
化学療法および/または放射線療法の副作用を処置するためのRARアンタゴニストまたはRARインバースアゴニストの使用
化学療法および/または放射線療法を受ける哺乳動物において化学療法および/または放射線療法の副作用を処置するための方法であって、RARα、RARβおよびRARγサブタイプの受容体に結合する少なくとも1つのレチノイン酸受容体(RAR)アンタゴニストまたはRARインバースアゴニストの治療有効量を哺乳動物に投与する工程を含む方法を開示する。このような副作用としては、化学放射線療法誘導脱毛症、化学放射線療法誘導血小板減少症、化学放射線療法誘導白血球減少症および化学放射線療法誘導好中球減少症が挙げられる。
【発明の詳細な説明】
【技術分野】
【0001】
関連出願
この出願は、2006年5月16日に出願された米国仮出願第60/800,773号(この全体の教示は、参考として本明細書に援用される)の利益を主張する。
【0002】
本発明は一般に、哺乳動物における化学療法および放射線療法の副作用を処置する方法に関する。
【背景技術】
【0003】
関連技術の説明
一般に、哺乳動物における正常細胞は、秩序正しく制御された様式で増殖し分裂する。癌は、細胞が異常になって、制御されずに増え始め、腫瘍と呼ばれる余分な組織塊へと発達する疾患である。これらの癌細胞は、近傍の組織に浸潤し、血流およびリンパ系を介して身体の他の部分へ拡散する。
【0004】
現在、4つの主な癌治療のタイプは、免疫療法、外科手術、放射線療法、および化学療法である。これらの癌治療は、単独で、または他の治療と関連させて適用することができる。したがって、癌患者は、一度に1つ又は複数の治療を受けることができる。単独治療は、種々の間隔で送達される療法によって、一定の期間にわたり得る。免疫療法は、該疾患と闘うために、免疫系の能力を刺激するか、または回復させることを意図する。免疫療法はまた、いくつかの癌治療によって引き起こされ得る免疫系に関連した副作用を減少させるために用いることもできる。外科手術は、身体から腫瘍を直接除去しようとする。
【0005】
照射療法としても知られている放射線療法は、該細胞の遺伝子材料を損傷させることによって癌細胞を殺して腫瘍を縮小させるために、x線、ガンマ線、ニュートロン、および他の供給源からの高エネルギー照射を用いる。癌細胞は永久的に損傷を受け、その結果死滅するが、放射線療法で損傷を受ける正常細胞は、それら自身を修復することができる。放射線療法中に生じ得る副作用としては、治療を受けている部位の皮膚刺激および毛髪喪失、ならびに骨髄に対する損傷が挙げられる。
【0006】
化学療法では、癌細胞を破壊するために、細胞毒性薬を単独で、または組み合わせて用いる。放射線療法でのように、癌細胞には損傷を与えて結果的に死滅させることができるが、その過程で影響を受けた健常細胞は、化学療法後にそれら自身を修復することができる。細胞毒性薬は、増殖細胞が分裂しそれ自身を複製する能力を妨害することによって作用する。したがって、癌細胞に加えて、他の正常な、速やかに分裂し増殖している細胞もまた影響を受ける可能性がある。例えば、骨髄中に形成される血液細胞に影響を与えて、骨髄抑制を引き起こす可能性がある。また、例えば、消化管内、口腔の内層および生殖系の細胞に影響を与えて、下痢および口腔のひりひり感を起こしたり、また、毛嚢に影響を与えて毛髪喪失を引き起こす可能性がある。
【0007】
骨髄抑制は、化学療法および放射線療法の多くの副作用のうちの1つである。それによって、赤血球、白血球、および血小板などの血液細胞の産生減少がもたらされる。その結果、患者は、貧血から、疲労を経験し、白血病から、より感染しやすくなり、血小板減少症から、打撲傷を受けやすく、切り傷の際に出血が多くなる可能性がある。骨髄抑制の副作用に対抗するために、薬剤が一般的に用いられる。例えば、癌の化学療法における貧血の副作用に対抗するために、Epogen(登録商標)(エポイエチン)が用いられており、血小板減少症の副作用に対抗するために、WinRho(登録商標)SDF(Rho(D)免疫グロブリン)が用いられている。
【0008】
化学療法および放射線療法の副作用の予防、またはそれらからの保護は、癌患者にとって大きな利益になると考えられる。これらの副作用を減少させる以前の多くの努力は概して不首尾に終わっている。命を脅かす副作用に関しては、副作用を減少させるために、化学療法剤および放射線療法剤の用量およびスケジュールを変更することに努力が集中されてきた。化学放射線療法を開始する前に、種々の組織における正常細胞の数を増加させるために、顆粒球コロニー刺激因子(G−CSF)、顆粒球−マクロファージ−CSF(GM−CSF)、上皮成長因子(EGF)、インターロイキン11(il−11)、エリスロポイエチン、トロンボポイエチン、巨核球発達および増殖因子、ピキシキナーゼ、幹細胞因子、FLT−リガンド、ならびにインターロイキン1、3、6、および7の使用など、他の選択肢が利用できるようになってきている。十分に理解されてはいないが、これらの因子による保護の機序は、細胞毒性剤または放射線療法による処置前の正常な危険標的細胞数の増加に関連しており、化学放射線療法後の細胞生存率の増加に関連していない可能性が高い。
【0009】
一般に、多形核白血球とも呼ばれる好中球は、顆粒球として知られる血球のうちで最も多い。好中球は急性炎症応答に関与する最も大きな細胞集団である。したがってそれらは、自然免疫の重要な要素であり、化学走化性刺激に速やかに応答する。好中球は、食作用と呼ばれる、包囲し消化する過程によって、細菌などの外来粒子を破壊する。好中球は、細菌感染に応答して増加し得る。多くの好中球が必要な場合、それらはバンドまたは杵核球と呼ばれる未成熟細胞として骨髄から放出される。好中球減少症は、異常に少数の好中顆粒球を特徴とする血液病である。したがって、好中球減少症に罹っている患者は、細菌感染により罹りやすくなり、これらの状態は命を脅かすものとなり得る。
【0010】
好中球減少症は、癌または後天性免疫不全症候群(AIDS)などの他の状態に副次的に生じ得る。好中球減少症はまた、薬剤療法などの事象に副次的に生じ得る。したがって好中球減少症は、免疫系に直接影響を及ぼす生理学的障害に起因し得る。例えば、好中球産生の減少は、白血病、骨髄腫、リンパ腫、または例えば、乳癌または前立腺癌などの転移性固形腫瘍が、骨髄に浸潤しそれに取って代る場合に生じる。一過性好中球減少症はウィルス感染に関連していることが多い。慢性好中球減少症は、ウィルス感染、例えば、ヒト免疫不全ウィルス(HIV)の感染に起因するAIDSに起因する免疫不全に関連していることが多い。自己免疫好中球減少症は、循環抗好中球抗体に関連している可能性がある。
【0011】
好中球減少症のはるかに一般的な原因は、薬物療法、特に、癌ならびに癌療法に関連した骨髄移植に対する化学療法および放射線療法の副作用である。したがって、薬物療法に副次的な好中球減少症は2つの群に分けることができる。第1の群は、抗体形成を刺激するためのハプテンとして作用する薬物から生じ得る免疫媒介好中球減少症を含む。ジフェニルヒダントインおよびフェノバルビタールによって引き起こされるものなどの急性過敏症反応は数日間継続する。しかし、慢性過敏症反応は数ヶ月または数年継続し得る。
【0012】
薬物に誘導された好中球減少症の第2の範囲は、高用量の細胞減少性癌薬剤または電離放射線療法後に生じることが予想される重症の好中球減少症を含む。これらの細胞毒性療法は、好中球前駆細胞の増殖的性質および循環好中球の通常の速い転換率により好中球減少症を誘導する。癌の化学療法または放射線療法に副次的な好中球減少症の危険性は、癌のタイプおよび病期、ならびに癌治療のタイプ、用量およびスケジュールなどの要因に依存する。
【0013】
好中球レベルを増加させるために現在存在する療法は、主に、フィルグラスチム(Neulasta(登録商標))、より最近では、フィルグラスチムのより長時間作用性の誘導体であるペグフィルグラスチム(Neulasta(登録商標))からなる。フィルグラスチムは、白血球の産生を選択的に刺激するヒトタンパク質、G−CSFの組換え型である。G−CSFは、好中球減少症の現在の選択薬剤である。これらの薬剤は双方とも、組換えタンパク質であるため、経口で活性ではなく、注射によって投与しなければならない。また、タンパク質ベースの薬剤は、しばしば急速な代謝を受ける。
【0014】
化学療法および放射線療法の分野における進歩にも関わらず、先行技術の薬剤および方法は、化学療法誘導脱毛症、放射線療法誘導脱毛症、化学療法誘導血小板減少症、放射線療法誘導血小板減少症、化学療法誘導白血球減少症、放射線療法誘導白血球減少症、化学療法誘導好中球減少症および放射線療法誘導好中球減少症などの、化学療法および放射線療法に起因する副作用の最少化において利用が限定されることが判明している。したがって、哺乳動物における化学放射線療法のこのような副作用を処置するための改善された方法を提供することが望ましいと考えられる。
【発明の概要】
【課題を解決するための手段】
【0015】
本発明の一実施形態により、化学療法および/または放射線療法を受けている哺乳動物における化学療法および/または放射線療法の副作用を処置するための方法が提供され、該方法は、RARα、RARβ および RARγサブタイプの受容体に結合するレチン酸受容体(RAR)アンタゴニストまたはRARインバースアゴニストの治療有効量を該哺乳動物に投与する工程を含んでなる。
【0016】
本発明の第2の実施形態により、哺乳動物における血小板産生を増加させる方法が提供され、該方法は、RARα、RARβおよびd RARγサブタイプの受容体に結合するRARアンタゴニストまたはRARインバースアゴニストの治療有効量を該哺乳動物に投与する工程を含んでなる。
【0017】
本発明の第3の実施形態により、血小板減少症を患っている哺乳動物を処置する方法が提供され、該方法は、RARα、RARβおよびd RARγサブタイプの受容体に結合するRARアンタゴニストまたはRARインバースアゴニストの治療有効量を該哺乳動物に投与する工程を含んでなる。
【0018】
本発明の第4の実施形態により、造血関連状態を患っている哺乳動物を処置する方法が提供され、該方法は、RARα、RARβおよびd RARγサブタイプの受容体に結合するRARアンタゴニストまたはRARインバースアゴニストの治療有効量を該哺乳動物に投与する工程を含んでなる。このような状態としては、限定はしないが、造血機能低下、免疫機能低下、好中球数減少、好中球動員減少、末梢血始原細胞の動員、敗血症、重症の慢性好中球数減少症、骨髄移植、感染性疾患、白血球減少症、血小板減少症、貧血、移植時の骨髄移植増大、放射線処置における骨髄回復の増大、化学物質または化学療法に誘導された骨髄形成不全または骨髄抑制、後天性免疫不全症候群など、ならびにそれらの組み合わせが挙げられる。
【図面の簡単な説明】
【0019】
【図1】図1A〜1Cは、シクロホスファミドに誘導された白血球減少症マウスモデルにおける白血球、好中球およびリンパ球の数に及ぼすVTP 194310の効果のグラフを示す図である。
【図2】図2A〜2Dは、HL60細胞のATRAに駆動された好中球の分化に対する全RARアンタゴニストVTP194310のブロック効果のグラフを示す図である。2.5×105細胞/mlに設定されたHL60細胞を、100nMのパン−RARアンタゴニストVTP194310と共に、100nMのATRAまたは100 nMのATRA で5日間処置した。Aは、位相差顕微鏡によって数え上げた生存細胞の総数を示す。分化は、ニトロブルーテトラゾリウムを減少させる細胞の能力により測定し(B)、CD11bの発現は、モノクローナル抗CD11bおよびFACSを用いて測定し(C)、酵素ステロイドスルファターゼの活性増加は、[3H]硫酸エストロンを用いた細胞超音波処理、続いて[3H]エストロンの抽出で測定した。
【図3】図3A〜3Bは、好中球分化に及ぼすVTP 194310 および G−CSFの単独、および組み合わせ効果のグラフを示す図である。ヒトCD34+ve 造血始原細胞(CD34−HPC)を均質にカラム精製し、幹細胞因子(100 ng/ml)およびIL3(20 ng/ml)中で(●)、ならびにパン特異的RARアンタゴニストVTP 194310の10nM(□)および100 nM(■)と共に培養した。好中球および単球の最適な産生を促進するために、CD34−HPCを、幹細胞因子(100 ng/ml)およびIL3(5 ng/ml)およびG−CSF(30 ng/ml)中で培養した(▲)。細胞表現型を、時々多色FACSアッセイにより判定した。培養は三重に設定し、各マーカーに関する多色FACS分析および単色分析は二重に行なった。好中球は、CD11b+ve/CD65+ve 細胞およびCD15+ve細胞として同定した。単球は、CD11b+ve/CD14+ve細胞として同定した。データは平均値±SDとして示されている。
【図4】図4A〜4Cは、シクロホスファミドに誘導された白血球減少症マウスモデルにおける好中球数および他のパラメーターに及ぼすVTP 194310の効果のグラフを示す図である。
【図5】黄色ブドウ球菌(Staphylococcus aureus)に致死的に感染した白血球減少症マウスの生存率に及ぼすVTP 194310の効果のグラフを示す図である。
【図6】図6A〜6Eは、VTP 194310または/およびPEG−G−CSFを投与されたマウスにおける総白血球(WBC)、好中球、リンパ球、赤血球(RBC)および血小板の数の全体の変化における変化のグラフを示す図である。1mg/kg/日または3mg/kg/日での媒体またはVTP 194310は、−1日目から1日目まで経口投与し、150mg/kgのCPMは、0日目に腹腔内投与し、PEG−G−CSFは、2日目に皮下投与した。血液サンプルはスケジュール通り採取し、血球数は、Abbott Cell−DYN 3700により測定した。データは、7〜8匹のマウスの平均値±SEを表す。単位は、1μl当たりの細胞数または血小板数である。
【図7】VTP 194310、PEG−G−CSFの各々単独に比較して、好中球減少症マウスにおける好中球回復率をさらに増大させるVTP 194310およびPEG−G−CSFの療法による好中球減少症処置の効果のグラフを示す図である。0日目、150 mg/kg CPMによって好中球減少症にしたマウスに、VTP194310(−1日目から1日目まで3mg/kg/日)または PEG−G−CSF(2日目に10μg/kg)による単独療法ならびにこれらの用量の組み合わせ処置を行なった。データは、7〜8匹のマウスの平均値±SEを表す。処置群間の統計的有意性に関するp値が示されている。単位は、1μl当たりの細胞数または血小板数である。
【図8】図8A〜8Eは、好中球減少症の5−FU誘導マウスモデルにおけるVTP 194310または/およびPEG−G−CSFを投与されたマウスにおける総白血球(WBC)、好中球、リンパ球、赤血球(RBC)および血小板の数の変化のグラフを示す図である。示されているとおり、3mg/kg/日の媒体またはVTP 194310は、2日目から4日目まで経口投与し、150mg/kgの5−FUは、0日目に静脈内投与し、PEG−G−CSFは、5、6、または7日目に皮下投与した。血液サンプルはスケジュール通り採取し、血球数は、Abbott Cell−DYN 3700により測定した。データは、8匹のマウスの平均値±SEを表す。単位は、1μl当たりの細胞数または血小板数である。
【図9】好中球減少症の5−FU誘導マウスモデルにおける好中球の回復に及ぼすVTP 194310またはPEG−G−CSFによる単独療法の効果と組み合わせ処置の効果の比較のグラフを示す図である。0日目、150 mg/kgの5−FUによって好中球減少症にしたマウスに、VTP194310(2日目から4日目まで3mg/kg/日)または PEG−G−CSF(5、6、または7日目に10μg/kg)による単独療法ならびにこれらの用量の組み合わせ処置を行なった。データは、8匹のマウスの平均値±SEを表す。
【発明を実施するための形態】
【0020】
発明の詳細な説明
本発明は、RARα、RARβおよび RARγサブタイプの受容体に結合するレチン酸受容体(RAR)の少なくとも1種のアンタゴニストおよび/またはインバースアゴニスト、すなわち、RARα、RARβ および RARγサブタイプの全てに結合するRARアンタゴニストまたはRARインバースアゴニストを用いる化学療法および/または放射線療法を受けている哺乳動物における、化学療法および/または放射線療法(すなわち、化学放射線療法)の副作用の処置のための方法に関する。このような副作用としては、限定はしないが、化学療法誘導脱毛症、放射線療法誘導脱毛症、化学療法誘導血小板減少症、放射線療法誘導血小板減少症、化学療法誘導白血球減少症、放射線療法誘導白血球減少症、化学療法誘導好中球減少症および放射線療法誘導好中球減少症など、ならびにそれらの組み合わせが挙げられる。
【0021】
RARα、RARβおよび RARγ受容体サブタイプにおけるアゴニスト活性を判定するために使用できるアッセイは、Feigner,P.L.、およびd Holm,M.、Focus、11:2頁、21頁+(1989)ならびに米国特許第5,455,265号および米国特許第7,166,726号に記載されており、これらは参照として、それらの全体が本明細書に援用されている。
【0022】
レチノイドインバースアゴニストの活性は、Kleinら、J.Biol.Chem.271,22692〜22696頁(1996)の手法によって試験することができ、これは特に参照として、本明細書に援用されている。
【0023】
レチノイド類、特に全てのトランスレチン酸(ATRA)は、広範囲の細胞型の生存、増殖および分化に重要な役割を果たすため、正常な哺乳動物の発達にとって必須である。ATRAおよび合成レチノイド類は、受容体の2つの別個の細胞内ファミリー、RAR類およびレチノイドX受容体(RXR類)に結合し、それらを活性化することができ、その結果、遺伝子発現の調節をもたらす。RARαと称される、最初に同定されたレチン酸受容体は、ステロイド/甲状腺ホルモン細胞内受容体スーパーファミリーの多くのメンバーの場合に示されたようなリガンド依存的な様式で、特定の標的遺伝子の転写を調節するように作用する。RARαの転写調節活性が依存する内因性低分子量リガンドは、ATRAである。レチン酸受容体に媒介された遺伝子発現の変化によって、細胞表現型の特徴的な変化がもたらされ、その結果、多くの組織にATRAに対する生物学的応答が現れる。RARαに密接に関連したさらに2つの遺伝子は、RARβおよびRARγと称される。RARsのように、RXRsもまた、少なくとも3つのサブタイプまたはイソ型、すなわち、RXRα、RXRβ、およびRXRγを有し、対応するユニークな発現パターンを有することが知られている(Manglesdorfら、Genes & Devel.、6:329〜44頁(1992))。
【0024】
RARα、RARβおよび RARγサブタイプの受容体に結合するRARアンタゴニストおよび/またはRARインバースアゴニストを含んでなる組成物を哺乳動物に投与することにより、血液の好中球および血小板の産生が向上し、化学療法に誘導された好中球減少症および/または血小板減少症など、化学療法の副作用の処置がもたらされると考えられる。
【0025】
RARα、RARβおよびRARγサブタイプの受容体に結合するRARのアンタゴニストおよびインバースアゴニストの代表例ならびにそれらを調製するための方法は、米国特許第5,776,699号および米国特許第5,958,954号ならびに米国特許出願公開2002/0193403号において、当業界によく知られており、それら各々の内容は参照として、それらの全体が本明細書に援用されている。以下の化合物の多くは、これらの出願および/または特許の1つまたは複数に含まれている。
【0026】
本発明の特定の実施形態は、一般式I:
【0027】
【化1】

により表され、使用することができる一クラスの化合物または薬学的に許容できるその塩であり;
式中、Xは、S、O、RがHまたは1個から6個の炭素のアルキルであるNRか、または
Xは、R1が独立してHまたは1個から6個の炭素のアルキルである[C(R1)2]nであり、nは、0と2の間でかつ0と2を含む整数であり;
R2は、独立して水素、1個から6個の炭素の低級アルキル、F、Cl、Br、I、CF3、1個から6個の炭素のフルオロ置換アルキル、OH、SH、1個から6個の炭素のアルコキシ、または1個から6個の炭素のアルキルチオであり;
R3は、独立して水素、1個から6個の炭素の低級アルキルまたはFであり;
mは、0〜3の値を有する整数であり;
nは、0〜4の値を有する整数であり;
oは、0〜3の値を有する整数であり;
Zは、−CONR1−、−CSNR1−、−NR1CO−、−NR1CS−、−C≡C−、−C=C−、−N=N−、−N=CR1−、−CR1=N−、
−COO−、−OCO−;−OSO−;−OCS−、またはn’が0から5の整数である−(CR1=CR1)n’−であり、;
Yは、フェニル基またはナフチル基、またはピリジル、チエニル、フリル、ピリダジニル、ピリミジニル、ピラジニル、チアゾリル、オキサゾリル、イミダゾリルおよびピラゾリルからなる群より選択されるヘテロアリールであり、前記フェニル基およびヘテロアリール基は、1つまたは2つのR2基で任意に置換されているか、またはZが−(CR1=CR1)n’−であり、n’が3、4または5である場合、Yは、前記(CR2=CR2)n’基とBとの間の直接的原子価結合を表し;
Aは、qが0〜5である(CH2)q、3個から6個の炭素を有する低級分枝鎖アルキル、3個から6個の炭素を有するシクロアルキル、2個から6個の炭素および1つまたは2つの二重結合を有するアルケニル、2個から6個の炭素および1つまたは2つの三重結合を有するアルキニルであり;
Bは、水素、COOHまたは薬学的に許容できるその塩、COOR8、CONR9R10、CH2OH、CH2OR11、CH2OCOR11、CHO、CH(OR12)2、CHOR13O、−COR7、CR7(OR12)2、CR7OR13O、またはトリ−低級アルキルシリルであり;
R7は、1個から5個の炭素を含有するアルキル基、シクロアルキル基またはアルケニル基であり;
R8は、1個から10個の炭素のアルキル基、またはアルキル基が1個から10個の炭素を有するトリメチルアルキル、1個から10個の炭素、3個から10個の炭素のシクロアルキル基、フェニルまたは低級アルキルフェニルであり;
R9およびR10は、独立して水素、1個から10個の炭素のアルキル基、3個から10個の炭素のシクロアルキル基、フェニルまたは低級アルキルフェニルであり;
R11は、低級アルキル、フェニルまたは低級アルキルフェニルであり;
R12は、低級アルキルであり;
R13は、2個から5個の炭素の二価アルキル基であり;
R14は、(R15)r−フェニル、(R15)r−ナフチル、または(R15)r−ヘテロアリールであり、前記ヘテロアリール基が、O、SおよびNからなる群より選択される1個から3個のヘテロ原子を有し、rが、0〜5の値を有する整数であり;
R15は、独立してH、F、Cl、Br、I、NO2、N(R8)2、N(R8)COR8、NR8CON(R8)2、OH、OCOR8、OR8、CN、1個から10個の炭素を有するアルキル基、1個から10個の炭素を有するフルオロ置換アルキル基、2個から10個の炭素および1つから3つの二重結合を有するアルケニル基、2個から10個の炭素および1つから3つの三重結合を有するアルキニル基、またはアルキル基が独立して1個から6個の炭素を有するトリアルキルシリル基またはトリアルキルシリルオキシ基である。
【0028】
本発明の別の特定の実施形態は、式Iにより表され、使用することができる一クラスの化合物であり、Zは、-CONR1−、−CSNR1−、−NR1CO−、または−NR1CS−であり;全て他の可変部分は、上記に定義されたとおりである。
【0029】
本発明の別の特定の実施形態は、式Iにより表され、使用することができる一クラスの化合物であり;式中、
R8は、1個から10個の炭素のアルキル基ま、またはアルキル基が1個から10個の炭素を有するトリメチルアルキル、5個から10個の炭素のシクロアルキル基、フェニルまたは低級アルキルフェニルであり;
R9およびR10は、独立して水素、1個から10個の炭素のアルキル基、5個から10個の炭素のシクロアルキル基、フェニルまたは低級アルキルフェニルであり;全て他の可変部分は、上記に定義されたとおりである。
【0030】
本発明の別の特定の実施形態は、一般式II:
【0031】
【化2】
により表され、使用することができる一クラスの化合物または該化合物の薬学的に許容できるその塩であり;
式中、Xは、−C(R1)2または−O−であり;R1は、HまたはC1〜C6であり;
R2は、C1〜C6低級アルキル、−F、−Cl、−Br、−I、−CF3、フルオロ置換C1〜C6アルキル、−OH、−SH、C1〜C6アルコキシ、またはC1〜C6アルキルチオであり;
mは、0から3の値を有する整数であり;
nは、0から4の値を有する整数であり;
oは、0から3の値を有する整数であり;
R3は、C1〜C6低級アルキルまたはFであり;
R8は、C1〜C10アルキル基、C1〜C10トリメチルシリルアルキルであり、C3〜C10シクロアルキル基、フェニルまたは低級アルキルフェニルであり;
R15は、独立して−H、−F、−Cl、−Br、−I、−NO2、−N(R8)2、−COR8、−NR8CON(R8)2、−OCOR8、−OR8、−CN、C1〜C10アルキル、フルオロ置換C1〜C10アルキル、1つから3つの二重結合を有するC2〜C10アルケニル、1つから3つの三重結合を有するC2〜C10アルキニル、またはC1〜C6トリアルキルシリルまたはトリアルキルシリルオキシであり;
tは、0〜5の整数であり;
前記−CONH基は、ベンゾピランおよびジヒドロナフタレン環の6位または7位にある。
【0032】
本発明の別の特定の実施形態は、一般式IIにより表され、使用することができる一クラスの化合物であり、式中:
R2はFであり;R8は、1個から10個の炭素のアルキル基、該アルキル基が1個から10個の炭素を有するトリメチルシリルアルキル、5個から10個の炭素のシクロアルキル基、フェニルまたは低級アルキルフェニルであり;
R15は、独立してH、F、Cl、Br、I、NO2、N(R8)2、COR8、NR8CON(R8)2、OCOR8、OR8、CN、1個から10個の炭素を有するアルキル基、1個から10個の炭素を有するフルオロ置換アルキル基、1個から10個の炭素および1つから3つの二重結合を有するアルケニル基、1個から10個の炭素および1つから3つの三重結合を有するアルキニル基、またはアルキル基が独立して1個から6個の炭素を有するトリアルキルシリル基またはトリアルキルシリルオキシ基であり;および他の全ての可変部分は、上記に定義されているとおりである。
【0033】
本発明の別の特定の実施形態は、一般式III:
【0034】
【化3】
により表され、使用することができる一クラスの化合物または薬学的に許容できるその塩であり;
式中、Xは、−C(CH3)2−または−O−であり;
R2は、−Hまたは−Brであり;
R2’およびR2’’は、独立して−Hまたは−Fであり;
R3の各々は、独立して−Hまたは−CH3であり;
R8は−Hまたは、C1〜C6 アルキルである。
【0035】
本発明の別の特定の実施形態は、一般式IV:
【0036】
【化4】
により表され、使用することができる一クラスの化合物または薬学的に許容できるその塩であり;
式中、X1は、−S−または−O−であり;
X2は、−CH−または−N−であり;
R2は、−H、−F、−CF3またはC1〜C6アルコキシであり;
R2’’は−H、−Fまたは−CF3であり;
R8は、−H、またはC1〜C6アルキルであり;
R14は、非置換フェニル、非置換チエニルもしくは非置換ピリジル、または1〜3つのR15基で置換されているフェニル、チエニルまたはピリジルであり;
R15の各々は、独立してC1〜C6アルキル、−Cl、−CF3、またはC1〜C6 アルコキシである。
【0037】
本発明の別の特定の実施形態は、一般式V:
【0038】
【化5】
により表され、使用することができる一クラスの化合物または薬学的に許容できるその塩であり;
式中、X2は、−CH−または−N−であり;
R2は、−H、−F、または−OCH3であり;
R2*は、−Hまたは−Fであり;
R8は、−H、またはC1〜C6アルキルであり;
R14は、フェニル、低級アルキルが1個から6個の炭素を有する4−(低級アルキル)フェニル、5−(低級アルキル)−2−チエニル、および6−(低級アルキル)−3−ピリジルからなる群より選択される
本発明の別の特定の実施形態は、一般式VI:
【0039】
【化6】
により表され、使用することができる一クラスの化合物または薬学的に許容できるその塩であり;
式中、R8は、−H、またはC1−C6−アルキルである。
【0040】
本発明の別の特定の実施形態は、一般式VII:
【0041】
【化7】
により表され、使用することができる一クラスの化合物または薬学的に許容できるその塩であり;
式中、R2*は、−Hまたは−Fであり;
R8は、−H、またはC1〜C6−アルキルであり;および
R14は、フェニルおよび4−(C1〜C6−アルキル)フェニルからなる群より選択される。
【0042】
本発明の別の特定の実施形態は、一般式VIII:
【0043】
【化8】
により表され、使用することができる一クラスの化合物または薬学的に許容できるその塩であり;
式中、R8は、−H、またはC1〜C6−アルキルである。R8がHである場合、この化合物は、AGN 193109と呼ばれる。
【0044】
本発明の別の特定の実施形態は、一般式IX:
【0045】
【化9】
により表され、使用することができる一クラスの化合物または薬学的に許容できるその塩であり;
式中、X1の各々は、R1の各々が独立してHまたはC1〜C6−アルキルである、−C(R1)2−、−C(R1)2−C(R1)2−、−S−、−O−、
−NR1−、−C(R1)2−O−、−(C(R1)2−S−、または−C(R1)2−NR1−;であり;
R2の各々は、独立してC1〜C6−アルキル、−F、−Cl、−Br、−I、−CF3、フルオロ置換C1〜C6−アルキル、−OH、−SH、C1〜C6−アルコキシ、またはC1〜C6−アルキルチオであり;
mは、0から4の整数であり;
nは、0から2の整数であり;
oは、0から3の整数であり;
R3は、−H、C1〜C6−アルコキシ、−F、−Cl、−Brまたは−Iであり;
R4は、(R5)p−フェニル、(R5)p−ナフチル、または(R5)p−ヘテロアリールであり、前記ヘテロアリール基は、5員または6員であり、酸素、硫黄、および窒素からなる群より選択される1個から3個のヘテロ原子を有し;pは、0から5の整数であり;
R5の各々の場合は、独立して−F、−Cl、−Br、−I、−NO2、−N(R8)2、−N(R8)COR8、−N(R8)CON(R8)2、−OH、−OCOR8、−OR8、−CN、−COOH、−COOR8、C1〜C10−アルキル、1つから3つの二重結合を有するC1〜C10アルケニル基、1つから3つの三重結合を有するC1〜C10アルキニル基、C1〜C6(トリアルキル)シリルまたはC1〜C6(トリアルキル)シリルオキシであり;
