
化学的方法
【課題】抗腫瘍性を有するある種のキナゾリン誘導体またはそれらの薬学的に許容しうる塩の製造において有用な改善された化学的方法および中間体を提供する。
【解決手段】抗腫瘍性を有するある種のキナゾリン誘導体が化合物4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン(AZD0530中の活性物質)で有り、更にジフマレート塩および三水和物などのその化合物の具体的な結晶形、およびこのような結晶形を含有する医薬組成物である。
【解決手段】抗腫瘍性を有するある種のキナゾリン誘導体が化合物4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン(AZD0530中の活性物質)で有り、更にジフマレート塩および三水和物などのその化合物の具体的な結晶形、およびこのような結晶形を含有する医薬組成物である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、抗腫瘍性を有するある種のキナゾリン誘導体またはそれらの薬学的に許容しうる塩の製造において有用な改善された化学的方法および中間体に関する。本発明は、更に、それら中間体の製造方法、およびそれら中間体を利用したこのようなキナゾリン誘導体の製造方法に関する。本発明は、更に、抗腫瘍性を各々有する、ある種のキナゾリン誘導体の特定の結晶形およびそれらの特定の結晶性の薬学的に許容しうる塩に関する。
【背景技術】
【0002】
具体的には、本発明は、化合物4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンの製造に有用な化学的方法および中間体に関するが、この化合物は、国際特許出願WO01/94341号の実施例14の表中に化合物番号73として開示されている。
【0003】
その化合物は、本明細書中に、式I
【0004】
【化1】

【0005】
によって、および化合物が知られているコード番号AZD0530として記載されている。
AZD0530は、非受容体チロシンキナーゼ酵素のSrcファミリーの阻害剤であり、したがって、腫瘍細胞の運動性の選択的阻害剤および哺乳動物癌細胞の播種および浸潤性の選択的阻害剤であって、転移性腫瘍成長の阻害をもたらす。具体的には、化合物AZD0530は、c−Src非受容体チロシンキナーゼの阻害剤であるので、ヒトまたは動物体の充実性腫瘍疾患の封じ込めおよび/または処置に用いるための抗浸潤薬として価値があるはずである。
【0006】
国際特許出願WO01/94341号に開示されている式Iの化合物を製造する経路は、化合物4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−ヒドロキシ−5−テトラヒドロピラン−4−イルオキシキナゾリンとアルキル化剤との、7位に2−(4−メチルピペラジン−1−イル)エトキシ側鎖を形成する反応を包含する。その反応生成物は、WO01/94341号に、二塩酸塩の形でおよび遊離塩基の形で開示されている。
【0007】
この既存の経路は、比較的少量の式Iの化合物の合成には十分であるが、その経路は、収束的よりもむしろ直線的な合成を包含し、多数のクロマトグラフィー精製工程の使用およびかなり多数の中間体の単離を必要とする。それだけでは、全体の合成収率は高くない。したがって、一層多量の式Iの化合物を製造するのに用いるのに適する一層有効なその
化合物の合成が要求されている。好ましくは、新しい合成は、高価で且つ時間のかかるクロマトグラフィー精製法を包含すべきではない。
【発明の開示】
【0008】
本発明により、本発明者は、ここで、式Iの化合物であるAZD0530の製造に適する方法を発明した。それら新しい方法は、最終生成物を高品質で且つ十分な収率で一層大規模に製造することを可能にするという点で好都合である。それら方法は、単離されるべき中間体の数の実質的な減少を可能にし、そして概して、従来の経路より収束的である。このような変化は、時間および費用について有意の利点を与える。好都合には、クロマトグラフィー精製法を必要としない。
【0009】
本発明により、更に、AZD0530の製造に用いることができる不可欠な中間体の製造方法を提供する。
本発明のもう一つの側面により、更に、抗腫瘍性を各々有する、AZD0530の特定の結晶形およびそれらの特定の結晶性の薬学的に許容しうる塩を提供する。
【0010】
本発明のもう一つの側面により、更に、式I
【0011】
【化2】
【0012】
を有する化合物であるAZD0530の製造方法であって、式II
【0013】
【化3】
【0014】
(式中、Lは、置換可能な基であり、そしてNH官能基は、必要ならば保護されている)を有するキナゾリンと、1−(2−ヒドロキシエチル)−4−メチルピペラジンとの、好都合には、適する塩基の存在下における反応を含み;その後、存在するいずれの保護基も、慣用的な手段によって除去し;そしてその後、必要ならば、遊離塩基の形で得られた式Iの化合物は、薬学的に許容しうる塩へと変換することができるし、塩の形で得られた式Iの化合物は、遊離塩基へと変換することができる方法を提供する。
【0015】
その反応は、好都合には、適する塩基、例えば、有機アミン塩基、例えば、ピリジン、
2,6−ルチジン、コリジン、4−ジメチルアミノピリジン、トリエチルアミン、N−メチルモルホリン、N−メチルモルホリンまたはジアザビシクロ[5.4.0]ウンデカ−7−エンなど;または例えば、アルカリ金属またはアルカリ土類金属の炭酸塩または水酸化物、例えば、炭酸ナトリウム、炭酸カリウム、炭酸カルシウム、水酸化ナトリウムまたは水酸化カリウム;または例えば、アルカリ金属アミド、例えば、ナトリウムヘキサメチルジシラザン;または例えば、アルカリ金属水素化物、例えば、水素化ナトリウム;または例えば、アルカリ金属またはアルカリ土類金属の(1−12C)アルコキシド、例えば、ナトリウムまたはカリウム tert−ブトキシド、ナトリウムまたはカリウム tert−ペントキシド、またはナトリウムまたはカリウム3,7−ジメチルオクトキシドの存在下で行うことができる。好都合には、適する塩基は、例えば、アルカリ金属水酸化物、例えば、水酸化ナトリウムまたは水酸化カリウム;または例えば、アルカリ金属(1−6C)アルコキシド、例えば、ナトリウムまたはカリウム tert−ブトキシド、またはナトリウムま
たはカリウム tert−ペントキシドである。より好都合には、適する塩基は、例えば、ア
ルカリ金属(1−6C)アルコキシド、例えば、ナトリウムまたはカリウム tert−ブト
キシド、またはナトリウムまたはカリウム tert−ペントキシドである。
【0016】
適する置換可能な基Lは、例えば、ハロゲノ基、(1−6C)アルコキシ基、アリールオキシ基またはスルホニルオキシ基、例えば、フルオロ基、クロロ基、ブロモ基、メトキシ基、エトキシ基、フェノキシ基、ペンタフルオロフェノキシ基、メタンスルホニルオキシ基またはトルエン−4−スルホニルオキシ基である。好都合には、置換可能な基Lは、ハロゲノ基である。より好都合には、置換可能な基Lは、フルオロ基である。
【0017】
その反応は、好都合には、適する不活性溶媒または希釈剤、または適する不活性溶媒または希釈剤の混合物の存在下で、例えば、ジブチルエーテル、メチル tert−ブチルエー
テル、ジ−(2−メトキシエチル)エーテル、1,2−ジメトキシエタン、1,2−ジエトキシエタン、テトラヒドロフランまたは1,4−ジオキサンなどの置換されていてよいジ−(1−6C)アルキルエーテルまたは環状アルキルエーテル;またはN,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリジン−2−オンまたはジメチルスルホキシドなどの双極性非プロトン性溶媒中で行われる。好都合には、50℃を超える沸点を有する適する不活性溶媒または希釈剤、例えば、ジ−(2−メトキシエチル)エーテルまたは1,2−ジエトキシエタンなどの置換されていてよいジ−(1−6C)アルキルエーテルを用いる。
【0018】
反応は、例えば、0〜250℃の範囲内、好都合には、50〜150℃の範囲内、より好都合には、75〜130℃の範囲内の温度で行われる。
