
化合物
【課題】一本鎖抗体部を含む化合物を用いて、病変部位に存在する抗原(病変マーカー)を検出等する際、病変部位以外に存在する抗原と一本鎖抗体部との結合により、病変部位へ到達する化合物の量が低下し、また、病変部位以外で抗原に結合した化合物からの信号(バックグラウンド)が発生する。その結果、高感度、高コントラストな病変部位の検出が困難であり、これを解消する化合物が求められている。
【解決手段】一般式(1)で表されるポリマーからなる化合物。L−Y−A (1)
(式中、Aは一本鎖抗体部であって、抗原結合部位からなるポリペプチドである。Lはリンカー部であってプロテアーゼ切断部位からなるポリペプチドである。Yはペプチド部であって、一本鎖抗体部Aとリンカー部Lとを連結する0個以上のアミノ酸からなる。リンカー部Lはペプチド部YのN末端または一本鎖抗体部AのN末端に結合している)
【解決手段】一般式(1)で表されるポリマーからなる化合物。L−Y−A (1)
(式中、Aは一本鎖抗体部であって、抗原結合部位からなるポリペプチドである。Lはリンカー部であってプロテアーゼ切断部位からなるポリペプチドである。Yはペプチド部であって、一本鎖抗体部Aとリンカー部Lとを連結する0個以上のアミノ酸からなる。リンカー部Lはペプチド部YのN末端または一本鎖抗体部AのN末端に結合している)
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、化合物および前記化合物の製造方法に関する。
【背景技術】
【0002】
病変部位に存在する抗原(病変マーカー)を検出するために、病変マーカーに結合する抗体などの病変マーカー結合分子に、放射性核種、MRI(Magnetic Resonance Imaging)信号を発信する分子、超音波信号を発信する分子、蛍光信号を発信する分子などの物理的な信号を発生する分子(以下、信号発信分子と略すことがある)を標識した体内診断用造影剤の開発がなされている。このような化合物を体内へ投与し、その化合物の信号発信分子からの信号を体外から検出することにより、病変部位、及び病変マーカーを検出することが可能である。
【0003】
上記のような化合物を血中へ投与した場合、病変部位から漏れ出て血中に存在する病変マーカーや正常部位に存在する病変マーカーなど、病変部位以外に存在する病変マーカーとの結合により、病変部位へ到達する化合物の量が低下する場合がある。また、そのような場合は、病変部位以外に結合した化合物からの信号(バックグラウンド)の発生により、病変部位の高コントラストな検出が困難になると考えられる。
【0004】
上記問題を解決する化合物として、ローファーらは、特定の生物活性の存在下にて、インビボで生物活性化されるプロドラッグ造影剤を報告している(特許文献1)。この文献のプロドラッグ造影は信号発信分子を有する標的化分子に、酵素で開裂可能なリンカーを介して、標的化分子の結合能力を低下させる分子を結合したものである。この造影剤は、アルカリフォスファターゼによる反応を受けてリンカーが開裂し、その結果標的化分子として用いた低分子化合物のヒト血清アルブミンへの結合能力が開裂前と比較して向上する。また、リンカーの開裂により、信号発信分子として用いたMRI信号を発生する分子の信号が変化し、病変部位のより高コントラストな検出が可能になることもローファーらは報告している。
【0005】
抗体は抗原を特異的に認識する分子であり、病変部位の検出、あるいは治療に好適に用いられる。しかし、特許文献1は、低分子化合物を用いた造影剤の利用可能性を記載しているのみで、抗体を用いた造影剤の分子設計ならびに調製方法は開示されておらず、抗体を用いたインビボで生物活性化される造影剤の実用性については不明である。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特表2000‐507577号公報
【発明の概要】
【発明が解決しようとする課題】
【0007】
抗体を含む化合物を用いて、病変部位に存在する抗原(病変マーカー)抗体を検出等する際、病変部位以外に存在する抗原と抗体との結合により、病変部位へ到達する抗体化合物の量が低下し、また、病変部位以外で抗原に結合した抗体化合物からの信号(バックグラウンド)が発生する。その結果、高感度、高コントラストな病変部位の検出が困難であり、これを解消する化合物が求められている。
【課題を解決するための手段】
【0008】
第一の本発明に係る化合物は、一般式(1)で表されるポリマーからなる。
L−Y−A (1)
(式中、Aは一本鎖抗体部であって、抗原結合部位からなるポリペプチドである。Lはリンカー部であってプロテアーゼ切断部位からなるポリペプチドである。Yはペプチド部であって、一本鎖抗体部Aとリンカー部Lとを連結する0個以上のアミノ酸からなる。リンカー部Lはペプチド部YのN末端または一本鎖抗体部AのN末端に結合している)
【0009】
第二の本発明に係る化合物は、一般式(2)で表されるポリマーからなる化合物。
X−L−Y−A (2)
(式中、Aは一本鎖抗体部であって、抗原結合部位からなるポリペプチドである。Lはリンカー部であってプロテアーゼ切断部位からなるポリペプチドである。Yはペプチド部であって、一本鎖抗体部Aとリンカー部Lとを連結する0個以上のアミノ酸からなる。リンカー部Lはペプチド部YのN末端または一本鎖抗体部AのN末端に結合している。Xはポリエチレングリコール、有機色素、有機ポリマー、粒子のいずれかである。)
【0010】
第三の本発明に係る化合物の製造方法は、一般式(1)で表されるポリマーからなる化合物の製造方法であって、一本鎖抗体部Aをコードする塩基配列と、リンカー部Lをコードする塩基配列と、ペプチド部Yをコードする塩基配列と、を有する核酸をプラスミドに挿入する工程と、前記核酸が挿入されたプラスミドを菌に導入する工程と、前記プラスミドを導入した菌が発現した化合物を回収する工程と、を有することを特徴とする。
L−Y−A (1)
(式中、Aは一本鎖抗体部であって、抗原結合部位からなるポリペプチドである。Lはリンカー部であってプロテアーゼ切断部位からなるポリペプチドである。Yはペプチド部であって、一本鎖抗体部Aとリンカー部Lとを連結する0個以上のアミノ酸からなる。リンカー部Lはペプチド部YのN末端または一本鎖抗体部AのN末端に結合している。)
【発明の効果】
【0011】
本発明の化合物は、病変部位以外では、一本鎖抗体部の抗原との結合能力を低下させた状態であり、病変部位においては、抗原との結合能力を向上させた状態となることにより、病変部位以外に存在する抗原との結合による病変部位へ到達する一本鎖抗体部の量の低下と、病変部位以外で抗原に結合した一本鎖抗体部からの信号(バックグラウンド)を抑えることが可能である。これにより、高コントラストな病変部位の検出が可能となる。
【図面の簡単な説明】
【0012】
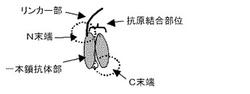
【図1】本発明で提供される化合物の模式図を示す。
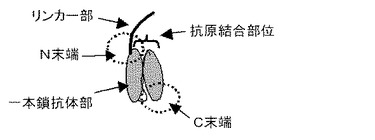
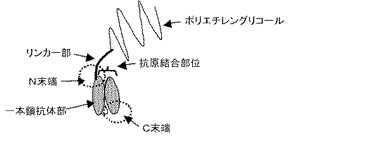
【図2】一本鎖抗体部のN末端にリンカー部を介してポリエチレングリコール(PEG)を結合させた化合物の模式図を示す。
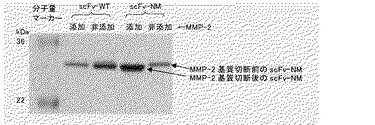
【図3】作製したscFv−NMとscFv−WTについて、MMP−2添加、非添加での還元型SDS−PAGEの結果を示す。
【図4】作製したscFv−NMのMMP−2添加前後における、HER2との相互作用をBiacore Xシステムを用いて測定した結果を示す。
【図5】作製したN末端PEG化scFv−NMのMMP−2添加前後での分子状態をゲルろ過クロマトグラフィーで検出した結果を示す。
【図6】2 kDa、5 kDa、12 kDa、20 kDa、40 kDaの PEG−マレイミドを反応させたscFv−CMの還元型SDS−PAGEの結果を示す。
【図7】作製したC末端PEG化scFv−CMにMMP−2を添加、非添加、及びMMP−2活性阻害剤存在下での還元型SDS−PAGEの結果を示す。
【図8】作製したscFv−WT、scFv−NM、scFv−NPMについて、MMP−2添加、非添加での還元型SDS−PAGEの結果を示す。
【図9】作製したN末端PEG化scFv−NM、C末端PEG化scFv−CMのMMP−2添加、非添加での、HER2との相互作用をBiacore Xシステムを用いて測定した結果を示す。
【発明を実施するための形態】
【0013】
本発明の実施形態について説明する。
【0014】
(第一の実施形態)
本発明の第一の実施形態に係る化合物は、一般式(1)で表されるポリマーからなる。
L−Y−A (1)
(式中、Aは一本鎖抗体部であって、抗原結合部位からなるポリペプチドである。Lはリンカー部であってプロテアーゼ切断部位からなるポリペプチドである。Yはペプチド部であって、一本鎖抗体部Aとリンカー部Lとを連結する0個以上のアミノ酸からなる。リンカー部Lはペプチド部YのN末端または一本鎖抗体部AのN末端に結合している)
【0015】
本実施形態に係る化合物は一本鎖抗体部とリンカー部とペプチド部とを有する。リンカー部はペプチド部のN末端に結合し、ペプチド部は一本鎖抗体部のN末端に結合している。または、リンカー部は一本鎖抗体部のN末端に直接結合している(ペプチド部が0個のアミノ酸からなる場合)。このような構成となっているため、一本鎖抗体部の抗原結合能力が阻害あるいは低減されている。そして、リンカー部がプロテアーゼによって切断されることにより、一本鎖抗体部の抗原結合能力が回復する。
本実施形態において、抗原結合能力が阻害あるいは低減される、とは、一本鎖抗体部が修飾等をされていない状態で抗原と結合する能力が、なんらかの要因により、失われ、あるいは、弱められることを指す。また、抗原結合能力が回復するとは、失われ、あるいは弱められた抗原結合能力が、向上し、修飾等をされていない状態の一本鎖抗体部と、同じ、または近い状態になることをいう。
【0016】
(一本鎖抗体部)
本発明において、一本鎖抗体部とは、抗体の重鎖可変(VH)領域と軽鎖可変(VL)領域とをペプチドからなるリンカーで連結した部位(抗原結合部位)からなるポリペプチドである。
ここで、抗体とは、ヒト、マウス、ラット、ラクダ、鳥、等、由来は問わず、さらにはキメラ体であってもよく、また、ポリクローナル抗体であってもモノクローナル抗体であってもよい。
【0017】
また上記抗体は限定なく用いられるが、本発明の目的に照らし合わせれば、例えば、EGFRファミリー、VEGFファミリー、VEGFRファミリー、PSA、CEA、マトリックスメタロプロテアーゼファミリー、EGFファミリー、インテグリンファミリー、セレクチンファミリー、エンドグリン、又はMUCファミリーからなる群より選ばれた病変マーカーに結合する抗体、さらにはHER2に結合する抗体を例としてあげることができる。
【0018】
一本鎖抗体部は、各種抗原に対応して安価に簡便に作製することができ、なおかつ、通常の抗体(全抗体など)と比べて分子量が小さいため、体外へ速やかに排泄されやすく、又病変部位に到達しやすい。そのため、一本鎖抗体部は病変部位の検出、又は治療に好適に用いられる。
【0019】
一本鎖抗体部としては、例えば以下のアミノ酸配列を含むポリペプチドからなる。
