
効率的な人工多能性幹細胞の樹立方法
本発明は、(a) Oct3/4もしくはそれをコードする核酸、(b) Klf4もしくはそれをコードする核酸、および(c) Sox2もしくはそれをコードする核酸、並びに(d1) L-Mycもしくはそれをコードする核酸および/または(d2) p53の機能阻害物質を、体細胞に接触させることを含む、iPS細胞の製造方法を提供する。好ましくは、(a) Oct3/4をコードする核酸、(b) Klf4をコードする核酸、(c) Sox2をコードする核酸、(d1) L-Mycをコードする核酸および、(e) Lin28もしくはLin28bをコードする核酸を、ベクターの複製に必要なベクター要素の5’側および3’側にloxP配列を同方向に配置したエピソーマルベクター中に挿入し、(d2) p53に対するshRNAをコードする核酸を一過的な発現を提供するベクター(プラスミドベクター等)中に挿入して、これらすべての核酸を体細胞に導入する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、効率的な人工多能性幹(以下、iPSという)細胞の樹立方法およびそのための薬剤に関する。より詳細には、本発明は、(a) Oct3/4もしくはそれをコードする核酸、(b) Klf4もしくはそれをコードする核酸、および(c) Sox2もしくはそれをコードする核酸、並びに(d1) L-Mycもしくはそれをコードする核酸および/または(d2) p53の機能阻害物質を、体細胞に接触させることによる、iPS細胞の樹立方法、上記(a)-(c)並びに(d1)および/または(d2)からなるiPS細胞誘導剤に関する。本発明はまた、上記(a)-(c)並びに(d1)および/または(d2)の核酸因子を含むエピソーマルベクターあるいは該エピソーマルベクターとloxP配列とを組み合わせたベクター、特に早期自己消失型であるエピソーマルベクター、およびそれを用いて、外来核酸因子のゲノムへの組込みを介さずに、該核酸因子の消失したiPS細胞を迅速に樹立する方法に関する。
【背景技術】
【0002】
近年、マウスおよびヒトのiPS細胞が相次いで樹立された。Yamanakaらは、マウス由来の線維芽細胞に、Oct3/4, Sox2, Klf4及びc-Myc遺伝子を導入し強制発現させることによって、iPS細胞を誘導した [WO 2007/069666 A1; Takahashi, K. and Yamanaka, S., Cell, 126: 663-676 (2006)]。その後、c-Myc遺伝子を除いた3因子によってもiPS細胞を作製できることが明らかとなった [Nakagawa, M. et al., Nat. Biotechnol., 26: 101-106 (2008)]。さらに、Yamanakaらは、ヒトの皮膚由来線維芽細胞にマウスと同様の4遺伝子を導入することにより、iPS細胞を樹立することに成功した [WO 2007/069666 A1; Takahashi, K. et al., Cell, 131:861-872 (2007)]。一方、Thomsonらのグループは、Klf4とc-Mycの代わりにNanogとLin28を使用してヒトiPS細胞を作製した[WO 2008/118820 A2; Yu, J. et al., Science, 318: 1917-1920 (2007)]。
【0003】
しかし、iPS細胞の樹立効率は1%以下と低く、特に、iPS細胞から分化した組織や個体において腫瘍化が懸念されるc-Mycを除く3因子(Oct3/4, Sox2, Klf4)を体細胞に導入してiPS細胞を作製した場合、その樹立効率が極めて低いという問題点がある。
【0004】
レトロウイルスやレンチウイルスなどのウイルス性ベクターは、非ウイルス性ベクターに比べて遺伝子導入効率が高く、そのためiPS細胞を容易に作製することができるという点で優れたベクターである。しかし、レトロウイルスやレンチウイルスは染色体中に組み込まれてしまうため、iPS細胞の臨床応用を考慮すると安全面で問題がある。そのため、アデノウイルスベクターやプラスミドなどの非ウイルス性ベクターを用いて染色体への組込みがないiPS細胞が報告されているが [Stadtfeld, M. et al., Science, 322: 945-949 (2008); Okita, K. et al., Science, 322: 949-953 (2008); Yu, J. et al., Science, 324: 797-801 (2009)]、レトロウイルスやレンチウイルスに比べると樹立効率は低い。また、iPS細胞の選択下では初期化因子の持続的な高発現が要求されるためか、一般的に組込みが起こりにくいとされているプラスミドベクターを用いても、ある頻度で初期化因子が染色体に組み込まれた安定発現株が得られる場合がある [Okita, K. et al., Science, 322: 949-953 (2008); Kaji, K. et al., Nature, 458: 771-775 (2009)]。
【0005】
そこで、まずレトロウイルスやレンチウイルスを用いてiPS細胞を樹立した後で、外来遺伝子を染色体から除去することにより、樹立効率と安全面を両立させようとする試みがなされている。例えば、レンチウイルスとCre-loxPシステムとを組み合わせた手法が報告されている [Chang, C.W. et al., Stem Cells, 27: 1042-1049 (2009); Soldner, F. et al., Cell, 136: 964-977(2009)]。しかし、これらの報告では、Creリコンビナーゼ処理後に残るloxP配列より外側のLTR配列が近傍の癌遺伝子を活性化するリスクを最小限にするため、LTR内部にloxP配列が挿入され、初期化因子の転写のためにCMVやEF1αといった別のプロモーターが挿入された複雑なコンストラクトが使用されており、より構築が容易なベクターの開発が望まれる。piggyBacトランスポゾンを用いて外来核酸因子を完全に消失させることもできるが [Kaji, K. et al., Nature, 458: 771-775 (2009)]、一過的にもゲノムへの組込みを介するという点で、内因性遺伝子の擾乱を引き起こす可能性を否定できない。
【0006】
一方、染色体外で安定に自律複製可能なエピソーマルベクターを用いる方法では、上記したiPS細胞樹立効率の低さに加え、薬剤選択の中止によるベクターの自然消失効率も低頻度で、かつ時間を要するため [Yu, J. et al., Science, 324: 797-801 (2009)]、iPS細胞の樹立効率の改善とともに、短時間で効率よくベクターを除去する方法が求められる。
【0007】
さらに、ヒトiPS細胞の臨床応用を念頭におく場合の別の問題点として、iPS細胞の樹立および維持培養に際し、血清やフィーダー細胞などの異種動物由来成分の混入による汚染が挙げられる。そのため、初期化因子の導入からヒトiPS細胞の樹立および維持培養までをすべて異種成分不含(Xeno-free)の条件下で行うことが望ましいが、これまでvirus-freeで樹立されたヒトiPS細胞では、初期化因子の導入からiPS細胞の樹立・維持培養までの少なくとも一部において、異種成分が使用されている [Okita, K. et al., Science, 322: 949-953(2008); Yu, J. et al., Science, 324: 797-801 (2009); Kaji, K. et al., Nature, 458: 771-775 (2009)]。一方、Xeno-freeの条件下で樹立されたヒトiPS細胞は、いずれもレトロウイルスやレンチウイルスにより初期化遺伝子が導入されたものであり、virus-freeで作製されたものではない [Rodoriguez-Piza, I. et al., Stem Cells, 28: 36-44 (2010); Ross, P.J. et al., Stem Cells Dev., 2009 Dec 23. (Epub ahead of print)]。
【発明の概要】
【発明が解決しようとする課題】
【0008】
本発明の目的は、臨床応用に適した安全なヒトiPS細胞を効率よく樹立することである。したがって、本発明の第一の課題は、iPS細胞、特にヒトiPS細胞の樹立効率を改善する手段を提供することであり、それを用いた効率的なiPS細胞の製造方法を提供することである。また、本発明の第二の課題は、外来核酸因子のゲノムへの組込みを一切介さずに、該核酸因子の消失したiPS細胞を迅速に樹立する方法を提供することである。さらに、本発明の第三の課題は、初期化因子の導入時からiPS細胞の樹立および維持培養に至るまで、ウイルスおよび異種成分を一切用いることなく(即ち、virus-freeかつxeno-freeの条件下で)ヒトiPS細胞を作製し、以ってヒト臨床に安全に使用することができるヒトiPS細胞を提供することである。
【課題を解決するための手段】
【0009】
本発明者らは、上記の課題を解決すべく、まずレトロウイルスベクターを用いて初期化遺伝子の好適な組み合わせを検討した。エピソーマルベクターを用いてヒトiPS細胞を樹立したYu, J. et al., Science, 324: 797-801 (2009)で使用された6因子(Oct3/4, Klf4, c-Myc, Sox2, Nanog, Lin28 (SV40 Large T抗原は樹立効率改善物質と位置づけ、初期化遺伝子から除外した))を基に、Nanogを除いた5因子および、さらにc-MycをL-Mycに置換した5因子を用いてヒト皮膚線維芽細胞(HDF)からヒトiPS細胞を誘導したところ、意外にもNanogを除いた5因子の方が、6因子の場合よりも効率よくヒトiPS細胞が樹立された。さらにc-MycをL-Mycに置換することにより、樹立効率は顕著に改善された。そこで、実際にエピソーマルベクターを用いて、6因子とc-MycをL-Mycに置換した5因子とでヒトiPS細胞樹立効率を比較したところ、同様にc-MycをL-Mycに置換した5因子を用いることにより、6因子と比較して著しく樹立効率が上昇した。
【0010】
次に、6因子またはc-MycをL-Mycに置換した5因子に加えて、p53に対するshRNAをコードするエピソーマルベクターをHDFに導入したところ、p53の機能を阻害することにより、6因子でもc-MycをL-Mycに置換した5因子と同程度のiPS細胞が得られ、該5因子にp53機能阻害を組み合わせた場合には、さらに顕著に樹立効率が上昇した。また、遺伝子導入後のHDFを継代する際に、SNL細胞でなく胎仔マウス線維芽細胞(MEF)をフィーダー細胞として用いることにより、ヒトiPS細胞の樹立効率が格段に向上することが見出された。
【0011】
本発明者らは、導入したエピソーマルベクターをiPS細胞樹立後に速やかに細胞から脱落させ得るように、該ベクターの自律複製に必要なベクター要素の両端にloxP配列を配置し、Creリコンビナーゼを作用させることにより該ベクター要素をベクターから切り出せるよう設計していたが、ヒトiPS細胞樹立後に細胞のゲノムDNAと染色体外DNAをそれぞれ単離して導入遺伝子の存在を調べたところ、いずれのDNA中にも導入したベクターは検出されず、意外にも該ベクターは、Creリコンビナーゼを用いるまでもなく速やかに細胞から脱落する、早期自己消失型ベクターであることが見出された。
【0012】
本発明者らはさらに、上記エピソーマルベクターを用いることにより、初期化因子の導入時からiPS細胞の樹立および維持培養に至るまで、完全にvirus-freeかつxeno-freeの条件下で、ヒトiPS細胞を作製することに成功した。
【0013】
以上の結果から、本発明者らは、初期化因子からNanogを除去するとともに、c-Mycに代えてL-Mycを用いるか、その代わりに、あるいはそれに加えて、p53機能阻害物質を用いることによりヒトiPS細胞の樹立効率を顕著に改善し得ること、エピソーマルベクターの設計を工夫することにより、外来核酸因子がゲノムに組み込まれることなく細胞から消失したiPS細胞を迅速に取得し得ること、並びに上記初期化因子とエピソーマルベクターとを組み合わせることにより、virus-freeかつxeno-freeの条件下で、ヒトiPS細胞を作製し得ることを見出し、本発明を完成するに至った。
【0014】
すなわち、本発明は以下の通りのものである。
[1] (a) Oct3/4もしくはそれをコードする核酸、(b) Klf4もしくはそれをコードする核酸、および(c) Sox2もしくはそれをコードする核酸、並びに(d1) L-Mycもしくはそれをコードする核酸および/または(d2) p53の機能阻害物質を、体細胞に接触させることを含む、iPS細胞の製造方法。
[2] さらに(e) Lin28もしくはLin28bまたはそれをコードする核酸を体細胞に接触させることを含む、上記[1]記載の方法。
[3] p53の機能阻害物質が、p53に対するsiRNA、shRNAおよびそれらをコードするDNAからなる群より選択される核酸である、上記[1]または[2]記載の方法。
[4] (a) Oct3/4をコードする核酸、(b) Klf4をコードする核酸、(c) Sox2をコードする核酸、(d1) L-Mycをコードする核酸および/または(d2) p53に対するshRNAをコードする核酸、並びに(e) Lin28もしくはLin28bをコードする核酸を、体細胞に導入することを含む、上記[2]または[3]記載の方法。
[5] 前記の(a)、(b)、(c)、(d1)および(e)からなる群より選択される少なくとも1つの核酸がエピソーマルベクターの形態で導入される、上記[4]記載の方法。
[6] (d2) p53に対するshRNAをコードする核酸が細胞内で自律複製できないプラスミドベクターの形態で導入される、上記[4]または[5]記載の方法。
[7] エピソーマルベクターが、50%以上の頻度で、5継代までにiPS細胞から脱落する自己消失型ベクターである、上記[5]または[6]記載の方法。
[8] エピソーマルベクターが、前記の核酸の複製に必要なベクター要素の5’側および3’側にloxP配列を同方向に配置したものである、上記[5]〜[7]のいずれかに記載の方法。
[9] 細胞をCreリコンビナーゼで処理する工程を含まない、上記[8]記載の方法。
[10] 体細胞がヒト由来である、上記[1]〜[9]のいずれかに記載の方法。
[11] 前記の(a)、(b)、(c)並びに(d1)および/または(d2)、あるいはさらに(e)の因子を体細胞に接触させる時点から、iPS細胞が樹立されるまでの間、細胞を非ヒト動物由来の成分の非存在下で培養することを含む、上記[10]記載の方法。
[12] ヒト細胞をフィーダー細胞として用いるか、フィーダー細胞を用いないことを特徴とする、上記[11]記載の方法。
[13] フィーダー細胞が体細胞と同一個体に由来する、上記[12]記載の方法。
[14] (a) Oct3/4をコードする核酸、(b) Klf4をコードする核酸、(c) Sox2をコードする核酸、(d1) L-Mycをコードする核酸および/または(d2) p53に対するshRNAをコードする核酸、並びに(e) Lin28もしくはLin28bをコードする核酸を含有してなる、iPS細胞誘導促進剤。
[15] 前記の(a)、(b)、(c)、(d1)および(e)からなる群より選択される少なくとも1つの核酸がエピソーマルベクターの形態である、上記[14]記載の剤。
[16] (d2) p53に対するshRNAをコードする核酸が細胞内で自律複製できないプラスミドベクターの形態である、上記[14]または[15]記載の剤。
[17] エピソーマルベクターが、50%以上の頻度で、5継代までにiPS細胞から脱落する自己消失型ベクターである、上記[15]または[16]記載の剤。
[18] エピソーマルベクターが、前記の各核酸の複製に必要なベクター要素の5’側および3’側にloxP配列を同方向に配置したものである、上記[15]〜[17]のいずれかに記載の剤。
[19] 上記[5]〜[9]のいずれかに記載の方法により得られる、前記の各核酸がゲノムに組み込まれることなく除去されたiPS細胞。
[20] ヒト由来である、上記[19]記載のiPS細胞。
[21] 上記[11]〜[13]のいずれかの方法により得られる、非ヒト動物由来の成分の混入のないヒトiPS細胞。
[22] 体細胞の製造における、上記[19]〜[21]のいずれかに記載のiPS細胞の使用。
[23] 体細胞の製造における細胞ソースとしての、上記[19]〜[21]のいずれかに記載のiPS細胞。
【発明の効果】
【0015】
c-Mycに代わるL-Mycの使用および/またはp53の機能阻害物質の使用、並びにNanogの不使用は、iPS細胞の樹立効率を顕著に増大させることができるので、従来きわめて樹立効率の低かった6因子(Oct3/4、Klf4、Sox2、c-Myc、Nanog、Lin28)によるヒトiPS細胞、特に初期化遺伝子がゲノムに組み込まれないタイプのヒトiPS細胞の作製に特に有用である。また、独自のエピソーマルベクターを使用することで、外来核酸因子のゲノムへの組込みを介さずにiPS細胞を樹立することができ、かつiPS細胞樹立後速やかにエピソームが脱落し、従来のエピソーマルベクターに比べて、早期にベクターの脱落が起こり得る。さらに、初期化因子の導入からiPS細胞の樹立および維持までの間、完全にvirus-freeかつxeno-freeの条件でヒトiPS細胞を作製できるため、ヒトiPS細胞の再生医療への応用において極めて有用である。
【0016】
以下、本明細書において、エピソーマルベクターを用いて樹立されたiPS細胞を「epi-iPS cells」または「epi-iPSCs」と略することがある。
また導入遺伝子の組み合わせを「Y1, Y2, Y3, Y4, T1, T2, T3」と略する場合、これらの組合せは以下の表1に示すとおりである。
【0017】
【表1】

【図面の簡単な説明】
【0018】
【図1】図1は、レトロウイルスで種々の遺伝子をヒト新生児皮膚線維芽細胞に導入し、ヒトiPS細胞を樹立した結果を示すコロニーの写真である。図中、OはOct3/4を、SはSox2を、KはKlf4を、LはLin28を、NはNanogを、Mはc-Mycを、UはL-Mycを、それぞれ示す。また「O-M-L」のような因子の連結表示は、各因子の翻訳領域を2A配列を挟んで繋げたコンストラクトを指す。各写真下の数字は、非ES様コロニー数/ES様コロニー数を示す。
【図2】図2は、図1の結果の一部をグラフ化したものである。ES様コロニーの数は黒棒、非ES様コロニーの数は白棒で示す。図中「6 factors」はOct3/4, Sox2, Klf4, Lin28, Nanog、およびc-MycまたはL-Mycの各遺伝子を導入した場合、「ctrl」は前記の6遺伝子からNanogを除いた5遺伝子を導入した場合、「L-M」、「M-L」、「L-U」、「U-L」は、それぞれの因子(LはLin28、Mはc-Myc、UはL-Myc)を連結したコンストラクトとOct3/4、Sox2およびKlf4とを導入した場合を、それぞれ示す。
【図3】図3は、pCXLE-hOct4、pCXLE-hSK、pCXLE-hKSO、pCXLE-hMLN、pCX-SV40LTの5種類のプラスミドをヒト成人皮膚由来の線維芽細胞に導入して樹立したiPS細胞コロニーの写真である。左は樹立時、右は3継代目(p3)の写真を示す。
【図4】図4は、pCXLE-hOct4、pCXLE-hSK、pCXLE-hKSO、pCXLE-hMLNの4種類のプラスミドをヒト成人皮膚由来の線維芽細胞に導入して樹立したiPS細胞コロニーの写真である。左は樹立時、右は2継代目(p2)の写真を示す。
【図5】図5は、pCXLE-hOct4、pCXLE-hSK、pCXLE-hULの3種類のプラスミド(左上パネル)、pCXLE-hOct4、pCXLE-hSK、pCXLE-hMLNの3種類のプラスミド(右上パネル)、並びにpCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hULの3種類のプラスミド(左下パネル)を、ヒト成人皮膚由来の線維芽細胞に導入して樹立したiPS細胞コロニーの写真である。各パネル上段はMEF、下段はMSTO細胞をフィーダー細胞として用いたものである。なお、いちばん右側の左肩にコロニー番号の記載のない写真は非ES様コロニーの写真である。
【図6】図6は、表2の結果をグラフ化したものである。ES様コロニーの数は黒棒、非ES様コロニーの数は白棒で示す。図中左から、(1)pCXLE-hOct4、pCXLE-hSK、pCXLE-hMLN、(2)pCXLE-hOct4、pCXLE-hSK、pCXLE-hUL、(3)pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hUL、(4)pCXLE-GFP、をそれぞれ導入した結果を示す。
【図7】図7は、pCXLE-hOct4、pCXLE-hSK、pCXLE-hMLNの3種類のプラスミド(左上パネル)、pCXLE-hOct4、pCXLE-hSK、pCXLE-hULの3種類のプラスミド(左下パネル)、pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hMLNの3種類のプラスミド(右上パネル)、pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hULの3種類のプラスミド(右下パネル)を、6歳のヒト皮膚由来線維芽細胞に導入して樹立したiPS細胞コロニーの写真である。各パネル上段はMEF、下段はMSTO細胞をフィーダー細胞として用いたものである。
【図8】図8は、pCXLE-hOct4、pCXLE-hSK、pCXLE-hMLNの3種類のプラスミド(左上パネル)、pCXLE-hOct4、pCXLE-hSK、pCXLE-hULの3種類のプラスミド(左下パネル)、pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hMLNの3種類のプラスミド(右上パネル)、pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hULの3種類のプラスミド(右下パネル)を、8カ月のヒト皮膚由来線維芽細胞に導入して樹立したiPS細胞コロニーの写真である。各パネル上段はMEF、下段はMSTO細胞をフィーダー細胞として用いたものである。
【図9】図9は、表3の結果(TIG120を用いた場合)をグラフ化したものである。ES様コロニーの数は黒棒、非ES様コロニーの数は白棒で示す。図中左から、(1)pCXLE-hOct4、pCXLE-hSK、pCXLE-hMLN、(2)pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hMLN、(3)pCXLE-hOct4、pCXLE-hSK、pCXLE-hUL、(4)pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hUL、(5)pCXLE-GFP、をそれぞれ導入した結果を示す。
【図10】図10は、表3の結果(TIG121を用いた場合)をグラフ化したものである。ES様コロニーの数は黒棒、非ES様コロニーの数は白棒で示す。図中左から、Fig. 9の説明における(1)〜(5)と同じである。
【図11】図11は、表3の結果(TIG120を用いた場合)をフィーダー細胞ごとにグラフ化したものである。図中左から、Fig. 9の説明における(1)〜(5)と同じである。
【図12】図12は、5種類の異なるiPS細胞を用いて、外来遺伝子 (Klf4、c-Myc、OriP) のゲノム中への組み込みの有無等を調べたGenomic PCRの結果を示す図である。図中「L」はlong DNAでの結果を、「S」はshort DNAでの結果を示す。「347A1」は実施例2で樹立されたiPSコロニーについての結果を、「349A1」は実施例3で樹立されたiPSコロニーについての結果を、「341A5」はOct3/4, Sox2, Klf4, c-Myc, Lin28, Nanogの6遺伝子をヒト胎児由来線維芽細胞に導入して樹立したiPSコロニーについての結果を、「345A1」は「347A1」と同じ遺伝子を導入して得られた別のiPSコロニーについての結果を、「352A3」は「341A5」と同じ遺伝子を導入して得られた別のiPSコロニーについての結果を、それぞれ示す。尚、HDFは遺伝子未導入のヒト胎児由来線維芽細胞ゲノムでの結果を、retroはOct3/4, Sox2, Klf4, c-Mycの4遺伝子をレトロウイルスベクターを用いてヒト胎児由来線維芽細胞に導入して樹立したiPSコロニーについての結果を、それぞれ示す。図中、endo (白矢頭) は内在性遺伝子を、Tg(黒矢頭)は外来遺伝子をそれぞれ示す。
【図13】図13は、pCX-EGFP、pCXE-EGFPおよびpCXLE-EGFPをHDFに導入後6日目および14日目の細胞の蛍光写真(GFP観察像)である。
【図14】図14は、p53shRNAをプラスミドベクター(EBNA-1とoriPを有さないプラスミドベクター)で導入した場合にもiPS細胞が樹立できるかどうかを調べた結果を示すグラフである。ES様コロニーの数は黒棒、非ES様コロニーの数は白棒で示す。図中左から、(1) pCXLE-hOct4、pCXLE-hSK、pCXLE-hMLN、(2) pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hMLN、(3) pCXLE-hOct4、pCXLE-hSK、pCXLE-hMLN、pSilencer-shp53、(4) pCXLE-hOct4、pCXLE-hSK、pCXLE-hUL、(5) pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hUL、(6) pCXLE-hOct4、pCXLE-hSK、pCXLE-hUL、pSilencer-shp53、(7) pCXLE-EGFP、をそれぞれ導入した結果を示す。
【図15】図15は、pCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hULをHLA 4座ホモの健常人歯髄幹細胞DP74株およびDP94株に導入して樹立したiPS細胞コロニーの写真である。
【図16】図16は、歯髄幹細胞DP74株に種々の初期化遺伝子を導入して得られたES様コロニーをカウントした結果を示すグラフである。ES様コロニーの数は黒棒、非ES様コロニーの数は白棒で示す。図中左から、(1)pCXLE-hOct4、pCXLE-hSK、pCXLE-hMLN、(2)pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hMLN、(3)pCXLE-hOct4、pCXLE-hSK、pCXLE-hUL、(4)pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hUL、(5)pEP4-EO2S-ET2K、pEP4-EO2S-EN2K、pCEP4-M2L(図中Thomson’s mix1)、(6)pEP4-EO2S-ET2K、pEP4-EO2S-Ck2M-EN2L(図中Thomson’s mix2)、(7)pEP4-EO2S-ET2K、pEP4-EO2S-EN2L、pEP4-EO2S-EM2K(図中Thomson’s mix3)、(8)pCXLE-GFP、をそれぞれ導入した結果を示す。
【図17】図17は、図16と同じ実験をDP94株を用いて行った結果を示すグラフである。
【図18】図18は、エピソーマルベクターで導入したEGFPの発現量(蛍光強度)を週ごとに測定した結果を示す図である。上図:蛍光強度をFACS解析した図。横軸は蛍光強度を、縦軸は細胞数を示す。また、チャート内の%値はGFP陽性細胞の割合を示す。下図:細胞の蛍光写真(GFP陽性像)。
【図19】図19は、樹立したiPS細胞18クローンが有するエピソーマルベクターのコピー数をリアルタイムPCRで調べた結果を示すグラフである。縦軸は1×104細胞あたりのエピソーマルベクターのコピー数を示す。「elepo 6d」は初期化遺伝子導入後6日目の細胞を示す。また「origin」は導入に用いたHDFを示す。
【図20】図20は、樹立したiPS細胞9クローンにおけるOct3/4の発現量を定量PCRで調べた結果を示すグラフである。白棒は外来性および内在性の遺伝子発現量(トータル発現量)を、黒棒は外来性の遺伝子発現量を、それぞれ示す。図中「elepoD4」は遺伝子導入後4日目の細胞を、また「KhES1」および「KhES3」はヒトES細胞を、それぞれ示す。
【図21】図21は、図20と同じ実験をSox2、Klf4、L-MycおよびLin28について行った結果を示すグラフである。
【図22】図22は、皮膚由来線維芽細胞(TIGおよびHDF)より樹立したヒトiPS細胞、もとのTIGおよびHDF、並びにヒトES細胞のDNAマイクロアレイ解析による各遺伝子間の発現量の相違に基づきクラスタリング解析を行った結果を示す図である。
【図23】図23は、ヒトES細胞(KhES3)とHDF(左図)、およびヒトES細胞(KhES3)とエピソーマルベクターを用いて樹立したヒトiPS細胞(右図)との間で遺伝子発現パターンに違いがあるかどうかを調べるために、DNAマイクロアレイの結果をScatter Plot解析した結果を示す図である。
【図24】図24は、エピソーマルベクターを用いて樹立したヒトiPS細胞のCGHアレイ解析の結果を示す図である。遺伝子導入に用いたTIG細胞(TIG120)に対する変動で示した。
【図25】図25は、DP74株をフィーダー細胞として用い、エピソーマルベクターで樹立したiPS細胞をリプロセル培地を用いて培養した1継代目の細胞の形態を示す写真(右)である。左はMSTOをフィーダー細胞として用いた場合の写真である。
【図26】図26は、Xeno-free(Xeno-free培地使用、およびフィーダー細胞不使用)の条件下で、エピソーマルベクターで樹立したiPS細胞を培養した1継代目の細胞の形態を示す写真である。左は通常のリプロセル培地を用いた場合の写真である。
【図27】図27は、Xeno-free(Xeno-free培地使用、およびDP74株をフィーダー細胞として使用)の条件下で、エピソーマルベクターで樹立したiPS細胞を培養した1継代目の細胞の形態を示す写真である。
【図28】図28は、Xeno-free条件下でpCXLE-EGFPをDP74に導入した結果を示す図である。左側は通常の条件で導入した結果(コントロール)を示す。上図:位相差像、下図:GFP観察像。
【図29】図29は、Xeno-free条件下で遺伝子導入後、表9に示す6種の条件下で細胞培養し、導入から26日目に出現したES細胞様コロニーの形態を示す写真である。
【図30】図30は、エピソーマル発現ベクターの構造を示す。Y4混合物には、3つのプラスミドを用いた(図中、pCXLE-hOct4-shp53はpCXLE-hOct3/4-shp53と記載される)。初期化因子(OCT3/4, SOX2, KLF4, L-MYC, LIN28およびp53に対するshRNA (shRNA for p53))は黒で示している。プロモーター(CAG)、WPRE、ポリアデニレーションシグナル(pA)、EBNA-1、OriPおよび2つのloxP部位も示している。
【図31】図31は、epi-iPSCクローンに残存するエピソーマルベクターのコピー数を示す。A: DP由来epip-iPS細胞の結果。B: 線維芽細胞由来epi-iPS細胞の結果。括弧内の数字は各クローンの継代数を示す。各クローンについて使用した細胞数も示している。陽性コントロールとして、レトロウイルスにより誘導したiPSクローン(253G-4)およびY4混合物のエレクトロポレーションから6日後の線維芽細胞(fibro-d6)を分析した。
【図32】図32は、RT-PCR解析による多能性細胞マーカー遺伝子の発現を示す。A: DP由来epip-iPS細胞の結果。B: 線維芽細胞由来epi-iPS細胞の結果。Y1 (454B-1)、Y2 (454C-2)、Y3 (454D-1) およびY4 (454E-2, 451F-3, 457C-1, 453F-2, 404C-2, 409B-2, 414C-2, 418C-1, 421C-1, 426C-2, 427D-4および428C-2) の各組合せにより樹立されたepi-iPSクローンから全RNAを単離した。また、レトロウイルスにより誘導したiPSCクローン (201B-7および253G-4) と、hESC株 (KhES-3およびH9) も試験した。OCT3/4およびSOX2と表示されたレーンにおいて、PCRプライマーは内在遺伝子のみを増幅したのに対し、Ret-Octレーンにおいては、PCRプライマーはレトロウイルスのOct3/4導入遺伝子を特異的に増幅した。ローディングコントロールとしてG3PDHを分析した。陰性コントロールとして、Y4混合物のエレクトロポレーションから4日後のヒト皮膚線維芽細胞 (HDF-elepo) から、全RNAを単離した。
【図33】図33は、NANOGプロモーター領域のDNAのメチル化状態を示す。白丸および黒丸は、それぞれ非メチル化CpGおよびメチル化CpGを示す。
【図34】図34は、epi-iPSCクローンから誘導したテラトーマを示す。A: 454E-2の結果。神経組織 (A)、軟骨 (B)、筋肉 (C) および腸管様上皮 (D) のヘマトキシリン-エオシン染色を示す。スケールバー=50 μm。B: 404C-2、409B-2、418C-1、421C-1、428C-2および454D-1の結果。神経組織、軟骨および腸管様上皮のヘマトキシリン-エオシン染色を示す。スケールバー=50 μm。
【図35】図35は、epi-iPSCクローンからドーパミン産生ニューロンへの分化を示す(クローン454E-2)。A: Tuj1 (緑, E) およびTH (赤, F) についての免疫染色像、並びにHoechst 33342 (青, G) を用いた核染色と合わせた像(Merge)を示す。スケールバー=20 μm。B: (A-C) Nestin (緑) およびKi67 (赤)についての二重免疫染色とDAPI核染色 (青)。高倍率像を (D) に示す。(EおよびF) Pax6 (赤) およびTH (緑) についての二重免疫染色。(G) TH (赤) およびTuJ1 (緑) についての二重免疫染色とDAPI。(HおよびI) TH (赤) およびMAP2ab (緑) についての二重免疫染色。(JおよびK) TH (赤) および小胞モノアミントランスポーター2 (VMAT2, 緑) についての二重染色。スケールバー: (A-C, E-G) においては100 μm; (D, H-K) においては20 μm。
【図36】図36は、ヒト末梢血単核細胞からのiPS細胞の樹立を示す。図36aは、実験手順の概要を示す。図36bは、3つの異なるプラスミドpCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hULをヒト末梢血単核細胞に導入することにより樹立されたiPS細胞コロニーの代表的な写真である。
【発明を実施するための形態】
【0019】
本発明は、(a) Oct3/4もしくはそれをコードする核酸、(b) Klf4もしくはそれをコードする核酸、および(c) Sox2もしくはそれをコードする核酸、並びに(d1) L-Mycもしくはそれをコードする核酸および/または(d2) p53の機能阻害物質を、体細胞に接触させることを含む、iPS細胞の製造方法を提供する。
【0020】
(A) 体細胞ソース
iPS細胞作製のための出発材料として用いることのできる体細胞は、哺乳動物(例えば、ヒト、マウス、サル、ブタ、ラット等)由来の生殖細胞以外のいかなる細胞であってもよく、例えば、角質化する上皮細胞(例、角質化表皮細胞)、粘膜上皮細胞(例、舌表層の上皮細胞)、外分泌腺上皮細胞(例、乳腺細胞)、ホルモン分泌細胞(例、副腎髄質細胞)、代謝・貯蔵用の細胞(例、肝細胞)、境界面を構成する内腔上皮細胞(例、I型肺胞細胞)、内鎖管の内腔上皮細胞(例、血管内皮細胞)、運搬能をもつ繊毛のある細胞(例、気道上皮細胞)、細胞外マトリックス分泌用細胞(例、線維芽細胞)、収縮性細胞(例、平滑筋細胞)、血液と免疫系の細胞(例、Tリンパ球)、感覚に関する細胞(例、桿細胞)、自律神経系ニューロン(例、コリン作動性ニューロン)、感覚器と末梢ニューロンの支持細胞(例、随伴細胞)、中枢神経系の神経細胞とグリア細胞(例、星状グリア細胞)、色素細胞(例、網膜色素上皮細胞)、およびそれらの前駆細胞(組織前駆細胞)等が挙げられる。細胞の分化の程度や細胞を採取する動物の齢などに特に制限はなく、未分化な前駆細胞 (体性幹細胞も含む) であっても、最終分化した成熟細胞であっても、同様に本発明における体細胞の起源として使用することができる。ここで未分化な前駆細胞としては、たとえば神経幹細胞、造血幹細胞、間葉系幹細胞、脂肪由来ストローマ(幹)細胞および歯髄幹細胞等の組織幹細胞(体性幹細胞)が挙げられる。造血幹細胞および間葉系幹細胞は骨髄、臍帯血および胎盤中に豊富に含まれる。骨髄、臍帯血および胎盤は、多くの公的および私的な血液バンクに寄託され、白血病等の血液疾患の治療に用いられているので、このような寄託された骨髄、臍帯血および胎盤もまた、体細胞ソースとして利用することができる。特に、臍帯血は出産時に得られる臍帯から容易に採取できるので、臍帯血から得られる造血幹細胞および間葉系幹細胞は、iPS細胞バンクのための好適な体細胞ソースである。また、歯髄幹細胞も、智歯や歯周病等で抜いた歯から単離、調製することができるため、入手が容易であり、今後、iPS細胞バンクのための体細胞ソースとしての利用が期待される。
【0021】
最終分化した成熟細胞としては、例えばT細胞やB細胞等の末梢血単核細胞が挙げられる。末梢血の採取は、非常に低侵襲性であり、且つ臨床検査において常套的に実施されている。臨床検査後の少量の余剰末梢血サンプルは通常廃棄されるので、それらはiPS細胞バンクのための好適な体細胞ソースである。特に、T細胞は比較的容易にインビトロで増幅させることができるので、少量の末梢血サンプルからでもiPS細胞を樹立することができる。最近、ヒト末梢血T細胞からのiPS細胞の作製が、いくつかのグループにより報告されている(Seki et al., Cell Stem Cell, 7: 11-14 (2010); Loh et al., Cell Stem Cell, 7: 15-19 (2010); Staerk et al., Cell Stem Cell, 7: 20-24 (2010))。
【0022】
体細胞を採取するソースとなる哺乳動物個体は特に制限されないが、得られるiPS細胞がヒトの再生医療用途に使用される場合には、拒絶反応が起こらないという観点から、患者本人またはHLAの型が同一もしくは実質的に同一である他人から体細胞を採取することが好ましい。ここでHLAの型が「実質的に同一」とは、免疫抑制剤などの使用により、該体細胞由来のiPS細胞から分化誘導することにより得られた細胞を患者に移植した場合に移植細胞が生着可能な程度にHLAの型が一致していることをいう。たとえば主たるHLA(例えばHLA-A、HLA-BおよびHLA-DRの3遺伝子座、さらにHLA-Cwを含む4遺伝子座)が同一である場合などが挙げられる(以下同じ)。また、ヒトに投与(移植)しない場合、例えば、患者の薬剤感受性や副作用の有無を評価するためのスクリーニング用の細胞のソースとしてiPS細胞を使用する場合には、同様に患者本人または薬剤感受性や副作用と相関する遺伝子多型が同一である他人から体細胞を採取することが望ましい。
【0023】
哺乳動物から分離した体細胞は、核初期化工程に供するに先立って、細胞の種類に応じてその培養に適した自体公知の培地で前培養することができる。そのような培地としては、例えば、約5〜20%の胎仔ウシ血清(FCS)を含む最小必須培地(MEM)、ダルベッコ改変イーグル培地(DMEM)、RPMI1640培地、199培地、F12培地などが挙げられるが、それらに限定されない。例えば、体細胞として歯髄幹細胞を用いる場合には、Mesenchymal stem cells basal medium (Lonza社) などの間葉系幹細胞用培地を用いることが好ましい。核初期化物質及びp53の機能阻害物質(さらに必要に応じて、他のiPS細胞の樹立効率改善物質)との接触に際し、例えば、カチオニックリポソームなど導入試薬を用いる場合には、導入効率の低下を防ぐため、無血清培地に交換しておくことが好ましい場合がある。
【0024】
ヒト臨床応用に適した完全xeno-freeのヒトiPS細胞を得る目的においては、FCSなどの非ヒト動物由来成分を含有しない培地を用いることがより望ましい。基本培地に種々の体細胞の培養に適したヒト由来成分(特に、増殖因子などの組換えヒトタンパク質)や非必須アミノ酸、ビタミン類等を補充した培地が市販されており、当業者は体細胞ソースに応じて適切なxeno-free培地を選択することができる。xeno-free培地で前培養された体細胞は、適当なxeno-freeの細胞剥離液を用いて培養容器から剥離し、回収後、核初期化物質及びp53の機能阻害物質との接触に供される。
【0025】
(B) 核初期化物質
本発明において「核初期化物質」とは、体細胞からiPS細胞を誘導することができるタンパク性因子(群)またはそれをコードする核酸(ベクターに組み込まれた形態を含む)でありうる。本発明に用いられる核初期化物質は、少なくともOct3/4、Klf4およびSox2またはそれらをコードする核酸からなり(但し、Klf4および/またはSox2はそれらの機能を代替し得ることが報告されている他の因子に置換してもよい)、p53の機能阻害物質を併用しない場合は、核初期化物質としてL-Mycまたはそれをコードする核酸、さらにLin28もしくはLin28bまたはそれをコードする核酸が組み合わされる。また、本発明に用いられる核初期化物質は、Nanogまたはそれをコードする核酸を含まないことを特徴とする。具体的には、以下の組み合わせが例示される(以下においては、タンパク性因子の名称のみを記載する)。
(1) Oct3/4, Klf4, Sox2, L-Myc(ここで、Sox2はSox1, Sox3, Sox15, Sox17またはSox18で置換可能である。また、Klf4はKlf1, Klf2またはKlf5で置換可能である。)
(2) Oct3/4, Klf4, Sox2, L-Myc, TERT, SV40 Large T antigen(以下、SV40LT)
(3) Oct3/4, Klf4, Sox2, L-Myc, TERT, HPV16 E6
(4) Oct3/4, Klf4, Sox2, L-Myc, TERT, HPV16 E7
(5) Oct3/4, Klf4, Sox2, L-Myc, TERT, HPV16 E6, HPV16 E7
(6) Oct3/4, Klf4, Sox2, L-Myc, TERT, Bmi1
