
加齢黄斑変性症を治療する方法および組成物ならびに加齢黄斑変性症を治療する化合物を識別する方法
本発明は、加齢黄斑変性症を治療または加齢黄斑変性症の進行を制限する方法と、かかる使用に適した化合物を識別する方法とを提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、加齢黄斑変性症を治療する方法および組成物ならびに加齢黄斑変性症を治療する化合物を識別する方法に関する。
(関連出願)
本願は、その全体を本明細書に援用する2008年4月18日に出願された米国仮特許出願番号第61/124,624号の優先権を主張する。
(政府の権利の記述)
本発明は米国国立衛生研究所(NIH)の助成番号R03 EY014403の下、政府支援で成されたものである。米国政府は本発明の一定の権利を有する。
【背景技術】
【0002】
加齢黄斑変性症(「AMD」)は、網膜の損傷により黄斑(視野中心)の視力喪失を生じさせる加齢に関連する疾病である。AMDは高齢者に一般的な疾患であり、66−74歳の患者の約10%、75−85歳の患者の30%が何らかのレベルの黄斑変性を有している。
【発明の概要】
【発明が解決しようとする課題】
【0003】
現在、大部分のAMD患者に有効な治療は存在せず、初期段階の介入法もない。
【課題を解決するための手段】
【0004】
1態様では、本発明は、加齢黄斑変性症(AMD)の治療に有効な量のOA1受容体のアゴニストをAMD患者に投与することを含む、AMDを治療する方法を提供する。第2の態様では、本発明は、AMDの進行の制限に有効な量のOA1受容体のアゴニストをAMDが進行する危険性のある患者に投与することを含む、AMDの進行を制限する方法を提供する。本発明のこれらの態様のいずれかの1つの好ましい実施形態では、OA1受容体のアゴニストは、L−DOPAおよびL−DOPA類似体からなるグループから選択される。
【0005】
別の態様では、本発明は、AMDを治療する化合物を識別する方法であって、細胞を試験化合物と接触させることを含み、該細胞は、
(a)OA1を発現している第1の細胞集団;および任意選択で、
(b)OA1を発現していない第2の細胞集団;を含み、
(c)以下の(i)および(ii)うちの一方または両方を増大させる試験化合物を陽性試験化合物と識別し;
(i)(A)試験化合物と接触させない第1の細胞集団における色素上皮誘導因子(PEDF)発現および(B)第2の細胞集団におけるPEDF発現の一方又は両方に対する、第1の細胞集団におけるPEDF発現;
(ii)(A)試験化合物と接触させない第1の細胞集団における細胞内カルシウム濃度および(B)第2の細胞集団;
陽性試験化合物は、AMDの治療およびAMDの進行の制限の少なくとも一方を行うための候補化合物である方法を提供する。
【0006】
さらなる態様では、本発明は、以下の工程を含む、AMDを治療する化合物を識別する方法を提供する:
(a)チロシナーゼ欠損の妊娠している雌の非ヒト哺乳動物に試験化合物を投与する工
程であって、試験化合物は胚の光受容体および/または網膜神経節の発達中に投与される工程、
(b)胚または出生後の非ヒト哺乳動物における光受容体および/または網膜神経節の発達に対する試験化合物の効果を、試験化合物を投与しなかった胚または出生後における光受容体および/または非ヒト哺乳動物網膜神経節の発達と比較する工程であって、光受容体および/または網膜神経節の発達を増大させる試験化合物が、AMDの進行を治療および/または制限する候補化合物である工程。
【0007】
またさらなる態様では、本発明は以下を含む組成物を提供する:
(a)AMDの進行を治療または制限するのに有効な量のL−DOPAまたはL−DOPA類似体;
(b)AMDの進行を治療または制限するのに有効な量のビタミンC源、ビタミンE源、ビタミンA源、亜鉛源および銅源を含む組成物。
【図面の簡単な説明】
【0008】
【図1A】in situでRPEをビオチン化(A)した後、ストレプトアビジン結合ビーズに(B)結合させるか、または(C)結合させないタンパク質のウェスタンブロット解析。(B)培養RPE、(C)OA1−GFPを発現するようトランスフェクトしたCOS細胞。ブロットはストレプトアビジン結合ビーズを使用して細胞表面をビオチン化および細分化した後にOA1およびアクチンを可視化するようブローブ標識した。培養細胞(B、C)について、細胞は分析に先立ち500μM(通常DMEM)または1μMのチロシンで3日間維持した。
【図1B】ストレプトアビジン結合ビーズに結合させたタンパク質のウェスタンブロット解析。
【図1C】ストレプトアビジン結合ビーズに結合させないタンパク質のウェスタンブロット解析。
【図1D】デンシトメトリーによるウェスタンブロット解析の定量化。OA1デンシトメトリーはペアになった細胞培養物対照の%として示される。ペアになった細胞培養物は、トランスフェクト後2つの等しい群へ分割したものであり、一方は通常DMEM(対照)に維持した対照であり、他方の群は収集するまで1μMチロシンDMEM(LT)で維持した。Paired−t検定分析を使用して、差が有意であるか否か試験した。*はp<0.001を示す。同じ方法を分析したアクチンは差を示さず、p=0.724であった。
【図1E】通常DMEMに維持し、その後抗OA1抗体で染色し、20倍で画像化した色素RPE細胞の複合焦点顕微鏡検査法。バー=25μm。
【図1F】1μMチロシンに維持し、その後抗OA1抗体で染色し、20倍で画像化した色素RPE細胞の複合焦点顕微鏡検査法。バー=25μm。
【図2A】トランスフェクトしたまたはトランスフェクトしていないCHO細胞における標準実験プロトコルの時間経過中の[Ca2+]iのトレース例。3分間の安定な基準線の確立後、試験薬を1μMで加えた。5分目に、対照として機能するようKClを加え、細胞にFura−2を装填し、利用できるようにした。トランスフェクト細胞とペアにした非トランスフェクト細胞の両方に同一のプロトコルを行なった。
【図2B】トランスフェクトCHO細胞および非トランスフェクトCHO細胞におけるチロシン、ドーパミンおよびL−DOPAに応じた[Ca2+]iの概要データ。非トランスフェクト細胞を、L−DOPA処理で示す。KClによる膜脱分極の実験対照も示される。バーはそれぞれ少なくとも10回の実験から集めたデータを表わし、検査薬添加後の基準線[Ca2+]iからの平均変化として示される。エラーバーは標準偏差を表し、有意差試験にt検定分析を使用した。*はp<0.01を示す。天然タンパク質を発現するOA1またはRPEを発現するようトランスフェクト細胞の[Ca2+]i増加の百日咳毒素感受性分析。データは少なくとも6回の実験の平均を表わす。
【図2C】天然タンパク質を発現するOA1またはRPEを発現するようトランスフェクト細胞の[Ca2+]i増加の百日咳毒素感度分析。データはトランスフェクト細胞の各群に対する少なくとも6回の実験および内因性OA1発現した処理済みおよび未処理RPEの20回の個々の実験の平均を示す。t検定分析を使用して有意差を試験した。*はp<0.01を示す。
【図2D】OA1を発現するようトランスフェクトされたCHOにおけるcAMPを測定した。対照群はトランスフェクトされたが未処理のCHO細胞であり、かかる細胞におけるcAMPの基準レベルを表わす。細胞を1.0μM L−DOPA、0.1μMホルスコリン、L−DOPA+0.1μMホルスコリン、および陽性対照として1μMホルスコリンでを処理した。結果は、同じ培養プレートでレプリカ単層を使用して、すべての実験群をすべてペアにして分析した少なくとも6回の実験で観察された平均cAMPレベルに相当する。エラーバーは各群の標準偏差を示し、観察された唯一の有意差は、ホルスコリン処理後のcAMPレベルの増加であった。
【図3A】OA1とL−DOPAの間の結合反応速度を放射性標識リガンド結合アッセイを使用して決定した。結果は、5回のかかる実験から集めたデータを表わし、平均特異的結合+/−SEMとして示す。双曲線適合は0.994のR2値を示し、Kdは9.34×10-6M+/1.14x10-6Mと決定された。
【図3B】OA1トランスフェクトCHO細胞に対する5μM[H3]L−DOPAの相対的結合を、1.0mMドーパミン、チロシンまたはL−DOPAの存在下で比較した。データは各群の平均合計結合+/−標準偏差を示す。*は、対照群間の結果を、潜在的な競争リガンドの存在下での結合と比較した場合のp<0.05を示す。
【図3C】ドーパミンがOA1活性アンタゴニストとして機能するか否かを決定するために、5μM[H3]L−DOPAおよびドーパミン間の競争相互作用を評価した。結果は、ドーパミンおよびL−DOPAが同じOA1結合部位に競争することを示し、データはr2値が0.95の結合モデルに適合する。ドーパミンのKiは2.388+/−0.266μM(平均+/−SEM)であり、L−DOPAのKdと同様であった。
【図3D】OA1による用量依存的OA1シグナリング。データは所与の濃度(各用量n=6)での細胞のL−DOPA処理により誘発された[Ca2+]iの平均増加量を示す。t検定を使用して、各用量で達成された応答を比較した。*は1および10μMでの比較が<0.01であることを示す。
【図3E】図3(a)に基づく単一部位の結合関係の動力学を示すScatchardプロット。
【図4】(A)−(H)。画像はすべて、OA1−GFPを発現するようトランスフェクトされたCHO細胞の2μm厚の共焦切断片を示す。β−アレスチンは免疫蛍光法を使用して可視化した。L−DOPA(A−C)の追加前、1μM L−DOPA(D−F)の処理後、および合一画像(C,F)は、白色光イメージングの解像度で2つのタンパク質が同時局在化する領域を示す。(G、H)はトランスフェクトCHO細胞の視野の低倍率であり、2個のトランスフェクト細胞が見える(矢印)(G)。残りの細胞集団は膜へのβ−アレスチンの集合がOA1発現細胞でのみ生じている(矢印)を示すためにβ−アレスチン抗体を使用して可視化している(H)。
【図5A】PEDF濃度を細胞馴化培地のELISAにより決定した。RPE(網膜色素上皮)細胞は、L−DOPA処理をしない対照細胞か、または通常DMEMで3日間維持する前に1μM L−DOPAで処理したOA1刺激細胞である。データは3連で行った3回の実験の平均として示され、エラーバーは標準偏差を示し、*はpaired t検定を使用してP<0.01を示す。
【図5B】ELISAにより決定された色素RPEからの馴化培地中のPEDF濃度。細胞は、対照色素RPE培養物または200μMのフェニルチオ尿素(PTU)で処理したペアになった培養物とした。データは3連で行った3回の実験の平均として示され、エラーバーは標準偏差を示し、*はpaired t検定を使用してP<0.01を示す。
【図5C】OA1シグナリングを刺激するための、PTUで処理し、次にL−DOPAで処理した色素RPE細胞の馴化培地におけるPEDF濃度。PTU処理前、次にPTU処理後、L−DOPA刺激後の同じ培養物から、ELISA分析を行った。結果を細胞培養物に関連して達成された値の平均+/−標準偏差として示す。*は、PTUを対照(PTUを対照前に試験した同じ培養物)と比較した場合ならびに同じ培養物からのPTUサンプルとL−DOPA/PTUを比較した場合に、p<0.01であることを示す。
【図6A】データは、全量、特異的、および非特異的のすべての分画における平均+/−標準誤差の結合[3H]−L−DOPAを示す。非特異的結合は、超過の未標識L−DOPA(1mM)の存在下で結合した放射性標識L−DOPAの測定により決定された。各所与濃度の特異的結合は、測定した結合合計から測定した非特異的結合を減算することにより決定される。
【図6B】チロシンおよび5μM[H3]L−DOPAの濃度の増大を使用して測定した、チロシンとL−DOPAの間の競争的相互作用を示す。各データポイントは、5つの複製ウェルからの平均データを示し、エラーバーは標準偏差である。データは、チロシンがL−DOPAとの結合に競争するが、親和性は低いことを示している。結果は、チロシンが52.9μMのKiを有し、r2値が0.85の単一部位結合モデルに適合する。チロシンの溶解度が制限されているため、飽和に達しない可能性がある。
【図7】野性型対OA欠損マウスにおけるウェスタンブロットおよびPEDF分泌のグラフ図。
【図8A】正常野性型マウスで予想されるのと比較して、L−DOPAの補給が網膜神経節細胞数を増加させることを示すデータのグラフ図。
【図8B】正常野性型マウスで予想されるのと比較して、L−DOPAの補給が光受容体数を増加することを示すデータのグラフ図。
【図8C】2匹の野性型および2匹のOA1−/yマウスにおけるPEDF検出を示すウェスタンブロット。
【発明を実施するための形態】
【0009】
本明細書で引用した文献はすべてその全体が本明細書に組み込まれる。
本願では、他に別記されていなければ、使用される技術は以下のいくつかの周知の参照文献に見い出されてもよい:Molecular Cloning: A Laboratory Manual (Sambrook, et al., 1989, Cold Spring Harbor Laboratory Press), Gene Expression Technology (Methods in Enzymology, Vol. 185, edited by D. Goeddel, 1991. Academic Press, placeCitySan Diego, StateCA), 敵uide to Protein Purification・in Methods in Enzymology
(M.P. Deutshcer, ed., (1990) Academic Press, Inc.); stocktickerPCR Protocols: A
Guide to Methods and Applications (Innis, et al. 1990. Academic Press, placeCitySan Diego, StateCA), Culture of Animal Cells: A Manual of Basic Technique, 2nd
Ed. (R.I. Freshney. 1987. Liss, Inc. New York, NY), Gene Transfer and Expression Protocols, pp. 109-128, ed. E.J. Murray, The Humana Press Inc., Clifton, N.J.), and the Ambion 1998 Catalog (Ambion, Austin, TX).
本明細書に使用する場合、単数形(英語で「a」、「an」および「the」)は文脈が別段指示していない限り、複数の参照物を含む。
【0010】
第1態様では、本発明は、加齢黄斑変性症(AMD)を治療する方法であって、AMDの治療に有効な量のOA1受容体のアゴニストをAMD患者に投与することからなる方法を提供する。
【0011】
第2の態様では、本発明は、AMDの進行を制限する方法であって、AMDの進行の制限に有効な量のOA1受容体のアゴニストを、AMDが進行する危険性のある患者に投与することからなる方法を提供する。
【0012】
ヒトOa1遺伝子はX染色体上に見出され、配列分析[14]に基づけばGタンパク質共役受容体(GPCR)[12,13]であると考えられる404個のアミノ酸からなるタンパク質OA1(配列番号2)をコードすることが示されている。本明細書に詳細に開示するように、本願発明者は、神経感覚網膜生存の重要な決定因子としてOA1シグナル伝達経路を同定した。OA1シグナル伝達経路の刺激は、AMDの治療を提供するとともに、潜在的リスクのあるAMD進行を制限する手段も提供する。いかなる機構によっても拘束されないが、本願発明者は少なくとも1つの効き目のある神経栄養因子であるPEDFの分泌を規制するL−DOPAによる自己分泌ループにOA1およびチロシナーゼが関与していると考える。したがって、L−DOPAの投与を使用して、OA1活性を刺激し、したがってPEDF発現をアップレギュレートし、これをAMDの治療およびAMD進行の制限のための有用な治療手段とすることが可能である。
【0013】
以下に詳細に論ずるように、そのようなOA1アゴニストは、例えば本発明の第3および第4の態様の新薬発見方法を使用して識別することが可能である。OA1アゴニストの例は以下に詳細に論ずる。
【0014】
好適には、患者はヒトである。
本明細書に使用する場合、本発明のすべての態様および実施形態について、「AMD」は、加齢黄斑変性症として知られる、網膜に対する損傷のために黄斑(視野中心)の視力が喪失する加齢に関連する疾患を意味する。本明細書に使用する場合、AMDは、以下により詳細に説明する滲出型AMDと萎縮型AMDの両方を包含する。
【0015】
AMDは、網膜色素上皮とその下の脈絡膜との間の黄斑における特徴的なドルーセン(黄色沈着物)から始まる。これらの初期の変化(加齢性黄斑変性症と呼ばれる)を有する人の大半は、良好な視力を維持している。ドルーセンのある人は、引き続き進行したAMDに進展する可能性がある。この危険性は、ドルーセンが大きく、多く、かつ黄斑の下の色素細胞層における妨害に関連している場合には、かなり高くなる。
【0016】
加齢性黄斑変性症の患者は進行したAMDの2つの主な形式のうちのいずれかに進み得るが、そのいずれも本発明の方法を使用して、治療可能であるか、またはその進行を制限することが可能である。「滲出型」AMDは、Bruch膜中の脈絡膜毛細血管の以上な血管
増殖による視力喪失を引き起こし、究極的には黄斑の下の血液およびタンパク質の漏出につながる。このような血管からの出血、漏出、および瘢痕は、もし治療していないで放置されると、光受容体への不可逆的な損傷および急速な視力喪失をもたらす。「萎縮型」AMDは黄斑中の感光性細胞がゆっくり分解したときに起こり、次第に罹患した目の視力喪失を徐々に引き起こす。AMDにおける目のかすみは、網膜色素上皮(RPE)の下のドルーセンの蓄積によるものであり、これは光受容体を焦点平面から異動させることにより光受容体の焦点特性を変化させる。
【0017】
萎縮型AMDは一方又は両方の目に生じ、加齢性黄斑変性症から、萎縮型AMDの中間期または進行期へと進行する可能性がある。
中間期の萎縮型AMD:中間サイズの多くのドルーセンがあるか、または1つまたは複数の大きなドルーセンがある。人によっては自身の視野中心にぼやけた点が見える。読書や他の作業のためにはより多くの光が必要され得る。
【0018】
進行期の萎縮型AMD:ドルーセンに加え、網膜中心領域の感光性細胞および支持組織の分解。この分解により視野中心にぼやけた点が生じ得る。時間が経つにつれ、ぼやけた点はより大きく暗くなり、中心視力のより多くを占めるようになり、読書することや、顔が非常に近づくまで顔を認識するのが困難となり得る。
【0019】
AMD症状には、中心視野のぼやけ/減少、中心暗点(影または視力の喪失領域)、ある暗色を別の暗色と区別し、かつ/または、ある光の色を別の光の色と区別する困難さ、明るい光に曝された後の視覚機能の回復の遅さ、コントラスト感度の喪失(その結果、環郭、影および色覚がより鮮明でなくなる)、網膜色素上皮(RPE)妨害(色素の凝集および/または脱落を含む)、RPE剥離、地図状萎縮、網膜下血管新生、円盤形瘢痕、および乱視(変形視)(複数の直線からなるグリッドが波状に見え、グリッドの部分がブランクに見える)が含まれるがこれらに限定されない。萎縮型AMDおよび滲出型AMDの症状は、一般に、疾患の進行の間同様に早いため、どの初期の患者でAMDの萎縮型対滲出型が進行しているかを決定するのは可能でない場合がある。萎縮型AMDは「地図状萎縮」として発達し、新しい血管が発生した場合、初期AMDは「滲出型」AMDとなる。
【0020】
本明細書に使用する場合、AMDを「治療する」とは、以下の1つまたは複数を遂行することを意味する:(a)AMDの重篤度の低下;(b)上述したAMDに特徴的な1つまたは複数の症状の進行の制限または予防;(c)上述したAMDに特徴的な1つまたは複数の症状の悪化の抑制;(d)以前に障害のあった患者のAMD再発の制限または予防;および(e)以前にAMDの症状があった患者の1つまたは複数の症状の再発の制限または予防。そのような治療は、滲出型AMDと萎縮型AMDの治療を含む。
【0021】
本明細書に使用する場合、AMDの「進行の制限」という用語は、AMDが進行する危険性のある個体におけるAMDの進行を予防するかまたは最小限にすること、ならびに加齢性黄斑変性症のAMD(滲出型または萎縮型)への進行または中間期の萎縮型AMDから進行期の萎縮型または「滲出型」AMDへの進行を制限すること、を意味する。1つの好ましい実施形態では、方法は、AMDの進行を制限するために、ドルーセン蓄積(加齢性黄斑変性症)のある患者を治療することを含む。別の好ましい実施形態では、方法は、治療していないAMD患者またはAMDの危険性のある患者と比較した視力喪失のラインの割合を減少させるのに有効な量のOA1アゴニストで患者を治療することを含む。別の好ましい実施形態では、方法は、滲出型AMD患者または滲出型AMDが進行する危険性のある患者を、新たな血管形成の速度および数を減少させるのに有効な量のOA1アゴニストで治療することを含む。より以下に詳細に論ずるように、OA1刺激はRPEにPEDF分泌を増大させ、PEDFは効き目のある抗血管新生因子である。したがって、OA1刺激戦略は、本明細書で論じた網膜の進行に対するその効果に加えて、「滲出型」AMDでの新たな血管の発達を止めることが可能である。
【0022】
別の好ましい実施形態では、方法は、中心視力がぼけているか減少している患者を、一方又は両方の目の視力のラインを増加させるのに有効な量のOA1アゴニストで治療することを含む。この実施形態では、視力のラインは標準Snellen試験により測定されるのと同じものであり、視力「ライン」の増大または減少はSnellen視力表上のどの最も細いラインを患者がはっきりと読むことができるかに基づく。
【0023】
「AMDが進行する危険性のある患者」とは、50歳以上(種々の好ましい実施形態では60歳以上、65歳以上、70歳以上、または75歳以上)で、ドルーセン沈着物の存在、白色人種、AMDを現在有しているか過去に有していた血族の存在、(Tyr402His)の補体因子H遺伝子(CFH)の突然変異、補体タンパク質C3遺伝子のArg80Gly変異型、高血圧、高いコレステロール値、肥満、喫煙、高脂肪摂取、およびフィブリン5遺伝子中の突然変異を含むがそれに限定されないAMDを進行させる任意の危険因子を有する任意の者を意味する。したがって、好ましい実施形態では、治療される患者は、特にAMDの進行を制限する方法において、これらの危険因子の1つまたは複数を有している。
【0024】
「治療上有効量」という語句は、本明細書に使用する場合、AMDの進行を制限するか
またはAMDを治療する(進行を防ぐか、または逆行させる)のに十分または有効な量を指す。適切な用量の範囲は、化合物の選択、投与経路、製剤の性質、患者の症状の状態、および関与する医師の判断によって決まる。例えば、経口投与は、静脈内注射による投与よりも高用量が必要であると予想される。当該技術分野でよく理解されるように、かかる投薬レベルの違いは、最適化のための標準の経験的ルーチンを使用して調節することが可能である。
【0025】
好ましい実施形態では、OA1受容体ゴニストは、L−DOPAおよびL−DOPA類似体からなるグループから選択された化合物を含む。
L−DOPAはパーキンソン病の治療の際に使用されることが知られている[2−アミノ−3−(3,4−ジヒドロキシフェニル)プロパン酸]であり、以下の構造を有している。
【0026】
【化1】

L−DOPAは市販されており、またその合成方法は当業者に周知である。
【0027】
本明細書に使用する場合、「L−DOPA類似体」は、その多くが当該技術分野で周知であるL−DOPAプロドラッグを含む、OA1刺激活性を保持しているL−DOPAの変異型であり、かかる類似体の例については以下に開示する。特定の作用機序によって拘束されるわけではないが、本願発明者は、OA1へのL−DOPAの結合には2つの結合部位が関与し、1つは一方又は両方の水酸基が関与し、1つはカルボン酸基が関与すると考えている。1実施形態では、L−DOPA類似体は、投与後(かつ一般に細胞表面上のOA1への結合前)にL−DOPAに代謝され、そのためOA1刺激活性を保持すると予想されるL−DOPAプロドラッグである。別の実施形態では、一方又は両方の水酸基および/またはカルボキシル基は、本発明の方法に使用される種々の類似体(プロドラッグまたは別のもの)を生産するために置換可能である。
【0028】
別の実施形態では、L−DOPA類似体はL−DOPAエステルを含む。L−DOPAエステルの例およびそれらを製造する方法は、WO/1997/016181;米国特許第4663349号;米国特許第4873263号;米国特許第4873263号;米国特許第5345885号、および米国特許第4771073号に開示されている。種々の好ましい実施形態では、L−DOPAエステルは、L−DOPAメチルエステル、L−DOPAブチルエステル、L−DOPAペンチルエステル、L−DOPAシクロヘキシルエステル、L−DOPAベンジルエステル、およびL−DOPAエチルエステルからなるグループから選択される。種々のさらなる好ましい実施形態では、L−DOPAエステルは、L−DOPAのアルキル、アリール、および置換および非置換のアラルキルエステルからなるグループから選択される。さらに好ましい実施形態では、L−DOPAエステルは以下の式により表わされる:
【0029】
【化2】
式中、Rは、
直鎖または分岐鎖のアルキル基(C1−C20)、例えばメチル、エチル、プロピル、ブ
チル、ミリスチル、パルミチル、ペンチル、テトラデシル、ヘキサデシル等;
アリール基(C6−C9)、例えばフェニル、トリル等;
アルコキシ(C1−5)[メトキシ、エトキシ、ブトキシ等]等の置換基を任意選択で有する置換または非置換のモノ、ジ、またはポリヒドロキシアルキル基(C1−C20)、例
えばベンジル、アルコキシベンジル、4−ヒドロキシブチル、2−ヒドロキシプロピル、2,3−ジヒドロキシプロピル、1,3−ジヒドロキシプロピル、6−ヒドロキシヘキシルおよび5−ヒドロキシペンチル等;
カルボアルコキシ(C1−5)[メトキシカルボニル、エトキシカルボニル、プロポキシカルボニル、ブトキシカルボニル等];
アミノ;
モノまたはジアルキルアミノ(C1−10)[メチルアミノ、メチルエチルアミノ、ジエ
チルアミノ等];
アシルアミノ(C1−5)[アセタミド、ブチルアミド];
ケトアルキル(C1−5)[メチルケト、エチルケト、ブチルケト等];
ハロ[クロロ、ブロモ等]またはカルボキサミド;
置換および非置換のアラルキル(C7−20)、例えばベンジル、アルコキシベンジルC8−14[メトキシ、エトキシ、イソブトキシ等];
フェニルエチル;
フェニルプロピル;
フェニルブチル;
フェニルヘキシル;
フェニルオクチル等;および
医薬として許容される有機または無機のカウンターイオン塩類。
【0030】
L−DOPAのエステルおよびその塩を製造するための合成法は当該技術分野で周知であり、例えばその各々を本明細書に援用する米国特許第3,891,696号、米国特許第4,035,507号;および米国特許第5,354,885号;ならびにJournal of
Pharmaceutical Sciences, 62, p.510(1973)に記載されている。
【0031】
別の実施形態では、L−DOPA類似体は、当該技術分野で周知の胆汁酸抱合体を含む。L−DOPA胆汁酸抱合体の例およびそれらを製造する方法はWO/2002/028882および米国特許出願公開第20020151526号に開示されている。経口投与に際して、これらのプロドラッグは腸肝系内で開裂し、ペアレント薬および/または胆汁酸からの活性代謝物を体循環へ放出する。重要なことに、プロドラッグの一部のみ(通常<50%)しか腸肝サイクルの各通過間に開裂されない。したがって、腸肝循環は、持続的な全身の薬物濃度の達成を可能にする薬物の貯蔵庫として作用する。コール酸、ケノデオキシコール酸、ウルソデオキシコール酸、デオキシコール酸、ウルソコラン酸およびリトコール酸のような天然の胆汁酸が特に好ましい。L−DOPAまたは他のL−DOPA類似体へのこれらの胆汁酸の接合部位は、好ましくは3−ヒドロキシ基またはC−24カルボキシル部分を介してである。任意選択で、薬物と胆汁酸の間に開裂リンカー官能基を導入し、かかるリンカーが選択されてもよい。好ましい実施形態では、そのようなL−DOPA胆汁酸抱合体は以下の式またはその医薬として許容される塩により表わされる:
【0032】
【化3】
式中、R1は水素およびOHからなるグループから選択され;
R2は水素およびOHからなるグループから選択され;
XはOHおよびD−Y−からなるグループから選択され、YはDをステロイドに共有結合させる共有結合および開裂リンカー基からなるグループから選択され;
DはL−DOPAおよび他のL−DOPA類似体からなるグループから選択されたメンバーであり;
Wは以下の(a)および(b)からなるグループから選択され、
(a)生理的pHで負に帯電する部分(かかる部分は−COOH、−SO3H、−−S
O2H、−−P(O)(OR6)(OH)、−−OP(O)(OR6)(OH)、−−O
SO3H等からなるグループから選択される)を含む置換アルキル基およびその医薬とし
て許容される塩類、R6はアルキル、置換アルキル、アリール、および置換アリールからなるグループから選択される;および
(b)式−M−Y’−D’の基、式中Mは−CH2OC(O)−−および−CH2CH2
C(O)−−からなるグループから選択され;Y’はD’をMに共有結合させる共有結合または開裂リンカー基であり、;D’はL−DOPAおよび他のL−DOPA類似体からなるグループから選択されたメンバーである;
Xが−Y−Dであるか、Wが−M−Y’−D’であるかの少なくとも一方を満たし、
式(I)の化合物は腸胆汁酸輸送体の基質である。
【0033】
別の実施形態では、L−DOPA類似体はジまたはトリペプチド誘導体を含む。L−DOPAジペプチドまたはトリペプチド類似体の例およびそれらを製造する方法は、米国特許第3803120号および米国特許第5686423号に開示されている。ジペプチドおよびトリペプチドL−DOPAプロドラッグの経口吸収は高い経口生物学的利用能を示し、いくつかの化合物はL−DOPAの60−100倍の血漿濃度を有する。好ましい実施形態では、かかるL−DOPAプロドラッグは以下の式により表わされる:
【0034】
【化4】
式中、nは0または1であり;
Rは水素またはヒドロキシル、好ましくはRはヒドロキシルであり;
R1は水素であり;
R2は水素、1−4個の炭素原子を有するアルキル、1つの−−OH、−−SH、−−SCH3、−−NH2、−−NHC(=NH)NH2、−−COOH、フェニル、ヒドロキシ
フェニル、インドリルまたはイミダゾリル基で置換された1−4個の炭素原子を有するアルキル、1−6個の炭素原子を有する1つのカルボアルコキシル基で置換された1−4個の炭素原子を有するアルキル、好ましくはR2は水素、メチルまたはヒドロキシメチルであり;または
R1およびR2はともにトリメチレンである。
【0035】
好ましくは、式(I)のL−DOPA(2−アミノ−3−(3,4−ジヒドロキシフェニル−)プロパン酸)のジまたはトリペプチド誘導体は共にトリメチレンである。
別の実施形態では、式(I)のL−DOPA[2−アミノ−3−(3,4−ジヒドロキシフェニル−)プロパン酸]のジペプチド誘導体は、以下の式により表される:
【0036】
【化5】
式中、R3は水素であり;R4はフェニルまたはヒドロキシフェニルであり;またはR3およびR4はともにトリメチレンである。
【0037】
別の実施形態では、L−DOPA類似体は当該技術分野で周知のアミンプロドラッグを含む。L−DOPAアミン類似体の例およびそれらを製造する方法は米国特許出願公開第20060025385号およびWO/2004/069146に開示されている。1つの好ましい実施形態では、かかるL−DOPAアミン類似体は以下の式またはその医薬として許容される塩により表わされる:
【0038】
【化6】
式中、*Cは不斉炭素を示し;
R1、R2、R3およびR4は各々独立して、水素、1−30個の炭素原子を有するアルキル、1−30個の炭素原子を有するアルケニル、1−30個の炭素原子を有するアルキニル、シクロアルキル、アリール、O−カルボキシ、C−カルボキシ、カルボニル、チオカルボニル、O−カルバミル、O−チオカルバミルおよび脂肪酸アシルから成るグループから選択されるか、または
他の選択肢としてR1およびR2と、R3およびR4との少なくとも一方が五員環または六員環を形成し;
R5およびR6は各々独立して、水素、アルキル、シクロアルキル、アリールおよびホスホニルから成るグループから選択される。
【0039】
好ましいL−DOPAアミン類似体には以下のものが含まれる:R5およびR6が各々水素である化合物;R1およびR2が各々水素である化合物;R3およびR4が各々水素である化合物;R1、R2、R3およびR4の少なくとも1つ、好ましくはR3および/
またはR4がカルボニル(例えばアセチル)である化合物。現在の実施形態によるさらなる好ましい化合物は、R1、R2、R3およびR4の少なくとも1つが1−30個の炭素原子を有するアルキル、アルケニル、またはアルキニルである化合物、または他の選択肢として、R1、R2、R3およびR4の少なくとも1つが脂肪酸アシル、例えばミリスチン酸、ラウリン酸、パルミチン酸、ステアリン酸、オレイン酸、アラキドン酸、リノール酸またはリノレン酸に由来する脂肪酸アシルである。現在の実施形態によるL−DOPAアミン類似体のさらに好ましい例はα−アミノ−3,4−ジヒドロキシ−ベンゼンプロパンアミド、α−N−アセチル−3,4−ジヒドロキシ−ベンゼンプロパンアミドおよびその医薬として許容される塩類を含む。
【0040】
さらに好ましい実施形態では、本発明で使用されるL−DOPAプロドラッグおよびそれらの合成方法は、米国特許第4065566号および第4035507号に開示され、以下の式により表わされる:
【0041】
【化7】
各Rは独立して、水素原子、アシル基、
【0042】
【化8】
基、−−COピリジン基、−−CO−−R3基(R3はN,N−C1−C2ジアルキルアミノ酸またはC4−C6シクロアルキルアミノ酸の残基を表わす)から選択され、
【0043】
【化9】
R1は水酸基および−−OM基から成るグループから選択されたメンバーであり、−−Mはアルカリ金属(Na、K等)またはアンモニウムイオンであり
;
R2は
【0044】
【化10】
基、−−CO−ピリジン基、および−−CO−−R3基から選択されたメンバーであり、R3はN,N−−(C1−C2)ジアルキルアミノ酸またはC4 −C6−シクロアルキルアミノ酸
【0045】
【化11】
の残基を示す。
【0046】
本発明で使用されるさらなるL−DOPAプロドラッグおよびそれらの合成方法は米国特許第4065566号および第4035507号に開示され、以下の式およびそのHX塩(Xは以前の医薬として許容される酸付加塩陰イオン、例えば塩化物、臭化物、過塩素酸塩、メタンスルフォナート、コハク酸塩等である)により表わされる:
【0047】
【化12】
式中、Rはアシル基を表わし;
R2は水素原子を表わし;
R1は−NHCH(R4)COOR5基を表し、R4は任意の天然アミノ酸の残基を表わし、R5は水素原子、C1−C5アルキル基(例えばメチル、エチル、プロピル、ブチル、ペンチル)およびC1−C5アルキルアリール基(例えば−−CH2、−C6H5、−−
CH2−−CH2−C6H5等)から成るグループから選択されたメンバーである。
【0048】
米国特許第4065566号および第4035507号に開示された好ましいL−DOPAプロドラッグの例には、以下が含まれる:
1.グリシル−3,4−ジアセチルオキシ−L−フェニルアラニンおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
2.グリシル−3,4−ジアセチルオキシ−L−フェニルアラニン−メチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
3.3,4−ジアセチルオキシ−L−フェニルアラニル−グリシンおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
4.N−ニコチノイル−3,4−ジヒドロキシ−L−フェニルアラニンおよびそのM塩(Mはアルカリ金属を表わす)。
5.N−ニコチノイル−3,4−ジアセチルオキシ−L−フェニルアラニンおよびそのM塩(Mはアルカリ金属を表わす)
6.N−ニコチノイル−3,4−ジピバリルオキシ−L−フェニルアラニンおよびそのM塩(Mはアルカリ金属を表わす)。
7.3,4−ジアセチルオキシ−L−フェニルアラニル−グリシンおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
8.3,4−ジアセチルオキシ−L−フェニルアラニル−グリシン−メチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
9.3,4−ジアセチルオキシ−L−フェニルアラニル−グリシン−エチルエステル およびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
10.3,4−ジアセチルオキシ−L−フェニルアラニル−グリシン−ベンジルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
11.3,4−ジアセチルオキシ−L−フェニルアラニル−L−ロイシンおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
12.3,4−ジアセチルオキシ−L−フェニルアラニル−L−ロイシン−メチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
13.3,4−ジアセチルオキシ−L−フェニルアラニル−L−ロイシン−エチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
14.3,4−ジアセチルオキシ−L−フェニルアラニル−L−ロイシン−ベンジルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
15.3,4−ジアセチルオキシ−L−フェニルアラニル−L−イソロイシンおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
16.3,4−ジアセチルオキシ−L−フェニルアラニル−L−イソロイシン−メチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
17.3,4−ジアセチルオキシ−L−フェニルアラニル−L−イソロイシン−エチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
18.3,4−ジアセチルオキシ−L−フェニルアラニル−L−イソロイシン−ベンジルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
19.3,4−ジアセチルオキシ−L−フェニルアラニル−フェニルアラニンおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
20.3,4−ジアセチルオキシ−L−フェニルアラニル−フェニルアラニン−メチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
21.3,4−ジアセチルオキシ−L−フェニルアラニル−フェニルアラニン−エチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
22.3,4−ジアセチルオキシ−L−フェニルアラニル−フェニルアラニン−ベンジルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
23.グリシル−3,4−ジアセチルオキシ−L−フェニルアラニンおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
24.グリシル−3,4−ジピバリルオキシ−L−フェニルアラニンおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
25.グリシル−3,4−ジアセチルオキシ−L−フェニルアラニン−メチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
26.グリシル−3,4−ジアセチルオキシ−L−フェニルアラニン−エチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
27.グリシル−3,4−ジアセチルオキシ−L−フェニルアラニン−ベンジルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
28.L−ロイシル−3,4−ジアセチルオキシ−L−フェニルアラニンおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
29.L−ロイシル−3,4−ジアセチルオキシ−L−フェニルアラニン−メチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
30.L−ロイシル−3,4−ジアセチルオキシ−L−フェニルアラニン−エチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
31.L−ロイシル−3,4−ジアセチルオキシ−L−フェニルアラニン−ベンジルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
32.L−イソロイシル−3,4−ジアセチルオキシ−L−フェニルアラニンおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
33.L−イソロイシル−3,4−ジアセチルオキシ−L−フェニルアラニン−メチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
34.L−イソロイシル−3,4−ジアセチルオキシ−L−フェニルアラニン−エチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
35.L−イソロイシル−3,4−ジアセチルオキシ−L−フェニルアラニン−ベンジルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
36.フェニルアラニル−3,4−ジアセチルオキシ−L−フェニルアラニンおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
