
化合物
【課題】 リーシュマニアに対する生理活性を有する化合物を提供する。
【解決手段】 一般式(1)
【化18】

(上記式(1)中、nは1〜3の整数を示す。)
で表される化合物。
【解決手段】 一般式(1)
【化18】
(上記式(1)中、nは1〜3の整数を示す。)
で表される化合物。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、生理活性物質として新規な化合物に関するものである。
【背景技術】
【0002】
リーシュマニア症は、原虫リーシュマニアに感染することを原因として発症する感染症である。症状は軽いものから重いものまであるが、主に治療薬として用いられている5価のアンチモンは副作用を引き起こす懸念がある。従って、副作用のリスクの低い新薬が求められる。
【0003】
非特許文献1によれば、リーシュマニアの治療に用いる薬として有用である新規な環状デプシペプチドの創出につき鋭意検討する中で、海綿動物であるAapptos Cliataに着目し、これらの抽出物の分画を進めたところ、純粋な活性ペプチドの単離に成功し、これらをCiliatamidesA−Cと命名した。また、非特許文献2には、CliatamidesA−Cの全合成に成功したことが報告されている。
【先行技術文献】
【非特許文献】
【0004】
【非特許文献1】Yoichi Nakao, Shizuka Kawatsu, Chikane Okamoto, Masaaki Okamoto, Yoshitsugu Matsumoto, Shigeki Matsunaga,Rob. W. M. van Soest, and Nobuhiro Fusetani, “Ciliatamides A−C, Bioactive Lipopeptides from the Deep-Sea Sponge Aaptos ciliate”, J. Nat. Prod. 2008, 71, p469−472.
【非特許文献2】Jana A. Lewis, R. Nathan Daniels, and Craig W. Lindsley, “Total Synthesis of Ciliatamides A-C: Stereochemical Revision and the Natural Product-Guided Synthesis of Unnatural Analogs, Org. Lett., 2008, 10, p4545-4548.
【発明の概要】
【発明が解決しようとする課題】
【0005】
ところで、一般的に医薬等の分野においては、ある化合物に有用な生理活性が見出された場合には、その生理活性に係る作用機構、活性部位等を解明するために、その化合物の類縁体を合成し、それらの生理活性を評価している。本発明者らはCiliatamidesA−Cの類縁体の合成を進めたところ、本発明に係る生理活性を有する化合物を見出した。
【課題を解決するための手段】
【0006】
即ち、本発明に係る化合物は、一般式(1)
【化1】
(上記式(1)中、nは1〜3の整数を示す。)
で表される。
【0007】
また、本発明に係る化合物は、化学式(2)
【化2】
で表される。
【0008】
また、本発明に係る化合物は、化学式(3)
【化3】
で表される。
【発明の効果】
【0009】
本発明に係る化合物は、リーシュマニアに対する生理活性を有する。
【図面の簡単な説明】
【0010】
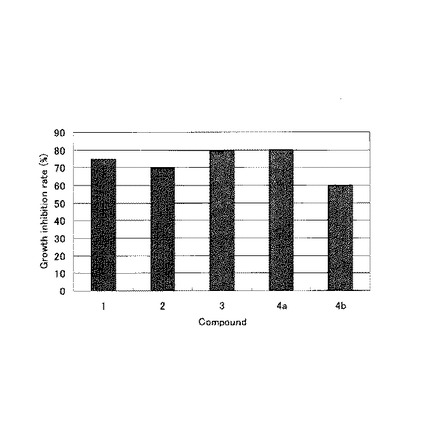
【図1】本発明の実施例に係る化合物の成長阻害率を示すグラフである。
【発明を実施するための形態】
【0011】
以下、本発明の実施の形態について説明する。本発明に係る化学物質は、一般式(4)または(5)で示すアミドを中間物質として合成することができる。
【0012】
また、本発明に係るアミドは、一般式(4)
【化4】
【0013】
(上記一般式(4)中、nは1〜3の整数、R1は水素原子、ヒドロキシ基、メトキシ基、または塩素原子、R2は水素原子、メチル基またはエチル基、R3はベンジルオキシカルボニル基、tert-ブトキシカルボニル基、フルオレニルメトキシカルボニル基、2,2,2-トリクロロエトキシカルボニル基またはアリルオキシカルボニル基を示す。)
【化5】
【0014】
(上記一般式(5)中、nは1〜3の整数、R1は水素原子、ヒドロキシ基、メトキシ基、または塩素原子、R2は水素原子、メチル基またはエチル基、R3はベンジルオキシカルボニル基、tert-ブトキシカルボニル基、フルオレニルメトキシカルボニル基、2,2,2-トリクロロエトキシカルボニル基またはアリルオキシカルボニル基を示す。)
上記一般式(4)または(5)で表されるアミドは、アミノシクロアルカンとN末端が保護されたフェニルアラニン化合物とを有機溶媒に溶解させ、縮合剤及び添加剤の存在下で反応させることにより得られる。
【0015】
アミノシクロアルカンとしては、シクロヘキシルアミン、シクロヘプチルアミン、シクロオクチルアミンなどが挙げられる。
【0016】
N末端を保護する官能基としては、ベンジルオキシカルボニル基、tert-ブトキシカルボニル基(Boc基)、フルオレニルメトキシカルボニル基、2,2,2-トリクロロエトキシカルボニル基、アリルオキシカルボニル基などを挙げることができる。
【0017】
