
動的遺伝子発現プロファイリング方法及びシステム
【課題】複数の試料において遺伝子発現プロファイルを同時に定量的に検出するための簡単で感受性の高い方法を提供する。
【解決手段】cDNAの合成のため、及び合成したcDNAのその後の増幅のために、試料特異的配列タグを含む試料特異的プライマーを用いること、cDNAのサブセットを選択的に増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、異なって発現された遺伝子の確認のために、試料間において、遺伝子の存在量の水準を比較することからなる。
【解決手段】cDNAの合成のため、及び合成したcDNAのその後の増幅のために、試料特異的配列タグを含む試料特異的プライマーを用いること、cDNAのサブセットを選択的に増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、異なって発現された遺伝子の確認のために、試料間において、遺伝子の存在量の水準を比較することからなる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、転写プロファイリング技術に関する。
【背景技術】
【0002】
ゲノム科学の導入は、薬剤発見の速度の加速に役立ってきた。ゲノム技術は、新規薬剤標的の発見においてその価値を証明した。この領域のさらなる改良は、有用な薬剤のより迅速でより費用効率の高い結果を得る、より効率的なツールを提供する。
【0003】
薬剤発見プロセスは幾つかのステップ:病気に関わる潜在的生化学的標的の確認、活性化合物及びさらなる化学的設計のスクリーニング、前臨床テスト及び最終的臨床試行、を含んでいる。このプロセスの効果はなお完全からは遠く、R&Dプロセスにおいて費やされた金額の約75%が資金的に失敗していると推測される。さらに、産物開発の晩期において失敗が発生し、このプロジェクトに関わる損失がより大きくなる。従って、将来の失敗を初期に取り除いて、全薬剤開発プロセスの費用を大幅に節減する必要がある。すなわち、最初の分子標的の質が、費用効果的薬剤開発のための決定的因子となる。
【0004】
標的を確認し実証するプロセスに有効な見込みのある一つの手段は、転写プロファイリングである。この方法は、特定の状況における遺伝子の発現を比較する:例えば、疾患細胞と正常細胞との間、対照細胞と薬剤処理細胞との間、または処理に反応する細胞と処理に抵抗性の細胞との間で比較する。この手段により生じる情報は、治療により標的とされる特定の遺伝子を直接確認することができ、重要なことに、疾患及び治療に含まれる生化学的経路を明らかにする。簡単に言えば、これは、生化学的標的を提供するのみならず、同時に、これらの標的の質を評定する方法も提供する。さらに、細胞に基づくスクリーニングと組み合わせて、薬剤発見の分野を劇的に変化させるために転写プロファイリングが位置付けられる。歴史的には、有効な薬剤のスクリーニングが、機能的細胞系におけるマーカーとして表現型変化を用いてうまく行われた。例えば、培地における腫瘍細胞の成長をモニターして、抗癌薬を確認した。同様に、抗生物質確認を目指すアッセイにおいて、バクテリア生存能力を用いた。そのようなスクリーンは、典型的に、標的の生化学的経路を予め知ることなく行われた。実際、確認された効果的化合物がそのような経路を明らかにし分子標的を指摘して、その後の次世代薬剤の合理的設計を可能にする。
【0005】
転写プロファイリングの最近のツールを用いて、薬剤の効果を評定するために表現型の変化の代わりに遺伝子発現を利用する新規スクリーニング法を設計することができる。例えば、これらの方法は、米国特許第5,262,311、5,665,547、5,599,672、5,580,726、6,045,988及び5,994,076号並びにLuehrsenら著(1997年、Biotechniques,第22巻:168〜74頁);Liang及びPardee著(1998年、Mol Biotechnol.第10巻:261〜7頁)に記載されている。そのような手段は、表現型スクリーニングが適用不可能であるが所望の転写プロフィールを容易に達成して特定の疾患に結びつけることができる、軽度認識生涯、抑鬱等のような中枢神経系(CNS)疾患の分野における薬剤発見に非常に貴重である。また、確認された効果的化合物は、根元的な分子プロセスを明らかにする。さらに、この方法は、複数の生化学的標的において同時に作用して所望の薬理学的効果を生む現存する薬剤の改良に役立ち得る。そのような場合、転写反応における変化は、複数標的への結合の最適化に基づく選択よりも、薬剤の作用についての優れたマーカーであり得る。
【0006】
本発明の前に、転写プロファイリングの最も進んだ方法は、DNAマイクロアレイを用いる技術に基づき、例えば、Greenberg,2001年,Neurology第57巻:755〜61頁;Wu,2001年,J Pathol.第195巻:53〜65頁;Dhimanら著、2001年,Vaccine,第20巻:22〜30頁;Bierら著、2001年Fresenius J Anal Chem.第371巻:151〜6頁;Millsら著、2001年,Nat Cell Biol.第3巻:E175〜8頁に考察され、米国特許第5,593,839、5,837,832、5,856,101、6,203,989、6,271,957及び6,287,778号に記載されている。DNAマイクロアレイは、試験アレイの表面に付着しているDNA分子への、mRNAの逆転写により得られる標識されたポリヌクレオチド試料のハイブリッド形成を評価することにより、所定の試料における数千の遺伝子の発現を同時に行う方法である。この技術は、転写の変換について価値のある情報を提供するが、完全とは程遠い。
【0007】
最初に、この技術は、マイクロアレイに在る遺伝子のプールに限定される。現在の印刷法は、単一チップ上に10,000〜15,000の遺伝子を乗せることができ、これは、本質的に特定の細胞型において発現される遺伝子の数である。種々の細胞型があるとすれば、特定の細胞型のために特定のアレイを開発する必要がある。理論的に可能であるが、この作業は達成困難である。それは、マイクロアレイ製造前にこれらの細胞において発現される遺伝子プールについての知識が必要だからである。
【0008】
さらに、組織試料における転写の数は、細胞試料における数よりかなり多く、マイクロアレイの現在の性能を超えている。さらに、遺伝子発現の一部の変化は択一的スプライシングから生じ、さらに、これが、評定すべき転写の数をさらに増やす。これらの困難を克服する唯一の可能性は、択一的にスプライスされた遺伝子を含む全ゲノムを覆う複数のアレイを開発することである。この手段は、単一実験の費用を著しく増加させ、大きな生物学的試料、おそらく、合理的に利用できるよりも大きな試料を必要とする。
【0009】
第2に、現在では、DNAマイクロアレイは定量的に正確なデータを提供せず、遺伝子発現において観察される変化は、独立した方法、例えば、定量的PCR(Q−PCR)により確認しなくてはならない。
【0010】
さらに、典型的マイクロアレイ実験は、この方法の再現性に影響を与える幾つかのマニュアルステップを含む。
【0011】
最後に、特に興味深い稀転写の発現は、現在の検出技術を用いて、マイクロアレイにより正確に測定することができない。これらの制限は、転写プロファイリングを行うための別の方法、好ましくは、1)アッセイ前に発現された遺伝子プールの配列の予備知識を必要としないが、それ自体、アッセイ中/後にこの情報を提供する方法;2)発現された転写の水準の定量的変化を測定する方法;3)稀遺伝子の発現を検出し得る方法;及び4)自動化できる方法を開発する必要性を示している。
【0012】
遺伝子発現を定量的に検出するためにキャピラリー電気泳動が用いられていた。Rajevicら著(2001年、Pflugers Arch.442頁(6増刊1):R190〜2頁)は、多くの腫瘍遺伝子の同時の発現の相違を検出するために7対のプライマーを用いることにより腫瘍遺伝子の異なる発現を検出する方法を開示している。センスプライマーを、蛍光染料で5’末端標識した。複数の蛍光RT−PCRの結果を、ABI−PRISM 310 Genetic Analyzerでのキャピラリー電気泳動により分析した。Borsonら(1998年,Biotechniques,第25巻:130〜7頁)は、産物の迅速な分離及び検出のためにキャピラリー電気泳動(CE)に組み合わせた定量的競合的逆転写PCR(QC−RT−PCR)に基づく低存在量mRNA転写体の依存的定量用の手法を記載している。Georgeら(1997年,J Chromatogr B Biomed Sci Appl,第695巻:93〜102頁)は、キャピラリー電気泳動系(ABI310)を、蛍光相違表示が発生するESTパターンの確認に適用することを記載している。Odinら(1999年,J Chromatogr B Biomed Sci Appl,第734巻:47〜53頁)は、PCR増幅cDNAの分離及び定量のために、多色を検出する自動キャピラリーゲル電気泳動を記載している。
【0013】
Omoriら(2000年、Genomics第67巻:140〜5頁)は、オリゴ(dT)またはオリゴ(dU)プライマーを用いて2つの独立した逆転写したcDNA試料における競合的PCRにより、市場で購入したα−グロビンmRNAの量を測定し比較している。オリゴ(dT)またはオリゴ(dU)プライマーは、3’オリゴ(dT)またはオリゴ(dU)配列及び5’共通配列を共有する。さらに、各試料についてオリゴ(dT)またはオリゴ(dU)プライマーは、3’オリゴ(dT)またはオリゴ(dU)配列と5’共通配列との間に独自の29個のヌクレオチド配列も含む。第1cDNA鎖の合成後、独自標識で標識された共通配列に相補的なプライマー及び遺伝子特異的プライマーを用いて、PCRを行ってcDNAを増幅する。増幅されたPCR産物を、次に、蛍光スキャナーの検出板上に点在させることにより分析する。
【発明の概要】
【発明が解決しようとする課題】
【0014】
当該分野において、複数の試料において遺伝子発現プロファイルを同時に定量的に検出するための簡単で感受性の高い方法が必要とされている。
【課題を解決するための手段】
【0015】
本発明は、二つまたはそれ以上の試料の発現プロファイリングの方法及び組成物を提供する。
【0016】
本発明は、2種以上の試料の遺伝子発現プロフィールを比較する方法であって、
(a)試料特異的配列タグを含む第1のオリゴヌクレオチドプライマーを用いて第1の試料から複数の第1cDNA鎖を合成し、試料特異的配列タグはその5’末端においてGCに富みその3’末端においてAtに富むこと、
(b)少なくともcDNAのサブセットを選択的に増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(c)一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、存在量は第1試料中の一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(d)第1試料中の一つまたは二つ以上の遺伝子の発現プロフィールを第2試料中の一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違は2つの試料中の一つまたは二つ以上の遺伝子の異なる発現を示すこと
を含んでなる方法を提供する。
【0017】
本発明は、二つまたはそれ以上の試料の遺伝子発現プロフィールを比較する方法であって、
(a)試料特異的配列タグを含む第1のオリゴヌクレオチドプライマーを用いて第1の試料から複数の第1cDNA鎖を合成し、第1オリゴヌクレオチドプライマーは少なくとも一つの変性ヌクレオチドを含むこと、
(b)少なくともcDNAのサブセットを選択的に増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(c)一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、存在量は第1試料中の一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(d)第1試料中の一つまたは二つ以上の遺伝子の発現プロフィールを第2試料中の一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違は2つの試料中の一つまたは二つ以上の遺伝子の異なる発現を示すこと
を含んでなる方法も提供する。
【0018】
本発明は、二つまたはそれ以上の試料の遺伝子発現プロフィールを比較する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、第1試料から複数の第1cDNA鎖を合成し、試料特異的配列タグが、従来のヌクレオチドを超える、もう一つの人工ヌクレオチドとの塩基対選択性を示す少なくとも一つの人工ヌクレオチドを含むこと、
(b)少なくともcDNAのサブセットを選択的に増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(c)一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、存在量が第1試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(d)第1試料中の一つまたは二つ以上の遺伝子の発現プロフィールを第2試料中の一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、二つの試料における一つまたは二つ以上の遺伝子の異なる発現を示すこと
を含んでなる方法も提供する。
【0019】
本発明は、二つまたはそれ以上の試料の遺伝子発現プロフィールを比較する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、第1試料から複数の第1cDNA鎖を合成し、試料特異的配列タグがその5’末端においてGCに富みその3’末端においてATに富むこと、
(b)第1任意配列タグを含む第2オリゴヌクレオチドプライマーを用いて、第1cDNA鎖に相補的な一つまたは二つ以上の第2cDNA鎖を選択的に合成すること、
(c)一つまたは二つ以上の第2cDNA鎖を増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(d)一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、存在量が第1試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(e)第1試料中の一つまたは二つ以上の遺伝子の発現プロフィールを第2試料中の一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、二つの試料における一つまたは二つ以上の遺伝子の異なる発現を示すこと
を含んでなる方法も提供する。
【0020】
本発明は、さらに、二つまたはそれ以上の試料の遺伝子発現プロフィールを比較する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、第1試料から複数の第1cDNA鎖を合成し、第1オリゴヌクレオチドプライマーが少なくとも一つの変性ヌクレオチドを含むこと、
(b)第1任意配列タグを含む第2オリゴヌクレオチドプライマーを用いて、第1cDNA鎖に相補的な一つまたは二つ以上の第2cDNA鎖を選択的に合成すること、
(c)一つまたは二つ以上の第2cDNA鎖を増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(d)一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、存在量が第1試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(e)第1試料中の一つまたは二つ以上の遺伝子の発現プロフィールを第2試料中の一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、二つの試料における一つまたは二つ以上の遺伝子の異なる発現を示すこと
を含んでなる方法を提供する。
【0021】
本発明は、さらに、二つまたはそれ以上の試料の遺伝子発現プロフィールを比較する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、第1試料から複数の第1cDNA鎖を合成し、試料特異的配列タグが、従来のヌクレオチドを超える、もう一つの人工ヌクレオチドとの塩基対選択性を示す少なくとも一つの人工ヌクレオチドを含むこと、
(b)第1任意配列タグを含む第2オリゴヌクレオチドプライマーを用いて、第1cDNA鎖に相補的な一つまたは二つ以上の第2cDNA鎖を選択的に合成すること、
(c)一つまたは二つ以上の第2cDNA鎖を増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(d)一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、存在量が第1試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(e)第1試料中の一つまたは二つ以上の遺伝子の発現プロフィールを第2試料中の一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、二つの試料における一つまたは二つ以上の遺伝子の異なる発現を示すこと
を含んでなる方法を提供する。
【0022】
本発明は、試料における一つまたは二つ以上の遺伝子の発現を調節するモジュレーターを確認する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、試料をモジュレーターに接触させる前に、複数の第1cDNA鎖を合成し、試料特異的配列タグがその5’末端においてGCに富みその3’末端においてATに富むこと、
(b)少なくともcDNAのサブセットを選択的に増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(c)一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、存在量が試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(d)モジュレーターとの接触前の試料中の一つまたは二つ以上の遺伝子の発現プロフィールを、モジュレーター接触後の試料中の一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、試料における一つまたは二つ以上の遺伝子発現を調節するモジュレーターを示すこと
を含んでなる方法を提供する。
【0023】
本発明は、試料における一つまたは二つ以上の遺伝子の発現を調節するモジュレーターを確認する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、試料をモジュレーターに接触させる前に、複数の第1cDNA鎖を合成し、第1オリゴヌクレオチドプライマーが少なくとも一つの変性ヌクレオチドを含むこと、
(b)少なくともcDNAのサブセットを選択的に増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(c)一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、存在量が試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(d)モジュレーターとの接触前の試料中の一つまたは二つ以上の遺伝子の発現プロフィールを、モジュレーター接触後の試料中の一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、試料における一つまたは二つ以上の遺伝子発現を調節するモジュレーターを示すこと
を含んでなる方法も提供する。
【0024】
本発明は、さらに、試料における一つまたは二つ以上の遺伝子の発現を調節するモジュレーターを確認する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、試料をモジュレーターに接触させる前に、複数の第1cDNA鎖を合成し、試料特異的配列タグがその5’末端においてGCに富みその3’末端においてATに富むこと、
(b)第1任意配列タグを含む第2オリゴヌクレオチドプライマーを用いて、一つまたは二つ以上の第2cDNA鎖を合成すること、
(c)第2cDNA鎖を増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(d)一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、存在量が試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(e)モジュレーターとの接触前の試料中の一つまたは二つ以上の遺伝子の発現プロフィールを、モジュレーター接触後の試料中の一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、試料における一つまたは二つ以上の遺伝子発現を調節するモジュレーターを示すこと
を含んでなる方法を提供する。
【0025】
本発明は、さらに、試料における一つまたは二つ以上の遺伝子の発現を調節するモジュレーターを確認する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、試料をモジュレーターに接触させる前に、複数の第1cDNA鎖を合成し、第1オリゴヌクレオチドプライマーが少なくとも一つの変性ヌクレオチドを含むこと、
(b)第1任意配列タグを含む第2オリゴヌクレオチドプライマーを用いて、一つまたは二つ以上の第2cDNA鎖を合成すること、
(c)第2cDNA鎖を増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(d)一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、存在量が試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(e)モジュレーターとの接触前の試料中の一つまたは二つ以上の遺伝子の発現プロフィールを、モジュレーター接触後の試料中の一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、試料における一つまたは二つ以上の遺伝子発現を調節するモジュレーターを示すこと
を含んでなる方法を提供する。
【0026】
好ましい態様において、本方法のステップ(a)が、二つまたはそれ以上の試料供給源からのRNAを第1cDNA鎖に逆転写することを含み、そのcDNAはそれらの供給源に従って異なって標識される。
【0027】
好ましくは、第1cDNA鎖が、第1試料由来の全RNAまたはmRNAを用いる逆転写により合成される。
【0028】
好ましくは、第2オリゴヌクレオチドプライマー中の第2配列が、遺伝子ファミリー特異的である。
【0029】
より好ましくは、第2オリゴヌクレオチドプライマー中の第2配列が、蛋白ファミリーに特異的なペプチドをコードする配列である。
【0030】
さらにより好ましくは、第2配列が、特異的蛋白ファミリー用のサイン配列モチーフをコードする配列を含む。
【0031】
好ましくは、蛋白ファミリーは、受容体チロシンキナーゼ、G蛋白共役受容体、7回膜通過受容体、イオンチャンネル、サイトカイン受容体、腫瘍マーカー、MAPKカスケードキナーゼ、転写因子、GTPアーゼ、ATPアーゼ、及び発生蛋白マーカーからなる群より選択される。
【0032】
好ましくは、第1オリゴヌクレオチドプライマーの配列特異的配列タグを含む第3オリゴヌクレオチドプライマーを増幅のために用いて一つまたは二つ以上の試料特異的増幅産物を産生する。
【0033】
また好ましくは、二つまたはそれ以上の試料の少なくとも一つが、正常試料、疾患試料、所定の発生段階または状態における試料、所定の処理段階または状態の前の試料、所定の処理段階または状態の後の試料及び所定の培養段階または状態における試料からなる群より誘導される。
【0034】
なお好ましくは、二つまたはそれ以上の試料の少なくとも一つが、動物、器官、組織型及び細胞型からなる群より誘導される。
【0035】
一つの態様において、少なくとも一つの試料が正常個体から誘導され、少なくとももう一つの試料が疾患個体から誘導される。
【0036】
もう一つの態様において、少なくとも一つの試料が個体の発生段階から誘導され、少なくとももう一つの試料が同じ個体の異なる発生段階から誘導される。
【0037】
さらにもう一つの態様において、少なくとも一つの試料が個体の疾患段階から誘導され、少なくとももう一つの試料が同じ個体の異なる疾患段階から誘導される。
【0038】
なおもう一つの態様において、少なくとも一つの試料が個体の疾患処理の段階から誘導され、少なくとももう一つの試料が同じ個体の異なる疾患処理の異なる段階から誘導される。
【0039】
もう一つの態様において、少なくとも一つの試料が環境因子に露出された個体から誘導され、少なくとももう一つの試料が同じ環境因子に露出されなかった個体または異なる濃度で環境因子に露出された個体から誘導される。
【0040】
一つの態様において、一つまたは二つ以上の第2cDNA鎖がPCRで増幅されて一つまたは二つ以上の増幅PCR産物が生じる。
【0041】
好ましくは、一つまたは二つ以上の増幅産物を、増幅中、所定の時間またはサイクル間隔でサンプリングする。
【0042】
一つの態様において、一つまたは二つ以上の増幅産物を、増幅の各サイクル後にサンプリングする。
【0043】
もう一つの態様において、一つまたは二つ以上の増幅産物を、一つまたは二つ以上の予定のサイクル後、例えば、2、5、10、25、30または45サイクル後にサンプリングする。
【0044】
もう一つの態様において、一つまたは二つ以上の増幅産物を、反応混合物の1%〜40%(v/v)を引き出す、好ましくは反応混合物の1%〜30%(v/v)を引き出すことによりサンプリングする。
【0045】
もう一つの態様において、反応混合物に、各サンプリングの後に、dNTP、プライマー、必要な試薬及びDNAポリメラーゼを出発反応混合物と同じ濃度で含む等体積の混合物を補給する。
【0046】
好ましくは、各サンプリングした増幅産物について存在量を検出する。
【0047】
好ましくは、本方法は、さらに、一つまたは二つ以上の増幅産物の存在量の検出前に、一つまたは二つ以上の増幅産物を分離することを含む。
【0048】
もう一つの態様において、一つまたは二つ以上の増幅産物を分離し、その存在量をクロマトグラフィーにより検出する。
【0049】
もう一つの態様において、一つまたは二つ以上の増幅産物を分離し、その存在量を質量分析により検出する。
【0050】
さらにもう一つの態様において、一つまたは二つ以上の増幅産物を分離し、その存在量を電気泳動により検出する。
【0051】
好ましくは、一つまたは二つ以上の増幅産物を分離し、その存在量をキャピラリー電気泳動により検出する。
【0052】
一つの態様において、第1オリゴヌクレオチドプライマー中の試料特異的配列は、15〜30ヌクレオチド長、より好ましくは20〜24ヌクレオチド長である。
【0053】
好ましい態様において、第1オリゴヌクレオチドプライマーは、さらに、5’オリゴ(dT)nVN3’の配列を含み、nは少なくとも5であり、VがdATP、dGTPまたはdCTPであり、NがdTTP(またはdUTP)、dATP、dGTPまたはdCTPである。
【0054】
好ましくは、5’オリゴ(dT)nVN3’においてnは12〜16である。
【0055】
また好ましくは、第1オリゴヌクレオチドプライマーにおいて、試料特異的配列タグは、オリゴ(dT)nVNの5’に配される。
【0056】
好ましくは、本方法の第2オリゴヌクレオチドプライマーが、さらに、第1cDNA鎖のサブセットに相補的な第2配列を含み、それにより一つまたは二つ以上の第2cDNA鎖を合成させる。
【0057】
より好ましくは、第2オリゴヌクレオチドプライマーにおいて、第2配列が第1任意配列の3’に配される。
【0058】
また、より好ましくは、第2オリゴヌクレオチドが、さらに、第1及び第2配列の間に(Z)mの配列を含み、Zが、A、T、GまたはCのいずれかと塩基対を形成することがでるヌクレオチドであり、mが少なくとも2である。好ましくは、mは4である。
【0059】
一つの態様において、第2オリゴヌクレオチドプライマー中の第2配列は5〜10ヌクレオチド長である。
【0060】
もう一つの態様において、第2オリゴヌクレオチドプライマー中の第2配列は6〜7ヌクレオチド長である。
【0061】
好ましくは、第2オリゴヌクレオチドプライマー中の第2配列はパリンドローム配列である。
【0062】
一つの態様において、第2オリゴヌクレオチドプライマー中の第1任意配列は15〜30ヌクレオチド長、好ましくは20ヌクレオチド長である。
【0063】
もう一つの態様において、第2オリゴヌクレオチドプライマー中の第1任意配列がA−Tに富む領域とG−Cに富む。
【0064】
好ましくは、G−Cに富む領域が、A−Tに富む領域の5’に配される。
【0065】
好ましくは、用いられる第2オリゴヌクレオチドプライマーが、比較すべき二つまたはそれ以上の試料について同じである。
【0066】
好ましい態様において、本方法の増幅ステップは、さらに、第2オリゴヌクレオチドプライマーの第1任意配列タグを含む第4オリゴヌクレオチドプライマーを用いることを含む。
【0067】
好ましくは、用いられる第4オリゴヌクレオチドプライマーが、比較すべき二つまたはそれ以上の試料について同じである。
【0068】
一つの態様において、第1cDNA鎖が、固体支持体に付着することなく溶液中で合成される。
【0069】
もう一つの態様において、第1cDNA鎖が、固体支持体に付着されて合成される。
【0070】
好ましくは、固体支持体が微粒子または反応管の内壁である。
【0071】
好ましい態様において、本方法が、さらに、一つまたは二つ以上の第2cDNA鎖の増幅前に、複数の第1cDNA鎖から一つまたは二つ以上の第2cDNA鎖を分離することを含む。
【0072】
一つの態様において、本方法で用いられる第3オリゴヌクレオチドプライマーが検出可能な標識に結合される。
【0073】
好ましくは、検出可能な標識が、蛍光標識、放射性標識、比色標識、磁気標識及び酵素標識からなる群より選択される。
【0074】
より好ましくは、検出可能な標識が、蛍光標識である。
【0075】
好ましい態様において、二つまたはそれ以上の試料の各々について用いられる第3オリゴヌクレオチドプライマーが試料特異的標識で標識される。
【0076】
本方法による一つの態様において、一つまたは二つ以上の遺伝子の発現プロフィールの相違が、二つまたはそれ以上の試料間の遺伝子からの増幅産物上の試料特異的検出可能標識の比により測定する。
【0077】
好ましくは、この方法が、さらに、増幅プロット(増幅サイクル数の関数としての信号強度)を発生させること、各PCRフラグメントの信号強度に基づいて一つまたは二つ以上の遺伝子の各々についての増幅の閾値サイクル数(Ct)を計算することを含む。特定の遺伝子の機能的に異なる発現は、2以上のサイクルの二つ(またはそれ以上)の試料におけるこの遺伝子についての閾値サイクル数(Ct)の相違の値として決められる。さらに、閾値サイクル数は、各遺伝子についての複製数を導き出し、二つまたはそれ以上の試料における遺伝子についての複製数の比により発現の相違を測定するために用いられる。
【0078】
この方法は、各増幅遺伝子について増幅サイクルの数の関数[サイクル数の関数としての信号強度の導関数、d(信号強度)/d(サイクル数)]としての信号強度変化の率のプロットを発生させることも含む。一つの試料からの各増幅遺伝子についてのd(信号強度)/d(サイクル数)の最大値に相当するサイクル数として決められる択一的閾値サイクル(aCt)を、もう一つの試料からの同じ遺伝子についてのaCtと比較する。二つまたはそれ以上の試料中の同じ遺伝子についてのaCt値間の一つのサイクルの相違は、択一的機能的相違発現として定義される。
【0079】
また好ましくは、本方法は、さらに、機能的相違発現または択一的機能的相違発現を示す一つまたは二つ以上の遺伝子に相当するPCRフラグメントを集めること、及び一つまたは二つ以上の遺伝子の配列を確認することを含む。
【0080】
一つの態様において、異なって発現される一つまたは二つ以上の遺伝子の配列同一性は、DNA塩基配列決定により確認される。
【0081】
一つの態様において、本方法は、さらに、第1増幅からの一つまたは二つ以上の増幅産物を用いて一つまたは二つ以上の第2増幅産物を発生させ、一つまたは二つ以上の第2増幅産物の存在量を検出する第2増幅反応を含む。
【0082】
好ましくは、本方法の増幅ステップは、PCRにより行われる。
【0083】
本発明の方法は、さらに、第2増幅反応としてネステッドPCR反応を含む。
【0084】
本発明は、遺伝子発現の水準を検出するための組成物であって、第1オリゴヌクレオチドプライマーを含んでなり、第1オリゴヌクレオチドプライマーが試料特異的配列タグを含み、第1オリゴヌクレオチドプライマーが少なくとも一つの変性ヌクレオチドを含む組成物を提供する。
【0085】
一つの態様において、第1オリゴヌクレオチドプライマーが、[5’−(特異的配列タグ)20−24T12−16AN−3’,5’−(特異的配列タグ)20−24T12−16CN−3’及び5’−(特異的配列タグ)20−24T12−16GN−3’]を含むプライマーの混合物として提供される。
【0086】
本発明は、遺伝子発現の水準を検出するための組成物であって、第1オリゴヌクレオチドプライマーを含んでなり、第1オリゴヌクレオチドプライマーが試料特異的配列タグを含み、試料特異的配列タグがその5’末端においてGCに富みその3’末端においてATに富む組成物も提供する。
【0087】
好ましくは、本組成物は、さらに、第2オリゴヌクレオチドプライマーを含む。
【0088】
より好ましくは、第2オリゴヌクレオチドプライマーは、第1任意配列タグを含む。
【0089】
好ましくは、第2プライマーは、さらに、第1cDNA鎖の配列に相補的な第2配列を含む。
【0090】
本組成物は、さらに、第1オリゴヌクレオチドプライマーの配列特異的配列タグを含む第3オリゴヌクレオチドプライマーを含み得る。
【0091】
本組成物は、さらに、第1任意配列タグを含む第4オリゴヌクレオチドプライマーを含み得る。
【0092】
本組成物は、さらに、逆転写酵素、DNAポリメラーゼ、逆転写酵素用の反応緩衝剤、DNAポリメラーゼ用の反応緩衝剤、及びdNTPからなる群より選択される一つまたは二つ以上の成分を含む。
【0093】
本発明は、遺伝子発現の水準を検出するためのキットであって、第1オリゴヌクレオチドプライマーを含んでなり、第1オリゴヌクレオチドプライマーが試料特異的配列タグを含み、第1オリゴヌクレオチドプライマーが少なくとも一つの変性ヌクレオチドを含み、また、その包装材料を含んでなるキットを提供する。
【0094】
本発明は、遺伝子発現の水準を検出するためのキットであって、第1オリゴヌクレオチドプライマーを含んでなり、第1オリゴヌクレオチドプライマーが試料特異的配列タグを含み、配列特異的配列タグがその5’末端においてGCに富みその3’末端においてATに富み、また、その包装材料を含んでなるキットも提供する。
【0095】
本発明のキットは、第2オリゴヌクレオチドプライマーも含み得る。
【0096】
好ましくは、第2オリゴヌクレオチドプライマーは、第1任意配列タグを含む。
【0097】
本発明のキットは、さらに、第1オリゴヌクレオチドプライマーの配列特異的配列タグを含む第3オリゴヌクレオチドプライマーを含み得る。
【0098】
本発明のキットは、なおさらに、第1任意配列タグを含む第4オリゴヌクレオチドプライマーを含み得る。
【0099】
好ましくは、第2プライマーは、さらに、第1cDNA鎖の配列に相補的な第2配列を含む。
【0100】
また好ましくは、本発明のキットは、さらに、逆転写酵素、DNAポリメラーゼ、逆転写酵素用の反応緩衝剤、DNAポリメラーゼ用の反応緩衝剤、及びdNTPからなる群より選択される一つまたは二つ以上の成分を含む。
【図面の簡単な説明】
【0101】
【図1】本発明の一つの形態による、試料特異的配列タグを用いるオリゴ−dTプライマーを用いる二つの試料からのmRNAの逆転写を示す図である。逆転写の結果となる各試料からのmRNAは試料特異タグで標識される。
【図2】本発明の一つの形態による、遺伝子ファミリー特異的配列を含むプライマーを用いる選択された遺伝子の第2cDNA鎖合成を示す図である。
【図3】本発明の一つの形態による、試料特異的タグを用いる増幅産物をPCR増幅により得ることを示す図である。
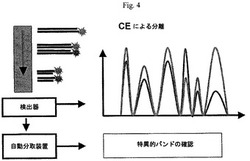
【図4】本発明の一つの形態による、PCR産物の分離及び分析を示す図である。
【図5】本発明の一つの形態による、PCR産物蓄積の典型的曲線を示す図である。異なる試料間の相違が最も容易に検出されるサイクルの範囲が狭いことが明らかである。a)遺伝子発現(Ct)の定量的測定は、信号強度が選択された閾値限界(通常、ベースラインの標準偏差の10倍に設定)を超える点に相当するサイクル数として定義される。機能的相違発現(ΔCt)は、二つのPCRフラグメントについてのCt値の相違として定義される。b)サイクル数の関数としてのd(信号強度)/d(サイクル数)のプロットに基づく閾値サイクルの択一的決定。閾値サイクル(aCt)の択一的決定は、d(信号強度)/d(サイクル数)の最大値に対応するサイクル数として定義される。閾値数と同様に、aCtを用いて、各遺伝子についての絶対複製数を決めることができる(log(複製数)=AaCt+B)。択一的機能的相違発現(ΔaCt)は、二つのPCRフラグメントについてのaCt値の相違として定義される。
【図6】本発明の一つの形態による、正規化PCR増幅スキームを示す図である。
【図7】本発明の好ましい形態による、転写プロファイリングの方法を示す図である。
【発明を実施するための形態】
【0102】
(定義)
ここで用いられる「試料」という用語は、その天然環境から単離されポリヌクレオチドを含む生物学的材料を意味する。本発明による「試料」は、精製または単離されたポリヌクレオチドからなる、またはこれは、組織試料のような生物学的試料、生物学的流体試料もしくはポリヌクレオチドを含む細胞試料を含み得る。生物学的流体試料は、血液、血漿、唾液、尿、脳脊髄液、洗浄液及び白血球泳動試料を含む。本発明の試料は、ポリヌクレオチドを含むいかなる植物、動物、バクテリアまたはウイルス材料であってもよい。
【0103】
ここで定義される「組織」は、生体において特別の機能を果たす細胞の集合体である。ここで用いられる「組織」という用語は、特別の生理学的領域からの細胞材料を意味する。特別の組織における細胞は、幾つかの異なる細胞型を含み得る。この非制限的例は、さらに、神経細胞及びグリア細胞、並びにキャピラリー内皮細胞及び血液細胞を含む脳組織である。「組織」という用語は、生体において独立または非接着性細胞、例えば、免疫細胞または血液細胞として通常存在し得る組織マイクロアレイ上の下位位置に含まれる複数の細胞を含むことも意図される。この用語は、さらに、細胞系及び、(例えば、特定組織型の生体分子特性の発現故に)特定の組織型を表す現在存在する細胞材料の他の供給源を含むことを意図する。
【0104】
ここで用いられる「複数」という用語は、2を超えることを意味する。本発明によれば、複数は3以上、100以上、1,000以上、例えば、試料中の全てのmRNAに相当するcDNAの数までであり得る。
【0105】
ここで用いられる「異なるタイプの組織」という用語は、好ましくは異なる器官からの組織、または、少なくとも、同じ器官中の解剖学的及び組織学的に異なる部位からの組織を意味する。
【0106】
ここで用いられる「細胞試料」は、他の細胞から分離される細胞を含む点において組織試料から区別される。
【0107】
ここで用いられる「個体」は単一の生物であり、ヒト、動物、植物、多細胞及び単細胞生物を含む。
【0108】
ここで用いられる「試料特異的配列」という用語は、特異的試料供給源から誘導されるポリヌクレオチド分子を確認するために用いられるポリヌクレオチド配列を意味する。本発明の「試料特異的配列」という用語は、単離または合成されたポリヌクレオチドの試料供給源を意味し、一つの試料の単離または合成されたポリヌクレオチドを他の試料のポリヌクレオチドから区別する。従って、試料特異的配列は、確認することができる独自の特徴を有する。試料特異的配列の独自の特徴は、特定の配列同一性または特定の配列長さであり得る。特定の配列同一性を用いる場合、一つの試料特異的配列は、少なくとも一つのヌクレオチド、例えば、少なくとも、2、3、4、5、10、15、20またはそれ以上、60個までのヌクレオチドにおいて、もう一つの試料特異的配列から異なるべきである。特定の配列長さが用いられる場合、一つの試料特異的配列は、少なくとも一つのヌクレオチド、例えば、少なくとも、2、3、4、5、10、15、20またはそれ以上、50個までのヌクレオチドにおいて、もう一つの試料特異的配列から長さが異なるべきである。
【0109】
ここで用いられる「特定試料由来のポリヌクレオチド分子」は、特定試料から単離されたポリヌクレオチドであり得る、または、例えば、逆転写またはポリメラーゼ連鎖反応(PCR)、リガーゼ連鎖反応(LCR)、ポリヌクレオチド特異的増幅(NSBA)、鎖移動増幅(SDA)及び当該分野で知られている任意の他の技術により特定試料から合成されたポリヌクレオチドであり得る。
【0110】
ここで用いられる「異なる試料」という用語は、異なる供給源からの同じ組織または試料を含んでも含まなくても、本発明の方法により比較される二つまたはそれ以上の試料を意味する。異なる供給源は、疾患及び正常供給源;異なる細胞型、異なる組織または器官型;異なる個体;異なる環境に曝される試料;異なる発生段階;異なる疾患段階;及び異なる処理段階であり得るが、これらに限定されない。
【0111】
ここで用いられる「増幅産物」という用語は、ヌクレオチド配列において鋳型ポリヌクレオチド配列及びその相補的配列に相当する、特定のポリヌクレオチド配列及び/またはその相補的配列の一部の複製であるポリヌクレオチドを意味する。本発明による「増幅産物」はDNAまたはRNAであり得、二本鎖または一本鎖であり得る。
【0112】
ここで用いられる「合成」及び「増幅」という用語は交換可能に用いられ、特定のポリヌクレオチド配列の複製を発生するためまたは特定のポリヌクレオチド配列の複製数または量を増加させるための反応を意味する。これは、限定はされないが、試験管内方法であるポリメラーゼ連鎖反応(PCR)、リガーゼ連鎖反応(LCR)、ポリヌクレオチド特異的増幅(NSBA)、及び当該分野で知られている任意の他の技術により達成することができる。例えば、ポリヌクレオチド増幅は、任意の特定ポリヌクレオチド配列を製造するためにポリメラーゼ及び対のポリヌクレオチドプライマー、すなわち標的ポリヌクレオチド配列または標的ポリヌクレオチドを、最初に存在する量より多い量で用いる方法である。
【0113】
ここで用いられる「選択的」という用語は、ポリヌクレオチドの増幅または合成の場合、相補的配列を含むポリヌクレオチドの選択群を増幅または合成することを意味する。選択は、増幅または合成反応において特定のオリゴヌクレオチドプライマーを用いることにより達成される。例えば、第2cDNA鎖の群は、遺伝子ファミリー特異的配列に相補的な配列(例えば、後でここに記載の第2配列)を含む第2オリゴヌクレオチドを用いて選択的合成することができる。
【0114】
ここで用いられる「少なくともサブセット」という用語は、反応における全てのポリヌクレオチドまたは増幅または合成反応における全てではないポリヌクレオチド鋳型の増幅または合成を意味する。例えば、ポリヌクレオチドのサブセット(例えば、第1cDNA鎖)は、全ての第1cDNA鎖の集合からのポリヌクレオチドの群(例えば、遺伝子ファミリー)を選択的に増幅または合成する特定のオリゴヌクレオチドプライマーの使用により増幅または合成することができる。
【0115】
ここで用いられる「標的ポリヌクレオチド」は、その発現水準を分析すべきポリヌクレオチド配列である。標的ポリヌクレオチドは、その発現水準が分析される前に単離または増幅することができる。例えば、標的ポリヌクレオチドは、その増幅のために用いられる対のオリゴヌクレオチドプライマーの2つの構成員のハイブリッド領域の間にある配列であり得る。標的ポリヌクレオチドはRNAまたはDNAであり得、例えば、これは、mRNAまたはcDNA、遺伝子のコード化領域またはその一部であり得る。標的ポリヌクレオチド配列は、通常、大きな「鋳型」配列の一部として存在するが、ある場合には、標的配列と鋳型は同じである。「鋳型配列」は通常、最初に存在するポリヌクレオチド配列を意味するが、増幅反応からの産物は、その後の増幅反応において鋳型配列として用いることもできる。「標的ポリヌクレオチド」または「鋳型配列」は、特定の配列であるまたはそれを含む正常(例えば、野生型)または突然変異ポリヌクレオチドであり得る。
【0116】
ここで用いられる「RT−PCR」という用語は、組み合わされた逆転写及びポリメラーゼ連鎖反応を意味する。この増幅方法は初期ステップを用い、初期ステップにおいては、特定のオリゴヌクレオチド、オリゴdTまたは、ランダムプライマーの混合物を用いて、RNAを第1一本鎖cDNAに逆転写させ;このcDNAは、次に、標準的増幅技術、例えばPCRを用いて増幅されて、第2の相補鎖及び二本鎖cDNAを産生する。
【0117】
ここで用いられる「オリゴヌクレオチドプライマー」という用語は、ポリヌクレオチド鋳型にアニーリングすることができると共に3’末端を提供してポリヌクレオチド鋳型に相補的な拡張産物を得ることができるポリヌクレオチド分子(すなわち、DNAまたはRNA)を意味する。開始及び拡張用の条件は、通常、四つの異なるデオキシリボヌクレオシド三リン酸及び、DNAポリメラーゼまたは逆転写酵素のような重合誘発剤が適当な緩衝剤(「緩衝剤」は、共因子であるまたは、pH、イオン強度等に影響を与える置換物質を含む)の存在及び、適当な温度を含む。本発明によるプライマーは、一本鎖または二本鎖であってよい。プライマーは増幅の最大高率のためには一本鎖であり、プライマーとその補体が、二本鎖ポリヌクレオチドを形成する。しかしながら、これは、二本鎖であってもよい。本発明で有用な「プライマー」は、長さが100ヌクレオチド以下、例えば、90、80、70、60、50、40、30、20、15以下または10である。
【0118】
ここで用いられる「任意配列」という用語は、個人の判断または裁量に基づくまたは付されるものと定義される。ある場合には、任意配列は、一部の塩基について完全にランダムまたは部分的にランダムであり得る。他の例において、特定の割合の各デオキシヌクレオチド、例えば、略同じ割合の各デオキシヌクレオチドまたは主に一つのデオキシヌクレオチドを含むように、または特定のデオキシヌクレオチドを含まないように、任意配列を選択することができる。特定の制限エンドヌクレアーゼ用の認識部位を含むまたは含まないように、任意配列を選択することができる。既知の配列のmRNAまたはcDNAに相補的な配列を含むように、または、既知の配列のmRNAまたはcDNAからの配列を含まないように、任意配列を選択することができる。
【0119】
ここで用いられる「GCに富む」という用語は、GC含量が少なくとも60%GC(例えば、5塩基伸長部におけるGまたはCの3塩基、6塩基伸長部におけるGまたはCの4塩基、7〜8塩基伸長部におけるGまたはCの5塩基、9〜10塩基伸長部におけるGまたはCの6塩基、11塩基伸長部におけるGまたはCの7塩基、12〜13塩基伸長部におけるGまたはCの8塩基、14〜15塩基伸長部におけるGまたはCの9塩基、16塩基伸長部におけるGまたはCの10塩基、17〜18塩基伸長部におけるGまたはCの11塩基、19〜20塩基伸長部におけるGまたはCの12塩基、21塩基伸長部におけるGまたはCの13塩基、22〜23塩基伸長部におけるGまたはCの14塩基、24塩基伸長部におけるGまたはCの15塩基、25〜26塩基伸長部におけるGまたはCの16塩基)、好ましくは少なくとも70%GC、少なくとも80%GCまたは少なくとも90%GCあるいは100%GCまでであるヌクレオチド(3’末端ヌクレオチド)の連続伸長部を意味する。
【0120】
ここで用いられる「ATに富む」という用語は、AT含量が少なくとも60%GC(例えば、5塩基伸長部におけるAまたはTの3塩基、6塩基伸長部におけるAまたはTの4塩基、7〜8塩基伸長部におけるAまたはTの5塩基、9〜10塩基伸長部におけるAまたはTの6塩基、11塩基伸長部におけるAまたはTの7塩基、12〜13塩基伸長部におけるAまたはTの8塩基、14〜15塩基伸長部におけるAまたはTの9塩基、16塩基伸長部におけるAまたはTの10塩基、17〜18塩基伸長部におけるAまたはTの11塩基、19〜20塩基伸長部におけるAまたはTの12塩基、21塩基伸長部におけるAまたはTの13塩基、22〜23塩基伸長部におけるAまたはTの14塩基、24塩基伸長部におけるAまたはTの15塩基、25〜26塩基伸長部におけるAまたはTの16塩基)、好ましくは少なくとも70%AT、少なくとも80%ATまたは少なくとも90%ATあるいは100%ATまでであるヌクレオチド(すなわち、5’または3’末端ヌクレオチドを含む)の連続伸長部を意味する。
【0121】
ここで用いられる「遺伝子ファミリー特異的」という用語は、増幅反応において二つ以上のポリヌクレオチド鋳型にアニーリングするヌクレオチドの配列またはオリゴヌクレオチドプライマーを意味する。「遺伝子ファミリー特異的」プライマーは、鋳型に完全に相補的である必要がない。通常、遺伝子ファミリー特異的配列を含むプライマーは、試料中の異なるmRNAまたはcDNAにより示される少なくとも2、5または20、通常少なくとも50以上、または通常少なくとも75の異なる遺伝子にアニーリングする。「異なる」という用語は、遺伝子の説明で用いる場合、FASTA(デフォルト設定)により決められる配列類似性が98%を超えないRNAコード領域において少なくとも100ntの伸長部を含む場合、任意の二つの遺伝子は異なると考えられる。「遺伝子ファミリー特異的配列」は4個以上のヌクレオチド、少なくとも5、6、7、8、9、10個以上で50個までのヌクレオチド長である。
【0122】
ここで用いられる「標識」または「検出可能標識」という用語は、検出可能(好ましくは定量可能な)な信号を提供するために用いることができると共に、ポリヌクレオチドの機能的に結合させることができる任意の原子または分子を意味する。標識は、蛍光、放射能、比色、重量測定、X線回折または吸収、磁気、酵素活性、質量分析、結合親和性、ハイブリッド形成高周波、ナノ結晶等により検出可能な信号を提供することができる。本発明のプライマーは、検出可能な標識を「検出」することにより増幅反応産物を「検出」することができるように標識することができる。「定性的または定量的」検出は、標識により発生する信号の大きさ(強度)または数に基づく視覚的または自動評価を意味する。本発明の方法により標識されたポリヌクレオチド(例えば、オリゴヌクレオチドプライマー)は、5’末端、3’末端または両末端、または内部において標識される。標識は、「直接」、例えば染料、または「間接」、例えば、ビオチン、ジゴキシン、アルカリホスファターゼ(AP)、ホースラディッシュペルオキシダーゼ(HRP)であり得る。「間接標識」の検出のために、捕捉され、放出され、標識されたポリヌクレオチドフラグメントを視覚化するために、標識された抗体または酵素基質のようなさらなる成分を加えることが必要である。好ましい態様において、オリゴヌクレオチドプライマーを蛍光標識で標識する。適当な蛍光標識は、蛍光色素、例えば、ローダミン及び誘導体(テキサスレッド等)、フルオレセイン及び誘導体(5−ブロモメチルフルオロセイン等)、ルシファーイエロー、IAEDANS、7−Me2N−クマリン−4−酢酸、7−OH−4−CH3−クマリン−3−酢酸、7−NH2−4−CH3−クマリン−3−酢酸(AMCA)、モノブロモバイマン、ピレントリスルホネート、例えば、Cascade Blue、及びモノブロモリメチル−アンモニオバイマン(例えば、DeLuca,Immunofluorescence Analysis,Antibody As a Tool,Marchalonisら編、John Wiley&Sons,Ltd.(1982年)参照、ここで参考のために取り込む)を含む。
【0123】