Yは、フェニル、ナフチル、またはピリジル、チエニル、フリル、ピリダジニル、ピリミジニル、ピラジニル、チアゾリル、オキサゾリル、イミダゾリルおよびピラゾリルからなる群より選択されるヘテロアリールであり;前記フェニル基およびヘテロアリール基は、任意にかつ独立して1つまたは2つのR2基で置換されており、またはYは、−(CR3=CR3)r−であり;
rは、1から3の整数であり;
Aは、(CH2)q、低級C3〜C6分枝鎖アルキル、C3〜C6シクロアルキル、1つまたは2つの二重結合を有するC2〜C6 アルケニル、1つまたは2つの三重結合を有するC2〜C6 アルキニルであり;Yが−(CR3=CR3)r−の場合、Aが(CH2)qであり、qが0であるという条件で、qは0〜5の整数であり;
Bは、−H、−COOH、−COOR8、−CONR9R10、−CH2OH,−CH2OR11、−CH2OCOR11、−CHO、−CH(OR12)2、−CHOR13O、−COR7、−CR7(OR12)2、−CR7OR13O、または−Si(C1〜6アルキル)3であり;
R7は、1個から5個の炭素を含有するアルキル基、シクロアルキル基またはアルケニル基であり;
R8は、C1〜C10アルキル、C1〜C10(トリメチルシリル)アルキル、C5〜C10シクロアルキル、フェニルまたは低級アルキルフェニルであり;
R9およびR10は、各々独立して−H、C1〜C10アルキル、C5〜C10シクロアルキル、フェニルまたは低級アルキルフェニルであり;
R11は、低級アルキル、フェニルまたは低級アルキルフェニルであり;
R12は、低級アルキルであり;
R13は、2個から5個の炭素の二価アルキル基である。
【0046】
本発明の別の特定の実施形態は、一般式X:
Y3(R4)−X−Y1(R1)(R2)−Z−Y2(R2)−A−B (X)
により表され、使用することができる一クラスの化合物または薬学的に許容できるその塩であり;
式中、Y1は、フェニル、ナフチル、またはピリジル、チエニル、フリル、ピリダジニル、ピリミジニル、ピラジニル、チアゾニル、オキサゾリル、イミダゾリルおよびピラゾリルからなる群より選択されるヘテロアリールであり、前記フェニル基、ナフチル基、およびヘテロアリール基が、任意にR1基で置換されており、1つまたは2つのR2基で任意にさらに置換されており;
R1は、C1〜C10アルキル、1−アデマンチル、2−テトラヒドロピラノキシ、C1〜C6トリアルキルシラニルオキシ、−OH、C1〜C10アルコキシ、C1〜C10アルキルチオ、または−OCH2O−(C1〜C6アルキル)であり;
R2は、C1〜C6−アルキル、−F、−Cl、−Br、−I、−CF3、−CF2CF3、−OH、−OR3、−NO2、−N(R3)2、−CN、−N3、−COR3、−NHCOR3、−COOH、または−COOR3であり;
Xは、−(C(R3)2)−、−S−、−SO−、−SO2−、−O−、−C(=O)−、−C(=S)−、−C(=NR1)−、−C(=C(R1)2)−または−NR3−であり;
Zは、−C≡C−、−N=N−、−N(O)=N−、−N=N(O)−、−N=CR3−、−CR3=N−、−(CR3=CR3)n−、−OCO−、−CSO−、−OCS−、
−COCR3=R3O−、−CO−NR3−、−CS−NR3、−NR3−CO−、または−NR3−CS−であり;
nは、0〜5の値を有する整数であり;
R3の各々は、独立して−HまたはC1−C6アルキルであり;
Y2は、フェニル基またはナフチル基、またはピリジル、チエニル、フリル、ピリダジニル、ピリミジニル、ピラジニル、チアゾリル、オキサゾリル、イミダゾリルおよびピラゾリルからなる群より選択されるヘテロアリールであり;前記フェニル基、ナフチル基およびヘテロアリール基は、非置換であっても1つまたは2つのR2基で置換されていてもよく、またはZが、−(CR3=CR3)n−であり、nが、3、4、または5である場合、Y2は、該−(CR3=CR3)n−基とBとの間の直接的原子価結合を表し;
Y3は、フェニル、ナフチル、またはピリジル、チエニル、フリル、ピリダジニル、ピリミジニル、ピラジニル、チアゾリル、オキサゾリル、イミダゾリルおよびピラゾリルからなる群より選択されるヘテロアリールであり;前記フェニル基、ナフチル基およびヘテロアリール基は、非置換であっても1つから5つのR4基で置換されていてもよく;
R4の各々は、独立してC1〜C10アルキル、1つから3つの三重結合を有するC2〜C10アルケニル、−F、−Cl、−Br、−I、−NO2、−N(R3)2、−N3、−COOH、−COO−(C1〜C6アルキル)、−OH、−SH、−O−C1〜C6アルキル、または-S−C1〜C6アルキルであり;
Aは、(CH2)q、低級C3〜C6分枝状アルキル、C3〜C6シクロアルキル、1つから2つの二重結合を有するC2〜C6アルケニル、1つから2つの三重結合を有するC2〜C6アルキニルであり;qが0から5であり;
Bは、−H、−COOH、−COOR8、−CONR9R10、−CH2OH,−CH2OR11、−CH2OCOR11、−CHO、−CH(OR12)2、−CHOR13O、−COR7、−CR7(OR12)2、−CR7OR13O、または−Si(C1〜C 6アルキル)3であり;
R7は、1個から5個の炭素を含有するアルキル基、シクロアルキル基またはアルケニル基であり;
R8は、C1〜C10アルキル、C1〜C10トリメチルシリルアルキル、C3〜C10シクロアルキル、フェニルまたは低級アルキルフェニルであり;
R9およびR10は、独立して水素、C1〜C10アルキル、C3〜C10シクロアルキル、フェニルまたは低級アルキルフェニルであり;
R11は、低級アルキル、フェニルまたは低級アルキルフェニルであり;
R12は、低級アルキルであり;
R13は、2個から5個の炭素の二価アルキル基である。
【0047】
本発明の別の特定の実施形態は、一般式Xにより表され、使用することができる一クラスの化合物であり、X が、−(C(R3)2)−、−S−、−SO−、−SO2−、−O−または−NR3−であり;Zが、−CO−NR3−,−CS−NR3−,−NR3−CO−、または−NR3−CS−であり;全て他の可変部分は、上記に定義されたとおりである。
【0048】
本発明の別の特定の実施形態は、一般式Xにより表され、使用することができる一クラスの化合物であり、X が、−C(=O)−、−C(=S)−、−C(=NR1)−、または−(C=C(R1)2)−であり;Zが、−CO−NR3−,−CS−NR3−,−NR3−CO−、または−NR3−CS−であり;全て他の可変部分は、上記に定義されたとおりである。
【0049】
本発明の別の特定の実施形態は、一般式Xにより表され、使用することができる一クラスの化合物であり、Y3により表される前記フェニル基、ナフチル基、またはヘテロアリール基が、非置換であるか、または1つから3つのR4基により置換されており、全て他の可変部分は、上記に定義されたとおりである。
【0050】
本発明は、RARα、RARβおよびRARγサブタイプの受容体に結合するRARアンタゴニストまたはインバースアゴニストである任意の化合物を使用することをを考慮しており、該化合物としては、米国特許番号:5,728,846、5,739,338、5,763,635、5,773,594、5,877,207、5,952,345、5,958,954、5,998,655、6,008,204、6,037,488、6,043,381、6,087,505、6,090,810、6,117,987、6,211,385、6,218,128、6,225,494、6,228,848、6,235,923、6,313,168、6,521,624、6,521,641、6,538,149、6,555,690、6,653,483、6,720,425、6,818,775、6,942,980、7,105,566、および7,166,726ならびに米国特許出願番号:10/446,580、11/016,534、11/500,277、11/503,635、11/607,406、および11/643,754に記載されているか、またはそれらに請求されているものが挙げられる。上記に引用された特許および特許出願の全ては、参照としてその全体が本明細書に援用されている。
【0051】
本明細書明細書内に入る非排他的リストの化合物およびこのクラスの化合物を製造する方法は、米国特許第5,728,846号に開示されており、それらの内容は、参照として本明細書に援用されている。さらに、これらの化合物は、Songらの米国特許出願第08/840,040号に開示されており、その出願は、本出願と共通の所有権を共有しており、参照としてその全体が本明細書に援用されている。
【0052】
本発明の方法に使用する好ましい化合物または薬学的に許容できるその塩は、以下の構造:
【0053】
【化10】
により表される。この化合物は、VTP 194310 と称される(以前にはAGN 194310と称されていた)。
【0054】
本発明の方法に使用する好ましい化合物または薬学的に許容できるその塩は、以下の構造:
【0055】
【化11】
により表される。この化合物は、VTP 196996と称される。
【0056】
さらなるRARアンタゴニストまたはインバースアゴニストは、参照としてその全体が本明細書に援用されるSongらの米国特許出願第08/845,019号に記載されており;本出願と共通の所有権を共有している。また、本発明の方法に有用な化合物は、参照としてその全体が本明細書に援用されるYoshimura らの国際特許出願公開WO 第94/1477号に開示されている。後者の出願は、PARアンタゴニストを開示している。
【0057】
さらに、本発明の方法に有用なさらなる化合物または薬学的に許容できるその塩の構造は、以下のとおりであり:
【0058】
【化12】
式中、nは、1 から10の整数であり;
本発明の方法に有用なさらなる化合物または薬学的に許容できるその塩の構造は、以下のとおりであり:
【0059】
【化13】
式中、nは、1 から10の整数であり;
本発明の方法に有用なさらなる化合物または薬学的に許容できるその塩の構造は、以下のとおりであり:
【0060】
【化14】
本発明の方法に有用なさらなる化合物または薬学的に許容できるその塩の構造は、以下のとおりであり:
【0061】
【化15】
本発明の方法に有用なさらなる化合物または薬学的に許容できるその塩の構造は、以下のとおりである。
【0062】
【化16】
本明細書に用いられる用語「アゴニスト」とは、受容体に結合してそれを活性化する化合物を意味すると解され、薬理学的応答(例えば、収縮、弛緩、分泌、酵素活性化など)を生じる。
【0063】
本明細書に用いられる用語「インバースアゴニスト」は、アゴニストの効果とは反対の効果を生じるが、同じ受容体において作用する化合物を意味するものとして理解することとする。用語「インバースアゴニスト」は、用語「ネガティブアンタゴニスト」と同義である。
【0064】
本明細書に用いられる用語「アンタゴニスト」は、該受容体を活性化することなくアゴニストと同じ部位に結合することにより、アゴニストの効果を減弱させる化合物を意味するものとして理解することとする。
【0065】
本明細書に用いられる用語「化学放射線療法」は、化学療法、放射線療法または双方を意味するものとして理解することとする。
【0066】
本明細書に用いられる用語「処置すること」または「処置」は、(1)ある状態、疾患、障害、傷害または病態に罹り得るか、または罹りやすくなり得るが、前記状態、疾患、障害、傷害または病態の臨床的または準臨床的症状をまだ経験していないか、または示していない哺乳動物において発現する前記状態、疾患、障害、傷害または病態の臨床的症状出現を、部分的または完全に、予防すること、重症度を減少させること、または遅延すること、(2)前記状態、疾患、障害、傷害または病態を、部分的に、または完全に抑制すること、すなわち、前記状態、疾患、障害、傷害または病態、またはそれらの少なくとも1つの臨床的または準臨床的症状の発現を阻止すること、または減少させること、または(3)部分的または完全に、前記状態を軽減すること、または前記疾患、障害、損傷または状態の重症度を低下させること、すなわち、前記状態、疾患、障害、傷害または病態、もしくはそれらの少なくとも1つの臨床的または準臨床的症状の後退をもたらすこと、を意味するものとして理解することとする。
【0067】
本明細書に用いられる用語「送達」は、RARα、RARβおよびRARγタイプの受容体に結合することのできるRARアンタゴニストまたはRARインバースアゴニストの治療有効量を哺乳動物の特定部位に提供して、該特定部位におけるRARα、RARβおよび RARγタイプの受容体に結合するRARアンタゴニストまたはRARインバースアゴニストの治療的有効濃度を生じさせることを意味するものとして理解することとする。
【0068】
本明細書に用いられる用語「対象」または「患者」または「宿主」または「哺乳動物」は、ヒトを含む哺乳動物を称する。
【0069】
「アルコキシ」および「ヒドロキシアルキル」など、単独で、またはより大きな部分の一部として用いられる用語「アルキル」とは、1個から10個の炭素原子を含有する飽和脂肪族を意味する。代表的な飽和直鎖状アルキル類としては、メチル、エチル、n−プロピル、n−ブチル、n−ペンチルなどが挙げられ、一方、飽和分枝状アルキル類としては、イソプロピル、sec−ブチル、イソブチル、tert−ブチル、イソペンチルなどが挙げられる。用語「低級アルキル」とは、1個から6個の炭素を含有するアルキルを称する。アルケニル基およびアルキニル基は、不飽和脂肪族基であり、隣接炭素原子間に少なくとも1つの二重結合または三重結合を含有する。代表的な直鎖状および分枝状のアルケニル類としては、エチレニル、プロピレニル、1−ブテニル、2−ブテニル、イソブチレニル、1−ペンテニル、2−ペンテニル、3−メチル−1−ブテニル、2−メチル−2−ブテニルなどが挙げられ、一方、代表的な直鎖状および分枝状のアルキニル類としては、アセチレニル、プロピニル、1−ブチニル、2−ブチニル、1−ペンチニル、2−ペンチニル、3−メチル−1−ブチニルなどが挙げられる。
【0070】
用語「シクロアルキル」とは、飽和環式炭化水素部分を意味し、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシルが挙げられる。また、シクロアルキル類には、1つ又は複数の芳香族(フェニルなど)または非芳香族(シクロヘキサンなど)の環状炭素に縮合したシクロアルキル(例えば、シクロペンタンまたはシクロヘキサン)など、8個から10個の炭素原子を有する二環系および三環系などの環状炭素系も含まれる。
【0071】
単独で、またはより大きな部分の一部として用いられる用語「ヘテロアリール」、「ヘテロ芳香族」、「ヘテロ芳香族環」、および「ヘテロアリール基」とは、単環式ヘテロ芳香族環が1つ又は複数の他の炭素環式またはヘテロ芳香族の芳香族環に縮合している単環式ヘテロ芳香族環および多環式芳香族環など、一般に5員から14員を有するヘテロ芳香族環基を称する。ヘテロアリール基は、1つ又は複数、一般に1個、2個、または3個の、窒素、酸素およびイオウなどの環ヘテロ原子を有する。
【0072】
本明細書に記載された化合物の「製薬組成物」およびそれらの薬学的に許容できる塩、溶媒和物および水和物は、薬学的に許容できる担体または希釈剤と組み合わせて製薬製剤中に使用することができる。好適な薬学的に許容できる担体としては、不活性な固体増量剤または希釈剤および滅菌の水性溶液または有機溶液が挙げられる。SARM化合物は、本明細書に記載された範囲の所望の用量を提供する上で十分な量で、このような製薬組成物中に存在する。本発明の化合物の製剤化および投与に関する技法は、Remington:the Science and Practice of Pharmacy、第19版、Mack Publishing 社、ペンシルベニア州、イーストン(1995)に見ることができる。
【0073】
薬学的に許容できる塩もまた、本発明の実施形態として含まれる。
【0074】
「薬学的に許容できる塩」は、何らかの酸性または塩基性の官能基を含有する化合物の塩である。例えば、アミンまたは他の塩基性基の薬学的に許容できる塩は、該化合物を、塩化水素、臭化水素、酢酸、過塩素酸などの好適な有機酸または無機酸と反応させることによって得ることができる。このような塩の他の例としては、塩酸塩、臭化水素酸塩、硫酸塩、メタンスルホン酸塩、硝酸塩、マレイン酸塩、酢酸塩、クエン酸塩、フマル酸塩、酒石酸塩(例えば、(+)−酒石酸塩、(−)−酒石酸塩またはラセミ混合物などのそれらの混合物)、コハク酸塩、安息香酸塩およびグルタミン酸などのアミノ酸との塩が挙げられる。
【0075】
カルボン酸または他の酸性官能基を含有する化合物の薬学的に許容できる塩は、好適な塩基と反応させることによって調製することができる。このような薬学的に許容できる塩は、薬学的に許容できるカチオンを与える塩基によって作製することができ、アルカリ金属塩(特に、ナトリウムおよびカリウム)、アルカリ土類金属塩(特に、カルシウムおよびマグネシウム)、アルミニウム塩およびアンモニウム塩、ならびに、トリメチルアミン、トリエチルアミン、モルホリン、ピリジン、ピペリジン、ピコリン、ジシクロヘキシルアミン、N,N−ジベンジルエチレンジアミン、2−ヒドロキシエチルアミン、ビス−(2−ヒドロキシエチル)アミン、トリ−(2−ヒドロキシエチル)アミン、プロカイン、ジベンジルピペリジン、N−ベンジル−β−フェネチルアミン、デヒドロアビエチルアミン、N,N’−ビスデヒドロアビエチルアミン、グルカミン、N−メチルグルカミン、コリジン、キニン、キノリン、ならびにリシンおよびアルギニンなどの塩基性アミノ酸などの生理学的に許容できる有機塩基から作製された塩が挙げられる。
【0076】
本発明の方法に使用するための、RARα、RARβ および RARγサブタイプの受容体に結合するRARアンタゴニストおよびRARインバースアゴニスト化合物を、製薬組成物中に組み込むことができる。全ての投与様式、例えば、経口、経直腸、非経口、局所、または静脈内注射、筋肉内注射、イントラステマル(intrastemal)注射または皮下注射、もしくは、吸入などの好適な形態が考慮されている。該製剤は、適切な場合は、個別の用量単位で簡便に提供することができ、製薬業界においてよく知られたいずれかの方法によって調製することができる。該化合物は通常、公知の確立された慣例に従って、1つ又は複数の薬学的に許容できる成分と共に製剤化される。したがって、該製薬組成物は、液剤、散剤、エリキシル剤、注射用液剤、懸濁剤、座剤などとして製剤化することができる。
【0077】
経口使用のための製剤は、該化合物が炭酸カルシウム、リン酸カルシウムまたはカオリンなどの不活性の固体希釈剤と共に混合されている錠剤または硬カプセル剤として、または該活性成分が、水またはプロピレングリコール、PEG類およびエタノール、または油脂性媒体、例えば、落花生油、液体パラフィンまたはオリーブ油などの混和性溶媒と共に混合されている軟ゼラチンカプセル剤として提供することができる。
【0078】
口腔における局所投与用に、該製薬組成物は、従来の様式で製剤化された頬側錠または舌下錠、滴剤または舐剤の形態をとることができる。
【0079】
表皮への局所投与のため、該化合物は、クリーム剤、ゲル剤、軟膏剤またはローション剤として、もしくは経皮パッチとして製剤化することができる。このような組成物は、例えば、好適な増粘剤、ゲル化剤、乳化剤、安定化剤、分散剤、懸濁化剤、および/または着色剤を添加して、水性または油性の基剤と共に製剤化することができる。
【0080】
該化合物はまた、デポー製剤として製剤化することもできる。このような長時間作用性の製剤は、移植(例えば、皮下または筋内に)により、または筋内注射により投与することができる。したがって、例えば、該化合物は、好適なポリマー材料または疎水性材料(例えば、許容できる油中に乳剤として)、またはイオン交換樹脂と共に、またはわずかに溶解性の誘導体として、例えば、わずかに溶解性の塩として製剤化することができる。
【0081】
該化合物は、注射による、簡便に、静脈内、筋内または皮下注射による、例えば、ボーラス投与または連続的静脈内注入による非経口投与のために製剤化することができる。注射用製剤は、単位用量形態、例えば、保存剤を添加したアンプルまたは多用量容器で提供することができる。該製薬組成物は、油性または水性媒体中、懸濁剤、液剤または乳剤などの形態をとることができ、懸濁化剤、安定化剤または分散化剤などの製剤化剤を含有することができる。あるいは、該化合物は、使用前に、好適な媒体、例えば滅菌したパイロジェンの無い水と共に構成するための粉末形態であってもよい。
【0082】
また、該化合物は、座剤または本発明の保持浣腸剤、例えばカカオ脂または他のグリセリドなどの慣例的な座剤用基剤を含有するものなど、直腸用組成物においても製剤化できる。
【0083】
鼻腔内投与のために、該化合物は、例えば、液体スプレーとして、散剤として、または滴剤の形態で用いることができる。
【0084】
吸入による投与のために、該化合物は、好適な噴射剤、例えば、ジクロロジフルオロメタン、トリクロロフルオロメタン、ジクロロテトラフルオロエタン、テトラフルオロエタン、ヘプタフルオロプロパン、二酸化炭素または他の好適な気体を使用して、加圧パックまたはネブライザーからのエアロゾルスプレー法の形態で、簡便に送達できる。加圧エアロゾルの場合、用量単位は、計量された量を送達するためのバルブを提供することによって判定できる。吸入器またはインシュレーターにおいて使用するための、例えばゼラチンのカプセルおよびカートリッジは、レチノイド化合物と、乳糖または澱粉などの好適な粉末基剤との粉末混合物を含有して製剤化することができる。
【0085】
水性懸濁剤は、懸濁化剤、例えば、カルボキシメチルセルロースナトリウム、メチルセルロース、ヒドロキシプロピルメチルセルロース、アルギン酸ナトリウム、ポリビニルピロリドン、トラガンカントゴムおよびアラビアゴム;天然ホスファチド、例えばリシチン、またはアルキレン酸化物と脂肪酸との縮合産物、例えば、ポリオキシエチレンステアレート、またはエチレンオキシドと長鎖脂肪族アルコールとの縮合産物、例えばヘプタデカエチレン−オキシセタノール、またはエチレンオキシドと脂肪酸由来の部分的エステルとヘキシトールとの縮合産物、例えばポリオキシエチレンソルビトールモノレエート、またはエチレンオキシドと脂肪酸由来の部分的エステルと無水ヘキシトールとの縮合産物、例えばポリオキシエチレンソルビタンモノレエートなどの分散化剤または湿潤化剤などの薬学的に許容できる賦形剤を含むことができる。水性懸濁剤はまた、一種または複数種の保存剤、例えばエチルまたは−n−プロピル−p−ヒドロキシベンゾエート、一種または複数種の着色剤、一種または複数種の芳香剤およびスクロース、サッカリンまたはシクラミン酸ナトリウムまたはシクラミン酸カルシウムなどの一種または複数種の甘味剤を含有することもできる。
【0086】
RARα、RARβおよび RARγサブタイプの受容体に結合するRARアンタゴニストおよびRARインバースアゴニスト化合物に加えて、少なくとも1種の他の薬理学的活性物質、例えば、トラマドール、アセトアミノフェン、アスピリン、ジクロフェナク、ジフルシナール、エトドラク、フェンブフェン、フェノプロフェン、フルフェニサール、フルルビプロフェン、イブプロフェン、インドメタシン、ケトプロフェン、ケトロラク、メクロフェナミン酸、メフェナム酸、ナブメトン、ナプロキセン、オキサプロジン、フェニルブタゾン、ピロキシカム、スリンダク、トルメチン、ゾメピラクなどおよびそれらの組み合わせなどの非麻薬性鎮痛薬、またはコデイン、オキシコドン、ジヒドロコデイン、ヒドロコドン、レボルファノール、モルフィンなどおよびそれらの組み合わせなどの麻薬性鎮痛薬、または例えば、G−CSF、GM−CSF、EGF、インターロイキン11、エリスロポイエチン、トロンボポイエチン、巨核球の発達および成長因子、ピキシキン類、幹細胞因子、FLT−リガンドならびにインターロイキン1、3、6および7などおよびそれらの組み合わせなどの他の薬剤を、RARα、RARβおよび RARγサブタイプの受容体に結合するRARアンタゴニストおよびRARインバースアゴニスト化合物と共に投与することができる。
【0087】
該化合物は、本発明に従って、治療有効量で投与される。治療的濃度は、哺乳動物、好ましくはヒトにおける、例えば化学放射線療法の副作用を処置する上で有効な濃度である。これらの量は、当業者によって決定できる。
【0088】
以下は、本発明の非限定例である。これらの例は、請求項によって規定される本発明の範囲を限定するものとして解釈すべきではない。
【0089】
以下の実施例の各々で、Allergan社(Irvine,CA)で合成され、特異的なパン−RARアンタゴニストである4−2[6−(2,2−ジメチル−(1H)−4−(4−エチルフェニル)−1−ベンゾチオピラン))エチニル]安息香酸(VTP 194310、以前AGN 194310と称された)を用いる。VTP 194310の構造は、本明細書上記に示されている。RARα、β および γに対する結合に関するVTP 194310のKiは、それぞれ、3、2および5nMである。VTP 194310は、トランス活性化アッセイにおいて、活性を示さないが、その代わり、ATRAおよび他のRARアゴニストによって誘導された遺伝子転写活性をブロックする。VTP 194310およびATRAは、−20℃で50%エタノール/50%ジメチルスルホキシド(DMSO)中、10mMの原液として保存された。
【実施例】
【0090】
実施例1
白血球減少症のシクロホスファミド誘導マウスにおける好中球およびリンパ球の回復
マウス
Charles River Laboratories(マサチューセッツ州、ウィルミントン)から購入したマウスを、12時間の明/暗サイクルで、マイクロアイソレーターケージ内に、個々に収容した。それらは、病原体の無い条件下で収容し、通常の食餌および水を自由に取らせた。それらを、実験前に、実験動物管理の評価および認定協会(AAALAC)に認定された動物施設(Allergan社、アービン、カリフォルニア州)で、1週間順化させた。試験設計は、施設内動物の管理および使用委員会によって承認された。体重は各試験を通してモニターした。マウスの体重は、処置開始日に22〜27gの範囲であった。該マウスは健常で、以前、他の実験操作に用いられなかった。
【0091】
インビボ白血球減少症モデル
0.2mlの生理食塩水中、200mg/kgのシクロホスファミド(CPM、Sigma−Aldrich、ミズーリ州、セントルイス)の腹腔内(i.p.)注射により、オスBDF1マウス(C57Bl×DBA2,7.5-12 週)に白血球減少症を誘導した。白血球の回復に及ぼすVTP 194310の効果を、結果の節に示したとおり、1mg/kgのVTP 194310によるマウスの経口胃管法によって評価した。VTP 194310(DMSO中に溶解)は、媒体としての落花生油により希釈した(5ml/kgの用量)。対照マウスには、DMSOおよび媒体のみを与えた。陽性対照として、2日目に、CPM処置マウスにおいて、10μg/kgのペギル化組換えメチオニルヒト顆粒球コロニー刺激因子(PEG−r−metHuG−CSF)(Neulasta、Pegfilgrastim;Amgen、サウザンドオークス、カリフォルニア州)の単回皮下注射により、顆粒球生成を刺激した。3つの別個の実験を実施した。
【0092】
麻酔下、該マウスの後眼窩洞から末梢血(60μl)を、ヘパリン化毛細管により採取し、EDTAでコーティングしたマイクロタイター管(Becton Dickinson、フランクリンレイクス、ニュージャージー州)に移した。5%のウシ血清アルブミン(フラクションV;Sigma、セントルイス、ミズーリ州)を含有するPBSにより該血液を1:4に希釈し、Advia 120 Hematology System(Bayer HealthCare Diagnostics Division、ニューヨーク州、タリータウン)を用いることにより、1群当たり少なくとも3匹のマウスに関して、白血球、好中球、およびリンパ球の数を得た。
【0093】
PE結合およびFITC結合モノクローナル抗体(Pharmingen、サンディエゴ、カリフォルニア州)を用いて、二重免疫染色により、脾臓および骨髄(大腿骨)吸引液から調製した単一の細胞懸濁液において、未成熟(Gr−1low/CD11b+ve)および成熟(Gr−1+ve/CD11b+ve)好中球を同定した。CellQuest Proスフトウェアプログラム(Becton Dickinson、サンホセ、カリフォルニア州)にインターフェースしたFACS Caliburで細胞を解析した。
【0094】
結果
VTP 194310の使用によりデータが示すように、白血球減少症のシクロホスファミド誘導マウスモデルにおいて白血球の回復を改善した。200 mg/kgでCPMの単回投与によるマウスの処置では、4日目に重大な白血球減少症および好中球減少症を生じた。CPM処置マウスの血中白血球数および好中球数は、正常なマウス(正常な対照群のデータは示していない)に関して、それぞれ8.98±0.33×103/μlおよび1.34±0.09×103/μlの値と比較して、0.69±0.09(SEM)x 103/μl および0.09±0.02×103/μl であった。図1に示されるように、白血球数、好中球数およびリンパ球数は、CPM処置マウスにおいて着実に上昇し、8日目にプラトー値に達した。
【0095】
CPMで処置した(0日目に)マウスに対して、処置4日前から1日前および処置0日目から3日目後にVTP 194310の投与による2つの方法で白血球の回復を改善した。白血球減少症のマウスの対照群と比較して、白血球数および好中球数は、VT P194310処置マウスにおいてより迅速に上昇した。回復の有意差は、CPM投与5日後という早い期間に観察された。8日目に、VT P194310処置の好中球減少症マウスの血中白血球数および好中球数は、CPM単独で処置されたマウスにおける数よりも約3倍高かった(図1を参照)。4日目から7日目にVTP 194310の白血球減少症マウスへの投与は、産生した白血球数および好中球数を改善したが、VTP 194310 を早期に与えた場合よりもかなりの程度で低かった。白血球減少症マウスのPeg−r−metHuG−CSF(2日目に)による処置により、血中好中球数を迅速に上昇させた。これらの細胞レベルは7日目にピークに達し、8日目までに白血球減少症マウスの対照群に見られた数に下降した(図1を参照)。
【0096】
処置4日前から1日前および処置0日目から3日目後に、VTP 194310が投与された白血球減少症マウスでは、対照白血球減少症マウスおよびPeg−r−metHuG−CSFが与えられたこれらのマウスと比較して、血中リンパ球数の増加を示した。8日目において、the lymphocyte counts for the VTP194310処置マウスに関するリンパ球数は、対照の回復マウスに関する2.38±0.26×103/μl(p値<0.01)と比較して、4.53±0.39×103/μl(VTP 194310,4日前から1日前)および 4.49±0.52×103/μl(VTP 194310、0日目から3日目)であった。
【0097】
実施例2
ヒトCD34+ve始原細胞の培養における好中球の回復
HL60細胞増殖および分化の評価
