)検体量が測定できる。
【0089】
一般的に、標準抽出溶媒を準備する。これは、既知濃度の対照検体を含む抽出溶媒であり(代表的にはアセトニトリル系)、その標準量を抽出後にHPLCへ噴射できる。上述のように、好適には、標準抽出溶媒は、較正抽出溶媒と同じ濃度の対照検体を含む。このことは一般的に、標準抽出溶媒および較正抽出溶媒をともに作るために、同じ貯蔵液を使用することで達成する。このように、試験サンプルの分析に使用するものと同じ濃度の内部標準を用いて、較正曲線を作る。
【0090】
従って、試験サンプルに対して、既知の正確な量の対照検体を加える。これは対照検体として機能し、また有利なことに、上述の理由により、サンプル内の検体および対照検体の濃度の合計を増加させる。さらに代表的には、存在する検体の推定量をはるかに超えた量であって、代表的には本発明の較正方法において各サンプルに加えた対照検体と同じ量の、対照検体を加える。試験サンプルに用いた検体および対照検体の総量が、(a)抽出および精製中の損失を十分に抑制するため、また(b)検体および対照検体の合計と同量を、較正方法におけるサンプルおよび試験サンプルに使用するときには、試験サンプルについて得られたデータを摂動する濃度効果は存在しないために、濃度依存効果が抑制される点で、このことは有利である。
【0091】
抽出後、抽出した試験サンプルを例えばHPLCにより精製する。検体および対照検体を表す分率を、本発明の較正方法により得られる値と全く類似の方法で、(検体のための)AMS、および(対照検体のための)分離技術により分析する。
【0092】
精製試験サンプルにおいて得られた比率A:Bを算出することで、未処理の試験サンプル内に存在する初期濃度Cを、標準曲線から外挿することができる。
【0093】
量Bを測定するときに、紫外線応答を確認するため、HPLCの移動相に溶解した標準対照検体の少量のアリコートを、HPLC−紫外線により繰り返し(代表的には1日に2回)分析する。代案として、任意の簡便なHPLC較正は、効果的な、例えば製造者の特許もしくは推奨の方法になりうる。AMSは長期にわたり一貫する(そして実際に、データは同位体比率に基づくため、装置間で一貫する)ことが知られている。しかし、紫外線応答がドリフトするとき、対照検体濃度の計算において、A:Bの比率がエラーを生じる方へ変わってしまう。代表的には、20%以下かもしくは同等のCVは容認できる。
【0094】
当然のことながら、本発明の方法を実行するとき、および実際には本発明の較正方法により複数のサンプルを準備するとき、異なる濃度の対照検体を使用できる。主に、より多くのデータ操作を含むため、異なる濃度の対照検体を用いることはより不便であり、特定の条件下では信頼性の劣る結果を生じるかもしれないが、同じ濃度(もしくは量)の対照検体を使用する必要はない。例えば、較正曲線を作るときにxμgの対照検体を使用する場合、精製した較正サンプルに対して行う定量化ステップ(v)の間、「B」の値を与えると考えられる。2xμgの対照検体を試験サンプルへ加えるとき、対応する定量化ステップ(iv)の間、「2B」の値を与えると考えられる。次にそのような試験サンプルに対して、較正グラフから試験サンプル内の初期検体濃度を読み取ることを可能とするために、A:Bの比率は半分となるべきである。しかし、そのような試験サンプルから得られるA:Bの比率を関連付けるために必要なことは、ただこの比率を2倍することである。
【0095】
しかし当然のことながら、この方法の条件を(異なる量の内部標準を用いることで)変えることで、較正曲線が試験サンプルに対して妥当であるという仮定は、必ずしも全ての場合で正しくない。実際には、「B」の値は正確に「2B」とはならず、そのために較正曲線を参照して算出する検体濃度は、絶対的に真の値ではない。それにもかかわらず、好適ではないが、較正データを作るときに用いることのできる試験サンプル分析を行うときに、同量の内部標準を使用する必要はない。初期の試験サンプル内に存在した検体の算出量は、おそらく特定の場合に、より少量の内部標準化を受けている可能性があり、より正確ではない。
【0096】
本発明の較正方法により、複数のサンプルをそれぞれ準備するとき、同量の対照検体を使用する必要がないという点で、同様のコメントを必要な変更を加えて適用する。従って、xμgの対照検体が1つのサンプル内に存在し、2xμgの対照検体が他のサンプル内に存在するとき、「B」および「2B」の値がステップ(v)により得られる。そのような場合、xμgの対照検体にもとづいてA:B(もしくはB:A)の比率を準備するために、「2B」の値を2で割ることができる。
【0097】
前述の議論は、より一般的な点を示す。すなわち、本明細書内で前述した、本発明の第1および第2の態様は、本発明の異なる態様に関連する。第1の態様は、試験サンプル内の検体量を測定するための、次の方法で使用するデータを提供する。
【0098】
本発明の第2の態様による好適な実施例において、分離ステップおよび接触ステップの全ての態様は、本発明の較正方法による対応する分離ステップと、同じ条件で実行する。このことは、本発明の第1の態様を実行するときに行う対応するステップと、これらの条件が実用的に近く類似であることを意味する。同様に、好適には、本発明の較正方法による対応の測定ステップと同じ条件で、測定ステップを実行する。このように、本発明の第2の態様により実行するものと同じ条件、もしくは実用的な類似の条件において、本発明の第1の態様の実行により得られるデータを得る。従って、本発明の第1の態様で得られるデータによって、本発明の第2の態様の実行により得られるデータから検体を定量化することが可能となる。実用的な類似の条件において両データを得るため、後者のデータを、前者の正確性をもとに修正できる。例えば、本発明の第2の態様による分離ステップの一部としての抽出ステップが、乾燥ステップを含むようなとき、乾燥ステップ中に生じうる検体の損失を確認するために、好適には、乾燥の前後で例えばLSC分析を行うために、アリコートを採取する。
【0099】