好都合には、NH官能基を保護する必要はない。しかしながら、保護基を用いることが望まれる場合、このような基は、概して、問題の基の保護について適宜、参考文献に記載されているまたは当該化学者に知られているいずれかの基より選択することができるし、慣用法によって導入することができる。保護基は、問題の保護基の除去について適宜、参考文献に記載されているまたは当該化学者に知られているいずれかの好都合な方法によって除去することができるし、このような方法は、分子中のどこか他の基の妨害を最小限にして保護基の除去を行うように選択される。
【0019】
保護基の具体的な例を、便宜上、下に与えるが、ここにおいて、「低級」は、例えば、低級アルキルの場合のように、それが付けられている基が、好ましくは、1〜4個の炭素原子を有するということを意味する。これら例が、完全なものではないということは理解されるであろう。保護基の除去方法の具体例が下に与えられている場合、これらは、同様に完全なものではない。具体的に述べられていない保護基および脱保護方法の使用は、当然ながら、本発明の範囲内である。
【0020】
NH官能基のための保護基の例には、ホルミル基、アリール−低級アルキル基(例えば、ベンジル、および4−メトキシベンジル、2−ニトロベンジルおよび2,4−ジメトキシベンジルなどの置換ベンジル、およびトリフェニルメチル);ジ−4−アニシルメチル基およびフリルメチル基;低級アルコキシカルボニル基(例えば、tert−ブトキシカルボニル);低級アルケニルオキシカルボニル基(例えば、アリルオキシカルボニル);アリール−低級アルコキシカルボニル基(例えば、ベンジルオキシカルボニル、4−メトキシベンジルオキシカルボニル、2−ニトロベンジルオキシカルボニルおよび4−ニトロベンジルオキシカルボニル);トリアルキルシリル基(例えば、トリメチルシリルおよび tert−ブチルジメチルシリル);アルキリデン基(例えば、メチリデン)およびベンジリデ
ン基および置換ベンジリデン基が含まれる。
【0021】
NH官能基のための保護基の除去に適当な方法には、例えば、2−ニトロベンジルオキシカルボニルなどの基の酸、塩基、金属または酵素に触媒された加水分解;ベンジルなどの基の水素化;および2−ニトロベンジルオキシカルボニルなどの基の光分解が含まれる。
【0022】
読者は、反応条件および試薬の一般的な指針については、Advanced Organic Chemistry, 4th Edition, by J. March, published by John Wiley & Sons 1992 を、そして保護基の一般的な指針については、Protective Groups in Organic Synthesis, 2nd Edition, by T. Green et al., also published by John Wiley & Son を参照する。
【0023】
式Iの化合物は、この方法から遊離塩基の形で得ることができるし、または或いは、ハロゲン化水素酸塩などの酸付加塩の形で得ることができる。塩から遊離塩基を得ることが望まれる場合、その塩は、適する塩基、例えば、有機アミン塩基、例えば、ピリジン、2,6−ルチジン、コリジン、4−ジメチルアミノピリジン、トリエチルアミン、N−メチルモルホリンまたはジアザビシクロ[5.4.0]ウンデカ−7−エンなど;または例えば、アルカリ金属またはアルカリ土類金属の炭酸塩または水酸化物、例えば、炭酸ナトリウム、炭酸カリウム、炭酸カルシウム、水酸化ナトリウムまたは水酸化カリウムで処理することができる。式Iの化合物を薬学的に許容しうる塩の形で得ることが望まれる場合、その遊離塩基形を、慣用法を用いて適する酸と反応させて、例えば、塩酸、臭化水素酸、硫酸、トリフルオロ酢酸、クエン酸またはマレイン酸などの無機酸または有機酸との酸付加塩を形成することができる。
【0024】
Lが、本明細書中に前に定義の置換可能な基である式IIのキナゾリン出発物質は、国際特許出願WO01/94341号に開示されたものなどの慣用法によって得ることができる。具体的には、Lがフルオロ基である式IIのキナゾリン出発物質は、国際特許出願WO01/94341号に、例えば、実施例4の表中の化合物番号5の製造について開示されたものなどの慣用法によって得ることができる。
【0025】
本発明のもう一つの特徴により、式III
【0026】
【化4】
【0027】
を有するキナゾリンの製造方法であって、
(a)式IV
【0028】
【化5】
【0029】
を有するキナゾリノンと活性化剤との、好都合には、適する塩基の存在下における、式V
【0030】
【化6】
【0031】
(式中、L1は置換可能な基である)
を有するキナゾリンを形成する反応;
(b)式Vのキナゾリンと、6−クロロ−2,3−メチレンジオキシアニリンとの、好都合には、適する塩基の存在下における、式VI
【0032】
【化7】
【0033】
を有するキナゾリノンを形成する置換反応であって、その後、遊離塩基の形で得られた式
VIの化合物は、塩へと変換することができるし、塩の形で得られた式VIの化合物は、遊離塩基へと変換することができる置換反応;および
(c)式VIのキナゾリンと、4−ヒドロキシテトラヒドロピランとの、好都合には、適する塩基の存在下における、式IIIのキナゾリンを形成する反応
を含み;その後、遊離塩基の形で得られた式IIIの化合物は、塩へと変換することができ
るし、塩の形で得られた式IIIの化合物は、遊離塩基へと変換することができる方法を提
供する。
【0034】
作業工程(a)について、脱離基L1を形成するであろう適する活性化剤は、例えば、塩化ホスホリルまたは臭化ホスホリルなどのハロゲン化ホスホリル;または塩化チオニルなどのハロゲン化剤;または四塩化炭素およびトリフェニルホスフィンの混合物によって形成されるハロゲン化剤;または四塩化炭素およびトリフェニルホスフィンの混合物によって形成されるハロゲン化剤である。或いは、そのようにして得られるいずれかの4−ハロキナゾリンは、必要ならば、ペンタフルオロフェノールとの反応により、炭酸カリウムなどの適する塩基の存在下およびN,N−ジメチルホルムアミドなどの適する溶媒の存在下において4−ペンタフルオロフェノキシキナゾリンへと変換することができる。作業工程(a)の際に用いることができる適する塩基は、例えば、有機アミン塩基、例えば、ピリジン、2,6−ルチジン、コリジン、4−ジメチルアミノピリジン、トリエチルアミン、ジイソプロピルエチルアミン、N−メチルモルホリンまたはジアザビシクロ[5.4.0]ウンデカ−7−エンなどである。作業工程(a)に適する溶媒または希釈剤は、例えば、トルエン、キシレン、クメン、クロロベンゼン、アニソールまたはフェネトールなどの芳香族溶媒である。更に適する溶媒または希釈剤は、アセトニトリル、プロピオニトリル、ブチロニトリル、酢酸エチル、テトラヒドロフランまたは1,4−ジオキサンなどの極性非プロトン性溶媒;またはN,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリジン−2−オンまたはジメチルスルホキシドなどの双極性非プロトン性溶媒である。更に適する溶媒または希釈剤は、水;または第一級、第二級または第三級の(1−6C)アルキルアルコールなどの極性プロトン性溶媒、例えば、メタノール、エタノール、ブタノールまたはペンタノールである。このような適する溶媒または希釈剤の混合物を用いることができる。その反応は、例えば、10〜250℃の範囲内、好都合には、40〜160℃の範囲内の温度で行うことができる。
【0035】
好都合には、作業工程(a)について、適する活性化剤は、例えば、塩化ホスホリルなどのハロゲン化ホスホリルであり、そして反応は、トリエチルアミンまたはジイソプロピルエチルアミンなどの有機アミン塩基の存在下において、トルエン、クロロベンゼン、アニソールまたはアセトニトリルなどの溶媒または希釈剤を用いて、70〜160℃の範囲内、より好都合には、70〜120℃の範囲内の温度で行われる。
【0036】
作業工程(b)の置換反応は、適する酸の存在下または適する塩基の存在下で行うことができる。適する酸は、例えば、無機酸、例えば、塩化水素または臭化水素などである。適する塩基は、例えば、有機アミン塩基、例えば、ピリジン、2,6−ルチジン、コリジン、4−ジメチルアミノピリジン、トリエチルアミン、ジイソプロピルエチルアミン、N−メチルモルホリンまたはジアザビシクロ[5.4.0]ウンデカ−7−エンなど;または例えば、アルカリ金属またはアルカリ土類金属の炭酸塩または水酸化物、例えば、炭酸ナトリウム、炭酸カリウム、炭酸カルシウム、水酸化ナトリウムまたは水酸化カリウム;または例えば、アルカリ金属水素化物、例えば、水素化ナトリウムである。