37.フェニルアラニル−3,4−ジアセチルオキシ−L−フェニルアラニン−メチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
38.フェニルアラニル−3,4−ジアセチルオキシ−L−フェニルアラニン−エチル およびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
39.フェニルアラニル−3,4−ジアセチルオキシ−L−フェニルアラニン−ベンジルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
40.3,4−ジアセチルオキシ−L−フェニルアラニル−3,4−ジアセチルオキシ−L−フェニルアラニンおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
41.3,4−ジアセチルオキシ−L−フェニルアラニル−3,4−ジアセチルオキシ−L−フェニルアラニン−メチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
42.3,4−ジアセチルオキシ−L−フェニルアラニル−3,4−ジアセチルオキシ−L−フェニルアラニン−エチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
43.3,4−ジアセチルオキシ−L−フェニルアラニル−3,4−ジアセチルオキシ−L−フェニルアラニン−ベンジルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
44.N−[N,N−ジメチルアミノ基]−グリシル−3,4−ジアセチルオキシ−L−フェニルアラニンおよびそのM塩(Mはアルカリ金属を表わす)。
45.N−ニコチノイル−3,4−ジニコチノイルオキシ−L−フェニルアラニンおよびそのM塩(Mはアルカリ金属を表わす)。
46.N−3−ピリジルアセチル−3,4−ジヒドロキシ−L−フェニルアラニンおよびそのM塩(Mはアルカリ金属を表わす)。
47.N−3−ピリジルアセチル−3,4−ジアセチルオキシ−L−フェニルアラニン およびそのM塩(Mはアルカリ金属を表わす)。
48.3,4−N,N−ジメチルアミノグリシル−L−フェニルアラニンメチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
49.N−[N,N−ジメチルアミノ基]グリシル−3,4−[N,N−ジメチルアミノグリシル]−L−フェニルアラニンおよびそのM塩(Mはアルカリ金属を表わす)。
50.N−[N,N−ジエチルアミノグリシル]−3,4−ジアセチルオキシ−L−フェニルアラニンおよびそのM塩(Mはアルカリ金属を表わす)。
【0049】
本明細書に使用する場合、用語「アルキル」は、直鎖および分岐鎖の基を有する脂肪族飽和炭化水素を指す。アルキル基は好ましくは1−30個の炭素原子、より好ましくは1−20個の炭素原子を有する。より低級のアルキル、例えば、1−6個の炭素原子のアル
キルは化合物の処方を容易にし、より高級のアルキルはBBB(脳血管関門)を通るその透過性の増強を提供する。
【0050】
アルキル基は、本発明によれば、置換されてもよいし、非置換でもよい。置換される場合、置換基は例えばシクロアルキル、アルケニル、アリール、ヘテロアリール、ヘテロアリサイクリック、ヒドロキシ、アルコキシ、アリールオキシ、チオヒドロキシ、チオアルコキシ、チオアリールオキシ、ハロ、カルボキシ、アルコキシカルボニル、チオカルボキシ、カルバミルおよびアミノであってよく、これらの用語については本明細書に定義した通りである。
【0051】
本明細書に使用する場合、用語「シクロアルキル」は、環の1つまたは複数が完全に共役したπ電子系を有していないすべてが炭素からなる単環または縮合環(つまり隣接した炭素原子対を共有する環)からなる基を指す。非制限的なシクロアルキル基の例には、シクロプロパン、シクロブタン、シクロペンタン、シクロペンテン、シクロヘキサン、シクロヘキサジエン、シクロヘプタン、シクロヘプタトリエンおよびアダマンタンである。シクロアルキル基は、本発明によれば、置換されてもよいし、非置換でもよい。置換される場合、置換基は例えばアルキル、シクロアルキル、アルケニル、アリール、ヘテロアリール、ヘテロアリサイクリック、ヒドロキシ、アルコキシ、アリールオキシ、チオヒドロキシ、チオアルコキシ、チオアリールオキシ、ハロ、カルボキシ、アルコキシカルボニル、チオカルボキシ、カルバミルおよびアミノであってよく、これらの用語については本明細書に定義した通りである。
【0052】
用語「アルケニル」は、少なくとも2つの炭素原子と少なくとも1つの炭素−炭素二重結合とから成るアルキル基を指す。
用語「アルキニル」は、少なくとも2つの炭素原子と少なくとも1つの炭素−炭素三重結合とから成るアルキル基を指す。
【0053】
上述したように、アルケニル基およびアルキニル基はいずれも好ましくは1−30個の炭素原子を有する。
「アリール」基は、完全に共役したπ電子系を有するすべてが炭素からなる単環式または縮合環の多環式(つまり隣接した炭素原子対を共有する環)からなる基を指す。非制限的なアリールの例は、フェニル、ナフタレニルおよびアントラセニルである。アリール基は、本発明によれば、置換されてもよいし、非置換でもよい。置換される場合、置換基は例えばアルキル、シクロアルキル、アルケニル、アリール、ヘテロアリール、ヘテロアリサイクリック、ヒドロキシ、アルコキシ、アリールオキシ、チオヒドロキシ、チオアルコキシ、チオアリールオキシ、ハロ、カルボキシ、アルコキシカルボニル、チオカルボキシ、カルバミルおよびアミノであってよく、これらの用語については本明細書に定義した通りである。
【0054】
用語「C−カルボキシ」は+C(=O)−OR’基 を指し、R’は、本明細書で定義
される、水素、アルキル、シクロアルキル、アルケニル、アリール、ヘテロアリール(環炭素を介して結合)またはヘテロアリサイクリック(環炭素を介して結合)である。
【0055】
用語「O−カルボキシ」はR’−−C(=O)−O−基を指し、R’は、本明細書で定義される、水素、アルキル、シクロアルキル、アルケニル、アリール、ヘテロアリール(環炭素を介して結合)またはヘテロアリサイクリック(環炭素を介して結合)である。
【0056】
用語「カルボニル」は−C(=O)−R’基を指し、R’は上記に定義した通りである。
用語「チオカルボニル」は−C(=S)−R’基を指し、R’は上記に定義した通りで
ある。
【0057】
「O−カルバミル」基は−OC(=O)−NR’R”基を指し、R’は上記に定義した通りであり、R”はR’に対して定義された通りである。
「O−チオカルバミル」基は−OC(=S)−NR’R”基を指し、R’およびR”は上記に定義した通りである。
【0058】
「脂肪酸アシル」は、R'''C(=O)−(O)−基を指し、R'''は少なくとも10個の炭素原子を有する飽和または不飽和の炭化水素鎖である。
用語「アルコキシ」は、−O−アルキルおよび−O−シクロアルキル基の両方を指し、上記に定義した通りである。アルコキシ基の代表的な例はメトキシ、エトキシ、プロポキシおよびtert−ブトキシを含む。
【0059】
−O−アルキルおよびO−シクロアルキル基は、本発明によれば、置換されてもよいし、非置換でもよい。置換される場合、置換基は、例えばシクロアルキル、アルケニル、アリール、ヘテロアリール、ヘテロアリサイクリック、ヒドロキシ、アルコキシ、アリールオキシ、チオヒドロキシ、チオアルコキシ、チオアリールオキシ、ハロ、カルボキシ、アルコキシカルボニル、チオカルボキシ、カルバミルおよびアミノであってよく、これらの用語については本明細書に定義した通りである。
【0060】
用語「チオアルコキシ」は−S−アルキル基および−S−シクロアルキル基の両方を指し、本明細書に定義した通りである。
用語「ヒドロキシ」は−OHグループを指す。
【0061】
用語「チオヒドロキシ」は−SH基を指す。
「アリールオキシ」基は−O−アリール基および−O−ヘテロアリール基の両方を指し、本明細書に定義した通りである。
【0062】
「チオアリールオキシ」基は−S−アリール基および−S−ヘテロアリール基の両方を指し、本明細書に定義した通りである。
用語「アミノ」は−NR’R”基を指し、R’およびR”は上記に定義した通りである。
【0063】
用語「アルコキシカルボニル」は、本明細書では互換的に「カルバルコキシ」とも呼ばれるが、上記に定義した通りカルボキシ基を指し、R’は水素ではない。
用語「ヘテロアリール」基は、環内に、例えば窒素、酸素および硫黄等の1つまたは複数の原子を有し、さらには完全に共役したπ電子系を有する単環または縮合環(つまり隣接した原子対を共有する環)を含む。ヘテロアリール基の非制限的な例には、ピロール、フラン、チオフェン、イミダゾール、オキサゾール、チアゾール、ピラゾール、ピリジン、ピリミジン、キノリン、イソキノリンおよびプリンが含まれる。
【0064】
「ヘテロアリサイクリック」基は、環内に、例えば窒素、酸素および硫黄等の1つまたは複数の原子を有する単環または縮合環を指す。環はさらに1つまたは複数の二重結合を有してもよいが、環は完全に共役したπ電子系を有しない。
【0065】
用語「ハロ」はフッ素、塩素、臭素またはヨウ素原子を指す。
用語「ホスホニル」は−P(=O)(OR’)2基を指し、R’は上記に定義した通り
である。
【0066】
本発明の第1または第2態様の任意の実施形態では、方法は、L−DOPAおよびL−
DOPA類似体からなるグループから選択された2つ以上の化合物を投与することを含んでもよい。別の好ましい実施形態では、方法はさらに、カルビドパまたはベンセラジド等のL−アミノ酸脱炭酸酵素阻害剤を含むがそれらに限定されるわけではないさらなる治療化合物を患者へ投与することを含んでもよい。かかるL−アミノ酸脱炭酸酵素阻害剤は、例えば、L−DOPAの血漿半減期を増大させ、かつL−DOPAからドーパミンへの末梢での変換を減少させるために使用することが可能であり、これはL−DOPA処理の副作用を低減する。別の実施形態では、方法は、AMDを治療またはAMDの進行を制限するのに有効な1つまたは複数の別の化合物を投与することをさらに含んでもよく、かかる化合物には、抗血管内皮細胞増殖因子(VEGF)剤等の抗血管新生治療薬、ラニビツマブまたはベバシツマブ等のVEGF抗体(またはその断片)、またはペガプタニブ等のVEGFアプタマーが含まれるが、それらに限定されるわけではない。別の実施形態では、L−DOPAまたはL−DOPA類似体は、L−DOPAまたはL−DOPA類似体を含有する栄養剤のような、より複雑な混合物中に存在してもよい。
【0067】
好ましい実施形態では、本明細書で説明している任意の1つまたは複数L−DOPAおよび/またはL−DOPA類似体を、栄養補助食品(ダイエタリーサプリメント)の形で使用してもよい。かかるサプリメントは、AMDの治療またはAMDの進行の制限に有益であり得る1つまたは複数のさらなる成分と組み合わせてもよい。1つの好ましい実施形態では、L−DOPAおよび/またはL−DOPA類似体は、ビタミンC源、ビタミンE源、ビタミンA源、亜鉛源、および銅源の組み合わせと組み合わされ、それらはAMDの治療に有用なものとして米国特許第6,660,297号に開示されており、この米国特許第6,660,297号全体は本明細書に援用する。任意の適切な量のかかる追加の成分の各々を、本発明の方法を実行する際にL−DOPAおよび/またはL−DOPA類似体と組み合わせて使用することが可能である。さらに好ましい実施形態では、かかる組み合わせはさらに、好ましくは1日当たり1mg−100mgの間;1mg−50mgの間、2mg−25mgの間、または2mg−10mgの間の、網膜保護効果を提供するのに適切な量のルテインおよび/またはゼアキサンシンをさらに含む。上記の好ましい実施形態のうちのいずれかのさらに好ましい実施形態では、かかる組み合わせはさらに、好ましくは1日当たり250mg−1000mgの間、300mg−750mgの間、350mg−750mgの間、または350mg−650mgの間の、網膜保護効果を提供するのに適切な量にドコサヘキサエン酸(DHA)および/またはエイコサペンタエン酸(EPA)をさらに含む。AMD患者を治療するかかる組成物の使用方法については、例えばウェブサイトwww.areds2.org/およびその中のリンクで検討されている。
【0068】
アスコルビン酸はビタミンCの好ましい供給源であるが、例えばアスコルビン酸ナトリウムのような他の供給源を代わりに使用してもよい。
D1−α酢酸トコフェロールはビタミンEの好ましい供給源であるが、例えばトリメチル酢酸トコフェリルおよび/またはコハク酸ビタミンEのような他の供給源を代わりに使用してもよい。
【0069】
β−カロチンは容易に商業的に入手できるため対象組成物に好ましいが、ビタミンAの代替カロテノイド代用形も同様に使用することが可能である。
亜鉛は、亜鉛元素を最も濃縮した形で供給し、消化器系によく許容されるため対象錠剤には酸化亜鉛の形が好まれるが、例えばグルコン酸亜鉛のような他の亜鉛の形式も代わりに使用でき、または対象組成物中の酸化亜鉛と組み合わせて使用してもよい。
【0070】
酸化第二銅の形をした銅は、亜鉛誘導性の銅欠乏貧血の予防を促すのに対象錠剤に好ましいが、例えばグルコン酸銅のような他の銅の形式も代わりに使用でき、または対象組成物中の酸化第二銅と組み合わせて使用してもよい。
【0071】
好ましい実施形態では、これらの他の成分の各々の量(1日当たりに基づく)は、以下の通りである:
450mg−600mgの間のビタミンC(一日摂取許容量(RDA)の約7−10倍);
400IU−540IUの間のビタミンE(RDAの約13−18倍);
17.2mg−28mgの間のβカロチン(ビタミンAのRDAの約6−10倍;βカロチンはビタミンAのプロドラッグである。);
68mg−100mgの間の亜鉛(亜鉛のRDAの約4−7倍);および
1.6mg−2.4mgの間の銅。
【0072】
さらに好ましい実施形態では、これらの他の成分の各々の量(1日当たりに基づく)は、以下の通りである:
500mgのビタミンC;
400IUビタミンE;
0mgまたは15mgのβカロチン;
25mgまたは80mgの酸化亜鉛;
2mgの酸化第二銅。
【0073】
本明細書の別の実施形態と組み合わせてもよいさらに好ましい実施形態では、目の健康を維持するのに有用であると考えられている他の成分を同様にL−DOPAおよび/またはL−DOPA類似体と組み合わせても良く、それには例えば、保護網膜効果をさらに提供するのに適切な量、好ましくは1日当たり1mg−100mgの間、1mg−50mgの間、2mg−25mgの間、または2mg−10mgの間のルテインおよび/またはゼアキサンシン;保護網膜効果をさらに提供するのに適切な量、好ましくは1日当たり250mg−1000mgの間、300mg−750mgの間、350mg−750mgの間、または350mg−650mgの間のドコサヘキサエン酸(DHA)および/またはエイコサペンタエン酸(EPA)が含まれるがそれらに限定されない。任意選択で使用可能なさらなる化合物の例には、α−リポ酸や、オリゴマーのアントシアニジン、アントシアノシドおよびその組み合わせを含むがそれらに限定されないフェノール化合物が含まれるが、それらに限定されない。
【0074】
L−DOPAおよび/またはL−DOPA類似体は、通常、医薬組成物の形で個別にまたは組み合わせて投与することが可能である。かかる組成物は医薬の分野で周知の方法で調製される。L−DOPAおよび/またはL−DOPA類似体は、唯一の活性薬物として投与してもよいし、または本発明の方法を実施するのに有用な1つまたは複数の別の化合物と組み合わせて使用してもよく、かかる別の化合物にはVEG−F等の抗血管治療薬や、カルビドパおよびベンセラジド等のL−アミノ酸脱炭酸酵素阻害剤が含まれるが、それらに限定されるわけではない。併用投与される場合、組み合わせは、同時または異なる時期に与えられる別々の組成物として処方してもよいし、単一の組成物として与えてもよい。
【0075】
L−DOPAおよび/またはL−DOPA類似体は、固体形式(顆粒剤、粉末剤または坐剤を含む)または液体形式(例えば溶液、懸濁剤または乳剤)に構成されてよい。L−DOPAおよび/またはL−DOPA類似体は種々の溶液に適用され、滅菌等の従来の医薬操作に曝されてもよく、および/または防腐剤、安定剤、湿潤剤、乳化剤、緩衝液等の以前の佐剤(アジュバント)を含んでもよい。
【0076】
L−DOPAおよび/またはL−DOPA類似体は、任意の適切な経路により投与されてよく、それには経口、局所(点眼薬および眼軟膏剤を含むが、それらに限定されない)、非経口、鼻腔内、肺、または直腸が含まれるが、それらに限定されるわけではなく、以
前の非毒性の医薬として許容される担体、佐剤および賦形剤を含む投薬単位製剤として投与される。本明細書に使用する場合、用語「非経口」は経皮、皮下、血管内(例えば静脈内)、筋肉内、または髄腔内注射または注入技術等を含む。さらに、本発明の化合物と医薬として許容される担体とを含む医薬製剤が提供される。L−DOPAおよび/またはL−DOPA類似体は、1つまたは複数の非毒性医薬として許容される担体、希釈剤および/または佐剤、ならびに所望の場合には別の有効成分と共に存在してよい、L−DOPAおよび/またはL−DOPA類似体を含む医薬組成物は、例えば錠剤、トローチ、菱形錠剤、水性または油性の懸濁剤、分散粉末剤または顆粒剤、乳剤(エマルション)、硬質または軟質カプセル剤、またはシロップ剤もしくはエリキシル剤として、経口使用に適した形式であってもよい。
【0077】
点眼薬は当該技術分野の任意の技術を使用して調製することが可能であり、それには塩化ナトリウムまたは濃グリセリン等の等張化剤、リン酸ナトリウムまたは酢酸ナトリウム等の緩衝液、ポリオキシエチレンソルビタンモノオレイン酸等の界面活性剤、ポリオキシル40ステアリン酸またはポリオキシエチレン硬化ヒマシ油、クエン酸ナトリウムまたはエデト酸ナトリウム等の安定化剤、塩化ベンザルコニウムまたはパラベン等の防腐剤の必要に応じた使用が含まれるが、それらに限定されるわけではない。点眼薬のpHは好ましくは4−8の範囲である。眼軟膏剤は、白色ワセリンまたは流動パラフィン等の一般に使用されている基剤と共に調製することが可能である。
【0078】
経口使用向けのL−DOPAおよび/またはL−DOPA類似体は、医薬組成物の製造ための当該技術分野の任意の周知方法により調製可能であり、かかる組成物は、口当たりのよい調製物を提供すべく甘味剤、香料、着色剤および防腐剤からグループから選択された1つまたは複数の薬物を含んでもよい。錠剤は、錠剤の製造に適した非毒性の医薬として許容される添加剤と組み合わせて、L−DOPAおよび/またはL−DOPA類似体を含む。これらの添加剤は、例えば炭酸カルシウム、炭酸ナトリウム、ラクトース、リン酸カルシウムまたはリン酸ナトリウムのような不活性希釈剤;例えばコーンスターチまたはアルギン酸等の顆粒および崩壊剤;例えばデンプン、ゼラチン、アラビアゴム等の結合剤、例えばステアリン酸マグネシウム、ステアリン酸、または滑石等の潤滑剤であってよい。錠剤はコーティングされなくてもよいし、既知の技術によりコーティングされてもよい。ある場合には、かかるコーティングが、胃腸管での崩壊および吸収を遅らせる既知の技術により調製され、それにより長期間にわたる持続作用を提供する。例えば、グリセリルモノステアリン酸またはグリセリルジステアリン酸のような時間遅延材料を使用することが可能である。
【0079】
経口使用のための製剤は、L−DOPAおよび/またはL−DOPA類似体が不活性の固体希釈剤(例えば炭酸カルシウム、リン酸カルシウム、カオリン)と混合された硬質カプセル剤として、または有効成分が水性または油性媒体(例えばピーナッツ油、流動パラフィン、オリーブ油)と混合された軟質ゼラチンカプセル剤として与えられてもよい。
【0080】
水性懸濁液は、水性懸濁液の製造に適した賦形剤と混合されたL−DOPAおよび/またはL−DOPA類似体を含む。かかる賦形剤は、懸濁化剤(例えばナトリウムカルボキシメチルセルロース、メチルセルロース、ヒドロプロピル−メチルセルロース、アルギン酸ナトリウム、ポリビニルピロリドン、トラガカントゴム、およびアラビアゴム)であり、分散剤または湿潤剤は天然のリン脂質(例えばレシチン)または脂肪酸を有するアルキレンオキシド縮合物(例えばポリオキシエチレンステアリン酸)、長鎖脂肪族アルコールを有するエチレンオキシド縮合物(例えばヘプタデカエチレンオキシセタノール)、または脂肪酸およびヘキシトールに由来する部分エステルを有するエチレンオキシド縮合物(例えばポリオキシエチレンソルビトールモノオレイン酸)、または脂肪酸およびヘキシトール無水物に由来する部分エステルを有するエチレンオキシド縮合物(例えばポリエチレ
ンソルビタンモノオレイン酸)であってよい。水性懸濁液は、1つまたは複数の防腐剤、(例えばエチルまたはn−プロピル p−オキシ安息香酸)、1つまたは複数の着色剤)、1つまたは複数の香料、および1つまたは複数の甘味剤(例えばスクロースまたはサッカリン)を含んでよい。
【0081】
油性懸濁液は、のL−DOPAおよび/またはL−DOPA類似体を、植物油(例えばピーナッツ油、オリーブ油、胡麻油、ココナツオイル)または鉱油(例えば流動パラフィン)に懸濁させることにより調製可能である。油性懸濁液は、増粘剤(例えば密蝋、固形パラフィン、またはセチルアルコール)を含んでもよい。口当たりのよい経口用製剤を与えるために、甘味剤と香料が添加されてもよい。これらの組成物は、アスコルビン酸等の抗酸化剤の添加により保存されてもよい。
【0082】
水の添加による水性懸濁液の調製に適した分散可能な粉末剤および顆粒剤は、分散もしくは湿潤剤、懸濁化剤、および1つまたは複数の防腐剤と混合して、有効成分を提供する。適切な分散もしくは湿潤剤または懸濁化剤は、既に上述したものにより例示されている。追加の添加剤(例えば甘味剤、香料、および着色剤)がさらに存在してもよい。
【0083】
本発明の方法に使用される医薬組成物は、水中油型エマルションの形をしていてもよい。油相は、植物油、鉱油、またはそれらの混合物であってよい。適切な乳化剤は天然ゴム(例えばアラビアゴムまたはトラガカントゴム)、天然のリン脂質(例えば、大豆、レシチン、および脂肪酸とヘキシトール、無水物とに由来するエステルまたは部分エステル、例えばソルビタンモノオレイン酸)、および該部分エステルのエチレンオキシドとの縮合物(例えばポリオキシエチレンソルビタンモノオレイン酸)であってよい。エマルションは甘味剤および香料をさらに含んでもよい。
【0084】
シロップ剤とエリキシル剤は、甘味剤(例えばグリセリン、プロピレングリコール、ソルビトール、グルコース、スクロース)と共に調製されてもよい。かかる調製物は解乳化剤、防腐剤、香料、および着色剤をさらに含んでもよい。医薬組成物は、無菌の注射可能な水性または油性の懸濁剤の形をしていてもよい。かかる懸濁剤は、上述した適切な分散または湿潤剤と懸濁剤とを使用して周知技術に従って製剤化されてよい。無菌の注射可能な調製物は。非毒性・非経口の許容される希釈剤または溶剤の形をした無菌注射剤溶液または懸濁剤であってもよい(例えば1,3−ブタンジオールの溶液として)。使用可能な許容される賦形剤および溶剤には、水、リンゲル液および生理食塩液がある。さらに、無菌の固定油が溶剤または懸濁剤として従来通りに使用される。この目的のためには、合成モノグリセリドまたはジグリセリドを含めた任意無味無臭の固定油が使用されてもよい。さらに、オレイン酸等の脂肪酸は注射物質の調製に用途がある。
【0085】
L−DOPAおよびL−DOPA類似体の鼻腔内投与のための特定方法が当該技術分野で周知であり;例えばKao et al., Pharmaceutical Research 17(8):978-984 (2000)を参照されたい。
【0086】
用量域は、化合物の選択、投与経路、製剤の性質、患者の症状の性質、および関与する医師の判断によって決まる。例えば、経口投与は、静脈内注射による投与よりも高用量が必要であると予想される。当該技術分野でよく理解されるように、かかる投薬レベルの違いは、最適化のための標準の経験的ルーチンを使用して調節することが可能である。ある実施形態では、L−DOPAおよび/またはL−DOPAS類似体は、10mg/日から1500mg/日の間の用量で投与することが可能であり、種々の好ましい実施形態では、投与が20mgと1200mg/日の間、50mgと1000mg/日の間、100mgと500mg/日の間、および200mgと400mg/日の間であってよい。
【0087】
本明細書で説明している化合物を含む医薬組成物は、それを必要とする個体に投与される。好ましい実施形態では患者は哺乳動物であり、より好ましい実施形態では患者はヒトである。治療の用途では、組成物は本発明の方法を実施するのに十分な量で投与される。これらの使用に有効な量は、化合物の性質(比放射能等)、投与経路、疾患の段階ならびに重篤度、患者の体重ならびに健康の全身状態、および規定する医師の判断を含むがこれらに限定されない要因によって決まる。活性化合物は広い用量域にわたって有効であるが、実際に投与される化合物の量は上記の関連する状況に照らして医師により決定されるであろうことが理解される。したがって、上記の用量域はいかようにも本発明の範囲を制限することを意図しない。
【0088】
第3態様では、本発明は以下の(a)および(b)を含む組成物を提供する:
(a)AMDの治療またはAMDの進行の制限に有効な量のL−DOPAまたはL−DOPA類似体;
(b)AMDの治療またはAMDの進行の制限に有効な量の、ビタミンC源、ビタミンE源、ビタミンA源、亜鉛源および銅源を含む組成物。
【0089】
組成物中のL−DOPAおよび/またはL−DOPAS類似体の量は、10mg/日から1500mg/日の間の用量での投与を与えるのに適しており、種々の好ましい実施形態では、投与は20mgと1200mg/日の間、50mgと1000mg/日の間、100mgと500mg/日の間、および200mgと400mg/日の間である。
【0090】
アスコルビン酸は対象錠剤中のビタミンCの好ましい供給源であるが、例えばアスコルビン酸ナトリウムのような他の供給源を代わりに使用してもよい。
D1−α酢酸トコフェロールはビタミンEの好ましい供給源であるが、例えばトリメチル酢酸トコフェリルおよび/またはコハク酸ビタミンEのような他の供給源を代わりに使用してもよい。
【0091】
β−カロチンは容易に商業的に入手できるため対象組成物に好ましいが、ビタミンAの代替カロテノイド代用形も同様に使用することが可能である。
亜鉛は、亜鉛元素を最も濃縮した形で供給し、消化器系によく許容されるため対象錠剤には酸化亜鉛の形が好まれるが、例えばグルコン酸亜鉛のような他の亜鉛の形式も代わりに使用でき、または対象組成物中の酸化亜鉛と組み合わせて使用してもよい。
【0092】
酸化第二銅の形をした銅は、亜鉛誘導性の銅欠乏貧血の予防を促すのに対象錠剤に好ましいが、例えばグルコン酸銅のような他の銅の形式も代わりに使用でき、または対象組成物中の酸化第二銅と組み合わせて使用してもよい。
【0093】
本発明のこの第3態様の1つの好ましい実施形態では、組成物(b)は以下の量の各成分の消化摂取を許容するのに適した処方を与える:
アスコルビン酸:少なくとも450mg;
dIαトコフェリル酢酸:400IU;
βカロチン:17.2mg;
酸化亜鉛:68mg;
酸化第二銅:1.6mg
本発明のこの第3態様の1つの好ましい実施形態では、組成物(b)は以下の量の各成分の摂取を許容するのに適した処方を与える:
500mgのビタミンC;
400 IUのビタミンE;
0mgまたは15mgのβカロチン;
25mgまたは80mgの酸化亜鉛;
2mgの酸化第二銅。
【0094】
上記に特定したような対象組成物の好ましい毎日用量が、上記に開示したような任意の適切な投与経路を介して、1回、2回、3回または4回以上の投薬形式で投与されてよい。好ましい実施形態では、投薬形式は本明細書で説明しているかかる投薬形式の任意の実施形態によれば、経口または局所の投薬形式である。別の好ましい実施形態では、対象組成物の毎日用量は、1日2回の1回分の投与形式、1日2回の2回分の投与形式、または1日合計4回分の投与形式で与えられる。1日1回の日用量合計をとるのと比較して、1回の用量当たりの1つまたは複数の投薬形式中、日用量合計の半分の1日2回投与は、主成分の吸収を改善すると共に、主成分の血中濃度をより良好に維持する。従って、対象組成物の好ましい製剤の1日に2回の投薬形式が毎日摂取される場合、各投薬形式は好まし
くは経口投与時に少なくとも約225mgのアスコルビン酸、約200IUのdl−αトコフェリル酢酸、約8.6mgのβ−カロチン、約34mgの酸化亜鉛および約0.8mgの酸化第二銅を提供するよう調剤される。対象組成物の好ましい製剤の4つの錠剤が毎日摂取される場合、各錠剤は好ましくは少なくとも約112.5mgのアスコルビン酸、約100IUのdl−αトコフェリル酢酸、約4.3mgのβ−カロチン、約17mgの酸化亜鉛、約0.4mgの酸化第二銅、および5mg−750mgの間のL−DOPAおよび/またはL−DOPA類似体を提供するように調剤される。
【0095】
別の好ましい実施形態では、組成物は以下を含む:
(a)5mgから1500mgの間のL−DOPAまたはL−DOPA類似体;
(b)450mgから600mgの間のビタミンC(一日摂取許容量(RDA)の約7−10倍);
(c)400IUから540IUの間のビタミンE(RDAの約13−18倍);
(d)17.2mgから28mgの間のβカロチン(ビタミンAのRDAの約6−10倍;βカロチンはビタミンAのプロドラッグである。);
(e)68mgから100mgの間の亜鉛(亜鉛用のRDAの約4−7倍);および
(f)少なくとも1.6mgの銅。
【0096】
種々の好ましい実施形態では、組成物は10mgから1200mgの間;25mgから1000mgの間;50mgから500mgの間;または100mgから400mgの間のL−DOPAまたはL−DOPA類似体を含んでもよい。
【0097】
本明細書の別の実施形態と組み合わせてもよいさらに好ましい実施形態では、目の健康を維持するのに有用であると考えられている他の成分を同様にL−DOPAおよび/またはL−DOPA類似体と組み合わせても良く、例えば、保護網膜効果をさらに提供するのに適切な量、好ましくは1日当たり1mg−100mgの間、1mg−50mgの間、2mg−25mgの間、または2mg−10mgの間のルテインおよび/またはゼアキサンシン;保護網膜効果をさらに提供するのに適切な量、好ましくは1日当たり250mg−1000mgの間、300mg−750mgの間、350mg−750mgの間、または350mg−650mgの間のドコサヘキサエン酸(DHA)および/またはエイコサペンタエン酸(EPA)が含まれるがそれらに限定されない。記載された量を提供する特定の投薬形式に必要な量は、本明細書の教示および1日当たりに投与される剤形の数基づいて当業者には決定することが可能である。
【0098】
第4の態様では、本発明は、AMDを治療する化合物を識別するin vitroの方法であって、細胞を試験化合物と接触させることからなり、細胞は、
(a)OA1を発現している第1の細胞集団;および任意選択で
(b)OA1を発現していない第2の細胞集団;を含み、
(c)以下の(i)および(ii)のうちの一方又は両方を増大させる試験化合物を陽
性試験化合物と識別し;
(i)(A)試験化合物と接触させない第1の細胞集団における色素上皮誘導因子(PEDF)発現および(B)第2の細胞集団におけるPEDF発現におけるPEDF発現のうちの一方又は両方に対する、第1の細胞集団におけるPEDF発現;
(ii)(A)試験化合物と接触させない第1の細胞集団における細胞内カルシウム濃度および(B)第2の細胞集団における細胞内カルシウム濃度のうちの一方又は両方に対する、第1の細胞集団における細胞内カルシウム濃度;
陽性試験化合物は、AMDの治療およびAMDの進行の制限のうちの少なくとも一方を行う候補化合物である方法を提供する。
【0099】
上述したように、ヒトOA1(配列番号1−2、NP000264.1)はGタンパク質共役受容体であり、本願発明者は本明細書でL−DOPAをOA1リガンドと確認した。より詳細に以下に開示するように、本願発明者はL−DOPAを通じてリンクされたOA1とチロシナーゼの間の自己分泌ループの存在を発見した。このループは、少なくとも1つの非常に効き目のある網膜神経栄養因子(PEDF)の分泌と、細胞内カルシウム濃度の増加とを含む。OA1は、その下流での作用が進行中の網膜の空間のパターニングを左右する選択的L−DOPA受容体である。したがって、OA1経路の刺激を介してPEDF発現および/または細胞内カルシウム濃度を選択的にアップレギュレートする試験化合物は、AMDの治療および/またはAMDの進行の制限を行うための候補化合物である。本発明のこの態様の方法は、以下のものを含むが、それらに限定されない任意のOA1同族体を用いて実施することが可能である:
マウス:配列番号3−4(NM_010951);
アフリカツメガエル(Xenopus tropicalis):配列番号5−6(NM_001011018);
雌ウシ:配列番号7−8(XM_001506318);
ラット:配列番号9−10(NM_001106958);
カモノハシ:配列番号11−12(XM_001506318);
アフリカツメガエル(Xenopus laevis):配列番号13−14 (NM_001096842)
ニワトリ:配列番号15−16(XM_416848);
ゼブラフィッシュ:配列番号17−18(NM_200822);
チンパンジー:配列番号19(XR_025625);
アカゲザル:配列番号21−22(XM_001090139);および
マカクザル:配列番号23(BV209253)。
【0100】
PEDFは色素上皮誘導因子であり(Exp Eye Res 53:411-414)、神経感覚網膜の発達を変更し、かつ血管増殖を阻害する可能性を備えた公知の神経栄養因子である。本発明のこの態様の方法は、以下のものを含むが、それらに限定されない任意のPEDF同族体を用いて実施することが可能である:
ヒト:配列番号25−26(NM_002615);
ラット:配列番号27−28(NM_031356);
キンカチョウ:配列番号29−30(XM_002197419);
ウマ:配列番号31−32(NM_001143954);
アフリカツメガエル(Xenopus tropicalis):配列番号33−34(NM_203755);
マウス:配列番号35−36(NM_011340);
大西洋サケ:配列番号37−38(NM_001140334);
ヒツジ:配列番号39−40(NM_001139447);
モルモット:配列番号41−42(EF679792);
雌ウシ:配列番号43−44(NM_174140);
イノシシ:配列番号45−46(NM_001078662);
カモノハシ:配列番号47−48(XM_001507128);
オオカミ:配列番号49−50(NM_001077588);
マカクザル:配列番号51−52(AB174277);
チンパンジー:配列番号53−54(XM_001154665);
アカゲザル:配列番号55−56(XM_001117361);および
ヒラメ:配列番号57−58(DQ115406)。
【0101】
細胞の第1集団および第2集団は任意の適切な真核細胞の種類であってよく、第1の細胞集団は細胞表面受容体タンパク質としてOA1を発現することが可能である。1つの好ましい実施形態では、細胞の第1集団および第2集団は、マウス、ラット、ハムスターまたはヒト細胞等の哺乳類由来の細胞である。特に細胞内カルシウム濃度の分析に関する実施形態に使用される場合、現在までに試験されているすべての真核細胞は、本発明の方法を実施するのに適していることが分かっている。細胞内カルシウム信号および/またはPEDF分泌のうちの一方又は両方に関連して本明細書に開示されたOA1シグナル伝達経路の保存に関して現在までに試験されている細胞の種類には、MCF7(乳癌上皮細胞)、COS細胞(腎臓繊維芽細胞)、MDCK細胞(腎臓上皮)、CHO(チャイニーズハムスター卵巣)、マウスRPEおよび3T3(マウス繊維芽細胞)、ならびに以下の実施例で開示されたものが含まれる。かかる細胞は種々の供給源(メリーランド州Walkersville所在のLifeLine Cell Technology社;ATCC(アメリカンタイプカルチャーコレクション)から入手可能であるか、または当該技術分野で周知かつ以下に説明する方法を用いて単離可能である。
【0102】
1実施形態では、細胞表面受容体タンパク質としてOA1を発現している第1の細胞集団の第1部分を試験化合物と接触させ、第1の細胞集団の第2部分は試験化合物と接触させず、第2部分と比較して第1部分におけるPEDFの発現および/または細胞内カルシウム濃度を増加させる化合物は、AMDの治療および/またはAMDの進行の制限を行う候補化合物である。
【0103】
代わりに、方法は、細胞表面受容体タンパク質としてOA1を発現しない第2の細胞集団の使用を含んでもよく、第2の細胞集団と比較して第1の細胞集団におけるPEDFの発現および/または細胞内カルシウム濃度を増加させる化合物は、AMDの治療および/またはAMDの進行の制限を行う候補化合物である。好ましい実施形態では、第1および第2の細胞集団は同じ細胞の種類であり、第1の細胞集団はOA1を発現するよう遺伝子組み換えされ、第2の細胞集団は遺伝子組み換えされない。この実施形態では、第2の細胞集団は第1の細胞集団と同様の発現ベクターでトランスフェクトされてよく、かかるトランスフェクションは、空の発現ベクター(つまりトランスフェクト細胞中のベクターから得られる発現タンパク質がない)または細胞膜へ適切に挿入されない切断型または変異型OA1を発現することが可能な発現ベクターでのトランスフェクションを含む。代わりに、細胞はOA1シグナル伝達に不活性であることが知られているOA1変異型をコードする発現ベクターまたは異なるGPCR経路(例えばcAMP)を介してシグナル伝達することが可能な遺伝子組み換えされたOA1をコードする発現ベクターでトランスフェクトすることも可能である。
【0104】
例えば、リガンド結合を変更せずにその活性を変更するために、OA1の7つの膜貫通領域を異なる細胞内c末端尾部と融合させることが可能である。
本明細書に使用する場合、「PEDF発現の増加」または「細胞内カルシウム濃度の増加」とは、第2の細胞集団で見られるよりも大きいアッセイの経過中の任意の第1の細胞集団における(または第1集団の第2部分に関する第1部分における)PEDF発現の増加または細胞内カルシウム濃度の増加である。この方法は、化合物が対照で見られるより
も大きいPEDF発現の増加または細胞内カルシウム濃度の増加を促進する限り、対照に対するPEDF発現の増加または細胞内カルシウム濃度の増加の特定量を要求しない。好ましい実施形態では、増加は標準的な統計学的測定により測定される統計学的に有意な増加である。
【0105】
細胞内カルシウム濃度の決定は当該技術分野で周知であり、Fura−2細胞ローディングおよびレシオメトリックイメージングを使用した方法の例が、以下の実施例に記載される。しかしながら、細胞内カルシウム濃度は当業者に周知のいかなる方法を使用して測定してもよく、それにはFu laTMI(以下を参照)およびFLIPerTMを用いたハイスループット法が含まれるが、それらに限定されるわけではない。
【0106】
細胞集団におけるPEDFの発現レベルの決定は、以下に説明するような当該技術分野の任意の技術を使用してもよく、それにはmRNAハイブリダイゼーション(ノーザンブロット、スロットブロット等)、適当なプライマー組を用いた逆転写ポリメラーゼ連鎖反応(PCR)技術、蛍光in situハイブリダイゼーション、およびPEDFを発現/分泌する馴化細胞培地での抗体検出(ウェスタンブロット、免疫細胞化学、ELISA)が含まれるが、これらに限定されない。PEDF抗体は市販されている(例えばマサチューセッツ州Cambridge所在のAbcam社)。タンパク質分析は(PEDFが発現タンパク質であるため)馴化細胞培地上で行われて良く、すべてのアッセイは細胞内PEDFタンパク質/mRNA生産で行うことが可能である。別の実施形態では、PEDFプロモータから検知信号(GFP、ルシフェラーゼ等)の発現を駆動する発現ベクターを有する組換え細胞を生成することが可能である。かかる細胞は、検知可能な蛍光強度の測定またはPEDFプロモータにより駆動される他の信号の測定を介して「PEDF発現」が測定される第1の細胞集団として使用することが可能である。
【0107】
この第4の態様に使用される場合、用語「接触」は、第1の細胞集団の細胞表面に発現されたOA1へのOA1リガンドの結合を促進するのに適した条件下でのin vitroの接触を意味する。本明細書に使用する場合、「接触」は培養の開始時に起こるか、または細胞集団の培養の開始後の任意の時間に起こる。PEDF発現および/または細胞内カルシウム濃度は、所与のアッセイのために適切に決定された試験化合物と接触した後にいつでも測定することが可能である。1実施形態では、経時変化を測定し、接触前に複数のレベルを測定し、接触後に種々の時間で測定する。種々の実施形態で、接触後のカルシウムシグナルの測定が5秒と60分の間でなされ;より好ましくは10秒と30分の間、10秒と10分の間、および10秒と5分の間、10秒と1分の間、および10秒と30秒の間になされる。種々の実施形態では、PEDF発現の測定は1分から72時間の間の範囲にわたり、EDF分泌の分析はPEDF mRNA発現、PEDFの細胞内タンパク質発現、またはPEDFプロモータにより駆動される検出信号の発現の分析よりも後の測定が必要とされる。
【0108】
所与のアッセイに対して、任意の適切な細胞培養条件が使用可能である。1つの好ましい実施形態では、細胞における内因性L−DOPAの生産を低減させ、かつ細胞表面に存在するOA1の量を維持するために(リガンド結合の際にOA1がエンドソームに内在化するため)、接触が非常に低濃度のチロシン(0.1μmと10μmの間のチロシン)を有する細胞培地またはチロシンがない細胞培養培地で起こる。1つの好ましい実施形態では、細胞表面でのOA1発現および局在化を最大限にするために、細胞は、試験化合物との接触前には低チロシン培地で培養され、次に試験化合物と接触させるためにチロシンを含まない培地に播かれる。別の好ましい実施形態では、接触が低チロシン培地で起こる。上記に開示された別の実施形態と組み合わせてもよい別の好ましい実施形態では、培地は、チロシンからのL−DOPAの細胞内生産を制限する、フェニルチオ尿素を含むがこれに限定されないチロシン阻害剤を含む。色素性細胞を使用する場合にこの実施形態は特に
好ましい。
【0109】