試験サンプルを分析するときに、本発明の第1の態様の方法で得られるデータを適用する最も直接的な方法は、いわゆるpMC法である。代案の方法に、いわゆるdpm法がある。AMS同位体が14Cである方法に関して、両者を以下に説明する。
【0100】
全ての生物は、大気中の天然存在度と同じ14Cを含む。「100%現在」もしくは100pMCとして任意に参照、もしくは定義する14Cのレベルは、1.18×1012個の炭素原子の中に1個の14C原子があること、もしくは1ミリグラムの炭素中に97.6アトモルの14C原子があることに対応する。必要ならばデータを標準化するために、正確に既知のpMC値のAMS標準を、装置による確認により入手することが可能である。2種類の最も広範に使用する14C標準は、米国のNational Institute of Standards Technology(NIST)の標準的なシュウ酸と、1960年代に収穫されAustralian National University(ANU)により認証された糖料作物である。NISTのシュウ酸のpMCは95、ANU砂糖のpMCは150.61である(後者は、原子爆弾からの放射性降下物が依然として比較的高い時期に収穫されたため、100を超える)。
【0101】
この「pMC」法により、直接的にデータを処理し計算することが可能となる。しかし、この方法を用いるとき、好適には、dpm法を用いるときよりも、より厳しい標準化方法をとる。例えば、好適には、本発明の2つの態様において、同じ実験的手順を実行する。
【0102】
基質抽出手順において変更を行うことは通常できず、そのため一般的には難しくない。しかし、サンプルを一度HPLC上に噴射したら、全てのサンプルを全く同様に扱うべきである。特に、
1.検体のHPLCピークにまたがって、同数の留分をためるべきである。
2.留分プールを形成するために、各留分から同量の体積を採取するべきである。
3.AMS分析のため、留分プールを同量の体積に等分するべきである。
4.同量の炭素キャリヤ(例えば液体パラフィン(LP))を加えるべきでる。
【0103】
項目4の観点から、また従来技術で知られるように、同位体の量が実質無視できるだけの量の物質(キャリヤ物質)を含み、AMSにより分析したいサンプルよりもその物質が十分に多量であることが、AMSサンプルの準備において通常である。代表的には、存在するキャリヤ物質の重量は、分析したい精製サンプル(蒸発により除去される精製溶媒を除く)の重量の、100から1000000倍、代表的には100から10000倍大きい。これは2つの点で有利である。
第1に、従来のAMS分析において、そうでなければ取り扱うことが難しい定量サンプル(例えばマイクログラム程度の量)の分析が可能となる。
第2に、本発明において、過剰な対照検体がサンプルおよび試験サンプル内に含まれるため、当業者に知られるように、これらの量は、AMS機器は同位体比率の算出にもとづくため、AMS機器により得られる値に影響する。
【0104】
そのため正確に検体の量を算出するために、検体を標識化するAMS同位体以外の、同位体の量を知る必要がある。実際には、使用するキャリヤ物質の正確な重量測定により達成する。キャリヤ物質の量は、サンプルよりも極めて過剰であるため(以下で議論する)、AMS同位体と検体の定量化のために使用する比率を得るための、非AMS同位体源のみをサンプルが構成することを、仮定することが妥当である。
【0105】
キャリヤ物質をキャリヤと呼び、測定下で希少同位体を無視できる量しか含んでいない任意の物質とすることができる。
【0106】
上記の条件1から4が満たされるとき、標準曲線を準備するときに、例えば、留分プールのアリコートの分析により得られるpMC値は、紫外線ピーク領域(A:Bの比率で)で割ることができ、x軸上のC値に対してy軸上にプロットすることができる。試験サンプルに対して、検体ピークのpMC値は、紫外線ピーク領域で割ることができ、次に、試験サンプル内の検体濃度を確立するためy軸からx軸へ修正できる。
【0107】
x軸は1mLサンプルあたりのdpmか、もしくは1mLサンプル辺りの重量とすることができる。例えば、投与する14C薬物の比活性度から、dpm/mLで割ることで、pgもしくはng/mLを算出できる。較正ラインを作るために使用する14C検体の比活性度は、高くなるだろう(例えばおよそ2GBqもしくは50mCi/mmol)。しかし、標準曲線から臨床サンプルのpgもしくはng値を算出するために、投与した薬物の比活性度を用いる必要がある。例えば、100μgおよび200nCiのマイクロドーズを投与したとき、比活性度は2nCi/μgである。
【0108】
pMC法を使用することで、pMC値をdpm値に変換する必要がなくなり、次に行う通常の算出を含まない。このため、これは好適な方法であるが、分析にpMC法が不可能である場合に、以下で説明するdpm法を使用できることがわかる。
【0109】
おそらくこの状況では、同位体比率に基づきAMSが1グラムの炭素あたりのdpm値を与え、生体サンプル1グラムあたりのdpmのような絶対値を与えないことを、理解することは有用である。これが、AMSとLSCの結果の根本的な違いである。AMSは、放射性崩壊の事象数ではなく、14C原子の数を測定する。同位体比率をdpm値に変換することは、単に生物医学研究者になじみの単位を与えるためである。サンプル値のdpm/gを算出するために、サンプル中の炭素比率を知る必要があり、CHN分析により容易に求めることができる。
【0110】
この「dpm」法では分析手順において変更が可能であるが、より多くのデータ抽出が必要である。例えば、HPLCカラムが劣化するにつれピーク形状は広がり、当初の想定で標準曲線を作るために使用するよりも多くの、ピークをまたがる留分をプールする必要が生じる。従ってpMC法が好適ではあるが、例えば以下すぐに例示するHPLC精製の実用性からは、dpm法の使用を意味するだろう。
【0111】