N末端をBoc基で保護したフェニルアラニン化合物としては、N-(tert-ブトキシカルボニル)-D-フェニルアラニン、N-(tert-ブトキシカルボニル)-L-フェニルアラニン、N-(tert-ブトキシカルボニル)-N-メチル-D-フェニルアラニン(以下、N-Boc-N-Me-D-Pheという。)、N-(tert-ブトキシカルボニル-N-メチル-L-フェニルアラニン(以下、N-Boc-N-Me-L-Pheという。)、N-(tert-ブトキシカルボニル)-N-エチル-D-フェニルアラニン、N-(tert-ブトキシカルボニル)-N-エチル-L-フェニルアラニン、N-(tert-ブトキシカルボニル)-N-メチル-D-p-ヒドロキシフェニルアラニン、N-(tert-ブトキシカルボニル)-N-メチル-L-p-ヒドロキシフェニルアラニン、N-(tert-ブトキシカルボニル)-N-メチル-L-p-メトキシフェニルアラニン、N-(tert-ブトキシカルボニル)-L-p-クロロフェニルアラニン等を挙げることができる。
【0018】
縮合剤としては、N-エチル-N’-3-ジメチルアミノプロピルカルボジイミド塩酸塩(EDCI・HCl)、ヘキサフルオロリン酸2-(1H-ベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウム(HBTU)、ベンゾトリアゾール-1-イル-トリス(ピロリジノ)-ホスホニウムヘキサフルオロホスフェイト(PyBOP)などを挙げることができ、1種を単独で用いてもよいし、2種以上を組み合わせて用いてもよい。
【0019】
また、添加剤としては、ジメチルアミノピリジン(DMAP)、ヒドロキシベンゾトリアゾール(HOBt)、1-ヒドロキシ-7-アザベンゾトリアゾール(HOAt)などを挙げることができ、1種を単独で用いてもよいし、2種以上を組み合わせて用いてもよい。
【0020】
有機溶媒としては、例えば、アセトニトリル等のニトリル、クロロホルム、ジクロロメタン等のハロゲン化溶媒、テトラヒドロフラン(THF)等のエーテル、ジメチルホルムアミド(DMF)等のアミド、ジメチルスルホキシド(DMSO)等のスルホキシド、アセトン等のケトン、メタノール等のアルコール等が挙げられる。これら溶媒は、1種を単独で用いてもよいし、2種類以上を組み合わせて用いてもよい。
【0021】
次に、得られたアミドとカルボン酸とを有機溶媒中において縮合剤及びアミン系塩基の存在下において反応させ本発明に係る化合物を得る。
【0022】
カルボン酸としては、飽和脂肪酸、不飽和脂肪酸の何れも用いることができる。飽和脂肪酸としては、直鎖の飽和脂肪酸を用いるのが好ましく、特にデカン酸を用いることが好ましい。
【0023】
また、不飽和脂肪酸としては、二重結合を1つ有するモノ不飽和脂肪酸を用いるのが好ましく、特に9-デセン酸を用いるのが好ましい。
【0024】
アミン系塩基としては、モノエチルアミン、ジエチルアミン、トリエチルアミン、ジイソプロピルアミン、ジイソプロピルエチルアミン等が挙げることができ、ジイソプロピルエチルアミンを用いることが特に好ましい。
【0025】
また、縮合剤としては、上述のEDCI・HCl、HBTU、PyBOP等を単独で、または2種以上を組み合わせて用いることができる。
【0026】
有機溶媒としては、上述のアミド合成の際に用いる有機溶媒を用いることができ、これらの溶媒を1種を単独で用いてもよいし、2種類以上を組み合わせて用いてもよい。
【実施例】
【0027】
以下、本発明について実施例を用いてより具体的に説明する。
【0028】
(アミド1の合成)
式(6)で示すアミド1を次の手順により得た。
【0029】
シクロヘキシルアミン100 μL (0.88 mmol)をDMF 2 mLに溶解させ、そこにN-Boc-N-Me-L-Phe 315.5 mg(1.13 mmol)、EDCI・HCl 270.2 mg (1.41 mmol)、HOBt 190.9 mg(1.41 mmol)を順に加え、最後にジクロロメタン2 mLを加えて、14 時間室温撹拌した。反応溶媒を減圧留去し、1M塩酸20 mL及び酢酸エチル20 mLを加え、その後酢酸エチル(2×20mL) により抽出を行った。5%の炭酸水素ナトリウム水溶液 (20 mL) を用いて有機層を洗浄し、さらにブライン(20 mL)で有機層を脱水後、無水硫酸ナトリウムで乾燥させ、乾燥剤をろ過し、ろ液を減圧濃縮した。残留物質をシリカゲルクロマトグラフィー(酢酸エチル:n-ヘキサン=3:7)で分離を行い、淡黄色油状のアミド1を得た。(320.0 mg, 収率89 %)
【化6】
【0030】
(アミド2の合成)
式(7)で示すアミド2を次の手順により得た。
【0031】
シクロヘプチルアミン94μL(0.74mmol)をDMF 4mLに溶解させ、そこにN-Boc-N-Me-L-Phe 200.0 mg(0.72 mmol)、EDCI・HCl 207.8 mg(1.08 mmol)、HOBt 146.7 mg (1.08 mmol)を順に加え、最後にジクロロメタン4 mLを加えて、4時間室温撹拌した。反応溶媒を減圧留去し、1M塩酸20 mL及び酢酸エチル20 mLを加え、その後酢酸エチル(2×20 mL)により抽出を行った。5%の炭酸水素ナトリウム水溶液(20 mL)を用いて有機層を洗浄し、さらにブライン(20 mL)で有機層を脱水後、無水硫酸ナトリウムで乾燥させ、乾燥剤をろ過し、ろ液を減圧濃縮した。さらに真空ポンプで乾燥をし、黄色油状のアミド2を得た。(291.6 mg, 収率96 %)
【化7】
【0032】
(アミド2’の合成)
式(8)で示すアミド2’を次の手順により得た。
【0033】
上述のアミド2の合成におけるN-Boc-N-Me-L-Pheの代わりにN-Boc-N-Me-D-Pheを用いた以外は、アミド2の合成と同様な反応、処理を行った。減圧濃縮後の油状物質をシリカゲルクロマトグラフィー(酢酸エチル:n-ヘキサン=3:7)で精製を行い、淡黄色油状のアミド2’を得た (223.1 mg, 収率83 %)。
【化8】
【0034】
(アミド3の合成)
式(9)で示すアミド3を次の手順により得た。