別の好ましい実施形態では、方法は、OA1への結合の競争相手として、L−DOPA、チロシンおよびドーパミンのうちの1つまたは複数の使用をさらに含んでもよい。この実施形態は、試験化合物をOA1リガンドであると識別した後に実行されてもよいし、またはOA1に結合する試験化合物の初期スクリーニングで実行されてもよい。以下の実施例で示されるように、1mM以上の濃度では、チロシンとドーパミンはOA1に対する結合に関しL−DOPAと競争し得る。したがって、1mMから100mMの間、好ましくは1 mMと50mMの間、または1mMと25mMの間の濃度のチロシンおよび/またはドーパミンを使用した競争アッセイを使用して、試験化合物がOA1経路経由で作用していることをさらに確認できると共に、L−DOPAの置換と比較したチロシンおよびドーパミンがOA1に結合する陽性試験化合物の置換能力を測定することが可能である。同様に、(試験されている試験化合物と同様なモル濃度の)L−DOPAと比較した競争的結合は、L−DOPAと比較してOA1への結合力が増加した化合物を識別するのを支援することが可能である。
【0110】
任意の適当な試験化合物を、小分子、ポリペプチドおよび核酸を含む本発明の第4および第5の態様の方法(以下を参照)を使用して評価することが可能である。試験化合物がポリペプチド配列を含む場合、かかるポリペプチドは化学合成されてもよいし、組み換え発現されてもよい。上記に開示したように、組換え発現は当該技術分野の標準的方法を使用して行うことが可能である。かかる発現ベクターには細菌またはウイルスの発現ベクターが含まれてよく、かかる宿主細胞は原核生物であってもよいし、真核生物であってもよい。固相法、液相法またはペプチド縮合法、またはそれらの任意の組み合わせの周知技術を使用して調製される合成ポリペプチドには、天然または非天然のアミノ酸が含まれ得る。ペプチド合成に使用されるアミノ酸は、標準的な脱保護、中和、カップリング、および洗浄プロトコルによる標準Boc(Nα−アミノ保護されたNα−t−ブチルオキシカルボニル)アミノ酸樹脂または標準の塩基性条件下で分解しやすいNα−アミノ保護9−フルオレニルメトキシカルボニル(Fmoc)アミノ酸であってよい。FmocおよびBoc Nα−アミノ保護アミノ酸は、いずれもSigma社、Cambridge Research Biochemical
社、または当業者に周知の他の化学企業から入手することが可能である。さらに、ポリペプチドは当該技術分野で周知の他のNα−保護基で合成することも可能である。固相ペプチド合成は当該技術分野の周知技術により達成され、自動シンセサイザの使用等により与えられる。
【0111】
試験化合物が抗体を含む場合、かかる抗体はポリクローナル抗体であってもよいし、またはモノクローナル抗体であってもよい。抗体は、ヒト化抗体であってもよいし、完全ヒト抗体であってもよいし、またはマウス型の抗体でもよい。かかる抗体は例えばHarlow and Lane, Antibodies; A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y., (1988).に記載されているような周知方法により作製することが可能である。
【0112】
試験化合物が核酸配列を含む場合、かかる核酸は化学合成されてもよいし、組み換え発現されてもよい。組換え発現技術は、当該技術分野で周知である(例えばSambrookら、1989、前掲参照)。核酸はDNAまたはRNAであってもよいし、一本鎖または二本鎖であってもよい。同様に、かかる核酸は、当該技術分野の標準的方法を用いて手動または自動反応により化学的または酵素的に合成することが可能である。化学的に合成されるか、またはin vitroの酵素合成により合成される場合、核酸は細胞へ導入される前に精製され得る。例えば、核酸は、溶媒または樹脂での抽出、沈殿、電気泳動、クロマトグラフィまたはその組み合わせにより、混合物から精製することが可能である。代わりに、核酸は、サンプル処理による損失を回避するため、精製を最低限にして使用してもよい。
【0113】
試験化合物がポリペプチド、抗体または核酸以外の化合物を含む場合、かかる化合物は有機化学合成を行なうための当該技術分野の種々の方法により製造することが可能である。
【0114】
第2の細胞集団に比べて第1の細胞集団におけるPEDFおよび/または細胞内カルシウム濃度の発現を増加させると識別された試験化合物は、以下に説明する本発明の第4の態様のインビボ法を含むがこれに限定されないさらなる任意の方法を使用して、AMDを治療またはAMDの進行を制限する候補化合物として使用するようさらに評価することが可能である。1つの好ましい実施形態では、方法はさらに、上述したように競争量のチロシンおよび/またはドーパミンの存在下でアッセイ中の陽性試験化合物を再試験することを含む。
【0115】
第5の態様では、本発明は、AMDを治療する化合物を識別する方法であって、
(a)チロシナーゼ欠損の妊娠している雌の非ヒト哺乳動物に試験化合物を投与する工程であって、試験化合物は胚光受容体および/または網膜神経節の発達中に投与される工程;および
(b)胚または出生後の非ヒト哺乳動物における光受容体および/または網膜神経節発達に対する試験化合物の効果を、試験化合物を投与していない胚または出生後の非ヒト哺乳動物における光受容体および/または網膜神経節の発達と比較する工程であって、光受容体および/または網膜神経節の発達を増大させる試験化合物は、AMDを治療および/またはAMDの進行を制限する候補化合物である工程;
からなる方法を提供する。
【0116】
本願発明者は、チロシナーゼ欠損動物における光受容体および神経節細胞の発達を助けるためにOA1シグナル伝達を使用できることを決定し、プロセス中にOA1シグナル伝達の神経組織栄養効果を確立した。したがって、OA1シグナル伝達を通じて神経網膜の発達を助ける化合物は、AMD治療の良い候補である。本発明は、AMD薬物スクリーニングのためのかかる動物モデルを第1に確立する。
【0117】
本明細書により詳細に記載するように、チロシナーゼはチロシンに作用してL−DOPAを生成する。したがって、チロシナーゼ欠損哺乳動物は、L−DOPAを生産せず、かかる哺乳動物を使用すると内因性L−DOPAがない状態で(網膜の進行および/またはPEDF発現の増加の救助を介して)OA1のアクチベータを識別することが可能となる。本明細書に使用する場合、「チロシナーゼ欠損」とは、妊娠している雌の非ヒト哺乳動物が正常な色素形成に適した量のL−DOPAを生成するのに適当な量のチロシナーゼを生産しないことを意味する。1つの好ましい実施形態では、妊娠している非ヒト哺乳動物は、機能的なチロシナーゼを発現またはやりとりする能力のないノックアウト動物(チロシナーゼ遺伝子の部分または全部が削除されているか、またはチロシナーゼ遺伝子またはチロシナーゼを制御、活性化するかもしくはメラノソームへとチロシナーゼをやりとりする付属遺伝子に自然突然変異を有する)である。かかるチロシナーゼノックアウトは当該技術分野で周知であり、市販で入手可能である(Lexicon Pharmaceuticals社、Jackson Laboratories社、Taconic Farms社)。別の実施形態では、チロシナーゼ欠乏は、チロシナーゼを標的とするsiRNAsの投与や、チロシナーゼ抗体/アプタマーによる治療等を含むがこれらに限定されない当該技術分野の周知方法により一過性に誘発されてもよい。
【0118】
非ヒト哺乳動物は、チロシナーゼ欠損(網膜アルビノ)の雌を得ることが可能であればいかなるものでもよく、すべての哺乳動物を含む。種々の好ましい実施形態では、非ヒト哺乳動物はマウス、ブタ、サルおよびラットである。
【0119】
1つの好ましい実施形態では、試験化合物の投与は、光受容体および/または網膜神経節の発達の出生後期間の間じゅう継続される。種々の非ヒト哺乳動物における胚および出生後の光受容体および/または網膜神経節の発達経路は、当業者によく理解されている。1つの例証的実施形態では、マウスの胚光受容体と網膜神経節の発達は胚の10日目(E10)に始まり、網膜の進行は、生後の14日目(P14)で完了し、この時には子マウスの目が開いている。したがって、種々の実施形態では、試験化合物は、まずE7、E8、E9またはE10の日に投与され(視覚の発達の最も初期の段階での存在を促進するため)、P1、P2、P3、P4、P5、P6、P7、P8、P9、P10、P11、P12、P13およびP14の日の間または所望に応じてその後に、所与のアッセイに所望される通りに投与を継続することが可能である(出生後1年以内)。当業者によって理解されるように、投与は、胚の時期に妊娠している雌の母体に行うか、出生後は子に行うことになる。別の実施形態では、色素細胞の発達は約日10.5E(OA1とチロシナーゼが現われる時期)に本格的に開始し、したがって1実施形態では、試験化合物の投与は約E10、E10.5またはE11日目に開始し、所望に応じてP1、P2、P3、P4、P5、P6、P7、P8、P9、P10、P11、P12、P13、P14日目または所望に応じてその後まで継続される。別の実施形態では、試験化合物の投与はE7と、E10またはE11との間に限定されてもよい。さらなる実施形態では、網膜神経節の発達は約E12日目に本格的に開始し、したがって1実施形態では、試験化合物の投与は約E12またはE13日に開始し、所望に応じてP1、P2、P3、P4、P5、P6、P7、P8、P9、P10、P11、P12、P13、P14日目または所望に応じてその後まで継続される。別の実施形態では、試験化合物の投与は約E7と、E12またはE13との間に限定されてもよい。ほとんどの好ましい実施形態では、試験化合物は、E7日からE14日まで毎日投与される。当業者に理解されるように、試験化合物の投与の正確なタイミングは、特定のアッセイの目的によって決まり、本明細書での教示に基づいて当業者が決定することが可能である。
【0120】
試験化合物は、L−DOPAまたはL−DOPA類似体の治療用投与のために上記に開示した投与経路を含む、実験動物への使用に適した任意の経路により投与されてよい。好ましい実施形態では、試験化合物は、動物の飲料水に入れて、非経口(上述)または局所(例えば点眼薬または眼軟膏剤で)敵に投与される。試験化合物の投与回数は、所与のアッセイに適した頻度としてよい。好ましい実施形態では、試験化合物は、治療の所望経過じゅう毎日投与される。他の実施形態では、投与は治療経過の間、1日、3日、4日、または5日おきである。投与回数は本明細書の教示および所与のアッセイの特定の目的に基づいて当業者が決定することが可能である。
【0121】
本明細書に使用する場合、「光受容体および/または網膜神経節の発達の増加」は、試験化合物で処理した胚/動物対未処理の胚/動物における光受容体および/または網膜神経節の発達の任意の増加である。この方法は、化合物が対照で見られるよりも大きな光受容体および/または網膜神経節の発達の増加を促進する限り、対照に対する光受容体および/または網膜神経節の発達の特定量の増加を要求しない。好ましい実施形態では、増加は標準的な統計学的測定で測定される統計学的に有意な増加である。1実施形態では、動物は適切な時点で安楽死させられ、網膜神経節細胞および/または光受容体は以下の実施例に開示されたものを含むがそれらに限定されない当該技術分野の標準的方法を使用して計数される。
【0122】
光受容体および/または網膜神経節の発達を増加させるとして識別された試験化合物を、AMDを治療またはAMDの発達を制限する候補化合物として使用するためにさらに評価することが可能であり、それには競争量のチロシンおよび/またはドーパミンの存在下で本発明の第3態様で開示したin vitroの方法を使用して陽性試験化合物を再試験することが含まれるが、それらに限定されるわけではない。以下の実施例に示すように
、1mM以上の濃度で、チロシンおよびドーパミンはOA1への結合に関しL−DOPAと競争することが可能である。したがって、1mMから100mMの間、好ましくは、1mMから50mMの間または1mMから25mMの間の濃度のチロシンおよび/またはドーパミンを使用した競争アッセイを使用して、試験化合物がOA1経路を介して作用していることをさらに確認し、かつすることが可能である、経由で作動している、またチロシンおよびドーパミンがL−DOPAの置換と比較してOA1へ結合する陽性試験化合物と置換する能力を測定することが可能である。
【0123】
実施例:L−DOPAはOA1の内因性リガンドである
(背景):白皮症は色素沈着の損失を特徴とする遺伝子異常である。神経感覚網膜は、色素が沈着せず、網膜色素上皮(RPE)の色素沈着の喪失の次に病理学的変化を示す。RPEの色素沈着の喪失がどのようにして隣接する神経感覚網膜の発達欠陥を引き起こすかはまだ決定されていないが、これらの重要な2つの組織間の相互作用を研究する固有の機会を与えている。白皮症を引き起こす遺伝子の一方は、RPEを含む色素沈着細胞にのみ発現されるオーファンGPCR(OA1)をコードしている。
【0124】
方法/原理的発見:OA1の機能およびシグナル電圧を、RPEおよびトランスフェクト細胞株で調べた。結果は、OA1が選択的L−DOPA受容体であり、2つの密接に関連する化合物であるチロシンおよびドーパミンからの測定可能なセカンドメッセンジャー活性はないことを示している。放射性標識リガンド結合により、OA1がL−DOPAの一つの飽和結合部位を示すことが確認された。ドーパミンは、その一つのOA1結合部位に関しL−DOPAと競争したが、これはドーパミンがOA1アンタゴニストとして機能し得ることを示唆している。L−DOPAに対するOA1応答を、細胞内カルシウムの流入およびβ−アレスチンの動員を含むいくつかの一般的なGPCR活性化の測定基準により定義した。さらに、L−DOPAを生産する酵素であるチロシナーゼの抑制により、RPEによるPEDF分泌が減少した。さらに、L−DOPAによるRPEでのOA1の刺激により、PEDF分泌が増大した。
【0125】
結論/有意性:まとめると、結果は、L−DOPAを介してリンクされたOA1とチロシナーゼとの間の自己分泌ループを示し、このループは少なくとも1つの非常に効き目のある網膜神経栄養因子の分泌を含んでいる。OA1は、その下流での作用が発達中の網膜の空間パターン形成を左右する選択的L−DOPA受容体である。結果は、メラニン合成機構の変化により引き起こされる網膜の白皮症の結果がL−DOPAの補給により治療され得ることを示唆している。
【0126】
導入:白皮症は、目、髪または皮膚の色素沈着の可変的な喪失がある、遺伝的遺伝子疾患のグループである。目が罹患した場合、弱視につながる神経感覚網膜発達に著しい変化がある[1−8]。白皮症には2つの大きな分類である眼皮膚白皮症(OCA)および眼白皮症(OA)がある。すべての色素化組織が色素沈着減少を示した場合にOCAが生じ、メラニン合成機構の欠陥を生じさせる遺伝子突然変異を伴う[3および7−9]。OAは皮膚組織に通常に色素がついている場合に生じるが、眼組織の色素沈着が減少している[10および11]。すべての組織で同じタンパク質は色素を生産するため、他の組織も通常着色するため、OAは、メラニンを合成する能力がないわけではなく、眼組織中のメラニン生成酵素の発現が不足していることに起因する可能性が高い。
【0127】
OAは、X染色体上に見出される少なくとも1つの遺伝子Oa1に結合され得る。Oa1は、配列決定分析に基づくと、オーファンGタンパク質共役受容体(GPCR)(Genbank GPR143)[12および13]であると考えられる404個のアミノ酸からなるタンパク質をコードする[14]。SchiaffinoらはOA1がGβのみならずいくつかのGαサブユニットと結合していることを実証しており、これはOA1がGPCRで
あるというさらなる証拠を加えている[14および15]。実際、Innamoratiらは、OA1由来のGPCR様活性とβ−アレスチン結合を示すために、組み合わせ発現法を使用した[16]。この研究は、OA1がホスホリパーゼCとイノシトール三リン酸のセカンドメッセンジャーによってGαqサブユニットを介してシグナルを送り得ることを示唆した。酵素発現系で、StalevaとOrlowは、メラノソーム区画中の成分により活性化されていると考えられるOA1からのGPCRシグナル伝達を実証した[17]。OA1がGPCRであるというかなりの量の情況証拠にもかかわらず、リガンドが同定されていないため、確認がなされていない。OA1がGPCRであるという考えに、他のデータは疑問を投げかけている。例えば、完全な細胞内タンパク質としてのOA1の局在性は、GPCRに一般的なことではなく、これはOA1がGPCRファミリーの固有のメンバーであろうことを示唆している[14]。OA1は細胞表面ではなくリソソーム内区画[14、15、18−21]およびメラノソーム[11、14、22]に主に局在化している。
【0128】
本研究では、OA1のエンドソームの局在性が培養細胞中の薬物に応答したOA1の内在化によるとの仮説に基づき、潜在的GPCRとしてのOA1の機能を調べた。さらに、すべての型のOCAおよびOAが同じ網膜の発現型を有し、これはチロシナーゼ活性とOA1シグナル伝達が網膜発達の上流で連結されていることを示唆しているという知見に基づいて、OA1のリガンドを求めた。したがって、チロシナーゼ活性がOA1のリガンドを生産するか否かの試験を行なった。メラニン合成の副産物はL−DOPAであり、これは網膜の発達の重大な時間にRPEでのメラニン合成の間に網膜に放出される[23および24]。データは、OA1が非常に選択的なL−DOPA受容体であり、L−DOPAがRPEによる神経栄養因子分泌の下流での作用によりOA1のシグナル伝達を引き起こすことを示唆している。したがって、第1の証拠がOA1のリガンドに対して示され、チロシナーゼまたはOA1の欠乏のいずれかにより網膜の発達に変更を生させる機構を提供する。
【0129】
結果:
OA1の細胞表面局在性
OA1は以前はin situで色素顆粒中に局在化していたが[22]、種々のトランスフェクト細胞を使用すると、OA1は原形質膜[16および17]および培養細胞のエンドソーム部分[14、16−18、20、21]の両方にも局地化していた。このような研究は、細胞表面ビオチン化/ウェスタンブロット法を使用してOA1がヒト組織のどこに存在するかを決定することから始まった。ヒトの目では、OA1はin situでRPEの頂端細胞表面に存在した(図1A)。細胞表面の、5つのヒトの目のビオチン化OA1の定量化により、全OA1のうちの少なくとも3.5+/−0.7%がin situでRPEの頂端細胞表面に存在することが示された。眼杯調製物を使用したビオチン化試薬へのアクセスは頂端面に限定されており、上皮中のOA1の極性は決定できない。さらに、基底細胞表面へのアクセスが欠如しているため、合計細胞表面OA1が過小評価されている可能性がある。細胞質タンパク質がビオチン化されなかったことを確認するため、ブロットを対照としてアクチンに対する抗体でプローブした。各実験で、アクチンは未結合の部分でのみ見つかった。
【0130】
組換えOA1およびOA1−GFPが、培養細胞のエンドソーム区画のみにほとんど排他的に局地化されていると報告したものもあった[14,15,17,18,20−22]。しかしながら、過剰発現された場合[16]や、またはエンドサイトーシスを阻害した場合[17]、OA1は細胞表面に蓄積する。OA1タンパク質がin situでRPEの頂端面に存在するという知見は、この問題を我々にさらに検討するに至らしめた。
【0131】
OA1発現および分布に対するチロシンの影響
GPCRのエンドソーム局在性は、通常リガンドへの接触後に生じる。したがって、受
容体のリガンドが、OA1の内在化を起こさせ得る標準インキュベーション培地に存在するか否かを調べた。標準培地は500μMチロシンを含み、チロシンは色素合成の出発物質であるため、受容体分布に対するチロシンの影響を評価した。チロシンが培養細胞中のOA1分布に影響するか否かを調べるため、DMEMをチロシンを含まずに調合し、透析したウシ胎仔血清を使用した。チロシンを含まない培地の存在下で、チロシンを含まない(図示しない)培地と低濃度のチロシン(1μM、図1B)を含む培地の両方で、培養RPE細胞の原形質膜上でOA1が検知された。5つの実験を平均し、合計OA1タンパク質の4.5+/−1%が、1μMチロシンに維持された培養RPEの表面で観察され、これはin situのRPEで観察されたのと類似していた。すべての実験で、未結合タンパク質分画でアクチンが観察されたが、これは細胞表面アッセイに細胞質タンパク質が存在しないことを実証している。同様に、COSで発現されたOA1−GFPは、チロシン感受性の細胞表面発現を示した(図1C)。6つのかかる実験の定量化により、一過性トランスフェクションを使用して細胞表面に見出されるOA1の量の重要な変化が示された。1μMチロシンに維持されたトランスフェクト細胞の未結合分画のOA1の範囲は5−40%に及び、再現性があって約5%という内因性OA1タンパク質の結果とは異なる。
【0132】
トランスフェクト細胞のOA1の分布が培地中のチロシンレベルに感受性であるのみならず、合計OA1−GFPの発現も1μMで維持した細胞で5倍に増大した。この差が細胞数ではなくOA1発現量に関連することを確認するために、アクチン発現をペアになったサンプルから評価した。光学濃度単位として示したデータ(図1D)は、アクチンに差がないことを示している。正常群と低チロシン群との間の細胞表面OA1の量も比較した。重要なことは、5つのRPE実験および6つのCOS中のOA1−GFP実験で、標準培地中の細胞の原形質膜部分のOA1は、他の部分に見出されるのと同様に、再現可能に検出されなかった。
【0133】
RPE細胞におけるOA1の分布を、共焦顕微鏡法によっても評価した。OA1は、以前に示されたように培養RPE細胞のエンドソームタンパク質であると特徴付けられたが(図1E)、対照的に、低チロシン培地中のOA1の分布は培養RPE細胞の原形質膜上で拡散し、エンドソーム蓄積はほとんどなく(図1F)、この知見は生化学法を使用して得られた結果と一致していた。
【0134】
OA1の天然アゴニストとしてのL−DOPA
メラニン形成におけるチロシナーゼの機能は、L−DOPAを生成するようチロシンに作用することから始まり、これはその後ドーパキノンを作成する第2の反応を経て、色素形成に至る[25]。チロシンとメラニンの間の中間体のうち、L−DOPAは最も大きな半減期を有すると共に、メラニン合成が起こった場合にL−DOPAはRPEの頂端の網膜下の空間へ放出される[23および24]。L−DOPAは、チロシンからドーパ作動性ニューロンにより生産された神経伝達物質であるドーパミンの前駆体でもある。細胞内貯蔵物からのカルシウムの放出はリガンドによるGPCR活性化の一般的な下流の作用である。細胞表面でのOA1の発現はチロシン感受性であるようであるため、チロシン、またはその代謝産物であるL−DOPAおよびドーパミンが、OA1依存的に細胞質へのCa2+流入を刺激することが可能か否かを調べた。CHO細胞にOA1発現ベクターをトランスフェクトし、該CHO細胞を1μMチロシンを含mDMEMで48時間維持し、その後チロシンを含まないDMEMで24時間維持し、OA1の細胞表面発現を促進した。細胞内Ca2+をFura−2を使用して評価し、[Ca2+]iをレシオメトリックイメージングで決定した[26]。リガンドが存在しない状態では、トランスフェクト細胞と非トランスフェクト細胞との間で[Ca2+]iの有意差はなかった(図2)。チロシンおよびいくつかのチロシン代謝産物を、1μMの濃度で、[Ca2+]iに対する効果を試験した。陽性対照として、各実験を20mM KClでの処理により終了し、細胞を脱分極さ
せると共に電位型チャネルの活性化により[Ca2+]iを増加させた。この方法は、Fura−2装填および試験されている細胞の反応性を確認する役割を果たした(図2)。L−DOPAだけが[Ca2+]iの有意な増加を誘発した(図2A)。チロシンとドーパミンは1mM(図示しない)まで細胞内[Ca2+]i濃度に陽性の効果がなかった。1μMドーパミンのわずかに陰性の効果は、統計学的に有意ではなかったが、ドーパミンを用いた11の実験で再現可能であった(図2B)。
【0135】
非天然細胞株におけるGPCRの過剰発現は、偽のシグナル伝達の結合につながり得る。L−DOPAに応じたOA1シグナル伝達が確かに天然の応答であったことを確認するために、OA1をRPE細胞で発現させた(図2C)。トランスフェクトRPE細胞を使用した結果は、トランスフェクトCHO細胞で達成された結果に類似していた。OA1を発現するようトランスフェクトされたRPE細胞は、[Ca2+]iの増加に伴い1.0μM L−DOPAに応答した。次に、内因性OA1受容体を発現しているRPE細胞が内因性レベルでL−DOPA反応を示すか否かを決定した。すべてのトランスフェクト細胞実験のように、OA1を発現するRPEは、1.0μM L−DOPAでの処理後に[Ca2+]iの増加を示した(図2C)。
【0136】
OA1のシグナル伝達活性をさらに特徴づけるために、百日咳毒素を使用してGq共役[Ca2+]iシグナル伝達とGiリンクシグナル伝達(図2C)とを区別した。研究されたすべての細胞で、百日咳毒素は[Ca2+]iの基準レベルを低下させ、これはGiサブユニット活性によるバックグラウンドシグナル伝達の阻害に対するその活性を示した。CHOとRPEの両方を含むOA1を発現するようトランスフェクトされた細胞ならびに天然レベルで内因性OA1タンパク質を発現するRPEで行なわれた実験に、百日咳毒素を使用した。試験したすべてのトランスフェクト細胞で、L−DOPAに対して測定した[Ca2+]i応答は毒素がない場合よりも大きく(図2)、この大部分は最初の[Ca2+]iが低いことによる。したがって、[Ca2+]iの増加が生じるL−DOPAに応答したOA1を介したシグナル伝達は、百日咳毒素感受性ではなく、Gqサブユニット媒介性である可能性がある。OA1を発現するようトランスフェクトされたCHO細胞でのセカンドメッセンジャーcAMPも測定した(図2D)。不活性細胞または最大未満のホルスコリン処理を使用して、L−DOPAに応じたcAMPの増加または減少のいずれかを測定するように実験を設定した。6回のかかる実験では、cAMPの変化は観察されず、これはGsサブユニットもGiサブユニットもOA1シグナル伝達に関与しないことを示唆している。
【0137】
放射性標識リガンド結合の標準的方法を使用して、OA1とL−DOPAの間の相互作用を特徴付けた(図3A)。CHO細胞をOA1を発現するようトランスフェクトし、次に、L−DOPAの結合を濃度依存的に定量し、その結果をさらにScatchardプロット分
析で特徴付けた(図3E)。結果は、9.35×10-6のKdのOA1発現細胞へのL−DOPAの飽和結合を示す。非トランスフェクトCHO細胞では特異的結合は観察されず、これは細胞が内因性L−DOPA受容体(図示しない)を有していないことを示している。すべての結合パラメータ、すなわち合計結合、特異的結合、および非特異的結合を、補助データ(図6A)として示す。チロシンは、OA1と相互作用する可能性を示した。が、チロシンもドーパミンもOA1シグナル伝達を刺激しなかった(図2参照)。競争リガンド結合を使用して、チロシンまたはドーパミンのいずれかがOA1に結合するためにL−DOPAと競争したか否かを判断した。高濃度(1mM)では、チロシンとドーパミンはいずれもOA1との結合のためにL−DOPAと競争した(図3B)。これをさらに特徴付けるために、L−DOPAとドーパミン(図3C)またはチロシン(図6B)のうちの一方との間の競争の動力学を調べた。ドーパミンは、2.33×10-6+/−0.2×10-6MのKiのL−DOPAとの単一部位への競争的結合を示した。チロシンによる同様の実験は、高濃度(図6B)でのみL−DOPA結合の阻害を実証した。チロシンは
高濃度でも親和性が低くおよび不溶性であるためにチロシンに関する飽和速度は不可能であった。
【0138】
L−DOPAに対するOA1の親和性が比較的低いことから、そのシグナル伝達活動がこの結合親和性の範囲で用量依存的であるかどうかを決定した。結合データをL−DOPAとOA1との関連で最も急激な上昇を示唆する濃度である1.0−10μMで試験し、結果を[Ca2+]iにより測定された濃度依存的GPCR応答を示す(図3C)。このように、L−DOPAおよびOA1の活性化速度は、放射性標識リガンド結合実験で観察された濃度範囲と一致した。
【0139】
リガンド結合に応じて、GPCRは、β−アレスチンを原形質膜へ動員し、これに続きリガンド受容体複合体が内在化した[27−33]。その後、β−アレスチン局在化に対するL−DOPAの効果を試験した(図4)。細胞をOA1を発現するようトランスフェクトし、次に1μMチロシンのDMEMで48時間培養し、それからタンパク質の細胞表面発現を可能にすべく分析を行った。その後、細胞を1μM L−DOPAで処理し、氷冷メタノールで迅速に固定した。最初、アゴニストが存在しない休止状態では、OA1−GFPは細胞表面で見つかり、β−アレスチンは細胞質中に拡散し(図4A−C)、タンパク質間の同時局在化はなかった。L−DOPAでの刺激後、OA1およびβ−アレスチンは原形質膜に同時局在化した(図4 D−F)。非トランスフェクト細胞はL−DOPA処理に対する反応を示さず(図4G,H)、これはβ−アレスチン分布に対するL−DOPAの効果が、[Ca2+]iのついて得られた結果と同様に、OA1依存的であることを示している。
【0140】
PEDF分泌に対する1−DOPAの効果
OA1の突然変異は、神経感覚網膜の発達の欠陥を引き起こす。以前の研究では、色素沈着RPEが非色素沈着RPEよりも有意に多くのPEDFを分泌すること[34]、およびPEDFは神経感覚網膜の発達を変化させる可能性のある神経栄養因子である[35−41]ことが示された。OA1の突然変異は、RPEの色素沈着の喪失を引き起こすが、これはOA1活性がRPEの色素沈着を制御していることを示唆している。したがって、色素化RPE細胞のL−DOPA刺激がPEDFの分泌の増加を引き起こすか否かを決定した(図5)。このアッセイは幾分困難であったが、その理由は色素沈着RPE細胞はL−DOPAを生産し、L−DOPAはOA1のアゴニストであるが、RPEの非色素沈着培養物ではOA1は容易に検知できないためである。したがって、色素化RPEを使用して、L−DOPA刺激がPEDF発現/分泌を増加させるか否かを決定した。RPE細胞をチロシンを含まない培地に24時間置き、次に1μM L−DOPAで1時間処理した。処理後、細胞を外因性L−DOPAを含まない標準培地に3日間戻した。対照細胞はL−DOPAで処理しないが培地を同じ時間に交換し、実験細胞を通常の培地に戻したものである。3日後に馴化培地を収集し、PEDFを測定した。結果は、ペアになった色素化RPEの対照単一層と比較すると、L−DOPAで処理した色素化細胞ではPEDF分泌が有意に増加することを示している(図5A)。重要なことには、この有意な増加は、着色し、従ってOA1を発現し、基本レベルのPEDF発現をしていた細胞で起こった。
【0141】
チロシナーゼ活性およびOA1シグナル伝達を伴う自己分泌ループを介して色素化RPE細胞がPEDFを分泌するか否かを決定するために、特異的チロシナーゼ阻害剤であるフェニルチオ尿素(PTU)を使用して、色素沈着およびL−DOPA生産を阻害した(図5B)。これらの実験では、色素化RPE細胞は、DMEMで維持するか、または200μM PTUを含むDMEMで3日間維持し、次にPEDF分泌を測定した。色素化RPEはかなりのPEDFを分泌したが、PTUは、PEDF分泌の有意な減少を引き起こし、これは色素化RPE細胞で観察される高レベルのPEDF分泌にチロシナーゼ活性が必要であることを示している。PEDF分泌の減少を引き起こしたのがPTU処理細胞に
おけるL−DOPAの欠如であることを確認するために、色素化RPEの3つの異なる培養物を使用して、48時間PTUに曝露し、その後引き続きPTUが存在する状態で1.0μM L−DOPAで処理し、72時間後にPEDFを測定した(図5C)。データはこの実験の対照に対する割合(パーセント)として示したが、その理由は、使用される培養物の色素沈着およびPEDF発現が実験開始前に変化したためである。PTU処理したRPEは、PEDF分泌を増加させることにより添加L−DOPAに応答したが、これはPEDF分泌に対するPTUの効果が、チロシナーゼが阻害された場合のL−DOPA生産の不足により引き起こされることを示している。
【0142】
考察:
RPEと神経感覚網膜の間には複雑な組織間関係がある。この関係の1態様は、RPE色素沈着および重大な神経感覚網膜の変化を生じさせるメラニン合成の欠如[8、23、42]を基軸とする。データは、少なくとも1つの効き目のある神経栄養因子であるPEDFの分泌を規制するL−DOPAを介して自己分泌ループにOA1とチロシナーゼが関与していることを示唆している。データはまた、白皮症で起こる網膜の発達における病理学的変化がOA1シグナル伝達経路の活性の変化に起因し得ることも示唆している。OA1シグナル伝達活性の減少は、OA1の突然変異により直接的に、またはチロシナーゼ活性によるL−DOPA生産の変化により間接的もたらされ得る。したがって、白皮症の種々の形式に伴う同様な網膜の発現型が一つの共通の経路であるOA1シグナル伝達と調整され得ると仮定される。
【0143】
研究では、in situでヒトRPEの先端面にOA1が観察された。以前の報告は、OA1がマウスではメラノソーム[22]に、および培養細胞ではエンドソーム区画[15−18、20−22、43]に局在化していることを示唆していた。in situRPE調製物からの結果は、OA1がRPEの頂端面に分布することを示している。RPE(合計OA1の約3.5%)の表面のOA1が限られた量であることは、免疫金電子顕微鏡法を使用した以前の研究におけるかかるOA1タンパク質の観察が不十分であることを説明している可能性がある。多くの細胞表面GPCRと同様、OA1は豊富なタンパク質ではない。
【0144】
培養細胞を使用した以前の研究で報告されたOA1のエンドソーム局在は、内因性タンパク質および遺伝子組換えタンパク質の両方に対する本研究で再現された。通常の培地で試験した場合、細胞表面に検出可能なOA1タンパク質はほとんど見あたらず、これは以前の研究と合致していた。しかしながら、培地でのチロシン減少により内因性および組換えOA1タンパク質の両方の細胞表面受容体の蓄積が増大した。これは、培養細胞における細胞表面へのOA1の分布がチロシン感受性であることを示唆している。以前の研究は、エンドサイトーシスが阻害され[17]、ヒトRPEの頂端面でOA1が観察された時に、OA1が細胞表面に局在化され得ることを実証した。データは、OA1が細胞表面GPCRであるが、チロシンまたはチロシンの代謝産物により刺激されてもよいエンドサイトーシスの標的でもあることを示唆している。この点で、本結果は、OA1を細胞内GPCRの固有の種類として分類したOA1局在化の過去の報告とは異なる。ほとんどのGPCRは種々の信号により内在化される細胞表面タンパク質であり、データはOA1がほとんどの他のGPCRと類似していることを示唆している。
【0145】
OA1シグナル伝達活性はL−DOPAにより刺激されるが、その前駆体であるチロシンによっても、そのニューロン代謝産物であるドーパミンでも刺激されない。この結果は、結局L−DOPAとチロシンが一つの水酸基だけ異なるため、密接に関連する分子を識別することが可能である非常に感度の高い受容体の活性を示唆している。チロシンが培養細胞でのOA1の細胞内局在化を引き起こすため、OA1はチロシン感受性である。しかしながら、チロシンに対するシグナル伝達応答がないことに注意すべきであり、競争結合
研究は、チロシンがOA1に対して低い親和性を有することを示唆している。データは、正常な培地中に存在する高濃度のチロシンへの細胞を連続的に曝露すると、OA1の内在化に十分であるが、測定可能なOA1活性化を生じさせる可能性は低いことを示唆している。L−DOPAとドーパミンの間の競争的相互作用は一つの部位であるという強い証拠が見出された。ドーパミンに対して観察されたKiは、L−DOPAに対して観察されたKdと類似しており、これは2つのチロシン代謝産物の親和性が類似していることを示唆している。結果は、ドーパミンからのOA1シグナル伝達がわずかではあるが再生可能に減少していることを示し、ドーパミンがOA1のための有能なアンタゴニストまたはインバースアゴニストであってもよいことを示唆している。
【0146】
オーファンGPCRとして、そのシグナル伝達経路が以前には同定されていない。本研究では、L−DOPAに応じたOA1シグナル伝達が[Ca2+]iの増加を引き起こすことが例証された。データは、L−DOPAに応答して観察された[Ca2+]iの増大が百日咳毒素には無反応であり、cAMPに対する効果は見出されず、OA1がGqサブユニットを介して恐らくシグナル伝達していることを示している。以前の研究は、OA1がG0、GiおよびGqサブユニットファミリーのメンバーを含むトランスフェクト細胞中の
多数のサブユニットと会合可能であることを示唆した。Innamoratiらは、過剰発現されたOA1の自発的活性がGqサブユニットを介して示される可能性が高いことを示した[16]。データは、RPEにおける内因性OA1からのリガンド依存性シグナル伝達が、Gqを介して起こる可能性が最も高く、RPE中で発現された天然OA1とCHOおよびRPEにおける過剰発現OA1とを比較すると乱雑な結合活性は観察されなかったことを示している。興味深いことに、Gqサブユニットの2つの過剰活性変異型が、皮膚および髪の過剰色素沈着を引き起こす[44]が、それらがRPEに影響を及ぼすかどうかは不明である。RPEおよび皮膚のメラノサイトは、色素沈着を生じさせるのに同じ酵素を使用するが、メラニン形成の制御は異なっている。最近の報告は、OA1-/-およびGαi3-/-の網膜発現型が同様であるため、OA1がGαi3を介してシグナル伝達することを示唆している[45]。この研究は、Gαi3とOA1の間の相互作用またはシグナル伝達に関するデータを提供せず、結果はGαi3を介したOA1のシグナル伝達をサポートしていない。しかしながら、OA1とGαi3はいずれも、網膜の発達の複合系のある部分を制御する、収束経路に活性を有し得る。
【0147】
L−DOPAに対するOA1の応答は、[Ca2+]iの増加、原形質膜OA1へのβ−アレスチンの動員、およびPEDF分泌の増加の3つの方法で測定した。さらに、色素化RPEにおけるチロシナーゼ活性の阻害はL−DOPA生産を抑制し、PEDF分泌の減少を生じさせる。まとめると、これらの研究は、L−DOPAとOA1の間の生産的なリガンド:受容体の関係に対する説得力のある論拠を提示している。さらにデータは、L−DOPAのみがOA1に対する生産的なリガンドであったため、チロシンとその代謝産物の間の選択性を示唆している。本願発明者は、OA1とL−DOPAの間の結合反応速度を決定し、2者の間の典型的な一部位の受容体:リガンド関係を観察した。μMの範囲のKdのOA1とL−DOPAの間の結合親和性は、内因性リガンド:受容体の関係では珍しくない。OA1に対する特異的で高親和性のアンタゴニストの将来の同定は、OA1とL−DOPAの間の相互作用のさらなる生化学的特徴付けを支援し、ドーパミンがインバースアゴニストか否かを決定するのに役立つだろう。
【0148】
本研究は、メラニン合成の中間生成物であるL−DOPAによる、オーファンGPCRであるOA1の選択的な活性化を示した。本研究は、OA1活性が、RPEによる、正常な網膜の発達を支援する能力のある分子であるPEDFの分泌を刺激することも示した[40および41]。ヒトでは、これはOA1活性化を介した薬理学的介入がメラニン生成機構(OCA 1−4)の欠損により引き起こされた白皮症に有用であり得ることを示唆している。不運にも、データはかかる薬理学的介入にOA1が必要であり、Oa1の突然
変異が白皮症の最も一般的な原因であることも示唆している。
方法:
細胞培養
RPE− 細胞を[46]に記載したように分離し、5%ウシ胎仔血清(FBS)を補給したDulbecco改変必須培地(DMEM)に維持した。チロシン濃度を低下させる実験では、JRH Biosciences社(カンザス州Lenexa所在)によりチロシンを含まないよ
うに特注製造したDMEMを使用した。透析FBSをInvitrogen社(カリフォルニア州San Diego所在)から購入した。
【0149】
COS−7およびCHO− 細胞はATCCから入手し、5%FBSを補給したDMEMで培養した。OA1分布の分析には、細胞を1μMチロシンと5%FBSを補給したチロシンを含まないDMEMで2−4日間培養し、次に実験のために上記のチロシンを含まない培地で培養した。
細胞表面ビオチン化
in situのヒトRPE 眼球赤道の約2mm前で切開し、前部を除去することによりヒト眼杯を作製した。硝子体および網膜を、その下にあるRPE単層を傷つけずに除去し、網膜を視神経頭で切断した。RPEが露出された得られた眼杯を反応緩衝液(100mM NaCl、50mM NaHCO3、pH8.0)で3回リンスし、スルホ−N
HS−LCビオチン(1mg/ml)を30分間に2回入れた。反応はをTG緩衝液(25mM トリス、192mM グリシン、pH8.3)で停止し、停止プロテアーゼ阻害剤カクテルを含む溶解緩衝液(2mM EDTA、1% トリトンXおよび1% トゥイーン20、Tris塩基性塩類緩衝液)中で細胞を収集した。インタクトな細胞と色素顆粒を20分間、14,000rpmで遠心分離により除去した。ビオチン化されたタンパク質を、固定化ストレプトアビジンビーズで一晩捕獲し、次に4×還元緩衝液(250mMトリス、pH 6.8、8% SDS、40% グリセリン、20% β−メルカプトエタノール、0.08% ブロモフェノールブルー)と混合した。OA1タンパク質を10% SDS−PAGEゲルで分離し、ウェスタンブロット解析用のポリクローナルウサギOA1抗体を用いて同定した。ペアにしたウェスタンブロットを、アクチンに対するモノクローナル抗体でプローブした。
【0150】
培養細胞− RPEおよびトランスフェクト細胞を、実験に関して説明したチロシン濃度を含むDMEMに維持した。培養物を反応緩衝液で3回リンスし、次に、in situ調製物のために上述したようにビオチン化した。
Oa1のクローニング
cDNAライブラリーを6人のヒトドナーの眼からプールされた組織から構築した。全RNAをTrizol試薬を使用して収集し、次にcDNAを、第1鎖の合成にポリチミジル酸プライマーを、第2鎖の合成にランダムヘキサマーを使用して合成した。cDNAの合成に続いて、リボヌクレアーゼAを使用してRNAを除去した。OA1のコーディング配列を5’および3’末端への制限部位を加え、かつ天然停止コドンを除去した末端プライマーを使用してPCRにより得た。PCR生成物をpEGFP N−1ベクター(Clontech社)中にGFPと共にフレーム内にライゲートした。配列は配列全体にわたり両方向で自動シーケンシングにより確認された。
免疫細胞化学