「結合」という用語は、例えば、水素結合、イオン結合またはファンデルワールス結合による共有結合または非共有結合を意味する。そのような結合は、これらの原子またはイオンの電子密度の最分布の結果として、同じまたは異なる原子またはイオンの少なくとも二つの間に形成することができる。本発明のポリヌクレオチド(例えば、オリゴヌクレオチドプライマー)は、検出可能な標識及び/または固体支持体に結合させることができる。
【0124】
ここで用いられる「反対方向」という用語は、プライマーに言及すると、一つのプライマーが、標的ポリヌクレオチド鋳型のセンス鎖に相補的なヌクレオチド配列を含み、もう一つのプライマーは、同じ標的ポリヌクレオチド鋳型のアンチセンス鎖に相補的なヌクレオチド配列を含むことを意味する。反対方向のプライマーは、それが補足する匹敵ポリヌクレオチド鋳型からPCR増幅産物を産生することができる。反対方向の2つのプライマーを、逆プライマー及び前方プライマーと呼ぶことができる。
【0125】
ここで用いられる「同じ方向」という用語は、プライマーが、標的ポリヌクレオチド鋳型の同じ鎖に相補的なヌクレオチド配列を含むことを意味する。同じ方向のプライマーは、それが補足する匹敵ポリヌクレオチド鋳型からPCR増幅産物を発生させない。
【0126】
ここで用いられる「ポリヌクレオチド」という用語は、通常、非修飾RNAまたはDNAまたは修飾RNAまたはDNAであり得る、任意のポリリボヌクレオチドまたはポリデオキシリボヌクレオチドを意味する。ここで用いられる「ポリヌクレオチド」という用語は、一つまたは二つ以上の修飾塩基を含む前述のDNAまたはRNAも含む。すなわち、安定性のためまたは他の理由のために修飾された背骨を有するDNAまたはRNAは「ポリヌクレオチド」である。ここで用いられる「ポリヌクレオチド」という用語は、ポリヌクレオチドのそのように化学的、酵素学的または代謝的に修飾された型、並びに、ウイルス及び例えば単純及び複合細胞を含む細胞のDNA及びRNA特性の化学的型を含む。本発明に有用なポリヌクレオチドは単離または精製されたポリヌクレオチド、あるいは増幅反応において増幅したポリヌクレオチドであり得る。
【0127】
ここで用いられる「単離」または「精製」という用語は、ポリヌクレオチドに言及する場合、自然発生配列が、その正常細胞(例えば、染色体)環境から除去されているまたは、非天然環境(例えば、人工合成)において合成されることを意味する。すなわち、「単離」または「精製」配列は、細胞非含有溶液である、または異なる細胞環境に置くことができる。「精製」という用語は、配列が存在する唯一のヌクレオチドであることを意味せず、天然にそれに付随する非ヌクレオチドまたはポリヌクレオチド材料は本質的に含まない(約90〜95%、99〜100%までの純度)ことを意味し、すなわち、単離された染色体から区別される。
【0128】
ここで用いられる「cDNA」という用語は、RNA依存DNAポリメラーゼ(例えば、逆転写酵素)の作用によりRNA鋳型から製造される相補的または複製ポリヌクレオチドを意味する。「cDNAクローン」は、クローニングベクター中に含まれる意図するRNA分子に相補的な二重DNA配列を意味する。
【0129】
ここで用いられる「ゲノムDNA」という用語は、RNA転写体から複製された相補的DNAに対して、染色体DNAを意味する。ここで用いられる「ゲノムDNA」という用語は、単一細胞中に存在するDNAの全て、または単一細胞中のDNAの一部であり得る。
【0130】
ここで用いられる「相補的」という用語は、ポリヌクレオチド(またはその一部)の単一鎖が、非平行ポリヌクレオチド単一鎖のヌクレオチド(いかなる不対ヌクレオチドによっても中断されていない)間の隣接塩基対形成により非平行ポリヌクレオチド鎖(またはその一部)にハイブリッド形成し、それにより、相補的鎖間に二本鎖ポリヌクレオチドを形成する性能を意味する。第1のポリヌクレオチドは、第1ポリヌクレオチドの各及びそれぞれのヌクレオチドが第2ポリヌクレオチドの相補的領域においてヌクレオチドと塩基対を形成すれば、第2ポリヌクレオチド鎖に「完全に相補的」であると言われる。第1のポリヌクレオチドは、第1ポリヌクレオチドの一つのヌクレオチドが第2ポリヌクレオチドの対応するヌクレオチドと塩基対を形成しない場合、第2ポリヌクレオチド鎖に完全に相補的ではない。ポリヌクレオチド鎖間の相補性の程度は、ポリヌクレオチド鎖間のアニーリングまたはハイブリッド形成の効率及び強度に著しい効果を有する。これは、ポリヌクレオチド鎖間の結合に依存する増幅反応において特に重要である。
【0131】
「発現」という用語は、細胞または細胞非含有系において蛋白またはヌクレオチド配列を生成することを意味し、RNA産物への転写、翻訳後修飾及び/またはその産物をコードするDNAから蛋白産物またはポリヌクレオチドへの翻訳、並びに可能な翻訳後修飾を誘発する。
【0132】
ここで用いられる「遺伝子発現プロフィールを比較する」という用語は、二つまたはそれ以上の試料中における一つまたは二つ以上のポリヌクレオチドの異なる発現を比較することを意味する。
【0133】
ここで用いられる「発現プロフィール」という用語は、試料中における一つまたは二つ以上の遺伝子の定量的(すなわち、存在量)及び定性的発現を意味する。
【0134】
ここで用いられる「発現プロフィールにおける相違」という用語は、遺伝子の発現における定量的(すなわち存在量)及び定性的相違を意味する。ポリヌクレオチド検出のための既知の方法(例えば、電気泳動)によれば、一つの試料において遺伝子発現を検出可能であるがもう一つの試料において検出されない場合、「発現プロフィールにおいて相違がある」。あるいは、二つの試料間の遺伝子発現の定量的相違(すなわち、増加または減少)が約20%、約30%、約50%、約70%、約90%、約100%(約2倍)以上から約1.2倍、2.5倍、5倍、10倍、20倍、50倍以上までである場合、「発現プロフィールにおいて相違がある」。二つの試料間の発現プロフィールの相違がある遺伝子は、二つの試料において異なって発現された遺伝子である。
【0135】
ここで用いられる「異なる発現」という用語は、二つ以上の試料間におけるポリヌクレオチド(例えば、遺伝子)の一時的及び/または細胞性発現パターンにおける定量的及び定性的な両相違、すなわち発現プロフィールの相違を意味する。ポリヌクレオチド検出のための既知の方法(例えば、電気泳動)によれば、一つの試料においてその発現を検出可能であるがもう一つの試料において検出されない場合、ポリヌクレオチドは「異なって発現される」と言われる。ポリヌクレオチドは、二つの試料間のその発現の定量的相違(すなわち、増加または減少)が約20%、約30%、約50%、約70%、約90%、約100%(約2倍)以上から約1.2倍、2.5倍、5倍、10倍、20倍、50倍以上までである場合も、「異なって発現される」と言われる。「異なって発現された」遺伝子転写体は、例えば、活性化及び不活性化状態において、一つの発生段階ともう一つの発生段階における個体の異なる細胞または組織型において、選択された疾患を有する個体の異なる細胞または組織において、健康生物の同じ細胞または組織において見られる遺伝子転写体の複製数または状態と比較して、二つまたはそれ以上の試料間において異なる数の複製が見つかるmRNA転写体を意味する。mRNA転写体複製の数は閾値サイクル(Ct)と比例しているので、後者を、異なる発現の定量的推定に用いることもできる。従って、二つの異なる試料において遺伝子のCt値の相違が2サイクル異常の場合、遺伝子は異なって発現されると考えることができる。
【0136】
ここで用いられる「存在量」という用語は、試料における標的ポリヌクレオチドの量(例えば、μg、μモルまたは複製数で測定される)を意味する。ポリヌクレオチドの「存在量」は、例えば、Basic Methods in Molecular Biology,(1986年,Davisら著,Elsevier,ニューヨーク)及びCurrent Protocols in Molecular Biology(1997年,Ausubelら著,John Weley&Sons,Inc.)に記載されているように、当該分野でよく知られた方法(例えば、UV吸収、既知の長さ及び量を参照しつつゲル上でのバンド強度を比較)により測定することができる。本発明においてポリヌクレオチドの存在量を測定する一つの方法は、そのようなポリヌクレオチドにより放出された蛍光強度を測定し、これを、参照ポリヌクレオチド、すなわち、既知の量のポリヌクレオチドにより放出された蛍光強度と比較することである。
【0137】
「遺伝子をコードするヌクレオチド配列を有するポリヌクレオチド」は、遺伝子のコード領域を含むポリヌクレオチド配列、すなわち、遺伝子産物をコードするポリヌクレオチド配列を意味する。コード領域は、cDNA、ゲノムDNAまたはRNA型で存在し得る。DNA型で存在する場合、オリゴヌクレオチドは一本鎖(すなわち、センス鎖)または二本鎖であってよい。適当な制御要素、例えば、エンハンサー/プロモーター、スプライス接合部、ポリアデニル化信号等を、要すれば、遺伝子のコード領域に近接して配して、一次RNA転写体の転写を適切に開始及び/または正確に加工することができる。あるいは、本発明のベクターにおいて利用されるコード領域は、内因性エンハンサー/プロモーター、スプライス接合部、介在配列、ポリアデニル化信号等を含む、または内因性と外因性の制御要素の両方を組み合わせて含むことができる。
【0138】
ここで用いられる「変性ヌクレオチド」という用語は、dA、dG、dC及びdTのいずれかである、またはdA、dG、dC及びdTの少なくとも二つの塩基と塩基対を形成することができるヌクレオチドを意味する。dA、dG、dC及びdTの少なくとも二つの塩基と塩基対を形成する変性ヌクレオチドの非限定的例には、イノシン、5−ニトロピロール、5−ニトロインドール、ハイポキサンチン、6H,8H,4−ジヒドロピリミド[4,5c][1,2]オキサシン−7−オン(P)、2−アミノ−6−メトキシアミノプリン、dPTP及び8−オキソ−dGTPがある。
【0139】
ここで用いられる「人工ヌクレオチド」という用語は、自然発生ヌクレオチドではないヌクレオチドを意味する。「自然発生」という用語は、ヒトが介在することなく自然に存在するヌクレオチドを意味する。対照的に、「人工ヌクレオチド」という用語は、ヒトが介在した場合にのみ存在するヌクレオチドを意味する。特に重要な人工ヌクレオチドは、従来のヌクレオチド(すなわち、dA、dG、dC及びdT)よりも、もう一つの人工ヌクレオチドと塩基対を形成する高い選択性を示すものである(例えば、Ohtsukiら著、2001年,Proc.Natl.Acad.Sci.第98巻:4922〜4925頁、ここで参考のために取り込む)。人工ヌクレオチドは、従来のいずれかのヌクレオチドと比較して、人口ヌクレオチドとの塩基対形成能が30%以上である場合に、「従来のヌクレオチドよりも、もう一つの人工ヌクレオチドと塩基対を形成する高い選択性を示す」。塩基対形成能は、Ohtsukiら著(前記書)に記載のようなT7転写アッセイにより測定することができる。「人工ヌクレオチド」の他の非限定的例を、Lutzら著、(1998年)Bioorg.Med.Chem.Lett.第8巻:1149〜1152頁);Voegal及びBenner,(1996年)Helv.Chim.Acta第76巻:1863〜1880頁;Horlacherら著、(1995年)Proc.Natl.Acad.Sci.第92巻:6329〜6333頁;Switzerら著、(1993年)、Biochemistry第32巻:10489〜10496頁;Tor及びDervan(1993年)J.Am.Chem.Soc.第115巻:4461〜4467頁;Piccirilliら著、(1991年)Biochemistry第30巻:10350〜10356頁;Switzerら著、(1989年)J.Am.Chem.Soc.第111巻:8322〜8323頁に見ることができ、これらの全てを参考のためにここに取り込む。「人工ヌクレオチド」は、前述のような変性ヌクレオチドでもよい。
【0140】
ここで用いられる「サイン配列モチーフ」という用語は、蛋白ファミリーの構成員、または蛋白の異なるファミリーの間に高度に保存されるアミノ酸配列を意味する。これらの保存残基は、「配列モチーフ」または「サイン配列」と呼ばれ、蛋白の機能と構造の両方を決めることができる。これらは、確認蛋白または活性部位及び結合部位のような重要な蛋白領域において一般的に用いられる。配列モチーフは、多くの蛋白ファミリーについてよく知られている。さらに、潜在的配列モチーフを、利用できるコンピュータープログラムを用いて、関連する蛋白配列を比較することにより見つけることができる。
【0141】
ここで用いられる「因子」という用語は、細胞が生存及び/または成長及び/または増殖するために必要とし、別の細胞により製造及び運び出すことができる任意の物質を意味する。そのような因子には、成長因子(例えば、インターロイキン、インスリン、トランスフェリン、ヒドロコルチゾン、線維芽細胞成長因子、神経成長因子、表皮成長因子)、アミノ酸、及びビタミン類があるが、これらに限定されない。
【0142】
ここで用いられる「固体支持体」という用語は、分子(例えば、オリゴヌクレオチドプライマー)が不可逆的に結合することができる表面を意味し、限定はされないが、膜、セファローズビーズ、磁気ビーズ、組織培養プレート、シリカ系マトリクス、膜系マトリクス、限定はされないがスチレン、ラテックスまたはシリカ系材料を含む表面を含むビーズがあり、材料として他のポリマー、例えば、酢酸セルロース、テフロン(登録商標)、ポリビニリデンジフルオライド、ナイロン、ニトロセルロース、ポリエステル、カーボネート、ポルスルホン、金属、ゼオライト、紙、アルミナ、ガラス、ポリプロピレン、塩化ポリビニル、塩化ポリビニリデン、ポリテトラフルオロエチレン、ポリエチレン、ポリアミド、プラスチック、濾紙、デキストラン、ゲルマニウム、珪素、(ポリ)テトラフルオロエチレン、ガリウムアルセニド、ガリウムホスフィド、酸化珪素、硝酸珪素及びそれらの組み合わせがある。本発明による固体支持体は、反応管の内壁を含む。
【0143】
「磁気ビーズ」は、磁場により引き寄せられる固体支持体を意味し;そのような固体支持体は、限定はされないが、Dynabeads、BioMag Streptavidin、MPG7 Streptavidin、Streptavidin Magnespherel、Streptavidin Magnetic Particles、AffiniTipJ、及び磁化性粒子の任意のMaga系、BioMag Superparamagnetic Particles、または分子(例えば、オリゴヌクレオチドプライマー)が付着または不動化される任意の他の磁気ビーズである。
【0144】
ここで用いられる「遺伝子発現を調節するモジュレーター」という用語は、その化合物または条件の不存在下における遺伝子の発現と比較して、遺伝子の発現(例えば、転写の水準)を増加または減少させることができる化合物または条件を意味する。ここで用いられる「条件」という用語は、個体の正常段階、疾患段階、疾患型または発生段階、または個体が曝される環境を意味する。相違が増加の場合、増加は約20%、約30%、約50%、約70%、約90%、約100%(約2倍)以上で、約5倍、10倍、20倍、50倍以上までである。相違が減少の場合、減少は約20%、30%、50%、70%、90%、100%(例えば、特定の蛋白またはRNAが存在しない)。遺伝子発現の水準(例えば、転写の水準)は、当該分野でよく知られた方法、例えば、Basic Methods in Molecular Biology,(1986年,Davisら著、Elsevier,ニューヨーク);及びCurrent Protocols in Molecular Biology(1997年,Ausubelら著、John Weley&Sons,Inc.)に記載のようなノーザンブロット、RT−PCRにより測定することができる。遺伝子発現の水準は、本発明により開示されるような本方法により検出することもできる。本発明による「モジュレーター」は、以下に定義のような薬剤、治療剤または有効薬剤も含む。
【0145】
ここで用いられる「ポリA部位」または「ポリA配列」という用語は、発生期RNA転写体の停止及びポリアデニル化の両方を導くDNA配列を意味する。真核生物細胞の組換えDNA配列の効果的発現は、得られる転写体の効率的転写及びポリアデニル化を導く信号の発現を必要とする。転写停止信号は、通常、ポリアデニル化信号の下流において見られ、数百ヌクレオチド長である。
【0146】
ここで用いられる「RNA転写体」という用語は、DNA配列のRNAポリメラーゼで触媒した転写から得られる産物を意味する。RNA転写体が、DNA配列の完全に相補的な複製である場合、これは、一次転写体と呼ばれる;またはこれは、一次転写体の転写後加工から導かれるRNA配列であり得、成熟RNAと呼ばれる。「メッセジャーRNA」(mRNA)は、イントロンを含まず、その細胞により蛋白に翻訳することができるRNAを意味する。
【0147】
「薬剤」または「治療剤」という用語は、薬剤の活性フラグメントまたは類似体、例えば、全寸法薬剤の活性の少なくとも50%を有する蛋白またはポリヌクレオチドを含む。 薬剤は、蛋白、ペプチドまたはポリヌクレオチドであり得る。
【0148】
ここで用いられる「薬剤の効果」または「治療剤の効果」という用語は、薬剤または治療剤が、診断用特性の発現を正常から著しく異ならない値に戻す性能と定義される(通常の統計的方法により、95%信頼水準内で決められる)。
【0149】
「疾患及び病状」は、細胞、組織及び/または個体の正常機能を損なう一つまたは二つ以上の生物学的特徴の変化である。
【0150】
ここで用いられる「疾患の進行」または「疾患段階」という用語は、疾患が進行し、徴候を引き起こし、過酷度を回復または継続及び/または減少させる一連の事象を意味する。
【0151】
本発明は、二つまたはそれ以上の試料において異なって発現される遺伝子を発現し、それらの発現の水準の相違を測定する方法を提供する。本発明は、試料特異的オリゴヌクレオチドプライマーを用いるRT−PCTに基づき、増幅産物を試料供給源によって識別することができる。
【0152】
本発明の実施には、特記しない限り、当該分野の技術範囲内である分子生物学、ミクロ生物学及び組換えDNA技術の従来技術を用いる。そのような技術は、文献に充分に説明されている。例えば、Sambrook,Fritsch&Maniatis,1989年,Molecular Cloning:A Laboratory Manual,第2版:Oligonucleotide Synthesis(M.J.Gait編、1984年);Polynucleotide Hybridization(B.D.Harnes&S.J.Higginsら編、1984年);A Practical Guide to Molecular Cloning(B.Perbal,1984年);及び連続出版物,Methods in Enzymology(Academic Press,Inc.);Short Protocols In Molecular Biology,(Ausubelら編、1995年)を参照されたい。本発明の実施には、米国特許第5,965,409;5,665,547;5,262,311;5,599,672;5,580,726;6,045,998;5,994,076;5,962,211;6,217,731;6,001,230;5,963,456;5,246,577;5,126,025;5,364,521;4,985,129号に開示の技術及び組成物も含み得る。以上及び以下に記載の全ての特許、特許出願及び公報をここで参考のために取り込む。
【0153】
(試料供給源)
本発明は、ここに定義のような二つまたはそれ以上の試料における標的ポリヌクレオチドの発現を検出、測定及び比較する方法を提供する。本発明の試料は、少なくとも一つのポリヌクレオチドを含む、または標的ポリヌクレオチドそれ自体であり得る。ポリヌクレオチドの配列情報の従来の知識は、特別の用途に依存して必要または必要でない。本発明による有用な試料は、限定はされないが、標的ポリヌクレオチド(ゲノムDNA、cDNAまたはRNA)の試料、細胞、有機体、組織、流体、血漿、血清、脊髄液、リンパ液、関節液、尿、涙、便;皮膚、気道、腸管及び尿生殖管の外分泌液;唾液、血球、腫瘍、器官、組織、試験管内細胞培養成分の試料、天然単離物(例えば、飲料水、海水、個体材料)、微生物検体、及びポリヌクレオチドトレーサー分子で「マークした」対象または検体を含む。
【0154】
本発明の有用な試料は、限定はされないが、例えば、異なる個体、同じまたは異なる個体の異なる発生段階、異なる疾患個体、正常個体、同じまたは異なる個体の異なる疾患段階、異なる疾患治療に付された個体、異なる環境因子に付された個体、病気の体質を有する個体、感染性疾患(例えば、HIV)に曝された固体を含む異なる供給源から得ることができる。有用な試料は、試験管内で培養した組織、細胞または他のポリヌクレオチド含有供給源から得ることもできる。培養試料は、限定はされないが、異なる培地及び条件(例えば、pH、圧力、または温度)において培養した培地(例えば、組織または細胞)、異なる期間培養した培地(例えば、組織または細胞)、異なる因子または試薬(例えば、薬剤候補、またはモジュレーター)で治療した培地(例えば、組織または細胞)、または異なる型の組織または細胞の培地を含む供給源から得ることができる。
【0155】
試料は、限定はされないが、血液疾患、血液脂質疾患、自己免疫疾患、骨または関節障害、心臓血管疾患、呼吸疾患、内分泌疾患、免疫疾患、感染性疾患、筋肉衰弱及び全身衰弱疾患、神経疾患、例えば、神経退化及び/または神経精神疾患;皮膚疾患、腎臓疾患、強皮症、発作、遺伝性出血性血管拡張症、糖尿病、糖尿病関連疾患(例えば、PVD)、高血圧、ゴシェ病、嚢胞性線維症、鎌状赤血球貧血、肝臓疾患、膵臓疾患;眼、耳、鼻及び/または喉疾患;生殖器官に影響を与える疾患、胃腸管疾患(例えば、結腸疾患、脾臓疾患、虫垂疾患、嚢胞疾患、及び他の疾患)等を含む疾患または病状を有する個体から得ることができる。ヒト疾患のさらなる説明は、Mendelian Inheritance in Man:A Catalog of Human Genes and Genetic Disorders by Victor A.McKusick(第12版(3巻セット)1998年6月,Johns Hopkins University Press,ISBN:0801857422)が参照され、その全体がここで取り込まれる。好ましくは、正常な人口統計的に適合した個体から及び/または疾患を有する患者からの非疾患組織からの試料が、対照を提供するための分析において用いられる。
【0156】
一つの局面において、試料は、特定の疾患を有するヒト及び正常なヒトから得られる組織または細胞試料である。組織試料は、死体または最近死んだ患者から(例えば、解剖により)得ることができる。組織は、外科検体、病理検体(例えば、生検)、または通常他の処理から廃棄される「臨床廃棄物」である試料から得ることができる。試料は、成人、子供及び/または胎児から(例えば、選択的堕胎または流産から)得ることができる。細胞は、組織からの細胞の懸濁液(例えば、廃棄組織のような切り刻んだ組織細胞の懸濁液、)体液(例えば、血液、血漿、血清等)、かきとった粘膜(例えば、頬からかきとった粘膜または乳頭塗抹)及び/または、気管支洗浄、羊水穿刺及び/または白血球泳動のような他の手順から得ることができる。
【0157】
ある局面において、分析のためにRNAを抽出する前にまず細胞を培養する。連続的に成長している細胞系、一次細胞系、及び/または二次細胞系からの細胞も用いることができる。
【0158】
もう一つの局面において、試料は、異なる疾患を有するヒト及び正常ヒトから得られる組織または細胞試料である。
【0159】
一つの局面において、単一個体からの複数の組織/細胞が得られる、すなわち、試料は個体の「全身」を表す。好ましくは、本発明による「全身」を表す試料は、単一個体からの少なくとも5つの異なる型の組織を含む。より好ましくは、本発明による「全身」を表す試料は、少なくとも10または少なくとも15の異なる組織を含む。組織は、皮膚、神経組織、心臓組織、肝臓組織、胃組織、大腸組織、結腸組織、小腸組織、食道組織、肺組織、心臓組織、脾臓組織、膵臓組織、腎臓組織、生殖器(男性または女性)からの組織、副腎組織等からなる群より選択される。単一器官の異なる解剖学的または組織学的位置、例えば、脳器官である小脳、大脳及び骨髄からの組織も得ることができる。本発明の一部の局面は、器官系、例えば、呼吸系、泌尿系、腎臓系、心臓血管系、消化系及び生殖系(男性または女性)を表す試料を含む(例えば、器官系中の複数の器官からの試料を含む)。
【0160】
好ましい局面において、「全身」を表す細胞は、前述のような組織から得ることができ、さらに、患者の体液から(例えば、血液試料から)の組織を含む。
【0161】
試料は、特徴を共有する個体からの複数の細胞を含むことができる。例えば、共有される特徴は、性、年齢、病状、疾病素質、感染性疾患(例えば、HIV)への露出、親戚、同じ疾患による死亡、同じ薬剤による処理、化学療法への露出、放射療法への露出、ホルモン療法への露出、手術への露出、同じ環境条件(例えば、発ガン物質、汚染物質、アスベスト、TCE、過塩素酸塩、ベンゼン、クロロホルム、ニコチン等)への露出、同じ遺伝的変化または変化群、同じ遺伝子または遺伝子組の発現(例えば、特別のHLA対立遺伝子のような共通のハプロタイプを有する個体から試料を得ることができる)、等であり得る。
【0162】
本発明の好ましい局面において、試料はヒトから誘導されるが、本発明の一つの局面において、他の器官からの試料も用いられる。一つの局面において、試料は、疾患または他の病状のモデルを提供する非ヒト動物からの組織を含む。試料が、慢性疾患の動物モデルからの検体を表す場合、試料は、疾患の異なる段階を表す検体、例えば、寛解期間または増悪期間における動物からのものを含むことができる。試料は、追加的または択一的に、疾患または病状を治療するための療法(例えば、薬剤、抗体、蛋白療法、遺伝子療法、アンチセンス療法、そららの組み合わせ、等)に曝された疾患または病状を有する非ヒト動物からの組織を含むことができる。一部の局面において、非ヒト動物試料は、外因性ポリヌクレオチドを含む少なくとも一種の細胞を含むことができる(例えば、動物はトランスジェニック動物、キメラ動物、ノックアウトまたはノックイン動物)。
【0163】
なおさらなる局面において、植物からの試料を用いることができる。好ましくは、そのような試料は、生活周期の異なる段階における植物を含む、及び/または異なる型の植物組織(例えば、少なくとも約5つの異なる植物組織)を含むことができる。一つの局面において、試料は、外因性ポリヌクレオチドを含む少なくとも一つの細胞を含む植物から得られる(例えば、植物はトランスジェニック植物であり得る)。
【0164】
(試料からのmRNAの単離)
本方法は、二つまたはそれ以上の試料中の遺伝子の発現を測定または比較する。本発明の一つの局面において、転写水準における遺伝子の発現を測定または比較する。
【0165】
比較すべき二つまたはそれ以上の試料(例えば、試料A及びB)からのRNAを抽出し、試料特異的オリゴヌクレオチドプライマー(例えば、プライマー1A及び1B、図7参照)を用いて、個々にcDNAに逆転写する。
【0166】
RNA(例えば、mRNA)を含むポリヌクレオチドを、当該分野で良く知られている方法(Ausubelら著、前記書)に従って細胞及び組織から単離し、以下に記載する。
【0167】
RNAは、以下の方法に従って組織から精製することができる。所望の組織の除去に続いて、2g以下の組織を切り出し、液体窒素中で急速冷凍してRNAの劣化を防止する。適当な体積のグアニジニウム溶液(例えば、組織2g当たり20mlのグアニジニウム溶液)の添加時に、組織試料を組織混合器において、10秒の激しい攪拌を2または3回行って粉砕する。組織グアニジニウム溶液(1L)を調製するために、イソチオシアン酸グアニジニウム590.8gを、約400mlのDEPC−処理H2O中に溶解する。2M Tris−HCl,pH7.5(最終濃度0.05M)の25mlとNA2EDTA(最終濃度0.01M)の20mlを添加し、溶液を一晩攪拌し、体積を950mlに調節し、50mlの2−MEを添加する。
【0168】
均質化組織試料を、12,000×gで12℃で10分間遠心分離する。得られる上澄みを、20%Sarkosylの0.1体積の存在下に65℃で2分間インキュベートし、5.7M CsCl溶液(0.1g CsCl/ml)の9mlの上に乗せ、113,000×gで22℃で一晩遠心分離する。上澄みを注意深く除去した後、試験管を逆さまにし、排液する。試験管(RNAペレットを含む)の底部を、50mlプラスチック管内に置き、3mlの組織再懸濁緩衝液(5mM EDTA,0.5%(v/v)Sarkosyl,5%(v/v)2−ME)の存在下に4℃で一晩(またはそれ以上)インキュベートして、RNAペレットを完全に再懸濁させる。得られるRNA溶液を、25:24:1のフェノール/クロロホルム/イソアミルアルコール、続いて24:1のクロロホルム/イソアミルアルコールで順次抽出し、3M酢酸ナトリウム,pH5.2及び100%エタノール2.5体積を添加して沈殿させ、DEPC水中で再懸濁させる(Chirgwinら著、1979年、Biochemistry,第18巻:5294頁)。
【0169】
あるいは、RNAを、以下の単一ステッププロトコルに従って組織から単離する。組織100mg当たり1mlの変性溶液(4Mチオ硫酸グアニジニウム、25mMクエン酸ナトリウム,pH7.0、0.1Mの2−ME、0.5%(w/v)N−ラウリルサルコシン)中でガラステフロンホモジナイザーにて均質化して、所望の組織を調製する。均質化物を5mlのポリプロピレン管に移した後、pH4の2M酢酸ナトリウム0.1ml、水飽和フェノール1ml、及び49:1のクロロホルム/イソアミルアルコール0.2mlを順次添加する。各成分の添加後、試料を混合し、全成分の添加後、0〜4℃で15分間インキュベートする。試料を、10,000×gで4℃で20分間遠心分離し、100%イソプロパノール1mlの添加により沈殿させ、−20℃で30分間インキュベートし、10,000×gで4℃で10分間遠心分離してペレット化する。得られるRNAペレットを0.3ml変性溶液に溶解し、遠心分離管に移し、100%イソプロパノール0.3mlの添加により−20℃で30分間沈殿させ、10,000×gで4℃で10分間遠心分離する。RNAペレットを70%エタノール中で洗い、乾燥し、100〜200μlのDEPC−処理水またはDEPC−処理0.5%SDS中に再懸濁させる(Chomczynski及びSacchi,1987年,Anal.Biochem.,第162巻:156頁)。
【0170】
全RNAを単離するためのキット及び試薬は、種々の会社から市販されており、例えば、RNA isolation kit(Stratagene,La Lola,カリフォルニア,Cat♯200345);PicoPure(登録商標)RNA Isolation Kit(Arcturus,Mountain View,カリフォルニア,Cat♯KIT0202);RNeasy Protect Mini,Midi,及びMaxi Kits(Qiagen,Cat♯74124)がある。
【0171】
一部の態様において、全RNAを、その後の分析、例えば、逆転写のための方法で用いる。他の態様において、mRNAを、全RNAから単離する、または試料から直接単離して、逆転写に用いる。mRNAの単離のためのキット及び試薬は市販されており、例えば、Oligotex mRNA Kits(Qiagen,Cat♯70022)がある。
【0172】
RNAを含むポリヌクレオチドを、試験管内転写の方法により製造することができる。
【0173】
試験管内転写の技術は、当業者によく知られている。簡単に言えば、所望の遺伝子を、SP6、T3またはT7プロモーターを含むベクターに挿入する。ベクターを、コード化配列の下流に配される単一部位においてベクターを消化する適当な制限酵素を用いて線状化する。フェノール/クロロホルム抽出に続いて、DNAをエタノールで沈殿させ、70%エタノール中で洗浄し、乾燥し、滅菌水中に再懸濁する。線状化DNAを、転写緩衝液(200mM Tris−HCl,pH8.0,40mM MgCl2,10mMスペルミジン,250NaCl[T7またはT3]または200mM Tris−HCl,pH7.5,30mM MgCl2,10mMスペルミジン[SP6])、ジチオスレイトール、RNaseインヒビター、4つのリボヌクレオシド三リン酸の各々、及びSP6、T7またはT3 RNAポリメラーゼと、37℃で30分間インキュベートすることにより、試験管内転写反応を行う。RNAを含む放射標識ポリヌクレオチドを調製するために、非標識UTPを除き、35S−UTPが反応混合物中に含まれる。次に、DNaseIとインキュベーションすることによりDNA鋳型を除去する。エタノール沈殿に続いて、シンチレーションカウンター中で放射標識RNAを計測してcpm/μlを決めた(Ausubelら著、前記書)。
【0174】
cDNAを合成し、発現の検出及び測定のための増幅産物を産生するために、試料から単離されたRNAを用いる。好ましい態様において、cDNAの合成及び増幅反応の両方が、オリゴヌクレオチドプライマーを用いる。
【0175】
(本発明のオリゴヌクレオチドプライマーの設計)
本発明による有用なオリゴヌクレオチドプライマーを、ここに記載の当該分野でよく知られている一般的指針、及び、本発明の方法の各ステップ用に以下に記載の特定の要求に従って設計することができる。
【0176】
1.プライマー設計の一般的手法
オリゴヌクレオチドプライマーは5〜100ヌクレオチド、好ましくは17〜45ヌクレオチド長であるが、異なる長さのプライマーが有用である。cDNAを合成するためのプライマーは、好ましくは10〜45ヌクレオチドであり、増幅用のプライマーは、好ましくは約17〜25ヌクレオチドである。本発明に有用なプライマーは、溶融温度推定の方法により特定の溶融温度(Tm)を有するようにも設計される。Oligo(登録商標),Primer Designを含む市販プログラム及び、Primer3及びOligo Calculatorを含むインターネットで利用できるプログラムを用いて、本発明で有用なポリヌクレオチド配列のTmを計算することができる。好ましくは、例えばOligo Calculatorにより計算される本発明で有用な増幅プライマーのTmは、好ましくは、約45〜65℃、より好ましくは約50〜60℃である。
【0177】
ポリヌクレオチドのTmは、もう一つのポリヌクレオチドへのハイブリッド形成(例えば、オリゴヌクレオチドプライマーの、鋳型ポリヌクレオチドへのアニーリング)に影響を与える。本発明の方法において、種々のステップで用いられるオリゴヌクレオチドプライマーが、標的鋳型または標的鋳型から誘導されるポリヌクレオチド(すなわち、第1及び2cDNA鎖及び増幅産物)に選択的にハイブリッド形成することが好ましい。典型的には、二つのポリヌクレオチド配列が実質的に相補的である場合(少なくとも14〜25ヌクレオチドの伸長部において少なくとも約65%相補的、好ましくは、約75%、より好ましくは少なくとも約90%相補的)、選択的ハイブリッド形成が起こる。ここに参考として取り込まれるKanehisa,M.,1984年,Polynucleotides Res.第12巻:203頁を参照のこと。結果として、開始部位におけるある程度のミスマッチは許容されることが予測される。そのようなミスマッチは、モノ、ジまたはトリヌクレオチドのような小さい。あるいは、ミスマッチの領域は、四以上のヌクレオチドの非中断列にミスマッチが存在する領域として定義されるループが含む。
【0178】
多くの因子が、プライマーが第2ポリヌクレオチド分子にハイブリッド形成する効率及び選択性に影響を与える。本発明によりオリゴヌクレオチドプライマーを設計する場合、プライマー長さ、ヌクレオチド配列及び/または組成、ハイブリッド形成温度、緩衝液組成及び、プライマーがそこにハイブリッド形成する必要がある領域における立体障害の可能性を含むこれらの因子が考慮される。
【0179】
プライマーが標的配列にアニーリングする効率及び正確さの両方とプライマー長さとの間に陽性関係が存在する。特に、より長い配列は、より短い配列よりも高い溶融温度(TM)を有し、所定の標的配列において繰り返される可能性が低く、それにより乱雑ハイブリッド形成が最小限になる。高G−C含量を有するまたはパリンドローム配列を含むプライマー配列は、所望の標的部位と同様に、自己ハイブリッド形成する傾向があるが、これは二分子よりも一分子のハイブリッド形成速度が溶液において通常好ましいからである。しかしながら、各G−C対は、A及びTの塩基対が標的配列に結合する場合に見られる二つの水素結合ではなく三つの水素結合により結合され、それにより一層堅く強い結合が形成されるので、充分な数のG−Cヌクレオチド対を含むプライマーを設計することも重要である。ハイブリッド形成温度は、初回処理反応またはハイブリッド形成混合物に含まれる有機溶媒、例えば、ホルムアミドの濃度と同様に、プライマーアニーリング効率と逆に変化するが、塩濃度の増加は結合を容易にする。厳格なアニーリング条件下では、より長いハイブリッド形成プローブまたは合成プライマーは、より許容的な条件下では充分であるより短いものよりも効率的にハイブリッド形成する。好ましくは、厳格なハイブリッド形成は、ポリヌクレオチド配列をオリゴヌクレオチドプライマーにハイブリッド形成させる条件(例えば、PCR増幅には95℃)下に適当な緩衝液(例えば、1×RT緩衝液,Stratagene Catalog♯600085,1×Pfu緩衝液,Stratagene Catalog♯200536;または1×クローンドPfu緩衝液,Stratagene Catalog♯200532、または、cDNA合成及び増幅に有用な他の酵素に適した他の緩衝液)中で行われる。長さ及び/またはポリヌクレオチド組成またはオリゴヌクレオチドプライマーに依り、厳格なハイブリッド形成条件は変化(例えば、約1M未満、より一般的には約500mM未満、好ましくは約200mM未満の塩濃度から)することができ、ハイブリッド形成温度は変化(例えば、0℃の低い温度から22℃超、約30℃超、(最も頻繁には)約37℃超に変化)することができる。より長いフラグメントは、特性のハイブリッド形成により高いハイブリッド形成温度を必要とす
る。複数の因子がハイブリッド形成の過酷度に影響を与えるので、パラメーターの組み合わせが、単一因子の絶対測定より、一層重要である。
【0180】
オリゴヌクレオチドプライマーはこれらを考慮しつつ設計し、以下の方法により合成することができる。
【0181】
2.オリゴヌクレオチド合成
オリゴヌクレオチドプライマー自体が、当該分野でも良く知られている技術を用いて合成される。特定の配列のオリゴヌクレオチドを調製する方法は当該分野で知られており、例えば、適当な配列のクローニング及び制限消化分析、及び直接化学的合成を含む。一旦設計すると、オリゴヌクレオチドは、例えば、Narangら著、1979年,Methods in Enzymology,第68巻:90頁に記載のホスホトリエステル法、Brownら著、1979年,Methods in Enzymology,第68巻:109頁に開示のホスホジエステル法、Beaucageら著、1981年,Tetrahedron Letters,第22巻:1859頁に開示のジエチルホスホルアミデート法、米国特許第4,458,066号に開示の固体支持体法を含む適当な化学的合成法、または市販の自動オリゴヌクレオチド合成器(市販されている)またはVLSIPS(登録商標)技術を用いる他の化学的方法により調製される。
【0182】
本発明のオリゴヌクレオチドは、一部の態様によれば、固体支持体に共有または非共有的に直接または間接的(例えば、結合部位を介して)に結合させることができる。オリゴヌクレオチドは、その上でそれらが合成される固相支持体と結合することができる、またはそれらは別途合成して、使用される固相支持体に付着させることができ、これらの方法は例えば、Lundら著、(1988年)Polynucleotides Research,第16巻:10861〜10880頁;Albretsenら著、(1990年),Anal.Biochem.,第189巻:40〜50頁;Wolfら著、(1987年)Polynucleotides Research,第15巻:2911〜2926頁;またはGhoshら著、(1987年),Polynucleotides Research,第15巻:5353〜5372頁;米国特許第5,427,779,5,512,429,5,589,586,5,716,854及び6,087,102号に開示されている。固体支持体上にポリヌクレオチド配列を固定する方法は、固体支持体の製造者によっても提供され、例えば、膜については:Pall Corporation,Schleicher&Schuell、磁気ビーズについては:Dyal、培養プレートについては:Costar,Nalgenunc、本発明で有用な他の支持体については:CPG,Inc.により提供される。好ましくは、オリゴヌクレオチドは、種々の型を含み種々の結合部位を含む同じ固相支持体を用いてその上で合成される。
【0183】
本発明の固体基材は、分子(例えば、捕捉要素)を不可逆的に結合することができる任意の表面であり、限定はされないが、膜、磁気ビーズ、組織培養プレート、シリカ系マトリクス、膜系マトリクス、及び限定はされないがスチレン、ラテックスまたはシリカ系材料及び他のポリマーを含む表面を含むビーズを含み、ポリマーの例には、酢酸セルロース、テフロン(登録商標)、ポリビニリデンジフルオライド、ナイロン、ニトロセルロース、ポリエステル、カーボネート、ポルスルホン、金属、ゼオライト、紙、アルミナ、ガラス、ポリプロピレン、塩化ポリビニル、塩化ポリビニリデン、ポリテトラフルオロエチレン、ポリエチレン、ポリアミド、プラスチック、濾紙、デキストラン、ゲルマニウム、珪素、(ポリ)テトラフルオロエチレン、ガリウムアルセニド、ガリウムホスフィド、酸化珪素、硝酸珪素及びそれらの組み合わせがある。本発明による有用な固体支持体は、Sambrookら著、(1989年)Molecular Cloning:A Laboratory Manual(第2版),第1〜3巻,Cold Spring Harbor Laboratory;Ausubelら著、前記書,米国特許第5,427,779,5,512,439,5,589,586,5,716,854及び6,087,102号,Southermら著、1999年,Nature Genetics Supplement,第21巻:5頁及びJoosら著,1997年,Analytical Biochemistry,第247巻:96頁に開示されている。本発明で用いるための固相支持体には、微粒子、ビーズ、膜、スライド、プレート、微小加工チップ等を含む種々の型がある。
【0184】
本発明の好ましい固体支持体は微粒子である。本発明で、制御孔ガラス(CPG)、高架橋ポリエステル、アクリルコポリマー、セルロース、ナイロン、デキストラン、ラテックス、ポリアクロレイン等からなる微粒子を含む種々の微粒子支持体を用いることができ、これらは以下の例示参考文献に開示されている:Meth.Enzymol.,Aセクション,11頁から147頁,第44巻(Academic Press,ニューヨーク,1976年);米国特許第4,678,814;4,413,070;及び4,046;720号;及びPon編,Agrawal第19章,Methods in Molecular Biology,第20巻,(Humana Press,Totowa,ニュージャージー,1993年)。微粒子支持体は、さらに、市販のヌクレオシド誘導CPG及びポリスチレンビーズ(例えば、Applied Biosystems,Foster City,Calif.から得られる);誘導された磁気ビーズ;ポリエチレングリコールをグラフトしたポリスチレン(例えば、TentaGel.TM.,Rapp Polymere,Tubingen ドイツ);等が含む。材料、多孔性、寸法、形状等のような支持体特性、及び用いられる結合部位の型の選択は、オリゴヌクレオチドを用いる条件に依存する。例えば、酵素(例えば、逆転写酵素またはDNAポリメラーゼ)を用いる連続加工を含む用途において、酵素の立体障害を最小化すると共に基材への接近を容易にする支持体及び結合材が好ましい。最も適当な微粒子支持体の選択において考慮すべき他の重要な因子は、寸法均一性、合成支持体としての効率、表面が知られる程度、及び光学的特性を含み、例えば、以下により詳細に説明するように、透明平滑ビーズが、表面で多数のビーズを扱う場合に、有用な利点を提供する。
【0185】
微粒子表面上にオリゴヌクレオチドを付着及び/または合成する結合部位の例には、例えば、Ponら著、(1988年)Biotechniques,第6巻:768〜775頁;Webb,米国特許第4,659,774号;Baranyら著,International patent application PCT/US91/06103;Brownら著、(1989年)J.Chem.Soc.Commun.,1989年:891〜893頁;Damhaら著、(1990年)Polynucleotides Research,第18巻:3813〜3821頁;Beattieら著、(1993年)Clinical Chemistry,第39巻:719〜722頁;Maskos及びSouthen,(1992年)Polynucleotides Research,第20巻:1679〜1684頁;等がある。
【0186】
本発明のもう一つの好ましい固体支持体は、反応管の内壁である。反応管は、任意の酢酸セルロース、テフロン(登録商標)、ポリビニリデンジフルオライド、ナイロン、ニトロセルロース、ポリエステル、カーボネート、ポルスルホン、金属、ゼオライト、紙、アルミナ、ガラス、ポリプロピレン、塩化ポリビニル、塩化ポリビニリデン、ポリテトラフルオロエチレン、ポリエチレン、ポリアミド、プラスチック、濾紙、デキストラン、ゲルマニウム、珪素、(ポリ)テトラフルオロエチレン、ガリウムアルセニド、ガリウムホスフィド、酸化珪素、硝酸珪素からなってよい。好ましくは、反応管の内壁はポリプロピレンからなる。
【0187】
オリゴヌクレオチドは、単一(または数個の)の固相支持体上で合成して、合成されたオリゴヌクレオチドで均一に被覆された領域の配列を形成することもできる。そのような配列を合成する技術が、McGallらの国際出願PCT/US93/03767;Peaseら著、(1994年)Proc.Natl.Acad.Sci.,第91巻:5022〜5026頁;Southern及びMaskos,International application PCT/GB89/01114;Maskos及びSouthern(前記書);Southernら著,(1992年)Genomics,第13巻:1008〜1017頁;及びMaskos及びSouthern,(1993年);Polynucleotides Research,第21巻:4663〜4669頁に開示されている。
【0188】
好ましくは、本発明は、微粒子またはビーズに結合されたオリゴヌクレオチドを用いて実施される。微粒子支持体及び、それらの表面にオリゴヌクレオチドを共有または非供給結合させる方法は良く知られており、以下の参考文献に例示されている:Beaucage及びIyer(前記書);Gait編,Oligonucleotide Synthesis:A Practical Approach(IRL Press,Oxford,1984年);及び先に引用の参考文献。通常、微粒子の寸法及び形状は重要でないが;寸法範囲は数μm、例えば、1〜2μmから数百μm、例えば、200〜1,000μmの直径の微粒子が好ましく、これらが、最少限の試薬及び試料使用量で、広範囲のオリゴヌクレオチドの製造及び取り扱いを容易にする。
【0189】
一部の好ましい態様において、本発明の固相支持体として、市販の孔制御ガラス(CPG)またはポリスチレン支持体が用いられる。そのような支持体は、塩基不安定性リンカー及び付着した初期ヌクレオシドを用いて利用できる(例えば、Applied Biosystems(Foster City,Calif.))。好ましくは、粒径が500〜5,000Åである微粒子が用いられる。
【0190】
別の好ましい態様において、非多孔質微粒子が光学的特性のために用いられ、これらは、顕微鏡スライドのような平坦支持体上で多数の微粒子を追跡する場合、有利に用いることができる。特に好ましい非多孔質微粒子は、Bangs Laboratories(Carmel,Ind.)から得られるグリシダルメタクリレート(GMA)ビーズである。そのような微粒子は、種々の寸法で有用であり、タグまたはタグ補体を合成するための種々の結合基を用いて誘導される。好ましくは、オリゴヌクレオチドの大規模平行操作のために、直径5μmGMAビーズの微粒子が用いられる。
【0191】
3.cDNA合成のためのオリゴヌクレオチドプライマー設計手法
本方法のcDNA合成及び増幅反応のための特定のオリゴヌクレオチドプライマーの設計は、標的配列を認識しアニーリングすることができる配列の選択を含む。オリゴヌクレオチドのTmは、オリゴヌクレオチド長及びGC含量の分析により最適化される。
【0192】
本発明で有用なプライマーの設計は、前述の幾つかのパラメーター及びプライマー配列の最適化の評価に役立てるために開発された容易に利用できるコンピュータープログラムを用いることにより容易にすることができる。そのようなプログラムの例には、「PrimerSelect」(DNAStar(登録商標)ソフトウェアパッケージ(DNAStar,Inc;Madison,ウィスコンシン))、OLIGO4.0(National Biosciences,Inc.)、PRIMER,Oligonucleotide Selection Program,PGEN and Amplify(Ausubelら著の中に記載、前記書)がある。
【0193】
A.オリゴヌクレオチドプライマーの組成
本発明で有用なオリゴヌクレオチドプライマーは、一つまたは二つ以上の変性塩基、例えば、前述の塩基からなる変性配列を含み得る。そのような変性オリゴヌクレオチドは、ポリヌクレオチドキナーゼ、DNAリガーゼ、及び他の修飾性酵素(Hill,F.Loakes,D.及びBrown D.M.,1998年,Proc Natl Acad Sci USA.,第95巻:4258〜4263頁)用の正常基材として働く。変性ヌクレオチドは、オリゴヌクレオチド配列中、任意の位置、すなわち、5’、3’または内部に組み込むことができる。変性した塩基は、当該分野で知られており、異なる変性を示すために異なるコードが用いられる(例えば、表I)。
【表1】

【0194】
また、変性塩基は、dA、dG、dC及びdTの少なくとも二つと塩基対を形成することができるヌクレオチドであってよい。そのような有用な変性塩基は、通常、ヌクレオチド類似体であり、当該分野で知られており、以下に記載されている。例えば、デオキシイノシン(dI)は、4つの天然DNA塩基のいずれかに結合するので自然発生変性塩基である。dIは、真に普遍的ではないが、4つの標準的塩基(すなわち、A、T、G及びC)を含むミスマッチよりは不安定性が低い。ここで用いられる「普遍的塩基」という用語は、隣接塩基対の相互作用を著しく不安定化することなく、または変性オリゴヌクレオチドの予想される機能的生化学的有用性を妨害することなく、4つの正常塩基をいずれかを置換する性能を示す塩基を意味する。dIとdA、dG、dC及びdTとの間の水素結合相互作用は弱く等しくなく、その結果、一部の塩基対偏向がある:dI:dCハイブリッド形成>dI:dA>dI:dG>dI:dT(Kawase,Yら著、1986年,Polynucleotides Res.,1919年:7727〜7736頁;Martin,F.H.ら著、1985年,Polynucleotides Res.,第13巻,8927〜8938頁;Case−Green,S.C.,Southern,E.M.,1994年,Polynucleotides Res.,第22巻,131〜136頁)。ポリヌクレオチド中に存在する場合、dIは、DNAポリヌクレオチドにより、選択的に、dCを発生期鎖に直接組み込む。
【0195】
より最近では、機能的に真の普遍的塩基であるがdA、dG、dCまたはdTのいずれかと対を形成した場合にWatson−Crick DNA二本鎖を不安定化しない非天然塩基が加工された。これらの普遍的DNA塩基類似体の適用が最近試験された(Loakes,2001年,Polynucleotides Res.,第29巻,2437〜2447頁)。2つの例は、3−ニトロピロール−2’−デオキシヌクレオシド及び5−ニトロインドール−2’−デオキシヌクレオシド(5−ニトロインドール)。これら前記2例は、真の普遍的塩基として作用する。より特異的な他の塩基修飾が合成された。dA、dG、dC及びdTの四つ全てではないが二つ以上と塩基対を形成する変性塩基も、本発明の方法に有用である。例には、「p」と表されるピリミジン(CまたはT)類似体6H,8H−3,4−ジヒドロピリミド[4,5−c][1,2]オキサジン−7−オン、及び「k」と表されるプリン(AまたはG)類似体N6−メトキシ−2,6−ジアミノプリンがある。「p」塩基は、dAまたはdGと対を形成し、「k」塩基はdTまたはdCと対を形成する(Bergstrom,D.E.,Zhang,P.,及びJohnson,W.T.,1997年,Polynucleotides Res.,第25巻1935〜1942頁)。