DIQMTQSPSSLSASVGDRVTITCRASQDVNTAVAWYQQKPGKAPKLLIYSASFLYSGVPSRFSGSRSGTDFTLTISSLQPEDFATYYCQQHYTTPPTFGQGTKVEIKGGGGSGGGGSGGGGSEVQLVESGGGLVQPGGSLRLSCAASGFNIKDTYIHWVRQAPGKGLEWVARIYPTNGYTRYADSVKGRFTISADTSKNTAYLQMNSLRAEDTAVYYCSRWGGDGFYAMDYWGQGTLVTVSSAAALEHHHHHHGGC(配列番号1)
なお、一本鎖抗体部のC末端には、任意のアミノ酸が結合していてもよい。
【0020】
(リンカー部)
本発明において、リンカー部とは、プロテアーゼ切断部位からなるポリペプチドである。ここで、プロテアーゼ切断部位とは、プロテアーゼに特異的に認識されるアミノ酸配列を有しており、そのアミノ酸配列が活性状態のプロテアーゼに認識され、特異的に加水分解されて切断される部位を意味する。
プロテアーゼとしては、特に限定はなく用いられるが、マトリックスメタロプロテアーゼファミリーまたはセリンプロテアーゼファミリーが好ましく、例えば、マトリックスメタロプロテアーゼー2(MMP−2)、マトリックスメタロプロテアーゼー9(MMP−9)、前立腺特異的抗原(PSA)、プラスミン、カテプシン、カスパーゼなどが標的プロテアーゼとして挙げられる。
プロテアーゼ切断部位については、それぞれのプロテアーゼが特異的に認識するアミノ酸配列であり、たとえば、MMP−2の切断部位としてアミノ酸配列PLGVR(配列番号2)を含むポリペプチドなどを利用できる。また、PSAの切断部位としてアミノ酸配列SSIYSQTEEQ(配列番号3)を含むポリペプチドなどを利用することができる。
リンカー部のN末端には任意のアミノ酸が結合していてもよい。
【0021】
(ペプチド部)
本発明において、ペプチド部とは、一本鎖抗体部とリンカー部とを連結する0個以上のアミノ酸からなる。そのアミノ酸の数は20個以下であることが好ましく、10個以下であることがさらに好ましい。アミノ酸の数が20個以上であると、そのアミノ酸配列が立体構造をとる可能性がある。その結果、プロテアーゼ切断部位がプロテアーゼによって切断された場合でも、一本鎖抗体部の抗原結合能力が阻害あるいは低減されたままとなってしまうおそれがあるためである。
【0022】
図1を用いて本実施形態をより詳細に説明する。図1は、本実施形態に係る化合物の模式図である。ただし、図1ではペプチド部は図示していない。
抗原結合能力を阻害あるいは低減する機構については、種々の機構が考えられる。第一に、ペプチド部または一本鎖抗体部のN末端に結合したリンカー部が一本鎖抗体部の抗原結合部位と抗原の間に存在することにより立体的障害を生じさせることが考えられる。この場合、リンカー部が一本鎖抗体部の抗原結合部位を覆うため、他の抗体が抗原に結合できなくなることなどが考えられる。
【0023】
リンカー部を一本鎖抗体部のN末端に結合させている理由は以下のとおりである。まず第一に、一本鎖抗体部における重鎖可変(VH)領域と軽鎖可変(VL)領域とを連結するリンカーにリンカー部を結合させると、一本鎖抗体部の構造自体が変わり、リンカー部切断後にも構造が戻らず、結果として、リンカー部切断後も抗原結合能力が回復しないおそれがある。一方、一本鎖抗体部のいずれかの末端にリンカー部を結合させた場合、構造自体には影響が少なく、リンカー部が切断された後に一本鎖抗体部がリンカー部を導入する前の構造に戻りやすく、リンカー部を切断後に抗原結合能力が回復しやすいと考えられる。そのため、まず、一本鎖抗体部のいずれかの末端にリンカー部を導入すれば、リンカー部が切断された後の抗原結合能力がリンカー部の導入前と同等までに回復することが可能であるという点で有効であると考えられる。次に、一本鎖抗体部のN末端はC末端よりも、抗原結合部位に近い。そのため、一本鎖抗体部のN末端にリンカー部を結合する場合は、C末端に結合するよりも、結合するリンカー部の長さが短くても、一本鎖抗体部の抗原結合部位への立体的障害作用などの効果をより効率よく生じさせられると考えられ、すなわち、より、強く抗原結合能力を低下させることが可能である。これにより、化合物の分子量を過剰に増大させることなく、抗原結合能力を阻害あるいは低減することが可能になると考えられる。化合物の分子量を過剰に増大させることを抑えることは、体外へ速やかに排泄されやすく、又病変部位に到達しやすいという一本鎖抗体部のみの性質に近い性質を化合物が維持できるため、有効である。さらに、一本鎖抗体部のN末端は抗原結合部位と近接しているため、一本鎖抗体部のN末端にリンカー部を導入すれば、C末端に導入するのに比べ抗原結合能力をより強く低下させることが可能であるという点で有効である。また、一本鎖抗体部のN末端にリンカー部を結合させることで、抗原結合部位から離れたC末端には他の分子を結合することができ、例えば、信号発信分子や治療用薬剤などを、抗原結合能力を低下させることなく結合させることが可能である。以上より、一本鎖抗体部のN末端にリンカー部を結合させることが最も有効であると考えられる。
以上のことは、リンカー部がペプチド部のN末端に結合し、ペプチド部が一本鎖抗体部のN末端に結合している場合についても同様のことが言える。
【0024】
(信号発信分子)
本実施形態に係る化合物には、一本鎖抗体部に信号発信分子を結合させることが可能である。
信号発信分子として、放射性核種、MRI信号を発信する分子、超音波信号を発信する分子、蛍光信号を発信する分子などの種々の分子を用いることができる。例えば、光を吸収し超音波信号や発光信号を発する信号発信分子としては、Alexa Fluor 680、Alaxa Fluor 700、Alexa Fluor 750、Alexa Fluor 790(以上商標、インビトロジェン株式会社)、Cy5、Cy5.5、Cy7(以上商標、GEヘルスケア)、HiLyte 647、HiLyte 680、HiLyte 750(以上商標、AnaSpec, Inc)、DY−680、DY−700、DY−730、DY−750、DY−782 (以上商標、Dyomics, Jena, Germany)などを利用することが可能である。また、光を吸収し超音波信号や発光信号を発する分子としては、生体透過性の優れた約600 nm以上約1000 nm以下の近赤外波長領域の光を吸収する近赤外光吸収色素を選択することが有効である。その他、微粒子としては、酸化鉄微粒子、金微粒子、金ナノロッド、白金、銀などの金属や金属酸化物の微粒子など種々の微粒子を利用することが可能であろう。また、本実施形態の一本鎖抗体部に抗癌剤などの治療用薬剤を結合し、病変部位へ治療用薬剤を送達することも可能であり、病変部位以外への治療用薬剤の送達を低減することによる副作用の低減が期待できる。
【0025】
また、本発明の化合物のうち、信号発信分子が結合したものは、病変マーカーの検出に用いることが可能である。すなわち、本発明は病変マーカーの検出方法において、信号発信分子が結合した本発明の化合物を個体に投与、又は個体より得られた試料に添加し、信号を検出することにより、該個体中もしくは個体より得られた試料中の病変マーカーの存否、又は病変マーカーを産生する病変部位の存否を検出する方法を提供する。病変マーカーの検出方法は以下の通りである。すなわち、信号発信分子が結合した本発明の化合物を個体に投与し、あるいは個体より得られた臓器等の試料に添加する。なお、個体とは、ヒト、実験動物やペット等の哺乳類、その他、特に限定されることなく、あらゆる生物を指し、個体中もしくは個体より得られた試料としては、臓器、組織、組織切片などを挙げることができる。投与あるいは添加後、信号発信分子を検出できる測定機器等を用いて、個体あるいは試料に含まれる信号発信分子の信号を検出する。その結果、信号が基準とする閾値以上で検出されれば、その個体に病変マーカー、あるいは、病変マーカーを産生する病変部位が存在すると推定され、または、試料に病変マーカーが存在する、あるいは、試料の由来となる個体に病変マーカーを産生する病変部位が存在すると推定することができる。
【0026】
(イメージング)
また、同様に、本発明の化合物のうち、信号発信分子が結合したものは、病変マーカーのイメージングに用いることが可能である。病変マーカーのイメージング方法は、以下の通りである。すなわち、本発明の化合物を個体に投与し、又は個体より得られた試料に添加し、信号を検出することにより、該個体中もしくは個体より得られた試料中の病変マーカーの存否、又は病変マーカーを産生する病変部位の存否をイメージングすることができる。
【0027】
(第二の実施形態)
以下、本発明の第二の実施形態に係る化合物の説明をするが、第一の実施形態と共通のことに関しては説明を省略する。
本発明の第二の実施形態に係る化合物は、一般式(2)で表されるポリマーからなる化合物。
X−L−Y−A (2)
(式中、Aは一本鎖抗体部であって、抗原結合部位からなるポリペプチドである。Lはリンカー部であってプロテアーゼ切断部位からなるポリペプチドである。Yはペプチド部であって、一本鎖抗体部Aとリンカー部Lとを連結する0個以上のアミノ酸からなる。リンカー部Lはペプチド部YのN末端または一本鎖抗体部AのN末端に結合している。Xはポリエチレングリコール、有機色素、有機ポリマー、粒子のいずれかである)
【0028】
本実施形態に係る化合物は、一本鎖抗体部と、リンカー部と、ペプチド部と、一本鎖抗体部の抗原結合部位を覆うことができる部位(一般式(2)のXに相当する)とを有する。リンカー部はペプチド部のN末端に結合し、ペプチド部は、一本鎖抗体部のN末端に結合している。または、リンカー部は一本鎖抗体部のN末端に直接結合している(ペプチド部が0個のアミノ酸からなる場合)。このような構成となっているため、一本鎖抗体部の抗原結合能力が阻害あるいは低減されている。そして、リンカ部が切断されることにより、前記一本鎖抗体部の抗原結合能力が回復する。
【0029】
抗原結合部位を覆うことができる部位としては、ポリエチレングリコール(以下、PEGと略すことがある)、有機色素、有機ポリマー、粒子のいずれかである。有機色素としては、Alexa Fluor 680、Alaxa Fluor 700、Alexa Fluor 750、Alexa Fluor 790(以上商標、インビトロジェン株式会社)、Cy5、Cy5.5、Cy7(以上商標、GEヘルスケア)、HiLyte 647、HiLyte 680、HiLyte 750(以上商標、AnaSpec, Inc)、DY−680、DY−700、DY−730、DY−750、DY−782 (以上商標、Dyomics, Jena, Germany)などを利用することが可能である。有機ポリマーとしては、ポリ乳酸(Poly lactic acid、PLA)、ポリ乳酸グリコール酸共重合体(Poly (DL−lactic−co−glycolic acid)、PLGA)などを利用することが可能である。
【0030】
図2にPEGを導入した場合の模式図を示す。ただし、図2ではペプチド部は図示していない。これらの分子を用いる場合であっても、リンカー部及び連結された抗原結合部位を覆うことができる部位は一本鎖抗体部のN末端に結合されていることが効果的と考えられる。例えば、PEGは抗原結合部位を覆うことができる部位として特に好ましく用いられるが、PEGは水溶液中で、立体的に広がりを持った状態で存在すると考えられる。そのため、一本鎖抗体部のN末端にリンカー部及び連結されたPEGを導入した場合、一本鎖抗体部の抗原結合部位と抗原の間にPEGが存在するため、例えば、C末端などのN末端部位以外に導入した場合と比較して、より大きな立体障害となり、抗原結合能力を阻害あるいは低減する作用が大きくなると考えられ、一本鎖抗体部の抗原結合能力をより強く低下させることが可能である。