上述のように、基質抽出方法において変更を行うことは通常できず、そのため、おそらくは難しくない。しかし、一度サンプルをHLPC上に噴射すると、
(a)検体のHPLCピークをまたいだ異なる数の留分をプールして標準曲線上で使用した留分と比較すること、
(b)AMS分析用のプールの投薬計画およびサンプルサイズを調整すること
が必要となる。
【0112】
条件(a)(b)が必要なとき、pMCデータをdpmデータに変換するべきである。この過程で、留分体積およびアリコート体積を算出に使用し、分析手順の相違を考慮に入れている。
【実施例】
【0113】
以下の例は、本発明の説明のためであり、限定するものではない。
【0114】
例1:較正曲線の作成
6個の1mLの対照血漿(分析する薬物に接触したことのない提供者からの人間血漿)サンプルを、個別のチューブに採取した。表1に示す標的濃度を達成するために、各チューブ内にいくらかの14C薬物を加えた。
【0115】
各チューブからアリコートを採取し、以下に説明するように、AMSを用いて直接分析した。表1に示すように達成した、各チューブの実際の濃度を、AMSの結果から測定した。
【0116】
【表1】
【0117】
次に、6個の血漿サンプル(それぞれ200μLのアリコート)を、標識化していない薬物を40μg/mL含む、正確な量の有機溶媒を用いて抽出した。その抽出は、窒素流上での乾燥により行い、200μLの溶媒内で再構成した。
【0118】
再構成した各サンプルのアリコート(50μL)を、HPLC上に噴射し、そのHPLC溶出液を、96wellプレート内に一連の留分として収集した。HPLCには、紫外線吸収検出機を備えた。薬物に対応するHPLCピークのピーク領域を、紫外線吸収により測定して記録した。
【0119】
分析する薬物を含む96wellプレート内の留分を特定するために、紫外線ピークの保持時間を使用した。この留分はAMSにより14Cを分析した。
【0120】
上記の分析により、以下の情報が得られた。
1)AMSにより直接測定した、dpm/mLでの、血漿サンプル内の実際の薬物濃度(全ての抽出もしくはHPLCに先立つ)
2)各サンプル内の、標識化していない薬物の濃度(血漿サンプルの抽出に、正確に等量が用いられる)
3)抽出およびHPLC分析に続く、各分析における標識化していない薬物に対応する紫外線ピーク領域(標識化していない薬物の量はμg程度の存在量であることに注意されたい。14C薬物もまた存在するが、pg程度の量であるため、紫外線ピーク領域に重大な寄与をしない)
4)AMS分析からの、HPLC留分用のdpm/mL値
【0121】
計算
まず、この結果は本発明により算出せず、従来の方法を参照する。従来の方法は単純に、HPLC画分(留分)内に存在する14C薬物の濃度をとり(以下の「試験サンプル測定」と題した章で説明する)、初めの血漿サンプルのdpm/mL濃度の推定値を算出する。この従来の方法は、多くの機会に文献で報告されている(RC Garnerら、薬物代謝排せつ,30(7),823−30.(2002);N Sarapaら,臨床薬理学,45(10),1198−205,(2005);JS Vogel,バイオテクノロジー,Suppl,25−9(2005);G Lappin ら,臨床薬理学,80(3),203−215(2006))。
【0122】
HPLC留分のAMSによる分析結果を表2に示す。
【0123】
【表2】
*抽出した200μLのうちの50μLを、HPLC上に噴射した。そのため、血漿内の濃度(dpm/mLと表記している)は、HPLC留分で測定される量の20倍である。
【0124】
分析が100%の効率(すなわち損失なし)で行われるならば、表2に記載の濃度は、初期血漿サンプルの分析結果(すなわち、表1で得た濃度)と同じはずである。表1と表2のデータの比較を、表3に示す。
【0125】
【表3】
【0126】
表3の結果は、チューブ1−5において、分析中におよそ58%にのぼる重大な損失が生じることを示す。しかし、表3に示す結果は、この分析において分析損失が重大で、従来のサンプル定量化方法はひどく間違っていることを、明確に示している。
【0127】
以下に、本発明により、分析結果が内部標準(標識化していない薬物)に関連することを仮定して算出し直した結果を示す。標識化していない薬物のピーク領域の結果を、表4に示す。
【0128】
【表4】
【0129】
表5は、留分中のdpmを紫外線ピーク領域(表4より)で割った値である。次にこれらの比率をグラフのy軸にプロットし、血漿中の実際の薬物濃度(表1で達成した濃度(dpm/mL))をx軸にプロットする。
【0130】
【表5】
【0131】
例2:試験サンプル測定
6.9498dpm/mLの薬物を含むように、血漿サンプルを準備した。40μg/mLの標識化していない薬物を含む抽出溶媒を用いて(200μLの)サンプルを抽出した。乾燥および再構成のため抽出物を200μLの溶媒内に採取し、50μLをHPLC上に噴射した。薬物のピーク領域を、紫外線吸収により記録した。一連の留分として溶出液を収集し、薬物に対応する留分の14CをAMSにより分析した。その分析を2回行った。このサンプルの分析結果を表6に示す。
【0132】
【表6】
*抽出した200μLのうちの50μLを、HPLC上に噴射した。そのため、血漿内の濃度は、HPLC留分で測定される量の20倍である。
【0133】
直接の方法(すなわち血漿濃度を、HPLC留分のAMSデータから直接算出し、較正曲線を用いない)による試験サンプルの分析により、4.306dpm/mLの平均値を得た。しかし、真の値は6.9498dpm/mLであった。従って、HPLCおよびAMS分析の結果は61%低かった。本発明を用いたサンプル分析により、(AMSにより測定した)留分中のdpm比率は、紫外線ピーク領域に対して0.000026567であった。較正曲線のy軸にこの値をとり(図1)、x軸の濃度により修正することで、7.04dpm/mLの平均濃度が得られ、真の値と1.3%のみ離れている。
【0134】
AMSによるサンプル分析
