
化学式A−R−Xの化合物、または薬学的に条件を満たす該化合物の塩を医療製剤の生産に使用する用法
腫瘍性疾患、アルコール摂取の濫用による病理学的作用、ウイルス性肝炎、脂肪肝、急性および慢性の膵炎、中毒性腎疾患、真性糖尿病の肝臓の抗インシュリン作用、ウィルソン病およびジデローシスの肝臓損傷、虚血性再灌流による疾患などの予防ないし治療に役立ち、また環境毒および薬物中毒に対する解毒作用に有効性を持ち、投与薬剤の臓器内在期間延長、または化学療法投薬時の有毒副作用克服に有効な医療用薬剤の生産に、化学式A−R−Xの化合物、または薬学的に条件を満たす該化合物の塩を用いることができる。化学式中のRは、親水性の端部基Aを有しているC6〜C40の脂肪族ないし芳香族炭化水素を表し、またXは炭素原子ないしヘテロ原子の自由電子ペアおよび/またはπ電子の基を表す。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、化学式A−R−Xの化合物または薬学的に条件を満たす該化合物の塩を用いて行う、疾病ないし特に人間にとっての不都合な状況の予防または治療のための医療用薬剤の生産およびそのような製剤そのものに関係している。
【背景技術】
【0002】
長期にわたってアルコールを過度に摂取すると肝臓障害いわゆる脂肪肝なることが多く、これは肝臓の炎症すなわち肝炎およびその後期段階としての肝硬変へと進むことがある。その場合に人命にかかわる危険性の有無とその度合いは、個人差はあるもののアルコールの摂取量と摂取期間の長さに直接関係する。アルコールに起因する肝臓の炎症(アルコール性肝炎)は生命を脅かす可能性のある病気であり、発熱、黄疸および白血球の増加を伴う。このようなアルコールに起因する肝臓の炎症はアルコールを全面的に絶つことにより治癒するが、肝硬変による瘢痕化の場合はその限りでない。
【0003】
このようなアルコールに起因するいわゆる脂肪肝またはアルコール性肝炎(ASH)に加えて、アルコール摂取が過度でない人あるいはアルコールを全くたしなまない人に起こる肝炎もある。このような肝炎は、例えば塗装現場で働いていたりする環境毒性であったり、または投薬治療をきっかけとして起こるものもある。
【0004】
物質代謝時には、チトクロームの助けを借りて酸化プロセスが起こることが知られている。チトクロームとは、その活動の中心がヘム構造をしている各種酵素のことをいう。この酵素は、数多くの酸化反応および水酸化反応に際して受容体に電子を運ぶ触媒の働きをする。
【0005】
例えばP450系チトクローム(CYP−450)は重要な働きをする。これは偏在するモノオキシゲナーゼであり、人体にとって異質な疎水性物質の新陳代謝および疎水性ホルモンであるステロイドの改質に際して重要な酵素の一つである。
【0006】
チトクロームP450酵素の中心的な働きは、人体にとって異質な物質を水酸化により可溶性とし、腎臓に送って排泄させることにある。このためチトクロームP450酵素は解毒に際して重要な働きをする。
【0007】
今日の医薬物質の約半数が、肝臓のチトクロームP450酵素により水酸化されるものとみなされている。このため医薬物質の人体内在期間は、チトクロームP450酵素の活動により部分的には相当に短縮される。哺乳動物の場合、チトクロームP450は中心的な解毒臓器である肝臓内に大半が存在する。チトクロームP450は通常、網状の内部細胞形質膜と結合した形で存在する。
【0008】
殺虫剤に対して昆虫が、また除草剤に対して植物が抵抗性を示すにあたりチトクロームP450酵素が介在し重要な役割を果たす。
【0009】
チトクロームP450の基本構造は6配位ヘム基であり、この構造にしたがって次の化学式にしたがった反応において触媒として働く。
【0010】
【化1】

【0011】
この場合、この反応に必要な2つの電子が、たとえば錯化合物酵素と関連するNADPH−チトクロームP450還元酵素により生成される。このようにP450には特に細胞に対する毒性を有する活性酸素種(ROS)も発生する。
【0012】
アルコール摂取の場合と同様に非アルコール性の脂肪肝、そして膵炎の場合にもチトクロームP450−2E1の合成が引き起こされることが知られている。他のチトクロームとは大きく異なるこのアイソフォームの機能と作用の仕組みについては、一例として生化学および生物物理学アーカイブ(1995年)Vol. 317、299〜304ページにM.H. ヴァンク他による記述がある。これによると、この酵素は長さ約15Åのカナル状であり、その先端は鉄原子を中心としたヘムリングになっており、反応の中心になる。
【0013】
癌治療に用いられるような化学療法もチトクロームP450酵素により分解できるものと長らく考えられてきた。
【0014】
しかしジァンク他が行った最近の研究(「新生物表現型の癌細胞をチトクロームP450−2J2が促進する、そしてこれは人間の腫瘍内で制御される」癌研究2005年、65:4707〜4715)により、チトクロームP450には腫瘍を促進する働きさえあり得ることが初めて明らかにされた。
【0015】
チトクロームP450−2J2の遺伝子構造が人間の腫瘍のなかで制御されることが明らかにされた。チトクロームP450−2J2とは、アラキドン酸の基部を4種類のエポキシエイコサトリエン酸(EET)異性体中に移す働きをするエポキシゲナーゼの一種である。さらにこの研究においては、EETにより腫瘍細胞が腫瘍壊死因子の作用から守られ、その結果として癌細胞の寿命が長くなったことから、EETにアポトーシスを阻止する働きがあることが分かった。その上にEETは癌細胞の有糸分裂と増殖を促進する。
【0016】
同じく、脈管形成すなわち新しい血管の生成をEETが促進することも明らかにすることができた。この過程は腫瘍の成長において重要な役割を果たす(ポツィ A他、「5,6−および8,9−エポキシエイコサトリエン酸(5,6−および8,9−EET)の「生体内アンギオゲニン脂質」中における能力の特性解析」J. Biol. Chem. 280:27138〜27146、2005年)。
【0017】
これに対してシャッテンベルク他の文献(「ヘパトサイトCYP2E1の過剰発現と脂肪性肝炎が肝臓におけるインスリン兆候を阻害」、J. Biol. Chem. 2005年280:9887〜9894)において、チトクロームP450の過剰発現と糖尿病とが初めて関連づけられた。
【0018】
ミュラー−エノッホ他は、ナトゥアーフォルシュング誌(2001年56c、1082〜1090ページ)に、リソホスファチジルコリン、リソホスファチジルイノシトールおよびアラキドン酸とオレイン酸ないしはモノアシルグリセロール、モノオレイルグリセロールおよびモノパルミトイルグリセロールを通してのラットのチトクロームP450 2B1の阻止作用について記している。
【0019】
さらにT.ヘーナー、D.ミュラー−エノッホ他は、ナトゥアーフォルシュング誌(2004年59c、599〜605ページ)に、アイソフォームのラットのチトクロームP450 2B1に対する単一鎖の脂質分子の影響について記している。
【非特許文献1】Archives of Biochemistry and Biophysics (1995) Vol. 317、299-304
【非特許文献2】Cancer Res. 2005, 65:4707-4715
【非特許文献3】J. Biol. Chem. 2005, 280:27138-27146
【非特許文献4】J. Biol. Chem. 2005, 280:9887-9894
【非特許文献5】Z. Naturforschung (2001) 56c, 1082-1090
【非特許文献6】Z. Naturforschung (2004) 59c, 599-605
【発明の開示】
【発明が解決しようとする課題】
【0020】
本発明に課された課題は、腫瘍性疾患、アルコール摂取の濫用による病理学的作用、ウイルス性肝炎、脂肪肝、急性および慢性の膵炎、中毒性腎疾患、真性糖尿病の肝臓の抗インシュリン作用、ウィルソン病およびジデローシスの場合の肝臓損傷、虚血性再灌流による疾患などの予防ないし治療に適し、また環境毒および薬物中毒に対する解毒作用に適し、投与薬剤の臓器内在期間延長、または化学療法投薬時の有毒副作用克服に適した医療用薬剤の生産手段を提供することにある。
【課題を解決するための手段】
【0021】
上記課題は、本発明に規定される特徴により満たされる。
【0022】
前記の疾病が下記の化合物により治療できるという驚くべきことが分かった。このような化合物により、反応性の細胞毒性種の酸素(ROS)、特にスーパーオキシド陰イオン(O2・−)および過酸化水素(H2O2)の生成が阻止され、これとチトクロームP450、特に第2遺伝子族、殊に2E1および2J2と直接酸化還元反応することがなくなる。
【0023】
このような化合物は同じく、アラキドン酸からエポキシエイコサトリエン酸の異性体への転換も阻止する。さらにそのような化合物の投薬に際して人体にとって異質な物質の水酸化が阻止されることも分かった。
【0024】
本発明にしたがって用いられる化合物は次の化学式により表される。
【0025】
【化2】
【0026】
この代わりに、薬学的に条件を満たすこの化合物の塩を用いることもできる。
【0027】
式中、Rは炭素原子数を6〜40個とすることが望ましい脂肪族ないし芳香族炭化水素を表し、端部基Aは親水性の基または水素であり、またXは炭素原子ないしヘテロ原子の自由電子ペアおよび/またはπ電子の基を表す。R基は特に脂肪親和性である。
【0028】
R基は通常アルキル基である。したがって直鎖であるかまたは枝分かれを有し、単一、2重または3重結合になっており、また置換することができる。通常これを形成するのは脂肪族の骨格であり、その炭素原子数は6〜26個、特に8〜22個である。炭化水素鎖の骨格の炭素原子数は10〜15個、特に10〜13個であるのが目的にかなっている。R基が、縮合化および/または脂肪親和性の置換が可能な脂肪族または芳香族炭化水素の基である場合は、炭素原子数を少なくとも5ないし6個、また最大で40ないし25個とするのが普通である。最小長さを炭素原子数にして7ないし8、また最大長さを炭素原子数にして22ないし20とすればさらに望ましい。
【0029】
X基は複素環状で、かつアルキニル基であるのが目的にかなっている。複素環には特に、窒素、酸素および/または硫黄が含まれる。この複素環は芳香族であってもなくてもよく、リング上の原子は5または6とするのが普通である。Xを縮合複素環状とするのが適している場合にはそのようにしてもよい。そのような複素環の例としては、イミダゾール、ピロール、ピラゾール、ピリジン、ピラジン、インドール、イソインドール、インダゾールがある。複素環は、リング上の原子を6個、特に5個とするのがよく、またヘテロ原子を1、2または3個とするのがよい。この他に適している複素環の例として、例えばチアゾール、トリアゾール、フランがある。
【0030】