【0031】
PEGの導入は、抗原結合能力を阻害等することに加えて、種々の効果が期待できる。例えば、PEGを導入することによる化合物の分子量の調節や化合物のタンパク質吸着能の調節が挙げられる。PEGを導入した化合物は病変部位へ到達する前の血中ではリンカー部が切断されずPEGが遊離しないため、化合物の分子量が大きく保たれ、かつ化合物のタンパク質吸着能が抑えられる。それにより、腎臓から排泄させる大きさより化合物の大きさを大きく保つことができ、腎臓からの排泄による化合物の損失を抑えることができる。また、タンパク質が化合物に吸着することに伴い働く化合物の体外への排泄による化合物の損失を抑えることができる。一方、病変部位では、リンカー部が切断され、PEGが遊離することにより、化合物の分子量が減少する。それにより、又病変部位では、それ以外の部位と比較して、化合物の浸透性が向上し、病変深部にまで化合物が浸透することで、化合物の病変部位への集積性を向上させることが可能であろう。それに対して、正常部位においては、リンカー部が切断されずPEGが遊離しないため、化合物は正常部位の深部にまで浸透しない。そのため、正常部位への化合物の集積により発生する信号(バックグラウンド)を低減させることが可能であろう。
PEGの分子量は20kDa以上であることが好ましい。また、PEGの分子量が40kDa以下であることが好ましい。
【0032】
リンカー部を切断する酵素と一本鎖抗体部が結合する病変マーカーが同一の場合、該酵素の存在とその活性を同時に検出することが可能であろう。また、リンカー部を切断する酵素が病変部位特異的であり、かつその酵素が、一本鎖抗体部が結合する病変マーカーと異なる場合には、病変部位の二つの病変マーカーが存在する場合にのみ、病変部位へ集積するため、位置精度を向上させること、および詳細な病変部位の情報を検出することが可能であろう。例えば、HER2に結合する一本鎖抗体部とMMP−2の切断部位を含むリンカー部を組み合わせた場合、癌の予後不良因子としてのHER2陽性であるということと、転移、浸潤などの性質を示すという指標としてのMMP−2活性が高いということの二つの性状を同時に示す腫瘍、つまり悪性度の高いと考えられる腫瘍を高い位置精度で検出することが可能になるであろう。また、この場合、血中等に存在する可溶性HER2やHER2陽性/MMP−2陰性の腫瘍部位は当該造影剤では検出されにくいため、バックグラウンドを低減させる事が可能になるであろう。また、血管内皮増殖因子VEGFに結合する一本鎖抗体部とMMP−2の切断部位を含むリンカー部を組み合わせた場合、血中へ漏れ出たVEGFには結合し難く、活性化したMMP−2が存在する病変部位で、高濃度に存在するVEGFに結合し易くなることで、VEGFを高濃度に存在する血管新生の盛んな病変部位を高い位置精度で検出することが可能になるであろう。
【0033】
(第三の実施形態)
本発明の第三の実施形態について説明する。本実施形態は、上記一般式(1)で表されるポリマーからなる化合物の製造方法に関する。具体的には以下の工程を有する。
(i)一本鎖抗体部Aをコードする塩基配列と、リンカー部Lをコードする塩基配列と、ペプチド部Yをコードする塩基配列と、を有する核酸をプラスミドに挿入する工程
(ii)前記核酸が挿入されたプラスミドを菌に導入する工程
(iii)前記プラスミドを導入した菌が発現した化合物を回収する工程
なお、本実施形態に係る化合物の製造方法は上記(i)から(iii)以外の工程を含んでいてもよい。
【実施例】
【0034】
以下に、本発明の特徴をさらに明らかにするために実施例に沿って本発明を説明するが、本発明に用いる一本鎖抗体部、リンカー部、抗原結合部位を覆うことができる部位の組合せは他の組合せを用いることも可能であり、本発明はこの実施例によって制限されるものではない。
【0035】
(実施例1 hu4D5−8 scFvの調製)
始めに、HER2へ結合するIgGの可変領域の遺伝子配列を基に、一本鎖抗体(scFv)部をコードする遺伝子断片を作製した。作製した遺伝子は発現産物のN末端に、MMP−2により特異的に切断されるアミノ酸配列PLGVR、C末端にはタンパク精製のためのヒスチジンが6残基連続した6 x Hisタグ、またそのさらに下流にスペーサーとしてグリシンを2残基挟み信号発信分子を導入するためのシステインが配置される様に設計した。比較として、N末端にMMP−2により特異的に切断されるアミノ酸配列PLGVRを含まない遺伝子についても同様に作製した。上記遺伝子断片をT7プロモーターの下流に挿入したプラスミドpET−22b (+)(Novagen社)を大腸菌(Escherichia coli BL21 (DE3))に導入し、発現用菌株を得た。得られた菌株をLB−Amp培地4 mlで一晩前培養後、全量を250 mlの2 x YT培地に添加し、28℃、120 rpmで8 時間振とう培養した。その後、終濃度1 mMでIPTGを添加し、28℃で一晩培養した。培養した大腸菌を8000x g、30 分、4℃で遠心分離し、その上清の培養液を回収した。得られた培養液の60%重量の硫酸アンモニウムを添加し、塩析によりタンパク質を沈殿させた。塩析操作した溶液を一晩4℃で静置後、8000xg、30 min、4℃で遠心分離することで沈殿物を回収した。得られた沈殿物を20 mM Tris・HCl / 500 mM NaClバッファーに溶解し、1 Lの同バッファーへ透析した。透析後のタンパク質溶液を、His・Bind(登録商標) Resin(Novagen社)を充填したカラムへ添加し、Niイオンを介した金属キレートアフィニティークロマトグラフィーによって精製した。精製したN末端MMP−2基質導入hu4D5−8 scFv (scFv−NM)と、N末端MMP−2基質未導入hu4D5−8 scFv (scFv−WT)は、SDS−PAGEによりシングルバンドを示し分子量は約28 kDaであることを確認した。
【0036】
(実施例2 scFv−NMのMMP−2添加によるリンカー部の切断の確認)
上記で調製したscFv−NMにMMP−2を添加することにより、ペプチドが切断されて生じる分子量の変化を測定した。TCNB buffer(50 mM Tris, 10 mM CaCl2, 150 mM NaCl, 0.05% Brij35, pH7.5)に透析したPEG化scFv 約4 μMに、活性型MMP−2(コスモ・バイオ株式会社)0.10 mg/mlを約15 nM添加し、約20時間反応を行い、分子量の変化を還元型SDS−PAGEにより測定した。その結果、図3に示すように、MMP−2を添加することにより、scFv−NMでは分子量の低下が検出された。一方、scFv−WTでは分子量の低下は検出されなかった。以上より、scFv−NMのペプチドがMMP−2により切断されることを確認した。
【0037】
(実施例3 scFv−NMのMMP−2添加による抗原結合能力の向上の確認)
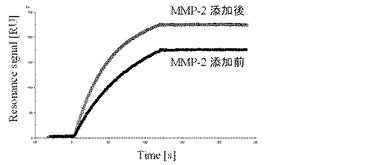
上記で作製したscFv−NM、scFv−WTと抗原であるHER2との相互作用をBiacore Xシステム(GEヘルスケア株式会社)を用いて測定し、MMP−2添加前と添加後でのscFvの結合能力の変化を測定した。抗原としては、Recombinant Human ErbB2 / Fc Chimera(R&D Systems,Inc.)を用いて、メーカーの推奨に従って、CM−5チップ表面のカルボキシメチルデキストラン鎖へのアミンカップリングにより固定した。固定化量は、約1000 RUであった。ランニングバッファーとしてはPBS−T(2.68 mM KCl / 137 mM NaCl / 1.47 mM KH2PO4 / 1mM Na2HPO4 / 0.005% Tween 20, pH7.4)を用いて、50 nMのscFvを流速20 μl/minの条件下でインジェクトし結合能力を評価した。その結果、図4に示すように、MMP−2添加前後でセンサーグラムの変化を確認でき、scFv−NMの抗原結合能力が向上することがわかった。一方、scFv−WTの抗原結合能力は、MMP−2添加前後ではほとんど変化しないことを確認した。
【0038】
(実施例4 N末端PEG化scFv−NMの作製)
実施例1で調製したscFv−NMを100 mMリン酸バッファー(pH 5.0)にバッファー置換後、scFv−NMに対して40倍モル量の5 kDa、20 kDa、又は40 kDaの PEG−アルデヒド(日油株式会社)を添加し、終濃度20 mMで2−ピコリンボランを添加し、4℃で約3日間反応させた。反応後、TCNB buffer(50 mM Tris, 10 mM CaCl2, 150 mM NaCl, 0.05% Brij35, pH7.5)にバッファー交換し、 Superdex 200 GL 10/300カラム(GEヘルスケア株式会社)を用いてゲルろ過クロマトグラフィーにより、PEGが1分子結合したと考えられる画分を分取することで、N末端にPEG化されたと考えられるscFv−NMを得た。
【0039】
(実施例5 N末端PEG化scFv−NMのMMP−2添加によるPEGの遊離の確認)
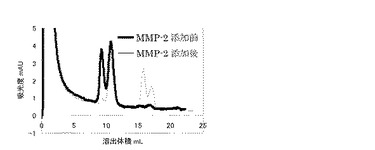
上記で調製したN末端がPEG化されたと考えられるscFv−NMについて、MMP−2を添加することにより、分子量の変化を評価した。TCNB bufferに溶解したN末端にPEG化されたと考えられるscFv−NMに、活性型MMP−2(コスモ・バイオ株式会社)0.10 mg/mlを終濃度約10 nM添加し、約20 h反応を行い、 Superdex 200 GL 10/300カラムを用いてゲルろ過クロマトグラフィーにより、分子量の変化を評価した。20 kDaのPEGを修飾したscFv−NMのMMP−2添加前後でのゲルろ過クロマトグラフィー(280 nmの吸収をモニタリング)の結果を図5に示す。図5に示すようにMMP−2を添加することにより、切断前にはなかった2つのピークが検出された。これらは、PEG未修飾のscFv−NMのモノマーとダイマーと考えられる。すなわち、この結果はN末端に導入したMMP−2基質をMMP−2が切断することにより、N末端に修飾されたPEGがscFv−NMより遊離したことを示している。つまり、N末端にPEGが修飾されていることを確認できた。また、MMP−2の添加により現れた、PEGが遊離したscFv−NMに相当すると考えられるピーク面積と、全体のピーク面積の割合を比較した結果、 PEG化scFv−NM全体のうちN末端がPEG化されていた割合は約50%程度であった。残りの50%はscFv−NM分子内のリジン残基のアミノ基に対してPEGが修飾されていたと考えられる。また、5 kDa、40 kDaのPEG−アルデヒドを用いた場合も同様に約50%程度がN末端にPEG化されていることを確認した。以上より、N末端PEG化scFv−NMの取得を確認した。
【0040】
(実施例6 C末端にMMP−2基質を導入したscFvの作製)