【0037】
その置換反応は、好都合には、適する溶媒または希釈剤、例えば、イソプロパノール、sec−ブタノールまたは tert−ブタノールなどの第一級、第二級または第三級の(1−6C)アルキルアルコール;塩化メチレン、クロロホルムまたは四塩化炭素などのハロゲン化溶媒;トルエン、キシレン、クメン、クロロベンゼン、アニソールまたはフェネトール
などの芳香族溶媒;アセトニトリル、プロピオニトリル、ブチロニトリル、酢酸エチル、テトラヒドロフランまたは1,4−ジオキサンなどの極性非プロトン性溶媒;またはN,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリジン−2−オンまたはジメチルスルホキシドなどの双極性非プロトン性溶媒の存在下で行われる。このような適する溶媒または希釈剤の混合物を用いることができる。その反応は、好都合には、例えば、10〜250℃の範囲内、好適には、40〜160℃の範囲内、より好都合には、70〜120℃の範囲内の温度で行われる。
【0038】
典型的に、作業工程(b)の置換反応は、イソプロパノールなどのプロトン性溶媒の存在下において、例えば、25〜150℃の範囲内の温度、好都合には、反応溶媒の還流温度でまたはその付近の温度で行うことができる。場合により、その置換反応は、酸の存在下、例えば、ジエチルエーテル中の塩化水素ガス;または式IVの化合物を、塩化チオニルまたは塩化ホスホリルなどのハロゲン化剤である活性化剤と反応させた場合に形成される塩化水素の存在下において行うことができる。
【0039】
作業工程(c)について、反応は、好都合には、適する塩基、例えば、有機アミン塩基、例えば、ピリジン、2,6−ルチジン、コリジン、4−ジメチルアミノピリジン、トリエチルアミン、N−メチルモルホリンまたはジアザビシクロ[5.4.0]ウンデカ−7−エンなど;または例えば、アルカリ金属またはアルカリ土類金属の炭酸塩または水酸化物、例えば、炭酸ナトリウム、炭酸カリウム、炭酸カルシウム、水酸化ナトリウムまたは水酸化カリウム;または例えば、アルカリ金属アミド、例えば、ナトリウムヘキサメチルジシラザン;または例えば、アルカリ金属水素化物、例えば、水素化ナトリウム;または例えば、アルカリ金属またはアルカリ土類金属の(1−12C)アルコキシド、例えば、ナトリウムまたはカリウム tert−ブトキシド、ナトリウムまたはカリウム tert−ペントキシド、またはナトリウムまたはカリウム3,7−ジメチルオクトキシドの存在下で行うことができる。好都合には、適する塩基は、例えば、アルカリ金属水酸化物、例えば、水酸化ナトリウムまたは水酸化カリウム;または例えば、アルカリ金属(1−6C)アルコキシド、例えば、ナトリウムまたはカリウム tert−ブトキシド、ナトリウムまたはカリ
ウム tert−ペントキシドである。より好都合には、適する塩基は、例えば、アルカリ金
属(1−6C)アルコキシド、例えば、ナトリウムまたはカリウム tert−ブトキシド、
またはナトリウムまたはカリウム tert−ペントキシドである。
【0040】
作業工程(c)について、反応は、好都合には、適する不活性溶媒または希釈剤、または適する不活性溶媒または希釈剤の混合物の存在下で、例えば、ジブチルエーテル、メチル tert−ブチルエーテル、ジ−(2−メトキシエチル)エーテル、1,2−ジメトキシ
エタン、1,2−ジエトキシエタン、テトラヒドロフランまたは1,4−ジオキサンなどの置換されていてよいジ−(1−6C)アルキルエーテルまたは環状アルキルエーテル;またはN,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリジン−2−オンまたはジメチルスルホキシドなどの双極性非プロトン性溶媒中で行われる。好都合には、50℃を超える沸点を有する適する不活性溶媒または希釈剤、例えば、テトラヒドロフランなどの環状アルキルエーテル;またはN−メチルピロリジン−2−オンなどの双極性非プロトン性溶媒を用いる。
【0041】
作業工程(c)について、反応は、例えば、0〜250℃の範囲内、好都合には、25〜125℃の範囲内、より好都合には、40〜80℃の範囲内の温度で行われる。
より好都合には、式Vの中間体は、そのままで単離しないが、有機溶媒中の溶液またはスラリーとして用いる。それによって、式VIの化合物は、式IVの化合物からワンポット法で製造することができる。この方式での式IVの化合物の式VIの化合物への変換は、以下の実施例5に詳しく説明されている。より一層好都合には、式Vの中間体は、6−クロロ−2,3−メチレンジオキシアニリンの存在下で形成され、それてワンポット法において直
接的に反応する。この方式での式IVの化合物の式VIの化合物への変換は、以下の実施例7に詳しく説明されている。
【0042】
式VIのキナゾリンは、本発明のもう一つの側面を形成する新規な化合物である。
本発明のもう一つの側面により、AZD0530の製造方法であって、式VIのキナゾリンを製造する直ぐ上の工程(a)および工程(b)と、本明細書中の前に定義のようなAZD0530へのその変換を含む方法を提供する。
【0043】
更に、式VIのキナゾリンの別の製造方法であって、
(a)式VII
【0044】
【化8】
【0045】
を有する2,4,6−トリフルオロベンゾニトリルと、6−クロロ−2,3−メチレンジオキシアニリンとの、好都合には、適する有機金属触媒の存在下における、式VIII
【0046】
【化9】
【0047】
を有するアミジンを形成する反応、および
(b)式VIIIのアミジンと、ホルムアミジンまたはその塩との、式VIのキナゾリノンを形成する反応
を含み;その後、遊離塩基の形で得られた式VIの化合物は、塩へと変換することができるし、塩の形で得られた式VIの化合物は、遊離塩基へと変換することができる方法を提供する。
【0048】
直ぐ上の作業工程(a)について、適する有機金属試薬は、例えば、トリメチルアルミニウムなどの有機アルミニウム化合物;ジフェニルホスフィノフェロセンなどの有機鉄化合物;またはテトラキス(トリフェニルホスフィン)パラジウム(0)などの有機パラジウム化合物である。その反応は、好都合には、本明細書中の前に定義の適する不活性溶媒または希釈剤の存在下で行われる。好都合には、トルエンまたはキシレン、クメンまたはクロロベンゼンなどの芳香族溶媒を、反応溶媒として用いる。その反応は、好都合には、例えば、10〜250℃の範囲内、好適には、75〜125℃の範囲内の温度で行われる。
【0049】
直ぐ上の作業工程(b)について、反応は、好都合には、本明細書中の前に定義の適する不活性溶媒または希釈剤の存在下で、例えば、トルエンまたはキシレン、クメン、クロロベンゼン、アニソールまたはフェネトールなどの芳香族溶媒中において、例えば、10〜250℃の範囲内、好適には、75〜125℃の範囲内の温度で行われる。
【0050】
この方式での式VIIの化合物の式VIの化合物への変換は、以下の実施例6に詳しく説明
されている。
更に、式III
【0051】
【化10】
【0052】
を有するキナゾリンの別の製造方法であって、
(a)式IV
【0053】
【化11】
【0054】
を有するキナゾリノンと、4−ヒドロキシテトラヒドロピランとの、好都合には、本明細書中の前に定義の適する塩基の存在下における、式IX
【0055】
【化12】
【0056】
を有するキナゾリンを形成する反応であって、その後、遊離塩基の形で得られた式IXの化合物は、塩へと変換することができるし、塩の形で得られた式IXの化合物は、遊離塩基へと変換することができる反応;
(b)式IXのキナゾリンと、本明細書中の前に定義の活性化剤との、好都合には、適する塩基の存在下における、式X
【0057】
【化13】
【0058】
(式中、L1は、本明細書中の前に定義の置換可能な基である)
を有するキナゾリノンを形成する反応;および
(c)式Xのキナゾリンと、6−クロロ−2,3−メチレンジオキシアニリンとの、好都合には、適する塩基の存在下における置換反応
を含み;その後、遊離塩基の形で得られた式IIIの化合物は、塩へと変換することができ
るし、塩の形で得られた式IIIの化合物は、遊離塩基へと変換することができる方法を提
供する。
【0059】