アルキン類は−C≡C−R12の構造とするのがよい。ここにR12は水素原子1個またはこれをC1−からC15−ないし最大C10−アルキル基で置換したものであり、ここにも2重または3重結合があってもよい。ただしR12には炭素原子を最大5個、特に最大3個とするのが通常である。本発明によるこの他の目的にかなったX基の形態として次のようなものがある。
− 1次、2次および3次のアミン、
− ヒドラジンおよびヒドラゾンのような置換済みないし非置換ジアゾ官能基、
− ニトリル、イソニトリル、
− チオ−およびイソチオシアン酸塩、アルキル硫化物、スルホ酸化物、チオール基などの硫黄を含む官能基、
− メチレンジオキシ官能基、
− アルキルエーテルおよびアルキルチオエーテル。
【0031】
X基は、補欠ヘム基と協調する基であるのが目的にかなっている。
【0032】
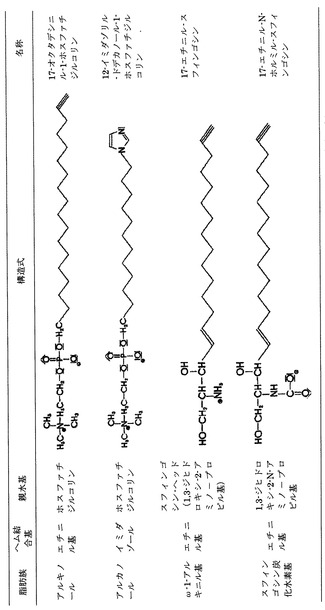
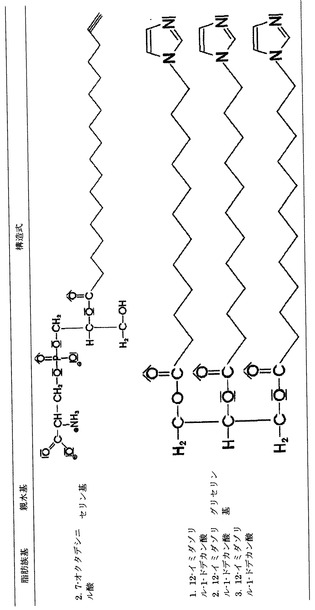
本発明にしたがって用いる分子としては、その末端基Aが医薬製剤に適した任意の親水基を有するものを用いることができ、また特にこれを−OH、−COOH、燐酸塩、燐酸塩エステル、硫酸基、アミノ基、SH、アミノ酸またはポリアルコール、カルニチン(γ−N−トリメチルアミノ−β−ヒドロキシ酪酸)、スフィンゴシン−先端基(1,3−ジヒドロキシ−2−アミノ−プロピル基)とすることができる。Aが水素原子1つであればAは親水性の末端ではないが、しかしこの化合物を本発明による医薬製剤としても用いることができる。アミノ酸としては、リジン、アルギニン、ヒスチジン、アスパラギン酸、グルタミン酸、グルタミン、アスパラギン、ホモシステイン、セリン、ホモセリンおよび/またはシトルリンなどのように基がプラスまたはマイナスに電離しているものが都合がよい。場合によってはグリセリンや蔗糖を置換したものとすることもできる。−OH基をアミノ基またはSH基または燐酸基ないし硫酸基に置き換える置換が目的にかなっている。最も目的にかなった形態は、Aが次の化学式によるグリセリンエステルの場合である。
【0033】
【化3】
【0034】
ここに、基R2〜R4は予め定義されたRx基であり、またBは酸素、硫黄、セレン、セレン酸塩、アミノ基または燐酸塩ないし硫酸基である。この他の望ましい本発明による形態として、この他の基R2〜R4を1つホスファチジルコリン基、ホスファチジルエタノールアミン基、ホスファチジルセリン基、またはホスファチジルイノシトール基としたものがある。この他には前記のアミノ酸の基が、特にプラスまたはマイナスに電離しているものが好適である。
【0035】
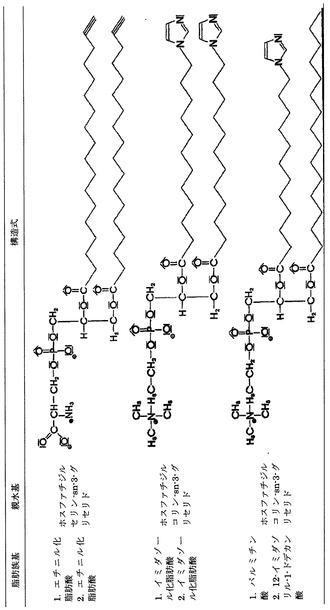
1,2−ジアシル−sn−グリセリン−3−燐酸誘導体の場合には、基本構造のケファリン、セラミド、レシチンおよびこれらに対応してリン脂質が上記において定義したXの末端を有するようにしたものも本発明の適用例に含まれる。
【0036】
枝分かれのない非置換の飽和X−RまたはBR2−4を代表するものとして例えば次のような化学式のものがある。
【0037】
a) アルカノール基 HO−CH2−(CH2)a−X
b) アルキル硫酸基 −O3SO−CH2−(CH2)a−X
c) アルキル−CoA−基 CoA−S−CO−(CH2)a−X
d) アルカン酸の基 HOOC−(CH2)a−X
【0038】
ここにaは、少なくとも6、特に少なくとも7を意味する。aを少なくとも8、特に少なくとも9とすることが特に望ましい。aの最大値は40とし、26であれば特に望ましく、さらに最大値を22とし、12が特に望ましい。最大値を11とし、10とすればさらに望ましい。
【0039】
これらに対応する、枝分かれした、置換済みまたは不飽和のものを代表する化学式は技術者であればたやすく導き出すことができ、したがってここに特に明記する必要はない。
【0040】
脂肪族基Rのこの他の例として、ドデカン基、オクタデカノール基、ウンデカニル硫酸基、パルミチル−CoA基またはラウリン酸基などがある。
【0041】
これに代わる1つの実施形態においてはX基のπ電子として、通常は末端にあるオレフィンの2重ないし3重結合、特にアセチレンのものが特に用いられることがある。イミダゾール基が窒素原子を介して結合した形のもの、もしくはエチニル基(−C≡C−)が特に基として目的にかなっている。
【0042】
上記において定義した化合物、特にイミダゾール化ないしエチニル化された脂肪酸をXとして含み、また試験管内では特別に効果的であることが示されたそのような化合物、例えば17−オクタデシニル−1−酸のような化合物を本発明に従って適用する場合は、数多くの物質の血液内在期間が大幅に延長されることが明らかにされたが、これは、そのような分子のカルボキシ末端が硫酸基により置換されたり、または芳香族の骨格のカルボキシ末端の近くにある炭素原子、すなわち1−3炭素原子(αβγ位)に、メチル基2個または脂肪族ないし芳香族のリング1個により置換される形で行われる。試験管内での活動に対応したこのような向上が生体内での活動にも起こる。
【0043】
この例として、2,2−ジメチル−11−ドデシニル酸と10−ウンデシニル硫酸塩は試験管内で、チトクロームP450の活動阻止の働きの点で10−ウンデシニル酸に匹敵するが、生体内では前者の働きが後者をはるかに上回る。
【0044】
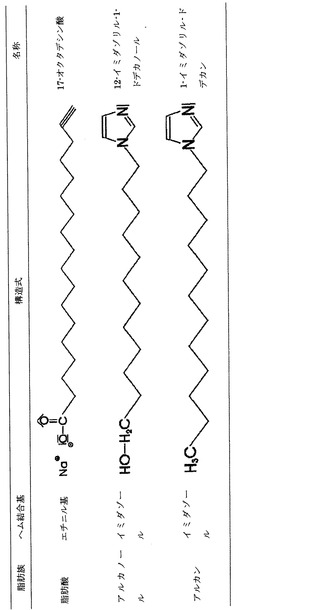
この場合、R1がイミダゾール基であるときは脂肪族の骨格の長さが炭素原子6〜26個分、特に9個分〜20ないし19個分であるのがよいと考えられる。この望ましい基を代表するものとして例えば12−イミダゾリル-ドデカノールまたは1−イミダゾリル−ドデカンがある。この物質およびその他の構造式に関しては任意の図面を参照のこと。
【0045】
本発明による形態のものとして、R1がエチニル基である場合には脂肪族の骨格の長さが炭素原子6〜26個分のものが考えられる。この望ましい基を代表するものとして例えば17−オクタデシニル−1−酸がある。
【0046】
この他の形態のものとして、R1がエチニル基である場合には脂肪族の骨格の長さが炭素原子9〜13個分のものがある。この望ましい基を代表するものとして例えば2,2−ジメチル−11−ドデシニル酸、10−ウンデシニル硫酸塩、10−ウンデシニル酸または10−ウンデシノールがある。
【0047】
これらの化合物には一連の長所がある。まずこれらの化合物はβ酸化物質代謝の酵素にとっては直接のアクセスの対象でなく、したがってこの酵素により急速に新陳代謝されることはない。
【0048】
グリセリン基の第2ポジションが化学式−R−Xの基、例えばエチニル化ないしイミダゾール化された脂肪族基により置換された上記の定義によるホスホグリセリドとトリグリセリドは、腸内で吸収された後にまず加水分解されることにより、2つの脂肪族基のうちの片方が切り離される。このようにしてエチニル化ないしイミダゾール化されたモノグリセリドが生成され、これは可溶性であるためにリソリポイドとも呼ばれるが、これから、脂質とタンパク質の非共有結合集合体であるリポプロテインの働きによりミセルに類似した粒子が形成され、非水溶性脂質の血液内での搬送に供され、体内の作用箇所まで運ばれる。
【0049】
ところで、上記の定義の通りエチニル化ないしイミダゾール化されそのまま投与されたたモノグリセリドにも同じことが当てはまる。
【0050】
腫瘍などの特定の病原組織においてはエネルギー転換が激しく、成長因子(VEGF,PDGF)が放出されることにより自己血管生成が促進されるため、上記のエチニル化ないしイミダゾール化されたモノグリセリドを積み込んだリポプロテインを血流と共にこの組織に移動させるとよい。すなわちエチニル化ないしイミダゾール化された脂肪族のリソリポイドの形の「梱包化」により、前記の病原組織を目的地とした搬送が可能となる。
【0051】
本発明の適用においては、癌治療に際して特にアラキドン酸からエポキシエイコサトリエン酸への転換が阻止されるという作用が化合物に発生する。後者は細胞の分裂と増殖を促進し、癌細胞のアポトーシスを阻止する。そのような化合物の適用により同じく、化学療法時に水酸基置換が阻止され、最終的にその排泄と不活性化につながらなくなる。このためそのような化合物は、直接癌治療および補助薬癌治療に用いることができる。このような理由から、病原組織を目的地とした搬送を可能とする上記の望ましい実施形態は特に有望である。
【0052】
本発明による化合物が薬学的に条件を満たす媒体に含めるようにした医療用薬剤は、同じく本発明によるものと考えられる。
【0053】
この他、アルコール摂取の濫用による病理学的作用の治療に本発明による化合物ないしその医療用薬剤が適応可能である。これは特に生命の危険と、その他のアルコールに起因する炎症プロセスとがある。純粋にアルコールに起因する生命の危険に加えて、脂肪過多症のような栄養摂取に起因したり内分泌性の要因であったり、また真性糖尿病や超脂肪過多によって起こる、脂肪性肝炎[非アルコール性肝炎(NASH)]から肝硬変に至るアルコールに無関係な重大な生命の危機もある。このようなアルコール性および非アルコール性の肝臓病は、肝臓のウイルス感染によることが多い。そのような場合は病状の進展が迅速である。上記の病気およびその原因ないし結果すべてが本発明による化合物を用いて治療可能なことが明らかになった。
【0054】
このような物質が膵臓の治療にも適していることが分かった。そのような炎症ないし膵炎も、アルコール摂取の濫用に加えて有毒物質により引き起こされることがある。これは特に、職業において取り扱う化学薬品または投薬のような環境毒に分類される。ウイルス感染またはメタボリックな内分泌性の要因によっても、そのような膵臓の炎症が起こることがあり、そのようなあらゆる場合に反応性の酸素が病気発症と病状進展に関与している。
【0055】
真性糖尿病の治療の場合、タイプ2とタイプ1のどちらにも本発明による薬剤が適していることが分かった。
【0056】
中毒性の腎臓病も、また化学療法投薬時の副作用によるその他の疾病、特にシスプラチン、カルボプラチン、二塩化チタノセンのような金属化合物または金化合物などによるものも、本発明による医薬品による治療の対象になる。この場合には特に、金属化合物またはハロゲン置換炭化水素、なかでもモノハロゲンおよびポリハロゲン置換炭化水素、そのなかでもハロタンタイプの蒸気麻酔薬、およびこれらに対応した芳香族炭化水素、ニトロソアミン、アクリルアミドのような他の毒性薬剤、またはパラセタモール、メトトレキサート、イソニアジドのような医薬品、またはアミノグリコリドまたはX線造影剤による臓器毒性が阻止されることが分かった。本発明による医薬品は、特に肝臓、腎臓、中枢神経系、膵臓などの臓器に対する解毒剤として、環境毒による臓器毒性の治療に適している。
【0057】
またこの医薬品は、癌治療における細胞活動停止剤の大量投与を可能とし、またそのような背景の下で化学療法における補助治療として成果を上げる見通しを向上させる。