HER2へ結合するIgGの可変領域の遺伝子配列を基に、一本鎖抗体(scFv)部をコードする遺伝子断片を作製した。作製した遺伝子のカルボキシ末端には、MMP−2により特異的に切断されるアミノ酸配列PLGVR、抗原結合能を低下させる分子を連結するためのシステイン残基、及びタンパク精製のためのヒスチジンが6残基連続した6 x His タグを配置した。上記遺伝子断片をT7プロモーターの下流に挿入したプラスミドpET−22b (+)(Novagen社)を大腸菌(Escherichia coli BL21 (DE3))に導入し、発現用菌株を得た。得られた菌株をLB−Amp培地4 mlで一晩前培養後、全量を250 mlの2 x YT培地に添加し、28℃、120 rpmで8時間振とう培養した。その後、終濃度1 mMでIPTGを添加し、28℃で一晩培養した。培養した大腸菌を8000 x g、30 min、4℃で遠心分離し、その上清の培養液を回収した。得られた培養液の60%重量の硫酸アンモニウムを添加し、塩析によりタンパク質を沈殿させた。塩析操作した溶液を一晩4℃で静置後、8000xg、30 min、4℃で遠心分離することで沈殿物を回収した。得られた沈殿物を20 mM Tris・HCl / 500 mM NaClバッファーに溶解し、1 Lの同バッファーへ透析した。透析後のタンパク質溶液を、His・Bind (登録商標)Resin(Novagen社)を充填したカラムへ添加し、Niイオンを介した金属キレートアフィニティークロマトグラフィーによって精製した。精製したC末端MMP−2基質導入scFv(scFv−CM)は、SDS−PAGEによりシングルバンドを示し分子量は約28 kDaであることを確認した。
【0041】
(実施例7 C末端PEG化scFv−CMの作製)
上記で調製したscFv−CMを5 mM EDTAを含むリン酸バッファー(2.68 mM KCl / 137 mM NaCl / 1.47 mM KH2PO4 / 1m M Na2HPO4 / 5 mM EDTA, pH7.4)にバッファー置換後、10〜20倍モル量のトリ(2−カルボキシエチル)ホスフィン塩酸塩(TCEP)によって、25℃で2〜4時間、還元処理した。この還元処理したscFv−CMを、25℃で2〜4時間、10倍モル量の2 kDa、5 kDa、12 kDa、20 kDa、又は40 kDaの PEG−マレイミド(日油株式会社)と反応させた。PEG化されていることは、還元型SDS−PAGEにより確認した(図6)。また、反応後、Superdex 200 GL 10/300カラムを用いてゲルろ過クロマトグラフィーにより、未反応のPEG−マレイミドを除去し、C末端PEG化scFv−CMを得た。
【0042】
(実施例8 C末端PEG化scFv−CMのMMP−2添加によるPEGの遊離の確認)
上記で調製したC末端PEG化scFv−CMのうち、20 kDa PEG−マレイミドを付加したものについて、MMP−2を添加することにより、分子量の変化を評価した。TCNB bufferに透析したC末端PEG化scFv−CMに、活性型MMP−2 (0.10 mg/ml)を終濃度約10 nM添加し、2時間、20時間と反応を行い、各時間での分子量の変化を還元型SDS−PAGEにより測定した。また、MMP−2活性阻害剤である1, 10−Phenanthroline(Sigma−aldrich)を1 mMで添加し、同様に20時間反応させた実験について行った。その結果、図7に示すように、MMP−2添加2時間でscFv−CMの分子量である28 kDa付近にバンドが検出され、20 時間でほぼscFv−CMがメインバンドとして検出された。また、このscFv−CMのバンドの検出は、1, 10−Phenanthroline添加により抑えられた。以上より、C末端PEG化scFv−CMのPEGがMMP−2特異的に切断されることにより、scFv−CMより遊離することを確認した。
【0043】
(実施例9 N末端にペプチドとMMP−2基質を導入したscFvの作製)
始めに、HER2へ結合するIgGの可変領域の遺伝子配列を基に、一本鎖抗体(scFv)部をコードする遺伝子断片を作製した。作製した遺伝子のN末端には、ペプチドGGSGGGS、その下流に
MMP−2により特異的に切断されるアミノ酸配列PLGVR、C末端にはタンパク精製のためのヒスチジンが6残基連続した6 x Hisタグ、またそのさらに下流にスペーサーとしてグリシンを2残基挟み信号発信分子を導入するためのシステインを配置した。上記遺伝子断片をT7プロモーターの下流に挿入したプラスミドpET−22b (+)(Novagen社)を大腸菌(Escherichia coli BL21 (DE3))に導入し、発現用菌株を得た。得られた菌株をLB−Amp培地4 mlで一晩前培養後、全量を250 mlの2 x YT培地に添加し、28℃、120 rpmで8 時間振とう培養した。その後、終濃度1 mMでIPTGを添加し、28℃で一晩培養した。培養した大腸菌を8000 x g、30 分、4℃で遠心分離し、その上清の培養液を回収した。得られた培養液の60%重量の硫酸アンモニウムを添加し、塩析によりタンパク質を沈殿させた。塩析操作した溶液を一晩4℃で静置後、8000 x g、30 min、4℃で遠心分離することで沈殿物を回収した。得られた沈殿物を20 mM Tris・HCl / 500 mM NaClバッファーに溶解し、1 Lの同バッファーへ透析した。透析後のタンパク質溶液を、His・Bind(商標) Resin(Novagen社)を充填したカラムへ添加し、Niイオンを介した金属キレートアフィニティークロマトグラフィーによって精製した。精製したN末端ペプチドとMMP−2基質を導入したhu4D5−8 scFv (scFv−NPM)は、SDS−PAGEによりシングルバンドを示し分子量は約28 kDaであることを確認した。
【0044】
(実施例10 scFv−NPMのMMP−2添加によるリンカー部の切断の確認)
上記で調製したscFv−NPMにMMP−2を添加することにより、ペプチドが切断されて生じる分子量の変化を測定した。TCNB bufferに透析したPEG化scFv 約4 μMに、活性型MMP−2 0.10 mg/mlを約10 nM添加し、約20時間反応を行い、分子量の変化を還元型SDS−PAGEにより測定した。その結果、図8に示すように、MMP−2を添加することにより、scFv−NPMでは分子量の低下が検出された。また、scFv−NM、scFv−CMも比較として示した。一方、scFv−WTでは分子量の低下は検出されなかった。以上より、scFv−NPMのペプチドがMMP−2により切断されることを確認した。
【0045】
(実施例11 scFvのMMP−2添加による結合能力の変化の確認)
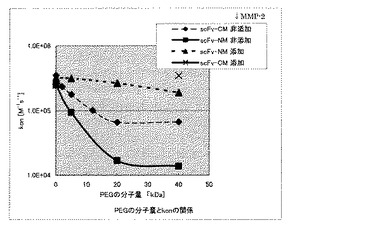
上記で作製した種々のscFvと抗原であるHER2との相互作用をBiacore Xシステム(GEヘルスケア株式会社)を用いて測定し、MMP−2添加前と添加後20時間での種々のscFvの結合能力の変化を測定した。抗原としては、Recombinant Human ErbB2 / Fc Chimera(R&D Systems, Inc.)を用いて、メーカーの推奨に従って、CM−5チップ表面のカルボキシメチルデキストラン鎖へのアミンカップリングにより固定した。固定化量は、約1000〜1300 RU程度であった。ランニングバッファーとしてはPBS−T(2.68 mM KCl / 137 mM NaCl / 1.47 mM KH2PO4 / 1m M Na2HPO4 / 0.005% Tween 20, pH7.4)を用いて、5〜100 nMに調製したサンプルを流速20 ul/minの条件下でインジェクトし結合能力を評価した。結合能力を評価したサンプルとしては、MMP−2非添加でのscFv−CM、MMP−2非添加でのscFv−CMのC末端に2、5、12、20、40 kDaのPEG−マレイミドを導入したサンプル、MMP−2添加後のscFv−CMのC末端に40 kDaのPEG−マレイミドを導入したサンプル、MMP−2を添加、非添加でのscFV−NM、MMP−2を添加、非添加でのscFv−NPM、MMP−2添加、非添加でのscFv−NMのN末端に5、20、40 kDaのPEG−アルデヒドを導入したサンプルについて結合能を評価した。図9には、作製したN末端PEG化scFv−NM、C末端PEG化scFv−CMのMMP−2添加、非添加での、HER2との相互作用をBiacore Xシステムを用いて測定して得られた結合速度定数konの対数を縦軸にPEGの分子量を横軸にとった結果を示す。図9に示すように、C末端に比べ、N末端にPEGを導入する方が、より大きくkonを低下させることが可能であり、MMP−2を添加することにより、konを大きく変化させることが可能であることを確認した。具体的には、N末端にPEG 5 kDaを導入したscFv−NMでは、C末端にPEG 12 kDaを導入したscFv−CMと同等のkonの値を示しており、C末端に比べN末端の方が、より低分子量のPEGで大きくkonを低下させることが可能であることを確認した。また、MMP−2添加によるkonの変化度はC末端にPEG 40 kDaを導入したscFv−CMでは約5倍程度であるのに対して、N末端にPEG 20 kDaを導入したscFv−NMでは約15倍であり、C末端に比べN末端の方が、より大きく結合能力を変化させることが可能であることを確認した。また、scFv−NMとscFv−NPMのMMP−2添加前後でのkonの変化についても比較したところ、ほぼ同等のkonの変化であることを確認した。
【技術分野】
【0001】
本発明は、化合物および前記化合物の製造方法に関する。
【背景技術】
【0002】
病変部位に存在する抗原(病変マーカー)を検出するために、病変マーカーに結合する抗体などの病変マーカー結合分子に、放射性核種、MRI(Magnetic Resonance Imaging)信号を発信する分子、超音波信号を発信する分子、蛍光信号を発信する分子などの物理的な信号を発生する分子(以下、信号発信分子と略すことがある)を標識した体内診断用造影剤の開発がなされている。このような化合物を体内へ投与し、その化合物の信号発信分子からの信号を体外から検出することにより、病変部位、及び病変マーカーを検出することが可能である。
【0003】
上記のような化合物を血中へ投与した場合、病変部位から漏れ出て血中に存在する病変マーカーや正常部位に存在する病変マーカーなど、病変部位以外に存在する病変マーカーとの結合により、病変部位へ到達する化合物の量が低下する場合がある。また、そのような場合は、病変部位以外に結合した化合物からの信号(バックグラウンド)の発生により、病変部位の高コントラストな検出が困難になると考えられる。
【0004】
上記問題を解決する化合物として、ローファーらは、特定の生物活性の存在下にて、インビボで生物活性化されるプロドラッグ造影剤を報告している(特許文献1)。この文献のプロドラッグ造影は信号発信分子を有する標的化分子に、酵素で開裂可能なリンカーを介して、標的化分子の結合能力を低下させる分子を結合したものである。この造影剤は、アルカリフォスファターゼによる反応を受けてリンカーが開裂し、その結果標的化分子として用いた低分子化合物のヒト血清アルブミンへの結合能力が開裂前と比較して向上する。また、リンカーの開裂により、信号発信分子として用いたMRI信号を発生する分子の信号が変化し、病変部位のより高コントラストな検出が可能になることもローファーらは報告している。
【0005】
抗体は抗原を特異的に認識する分子であり、病変部位の検出、あるいは治療に好適に用いられる。しかし、特許文献1は、低分子化合物を用いた造影剤の利用可能性を記載しているのみで、抗体を用いた造影剤の分子設計ならびに調製方法は開示されておらず、抗体を用いたインビボで生物活性化される造影剤の実用性については不明である。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特表2000‐507577号公報