黒鉛生成の方法は、Vogel, J.S.による「生物医学AMS用の、汚染を生じない黒鉛の迅速生産」Radiocarbon, 34 344−350(1992)に従った。
【0135】
血漿サンプルを、黒鉛化チューブ内で直接等分(60μL)した。HPLC留分(代表的には100μL)を黒鉛化チューブ内で炭素キャリヤ(液体パラフィン、2.5μL)とともに等分した。黒鉛化チューブを、前もって熱した酸化銅のワイヤ(50±10mg)を含むサンプルチューブ内に投入し、Savant AES2010 Speed Vac.を用いて、真空中で完全に乾燥した。黒鉛化する炭素の最終的な量は、各ケースでおよそ2mgであった。
【0136】
工程管理したANU砂糖(AMS装置の較正用のよく知られたAMS標準;5−7mg)および人造黒鉛(2−3mg)を、前もって熱した酸化銅のワイヤを含む個別のサンプルチューブに投入する。2.5μLの液体パラフィン制御もまた、酸化銅を有する個別のサンプルチューブ内に投入した。全ての標準および対照を、上述のように真空中で乾燥した。
【0137】
燃焼(酸化)
乾燥したサンプルおよび酸化銅を含むガラスのサンプルチューブを、より大きなガラスの燃焼チューブへ投入し、カーボライト(Carbolite)加熱機において真空下で熱遮蔽して900℃で2時間熱した。燃焼後、チューブを大気温度までゆっくりと冷却した。サンプルおよび対照基質の酸化により、遮蔽したチューブ内で二酸化炭素が生じた。
【0138】
黒鉛化(減少)
より大きな燃焼チューブの終端部をY字マニホールドに接続した。コバルト粉末を含む(6.5±1.5mg)ホウケイ酸ガラスチューブを、甚句粉末と水酸化チタンの混合物を25:3w/wの比率(120−200mg)で含むより大きなガラスの黒鉛化チューブ内に投入し、その黒鉛化チューブをY字マニホールドのもう一方の端部へ接続した。燃焼チューブを、イソプロパノール/乾燥氷浴内に浸し、黒鉛化チューブを液体窒素浴に浸す。全体の系を真空下で設置する。燃焼チューブの端部を破壊した後、酸化したサンプルから生じる二酸化炭素を、黒鉛化チューブへと低温で移す。一度移したら、黒鉛化チューブを真空化で熱遮蔽し、加熱機内に設置して500℃で4時間熱し、その後さらに550℃で6時間加熱したあと大気温度までゆっくりと冷却した。
【0139】
カソードの黒鉛による梱包
黒鉛化過程が完了したら、カソード内で梱包する準備ができるまで、その黒鉛を遮蔽した黒鉛化チューブ内に残した。カソードを梱包するために、黒鉛化チューブを開き、黒鉛を含むホウケイ酸ガラスチューブは、コバルト触媒上に吸収して除去した。そのコバルト/黒鉛を、慎重にアルミカソード内へ出し、Parr Pellet Press内で100−200psiで圧縮し、カソード内に黒鉛のタブレットを形成した。全ての圧縮後、メタノールで湿したティッシュで拭くことで、圧縮機を洗浄した。次に、サンプルおよび工程管理カソードを、室温での貯蔵のため標識化したプラスチックキャップのチューブ内に設置した。分析が必要な時、これらのカソードを、以下に示す他の機械の標準および対照により、134−位置AMSサンプルホイール内に設置する。
【0140】
【表7】
【0141】
AMS装置を調整するため、もしくは、データの正規化に使用するプールしたANU砂糖カソードを除いて工程の効率を測定するために、上述のカソードを使用した。
【0142】
AMS手順
5MV 15SDH−2 ペレトロン(Pelletron)AMSシステム(National Electrostatics社製)を用いて、AMS分析を行った。黒鉛含有カソードを設置したサンプルホイールを、AMS装置のイオン源内部に挿入した。マルチカソードの陰イオン源(MC−SNICS)は、黒鉛の表面上で加速されるセシウムイオン(Cs+)ビームを生じた。結果の炭素陰イオンビームは、12C-,13C−,および14C−、および16O−のような他のイオンを含んでいた。同重体14N−は不安定で、そのため14Cの測定を阻害しない。
【0143】
炭素イオンビームを前もって加速し、球面静電分析器を通し、次に入射磁石へ向け進ませた。12C−の出力は、代表的には1−100μAである。12C−(150μs)、13C−(600μs)、および14C−(0.1s)を通常68keVで順番に射出し、対応する1サイクルに対して各同位体の測定に1つを結合するように、磁石を設定した。タンデムペレトロン(Pelletron)加速器の中心正極へ向けて、エインゼル(Einzel)レンズを通して炭素イオンビームを加速した。およそ17.5から22.5MeVの粒子エネルギーをもつよう、この一連の分析に用いるターミナル電圧は3.5から4.5MVとした。中心電極では、正に帯電した炭素イオン(12,13,14C+1から+6)を生じるために、炭素原子から電子を奪った。測定のために、このエネルギーにおいて最も豊富となるように、C4+イオンを選択した。これらのイオンを中心電極から遠ざかる方向へ、静電四重極トリプレットおよび分析磁石へ向けて加速した。
【0144】
事後分析磁石を通過してすぐに、12C4+および13C4+イオンを、オフセットファラデーカップ内でイオン電流として計測した。14C4+イオンは、静電四重極ダブレットおよび円筒形静電分析器を通して、高エネルギービーム線上を通過させた。ここから、ガスイオン化検出器へイオンを入れ、それらのイオンを、各イオンのエネルギー損失および全エネルギーを測定する(全部で4個の)アノード上に集めた。一般的に、他の非干渉14C4+イオンは、静電分析器、磁石、スリット、および荷電分離の組み合わせにより、ガスイオン化検出器には入らなかった。ビーム線上でおよそ10−9Torrの真空圧力を、イオン源で10−6Torrの真空圧力を維持した。装置によるイオン転移は30−60%であった。
【0145】
データの処理
AMSデータを、準備したサンプルのdpm/mL値の算出に使用した。AMSの結果はpMCで表現した。100pMCは、
13.56dpm/g C もしくは 0.01356dpm/mg Cに等しいか、
もしくは、98フェムトモルの14C/g C(1フェムトモル=10−15モル)に等しいか、