スライド上の細胞を室温にて3% パラホルムアルデヒドで固定し、0.1% トリトンX−100の10%ミルクTBST溶液でリンスし、次に10%ミルクTBST溶液でブロッキングした。β−アレスチンをβ−アレスチンに対するポリクローナル抗体を使用して可視化し、4℃で一晩インキュベートした。50%のグリセリンを使用してカバースリップを載せ、Compix Confocal Imaging Systemsソフトウェア(Simple PCI Version 4.0.6.1605)を搭載したNikon Eclipse E800 レーザー走査共焦点顕微鏡を用いて光学切片
により免疫染色を分析した。OA1−GFPおよびβ−アレスチン分布の三次元分析はIm
age J 1.32で行なった。
[Ca2+]iの測定
ガラスカバースリップ上に配置したOA1−GFPを発現しているCHO細胞を、スリップする、Ca2+含有HEPES干渉ハンクス平衡塩類溶液(HBSS)(pH7.45)でリンスし、次に2.5μM Fura−2(無水ジメチルスルホキシドおよび0.002% プルロン酸で可溶化)で20分間、37℃、5% CO2でインキュベートした
。Fura−2で満たされた細胞をHBSSで15分間、37℃、5% CO2でインキ
ュベートし、染料をその活性型へ十分に開裂させた。各カバースリップを、40×1.35 NA 紫外線−蛍光対物レンズを備えた倒立型Olympus IX70顕微鏡のステージ上で37℃に維持したチャンバに入れて1mlのHBSS中でインキュベートした。
【0151】
フィルタホイールを使用して、200W Xe(キセノン)球からの励起光を340および380nmのフィルタに交互に通過させた。中心が510nmの10nmバンドパスフィルタを、CCDカメラに通過させる(Photometrics CH-250)放射蛍光のために選択
した。各実験に関し、画像の対を最初の3分間の間、毎分撮影し、これは安定した基準線を設定した。その後、L−DOPA(最終濃度1μM)を加え、画像の組を次の3分間の間、30秒毎に撮影した。最後に、細胞にFura−2が装填されていることを確認するために、KCl(最終濃度20mM)を各実験の終了の1分前に陽性対照として加えた。同じ作業を、チロシンおよびドーパミン(いずれも最終濃度1μM)に対して独立して繰り返した。Silicon Graphics Personal IRISコンピュータを使用して、細胞内の各ピクセルについて340/380nm比を計算し、次にMicrosoft Excel version 4.0を使用し
て分析した(Microsoft社、ワシントン州Redmond所在)。一旦340/380nmの比が決定されたら、各比を1(自身で割った時間0の比)で正規化し、遊離イオン濃度を以下の方程式を使用して計算した:
[Cai]#=Kd#*(R-Rmin#)/Rmax#−R)
式中、R、Rmin、およびRmaxはそれぞれ実測比、最小比および最大比である。Rmax
は、完全に脱プロトン化した条件下でのイオン感受性波長の蛍光強度の比を表わし、Rminは完全にプロトン化した場合の染料の比である。Fura−2の場合、RはCa2+が増
加するにつれて増加する。従って、RminはCa2+(Ca2+<1nM)が存在しない状態
のFura−2を表わし、Rmaxは以前に説明したCa2+−Fura−2キレートを表す
[26]。Rmin、RmaxおよびKdをFura−2装填細胞で独立した実験にて決定し、続いて実験手順の遊離Ca2+の計算のために利用した。
放射性標識リガンド結合
OA1−GFPを発現するようにトランスフェクトしたCHO細胞を24ウェルプレートに播いた。細胞を−2℃まで冷却し、次に、冷やした結合緩衝液(25mM トリス、150mM NaCl、5mM EDTA、5μM ジギトニン(pH 7.45))でリンスした。細胞を、10-4M〜10-9Mの間の濃度の[3H]−L−DOPA(Moravek
Biochemicals社、カリフォルニア州Brea所在)を含む結合緩衝液中で2時間インキュベ
ートした。分析のどの工程でも、温度を−2℃を超えないようにした。対照は、非トランスフェクトCHOに対して行った分析を含み、特異的結合を10-3Mの未標識の過剰L−DOPAとの競争により決定した。結合したL−DOPAをシンチレーション分光法で定量化された。
cAMPの測定
細胞をホルスコリン(15分)で前処理し、次に以前に説明したアッセイ設定を使用してL−DOPAで攻撃した[47]。リガンドへの曝露の1分後、細胞を氷冷緩衝液中にこすり落とし、ボイルし、遠心分離した。その後、それぞれ等量(50μl)の上澄液および3H cAMP(New England Nuclear社)を合一して100μl 冷PKAとした。2時間後、溶液を活性炭上に通過させ、上澄液をシンチレーション計数器で数える。結果を、50μlのcAMP(0.25−32.0pmole/50μl)を使用して生成した、細胞質ゾルの代わりに標準曲線を使用して達成されたものと比較する。
実施例2:生体内のOA1ループ機能
OA欠損マウスにおけるPEDF分泌を野性型マウスと比較したところ、野生型マウスはOA1−/yマウスよりも有意に多くのPEDFを分泌することが示された。使用される培地(C.M.)はPEDFを含み、OA1 −/yからのCM中のPEDFは、RPE由来ではなく使用される培地に由来する可能性が考えられる。結果(図7)を定量化し、グラフに要約する。両群のCM中のバックグラウンドPEDFを用いても、差は有意である。t検定分析結果を示す。
【0152】
チロシナーゼ欠損の妊娠マウスを、正常条件で(L−DOPAは無し)または妊娠マウスの仔の胚7日目から該マウスの飲料水に1.0mg/ml L−DOPAを補給して維持した。動物を、視覚の発達が終わり眼が開く出生後14日目まで、L−DOPAを補給して維持した。
【0153】
白皮症では2つの細胞の種類の数が減少している:網膜神経節細胞および光受容体。図8Aは、L−DOPAの補給が、正常な野性型マウスで予想されるものと比較して網膜神経節細胞数を増加させることを実証している。図8Bは、光受容体に関する同じ結果を示す。光受容体は密集しすぎているため、直接数えられない。逆に、光受容体の核により占められた領域を光受容体数の基準として測定する。L−DOPAの補給により光受容体の核領域が増加し、光受容体の数も増加した。繰り返すが、これはアルビノ動物を標準レベルに戻したかのように見えた。
【0154】
図8Cに示されるように、4匹のペアになった同腹仔マウス、2匹の野生型、および2匹のOA1 −/y(雌、OA1欠損)を安楽死させ、各動物からの網膜をレーンに独立して載せ、次にタンパク質をウェスタンブロットしてPEDFを検出した。PEDFは野生型マウス由来の網膜では容易に観察された。対照的にPEDFはOA1−/yマウス由来の網膜では容易に検出されない。
【0155】
要約すると、このデータは、OA1−/yマウスが野性型マウスよりもPEDFを少なく生産することを示している。チロシナーゼ欠損マウスにおけるL−DOPA刺激は、白皮症の2つの最も顕著な神経感覚網膜欠陥である光受容細胞および網膜神経節細胞の損失を助ける。最後に、PEDFレベルはOA1欠損マウスの網膜では低下するため、OA1自己分泌ループがin vivoで機能し、経口L−DOPAにより刺激可能であることが結論付けられる。
【0156】
データはまとめると、RPE色素沈着とAMDの間のつながり(リンケージ)が、OA1のシグナル伝達活性を介している可能性を示している。データは、OA1のリガンドがL−DOPAであり、L−DOPAから生じたOA1シグナル伝達がPEDFの発現を制御することを示している。PEDFはRPEにより作製される最も効き目のある神経栄養因子である。したがって、PEDF発現を制御するOA1のリガンドとしてのL−DOPAの同定は、L−DOPAとRPEにおける神経組織栄養活性とを結びつける。L−DOPAが色素形成の副産物として生産されるため、これは初めてRPE色素沈着と神経組織栄養活性との間のつながりを確立した。この系はOA1自己分泌ループとして定義される。チロシナーゼは色素を形成し、L−DOPAを放出する。放出されたL−DOPAはOA1に結合し、OA1を介したシグナル伝達を開始する。OA1シグナル伝達は、チロシナーゼとPEDFの両方の発現を制御する。
【0157】
これまで、データは上記モデルを生化学的に、培養細胞で、および生体内で例証している。食事のL−DOPAを使用してアルビノ動物における網膜の発達を助けることが可能であるという事実は、RPEの栄養要因発現を生体内で刺激するのに食事のL−DOPAが使用可能であることを示している。AMDは、色素沈着に幾分関連するRPE欠損と明
らかに関係している。青い目の個体は黒い目の個体よりはるかに大きな頻度でAMDになり、したがってRPEの色素沈着レベルはAMDプロセスを制御する。RPEの色素沈着レベルはOA1シグナル伝達により制御され、これは上述したのと同じOA1自己分泌ループの一部である。したがって、AMDはRPEにおけるOA1シグナル伝達と関連付けられる。したがって、RPEの色素沈着がより低い者は、チロシナーゼがより低く、L−DOPAがより低く、OA1がより低く、およびPEDF生産がより低いだろう。本願発明者はOA1のリガンドとして食事L−DOPAまたは関連化合物を使用し、その活性を刺激することが可能である。神経感覚網膜の健康の最終決定因子はPEDFであるが、本願発明者はOA1を使用して、OA1ループ活性を増加させると共に、RPEの神経組織栄養の活性を増加させることが可能である。OA1シグナル伝達の効果はニューロンの生存を促進するだろう。
【0158】
参考文献
1. Akeo K, Shirai S, Okisaka S, Shimizu H, Miyata H, et al. (1996)Histology of fetal eyes with oculocutaneous albinism. Arch Ophthalmol 114: 613-616.
2. Gregor Z (1978)The perifoveal vasculature in albinism. Br J Ophthalmol 62: 554-557.
3. Schraermeyer U, Heimann K (1999)Current understanding on the role of retinal
pigment epitheliμm and its pigmentation. Pigment Cell Res 12: 219-236.
4. Rachel RA, Mason CA, Beermann F (2002)Influence of tyrosinase levels on pigment accμmulation in the retinal pigment epitheliμm and on the uncrossed retinal projection. Pigment Cell Res 15: 273-281.
5. Okulicz JF, Shah RS, Schwartz RA, Janniger CK (2003)Oculocutaneous albinism.
J Eur Acad Dermatol Venereol 17: 251-256.
6. Donatien P, Jeffery G (2002)Correlation between rod photoreceptor nμmbers and levels of ocular pigmentation. Invest Ophthalmol Vis Sci 43: 1198-1203.
7. Russell-Eggitt I (2001)Albinism. Ophthalmol Clin North Am 14: 533-546.
8. Oetting WS (1999)Albinism. Curr Opin Pediatr 11: 565-571.
9. Oetting WS, King RA (1999)Molecular basis of albinism: mutations and polymorphisms of pigmentation genes associated with albinism. Hμm Mutat 13: 99-115.
10. Shen B, Samaraweera P, Rosenberg B, Orlow SJ (2001)Ocular albinism type 1: more than meets the eye. Pigment Cell Res 14: 243-248.
11. Incerti B, Cortese K, Pizzigoni A, Surace EM, Varani S, et al. (2000)Oa1 knock-out: new insights on the pathogenesis of ocular albinism type 1. Hμm Mol Genet 9: 2781-2788.
12. Bassi MT, Schiaffino MV, Renieri A, De Nigris F, Galli L, et al. (1995)Cloning of the gene for ocular albinism type 1 from the distal short arm of the X chromosome. Nat Genet 10: 13-19.
13. Schiaffino MV, Bassi MT, Galli L, Renieri A, Bruttini M, et al. (1995)Analysis of the OA1 gene reveals mutations in only one-third of patients with X-linked ocular albinism. Hμm Mol Genet 4: 2319-2325.
14. Schiaffino MV, d'Addio M, Alloni A, Baschirotto C, Valetti C, et al. (1999)Ocular albinism: evidence for a defect in an intracellular signal transduction system. Nat Genet 23: 108-112.
15. Schiaffino MV, Tacchetti C (2005)The ocular albinism type 1 (OA1)protein and the evidence for an intracellular signal transduction system involved in melanosome biogenesis. Pigment Cell Res 18: 227-233.
16. Innamorati G, Piccirillo R, Bagnato P, Palmisano I, Schiaffino MV (2006)The
melanosomal/lysosomal protein OA1 has properties of a G protein-coupled receptor. Pigment Cell Research 19: 125-135.
17. Staleva L, Orlow SJ (2006)Ocular albinism 1 protein: trafficking and function when expressed in Saccharomyces cerevisiae. Exp Eye Res 82: 311-318.
18. Shen B, Orlow SJ (2001)The ocular albinism type 1 gene product is an N-glycoprotein but glycosylation is not required for its subcellular distribution. Pigment Cell Res 14: 485-490.
19. d'Addio M, Pizzigoni A, Bassi MT, Baschirotto C, Valetti C, et al. (2000)Defective intracellular transport and processing of OA1 is a major cause of ocular
albinism type 1. Hμm Mol Genet 9: 3011-3018.
20. Shen B, Rosenberg B, Orlow SJ (2001)Intracellular distribution and late endosomal effects of the ocular albinism type 1 gene product: consequences of disease-causing mutations and implications for melanosome biogenesis. Traffic 2: 202-211.
21. Samaraweera P, Shen B, Newton JM, Barsh GS, Orlow SJ (2001)The mouse ocular
albinism 1 gene product is an endolysosomal protein. Exp Eye Res 72: 319-329.
22. Schiaffino MV, Baschirotto C, Pellegrini G, Montalti S, Tacchetti C, et al. (1996)The ocular albinism type 1 gene product is a membrane glycoprotein localized to melanosomes. Proc Natl Acad Sci U S A 93: 9055-9060.
23. Ilia M, Jeffery G (2000)Retinal cell addition and rod production depend on early stages of ocular melanin synthesis. J Comp Neurol 420: 437-444.
24. Ilia M, Jeffery G (1999)Retinal mitosis is regulated by dopa, a melanin precursor that may influence the time at which cells exit the cell cycle: analysis of patterns of cell production in pigmented and albino retinae. J Comp Neurol 405: 394-405.
25. Ito S (2003)The IFPCS presidential lecture: a chemist's view of melanogenesis. Pigment Cell Res 16: 230-236.
26. Martinez-Zaguilan R, Tompkins LS, Gillies RJ, Lynch RM (2006)Simultaneous analysis of intracellular pH and Ca2+ from cell populations. Methods Mol Biol 312: 269-287.
27. Ferguson SS, Caron MG (2004)Green fluorescent protein-tagged beta-arrestin translocation as a measure of G protein-coupled receptor activation. Methods in Molecular Biology 237: 121-126.
28. Barak LS, Warabi K, Feng X, Caron MG, Kwatra MM (1999)Real-time visualization of the cellular redistribution of G protein-coupled receptor kinase 2 and beta- arrestin 2 during homologous desensitization of the substance P receptor. J Biol Chem 274: 7565-7569.
29. Zhang J, Barak LS, Anborgh PH, Laporte SA, Caron MG, et al. (1999)Cellular trafficking of G protein-coupled receptor/beta- arrestin endocytic complexes. J Biol Chem 274: 10999-11006.
30. Tohgo A, Choy EW, Gesty-Palmer D, Pierce KL, Laporte S, et al. (2003)The stability of the G protein-coupled receptor-beta-arrestin interaction determines the mechanism and functional consequence of ERK activation. J Biol Chem 278: 6258-6267.
31. Ferguson SS, Zhang J, Barak LS, Caron MG (1998)Molecular mechanisms of G protein-coupled receptor desensitization and resensitization. Life Sci 62: 1561-1565.
32. Barak LS, Ferguson SS, Zhang J, Caron MG (1997)A beta-arrestin/green fluorescent protein biosensor for detecting G protein-coupled receptor activation. J Biol Chem 272: 27497-27500.
33. Barak LS, Ferguson SS, Zhang J, Martenson C, Meyer T, et al. (1997)Internal
trafficking and surface mobility of a functionally intact beta2-adrenergic rece
ptor-green fluorescent protein conjugate. Mol Pharmacol 51: 177-184.
34. McKay BS, Goodman B, Falk T, Sherman SJ (2006)Retinal pigment epithelial cell transplantation could provide trophic support in Parkinson's disease: Results
from an in vitro model system. Exp Neurol 201: 234-243.
35. Tombran-Tink J, Shivaram SM, Chader GJ, Johnson LV, Bok D (1995)Expression,
secretion, and age-related downregulation of pigment epitheliμm-derived factor, a serpin with neurotrophic activity. J Neurosci 15: 4992-5003.
3 6. Malchiodi-Albedi F, Feher J, Caiazza S, Formisano G, Perilli R, et al. (1998)PEDF (pigment epitheliμm-derived factor)promotes increase and maturation of pigment granules in pigment epithelial cells in neonatal albino rat retinal cultures. Int J Dev Neurosci 16: 423-432.
37. Behling KC, Surace EM, Bennett J (2002)Pigment epitheliμm-derived factor expression in the developing mouse eye. Mol placeVis 8: 449-454.
38. Aymerich MS, Alberdi EM, Martinez A, Becerra SP (2001)Evidence for pigment epitheliμm-derived factor receptors in the neural retina. Invest Ophthalmol Vis Sci 42: 3287-3293.
39. Tombran-Tink J, Chader GG, Johnson LV (1991)PEDF: a pigment epitheliμm-derived factor with potent neuronal differentiative activity. Exp Eye Res 53: 411-414.
40. Jablonski MM, Tombran-Tink J, Mrazek DA, Iannaccone A (2001)Pigment epitheliμm-derived factor supports normal Muller cell development and glutamine synthetase expression after removal of the retinal pigment epitheliμm. Glia 35: 14-25.
41. Jablonski MM, Tombran-Tink J, Mrazek DA, Iannaccone A (2000)Pigment epitheliμm-derived factor supports normal development of photoreceptor neurons and opsin expression after retinal pigment epitheliμm removal. J Neurosci 20: 7149-7157.
42. Jeffery G (1998)The retinal pigment epitheliμm as a developmental regulator of the neural retina. Eye 12 (Pt 3b): 499-503.
43. Piccirillo R, Palmisano I, Innamorati G, Bagnato P, Altimare D, et al. (2003)An unconventional dileucine-based motif and a novel cytosolic motif are required for the lysosomal and melanosomal targeting of OA1. Journal of Cell Science 119: 2003-2014.
44. Van Raamsdonk CD, Fitch KR, Fuchs H, de Angelis MH, Barsh GS (2004)Effects of G-protein mutations on skin color. Nat Genet 36: 961-968.
45. Young A, Powelson EB, Whitney IE, Raven MA, Nusinowitz S, et al. (2008)Involvement of OA1, an intracellular GPCR, and G alpha i3, its binding protein, in melanosomal biogenesis and optic pathway formation. Invest Ophthalmol Vis Sci 49:
3245-3252.
46. Hu J, Bok D (2001)A cell culture mediμm th at supports the differentiation of hμman retinal pigment epitheliμm into functionally polarized monolayers. Mol placeVis 7: 14-19.
47. Stamer WD, Golightly SF, Hosohata Y, Ryan EP, Porter AC, et al. (2001)Cannabinoid CB(1)receptor expression, activation and detection of endogenous ligand in trabecular meshwork and ciliary process tissues. Eur J Pharmacol 431: 277-286.
【技術分野】
【0001】
本発明は、加齢黄斑変性症を治療する方法および組成物ならびに加齢黄斑変性症を治療する化合物を識別する方法に関する。
(関連出願)
本願は、その全体を本明細書に援用する2008年4月18日に出願された米国仮特許出願番号第61/124,624号の優先権を主張する。
(政府の権利の記述)
本発明は米国国立衛生研究所(NIH)の助成番号R03 EY014403の下、政府支援で成されたものである。米国政府は本発明の一定の権利を有する。
【背景技術】
【0002】
加齢黄斑変性症(「AMD」)は、網膜の損傷により黄斑(視野中心)の視力喪失を生じさせる加齢に関連する疾病である。AMDは高齢者に一般的な疾患であり、66−74歳の患者の約10%、75−85歳の患者の30%が何らかのレベルの黄斑変性を有している。
【発明の概要】
【発明が解決しようとする課題】
【0003】
現在、大部分のAMD患者に有効な治療は存在せず、初期段階の介入法もない。
【課題を解決するための手段】
【0004】
1態様では、本発明は、加齢黄斑変性症(AMD)の治療に有効な量のOA1受容体のアゴニストをAMD患者に投与することを含む、AMDを治療する方法を提供する。第2の態様では、本発明は、AMDの進行の制限に有効な量のOA1受容体のアゴニストをAMDが進行する危険性のある患者に投与することを含む、AMDの進行を制限する方法を提供する。本発明のこれらの態様のいずれかの1つの好ましい実施形態では、OA1受容体のアゴニストは、L−DOPAおよびL−DOPA類似体からなるグループから選択される。
【0005】
別の態様では、本発明は、AMDを治療する化合物を識別する方法であって、細胞を試験化合物と接触させることを含み、該細胞は、
(a)OA1を発現している第1の細胞集団;および任意選択で、
(b)OA1を発現していない第2の細胞集団;を含み、
(c)以下の(i)および(ii)うちの一方または両方を増大させる試験化合物を陽性試験化合物と識別し;
(i)(A)試験化合物と接触させない第1の細胞集団における色素上皮誘導因子(PEDF)発現および(B)第2の細胞集団におけるPEDF発現の一方又は両方に対する、第1の細胞集団におけるPEDF発現;
(ii)(A)試験化合物と接触させない第1の細胞集団における細胞内カルシウム濃度および(B)第2の細胞集団;
陽性試験化合物は、AMDの治療およびAMDの進行の制限の少なくとも一方を行うための候補化合物である方法を提供する。
【0006】
さらなる態様では、本発明は、以下の工程を含む、AMDを治療する化合物を識別する方法を提供する:
(a)チロシナーゼ欠損の妊娠している雌の非ヒト哺乳動物に試験化合物を投与する工
程であって、試験化合物は胚の光受容体および/または網膜神経節の発達中に投与される工程、
(b)胚または出生後の非ヒト哺乳動物における光受容体および/または網膜神経節の発達に対する試験化合物の効果を、試験化合物を投与しなかった胚または出生後における光受容体および/または非ヒト哺乳動物網膜神経節の発達と比較する工程であって、光受容体および/または網膜神経節の発達を増大させる試験化合物が、AMDの進行を治療および/または制限する候補化合物である工程。
【0007】
またさらなる態様では、本発明は以下を含む組成物を提供する:
(a)AMDの進行を治療または制限するのに有効な量のL−DOPAまたはL−DOPA類似体;
(b)AMDの進行を治療または制限するのに有効な量のビタミンC源、ビタミンE源、ビタミンA源、亜鉛源および銅源を含む組成物。
【図面の簡単な説明】
【0008】
【図1A】in situでRPEをビオチン化(A)した後、ストレプトアビジン結合ビーズに(B)結合させるか、または(C)結合させないタンパク質のウェスタンブロット解析。(B)培養RPE、(C)OA1−GFPを発現するようトランスフェクトしたCOS細胞。ブロットはストレプトアビジン結合ビーズを使用して細胞表面をビオチン化および細分化した後にOA1およびアクチンを可視化するようブローブ標識した。培養細胞(B、C)について、細胞は分析に先立ち500μM(通常DMEM)または1μMのチロシンで3日間維持した。
【図1B】ストレプトアビジン結合ビーズに結合させたタンパク質のウェスタンブロット解析。
【図1C】ストレプトアビジン結合ビーズに結合させないタンパク質のウェスタンブロット解析。
【図1D】デンシトメトリーによるウェスタンブロット解析の定量化。OA1デンシトメトリーはペアになった細胞培養物対照の%として示される。ペアになった細胞培養物は、トランスフェクト後2つの等しい群へ分割したものであり、一方は通常DMEM(対照)に維持した対照であり、他方の群は収集するまで1μMチロシンDMEM(LT)で維持した。Paired−t検定分析を使用して、差が有意であるか否か試験した。*はp<0.001を示す。同じ方法を分析したアクチンは差を示さず、p=0.724であった。
【図1E】通常DMEMに維持し、その後抗OA1抗体で染色し、20倍で画像化した色素RPE細胞の複合焦点顕微鏡検査法。バー=25μm。
【図1F】1μMチロシンに維持し、その後抗OA1抗体で染色し、20倍で画像化した色素RPE細胞の複合焦点顕微鏡検査法。バー=25μm。
【図2A】トランスフェクトしたまたはトランスフェクトしていないCHO細胞における標準実験プロトコルの時間経過中の[Ca2+]iのトレース例。3分間の安定な基準線の確立後、試験薬を1μMで加えた。5分目に、対照として機能するようKClを加え、細胞にFura−2を装填し、利用できるようにした。トランスフェクト細胞とペアにした非トランスフェクト細胞の両方に同一のプロトコルを行なった。
【図2B】トランスフェクトCHO細胞および非トランスフェクトCHO細胞におけるチロシン、ドーパミンおよびL−DOPAに応じた[Ca2+]iの概要データ。非トランスフェクト細胞を、L−DOPA処理で示す。KClによる膜脱分極の実験対照も示される。バーはそれぞれ少なくとも10回の実験から集めたデータを表わし、検査薬添加後の基準線[Ca2+]iからの平均変化として示される。エラーバーは標準偏差を表し、有意差試験にt検定分析を使用した。*はp<0.01を示す。天然タンパク質を発現するOA1またはRPEを発現するようトランスフェクト細胞の[Ca2+]i増加の百日咳毒素感受性分析。データは少なくとも6回の実験の平均を表わす。
【図2C】天然タンパク質を発現するOA1またはRPEを発現するようトランスフェクト細胞の[Ca2+]i増加の百日咳毒素感度分析。データはトランスフェクト細胞の各群に対する少なくとも6回の実験および内因性OA1発現した処理済みおよび未処理RPEの20回の個々の実験の平均を示す。t検定分析を使用して有意差を試験した。*はp<0.01を示す。
【図2D】OA1を発現するようトランスフェクトされたCHOにおけるcAMPを測定した。対照群はトランスフェクトされたが未処理のCHO細胞であり、かかる細胞におけるcAMPの基準レベルを表わす。細胞を1.0μM L−DOPA、0.1μMホルスコリン、L−DOPA+0.1μMホルスコリン、および陽性対照として1μMホルスコリンでを処理した。結果は、同じ培養プレートでレプリカ単層を使用して、すべての実験群をすべてペアにして分析した少なくとも6回の実験で観察された平均cAMPレベルに相当する。エラーバーは各群の標準偏差を示し、観察された唯一の有意差は、ホルスコリン処理後のcAMPレベルの増加であった。
【図3A】OA1とL−DOPAの間の結合反応速度を放射性標識リガンド結合アッセイを使用して決定した。結果は、5回のかかる実験から集めたデータを表わし、平均特異的結合+/−SEMとして示す。双曲線適合は0.994のR2値を示し、Kdは9.34×10-6M+/1.14x10-6Mと決定された。
【図3B】OA1トランスフェクトCHO細胞に対する5μM[H3]L−DOPAの相対的結合を、1.0mMドーパミン、チロシンまたはL−DOPAの存在下で比較した。データは各群の平均合計結合+/−標準偏差を示す。*は、対照群間の結果を、潜在的な競争リガンドの存在下での結合と比較した場合のp<0.05を示す。
【図3C】ドーパミンがOA1活性アンタゴニストとして機能するか否かを決定するために、5μM[H3]L−DOPAおよびドーパミン間の競争相互作用を評価した。結果は、ドーパミンおよびL−DOPAが同じOA1結合部位に競争することを示し、データはr2値が0.95の結合モデルに適合する。ドーパミンのKiは2.388+/−0.266μM(平均+/−SEM)であり、L−DOPAのKdと同様であった。
【図3D】OA1による用量依存的OA1シグナリング。データは所与の濃度(各用量n=6)での細胞のL−DOPA処理により誘発された[Ca2+]iの平均増加量を示す。t検定を使用して、各用量で達成された応答を比較した。*は1および10μMでの比較が<0.01であることを示す。
【図3E】図3(a)に基づく単一部位の結合関係の動力学を示すScatchardプロット。
【図4】(A)−(H)。画像はすべて、OA1−GFPを発現するようトランスフェクトされたCHO細胞の2μm厚の共焦切断片を示す。β−アレスチンは免疫蛍光法を使用して可視化した。L−DOPA(A−C)の追加前、1μM L−DOPA(D−F)の処理後、および合一画像(C,F)は、白色光イメージングの解像度で2つのタンパク質が同時局在化する領域を示す。(G、H)はトランスフェクトCHO細胞の視野の低倍率であり、2個のトランスフェクト細胞が見える(矢印)(G)。残りの細胞集団は膜へのβ−アレスチンの集合がOA1発現細胞でのみ生じている(矢印)を示すためにβ−アレスチン抗体を使用して可視化している(H)。
【図5A】PEDF濃度を細胞馴化培地のELISAにより決定した。RPE(網膜色素上皮)細胞は、L−DOPA処理をしない対照細胞か、または通常DMEMで3日間維持する前に1μM L−DOPAで処理したOA1刺激細胞である。データは3連で行った3回の実験の平均として示され、エラーバーは標準偏差を示し、*はpaired t検定を使用してP<0.01を示す。
【図5B】ELISAにより決定された色素RPEからの馴化培地中のPEDF濃度。細胞は、対照色素RPE培養物または200μMのフェニルチオ尿素(PTU)で処理したペアになった培養物とした。データは3連で行った3回の実験の平均として示され、エラーバーは標準偏差を示し、*はpaired t検定を使用してP<0.01を示す。
【図5C】OA1シグナリングを刺激するための、PTUで処理し、次にL−DOPAで処理した色素RPE細胞の馴化培地におけるPEDF濃度。PTU処理前、次にPTU処理後、L−DOPA刺激後の同じ培養物から、ELISA分析を行った。結果を細胞培養物に関連して達成された値の平均+/−標準偏差として示す。*は、PTUを対照(PTUを対照前に試験した同じ培養物)と比較した場合ならびに同じ培養物からのPTUサンプルとL−DOPA/PTUを比較した場合に、p<0.01であることを示す。
【図6A】データは、全量、特異的、および非特異的のすべての分画における平均+/−標準誤差の結合[3H]−L−DOPAを示す。非特異的結合は、超過の未標識L−DOPA(1mM)の存在下で結合した放射性標識L−DOPAの測定により決定された。各所与濃度の特異的結合は、測定した結合合計から測定した非特異的結合を減算することにより決定される。
【図6B】チロシンおよび5μM[H3]L−DOPAの濃度の増大を使用して測定した、チロシンとL−DOPAの間の競争的相互作用を示す。各データポイントは、5つの複製ウェルからの平均データを示し、エラーバーは標準偏差である。データは、チロシンがL−DOPAとの結合に競争するが、親和性は低いことを示している。結果は、チロシンが52.9μMのKiを有し、r2値が0.85の単一部位結合モデルに適合する。チロシンの溶解度が制限されているため、飽和に達しない可能性がある。
【図7】野性型対OA欠損マウスにおけるウェスタンブロットおよびPEDF分泌のグラフ図。
【図8A】正常野性型マウスで予想されるのと比較して、L−DOPAの補給が網膜神経節細胞数を増加させることを示すデータのグラフ図。
【図8B】正常野性型マウスで予想されるのと比較して、L−DOPAの補給が光受容体数を増加することを示すデータのグラフ図。
【図8C】2匹の野性型および2匹のOA1−/yマウスにおけるPEDF検出を示すウェスタンブロット。
【発明を実施するための形態】
【0009】
本明細書で引用した文献はすべてその全体が本明細書に組み込まれる。
本願では、他に別記されていなければ、使用される技術は以下のいくつかの周知の参照文献に見い出されてもよい:Molecular Cloning: A Laboratory Manual (Sambrook, et al., 1989, Cold Spring Harbor Laboratory Press), Gene Expression Technology (Methods in Enzymology, Vol. 185, edited by D. Goeddel, 1991. Academic Press, placeCitySan Diego, StateCA), 敵uide to Protein Purification・in Methods in Enzymology
(M.P. Deutshcer, ed., (1990) Academic Press, Inc.); stocktickerPCR Protocols: A
Guide to Methods and Applications (Innis, et al. 1990. Academic Press, placeCitySan Diego, StateCA), Culture of Animal Cells: A Manual of Basic Technique, 2nd
Ed. (R.I. Freshney. 1987. Liss, Inc. New York, NY), Gene Transfer and Expression Protocols, pp. 109-128, ed. E.J. Murray, The Humana Press Inc., Clifton, N.J.), and the Ambion 1998 Catalog (Ambion, Austin, TX).