(7) Oct3/4, Klf4, Sox2, L-Myc, Lin28
(8) Oct3/4, Klf4, Sox2, L-Myc, Lin28, SV40LT
(9) Oct3/4, Klf4, Sox2, L-Myc, Lin28, TERT, SV40LT
(10) Oct3/4, Klf4, Sox2, L-Myc, SV40LT
(11) Oct3/4, Esrrb, Sox2, L-Myc (EsrrbはEsrrgで置換可能である。)
(12) Oct3/4, Klf4, Sox2
(13) Oct3/4, Klf4, Sox2, TERT, SV40LT
(14) Oct3/4, Klf4, Sox2, TERT, HPV16 E6
(15) Oct3/4, Klf4, Sox2, TERT, HPV16 E7
(16) Oct3/4, Klf4, Sox2, TERT, HPV16 E6, HPV16 E7
(17) Oct3/4, Klf4, Sox2, TERT, Bmil
(18) Oct3/4, Klf4, Sox2, Lin28
(19) Oct3/4, Klf4, Sox2, Lin28, SV40LT
(20) Oct3/4, Klf4, Sox2, Lin28, TERT, SV40LT
(21) Oct3/4, Klf4, Sox2, SV40LT
(22) Oct3/4, Esrrb, Sox2 (EsrrbはEsrrgで置換可能である。)
上記において、Lin28に代えてLin28bを用いることもできる。また、EsrrbもしくはEsrrgを用いる場合(上記(11)および(22))、Klf4を併用してもよい。
また、上記(1)-(22)には該当しないが、それらのいずれかにおける構成要素をすべて含み、且つ任意の他の物質をさらに含む組み合わせも、本発明における「核初期化物質」の範疇に含まれ得る。また、核初期化の対象となる体細胞が上記(1)-(22)のいずれかにおける構成要素の一部を、核初期化のために十分なレベルで内在的に発現している条件下にあっては、当該構成要素を除いた残りの構成要素のみの組み合わせもまた、本発明における「核初期化物質」の範疇に含まれ得る。
【0026】
これらの組み合わせの中で、Oct3/4, Sox2, Klf4、Lin28(Lin28b)およびL-Mycの5因子、並びにOct3/4, Sox2, Klf4およびLin28(Lin28b)の4因子が、好ましい核初期化物質の例として挙げられる。これらに、SV40 Large T antigenをさらに加えた6因子もしくは5因子も好ましい。
【0027】
上記の各核初期化物質のマウスおよびヒトcDNA配列情報は、WO 2007/069666に記載のNCBI accession numbersを参照することにより取得することができ(Nanogは当該公報中では「ECAT4」との名称で記載されている。尚、Lin28、Lin28b、Esrrb、Esrrg、L-MycのマウスおよびヒトcDNA配列情報は、それぞれ下記NCBI accession numbersを参照することにより取得できる。)、当業者は容易にこれらのcDNAを単離することができる。
遺伝子名 マウス ヒト
Lin28 NM_145833 NM_024674
Lin28b NM_001031772 NM_001004317
Esrrb NM_011934 NM_004452
Esrrg NM_011935 NM_001438
L-Myc NM_008506 NM_001033081
【0028】
核初期化物質としてタンパク性因子自体を用いる場合には、得られたcDNAを適当な発現ベクターに挿入して宿主細胞に導入し、該細胞を培養して得られる培養物から組換えタンパク性因子を回収することにより調製することができる。一方、核初期化物質としてタンパク性因子をコードする核酸を用いる場合、得られたcDNAを、ウイルスベクター、プラスミドベクター、エピソーマルベクター等に挿入して発現ベクターを構築し、核初期化工程に供される。
【0029】
(C) 核初期化物質の体細胞への導入方法
核初期化物質の体細胞への導入は、該物質がタンパク性因子である場合、自体公知の細胞へのタンパク質導入方法を用いて実施することができる。そのような方法としては、例えば、タンパク質導入試薬を用いる方法、タンパク質導入ドメイン(PTD)もしくは細胞透過性ペプチド(CPP)融合タンパク質を用いる方法、マイクロインジェクション法などが挙げられる。タンパク質導入試薬としては、カチオン性脂質をベースとしたBioPOTER Protein Delivery Reagent(Gene Therapy Systmes)、Pro-JectTM Protein Transfection Reagent(PIERCE)及びProVectin(IMGENEX)、脂質をベースとしたProfect-1(Targeting Systems)、膜透過性ペプチドをベースとしたPenetrain Peptide(Q biogene)及びChariot Kit(Active Motif)、HVJエンベロープ(不活化センダイウイルス)を利用したGenomONE(石原産業)等が市販されている。導入はこれらの試薬に添付のプロトコルに従って行うことができるが、一般的な手順は以下の通りである。核初期化物質を適当な溶媒(例えば、PBS、HEPES等の緩衝液)に希釈し、導入試薬を加えて室温で5-15分程度インキュベートして複合体を形成させ、これを無血清培地に交換した細胞に添加して37℃で1ないし数時間インキュベートする。その後培地を除去して血清含有培地に交換する。
【0030】
PTDとしては、ショウジョウバエ由来のAntP、HIV由来のTAT (Frankel, A. et al, Cell 55, 1189-93 (1988); Green, M. & Loewenstein, P.M. Cell 55, 1179-88 (1988))、Penetratin (Derossi, D. et al, J. Biol. Chem. 269, 10444-50 (1994))、Buforin II (Park, C. B. et al. Proc. Natl Acad. Sci. USA 97, 8245-50 (2000))、Transportan (Pooga, M. et al. FASEB J. 12, 67-77 (1998))、MAP (model amphipathic peptide) (Oehlke, J. et al. Biochim. Biophys. Acta. 1414, 127-39 (1998))、K-FGF (Lin, Y. Z. et al. J. Biol. Chem. 270, 14255-14258 (1995))、Ku70 (Sawada, M. et al. Nature Cell Biol. 5, 352-7 (2003))、Prion (Lundberg, P. et al. Biochem. Biophys. Res. Commun. 299, 85-90 (2002))、pVEC (Elmquist, A. et al. Exp. Cell Res. 269, 237-44 (2001))、Pep-1 (Morris, M. C. et al. Nature Biotechnol. 19, 1173-6 (2001))、Pep-7 (Gao, C. et al. Bioorg. Med. Chem. 10, 4057-65 (2002))、SynB1 (Rousselle, C. et al. MoI. Pharmacol. 57, 679-86 (2000))、HN-I (Hong, F. D. & Clayman, G L. Cancer Res. 60, 6551-6 (2000))、HSV由来のVP22等のタンパク質の細胞通過ドメインを用いたものが開発されている。PTD由来のCPPとしては、11R (Cell Stem Cell, 4:381-384(2009)) や9R (Cell Stem Cell, 4:472-476(2009))等のポリアルギニンが挙げられる。
【0031】
核初期化物質のcDNAとPTDもしくはCPP配列とを組み込んだ融合タンパク質発現ベクターを作製して組換え発現させ、融合タンパク質を回収して導入に用いる。導入は、タンパク質導入試薬を添加しない以外は上記と同様にして行うことができる。
【0032】
マイクロインジェクションは、先端径1 μm程度のガラス針にタンパク質溶液を入れ、細胞に穿刺導入する方法であり、確実に細胞内にタンパク質を導入することができる。
【0033】
タンパク質導入操作は1回以上の任意の回数(例えば、1回以上10回以下、又は1回以上5回以下等)行うことができ、好ましくは導入操作を2回以上(たとえば3回又は4回)繰り返して行うことができる。導入操作を繰り返し行う場合の間隔としては、例えば6〜48時間、好ましくは12〜24時間が挙げられる。
【0034】
iPS細胞の樹立効率を重視するのであれば、核初期化物質を、タンパク性因子自体としてではなく、それをコードする核酸の形態で用いることが好ましい。該核酸はDNAであってもRNAであってもよく、あるいはDNA/RNAキメラであってもよいが、また、該核酸は二本鎖であっても、一本鎖であってもよい。好ましくは該核酸は二本鎖DNA、特にcDNAである。
【0035】
核初期化物質のcDNAは、宿主となる体細胞で機能し得るプロモーターを含む適当な発現ベクターに挿入される。発現ベクターとしては、例えば、レトロウイルス、レンチウイルス、アデノウイルス、アデノ随伴ウイルス、ヘルペスウイルス、センダイウイルスなどのウイルスベクター、動物細胞発現プラスミド(例、pA1-11,pXT1,pRc/CMV,pRc/RSV,pcDNAI/Neo)などが用いられ得る。
【0036】
用いるベクターの種類は、得られるiPS細胞の用途に応じて適宜選択することができる。例えば、アデノウイルスベクター、プラスミドベクター、アデノ随伴ウイルスベクター、レトロウイルスベクター、レンチウイルスベクター、センダイウイルスベクター、エピソーマルベクターなどが使用され得る。
【0037】
発現ベクターにおいて使用されるプロモーターとしては、例えばEF1αプロモーター、CAGプロモーター、SRαプロモーター、SV40プロモーター、LTRプロモーター、CMV(サイトメガロウイルス)プロモーター、RSV(ラウス肉腫ウイルス)プロモーター、MoMuLV(モロニーマウス白血病ウイルス)LTR、HSV-TK(単純ヘルペスウイルスチミジンキナーゼ)プロモーターなどが用いられる。なかでも、EF1αプロモーター、CAGプロモーター、MoMuLV LTR、CMVプロモーター、SRαプロモーターなどが好ましい。
発現ベクターは、プロモーターの他に、所望によりエンハンサー、ポリA付加シグナル、選択マーカー遺伝子、SV40複製起点などを含有していてもよい。選択マーカー遺伝子としては、例えば、ジヒドロ葉酸還元酵素遺伝子、ネオマイシン耐性遺伝子、ピューロマイシン耐性遺伝子等が挙げられる。
【0038】
核初期化物質である核酸(初期化遺伝子)は、各々別個の発現ベクター上に組み込んでもよいし、1つの発現ベクターに2種類以上、好ましくは2〜3種類の遺伝子を組み込んでもよい。遺伝子導入効率の高いレトロウイルスやレンチウイルスベクターを用いる場合は前者が、プラスミド、アデノウイルス、エピソーマルベクターなどを用いる場合は後者を選択することが好ましい。さらに、2種類以上の遺伝子を組み込んだ発現ベクターと、1遺伝子のみを組み込んだ発現ベクターとを併用することもできる。
【0039】
上記において複数の初期化遺伝子(例えば、Oct3/4、Sox2、Klf4、L-Myc、Lin28(Lin28b)およびSV40LTから選択される2つ以上、好ましくは2〜3遺伝子)を1つの発現ベクターに組み込む場合、これら複数の遺伝子は、好ましくはポリシストロニック発現を可能にする配列を介して発現ベクターに組み込むことができる。ポリシストロニック発現を可能にする配列を用いることにより、1種類の発現ベクターに組み込まれている複数の遺伝子をより効率的に発現させることが可能になる。ポリシストロニック発現を可能にする配列としては、例えば、口蹄疫ウイルスの2A配列(配列番号63; PLoS ONE 3, e2532, 2008、Stem Cells 25, 1707, 2007)、IRES配列(U.S. Patent No. 4,937,190)など、好ましくは2A配列を用いることができる。複数の初期化遺伝子を1つの発現ベクターにポリシストロニックに連結して挿入する場合、初期化遺伝子の順序は特に制限はないが、例えば、5’から3’方向に(i) Sox2およびKlf4、(ii) L-MycおよびLin28(Lin28b)、(iii) Klf4、Sox2およびOct3/4をこの順序で連結することができる。
【0040】
初期化遺伝子を含む発現ベクターは、ベクターの種類に応じて、自体公知の手法により細胞に導入することができる。例えば、ウイルスベクターの場合、該核酸を含むプラスミドを適当なパッケージング細胞(例、Plat-E細胞)や相補細胞株(例、293細胞)に導入して、培養上清中に産生されるウイルスベクターを回収し、各ウイルスベクターに応じた適切な方法により、該ベクターを細胞に感染させる。例えば、ベクターとしてレトロウイルスベクターを用いる具体的手段が WO 2007/69666、Cell, 126, 663-676 (2006) 及び Cell, 131, 861-872 (2007) に開示されており、ベクターとしてレンチウイルスベクターを用いる場合については、Science, 318, 1917-1920 (2007) に開示がある。iPS細胞を再生医療のための細胞ソースとして利用する場合、初期化遺伝子の発現(再活性化)は、iPS細胞由来の分化細胞から再生された組織における発癌リスクを高める可能性があるので、初期化遺伝子は細胞の染色体に組み込まれず、一過的に発現することが好ましい。かかる観点からは、染色体への組込みが稀なアデノウイルスベクターの使用が好ましい。アデノウイルスベクターを用いる具体的手段は、Science, 322, 945-949 (2008) に開示されている。また、アデノ随伴ウイルスも染色体への組込み頻度が低く、アデノウイルスベクターと比べて細胞毒性や炎症惹起作用が低いので、別の好ましいベクターとして挙げられる。センダイウイルスベクターは染色体外で安定に存在することができ、必要に応じてsiRNAにより分解除去することができるので、同様に好ましく利用され得る。センダイウイルスベクターについては、J. Biol. Chem., 282, 27383-27391 (2007)、Proc. Jpn. Acad., Ser. B 85, 348-362 (2009)、または特許第3602058号に記載のものを用いることができる。
【0041】
レトロウイルスベクターやレンチウイルスベクターを用いる場合は、いったん導入遺伝子のサイレンシングが起こったとしても、後に再活性化される可能性があるので、例えば、Cre/loxPシステムを用いて、不要となった時点で核初期化物質をコードする核酸を切り出す方法が好ましく用いられ得る。即ち、該核酸の両端にloxP配列を配置しておき、iPS細胞が誘導された後で、プラスミドベクターもしくはアデノウイルスベクターを用いて細胞にCreリコンビナーゼを作用させ、loxP配列に挟まれた領域を切り出すことができる。また、LTR U3領域のエンハンサー−プロモーター配列は、挿入突然変異によって近傍の宿主遺伝子を上方制御する可能性があるので、当該配列を欠失、もしくはSV40などのポリアデニル化配列で置換した3’-自己不活性化(SIN)LTRを使用して、切り出されずゲノム中に残存するloxP配列より外側のLTRによる内因性遺伝子の発現制御を回避することがより好ましい。Cre-loxPシステムおよびSIN LTRを用いる具体的手段は、Chang et al., Stem Cells, 27: 1042-1049 (2009) に開示されている。
【0042】
一方、非ウイルスベクターであるプラスミドベクターの場合には、リポフェクション法、リポソーム法、エレクトロポレーション法、リン酸カルシウム共沈殿法、DEAEデキストラン法、マイクロインジェクション法、遺伝子銃法などを用いて該ベクターを細胞に導入することができる。ベクターとしてプラスミドを用いる具体的手段は、例えばScience, 322, 949-953 (2008) 等に記載されている。
【0043】
プラスミドベクターやアデノウイルスベクター等を用いる場合、遺伝子導入は1回以上の任意の回数(例えば、1回以上10回以下、又は1回以上5回以下など)行うことができる。2種以上の発現ベクターを体細胞に導入する場合には、これらの全ての種類の発現ベクターを同時に体細胞に導入することが好ましいが、この場合においても、導入操作は1回以上の任意の回数(例えば、1回以上10回以下、又は1回以上5回以下など)行うことができ、好ましくは導入操作を2回以上 (たとえば3回又は4回) 繰り返して行うことができる。
【0044】
尚、アデノウイルスやプラスミドを用いる場合でも、導入遺伝子が染色体に組み込まれることがあるので、結局はサザンブロットやPCRにより染色体への遺伝子挿入がないことを確認する必要がある。そのため、上記Cre-loxPシステムのように、いったん染色体に導入遺伝子を組み込んだ後に、該遺伝子を除去する手段を用いることは好都合であり得る。別の好ましい一実施態様においては、トランスポゾンを用いて染色体に導入遺伝子を組み込んだ後に、プラスミドベクターもしくはアデノウイルスベクターを用いて細胞に転移酵素を作用させ、導入遺伝子を完全に染色体から除去する方法が用いられ得る。好ましいトランスポゾンとしては、例えば、鱗翅目昆虫由来のトランスポゾンであるpiggyBac等が挙げられる。piggyBacトランスポゾンを用いる具体的手段は、Kaji, K. et al., Nature, 458: 771-775 (2009)、Woltjen et al., Nature, 458: 766-770 (2009) に開示されている。
【0045】
別の好ましい非組込み型ベクターとして、染色体外で自律複製可能なエピソーマルベクターが挙げられる。エピソーマルベクターを用いる具体的手段は、Yu et al., Science, 324, 797-801 (2009) に開示されている。本発明の特に好ましい一実施態様においては、エピソーマルベクターの複製に必要なベクター要素の5’側および3’側にloxP配列を同方向に配置したエピソーマルベクターが使用され得る。エピソーマルベクターは染色体外で自律複製可能なため、ゲノムに組み込まれなくとも宿主細胞内での安定な発現を提供し得るが、いったんiPS細胞が樹立された後は、当該ベクターは速やかに除かれることが望ましい。エピソーマルベクターの複製に必要なベクター要素を2つのloxP配列で挟み、これにCreリコンビナーゼを作用させて当該ベクター要素を切り出すことにより、エピソーマルベクターの自律複製能を喪失させることができ、該ベクターを早期にiPS細胞から脱落させることができる。
【0046】
本発明に用いられるエピソーマルベクターとしては、例えば、EBV、SV40等に由来する自律複製に必要な配列をベクター要素として含むベクターが挙げられる。自律複製に必要なベクター要素としては、具体的には、複製開始点と、複製開始点に結合して複製を制御するタンパク質をコードする遺伝子であり、例えば、EBVにあっては複製開始点oriPとEBNA-1遺伝子、SV40にあっては複製開始点oriとSV40 large T antigen遺伝子が挙げられる。
【0047】
また、エピソーマル発現ベクターは、初期化遺伝子の転写を制御するプロモーターを含む。該プロモーターとしては、前記と同様のプロモーターが用いられ得る。また、エピソーマル発現ベクターは、前記と同様に、所望によりエンハンサー、ポリA付加シグナル、選択マーカー遺伝子などをさらに含有していてもよい。選択マーカー遺伝子としては、例えば、ジヒドロ葉酸還元酵素遺伝子、ネオマイシン耐性遺伝子等が挙げられる。
【0048】
本発明で使用されるloxP配列としては、バクテリオファージP1由来の野生型loxP配列(配列番号29)の他、初期化遺伝子の複製に必要なベクター要素を挟む位置に同方向で配置された場合に、組換えを起こしてloxP配列間の配列を欠失させ得る任意の変異loxP配列が挙げられる。変異loxP配列としては、例えば、5’側反復配列に変異のあるlox71(配列番号30)、3’側反復配列に変異のあるlox66(配列番号31)、スペーサー部分に変異のあるlox2272やlox511などが挙げられる。該ベクター要素の5’側および3’側に配置される2つのloxP配列は、同一であっても異なっていてもよいが、スペーサー部分に変異のある変異loxP配列の場合は同一のもの(例、lox2272同士、lox511同士)が用いられる。好ましくは、5’側反復配列に変異のある変異loxP配列(例、lox71)と3’側反復配列に変異のある変異loxP配列(例、lox66)との組合せが挙げられる。この場合、組換えの結果染色体上に残るloxP配列は5’側および3’側の反復配列に二重変異を有するため、Creリコンビナーゼに認識されにくく、不必要な組換えにより染色体の欠失変異を起こすリスクが低減される。lox71とlox66とを用いる場合、前記ベクター要素の5’側および3’側にいずれの変異loxP配列を配置してもよいが、変異部位がloxP配列の外端に配置されるような向きで変異loxP配列を挿入する必要がある。尚、本発明の好ましいエピソーマルベクターはCreリコンビナーゼを作用させなくても、早期に細胞から脱落する自己消失型ベクターであるが、例外的に細胞からの脱落に時間がかかる場合も想定されるので、Creリコンビナーゼ処理による不必要な組換えなどのリスクに備えてloxP配列を設計しておくことが好ましい。
【0049】
2つのloxP配列は、初期化遺伝子の複製に必要なベクター要素(即ち、複製開始点、または複製開始点に結合して複製を制御するタンパク質をコードする遺伝子配列)の5’側および3’側に、同方向に配置される。loxP配列が挟むベクター要素は、複製開始点、または複製開始点に結合して複製を制御するタンパク質をコードする遺伝子配列のいずれか一方だけであってもよいし、両方であってもよい。
【0050】
エピソーマルベクターやプラスミドベクターには、RNAの安定性を向上させる目的で、例えば、初期化因子のコーディング領域とポリA付加シグナルとの間にウッドチャック肝炎ウイルス転写後調節エレメント(WPRE)配列を挿入してもよい。
【0051】
エピソーマルベクターは、例えばリポフェクション法、リポソーム法、エレクトロポレーション法、リン酸カルシウム共沈殿法、DEAEデキストラン法、マイクロインジェクション法、遺伝子銃法などを用いて該ベクターを細胞に導入することができる。具体的には、例えばScience, 324: 797-801 (2009) 等に記載される方法を用いることができる。
【0052】
iPS細胞から初期化遺伝子の複製に必要なベクター要素が除去されたか否かの確認は、該ベクター要素内部および/またはloxP配列近傍の塩基配列を含む核酸をプローブまたはプライマーとして用い、該iPS細胞から単離したエピソーム画分を鋳型としてサザンブロット分析またはPCR分析を行い、バンドの有無または検出バンドの長さを調べることにより実施することができる。エピソーム画分の調製は当該分野で周知の方法と用いて行えばよく、例えば、Science, 324: 797-801 (2009) 等に記載される方法を用いることができる。
【0053】
後述の実施例に示されるように、本発明で提供されるloxP配列を含むエピソーマルベクターの中には、体細胞に導入した際に、該ベクターを構成する外来核酸因子(初期化遺伝子を含む)が一過的にも細胞のゲノム中に組み込まれないというエピソーマルベクター本来の効果だけでなく、Creリコンビナーゼ処理を施すことなく、早い段階でエピソームとして存在する該ベクターがiPS細胞から脱落するという予期せぬ効果を奏するものがある。即ち、本発明はまた、iPS細胞の樹立に十分な初期化因子の発現を提供した後、早期に細胞から脱落する自己消失型のエピソーマルベクターを提供する。かかるベクターは、50%以上、好ましくは60%以上、より好ましくは70%以上の頻度で、5継代までにiPS細胞から脱落することを特徴とする。あるいは、該自己消失型エピソーマルベクターは、導入後1週間以内における1x104細胞あたりのコピー数が106個のオーダーであるのに対し、iPS細胞樹立時(例えば、ベクター導入から約4週間後)における1x104細胞あたりのコピー数が100個以下、好ましくは50個以下、より好ましくは30個以下にまで減衰する程度に細胞内で不安定であることを特徴とする。
【0054】
具体的には、本発明の早期自己消失型ベクターは以下の(i)〜(iv)のうちの少なくと1つ、好ましくは2つ以上、より好ましくは3つ以上、特に好ましくはすべての構造的特徴を有するものである。
(i) エピソーマルベクターの複製に必要なベクター要素(例えば、EBNA-1遺伝子やSV40 Large T antigen遺伝子、好ましくはEBNA-1遺伝子)の5’側および3’側にloxP配列が同方向に配置されている。
(ii) 初期化因子をコードする核酸がCAGプロモーターの制御下にある。
(iii) 初期化因子をコードする核酸がウサギβ-グロビンポリA付加シグナルの制御下にある。
(iv) 初期化因子をコードする核酸とポリA付加シグナルとの間にWPRE配列を含む。
【0055】
(D) p53の機能阻害物質
本発明は、上記の核初期化物質に加えて、p53の機能阻害物質を接触させることがより好ましい。本明細書において「p53の機能阻害物質」とは、(a)p53タンパク質の機能もしくは(b)p53遺伝子の発現を阻害し得る限り、いかなる物質であってもよい。すなわち、p53タンパク質に直接作用してその機能を阻害する物質や、p53遺伝子に直接作用してその発現を阻害する物質のみならず、p53のシグナル伝達に関与する因子に作用することにより、結果的にp53タンパク質の機能やp53遺伝子の発現を阻害する物質も、本明細書における「p53の機能阻害物質」に含まれる。好ましくは、p53の機能阻害物質は、p53遺伝子の発現を阻害する物質であり、より好ましくはp53に対するsiRNAやshRNAをコードする発現ベクターである。
【0056】
p53タンパク質の機能を阻害する物質としては、例えば、p53の化学的阻害物質、p53のドミナントネガティブ変異体もしくはそれをコードする核酸、抗p53アンタゴニスト抗体もしくはそれをコードする核酸、p53応答エレメントのコンセンサス配列を含むデコイ核酸、p53経路を阻害する物質などが挙げられるが、これらに限定されない。好ましくは、p53の化学的阻害物質、p53のドミナントネガティブ変異体もしくはそれをコードする核酸、p53経路阻害物質が挙げられる。
【0057】
(D1) p53の化学的阻害物質
p53の化学的阻害物質としては、例えば、WO 00/44364に開示されるpifithrin(PFT)-α及び-βに代表されるp53阻害剤、Stormら(Nat. Chem. Biol. 2, 474 (2006))に開示されるPFT-μ、それらの類縁体及びそれらの塩(例えば、塩酸酸、臭素酸塩等の酸付加塩など)等が挙げられるが、これらに限定されない。これらのうち、PFT-α及びその類縁体[2-(2-Imino-4,5,6,7-tetrahydrobenzothiazol-3-yl)-1-p-tolylethanone, HBr (製品名:Pifithrin-α)及び1-(4-Nitrophenyl)-2-(4,5,6,7-tetrahydro-2-imino-3(2H)-benzothiazolyl)ethanone, HBr(製品名:Pifithrin-α, p-Nitro)]、PFT-β及びその類縁体[2-(4-Methylphenyl)imidazo[2,1-b]-5,6,7,8-tetrahydrobenzothiazole, HBr(製品名:Pifithrin-α, Cyclic)及び2-(4-Nitrophenyl)imidazo[2,1-b]-5,6,7,8-tetrahydrobenzothiazole(製品名:Pifithrin-α, p-Nitro, Cyclic)]、PFT-μ[Phenylacetylenylsulfonamide(製品名:Pifithrin-μ)]は、Merck社より市販されている。
【0058】
体細胞へのp53の化学的阻害物質の接触は、該阻害物質を適当な濃度で水性もしくは非水性溶媒に溶解し、ヒトまたはマウスより単離した体細胞の培養に適した培地(例えば、約5〜20%の胎仔ウシ血清を含む最小必須培地(MEM)、ダルベッコ改変イーグル培地(DMEM)、RPMI1640培地、199培地、F12培地など)中に、阻害物質濃度がp53の機能阻害に十分で且つ細胞毒性がみられない範囲となるように該阻害物質溶液を添加して、細胞を一定期間培養することにより実施することができる。阻害物質濃度は用いる阻害物質の種類によって異なるが、約0.1nM〜約100nMの範囲で適宜選択される。接触期間は細胞の核初期化が達成されるのに十分な時間であれば特に制限はないが、通常は陽性コロニーが出現するまで培地に共存させておけばよい。
【0059】
p53遺伝子は癌抑制遺伝子として知られており、p53の恒常的な機能阻害は発癌のリスクを高める可能性がある。p53の化学的阻害物質は、培地に添加するだけで細胞への導入が可能であるという利点に加えて、iPS細胞の誘導後に該阻害物質を含む培地を除去することにより、容易かつ迅速にp53の機能阻害を解除できる点でも有用である。
【0060】
(D2) p53のドミナントネガティブ変異体
p53のドミナントネガティブ変異体としては、体細胞に内在する野生型p53タンパク質と競合的に作用して、その機能を阻害し得る限り特に制限はないが、例えば、マウスp53のDNA結合領域に位置する275位(ヒトの場合は278位)のプロリンをセリンに点変異させたp53P275S(de Vries, A., Proc. Natl. Acad. Sci. USA, 99, 2948-2953 (2002))、マウスp53の14-301位(ヒトp53では11-304位に対応)のアミノ酸を欠失させたp53DD(Bowman, T., Genes Develop., 10, 826-835 (1996))などが挙げられる。その他にも、例えば、マウスp53の58位(ヒトの場合は61位)のセリンをアラニンに点変異させたp53S58A、ヒトp53の135位(マウスの場合は132位)のシステインをチロシンに点変異させたp53C135Y、マウスp53の135位(ヒトの場合は138位)のアラニンをバリンに点変異させたp53A135V、172位(ヒトの場合は175位)のアルギニンをヒスチジンに点変異させたp53R172H、270位(ヒトの場合は273位)のアルギニンをヒスチジンに点変異させたp53R270H、マウスp53の278位(ヒトの場合は281位)のアスパラギン酸をアスパラギンに点変異させたp53D278Nなどが知られており、同様に使用することができる。
【0061】
p53のドミナントネガティブ変異体は、例えば、以下の手法により得ることができる。まず、配列番号1または3に示されるマウスまたはヒトのp53 cDNA配列情報に基づいて適当なオリゴヌクレオチドをプローブもしくはプライマーとして合成し、マウスまたはヒトの細胞・組織由来のmRNA、cDNAもしくはcDNAライブラリーから、ハイブリダイゼーション法や(RT-)PCR法を用いてマウスまたはヒトp53 cDNAをクローニングし、適当なプラスミドにサブクローニングする。変異を導入しようとする部位のコドン(例えば、p53P275Sの場合、配列番号1に示される塩基配列中塩基番号951-953で示されるcct)を所望の他のアミノ酸をコードするコドン(例えば、p53P275Sの場合、tct)に置換した形で、当該部位を含むプライマーを合成し、これを用いてp53 cDNAを挿入したプラスミドを鋳型とするインバースPCRを行うことにより、目的のドミナントネガティブ変異体をコードする核酸を取得する。p53DDのような欠失変異体の場合には、欠失させる部位の外側にプライマーを設計して、同様にインバースPCRを行えばよい。このようにして得られたドミナントネガティブ変異体をコードする核酸を宿主細胞に導入し、該細胞を培養して得られる培養物から組換えタンパク質を回収することにより、所望のドミナントネガティブ変異体を取得することができる。
【0062】
体細胞へのドミナントネガティブ変異体の接触は、上記タンパク性の核初期化物質の場合と同様に実施することができる。上述のように、p53の恒常的な機能阻害は発癌のリスクを高める可能性があるが、p53のドミナントネガティブ変異体は、導入された細胞内でプロテアーゼによる分解を受けて徐々に消失し、それに応じて細胞に内在するp53の機能が回復することから、該変異体タンパク質の使用は、得られるiPS細胞を治療用途で利用する場合のように、高度な安全性を要求される場合に好適であり得る。
【0063】
(D3) p53のドミナントネガティブ変異体をコードする核酸
本発明の別の好ましい実施態様において、p53機能阻害物質は、p53のドミナントネガティブ変異体をコードする核酸である。該核酸はDNAであってもRNAであってもよく、あるいはDNA/RNAキメラであってもよいが、好ましくはDNAである。また、該核酸は二本鎖であっても、一本鎖であってもよい。p53のドミナントネガティブ変異体をコードするcDNAは、該変異体タンパク質の作製について上記した手法によりクローニングすることができる。
単離されたcDNAは、前記核初期化物質である核酸(初期化遺伝子)の場合と同様に、適当な発現ベクターに挿入され、体細胞に導入され得る。
【0064】
(D4) p53経路阻害物質
ここでp53経路とは、p53を活性化し得るあらゆる上流のシグナルカスケードおよび活性化p53によって媒介されるあらゆる下流のシグナルカスケードを包含する意味で用いられる。したがって、p53経路阻害物質には、上記シグナル伝達経路のいずれかを阻害するいかなる物質も含まれるが、好ましい一実施態様においては、p53経路阻害物質はp53によりその転写が活性化されるp21の発現もしくは機能(Myc阻害活性)を阻害する物質であり、例えば、p21に対するsiRNA、shRNA、アンチセンス核酸、リボザイム等が挙げられる。p21の発現を阻害するこれらの核酸は、後記p53に対するsiRNA、shRNA、アンチセンス核酸、リボザイムと同様の方法により設計・合成し、体細胞に導入することができる。当該核酸は、それらを発現するベクターの形態で提供されてもよく、該ベクターは、後記p53に対するsiRNA、shRNA、アンチセンス核酸、リボザイムを発現するベクターと同様の方法により構築し、体細胞に導入することができる。
【0065】
別の好ましい一実施態様においては、p53経路阻害物質はARF-MDM2-p53経路を阻害する物質であり、例えば、ARF-MDM2-p53経路阻害物質として、p53に直接結合してその核外輸送やユビキチン化を促進するMDM2もしくはそれをコードする核酸、p53へのMDM2の作用を阻害するp19ARFやATM(ataxia-telangiectasia mutated)の発現もしくは機能を阻害する物質(例えば、これらの因子に対するsiRNAやshRNA)等が挙げられる。
【0066】
(D5) その他の物質
p53タンパク質の機能を阻害するその他の物質として、例えば、抗p53アンタゴニスト抗体もしくはそれをコードする核酸が挙げられる。抗p53アンタゴニスト抗体はポリクローナル抗体、モノクローナル抗体の何れであってもよい。抗体のアイソタイプは特に限定されないが、好ましくはIgG、IgMまたはIgA、特に好ましくはIgGが挙げられる。また、該抗体は、完全抗体分子の他、例えばFab、Fab'、F(ab’)2等のフラグメント、scFv、scFv-Fc、ミニボディー、ダイアボディー等の遺伝子工学的に作製されたコンジュゲート分子、あるいはポリエチレングリコール(PEG)等の蛋白質安定化作用を有する分子等で修飾されたそれらの誘導体などであってもよい。抗p53アンタゴニスト抗体は、p53またはその部分ペプチドを抗原として用い、自体公知の抗体または抗血清の製造法に従って製造することができる。また、公知の抗p53アンタゴニスト抗体として、例えば、PAb1801(Oncogene Science Ab-2)及びDO-1(Oncogene Science Ab-6)(Gire and Wynford-Thomas, Mol. Cell. Biol., 18, 1611-1621 (1998))等が挙げられる。抗p53アンタゴニスト抗体をコードする核酸は、抗p53モノクローナル抗体産生ハイブリドーマから常法により単離することができる。得られるH鎖及びL鎖遺伝子を連結して単鎖抗体をコードする核酸を作製することもできる。
【0067】
p53タンパク質の機能を阻害する別の物質として、抗p21アンタゴニスト抗体もしくはそれをコードする核酸が挙げられる。抗p21アンタゴニスト抗体及びそれをコードする核酸も、上記抗p53アンタゴニスト抗体及びそれをコードする核酸と同様にして作製することができる。
【0068】
p53タンパク質の機能を阻害するさらに別の物質は、p53応答エレメントのコンセンサス配列(例、Pu-Pu-Pu-G-A/T-T/A-C-Py-Py-Py (Pu: プリン塩基, Py: ピリミジン塩基); 配列番号27)を含むデコイ核酸である。このような核酸は上記塩基配列情報に基づいてDNA/RNA自動合成機で合成することができる。あるいはそのようなデコイ核酸は市販されている(例、p53 transcription factor decoy (GeneDetect.com))。
【0069】
抗p53アンタゴニスト抗体及び抗p21アンタゴニスト抗体はp53のドミナントネガティブ変異体と同様に、また、該抗体をコードする核酸は該変異体をコードする核酸と同様にして、それぞれ細胞に導入することができる。また、上記デコイ核酸は、リポフェクション法などにより細胞に導入することができる。
【0070】
一方、p53遺伝子の発現を阻害する物質としては、例えば、p53に対するsiRNAもしくはshRNA、p53に対するsiRNAもしくはshRNAを発現するベクター、p53に対するアンチセンス核酸及びp53に対するリボザイム等が挙げられるが、好ましくはp53に対するsiRNA、shRNA及びsiRNA、shRNAを発現するベクターである。
【0071】
(D6) p53に対するsiRNA及びshRNA
p53に対するsiRNAは、配列番号1または3に示されるマウスまたはヒトのp53 cDNA配列情報に基づいて、例えば、Elbashirら(Genes Dev., 15, 188-200 (2001))の提唱する規則に従って設計することができる。siRNAの標的配列としては、原則的にはAA+(N)19であるが、AA+(N)21もしくはNA+(N)21であってもよい。また、センス鎖の5’末端がAAである必要はない。標的配列の位置は特に制限されるわけではないが、5’-UTR及び開始コドンから約50塩基まで、並びに3’-UTR以外の領域から標的配列を選択することが望ましい。標的配列のGC含量も特に制限はないが、約30-約50%が好ましく、GC分布に偏りがなく繰り返しが少ない配列が望ましい。尚、下記(b2)のsiRNAもしくはshRNAを発現するベクターの設計において、プロモーターとしてpolIII系プロモーターを使用する場合、ポリメラーゼの転写が停止しないように、4塩基以上TまたはAが連続する配列は選択しないようにすべきである。
【0072】
上述の規則に基づいて選択された標的配列の候補群について、標的以外のmRNAにおいて16-17塩基の連続した配列に相同性がないかどうかを、BLAST(http://www.ncbi.nlm.nih.gov/BLAST/)等のホモロジー検索ソフトを用いて調べ、選択した標的配列の特異性を確認する。特異性の確認された標的配列について、AA(もしくはNA)以降の19-21塩基にTTもしくはUUの3’末端オーバーハングを有するセンス鎖と、該19-21塩基に相補的な配列及びTTもしくはUUの3’末端オーバーハングを有するアンチセンス鎖とからなる2本鎖RNAをsiRNAとして設計する。また、shRNAは、ループ構造を形成しうる任意のリンカー配列(例えば、8-25塩基程度)を適宜選択し、上記センス鎖とアンチセンス鎖とを該リンカー配列を介して連結することにより設計することができる。
【0073】
siRNA及び/又はshRNAの配列は、種々のwebサイト上に無料で提供される検索ソフトを用いて検索が可能である。このようなサイトとしては、例えば、Ambionが提供するsiRNA Target Finder(http://www.ambion.com/jp/techlib/misc/siRNA_finder.html)及びpSilencerTM Expression Vector用 インサート デザインツール(http://www.ambion.com/jp/techlib/misc/psilencer_converter.html)、RNAi Codexが提供するGeneSeer(http://codex.cshl.edu/scripts/newsearchhairpin.cgi)がこれらに限定されず、QIAGEN、タカラバイオ、SiSearch、Dharmacon、Whitehead Institute、Invitrogen、Promega等のwebサイト上でも同様に検索が可能である。
【0074】
以下に、Ambion(配列番号5-24)およびRNAi Codex(配列番号25及び26)のwebサイト上で提供されるソフトウェアを用いて設計した、マウスp53に対するshRNA配列を示す。下線で示した配列がdicerで切断された後に生じるdsRNAのセンス鎖(5’側)及びアンチセンス鎖(3’側)である(3’-オーバーハング「TT」を含まない)。小文字はミスマッチまたはループを示す。
[配列番号5]
5’-TTTGACTGGATGACTGCCATGGttcaagagaCCATGGCAGTCATCCAGTCTTTTTT-3’
[配列番号6]
5’-TTTGATATCCTGCCATCACCTCttcaagagaGAGGTGATGGCAGGATATCTTTTTT-3’
[配列番号7]
5’-TTTGGCCCAAGTGAAGCCCTCCttcaagagaGGAGGGCTTCACTTGGGCCTTTTTT-3’
[配列番号8]
5’-TTTGTGAAGCCCTCCGAGTGTCttcaagagaGACACTCGGAGGGCTTCACTTTTTT-3’
[配列番号9]
5’-TTTGCCCTCCGAGTGTCAGGAGttcaagagaCTCCTGACACTCGGAGGGCTTTTTT-3’
[配列番号10]