もしくは、98アトモルの14C/mg C(1アトモル=10−18モル)に等しい。
従って、サンプルの密度を1g/cm3と仮定すると、以下のようになる。
pMC×0.1356 = dpm14C/g C
および (dpm14C/g C)×(サンプル内の%w/v C)=dpm14C/mL
サンプルの[14C]/[12C]比率 =
全[14C](薬物+生体サンプル+キャリヤ1)
全[12C](薬物+生体サンプル+キャリヤ1)
1−適用可能な場合
【0146】
数学的処理
以下の議論は、本明細書内で上述した、本発明と同様のコンセプトの説明であるが、本発明により提供する内部標準化の数学的な表式に関する議論である。上述の議論と同様に、以下の議論では14Cで標識化した合成物により例示するが、当然のことながら、そのコンセプトは他のAMS同位体標識合成物に対して等しく適用できる。
【0147】
薬物の発展の間、例えば特定の研究形態における14C薬物濃度測定において、AMSがますます用いられ、生体サンプル内の全薬物および代謝物(例えば14Cもしくは他のAMS同位体の濃度)の測定のために式が十分に確立されている一方で、検体のクロマトグラフィ分離およびAMSによる分析の結果生じる損失を求める式は導かれていない。そのような式を導けば、本明細書の上述の議論から当然のこととして、正確に検体濃度を測定するために、分析中に生じるあらゆる質量損失を考慮することができる。本発明の恩恵により、分析中のあらゆる質量損失の見積もりに関する本発明の観点による方法において、14C−および他のAMS同位体含有検体のクロマトグラフィ分離およびAMS分析を説明する式を導くことができる。
【0148】
同位体比率の方法として、AMSは検体内に、代表的には12Cおよび14Cのような少なくとも2種の同位体を、薬剤物質として必要とする。同位体比率を測定するために、代表的にはサンプルを黒鉛に変換し、次に分析のためAMS内に設置する(J.S.Vogel,生物医学AMS用の、非汚染な黒鉛の迅速生産、Radiocarbon,34(1992)344−350)。AMSは極めて高感度で、14C薬物の特定の活性度に応じて、フェムトグラムもしくはアトグラムの範囲(10−15から10−18g)の14C薬物濃度の測定が可能である(G.Lappin,R.C.Garner, 加速質量分析器を用いた、放射性標識薬物およびその代謝物の超高感度検出,I.Wilson編,分析用分離のハンドブック,Elsevier,Amsterdam,2003,pp.331−349)。その超高感度のため、本明細書に上述のように、マイクロドージングおよび絶対バイオアビリティ研究のような技術に対して、AMSを使用している。共通に入手可能なHPLCとAMSとの間のインタフェースがないため、HPLCにより血漿抽出を分析し、対象の検体の保持時間に対応するHPLC溶離剤の留分を、12C:14Cの同位体比率を測定する前に「オフライン」で黒鉛化する(I.N.White,K.Brown,Techniques,薬学および毒物学への加速器質量分析の適用,Trends Pharamacol Sci 25(2004)442−7)。
【0149】
VogelおよびLoveは、生体サンプルで測定した同位体比率を薬物濃度へ変換する方法を説明し、式1により表わされる(J.Vogel,A.H.Love,加速器質量分析器による同位体分子標識の定量化,A.L.Burlingame編,Methods in Enzymology,Academic Press,New York,2005)。
K=(RM−RN).Ψ.W/L (式1)
この式において、Kは検体の濃度、RMは同位体比率、RNは生体サンプル中の自然バックグラウンドの同位体比率、Ψはサンプル中の炭素質量分率、Wは検体の分子量、そしてLは検体の比分子活性度である。
【0150】
同位体比率をAMSによりModernという単位で表し、1 Modern=98アトモル14C/mg 炭素 である。同位体比率は炭素の質量に対して表わすので、サンプル中の質量分率(Ψ)は、炭素の質量あたりの濃度を、サンプルの質量あたりの濃度へ変換する式中に含まれる。通常の炭素分析器により、質量分率を測定する。
【0151】
(RM−RN)をRnetとして表わして式1を簡略化することができ、また式2のように、モルの代わりに比活性度を質量(LMass)に対して表わす。
K=RnetΨ / Lmass (式2)
【0152】
式1および式2は一般的に確立されているが、薬物が代謝化されているとき、式の結果は親薬物と代謝物(すなわち質量等価物)の混合物の全14C(これより例として14Cを参照する)しか提供しない。存在する個別の検体の濃度を求めるために、本明細書に前述して説明したように、AMS分析の前に、代表的にはサンプルを抽出し、検体をHPLCのようなクロマトグラフィ技術により分離しなければならない。今までのところ、この過程を記述する式は報告されておらず、本発明の恩恵により、ここに初めて導かれる。
【0153】
検体がHPLCに続く全てのバックグラウンド炭素から分離されたと仮定すると、RN=0である。従って、分離検体の同位体比率からRnetを判別するために、後者をRAと指定する。実験動物もしくは人間へ投与した薬物の比活性度(Lmass)は生化学過程では変化せず、そのためHPLCにより分離した検体の比活性度は、親薬物の比活性度と等しいはずである(すなわちRA=L)。式2に代入すると、K=Ψであり、言い換えれば、HPLC留分内の検体量は、その留分内の検体からの炭素量と等しく、十分に同値である。そのため、分離した検体の濃度は、その同位体比率から求めることができないことがわかった。しかし、上述の説明のように、分離した検体へ14Cではなく12Cを加えて同位体希釈を行うことで、この制約を克服することが可能である。このような同位体希釈物(本明細書に上述したキャリヤ物質)は、かなり古くそのため14C(14Cのt1/2=5760年=106年あたり173半減期)が崩壊して失われた石油化学源からの炭素として入手可能である。この種の代表的な同位体希釈物は、液体パラフィンである。AMS分析における液体パラフィン使用は十分に確立され、液体パラフィンキャリヤ物質は、主に極めて小さなサンプルを扱えるサイズまで多くするために使用するため、「炭素キャリヤ」と呼ばれる(G.Lappin,S.Temple,加速器質量分析器,薬物発達における放射性追跡子,Tayler and Francis CRC Press,Florida,USA,2006)。しかし、HPLCにより分離した留分の場合には、これもまた本明細書で上述したが、炭素キャリヤは、同位体希釈という追加の目的を果たす。