【0058】
この医薬品は例えば、臓器、特に心臓および脳の梗塞(心筋梗塞、卒中の発作)の後に生物学的組織の再灌注により生じる損傷を防ぐのに特に適していることが分かった。この例として、組織破壊のうち60〜80%がこのような再灌注損傷によるものであったり、この医薬品により組織の死滅の広がりが同じ割合だけ減少したりといったことが動物実験により認められている。再灌注損傷の中心的な原因は、貧血中に生成される活性酸素であることは以前から知られている。
【0059】
本発明による医薬品は、臓器移植時の再灌注損傷の防止にも特に適している。このような臓器は、新しい被移植者の体内に移植されるまで冷却された培養液中で保管される。移植後、この培養液は被移植者の循環系につながれて体液に合流するが、これが再灌注損傷につながる。本発明による医薬品を、臓器保管前と保管中および被移植者への移植の直前にも投与することにより、移植に関係するこの重大な問題を解決することができる。
【0060】
遷移金属である鉄(ウィルソン病の場合には銅)が蓄積されると、チトクロームP450に起因する酸化損傷が起こる可能性が生じる。鉄沈着症に属する鉄過剰蓄積に加えて、貧血性の再灌注損傷の場合には細胞内遊離鉄濃度の上昇が起こることが知られている。細胞内鉄濃度の上昇は、シスプラチンないしハロゲン置換炭化水素のような有毒化合物またはウイルス性肝炎によっても起こる。これに該当するものとしては、肝臓に加えて、腎臓により除去されるシスプラチンのような有毒化合物の場合の腎臓がある。遷移金属である鉄(ウィルソン病の場合には銅)による酸化損傷の生化学的な根拠として次のフェントン反応がある。
【0061】
【化4】
【0062】
ここで、チトクロームP450反応サイクルの一環として生じるH2O2(酸化還元電位+0.32ボルト)は、極端に強力な水酸基置換基HO・(酸化還元電位+2.31ボルト)へと変化する。
【0063】
シスプラチンに起因する腎臓病に伴う遊離鉄濃度の上昇については、例えばバリガ他による文献(「シスプラチンに起因する腎臓毒性における鉄触媒の根源としてのチトクロームP450の役割」、キドニーインターナショナル、1998年、54、1562〜1569)がある。貧血性の再灌注損傷またはウイルス性肝炎の場合の鉄濃度の上昇については、例えばパラー他による文献(「肝臓における再酸素化中の組織破壊ヒドロキシル基形成をチトクロームP450により治療」、Proc. Natl. Acad. Sci.米国1994年、91、7002〜7006)ないしはチャポウトート他による文献(「ウイルス性C硬変が進行した肝硬変細胞癌の患者における肝臓内鉄分過剰」、Gut2000、46、711〜714)がある。
【0064】
本発明による物質には、特にチトクロームP450の第2遺伝子族の人間アイソフォーム、特にアイソフォーム2E1および2J2、そしてこれらに起因する疾病に対する抑止物質としての働きがあることが分かった。
【0065】
本発明の目的に特にかなった形態として、医薬製剤をリポソームで包むことが考えられる。製剤の根底にある化合物は長さの長い脂肪族の基であるという事実があるため、リポソームによる包み込みは投薬に非常に適した形態である。このようなリポソームは、静脈内、筋肉内、腹膜内、経皮または経口投与に適している。またエアロゾルの投与にも適している。
【0066】
ただし本発明による化合物はそのまま直接投与することもできる。そのような場合にも上記のような投与法が適している。
【発明を実施するための最良の形態】
【0067】
〔合成法〕
本発明による様々な化合物の合成法をいくつか以下に示す。
【0068】
1: 12−イミダゾリル−1−ドデカン酸
a) 12−イミダゾリル−1−ドデカン酸はアルターマン他の文献に記されている工程(「チトクロームP450 4A1およびチトクロームP450 4A1/NADPH−P450還元酵素溶解タンパク質による脂肪酸の区別とオメガヒドロキシル化」、生化学&生物物理学アーカイブ(1995年)Vol. 320、289〜296ページ)により合成される。
【0069】
このためには、12−ブロモ−1−ドデカノールがジョーンズの試薬により12−ブロモ−1−ドデカン酸へと酸化される。この白色の固形の酸は引き続いてジアゾメタンと共に、対応するメチルエステルへとエステル化される。このメチルエステルは、直接イミダソールと混ぜられ、80℃で5時間かけて12−イミダゾリル−1−ドデカン酸メチルエステルに転換される。このようにして得られた厚い塊が水とジクロルメタンとに分離され、有機相はNa2SO4により乾燥され蒸発濃縮される。油性の残滓はシリカゲルにかけてクロマトグラフ的に洗浄され、引き続いてメタノールとテトラヒドロフランの混合物(3:4)中で溶解され、LiOH・H2Oと混ぜられ、混合物は還流の下で2時間加熱される。溶剤を蒸発させた後、白色の残滓を再び水に解かして、ジクロルメタンにより抽出を行い、pH5〜6に酸性化した後、酢酸エチルにより改めて抽出を行う。酢酸エチル抽出物をNa2SO4により乾燥させ、フィルタリングして蒸発濃縮する。白色の固形の残滓がメタノール/エーテルにより結晶化されて12−イミダゾリル−1−ドデカン酸が得られる。
【0070】
b) ル(人名)他による文献に記載されている工程(「脂肪酸オメガヒドロキシル化に対する抑止物質としてのラウリン酸に類似したヘム調整」、生化学&生物物理学アーカイブ(1997年)Vol. 337、1〜7ページ)にしたがって12−イミダゾリル−1−ドデカノールと1−イミダゾリルドデカンが合成される。
【0071】
このためにはモル比1:3の12−ブロモ−1−ドデカノールとイミダゾールが80℃で5時間加熱される。この生成原料が水とジクロルメタンとに分離される。有機相はNa2SO4により乾燥され蒸発濃縮される。ベンゾル/n−ヘキサンにより12−イミダゾリル−1−ドデカノールが結晶化される。
【0072】
c) モル比1:3の1−ブロモドデカンとイミダゾールを撹拌して85℃で加熱することにより1−イミダゾリルドデカンが生成される。この生成原料がジクロルメタンに溶かされ、水と共に3回加振される。有機相はNa2SO4により乾燥され、フィルタリングされ、蒸発濃縮される。油性の蒸発残滓はn−ヘキサンにより結晶化され、1−イミダゾリルドデカンが得られる。
【0073】
2: 12−イミダゾリル−1−ホスファチジルコリンの合成
ホスファチジルコリンが、酸性条件下で、かつジシクロヘキシルカルボジイミドの存在の下でO−ホスホリルイソ尿素に転換される。反応混合物に12−イミダゾリル−1−ドデカノールが添加され、これがホスホリル基に対して求核置換作用をしてエステル結合が形成されることにより、12−イミダゾリル−1−ホスファチジルコリンが生成される。これによりジシクロヘキシル尿素が析出される。この反応がうまく行くには4−ジエチルアミノピリジンが必要とされる。
【0074】
この反応のメカニズムは、ジシクロヘキシルカルボジイミドを用いて有機酸をアルコールにエステル化する架橋エステル化に類似している。
【0075】
3: 1−パルミトイル−2−イミダゾイル−グリセロ−3−ホスファチジルコリンの合成
非改質脂肪酸とラベル化(この場合はエチニル化ないしイミダゾール化)された脂肪酸を含むホスファチジルコリン−ジグリセリド合成の原則については、エビル他の文献(「歩留りの高いホスホ脂質の合成」、メソッズ エンツィモル 1983年98号、623〜32)がある。
【0076】
3a: 1,2−ジパルミトイル−3−ベンジルグリセリドの合成
このためには1,2−イソプロピリデン−sn−グリセリンがp−キシレンに溶解され、これにカリウム−tert−ブチレートと塩化ベンジルを加えた上で撹拌する。反応終了後に等分の水とジイソプロピルエーテルが加えられ、相の分離が行われる。上の層に含まれる3−ベンジル−sn−グリセリンが蒸発により取り出され、さらに洗浄ステップが実施される。
【0077】
洗浄された3−ベンジル−sn−グリセリンは引き続いて、パルミチン酸のような脂肪酸と共に四塩化炭素に溶かされる。4−ジエチルアミノピリジンとジシクロヘキシルカルボジイミドを添加すると、3−ベンジル−sn−グリセリンのアルコール基と脂肪酸の間にエステル結合が生じてジシクロヘキシル尿素が析出される。この反応メカニズムが「架橋エステル化」と呼ばれる。
【0078】
析出したジシクロヘキシル尿素は除去され、溶剤は蒸発させられる。洗浄ステップをさらに追加した後、生成物として1,2−ジパルミトイル−3−ベンジル−sn−グリセリドが得られる。
【0079】
3b: 1,2−ジパルミトイル−sn−グリセリドの合成
1,2−ジパルミトイル−3−ベンジル−sn−グリセリドがテトラヒドロフランに溶かされ、触媒(10%のPd/C)の存在の下で、水素原子による水素添加分解が行われる。このときベンジル基が水素原子により置き換えられ、1,2−ジパルミトイル−sn−グリセリドが生成される。
【0080】
3c: 1,2−ジパルミトイル−sn−グリセリドの燐酸化
三塩化ホスホリルがテトラヒドロフランと混ぜられ、氷中で撹拌される。引き続いてテトラヒドロフランに溶かした1,2−ジパルミトイル−sn−グリセリドを滴下して添加する。このようにしてまず1,2−ジパルミトイル−sn−グリセリド−3−二塩化ホスホリルが発生する。
【0081】
引き続いてテトラヒドロフランに溶かしたトリエチルアミンを改めて添加し、テトラヒドロフランに溶かしたブロモエタノールを滴下により加え、温度を25℃に上昇させる。このとき主として発生するのは1,2−ジパルミトイル−sn−グリセリド−3−ホスホリル−ブロモエチルエステル−1塩化物であり、ジ−ブロモエチルエステルは副産物として少量発生するに過ぎない。
【0082】
この混合物が洗浄され、冷却され、炭酸ナトリウムとヘキサンと混ぜられ加振される。このとき、燐酸塩の基と塩化物の間の結合に水酸化が起こる。生成物として1,2−ジパルミトイル−sn−グリセリド−3−ホスホリル−ブロモエチルエステルのナトリウム塩が得られる。
【0083】
1,2−ジパルミトイル−sn−グリセリド−3−ホスホリル−(N−ブトキシカルボニル)−エタノールアミンエステルおよび1,2−ジパルミトイル−sn−グリセリド−3−ホスホリル−(N−ブトキシカルボニル)−tert−ブチルセリンエステルのナトリウム塩についても同様に説明される。
【0084】
3d: 1,2−ジパルミトイル−sn−グリセリド−3−ホスホアルキルエステルの加水分解
1,2−ジパルミトイル−sn−グリセリド−3−ホスホリル−ブロモエチルエステルまたは、この代わりに上記に挙げたホスホアルキルエステルが、ジエチルエーテルと蒸留水の混合物すなわちCaCl2・2H2O中で溶かされる。
【0085】
パリツシュ緩衝剤を添加することによりpH値が7.5に調整され、引き続いて酵素ホスホリパーゼA2が添加され、35℃で60分間撹拌された。このとき、グリセリン基の第2ポジションのエステル結合が加水分解され、これに対応して第2ポジションにOH基を有する1−パルミトイル−sn−グリセリド−3−ホスホアルキルエステルと遊離脂肪酸ができる。