5’-TTTGTCTGTTATGTGCACGTACttcaagagaGTACGTGCACATAACAGACTTTTTT-3’
[配列番号11]
5’-TTTGTACTCTCCTCCCCTCAATttcaagagaATTGAGGGGAGGAGAGTACTTTTTT-3’
[配列番号12]
5’-TTTGCTATTCTGCCAGCTGGCGttcaagagaCGCCAGCTGGCAGAATAGCTTTTTT-3’
[配列番号13]
5’-TTTGACGTGCCCTGTGCAGTTGttcaagagaCAACTGCACAGGGCACGTCTTTTTT-3’
[配列番号14]
5’-TTTGAAGTCACAGCACATGACGttcaagagaCGTCATGTGCTGTGACTTCTTTTTT-3’
[配列番号15]
5’-TTTGTCACAGCACATGACGGAGttcaagagaCTCCGTCATGTGCTGTGACTTTTTT-3’
[配列番号16]
5’-TTTGGAAATTTGTATCCCGAGTttcaagagaACTCGGGATACAAATTTCCTTTTTT-3’
[配列番号17]
5’-TTTGTACATGTGTAATAGCTCCttcaagagaGGAGCTATTACACATGTACTTTTTT-3’
[配列番号18]
5’-TTTGACTCCAGTGGGAACCTTCttcaagagaGAAGGTTCCCACTGGAGTCTTTTTT-3’
[配列番号19]
5’-TTTGTCCTTTGCCCTGAACTGCttcaagagaGCAGTTCAGGGCAAAGGACTTTTTT-3’
[配列番号20]
5’-TTTGATCCGCGGGCGTAAACGCttcaagagaGCGTTTACGCCCGCGGATCTTTTTT-3’
[配列番号21]
5’-TTTGACCAAGAAGGGCCAGTCTttcaagagaAGACTGGCCCTTCTTGGTCTTTTTT-3’
[配列番号22]
5’-TTTGAAAGTGGGGCCTGACTCAttcaagagaTGAGTCAGGCCCCACTTTCTTTTTT-3’
[配列番号23]
5’-TTTGTTGGGGAATAGGTTGATAttcaagagaTATCAACCTATTCCCCAACTTTTTT-3’
[配列番号24]
5’-TTTGATTCTATCTTGGGCCCTCttcaagagaGAGGGCCCAAGATAGAATCTTTTTT-3’
[配列番号25]
5’-TTTGCAuTACAgGTACgTGTGTAgtgtgctgtccTACACATGTACTTGTAGTGTTTTTT-3’
[配列番号26]
5’-TTTGCAGTuTACTTuCCGCCgTAgtgtgctgtccTATGGCGGGAAGTAGACTGTTTTTT-3’
【0075】
p53に対するsiRNAは、上記のようにして設計されたセンス鎖及びアンチセンス鎖オリゴヌクレオチドをDNA/RNA自動合成機でそれぞれ合成し、例えば、適当なアニーリング緩衝液中、約90〜約95℃で約1分程度変性させた後、約30〜約70℃で約1〜約8時間アニーリングさせることにより調製することができる。また、p53に対するshRNAは、上記のようにして設計されたshRNA配列を有するオリゴヌクレオチドをDNA/RNA自動合成機で合成し、上記と同様にしてセルフアニーリングさせることによって調製することができる。
【0076】
siRNA及びshRNAを構成するヌクレオチド分子は、天然型のRNAでもよいが、安定性(化学的および/または対酵素)や比活性(mRNAとの親和性)を向上させるために、種々の化学修飾を含むことができる。例えば、ヌクレアーゼなどの加水分解酵素による分解を防ぐために、siRNAまたはshRNAを構成する各ヌクレオチドのリン酸残基(ホスフェート)を、例えば、ホスホロチオエート(PS)、メチルホスホネート、ホスホロジチオネートなどの化学修飾リン酸残基に置換することができる。また、各ヌクレオチドの糖(リボース)の2'位の水酸基を、-OR(Rは、例えばCH3(2'-O-Me)、CH2CH2OCH3(2'-O-MOE)、CH2CH2NHC(NH)NH2、CH2CONHCH3、CH2CH2CN等を示す)に置換してもよい。さらに、塩基部分(ピリミジン、プリン)に化学修飾を施してもよく、例えば、ピリミジン塩基の5位へのメチル基やカチオン性官能基の導入、あるいは2位のカルボニル基のチオカルボニルへの置換などが挙げられる。
【0077】
RNAの糖部のコンフォーメーションはC2'-endo(S型)とC3'-endo(N型)の2つが支配的であり、一本鎖RNAではこの両者の平衡として存在するが、二本鎖を形成するとN型に固定される。したがって、標的RNAに対して強い結合能を付与するために、2'酸素と4’炭素を架橋することにより、糖部のコンフォーメーションをN型に固定したRNA誘導体であるBNA(LNA)(Imanishi, T. et al., Chem. Commun., 1653-9, 2002; Jepsen, J.S. et al., Oligonucleotides, 14, 130-46, 2004)やENA(Morita, K. et al., Nucleosides Nucleotides Nucleic Acids, 22, 1619-21, 2003)もまた、好ましく用いられ得る。
但し、天然型RNA中のすべてのリボヌクレオシド分子を修飾型で置換すると、RNAi活性が失われる場合があるので、RISC複合体が機能できる最小限の修飾ヌクレオシドの導入が必要である。
【0078】
p53に対するsiRNAは、例えば、Ambion(例、Ambion Cat# AM16708, siRNA ID# 69659, 69753, 69843, 187424, 187425, 187426)やSanta Cruz(例、Santa Cruz Cat# sc-29436, 44219)等から購入することもできる。
【0079】
また、ヒトp53に対するsiRNAおよびshRNAも、上記のいずれかの検索ソフトを用いて、配列番号3に示されるヒトp53 cDNAの配列もしくはRefseq. No.(NM_000546)等をクエリーとして入力することにより設計し、合成することができ、あるいはAmbion等から購入することもできる。具体的には、配列5’-GACTCCAGTGGTAATCTACTGctcgagCAGTAGATTACCACTGGAGTC-3’(配列番号28;下線部がp53のターゲット配列。大文字がdsRNAを形成する部分)を有するヒトp53に対するshRNA、Science, 296, 550-553 (2002) に記載されるp53に対するshRNAなどが例示される。
【0080】
p53に対するsiRNAもしくはshRNAの体細胞への接触は、プラスミドDNAの場合と同様に、リポソーム法、ポリアミン法、エレクトロポレーション法、ビーズ法等を用いて、該核酸を細胞内へ導入することにより実施することができる。カチオニックリポソームを用いた方法が最も一般的で、導入効率も高い。Lipofectamine2000やOligofectamine(Invitrogen)などの一般的な遺伝子導入試薬の他、例えば、GeneEraserTMsiRNA transfection reagent(Stratagene)等のsiRNA導入に適した導入試薬も市販されている。
【0081】
(D7) p53に対するsiRNAもしくはshRNAを発現するベクター
siRNAを発現するベクターには、タンデムタイプとステムループ(ヘアピン)タイプとがある。前者はsiRNAのセンス鎖の発現カセットとアンチセンス鎖の発現カセットをタンデムに連結したもので、細胞内で各鎖が発現してアニーリングすることにより2本鎖のsiRNA(dsRNA)を形成するというものである。一方、後者はshRNAの発現カセットをベクターに挿入したもので、細胞内でshRNAが発現しdicerによるプロセシングを受けてdsRNAを形成するというものである。プロモーターとしては、polII系プロモーター(例えば、CMV前初期プロモーター)を使用することもできるが、短いRNAの転写を正確に行わせるために、polIII系プロモーターを使用するのが一般的である。polIII系プロモーターとしては、マウスおよびヒトのU6-snRNAプロモーター、ヒトH1-RNase P RNAプロモーター、ヒトバリン-tRNAプロモーターなどが挙げられる。また、転写終結シグナルとして4個以上T残基が連続した配列が用いられる。
【0082】
このようにして構築したsiRNAもしくはshRNA発現カセットを、次いでプラスミドベクターやウイルスベクターに挿入する。このようなベクターとしては、核初期化物質である核酸(初期化遺伝子)について上記したと同様のものが、好ましく利用され得る(レトロウイルス、レンチウイルス、アデノウイルス、アデノ随伴ウイルス、ヘルペスウイルス、センダイウイルスなどのウイルスベクターや、動物細胞発現プラスミド、エピソーマルベクターなど)。使用するベクターは、初期化遺伝子の場合と同様、得られるiPS細胞の用途に応じて適宜選択され得る。即ち、p53遺伝子は癌抑制遺伝子として知られており、p53の恒常的な機能阻害は発癌のリスクを高める可能性があるので、作製されるiPS細胞が医療用途を念頭におく場合、p53に対するsiRNAやshRNAは細胞内で一過的に発現するようにデザインされることが望ましい。したがって、p53に対するsiRNAやshRNAをコードする核酸を担持するベクターとしては、自律複製できないプラスミドベクターなどがより好ましい。本発明の初期化因子と組み合わせた場合、p53に対するsiRNAやshRNAはプラスミドからの一過的な発現でも十分に高いiPS細胞樹立効率を与える。
【0083】
あるいは、p53に対するshRNAをコードする発現ベクターとして、市販のプラスミド(例えば、Addgene社から市販されるpMKO.1-puro p53 shRNA2: #10672等)をもとに作製したレトロウイルス等のウイルスベクター、プラスミドベクター、エピソーマルベクターなどを使用することもできる。必要に応じて、上記Cre-loxPシステムやpiggyBacトランスポゾンシステムを利用することもできる。
【0084】
p53に対するsiRNAもしくはshRNAを発現するベクターの体細胞への接触は、上記のようにして調製されるプラスミドベクター、エピソーマルベクターもしくはウイルスベクターを細胞に導入することにより行われる。これらの遺伝子導入は、初期化遺伝子について上記したと同様の手法で行うことができる。
【0085】
(D8) その他の物質
p53遺伝子の発現を阻害する他の物質として、p53に対するアンチセンス核酸やリボザイムが挙げられる。
アンチセンス核酸はDNAであってもRNAであってもよく、あるいはDNA/RNAキメラであってもよい。アンチセンス核酸がDNAの場合、標的RNAとアンチセンスDNAとによって形成されるRNA:DNAハイブリッドは、内在性RNase Hに認識されて標的RNAの選択的な分解を引き起こすことができる。したがって、RNase Hによる分解を指向するアンチセンスDNAの場合、標的配列は、p53 mRNA中の配列だけでなく、p53遺伝子の初期転写産物におけるイントロン領域の配列であってもよい。アンチセンス核酸の標的領域は、該アンチセンス核酸がハイブリダイズすることにより、結果としてp53蛋白質への翻訳が阻害されるものであればその長さに特に制限はなく、p53 mRNAの全配列であっても部分配列であってもよく、短いもので約15塩基程度、長いものでmRNAもしくは初期転写産物の全配列が挙げられる。合成の容易さや抗原性、細胞内移行性の問題等を考慮すれば、約15〜約40塩基、特に約18〜約30塩基からなるオリゴヌクレオチドが好ましい。標的配列の位置としては、5’-及び3’-UTR、開始コドン近傍などが挙げられるが、それらに限定されない。
【0086】
リボザイムとは、狭義には、核酸を切断する酵素活性を有するRNAをいうが、本明細書では配列特異的な核酸切断活性を有する限りDNAをも包含する概念として用いるものとする。リボザイムとして最も汎用性の高いものとしては、ウイロイドやウイルソイド等の感染性RNAに見られるセルフスプライシングRNAがあり、ハンマーヘッド型やヘアピン型等が知られている。ハンマーヘッド型は約40塩基程度で酵素活性を発揮し、ハンマーヘッド構造をとる部分に隣接する両端の数塩基ずつ(合わせて約10塩基程度)をmRNAの所望の切断部位と相補的な配列にすることにより、標的mRNAのみを特異的に切断することが可能である。
【0087】
アンチセンス核酸やリボザイムはDNA/RNA自動合成機を用いて合成することができる。これらを構成するヌクレオチド分子もまた、安定性、比活性などを向上させるために、上記のsiRNAの場合と同様の修飾を受けていてもよい。
あるいは、アンチセンス核酸やリボザイムは、siRNAの場合と同様に、それらをコードする核酸の形態で使用することもできる。
【0088】
p53の機能阻害物質は、体細胞の核初期化工程においてp53の機能を阻害するのに十分な様式で体細胞に接触させる必要がある。この条件が満たされる限り、核初期化物質とp53の機能阻害物質とは、同時に体細胞に接触させてもよいし、また、どちらかを先に接触させてもよい。一実施態様において、例えば、核初期化物質がタンパク性因子をコードする核酸であり、p53の機能阻害物質が化学的阻害物質である場合には、前者は遺伝子導入処理からタンパク性因子を大量発現するまでに一定期間のラグがあるのに対し、後者は速やかにp53の機能を阻害しうることから、遺伝子導入処理から一定期間細胞を培養した後に、p53の化学的阻害物質を培地に添加することができる。別の実施態様において、例えば、核初期化物質とp53の機能阻害物質とがいずれもウイルスベクター、プラスミドベクター、エピソーマルベクター等の形態で用いられる場合には、両者を同時に細胞に導入してもよい。
【0089】
(E) iPS細胞の樹立効率改善物質
p53の機能阻害物質に加え、公知の他のiPS細胞樹立効率改善物質を体細胞に接触させることにより、iPS細胞の樹立効率をさらに高めることが期待できる。そのようなiPS細胞の樹立効率改善物質としては、例えば、ヒストンデアセチラーゼ(HDAC)阻害剤[例えば、バルプロ酸 (VPA)(Nat. Biotechnol., 26(7): 795-797 (2008))、トリコスタチンA、酪酸ナトリウム、MC 1293、M344等の低分子阻害剤、HDACに対するsiRNAおよびshRNA(例、HDAC1 siRNA Smartpool(登録商標)(Millipore)、HuSH 29mer shRNA Constructs against HDAC1 (OriGene)等)等の核酸性発現阻害剤など]、G9aヒストンメチルトランスフェラーゼ阻害剤[例えば、BIX-01294 (Cell Stem Cell, 2: 525-528 (2008))等の低分子阻害剤、G9aに対するsiRNAおよびshRNA(例、G9a siRNA(human) (Santa Cruz Biotechnology)等)等の核酸性発現阻害剤など]、L-calcium channel agonist (例えばBayk8644) (Cell Stem Cell, 3, 568-574 (2008))、UTF1(Cell Stem Cell, 3, 475-479 (2008))、細胞内シグナル伝達の修飾剤[例えば、Wnt Signaling活性化剤(例えばsoluble Wnt3a)(Cell Stem Cell, 3, 132-135 (2008))、TGF-βの阻害剤、MEK阻害剤、2i/LIF (2iはmitogen-activated protein kinase signallingおよびglycogen synthase kinase-3の阻害剤、PloS Biology, 6(10), 2237-2247 (2008))]、他の天然もしくは合成低分子化合物(例、5’-azacytidine、thiazovivin、ビタミンC等)、ES細胞特異的miRNA(例えば、miR-302-367クラスター (Mol. Cell. Biol. doi:10.1128/MCB.00398-08、WO2009/075119)、miR-302 (RNA (2008) 14: 1-10)、miR-291-3p, miR-294およびmiR-295 (Nat. Biotechnol. 27: 459-461 (2009)))等が挙げられるが、それらに限定されない。前記において核酸性の発現阻害剤はsiRNAもしくはshRNAをコードするDNAを含む発現ベクターの形態であってもよい。
【0090】
尚、前記核初期化物質の構成要素のうち、例えば、SV40 Large T等は、体細胞の核初期化のために必須ではなく補助的な因子であるという点において、iPS細胞の樹立効率改善物質の範疇にも含まれ得る。核初期化の機序が明らかでない現状においては、核初期化に必須の因子以外の補助的な因子について、それらを核初期化物質として位置づけるか、あるいはiPS細胞の樹立効率改善物質として位置づけるかは便宜的であってもよい。即ち、体細胞の核初期化プロセスは、体細胞への核初期化物質およびiPS細胞の樹立効率改善物質の接触によって生じる全体的事象として捉えられるので、当業者にとって両者を必ずしも明確に区別する必要性はないであろう。
【0091】
これら他のiPS細胞樹立効率改善物質の体細胞への接触は、該物質が(a) タンパク性因子である場合、(b) 該タンパク性因子をコードする核酸である場合、あるいは(c) 低分子化合物である場合に応じて、p53の機能阻害物質についてそれぞれ上記したと同様の方法により、実施することができる。
他のiPS細胞の樹立効率改善物質は、該物質の非存在下と比較して体細胞からのiPS細胞樹立効率が有意に改善される限り、核初期化物質と同時に体細胞に接触させてもよいし、また、どちらかを先に接触させてもよく、該物質の物性に応じて、p53の機能阻害物質について上記したと同様のタイミングで体細胞と接触させることができる。
【0092】
(F) 培養条件による樹立効率の改善
体細胞の核初期化工程において低酸素条件下で細胞を培養することにより、iPS細胞の樹立効率をさらに改善することができる。本明細書において「低酸素条件」とは、細胞を培養する際の雰囲気中の酸素濃度が、大気中のそれよりも有意に低いことを意味する。具体的には、通常の細胞培養で一般的に使用される5-10% CO2/95-90%大気の雰囲気中の酸素濃度よりも低い酸素濃度の条件が挙げられ、例えば雰囲気中の酸素濃度が18%以下の条件が該当する。好ましくは、雰囲気中の酸素濃度は15%以下(例、14%以下、13%以下、12%以下、11%以下など)、10%以下(例、9%以下、8%以下、7%以下、6%以下など)、または5%以下(例、4%以下、3%以下、2%以下など)である。また、雰囲気中の酸素濃度は、好ましくは0.1%以上(例、0.2%以上、0.3%以上、0.4%以上など)、0.5%以上(例、0.6%以上、0.7%以上、0.8%以上、0.95%以上など)、または1%以上(例、1.1%以上、1.2%以上、1.3%以上、1.4%以上など)である。
【0093】
細胞の環境において低酸素状態を創出する手法は特に制限されないが、酸素濃度の調節可能なCO2インキュベーター内で細胞を培養する方法が最も容易であり、好適な例として挙げられる。酸素濃度の調節可能なCO2インキュベーターは、種々の機器メーカーから販売されている(例えば、Thermo scientific社、池本理化学工業、十慈フィールド、和研薬株式会社などのメーカー製の低酸素培養用CO2インキュベーターを用いることができる)。
【0094】
低酸素条件下で細胞培養を開始する時期は、iPS細胞の樹立効率が正常酸素濃度(20%)の場合に比して改善されることを妨げない限り特に限定されず、体細胞への核初期化物質の接触より前であっても、該接触と同時であっても、該接触より後であってもよいが、例えば、体細胞に核初期化物質を接触させた直後から、あるいは接触後一定期間(例えば、1ないし10(例、2,3,4,5,6,7,8または9)日)おいた後に低酸素条件下で培養することが好ましい。
【0095】
低酸素条件下で細胞を培養する期間も、iPS細胞の樹立効率が正常酸素濃度(20%)の場合に比して改善されることを妨げない限り特に限定されず、例えば3日以上、5日以上、7日以上または10日以上で、50日以下、40日以下、35日以下または30日以下の期間等が挙げられるが、それらに限定されない。低酸素条件下での好ましい培養期間は、雰囲気中の酸素濃度によっても変動し、当業者は用いる酸素濃度に応じて適宜当該培養期間を調整することができる。また、一実施態様において、iPS細胞の候補コロニーの選択を、薬剤耐性を指標にして行う場合には、薬剤選択を開始する迄に低酸素条件から正常酸素濃度に戻すことが好ましい。
【0096】
さらに、低酸素条件下で細胞培養を開始する好ましい時期および好ましい培養期間は、用いられる核初期化物質の種類、正常酸素濃度条件下でのiPS細胞樹立効率などによっても変動する。
【0097】
核初期化物質およびp53の機能阻害物質(さらに必要に応じて他のiPS細胞の樹立効率改善物質)を接触させた後、細胞を、例えばES細胞の培養に適した条件下で培養することができる。マウス細胞の場合、通常の培地に分化抑制因子としてLeukemia Inhibitory Factor(LIF)を添加して培養を行う。一方、ヒト細胞の場合には、LIFの代わりに塩基性線維芽細胞増殖因子(bFGF)および/または幹細胞因子(SCF)を添加することが望ましい。また通常、細胞は、フィーダー細胞として、放射線や抗生物質で処理して細胞分裂を停止させたマウス胎仔由来の線維芽細胞の共存下で培養される。マウス胎仔由来の線維芽細胞としては、通常STO細胞株(ATCC CRL-1503)等がフィーダーとしてよく使われるが、iPS細胞の誘導には、STO細胞にネオマイシン耐性遺伝子とLIF遺伝子を安定に組み込んだSNL細胞(SNL76/7 STO細胞; ECACC 07032801)(McMahon, A. P. & Bradley, A. Cell 62, 1073-1085 (1990))等がよく使われている。しかしながら、本発明においては、マウス胎仔由来の初代線維芽細胞(MEF)を用いた方がヒトiPS細胞の樹立効率がより改善されるので、MEFの使用がより好ましい。マイトマイシンC処理済のMEFは、ミリポア社やリプロセル社から市販されている。これらのフィーダー細胞との共培養は、核初期化物質の接触より前から開始してもよいし、該接触時から、あるいは該接触より後(例えば1-10日後)から開始してもよい。
【0098】
本発明者らは、本発明の核初期化物質およびp53の機能阻害物質と非ウイルス性ベクター、好ましくはエピソーマルベクター、とりわけ早期自己消失型エピソーマルベクターとを組み合わせたことにより、体細胞への核初期化物質およびp53の機能阻害物質の導入から、iPS細胞を樹立し、さらにiPS細胞として維持するまでの間、非ヒト動物由来成分を一切用いずに培養して(即ち、完全xeno-freeの条件下で)、ヒトiPS細胞を作製する方法を初めて提供することに成功した。xeno-freeの条件下でヒトiPS細胞を誘導する場合、核初期化物質およびp53の機能阻害物質(さらに必要に応じて他のiPS細胞の樹立効率改善物質)を接触させた後、細胞は、FCSや他の非ヒト動物由来成分を含まない培地中で培養される。分化抑制因子として培地に添加される物質(例、bFGF、SCF等)としては、ヒト由来の精製されたタンパク質、好ましくは組換えタンパク質が使用される。フィーダー細胞としては、ヒト由来の任意の体細胞を用いることができるが、例えばヒト皮膚線維芽細胞(HDF)やヒト歯髄幹細胞などが好ましく用いられ得る。また、フィーダー細胞を用いることなくヒトiPS細胞を誘導することも可能であり、この場合、細胞容器のコーティング剤として、マトリゲルやゼラチンに代えて市販のxeno-freeのコーティング剤を用いることができる。
【0099】
iPS細胞の候補コロニーの選択は、薬剤耐性とレポーター活性を指標とする方法と目視による形態観察による方法とが挙げられる。前者としては、例えば、分化多能性細胞において特異的に高発現する遺伝子(例えば、Fbx15、Nanog、Oct3/4など、好ましくはNanog又はOct3/4)の遺伝子座に、薬剤耐性遺伝子及び/又はレポーター遺伝子をターゲッティングした組換え体細胞を用い、薬剤耐性及び/又はレポーター活性陽性のコロニーを選択するというものである。そのような組換え体細胞としては、例えばFbx15遺伝子座にβgeo(β-ガラクトシダーゼとネオマイシンホスホトランスフェラーゼとの融合タンパク質をコードする)遺伝子をノックインしたマウス由来のMEF(Takahashi & Yamanaka, Cell, 126, 663-676 (2006))、あるいはNanog遺伝子座に緑色蛍光タンパク質(GFP)遺伝子とピューロマイシン耐性遺伝子を組み込んだトランスジェニックマウス由来のMEF(Okita et al., Nature, 448, 313-317 (2007))等が挙げられる。一方、目視による形態観察で候補コロニーを選択する方法としては、例えばTakahashi et al., Cell, 131, 861-872 (2007)に記載の方法が挙げられる。レポーター細胞を用いる方法は簡便で効率的ではあるが、iPS細胞がヒトの治療用途を目的として作製される場合、安全性の観点から目視によるコロニー選択が望ましい。
【0100】
選択されたコロニーの細胞がiPS細胞であることの確認は、上記したNanog(もしくはOct3/4)レポーター陽性(ピューロマイシン耐性、GFP陽性など)および目視によるES細胞様コロニーの形成によっても行い得るが、より正確を期すために、アルカリフォスファターゼ染色や、各種ES細胞特異的遺伝子の発現を解析したり、選択された細胞をマウスに移植してテラトーマ形成を確認する等の試験を実施することもできる。
【0101】
このようにして樹立されたiPS細胞は、種々の目的で使用することができる。例えば、ES細胞で報告されている分化誘導法を利用して、iPS細胞から種々の細胞(例、心筋細胞、血液細胞、神経細胞、血管内皮細胞、インスリン分泌細胞等)への分化を誘導することができる。したがって、患者本人やHLAの型が同一もしくは実質的に同一である他人から採取した体細胞を用いてiPS細胞を誘導すれば、そこから所望の細胞(即ち、該患者が罹病している臓器の細胞や疾患に対する治療効果を発揮する細胞など)に分化させて該患者に移植するという、自家もしくは同種異系移植による幹細胞療法が可能となる。さらに、iPS細胞から分化させた機能細胞(例、肝細胞)は、対応する既存の細胞株よりも実際の生体内での該機能細胞の状態をより反映していると考えられるので、医薬候補化合物の薬効や毒性のin vitroスクリーニング等にも好適に用いることができる。
【0102】
以下に実施例を挙げて本発明をより具体的に説明するが、本発明がこれらに限定されないことは言うまでもない。
【実施例】
【0103】
実施例1
レトロウイルスによるヒトiPS細胞の樹立
初期化に用いるレトロウイルスはpMXsプラスミドおよびPlat-Eパッケージング細胞(東京大学、北村俊夫博士より供与された, Morita, S. et al., Gene Ther. 7, 1063-1066(2000))を元に作製した。pMXsのマルチクローニングサイトへ種々のコンストラクトを挿入して初期化に用いるレトロウイルスベクターを作製した。挿入したコンストラクトはヒトOct3/4(図1中O)、ヒトSox2(図1中S)、ヒトKlf4(図1中K)、ヒトc-Myc(図1中M)、ヒトLin28(図1中L)、ヒトNanog(図1中N)、ヒトL-Myc(図1中U)と、それぞれの遺伝子の翻訳領域を口蹄疫ウイルスの2A配列をはさんで繋げたコンストラクト(Fig. 1では、各遺伝子の略号(上記)をハイフンで結んで記載(例: O-M-L))である。陰性コントロールとしてGFPを用いた。
初期化に使用するレトロウイルスは、前日に6 well培養プレート (Falcon) に1 well当り0.6x106細胞で播種したPlat-E細胞に、前述のレトロウイルスベクターを導入して作製した。培養液はDMEM/10% FCS (DMEM (Nacalai tesque) にウシ胎仔血清を10%加えたもの) を使用し、37℃、5% CO2で培養した。ベクターの導入のためにFuGene6 transfection reagent (Roche) 4.5 μLをOpti-MEM I Reduced-Serum Medium (Invitrogen) 100 μLに入れ、室温で5分間静置した。その後、各発現ベクターを1.5 μg加え、さらに室温で15分静置してからPlat-Eの培養液に加えた。2日目にPlat-Eの上清を新しい培地に換え、3日目に培養上清を回収して0.45 μm sterile filter (Whatman)で濾過し、polybrene (Nacalai) を4 μg/mLとなるように加えてウイルス液とした。
実験には新生児白人の皮膚より樹立した線維芽細胞(Cell Applications) にレンチウイルスでマウスSlc7a1を導入した細胞を使用した。この線維芽細胞はDMEM/10% FCSを培養液として使用し、100 mm培養ディッシュで37℃、5% CO2の条件で培養し、維持した。初期化因子導入の前日に、培地を除き、PBS 5 mLを加えて細胞を洗浄した。PBSを除いた後、0.25% Trypsin/1 mM EDTA (Invitrogen) を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらDMEM/10% FCSを加えて懸濁し、1.3x105細胞を6 well培養プレート (Falcon) の1 wellへ播いておいた。
翌日に、Plat-E細胞を用いて産生したレトロウイルスをそれぞれ混和して、感染させた。24時間後に培地を交換して感染を終了した。感染開始から6日目にプレートの培地を除き、PBS 1 mLを加えて細胞を洗浄した。PBSを除いた後、0.25% Trypsin/1 mM EDTA (Invitrogen) を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらDMEM/10% FCSを加えて懸濁し、5x105細胞を、あらかじめフィーダー細胞を蒔いておいた100 mm dishに蒔いた。フィーダー細胞にはマイトマイシンCで処理して、細胞分裂を止めたSNL細胞を用いた。翌日に霊長類ES細胞用培地(リプロセル)に4 ng/mLとなる様にbFGF (Wako) を加えた培地に交換し、以後2日おきに培地交換を続けた。35日目にヒトES細胞様のコロニーを数えた。コロニーの写真とコロニー数を図1に、またコロニー数をカウントした結果のまとめを図2に示す。いずれの遺伝子の組み合わせにおいても、c-Mycの代わりにL-Mycを用いたほうが、ヒトiPSコロニー(ES様コロニー)の樹立効率が格段に高かった。またc-Myc、L-Mycのいずれを用いた場合も、Nanogを含む6遺伝子(図2中6 factors)よりもNanogを除いた5遺伝子(Oct3/4, Sox2, Klf4, Lin28、およびc-MycまたはL-Myc;図2中ctrl))を用いたほうが樹立効率が高かった。またL-Mycを用いた場合は、L-Myc-Lin28の順でつないでも(図2中U-L)、つないでいない場合(5遺伝子別々に導入した場合:図2中ctrl)に比して樹立効率は低下しなかった。
【0104】
実施例2
エピソーマルプラスミドによるヒトiPS細胞の樹立(1)
初期化に用いるプラスミドはpCX-EGFP(大阪大学、岡部勝博士より供与された, FEBS Letters, 407, 313-319, 1997)を元に作製した。はじめにEGFPの下流にウッドチャック肝炎ウイルス転写後調節エレメント(WPRE)配列を挿入した。細胞内でこのベクターを複製させるためのカセットはpCEP4 (Invitrogen) のEBNA-1の両端にloxP配列を挿入して作製した。このEBNA-1とoriPを含むカセットを、前述のWPREを挿入したpCX-EGFPのBamHIサイトへ組み込み、これをpCXLE-EGFPと名付けた。このpCXLE-EGFPをEcoRIで処理し、EGFPの代わりに種々のコンストラクトを挿入して初期化に用いるプラスミドを作製した。挿入したコンストラクトは1)ヒトOct3/4、2)ヒトSox2およびヒトKlf4の各翻訳領域を口蹄疫ウイルスの2A配列をはさんで繋げたコンストラクト、3)ヒトKlf4、ヒトSox2およびヒトOct3/4の各翻訳領域を口蹄疫ウイルスの2A配列をはさんで繋げたコンストラクト、4)ヒトc-Myc、ヒトLin28およびNanogの各翻訳領域を口蹄疫ウイルスの2A配列をはさんで繋げたコンストラクラクト、および5)SV40 Large T antigenの5種であり(各コンストラクトはCAGプロモーターおよびウサギβ-グロビンpolyA配列の制御下にある)、それぞれpCXLE-hOct4、pCXLE-hSK、pCXLE-hKSO、pCXLE-hMLN、pCX-SV40LTと名付けた(pCX-SV40LTはWPRE配列、EBNA-1カセットおよびloxP配列のいずれも有してないプラスミドである)。
実験には36歳白人女性の顔の皮膚より樹立した線維芽細胞 (Cell Applications, Lot1388) を使用した。この線維芽細胞はDMEM/10% FCS (DMEM (Nacalai tesque) にウシ胎児血清を10%加えたもの) を培養液として使用し、100 mm 培養ディッシュで37℃、5% CO2の条件で培養し、維持した。プラスミド導入時には、培地を除き、PBS 5 mLを加えて細胞を洗浄した。PBSを除いた後、0.25% Trypsin/1 mM EDTA (Invitrogen) を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらDMEM/10% FCSを加えて懸濁し、6x105細胞を15 mL遠心チューブに回収した。800 rpmで5分間遠心して上清を除いた。プラスミド全量3 μg(pCXLE-hOct4 0.5 μg、pCXLE-hSK 1 μg、pCXLE-hKSO 0.5 μg、pCXLE-hMLN 0.5 μg、pCX-SV40LT 0.5 μg)を、Microporator(エアブラウン)を用いて細胞に導入した。導入時の条件は100 μLチップを用い、1650 V, 10 ms, 3回のパルスで行った。導入後の細胞はあらかじめDMEM/10% FCSを3 mL入れておいた6 well培養プレート (Falcon) へ移し、37℃、5% CO2の条件で6日間培養した。その後培地を除き、PBS 2 mLを加えて細胞を洗浄した。PBSを除いた後、0.25% Trypsin/1 mM EDTA (Invitrogen) を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらDMEM/10% FCSを加えて懸濁し、2x105の細胞を、あらかじめフィーダー細胞を蒔いておいた100 mm dishに蒔いた。フィーダー細胞としてはマイトマイシンC処理をしたMEFを使用した。翌日に霊長類ES細胞用培地(リプロセル)に4 ng/mLとなる様にbFGF (Wako) を加えた培地に交換し、以後2日おきに培地交換を続けた。26日目にヒトES細胞様コロニーが1個出現した。樹立時の写真を図3(左)に、3継代目のコロニーの写真を図3(右)に示す。2x105の細胞から1個のES様コロニーが樹立できた。
【0105】
実施例3
エピソーマルプラスミドによるヒトiPS細胞の樹立(2)
初期化に用いるプラスミドとして、実施例2におけるpCX-SV40LT以外の4種のプラスミド:pCXLE-hOct4、pCXLE-hSK、pCXLE-hKSO、pCXLE-hMLNを用いた。
実験には36歳白人女性の顔の皮膚より樹立した線維芽細胞 (Cell Applications, Lot1388) を使用した。この線維芽細胞はDMEM/10% FCS (DMEM (Nacalai tesque) にウシ胎児血清を10%加えたもの) を培養液として使用し、100 mm 培養ディッシュで37℃、5% CO2の条件で培養し、維持した。プラスミド導入時には、培地を除き、PBS 5 mLを加えて細胞を洗浄した。PBSを除いた後、0.25% Trypsin/1 mM EDTA (Invitrogen) を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらDMEM/10% FCSを加えて懸濁し、6x105細胞を15 mL遠心チューブに回収した。800 rpmで5分間遠心して上清を除いた。プラスミド全量3 μg(pCXLE-hOct4 1 μg、pCXLE-hSK 1 μg、pCXLE-hKSO 0.5 μg、pCXLE-hMLN 0.5 μg)をMicroporator(エアブラウン)を用いて細胞に導入した。導入時の条件は100 μLチップを用い、1650 V, 10 ms, 3回のパルスで行った。導入後の細胞はあらかじめDMEM/10% FCSを3 mL入れておいた6 well培養プレート (Falcon) へ移し、37℃、5% CO2の条件で6日間培養した。その後培地を除き、PBS 2 mLを加えて細胞を洗浄した。PBSを除いた後、0.25% Trypsin/1 mM EDTA (Invitrogen) を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらDMEM/10% FCSを加えて懸濁し、1x105の細胞をあらかじめフィーダー細胞を蒔いておいた100 mm dishに蒔いた。フィーダー細胞としてはマイトマイシンC処理をしたMEFまたはSNL76/7を使用した。翌日に霊長類ES細胞用培地(リプロセル)に4 ng/mLとなる様にbFGF (Wako) を加えた培地に交換し、以後2日おきに培地交換を続けた。フィーダーとしてMEFを用いた場合、24日目にヒトES細胞様コロニーが1個出現した。樹立時の写真を図4(左)に、2継代目のコロニーの写真を図4(右)に示す。1x105の細胞から1個のES様コロニーが樹立できた。なお、フィーダーとしてSNL76/7を用いた場合はiPSコロニーは樹立できなかった。
【0106】
実施例4
エピソーマルプラスミドによるヒトiPS細胞の樹立(3)
実施例2及び3に示したように、Oct3/4、Sox2、Klf4、c-Myc、Lin28およびNanogの6遺伝子、またはこれにSV40 large Tを加えた7遺伝子をエピソーマルプラスミドにて細胞に導入した場合は、1〜2x105の細胞から0個〜1個程度しかiPS細胞を樹立することができなかった。この誘導効率の低さに関しては、同じ7遺伝子を導入したScience, 324, 797-801 (2009) にも記載されている。そこで樹立効率の改善を目的として、実施例1の予備実験で良好な結果を得たL-Myc(L-Myc-Lin28連結コンストラクト)を使用し、また不要と判断されたNanogは用いないこととした。さらに、ヒトp53に対するshRNAをコードするDNA(5’-GACTCCAGTGGTAATCTACttcaagagaGTAGATTACCACTGGAGTC-3’(配列番号32):下線部がp53のターゲット配列。大文字がdsRNAを形成する部分。以下、単にp53 shRNAという)も使用した。
実施例2にて作製したpCXLE-hOct4、pCXLE-hSK、pCXLE-hMLNに加え、1)実施例2にて作製したpCXLE-EGFPをEcoRIで処理し、EGFPの代わりにヒトL-MycおよびヒトLin28の各翻訳領域を口蹄疫ウイルスの2A配列をはさんで繋げたコンストラクラクトを挿入したプラスミド(pCXLE-hUL)と、2)pCXLE-hOct4のBamHIサイトへp53に対するshRNA発現コンストラクト(U6プロモーターにより駆動される)を挿入したプラスミド(pCXLE-hOct4-shp53)とを作製し、使用した。またコントロールとしてpCXLE-EGFPを用いた。pCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hULの構造を図30に示す(図中、pCXLE-hOct4-shp53はpCXLE-hOct3/4-shp53と記載される)。
実験には21歳日本人女性の皮膚より樹立した線維芽細胞 (JCRB, TIG113) にレンチウイルスでマウスSlc7a1を導入した細胞を使用した。この線維芽細胞はDMEM/10% FCS (DMEM (Nacalai tesque) にウシ胎児血清を10%加えたもの) を培養液として使用し、100 mm 培養ディッシュで37℃、5% CO2の条件で培養し、維持した。プラスミド導入時には、培地を除き、PBS 5 mLを加えて細胞を洗浄した。PBSを除いた後、0.25% Trypsin/1 mM EDTA (Invitrogen)を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらDMEM/10% FCSを加えて懸濁し、6x105細胞を15 mL遠心チューブに回収した。800 rpmで5分間遠心して上清を除いた。表2に示す各プラスミド(トータル3 μg)をMicroporator(エアブラウン)を用いて細胞に導入した。導入時の条件は100 μLチップを用い、1650 V, 10 ms, 3回のパルスで行った。導入後の細胞はあらかじめDMEM/10% FCSを3 mL入れておいた6 well培養プレート (Falcon) へ移し、37℃、5% CO2の条件で7日間培養した。その後培地を除き、PBS 2 mLを加えて細胞を洗浄した。PBSを除いた後、0.25% Trypsin/1 mM EDTA (Invitrogen) を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらDMEM/10% FCSを加えて懸濁し、1.5x105の細胞をあらかじめフィーダー細胞を蒔いておいた100 mm dishに蒔いた。フィーダー細胞としてはマイトマイシンC処理をしたMEFまたはSNL76/7を使用した。翌日に、霊長類ES細胞用培地(リプロセル)に4 ng/mLとなる様にbFGF (Wako) を加えた培地に交換し、以後2日おきに培地交換を続けた。28日目に出現したヒトES細胞様のコロニーをカウントした。コロニーの写真を図5に、またコロニー数をカウントした結果を表2および図6(フィーダーがMEFの場合)に示す。
【0107】
【表2】
【0108】
いずれのフィーダーを用いた場合もiPSコロニー(ES様コロニー)を樹立することができたが、MEFを使ったほうがより多くのコロニーを樹立することができた。また、従来の6遺伝子(Oct3/4、Sox2、Klf4、c-Myc、Lin28およびNanog)を用いた場合に比べ、Oct3/4、Sox2、Klf4、L-Myc、Lin28の5遺伝子を用いたほうが樹立効率が上昇し、さらにそこへp53 shRNAを加えることにより樹立効率は格段に上昇した。