直ぐ上の作業工程(a)について、反応は、好都合には、上の作業工程(c)について定義の(式VIのキナゾリンから式IIIのキナゾリンの製造に関する)適する塩基の存在下
、上の作業工程(c)について定義の(式VIのキナゾリンから式IIIのキナゾリンの製造
に関する)適する不活性溶媒または希釈剤、または適する不活性溶媒または希釈剤の混合物の存在下における、例えば、0〜250℃の範囲内、好都合には、25〜125の範囲内、より好都合には、40〜80℃の範囲内の温度で行うことができる。
【0060】
直ぐ上の作業工程(b)について、脱離基L1を形成するであろう適する活性化剤は、例えば、塩化ホスホリルまたは臭化ホスホリルなどのハロゲン化ホスホリル;または塩化チオニルなどのハロゲン化剤;または四塩化炭素およびトリフェニルホスフィンの混合物によって形成されるハロゲン化剤;または四塩化炭素およびトリフェニルホスフィンの混合物によって形成されるハロゲン化剤である。直ぐ上の作業工程(b)の際に用いることができる適する塩基は、例えば、有機アミン塩基、例えば、ピリジン、2,6−ルチジン、コリジン、4−ジメチルアミノピリジン、トリエチルアミン、ジイソプロピルエチルアミン、N−メチルモルホリンまたはジアザビシクロ[5.4.0]ウンデカ−7−エンなどである。直ぐ上の作業工程(b)に適する溶媒または希釈剤は、例えば、トルエン、キシレン、クメン、クロロベンゼン、アニソールまたはフェネトールなどの芳香族溶媒である。更に適する溶媒または希釈剤は、アセトニトリル、プロピオニトリル、ブチロニトリル、酢酸エチル、テトラヒドロフランまたは1,4−ジオキサンなどの極性非プロトン性溶媒;またはN,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリジン−2−オンまたはジメチルスルホキシドなどの双極性非プロトン性溶媒である。このような適する溶媒または希釈剤の混合物を用いることができる。その反応は、例えば、10〜250℃の範囲内、好都合には、40〜120℃の範囲内の温度で行うことができる。
【0061】
好都合には、直ぐ上の作業工程(b)について、適する活性化剤は、例えば、塩化ホスホリルなどのハロゲン化ホスホリルであり、そして反応は、トリエチルアミンまたはジイソプロピルエチルアミンなどの有機アミン塩基の存在下において、トルエン、クロロベンゼンまたはアセトニトリルなどの溶媒または希釈剤を用いて、70〜100℃の範囲内の温度で行われる。
【0062】
直ぐ上の作業工程(c)の置換反応は、適する酸の存在下または適する塩基の存在下で
行うことができる。適する酸は、例えば、無機酸、例えば、塩化水素または臭化水素などである。適する塩基は、例えば、有機アミン塩基、例えば、ピリジン、2,6−ルチジン、コリジン、4−ジメチルアミノピリジン、トリエチルアミン、ジイソプロピルエチルアミン、N−メチルモルホリンまたはジアザビシクロ[5.4.0]ウンデカ−7−エンなど;または例えば、アルカリ金属またはアルカリ土類金属の炭酸塩または水酸化物、例えば、炭酸ナトリウム、炭酸カリウム、炭酸カルシウム、水酸化ナトリウムまたは水酸化カリウム;または例えば、アルカリ金属水素化物、例えば、水素化ナトリウムである。
【0063】
直ぐ上の作業工程(c)の置換反応は、好都合には、適する溶媒または希釈剤、例えば、イソプロパノール、sec−ブタノールまたは tert−ブタノールなどの第一級、第二級または第三級の(1−6C)アルキルアルコール;塩化メチレン、クロロホルムまたは四塩化炭素などのハロゲン化溶媒;トルエン、キシレン、クメン、クロロベンゼン、アニソールまたはフェネトールなどの芳香族溶媒;アセトニトリル、プロピオニトリル、ブチロニトリル、酢酸エチル、テトラヒドロフランまたは1,4−ジオキサンなどの極性非プロトン性溶媒;またはN,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリジン−2−オンまたはジメチルスルホキシドなどの双極性非プロトン性溶媒の存在下で行われる。このような適する溶媒または希釈剤の混合物を用いることができる。その反応は、好都合には、例えば、10〜250℃の範囲内、好適には、40〜120℃の範囲内の温度で行われる。
【0064】
好都合には、式Xの中間体は、そのままで単離しないが、有機溶媒中の溶液またはスラリーとして用いる。それによって、式IIIの化合物は、式IXの化合物からワンポット法で
製造することができる。この方式での式IXの化合物の式IIIの化合物への変換は、以下の
実施例8に詳しく説明されている。
【0065】
本発明のもう一つの側面により、AZD0530の製造方法であって、式IXのキナゾリンを製造する直ぐ上の工程(a)と、本明細書中の前に定義のようなAZD0530へのその変換を含む方法を提供する。
【0066】
式IVのキナゾリノンのような必要な出発物質は、有機化学の標準法によって得ることができる。式IVのキナゾリノンの製造は、以下の代表的な実施例中に記載されている(実施例1および実施例2)。或いは、このような必要な出発物質は、当該化学者の常套技術の範囲内である、示されているのに類似した手順によって入手可能である。
【0067】
本発明のもう一つの側面により、式I
【0068】
【化14】
【0069】
を有する化合物であるAZD0530の製造方法であって、
(a)式XI
【0070】
【化15】
【0071】
を有するキナゾリンと、本明細書中の前に定義の活性化剤との、好都合には、本明細書中の前に定義の適する塩基の存在下における、式XII
【0072】
【化16】
【0073】
(式中、L1は、本明細書中の前に定義の置換可能な基である)
を有するキナゾリノンを形成する反応;および
(b)式XIIのキナゾリンと、6−クロロ−2,3−メチレンジオキシアニリンとの、
好都合には、本明細書中の前に定義の適する塩基の存在下における置換反応
を含み;その後、遊離塩基の形で得られた式Iの化合物は、薬学的に許容しうる塩へと変換することができるし、塩の形で得られた式Iの化合物は、遊離塩基へと変換することができる方法を提供する。
【0074】
好都合には、直ぐ上の作業工程(a)について、適する活性化剤は、例えば、塩化ホスホリルなどのハロゲン化ホスホリルであり、そして反応は、トリエチルアミンまたはジイソプロピルエチルアミンなどの有機アミン塩基の存在下において、ブチロニトリルまたはトルエンなどの溶媒または希釈剤を用いて、70〜120℃の範囲内の温度で行われる。
【0075】
好都合には、直ぐ上の作業工程(b)について、置換反応は、好都合には、ブチロニトリルまたはトルエンなどの適する不活性溶媒または希釈剤の存在下において、70〜120℃の範囲内の温度で行われる。
【0076】
好都合には、式XIIの中間体は、そのままで単離しないが、有機溶媒中の溶液またはス
ラリーとして用いる。それによって、式Iの化合物は、式XIの化合物からワンポット法で製造することができる。この方式での式XIの化合物の式Iの化合物への変換は、以下の実施例11および実施例12に詳しく説明されている。
【0077】
本発明のもう一つの側面により、式XIのキナゾリンの製造方法であって、
(a)式IV
【0078】
【化17】
【0079】
を有するキナゾリノンと、4−ヒドロキシテトラヒドロピランとの、好都合には、本明細書中の前に定義の適する塩基の存在下における、式IX
【0080】
【化18】
【0081】
を有するキナゾリンを形成する反応であって、その後、遊離塩基の形で得られた式IXの化合物は、塩へと変換することができるし、塩の形で得られた式IXの化合物は、遊離塩基へと変換することができる反応;および
(b)NH官能基が、必要ならば保護されている式IXのキナゾリンと、1−(2−ヒドロキシエチル)−4−メチルピペラジンとの、好都合には、本明細書中の前に定義の適する塩基の存在下における反応を含み;その後、存在するいずれの保護基も、慣用的な手段によって除去し;そしてその後、必要ならば、遊離塩基の形で得られた式XIの化合物は、塩へと変換することができるし;塩の形で得られた式XIの化合物は、遊離塩基へと変換することができる方法を提供する。
【0082】
直ぐ上の作業工程(a)について、反応は、好都合には、上の作業工程(c)について定義の(式VIのキナゾリンから式IIIのキナゾリンの製造に関する)適する塩基の存在下