本明細書に使用する場合、単数形(英語で「a」、「an」および「the」)は文脈が別段指示していない限り、複数の参照物を含む。
【0010】
第1態様では、本発明は、加齢黄斑変性症(AMD)を治療する方法であって、AMDの治療に有効な量のOA1受容体のアゴニストをAMD患者に投与することからなる方法を提供する。
【0011】
第2の態様では、本発明は、AMDの進行を制限する方法であって、AMDの進行の制限に有効な量のOA1受容体のアゴニストを、AMDが進行する危険性のある患者に投与することからなる方法を提供する。
【0012】
ヒトOa1遺伝子はX染色体上に見出され、配列分析[14]に基づけばGタンパク質共役受容体(GPCR)[12,13]であると考えられる404個のアミノ酸からなるタンパク質OA1(配列番号2)をコードすることが示されている。本明細書に詳細に開示するように、本願発明者は、神経感覚網膜生存の重要な決定因子としてOA1シグナル伝達経路を同定した。OA1シグナル伝達経路の刺激は、AMDの治療を提供するとともに、潜在的リスクのあるAMD進行を制限する手段も提供する。いかなる機構によっても拘束されないが、本願発明者は少なくとも1つの効き目のある神経栄養因子であるPEDFの分泌を規制するL−DOPAによる自己分泌ループにOA1およびチロシナーゼが関与していると考える。したがって、L−DOPAの投与を使用して、OA1活性を刺激し、したがってPEDF発現をアップレギュレートし、これをAMDの治療およびAMD進行の制限のための有用な治療手段とすることが可能である。
【0013】
以下に詳細に論ずるように、そのようなOA1アゴニストは、例えば本発明の第3および第4の態様の新薬発見方法を使用して識別することが可能である。OA1アゴニストの例は以下に詳細に論ずる。
【0014】
好適には、患者はヒトである。
本明細書に使用する場合、本発明のすべての態様および実施形態について、「AMD」は、加齢黄斑変性症として知られる、網膜に対する損傷のために黄斑(視野中心)の視力が喪失する加齢に関連する疾患を意味する。本明細書に使用する場合、AMDは、以下により詳細に説明する滲出型AMDと萎縮型AMDの両方を包含する。
【0015】
AMDは、網膜色素上皮とその下の脈絡膜との間の黄斑における特徴的なドルーセン(黄色沈着物)から始まる。これらの初期の変化(加齢性黄斑変性症と呼ばれる)を有する人の大半は、良好な視力を維持している。ドルーセンのある人は、引き続き進行したAMDに進展する可能性がある。この危険性は、ドルーセンが大きく、多く、かつ黄斑の下の色素細胞層における妨害に関連している場合には、かなり高くなる。
【0016】
加齢性黄斑変性症の患者は進行したAMDの2つの主な形式のうちのいずれかに進み得るが、そのいずれも本発明の方法を使用して、治療可能であるか、またはその進行を制限することが可能である。「滲出型」AMDは、Bruch膜中の脈絡膜毛細血管の以上な血管
増殖による視力喪失を引き起こし、究極的には黄斑の下の血液およびタンパク質の漏出につながる。このような血管からの出血、漏出、および瘢痕は、もし治療していないで放置されると、光受容体への不可逆的な損傷および急速な視力喪失をもたらす。「萎縮型」AMDは黄斑中の感光性細胞がゆっくり分解したときに起こり、次第に罹患した目の視力喪失を徐々に引き起こす。AMDにおける目のかすみは、網膜色素上皮(RPE)の下のドルーセンの蓄積によるものであり、これは光受容体を焦点平面から異動させることにより光受容体の焦点特性を変化させる。
【0017】
萎縮型AMDは一方又は両方の目に生じ、加齢性黄斑変性症から、萎縮型AMDの中間期または進行期へと進行する可能性がある。
中間期の萎縮型AMD:中間サイズの多くのドルーセンがあるか、または1つまたは複数の大きなドルーセンがある。人によっては自身の視野中心にぼやけた点が見える。読書や他の作業のためにはより多くの光が必要され得る。
【0018】
進行期の萎縮型AMD:ドルーセンに加え、網膜中心領域の感光性細胞および支持組織の分解。この分解により視野中心にぼやけた点が生じ得る。時間が経つにつれ、ぼやけた点はより大きく暗くなり、中心視力のより多くを占めるようになり、読書することや、顔が非常に近づくまで顔を認識するのが困難となり得る。
【0019】
AMD症状には、中心視野のぼやけ/減少、中心暗点(影または視力の喪失領域)、ある暗色を別の暗色と区別し、かつ/または、ある光の色を別の光の色と区別する困難さ、明るい光に曝された後の視覚機能の回復の遅さ、コントラスト感度の喪失(その結果、環郭、影および色覚がより鮮明でなくなる)、網膜色素上皮(RPE)妨害(色素の凝集および/または脱落を含む)、RPE剥離、地図状萎縮、網膜下血管新生、円盤形瘢痕、および乱視(変形視)(複数の直線からなるグリッドが波状に見え、グリッドの部分がブランクに見える)が含まれるがこれらに限定されない。萎縮型AMDおよび滲出型AMDの症状は、一般に、疾患の進行の間同様に早いため、どの初期の患者でAMDの萎縮型対滲出型が進行しているかを決定するのは可能でない場合がある。萎縮型AMDは「地図状萎縮」として発達し、新しい血管が発生した場合、初期AMDは「滲出型」AMDとなる。
【0020】
本明細書に使用する場合、AMDを「治療する」とは、以下の1つまたは複数を遂行することを意味する:(a)AMDの重篤度の低下;(b)上述したAMDに特徴的な1つまたは複数の症状の進行の制限または予防;(c)上述したAMDに特徴的な1つまたは複数の症状の悪化の抑制;(d)以前に障害のあった患者のAMD再発の制限または予防;および(e)以前にAMDの症状があった患者の1つまたは複数の症状の再発の制限または予防。そのような治療は、滲出型AMDと萎縮型AMDの治療を含む。
【0021】
本明細書に使用する場合、AMDの「進行の制限」という用語は、AMDが進行する危険性のある個体におけるAMDの進行を予防するかまたは最小限にすること、ならびに加齢性黄斑変性症のAMD(滲出型または萎縮型)への進行または中間期の萎縮型AMDから進行期の萎縮型または「滲出型」AMDへの進行を制限すること、を意味する。1つの好ましい実施形態では、方法は、AMDの進行を制限するために、ドルーセン蓄積(加齢性黄斑変性症)のある患者を治療することを含む。別の好ましい実施形態では、方法は、治療していないAMD患者またはAMDの危険性のある患者と比較した視力喪失のラインの割合を減少させるのに有効な量のOA1アゴニストで患者を治療することを含む。別の好ましい実施形態では、方法は、滲出型AMD患者または滲出型AMDが進行する危険性のある患者を、新たな血管形成の速度および数を減少させるのに有効な量のOA1アゴニストで治療することを含む。より以下に詳細に論ずるように、OA1刺激はRPEにPEDF分泌を増大させ、PEDFは効き目のある抗血管新生因子である。したがって、OA1刺激戦略は、本明細書で論じた網膜の進行に対するその効果に加えて、「滲出型」AMDでの新たな血管の発達を止めることが可能である。
【0022】
別の好ましい実施形態では、方法は、中心視力がぼけているか減少している患者を、一方又は両方の目の視力のラインを増加させるのに有効な量のOA1アゴニストで治療することを含む。この実施形態では、視力のラインは標準Snellen試験により測定されるのと同じものであり、視力「ライン」の増大または減少はSnellen視力表上のどの最も細いラインを患者がはっきりと読むことができるかに基づく。
【0023】
「AMDが進行する危険性のある患者」とは、50歳以上(種々の好ましい実施形態では60歳以上、65歳以上、70歳以上、または75歳以上)で、ドルーセン沈着物の存在、白色人種、AMDを現在有しているか過去に有していた血族の存在、(Tyr402His)の補体因子H遺伝子(CFH)の突然変異、補体タンパク質C3遺伝子のArg80Gly変異型、高血圧、高いコレステロール値、肥満、喫煙、高脂肪摂取、およびフィブリン5遺伝子中の突然変異を含むがそれに限定されないAMDを進行させる任意の危険因子を有する任意の者を意味する。したがって、好ましい実施形態では、治療される患者は、特にAMDの進行を制限する方法において、これらの危険因子の1つまたは複数を有している。
【0024】
「治療上有効量」という語句は、本明細書に使用する場合、AMDの進行を制限するか
またはAMDを治療する(進行を防ぐか、または逆行させる)のに十分または有効な量を指す。適切な用量の範囲は、化合物の選択、投与経路、製剤の性質、患者の症状の状態、および関与する医師の判断によって決まる。例えば、経口投与は、静脈内注射による投与よりも高用量が必要であると予想される。当該技術分野でよく理解されるように、かかる投薬レベルの違いは、最適化のための標準の経験的ルーチンを使用して調節することが可能である。
【0025】
好ましい実施形態では、OA1受容体ゴニストは、L−DOPAおよびL−DOPA類似体からなるグループから選択された化合物を含む。
L−DOPAはパーキンソン病の治療の際に使用されることが知られている[2−アミノ−3−(3,4−ジヒドロキシフェニル)プロパン酸]であり、以下の構造を有している。
【0026】
【化1】
L−DOPAは市販されており、またその合成方法は当業者に周知である。
【0027】
本明細書に使用する場合、「L−DOPA類似体」は、その多くが当該技術分野で周知であるL−DOPAプロドラッグを含む、OA1刺激活性を保持しているL−DOPAの変異型であり、かかる類似体の例については以下に開示する。特定の作用機序によって拘束されるわけではないが、本願発明者は、OA1へのL−DOPAの結合には2つの結合部位が関与し、1つは一方又は両方の水酸基が関与し、1つはカルボン酸基が関与すると考えている。1実施形態では、L−DOPA類似体は、投与後(かつ一般に細胞表面上のOA1への結合前)にL−DOPAに代謝され、そのためOA1刺激活性を保持すると予想されるL−DOPAプロドラッグである。別の実施形態では、一方又は両方の水酸基および/またはカルボキシル基は、本発明の方法に使用される種々の類似体(プロドラッグまたは別のもの)を生産するために置換可能である。
【0028】
別の実施形態では、L−DOPA類似体はL−DOPAエステルを含む。L−DOPAエステルの例およびそれらを製造する方法は、WO/1997/016181;米国特許第4663349号;米国特許第4873263号;米国特許第4873263号;米国特許第5345885号、および米国特許第4771073号に開示されている。種々の好ましい実施形態では、L−DOPAエステルは、L−DOPAメチルエステル、L−DOPAブチルエステル、L−DOPAペンチルエステル、L−DOPAシクロヘキシルエステル、L−DOPAベンジルエステル、およびL−DOPAエチルエステルからなるグループから選択される。種々のさらなる好ましい実施形態では、L−DOPAエステルは、L−DOPAのアルキル、アリール、および置換および非置換のアラルキルエステルからなるグループから選択される。さらに好ましい実施形態では、L−DOPAエステルは以下の式により表わされる:
【0029】
【化2】
式中、Rは、
直鎖または分岐鎖のアルキル基(C1−C20)、例えばメチル、エチル、プロピル、ブ
チル、ミリスチル、パルミチル、ペンチル、テトラデシル、ヘキサデシル等;
アリール基(C6−C9)、例えばフェニル、トリル等;
アルコキシ(C1−5)[メトキシ、エトキシ、ブトキシ等]等の置換基を任意選択で有する置換または非置換のモノ、ジ、またはポリヒドロキシアルキル基(C1−C20)、例
えばベンジル、アルコキシベンジル、4−ヒドロキシブチル、2−ヒドロキシプロピル、2,3−ジヒドロキシプロピル、1,3−ジヒドロキシプロピル、6−ヒドロキシヘキシルおよび5−ヒドロキシペンチル等;
カルボアルコキシ(C1−5)[メトキシカルボニル、エトキシカルボニル、プロポキシカルボニル、ブトキシカルボニル等];
アミノ;
モノまたはジアルキルアミノ(C1−10)[メチルアミノ、メチルエチルアミノ、ジエ
チルアミノ等];
アシルアミノ(C1−5)[アセタミド、ブチルアミド];
ケトアルキル(C1−5)[メチルケト、エチルケト、ブチルケト等];
ハロ[クロロ、ブロモ等]またはカルボキサミド;
置換および非置換のアラルキル(C7−20)、例えばベンジル、アルコキシベンジルC8−14[メトキシ、エトキシ、イソブトキシ等];
フェニルエチル;
フェニルプロピル;
フェニルブチル;
フェニルヘキシル;
フェニルオクチル等;および
医薬として許容される有機または無機のカウンターイオン塩類。
【0030】
L−DOPAのエステルおよびその塩を製造するための合成法は当該技術分野で周知であり、例えばその各々を本明細書に援用する米国特許第3,891,696号、米国特許第4,035,507号;および米国特許第5,354,885号;ならびにJournal of
Pharmaceutical Sciences, 62, p.510(1973)に記載されている。
【0031】
別の実施形態では、L−DOPA類似体は、当該技術分野で周知の胆汁酸抱合体を含む。L−DOPA胆汁酸抱合体の例およびそれらを製造する方法はWO/2002/028882および米国特許出願公開第20020151526号に開示されている。経口投与に際して、これらのプロドラッグは腸肝系内で開裂し、ペアレント薬および/または胆汁酸からの活性代謝物を体循環へ放出する。重要なことに、プロドラッグの一部のみ(通常<50%)しか腸肝サイクルの各通過間に開裂されない。したがって、腸肝循環は、持続的な全身の薬物濃度の達成を可能にする薬物の貯蔵庫として作用する。コール酸、ケノデオキシコール酸、ウルソデオキシコール酸、デオキシコール酸、ウルソコラン酸およびリトコール酸のような天然の胆汁酸が特に好ましい。L−DOPAまたは他のL−DOPA類似体へのこれらの胆汁酸の接合部位は、好ましくは3−ヒドロキシ基またはC−24カルボキシル部分を介してである。任意選択で、薬物と胆汁酸の間に開裂リンカー官能基を導入し、かかるリンカーが選択されてもよい。好ましい実施形態では、そのようなL−DOPA胆汁酸抱合体は以下の式またはその医薬として許容される塩により表わされる:
【0032】
【化3】
式中、R1は水素およびOHからなるグループから選択され;
R2は水素およびOHからなるグループから選択され;
XはOHおよびD−Y−からなるグループから選択され、YはDをステロイドに共有結合させる共有結合および開裂リンカー基からなるグループから選択され;
DはL−DOPAおよび他のL−DOPA類似体からなるグループから選択されたメンバーであり;
Wは以下の(a)および(b)からなるグループから選択され、
(a)生理的pHで負に帯電する部分(かかる部分は−COOH、−SO3H、−−S
O2H、−−P(O)(OR6)(OH)、−−OP(O)(OR6)(OH)、−−O
SO3H等からなるグループから選択される)を含む置換アルキル基およびその医薬とし
て許容される塩類、R6はアルキル、置換アルキル、アリール、および置換アリールからなるグループから選択される;および
(b)式−M−Y’−D’の基、式中Mは−CH2OC(O)−−および−CH2CH2
C(O)−−からなるグループから選択され;Y’はD’をMに共有結合させる共有結合または開裂リンカー基であり、;D’はL−DOPAおよび他のL−DOPA類似体からなるグループから選択されたメンバーである;
Xが−Y−Dであるか、Wが−M−Y’−D’であるかの少なくとも一方を満たし、
式(I)の化合物は腸胆汁酸輸送体の基質である。
【0033】
別の実施形態では、L−DOPA類似体はジまたはトリペプチド誘導体を含む。L−DOPAジペプチドまたはトリペプチド類似体の例およびそれらを製造する方法は、米国特許第3803120号および米国特許第5686423号に開示されている。ジペプチドおよびトリペプチドL−DOPAプロドラッグの経口吸収は高い経口生物学的利用能を示し、いくつかの化合物はL−DOPAの60−100倍の血漿濃度を有する。好ましい実施形態では、かかるL−DOPAプロドラッグは以下の式により表わされる:
【0034】
【化4】
式中、nは0または1であり;
Rは水素またはヒドロキシル、好ましくはRはヒドロキシルであり;
R1は水素であり;
R2は水素、1−4個の炭素原子を有するアルキル、1つの−−OH、−−SH、−−SCH3、−−NH2、−−NHC(=NH)NH2、−−COOH、フェニル、ヒドロキシ
フェニル、インドリルまたはイミダゾリル基で置換された1−4個の炭素原子を有するアルキル、1−6個の炭素原子を有する1つのカルボアルコキシル基で置換された1−4個の炭素原子を有するアルキル、好ましくはR2は水素、メチルまたはヒドロキシメチルであり;または
R1およびR2はともにトリメチレンである。
【0035】
好ましくは、式(I)のL−DOPA(2−アミノ−3−(3,4−ジヒドロキシフェニル−)プロパン酸)のジまたはトリペプチド誘導体は共にトリメチレンである。
別の実施形態では、式(I)のL−DOPA[2−アミノ−3−(3,4−ジヒドロキシフェニル−)プロパン酸]のジペプチド誘導体は、以下の式により表される:
【0036】
【化5】
式中、R3は水素であり;R4はフェニルまたはヒドロキシフェニルであり;またはR3およびR4はともにトリメチレンである。
【0037】
別の実施形態では、L−DOPA類似体は当該技術分野で周知のアミンプロドラッグを含む。L−DOPAアミン類似体の例およびそれらを製造する方法は米国特許出願公開第20060025385号およびWO/2004/069146に開示されている。1つの好ましい実施形態では、かかるL−DOPAアミン類似体は以下の式またはその医薬として許容される塩により表わされる:
【0038】
【化6】
式中、*Cは不斉炭素を示し;
R1、R2、R3およびR4は各々独立して、水素、1−30個の炭素原子を有するアルキル、1−30個の炭素原子を有するアルケニル、1−30個の炭素原子を有するアルキニル、シクロアルキル、アリール、O−カルボキシ、C−カルボキシ、カルボニル、チオカルボニル、O−カルバミル、O−チオカルバミルおよび脂肪酸アシルから成るグループから選択されるか、または
他の選択肢としてR1およびR2と、R3およびR4との少なくとも一方が五員環または六員環を形成し;
R5およびR6は各々独立して、水素、アルキル、シクロアルキル、アリールおよびホスホニルから成るグループから選択される。
【0039】
好ましいL−DOPAアミン類似体には以下のものが含まれる:R5およびR6が各々水素である化合物;R1およびR2が各々水素である化合物;R3およびR4が各々水素である化合物;R1、R2、R3およびR4の少なくとも1つ、好ましくはR3および/
またはR4がカルボニル(例えばアセチル)である化合物。現在の実施形態によるさらなる好ましい化合物は、R1、R2、R3およびR4の少なくとも1つが1−30個の炭素原子を有するアルキル、アルケニル、またはアルキニルである化合物、または他の選択肢として、R1、R2、R3およびR4の少なくとも1つが脂肪酸アシル、例えばミリスチン酸、ラウリン酸、パルミチン酸、ステアリン酸、オレイン酸、アラキドン酸、リノール酸またはリノレン酸に由来する脂肪酸アシルである。現在の実施形態によるL−DOPAアミン類似体のさらに好ましい例はα−アミノ−3,4−ジヒドロキシ−ベンゼンプロパンアミド、α−N−アセチル−3,4−ジヒドロキシ−ベンゼンプロパンアミドおよびその医薬として許容される塩類を含む。
【0040】
さらに好ましい実施形態では、本発明で使用されるL−DOPAプロドラッグおよびそれらの合成方法は、米国特許第4065566号および第4035507号に開示され、以下の式により表わされる:
【0041】
【化7】
各Rは独立して、水素原子、アシル基、
【0042】
【化8】
基、−−COピリジン基、−−CO−−R3基(R3はN,N−C1−C2ジアルキルアミノ酸またはC4−C6シクロアルキルアミノ酸の残基を表わす)から選択され、
【0043】
【化9】
R1は水酸基および−−OM基から成るグループから選択されたメンバーであり、−−Mはアルカリ金属(Na、K等)またはアンモニウムイオンであり
;
R2は
【0044】
【化10】
基、−−CO−ピリジン基、および−−CO−−R3基から選択されたメンバーであり、R3はN,N−−(C1−C2)ジアルキルアミノ酸またはC4 −C6−シクロアルキルアミノ酸
【0045】
【化11】
の残基を示す。
【0046】
本発明で使用されるさらなるL−DOPAプロドラッグおよびそれらの合成方法は米国特許第4065566号および第4035507号に開示され、以下の式およびそのHX塩(Xは以前の医薬として許容される酸付加塩陰イオン、例えば塩化物、臭化物、過塩素酸塩、メタンスルフォナート、コハク酸塩等である)により表わされる:
【0047】
【化12】
式中、Rはアシル基を表わし;
R2は水素原子を表わし;
R1は−NHCH(R4)COOR5基を表し、R4は任意の天然アミノ酸の残基を表わし、R5は水素原子、C1−C5アルキル基(例えばメチル、エチル、プロピル、ブチル、ペンチル)およびC1−C5アルキルアリール基(例えば−−CH2、−C6H5、−−
CH2−−CH2−C6H5等)から成るグループから選択されたメンバーである。
【0048】
米国特許第4065566号および第4035507号に開示された好ましいL−DOPAプロドラッグの例には、以下が含まれる:
1.グリシル−3,4−ジアセチルオキシ−L−フェニルアラニンおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
2.グリシル−3,4−ジアセチルオキシ−L−フェニルアラニン−メチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
3.3,4−ジアセチルオキシ−L−フェニルアラニル−グリシンおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
4.N−ニコチノイル−3,4−ジヒドロキシ−L−フェニルアラニンおよびそのM塩(Mはアルカリ金属を表わす)。
5.N−ニコチノイル−3,4−ジアセチルオキシ−L−フェニルアラニンおよびそのM塩(Mはアルカリ金属を表わす)
6.N−ニコチノイル−3,4−ジピバリルオキシ−L−フェニルアラニンおよびそのM塩(Mはアルカリ金属を表わす)。
7.3,4−ジアセチルオキシ−L−フェニルアラニル−グリシンおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
8.3,4−ジアセチルオキシ−L−フェニルアラニル−グリシン−メチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
9.3,4−ジアセチルオキシ−L−フェニルアラニル−グリシン−エチルエステル およびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
10.3,4−ジアセチルオキシ−L−フェニルアラニル−グリシン−ベンジルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
11.3,4−ジアセチルオキシ−L−フェニルアラニル−L−ロイシンおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
12.3,4−ジアセチルオキシ−L−フェニルアラニル−L−ロイシン−メチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
13.3,4−ジアセチルオキシ−L−フェニルアラニル−L−ロイシン−エチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
14.3,4−ジアセチルオキシ−L−フェニルアラニル−L−ロイシン−ベンジルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
15.3,4−ジアセチルオキシ−L−フェニルアラニル−L−イソロイシンおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
16.3,4−ジアセチルオキシ−L−フェニルアラニル−L−イソロイシン−メチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
17.3,4−ジアセチルオキシ−L−フェニルアラニル−L−イソロイシン−エチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
18.3,4−ジアセチルオキシ−L−フェニルアラニル−L−イソロイシン−ベンジルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
19.3,4−ジアセチルオキシ−L−フェニルアラニル−フェニルアラニンおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
20.3,4−ジアセチルオキシ−L−フェニルアラニル−フェニルアラニン−メチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
21.3,4−ジアセチルオキシ−L−フェニルアラニル−フェニルアラニン−エチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
22.3,4−ジアセチルオキシ−L−フェニルアラニル−フェニルアラニン−ベンジルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
23.グリシル−3,4−ジアセチルオキシ−L−フェニルアラニンおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
24.グリシル−3,4−ジピバリルオキシ−L−フェニルアラニンおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
25.グリシル−3,4−ジアセチルオキシ−L−フェニルアラニン−メチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
26.グリシル−3,4−ジアセチルオキシ−L−フェニルアラニン−エチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
27.グリシル−3,4−ジアセチルオキシ−L−フェニルアラニン−ベンジルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
28.L−ロイシル−3,4−ジアセチルオキシ−L−フェニルアラニンおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
29.L−ロイシル−3,4−ジアセチルオキシ−L−フェニルアラニン−メチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
30.L−ロイシル−3,4−ジアセチルオキシ−L−フェニルアラニン−エチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
31.L−ロイシル−3,4−ジアセチルオキシ−L−フェニルアラニン−ベンジルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
32.L−イソロイシル−3,4−ジアセチルオキシ−L−フェニルアラニンおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
33.L−イソロイシル−3,4−ジアセチルオキシ−L−フェニルアラニン−メチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
34.L−イソロイシル−3,4−ジアセチルオキシ−L−フェニルアラニン−エチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
35.L−イソロイシル−3,4−ジアセチルオキシ−L−フェニルアラニン−ベンジルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
36.フェニルアラニル−3,4−ジアセチルオキシ−L−フェニルアラニンおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
37.フェニルアラニル−3,4−ジアセチルオキシ−L−フェニルアラニン−メチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
38.フェニルアラニル−3,4−ジアセチルオキシ−L−フェニルアラニン−エチル およびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
39.フェニルアラニル−3,4−ジアセチルオキシ−L−フェニルアラニン−ベンジルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
40.3,4−ジアセチルオキシ−L−フェニルアラニル−3,4−ジアセチルオキシ−L−フェニルアラニンおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
41.3,4−ジアセチルオキシ−L−フェニルアラニル−3,4−ジアセチルオキシ−L−フェニルアラニン−メチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
42.3,4−ジアセチルオキシ−L−フェニルアラニル−3,4−ジアセチルオキシ−L−フェニルアラニン−エチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
43.3,4−ジアセチルオキシ−L−フェニルアラニル−3,4−ジアセチルオキシ−L−フェニルアラニン−ベンジルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
44.N−[N,N−ジメチルアミノ基]−グリシル−3,4−ジアセチルオキシ−L−フェニルアラニンおよびそのM塩(Mはアルカリ金属を表わす)。
45.N−ニコチノイル−3,4−ジニコチノイルオキシ−L−フェニルアラニンおよびそのM塩(Mはアルカリ金属を表わす)。
46.N−3−ピリジルアセチル−3,4−ジヒドロキシ−L−フェニルアラニンおよびそのM塩(Mはアルカリ金属を表わす)。
47.N−3−ピリジルアセチル−3,4−ジアセチルオキシ−L−フェニルアラニン およびそのM塩(Mはアルカリ金属を表わす)。
48.3,4−N,N−ジメチルアミノグリシル−L−フェニルアラニンメチルエステルおよびそのHX塩(Xは医薬として許容される陰イオンを表わす)。
49.N−[N,N−ジメチルアミノ基]グリシル−3,4−[N,N−ジメチルアミノグリシル]−L−フェニルアラニンおよびそのM塩(Mはアルカリ金属を表わす)。
50.N−[N,N−ジエチルアミノグリシル]−3,4−ジアセチルオキシ−L−フェニルアラニンおよびそのM塩(Mはアルカリ金属を表わす)。
【0049】
本明細書に使用する場合、用語「アルキル」は、直鎖および分岐鎖の基を有する脂肪族飽和炭化水素を指す。アルキル基は好ましくは1−30個の炭素原子、より好ましくは1−20個の炭素原子を有する。より低級のアルキル、例えば、1−6個の炭素原子のアル
キルは化合物の処方を容易にし、より高級のアルキルはBBB(脳血管関門)を通るその透過性の増強を提供する。
【0050】
アルキル基は、本発明によれば、置換されてもよいし、非置換でもよい。置換される場合、置換基は例えばシクロアルキル、アルケニル、アリール、ヘテロアリール、ヘテロアリサイクリック、ヒドロキシ、アルコキシ、アリールオキシ、チオヒドロキシ、チオアルコキシ、チオアリールオキシ、ハロ、カルボキシ、アルコキシカルボニル、チオカルボキシ、カルバミルおよびアミノであってよく、これらの用語については本明細書に定義した通りである。
【0051】
本明細書に使用する場合、用語「シクロアルキル」は、環の1つまたは複数が完全に共役したπ電子系を有していないすべてが炭素からなる単環または縮合環(つまり隣接した炭素原子対を共有する環)からなる基を指す。非制限的なシクロアルキル基の例には、シクロプロパン、シクロブタン、シクロペンタン、シクロペンテン、シクロヘキサン、シクロヘキサジエン、シクロヘプタン、シクロヘプタトリエンおよびアダマンタンである。シクロアルキル基は、本発明によれば、置換されてもよいし、非置換でもよい。置換される場合、置換基は例えばアルキル、シクロアルキル、アルケニル、アリール、ヘテロアリール、ヘテロアリサイクリック、ヒドロキシ、アルコキシ、アリールオキシ、チオヒドロキシ、チオアルコキシ、チオアリールオキシ、ハロ、カルボキシ、アルコキシカルボニル、チオカルボキシ、カルバミルおよびアミノであってよく、これらの用語については本明細書に定義した通りである。
【0052】
用語「アルケニル」は、少なくとも2つの炭素原子と少なくとも1つの炭素−炭素二重結合とから成るアルキル基を指す。
用語「アルキニル」は、少なくとも2つの炭素原子と少なくとも1つの炭素−炭素三重結合とから成るアルキル基を指す。
【0053】
上述したように、アルケニル基およびアルキニル基はいずれも好ましくは1−30個の炭素原子を有する。
「アリール」基は、完全に共役したπ電子系を有するすべてが炭素からなる単環式または縮合環の多環式(つまり隣接した炭素原子対を共有する環)からなる基を指す。非制限的なアリールの例は、フェニル、ナフタレニルおよびアントラセニルである。アリール基は、本発明によれば、置換されてもよいし、非置換でもよい。置換される場合、置換基は例えばアルキル、シクロアルキル、アルケニル、アリール、ヘテロアリール、ヘテロアリサイクリック、ヒドロキシ、アルコキシ、アリールオキシ、チオヒドロキシ、チオアルコキシ、チオアリールオキシ、ハロ、カルボキシ、アルコキシカルボニル、チオカルボキシ、カルバミルおよびアミノであってよく、これらの用語については本明細書に定義した通りである。
【0054】
用語「C−カルボキシ」は+C(=O)−OR’基 を指し、R’は、本明細書で定義
される、水素、アルキル、シクロアルキル、アルケニル、アリール、ヘテロアリール(環炭素を介して結合)またはヘテロアリサイクリック(環炭素を介して結合)である。
【0055】
用語「O−カルボキシ」はR’−−C(=O)−O−基を指し、R’は、本明細書で定義される、水素、アルキル、シクロアルキル、アルケニル、アリール、ヘテロアリール(環炭素を介して結合)またはヘテロアリサイクリック(環炭素を介して結合)である。
【0056】
用語「カルボニル」は−C(=O)−R’基を指し、R’は上記に定義した通りである。
用語「チオカルボニル」は−C(=S)−R’基を指し、R’は上記に定義した通りで
ある。
【0057】
「O−カルバミル」基は−OC(=O)−NR’R”基を指し、R’は上記に定義した通りであり、R”はR’に対して定義された通りである。
「O−チオカルバミル」基は−OC(=S)−NR’R”基を指し、R’およびR”は上記に定義した通りである。
【0058】
「脂肪酸アシル」は、R'''C(=O)−(O)−基を指し、R'''は少なくとも10個の炭素原子を有する飽和または不飽和の炭化水素鎖である。
用語「アルコキシ」は、−O−アルキルおよび−O−シクロアルキル基の両方を指し、上記に定義した通りである。アルコキシ基の代表的な例はメトキシ、エトキシ、プロポキシおよびtert−ブトキシを含む。
【0059】
−O−アルキルおよびO−シクロアルキル基は、本発明によれば、置換されてもよいし、非置換でもよい。置換される場合、置換基は、例えばシクロアルキル、アルケニル、アリール、ヘテロアリール、ヘテロアリサイクリック、ヒドロキシ、アルコキシ、アリールオキシ、チオヒドロキシ、チオアルコキシ、チオアリールオキシ、ハロ、カルボキシ、アルコキシカルボニル、チオカルボキシ、カルバミルおよびアミノであってよく、これらの用語については本明細書に定義した通りである。
【0060】
用語「チオアルコキシ」は−S−アルキル基および−S−シクロアルキル基の両方を指し、本明細書に定義した通りである。
用語「ヒドロキシ」は−OHグループを指す。
【0061】
用語「チオヒドロキシ」は−SH基を指す。
「アリールオキシ」基は−O−アリール基および−O−ヘテロアリール基の両方を指し、本明細書に定義した通りである。
【0062】
「チオアリールオキシ」基は−S−アリール基および−S−ヘテロアリール基の両方を指し、本明細書に定義した通りである。
用語「アミノ」は−NR’R”基を指し、R’およびR”は上記に定義した通りである。
【0063】
用語「アルコキシカルボニル」は、本明細書では互換的に「カルバルコキシ」とも呼ばれるが、上記に定義した通りカルボキシ基を指し、R’は水素ではない。
用語「ヘテロアリール」基は、環内に、例えば窒素、酸素および硫黄等の1つまたは複数の原子を有し、さらには完全に共役したπ電子系を有する単環または縮合環(つまり隣接した原子対を共有する環)を含む。ヘテロアリール基の非制限的な例には、ピロール、フラン、チオフェン、イミダゾール、オキサゾール、チアゾール、ピラゾール、ピリジン、ピリミジン、キノリン、イソキノリンおよびプリンが含まれる。
【0064】
「ヘテロアリサイクリック」基は、環内に、例えば窒素、酸素および硫黄等の1つまたは複数の原子を有する単環または縮合環を指す。環はさらに1つまたは複数の二重結合を有してもよいが、環は完全に共役したπ電子系を有しない。
【0065】
用語「ハロ」はフッ素、塩素、臭素またはヨウ素原子を指す。
用語「ホスホニル」は−P(=O)(OR’)2基を指し、R’は上記に定義した通り
である。
【0066】
本発明の第1または第2態様の任意の実施形態では、方法は、L−DOPAおよびL−
DOPA類似体からなるグループから選択された2つ以上の化合物を投与することを含んでもよい。別の好ましい実施形態では、方法はさらに、カルビドパまたはベンセラジド等のL−アミノ酸脱炭酸酵素阻害剤を含むがそれらに限定されるわけではないさらなる治療化合物を患者へ投与することを含んでもよい。かかるL−アミノ酸脱炭酸酵素阻害剤は、例えば、L−DOPAの血漿半減期を増大させ、かつL−DOPAからドーパミンへの末梢での変換を減少させるために使用することが可能であり、これはL−DOPA処理の副作用を低減する。別の実施形態では、方法は、AMDを治療またはAMDの進行を制限するのに有効な1つまたは複数の別の化合物を投与することをさらに含んでもよく、かかる化合物には、抗血管内皮細胞増殖因子(VEGF)剤等の抗血管新生治療薬、ラニビツマブまたはベバシツマブ等のVEGF抗体(またはその断片)、またはペガプタニブ等のVEGFアプタマーが含まれるが、それらに限定されるわけではない。別の実施形態では、L−DOPAまたはL−DOPA類似体は、L−DOPAまたはL−DOPA類似体を含有する栄養剤のような、より複雑な混合物中に存在してもよい。
【0067】
好ましい実施形態では、本明細書で説明している任意の1つまたは複数L−DOPAおよび/またはL−DOPA類似体を、栄養補助食品(ダイエタリーサプリメント)の形で使用してもよい。かかるサプリメントは、AMDの治療またはAMDの進行の制限に有益であり得る1つまたは複数のさらなる成分と組み合わせてもよい。1つの好ましい実施形態では、L−DOPAおよび/またはL−DOPA類似体は、ビタミンC源、ビタミンE源、ビタミンA源、亜鉛源、および銅源の組み合わせと組み合わされ、それらはAMDの治療に有用なものとして米国特許第6,660,297号に開示されており、この米国特許第6,660,297号全体は本明細書に援用する。任意の適切な量のかかる追加の成分の各々を、本発明の方法を実行する際にL−DOPAおよび/またはL−DOPA類似体と組み合わせて使用することが可能である。さらに好ましい実施形態では、かかる組み合わせはさらに、好ましくは1日当たり1mg−100mgの間;1mg−50mgの間、2mg−25mgの間、または2mg−10mgの間の、網膜保護効果を提供するのに適切な量のルテインおよび/またはゼアキサンシンをさらに含む。上記の好ましい実施形態のうちのいずれかのさらに好ましい実施形態では、かかる組み合わせはさらに、好ましくは1日当たり250mg−1000mgの間、300mg−750mgの間、350mg−750mgの間、または350mg−650mgの間の、網膜保護効果を提供するのに適切な量にドコサヘキサエン酸(DHA)および/またはエイコサペンタエン酸(EPA)をさらに含む。AMD患者を治療するかかる組成物の使用方法については、例えばウェブサイトwww.areds2.org/およびその中のリンクで検討されている。
【0068】
アスコルビン酸はビタミンCの好ましい供給源であるが、例えばアスコルビン酸ナトリウムのような他の供給源を代わりに使用してもよい。
D1−α酢酸トコフェロールはビタミンEの好ましい供給源であるが、例えばトリメチル酢酸トコフェリルおよび/またはコハク酸ビタミンEのような他の供給源を代わりに使用してもよい。
【0069】
β−カロチンは容易に商業的に入手できるため対象組成物に好ましいが、ビタミンAの代替カロテノイド代用形も同様に使用することが可能である。
亜鉛は、亜鉛元素を最も濃縮した形で供給し、消化器系によく許容されるため対象錠剤には酸化亜鉛の形が好まれるが、例えばグルコン酸亜鉛のような他の亜鉛の形式も代わりに使用でき、または対象組成物中の酸化亜鉛と組み合わせて使用してもよい。