【0109】
実施例5
エピソーマルプラスミドによるヒトiPS細胞の樹立(4)
初期化に用いるプラスミドとして、実施例4と同じpCXLE-hOct4、pCXLE-hSK、pCXLE-hMLN、pCXLE-hUL、pCXLE-hOct4-shp53の5種類のプラスミドと、コントロールとしてpCXLE-EGFPを用いた。
実験には6歳日本人女性の皮膚より樹立した線維芽細胞 (JCRB, TIG120) および8カ月齢日本人男性の皮膚より樹立した線維芽細胞 (JCRB, TIG121) のそれぞれにレンチウイルスでマウスSlc7a1を導入した細胞を使用した。これらの線維芽細胞はDMEM/10% FCS (DMEM (Nacalai tesque) にウシ胎児血清を10%加えたもの) を培養液として使用し、100 mm 培養ディッシュで37℃、5% CO2の条件で培養し、維持した。プラスミド導入時には、培地を除き、PBS 5 mLを加えて細胞を洗浄した。PBSを除いた後、0.25% Trypsin/1 mM EDTA (Invitrogen)を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらDMEM/10% FCSを加えて懸濁し、6x105細胞を15 mL遠心チューブに回収した。800 rpmで5分間遠心して上清を除いた。表3に示すプラスミド(トータル3 μg)をMicroporator(エアブラウン)を用いて細胞に導入した。導入時の条件は100 μLチップを用い、1650 V, 10 ms, 3回のパルスで行った。導入後の細胞はあらかじめDMEM/10% FCSを3 mL入れておいた6 well培養プレート (Falcon) へ移し、37℃、5% CO2の条件で7日間培養した。その後培地を除き、PBS 2 mLを加えて細胞を洗浄した。PBSを除いた後、0.25% Trypsin/1 mM EDTA (Invitrogen) を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらDMEM/10% FCSを加えて懸濁し、1x105の細胞をあらかじめフィーダー細胞を蒔いておいた100 mm dishに蒔いた。フィーダー細胞としてはマイトマイシンC処理をしたMEFまたはSNL76/7を使用した。翌日に、霊長類ES細胞用培地(リプロセル)に4 ng/mLとなる様にbFGF (Wako) を加えた培地に交換し、以後2日おきに培地交換を続けた。28日目に出現したヒトES細胞様のコロニーをカウントした。コロニーの写真を図7(TIG120を用いた場合)および図8(TIG121を用いた場合)に、またコロニー数をカウントした結果を表3、図9(TIG120を用い、フィーダーとしてMEFを用いた場合)および図10(TIG121を用い、フィーダーとしてMEFを用いた場合)に示す。
【0110】
【表3】
【0111】
従来の6遺伝子(Oct3/4、Sox2、Klf4、c-Myc、Lin28およびNanog)を用いた場合に比べ、そこへp53 shRNAを加えた場合のほうが樹立効率が上昇した。さらに、従来の6遺伝子よりもOct3/4、Sox2、Klf4、L-Myc、Lin28の5遺伝子を用いたほうが樹立効率が上昇し、さらにそこへp53 shRNAを加えることにより樹立効率は格段に上昇した。
フィーダー細胞としてMEFを用いた場合とSNL76/7(MSTO)を用いた場合とを比較した結果を図11に示す(TIG120を用いた場合)。いずれのフィーダーを用いた場合もiPSコロニー(ES様コロニー)を樹立することができたが、MEFを使ったほうがより多くのコロニーを樹立することができた。
【0112】
実施例6
iPSコロニーにおける外来遺伝子の存在の有無の確認(1)
エピソーマルプラスミドで初期化遺伝子を導入して樹立した5種類の異なるiPS細胞を用いて、外来遺伝子のゲノム中への組み込みの有無等を調べた。
6 well培養プレート (Falcon) でほぼコンフルエントとなったiPS細胞を、PBS 2 mLを加えて洗浄した。PBSを除いた後、ゲノム回収バッファー(50 mM Tris-HCl, 20 mM EDTA, 100 mM NaCl, 1% SDS, 50 μg/mL proteinase K)を400 μL加えて細胞を溶かし、1.5 mLチューブに回収した後、細胞ライセートを55℃で一晩インキュベートした。ここにPCI(フェノール、クロロホルムおよびイソアミルアルコールを25:24:1で混和させたもの)を150 μL加えて、15分間混和した。これを13200 rpm、10分間遠心し、上層の水層を回収した。ここにエタノール1 mLを加えて、混和後に析出してきたものを別のチューブへ回収し、これをlong DNAとした。析出物を取り除いた後の液を13200 rpm、15分間遠心した。これで沈澱してきたものをshort DNAとした。いずれのDNAも70% エタノールで洗浄し、TEバッファーに溶かしてPCRの鋳型とした。PCRにはTaKaRa EX Taq (宝酒造) を使用し、一回の反応にlong DNA 50 ng、short DNA 250 ngをそれぞれ鋳型として用いた。プライマーは以下の組み合わせを使用した。
Klf4の検出; hKlf4-S1016 ACC CAT CCT TCC TGC CCG ATC AGA(配列番号33)
hKlf4-AS1170 ATC ACA AGT GTG GGT GGC GGT CCT(配列番号34)
c-Mycの検出; hMyc-S547 GCC GCC GCC TCA GAG TGC ATC GAC(配列番号35)
hMyc-AS947 CGA GTG GAG GGA GGC GCT GCG TAG(配列番号36)
pCEP4由来の; pEP4-SF1 TTC CAC GAG GGT AGT GAA CC(配列番号37)
OriPの検出 pEP4-SR1 TCG GGG GTG TTA GAG ACA AC(配列番号38)
OriPの検出時には元となった線維芽細胞のゲノム(50 ng)に導入したプラスミドを200, 20, 2 fg加えたもの(図12中「+plasmid」)をコントロールとして同時にPCRを行った。結果を図12に示す。いずれのiPS細胞においても、外来遺伝子(Tg)由来のKlf4およびc-Myc、並びにベクター成分のOriPは、long DNA(図12中L)およびshort DNA(図12中S)のどちらからも検出されなかったことから、導入したエピソーマルプラスミドは細胞から脱落したものと予想された。
【0113】
実施例7
エピソーマルプラスミドによるヒトiPS細胞の樹立(5)
様々な年齢の日本人及び白人男女の皮膚より樹立した線維芽細胞(HDF) を体細胞ソースとして用い、実施例5と同様にしてOct3/4、Sox2、Klf4、L-Myc、Lin28の5遺伝子およびp53 shRNAを、pCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hULを用いて導入した。フィーダー細胞はMSTOおよびMEFのいずれかを用いた。また線維芽細胞はマウスSlc7a1を導入した細胞、導入しなかった細胞のいずれも用意した。遺伝子導入27〜32日目に出現したヒトES細胞様のコロニーをカウントした結果(n=1〜5)をまとめて表4に示す。
【0114】
【表4】
【0115】
表中のコロニー数はHDF 1x105個あたりの樹立コロニー数の平均値を示す。表4に示されるように、年齢、性別、人種に関わりなく、iPS細胞を樹立できることが明らかになった。また表では分けて記載しなかったが、Slc7a1の導入の有無にかかわらずiPS細胞が樹立できることも分かった。
【0116】
実施例8
一過性発現プラスミドによるp53shRNAの導入
p53遺伝子は癌抑制遺伝子として知られており、p53の長期的な機能阻害は発癌のリスクを高める可能性がある。そこで、p53 shRNAを一過性の発現ベクター(EBNA-1とoriPを有さない通常のプラスミドベクター:以下プラスミドベクター)により発現させた場合でも、エピソーマルベクター同様にiPS細胞が樹立できるかどうかを検討した。
まず予備実験として、エピソーマルベクターとプラスミドベクターの遺伝子発現の消失の程度に差があるかどうかを検討した。実験には、pCX-EGFP(大阪大学、岡部勝博士より供与された, FEBS Letters, 407, 313-319, 1997)、実施例2で作製したpCXLE-EGFP、およびpCXLE-EGFPからloxP配列を除いたプラスミドであるpCXE-EGFPを用いた。各プラスミドをHDF1419に導入し、導入後6日目および14日目にGFPの発現を観察した。なお導入時の条件はMicroporatorにより、100 μLチップを用いて 1650 V, 10 ms, 3回のパルスで行った。結果を図13に示す。いずれのプラスミドでも6日目にGFPによる蛍光が認められたが、発現量はpCX-EGFPが他の2つに比べ低かった。14日目になるとまだ蛍光が認められるもののpCX-EGFPの発現量は他の2つに比較し顕著に低かった。以上によりプラスミドベクターはエピソーマルベクターと比べて遺伝子発現の消失が早いことが確認された。
次にp53 shRNAをプラスミドベクターで導入した場合にもiPS細胞が樹立できるかどうかを検討した。導入には以下のプラスミドベクターまたはエピソーマルベクターを用いた。
(a) pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hMLN
(b) pCXLE-hOct4、pCXLE-hSK、pCXLE-hMLN、pSilencer-shp53
(pSilencer-shp53はpSilencerTM(Ambion)のU6プロモーターの下流にp53 shRNAを組み込んだもの)
(c) pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hUL
(d) pCXLE-hOct4、pCXLE-hSK、pCXLE-hUL、pSilencer-shp53
導入にはそれぞれ0.75 μg((b)および(d))もしくは1 μg((a)および(c))のベクターを用い、合計3 μgを使用した。
体細胞として36歳白人女性の皮膚より樹立した線維芽細胞(HDF1388)を、またフィーダー細胞としてMEFを用い、実施例5と同様にしてiPS細胞の樹立を行った。なお導入時の条件はMicroporatorにより、100 μLチップを用いて 1650 V, 10 ms, 3回のパルスで行った。導入27日目に出現したヒトES細胞様のコロニーをカウントした結果を図14に示す。上記(a)と(b)(図14中、+M-L-NのEpiとTrans)、(c)と(d)(図14中、+U-LのEpiとTrans)のどちらの比較においても、p53 shRNAをエピソーマルベクターで導入した場合とプラスミドベクターで導入した場合とで樹立コロニー数に大差は無く、プラスミドベクターによる導入でも十分にiPS細胞が樹立できることが分かった。
【0117】
実施例9
HLAホモ健常人由来歯髄幹細胞からのiPS細胞の樹立
HLA-A、HLA-B、HLA-CwおよびHLA-DRB1の4遺伝子座がホモの健常人由来の歯髄幹細胞(DP74株およびDP94株)は、岐阜大学、國貞隆弘博士、手塚健一博士より提供を受けた。このDP74株およびDP94株は、J. Dent. Res., 87(7): 676-681 (2008) およびWO 2010/013359に記載の方法に基づき樹立された。両細胞株の上記4つのHLA遺伝子型と、そのアレル頻度(この4つのHLA遺伝子型を持つアレル(ハプロタイプ)の、日本人の全アレルにおける頻度)を表5に示す。
【0118】
【表5】
【0119】
初期化に用いるプラスミドとして、5種のプラスミド:pCXLE-hOct4、pCXLE-hSK、pCXLE-hMLN、pCXLE-hULおよびpCXLE-hOct4-shp53を用いて、実施例5と同様の手法にてiPS細胞を樹立した。その際、併せてScience, 324:797-801 (2009) に記載のプラスミド(Thomson’s mix)導入によるiPS細胞樹立も行った。導入にはそれぞれ合計3 μgのベクターを使用し、導入時の条件はMicroporatorにより、100 μLチップを用いて 1650 V, 10 ms, 3回のパルスで行った。フィーダー細胞はMSTOを用いた。
pCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hULをDP74およびDP94に導入して樹立したiPS細胞の写真を図15に示す。また、導入28日目に出現したヒトES細胞様のコロニーをカウントした結果を図16および図17に示す(数値は3回の実験の平均値と標準偏差を示す)。DP74株 (図16) およびDP94株 (図17) のいずれの場合もOct3/4、Sox2、Klf4、L-Myc、Lin28の5遺伝子およびp53 shRNA(pCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hUL)を導入した場合が最も樹立効率が高く、HDFと同様の良好な結果が得られた。
【0120】
実施例10
iPSコロニーにおける外来遺伝子の存在の有無の確認(2)
予備検討としてpCXLE-EGFPをHDFに導入し、EGFPの発現がどのように消失していくかを検討した。導入後1週目〜4週目まで毎週、1×104個の細胞におけるEGFPの発現量(蛍光強度)をFACS解析した。結果を図18に示す。EGFPの発現量および発現細胞数は週を追うごとに減少し、遺伝子導入から4週後にはGFP陽性細胞はわずか2.4%に減少していた。
次に、pCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hULをHDFに導入して作製した、樹立直後のiPSクローン18種類を用いて、外来遺伝子の存在の有無を確認した。
0.2 mLチューブにゲノム回収バッファー(TaKaRa Ex Taq用バッファーに167 μg/mL proteinase Kを加えたもの)を5 μL分注しておき、遺伝子導入から30日目の個々のiPS細胞のコロニーを物理的にディッシュから剥してこのチューブに回収した。回収後、チューブを55℃で3時間インキュベートして細胞ライセートを得た。H2Oを10 μL加えたのちに95℃で3分間加熱してProteinase Kを失活させた。これをそのままリアルタイムPCRの鋳型として用いた。PCRにはSYBR Premix EX Taq II (宝酒造) を使用した。エピソーマルベクターのコピー数を計算するためにEBNA-1用のPCRプライマー対を、また、細胞数を見積もるために内在のFBXO15遺伝子座用の別のプライマー対をデザインした。
EBNA-1プライマー(エピソーマルベクターの検出用)
EBNA-183F ATC AGG GCC AAG ACA TAG AGA TG(配列番号39)
EBNA-243R GCC AAT GCA ACT TGG ACG TT(配列番号40)
Fbx15プライマー(内在性アレルの検出用)
hFbx15-2F GCC AGG AGG TCT TCG CTG TA(配列番号41)
hFbx15-2R AAT GCA CGG CTA GGG TCA AA(配列番号42)
Fbx15の増幅量から反応液中の細胞数を求め、これでEBNA-1の増幅量を補正することで、1×104細胞あたりのエピソーマルベクターのコピー数を算出した。結果を図19に示す。18クローンの平均値は1×104細胞あたりわずか20.3±13.7コピーであったことから、遺伝子導入30日経過後は極めて僅かの外来遺伝子(エピソーマルベクター)しか残っていないことが明らかとなった。
次に、細胞を継代培養する以外は同じ実験を行った。
1× Ex Taqバッファー (Takara) および167 μg/ml プロテイナーゼKからなる新鮮な細胞溶解液を調製した。樹立したiPS細胞を解析するために、60 mmディッシュ中で培養した細胞を、CTK処理によるフィーダー細胞除去後に、セルスクレイパーを用いて集めた。該細胞をチューブに入れて遠心した後、細胞ペレットを200 μlの溶解液で溶解した。55℃で3時間インキュベーションした後、95℃でプロテイナーゼKを失活させ、ライセートを定量的PCR解析に用いた。増幅標準には、FBXO15とEBNA-1の両方のアンプリコンを有するpCXLE-hFbx15-cont2プラスミドを用いた。
DP由来epi-iPS細胞の結果を図31Aに示す。トランスフェクションの6日後に、細胞あたり約200コピーのエピソーマルベクターが検出された。対照的に、試験した7クローン中5クローンにおいて、EBNA-1 DNAは全く検出できなかった。残りの2クローンにおいては、それぞれ-0.001および2コピーが検出された(図31A)。後者のクローンは、おそらくプラスミドが染色体中に組み込まれているだろう。
線維芽細胞由来のepi-iPS細胞の結果を図31Bに示す。404C-2および409B-2を含むいくつかのクローンにおいて、エピソーマルベクターは全く検出できなかった。これらのデータから、epi-iPSCクローンの大部分においてエピソーマルベクターが自然消失していることが示された。
【0121】
実施例11
iPSコロニーにおける外来遺伝子の発現の有無の確認
pCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hULをHDFに導入して作製した、樹立後5から9継代目のiPSクローン9種類を用いて、外来遺伝子の発現の有無を確認した。
6-wellプレートの1ウェルのiPS細胞から全RNAを抽出し、このうち1 μgをプライマーdT(20) および逆転写酵素Rever Tra Ace(TOYOBO)を用いてmRNAを相補鎖DNAへと逆転写した。反応物の最終容量は、20 μLであった。この逆転写物にH2Oを60 μL加えた。そのうちの1 μL(12.5 ngのRNA量に相当)を鋳型として、以下の表6に記載した、外来性遺伝子の発現量と内在性遺伝子の発現量を合わせた発現量(トータル発現量)測定用、および外来性遺伝子発現量測定用のプライマーセットを用いて、SYBR Green IIを指標として導入遺伝子ごとに定量PCRを行った。増幅のための条件は以下の通りであった。95℃/30秒で1サイクル、次に(94℃/10秒、60℃/10秒、72℃/30秒)を50サイクル。
【0122】
【表6】
【0123】
次に、遺伝子導入に用いたpCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hULを106 copies/μLに調製した溶液およびこの溶液を10倍ずつ系列希釈した溶液を鋳型として同様に定量PCRを行い、各初期化遺伝子について、トータル発現量測定用プライマーセットおよび外来性遺伝子発現量測定用プライマーセットごとの検量線を作成した。この検量線を用いて、各iPS細胞における定量PCRの結果を変換して、全RNA 12.5ngに対する各遺伝子のcopy数を、トータル発現量および外来性遺伝子発現量として算出した。このcopy数を対数で縦軸に表したグラフを図20(Oct3/4)および図21(Sox2、Klf4、L-Myc、Lin28)に示す(灰色のゾーンは検出限界以下)。409B-2クローンや421C-1クローンなど、調べた全ての外来遺伝子の発現がいずれも検出限界以下のクローンが多数認められた。これらの細胞を選択することにより、より安全性の高いiPS細胞が利用できることが示された。
【0124】
実施例12
ヒトEpi-iPS細胞の特性解析
1) マイクロアレイ解析およびCGHアレイ解析
pCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hULを皮膚由来線維芽細胞(TIGおよびHDF)に導入して樹立したヒトiPS細胞、ヒトES細胞、および導入に用いたTIGおよびHDFの遺伝子発現パターンに違いがあるかどうか調べるために、DNAマイクロアレイ解析を行った。解析はCell, 131, 861-872 (2007) に記載の手法にて行った。各細胞間の相関係数を表7に示す。
【0125】
【表7】
【0126】
各遺伝子間の発現量の相違に基づきクラスタリング解析を行った結果を図22に、またScatter plot解析を行った結果を図23に示す。エピソーマルベクターにより樹立したiPS細胞は、ES細胞と良く似た発現パターンを示したことから、ES細胞に近いiPS細胞であることが明らかとなった。
また、CGHアレイ解析を行った結果を図24に示す。導入に用いたTIG細胞と比較した結果、特に大きな異常は認められなかった。また、幾つかについてはGバンド染色による核型解析を行ったが、異常は認められなかった。
なお、以上の実験には以下のアレイとスキャナを用いて行った。
・アレイ ヒトGE用:G4112F Whole Human Genome Microarray Kit, 4 x 44K(Agilent)
・アレイ ヒトCGH用:G4426B#14950 Human Genome CGH Microarray Kit 4x44K(Agilent)
・スキャナ:DNA Microarray Scanner, Model G2539A
2) RT-PCR解析
Epi-iPS細胞におけるOCT3/4、SOX2、NANOGおよびESG1等の多能性幹細胞マーカーの発現を調べた。DP由来epi-iPS細胞の結果を図32Aに示し、線維芽細胞由来epi-iPS細胞の結果を図32Bに示す。RT-PCR解析の結果、epi-iPSCクローンはこれらの遺伝子を、hESCやレトロウイルスにより誘導されたiPSCクローンに匹敵するレベルで発現していることが明らかとなった。
3) バイサルファイトシークエンス解析
フィーダー細胞を除去した後、iPS細胞からゲノミックDNAを抽出し、以前に記載したようにして解析した (K.Takahashi et al, Cell 131, 861 (Nov.30, 2007))。
NANOGのプロモーター領域中のCpG部位のDNAメチル化レベルは親HDFおよびDP細胞では高かったが、epi-iPSおよびES細胞では低かった(図33)。
4) テラトーマ形成
インビボでのepi-iPSCの分化能を調べた。CTK溶液を用いて細胞を集め、遠心した。細胞ペレットをDMEM/F12に再懸濁した。コンフルエントな60 mmディッシュから細胞の半分をSCIDマウス(CREA, 日本)の精巣に注射した。注射から8〜12週間後に腫瘍を切除し、PBS中で4% パラホルムアルデヒドを用いて固定した。パラフィン包埋した組織を切片化し、ヘマトキシリンおよびエオシンで染色した。結果を図34に示す。組織学的試験から、これらの腫瘍はテラトーマであり、神経上皮、軟骨、筋肉および腸管様上皮を含む、三胚葉系列の全ての組織を含んでいることが確認された(図34A, B)。
5) インビトロ分化
BMPアンタゴニストとアクチビン/Nodal阻害剤による二重SMAD阻害 (M.Eiraku et al, Cell Stem Cell 3, 519 (2008)) と組み合わせた胚様体様の凝集体の無血清培養(SFEB)法を用いて、ドーパミン産生ニューロンへのインビトロ指向性分化を実施した。
ドーパミン産生ニューロンの誘導のために、以下の分化培地を調製した: 5% Knockout Serum Replacement (KSR; Invitrogen)、2 mM グルタミン、0.1 mM 非必須アミノ酸および0.1 mM 2-メルカプトエタノールを補充したDMEM/F12。Accumax (Invitrogen) 中でiPS細胞を単一細胞に解離させ、96-ウェル低細胞接着プレート (LIPIDURE-COAT PLATE A-U96, NOF Corporation)を用いて、10 μM Y-27632、2 μM ドルソモルフィンおよび10 μM SB431542 (Sigma) を補充した分化培地中に、9000細胞/150 μl/ウェルの密度で再凝集させた。培養5日後に、細胞凝集体を100 ng/ml FGF-8および20 ng/ml Wnt1で刺激した。8日目から、200 ng/ml SHHを培地に加えた。12日目に、凝集した細胞を回収して、200 ng/ml SHHを含むNB培地 (B27サプリメントを含有するNeurobasal培地; Invitrogen) 中、ポリオルニチン/ラミニンでコーティングした60 mmディッシュに移した。その後15日目に、培地を1 ng/ml FGF-20および12.5 ng/ml bFGFを含むNB培地に変更した。22日目に細胞を集め、2 ng/ml GDNF、20 ng/ml BDNF、 400 μM dbcAMPおよび200 μM アスコルビン酸を補充したNB培地中、8-ウェルチャンバープレート上に、ウェルあたり2 × 105個の密度で播種した。29日目に、分化した細胞を免疫染色分析のために固定した。ネスチン (Chemicon)、Ki67 (Novocastra)、Pax6 (Covance)、βIII-チューブリン (Covance Research Products)、チロシンヒドロキシラーゼ(Chemicon)、MAP2ab (Sigma) およびVAMT-2 (PelFreeze) に対する1次抗体を用いた。Alexa488、Alexa594またはCy5を結合した2次抗体を適切に用いた。核を可視化するために、200 ng/mlの4’,6’-ジアミジノ-2-フェニルインドール (DAPI)を最後の洗浄の際に加えた。
結果を図35に示す。大部分の細胞が未熟な神経マーカーであるネスチンを発現していた(図35B-A)。まだ増殖中でKi67陽性の細胞もあった(図35B-B,C,D)。未熟な神経細胞集団はPax6陽性であったのに対し、より成熟した細胞集団は、SHHやFGF8等の誘導因子で処理した後、ドーパミン産生ニューロンのマーカーであるチロシンヒドロキシラーゼ(TH)を発現した(図35B-E, 35B-F)。また、TH陽性細胞は神経マーカーのTuj1およびMAP2ab、並びにドーパミントランスポーターであるVMAT2と共局在していた (図35A-E,F,G, 35B-G,H,I.J,K)。従って、epi-iPSCはドーパミン産生ニューロンに分化する能力を有している。Y3混合物(pCXLE-hOct4、pCXLE-hSK、pCXLE-hUL)を用いて樹立したepi-iPSCについても、同様の解析(RT-PCR、コピー数分析、核型分析およびテラトーマ形成)を行った。その結果、Y4混合物(pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hUL)を用いて樹立したepi-iPSCに比して遜色ないepi-iPSCであることが示された。各epi-iPSCクローンの解析リストを表8に示す。
【0127】
【表8】
【0128】
実施例13
異種成分不含(Xeno-free)条件下でのiPS細胞の樹立および培養
エピソーマルベクターを用いて樹立したiPS細胞を再生医療分野で利用するためには、Xeno-free条件下でのiPS細胞の樹立および維持が重要なポイントである。そこで以下の検討を行った。
1)歯髄幹細胞株のフィーダー細胞としての使用
遺伝子導入に用いたヒト皮膚由来線維芽細胞(HDF)をフィーダー細胞として用いた場合でも、ヒトiPS細胞を樹立・維持できることが知られている(Takahashi et al., PLoSone, vol.4, issue 12, e8067 (2009))。自己のフィーダー細胞を用いることで、Xeno-free条件下でのiPS細胞の維持が可能となるため、臨床応用における意義は大きい。そこで歯髄幹細胞株DP74をフィーダー細胞として用いることができるかどうかを検討した。iPS細胞はpCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hULをHDFに導入して樹立したヒトiPS細胞を用い、培養液は通常の霊長類ES細胞用培地(リプロセル)に4 ng/mLとなる様にbFGF (Wako) を加えた培地(以下リプロセル培地)を用いた。1継代後の細胞の写真を図25に示す。フィーダー細胞としてMSTOを用いた場合と比較して遜色なくES様形態が維持された。
2)Xeno-free条件下でのiPS細胞の維持(1)
Xeno-free培地であるTeSR2(Stemcell Technologies)およびXeno-freeのプレートコーティング剤であるCELLstart (GIBCO) を用い(feeder細胞は使わず)、完全なXeno-free条件下でiPS細胞が維持できるかどうかを検討した。iPS細胞はpCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hULをHDFに導入して樹立したヒトiPS細胞を用いた。1継代後の細胞の写真を図26に示す。リプロセル培地では細胞形態が変化し始めているが、TeSR2を用いた場合では未分化な形態を維持していた。
3)Xeno-free条件下でのiPS細胞の維持(2)
Xeno-free培地であるTeSR2(Stemcell Technologies)およびKSR-XF(KnockOut DMEM(Invitrogen)に15% KnockOut SR XenoFree(Invitrogen), 2 mM GlutaMAX-I(Invitrogen), 0.1 mM 非必須アミノ酸(Invitrogen), 0.1 mM 2-mercaptoethanol(Invitrogen)および8 ng/mL bFGF(Wako)を加えたもの)を用い、フィーダー細胞としてDP74株を用いてiPS細胞が維持できるかどうかを検討した。iPS細胞はpCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hULをHDFに導入して樹立したヒトiPS細胞を用いた。1継代後の細胞の写真を図27に示す。いずれのXeno-free培地を用いた場合も、ES様形態が維持された。
【0129】
実施例14
Xeno-free条件下での遺伝子導入
iPS細胞樹立後だけでなく、樹立前、すなわち体細胞に初期化遺伝子を導入する段階も、同様にXeno-free条件が可能かどうか検討を行った。
予備検討として、Microporatorを用いてDP74にpCXLE-EGFPを導入した。Xeno-free培地としてStemPro MSC-SFM (Invitrogen)を用いた。
DP74はStemPro MSC-SFM (Invitrogen) を培養液として使用し、CELLstartでコートした100 mm 培養ディッシュで37℃、5% CO2の条件で培養し、維持した。pCXLE-EGFP導入時には、培地を除き、PBS 5 mLを加えて細胞を洗浄した。PBSを除いた後、Xeno-freeの細胞剥離用液であるTripLE select (Invitrogen) を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらStemPro MSC-SFMを加えて懸濁し、15 mL遠心チューブに回収した。800 rpmで5分間遠心して上清を除いた。再びStemPro MSC-SFMに懸濁した後、6x105細胞を1.5 mLチューブに回収した。800 rpmで5分間遠心して上清を除いた。pCXLE-EGFP(3 μg)をMicroporatorを用いて細胞に導入した。導入時の条件は100 μLチップを用い、1850 V, 20 ms, 1回のパルスか、または1200 V, 30 ms, 2回のパルスで行った。導入後の細胞はあらかじめCELLstartでコートして、StemPro MSC SFMを3 mL入れておいた6 well培養プレート (Falcon) へ移し、37℃、5% CO2の条件で7日間培養した。結果を図28に示す。Xeno-free培地(StemPro MSC-SFM)を用いた場合は、1850 V, 20ms, 1回の条件では導入効率が低かった。しかし1200 V, 30 ms, 2回の条件に変更することにより、細胞生存率と導入効率は上昇した。従って、この条件で以降の遺伝子導入を行うことにした。
初期化に用いるプラスミドとして、pCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hULの3種類のプラスミドを用いた。実験には歯髄幹細胞DP74株を使用した。DP74株はStemPro MSC-SFMを培養液として使用し、CELLstartでコートした100 mm 培養ディッシュで37℃、5% CO2の条件で培養し、維持した。プラスミド導入時には、培地を除き、PBS 5 mLを加えて細胞を洗浄した。PBSを除いた後、TripLE select (Invitrogen) を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらStemPro MSC-SFMを加えて懸濁し、15 mL遠心チューブに回収した。800 rpmで5分間遠心して上清を除いた。再びStemPro MSC-SFMに懸濁した後、6x105細胞を1.5 mLチューブに回収した。800 rpmで5分間遠心して上清を除いた。3種のプラスミド(各1 μg、合計3 μg)をMicroporatorを用いて細胞に導入した。導入時の条件は100 μLチップを用い、1200 V, 30 ms, 2回のパルスで行った。導入後の細胞はあらかじめCELLstartでコートして、StemPro MSC-SFMを3 mL入れておいた6 well培養プレート (Falcon) へ移し、37℃、5% CO2の条件で7日間培養した。そののち培地を除き、PBS 2 mLを加えて細胞を洗浄した。PBSを除いた後、TripLE select (Invitrogen) を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらStemPro MSC-SFMを加えて懸濁し、15 mL遠心チューブに回収した。800 rpmで5分間遠心して上清を除いた。再びStemPro MSC-SFMに懸濁した後、1x105の細胞を、表9に示す各種条件で100 mm dishに蒔いた。翌日に表9に示す培地に交換し、以後2日おきに培地交換を続けた。導入後26日目に出現したヒトES細胞様のコロニーの写真を図29に示す。
【0130】
【表9】
【0131】
いずれの培地およびフィーダー細胞を用いた場合も、iPSコロニーが樹立できた。特に表9中のNo. 4〜No. 6は、いずれもXeno-freeの条件であることから、エピソーマルベクターを用いたvirus-freeの条件下、かつ遺伝子導入時から完全なXeno-freeの条件下で、歯髄幹細胞からiPS細胞を樹立及び維持できることが明らかになった。
【0132】
実施例15
ヒト末梢血単核細胞からのiPS細胞の樹立
3 μgの発現プラスミド (1 μgのpCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hUL) を、Nucleofector (Lonza) とHuman T cell kitを製造者の指示に従って用い、新たに単離した末梢血単核細胞 (5.0x106) に、エレクトロポレーションにより導入した。プログラムV-24の条件を用いた。6時間後、トランスフェクトした血液細胞をサイトカイン (IL-6、sIL-6R、SCF、TPO、Flit3/4リガンドおよびIL-2) および抗CD3/CD28抗体で刺激した。2日後に、細胞をMEFフィーダーで覆った6-ウェルプレート上に播種した。次いで、図36aに示すように、播種後培養培地を、IL-6、sIL-6R、SCF、TPO、Flit3/4リガンドおよびIL-2を補充したX-vivo 10から、hES培地に徐々に変更した。29日目にコロニーの写真を撮影した(図36b)。平坦なhESC様の形態を有するヒトiPSコロニーを樹立することができた。
【0133】
本発明を好ましい態様を強調して説明してきたが、好ましい態様が変更され得ることは当業者にとって自明であろう。本発明は、本発明が本明細書に詳細に記載された以外の方法で実施され得ることを意図する。したがって、本発明は添付の「請求の範囲」の精神および範囲に包含されるすべての変更を含むものである。
ここで述べられた特許および特許出願明細書を含む全ての刊行物に記載された内容は、ここに引用されたことによって、その全てが明示されたと同程度に本明細書に組み込まれるものである。
本願は、米国仮特許出願第61/232,402号および第61/307,306号を基礎としており、それらの内容は、ここで参照することにより本願に組み込まれるものである。
【技術分野】
【0001】
本発明は、効率的な人工多能性幹(以下、iPSという)細胞の樹立方法およびそのための薬剤に関する。より詳細には、本発明は、(a) Oct3/4もしくはそれをコードする核酸、(b) Klf4もしくはそれをコードする核酸、および(c) Sox2もしくはそれをコードする核酸、並びに(d1) L-Mycもしくはそれをコードする核酸および/または(d2) p53の機能阻害物質を、体細胞に接触させることによる、iPS細胞の樹立方法、上記(a)-(c)並びに(d1)および/または(d2)からなるiPS細胞誘導剤に関する。本発明はまた、上記(a)-(c)並びに(d1)および/または(d2)の核酸因子を含むエピソーマルベクターあるいは該エピソーマルベクターとloxP配列とを組み合わせたベクター、特に早期自己消失型であるエピソーマルベクター、およびそれを用いて、外来核酸因子のゲノムへの組込みを介さずに、該核酸因子の消失したiPS細胞を迅速に樹立する方法に関する。
【背景技術】
【0002】
近年、マウスおよびヒトのiPS細胞が相次いで樹立された。Yamanakaらは、マウス由来の線維芽細胞に、Oct3/4, Sox2, Klf4及びc-Myc遺伝子を導入し強制発現させることによって、iPS細胞を誘導した [WO 2007/069666 A1; Takahashi, K. and Yamanaka, S., Cell, 126: 663-676 (2006)]。その後、c-Myc遺伝子を除いた3因子によってもiPS細胞を作製できることが明らかとなった [Nakagawa, M. et al., Nat. Biotechnol., 26: 101-106 (2008)]。さらに、Yamanakaらは、ヒトの皮膚由来線維芽細胞にマウスと同様の4遺伝子を導入することにより、iPS細胞を樹立することに成功した [WO 2007/069666 A1; Takahashi, K. et al., Cell, 131:861-872 (2007)]。一方、Thomsonらのグループは、Klf4とc-Mycの代わりにNanogとLin28を使用してヒトiPS細胞を作製した[WO 2008/118820 A2; Yu, J. et al., Science, 318: 1917-1920 (2007)]。
【0003】
しかし、iPS細胞の樹立効率は1%以下と低く、特に、iPS細胞から分化した組織や個体において腫瘍化が懸念されるc-Mycを除く3因子(Oct3/4, Sox2, Klf4)を体細胞に導入してiPS細胞を作製した場合、その樹立効率が極めて低いという問題点がある。
【0004】
レトロウイルスやレンチウイルスなどのウイルス性ベクターは、非ウイルス性ベクターに比べて遺伝子導入効率が高く、そのためiPS細胞を容易に作製することができるという点で優れたベクターである。しかし、レトロウイルスやレンチウイルスは染色体中に組み込まれてしまうため、iPS細胞の臨床応用を考慮すると安全面で問題がある。そのため、アデノウイルスベクターやプラスミドなどの非ウイルス性ベクターを用いて染色体への組込みがないiPS細胞が報告されているが [Stadtfeld, M. et al., Science, 322: 945-949 (2008); Okita, K. et al., Science, 322: 949-953 (2008); Yu, J. et al., Science, 324: 797-801 (2009)]、レトロウイルスやレンチウイルスに比べると樹立効率は低い。また、iPS細胞の選択下では初期化因子の持続的な高発現が要求されるためか、一般的に組込みが起こりにくいとされているプラスミドベクターを用いても、ある頻度で初期化因子が染色体に組み込まれた安定発現株が得られる場合がある [Okita, K. et al., Science, 322: 949-953 (2008); Kaji, K. et al., Nature, 458: 771-775 (2009)]。
【0005】
そこで、まずレトロウイルスやレンチウイルスを用いてiPS細胞を樹立した後で、外来遺伝子を染色体から除去することにより、樹立効率と安全面を両立させようとする試みがなされている。例えば、レンチウイルスとCre-loxPシステムとを組み合わせた手法が報告されている [Chang, C.W. et al., Stem Cells, 27: 1042-1049 (2009); Soldner, F. et al., Cell, 136: 964-977(2009)]。しかし、これらの報告では、Creリコンビナーゼ処理後に残るloxP配列より外側のLTR配列が近傍の癌遺伝子を活性化するリスクを最小限にするため、LTR内部にloxP配列が挿入され、初期化因子の転写のためにCMVやEF1αといった別のプロモーターが挿入された複雑なコンストラクトが使用されており、より構築が容易なベクターの開発が望まれる。piggyBacトランスポゾンを用いて外来核酸因子を完全に消失させることもできるが [Kaji, K. et al., Nature, 458: 771-775 (2009)]、一過的にもゲノムへの組込みを介するという点で、内因性遺伝子の擾乱を引き起こす可能性を否定できない。
【0006】
一方、染色体外で安定に自律複製可能なエピソーマルベクターを用いる方法では、上記したiPS細胞樹立効率の低さに加え、薬剤選択の中止によるベクターの自然消失効率も低頻度で、かつ時間を要するため [Yu, J. et al., Science, 324: 797-801 (2009)]、iPS細胞の樹立効率の改善とともに、短時間で効率よくベクターを除去する方法が求められる。
【0007】