【0035】
シクロオクチルアミン100 μL (0.72 mmol)をDMF 2 mLに溶解させ、そこにN-Boc-N-Me-L-Phe 239.6 mg(0.86 mmol)、EDCI・HCl 221.9 mg (1.16 mmol)、HOBt 159.2 mg (1.17 mmol)を順に加え、最後にジクロロメタン2 mLを加えて、14時間室温撹拌した。反応溶媒を減圧留去し、1M塩酸20 mL及び酢酸エチル20 mLを加え、その後酢酸エチル(2×20 mL)により抽出を行った。5%炭酸水素ナトリウム水溶液(20 mL)を用いて有機層を洗浄し、さらにブライン(20 mL)で有機層を脱水後、無水硫酸ナトリウムで乾燥させた後、減圧濃縮した。残留物質をシリカゲルクロマトグラフィー(酢酸エチル:n-ヘキサン=3:7)で分離を行い、淡黄色油状のアミド3を得た。(255.7 mg, 収率92 %)
【化9】
【0036】
(化合物1の合成)
式(10)に示す化合物1を次の手順により得た。
【0037】
152.5 mg (0.42 mmol)のアミド1を4M塩酸/ジオキサン溶液2 mLに溶解させ、3時間室温撹拌を行い、溶媒を減圧留去し、Boc基を除去した白色粉末を得た。この白色粉末をDMF 2 mLに溶解させ、ジイソプロピルエチルアミン230 μL (1.35 mmol)、PyBOP 286.5mg (0.55 mmol)、9-デセン酸100 mL(0.54 mmol)を順に加え、さらにジクロロメタン2 mLを加えて、14時間室温において撹拌を行った。反応溶媒を減圧留去し、1M塩酸20 mL及び酢酸エチル20 mLを加え、その後酢酸エチル(2×20 mL)により抽出を行った。5%の炭酸水素ナトリウム水溶液(20 mL)を用いて有機層を洗浄し、さらにブライン(20 mL)で有機層を脱水後、無水硫酸ナトリウムで乾燥させ、ろ過、ろ液の減圧留去を行った。残留物質をシリカゲルクロマトグラフィー(酢酸エチル:n-ヘキサン=3:7)で精製し、透明油状の化合物1を得た。(138.4 mg、収率80%)、さらに順相高速液体クロマトグラフィー(HPLC)[Inertsil, 250×20 mm, 溶媒 酢酸エチル:n-ヘキサン=3:7, flow rate 3.0 mL/min, refractive detection]によりτR = 42 minを分取し、スペクトル試料とした。
【表1】
【化10】
【0038】
(化合物2の合成)
式(11)に示す化合物2を次の手順により得た。
【0039】
138.8 mg(0.37 mmol)のアミド2を4M 塩酸/ジオキサン溶液2 mLに溶解させ、2時間室温において撹拌を行い、溶媒を減圧留去し、Boc基を除去した白色粉末を得た。この白色粉末をDMF 3 mLに溶解させ、ジイソプロピルエチルアミン200 μL(1.18 mmol)、PyBOP 251.4 mg (0.48 mmol)、9-デセン酸68 μL (0.37 mmol)を順に加え、最後にジクロロメタン3 mLを加えて、14時間室温撹拌を行った。反応溶媒を減圧留去し、1M塩酸20 mL及び酢酸エチル20 mLを加え抽出し、反応溶媒を減圧留去し、1M塩酸20 mL及び酢酸エチル20 mLを加え、その後酢酸エチル(2×20 mL)により抽出を行った。5%の炭酸水素ナトリウム水溶液(20 mL)を用いて有機層を洗浄し、さらにブライン(20 mL)で有機層を脱水後、無水硫酸ナトリウムで乾燥させ、ろ過、ろ液の減圧留去を行った。そして、残留物質をシリカゲルクロマトグラフィー(酢酸エチル:n-ヘキサン=1:4)により単離精製し、化合物2を得た。(淡黄色油状:147.4 mg, 収率94 %)、さらにスペクトル試料として、順層HPLC[Inertsil, 250×20 mm, 溶媒 酢酸エチル:n-ヘキサン=1:4, flow rate 3.0mL/min, refractive detection]によりτR=39 minのピークを集め、再度精製を行った。
【表2】
【化11】
【0040】
(化合物3の合成)
式(12)に示す化合物3を次の手順により得た。
【0041】
50.4 mg(0.13 mmol)のアミド3を4M 塩酸/ジオキサン溶液2 mLに溶解させ、2時間室温において撹拌を行い、溶媒を減圧留去し、Boc基を除去した白色粉末を得た。この白色粉末をDMF2mLに溶解させ、ジイソプロピルエチルアミン240 μL (1.41 mmol)、PyBOP 319.0 mg(0.61 mmol)、9−デセン酸112μL(0.61 mmol)を順に加え、最後にジクロロメタン2mLを加えて、14時間室温において撹拌を行った。反応溶媒を減圧留去し、1M塩酸20 mL及び酢酸エチル20 mLを加え、その後酢酸エチル(2×20 mL)により抽出を行った。5%の炭酸水素ナトリウム水溶液(20 mL)を用いて有機層を洗浄し、さらにブライン(20 mL)で有機層を脱水後、無水硫酸ナトリウムで乾燥させ、乾燥剤をろ過し、ろ液を減圧濃縮した。残留物質をシリカゲルクロマトグラフィー(酢酸エチル:n-ヘキサン=2:3)で分離し、透明油状の化合物3を得た(99.4 mg, 収率48 %)。さらに順相HPLC[Inertsil, 250×20 mm, 溶媒 酢酸エチル:n-ヘキサン=3:7, flow rate 3.0 mL/min, refractive detection]によりτR=50 minを分取し、精製した。
【表3】
【化12】
【0042】
(化合物4aの合成)
式(2)に示す化合物4aを次の手順により得た。
【0043】