【発明の概要】
【発明が解決しようとする課題】
【0007】
抗体を含む化合物を用いて、病変部位に存在する抗原(病変マーカー)抗体を検出等する際、病変部位以外に存在する抗原と抗体との結合により、病変部位へ到達する抗体化合物の量が低下し、また、病変部位以外で抗原に結合した抗体化合物からの信号(バックグラウンド)が発生する。その結果、高感度、高コントラストな病変部位の検出が困難であり、これを解消する化合物が求められている。
【課題を解決するための手段】
【0008】
第一の本発明に係る化合物は、一般式(1)で表されるポリマーからなる。
L−Y−A (1)
(式中、Aは一本鎖抗体部であって、抗原結合部位からなるポリペプチドである。Lはリンカー部であってプロテアーゼ切断部位からなるポリペプチドである。Yはペプチド部であって、一本鎖抗体部Aとリンカー部Lとを連結する0個以上のアミノ酸からなる。リンカー部Lはペプチド部YのN末端または一本鎖抗体部AのN末端に結合している)
【0009】
第二の本発明に係る化合物は、一般式(2)で表されるポリマーからなる化合物。
X−L−Y−A (2)
(式中、Aは一本鎖抗体部であって、抗原結合部位からなるポリペプチドである。Lはリンカー部であってプロテアーゼ切断部位からなるポリペプチドである。Yはペプチド部であって、一本鎖抗体部Aとリンカー部Lとを連結する0個以上のアミノ酸からなる。リンカー部Lはペプチド部YのN末端または一本鎖抗体部AのN末端に結合している。Xはポリエチレングリコール、有機色素、有機ポリマー、粒子のいずれかである。)
【0010】
第三の本発明に係る化合物の製造方法は、一般式(1)で表されるポリマーからなる化合物の製造方法であって、一本鎖抗体部Aをコードする塩基配列と、リンカー部Lをコードする塩基配列と、ペプチド部Yをコードする塩基配列と、を有する核酸をプラスミドに挿入する工程と、前記核酸が挿入されたプラスミドを菌に導入する工程と、前記プラスミドを導入した菌が発現した化合物を回収する工程と、を有することを特徴とする。
L−Y−A (1)
(式中、Aは一本鎖抗体部であって、抗原結合部位からなるポリペプチドである。Lはリンカー部であってプロテアーゼ切断部位からなるポリペプチドである。Yはペプチド部であって、一本鎖抗体部Aとリンカー部Lとを連結する0個以上のアミノ酸からなる。リンカー部Lはペプチド部YのN末端または一本鎖抗体部AのN末端に結合している。)
【発明の効果】
【0011】
本発明の化合物は、病変部位以外では、一本鎖抗体部の抗原との結合能力を低下させた状態であり、病変部位においては、抗原との結合能力を向上させた状態となることにより、病変部位以外に存在する抗原との結合による病変部位へ到達する一本鎖抗体部の量の低下と、病変部位以外で抗原に結合した一本鎖抗体部からの信号(バックグラウンド)を抑えることが可能である。これにより、高コントラストな病変部位の検出が可能となる。
【図面の簡単な説明】
【0012】
【図1】本発明で提供される化合物の模式図を示す。
【図2】一本鎖抗体部のN末端にリンカー部を介してポリエチレングリコール(PEG)を結合させた化合物の模式図を示す。
【図3】作製したscFv−NMとscFv−WTについて、MMP−2添加、非添加での還元型SDS−PAGEの結果を示す。
【図4】作製したscFv−NMのMMP−2添加前後における、HER2との相互作用をBiacore Xシステムを用いて測定した結果を示す。
【図5】作製したN末端PEG化scFv−NMのMMP−2添加前後での分子状態をゲルろ過クロマトグラフィーで検出した結果を示す。
【図6】2 kDa、5 kDa、12 kDa、20 kDa、40 kDaの PEG−マレイミドを反応させたscFv−CMの還元型SDS−PAGEの結果を示す。
【図7】作製したC末端PEG化scFv−CMにMMP−2を添加、非添加、及びMMP−2活性阻害剤存在下での還元型SDS−PAGEの結果を示す。
【図8】作製したscFv−WT、scFv−NM、scFv−NPMについて、MMP−2添加、非添加での還元型SDS−PAGEの結果を示す。
【図9】作製したN末端PEG化scFv−NM、C末端PEG化scFv−CMのMMP−2添加、非添加での、HER2との相互作用をBiacore Xシステムを用いて測定した結果を示す。
【発明を実施するための形態】
【0013】
本発明の実施形態について説明する。
【0014】
(第一の実施形態)
本発明の第一の実施形態に係る化合物は、一般式(1)で表されるポリマーからなる。
L−Y−A (1)
(式中、Aは一本鎖抗体部であって、抗原結合部位からなるポリペプチドである。Lはリンカー部であってプロテアーゼ切断部位からなるポリペプチドである。Yはペプチド部であって、一本鎖抗体部Aとリンカー部Lとを連結する0個以上のアミノ酸からなる。リンカー部Lはペプチド部YのN末端または一本鎖抗体部AのN末端に結合している)
【0015】
本実施形態に係る化合物は一本鎖抗体部とリンカー部とペプチド部とを有する。リンカー部はペプチド部のN末端に結合し、ペプチド部は一本鎖抗体部のN末端に結合している。または、リンカー部は一本鎖抗体部のN末端に直接結合している(ペプチド部が0個のアミノ酸からなる場合)。このような構成となっているため、一本鎖抗体部の抗原結合能力が阻害あるいは低減されている。そして、リンカー部がプロテアーゼによって切断されることにより、一本鎖抗体部の抗原結合能力が回復する。
本実施形態において、抗原結合能力が阻害あるいは低減される、とは、一本鎖抗体部が修飾等をされていない状態で抗原と結合する能力が、なんらかの要因により、失われ、あるいは、弱められることを指す。また、抗原結合能力が回復するとは、失われ、あるいは弱められた抗原結合能力が、向上し、修飾等をされていない状態の一本鎖抗体部と、同じ、または近い状態になることをいう。
【0016】
(一本鎖抗体部)
本発明において、一本鎖抗体部とは、抗体の重鎖可変(VH)領域と軽鎖可変(VL)領域とをペプチドからなるリンカーで連結した部位(抗原結合部位)からなるポリペプチドである。
ここで、抗体とは、ヒト、マウス、ラット、ラクダ、鳥、等、由来は問わず、さらにはキメラ体であってもよく、また、ポリクローナル抗体であってもモノクローナル抗体であってもよい。
【0017】
また上記抗体は限定なく用いられるが、本発明の目的に照らし合わせれば、例えば、EGFRファミリー、VEGFファミリー、VEGFRファミリー、PSA、CEA、マトリックスメタロプロテアーゼファミリー、EGFファミリー、インテグリンファミリー、セレクチンファミリー、エンドグリン、又はMUCファミリーからなる群より選ばれた病変マーカーに結合する抗体、さらにはHER2に結合する抗体を例としてあげることができる。
【0018】
一本鎖抗体部は、各種抗原に対応して安価に簡便に作製することができ、なおかつ、通常の抗体(全抗体など)と比べて分子量が小さいため、体外へ速やかに排泄されやすく、又病変部位に到達しやすい。そのため、一本鎖抗体部は病変部位の検出、又は治療に好適に用いられる。
【0019】
一本鎖抗体部としては、例えば以下のアミノ酸配列を含むポリペプチドからなる。
DIQMTQSPSSLSASVGDRVTITCRASQDVNTAVAWYQQKPGKAPKLLIYSASFLYSGVPSRFSGSRSGTDFTLTISSLQPEDFATYYCQQHYTTPPTFGQGTKVEIKGGGGSGGGGSGGGGSEVQLVESGGGLVQPGGSLRLSCAASGFNIKDTYIHWVRQAPGKGLEWVARIYPTNGYTRYADSVKGRFTISADTSKNTAYLQMNSLRAEDTAVYYCSRWGGDGFYAMDYWGQGTLVTVSSAAALEHHHHHHGGC(配列番号1)
なお、一本鎖抗体部のC末端には、任意のアミノ酸が結合していてもよい。
【0020】
(リンカー部)
本発明において、リンカー部とは、プロテアーゼ切断部位からなるポリペプチドである。ここで、プロテアーゼ切断部位とは、プロテアーゼに特異的に認識されるアミノ酸配列を有しており、そのアミノ酸配列が活性状態のプロテアーゼに認識され、特異的に加水分解されて切断される部位を意味する。
プロテアーゼとしては、特に限定はなく用いられるが、マトリックスメタロプロテアーゼファミリーまたはセリンプロテアーゼファミリーが好ましく、例えば、マトリックスメタロプロテアーゼー2(MMP−2)、マトリックスメタロプロテアーゼー9(MMP−9)、前立腺特異的抗原(PSA)、プラスミン、カテプシン、カスパーゼなどが標的プロテアーゼとして挙げられる。
プロテアーゼ切断部位については、それぞれのプロテアーゼが特異的に認識するアミノ酸配列であり、たとえば、MMP−2の切断部位としてアミノ酸配列PLGVR(配列番号2)を含むポリペプチドなどを利用できる。また、PSAの切断部位としてアミノ酸配列SSIYSQTEEQ(配列番号3)を含むポリペプチドなどを利用することができる。
リンカー部のN末端には任意のアミノ酸が結合していてもよい。
【0021】
(ペプチド部)
本発明において、ペプチド部とは、一本鎖抗体部とリンカー部とを連結する0個以上のアミノ酸からなる。そのアミノ酸の数は20個以下であることが好ましく、10個以下であることがさらに好ましい。アミノ酸の数が20個以上であると、そのアミノ酸配列が立体構造をとる可能性がある。その結果、プロテアーゼ切断部位がプロテアーゼによって切断された場合でも、一本鎖抗体部の抗原結合能力が阻害あるいは低減されたままとなってしまうおそれがあるためである。
【0022】
図1を用いて本実施形態をより詳細に説明する。図1は、本実施形態に係る化合物の模式図である。ただし、図1ではペプチド部は図示していない。
抗原結合能力を阻害あるいは低減する機構については、種々の機構が考えられる。第一に、ペプチド部または一本鎖抗体部のN末端に結合したリンカー部が一本鎖抗体部の抗原結合部位と抗原の間に存在することにより立体的障害を生じさせることが考えられる。この場合、リンカー部が一本鎖抗体部の抗原結合部位を覆うため、他の抗体が抗原に結合できなくなることなどが考えられる。
【0023】
リンカー部を一本鎖抗体部のN末端に結合させている理由は以下のとおりである。まず第一に、一本鎖抗体部における重鎖可変(VH)領域と軽鎖可変(VL)領域とを連結するリンカーにリンカー部を結合させると、一本鎖抗体部の構造自体が変わり、リンカー部切断後にも構造が戻らず、結果として、リンカー部切断後も抗原結合能力が回復しないおそれがある。一方、一本鎖抗体部のいずれかの末端にリンカー部を結合させた場合、構造自体には影響が少なく、リンカー部が切断された後に一本鎖抗体部がリンカー部を導入する前の構造に戻りやすく、リンカー部を切断後に抗原結合能力が回復しやすいと考えられる。そのため、まず、一本鎖抗体部のいずれかの末端にリンカー部を導入すれば、リンカー部が切断された後の抗原結合能力がリンカー部の導入前と同等までに回復することが可能であるという点で有効であると考えられる。次に、一本鎖抗体部のN末端はC末端よりも、抗原結合部位に近い。そのため、一本鎖抗体部のN末端にリンカー部を結合する場合は、C末端に結合するよりも、結合するリンカー部の長さが短くても、一本鎖抗体部の抗原結合部位への立体的障害作用などの効果をより効率よく生じさせられると考えられ、すなわち、より、強く抗原結合能力を低下させることが可能である。これにより、化合物の分子量を過剰に増大させることなく、抗原結合能力を阻害あるいは低減することが可能になると考えられる。化合物の分子量を過剰に増大させることを抑えることは、体外へ速やかに排泄されやすく、又病変部位に到達しやすいという一本鎖抗体部のみの性質に近い性質を化合物が維持できるため、有効である。さらに、一本鎖抗体部のN末端は抗原結合部位と近接しているため、一本鎖抗体部のN末端にリンカー部を導入すれば、C末端に導入するのに比べ抗原結合能力をより強く低下させることが可能であるという点で有効である。また、一本鎖抗体部のN末端にリンカー部を結合させることで、抗原結合部位から離れたC末端には他の分子を結合することができ、例えば、信号発信分子や治療用薬剤などを、抗原結合能力を低下させることなく結合させることが可能である。以上より、一本鎖抗体部のN末端にリンカー部を結合させることが最も有効であると考えられる。