【0196】
例えば、dPTP(dP)は、その塩基が2つの互変異性体型のいずれかに存在することができるので、チミジン(T)またはデオキシシチジン(dC)として挙動することができる。イミノ型において、dPは、チミジンの塩基対形成特性を有し、そのためdAと塩基対を形成し;アミノ型においては、dCと同様に、dGと塩基対を形成する(Sekiguchi,M.,1996年,Genes to Cells,第1巻,139〜145頁;Pavlov,Y,ら著、1994年,Biochemistry,第33巻,4695〜4701頁)。8−オキソ−dGTPは、dCまたはdAのいずれかと塩基対を形成する(Sekiguchi,M.,前記書;Zaccolo,M.,ら著、1996年,J.Mol.Biol.,第255巻,589〜603頁)。
【0197】
本発明のオリゴヌクレオチドプライマーは、定義項目において先に定義した人工ヌクレオチドを含むことができる。人工ヌクレオチドは、本発明のオリゴヌクレオチドプライマーの5’、3’または内部に配することができる。「人工ヌクレオチド」は、オリゴヌクレオチド中に用いて、非特異的アニーリング及び背景増幅させて、ポリヌクレオチド増幅の特異性を増すことができる。この目的で、用いられる人工ヌクレオチドが、従来のヌクレオチド(すなわちdA,dT,dG,dC及びdU)を超える、もう一つの人工ヌクレオチドとの塩基対選択性を示すことが好ましい。一つの態様において、一つまたは二つ以上の人工ヌクレオチドXTP(2−アミノ−6−(N,N−ジメチルアミノ)プリン−5’−三リン酸)またはYTP(ピリジン−2−オン−リボヌクレオシド−5’−三リン酸)が用いられるが、これは、dXTP及びdYTPが、従来のヌクレオチドを超える、互いの塩基対形成選択性を示し、dUTPについての僅かな選択性も示されるからである(Ohtsukiら著、前記書)。
【0198】
本発明のオリゴヌクレオチドプライマー(例えば、第1オリゴヌクレオチドプライマー)は、その5’末端においてGCに富む(すなわち、5’末端ヌクレオチドを含むヌクレオチドの連続的伸長部)と共にその3’末端においてATに富む(すなわち、3’末端ヌクレオチドを含むヌクレオチドの連続的伸長部)配列(例えば、試料特異的配列タグ)を含むことができる。その5’末端においてGCに富みその3’末端においてAtに富む配列の使用は、ATがGCよりも弱い塩基対を形成するので、プライマーアニーリングの特異性を増加させる。従って、ポリヌクレオチドの合成及び増幅の特異性を増すことができる。
【0199】
B.第1cDNA鎖合成のための第1オリゴヌクレオチドプライマー
本発明の方法において、第1cDNA鎖の合成のために第1オリゴヌクレオチドプライマーが用いられる。一つの態様において、増幅産物を生成するための他のプライマー用の鋳型として作用する配列を有する第1オリゴヌクレオチドプライマーも設計される。第1オリゴヌクレオチドプライマーは、20〜100ヌクレオチド長、好ましくは30〜60ヌクレオチド長、より好ましくは30〜45ヌクレオチド長、さらにより好ましくは34〜42ヌクレオチド長であり得る。
【0200】
本発明の一つの独自の特徴は、同じ反応混合物中で二つまたはそれ以上の試料を分析できることである。この目的のために、試料供給源の起源を適当に確認する必要である。好ましくは、第1オリゴヌクレオチドプライマーは、試料特異的タグを含む。例えば、試料Aから第1cDNA鎖を合成するための第1オリゴヌクレオチドプライマーは、試料特異的配列タグAを含み;試料Bから第1cDNA鎖を合成するための第1オリゴヌクレオチドプライマーは、試料特異的配列タグBを含む。試料特異的タグを含むそのような第1オリゴヌクレオチドプライマーの使用は、得られるポリヌクレオチド合成及び増幅産物を同じ供給源に従って区別することができる機構を提供する。例えば、試料AからのcDNAまたは増幅産物は、試料特異的タグBを含む試料BからのcDNAまたは増幅産物と識別される、試料特異的タグAを含む。試料特異的配列タグは、15〜60ヌクレオチド長、好ましくは18〜40ヌクレオチド長、より好ましくは20〜30ヌクレオチド長、さらにより好ましくは20〜24ヌクレオチド長であり得る。
【0201】
本発明による試料特異的配列タグはポリヌクレオチド配列(すなわち、試料特異的配列タグ)であり得る、またはこれは、当該分野で知られている任意の他の確認可能なタグであり得る。異なる第1オリゴヌクレオチド(すなわち、異なる試料)用の試料特異的配列タグは、ヌクレオチド配列が相違する、または単に長さが相違し得る。
【0202】
試料特異的タグ(例えば、試料特異的配列タグ)は、第1オリゴヌクレオチド(すなわち、5’末端ヌクレオチド及び3’ヌクレオチドから離れた少なくとも一つのヌクレオチド)の5’末端、3’末端またはその両方、またはその中間部に配することができる。好ましい態様において、試料特異的タグが、第1オリゴヌクレオチドプライマーの5’末端に配される、すなわち、試料特異的配列の5’に他のヌクレオチドが無い。
【0203】
真核生物mRNAの大部分(ヒストンmRNAが顕著に除かれている)が、3’−末端「ポリA」尾部を有して合成される。ポリ(A)配列はDNA中にコードされず、転写後に核内のRNAに天下される。ポリ(A)の添加は、酵素ポリ(A)ポリメラーゼにより触媒され、200Aまでの残基がmRNAの遊離3’−OH末端に付加される。3’−末端ポリ(A)尾部の存在は、重要な実用的結果を提供する。mRNAのポリ(A)領域は、オリゴ(U)またはオリゴ(dT)と塩基対を形成することができ、この反応を用いて、ポリ(A)+mRNAを単離し、mRNAからcDNAを合成することができる。オリゴ(dT)またはオリゴ(dU)配列をプライマーとして用いて、逆転写酵素を用いる第1cDNA鎖の合成を始めることができる。
【0204】
第1オリゴヌクレオチドプライマーは、さらに、オリゴ(dT)またはオリゴ(dU)配列を含むことができる。好ましくは、オリゴ(dT)またはオリゴ(dU)配列は、試料特異的配列の3’に配される。オリゴ(dT)またはオリゴ(dU)配列は、少なくとも5ヌクレオチド長であり、5〜20ヌクレオチド長、好ましくは8〜18ヌクレオチド長、より好ましくは12〜16ヌクレオチド長であり得る。
【0205】
一つの態様において、試料特異的配列タグは、第1オリゴヌクレオチドプライマーの5’末端に、約20〜24個のヌクレオチドの一般的構造を含む。好ましい態様において、この約20ヌクレオチドの一般的構造には、3’末端においてオリゴ(dT)またはオリゴ(dU)伸長部(12〜16残基)が続く。好ましい態様において、オリゴ(dT)またはオリゴ(dU)伸長部は、試料特異的配列の3’の直ぐ隣である。しかしながら、試料特異的タグとオリゴ(dT)またはオリゴ(dU)伸長部との間に、非試料特異的配列、すなわち、試料Aと試料Bの両方に共通の配列(例えば、少なくとも一つのヌクレオチドまたは少なくとも2、3、5、6または10あるいは20までのヌクレオチド)がある。
【0206】
cDNA合成におけるオリゴ(dT)またはオリゴ(dU)の使用に関わる一つの潜在的問題がある。ポリA尾部はかなり長い可能性があるので、オリゴ(dT)またはオリゴ(dU)を単に用いることは、非ポリA領域の直前において逆転写を正確に開始することができない。実際、オリゴdTはランダムにポリA配列の伸長部にアニーリングするので、単一鋳型mRNAからの逆転写の最終産物であっても、各々が5’−末端において異なる長さのポリTを有する第1cDNA鎖の異種集合を得ることができる。この問題を克服するために、二つまたはそれ以上のデオキシヌクレオチド、例えば、VNを、オリゴ(dT)またはオリゴ(dU)プライマーの3’−末端に付加することができ、ここで、VはdTTPを除く任意のdNTPでありNは四つのdNTPのいずれかである。そのようにして、そのようなプライマーは、ポリA尾部と非尾部領域との結合部において安定にアニーリングし、所定の鋳型から合成された第1cDNA鎖の均一寸法が確保される。その意味において、第1cDNA鎖合成に用いられるプライマーは、実際、変性オリゴヌクレオチドである(Smithら著,1997年,Biotechniques第23巻:274〜279頁)。
【0207】
一つの態様において、第1プライマーの3’末端は、さらに、変性配列、すなわち、二つ以上のヌクレオチド組成を含む配列を含む。第1オリゴヌクレオチドプライマーは、任意の長さ、好ましくは5ヌクレオチド未満、より好ましくは2ヌクレオチド長の変性配列を含むことができる。一つの態様において、第1オリゴヌクレオチドプライマーにおける変性配列はVであり、ここで、Vは、dA,dCまたはdG及びNはdA,dT(またはdU)、dCまたはdGである。好ましい態様において、第1オリゴヌクレオチドプライマーは、5’(試料特異的配列タグ)20〜24(dT)12〜16VN3’の組成を含む。もう一つの態様において、第1オリゴヌクレオチドプライマーは、5’(試料特異的配列タグ)20〜24(dU)12〜16VN3’の組成を含む。
【0208】
第1オリゴヌクレオチドプライマー上のオリゴ(dT)またはオリゴ(dU)伸長部は、各試料中の相補的(ポリA)尾部mRNAにアリーリングされて、第1cDNA鎖合成の開始を可能にする。変性ヌクレオチドは、オリゴ(dT)またはオリゴ(dU)のアニーリングを容易にし、第1cDNA鎖合成の効率を上げる。プライマー特異的配列タグは、二つの試料の各々について独自であり、cDNAの起源を識別させる。
【0209】
変性塩基の使用により、第1cDNA鎖合成用の第1オリゴヌクレオチドプライマーの混合物が得られる。例えば、一つの態様において、各試料用の特定プライマーの混合物を用いて逆転写を行う。これらのプライマーは以下の構造を有する:試料Aについては5’−(特異的配列タグA)20〜24T12〜16AN−3’,5’−(特異的配列タグA)20〜24T12〜16CN−3’,5’−(特異的配列タグA)20〜24T12〜16GN−3’(Nは、A、T、C及びGの混合物を含む変性塩基);試料Bについては5’−(特異的配列タグB)20〜24T12〜16AN−3’,5’−(特異的配列タグB)20〜24T12〜16CN−3’,5’−(特異的配列タグB)20〜24T12〜16GN−3’。試料特異的配列タグは、混合物中の各プライマーについて同一である必要はない。例えば、一つの態様において、各試料についての特定プライマーの混合物を用いて逆転写が行う。これらのプライマーは、以下の構造を有する:試料Aについては5’−(特異的配列タグA1)20〜24T12〜16AN−3’,5’−(特異的配列タグA2)20〜24T12〜16CN−3’,5’−(特異的配列タグA3)20〜24T12〜16GN−3’(Nは、A、T、C及びGの混合物を含む変性塩基);試料Bについて5’−(特異的配列タグB1)20〜24T12〜16AN−3’,5’−(特
異的配列タグB2)20〜24T12〜16CN−3’,5’−(特異的配列タグB3)20〜24T12〜16GN−3’。
【0210】
当該分野で知られている他のヌクレオチドタグも、本発明の試料特異的タグとして用いることができ、これは例えば、Churchら著、(1988年,Science,第240巻:185〜188頁)、Dollinger,(1994年,265〜274頁,Mullisら編,The Polymerase Chain Reaction,Birkhauser,Boston,),Brenner及びLerner,(1992年,Proc.Natl.Acad.Sci.,第89巻:5381〜5383頁)、Alper,(1994年,Science,第264巻:1399〜1401頁)、Needelsら著,(1993年,Proc.Natl.Acad.Sci.,第90巻:10700〜10704頁)及び米国特許第6,280,935,6,172,218,6,150,516,5,846,719,6,172,214,6,235,475に開示されており、これら全てをここで参考のために取り込む。前記特許は、オリゴヌクレオチドタグの使用により分子のクラスまたは下位集合を追跡、確認及び/または分類する方法を開示している。ポリヌクレオチドに付着したタグを、固相支持体上の補体に特異的にハイブリッド形成することにより、最小限の交差ハイブリッド形成セットから選択されたオリゴヌクレオチドを含むオリゴヌクレオチドタグを、ポリヌクレオチドの分類に用いることができる。そのようなオリゴヌクレオチドは、各々、3〜9ヌクレオチド長の複数のサブユニットからなる。最小限の交差ハイブリッド形成セットのサブユニットは、同じセットの任意の他のサブユニットの補体との二つまたはそれ以上のミスマッチを有する二量体または三量体を形成する。特別の態様において利用できるオリゴヌクレオチドタグの数は、タグ当たりのサブユニットの数及びサブユニットの長さに依存する。もう一つの有用なヌクレオチドタグが、標的ポリヌクレオチドの末端への、コードされたアダプターの一つまたは二つ以上のセットのライゲーションに基づくポリヌクレオチド配列分析の方法を提供する米国特許第6,013,445号(ここで参考に取り込む)により開示されている。標的ポリヌクレオチド上の相補突起鎖と完全にマッチした二量体を、その突起鎖が形成するコードされたアダプターをライゲートし、突起鎖中のヌクレオチドの同一性を、
コードされたアダプターの有するオリゴヌクレオチドタグにより決める。
【0211】
好ましい態様において、第1オリゴヌクレオチドプライマーが、前述の固体支持体に供給結合する。この場合、逆転写反応が、支持体に永続的に結合された第1cDNA鎖を発生し、それにより、これら第1cDNA鎖を複数の反応に用い、第1cDNA鎖から、合成した第2cDNA鎖を容易に分離することができる。好ましくは、第1オリゴヌクレオチドプライマーの5’を固体支持体に結合する。
【0212】
もう一つの態様において、第1オリゴヌクレオチドプライマーが、固体支持体に付着させることなく、溶液中で合成される。
【0213】
C.第2cDNA鎖合成のための第2オリゴヌクレオチドプライマー
本発明の方法は、第1cDNA鎖を発生させた後、第2オリゴヌクレオチドプライマーを用いて第2cDNA鎖を合成することを含む。この場合、合成された第2cDNA鎖または二本鎖cDNAが、その後の増幅のための鋳型として用いられる。あるいは、合成された第1cDNA鎖を、増幅用の鋳型として直接用いて、第2cDNA鎖を合成することができる。
【0214】
一つの態様において、第2オリゴヌクレオチドプライマーは、増幅産物を生成するための他のプライマー用の鋳型として作用する配列を用いても設計される。第2オリゴヌクレオチドプライマーは、20〜100ヌクレオチド長、好ましくは17〜60ヌクレオチド長、より好ましくは20〜45ヌクレオチド長、さらにより好ましくは20〜25ヌクレオチド長であり得る。好ましくは、第2オリゴヌクレオチドプライマーは、第1任意配列タグを含む。また好ましくは、一つの試料(例えば、試料A)用の第2オリゴヌクレオチドプライマーは、もう一つの試料(例えば、試料B)用の第2のオリゴヌクレオチドプライマーと同じ第1任意配列を含む。異なる試料から第2cDNA鎖を合成するために用いられる第2オリゴヌクレオチドプライマー中に同じ第1任意配列タグが存在するので、異なる試料から誘導されるcDNAの増幅のために、共通の増幅オリゴヌクレオチドプライマー(例えば、以下に記載のような第3オリゴヌクレオチドプライマー)を用いることができる。
【0215】
第1任意配列タグを、第2オリゴヌクレオチドプライマーの5’または3’末端、あるいは内部に配する(すなわち、5’末端ヌクレオチド及び3’ヌクレオチドから離れた少なくとも一つのヌクレオチド)ことができる。好ましくは、第1任意配列タグが、5’末端に配される、すなわち、第2cDNA鎖合成用の第2プライマーの任意配列の5’に他のヌクレオチドが存在しない。第1任意配列は、5〜30ヌクレオチド長である。
【0216】
第2オリゴヌクレオチドプライマーは、さらに、第1cDNA鎖のサブセット(すなわち、複数)に相補的な第2配列を含み、それにより、試料から二つまたはそれ以上の異なる第2cDNA鎖を合成することができる。好ましくは、第2配列は短い配列、例えば、25ヌクレオチド未満の長さ、好ましくは20ヌクレオチド未満の長さ、より好ましくは15ヌクレオチド未満の長さ、さらにより好ましくは10ヌクレオチド未満の長さであり、それにより、試料から合成された第1cDNAのサブセットにアニーリングすることができる。一つの態様において、第2オリゴヌクレオチドプライマーの第2配列は6〜7ヌクレオチド長である。もう一つの態様において、第2配列は、3’末端においてランダムに選択された配列(例えば、6〜7塩基)を含み、それにより、第2配列に相補的な配列を含む遺伝子(すなわち、第1cDNA鎖)からcDNAのサブセットが合成される。
【0217】
通常、プライマーから延ばすためのポリメラーゼのために3’末端において完全または完全に近いマッチングを有すべきなので、第2オリゴヌクレオチドプライマーの3’末端は非常に重要である。好ましくは、第2の配列が、第1任意配列の3’に配される。一つの態様において、第2配列が、第1任意配列の3’の直ぐ近くに配される、すなわち、第2配列と第1任意配列との間に他のヌクレオチドが存在しない。
【0218】
好ましい態様において、第1任意配列と第2配列との間に第3配列が配される。好ましくは、第3配列は、前述のような一つまたは二つ以上の変性ヌクレオチドを含む。第3配列は、1〜15ヌクレオチド長、好ましくは1〜10ヌクレオチド長、より好ましくは2〜6ヌクレオチド長である。一つの態様において、第1任意配列と第2配列との間に配される第3配列は4ヌクレオチド長である(例えば、図7中のZ4)。第3配列は、全ての変性ヌクレオチドを含む、または、変性ヌクレオチドと非変性ヌクレオチドとの配列を含み得る。第3配列中の変性ヌクレオチドは、dA、dT、dG及びdCのいずれかである、または、dA、dT、dG及びdCの二つまたはそれ以上と塩基対を形成することができるヌクレオチドであり得る。好ましい態様において、第3配列は四つの変性ヌクレオチドを含み、その各々がdA、dT、dG及びdCの二つまたはそれ以上と塩基対を形成することができる。より好ましい態様において、変性ヌクレオチドはdIまたは5−ニトロピロールである。変性ヌクレオチドを含む一つの目的は、プライマーの全体的安定性を増すことである。DNAポリメラーゼがdITP鋳型を読み取ることができ、そのようなdITPをPCRにおける鋳型として用いる場合、四つのdNTPのいずれかをランダムに
組み込むことが知られている。
【0219】
第2及び第3配列の選択は、そこからcDNAが合成され増幅される特定の遺伝子サブセットを決める。第2及び/または第3配列を変得ることにより、合成/増幅産物の寸法を調節するのみでなく、増幅すべき特定の遺伝子ファミリーを選択することができる。例えば、小さいG蛋白は全て、GxGxxGのサインモチーフを有し、ここで、Gはグリシン及びxは任意のアミノ酸を表す。変性オリゴヌクレオチドを用い、このサインモチーフをマッチングさせることにより、全ての小さいG蛋白の発現プロフィールを研究することができる。キナーゼ、ホスファターゼのような同様の多くの蛋白ファミリーは、サインモチーフを有し、多くの機能的ドメインまたはモチーフが、サイン配列を有する(ジンクフィンガー等)。これらのモチーフまたはサイン配列は良好に記録され、これらのモチーフ/サイン配列の詳細な記載を含む検索自由なデータベースがある。例えば、Accelrysにより開発されたGCG Wisconsin Package配列分析ツール(その一部は以前はGCG)は、そのようなモチーフ検索及び記録を提供し、その全内容をここで参考のために取り込む。
【0220】
一つの態様において、第2オリゴヌクレオチドは、5’(第1任意配列)10−12(第3配列)4(第2配列)6−73’の一般的構造を含む。変性塩基(例えば、dA、dT、dG及びdCのいずれか)の使用により、第2cDNA鎖合成用の第2オリゴヌクレオチドプライマーの混合物が得られる。
【0221】
第2オリゴヌクレオチドプライマーは、前述のように固体支持体に結合してよい、または結合しなくてよい。好ましい態様において、第2オリゴヌクレオチドは固体支持体に結合しないが、第1オリゴヌクレオチドは結合し、それにより、合成後に固体支持体に結合する第1cDNA鎖から、合成された第2cDNA鎖を容易に分離することができる。
【0222】
第2オリゴヌクレオチドプライマーの第1任意配列を設計する際、cDNA合成及び増幅反応の特異性を増加させ背景を低下させるために、その配列が、GenBankデータベースまたは他の利用できるデータベースにおける任意の哺乳動物ゲノム配列における配列に強度にマッチングしないことが好ましい。「強度のマッチング」は、第1任意配列と、種、例えばGenBankデータベースまたは他のデータベースで利用できるヒトまたは全ての哺乳動物の配列との間の配列同一性が30%未満(例えば、20%未満、10%未満、5%未満、2%未満の配列同一性)であることを意味する。一部の態様において、試料供給源が知られている、例えば、ヒト、イヌまたは他の動物あるいは植物のような特定の種である場合、GenBankデータベースまたは他のデータベースにおける特定の種について、第1任意配列は、ゲノム配列中の配列に強度のマッチングを示さないことが好ましい。
【0223】
D.オリゴヌクレオチドプライマーの標識
本発明のオリゴヌクレオチドプライマーは、前述のように、分光的、光化学的、生化学的、免疫化学的、酵素学的または化学的手段により検出可能な部分を組み込むすることにより標識することができる。オリゴヌクレオチドプライマーに標識を結合または共役させる方法は、もちろん、用いられる標識の型及び、プライマー上の標識の位置に依存する。本発明で有用なプライマーは、5’末端、3’末端で標識、またはプライマーの長さ全体において標識することができる。
【0224】
本発明での使用に適当な種々の標識、及びそれらをプライマー中に含むための方法は当該分野で知られており、限定はされないが、互いに相互作用して信号を増加、変化または減衰させる得る、酵素(例えば、アルカリホスファターゼ及びホースラディッシュペルオキシダーゼ)及び酵素基質、放射活性原子、蛍光染料、発色団、化学発光水準、電気化学発酵水準、例えば、Origen(登録商標)(Igen)を含む。もちろん、熱サイクラー装置を用いて行われるPCR系アッセイにおいて標識分子を用いる場合、標識が、この自動プロセスで必要な温度サイクルに耐えなくてはならない。
【0225】
本発明の標識プライマーの組み立てにおける標識として用いられる蛍光体には、ローダミン及び誘導体(テキサスレッド等)、フルオレセイン及び誘導体(5−ブロモメチルフルオロセイン等)、ルシファーイエロー、IAEDANS、7−Me2N−クマリン−4−酢酸、7−OH−4−CH3−クマリン−3−酢酸、7−NH2−4−CH3−クマリン−3−酢酸(AMCA)、モノブロモバイマン、ピレントリスルホネート、例えば、Cascade Blue、及びモノブロモリメチル−アンモニオバイマンがある。通常、モノ色素計ではなくフィルターを有する蛍光計を用い、検出効率を増すためには、ストロークの移動が大きな蛍光体が好ましい。
【0226】
標識は、種々の技術により、オリゴヌクレオチドに直接または間接的に付着することができる。用いられる標識またはタグの正確な型に依存して、標識は、プライマーの5’末端に配する、またはプライマーの内部に配する、または種々の寸法及び組成のスペーサーアームに付着させて、信号相互作用を容易にすることができる。市販のホスホラミダイト試薬を用いて、適当に保護されたホスホラミダイトを介して5’末端において官能基(例えば、チオールまたは一次アミン)を含むオリゴマーを製造することができ、例えば、PCR Protocols:A Guide to Medhods and Applications,Innisら編,Academic Press,Ind.,1990年に記載のプロトコルを用いてそれらを標識することができる。
【0227】
典型的には5’末端において、オリゴヌクレオチドプライマー配列中に、一つまたは二つ以上のスルフヒドリル、アミノまたはヒドロキシル基を導入するためにオリゴヌクレオチド官能化試薬を導入するための方法が、米国特許第4,914,210号に記載されている。ポリヌクレオチドキナーゼ及びガンマ−32P−ATPまたはガンマ−33P−ATPを用いることにより5’リン酸基を放射性同位体として導入して、レポーター基を提供することができる。合成中に導入されたアミノチミジン残基、6−アミノヘキシル残基を、ビオチンのN−ヒドロキシスクシンイミドエステルと反応させることにより、ビオチンを5’末端に付加することができる。
【0228】
(cDNAの合成)
本発明の方法により、cDNAを調製し増幅に用いることができる。一部の態様において、第1のcDNA鎖を調製し、その後の増幅反応及び分析に直接用いる。本発明の好ましい態様において、第1cDNAと第2cDNAの両方が合成される。合成された第1及び第2cDNA鎖をその後の増幅反応に用い、第2cDNA鎖を、第1cDNA鎖から分離して増幅反応に用いることができる。
【0229】
cDNAの調製は良く知られており、文献(例えば、Ausubelら著、前記書)に充分に記載されており、以下に記載する。
【0230】
cDNAは、以下の方法により調製することができる。全細胞RNAを単離(前述のように)し、オリゴ(dT)またはオリゴ(dU)−セルロースのカラムを通してポリA RNAを単離する。結合したポリA mRNAを、低イオン強度緩衝材を用いてカラムから溶離する。cDNA分子を製造するために、前述のようなオリゴ(dT)nまたはオリゴ(dU)n(ここで、nは好ましくは12〜16ヌクレオチド長)を含む第1オリゴヌクレオチドプライマーを、DNA合成用の鋳型としてRNAを用いる酵素である逆転写酵素用のプライマーとして用いられるポリA尾部にハイブリッド形成する。あるいは、mRNA種を、cDNA合成用のプライマーとして、所望のmRNAに相補的な多くの配列を含む短いオリゴヌクレオチドフラグメントを用いることにより、多くの位置から用意する。得られるRNA−RNAハイブリッド(すなわち、RNA及び第1cDNA鎖)は、当該分野で知られている種々の酵素的ステップ(Watsonら著、1992年、Recombinant DNA、第2版、Scientific American Books,ニューヨーク)により二本鎖DNA分子(すなわち、第1及び第2cDNA鎖)に転化される。
【0231】
本発明の一つの態様において、第1cDNA鎖が、試料特異的配列を含むオリゴヌクレオチドプライマーを用いて合成され、それにより、合成された第1cDNA鎖をその試料供給源について確認することができる。従って、第1cDNA鎖合成に用いられるオリゴヌクレオチドプライマーは、少なくともオリゴ(dT)またはオリゴ(dU)配列及び試料特異的配列を含む。
【0232】
一つの態様において、試料Aから単離されたmRNA及び、試料A特異的配列を含むオリゴヌクレオチドプライマーを用いて、試料A用の第1cDNA鎖が合成される。試料Bから単離されたmRNA及び、試料B特異的配列を含むオリゴヌクレオチドプライマーを用いて、試料B用の第1cDNA鎖が合成される。試料A特異的配列は、異なるヌクレオチド同一性を含むことにより試料B特異的配列から異なる、及び/または、これは、異なる長さのヌクレオチドを含むことにより試料B特異的配列から異なる。
【0233】
好ましい態様において、第1オリゴヌクレオチドプライマーは、例えばビーズ上で共有結合により、固体支持体に結合される。このことが有利であるのは、一旦ビーズ上で合成されると、これら第1cDNA鎖を過剰の試薬から容易に洗浄し精製することができ、それにより、これらのビーズを別の反応で直接用いることが可能であるからである。第2に、第2cDNA鎖合成(以下参照)後、これらの結合した第1cDNA鎖を、二本鎖DNAを変性することにより第2cDNA鎖から分離することができ、それにより、他の関連または関連しない実験において用いることができ、例えば、分離された第2cDNA鎖を、その後の増幅反応により増幅することができる。
【0234】
好ましくは、第1cDNA鎖の合成は、逆転写反応による。第1cDNA鎖は、RNA試料を、試料中のRNA試料の逆転写に充分な条件下に第1オリゴヌクレオチドプライマー及び必要な試薬と接触させることにより調製される。プライマー及びRNAと接触される必要な試薬は、当業者に知られており、通常、少なくとも、逆転写活性を有する酵素及び、dNTPを適当な緩衝媒体中に含む。
【0235】
第1cDNA鎖合成ステップのために、逆転写活性を有する種々の酵素、通常、DNAポリメラーゼを用いることができる。適当なDNAポリメラーゼの例には、好熱性バクテリア及び始原菌、レトロウイルス、酵母、アカパンカビ、ショウジョウバエ、霊長類及びげっ歯類からなる群より選択される有機体から誘導されるDNAポリメラーゼが含まれる。好ましくは、DNAポリメラーゼは、米国特許第4,943,531号に記載のモロニーネズミ白血病ウイルス(M−MLV)及び米国特許第5,405,776号(この開示をここで参考のために取り込む)に記載のRNaseH活性を欠くM−MLV逆転写酵素、鳥類骨髄芽球症ウイルス(AMV)、ヒトT細胞白血病ウイルスI型(HTLV−I)、ウシ白血病ウイルス(BLV)、ラウス肉腫ウイルス(RSV)、ヒト免疫不全ウイルス(HIV)及び、米国特許第5,322,770号に記載のThermus aquaticus(Taq)またはThermus thermophilus(Tth)(その開示をここで参考のために取り込む)、並びに米国特許第5,436,149号に記載のBcaBEST(登録商標)DNA Polymerase(その開示をここで参考のために取り込む)からなる群より選択される。逆転写酵素活性を有する適当なDNAポリメラーゼは、市販の有機体、または当業者に知られている方法によりポリメラーゼをコードするクローンド遺伝子を高水準で発現する細胞から得られる有機体から単離することができ、ポリメラーゼを得る特別の方法は、主に、便利さ、費用、入手性等のような因子に基づいて選択される。
【0236】
初回処理RNAの逆転写による第1cDNA鎖合成に必要な種々のdNTP及び緩衝培地は、Clontech、Sigma、Life Technologies、Amersham、Boehringer−Mannheimを含む種々の供給源から市場で購入することができる。第1鎖合成に適した緩衝培地は、通常、Tris−HCl、HEPES−KOH等のような緩衝剤を、通常、典型的に6〜9の範囲のpHを支持する10〜100μMの範囲の濃度;KCl、NaCl等のような一価イオンを含む塩を0〜200mMの範囲の濃度で;MgCl2、Mg(OAc)等のような二価カチオンを含む塩を、通常1〜10mMの範囲の濃度で;及び緩衝剤のようなさらなる試薬、例えば、DDT、洗剤、アルブミン、ポリアルコール(グリセロール)等を含む。試薬混合物の条件は、効率的な第1鎖合成を促進するように選択される。典型的には、プライマーは最初に、高温、通常50〜95℃でRNA試料と組み合わされ、続いて、温度を約0〜60℃の範囲に下げて、試料中の対応するRNAへのプライマーの特異的アニーリングを確保する。このアニーリングステップに続いて、初回処理したRNAを次に、逆転写及び、初回処理RNAの第1cDNA鎖合成を促進するのに充分な条件下にdNTP及び逆転写酵素と組み合わされる。プライマーのRNAへのアニーリングの後、ポリメラーゼの活性を遅らせるまたは開始時間を調節することができ場合、適当なタイプの試薬を用いることにより、試薬の全てを一度に組み合わせることができる。
【0237】
一部の態様において、第2cDNA鎖の合成のために、第1cDNA鎖を鋳型として用いる。
【0238】
任意に、RNase H消化によりまたは0.1〜1M NaOHでの処理により、第2cDNA鎖の合成前に、RNAを除去することができる。
【0239】
所定の試料の発現プロフィールをかなり複雑にすることができ、任意の系の溶解がある程度に限定されるので、発現された遺伝子の、全セットよりも、サブセットを選択的に増幅させることが有利である。このことも有用であり得るのは、ある状況において、所定のサブセットの遺伝子のみが有益であり、有益でない他の遺伝子は濾去して信号−ノイズ比を高めることが有利であるからである。しかしながら、完全ゲノムの分析が望まれる場合、第1cDNA鎖を固体支持体(前述)に結合すると、異なるプライマーを用いる複数ランを容易に達成することができる。従って、第2鎖プライマー(すなわち、第2オリゴヌクレオチドプライマー)の同一性は、発現遺伝子のどのサブセットが増幅するか定める。
【0240】
第2cDNA鎖は第1cDNA鎖にアニーリングされ、最初のmRNAの完全二本鎖DNA複製を形成する。
【0241】
第2オリゴヌクレオチドプライマーの組成は、合成される全ての発現遺伝子のサブセットを定める。通常、DNAポリメラーゼ初回処理に最も重要である前述のプライマーの3’末端は、合成すべきcDNA分子の選択に役立つ短い配列(例えば、6〜7bp配列)を含む。
【0242】
哺乳動物ゲノムの発現部分におけるそのような短い3’末端初回処理配列の発生を推定することができる。例えば、6bpパリンドローム配列を用いる場合、用いられる特定の配列に依存して、現在のマウス配列データ(例えば、配列マウスcDNA中に1.5×108塩基)を用いて約5,000〜10,000回の発生が予測される。特定の細胞及び組織が、全ゲノム中の全ての遺伝子の一部(10〜30%)のみを発現するので、及び、一般的に用いられるPCR条件下では、2,000塩基より長い転写体が増幅する可能性が低いので、500〜2,000の個体の転写体が検出されることが予想される。そのようなプライマーを用いる単一反応において、発現遺伝子の約5〜10%(5,000/1.5×108(頻度)×2,000(増幅可能フラグメントの寸法)×100=6.7%)が覆われることが推定される。従って、約20の別々の反応(同じ遺伝子試料を用いる)が、単一哺乳動物ゲノム中の全ての転写配列のかなりの部分を覆うことが予測される。
【0243】
天然ポリヌクレオチドには、一般的に見られない塩基が、本発明のポリヌクレオチドに含まれてよく、その例には、イノシン及び7−デアザグアニンがある。相補性は完全である必要がなく、適当な二量体はミスマッチ塩基対またはマッチしていない塩基を含み得る。ポリヌクレオチド技術の当業者は、例えば、オリゴヌクレオチド長、オリゴヌクレオチドの塩基組成及び配列、イオン強度、及びミスマッチ塩基対の発生率を含む変数の数を考慮して、二量体安定性を経験的に決めることができる。
【0244】
ポリヌクレオチド二量体の安定性を、溶融温度すなわち「Tm」により測定する。特定条件下での特定のポリヌクレオチドのTmは、塩基対の半分が解離する温度である。二本鎖DNA分子の溶融温度は、顕著にその塩基組成に依存する。GC塩基対に富むDNA分子は、AT塩基対に富むものよりTmが高い。Tmの塩基組成への依存は直線的であり、G−C含量の1%増加毎に約0.4℃増加する。GC塩基対は、二つの水素結合ではなく三つの水素結合により塩基が一緒に保持されるので、AT対よりも安定である。さらに、隣接GC塩基対は、隣接AT塩基対よりも、より強度に互いに相互作用する。ここで、DNAのATに富む領域はより容易に溶融する。
【0245】
Tmへの主要な効果は、溶液のイオン強度により発揮される。一価カチオン濃度の10倍増加毎にTmが16.6℃増加する。最も一般的に用いられる条件は、一価Na+濃度が0.18MとなりTmが90℃台となる0.12Mリン酸緩衝液中でDNAを操作することである。Tmは、水素結合を不安定化させるホルムアミドのような試薬の存在下に反応を行うことにより大きく変化させることができる。これによりTmが40℃の低い温度まで下げられ、高温に曝すことから生じ得る損害(例えば、鎖破損)をDNAが被らないという利点が得られる(Stryer,Biochemistry,1998年,第3版,W.H.Freeman及びCo.,81〜82頁及びLewin,Genes II,1985年,John Wiley&Sons,63〜64頁)。
【0246】
合成された第2cDNA鎖を、任意に、固体支持体に結合された合成された第1cDNA鎖から分離することができる。次に、結合した第1cDNA鎖を単離し、後に他の反応で用いることができる。あるいは、これらの結合二本鎖cDNAを、その後のPCRに直接用いることができる。
【0247】
一つの態様において、新しく合成した第2cDNA鎖が、例えば、cDNAを変性温度、すなわち、Tmより高い温度に曝すことにより、結合した第1cDNA鎖から分離される。結合した第1cDNA鎖は、次に、分析用の第2cDNA鎖の新しいプールを産生するために異なるオリゴヌクレオチドプライマーを用いることにより、さらなる分析に再利用することができる。
【0248】
あるいは、cDNAフラグメントの特定のプールを、適当な制限酵素(例えば、導入したパリンドローム部位の確認)の作用による二本鎖DNAの酵素的消化により、及び特異的配列「C」及び5’末端及び、制限酵素により発生したオーバーハンドと適合する一本鎖オーバーハンドを含む特異的アダプターのライゲーションにより、産生することができる。
【0249】
(増幅)
合成したcDNA(例えば、第1鎖、第2鎖または二本鎖)を用いて、分析用の増幅産物を産生する。本発明において、当該分野で知られている他の増幅方法(例えば、LCR及びNSBA)も用いることができるが、PCR増幅が好ましい。
【0250】
PCR法は、当業者に良く知られており、例えば、Mullis及びFaloona,1987年,Methods Enzymol.,第155巻:335頁,Saikiら著、1985年,Science第230巻:1350頁,及び米国特許第4,683,202,4,683,195及び4,800,159号に記載されており、ここに参考として取り込む。その最も単純な型において、PCRは、反対側の鎖にハイブリッド形成すると共に標的DNAの所望の領域に隣接する二つのオリゴヌクレオチドプライマーを用いて、特定のDNA配列を酵素的に合成する試験管内方法である。鋳型変性、プライマーアニーリング及びDNAポリメラーゼによるアニーリングしたプライマーの延長を含む反応ステップの繰り返しにより、その末端がプライマーの5’末端により定められる特性のフラグメントが指数関数的に蓄積する。PCRは、109の係数で特定のDNA配列の選択的豊富化を発生させることができると報告されている。
【0251】
本発明において、PCRは、鋳型DNA、すなわちcDNA(少なくとも1fg;より有用には、1〜1,000ng)及び少なくとも25pmolのオリゴヌクレオチドプライマー(すなわち、第3及び第4オリゴヌクレオチドプライマー)を用いて行われる。例えば、典型的反応混合物は:cDNAの1〜1,000pg、オリゴヌクレオチドプライマー25〜100pmol、適当な10×緩衝液2.5〜10μl、10μM dNTP0.4〜2μl、Taq DNAポリメラーゼ2.5単位、及び合計体積25〜100μlとする脱イオン水を含む。鉱油を上に乗せ、PCRを、プログラム可能な熱サイクラーを用いて行う。
【0252】
好ましくは、第3オリゴヌクレオチドプライマーは、第1オリゴヌクレオチドプライマーの試料特異的配列を含む。好ましい態様において、第3オリゴヌクレオチドプライマーは、試料特異的配列の全体または一部を含み、その相補的配列(すなわち、第2cDNA)にアニーリングすることができる。この態様は、好ましくは、第4オリゴヌクレオチドプライマー(すなわち、第3オリゴヌクレオチドプライマーに反対の方向)も含む。好ましくは、この第4オリゴヌクレオチドプライマーは、第2オリゴヌクレオチドプライマーの第1任意配列を含む。異なる試料の第2cDNA鎖を、同じ第2オリゴヌクレオチドを用いて合成する場合、同じ第4オリゴヌクレオチドプライマーを用いて、PCRによりcDNAを増幅させることができる。
【0253】
試料特異的配列を含む第3オリゴヌクレオチドプライマーの使用により、増幅産物を、それらの同一性の痕跡を失うことなく、同じ起源に従って確認できることが確保される。
【0254】
PCRサイクルの各ステップの長さ及び温度、並びにサイクル数は、効果の厳格な要求に従って調節される。アニーリング温度及びタイミングは、プライマーが鋳型にアニーリングすることが予想される効率と、許容すべきミスマッチの程度との両方により決められる。プライマーアニーリング条件の過酷度を最適化する性能は、当業者の知識に充分収まるものである。30℃〜72℃のアニーリング温度が用いられる。鋳型分子の初期変性が、92℃〜99℃で4分間起こり、その後に、変性(94〜99℃で15秒〜1分間)、アニーリング(温度は前述のように決められる;1〜2分)及び伸長(72℃で1〜3分間)が20〜40サイクル行われる。好ましくは、増幅産物が、検出可能な標識で標識され、それによりそれらの同一性及び存在量を検出することができる。先に定義の検出可能な標識(例えば、蛍光、放射能または比色標識)を、種々の手段により増幅産物に結合させることができる。例えば、dNTPが標識され、dNTPが一旦ポリヌクレオチド中に組み込まれると、増幅ポリヌクレオチドが標識されることになる。あるいは、増幅に用いるプライマーを標識して、同様に増幅ポリヌクレオチドを標識することができる。さらに、標識されたプローブ(例えば、増幅産物に相補的なオリゴヌクレオチド)を用いて増幅産物にハイブリッド形成し、それにより、増幅産物に検出可能なサインを与える(すなわち、ハイブリッド形成アッセイ)。
【0255】
好ましい態様において、各試料特異的PCTプライマー(すなわち、第3オリゴヌクレオチドプライマー)の5’末端が、特定の蛍光標識に結合し、それにより、PCR産物の系列を、試料起源に従って、それらの蛍光マーカーにより容易に追跡することができる。さらに、蛍光信号の強度は、PCR産物の量に正比例する。所定の産物の蛍光強度を記録することにより、異なる起源のPCR産物間の比を得ることができる。
【0256】
各試料は、好ましくは、その特異的蛍光標識を有するが、同じ蛍光標識を、二つ以上の試料で用いることができる。例えば、試料A用のフルオレセインでタグした第3オリゴヌクレオチドプライマーが、試料Bのものより1塩基短く(または長い)、分離手段が1bpの相違を検出するのに充分に感受性である場合、これらの二つの試料から生じる「同じ」PCRフラグメントが、1bp寸法が異なる二つの近いピークとして分析される(例えば、変性高性能液体クロマトグラフィー(DHPLC)により)。寸法の相違が説明される場合、同じ記録及び計算を実現することができる。2以上の試料について同じ手法を用いることができる。
【0257】
蛍光標識は好ましい標識であるが、同じ目的の達成のために他の標識も用いることができる。labが異なる分子量の同位体である場合(すなわち、P31対P32対P33;O16対O18、等)、試料A用のプライマーは、試料B用のプライマーより「重い」。そのような相違により、検出され得る閾値、例えば分光測定により分けられる異なる起源のPCR産物が得られ、これらの密に関連するピークに基づいて比を計算することができる。
【0258】
一つの態様において、二つまたはそれ以上の試料により調製される集められたcDNAは組み合わされて、二つのプライマー対を用いるPCR増幅に付される(図3)。プライマー4は共通プライマーであり、プライマー2と同じであるか、プライマー2中の5’末端独自配列に同一である。プライマー3A及び3Bは、逆転写中にDNAに組み込まれる試料特異的タグ配列と同じである。さらに、これらのプライマーの各々が、プライマーの5’末端において特定の蛍光標識を含む。これは、これらの二つの別々の試料から生じるPCR産物が、PCRを同じ反応混合物中で行っても、異なる試料特異的蛍光体により別々に標識されることを確保する。プライマー3A及び3Bは、逆転写反応中に導入された特異的配列A1、A2、A3及びB1、B2、B3に同一の対応するプライマー3A1、3A2、3A3及び3B1、3B2、3B3の混合物を表す。これらのプライマーの各々が、独自の蛍光染料を含むことができる。
【0259】
各試料についての単一プライマーの代わりにプライマー混合物を用いることにより、単一反応中で分析することができる遺伝子の数が増加する。mRNA中のポリAの前にあるヌクレオチドに依存して特定の配列A1〜A3及びB1〜B3が組み込まれると共にそれらの増幅の産物が異なる蛍光チャンネルにおいて現れるので、この方法は、類似の寸法を有するがポリA配列の前にあるヌクレオチドが異なるDNAフラグメント間において識別することができる。
【0260】
本発明の一部の態様において、鋳型として先行する増幅反応における増幅産物を用いてネステッド増幅が行われる。ネステッドPCRの使用は、種特異的産物の収率を大幅に高めることもでき、それにより、単一プライマー対がそれ自体失敗する場合、アッセイの感受性も高められる。好ましくは、ネステッドPCRプライマーの一つは、試料特異的配列を含み、それにより、増幅産物の同じ起源の追跡が維持される。また好ましくは、試料特異的配列を含むプライマーが、特定の検出可能な標識で標識されて、増幅産物の検出及び分析が可能となる。例えば、ネステッドPCRを含む方法は、二つの連続PCR反応を含む。第1のプライマー対を用いる(例えば、第3及び第4のオリゴヌクレオチドプライマーを用いる)複数サイクルのPCR(例えば、10〜40、または10〜30、または10〜20サイクル)の後、少量の第1反応(例えば、50μl反応の1μl)が、第1対の内部にあるまたはその間に入っている配列にアニーリングする新規プライマーセットを用いる第2の複数サイクルのPCR反応(例えば、10〜40、または10〜30、または10〜20サイクル)用の鋳型として作用する。
【0261】
ネステッドプライマーを設計しネステッドPCRを実施するための方法は、当該分野で知られている(Current Protocol in Molecular Biology、前記書、を参照)。前述のようなプライマーを選択するための一般的基準は、ネステッドプライマーの設計にも適用される。両方のネステッドプライマーが、第1のプライマー対及びネステッドプライマーの少なくとも一つの内部(例えば、その中)の配列にアニーリングする必要があるが、本発明によれば、試料特異的配列を含む必要がある。
【0262】
(増幅産物の分離及び検出)
予定の時間またはサイクル(例えば、第5サイクル、第8サイクル、第10サイクル、または他のサイクル)から出発するPCR増幅中、少量、例えば、1%〜40%(v/v)の反応混合物が、各サイクル後に自動的に引き出され、反応混合物には、dNTP、蛍光標識したプライマー及びDNAポリメラーゼのような等体積の新しい成分が補給される。引き出された試料は、次に、分離され分析される。ポリヌクレオチドの存在または存在量を検出する方法は、当該分野でよく知られており、定量的分析を同時に行い得ることが好ましいが個々のポリヌクレオチドを分離することができる限り、それらのいずれかを、本発明の方法で用いることができる。増幅産物の分離及び分析のために有用な方法は、限定はされないが、電気泳動(例えば、キャピラリー電気泳動(CE))、クロマトグラフィー(dHPCL)及び質量分析を含む。
【0263】
一つの態様において、CEは好ましい分離手段であるが、それは、少なくとも10〜1,000塩基対の範囲のポリヌクレオチドの例外的分離を提供し単一塩基対を分離するからである。CEは、ここで参考として取り込まれる米国特許第6,217,731;6,001,230及び5,963,456号に開示のような、当該分野でよく知られている方法により行うことができる。最近開発された処理CE装置は市販されており、例えば、Spectrumedix Corporation(State College,PA)からのHTS9610高処理量分析システム及びSCE9610完全自動化96キャピラリー電気泳動遺伝子分析システム;Beckman Instruments Inc(Fullerton,カリフォルニア)からのP/ACE 5,000系及びCEQ系;及びABI PRISM 3100系分析器(Applied Biosystems,Foster City,カリフォルニア)がある。CEカラムの最終近くに、増幅DNAフラグメントは、両方の蛍光標識の信号を測定する蛍光検出器を通過する。これらの装置は、蛍光標識したPCR産物の検出のための自動化高処理量を提供する。
【0264】
本方法におけるCEの使用により、従来のスラブゲル電気泳動と比較して、より高い生産性が得られる。分離速度は、高い電場がゲルに適用された場合に発生する熱故に、スラブゲル電気泳動において制限される。キャピラリーの大きな表面積からの熱の除去が迅速であるので、キャピラリー電気泳動に、より高い電場を適用することができ、それにより、分離プロセスが速くなる。キャピラリーゲルを用いることにより、分離速度が、従来のスラブゲルシステムよりも約10倍増加する。
【0265】
CEを用いると、複数の試料を同時に分析することができ、これは高処理量に必須である。これは、本発明の一つの態様において複数キャピラリーシステムを用いることにより達成される。しかしながら、DNA塩基からの蛍光の検出は、多孔質マトリクス及びキャピラリー壁からの光の散乱により複雑になり得る。光の散乱を避けるために、共焦蛍光スキャナーを用いることができる(Quesadaら著,1991年,Biotechniques第10巻:616〜25頁)。
【0266】
一つの態様において、本方法は、元の試料に含まれる特定のcDNA(すなわち、mRNA)の幾つの複製がPCR増幅用の鋳型として用いられるか測定する。元の複製の数を決めるために、核酸抽出の効率及び、各PCR反応の効率を知らなくてはならない。さらに、検出ステップにより、標的配列の幾つの複製が作られたか示されるが、元の試料に幾つの複製が含まれていたかは示されない。
【0267】
好ましい態様において、試料に含まれる標的配列の複製の正確な数ではなく、遺伝子発現の相違が測定される。検出された蛍光信号強度(例えば、その後のCE分離)を記録することができ、二つの試料からの各ピークの相対比を決めるために用いることができる(図4)。好ましい態様において、二つまたはそれ以上の試料から誘導されるcDNAが、同じPCR反応において増幅される。各試料は、共通のプライマー(例えば、第4オリゴヌクレオチドプライマー)及び試料特異的プライマーにより増幅され、それにより、異なる試料からのcDNAが、同じ共通プライマーについて競合する。この競合故に、二つの試料からの増幅産物の量の比が、二つの試料の各々における初期標的ポリヌクレオチドの量の比を反映する。例えば、1の比(例えば、試料A/試料B)は、試料A及びB中の標的ポリヌクレオチドの同じ初期量、すなわち、標的ポリヌクレオチドが二つの試料中で異なって発現されないことを示す。1を超える比(例えば、試料A/試料B)は、試料B中よりも、試料A中の標的ポリヌクレオチドの量が高いことを示す。1より小さい比(例えば、試料A/試料B)は、試料B中よりも、試料A中において標的ポリヌクレオチドの量が少ないことを示す。前述の両方の場合において、標的ポリヌクレオチドが、二つの試料中において異なって発現される。2つの試料中に存在する主要なポリヌクレオチドの量(すなわち、これらのポリヌクレオチドの発現水準)が略同じであり、従って、増幅の比が一定(例えば、約1)に維持されることが予測される。
【0268】
もう一つの好ましい態様において、CEにより分離される各PCRフラグメントについて(及び、従って、各遺伝子について)の信号強度を、サイクル数の関数としてプロットする。信号強度は、電気泳動図のピークの合計面積により表すことができる。閾値サイクル数(Ct)は、各増幅遺伝子についてPCRフラグメントの信号強度が設定された閾値数(例えば、信号強度の背景値の10の標準偏差)に達したときにサイクル数として計算される。特定遺伝子の操作が異なる発現は、一つのサイクルにおけるよりも、二つ(またはそれ以上)の試料におけるこの遺伝子についての閾値サイクル数(Ct)における相違として決められる。さらに閾値サイクル数を用いて、各遺伝子についての複製数を誘導し、二つまたはそれ以上の試料における遺伝子についての複製数の比による発現の相違を測定する(図5a)。
【0269】
この方法は、各増幅遺伝子についての増幅サイクルの数の関数としての信号強度変化の割合[サイクル数の関数としての信号強度の導関数、d(信号強度)/d(サイクル数)]をプロットすることも含む。一つの試料からの各増幅遺伝子についてのd(信号強度)/d(サイクル数)の最大値に対応するサイクル数として決められる択一的閾値サイクル(aCt)を、もう一つの試料からの同じ遺伝子についてのaCtと比較する。二つまたはそれ以上の試料における同じ遺伝子についてのaCt値間の一サイクルの相違を、択一的操作相違発現と定義する(図5b)。
【0270】