152.5mg(0.42mmol)のアミド2 を4M塩酸/ジオキサン溶液2 mLに溶解させ、3時間室温において撹拌を行い、溶媒を減圧留去し、Boc基を除去した透明油状物質を得た。この透明油状物質をDMF 2mLに溶解させ、ジイソプロピルエチルアミン225 μL (1.32 mmol)、PyBOP 319.2 mg (0.61 mmol)、デカン酸111.5 mg (0.65 mmol)を順に、そしてジクロロメタン2 mLを加えて、16時間室温で撹拌を行った。反応溶媒を減圧留去し、1M塩酸20 mL及び酢酸エチル20 mLを加え、その後酢酸エチル(2×20 mL)により抽出を行った。5%の炭酸水素ナトリウム水溶液(20 mL)を用いて有機層を洗浄し、さらにブライン(20 mL)で有機層を脱水後、無水硫酸ナトリウムで乾燥させ、ろ過、ろ液の減圧留去を行った。残留物質をシリカゲルクロマトグラフィー(酢酸エチル:n-ヘキサン=2:3)で精製し、化合物4aを得た(透明油状:156.2 mg, 収率88 %)。さらに順相HPLC[Inertsil, 250×20 mm, 溶媒 酢酸エチル:n-ヘキサン=2:3, flow rate 3.0 mL/min, refractive detection]によりτR=27 minの部分を分取し、スペクトル用試料を得た。
【表4】
【化13】
【0044】
(化合物4bの合成)
式(3)で示す化合物4bを次の手順によって得た。
【0045】
10.2mg(0.03 mmol)のアミド2’を4M塩酸/ジオキサン2 mLに溶解させ、1時間室温撹拌し、反応溶媒を減圧留去させ、Boc基を除去した透明油状物質を得た。保護基を除去したアミド2’をDMF 2mLに溶解し、トリエチルアミン4μL (0.03 mmol)、EDCI・HCl 15.4 mg (0.08 mmol)、HOAt 9.4 mg (0.07 mmol)、デカン酸10.6 mg (0.06 mmol)、ジクロロメタン2 mLを順に加え、19時間室温で撹拌を行った。反応溶媒を減圧留去し、1M塩酸10 mL及び酢酸エチル10 mLを加え、その後酢酸エチル(3×10 mL) により抽出を行った。5%炭酸水素ナトリウム水溶液(20 mL)を用いて有機層を洗浄し、さらにブライン(20 mL)で有機層を脱水後、無水硫酸ナトリウムで乾燥させ、乾燥剤をろ過し、ろ液を減圧濃縮した。残渣をシリカゲルクロマトグラフィー(酢酸エチル:n-ヘキサン=2:3)にて分離精製し透明油状の化合物4bを得た(12.1 mg, 収率94%)。さらに順相HPLCにて[Inertsil, 250×20 mm, 溶媒 酢酸エチル:n-ヘキサン=3:7, flow rate 3.0 mL/min, refractive detection] τR=32 min の部分を集め再度精製した。
【表5】
【化14】
【0046】
(生理活性測定)
上述した手順で合成した化合物1〜3、4a及び4bについて下記に示す条件の下、生理活性測定を行った。リーシュマニア原虫として、L.major promastigotesを選択し、蛍光蛋白egfpを導入したL.major/egfp promastigotesを用いて、上記化合物をサンプルとして下記の条件で成長阻害率を求めた。
【0047】
(測定手順)
25oC で、96-穴 プレートにL.major/promastigotesを10%ウシ胎児血清と25 mMのHepes Buffer(ICN Biomedeivals Inc., Aurora, OH)で栄養を補った199培地(日水製薬)で培養したL.major/egfp promastigotsの蛍光シグナルを蛍光ミクロプレート読み取り装置(Fluoroscan Ascent FL, 大日本製薬)で励起光を485 nm、放射光538 nmで測定した。96穴プレートの各ウェルにL.major/egfp原虫100μL(1×106 cell/mL)、サンプル溶液10μL (メタノールに溶解)し、25℃で72時間培養を行った。標準物質として、Amphotericin の成長阻害率を100%として、各試料の阻害率を示した。
【0048】
測定の結果、求められた成長阻害率を図1に示す。
【0049】
これらの結果により、化合物1〜3、4a及び4bはリーシュマニア原虫の成長を阻害する生理活性特性を有することが示された。
【技術分野】
【0001】
本発明は、生理活性物質として新規な化合物に関するものである。
【背景技術】
【0002】
リーシュマニア症は、原虫リーシュマニアに感染することを原因として発症する感染症である。症状は軽いものから重いものまであるが、主に治療薬として用いられている5価のアンチモンは副作用を引き起こす懸念がある。従って、副作用のリスクの低い新薬が求められる。
【0003】
非特許文献1によれば、リーシュマニアの治療に用いる薬として有用である新規な環状デプシペプチドの創出につき鋭意検討する中で、海綿動物であるAapptos Cliataに着目し、これらの抽出物の分画を進めたところ、純粋な活性ペプチドの単離に成功し、これらをCiliatamidesA−Cと命名した。また、非特許文献2には、CliatamidesA−Cの全合成に成功したことが報告されている。
【先行技術文献】
【非特許文献】
【0004】
【非特許文献1】Yoichi Nakao, Shizuka Kawatsu, Chikane Okamoto, Masaaki Okamoto, Yoshitsugu Matsumoto, Shigeki Matsunaga,Rob. W. M. van Soest, and Nobuhiro Fusetani, “Ciliatamides A−C, Bioactive Lipopeptides from the Deep-Sea Sponge Aaptos ciliate”, J. Nat. Prod. 2008, 71, p469−472.