以上のことは、リンカー部がペプチド部のN末端に結合し、ペプチド部が一本鎖抗体部のN末端に結合している場合についても同様のことが言える。
【0024】
(信号発信分子)
本実施形態に係る化合物には、一本鎖抗体部に信号発信分子を結合させることが可能である。
信号発信分子として、放射性核種、MRI信号を発信する分子、超音波信号を発信する分子、蛍光信号を発信する分子などの種々の分子を用いることができる。例えば、光を吸収し超音波信号や発光信号を発する信号発信分子としては、Alexa Fluor 680、Alaxa Fluor 700、Alexa Fluor 750、Alexa Fluor 790(以上商標、インビトロジェン株式会社)、Cy5、Cy5.5、Cy7(以上商標、GEヘルスケア)、HiLyte 647、HiLyte 680、HiLyte 750(以上商標、AnaSpec, Inc)、DY−680、DY−700、DY−730、DY−750、DY−782 (以上商標、Dyomics, Jena, Germany)などを利用することが可能である。また、光を吸収し超音波信号や発光信号を発する分子としては、生体透過性の優れた約600 nm以上約1000 nm以下の近赤外波長領域の光を吸収する近赤外光吸収色素を選択することが有効である。その他、微粒子としては、酸化鉄微粒子、金微粒子、金ナノロッド、白金、銀などの金属や金属酸化物の微粒子など種々の微粒子を利用することが可能であろう。また、本実施形態の一本鎖抗体部に抗癌剤などの治療用薬剤を結合し、病変部位へ治療用薬剤を送達することも可能であり、病変部位以外への治療用薬剤の送達を低減することによる副作用の低減が期待できる。
【0025】
また、本発明の化合物のうち、信号発信分子が結合したものは、病変マーカーの検出に用いることが可能である。すなわち、本発明は病変マーカーの検出方法において、信号発信分子が結合した本発明の化合物を個体に投与、又は個体より得られた試料に添加し、信号を検出することにより、該個体中もしくは個体より得られた試料中の病変マーカーの存否、又は病変マーカーを産生する病変部位の存否を検出する方法を提供する。病変マーカーの検出方法は以下の通りである。すなわち、信号発信分子が結合した本発明の化合物を個体に投与し、あるいは個体より得られた臓器等の試料に添加する。なお、個体とは、ヒト、実験動物やペット等の哺乳類、その他、特に限定されることなく、あらゆる生物を指し、個体中もしくは個体より得られた試料としては、臓器、組織、組織切片などを挙げることができる。投与あるいは添加後、信号発信分子を検出できる測定機器等を用いて、個体あるいは試料に含まれる信号発信分子の信号を検出する。その結果、信号が基準とする閾値以上で検出されれば、その個体に病変マーカー、あるいは、病変マーカーを産生する病変部位が存在すると推定され、または、試料に病変マーカーが存在する、あるいは、試料の由来となる個体に病変マーカーを産生する病変部位が存在すると推定することができる。
【0026】
(イメージング)
また、同様に、本発明の化合物のうち、信号発信分子が結合したものは、病変マーカーのイメージングに用いることが可能である。病変マーカーのイメージング方法は、以下の通りである。すなわち、本発明の化合物を個体に投与し、又は個体より得られた試料に添加し、信号を検出することにより、該個体中もしくは個体より得られた試料中の病変マーカーの存否、又は病変マーカーを産生する病変部位の存否をイメージングすることができる。
【0027】
(第二の実施形態)
以下、本発明の第二の実施形態に係る化合物の説明をするが、第一の実施形態と共通のことに関しては説明を省略する。
本発明の第二の実施形態に係る化合物は、一般式(2)で表されるポリマーからなる化合物。
X−L−Y−A (2)
(式中、Aは一本鎖抗体部であって、抗原結合部位からなるポリペプチドである。Lはリンカー部であってプロテアーゼ切断部位からなるポリペプチドである。Yはペプチド部であって、一本鎖抗体部Aとリンカー部Lとを連結する0個以上のアミノ酸からなる。リンカー部Lはペプチド部YのN末端または一本鎖抗体部AのN末端に結合している。Xはポリエチレングリコール、有機色素、有機ポリマー、粒子のいずれかである)
【0028】
本実施形態に係る化合物は、一本鎖抗体部と、リンカー部と、ペプチド部と、一本鎖抗体部の抗原結合部位を覆うことができる部位(一般式(2)のXに相当する)とを有する。リンカー部はペプチド部のN末端に結合し、ペプチド部は、一本鎖抗体部のN末端に結合している。または、リンカー部は一本鎖抗体部のN末端に直接結合している(ペプチド部が0個のアミノ酸からなる場合)。このような構成となっているため、一本鎖抗体部の抗原結合能力が阻害あるいは低減されている。そして、リンカ部が切断されることにより、前記一本鎖抗体部の抗原結合能力が回復する。
【0029】
抗原結合部位を覆うことができる部位としては、ポリエチレングリコール(以下、PEGと略すことがある)、有機色素、有機ポリマー、粒子のいずれかである。有機色素としては、Alexa Fluor 680、Alaxa Fluor 700、Alexa Fluor 750、Alexa Fluor 790(以上商標、インビトロジェン株式会社)、Cy5、Cy5.5、Cy7(以上商標、GEヘルスケア)、HiLyte 647、HiLyte 680、HiLyte 750(以上商標、AnaSpec, Inc)、DY−680、DY−700、DY−730、DY−750、DY−782 (以上商標、Dyomics, Jena, Germany)などを利用することが可能である。有機ポリマーとしては、ポリ乳酸(Poly lactic acid、PLA)、ポリ乳酸グリコール酸共重合体(Poly (DL−lactic−co−glycolic acid)、PLGA)などを利用することが可能である。
【0030】
図2にPEGを導入した場合の模式図を示す。ただし、図2ではペプチド部は図示していない。これらの分子を用いる場合であっても、リンカー部及び連結された抗原結合部位を覆うことができる部位は一本鎖抗体部のN末端に結合されていることが効果的と考えられる。例えば、PEGは抗原結合部位を覆うことができる部位として特に好ましく用いられるが、PEGは水溶液中で、立体的に広がりを持った状態で存在すると考えられる。そのため、一本鎖抗体部のN末端にリンカー部及び連結されたPEGを導入した場合、一本鎖抗体部の抗原結合部位と抗原の間にPEGが存在するため、例えば、C末端などのN末端部位以外に導入した場合と比較して、より大きな立体障害となり、抗原結合能力を阻害あるいは低減する作用が大きくなると考えられ、一本鎖抗体部の抗原結合能力をより強く低下させることが可能である。
【0031】
PEGの導入は、抗原結合能力を阻害等することに加えて、種々の効果が期待できる。例えば、PEGを導入することによる化合物の分子量の調節や化合物のタンパク質吸着能の調節が挙げられる。PEGを導入した化合物は病変部位へ到達する前の血中ではリンカー部が切断されずPEGが遊離しないため、化合物の分子量が大きく保たれ、かつ化合物のタンパク質吸着能が抑えられる。それにより、腎臓から排泄させる大きさより化合物の大きさを大きく保つことができ、腎臓からの排泄による化合物の損失を抑えることができる。また、タンパク質が化合物に吸着することに伴い働く化合物の体外への排泄による化合物の損失を抑えることができる。一方、病変部位では、リンカー部が切断され、PEGが遊離することにより、化合物の分子量が減少する。それにより、又病変部位では、それ以外の部位と比較して、化合物の浸透性が向上し、病変深部にまで化合物が浸透することで、化合物の病変部位への集積性を向上させることが可能であろう。それに対して、正常部位においては、リンカー部が切断されずPEGが遊離しないため、化合物は正常部位の深部にまで浸透しない。そのため、正常部位への化合物の集積により発生する信号(バックグラウンド)を低減させることが可能であろう。
PEGの分子量は20kDa以上であることが好ましい。また、PEGの分子量が40kDa以下であることが好ましい。
【0032】
リンカー部を切断する酵素と一本鎖抗体部が結合する病変マーカーが同一の場合、該酵素の存在とその活性を同時に検出することが可能であろう。また、リンカー部を切断する酵素が病変部位特異的であり、かつその酵素が、一本鎖抗体部が結合する病変マーカーと異なる場合には、病変部位の二つの病変マーカーが存在する場合にのみ、病変部位へ集積するため、位置精度を向上させること、および詳細な病変部位の情報を検出することが可能であろう。例えば、HER2に結合する一本鎖抗体部とMMP−2の切断部位を含むリンカー部を組み合わせた場合、癌の予後不良因子としてのHER2陽性であるということと、転移、浸潤などの性質を示すという指標としてのMMP−2活性が高いということの二つの性状を同時に示す腫瘍、つまり悪性度の高いと考えられる腫瘍を高い位置精度で検出することが可能になるであろう。また、この場合、血中等に存在する可溶性HER2やHER2陽性/MMP−2陰性の腫瘍部位は当該造影剤では検出されにくいため、バックグラウンドを低減させる事が可能になるであろう。また、血管内皮増殖因子VEGFに結合する一本鎖抗体部とMMP−2の切断部位を含むリンカー部を組み合わせた場合、血中へ漏れ出たVEGFには結合し難く、活性化したMMP−2が存在する病変部位で、高濃度に存在するVEGFに結合し易くなることで、VEGFを高濃度に存在する血管新生の盛んな病変部位を高い位置精度で検出することが可能になるであろう。
【0033】
(第三の実施形態)
本発明の第三の実施形態について説明する。本実施形態は、上記一般式(1)で表されるポリマーからなる化合物の製造方法に関する。具体的には以下の工程を有する。
(i)一本鎖抗体部Aをコードする塩基配列と、リンカー部Lをコードする塩基配列と、ペプチド部Yをコードする塩基配列と、を有する核酸をプラスミドに挿入する工程
(ii)前記核酸が挿入されたプラスミドを菌に導入する工程
(iii)前記プラスミドを導入した菌が発現した化合物を回収する工程
なお、本実施形態に係る化合物の製造方法は上記(i)から(iii)以外の工程を含んでいてもよい。
【実施例】
【0034】
以下に、本発明の特徴をさらに明らかにするために実施例に沿って本発明を説明するが、本発明に用いる一本鎖抗体部、リンカー部、抗原結合部位を覆うことができる部位の組合せは他の組合せを用いることも可能であり、本発明はこの実施例によって制限されるものではない。
【0035】
(実施例1 hu4D5−8 scFvの調製)