また好ましくは、この方法は、さらに、操作相違発現または択一的操作相違発現を示す一つまたは二つ以上の遺伝子に対応するPCRフラグメントを集め、一つまたは二つ以上の遺伝子の配列を確認することを含む。
【0271】
二つの試料における特定のポリヌクレオチドの比を、二つの試料間で異なって発現されるかどうか決めるための公比に対して測定する。ここで用いられる「公比」という用語は、二つの試料間で発現される全ての遺伝子の相対的一定比を意味する。これは、非処理試料と比較される処理試料中の特定の信号導入経路の活性化のような特定の事象により引き起こされる特定の変化よりも、全体的変化(全出発材料の量)を反映する。特定遺伝子の発現の比をこの公比と比較することにより、比較される試料間で特定遺伝子の発現が異なるかどうか直ちに明らかになる。
【0272】
別々のPCR反応において二つの試料を増幅する場合、各PCR増幅について内部制御が提供され、比が計算される前に内部制御によって、各試料の増幅が、まず正常化される。定量的PCRのために内部制御を使用することは当該分野でよく知られており、例えば、Ausubelらにより記載されている。二つの基本的タイプの制御がある:第1のものは、外因性制御として一般的に知られており((Gillilandら著、(1990年)PCR Protocols,Innisら著、60〜69頁,Academic Press;Wangら著、(1989年)Proc.Natl.Acad.Sci.USA第86巻:9717〜9721頁、その両方がここで参考のために特に取り込まれる)、第2のものは、内因性制御として知られている(Dvekslerら著、(1992年)PCR Methods 及びApplications第6巻:283頁から285頁;Spanakis(1993年)Nucleic Acids Research第21巻:3809〜3819頁、その両方がここで参考のために特に取り込まれる)。
【0273】
外因性制御は、既知の濃度で抽出ステップまたはPCRステップのいずれかに添加される人工的導入核酸分子の使用を含む。定量化のための内部標準として作用するために既知の濃度で外因性核酸を添加する概念は、Chellyら著、(1988年)Nature第333巻:858〜860頁により導入され、これをここで特に参考のために取り込まれる。従って、標的配列として同じプライマーを用いて増幅される制御フラグメントを利用することは、内部標準に対する、標的配列増幅効率をより正確に反映する(例えば、WO93/02215;WO92/11273;米国特許第5,213,961及び5,219,727号を参照のこと:全てここで参考のために取り込む)。同様の手法が、NASBA(Kievitsら著、1991年,J Virol Methods.第35巻:273〜86頁)またはSDA(Walker,1994年,Nucleic Acids Res.第22巻:2670〜7頁)のような等温増幅反応を利用する核酸の定量的測定のために効果的であるとわかった。
【0274】
内因性制御の使用は、抽出効率の変化を調整する。制御の選択は、機能するために幾つかの要求事項を満たすべきことにおいて重要である。第1の要求は、制御の複製数が一定でなくてはならないことであり:第2の要求は、制御が、モニターされる配列を同様の効率で増幅させるべきことである。幾つかの構成的に発現された遺伝子は、制御候補として考えられてきた。これらの遺伝子の発現は、種々の条件において比較的一定だからである。その例には、限定はされないが、β−アクチン遺伝子、グリセルアルデヒド−3−ホスフェートデヒドロゲナーゼ遺伝子(GAPDH)及び16SリボソーマルRNA遺伝子がある。これらの遺伝子は構成的に発現されていると考えられる。
【0275】
異なって発現されたポリヌクレオチドの分類について、任意に閾値を設定することができる。例えば、1.2より大きく0.5より小さい比を有するポリヌクレオチドは、一つの態様による二つの試料における異なって発現されたポリヌクレオチド(すなわち、遺伝子)とみなされる。異なって発現されたと確認されるポリヌクレオチドは、例えば、自動分取装置により集めることができ、遺伝子の同一性は、通常のDNA塩基配列決定により達成することができる。自動分取装置は、例えば、Bio−Rad Laboratories(Hercules,カリフォルニア)から市販されている。
【0276】
もう一つの態様において、溶出PCRフラグメントの分子量の決めるためにCEを校正することができ、第2cDNA鎖を選択的に合成するために用いられる正確な配列が知られているので、所望の各PCRフラグメントの同一性を、利用できるゲノム配列データベース情報に基づいて容易に決めることができる。ヒトゲノムは完全に塩基配列決定され、大腸菌、酵母、C.elegan及びショウジョウバエのような他の有機体も塩基配列決定されている。DNA塩基配列決定技術が迅速に発達したので、大部分の所望の他の有機体の全ゲノムが、まもなく完全に塩基配列決定されるであろう。
【0277】
プロファイリングするこの方法の一つの独自の特徴は、全増幅プロセスにおいてPCRをモニターするその性能である。これに対して、相違ディスプレイのような現存の方法は、PCR産物の最終量しか測定しない。この方法の利点は、さもなければ他の従来法を用いて見逃される遺伝子発現の変化を検出し得ることである。この局面は、PCR産物蓄積の典型的曲線により示すことができる(図5を参照)。
【0278】
PCR増幅反応の開始時に、PCR産物の量は、大部分の装置の検出限界より低く、定量的相違を観察することができない。細胞当たり数個の複製の水準で、通常存在する希少遺伝子転写を検出するために、非常に遅い段階でPCR産物をモニターすることが必要である。典型的に、これらの遺伝子の検出は、これらの希少転写体が検出可能な水準まで増幅されるかなり前に反応が典型的に停止されるので、困難である。増幅曲線の中間部は、信号が検出限界を超え対数相に入ると、遺伝子発現における定量的相違を検出するための最良の信号を構成する。しかしながら、反応の指数的性質故に、この相は比較的短く、反応が後期静止期に移る前に数サイクルしか続かない。PCR増幅のこの後期静止期において、PCR産物の蓄積は、さらなる基質の不足、ポリメラーゼの不足、産物によるポリメラーゼ活性の阻害、またはそれらの組み合わせのような幾つかの因子により飽和する。明らかに、この後期静止期は、やはり、遺伝子発現における定量的相違を検出するための機会をほとんど提供しない。従って、予定数のサイクル後のPCR産物を定量する方法は、増幅の対数相に偶然存在する遺伝子しか確認できず、増幅プロセスの早期または後期において異なってしか検出されない遺伝子を見逃す。
【0279】
本発明は、各々の個々の増幅フラグメントについての完全増幅極性を定めるので、この制限を克服する。さらに、発現相違を測定するための定量的基礎が提供される。実時間定量的PCRの実施において、パラメーターCtが実験的に定義された。ここで用いられる「Ct」という用語は、定量的PCR反応により発生する信号が始めて「閾値」を超える、すなわち、標的核酸配列の増幅の最初の信頼性ある検出がなされるときのサイクル数を意味する。「信頼性ある」という用語は、信号が、PCR中に増幅された産物の検出可能な水準を反映することを意味する。Ctは、通常、標的核酸の未知量の出発量に関連し、例えば、標的の量が少ないとCtが遅れる。Ctは、簡単な数式により、初期複製数または出発DNAの濃度に関連する:
Log(複製数)=aCt+b ここで、a及びbは定数である。
【0280】
従って、二つの異なる試料から生じる同じ遺伝子のフラグメントについてのCtを測定することにより、三つの試料におけるこの遺伝子の最初の濃度を容易に評価することができる。
【0281】
発現プロフィールのためにPCR増幅を用いる一般的関心事は増幅が偏る可能性があることである。特に、一部の配列が、他のものよりも優れた効率で増幅される。この偏りは、出発試料と比較して、PCR産物の最終的状態を変化させることができる。しかしながら、そのような偏りが本発明に影響を与えないのは、本発明が、異なる試料からのcDNA標的の増幅が、同じ反応混合物中で共通のPCRプライマーを用いて行われるという態様を提供するからである。従って、異なる試料から生じる増幅PCR産物の比は、各試料中のcDNAの最初の量によってしか影響されず、増幅の効率によって影響されない。所定のPCR反応において、一つのPCR標的の増幅がもう一つの標的に対して偏るが、この比は、各PCR産物の寸法または組成に関わらず一定に維持される。すなわち、この方法は、同じPCR反応における二つの試料の絶対的ではなく相対的な増幅を測定することによりそのような問題を回避する態様を提供する。
【0282】
他の潜在的問題が、DNAポリメラーゼの利用性が増幅の制限要素となる場合、増幅の後期において生じ得る。その結果、より豊富なフラグメントが、豊富でないフラグメントの増幅を速度論的に阻害する。この問題の重要性は、検出装置の感受性に依存するので、実験的に予測することはできない。この問題を緩和する一つの方法は、増幅の後期サイクルにおいてDNAポリメラーゼの濃度を徐々に増加させることである。
【0283】
PCRの速度論的偏りの問題を扱うもう一つの方法は新規概念である(正規化増幅または定常状態への増幅)。一つの態様において、10〜20サイクルで出発する増幅の各サイクルにさらなるステップを含むことが提案される。このステップは、増幅混合物を、プライマー2に含まれるパリンドローム配列に対して提供される制限酵素で処理することからなる(図6)。より豊富なPCRフラグメントは、単にそれが相対的に豊富であることにより、制限酵素により選択的に消化される。消化により、DNAポリメラーゼ用の初回処理部位が除去され、それにより、消化されたフラグメントのさらなる増幅が防止される。あまり豊富でないDNAフラグメントを含む未消化PCRフラグメントは、増幅を続け、反応中、各フラグメントの2つの複製を生じる。制限酵素の濃度及びこの処理の時間を調節することにより、この反応を、特定の許容できる濃度に達した後のPCRフラグメントのさらなる増幅を制限するように調節することができるべきである。同じ遺伝子に対応する消化フラグメントと未消化フラグメントとの間の寸法の相違を無くすために、反応混合物が、過剰量の制限酵素で処理される。同様に、DNAの反対側鎖の初回処理から生じる一本鎖DNA系を、一本鎖DNase(例えば、エキソヌクレアーゼI及びVII)で処理することにより除去することができる。
【0284】
前記記載は、二つの元の試料間の相違を測定する対応に適用される。しかしながら、二つまたはそれ以上の試料を、前述と同じ方法で少し修正して用い得ることも理解すべきである。例えば、第3試料特異的プライマー及び第3蛍光標識を用いることにより、三つの試料に対して同じ方法を用いることができる。同様に、それ以上の試料も同様の手法を用いて分析することができる。
【0285】
(本発明の方法を実施するためのキット)
本発明は、本発明の種々の態様を実施するための組成及びキットを含む。好ましくは、本発明のキットは第1オリゴヌクレオチドプライマーを含み、第1オリゴヌクレオチドプライマーは試料特異的配列タグを含み、試料特異的配列タグはその5末端においてGCに富みその3末端においてAtに富む。好ましくは、第1オリゴヌクレオチドは固体支持体に付着される。さらに、本発明のキットは、さらに、第2オリゴヌクレオチドプライマー、第3オリゴヌクレオチドプライマーまたは第4オリゴヌクレオチドプライマーを含むことができ、第2オリゴヌクレオチドプライマーは任意配列タグを含むことができる。キットは、さらに、逆転写酵素、DNAポリメラーゼ、反応緩衝剤、及びdNTPからなる群より選択される一つまたは二つ以上の成分を含み得る。
【0286】
(本方法の適用例)
1.発生及び信号導入経路
異なる生物学的試料の発現プロフィールの比較は、正常発生プロセスの研究に非常に重要である。
【0287】
例えば、幹細胞分化は、特定の機能を有する細胞を産生する幹細胞への一連の特化により特徴付けられる。全能胚幹細胞を部分的に多能幹細胞に分化させ、それから特定の条件下に血液幹細胞を産生することができる。これらの委任血液幹細胞は、サイトカインの宿主または「刺激因子」に反応して、さらに分解してより特化した血液細胞、例えば、赤血球、血小板及び白血球が得られる。この複雑なプロセルの各ステップ中、特定のサイトカインに反応して全遺伝子発現プロフィールにおいて劇的変化が生じる。幹細胞分解中のこれらの種類の宿命決定に支配的な因子が何であるか決めることが非常に重要であるのは、これらのステップの部分的または完全逆転が、分化中に失われる望ましい特徴を取り戻すのに有益であるからである。多くの他の発生プロセスにおいて同様の手法が望ましい。本発明は、発生中の遺伝子発現プロフィールにおけるそのような変化を研究するためのツールを提供し、そのような研究に非常に価値がある。
【0288】
2.治療的使用及び診断マーカー
本発明は、異なる生物学的試料の発現プロフィールを比較する方法を提供し、これはさらなる研究及び発展のために有能な薬剤標的を確認するための非常に価値のある手段、及び特定の病状のための有用な診断マーカーを提供する。
【0289】
発現プロフィールが疾患試料と正常試料とで変化し得る少なくとも二つのタイプの遺伝子がある。一つのタイプの遺伝子の発現プロフィールの変化は、病状を引き起こす。これらの遺伝子のアップまたはダウンレギュレーションにより病状を進行させる一連の事象が引き起こされる。これらの「病因遺伝子」の活性を調節することにより、病状を逆転させ、それにより効果的に治療するまたは病状を軽減することができる。これらの遺伝子及びそれらの産物は、価値のある薬剤標的を構成し、それを単に確認することが、疾患の治療の長期目的に有益である。そのような遺伝子の例には、制限はされないが、腫瘍遺伝子(Ras等)及び腫瘍抑制遺伝子(Rb、NF1等)がある。
【0290】
発現プロフィールが変化する第2のタイプの遺伝子は、これらの変化が、そのような病状の原因ではなく結果である点において異なっている。疾患表現型が逆転することを期待してこれらの遺伝子の活性を調節することは不可能であるが、これらの遺伝子の確認より、それにも関わらず、そのような病状の初期の正確な診断が助けられ、それにより、そのような病状の初期活効果的治療が容易になる。そのような遺伝子の例には、限定はされないが、腫瘍抗原CA125、α胎児蛋白などがある。
【0291】
さらに、本発明を、細胞、組織または個体に対する特定の治療の効果を研究するために用いることもできる。これは、どの信号導入経路が特定の治療により影響を受けるかに基づいて有能な薬剤の効果を研究及び/または予想した場合、基本的かつ薬学的研究に有用である。そのような薬剤の潜在的標的を確認することにより、他の意図しない標的は放置して所望の標的のみに影響を与える優れた薬剤をさらにスクリーニングすることにより、ある望ましくない副作用を除去することができる。本発明は、意図する標的において大きな望ましい変化を引き起こす改良された薬剤を確認するために高処理率スクリーニングを行う手段を提供するので、薬剤最適化も可能である。
【0292】
3.他の用途
転写プロファイリングの単純な方法が非常に有用である他の領域は、遺伝的に修飾された細胞及び有機体の特徴解析である(例えば、修飾された遺伝子、過発現遺伝子またはミス遺伝子(ノックアウト)を有するトランスジェニック動物。そのような細胞及び有機体は、標的遺伝子の修飾された機能の結果、あるいは補償効果として、しばしば、変化し転写プロフィールを示す。そのような変化は、異なって発現された遺伝子の同一性により定められる特別の経路に置くことにより、遺伝子の機能を示すことができる。これは、特別の遺伝子における変化の転写サインを定めること及び、転写サインのデータベースに対して、疾患の影響を受けている有機体から得られる細胞または組織における転写変化を比較することにより特別の疾患における遺伝子修飾を決めるためにそのようなサインを用いることに役立つ。
【0293】
4.ビジネス法
本発明は、薬学的ビジネスを行うためのビジネス法も提供する。薬剤標的の確認及び実証は、薬剤開発プロセスにおける重要な速度制限ステップである。本発明は、疾患の発現プロフィールを正常組織と迅速に比較して、それにより、薬剤設計のための有能なリード分子を確認するプロセスの速度を著しく上昇させる方法を提供する。関連する高処理手段は、薬剤スクリーニング及び薬剤最適化プロセスの速度を上昇させることにも役立つ。さらに、多くの信頼性のある診断マーカーを確認し、さらに、診断目的で開発することができる。これは、これらの薬剤標的またはマーカーの確認及び開発を行うために薬学ビジネスの基礎のみならず、これらの初期の発見の権利を第三者に許可し、選択された標的をさらに研究及び開発させる機会も提供する。さらに、本発明の専売技術を用いて、そのような標的またはマーカーを確認及び開発する業務を提供することも可能である。
【0294】
本発明は、ゲノム発見用の新しい基礎として転写プロファイリング用の有用なツールになる可能性がある。このシステムは、改良及びさらなる開発の可能性を有する(例えば、試料の数の増加、塩基配列決定の必要性を除くバンドIDデータベースの作成、等)。これは、下流での適用のために特定の遺伝子セットを選択するために初期データを提供することによりDNA診断(特に、低密度及び中密度マイクロアレイの開発)の全プロセスの加速も行う。
【0295】
本発明の実施は、特記しない限り、当該技術分野に含まれる分子生物学、細胞生物学、細胞培養、ミクロ生物学及び組換えDNAの従来技術を用いる。そのような技術は文献に充分に説明されている。例えば、Molecular Cloning:A Laboratory Manual,第2版,Sambrook,Fritsch及びManiatis編(Cold Spring Harbor Laboratory Press:1989年);DNA Cloning,第I及びII巻(D.N.Glover ed.,1985年);Oligonucleotide Synthesis(M.J.Gait ed.,1984年);Mullisら著、;米国特許第4,683,195;Polynucleotide Hybridization(B.D.Hames&S.J.Higgins編,1984年);Transcription And Translation(B.D.Hames&S.J.Higgins編,1984年);B.Oerbal,A Practical Guide To Molecular Cloning(1984年);Methods In Enzymology(Academic Press,Inc.,ニューヨーク);Methods In Enzymology,第154及び155巻(Wuら編);Immunochemical Methods In Cell And Molecular Biology(Mayer&Walker編,Academic Press,ロンドン,1987年)を参照されたい。全ての引用された参考資料(本出願全体で引用された参考文献、発行特許、公開特許出願を含む)の内容をここで参考のために取り込む。
【0296】
特に、生物学的試料からの全RNAの単離及びcDNA用のmRNAのその後の精製は良く知られている分子生物学技術である。実験手順の詳細が、前記一つまたは二つ以上の実験用参考書物及び他の化学的文献に記載されている。市販キットも、そのような目的で広く利用できる(例えば、QiagenがmRNA単離用キットを販売し、GIBCO BRLが逆転写酵素を用いるcDNA合成用キットを販売)。PCR増幅、クロマトグラフィー、キャピラリー電気泳動(CE)は、全て、一般的分子生物学技術であり、さらに加工することはない。
【0297】
本発明を、以下の材料及び方法を用いる以下の非限定的実施例により説明する。
【実施例】
【0298】
(実施例1 RNAの調製)
RNAを、以下の手順に従って、Trizol試薬及びQiagen製のRNeasy Midi/Maxi Kitを用いて製造することができる。
【0299】
組織をホモジナイザー中で、組織50〜100mg当たり1mlのTIZOLを用いて30分間均質化し、続いて、最終均質化を1分間行った。試料の体積は、均質化に用いたTrizol試薬の体積の10%を超えるべきでない。均質化組織を室温で少なくとも15分から1時間まで放置する、または、必要になるまで−70℃で貯蔵した。Trizolの1ml当たり0.2mlのクロロホルムを添加し、振り混ぜた。混合物を室温で5分間インキュベートし、次に、4,000rpmで5分間遠心分離した。上相を別の管に集めた。Trizolの1ml当たり0.5mlのイソプロピルアルコールを添加してRNAを沈殿させた。混合物を氷上に5分間乗せ、4,000rpmで10分間遠心分離した。上澄みを除去し、ペレットをTrizolの1ml当たり1mlの75%EtOHで洗い、混合し、4,000rpmで5分間遠心分離した。上澄みを除去し、ペレットを30分〜1時間風乾した。ペレットを風乾後、ペレットをRNaseを含まない水中に所望の濃度に再懸濁させた。
【0300】
抽出されたRNAは、適当な体積の緩衝剤RLTを添加し、充分に混合することにより清浄化することができ。適当な体積のエタノール(96〜100%)を、希釈したRNAに添加し、激しく振動させることにより充分に混合した。試料を、RNeasy MidiスピンカラムまたはRNeasy Maxiスピンカラムに適用し、15mlまたは50ml遠心分離管に入れ、3,000〜5,000×gで5分間遠心分離した。流動部を廃棄した。
【0301】
一般的に、RNeasyシリカ−膜技術が、DNaseで処理することなくDNAの大部分を効率的に除去するので、DNase消化は必要ない。しかしながら、非常に少量のDNAに感受性の特定のRNA用途には、さらなるDNA除去が必要であることがある。DNaseでDNAを除去するために、2.0mlの緩衝剤RW1をピペットでスピンカラムに入れ、3,000〜5,000×gで5分間遠心分離して洗った。流動部を廃棄し、遠心分離管を再利用した。20μlのDNase1貯蔵溶液を、緩衝剤RDD140μlに添加した。管を軽く打つことにより混合し、短時間遠心分離して、管の残渣から残留液を圧桁。DNase1インキュベーション混合物(160μl)を、スピンカラム膜上にピペットで直接乗せ、作業台(20〜30℃)上に15分間乗せた。RW1緩衝剤2.0mlを、ピペットでスピンカラムに入れ、作業台上に5分間乗せた。次に、3,000〜5,000×gで5分間遠心分離した。流動部分を廃棄した。以下のRPE緩衝剤洗浄ステップにおいて遠心分離管を再利用した。製造者のマニュアルの指示に従って、プロトコルの残りのためにRNeasy Kitを用いる。
【0302】
(実施例2 溶液中の逆転写)
RNA試料(1〜5μl)を、dNTP溶液(10mM)1μl及び第1オリゴヌクレオチド0.0005〜0.5μM(20μl混合物中の最終濃度)と混合し、70℃で7分間加熱し、4℃で2分間冷却した。前記混合物を、次に、反応混合物(RT緩衝剤(250mM Tris−HCl(pH8.3:25℃),375mM KCl,15mM MgCl2)4μl、0.1M DTT 2μL,RNAse阻害剤(Ambion)1μL及びSuperScriptII逆転写酵素(Invitrogen)1μl及び水5〜10μl)と混合し、合計体積20μlとした。逆転写反応液を45℃で1〜2時間インキュベートし、65℃で10分間加熱することにより完了させた。試料(5〜20μl)を、PCRにより直接分析した。任意に、PCR増幅の前に、RNAse H酵素(Invitrogen)を用いてインキュベーションすることによりRNA鋳型を変性させた。
【0303】
(実施例3 ビーズ上の逆転写)
a.ビーズへのオリゴヌクレオチドのカップリング
Ultralink(登録商標)ヨードアセチルビーズ(Pierce)(100〜1,000μl)を、5×TE緩衝剤(50mM Tris,5mM EDTA,pH8.0)で4回洗い、チオール化(5’チオール)オリゴヌクレオチド(1〜10μM)の溶液と混合した。カップリング反応を、還元剤TCEP(Tris(2−カルボキシエチル)ホスフィン)(100〜500μM)を添加することにより開始し、室温で連続的に混合しつつ1〜2時間行った。ビーズ上の未反応活性基を、βメルカプトエタノール1%を添加することにより10分間急冷した。オリゴビーズを、各10倍体積の5×TE緩衝剤で4回、5×TE緩衝剤で2回(75℃)、RT緩衝剤で2回(85℃)、2×RT緩衝剤及びRT緩衝剤(95℃)で順次洗った。調製したビーズを、RT緩衝剤中4℃で維持した。
【0304】
b.逆転写
RNA試料(1〜5μl)を、dNTP溶液(10mM)1μl及びオリゴ−ビーズ6〜10μlと混合し、70℃で7分間加熱し、4℃で2分間冷却し、反応混合物(RT緩衝剤(250mM Tris−HCl(pH8.3:25℃),375mM KCl,15mM MgCl2)4μl、0.1M DTT2μL,RNAse阻害剤(Ambion)1μL及びSuperScriptII逆転写酵素(Invitrogen)1μl及び水0〜4μl))と混合した。逆転写反応液を45℃で1〜2時間インキュベートした。65℃で10分間加熱することにより反応を完了させた。ビーズをPCR緩衝剤で2回洗った。RNA鋳型を、RNAse H酵素(Invitrogen)とのインキュベーション、またはアルカリ加水分解により破壊した。後者の反応は、反応混合物に0.5M NaOH3.5μlを添加し、65℃で5分間インキュベーションし、1M Tris(pH7.5)3.5μlで中和することにより行った。ビーズを、PCR緩衝剤で2回洗った。
【0305】
(実施例4 第2鎖合成)
結合したDNAの第2鎖の合成を、Taqポリメラーゼ(Hot−start Taq、Qiagen製)(0.5〜1.5μ)及びPwo DNAポリメラーゼ(Roche製)(0.25〜0.5μ)の混合物を用いて、PCR熱サイクラーにおいて、95℃で30秒、56℃で30秒及び72℃で2分間行った。反応混合物は、RT反応からのオリゴビーズ上のcDNA6〜10μl、10×PCR緩衝剤(Hot−start Taq,Qiagan製)または10×RT−PCR緩衝剤(500mM Tris,200mM KCl,100mM (NH4)2SO4,2.5mM Mg2Cl,pH8.5)5μl、dNTP0.1mM、第2プライマー0.5〜1μl(100μM)を合計体積50μlで含んだ。合成した第2DNA鎖を、96℃でビーズから除去し、さらなる増幅に用いた。
【0306】
あるいは、Tris(pH7.5)50mM、10mM MgCl2、1mM DTT、及び0.05mg/ml BSA、0.1mM dNTP、7mM MgCl2及び1μM第2プライマーの存在下に、DNAポリメラーゼ1またはそのKlenowフラグメントの存在下に37℃で30分間、第2鎖DNAを合成した。合成した第2DNA鎖を前述のように除去した。
【0307】
(実施例5 PCR増幅)
合成されたcDNA(5〜20μl)のPCR増幅を、10×PCR緩衝剤または10×RT−PCR緩衝剤(前記参照)10μl、2〜3mM MgCl2、0.05〜0.2mM dNTPs、FAM、RoxまたはHexで標識した0.1〜1μM第3プライマー、0〜1μM非標識第3プライマー、第4プライマー1〜2μl、DMSO 0〜5%、校正DNAポリメラーゼ(PwoまたはTgo(Roche))0.5〜2μ、及びhot−start DNAポリメラーゼ(例えば、Hot−Start Taqポリメラーゼ(Qiagen製))1.5〜3μの存在下に行った。増幅を、「I−サイクラー」(BioRad)または「PCR Express」(Thermo Hybaid)熱サイクラーを使用して、以下のサイクルプログラムを用いて行った:95℃で30秒、56〜60℃で15〜30℃、72℃で1分30秒、合計30〜40サイクル。10〜15回目のサイクルで出発して延長ステップ(72℃)の最後に各サイクル後に少量の試料(典型的に25μl)を引き出した。プライマー、ポリメラーゼ及びdNTPを含む等体積のPCR混合物を、各試料除去後に反応混合物に入れた。集めた試料を、Spectrumedix(SCE−9610 Genetic Analysis System)またはABI(3700 Prism System)からのCEシステムを用いて分析
した。
【0308】
(実施例6 キャピラリー電気泳動)
キャピラリー電気泳動を、製造者の指示に従ってSpectrumedix Corporation製のSCE9610完全自動化96−キャピラリー電気泳動遺伝子分析システムで行った。
【0309】
(その他の態様)
前述の態様は、実施した実験及び、本発明を製造し実施する際に本発明者が考えた技術を示す。これらの態様は、本発明の実施技術を示しその有用性を示すために役立つ技術の開示を含むと考えられる。当業者は、ここに記載の技術及び態様は、好ましい技術であり、通常、種々の同様の方法及び技術を用いて同じ結果を達成し得ると考える。
【0310】
前述の全ての参考文献の全体をここで参考のために取り込む。
【技術分野】
【0001】
本発明は、転写プロファイリング技術に関する。
【背景技術】
【0002】
ゲノム科学の導入は、薬剤発見の速度の加速に役立ってきた。ゲノム技術は、新規薬剤標的の発見においてその価値を証明した。この領域のさらなる改良は、有用な薬剤のより迅速でより費用効率の高い結果を得る、より効率的なツールを提供する。
【0003】
薬剤発見プロセスは幾つかのステップ:病気に関わる潜在的生化学的標的の確認、活性化合物及びさらなる化学的設計のスクリーニング、前臨床テスト及び最終的臨床試行、を含んでいる。このプロセスの効果はなお完全からは遠く、R&Dプロセスにおいて費やされた金額の約75%が資金的に失敗していると推測される。さらに、産物開発の晩期において失敗が発生し、このプロジェクトに関わる損失がより大きくなる。従って、将来の失敗を初期に取り除いて、全薬剤開発プロセスの費用を大幅に節減する必要がある。すなわち、最初の分子標的の質が、費用効果的薬剤開発のための決定的因子となる。
【0004】
標的を確認し実証するプロセスに有効な見込みのある一つの手段は、転写プロファイリングである。この方法は、特定の状況における遺伝子の発現を比較する:例えば、疾患細胞と正常細胞との間、対照細胞と薬剤処理細胞との間、または処理に反応する細胞と処理に抵抗性の細胞との間で比較する。この手段により生じる情報は、治療により標的とされる特定の遺伝子を直接確認することができ、重要なことに、疾患及び治療に含まれる生化学的経路を明らかにする。簡単に言えば、これは、生化学的標的を提供するのみならず、同時に、これらの標的の質を評定する方法も提供する。さらに、細胞に基づくスクリーニングと組み合わせて、薬剤発見の分野を劇的に変化させるために転写プロファイリングが位置付けられる。歴史的には、有効な薬剤のスクリーニングが、機能的細胞系におけるマーカーとして表現型変化を用いてうまく行われた。例えば、培地における腫瘍細胞の成長をモニターして、抗癌薬を確認した。同様に、抗生物質確認を目指すアッセイにおいて、バクテリア生存能力を用いた。そのようなスクリーンは、典型的に、標的の生化学的経路を予め知ることなく行われた。実際、確認された効果的化合物がそのような経路を明らかにし分子標的を指摘して、その後の次世代薬剤の合理的設計を可能にする。
【0005】
転写プロファイリングの最近のツールを用いて、薬剤の効果を評定するために表現型の変化の代わりに遺伝子発現を利用する新規スクリーニング法を設計することができる。例えば、これらの方法は、米国特許第5,262,311、5,665,547、5,599,672、5,580,726、6,045,988及び5,994,076号並びにLuehrsenら著(1997年、Biotechniques,第22巻:168〜74頁);Liang及びPardee著(1998年、Mol Biotechnol.第10巻:261〜7頁)に記載されている。そのような手段は、表現型スクリーニングが適用不可能であるが所望の転写プロフィールを容易に達成して特定の疾患に結びつけることができる、軽度認識生涯、抑鬱等のような中枢神経系(CNS)疾患の分野における薬剤発見に非常に貴重である。また、確認された効果的化合物は、根元的な分子プロセスを明らかにする。さらに、この方法は、複数の生化学的標的において同時に作用して所望の薬理学的効果を生む現存する薬剤の改良に役立ち得る。そのような場合、転写反応における変化は、複数標的への結合の最適化に基づく選択よりも、薬剤の作用についての優れたマーカーであり得る。
【0006】
本発明の前に、転写プロファイリングの最も進んだ方法は、DNAマイクロアレイを用いる技術に基づき、例えば、Greenberg,2001年,Neurology第57巻:755〜61頁;Wu,2001年,J Pathol.第195巻:53〜65頁;Dhimanら著、2001年,Vaccine,第20巻:22〜30頁;Bierら著、2001年Fresenius J Anal Chem.第371巻:151〜6頁;Millsら著、2001年,Nat Cell Biol.第3巻:E175〜8頁に考察され、米国特許第5,593,839、5,837,832、5,856,101、6,203,989、6,271,957及び6,287,778号に記載されている。DNAマイクロアレイは、試験アレイの表面に付着しているDNA分子への、mRNAの逆転写により得られる標識されたポリヌクレオチド試料のハイブリッド形成を評価することにより、所定の試料における数千の遺伝子の発現を同時に行う方法である。この技術は、転写の変換について価値のある情報を提供するが、完全とは程遠い。
【0007】
最初に、この技術は、マイクロアレイに在る遺伝子のプールに限定される。現在の印刷法は、単一チップ上に10,000〜15,000の遺伝子を乗せることができ、これは、本質的に特定の細胞型において発現される遺伝子の数である。種々の細胞型があるとすれば、特定の細胞型のために特定のアレイを開発する必要がある。理論的に可能であるが、この作業は達成困難である。それは、マイクロアレイ製造前にこれらの細胞において発現される遺伝子プールについての知識が必要だからである。
【0008】
さらに、組織試料における転写の数は、細胞試料における数よりかなり多く、マイクロアレイの現在の性能を超えている。さらに、遺伝子発現の一部の変化は択一的スプライシングから生じ、さらに、これが、評定すべき転写の数をさらに増やす。これらの困難を克服する唯一の可能性は、択一的にスプライスされた遺伝子を含む全ゲノムを覆う複数のアレイを開発することである。この手段は、単一実験の費用を著しく増加させ、大きな生物学的試料、おそらく、合理的に利用できるよりも大きな試料を必要とする。
【0009】
第2に、現在では、DNAマイクロアレイは定量的に正確なデータを提供せず、遺伝子発現において観察される変化は、独立した方法、例えば、定量的PCR(Q−PCR)により確認しなくてはならない。
【0010】
さらに、典型的マイクロアレイ実験は、この方法の再現性に影響を与える幾つかのマニュアルステップを含む。
【0011】
最後に、特に興味深い稀転写の発現は、現在の検出技術を用いて、マイクロアレイにより正確に測定することができない。これらの制限は、転写プロファイリングを行うための別の方法、好ましくは、1)アッセイ前に発現された遺伝子プールの配列の予備知識を必要としないが、それ自体、アッセイ中/後にこの情報を提供する方法;2)発現された転写の水準の定量的変化を測定する方法;3)稀遺伝子の発現を検出し得る方法;及び4)自動化できる方法を開発する必要性を示している。
【0012】
遺伝子発現を定量的に検出するためにキャピラリー電気泳動が用いられていた。Rajevicら著(2001年、Pflugers Arch.442頁(6増刊1):R190〜2頁)は、多くの腫瘍遺伝子の同時の発現の相違を検出するために7対のプライマーを用いることにより腫瘍遺伝子の異なる発現を検出する方法を開示している。センスプライマーを、蛍光染料で5’末端標識した。複数の蛍光RT−PCRの結果を、ABI−PRISM 310 Genetic Analyzerでのキャピラリー電気泳動により分析した。Borsonら(1998年,Biotechniques,第25巻:130〜7頁)は、産物の迅速な分離及び検出のためにキャピラリー電気泳動(CE)に組み合わせた定量的競合的逆転写PCR(QC−RT−PCR)に基づく低存在量mRNA転写体の依存的定量用の手法を記載している。Georgeら(1997年,J Chromatogr B Biomed Sci Appl,第695巻:93〜102頁)は、キャピラリー電気泳動系(ABI310)を、蛍光相違表示が発生するESTパターンの確認に適用することを記載している。Odinら(1999年,J Chromatogr B Biomed Sci Appl,第734巻:47〜53頁)は、PCR増幅cDNAの分離及び定量のために、多色を検出する自動キャピラリーゲル電気泳動を記載している。
【0013】
Omoriら(2000年、Genomics第67巻:140〜5頁)は、オリゴ(dT)またはオリゴ(dU)プライマーを用いて2つの独立した逆転写したcDNA試料における競合的PCRにより、市場で購入したα−グロビンmRNAの量を測定し比較している。オリゴ(dT)またはオリゴ(dU)プライマーは、3’オリゴ(dT)またはオリゴ(dU)配列及び5’共通配列を共有する。さらに、各試料についてオリゴ(dT)またはオリゴ(dU)プライマーは、3’オリゴ(dT)またはオリゴ(dU)配列と5’共通配列との間に独自の29個のヌクレオチド配列も含む。第1cDNA鎖の合成後、独自標識で標識された共通配列に相補的なプライマー及び遺伝子特異的プライマーを用いて、PCRを行ってcDNAを増幅する。増幅されたPCR産物を、次に、蛍光スキャナーの検出板上に点在させることにより分析する。
【発明の概要】
【発明が解決しようとする課題】
【0014】
当該分野において、複数の試料において遺伝子発現プロファイルを同時に定量的に検出するための簡単で感受性の高い方法が必要とされている。
【課題を解決するための手段】
【0015】
本発明は、二つまたはそれ以上の試料の発現プロファイリングの方法及び組成物を提供する。
【0016】
本発明は、2種以上の試料の遺伝子発現プロフィールを比較する方法であって、
(a)試料特異的配列タグを含む第1のオリゴヌクレオチドプライマーを用いて第1の試料から複数の第1cDNA鎖を合成し、試料特異的配列タグはその5’末端においてGCに富みその3’末端においてAtに富むこと、
(b)少なくともcDNAのサブセットを選択的に増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(c)一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、存在量は第1試料中の一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(d)第1試料中の一つまたは二つ以上の遺伝子の発現プロフィールを第2試料中の一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違は2つの試料中の一つまたは二つ以上の遺伝子の異なる発現を示すこと
を含んでなる方法を提供する。
【0017】
本発明は、二つまたはそれ以上の試料の遺伝子発現プロフィールを比較する方法であって、
(a)試料特異的配列タグを含む第1のオリゴヌクレオチドプライマーを用いて第1の試料から複数の第1cDNA鎖を合成し、第1オリゴヌクレオチドプライマーは少なくとも一つの変性ヌクレオチドを含むこと、
(b)少なくともcDNAのサブセットを選択的に増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(c)一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、存在量は第1試料中の一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(d)第1試料中の一つまたは二つ以上の遺伝子の発現プロフィールを第2試料中の一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違は2つの試料中の一つまたは二つ以上の遺伝子の異なる発現を示すこと
を含んでなる方法も提供する。
【0018】
本発明は、二つまたはそれ以上の試料の遺伝子発現プロフィールを比較する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、第1試料から複数の第1cDNA鎖を合成し、試料特異的配列タグが、従来のヌクレオチドを超える、もう一つの人工ヌクレオチドとの塩基対選択性を示す少なくとも一つの人工ヌクレオチドを含むこと、
(b)少なくともcDNAのサブセットを選択的に増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(c)一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、存在量が第1試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(d)第1試料中の一つまたは二つ以上の遺伝子の発現プロフィールを第2試料中の一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、二つの試料における一つまたは二つ以上の遺伝子の異なる発現を示すこと
を含んでなる方法も提供する。
【0019】
本発明は、二つまたはそれ以上の試料の遺伝子発現プロフィールを比較する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、第1試料から複数の第1cDNA鎖を合成し、試料特異的配列タグがその5’末端においてGCに富みその3’末端においてATに富むこと、
(b)第1任意配列タグを含む第2オリゴヌクレオチドプライマーを用いて、第1cDNA鎖に相補的な一つまたは二つ以上の第2cDNA鎖を選択的に合成すること、
(c)一つまたは二つ以上の第2cDNA鎖を増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(d)一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、存在量が第1試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(e)第1試料中の一つまたは二つ以上の遺伝子の発現プロフィールを第2試料中の一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、二つの試料における一つまたは二つ以上の遺伝子の異なる発現を示すこと
を含んでなる方法も提供する。
【0020】
本発明は、さらに、二つまたはそれ以上の試料の遺伝子発現プロフィールを比較する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、第1試料から複数の第1cDNA鎖を合成し、第1オリゴヌクレオチドプライマーが少なくとも一つの変性ヌクレオチドを含むこと、
(b)第1任意配列タグを含む第2オリゴヌクレオチドプライマーを用いて、第1cDNA鎖に相補的な一つまたは二つ以上の第2cDNA鎖を選択的に合成すること、
(c)一つまたは二つ以上の第2cDNA鎖を増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(d)一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、存在量が第1試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(e)第1試料中の一つまたは二つ以上の遺伝子の発現プロフィールを第2試料中の一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、二つの試料における一つまたは二つ以上の遺伝子の異なる発現を示すこと
を含んでなる方法を提供する。
【0021】
本発明は、さらに、二つまたはそれ以上の試料の遺伝子発現プロフィールを比較する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、第1試料から複数の第1cDNA鎖を合成し、試料特異的配列タグが、従来のヌクレオチドを超える、もう一つの人工ヌクレオチドとの塩基対選択性を示す少なくとも一つの人工ヌクレオチドを含むこと、
(b)第1任意配列タグを含む第2オリゴヌクレオチドプライマーを用いて、第1cDNA鎖に相補的な一つまたは二つ以上の第2cDNA鎖を選択的に合成すること、
(c)一つまたは二つ以上の第2cDNA鎖を増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(d)一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、存在量が第1試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(e)第1試料中の一つまたは二つ以上の遺伝子の発現プロフィールを第2試料中の一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、二つの試料における一つまたは二つ以上の遺伝子の異なる発現を示すこと
を含んでなる方法を提供する。
【0022】
本発明は、試料における一つまたは二つ以上の遺伝子の発現を調節するモジュレーターを確認する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、試料をモジュレーターに接触させる前に、複数の第1cDNA鎖を合成し、試料特異的配列タグがその5’末端においてGCに富みその3’末端においてATに富むこと、
(b)少なくともcDNAのサブセットを選択的に増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(c)一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、存在量が試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(d)モジュレーターとの接触前の試料中の一つまたは二つ以上の遺伝子の発現プロフィールを、モジュレーター接触後の試料中の一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、試料における一つまたは二つ以上の遺伝子発現を調節するモジュレーターを示すこと
を含んでなる方法を提供する。
【0023】
本発明は、試料における一つまたは二つ以上の遺伝子の発現を調節するモジュレーターを確認する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、試料をモジュレーターに接触させる前に、複数の第1cDNA鎖を合成し、第1オリゴヌクレオチドプライマーが少なくとも一つの変性ヌクレオチドを含むこと、
(b)少なくともcDNAのサブセットを選択的に増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(c)一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、存在量が試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(d)モジュレーターとの接触前の試料中の一つまたは二つ以上の遺伝子の発現プロフィールを、モジュレーター接触後の試料中の一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、試料における一つまたは二つ以上の遺伝子発現を調節するモジュレーターを示すこと
を含んでなる方法も提供する。
【0024】
本発明は、さらに、試料における一つまたは二つ以上の遺伝子の発現を調節するモジュレーターを確認する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、試料をモジュレーターに接触させる前に、複数の第1cDNA鎖を合成し、試料特異的配列タグがその5’末端においてGCに富みその3’末端においてATに富むこと、
(b)第1任意配列タグを含む第2オリゴヌクレオチドプライマーを用いて、一つまたは二つ以上の第2cDNA鎖を合成すること、
(c)第2cDNA鎖を増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(d)一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、存在量が試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(e)モジュレーターとの接触前の試料中の一つまたは二つ以上の遺伝子の発現プロフィールを、モジュレーター接触後の試料中の一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、試料における一つまたは二つ以上の遺伝子発現を調節するモジュレーターを示すこと
を含んでなる方法を提供する。
【0025】
本発明は、さらに、試料における一つまたは二つ以上の遺伝子の発現を調節するモジュレーターを確認する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、試料をモジュレーターに接触させる前に、複数の第1cDNA鎖を合成し、第1オリゴヌクレオチドプライマーが少なくとも一つの変性ヌクレオチドを含むこと、
(b)第1任意配列タグを含む第2オリゴヌクレオチドプライマーを用いて、一つまたは二つ以上の第2cDNA鎖を合成すること、
(c)第2cDNA鎖を増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(d)一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、存在量が試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(e)モジュレーターとの接触前の試料中の一つまたは二つ以上の遺伝子の発現プロフィールを、モジュレーター接触後の試料中の一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、試料における一つまたは二つ以上の遺伝子発現を調節するモジュレーターを示すこと
を含んでなる方法を提供する。
【0026】
好ましい態様において、本方法のステップ(a)が、二つまたはそれ以上の試料供給源からのRNAを第1cDNA鎖に逆転写することを含み、そのcDNAはそれらの供給源に従って異なって標識される。
【0027】
好ましくは、第1cDNA鎖が、第1試料由来の全RNAまたはmRNAを用いる逆転写により合成される。
【0028】
好ましくは、第2オリゴヌクレオチドプライマー中の第2配列が、遺伝子ファミリー特異的である。
【0029】
より好ましくは、第2オリゴヌクレオチドプライマー中の第2配列が、蛋白ファミリーに特異的なペプチドをコードする配列である。
【0030】
さらにより好ましくは、第2配列が、特異的蛋白ファミリー用のサイン配列モチーフをコードする配列を含む。
【0031】
好ましくは、蛋白ファミリーは、受容体チロシンキナーゼ、G蛋白共役受容体、7回膜通過受容体、イオンチャンネル、サイトカイン受容体、腫瘍マーカー、MAPKカスケードキナーゼ、転写因子、GTPアーゼ、ATPアーゼ、及び発生蛋白マーカーからなる群より選択される。
【0032】
好ましくは、第1オリゴヌクレオチドプライマーの配列特異的配列タグを含む第3オリゴヌクレオチドプライマーを増幅のために用いて一つまたは二つ以上の試料特異的増幅産物を産生する。
【0033】
また好ましくは、二つまたはそれ以上の試料の少なくとも一つが、正常試料、疾患試料、所定の発生段階または状態における試料、所定の処理段階または状態の前の試料、所定の処理段階または状態の後の試料及び所定の培養段階または状態における試料からなる群より誘導される。
【0034】
なお好ましくは、二つまたはそれ以上の試料の少なくとも一つが、動物、器官、組織型及び細胞型からなる群より誘導される。
【0035】
一つの態様において、少なくとも一つの試料が正常個体から誘導され、少なくとももう一つの試料が疾患個体から誘導される。
【0036】
もう一つの態様において、少なくとも一つの試料が個体の発生段階から誘導され、少なくとももう一つの試料が同じ個体の異なる発生段階から誘導される。
【0037】
さらにもう一つの態様において、少なくとも一つの試料が個体の疾患段階から誘導され、少なくとももう一つの試料が同じ個体の異なる疾患段階から誘導される。
【0038】
なおもう一つの態様において、少なくとも一つの試料が個体の疾患処理の段階から誘導され、少なくとももう一つの試料が同じ個体の異なる疾患処理の異なる段階から誘導される。
【0039】