さらに、ヒトiPS細胞の臨床応用を念頭におく場合の別の問題点として、iPS細胞の樹立および維持培養に際し、血清やフィーダー細胞などの異種動物由来成分の混入による汚染が挙げられる。そのため、初期化因子の導入からヒトiPS細胞の樹立および維持培養までをすべて異種成分不含(Xeno-free)の条件下で行うことが望ましいが、これまでvirus-freeで樹立されたヒトiPS細胞では、初期化因子の導入からiPS細胞の樹立・維持培養までの少なくとも一部において、異種成分が使用されている [Okita, K. et al., Science, 322: 949-953(2008); Yu, J. et al., Science, 324: 797-801 (2009); Kaji, K. et al., Nature, 458: 771-775 (2009)]。一方、Xeno-freeの条件下で樹立されたヒトiPS細胞は、いずれもレトロウイルスやレンチウイルスにより初期化遺伝子が導入されたものであり、virus-freeで作製されたものではない [Rodoriguez-Piza, I. et al., Stem Cells, 28: 36-44 (2010); Ross, P.J. et al., Stem Cells Dev., 2009 Dec 23. (Epub ahead of print)]。
【発明の概要】
【発明が解決しようとする課題】
【0008】
本発明の目的は、臨床応用に適した安全なヒトiPS細胞を効率よく樹立することである。したがって、本発明の第一の課題は、iPS細胞、特にヒトiPS細胞の樹立効率を改善する手段を提供することであり、それを用いた効率的なiPS細胞の製造方法を提供することである。また、本発明の第二の課題は、外来核酸因子のゲノムへの組込みを一切介さずに、該核酸因子の消失したiPS細胞を迅速に樹立する方法を提供することである。さらに、本発明の第三の課題は、初期化因子の導入時からiPS細胞の樹立および維持培養に至るまで、ウイルスおよび異種成分を一切用いることなく(即ち、virus-freeかつxeno-freeの条件下で)ヒトiPS細胞を作製し、以ってヒト臨床に安全に使用することができるヒトiPS細胞を提供することである。
【課題を解決するための手段】
【0009】
本発明者らは、上記の課題を解決すべく、まずレトロウイルスベクターを用いて初期化遺伝子の好適な組み合わせを検討した。エピソーマルベクターを用いてヒトiPS細胞を樹立したYu, J. et al., Science, 324: 797-801 (2009)で使用された6因子(Oct3/4, Klf4, c-Myc, Sox2, Nanog, Lin28 (SV40 Large T抗原は樹立効率改善物質と位置づけ、初期化遺伝子から除外した))を基に、Nanogを除いた5因子および、さらにc-MycをL-Mycに置換した5因子を用いてヒト皮膚線維芽細胞(HDF)からヒトiPS細胞を誘導したところ、意外にもNanogを除いた5因子の方が、6因子の場合よりも効率よくヒトiPS細胞が樹立された。さらにc-MycをL-Mycに置換することにより、樹立効率は顕著に改善された。そこで、実際にエピソーマルベクターを用いて、6因子とc-MycをL-Mycに置換した5因子とでヒトiPS細胞樹立効率を比較したところ、同様にc-MycをL-Mycに置換した5因子を用いることにより、6因子と比較して著しく樹立効率が上昇した。
【0010】
次に、6因子またはc-MycをL-Mycに置換した5因子に加えて、p53に対するshRNAをコードするエピソーマルベクターをHDFに導入したところ、p53の機能を阻害することにより、6因子でもc-MycをL-Mycに置換した5因子と同程度のiPS細胞が得られ、該5因子にp53機能阻害を組み合わせた場合には、さらに顕著に樹立効率が上昇した。また、遺伝子導入後のHDFを継代する際に、SNL細胞でなく胎仔マウス線維芽細胞(MEF)をフィーダー細胞として用いることにより、ヒトiPS細胞の樹立効率が格段に向上することが見出された。
【0011】
本発明者らは、導入したエピソーマルベクターをiPS細胞樹立後に速やかに細胞から脱落させ得るように、該ベクターの自律複製に必要なベクター要素の両端にloxP配列を配置し、Creリコンビナーゼを作用させることにより該ベクター要素をベクターから切り出せるよう設計していたが、ヒトiPS細胞樹立後に細胞のゲノムDNAと染色体外DNAをそれぞれ単離して導入遺伝子の存在を調べたところ、いずれのDNA中にも導入したベクターは検出されず、意外にも該ベクターは、Creリコンビナーゼを用いるまでもなく速やかに細胞から脱落する、早期自己消失型ベクターであることが見出された。
【0012】
本発明者らはさらに、上記エピソーマルベクターを用いることにより、初期化因子の導入時からiPS細胞の樹立および維持培養に至るまで、完全にvirus-freeかつxeno-freeの条件下で、ヒトiPS細胞を作製することに成功した。
【0013】
以上の結果から、本発明者らは、初期化因子からNanogを除去するとともに、c-Mycに代えてL-Mycを用いるか、その代わりに、あるいはそれに加えて、p53機能阻害物質を用いることによりヒトiPS細胞の樹立効率を顕著に改善し得ること、エピソーマルベクターの設計を工夫することにより、外来核酸因子がゲノムに組み込まれることなく細胞から消失したiPS細胞を迅速に取得し得ること、並びに上記初期化因子とエピソーマルベクターとを組み合わせることにより、virus-freeかつxeno-freeの条件下で、ヒトiPS細胞を作製し得ることを見出し、本発明を完成するに至った。
【0014】
すなわち、本発明は以下の通りのものである。
[1] (a) Oct3/4もしくはそれをコードする核酸、(b) Klf4もしくはそれをコードする核酸、および(c) Sox2もしくはそれをコードする核酸、並びに(d1) L-Mycもしくはそれをコードする核酸および/または(d2) p53の機能阻害物質を、体細胞に接触させることを含む、iPS細胞の製造方法。
[2] さらに(e) Lin28もしくはLin28bまたはそれをコードする核酸を体細胞に接触させることを含む、上記[1]記載の方法。
[3] p53の機能阻害物質が、p53に対するsiRNA、shRNAおよびそれらをコードするDNAからなる群より選択される核酸である、上記[1]または[2]記載の方法。
[4] (a) Oct3/4をコードする核酸、(b) Klf4をコードする核酸、(c) Sox2をコードする核酸、(d1) L-Mycをコードする核酸および/または(d2) p53に対するshRNAをコードする核酸、並びに(e) Lin28もしくはLin28bをコードする核酸を、体細胞に導入することを含む、上記[2]または[3]記載の方法。
[5] 前記の(a)、(b)、(c)、(d1)および(e)からなる群より選択される少なくとも1つの核酸がエピソーマルベクターの形態で導入される、上記[4]記載の方法。
[6] (d2) p53に対するshRNAをコードする核酸が細胞内で自律複製できないプラスミドベクターの形態で導入される、上記[4]または[5]記載の方法。
[7] エピソーマルベクターが、50%以上の頻度で、5継代までにiPS細胞から脱落する自己消失型ベクターである、上記[5]または[6]記載の方法。
[8] エピソーマルベクターが、前記の核酸の複製に必要なベクター要素の5’側および3’側にloxP配列を同方向に配置したものである、上記[5]〜[7]のいずれかに記載の方法。
[9] 細胞をCreリコンビナーゼで処理する工程を含まない、上記[8]記載の方法。
[10] 体細胞がヒト由来である、上記[1]〜[9]のいずれかに記載の方法。
[11] 前記の(a)、(b)、(c)並びに(d1)および/または(d2)、あるいはさらに(e)の因子を体細胞に接触させる時点から、iPS細胞が樹立されるまでの間、細胞を非ヒト動物由来の成分の非存在下で培養することを含む、上記[10]記載の方法。
[12] ヒト細胞をフィーダー細胞として用いるか、フィーダー細胞を用いないことを特徴とする、上記[11]記載の方法。
[13] フィーダー細胞が体細胞と同一個体に由来する、上記[12]記載の方法。
[14] (a) Oct3/4をコードする核酸、(b) Klf4をコードする核酸、(c) Sox2をコードする核酸、(d1) L-Mycをコードする核酸および/または(d2) p53に対するshRNAをコードする核酸、並びに(e) Lin28もしくはLin28bをコードする核酸を含有してなる、iPS細胞誘導促進剤。
[15] 前記の(a)、(b)、(c)、(d1)および(e)からなる群より選択される少なくとも1つの核酸がエピソーマルベクターの形態である、上記[14]記載の剤。
[16] (d2) p53に対するshRNAをコードする核酸が細胞内で自律複製できないプラスミドベクターの形態である、上記[14]または[15]記載の剤。
[17] エピソーマルベクターが、50%以上の頻度で、5継代までにiPS細胞から脱落する自己消失型ベクターである、上記[15]または[16]記載の剤。
[18] エピソーマルベクターが、前記の各核酸の複製に必要なベクター要素の5’側および3’側にloxP配列を同方向に配置したものである、上記[15]〜[17]のいずれかに記載の剤。
[19] 上記[5]〜[9]のいずれかに記載の方法により得られる、前記の各核酸がゲノムに組み込まれることなく除去されたiPS細胞。
[20] ヒト由来である、上記[19]記載のiPS細胞。
[21] 上記[11]〜[13]のいずれかの方法により得られる、非ヒト動物由来の成分の混入のないヒトiPS細胞。
[22] 体細胞の製造における、上記[19]〜[21]のいずれかに記載のiPS細胞の使用。
[23] 体細胞の製造における細胞ソースとしての、上記[19]〜[21]のいずれかに記載のiPS細胞。
【発明の効果】
【0015】
c-Mycに代わるL-Mycの使用および/またはp53の機能阻害物質の使用、並びにNanogの不使用は、iPS細胞の樹立効率を顕著に増大させることができるので、従来きわめて樹立効率の低かった6因子(Oct3/4、Klf4、Sox2、c-Myc、Nanog、Lin28)によるヒトiPS細胞、特に初期化遺伝子がゲノムに組み込まれないタイプのヒトiPS細胞の作製に特に有用である。また、独自のエピソーマルベクターを使用することで、外来核酸因子のゲノムへの組込みを介さずにiPS細胞を樹立することができ、かつiPS細胞樹立後速やかにエピソームが脱落し、従来のエピソーマルベクターに比べて、早期にベクターの脱落が起こり得る。さらに、初期化因子の導入からiPS細胞の樹立および維持までの間、完全にvirus-freeかつxeno-freeの条件でヒトiPS細胞を作製できるため、ヒトiPS細胞の再生医療への応用において極めて有用である。
【0016】
以下、本明細書において、エピソーマルベクターを用いて樹立されたiPS細胞を「epi-iPS cells」または「epi-iPSCs」と略することがある。
また導入遺伝子の組み合わせを「Y1, Y2, Y3, Y4, T1, T2, T3」と略する場合、これらの組合せは以下の表1に示すとおりである。
【0017】
【表1】
【図面の簡単な説明】
【0018】
【図1】図1は、レトロウイルスで種々の遺伝子をヒト新生児皮膚線維芽細胞に導入し、ヒトiPS細胞を樹立した結果を示すコロニーの写真である。図中、OはOct3/4を、SはSox2を、KはKlf4を、LはLin28を、NはNanogを、Mはc-Mycを、UはL-Mycを、それぞれ示す。また「O-M-L」のような因子の連結表示は、各因子の翻訳領域を2A配列を挟んで繋げたコンストラクトを指す。各写真下の数字は、非ES様コロニー数/ES様コロニー数を示す。
【図2】図2は、図1の結果の一部をグラフ化したものである。ES様コロニーの数は黒棒、非ES様コロニーの数は白棒で示す。図中「6 factors」はOct3/4, Sox2, Klf4, Lin28, Nanog、およびc-MycまたはL-Mycの各遺伝子を導入した場合、「ctrl」は前記の6遺伝子からNanogを除いた5遺伝子を導入した場合、「L-M」、「M-L」、「L-U」、「U-L」は、それぞれの因子(LはLin28、Mはc-Myc、UはL-Myc)を連結したコンストラクトとOct3/4、Sox2およびKlf4とを導入した場合を、それぞれ示す。
【図3】図3は、pCXLE-hOct4、pCXLE-hSK、pCXLE-hKSO、pCXLE-hMLN、pCX-SV40LTの5種類のプラスミドをヒト成人皮膚由来の線維芽細胞に導入して樹立したiPS細胞コロニーの写真である。左は樹立時、右は3継代目(p3)の写真を示す。
【図4】図4は、pCXLE-hOct4、pCXLE-hSK、pCXLE-hKSO、pCXLE-hMLNの4種類のプラスミドをヒト成人皮膚由来の線維芽細胞に導入して樹立したiPS細胞コロニーの写真である。左は樹立時、右は2継代目(p2)の写真を示す。
【図5】図5は、pCXLE-hOct4、pCXLE-hSK、pCXLE-hULの3種類のプラスミド(左上パネル)、pCXLE-hOct4、pCXLE-hSK、pCXLE-hMLNの3種類のプラスミド(右上パネル)、並びにpCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hULの3種類のプラスミド(左下パネル)を、ヒト成人皮膚由来の線維芽細胞に導入して樹立したiPS細胞コロニーの写真である。各パネル上段はMEF、下段はMSTO細胞をフィーダー細胞として用いたものである。なお、いちばん右側の左肩にコロニー番号の記載のない写真は非ES様コロニーの写真である。
【図6】図6は、表2の結果をグラフ化したものである。ES様コロニーの数は黒棒、非ES様コロニーの数は白棒で示す。図中左から、(1)pCXLE-hOct4、pCXLE-hSK、pCXLE-hMLN、(2)pCXLE-hOct4、pCXLE-hSK、pCXLE-hUL、(3)pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hUL、(4)pCXLE-GFP、をそれぞれ導入した結果を示す。
【図7】図7は、pCXLE-hOct4、pCXLE-hSK、pCXLE-hMLNの3種類のプラスミド(左上パネル)、pCXLE-hOct4、pCXLE-hSK、pCXLE-hULの3種類のプラスミド(左下パネル)、pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hMLNの3種類のプラスミド(右上パネル)、pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hULの3種類のプラスミド(右下パネル)を、6歳のヒト皮膚由来線維芽細胞に導入して樹立したiPS細胞コロニーの写真である。各パネル上段はMEF、下段はMSTO細胞をフィーダー細胞として用いたものである。
【図8】図8は、pCXLE-hOct4、pCXLE-hSK、pCXLE-hMLNの3種類のプラスミド(左上パネル)、pCXLE-hOct4、pCXLE-hSK、pCXLE-hULの3種類のプラスミド(左下パネル)、pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hMLNの3種類のプラスミド(右上パネル)、pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hULの3種類のプラスミド(右下パネル)を、8カ月のヒト皮膚由来線維芽細胞に導入して樹立したiPS細胞コロニーの写真である。各パネル上段はMEF、下段はMSTO細胞をフィーダー細胞として用いたものである。
【図9】図9は、表3の結果(TIG120を用いた場合)をグラフ化したものである。ES様コロニーの数は黒棒、非ES様コロニーの数は白棒で示す。図中左から、(1)pCXLE-hOct4、pCXLE-hSK、pCXLE-hMLN、(2)pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hMLN、(3)pCXLE-hOct4、pCXLE-hSK、pCXLE-hUL、(4)pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hUL、(5)pCXLE-GFP、をそれぞれ導入した結果を示す。
【図10】図10は、表3の結果(TIG121を用いた場合)をグラフ化したものである。ES様コロニーの数は黒棒、非ES様コロニーの数は白棒で示す。図中左から、Fig. 9の説明における(1)〜(5)と同じである。
【図11】図11は、表3の結果(TIG120を用いた場合)をフィーダー細胞ごとにグラフ化したものである。図中左から、Fig. 9の説明における(1)〜(5)と同じである。
【図12】図12は、5種類の異なるiPS細胞を用いて、外来遺伝子 (Klf4、c-Myc、OriP) のゲノム中への組み込みの有無等を調べたGenomic PCRの結果を示す図である。図中「L」はlong DNAでの結果を、「S」はshort DNAでの結果を示す。「347A1」は実施例2で樹立されたiPSコロニーについての結果を、「349A1」は実施例3で樹立されたiPSコロニーについての結果を、「341A5」はOct3/4, Sox2, Klf4, c-Myc, Lin28, Nanogの6遺伝子をヒト胎児由来線維芽細胞に導入して樹立したiPSコロニーについての結果を、「345A1」は「347A1」と同じ遺伝子を導入して得られた別のiPSコロニーについての結果を、「352A3」は「341A5」と同じ遺伝子を導入して得られた別のiPSコロニーについての結果を、それぞれ示す。尚、HDFは遺伝子未導入のヒト胎児由来線維芽細胞ゲノムでの結果を、retroはOct3/4, Sox2, Klf4, c-Mycの4遺伝子をレトロウイルスベクターを用いてヒト胎児由来線維芽細胞に導入して樹立したiPSコロニーについての結果を、それぞれ示す。図中、endo (白矢頭) は内在性遺伝子を、Tg(黒矢頭)は外来遺伝子をそれぞれ示す。
【図13】図13は、pCX-EGFP、pCXE-EGFPおよびpCXLE-EGFPをHDFに導入後6日目および14日目の細胞の蛍光写真(GFP観察像)である。
【図14】図14は、p53shRNAをプラスミドベクター(EBNA-1とoriPを有さないプラスミドベクター)で導入した場合にもiPS細胞が樹立できるかどうかを調べた結果を示すグラフである。ES様コロニーの数は黒棒、非ES様コロニーの数は白棒で示す。図中左から、(1) pCXLE-hOct4、pCXLE-hSK、pCXLE-hMLN、(2) pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hMLN、(3) pCXLE-hOct4、pCXLE-hSK、pCXLE-hMLN、pSilencer-shp53、(4) pCXLE-hOct4、pCXLE-hSK、pCXLE-hUL、(5) pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hUL、(6) pCXLE-hOct4、pCXLE-hSK、pCXLE-hUL、pSilencer-shp53、(7) pCXLE-EGFP、をそれぞれ導入した結果を示す。
【図15】図15は、pCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hULをHLA 4座ホモの健常人歯髄幹細胞DP74株およびDP94株に導入して樹立したiPS細胞コロニーの写真である。
【図16】図16は、歯髄幹細胞DP74株に種々の初期化遺伝子を導入して得られたES様コロニーをカウントした結果を示すグラフである。ES様コロニーの数は黒棒、非ES様コロニーの数は白棒で示す。図中左から、(1)pCXLE-hOct4、pCXLE-hSK、pCXLE-hMLN、(2)pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hMLN、(3)pCXLE-hOct4、pCXLE-hSK、pCXLE-hUL、(4)pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hUL、(5)pEP4-EO2S-ET2K、pEP4-EO2S-EN2K、pCEP4-M2L(図中Thomson’s mix1)、(6)pEP4-EO2S-ET2K、pEP4-EO2S-Ck2M-EN2L(図中Thomson’s mix2)、(7)pEP4-EO2S-ET2K、pEP4-EO2S-EN2L、pEP4-EO2S-EM2K(図中Thomson’s mix3)、(8)pCXLE-GFP、をそれぞれ導入した結果を示す。
【図17】図17は、図16と同じ実験をDP94株を用いて行った結果を示すグラフである。
【図18】図18は、エピソーマルベクターで導入したEGFPの発現量(蛍光強度)を週ごとに測定した結果を示す図である。上図:蛍光強度をFACS解析した図。横軸は蛍光強度を、縦軸は細胞数を示す。また、チャート内の%値はGFP陽性細胞の割合を示す。下図:細胞の蛍光写真(GFP陽性像)。
【図19】図19は、樹立したiPS細胞18クローンが有するエピソーマルベクターのコピー数をリアルタイムPCRで調べた結果を示すグラフである。縦軸は1×104細胞あたりのエピソーマルベクターのコピー数を示す。「elepo 6d」は初期化遺伝子導入後6日目の細胞を示す。また「origin」は導入に用いたHDFを示す。
【図20】図20は、樹立したiPS細胞9クローンにおけるOct3/4の発現量を定量PCRで調べた結果を示すグラフである。白棒は外来性および内在性の遺伝子発現量(トータル発現量)を、黒棒は外来性の遺伝子発現量を、それぞれ示す。図中「elepoD4」は遺伝子導入後4日目の細胞を、また「KhES1」および「KhES3」はヒトES細胞を、それぞれ示す。
【図21】図21は、図20と同じ実験をSox2、Klf4、L-MycおよびLin28について行った結果を示すグラフである。
【図22】図22は、皮膚由来線維芽細胞(TIGおよびHDF)より樹立したヒトiPS細胞、もとのTIGおよびHDF、並びにヒトES細胞のDNAマイクロアレイ解析による各遺伝子間の発現量の相違に基づきクラスタリング解析を行った結果を示す図である。
【図23】図23は、ヒトES細胞(KhES3)とHDF(左図)、およびヒトES細胞(KhES3)とエピソーマルベクターを用いて樹立したヒトiPS細胞(右図)との間で遺伝子発現パターンに違いがあるかどうかを調べるために、DNAマイクロアレイの結果をScatter Plot解析した結果を示す図である。
【図24】図24は、エピソーマルベクターを用いて樹立したヒトiPS細胞のCGHアレイ解析の結果を示す図である。遺伝子導入に用いたTIG細胞(TIG120)に対する変動で示した。
【図25】図25は、DP74株をフィーダー細胞として用い、エピソーマルベクターで樹立したiPS細胞をリプロセル培地を用いて培養した1継代目の細胞の形態を示す写真(右)である。左はMSTOをフィーダー細胞として用いた場合の写真である。
【図26】図26は、Xeno-free(Xeno-free培地使用、およびフィーダー細胞不使用)の条件下で、エピソーマルベクターで樹立したiPS細胞を培養した1継代目の細胞の形態を示す写真である。左は通常のリプロセル培地を用いた場合の写真である。
【図27】図27は、Xeno-free(Xeno-free培地使用、およびDP74株をフィーダー細胞として使用)の条件下で、エピソーマルベクターで樹立したiPS細胞を培養した1継代目の細胞の形態を示す写真である。
【図28】図28は、Xeno-free条件下でpCXLE-EGFPをDP74に導入した結果を示す図である。左側は通常の条件で導入した結果(コントロール)を示す。上図:位相差像、下図:GFP観察像。
【図29】図29は、Xeno-free条件下で遺伝子導入後、表9に示す6種の条件下で細胞培養し、導入から26日目に出現したES細胞様コロニーの形態を示す写真である。
【図30】図30は、エピソーマル発現ベクターの構造を示す。Y4混合物には、3つのプラスミドを用いた(図中、pCXLE-hOct4-shp53はpCXLE-hOct3/4-shp53と記載される)。初期化因子(OCT3/4, SOX2, KLF4, L-MYC, LIN28およびp53に対するshRNA (shRNA for p53))は黒で示している。プロモーター(CAG)、WPRE、ポリアデニレーションシグナル(pA)、EBNA-1、OriPおよび2つのloxP部位も示している。
【図31】図31は、epi-iPSCクローンに残存するエピソーマルベクターのコピー数を示す。A: DP由来epip-iPS細胞の結果。B: 線維芽細胞由来epi-iPS細胞の結果。括弧内の数字は各クローンの継代数を示す。各クローンについて使用した細胞数も示している。陽性コントロールとして、レトロウイルスにより誘導したiPSクローン(253G-4)およびY4混合物のエレクトロポレーションから6日後の線維芽細胞(fibro-d6)を分析した。
【図32】図32は、RT-PCR解析による多能性細胞マーカー遺伝子の発現を示す。A: DP由来epip-iPS細胞の結果。B: 線維芽細胞由来epi-iPS細胞の結果。Y1 (454B-1)、Y2 (454C-2)、Y3 (454D-1) およびY4 (454E-2, 451F-3, 457C-1, 453F-2, 404C-2, 409B-2, 414C-2, 418C-1, 421C-1, 426C-2, 427D-4および428C-2) の各組合せにより樹立されたepi-iPSクローンから全RNAを単離した。また、レトロウイルスにより誘導したiPSCクローン (201B-7および253G-4) と、hESC株 (KhES-3およびH9) も試験した。OCT3/4およびSOX2と表示されたレーンにおいて、PCRプライマーは内在遺伝子のみを増幅したのに対し、Ret-Octレーンにおいては、PCRプライマーはレトロウイルスのOct3/4導入遺伝子を特異的に増幅した。ローディングコントロールとしてG3PDHを分析した。陰性コントロールとして、Y4混合物のエレクトロポレーションから4日後のヒト皮膚線維芽細胞 (HDF-elepo) から、全RNAを単離した。
【図33】図33は、NANOGプロモーター領域のDNAのメチル化状態を示す。白丸および黒丸は、それぞれ非メチル化CpGおよびメチル化CpGを示す。
【図34】図34は、epi-iPSCクローンから誘導したテラトーマを示す。A: 454E-2の結果。神経組織 (A)、軟骨 (B)、筋肉 (C) および腸管様上皮 (D) のヘマトキシリン-エオシン染色を示す。スケールバー=50 μm。B: 404C-2、409B-2、418C-1、421C-1、428C-2および454D-1の結果。神経組織、軟骨および腸管様上皮のヘマトキシリン-エオシン染色を示す。スケールバー=50 μm。
【図35】図35は、epi-iPSCクローンからドーパミン産生ニューロンへの分化を示す(クローン454E-2)。A: Tuj1 (緑, E) およびTH (赤, F) についての免疫染色像、並びにHoechst 33342 (青, G) を用いた核染色と合わせた像(Merge)を示す。スケールバー=20 μm。B: (A-C) Nestin (緑) およびKi67 (赤)についての二重免疫染色とDAPI核染色 (青)。高倍率像を (D) に示す。(EおよびF) Pax6 (赤) およびTH (緑) についての二重免疫染色。(G) TH (赤) およびTuJ1 (緑) についての二重免疫染色とDAPI。(HおよびI) TH (赤) およびMAP2ab (緑) についての二重免疫染色。(JおよびK) TH (赤) および小胞モノアミントランスポーター2 (VMAT2, 緑) についての二重染色。スケールバー: (A-C, E-G) においては100 μm; (D, H-K) においては20 μm。
【図36】図36は、ヒト末梢血単核細胞からのiPS細胞の樹立を示す。図36aは、実験手順の概要を示す。図36bは、3つの異なるプラスミドpCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hULをヒト末梢血単核細胞に導入することにより樹立されたiPS細胞コロニーの代表的な写真である。
【発明を実施するための形態】
【0019】
本発明は、(a) Oct3/4もしくはそれをコードする核酸、(b) Klf4もしくはそれをコードする核酸、および(c) Sox2もしくはそれをコードする核酸、並びに(d1) L-Mycもしくはそれをコードする核酸および/または(d2) p53の機能阻害物質を、体細胞に接触させることを含む、iPS細胞の製造方法を提供する。
【0020】
(A) 体細胞ソース
iPS細胞作製のための出発材料として用いることのできる体細胞は、哺乳動物(例えば、ヒト、マウス、サル、ブタ、ラット等)由来の生殖細胞以外のいかなる細胞であってもよく、例えば、角質化する上皮細胞(例、角質化表皮細胞)、粘膜上皮細胞(例、舌表層の上皮細胞)、外分泌腺上皮細胞(例、乳腺細胞)、ホルモン分泌細胞(例、副腎髄質細胞)、代謝・貯蔵用の細胞(例、肝細胞)、境界面を構成する内腔上皮細胞(例、I型肺胞細胞)、内鎖管の内腔上皮細胞(例、血管内皮細胞)、運搬能をもつ繊毛のある細胞(例、気道上皮細胞)、細胞外マトリックス分泌用細胞(例、線維芽細胞)、収縮性細胞(例、平滑筋細胞)、血液と免疫系の細胞(例、Tリンパ球)、感覚に関する細胞(例、桿細胞)、自律神経系ニューロン(例、コリン作動性ニューロン)、感覚器と末梢ニューロンの支持細胞(例、随伴細胞)、中枢神経系の神経細胞とグリア細胞(例、星状グリア細胞)、色素細胞(例、網膜色素上皮細胞)、およびそれらの前駆細胞(組織前駆細胞)等が挙げられる。細胞の分化の程度や細胞を採取する動物の齢などに特に制限はなく、未分化な前駆細胞 (体性幹細胞も含む) であっても、最終分化した成熟細胞であっても、同様に本発明における体細胞の起源として使用することができる。ここで未分化な前駆細胞としては、たとえば神経幹細胞、造血幹細胞、間葉系幹細胞、脂肪由来ストローマ(幹)細胞および歯髄幹細胞等の組織幹細胞(体性幹細胞)が挙げられる。造血幹細胞および間葉系幹細胞は骨髄、臍帯血および胎盤中に豊富に含まれる。骨髄、臍帯血および胎盤は、多くの公的および私的な血液バンクに寄託され、白血病等の血液疾患の治療に用いられているので、このような寄託された骨髄、臍帯血および胎盤もまた、体細胞ソースとして利用することができる。特に、臍帯血は出産時に得られる臍帯から容易に採取できるので、臍帯血から得られる造血幹細胞および間葉系幹細胞は、iPS細胞バンクのための好適な体細胞ソースである。また、歯髄幹細胞も、智歯や歯周病等で抜いた歯から単離、調製することができるため、入手が容易であり、今後、iPS細胞バンクのための体細胞ソースとしての利用が期待される。
【0021】
最終分化した成熟細胞としては、例えばT細胞やB細胞等の末梢血単核細胞が挙げられる。末梢血の採取は、非常に低侵襲性であり、且つ臨床検査において常套的に実施されている。臨床検査後の少量の余剰末梢血サンプルは通常廃棄されるので、それらはiPS細胞バンクのための好適な体細胞ソースである。特に、T細胞は比較的容易にインビトロで増幅させることができるので、少量の末梢血サンプルからでもiPS細胞を樹立することができる。最近、ヒト末梢血T細胞からのiPS細胞の作製が、いくつかのグループにより報告されている(Seki et al., Cell Stem Cell, 7: 11-14 (2010); Loh et al., Cell Stem Cell, 7: 15-19 (2010); Staerk et al., Cell Stem Cell, 7: 20-24 (2010))。
【0022】
体細胞を採取するソースとなる哺乳動物個体は特に制限されないが、得られるiPS細胞がヒトの再生医療用途に使用される場合には、拒絶反応が起こらないという観点から、患者本人またはHLAの型が同一もしくは実質的に同一である他人から体細胞を採取することが好ましい。ここでHLAの型が「実質的に同一」とは、免疫抑制剤などの使用により、該体細胞由来のiPS細胞から分化誘導することにより得られた細胞を患者に移植した場合に移植細胞が生着可能な程度にHLAの型が一致していることをいう。たとえば主たるHLA(例えばHLA-A、HLA-BおよびHLA-DRの3遺伝子座、さらにHLA-Cwを含む4遺伝子座)が同一である場合などが挙げられる(以下同じ)。また、ヒトに投与(移植)しない場合、例えば、患者の薬剤感受性や副作用の有無を評価するためのスクリーニング用の細胞のソースとしてiPS細胞を使用する場合には、同様に患者本人または薬剤感受性や副作用と相関する遺伝子多型が同一である他人から体細胞を採取することが望ましい。
【0023】
哺乳動物から分離した体細胞は、核初期化工程に供するに先立って、細胞の種類に応じてその培養に適した自体公知の培地で前培養することができる。そのような培地としては、例えば、約5〜20%の胎仔ウシ血清(FCS)を含む最小必須培地(MEM)、ダルベッコ改変イーグル培地(DMEM)、RPMI1640培地、199培地、F12培地などが挙げられるが、それらに限定されない。例えば、体細胞として歯髄幹細胞を用いる場合には、Mesenchymal stem cells basal medium (Lonza社) などの間葉系幹細胞用培地を用いることが好ましい。核初期化物質及びp53の機能阻害物質(さらに必要に応じて、他のiPS細胞の樹立効率改善物質)との接触に際し、例えば、カチオニックリポソームなど導入試薬を用いる場合には、導入効率の低下を防ぐため、無血清培地に交換しておくことが好ましい場合がある。
【0024】
ヒト臨床応用に適した完全xeno-freeのヒトiPS細胞を得る目的においては、FCSなどの非ヒト動物由来成分を含有しない培地を用いることがより望ましい。基本培地に種々の体細胞の培養に適したヒト由来成分(特に、増殖因子などの組換えヒトタンパク質)や非必須アミノ酸、ビタミン類等を補充した培地が市販されており、当業者は体細胞ソースに応じて適切なxeno-free培地を選択することができる。xeno-free培地で前培養された体細胞は、適当なxeno-freeの細胞剥離液を用いて培養容器から剥離し、回収後、核初期化物質及びp53の機能阻害物質との接触に供される。
【0025】
(B) 核初期化物質
本発明において「核初期化物質」とは、体細胞からiPS細胞を誘導することができるタンパク性因子(群)またはそれをコードする核酸(ベクターに組み込まれた形態を含む)でありうる。本発明に用いられる核初期化物質は、少なくともOct3/4、Klf4およびSox2またはそれらをコードする核酸からなり(但し、Klf4および/またはSox2はそれらの機能を代替し得ることが報告されている他の因子に置換してもよい)、p53の機能阻害物質を併用しない場合は、核初期化物質としてL-Mycまたはそれをコードする核酸、さらにLin28もしくはLin28bまたはそれをコードする核酸が組み合わされる。また、本発明に用いられる核初期化物質は、Nanogまたはそれをコードする核酸を含まないことを特徴とする。具体的には、以下の組み合わせが例示される(以下においては、タンパク性因子の名称のみを記載する)。
(1) Oct3/4, Klf4, Sox2, L-Myc(ここで、Sox2はSox1, Sox3, Sox15, Sox17またはSox18で置換可能である。また、Klf4はKlf1, Klf2またはKlf5で置換可能である。)
(2) Oct3/4, Klf4, Sox2, L-Myc, TERT, SV40 Large T antigen(以下、SV40LT)
(3) Oct3/4, Klf4, Sox2, L-Myc, TERT, HPV16 E6
(4) Oct3/4, Klf4, Sox2, L-Myc, TERT, HPV16 E7
(5) Oct3/4, Klf4, Sox2, L-Myc, TERT, HPV16 E6, HPV16 E7
(6) Oct3/4, Klf4, Sox2, L-Myc, TERT, Bmi1
(7) Oct3/4, Klf4, Sox2, L-Myc, Lin28
(8) Oct3/4, Klf4, Sox2, L-Myc, Lin28, SV40LT
(9) Oct3/4, Klf4, Sox2, L-Myc, Lin28, TERT, SV40LT
(10) Oct3/4, Klf4, Sox2, L-Myc, SV40LT
(11) Oct3/4, Esrrb, Sox2, L-Myc (EsrrbはEsrrgで置換可能である。)
(12) Oct3/4, Klf4, Sox2
(13) Oct3/4, Klf4, Sox2, TERT, SV40LT
(14) Oct3/4, Klf4, Sox2, TERT, HPV16 E6
(15) Oct3/4, Klf4, Sox2, TERT, HPV16 E7
(16) Oct3/4, Klf4, Sox2, TERT, HPV16 E6, HPV16 E7
(17) Oct3/4, Klf4, Sox2, TERT, Bmil
(18) Oct3/4, Klf4, Sox2, Lin28
(19) Oct3/4, Klf4, Sox2, Lin28, SV40LT
(20) Oct3/4, Klf4, Sox2, Lin28, TERT, SV40LT
(21) Oct3/4, Klf4, Sox2, SV40LT
(22) Oct3/4, Esrrb, Sox2 (EsrrbはEsrrgで置換可能である。)
上記において、Lin28に代えてLin28bを用いることもできる。また、EsrrbもしくはEsrrgを用いる場合(上記(11)および(22))、Klf4を併用してもよい。
また、上記(1)-(22)には該当しないが、それらのいずれかにおける構成要素をすべて含み、且つ任意の他の物質をさらに含む組み合わせも、本発明における「核初期化物質」の範疇に含まれ得る。また、核初期化の対象となる体細胞が上記(1)-(22)のいずれかにおける構成要素の一部を、核初期化のために十分なレベルで内在的に発現している条件下にあっては、当該構成要素を除いた残りの構成要素のみの組み合わせもまた、本発明における「核初期化物質」の範疇に含まれ得る。
【0026】
これらの組み合わせの中で、Oct3/4, Sox2, Klf4、Lin28(Lin28b)およびL-Mycの5因子、並びにOct3/4, Sox2, Klf4およびLin28(Lin28b)の4因子が、好ましい核初期化物質の例として挙げられる。これらに、SV40 Large T antigenをさらに加えた6因子もしくは5因子も好ましい。
【0027】
上記の各核初期化物質のマウスおよびヒトcDNA配列情報は、WO 2007/069666に記載のNCBI accession numbersを参照することにより取得することができ(Nanogは当該公報中では「ECAT4」との名称で記載されている。尚、Lin28、Lin28b、Esrrb、Esrrg、L-MycのマウスおよびヒトcDNA配列情報は、それぞれ下記NCBI accession numbersを参照することにより取得できる。)、当業者は容易にこれらのcDNAを単離することができる。
遺伝子名 マウス ヒト
Lin28 NM_145833 NM_024674
Lin28b NM_001031772 NM_001004317
Esrrb NM_011934 NM_004452
Esrrg NM_011935 NM_001438
L-Myc NM_008506 NM_001033081
【0028】
核初期化物質としてタンパク性因子自体を用いる場合には、得られたcDNAを適当な発現ベクターに挿入して宿主細胞に導入し、該細胞を培養して得られる培養物から組換えタンパク性因子を回収することにより調製することができる。一方、核初期化物質としてタンパク性因子をコードする核酸を用いる場合、得られたcDNAを、ウイルスベクター、プラスミドベクター、エピソーマルベクター等に挿入して発現ベクターを構築し、核初期化工程に供される。
【0029】
(C) 核初期化物質の体細胞への導入方法
核初期化物質の体細胞への導入は、該物質がタンパク性因子である場合、自体公知の細胞へのタンパク質導入方法を用いて実施することができる。そのような方法としては、例えば、タンパク質導入試薬を用いる方法、タンパク質導入ドメイン(PTD)もしくは細胞透過性ペプチド(CPP)融合タンパク質を用いる方法、マイクロインジェクション法などが挙げられる。タンパク質導入試薬としては、カチオン性脂質をベースとしたBioPOTER Protein Delivery Reagent(Gene Therapy Systmes)、Pro-JectTM Protein Transfection Reagent(PIERCE)及びProVectin(IMGENEX)、脂質をベースとしたProfect-1(Targeting Systems)、膜透過性ペプチドをベースとしたPenetrain Peptide(Q biogene)及びChariot Kit(Active Motif)、HVJエンベロープ(不活化センダイウイルス)を利用したGenomONE(石原産業)等が市販されている。導入はこれらの試薬に添付のプロトコルに従って行うことができるが、一般的な手順は以下の通りである。核初期化物質を適当な溶媒(例えば、PBS、HEPES等の緩衝液)に希釈し、導入試薬を加えて室温で5-15分程度インキュベートして複合体を形成させ、これを無血清培地に交換した細胞に添加して37℃で1ないし数時間インキュベートする。その後培地を除去して血清含有培地に交換する。
【0030】
PTDとしては、ショウジョウバエ由来のAntP、HIV由来のTAT (Frankel, A. et al, Cell 55, 1189-93 (1988); Green, M. & Loewenstein, P.M. Cell 55, 1179-88 (1988))、Penetratin (Derossi, D. et al, J. Biol. Chem. 269, 10444-50 (1994))、Buforin II (Park, C. B. et al. Proc. Natl Acad. Sci. USA 97, 8245-50 (2000))、Transportan (Pooga, M. et al. FASEB J. 12, 67-77 (1998))、MAP (model amphipathic peptide) (Oehlke, J. et al. Biochim. Biophys. Acta. 1414, 127-39 (1998))、K-FGF (Lin, Y. Z. et al. J. Biol. Chem. 270, 14255-14258 (1995))、Ku70 (Sawada, M. et al. Nature Cell Biol. 5, 352-7 (2003))、Prion (Lundberg, P. et al. Biochem. Biophys. Res. Commun. 299, 85-90 (2002))、pVEC (Elmquist, A. et al. Exp. Cell Res. 269, 237-44 (2001))、Pep-1 (Morris, M. C. et al. Nature Biotechnol. 19, 1173-6 (2001))、Pep-7 (Gao, C. et al. Bioorg. Med. Chem. 10, 4057-65 (2002))、SynB1 (Rousselle, C. et al. MoI. Pharmacol. 57, 679-86 (2000))、HN-I (Hong, F. D. & Clayman, G L. Cancer Res. 60, 6551-6 (2000))、HSV由来のVP22等のタンパク質の細胞通過ドメインを用いたものが開発されている。PTD由来のCPPとしては、11R (Cell Stem Cell, 4:381-384(2009)) や9R (Cell Stem Cell, 4:472-476(2009))等のポリアルギニンが挙げられる。