、上の作業工程(c)について定義の(式VIのキナゾリンから式IIIのキナゾリンの製造
に関する)適する不活性溶媒または希釈剤、または適する不活性溶媒または希釈剤の混合物の存在下における、例えば、0〜250℃の範囲内、好都合には、25〜125の範囲内、より好都合には、40〜80℃の範囲内の温度で行うことができる。
【0083】
直ぐ上の作業工程(b)について、適する塩基は、例えば、アルカリ金属(1−6C)アルコキシド、例えば、ナトリウムまたはカリウム tert−ブトキシド、ナトリウムまた
はカリウム tert−ペントキシド、またはナトリウムまたはカリウム3,7−ジメチルオ
クトキシドである。その反応は、好都合には、適する不活性溶媒または希釈剤、または適する不活性溶媒または希釈剤の混合物の存在下で、例えば、ジブチルエーテル、メチル tert−ブチルエーテル、ジ−(2−メトキシエチル)エーテル、1,2−ジメトキシエタ
ン、1,2−ジエトキシエタン、テトラヒドロフランまたは1,4−ジオキサンなどの置換されていてよいジ−(1−6C)アルキルエーテルまたは環状アルキルエーテル;またはN,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリジン−2−オンまたはジメチルスルホキシドなどの双極性非プロトン性溶媒中で行われる。好都合には、50℃を超える沸点を有する適する不活性溶媒または希釈剤、例えば、テトラヒドロフランまたは1,4−ジオキサンなどの環状アルキルエーテル;またはジ−(2
−メトキシエチル)エーテルまたは1,2−ジエトキシエタンなどの置換されていてよいジ−(1−6C)アルキルエーテルを用いる。好都合には、その反応は、例えば、50〜150℃の範囲内の温度で、より好都合には、約70℃で行われる。
【0084】
式XIのキナゾリノンは、本発明のもう一つの側面を形成する新規な化合物である。
AZD0530結晶形
本明細書中の前に述べられたように、AZD0530として現在知られている化合物4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンは、非受容体チロシンキナーゼ酵素のSrcファミリーの阻害剤であり、したがって、腫瘍細胞の運動性の選択的阻害剤および哺乳動物癌細胞の播種および浸潤性の選択的阻害剤であって、転移性腫瘍成長の阻害をもたらす。具体的には、化合物AZD0530は、c−Src非受容体チロシンキナーゼの阻害剤であるので、ヒトまたは動物体の充実性腫瘍疾患の封じ込めおよび/または処置に用いるための抗浸潤薬として価値があるはずである。
【0085】
その化合物は、国際特許出願WO01/94341号の実施例14の表中に化合物番号73として開示された。その化合物は、二塩酸塩の形または遊離塩基の形で得られたということが述べられた。AZD0530の二塩酸塩形およびAZD0530の遊離塩基形の結晶化度は、述べられなかった。
【0086】
国際特許出願WO01/94341号には、そこに開示されたキナゾリン誘導体が、溶媒和した形で、更には、非溶媒和の形で存在しうるということは、特に述べられなかった。具体的には、特定の水和した形のAZD0530は開示されなかった。
【0087】
AZD0530の遊離塩基形についての引き続きの分析は、X線粉末回折分析、示差走査熱量測定または熱重量分析を用いて行った。AZD0530の遊離塩基形は、結晶形および非晶形の混合物であることが確かめられた。熱量測定は、約30〜85℃の広範な吸熱を示した。約65℃に開始し且つピークを約79℃に有する単一の広範な融解吸熱が存在した。重量分析は、約25〜120℃の温度範囲において原試料重量の約10%の損失重量を示した。
【0088】
薬学的に許容しうる塩に関して、国際特許出願WO01/94341号には、その中の式Iの化合物の適する薬学的に許容しうる塩は、例えば、その式Iの化合物の酸付加塩、例えば、塩酸、臭化水素酸、硫酸、トリフルオロ酢酸、クエン酸またはマレイン酸などの無機酸または有機酸との酸付加塩;または例えば、十分に酸性であるその式Iの化合物の塩、例えば、カルシウム塩またはマグネシウム塩などのアルカリ金属またはアルカリ土類金属の塩;またはアンモニウム塩;またはメチルアミン、ジメチルアミン、トリメチルアミン、ピペリジン、モルホリンまたはトリス−(2−ヒドロキシエチル)アミンなどの有機塩基との塩であるということが述べられた。
【0089】
国際特許出願WO01/94341号には、その中の式Iを有するいずれか特定の化合物またはそのいずれか特定の薬学的に許容しうる塩が、結晶性物理的形態のような驚くほど有益な物理的形態を有するということは述べられていない。
【0090】
多数の薬学的に活性な化合物は、製造および/または製剤化の方法中の単離および取扱いに適する物理的形態を有することがない。物理的形態のこのような欠陥を克服する一つの方法は、その適する薬学的に許容しうる塩が存在するか否かを決定することである。物理的形態のこのような欠陥を克服するもう一つの方法は、適する薬学的に許容しうる多形が存在するか否かを決定することである。物理的形態のこのような欠陥を克服するもう一つの方法は、適する形を有する溶媒和化合物または水和物を形成することである。好都合
には、このような形は、適度な融点を有するさらさらした結晶性固体を構成する。
【0091】
本発明者は、ここで、本明細書中の式Iの化合物であるAZD0530のある種の形であって、ある種のその薬学的に許容しうる塩を含めた形が、好都合な性質を有する結晶質であるということを発見した。このような結晶質は、非晶質を実質的に含まない。
【0092】
特定の結晶形の化合物は、いずれか他の結晶形または非晶形のものとは異なる物理的性質を有することがありうるが、このような性質は、特に、その化合物を商業規模で製造するまたは用いる場合に、その化合物の化学的および薬学的加工に顕著に影響を与えることがありうる。例えば、結晶形の化合物は各々、結晶のサイズおよび形状、融点、密度、吸湿性および安定性などの物理的性質の相違を示すことがありうる。このような相違は、化合物の機械的取扱適性(固体材料の流動性など)および化合物の圧縮特性を変更することがありうる。異なった結晶形の化合物は、異なった熱力学的安定性を有することがありうる。概して、より安定な形、例えば、より安定な多形形は、商業規模での製剤化および加工に一層適する物理的形態である。
【0093】
例えば、安定性に乏しい形、例えば、安定性に乏しい多形の加工において、問題が生じることがありうる。錠剤成形方法において用いられるような圧縮力は、若干の安定性に乏しい形をより安定な形へと変換して、製剤化された製品中においてより安定な形の結晶生長を引き起こすことがありうる。これは、いずれかこのような結晶化方法が、錠剤の保全性を破壊して、減少した錠剤強度を有する脆い錠剤を生じることがありうるので、望ましくないことがありうる。更に、二つのこのような形の可変混合物を与えようとした場合、一つまたは双方の活性化合物の溶解速度およびバイオアベイラビリティーは、例えば、各々の形が異なった粒度有することがありうるように変動することがありうる。粒度が、薬学的に活性な化合物の溶解速度およびバイオアベイラビリティーに影響することがありうるということは周知である。したがって、製品の品質は、不都合に影響を受けることがありうるし、投薬への生物学的作用について再現不可能の問題が起こりうる。
【0094】
更に、カプセル剤または錠剤の形での医薬化合物は、化合物の組成が調節されている且つ安定であるという適当な調節根拠を示す必要があるような準安定な形またはそのような形の混合物ではなく、安定な形、例えば、安定な塩または最も安定な多形を用いて製造されることが好ましい。熱力学的に安定性に乏しい形、例えば、安定性に乏しい多形を、錠剤中に単独でまたは熱力学的により安定な形との混合物で与えた場合、その熱力学的により安定な形の量は、貯蔵時に増加する傾向がありうるので、錠剤の組成、例えば、錠剤の多形組成を調節することはきわめて難しいであろう。
【0095】
したがって、これら因子は、固相、すなわち、化合物の錠剤またはカプセル剤に、およびそれらの懸濁製剤に影響力を有することがありうる。