【0154】
クロマトグラフィによりサンプルから精製した検体内の14C:12Cの比率をRAとする(上述のように)。同位体希釈のためにRAに加える12Cの量(すなわち炭素キャリヤ内の12Cの量)をΦとする。同位体希釈後の生成物の14C:12Cの比率をRDとする。式2に代入することで、式3を得る。
K=RDΦ / Lmass (式3)
【0155】
式3は、同位体希釈が正確に既知であることが好適であることを示す。すなわち、あらゆる誤差は、最終結果の誤差へ直接影響する。炭素キャリヤの量を正確に投与し、それにより既知とするが、例えば抽出中やHPLCカラム上での、あらゆる検体の分析損失もまたKの測定に対して誤差を導く。従って、分析回復を表す他のパラメータ(θ)を、式3に導入し、式4を得る。
K=RDΦ/ Lmassθ (式4)
【0156】
一般的な実験的回復を簡単に行うことでθを計測することが可能であり、またその回復が再現可能であることが示されているケースではその計測は容認できる一方で、分析する各サンプルに対して個別に回復を測定することがより望ましい。このことは、本明細書に上述したように、内部標準を使用する定量的HPLC分析で通常用いるものと同様の方法で達成できる。これらの合成物は限定的なアベイラビリティを有し、しばしば製造が比較的高価であるため、一般的に、14Cで標識化した内部標準の使用は実用的ではない。しかし、その代わりに、内部標準として放射性標識化をしない検体(対照検体)を使用し、紫外線吸収のような通常の検出方法により各サンプル内の濃度を測定することが可能である。
【0157】
対照検体をサンプルへ追加しても、代表的には14CであるAMS同位体の存在により識別可能であるため、検体の測定に支障は生じない。対照検体からサンプルへ加えた少量の12Cは、炭素キャリヤ内の12Cと比べて多くない(上述のように、また当業者によく知られるように、代表的にはμg程度の量の対照検体を加え、mg程度の量の炭素キャリヤを用いる)。さらに、一定に等量に正確に既知の量の対照検体を加えるとき(代表的にはそうであるが必ずしも必要ではない)、全ての有効なサンプルは同じ全濃度の検体を含み、そのため濃度に依存する影響は最小限となる。AMS分析において、サンプル中の検体濃度はしばしば極めて小さく、非特異結合による損失が大きくなりうることに留意しておくことは有利である。余剰の対照検体を追加することで、これらの非特異結合の効果を克服し、それにより回復を改善する。
【0158】
上述のように、内部標準として対照検体を使用する手順は、HPLC分析で用いるものと類似の手順に従う。まず標準曲線を作り、それにより一連の血漿サンプルを混合して、内部標準となる既知で一般的には等量の対照検体とともに、14C−もしくは他のAMS同位体標識化薬物の濃度を上昇させる。各混合較正剤内の、14C−もしくは他のAMS同位体標識化薬物の「真の」濃度を、正確に投与した量から求める。さらに、この環境下では、14Cの全濃度は14C薬物の濃度と等しいため、実際の測定値(本明細書内で上述の量「C」)を得るために、較正剤のアリコートを直接AMS(式2)により分析することができる。各血漿サンプルを抽出し、その抽出はHPLC上で行い、検体の保持時間に対応する留分を収集して(代表的には)黒鉛化して、12C:14Cの比率(代表的にはModernで表す)をAMSにより測定する。さらに、対照検体の紫外線応答のピーク(内部標準)を測定する(上述のように、例示として紫外線を使用するが、任意の適切な測定技術を用いることができる)。次に、真の濃度(「C」)をx軸に、HPLC留分のModern値(「B」)をその紫外線応答(「A」)で割った値をy軸にとって、標準曲線を作る。AMSは、14C検出が飽和する点まで線形に応答する。飽和により検出器は損傷してしまうが、飽和することは避けるため、実際にはその応答は常に線形である(L.K.Fifield,加速器質量分析およびその応用,Rep Prog Phys62(1999)1223−1274)。そのため、ラインは線形回帰による較正データに一致する。ここに説明する標準曲線は、装置応答を較正しないため、通常のHPLC較正ラインとは異なることを、理解しておくことは重要である。当業者によく知られるように、例えばAustralian National University砂糖もしくはシュウ酸のような、正確な量の12C:14Cの比率の分離標準を用いて、AMS装置を較正できる(G.Lappin,S.Temple,加速器質量分析,薬物発達における放射性追跡子,Taylor and Francis CRC Press,Florida,USA,2006)。
【0159】
分析下の各サンプルに対して、既知の(通常は上述した較正方法で使用するものと同様の)量の対照検体を内部標準として加える。サンプルを抽出し、HPLC上で実行し、検体の保持時間に対応する留分を収集し、黒鉛化し、Modern値をAMSにより測定する。上述の標準曲線と同じ方法において、紫外線応答もまた測定する。次に、y軸上の紫外線応答で割った留分のModern値は、x軸上の対応値から薬物濃度を決定する。y軸とx軸の間の対応は、曲線の傾きを規定し(線形のとき、その式はy=mx+c)、次に式3に代入して式5を得る。
K= (RA/mU+C)Φ/Lmass (式5)
式中、KはHPLC留分中の検体量、RAは同位体希釈後の検体のModern値、Φは同位体希釈物である12Cの量、Lmassは検体の質量比活性度、mは標準曲線の傾き、Uは紫外線応答(もしくは任意の適切な検出方法の検出応答)、およびCは標準曲線のy切片である。