【0070】
酸化第二銅の形をした銅は、亜鉛誘導性の銅欠乏貧血の予防を促すのに対象錠剤に好ましいが、例えばグルコン酸銅のような他の銅の形式も代わりに使用でき、または対象組成物中の酸化第二銅と組み合わせて使用してもよい。
【0071】
好ましい実施形態では、これらの他の成分の各々の量(1日当たりに基づく)は、以下の通りである:
450mg−600mgの間のビタミンC(一日摂取許容量(RDA)の約7−10倍);
400IU−540IUの間のビタミンE(RDAの約13−18倍);
17.2mg−28mgの間のβカロチン(ビタミンAのRDAの約6−10倍;βカロチンはビタミンAのプロドラッグである。);
68mg−100mgの間の亜鉛(亜鉛のRDAの約4−7倍);および
1.6mg−2.4mgの間の銅。
【0072】
さらに好ましい実施形態では、これらの他の成分の各々の量(1日当たりに基づく)は、以下の通りである:
500mgのビタミンC;
400IUビタミンE;
0mgまたは15mgのβカロチン;
25mgまたは80mgの酸化亜鉛;
2mgの酸化第二銅。
【0073】
本明細書の別の実施形態と組み合わせてもよいさらに好ましい実施形態では、目の健康を維持するのに有用であると考えられている他の成分を同様にL−DOPAおよび/またはL−DOPA類似体と組み合わせても良く、それには例えば、保護網膜効果をさらに提供するのに適切な量、好ましくは1日当たり1mg−100mgの間、1mg−50mgの間、2mg−25mgの間、または2mg−10mgの間のルテインおよび/またはゼアキサンシン;保護網膜効果をさらに提供するのに適切な量、好ましくは1日当たり250mg−1000mgの間、300mg−750mgの間、350mg−750mgの間、または350mg−650mgの間のドコサヘキサエン酸(DHA)および/またはエイコサペンタエン酸(EPA)が含まれるがそれらに限定されない。任意選択で使用可能なさらなる化合物の例には、α−リポ酸や、オリゴマーのアントシアニジン、アントシアノシドおよびその組み合わせを含むがそれらに限定されないフェノール化合物が含まれるが、それらに限定されない。
【0074】
L−DOPAおよび/またはL−DOPA類似体は、通常、医薬組成物の形で個別にまたは組み合わせて投与することが可能である。かかる組成物は医薬の分野で周知の方法で調製される。L−DOPAおよび/またはL−DOPA類似体は、唯一の活性薬物として投与してもよいし、または本発明の方法を実施するのに有用な1つまたは複数の別の化合物と組み合わせて使用してもよく、かかる別の化合物にはVEG−F等の抗血管治療薬や、カルビドパおよびベンセラジド等のL−アミノ酸脱炭酸酵素阻害剤が含まれるが、それらに限定されるわけではない。併用投与される場合、組み合わせは、同時または異なる時期に与えられる別々の組成物として処方してもよいし、単一の組成物として与えてもよい。
【0075】
L−DOPAおよび/またはL−DOPA類似体は、固体形式(顆粒剤、粉末剤または坐剤を含む)または液体形式(例えば溶液、懸濁剤または乳剤)に構成されてよい。L−DOPAおよび/またはL−DOPA類似体は種々の溶液に適用され、滅菌等の従来の医薬操作に曝されてもよく、および/または防腐剤、安定剤、湿潤剤、乳化剤、緩衝液等の以前の佐剤(アジュバント)を含んでもよい。
【0076】
L−DOPAおよび/またはL−DOPA類似体は、任意の適切な経路により投与されてよく、それには経口、局所(点眼薬および眼軟膏剤を含むが、それらに限定されない)、非経口、鼻腔内、肺、または直腸が含まれるが、それらに限定されるわけではなく、以
前の非毒性の医薬として許容される担体、佐剤および賦形剤を含む投薬単位製剤として投与される。本明細書に使用する場合、用語「非経口」は経皮、皮下、血管内(例えば静脈内)、筋肉内、または髄腔内注射または注入技術等を含む。さらに、本発明の化合物と医薬として許容される担体とを含む医薬製剤が提供される。L−DOPAおよび/またはL−DOPA類似体は、1つまたは複数の非毒性医薬として許容される担体、希釈剤および/または佐剤、ならびに所望の場合には別の有効成分と共に存在してよい、L−DOPAおよび/またはL−DOPA類似体を含む医薬組成物は、例えば錠剤、トローチ、菱形錠剤、水性または油性の懸濁剤、分散粉末剤または顆粒剤、乳剤(エマルション)、硬質または軟質カプセル剤、またはシロップ剤もしくはエリキシル剤として、経口使用に適した形式であってもよい。
【0077】
点眼薬は当該技術分野の任意の技術を使用して調製することが可能であり、それには塩化ナトリウムまたは濃グリセリン等の等張化剤、リン酸ナトリウムまたは酢酸ナトリウム等の緩衝液、ポリオキシエチレンソルビタンモノオレイン酸等の界面活性剤、ポリオキシル40ステアリン酸またはポリオキシエチレン硬化ヒマシ油、クエン酸ナトリウムまたはエデト酸ナトリウム等の安定化剤、塩化ベンザルコニウムまたはパラベン等の防腐剤の必要に応じた使用が含まれるが、それらに限定されるわけではない。点眼薬のpHは好ましくは4−8の範囲である。眼軟膏剤は、白色ワセリンまたは流動パラフィン等の一般に使用されている基剤と共に調製することが可能である。
【0078】
経口使用向けのL−DOPAおよび/またはL−DOPA類似体は、医薬組成物の製造ための当該技術分野の任意の周知方法により調製可能であり、かかる組成物は、口当たりのよい調製物を提供すべく甘味剤、香料、着色剤および防腐剤からグループから選択された1つまたは複数の薬物を含んでもよい。錠剤は、錠剤の製造に適した非毒性の医薬として許容される添加剤と組み合わせて、L−DOPAおよび/またはL−DOPA類似体を含む。これらの添加剤は、例えば炭酸カルシウム、炭酸ナトリウム、ラクトース、リン酸カルシウムまたはリン酸ナトリウムのような不活性希釈剤;例えばコーンスターチまたはアルギン酸等の顆粒および崩壊剤;例えばデンプン、ゼラチン、アラビアゴム等の結合剤、例えばステアリン酸マグネシウム、ステアリン酸、または滑石等の潤滑剤であってよい。錠剤はコーティングされなくてもよいし、既知の技術によりコーティングされてもよい。ある場合には、かかるコーティングが、胃腸管での崩壊および吸収を遅らせる既知の技術により調製され、それにより長期間にわたる持続作用を提供する。例えば、グリセリルモノステアリン酸またはグリセリルジステアリン酸のような時間遅延材料を使用することが可能である。
【0079】
経口使用のための製剤は、L−DOPAおよび/またはL−DOPA類似体が不活性の固体希釈剤(例えば炭酸カルシウム、リン酸カルシウム、カオリン)と混合された硬質カプセル剤として、または有効成分が水性または油性媒体(例えばピーナッツ油、流動パラフィン、オリーブ油)と混合された軟質ゼラチンカプセル剤として与えられてもよい。
【0080】
水性懸濁液は、水性懸濁液の製造に適した賦形剤と混合されたL−DOPAおよび/またはL−DOPA類似体を含む。かかる賦形剤は、懸濁化剤(例えばナトリウムカルボキシメチルセルロース、メチルセルロース、ヒドロプロピル−メチルセルロース、アルギン酸ナトリウム、ポリビニルピロリドン、トラガカントゴム、およびアラビアゴム)であり、分散剤または湿潤剤は天然のリン脂質(例えばレシチン)または脂肪酸を有するアルキレンオキシド縮合物(例えばポリオキシエチレンステアリン酸)、長鎖脂肪族アルコールを有するエチレンオキシド縮合物(例えばヘプタデカエチレンオキシセタノール)、または脂肪酸およびヘキシトールに由来する部分エステルを有するエチレンオキシド縮合物(例えばポリオキシエチレンソルビトールモノオレイン酸)、または脂肪酸およびヘキシトール無水物に由来する部分エステルを有するエチレンオキシド縮合物(例えばポリエチレ
ンソルビタンモノオレイン酸)であってよい。水性懸濁液は、1つまたは複数の防腐剤、(例えばエチルまたはn−プロピル p−オキシ安息香酸)、1つまたは複数の着色剤)、1つまたは複数の香料、および1つまたは複数の甘味剤(例えばスクロースまたはサッカリン)を含んでよい。
【0081】
油性懸濁液は、のL−DOPAおよび/またはL−DOPA類似体を、植物油(例えばピーナッツ油、オリーブ油、胡麻油、ココナツオイル)または鉱油(例えば流動パラフィン)に懸濁させることにより調製可能である。油性懸濁液は、増粘剤(例えば密蝋、固形パラフィン、またはセチルアルコール)を含んでもよい。口当たりのよい経口用製剤を与えるために、甘味剤と香料が添加されてもよい。これらの組成物は、アスコルビン酸等の抗酸化剤の添加により保存されてもよい。
【0082】
水の添加による水性懸濁液の調製に適した分散可能な粉末剤および顆粒剤は、分散もしくは湿潤剤、懸濁化剤、および1つまたは複数の防腐剤と混合して、有効成分を提供する。適切な分散もしくは湿潤剤または懸濁化剤は、既に上述したものにより例示されている。追加の添加剤(例えば甘味剤、香料、および着色剤)がさらに存在してもよい。
【0083】
本発明の方法に使用される医薬組成物は、水中油型エマルションの形をしていてもよい。油相は、植物油、鉱油、またはそれらの混合物であってよい。適切な乳化剤は天然ゴム(例えばアラビアゴムまたはトラガカントゴム)、天然のリン脂質(例えば、大豆、レシチン、および脂肪酸とヘキシトール、無水物とに由来するエステルまたは部分エステル、例えばソルビタンモノオレイン酸)、および該部分エステルのエチレンオキシドとの縮合物(例えばポリオキシエチレンソルビタンモノオレイン酸)であってよい。エマルションは甘味剤および香料をさらに含んでもよい。
【0084】
シロップ剤とエリキシル剤は、甘味剤(例えばグリセリン、プロピレングリコール、ソルビトール、グルコース、スクロース)と共に調製されてもよい。かかる調製物は解乳化剤、防腐剤、香料、および着色剤をさらに含んでもよい。医薬組成物は、無菌の注射可能な水性または油性の懸濁剤の形をしていてもよい。かかる懸濁剤は、上述した適切な分散または湿潤剤と懸濁剤とを使用して周知技術に従って製剤化されてよい。無菌の注射可能な調製物は。非毒性・非経口の許容される希釈剤または溶剤の形をした無菌注射剤溶液または懸濁剤であってもよい(例えば1,3−ブタンジオールの溶液として)。使用可能な許容される賦形剤および溶剤には、水、リンゲル液および生理食塩液がある。さらに、無菌の固定油が溶剤または懸濁剤として従来通りに使用される。この目的のためには、合成モノグリセリドまたはジグリセリドを含めた任意無味無臭の固定油が使用されてもよい。さらに、オレイン酸等の脂肪酸は注射物質の調製に用途がある。
【0085】
L−DOPAおよびL−DOPA類似体の鼻腔内投与のための特定方法が当該技術分野で周知であり;例えばKao et al., Pharmaceutical Research 17(8):978-984 (2000)を参照されたい。
【0086】
用量域は、化合物の選択、投与経路、製剤の性質、患者の症状の性質、および関与する医師の判断によって決まる。例えば、経口投与は、静脈内注射による投与よりも高用量が必要であると予想される。当該技術分野でよく理解されるように、かかる投薬レベルの違いは、最適化のための標準の経験的ルーチンを使用して調節することが可能である。ある実施形態では、L−DOPAおよび/またはL−DOPAS類似体は、10mg/日から1500mg/日の間の用量で投与することが可能であり、種々の好ましい実施形態では、投与が20mgと1200mg/日の間、50mgと1000mg/日の間、100mgと500mg/日の間、および200mgと400mg/日の間であってよい。
【0087】
本明細書で説明している化合物を含む医薬組成物は、それを必要とする個体に投与される。好ましい実施形態では患者は哺乳動物であり、より好ましい実施形態では患者はヒトである。治療の用途では、組成物は本発明の方法を実施するのに十分な量で投与される。これらの使用に有効な量は、化合物の性質(比放射能等)、投与経路、疾患の段階ならびに重篤度、患者の体重ならびに健康の全身状態、および規定する医師の判断を含むがこれらに限定されない要因によって決まる。活性化合物は広い用量域にわたって有効であるが、実際に投与される化合物の量は上記の関連する状況に照らして医師により決定されるであろうことが理解される。したがって、上記の用量域はいかようにも本発明の範囲を制限することを意図しない。
【0088】
第3態様では、本発明は以下の(a)および(b)を含む組成物を提供する:
(a)AMDの治療またはAMDの進行の制限に有効な量のL−DOPAまたはL−DOPA類似体;
(b)AMDの治療またはAMDの進行の制限に有効な量の、ビタミンC源、ビタミンE源、ビタミンA源、亜鉛源および銅源を含む組成物。
【0089】
組成物中のL−DOPAおよび/またはL−DOPAS類似体の量は、10mg/日から1500mg/日の間の用量での投与を与えるのに適しており、種々の好ましい実施形態では、投与は20mgと1200mg/日の間、50mgと1000mg/日の間、100mgと500mg/日の間、および200mgと400mg/日の間である。
【0090】
アスコルビン酸は対象錠剤中のビタミンCの好ましい供給源であるが、例えばアスコルビン酸ナトリウムのような他の供給源を代わりに使用してもよい。
D1−α酢酸トコフェロールはビタミンEの好ましい供給源であるが、例えばトリメチル酢酸トコフェリルおよび/またはコハク酸ビタミンEのような他の供給源を代わりに使用してもよい。
【0091】
β−カロチンは容易に商業的に入手できるため対象組成物に好ましいが、ビタミンAの代替カロテノイド代用形も同様に使用することが可能である。
亜鉛は、亜鉛元素を最も濃縮した形で供給し、消化器系によく許容されるため対象錠剤には酸化亜鉛の形が好まれるが、例えばグルコン酸亜鉛のような他の亜鉛の形式も代わりに使用でき、または対象組成物中の酸化亜鉛と組み合わせて使用してもよい。
【0092】
酸化第二銅の形をした銅は、亜鉛誘導性の銅欠乏貧血の予防を促すのに対象錠剤に好ましいが、例えばグルコン酸銅のような他の銅の形式も代わりに使用でき、または対象組成物中の酸化第二銅と組み合わせて使用してもよい。
【0093】
本発明のこの第3態様の1つの好ましい実施形態では、組成物(b)は以下の量の各成分の消化摂取を許容するのに適した処方を与える:
アスコルビン酸:少なくとも450mg;
dIαトコフェリル酢酸:400IU;
βカロチン:17.2mg;
酸化亜鉛:68mg;
酸化第二銅:1.6mg
本発明のこの第3態様の1つの好ましい実施形態では、組成物(b)は以下の量の各成分の摂取を許容するのに適した処方を与える:
500mgのビタミンC;
400 IUのビタミンE;
0mgまたは15mgのβカロチン;
25mgまたは80mgの酸化亜鉛;
2mgの酸化第二銅。
【0094】
上記に特定したような対象組成物の好ましい毎日用量が、上記に開示したような任意の適切な投与経路を介して、1回、2回、3回または4回以上の投薬形式で投与されてよい。好ましい実施形態では、投薬形式は本明細書で説明しているかかる投薬形式の任意の実施形態によれば、経口または局所の投薬形式である。別の好ましい実施形態では、対象組成物の毎日用量は、1日2回の1回分の投与形式、1日2回の2回分の投与形式、または1日合計4回分の投与形式で与えられる。1日1回の日用量合計をとるのと比較して、1回の用量当たりの1つまたは複数の投薬形式中、日用量合計の半分の1日2回投与は、主成分の吸収を改善すると共に、主成分の血中濃度をより良好に維持する。従って、対象組成物の好ましい製剤の1日に2回の投薬形式が毎日摂取される場合、各投薬形式は好まし
くは経口投与時に少なくとも約225mgのアスコルビン酸、約200IUのdl−αトコフェリル酢酸、約8.6mgのβ−カロチン、約34mgの酸化亜鉛および約0.8mgの酸化第二銅を提供するよう調剤される。対象組成物の好ましい製剤の4つの錠剤が毎日摂取される場合、各錠剤は好ましくは少なくとも約112.5mgのアスコルビン酸、約100IUのdl−αトコフェリル酢酸、約4.3mgのβ−カロチン、約17mgの酸化亜鉛、約0.4mgの酸化第二銅、および5mg−750mgの間のL−DOPAおよび/またはL−DOPA類似体を提供するように調剤される。
【0095】
別の好ましい実施形態では、組成物は以下を含む:
(a)5mgから1500mgの間のL−DOPAまたはL−DOPA類似体;
(b)450mgから600mgの間のビタミンC(一日摂取許容量(RDA)の約7−10倍);
(c)400IUから540IUの間のビタミンE(RDAの約13−18倍);
(d)17.2mgから28mgの間のβカロチン(ビタミンAのRDAの約6−10倍;βカロチンはビタミンAのプロドラッグである。);
(e)68mgから100mgの間の亜鉛(亜鉛用のRDAの約4−7倍);および
(f)少なくとも1.6mgの銅。
【0096】
種々の好ましい実施形態では、組成物は10mgから1200mgの間;25mgから1000mgの間;50mgから500mgの間;または100mgから400mgの間のL−DOPAまたはL−DOPA類似体を含んでもよい。
【0097】
本明細書の別の実施形態と組み合わせてもよいさらに好ましい実施形態では、目の健康を維持するのに有用であると考えられている他の成分を同様にL−DOPAおよび/またはL−DOPA類似体と組み合わせても良く、例えば、保護網膜効果をさらに提供するのに適切な量、好ましくは1日当たり1mg−100mgの間、1mg−50mgの間、2mg−25mgの間、または2mg−10mgの間のルテインおよび/またはゼアキサンシン;保護網膜効果をさらに提供するのに適切な量、好ましくは1日当たり250mg−1000mgの間、300mg−750mgの間、350mg−750mgの間、または350mg−650mgの間のドコサヘキサエン酸(DHA)および/またはエイコサペンタエン酸(EPA)が含まれるがそれらに限定されない。記載された量を提供する特定の投薬形式に必要な量は、本明細書の教示および1日当たりに投与される剤形の数基づいて当業者には決定することが可能である。
【0098】
第4の態様では、本発明は、AMDを治療する化合物を識別するin vitroの方法であって、細胞を試験化合物と接触させることからなり、細胞は、
(a)OA1を発現している第1の細胞集団;および任意選択で
(b)OA1を発現していない第2の細胞集団;を含み、
(c)以下の(i)および(ii)のうちの一方又は両方を増大させる試験化合物を陽
性試験化合物と識別し;
(i)(A)試験化合物と接触させない第1の細胞集団における色素上皮誘導因子(PEDF)発現および(B)第2の細胞集団におけるPEDF発現におけるPEDF発現のうちの一方又は両方に対する、第1の細胞集団におけるPEDF発現;
(ii)(A)試験化合物と接触させない第1の細胞集団における細胞内カルシウム濃度および(B)第2の細胞集団における細胞内カルシウム濃度のうちの一方又は両方に対する、第1の細胞集団における細胞内カルシウム濃度;
陽性試験化合物は、AMDの治療およびAMDの進行の制限のうちの少なくとも一方を行う候補化合物である方法を提供する。
【0099】
上述したように、ヒトOA1(配列番号1−2、NP000264.1)はGタンパク質共役受容体であり、本願発明者は本明細書でL−DOPAをOA1リガンドと確認した。より詳細に以下に開示するように、本願発明者はL−DOPAを通じてリンクされたOA1とチロシナーゼの間の自己分泌ループの存在を発見した。このループは、少なくとも1つの非常に効き目のある網膜神経栄養因子(PEDF)の分泌と、細胞内カルシウム濃度の増加とを含む。OA1は、その下流での作用が進行中の網膜の空間のパターニングを左右する選択的L−DOPA受容体である。したがって、OA1経路の刺激を介してPEDF発現および/または細胞内カルシウム濃度を選択的にアップレギュレートする試験化合物は、AMDの治療および/またはAMDの進行の制限を行うための候補化合物である。本発明のこの態様の方法は、以下のものを含むが、それらに限定されない任意のOA1同族体を用いて実施することが可能である:
マウス:配列番号3−4(NM_010951);
アフリカツメガエル(Xenopus tropicalis):配列番号5−6(NM_001011018);
雌ウシ:配列番号7−8(XM_001506318);
ラット:配列番号9−10(NM_001106958);
カモノハシ:配列番号11−12(XM_001506318);
アフリカツメガエル(Xenopus laevis):配列番号13−14 (NM_001096842)
ニワトリ:配列番号15−16(XM_416848);
ゼブラフィッシュ:配列番号17−18(NM_200822);
チンパンジー:配列番号19(XR_025625);
アカゲザル:配列番号21−22(XM_001090139);および
マカクザル:配列番号23(BV209253)。
【0100】
PEDFは色素上皮誘導因子であり(Exp Eye Res 53:411-414)、神経感覚網膜の発達を変更し、かつ血管増殖を阻害する可能性を備えた公知の神経栄養因子である。本発明のこの態様の方法は、以下のものを含むが、それらに限定されない任意のPEDF同族体を用いて実施することが可能である:
ヒト:配列番号25−26(NM_002615);
ラット:配列番号27−28(NM_031356);
キンカチョウ:配列番号29−30(XM_002197419);
ウマ:配列番号31−32(NM_001143954);
アフリカツメガエル(Xenopus tropicalis):配列番号33−34(NM_203755);
マウス:配列番号35−36(NM_011340);
大西洋サケ:配列番号37−38(NM_001140334);
ヒツジ:配列番号39−40(NM_001139447);
モルモット:配列番号41−42(EF679792);
雌ウシ:配列番号43−44(NM_174140);
イノシシ:配列番号45−46(NM_001078662);
カモノハシ:配列番号47−48(XM_001507128);
オオカミ:配列番号49−50(NM_001077588);
マカクザル:配列番号51−52(AB174277);
チンパンジー:配列番号53−54(XM_001154665);
アカゲザル:配列番号55−56(XM_001117361);および
ヒラメ:配列番号57−58(DQ115406)。
【0101】
細胞の第1集団および第2集団は任意の適切な真核細胞の種類であってよく、第1の細胞集団は細胞表面受容体タンパク質としてOA1を発現することが可能である。1つの好ましい実施形態では、細胞の第1集団および第2集団は、マウス、ラット、ハムスターまたはヒト細胞等の哺乳類由来の細胞である。特に細胞内カルシウム濃度の分析に関する実施形態に使用される場合、現在までに試験されているすべての真核細胞は、本発明の方法を実施するのに適していることが分かっている。細胞内カルシウム信号および/またはPEDF分泌のうちの一方又は両方に関連して本明細書に開示されたOA1シグナル伝達経路の保存に関して現在までに試験されている細胞の種類には、MCF7(乳癌上皮細胞)、COS細胞(腎臓繊維芽細胞)、MDCK細胞(腎臓上皮)、CHO(チャイニーズハムスター卵巣)、マウスRPEおよび3T3(マウス繊維芽細胞)、ならびに以下の実施例で開示されたものが含まれる。かかる細胞は種々の供給源(メリーランド州Walkersville所在のLifeLine Cell Technology社;ATCC(アメリカンタイプカルチャーコレクション)から入手可能であるか、または当該技術分野で周知かつ以下に説明する方法を用いて単離可能である。
【0102】
1実施形態では、細胞表面受容体タンパク質としてOA1を発現している第1の細胞集団の第1部分を試験化合物と接触させ、第1の細胞集団の第2部分は試験化合物と接触させず、第2部分と比較して第1部分におけるPEDFの発現および/または細胞内カルシウム濃度を増加させる化合物は、AMDの治療および/またはAMDの進行の制限を行う候補化合物である。
【0103】
代わりに、方法は、細胞表面受容体タンパク質としてOA1を発現しない第2の細胞集団の使用を含んでもよく、第2の細胞集団と比較して第1の細胞集団におけるPEDFの発現および/または細胞内カルシウム濃度を増加させる化合物は、AMDの治療および/またはAMDの進行の制限を行う候補化合物である。好ましい実施形態では、第1および第2の細胞集団は同じ細胞の種類であり、第1の細胞集団はOA1を発現するよう遺伝子組み換えされ、第2の細胞集団は遺伝子組み換えされない。この実施形態では、第2の細胞集団は第1の細胞集団と同様の発現ベクターでトランスフェクトされてよく、かかるトランスフェクションは、空の発現ベクター(つまりトランスフェクト細胞中のベクターから得られる発現タンパク質がない)または細胞膜へ適切に挿入されない切断型または変異型OA1を発現することが可能な発現ベクターでのトランスフェクションを含む。代わりに、細胞はOA1シグナル伝達に不活性であることが知られているOA1変異型をコードする発現ベクターまたは異なるGPCR経路(例えばcAMP)を介してシグナル伝達することが可能な遺伝子組み換えされたOA1をコードする発現ベクターでトランスフェクトすることも可能である。
【0104】
例えば、リガンド結合を変更せずにその活性を変更するために、OA1の7つの膜貫通領域を異なる細胞内c末端尾部と融合させることが可能である。
本明細書に使用する場合、「PEDF発現の増加」または「細胞内カルシウム濃度の増加」とは、第2の細胞集団で見られるよりも大きいアッセイの経過中の任意の第1の細胞集団における(または第1集団の第2部分に関する第1部分における)PEDF発現の増加または細胞内カルシウム濃度の増加である。この方法は、化合物が対照で見られるより
も大きいPEDF発現の増加または細胞内カルシウム濃度の増加を促進する限り、対照に対するPEDF発現の増加または細胞内カルシウム濃度の増加の特定量を要求しない。好ましい実施形態では、増加は標準的な統計学的測定により測定される統計学的に有意な増加である。
【0105】
細胞内カルシウム濃度の決定は当該技術分野で周知であり、Fura−2細胞ローディングおよびレシオメトリックイメージングを使用した方法の例が、以下の実施例に記載される。しかしながら、細胞内カルシウム濃度は当業者に周知のいかなる方法を使用して測定してもよく、それにはFu laTMI(以下を参照)およびFLIPerTMを用いたハイスループット法が含まれるが、それらに限定されるわけではない。
【0106】
細胞集団におけるPEDFの発現レベルの決定は、以下に説明するような当該技術分野の任意の技術を使用してもよく、それにはmRNAハイブリダイゼーション(ノーザンブロット、スロットブロット等)、適当なプライマー組を用いた逆転写ポリメラーゼ連鎖反応(PCR)技術、蛍光in situハイブリダイゼーション、およびPEDFを発現/分泌する馴化細胞培地での抗体検出(ウェスタンブロット、免疫細胞化学、ELISA)が含まれるが、これらに限定されない。PEDF抗体は市販されている(例えばマサチューセッツ州Cambridge所在のAbcam社)。タンパク質分析は(PEDFが発現タンパク質であるため)馴化細胞培地上で行われて良く、すべてのアッセイは細胞内PEDFタンパク質/mRNA生産で行うことが可能である。別の実施形態では、PEDFプロモータから検知信号(GFP、ルシフェラーゼ等)の発現を駆動する発現ベクターを有する組換え細胞を生成することが可能である。かかる細胞は、検知可能な蛍光強度の測定またはPEDFプロモータにより駆動される他の信号の測定を介して「PEDF発現」が測定される第1の細胞集団として使用することが可能である。
【0107】
この第4の態様に使用される場合、用語「接触」は、第1の細胞集団の細胞表面に発現されたOA1へのOA1リガンドの結合を促進するのに適した条件下でのin vitroの接触を意味する。本明細書に使用する場合、「接触」は培養の開始時に起こるか、または細胞集団の培養の開始後の任意の時間に起こる。PEDF発現および/または細胞内カルシウム濃度は、所与のアッセイのために適切に決定された試験化合物と接触した後にいつでも測定することが可能である。1実施形態では、経時変化を測定し、接触前に複数のレベルを測定し、接触後に種々の時間で測定する。種々の実施形態で、接触後のカルシウムシグナルの測定が5秒と60分の間でなされ;より好ましくは10秒と30分の間、10秒と10分の間、および10秒と5分の間、10秒と1分の間、および10秒と30秒の間になされる。種々の実施形態では、PEDF発現の測定は1分から72時間の間の範囲にわたり、EDF分泌の分析はPEDF mRNA発現、PEDFの細胞内タンパク質発現、またはPEDFプロモータにより駆動される検出信号の発現の分析よりも後の測定が必要とされる。
【0108】
所与のアッセイに対して、任意の適切な細胞培養条件が使用可能である。1つの好ましい実施形態では、細胞における内因性L−DOPAの生産を低減させ、かつ細胞表面に存在するOA1の量を維持するために(リガンド結合の際にOA1がエンドソームに内在化するため)、接触が非常に低濃度のチロシン(0.1μmと10μmの間のチロシン)を有する細胞培地またはチロシンがない細胞培養培地で起こる。1つの好ましい実施形態では、細胞表面でのOA1発現および局在化を最大限にするために、細胞は、試験化合物との接触前には低チロシン培地で培養され、次に試験化合物と接触させるためにチロシンを含まない培地に播かれる。別の好ましい実施形態では、接触が低チロシン培地で起こる。上記に開示された別の実施形態と組み合わせてもよい別の好ましい実施形態では、培地は、チロシンからのL−DOPAの細胞内生産を制限する、フェニルチオ尿素を含むがこれに限定されないチロシン阻害剤を含む。色素性細胞を使用する場合にこの実施形態は特に
好ましい。
【0109】
別の好ましい実施形態では、方法は、OA1への結合の競争相手として、L−DOPA、チロシンおよびドーパミンのうちの1つまたは複数の使用をさらに含んでもよい。この実施形態は、試験化合物をOA1リガンドであると識別した後に実行されてもよいし、またはOA1に結合する試験化合物の初期スクリーニングで実行されてもよい。以下の実施例で示されるように、1mM以上の濃度では、チロシンとドーパミンはOA1に対する結合に関しL−DOPAと競争し得る。したがって、1mMから100mMの間、好ましくは1 mMと50mMの間、または1mMと25mMの間の濃度のチロシンおよび/またはドーパミンを使用した競争アッセイを使用して、試験化合物がOA1経路経由で作用していることをさらに確認できると共に、L−DOPAの置換と比較したチロシンおよびドーパミンがOA1に結合する陽性試験化合物の置換能力を測定することが可能である。同様に、(試験されている試験化合物と同様なモル濃度の)L−DOPAと比較した競争的結合は、L−DOPAと比較してOA1への結合力が増加した化合物を識別するのを支援することが可能である。
【0110】
任意の適当な試験化合物を、小分子、ポリペプチドおよび核酸を含む本発明の第4および第5の態様の方法(以下を参照)を使用して評価することが可能である。試験化合物がポリペプチド配列を含む場合、かかるポリペプチドは化学合成されてもよいし、組み換え発現されてもよい。上記に開示したように、組換え発現は当該技術分野の標準的方法を使用して行うことが可能である。かかる発現ベクターには細菌またはウイルスの発現ベクターが含まれてよく、かかる宿主細胞は原核生物であってもよいし、真核生物であってもよい。固相法、液相法またはペプチド縮合法、またはそれらの任意の組み合わせの周知技術を使用して調製される合成ポリペプチドには、天然または非天然のアミノ酸が含まれ得る。ペプチド合成に使用されるアミノ酸は、標準的な脱保護、中和、カップリング、および洗浄プロトコルによる標準Boc(Nα−アミノ保護されたNα−t−ブチルオキシカルボニル)アミノ酸樹脂または標準の塩基性条件下で分解しやすいNα−アミノ保護9−フルオレニルメトキシカルボニル(Fmoc)アミノ酸であってよい。FmocおよびBoc Nα−アミノ保護アミノ酸は、いずれもSigma社、Cambridge Research Biochemical
社、または当業者に周知の他の化学企業から入手することが可能である。さらに、ポリペプチドは当該技術分野で周知の他のNα−保護基で合成することも可能である。固相ペプチド合成は当該技術分野の周知技術により達成され、自動シンセサイザの使用等により与えられる。
【0111】
試験化合物が抗体を含む場合、かかる抗体はポリクローナル抗体であってもよいし、またはモノクローナル抗体であってもよい。抗体は、ヒト化抗体であってもよいし、完全ヒト抗体であってもよいし、またはマウス型の抗体でもよい。かかる抗体は例えばHarlow and Lane, Antibodies; A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y., (1988).に記載されているような周知方法により作製することが可能である。
【0112】
試験化合物が核酸配列を含む場合、かかる核酸は化学合成されてもよいし、組み換え発現されてもよい。組換え発現技術は、当該技術分野で周知である(例えばSambrookら、1989、前掲参照)。核酸はDNAまたはRNAであってもよいし、一本鎖または二本鎖であってもよい。同様に、かかる核酸は、当該技術分野の標準的方法を用いて手動または自動反応により化学的または酵素的に合成することが可能である。化学的に合成されるか、またはin vitroの酵素合成により合成される場合、核酸は細胞へ導入される前に精製され得る。例えば、核酸は、溶媒または樹脂での抽出、沈殿、電気泳動、クロマトグラフィまたはその組み合わせにより、混合物から精製することが可能である。代わりに、核酸は、サンプル処理による損失を回避するため、精製を最低限にして使用してもよい。
【0113】
試験化合物がポリペプチド、抗体または核酸以外の化合物を含む場合、かかる化合物は有機化学合成を行なうための当該技術分野の種々の方法により製造することが可能である。
【0114】
第2の細胞集団に比べて第1の細胞集団におけるPEDFおよび/または細胞内カルシウム濃度の発現を増加させると識別された試験化合物は、以下に説明する本発明の第4の態様のインビボ法を含むがこれに限定されないさらなる任意の方法を使用して、AMDを治療またはAMDの進行を制限する候補化合物として使用するようさらに評価することが可能である。1つの好ましい実施形態では、方法はさらに、上述したように競争量のチロシンおよび/またはドーパミンの存在下でアッセイ中の陽性試験化合物を再試験することを含む。
【0115】
第5の態様では、本発明は、AMDを治療する化合物を識別する方法であって、
(a)チロシナーゼ欠損の妊娠している雌の非ヒト哺乳動物に試験化合物を投与する工程であって、試験化合物は胚光受容体および/または網膜神経節の発達中に投与される工程;および
(b)胚または出生後の非ヒト哺乳動物における光受容体および/または網膜神経節発達に対する試験化合物の効果を、試験化合物を投与していない胚または出生後の非ヒト哺乳動物における光受容体および/または網膜神経節の発達と比較する工程であって、光受容体および/または網膜神経節の発達を増大させる試験化合物は、AMDを治療および/またはAMDの進行を制限する候補化合物である工程;
からなる方法を提供する。
【0116】
本願発明者は、チロシナーゼ欠損動物における光受容体および神経節細胞の発達を助けるためにOA1シグナル伝達を使用できることを決定し、プロセス中にOA1シグナル伝達の神経組織栄養効果を確立した。したがって、OA1シグナル伝達を通じて神経網膜の発達を助ける化合物は、AMD治療の良い候補である。本発明は、AMD薬物スクリーニングのためのかかる動物モデルを第1に確立する。
【0117】
本明細書により詳細に記載するように、チロシナーゼはチロシンに作用してL−DOPAを生成する。したがって、チロシナーゼ欠損哺乳動物は、L−DOPAを生産せず、かかる哺乳動物を使用すると内因性L−DOPAがない状態で(網膜の進行および/またはPEDF発現の増加の救助を介して)OA1のアクチベータを識別することが可能となる。本明細書に使用する場合、「チロシナーゼ欠損」とは、妊娠している雌の非ヒト哺乳動物が正常な色素形成に適した量のL−DOPAを生成するのに適当な量のチロシナーゼを生産しないことを意味する。1つの好ましい実施形態では、妊娠している非ヒト哺乳動物は、機能的なチロシナーゼを発現またはやりとりする能力のないノックアウト動物(チロシナーゼ遺伝子の部分または全部が削除されているか、またはチロシナーゼ遺伝子またはチロシナーゼを制御、活性化するかもしくはメラノソームへとチロシナーゼをやりとりする付属遺伝子に自然突然変異を有する)である。かかるチロシナーゼノックアウトは当該技術分野で周知であり、市販で入手可能である(Lexicon Pharmaceuticals社、Jackson Laboratories社、Taconic Farms社)。別の実施形態では、チロシナーゼ欠乏は、チロシナーゼを標的とするsiRNAsの投与や、チロシナーゼ抗体/アプタマーによる治療等を含むがこれらに限定されない当該技術分野の周知方法により一過性に誘発されてもよい。
【0118】
非ヒト哺乳動物は、チロシナーゼ欠損(網膜アルビノ)の雌を得ることが可能であればいかなるものでもよく、すべての哺乳動物を含む。種々の好ましい実施形態では、非ヒト哺乳動物はマウス、ブタ、サルおよびラットである。
【0119】
1つの好ましい実施形態では、試験化合物の投与は、光受容体および/または網膜神経節の発達の出生後期間の間じゅう継続される。種々の非ヒト哺乳動物における胚および出生後の光受容体および/または網膜神経節の発達経路は、当業者によく理解されている。1つの例証的実施形態では、マウスの胚光受容体と網膜神経節の発達は胚の10日目(E10)に始まり、網膜の進行は、生後の14日目(P14)で完了し、この時には子マウスの目が開いている。したがって、種々の実施形態では、試験化合物は、まずE7、E8、E9またはE10の日に投与され(視覚の発達の最も初期の段階での存在を促進するため)、P1、P2、P3、P4、P5、P6、P7、P8、P9、P10、P11、P12、P13およびP14の日の間または所望に応じてその後に、所与のアッセイに所望される通りに投与を継続することが可能である(出生後1年以内)。当業者によって理解されるように、投与は、胚の時期に妊娠している雌の母体に行うか、出生後は子に行うことになる。別の実施形態では、色素細胞の発達は約日10.5E(OA1とチロシナーゼが現われる時期)に本格的に開始し、したがって1実施形態では、試験化合物の投与は約E10、E10.5またはE11日目に開始し、所望に応じてP1、P2、P3、P4、P5、P6、P7、P8、P9、P10、P11、P12、P13、P14日目または所望に応じてその後まで継続される。別の実施形態では、試験化合物の投与はE7と、E10またはE11との間に限定されてもよい。さらなる実施形態では、網膜神経節の発達は約E12日目に本格的に開始し、したがって1実施形態では、試験化合物の投与は約E12またはE13日に開始し、所望に応じてP1、P2、P3、P4、P5、P6、P7、P8、P9、P10、P11、P12、P13、P14日目または所望に応じてその後まで継続される。別の実施形態では、試験化合物の投与は約E7と、E12またはE13との間に限定されてもよい。ほとんどの好ましい実施形態では、試験化合物は、E7日からE14日まで毎日投与される。当業者に理解されるように、試験化合物の投与の正確なタイミングは、特定のアッセイの目的によって決まり、本明細書での教示に基づいて当業者が決定することが可能である。
【0120】
試験化合物は、L−DOPAまたはL−DOPA類似体の治療用投与のために上記に開示した投与経路を含む、実験動物への使用に適した任意の経路により投与されてよい。好ましい実施形態では、試験化合物は、動物の飲料水に入れて、非経口(上述)または局所(例えば点眼薬または眼軟膏剤で)敵に投与される。試験化合物の投与回数は、所与のアッセイに適した頻度としてよい。好ましい実施形態では、試験化合物は、治療の所望経過じゅう毎日投与される。他の実施形態では、投与は治療経過の間、1日、3日、4日、または5日おきである。投与回数は本明細書の教示および所与のアッセイの特定の目的に基づいて当業者が決定することが可能である。
【0121】
本明細書に使用する場合、「光受容体および/または網膜神経節の発達の増加」は、試験化合物で処理した胚/動物対未処理の胚/動物における光受容体および/または網膜神経節の発達の任意の増加である。この方法は、化合物が対照で見られるよりも大きな光受容体および/または網膜神経節の発達の増加を促進する限り、対照に対する光受容体および/または網膜神経節の発達の特定量の増加を要求しない。好ましい実施形態では、増加は標準的な統計学的測定で測定される統計学的に有意な増加である。1実施形態では、動物は適切な時点で安楽死させられ、網膜神経節細胞および/または光受容体は以下の実施例に開示されたものを含むがそれらに限定されない当該技術分野の標準的方法を使用して計数される。
【0122】
光受容体および/または網膜神経節の発達を増加させるとして識別された試験化合物を、AMDを治療またはAMDの発達を制限する候補化合物として使用するためにさらに評価することが可能であり、それには競争量のチロシンおよび/またはドーパミンの存在下で本発明の第3態様で開示したin vitroの方法を使用して陽性試験化合物を再試験することが含まれるが、それらに限定されるわけではない。以下の実施例に示すように