前骨髄性細胞系HL60の培養は、10%の胎仔ウシ血清(FBS、Invitrogen,ペイスリー、英国)ペニシリン(100 U/ml)、およびストレプトマイシン(100 μg/ml)を含有する4 mlのRPMI1640培地(Invitrogen、ペイスリー、英国)中、2.5×105細胞/mlを接種した。ニトロブルーテトラゾリウムを減少させる細胞の能力、早期成熟マーカーCD11bの獲得発現およびステロイドスルファターゼ(骨髄性成熟のマーカー)の活性増加により、分化を測定した。
【0098】
ヒトCD34+ve造血始原細胞の単離および培養
動員幹細胞を有する成人からの倫理的に許容された血中白血球泳動細胞は、地方の国立血液サービス幹細胞ラボラトリー(National Blood Service Stem Cell Laboratory)、バーミンガムよりインフォームドコンセント後に提供された。CliniMACS磁気分離器上で免疫磁気ビーズと共に抗CD34モノクローナル抗体の使用により、CD34+ve細胞を精製した。
【0099】
10% FBS、抗体(100 U/mlのペニシリンおよび100 μg/mlのストレプトマイシン)および組換えヒト幹細胞因子(SCF)の量、組換えヒトインターロイキン3(IL3)、組換えヒト顆粒球コロニー刺激因子(G−CSF)およびパン−RARアンタゴニストVTP194310を含有する200 μlのRPMI 1640培地中、細胞を5×105細胞/mlで、96ウェルマイクロタイタープレート内で平板培養し、その結果が示された。SCF、IL3およびG−CSFは、R and D Systems、アビングドン、英国から入手した。三通りの培養物により各々の条件が確立された。培養物を、最初にマイクロタイターウェルに、次に2 mlの培養物をCostarウェルに入れて分割し、2.5×105細胞/mlから10×105細胞/mlの間の細胞密度を維持した。細胞を、加湿インキュベーターおよび5% CO2雰囲気中、37℃で増殖させた。
【0100】
100 nMのパン−RARアンタゴニストVTP194310は、100 nMのATRAの能力を完全に遮断し、HL60細胞の好中球分化を誘導した。HL60細胞を100 nMのATRAにより5日間処置することにより、増殖停止、ニトロブルーテトラゾリウムを減少させる能力の獲得、CD11bの発現、およびステロイドスルファターゼ活性の増大をもたらした。100 nMの VTP194310がATRAと共投与された場合、これら事象の全ては、抑止された(図2を参照)。
【0101】
図3は、増殖および精製CD34+ve(>99%)造血始原細胞の自然発生的分化に対して10 nmおよび100 nMのVTP194310の効果を示している。CD34+ve細胞は、生存を保証するSCF(100 ng/ml)およびIL3(20 ng/ml)の量で培養した。液体懸濁液の培養中、骨髄始原細胞から好中球および単球の最適数を産生するために、我々はまた、実験室内でルーチンに使用する条件を使用した。これらは、100 ng/mlの SCF、5 ng/mlのIL3および30 ng/mlのG−CSFである。
【0102】
VTP194310が、RAR類および血清中レチノイド類のいずれの効果も遮断したかどうかを確認するために、我々は、膜貫通糖タンパク質CD38の発現のために細胞を染色した。RARαの直接制御下、CD38は、レチノイドを誘導し、またCD38は、CD34+ve細胞を介して骨髄分化中に増加し、CD38の誘導および分化は、機能的には関連していない。VTP194310が、インビトロでヒト造血細胞によりinhibits expression of CD38の発現を阻害することを、Prusらは示している(Prusら、「Retinoic Acid Receptor Anatgonist Inhibits CD38 Antigen Expression on Human Hematopoeitic Cells In Vitro」、Leukaemia & Lymphoma、45、1025−1035頁(2004))。細胞が、これらの条件プラスVTP194310(10 nMおよび100 nM)で増殖された場合の39±4%(n = 4)と比較して、6日目に、35±2%(n = 4)の細胞が、SCF/IL3(20 ng/mlで)において増殖された培養物中にCD38を発現した。VTP194310の不在下では、CD38の発現は、15日目までに50±2%(n = 3)、および27日目までに72±1%(n = 4)に上昇した。VTP194310(10 nMおよび100 nM)の存在下、CD38の発現は、15日目までに15±3%(n = 4)、および27日目までに6±1%(n = 4)に降下した。
【0103】
VTP194310(10 nMおよび100 nM)およびG−CSFは、培養物および産生した細胞数の寿命の双方を実質的に増加させた。SCF/IL3で処理された培養物は、33日目までに殆どが終了したが(細胞密度0.6×105/ml)、一方、VTP194310(10 nMおよび100 nM)/SCF/IL3またはG−CSF/SCF/IL3で処理された培養物は、依然として大多数の細胞を産生した(図2A、上部パネル)。細胞密度は、55日目にこれらの培養物が低レベル(0.5×105 cells/mlから1.5×105 cells/mlまで)であった。100 nMのVTP194310で補足された培養物は、他の培養物よりも僅かに低い率で細胞を産生し(図3Aを参照)、33日目に、10 nMのVTP194310(4.0×107±0.2、n = 4)またはG−CSF(3.8×107±0.5、n = 4)で補足された培養物よりも総数で僅かに低く産生(2.9×107±0.2、n = 4)した。
【0104】
各培養条件および実験全般にわたって産生された細胞は、主として骨髄であった(図3B、下部パネル)。
【0105】
赤血球コンパートメント(グリコホリン+ve/CD45+ve)は、小さく(6日目に9%から14%まで)、適切な増殖およびエリスロポイエチンなどの生存因子を欠くと徐々に消失した(図3B)。VTP194310は、赤血球細胞の罹患率に対して有意な効果はなかった。リンパ様始原細胞および成熟リンパ球は、培養物中の重要な範囲のいずれにも見られなかった。
【0106】
SCF/IL3中で培養されたCD34+ve細胞は、22日目までに完全に成熟し、それによって細胞の49±4%が好中球(CD65+ve/CD11b+veおよびCD15+veとして)であり、細胞の53±8%が単球(CD14+ve/CD11b+ve)であった。CD33+ve/CD15−ve/CD14−veとして同定された未熟骨髄細胞は、19日目までに培養物から無くなった。細胞が、G−CSF/SCF/IL3中で培養された場合、等しい数の好中球および単球が産生されたが、SCF/IL3が使用された場合よりもはるかに数が多かった(図3A)。22日までに、G−SCF/SCF/IL3の培養物は、成熟好中球および単球を大量に含有した(図3Aにおける総細胞数と好中球数と単球数とを比較する)。
【0107】
CD34+ve細胞の培養においてSCF/IL3の使用に対して10 nMおよび100 nMのVTP194310の添加効果は、産生された好中球数を増加させることであった(図3A下部の2つのパネルを比較する)。これに関して、10 nMのVTP194310は、100 nMのVTP194310よりも有効であった。単球の累積数は、VTP194310の存在により有意に影響を及ぼさなかった(図3A)。20日目から30日目の間のVTP194310処理培養物の視覚検査により、芽細胞は、該培養物の至る所にあることが明らかとなり、マーカー分析により、細胞の実在する割合は、好中球または単球のいずれにも成熟していなかったことを示した。19日目と29日目との間、好中球は、33%±2(10 nMのVTP194310)および30%±5(100nMの VTP194310)のレベルで、単球は、32%±4(10 nMのVTP194310)および21%±5(100 nMのVTP194310)のレベルで存在した。二重および三重のFACS分析による全ての検査により、VTP194310処理培養物に持続していた2つの集団の細胞を確認することができた。これらは、SCFに対する受容体を発現し、CD34−ve、およびCD33+ve/CD15−ve/CD14−veとして同定された未熟骨髄性細胞である細胞であった(図3B)。これらの細胞集団は10 nMのVTP194310処理培養物と比較して、100 nMのVTP194310処理培養物中に広範囲で存在した。VTP194310は、培養中のCD34+ve細胞の数および持続性に影響を及ぼさなかった(図3B)。これらのマーカー分析は、VTP194310処理培養物中の好中球の産生増加が、骨髄前駆体集団の拡大および好中球分化の減速に関連する概念と釣り合っている。
【0108】
VTP194310処理からの成熟顆粒球および対照マウスは、エキソビボで同様の率でアポトーシスを受ける。同様に我々は、ヒト末梢血白血球(デキストラン沈降法により調製)が、10% FBSおよび10 nMおよび100 nMのVTP194310を含有する培地で補足されたRPMI1640培地中で培養された場合、好中球は同様の率でアポトーシスを受けることを観察した。これら条件の各々における1日目において、核物質のアクリジンオレンジ染色および蛍光顕微鏡により観察されるように、65%から70%の好中球はアポトーシスであった。好中球の全ては、2日目までに死滅した。
【0109】
実施例3
白血球減少症のシクロホスファミド誘導マウスモデルにおいて末梢および全身の好中球およびリンパ球の回復
白血球減少症マウスにおけるVTP 194310駆動好中球回復が、脾臓および骨髄中の好中球(Gr1+ve/CD 11b+ve細胞)のレベルを見ることにより全身的であるかどうかを調べた。これらのマウスは、CPM処置に関して1日前から2日目にVTP 194310 により胃管栄養を行った。
【0110】
結果
データが示すように、対照の回復マウスと比較して、好中球数およびリンパ球数は、VTP 194310処理マウスにおいて6日目から8日目に有意に上昇した(図4Aを参照)。対照と比較して、脾臓重量は、VTP 194310処理マウスにおいて7日目と8日目に増加した。Gr−1+ve/CD1lb+ve細胞の絶対数は、VTP 194310処理マウスの脾臓において6日目と7日目に有意に増加した(see Figure 図4Bを参照)。さらに、Gr−1low/CD 1lb+veとして同定された未熟顆粒球は、VTP 194310処理マウスの脾臓において6日目と7日目に上昇した。7日目におけるこられの細胞の絶対数は、対照マウスの0.46±0.13×107と比較して、VTP 194310処理マウスにおいて0.93±0.02 x107であった(p = 0.02)。また、VTP 194310処理マウスの骨髄においてGr−1+ve/CD1lb+ve細胞の広がりが増加した(図4Cを参照)。それゆえ、VTP 194310により引き起こされた好中球の回復は全身的である。
【0111】
実施例4
白血球減少症マウスにおいて黄色ブドウ球菌に対するVTP194310の防護効果の測定
American Type Culture Collection(ATCC)(ロックヴィレ、メリーランド州)から入手された黄色ブドウ球菌(S.aureus)29213 を、白血球減少症メスBDF1マウス(20〜22 g)において感染症を誘導させるために用いた。黄色ブドウ球菌を、中間対数増殖期(600 nm = 0.3での光学密度)までトリプシンダイズブロス中37℃で培養し、収穫し、PBSで洗浄した。細菌数を、PBSとの連続希釈により数え、血液寒天上で平板培養し、37℃でインキュベーション48時間後にコロニー形成単位(CFU)をカウントした。この懸濁液を10° CFU/mlに調整した。
【0112】
マウスを200 mg/kgでCPMのi.p.注射により白血球減少症にした。黄色ブドウ球菌の致死量(LD)は、CPM処置4日後、10匹のマウス群に1×103から1×108 CFUまでの腹腔内注射により判定した。10日の生存動物を数え、黄色ブドウ球菌のLD95をプロビット解析により算出した。VTP 194310(1 mg/kgで)による処置は、CPM投与1日前に開始し、さらに3日間続けた。CPM投与4日後、この動物を、200μlのPBS中、4.1および2.5×106 CFUの黄色ブドウ球菌により静脈内に感染させた。生存動物は、誘発後14日間、毎日記録した。1条件当り12匹のマウスであり、VTP 194310の防護効果の有意性を、Logrank Test(GraphPad Prismバージョン3.0、GraphPad Software社、サンディエゴ、カリフォルニア州)により測定した。全ての実験は、関連する法律および施設のガイドラインに準拠して実施し、the Animal Care and Use Committeeにより承認された。
【0113】
結果
本実施例において、VTP 194310処置白血球減少症マウスにおける好中球回復の改善が、黄色ブドウ球菌の致死量を投与することにより、マウスを感染症から防止できるかどうかを調査した(図5を参照)。VTP 194310は、CPM投与1日前、CPM投与と同時に、CPM投与2日後に投与され、黄色ブドウ球菌は、CPM投与4日後に注射された。CPMおよび4.1×106 CFUの黄色ブドウ球菌を投与されたマウスは、10日目に全て死亡した。低用量の黄色ブドウ球菌を受けたマウスの10%だけが生存し、これらのマウスは、11日目までに死亡した。10日目において、VTP 194310を受けたマウスの83%、および黄色ブドウ球菌の各用量では生存した。この実験は14日目に終了し、VTP194310および2.5×106 CFUを受けたマウスの67%は生存したが、一方、VTP 194310およびより高い用量の細菌を受けたマウスの42%は生存した。したがって、対照群と比較してVTP 194310による処置により、黄色ブドウ球菌に対して極めて有意な防護効果があった(4.1×106 CFUの黄色ブドウ球菌により感染したマウスに関してp値= 0.0031;2.5×106 CFUの黄色ブドウ球菌により感染したマウスに関してp値< 0.0001)。
【0114】
黄色ブドウ球菌を与えたVTP194310処置マウスの生存は、黄色ブドウ球菌に対するVTP194310の活性によるものではなかった。この化合物は、黄色ブドウ球菌EMRSA−16 252および黄色ブドウ球菌MSSA 476に対する活性関する幾つかのアッセイにおいて適切な濃度で分析した。寒天上の細菌増殖は、寒天に組み込まれた2μMのVTP194310の存在により、または50μlの2μMのVTP194310が、寒天ウェル拡散アッセイに添加された場合に阻止されなかった。さらに、2μMのVTP194310の存在下、液体培養中での増殖期間の増殖率に変化を示す株は無かった。
【0115】
実施例5
CPM誘導好中球減少症マウスにおける好中球回復に対する単一療法およびVTP 194310およびペギル化G−CSFの併用療法効果の比較
顆粒球コロニー刺激因子(G−CSF)は、好中球減少症における好中球の回復を駆動する重要な因子であり、化学療法誘導性の重症の好中球減少症の処置のために臨床的に使用されている。したがって、単一療法のVTP194310、単一療法G−CSFおよびVTP194310とG−CSFとの併用使用の効果を比較することは重要である。VTP194310による単一療法(3mg/kg、1日前から1日目)およびペギル化G−CSF(10μg/kg、2日目)は、好中球減少症マウス(150 mg/kgのCPM)の対照に比して好中球の回復を増強した。VTP194310およびペギル化G−CSFの用量および措置による併用処置により、好中球の回復率をさらに増加した。血中好中球の上昇を刺激するVTP194310およびペギル化G−CSFの併用効果は、相加的効果よりも僅かに大きかった。さらに、VTP194310と一緒のG−CSFは、G−CSF単独で使用された場合よりも長く持続する好中球を生じた。これらの知見は、VTP194310が回復期の間G−CSFと共に作用する提案と釣り合っている。
【0116】
製剤
VTP194310のDMSOストック溶液を、DMSOに溶解されたVTP194310を計量することにより作製した(25 mg/1.33 mlのDMSO)。このストック溶液を次に、媒体(39.2 mlのトウモロコシ油+0.8 mlのDMSO)と混合した。
【0117】
3.0 mg/kgのVTP194310溶液を、39.2 mlのトウモロコシ油+0.8 mlのVTP 194310 DMSOストックを混合することにより作製した。
【0118】
1.0 mg/kgのVTP194310溶液は、VTP194310のDMSOストック1:3をDMSOで希釈する(例えば、1 mlのDMSO + 0.5 mlの4310 Stock)ことにより作製され、次いで39.2 mlのトウモロコシ油 + 0.8 mlの希釈VTP194310ストックを組合わせた。
【0119】
CPM溶液の調製:
150 mg/kgのCPM溶液を、187.5 mgの5−FUを計量し、10 gの生理食塩水と混合することにより作製した。
【0120】
1μg/kgおよび10μg/kgのPEG−G−CSF溶液は、6 mg/mlのストックをPBSで希釈することにより作製した。
【0121】
マウス:
オスBDF1マウス(C57Bl×DBA2)、7〜8週齢、DOB:2006年7月1日)をこの実験に用いた。
【0122】
血液サンプル:
35μlの血液(顎下静脈を介して)を、スケジュールされた時点に従ってまたは血液細胞および血小板の完全回復まで採血した。血液サンプルを、2% BSA(フラクションV)および0.2 mMのEDTAを含有するPBS生理食塩水で1:5に希釈後、Hematology Analyzer Cell−DYN 3700(Abbott Diagnostics)上で分析した。
【0123】
結果
各群のマウスを、媒体、3 mg/kgでのVTP194310、1 μg/kgまたは10 μg/kgでのPEG−G−CSF、および150 mg/kgでのCPMにより処置した。総白血球数(WBC)、好中球、リンパ球、赤血球(RBC)および血小板の平均数、ならびに各処置群に対して得られたデータに関する標準誤差(SE)を図6に示す。図6Bおよび図7に示されるように、1日前および1日目に1 mg/kg/日または3 mg/kg/日でのVTP194310による単一療法および2日目に1 μg/kg または10 μg/kg でのPEG−G−CSFによる単一療法は、好中球減少症マウス(0日目に150 mg/kgのCPMにより誘導された)の対照に比して、好中球の回復を増強した。VTP194310またはPEG−G−CSFのいずれかによる単一療法と比較して、これらVTP194310およびPEG−G−CSFの用量および措置による併用処置により、好中球の回復率をさらに増加した。血中好中球の上昇を刺激する上でVTP194310 およびPEG−G−CSFの併用効果は、相加的以上であった。さらに、VTP194310と一緒の PEG−G−CSFは、PEG−G−CSF単独で使用された場合よりも長く持続する好中球を生じた。結論として、VTP194310は、好中球減少症のCPM誘導マウスモデルにおいて好中球の回復を増強する上で10 μg/kgでのPEG−G−CSFと同様に有効であると思われる。VTP194310およびPEG−G−CSFによる併用処置は、さらに回復を増加した。
【0124】
実施例6
5−FU誘導好中球減少症マウスにおける好中球回復に対する単一療法およびVTP 194310およびペギル化G−CSFの併用療法効果の比較
顆粒球コロニー刺激因子(G−CSF)は、好中球減少症における好中球の回復を駆動する重要な因子であり、したがって、患者における化学療法誘導性の重症好中球減少症の処置のために使用されている。単一療法のVTP194310、単一療法G−CSFの効果およびVTP194310とG−CSFとの併用効果を比較することは重要である。ここに我々は、好中球減少症、すなわち150 mg/kgの5−FUの投与により誘導された異なるマウスモデルを用いて実験を実施した。この目的は、5−FUマウスモデルにおけるVTP 194310およびPEG−G−CSFの効果を調査することであった。VTP194310による単一療法(3mg/kg、2日目から4日目)は、我々が以前に観察した好中球の回復を改善した。異なる日(5日目、6日目、または7日目)に投与された10μg/kgでのPEG−G−CSFによる単一療法は、好中球の回復を増強する効果は殆どなかった。しかしながら、VTP194310およびPEG−G−CSFの同じ用量および措置による併用処置は、有意に好中球の回復率を増加した。
【0125】
製剤:
VTP194310のDMSOストック溶液を、VTP194310を計量し、DMSO中に溶解することによって作製した(25 mg/1.33 mlのDMSO)。
【0126】
この媒体溶液を、39.2 mlのトウモロコシ油+0.8 mlのDMSOを混合することにより作製した。
【0127】
3.0 mg/kgのVTP194310溶液を、39.2 mlのトウモロコシ油+0.8 mlのVTP 194310 DMSOストック溶液を混合することにより作製した。
【0128】
150 mg/kgの5−FU溶液を、187.5 mgの5−FUを計量し、10 gの生理食塩水と混合することにより作製した。
【0129】
10μg/kgのPEG−G−CSF溶液は、6 mg/mlのストックをPBSで希釈することにより作製した。
【0130】
マウス:
オスBDF1マウス(C57Bl×DBA2)、9週齢、DOB:2006年6月26日)を本実験に用いた。
【0131】
結果
各群のマウスを表に掲げた措置に従って、0日目に150 mg/kgの5−FUで静脈内で処置し、2日目から4日目に媒体、3 mg/kgでのVTP194310を胃管栄養法により経口的に、および/または5日目、6日目、または7日目に10 μg/kgでのPEG−G−CSFにより皮下的に処置した。総白血球数(WBC)、好中球、リンパ球、赤血球(RBC)および血小板の平均数、ならびに各処置群に対して得られたデータに関する標準誤差(SE)を図8に示す。好中球減少症の5−FU誘導マウスモデルにおいて好中球の回復を増強する上で、2日目から4日目の3日間で3 mg/kg/日でのVTP194310の投与の方が、5日目、6日目、または7日目に投与された10 μg/kgでのPEG−G−CSFよりも有効である。この5−FU誘導好中球減少症モデルにおいて好中球回復を刺激する上でVTP194310とPEG−G−CSFとの併用処置は、VTP194310による単一療法またはPEG−G−CSFによる単一療法よりも有効であると思われる。
【0132】
図8Bおよび図9に示されるように、2日目から4日目に3 mg/kg/日でのVTP194310による単一療法は、以前に得られた結果(RT−06−34)と一致する好中球の回復を改善した。5日目、6日目、または7日目に10μg/kgでのPEG−G−CSFによる単一療法は、5−FU好中球減少症マウスの対照に比して好中球回復を増強する効果が殆ど無いことを示した。VTP194310 およびPEG−G−CSFの同じ用量および措置による併用処置は、VTP194310による単一療法と比較して好中球の回復率をさらに増加した。
【0133】
実施例7
VTP 194310(以前にAGN 194310と呼ばれた)の合成
VTP 194310は、以下の化学構造:
【0134】
【化17】
を有する。
【0135】
この化合物、4−[[4−(4−エチルフェニル)−2,2−ジメチル−(2H)−チオクロメン−6−イル]−エチニル]−安息香酸は、従来の有機合成手段を用いて合成することができる。以下の反応スキームは、現在この化合物を製造するのに好ましい方法である。
【0136】
ステップ1:厚肉スクリューキャップチューブに、3−メチル−2−ブテン酸(13.86 g、138.4 mmol)、4−メトキシチオフェノール(20.0 g、138.4 mmol)、およびピペリジン(3.45 g、41.6 mmol)を充填した。この混合物を、32時間10℃に加熱し、室温に冷却し、EtOAc(700 mL)に溶解した。生じた溶液を、1M水性HCl、H2O、および飽和水性NaClで洗浄してから、Na2SO4で乾燥した。減圧下、乾燥溶液の濃縮によりオイルが得られ、これをフリーザー内で放置すると結晶性固体を得た。この結晶性固体を、ペンタンで洗浄することにより3−(4−メトキシ−フェニルスルファニル)−3−メチル−酪酸を淡黄色結晶として単離した(27.33 g、82%)。1H NMR(300 MHz,CDCl3)δ:7.48(2H,d,J=9.0 Hz),6.89(2H,d,J=8.9 Hz),3.83(3H,s),2.54(2H,s),1.40(6H,s)。
【0137】
ステップ2:3−(4−メトキシ−フェニルスルファニル)−3−メチル−酪酸(20.0 g,83.2 mmol)の250 mLのベンゼン溶液に、室温で10 mLのベンゼン中の塩化オキサリル(15.84 g、124.8 mmol)溶液を30分かけて加えた。4時間後、この溶液を、氷冷5%水性NaOH(注意:この手法中、大量のガス放出される)で洗浄し、次いで氷冷H2O、最後に飽和水性NaClで洗浄した。この溶液を乾燥し(Na2SO4)、減圧濃縮すると透明な黄色オイルを得た。この物質を、さらに精製することなく以下のステップに用いた。1H NMR(300 MHz,CDCl3)δ:7.45(2H,d,J=8.8 Hz),6.90(2H,d,J=8.8 Hz),3.84(3H,s),3.12(2H,s),1.41(6H,s)。
【0138】
ステップ3:ステップ2(21.5 g、83.2 mmol)のアシルクロリド生成物の250 mLのCH2Cl2溶液に、0℃で SnCl4(21.7 g、83.2 mmol)の30 mLのCH2Cl2溶液を滴下しながら加えた。2時間後、反応を150 mLのH2Oの緩徐な添加によりクエンチした。有機層を、1M水性HCl、5%水性NaOH、H2O、および最後に飽和水性NaClで洗浄してから、MgSO4で乾燥した。減圧濃縮および残渣オイルの減圧蒸留(バルブからバルブ、125−135℃、5 mm/Hg)により、14.48 g(78%)の6−メトキシ−2,2−ジメチル−チオクロマン−4−オンを黄色オイルとして得た。1H NMR(300 MHz,CDCl3)δ:7.62(1H,d,J=2.9 Hz),7.14(1H,d,J=8.6 Hz),7.03(1H,dd,J=2.8,8.3 Hz),3.83(3H,s),2.87(2H,s),1.46(6H,s)。
【0139】
ステップ4:6−メトキシ−2,2−ジメチル−チオクロマン−4−オン(6.0 g、27 mmol)の−23℃に冷却された50 mLのCH2Cl2溶液に、BBr3(20.0 g、80.0 mmol;80.0 mLの中CH2Cl2 1M溶液)を20分間かけて加えた。−23℃で5時間攪拌後、この溶液を−78℃に冷却し、50 mLのH2Oの緩徐な添加によりクエンチした。室温に暖めてから水層をCH2Cl2で抽出し、有機層を合わせて、飽和水性NaHCO3、H2O、および飽和水性NaClで洗浄してから、MgSO4で乾燥した。溶媒の減圧留去により緑褐色固体が得られ、これを結晶化(Et2O/ヘキサン類)させると、2.25 g(40%)の6−ヒドロキシ−2,2−ジメチルチオクロマン−4−オンを淡褐色固体として得た。1H NMR(300 MHz,CDCl3)δ:7.63(1H,d,J=2.8 Hz),7.15(1H,d,J=8.5 Hz),7.01(1H,dd,J=2.8,8.5 Hz),2.87(2H,s),1.46(6H,s)。
【0140】
ステップ5:6−ヒドロキシ−2,2−ジメチルチオクロマン−4−オン(165.0 mg、0.79 mmol)の5.0 mLの無水ピリジン溶液に、無水トリフルオロメタンスルホン酸(245.0 mg、0.87 mmol)を0℃で加えた。0℃で4時間後、この溶液を濃縮し、残渣オイルをEt2Oに溶解し、H2Oに次いで飽和水性NaClで洗浄し、MgSO4で乾燥した。溶媒の減圧留去およびカラムクロマトグラフィー(5% EtOAc/ヘキサン類)により、126.0 mg(47%)の2,2−ジメチル−4−オキソ−チオクロマン−6−イル トリフルオロメタンスルホン酸塩を無色固体として得た。1H NMR(300 MHz,CDCl3)δ:7.97(1H,s),7.32(2H,s),2.90(2H,s),1.49(6H,s)。
【0141】
ステップ6:2,2−ジメチル−4−オキソ−チオクロマン−6−イル トリフルオロメタンスルホン酸塩(2.88 g、8.50 mmol)の10 mLのEt3Nおよび20.0 mLのDMF溶液を、アルゴンにより10分間パージした。この溶液に、トリメチルシリルアセチレン(4.15 g、42.0 mmol)およびビス(トリフェニルホスフィン)−パラジウム(II)クロリド(298.0 mg、0.425 mmol)を加えた。この溶液を、95℃で5時間加熱し、室温に冷却し、H2Oで希釈した。EtOAcによる抽出に次いで、有機層を合わせてH2Oおよび飽和水性NaClで洗浄し、MgSO4で乾燥した。乾燥溶液の減圧濃縮およびカラムクロマトグラフィー(3% EtOAc/ヘキサン類)による生成物の単離により、2.23 g(91%)の2,2−ジメチル−6−トリメチルシラニルエチニル−チオクロマン−4−オンを橙色オイルとして得た。1H NMR(300 MHz,CDCl3)δ:8.18(1H,d,J=1.9 Hz),7.34(1H,dd,J=1.9,8.1 Hz),7.15(1H,d,J=8.1 Hz),2.85(2H,s),1.45(6H,s),0.23(9H,s)。
【0142】
ステップ7:2,2−ジメチル−6−トリメチルシラニルエチニル−チオクロマン−4−オンの(110.0 mg、0.38 mmol)およびK2CO3(40.0 mg、0.29 mmol)の10.0 mLのMeOH溶液を、室温で一晩攪拌した。該溶液をH2Oで希釈し、Et2Oで抽出した。有機層を合わせて、H2Oおよび飽和水性NaClで洗浄し、MgSO4で乾燥した。溶媒の減圧留去により、81 mg(99%)の6−エチニル−2,2−ジメチルチオクロマン−4−オンを橙色オイルとして得た。1H NMR(300 MHz,CDCl3)δ:8.20(1H,d,J=1.9 Hz),7.46(1H,dd,J=1.9,8.1 Hz),7.18(1H,d,J=8.1 Hz),3.08(1H,s),2.86(2H,s),1.46(6H,s)。
【0143】
ステップ8:6−エチニル−2,2−ジメチルチオクロマン−4−オン(82.0 mg、0.38 mmol)およびエチル4−ヨードベンゾエート(104.9 mg、0.38 mmol)の5.0 mLのEt3N溶液を、アルゴンで10分間パージした。この溶液に、ビス(トリフェニルホスフィン)−パラジウム(II)クロリド(88.0 mg、0.12 mmol)およびヨウ化銅(I)(22.9 mg、0.12 mmol)を加えた。さらに5分間アルゴンでパージ後、該溶液を、室温で一晩攪拌した。反応混合物を、Et2O洗浄を用いたセライトパッドを通してろ過した。ろ液の減圧濃縮、次いで残渣の固体のカラムクロマトグラフィーにより、100 mg(72%)のエチル4−[(2,2−ジメチル−4−オキソ−チオクロマン−6−イル)エチニル]−ベンゾエートを黄色固体として得た。1H NMR(300 MHz,CDCl3)δ:8.25(1H,d,J=1.8 Hz),8.00(2H,d,J=8.4 Hz),7.55(2H,d,J=8.4 Hz),7.53(1H,dd,J=1.8,8.2 Hz),7.21(1H,d,J=8.2 Hz),4.37(2H,q,J=7.1 Hz),2.88(2H,s),1.47(6H,s),1.39(3H,t,J=7.1 Hz)。