AZD0530の化合物の性質の研究は、結晶性塩および/または結晶性の溶媒和化合物または水和物が形成されうるか否か、および多形現象が起こるか否かを発見するために行った。例えば、次の薬学的に許容しうる酸を、AZD0530のメタノール性溶液に個々に加えて、いずれの結晶性塩が形成されるか否かを確かめた(塩酸、クエン酸、マレイン酸、コハク酸、リンゴ酸、アジピン酸、マロン酸、4−トルエンスルホン酸、メタンスルホン酸、サリチル酸、酒石酸、アスコルビン酸、フマル酸、グリコール酸およびリン酸)。
【0096】
本発明者は、ここで、驚くべきことに、化合物の医薬加工に価値があるように十分に安定であり且つ結晶性であるAZD0530の薬学的に許容しうる塩および/または溶媒和した形が、比較的少数しか存在しないということを発見した。具体的には、AZD0530結晶性塩形成は、最初は、リンゴ酸、メタンスルホン酸、フマル酸およびリン酸でのみ
示された。引き続きの研究は、リン酸との塩が非晶質であるということを示した。本発明者は、ここで、フマル酸で形成された塩が、好ましい性質を有するということを発見した。
【0097】
AZD0530の一つまたはそれを超える特定の結晶形の試料を、X線粉末回折(以下、XRPD)分析、示差走査熱量測定(以下、DSC)、熱重量分析(以下、TGA)、拡散反射赤外フーリエ変換(DRIFT)分光法、近赤外(NIR)分光法、溶液および/または固相核磁気共鳴分光法、および/またはカール・フィッシャー分析による水分定量法の組合せを用いて分析した。
【0098】
ジフマレート塩結晶形
本発明者は、AZD0530およびフマル酸が、以下、AZD0530ジフマレートと称されるジフマル酸塩の形で結晶性塩を形成するということを発見した。AZD0530ジフマレート塩は、それが、容易に単離されるし且つ十分に安定でもあって、商業規模において高レベルの純度および高収率で容易に製造することができる結晶性物理的形態を有するという点で異例である。
【0099】
本発明のこの側面により、実質的に均一な結晶形の、実質的にジフマレート塩の形の式Iの化合物である4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン(AZD0530ジフマレート)を提供する。
【0100】
本発明が、実質的に均一な結晶形の式Iの化合物に関すると述べられている場合、(XRPD手段によって決定することができる)結晶化度は、好都合には、約60%を超える、より好都合には、約80%を超える、好ましくは、約90%を超える、より好ましくは、約95%を超える。
【0101】
本発明のこの側面が、AZD0530ジフマレートに関すると述べられている場合、各々のAZD0530分子対各々のフマル酸分子のモル比は、1:1.7〜1:2.5の範囲内、好都合には、1:1.8〜1:2.3の範囲内、より好都合には、1:1.9〜1:2.1の範囲内にあり、好ましくは、約1当量のAZD0530対約2当量のフマル酸を有する。
【0102】
AZD0530ジフマレートは、式Iを有する安定な形の化合物である。具体的には、AZD0530ジフマレートは、実質的に非吸湿性であり、したがって、非晶形のAZD0530とは異なり、水蒸気に暴露されたとしても、貯蔵中に容易に形態変化することはない。このような形態変化はいずれも、熱力学的に安定性に乏しい形の熱力学的により安定な形への変換が、溶解速度の減少を引き起こすことがありうることから、問題でありうる。式Iを有する二つのこのような形の化合物の可変混合物を与えようとした場合、一つまたは双方の活性化合物の溶解速度およびバイオアベイラビリティーは、それら二つの形の異なった特性の結果として変動することがありうる。
【0103】
AZD0530ジフマレートは、結晶のサイズおよび形状、融点、密度および吸湿性などの、式Iを有する他の既知の形の化合物と比較した場合に異なる他の物理的性質を示す。このような相違は、製造の際に、改善された固体材料流動性および/または改善された濾過のような、好都合な化合物取扱適性を提供することができる。このような利点は、商業規模での式Iの化合物の改善された製剤化および加工を提供することができる。具体的には、AZD0530ジフマレートの晶癖は、好都合な濾過特性を有する材料を提供する。
【0104】
更に、AZD0530ジフマレートは、商業規模において高レベルの純度および高収率で容易に製造することができる。
AZD0530ジフマレートは、実質的には以下の図1に示されるような、下の表1に示される2θスケール上のピーク(最初の4個のピークと、より強い他のピークの内の6個を挙げている)を包含するX線回折図形を有する。
【0105】
【表1】
【0106】
具体的には、表1中の約7.1°、9.1°および10.6°にある一つまたはそれを超えるピークは、AZD0530ジフマレートについて顕著であると考えられる。
以下に述べられるように、XRPDスペクトル中のピーク位置の測定誤差は、約±0.3°2θであろう。このような程度の測定誤差は、XRPDスペクトルが同じ物理的形態に由来したか否かを評価する場合に考慮されるべきである。当業者は、個々のピーク位置よりもむしろ、それらピークの相対位置が、AZD0530ジフマレートの試料が実質的に同じであるか否かについてのより信頼できる指標であるということを理解するであろう。
【0107】
以下に述べられるように、XRPDディフラクトグラム中のピークの強さは、用いられる測定条件に依存して、若干の変動性を示すこともありうる。したがって、表1においておよび以下に引用されるように、相対強さは、数字で記述されていない。その代わり、強さについて次の定義を用いる。
【0108】
【表2】
【0109】
AZD0530ジフマレートのDSCサーモグラム分析は、その塩が、約231〜240℃の範囲内の融点を有する;言い換えると、融解の開始は約231℃であり、融点ピークは約237℃であるということを示した。具体的には、融点は、約233〜239℃の範囲内である。より具体的には、融点は、約234〜238℃の範囲内である。より具体的には、融点は、約237℃である。典型的に、DSC分析は、AZD0530ジフマレートが、融解開始を約235℃におよび融点ピークを約237℃に有する高融点固体であるということを示している。
【0110】
AZD0530ジフマレートのDRIFT分光法トレースは、以下の図5に示されるが、それは、約3359cm−1(N−H)、3100〜2700cm−1、1719cm−1(C=O)、1662cm−1、1616cm−1、1586cm−1、1523cm−1、1501cm−1、1360〜1200cm−1、1200〜1000cm−1および979cm−1にあるピークを包含する。具体的には、約3359cm−1および1719cm−1にある一つまたは双方のピークは、AZD0530ジフマレートについて顕著であると考えられる。
【0111】
非晶形のAZD0530ジフマレートは、その材料の試料をグラインダー中に入れ且つ約10分間またはそれを超えて粉砕した場合に得ることができる。粉砕された材料の非晶性は、XRPDスペクトル中の明瞭なピークの不存在によって示された。
【0112】
セスキフマレート塩結晶形
本発明者は、更に、AZD0530ジフマレートを水中でスラリーにした場合、またはAZD0530フマル酸塩製造の際に2当量未満のフマル酸を用いた場合、より少ないフマル酸含量を有するAZD0530フマル酸塩が形成されるということを発見した。本発明者は、このような塩が、結晶水を収容するのに十分な空間を有する結晶格子を形成するということに注目した。それによって、かなり均一な結晶性塩を、2〜6当量の水を含有するセスキフマル酸塩の形で得ることができる。
【0113】
具体的には、実質的に均一な結晶性塩を、以下、AZD0530セスキフマレート四水和物と称されるセスキフマル酸塩四水和物の形で得ることができる。AZD0530セスキフマレート四水和物は、単離可能であり且つ適度な安定性を有する結晶性物理的形態を有する。
【0114】
この実質的に均一な結晶形の(XRPD手段によって決定することができる)結晶化度は、好都合には、約60%を超える、より好都合には、約80%を超える、好ましくは、約90%を超える。最も好ましくは、結晶化度は、約95%を超える。
【0115】
AZD0530セスキフマレート四水和物の場合、各々のAZD0530分子対各々のフマル酸分子のモル比は、1:1.3〜1:1.7の範囲内、好都合には、1:1.4〜1:1.6の範囲内にあり、より好都合には、約1当量のAZD0530対約1.5当量のフマル酸を有する。
【0116】