、1mM以上の濃度で、チロシンおよびドーパミンはOA1への結合に関しL−DOPAと競争することが可能である。したがって、1mMから100mMの間、好ましくは、1mMから50mMの間または1mMから25mMの間の濃度のチロシンおよび/またはドーパミンを使用した競争アッセイを使用して、試験化合物がOA1経路を介して作用していることをさらに確認し、かつすることが可能である、経由で作動している、またチロシンおよびドーパミンがL−DOPAの置換と比較してOA1へ結合する陽性試験化合物と置換する能力を測定することが可能である。
【0123】
実施例:L−DOPAはOA1の内因性リガンドである
(背景):白皮症は色素沈着の損失を特徴とする遺伝子異常である。神経感覚網膜は、色素が沈着せず、網膜色素上皮(RPE)の色素沈着の喪失の次に病理学的変化を示す。RPEの色素沈着の喪失がどのようにして隣接する神経感覚網膜の発達欠陥を引き起こすかはまだ決定されていないが、これらの重要な2つの組織間の相互作用を研究する固有の機会を与えている。白皮症を引き起こす遺伝子の一方は、RPEを含む色素沈着細胞にのみ発現されるオーファンGPCR(OA1)をコードしている。
【0124】
方法/原理的発見:OA1の機能およびシグナル電圧を、RPEおよびトランスフェクト細胞株で調べた。結果は、OA1が選択的L−DOPA受容体であり、2つの密接に関連する化合物であるチロシンおよびドーパミンからの測定可能なセカンドメッセンジャー活性はないことを示している。放射性標識リガンド結合により、OA1がL−DOPAの一つの飽和結合部位を示すことが確認された。ドーパミンは、その一つのOA1結合部位に関しL−DOPAと競争したが、これはドーパミンがOA1アンタゴニストとして機能し得ることを示唆している。L−DOPAに対するOA1応答を、細胞内カルシウムの流入およびβ−アレスチンの動員を含むいくつかの一般的なGPCR活性化の測定基準により定義した。さらに、L−DOPAを生産する酵素であるチロシナーゼの抑制により、RPEによるPEDF分泌が減少した。さらに、L−DOPAによるRPEでのOA1の刺激により、PEDF分泌が増大した。
【0125】
結論/有意性:まとめると、結果は、L−DOPAを介してリンクされたOA1とチロシナーゼとの間の自己分泌ループを示し、このループは少なくとも1つの非常に効き目のある網膜神経栄養因子の分泌を含んでいる。OA1は、その下流での作用が発達中の網膜の空間パターン形成を左右する選択的L−DOPA受容体である。結果は、メラニン合成機構の変化により引き起こされる網膜の白皮症の結果がL−DOPAの補給により治療され得ることを示唆している。
【0126】
導入:白皮症は、目、髪または皮膚の色素沈着の可変的な喪失がある、遺伝的遺伝子疾患のグループである。目が罹患した場合、弱視につながる神経感覚網膜発達に著しい変化がある[1−8]。白皮症には2つの大きな分類である眼皮膚白皮症(OCA)および眼白皮症(OA)がある。すべての色素化組織が色素沈着減少を示した場合にOCAが生じ、メラニン合成機構の欠陥を生じさせる遺伝子突然変異を伴う[3および7−9]。OAは皮膚組織に通常に色素がついている場合に生じるが、眼組織の色素沈着が減少している[10および11]。すべての組織で同じタンパク質は色素を生産するため、他の組織も通常着色するため、OAは、メラニンを合成する能力がないわけではなく、眼組織中のメラニン生成酵素の発現が不足していることに起因する可能性が高い。
【0127】
OAは、X染色体上に見出される少なくとも1つの遺伝子Oa1に結合され得る。Oa1は、配列決定分析に基づくと、オーファンGタンパク質共役受容体(GPCR)(Genbank GPR143)[12および13]であると考えられる404個のアミノ酸からなるタンパク質をコードする[14]。SchiaffinoらはOA1がGβのみならずいくつかのGαサブユニットと結合していることを実証しており、これはOA1がGPCRで
あるというさらなる証拠を加えている[14および15]。実際、Innamoratiらは、OA1由来のGPCR様活性とβ−アレスチン結合を示すために、組み合わせ発現法を使用した[16]。この研究は、OA1がホスホリパーゼCとイノシトール三リン酸のセカンドメッセンジャーによってGαqサブユニットを介してシグナルを送り得ることを示唆した。酵素発現系で、StalevaとOrlowは、メラノソーム区画中の成分により活性化されていると考えられるOA1からのGPCRシグナル伝達を実証した[17]。OA1がGPCRであるというかなりの量の情況証拠にもかかわらず、リガンドが同定されていないため、確認がなされていない。OA1がGPCRであるという考えに、他のデータは疑問を投げかけている。例えば、完全な細胞内タンパク質としてのOA1の局在性は、GPCRに一般的なことではなく、これはOA1がGPCRファミリーの固有のメンバーであろうことを示唆している[14]。OA1は細胞表面ではなくリソソーム内区画[14、15、18−21]およびメラノソーム[11、14、22]に主に局在化している。
【0128】
本研究では、OA1のエンドソームの局在性が培養細胞中の薬物に応答したOA1の内在化によるとの仮説に基づき、潜在的GPCRとしてのOA1の機能を調べた。さらに、すべての型のOCAおよびOAが同じ網膜の発現型を有し、これはチロシナーゼ活性とOA1シグナル伝達が網膜発達の上流で連結されていることを示唆しているという知見に基づいて、OA1のリガンドを求めた。したがって、チロシナーゼ活性がOA1のリガンドを生産するか否かの試験を行なった。メラニン合成の副産物はL−DOPAであり、これは網膜の発達の重大な時間にRPEでのメラニン合成の間に網膜に放出される[23および24]。データは、OA1が非常に選択的なL−DOPA受容体であり、L−DOPAがRPEによる神経栄養因子分泌の下流での作用によりOA1のシグナル伝達を引き起こすことを示唆している。したがって、第1の証拠がOA1のリガンドに対して示され、チロシナーゼまたはOA1の欠乏のいずれかにより網膜の発達に変更を生させる機構を提供する。
【0129】
結果:
OA1の細胞表面局在性
OA1は以前はin situで色素顆粒中に局在化していたが[22]、種々のトランスフェクト細胞を使用すると、OA1は原形質膜[16および17]および培養細胞のエンドソーム部分[14、16−18、20、21]の両方にも局地化していた。このような研究は、細胞表面ビオチン化/ウェスタンブロット法を使用してOA1がヒト組織のどこに存在するかを決定することから始まった。ヒトの目では、OA1はin situでRPEの頂端細胞表面に存在した(図1A)。細胞表面の、5つのヒトの目のビオチン化OA1の定量化により、全OA1のうちの少なくとも3.5+/−0.7%がin situでRPEの頂端細胞表面に存在することが示された。眼杯調製物を使用したビオチン化試薬へのアクセスは頂端面に限定されており、上皮中のOA1の極性は決定できない。さらに、基底細胞表面へのアクセスが欠如しているため、合計細胞表面OA1が過小評価されている可能性がある。細胞質タンパク質がビオチン化されなかったことを確認するため、ブロットを対照としてアクチンに対する抗体でプローブした。各実験で、アクチンは未結合の部分でのみ見つかった。
【0130】
組換えOA1およびOA1−GFPが、培養細胞のエンドソーム区画のみにほとんど排他的に局地化されていると報告したものもあった[14,15,17,18,20−22]。しかしながら、過剰発現された場合[16]や、またはエンドサイトーシスを阻害した場合[17]、OA1は細胞表面に蓄積する。OA1タンパク質がin situでRPEの頂端面に存在するという知見は、この問題を我々にさらに検討するに至らしめた。
【0131】
OA1発現および分布に対するチロシンの影響
GPCRのエンドソーム局在性は、通常リガンドへの接触後に生じる。したがって、受
容体のリガンドが、OA1の内在化を起こさせ得る標準インキュベーション培地に存在するか否かを調べた。標準培地は500μMチロシンを含み、チロシンは色素合成の出発物質であるため、受容体分布に対するチロシンの影響を評価した。チロシンが培養細胞中のOA1分布に影響するか否かを調べるため、DMEMをチロシンを含まずに調合し、透析したウシ胎仔血清を使用した。チロシンを含まない培地の存在下で、チロシンを含まない(図示しない)培地と低濃度のチロシン(1μM、図1B)を含む培地の両方で、培養RPE細胞の原形質膜上でOA1が検知された。5つの実験を平均し、合計OA1タンパク質の4.5+/−1%が、1μMチロシンに維持された培養RPEの表面で観察され、これはin situのRPEで観察されたのと類似していた。すべての実験で、未結合タンパク質分画でアクチンが観察されたが、これは細胞表面アッセイに細胞質タンパク質が存在しないことを実証している。同様に、COSで発現されたOA1−GFPは、チロシン感受性の細胞表面発現を示した(図1C)。6つのかかる実験の定量化により、一過性トランスフェクションを使用して細胞表面に見出されるOA1の量の重要な変化が示された。1μMチロシンに維持されたトランスフェクト細胞の未結合分画のOA1の範囲は5−40%に及び、再現性があって約5%という内因性OA1タンパク質の結果とは異なる。
【0132】
トランスフェクト細胞のOA1の分布が培地中のチロシンレベルに感受性であるのみならず、合計OA1−GFPの発現も1μMで維持した細胞で5倍に増大した。この差が細胞数ではなくOA1発現量に関連することを確認するために、アクチン発現をペアになったサンプルから評価した。光学濃度単位として示したデータ(図1D)は、アクチンに差がないことを示している。正常群と低チロシン群との間の細胞表面OA1の量も比較した。重要なことは、5つのRPE実験および6つのCOS中のOA1−GFP実験で、標準培地中の細胞の原形質膜部分のOA1は、他の部分に見出されるのと同様に、再現可能に検出されなかった。
【0133】
RPE細胞におけるOA1の分布を、共焦顕微鏡法によっても評価した。OA1は、以前に示されたように培養RPE細胞のエンドソームタンパク質であると特徴付けられたが(図1E)、対照的に、低チロシン培地中のOA1の分布は培養RPE細胞の原形質膜上で拡散し、エンドソーム蓄積はほとんどなく(図1F)、この知見は生化学法を使用して得られた結果と一致していた。
【0134】
OA1の天然アゴニストとしてのL−DOPA
メラニン形成におけるチロシナーゼの機能は、L−DOPAを生成するようチロシンに作用することから始まり、これはその後ドーパキノンを作成する第2の反応を経て、色素形成に至る[25]。チロシンとメラニンの間の中間体のうち、L−DOPAは最も大きな半減期を有すると共に、メラニン合成が起こった場合にL−DOPAはRPEの頂端の網膜下の空間へ放出される[23および24]。L−DOPAは、チロシンからドーパ作動性ニューロンにより生産された神経伝達物質であるドーパミンの前駆体でもある。細胞内貯蔵物からのカルシウムの放出はリガンドによるGPCR活性化の一般的な下流の作用である。細胞表面でのOA1の発現はチロシン感受性であるようであるため、チロシン、またはその代謝産物であるL−DOPAおよびドーパミンが、OA1依存的に細胞質へのCa2+流入を刺激することが可能か否かを調べた。CHO細胞にOA1発現ベクターをトランスフェクトし、該CHO細胞を1μMチロシンを含mDMEMで48時間維持し、その後チロシンを含まないDMEMで24時間維持し、OA1の細胞表面発現を促進した。細胞内Ca2+をFura−2を使用して評価し、[Ca2+]iをレシオメトリックイメージングで決定した[26]。リガンドが存在しない状態では、トランスフェクト細胞と非トランスフェクト細胞との間で[Ca2+]iの有意差はなかった(図2)。チロシンおよびいくつかのチロシン代謝産物を、1μMの濃度で、[Ca2+]iに対する効果を試験した。陽性対照として、各実験を20mM KClでの処理により終了し、細胞を脱分極さ
せると共に電位型チャネルの活性化により[Ca2+]iを増加させた。この方法は、Fura−2装填および試験されている細胞の反応性を確認する役割を果たした(図2)。L−DOPAだけが[Ca2+]iの有意な増加を誘発した(図2A)。チロシンとドーパミンは1mM(図示しない)まで細胞内[Ca2+]i濃度に陽性の効果がなかった。1μMドーパミンのわずかに陰性の効果は、統計学的に有意ではなかったが、ドーパミンを用いた11の実験で再現可能であった(図2B)。
【0135】
非天然細胞株におけるGPCRの過剰発現は、偽のシグナル伝達の結合につながり得る。L−DOPAに応じたOA1シグナル伝達が確かに天然の応答であったことを確認するために、OA1をRPE細胞で発現させた(図2C)。トランスフェクトRPE細胞を使用した結果は、トランスフェクトCHO細胞で達成された結果に類似していた。OA1を発現するようトランスフェクトされたRPE細胞は、[Ca2+]iの増加に伴い1.0μM L−DOPAに応答した。次に、内因性OA1受容体を発現しているRPE細胞が内因性レベルでL−DOPA反応を示すか否かを決定した。すべてのトランスフェクト細胞実験のように、OA1を発現するRPEは、1.0μM L−DOPAでの処理後に[Ca2+]iの増加を示した(図2C)。
【0136】
OA1のシグナル伝達活性をさらに特徴づけるために、百日咳毒素を使用してGq共役[Ca2+]iシグナル伝達とGiリンクシグナル伝達(図2C)とを区別した。研究されたすべての細胞で、百日咳毒素は[Ca2+]iの基準レベルを低下させ、これはGiサブユニット活性によるバックグラウンドシグナル伝達の阻害に対するその活性を示した。CHOとRPEの両方を含むOA1を発現するようトランスフェクトされた細胞ならびに天然レベルで内因性OA1タンパク質を発現するRPEで行なわれた実験に、百日咳毒素を使用した。試験したすべてのトランスフェクト細胞で、L−DOPAに対して測定した[Ca2+]i応答は毒素がない場合よりも大きく(図2)、この大部分は最初の[Ca2+]iが低いことによる。したがって、[Ca2+]iの増加が生じるL−DOPAに応答したOA1を介したシグナル伝達は、百日咳毒素感受性ではなく、Gqサブユニット媒介性である可能性がある。OA1を発現するようトランスフェクトされたCHO細胞でのセカンドメッセンジャーcAMPも測定した(図2D)。不活性細胞または最大未満のホルスコリン処理を使用して、L−DOPAに応じたcAMPの増加または減少のいずれかを測定するように実験を設定した。6回のかかる実験では、cAMPの変化は観察されず、これはGsサブユニットもGiサブユニットもOA1シグナル伝達に関与しないことを示唆している。
【0137】
放射性標識リガンド結合の標準的方法を使用して、OA1とL−DOPAの間の相互作用を特徴付けた(図3A)。CHO細胞をOA1を発現するようトランスフェクトし、次に、L−DOPAの結合を濃度依存的に定量し、その結果をさらにScatchardプロット分
析で特徴付けた(図3E)。結果は、9.35×10-6のKdのOA1発現細胞へのL−DOPAの飽和結合を示す。非トランスフェクトCHO細胞では特異的結合は観察されず、これは細胞が内因性L−DOPA受容体(図示しない)を有していないことを示している。すべての結合パラメータ、すなわち合計結合、特異的結合、および非特異的結合を、補助データ(図6A)として示す。チロシンは、OA1と相互作用する可能性を示した。が、チロシンもドーパミンもOA1シグナル伝達を刺激しなかった(図2参照)。競争リガンド結合を使用して、チロシンまたはドーパミンのいずれかがOA1に結合するためにL−DOPAと競争したか否かを判断した。高濃度(1mM)では、チロシンとドーパミンはいずれもOA1との結合のためにL−DOPAと競争した(図3B)。これをさらに特徴付けるために、L−DOPAとドーパミン(図3C)またはチロシン(図6B)のうちの一方との間の競争の動力学を調べた。ドーパミンは、2.33×10-6+/−0.2×10-6MのKiのL−DOPAとの単一部位への競争的結合を示した。チロシンによる同様の実験は、高濃度(図6B)でのみL−DOPA結合の阻害を実証した。チロシンは
高濃度でも親和性が低くおよび不溶性であるためにチロシンに関する飽和速度は不可能であった。
【0138】
L−DOPAに対するOA1の親和性が比較的低いことから、そのシグナル伝達活動がこの結合親和性の範囲で用量依存的であるかどうかを決定した。結合データをL−DOPAとOA1との関連で最も急激な上昇を示唆する濃度である1.0−10μMで試験し、結果を[Ca2+]iにより測定された濃度依存的GPCR応答を示す(図3C)。このように、L−DOPAおよびOA1の活性化速度は、放射性標識リガンド結合実験で観察された濃度範囲と一致した。
【0139】
リガンド結合に応じて、GPCRは、β−アレスチンを原形質膜へ動員し、これに続きリガンド受容体複合体が内在化した[27−33]。その後、β−アレスチン局在化に対するL−DOPAの効果を試験した(図4)。細胞をOA1を発現するようトランスフェクトし、次に1μMチロシンのDMEMで48時間培養し、それからタンパク質の細胞表面発現を可能にすべく分析を行った。その後、細胞を1μM L−DOPAで処理し、氷冷メタノールで迅速に固定した。最初、アゴニストが存在しない休止状態では、OA1−GFPは細胞表面で見つかり、β−アレスチンは細胞質中に拡散し(図4A−C)、タンパク質間の同時局在化はなかった。L−DOPAでの刺激後、OA1およびβ−アレスチンは原形質膜に同時局在化した(図4 D−F)。非トランスフェクト細胞はL−DOPA処理に対する反応を示さず(図4G,H)、これはβ−アレスチン分布に対するL−DOPAの効果が、[Ca2+]iのついて得られた結果と同様に、OA1依存的であることを示している。
【0140】
PEDF分泌に対する1−DOPAの効果
OA1の突然変異は、神経感覚網膜の発達の欠陥を引き起こす。以前の研究では、色素沈着RPEが非色素沈着RPEよりも有意に多くのPEDFを分泌すること[34]、およびPEDFは神経感覚網膜の発達を変化させる可能性のある神経栄養因子である[35−41]ことが示された。OA1の突然変異は、RPEの色素沈着の喪失を引き起こすが、これはOA1活性がRPEの色素沈着を制御していることを示唆している。したがって、色素化RPE細胞のL−DOPA刺激がPEDFの分泌の増加を引き起こすか否かを決定した(図5)。このアッセイは幾分困難であったが、その理由は色素沈着RPE細胞はL−DOPAを生産し、L−DOPAはOA1のアゴニストであるが、RPEの非色素沈着培養物ではOA1は容易に検知できないためである。したがって、色素化RPEを使用して、L−DOPA刺激がPEDF発現/分泌を増加させるか否かを決定した。RPE細胞をチロシンを含まない培地に24時間置き、次に1μM L−DOPAで1時間処理した。処理後、細胞を外因性L−DOPAを含まない標準培地に3日間戻した。対照細胞はL−DOPAで処理しないが培地を同じ時間に交換し、実験細胞を通常の培地に戻したものである。3日後に馴化培地を収集し、PEDFを測定した。結果は、ペアになった色素化RPEの対照単一層と比較すると、L−DOPAで処理した色素化細胞ではPEDF分泌が有意に増加することを示している(図5A)。重要なことには、この有意な増加は、着色し、従ってOA1を発現し、基本レベルのPEDF発現をしていた細胞で起こった。
【0141】
チロシナーゼ活性およびOA1シグナル伝達を伴う自己分泌ループを介して色素化RPE細胞がPEDFを分泌するか否かを決定するために、特異的チロシナーゼ阻害剤であるフェニルチオ尿素(PTU)を使用して、色素沈着およびL−DOPA生産を阻害した(図5B)。これらの実験では、色素化RPE細胞は、DMEMで維持するか、または200μM PTUを含むDMEMで3日間維持し、次にPEDF分泌を測定した。色素化RPEはかなりのPEDFを分泌したが、PTUは、PEDF分泌の有意な減少を引き起こし、これは色素化RPE細胞で観察される高レベルのPEDF分泌にチロシナーゼ活性が必要であることを示している。PEDF分泌の減少を引き起こしたのがPTU処理細胞に
おけるL−DOPAの欠如であることを確認するために、色素化RPEの3つの異なる培養物を使用して、48時間PTUに曝露し、その後引き続きPTUが存在する状態で1.0μM L−DOPAで処理し、72時間後にPEDFを測定した(図5C)。データはこの実験の対照に対する割合(パーセント)として示したが、その理由は、使用される培養物の色素沈着およびPEDF発現が実験開始前に変化したためである。PTU処理したRPEは、PEDF分泌を増加させることにより添加L−DOPAに応答したが、これはPEDF分泌に対するPTUの効果が、チロシナーゼが阻害された場合のL−DOPA生産の不足により引き起こされることを示している。
【0142】
考察:
RPEと神経感覚網膜の間には複雑な組織間関係がある。この関係の1態様は、RPE色素沈着および重大な神経感覚網膜の変化を生じさせるメラニン合成の欠如[8、23、42]を基軸とする。データは、少なくとも1つの効き目のある神経栄養因子であるPEDFの分泌を規制するL−DOPAを介して自己分泌ループにOA1とチロシナーゼが関与していることを示唆している。データはまた、白皮症で起こる網膜の発達における病理学的変化がOA1シグナル伝達経路の活性の変化に起因し得ることも示唆している。OA1シグナル伝達活性の減少は、OA1の突然変異により直接的に、またはチロシナーゼ活性によるL−DOPA生産の変化により間接的もたらされ得る。したがって、白皮症の種々の形式に伴う同様な網膜の発現型が一つの共通の経路であるOA1シグナル伝達と調整され得ると仮定される。
【0143】
研究では、in situでヒトRPEの先端面にOA1が観察された。以前の報告は、OA1がマウスではメラノソーム[22]に、および培養細胞ではエンドソーム区画[15−18、20−22、43]に局在化していることを示唆していた。in situRPE調製物からの結果は、OA1がRPEの頂端面に分布することを示している。RPE(合計OA1の約3.5%)の表面のOA1が限られた量であることは、免疫金電子顕微鏡法を使用した以前の研究におけるかかるOA1タンパク質の観察が不十分であることを説明している可能性がある。多くの細胞表面GPCRと同様、OA1は豊富なタンパク質ではない。
【0144】
培養細胞を使用した以前の研究で報告されたOA1のエンドソーム局在は、内因性タンパク質および遺伝子組換えタンパク質の両方に対する本研究で再現された。通常の培地で試験した場合、細胞表面に検出可能なOA1タンパク質はほとんど見あたらず、これは以前の研究と合致していた。しかしながら、培地でのチロシン減少により内因性および組換えOA1タンパク質の両方の細胞表面受容体の蓄積が増大した。これは、培養細胞における細胞表面へのOA1の分布がチロシン感受性であることを示唆している。以前の研究は、エンドサイトーシスが阻害され[17]、ヒトRPEの頂端面でOA1が観察された時に、OA1が細胞表面に局在化され得ることを実証した。データは、OA1が細胞表面GPCRであるが、チロシンまたはチロシンの代謝産物により刺激されてもよいエンドサイトーシスの標的でもあることを示唆している。この点で、本結果は、OA1を細胞内GPCRの固有の種類として分類したOA1局在化の過去の報告とは異なる。ほとんどのGPCRは種々の信号により内在化される細胞表面タンパク質であり、データはOA1がほとんどの他のGPCRと類似していることを示唆している。
【0145】
OA1シグナル伝達活性はL−DOPAにより刺激されるが、その前駆体であるチロシンによっても、そのニューロン代謝産物であるドーパミンでも刺激されない。この結果は、結局L−DOPAとチロシンが一つの水酸基だけ異なるため、密接に関連する分子を識別することが可能である非常に感度の高い受容体の活性を示唆している。チロシンが培養細胞でのOA1の細胞内局在化を引き起こすため、OA1はチロシン感受性である。しかしながら、チロシンに対するシグナル伝達応答がないことに注意すべきであり、競争結合
研究は、チロシンがOA1に対して低い親和性を有することを示唆している。データは、正常な培地中に存在する高濃度のチロシンへの細胞を連続的に曝露すると、OA1の内在化に十分であるが、測定可能なOA1活性化を生じさせる可能性は低いことを示唆している。L−DOPAとドーパミンの間の競争的相互作用は一つの部位であるという強い証拠が見出された。ドーパミンに対して観察されたKiは、L−DOPAに対して観察されたKdと類似しており、これは2つのチロシン代謝産物の親和性が類似していることを示唆している。結果は、ドーパミンからのOA1シグナル伝達がわずかではあるが再生可能に減少していることを示し、ドーパミンがOA1のための有能なアンタゴニストまたはインバースアゴニストであってもよいことを示唆している。
【0146】
オーファンGPCRとして、そのシグナル伝達経路が以前には同定されていない。本研究では、L−DOPAに応じたOA1シグナル伝達が[Ca2+]iの増加を引き起こすことが例証された。データは、L−DOPAに応答して観察された[Ca2+]iの増大が百日咳毒素には無反応であり、cAMPに対する効果は見出されず、OA1がGqサブユニットを介して恐らくシグナル伝達していることを示している。以前の研究は、OA1がG0、GiおよびGqサブユニットファミリーのメンバーを含むトランスフェクト細胞中の
多数のサブユニットと会合可能であることを示唆した。Innamoratiらは、過剰発現されたOA1の自発的活性がGqサブユニットを介して示される可能性が高いことを示した[16]。データは、RPEにおける内因性OA1からのリガンド依存性シグナル伝達が、Gqを介して起こる可能性が最も高く、RPE中で発現された天然OA1とCHOおよびRPEにおける過剰発現OA1とを比較すると乱雑な結合活性は観察されなかったことを示している。興味深いことに、Gqサブユニットの2つの過剰活性変異型が、皮膚および髪の過剰色素沈着を引き起こす[44]が、それらがRPEに影響を及ぼすかどうかは不明である。RPEおよび皮膚のメラノサイトは、色素沈着を生じさせるのに同じ酵素を使用するが、メラニン形成の制御は異なっている。最近の報告は、OA1-/-およびGαi3-/-の網膜発現型が同様であるため、OA1がGαi3を介してシグナル伝達することを示唆している[45]。この研究は、Gαi3とOA1の間の相互作用またはシグナル伝達に関するデータを提供せず、結果はGαi3を介したOA1のシグナル伝達をサポートしていない。しかしながら、OA1とGαi3はいずれも、網膜の発達の複合系のある部分を制御する、収束経路に活性を有し得る。
【0147】
L−DOPAに対するOA1の応答は、[Ca2+]iの増加、原形質膜OA1へのβ−アレスチンの動員、およびPEDF分泌の増加の3つの方法で測定した。さらに、色素化RPEにおけるチロシナーゼ活性の阻害はL−DOPA生産を抑制し、PEDF分泌の減少を生じさせる。まとめると、これらの研究は、L−DOPAとOA1の間の生産的なリガンド:受容体の関係に対する説得力のある論拠を提示している。さらにデータは、L−DOPAのみがOA1に対する生産的なリガンドであったため、チロシンとその代謝産物の間の選択性を示唆している。本願発明者は、OA1とL−DOPAの間の結合反応速度を決定し、2者の間の典型的な一部位の受容体:リガンド関係を観察した。μMの範囲のKdのOA1とL−DOPAの間の結合親和性は、内因性リガンド:受容体の関係では珍しくない。OA1に対する特異的で高親和性のアンタゴニストの将来の同定は、OA1とL−DOPAの間の相互作用のさらなる生化学的特徴付けを支援し、ドーパミンがインバースアゴニストか否かを決定するのに役立つだろう。
【0148】
本研究は、メラニン合成の中間生成物であるL−DOPAによる、オーファンGPCRであるOA1の選択的な活性化を示した。本研究は、OA1活性が、RPEによる、正常な網膜の発達を支援する能力のある分子であるPEDFの分泌を刺激することも示した[40および41]。ヒトでは、これはOA1活性化を介した薬理学的介入がメラニン生成機構(OCA 1−4)の欠損により引き起こされた白皮症に有用であり得ることを示唆している。不運にも、データはかかる薬理学的介入にOA1が必要であり、Oa1の突然
変異が白皮症の最も一般的な原因であることも示唆している。
方法:
細胞培養
RPE− 細胞を[46]に記載したように分離し、5%ウシ胎仔血清(FBS)を補給したDulbecco改変必須培地(DMEM)に維持した。チロシン濃度を低下させる実験では、JRH Biosciences社(カンザス州Lenexa所在)によりチロシンを含まないよ
うに特注製造したDMEMを使用した。透析FBSをInvitrogen社(カリフォルニア州San Diego所在)から購入した。
【0149】
COS−7およびCHO− 細胞はATCCから入手し、5%FBSを補給したDMEMで培養した。OA1分布の分析には、細胞を1μMチロシンと5%FBSを補給したチロシンを含まないDMEMで2−4日間培養し、次に実験のために上記のチロシンを含まない培地で培養した。
細胞表面ビオチン化
in situのヒトRPE 眼球赤道の約2mm前で切開し、前部を除去することによりヒト眼杯を作製した。硝子体および網膜を、その下にあるRPE単層を傷つけずに除去し、網膜を視神経頭で切断した。RPEが露出された得られた眼杯を反応緩衝液(100mM NaCl、50mM NaHCO3、pH8.0)で3回リンスし、スルホ−N
HS−LCビオチン(1mg/ml)を30分間に2回入れた。反応はをTG緩衝液(25mM トリス、192mM グリシン、pH8.3)で停止し、停止プロテアーゼ阻害剤カクテルを含む溶解緩衝液(2mM EDTA、1% トリトンXおよび1% トゥイーン20、Tris塩基性塩類緩衝液)中で細胞を収集した。インタクトな細胞と色素顆粒を20分間、14,000rpmで遠心分離により除去した。ビオチン化されたタンパク質を、固定化ストレプトアビジンビーズで一晩捕獲し、次に4×還元緩衝液(250mMトリス、pH 6.8、8% SDS、40% グリセリン、20% β−メルカプトエタノール、0.08% ブロモフェノールブルー)と混合した。OA1タンパク質を10% SDS−PAGEゲルで分離し、ウェスタンブロット解析用のポリクローナルウサギOA1抗体を用いて同定した。ペアにしたウェスタンブロットを、アクチンに対するモノクローナル抗体でプローブした。
【0150】
培養細胞− RPEおよびトランスフェクト細胞を、実験に関して説明したチロシン濃度を含むDMEMに維持した。培養物を反応緩衝液で3回リンスし、次に、in situ調製物のために上述したようにビオチン化した。
Oa1のクローニング
cDNAライブラリーを6人のヒトドナーの眼からプールされた組織から構築した。全RNAをTrizol試薬を使用して収集し、次にcDNAを、第1鎖の合成にポリチミジル酸プライマーを、第2鎖の合成にランダムヘキサマーを使用して合成した。cDNAの合成に続いて、リボヌクレアーゼAを使用してRNAを除去した。OA1のコーディング配列を5’および3’末端への制限部位を加え、かつ天然停止コドンを除去した末端プライマーを使用してPCRにより得た。PCR生成物をpEGFP N−1ベクター(Clontech社)中にGFPと共にフレーム内にライゲートした。配列は配列全体にわたり両方向で自動シーケンシングにより確認された。
免疫細胞化学
スライド上の細胞を室温にて3% パラホルムアルデヒドで固定し、0.1% トリトンX−100の10%ミルクTBST溶液でリンスし、次に10%ミルクTBST溶液でブロッキングした。β−アレスチンをβ−アレスチンに対するポリクローナル抗体を使用して可視化し、4℃で一晩インキュベートした。50%のグリセリンを使用してカバースリップを載せ、Compix Confocal Imaging Systemsソフトウェア(Simple PCI Version 4.0.6.1605)を搭載したNikon Eclipse E800 レーザー走査共焦点顕微鏡を用いて光学切片
により免疫染色を分析した。OA1−GFPおよびβ−アレスチン分布の三次元分析はIm
age J 1.32で行なった。
[Ca2+]iの測定
ガラスカバースリップ上に配置したOA1−GFPを発現しているCHO細胞を、スリップする、Ca2+含有HEPES干渉ハンクス平衡塩類溶液(HBSS)(pH7.45)でリンスし、次に2.5μM Fura−2(無水ジメチルスルホキシドおよび0.002% プルロン酸で可溶化)で20分間、37℃、5% CO2でインキュベートした
。Fura−2で満たされた細胞をHBSSで15分間、37℃、5% CO2でインキ
ュベートし、染料をその活性型へ十分に開裂させた。各カバースリップを、40×1.35 NA 紫外線−蛍光対物レンズを備えた倒立型Olympus IX70顕微鏡のステージ上で37℃に維持したチャンバに入れて1mlのHBSS中でインキュベートした。
【0151】
フィルタホイールを使用して、200W Xe(キセノン)球からの励起光を340および380nmのフィルタに交互に通過させた。中心が510nmの10nmバンドパスフィルタを、CCDカメラに通過させる(Photometrics CH-250)放射蛍光のために選択
した。各実験に関し、画像の対を最初の3分間の間、毎分撮影し、これは安定した基準線を設定した。その後、L−DOPA(最終濃度1μM)を加え、画像の組を次の3分間の間、30秒毎に撮影した。最後に、細胞にFura−2が装填されていることを確認するために、KCl(最終濃度20mM)を各実験の終了の1分前に陽性対照として加えた。同じ作業を、チロシンおよびドーパミン(いずれも最終濃度1μM)に対して独立して繰り返した。Silicon Graphics Personal IRISコンピュータを使用して、細胞内の各ピクセルについて340/380nm比を計算し、次にMicrosoft Excel version 4.0を使用し
て分析した(Microsoft社、ワシントン州Redmond所在)。一旦340/380nmの比が決定されたら、各比を1(自身で割った時間0の比)で正規化し、遊離イオン濃度を以下の方程式を使用して計算した:
[Cai]#=Kd#*(R-Rmin#)/Rmax#−R)
式中、R、Rmin、およびRmaxはそれぞれ実測比、最小比および最大比である。Rmax
は、完全に脱プロトン化した条件下でのイオン感受性波長の蛍光強度の比を表わし、Rminは完全にプロトン化した場合の染料の比である。Fura−2の場合、RはCa2+が増
加するにつれて増加する。従って、RminはCa2+(Ca2+<1nM)が存在しない状態
のFura−2を表わし、Rmaxは以前に説明したCa2+−Fura−2キレートを表す
[26]。Rmin、RmaxおよびKdをFura−2装填細胞で独立した実験にて決定し、続いて実験手順の遊離Ca2+の計算のために利用した。
放射性標識リガンド結合
OA1−GFPを発現するようにトランスフェクトしたCHO細胞を24ウェルプレートに播いた。細胞を−2℃まで冷却し、次に、冷やした結合緩衝液(25mM トリス、150mM NaCl、5mM EDTA、5μM ジギトニン(pH 7.45))でリンスした。細胞を、10-4M〜10-9Mの間の濃度の[3H]−L−DOPA(Moravek
Biochemicals社、カリフォルニア州Brea所在)を含む結合緩衝液中で2時間インキュベ
ートした。分析のどの工程でも、温度を−2℃を超えないようにした。対照は、非トランスフェクトCHOに対して行った分析を含み、特異的結合を10-3Mの未標識の過剰L−DOPAとの競争により決定した。結合したL−DOPAをシンチレーション分光法で定量化された。
cAMPの測定
細胞をホルスコリン(15分)で前処理し、次に以前に説明したアッセイ設定を使用してL−DOPAで攻撃した[47]。リガンドへの曝露の1分後、細胞を氷冷緩衝液中にこすり落とし、ボイルし、遠心分離した。その後、それぞれ等量(50μl)の上澄液および3H cAMP(New England Nuclear社)を合一して100μl 冷PKAとした。2時間後、溶液を活性炭上に通過させ、上澄液をシンチレーション計数器で数える。結果を、50μlのcAMP(0.25−32.0pmole/50μl)を使用して生成した、細胞質ゾルの代わりに標準曲線を使用して達成されたものと比較する。
実施例2:生体内のOA1ループ機能
OA欠損マウスにおけるPEDF分泌を野性型マウスと比較したところ、野生型マウスはOA1−/yマウスよりも有意に多くのPEDFを分泌することが示された。使用される培地(C.M.)はPEDFを含み、OA1 −/yからのCM中のPEDFは、RPE由来ではなく使用される培地に由来する可能性が考えられる。結果(図7)を定量化し、グラフに要約する。両群のCM中のバックグラウンドPEDFを用いても、差は有意である。t検定分析結果を示す。
【0152】
チロシナーゼ欠損の妊娠マウスを、正常条件で(L−DOPAは無し)または妊娠マウスの仔の胚7日目から該マウスの飲料水に1.0mg/ml L−DOPAを補給して維持した。動物を、視覚の発達が終わり眼が開く出生後14日目まで、L−DOPAを補給して維持した。
【0153】
白皮症では2つの細胞の種類の数が減少している:網膜神経節細胞および光受容体。図8Aは、L−DOPAの補給が、正常な野性型マウスで予想されるものと比較して網膜神経節細胞数を増加させることを実証している。図8Bは、光受容体に関する同じ結果を示す。光受容体は密集しすぎているため、直接数えられない。逆に、光受容体の核により占められた領域を光受容体数の基準として測定する。L−DOPAの補給により光受容体の核領域が増加し、光受容体の数も増加した。繰り返すが、これはアルビノ動物を標準レベルに戻したかのように見えた。
【0154】
図8Cに示されるように、4匹のペアになった同腹仔マウス、2匹の野生型、および2匹のOA1 −/y(雌、OA1欠損)を安楽死させ、各動物からの網膜をレーンに独立して載せ、次にタンパク質をウェスタンブロットしてPEDFを検出した。PEDFは野生型マウス由来の網膜では容易に観察された。対照的にPEDFはOA1−/yマウス由来の網膜では容易に検出されない。
【0155】
要約すると、このデータは、OA1−/yマウスが野性型マウスよりもPEDFを少なく生産することを示している。チロシナーゼ欠損マウスにおけるL−DOPA刺激は、白皮症の2つの最も顕著な神経感覚網膜欠陥である光受容細胞および網膜神経節細胞の損失を助ける。最後に、PEDFレベルはOA1欠損マウスの網膜では低下するため、OA1自己分泌ループがin vivoで機能し、経口L−DOPAにより刺激可能であることが結論付けられる。
【0156】
データはまとめると、RPE色素沈着とAMDの間のつながり(リンケージ)が、OA1のシグナル伝達活性を介している可能性を示している。データは、OA1のリガンドがL−DOPAであり、L−DOPAから生じたOA1シグナル伝達がPEDFの発現を制御することを示している。PEDFはRPEにより作製される最も効き目のある神経栄養因子である。したがって、PEDF発現を制御するOA1のリガンドとしてのL−DOPAの同定は、L−DOPAとRPEにおける神経組織栄養活性とを結びつける。L−DOPAが色素形成の副産物として生産されるため、これは初めてRPE色素沈着と神経組織栄養活性との間のつながりを確立した。この系はOA1自己分泌ループとして定義される。チロシナーゼは色素を形成し、L−DOPAを放出する。放出されたL−DOPAはOA1に結合し、OA1を介したシグナル伝達を開始する。OA1シグナル伝達は、チロシナーゼとPEDFの両方の発現を制御する。
【0157】
これまで、データは上記モデルを生化学的に、培養細胞で、および生体内で例証している。食事のL−DOPAを使用してアルビノ動物における網膜の発達を助けることが可能であるという事実は、RPEの栄養要因発現を生体内で刺激するのに食事のL−DOPAが使用可能であることを示している。AMDは、色素沈着に幾分関連するRPE欠損と明
らかに関係している。青い目の個体は黒い目の個体よりはるかに大きな頻度でAMDになり、したがってRPEの色素沈着レベルはAMDプロセスを制御する。RPEの色素沈着レベルはOA1シグナル伝達により制御され、これは上述したのと同じOA1自己分泌ループの一部である。したがって、AMDはRPEにおけるOA1シグナル伝達と関連付けられる。したがって、RPEの色素沈着がより低い者は、チロシナーゼがより低く、L−DOPAがより低く、OA1がより低く、およびPEDF生産がより低いだろう。本願発明者はOA1のリガンドとして食事L−DOPAまたは関連化合物を使用し、その活性を刺激することが可能である。神経感覚網膜の健康の最終決定因子はPEDFであるが、本願発明者はOA1を使用して、OA1ループ活性を増加させると共に、RPEの神経組織栄養の活性を増加させることが可能である。OA1シグナル伝達の効果はニューロンの生存を促進するだろう。
【0158】
参考文献
1. Akeo K, Shirai S, Okisaka S, Shimizu H, Miyata H, et al. (1996)Histology of fetal eyes with oculocutaneous albinism. Arch Ophthalmol 114: 613-616.