【0086】
こうして得られた分子のグリセリン基の第2ポジションは、イミダゾール化ないしエチニル化された脂肪酸のようなラベル化脂肪酸により狙い通りにエステル化を行うことができる。第3ポジションのホスホアルキルエステルも、コリン、セリン、エタノールアミンまたはイノシトールなど適切なアルコールによりエステル転換することができる。
【0087】
3e: ラベル化脂肪酸による第2ポジションのエステル化
得られた1−パルミトイル−sn−グリセリド−3−ホスホアルキルエステルをテトラクロルメタンに溶かし、イミダゾール化ないしエチニル化された脂肪酸を添加し、混合物を撹拌する。
【0088】
添加される脂肪酸は、例えばシグマ・アルドリッチ社から入手できる17−オクタデシン酸でよい。これは12−イミダゾリル−1−ドデカン酸であり、上記1項で述べた要領でも同様に合成することができる。
【0089】
引き続いて改めて「架橋エステル化」が行われ、混合物に4−ジエチルアミノピリジンとジシクロヘキシルカルボジイミドが添加される。このとき、グリセリン基に残存するOH基とラベル化脂肪酸のカルボキシル基の間にエステル結合が生じる。
【0090】
析出したジシクロヘキシル尿素は除去され、溶剤は蒸発させられる。洗浄ステップをさらに追加した後、生成物として1−パルミトイル−2−アシル−sn−グリセリド―3−ホスホアルキルエステルが得られる。
【0091】
3f: グリセリン基の第3ポジションのホスホアルキルエステルのエステル転換
1−パルミトイル−2−アシル−sn−グリセリド−3−ホスホリル−ブロモエチルエステルをクロロフォルムに溶かす。引き続いて2−プロパノール−トリメチルアミンが添加される。反応容器を50℃で培養状態にして、続いて溶剤を窒素と共に蒸発させる。反応生成物を洗浄するとラベル化1−パルミトイル−2−アシル−sn−グリセリド−3−ホスファチジルコリンが得られる。
【0092】
ラベル化1−パルミトイル−2−アシル−sn−グリセリド−3−ホスファチジルセリンを作り出すために、前記の1−パルミトイル−2−アシル−sn−グリセリド−3−ホスホリル−(N−ブトキシカルボニル)−エタノールアミンエステルをCH2Cl2に溶かし、トリフルオロ酢酸と過クロム酸が添加される。引き続いて冷やした状態で撹拌し、水とメタノールにより洗う。相を分離した後、下側の相についてNa2CO3により抽出を行い、蒸発濃縮を行う。メタノール添加後に生成される結晶が1−パルミトイル−2−アシル−sn−グリセリド−3−ホスファチジル−エタノールアミンである。
【0093】
ラベル化1−パルミトイル−2−アシル−sn−グリセリド−3−ホスファチジルセリンを作り出すための工程も類似している。この場合の出発材料は、上記の1−パルミトイル−2−アシル−sn−グリセリド−3−ホスホリル−(N−ブトキシカルボニル)−tert−ブチルセリンエステルである。
【0094】
〔図面について〕
添付する図面には、本発明による化合物の例がいくつか例示されている。
【0095】
図1〜3は、本発明にしたがって生産される、化学式がA−R−Xの化合物の例を示す。
【0096】
技術者であれば、挙げられている請求の範囲にある以外の数多くの化合物をまとめられることがすぐに分かる。したがって脂肪族の基は、直鎖のことも枝分かれしていることもあり、単結合、二重結合、三重結合のいずれの場合もあって置換可能であり、骨格の炭素原子数は9〜19となる。炭化水素の骨格は同じく脂環および/または芳香族炭化水素により形成することができ、その場合リング構造のために最大40個の炭素原子を必要とすることがある。
親水基としては、イノシトールやエタノールアミン、ないしはこれらのグリセリドなど他のアルコールも問題になる。
【0097】
〔毒性〕
12−イミダゾリル−1−ドデカノール(物質1)および12−(1)−イミダゾリル-ドデカン(物質2)の急性毒性についての試験が、オスのCDラットを使って行われた。その結果12−イミダゾリル−1−ドデカノールのLD50(14日)が1000mg/kg b.w., p.o.であり、12−(1)−イミダゾリル−ドデカンのLD50(14日)は1000mg/kg b.w., p.o.であった。
【0098】
最初の不耐性反応:物質1: 1000mg/kg b.w., p.o.
物質2: 500mg/kg b.w., p.o.
投与量の影響なし:物質1: 500mg/kg b.w., p.o.
物質2: 250mg/kg b.w., p.o.
【0099】
〔12−イミダゾリル−1−ドデカノールの抗腫瘍作用の測定〕
このためには「24−ウェルプレート」に癌細胞株を4本引き、24時間放置して成長させた。続いて供試物質である12−イミダゾリル−1−ドデカノールを様々な濃度で浮遊細胞に加え、DMSOに溶かした。細胞媒質中のDMSOの濃度はそれぞれ0.1%であった。このDMSO濃度は対照試験により毒性がないことが分かり、細胞計数はそれぞれ4日間の培養期間の後に行われた。すべての場合に、癌細胞の増殖が強力に阻止されることが分かった。
【0100】
12−イミダゾリル−1−ドデカノールの、抑制剤の最大半減濃度値IC50は次のように求められた。
【0101】
HepG2(肝細胞) 50nM
Panc-1(膵臓細胞) 50nM
PC-3(前立腺細胞) 50nM
SW620(大腸細胞) 100nM
【0102】
癌細胞株すべてに抑制剤の強力な抗抗腫瘍効果が認められた。
【0103】
〔12−イミダゾリル−1−ドデカノールの細胞毒性の評価〕
12−イミダゾリル−1−ドデカノールの細胞毒性値の最大半減値がLDH細胞毒性試験により求められた。
この測定値は次の通りとなっている:
MRC-5(肺繊維芽細胞) =500μM
Panc-1(膵臓細胞) =500μM
【0104】
細胞毒性値の最大半減値500μMを、Panc−1に対する抑制剤の最大半減濃度値IC50=50μMと比較してみると10,000倍の開きがある。このことから12−イミダゾリル−1−ドデカノールは、ガン細胞の増殖に対して非常に効果的で細胞毒性が比較的低い抑制剤であると推定される。
言い換えれば本発明による物質の治療面での窓口の幅は広い。
【0105】
〔12−イミダゾリル−1−ドデカノールによるHepG2細胞の活動性抑制〕
遊走試験条件: HepG2肝臓癌細胞がコラーゲンマトリクスに埋め込まれた。30の個別細胞の写真による監視が900分にわたって連続的に行われた。遊走する細胞の割合の平均値が求められた。
【0106】
結果1: 非治療の対照HepG2細胞の遊走の平均値は15%だった。12−イミダゾリル−1−ドデカノール1μMないし10μMをマトリクス媒質に加えた場合、細胞の遊走は50%ないし75%低下した。
【0107】
結果2: HepG2細胞はインスリン受容体を明確に表わすため、12−イミダゾリル−1−ドデカノールありとなしの場合の遊走に対するインスリンの影響が求められた。
【0108】
【表1】
【0109】
インスリンは遊走を2.5倍に増大させる。作用物質により450分の観察時間(観察時間の前半)後にはこの増大が全面的にブロックされた。900分後(観察時間の後半)には増大がマイナスになった。遊走の平均値は対照以下の13%となった。
【図面の簡単な説明】
【0110】
【図1】本発明にしたがいイミダゾール化ないしエチニル化された脂肪族の例を示す表である。
【図2】本発明にしたがいイミダゾール化ないしエチニル化され、親水基により改質された脂肪族の例を示す表である。
【図3】本発明にしたがいイミダゾール化ないしエチニル化されたグリセリドの例を示す表である。
【図4】本発明にしたがいイミダゾール化ないしエチニル化されたホスホグリセリドの例を示す表である。
【技術分野】
【0001】
本発明は、化学式A−R−Xの化合物または薬学的に条件を満たす該化合物の塩を用いて行う、疾病ないし特に人間にとっての不都合な状況の予防または治療のための医療用薬剤の生産およびそのような製剤そのものに関係している。
【背景技術】
【0002】
長期にわたってアルコールを過度に摂取すると肝臓障害いわゆる脂肪肝なることが多く、これは肝臓の炎症すなわち肝炎およびその後期段階としての肝硬変へと進むことがある。その場合に人命にかかわる危険性の有無とその度合いは、個人差はあるもののアルコールの摂取量と摂取期間の長さに直接関係する。アルコールに起因する肝臓の炎症(アルコール性肝炎)は生命を脅かす可能性のある病気であり、発熱、黄疸および白血球の増加を伴う。このようなアルコールに起因する肝臓の炎症はアルコールを全面的に絶つことにより治癒するが、肝硬変による瘢痕化の場合はその限りでない。
【0003】
このようなアルコールに起因するいわゆる脂肪肝またはアルコール性肝炎(ASH)に加えて、アルコール摂取が過度でない人あるいはアルコールを全くたしなまない人に起こる肝炎もある。このような肝炎は、例えば塗装現場で働いていたりする環境毒性であったり、または投薬治療をきっかけとして起こるものもある。
【0004】
物質代謝時には、チトクロームの助けを借りて酸化プロセスが起こることが知られている。チトクロームとは、その活動の中心がヘム構造をしている各種酵素のことをいう。この酵素は、数多くの酸化反応および水酸化反応に際して受容体に電子を運ぶ触媒の働きをする。
【0005】
例えばP450系チトクローム(CYP−450)は重要な働きをする。これは偏在するモノオキシゲナーゼであり、人体にとって異質な疎水性物質の新陳代謝および疎水性ホルモンであるステロイドの改質に際して重要な酵素の一つである。
【0006】
チトクロームP450酵素の中心的な働きは、人体にとって異質な物質を水酸化により可溶性とし、腎臓に送って排泄させることにある。このためチトクロームP450酵素は解毒に際して重要な働きをする。
【0007】
今日の医薬物質の約半数が、肝臓のチトクロームP450酵素により水酸化されるものとみなされている。このため医薬物質の人体内在期間は、チトクロームP450酵素の活動により部分的には相当に短縮される。哺乳動物の場合、チトクロームP450は中心的な解毒臓器である肝臓内に大半が存在する。チトクロームP450は通常、網状の内部細胞形質膜と結合した形で存在する。
【0008】
殺虫剤に対して昆虫が、また除草剤に対して植物が抵抗性を示すにあたりチトクロームP450酵素が介在し重要な役割を果たす。
【0009】
チトクロームP450の基本構造は6配位ヘム基であり、この構造にしたがって次の化学式にしたがった反応において触媒として働く。
【0010】
【化1】
【0011】
この場合、この反応に必要な2つの電子が、たとえば錯化合物酵素と関連するNADPH−チトクロームP450還元酵素により生成される。このようにP450には特に細胞に対する毒性を有する活性酸素種(ROS)も発生する。
【0012】