【0144】
ステップ9:ナトリムビス(トリメチルシリル)アミド(1.12 g、6.13 mmol)の16.2 mLのTHF溶液を、−78℃に冷却し、この溶液にエチル4−[(2,2−ジメチル−4−オキソ−チオクロマン−6−イル)エチニル]−ベンゾエート(1.86 g、5.10 mmol)の15.0 mLのTHF溶液を徐々に加えた。30分後、2−[N,N−ビス(トリフルオロメタンスルホニル)アミノ]−5−ピリジン(2.40 g、6.13 mmol)の10 mLのTHF溶液を加えた。5 分後、この溶液を室温に温め、一晩攪拌した。反応を、飽和水性NH4Clの添加によりクエンチし、EtOAcで抽出した。有機層を合わせて、5%水性NaOHおよびH2Oで洗浄してから、乾燥(MgSO4)および減圧濃縮した。,1.53 g(61%)のエチル4−((2,2−ジメチル−4−トリフルオロメタンスルホニルオキシ−(2H)−チオクロメン−6−イル)エチニル)−ベンゾエートを、カラムクロマトグラフィー(2% EtOAc/ヘキサン類)により黄色固体として単離した。1H NMR(300 MHz,CDCl3)δ:8.03(2H,d,J=8.4 Hz),7.61(1H,d,J=1.8 Hz),7.59(2H,d,J=8.4 Hz),7.41(1H,dd,J=1.8,8.1 Hz),7.29(1H,d,J=8.1 Hz),5.91(1H,s),4.39(2H,q,J=7.1 Hz),1.53(6H,s),1.41(3H,t,J=7.1 Hz)。
【0145】
ステップ10:4−エチルブロモベンゼン(670.9 mg、3.63 mmol)の4.0 mLのTHF溶液を−78℃に冷却し;t−ブチルリチウム(464.5 mg、7.25 mmol,4.26 mLのペンタン中1.7M溶液)を加えると黄色溶液を得た。30分後、ZnCl2(658.7 mg、4.83 mmol)の8.0 mLのTHF溶液はカニューレを介して徐々に加えた。生じた溶液を室温に温め、カニューレを通してエチル4−(2,2−ジメチル−4−トリフルオロメタンスルホニルオキシ−(2H)−チオ−クロメン−6−イル)エチニル)−ベンゾエート(1.20 g、2.42 mmol)および テトラキス(トリフェニルホスフィン)パラジウム(0)(111.7 mg、0.097 mmol)の8.0 mLのTHF溶液に移した。この溶液を50℃に1時間加熱し、室温に冷却し、反応を飽和水性NH4Clの添加によりクエンチした。この溶液をEtOAcで抽出し、有機層を合わせてH2Oおよび飽和水性NaClで洗浄してから、乾燥し(MgSO4)、減圧濃縮した。カラムクロマトグラフィー(5% EtOAc/ヘキサン類)により、エチル4−[[4−(4−エチルフェニル)−2,2−ジメチル−(2H)−チオクロメン−6−イル]−エチニル]−ベンゾエートを無色オイルとして単離した。1H NMR(300 MHz,CDCl3)δ:7.99(2H,d,J=8.2 Hz),7.52(2H,d,J=8.4 Hz),7.40(5H,m),7.35(2H,m),5.85(1H,s),4.38(2H,q,J=7.1 Hz),2.72(2H,q,J=7.6 Hz),1.48(6H,s),1.40(3H,t,J=7.1 Hz),1.30(3H,t,J=7.6 Hz)。
【0146】
ステップ11:エチル4−[[4−(4−エチルフェニル)−2,2−ジメチル−(2H)−チオクロメン−6−イル]−エチニル]−ベンゾエート(940.0 mg、2.08 mmol)の10.0 mLのTHFおよび5.0 mLのEtOH溶液に、NaOH(416.0 mg、10.4 mmol、5.2 mLの2M水溶液)を加えた。生じた溶液を室温で一晩攪拌した。反応混合物を10%水性HClで酸性にし、EtOAcで抽出した。有機層を合わせて、H2O、飽和水性NaClで洗浄し、乾燥(Na2SO4)してから、溶媒を減圧留去した。残渣の固体を、CH3CNから再結晶して、786.0 mg(89%)の4−[[4−(4−エチルフェニル)−2,2−ジメチル−(2H)−チオクロメン−6−イル]−エチニル]−安息香酸を無色固体として得た。1H NMR(300 MHz,d6−acetone)δ:8.01(2H,d,J=8.3 Hz),7.60(2H,d,J=8.5 Hz),7.42(2H,m),7.29(2H,m),7.22(3H,m),5.94(1H,s),2.69(2H,q,J=7.7 Hz),1.47(6H,s),1.25(3H,t,J=7.7 Hz)。この化合物、すなわち、最後の所望の生成物は、VTP 194310と称された。
【0147】
上記の説明は、多くの明細を含有するが、これらの明細は、本発明を限定するものとして解釈してはならないが、単にそれらの好ましい実施形態の例証として解釈スべきである。当業者は、本明細書に添付された請求項により定義されている範囲内の他の多くの実施形態と本発明の趣旨とを想像するであろう。
【0148】
本出願に引用されるいずれの特許、特許出願、および他の刊行物は、参照としてその全体が本明細書に援用されている。
【技術分野】
【0001】
関連出願
この出願は、2006年5月16日に出願された米国仮出願第60/800,773号(この全体の教示は、参考として本明細書に援用される)の利益を主張する。
【0002】
本発明は一般に、哺乳動物における化学療法および放射線療法の副作用を処置する方法に関する。
【背景技術】
【0003】
関連技術の説明
一般に、哺乳動物における正常細胞は、秩序正しく制御された様式で増殖し分裂する。癌は、細胞が異常になって、制御されずに増え始め、腫瘍と呼ばれる余分な組織塊へと発達する疾患である。これらの癌細胞は、近傍の組織に浸潤し、血流およびリンパ系を介して身体の他の部分へ拡散する。
【0004】
現在、4つの主な癌治療のタイプは、免疫療法、外科手術、放射線療法、および化学療法である。これらの癌治療は、単独で、または他の治療と関連させて適用することができる。したがって、癌患者は、一度に1つ又は複数の治療を受けることができる。単独治療は、種々の間隔で送達される療法によって、一定の期間にわたり得る。免疫療法は、該疾患と闘うために、免疫系の能力を刺激するか、または回復させることを意図する。免疫療法はまた、いくつかの癌治療によって引き起こされ得る免疫系に関連した副作用を減少させるために用いることもできる。外科手術は、身体から腫瘍を直接除去しようとする。
【0005】
照射療法としても知られている放射線療法は、該細胞の遺伝子材料を損傷させることによって癌細胞を殺して腫瘍を縮小させるために、x線、ガンマ線、ニュートロン、および他の供給源からの高エネルギー照射を用いる。癌細胞は永久的に損傷を受け、その結果死滅するが、放射線療法で損傷を受ける正常細胞は、それら自身を修復することができる。放射線療法中に生じ得る副作用としては、治療を受けている部位の皮膚刺激および毛髪喪失、ならびに骨髄に対する損傷が挙げられる。
【0006】
化学療法では、癌細胞を破壊するために、細胞毒性薬を単独で、または組み合わせて用いる。放射線療法でのように、癌細胞には損傷を与えて結果的に死滅させることができるが、その過程で影響を受けた健常細胞は、化学療法後にそれら自身を修復することができる。細胞毒性薬は、増殖細胞が分裂しそれ自身を複製する能力を妨害することによって作用する。したがって、癌細胞に加えて、他の正常な、速やかに分裂し増殖している細胞もまた影響を受ける可能性がある。例えば、骨髄中に形成される血液細胞に影響を与えて、骨髄抑制を引き起こす可能性がある。また、例えば、消化管内、口腔の内層および生殖系の細胞に影響を与えて、下痢および口腔のひりひり感を起こしたり、また、毛嚢に影響を与えて毛髪喪失を引き起こす可能性がある。
【0007】
骨髄抑制は、化学療法および放射線療法の多くの副作用のうちの1つである。それによって、赤血球、白血球、および血小板などの血液細胞の産生減少がもたらされる。その結果、患者は、貧血から、疲労を経験し、白血病から、より感染しやすくなり、血小板減少症から、打撲傷を受けやすく、切り傷の際に出血が多くなる可能性がある。骨髄抑制の副作用に対抗するために、薬剤が一般的に用いられる。例えば、癌の化学療法における貧血の副作用に対抗するために、Epogen(登録商標)(エポイエチン)が用いられており、血小板減少症の副作用に対抗するために、WinRho(登録商標)SDF(Rho(D)免疫グロブリン)が用いられている。
【0008】
化学療法および放射線療法の副作用の予防、またはそれらからの保護は、癌患者にとって大きな利益になると考えられる。これらの副作用を減少させる以前の多くの努力は概して不首尾に終わっている。命を脅かす副作用に関しては、副作用を減少させるために、化学療法剤および放射線療法剤の用量およびスケジュールを変更することに努力が集中されてきた。化学放射線療法を開始する前に、種々の組織における正常細胞の数を増加させるために、顆粒球コロニー刺激因子(G−CSF)、顆粒球−マクロファージ−CSF(GM−CSF)、上皮成長因子(EGF)、インターロイキン11(il−11)、エリスロポイエチン、トロンボポイエチン、巨核球発達および増殖因子、ピキシキナーゼ、幹細胞因子、FLT−リガンド、ならびにインターロイキン1、3、6、および7の使用など、他の選択肢が利用できるようになってきている。十分に理解されてはいないが、これらの因子による保護の機序は、細胞毒性剤または放射線療法による処置前の正常な危険標的細胞数の増加に関連しており、化学放射線療法後の細胞生存率の増加に関連していない可能性が高い。
【0009】
一般に、多形核白血球とも呼ばれる好中球は、顆粒球として知られる血球のうちで最も多い。好中球は急性炎症応答に関与する最も大きな細胞集団である。したがってそれらは、自然免疫の重要な要素であり、化学走化性刺激に速やかに応答する。好中球は、食作用と呼ばれる、包囲し消化する過程によって、細菌などの外来粒子を破壊する。好中球は、細菌感染に応答して増加し得る。多くの好中球が必要な場合、それらはバンドまたは杵核球と呼ばれる未成熟細胞として骨髄から放出される。好中球減少症は、異常に少数の好中顆粒球を特徴とする血液病である。したがって、好中球減少症に罹っている患者は、細菌感染により罹りやすくなり、これらの状態は命を脅かすものとなり得る。
【0010】
好中球減少症は、癌または後天性免疫不全症候群(AIDS)などの他の状態に副次的に生じ得る。好中球減少症はまた、薬剤療法などの事象に副次的に生じ得る。したがって好中球減少症は、免疫系に直接影響を及ぼす生理学的障害に起因し得る。例えば、好中球産生の減少は、白血病、骨髄腫、リンパ腫、または例えば、乳癌または前立腺癌などの転移性固形腫瘍が、骨髄に浸潤しそれに取って代る場合に生じる。一過性好中球減少症はウィルス感染に関連していることが多い。慢性好中球減少症は、ウィルス感染、例えば、ヒト免疫不全ウィルス(HIV)の感染に起因するAIDSに起因する免疫不全に関連していることが多い。自己免疫好中球減少症は、循環抗好中球抗体に関連している可能性がある。
【0011】
好中球減少症のはるかに一般的な原因は、薬物療法、特に、癌ならびに癌療法に関連した骨髄移植に対する化学療法および放射線療法の副作用である。したがって、薬物療法に副次的な好中球減少症は2つの群に分けることができる。第1の群は、抗体形成を刺激するためのハプテンとして作用する薬物から生じ得る免疫媒介好中球減少症を含む。ジフェニルヒダントインおよびフェノバルビタールによって引き起こされるものなどの急性過敏症反応は数日間継続する。しかし、慢性過敏症反応は数ヶ月または数年継続し得る。
【0012】
薬物に誘導された好中球減少症の第2の範囲は、高用量の細胞減少性癌薬剤または電離放射線療法後に生じることが予想される重症の好中球減少症を含む。これらの細胞毒性療法は、好中球前駆細胞の増殖的性質および循環好中球の通常の速い転換率により好中球減少症を誘導する。癌の化学療法または放射線療法に副次的な好中球減少症の危険性は、癌のタイプおよび病期、ならびに癌治療のタイプ、用量およびスケジュールなどの要因に依存する。
【0013】
好中球レベルを増加させるために現在存在する療法は、主に、フィルグラスチム(Neulasta(登録商標))、より最近では、フィルグラスチムのより長時間作用性の誘導体であるペグフィルグラスチム(Neulasta(登録商標))からなる。フィルグラスチムは、白血球の産生を選択的に刺激するヒトタンパク質、G−CSFの組換え型である。G−CSFは、好中球減少症の現在の選択薬剤である。これらの薬剤は双方とも、組換えタンパク質であるため、経口で活性ではなく、注射によって投与しなければならない。また、タンパク質ベースの薬剤は、しばしば急速な代謝を受ける。
【0014】
化学療法および放射線療法の分野における進歩にも関わらず、先行技術の薬剤および方法は、化学療法誘導脱毛症、放射線療法誘導脱毛症、化学療法誘導血小板減少症、放射線療法誘導血小板減少症、化学療法誘導白血球減少症、放射線療法誘導白血球減少症、化学療法誘導好中球減少症および放射線療法誘導好中球減少症などの、化学療法および放射線療法に起因する副作用の最少化において利用が限定されることが判明している。したがって、哺乳動物における化学放射線療法のこのような副作用を処置するための改善された方法を提供することが望ましいと考えられる。
【発明の概要】
【課題を解決するための手段】
【0015】
本発明の一実施形態により、化学療法および/または放射線療法を受けている哺乳動物における化学療法および/または放射線療法の副作用を処置するための方法が提供され、該方法は、RARα、RARβ および RARγサブタイプの受容体に結合するレチン酸受容体(RAR)アンタゴニストまたはRARインバースアゴニストの治療有効量を該哺乳動物に投与する工程を含んでなる。
【0016】
本発明の第2の実施形態により、哺乳動物における血小板産生を増加させる方法が提供され、該方法は、RARα、RARβおよびd RARγサブタイプの受容体に結合するRARアンタゴニストまたはRARインバースアゴニストの治療有効量を該哺乳動物に投与する工程を含んでなる。
【0017】
本発明の第3の実施形態により、血小板減少症を患っている哺乳動物を処置する方法が提供され、該方法は、RARα、RARβおよびd RARγサブタイプの受容体に結合するRARアンタゴニストまたはRARインバースアゴニストの治療有効量を該哺乳動物に投与する工程を含んでなる。
【0018】
本発明の第4の実施形態により、造血関連状態を患っている哺乳動物を処置する方法が提供され、該方法は、RARα、RARβおよびd RARγサブタイプの受容体に結合するRARアンタゴニストまたはRARインバースアゴニストの治療有効量を該哺乳動物に投与する工程を含んでなる。このような状態としては、限定はしないが、造血機能低下、免疫機能低下、好中球数減少、好中球動員減少、末梢血始原細胞の動員、敗血症、重症の慢性好中球数減少症、骨髄移植、感染性疾患、白血球減少症、血小板減少症、貧血、移植時の骨髄移植増大、放射線処置における骨髄回復の増大、化学物質または化学療法に誘導された骨髄形成不全または骨髄抑制、後天性免疫不全症候群など、ならびにそれらの組み合わせが挙げられる。
【図面の簡単な説明】
【0019】
【図1】図1A〜1Cは、シクロホスファミドに誘導された白血球減少症マウスモデルにおける白血球、好中球およびリンパ球の数に及ぼすVTP 194310の効果のグラフを示す図である。
【図2】図2A〜2Dは、HL60細胞のATRAに駆動された好中球の分化に対する全RARアンタゴニストVTP194310のブロック効果のグラフを示す図である。2.5×105細胞/mlに設定されたHL60細胞を、100nMのパン−RARアンタゴニストVTP194310と共に、100nMのATRAまたは100 nMのATRA で5日間処置した。Aは、位相差顕微鏡によって数え上げた生存細胞の総数を示す。分化は、ニトロブルーテトラゾリウムを減少させる細胞の能力により測定し(B)、CD11bの発現は、モノクローナル抗CD11bおよびFACSを用いて測定し(C)、酵素ステロイドスルファターゼの活性増加は、[3H]硫酸エストロンを用いた細胞超音波処理、続いて[3H]エストロンの抽出で測定した。
【図3】図3A〜3Bは、好中球分化に及ぼすVTP 194310 および G−CSFの単独、および組み合わせ効果のグラフを示す図である。ヒトCD34+ve 造血始原細胞(CD34−HPC)を均質にカラム精製し、幹細胞因子(100 ng/ml)およびIL3(20 ng/ml)中で(●)、ならびにパン特異的RARアンタゴニストVTP 194310の10nM(□)および100 nM(■)と共に培養した。好中球および単球の最適な産生を促進するために、CD34−HPCを、幹細胞因子(100 ng/ml)およびIL3(5 ng/ml)およびG−CSF(30 ng/ml)中で培養した(▲)。細胞表現型を、時々多色FACSアッセイにより判定した。培養は三重に設定し、各マーカーに関する多色FACS分析および単色分析は二重に行なった。好中球は、CD11b+ve/CD65+ve 細胞およびCD15+ve細胞として同定した。単球は、CD11b+ve/CD14+ve細胞として同定した。データは平均値±SDとして示されている。
【図4】図4A〜4Cは、シクロホスファミドに誘導された白血球減少症マウスモデルにおける好中球数および他のパラメーターに及ぼすVTP 194310の効果のグラフを示す図である。
【図5】黄色ブドウ球菌(Staphylococcus aureus)に致死的に感染した白血球減少症マウスの生存率に及ぼすVTP 194310の効果のグラフを示す図である。
【図6】図6A〜6Eは、VTP 194310または/およびPEG−G−CSFを投与されたマウスにおける総白血球(WBC)、好中球、リンパ球、赤血球(RBC)および血小板の数の全体の変化における変化のグラフを示す図である。1mg/kg/日または3mg/kg/日での媒体またはVTP 194310は、−1日目から1日目まで経口投与し、150mg/kgのCPMは、0日目に腹腔内投与し、PEG−G−CSFは、2日目に皮下投与した。血液サンプルはスケジュール通り採取し、血球数は、Abbott Cell−DYN 3700により測定した。データは、7〜8匹のマウスの平均値±SEを表す。単位は、1μl当たりの細胞数または血小板数である。
【図7】VTP 194310、PEG−G−CSFの各々単独に比較して、好中球減少症マウスにおける好中球回復率をさらに増大させるVTP 194310およびPEG−G−CSFの療法による好中球減少症処置の効果のグラフを示す図である。0日目、150 mg/kg CPMによって好中球減少症にしたマウスに、VTP194310(−1日目から1日目まで3mg/kg/日)または PEG−G−CSF(2日目に10μg/kg)による単独療法ならびにこれらの用量の組み合わせ処置を行なった。データは、7〜8匹のマウスの平均値±SEを表す。処置群間の統計的有意性に関するp値が示されている。単位は、1μl当たりの細胞数または血小板数である。
【図8】図8A〜8Eは、好中球減少症の5−FU誘導マウスモデルにおけるVTP 194310または/およびPEG−G−CSFを投与されたマウスにおける総白血球(WBC)、好中球、リンパ球、赤血球(RBC)および血小板の数の変化のグラフを示す図である。示されているとおり、3mg/kg/日の媒体またはVTP 194310は、2日目から4日目まで経口投与し、150mg/kgの5−FUは、0日目に静脈内投与し、PEG−G−CSFは、5、6、または7日目に皮下投与した。血液サンプルはスケジュール通り採取し、血球数は、Abbott Cell−DYN 3700により測定した。データは、8匹のマウスの平均値±SEを表す。単位は、1μl当たりの細胞数または血小板数である。
【図9】好中球減少症の5−FU誘導マウスモデルにおける好中球の回復に及ぼすVTP 194310またはPEG−G−CSFによる単独療法の効果と組み合わせ処置の効果の比較のグラフを示す図である。0日目、150 mg/kgの5−FUによって好中球減少症にしたマウスに、VTP194310(2日目から4日目まで3mg/kg/日)または PEG−G−CSF(5、6、または7日目に10μg/kg)による単独療法ならびにこれらの用量の組み合わせ処置を行なった。データは、8匹のマウスの平均値±SEを表す。
【発明を実施するための形態】
【0020】
発明の詳細な説明
本発明は、RARα、RARβおよび RARγサブタイプの受容体に結合するレチン酸受容体(RAR)の少なくとも1種のアンタゴニストおよび/またはインバースアゴニスト、すなわち、RARα、RARβ および RARγサブタイプの全てに結合するRARアンタゴニストまたはRARインバースアゴニストを用いる化学療法および/または放射線療法を受けている哺乳動物における、化学療法および/または放射線療法(すなわち、化学放射線療法)の副作用の処置のための方法に関する。このような副作用としては、限定はしないが、化学療法誘導脱毛症、放射線療法誘導脱毛症、化学療法誘導血小板減少症、放射線療法誘導血小板減少症、化学療法誘導白血球減少症、放射線療法誘導白血球減少症、化学療法誘導好中球減少症および放射線療法誘導好中球減少症など、ならびにそれらの組み合わせが挙げられる。
【0021】
RARα、RARβおよび RARγ受容体サブタイプにおけるアゴニスト活性を判定するために使用できるアッセイは、Feigner,P.L.、およびd Holm,M.、Focus、11:2頁、21頁+(1989)ならびに米国特許第5,455,265号および米国特許第7,166,726号に記載されており、これらは参照として、それらの全体が本明細書に援用されている。
【0022】
レチノイドインバースアゴニストの活性は、Kleinら、J.Biol.Chem.271,22692〜22696頁(1996)の手法によって試験することができ、これは特に参照として、本明細書に援用されている。
【0023】
レチノイド類、特に全てのトランスレチン酸(ATRA)は、広範囲の細胞型の生存、増殖および分化に重要な役割を果たすため、正常な哺乳動物の発達にとって必須である。ATRAおよび合成レチノイド類は、受容体の2つの別個の細胞内ファミリー、RAR類およびレチノイドX受容体(RXR類)に結合し、それらを活性化することができ、その結果、遺伝子発現の調節をもたらす。RARαと称される、最初に同定されたレチン酸受容体は、ステロイド/甲状腺ホルモン細胞内受容体スーパーファミリーの多くのメンバーの場合に示されたようなリガンド依存的な様式で、特定の標的遺伝子の転写を調節するように作用する。RARαの転写調節活性が依存する内因性低分子量リガンドは、ATRAである。レチン酸受容体に媒介された遺伝子発現の変化によって、細胞表現型の特徴的な変化がもたらされ、その結果、多くの組織にATRAに対する生物学的応答が現れる。RARαに密接に関連したさらに2つの遺伝子は、RARβおよびRARγと称される。RARsのように、RXRsもまた、少なくとも3つのサブタイプまたはイソ型、すなわち、RXRα、RXRβ、およびRXRγを有し、対応するユニークな発現パターンを有することが知られている(Manglesdorfら、Genes & Devel.、6:329〜44頁(1992))。
【0024】
RARα、RARβおよび RARγサブタイプの受容体に結合するRARアンタゴニストおよび/またはRARインバースアゴニストを含んでなる組成物を哺乳動物に投与することにより、血液の好中球および血小板の産生が向上し、化学療法に誘導された好中球減少症および/または血小板減少症など、化学療法の副作用の処置がもたらされると考えられる。
【0025】
RARα、RARβおよびRARγサブタイプの受容体に結合するRARのアンタゴニストおよびインバースアゴニストの代表例ならびにそれらを調製するための方法は、米国特許第5,776,699号および米国特許第5,958,954号ならびに米国特許出願公開2002/0193403号において、当業界によく知られており、それら各々の内容は参照として、それらの全体が本明細書に援用されている。以下の化合物の多くは、これらの出願および/または特許の1つまたは複数に含まれている。
【0026】
本発明の特定の実施形態は、一般式I:
【0027】
【化1】
により表され、使用することができる一クラスの化合物または薬学的に許容できるその塩であり;
式中、Xは、S、O、RがHまたは1個から6個の炭素のアルキルであるNRか、または
Xは、R1が独立してHまたは1個から6個の炭素のアルキルである[C(R1)2]nであり、nは、0と2の間でかつ0と2を含む整数であり;
R2は、独立して水素、1個から6個の炭素の低級アルキル、F、Cl、Br、I、CF3、1個から6個の炭素のフルオロ置換アルキル、OH、SH、1個から6個の炭素のアルコキシ、または1個から6個の炭素のアルキルチオであり;
R3は、独立して水素、1個から6個の炭素の低級アルキルまたはFであり;
mは、0〜3の値を有する整数であり;
nは、0〜4の値を有する整数であり;
oは、0〜3の値を有する整数であり;
Zは、−CONR1−、−CSNR1−、−NR1CO−、−NR1CS−、−C≡C−、−C=C−、−N=N−、−N=CR1−、−CR1=N−、
−COO−、−OCO−;−OSO−;−OCS−、またはn’が0から5の整数である−(CR1=CR1)n’−であり、;
Yは、フェニル基またはナフチル基、またはピリジル、チエニル、フリル、ピリダジニル、ピリミジニル、ピラジニル、チアゾリル、オキサゾリル、イミダゾリルおよびピラゾリルからなる群より選択されるヘテロアリールであり、前記フェニル基およびヘテロアリール基は、1つまたは2つのR2基で任意に置換されているか、またはZが−(CR1=CR1)n’−であり、n’が3、4または5である場合、Yは、前記(CR2=CR2)n’基とBとの間の直接的原子価結合を表し;
Aは、qが0〜5である(CH2)q、3個から6個の炭素を有する低級分枝鎖アルキル、3個から6個の炭素を有するシクロアルキル、2個から6個の炭素および1つまたは2つの二重結合を有するアルケニル、2個から6個の炭素および1つまたは2つの三重結合を有するアルキニルであり;
Bは、水素、COOHまたは薬学的に許容できるその塩、COOR8、CONR9R10、CH2OH、CH2OR11、CH2OCOR11、CHO、CH(OR12)2、CHOR13O、−COR7、CR7(OR12)2、CR7OR13O、またはトリ−低級アルキルシリルであり;
R7は、1個から5個の炭素を含有するアルキル基、シクロアルキル基またはアルケニル基であり;
R8は、1個から10個の炭素のアルキル基、またはアルキル基が1個から10個の炭素を有するトリメチルアルキル、1個から10個の炭素、3個から10個の炭素のシクロアルキル基、フェニルまたは低級アルキルフェニルであり;
R9およびR10は、独立して水素、1個から10個の炭素のアルキル基、3個から10個の炭素のシクロアルキル基、フェニルまたは低級アルキルフェニルであり;
R11は、低級アルキル、フェニルまたは低級アルキルフェニルであり;
R12は、低級アルキルであり;
R13は、2個から5個の炭素の二価アルキル基であり;
R14は、(R15)r−フェニル、(R15)r−ナフチル、または(R15)r−ヘテロアリールであり、前記ヘテロアリール基が、O、SおよびNからなる群より選択される1個から3個のヘテロ原子を有し、rが、0〜5の値を有する整数であり;
R15は、独立してH、F、Cl、Br、I、NO2、N(R8)2、N(R8)COR8、NR8CON(R8)2、OH、OCOR8、OR8、CN、1個から10個の炭素を有するアルキル基、1個から10個の炭素を有するフルオロ置換アルキル基、2個から10個の炭素および1つから3つの二重結合を有するアルケニル基、2個から10個の炭素および1つから3つの三重結合を有するアルキニル基、またはアルキル基が独立して1個から6個の炭素を有するトリアルキルシリル基またはトリアルキルシリルオキシ基である。
【0028】
本発明の別の特定の実施形態は、式Iにより表され、使用することができる一クラスの化合物であり、Zは、-CONR1−、−CSNR1−、−NR1CO−、または−NR1CS−であり;全て他の可変部分は、上記に定義されたとおりである。
【0029】
本発明の別の特定の実施形態は、式Iにより表され、使用することができる一クラスの化合物であり;式中、
R8は、1個から10個の炭素のアルキル基ま、またはアルキル基が1個から10個の炭素を有するトリメチルアルキル、5個から10個の炭素のシクロアルキル基、フェニルまたは低級アルキルフェニルであり;
R9およびR10は、独立して水素、1個から10個の炭素のアルキル基、5個から10個の炭素のシクロアルキル基、フェニルまたは低級アルキルフェニルであり;全て他の可変部分は、上記に定義されたとおりである。
【0030】
本発明の別の特定の実施形態は、一般式II:
【0031】
【化2】
により表され、使用することができる一クラスの化合物または該化合物の薬学的に許容できるその塩であり;
式中、Xは、−C(R1)2または−O−であり;R1は、HまたはC1〜C6であり;
R2は、C1〜C6低級アルキル、−F、−Cl、−Br、−I、−CF3、フルオロ置換C1〜C6アルキル、−OH、−SH、C1〜C6アルコキシ、またはC1〜C6アルキルチオであり;
mは、0から3の値を有する整数であり;
nは、0から4の値を有する整数であり;
oは、0から3の値を有する整数であり;
R3は、C1〜C6低級アルキルまたはFであり;
R8は、C1〜C10アルキル基、C1〜C10トリメチルシリルアルキルであり、C3〜C10シクロアルキル基、フェニルまたは低級アルキルフェニルであり;
R15は、独立して−H、−F、−Cl、−Br、−I、−NO2、−N(R8)2、−COR8、−NR8CON(R8)2、−OCOR8、−OR8、−CN、C1〜C10アルキル、フルオロ置換C1〜C10アルキル、1つから3つの二重結合を有するC2〜C10アルケニル、1つから3つの三重結合を有するC2〜C10アルキニル、またはC1〜C6トリアルキルシリルまたはトリアルキルシリルオキシであり;
tは、0〜5の整数であり;
前記−CONH基は、ベンゾピランおよびジヒドロナフタレン環の6位または7位にある。
【0032】
本発明の別の特定の実施形態は、一般式IIにより表され、使用することができる一クラスの化合物であり、式中:
R2はFであり;R8は、1個から10個の炭素のアルキル基、該アルキル基が1個から10個の炭素を有するトリメチルシリルアルキル、5個から10個の炭素のシクロアルキル基、フェニルまたは低級アルキルフェニルであり;