【非特許文献2】Jana A. Lewis, R. Nathan Daniels, and Craig W. Lindsley, “Total Synthesis of Ciliatamides A-C: Stereochemical Revision and the Natural Product-Guided Synthesis of Unnatural Analogs, Org. Lett., 2008, 10, p4545-4548.
【発明の概要】
【発明が解決しようとする課題】
【0005】
ところで、一般的に医薬等の分野においては、ある化合物に有用な生理活性が見出された場合には、その生理活性に係る作用機構、活性部位等を解明するために、その化合物の類縁体を合成し、それらの生理活性を評価している。本発明者らはCiliatamidesA−Cの類縁体の合成を進めたところ、本発明に係る生理活性を有する化合物を見出した。
【課題を解決するための手段】
【0006】
即ち、本発明に係る化合物は、一般式(1)
【化1】
(上記式(1)中、nは1〜3の整数を示す。)
で表される。
【0007】
また、本発明に係る化合物は、化学式(2)
【化2】
で表される。
【0008】
また、本発明に係る化合物は、化学式(3)
【化3】
で表される。
【発明の効果】
【0009】
本発明に係る化合物は、リーシュマニアに対する生理活性を有する。
【図面の簡単な説明】
【0010】
【図1】本発明の実施例に係る化合物の成長阻害率を示すグラフである。
【発明を実施するための形態】
【0011】
以下、本発明の実施の形態について説明する。本発明に係る化学物質は、一般式(4)または(5)で示すアミドを中間物質として合成することができる。
【0012】
また、本発明に係るアミドは、一般式(4)
【化4】
【0013】
(上記一般式(4)中、nは1〜3の整数、R1は水素原子、ヒドロキシ基、メトキシ基、または塩素原子、R2は水素原子、メチル基またはエチル基、R3はベンジルオキシカルボニル基、tert-ブトキシカルボニル基、フルオレニルメトキシカルボニル基、2,2,2-トリクロロエトキシカルボニル基またはアリルオキシカルボニル基を示す。)
【化5】
【0014】
(上記一般式(5)中、nは1〜3の整数、R1は水素原子、ヒドロキシ基、メトキシ基、または塩素原子、R2は水素原子、メチル基またはエチル基、R3はベンジルオキシカルボニル基、tert-ブトキシカルボニル基、フルオレニルメトキシカルボニル基、2,2,2-トリクロロエトキシカルボニル基またはアリルオキシカルボニル基を示す。)
上記一般式(4)または(5)で表されるアミドは、アミノシクロアルカンとN末端が保護されたフェニルアラニン化合物とを有機溶媒に溶解させ、縮合剤及び添加剤の存在下で反応させることにより得られる。
【0015】
アミノシクロアルカンとしては、シクロヘキシルアミン、シクロヘプチルアミン、シクロオクチルアミンなどが挙げられる。
【0016】
N末端を保護する官能基としては、ベンジルオキシカルボニル基、tert-ブトキシカルボニル基(Boc基)、フルオレニルメトキシカルボニル基、2,2,2-トリクロロエトキシカルボニル基、アリルオキシカルボニル基などを挙げることができる。
【0017】
N末端をBoc基で保護したフェニルアラニン化合物としては、N-(tert-ブトキシカルボニル)-D-フェニルアラニン、N-(tert-ブトキシカルボニル)-L-フェニルアラニン、N-(tert-ブトキシカルボニル)-N-メチル-D-フェニルアラニン(以下、N-Boc-N-Me-D-Pheという。)、N-(tert-ブトキシカルボニル-N-メチル-L-フェニルアラニン(以下、N-Boc-N-Me-L-Pheという。)、N-(tert-ブトキシカルボニル)-N-エチル-D-フェニルアラニン、N-(tert-ブトキシカルボニル)-N-エチル-L-フェニルアラニン、N-(tert-ブトキシカルボニル)-N-メチル-D-p-ヒドロキシフェニルアラニン、N-(tert-ブトキシカルボニル)-N-メチル-L-p-ヒドロキシフェニルアラニン、N-(tert-ブトキシカルボニル)-N-メチル-L-p-メトキシフェニルアラニン、N-(tert-ブトキシカルボニル)-L-p-クロロフェニルアラニン等を挙げることができる。
【0018】
縮合剤としては、N-エチル-N’-3-ジメチルアミノプロピルカルボジイミド塩酸塩(EDCI・HCl)、ヘキサフルオロリン酸2-(1H-ベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウム(HBTU)、ベンゾトリアゾール-1-イル-トリス(ピロリジノ)-ホスホニウムヘキサフルオロホスフェイト(PyBOP)などを挙げることができ、1種を単独で用いてもよいし、2種以上を組み合わせて用いてもよい。
【0019】
また、添加剤としては、ジメチルアミノピリジン(DMAP)、ヒドロキシベンゾトリアゾール(HOBt)、1-ヒドロキシ-7-アザベンゾトリアゾール(HOAt)などを挙げることができ、1種を単独で用いてもよいし、2種以上を組み合わせて用いてもよい。
【0020】