アルコール摂取の場合と同様に非アルコール性の脂肪肝、そして膵炎の場合にもチトクロームP450−2E1の合成が引き起こされることが知られている。他のチトクロームとは大きく異なるこのアイソフォームの機能と作用の仕組みについては、一例として生化学および生物物理学アーカイブ(1995年)Vol. 317、299〜304ページにM.H. ヴァンク他による記述がある。これによると、この酵素は長さ約15Åのカナル状であり、その先端は鉄原子を中心としたヘムリングになっており、反応の中心になる。
【0013】
癌治療に用いられるような化学療法もチトクロームP450酵素により分解できるものと長らく考えられてきた。
【0014】
しかしジァンク他が行った最近の研究(「新生物表現型の癌細胞をチトクロームP450−2J2が促進する、そしてこれは人間の腫瘍内で制御される」癌研究2005年、65:4707〜4715)により、チトクロームP450には腫瘍を促進する働きさえあり得ることが初めて明らかにされた。
【0015】
チトクロームP450−2J2の遺伝子構造が人間の腫瘍のなかで制御されることが明らかにされた。チトクロームP450−2J2とは、アラキドン酸の基部を4種類のエポキシエイコサトリエン酸(EET)異性体中に移す働きをするエポキシゲナーゼの一種である。さらにこの研究においては、EETにより腫瘍細胞が腫瘍壊死因子の作用から守られ、その結果として癌細胞の寿命が長くなったことから、EETにアポトーシスを阻止する働きがあることが分かった。その上にEETは癌細胞の有糸分裂と増殖を促進する。
【0016】
同じく、脈管形成すなわち新しい血管の生成をEETが促進することも明らかにすることができた。この過程は腫瘍の成長において重要な役割を果たす(ポツィ A他、「5,6−および8,9−エポキシエイコサトリエン酸(5,6−および8,9−EET)の「生体内アンギオゲニン脂質」中における能力の特性解析」J. Biol. Chem. 280:27138〜27146、2005年)。
【0017】
これに対してシャッテンベルク他の文献(「ヘパトサイトCYP2E1の過剰発現と脂肪性肝炎が肝臓におけるインスリン兆候を阻害」、J. Biol. Chem. 2005年280:9887〜9894)において、チトクロームP450の過剰発現と糖尿病とが初めて関連づけられた。
【0018】
ミュラー−エノッホ他は、ナトゥアーフォルシュング誌(2001年56c、1082〜1090ページ)に、リソホスファチジルコリン、リソホスファチジルイノシトールおよびアラキドン酸とオレイン酸ないしはモノアシルグリセロール、モノオレイルグリセロールおよびモノパルミトイルグリセロールを通してのラットのチトクロームP450 2B1の阻止作用について記している。
【0019】
さらにT.ヘーナー、D.ミュラー−エノッホ他は、ナトゥアーフォルシュング誌(2004年59c、599〜605ページ)に、アイソフォームのラットのチトクロームP450 2B1に対する単一鎖の脂質分子の影響について記している。
【非特許文献1】Archives of Biochemistry and Biophysics (1995) Vol. 317、299-304
【非特許文献2】Cancer Res. 2005, 65:4707-4715
【非特許文献3】J. Biol. Chem. 2005, 280:27138-27146
【非特許文献4】J. Biol. Chem. 2005, 280:9887-9894
【非特許文献5】Z. Naturforschung (2001) 56c, 1082-1090
【非特許文献6】Z. Naturforschung (2004) 59c, 599-605
【発明の開示】
【発明が解決しようとする課題】
【0020】
本発明に課された課題は、腫瘍性疾患、アルコール摂取の濫用による病理学的作用、ウイルス性肝炎、脂肪肝、急性および慢性の膵炎、中毒性腎疾患、真性糖尿病の肝臓の抗インシュリン作用、ウィルソン病およびジデローシスの場合の肝臓損傷、虚血性再灌流による疾患などの予防ないし治療に適し、また環境毒および薬物中毒に対する解毒作用に適し、投与薬剤の臓器内在期間延長、または化学療法投薬時の有毒副作用克服に適した医療用薬剤の生産手段を提供することにある。
【課題を解決するための手段】
【0021】
上記課題は、本発明に規定される特徴により満たされる。
【0022】
前記の疾病が下記の化合物により治療できるという驚くべきことが分かった。このような化合物により、反応性の細胞毒性種の酸素(ROS)、特にスーパーオキシド陰イオン(O2・−)および過酸化水素(H2O2)の生成が阻止され、これとチトクロームP450、特に第2遺伝子族、殊に2E1および2J2と直接酸化還元反応することがなくなる。
【0023】
このような化合物は同じく、アラキドン酸からエポキシエイコサトリエン酸の異性体への転換も阻止する。さらにそのような化合物の投薬に際して人体にとって異質な物質の水酸化が阻止されることも分かった。
【0024】
本発明にしたがって用いられる化合物は次の化学式により表される。
【0025】
【化2】
【0026】
この代わりに、薬学的に条件を満たすこの化合物の塩を用いることもできる。
【0027】
式中、Rは炭素原子数を6〜40個とすることが望ましい脂肪族ないし芳香族炭化水素を表し、端部基Aは親水性の基または水素であり、またXは炭素原子ないしヘテロ原子の自由電子ペアおよび/またはπ電子の基を表す。R基は特に脂肪親和性である。
【0028】
R基は通常アルキル基である。したがって直鎖であるかまたは枝分かれを有し、単一、2重または3重結合になっており、また置換することができる。通常これを形成するのは脂肪族の骨格であり、その炭素原子数は6〜26個、特に8〜22個である。炭化水素鎖の骨格の炭素原子数は10〜15個、特に10〜13個であるのが目的にかなっている。R基が、縮合化および/または脂肪親和性の置換が可能な脂肪族または芳香族炭化水素の基である場合は、炭素原子数を少なくとも5ないし6個、また最大で40ないし25個とするのが普通である。最小長さを炭素原子数にして7ないし8、また最大長さを炭素原子数にして22ないし20とすればさらに望ましい。
【0029】
X基は複素環状で、かつアルキニル基であるのが目的にかなっている。複素環には特に、窒素、酸素および/または硫黄が含まれる。この複素環は芳香族であってもなくてもよく、リング上の原子は5または6とするのが普通である。Xを縮合複素環状とするのが適している場合にはそのようにしてもよい。そのような複素環の例としては、イミダゾール、ピロール、ピラゾール、ピリジン、ピラジン、インドール、イソインドール、インダゾールがある。複素環は、リング上の原子を6個、特に5個とするのがよく、またヘテロ原子を1、2または3個とするのがよい。この他に適している複素環の例として、例えばチアゾール、トリアゾール、フランがある。
【0030】
アルキン類は−C≡C−R12の構造とするのがよい。ここにR12は水素原子1個またはこれをC1−からC15−ないし最大C10−アルキル基で置換したものであり、ここにも2重または3重結合があってもよい。ただしR12には炭素原子を最大5個、特に最大3個とするのが通常である。本発明によるこの他の目的にかなったX基の形態として次のようなものがある。
− 1次、2次および3次のアミン、
− ヒドラジンおよびヒドラゾンのような置換済みないし非置換ジアゾ官能基、
− ニトリル、イソニトリル、
− チオ−およびイソチオシアン酸塩、アルキル硫化物、スルホ酸化物、チオール基などの硫黄を含む官能基、
− メチレンジオキシ官能基、
− アルキルエーテルおよびアルキルチオエーテル。
【0031】
X基は、補欠ヘム基と協調する基であるのが目的にかなっている。
【0032】
本発明にしたがって用いる分子としては、その末端基Aが医薬製剤に適した任意の親水基を有するものを用いることができ、また特にこれを−OH、−COOH、燐酸塩、燐酸塩エステル、硫酸基、アミノ基、SH、アミノ酸またはポリアルコール、カルニチン(γ−N−トリメチルアミノ−β−ヒドロキシ酪酸)、スフィンゴシン−先端基(1,3−ジヒドロキシ−2−アミノ−プロピル基)とすることができる。Aが水素原子1つであればAは親水性の末端ではないが、しかしこの化合物を本発明による医薬製剤としても用いることができる。アミノ酸としては、リジン、アルギニン、ヒスチジン、アスパラギン酸、グルタミン酸、グルタミン、アスパラギン、ホモシステイン、セリン、ホモセリンおよび/またはシトルリンなどのように基がプラスまたはマイナスに電離しているものが都合がよい。場合によってはグリセリンや蔗糖を置換したものとすることもできる。−OH基をアミノ基またはSH基または燐酸基ないし硫酸基に置き換える置換が目的にかなっている。最も目的にかなった形態は、Aが次の化学式によるグリセリンエステルの場合である。
【0033】
【化3】
【0034】
ここに、基R2〜R4は予め定義されたRx基であり、またBは酸素、硫黄、セレン、セレン酸塩、アミノ基または燐酸塩ないし硫酸基である。この他の望ましい本発明による形態として、この他の基R2〜R4を1つホスファチジルコリン基、ホスファチジルエタノールアミン基、ホスファチジルセリン基、またはホスファチジルイノシトール基としたものがある。この他には前記のアミノ酸の基が、特にプラスまたはマイナスに電離しているものが好適である。
【0035】
1,2−ジアシル−sn−グリセリン−3−燐酸誘導体の場合には、基本構造のケファリン、セラミド、レシチンおよびこれらに対応してリン脂質が上記において定義したXの末端を有するようにしたものも本発明の適用例に含まれる。
【0036】
枝分かれのない非置換の飽和X−RまたはBR2−4を代表するものとして例えば次のような化学式のものがある。
【0037】
a) アルカノール基 HO−CH2−(CH2)a−X
b) アルキル硫酸基 −O3SO−CH2−(CH2)a−X
c) アルキル−CoA−基 CoA−S−CO−(CH2)a−X
d) アルカン酸の基 HOOC−(CH2)a−X
【0038】
ここにaは、少なくとも6、特に少なくとも7を意味する。aを少なくとも8、特に少なくとも9とすることが特に望ましい。aの最大値は40とし、26であれば特に望ましく、さらに最大値を22とし、12が特に望ましい。最大値を11とし、10とすればさらに望ましい。
【0039】
これらに対応する、枝分かれした、置換済みまたは不飽和のものを代表する化学式は技術者であればたやすく導き出すことができ、したがってここに特に明記する必要はない。
【0040】
脂肪族基Rのこの他の例として、ドデカン基、オクタデカノール基、ウンデカニル硫酸基、パルミチル−CoA基またはラウリン酸基などがある。
【0041】
これに代わる1つの実施形態においてはX基のπ電子として、通常は末端にあるオレフィンの2重ないし3重結合、特にアセチレンのものが特に用いられることがある。イミダゾール基が窒素原子を介して結合した形のもの、もしくはエチニル基(−C≡C−)が特に基として目的にかなっている。