R15は、独立してH、F、Cl、Br、I、NO2、N(R8)2、COR8、NR8CON(R8)2、OCOR8、OR8、CN、1個から10個の炭素を有するアルキル基、1個から10個の炭素を有するフルオロ置換アルキル基、1個から10個の炭素および1つから3つの二重結合を有するアルケニル基、1個から10個の炭素および1つから3つの三重結合を有するアルキニル基、またはアルキル基が独立して1個から6個の炭素を有するトリアルキルシリル基またはトリアルキルシリルオキシ基であり;および他の全ての可変部分は、上記に定義されているとおりである。
【0033】
本発明の別の特定の実施形態は、一般式III:
【0034】
【化3】
により表され、使用することができる一クラスの化合物または薬学的に許容できるその塩であり;
式中、Xは、−C(CH3)2−または−O−であり;
R2は、−Hまたは−Brであり;
R2’およびR2’’は、独立して−Hまたは−Fであり;
R3の各々は、独立して−Hまたは−CH3であり;
R8は−Hまたは、C1〜C6 アルキルである。
【0035】
本発明の別の特定の実施形態は、一般式IV:
【0036】
【化4】
により表され、使用することができる一クラスの化合物または薬学的に許容できるその塩であり;
式中、X1は、−S−または−O−であり;
X2は、−CH−または−N−であり;
R2は、−H、−F、−CF3またはC1〜C6アルコキシであり;
R2’’は−H、−Fまたは−CF3であり;
R8は、−H、またはC1〜C6アルキルであり;
R14は、非置換フェニル、非置換チエニルもしくは非置換ピリジル、または1〜3つのR15基で置換されているフェニル、チエニルまたはピリジルであり;
R15の各々は、独立してC1〜C6アルキル、−Cl、−CF3、またはC1〜C6 アルコキシである。
【0037】
本発明の別の特定の実施形態は、一般式V:
【0038】
【化5】
により表され、使用することができる一クラスの化合物または薬学的に許容できるその塩であり;
式中、X2は、−CH−または−N−であり;
R2は、−H、−F、または−OCH3であり;
R2*は、−Hまたは−Fであり;
R8は、−H、またはC1〜C6アルキルであり;
R14は、フェニル、低級アルキルが1個から6個の炭素を有する4−(低級アルキル)フェニル、5−(低級アルキル)−2−チエニル、および6−(低級アルキル)−3−ピリジルからなる群より選択される
本発明の別の特定の実施形態は、一般式VI:
【0039】
【化6】
により表され、使用することができる一クラスの化合物または薬学的に許容できるその塩であり;
式中、R8は、−H、またはC1−C6−アルキルである。
【0040】
本発明の別の特定の実施形態は、一般式VII:
【0041】
【化7】
により表され、使用することができる一クラスの化合物または薬学的に許容できるその塩であり;
式中、R2*は、−Hまたは−Fであり;
R8は、−H、またはC1〜C6−アルキルであり;および
R14は、フェニルおよび4−(C1〜C6−アルキル)フェニルからなる群より選択される。
【0042】
本発明の別の特定の実施形態は、一般式VIII:
【0043】
【化8】
により表され、使用することができる一クラスの化合物または薬学的に許容できるその塩であり;
式中、R8は、−H、またはC1〜C6−アルキルである。R8がHである場合、この化合物は、AGN 193109と呼ばれる。
【0044】
本発明の別の特定の実施形態は、一般式IX:
【0045】
【化9】
により表され、使用することができる一クラスの化合物または薬学的に許容できるその塩であり;
式中、X1の各々は、R1の各々が独立してHまたはC1〜C6−アルキルである、−C(R1)2−、−C(R1)2−C(R1)2−、−S−、−O−、
−NR1−、−C(R1)2−O−、−(C(R1)2−S−、または−C(R1)2−NR1−;であり;
R2の各々は、独立してC1〜C6−アルキル、−F、−Cl、−Br、−I、−CF3、フルオロ置換C1〜C6−アルキル、−OH、−SH、C1〜C6−アルコキシ、またはC1〜C6−アルキルチオであり;
mは、0から4の整数であり;
nは、0から2の整数であり;
oは、0から3の整数であり;
R3は、−H、C1〜C6−アルコキシ、−F、−Cl、−Brまたは−Iであり;
R4は、(R5)p−フェニル、(R5)p−ナフチル、または(R5)p−ヘテロアリールであり、前記ヘテロアリール基は、5員または6員であり、酸素、硫黄、および窒素からなる群より選択される1個から3個のヘテロ原子を有し;pは、0から5の整数であり;
R5の各々の場合は、独立して−F、−Cl、−Br、−I、−NO2、−N(R8)2、−N(R8)COR8、−N(R8)CON(R8)2、−OH、−OCOR8、−OR8、−CN、−COOH、−COOR8、C1〜C10−アルキル、1つから3つの二重結合を有するC1〜C10アルケニル基、1つから3つの三重結合を有するC1〜C10アルキニル基、C1〜C6(トリアルキル)シリルまたはC1〜C6(トリアルキル)シリルオキシであり;
Yは、フェニル、ナフチル、またはピリジル、チエニル、フリル、ピリダジニル、ピリミジニル、ピラジニル、チアゾリル、オキサゾリル、イミダゾリルおよびピラゾリルからなる群より選択されるヘテロアリールであり;前記フェニル基およびヘテロアリール基は、任意にかつ独立して1つまたは2つのR2基で置換されており、またはYは、−(CR3=CR3)r−であり;
rは、1から3の整数であり;
Aは、(CH2)q、低級C3〜C6分枝鎖アルキル、C3〜C6シクロアルキル、1つまたは2つの二重結合を有するC2〜C6 アルケニル、1つまたは2つの三重結合を有するC2〜C6 アルキニルであり;Yが−(CR3=CR3)r−の場合、Aが(CH2)qであり、qが0であるという条件で、qは0〜5の整数であり;
Bは、−H、−COOH、−COOR8、−CONR9R10、−CH2OH,−CH2OR11、−CH2OCOR11、−CHO、−CH(OR12)2、−CHOR13O、−COR7、−CR7(OR12)2、−CR7OR13O、または−Si(C1〜6アルキル)3であり;
R7は、1個から5個の炭素を含有するアルキル基、シクロアルキル基またはアルケニル基であり;
R8は、C1〜C10アルキル、C1〜C10(トリメチルシリル)アルキル、C5〜C10シクロアルキル、フェニルまたは低級アルキルフェニルであり;
R9およびR10は、各々独立して−H、C1〜C10アルキル、C5〜C10シクロアルキル、フェニルまたは低級アルキルフェニルであり;
R11は、低級アルキル、フェニルまたは低級アルキルフェニルであり;
R12は、低級アルキルであり;
R13は、2個から5個の炭素の二価アルキル基である。
【0046】
本発明の別の特定の実施形態は、一般式X:
Y3(R4)−X−Y1(R1)(R2)−Z−Y2(R2)−A−B (X)
により表され、使用することができる一クラスの化合物または薬学的に許容できるその塩であり;
式中、Y1は、フェニル、ナフチル、またはピリジル、チエニル、フリル、ピリダジニル、ピリミジニル、ピラジニル、チアゾニル、オキサゾリル、イミダゾリルおよびピラゾリルからなる群より選択されるヘテロアリールであり、前記フェニル基、ナフチル基、およびヘテロアリール基が、任意にR1基で置換されており、1つまたは2つのR2基で任意にさらに置換されており;
R1は、C1〜C10アルキル、1−アデマンチル、2−テトラヒドロピラノキシ、C1〜C6トリアルキルシラニルオキシ、−OH、C1〜C10アルコキシ、C1〜C10アルキルチオ、または−OCH2O−(C1〜C6アルキル)であり;
R2は、C1〜C6−アルキル、−F、−Cl、−Br、−I、−CF3、−CF2CF3、−OH、−OR3、−NO2、−N(R3)2、−CN、−N3、−COR3、−NHCOR3、−COOH、または−COOR3であり;
Xは、−(C(R3)2)−、−S−、−SO−、−SO2−、−O−、−C(=O)−、−C(=S)−、−C(=NR1)−、−C(=C(R1)2)−または−NR3−であり;
Zは、−C≡C−、−N=N−、−N(O)=N−、−N=N(O)−、−N=CR3−、−CR3=N−、−(CR3=CR3)n−、−OCO−、−CSO−、−OCS−、
−COCR3=R3O−、−CO−NR3−、−CS−NR3、−NR3−CO−、または−NR3−CS−であり;
nは、0〜5の値を有する整数であり;
R3の各々は、独立して−HまたはC1−C6アルキルであり;
Y2は、フェニル基またはナフチル基、またはピリジル、チエニル、フリル、ピリダジニル、ピリミジニル、ピラジニル、チアゾリル、オキサゾリル、イミダゾリルおよびピラゾリルからなる群より選択されるヘテロアリールであり;前記フェニル基、ナフチル基およびヘテロアリール基は、非置換であっても1つまたは2つのR2基で置換されていてもよく、またはZが、−(CR3=CR3)n−であり、nが、3、4、または5である場合、Y2は、該−(CR3=CR3)n−基とBとの間の直接的原子価結合を表し;
Y3は、フェニル、ナフチル、またはピリジル、チエニル、フリル、ピリダジニル、ピリミジニル、ピラジニル、チアゾリル、オキサゾリル、イミダゾリルおよびピラゾリルからなる群より選択されるヘテロアリールであり;前記フェニル基、ナフチル基およびヘテロアリール基は、非置換であっても1つから5つのR4基で置換されていてもよく;
R4の各々は、独立してC1〜C10アルキル、1つから3つの三重結合を有するC2〜C10アルケニル、−F、−Cl、−Br、−I、−NO2、−N(R3)2、−N3、−COOH、−COO−(C1〜C6アルキル)、−OH、−SH、−O−C1〜C6アルキル、または-S−C1〜C6アルキルであり;
Aは、(CH2)q、低級C3〜C6分枝状アルキル、C3〜C6シクロアルキル、1つから2つの二重結合を有するC2〜C6アルケニル、1つから2つの三重結合を有するC2〜C6アルキニルであり;qが0から5であり;
Bは、−H、−COOH、−COOR8、−CONR9R10、−CH2OH,−CH2OR11、−CH2OCOR11、−CHO、−CH(OR12)2、−CHOR13O、−COR7、−CR7(OR12)2、−CR7OR13O、または−Si(C1〜C 6アルキル)3であり;
R7は、1個から5個の炭素を含有するアルキル基、シクロアルキル基またはアルケニル基であり;
R8は、C1〜C10アルキル、C1〜C10トリメチルシリルアルキル、C3〜C10シクロアルキル、フェニルまたは低級アルキルフェニルであり;
R9およびR10は、独立して水素、C1〜C10アルキル、C3〜C10シクロアルキル、フェニルまたは低級アルキルフェニルであり;
R11は、低級アルキル、フェニルまたは低級アルキルフェニルであり;
R12は、低級アルキルであり;
R13は、2個から5個の炭素の二価アルキル基である。
【0047】
本発明の別の特定の実施形態は、一般式Xにより表され、使用することができる一クラスの化合物であり、X が、−(C(R3)2)−、−S−、−SO−、−SO2−、−O−または−NR3−であり;Zが、−CO−NR3−,−CS−NR3−,−NR3−CO−、または−NR3−CS−であり;全て他の可変部分は、上記に定義されたとおりである。
【0048】
本発明の別の特定の実施形態は、一般式Xにより表され、使用することができる一クラスの化合物であり、X が、−C(=O)−、−C(=S)−、−C(=NR1)−、または−(C=C(R1)2)−であり;Zが、−CO−NR3−,−CS−NR3−,−NR3−CO−、または−NR3−CS−であり;全て他の可変部分は、上記に定義されたとおりである。
【0049】
本発明の別の特定の実施形態は、一般式Xにより表され、使用することができる一クラスの化合物であり、Y3により表される前記フェニル基、ナフチル基、またはヘテロアリール基が、非置換であるか、または1つから3つのR4基により置換されており、全て他の可変部分は、上記に定義されたとおりである。
【0050】
本発明は、RARα、RARβおよびRARγサブタイプの受容体に結合するRARアンタゴニストまたはインバースアゴニストである任意の化合物を使用することをを考慮しており、該化合物としては、米国特許番号:5,728,846、5,739,338、5,763,635、5,773,594、5,877,207、5,952,345、5,958,954、5,998,655、6,008,204、6,037,488、6,043,381、6,087,505、6,090,810、6,117,987、6,211,385、6,218,128、6,225,494、6,228,848、6,235,923、6,313,168、6,521,624、6,521,641、6,538,149、6,555,690、6,653,483、6,720,425、6,818,775、6,942,980、7,105,566、および7,166,726ならびに米国特許出願番号:10/446,580、11/016,534、11/500,277、11/503,635、11/607,406、および11/643,754に記載されているか、またはそれらに請求されているものが挙げられる。上記に引用された特許および特許出願の全ては、参照としてその全体が本明細書に援用されている。
【0051】
本明細書明細書内に入る非排他的リストの化合物およびこのクラスの化合物を製造する方法は、米国特許第5,728,846号に開示されており、それらの内容は、参照として本明細書に援用されている。さらに、これらの化合物は、Songらの米国特許出願第08/840,040号に開示されており、その出願は、本出願と共通の所有権を共有しており、参照としてその全体が本明細書に援用されている。
【0052】
本発明の方法に使用する好ましい化合物または薬学的に許容できるその塩は、以下の構造:
【0053】
【化10】
により表される。この化合物は、VTP 194310 と称される(以前にはAGN 194310と称されていた)。
【0054】
本発明の方法に使用する好ましい化合物または薬学的に許容できるその塩は、以下の構造:
【0055】
【化11】
により表される。この化合物は、VTP 196996と称される。
【0056】
さらなるRARアンタゴニストまたはインバースアゴニストは、参照としてその全体が本明細書に援用されるSongらの米国特許出願第08/845,019号に記載されており;本出願と共通の所有権を共有している。また、本発明の方法に有用な化合物は、参照としてその全体が本明細書に援用されるYoshimura らの国際特許出願公開WO 第94/1477号に開示されている。後者の出願は、PARアンタゴニストを開示している。
【0057】
さらに、本発明の方法に有用なさらなる化合物または薬学的に許容できるその塩の構造は、以下のとおりであり:
【0058】
【化12】
式中、nは、1 から10の整数であり;
本発明の方法に有用なさらなる化合物または薬学的に許容できるその塩の構造は、以下のとおりであり:
【0059】
【化13】
式中、nは、1 から10の整数であり;
本発明の方法に有用なさらなる化合物または薬学的に許容できるその塩の構造は、以下のとおりであり:
【0060】
【化14】
本発明の方法に有用なさらなる化合物または薬学的に許容できるその塩の構造は、以下のとおりであり:
【0061】
【化15】
本発明の方法に有用なさらなる化合物または薬学的に許容できるその塩の構造は、以下のとおりである。
【0062】
【化16】
本明細書に用いられる用語「アゴニスト」とは、受容体に結合してそれを活性化する化合物を意味すると解され、薬理学的応答(例えば、収縮、弛緩、分泌、酵素活性化など)を生じる。
【0063】
本明細書に用いられる用語「インバースアゴニスト」は、アゴニストの効果とは反対の効果を生じるが、同じ受容体において作用する化合物を意味するものとして理解することとする。用語「インバースアゴニスト」は、用語「ネガティブアンタゴニスト」と同義である。
【0064】
本明細書に用いられる用語「アンタゴニスト」は、該受容体を活性化することなくアゴニストと同じ部位に結合することにより、アゴニストの効果を減弱させる化合物を意味するものとして理解することとする。
【0065】
本明細書に用いられる用語「化学放射線療法」は、化学療法、放射線療法または双方を意味するものとして理解することとする。
【0066】
本明細書に用いられる用語「処置すること」または「処置」は、(1)ある状態、疾患、障害、傷害または病態に罹り得るか、または罹りやすくなり得るが、前記状態、疾患、障害、傷害または病態の臨床的または準臨床的症状をまだ経験していないか、または示していない哺乳動物において発現する前記状態、疾患、障害、傷害または病態の臨床的症状出現を、部分的または完全に、予防すること、重症度を減少させること、または遅延すること、(2)前記状態、疾患、障害、傷害または病態を、部分的に、または完全に抑制すること、すなわち、前記状態、疾患、障害、傷害または病態、またはそれらの少なくとも1つの臨床的または準臨床的症状の発現を阻止すること、または減少させること、または(3)部分的または完全に、前記状態を軽減すること、または前記疾患、障害、損傷または状態の重症度を低下させること、すなわち、前記状態、疾患、障害、傷害または病態、もしくはそれらの少なくとも1つの臨床的または準臨床的症状の後退をもたらすこと、を意味するものとして理解することとする。
【0067】
本明細書に用いられる用語「送達」は、RARα、RARβおよびRARγタイプの受容体に結合することのできるRARアンタゴニストまたはRARインバースアゴニストの治療有効量を哺乳動物の特定部位に提供して、該特定部位におけるRARα、RARβおよび RARγタイプの受容体に結合するRARアンタゴニストまたはRARインバースアゴニストの治療的有効濃度を生じさせることを意味するものとして理解することとする。
【0068】
本明細書に用いられる用語「対象」または「患者」または「宿主」または「哺乳動物」は、ヒトを含む哺乳動物を称する。
【0069】
「アルコキシ」および「ヒドロキシアルキル」など、単独で、またはより大きな部分の一部として用いられる用語「アルキル」とは、1個から10個の炭素原子を含有する飽和脂肪族を意味する。代表的な飽和直鎖状アルキル類としては、メチル、エチル、n−プロピル、n−ブチル、n−ペンチルなどが挙げられ、一方、飽和分枝状アルキル類としては、イソプロピル、sec−ブチル、イソブチル、tert−ブチル、イソペンチルなどが挙げられる。用語「低級アルキル」とは、1個から6個の炭素を含有するアルキルを称する。アルケニル基およびアルキニル基は、不飽和脂肪族基であり、隣接炭素原子間に少なくとも1つの二重結合または三重結合を含有する。代表的な直鎖状および分枝状のアルケニル類としては、エチレニル、プロピレニル、1−ブテニル、2−ブテニル、イソブチレニル、1−ペンテニル、2−ペンテニル、3−メチル−1−ブテニル、2−メチル−2−ブテニルなどが挙げられ、一方、代表的な直鎖状および分枝状のアルキニル類としては、アセチレニル、プロピニル、1−ブチニル、2−ブチニル、1−ペンチニル、2−ペンチニル、3−メチル−1−ブチニルなどが挙げられる。
【0070】
用語「シクロアルキル」とは、飽和環式炭化水素部分を意味し、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシルが挙げられる。また、シクロアルキル類には、1つ又は複数の芳香族(フェニルなど)または非芳香族(シクロヘキサンなど)の環状炭素に縮合したシクロアルキル(例えば、シクロペンタンまたはシクロヘキサン)など、8個から10個の炭素原子を有する二環系および三環系などの環状炭素系も含まれる。
【0071】
単独で、またはより大きな部分の一部として用いられる用語「ヘテロアリール」、「ヘテロ芳香族」、「ヘテロ芳香族環」、および「ヘテロアリール基」とは、単環式ヘテロ芳香族環が1つ又は複数の他の炭素環式またはヘテロ芳香族の芳香族環に縮合している単環式ヘテロ芳香族環および多環式芳香族環など、一般に5員から14員を有するヘテロ芳香族環基を称する。ヘテロアリール基は、1つ又は複数、一般に1個、2個、または3個の、窒素、酸素およびイオウなどの環ヘテロ原子を有する。
【0072】
本明細書に記載された化合物の「製薬組成物」およびそれらの薬学的に許容できる塩、溶媒和物および水和物は、薬学的に許容できる担体または希釈剤と組み合わせて製薬製剤中に使用することができる。好適な薬学的に許容できる担体としては、不活性な固体増量剤または希釈剤および滅菌の水性溶液または有機溶液が挙げられる。SARM化合物は、本明細書に記載された範囲の所望の用量を提供する上で十分な量で、このような製薬組成物中に存在する。本発明の化合物の製剤化および投与に関する技法は、Remington:the Science and Practice of Pharmacy、第19版、Mack Publishing 社、ペンシルベニア州、イーストン(1995)に見ることができる。
【0073】
薬学的に許容できる塩もまた、本発明の実施形態として含まれる。
【0074】
「薬学的に許容できる塩」は、何らかの酸性または塩基性の官能基を含有する化合物の塩である。例えば、アミンまたは他の塩基性基の薬学的に許容できる塩は、該化合物を、塩化水素、臭化水素、酢酸、過塩素酸などの好適な有機酸または無機酸と反応させることによって得ることができる。このような塩の他の例としては、塩酸塩、臭化水素酸塩、硫酸塩、メタンスルホン酸塩、硝酸塩、マレイン酸塩、酢酸塩、クエン酸塩、フマル酸塩、酒石酸塩(例えば、(+)−酒石酸塩、(−)−酒石酸塩またはラセミ混合物などのそれらの混合物)、コハク酸塩、安息香酸塩およびグルタミン酸などのアミノ酸との塩が挙げられる。
【0075】
カルボン酸または他の酸性官能基を含有する化合物の薬学的に許容できる塩は、好適な塩基と反応させることによって調製することができる。このような薬学的に許容できる塩は、薬学的に許容できるカチオンを与える塩基によって作製することができ、アルカリ金属塩(特に、ナトリウムおよびカリウム)、アルカリ土類金属塩(特に、カルシウムおよびマグネシウム)、アルミニウム塩およびアンモニウム塩、ならびに、トリメチルアミン、トリエチルアミン、モルホリン、ピリジン、ピペリジン、ピコリン、ジシクロヘキシルアミン、N,N−ジベンジルエチレンジアミン、2−ヒドロキシエチルアミン、ビス−(2−ヒドロキシエチル)アミン、トリ−(2−ヒドロキシエチル)アミン、プロカイン、ジベンジルピペリジン、N−ベンジル−β−フェネチルアミン、デヒドロアビエチルアミン、N,N’−ビスデヒドロアビエチルアミン、グルカミン、N−メチルグルカミン、コリジン、キニン、キノリン、ならびにリシンおよびアルギニンなどの塩基性アミノ酸などの生理学的に許容できる有機塩基から作製された塩が挙げられる。
【0076】
本発明の方法に使用するための、RARα、RARβ および RARγサブタイプの受容体に結合するRARアンタゴニストおよびRARインバースアゴニスト化合物を、製薬組成物中に組み込むことができる。全ての投与様式、例えば、経口、経直腸、非経口、局所、または静脈内注射、筋肉内注射、イントラステマル(intrastemal)注射または皮下注射、もしくは、吸入などの好適な形態が考慮されている。該製剤は、適切な場合は、個別の用量単位で簡便に提供することができ、製薬業界においてよく知られたいずれかの方法によって調製することができる。該化合物は通常、公知の確立された慣例に従って、1つ又は複数の薬学的に許容できる成分と共に製剤化される。したがって、該製薬組成物は、液剤、散剤、エリキシル剤、注射用液剤、懸濁剤、座剤などとして製剤化することができる。
【0077】
経口使用のための製剤は、該化合物が炭酸カルシウム、リン酸カルシウムまたはカオリンなどの不活性の固体希釈剤と共に混合されている錠剤または硬カプセル剤として、または該活性成分が、水またはプロピレングリコール、PEG類およびエタノール、または油脂性媒体、例えば、落花生油、液体パラフィンまたはオリーブ油などの混和性溶媒と共に混合されている軟ゼラチンカプセル剤として提供することができる。
【0078】
口腔における局所投与用に、該製薬組成物は、従来の様式で製剤化された頬側錠または舌下錠、滴剤または舐剤の形態をとることができる。
【0079】
表皮への局所投与のため、該化合物は、クリーム剤、ゲル剤、軟膏剤またはローション剤として、もしくは経皮パッチとして製剤化することができる。このような組成物は、例えば、好適な増粘剤、ゲル化剤、乳化剤、安定化剤、分散剤、懸濁化剤、および/または着色剤を添加して、水性または油性の基剤と共に製剤化することができる。
【0080】
該化合物はまた、デポー製剤として製剤化することもできる。このような長時間作用性の製剤は、移植(例えば、皮下または筋内に)により、または筋内注射により投与することができる。したがって、例えば、該化合物は、好適なポリマー材料または疎水性材料(例えば、許容できる油中に乳剤として)、またはイオン交換樹脂と共に、またはわずかに溶解性の誘導体として、例えば、わずかに溶解性の塩として製剤化することができる。
【0081】
該化合物は、注射による、簡便に、静脈内、筋内または皮下注射による、例えば、ボーラス投与または連続的静脈内注入による非経口投与のために製剤化することができる。注射用製剤は、単位用量形態、例えば、保存剤を添加したアンプルまたは多用量容器で提供することができる。該製薬組成物は、油性または水性媒体中、懸濁剤、液剤または乳剤などの形態をとることができ、懸濁化剤、安定化剤または分散化剤などの製剤化剤を含有することができる。あるいは、該化合物は、使用前に、好適な媒体、例えば滅菌したパイロジェンの無い水と共に構成するための粉末形態であってもよい。
【0082】
また、該化合物は、座剤または本発明の保持浣腸剤、例えばカカオ脂または他のグリセリドなどの慣例的な座剤用基剤を含有するものなど、直腸用組成物においても製剤化できる。
【0083】
鼻腔内投与のために、該化合物は、例えば、液体スプレーとして、散剤として、または滴剤の形態で用いることができる。
【0084】
吸入による投与のために、該化合物は、好適な噴射剤、例えば、ジクロロジフルオロメタン、トリクロロフルオロメタン、ジクロロテトラフルオロエタン、テトラフルオロエタン、ヘプタフルオロプロパン、二酸化炭素または他の好適な気体を使用して、加圧パックまたはネブライザーからのエアロゾルスプレー法の形態で、簡便に送達できる。加圧エアロゾルの場合、用量単位は、計量された量を送達するためのバルブを提供することによって判定できる。吸入器またはインシュレーターにおいて使用するための、例えばゼラチンのカプセルおよびカートリッジは、レチノイド化合物と、乳糖または澱粉などの好適な粉末基剤との粉末混合物を含有して製剤化することができる。
【0085】
水性懸濁剤は、懸濁化剤、例えば、カルボキシメチルセルロースナトリウム、メチルセルロース、ヒドロキシプロピルメチルセルロース、アルギン酸ナトリウム、ポリビニルピロリドン、トラガンカントゴムおよびアラビアゴム;天然ホスファチド、例えばリシチン、またはアルキレン酸化物と脂肪酸との縮合産物、例えば、ポリオキシエチレンステアレート、またはエチレンオキシドと長鎖脂肪族アルコールとの縮合産物、例えばヘプタデカエチレン−オキシセタノール、またはエチレンオキシドと脂肪酸由来の部分的エステルとヘキシトールとの縮合産物、例えばポリオキシエチレンソルビトールモノレエート、またはエチレンオキシドと脂肪酸由来の部分的エステルと無水ヘキシトールとの縮合産物、例えばポリオキシエチレンソルビタンモノレエートなどの分散化剤または湿潤化剤などの薬学的に許容できる賦形剤を含むことができる。水性懸濁剤はまた、一種または複数種の保存剤、例えばエチルまたは−n−プロピル−p−ヒドロキシベンゾエート、一種または複数種の着色剤、一種または複数種の芳香剤およびスクロース、サッカリンまたはシクラミン酸ナトリウムまたはシクラミン酸カルシウムなどの一種または複数種の甘味剤を含有することもできる。
【0086】
RARα、RARβおよび RARγサブタイプの受容体に結合するRARアンタゴニストおよびRARインバースアゴニスト化合物に加えて、少なくとも1種の他の薬理学的活性物質、例えば、トラマドール、アセトアミノフェン、アスピリン、ジクロフェナク、ジフルシナール、エトドラク、フェンブフェン、フェノプロフェン、フルフェニサール、フルルビプロフェン、イブプロフェン、インドメタシン、ケトプロフェン、ケトロラク、メクロフェナミン酸、メフェナム酸、ナブメトン、ナプロキセン、オキサプロジン、フェニルブタゾン、ピロキシカム、スリンダク、トルメチン、ゾメピラクなどおよびそれらの組み合わせなどの非麻薬性鎮痛薬、またはコデイン、オキシコドン、ジヒドロコデイン、ヒドロコドン、レボルファノール、モルフィンなどおよびそれらの組み合わせなどの麻薬性鎮痛薬、または例えば、G−CSF、GM−CSF、EGF、インターロイキン11、エリスロポイエチン、トロンボポイエチン、巨核球の発達および成長因子、ピキシキン類、幹細胞因子、FLT−リガンドならびにインターロイキン1、3、6および7などおよびそれらの組み合わせなどの他の薬剤を、RARα、RARβおよび RARγサブタイプの受容体に結合するRARアンタゴニストおよびRARインバースアゴニスト化合物と共に投与することができる。
【0087】
該化合物は、本発明に従って、治療有効量で投与される。治療的濃度は、哺乳動物、好ましくはヒトにおける、例えば化学放射線療法の副作用を処置する上で有効な濃度である。これらの量は、当業者によって決定できる。
【0088】
以下は、本発明の非限定例である。これらの例は、請求項によって規定される本発明の範囲を限定するものとして解釈すべきではない。
【0089】
以下の実施例の各々で、Allergan社(Irvine,CA)で合成され、特異的なパン−RARアンタゴニストである4−2[6−(2,2−ジメチル−(1H)−4−(4−エチルフェニル)−1−ベンゾチオピラン))エチニル]安息香酸(VTP 194310、以前AGN 194310と称された)を用いる。VTP 194310の構造は、本明細書上記に示されている。RARα、β および γに対する結合に関するVTP 194310のKiは、それぞれ、3、2および5nMである。VTP 194310は、トランス活性化アッセイにおいて、活性を示さないが、その代わり、ATRAおよび他のRARアゴニストによって誘導された遺伝子転写活性をブロックする。VTP 194310およびATRAは、−20℃で50%エタノール/50%ジメチルスルホキシド(DMSO)中、10mMの原液として保存された。
【実施例】
【0090】
実施例1
白血球減少症のシクロホスファミド誘導マウスにおける好中球およびリンパ球の回復
マウス
Charles River Laboratories(マサチューセッツ州、ウィルミントン)から購入したマウスを、12時間の明/暗サイクルで、マイクロアイソレーターケージ内に、個々に収容した。それらは、病原体の無い条件下で収容し、通常の食餌および水を自由に取らせた。それらを、実験前に、実験動物管理の評価および認定協会(AAALAC)に認定された動物施設(Allergan社、アービン、カリフォルニア州)で、1週間順化させた。試験設計は、施設内動物の管理および使用委員会によって承認された。体重は各試験を通してモニターした。マウスの体重は、処置開始日に22〜27gの範囲であった。該マウスは健常で、以前、他の実験操作に用いられなかった。
【0091】
インビボ白血球減少症モデル