AZD0530セスキフマレート四水和物の場合、各々のAZD0530分子対各々の水分子のモル比は、1:3.5〜1:4.5の範囲内、好都合には、1:3.7〜1:4.3の範囲内にあり、より好都合には、約1当量のAZD0530対約4当量の水を有する。
【0117】
AZD0530セスキフマレート四水和物は、実質的には図2に示されるような、下の表2に示される2θスケール上のピーク(最も強いXRPDピークの内の10個を挙げている)を包含するX線回折図形を有する。
【0118】
【表3】
【0119】
具体的には、表2中の約2.8°、10.3°および22.6°にある一つまたはそれを超えるピークは、AZD0530セスキフマレート四水和物に特有であると考えられる。
【0120】
AZD0530セスキフマレート四水和物のDSCサーモグラム分析は、その塩が、約25〜100℃の最初の熱イベントを有するということを示したが、それは、水和水の損失のためであると考えられる。発熱は、AZD0530ジフマレートの結晶化に対応して、約150℃を超えて生じる。引き続きの加熱で、更なる熱イベントは、AZD0530ジフマレートの融点に対応して、約230〜240℃で生じる。
【0121】
AZD0530セスキフマレート四水和物のTGAは、約4当量の水の損失に対応して、約30〜130℃で約8%〜10%の損失重量を示した。
AZD0530セスキフマレート四水和物のDRIFT分光法トレースは、以下の図6に示されるが、それは、約3345cm−1(N−H)、3100〜2700cm−1、1698cm−1(C=O)、1660〜1450cm−1および1400〜1000cm−1にあるピークを包含する。具体的には、約3345cm−1および1698cm−1にある一つまたは双方のピークは、AZD0530セスキフマレート四水和物について顕著であると考えられる。
【0122】
三水和物遊離塩基結晶形
本発明者は、更に、AZD0530を、以下、AZD0530三水和物と称される結晶性三水和物の形で、湿潤している有機溶媒から結晶化させることができるということを発見した。AZD0530三水和物は、それが、容易に単離されるし且つ十分に安定でもあって、商業規模において高レベルの純度および高収率で容易に製造することができる結晶性物理的形態を有するという点で異例である。
【0123】
本発明のこの側面により、実質的に均一な結晶形の、実質的に三水和物の形の式Iの化合物である4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン(AZD0530三水和物)を提供する。
【0124】
本発明が、実質的に均一な三水和物結晶形の式Iの化合物に関すると述べられている場
合、(XRPD手段によって決定することができる)結晶化度は、好都合には、約90%を超える、より好都合には、約95%を超える。
【0125】
本発明のこの側面が、AZD0530三水和物に関すると述べられている場合、各々のAZD0530分子対各々の水分子のモル比は、1:2〜1:4の範囲内、好都合には、1:2.5〜1:3.5の範囲内、より好都合には、1:2.75〜1:3.25の範囲内にあり、好ましくは、約1当量のAZD0530対約3当量の水を有する。
【0126】
AZD0530三水和物は、式Iを有する安定な形の化合物である。具体的には、AZD0530三水和物は、水の存在下で安定である。例えば、AZD0530を、水性懸濁液として製造する場合、得られた懸濁液は安定であるが、式Iを有する他の形の化合物を用いて製造された水性懸濁液は、水和した形のAZD0530へと部分的にまたは完全に変換する傾向がありうる。
【0127】
AZD0530三水和物は、商業規模において高レベルの純度および高収率で容易に製造することができる。更に、AZD0530三水和物は、無水物結晶形のAZD0530へと、およびAZD0530のある種の薬学的に許容しうる塩結晶形へと変換することができる。AZD0530三水和物の製造、その精製および他の結晶形への変換は、収率および純度の点で有益である。
【0128】
AZD0530三水和物は、実質的には以下の図3に示されるような、下の表3に示される2シータ(θ)スケール上のピーク(最も強いXRPDピークの内の10個を挙げている)を包含するX線回折図形を有する。
【0129】
【表4】
【0130】
具体的には、表3中の約16.0°にあるピークは、無水物結晶形と比較して(下を参照されたい)、AZD0530三水和物について顕著であると考えられる。
以下に述べられるように、XRPDスペクトル中のピーク位置の測定誤差は、約±0.3°2θであろう。このような程度の測定誤差は、XRPDスペクトルが同じ物理的形態に由来したか否かを評価する場合に考慮されるべきである。当業者は、個々のピーク位置よりもむしろ、それらピークの相対位置が、AZD0530三水和物の試料が実質的に同じであるか否かについてのより信頼できる指標であるということを理解するであろう。
【0131】
AZD0530三水和物のDSCサーモグラム分析は、約50〜94℃の広範な吸熱を示したが、それは、水の損失のためであると考えられる。その吸熱は、約75℃にピーク
を有する約65℃での開始を示した。
【0132】
AZD0530三水和物のTGAは、約3当量の水の損失に対応して、約30〜110℃で約9%の損失重量を示した。
無水遊離塩基結晶形
本発明者は、更に、AZD0530が、二つの無水物の形で、すなわち、一定の融点を有していない非晶質の非結晶形と、十分に規定された狭い融点を有する高結晶形とで得ることができるということを発見した。本発明者は、AZD0530三水和物を、以下、無水AZD0530と称される実質的に均一な無水物結晶形へと容易に変換することができるということを発見した。したがって、AZD0530三水和物の結晶化およびその後の無水AZD0530への変換は、無水AZD0530を高純度で製造する手段を提供する。無水AZD0530は、それが、容易に単離されるし且つ実質的無水条件下において十分に安定でもあって、商業規模において高レベルの純度および高収率で容易に製造することができる結晶性物理的形態を有するという点で異例である。
【0133】
この実質的に均一な結晶形の(XRPD手段によって決定することができる)結晶化度は、好都合には、約60%を超える、より好都合には、約80%を超える、好ましくは、約90%を超える、そしてより好ましくは、約95%を超える。
【0134】
無水AZD0530は、式Iを有する安定な形の化合物である。具体的には、無水AZD0530は、水の不存在下においてきわめて安定である。しかしながら、無水AZD0530は、実質的無水貯蔵条件が維持されない場合、貯蔵中にAZD0530三水和物へと変換する傾向がある。
【0135】
無水AZD0530は、商業規模において高レベルの純度および高収率で容易に製造することができる。更に、無水AZD0530は、AZD0530のある種の薬学的に許容しうる塩結晶形へと変換することができる。
【0136】
無水AZD0530は、実質的には図4に示されるような、下の表4に示される2θスケール上のピーク(最も強いXRPDピークの内の10個を挙げている)を包含するX線回折図形を有する。
【0137】
【表5】
【0138】
表4中の約15.1°、17.0°および18.0°にある一つまたはそれを超える強いピーク、具体的には、約20.4°および23.3°にある一つまたは双方のきわめて
強いピークは、三水和物形と比較して(上を参照されたい)、無水AZD0530について顕著であると考えられる。
【0139】
以下に述べられるように、XRPDスペクトル中のピーク位置の測定誤差は、約±0.3°2θであろう。このような程度の測定誤差は、XRPDスペクトルが同じ物理的形態に由来したか否かを評価する場合に考慮されるべきである。当業者は、個々のピーク位置よりもむしろ、それらピークの相対位置が、無水AZD0530の試料が実質的に同じであるか否かについてのより信頼できる指標であるということを理解するであろう。
【0140】
無水AZD0530のDSCサーモグラム分析は、約133〜152℃の熱イベントを示した。融解の開始は約142℃であり、約144℃に融点ピークを有した。
式Iの化合物の次の具体的な結晶形は、本明細書中に開示されている。
【0141】
(i)AZD0530ジフマレート;
(ii)AZD0530セスキフマレート四水和物;
(iii)AZD0530三水和物;および
(iv)無水AZD0530。