【0042】
上記において定義した化合物、特にイミダゾール化ないしエチニル化された脂肪酸をXとして含み、また試験管内では特別に効果的であることが示されたそのような化合物、例えば17−オクタデシニル−1−酸のような化合物を本発明に従って適用する場合は、数多くの物質の血液内在期間が大幅に延長されることが明らかにされたが、これは、そのような分子のカルボキシ末端が硫酸基により置換されたり、または芳香族の骨格のカルボキシ末端の近くにある炭素原子、すなわち1−3炭素原子(αβγ位)に、メチル基2個または脂肪族ないし芳香族のリング1個により置換される形で行われる。試験管内での活動に対応したこのような向上が生体内での活動にも起こる。
【0043】
この例として、2,2−ジメチル−11−ドデシニル酸と10−ウンデシニル硫酸塩は試験管内で、チトクロームP450の活動阻止の働きの点で10−ウンデシニル酸に匹敵するが、生体内では前者の働きが後者をはるかに上回る。
【0044】
この場合、R1がイミダゾール基であるときは脂肪族の骨格の長さが炭素原子6〜26個分、特に9個分〜20ないし19個分であるのがよいと考えられる。この望ましい基を代表するものとして例えば12−イミダゾリル-ドデカノールまたは1−イミダゾリル−ドデカンがある。この物質およびその他の構造式に関しては任意の図面を参照のこと。
【0045】
本発明による形態のものとして、R1がエチニル基である場合には脂肪族の骨格の長さが炭素原子6〜26個分のものが考えられる。この望ましい基を代表するものとして例えば17−オクタデシニル−1−酸がある。
【0046】
この他の形態のものとして、R1がエチニル基である場合には脂肪族の骨格の長さが炭素原子9〜13個分のものがある。この望ましい基を代表するものとして例えば2,2−ジメチル−11−ドデシニル酸、10−ウンデシニル硫酸塩、10−ウンデシニル酸または10−ウンデシノールがある。
【0047】
これらの化合物には一連の長所がある。まずこれらの化合物はβ酸化物質代謝の酵素にとっては直接のアクセスの対象でなく、したがってこの酵素により急速に新陳代謝されることはない。
【0048】
グリセリン基の第2ポジションが化学式−R−Xの基、例えばエチニル化ないしイミダゾール化された脂肪族基により置換された上記の定義によるホスホグリセリドとトリグリセリドは、腸内で吸収された後にまず加水分解されることにより、2つの脂肪族基のうちの片方が切り離される。このようにしてエチニル化ないしイミダゾール化されたモノグリセリドが生成され、これは可溶性であるためにリソリポイドとも呼ばれるが、これから、脂質とタンパク質の非共有結合集合体であるリポプロテインの働きによりミセルに類似した粒子が形成され、非水溶性脂質の血液内での搬送に供され、体内の作用箇所まで運ばれる。
【0049】
ところで、上記の定義の通りエチニル化ないしイミダゾール化されそのまま投与されたたモノグリセリドにも同じことが当てはまる。
【0050】
腫瘍などの特定の病原組織においてはエネルギー転換が激しく、成長因子(VEGF,PDGF)が放出されることにより自己血管生成が促進されるため、上記のエチニル化ないしイミダゾール化されたモノグリセリドを積み込んだリポプロテインを血流と共にこの組織に移動させるとよい。すなわちエチニル化ないしイミダゾール化された脂肪族のリソリポイドの形の「梱包化」により、前記の病原組織を目的地とした搬送が可能となる。
【0051】
本発明の適用においては、癌治療に際して特にアラキドン酸からエポキシエイコサトリエン酸への転換が阻止されるという作用が化合物に発生する。後者は細胞の分裂と増殖を促進し、癌細胞のアポトーシスを阻止する。そのような化合物の適用により同じく、化学療法時に水酸基置換が阻止され、最終的にその排泄と不活性化につながらなくなる。このためそのような化合物は、直接癌治療および補助薬癌治療に用いることができる。このような理由から、病原組織を目的地とした搬送を可能とする上記の望ましい実施形態は特に有望である。
【0052】
本発明による化合物が薬学的に条件を満たす媒体に含めるようにした医療用薬剤は、同じく本発明によるものと考えられる。
【0053】
この他、アルコール摂取の濫用による病理学的作用の治療に本発明による化合物ないしその医療用薬剤が適応可能である。これは特に生命の危険と、その他のアルコールに起因する炎症プロセスとがある。純粋にアルコールに起因する生命の危険に加えて、脂肪過多症のような栄養摂取に起因したり内分泌性の要因であったり、また真性糖尿病や超脂肪過多によって起こる、脂肪性肝炎[非アルコール性肝炎(NASH)]から肝硬変に至るアルコールに無関係な重大な生命の危機もある。このようなアルコール性および非アルコール性の肝臓病は、肝臓のウイルス感染によることが多い。そのような場合は病状の進展が迅速である。上記の病気およびその原因ないし結果すべてが本発明による化合物を用いて治療可能なことが明らかになった。
【0054】
このような物質が膵臓の治療にも適していることが分かった。そのような炎症ないし膵炎も、アルコール摂取の濫用に加えて有毒物質により引き起こされることがある。これは特に、職業において取り扱う化学薬品または投薬のような環境毒に分類される。ウイルス感染またはメタボリックな内分泌性の要因によっても、そのような膵臓の炎症が起こることがあり、そのようなあらゆる場合に反応性の酸素が病気発症と病状進展に関与している。
【0055】
真性糖尿病の治療の場合、タイプ2とタイプ1のどちらにも本発明による薬剤が適していることが分かった。
【0056】
中毒性の腎臓病も、また化学療法投薬時の副作用によるその他の疾病、特にシスプラチン、カルボプラチン、二塩化チタノセンのような金属化合物または金化合物などによるものも、本発明による医薬品による治療の対象になる。この場合には特に、金属化合物またはハロゲン置換炭化水素、なかでもモノハロゲンおよびポリハロゲン置換炭化水素、そのなかでもハロタンタイプの蒸気麻酔薬、およびこれらに対応した芳香族炭化水素、ニトロソアミン、アクリルアミドのような他の毒性薬剤、またはパラセタモール、メトトレキサート、イソニアジドのような医薬品、またはアミノグリコリドまたはX線造影剤による臓器毒性が阻止されることが分かった。本発明による医薬品は、特に肝臓、腎臓、中枢神経系、膵臓などの臓器に対する解毒剤として、環境毒による臓器毒性の治療に適している。
【0057】
またこの医薬品は、癌治療における細胞活動停止剤の大量投与を可能とし、またそのような背景の下で化学療法における補助治療として成果を上げる見通しを向上させる。
【0058】
この医薬品は例えば、臓器、特に心臓および脳の梗塞(心筋梗塞、卒中の発作)の後に生物学的組織の再灌注により生じる損傷を防ぐのに特に適していることが分かった。この例として、組織破壊のうち60〜80%がこのような再灌注損傷によるものであったり、この医薬品により組織の死滅の広がりが同じ割合だけ減少したりといったことが動物実験により認められている。再灌注損傷の中心的な原因は、貧血中に生成される活性酸素であることは以前から知られている。
【0059】
本発明による医薬品は、臓器移植時の再灌注損傷の防止にも特に適している。このような臓器は、新しい被移植者の体内に移植されるまで冷却された培養液中で保管される。移植後、この培養液は被移植者の循環系につながれて体液に合流するが、これが再灌注損傷につながる。本発明による医薬品を、臓器保管前と保管中および被移植者への移植の直前にも投与することにより、移植に関係するこの重大な問題を解決することができる。
【0060】
遷移金属である鉄(ウィルソン病の場合には銅)が蓄積されると、チトクロームP450に起因する酸化損傷が起こる可能性が生じる。鉄沈着症に属する鉄過剰蓄積に加えて、貧血性の再灌注損傷の場合には細胞内遊離鉄濃度の上昇が起こることが知られている。細胞内鉄濃度の上昇は、シスプラチンないしハロゲン置換炭化水素のような有毒化合物またはウイルス性肝炎によっても起こる。これに該当するものとしては、肝臓に加えて、腎臓により除去されるシスプラチンのような有毒化合物の場合の腎臓がある。遷移金属である鉄(ウィルソン病の場合には銅)による酸化損傷の生化学的な根拠として次のフェントン反応がある。
【0061】
【化4】
【0062】
ここで、チトクロームP450反応サイクルの一環として生じるH2O2(酸化還元電位+0.32ボルト)は、極端に強力な水酸基置換基HO・(酸化還元電位+2.31ボルト)へと変化する。
【0063】
シスプラチンに起因する腎臓病に伴う遊離鉄濃度の上昇については、例えばバリガ他による文献(「シスプラチンに起因する腎臓毒性における鉄触媒の根源としてのチトクロームP450の役割」、キドニーインターナショナル、1998年、54、1562〜1569)がある。貧血性の再灌注損傷またはウイルス性肝炎の場合の鉄濃度の上昇については、例えばパラー他による文献(「肝臓における再酸素化中の組織破壊ヒドロキシル基形成をチトクロームP450により治療」、Proc. Natl. Acad. Sci.米国1994年、91、7002〜7006)ないしはチャポウトート他による文献(「ウイルス性C硬変が進行した肝硬変細胞癌の患者における肝臓内鉄分過剰」、Gut2000、46、711〜714)がある。
【0064】
本発明による物質には、特にチトクロームP450の第2遺伝子族の人間アイソフォーム、特にアイソフォーム2E1および2J2、そしてこれらに起因する疾病に対する抑止物質としての働きがあることが分かった。
【0065】
本発明の目的に特にかなった形態として、医薬製剤をリポソームで包むことが考えられる。製剤の根底にある化合物は長さの長い脂肪族の基であるという事実があるため、リポソームによる包み込みは投薬に非常に適した形態である。このようなリポソームは、静脈内、筋肉内、腹膜内、経皮または経口投与に適している。またエアロゾルの投与にも適している。
【0066】
ただし本発明による化合物はそのまま直接投与することもできる。そのような場合にも上記のような投与法が適している。
【発明を実施するための最良の形態】
【0067】
〔合成法〕
本発明による様々な化合物の合成法をいくつか以下に示す。
【0068】
1: 12−イミダゾリル−1−ドデカン酸
a) 12−イミダゾリル−1−ドデカン酸はアルターマン他の文献に記されている工程(「チトクロームP450 4A1およびチトクロームP450 4A1/NADPH−P450還元酵素溶解タンパク質による脂肪酸の区別とオメガヒドロキシル化」、生化学&生物物理学アーカイブ(1995年)Vol. 320、289〜296ページ)により合成される。
【0069】