【0160】
上述のように作る代表的な標準曲線を図2に示す。図2は、それぞれが5つの分離複製からなる14Cで標識化した薬物の7つの異なる濃度に対して、C値に対するB:Aの比率をプロットして得られたさらなる較正曲線である。この特定の標準曲線において、5つの分離複製からなる14C薬物の7つの濃度を使用した。炭素キャリヤとして液体パラフィンを使用した(1.6266mg 12C)。内部標準である放射性標識化していない検体の濃度は、35μg/mLであった。ラインの傾きは0.1359で、cの値は−0.0866であった。実際には、cの値はしばしばわずかで無視できる。図2に示す標準曲線の範囲は、1fgから160pg/HPLC留分であった。
【0161】
4fgから120pg/HPLC留分と等価な範囲を達成するために、14C薬物を混合して正確に希釈した血漿を、HPLCおよびAMSにより抽出した。標準曲線から求めた濃度は、平均で真の値の97.2%(n=25)であった(84.2−118.8%の範囲)。標準曲線を用いず、分析損失を考慮しないとき(すなわち式3)、混合血漿サンプルの濃度は、真の値の52.8−92.3%の範囲で、平均で69.1%(n=25)と求まった。従って、平均分析回復はおよそ72%であった。そのため、全く回復を考慮しないと(すなわち式4のθを1と仮定する)、結果は平均の誤差が−28%を示した。この誤差は、本発明による上述の内部標準方法を用いて、平均で2.8%減少した。
【特許請求の範囲】
【請求項1】
試験サンプルにおいてAMS同位体で標識化された検体の量を定める方法に用いるための較正方法であって、
(i)複数の較正サンプルを提供するために、前記検体または標識化されていない対照検体のいずれにも汚染されていない複数のサンプルを、既知量の前記対照検体および量Cの検体と接触させることであり、前記較正サンプルの各々が、既知量の対照検体ではあるがCと異なる量を含むもの、
(ii)複数のサンプルの各々に加えられた検体の量CをAMSにより測定すること、
(iii)複数の精製されたサンプルを提供するために、複数のサンプルにおいて、検体および対照検体を、他の種から分離すること、
(iv)前記精製されたサンプルにおいて検体の量AをAMSにより測定すること、および
(v)前記精製されたサンプルにおいて対照検体の量BをAMSにより測定すること
を含む、較正方法。
【請求項2】
同じ量の対照検体は前記複数の前記較正例の各々に加えられる、請求項1の較正方法。
【請求項3】
前記量Cを測定することは対照検体を前記サンプルの各々に接触させる前に行われる、請求項1または請求項2の較正方法。
【請求項4】
サンプルはヒトまたは動物由来である、請求項1から3までのいずれか1項の較正方法。
【請求項5】
サンプルはヒト由来である、先行する請求項のいずれか1項の較正方法。
【請求項6】
サンプルは、尿、糞または血液である、先行する請求項のいずれか1項の較正方法。
【請求項7】
サンプルは血液である、請求項6の較正方法。
【請求項8】
サンプルは血漿である、請求項6または請求項7の較正方法。
【請求項9】
試験サンプルにおいてAMS同位体で標識化された検体の量を定めることに用いるための方法であって、
(i)前記試験サンプルを、既知量の標識化していない対照検体と接触させること、
(ii)精製された試験サンプルを提供するために、試験サンプルにおいて、検体、もし存在すればおよび対照検体を、他の種から分離すること、
(iii)前記精製された試験サンプルにおいて検体の量AをAMSにより測定すること、および
(iv)前記精製された試験サンプルにおいて対照検体の量Bを測定すること
を含む、方法。
【請求項10】
試験サンプルはヒトまたは動物由来である、請求項9の方法。
【請求項11】
試験サンプルはヒト由来である、請求項9の方法。
【請求項12】
試験サンプルは、検体および対照検体のマイクロドーズを前もって投与された対象から得られる、請求項10または請求項11の方法。
【請求項13】
試験サンプルは、尿、糞または血液である、請求項9から12までのいずれか1項の方法。
【請求項14】
試験サンプルは血液である、請求項12の方法。
【請求項15】
試験サンプルは血漿である、請求項13または請求項14の方法。
【請求項16】
さらに、請求項1から8までのいずれか1項の較正方法を含み、請求項8から15までのいずれか1項の方法および請求項1から8までのいずれか1項に記載の較正方法の双方において、同じ検体および対照検体を採用する、請求項9から15までのいずれか1項の方法。
【請求項17】
請求項2から8までのいずれか1項に記載の較正方法において、試験サンプルは、前記試験サンプルと同じ量の対照検体と接触される、請求項14の方法。
【請求項18】
請求項9から15までのいずれか1項の方法での接触させること、分離すること、および測定することのステップ(i)−(iv)は、請求項1から8までのいずれか1項に記載の較正方法と、接触させること、分離すること、および測定することのステップ(i)および(iii)−(v)で同じ条件下に行われる、請求項16または請求項17のいずれか1項の方法。
【請求項19】
AMS同位体は、3H、7Be、10Be、14C、17O、18O、26Mg、26Al、32Si、35S、36Cl、41Ca、55Fe、60Fe、53Mn、79Se、59Ni、および129Iを含む群から選ばれる、先行する請求項のいずれか1項の方法。
【請求項20】
AMS同位体は3Hおよび14Cを含む群から選ばれる、先行する請求項のいずれか1項の方法。
【請求項21】
AMS同位体は14Cである、先行するいずれか1項の方法。
【請求項22】
検体は薬物候補である、先行する請求項のいずれか1項の方法。
【請求項23】
検体は薬物候補の代謝物である、請求項1から21までのいずれか1項の方法。
【請求項24】
分離することには、HPLCによって分離することが含まれる、先行する請求項のいずれか1項の方法。