もう一つの態様において、少なくとも一つの試料が環境因子に露出された個体から誘導され、少なくとももう一つの試料が同じ環境因子に露出されなかった個体または異なる濃度で環境因子に露出された個体から誘導される。
【0040】
一つの態様において、一つまたは二つ以上の第2cDNA鎖がPCRで増幅されて一つまたは二つ以上の増幅PCR産物が生じる。
【0041】
好ましくは、一つまたは二つ以上の増幅産物を、増幅中、所定の時間またはサイクル間隔でサンプリングする。
【0042】
一つの態様において、一つまたは二つ以上の増幅産物を、増幅の各サイクル後にサンプリングする。
【0043】
もう一つの態様において、一つまたは二つ以上の増幅産物を、一つまたは二つ以上の予定のサイクル後、例えば、2、5、10、25、30または45サイクル後にサンプリングする。
【0044】
もう一つの態様において、一つまたは二つ以上の増幅産物を、反応混合物の1%〜40%(v/v)を引き出す、好ましくは反応混合物の1%〜30%(v/v)を引き出すことによりサンプリングする。
【0045】
もう一つの態様において、反応混合物に、各サンプリングの後に、dNTP、プライマー、必要な試薬及びDNAポリメラーゼを出発反応混合物と同じ濃度で含む等体積の混合物を補給する。
【0046】
好ましくは、各サンプリングした増幅産物について存在量を検出する。
【0047】
好ましくは、本方法は、さらに、一つまたは二つ以上の増幅産物の存在量の検出前に、一つまたは二つ以上の増幅産物を分離することを含む。
【0048】
もう一つの態様において、一つまたは二つ以上の増幅産物を分離し、その存在量をクロマトグラフィーにより検出する。
【0049】
もう一つの態様において、一つまたは二つ以上の増幅産物を分離し、その存在量を質量分析により検出する。
【0050】
さらにもう一つの態様において、一つまたは二つ以上の増幅産物を分離し、その存在量を電気泳動により検出する。
【0051】
好ましくは、一つまたは二つ以上の増幅産物を分離し、その存在量をキャピラリー電気泳動により検出する。
【0052】
一つの態様において、第1オリゴヌクレオチドプライマー中の試料特異的配列は、15〜30ヌクレオチド長、より好ましくは20〜24ヌクレオチド長である。
【0053】
好ましい態様において、第1オリゴヌクレオチドプライマーは、さらに、5’オリゴ(dT)nVN3’の配列を含み、nは少なくとも5であり、VがdATP、dGTPまたはdCTPであり、NがdTTP(またはdUTP)、dATP、dGTPまたはdCTPである。
【0054】
好ましくは、5’オリゴ(dT)nVN3’においてnは12〜16である。
【0055】
また好ましくは、第1オリゴヌクレオチドプライマーにおいて、試料特異的配列タグは、オリゴ(dT)nVNの5’に配される。
【0056】
好ましくは、本方法の第2オリゴヌクレオチドプライマーが、さらに、第1cDNA鎖のサブセットに相補的な第2配列を含み、それにより一つまたは二つ以上の第2cDNA鎖を合成させる。
【0057】
より好ましくは、第2オリゴヌクレオチドプライマーにおいて、第2配列が第1任意配列の3’に配される。
【0058】
また、より好ましくは、第2オリゴヌクレオチドが、さらに、第1及び第2配列の間に(Z)mの配列を含み、Zが、A、T、GまたはCのいずれかと塩基対を形成することがでるヌクレオチドであり、mが少なくとも2である。好ましくは、mは4である。
【0059】
一つの態様において、第2オリゴヌクレオチドプライマー中の第2配列は5〜10ヌクレオチド長である。
【0060】
もう一つの態様において、第2オリゴヌクレオチドプライマー中の第2配列は6〜7ヌクレオチド長である。
【0061】
好ましくは、第2オリゴヌクレオチドプライマー中の第2配列はパリンドローム配列である。
【0062】
一つの態様において、第2オリゴヌクレオチドプライマー中の第1任意配列は15〜30ヌクレオチド長、好ましくは20ヌクレオチド長である。
【0063】
もう一つの態様において、第2オリゴヌクレオチドプライマー中の第1任意配列がA−Tに富む領域とG−Cに富む。
【0064】
好ましくは、G−Cに富む領域が、A−Tに富む領域の5’に配される。
【0065】
好ましくは、用いられる第2オリゴヌクレオチドプライマーが、比較すべき二つまたはそれ以上の試料について同じである。
【0066】
好ましい態様において、本方法の増幅ステップは、さらに、第2オリゴヌクレオチドプライマーの第1任意配列タグを含む第4オリゴヌクレオチドプライマーを用いることを含む。
【0067】
好ましくは、用いられる第4オリゴヌクレオチドプライマーが、比較すべき二つまたはそれ以上の試料について同じである。
【0068】
一つの態様において、第1cDNA鎖が、固体支持体に付着することなく溶液中で合成される。
【0069】
もう一つの態様において、第1cDNA鎖が、固体支持体に付着されて合成される。
【0070】
好ましくは、固体支持体が微粒子または反応管の内壁である。
【0071】
好ましい態様において、本方法が、さらに、一つまたは二つ以上の第2cDNA鎖の増幅前に、複数の第1cDNA鎖から一つまたは二つ以上の第2cDNA鎖を分離することを含む。
【0072】
一つの態様において、本方法で用いられる第3オリゴヌクレオチドプライマーが検出可能な標識に結合される。
【0073】
好ましくは、検出可能な標識が、蛍光標識、放射性標識、比色標識、磁気標識及び酵素標識からなる群より選択される。
【0074】
より好ましくは、検出可能な標識が、蛍光標識である。
【0075】
好ましい態様において、二つまたはそれ以上の試料の各々について用いられる第3オリゴヌクレオチドプライマーが試料特異的標識で標識される。
【0076】
本方法による一つの態様において、一つまたは二つ以上の遺伝子の発現プロフィールの相違が、二つまたはそれ以上の試料間の遺伝子からの増幅産物上の試料特異的検出可能標識の比により測定する。
【0077】
好ましくは、この方法が、さらに、増幅プロット(増幅サイクル数の関数としての信号強度)を発生させること、各PCRフラグメントの信号強度に基づいて一つまたは二つ以上の遺伝子の各々についての増幅の閾値サイクル数(Ct)を計算することを含む。特定の遺伝子の機能的に異なる発現は、2以上のサイクルの二つ(またはそれ以上)の試料におけるこの遺伝子についての閾値サイクル数(Ct)の相違の値として決められる。さらに、閾値サイクル数は、各遺伝子についての複製数を導き出し、二つまたはそれ以上の試料における遺伝子についての複製数の比により発現の相違を測定するために用いられる。
【0078】
この方法は、各増幅遺伝子について増幅サイクルの数の関数[サイクル数の関数としての信号強度の導関数、d(信号強度)/d(サイクル数)]としての信号強度変化の率のプロットを発生させることも含む。一つの試料からの各増幅遺伝子についてのd(信号強度)/d(サイクル数)の最大値に相当するサイクル数として決められる択一的閾値サイクル(aCt)を、もう一つの試料からの同じ遺伝子についてのaCtと比較する。二つまたはそれ以上の試料中の同じ遺伝子についてのaCt値間の一つのサイクルの相違は、択一的機能的相違発現として定義される。
【0079】
また好ましくは、本方法は、さらに、機能的相違発現または択一的機能的相違発現を示す一つまたは二つ以上の遺伝子に相当するPCRフラグメントを集めること、及び一つまたは二つ以上の遺伝子の配列を確認することを含む。
【0080】
一つの態様において、異なって発現される一つまたは二つ以上の遺伝子の配列同一性は、DNA塩基配列決定により確認される。
【0081】
一つの態様において、本方法は、さらに、第1増幅からの一つまたは二つ以上の増幅産物を用いて一つまたは二つ以上の第2増幅産物を発生させ、一つまたは二つ以上の第2増幅産物の存在量を検出する第2増幅反応を含む。
【0082】
好ましくは、本方法の増幅ステップは、PCRにより行われる。
【0083】
本発明の方法は、さらに、第2増幅反応としてネステッドPCR反応を含む。
【0084】
本発明は、遺伝子発現の水準を検出するための組成物であって、第1オリゴヌクレオチドプライマーを含んでなり、第1オリゴヌクレオチドプライマーが試料特異的配列タグを含み、第1オリゴヌクレオチドプライマーが少なくとも一つの変性ヌクレオチドを含む組成物を提供する。
【0085】
一つの態様において、第1オリゴヌクレオチドプライマーが、[5’−(特異的配列タグ)20−24T12−16AN−3’,5’−(特異的配列タグ)20−24T12−16CN−3’及び5’−(特異的配列タグ)20−24T12−16GN−3’]を含むプライマーの混合物として提供される。
【0086】
本発明は、遺伝子発現の水準を検出するための組成物であって、第1オリゴヌクレオチドプライマーを含んでなり、第1オリゴヌクレオチドプライマーが試料特異的配列タグを含み、試料特異的配列タグがその5’末端においてGCに富みその3’末端においてATに富む組成物も提供する。
【0087】
好ましくは、本組成物は、さらに、第2オリゴヌクレオチドプライマーを含む。
【0088】
より好ましくは、第2オリゴヌクレオチドプライマーは、第1任意配列タグを含む。
【0089】
好ましくは、第2プライマーは、さらに、第1cDNA鎖の配列に相補的な第2配列を含む。
【0090】
本組成物は、さらに、第1オリゴヌクレオチドプライマーの配列特異的配列タグを含む第3オリゴヌクレオチドプライマーを含み得る。
【0091】
本組成物は、さらに、第1任意配列タグを含む第4オリゴヌクレオチドプライマーを含み得る。
【0092】
本組成物は、さらに、逆転写酵素、DNAポリメラーゼ、逆転写酵素用の反応緩衝剤、DNAポリメラーゼ用の反応緩衝剤、及びdNTPからなる群より選択される一つまたは二つ以上の成分を含む。
【0093】
本発明は、遺伝子発現の水準を検出するためのキットであって、第1オリゴヌクレオチドプライマーを含んでなり、第1オリゴヌクレオチドプライマーが試料特異的配列タグを含み、第1オリゴヌクレオチドプライマーが少なくとも一つの変性ヌクレオチドを含み、また、その包装材料を含んでなるキットを提供する。
【0094】
本発明は、遺伝子発現の水準を検出するためのキットであって、第1オリゴヌクレオチドプライマーを含んでなり、第1オリゴヌクレオチドプライマーが試料特異的配列タグを含み、配列特異的配列タグがその5’末端においてGCに富みその3’末端においてATに富み、また、その包装材料を含んでなるキットも提供する。
【0095】
本発明のキットは、第2オリゴヌクレオチドプライマーも含み得る。
【0096】
好ましくは、第2オリゴヌクレオチドプライマーは、第1任意配列タグを含む。
【0097】
本発明のキットは、さらに、第1オリゴヌクレオチドプライマーの配列特異的配列タグを含む第3オリゴヌクレオチドプライマーを含み得る。
【0098】
本発明のキットは、なおさらに、第1任意配列タグを含む第4オリゴヌクレオチドプライマーを含み得る。
【0099】
好ましくは、第2プライマーは、さらに、第1cDNA鎖の配列に相補的な第2配列を含む。
【0100】
また好ましくは、本発明のキットは、さらに、逆転写酵素、DNAポリメラーゼ、逆転写酵素用の反応緩衝剤、DNAポリメラーゼ用の反応緩衝剤、及びdNTPからなる群より選択される一つまたは二つ以上の成分を含む。
【図面の簡単な説明】
【0101】
【図1】本発明の一つの形態による、試料特異的配列タグを用いるオリゴ−dTプライマーを用いる二つの試料からのmRNAの逆転写を示す図である。逆転写の結果となる各試料からのmRNAは試料特異タグで標識される。
【図2】本発明の一つの形態による、遺伝子ファミリー特異的配列を含むプライマーを用いる選択された遺伝子の第2cDNA鎖合成を示す図である。
【図3】本発明の一つの形態による、試料特異的タグを用いる増幅産物をPCR増幅により得ることを示す図である。
【図4】本発明の一つの形態による、PCR産物の分離及び分析を示す図である。
【図5】本発明の一つの形態による、PCR産物蓄積の典型的曲線を示す図である。異なる試料間の相違が最も容易に検出されるサイクルの範囲が狭いことが明らかである。a)遺伝子発現(Ct)の定量的測定は、信号強度が選択された閾値限界(通常、ベースラインの標準偏差の10倍に設定)を超える点に相当するサイクル数として定義される。機能的相違発現(ΔCt)は、二つのPCRフラグメントについてのCt値の相違として定義される。b)サイクル数の関数としてのd(信号強度)/d(サイクル数)のプロットに基づく閾値サイクルの択一的決定。閾値サイクル(aCt)の択一的決定は、d(信号強度)/d(サイクル数)の最大値に対応するサイクル数として定義される。閾値数と同様に、aCtを用いて、各遺伝子についての絶対複製数を決めることができる(log(複製数)=AaCt+B)。択一的機能的相違発現(ΔaCt)は、二つのPCRフラグメントについてのaCt値の相違として定義される。
【図6】本発明の一つの形態による、正規化PCR増幅スキームを示す図である。
【図7】本発明の好ましい形態による、転写プロファイリングの方法を示す図である。
【発明を実施するための形態】
【0102】
(定義)
ここで用いられる「試料」という用語は、その天然環境から単離されポリヌクレオチドを含む生物学的材料を意味する。本発明による「試料」は、精製または単離されたポリヌクレオチドからなる、またはこれは、組織試料のような生物学的試料、生物学的流体試料もしくはポリヌクレオチドを含む細胞試料を含み得る。生物学的流体試料は、血液、血漿、唾液、尿、脳脊髄液、洗浄液及び白血球泳動試料を含む。本発明の試料は、ポリヌクレオチドを含むいかなる植物、動物、バクテリアまたはウイルス材料であってもよい。
【0103】
ここで定義される「組織」は、生体において特別の機能を果たす細胞の集合体である。ここで用いられる「組織」という用語は、特別の生理学的領域からの細胞材料を意味する。特別の組織における細胞は、幾つかの異なる細胞型を含み得る。この非制限的例は、さらに、神経細胞及びグリア細胞、並びにキャピラリー内皮細胞及び血液細胞を含む脳組織である。「組織」という用語は、生体において独立または非接着性細胞、例えば、免疫細胞または血液細胞として通常存在し得る組織マイクロアレイ上の下位位置に含まれる複数の細胞を含むことも意図される。この用語は、さらに、細胞系及び、(例えば、特定組織型の生体分子特性の発現故に)特定の組織型を表す現在存在する細胞材料の他の供給源を含むことを意図する。
【0104】
ここで用いられる「複数」という用語は、2を超えることを意味する。本発明によれば、複数は3以上、100以上、1,000以上、例えば、試料中の全てのmRNAに相当するcDNAの数までであり得る。
【0105】
ここで用いられる「異なるタイプの組織」という用語は、好ましくは異なる器官からの組織、または、少なくとも、同じ器官中の解剖学的及び組織学的に異なる部位からの組織を意味する。
【0106】
ここで用いられる「細胞試料」は、他の細胞から分離される細胞を含む点において組織試料から区別される。
【0107】
ここで用いられる「個体」は単一の生物であり、ヒト、動物、植物、多細胞及び単細胞生物を含む。
【0108】
ここで用いられる「試料特異的配列」という用語は、特異的試料供給源から誘導されるポリヌクレオチド分子を確認するために用いられるポリヌクレオチド配列を意味する。本発明の「試料特異的配列」という用語は、単離または合成されたポリヌクレオチドの試料供給源を意味し、一つの試料の単離または合成されたポリヌクレオチドを他の試料のポリヌクレオチドから区別する。従って、試料特異的配列は、確認することができる独自の特徴を有する。試料特異的配列の独自の特徴は、特定の配列同一性または特定の配列長さであり得る。特定の配列同一性を用いる場合、一つの試料特異的配列は、少なくとも一つのヌクレオチド、例えば、少なくとも、2、3、4、5、10、15、20またはそれ以上、60個までのヌクレオチドにおいて、もう一つの試料特異的配列から異なるべきである。特定の配列長さが用いられる場合、一つの試料特異的配列は、少なくとも一つのヌクレオチド、例えば、少なくとも、2、3、4、5、10、15、20またはそれ以上、50個までのヌクレオチドにおいて、もう一つの試料特異的配列から長さが異なるべきである。
【0109】
ここで用いられる「特定試料由来のポリヌクレオチド分子」は、特定試料から単離されたポリヌクレオチドであり得る、または、例えば、逆転写またはポリメラーゼ連鎖反応(PCR)、リガーゼ連鎖反応(LCR)、ポリヌクレオチド特異的増幅(NSBA)、鎖移動増幅(SDA)及び当該分野で知られている任意の他の技術により特定試料から合成されたポリヌクレオチドであり得る。
【0110】
ここで用いられる「異なる試料」という用語は、異なる供給源からの同じ組織または試料を含んでも含まなくても、本発明の方法により比較される二つまたはそれ以上の試料を意味する。異なる供給源は、疾患及び正常供給源;異なる細胞型、異なる組織または器官型;異なる個体;異なる環境に曝される試料;異なる発生段階;異なる疾患段階;及び異なる処理段階であり得るが、これらに限定されない。
【0111】
ここで用いられる「増幅産物」という用語は、ヌクレオチド配列において鋳型ポリヌクレオチド配列及びその相補的配列に相当する、特定のポリヌクレオチド配列及び/またはその相補的配列の一部の複製であるポリヌクレオチドを意味する。本発明による「増幅産物」はDNAまたはRNAであり得、二本鎖または一本鎖であり得る。
【0112】
ここで用いられる「合成」及び「増幅」という用語は交換可能に用いられ、特定のポリヌクレオチド配列の複製を発生するためまたは特定のポリヌクレオチド配列の複製数または量を増加させるための反応を意味する。これは、限定はされないが、試験管内方法であるポリメラーゼ連鎖反応(PCR)、リガーゼ連鎖反応(LCR)、ポリヌクレオチド特異的増幅(NSBA)、及び当該分野で知られている任意の他の技術により達成することができる。例えば、ポリヌクレオチド増幅は、任意の特定ポリヌクレオチド配列を製造するためにポリメラーゼ及び対のポリヌクレオチドプライマー、すなわち標的ポリヌクレオチド配列または標的ポリヌクレオチドを、最初に存在する量より多い量で用いる方法である。
【0113】
ここで用いられる「選択的」という用語は、ポリヌクレオチドの増幅または合成の場合、相補的配列を含むポリヌクレオチドの選択群を増幅または合成することを意味する。選択は、増幅または合成反応において特定のオリゴヌクレオチドプライマーを用いることにより達成される。例えば、第2cDNA鎖の群は、遺伝子ファミリー特異的配列に相補的な配列(例えば、後でここに記載の第2配列)を含む第2オリゴヌクレオチドを用いて選択的合成することができる。
【0114】
ここで用いられる「少なくともサブセット」という用語は、反応における全てのポリヌクレオチドまたは増幅または合成反応における全てではないポリヌクレオチド鋳型の増幅または合成を意味する。例えば、ポリヌクレオチドのサブセット(例えば、第1cDNA鎖)は、全ての第1cDNA鎖の集合からのポリヌクレオチドの群(例えば、遺伝子ファミリー)を選択的に増幅または合成する特定のオリゴヌクレオチドプライマーの使用により増幅または合成することができる。
【0115】
ここで用いられる「標的ポリヌクレオチド」は、その発現水準を分析すべきポリヌクレオチド配列である。標的ポリヌクレオチドは、その発現水準が分析される前に単離または増幅することができる。例えば、標的ポリヌクレオチドは、その増幅のために用いられる対のオリゴヌクレオチドプライマーの2つの構成員のハイブリッド領域の間にある配列であり得る。標的ポリヌクレオチドはRNAまたはDNAであり得、例えば、これは、mRNAまたはcDNA、遺伝子のコード化領域またはその一部であり得る。標的ポリヌクレオチド配列は、通常、大きな「鋳型」配列の一部として存在するが、ある場合には、標的配列と鋳型は同じである。「鋳型配列」は通常、最初に存在するポリヌクレオチド配列を意味するが、増幅反応からの産物は、その後の増幅反応において鋳型配列として用いることもできる。「標的ポリヌクレオチド」または「鋳型配列」は、特定の配列であるまたはそれを含む正常(例えば、野生型)または突然変異ポリヌクレオチドであり得る。
【0116】
ここで用いられる「RT−PCR」という用語は、組み合わされた逆転写及びポリメラーゼ連鎖反応を意味する。この増幅方法は初期ステップを用い、初期ステップにおいては、特定のオリゴヌクレオチド、オリゴdTまたは、ランダムプライマーの混合物を用いて、RNAを第1一本鎖cDNAに逆転写させ;このcDNAは、次に、標準的増幅技術、例えばPCRを用いて増幅されて、第2の相補鎖及び二本鎖cDNAを産生する。
【0117】
ここで用いられる「オリゴヌクレオチドプライマー」という用語は、ポリヌクレオチド鋳型にアニーリングすることができると共に3’末端を提供してポリヌクレオチド鋳型に相補的な拡張産物を得ることができるポリヌクレオチド分子(すなわち、DNAまたはRNA)を意味する。開始及び拡張用の条件は、通常、四つの異なるデオキシリボヌクレオシド三リン酸及び、DNAポリメラーゼまたは逆転写酵素のような重合誘発剤が適当な緩衝剤(「緩衝剤」は、共因子であるまたは、pH、イオン強度等に影響を与える置換物質を含む)の存在及び、適当な温度を含む。本発明によるプライマーは、一本鎖または二本鎖であってよい。プライマーは増幅の最大高率のためには一本鎖であり、プライマーとその補体が、二本鎖ポリヌクレオチドを形成する。しかしながら、これは、二本鎖であってもよい。本発明で有用な「プライマー」は、長さが100ヌクレオチド以下、例えば、90、80、70、60、50、40、30、20、15以下または10である。
【0118】
ここで用いられる「任意配列」という用語は、個人の判断または裁量に基づくまたは付されるものと定義される。ある場合には、任意配列は、一部の塩基について完全にランダムまたは部分的にランダムであり得る。他の例において、特定の割合の各デオキシヌクレオチド、例えば、略同じ割合の各デオキシヌクレオチドまたは主に一つのデオキシヌクレオチドを含むように、または特定のデオキシヌクレオチドを含まないように、任意配列を選択することができる。特定の制限エンドヌクレアーゼ用の認識部位を含むまたは含まないように、任意配列を選択することができる。既知の配列のmRNAまたはcDNAに相補的な配列を含むように、または、既知の配列のmRNAまたはcDNAからの配列を含まないように、任意配列を選択することができる。
【0119】
ここで用いられる「GCに富む」という用語は、GC含量が少なくとも60%GC(例えば、5塩基伸長部におけるGまたはCの3塩基、6塩基伸長部におけるGまたはCの4塩基、7〜8塩基伸長部におけるGまたはCの5塩基、9〜10塩基伸長部におけるGまたはCの6塩基、11塩基伸長部におけるGまたはCの7塩基、12〜13塩基伸長部におけるGまたはCの8塩基、14〜15塩基伸長部におけるGまたはCの9塩基、16塩基伸長部におけるGまたはCの10塩基、17〜18塩基伸長部におけるGまたはCの11塩基、19〜20塩基伸長部におけるGまたはCの12塩基、21塩基伸長部におけるGまたはCの13塩基、22〜23塩基伸長部におけるGまたはCの14塩基、24塩基伸長部におけるGまたはCの15塩基、25〜26塩基伸長部におけるGまたはCの16塩基)、好ましくは少なくとも70%GC、少なくとも80%GCまたは少なくとも90%GCあるいは100%GCまでであるヌクレオチド(3’末端ヌクレオチド)の連続伸長部を意味する。
【0120】
ここで用いられる「ATに富む」という用語は、AT含量が少なくとも60%GC(例えば、5塩基伸長部におけるAまたはTの3塩基、6塩基伸長部におけるAまたはTの4塩基、7〜8塩基伸長部におけるAまたはTの5塩基、9〜10塩基伸長部におけるAまたはTの6塩基、11塩基伸長部におけるAまたはTの7塩基、12〜13塩基伸長部におけるAまたはTの8塩基、14〜15塩基伸長部におけるAまたはTの9塩基、16塩基伸長部におけるAまたはTの10塩基、17〜18塩基伸長部におけるAまたはTの11塩基、19〜20塩基伸長部におけるAまたはTの12塩基、21塩基伸長部におけるAまたはTの13塩基、22〜23塩基伸長部におけるAまたはTの14塩基、24塩基伸長部におけるAまたはTの15塩基、25〜26塩基伸長部におけるAまたはTの16塩基)、好ましくは少なくとも70%AT、少なくとも80%ATまたは少なくとも90%ATあるいは100%ATまでであるヌクレオチド(すなわち、5’または3’末端ヌクレオチドを含む)の連続伸長部を意味する。
【0121】
ここで用いられる「遺伝子ファミリー特異的」という用語は、増幅反応において二つ以上のポリヌクレオチド鋳型にアニーリングするヌクレオチドの配列またはオリゴヌクレオチドプライマーを意味する。「遺伝子ファミリー特異的」プライマーは、鋳型に完全に相補的である必要がない。通常、遺伝子ファミリー特異的配列を含むプライマーは、試料中の異なるmRNAまたはcDNAにより示される少なくとも2、5または20、通常少なくとも50以上、または通常少なくとも75の異なる遺伝子にアニーリングする。「異なる」という用語は、遺伝子の説明で用いる場合、FASTA(デフォルト設定)により決められる配列類似性が98%を超えないRNAコード領域において少なくとも100ntの伸長部を含む場合、任意の二つの遺伝子は異なると考えられる。「遺伝子ファミリー特異的配列」は4個以上のヌクレオチド、少なくとも5、6、7、8、9、10個以上で50個までのヌクレオチド長である。
【0122】
ここで用いられる「標識」または「検出可能標識」という用語は、検出可能(好ましくは定量可能な)な信号を提供するために用いることができると共に、ポリヌクレオチドの機能的に結合させることができる任意の原子または分子を意味する。標識は、蛍光、放射能、比色、重量測定、X線回折または吸収、磁気、酵素活性、質量分析、結合親和性、ハイブリッド形成高周波、ナノ結晶等により検出可能な信号を提供することができる。本発明のプライマーは、検出可能な標識を「検出」することにより増幅反応産物を「検出」することができるように標識することができる。「定性的または定量的」検出は、標識により発生する信号の大きさ(強度)または数に基づく視覚的または自動評価を意味する。本発明の方法により標識されたポリヌクレオチド(例えば、オリゴヌクレオチドプライマー)は、5’末端、3’末端または両末端、または内部において標識される。標識は、「直接」、例えば染料、または「間接」、例えば、ビオチン、ジゴキシン、アルカリホスファターゼ(AP)、ホースラディッシュペルオキシダーゼ(HRP)であり得る。「間接標識」の検出のために、捕捉され、放出され、標識されたポリヌクレオチドフラグメントを視覚化するために、標識された抗体または酵素基質のようなさらなる成分を加えることが必要である。好ましい態様において、オリゴヌクレオチドプライマーを蛍光標識で標識する。適当な蛍光標識は、蛍光色素、例えば、ローダミン及び誘導体(テキサスレッド等)、フルオレセイン及び誘導体(5−ブロモメチルフルオロセイン等)、ルシファーイエロー、IAEDANS、7−Me2N−クマリン−4−酢酸、7−OH−4−CH3−クマリン−3−酢酸、7−NH2−4−CH3−クマリン−3−酢酸(AMCA)、モノブロモバイマン、ピレントリスルホネート、例えば、Cascade Blue、及びモノブロモリメチル−アンモニオバイマン(例えば、DeLuca,Immunofluorescence Analysis,Antibody As a Tool,Marchalonisら編、John Wiley&Sons,Ltd.(1982年)参照、ここで参考のために取り込む)を含む。
【0123】
「結合」という用語は、例えば、水素結合、イオン結合またはファンデルワールス結合による共有結合または非共有結合を意味する。そのような結合は、これらの原子またはイオンの電子密度の最分布の結果として、同じまたは異なる原子またはイオンの少なくとも二つの間に形成することができる。本発明のポリヌクレオチド(例えば、オリゴヌクレオチドプライマー)は、検出可能な標識及び/または固体支持体に結合させることができる。
【0124】
ここで用いられる「反対方向」という用語は、プライマーに言及すると、一つのプライマーが、標的ポリヌクレオチド鋳型のセンス鎖に相補的なヌクレオチド配列を含み、もう一つのプライマーは、同じ標的ポリヌクレオチド鋳型のアンチセンス鎖に相補的なヌクレオチド配列を含むことを意味する。反対方向のプライマーは、それが補足する匹敵ポリヌクレオチド鋳型からPCR増幅産物を産生することができる。反対方向の2つのプライマーを、逆プライマー及び前方プライマーと呼ぶことができる。
【0125】
ここで用いられる「同じ方向」という用語は、プライマーが、標的ポリヌクレオチド鋳型の同じ鎖に相補的なヌクレオチド配列を含むことを意味する。同じ方向のプライマーは、それが補足する匹敵ポリヌクレオチド鋳型からPCR増幅産物を発生させない。
【0126】
ここで用いられる「ポリヌクレオチド」という用語は、通常、非修飾RNAまたはDNAまたは修飾RNAまたはDNAであり得る、任意のポリリボヌクレオチドまたはポリデオキシリボヌクレオチドを意味する。ここで用いられる「ポリヌクレオチド」という用語は、一つまたは二つ以上の修飾塩基を含む前述のDNAまたはRNAも含む。すなわち、安定性のためまたは他の理由のために修飾された背骨を有するDNAまたはRNAは「ポリヌクレオチド」である。ここで用いられる「ポリヌクレオチド」という用語は、ポリヌクレオチドのそのように化学的、酵素学的または代謝的に修飾された型、並びに、ウイルス及び例えば単純及び複合細胞を含む細胞のDNA及びRNA特性の化学的型を含む。本発明に有用なポリヌクレオチドは単離または精製されたポリヌクレオチド、あるいは増幅反応において増幅したポリヌクレオチドであり得る。
【0127】
ここで用いられる「単離」または「精製」という用語は、ポリヌクレオチドに言及する場合、自然発生配列が、その正常細胞(例えば、染色体)環境から除去されているまたは、非天然環境(例えば、人工合成)において合成されることを意味する。すなわち、「単離」または「精製」配列は、細胞非含有溶液である、または異なる細胞環境に置くことができる。「精製」という用語は、配列が存在する唯一のヌクレオチドであることを意味せず、天然にそれに付随する非ヌクレオチドまたはポリヌクレオチド材料は本質的に含まない(約90〜95%、99〜100%までの純度)ことを意味し、すなわち、単離された染色体から区別される。
【0128】
ここで用いられる「cDNA」という用語は、RNA依存DNAポリメラーゼ(例えば、逆転写酵素)の作用によりRNA鋳型から製造される相補的または複製ポリヌクレオチドを意味する。「cDNAクローン」は、クローニングベクター中に含まれる意図するRNA分子に相補的な二重DNA配列を意味する。
【0129】
ここで用いられる「ゲノムDNA」という用語は、RNA転写体から複製された相補的DNAに対して、染色体DNAを意味する。ここで用いられる「ゲノムDNA」という用語は、単一細胞中に存在するDNAの全て、または単一細胞中のDNAの一部であり得る。
【0130】
ここで用いられる「相補的」という用語は、ポリヌクレオチド(またはその一部)の単一鎖が、非平行ポリヌクレオチド単一鎖のヌクレオチド(いかなる不対ヌクレオチドによっても中断されていない)間の隣接塩基対形成により非平行ポリヌクレオチド鎖(またはその一部)にハイブリッド形成し、それにより、相補的鎖間に二本鎖ポリヌクレオチドを形成する性能を意味する。第1のポリヌクレオチドは、第1ポリヌクレオチドの各及びそれぞれのヌクレオチドが第2ポリヌクレオチドの相補的領域においてヌクレオチドと塩基対を形成すれば、第2ポリヌクレオチド鎖に「完全に相補的」であると言われる。第1のポリヌクレオチドは、第1ポリヌクレオチドの一つのヌクレオチドが第2ポリヌクレオチドの対応するヌクレオチドと塩基対を形成しない場合、第2ポリヌクレオチド鎖に完全に相補的ではない。ポリヌクレオチド鎖間の相補性の程度は、ポリヌクレオチド鎖間のアニーリングまたはハイブリッド形成の効率及び強度に著しい効果を有する。これは、ポリヌクレオチド鎖間の結合に依存する増幅反応において特に重要である。
【0131】
「発現」という用語は、細胞または細胞非含有系において蛋白またはヌクレオチド配列を生成することを意味し、RNA産物への転写、翻訳後修飾及び/またはその産物をコードするDNAから蛋白産物またはポリヌクレオチドへの翻訳、並びに可能な翻訳後修飾を誘発する。
【0132】
ここで用いられる「遺伝子発現プロフィールを比較する」という用語は、二つまたはそれ以上の試料中における一つまたは二つ以上のポリヌクレオチドの異なる発現を比較することを意味する。
【0133】
ここで用いられる「発現プロフィール」という用語は、試料中における一つまたは二つ以上の遺伝子の定量的(すなわち、存在量)及び定性的発現を意味する。
【0134】
ここで用いられる「発現プロフィールにおける相違」という用語は、遺伝子の発現における定量的(すなわち存在量)及び定性的相違を意味する。ポリヌクレオチド検出のための既知の方法(例えば、電気泳動)によれば、一つの試料において遺伝子発現を検出可能であるがもう一つの試料において検出されない場合、「発現プロフィールにおいて相違がある」。あるいは、二つの試料間の遺伝子発現の定量的相違(すなわち、増加または減少)が約20%、約30%、約50%、約70%、約90%、約100%(約2倍)以上から約1.2倍、2.5倍、5倍、10倍、20倍、50倍以上までである場合、「発現プロフィールにおいて相違がある」。二つの試料間の発現プロフィールの相違がある遺伝子は、二つの試料において異なって発現された遺伝子である。
【0135】
ここで用いられる「異なる発現」という用語は、二つ以上の試料間におけるポリヌクレオチド(例えば、遺伝子)の一時的及び/または細胞性発現パターンにおける定量的及び定性的な両相違、すなわち発現プロフィールの相違を意味する。ポリヌクレオチド検出のための既知の方法(例えば、電気泳動)によれば、一つの試料においてその発現を検出可能であるがもう一つの試料において検出されない場合、ポリヌクレオチドは「異なって発現される」と言われる。ポリヌクレオチドは、二つの試料間のその発現の定量的相違(すなわち、増加または減少)が約20%、約30%、約50%、約70%、約90%、約100%(約2倍)以上から約1.2倍、2.5倍、5倍、10倍、20倍、50倍以上までである場合も、「異なって発現される」と言われる。「異なって発現された」遺伝子転写体は、例えば、活性化及び不活性化状態において、一つの発生段階ともう一つの発生段階における個体の異なる細胞または組織型において、選択された疾患を有する個体の異なる細胞または組織において、健康生物の同じ細胞または組織において見られる遺伝子転写体の複製数または状態と比較して、二つまたはそれ以上の試料間において異なる数の複製が見つかるmRNA転写体を意味する。mRNA転写体複製の数は閾値サイクル(Ct)と比例しているので、後者を、異なる発現の定量的推定に用いることもできる。従って、二つの異なる試料において遺伝子のCt値の相違が2サイクル異常の場合、遺伝子は異なって発現されると考えることができる。
【0136】
ここで用いられる「存在量」という用語は、試料における標的ポリヌクレオチドの量(例えば、μg、μモルまたは複製数で測定される)を意味する。ポリヌクレオチドの「存在量」は、例えば、Basic Methods in Molecular Biology,(1986年,Davisら著,Elsevier,ニューヨーク)及びCurrent Protocols in Molecular Biology(1997年,Ausubelら著,John Weley&Sons,Inc.)に記載されているように、当該分野でよく知られた方法(例えば、UV吸収、既知の長さ及び量を参照しつつゲル上でのバンド強度を比較)により測定することができる。本発明においてポリヌクレオチドの存在量を測定する一つの方法は、そのようなポリヌクレオチドにより放出された蛍光強度を測定し、これを、参照ポリヌクレオチド、すなわち、既知の量のポリヌクレオチドにより放出された蛍光強度と比較することである。
【0137】
「遺伝子をコードするヌクレオチド配列を有するポリヌクレオチド」は、遺伝子のコード領域を含むポリヌクレオチド配列、すなわち、遺伝子産物をコードするポリヌクレオチド配列を意味する。コード領域は、cDNA、ゲノムDNAまたはRNA型で存在し得る。DNA型で存在する場合、オリゴヌクレオチドは一本鎖(すなわち、センス鎖)または二本鎖であってよい。適当な制御要素、例えば、エンハンサー/プロモーター、スプライス接合部、ポリアデニル化信号等を、要すれば、遺伝子のコード領域に近接して配して、一次RNA転写体の転写を適切に開始及び/または正確に加工することができる。あるいは、本発明のベクターにおいて利用されるコード領域は、内因性エンハンサー/プロモーター、スプライス接合部、介在配列、ポリアデニル化信号等を含む、または内因性と外因性の制御要素の両方を組み合わせて含むことができる。
【0138】
ここで用いられる「変性ヌクレオチド」という用語は、dA、dG、dC及びdTのいずれかである、またはdA、dG、dC及びdTの少なくとも二つの塩基と塩基対を形成することができるヌクレオチドを意味する。dA、dG、dC及びdTの少なくとも二つの塩基と塩基対を形成する変性ヌクレオチドの非限定的例には、イノシン、5−ニトロピロール、5−ニトロインドール、ハイポキサンチン、6H,8H,4−ジヒドロピリミド[4,5c][1,2]オキサシン−7−オン(P)、2−アミノ−6−メトキシアミノプリン、dPTP及び8−オキソ−dGTPがある。
【0139】
ここで用いられる「人工ヌクレオチド」という用語は、自然発生ヌクレオチドではないヌクレオチドを意味する。「自然発生」という用語は、ヒトが介在することなく自然に存在するヌクレオチドを意味する。対照的に、「人工ヌクレオチド」という用語は、ヒトが介在した場合にのみ存在するヌクレオチドを意味する。特に重要な人工ヌクレオチドは、従来のヌクレオチド(すなわち、dA、dG、dC及びdT)よりも、もう一つの人工ヌクレオチドと塩基対を形成する高い選択性を示すものである(例えば、Ohtsukiら著、2001年,Proc.Natl.Acad.Sci.第98巻:4922〜4925頁、ここで参考のために取り込む)。人工ヌクレオチドは、従来のいずれかのヌクレオチドと比較して、人口ヌクレオチドとの塩基対形成能が30%以上である場合に、「従来のヌクレオチドよりも、もう一つの人工ヌクレオチドと塩基対を形成する高い選択性を示す」。塩基対形成能は、Ohtsukiら著(前記書)に記載のようなT7転写アッセイにより測定することができる。「人工ヌクレオチド」の他の非限定的例を、Lutzら著、(1998年)Bioorg.Med.Chem.Lett.第8巻:1149〜1152頁);Voegal及びBenner,(1996年)Helv.Chim.Acta第76巻:1863〜1880頁;Horlacherら著、(1995年)Proc.Natl.Acad.Sci.第92巻:6329〜6333頁;Switzerら著、(1993年)、Biochemistry第32巻:10489〜10496頁;Tor及びDervan(1993年)J.Am.Chem.Soc.第115巻:4461〜4467頁;Piccirilliら著、(1991年)Biochemistry第30巻:10350〜10356頁;Switzerら著、(1989年)J.Am.Chem.Soc.第111巻:8322〜8323頁に見ることができ、これらの全てを参考のためにここに取り込む。「人工ヌクレオチド」は、前述のような変性ヌクレオチドでもよい。
【0140】
ここで用いられる「サイン配列モチーフ」という用語は、蛋白ファミリーの構成員、または蛋白の異なるファミリーの間に高度に保存されるアミノ酸配列を意味する。これらの保存残基は、「配列モチーフ」または「サイン配列」と呼ばれ、蛋白の機能と構造の両方を決めることができる。これらは、確認蛋白または活性部位及び結合部位のような重要な蛋白領域において一般的に用いられる。配列モチーフは、多くの蛋白ファミリーについてよく知られている。さらに、潜在的配列モチーフを、利用できるコンピュータープログラムを用いて、関連する蛋白配列を比較することにより見つけることができる。
【0141】
ここで用いられる「因子」という用語は、細胞が生存及び/または成長及び/または増殖するために必要とし、別の細胞により製造及び運び出すことができる任意の物質を意味する。そのような因子には、成長因子(例えば、インターロイキン、インスリン、トランスフェリン、ヒドロコルチゾン、線維芽細胞成長因子、神経成長因子、表皮成長因子)、アミノ酸、及びビタミン類があるが、これらに限定されない。
【0142】
ここで用いられる「固体支持体」という用語は、分子(例えば、オリゴヌクレオチドプライマー)が不可逆的に結合することができる表面を意味し、限定はされないが、膜、セファローズビーズ、磁気ビーズ、組織培養プレート、シリカ系マトリクス、膜系マトリクス、限定はされないがスチレン、ラテックスまたはシリカ系材料を含む表面を含むビーズがあり、材料として他のポリマー、例えば、酢酸セルロース、テフロン(登録商標)、ポリビニリデンジフルオライド、ナイロン、ニトロセルロース、ポリエステル、カーボネート、ポルスルホン、金属、ゼオライト、紙、アルミナ、ガラス、ポリプロピレン、塩化ポリビニル、塩化ポリビニリデン、ポリテトラフルオロエチレン、ポリエチレン、ポリアミド、プラスチック、濾紙、デキストラン、ゲルマニウム、珪素、(ポリ)テトラフルオロエチレン、ガリウムアルセニド、ガリウムホスフィド、酸化珪素、硝酸珪素及びそれらの組み合わせがある。本発明による固体支持体は、反応管の内壁を含む。
【0143】
「磁気ビーズ」は、磁場により引き寄せられる固体支持体を意味し;そのような固体支持体は、限定はされないが、Dynabeads、BioMag Streptavidin、MPG7 Streptavidin、Streptavidin Magnespherel、Streptavidin Magnetic Particles、AffiniTipJ、及び磁化性粒子の任意のMaga系、BioMag Superparamagnetic Particles、または分子(例えば、オリゴヌクレオチドプライマー)が付着または不動化される任意の他の磁気ビーズである。
【0144】
ここで用いられる「遺伝子発現を調節するモジュレーター」という用語は、その化合物または条件の不存在下における遺伝子の発現と比較して、遺伝子の発現(例えば、転写の水準)を増加または減少させることができる化合物または条件を意味する。ここで用いられる「条件」という用語は、個体の正常段階、疾患段階、疾患型または発生段階、または個体が曝される環境を意味する。相違が増加の場合、増加は約20%、約30%、約50%、約70%、約90%、約100%(約2倍)以上で、約5倍、10倍、20倍、50倍以上までである。相違が減少の場合、減少は約20%、30%、50%、70%、90%、100%(例えば、特定の蛋白またはRNAが存在しない)。遺伝子発現の水準(例えば、転写の水準)は、当該分野でよく知られた方法、例えば、Basic Methods in Molecular Biology,(1986年,Davisら著、Elsevier,ニューヨーク);及びCurrent Protocols in Molecular Biology(1997年,Ausubelら著、John Weley&Sons,Inc.)に記載のようなノーザンブロット、RT−PCRにより測定することができる。遺伝子発現の水準は、本発明により開示されるような本方法により検出することもできる。本発明による「モジュレーター」は、以下に定義のような薬剤、治療剤または有効薬剤も含む。
【0145】
ここで用いられる「ポリA部位」または「ポリA配列」という用語は、発生期RNA転写体の停止及びポリアデニル化の両方を導くDNA配列を意味する。真核生物細胞の組換えDNA配列の効果的発現は、得られる転写体の効率的転写及びポリアデニル化を導く信号の発現を必要とする。転写停止信号は、通常、ポリアデニル化信号の下流において見られ、数百ヌクレオチド長である。
【0146】
ここで用いられる「RNA転写体」という用語は、DNA配列のRNAポリメラーゼで触媒した転写から得られる産物を意味する。RNA転写体が、DNA配列の完全に相補的な複製である場合、これは、一次転写体と呼ばれる;またはこれは、一次転写体の転写後加工から導かれるRNA配列であり得、成熟RNAと呼ばれる。「メッセジャーRNA」(mRNA)は、イントロンを含まず、その細胞により蛋白に翻訳することができるRNAを意味する。
【0147】
「薬剤」または「治療剤」という用語は、薬剤の活性フラグメントまたは類似体、例えば、全寸法薬剤の活性の少なくとも50%を有する蛋白またはポリヌクレオチドを含む。 薬剤は、蛋白、ペプチドまたはポリヌクレオチドであり得る。
【0148】
ここで用いられる「薬剤の効果」または「治療剤の効果」という用語は、薬剤または治療剤が、診断用特性の発現を正常から著しく異ならない値に戻す性能と定義される(通常の統計的方法により、95%信頼水準内で決められる)。
【0149】
「疾患及び病状」は、細胞、組織及び/または個体の正常機能を損なう一つまたは二つ以上の生物学的特徴の変化である。
【0150】
ここで用いられる「疾患の進行」または「疾患段階」という用語は、疾患が進行し、徴候を引き起こし、過酷度を回復または継続及び/または減少させる一連の事象を意味する。
【0151】
本発明は、二つまたはそれ以上の試料において異なって発現される遺伝子を発現し、それらの発現の水準の相違を測定する方法を提供する。本発明は、試料特異的オリゴヌクレオチドプライマーを用いるRT−PCTに基づき、増幅産物を試料供給源によって識別することができる。
【0152】
本発明の実施には、特記しない限り、当該分野の技術範囲内である分子生物学、ミクロ生物学及び組換えDNA技術の従来技術を用いる。そのような技術は、文献に充分に説明されている。例えば、Sambrook,Fritsch&Maniatis,1989年,Molecular Cloning:A Laboratory Manual,第2版:Oligonucleotide Synthesis(M.J.Gait編、1984年);Polynucleotide Hybridization(B.D.Harnes&S.J.Higginsら編、1984年);A Practical Guide to Molecular Cloning(B.Perbal,1984年);及び連続出版物,Methods in Enzymology(Academic Press,Inc.);Short Protocols In Molecular Biology,(Ausubelら編、1995年)を参照されたい。本発明の実施には、米国特許第5,965,409;5,665,547;5,262,311;5,599,672;5,580,726;6,045,998;5,994,076;5,962,211;6,217,731;6,001,230;5,963,456;5,246,577;5,126,025;5,364,521;4,985,129号に開示の技術及び組成物も含み得る。以上及び以下に記載の全ての特許、特許出願及び公報をここで参考のために取り込む。
【0153】
(試料供給源)
本発明は、ここに定義のような二つまたはそれ以上の試料における標的ポリヌクレオチドの発現を検出、測定及び比較する方法を提供する。本発明の試料は、少なくとも一つのポリヌクレオチドを含む、または標的ポリヌクレオチドそれ自体であり得る。ポリヌクレオチドの配列情報の従来の知識は、特別の用途に依存して必要または必要でない。本発明による有用な試料は、限定はされないが、標的ポリヌクレオチド(ゲノムDNA、cDNAまたはRNA)の試料、細胞、有機体、組織、流体、血漿、血清、脊髄液、リンパ液、関節液、尿、涙、便;皮膚、気道、腸管及び尿生殖管の外分泌液;唾液、血球、腫瘍、器官、組織、試験管内細胞培養成分の試料、天然単離物(例えば、飲料水、海水、個体材料)、微生物検体、及びポリヌクレオチドトレーサー分子で「マークした」対象または検体を含む。
【0154】
本発明の有用な試料は、限定はされないが、例えば、異なる個体、同じまたは異なる個体の異なる発生段階、異なる疾患個体、正常個体、同じまたは異なる個体の異なる疾患段階、異なる疾患治療に付された個体、異なる環境因子に付された個体、病気の体質を有する個体、感染性疾患(例えば、HIV)に曝された固体を含む異なる供給源から得ることができる。有用な試料は、試験管内で培養した組織、細胞または他のポリヌクレオチド含有供給源から得ることもできる。培養試料は、限定はされないが、異なる培地及び条件(例えば、pH、圧力、または温度)において培養した培地(例えば、組織または細胞)、異なる期間培養した培地(例えば、組織または細胞)、異なる因子または試薬(例えば、薬剤候補、またはモジュレーター)で治療した培地(例えば、組織または細胞)、または異なる型の組織または細胞の培地を含む供給源から得ることができる。
【0155】
試料は、限定はされないが、血液疾患、血液脂質疾患、自己免疫疾患、骨または関節障害、心臓血管疾患、呼吸疾患、内分泌疾患、免疫疾患、感染性疾患、筋肉衰弱及び全身衰弱疾患、神経疾患、例えば、神経退化及び/または神経精神疾患;皮膚疾患、腎臓疾患、強皮症、発作、遺伝性出血性血管拡張症、糖尿病、糖尿病関連疾患(例えば、PVD)、高血圧、ゴシェ病、嚢胞性線維症、鎌状赤血球貧血、肝臓疾患、膵臓疾患;眼、耳、鼻及び/または喉疾患;生殖器官に影響を与える疾患、胃腸管疾患(例えば、結腸疾患、脾臓疾患、虫垂疾患、嚢胞疾患、及び他の疾患)等を含む疾患または病状を有する個体から得ることができる。ヒト疾患のさらなる説明は、Mendelian Inheritance in Man:A Catalog of Human Genes and Genetic Disorders by Victor A.McKusick(第12版(3巻セット)1998年6月,Johns Hopkins University Press,ISBN:0801857422)が参照され、その全体がここで取り込まれる。好ましくは、正常な人口統計的に適合した個体から及び/または疾患を有する患者からの非疾患組織からの試料が、対照を提供するための分析において用いられる。
【0156】
一つの局面において、試料は、特定の疾患を有するヒト及び正常なヒトから得られる組織または細胞試料である。組織試料は、死体または最近死んだ患者から(例えば、解剖により)得ることができる。組織は、外科検体、病理検体(例えば、生検)、または通常他の処理から廃棄される「臨床廃棄物」である試料から得ることができる。試料は、成人、子供及び/または胎児から(例えば、選択的堕胎または流産から)得ることができる。細胞は、組織からの細胞の懸濁液(例えば、廃棄組織のような切り刻んだ組織細胞の懸濁液、)体液(例えば、血液、血漿、血清等)、かきとった粘膜(例えば、頬からかきとった粘膜または乳頭塗抹)及び/または、気管支洗浄、羊水穿刺及び/または白血球泳動のような他の手順から得ることができる。
【0157】
ある局面において、分析のためにRNAを抽出する前にまず細胞を培養する。連続的に成長している細胞系、一次細胞系、及び/または二次細胞系からの細胞も用いることができる。
【0158】
もう一つの局面において、試料は、異なる疾患を有するヒト及び正常ヒトから得られる組織または細胞試料である。
【0159】
一つの局面において、単一個体からの複数の組織/細胞が得られる、すなわち、試料は個体の「全身」を表す。好ましくは、本発明による「全身」を表す試料は、単一個体からの少なくとも5つの異なる型の組織を含む。より好ましくは、本発明による「全身」を表す試料は、少なくとも10または少なくとも15の異なる組織を含む。組織は、皮膚、神経組織、心臓組織、肝臓組織、胃組織、大腸組織、結腸組織、小腸組織、食道組織、肺組織、心臓組織、脾臓組織、膵臓組織、腎臓組織、生殖器(男性または女性)からの組織、副腎組織等からなる群より選択される。単一器官の異なる解剖学的または組織学的位置、例えば、脳器官である小脳、大脳及び骨髄からの組織も得ることができる。本発明の一部の局面は、器官系、例えば、呼吸系、泌尿系、腎臓系、心臓血管系、消化系及び生殖系(男性または女性)を表す試料を含む(例えば、器官系中の複数の器官からの試料を含む)。
【0160】
好ましい局面において、「全身」を表す細胞は、前述のような組織から得ることができ、さらに、患者の体液から(例えば、血液試料から)の組織を含む。
【0161】
試料は、特徴を共有する個体からの複数の細胞を含むことができる。例えば、共有される特徴は、性、年齢、病状、疾病素質、感染性疾患(例えば、HIV)への露出、親戚、同じ疾患による死亡、同じ薬剤による処理、化学療法への露出、放射療法への露出、ホルモン療法への露出、手術への露出、同じ環境条件(例えば、発ガン物質、汚染物質、アスベスト、TCE、過塩素酸塩、ベンゼン、クロロホルム、ニコチン等)への露出、同じ遺伝的変化または変化群、同じ遺伝子または遺伝子組の発現(例えば、特別のHLA対立遺伝子のような共通のハプロタイプを有する個体から試料を得ることができる)、等であり得る。