このためには、12−ブロモ−1−ドデカノールがジョーンズの試薬により12−ブロモ−1−ドデカン酸へと酸化される。この白色の固形の酸は引き続いてジアゾメタンと共に、対応するメチルエステルへとエステル化される。このメチルエステルは、直接イミダソールと混ぜられ、80℃で5時間かけて12−イミダゾリル−1−ドデカン酸メチルエステルに転換される。このようにして得られた厚い塊が水とジクロルメタンとに分離され、有機相はNa2SO4により乾燥され蒸発濃縮される。油性の残滓はシリカゲルにかけてクロマトグラフ的に洗浄され、引き続いてメタノールとテトラヒドロフランの混合物(3:4)中で溶解され、LiOH・H2Oと混ぜられ、混合物は還流の下で2時間加熱される。溶剤を蒸発させた後、白色の残滓を再び水に解かして、ジクロルメタンにより抽出を行い、pH5〜6に酸性化した後、酢酸エチルにより改めて抽出を行う。酢酸エチル抽出物をNa2SO4により乾燥させ、フィルタリングして蒸発濃縮する。白色の固形の残滓がメタノール/エーテルにより結晶化されて12−イミダゾリル−1−ドデカン酸が得られる。
【0070】
b) ル(人名)他による文献に記載されている工程(「脂肪酸オメガヒドロキシル化に対する抑止物質としてのラウリン酸に類似したヘム調整」、生化学&生物物理学アーカイブ(1997年)Vol. 337、1〜7ページ)にしたがって12−イミダゾリル−1−ドデカノールと1−イミダゾリルドデカンが合成される。
【0071】
このためにはモル比1:3の12−ブロモ−1−ドデカノールとイミダゾールが80℃で5時間加熱される。この生成原料が水とジクロルメタンとに分離される。有機相はNa2SO4により乾燥され蒸発濃縮される。ベンゾル/n−ヘキサンにより12−イミダゾリル−1−ドデカノールが結晶化される。
【0072】
c) モル比1:3の1−ブロモドデカンとイミダゾールを撹拌して85℃で加熱することにより1−イミダゾリルドデカンが生成される。この生成原料がジクロルメタンに溶かされ、水と共に3回加振される。有機相はNa2SO4により乾燥され、フィルタリングされ、蒸発濃縮される。油性の蒸発残滓はn−ヘキサンにより結晶化され、1−イミダゾリルドデカンが得られる。
【0073】
2: 12−イミダゾリル−1−ホスファチジルコリンの合成
ホスファチジルコリンが、酸性条件下で、かつジシクロヘキシルカルボジイミドの存在の下でO−ホスホリルイソ尿素に転換される。反応混合物に12−イミダゾリル−1−ドデカノールが添加され、これがホスホリル基に対して求核置換作用をしてエステル結合が形成されることにより、12−イミダゾリル−1−ホスファチジルコリンが生成される。これによりジシクロヘキシル尿素が析出される。この反応がうまく行くには4−ジエチルアミノピリジンが必要とされる。
【0074】
この反応のメカニズムは、ジシクロヘキシルカルボジイミドを用いて有機酸をアルコールにエステル化する架橋エステル化に類似している。
【0075】
3: 1−パルミトイル−2−イミダゾイル−グリセロ−3−ホスファチジルコリンの合成
非改質脂肪酸とラベル化(この場合はエチニル化ないしイミダゾール化)された脂肪酸を含むホスファチジルコリン−ジグリセリド合成の原則については、エビル他の文献(「歩留りの高いホスホ脂質の合成」、メソッズ エンツィモル 1983年98号、623〜32)がある。
【0076】
3a: 1,2−ジパルミトイル−3−ベンジルグリセリドの合成
このためには1,2−イソプロピリデン−sn−グリセリンがp−キシレンに溶解され、これにカリウム−tert−ブチレートと塩化ベンジルを加えた上で撹拌する。反応終了後に等分の水とジイソプロピルエーテルが加えられ、相の分離が行われる。上の層に含まれる3−ベンジル−sn−グリセリンが蒸発により取り出され、さらに洗浄ステップが実施される。
【0077】
洗浄された3−ベンジル−sn−グリセリンは引き続いて、パルミチン酸のような脂肪酸と共に四塩化炭素に溶かされる。4−ジエチルアミノピリジンとジシクロヘキシルカルボジイミドを添加すると、3−ベンジル−sn−グリセリンのアルコール基と脂肪酸の間にエステル結合が生じてジシクロヘキシル尿素が析出される。この反応メカニズムが「架橋エステル化」と呼ばれる。
【0078】
析出したジシクロヘキシル尿素は除去され、溶剤は蒸発させられる。洗浄ステップをさらに追加した後、生成物として1,2−ジパルミトイル−3−ベンジル−sn−グリセリドが得られる。
【0079】
3b: 1,2−ジパルミトイル−sn−グリセリドの合成
1,2−ジパルミトイル−3−ベンジル−sn−グリセリドがテトラヒドロフランに溶かされ、触媒(10%のPd/C)の存在の下で、水素原子による水素添加分解が行われる。このときベンジル基が水素原子により置き換えられ、1,2−ジパルミトイル−sn−グリセリドが生成される。
【0080】
3c: 1,2−ジパルミトイル−sn−グリセリドの燐酸化
三塩化ホスホリルがテトラヒドロフランと混ぜられ、氷中で撹拌される。引き続いてテトラヒドロフランに溶かした1,2−ジパルミトイル−sn−グリセリドを滴下して添加する。このようにしてまず1,2−ジパルミトイル−sn−グリセリド−3−二塩化ホスホリルが発生する。
【0081】
引き続いてテトラヒドロフランに溶かしたトリエチルアミンを改めて添加し、テトラヒドロフランに溶かしたブロモエタノールを滴下により加え、温度を25℃に上昇させる。このとき主として発生するのは1,2−ジパルミトイル−sn−グリセリド−3−ホスホリル−ブロモエチルエステル−1塩化物であり、ジ−ブロモエチルエステルは副産物として少量発生するに過ぎない。
【0082】
この混合物が洗浄され、冷却され、炭酸ナトリウムとヘキサンと混ぜられ加振される。このとき、燐酸塩の基と塩化物の間の結合に水酸化が起こる。生成物として1,2−ジパルミトイル−sn−グリセリド−3−ホスホリル−ブロモエチルエステルのナトリウム塩が得られる。
【0083】
1,2−ジパルミトイル−sn−グリセリド−3−ホスホリル−(N−ブトキシカルボニル)−エタノールアミンエステルおよび1,2−ジパルミトイル−sn−グリセリド−3−ホスホリル−(N−ブトキシカルボニル)−tert−ブチルセリンエステルのナトリウム塩についても同様に説明される。
【0084】
3d: 1,2−ジパルミトイル−sn−グリセリド−3−ホスホアルキルエステルの加水分解
1,2−ジパルミトイル−sn−グリセリド−3−ホスホリル−ブロモエチルエステルまたは、この代わりに上記に挙げたホスホアルキルエステルが、ジエチルエーテルと蒸留水の混合物すなわちCaCl2・2H2O中で溶かされる。
【0085】
パリツシュ緩衝剤を添加することによりpH値が7.5に調整され、引き続いて酵素ホスホリパーゼA2が添加され、35℃で60分間撹拌された。このとき、グリセリン基の第2ポジションのエステル結合が加水分解され、これに対応して第2ポジションにOH基を有する1−パルミトイル−sn−グリセリド−3−ホスホアルキルエステルと遊離脂肪酸ができる。
【0086】
こうして得られた分子のグリセリン基の第2ポジションは、イミダゾール化ないしエチニル化された脂肪酸のようなラベル化脂肪酸により狙い通りにエステル化を行うことができる。第3ポジションのホスホアルキルエステルも、コリン、セリン、エタノールアミンまたはイノシトールなど適切なアルコールによりエステル転換することができる。
【0087】
3e: ラベル化脂肪酸による第2ポジションのエステル化
得られた1−パルミトイル−sn−グリセリド−3−ホスホアルキルエステルをテトラクロルメタンに溶かし、イミダゾール化ないしエチニル化された脂肪酸を添加し、混合物を撹拌する。
【0088】
添加される脂肪酸は、例えばシグマ・アルドリッチ社から入手できる17−オクタデシン酸でよい。これは12−イミダゾリル−1−ドデカン酸であり、上記1項で述べた要領でも同様に合成することができる。
【0089】
引き続いて改めて「架橋エステル化」が行われ、混合物に4−ジエチルアミノピリジンとジシクロヘキシルカルボジイミドが添加される。このとき、グリセリン基に残存するOH基とラベル化脂肪酸のカルボキシル基の間にエステル結合が生じる。
【0090】
析出したジシクロヘキシル尿素は除去され、溶剤は蒸発させられる。洗浄ステップをさらに追加した後、生成物として1−パルミトイル−2−アシル−sn−グリセリド―3−ホスホアルキルエステルが得られる。
【0091】
3f: グリセリン基の第3ポジションのホスホアルキルエステルのエステル転換
1−パルミトイル−2−アシル−sn−グリセリド−3−ホスホリル−ブロモエチルエステルをクロロフォルムに溶かす。引き続いて2−プロパノール−トリメチルアミンが添加される。反応容器を50℃で培養状態にして、続いて溶剤を窒素と共に蒸発させる。反応生成物を洗浄するとラベル化1−パルミトイル−2−アシル−sn−グリセリド−3−ホスファチジルコリンが得られる。
【0092】
ラベル化1−パルミトイル−2−アシル−sn−グリセリド−3−ホスファチジルセリンを作り出すために、前記の1−パルミトイル−2−アシル−sn−グリセリド−3−ホスホリル−(N−ブトキシカルボニル)−エタノールアミンエステルをCH2Cl2に溶かし、トリフルオロ酢酸と過クロム酸が添加される。引き続いて冷やした状態で撹拌し、水とメタノールにより洗う。相を分離した後、下側の相についてNa2CO3により抽出を行い、蒸発濃縮を行う。メタノール添加後に生成される結晶が1−パルミトイル−2−アシル−sn−グリセリド−3−ホスファチジル−エタノールアミンである。
【0093】
ラベル化1−パルミトイル−2−アシル−sn−グリセリド−3−ホスファチジルセリンを作り出すための工程も類似している。この場合の出発材料は、上記の1−パルミトイル−2−アシル−sn−グリセリド−3−ホスホリル−(N−ブトキシカルボニル)−tert−ブチルセリンエステルである。
【0094】
〔図面について〕
添付する図面には、本発明による化合物の例がいくつか例示されている。
【0095】
図1〜3は、本発明にしたがって生産される、化学式がA−R−Xの化合物の例を示す。
【0096】
技術者であれば、挙げられている請求の範囲にある以外の数多くの化合物をまとめられることがすぐに分かる。したがって脂肪族の基は、直鎖のことも枝分かれしていることもあり、単結合、二重結合、三重結合のいずれの場合もあって置換可能であり、骨格の炭素原子数は9〜19となる。炭化水素の骨格は同じく脂環および/または芳香族炭化水素により形成することができ、その場合リング構造のために最大40個の炭素原子を必要とすることがある。
親水基としては、イノシトールやエタノールアミン、ないしはこれらのグリセリドなど他のアルコールも問題になる。
【0097】
〔毒性〕
12−イミダゾリル−1−ドデカノール(物質1)および12−(1)−イミダゾリル-ドデカン(物質2)の急性毒性についての試験が、オスのCDラットを使って行われた。その結果12−イミダゾリル−1−ドデカノールのLD50(14日)が1000mg/kg b.w., p.o.であり、12−(1)−イミダゾリル−ドデカンのLD50(14日)は1000mg/kg b.w., p.o.であった。
【0098】
最初の不耐性反応:物質1: 1000mg/kg b.w., p.o.