有機溶媒としては、例えば、アセトニトリル等のニトリル、クロロホルム、ジクロロメタン等のハロゲン化溶媒、テトラヒドロフラン(THF)等のエーテル、ジメチルホルムアミド(DMF)等のアミド、ジメチルスルホキシド(DMSO)等のスルホキシド、アセトン等のケトン、メタノール等のアルコール等が挙げられる。これら溶媒は、1種を単独で用いてもよいし、2種類以上を組み合わせて用いてもよい。
【0021】
次に、得られたアミドとカルボン酸とを有機溶媒中において縮合剤及びアミン系塩基の存在下において反応させ本発明に係る化合物を得る。
【0022】
カルボン酸としては、飽和脂肪酸、不飽和脂肪酸の何れも用いることができる。飽和脂肪酸としては、直鎖の飽和脂肪酸を用いるのが好ましく、特にデカン酸を用いることが好ましい。
【0023】
また、不飽和脂肪酸としては、二重結合を1つ有するモノ不飽和脂肪酸を用いるのが好ましく、特に9-デセン酸を用いるのが好ましい。
【0024】
アミン系塩基としては、モノエチルアミン、ジエチルアミン、トリエチルアミン、ジイソプロピルアミン、ジイソプロピルエチルアミン等が挙げることができ、ジイソプロピルエチルアミンを用いることが特に好ましい。
【0025】
また、縮合剤としては、上述のEDCI・HCl、HBTU、PyBOP等を単独で、または2種以上を組み合わせて用いることができる。
【0026】
有機溶媒としては、上述のアミド合成の際に用いる有機溶媒を用いることができ、これらの溶媒を1種を単独で用いてもよいし、2種類以上を組み合わせて用いてもよい。
【実施例】
【0027】
以下、本発明について実施例を用いてより具体的に説明する。
【0028】
(アミド1の合成)
式(6)で示すアミド1を次の手順により得た。
【0029】
シクロヘキシルアミン100 μL (0.88 mmol)をDMF 2 mLに溶解させ、そこにN-Boc-N-Me-L-Phe 315.5 mg(1.13 mmol)、EDCI・HCl 270.2 mg (1.41 mmol)、HOBt 190.9 mg(1.41 mmol)を順に加え、最後にジクロロメタン2 mLを加えて、14 時間室温撹拌した。反応溶媒を減圧留去し、1M塩酸20 mL及び酢酸エチル20 mLを加え、その後酢酸エチル(2×20mL) により抽出を行った。5%の炭酸水素ナトリウム水溶液 (20 mL) を用いて有機層を洗浄し、さらにブライン(20 mL)で有機層を脱水後、無水硫酸ナトリウムで乾燥させ、乾燥剤をろ過し、ろ液を減圧濃縮した。残留物質をシリカゲルクロマトグラフィー(酢酸エチル:n-ヘキサン=3:7)で分離を行い、淡黄色油状のアミド1を得た。(320.0 mg, 収率89 %)
【化6】
【0030】
(アミド2の合成)
式(7)で示すアミド2を次の手順により得た。
【0031】
シクロヘプチルアミン94μL(0.74mmol)をDMF 4mLに溶解させ、そこにN-Boc-N-Me-L-Phe 200.0 mg(0.72 mmol)、EDCI・HCl 207.8 mg(1.08 mmol)、HOBt 146.7 mg (1.08 mmol)を順に加え、最後にジクロロメタン4 mLを加えて、4時間室温撹拌した。反応溶媒を減圧留去し、1M塩酸20 mL及び酢酸エチル20 mLを加え、その後酢酸エチル(2×20 mL)により抽出を行った。5%の炭酸水素ナトリウム水溶液(20 mL)を用いて有機層を洗浄し、さらにブライン(20 mL)で有機層を脱水後、無水硫酸ナトリウムで乾燥させ、乾燥剤をろ過し、ろ液を減圧濃縮した。さらに真空ポンプで乾燥をし、黄色油状のアミド2を得た。(291.6 mg, 収率96 %)
【化7】
【0032】
(アミド2’の合成)
式(8)で示すアミド2’を次の手順により得た。
【0033】
上述のアミド2の合成におけるN-Boc-N-Me-L-Pheの代わりにN-Boc-N-Me-D-Pheを用いた以外は、アミド2の合成と同様な反応、処理を行った。減圧濃縮後の油状物質をシリカゲルクロマトグラフィー(酢酸エチル:n-ヘキサン=3:7)で精製を行い、淡黄色油状のアミド2’を得た (223.1 mg, 収率83 %)。
【化8】
【0034】
(アミド3の合成)
式(9)で示すアミド3を次の手順により得た。
【0035】
シクロオクチルアミン100 μL (0.72 mmol)をDMF 2 mLに溶解させ、そこにN-Boc-N-Me-L-Phe 239.6 mg(0.86 mmol)、EDCI・HCl 221.9 mg (1.16 mmol)、HOBt 159.2 mg (1.17 mmol)を順に加え、最後にジクロロメタン2 mLを加えて、14時間室温撹拌した。反応溶媒を減圧留去し、1M塩酸20 mL及び酢酸エチル20 mLを加え、その後酢酸エチル(2×20 mL)により抽出を行った。5%炭酸水素ナトリウム水溶液(20 mL)を用いて有機層を洗浄し、さらにブライン(20 mL)で有機層を脱水後、無水硫酸ナトリウムで乾燥させた後、減圧濃縮した。残留物質をシリカゲルクロマトグラフィー(酢酸エチル:n-ヘキサン=3:7)で分離を行い、淡黄色油状のアミド3を得た。(255.7 mg, 収率92 %)
【化9】
【0036】
(化合物1の合成)
式(10)に示す化合物1を次の手順により得た。
【0037】