0.2mlの生理食塩水中、200mg/kgのシクロホスファミド(CPM、Sigma−Aldrich、ミズーリ州、セントルイス)の腹腔内(i.p.)注射により、オスBDF1マウス(C57Bl×DBA2,7.5-12 週)に白血球減少症を誘導した。白血球の回復に及ぼすVTP 194310の効果を、結果の節に示したとおり、1mg/kgのVTP 194310によるマウスの経口胃管法によって評価した。VTP 194310(DMSO中に溶解)は、媒体としての落花生油により希釈した(5ml/kgの用量)。対照マウスには、DMSOおよび媒体のみを与えた。陽性対照として、2日目に、CPM処置マウスにおいて、10μg/kgのペギル化組換えメチオニルヒト顆粒球コロニー刺激因子(PEG−r−metHuG−CSF)(Neulasta、Pegfilgrastim;Amgen、サウザンドオークス、カリフォルニア州)の単回皮下注射により、顆粒球生成を刺激した。3つの別個の実験を実施した。
【0092】
麻酔下、該マウスの後眼窩洞から末梢血(60μl)を、ヘパリン化毛細管により採取し、EDTAでコーティングしたマイクロタイター管(Becton Dickinson、フランクリンレイクス、ニュージャージー州)に移した。5%のウシ血清アルブミン(フラクションV;Sigma、セントルイス、ミズーリ州)を含有するPBSにより該血液を1:4に希釈し、Advia 120 Hematology System(Bayer HealthCare Diagnostics Division、ニューヨーク州、タリータウン)を用いることにより、1群当たり少なくとも3匹のマウスに関して、白血球、好中球、およびリンパ球の数を得た。
【0093】
PE結合およびFITC結合モノクローナル抗体(Pharmingen、サンディエゴ、カリフォルニア州)を用いて、二重免疫染色により、脾臓および骨髄(大腿骨)吸引液から調製した単一の細胞懸濁液において、未成熟(Gr−1low/CD11b+ve)および成熟(Gr−1+ve/CD11b+ve)好中球を同定した。CellQuest Proスフトウェアプログラム(Becton Dickinson、サンホセ、カリフォルニア州)にインターフェースしたFACS Caliburで細胞を解析した。
【0094】
結果
VTP 194310の使用によりデータが示すように、白血球減少症のシクロホスファミド誘導マウスモデルにおいて白血球の回復を改善した。200 mg/kgでCPMの単回投与によるマウスの処置では、4日目に重大な白血球減少症および好中球減少症を生じた。CPM処置マウスの血中白血球数および好中球数は、正常なマウス(正常な対照群のデータは示していない)に関して、それぞれ8.98±0.33×103/μlおよび1.34±0.09×103/μlの値と比較して、0.69±0.09(SEM)x 103/μl および0.09±0.02×103/μl であった。図1に示されるように、白血球数、好中球数およびリンパ球数は、CPM処置マウスにおいて着実に上昇し、8日目にプラトー値に達した。
【0095】
CPMで処置した(0日目に)マウスに対して、処置4日前から1日前および処置0日目から3日目後にVTP 194310の投与による2つの方法で白血球の回復を改善した。白血球減少症のマウスの対照群と比較して、白血球数および好中球数は、VT P194310処置マウスにおいてより迅速に上昇した。回復の有意差は、CPM投与5日後という早い期間に観察された。8日目に、VT P194310処置の好中球減少症マウスの血中白血球数および好中球数は、CPM単独で処置されたマウスにおける数よりも約3倍高かった(図1を参照)。4日目から7日目にVTP 194310の白血球減少症マウスへの投与は、産生した白血球数および好中球数を改善したが、VTP 194310 を早期に与えた場合よりもかなりの程度で低かった。白血球減少症マウスのPeg−r−metHuG−CSF(2日目に)による処置により、血中好中球数を迅速に上昇させた。これらの細胞レベルは7日目にピークに達し、8日目までに白血球減少症マウスの対照群に見られた数に下降した(図1を参照)。
【0096】
処置4日前から1日前および処置0日目から3日目後に、VTP 194310が投与された白血球減少症マウスでは、対照白血球減少症マウスおよびPeg−r−metHuG−CSFが与えられたこれらのマウスと比較して、血中リンパ球数の増加を示した。8日目において、the lymphocyte counts for the VTP194310処置マウスに関するリンパ球数は、対照の回復マウスに関する2.38±0.26×103/μl(p値<0.01)と比較して、4.53±0.39×103/μl(VTP 194310,4日前から1日前)および 4.49±0.52×103/μl(VTP 194310、0日目から3日目)であった。
【0097】
実施例2
ヒトCD34+ve始原細胞の培養における好中球の回復
HL60細胞増殖および分化の評価
前骨髄性細胞系HL60の培養は、10%の胎仔ウシ血清(FBS、Invitrogen,ペイスリー、英国)ペニシリン(100 U/ml)、およびストレプトマイシン(100 μg/ml)を含有する4 mlのRPMI1640培地(Invitrogen、ペイスリー、英国)中、2.5×105細胞/mlを接種した。ニトロブルーテトラゾリウムを減少させる細胞の能力、早期成熟マーカーCD11bの獲得発現およびステロイドスルファターゼ(骨髄性成熟のマーカー)の活性増加により、分化を測定した。
【0098】
ヒトCD34+ve造血始原細胞の単離および培養
動員幹細胞を有する成人からの倫理的に許容された血中白血球泳動細胞は、地方の国立血液サービス幹細胞ラボラトリー(National Blood Service Stem Cell Laboratory)、バーミンガムよりインフォームドコンセント後に提供された。CliniMACS磁気分離器上で免疫磁気ビーズと共に抗CD34モノクローナル抗体の使用により、CD34+ve細胞を精製した。
【0099】
10% FBS、抗体(100 U/mlのペニシリンおよび100 μg/mlのストレプトマイシン)および組換えヒト幹細胞因子(SCF)の量、組換えヒトインターロイキン3(IL3)、組換えヒト顆粒球コロニー刺激因子(G−CSF)およびパン−RARアンタゴニストVTP194310を含有する200 μlのRPMI 1640培地中、細胞を5×105細胞/mlで、96ウェルマイクロタイタープレート内で平板培養し、その結果が示された。SCF、IL3およびG−CSFは、R and D Systems、アビングドン、英国から入手した。三通りの培養物により各々の条件が確立された。培養物を、最初にマイクロタイターウェルに、次に2 mlの培養物をCostarウェルに入れて分割し、2.5×105細胞/mlから10×105細胞/mlの間の細胞密度を維持した。細胞を、加湿インキュベーターおよび5% CO2雰囲気中、37℃で増殖させた。
【0100】
100 nMのパン−RARアンタゴニストVTP194310は、100 nMのATRAの能力を完全に遮断し、HL60細胞の好中球分化を誘導した。HL60細胞を100 nMのATRAにより5日間処置することにより、増殖停止、ニトロブルーテトラゾリウムを減少させる能力の獲得、CD11bの発現、およびステロイドスルファターゼ活性の増大をもたらした。100 nMの VTP194310がATRAと共投与された場合、これら事象の全ては、抑止された(図2を参照)。
【0101】
図3は、増殖および精製CD34+ve(>99%)造血始原細胞の自然発生的分化に対して10 nmおよび100 nMのVTP194310の効果を示している。CD34+ve細胞は、生存を保証するSCF(100 ng/ml)およびIL3(20 ng/ml)の量で培養した。液体懸濁液の培養中、骨髄始原細胞から好中球および単球の最適数を産生するために、我々はまた、実験室内でルーチンに使用する条件を使用した。これらは、100 ng/mlの SCF、5 ng/mlのIL3および30 ng/mlのG−CSFである。
【0102】
VTP194310が、RAR類および血清中レチノイド類のいずれの効果も遮断したかどうかを確認するために、我々は、膜貫通糖タンパク質CD38の発現のために細胞を染色した。RARαの直接制御下、CD38は、レチノイドを誘導し、またCD38は、CD34+ve細胞を介して骨髄分化中に増加し、CD38の誘導および分化は、機能的には関連していない。VTP194310が、インビトロでヒト造血細胞によりinhibits expression of CD38の発現を阻害することを、Prusらは示している(Prusら、「Retinoic Acid Receptor Anatgonist Inhibits CD38 Antigen Expression on Human Hematopoeitic Cells In Vitro」、Leukaemia & Lymphoma、45、1025−1035頁(2004))。細胞が、これらの条件プラスVTP194310(10 nMおよび100 nM)で増殖された場合の39±4%(n = 4)と比較して、6日目に、35±2%(n = 4)の細胞が、SCF/IL3(20 ng/mlで)において増殖された培養物中にCD38を発現した。VTP194310の不在下では、CD38の発現は、15日目までに50±2%(n = 3)、および27日目までに72±1%(n = 4)に上昇した。VTP194310(10 nMおよび100 nM)の存在下、CD38の発現は、15日目までに15±3%(n = 4)、および27日目までに6±1%(n = 4)に降下した。
【0103】
VTP194310(10 nMおよび100 nM)およびG−CSFは、培養物および産生した細胞数の寿命の双方を実質的に増加させた。SCF/IL3で処理された培養物は、33日目までに殆どが終了したが(細胞密度0.6×105/ml)、一方、VTP194310(10 nMおよび100 nM)/SCF/IL3またはG−CSF/SCF/IL3で処理された培養物は、依然として大多数の細胞を産生した(図2A、上部パネル)。細胞密度は、55日目にこれらの培養物が低レベル(0.5×105 cells/mlから1.5×105 cells/mlまで)であった。100 nMのVTP194310で補足された培養物は、他の培養物よりも僅かに低い率で細胞を産生し(図3Aを参照)、33日目に、10 nMのVTP194310(4.0×107±0.2、n = 4)またはG−CSF(3.8×107±0.5、n = 4)で補足された培養物よりも総数で僅かに低く産生(2.9×107±0.2、n = 4)した。
【0104】
各培養条件および実験全般にわたって産生された細胞は、主として骨髄であった(図3B、下部パネル)。
【0105】
赤血球コンパートメント(グリコホリン+ve/CD45+ve)は、小さく(6日目に9%から14%まで)、適切な増殖およびエリスロポイエチンなどの生存因子を欠くと徐々に消失した(図3B)。VTP194310は、赤血球細胞の罹患率に対して有意な効果はなかった。リンパ様始原細胞および成熟リンパ球は、培養物中の重要な範囲のいずれにも見られなかった。
【0106】
SCF/IL3中で培養されたCD34+ve細胞は、22日目までに完全に成熟し、それによって細胞の49±4%が好中球(CD65+ve/CD11b+veおよびCD15+veとして)であり、細胞の53±8%が単球(CD14+ve/CD11b+ve)であった。CD33+ve/CD15−ve/CD14−veとして同定された未熟骨髄細胞は、19日目までに培養物から無くなった。細胞が、G−CSF/SCF/IL3中で培養された場合、等しい数の好中球および単球が産生されたが、SCF/IL3が使用された場合よりもはるかに数が多かった(図3A)。22日までに、G−SCF/SCF/IL3の培養物は、成熟好中球および単球を大量に含有した(図3Aにおける総細胞数と好中球数と単球数とを比較する)。
【0107】
CD34+ve細胞の培養においてSCF/IL3の使用に対して10 nMおよび100 nMのVTP194310の添加効果は、産生された好中球数を増加させることであった(図3A下部の2つのパネルを比較する)。これに関して、10 nMのVTP194310は、100 nMのVTP194310よりも有効であった。単球の累積数は、VTP194310の存在により有意に影響を及ぼさなかった(図3A)。20日目から30日目の間のVTP194310処理培養物の視覚検査により、芽細胞は、該培養物の至る所にあることが明らかとなり、マーカー分析により、細胞の実在する割合は、好中球または単球のいずれにも成熟していなかったことを示した。19日目と29日目との間、好中球は、33%±2(10 nMのVTP194310)および30%±5(100nMの VTP194310)のレベルで、単球は、32%±4(10 nMのVTP194310)および21%±5(100 nMのVTP194310)のレベルで存在した。二重および三重のFACS分析による全ての検査により、VTP194310処理培養物に持続していた2つの集団の細胞を確認することができた。これらは、SCFに対する受容体を発現し、CD34−ve、およびCD33+ve/CD15−ve/CD14−veとして同定された未熟骨髄性細胞である細胞であった(図3B)。これらの細胞集団は10 nMのVTP194310処理培養物と比較して、100 nMのVTP194310処理培養物中に広範囲で存在した。VTP194310は、培養中のCD34+ve細胞の数および持続性に影響を及ぼさなかった(図3B)。これらのマーカー分析は、VTP194310処理培養物中の好中球の産生増加が、骨髄前駆体集団の拡大および好中球分化の減速に関連する概念と釣り合っている。
【0108】
VTP194310処理からの成熟顆粒球および対照マウスは、エキソビボで同様の率でアポトーシスを受ける。同様に我々は、ヒト末梢血白血球(デキストラン沈降法により調製)が、10% FBSおよび10 nMおよび100 nMのVTP194310を含有する培地で補足されたRPMI1640培地中で培養された場合、好中球は同様の率でアポトーシスを受けることを観察した。これら条件の各々における1日目において、核物質のアクリジンオレンジ染色および蛍光顕微鏡により観察されるように、65%から70%の好中球はアポトーシスであった。好中球の全ては、2日目までに死滅した。
【0109】
実施例3
白血球減少症のシクロホスファミド誘導マウスモデルにおいて末梢および全身の好中球およびリンパ球の回復
白血球減少症マウスにおけるVTP 194310駆動好中球回復が、脾臓および骨髄中の好中球(Gr1+ve/CD 11b+ve細胞)のレベルを見ることにより全身的であるかどうかを調べた。これらのマウスは、CPM処置に関して1日前から2日目にVTP 194310 により胃管栄養を行った。
【0110】
結果
データが示すように、対照の回復マウスと比較して、好中球数およびリンパ球数は、VTP 194310処理マウスにおいて6日目から8日目に有意に上昇した(図4Aを参照)。対照と比較して、脾臓重量は、VTP 194310処理マウスにおいて7日目と8日目に増加した。Gr−1+ve/CD1lb+ve細胞の絶対数は、VTP 194310処理マウスの脾臓において6日目と7日目に有意に増加した(see Figure 図4Bを参照)。さらに、Gr−1low/CD 1lb+veとして同定された未熟顆粒球は、VTP 194310処理マウスの脾臓において6日目と7日目に上昇した。7日目におけるこられの細胞の絶対数は、対照マウスの0.46±0.13×107と比較して、VTP 194310処理マウスにおいて0.93±0.02 x107であった(p = 0.02)。また、VTP 194310処理マウスの骨髄においてGr−1+ve/CD1lb+ve細胞の広がりが増加した(図4Cを参照)。それゆえ、VTP 194310により引き起こされた好中球の回復は全身的である。
【0111】
実施例4
白血球減少症マウスにおいて黄色ブドウ球菌に対するVTP194310の防護効果の測定
American Type Culture Collection(ATCC)(ロックヴィレ、メリーランド州)から入手された黄色ブドウ球菌(S.aureus)29213 を、白血球減少症メスBDF1マウス(20〜22 g)において感染症を誘導させるために用いた。黄色ブドウ球菌を、中間対数増殖期(600 nm = 0.3での光学密度)までトリプシンダイズブロス中37℃で培養し、収穫し、PBSで洗浄した。細菌数を、PBSとの連続希釈により数え、血液寒天上で平板培養し、37℃でインキュベーション48時間後にコロニー形成単位(CFU)をカウントした。この懸濁液を10° CFU/mlに調整した。
【0112】
マウスを200 mg/kgでCPMのi.p.注射により白血球減少症にした。黄色ブドウ球菌の致死量(LD)は、CPM処置4日後、10匹のマウス群に1×103から1×108 CFUまでの腹腔内注射により判定した。10日の生存動物を数え、黄色ブドウ球菌のLD95をプロビット解析により算出した。VTP 194310(1 mg/kgで)による処置は、CPM投与1日前に開始し、さらに3日間続けた。CPM投与4日後、この動物を、200μlのPBS中、4.1および2.5×106 CFUの黄色ブドウ球菌により静脈内に感染させた。生存動物は、誘発後14日間、毎日記録した。1条件当り12匹のマウスであり、VTP 194310の防護効果の有意性を、Logrank Test(GraphPad Prismバージョン3.0、GraphPad Software社、サンディエゴ、カリフォルニア州)により測定した。全ての実験は、関連する法律および施設のガイドラインに準拠して実施し、the Animal Care and Use Committeeにより承認された。
【0113】
結果
本実施例において、VTP 194310処置白血球減少症マウスにおける好中球回復の改善が、黄色ブドウ球菌の致死量を投与することにより、マウスを感染症から防止できるかどうかを調査した(図5を参照)。VTP 194310は、CPM投与1日前、CPM投与と同時に、CPM投与2日後に投与され、黄色ブドウ球菌は、CPM投与4日後に注射された。CPMおよび4.1×106 CFUの黄色ブドウ球菌を投与されたマウスは、10日目に全て死亡した。低用量の黄色ブドウ球菌を受けたマウスの10%だけが生存し、これらのマウスは、11日目までに死亡した。10日目において、VTP 194310を受けたマウスの83%、および黄色ブドウ球菌の各用量では生存した。この実験は14日目に終了し、VTP194310および2.5×106 CFUを受けたマウスの67%は生存したが、一方、VTP 194310およびより高い用量の細菌を受けたマウスの42%は生存した。したがって、対照群と比較してVTP 194310による処置により、黄色ブドウ球菌に対して極めて有意な防護効果があった(4.1×106 CFUの黄色ブドウ球菌により感染したマウスに関してp値= 0.0031;2.5×106 CFUの黄色ブドウ球菌により感染したマウスに関してp値< 0.0001)。
【0114】
黄色ブドウ球菌を与えたVTP194310処置マウスの生存は、黄色ブドウ球菌に対するVTP194310の活性によるものではなかった。この化合物は、黄色ブドウ球菌EMRSA−16 252および黄色ブドウ球菌MSSA 476に対する活性関する幾つかのアッセイにおいて適切な濃度で分析した。寒天上の細菌増殖は、寒天に組み込まれた2μMのVTP194310の存在により、または50μlの2μMのVTP194310が、寒天ウェル拡散アッセイに添加された場合に阻止されなかった。さらに、2μMのVTP194310の存在下、液体培養中での増殖期間の増殖率に変化を示す株は無かった。
【0115】
実施例5
CPM誘導好中球減少症マウスにおける好中球回復に対する単一療法およびVTP 194310およびペギル化G−CSFの併用療法効果の比較
顆粒球コロニー刺激因子(G−CSF)は、好中球減少症における好中球の回復を駆動する重要な因子であり、化学療法誘導性の重症の好中球減少症の処置のために臨床的に使用されている。したがって、単一療法のVTP194310、単一療法G−CSFおよびVTP194310とG−CSFとの併用使用の効果を比較することは重要である。VTP194310による単一療法(3mg/kg、1日前から1日目)およびペギル化G−CSF(10μg/kg、2日目)は、好中球減少症マウス(150 mg/kgのCPM)の対照に比して好中球の回復を増強した。VTP194310およびペギル化G−CSFの用量および措置による併用処置により、好中球の回復率をさらに増加した。血中好中球の上昇を刺激するVTP194310およびペギル化G−CSFの併用効果は、相加的効果よりも僅かに大きかった。さらに、VTP194310と一緒のG−CSFは、G−CSF単独で使用された場合よりも長く持続する好中球を生じた。これらの知見は、VTP194310が回復期の間G−CSFと共に作用する提案と釣り合っている。
【0116】
製剤
VTP194310のDMSOストック溶液を、DMSOに溶解されたVTP194310を計量することにより作製した(25 mg/1.33 mlのDMSO)。このストック溶液を次に、媒体(39.2 mlのトウモロコシ油+0.8 mlのDMSO)と混合した。
【0117】
3.0 mg/kgのVTP194310溶液を、39.2 mlのトウモロコシ油+0.8 mlのVTP 194310 DMSOストックを混合することにより作製した。
【0118】
1.0 mg/kgのVTP194310溶液は、VTP194310のDMSOストック1:3をDMSOで希釈する(例えば、1 mlのDMSO + 0.5 mlの4310 Stock)ことにより作製され、次いで39.2 mlのトウモロコシ油 + 0.8 mlの希釈VTP194310ストックを組合わせた。
【0119】
CPM溶液の調製:
150 mg/kgのCPM溶液を、187.5 mgの5−FUを計量し、10 gの生理食塩水と混合することにより作製した。
【0120】
1μg/kgおよび10μg/kgのPEG−G−CSF溶液は、6 mg/mlのストックをPBSで希釈することにより作製した。
【0121】
マウス:
オスBDF1マウス(C57Bl×DBA2)、7〜8週齢、DOB:2006年7月1日)をこの実験に用いた。
【0122】
血液サンプル:
35μlの血液(顎下静脈を介して)を、スケジュールされた時点に従ってまたは血液細胞および血小板の完全回復まで採血した。血液サンプルを、2% BSA(フラクションV)および0.2 mMのEDTAを含有するPBS生理食塩水で1:5に希釈後、Hematology Analyzer Cell−DYN 3700(Abbott Diagnostics)上で分析した。
【0123】
結果
各群のマウスを、媒体、3 mg/kgでのVTP194310、1 μg/kgまたは10 μg/kgでのPEG−G−CSF、および150 mg/kgでのCPMにより処置した。総白血球数(WBC)、好中球、リンパ球、赤血球(RBC)および血小板の平均数、ならびに各処置群に対して得られたデータに関する標準誤差(SE)を図6に示す。図6Bおよび図7に示されるように、1日前および1日目に1 mg/kg/日または3 mg/kg/日でのVTP194310による単一療法および2日目に1 μg/kg または10 μg/kg でのPEG−G−CSFによる単一療法は、好中球減少症マウス(0日目に150 mg/kgのCPMにより誘導された)の対照に比して、好中球の回復を増強した。VTP194310またはPEG−G−CSFのいずれかによる単一療法と比較して、これらVTP194310およびPEG−G−CSFの用量および措置による併用処置により、好中球の回復率をさらに増加した。血中好中球の上昇を刺激する上でVTP194310 およびPEG−G−CSFの併用効果は、相加的以上であった。さらに、VTP194310と一緒の PEG−G−CSFは、PEG−G−CSF単独で使用された場合よりも長く持続する好中球を生じた。結論として、VTP194310は、好中球減少症のCPM誘導マウスモデルにおいて好中球の回復を増強する上で10 μg/kgでのPEG−G−CSFと同様に有効であると思われる。VTP194310およびPEG−G−CSFによる併用処置は、さらに回復を増加した。
【0124】
実施例6
5−FU誘導好中球減少症マウスにおける好中球回復に対する単一療法およびVTP 194310およびペギル化G−CSFの併用療法効果の比較
顆粒球コロニー刺激因子(G−CSF)は、好中球減少症における好中球の回復を駆動する重要な因子であり、したがって、患者における化学療法誘導性の重症好中球減少症の処置のために使用されている。単一療法のVTP194310、単一療法G−CSFの効果およびVTP194310とG−CSFとの併用効果を比較することは重要である。ここに我々は、好中球減少症、すなわち150 mg/kgの5−FUの投与により誘導された異なるマウスモデルを用いて実験を実施した。この目的は、5−FUマウスモデルにおけるVTP 194310およびPEG−G−CSFの効果を調査することであった。VTP194310による単一療法(3mg/kg、2日目から4日目)は、我々が以前に観察した好中球の回復を改善した。異なる日(5日目、6日目、または7日目)に投与された10μg/kgでのPEG−G−CSFによる単一療法は、好中球の回復を増強する効果は殆どなかった。しかしながら、VTP194310およびPEG−G−CSFの同じ用量および措置による併用処置は、有意に好中球の回復率を増加した。
【0125】
製剤:
VTP194310のDMSOストック溶液を、VTP194310を計量し、DMSO中に溶解することによって作製した(25 mg/1.33 mlのDMSO)。
【0126】
この媒体溶液を、39.2 mlのトウモロコシ油+0.8 mlのDMSOを混合することにより作製した。
【0127】
3.0 mg/kgのVTP194310溶液を、39.2 mlのトウモロコシ油+0.8 mlのVTP 194310 DMSOストック溶液を混合することにより作製した。
【0128】
150 mg/kgの5−FU溶液を、187.5 mgの5−FUを計量し、10 gの生理食塩水と混合することにより作製した。
【0129】
10μg/kgのPEG−G−CSF溶液は、6 mg/mlのストックをPBSで希釈することにより作製した。
【0130】
マウス:
オスBDF1マウス(C57Bl×DBA2)、9週齢、DOB:2006年6月26日)を本実験に用いた。
【0131】
結果
各群のマウスを表に掲げた措置に従って、0日目に150 mg/kgの5−FUで静脈内で処置し、2日目から4日目に媒体、3 mg/kgでのVTP194310を胃管栄養法により経口的に、および/または5日目、6日目、または7日目に10 μg/kgでのPEG−G−CSFにより皮下的に処置した。総白血球数(WBC)、好中球、リンパ球、赤血球(RBC)および血小板の平均数、ならびに各処置群に対して得られたデータに関する標準誤差(SE)を図8に示す。好中球減少症の5−FU誘導マウスモデルにおいて好中球の回復を増強する上で、2日目から4日目の3日間で3 mg/kg/日でのVTP194310の投与の方が、5日目、6日目、または7日目に投与された10 μg/kgでのPEG−G−CSFよりも有効である。この5−FU誘導好中球減少症モデルにおいて好中球回復を刺激する上でVTP194310とPEG−G−CSFとの併用処置は、VTP194310による単一療法またはPEG−G−CSFによる単一療法よりも有効であると思われる。
【0132】
図8Bおよび図9に示されるように、2日目から4日目に3 mg/kg/日でのVTP194310による単一療法は、以前に得られた結果(RT−06−34)と一致する好中球の回復を改善した。5日目、6日目、または7日目に10μg/kgでのPEG−G−CSFによる単一療法は、5−FU好中球減少症マウスの対照に比して好中球回復を増強する効果が殆ど無いことを示した。VTP194310 およびPEG−G−CSFの同じ用量および措置による併用処置は、VTP194310による単一療法と比較して好中球の回復率をさらに増加した。
【0133】
実施例7
VTP 194310(以前にAGN 194310と呼ばれた)の合成
VTP 194310は、以下の化学構造:
【0134】
【化17】
を有する。
【0135】
この化合物、4−[[4−(4−エチルフェニル)−2,2−ジメチル−(2H)−チオクロメン−6−イル]−エチニル]−安息香酸は、従来の有機合成手段を用いて合成することができる。以下の反応スキームは、現在この化合物を製造するのに好ましい方法である。
【0136】
ステップ1:厚肉スクリューキャップチューブに、3−メチル−2−ブテン酸(13.86 g、138.4 mmol)、4−メトキシチオフェノール(20.0 g、138.4 mmol)、およびピペリジン(3.45 g、41.6 mmol)を充填した。この混合物を、32時間10℃に加熱し、室温に冷却し、EtOAc(700 mL)に溶解した。生じた溶液を、1M水性HCl、H2O、および飽和水性NaClで洗浄してから、Na2SO4で乾燥した。減圧下、乾燥溶液の濃縮によりオイルが得られ、これをフリーザー内で放置すると結晶性固体を得た。この結晶性固体を、ペンタンで洗浄することにより3−(4−メトキシ−フェニルスルファニル)−3−メチル−酪酸を淡黄色結晶として単離した(27.33 g、82%)。1H NMR(300 MHz,CDCl3)δ:7.48(2H,d,J=9.0 Hz),6.89(2H,d,J=8.9 Hz),3.83(3H,s),2.54(2H,s),1.40(6H,s)。
【0137】
ステップ2:3−(4−メトキシ−フェニルスルファニル)−3−メチル−酪酸(20.0 g,83.2 mmol)の250 mLのベンゼン溶液に、室温で10 mLのベンゼン中の塩化オキサリル(15.84 g、124.8 mmol)溶液を30分かけて加えた。4時間後、この溶液を、氷冷5%水性NaOH(注意:この手法中、大量のガス放出される)で洗浄し、次いで氷冷H2O、最後に飽和水性NaClで洗浄した。この溶液を乾燥し(Na2SO4)、減圧濃縮すると透明な黄色オイルを得た。この物質を、さらに精製することなく以下のステップに用いた。1H NMR(300 MHz,CDCl3)δ:7.45(2H,d,J=8.8 Hz),6.90(2H,d,J=8.8 Hz),3.84(3H,s),3.12(2H,s),1.41(6H,s)。
【0138】
ステップ3:ステップ2(21.5 g、83.2 mmol)のアシルクロリド生成物の250 mLのCH2Cl2溶液に、0℃で SnCl4(21.7 g、83.2 mmol)の30 mLのCH2Cl2溶液を滴下しながら加えた。2時間後、反応を150 mLのH2Oの緩徐な添加によりクエンチした。有機層を、1M水性HCl、5%水性NaOH、H2O、および最後に飽和水性NaClで洗浄してから、MgSO4で乾燥した。減圧濃縮および残渣オイルの減圧蒸留(バルブからバルブ、125−135℃、5 mm/Hg)により、14.48 g(78%)の6−メトキシ−2,2−ジメチル−チオクロマン−4−オンを黄色オイルとして得た。1H NMR(300 MHz,CDCl3)δ:7.62(1H,d,J=2.9 Hz),7.14(1H,d,J=8.6 Hz),7.03(1H,dd,J=2.8,8.3 Hz),3.83(3H,s),2.87(2H,s),1.46(6H,s)。