物質2: 500mg/kg b.w., p.o.
投与量の影響なし:物質1: 500mg/kg b.w., p.o.
物質2: 250mg/kg b.w., p.o.
【0099】
〔12−イミダゾリル−1−ドデカノールの抗腫瘍作用の測定〕
このためには「24−ウェルプレート」に癌細胞株を4本引き、24時間放置して成長させた。続いて供試物質である12−イミダゾリル−1−ドデカノールを様々な濃度で浮遊細胞に加え、DMSOに溶かした。細胞媒質中のDMSOの濃度はそれぞれ0.1%であった。このDMSO濃度は対照試験により毒性がないことが分かり、細胞計数はそれぞれ4日間の培養期間の後に行われた。すべての場合に、癌細胞の増殖が強力に阻止されることが分かった。
【0100】
12−イミダゾリル−1−ドデカノールの、抑制剤の最大半減濃度値IC50は次のように求められた。
【0101】
HepG2(肝細胞) 50nM
Panc-1(膵臓細胞) 50nM
PC-3(前立腺細胞) 50nM
SW620(大腸細胞) 100nM
【0102】
癌細胞株すべてに抑制剤の強力な抗抗腫瘍効果が認められた。
【0103】
〔12−イミダゾリル−1−ドデカノールの細胞毒性の評価〕
12−イミダゾリル−1−ドデカノールの細胞毒性値の最大半減値がLDH細胞毒性試験により求められた。
この測定値は次の通りとなっている:
MRC-5(肺繊維芽細胞) =500μM
Panc-1(膵臓細胞) =500μM
【0104】
細胞毒性値の最大半減値500μMを、Panc−1に対する抑制剤の最大半減濃度値IC50=50μMと比較してみると10,000倍の開きがある。このことから12−イミダゾリル−1−ドデカノールは、ガン細胞の増殖に対して非常に効果的で細胞毒性が比較的低い抑制剤であると推定される。
言い換えれば本発明による物質の治療面での窓口の幅は広い。
【0105】
〔12−イミダゾリル−1−ドデカノールによるHepG2細胞の活動性抑制〕
遊走試験条件: HepG2肝臓癌細胞がコラーゲンマトリクスに埋め込まれた。30の個別細胞の写真による監視が900分にわたって連続的に行われた。遊走する細胞の割合の平均値が求められた。
【0106】
結果1: 非治療の対照HepG2細胞の遊走の平均値は15%だった。12−イミダゾリル−1−ドデカノール1μMないし10μMをマトリクス媒質に加えた場合、細胞の遊走は50%ないし75%低下した。
【0107】
結果2: HepG2細胞はインスリン受容体を明確に表わすため、12−イミダゾリル−1−ドデカノールありとなしの場合の遊走に対するインスリンの影響が求められた。
【0108】
【表1】
【0109】
インスリンは遊走を2.5倍に増大させる。作用物質により450分の観察時間(観察時間の前半)後にはこの増大が全面的にブロックされた。900分後(観察時間の後半)には増大がマイナスになった。遊走の平均値は対照以下の13%となった。
【図面の簡単な説明】
【0110】
【図1】本発明にしたがいイミダゾール化ないしエチニル化された脂肪族の例を示す表である。
【図2】本発明にしたがいイミダゾール化ないしエチニル化され、親水基により改質された脂肪族の例を示す表である。
【図3】本発明にしたがいイミダゾール化ないしエチニル化されたグリセリドの例を示す表である。
【図4】本発明にしたがいイミダゾール化ないしエチニル化されたホスホグリセリドの例を示す表である。
【特許請求の範囲】
【請求項1】
下記化学式の化合物、または薬学的に条件を満たす前記化合物の塩を、腫瘍性疾患、アルコール摂取の濫用による病理学的作用、ウイルス性肝炎、脂肪肝、急性および慢性の膵炎、中毒性腎疾患、真性糖尿病の場合の肝臓の抗インシュリン作用、ウィルソン病およびジデローシスの場合の肝臓損傷、もしくは虚血性再灌流による疾患の予防ないし治療のため、環境毒および薬物中毒に対する解毒用途のため、投薬薬剤の臓器内在期間延長、または化学療法投薬時の有毒副作用克服のための医薬製剤の生産に使用する用法。
【化1】
(式中、RはC6〜C40の脂肪族ないし芳香族炭化水素を表し、端部基Aは親水性の基または水素であり、Xは炭素原子ないしヘテロ原子の自由電子ペアおよび/またはπ電子の基を表す。)
【請求項2】
Xが、アミン、ジアゾ基、ニトリル、イソニトリル、メチレンジオキシ基、Sを含む基、複素環および/またはアルキニル基である、請求項1に記載の用法。
【請求項3】
親水性の端部基Aが、カルボン酸基、硫酸基、燐酸基、燐酸塩エステル、配糖体、ポリオール基、ポリサッカライドまたはアミノ酸基である、請求項1または2に記載の用法。
【請求項4】
Xが、イミダゾール基、ニトリル基、イソニトリル基、チオール基またはエチニル基(−C≡CR12)であり、式中、R12はHまたは場合によって置換されたC1〜C15炭化水素である、請求項1〜3のいずれかに記載の用法。
【請求項5】
A−R−が、アルカノール基、アルキル硫酸塩の基、アルキル−CoA−基であるか、または主鎖の炭素原子数6〜26個で直鎖ないし枝分かれしており、単結合、二重結合、または三重結合があり、置換されているアルカン酸の基である、請求項1〜4のいずれかに記載の用法。
【請求項6】
ポリオール基が下記化学式のグリセリン基である、請求項3に記載の用法。
【化2】
(式中、少なくともR2〜R4基はRX基であり、Bは酸素、硫黄、セレンおよび/またはセレン酸塩、アミノ基、リンおよび/または硫酸基を表す。)
【請求項7】
R2〜R4基のうちの一つが、ホスファチジルコリン基、ホスファチジルエタノールアミン基、ホスファチジルセリン基またはホスファチジルイノシトール基である、請求項6に記載の用法。
【請求項8】
請求項1〜7のいずれかに記載の用法における化合物を、薬学的に条件を満たす媒体中に含む医薬製剤。
【請求項9】
リポソームに包まれている、請求項8に記載の医薬製剤。
【請求項1】
下記化学式の化合物、または薬学的に条件を満たす前記化合物の塩を、腫瘍性疾患、アルコール摂取の濫用による病理学的作用、ウイルス性肝炎、脂肪肝、急性および慢性の膵炎、中毒性腎疾患、真性糖尿病の場合の肝臓の抗インシュリン作用、ウィルソン病およびジデローシスの場合の肝臓損傷、もしくは虚血性再灌流による疾患の予防ないし治療のため、環境毒および薬物中毒に対する解毒用途のため、投薬薬剤の臓器内在期間延長、または化学療法投薬時の有毒副作用克服のための医薬製剤の生産に使用する用法。
【化1】
(式中、RはC6〜C40の脂肪族ないし芳香族炭化水素を表し、端部基Aは親水性の基または水素であり、Xは炭素原子ないしヘテロ原子の自由電子ペアおよび/またはπ電子の基を表す。)
【請求項2】
Xが、アミン、ジアゾ基、ニトリル、イソニトリル、メチレンジオキシ基、Sを含む基、複素環および/またはアルキニル基である、請求項1に記載の用法。
【請求項3】
親水性の端部基Aが、カルボン酸基、硫酸基、燐酸基、燐酸塩エステル、配糖体、ポリオール基、ポリサッカライドまたはアミノ酸基である、請求項1または2に記載の用法。
【請求項4】
Xが、イミダゾール基、ニトリル基、イソニトリル基、チオール基またはエチニル基(−C≡CR12)であり、式中、R12はHまたは場合によって置換されたC1〜C15炭化水素である、請求項1〜3のいずれかに記載の用法。
【請求項5】
A−R−が、アルカノール基、アルキル硫酸塩の基、アルキル−CoA−基であるか、または主鎖の炭素原子数6〜26個で直鎖ないし枝分かれしており、単結合、二重結合、または三重結合があり、置換されているアルカン酸の基である、請求項1〜4のいずれかに記載の用法。
【請求項6】
ポリオール基が下記化学式のグリセリン基である、請求項3に記載の用法。
【化2】
(式中、少なくともR2〜R4基はRX基であり、Bは酸素、硫黄、セレンおよび/またはセレン酸塩、アミノ基、リンおよび/または硫酸基を表す。)
【請求項7】
R2〜R4基のうちの一つが、ホスファチジルコリン基、ホスファチジルエタノールアミン基、ホスファチジルセリン基またはホスファチジルイノシトール基である、請求項6に記載の用法。
【請求項8】
請求項1〜7のいずれかに記載の用法における化合物を、薬学的に条件を満たす媒体中に含む医薬製剤。
【請求項9】
リポソームに包まれている、請求項8に記載の医薬製剤。
【図1】

【図2】
【図3】
【図4】
【図2】
【図3】
【図4】
【公表番号】特表2009−535309(P2009−535309A)
【公表日】平成21年10月1日(2009.10.1)
【国際特許分類】
【出願番号】特願2009−506914(P2009−506914)
【出願日】平成19年4月27日(2007.4.27)
【国際出願番号】PCT/DE2007/000768
【国際公開番号】WO2007/124734
【国際公開日】平成19年11月8日(2007.11.8)
【出願人】(508321281)
【氏名又は名称原語表記】MUELLER−ENOCH,Dieter
【住所又は居所原語表記】Lisztweg 5,89160 Dornstadt,Deutschland
【出願人】(508321270)
【氏名又は名称原語表記】HAEHNER,Thomas
【住所又は居所原語表記】Rathausgasse 9,89160 Dornstadt,Deutschland
【Fターム(参考)】
【公表日】平成21年10月1日(2009.10.1)
【国際特許分類】
【出願日】平成19年4月27日(2007.4.27)
【国際出願番号】PCT/DE2007/000768
【国際公開番号】WO2007/124734
【国際公開日】平成19年11月8日(2007.11.8)
【出願人】(508321281)
【氏名又は名称原語表記】MUELLER−ENOCH,Dieter
【住所又は居所原語表記】Lisztweg 5,89160 Dornstadt,Deutschland
【出願人】(508321270)
【氏名又は名称原語表記】HAEHNER,Thomas
【住所又は居所原語表記】Rathausgasse 9,89160 Dornstadt,Deutschland
【Fターム(参考)】
[ Back to top ]