152.5 mg (0.42 mmol)のアミド1を4M塩酸/ジオキサン溶液2 mLに溶解させ、3時間室温撹拌を行い、溶媒を減圧留去し、Boc基を除去した白色粉末を得た。この白色粉末をDMF 2 mLに溶解させ、ジイソプロピルエチルアミン230 μL (1.35 mmol)、PyBOP 286.5mg (0.55 mmol)、9-デセン酸100 mL(0.54 mmol)を順に加え、さらにジクロロメタン2 mLを加えて、14時間室温において撹拌を行った。反応溶媒を減圧留去し、1M塩酸20 mL及び酢酸エチル20 mLを加え、その後酢酸エチル(2×20 mL)により抽出を行った。5%の炭酸水素ナトリウム水溶液(20 mL)を用いて有機層を洗浄し、さらにブライン(20 mL)で有機層を脱水後、無水硫酸ナトリウムで乾燥させ、ろ過、ろ液の減圧留去を行った。残留物質をシリカゲルクロマトグラフィー(酢酸エチル:n-ヘキサン=3:7)で精製し、透明油状の化合物1を得た。(138.4 mg、収率80%)、さらに順相高速液体クロマトグラフィー(HPLC)[Inertsil, 250×20 mm, 溶媒 酢酸エチル:n-ヘキサン=3:7, flow rate 3.0 mL/min, refractive detection]によりτR = 42 minを分取し、スペクトル試料とした。
【表1】
【化10】
【0038】
(化合物2の合成)
式(11)に示す化合物2を次の手順により得た。
【0039】
138.8 mg(0.37 mmol)のアミド2を4M 塩酸/ジオキサン溶液2 mLに溶解させ、2時間室温において撹拌を行い、溶媒を減圧留去し、Boc基を除去した白色粉末を得た。この白色粉末をDMF 3 mLに溶解させ、ジイソプロピルエチルアミン200 μL(1.18 mmol)、PyBOP 251.4 mg (0.48 mmol)、9-デセン酸68 μL (0.37 mmol)を順に加え、最後にジクロロメタン3 mLを加えて、14時間室温撹拌を行った。反応溶媒を減圧留去し、1M塩酸20 mL及び酢酸エチル20 mLを加え抽出し、反応溶媒を減圧留去し、1M塩酸20 mL及び酢酸エチル20 mLを加え、その後酢酸エチル(2×20 mL)により抽出を行った。5%の炭酸水素ナトリウム水溶液(20 mL)を用いて有機層を洗浄し、さらにブライン(20 mL)で有機層を脱水後、無水硫酸ナトリウムで乾燥させ、ろ過、ろ液の減圧留去を行った。そして、残留物質をシリカゲルクロマトグラフィー(酢酸エチル:n-ヘキサン=1:4)により単離精製し、化合物2を得た。(淡黄色油状:147.4 mg, 収率94 %)、さらにスペクトル試料として、順層HPLC[Inertsil, 250×20 mm, 溶媒 酢酸エチル:n-ヘキサン=1:4, flow rate 3.0mL/min, refractive detection]によりτR=39 minのピークを集め、再度精製を行った。
【表2】
【化11】
【0040】
(化合物3の合成)
式(12)に示す化合物3を次の手順により得た。
【0041】
50.4 mg(0.13 mmol)のアミド3を4M 塩酸/ジオキサン溶液2 mLに溶解させ、2時間室温において撹拌を行い、溶媒を減圧留去し、Boc基を除去した白色粉末を得た。この白色粉末をDMF2mLに溶解させ、ジイソプロピルエチルアミン240 μL (1.41 mmol)、PyBOP 319.0 mg(0.61 mmol)、9−デセン酸112μL(0.61 mmol)を順に加え、最後にジクロロメタン2mLを加えて、14時間室温において撹拌を行った。反応溶媒を減圧留去し、1M塩酸20 mL及び酢酸エチル20 mLを加え、その後酢酸エチル(2×20 mL)により抽出を行った。5%の炭酸水素ナトリウム水溶液(20 mL)を用いて有機層を洗浄し、さらにブライン(20 mL)で有機層を脱水後、無水硫酸ナトリウムで乾燥させ、乾燥剤をろ過し、ろ液を減圧濃縮した。残留物質をシリカゲルクロマトグラフィー(酢酸エチル:n-ヘキサン=2:3)で分離し、透明油状の化合物3を得た(99.4 mg, 収率48 %)。さらに順相HPLC[Inertsil, 250×20 mm, 溶媒 酢酸エチル:n-ヘキサン=3:7, flow rate 3.0 mL/min, refractive detection]によりτR=50 minを分取し、精製した。
【表3】
【化12】
【0042】
(化合物4aの合成)
式(2)に示す化合物4aを次の手順により得た。
【0043】
152.5mg(0.42mmol)のアミド2 を4M塩酸/ジオキサン溶液2 mLに溶解させ、3時間室温において撹拌を行い、溶媒を減圧留去し、Boc基を除去した透明油状物質を得た。この透明油状物質をDMF 2mLに溶解させ、ジイソプロピルエチルアミン225 μL (1.32 mmol)、PyBOP 319.2 mg (0.61 mmol)、デカン酸111.5 mg (0.65 mmol)を順に、そしてジクロロメタン2 mLを加えて、16時間室温で撹拌を行った。反応溶媒を減圧留去し、1M塩酸20 mL及び酢酸エチル20 mLを加え、その後酢酸エチル(2×20 mL)により抽出を行った。5%の炭酸水素ナトリウム水溶液(20 mL)を用いて有機層を洗浄し、さらにブライン(20 mL)で有機層を脱水後、無水硫酸ナトリウムで乾燥させ、ろ過、ろ液の減圧留去を行った。残留物質をシリカゲルクロマトグラフィー(酢酸エチル:n-ヘキサン=2:3)で精製し、化合物4aを得た(透明油状:156.2 mg, 収率88 %)。さらに順相HPLC[Inertsil, 250×20 mm, 溶媒 酢酸エチル:n-ヘキサン=2:3, flow rate 3.0 mL/min, refractive detection]によりτR=27 minの部分を分取し、スペクトル用試料を得た。