始めに、HER2へ結合するIgGの可変領域の遺伝子配列を基に、一本鎖抗体(scFv)部をコードする遺伝子断片を作製した。作製した遺伝子は発現産物のN末端に、MMP−2により特異的に切断されるアミノ酸配列PLGVR、C末端にはタンパク精製のためのヒスチジンが6残基連続した6 x Hisタグ、またそのさらに下流にスペーサーとしてグリシンを2残基挟み信号発信分子を導入するためのシステインが配置される様に設計した。比較として、N末端にMMP−2により特異的に切断されるアミノ酸配列PLGVRを含まない遺伝子についても同様に作製した。上記遺伝子断片をT7プロモーターの下流に挿入したプラスミドpET−22b (+)(Novagen社)を大腸菌(Escherichia coli BL21 (DE3))に導入し、発現用菌株を得た。得られた菌株をLB−Amp培地4 mlで一晩前培養後、全量を250 mlの2 x YT培地に添加し、28℃、120 rpmで8 時間振とう培養した。その後、終濃度1 mMでIPTGを添加し、28℃で一晩培養した。培養した大腸菌を8000x g、30 分、4℃で遠心分離し、その上清の培養液を回収した。得られた培養液の60%重量の硫酸アンモニウムを添加し、塩析によりタンパク質を沈殿させた。塩析操作した溶液を一晩4℃で静置後、8000xg、30 min、4℃で遠心分離することで沈殿物を回収した。得られた沈殿物を20 mM Tris・HCl / 500 mM NaClバッファーに溶解し、1 Lの同バッファーへ透析した。透析後のタンパク質溶液を、His・Bind(登録商標) Resin(Novagen社)を充填したカラムへ添加し、Niイオンを介した金属キレートアフィニティークロマトグラフィーによって精製した。精製したN末端MMP−2基質導入hu4D5−8 scFv (scFv−NM)と、N末端MMP−2基質未導入hu4D5−8 scFv (scFv−WT)は、SDS−PAGEによりシングルバンドを示し分子量は約28 kDaであることを確認した。
【0036】
(実施例2 scFv−NMのMMP−2添加によるリンカー部の切断の確認)
上記で調製したscFv−NMにMMP−2を添加することにより、ペプチドが切断されて生じる分子量の変化を測定した。TCNB buffer(50 mM Tris, 10 mM CaCl2, 150 mM NaCl, 0.05% Brij35, pH7.5)に透析したPEG化scFv 約4 μMに、活性型MMP−2(コスモ・バイオ株式会社)0.10 mg/mlを約15 nM添加し、約20時間反応を行い、分子量の変化を還元型SDS−PAGEにより測定した。その結果、図3に示すように、MMP−2を添加することにより、scFv−NMでは分子量の低下が検出された。一方、scFv−WTでは分子量の低下は検出されなかった。以上より、scFv−NMのペプチドがMMP−2により切断されることを確認した。
【0037】
(実施例3 scFv−NMのMMP−2添加による抗原結合能力の向上の確認)
上記で作製したscFv−NM、scFv−WTと抗原であるHER2との相互作用をBiacore Xシステム(GEヘルスケア株式会社)を用いて測定し、MMP−2添加前と添加後でのscFvの結合能力の変化を測定した。抗原としては、Recombinant Human ErbB2 / Fc Chimera(R&D Systems,Inc.)を用いて、メーカーの推奨に従って、CM−5チップ表面のカルボキシメチルデキストラン鎖へのアミンカップリングにより固定した。固定化量は、約1000 RUであった。ランニングバッファーとしてはPBS−T(2.68 mM KCl / 137 mM NaCl / 1.47 mM KH2PO4 / 1mM Na2HPO4 / 0.005% Tween 20, pH7.4)を用いて、50 nMのscFvを流速20 μl/minの条件下でインジェクトし結合能力を評価した。その結果、図4に示すように、MMP−2添加前後でセンサーグラムの変化を確認でき、scFv−NMの抗原結合能力が向上することがわかった。一方、scFv−WTの抗原結合能力は、MMP−2添加前後ではほとんど変化しないことを確認した。
【0038】
(実施例4 N末端PEG化scFv−NMの作製)
実施例1で調製したscFv−NMを100 mMリン酸バッファー(pH 5.0)にバッファー置換後、scFv−NMに対して40倍モル量の5 kDa、20 kDa、又は40 kDaの PEG−アルデヒド(日油株式会社)を添加し、終濃度20 mMで2−ピコリンボランを添加し、4℃で約3日間反応させた。反応後、TCNB buffer(50 mM Tris, 10 mM CaCl2, 150 mM NaCl, 0.05% Brij35, pH7.5)にバッファー交換し、 Superdex 200 GL 10/300カラム(GEヘルスケア株式会社)を用いてゲルろ過クロマトグラフィーにより、PEGが1分子結合したと考えられる画分を分取することで、N末端にPEG化されたと考えられるscFv−NMを得た。
【0039】
(実施例5 N末端PEG化scFv−NMのMMP−2添加によるPEGの遊離の確認)
上記で調製したN末端がPEG化されたと考えられるscFv−NMについて、MMP−2を添加することにより、分子量の変化を評価した。TCNB bufferに溶解したN末端にPEG化されたと考えられるscFv−NMに、活性型MMP−2(コスモ・バイオ株式会社)0.10 mg/mlを終濃度約10 nM添加し、約20 h反応を行い、 Superdex 200 GL 10/300カラムを用いてゲルろ過クロマトグラフィーにより、分子量の変化を評価した。20 kDaのPEGを修飾したscFv−NMのMMP−2添加前後でのゲルろ過クロマトグラフィー(280 nmの吸収をモニタリング)の結果を図5に示す。図5に示すようにMMP−2を添加することにより、切断前にはなかった2つのピークが検出された。これらは、PEG未修飾のscFv−NMのモノマーとダイマーと考えられる。すなわち、この結果はN末端に導入したMMP−2基質をMMP−2が切断することにより、N末端に修飾されたPEGがscFv−NMより遊離したことを示している。つまり、N末端にPEGが修飾されていることを確認できた。また、MMP−2の添加により現れた、PEGが遊離したscFv−NMに相当すると考えられるピーク面積と、全体のピーク面積の割合を比較した結果、 PEG化scFv−NM全体のうちN末端がPEG化されていた割合は約50%程度であった。残りの50%はscFv−NM分子内のリジン残基のアミノ基に対してPEGが修飾されていたと考えられる。また、5 kDa、40 kDaのPEG−アルデヒドを用いた場合も同様に約50%程度がN末端にPEG化されていることを確認した。以上より、N末端PEG化scFv−NMの取得を確認した。
【0040】
(実施例6 C末端にMMP−2基質を導入したscFvの作製)
HER2へ結合するIgGの可変領域の遺伝子配列を基に、一本鎖抗体(scFv)部をコードする遺伝子断片を作製した。作製した遺伝子のカルボキシ末端には、MMP−2により特異的に切断されるアミノ酸配列PLGVR、抗原結合能を低下させる分子を連結するためのシステイン残基、及びタンパク精製のためのヒスチジンが6残基連続した6 x His タグを配置した。上記遺伝子断片をT7プロモーターの下流に挿入したプラスミドpET−22b (+)(Novagen社)を大腸菌(Escherichia coli BL21 (DE3))に導入し、発現用菌株を得た。得られた菌株をLB−Amp培地4 mlで一晩前培養後、全量を250 mlの2 x YT培地に添加し、28℃、120 rpmで8時間振とう培養した。その後、終濃度1 mMでIPTGを添加し、28℃で一晩培養した。培養した大腸菌を8000 x g、30 min、4℃で遠心分離し、その上清の培養液を回収した。得られた培養液の60%重量の硫酸アンモニウムを添加し、塩析によりタンパク質を沈殿させた。塩析操作した溶液を一晩4℃で静置後、8000xg、30 min、4℃で遠心分離することで沈殿物を回収した。得られた沈殿物を20 mM Tris・HCl / 500 mM NaClバッファーに溶解し、1 Lの同バッファーへ透析した。透析後のタンパク質溶液を、His・Bind (登録商標)Resin(Novagen社)を充填したカラムへ添加し、Niイオンを介した金属キレートアフィニティークロマトグラフィーによって精製した。精製したC末端MMP−2基質導入scFv(scFv−CM)は、SDS−PAGEによりシングルバンドを示し分子量は約28 kDaであることを確認した。
【0041】
(実施例7 C末端PEG化scFv−CMの作製)
上記で調製したscFv−CMを5 mM EDTAを含むリン酸バッファー(2.68 mM KCl / 137 mM NaCl / 1.47 mM KH2PO4 / 1m M Na2HPO4 / 5 mM EDTA, pH7.4)にバッファー置換後、10〜20倍モル量のトリ(2−カルボキシエチル)ホスフィン塩酸塩(TCEP)によって、25℃で2〜4時間、還元処理した。この還元処理したscFv−CMを、25℃で2〜4時間、10倍モル量の2 kDa、5 kDa、12 kDa、20 kDa、又は40 kDaの PEG−マレイミド(日油株式会社)と反応させた。PEG化されていることは、還元型SDS−PAGEにより確認した(図6)。また、反応後、Superdex 200 GL 10/300カラムを用いてゲルろ過クロマトグラフィーにより、未反応のPEG−マレイミドを除去し、C末端PEG化scFv−CMを得た。
【0042】
(実施例8 C末端PEG化scFv−CMのMMP−2添加によるPEGの遊離の確認)
上記で調製したC末端PEG化scFv−CMのうち、20 kDa PEG−マレイミドを付加したものについて、MMP−2を添加することにより、分子量の変化を評価した。TCNB bufferに透析したC末端PEG化scFv−CMに、活性型MMP−2 (0.10 mg/ml)を終濃度約10 nM添加し、2時間、20時間と反応を行い、各時間での分子量の変化を還元型SDS−PAGEにより測定した。また、MMP−2活性阻害剤である1, 10−Phenanthroline(Sigma−aldrich)を1 mMで添加し、同様に20時間反応させた実験について行った。その結果、図7に示すように、MMP−2添加2時間でscFv−CMの分子量である28 kDa付近にバンドが検出され、20 時間でほぼscFv−CMがメインバンドとして検出された。また、このscFv−CMのバンドの検出は、1, 10−Phenanthroline添加により抑えられた。以上より、C末端PEG化scFv−CMのPEGがMMP−2特異的に切断されることにより、scFv−CMより遊離することを確認した。
【0043】
(実施例9 N末端にペプチドとMMP−2基質を導入したscFvの作製)
始めに、HER2へ結合するIgGの可変領域の遺伝子配列を基に、一本鎖抗体(scFv)部をコードする遺伝子断片を作製した。作製した遺伝子のN末端には、ペプチドGGSGGGS、その下流に