【0031】
核初期化物質のcDNAとPTDもしくはCPP配列とを組み込んだ融合タンパク質発現ベクターを作製して組換え発現させ、融合タンパク質を回収して導入に用いる。導入は、タンパク質導入試薬を添加しない以外は上記と同様にして行うことができる。
【0032】
マイクロインジェクションは、先端径1 μm程度のガラス針にタンパク質溶液を入れ、細胞に穿刺導入する方法であり、確実に細胞内にタンパク質を導入することができる。
【0033】
タンパク質導入操作は1回以上の任意の回数(例えば、1回以上10回以下、又は1回以上5回以下等)行うことができ、好ましくは導入操作を2回以上(たとえば3回又は4回)繰り返して行うことができる。導入操作を繰り返し行う場合の間隔としては、例えば6〜48時間、好ましくは12〜24時間が挙げられる。
【0034】
iPS細胞の樹立効率を重視するのであれば、核初期化物質を、タンパク性因子自体としてではなく、それをコードする核酸の形態で用いることが好ましい。該核酸はDNAであってもRNAであってもよく、あるいはDNA/RNAキメラであってもよいが、また、該核酸は二本鎖であっても、一本鎖であってもよい。好ましくは該核酸は二本鎖DNA、特にcDNAである。
【0035】
核初期化物質のcDNAは、宿主となる体細胞で機能し得るプロモーターを含む適当な発現ベクターに挿入される。発現ベクターとしては、例えば、レトロウイルス、レンチウイルス、アデノウイルス、アデノ随伴ウイルス、ヘルペスウイルス、センダイウイルスなどのウイルスベクター、動物細胞発現プラスミド(例、pA1-11,pXT1,pRc/CMV,pRc/RSV,pcDNAI/Neo)などが用いられ得る。
【0036】
用いるベクターの種類は、得られるiPS細胞の用途に応じて適宜選択することができる。例えば、アデノウイルスベクター、プラスミドベクター、アデノ随伴ウイルスベクター、レトロウイルスベクター、レンチウイルスベクター、センダイウイルスベクター、エピソーマルベクターなどが使用され得る。
【0037】
発現ベクターにおいて使用されるプロモーターとしては、例えばEF1αプロモーター、CAGプロモーター、SRαプロモーター、SV40プロモーター、LTRプロモーター、CMV(サイトメガロウイルス)プロモーター、RSV(ラウス肉腫ウイルス)プロモーター、MoMuLV(モロニーマウス白血病ウイルス)LTR、HSV-TK(単純ヘルペスウイルスチミジンキナーゼ)プロモーターなどが用いられる。なかでも、EF1αプロモーター、CAGプロモーター、MoMuLV LTR、CMVプロモーター、SRαプロモーターなどが好ましい。
発現ベクターは、プロモーターの他に、所望によりエンハンサー、ポリA付加シグナル、選択マーカー遺伝子、SV40複製起点などを含有していてもよい。選択マーカー遺伝子としては、例えば、ジヒドロ葉酸還元酵素遺伝子、ネオマイシン耐性遺伝子、ピューロマイシン耐性遺伝子等が挙げられる。
【0038】
核初期化物質である核酸(初期化遺伝子)は、各々別個の発現ベクター上に組み込んでもよいし、1つの発現ベクターに2種類以上、好ましくは2〜3種類の遺伝子を組み込んでもよい。遺伝子導入効率の高いレトロウイルスやレンチウイルスベクターを用いる場合は前者が、プラスミド、アデノウイルス、エピソーマルベクターなどを用いる場合は後者を選択することが好ましい。さらに、2種類以上の遺伝子を組み込んだ発現ベクターと、1遺伝子のみを組み込んだ発現ベクターとを併用することもできる。
【0039】
上記において複数の初期化遺伝子(例えば、Oct3/4、Sox2、Klf4、L-Myc、Lin28(Lin28b)およびSV40LTから選択される2つ以上、好ましくは2〜3遺伝子)を1つの発現ベクターに組み込む場合、これら複数の遺伝子は、好ましくはポリシストロニック発現を可能にする配列を介して発現ベクターに組み込むことができる。ポリシストロニック発現を可能にする配列を用いることにより、1種類の発現ベクターに組み込まれている複数の遺伝子をより効率的に発現させることが可能になる。ポリシストロニック発現を可能にする配列としては、例えば、口蹄疫ウイルスの2A配列(配列番号63; PLoS ONE 3, e2532, 2008、Stem Cells 25, 1707, 2007)、IRES配列(U.S. Patent No. 4,937,190)など、好ましくは2A配列を用いることができる。複数の初期化遺伝子を1つの発現ベクターにポリシストロニックに連結して挿入する場合、初期化遺伝子の順序は特に制限はないが、例えば、5’から3’方向に(i) Sox2およびKlf4、(ii) L-MycおよびLin28(Lin28b)、(iii) Klf4、Sox2およびOct3/4をこの順序で連結することができる。
【0040】
初期化遺伝子を含む発現ベクターは、ベクターの種類に応じて、自体公知の手法により細胞に導入することができる。例えば、ウイルスベクターの場合、該核酸を含むプラスミドを適当なパッケージング細胞(例、Plat-E細胞)や相補細胞株(例、293細胞)に導入して、培養上清中に産生されるウイルスベクターを回収し、各ウイルスベクターに応じた適切な方法により、該ベクターを細胞に感染させる。例えば、ベクターとしてレトロウイルスベクターを用いる具体的手段が WO 2007/69666、Cell, 126, 663-676 (2006) 及び Cell, 131, 861-872 (2007) に開示されており、ベクターとしてレンチウイルスベクターを用いる場合については、Science, 318, 1917-1920 (2007) に開示がある。iPS細胞を再生医療のための細胞ソースとして利用する場合、初期化遺伝子の発現(再活性化)は、iPS細胞由来の分化細胞から再生された組織における発癌リスクを高める可能性があるので、初期化遺伝子は細胞の染色体に組み込まれず、一過的に発現することが好ましい。かかる観点からは、染色体への組込みが稀なアデノウイルスベクターの使用が好ましい。アデノウイルスベクターを用いる具体的手段は、Science, 322, 945-949 (2008) に開示されている。また、アデノ随伴ウイルスも染色体への組込み頻度が低く、アデノウイルスベクターと比べて細胞毒性や炎症惹起作用が低いので、別の好ましいベクターとして挙げられる。センダイウイルスベクターは染色体外で安定に存在することができ、必要に応じてsiRNAにより分解除去することができるので、同様に好ましく利用され得る。センダイウイルスベクターについては、J. Biol. Chem., 282, 27383-27391 (2007)、Proc. Jpn. Acad., Ser. B 85, 348-362 (2009)、または特許第3602058号に記載のものを用いることができる。
【0041】
レトロウイルスベクターやレンチウイルスベクターを用いる場合は、いったん導入遺伝子のサイレンシングが起こったとしても、後に再活性化される可能性があるので、例えば、Cre/loxPシステムを用いて、不要となった時点で核初期化物質をコードする核酸を切り出す方法が好ましく用いられ得る。即ち、該核酸の両端にloxP配列を配置しておき、iPS細胞が誘導された後で、プラスミドベクターもしくはアデノウイルスベクターを用いて細胞にCreリコンビナーゼを作用させ、loxP配列に挟まれた領域を切り出すことができる。また、LTR U3領域のエンハンサー−プロモーター配列は、挿入突然変異によって近傍の宿主遺伝子を上方制御する可能性があるので、当該配列を欠失、もしくはSV40などのポリアデニル化配列で置換した3’-自己不活性化(SIN)LTRを使用して、切り出されずゲノム中に残存するloxP配列より外側のLTRによる内因性遺伝子の発現制御を回避することがより好ましい。Cre-loxPシステムおよびSIN LTRを用いる具体的手段は、Chang et al., Stem Cells, 27: 1042-1049 (2009) に開示されている。
【0042】
一方、非ウイルスベクターであるプラスミドベクターの場合には、リポフェクション法、リポソーム法、エレクトロポレーション法、リン酸カルシウム共沈殿法、DEAEデキストラン法、マイクロインジェクション法、遺伝子銃法などを用いて該ベクターを細胞に導入することができる。ベクターとしてプラスミドを用いる具体的手段は、例えばScience, 322, 949-953 (2008) 等に記載されている。
【0043】
プラスミドベクターやアデノウイルスベクター等を用いる場合、遺伝子導入は1回以上の任意の回数(例えば、1回以上10回以下、又は1回以上5回以下など)行うことができる。2種以上の発現ベクターを体細胞に導入する場合には、これらの全ての種類の発現ベクターを同時に体細胞に導入することが好ましいが、この場合においても、導入操作は1回以上の任意の回数(例えば、1回以上10回以下、又は1回以上5回以下など)行うことができ、好ましくは導入操作を2回以上 (たとえば3回又は4回) 繰り返して行うことができる。
【0044】
尚、アデノウイルスやプラスミドを用いる場合でも、導入遺伝子が染色体に組み込まれることがあるので、結局はサザンブロットやPCRにより染色体への遺伝子挿入がないことを確認する必要がある。そのため、上記Cre-loxPシステムのように、いったん染色体に導入遺伝子を組み込んだ後に、該遺伝子を除去する手段を用いることは好都合であり得る。別の好ましい一実施態様においては、トランスポゾンを用いて染色体に導入遺伝子を組み込んだ後に、プラスミドベクターもしくはアデノウイルスベクターを用いて細胞に転移酵素を作用させ、導入遺伝子を完全に染色体から除去する方法が用いられ得る。好ましいトランスポゾンとしては、例えば、鱗翅目昆虫由来のトランスポゾンであるpiggyBac等が挙げられる。piggyBacトランスポゾンを用いる具体的手段は、Kaji, K. et al., Nature, 458: 771-775 (2009)、Woltjen et al., Nature, 458: 766-770 (2009) に開示されている。
【0045】
別の好ましい非組込み型ベクターとして、染色体外で自律複製可能なエピソーマルベクターが挙げられる。エピソーマルベクターを用いる具体的手段は、Yu et al., Science, 324, 797-801 (2009) に開示されている。本発明の特に好ましい一実施態様においては、エピソーマルベクターの複製に必要なベクター要素の5’側および3’側にloxP配列を同方向に配置したエピソーマルベクターが使用され得る。エピソーマルベクターは染色体外で自律複製可能なため、ゲノムに組み込まれなくとも宿主細胞内での安定な発現を提供し得るが、いったんiPS細胞が樹立された後は、当該ベクターは速やかに除かれることが望ましい。エピソーマルベクターの複製に必要なベクター要素を2つのloxP配列で挟み、これにCreリコンビナーゼを作用させて当該ベクター要素を切り出すことにより、エピソーマルベクターの自律複製能を喪失させることができ、該ベクターを早期にiPS細胞から脱落させることができる。
【0046】
本発明に用いられるエピソーマルベクターとしては、例えば、EBV、SV40等に由来する自律複製に必要な配列をベクター要素として含むベクターが挙げられる。自律複製に必要なベクター要素としては、具体的には、複製開始点と、複製開始点に結合して複製を制御するタンパク質をコードする遺伝子であり、例えば、EBVにあっては複製開始点oriPとEBNA-1遺伝子、SV40にあっては複製開始点oriとSV40 large T antigen遺伝子が挙げられる。
【0047】
また、エピソーマル発現ベクターは、初期化遺伝子の転写を制御するプロモーターを含む。該プロモーターとしては、前記と同様のプロモーターが用いられ得る。また、エピソーマル発現ベクターは、前記と同様に、所望によりエンハンサー、ポリA付加シグナル、選択マーカー遺伝子などをさらに含有していてもよい。選択マーカー遺伝子としては、例えば、ジヒドロ葉酸還元酵素遺伝子、ネオマイシン耐性遺伝子等が挙げられる。
【0048】
本発明で使用されるloxP配列としては、バクテリオファージP1由来の野生型loxP配列(配列番号29)の他、初期化遺伝子の複製に必要なベクター要素を挟む位置に同方向で配置された場合に、組換えを起こしてloxP配列間の配列を欠失させ得る任意の変異loxP配列が挙げられる。変異loxP配列としては、例えば、5’側反復配列に変異のあるlox71(配列番号30)、3’側反復配列に変異のあるlox66(配列番号31)、スペーサー部分に変異のあるlox2272やlox511などが挙げられる。該ベクター要素の5’側および3’側に配置される2つのloxP配列は、同一であっても異なっていてもよいが、スペーサー部分に変異のある変異loxP配列の場合は同一のもの(例、lox2272同士、lox511同士)が用いられる。好ましくは、5’側反復配列に変異のある変異loxP配列(例、lox71)と3’側反復配列に変異のある変異loxP配列(例、lox66)との組合せが挙げられる。この場合、組換えの結果染色体上に残るloxP配列は5’側および3’側の反復配列に二重変異を有するため、Creリコンビナーゼに認識されにくく、不必要な組換えにより染色体の欠失変異を起こすリスクが低減される。lox71とlox66とを用いる場合、前記ベクター要素の5’側および3’側にいずれの変異loxP配列を配置してもよいが、変異部位がloxP配列の外端に配置されるような向きで変異loxP配列を挿入する必要がある。尚、本発明の好ましいエピソーマルベクターはCreリコンビナーゼを作用させなくても、早期に細胞から脱落する自己消失型ベクターであるが、例外的に細胞からの脱落に時間がかかる場合も想定されるので、Creリコンビナーゼ処理による不必要な組換えなどのリスクに備えてloxP配列を設計しておくことが好ましい。
【0049】
2つのloxP配列は、初期化遺伝子の複製に必要なベクター要素(即ち、複製開始点、または複製開始点に結合して複製を制御するタンパク質をコードする遺伝子配列)の5’側および3’側に、同方向に配置される。loxP配列が挟むベクター要素は、複製開始点、または複製開始点に結合して複製を制御するタンパク質をコードする遺伝子配列のいずれか一方だけであってもよいし、両方であってもよい。
【0050】
エピソーマルベクターやプラスミドベクターには、RNAの安定性を向上させる目的で、例えば、初期化因子のコーディング領域とポリA付加シグナルとの間にウッドチャック肝炎ウイルス転写後調節エレメント(WPRE)配列を挿入してもよい。
【0051】
エピソーマルベクターは、例えばリポフェクション法、リポソーム法、エレクトロポレーション法、リン酸カルシウム共沈殿法、DEAEデキストラン法、マイクロインジェクション法、遺伝子銃法などを用いて該ベクターを細胞に導入することができる。具体的には、例えばScience, 324: 797-801 (2009) 等に記載される方法を用いることができる。
【0052】
iPS細胞から初期化遺伝子の複製に必要なベクター要素が除去されたか否かの確認は、該ベクター要素内部および/またはloxP配列近傍の塩基配列を含む核酸をプローブまたはプライマーとして用い、該iPS細胞から単離したエピソーム画分を鋳型としてサザンブロット分析またはPCR分析を行い、バンドの有無または検出バンドの長さを調べることにより実施することができる。エピソーム画分の調製は当該分野で周知の方法と用いて行えばよく、例えば、Science, 324: 797-801 (2009) 等に記載される方法を用いることができる。
【0053】
後述の実施例に示されるように、本発明で提供されるloxP配列を含むエピソーマルベクターの中には、体細胞に導入した際に、該ベクターを構成する外来核酸因子(初期化遺伝子を含む)が一過的にも細胞のゲノム中に組み込まれないというエピソーマルベクター本来の効果だけでなく、Creリコンビナーゼ処理を施すことなく、早い段階でエピソームとして存在する該ベクターがiPS細胞から脱落するという予期せぬ効果を奏するものがある。即ち、本発明はまた、iPS細胞の樹立に十分な初期化因子の発現を提供した後、早期に細胞から脱落する自己消失型のエピソーマルベクターを提供する。かかるベクターは、50%以上、好ましくは60%以上、より好ましくは70%以上の頻度で、5継代までにiPS細胞から脱落することを特徴とする。あるいは、該自己消失型エピソーマルベクターは、導入後1週間以内における1x104細胞あたりのコピー数が106個のオーダーであるのに対し、iPS細胞樹立時(例えば、ベクター導入から約4週間後)における1x104細胞あたりのコピー数が100個以下、好ましくは50個以下、より好ましくは30個以下にまで減衰する程度に細胞内で不安定であることを特徴とする。
【0054】
具体的には、本発明の早期自己消失型ベクターは以下の(i)〜(iv)のうちの少なくと1つ、好ましくは2つ以上、より好ましくは3つ以上、特に好ましくはすべての構造的特徴を有するものである。
(i) エピソーマルベクターの複製に必要なベクター要素(例えば、EBNA-1遺伝子やSV40 Large T antigen遺伝子、好ましくはEBNA-1遺伝子)の5’側および3’側にloxP配列が同方向に配置されている。
(ii) 初期化因子をコードする核酸がCAGプロモーターの制御下にある。
(iii) 初期化因子をコードする核酸がウサギβ-グロビンポリA付加シグナルの制御下にある。
(iv) 初期化因子をコードする核酸とポリA付加シグナルとの間にWPRE配列を含む。
【0055】
(D) p53の機能阻害物質
本発明は、上記の核初期化物質に加えて、p53の機能阻害物質を接触させることがより好ましい。本明細書において「p53の機能阻害物質」とは、(a)p53タンパク質の機能もしくは(b)p53遺伝子の発現を阻害し得る限り、いかなる物質であってもよい。すなわち、p53タンパク質に直接作用してその機能を阻害する物質や、p53遺伝子に直接作用してその発現を阻害する物質のみならず、p53のシグナル伝達に関与する因子に作用することにより、結果的にp53タンパク質の機能やp53遺伝子の発現を阻害する物質も、本明細書における「p53の機能阻害物質」に含まれる。好ましくは、p53の機能阻害物質は、p53遺伝子の発現を阻害する物質であり、より好ましくはp53に対するsiRNAやshRNAをコードする発現ベクターである。
【0056】
p53タンパク質の機能を阻害する物質としては、例えば、p53の化学的阻害物質、p53のドミナントネガティブ変異体もしくはそれをコードする核酸、抗p53アンタゴニスト抗体もしくはそれをコードする核酸、p53応答エレメントのコンセンサス配列を含むデコイ核酸、p53経路を阻害する物質などが挙げられるが、これらに限定されない。好ましくは、p53の化学的阻害物質、p53のドミナントネガティブ変異体もしくはそれをコードする核酸、p53経路阻害物質が挙げられる。
【0057】
(D1) p53の化学的阻害物質
p53の化学的阻害物質としては、例えば、WO 00/44364に開示されるpifithrin(PFT)-α及び-βに代表されるp53阻害剤、Stormら(Nat. Chem. Biol. 2, 474 (2006))に開示されるPFT-μ、それらの類縁体及びそれらの塩(例えば、塩酸酸、臭素酸塩等の酸付加塩など)等が挙げられるが、これらに限定されない。これらのうち、PFT-α及びその類縁体[2-(2-Imino-4,5,6,7-tetrahydrobenzothiazol-3-yl)-1-p-tolylethanone, HBr (製品名:Pifithrin-α)及び1-(4-Nitrophenyl)-2-(4,5,6,7-tetrahydro-2-imino-3(2H)-benzothiazolyl)ethanone, HBr(製品名:Pifithrin-α, p-Nitro)]、PFT-β及びその類縁体[2-(4-Methylphenyl)imidazo[2,1-b]-5,6,7,8-tetrahydrobenzothiazole, HBr(製品名:Pifithrin-α, Cyclic)及び2-(4-Nitrophenyl)imidazo[2,1-b]-5,6,7,8-tetrahydrobenzothiazole(製品名:Pifithrin-α, p-Nitro, Cyclic)]、PFT-μ[Phenylacetylenylsulfonamide(製品名:Pifithrin-μ)]は、Merck社より市販されている。
【0058】
体細胞へのp53の化学的阻害物質の接触は、該阻害物質を適当な濃度で水性もしくは非水性溶媒に溶解し、ヒトまたはマウスより単離した体細胞の培養に適した培地(例えば、約5〜20%の胎仔ウシ血清を含む最小必須培地(MEM)、ダルベッコ改変イーグル培地(DMEM)、RPMI1640培地、199培地、F12培地など)中に、阻害物質濃度がp53の機能阻害に十分で且つ細胞毒性がみられない範囲となるように該阻害物質溶液を添加して、細胞を一定期間培養することにより実施することができる。阻害物質濃度は用いる阻害物質の種類によって異なるが、約0.1nM〜約100nMの範囲で適宜選択される。接触期間は細胞の核初期化が達成されるのに十分な時間であれば特に制限はないが、通常は陽性コロニーが出現するまで培地に共存させておけばよい。
【0059】
p53遺伝子は癌抑制遺伝子として知られており、p53の恒常的な機能阻害は発癌のリスクを高める可能性がある。p53の化学的阻害物質は、培地に添加するだけで細胞への導入が可能であるという利点に加えて、iPS細胞の誘導後に該阻害物質を含む培地を除去することにより、容易かつ迅速にp53の機能阻害を解除できる点でも有用である。
【0060】
(D2) p53のドミナントネガティブ変異体
p53のドミナントネガティブ変異体としては、体細胞に内在する野生型p53タンパク質と競合的に作用して、その機能を阻害し得る限り特に制限はないが、例えば、マウスp53のDNA結合領域に位置する275位(ヒトの場合は278位)のプロリンをセリンに点変異させたp53P275S(de Vries, A., Proc. Natl. Acad. Sci. USA, 99, 2948-2953 (2002))、マウスp53の14-301位(ヒトp53では11-304位に対応)のアミノ酸を欠失させたp53DD(Bowman, T., Genes Develop., 10, 826-835 (1996))などが挙げられる。その他にも、例えば、マウスp53の58位(ヒトの場合は61位)のセリンをアラニンに点変異させたp53S58A、ヒトp53の135位(マウスの場合は132位)のシステインをチロシンに点変異させたp53C135Y、マウスp53の135位(ヒトの場合は138位)のアラニンをバリンに点変異させたp53A135V、172位(ヒトの場合は175位)のアルギニンをヒスチジンに点変異させたp53R172H、270位(ヒトの場合は273位)のアルギニンをヒスチジンに点変異させたp53R270H、マウスp53の278位(ヒトの場合は281位)のアスパラギン酸をアスパラギンに点変異させたp53D278Nなどが知られており、同様に使用することができる。
【0061】
p53のドミナントネガティブ変異体は、例えば、以下の手法により得ることができる。まず、配列番号1または3に示されるマウスまたはヒトのp53 cDNA配列情報に基づいて適当なオリゴヌクレオチドをプローブもしくはプライマーとして合成し、マウスまたはヒトの細胞・組織由来のmRNA、cDNAもしくはcDNAライブラリーから、ハイブリダイゼーション法や(RT-)PCR法を用いてマウスまたはヒトp53 cDNAをクローニングし、適当なプラスミドにサブクローニングする。変異を導入しようとする部位のコドン(例えば、p53P275Sの場合、配列番号1に示される塩基配列中塩基番号951-953で示されるcct)を所望の他のアミノ酸をコードするコドン(例えば、p53P275Sの場合、tct)に置換した形で、当該部位を含むプライマーを合成し、これを用いてp53 cDNAを挿入したプラスミドを鋳型とするインバースPCRを行うことにより、目的のドミナントネガティブ変異体をコードする核酸を取得する。p53DDのような欠失変異体の場合には、欠失させる部位の外側にプライマーを設計して、同様にインバースPCRを行えばよい。このようにして得られたドミナントネガティブ変異体をコードする核酸を宿主細胞に導入し、該細胞を培養して得られる培養物から組換えタンパク質を回収することにより、所望のドミナントネガティブ変異体を取得することができる。
【0062】
体細胞へのドミナントネガティブ変異体の接触は、上記タンパク性の核初期化物質の場合と同様に実施することができる。上述のように、p53の恒常的な機能阻害は発癌のリスクを高める可能性があるが、p53のドミナントネガティブ変異体は、導入された細胞内でプロテアーゼによる分解を受けて徐々に消失し、それに応じて細胞に内在するp53の機能が回復することから、該変異体タンパク質の使用は、得られるiPS細胞を治療用途で利用する場合のように、高度な安全性を要求される場合に好適であり得る。
【0063】
(D3) p53のドミナントネガティブ変異体をコードする核酸
本発明の別の好ましい実施態様において、p53機能阻害物質は、p53のドミナントネガティブ変異体をコードする核酸である。該核酸はDNAであってもRNAであってもよく、あるいはDNA/RNAキメラであってもよいが、好ましくはDNAである。また、該核酸は二本鎖であっても、一本鎖であってもよい。p53のドミナントネガティブ変異体をコードするcDNAは、該変異体タンパク質の作製について上記した手法によりクローニングすることができる。
単離されたcDNAは、前記核初期化物質である核酸(初期化遺伝子)の場合と同様に、適当な発現ベクターに挿入され、体細胞に導入され得る。
【0064】
(D4) p53経路阻害物質
ここでp53経路とは、p53を活性化し得るあらゆる上流のシグナルカスケードおよび活性化p53によって媒介されるあらゆる下流のシグナルカスケードを包含する意味で用いられる。したがって、p53経路阻害物質には、上記シグナル伝達経路のいずれかを阻害するいかなる物質も含まれるが、好ましい一実施態様においては、p53経路阻害物質はp53によりその転写が活性化されるp21の発現もしくは機能(Myc阻害活性)を阻害する物質であり、例えば、p21に対するsiRNA、shRNA、アンチセンス核酸、リボザイム等が挙げられる。p21の発現を阻害するこれらの核酸は、後記p53に対するsiRNA、shRNA、アンチセンス核酸、リボザイムと同様の方法により設計・合成し、体細胞に導入することができる。当該核酸は、それらを発現するベクターの形態で提供されてもよく、該ベクターは、後記p53に対するsiRNA、shRNA、アンチセンス核酸、リボザイムを発現するベクターと同様の方法により構築し、体細胞に導入することができる。
【0065】
別の好ましい一実施態様においては、p53経路阻害物質はARF-MDM2-p53経路を阻害する物質であり、例えば、ARF-MDM2-p53経路阻害物質として、p53に直接結合してその核外輸送やユビキチン化を促進するMDM2もしくはそれをコードする核酸、p53へのMDM2の作用を阻害するp19ARFやATM(ataxia-telangiectasia mutated)の発現もしくは機能を阻害する物質(例えば、これらの因子に対するsiRNAやshRNA)等が挙げられる。
【0066】
(D5) その他の物質
p53タンパク質の機能を阻害するその他の物質として、例えば、抗p53アンタゴニスト抗体もしくはそれをコードする核酸が挙げられる。抗p53アンタゴニスト抗体はポリクローナル抗体、モノクローナル抗体の何れであってもよい。抗体のアイソタイプは特に限定されないが、好ましくはIgG、IgMまたはIgA、特に好ましくはIgGが挙げられる。また、該抗体は、完全抗体分子の他、例えばFab、Fab'、F(ab’)2等のフラグメント、scFv、scFv-Fc、ミニボディー、ダイアボディー等の遺伝子工学的に作製されたコンジュゲート分子、あるいはポリエチレングリコール(PEG)等の蛋白質安定化作用を有する分子等で修飾されたそれらの誘導体などであってもよい。抗p53アンタゴニスト抗体は、p53またはその部分ペプチドを抗原として用い、自体公知の抗体または抗血清の製造法に従って製造することができる。また、公知の抗p53アンタゴニスト抗体として、例えば、PAb1801(Oncogene Science Ab-2)及びDO-1(Oncogene Science Ab-6)(Gire and Wynford-Thomas, Mol. Cell. Biol., 18, 1611-1621 (1998))等が挙げられる。抗p53アンタゴニスト抗体をコードする核酸は、抗p53モノクローナル抗体産生ハイブリドーマから常法により単離することができる。得られるH鎖及びL鎖遺伝子を連結して単鎖抗体をコードする核酸を作製することもできる。
【0067】
p53タンパク質の機能を阻害する別の物質として、抗p21アンタゴニスト抗体もしくはそれをコードする核酸が挙げられる。抗p21アンタゴニスト抗体及びそれをコードする核酸も、上記抗p53アンタゴニスト抗体及びそれをコードする核酸と同様にして作製することができる。
【0068】
p53タンパク質の機能を阻害するさらに別の物質は、p53応答エレメントのコンセンサス配列(例、Pu-Pu-Pu-G-A/T-T/A-C-Py-Py-Py (Pu: プリン塩基, Py: ピリミジン塩基); 配列番号27)を含むデコイ核酸である。このような核酸は上記塩基配列情報に基づいてDNA/RNA自動合成機で合成することができる。あるいはそのようなデコイ核酸は市販されている(例、p53 transcription factor decoy (GeneDetect.com))。
【0069】
抗p53アンタゴニスト抗体及び抗p21アンタゴニスト抗体はp53のドミナントネガティブ変異体と同様に、また、該抗体をコードする核酸は該変異体をコードする核酸と同様にして、それぞれ細胞に導入することができる。また、上記デコイ核酸は、リポフェクション法などにより細胞に導入することができる。
【0070】
一方、p53遺伝子の発現を阻害する物質としては、例えば、p53に対するsiRNAもしくはshRNA、p53に対するsiRNAもしくはshRNAを発現するベクター、p53に対するアンチセンス核酸及びp53に対するリボザイム等が挙げられるが、好ましくはp53に対するsiRNA、shRNA及びsiRNA、shRNAを発現するベクターである。
【0071】
(D6) p53に対するsiRNA及びshRNA
p53に対するsiRNAは、配列番号1または3に示されるマウスまたはヒトのp53 cDNA配列情報に基づいて、例えば、Elbashirら(Genes Dev., 15, 188-200 (2001))の提唱する規則に従って設計することができる。siRNAの標的配列としては、原則的にはAA+(N)19であるが、AA+(N)21もしくはNA+(N)21であってもよい。また、センス鎖の5’末端がAAである必要はない。標的配列の位置は特に制限されるわけではないが、5’-UTR及び開始コドンから約50塩基まで、並びに3’-UTR以外の領域から標的配列を選択することが望ましい。標的配列のGC含量も特に制限はないが、約30-約50%が好ましく、GC分布に偏りがなく繰り返しが少ない配列が望ましい。尚、下記(b2)のsiRNAもしくはshRNAを発現するベクターの設計において、プロモーターとしてpolIII系プロモーターを使用する場合、ポリメラーゼの転写が停止しないように、4塩基以上TまたはAが連続する配列は選択しないようにすべきである。
【0072】
上述の規則に基づいて選択された標的配列の候補群について、標的以外のmRNAにおいて16-17塩基の連続した配列に相同性がないかどうかを、BLAST(http://www.ncbi.nlm.nih.gov/BLAST/)等のホモロジー検索ソフトを用いて調べ、選択した標的配列の特異性を確認する。特異性の確認された標的配列について、AA(もしくはNA)以降の19-21塩基にTTもしくはUUの3’末端オーバーハングを有するセンス鎖と、該19-21塩基に相補的な配列及びTTもしくはUUの3’末端オーバーハングを有するアンチセンス鎖とからなる2本鎖RNAをsiRNAとして設計する。また、shRNAは、ループ構造を形成しうる任意のリンカー配列(例えば、8-25塩基程度)を適宜選択し、上記センス鎖とアンチセンス鎖とを該リンカー配列を介して連結することにより設計することができる。
【0073】
siRNA及び/又はshRNAの配列は、種々のwebサイト上に無料で提供される検索ソフトを用いて検索が可能である。このようなサイトとしては、例えば、Ambionが提供するsiRNA Target Finder(http://www.ambion.com/jp/techlib/misc/siRNA_finder.html)及びpSilencerTM Expression Vector用 インサート デザインツール(http://www.ambion.com/jp/techlib/misc/psilencer_converter.html)、RNAi Codexが提供するGeneSeer(http://codex.cshl.edu/scripts/newsearchhairpin.cgi)がこれらに限定されず、QIAGEN、タカラバイオ、SiSearch、Dharmacon、Whitehead Institute、Invitrogen、Promega等のwebサイト上でも同様に検索が可能である。
【0074】
以下に、Ambion(配列番号5-24)およびRNAi Codex(配列番号25及び26)のwebサイト上で提供されるソフトウェアを用いて設計した、マウスp53に対するshRNA配列を示す。下線で示した配列がdicerで切断された後に生じるdsRNAのセンス鎖(5’側)及びアンチセンス鎖(3’側)である(3’-オーバーハング「TT」を含まない)。小文字はミスマッチまたはループを示す。
[配列番号5]
5’-TTTGACTGGATGACTGCCATGGttcaagagaCCATGGCAGTCATCCAGTCTTTTTT-3’
[配列番号6]
5’-TTTGATATCCTGCCATCACCTCttcaagagaGAGGTGATGGCAGGATATCTTTTTT-3’
[配列番号7]
5’-TTTGGCCCAAGTGAAGCCCTCCttcaagagaGGAGGGCTTCACTTGGGCCTTTTTT-3’
[配列番号8]
5’-TTTGTGAAGCCCTCCGAGTGTCttcaagagaGACACTCGGAGGGCTTCACTTTTTT-3’
[配列番号9]
5’-TTTGCCCTCCGAGTGTCAGGAGttcaagagaCTCCTGACACTCGGAGGGCTTTTTT-3’
[配列番号10]
5’-TTTGTCTGTTATGTGCACGTACttcaagagaGTACGTGCACATAACAGACTTTTTT-3’
[配列番号11]
5’-TTTGTACTCTCCTCCCCTCAATttcaagagaATTGAGGGGAGGAGAGTACTTTTTT-3’
[配列番号12]
5’-TTTGCTATTCTGCCAGCTGGCGttcaagagaCGCCAGCTGGCAGAATAGCTTTTTT-3’
[配列番号13]
5’-TTTGACGTGCCCTGTGCAGTTGttcaagagaCAACTGCACAGGGCACGTCTTTTTT-3’
[配列番号14]
5’-TTTGAAGTCACAGCACATGACGttcaagagaCGTCATGTGCTGTGACTTCTTTTTT-3’
[配列番号15]
5’-TTTGTCACAGCACATGACGGAGttcaagagaCTCCGTCATGTGCTGTGACTTTTTT-3’
[配列番号16]
5’-TTTGGAAATTTGTATCCCGAGTttcaagagaACTCGGGATACAAATTTCCTTTTTT-3’
[配列番号17]
5’-TTTGTACATGTGTAATAGCTCCttcaagagaGGAGCTATTACACATGTACTTTTTT-3’
[配列番号18]
5’-TTTGACTCCAGTGGGAACCTTCttcaagagaGAAGGTTCCCACTGGAGTCTTTTTT-3’
[配列番号19]
5’-TTTGTCCTTTGCCCTGAACTGCttcaagagaGCAGTTCAGGGCAAAGGACTTTTTT-3’
[配列番号20]
5’-TTTGATCCGCGGGCGTAAACGCttcaagagaGCGTTTACGCCCGCGGATCTTTTTT-3’
[配列番号21]
5’-TTTGACCAAGAAGGGCCAGTCTttcaagagaAGACTGGCCCTTCTTGGTCTTTTTT-3’
[配列番号22]
5’-TTTGAAAGTGGGGCCTGACTCAttcaagagaTGAGTCAGGCCCCACTTTCTTTTTT-3’
[配列番号23]
5’-TTTGTTGGGGAATAGGTTGATAttcaagagaTATCAACCTATTCCCCAACTTTTTT-3’
[配列番号24]
5’-TTTGATTCTATCTTGGGCCCTCttcaagagaGAGGGCCCAAGATAGAATCTTTTTT-3’
[配列番号25]
5’-TTTGCAuTACAgGTACgTGTGTAgtgtgctgtccTACACATGTACTTGTAGTGTTTTTT-3’
[配列番号26]
5’-TTTGCAGTuTACTTuCCGCCgTAgtgtgctgtccTATGGCGGGAAGTAGACTGTTTTTT-3’
【0075】
p53に対するsiRNAは、上記のようにして設計されたセンス鎖及びアンチセンス鎖オリゴヌクレオチドをDNA/RNA自動合成機でそれぞれ合成し、例えば、適当なアニーリング緩衝液中、約90〜約95℃で約1分程度変性させた後、約30〜約70℃で約1〜約8時間アニーリングさせることにより調製することができる。また、p53に対するshRNAは、上記のようにして設計されたshRNA配列を有するオリゴヌクレオチドをDNA/RNA自動合成機で合成し、上記と同様にしてセルフアニーリングさせることによって調製することができる。
【0076】
siRNA及びshRNAを構成するヌクレオチド分子は、天然型のRNAでもよいが、安定性(化学的および/または対酵素)や比活性(mRNAとの親和性)を向上させるために、種々の化学修飾を含むことができる。例えば、ヌクレアーゼなどの加水分解酵素による分解を防ぐために、siRNAまたはshRNAを構成する各ヌクレオチドのリン酸残基(ホスフェート)を、例えば、ホスホロチオエート(PS)、メチルホスホネート、ホスホロジチオネートなどの化学修飾リン酸残基に置換することができる。また、各ヌクレオチドの糖(リボース)の2'位の水酸基を、-OR(Rは、例えばCH3(2'-O-Me)、CH2CH2OCH3(2'-O-MOE)、CH2CH2NHC(NH)NH2、CH2CONHCH3、CH2CH2CN等を示す)に置換してもよい。さらに、塩基部分(ピリミジン、プリン)に化学修飾を施してもよく、例えば、ピリミジン塩基の5位へのメチル基やカチオン性官能基の導入、あるいは2位のカルボニル基のチオカルボニルへの置換などが挙げられる。
【0077】
RNAの糖部のコンフォーメーションはC2'-endo(S型)とC3'-endo(N型)の2つが支配的であり、一本鎖RNAではこの両者の平衡として存在するが、二本鎖を形成するとN型に固定される。したがって、標的RNAに対して強い結合能を付与するために、2'酸素と4’炭素を架橋することにより、糖部のコンフォーメーションをN型に固定したRNA誘導体であるBNA(LNA)(Imanishi, T. et al., Chem. Commun., 1653-9, 2002; Jepsen, J.S. et al., Oligonucleotides, 14, 130-46, 2004)やENA(Morita, K. et al., Nucleosides Nucleotides Nucleic Acids, 22, 1619-21, 2003)もまた、好ましく用いられ得る。
但し、天然型RNA中のすべてのリボヌクレオシド分子を修飾型で置換すると、RNAi活性が失われる場合があるので、RISC複合体が機能できる最小限の修飾ヌクレオシドの導入が必要である。
【0078】
p53に対するsiRNAは、例えば、Ambion(例、Ambion Cat# AM16708, siRNA ID# 69659, 69753, 69843, 187424, 187425, 187426)やSanta Cruz(例、Santa Cruz Cat# sc-29436, 44219)等から購入することもできる。
【0079】
また、ヒトp53に対するsiRNAおよびshRNAも、上記のいずれかの検索ソフトを用いて、配列番号3に示されるヒトp53 cDNAの配列もしくはRefseq. No.(NM_000546)等をクエリーとして入力することにより設計し、合成することができ、あるいはAmbion等から購入することもできる。具体的には、配列5’-GACTCCAGTGGTAATCTACTGctcgagCAGTAGATTACCACTGGAGTC-3’(配列番号28;下線部がp53のターゲット配列。大文字がdsRNAを形成する部分)を有するヒトp53に対するshRNA、Science, 296, 550-553 (2002) に記載されるp53に対するshRNAなどが例示される。
【0080】
p53に対するsiRNAもしくはshRNAの体細胞への接触は、プラスミドDNAの場合と同様に、リポソーム法、ポリアミン法、エレクトロポレーション法、ビーズ法等を用いて、該核酸を細胞内へ導入することにより実施することができる。カチオニックリポソームを用いた方法が最も一般的で、導入効率も高い。Lipofectamine2000やOligofectamine(Invitrogen)などの一般的な遺伝子導入試薬の他、例えば、GeneEraserTMsiRNA transfection reagent(Stratagene)等のsiRNA導入に適した導入試薬も市販されている。
【0081】
(D7) p53に対するsiRNAもしくはshRNAを発現するベクター