【0162】
本発明の好ましい局面において、試料はヒトから誘導されるが、本発明の一つの局面において、他の器官からの試料も用いられる。一つの局面において、試料は、疾患または他の病状のモデルを提供する非ヒト動物からの組織を含む。試料が、慢性疾患の動物モデルからの検体を表す場合、試料は、疾患の異なる段階を表す検体、例えば、寛解期間または増悪期間における動物からのものを含むことができる。試料は、追加的または択一的に、疾患または病状を治療するための療法(例えば、薬剤、抗体、蛋白療法、遺伝子療法、アンチセンス療法、そららの組み合わせ、等)に曝された疾患または病状を有する非ヒト動物からの組織を含むことができる。一部の局面において、非ヒト動物試料は、外因性ポリヌクレオチドを含む少なくとも一種の細胞を含むことができる(例えば、動物はトランスジェニック動物、キメラ動物、ノックアウトまたはノックイン動物)。
【0163】
なおさらなる局面において、植物からの試料を用いることができる。好ましくは、そのような試料は、生活周期の異なる段階における植物を含む、及び/または異なる型の植物組織(例えば、少なくとも約5つの異なる植物組織)を含むことができる。一つの局面において、試料は、外因性ポリヌクレオチドを含む少なくとも一つの細胞を含む植物から得られる(例えば、植物はトランスジェニック植物であり得る)。
【0164】
(試料からのmRNAの単離)
本方法は、二つまたはそれ以上の試料中の遺伝子の発現を測定または比較する。本発明の一つの局面において、転写水準における遺伝子の発現を測定または比較する。
【0165】
比較すべき二つまたはそれ以上の試料(例えば、試料A及びB)からのRNAを抽出し、試料特異的オリゴヌクレオチドプライマー(例えば、プライマー1A及び1B、図7参照)を用いて、個々にcDNAに逆転写する。
【0166】
RNA(例えば、mRNA)を含むポリヌクレオチドを、当該分野で良く知られている方法(Ausubelら著、前記書)に従って細胞及び組織から単離し、以下に記載する。
【0167】
RNAは、以下の方法に従って組織から精製することができる。所望の組織の除去に続いて、2g以下の組織を切り出し、液体窒素中で急速冷凍してRNAの劣化を防止する。適当な体積のグアニジニウム溶液(例えば、組織2g当たり20mlのグアニジニウム溶液)の添加時に、組織試料を組織混合器において、10秒の激しい攪拌を2または3回行って粉砕する。組織グアニジニウム溶液(1L)を調製するために、イソチオシアン酸グアニジニウム590.8gを、約400mlのDEPC−処理H2O中に溶解する。2M Tris−HCl,pH7.5(最終濃度0.05M)の25mlとNA2EDTA(最終濃度0.01M)の20mlを添加し、溶液を一晩攪拌し、体積を950mlに調節し、50mlの2−MEを添加する。
【0168】
均質化組織試料を、12,000×gで12℃で10分間遠心分離する。得られる上澄みを、20%Sarkosylの0.1体積の存在下に65℃で2分間インキュベートし、5.7M CsCl溶液(0.1g CsCl/ml)の9mlの上に乗せ、113,000×gで22℃で一晩遠心分離する。上澄みを注意深く除去した後、試験管を逆さまにし、排液する。試験管(RNAペレットを含む)の底部を、50mlプラスチック管内に置き、3mlの組織再懸濁緩衝液(5mM EDTA,0.5%(v/v)Sarkosyl,5%(v/v)2−ME)の存在下に4℃で一晩(またはそれ以上)インキュベートして、RNAペレットを完全に再懸濁させる。得られるRNA溶液を、25:24:1のフェノール/クロロホルム/イソアミルアルコール、続いて24:1のクロロホルム/イソアミルアルコールで順次抽出し、3M酢酸ナトリウム,pH5.2及び100%エタノール2.5体積を添加して沈殿させ、DEPC水中で再懸濁させる(Chirgwinら著、1979年、Biochemistry,第18巻:5294頁)。
【0169】
あるいは、RNAを、以下の単一ステッププロトコルに従って組織から単離する。組織100mg当たり1mlの変性溶液(4Mチオ硫酸グアニジニウム、25mMクエン酸ナトリウム,pH7.0、0.1Mの2−ME、0.5%(w/v)N−ラウリルサルコシン)中でガラステフロンホモジナイザーにて均質化して、所望の組織を調製する。均質化物を5mlのポリプロピレン管に移した後、pH4の2M酢酸ナトリウム0.1ml、水飽和フェノール1ml、及び49:1のクロロホルム/イソアミルアルコール0.2mlを順次添加する。各成分の添加後、試料を混合し、全成分の添加後、0〜4℃で15分間インキュベートする。試料を、10,000×gで4℃で20分間遠心分離し、100%イソプロパノール1mlの添加により沈殿させ、−20℃で30分間インキュベートし、10,000×gで4℃で10分間遠心分離してペレット化する。得られるRNAペレットを0.3ml変性溶液に溶解し、遠心分離管に移し、100%イソプロパノール0.3mlの添加により−20℃で30分間沈殿させ、10,000×gで4℃で10分間遠心分離する。RNAペレットを70%エタノール中で洗い、乾燥し、100〜200μlのDEPC−処理水またはDEPC−処理0.5%SDS中に再懸濁させる(Chomczynski及びSacchi,1987年,Anal.Biochem.,第162巻:156頁)。
【0170】
全RNAを単離するためのキット及び試薬は、種々の会社から市販されており、例えば、RNA isolation kit(Stratagene,La Lola,カリフォルニア,Cat♯200345);PicoPure(登録商標)RNA Isolation Kit(Arcturus,Mountain View,カリフォルニア,Cat♯KIT0202);RNeasy Protect Mini,Midi,及びMaxi Kits(Qiagen,Cat♯74124)がある。
【0171】
一部の態様において、全RNAを、その後の分析、例えば、逆転写のための方法で用いる。他の態様において、mRNAを、全RNAから単離する、または試料から直接単離して、逆転写に用いる。mRNAの単離のためのキット及び試薬は市販されており、例えば、Oligotex mRNA Kits(Qiagen,Cat♯70022)がある。
【0172】
RNAを含むポリヌクレオチドを、試験管内転写の方法により製造することができる。
【0173】
試験管内転写の技術は、当業者によく知られている。簡単に言えば、所望の遺伝子を、SP6、T3またはT7プロモーターを含むベクターに挿入する。ベクターを、コード化配列の下流に配される単一部位においてベクターを消化する適当な制限酵素を用いて線状化する。フェノール/クロロホルム抽出に続いて、DNAをエタノールで沈殿させ、70%エタノール中で洗浄し、乾燥し、滅菌水中に再懸濁する。線状化DNAを、転写緩衝液(200mM Tris−HCl,pH8.0,40mM MgCl2,10mMスペルミジン,250NaCl[T7またはT3]または200mM Tris−HCl,pH7.5,30mM MgCl2,10mMスペルミジン[SP6])、ジチオスレイトール、RNaseインヒビター、4つのリボヌクレオシド三リン酸の各々、及びSP6、T7またはT3 RNAポリメラーゼと、37℃で30分間インキュベートすることにより、試験管内転写反応を行う。RNAを含む放射標識ポリヌクレオチドを調製するために、非標識UTPを除き、35S−UTPが反応混合物中に含まれる。次に、DNaseIとインキュベーションすることによりDNA鋳型を除去する。エタノール沈殿に続いて、シンチレーションカウンター中で放射標識RNAを計測してcpm/μlを決めた(Ausubelら著、前記書)。
【0174】
cDNAを合成し、発現の検出及び測定のための増幅産物を産生するために、試料から単離されたRNAを用いる。好ましい態様において、cDNAの合成及び増幅反応の両方が、オリゴヌクレオチドプライマーを用いる。
【0175】
(本発明のオリゴヌクレオチドプライマーの設計)
本発明による有用なオリゴヌクレオチドプライマーを、ここに記載の当該分野でよく知られている一般的指針、及び、本発明の方法の各ステップ用に以下に記載の特定の要求に従って設計することができる。
【0176】
1.プライマー設計の一般的手法
オリゴヌクレオチドプライマーは5〜100ヌクレオチド、好ましくは17〜45ヌクレオチド長であるが、異なる長さのプライマーが有用である。cDNAを合成するためのプライマーは、好ましくは10〜45ヌクレオチドであり、増幅用のプライマーは、好ましくは約17〜25ヌクレオチドである。本発明に有用なプライマーは、溶融温度推定の方法により特定の溶融温度(Tm)を有するようにも設計される。Oligo(登録商標),Primer Designを含む市販プログラム及び、Primer3及びOligo Calculatorを含むインターネットで利用できるプログラムを用いて、本発明で有用なポリヌクレオチド配列のTmを計算することができる。好ましくは、例えばOligo Calculatorにより計算される本発明で有用な増幅プライマーのTmは、好ましくは、約45〜65℃、より好ましくは約50〜60℃である。
【0177】
ポリヌクレオチドのTmは、もう一つのポリヌクレオチドへのハイブリッド形成(例えば、オリゴヌクレオチドプライマーの、鋳型ポリヌクレオチドへのアニーリング)に影響を与える。本発明の方法において、種々のステップで用いられるオリゴヌクレオチドプライマーが、標的鋳型または標的鋳型から誘導されるポリヌクレオチド(すなわち、第1及び2cDNA鎖及び増幅産物)に選択的にハイブリッド形成することが好ましい。典型的には、二つのポリヌクレオチド配列が実質的に相補的である場合(少なくとも14〜25ヌクレオチドの伸長部において少なくとも約65%相補的、好ましくは、約75%、より好ましくは少なくとも約90%相補的)、選択的ハイブリッド形成が起こる。ここに参考として取り込まれるKanehisa,M.,1984年,Polynucleotides Res.第12巻:203頁を参照のこと。結果として、開始部位におけるある程度のミスマッチは許容されることが予測される。そのようなミスマッチは、モノ、ジまたはトリヌクレオチドのような小さい。あるいは、ミスマッチの領域は、四以上のヌクレオチドの非中断列にミスマッチが存在する領域として定義されるループが含む。
【0178】
多くの因子が、プライマーが第2ポリヌクレオチド分子にハイブリッド形成する効率及び選択性に影響を与える。本発明によりオリゴヌクレオチドプライマーを設計する場合、プライマー長さ、ヌクレオチド配列及び/または組成、ハイブリッド形成温度、緩衝液組成及び、プライマーがそこにハイブリッド形成する必要がある領域における立体障害の可能性を含むこれらの因子が考慮される。
【0179】
プライマーが標的配列にアニーリングする効率及び正確さの両方とプライマー長さとの間に陽性関係が存在する。特に、より長い配列は、より短い配列よりも高い溶融温度(TM)を有し、所定の標的配列において繰り返される可能性が低く、それにより乱雑ハイブリッド形成が最小限になる。高G−C含量を有するまたはパリンドローム配列を含むプライマー配列は、所望の標的部位と同様に、自己ハイブリッド形成する傾向があるが、これは二分子よりも一分子のハイブリッド形成速度が溶液において通常好ましいからである。しかしながら、各G−C対は、A及びTの塩基対が標的配列に結合する場合に見られる二つの水素結合ではなく三つの水素結合により結合され、それにより一層堅く強い結合が形成されるので、充分な数のG−Cヌクレオチド対を含むプライマーを設計することも重要である。ハイブリッド形成温度は、初回処理反応またはハイブリッド形成混合物に含まれる有機溶媒、例えば、ホルムアミドの濃度と同様に、プライマーアニーリング効率と逆に変化するが、塩濃度の増加は結合を容易にする。厳格なアニーリング条件下では、より長いハイブリッド形成プローブまたは合成プライマーは、より許容的な条件下では充分であるより短いものよりも効率的にハイブリッド形成する。好ましくは、厳格なハイブリッド形成は、ポリヌクレオチド配列をオリゴヌクレオチドプライマーにハイブリッド形成させる条件(例えば、PCR増幅には95℃)下に適当な緩衝液(例えば、1×RT緩衝液,Stratagene Catalog♯600085,1×Pfu緩衝液,Stratagene Catalog♯200536;または1×クローンドPfu緩衝液,Stratagene Catalog♯200532、または、cDNA合成及び増幅に有用な他の酵素に適した他の緩衝液)中で行われる。長さ及び/またはポリヌクレオチド組成またはオリゴヌクレオチドプライマーに依り、厳格なハイブリッド形成条件は変化(例えば、約1M未満、より一般的には約500mM未満、好ましくは約200mM未満の塩濃度から)することができ、ハイブリッド形成温度は変化(例えば、0℃の低い温度から22℃超、約30℃超、(最も頻繁には)約37℃超に変化)することができる。より長いフラグメントは、特性のハイブリッド形成により高いハイブリッド形成温度を必要とす
る。複数の因子がハイブリッド形成の過酷度に影響を与えるので、パラメーターの組み合わせが、単一因子の絶対測定より、一層重要である。
【0180】
オリゴヌクレオチドプライマーはこれらを考慮しつつ設計し、以下の方法により合成することができる。
【0181】
2.オリゴヌクレオチド合成
オリゴヌクレオチドプライマー自体が、当該分野でも良く知られている技術を用いて合成される。特定の配列のオリゴヌクレオチドを調製する方法は当該分野で知られており、例えば、適当な配列のクローニング及び制限消化分析、及び直接化学的合成を含む。一旦設計すると、オリゴヌクレオチドは、例えば、Narangら著、1979年,Methods in Enzymology,第68巻:90頁に記載のホスホトリエステル法、Brownら著、1979年,Methods in Enzymology,第68巻:109頁に開示のホスホジエステル法、Beaucageら著、1981年,Tetrahedron Letters,第22巻:1859頁に開示のジエチルホスホルアミデート法、米国特許第4,458,066号に開示の固体支持体法を含む適当な化学的合成法、または市販の自動オリゴヌクレオチド合成器(市販されている)またはVLSIPS(登録商標)技術を用いる他の化学的方法により調製される。
【0182】
本発明のオリゴヌクレオチドは、一部の態様によれば、固体支持体に共有または非共有的に直接または間接的(例えば、結合部位を介して)に結合させることができる。オリゴヌクレオチドは、その上でそれらが合成される固相支持体と結合することができる、またはそれらは別途合成して、使用される固相支持体に付着させることができ、これらの方法は例えば、Lundら著、(1988年)Polynucleotides Research,第16巻:10861〜10880頁;Albretsenら著、(1990年),Anal.Biochem.,第189巻:40〜50頁;Wolfら著、(1987年)Polynucleotides Research,第15巻:2911〜2926頁;またはGhoshら著、(1987年),Polynucleotides Research,第15巻:5353〜5372頁;米国特許第5,427,779,5,512,429,5,589,586,5,716,854及び6,087,102号に開示されている。固体支持体上にポリヌクレオチド配列を固定する方法は、固体支持体の製造者によっても提供され、例えば、膜については:Pall Corporation,Schleicher&Schuell、磁気ビーズについては:Dyal、培養プレートについては:Costar,Nalgenunc、本発明で有用な他の支持体については:CPG,Inc.により提供される。好ましくは、オリゴヌクレオチドは、種々の型を含み種々の結合部位を含む同じ固相支持体を用いてその上で合成される。
【0183】
本発明の固体基材は、分子(例えば、捕捉要素)を不可逆的に結合することができる任意の表面であり、限定はされないが、膜、磁気ビーズ、組織培養プレート、シリカ系マトリクス、膜系マトリクス、及び限定はされないがスチレン、ラテックスまたはシリカ系材料及び他のポリマーを含む表面を含むビーズを含み、ポリマーの例には、酢酸セルロース、テフロン(登録商標)、ポリビニリデンジフルオライド、ナイロン、ニトロセルロース、ポリエステル、カーボネート、ポルスルホン、金属、ゼオライト、紙、アルミナ、ガラス、ポリプロピレン、塩化ポリビニル、塩化ポリビニリデン、ポリテトラフルオロエチレン、ポリエチレン、ポリアミド、プラスチック、濾紙、デキストラン、ゲルマニウム、珪素、(ポリ)テトラフルオロエチレン、ガリウムアルセニド、ガリウムホスフィド、酸化珪素、硝酸珪素及びそれらの組み合わせがある。本発明による有用な固体支持体は、Sambrookら著、(1989年)Molecular Cloning:A Laboratory Manual(第2版),第1〜3巻,Cold Spring Harbor Laboratory;Ausubelら著、前記書,米国特許第5,427,779,5,512,439,5,589,586,5,716,854及び6,087,102号,Southermら著、1999年,Nature Genetics Supplement,第21巻:5頁及びJoosら著,1997年,Analytical Biochemistry,第247巻:96頁に開示されている。本発明で用いるための固相支持体には、微粒子、ビーズ、膜、スライド、プレート、微小加工チップ等を含む種々の型がある。
【0184】
本発明の好ましい固体支持体は微粒子である。本発明で、制御孔ガラス(CPG)、高架橋ポリエステル、アクリルコポリマー、セルロース、ナイロン、デキストラン、ラテックス、ポリアクロレイン等からなる微粒子を含む種々の微粒子支持体を用いることができ、これらは以下の例示参考文献に開示されている:Meth.Enzymol.,Aセクション,11頁から147頁,第44巻(Academic Press,ニューヨーク,1976年);米国特許第4,678,814;4,413,070;及び4,046;720号;及びPon編,Agrawal第19章,Methods in Molecular Biology,第20巻,(Humana Press,Totowa,ニュージャージー,1993年)。微粒子支持体は、さらに、市販のヌクレオシド誘導CPG及びポリスチレンビーズ(例えば、Applied Biosystems,Foster City,Calif.から得られる);誘導された磁気ビーズ;ポリエチレングリコールをグラフトしたポリスチレン(例えば、TentaGel.TM.,Rapp Polymere,Tubingen ドイツ);等が含む。材料、多孔性、寸法、形状等のような支持体特性、及び用いられる結合部位の型の選択は、オリゴヌクレオチドを用いる条件に依存する。例えば、酵素(例えば、逆転写酵素またはDNAポリメラーゼ)を用いる連続加工を含む用途において、酵素の立体障害を最小化すると共に基材への接近を容易にする支持体及び結合材が好ましい。最も適当な微粒子支持体の選択において考慮すべき他の重要な因子は、寸法均一性、合成支持体としての効率、表面が知られる程度、及び光学的特性を含み、例えば、以下により詳細に説明するように、透明平滑ビーズが、表面で多数のビーズを扱う場合に、有用な利点を提供する。
【0185】
微粒子表面上にオリゴヌクレオチドを付着及び/または合成する結合部位の例には、例えば、Ponら著、(1988年)Biotechniques,第6巻:768〜775頁;Webb,米国特許第4,659,774号;Baranyら著,International patent application PCT/US91/06103;Brownら著、(1989年)J.Chem.Soc.Commun.,1989年:891〜893頁;Damhaら著、(1990年)Polynucleotides Research,第18巻:3813〜3821頁;Beattieら著、(1993年)Clinical Chemistry,第39巻:719〜722頁;Maskos及びSouthen,(1992年)Polynucleotides Research,第20巻:1679〜1684頁;等がある。
【0186】
本発明のもう一つの好ましい固体支持体は、反応管の内壁である。反応管は、任意の酢酸セルロース、テフロン(登録商標)、ポリビニリデンジフルオライド、ナイロン、ニトロセルロース、ポリエステル、カーボネート、ポルスルホン、金属、ゼオライト、紙、アルミナ、ガラス、ポリプロピレン、塩化ポリビニル、塩化ポリビニリデン、ポリテトラフルオロエチレン、ポリエチレン、ポリアミド、プラスチック、濾紙、デキストラン、ゲルマニウム、珪素、(ポリ)テトラフルオロエチレン、ガリウムアルセニド、ガリウムホスフィド、酸化珪素、硝酸珪素からなってよい。好ましくは、反応管の内壁はポリプロピレンからなる。
【0187】
オリゴヌクレオチドは、単一(または数個の)の固相支持体上で合成して、合成されたオリゴヌクレオチドで均一に被覆された領域の配列を形成することもできる。そのような配列を合成する技術が、McGallらの国際出願PCT/US93/03767;Peaseら著、(1994年)Proc.Natl.Acad.Sci.,第91巻:5022〜5026頁;Southern及びMaskos,International application PCT/GB89/01114;Maskos及びSouthern(前記書);Southernら著,(1992年)Genomics,第13巻:1008〜1017頁;及びMaskos及びSouthern,(1993年);Polynucleotides Research,第21巻:4663〜4669頁に開示されている。
【0188】
好ましくは、本発明は、微粒子またはビーズに結合されたオリゴヌクレオチドを用いて実施される。微粒子支持体及び、それらの表面にオリゴヌクレオチドを共有または非供給結合させる方法は良く知られており、以下の参考文献に例示されている:Beaucage及びIyer(前記書);Gait編,Oligonucleotide Synthesis:A Practical Approach(IRL Press,Oxford,1984年);及び先に引用の参考文献。通常、微粒子の寸法及び形状は重要でないが;寸法範囲は数μm、例えば、1〜2μmから数百μm、例えば、200〜1,000μmの直径の微粒子が好ましく、これらが、最少限の試薬及び試料使用量で、広範囲のオリゴヌクレオチドの製造及び取り扱いを容易にする。
【0189】
一部の好ましい態様において、本発明の固相支持体として、市販の孔制御ガラス(CPG)またはポリスチレン支持体が用いられる。そのような支持体は、塩基不安定性リンカー及び付着した初期ヌクレオシドを用いて利用できる(例えば、Applied Biosystems(Foster City,Calif.))。好ましくは、粒径が500〜5,000Åである微粒子が用いられる。
【0190】
別の好ましい態様において、非多孔質微粒子が光学的特性のために用いられ、これらは、顕微鏡スライドのような平坦支持体上で多数の微粒子を追跡する場合、有利に用いることができる。特に好ましい非多孔質微粒子は、Bangs Laboratories(Carmel,Ind.)から得られるグリシダルメタクリレート(GMA)ビーズである。そのような微粒子は、種々の寸法で有用であり、タグまたはタグ補体を合成するための種々の結合基を用いて誘導される。好ましくは、オリゴヌクレオチドの大規模平行操作のために、直径5μmGMAビーズの微粒子が用いられる。
【0191】
3.cDNA合成のためのオリゴヌクレオチドプライマー設計手法
本方法のcDNA合成及び増幅反応のための特定のオリゴヌクレオチドプライマーの設計は、標的配列を認識しアニーリングすることができる配列の選択を含む。オリゴヌクレオチドのTmは、オリゴヌクレオチド長及びGC含量の分析により最適化される。
【0192】
本発明で有用なプライマーの設計は、前述の幾つかのパラメーター及びプライマー配列の最適化の評価に役立てるために開発された容易に利用できるコンピュータープログラムを用いることにより容易にすることができる。そのようなプログラムの例には、「PrimerSelect」(DNAStar(登録商標)ソフトウェアパッケージ(DNAStar,Inc;Madison,ウィスコンシン))、OLIGO4.0(National Biosciences,Inc.)、PRIMER,Oligonucleotide Selection Program,PGEN and Amplify(Ausubelら著の中に記載、前記書)がある。
【0193】
A.オリゴヌクレオチドプライマーの組成
本発明で有用なオリゴヌクレオチドプライマーは、一つまたは二つ以上の変性塩基、例えば、前述の塩基からなる変性配列を含み得る。そのような変性オリゴヌクレオチドは、ポリヌクレオチドキナーゼ、DNAリガーゼ、及び他の修飾性酵素(Hill,F.Loakes,D.及びBrown D.M.,1998年,Proc Natl Acad Sci USA.,第95巻:4258〜4263頁)用の正常基材として働く。変性ヌクレオチドは、オリゴヌクレオチド配列中、任意の位置、すなわち、5’、3’または内部に組み込むことができる。変性した塩基は、当該分野で知られており、異なる変性を示すために異なるコードが用いられる(例えば、表I)。
【表1】
【0194】
また、変性塩基は、dA、dG、dC及びdTの少なくとも二つと塩基対を形成することができるヌクレオチドであってよい。そのような有用な変性塩基は、通常、ヌクレオチド類似体であり、当該分野で知られており、以下に記載されている。例えば、デオキシイノシン(dI)は、4つの天然DNA塩基のいずれかに結合するので自然発生変性塩基である。dIは、真に普遍的ではないが、4つの標準的塩基(すなわち、A、T、G及びC)を含むミスマッチよりは不安定性が低い。ここで用いられる「普遍的塩基」という用語は、隣接塩基対の相互作用を著しく不安定化することなく、または変性オリゴヌクレオチドの予想される機能的生化学的有用性を妨害することなく、4つの正常塩基をいずれかを置換する性能を示す塩基を意味する。dIとdA、dG、dC及びdTとの間の水素結合相互作用は弱く等しくなく、その結果、一部の塩基対偏向がある:dI:dCハイブリッド形成>dI:dA>dI:dG>dI:dT(Kawase,Yら著、1986年,Polynucleotides Res.,1919年:7727〜7736頁;Martin,F.H.ら著、1985年,Polynucleotides Res.,第13巻,8927〜8938頁;Case−Green,S.C.,Southern,E.M.,1994年,Polynucleotides Res.,第22巻,131〜136頁)。ポリヌクレオチド中に存在する場合、dIは、DNAポリヌクレオチドにより、選択的に、dCを発生期鎖に直接組み込む。
【0195】
より最近では、機能的に真の普遍的塩基であるがdA、dG、dCまたはdTのいずれかと対を形成した場合にWatson−Crick DNA二本鎖を不安定化しない非天然塩基が加工された。これらの普遍的DNA塩基類似体の適用が最近試験された(Loakes,2001年,Polynucleotides Res.,第29巻,2437〜2447頁)。2つの例は、3−ニトロピロール−2’−デオキシヌクレオシド及び5−ニトロインドール−2’−デオキシヌクレオシド(5−ニトロインドール)。これら前記2例は、真の普遍的塩基として作用する。より特異的な他の塩基修飾が合成された。dA、dG、dC及びdTの四つ全てではないが二つ以上と塩基対を形成する変性塩基も、本発明の方法に有用である。例には、「p」と表されるピリミジン(CまたはT)類似体6H,8H−3,4−ジヒドロピリミド[4,5−c][1,2]オキサジン−7−オン、及び「k」と表されるプリン(AまたはG)類似体N6−メトキシ−2,6−ジアミノプリンがある。「p」塩基は、dAまたはdGと対を形成し、「k」塩基はdTまたはdCと対を形成する(Bergstrom,D.E.,Zhang,P.,及びJohnson,W.T.,1997年,Polynucleotides Res.,第25巻1935〜1942頁)。
【0196】
例えば、dPTP(dP)は、その塩基が2つの互変異性体型のいずれかに存在することができるので、チミジン(T)またはデオキシシチジン(dC)として挙動することができる。イミノ型において、dPは、チミジンの塩基対形成特性を有し、そのためdAと塩基対を形成し;アミノ型においては、dCと同様に、dGと塩基対を形成する(Sekiguchi,M.,1996年,Genes to Cells,第1巻,139〜145頁;Pavlov,Y,ら著、1994年,Biochemistry,第33巻,4695〜4701頁)。8−オキソ−dGTPは、dCまたはdAのいずれかと塩基対を形成する(Sekiguchi,M.,前記書;Zaccolo,M.,ら著、1996年,J.Mol.Biol.,第255巻,589〜603頁)。
【0197】
本発明のオリゴヌクレオチドプライマーは、定義項目において先に定義した人工ヌクレオチドを含むことができる。人工ヌクレオチドは、本発明のオリゴヌクレオチドプライマーの5’、3’または内部に配することができる。「人工ヌクレオチド」は、オリゴヌクレオチド中に用いて、非特異的アニーリング及び背景増幅させて、ポリヌクレオチド増幅の特異性を増すことができる。この目的で、用いられる人工ヌクレオチドが、従来のヌクレオチド(すなわちdA,dT,dG,dC及びdU)を超える、もう一つの人工ヌクレオチドとの塩基対選択性を示すことが好ましい。一つの態様において、一つまたは二つ以上の人工ヌクレオチドXTP(2−アミノ−6−(N,N−ジメチルアミノ)プリン−5’−三リン酸)またはYTP(ピリジン−2−オン−リボヌクレオシド−5’−三リン酸)が用いられるが、これは、dXTP及びdYTPが、従来のヌクレオチドを超える、互いの塩基対形成選択性を示し、dUTPについての僅かな選択性も示されるからである(Ohtsukiら著、前記書)。
【0198】
本発明のオリゴヌクレオチドプライマー(例えば、第1オリゴヌクレオチドプライマー)は、その5’末端においてGCに富む(すなわち、5’末端ヌクレオチドを含むヌクレオチドの連続的伸長部)と共にその3’末端においてATに富む(すなわち、3’末端ヌクレオチドを含むヌクレオチドの連続的伸長部)配列(例えば、試料特異的配列タグ)を含むことができる。その5’末端においてGCに富みその3’末端においてAtに富む配列の使用は、ATがGCよりも弱い塩基対を形成するので、プライマーアニーリングの特異性を増加させる。従って、ポリヌクレオチドの合成及び増幅の特異性を増すことができる。
【0199】
B.第1cDNA鎖合成のための第1オリゴヌクレオチドプライマー
本発明の方法において、第1cDNA鎖の合成のために第1オリゴヌクレオチドプライマーが用いられる。一つの態様において、増幅産物を生成するための他のプライマー用の鋳型として作用する配列を有する第1オリゴヌクレオチドプライマーも設計される。第1オリゴヌクレオチドプライマーは、20〜100ヌクレオチド長、好ましくは30〜60ヌクレオチド長、より好ましくは30〜45ヌクレオチド長、さらにより好ましくは34〜42ヌクレオチド長であり得る。
【0200】
本発明の一つの独自の特徴は、同じ反応混合物中で二つまたはそれ以上の試料を分析できることである。この目的のために、試料供給源の起源を適当に確認する必要である。好ましくは、第1オリゴヌクレオチドプライマーは、試料特異的タグを含む。例えば、試料Aから第1cDNA鎖を合成するための第1オリゴヌクレオチドプライマーは、試料特異的配列タグAを含み;試料Bから第1cDNA鎖を合成するための第1オリゴヌクレオチドプライマーは、試料特異的配列タグBを含む。試料特異的タグを含むそのような第1オリゴヌクレオチドプライマーの使用は、得られるポリヌクレオチド合成及び増幅産物を同じ供給源に従って区別することができる機構を提供する。例えば、試料AからのcDNAまたは増幅産物は、試料特異的タグBを含む試料BからのcDNAまたは増幅産物と識別される、試料特異的タグAを含む。試料特異的配列タグは、15〜60ヌクレオチド長、好ましくは18〜40ヌクレオチド長、より好ましくは20〜30ヌクレオチド長、さらにより好ましくは20〜24ヌクレオチド長であり得る。
【0201】
本発明による試料特異的配列タグはポリヌクレオチド配列(すなわち、試料特異的配列タグ)であり得る、またはこれは、当該分野で知られている任意の他の確認可能なタグであり得る。異なる第1オリゴヌクレオチド(すなわち、異なる試料)用の試料特異的配列タグは、ヌクレオチド配列が相違する、または単に長さが相違し得る。
【0202】
試料特異的タグ(例えば、試料特異的配列タグ)は、第1オリゴヌクレオチド(すなわち、5’末端ヌクレオチド及び3’ヌクレオチドから離れた少なくとも一つのヌクレオチド)の5’末端、3’末端またはその両方、またはその中間部に配することができる。好ましい態様において、試料特異的タグが、第1オリゴヌクレオチドプライマーの5’末端に配される、すなわち、試料特異的配列の5’に他のヌクレオチドが無い。
【0203】
真核生物mRNAの大部分(ヒストンmRNAが顕著に除かれている)が、3’−末端「ポリA」尾部を有して合成される。ポリ(A)配列はDNA中にコードされず、転写後に核内のRNAに天下される。ポリ(A)の添加は、酵素ポリ(A)ポリメラーゼにより触媒され、200Aまでの残基がmRNAの遊離3’−OH末端に付加される。3’−末端ポリ(A)尾部の存在は、重要な実用的結果を提供する。mRNAのポリ(A)領域は、オリゴ(U)またはオリゴ(dT)と塩基対を形成することができ、この反応を用いて、ポリ(A)+mRNAを単離し、mRNAからcDNAを合成することができる。オリゴ(dT)またはオリゴ(dU)配列をプライマーとして用いて、逆転写酵素を用いる第1cDNA鎖の合成を始めることができる。
【0204】
第1オリゴヌクレオチドプライマーは、さらに、オリゴ(dT)またはオリゴ(dU)配列を含むことができる。好ましくは、オリゴ(dT)またはオリゴ(dU)配列は、試料特異的配列の3’に配される。オリゴ(dT)またはオリゴ(dU)配列は、少なくとも5ヌクレオチド長であり、5〜20ヌクレオチド長、好ましくは8〜18ヌクレオチド長、より好ましくは12〜16ヌクレオチド長であり得る。
【0205】
一つの態様において、試料特異的配列タグは、第1オリゴヌクレオチドプライマーの5’末端に、約20〜24個のヌクレオチドの一般的構造を含む。好ましい態様において、この約20ヌクレオチドの一般的構造には、3’末端においてオリゴ(dT)またはオリゴ(dU)伸長部(12〜16残基)が続く。好ましい態様において、オリゴ(dT)またはオリゴ(dU)伸長部は、試料特異的配列の3’の直ぐ隣である。しかしながら、試料特異的タグとオリゴ(dT)またはオリゴ(dU)伸長部との間に、非試料特異的配列、すなわち、試料Aと試料Bの両方に共通の配列(例えば、少なくとも一つのヌクレオチドまたは少なくとも2、3、5、6または10あるいは20までのヌクレオチド)がある。
【0206】
cDNA合成におけるオリゴ(dT)またはオリゴ(dU)の使用に関わる一つの潜在的問題がある。ポリA尾部はかなり長い可能性があるので、オリゴ(dT)またはオリゴ(dU)を単に用いることは、非ポリA領域の直前において逆転写を正確に開始することができない。実際、オリゴdTはランダムにポリA配列の伸長部にアニーリングするので、単一鋳型mRNAからの逆転写の最終産物であっても、各々が5’−末端において異なる長さのポリTを有する第1cDNA鎖の異種集合を得ることができる。この問題を克服するために、二つまたはそれ以上のデオキシヌクレオチド、例えば、VNを、オリゴ(dT)またはオリゴ(dU)プライマーの3’−末端に付加することができ、ここで、VはdTTPを除く任意のdNTPでありNは四つのdNTPのいずれかである。そのようにして、そのようなプライマーは、ポリA尾部と非尾部領域との結合部において安定にアニーリングし、所定の鋳型から合成された第1cDNA鎖の均一寸法が確保される。その意味において、第1cDNA鎖合成に用いられるプライマーは、実際、変性オリゴヌクレオチドである(Smithら著,1997年,Biotechniques第23巻:274〜279頁)。
【0207】
一つの態様において、第1プライマーの3’末端は、さらに、変性配列、すなわち、二つ以上のヌクレオチド組成を含む配列を含む。第1オリゴヌクレオチドプライマーは、任意の長さ、好ましくは5ヌクレオチド未満、より好ましくは2ヌクレオチド長の変性配列を含むことができる。一つの態様において、第1オリゴヌクレオチドプライマーにおける変性配列はVであり、ここで、Vは、dA,dCまたはdG及びNはdA,dT(またはdU)、dCまたはdGである。好ましい態様において、第1オリゴヌクレオチドプライマーは、5’(試料特異的配列タグ)20〜24(dT)12〜16VN3’の組成を含む。もう一つの態様において、第1オリゴヌクレオチドプライマーは、5’(試料特異的配列タグ)20〜24(dU)12〜16VN3’の組成を含む。
【0208】
第1オリゴヌクレオチドプライマー上のオリゴ(dT)またはオリゴ(dU)伸長部は、各試料中の相補的(ポリA)尾部mRNAにアリーリングされて、第1cDNA鎖合成の開始を可能にする。変性ヌクレオチドは、オリゴ(dT)またはオリゴ(dU)のアニーリングを容易にし、第1cDNA鎖合成の効率を上げる。プライマー特異的配列タグは、二つの試料の各々について独自であり、cDNAの起源を識別させる。
【0209】
変性塩基の使用により、第1cDNA鎖合成用の第1オリゴヌクレオチドプライマーの混合物が得られる。例えば、一つの態様において、各試料用の特定プライマーの混合物を用いて逆転写を行う。これらのプライマーは以下の構造を有する:試料Aについては5’−(特異的配列タグA)20〜24T12〜16AN−3’,5’−(特異的配列タグA)20〜24T12〜16CN−3’,5’−(特異的配列タグA)20〜24T12〜16GN−3’(Nは、A、T、C及びGの混合物を含む変性塩基);試料Bについては5’−(特異的配列タグB)20〜24T12〜16AN−3’,5’−(特異的配列タグB)20〜24T12〜16CN−3’,5’−(特異的配列タグB)20〜24T12〜16GN−3’。試料特異的配列タグは、混合物中の各プライマーについて同一である必要はない。例えば、一つの態様において、各試料についての特定プライマーの混合物を用いて逆転写が行う。これらのプライマーは、以下の構造を有する:試料Aについては5’−(特異的配列タグA1)20〜24T12〜16AN−3’,5’−(特異的配列タグA2)20〜24T12〜16CN−3’,5’−(特異的配列タグA3)20〜24T12〜16GN−3’(Nは、A、T、C及びGの混合物を含む変性塩基);試料Bについて5’−(特異的配列タグB1)20〜24T12〜16AN−3’,5’−(特
異的配列タグB2)20〜24T12〜16CN−3’,5’−(特異的配列タグB3)20〜24T12〜16GN−3’。
【0210】
当該分野で知られている他のヌクレオチドタグも、本発明の試料特異的タグとして用いることができ、これは例えば、Churchら著、(1988年,Science,第240巻:185〜188頁)、Dollinger,(1994年,265〜274頁,Mullisら編,The Polymerase Chain Reaction,Birkhauser,Boston,),Brenner及びLerner,(1992年,Proc.Natl.Acad.Sci.,第89巻:5381〜5383頁)、Alper,(1994年,Science,第264巻:1399〜1401頁)、Needelsら著,(1993年,Proc.Natl.Acad.Sci.,第90巻:10700〜10704頁)及び米国特許第6,280,935,6,172,218,6,150,516,5,846,719,6,172,214,6,235,475に開示されており、これら全てをここで参考のために取り込む。前記特許は、オリゴヌクレオチドタグの使用により分子のクラスまたは下位集合を追跡、確認及び/または分類する方法を開示している。ポリヌクレオチドに付着したタグを、固相支持体上の補体に特異的にハイブリッド形成することにより、最小限の交差ハイブリッド形成セットから選択されたオリゴヌクレオチドを含むオリゴヌクレオチドタグを、ポリヌクレオチドの分類に用いることができる。そのようなオリゴヌクレオチドは、各々、3〜9ヌクレオチド長の複数のサブユニットからなる。最小限の交差ハイブリッド形成セットのサブユニットは、同じセットの任意の他のサブユニットの補体との二つまたはそれ以上のミスマッチを有する二量体または三量体を形成する。特別の態様において利用できるオリゴヌクレオチドタグの数は、タグ当たりのサブユニットの数及びサブユニットの長さに依存する。もう一つの有用なヌクレオチドタグが、標的ポリヌクレオチドの末端への、コードされたアダプターの一つまたは二つ以上のセットのライゲーションに基づくポリヌクレオチド配列分析の方法を提供する米国特許第6,013,445号(ここで参考に取り込む)により開示されている。標的ポリヌクレオチド上の相補突起鎖と完全にマッチした二量体を、その突起鎖が形成するコードされたアダプターをライゲートし、突起鎖中のヌクレオチドの同一性を、
コードされたアダプターの有するオリゴヌクレオチドタグにより決める。
【0211】
好ましい態様において、第1オリゴヌクレオチドプライマーが、前述の固体支持体に供給結合する。この場合、逆転写反応が、支持体に永続的に結合された第1cDNA鎖を発生し、それにより、これら第1cDNA鎖を複数の反応に用い、第1cDNA鎖から、合成した第2cDNA鎖を容易に分離することができる。好ましくは、第1オリゴヌクレオチドプライマーの5’を固体支持体に結合する。
【0212】
もう一つの態様において、第1オリゴヌクレオチドプライマーが、固体支持体に付着させることなく、溶液中で合成される。
【0213】
C.第2cDNA鎖合成のための第2オリゴヌクレオチドプライマー
本発明の方法は、第1cDNA鎖を発生させた後、第2オリゴヌクレオチドプライマーを用いて第2cDNA鎖を合成することを含む。この場合、合成された第2cDNA鎖または二本鎖cDNAが、その後の増幅のための鋳型として用いられる。あるいは、合成された第1cDNA鎖を、増幅用の鋳型として直接用いて、第2cDNA鎖を合成することができる。
【0214】
一つの態様において、第2オリゴヌクレオチドプライマーは、増幅産物を生成するための他のプライマー用の鋳型として作用する配列を用いても設計される。第2オリゴヌクレオチドプライマーは、20〜100ヌクレオチド長、好ましくは17〜60ヌクレオチド長、より好ましくは20〜45ヌクレオチド長、さらにより好ましくは20〜25ヌクレオチド長であり得る。好ましくは、第2オリゴヌクレオチドプライマーは、第1任意配列タグを含む。また好ましくは、一つの試料(例えば、試料A)用の第2オリゴヌクレオチドプライマーは、もう一つの試料(例えば、試料B)用の第2のオリゴヌクレオチドプライマーと同じ第1任意配列を含む。異なる試料から第2cDNA鎖を合成するために用いられる第2オリゴヌクレオチドプライマー中に同じ第1任意配列タグが存在するので、異なる試料から誘導されるcDNAの増幅のために、共通の増幅オリゴヌクレオチドプライマー(例えば、以下に記載のような第3オリゴヌクレオチドプライマー)を用いることができる。
【0215】
第1任意配列タグを、第2オリゴヌクレオチドプライマーの5’または3’末端、あるいは内部に配する(すなわち、5’末端ヌクレオチド及び3’ヌクレオチドから離れた少なくとも一つのヌクレオチド)ことができる。好ましくは、第1任意配列タグが、5’末端に配される、すなわち、第2cDNA鎖合成用の第2プライマーの任意配列の5’に他のヌクレオチドが存在しない。第1任意配列は、5〜30ヌクレオチド長である。
【0216】
第2オリゴヌクレオチドプライマーは、さらに、第1cDNA鎖のサブセット(すなわち、複数)に相補的な第2配列を含み、それにより、試料から二つまたはそれ以上の異なる第2cDNA鎖を合成することができる。好ましくは、第2配列は短い配列、例えば、25ヌクレオチド未満の長さ、好ましくは20ヌクレオチド未満の長さ、より好ましくは15ヌクレオチド未満の長さ、さらにより好ましくは10ヌクレオチド未満の長さであり、それにより、試料から合成された第1cDNAのサブセットにアニーリングすることができる。一つの態様において、第2オリゴヌクレオチドプライマーの第2配列は6〜7ヌクレオチド長である。もう一つの態様において、第2配列は、3’末端においてランダムに選択された配列(例えば、6〜7塩基)を含み、それにより、第2配列に相補的な配列を含む遺伝子(すなわち、第1cDNA鎖)からcDNAのサブセットが合成される。
【0217】
通常、プライマーから延ばすためのポリメラーゼのために3’末端において完全または完全に近いマッチングを有すべきなので、第2オリゴヌクレオチドプライマーの3’末端は非常に重要である。好ましくは、第2の配列が、第1任意配列の3’に配される。一つの態様において、第2配列が、第1任意配列の3’の直ぐ近くに配される、すなわち、第2配列と第1任意配列との間に他のヌクレオチドが存在しない。
【0218】
好ましい態様において、第1任意配列と第2配列との間に第3配列が配される。好ましくは、第3配列は、前述のような一つまたは二つ以上の変性ヌクレオチドを含む。第3配列は、1〜15ヌクレオチド長、好ましくは1〜10ヌクレオチド長、より好ましくは2〜6ヌクレオチド長である。一つの態様において、第1任意配列と第2配列との間に配される第3配列は4ヌクレオチド長である(例えば、図7中のZ4)。第3配列は、全ての変性ヌクレオチドを含む、または、変性ヌクレオチドと非変性ヌクレオチドとの配列を含み得る。第3配列中の変性ヌクレオチドは、dA、dT、dG及びdCのいずれかである、または、dA、dT、dG及びdCの二つまたはそれ以上と塩基対を形成することができるヌクレオチドであり得る。好ましい態様において、第3配列は四つの変性ヌクレオチドを含み、その各々がdA、dT、dG及びdCの二つまたはそれ以上と塩基対を形成することができる。より好ましい態様において、変性ヌクレオチドはdIまたは5−ニトロピロールである。変性ヌクレオチドを含む一つの目的は、プライマーの全体的安定性を増すことである。DNAポリメラーゼがdITP鋳型を読み取ることができ、そのようなdITPをPCRにおける鋳型として用いる場合、四つのdNTPのいずれかをランダムに
組み込むことが知られている。
【0219】
第2及び第3配列の選択は、そこからcDNAが合成され増幅される特定の遺伝子サブセットを決める。第2及び/または第3配列を変得ることにより、合成/増幅産物の寸法を調節するのみでなく、増幅すべき特定の遺伝子ファミリーを選択することができる。例えば、小さいG蛋白は全て、GxGxxGのサインモチーフを有し、ここで、Gはグリシン及びxは任意のアミノ酸を表す。変性オリゴヌクレオチドを用い、このサインモチーフをマッチングさせることにより、全ての小さいG蛋白の発現プロフィールを研究することができる。キナーゼ、ホスファターゼのような同様の多くの蛋白ファミリーは、サインモチーフを有し、多くの機能的ドメインまたはモチーフが、サイン配列を有する(ジンクフィンガー等)。これらのモチーフまたはサイン配列は良好に記録され、これらのモチーフ/サイン配列の詳細な記載を含む検索自由なデータベースがある。例えば、Accelrysにより開発されたGCG Wisconsin Package配列分析ツール(その一部は以前はGCG)は、そのようなモチーフ検索及び記録を提供し、その全内容をここで参考のために取り込む。
【0220】
一つの態様において、第2オリゴヌクレオチドは、5’(第1任意配列)10−12(第3配列)4(第2配列)6−73’の一般的構造を含む。変性塩基(例えば、dA、dT、dG及びdCのいずれか)の使用により、第2cDNA鎖合成用の第2オリゴヌクレオチドプライマーの混合物が得られる。
【0221】
第2オリゴヌクレオチドプライマーは、前述のように固体支持体に結合してよい、または結合しなくてよい。好ましい態様において、第2オリゴヌクレオチドは固体支持体に結合しないが、第1オリゴヌクレオチドは結合し、それにより、合成後に固体支持体に結合する第1cDNA鎖から、合成された第2cDNA鎖を容易に分離することができる。
【0222】
第2オリゴヌクレオチドプライマーの第1任意配列を設計する際、cDNA合成及び増幅反応の特異性を増加させ背景を低下させるために、その配列が、GenBankデータベースまたは他の利用できるデータベースにおける任意の哺乳動物ゲノム配列における配列に強度にマッチングしないことが好ましい。「強度のマッチング」は、第1任意配列と、種、例えばGenBankデータベースまたは他のデータベースで利用できるヒトまたは全ての哺乳動物の配列との間の配列同一性が30%未満(例えば、20%未満、10%未満、5%未満、2%未満の配列同一性)であることを意味する。一部の態様において、試料供給源が知られている、例えば、ヒト、イヌまたは他の動物あるいは植物のような特定の種である場合、GenBankデータベースまたは他のデータベースにおける特定の種について、第1任意配列は、ゲノム配列中の配列に強度のマッチングを示さないことが好ましい。
【0223】
D.オリゴヌクレオチドプライマーの標識
本発明のオリゴヌクレオチドプライマーは、前述のように、分光的、光化学的、生化学的、免疫化学的、酵素学的または化学的手段により検出可能な部分を組み込むすることにより標識することができる。オリゴヌクレオチドプライマーに標識を結合または共役させる方法は、もちろん、用いられる標識の型及び、プライマー上の標識の位置に依存する。本発明で有用なプライマーは、5’末端、3’末端で標識、またはプライマーの長さ全体において標識することができる。
【0224】
本発明での使用に適当な種々の標識、及びそれらをプライマー中に含むための方法は当該分野で知られており、限定はされないが、互いに相互作用して信号を増加、変化または減衰させる得る、酵素(例えば、アルカリホスファターゼ及びホースラディッシュペルオキシダーゼ)及び酵素基質、放射活性原子、蛍光染料、発色団、化学発光水準、電気化学発酵水準、例えば、Origen(登録商標)(Igen)を含む。もちろん、熱サイクラー装置を用いて行われるPCR系アッセイにおいて標識分子を用いる場合、標識が、この自動プロセスで必要な温度サイクルに耐えなくてはならない。
【0225】
本発明の標識プライマーの組み立てにおける標識として用いられる蛍光体には、ローダミン及び誘導体(テキサスレッド等)、フルオレセイン及び誘導体(5−ブロモメチルフルオロセイン等)、ルシファーイエロー、IAEDANS、7−Me2N−クマリン−4−酢酸、7−OH−4−CH3−クマリン−3−酢酸、7−NH2−4−CH3−クマリン−3−酢酸(AMCA)、モノブロモバイマン、ピレントリスルホネート、例えば、Cascade Blue、及びモノブロモリメチル−アンモニオバイマンがある。通常、モノ色素計ではなくフィルターを有する蛍光計を用い、検出効率を増すためには、ストロークの移動が大きな蛍光体が好ましい。
【0226】
標識は、種々の技術により、オリゴヌクレオチドに直接または間接的に付着することができる。用いられる標識またはタグの正確な型に依存して、標識は、プライマーの5’末端に配する、またはプライマーの内部に配する、または種々の寸法及び組成のスペーサーアームに付着させて、信号相互作用を容易にすることができる。市販のホスホラミダイト試薬を用いて、適当に保護されたホスホラミダイトを介して5’末端において官能基(例えば、チオールまたは一次アミン)を含むオリゴマーを製造することができ、例えば、PCR Protocols:A Guide to Medhods and Applications,Innisら編,Academic Press,Ind.,1990年に記載のプロトコルを用いてそれらを標識することができる。
【0227】
典型的には5’末端において、オリゴヌクレオチドプライマー配列中に、一つまたは二つ以上のスルフヒドリル、アミノまたはヒドロキシル基を導入するためにオリゴヌクレオチド官能化試薬を導入するための方法が、米国特許第4,914,210号に記載されている。ポリヌクレオチドキナーゼ及びガンマ−32P−ATPまたはガンマ−33P−ATPを用いることにより5’リン酸基を放射性同位体として導入して、レポーター基を提供することができる。合成中に導入されたアミノチミジン残基、6−アミノヘキシル残基を、ビオチンのN−ヒドロキシスクシンイミドエステルと反応させることにより、ビオチンを5’末端に付加することができる。
【0228】
(cDNAの合成)
本発明の方法により、cDNAを調製し増幅に用いることができる。一部の態様において、第1のcDNA鎖を調製し、その後の増幅反応及び分析に直接用いる。本発明の好ましい態様において、第1cDNAと第2cDNAの両方が合成される。合成された第1及び第2cDNA鎖をその後の増幅反応に用い、第2cDNA鎖を、第1cDNA鎖から分離して増幅反応に用いることができる。