【0139】
ステップ4:6−メトキシ−2,2−ジメチル−チオクロマン−4−オン(6.0 g、27 mmol)の−23℃に冷却された50 mLのCH2Cl2溶液に、BBr3(20.0 g、80.0 mmol;80.0 mLの中CH2Cl2 1M溶液)を20分間かけて加えた。−23℃で5時間攪拌後、この溶液を−78℃に冷却し、50 mLのH2Oの緩徐な添加によりクエンチした。室温に暖めてから水層をCH2Cl2で抽出し、有機層を合わせて、飽和水性NaHCO3、H2O、および飽和水性NaClで洗浄してから、MgSO4で乾燥した。溶媒の減圧留去により緑褐色固体が得られ、これを結晶化(Et2O/ヘキサン類)させると、2.25 g(40%)の6−ヒドロキシ−2,2−ジメチルチオクロマン−4−オンを淡褐色固体として得た。1H NMR(300 MHz,CDCl3)δ:7.63(1H,d,J=2.8 Hz),7.15(1H,d,J=8.5 Hz),7.01(1H,dd,J=2.8,8.5 Hz),2.87(2H,s),1.46(6H,s)。
【0140】
ステップ5:6−ヒドロキシ−2,2−ジメチルチオクロマン−4−オン(165.0 mg、0.79 mmol)の5.0 mLの無水ピリジン溶液に、無水トリフルオロメタンスルホン酸(245.0 mg、0.87 mmol)を0℃で加えた。0℃で4時間後、この溶液を濃縮し、残渣オイルをEt2Oに溶解し、H2Oに次いで飽和水性NaClで洗浄し、MgSO4で乾燥した。溶媒の減圧留去およびカラムクロマトグラフィー(5% EtOAc/ヘキサン類)により、126.0 mg(47%)の2,2−ジメチル−4−オキソ−チオクロマン−6−イル トリフルオロメタンスルホン酸塩を無色固体として得た。1H NMR(300 MHz,CDCl3)δ:7.97(1H,s),7.32(2H,s),2.90(2H,s),1.49(6H,s)。
【0141】
ステップ6:2,2−ジメチル−4−オキソ−チオクロマン−6−イル トリフルオロメタンスルホン酸塩(2.88 g、8.50 mmol)の10 mLのEt3Nおよび20.0 mLのDMF溶液を、アルゴンにより10分間パージした。この溶液に、トリメチルシリルアセチレン(4.15 g、42.0 mmol)およびビス(トリフェニルホスフィン)−パラジウム(II)クロリド(298.0 mg、0.425 mmol)を加えた。この溶液を、95℃で5時間加熱し、室温に冷却し、H2Oで希釈した。EtOAcによる抽出に次いで、有機層を合わせてH2Oおよび飽和水性NaClで洗浄し、MgSO4で乾燥した。乾燥溶液の減圧濃縮およびカラムクロマトグラフィー(3% EtOAc/ヘキサン類)による生成物の単離により、2.23 g(91%)の2,2−ジメチル−6−トリメチルシラニルエチニル−チオクロマン−4−オンを橙色オイルとして得た。1H NMR(300 MHz,CDCl3)δ:8.18(1H,d,J=1.9 Hz),7.34(1H,dd,J=1.9,8.1 Hz),7.15(1H,d,J=8.1 Hz),2.85(2H,s),1.45(6H,s),0.23(9H,s)。
【0142】
ステップ7:2,2−ジメチル−6−トリメチルシラニルエチニル−チオクロマン−4−オンの(110.0 mg、0.38 mmol)およびK2CO3(40.0 mg、0.29 mmol)の10.0 mLのMeOH溶液を、室温で一晩攪拌した。該溶液をH2Oで希釈し、Et2Oで抽出した。有機層を合わせて、H2Oおよび飽和水性NaClで洗浄し、MgSO4で乾燥した。溶媒の減圧留去により、81 mg(99%)の6−エチニル−2,2−ジメチルチオクロマン−4−オンを橙色オイルとして得た。1H NMR(300 MHz,CDCl3)δ:8.20(1H,d,J=1.9 Hz),7.46(1H,dd,J=1.9,8.1 Hz),7.18(1H,d,J=8.1 Hz),3.08(1H,s),2.86(2H,s),1.46(6H,s)。
【0143】
ステップ8:6−エチニル−2,2−ジメチルチオクロマン−4−オン(82.0 mg、0.38 mmol)およびエチル4−ヨードベンゾエート(104.9 mg、0.38 mmol)の5.0 mLのEt3N溶液を、アルゴンで10分間パージした。この溶液に、ビス(トリフェニルホスフィン)−パラジウム(II)クロリド(88.0 mg、0.12 mmol)およびヨウ化銅(I)(22.9 mg、0.12 mmol)を加えた。さらに5分間アルゴンでパージ後、該溶液を、室温で一晩攪拌した。反応混合物を、Et2O洗浄を用いたセライトパッドを通してろ過した。ろ液の減圧濃縮、次いで残渣の固体のカラムクロマトグラフィーにより、100 mg(72%)のエチル4−[(2,2−ジメチル−4−オキソ−チオクロマン−6−イル)エチニル]−ベンゾエートを黄色固体として得た。1H NMR(300 MHz,CDCl3)δ:8.25(1H,d,J=1.8 Hz),8.00(2H,d,J=8.4 Hz),7.55(2H,d,J=8.4 Hz),7.53(1H,dd,J=1.8,8.2 Hz),7.21(1H,d,J=8.2 Hz),4.37(2H,q,J=7.1 Hz),2.88(2H,s),1.47(6H,s),1.39(3H,t,J=7.1 Hz)。
【0144】
ステップ9:ナトリムビス(トリメチルシリル)アミド(1.12 g、6.13 mmol)の16.2 mLのTHF溶液を、−78℃に冷却し、この溶液にエチル4−[(2,2−ジメチル−4−オキソ−チオクロマン−6−イル)エチニル]−ベンゾエート(1.86 g、5.10 mmol)の15.0 mLのTHF溶液を徐々に加えた。30分後、2−[N,N−ビス(トリフルオロメタンスルホニル)アミノ]−5−ピリジン(2.40 g、6.13 mmol)の10 mLのTHF溶液を加えた。5 分後、この溶液を室温に温め、一晩攪拌した。反応を、飽和水性NH4Clの添加によりクエンチし、EtOAcで抽出した。有機層を合わせて、5%水性NaOHおよびH2Oで洗浄してから、乾燥(MgSO4)および減圧濃縮した。,1.53 g(61%)のエチル4−((2,2−ジメチル−4−トリフルオロメタンスルホニルオキシ−(2H)−チオクロメン−6−イル)エチニル)−ベンゾエートを、カラムクロマトグラフィー(2% EtOAc/ヘキサン類)により黄色固体として単離した。1H NMR(300 MHz,CDCl3)δ:8.03(2H,d,J=8.4 Hz),7.61(1H,d,J=1.8 Hz),7.59(2H,d,J=8.4 Hz),7.41(1H,dd,J=1.8,8.1 Hz),7.29(1H,d,J=8.1 Hz),5.91(1H,s),4.39(2H,q,J=7.1 Hz),1.53(6H,s),1.41(3H,t,J=7.1 Hz)。
【0145】
ステップ10:4−エチルブロモベンゼン(670.9 mg、3.63 mmol)の4.0 mLのTHF溶液を−78℃に冷却し;t−ブチルリチウム(464.5 mg、7.25 mmol,4.26 mLのペンタン中1.7M溶液)を加えると黄色溶液を得た。30分後、ZnCl2(658.7 mg、4.83 mmol)の8.0 mLのTHF溶液はカニューレを介して徐々に加えた。生じた溶液を室温に温め、カニューレを通してエチル4−(2,2−ジメチル−4−トリフルオロメタンスルホニルオキシ−(2H)−チオ−クロメン−6−イル)エチニル)−ベンゾエート(1.20 g、2.42 mmol)および テトラキス(トリフェニルホスフィン)パラジウム(0)(111.7 mg、0.097 mmol)の8.0 mLのTHF溶液に移した。この溶液を50℃に1時間加熱し、室温に冷却し、反応を飽和水性NH4Clの添加によりクエンチした。この溶液をEtOAcで抽出し、有機層を合わせてH2Oおよび飽和水性NaClで洗浄してから、乾燥し(MgSO4)、減圧濃縮した。カラムクロマトグラフィー(5% EtOAc/ヘキサン類)により、エチル4−[[4−(4−エチルフェニル)−2,2−ジメチル−(2H)−チオクロメン−6−イル]−エチニル]−ベンゾエートを無色オイルとして単離した。1H NMR(300 MHz,CDCl3)δ:7.99(2H,d,J=8.2 Hz),7.52(2H,d,J=8.4 Hz),7.40(5H,m),7.35(2H,m),5.85(1H,s),4.38(2H,q,J=7.1 Hz),2.72(2H,q,J=7.6 Hz),1.48(6H,s),1.40(3H,t,J=7.1 Hz),1.30(3H,t,J=7.6 Hz)。
【0146】
ステップ11:エチル4−[[4−(4−エチルフェニル)−2,2−ジメチル−(2H)−チオクロメン−6−イル]−エチニル]−ベンゾエート(940.0 mg、2.08 mmol)の10.0 mLのTHFおよび5.0 mLのEtOH溶液に、NaOH(416.0 mg、10.4 mmol、5.2 mLの2M水溶液)を加えた。生じた溶液を室温で一晩攪拌した。反応混合物を10%水性HClで酸性にし、EtOAcで抽出した。有機層を合わせて、H2O、飽和水性NaClで洗浄し、乾燥(Na2SO4)してから、溶媒を減圧留去した。残渣の固体を、CH3CNから再結晶して、786.0 mg(89%)の4−[[4−(4−エチルフェニル)−2,2−ジメチル−(2H)−チオクロメン−6−イル]−エチニル]−安息香酸を無色固体として得た。1H NMR(300 MHz,d6−acetone)δ:8.01(2H,d,J=8.3 Hz),7.60(2H,d,J=8.5 Hz),7.42(2H,m),7.29(2H,m),7.22(3H,m),5.94(1H,s),2.69(2H,q,J=7.7 Hz),1.47(6H,s),1.25(3H,t,J=7.7 Hz)。この化合物、すなわち、最後の所望の生成物は、VTP 194310と称された。
【0147】
上記の説明は、多くの明細を含有するが、これらの明細は、本発明を限定するものとして解釈してはならないが、単にそれらの好ましい実施形態の例証として解釈スべきである。当業者は、本明細書に添付された請求項により定義されている範囲内の他の多くの実施形態と本発明の趣旨とを想像するであろう。
【0148】
本出願に引用されるいずれの特許、特許出願、および他の刊行物は、参照としてその全体が本明細書に援用されている。
【特許請求の範囲】
【請求項1】
化学療法および/または放射線療法を受ける哺乳動物において化学療法および/または放射線療法の副作用を処置するための方法であって、該方法は、RARα、RARβおよびRARγサブタイプの受容体に結合する少なくとも1つのレチノイン酸受容体(RAR)アンタゴニストまたはRARインバースアゴニストの治療有効量を該哺乳動物に投与する工程を含む、方法。
【請求項2】
前記RARアンタゴニストまたはRARインバースアゴニストが、前記哺乳動物における好中球産生を増加させるのに有効である、請求項1に記載の方法。
【請求項3】
血小板産生を必要とする哺乳動物において血小板産生を増加させるための方法であって、該方法は、RARα、RARβおよびRARγサブタイプの受容体に結合する少なくとも1つのRARアンタゴニストまたはRARインバースアゴニストの有効量を該哺乳動物に投与する工程を含む、方法。
【請求項4】
前記RARアンタゴニストまたはRARインバースアゴニストが、化学構造:
【化18】
または薬学的に許容できるその塩を有し;
式中、Xは、S、O、RがHまたは1個から6個の炭素のアルキルであるNRか、または
Xは、R1が、独立してHまたは1個から6個の炭素のアルキルである[C(R1)2]nであり、nは、0と2の間でかつ0と2を含む整数であり;
R2は、独立して水素、1個から6個の炭素の低級アルキル、F、Cl、Br、I、CF3、1個から6個の炭素のフルオロ置換アルキル、OH、SH、1個から6個の炭素のアルコキシ、または1個から6個の炭素のアルキルチオであり;
R3は、独立して水素、1個から6個の炭素の低級アルキルまたはFであり;
mは、0〜3の値を有する整数であり;
nは、0〜4の値を有する整数であり;
oは、0〜3の値を有する整数であり;
Zは、−CONR1−、−CSNR1−、−NR1CO−、−NR1CS−、−C≡C−、−C=C−、−N=N−、−N=CR1−、−CR1=N−、
−COO−、−OCO−;−OSO−;−OCS−、またはn’が0から5の整数である−(CR1=CR1)n’−であり、;
Yは、フェニル基またはナフチル基、またはピリジル、チエニル、フリル、ピリダジニル、ピリミジニル、ピラジニル、チアゾリル、オキサゾリル、イミダゾリルおよびピラゾリルからなる群より選択されるヘテロアリールであり、該フェニル基およびヘテロアリール基は、1つまたは2つのR2基で任意に置換されているか、またはZが(CR1=CR1)n’-であり、n’が3、4または5である場合、Yは、前記(CR2=CR2)n’基とBとの間の直接的原子価結合を表し;
Aは、qが0〜5である(CH2)q、3個から6個の炭素を有する低級分枝鎖アルキル、3個から6個の炭素を有するシクロアルキル、2個から6個の炭素および1つまたは2つの二重結合を有するアルケニル、2個から6個の炭素および1つまたは2つの三重結合を有するアルキニルであり;
Bは、水素、COOHまたは薬学的に許容できるその塩、COOR8、CONR9R10、CH2OH、CH2OR11、CH2OCOR11、CHO、CH(OR12)2、CHOR13O、−COR7、CR7(OR12)2、CR7OR13O、またはトリ−低級アルキルシリルであり;
R7は、1個から5個の炭素を含有するアルキル基、シクロアルキル基またはアルケニル基であり;
R8は、1個から10個の炭素のアルキル基、またはアルキル基が1個から10個の炭素を有するトリメチルアルキル、3個から10個の炭素のシクロアルキル基、フェニルまたは低級アルキルフェニルであり;
R9およびR10は、独立して水素、1個から10個の炭素のアルキル基、3個から10個の炭素のシクロアルキル基、フェニルまたは低級アルキルフェニルであり;
R11は、低級アルキル、フェニルまたは低級アルキルフェニルであり;
R12は、低級アルキルであり;
R13は、2個から5個の炭素の二価アルキル基であり;
R14は、(R15)r−フェニル、(R15)r−ナフチル、または(R15)r−ヘテロアリールであり、該ヘテロアリール基が、O、SおよびNからなる群より選択される1個から3個のヘテロ原子を有し、rが、0〜5の値を有する整数であり;
R15は、独立してH、F、Cl、Br、I、NO2、N(R8)2、N(R8)COR8、NR8CON(R8)2、OH、OCOR8、OR8、CN、1個から10個の炭素を有するアルキル基、1個から10個の炭素を有するフルオロ置換アルキル基、2個から10個の炭素および1つから3つの二重結合を有するアルケニル基、2個から10個の炭素および1つから3つの三重結合を有するアルキニル基、またはアルキル基が独立して1個から6個の炭素を有するトリアルキルシリル基またはトリアルキルシリルオキシ基である、請求項1から3に記載の方法。
【請求項5】
R8は、1個から10個の炭素のアルキル基、またはアルキル基が1個から10個の炭素を有するトリメチルアルキル、5個から10個の炭素のシクロアルキル基、フェニルまたは低級アルキルフェニルであり;および
R9およびR10は、独立して水素、1個から10個の炭素のアルキル基、5個から10個の炭素のシクロアルキル基、フェニルまたは低級アルキルフェニルである、請求項4に記載の方法。
【請求項6】
Zが、−CONR1−、−CSNR1−、−NR1CO−、または−NR1CS−である、請求項4または5に記載の方法。
【請求項7】
前記RARアンタゴニストまたはRARインバースアゴニストが、式II:
【化19】
による化学構造または薬学的に許容できるその塩を有し;
式中、Xは、C(R1)2またはOであり;R1は、Hまたは1個から6個の炭素のアルキルであり;
R2は、1個から6個の炭素の低級アルキル、F、Cl、Br、I、CF3、1個から6個の炭素のフルオロ置換アルキル、OH、SH、1個から6個の炭素のアルコキシ、または1個から6個の炭素のアルキルチオであり;
nは、0〜4の値を有する整数であり;
mは、0〜3の値を有する整数であり;
oは、0〜3の値を有する整数であり;
R3は、1個から6個の炭素の低級アルキルまたはFであり;
R8は、1個から10個の炭素のアルキル基、アルキル基が1個から10個の炭素を有するトリメチルシリルアルキル、または3個から10個の炭素のシクロアルキル基、フェニルまたは低級アルキルフェニルであり;
R15は、独立してH、F、Cl、Br、I、NO2、N(R8)2、COR8、NR8CON(R8)2、OCOR8、OR8、CN、1個から10個の炭素を有するアルキル基、1個から10個の炭素を有するフルオロ置換アルキル基、2個から10個の炭素および1つから3つの二重結合を有するアルケニル基、2個から10個の炭素および1つから3つの三重結合を有するアルキニル基、またはアルキル基が独立して1個から6個の炭素を有するトリアルキルシリル基またはトリアルキルシリルオキシ基であり;
tは、0〜5の値を有する整数であり;および
前記CONH基は、前記ベンゾピランおよびジヒドロナフタレン環の6位または7位にある、請求項1から3に記載の方法。
【請求項8】
R2がFであり;
R8は、1個から10個の炭素のアルキル基、アルキル基が1個から10個の炭素を有するトリメチルシリルアルキル、または5個から10個の炭素のシクロアルキル基、フェニルまたは低級アルキルフェニルであり;
R15は、独立してH、F、Cl、Br、I、NO2、N(R8)2、COR8、NR8CON(R8)2、OCOR8、OR8、CN、1個から10個の炭素を有するアルキル基、1個から10個の炭素を有するフルオロ置換アルキル基、1個から10個の炭素および1つから3つの二重結合を有するアルケニル基、1個から10個の炭素および1つから3つの三重結合を有するアルキニル基、またはアルキル基が独立して1個から6個の炭素を有するトリアルキルシリル基またはトリアルキルシリルオキシ基である、請求項7に記載の方法。
【請求項9】
前記RARアンタゴニストまたはRARインバースアゴニストが、式III:
【化20】
による化学構造または薬学的に許容できるその塩を有し;
式中、Xは、−C(CH3)2−または−O−であり;
R2は、−Hまたは−Brであり;
R2’およびR2’’は、独立して−Hまたは−Fであり;
R3の各々は、独立して−Hまたは−CH3であり;および
R8は−Hまたは、C1〜C6 アルキルである、請求項1から3に記載の方法。
【請求項10】
前記RARアンタゴニストまたはRARインバースアゴニストが、式IV:
【化21】
による化学構造または薬学的に許容できるその塩を有し;
式中、X1は、−S−または-O−であり;
X2は、−CH−または-N−であり;
R2は、−H、−F、−CF3またはC1〜C6アルコキシであり;
R2’’は−H、−Fまたは−CF3であり;
R8は、−H、またはC1〜C6アルキルであり;および
R14は、非置換フェニル、非置換チエニルもしくは非置換ピリジル、または1〜3つのR15基で置換されているフェニル、チエニルまたはピリジルであり;および
R15の各々は、独立してC1〜C6アルキル、塩素、−CF3、またはC1〜C6 アルコキシである、請求項1から3に記載の方法。
【請求項11】
前記RARアンタゴニストまたはRARインバースアゴニストが、式V:
【化22】
による化学構造または薬学的に許容できるその塩を有し;
式中、X2は、-CH−または-N−であり;
R2は、−H、−F、または−OCH3であり;
R2*は、−Hまたは−Fであり;
R8は、−H、またはC1〜C6アルキルであり;および
R14は、フェニル、低級アルキルが1個から6個の炭素を有する4−(低級アルキル)フェニル、5−(低級アルキル)−2−チエニル、および6−(低級アルキル)−3−ピリジルからなる群より選択される、請求項1から3に記載の方法。
【請求項12】
前記RARアンタゴニストまたはRARインバースアゴニストが、式VI:
【化23】
による化学構造または薬学的に許容できるその塩を有し、
式中、R8が、−H、またはC1〜C6−アルキルである、請求項1から3に記載の方法。
【請求項13】
前記RARアンタゴニストまたはRARインバースアゴニストが、化学構造:
【化24】
または薬学的に許容できるその塩を有する、請求項1から3に記載の方法。
【請求項14】
前記RARアンタゴニストまたはRARインバースアゴニストが、式VII:
【化25】
による化学構造または薬学的に許容できるその塩を有し;
式中、R2*は、−Hまたは−Fであり;
R8は、−H、またはC1〜C6−アルキルであり;および
R14は、フェニルおよび4−(C1〜C6−アルキル)フェニルからなる群より選択される、請求項1から3に記載の方法。
【請求項15】
前記RARアンタゴニストまたはRARインバースアゴニストが、式VIII:
【化26】
による化学構造または薬学的に許容できるその塩を有し;
式中、R8は、−H、またはC1〜C6−アルキルである、請求項1から3に記載の方法。
【請求項16】
前記RARアンタゴニストまたはRARインバースアゴニストが、式IX:
【化27】
による化学構造または薬学的に許容できるその塩を有し;
式中、X1は、R1が独立して−HまたはC1〜C6−アルキルである−C(R1)2−、−C(R1)2−C(R1)2−、−S−、−O−、
−NR1−、−C(R1)2−O−、−(C(R1)2−S−、または−C(R1)2−NR1−;であり;
R2の各々は、独立してC1〜C6−アルキル、-F、-Cl、-Br、-I、-CF3、フルオロ置換C1〜C6−アルキル、−OH、−SH、C1〜C6−アルコキシ、またはC1〜C6−アルキルチオであり;
mは、0から4の整数であり;
nは、0から2の整数であり;
oは、0から3の整数であり;
R3は、−H、C1〜C6−アルコキシ、−F、−Cl、−Brまたは−Iであり;
R4は、(R5)p−フェニル、(R5)p−ナフチル、または(R5)p−ヘテロアリールであり;該ヘテロアリール基は、5員または6員であり、酸素、硫黄、および窒素からなる群より選択される1個から3個のヘテロ原子を有し;pは、0から5の整数であり;
R5の各々は、独立して−F、−Cl、−Br、−I、−NO2、−N(R8)2、−N(R8)COR8、−N(R8)CON(R8)2、−OH、−OCOR8、−OR8、−CN、−COOH、−COOR8、C1〜C10−アルキル、1つから3つの二重結合を有するC1〜C10アルケニル基、1つから3つの三重結合を有するC1〜C10アルキニル基、C1〜C6(トリアルキル)シリルまたはC1〜C6(トリアルキル)シリルオキシであり;
Yは、フェニル、ナフチル、またはピリジル、チエニル、フリル、ピリダジニル、ピリミジニル、ピラジニル、チアゾリル、オキサゾリル、イミダゾリルおよびピラゾリルからなる群より選択されるヘテロアリールであり;該フェニル基およびヘテロアリール基は、任意にかつ独立して1つまたは2つのR2基で置換されており、またはYは、−(CR3=CR3)r−であり;
rは、1から3の整数であり;
Aは、(CH2)q、低級C3〜C6分枝鎖アルキル、C3〜C6シクロアルキル、1つまたは2つの二重結合を有するC2〜C6アルケニル、1つまたは2つの三重結合を有するC2〜C6アルケニルであり;qは0〜5の整数であるが、ただし、Yが−(CR3=CR3)r−の場合、Aが(CH2)qであり、かつqが0であり;
Bは、−H、−COOH、−COOR8、−CONR9R10、−CH2OH,−CH2OR11、−CH2OCOR11、−CHO、−CH(OR12)2、−CHOR13O、−COR7、−CR7(OR12)2、−CR7OR13O、または−Si(C1〜6アルキル)3であり;
R7は、1個から5個の炭素を含有するアルキル基、シクロアルキル基またはアルケニル基であり;
R8は、C1〜C10アルキル、C1〜C10(トリメチルシリル)アルキル、C5〜C10シクロアルキル、フェニルまたは低級アルキルフェニルであり;
R9およびR10は、各々独立して−H、C1〜C10アルキル、C5〜C10シクロアルキル、フェニルまたは低級アルキルフェニルであり;
R11は、低級アルキル、フェニルまたは低級アルキルフェニルであり;
R12は、低級アルキルであり;
R13は、2個から5個の炭素の二価アルキル基である、請求項1から3に記載の方法。
【請求項17】
前記RARアンタゴニストまたはRARインバースアゴニストが、式X:
Y3(R4)−X−Y1(R1)(R2)−Z−Y2(R2)−A−B (X);
による化学構造または薬学的に許容できるその塩を有し;
式中、Y1は、フェニル、ナフチル、またはピリジル、チエニル、フリル、ピリダジニル、ピリミジニル、ピラジニル、チアゾリル、オキサゾリル、イミダゾリルおよびピラゾリルからなる群より選択されるヘテロアリールであり、該フェニル基、ナフチル基、およびヘテロアリール基が、任意にかつ独立してR1基で置換されており、さらに1つまたは2つのR2基で任意に置換されており;
R1は、C1〜C10アルキル、1−アデマンチル、2−テトラヒドロピラノキシ、C1〜C6トリアルキルシラニルオキシ、−OH、C1〜C10アルコキシ、C1〜C10アルキルチオ、または−OCH2O−(C1〜C6アルキル)であり;
R2の各々は、独立してC1〜C6−アルキル、−F、−Cl、−Br、−I、−CF3、−CF2CF3、−OH、−OR3、−NO2、−N(R3)2、−CN、−N3、−COR3、−NHCOR3、−COOH、または−COOR3であり;
Xは、−(C(R3)2)−、−S−、−SO−、−SO2−、−O−、−C(=O)−、−C(=S)−、−C(=NR1)−、−C(=C(R1)2)−または−NR3−であり;
Zは、−C≡C−、−N=N−、−N(O)=N−、−N=N(O)−、−N=CR3−、−CR3=N−、−(CR3=CR3)n−、−OCO−、−CSO−、−OCS−、−COCR3=R3O−、−CO−NR3−、−CS−NR3、−NR3−CO−、または−NR3−CS−であり;
nは、0〜5の値を有する整数であり;
R3の各々は、独立して−HまたはC1−C6アルキルであり;
Y2は、フェニル、ナフチル、またはピリジル、チエニル、フリル、ピリダジニル、ピリミジニル、ピラジニル、チアゾニル、オキサゾリル、イミダゾリルおよびピラゾリルからなる群より選択されるヘテロアリールであり;該フェニル基、ナフチル基およびヘテロアリール基は、非置換であっても1つまたは2つのR2基で置換されていてもよく、またはZが、−(CR3=CR3)n−であり、nが、3、4、または5である場合、Y2は、該−(CR3=CR3)n−基とBとの間の直接的原子価結合を表し;
Y3は、フェニル、ナフチル、またはピリジル、チエニル、フリル、ピリダジニル、ピリミジニル、ピラジニル、チアゾリル、オキサゾリル、イミダゾリルおよびピラゾリルからなる群より選択されるヘテロアリールであり;前記フェニル基、ナフチル基およびヘテロアリール基は、非置換であっても1つから5つのR4基で置換されていてもよく;
R4の各々は、独立してC1〜C10アルキル、1つから3つの三重結合を有するC2〜C10アルケニル、−F、−Cl、−Br、−I、−NO2、−N(R3)2、−N3、−COOH、−COO−(C1〜C6アルキル)、−OH、−SH、−O−C1〜C6アルキル、または-S−C1〜C6アルキルであり;
Aは、(CH2)q、低級C3〜C6分枝状アルキル、C3〜C6シクロアルキル、1つから2つの二重結合を有するC2〜C6アルケニル、1つから2つの三重結合を有するC2〜C6アルキニルであり;qが0から5であり;
Bは、−H、−COOH、−COOR8、−CONR9R10、−CH2OH,−CH2OR11、−CH2OCOR11、−CHO、−CH(OR12)2、−CHOR13O、−COR7、−CR7(OR12)2、−CR7OR13O、または−Si(C1〜C 6アルキル)3であり;
R7は、1個から5個の炭素を含有するアルキル基、シクロアルキル基またはアルケニル基であり;
R8は、C1〜C10アルキル、C1〜C10トリメチルシリルアルキル、C3〜C10シクロアルキル、フェニルまたは低級アルキルフェニルであり;
R9およびR10は、独立して水素、C1〜C10アルキル基、C3〜C10シクロアルキル、フェニルまたは低級アルキルフェニルであり;
R11は、低級アルキル、フェニルまたは低級アルキルフェニルであり;
R12は、低級アルキルであり;
R13は、2個から5個の炭素の二価アルキル基である、請求項1から3に記載の方法。
【請求項18】
Xが、−(C(R3)2)−、−S−、−SO−、−SO2−、−O−または−NR3−であり;および
Zが、−CO−NR3−,−CS−NR3−,−NR3−CO−、または−NR3−CS−である、請求項17に記載の方法。
【請求項19】
Xが、−C(=O)−、−C(=S)−、−C(=NR1)−、または−C(=C(R1)2)−であり;および
Zが、−CO−NR3−、−CS−NR3−、−NR3−CO−、または−NR3−CS−である、請求項17に記載の方法。
【請求項20】
Y3により表される前記フェニル、ナフチル、またはヘテロアリールが、非置換であるか、または1つから3つのR4基により置換されている、請求項17から19のいずれか一項に記載の方法。
【請求項21】
前記RARアンタゴニストまたはRARインバースアゴニストが、化学構造:
【化28】
または薬学的に許容できるその塩を有する、請求項1から3に記載の方法。
【請求項22】
前記RARアンタゴニストまたはRARインバースアゴニストが、化学構造:
【化29】
または薬学的に許容できるその塩を有し;
式中nが、1から10の整数である、請求項1から3に記載の方法。
【請求項23】
前記RARアンタゴニストまたはRARインバースアゴニストが、化学構造:
【化30】
または薬学的に許容できるその塩を有し;
式中nが、1から10の整数である、請求項1から3に記載の方法。
【請求項24】
前記RARアンタゴニストまたはRARインバースアゴニストが、化学構造:
【化31】
または薬学的に許容できるその塩を有する、請求項1から3に記載の方法。
【請求項25】
前記RARアンタゴニストまたはRARインバースアゴニストが、化学構造:
【化32】
または薬学的に許容できるその塩を有する、請求項1から3に記載の方法。
【請求項26】
前記RARアンタゴニストまたはRARインバースアゴニストが、化学構造:
【化33】
または薬学的に許容できるその塩を有する、請求項1から3に記載の方法。
【請求項27】
前記RARアンタゴニストまたはRARインバースアゴニストが、経口投与、局所投与、全身投与、非経口投与、皮下投与、または静脈内投与される、請求項1から26に記載の方法。
【請求項28】
前記副作用が、造血毒性、造血始原細胞の骨髄から末梢血への動員減少、貧血、骨髄抑制、汎血球減少症、血小板減少症、好中球減少症、リンパ球減少症、白血球減少症、口内炎、脱毛症、頭痛、筋肉痛およびそれらの組合わせからなる群より選択される、請求項1に記載の方法。
【請求項29】
前記副作用が、化学療法誘導性の好中球減少症および/または放射線療法誘導性の好中球減少症である、請求項1に記載の方法。
【請求項30】
前記副作用が、化学療法誘導性の好中球減少症および/または放射線療法誘導性の好中球減少症である、請求項13、21から26のいずれか一項に記載の方法。
【請求項31】