siRNAを発現するベクターには、タンデムタイプとステムループ(ヘアピン)タイプとがある。前者はsiRNAのセンス鎖の発現カセットとアンチセンス鎖の発現カセットをタンデムに連結したもので、細胞内で各鎖が発現してアニーリングすることにより2本鎖のsiRNA(dsRNA)を形成するというものである。一方、後者はshRNAの発現カセットをベクターに挿入したもので、細胞内でshRNAが発現しdicerによるプロセシングを受けてdsRNAを形成するというものである。プロモーターとしては、polII系プロモーター(例えば、CMV前初期プロモーター)を使用することもできるが、短いRNAの転写を正確に行わせるために、polIII系プロモーターを使用するのが一般的である。polIII系プロモーターとしては、マウスおよびヒトのU6-snRNAプロモーター、ヒトH1-RNase P RNAプロモーター、ヒトバリン-tRNAプロモーターなどが挙げられる。また、転写終結シグナルとして4個以上T残基が連続した配列が用いられる。
【0082】
このようにして構築したsiRNAもしくはshRNA発現カセットを、次いでプラスミドベクターやウイルスベクターに挿入する。このようなベクターとしては、核初期化物質である核酸(初期化遺伝子)について上記したと同様のものが、好ましく利用され得る(レトロウイルス、レンチウイルス、アデノウイルス、アデノ随伴ウイルス、ヘルペスウイルス、センダイウイルスなどのウイルスベクターや、動物細胞発現プラスミド、エピソーマルベクターなど)。使用するベクターは、初期化遺伝子の場合と同様、得られるiPS細胞の用途に応じて適宜選択され得る。即ち、p53遺伝子は癌抑制遺伝子として知られており、p53の恒常的な機能阻害は発癌のリスクを高める可能性があるので、作製されるiPS細胞が医療用途を念頭におく場合、p53に対するsiRNAやshRNAは細胞内で一過的に発現するようにデザインされることが望ましい。したがって、p53に対するsiRNAやshRNAをコードする核酸を担持するベクターとしては、自律複製できないプラスミドベクターなどがより好ましい。本発明の初期化因子と組み合わせた場合、p53に対するsiRNAやshRNAはプラスミドからの一過的な発現でも十分に高いiPS細胞樹立効率を与える。
【0083】
あるいは、p53に対するshRNAをコードする発現ベクターとして、市販のプラスミド(例えば、Addgene社から市販されるpMKO.1-puro p53 shRNA2: #10672等)をもとに作製したレトロウイルス等のウイルスベクター、プラスミドベクター、エピソーマルベクターなどを使用することもできる。必要に応じて、上記Cre-loxPシステムやpiggyBacトランスポゾンシステムを利用することもできる。
【0084】
p53に対するsiRNAもしくはshRNAを発現するベクターの体細胞への接触は、上記のようにして調製されるプラスミドベクター、エピソーマルベクターもしくはウイルスベクターを細胞に導入することにより行われる。これらの遺伝子導入は、初期化遺伝子について上記したと同様の手法で行うことができる。
【0085】
(D8) その他の物質
p53遺伝子の発現を阻害する他の物質として、p53に対するアンチセンス核酸やリボザイムが挙げられる。
アンチセンス核酸はDNAであってもRNAであってもよく、あるいはDNA/RNAキメラであってもよい。アンチセンス核酸がDNAの場合、標的RNAとアンチセンスDNAとによって形成されるRNA:DNAハイブリッドは、内在性RNase Hに認識されて標的RNAの選択的な分解を引き起こすことができる。したがって、RNase Hによる分解を指向するアンチセンスDNAの場合、標的配列は、p53 mRNA中の配列だけでなく、p53遺伝子の初期転写産物におけるイントロン領域の配列であってもよい。アンチセンス核酸の標的領域は、該アンチセンス核酸がハイブリダイズすることにより、結果としてp53蛋白質への翻訳が阻害されるものであればその長さに特に制限はなく、p53 mRNAの全配列であっても部分配列であってもよく、短いもので約15塩基程度、長いものでmRNAもしくは初期転写産物の全配列が挙げられる。合成の容易さや抗原性、細胞内移行性の問題等を考慮すれば、約15〜約40塩基、特に約18〜約30塩基からなるオリゴヌクレオチドが好ましい。標的配列の位置としては、5’-及び3’-UTR、開始コドン近傍などが挙げられるが、それらに限定されない。
【0086】
リボザイムとは、狭義には、核酸を切断する酵素活性を有するRNAをいうが、本明細書では配列特異的な核酸切断活性を有する限りDNAをも包含する概念として用いるものとする。リボザイムとして最も汎用性の高いものとしては、ウイロイドやウイルソイド等の感染性RNAに見られるセルフスプライシングRNAがあり、ハンマーヘッド型やヘアピン型等が知られている。ハンマーヘッド型は約40塩基程度で酵素活性を発揮し、ハンマーヘッド構造をとる部分に隣接する両端の数塩基ずつ(合わせて約10塩基程度)をmRNAの所望の切断部位と相補的な配列にすることにより、標的mRNAのみを特異的に切断することが可能である。
【0087】
アンチセンス核酸やリボザイムはDNA/RNA自動合成機を用いて合成することができる。これらを構成するヌクレオチド分子もまた、安定性、比活性などを向上させるために、上記のsiRNAの場合と同様の修飾を受けていてもよい。
あるいは、アンチセンス核酸やリボザイムは、siRNAの場合と同様に、それらをコードする核酸の形態で使用することもできる。
【0088】
p53の機能阻害物質は、体細胞の核初期化工程においてp53の機能を阻害するのに十分な様式で体細胞に接触させる必要がある。この条件が満たされる限り、核初期化物質とp53の機能阻害物質とは、同時に体細胞に接触させてもよいし、また、どちらかを先に接触させてもよい。一実施態様において、例えば、核初期化物質がタンパク性因子をコードする核酸であり、p53の機能阻害物質が化学的阻害物質である場合には、前者は遺伝子導入処理からタンパク性因子を大量発現するまでに一定期間のラグがあるのに対し、後者は速やかにp53の機能を阻害しうることから、遺伝子導入処理から一定期間細胞を培養した後に、p53の化学的阻害物質を培地に添加することができる。別の実施態様において、例えば、核初期化物質とp53の機能阻害物質とがいずれもウイルスベクター、プラスミドベクター、エピソーマルベクター等の形態で用いられる場合には、両者を同時に細胞に導入してもよい。
【0089】
(E) iPS細胞の樹立効率改善物質
p53の機能阻害物質に加え、公知の他のiPS細胞樹立効率改善物質を体細胞に接触させることにより、iPS細胞の樹立効率をさらに高めることが期待できる。そのようなiPS細胞の樹立効率改善物質としては、例えば、ヒストンデアセチラーゼ(HDAC)阻害剤[例えば、バルプロ酸 (VPA)(Nat. Biotechnol., 26(7): 795-797 (2008))、トリコスタチンA、酪酸ナトリウム、MC 1293、M344等の低分子阻害剤、HDACに対するsiRNAおよびshRNA(例、HDAC1 siRNA Smartpool(登録商標)(Millipore)、HuSH 29mer shRNA Constructs against HDAC1 (OriGene)等)等の核酸性発現阻害剤など]、G9aヒストンメチルトランスフェラーゼ阻害剤[例えば、BIX-01294 (Cell Stem Cell, 2: 525-528 (2008))等の低分子阻害剤、G9aに対するsiRNAおよびshRNA(例、G9a siRNA(human) (Santa Cruz Biotechnology)等)等の核酸性発現阻害剤など]、L-calcium channel agonist (例えばBayk8644) (Cell Stem Cell, 3, 568-574 (2008))、UTF1(Cell Stem Cell, 3, 475-479 (2008))、細胞内シグナル伝達の修飾剤[例えば、Wnt Signaling活性化剤(例えばsoluble Wnt3a)(Cell Stem Cell, 3, 132-135 (2008))、TGF-βの阻害剤、MEK阻害剤、2i/LIF (2iはmitogen-activated protein kinase signallingおよびglycogen synthase kinase-3の阻害剤、PloS Biology, 6(10), 2237-2247 (2008))]、他の天然もしくは合成低分子化合物(例、5’-azacytidine、thiazovivin、ビタミンC等)、ES細胞特異的miRNA(例えば、miR-302-367クラスター (Mol. Cell. Biol. doi:10.1128/MCB.00398-08、WO2009/075119)、miR-302 (RNA (2008) 14: 1-10)、miR-291-3p, miR-294およびmiR-295 (Nat. Biotechnol. 27: 459-461 (2009)))等が挙げられるが、それらに限定されない。前記において核酸性の発現阻害剤はsiRNAもしくはshRNAをコードするDNAを含む発現ベクターの形態であってもよい。
【0090】
尚、前記核初期化物質の構成要素のうち、例えば、SV40 Large T等は、体細胞の核初期化のために必須ではなく補助的な因子であるという点において、iPS細胞の樹立効率改善物質の範疇にも含まれ得る。核初期化の機序が明らかでない現状においては、核初期化に必須の因子以外の補助的な因子について、それらを核初期化物質として位置づけるか、あるいはiPS細胞の樹立効率改善物質として位置づけるかは便宜的であってもよい。即ち、体細胞の核初期化プロセスは、体細胞への核初期化物質およびiPS細胞の樹立効率改善物質の接触によって生じる全体的事象として捉えられるので、当業者にとって両者を必ずしも明確に区別する必要性はないであろう。
【0091】
これら他のiPS細胞樹立効率改善物質の体細胞への接触は、該物質が(a) タンパク性因子である場合、(b) 該タンパク性因子をコードする核酸である場合、あるいは(c) 低分子化合物である場合に応じて、p53の機能阻害物質についてそれぞれ上記したと同様の方法により、実施することができる。
他のiPS細胞の樹立効率改善物質は、該物質の非存在下と比較して体細胞からのiPS細胞樹立効率が有意に改善される限り、核初期化物質と同時に体細胞に接触させてもよいし、また、どちらかを先に接触させてもよく、該物質の物性に応じて、p53の機能阻害物質について上記したと同様のタイミングで体細胞と接触させることができる。
【0092】
(F) 培養条件による樹立効率の改善
体細胞の核初期化工程において低酸素条件下で細胞を培養することにより、iPS細胞の樹立効率をさらに改善することができる。本明細書において「低酸素条件」とは、細胞を培養する際の雰囲気中の酸素濃度が、大気中のそれよりも有意に低いことを意味する。具体的には、通常の細胞培養で一般的に使用される5-10% CO2/95-90%大気の雰囲気中の酸素濃度よりも低い酸素濃度の条件が挙げられ、例えば雰囲気中の酸素濃度が18%以下の条件が該当する。好ましくは、雰囲気中の酸素濃度は15%以下(例、14%以下、13%以下、12%以下、11%以下など)、10%以下(例、9%以下、8%以下、7%以下、6%以下など)、または5%以下(例、4%以下、3%以下、2%以下など)である。また、雰囲気中の酸素濃度は、好ましくは0.1%以上(例、0.2%以上、0.3%以上、0.4%以上など)、0.5%以上(例、0.6%以上、0.7%以上、0.8%以上、0.95%以上など)、または1%以上(例、1.1%以上、1.2%以上、1.3%以上、1.4%以上など)である。
【0093】
細胞の環境において低酸素状態を創出する手法は特に制限されないが、酸素濃度の調節可能なCO2インキュベーター内で細胞を培養する方法が最も容易であり、好適な例として挙げられる。酸素濃度の調節可能なCO2インキュベーターは、種々の機器メーカーから販売されている(例えば、Thermo scientific社、池本理化学工業、十慈フィールド、和研薬株式会社などのメーカー製の低酸素培養用CO2インキュベーターを用いることができる)。
【0094】
低酸素条件下で細胞培養を開始する時期は、iPS細胞の樹立効率が正常酸素濃度(20%)の場合に比して改善されることを妨げない限り特に限定されず、体細胞への核初期化物質の接触より前であっても、該接触と同時であっても、該接触より後であってもよいが、例えば、体細胞に核初期化物質を接触させた直後から、あるいは接触後一定期間(例えば、1ないし10(例、2,3,4,5,6,7,8または9)日)おいた後に低酸素条件下で培養することが好ましい。
【0095】
低酸素条件下で細胞を培養する期間も、iPS細胞の樹立効率が正常酸素濃度(20%)の場合に比して改善されることを妨げない限り特に限定されず、例えば3日以上、5日以上、7日以上または10日以上で、50日以下、40日以下、35日以下または30日以下の期間等が挙げられるが、それらに限定されない。低酸素条件下での好ましい培養期間は、雰囲気中の酸素濃度によっても変動し、当業者は用いる酸素濃度に応じて適宜当該培養期間を調整することができる。また、一実施態様において、iPS細胞の候補コロニーの選択を、薬剤耐性を指標にして行う場合には、薬剤選択を開始する迄に低酸素条件から正常酸素濃度に戻すことが好ましい。
【0096】
さらに、低酸素条件下で細胞培養を開始する好ましい時期および好ましい培養期間は、用いられる核初期化物質の種類、正常酸素濃度条件下でのiPS細胞樹立効率などによっても変動する。
【0097】
核初期化物質およびp53の機能阻害物質(さらに必要に応じて他のiPS細胞の樹立効率改善物質)を接触させた後、細胞を、例えばES細胞の培養に適した条件下で培養することができる。マウス細胞の場合、通常の培地に分化抑制因子としてLeukemia Inhibitory Factor(LIF)を添加して培養を行う。一方、ヒト細胞の場合には、LIFの代わりに塩基性線維芽細胞増殖因子(bFGF)および/または幹細胞因子(SCF)を添加することが望ましい。また通常、細胞は、フィーダー細胞として、放射線や抗生物質で処理して細胞分裂を停止させたマウス胎仔由来の線維芽細胞の共存下で培養される。マウス胎仔由来の線維芽細胞としては、通常STO細胞株(ATCC CRL-1503)等がフィーダーとしてよく使われるが、iPS細胞の誘導には、STO細胞にネオマイシン耐性遺伝子とLIF遺伝子を安定に組み込んだSNL細胞(SNL76/7 STO細胞; ECACC 07032801)(McMahon, A. P. & Bradley, A. Cell 62, 1073-1085 (1990))等がよく使われている。しかしながら、本発明においては、マウス胎仔由来の初代線維芽細胞(MEF)を用いた方がヒトiPS細胞の樹立効率がより改善されるので、MEFの使用がより好ましい。マイトマイシンC処理済のMEFは、ミリポア社やリプロセル社から市販されている。これらのフィーダー細胞との共培養は、核初期化物質の接触より前から開始してもよいし、該接触時から、あるいは該接触より後(例えば1-10日後)から開始してもよい。
【0098】
本発明者らは、本発明の核初期化物質およびp53の機能阻害物質と非ウイルス性ベクター、好ましくはエピソーマルベクター、とりわけ早期自己消失型エピソーマルベクターとを組み合わせたことにより、体細胞への核初期化物質およびp53の機能阻害物質の導入から、iPS細胞を樹立し、さらにiPS細胞として維持するまでの間、非ヒト動物由来成分を一切用いずに培養して(即ち、完全xeno-freeの条件下で)、ヒトiPS細胞を作製する方法を初めて提供することに成功した。xeno-freeの条件下でヒトiPS細胞を誘導する場合、核初期化物質およびp53の機能阻害物質(さらに必要に応じて他のiPS細胞の樹立効率改善物質)を接触させた後、細胞は、FCSや他の非ヒト動物由来成分を含まない培地中で培養される。分化抑制因子として培地に添加される物質(例、bFGF、SCF等)としては、ヒト由来の精製されたタンパク質、好ましくは組換えタンパク質が使用される。フィーダー細胞としては、ヒト由来の任意の体細胞を用いることができるが、例えばヒト皮膚線維芽細胞(HDF)やヒト歯髄幹細胞などが好ましく用いられ得る。また、フィーダー細胞を用いることなくヒトiPS細胞を誘導することも可能であり、この場合、細胞容器のコーティング剤として、マトリゲルやゼラチンに代えて市販のxeno-freeのコーティング剤を用いることができる。
【0099】
iPS細胞の候補コロニーの選択は、薬剤耐性とレポーター活性を指標とする方法と目視による形態観察による方法とが挙げられる。前者としては、例えば、分化多能性細胞において特異的に高発現する遺伝子(例えば、Fbx15、Nanog、Oct3/4など、好ましくはNanog又はOct3/4)の遺伝子座に、薬剤耐性遺伝子及び/又はレポーター遺伝子をターゲッティングした組換え体細胞を用い、薬剤耐性及び/又はレポーター活性陽性のコロニーを選択するというものである。そのような組換え体細胞としては、例えばFbx15遺伝子座にβgeo(β-ガラクトシダーゼとネオマイシンホスホトランスフェラーゼとの融合タンパク質をコードする)遺伝子をノックインしたマウス由来のMEF(Takahashi & Yamanaka, Cell, 126, 663-676 (2006))、あるいはNanog遺伝子座に緑色蛍光タンパク質(GFP)遺伝子とピューロマイシン耐性遺伝子を組み込んだトランスジェニックマウス由来のMEF(Okita et al., Nature, 448, 313-317 (2007))等が挙げられる。一方、目視による形態観察で候補コロニーを選択する方法としては、例えばTakahashi et al., Cell, 131, 861-872 (2007)に記載の方法が挙げられる。レポーター細胞を用いる方法は簡便で効率的ではあるが、iPS細胞がヒトの治療用途を目的として作製される場合、安全性の観点から目視によるコロニー選択が望ましい。
【0100】
選択されたコロニーの細胞がiPS細胞であることの確認は、上記したNanog(もしくはOct3/4)レポーター陽性(ピューロマイシン耐性、GFP陽性など)および目視によるES細胞様コロニーの形成によっても行い得るが、より正確を期すために、アルカリフォスファターゼ染色や、各種ES細胞特異的遺伝子の発現を解析したり、選択された細胞をマウスに移植してテラトーマ形成を確認する等の試験を実施することもできる。
【0101】
このようにして樹立されたiPS細胞は、種々の目的で使用することができる。例えば、ES細胞で報告されている分化誘導法を利用して、iPS細胞から種々の細胞(例、心筋細胞、血液細胞、神経細胞、血管内皮細胞、インスリン分泌細胞等)への分化を誘導することができる。したがって、患者本人やHLAの型が同一もしくは実質的に同一である他人から採取した体細胞を用いてiPS細胞を誘導すれば、そこから所望の細胞(即ち、該患者が罹病している臓器の細胞や疾患に対する治療効果を発揮する細胞など)に分化させて該患者に移植するという、自家もしくは同種異系移植による幹細胞療法が可能となる。さらに、iPS細胞から分化させた機能細胞(例、肝細胞)は、対応する既存の細胞株よりも実際の生体内での該機能細胞の状態をより反映していると考えられるので、医薬候補化合物の薬効や毒性のin vitroスクリーニング等にも好適に用いることができる。
【0102】
以下に実施例を挙げて本発明をより具体的に説明するが、本発明がこれらに限定されないことは言うまでもない。
【実施例】
【0103】
実施例1
レトロウイルスによるヒトiPS細胞の樹立
初期化に用いるレトロウイルスはpMXsプラスミドおよびPlat-Eパッケージング細胞(東京大学、北村俊夫博士より供与された, Morita, S. et al., Gene Ther. 7, 1063-1066(2000))を元に作製した。pMXsのマルチクローニングサイトへ種々のコンストラクトを挿入して初期化に用いるレトロウイルスベクターを作製した。挿入したコンストラクトはヒトOct3/4(図1中O)、ヒトSox2(図1中S)、ヒトKlf4(図1中K)、ヒトc-Myc(図1中M)、ヒトLin28(図1中L)、ヒトNanog(図1中N)、ヒトL-Myc(図1中U)と、それぞれの遺伝子の翻訳領域を口蹄疫ウイルスの2A配列をはさんで繋げたコンストラクト(Fig. 1では、各遺伝子の略号(上記)をハイフンで結んで記載(例: O-M-L))である。陰性コントロールとしてGFPを用いた。
初期化に使用するレトロウイルスは、前日に6 well培養プレート (Falcon) に1 well当り0.6x106細胞で播種したPlat-E細胞に、前述のレトロウイルスベクターを導入して作製した。培養液はDMEM/10% FCS (DMEM (Nacalai tesque) にウシ胎仔血清を10%加えたもの) を使用し、37℃、5% CO2で培養した。ベクターの導入のためにFuGene6 transfection reagent (Roche) 4.5 μLをOpti-MEM I Reduced-Serum Medium (Invitrogen) 100 μLに入れ、室温で5分間静置した。その後、各発現ベクターを1.5 μg加え、さらに室温で15分静置してからPlat-Eの培養液に加えた。2日目にPlat-Eの上清を新しい培地に換え、3日目に培養上清を回収して0.45 μm sterile filter (Whatman)で濾過し、polybrene (Nacalai) を4 μg/mLとなるように加えてウイルス液とした。
実験には新生児白人の皮膚より樹立した線維芽細胞(Cell Applications) にレンチウイルスでマウスSlc7a1を導入した細胞を使用した。この線維芽細胞はDMEM/10% FCSを培養液として使用し、100 mm培養ディッシュで37℃、5% CO2の条件で培養し、維持した。初期化因子導入の前日に、培地を除き、PBS 5 mLを加えて細胞を洗浄した。PBSを除いた後、0.25% Trypsin/1 mM EDTA (Invitrogen) を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらDMEM/10% FCSを加えて懸濁し、1.3x105細胞を6 well培養プレート (Falcon) の1 wellへ播いておいた。
翌日に、Plat-E細胞を用いて産生したレトロウイルスをそれぞれ混和して、感染させた。24時間後に培地を交換して感染を終了した。感染開始から6日目にプレートの培地を除き、PBS 1 mLを加えて細胞を洗浄した。PBSを除いた後、0.25% Trypsin/1 mM EDTA (Invitrogen) を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらDMEM/10% FCSを加えて懸濁し、5x105細胞を、あらかじめフィーダー細胞を蒔いておいた100 mm dishに蒔いた。フィーダー細胞にはマイトマイシンCで処理して、細胞分裂を止めたSNL細胞を用いた。翌日に霊長類ES細胞用培地(リプロセル)に4 ng/mLとなる様にbFGF (Wako) を加えた培地に交換し、以後2日おきに培地交換を続けた。35日目にヒトES細胞様のコロニーを数えた。コロニーの写真とコロニー数を図1に、またコロニー数をカウントした結果のまとめを図2に示す。いずれの遺伝子の組み合わせにおいても、c-Mycの代わりにL-Mycを用いたほうが、ヒトiPSコロニー(ES様コロニー)の樹立効率が格段に高かった。またc-Myc、L-Mycのいずれを用いた場合も、Nanogを含む6遺伝子(図2中6 factors)よりもNanogを除いた5遺伝子(Oct3/4, Sox2, Klf4, Lin28、およびc-MycまたはL-Myc;図2中ctrl))を用いたほうが樹立効率が高かった。またL-Mycを用いた場合は、L-Myc-Lin28の順でつないでも(図2中U-L)、つないでいない場合(5遺伝子別々に導入した場合:図2中ctrl)に比して樹立効率は低下しなかった。
【0104】
実施例2
エピソーマルプラスミドによるヒトiPS細胞の樹立(1)
初期化に用いるプラスミドはpCX-EGFP(大阪大学、岡部勝博士より供与された, FEBS Letters, 407, 313-319, 1997)を元に作製した。はじめにEGFPの下流にウッドチャック肝炎ウイルス転写後調節エレメント(WPRE)配列を挿入した。細胞内でこのベクターを複製させるためのカセットはpCEP4 (Invitrogen) のEBNA-1の両端にloxP配列を挿入して作製した。このEBNA-1とoriPを含むカセットを、前述のWPREを挿入したpCX-EGFPのBamHIサイトへ組み込み、これをpCXLE-EGFPと名付けた。このpCXLE-EGFPをEcoRIで処理し、EGFPの代わりに種々のコンストラクトを挿入して初期化に用いるプラスミドを作製した。挿入したコンストラクトは1)ヒトOct3/4、2)ヒトSox2およびヒトKlf4の各翻訳領域を口蹄疫ウイルスの2A配列をはさんで繋げたコンストラクト、3)ヒトKlf4、ヒトSox2およびヒトOct3/4の各翻訳領域を口蹄疫ウイルスの2A配列をはさんで繋げたコンストラクト、4)ヒトc-Myc、ヒトLin28およびNanogの各翻訳領域を口蹄疫ウイルスの2A配列をはさんで繋げたコンストラクラクト、および5)SV40 Large T antigenの5種であり(各コンストラクトはCAGプロモーターおよびウサギβ-グロビンpolyA配列の制御下にある)、それぞれpCXLE-hOct4、pCXLE-hSK、pCXLE-hKSO、pCXLE-hMLN、pCX-SV40LTと名付けた(pCX-SV40LTはWPRE配列、EBNA-1カセットおよびloxP配列のいずれも有してないプラスミドである)。
実験には36歳白人女性の顔の皮膚より樹立した線維芽細胞 (Cell Applications, Lot1388) を使用した。この線維芽細胞はDMEM/10% FCS (DMEM (Nacalai tesque) にウシ胎児血清を10%加えたもの) を培養液として使用し、100 mm 培養ディッシュで37℃、5% CO2の条件で培養し、維持した。プラスミド導入時には、培地を除き、PBS 5 mLを加えて細胞を洗浄した。PBSを除いた後、0.25% Trypsin/1 mM EDTA (Invitrogen) を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらDMEM/10% FCSを加えて懸濁し、6x105細胞を15 mL遠心チューブに回収した。800 rpmで5分間遠心して上清を除いた。プラスミド全量3 μg(pCXLE-hOct4 0.5 μg、pCXLE-hSK 1 μg、pCXLE-hKSO 0.5 μg、pCXLE-hMLN 0.5 μg、pCX-SV40LT 0.5 μg)を、Microporator(エアブラウン)を用いて細胞に導入した。導入時の条件は100 μLチップを用い、1650 V, 10 ms, 3回のパルスで行った。導入後の細胞はあらかじめDMEM/10% FCSを3 mL入れておいた6 well培養プレート (Falcon) へ移し、37℃、5% CO2の条件で6日間培養した。その後培地を除き、PBS 2 mLを加えて細胞を洗浄した。PBSを除いた後、0.25% Trypsin/1 mM EDTA (Invitrogen) を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらDMEM/10% FCSを加えて懸濁し、2x105の細胞を、あらかじめフィーダー細胞を蒔いておいた100 mm dishに蒔いた。フィーダー細胞としてはマイトマイシンC処理をしたMEFを使用した。翌日に霊長類ES細胞用培地(リプロセル)に4 ng/mLとなる様にbFGF (Wako) を加えた培地に交換し、以後2日おきに培地交換を続けた。26日目にヒトES細胞様コロニーが1個出現した。樹立時の写真を図3(左)に、3継代目のコロニーの写真を図3(右)に示す。2x105の細胞から1個のES様コロニーが樹立できた。
【0105】
実施例3
エピソーマルプラスミドによるヒトiPS細胞の樹立(2)
初期化に用いるプラスミドとして、実施例2におけるpCX-SV40LT以外の4種のプラスミド:pCXLE-hOct4、pCXLE-hSK、pCXLE-hKSO、pCXLE-hMLNを用いた。
実験には36歳白人女性の顔の皮膚より樹立した線維芽細胞 (Cell Applications, Lot1388) を使用した。この線維芽細胞はDMEM/10% FCS (DMEM (Nacalai tesque) にウシ胎児血清を10%加えたもの) を培養液として使用し、100 mm 培養ディッシュで37℃、5% CO2の条件で培養し、維持した。プラスミド導入時には、培地を除き、PBS 5 mLを加えて細胞を洗浄した。PBSを除いた後、0.25% Trypsin/1 mM EDTA (Invitrogen) を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらDMEM/10% FCSを加えて懸濁し、6x105細胞を15 mL遠心チューブに回収した。800 rpmで5分間遠心して上清を除いた。プラスミド全量3 μg(pCXLE-hOct4 1 μg、pCXLE-hSK 1 μg、pCXLE-hKSO 0.5 μg、pCXLE-hMLN 0.5 μg)をMicroporator(エアブラウン)を用いて細胞に導入した。導入時の条件は100 μLチップを用い、1650 V, 10 ms, 3回のパルスで行った。導入後の細胞はあらかじめDMEM/10% FCSを3 mL入れておいた6 well培養プレート (Falcon) へ移し、37℃、5% CO2の条件で6日間培養した。その後培地を除き、PBS 2 mLを加えて細胞を洗浄した。PBSを除いた後、0.25% Trypsin/1 mM EDTA (Invitrogen) を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらDMEM/10% FCSを加えて懸濁し、1x105の細胞をあらかじめフィーダー細胞を蒔いておいた100 mm dishに蒔いた。フィーダー細胞としてはマイトマイシンC処理をしたMEFまたはSNL76/7を使用した。翌日に霊長類ES細胞用培地(リプロセル)に4 ng/mLとなる様にbFGF (Wako) を加えた培地に交換し、以後2日おきに培地交換を続けた。フィーダーとしてMEFを用いた場合、24日目にヒトES細胞様コロニーが1個出現した。樹立時の写真を図4(左)に、2継代目のコロニーの写真を図4(右)に示す。1x105の細胞から1個のES様コロニーが樹立できた。なお、フィーダーとしてSNL76/7を用いた場合はiPSコロニーは樹立できなかった。
【0106】
実施例4
エピソーマルプラスミドによるヒトiPS細胞の樹立(3)
実施例2及び3に示したように、Oct3/4、Sox2、Klf4、c-Myc、Lin28およびNanogの6遺伝子、またはこれにSV40 large Tを加えた7遺伝子をエピソーマルプラスミドにて細胞に導入した場合は、1〜2x105の細胞から0個〜1個程度しかiPS細胞を樹立することができなかった。この誘導効率の低さに関しては、同じ7遺伝子を導入したScience, 324, 797-801 (2009) にも記載されている。そこで樹立効率の改善を目的として、実施例1の予備実験で良好な結果を得たL-Myc(L-Myc-Lin28連結コンストラクト)を使用し、また不要と判断されたNanogは用いないこととした。さらに、ヒトp53に対するshRNAをコードするDNA(5’-GACTCCAGTGGTAATCTACttcaagagaGTAGATTACCACTGGAGTC-3’(配列番号32):下線部がp53のターゲット配列。大文字がdsRNAを形成する部分。以下、単にp53 shRNAという)も使用した。
実施例2にて作製したpCXLE-hOct4、pCXLE-hSK、pCXLE-hMLNに加え、1)実施例2にて作製したpCXLE-EGFPをEcoRIで処理し、EGFPの代わりにヒトL-MycおよびヒトLin28の各翻訳領域を口蹄疫ウイルスの2A配列をはさんで繋げたコンストラクラクトを挿入したプラスミド(pCXLE-hUL)と、2)pCXLE-hOct4のBamHIサイトへp53に対するshRNA発現コンストラクト(U6プロモーターにより駆動される)を挿入したプラスミド(pCXLE-hOct4-shp53)とを作製し、使用した。またコントロールとしてpCXLE-EGFPを用いた。pCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hULの構造を図30に示す(図中、pCXLE-hOct4-shp53はpCXLE-hOct3/4-shp53と記載される)。
実験には21歳日本人女性の皮膚より樹立した線維芽細胞 (JCRB, TIG113) にレンチウイルスでマウスSlc7a1を導入した細胞を使用した。この線維芽細胞はDMEM/10% FCS (DMEM (Nacalai tesque) にウシ胎児血清を10%加えたもの) を培養液として使用し、100 mm 培養ディッシュで37℃、5% CO2の条件で培養し、維持した。プラスミド導入時には、培地を除き、PBS 5 mLを加えて細胞を洗浄した。PBSを除いた後、0.25% Trypsin/1 mM EDTA (Invitrogen)を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらDMEM/10% FCSを加えて懸濁し、6x105細胞を15 mL遠心チューブに回収した。800 rpmで5分間遠心して上清を除いた。表2に示す各プラスミド(トータル3 μg)をMicroporator(エアブラウン)を用いて細胞に導入した。導入時の条件は100 μLチップを用い、1650 V, 10 ms, 3回のパルスで行った。導入後の細胞はあらかじめDMEM/10% FCSを3 mL入れておいた6 well培養プレート (Falcon) へ移し、37℃、5% CO2の条件で7日間培養した。その後培地を除き、PBS 2 mLを加えて細胞を洗浄した。PBSを除いた後、0.25% Trypsin/1 mM EDTA (Invitrogen) を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらDMEM/10% FCSを加えて懸濁し、1.5x105の細胞をあらかじめフィーダー細胞を蒔いておいた100 mm dishに蒔いた。フィーダー細胞としてはマイトマイシンC処理をしたMEFまたはSNL76/7を使用した。翌日に、霊長類ES細胞用培地(リプロセル)に4 ng/mLとなる様にbFGF (Wako) を加えた培地に交換し、以後2日おきに培地交換を続けた。28日目に出現したヒトES細胞様のコロニーをカウントした。コロニーの写真を図5に、またコロニー数をカウントした結果を表2および図6(フィーダーがMEFの場合)に示す。
【0107】
【表2】
【0108】
いずれのフィーダーを用いた場合もiPSコロニー(ES様コロニー)を樹立することができたが、MEFを使ったほうがより多くのコロニーを樹立することができた。また、従来の6遺伝子(Oct3/4、Sox2、Klf4、c-Myc、Lin28およびNanog)を用いた場合に比べ、Oct3/4、Sox2、Klf4、L-Myc、Lin28の5遺伝子を用いたほうが樹立効率が上昇し、さらにそこへp53 shRNAを加えることにより樹立効率は格段に上昇した。
【0109】
実施例5
エピソーマルプラスミドによるヒトiPS細胞の樹立(4)
初期化に用いるプラスミドとして、実施例4と同じpCXLE-hOct4、pCXLE-hSK、pCXLE-hMLN、pCXLE-hUL、pCXLE-hOct4-shp53の5種類のプラスミドと、コントロールとしてpCXLE-EGFPを用いた。
実験には6歳日本人女性の皮膚より樹立した線維芽細胞 (JCRB, TIG120) および8カ月齢日本人男性の皮膚より樹立した線維芽細胞 (JCRB, TIG121) のそれぞれにレンチウイルスでマウスSlc7a1を導入した細胞を使用した。これらの線維芽細胞はDMEM/10% FCS (DMEM (Nacalai tesque) にウシ胎児血清を10%加えたもの) を培養液として使用し、100 mm 培養ディッシュで37℃、5% CO2の条件で培養し、維持した。プラスミド導入時には、培地を除き、PBS 5 mLを加えて細胞を洗浄した。PBSを除いた後、0.25% Trypsin/1 mM EDTA (Invitrogen)を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらDMEM/10% FCSを加えて懸濁し、6x105細胞を15 mL遠心チューブに回収した。800 rpmで5分間遠心して上清を除いた。表3に示すプラスミド(トータル3 μg)をMicroporator(エアブラウン)を用いて細胞に導入した。導入時の条件は100 μLチップを用い、1650 V, 10 ms, 3回のパルスで行った。導入後の細胞はあらかじめDMEM/10% FCSを3 mL入れておいた6 well培養プレート (Falcon) へ移し、37℃、5% CO2の条件で7日間培養した。その後培地を除き、PBS 2 mLを加えて細胞を洗浄した。PBSを除いた後、0.25% Trypsin/1 mM EDTA (Invitrogen) を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらDMEM/10% FCSを加えて懸濁し、1x105の細胞をあらかじめフィーダー細胞を蒔いておいた100 mm dishに蒔いた。フィーダー細胞としてはマイトマイシンC処理をしたMEFまたはSNL76/7を使用した。翌日に、霊長類ES細胞用培地(リプロセル)に4 ng/mLとなる様にbFGF (Wako) を加えた培地に交換し、以後2日おきに培地交換を続けた。28日目に出現したヒトES細胞様のコロニーをカウントした。コロニーの写真を図7(TIG120を用いた場合)および図8(TIG121を用いた場合)に、またコロニー数をカウントした結果を表3、図9(TIG120を用い、フィーダーとしてMEFを用いた場合)および図10(TIG121を用い、フィーダーとしてMEFを用いた場合)に示す。
【0110】
【表3】
【0111】
従来の6遺伝子(Oct3/4、Sox2、Klf4、c-Myc、Lin28およびNanog)を用いた場合に比べ、そこへp53 shRNAを加えた場合のほうが樹立効率が上昇した。さらに、従来の6遺伝子よりもOct3/4、Sox2、Klf4、L-Myc、Lin28の5遺伝子を用いたほうが樹立効率が上昇し、さらにそこへp53 shRNAを加えることにより樹立効率は格段に上昇した。
フィーダー細胞としてMEFを用いた場合とSNL76/7(MSTO)を用いた場合とを比較した結果を図11に示す(TIG120を用いた場合)。いずれのフィーダーを用いた場合もiPSコロニー(ES様コロニー)を樹立することができたが、MEFを使ったほうがより多くのコロニーを樹立することができた。
【0112】
実施例6
iPSコロニーにおける外来遺伝子の存在の有無の確認(1)
エピソーマルプラスミドで初期化遺伝子を導入して樹立した5種類の異なるiPS細胞を用いて、外来遺伝子のゲノム中への組み込みの有無等を調べた。
6 well培養プレート (Falcon) でほぼコンフルエントとなったiPS細胞を、PBS 2 mLを加えて洗浄した。PBSを除いた後、ゲノム回収バッファー(50 mM Tris-HCl, 20 mM EDTA, 100 mM NaCl, 1% SDS, 50 μg/mL proteinase K)を400 μL加えて細胞を溶かし、1.5 mLチューブに回収した後、細胞ライセートを55℃で一晩インキュベートした。ここにPCI(フェノール、クロロホルムおよびイソアミルアルコールを25:24:1で混和させたもの)を150 μL加えて、15分間混和した。これを13200 rpm、10分間遠心し、上層の水層を回収した。ここにエタノール1 mLを加えて、混和後に析出してきたものを別のチューブへ回収し、これをlong DNAとした。析出物を取り除いた後の液を13200 rpm、15分間遠心した。これで沈澱してきたものをshort DNAとした。いずれのDNAも70% エタノールで洗浄し、TEバッファーに溶かしてPCRの鋳型とした。PCRにはTaKaRa EX Taq (宝酒造) を使用し、一回の反応にlong DNA 50 ng、short DNA 250 ngをそれぞれ鋳型として用いた。プライマーは以下の組み合わせを使用した。
Klf4の検出; hKlf4-S1016 ACC CAT CCT TCC TGC CCG ATC AGA(配列番号33)
hKlf4-AS1170 ATC ACA AGT GTG GGT GGC GGT CCT(配列番号34)
c-Mycの検出; hMyc-S547 GCC GCC GCC TCA GAG TGC ATC GAC(配列番号35)
hMyc-AS947 CGA GTG GAG GGA GGC GCT GCG TAG(配列番号36)
pCEP4由来の; pEP4-SF1 TTC CAC GAG GGT AGT GAA CC(配列番号37)
OriPの検出 pEP4-SR1 TCG GGG GTG TTA GAG ACA AC(配列番号38)
OriPの検出時には元となった線維芽細胞のゲノム(50 ng)に導入したプラスミドを200, 20, 2 fg加えたもの(図12中「+plasmid」)をコントロールとして同時にPCRを行った。結果を図12に示す。いずれのiPS細胞においても、外来遺伝子(Tg)由来のKlf4およびc-Myc、並びにベクター成分のOriPは、long DNA(図12中L)およびshort DNA(図12中S)のどちらからも検出されなかったことから、導入したエピソーマルプラスミドは細胞から脱落したものと予想された。
【0113】
実施例7
エピソーマルプラスミドによるヒトiPS細胞の樹立(5)
様々な年齢の日本人及び白人男女の皮膚より樹立した線維芽細胞(HDF) を体細胞ソースとして用い、実施例5と同様にしてOct3/4、Sox2、Klf4、L-Myc、Lin28の5遺伝子およびp53 shRNAを、pCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hULを用いて導入した。フィーダー細胞はMSTOおよびMEFのいずれかを用いた。また線維芽細胞はマウスSlc7a1を導入した細胞、導入しなかった細胞のいずれも用意した。遺伝子導入27〜32日目に出現したヒトES細胞様のコロニーをカウントした結果(n=1〜5)をまとめて表4に示す。