【0229】
cDNAの調製は良く知られており、文献(例えば、Ausubelら著、前記書)に充分に記載されており、以下に記載する。
【0230】
cDNAは、以下の方法により調製することができる。全細胞RNAを単離(前述のように)し、オリゴ(dT)またはオリゴ(dU)−セルロースのカラムを通してポリA RNAを単離する。結合したポリA mRNAを、低イオン強度緩衝材を用いてカラムから溶離する。cDNA分子を製造するために、前述のようなオリゴ(dT)nまたはオリゴ(dU)n(ここで、nは好ましくは12〜16ヌクレオチド長)を含む第1オリゴヌクレオチドプライマーを、DNA合成用の鋳型としてRNAを用いる酵素である逆転写酵素用のプライマーとして用いられるポリA尾部にハイブリッド形成する。あるいは、mRNA種を、cDNA合成用のプライマーとして、所望のmRNAに相補的な多くの配列を含む短いオリゴヌクレオチドフラグメントを用いることにより、多くの位置から用意する。得られるRNA−RNAハイブリッド(すなわち、RNA及び第1cDNA鎖)は、当該分野で知られている種々の酵素的ステップ(Watsonら著、1992年、Recombinant DNA、第2版、Scientific American Books,ニューヨーク)により二本鎖DNA分子(すなわち、第1及び第2cDNA鎖)に転化される。
【0231】
本発明の一つの態様において、第1cDNA鎖が、試料特異的配列を含むオリゴヌクレオチドプライマーを用いて合成され、それにより、合成された第1cDNA鎖をその試料供給源について確認することができる。従って、第1cDNA鎖合成に用いられるオリゴヌクレオチドプライマーは、少なくともオリゴ(dT)またはオリゴ(dU)配列及び試料特異的配列を含む。
【0232】
一つの態様において、試料Aから単離されたmRNA及び、試料A特異的配列を含むオリゴヌクレオチドプライマーを用いて、試料A用の第1cDNA鎖が合成される。試料Bから単離されたmRNA及び、試料B特異的配列を含むオリゴヌクレオチドプライマーを用いて、試料B用の第1cDNA鎖が合成される。試料A特異的配列は、異なるヌクレオチド同一性を含むことにより試料B特異的配列から異なる、及び/または、これは、異なる長さのヌクレオチドを含むことにより試料B特異的配列から異なる。
【0233】
好ましい態様において、第1オリゴヌクレオチドプライマーは、例えばビーズ上で共有結合により、固体支持体に結合される。このことが有利であるのは、一旦ビーズ上で合成されると、これら第1cDNA鎖を過剰の試薬から容易に洗浄し精製することができ、それにより、これらのビーズを別の反応で直接用いることが可能であるからである。第2に、第2cDNA鎖合成(以下参照)後、これらの結合した第1cDNA鎖を、二本鎖DNAを変性することにより第2cDNA鎖から分離することができ、それにより、他の関連または関連しない実験において用いることができ、例えば、分離された第2cDNA鎖を、その後の増幅反応により増幅することができる。
【0234】
好ましくは、第1cDNA鎖の合成は、逆転写反応による。第1cDNA鎖は、RNA試料を、試料中のRNA試料の逆転写に充分な条件下に第1オリゴヌクレオチドプライマー及び必要な試薬と接触させることにより調製される。プライマー及びRNAと接触される必要な試薬は、当業者に知られており、通常、少なくとも、逆転写活性を有する酵素及び、dNTPを適当な緩衝媒体中に含む。
【0235】
第1cDNA鎖合成ステップのために、逆転写活性を有する種々の酵素、通常、DNAポリメラーゼを用いることができる。適当なDNAポリメラーゼの例には、好熱性バクテリア及び始原菌、レトロウイルス、酵母、アカパンカビ、ショウジョウバエ、霊長類及びげっ歯類からなる群より選択される有機体から誘導されるDNAポリメラーゼが含まれる。好ましくは、DNAポリメラーゼは、米国特許第4,943,531号に記載のモロニーネズミ白血病ウイルス(M−MLV)及び米国特許第5,405,776号(この開示をここで参考のために取り込む)に記載のRNaseH活性を欠くM−MLV逆転写酵素、鳥類骨髄芽球症ウイルス(AMV)、ヒトT細胞白血病ウイルスI型(HTLV−I)、ウシ白血病ウイルス(BLV)、ラウス肉腫ウイルス(RSV)、ヒト免疫不全ウイルス(HIV)及び、米国特許第5,322,770号に記載のThermus aquaticus(Taq)またはThermus thermophilus(Tth)(その開示をここで参考のために取り込む)、並びに米国特許第5,436,149号に記載のBcaBEST(登録商標)DNA Polymerase(その開示をここで参考のために取り込む)からなる群より選択される。逆転写酵素活性を有する適当なDNAポリメラーゼは、市販の有機体、または当業者に知られている方法によりポリメラーゼをコードするクローンド遺伝子を高水準で発現する細胞から得られる有機体から単離することができ、ポリメラーゼを得る特別の方法は、主に、便利さ、費用、入手性等のような因子に基づいて選択される。
【0236】
初回処理RNAの逆転写による第1cDNA鎖合成に必要な種々のdNTP及び緩衝培地は、Clontech、Sigma、Life Technologies、Amersham、Boehringer−Mannheimを含む種々の供給源から市場で購入することができる。第1鎖合成に適した緩衝培地は、通常、Tris−HCl、HEPES−KOH等のような緩衝剤を、通常、典型的に6〜9の範囲のpHを支持する10〜100μMの範囲の濃度;KCl、NaCl等のような一価イオンを含む塩を0〜200mMの範囲の濃度で;MgCl2、Mg(OAc)等のような二価カチオンを含む塩を、通常1〜10mMの範囲の濃度で;及び緩衝剤のようなさらなる試薬、例えば、DDT、洗剤、アルブミン、ポリアルコール(グリセロール)等を含む。試薬混合物の条件は、効率的な第1鎖合成を促進するように選択される。典型的には、プライマーは最初に、高温、通常50〜95℃でRNA試料と組み合わされ、続いて、温度を約0〜60℃の範囲に下げて、試料中の対応するRNAへのプライマーの特異的アニーリングを確保する。このアニーリングステップに続いて、初回処理したRNAを次に、逆転写及び、初回処理RNAの第1cDNA鎖合成を促進するのに充分な条件下にdNTP及び逆転写酵素と組み合わされる。プライマーのRNAへのアニーリングの後、ポリメラーゼの活性を遅らせるまたは開始時間を調節することができ場合、適当なタイプの試薬を用いることにより、試薬の全てを一度に組み合わせることができる。
【0237】
一部の態様において、第2cDNA鎖の合成のために、第1cDNA鎖を鋳型として用いる。
【0238】
任意に、RNase H消化によりまたは0.1〜1M NaOHでの処理により、第2cDNA鎖の合成前に、RNAを除去することができる。
【0239】
所定の試料の発現プロフィールをかなり複雑にすることができ、任意の系の溶解がある程度に限定されるので、発現された遺伝子の、全セットよりも、サブセットを選択的に増幅させることが有利である。このことも有用であり得るのは、ある状況において、所定のサブセットの遺伝子のみが有益であり、有益でない他の遺伝子は濾去して信号−ノイズ比を高めることが有利であるからである。しかしながら、完全ゲノムの分析が望まれる場合、第1cDNA鎖を固体支持体(前述)に結合すると、異なるプライマーを用いる複数ランを容易に達成することができる。従って、第2鎖プライマー(すなわち、第2オリゴヌクレオチドプライマー)の同一性は、発現遺伝子のどのサブセットが増幅するか定める。
【0240】
第2cDNA鎖は第1cDNA鎖にアニーリングされ、最初のmRNAの完全二本鎖DNA複製を形成する。
【0241】
第2オリゴヌクレオチドプライマーの組成は、合成される全ての発現遺伝子のサブセットを定める。通常、DNAポリメラーゼ初回処理に最も重要である前述のプライマーの3’末端は、合成すべきcDNA分子の選択に役立つ短い配列(例えば、6〜7bp配列)を含む。
【0242】
哺乳動物ゲノムの発現部分におけるそのような短い3’末端初回処理配列の発生を推定することができる。例えば、6bpパリンドローム配列を用いる場合、用いられる特定の配列に依存して、現在のマウス配列データ(例えば、配列マウスcDNA中に1.5×108塩基)を用いて約5,000〜10,000回の発生が予測される。特定の細胞及び組織が、全ゲノム中の全ての遺伝子の一部(10〜30%)のみを発現するので、及び、一般的に用いられるPCR条件下では、2,000塩基より長い転写体が増幅する可能性が低いので、500〜2,000の個体の転写体が検出されることが予想される。そのようなプライマーを用いる単一反応において、発現遺伝子の約5〜10%(5,000/1.5×108(頻度)×2,000(増幅可能フラグメントの寸法)×100=6.7%)が覆われることが推定される。従って、約20の別々の反応(同じ遺伝子試料を用いる)が、単一哺乳動物ゲノム中の全ての転写配列のかなりの部分を覆うことが予測される。
【0243】
天然ポリヌクレオチドには、一般的に見られない塩基が、本発明のポリヌクレオチドに含まれてよく、その例には、イノシン及び7−デアザグアニンがある。相補性は完全である必要がなく、適当な二量体はミスマッチ塩基対またはマッチしていない塩基を含み得る。ポリヌクレオチド技術の当業者は、例えば、オリゴヌクレオチド長、オリゴヌクレオチドの塩基組成及び配列、イオン強度、及びミスマッチ塩基対の発生率を含む変数の数を考慮して、二量体安定性を経験的に決めることができる。
【0244】
ポリヌクレオチド二量体の安定性を、溶融温度すなわち「Tm」により測定する。特定条件下での特定のポリヌクレオチドのTmは、塩基対の半分が解離する温度である。二本鎖DNA分子の溶融温度は、顕著にその塩基組成に依存する。GC塩基対に富むDNA分子は、AT塩基対に富むものよりTmが高い。Tmの塩基組成への依存は直線的であり、G−C含量の1%増加毎に約0.4℃増加する。GC塩基対は、二つの水素結合ではなく三つの水素結合により塩基が一緒に保持されるので、AT対よりも安定である。さらに、隣接GC塩基対は、隣接AT塩基対よりも、より強度に互いに相互作用する。ここで、DNAのATに富む領域はより容易に溶融する。
【0245】
Tmへの主要な効果は、溶液のイオン強度により発揮される。一価カチオン濃度の10倍増加毎にTmが16.6℃増加する。最も一般的に用いられる条件は、一価Na+濃度が0.18MとなりTmが90℃台となる0.12Mリン酸緩衝液中でDNAを操作することである。Tmは、水素結合を不安定化させるホルムアミドのような試薬の存在下に反応を行うことにより大きく変化させることができる。これによりTmが40℃の低い温度まで下げられ、高温に曝すことから生じ得る損害(例えば、鎖破損)をDNAが被らないという利点が得られる(Stryer,Biochemistry,1998年,第3版,W.H.Freeman及びCo.,81〜82頁及びLewin,Genes II,1985年,John Wiley&Sons,63〜64頁)。
【0246】
合成された第2cDNA鎖を、任意に、固体支持体に結合された合成された第1cDNA鎖から分離することができる。次に、結合した第1cDNA鎖を単離し、後に他の反応で用いることができる。あるいは、これらの結合二本鎖cDNAを、その後のPCRに直接用いることができる。
【0247】
一つの態様において、新しく合成した第2cDNA鎖が、例えば、cDNAを変性温度、すなわち、Tmより高い温度に曝すことにより、結合した第1cDNA鎖から分離される。結合した第1cDNA鎖は、次に、分析用の第2cDNA鎖の新しいプールを産生するために異なるオリゴヌクレオチドプライマーを用いることにより、さらなる分析に再利用することができる。
【0248】
あるいは、cDNAフラグメントの特定のプールを、適当な制限酵素(例えば、導入したパリンドローム部位の確認)の作用による二本鎖DNAの酵素的消化により、及び特異的配列「C」及び5’末端及び、制限酵素により発生したオーバーハンドと適合する一本鎖オーバーハンドを含む特異的アダプターのライゲーションにより、産生することができる。
【0249】
(増幅)
合成したcDNA(例えば、第1鎖、第2鎖または二本鎖)を用いて、分析用の増幅産物を産生する。本発明において、当該分野で知られている他の増幅方法(例えば、LCR及びNSBA)も用いることができるが、PCR増幅が好ましい。
【0250】
PCR法は、当業者に良く知られており、例えば、Mullis及びFaloona,1987年,Methods Enzymol.,第155巻:335頁,Saikiら著、1985年,Science第230巻:1350頁,及び米国特許第4,683,202,4,683,195及び4,800,159号に記載されており、ここに参考として取り込む。その最も単純な型において、PCRは、反対側の鎖にハイブリッド形成すると共に標的DNAの所望の領域に隣接する二つのオリゴヌクレオチドプライマーを用いて、特定のDNA配列を酵素的に合成する試験管内方法である。鋳型変性、プライマーアニーリング及びDNAポリメラーゼによるアニーリングしたプライマーの延長を含む反応ステップの繰り返しにより、その末端がプライマーの5’末端により定められる特性のフラグメントが指数関数的に蓄積する。PCRは、109の係数で特定のDNA配列の選択的豊富化を発生させることができると報告されている。
【0251】
本発明において、PCRは、鋳型DNA、すなわちcDNA(少なくとも1fg;より有用には、1〜1,000ng)及び少なくとも25pmolのオリゴヌクレオチドプライマー(すなわち、第3及び第4オリゴヌクレオチドプライマー)を用いて行われる。例えば、典型的反応混合物は:cDNAの1〜1,000pg、オリゴヌクレオチドプライマー25〜100pmol、適当な10×緩衝液2.5〜10μl、10μM dNTP0.4〜2μl、Taq DNAポリメラーゼ2.5単位、及び合計体積25〜100μlとする脱イオン水を含む。鉱油を上に乗せ、PCRを、プログラム可能な熱サイクラーを用いて行う。
【0252】
好ましくは、第3オリゴヌクレオチドプライマーは、第1オリゴヌクレオチドプライマーの試料特異的配列を含む。好ましい態様において、第3オリゴヌクレオチドプライマーは、試料特異的配列の全体または一部を含み、その相補的配列(すなわち、第2cDNA)にアニーリングすることができる。この態様は、好ましくは、第4オリゴヌクレオチドプライマー(すなわち、第3オリゴヌクレオチドプライマーに反対の方向)も含む。好ましくは、この第4オリゴヌクレオチドプライマーは、第2オリゴヌクレオチドプライマーの第1任意配列を含む。異なる試料の第2cDNA鎖を、同じ第2オリゴヌクレオチドを用いて合成する場合、同じ第4オリゴヌクレオチドプライマーを用いて、PCRによりcDNAを増幅させることができる。
【0253】
試料特異的配列を含む第3オリゴヌクレオチドプライマーの使用により、増幅産物を、それらの同一性の痕跡を失うことなく、同じ起源に従って確認できることが確保される。
【0254】
PCRサイクルの各ステップの長さ及び温度、並びにサイクル数は、効果の厳格な要求に従って調節される。アニーリング温度及びタイミングは、プライマーが鋳型にアニーリングすることが予想される効率と、許容すべきミスマッチの程度との両方により決められる。プライマーアニーリング条件の過酷度を最適化する性能は、当業者の知識に充分収まるものである。30℃〜72℃のアニーリング温度が用いられる。鋳型分子の初期変性が、92℃〜99℃で4分間起こり、その後に、変性(94〜99℃で15秒〜1分間)、アニーリング(温度は前述のように決められる;1〜2分)及び伸長(72℃で1〜3分間)が20〜40サイクル行われる。好ましくは、増幅産物が、検出可能な標識で標識され、それによりそれらの同一性及び存在量を検出することができる。先に定義の検出可能な標識(例えば、蛍光、放射能または比色標識)を、種々の手段により増幅産物に結合させることができる。例えば、dNTPが標識され、dNTPが一旦ポリヌクレオチド中に組み込まれると、増幅ポリヌクレオチドが標識されることになる。あるいは、増幅に用いるプライマーを標識して、同様に増幅ポリヌクレオチドを標識することができる。さらに、標識されたプローブ(例えば、増幅産物に相補的なオリゴヌクレオチド)を用いて増幅産物にハイブリッド形成し、それにより、増幅産物に検出可能なサインを与える(すなわち、ハイブリッド形成アッセイ)。
【0255】
好ましい態様において、各試料特異的PCTプライマー(すなわち、第3オリゴヌクレオチドプライマー)の5’末端が、特定の蛍光標識に結合し、それにより、PCR産物の系列を、試料起源に従って、それらの蛍光マーカーにより容易に追跡することができる。さらに、蛍光信号の強度は、PCR産物の量に正比例する。所定の産物の蛍光強度を記録することにより、異なる起源のPCR産物間の比を得ることができる。
【0256】
各試料は、好ましくは、その特異的蛍光標識を有するが、同じ蛍光標識を、二つ以上の試料で用いることができる。例えば、試料A用のフルオレセインでタグした第3オリゴヌクレオチドプライマーが、試料Bのものより1塩基短く(または長い)、分離手段が1bpの相違を検出するのに充分に感受性である場合、これらの二つの試料から生じる「同じ」PCRフラグメントが、1bp寸法が異なる二つの近いピークとして分析される(例えば、変性高性能液体クロマトグラフィー(DHPLC)により)。寸法の相違が説明される場合、同じ記録及び計算を実現することができる。2以上の試料について同じ手法を用いることができる。
【0257】
蛍光標識は好ましい標識であるが、同じ目的の達成のために他の標識も用いることができる。labが異なる分子量の同位体である場合(すなわち、P31対P32対P33;O16対O18、等)、試料A用のプライマーは、試料B用のプライマーより「重い」。そのような相違により、検出され得る閾値、例えば分光測定により分けられる異なる起源のPCR産物が得られ、これらの密に関連するピークに基づいて比を計算することができる。
【0258】
一つの態様において、二つまたはそれ以上の試料により調製される集められたcDNAは組み合わされて、二つのプライマー対を用いるPCR増幅に付される(図3)。プライマー4は共通プライマーであり、プライマー2と同じであるか、プライマー2中の5’末端独自配列に同一である。プライマー3A及び3Bは、逆転写中にDNAに組み込まれる試料特異的タグ配列と同じである。さらに、これらのプライマーの各々が、プライマーの5’末端において特定の蛍光標識を含む。これは、これらの二つの別々の試料から生じるPCR産物が、PCRを同じ反応混合物中で行っても、異なる試料特異的蛍光体により別々に標識されることを確保する。プライマー3A及び3Bは、逆転写反応中に導入された特異的配列A1、A2、A3及びB1、B2、B3に同一の対応するプライマー3A1、3A2、3A3及び3B1、3B2、3B3の混合物を表す。これらのプライマーの各々が、独自の蛍光染料を含むことができる。
【0259】
各試料についての単一プライマーの代わりにプライマー混合物を用いることにより、単一反応中で分析することができる遺伝子の数が増加する。mRNA中のポリAの前にあるヌクレオチドに依存して特定の配列A1〜A3及びB1〜B3が組み込まれると共にそれらの増幅の産物が異なる蛍光チャンネルにおいて現れるので、この方法は、類似の寸法を有するがポリA配列の前にあるヌクレオチドが異なるDNAフラグメント間において識別することができる。
【0260】
本発明の一部の態様において、鋳型として先行する増幅反応における増幅産物を用いてネステッド増幅が行われる。ネステッドPCRの使用は、種特異的産物の収率を大幅に高めることもでき、それにより、単一プライマー対がそれ自体失敗する場合、アッセイの感受性も高められる。好ましくは、ネステッドPCRプライマーの一つは、試料特異的配列を含み、それにより、増幅産物の同じ起源の追跡が維持される。また好ましくは、試料特異的配列を含むプライマーが、特定の検出可能な標識で標識されて、増幅産物の検出及び分析が可能となる。例えば、ネステッドPCRを含む方法は、二つの連続PCR反応を含む。第1のプライマー対を用いる(例えば、第3及び第4のオリゴヌクレオチドプライマーを用いる)複数サイクルのPCR(例えば、10〜40、または10〜30、または10〜20サイクル)の後、少量の第1反応(例えば、50μl反応の1μl)が、第1対の内部にあるまたはその間に入っている配列にアニーリングする新規プライマーセットを用いる第2の複数サイクルのPCR反応(例えば、10〜40、または10〜30、または10〜20サイクル)用の鋳型として作用する。
【0261】
ネステッドプライマーを設計しネステッドPCRを実施するための方法は、当該分野で知られている(Current Protocol in Molecular Biology、前記書、を参照)。前述のようなプライマーを選択するための一般的基準は、ネステッドプライマーの設計にも適用される。両方のネステッドプライマーが、第1のプライマー対及びネステッドプライマーの少なくとも一つの内部(例えば、その中)の配列にアニーリングする必要があるが、本発明によれば、試料特異的配列を含む必要がある。
【0262】
(増幅産物の分離及び検出)
予定の時間またはサイクル(例えば、第5サイクル、第8サイクル、第10サイクル、または他のサイクル)から出発するPCR増幅中、少量、例えば、1%〜40%(v/v)の反応混合物が、各サイクル後に自動的に引き出され、反応混合物には、dNTP、蛍光標識したプライマー及びDNAポリメラーゼのような等体積の新しい成分が補給される。引き出された試料は、次に、分離され分析される。ポリヌクレオチドの存在または存在量を検出する方法は、当該分野でよく知られており、定量的分析を同時に行い得ることが好ましいが個々のポリヌクレオチドを分離することができる限り、それらのいずれかを、本発明の方法で用いることができる。増幅産物の分離及び分析のために有用な方法は、限定はされないが、電気泳動(例えば、キャピラリー電気泳動(CE))、クロマトグラフィー(dHPCL)及び質量分析を含む。
【0263】
一つの態様において、CEは好ましい分離手段であるが、それは、少なくとも10〜1,000塩基対の範囲のポリヌクレオチドの例外的分離を提供し単一塩基対を分離するからである。CEは、ここで参考として取り込まれる米国特許第6,217,731;6,001,230及び5,963,456号に開示のような、当該分野でよく知られている方法により行うことができる。最近開発された処理CE装置は市販されており、例えば、Spectrumedix Corporation(State College,PA)からのHTS9610高処理量分析システム及びSCE9610完全自動化96キャピラリー電気泳動遺伝子分析システム;Beckman Instruments Inc(Fullerton,カリフォルニア)からのP/ACE 5,000系及びCEQ系;及びABI PRISM 3100系分析器(Applied Biosystems,Foster City,カリフォルニア)がある。CEカラムの最終近くに、増幅DNAフラグメントは、両方の蛍光標識の信号を測定する蛍光検出器を通過する。これらの装置は、蛍光標識したPCR産物の検出のための自動化高処理量を提供する。
【0264】
本方法におけるCEの使用により、従来のスラブゲル電気泳動と比較して、より高い生産性が得られる。分離速度は、高い電場がゲルに適用された場合に発生する熱故に、スラブゲル電気泳動において制限される。キャピラリーの大きな表面積からの熱の除去が迅速であるので、キャピラリー電気泳動に、より高い電場を適用することができ、それにより、分離プロセスが速くなる。キャピラリーゲルを用いることにより、分離速度が、従来のスラブゲルシステムよりも約10倍増加する。
【0265】
CEを用いると、複数の試料を同時に分析することができ、これは高処理量に必須である。これは、本発明の一つの態様において複数キャピラリーシステムを用いることにより達成される。しかしながら、DNA塩基からの蛍光の検出は、多孔質マトリクス及びキャピラリー壁からの光の散乱により複雑になり得る。光の散乱を避けるために、共焦蛍光スキャナーを用いることができる(Quesadaら著,1991年,Biotechniques第10巻:616〜25頁)。
【0266】
一つの態様において、本方法は、元の試料に含まれる特定のcDNA(すなわち、mRNA)の幾つの複製がPCR増幅用の鋳型として用いられるか測定する。元の複製の数を決めるために、核酸抽出の効率及び、各PCR反応の効率を知らなくてはならない。さらに、検出ステップにより、標的配列の幾つの複製が作られたか示されるが、元の試料に幾つの複製が含まれていたかは示されない。
【0267】
好ましい態様において、試料に含まれる標的配列の複製の正確な数ではなく、遺伝子発現の相違が測定される。検出された蛍光信号強度(例えば、その後のCE分離)を記録することができ、二つの試料からの各ピークの相対比を決めるために用いることができる(図4)。好ましい態様において、二つまたはそれ以上の試料から誘導されるcDNAが、同じPCR反応において増幅される。各試料は、共通のプライマー(例えば、第4オリゴヌクレオチドプライマー)及び試料特異的プライマーにより増幅され、それにより、異なる試料からのcDNAが、同じ共通プライマーについて競合する。この競合故に、二つの試料からの増幅産物の量の比が、二つの試料の各々における初期標的ポリヌクレオチドの量の比を反映する。例えば、1の比(例えば、試料A/試料B)は、試料A及びB中の標的ポリヌクレオチドの同じ初期量、すなわち、標的ポリヌクレオチドが二つの試料中で異なって発現されないことを示す。1を超える比(例えば、試料A/試料B)は、試料B中よりも、試料A中の標的ポリヌクレオチドの量が高いことを示す。1より小さい比(例えば、試料A/試料B)は、試料B中よりも、試料A中において標的ポリヌクレオチドの量が少ないことを示す。前述の両方の場合において、標的ポリヌクレオチドが、二つの試料中において異なって発現される。2つの試料中に存在する主要なポリヌクレオチドの量(すなわち、これらのポリヌクレオチドの発現水準)が略同じであり、従って、増幅の比が一定(例えば、約1)に維持されることが予測される。
【0268】
もう一つの好ましい態様において、CEにより分離される各PCRフラグメントについて(及び、従って、各遺伝子について)の信号強度を、サイクル数の関数としてプロットする。信号強度は、電気泳動図のピークの合計面積により表すことができる。閾値サイクル数(Ct)は、各増幅遺伝子についてPCRフラグメントの信号強度が設定された閾値数(例えば、信号強度の背景値の10の標準偏差)に達したときにサイクル数として計算される。特定遺伝子の操作が異なる発現は、一つのサイクルにおけるよりも、二つ(またはそれ以上)の試料におけるこの遺伝子についての閾値サイクル数(Ct)における相違として決められる。さらに閾値サイクル数を用いて、各遺伝子についての複製数を誘導し、二つまたはそれ以上の試料における遺伝子についての複製数の比による発現の相違を測定する(図5a)。
【0269】
この方法は、各増幅遺伝子についての増幅サイクルの数の関数としての信号強度変化の割合[サイクル数の関数としての信号強度の導関数、d(信号強度)/d(サイクル数)]をプロットすることも含む。一つの試料からの各増幅遺伝子についてのd(信号強度)/d(サイクル数)の最大値に対応するサイクル数として決められる択一的閾値サイクル(aCt)を、もう一つの試料からの同じ遺伝子についてのaCtと比較する。二つまたはそれ以上の試料における同じ遺伝子についてのaCt値間の一サイクルの相違を、択一的操作相違発現と定義する(図5b)。
【0270】
また好ましくは、この方法は、さらに、操作相違発現または択一的操作相違発現を示す一つまたは二つ以上の遺伝子に対応するPCRフラグメントを集め、一つまたは二つ以上の遺伝子の配列を確認することを含む。
【0271】
二つの試料における特定のポリヌクレオチドの比を、二つの試料間で異なって発現されるかどうか決めるための公比に対して測定する。ここで用いられる「公比」という用語は、二つの試料間で発現される全ての遺伝子の相対的一定比を意味する。これは、非処理試料と比較される処理試料中の特定の信号導入経路の活性化のような特定の事象により引き起こされる特定の変化よりも、全体的変化(全出発材料の量)を反映する。特定遺伝子の発現の比をこの公比と比較することにより、比較される試料間で特定遺伝子の発現が異なるかどうか直ちに明らかになる。
【0272】
別々のPCR反応において二つの試料を増幅する場合、各PCR増幅について内部制御が提供され、比が計算される前に内部制御によって、各試料の増幅が、まず正常化される。定量的PCRのために内部制御を使用することは当該分野でよく知られており、例えば、Ausubelらにより記載されている。二つの基本的タイプの制御がある:第1のものは、外因性制御として一般的に知られており((Gillilandら著、(1990年)PCR Protocols,Innisら著、60〜69頁,Academic Press;Wangら著、(1989年)Proc.Natl.Acad.Sci.USA第86巻:9717〜9721頁、その両方がここで参考のために特に取り込まれる)、第2のものは、内因性制御として知られている(Dvekslerら著、(1992年)PCR Methods 及びApplications第6巻:283頁から285頁;Spanakis(1993年)Nucleic Acids Research第21巻:3809〜3819頁、その両方がここで参考のために特に取り込まれる)。
【0273】
外因性制御は、既知の濃度で抽出ステップまたはPCRステップのいずれかに添加される人工的導入核酸分子の使用を含む。定量化のための内部標準として作用するために既知の濃度で外因性核酸を添加する概念は、Chellyら著、(1988年)Nature第333巻:858〜860頁により導入され、これをここで特に参考のために取り込まれる。従って、標的配列として同じプライマーを用いて増幅される制御フラグメントを利用することは、内部標準に対する、標的配列増幅効率をより正確に反映する(例えば、WO93/02215;WO92/11273;米国特許第5,213,961及び5,219,727号を参照のこと:全てここで参考のために取り込む)。同様の手法が、NASBA(Kievitsら著、1991年,J Virol Methods.第35巻:273〜86頁)またはSDA(Walker,1994年,Nucleic Acids Res.第22巻:2670〜7頁)のような等温増幅反応を利用する核酸の定量的測定のために効果的であるとわかった。
【0274】
内因性制御の使用は、抽出効率の変化を調整する。制御の選択は、機能するために幾つかの要求事項を満たすべきことにおいて重要である。第1の要求は、制御の複製数が一定でなくてはならないことであり:第2の要求は、制御が、モニターされる配列を同様の効率で増幅させるべきことである。幾つかの構成的に発現された遺伝子は、制御候補として考えられてきた。これらの遺伝子の発現は、種々の条件において比較的一定だからである。その例には、限定はされないが、β−アクチン遺伝子、グリセルアルデヒド−3−ホスフェートデヒドロゲナーゼ遺伝子(GAPDH)及び16SリボソーマルRNA遺伝子がある。これらの遺伝子は構成的に発現されていると考えられる。
【0275】
異なって発現されたポリヌクレオチドの分類について、任意に閾値を設定することができる。例えば、1.2より大きく0.5より小さい比を有するポリヌクレオチドは、一つの態様による二つの試料における異なって発現されたポリヌクレオチド(すなわち、遺伝子)とみなされる。異なって発現されたと確認されるポリヌクレオチドは、例えば、自動分取装置により集めることができ、遺伝子の同一性は、通常のDNA塩基配列決定により達成することができる。自動分取装置は、例えば、Bio−Rad Laboratories(Hercules,カリフォルニア)から市販されている。
【0276】
もう一つの態様において、溶出PCRフラグメントの分子量の決めるためにCEを校正することができ、第2cDNA鎖を選択的に合成するために用いられる正確な配列が知られているので、所望の各PCRフラグメントの同一性を、利用できるゲノム配列データベース情報に基づいて容易に決めることができる。ヒトゲノムは完全に塩基配列決定され、大腸菌、酵母、C.elegan及びショウジョウバエのような他の有機体も塩基配列決定されている。DNA塩基配列決定技術が迅速に発達したので、大部分の所望の他の有機体の全ゲノムが、まもなく完全に塩基配列決定されるであろう。
【0277】
プロファイリングするこの方法の一つの独自の特徴は、全増幅プロセスにおいてPCRをモニターするその性能である。これに対して、相違ディスプレイのような現存の方法は、PCR産物の最終量しか測定しない。この方法の利点は、さもなければ他の従来法を用いて見逃される遺伝子発現の変化を検出し得ることである。この局面は、PCR産物蓄積の典型的曲線により示すことができる(図5を参照)。
【0278】
PCR増幅反応の開始時に、PCR産物の量は、大部分の装置の検出限界より低く、定量的相違を観察することができない。細胞当たり数個の複製の水準で、通常存在する希少遺伝子転写を検出するために、非常に遅い段階でPCR産物をモニターすることが必要である。典型的に、これらの遺伝子の検出は、これらの希少転写体が検出可能な水準まで増幅されるかなり前に反応が典型的に停止されるので、困難である。増幅曲線の中間部は、信号が検出限界を超え対数相に入ると、遺伝子発現における定量的相違を検出するための最良の信号を構成する。しかしながら、反応の指数的性質故に、この相は比較的短く、反応が後期静止期に移る前に数サイクルしか続かない。PCR増幅のこの後期静止期において、PCR産物の蓄積は、さらなる基質の不足、ポリメラーゼの不足、産物によるポリメラーゼ活性の阻害、またはそれらの組み合わせのような幾つかの因子により飽和する。明らかに、この後期静止期は、やはり、遺伝子発現における定量的相違を検出するための機会をほとんど提供しない。従って、予定数のサイクル後のPCR産物を定量する方法は、増幅の対数相に偶然存在する遺伝子しか確認できず、増幅プロセスの早期または後期において異なってしか検出されない遺伝子を見逃す。
【0279】
本発明は、各々の個々の増幅フラグメントについての完全増幅極性を定めるので、この制限を克服する。さらに、発現相違を測定するための定量的基礎が提供される。実時間定量的PCRの実施において、パラメーターCtが実験的に定義された。ここで用いられる「Ct」という用語は、定量的PCR反応により発生する信号が始めて「閾値」を超える、すなわち、標的核酸配列の増幅の最初の信頼性ある検出がなされるときのサイクル数を意味する。「信頼性ある」という用語は、信号が、PCR中に増幅された産物の検出可能な水準を反映することを意味する。Ctは、通常、標的核酸の未知量の出発量に関連し、例えば、標的の量が少ないとCtが遅れる。Ctは、簡単な数式により、初期複製数または出発DNAの濃度に関連する:
Log(複製数)=aCt+b ここで、a及びbは定数である。
【0280】
従って、二つの異なる試料から生じる同じ遺伝子のフラグメントについてのCtを測定することにより、三つの試料におけるこの遺伝子の最初の濃度を容易に評価することができる。
【0281】
発現プロフィールのためにPCR増幅を用いる一般的関心事は増幅が偏る可能性があることである。特に、一部の配列が、他のものよりも優れた効率で増幅される。この偏りは、出発試料と比較して、PCR産物の最終的状態を変化させることができる。しかしながら、そのような偏りが本発明に影響を与えないのは、本発明が、異なる試料からのcDNA標的の増幅が、同じ反応混合物中で共通のPCRプライマーを用いて行われるという態様を提供するからである。従って、異なる試料から生じる増幅PCR産物の比は、各試料中のcDNAの最初の量によってしか影響されず、増幅の効率によって影響されない。所定のPCR反応において、一つのPCR標的の増幅がもう一つの標的に対して偏るが、この比は、各PCR産物の寸法または組成に関わらず一定に維持される。すなわち、この方法は、同じPCR反応における二つの試料の絶対的ではなく相対的な増幅を測定することによりそのような問題を回避する態様を提供する。
【0282】
他の潜在的問題が、DNAポリメラーゼの利用性が増幅の制限要素となる場合、増幅の後期において生じ得る。その結果、より豊富なフラグメントが、豊富でないフラグメントの増幅を速度論的に阻害する。この問題の重要性は、検出装置の感受性に依存するので、実験的に予測することはできない。この問題を緩和する一つの方法は、増幅の後期サイクルにおいてDNAポリメラーゼの濃度を徐々に増加させることである。
【0283】
PCRの速度論的偏りの問題を扱うもう一つの方法は新規概念である(正規化増幅または定常状態への増幅)。一つの態様において、10〜20サイクルで出発する増幅の各サイクルにさらなるステップを含むことが提案される。このステップは、増幅混合物を、プライマー2に含まれるパリンドローム配列に対して提供される制限酵素で処理することからなる(図6)。より豊富なPCRフラグメントは、単にそれが相対的に豊富であることにより、制限酵素により選択的に消化される。消化により、DNAポリメラーゼ用の初回処理部位が除去され、それにより、消化されたフラグメントのさらなる増幅が防止される。あまり豊富でないDNAフラグメントを含む未消化PCRフラグメントは、増幅を続け、反応中、各フラグメントの2つの複製を生じる。制限酵素の濃度及びこの処理の時間を調節することにより、この反応を、特定の許容できる濃度に達した後のPCRフラグメントのさらなる増幅を制限するように調節することができるべきである。同じ遺伝子に対応する消化フラグメントと未消化フラグメントとの間の寸法の相違を無くすために、反応混合物が、過剰量の制限酵素で処理される。同様に、DNAの反対側鎖の初回処理から生じる一本鎖DNA系を、一本鎖DNase(例えば、エキソヌクレアーゼI及びVII)で処理することにより除去することができる。
【0284】
前記記載は、二つの元の試料間の相違を測定する対応に適用される。しかしながら、二つまたはそれ以上の試料を、前述と同じ方法で少し修正して用い得ることも理解すべきである。例えば、第3試料特異的プライマー及び第3蛍光標識を用いることにより、三つの試料に対して同じ方法を用いることができる。同様に、それ以上の試料も同様の手法を用いて分析することができる。
【0285】
(本発明の方法を実施するためのキット)
本発明は、本発明の種々の態様を実施するための組成及びキットを含む。好ましくは、本発明のキットは第1オリゴヌクレオチドプライマーを含み、第1オリゴヌクレオチドプライマーは試料特異的配列タグを含み、試料特異的配列タグはその5末端においてGCに富みその3末端においてAtに富む。好ましくは、第1オリゴヌクレオチドは固体支持体に付着される。さらに、本発明のキットは、さらに、第2オリゴヌクレオチドプライマー、第3オリゴヌクレオチドプライマーまたは第4オリゴヌクレオチドプライマーを含むことができ、第2オリゴヌクレオチドプライマーは任意配列タグを含むことができる。キットは、さらに、逆転写酵素、DNAポリメラーゼ、反応緩衝剤、及びdNTPからなる群より選択される一つまたは二つ以上の成分を含み得る。
【0286】
(本方法の適用例)
1.発生及び信号導入経路
異なる生物学的試料の発現プロフィールの比較は、正常発生プロセスの研究に非常に重要である。
【0287】
例えば、幹細胞分化は、特定の機能を有する細胞を産生する幹細胞への一連の特化により特徴付けられる。全能胚幹細胞を部分的に多能幹細胞に分化させ、それから特定の条件下に血液幹細胞を産生することができる。これらの委任血液幹細胞は、サイトカインの宿主または「刺激因子」に反応して、さらに分解してより特化した血液細胞、例えば、赤血球、血小板及び白血球が得られる。この複雑なプロセルの各ステップ中、特定のサイトカインに反応して全遺伝子発現プロフィールにおいて劇的変化が生じる。幹細胞分解中のこれらの種類の宿命決定に支配的な因子が何であるか決めることが非常に重要であるのは、これらのステップの部分的または完全逆転が、分化中に失われる望ましい特徴を取り戻すのに有益であるからである。多くの他の発生プロセスにおいて同様の手法が望ましい。本発明は、発生中の遺伝子発現プロフィールにおけるそのような変化を研究するためのツールを提供し、そのような研究に非常に価値がある。
【0288】
2.治療的使用及び診断マーカー
本発明は、異なる生物学的試料の発現プロフィールを比較する方法を提供し、これはさらなる研究及び発展のために有能な薬剤標的を確認するための非常に価値のある手段、及び特定の病状のための有用な診断マーカーを提供する。
【0289】
発現プロフィールが疾患試料と正常試料とで変化し得る少なくとも二つのタイプの遺伝子がある。一つのタイプの遺伝子の発現プロフィールの変化は、病状を引き起こす。これらの遺伝子のアップまたはダウンレギュレーションにより病状を進行させる一連の事象が引き起こされる。これらの「病因遺伝子」の活性を調節することにより、病状を逆転させ、それにより効果的に治療するまたは病状を軽減することができる。これらの遺伝子及びそれらの産物は、価値のある薬剤標的を構成し、それを単に確認することが、疾患の治療の長期目的に有益である。そのような遺伝子の例には、制限はされないが、腫瘍遺伝子(Ras等)及び腫瘍抑制遺伝子(Rb、NF1等)がある。
【0290】
発現プロフィールが変化する第2のタイプの遺伝子は、これらの変化が、そのような病状の原因ではなく結果である点において異なっている。疾患表現型が逆転することを期待してこれらの遺伝子の活性を調節することは不可能であるが、これらの遺伝子の確認より、それにも関わらず、そのような病状の初期の正確な診断が助けられ、それにより、そのような病状の初期活効果的治療が容易になる。そのような遺伝子の例には、限定はされないが、腫瘍抗原CA125、α胎児蛋白などがある。
【0291】
さらに、本発明を、細胞、組織または個体に対する特定の治療の効果を研究するために用いることもできる。これは、どの信号導入経路が特定の治療により影響を受けるかに基づいて有能な薬剤の効果を研究及び/または予想した場合、基本的かつ薬学的研究に有用である。そのような薬剤の潜在的標的を確認することにより、他の意図しない標的は放置して所望の標的のみに影響を与える優れた薬剤をさらにスクリーニングすることにより、ある望ましくない副作用を除去することができる。本発明は、意図する標的において大きな望ましい変化を引き起こす改良された薬剤を確認するために高処理率スクリーニングを行う手段を提供するので、薬剤最適化も可能である。
【0292】
3.他の用途
転写プロファイリングの単純な方法が非常に有用である他の領域は、遺伝的に修飾された細胞及び有機体の特徴解析である(例えば、修飾された遺伝子、過発現遺伝子またはミス遺伝子(ノックアウト)を有するトランスジェニック動物。そのような細胞及び有機体は、標的遺伝子の修飾された機能の結果、あるいは補償効果として、しばしば、変化し転写プロフィールを示す。そのような変化は、異なって発現された遺伝子の同一性により定められる特別の経路に置くことにより、遺伝子の機能を示すことができる。これは、特別の遺伝子における変化の転写サインを定めること及び、転写サインのデータベースに対して、疾患の影響を受けている有機体から得られる細胞または組織における転写変化を比較することにより特別の疾患における遺伝子修飾を決めるためにそのようなサインを用いることに役立つ。
【0293】
4.ビジネス法
本発明は、薬学的ビジネスを行うためのビジネス法も提供する。薬剤標的の確認及び実証は、薬剤開発プロセスにおける重要な速度制限ステップである。本発明は、疾患の発現プロフィールを正常組織と迅速に比較して、それにより、薬剤設計のための有能なリード分子を確認するプロセスの速度を著しく上昇させる方法を提供する。関連する高処理手段は、薬剤スクリーニング及び薬剤最適化プロセスの速度を上昇させることにも役立つ。さらに、多くの信頼性のある診断マーカーを確認し、さらに、診断目的で開発することができる。これは、これらの薬剤標的またはマーカーの確認及び開発を行うために薬学ビジネスの基礎のみならず、これらの初期の発見の権利を第三者に許可し、選択された標的をさらに研究及び開発させる機会も提供する。さらに、本発明の専売技術を用いて、そのような標的またはマーカーを確認及び開発する業務を提供することも可能である。
【0294】
本発明は、ゲノム発見用の新しい基礎として転写プロファイリング用の有用なツールになる可能性がある。このシステムは、改良及びさらなる開発の可能性を有する(例えば、試料の数の増加、塩基配列決定の必要性を除くバンドIDデータベースの作成、等)。これは、下流での適用のために特定の遺伝子セットを選択するために初期データを提供することによりDNA診断(特に、低密度及び中密度マイクロアレイの開発)の全プロセスの加速も行う。
【0295】
本発明の実施は、特記しない限り、当該技術分野に含まれる分子生物学、細胞生物学、細胞培養、ミクロ生物学及び組換えDNAの従来技術を用いる。そのような技術は文献に充分に説明されている。例えば、Molecular Cloning:A Laboratory Manual,第2版,Sambrook,Fritsch及びManiatis編(Cold Spring Harbor Laboratory Press:1989年);DNA Cloning,第I及びII巻(D.N.Glover ed.,1985年);Oligonucleotide Synthesis(M.J.Gait ed.,1984年);Mullisら著、;米国特許第4,683,195;Polynucleotide Hybridization(B.D.Hames&S.J.Higgins編,1984年);Transcription And Translation(B.D.Hames&S.J.Higgins編,1984年);B.Oerbal,A Practical Guide To Molecular Cloning(1984年);Methods In Enzymology(Academic Press,Inc.,ニューヨーク);Methods In Enzymology,第154及び155巻(Wuら編);Immunochemical Methods In Cell And Molecular Biology(Mayer&Walker編,Academic Press,ロンドン,1987年)を参照されたい。全ての引用された参考資料(本出願全体で引用された参考文献、発行特許、公開特許出願を含む)の内容をここで参考のために取り込む。
【0296】
特に、生物学的試料からの全RNAの単離及びcDNA用のmRNAのその後の精製は良く知られている分子生物学技術である。実験手順の詳細が、前記一つまたは二つ以上の実験用参考書物及び他の化学的文献に記載されている。市販キットも、そのような目的で広く利用できる(例えば、QiagenがmRNA単離用キットを販売し、GIBCO BRLが逆転写酵素を用いるcDNA合成用キットを販売)。PCR増幅、クロマトグラフィー、キャピラリー電気泳動(CE)は、全て、一般的分子生物学技術であり、さらに加工することはない。
【0297】
本発明を、以下の材料及び方法を用いる以下の非限定的実施例により説明する。
【実施例】
【0298】
(実施例1 RNAの調製)
RNAを、以下の手順に従って、Trizol試薬及びQiagen製のRNeasy Midi/Maxi Kitを用いて製造することができる。
【0299】
組織をホモジナイザー中で、組織50〜100mg当たり1mlのTIZOLを用いて30分間均質化し、続いて、最終均質化を1分間行った。試料の体積は、均質化に用いたTrizol試薬の体積の10%を超えるべきでない。均質化組織を室温で少なくとも15分から1時間まで放置する、または、必要になるまで−70℃で貯蔵した。Trizolの1ml当たり0.2mlのクロロホルムを添加し、振り混ぜた。混合物を室温で5分間インキュベートし、次に、4,000rpmで5分間遠心分離した。上相を別の管に集めた。Trizolの1ml当たり0.5mlのイソプロピルアルコールを添加してRNAを沈殿させた。混合物を氷上に5分間乗せ、4,000rpmで10分間遠心分離した。上澄みを除去し、ペレットをTrizolの1ml当たり1mlの75%EtOHで洗い、混合し、4,000rpmで5分間遠心分離した。上澄みを除去し、ペレットを30分〜1時間風乾した。ペレットを風乾後、ペレットをRNaseを含まない水中に所望の濃度に再懸濁させた。
【0300】
抽出されたRNAは、適当な体積の緩衝剤RLTを添加し、充分に混合することにより清浄化することができ。適当な体積のエタノール(96〜100%)を、希釈したRNAに添加し、激しく振動させることにより充分に混合した。試料を、RNeasy MidiスピンカラムまたはRNeasy Maxiスピンカラムに適用し、15mlまたは50ml遠心分離管に入れ、3,000〜5,000×gで5分間遠心分離した。流動部を廃棄した。
【0301】
一般的に、RNeasyシリカ−膜技術が、DNaseで処理することなくDNAの大部分を効率的に除去するので、DNase消化は必要ない。しかしながら、非常に少量のDNAに感受性の特定のRNA用途には、さらなるDNA除去が必要であることがある。DNaseでDNAを除去するために、2.0mlの緩衝剤RW1をピペットでスピンカラムに入れ、3,000〜5,000×gで5分間遠心分離して洗った。流動部を廃棄し、遠心分離管を再利用した。20μlのDNase1貯蔵溶液を、緩衝剤RDD140μlに添加した。管を軽く打つことにより混合し、短時間遠心分離して、管の残渣から残留液を圧桁。DNase1インキュベーション混合物(160μl)を、スピンカラム膜上にピペットで直接乗せ、作業台(20〜30℃)上に15分間乗せた。RW1緩衝剤2.0mlを、ピペットでスピンカラムに入れ、作業台上に5分間乗せた。次に、3,000〜5,000×gで5分間遠心分離した。流動部分を廃棄した。以下のRPE緩衝剤洗浄ステップにおいて遠心分離管を再利用した。製造者のマニュアルの指示に従って、プロトコルの残りのためにRNeasy Kitを用いる。
【0302】
(実施例2 溶液中の逆転写)
RNA試料(1〜5μl)を、dNTP溶液(10mM)1μl及び第1オリゴヌクレオチド0.0005〜0.5μM(20μl混合物中の最終濃度)と混合し、70℃で7分間加熱し、4℃で2分間冷却した。前記混合物を、次に、反応混合物(RT緩衝剤(250mM Tris−HCl(pH8.3:25℃),375mM KCl,15mM MgCl2)4μl、0.1M DTT 2μL,RNAse阻害剤(Ambion)1μL及びSuperScriptII逆転写酵素(Invitrogen)1μl及び水5〜10μl)と混合し、合計体積20μlとした。逆転写反応液を45℃で1〜2時間インキュベートし、65℃で10分間加熱することにより完了させた。試料(5〜20μl)を、PCRにより直接分析した。任意に、PCR増幅の前に、RNAse H酵素(Invitrogen)を用いてインキュベーションすることによりRNA鋳型を変性させた。
【0303】
(実施例3 ビーズ上の逆転写)
a.ビーズへのオリゴヌクレオチドのカップリング
Ultralink(登録商標)ヨードアセチルビーズ(Pierce)(100〜1,000μl)を、5×TE緩衝剤(50mM Tris,5mM EDTA,pH8.0)で4回洗い、チオール化(5’チオール)オリゴヌクレオチド(1〜10μM)の溶液と混合した。カップリング反応を、還元剤TCEP(Tris(2−カルボキシエチル)ホスフィン)(100〜500μM)を添加することにより開始し、室温で連続的に混合しつつ1〜2時間行った。ビーズ上の未反応活性基を、βメルカプトエタノール1%を添加することにより10分間急冷した。オリゴビーズを、各10倍体積の5×TE緩衝剤で4回、5×TE緩衝剤で2回(75℃)、RT緩衝剤で2回(85℃)、2×RT緩衝剤及びRT緩衝剤(95℃)で順次洗った。調製したビーズを、RT緩衝剤中4℃で維持した。
【0304】
b.逆転写
RNA試料(1〜5μl)を、dNTP溶液(10mM)1μl及びオリゴ−ビーズ6〜10μlと混合し、70℃で7分間加熱し、4℃で2分間冷却し、反応混合物(RT緩衝剤(250mM Tris−HCl(pH8.3:25℃),375mM KCl,15mM MgCl2)4μl、0.1M DTT2μL,RNAse阻害剤(Ambion)1μL及びSuperScriptII逆転写酵素(Invitrogen)1μl及び水0〜4μl))と混合した。逆転写反応液を45℃で1〜2時間インキュベートした。65℃で10分間加熱することにより反応を完了させた。ビーズをPCR緩衝剤で2回洗った。RNA鋳型を、RNAse H酵素(Invitrogen)とのインキュベーション、またはアルカリ加水分解により破壊した。後者の反応は、反応混合物に0.5M NaOH3.5μlを添加し、65℃で5分間インキュベーションし、1M Tris(pH7.5)3.5μlで中和することにより行った。ビーズを、PCR緩衝剤で2回洗った。