MMP−2により特異的に切断されるアミノ酸配列PLGVR、C末端にはタンパク精製のためのヒスチジンが6残基連続した6 x Hisタグ、またそのさらに下流にスペーサーとしてグリシンを2残基挟み信号発信分子を導入するためのシステインを配置した。上記遺伝子断片をT7プロモーターの下流に挿入したプラスミドpET−22b (+)(Novagen社)を大腸菌(Escherichia coli BL21 (DE3))に導入し、発現用菌株を得た。得られた菌株をLB−Amp培地4 mlで一晩前培養後、全量を250 mlの2 x YT培地に添加し、28℃、120 rpmで8 時間振とう培養した。その後、終濃度1 mMでIPTGを添加し、28℃で一晩培養した。培養した大腸菌を8000 x g、30 分、4℃で遠心分離し、その上清の培養液を回収した。得られた培養液の60%重量の硫酸アンモニウムを添加し、塩析によりタンパク質を沈殿させた。塩析操作した溶液を一晩4℃で静置後、8000 x g、30 min、4℃で遠心分離することで沈殿物を回収した。得られた沈殿物を20 mM Tris・HCl / 500 mM NaClバッファーに溶解し、1 Lの同バッファーへ透析した。透析後のタンパク質溶液を、His・Bind(商標) Resin(Novagen社)を充填したカラムへ添加し、Niイオンを介した金属キレートアフィニティークロマトグラフィーによって精製した。精製したN末端ペプチドとMMP−2基質を導入したhu4D5−8 scFv (scFv−NPM)は、SDS−PAGEによりシングルバンドを示し分子量は約28 kDaであることを確認した。
【0044】
(実施例10 scFv−NPMのMMP−2添加によるリンカー部の切断の確認)
上記で調製したscFv−NPMにMMP−2を添加することにより、ペプチドが切断されて生じる分子量の変化を測定した。TCNB bufferに透析したPEG化scFv 約4 μMに、活性型MMP−2 0.10 mg/mlを約10 nM添加し、約20時間反応を行い、分子量の変化を還元型SDS−PAGEにより測定した。その結果、図8に示すように、MMP−2を添加することにより、scFv−NPMでは分子量の低下が検出された。また、scFv−NM、scFv−CMも比較として示した。一方、scFv−WTでは分子量の低下は検出されなかった。以上より、scFv−NPMのペプチドがMMP−2により切断されることを確認した。
【0045】
(実施例11 scFvのMMP−2添加による結合能力の変化の確認)
上記で作製した種々のscFvと抗原であるHER2との相互作用をBiacore Xシステム(GEヘルスケア株式会社)を用いて測定し、MMP−2添加前と添加後20時間での種々のscFvの結合能力の変化を測定した。抗原としては、Recombinant Human ErbB2 / Fc Chimera(R&D Systems, Inc.)を用いて、メーカーの推奨に従って、CM−5チップ表面のカルボキシメチルデキストラン鎖へのアミンカップリングにより固定した。固定化量は、約1000〜1300 RU程度であった。ランニングバッファーとしてはPBS−T(2.68 mM KCl / 137 mM NaCl / 1.47 mM KH2PO4 / 1m M Na2HPO4 / 0.005% Tween 20, pH7.4)を用いて、5〜100 nMに調製したサンプルを流速20 ul/minの条件下でインジェクトし結合能力を評価した。結合能力を評価したサンプルとしては、MMP−2非添加でのscFv−CM、MMP−2非添加でのscFv−CMのC末端に2、5、12、20、40 kDaのPEG−マレイミドを導入したサンプル、MMP−2添加後のscFv−CMのC末端に40 kDaのPEG−マレイミドを導入したサンプル、MMP−2を添加、非添加でのscFV−NM、MMP−2を添加、非添加でのscFv−NPM、MMP−2添加、非添加でのscFv−NMのN末端に5、20、40 kDaのPEG−アルデヒドを導入したサンプルについて結合能を評価した。図9には、作製したN末端PEG化scFv−NM、C末端PEG化scFv−CMのMMP−2添加、非添加での、HER2との相互作用をBiacore Xシステムを用いて測定して得られた結合速度定数konの対数を縦軸にPEGの分子量を横軸にとった結果を示す。図9に示すように、C末端に比べ、N末端にPEGを導入する方が、より大きくkonを低下させることが可能であり、MMP−2を添加することにより、konを大きく変化させることが可能であることを確認した。具体的には、N末端にPEG 5 kDaを導入したscFv−NMでは、C末端にPEG 12 kDaを導入したscFv−CMと同等のkonの値を示しており、C末端に比べN末端の方が、より低分子量のPEGで大きくkonを低下させることが可能であることを確認した。また、MMP−2添加によるkonの変化度はC末端にPEG 40 kDaを導入したscFv−CMでは約5倍程度であるのに対して、N末端にPEG 20 kDaを導入したscFv−NMでは約15倍であり、C末端に比べN末端の方が、より大きく結合能力を変化させることが可能であることを確認した。また、scFv−NMとscFv−NPMのMMP−2添加前後でのkonの変化についても比較したところ、ほぼ同等のkonの変化であることを確認した。
【特許請求の範囲】
【請求項1】
一般式(1)で表されるポリマーからなる化合物。
L−Y−A (1)
(式中、Aは一本鎖抗体部であって、抗原結合部位からなるポリペプチドである。Lはリンカー部であってプロテアーゼ切断部位からなるポリペプチドである。Yはペプチド部であって、一本鎖抗体部Aとリンカー部Lとを連結する0個以上のアミノ酸からなる。リンカー部Lはペプチド部YのN末端または一本鎖抗体部AのN末端に結合している)
【請求項2】
一般式(2)で表されるポリマーからなる化合物。
X−L−Y−A (2)
(式中、Aは一本鎖抗体部であって、抗原結合部位からなるポリペプチドである。Lはリンカー部であってプロテアーゼ切断部位からなるポリペプチドである。Yはペプチド部であって、一本鎖抗体部Aとリンカー部Lとを連結する0個以上のアミノ酸からなる。リンカー部Lはペプチド部YのN末端または一本鎖抗体部AのN末端に結合している。Xはポリエチレングリコール、有機色素、有機ポリマー、粒子のいずれかである)
【請求項3】
前記ポリエチレングリコールの分子量が20kDa以上であることを特徴とする請求項2に記載の化合物。
【請求項4】
前記ポリエチレングリコールの分子量が40kDa以下であることを特徴とする請求項2に記載の化合物。
【請求項5】
前記リンカー部が、配列番号2または配列番号3のいずれか一方のアミノ酸配列からなるポリペプチドであることを特徴とする請求項1乃至4のいずれか一項に記載の化合物。
【請求項6】
前記一本鎖抗体部が、配列番号1のアミノ酸配列からなるポリペプチドであることを特徴とする請求項1乃至5のいずれか一項に記載の化合物。
【請求項7】
前記一本鎖抗体部に信号発信分子が結合していることを特徴とする請求項1乃至6のいずれか一項に記載の化合物。
【請求項8】
前記信号発信分子が酸化鉄微粒子であることを特徴とする請求項7に記載の化合物。
【請求項9】
病変マーカーの検出方法において、請求項7又は8に記載の化合物を個体に投与、又は個体より得られた試料に添加し、信号を検出することにより、該個体中もしくは個体より得られた試料中の病変マーカーの存否、又は病変マーカーを産生する病変部位の存否を検出する方法。
【請求項10】
一般式(1)で表されるポリマーからなる化合物の製造方法であって、
一本鎖抗体部Aをコードする塩基配列と、リンカー部Lをコードする塩基配列と、ペプチド部Yをコードする塩基配列と、を有する核酸をプラスミドに挿入する工程と、
前記核酸が挿入されたプラスミドを菌に導入する工程と、
前記プラスミドを導入した菌が発現した化合物を回収する工程と、
を有することを特徴とする製造方法。
L−Y−A (1)
(式中、Aは一本鎖抗体部であって、抗原結合部位からなるポリペプチドである。Lはリンカー部であってプロテアーゼ切断部位からなるポリペプチドである。Yはペプチド部であって、一本鎖抗体部Aとリンカー部Lとを連結する0個以上のアミノ酸からなる。リンカー部Lはペプチド部YのN末端または一本鎖抗体部AのN末端に結合している)
【請求項1】
一般式(1)で表されるポリマーからなる化合物。
L−Y−A (1)
(式中、Aは一本鎖抗体部であって、抗原結合部位からなるポリペプチドである。Lはリンカー部であってプロテアーゼ切断部位からなるポリペプチドである。Yはペプチド部であって、一本鎖抗体部Aとリンカー部Lとを連結する0個以上のアミノ酸からなる。リンカー部Lはペプチド部YのN末端または一本鎖抗体部AのN末端に結合している)
【請求項2】
一般式(2)で表されるポリマーからなる化合物。
X−L−Y−A (2)
(式中、Aは一本鎖抗体部であって、抗原結合部位からなるポリペプチドである。Lはリンカー部であってプロテアーゼ切断部位からなるポリペプチドである。Yはペプチド部であって、一本鎖抗体部Aとリンカー部Lとを連結する0個以上のアミノ酸からなる。リンカー部Lはペプチド部YのN末端または一本鎖抗体部AのN末端に結合している。Xはポリエチレングリコール、有機色素、有機ポリマー、粒子のいずれかである)
【請求項3】
前記ポリエチレングリコールの分子量が20kDa以上であることを特徴とする請求項2に記載の化合物。
【請求項4】
前記ポリエチレングリコールの分子量が40kDa以下であることを特徴とする請求項2に記載の化合物。
【請求項5】
前記リンカー部が、配列番号2または配列番号3のいずれか一方のアミノ酸配列からなるポリペプチドであることを特徴とする請求項1乃至4のいずれか一項に記載の化合物。
【請求項6】
前記一本鎖抗体部が、配列番号1のアミノ酸配列からなるポリペプチドであることを特徴とする請求項1乃至5のいずれか一項に記載の化合物。
【請求項7】
前記一本鎖抗体部に信号発信分子が結合していることを特徴とする請求項1乃至6のいずれか一項に記載の化合物。
【請求項8】
前記信号発信分子が酸化鉄微粒子であることを特徴とする請求項7に記載の化合物。
【請求項9】
病変マーカーの検出方法において、請求項7又は8に記載の化合物を個体に投与、又は個体より得られた試料に添加し、信号を検出することにより、該個体中もしくは個体より得られた試料中の病変マーカーの存否、又は病変マーカーを産生する病変部位の存否を検出する方法。
【請求項10】
一般式(1)で表されるポリマーからなる化合物の製造方法であって、
一本鎖抗体部Aをコードする塩基配列と、リンカー部Lをコードする塩基配列と、ペプチド部Yをコードする塩基配列と、を有する核酸をプラスミドに挿入する工程と、
前記核酸が挿入されたプラスミドを菌に導入する工程と、
前記プラスミドを導入した菌が発現した化合物を回収する工程と、
を有することを特徴とする製造方法。
L−Y−A (1)
(式中、Aは一本鎖抗体部であって、抗原結合部位からなるポリペプチドである。Lはリンカー部であってプロテアーゼ切断部位からなるポリペプチドである。Yはペプチド部であって、一本鎖抗体部Aとリンカー部Lとを連結する0個以上のアミノ酸からなる。リンカー部Lはペプチド部YのN末端または一本鎖抗体部AのN末端に結合している)
【図1】


【図2】
【図3】
【図4】
【図5】
【図6】

【図7】

【図8】

【図9】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公開番号】特開2011−26294(P2011−26294A)
【公開日】平成23年2月10日(2011.2.10)
【国際特許分類】
【出願番号】特願2010−109288(P2010−109288)
【出願日】平成22年5月11日(2010.5.11)
【出願人】(000001007)キヤノン株式会社 (59,756)
【Fターム(参考)】
【公開日】平成23年2月10日(2011.2.10)
【国際特許分類】
【出願日】平成22年5月11日(2010.5.11)
【出願人】(000001007)キヤノン株式会社 (59,756)
【Fターム(参考)】
[ Back to top ]