【表4】
【化13】
【0044】
(化合物4bの合成)
式(3)で示す化合物4bを次の手順によって得た。
【0045】
10.2mg(0.03 mmol)のアミド2’を4M塩酸/ジオキサン2 mLに溶解させ、1時間室温撹拌し、反応溶媒を減圧留去させ、Boc基を除去した透明油状物質を得た。保護基を除去したアミド2’をDMF 2mLに溶解し、トリエチルアミン4μL (0.03 mmol)、EDCI・HCl 15.4 mg (0.08 mmol)、HOAt 9.4 mg (0.07 mmol)、デカン酸10.6 mg (0.06 mmol)、ジクロロメタン2 mLを順に加え、19時間室温で撹拌を行った。反応溶媒を減圧留去し、1M塩酸10 mL及び酢酸エチル10 mLを加え、その後酢酸エチル(3×10 mL) により抽出を行った。5%炭酸水素ナトリウム水溶液(20 mL)を用いて有機層を洗浄し、さらにブライン(20 mL)で有機層を脱水後、無水硫酸ナトリウムで乾燥させ、乾燥剤をろ過し、ろ液を減圧濃縮した。残渣をシリカゲルクロマトグラフィー(酢酸エチル:n-ヘキサン=2:3)にて分離精製し透明油状の化合物4bを得た(12.1 mg, 収率94%)。さらに順相HPLCにて[Inertsil, 250×20 mm, 溶媒 酢酸エチル:n-ヘキサン=3:7, flow rate 3.0 mL/min, refractive detection] τR=32 min の部分を集め再度精製した。
【表5】
【化14】
【0046】
(生理活性測定)
上述した手順で合成した化合物1〜3、4a及び4bについて下記に示す条件の下、生理活性測定を行った。リーシュマニア原虫として、L.major promastigotesを選択し、蛍光蛋白egfpを導入したL.major/egfp promastigotesを用いて、上記化合物をサンプルとして下記の条件で成長阻害率を求めた。
【0047】
(測定手順)
25oC で、96-穴 プレートにL.major/promastigotesを10%ウシ胎児血清と25 mMのHepes Buffer(ICN Biomedeivals Inc., Aurora, OH)で栄養を補った199培地(日水製薬)で培養したL.major/egfp promastigotsの蛍光シグナルを蛍光ミクロプレート読み取り装置(Fluoroscan Ascent FL, 大日本製薬)で励起光を485 nm、放射光538 nmで測定した。96穴プレートの各ウェルにL.major/egfp原虫100μL(1×106 cell/mL)、サンプル溶液10μL (メタノールに溶解)し、25℃で72時間培養を行った。標準物質として、Amphotericin の成長阻害率を100%として、各試料の阻害率を示した。
【0048】
測定の結果、求められた成長阻害率を図1に示す。
【0049】
これらの結果により、化合物1〜3、4a及び4bはリーシュマニア原虫の成長を阻害する生理活性特性を有することが示された。
【特許請求の範囲】
【請求項1】
一般式(1)
【化15】
(上記式(1)中、nは1〜3の整数を示す。)
で表される化合物。
【請求項2】
化学式(2)
【化16】
で表される化合物。
【請求項3】
化学式(3)
【化17】
で表される化合物。
【請求項1】
一般式(1)
【化15】
(上記式(1)中、nは1〜3の整数を示す。)
で表される化合物。
【請求項2】
化学式(2)
【化16】
で表される化合物。
【請求項3】
化学式(3)
【化17】
で表される化合物。
【図1】

【公開番号】特開2011−184342(P2011−184342A)
【公開日】平成23年9月22日(2011.9.22)
【国際特許分類】
【出願番号】特願2010−50282(P2010−50282)
【出願日】平成22年3月8日(2010.3.8)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成22年3月1日 青山学院大学大学院理工学研究科発行の「2009年度 生命科学コース 修士論文発表会プログラム」に発表
【出願人】(399109333)学校法人青山学院 (12)
【Fターム(参考)】
【公開日】平成23年9月22日(2011.9.22)
【国際特許分類】
【出願日】平成22年3月8日(2010.3.8)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成22年3月1日 青山学院大学大学院理工学研究科発行の「2009年度 生命科学コース 修士論文発表会プログラム」に発表
【出願人】(399109333)学校法人青山学院 (12)
【Fターム(参考)】
[ Back to top ]