2. Gregor Z (1978)The perifoveal vasculature in albinism. Br J Ophthalmol 62: 554-557.
3. Schraermeyer U, Heimann K (1999)Current understanding on the role of retinal
pigment epitheliμm and its pigmentation. Pigment Cell Res 12: 219-236.
4. Rachel RA, Mason CA, Beermann F (2002)Influence of tyrosinase levels on pigment accμmulation in the retinal pigment epitheliμm and on the uncrossed retinal projection. Pigment Cell Res 15: 273-281.
5. Okulicz JF, Shah RS, Schwartz RA, Janniger CK (2003)Oculocutaneous albinism.
J Eur Acad Dermatol Venereol 17: 251-256.
6. Donatien P, Jeffery G (2002)Correlation between rod photoreceptor nμmbers and levels of ocular pigmentation. Invest Ophthalmol Vis Sci 43: 1198-1203.
7. Russell-Eggitt I (2001)Albinism. Ophthalmol Clin North Am 14: 533-546.
8. Oetting WS (1999)Albinism. Curr Opin Pediatr 11: 565-571.
9. Oetting WS, King RA (1999)Molecular basis of albinism: mutations and polymorphisms of pigmentation genes associated with albinism. Hμm Mutat 13: 99-115.
10. Shen B, Samaraweera P, Rosenberg B, Orlow SJ (2001)Ocular albinism type 1: more than meets the eye. Pigment Cell Res 14: 243-248.
11. Incerti B, Cortese K, Pizzigoni A, Surace EM, Varani S, et al. (2000)Oa1 knock-out: new insights on the pathogenesis of ocular albinism type 1. Hμm Mol Genet 9: 2781-2788.
12. Bassi MT, Schiaffino MV, Renieri A, De Nigris F, Galli L, et al. (1995)Cloning of the gene for ocular albinism type 1 from the distal short arm of the X chromosome. Nat Genet 10: 13-19.
13. Schiaffino MV, Bassi MT, Galli L, Renieri A, Bruttini M, et al. (1995)Analysis of the OA1 gene reveals mutations in only one-third of patients with X-linked ocular albinism. Hμm Mol Genet 4: 2319-2325.
14. Schiaffino MV, d'Addio M, Alloni A, Baschirotto C, Valetti C, et al. (1999)Ocular albinism: evidence for a defect in an intracellular signal transduction system. Nat Genet 23: 108-112.
15. Schiaffino MV, Tacchetti C (2005)The ocular albinism type 1 (OA1)protein and the evidence for an intracellular signal transduction system involved in melanosome biogenesis. Pigment Cell Res 18: 227-233.
16. Innamorati G, Piccirillo R, Bagnato P, Palmisano I, Schiaffino MV (2006)The
melanosomal/lysosomal protein OA1 has properties of a G protein-coupled receptor. Pigment Cell Research 19: 125-135.
17. Staleva L, Orlow SJ (2006)Ocular albinism 1 protein: trafficking and function when expressed in Saccharomyces cerevisiae. Exp Eye Res 82: 311-318.
18. Shen B, Orlow SJ (2001)The ocular albinism type 1 gene product is an N-glycoprotein but glycosylation is not required for its subcellular distribution. Pigment Cell Res 14: 485-490.
19. d'Addio M, Pizzigoni A, Bassi MT, Baschirotto C, Valetti C, et al. (2000)Defective intracellular transport and processing of OA1 is a major cause of ocular
albinism type 1. Hμm Mol Genet 9: 3011-3018.
20. Shen B, Rosenberg B, Orlow SJ (2001)Intracellular distribution and late endosomal effects of the ocular albinism type 1 gene product: consequences of disease-causing mutations and implications for melanosome biogenesis. Traffic 2: 202-211.
21. Samaraweera P, Shen B, Newton JM, Barsh GS, Orlow SJ (2001)The mouse ocular
albinism 1 gene product is an endolysosomal protein. Exp Eye Res 72: 319-329.
22. Schiaffino MV, Baschirotto C, Pellegrini G, Montalti S, Tacchetti C, et al. (1996)The ocular albinism type 1 gene product is a membrane glycoprotein localized to melanosomes. Proc Natl Acad Sci U S A 93: 9055-9060.
23. Ilia M, Jeffery G (2000)Retinal cell addition and rod production depend on early stages of ocular melanin synthesis. J Comp Neurol 420: 437-444.
24. Ilia M, Jeffery G (1999)Retinal mitosis is regulated by dopa, a melanin precursor that may influence the time at which cells exit the cell cycle: analysis of patterns of cell production in pigmented and albino retinae. J Comp Neurol 405: 394-405.
25. Ito S (2003)The IFPCS presidential lecture: a chemist's view of melanogenesis. Pigment Cell Res 16: 230-236.
26. Martinez-Zaguilan R, Tompkins LS, Gillies RJ, Lynch RM (2006)Simultaneous analysis of intracellular pH and Ca2+ from cell populations. Methods Mol Biol 312: 269-287.
27. Ferguson SS, Caron MG (2004)Green fluorescent protein-tagged beta-arrestin translocation as a measure of G protein-coupled receptor activation. Methods in Molecular Biology 237: 121-126.
28. Barak LS, Warabi K, Feng X, Caron MG, Kwatra MM (1999)Real-time visualization of the cellular redistribution of G protein-coupled receptor kinase 2 and beta- arrestin 2 during homologous desensitization of the substance P receptor. J Biol Chem 274: 7565-7569.
29. Zhang J, Barak LS, Anborgh PH, Laporte SA, Caron MG, et al. (1999)Cellular trafficking of G protein-coupled receptor/beta- arrestin endocytic complexes. J Biol Chem 274: 10999-11006.
30. Tohgo A, Choy EW, Gesty-Palmer D, Pierce KL, Laporte S, et al. (2003)The stability of the G protein-coupled receptor-beta-arrestin interaction determines the mechanism and functional consequence of ERK activation. J Biol Chem 278: 6258-6267.
31. Ferguson SS, Zhang J, Barak LS, Caron MG (1998)Molecular mechanisms of G protein-coupled receptor desensitization and resensitization. Life Sci 62: 1561-1565.
32. Barak LS, Ferguson SS, Zhang J, Caron MG (1997)A beta-arrestin/green fluorescent protein biosensor for detecting G protein-coupled receptor activation. J Biol Chem 272: 27497-27500.
33. Barak LS, Ferguson SS, Zhang J, Martenson C, Meyer T, et al. (1997)Internal
trafficking and surface mobility of a functionally intact beta2-adrenergic rece
ptor-green fluorescent protein conjugate. Mol Pharmacol 51: 177-184.
34. McKay BS, Goodman B, Falk T, Sherman SJ (2006)Retinal pigment epithelial cell transplantation could provide trophic support in Parkinson's disease: Results
from an in vitro model system. Exp Neurol 201: 234-243.
35. Tombran-Tink J, Shivaram SM, Chader GJ, Johnson LV, Bok D (1995)Expression,
secretion, and age-related downregulation of pigment epitheliμm-derived factor, a serpin with neurotrophic activity. J Neurosci 15: 4992-5003.
3 6. Malchiodi-Albedi F, Feher J, Caiazza S, Formisano G, Perilli R, et al. (1998)PEDF (pigment epitheliμm-derived factor)promotes increase and maturation of pigment granules in pigment epithelial cells in neonatal albino rat retinal cultures. Int J Dev Neurosci 16: 423-432.
37. Behling KC, Surace EM, Bennett J (2002)Pigment epitheliμm-derived factor expression in the developing mouse eye. Mol placeVis 8: 449-454.
38. Aymerich MS, Alberdi EM, Martinez A, Becerra SP (2001)Evidence for pigment epitheliμm-derived factor receptors in the neural retina. Invest Ophthalmol Vis Sci 42: 3287-3293.
39. Tombran-Tink J, Chader GG, Johnson LV (1991)PEDF: a pigment epitheliμm-derived factor with potent neuronal differentiative activity. Exp Eye Res 53: 411-414.
40. Jablonski MM, Tombran-Tink J, Mrazek DA, Iannaccone A (2001)Pigment epitheliμm-derived factor supports normal Muller cell development and glutamine synthetase expression after removal of the retinal pigment epitheliμm. Glia 35: 14-25.
41. Jablonski MM, Tombran-Tink J, Mrazek DA, Iannaccone A (2000)Pigment epitheliμm-derived factor supports normal development of photoreceptor neurons and opsin expression after retinal pigment epitheliμm removal. J Neurosci 20: 7149-7157.
42. Jeffery G (1998)The retinal pigment epitheliμm as a developmental regulator of the neural retina. Eye 12 (Pt 3b): 499-503.
43. Piccirillo R, Palmisano I, Innamorati G, Bagnato P, Altimare D, et al. (2003)An unconventional dileucine-based motif and a novel cytosolic motif are required for the lysosomal and melanosomal targeting of OA1. Journal of Cell Science 119: 2003-2014.
44. Van Raamsdonk CD, Fitch KR, Fuchs H, de Angelis MH, Barsh GS (2004)Effects of G-protein mutations on skin color. Nat Genet 36: 961-968.
45. Young A, Powelson EB, Whitney IE, Raven MA, Nusinowitz S, et al. (2008)Involvement of OA1, an intracellular GPCR, and G alpha i3, its binding protein, in melanosomal biogenesis and optic pathway formation. Invest Ophthalmol Vis Sci 49:
3245-3252.
46. Hu J, Bok D (2001)A cell culture mediμm th at supports the differentiation of hμman retinal pigment epitheliμm into functionally polarized monolayers. Mol placeVis 7: 14-19.
47. Stamer WD, Golightly SF, Hosohata Y, Ryan EP, Porter AC, et al. (2001)Cannabinoid CB(1)receptor expression, activation and detection of endogenous ligand in trabecular meshwork and ciliary process tissues. Eur J Pharmacol 431: 277-286.
【特許請求の範囲】
【請求項1】
加齢黄斑変性症(AMD)を治療する方法であって、
AMDの治療に有効な量のOA1受容体のアゴニストをAMD患者に投与することからなる方法。
【請求項2】
加齢黄斑変性症(AMD)の進行を制限する方法であって、
AMDの進行の制限に有効な量のOA1受容体のアゴニストをAMDが進行する危険性のある患者に投与することからなる方法。
【請求項3】
OA1受容体のアゴニストはL−DOPAおよびL−DOPA類似体からなるグループから選択される請求項1または2に記載の方法。
【請求項4】
前記OA1受容体のアゴニストはL−DOPAを含む請求項3に記載の方法。
【請求項5】
前記OA1受容体のアゴニストはL−DOPAプロドラッグを含む請求項3に記載の方法。
【請求項6】
L−DOPAプロドラッグはL−DOPAエステルを含む請求項5に記載の方法。
【請求項7】
L−DOPAプロドラッグはL−DOPAの胆汁酸抱合体を含む請求項5に記載の方法。
【請求項8】
L−DOPAプロドラッグはジペプチドまたはトリペプチドのL−DOPA類似体を含む請求項5に記載に記載の方法。
【請求項9】
L−DOPAプロドラッグはアミドL−DOPA類似体を含む請求項5に記載に記載の方法。
【請求項10】
患者が60歳以上である請求項1−9のいずれか一項に記載の方法。
【請求項11】
前記方法がAMDを治療する方法であり、患者は滲出型AMDを有する請求項1−10のいずれか一項に記載の方法。
【請求項12】
前記方法がAMDを治療する方法であり、患者は萎縮型AMDを有する請求項1−10のいずれか一項に記載の方法。
【請求項13】
前記方法がAMDの進行を制限する方法であり、患者はドルーセン沈着物を有する請求項1−10のいずれか一項に記載の方法。
【請求項14】
患者は白色人種である請求項13に記載に記載の方法。
【請求項15】
AMDを治療する化合物を識別する方法であって、
細胞を試験化合物と接触させることからなり、該細胞は、
(a)OA1を発現している第1の細胞集団;および任意選択で
(b)OA1を発現していない第2の細胞集団;を含み、
(c)以下の(i)および(ii)のうちの一方または両方を増大させる試験化合物を陽性試験化合物と識別し;
(i)(A)試験化合物と接触させない第1の細胞集団における色素上皮誘導因子(PEDF)発現および(B)第2の細胞集団におけるPEDF発現のうちの一方又は両方に対する、第1の細胞集団におけるPEDF発現;
(ii)(A)試験化合物と接触させない第1の細胞集団における細胞内カルシウム濃度および(B)第2の細胞集団における細胞内カルシウム濃度のうちの一方又は両方に対する、第1の細胞集団における細胞内カルシウム濃度;
陽性試験化合物は、AMDの治療およびAMDの進行の制限のうちの少なくとも一方を行う候補化合物である、方法。
【請求項16】
工程(c)は、
(a)試験化合物と接触させない第1の細胞集団における色素上皮誘導因子(PEDF)発現、および
(b)第2の細胞集団におけるPEDF発現
のうちの一方又は両方に対する、第1の細胞集団におけるPEDF発現を増加させる試験化合物を陽性試験化合物であると識別することを含む請求項15に記載に記載の方法。
【請求項17】
工程(c)は、
(a)試験化合物と接触させない第1の細胞集団における細胞内カルシウム濃度、および
(b)第2の細胞集団における細胞内カルシウム濃度
のうちの一方又は両方に対する、第1の細胞集団における細胞内カルシウム濃度を増加させる試験化合物を陽性試験化合物であると識別することを含む請求項15に記載に記載の方法。
【請求項18】
第1の細胞集団および第2の細胞集団は、マウス、ラット、ハムスターおよびヒト細胞からなるグループから選択される請求項15−17のいずれか一項に記載の方法。
【請求項19】
第1の細胞集団および第2の細胞集団は、網膜色素上皮細胞である請求項15−18のいずれか一項に記載の方法。
【請求項20】
接触が0μm−10μmの間のチロシンの存在下で起こる請求項15−19のいずれか一項に記載の方法。
【請求項21】
接触がチロシナーゼ阻害剤の存在下で起こる請求項15−20のいずれか一項に記載の方法。
【請求項22】
AMDを治療する化合物を識別する方法であって、
(a)チロシナーゼ欠損の妊娠している雌の非ヒト哺乳動物に試験化合物を投与する工程であって、試験化合物は胚の光受容体および網膜神経節のうちの少なくとも一方の発達中に投与される工程;および
(b)胚または出生後の非ヒト哺乳動物における光受容体および網膜神経節のうちの少なくとも一方の発達に対する試験化合物の効果を、試験化合物を投与しなかった胚または出生後非ヒト哺乳動物における光受容体および網膜神経節のうちの少なくとも一方の発達と比較する工程であって、光受容体および網膜神経節のうちの少なくとも一方の発達を増加させる試験化合物が、AMDの治療およびAMDの進行の制限のうちの少なくとも一方を行う候補化合物である工程;からなる方法。
【請求項23】
試験化合物の投与を出生後に継続する工程をさらに含む請求項22に記載の方法。
【請求項24】
哺乳動物はマウスである請求項22または23に記載に記載の方法。
【請求項25】
試験化合物の投与は、胚7日目と胚12日目の間に開始される請求項22−24のいずれか一項に記載の方法。
【請求項26】
試験化合物の投与を生後1日目から生後14日目まで出生後に継続することをさらに含む請求項22−25のいずれか一項に記載の方法。
【請求項27】
測定される結果は、網膜ニューロンの数を測定することを含む請求項22−26のいずれか一項に記載の方法。
【請求項28】
測定される結果は、網膜神経節細胞の数を測定することを含む請求項22−27のいずれか一項に記載の方法。
【請求項29】
測定される結果は、光受容体の数を測定することを含む請求項22−28のいずれか一項に記載の方法。
【請求項30】
ビタミンC源、ビタミンE源、β−カロチン源、亜鉛源、および銅源の組み合わせを患者に投与することをさらに含む請求項1−14のいずれか一項に記載の方法。
【請求項31】
450mgから600mgの間のビタミンC;
400IUから540IUの間のビタミンE;
17.2mgから28mgの間のβカロチン;
68mgから100mgの間の亜鉛;および
1.6mgから2.4mgの間の銅;
を投与することを含む請求項30に記載の方法。
【請求項32】
(a)AMDの進行を治療または制限するのに有効な量のL−DOPAまたはL−DOPA類似体;および
(b)ビタミンC源、ビタミンE源、ビタミンA源、亜鉛源および銅源を含む、AMDの進行を治療または制限するのに有効な量の組成物;
を含む組成物。
【請求項33】
5mgから1500mgの間のL−DOPAまたはL−DOPA類似体;
450mgから600mgの間のビタミンC;
400IUから540IUの間のビタミンE;
17.2mgから28mgの間のβカロチン;
68mgから100mgの間の亜鉛;および
少なくとも1.6mgの銅;
を含む請求項32に記載の組成物。
【請求項1】
加齢黄斑変性症(AMD)を治療する方法であって、
AMDの治療に有効な量のOA1受容体のアゴニストをAMD患者に投与することからなる方法。
【請求項2】
加齢黄斑変性症(AMD)の進行を制限する方法であって、
AMDの進行の制限に有効な量のOA1受容体のアゴニストをAMDが進行する危険性のある患者に投与することからなる方法。
【請求項3】
OA1受容体のアゴニストはL−DOPAおよびL−DOPA類似体からなるグループから選択される請求項1または2に記載の方法。
【請求項4】
前記OA1受容体のアゴニストはL−DOPAを含む請求項3に記載の方法。
【請求項5】
前記OA1受容体のアゴニストはL−DOPAプロドラッグを含む請求項3に記載の方法。
【請求項6】
L−DOPAプロドラッグはL−DOPAエステルを含む請求項5に記載の方法。
【請求項7】
L−DOPAプロドラッグはL−DOPAの胆汁酸抱合体を含む請求項5に記載の方法。
【請求項8】
L−DOPAプロドラッグはジペプチドまたはトリペプチドのL−DOPA類似体を含む請求項5に記載に記載の方法。
【請求項9】
L−DOPAプロドラッグはアミドL−DOPA類似体を含む請求項5に記載に記載の方法。
【請求項10】
患者が60歳以上である請求項1−9のいずれか一項に記載の方法。
【請求項11】
前記方法がAMDを治療する方法であり、患者は滲出型AMDを有する請求項1−10のいずれか一項に記載の方法。
【請求項12】
前記方法がAMDを治療する方法であり、患者は萎縮型AMDを有する請求項1−10のいずれか一項に記載の方法。
【請求項13】
前記方法がAMDの進行を制限する方法であり、患者はドルーセン沈着物を有する請求項1−10のいずれか一項に記載の方法。
【請求項14】
患者は白色人種である請求項13に記載に記載の方法。
【請求項15】
AMDを治療する化合物を識別する方法であって、
細胞を試験化合物と接触させることからなり、該細胞は、
(a)OA1を発現している第1の細胞集団;および任意選択で
(b)OA1を発現していない第2の細胞集団;を含み、
(c)以下の(i)および(ii)のうちの一方または両方を増大させる試験化合物を陽性試験化合物と識別し;
(i)(A)試験化合物と接触させない第1の細胞集団における色素上皮誘導因子(PEDF)発現および(B)第2の細胞集団におけるPEDF発現のうちの一方又は両方に対する、第1の細胞集団におけるPEDF発現;
(ii)(A)試験化合物と接触させない第1の細胞集団における細胞内カルシウム濃度および(B)第2の細胞集団における細胞内カルシウム濃度のうちの一方又は両方に対する、第1の細胞集団における細胞内カルシウム濃度;
陽性試験化合物は、AMDの治療およびAMDの進行の制限のうちの少なくとも一方を行う候補化合物である、方法。
【請求項16】
工程(c)は、
(a)試験化合物と接触させない第1の細胞集団における色素上皮誘導因子(PEDF)発現、および
(b)第2の細胞集団におけるPEDF発現
のうちの一方又は両方に対する、第1の細胞集団におけるPEDF発現を増加させる試験化合物を陽性試験化合物であると識別することを含む請求項15に記載に記載の方法。
【請求項17】
工程(c)は、
(a)試験化合物と接触させない第1の細胞集団における細胞内カルシウム濃度、および
(b)第2の細胞集団における細胞内カルシウム濃度
のうちの一方又は両方に対する、第1の細胞集団における細胞内カルシウム濃度を増加させる試験化合物を陽性試験化合物であると識別することを含む請求項15に記載に記載の方法。
【請求項18】
第1の細胞集団および第2の細胞集団は、マウス、ラット、ハムスターおよびヒト細胞からなるグループから選択される請求項15−17のいずれか一項に記載の方法。
【請求項19】
第1の細胞集団および第2の細胞集団は、網膜色素上皮細胞である請求項15−18のいずれか一項に記載の方法。
【請求項20】
接触が0μm−10μmの間のチロシンの存在下で起こる請求項15−19のいずれか一項に記載の方法。
【請求項21】
接触がチロシナーゼ阻害剤の存在下で起こる請求項15−20のいずれか一項に記載の方法。
【請求項22】
AMDを治療する化合物を識別する方法であって、
(a)チロシナーゼ欠損の妊娠している雌の非ヒト哺乳動物に試験化合物を投与する工程であって、試験化合物は胚の光受容体および網膜神経節のうちの少なくとも一方の発達中に投与される工程;および
(b)胚または出生後の非ヒト哺乳動物における光受容体および網膜神経節のうちの少なくとも一方の発達に対する試験化合物の効果を、試験化合物を投与しなかった胚または出生後非ヒト哺乳動物における光受容体および網膜神経節のうちの少なくとも一方の発達と比較する工程であって、光受容体および網膜神経節のうちの少なくとも一方の発達を増加させる試験化合物が、AMDの治療およびAMDの進行の制限のうちの少なくとも一方を行う候補化合物である工程;からなる方法。
【請求項23】
試験化合物の投与を出生後に継続する工程をさらに含む請求項22に記載の方法。
【請求項24】
哺乳動物はマウスである請求項22または23に記載に記載の方法。
【請求項25】
試験化合物の投与は、胚7日目と胚12日目の間に開始される請求項22−24のいずれか一項に記載の方法。
【請求項26】
試験化合物の投与を生後1日目から生後14日目まで出生後に継続することをさらに含む請求項22−25のいずれか一項に記載の方法。
【請求項27】
測定される結果は、網膜ニューロンの数を測定することを含む請求項22−26のいずれか一項に記載の方法。
【請求項28】
測定される結果は、網膜神経節細胞の数を測定することを含む請求項22−27のいずれか一項に記載の方法。
【請求項29】
測定される結果は、光受容体の数を測定することを含む請求項22−28のいずれか一項に記載の方法。
【請求項30】
ビタミンC源、ビタミンE源、β−カロチン源、亜鉛源、および銅源の組み合わせを患者に投与することをさらに含む請求項1−14のいずれか一項に記載の方法。
【請求項31】
450mgから600mgの間のビタミンC;
400IUから540IUの間のビタミンE;
17.2mgから28mgの間のβカロチン;
68mgから100mgの間の亜鉛;および
1.6mgから2.4mgの間の銅;
を投与することを含む請求項30に記載の方法。
【請求項32】
(a)AMDの進行を治療または制限するのに有効な量のL−DOPAまたはL−DOPA類似体;および
(b)ビタミンC源、ビタミンE源、ビタミンA源、亜鉛源および銅源を含む、AMDの進行を治療または制限するのに有効な量の組成物;
を含む組成物。
【請求項33】
5mgから1500mgの間のL−DOPAまたはL−DOPA類似体;
450mgから600mgの間のビタミンC;
400IUから540IUの間のビタミンE;
17.2mgから28mgの間のβカロチン;
68mgから100mgの間の亜鉛;および
少なくとも1.6mgの銅;
を含む請求項32に記載の組成物。
【図1E】

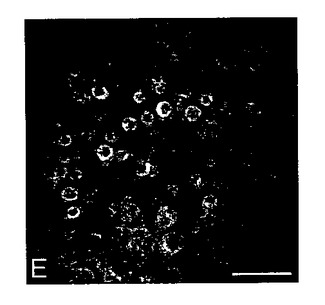
【図1F】
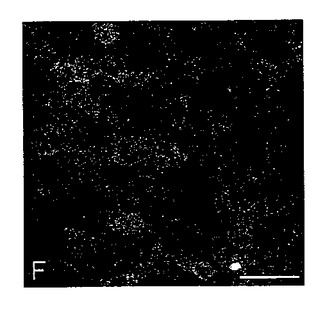
【図3E】
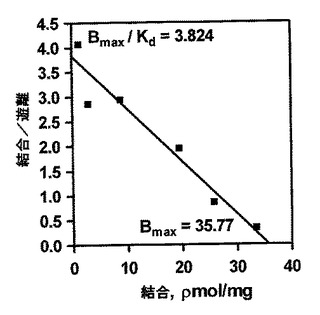
【図6A】
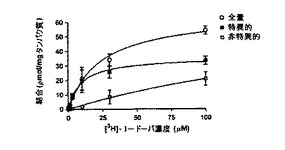
【図6B】
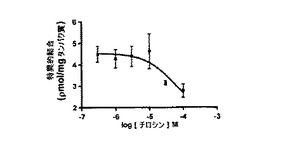
【図1A】
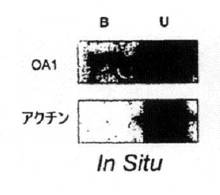
【図1B】
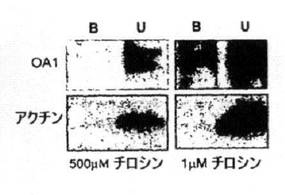
【図1C】
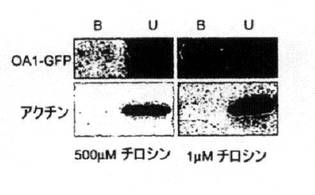
【図1D】
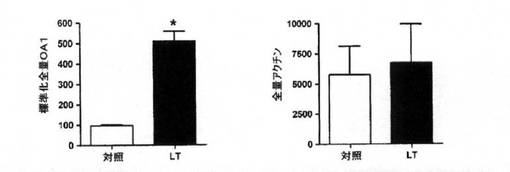
【図2A】
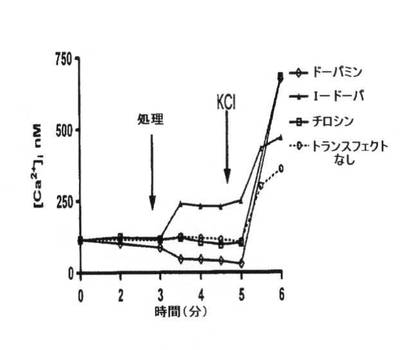
【図2B】
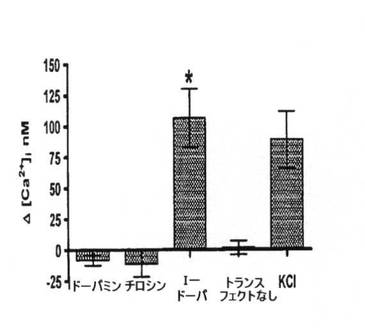
【図2C】
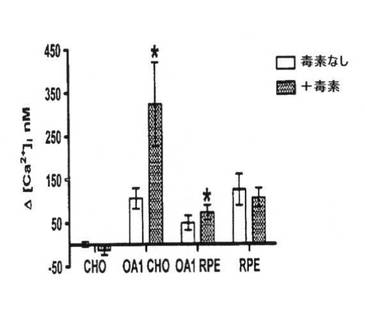
【図2D】
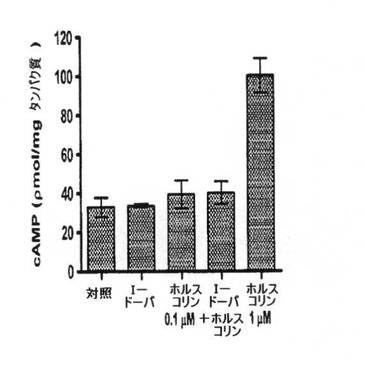
【図3A】
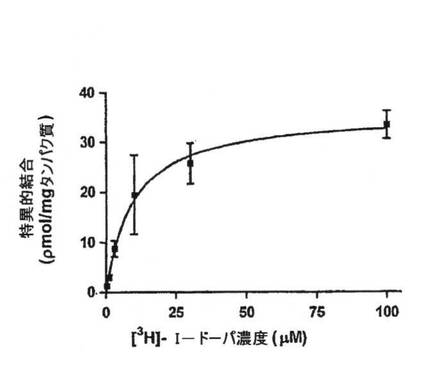
【図3B】
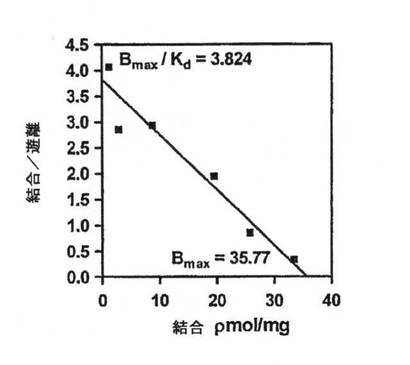
【図3C】
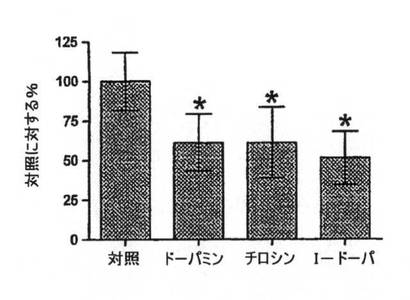
【図3D】
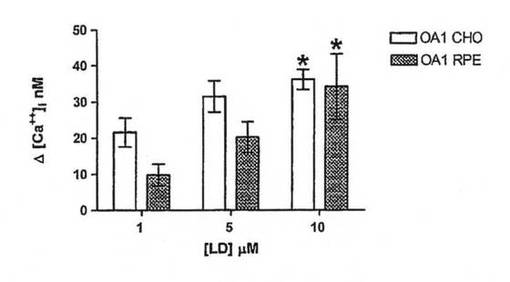
【図4】
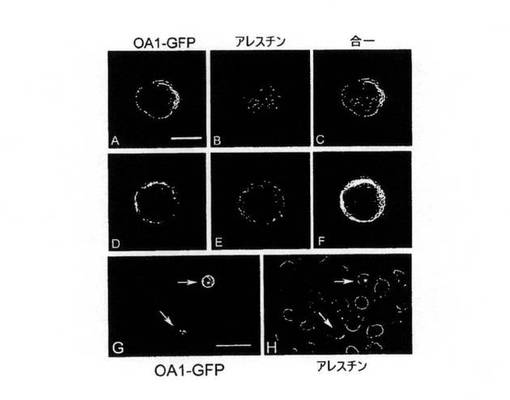
【図5A】
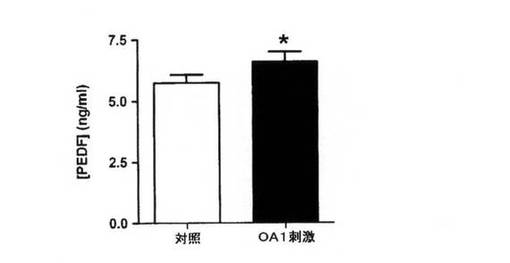
【図5B】
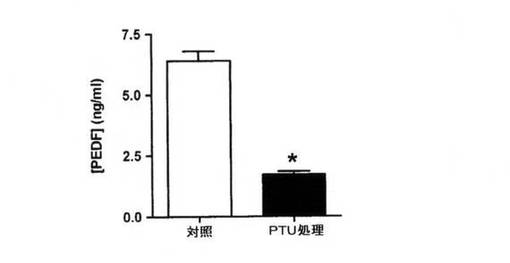
【図5C】
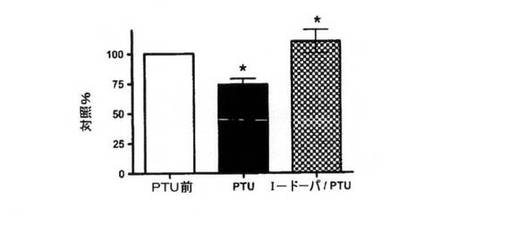
【図7】
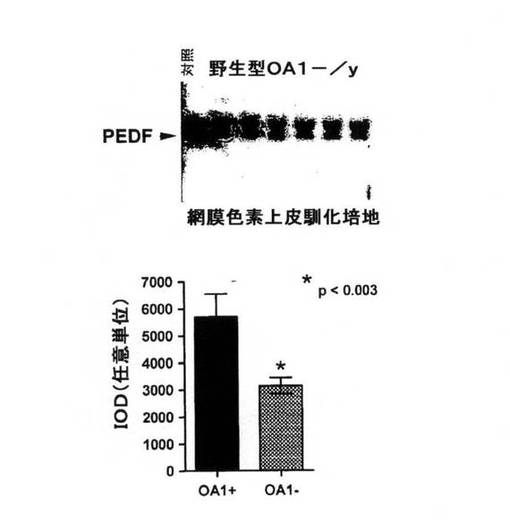
【図8A】
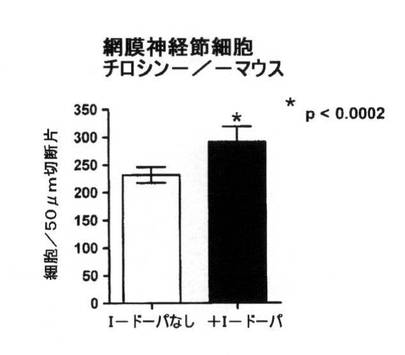
【図8B】
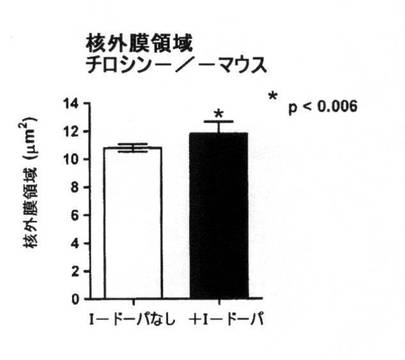
【図8C】

【図1F】
【図3E】
【図6A】
【図6B】
【図1A】
【図1B】
【図1C】
【図1D】
【図2A】
【図2B】
【図2C】
【図2D】
【図3A】
【図3B】
【図3C】
【図3D】
【図4】
【図5A】
【図5B】
【図5C】
【図7】
【図8A】
【図8B】
【図8C】
【公表番号】特表2011−520788(P2011−520788A)
【公表日】平成23年7月21日(2011.7.21)
【国際特許分類】
【出願番号】特願2011−505240(P2011−505240)
【出願日】平成21年4月17日(2009.4.17)
【国際出願番号】PCT/US2009/041021
【国際公開番号】WO2009/129497
【国際公開日】平成21年10月22日(2009.10.22)
【出願人】(510273732)アリゾナ ボード オブ リージェンツ ア ボディー コープ.オブ ザ ステイト オブ アリゾナ アクティング フォー アンド オン ビハーフ オブ ザ ユニバーシティ オブ アリゾナ (1)
【氏名又は名称原語表記】ARIZONA BOARD OF REGENTS,A BODY CORP.OF THE STATE OF ARIZONA,ACTING FOR AND ON BEHALF OF THE UNIVERSITY OF ARIZONA
【Fターム(参考)】
【公表日】平成23年7月21日(2011.7.21)
【国際特許分類】
【出願日】平成21年4月17日(2009.4.17)
【国際出願番号】PCT/US2009/041021
【国際公開番号】WO2009/129497
【国際公開日】平成21年10月22日(2009.10.22)
【出願人】(510273732)アリゾナ ボード オブ リージェンツ ア ボディー コープ.オブ ザ ステイト オブ アリゾナ アクティング フォー アンド オン ビハーフ オブ ザ ユニバーシティ オブ アリゾナ (1)
【氏名又は名称原語表記】ARIZONA BOARD OF REGENTS,A BODY CORP.OF THE STATE OF ARIZONA,ACTING FOR AND ON BEHALF OF THE UNIVERSITY OF ARIZONA
【Fターム(参考)】
[ Back to top ]