【請求項25】
量Bを測定することは、対照検体のUV(紫外線)吸収を測定することによって達成される、先行する請求項のいずれか1項の較正方法。
【請求項1】
試験サンプルにおいてAMS同位体で標識化された検体の量を定める方法に用いるための較正方法であって、
(i)複数の較正サンプルを提供するために、前記検体または標識化されていない対照検体のいずれにも汚染されていない複数のサンプルを、既知量の前記対照検体および量Cの検体と接触させることであり、前記較正サンプルの各々が、既知量の対照検体ではあるがCと異なる量を含むもの、
(ii)複数のサンプルの各々に加えられた検体の量CをAMSにより測定すること、
(iii)複数の精製されたサンプルを提供するために、複数のサンプルにおいて、検体および対照検体を、他の種から分離すること、
(iv)前記精製されたサンプルにおいて検体の量AをAMSにより測定すること、および
(v)前記精製されたサンプルにおいて対照検体の量BをAMSにより測定すること
を含む、較正方法。
【請求項2】
同じ量の対照検体は前記複数の前記較正例の各々に加えられる、請求項1の較正方法。
【請求項3】
前記量Cを測定することは対照検体を前記サンプルの各々に接触させる前に行われる、請求項1または請求項2の較正方法。
【請求項4】
サンプルはヒトまたは動物由来である、請求項1から3までのいずれか1項の較正方法。
【請求項5】
サンプルはヒト由来である、先行する請求項のいずれか1項の較正方法。
【請求項6】
サンプルは、尿、糞または血液である、先行する請求項のいずれか1項の較正方法。
【請求項7】
サンプルは血液である、請求項6の較正方法。
【請求項8】
サンプルは血漿である、請求項6または請求項7の較正方法。
【請求項9】
試験サンプルにおいてAMS同位体で標識化された検体の量を定めることに用いるための方法であって、
(i)前記試験サンプルを、既知量の標識化していない対照検体と接触させること、
(ii)精製された試験サンプルを提供するために、試験サンプルにおいて、検体、もし存在すればおよび対照検体を、他の種から分離すること、
(iii)前記精製された試験サンプルにおいて検体の量AをAMSにより測定すること、および
(iv)前記精製された試験サンプルにおいて対照検体の量Bを測定すること
を含む、方法。
【請求項10】
試験サンプルはヒトまたは動物由来である、請求項9の方法。
【請求項11】
試験サンプルはヒト由来である、請求項9の方法。
【請求項12】
試験サンプルは、検体および対照検体のマイクロドーズを前もって投与された対象から得られる、請求項10または請求項11の方法。
【請求項13】
試験サンプルは、尿、糞または血液である、請求項9から12までのいずれか1項の方法。
【請求項14】
試験サンプルは血液である、請求項12の方法。
【請求項15】
試験サンプルは血漿である、請求項13または請求項14の方法。
【請求項16】
さらに、請求項1から8までのいずれか1項の較正方法を含み、請求項8から15までのいずれか1項の方法および請求項1から8までのいずれか1項に記載の較正方法の双方において、同じ検体および対照検体を採用する、請求項9から15までのいずれか1項の方法。
【請求項17】
請求項2から8までのいずれか1項に記載の較正方法において、試験サンプルは、前記試験サンプルと同じ量の対照検体と接触される、請求項14の方法。
【請求項18】
請求項9から15までのいずれか1項の方法での接触させること、分離すること、および測定することのステップ(i)−(iv)は、請求項1から8までのいずれか1項に記載の較正方法と、接触させること、分離すること、および測定することのステップ(i)および(iii)−(v)で同じ条件下に行われる、請求項16または請求項17のいずれか1項の方法。
【請求項19】
AMS同位体は、3H、7Be、10Be、14C、17O、18O、26Mg、26Al、32Si、35S、36Cl、41Ca、55Fe、60Fe、53Mn、79Se、59Ni、および129Iを含む群から選ばれる、先行する請求項のいずれか1項の方法。
【請求項20】
AMS同位体は3Hおよび14Cを含む群から選ばれる、先行する請求項のいずれか1項の方法。
【請求項21】
AMS同位体は14Cである、先行するいずれか1項の方法。
【請求項22】
検体は薬物候補である、先行する請求項のいずれか1項の方法。
【請求項23】
検体は薬物候補の代謝物である、請求項1から21までのいずれか1項の方法。
【請求項24】
分離することには、HPLCによって分離することが含まれる、先行する請求項のいずれか1項の方法。
【請求項25】
量Bを測定することは、対照検体のUV(紫外線)吸収を測定することによって達成される、先行する請求項のいずれか1項の較正方法。
【図1】

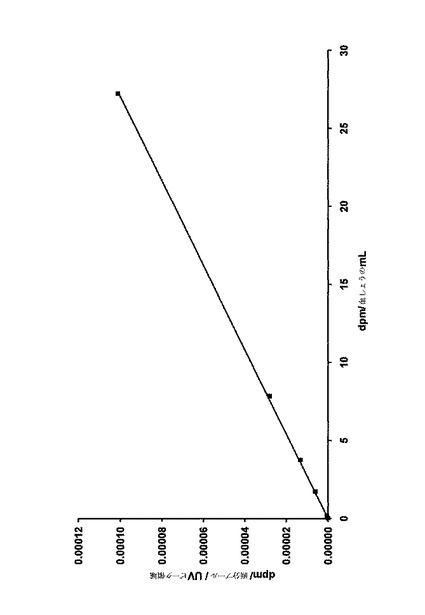
【図2】
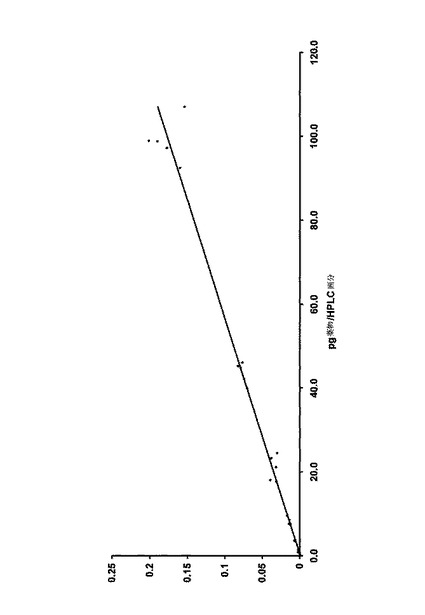
【図2】
【公表番号】特表2010−533857(P2010−533857A)
【公表日】平成22年10月28日(2010.10.28)
【国際特許分類】
【出願番号】特願2010−516582(P2010−516582)
【出願日】平成20年7月18日(2008.7.18)
【国際出願番号】PCT/GB2008/002464
【国際公開番号】WO2009/010764
【国際公開日】平成21年1月22日(2009.1.22)
【出願人】(510016667)エクセレロン リミテッド (1)
【Fターム(参考)】
【公表日】平成22年10月28日(2010.10.28)
【国際特許分類】
【出願日】平成20年7月18日(2008.7.18)
【国際出願番号】PCT/GB2008/002464
【国際公開番号】WO2009/010764
【国際公開日】平成21年1月22日(2009.1.22)
【出願人】(510016667)エクセレロン リミテッド (1)
【Fターム(参考)】
[ Back to top ]