前記副作用が、化学療法誘導性の血小板減少症および/または放射線療法誘導性の血小板減少症である、請求項13、21から26のいずれか一項に記載の方法。
【請求項32】
化学療法および/または放射線療法の開始前かまたはそれと同時に、もしくは化学療法および/または放射線療法の開始後に、前記RARアンタゴニストまたはRARインバースアゴニストの投与が開始される、請求項1に記載の方法。
【請求項33】
前記RARアンタゴニストまたはRARインバースアゴニストが、1日、1回投与される、請求項1から3に記載の方法。
【請求項34】
血小板減少症に罹っている哺乳動物を処置するための方法であって、該方法は、RARα、RARβおよびRARγサブタイプの受容体に結合するRARアンタゴニストまたはRARインバースアゴニストの治療有効量を、処置を必要とする該哺乳動物に投与することを含む、方法。
【請求項35】
前記血小板減少症が、化学療法または放射線療法が原因である、請求項34に記載の方法。
【請求項36】
前記RARアンタゴニストまたはRARインバースアゴニストが、化学療法または放射線療法の前に前記哺乳動物に投与される、請求項34に記載の方法。
【請求項37】
前記血小板減少症が、骨髄輸血または血液疾患が原因である、請求項34に記載の方法。
【請求項38】
前記RARアンタゴニストまたはRARインバースアゴニストが、4−[[4−(4−エチルフェニル)−2,2−ジメチル−(2H)−チオクロメン−6−イル]−エチニル]−安息香酸(VTP 194310)または薬学的に許容できるその塩である、請求項34に記載の方法。
【請求項39】
前記RARアンタゴニストまたはRARインバースアゴニストが、4−(3−ブロモ−4−エトキシ−5−(4−メチルベンゾイル)ベンズアミド)安息香酸(VTP 196996)、または薬学的に許容できるその塩である、請求項34に記載の方法。
【請求項40】
造血関連状態に罹っている哺乳動物を処置する方法であって、該方法は、RARα、RARβおよびRARγサブタイプの受容体に結合するRARアンタゴニストまたはRARインバースアゴニストの治療有効量を、該哺乳動物に投与することを含む、方法。
【請求項41】
前記状態が、造血機能の低下、免疫機能の低下、好中球数の減少、好中球動員の低下、末梢血始原細胞の動員、敗血症、重症の慢性好中球減少症、骨髄移植、感染症、白血球減少症、血小板減少症、貧血、移植の間の骨髄生着の亢進、放射線処置における骨髄回復亢進、化学物質または化学療法誘導性の骨髄形成不全症または骨髄抑制、後天性免疫不全症候群およびそれらの組合わせからなる群より選択される、請求項40に記載の方法。
【請求項42】
前記RARアンタゴニストまたはRARインバースアゴニストが、4−[[4−(4−エチルフェニル)−2,2−ジメチル−(2H)−チオクロメン−6−イル]−エチニル]−安息香酸(VTP 194310)または薬学的に許容できるその塩である、請求項40に記載の方法。
【請求項43】
前記RARアンタゴニストまたはRARインバースアゴニストが、4−(3−ブロモ−4−エトキシ−5−(4−メチルベンゾイル)ベンズアミド)安息香酸(VTP 196996)、または薬学的に許容できるその塩である、請求項40に記載の方法。
【請求項44】
処置を必要とする哺乳動物において好中球および/または血小板の減少または抑制に関連する状態、疾患、障害、傷害または病態を処置するための方法であって、該方法は、RARα、RARβおよびRARγサブタイプの受容体に結合するRARアンタゴニストまたはRARインバースアゴニストの治療有効量を該哺乳動物に投与することを含む、方法。
【請求項45】
前記RARアンタゴニストまたはRARインバースアゴニストが、4−[[4−(4−エチルフェニル)−2,2−ジメチル−(2H)−チオクロメン−6−イル]−エチニル]−安息香酸(VTP 194310)または薬学的に許容できるその塩である、請求項44に記載の方法。
【請求項46】
前記RARアンタゴニストまたはRARインバースアゴニストが、4−(3−ブロモ−4−エトキシ−5−(4−メチルベンゾイル)ベンズアミド)安息香酸(VTP 196996)、または薬学的に許容できるその塩である、請求項44に記載の方法。
【請求項1】
化学療法および/または放射線療法を受ける哺乳動物において化学療法および/または放射線療法の副作用を処置するための方法であって、該方法は、RARα、RARβおよびRARγサブタイプの受容体に結合する少なくとも1つのレチノイン酸受容体(RAR)アンタゴニストまたはRARインバースアゴニストの治療有効量を該哺乳動物に投与する工程を含む、方法。
【請求項2】
前記RARアンタゴニストまたはRARインバースアゴニストが、前記哺乳動物における好中球産生を増加させるのに有効である、請求項1に記載の方法。
【請求項3】
血小板産生を必要とする哺乳動物において血小板産生を増加させるための方法であって、該方法は、RARα、RARβおよびRARγサブタイプの受容体に結合する少なくとも1つのRARアンタゴニストまたはRARインバースアゴニストの有効量を該哺乳動物に投与する工程を含む、方法。
【請求項4】
前記RARアンタゴニストまたはRARインバースアゴニストが、化学構造:
【化18】
または薬学的に許容できるその塩を有し;
式中、Xは、S、O、RがHまたは1個から6個の炭素のアルキルであるNRか、または
Xは、R1が、独立してHまたは1個から6個の炭素のアルキルである[C(R1)2]nであり、nは、0と2の間でかつ0と2を含む整数であり;
R2は、独立して水素、1個から6個の炭素の低級アルキル、F、Cl、Br、I、CF3、1個から6個の炭素のフルオロ置換アルキル、OH、SH、1個から6個の炭素のアルコキシ、または1個から6個の炭素のアルキルチオであり;
R3は、独立して水素、1個から6個の炭素の低級アルキルまたはFであり;
mは、0〜3の値を有する整数であり;
nは、0〜4の値を有する整数であり;
oは、0〜3の値を有する整数であり;
Zは、−CONR1−、−CSNR1−、−NR1CO−、−NR1CS−、−C≡C−、−C=C−、−N=N−、−N=CR1−、−CR1=N−、
−COO−、−OCO−;−OSO−;−OCS−、またはn’が0から5の整数である−(CR1=CR1)n’−であり、;
Yは、フェニル基またはナフチル基、またはピリジル、チエニル、フリル、ピリダジニル、ピリミジニル、ピラジニル、チアゾリル、オキサゾリル、イミダゾリルおよびピラゾリルからなる群より選択されるヘテロアリールであり、該フェニル基およびヘテロアリール基は、1つまたは2つのR2基で任意に置換されているか、またはZが(CR1=CR1)n’-であり、n’が3、4または5である場合、Yは、前記(CR2=CR2)n’基とBとの間の直接的原子価結合を表し;
Aは、qが0〜5である(CH2)q、3個から6個の炭素を有する低級分枝鎖アルキル、3個から6個の炭素を有するシクロアルキル、2個から6個の炭素および1つまたは2つの二重結合を有するアルケニル、2個から6個の炭素および1つまたは2つの三重結合を有するアルキニルであり;
Bは、水素、COOHまたは薬学的に許容できるその塩、COOR8、CONR9R10、CH2OH、CH2OR11、CH2OCOR11、CHO、CH(OR12)2、CHOR13O、−COR7、CR7(OR12)2、CR7OR13O、またはトリ−低級アルキルシリルであり;
R7は、1個から5個の炭素を含有するアルキル基、シクロアルキル基またはアルケニル基であり;
R8は、1個から10個の炭素のアルキル基、またはアルキル基が1個から10個の炭素を有するトリメチルアルキル、3個から10個の炭素のシクロアルキル基、フェニルまたは低級アルキルフェニルであり;
R9およびR10は、独立して水素、1個から10個の炭素のアルキル基、3個から10個の炭素のシクロアルキル基、フェニルまたは低級アルキルフェニルであり;
R11は、低級アルキル、フェニルまたは低級アルキルフェニルであり;
R12は、低級アルキルであり;
R13は、2個から5個の炭素の二価アルキル基であり;
R14は、(R15)r−フェニル、(R15)r−ナフチル、または(R15)r−ヘテロアリールであり、該ヘテロアリール基が、O、SおよびNからなる群より選択される1個から3個のヘテロ原子を有し、rが、0〜5の値を有する整数であり;
R15は、独立してH、F、Cl、Br、I、NO2、N(R8)2、N(R8)COR8、NR8CON(R8)2、OH、OCOR8、OR8、CN、1個から10個の炭素を有するアルキル基、1個から10個の炭素を有するフルオロ置換アルキル基、2個から10個の炭素および1つから3つの二重結合を有するアルケニル基、2個から10個の炭素および1つから3つの三重結合を有するアルキニル基、またはアルキル基が独立して1個から6個の炭素を有するトリアルキルシリル基またはトリアルキルシリルオキシ基である、請求項1から3に記載の方法。
【請求項5】
R8は、1個から10個の炭素のアルキル基、またはアルキル基が1個から10個の炭素を有するトリメチルアルキル、5個から10個の炭素のシクロアルキル基、フェニルまたは低級アルキルフェニルであり;および
R9およびR10は、独立して水素、1個から10個の炭素のアルキル基、5個から10個の炭素のシクロアルキル基、フェニルまたは低級アルキルフェニルである、請求項4に記載の方法。
【請求項6】
Zが、−CONR1−、−CSNR1−、−NR1CO−、または−NR1CS−である、請求項4または5に記載の方法。
【請求項7】
前記RARアンタゴニストまたはRARインバースアゴニストが、式II:
【化19】
による化学構造または薬学的に許容できるその塩を有し;
式中、Xは、C(R1)2またはOであり;R1は、Hまたは1個から6個の炭素のアルキルであり;
R2は、1個から6個の炭素の低級アルキル、F、Cl、Br、I、CF3、1個から6個の炭素のフルオロ置換アルキル、OH、SH、1個から6個の炭素のアルコキシ、または1個から6個の炭素のアルキルチオであり;
nは、0〜4の値を有する整数であり;
mは、0〜3の値を有する整数であり;
oは、0〜3の値を有する整数であり;
R3は、1個から6個の炭素の低級アルキルまたはFであり;
R8は、1個から10個の炭素のアルキル基、アルキル基が1個から10個の炭素を有するトリメチルシリルアルキル、または3個から10個の炭素のシクロアルキル基、フェニルまたは低級アルキルフェニルであり;
R15は、独立してH、F、Cl、Br、I、NO2、N(R8)2、COR8、NR8CON(R8)2、OCOR8、OR8、CN、1個から10個の炭素を有するアルキル基、1個から10個の炭素を有するフルオロ置換アルキル基、2個から10個の炭素および1つから3つの二重結合を有するアルケニル基、2個から10個の炭素および1つから3つの三重結合を有するアルキニル基、またはアルキル基が独立して1個から6個の炭素を有するトリアルキルシリル基またはトリアルキルシリルオキシ基であり;
tは、0〜5の値を有する整数であり;および
前記CONH基は、前記ベンゾピランおよびジヒドロナフタレン環の6位または7位にある、請求項1から3に記載の方法。
【請求項8】
R2がFであり;
R8は、1個から10個の炭素のアルキル基、アルキル基が1個から10個の炭素を有するトリメチルシリルアルキル、または5個から10個の炭素のシクロアルキル基、フェニルまたは低級アルキルフェニルであり;
R15は、独立してH、F、Cl、Br、I、NO2、N(R8)2、COR8、NR8CON(R8)2、OCOR8、OR8、CN、1個から10個の炭素を有するアルキル基、1個から10個の炭素を有するフルオロ置換アルキル基、1個から10個の炭素および1つから3つの二重結合を有するアルケニル基、1個から10個の炭素および1つから3つの三重結合を有するアルキニル基、またはアルキル基が独立して1個から6個の炭素を有するトリアルキルシリル基またはトリアルキルシリルオキシ基である、請求項7に記載の方法。
【請求項9】
前記RARアンタゴニストまたはRARインバースアゴニストが、式III:
【化20】
による化学構造または薬学的に許容できるその塩を有し;
式中、Xは、−C(CH3)2−または−O−であり;
R2は、−Hまたは−Brであり;
R2’およびR2’’は、独立して−Hまたは−Fであり;
R3の各々は、独立して−Hまたは−CH3であり;および
R8は−Hまたは、C1〜C6 アルキルである、請求項1から3に記載の方法。
【請求項10】
前記RARアンタゴニストまたはRARインバースアゴニストが、式IV:
【化21】
による化学構造または薬学的に許容できるその塩を有し;
式中、X1は、−S−または-O−であり;
X2は、−CH−または-N−であり;
R2は、−H、−F、−CF3またはC1〜C6アルコキシであり;
R2’’は−H、−Fまたは−CF3であり;
R8は、−H、またはC1〜C6アルキルであり;および
R14は、非置換フェニル、非置換チエニルもしくは非置換ピリジル、または1〜3つのR15基で置換されているフェニル、チエニルまたはピリジルであり;および
R15の各々は、独立してC1〜C6アルキル、塩素、−CF3、またはC1〜C6 アルコキシである、請求項1から3に記載の方法。
【請求項11】
前記RARアンタゴニストまたはRARインバースアゴニストが、式V:
【化22】
による化学構造または薬学的に許容できるその塩を有し;
式中、X2は、-CH−または-N−であり;
R2は、−H、−F、または−OCH3であり;
R2*は、−Hまたは−Fであり;
R8は、−H、またはC1〜C6アルキルであり;および
R14は、フェニル、低級アルキルが1個から6個の炭素を有する4−(低級アルキル)フェニル、5−(低級アルキル)−2−チエニル、および6−(低級アルキル)−3−ピリジルからなる群より選択される、請求項1から3に記載の方法。
【請求項12】
前記RARアンタゴニストまたはRARインバースアゴニストが、式VI:
【化23】
による化学構造または薬学的に許容できるその塩を有し、
式中、R8が、−H、またはC1〜C6−アルキルである、請求項1から3に記載の方法。
【請求項13】
前記RARアンタゴニストまたはRARインバースアゴニストが、化学構造:
【化24】
または薬学的に許容できるその塩を有する、請求項1から3に記載の方法。
【請求項14】
前記RARアンタゴニストまたはRARインバースアゴニストが、式VII:
【化25】
による化学構造または薬学的に許容できるその塩を有し;
式中、R2*は、−Hまたは−Fであり;
R8は、−H、またはC1〜C6−アルキルであり;および
R14は、フェニルおよび4−(C1〜C6−アルキル)フェニルからなる群より選択される、請求項1から3に記載の方法。
【請求項15】
前記RARアンタゴニストまたはRARインバースアゴニストが、式VIII:
【化26】
による化学構造または薬学的に許容できるその塩を有し;
式中、R8は、−H、またはC1〜C6−アルキルである、請求項1から3に記載の方法。
【請求項16】
前記RARアンタゴニストまたはRARインバースアゴニストが、式IX:
【化27】
による化学構造または薬学的に許容できるその塩を有し;
式中、X1は、R1が独立して−HまたはC1〜C6−アルキルである−C(R1)2−、−C(R1)2−C(R1)2−、−S−、−O−、
−NR1−、−C(R1)2−O−、−(C(R1)2−S−、または−C(R1)2−NR1−;であり;
R2の各々は、独立してC1〜C6−アルキル、-F、-Cl、-Br、-I、-CF3、フルオロ置換C1〜C6−アルキル、−OH、−SH、C1〜C6−アルコキシ、またはC1〜C6−アルキルチオであり;
mは、0から4の整数であり;
nは、0から2の整数であり;
oは、0から3の整数であり;
R3は、−H、C1〜C6−アルコキシ、−F、−Cl、−Brまたは−Iであり;
R4は、(R5)p−フェニル、(R5)p−ナフチル、または(R5)p−ヘテロアリールであり;該ヘテロアリール基は、5員または6員であり、酸素、硫黄、および窒素からなる群より選択される1個から3個のヘテロ原子を有し;pは、0から5の整数であり;
R5の各々は、独立して−F、−Cl、−Br、−I、−NO2、−N(R8)2、−N(R8)COR8、−N(R8)CON(R8)2、−OH、−OCOR8、−OR8、−CN、−COOH、−COOR8、C1〜C10−アルキル、1つから3つの二重結合を有するC1〜C10アルケニル基、1つから3つの三重結合を有するC1〜C10アルキニル基、C1〜C6(トリアルキル)シリルまたはC1〜C6(トリアルキル)シリルオキシであり;
Yは、フェニル、ナフチル、またはピリジル、チエニル、フリル、ピリダジニル、ピリミジニル、ピラジニル、チアゾリル、オキサゾリル、イミダゾリルおよびピラゾリルからなる群より選択されるヘテロアリールであり;該フェニル基およびヘテロアリール基は、任意にかつ独立して1つまたは2つのR2基で置換されており、またはYは、−(CR3=CR3)r−であり;
rは、1から3の整数であり;
Aは、(CH2)q、低級C3〜C6分枝鎖アルキル、C3〜C6シクロアルキル、1つまたは2つの二重結合を有するC2〜C6アルケニル、1つまたは2つの三重結合を有するC2〜C6アルケニルであり;qは0〜5の整数であるが、ただし、Yが−(CR3=CR3)r−の場合、Aが(CH2)qであり、かつqが0であり;
Bは、−H、−COOH、−COOR8、−CONR9R10、−CH2OH,−CH2OR11、−CH2OCOR11、−CHO、−CH(OR12)2、−CHOR13O、−COR7、−CR7(OR12)2、−CR7OR13O、または−Si(C1〜6アルキル)3であり;
R7は、1個から5個の炭素を含有するアルキル基、シクロアルキル基またはアルケニル基であり;
R8は、C1〜C10アルキル、C1〜C10(トリメチルシリル)アルキル、C5〜C10シクロアルキル、フェニルまたは低級アルキルフェニルであり;
R9およびR10は、各々独立して−H、C1〜C10アルキル、C5〜C10シクロアルキル、フェニルまたは低級アルキルフェニルであり;
R11は、低級アルキル、フェニルまたは低級アルキルフェニルであり;
R12は、低級アルキルであり;
R13は、2個から5個の炭素の二価アルキル基である、請求項1から3に記載の方法。
【請求項17】
前記RARアンタゴニストまたはRARインバースアゴニストが、式X:
Y3(R4)−X−Y1(R1)(R2)−Z−Y2(R2)−A−B (X);
による化学構造または薬学的に許容できるその塩を有し;
式中、Y1は、フェニル、ナフチル、またはピリジル、チエニル、フリル、ピリダジニル、ピリミジニル、ピラジニル、チアゾリル、オキサゾリル、イミダゾリルおよびピラゾリルからなる群より選択されるヘテロアリールであり、該フェニル基、ナフチル基、およびヘテロアリール基が、任意にかつ独立してR1基で置換されており、さらに1つまたは2つのR2基で任意に置換されており;
R1は、C1〜C10アルキル、1−アデマンチル、2−テトラヒドロピラノキシ、C1〜C6トリアルキルシラニルオキシ、−OH、C1〜C10アルコキシ、C1〜C10アルキルチオ、または−OCH2O−(C1〜C6アルキル)であり;
R2の各々は、独立してC1〜C6−アルキル、−F、−Cl、−Br、−I、−CF3、−CF2CF3、−OH、−OR3、−NO2、−N(R3)2、−CN、−N3、−COR3、−NHCOR3、−COOH、または−COOR3であり;
Xは、−(C(R3)2)−、−S−、−SO−、−SO2−、−O−、−C(=O)−、−C(=S)−、−C(=NR1)−、−C(=C(R1)2)−または−NR3−であり;
Zは、−C≡C−、−N=N−、−N(O)=N−、−N=N(O)−、−N=CR3−、−CR3=N−、−(CR3=CR3)n−、−OCO−、−CSO−、−OCS−、−COCR3=R3O−、−CO−NR3−、−CS−NR3、−NR3−CO−、または−NR3−CS−であり;
nは、0〜5の値を有する整数であり;
R3の各々は、独立して−HまたはC1−C6アルキルであり;
Y2は、フェニル、ナフチル、またはピリジル、チエニル、フリル、ピリダジニル、ピリミジニル、ピラジニル、チアゾニル、オキサゾリル、イミダゾリルおよびピラゾリルからなる群より選択されるヘテロアリールであり;該フェニル基、ナフチル基およびヘテロアリール基は、非置換であっても1つまたは2つのR2基で置換されていてもよく、またはZが、−(CR3=CR3)n−であり、nが、3、4、または5である場合、Y2は、該−(CR3=CR3)n−基とBとの間の直接的原子価結合を表し;
Y3は、フェニル、ナフチル、またはピリジル、チエニル、フリル、ピリダジニル、ピリミジニル、ピラジニル、チアゾリル、オキサゾリル、イミダゾリルおよびピラゾリルからなる群より選択されるヘテロアリールであり;前記フェニル基、ナフチル基およびヘテロアリール基は、非置換であっても1つから5つのR4基で置換されていてもよく;
R4の各々は、独立してC1〜C10アルキル、1つから3つの三重結合を有するC2〜C10アルケニル、−F、−Cl、−Br、−I、−NO2、−N(R3)2、−N3、−COOH、−COO−(C1〜C6アルキル)、−OH、−SH、−O−C1〜C6アルキル、または-S−C1〜C6アルキルであり;
Aは、(CH2)q、低級C3〜C6分枝状アルキル、C3〜C6シクロアルキル、1つから2つの二重結合を有するC2〜C6アルケニル、1つから2つの三重結合を有するC2〜C6アルキニルであり;qが0から5であり;
Bは、−H、−COOH、−COOR8、−CONR9R10、−CH2OH,−CH2OR11、−CH2OCOR11、−CHO、−CH(OR12)2、−CHOR13O、−COR7、−CR7(OR12)2、−CR7OR13O、または−Si(C1〜C 6アルキル)3であり;
R7は、1個から5個の炭素を含有するアルキル基、シクロアルキル基またはアルケニル基であり;
R8は、C1〜C10アルキル、C1〜C10トリメチルシリルアルキル、C3〜C10シクロアルキル、フェニルまたは低級アルキルフェニルであり;
R9およびR10は、独立して水素、C1〜C10アルキル基、C3〜C10シクロアルキル、フェニルまたは低級アルキルフェニルであり;
R11は、低級アルキル、フェニルまたは低級アルキルフェニルであり;
R12は、低級アルキルであり;
R13は、2個から5個の炭素の二価アルキル基である、請求項1から3に記載の方法。
【請求項18】
Xが、−(C(R3)2)−、−S−、−SO−、−SO2−、−O−または−NR3−であり;および
Zが、−CO−NR3−,−CS−NR3−,−NR3−CO−、または−NR3−CS−である、請求項17に記載の方法。
【請求項19】
Xが、−C(=O)−、−C(=S)−、−C(=NR1)−、または−C(=C(R1)2)−であり;および
Zが、−CO−NR3−、−CS−NR3−、−NR3−CO−、または−NR3−CS−である、請求項17に記載の方法。
【請求項20】
Y3により表される前記フェニル、ナフチル、またはヘテロアリールが、非置換であるか、または1つから3つのR4基により置換されている、請求項17から19のいずれか一項に記載の方法。
【請求項21】
前記RARアンタゴニストまたはRARインバースアゴニストが、化学構造:
【化28】
または薬学的に許容できるその塩を有する、請求項1から3に記載の方法。
【請求項22】
前記RARアンタゴニストまたはRARインバースアゴニストが、化学構造:
【化29】
または薬学的に許容できるその塩を有し;
式中nが、1から10の整数である、請求項1から3に記載の方法。
【請求項23】
前記RARアンタゴニストまたはRARインバースアゴニストが、化学構造:
【化30】
または薬学的に許容できるその塩を有し;
式中nが、1から10の整数である、請求項1から3に記載の方法。
【請求項24】
前記RARアンタゴニストまたはRARインバースアゴニストが、化学構造:
【化31】
または薬学的に許容できるその塩を有する、請求項1から3に記載の方法。
【請求項25】
前記RARアンタゴニストまたはRARインバースアゴニストが、化学構造:
【化32】
または薬学的に許容できるその塩を有する、請求項1から3に記載の方法。
【請求項26】
前記RARアンタゴニストまたはRARインバースアゴニストが、化学構造:
【化33】
または薬学的に許容できるその塩を有する、請求項1から3に記載の方法。
【請求項27】
前記RARアンタゴニストまたはRARインバースアゴニストが、経口投与、局所投与、全身投与、非経口投与、皮下投与、または静脈内投与される、請求項1から26に記載の方法。
【請求項28】
前記副作用が、造血毒性、造血始原細胞の骨髄から末梢血への動員減少、貧血、骨髄抑制、汎血球減少症、血小板減少症、好中球減少症、リンパ球減少症、白血球減少症、口内炎、脱毛症、頭痛、筋肉痛およびそれらの組合わせからなる群より選択される、請求項1に記載の方法。
【請求項29】
前記副作用が、化学療法誘導性の好中球減少症および/または放射線療法誘導性の好中球減少症である、請求項1に記載の方法。
【請求項30】
前記副作用が、化学療法誘導性の好中球減少症および/または放射線療法誘導性の好中球減少症である、請求項13、21から26のいずれか一項に記載の方法。
【請求項31】
前記副作用が、化学療法誘導性の血小板減少症および/または放射線療法誘導性の血小板減少症である、請求項13、21から26のいずれか一項に記載の方法。
【請求項32】
化学療法および/または放射線療法の開始前かまたはそれと同時に、もしくは化学療法および/または放射線療法の開始後に、前記RARアンタゴニストまたはRARインバースアゴニストの投与が開始される、請求項1に記載の方法。
【請求項33】
前記RARアンタゴニストまたはRARインバースアゴニストが、1日、1回投与される、請求項1から3に記載の方法。
【請求項34】
血小板減少症に罹っている哺乳動物を処置するための方法であって、該方法は、RARα、RARβおよびRARγサブタイプの受容体に結合するRARアンタゴニストまたはRARインバースアゴニストの治療有効量を、処置を必要とする該哺乳動物に投与することを含む、方法。
【請求項35】
前記血小板減少症が、化学療法または放射線療法が原因である、請求項34に記載の方法。
【請求項36】
前記RARアンタゴニストまたはRARインバースアゴニストが、化学療法または放射線療法の前に前記哺乳動物に投与される、請求項34に記載の方法。
【請求項37】
前記血小板減少症が、骨髄輸血または血液疾患が原因である、請求項34に記載の方法。
【請求項38】
前記RARアンタゴニストまたはRARインバースアゴニストが、4−[[4−(4−エチルフェニル)−2,2−ジメチル−(2H)−チオクロメン−6−イル]−エチニル]−安息香酸(VTP 194310)または薬学的に許容できるその塩である、請求項34に記載の方法。
【請求項39】
前記RARアンタゴニストまたはRARインバースアゴニストが、4−(3−ブロモ−4−エトキシ−5−(4−メチルベンゾイル)ベンズアミド)安息香酸(VTP 196996)、または薬学的に許容できるその塩である、請求項34に記載の方法。
【請求項40】
造血関連状態に罹っている哺乳動物を処置する方法であって、該方法は、RARα、RARβおよびRARγサブタイプの受容体に結合するRARアンタゴニストまたはRARインバースアゴニストの治療有効量を、該哺乳動物に投与することを含む、方法。
【請求項41】
前記状態が、造血機能の低下、免疫機能の低下、好中球数の減少、好中球動員の低下、末梢血始原細胞の動員、敗血症、重症の慢性好中球減少症、骨髄移植、感染症、白血球減少症、血小板減少症、貧血、移植の間の骨髄生着の亢進、放射線処置における骨髄回復亢進、化学物質または化学療法誘導性の骨髄形成不全症または骨髄抑制、後天性免疫不全症候群およびそれらの組合わせからなる群より選択される、請求項40に記載の方法。
【請求項42】
前記RARアンタゴニストまたはRARインバースアゴニストが、4−[[4−(4−エチルフェニル)−2,2−ジメチル−(2H)−チオクロメン−6−イル]−エチニル]−安息香酸(VTP 194310)または薬学的に許容できるその塩である、請求項40に記載の方法。
【請求項43】
前記RARアンタゴニストまたはRARインバースアゴニストが、4−(3−ブロモ−4−エトキシ−5−(4−メチルベンゾイル)ベンズアミド)安息香酸(VTP 196996)、または薬学的に許容できるその塩である、請求項40に記載の方法。
【請求項44】
処置を必要とする哺乳動物において好中球および/または血小板の減少または抑制に関連する状態、疾患、障害、傷害または病態を処置するための方法であって、該方法は、RARα、RARβおよびRARγサブタイプの受容体に結合するRARアンタゴニストまたはRARインバースアゴニストの治療有効量を該哺乳動物に投与することを含む、方法。
【請求項45】
前記RARアンタゴニストまたはRARインバースアゴニストが、4−[[4−(4−エチルフェニル)−2,2−ジメチル−(2H)−チオクロメン−6−イル]−エチニル]−安息香酸(VTP 194310)または薬学的に許容できるその塩である、請求項44に記載の方法。
【請求項46】
前記RARアンタゴニストまたはRARインバースアゴニストが、4−(3−ブロモ−4−エトキシ−5−(4−メチルベンゾイル)ベンズアミド)安息香酸(VTP 196996)、または薬学的に許容できるその塩である、請求項44に記載の方法。
【図1】

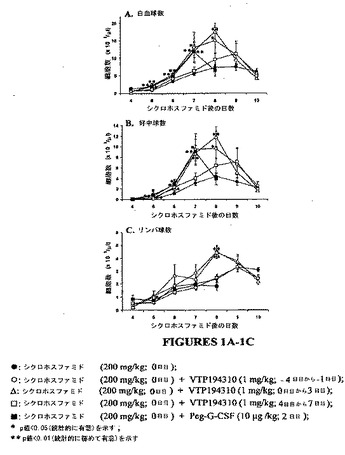
【図2】
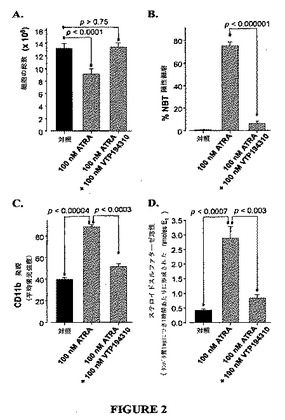
【図3】
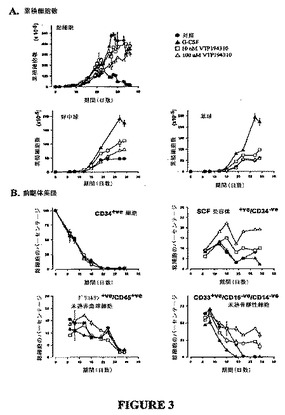
【図4】
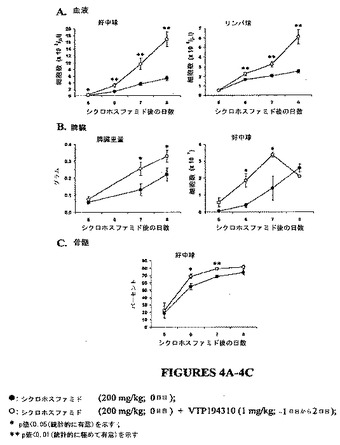
【図5】
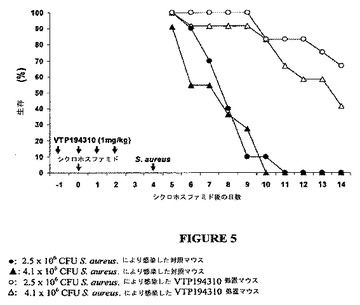
【図6】
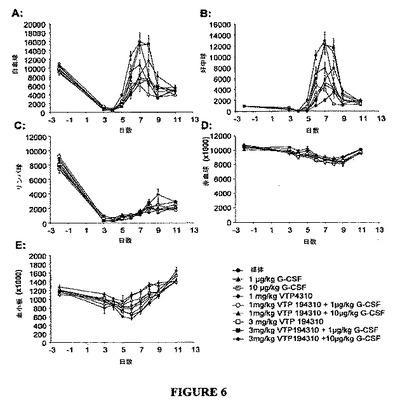
【図7】

【図8】
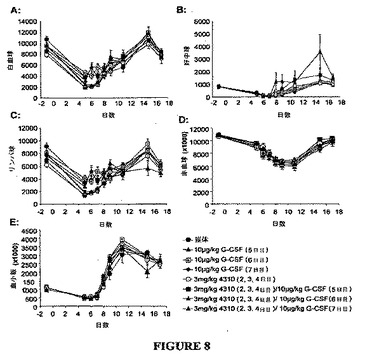
【図9】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公表番号】特表2009−537540(P2009−537540A)
【公表日】平成21年10月29日(2009.10.29)
【国際特許分類】
【出願番号】特願2009−511053(P2009−511053)
【出願日】平成19年5月16日(2007.5.16)
【国際出願番号】PCT/US2007/011730
【国際公開番号】WO2007/136653
【国際公開日】平成19年11月29日(2007.11.29)
【出願人】(507114738)ビテ ファーマシューティカルズ, インコーポレイテッド (11)
【Fターム(参考)】
【公表日】平成21年10月29日(2009.10.29)
【国際特許分類】
【出願日】平成19年5月16日(2007.5.16)
【国際出願番号】PCT/US2007/011730
【国際公開番号】WO2007/136653
【国際公開日】平成19年11月29日(2007.11.29)
【出願人】(507114738)ビテ ファーマシューティカルズ, インコーポレイテッド (11)
【Fターム(参考)】
[ Back to top ]