【0305】
(実施例4 第2鎖合成)
結合したDNAの第2鎖の合成を、Taqポリメラーゼ(Hot−start Taq、Qiagen製)(0.5〜1.5μ)及びPwo DNAポリメラーゼ(Roche製)(0.25〜0.5μ)の混合物を用いて、PCR熱サイクラーにおいて、95℃で30秒、56℃で30秒及び72℃で2分間行った。反応混合物は、RT反応からのオリゴビーズ上のcDNA6〜10μl、10×PCR緩衝剤(Hot−start Taq,Qiagan製)または10×RT−PCR緩衝剤(500mM Tris,200mM KCl,100mM (NH4)2SO4,2.5mM Mg2Cl,pH8.5)5μl、dNTP0.1mM、第2プライマー0.5〜1μl(100μM)を合計体積50μlで含んだ。合成した第2DNA鎖を、96℃でビーズから除去し、さらなる増幅に用いた。
【0306】
あるいは、Tris(pH7.5)50mM、10mM MgCl2、1mM DTT、及び0.05mg/ml BSA、0.1mM dNTP、7mM MgCl2及び1μM第2プライマーの存在下に、DNAポリメラーゼ1またはそのKlenowフラグメントの存在下に37℃で30分間、第2鎖DNAを合成した。合成した第2DNA鎖を前述のように除去した。
【0307】
(実施例5 PCR増幅)
合成されたcDNA(5〜20μl)のPCR増幅を、10×PCR緩衝剤または10×RT−PCR緩衝剤(前記参照)10μl、2〜3mM MgCl2、0.05〜0.2mM dNTPs、FAM、RoxまたはHexで標識した0.1〜1μM第3プライマー、0〜1μM非標識第3プライマー、第4プライマー1〜2μl、DMSO 0〜5%、校正DNAポリメラーゼ(PwoまたはTgo(Roche))0.5〜2μ、及びhot−start DNAポリメラーゼ(例えば、Hot−Start Taqポリメラーゼ(Qiagen製))1.5〜3μの存在下に行った。増幅を、「I−サイクラー」(BioRad)または「PCR Express」(Thermo Hybaid)熱サイクラーを使用して、以下のサイクルプログラムを用いて行った:95℃で30秒、56〜60℃で15〜30℃、72℃で1分30秒、合計30〜40サイクル。10〜15回目のサイクルで出発して延長ステップ(72℃)の最後に各サイクル後に少量の試料(典型的に25μl)を引き出した。プライマー、ポリメラーゼ及びdNTPを含む等体積のPCR混合物を、各試料除去後に反応混合物に入れた。集めた試料を、Spectrumedix(SCE−9610 Genetic Analysis System)またはABI(3700 Prism System)からのCEシステムを用いて分析
した。
【0308】
(実施例6 キャピラリー電気泳動)
キャピラリー電気泳動を、製造者の指示に従ってSpectrumedix Corporation製のSCE9610完全自動化96−キャピラリー電気泳動遺伝子分析システムで行った。
【0309】
(その他の態様)
前述の態様は、実施した実験及び、本発明を製造し実施する際に本発明者が考えた技術を示す。これらの態様は、本発明の実施技術を示しその有用性を示すために役立つ技術の開示を含むと考えられる。当業者は、ここに記載の技術及び態様は、好ましい技術であり、通常、種々の同様の方法及び技術を用いて同じ結果を達成し得ると考える。
【0310】
前述の全ての参考文献の全体をここで参考のために取り込む。
【特許請求の範囲】
【請求項1】
2種以上の試料の遺伝子発現プロフィールを比較する方法であって、
(a)試料特異的配列タグを含む第1のオリゴヌクレオチドプライマーを用いて第1の試料から複数の第1cDNA鎖を合成し、前記試料特異的配列タグはその5’末端においてGCに富みその3’末端においてAtに富むこと、
(b)少なくとも前記cDNAのサブセットを選択的に増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(c)前記一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、存在量は前記第1試料中の一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(d)前記第1試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールを第2試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違は2つの試料中の前記一つまたは二つ以上の遺伝子の異なる発現を示すことを含んでなる方法。
【請求項2】
前記ステップ(a)が、二つまたはそれ以上の試料供給源からのRNAを第1cDNA鎖に逆転写することを含み、そのcDNAはそれらの供給源に従って異なって標識される請求項1の方法。
【請求項3】
前記複数の第1cDNA鎖が、前記第1試料由来の全RNAまたはmRNAを用いる逆転写により合成される請求項1の方法。
【請求項4】
前記第1オリゴヌクレオチドプライマーの前記配列特異的配列タグを含む第3オリゴヌクレオチドプライマーを、前記増幅のために用いて一つまたは二つ以上の試料特異的増幅産物を産生する請求項1の方法。
【請求項5】
前記二つまたはそれ以上の試料の少なくとも一つが、正常試料、疾患試料、所定の発生段階または状態における試料、所定の処理段階または状態の前の試料、所定の処理段階または状態の後の試料及び所定の培養段階または状態における試料からなる群より誘導される請求項1の方法。
【請求項6】
前記二つまたはそれ以上の試料の少なくとも一つが、動物、器官、組織型及び細胞型からなる群より誘導される請求項1の方法。
【請求項7】
前記第1オリゴヌクレオチドプライマーにおける前記試料特異的配列が15〜30ヌクレオチド長である請求項1の方法。
【請求項8】
前記試料特異的配列が20〜24ヌクレオチド長である請求項1の方法。
【請求項9】
前記第1オリゴヌクレオチドプライマーが、さらに、5’オリゴ(dT)nVN3’の配列を含み、nは少なくとも5であり、VがdATP、dGTPまたはdCTPであり、NがdTTP(またはdUTP)、dATP、dGTPまたはdCTPである請求項1の方法。
【請求項10】
前記第1オリゴヌクレオチドプライマーが、[5’−(特異的配列タグ)20−24T12−16AN−3’、5’−(特異的配列タグ)20−24T12−16CN−3’及び5’−(特異的配列タグ)20−24T12−16GN−3’]を含むプライマーの混合物として提供され、前記特異的配列タグが前記混合物中の各プライマーについて同一または異なる請求項1の方法。
【請求項11】
nが12〜16である請求項10の方法。
【請求項12】
前記第1オリゴヌクレオチドプライマーにおいて、前記試料特異的配列タグがオリゴ(dT)nVNの5’に配される請求項10の方法。
【請求項13】
第1任意配列タグを含む第2オリゴヌクレオチドプライマーを用いて、前記第1cDNA鎖に相補的な一つまたは二つ以上の第2cDNA鎖を合成することをさらに含んでなり、ステップ(b)が少なくとも前記第2cDNA鎖のサブセットを増幅させて一つまたは二つ以上の試料特異的増幅産物を産生する請求項1の方法。
【請求項14】
前記第2オリゴヌクレオチドプライマーが、さらに、前記第1cDNA鎖のサブセットに相補的な第2配列を含み、それにより一つまたは二つ以上の第2cDNA鎖を合成させる請求項13の方法。
【請求項15】
前記第2オリゴヌクレオチドプライマーにおいて、前記第2配列が前記第1任意配列の3’に配される請求項14の方法。
【請求項16】
前記第2オリゴヌクレオチドが、さらに、前記第1及び第2配列の間に(Z)mの配列を含み、Zが、A、T、GまたはCのいずれかと塩基対を形成することができるヌクレオチドであり、mが少なくとも2である請求項14の方法。
【請求項17】
mが4である請求項16の方法。
【請求項18】
前記第2配列が5〜10ヌクレオチド長である請求項14の方法。
【請求項19】
前記第2配列が6〜7ヌクレオチド長である請求項18の方法。
【請求項20】
前記第2オリゴヌクレオチドプライマー中の前記第1任意配列が15〜30ヌクレオチド長である請求項13の方法。
【請求項21】
前記第2オリゴヌクレオチドプライマー中の前記第1任意配列がA−Tに富む領域とG−Cに富む領域とを含む請求項13の方法。
【請求項22】
前記G−Cに富む領域が、前記A−Tに富む領域の5’に配される請求項21の方法。
【請求項23】
前記用いられる第2オリゴヌクレオチドプライマーが、前記比較すべき二つまたはそれ以上の試料について同じである請求項13の方法。
【請求項24】
前記増幅が、さらに、前記第2オリゴヌクレオチドプライマーの前記第1任意配列タグを含む第4オリゴヌクレオチドプライマーを用いることを含む請求項4の方法。
【請求項25】
前記用いられる第4オリゴヌクレオチドプライマーが、前記比較すべき二つまたはそれ以上の試料について同じである請求項24の方法。
【請求項26】
前記第2オリゴヌクレオチドプライマー中の前記第2配列が遺伝子ファミリー特異的である請求項14の方法。
【請求項27】
前記第2オリゴヌクレオチドプライマー中の前記第2配列が、蛋白ファミリーに特異的であるペプチドをコードする配列である請求項14の方法。
【請求項28】
前記第2配列が、特異的蛋白ファミリー用のサイン配列モチーフをコードする配列を含む請求項27の方法。
【請求項29】
前記蛋白ファミリーが、受容体チロシンキナーゼ、G蛋白共役受容体、7回膜通過受容体、イオンチャンネル、サイトカイン受容体、腫瘍マーカー、MAPKカスケードキナーゼ、転写因子、GTPアーゼ、ATPアーゼ、及び発生蛋白マーカーからなる群より選択される請求項28の方法。
【請求項30】
前記第1cDNA鎖が、固体支持体に付着することなく溶液中で合成される請求項1の方法。
【請求項31】
前記第1cDNA鎖が、固体支持体に付着されて合成される請求項1の方法。
【請求項32】
前記固体支持体が微粒子または反応管の内壁である請求項31の方法。
【請求項33】
前記方法が、さらに、前記一つまたは二つ以上の第2cDNA鎖の増幅前に、前記複数の第1cDNA鎖から前記一つまたは二つ以上の第2cDNA鎖を分離することを含む請求項13の方法。
【請求項34】
前記第3オリゴヌクレオチドプライマーが検出可能な標識に結合される請求項4の方法。
【請求項35】
前記検出可能な標識が、蛍光標識、放射性標識、比色標識、磁気標識及び酵素標識からなる群より選択される請求項34の方法。
【請求項36】
前記検出可能な標識が、蛍光標識である請求項35の方法。
【請求項37】
前記二つまたはそれ以上の試料の各々について用いられる前記第3オリゴヌクレオチドプライマーが試料特異的標識で標識される請求項34の方法。
【請求項38】
前記一つまたは二つ以上の増幅産物が、増幅中、所定の時間にまたは周期的間隔をおいてサンプリングされる請求項1の方法。
【請求項39】
各サンプリングした増幅産物について存在量を検出する請求項38の方法。
【請求項40】
前記方法が、さらに、前記一つまたは二つ以上の増幅産物の存在量を検出する前に、前記一つまたは二つ以上の増幅産物を分離することを含む1の方法。
【請求項41】
前記一つまたは二つ以上の増幅産物を分離し、それらの存在量をクロマトグラフィーにより検出する請求項40の方法。
【請求項42】
前記一つまたは二つ以上の増幅産物を分離し、それらの存在量を蛍光測定により検出する請求項40の方法。
【請求項43】
前記一つまたは二つ以上の増幅産物を分離し、それらの存在量を光学的濃度測定により検出する請求項40の方法。
【請求項44】
前記一つまたは二つ以上の増幅産物を分離し、それらの存在量を質量分析により検出する請求項40の方法。
【請求項45】
前記一つまたは二つ以上の増幅産物を電気泳動により分離する請求項40の方法。
【請求項46】
前記一つまたは二つ以上の増幅産物をキャピラリー電気泳動により分離する請求項45の方法。
【請求項47】
前記一つまたは二つ以上の遺伝子の発現プロフィールにおける前記相違を、二つまたはそれ以上の試料間の前記遺伝子からの増幅産物上の試料特異的検出可能標識の比により測定する請求項1の方法。
【請求項48】
前記方法が、さらに、増幅プロットを発生させること、前記一つまたは二つ以上の遺伝子の各々について増幅のCtを計算すること、及び前記Ctの比により発現プロフィールにおける相違を測定することを含む請求項1の方法。
【請求項49】
前記方法が、さらに、異なって発現される一つまたは二つ以上の遺伝子を集めること、及びDNA塩基配列決定により前記一つまたは二つ以上の遺伝子の配列同一性を確認することを含む請求項1の方法。
【請求項50】
前記増幅をPCRにより行う請求項1の方法。
【請求項51】
二つまたはそれ以上の試料の遺伝子発現プロフィールを比較する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、第1試料から複数の第1cDNA鎖を合成し、前記第1オリゴヌクレオチドプライマーが少なくとも一つの変性ヌクレオチドを含むこと、
(b)少なくとも前記cDNAのサブセットを選択的に増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(c)前記一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、前記存在量が前記第1試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(d)前記第1試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールを第2試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、二つの試料における前記一つまたは二つ以上の遺伝子の異なる発現を示すこと
を含んでなる方法。
【請求項52】
二つまたはそれ以上の試料の遺伝子発現プロフィールを比較する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、第1試料から複数の第1cDNA鎖を合成し、前記試料特異的配列タグが少なくとも一つの人工ヌクレオチドを含むこと、
(b)少なくとも前記cDNAのサブセットを選択的に増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(c)前記一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、前記存在量が前記第1試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(d)前記第1試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールを第2試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、二つの試料における前記一つまたは二つ以上の遺伝子の異なる発現を示すこと
を含んでなる方法。
【請求項53】
二つまたはそれ以上の試料の遺伝子発現プロフィールを比較する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、第1試料から複数の第1cDNA鎖を合成し、前記試料特異的配列タグがその5’末端においてGCに富みその3’末端においてATに富むこと、
(b)第1任意配列タグを含む第2オリゴヌクレオチドプライマーを用いて、前記第1cDNA鎖に相補的な一つまたは二つ以上の第2cDNA鎖を選択的に合成すること、
(c)前記一つまたは二つ以上の第2cDNA鎖を増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(d)前記一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、前記存在量が前記第1試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(e)前記第1試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールを第2試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、二つの試料における前記一つまたは二つ以上の遺伝子の異なる発現を示すこと
を含んでなる方法。
【請求項54】
二つまたはそれ以上の試料の遺伝子発現プロフィールを比較する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、第1試料から複数の第1cDNA鎖を合成し、前記第1オリゴヌクレオチドプライマーが少なくとも一つの変性ヌクレオチドを含むこと、
(b)第1任意配列タグを含む第2オリゴヌクレオチドプライマーを用いて、前記第1cDNA鎖に相補的な一つまたは二つ以上の第2cDNA鎖を選択的に合成すること、
(c)前記一つまたは二つ以上の第2cDNA鎖を増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(d)前記一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、前記存在量が前記第1試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(e)前記第1試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールを第2試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、二つの試料における前記一つまたは二つ以上の遺伝子の異なる発現を示すこと
を含んでなる方法。
【請求項55】
二つまたはそれ以上の試料の遺伝子発現プロフィールを比較する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、第1試料から複数の第1cDNA鎖を合成し、前記試料特異的配列タグが少なくとも一つの人工ヌクレオチドを含むこと、
(b)第1任意配列タグを含む第2オリゴヌクレオチドプライマーを用いて、前記第1cDNA鎖に相補的な一つまたは二つ以上の第2cDNA鎖を選択的に合成すること、
(c)前記一つまたは二つ以上の第2cDNA鎖を増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(d)前記一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、前記存在量が前記第1試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(e)前記第1試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールを第2試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、二つの試料における前記一つまたは二つ以上の遺伝子の異なる発現を示すこと
を含んでなる方法。
【請求項56】
試料における一つまたは二つ以上の遺伝子の発現を調節するモジュレーターを確認する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、前記試料を前記モジュレーターに接触させる前に、複数の第1cDNA鎖を合成し、前記試料特異的配列タグがその5’末端においてGCに富みその3’末端においてATに富むこと、
(b)少なくとも前記cDNAのサブセットを選択的に増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(c)前記一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、前記存在量が前記試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(d)前記モジュレーターとの接触前の前記試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールを、前記モジュレーター接触後の前記試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、前記試料における一つまたは二つ以上の遺伝子発現を調節する前記モジュレーターを示すこと
を含んでなる方法。
【請求項57】
試料における一つまたは二つ以上の遺伝子の発現を調節するモジュレーターを確認する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、前記試料を前記モジュレーターに接触させる前に、複数の第1cDNA鎖を合成し、前記第1オリゴヌクレオチドプライマーが少なくとも一つの変性ヌクレオチドを含むこと、
(b)少なくとも前記cDNAのサブセットを選択的に増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(c)前記一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、前記存在量が前記試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(d)前記モジュレーターとの接触前の前記試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールを、前記モジュレーター接触後の前記試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、前記試料における一つまたは二つ以上の遺伝子発現を調節する前記モジュレーターを示すこと
を含んでなる方法。
【請求項58】
試料における一つまたは二つ以上の遺伝子の発現を調節するモジュレーターを確認する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、前記試料を前記モジュレーターに接触させる前に、複数の第1cDNA鎖を合成し、前記試料特異的配列タグがその5’末端においてGCに富みその3’末端においてATに富むこと、
(b)第1任意配列タグを含む第2オリゴヌクレオチドプライマーを用いて、一つまたは二つ以上の第2cDNA鎖を合成すること、
(c)前記第2cDNA鎖を増幅させて一つまたは二つ以上の試料特異的増幅産物を産
生すること、
(d)前記一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、前記存在量が前記試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(e)前記モジュレーターとの接触前の前記試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールを、前記モジュレーター接触後の前記試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、前記試料における一つまたは二つ以上の遺伝子発現を調節する前記モジュレーターを示すこと
を含んでなる方法。
【請求項59】
試料における一つまたは二つ以上の遺伝子の発現を調節するモジュレーターを確認する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、前記試料を前記モジュレーターに接触させる前に、複数の第1cDNA鎖を合成し、前記第1オリゴヌクレオチドプライマーが少なくとも一つの変性ヌクレオチドを含むこと、
(b)第1任意配列タグを含む第2オリゴヌクレオチドプライマーを用いて、一つまたは二つ以上の第2cDNA鎖を合成すること、
(c)前記第2cDNA鎖を増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(d)前記一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、前記存在量が前記試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(e)前記モジュレーターとの接触前の前記試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールを、前記モジュレーター接触後の前記試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、前記試料における一つまたは二つ以上の遺伝子発現を調節する前記モジュレーターを示すこと
を含んでなる方法。
【請求項60】
遺伝子発現の水準を検出するための組成物であって、第1オリゴヌクレオチドプライマーを含んでなり、前記第1オリゴヌクレオチドプライマーが試料特異的配列タグを含み、前記試料特異的配列タグがその5’末端においてGCに富みその3’末端においてATに富む組成物。
【請求項61】
第1任意配列タグを含む第2オリゴヌクレオチドプライマーをさらに含む請求項60の組成物。
【請求項62】
前記第1オリゴヌクレオチドプライマーの前記配列特異的配列タグを含む第3オリゴヌクレオチドプライマーをさらに含む請求項61の組成物。
【請求項63】
前記第1任意配列タグを含む第4オリゴヌクレオチドプライマーをさらに含む請求項62の組成物。
【請求項64】
前記第2プライマーが、前記第1cDNA鎖の配列に相補的な第2配列をさらに含む請求項60の組成物。
【請求項65】
逆転写酵素、DNAポリメラーゼ、前記逆転写酵素用の反応緩衝剤、前記DNAポリメラーゼ用の反応緩衝剤、及びdNTPからなる群より選択される一つまたは二つ以上の成分をさらに含む請求項60の組成物。
【請求項66】
遺伝子発現の水準を検出するための組成物であって、第1オリゴヌクレオチドプライマーを含んでなり、前記第1オリゴヌクレオチドプライマーが試料特異的配列タグを含み、前記第1オリゴヌクレオチドプライマーが少なくとも一つの変性ヌクレオチドを含む組成
物。
【請求項67】
遺伝子発現の水準を検出するためのキットであって、第1オリゴヌクレオチドプライマーを含んでなり、前記第1オリゴヌクレオチドプライマーが試料特異的配列タグを含み、前記試料特異的配列タグがその5’末端においてGCに富みその3’末端においてATに富み、また、その包装材料を含んでなるキット。
【請求項68】
第1任意配列タグを含む第2オリゴヌクレオチドプライマーをさらに含む請求項67のキット。
【請求項69】
前記第1オリゴヌクレオチドプライマーの前記配列特異的配列タグを含む第3オリゴヌクレオチドプライマーをさらに含む請求項67のキット。
【請求項70】
前記第1任意配列タグを含む第4オリゴヌクレオチドプライマーをさらに含む請求項68のキット。
【請求項71】
前記第2プライマーが、前記第1cDNA鎖の配列に相補的な第2配列をさらに含む請求項68の組成物。
【請求項72】
逆転写酵素、DNAポリメラーゼ、前記逆転写酵素用の反応緩衝剤、前記DNAポリメラーゼ用の反応緩衝剤、及びdNTPからなる群より選択される一つまたは二つ以上の成分をさらに含む請求項67の組成物。
【請求項73】
遺伝子発現の水準を検出するためのキットであって、第1オリゴヌクレオチドプライマーを含んでなり、前記第1オリゴヌクレオチドプライマーが試料特異的配列タグを含み、前記第1オリゴヌクレオチドプライマーが少なくとも一つの変性ヌクレオチドを含み、また、その包装材料を含んでなるキット。
【請求項1】
2種以上の試料の遺伝子発現プロフィールを比較する方法であって、
(a)試料特異的配列タグを含む第1のオリゴヌクレオチドプライマーを用いて第1の試料から複数の第1cDNA鎖を合成し、前記試料特異的配列タグはその5’末端においてGCに富みその3’末端においてAtに富むこと、
(b)少なくとも前記cDNAのサブセットを選択的に増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(c)前記一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、存在量は前記第1試料中の一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(d)前記第1試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールを第2試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違は2つの試料中の前記一つまたは二つ以上の遺伝子の異なる発現を示すことを含んでなる方法。
【請求項2】
前記ステップ(a)が、二つまたはそれ以上の試料供給源からのRNAを第1cDNA鎖に逆転写することを含み、そのcDNAはそれらの供給源に従って異なって標識される請求項1の方法。
【請求項3】
前記複数の第1cDNA鎖が、前記第1試料由来の全RNAまたはmRNAを用いる逆転写により合成される請求項1の方法。
【請求項4】
前記第1オリゴヌクレオチドプライマーの前記配列特異的配列タグを含む第3オリゴヌクレオチドプライマーを、前記増幅のために用いて一つまたは二つ以上の試料特異的増幅産物を産生する請求項1の方法。
【請求項5】
前記二つまたはそれ以上の試料の少なくとも一つが、正常試料、疾患試料、所定の発生段階または状態における試料、所定の処理段階または状態の前の試料、所定の処理段階または状態の後の試料及び所定の培養段階または状態における試料からなる群より誘導される請求項1の方法。
【請求項6】
前記二つまたはそれ以上の試料の少なくとも一つが、動物、器官、組織型及び細胞型からなる群より誘導される請求項1の方法。
【請求項7】
前記第1オリゴヌクレオチドプライマーにおける前記試料特異的配列が15〜30ヌクレオチド長である請求項1の方法。
【請求項8】
前記試料特異的配列が20〜24ヌクレオチド長である請求項1の方法。
【請求項9】
前記第1オリゴヌクレオチドプライマーが、さらに、5’オリゴ(dT)nVN3’の配列を含み、nは少なくとも5であり、VがdATP、dGTPまたはdCTPであり、NがdTTP(またはdUTP)、dATP、dGTPまたはdCTPである請求項1の方法。
【請求項10】
前記第1オリゴヌクレオチドプライマーが、[5’−(特異的配列タグ)20−24T12−16AN−3’、5’−(特異的配列タグ)20−24T12−16CN−3’及び5’−(特異的配列タグ)20−24T12−16GN−3’]を含むプライマーの混合物として提供され、前記特異的配列タグが前記混合物中の各プライマーについて同一または異なる請求項1の方法。
【請求項11】
nが12〜16である請求項10の方法。
【請求項12】
前記第1オリゴヌクレオチドプライマーにおいて、前記試料特異的配列タグがオリゴ(dT)nVNの5’に配される請求項10の方法。
【請求項13】
第1任意配列タグを含む第2オリゴヌクレオチドプライマーを用いて、前記第1cDNA鎖に相補的な一つまたは二つ以上の第2cDNA鎖を合成することをさらに含んでなり、ステップ(b)が少なくとも前記第2cDNA鎖のサブセットを増幅させて一つまたは二つ以上の試料特異的増幅産物を産生する請求項1の方法。
【請求項14】
前記第2オリゴヌクレオチドプライマーが、さらに、前記第1cDNA鎖のサブセットに相補的な第2配列を含み、それにより一つまたは二つ以上の第2cDNA鎖を合成させる請求項13の方法。
【請求項15】
前記第2オリゴヌクレオチドプライマーにおいて、前記第2配列が前記第1任意配列の3’に配される請求項14の方法。
【請求項16】
前記第2オリゴヌクレオチドが、さらに、前記第1及び第2配列の間に(Z)mの配列を含み、Zが、A、T、GまたはCのいずれかと塩基対を形成することができるヌクレオチドであり、mが少なくとも2である請求項14の方法。
【請求項17】
mが4である請求項16の方法。
【請求項18】
前記第2配列が5〜10ヌクレオチド長である請求項14の方法。
【請求項19】
前記第2配列が6〜7ヌクレオチド長である請求項18の方法。
【請求項20】
前記第2オリゴヌクレオチドプライマー中の前記第1任意配列が15〜30ヌクレオチド長である請求項13の方法。
【請求項21】
前記第2オリゴヌクレオチドプライマー中の前記第1任意配列がA−Tに富む領域とG−Cに富む領域とを含む請求項13の方法。
【請求項22】
前記G−Cに富む領域が、前記A−Tに富む領域の5’に配される請求項21の方法。
【請求項23】
前記用いられる第2オリゴヌクレオチドプライマーが、前記比較すべき二つまたはそれ以上の試料について同じである請求項13の方法。
【請求項24】
前記増幅が、さらに、前記第2オリゴヌクレオチドプライマーの前記第1任意配列タグを含む第4オリゴヌクレオチドプライマーを用いることを含む請求項4の方法。
【請求項25】
前記用いられる第4オリゴヌクレオチドプライマーが、前記比較すべき二つまたはそれ以上の試料について同じである請求項24の方法。
【請求項26】
前記第2オリゴヌクレオチドプライマー中の前記第2配列が遺伝子ファミリー特異的である請求項14の方法。
【請求項27】
前記第2オリゴヌクレオチドプライマー中の前記第2配列が、蛋白ファミリーに特異的であるペプチドをコードする配列である請求項14の方法。
【請求項28】
前記第2配列が、特異的蛋白ファミリー用のサイン配列モチーフをコードする配列を含む請求項27の方法。
【請求項29】
前記蛋白ファミリーが、受容体チロシンキナーゼ、G蛋白共役受容体、7回膜通過受容体、イオンチャンネル、サイトカイン受容体、腫瘍マーカー、MAPKカスケードキナーゼ、転写因子、GTPアーゼ、ATPアーゼ、及び発生蛋白マーカーからなる群より選択される請求項28の方法。
【請求項30】
前記第1cDNA鎖が、固体支持体に付着することなく溶液中で合成される請求項1の方法。
【請求項31】
前記第1cDNA鎖が、固体支持体に付着されて合成される請求項1の方法。
【請求項32】
前記固体支持体が微粒子または反応管の内壁である請求項31の方法。
【請求項33】
前記方法が、さらに、前記一つまたは二つ以上の第2cDNA鎖の増幅前に、前記複数の第1cDNA鎖から前記一つまたは二つ以上の第2cDNA鎖を分離することを含む請求項13の方法。
【請求項34】
前記第3オリゴヌクレオチドプライマーが検出可能な標識に結合される請求項4の方法。
【請求項35】
前記検出可能な標識が、蛍光標識、放射性標識、比色標識、磁気標識及び酵素標識からなる群より選択される請求項34の方法。
【請求項36】
前記検出可能な標識が、蛍光標識である請求項35の方法。
【請求項37】
前記二つまたはそれ以上の試料の各々について用いられる前記第3オリゴヌクレオチドプライマーが試料特異的標識で標識される請求項34の方法。
【請求項38】
前記一つまたは二つ以上の増幅産物が、増幅中、所定の時間にまたは周期的間隔をおいてサンプリングされる請求項1の方法。
【請求項39】
各サンプリングした増幅産物について存在量を検出する請求項38の方法。
【請求項40】
前記方法が、さらに、前記一つまたは二つ以上の増幅産物の存在量を検出する前に、前記一つまたは二つ以上の増幅産物を分離することを含む1の方法。
【請求項41】
前記一つまたは二つ以上の増幅産物を分離し、それらの存在量をクロマトグラフィーにより検出する請求項40の方法。
【請求項42】
前記一つまたは二つ以上の増幅産物を分離し、それらの存在量を蛍光測定により検出する請求項40の方法。
【請求項43】
前記一つまたは二つ以上の増幅産物を分離し、それらの存在量を光学的濃度測定により検出する請求項40の方法。
【請求項44】
前記一つまたは二つ以上の増幅産物を分離し、それらの存在量を質量分析により検出する請求項40の方法。
【請求項45】
前記一つまたは二つ以上の増幅産物を電気泳動により分離する請求項40の方法。
【請求項46】
前記一つまたは二つ以上の増幅産物をキャピラリー電気泳動により分離する請求項45の方法。
【請求項47】
前記一つまたは二つ以上の遺伝子の発現プロフィールにおける前記相違を、二つまたはそれ以上の試料間の前記遺伝子からの増幅産物上の試料特異的検出可能標識の比により測定する請求項1の方法。
【請求項48】
前記方法が、さらに、増幅プロットを発生させること、前記一つまたは二つ以上の遺伝子の各々について増幅のCtを計算すること、及び前記Ctの比により発現プロフィールにおける相違を測定することを含む請求項1の方法。
【請求項49】
前記方法が、さらに、異なって発現される一つまたは二つ以上の遺伝子を集めること、及びDNA塩基配列決定により前記一つまたは二つ以上の遺伝子の配列同一性を確認することを含む請求項1の方法。
【請求項50】
前記増幅をPCRにより行う請求項1の方法。
【請求項51】
二つまたはそれ以上の試料の遺伝子発現プロフィールを比較する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、第1試料から複数の第1cDNA鎖を合成し、前記第1オリゴヌクレオチドプライマーが少なくとも一つの変性ヌクレオチドを含むこと、
(b)少なくとも前記cDNAのサブセットを選択的に増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(c)前記一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、前記存在量が前記第1試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(d)前記第1試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールを第2試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、二つの試料における前記一つまたは二つ以上の遺伝子の異なる発現を示すこと
を含んでなる方法。
【請求項52】
二つまたはそれ以上の試料の遺伝子発現プロフィールを比較する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、第1試料から複数の第1cDNA鎖を合成し、前記試料特異的配列タグが少なくとも一つの人工ヌクレオチドを含むこと、
(b)少なくとも前記cDNAのサブセットを選択的に増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(c)前記一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、前記存在量が前記第1試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(d)前記第1試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールを第2試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、二つの試料における前記一つまたは二つ以上の遺伝子の異なる発現を示すこと
を含んでなる方法。
【請求項53】
二つまたはそれ以上の試料の遺伝子発現プロフィールを比較する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、第1試料から複数の第1cDNA鎖を合成し、前記試料特異的配列タグがその5’末端においてGCに富みその3’末端においてATに富むこと、
(b)第1任意配列タグを含む第2オリゴヌクレオチドプライマーを用いて、前記第1cDNA鎖に相補的な一つまたは二つ以上の第2cDNA鎖を選択的に合成すること、
(c)前記一つまたは二つ以上の第2cDNA鎖を増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(d)前記一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、前記存在量が前記第1試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(e)前記第1試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールを第2試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、二つの試料における前記一つまたは二つ以上の遺伝子の異なる発現を示すこと
を含んでなる方法。
【請求項54】
二つまたはそれ以上の試料の遺伝子発現プロフィールを比較する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、第1試料から複数の第1cDNA鎖を合成し、前記第1オリゴヌクレオチドプライマーが少なくとも一つの変性ヌクレオチドを含むこと、
(b)第1任意配列タグを含む第2オリゴヌクレオチドプライマーを用いて、前記第1cDNA鎖に相補的な一つまたは二つ以上の第2cDNA鎖を選択的に合成すること、
(c)前記一つまたは二つ以上の第2cDNA鎖を増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(d)前記一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、前記存在量が前記第1試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(e)前記第1試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールを第2試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、二つの試料における前記一つまたは二つ以上の遺伝子の異なる発現を示すこと
を含んでなる方法。
【請求項55】
二つまたはそれ以上の試料の遺伝子発現プロフィールを比較する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、第1試料から複数の第1cDNA鎖を合成し、前記試料特異的配列タグが少なくとも一つの人工ヌクレオチドを含むこと、
(b)第1任意配列タグを含む第2オリゴヌクレオチドプライマーを用いて、前記第1cDNA鎖に相補的な一つまたは二つ以上の第2cDNA鎖を選択的に合成すること、
(c)前記一つまたは二つ以上の第2cDNA鎖を増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(d)前記一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、前記存在量が前記第1試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(e)前記第1試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールを第2試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、二つの試料における前記一つまたは二つ以上の遺伝子の異なる発現を示すこと
を含んでなる方法。
【請求項56】
試料における一つまたは二つ以上の遺伝子の発現を調節するモジュレーターを確認する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、前記試料を前記モジュレーターに接触させる前に、複数の第1cDNA鎖を合成し、前記試料特異的配列タグがその5’末端においてGCに富みその3’末端においてATに富むこと、
(b)少なくとも前記cDNAのサブセットを選択的に増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(c)前記一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、前記存在量が前記試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(d)前記モジュレーターとの接触前の前記試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールを、前記モジュレーター接触後の前記試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、前記試料における一つまたは二つ以上の遺伝子発現を調節する前記モジュレーターを示すこと
を含んでなる方法。
【請求項57】
試料における一つまたは二つ以上の遺伝子の発現を調節するモジュレーターを確認する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、前記試料を前記モジュレーターに接触させる前に、複数の第1cDNA鎖を合成し、前記第1オリゴヌクレオチドプライマーが少なくとも一つの変性ヌクレオチドを含むこと、
(b)少なくとも前記cDNAのサブセットを選択的に増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(c)前記一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、前記存在量が前記試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(d)前記モジュレーターとの接触前の前記試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールを、前記モジュレーター接触後の前記試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、前記試料における一つまたは二つ以上の遺伝子発現を調節する前記モジュレーターを示すこと
を含んでなる方法。
【請求項58】
試料における一つまたは二つ以上の遺伝子の発現を調節するモジュレーターを確認する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、前記試料を前記モジュレーターに接触させる前に、複数の第1cDNA鎖を合成し、前記試料特異的配列タグがその5’末端においてGCに富みその3’末端においてATに富むこと、
(b)第1任意配列タグを含む第2オリゴヌクレオチドプライマーを用いて、一つまたは二つ以上の第2cDNA鎖を合成すること、
(c)前記第2cDNA鎖を増幅させて一つまたは二つ以上の試料特異的増幅産物を産
生すること、
(d)前記一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、前記存在量が前記試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(e)前記モジュレーターとの接触前の前記試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールを、前記モジュレーター接触後の前記試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、前記試料における一つまたは二つ以上の遺伝子発現を調節する前記モジュレーターを示すこと
を含んでなる方法。
【請求項59】
試料における一つまたは二つ以上の遺伝子の発現を調節するモジュレーターを確認する方法であって、
(a)試料特異的配列タグを含む第1オリゴヌクレオチドプライマーを用いて、前記試料を前記モジュレーターに接触させる前に、複数の第1cDNA鎖を合成し、前記第1オリゴヌクレオチドプライマーが少なくとも一つの変性ヌクレオチドを含むこと、
(b)第1任意配列タグを含む第2オリゴヌクレオチドプライマーを用いて、一つまたは二つ以上の第2cDNA鎖を合成すること、
(c)前記第2cDNA鎖を増幅させて一つまたは二つ以上の試料特異的増幅産物を産生すること、
(d)前記一つまたは二つ以上の試料特異的増幅産物の存在量を検出し、前記存在量が前記試料における一つまたは二つ以上の遺伝子の発現プロフィールを決めること、及び
(e)前記モジュレーターとの接触前の前記試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールを、前記モジュレーター接触後の前記試料中の前記一つまたは二つ以上の遺伝子の発現プロフィールと比較し、発現プロフィールにおける相違が、前記試料における一つまたは二つ以上の遺伝子発現を調節する前記モジュレーターを示すこと
を含んでなる方法。
【請求項60】
遺伝子発現の水準を検出するための組成物であって、第1オリゴヌクレオチドプライマーを含んでなり、前記第1オリゴヌクレオチドプライマーが試料特異的配列タグを含み、前記試料特異的配列タグがその5’末端においてGCに富みその3’末端においてATに富む組成物。
【請求項61】
第1任意配列タグを含む第2オリゴヌクレオチドプライマーをさらに含む請求項60の組成物。
【請求項62】
前記第1オリゴヌクレオチドプライマーの前記配列特異的配列タグを含む第3オリゴヌクレオチドプライマーをさらに含む請求項61の組成物。
【請求項63】
前記第1任意配列タグを含む第4オリゴヌクレオチドプライマーをさらに含む請求項62の組成物。
【請求項64】
前記第2プライマーが、前記第1cDNA鎖の配列に相補的な第2配列をさらに含む請求項60の組成物。
【請求項65】
逆転写酵素、DNAポリメラーゼ、前記逆転写酵素用の反応緩衝剤、前記DNAポリメラーゼ用の反応緩衝剤、及びdNTPからなる群より選択される一つまたは二つ以上の成分をさらに含む請求項60の組成物。
【請求項66】
遺伝子発現の水準を検出するための組成物であって、第1オリゴヌクレオチドプライマーを含んでなり、前記第1オリゴヌクレオチドプライマーが試料特異的配列タグを含み、前記第1オリゴヌクレオチドプライマーが少なくとも一つの変性ヌクレオチドを含む組成
物。
【請求項67】
遺伝子発現の水準を検出するためのキットであって、第1オリゴヌクレオチドプライマーを含んでなり、前記第1オリゴヌクレオチドプライマーが試料特異的配列タグを含み、前記試料特異的配列タグがその5’末端においてGCに富みその3’末端においてATに富み、また、その包装材料を含んでなるキット。
【請求項68】
第1任意配列タグを含む第2オリゴヌクレオチドプライマーをさらに含む請求項67のキット。
【請求項69】
前記第1オリゴヌクレオチドプライマーの前記配列特異的配列タグを含む第3オリゴヌクレオチドプライマーをさらに含む請求項67のキット。
【請求項70】
前記第1任意配列タグを含む第4オリゴヌクレオチドプライマーをさらに含む請求項68のキット。
【請求項71】
前記第2プライマーが、前記第1cDNA鎖の配列に相補的な第2配列をさらに含む請求項68の組成物。
【請求項72】
逆転写酵素、DNAポリメラーゼ、前記逆転写酵素用の反応緩衝剤、前記DNAポリメラーゼ用の反応緩衝剤、及びdNTPからなる群より選択される一つまたは二つ以上の成分をさらに含む請求項67の組成物。
【請求項73】
遺伝子発現の水準を検出するためのキットであって、第1オリゴヌクレオチドプライマーを含んでなり、前記第1オリゴヌクレオチドプライマーが試料特異的配列タグを含み、前記第1オリゴヌクレオチドプライマーが少なくとも一つの変性ヌクレオチドを含み、また、その包装材料を含んでなるキット。
【図1】

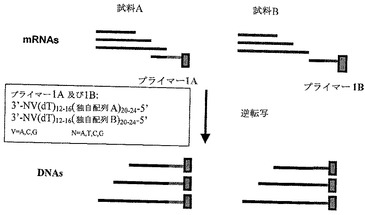
【図2】
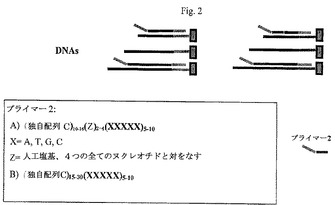
【図3】
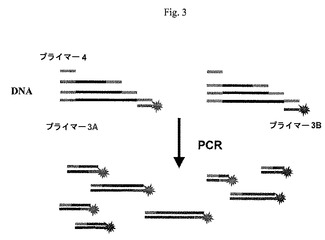
【図4】
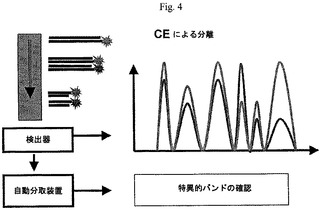
【図5】
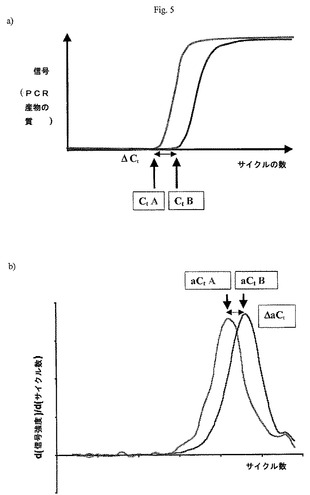
【図6】
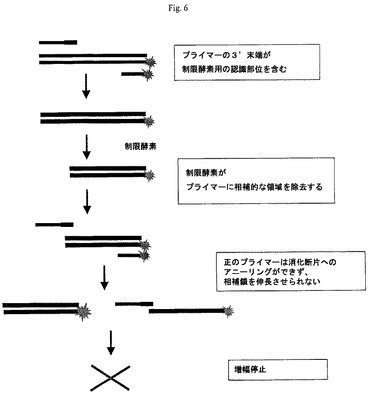
【図7】
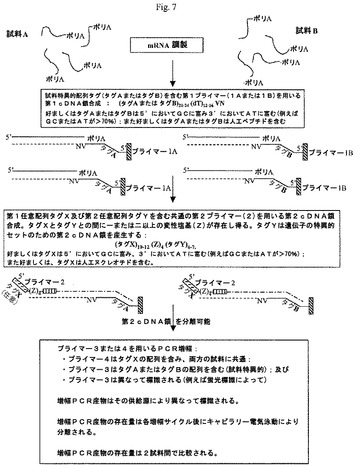
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2010−46073(P2010−46073A)
【公開日】平成22年3月4日(2010.3.4)
【国際特許分類】
【外国語出願】
【出願番号】特願2009−237655(P2009−237655)
【出願日】平成21年10月14日(2009.10.14)
【分割の表示】特願2003−538342(P2003−538342)の分割
【原出願日】平成14年10月24日(2002.10.24)
【出願人】(504161869)センション インコーポレイテッド (1)
【Fターム(参考)】
【公開日】平成22年3月4日(2010.3.4)
【国際特許分類】
【出願番号】特願2009−237655(P2009−237655)
【出願日】平成21年10月14日(2009.10.14)
【分割の表示】特願2003−538342(P2003−538342)の分割
【原出願日】平成14年10月24日(2002.10.24)
【出願人】(504161869)センション インコーポレイテッド (1)
【Fターム(参考)】
[ Back to top ]