【0114】
【表4】
【0115】
表中のコロニー数はHDF 1x105個あたりの樹立コロニー数の平均値を示す。表4に示されるように、年齢、性別、人種に関わりなく、iPS細胞を樹立できることが明らかになった。また表では分けて記載しなかったが、Slc7a1の導入の有無にかかわらずiPS細胞が樹立できることも分かった。
【0116】
実施例8
一過性発現プラスミドによるp53shRNAの導入
p53遺伝子は癌抑制遺伝子として知られており、p53の長期的な機能阻害は発癌のリスクを高める可能性がある。そこで、p53 shRNAを一過性の発現ベクター(EBNA-1とoriPを有さない通常のプラスミドベクター:以下プラスミドベクター)により発現させた場合でも、エピソーマルベクター同様にiPS細胞が樹立できるかどうかを検討した。
まず予備実験として、エピソーマルベクターとプラスミドベクターの遺伝子発現の消失の程度に差があるかどうかを検討した。実験には、pCX-EGFP(大阪大学、岡部勝博士より供与された, FEBS Letters, 407, 313-319, 1997)、実施例2で作製したpCXLE-EGFP、およびpCXLE-EGFPからloxP配列を除いたプラスミドであるpCXE-EGFPを用いた。各プラスミドをHDF1419に導入し、導入後6日目および14日目にGFPの発現を観察した。なお導入時の条件はMicroporatorにより、100 μLチップを用いて 1650 V, 10 ms, 3回のパルスで行った。結果を図13に示す。いずれのプラスミドでも6日目にGFPによる蛍光が認められたが、発現量はpCX-EGFPが他の2つに比べ低かった。14日目になるとまだ蛍光が認められるもののpCX-EGFPの発現量は他の2つに比較し顕著に低かった。以上によりプラスミドベクターはエピソーマルベクターと比べて遺伝子発現の消失が早いことが確認された。
次にp53 shRNAをプラスミドベクターで導入した場合にもiPS細胞が樹立できるかどうかを検討した。導入には以下のプラスミドベクターまたはエピソーマルベクターを用いた。
(a) pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hMLN
(b) pCXLE-hOct4、pCXLE-hSK、pCXLE-hMLN、pSilencer-shp53
(pSilencer-shp53はpSilencerTM(Ambion)のU6プロモーターの下流にp53 shRNAを組み込んだもの)
(c) pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hUL
(d) pCXLE-hOct4、pCXLE-hSK、pCXLE-hUL、pSilencer-shp53
導入にはそれぞれ0.75 μg((b)および(d))もしくは1 μg((a)および(c))のベクターを用い、合計3 μgを使用した。
体細胞として36歳白人女性の皮膚より樹立した線維芽細胞(HDF1388)を、またフィーダー細胞としてMEFを用い、実施例5と同様にしてiPS細胞の樹立を行った。なお導入時の条件はMicroporatorにより、100 μLチップを用いて 1650 V, 10 ms, 3回のパルスで行った。導入27日目に出現したヒトES細胞様のコロニーをカウントした結果を図14に示す。上記(a)と(b)(図14中、+M-L-NのEpiとTrans)、(c)と(d)(図14中、+U-LのEpiとTrans)のどちらの比較においても、p53 shRNAをエピソーマルベクターで導入した場合とプラスミドベクターで導入した場合とで樹立コロニー数に大差は無く、プラスミドベクターによる導入でも十分にiPS細胞が樹立できることが分かった。
【0117】
実施例9
HLAホモ健常人由来歯髄幹細胞からのiPS細胞の樹立
HLA-A、HLA-B、HLA-CwおよびHLA-DRB1の4遺伝子座がホモの健常人由来の歯髄幹細胞(DP74株およびDP94株)は、岐阜大学、國貞隆弘博士、手塚健一博士より提供を受けた。このDP74株およびDP94株は、J. Dent. Res., 87(7): 676-681 (2008) およびWO 2010/013359に記載の方法に基づき樹立された。両細胞株の上記4つのHLA遺伝子型と、そのアレル頻度(この4つのHLA遺伝子型を持つアレル(ハプロタイプ)の、日本人の全アレルにおける頻度)を表5に示す。
【0118】
【表5】
【0119】
初期化に用いるプラスミドとして、5種のプラスミド:pCXLE-hOct4、pCXLE-hSK、pCXLE-hMLN、pCXLE-hULおよびpCXLE-hOct4-shp53を用いて、実施例5と同様の手法にてiPS細胞を樹立した。その際、併せてScience, 324:797-801 (2009) に記載のプラスミド(Thomson’s mix)導入によるiPS細胞樹立も行った。導入にはそれぞれ合計3 μgのベクターを使用し、導入時の条件はMicroporatorにより、100 μLチップを用いて 1650 V, 10 ms, 3回のパルスで行った。フィーダー細胞はMSTOを用いた。
pCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hULをDP74およびDP94に導入して樹立したiPS細胞の写真を図15に示す。また、導入28日目に出現したヒトES細胞様のコロニーをカウントした結果を図16および図17に示す(数値は3回の実験の平均値と標準偏差を示す)。DP74株 (図16) およびDP94株 (図17) のいずれの場合もOct3/4、Sox2、Klf4、L-Myc、Lin28の5遺伝子およびp53 shRNA(pCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hUL)を導入した場合が最も樹立効率が高く、HDFと同様の良好な結果が得られた。
【0120】
実施例10
iPSコロニーにおける外来遺伝子の存在の有無の確認(2)
予備検討としてpCXLE-EGFPをHDFに導入し、EGFPの発現がどのように消失していくかを検討した。導入後1週目〜4週目まで毎週、1×104個の細胞におけるEGFPの発現量(蛍光強度)をFACS解析した。結果を図18に示す。EGFPの発現量および発現細胞数は週を追うごとに減少し、遺伝子導入から4週後にはGFP陽性細胞はわずか2.4%に減少していた。
次に、pCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hULをHDFに導入して作製した、樹立直後のiPSクローン18種類を用いて、外来遺伝子の存在の有無を確認した。
0.2 mLチューブにゲノム回収バッファー(TaKaRa Ex Taq用バッファーに167 μg/mL proteinase Kを加えたもの)を5 μL分注しておき、遺伝子導入から30日目の個々のiPS細胞のコロニーを物理的にディッシュから剥してこのチューブに回収した。回収後、チューブを55℃で3時間インキュベートして細胞ライセートを得た。H2Oを10 μL加えたのちに95℃で3分間加熱してProteinase Kを失活させた。これをそのままリアルタイムPCRの鋳型として用いた。PCRにはSYBR Premix EX Taq II (宝酒造) を使用した。エピソーマルベクターのコピー数を計算するためにEBNA-1用のPCRプライマー対を、また、細胞数を見積もるために内在のFBXO15遺伝子座用の別のプライマー対をデザインした。
EBNA-1プライマー(エピソーマルベクターの検出用)
EBNA-183F ATC AGG GCC AAG ACA TAG AGA TG(配列番号39)
EBNA-243R GCC AAT GCA ACT TGG ACG TT(配列番号40)
Fbx15プライマー(内在性アレルの検出用)
hFbx15-2F GCC AGG AGG TCT TCG CTG TA(配列番号41)
hFbx15-2R AAT GCA CGG CTA GGG TCA AA(配列番号42)
Fbx15の増幅量から反応液中の細胞数を求め、これでEBNA-1の増幅量を補正することで、1×104細胞あたりのエピソーマルベクターのコピー数を算出した。結果を図19に示す。18クローンの平均値は1×104細胞あたりわずか20.3±13.7コピーであったことから、遺伝子導入30日経過後は極めて僅かの外来遺伝子(エピソーマルベクター)しか残っていないことが明らかとなった。
次に、細胞を継代培養する以外は同じ実験を行った。
1× Ex Taqバッファー (Takara) および167 μg/ml プロテイナーゼKからなる新鮮な細胞溶解液を調製した。樹立したiPS細胞を解析するために、60 mmディッシュ中で培養した細胞を、CTK処理によるフィーダー細胞除去後に、セルスクレイパーを用いて集めた。該細胞をチューブに入れて遠心した後、細胞ペレットを200 μlの溶解液で溶解した。55℃で3時間インキュベーションした後、95℃でプロテイナーゼKを失活させ、ライセートを定量的PCR解析に用いた。増幅標準には、FBXO15とEBNA-1の両方のアンプリコンを有するpCXLE-hFbx15-cont2プラスミドを用いた。
DP由来epi-iPS細胞の結果を図31Aに示す。トランスフェクションの6日後に、細胞あたり約200コピーのエピソーマルベクターが検出された。対照的に、試験した7クローン中5クローンにおいて、EBNA-1 DNAは全く検出できなかった。残りの2クローンにおいては、それぞれ-0.001および2コピーが検出された(図31A)。後者のクローンは、おそらくプラスミドが染色体中に組み込まれているだろう。
線維芽細胞由来のepi-iPS細胞の結果を図31Bに示す。404C-2および409B-2を含むいくつかのクローンにおいて、エピソーマルベクターは全く検出できなかった。これらのデータから、epi-iPSCクローンの大部分においてエピソーマルベクターが自然消失していることが示された。
【0121】
実施例11
iPSコロニーにおける外来遺伝子の発現の有無の確認
pCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hULをHDFに導入して作製した、樹立後5から9継代目のiPSクローン9種類を用いて、外来遺伝子の発現の有無を確認した。
6-wellプレートの1ウェルのiPS細胞から全RNAを抽出し、このうち1 μgをプライマーdT(20) および逆転写酵素Rever Tra Ace(TOYOBO)を用いてmRNAを相補鎖DNAへと逆転写した。反応物の最終容量は、20 μLであった。この逆転写物にH2Oを60 μL加えた。そのうちの1 μL(12.5 ngのRNA量に相当)を鋳型として、以下の表6に記載した、外来性遺伝子の発現量と内在性遺伝子の発現量を合わせた発現量(トータル発現量)測定用、および外来性遺伝子発現量測定用のプライマーセットを用いて、SYBR Green IIを指標として導入遺伝子ごとに定量PCRを行った。増幅のための条件は以下の通りであった。95℃/30秒で1サイクル、次に(94℃/10秒、60℃/10秒、72℃/30秒)を50サイクル。
【0122】
【表6】
【0123】
次に、遺伝子導入に用いたpCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hULを106 copies/μLに調製した溶液およびこの溶液を10倍ずつ系列希釈した溶液を鋳型として同様に定量PCRを行い、各初期化遺伝子について、トータル発現量測定用プライマーセットおよび外来性遺伝子発現量測定用プライマーセットごとの検量線を作成した。この検量線を用いて、各iPS細胞における定量PCRの結果を変換して、全RNA 12.5ngに対する各遺伝子のcopy数を、トータル発現量および外来性遺伝子発現量として算出した。このcopy数を対数で縦軸に表したグラフを図20(Oct3/4)および図21(Sox2、Klf4、L-Myc、Lin28)に示す(灰色のゾーンは検出限界以下)。409B-2クローンや421C-1クローンなど、調べた全ての外来遺伝子の発現がいずれも検出限界以下のクローンが多数認められた。これらの細胞を選択することにより、より安全性の高いiPS細胞が利用できることが示された。
【0124】
実施例12
ヒトEpi-iPS細胞の特性解析
1) マイクロアレイ解析およびCGHアレイ解析
pCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hULを皮膚由来線維芽細胞(TIGおよびHDF)に導入して樹立したヒトiPS細胞、ヒトES細胞、および導入に用いたTIGおよびHDFの遺伝子発現パターンに違いがあるかどうか調べるために、DNAマイクロアレイ解析を行った。解析はCell, 131, 861-872 (2007) に記載の手法にて行った。各細胞間の相関係数を表7に示す。
【0125】
【表7】
【0126】
各遺伝子間の発現量の相違に基づきクラスタリング解析を行った結果を図22に、またScatter plot解析を行った結果を図23に示す。エピソーマルベクターにより樹立したiPS細胞は、ES細胞と良く似た発現パターンを示したことから、ES細胞に近いiPS細胞であることが明らかとなった。
また、CGHアレイ解析を行った結果を図24に示す。導入に用いたTIG細胞と比較した結果、特に大きな異常は認められなかった。また、幾つかについてはGバンド染色による核型解析を行ったが、異常は認められなかった。
なお、以上の実験には以下のアレイとスキャナを用いて行った。
・アレイ ヒトGE用:G4112F Whole Human Genome Microarray Kit, 4 x 44K(Agilent)
・アレイ ヒトCGH用:G4426B#14950 Human Genome CGH Microarray Kit 4x44K(Agilent)
・スキャナ:DNA Microarray Scanner, Model G2539A
2) RT-PCR解析
Epi-iPS細胞におけるOCT3/4、SOX2、NANOGおよびESG1等の多能性幹細胞マーカーの発現を調べた。DP由来epi-iPS細胞の結果を図32Aに示し、線維芽細胞由来epi-iPS細胞の結果を図32Bに示す。RT-PCR解析の結果、epi-iPSCクローンはこれらの遺伝子を、hESCやレトロウイルスにより誘導されたiPSCクローンに匹敵するレベルで発現していることが明らかとなった。
3) バイサルファイトシークエンス解析
フィーダー細胞を除去した後、iPS細胞からゲノミックDNAを抽出し、以前に記載したようにして解析した (K.Takahashi et al, Cell 131, 861 (Nov.30, 2007))。
NANOGのプロモーター領域中のCpG部位のDNAメチル化レベルは親HDFおよびDP細胞では高かったが、epi-iPSおよびES細胞では低かった(図33)。
4) テラトーマ形成
インビボでのepi-iPSCの分化能を調べた。CTK溶液を用いて細胞を集め、遠心した。細胞ペレットをDMEM/F12に再懸濁した。コンフルエントな60 mmディッシュから細胞の半分をSCIDマウス(CREA, 日本)の精巣に注射した。注射から8〜12週間後に腫瘍を切除し、PBS中で4% パラホルムアルデヒドを用いて固定した。パラフィン包埋した組織を切片化し、ヘマトキシリンおよびエオシンで染色した。結果を図34に示す。組織学的試験から、これらの腫瘍はテラトーマであり、神経上皮、軟骨、筋肉および腸管様上皮を含む、三胚葉系列の全ての組織を含んでいることが確認された(図34A, B)。
5) インビトロ分化
BMPアンタゴニストとアクチビン/Nodal阻害剤による二重SMAD阻害 (M.Eiraku et al, Cell Stem Cell 3, 519 (2008)) と組み合わせた胚様体様の凝集体の無血清培養(SFEB)法を用いて、ドーパミン産生ニューロンへのインビトロ指向性分化を実施した。
ドーパミン産生ニューロンの誘導のために、以下の分化培地を調製した: 5% Knockout Serum Replacement (KSR; Invitrogen)、2 mM グルタミン、0.1 mM 非必須アミノ酸および0.1 mM 2-メルカプトエタノールを補充したDMEM/F12。Accumax (Invitrogen) 中でiPS細胞を単一細胞に解離させ、96-ウェル低細胞接着プレート (LIPIDURE-COAT PLATE A-U96, NOF Corporation)を用いて、10 μM Y-27632、2 μM ドルソモルフィンおよび10 μM SB431542 (Sigma) を補充した分化培地中に、9000細胞/150 μl/ウェルの密度で再凝集させた。培養5日後に、細胞凝集体を100 ng/ml FGF-8および20 ng/ml Wnt1で刺激した。8日目から、200 ng/ml SHHを培地に加えた。12日目に、凝集した細胞を回収して、200 ng/ml SHHを含むNB培地 (B27サプリメントを含有するNeurobasal培地; Invitrogen) 中、ポリオルニチン/ラミニンでコーティングした60 mmディッシュに移した。その後15日目に、培地を1 ng/ml FGF-20および12.5 ng/ml bFGFを含むNB培地に変更した。22日目に細胞を集め、2 ng/ml GDNF、20 ng/ml BDNF、 400 μM dbcAMPおよび200 μM アスコルビン酸を補充したNB培地中、8-ウェルチャンバープレート上に、ウェルあたり2 × 105個の密度で播種した。29日目に、分化した細胞を免疫染色分析のために固定した。ネスチン (Chemicon)、Ki67 (Novocastra)、Pax6 (Covance)、βIII-チューブリン (Covance Research Products)、チロシンヒドロキシラーゼ(Chemicon)、MAP2ab (Sigma) およびVAMT-2 (PelFreeze) に対する1次抗体を用いた。Alexa488、Alexa594またはCy5を結合した2次抗体を適切に用いた。核を可視化するために、200 ng/mlの4’,6’-ジアミジノ-2-フェニルインドール (DAPI)を最後の洗浄の際に加えた。
結果を図35に示す。大部分の細胞が未熟な神経マーカーであるネスチンを発現していた(図35B-A)。まだ増殖中でKi67陽性の細胞もあった(図35B-B,C,D)。未熟な神経細胞集団はPax6陽性であったのに対し、より成熟した細胞集団は、SHHやFGF8等の誘導因子で処理した後、ドーパミン産生ニューロンのマーカーであるチロシンヒドロキシラーゼ(TH)を発現した(図35B-E, 35B-F)。また、TH陽性細胞は神経マーカーのTuj1およびMAP2ab、並びにドーパミントランスポーターであるVMAT2と共局在していた (図35A-E,F,G, 35B-G,H,I.J,K)。従って、epi-iPSCはドーパミン産生ニューロンに分化する能力を有している。Y3混合物(pCXLE-hOct4、pCXLE-hSK、pCXLE-hUL)を用いて樹立したepi-iPSCについても、同様の解析(RT-PCR、コピー数分析、核型分析およびテラトーマ形成)を行った。その結果、Y4混合物(pCXLE-hOct4-shp53、pCXLE-hSK、pCXLE-hUL)を用いて樹立したepi-iPSCに比して遜色ないepi-iPSCであることが示された。各epi-iPSCクローンの解析リストを表8に示す。
【0127】
【表8】
【0128】
実施例13
異種成分不含(Xeno-free)条件下でのiPS細胞の樹立および培養
エピソーマルベクターを用いて樹立したiPS細胞を再生医療分野で利用するためには、Xeno-free条件下でのiPS細胞の樹立および維持が重要なポイントである。そこで以下の検討を行った。
1)歯髄幹細胞株のフィーダー細胞としての使用
遺伝子導入に用いたヒト皮膚由来線維芽細胞(HDF)をフィーダー細胞として用いた場合でも、ヒトiPS細胞を樹立・維持できることが知られている(Takahashi et al., PLoSone, vol.4, issue 12, e8067 (2009))。自己のフィーダー細胞を用いることで、Xeno-free条件下でのiPS細胞の維持が可能となるため、臨床応用における意義は大きい。そこで歯髄幹細胞株DP74をフィーダー細胞として用いることができるかどうかを検討した。iPS細胞はpCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hULをHDFに導入して樹立したヒトiPS細胞を用い、培養液は通常の霊長類ES細胞用培地(リプロセル)に4 ng/mLとなる様にbFGF (Wako) を加えた培地(以下リプロセル培地)を用いた。1継代後の細胞の写真を図25に示す。フィーダー細胞としてMSTOを用いた場合と比較して遜色なくES様形態が維持された。
2)Xeno-free条件下でのiPS細胞の維持(1)
Xeno-free培地であるTeSR2(Stemcell Technologies)およびXeno-freeのプレートコーティング剤であるCELLstart (GIBCO) を用い(feeder細胞は使わず)、完全なXeno-free条件下でiPS細胞が維持できるかどうかを検討した。iPS細胞はpCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hULをHDFに導入して樹立したヒトiPS細胞を用いた。1継代後の細胞の写真を図26に示す。リプロセル培地では細胞形態が変化し始めているが、TeSR2を用いた場合では未分化な形態を維持していた。
3)Xeno-free条件下でのiPS細胞の維持(2)
Xeno-free培地であるTeSR2(Stemcell Technologies)およびKSR-XF(KnockOut DMEM(Invitrogen)に15% KnockOut SR XenoFree(Invitrogen), 2 mM GlutaMAX-I(Invitrogen), 0.1 mM 非必須アミノ酸(Invitrogen), 0.1 mM 2-mercaptoethanol(Invitrogen)および8 ng/mL bFGF(Wako)を加えたもの)を用い、フィーダー細胞としてDP74株を用いてiPS細胞が維持できるかどうかを検討した。iPS細胞はpCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hULをHDFに導入して樹立したヒトiPS細胞を用いた。1継代後の細胞の写真を図27に示す。いずれのXeno-free培地を用いた場合も、ES様形態が維持された。
【0129】
実施例14
Xeno-free条件下での遺伝子導入
iPS細胞樹立後だけでなく、樹立前、すなわち体細胞に初期化遺伝子を導入する段階も、同様にXeno-free条件が可能かどうか検討を行った。
予備検討として、Microporatorを用いてDP74にpCXLE-EGFPを導入した。Xeno-free培地としてStemPro MSC-SFM (Invitrogen)を用いた。
DP74はStemPro MSC-SFM (Invitrogen) を培養液として使用し、CELLstartでコートした100 mm 培養ディッシュで37℃、5% CO2の条件で培養し、維持した。pCXLE-EGFP導入時には、培地を除き、PBS 5 mLを加えて細胞を洗浄した。PBSを除いた後、Xeno-freeの細胞剥離用液であるTripLE select (Invitrogen) を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらStemPro MSC-SFMを加えて懸濁し、15 mL遠心チューブに回収した。800 rpmで5分間遠心して上清を除いた。再びStemPro MSC-SFMに懸濁した後、6x105細胞を1.5 mLチューブに回収した。800 rpmで5分間遠心して上清を除いた。pCXLE-EGFP(3 μg)をMicroporatorを用いて細胞に導入した。導入時の条件は100 μLチップを用い、1850 V, 20 ms, 1回のパルスか、または1200 V, 30 ms, 2回のパルスで行った。導入後の細胞はあらかじめCELLstartでコートして、StemPro MSC SFMを3 mL入れておいた6 well培養プレート (Falcon) へ移し、37℃、5% CO2の条件で7日間培養した。結果を図28に示す。Xeno-free培地(StemPro MSC-SFM)を用いた場合は、1850 V, 20ms, 1回の条件では導入効率が低かった。しかし1200 V, 30 ms, 2回の条件に変更することにより、細胞生存率と導入効率は上昇した。従って、この条件で以降の遺伝子導入を行うことにした。
初期化に用いるプラスミドとして、pCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hULの3種類のプラスミドを用いた。実験には歯髄幹細胞DP74株を使用した。DP74株はStemPro MSC-SFMを培養液として使用し、CELLstartでコートした100 mm 培養ディッシュで37℃、5% CO2の条件で培養し、維持した。プラスミド導入時には、培地を除き、PBS 5 mLを加えて細胞を洗浄した。PBSを除いた後、TripLE select (Invitrogen) を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらStemPro MSC-SFMを加えて懸濁し、15 mL遠心チューブに回収した。800 rpmで5分間遠心して上清を除いた。再びStemPro MSC-SFMに懸濁した後、6x105細胞を1.5 mLチューブに回収した。800 rpmで5分間遠心して上清を除いた。3種のプラスミド(各1 μg、合計3 μg)をMicroporatorを用いて細胞に導入した。導入時の条件は100 μLチップを用い、1200 V, 30 ms, 2回のパルスで行った。導入後の細胞はあらかじめCELLstartでコートして、StemPro MSC-SFMを3 mL入れておいた6 well培養プレート (Falcon) へ移し、37℃、5% CO2の条件で7日間培養した。そののち培地を除き、PBS 2 mLを加えて細胞を洗浄した。PBSを除いた後、TripLE select (Invitrogen) を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらStemPro MSC-SFMを加えて懸濁し、15 mL遠心チューブに回収した。800 rpmで5分間遠心して上清を除いた。再びStemPro MSC-SFMに懸濁した後、1x105の細胞を、表9に示す各種条件で100 mm dishに蒔いた。翌日に表9に示す培地に交換し、以後2日おきに培地交換を続けた。導入後26日目に出現したヒトES細胞様のコロニーの写真を図29に示す。
【0130】
【表9】
【0131】
いずれの培地およびフィーダー細胞を用いた場合も、iPSコロニーが樹立できた。特に表9中のNo. 4〜No. 6は、いずれもXeno-freeの条件であることから、エピソーマルベクターを用いたvirus-freeの条件下、かつ遺伝子導入時から完全なXeno-freeの条件下で、歯髄幹細胞からiPS細胞を樹立及び維持できることが明らかになった。
【0132】
実施例15
ヒト末梢血単核細胞からのiPS細胞の樹立
3 μgの発現プラスミド (1 μgのpCXLE-hOct4-shp53、pCXLE-hSKおよびpCXLE-hUL) を、Nucleofector (Lonza) とHuman T cell kitを製造者の指示に従って用い、新たに単離した末梢血単核細胞 (5.0x106) に、エレクトロポレーションにより導入した。プログラムV-24の条件を用いた。6時間後、トランスフェクトした血液細胞をサイトカイン (IL-6、sIL-6R、SCF、TPO、Flit3/4リガンドおよびIL-2) および抗CD3/CD28抗体で刺激した。2日後に、細胞をMEFフィーダーで覆った6-ウェルプレート上に播種した。次いで、図36aに示すように、播種後培養培地を、IL-6、sIL-6R、SCF、TPO、Flit3/4リガンドおよびIL-2を補充したX-vivo 10から、hES培地に徐々に変更した。29日目にコロニーの写真を撮影した(図36b)。平坦なhESC様の形態を有するヒトiPSコロニーを樹立することができた。
【0133】
本発明を好ましい態様を強調して説明してきたが、好ましい態様が変更され得ることは当業者にとって自明であろう。本発明は、本発明が本明細書に詳細に記載された以外の方法で実施され得ることを意図する。したがって、本発明は添付の「請求の範囲」の精神および範囲に包含されるすべての変更を含むものである。
ここで述べられた特許および特許出願明細書を含む全ての刊行物に記載された内容は、ここに引用されたことによって、その全てが明示されたと同程度に本明細書に組み込まれるものである。
本願は、米国仮特許出願第61/232,402号および第61/307,306号を基礎としており、それらの内容は、ここで参照することにより本願に組み込まれるものである。
【特許請求の範囲】
【請求項1】
(a) Oct3/4もしくはそれをコードする核酸、(b) Klf4もしくはそれをコードする核酸、および(c) Sox2もしくはそれをコードする核酸、並びに(d1) L-Mycもしくはそれをコードする核酸および/または(d2) p53の機能阻害物質を、体細胞に接触させることを含む、iPS細胞の製造方法。
【請求項2】
さらに(e) Lin28もしくはLin28bまたはそれをコードする核酸を体細胞に接触させることを含む、請求項1記載の方法。
【請求項3】
p53の機能阻害物質が、p53に対するsiRNA、shRNAおよびそれらをコードするDNAからなる群より選択される核酸である、請求項1または2記載の方法。
【請求項4】
(a) Oct3/4をコードする核酸、(b) Klf4をコードする核酸、(c) Sox2をコードする核酸、(d1) L-Mycをコードする核酸および/または(d2) p53に対するshRNAをコードする核酸、並びに(e) Lin28もしくはLin28bをコードする核酸を、体細胞に導入することを含む、請求項2または3記載の方法。
【請求項5】
前記の(a)、(b)、(c)、(d1)および(e)からなる群より選択される少なくとも1つの核酸がエピソーマルベクターの形態で導入される、請求項4記載の方法。
【請求項6】
(d2) p53に対するshRNAをコードする核酸が細胞内で自律複製できないプラスミドベクターの形態で導入される、請求項4または5記載の方法。
【請求項7】
エピソーマルベクターが、50%以上の頻度で、5継代までにiPS細胞から脱落する自己消失型ベクターである、請求項5または6記載の方法。
【請求項8】
エピソーマルベクターが、前記の核酸の複製に必要なベクター要素の5’側および3’側にloxP配列を同方向に配置したものである、請求項5〜7のいずれか1項に記載の方法。
【請求項9】
細胞をCreリコンビナーゼで処理する工程を含まない、請求項8記載の方法。
【請求項10】
体細胞がヒト由来である、請求項1〜9のいずれか1項に記載の方法。
【請求項11】
前記の(a)、(b)、(c)並びに(d1)および/または(d2)、あるいはさらに(e)の因子を体細胞に接触させる時点から、iPS細胞が樹立されるまでの間、細胞を非ヒト動物由来の成分の非存在下で培養することを含む、請求項10記載の方法。
【請求項12】
ヒト細胞をフィーダー細胞として用いるか、フィーダー細胞を用いないことを特徴とする、請求項11記載の方法。
【請求項13】
フィーダー細胞が体細胞と同一個体に由来する、請求項12記載の方法。
【請求項14】
(a) Oct3/4をコードする核酸、(b) Klf4をコードする核酸、(c) Sox2をコードする核酸、(d1) L-Mycをコードする核酸および/または(d2) p53に対するshRNAをコードする核酸、並びに(e) Lin28もしくはLin28bまたはそれをコードする核酸を含有してなる、iPS細胞誘導促進剤。
【請求項15】
前記の(a)、(b)、(c)、(d1)および(e)からなる群より選択される少なくとも1つの核酸がエピソーマルベクターの形態である、請求項14記載の剤。
【請求項16】
(d2) p53に対するshRNAをコードする核酸が細胞内で自律複製できないプラスミドベクターの形態である、請求項14または15記載の剤。
【請求項17】
エピソーマルベクターが、50%以上の頻度で、5継代までにiPS細胞から脱落する自己消失型ベクターである、請求項15または16記載の剤。
【請求項18】
エピソーマルベクターが、前記の各核酸の複製に必要なベクター要素の5’側および3’側にloxP配列を同方向に配置したものである、請求項15〜17のいずれか1項に記載の剤。
【請求項19】
請求項5〜9のいずれか1項に記載の方法により得られる、前記の各核酸がゲノムに組み込まれることなく除去されたiPS細胞。
【請求項20】
ヒト由来である、請求項19記載のiPS細胞。
【請求項21】
請求項11〜13のいずれか1項に記載の方法により得られる、非ヒト動物由来の成分の混入のないヒトiPS細胞。
【請求項22】
体細胞の製造における、請求項19〜21のいずれか1項に記載のiPS細胞の使用。
【請求項23】
体細胞の製造における細胞ソースとしての、請求項19〜21のいずれか1項に記載のiPS細胞。
【請求項1】
(a) Oct3/4もしくはそれをコードする核酸、(b) Klf4もしくはそれをコードする核酸、および(c) Sox2もしくはそれをコードする核酸、並びに(d1) L-Mycもしくはそれをコードする核酸および/または(d2) p53の機能阻害物質を、体細胞に接触させることを含む、iPS細胞の製造方法。
【請求項2】
さらに(e) Lin28もしくはLin28bまたはそれをコードする核酸を体細胞に接触させることを含む、請求項1記載の方法。
【請求項3】
p53の機能阻害物質が、p53に対するsiRNA、shRNAおよびそれらをコードするDNAからなる群より選択される核酸である、請求項1または2記載の方法。
【請求項4】
(a) Oct3/4をコードする核酸、(b) Klf4をコードする核酸、(c) Sox2をコードする核酸、(d1) L-Mycをコードする核酸および/または(d2) p53に対するshRNAをコードする核酸、並びに(e) Lin28もしくはLin28bをコードする核酸を、体細胞に導入することを含む、請求項2または3記載の方法。
【請求項5】
前記の(a)、(b)、(c)、(d1)および(e)からなる群より選択される少なくとも1つの核酸がエピソーマルベクターの形態で導入される、請求項4記載の方法。
【請求項6】
(d2) p53に対するshRNAをコードする核酸が細胞内で自律複製できないプラスミドベクターの形態で導入される、請求項4または5記載の方法。
【請求項7】
エピソーマルベクターが、50%以上の頻度で、5継代までにiPS細胞から脱落する自己消失型ベクターである、請求項5または6記載の方法。
【請求項8】
エピソーマルベクターが、前記の核酸の複製に必要なベクター要素の5’側および3’側にloxP配列を同方向に配置したものである、請求項5〜7のいずれか1項に記載の方法。
【請求項9】
細胞をCreリコンビナーゼで処理する工程を含まない、請求項8記載の方法。
【請求項10】
体細胞がヒト由来である、請求項1〜9のいずれか1項に記載の方法。
【請求項11】
前記の(a)、(b)、(c)並びに(d1)および/または(d2)、あるいはさらに(e)の因子を体細胞に接触させる時点から、iPS細胞が樹立されるまでの間、細胞を非ヒト動物由来の成分の非存在下で培養することを含む、請求項10記載の方法。
【請求項12】
ヒト細胞をフィーダー細胞として用いるか、フィーダー細胞を用いないことを特徴とする、請求項11記載の方法。
【請求項13】
フィーダー細胞が体細胞と同一個体に由来する、請求項12記載の方法。
【請求項14】
(a) Oct3/4をコードする核酸、(b) Klf4をコードする核酸、(c) Sox2をコードする核酸、(d1) L-Mycをコードする核酸および/または(d2) p53に対するshRNAをコードする核酸、並びに(e) Lin28もしくはLin28bまたはそれをコードする核酸を含有してなる、iPS細胞誘導促進剤。
【請求項15】
前記の(a)、(b)、(c)、(d1)および(e)からなる群より選択される少なくとも1つの核酸がエピソーマルベクターの形態である、請求項14記載の剤。
【請求項16】
(d2) p53に対するshRNAをコードする核酸が細胞内で自律複製できないプラスミドベクターの形態である、請求項14または15記載の剤。
【請求項17】
エピソーマルベクターが、50%以上の頻度で、5継代までにiPS細胞から脱落する自己消失型ベクターである、請求項15または16記載の剤。
【請求項18】
エピソーマルベクターが、前記の各核酸の複製に必要なベクター要素の5’側および3’側にloxP配列を同方向に配置したものである、請求項15〜17のいずれか1項に記載の剤。
【請求項19】
請求項5〜9のいずれか1項に記載の方法により得られる、前記の各核酸がゲノムに組み込まれることなく除去されたiPS細胞。
【請求項20】
ヒト由来である、請求項19記載のiPS細胞。
【請求項21】
請求項11〜13のいずれか1項に記載の方法により得られる、非ヒト動物由来の成分の混入のないヒトiPS細胞。
【請求項22】
体細胞の製造における、請求項19〜21のいずれか1項に記載のiPS細胞の使用。
【請求項23】
体細胞の製造における細胞ソースとしての、請求項19〜21のいずれか1項に記載のiPS細胞。
【図2】

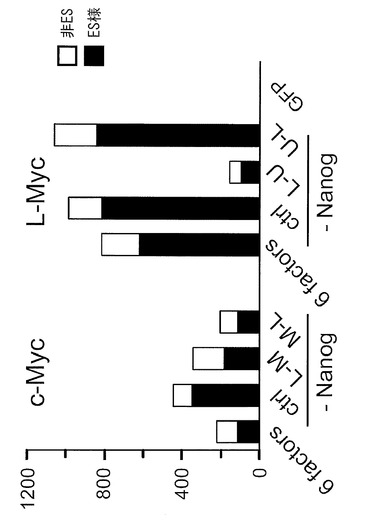
【図6】
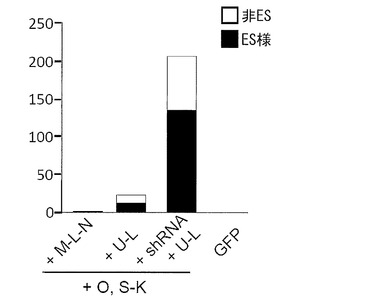
【図9】
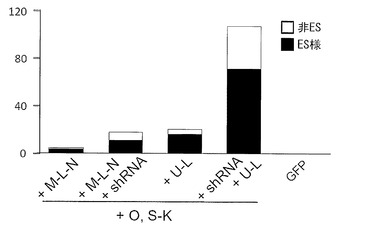
【図10】
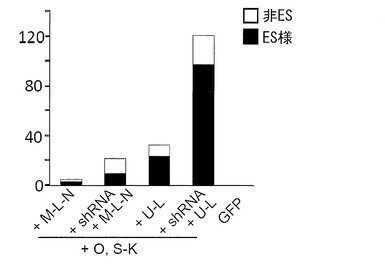
【図11】
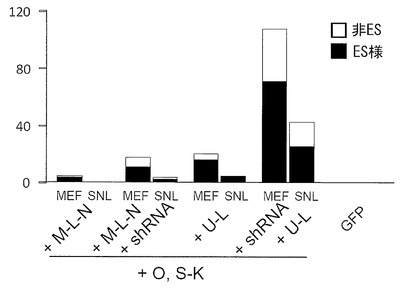
【図14】
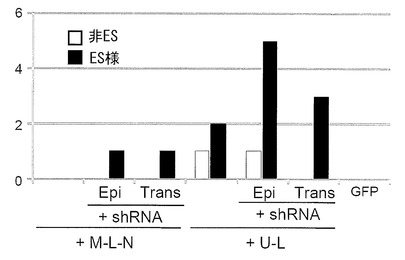
【図16】
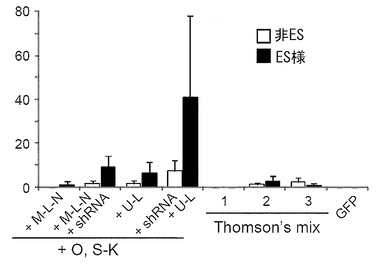
【図17】
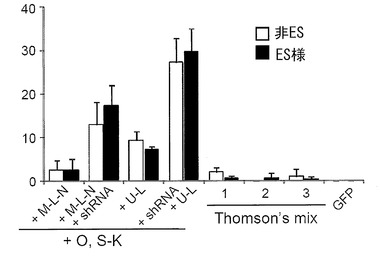
【図19】
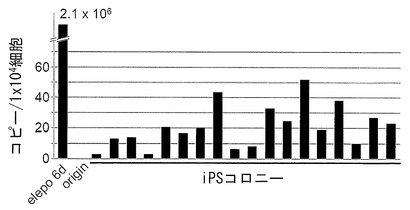
【図30】
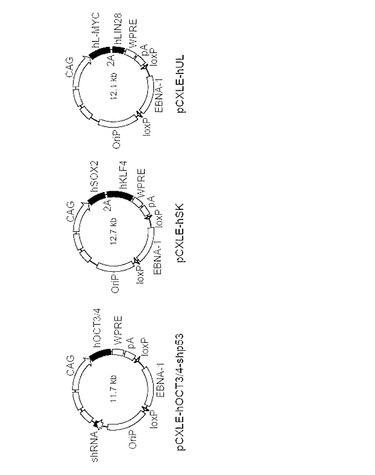
【図31】
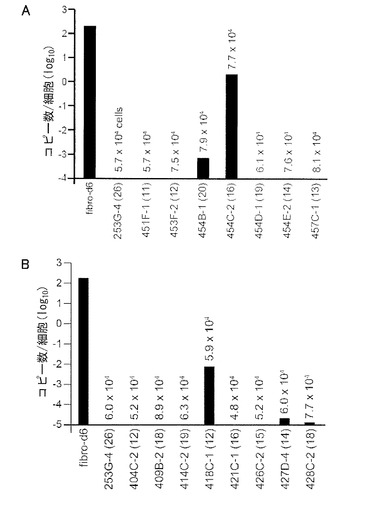
【図33】
【図1】
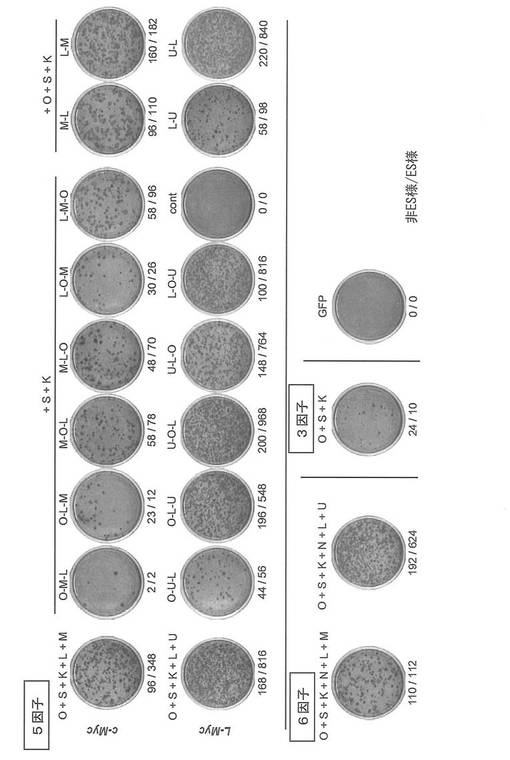
【図3】
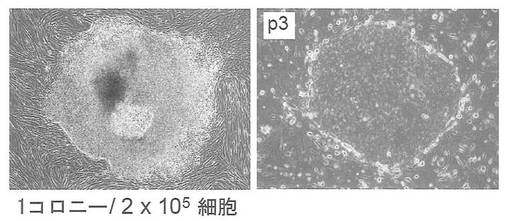
【図4】
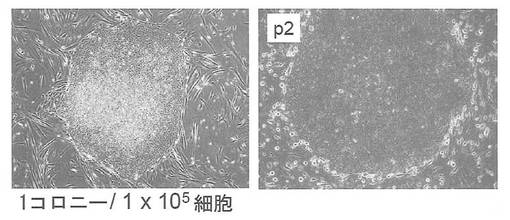
【図5】
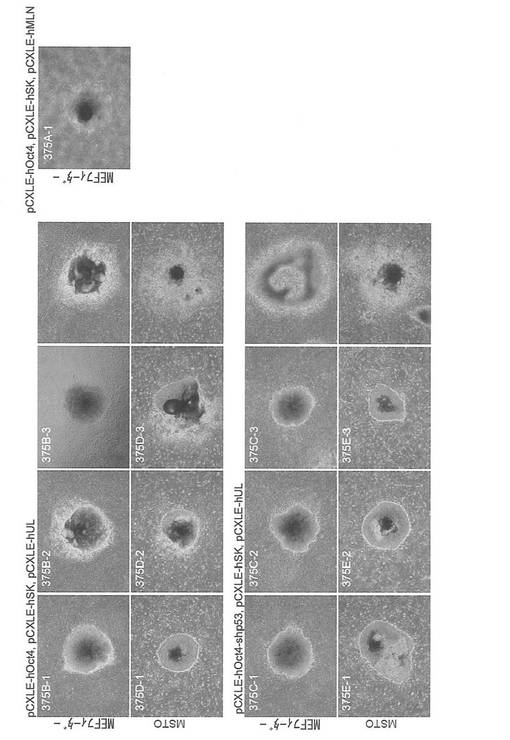
【図7】
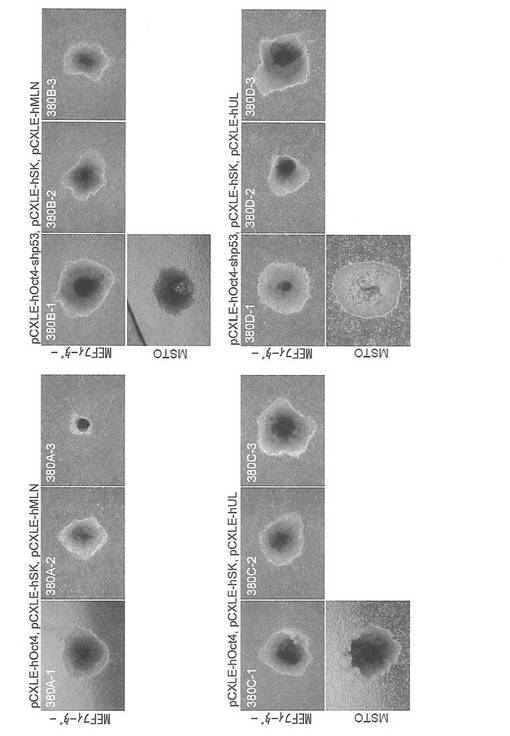
【図8】
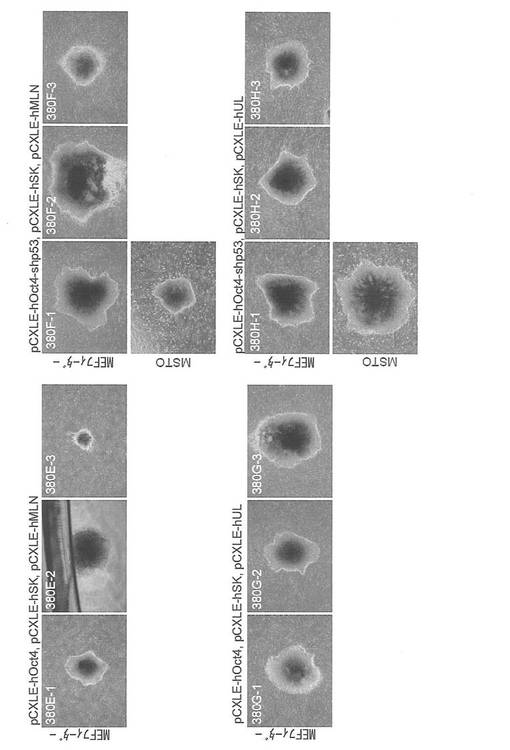
【図12】
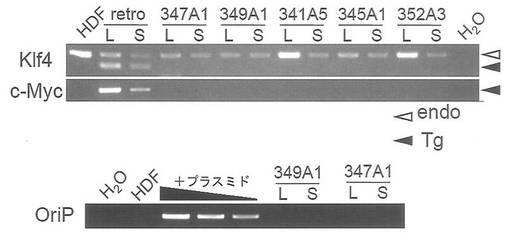
【図13】
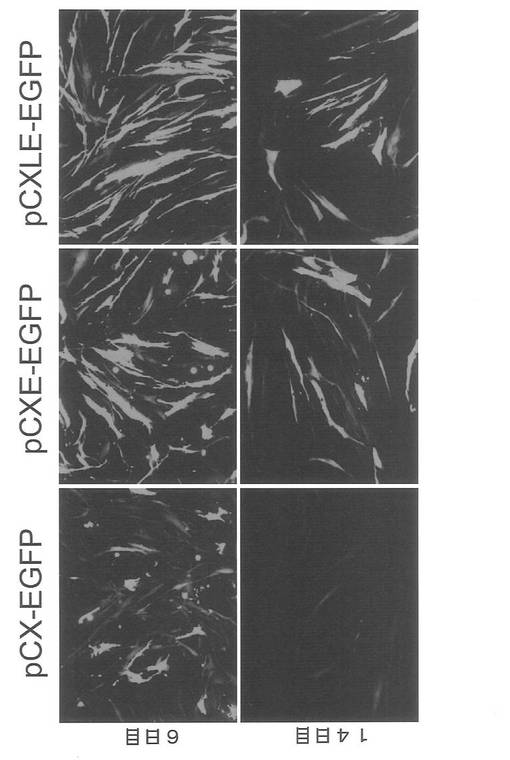
【図15】
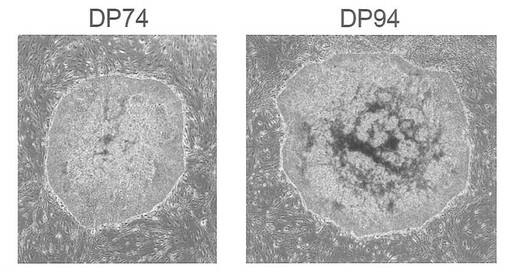
【図18】
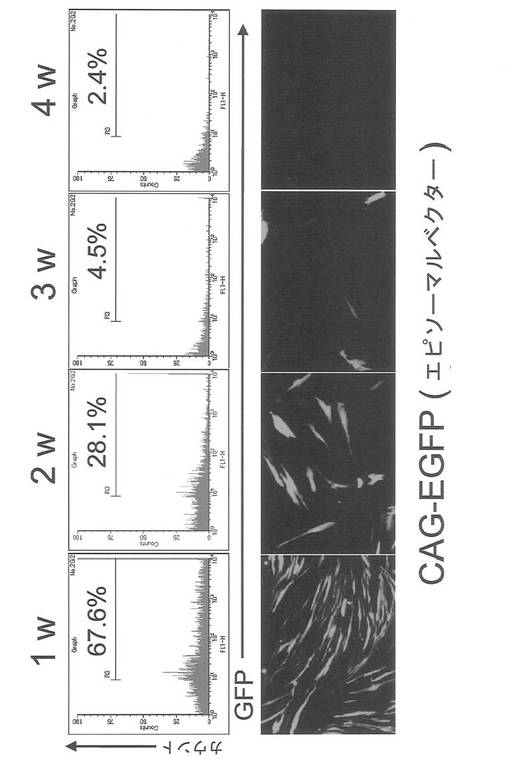
【図20】
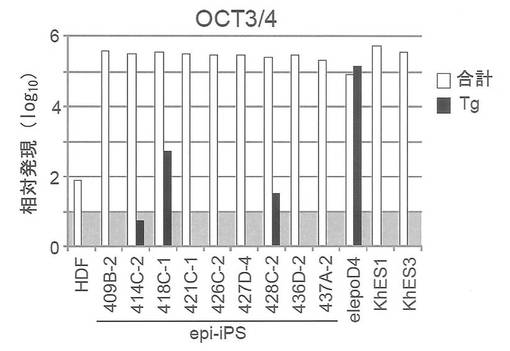
【図21】
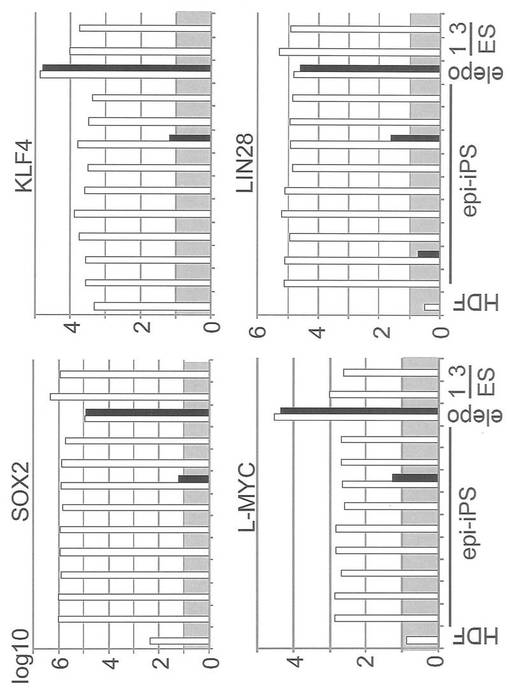
【図22】

【図23】
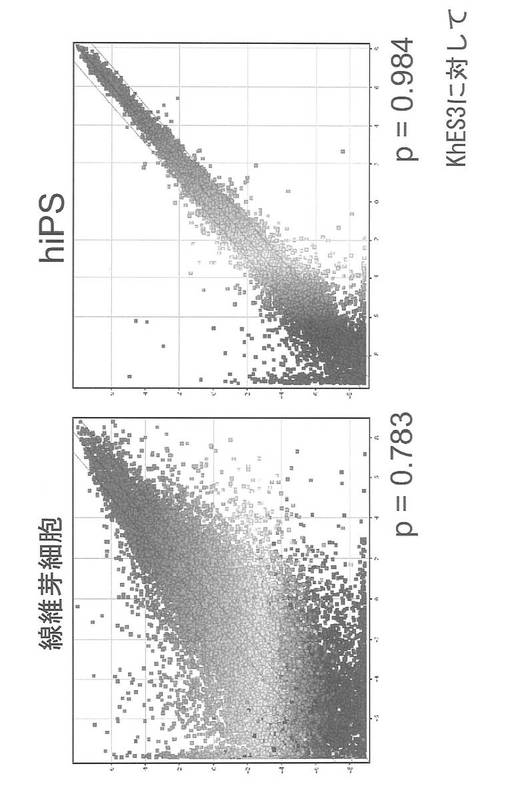
【図24】

【図25】
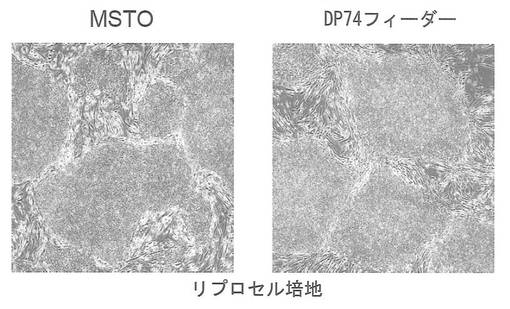
【図26】
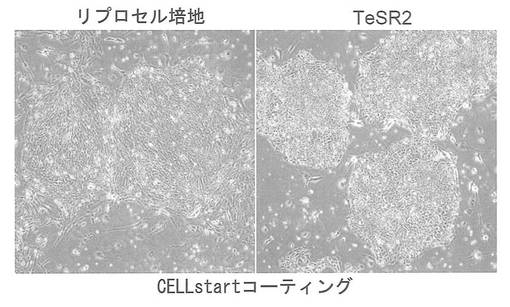
【図27】
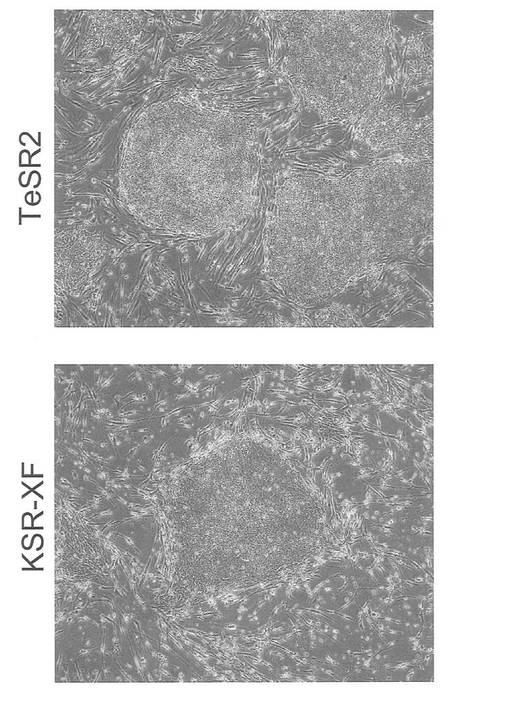
【図28】
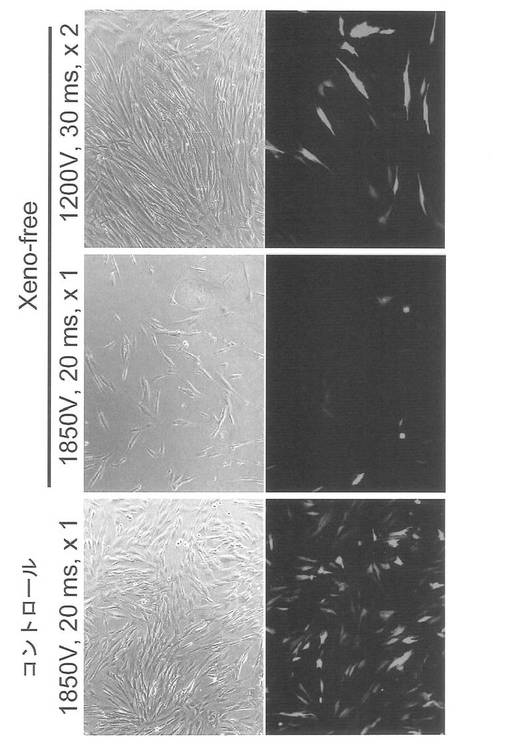
【図29】
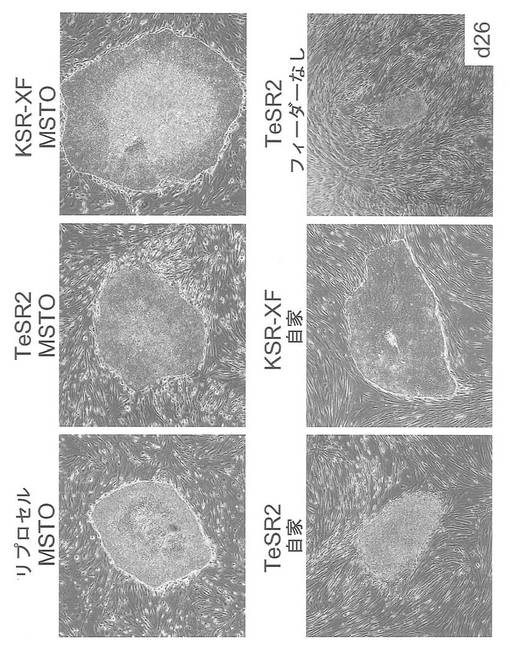
【図32】

【図34A】
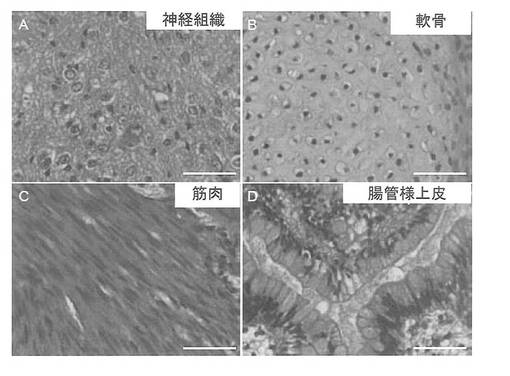
【図34B】
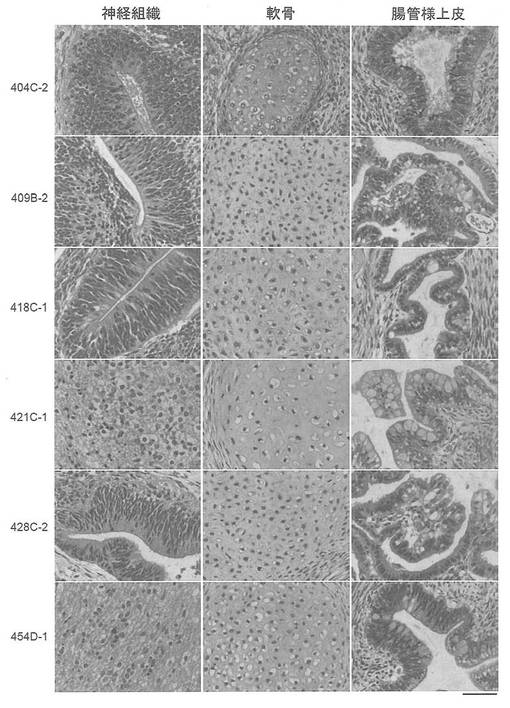
【図35A】
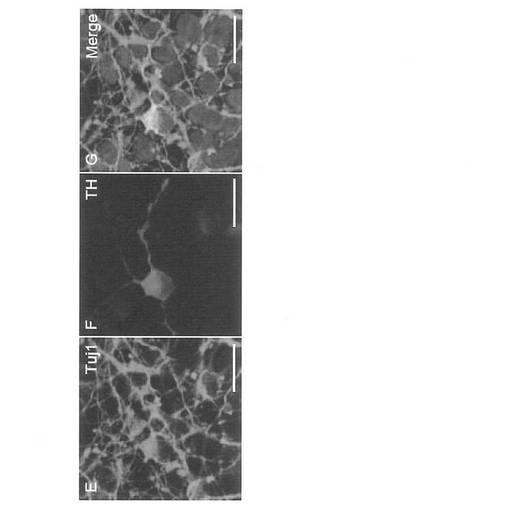
【図35B】
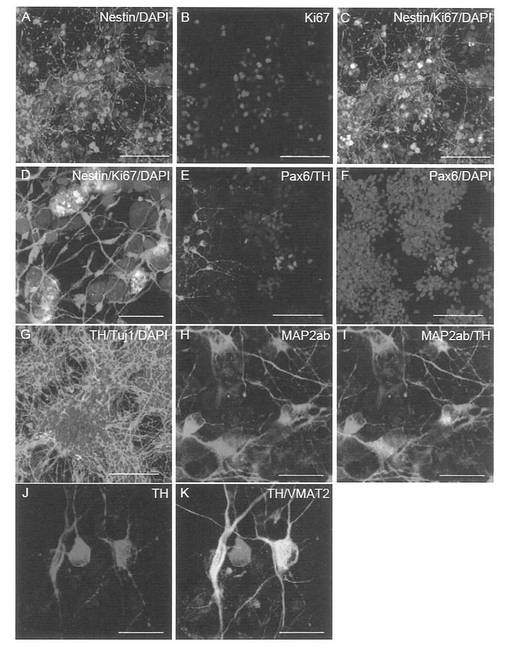
【図36】
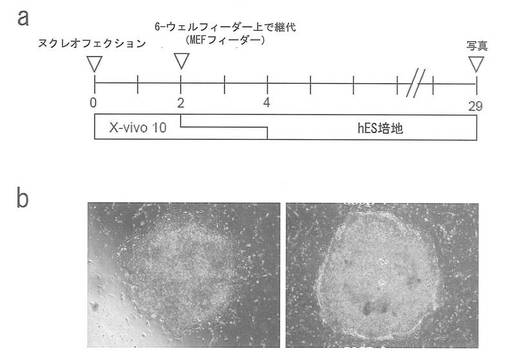
【図6】
【図9】
【図10】
【図11】
【図14】
【図16】
【図17】
【図19】
【図30】
【図31】
【図33】
【図1】
【図3】
【図4】
【図5】
【図7】
【図8】
【図12】
【図13】
【図15】
【図18】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図32】
【図34A】
【図34B】
【図35A】
【図35B】
【図36】
【公表番号】特表2013−501505(P2013−501505A)
【公表日】平成25年1月17日(2013.1.17)
【国際特許分類】
【出願番号】特願2012−523493(P2012−523493)
【出願日】平成22年8月6日(2010.8.6)
【国際出願番号】PCT/JP2010/063733
【国際公開番号】WO2011/016588
【国際公開日】平成23年2月10日(2011.2.10)
【出願人】(504132272)国立大学法人京都大学 (1,269)
【Fターム(参考)】
【公表日】平成25年1月17日(2013.1.17)
【国際特許分類】
【出願日】平成22年8月6日(2010.8.6)
【国際出願番号】PCT/JP2010/063733
【国際公開番号】WO2011/016588
【国際公開日】平成23年2月10日(2011.2.10)
【出願人】(504132272)国立大学法人京都大学 (1,269)
【Fターム(参考)】
[ Back to top ]