【0142】
これら物質は各々、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン(AZD0530)などの化合物について国際特許出願WO01/94341号に開示されたものと同じ薬理学的性質を有する。具体的には、これら物質は各々、c−Srcなどの非受容体チロシンキナーゼの阻害剤であり、それは、腫瘍細胞の運動性の選択的阻害および哺乳動物癌細胞の播種および浸潤性の選択的阻害を与えて、転移性腫瘍成長の阻害をもたらす。具体的には、これら物質は各々、充実性腫瘍疾患の封じ込めおよび/または処置に用いるための抗浸潤薬として価値があるはずである。式Iの化合物のこれら結晶形は、以下、集合的に、「本発明の活性物質」と記載される。
【0143】
ヒトを含めた哺乳動物の処置に本発明の活性物質を用いるために、それを、通常は、標準的な医薬慣例にしたがって医薬組成物として製剤化する。
本発明のもう一つの側面により、医薬組成物であって、本発明の活性物質を薬学的に許容しうる希釈剤または担体と一緒に含む医薬組成物を提供する。
【0144】
例えば、本発明の組成物は、経口投与に(例えば、錠剤、ロゼンジ、硬または軟カプセル剤、水性または油状懸濁剤、乳剤、分散性散剤または顆粒剤、シロップ剤またはエリキシル剤として)、局所投与に(例えば、クリーム剤、軟膏剤、ゲル剤、または水性または油状液剤または懸濁剤として)、吹入に(例えば、水性懸濁剤として)または非経口投与に(例えば、静脈内、皮下、腹腔内または筋肉内投与用の滅菌水性または油状液剤として、または直腸投与用の坐剤として)適合した形であってよい。
【0145】
好ましい投与方法は、経口投与である。本発明の活性物質は、好都合には、錠剤の形で経口投与される。
本発明のそれら組成物は、当該技術分野において周知の慣用的な医薬賦形剤を用いて慣用法によって得ることができる。したがって、経口使用を予定した組成物は、例えば、一つまたはそれを超える着色剤、甘味剤、着香剤および/または保存剤を含有してよい。例えば、その組成物は、一つまたはそれを超える増量剤、結合剤、崩壊剤および/または滑沢剤を含有してよい。適する増量剤には、例えば、ラクトース、糖、デンプン、化工デンプン、マンニトール、ソルビトール、無機塩類、セルロース誘導体(例えば、微結晶性セルロース、セルロース)、硫酸カルシウム、キシリトールおよびラクチトール(lactitol)が含まれる。適する結合剤には、例えば、ポリビニルピロリドン、ラクトース、デンプ
ン、化工デンプン、糖類、アラビアゴム、トラガカントゴム、グアーゴム、ペクチン、ワックスバインダー、微結晶性セルロース、メチルセルロース、カルボキシメチルセルロース、ヒドロキシプロピルメチルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース、コポリビドン、ゼラチンおよびアルギン酸ナトリウムが含まれる。適する崩壊剤には、例えば、クロスカルメロースナトリウム、クロスポビドン、ポリビニルピロリドン、ナトリウムデンプングリコラート、トウモロコシデンプン、微結晶性セルロース、ヒドロキシプロピルメチルセルロースおよびヒドロキシプロピルセルロースが含まれる。適する滑沢剤には、例えば、ステアリン酸マグネシウム、ステアリン酸、パルミチン酸、ステアリン酸カルシウム、タルク、カルナウバロウ(carnuba wax)、水素添加植
物油、鉱油、ポリエチレングリコールおよびステアリルフマル酸ナトリウムが含まれる。加えることができる追加の慣用的な賦形剤には、保存剤、安定化剤、酸化防止剤、シリカフローコンディショナー、付着防止剤または滑剤が含まれる。用いることができる他の適する増量剤、結合剤、崩壊剤、滑沢剤および追加の賦形剤は、次の参考文献に記載されている。Handbook of Pharmaceutical Excipients, 3rd Edition;The Theory and Practice of Industrial Pharmacy, 3rd Edition 1986;Pharmaceutical Dosage Forms 1998;Modern Pharmaceutics, 3rd Edition 1995;および Remington’s Pharmaceutical Sciences, 20th Edition 2000。
【0146】
一つまたはそれを超える賦形剤と混合されて単一剤形を生じる本発明の活性物質の量は、必然的に、宿主処置および具体的な投与経路に依存して異なるであろう。例えば、ヒトへの経口投与を予定した製剤は、好都合には、全組成物の約5〜約98重量%であってよい適切且つ好都合な量の賦形剤と配合される、例えば、0.5mg〜0.5gの活性剤(好都合には、1〜250mg、より好都合には、10〜200mgまたは25〜100mg)を含有するであろう。好ましくは、その製剤は、例えば、50mg〜500mgの活性物質を含むであろう。より好ましくは、その製剤は、例えば、100mg〜250mgの活性物質、特に、125mg〜225mgの活性物質を含むであろう。
【0147】
本発明の活性物質を治療目的または予防目的に用いる場合、それは、概して、必要ならば分割用量で与えられる、例えば、0.1mg/kg〜20mg/kg体重の範囲内の1日経口用量が与えられるように投与されるであろう。好ましくは、例えば、1mg/kg〜10mg/kg体重の範囲内の1日経口用量が与えられる。より好ましくは、例えば、2mg/kg〜8mg/kg体重の範囲内の1日用量が与えられる。
【0148】
本発明の活性物質は、許容しうる毒性プロフィールを示す。
本発明の活性物質は、国際特許出願WO01/94341号に、その中の式Iの化合物について開示されたものと同じ薬理学的性質を有する。具体的には、本発明の活性物質は、c−Srcなどの非受容体チロシンキナーゼの阻害剤であり、それは、腫瘍細胞の運動性の選択的阻害および哺乳動物癌細胞の播種および浸潤性の選択的阻害を与えて、転移性腫瘍成長の阻害をもたらす。具体的には、本発明の活性物質は、充実性腫瘍疾患の封じ込めおよび/または処置に用いるための抗浸潤薬として価値があるはずである。例えば、本発明の活性物質は、肺癌(小細胞性肺癌および非小細胞性肺癌を含めた)、乳癌、前立腺癌、卵巣癌、結腸直腸癌、胃癌、脳癌(グリオーマおよび下垂体腺腫を含めた)、頭頸部癌、膀胱癌、膵臓癌、食道癌、胃癌、腎癌、皮膚癌(悪性黒色腫を含めた)、婦人科学的癌(子宮頸部、子宮内膜、膣、外陰部および子宮を含めた)および甲状腺癌などの多数の一般的なヒト癌の処置に、およびCMLおよびALLなどの一定範囲の白血病およびリンパ系悪性疾患の処置に、および癌腫および肉腫などの充実性腫瘍の処置に有用である。
【0149】
本発明の活性物質の薬理学的性質は、例えば、国際特許出願WO01/94341号に開示された試験手順、または十分に当業者の領域の範囲内である同等の試験手順の一つまたはそれを超えるものを用いて評価することができる。その特許出願によるこのような試
験手順は、本明細書中に援用される。
【0150】
本発明のもう一つの側面により、ヒトまたは動物体の療法による処置方法に用いるための、本明細書中の前に定義の本発明の活性物質を提供する。
上述のように、c−Src非受容体チロシンキナーゼの主な役割は、限局性腫瘍が、血流中への播種、他の組織の浸潤および転移性腫瘍成長の開始の時期を経て進行するのに必然的に要求される細胞運動性を調節することである。本発明者は、本発明の活性物質が、転移する腫瘍細胞の浸潤性および移動能力をもたらすシグナル伝達段階に関与するc−Srcキナーゼなどの一つまたはそれを超える非受容体チロシン特異的プロテインキナーゼの阻害によって得られると考えられる強力な抗腫瘍活性を有するということを発見した。
【0151】
したがって、本発明の活性物質は、抗腫瘍薬として、具体的には、転移性腫瘍成長の阻害をもたらす哺乳動物癌細胞の運動性、播種および浸潤性の選択的阻害剤として価値がある。具体的には、本発明の活性物質は、充実性腫瘍疾患の封じ込めおよび/または処置において抗浸潤薬として価値がある。具体的には、本発明の活性物質は、転移する腫瘍細胞の浸潤性および移動能力をもたらすシグナル伝達段階に関与するc−Srcキナーゼなどの一つまたはそれを超える多数の非受容体チロシンキナーゼの阻害に感受性である腫瘍の予防または処置に有用であると考えられる。更に、本発明の活性物質は、酵素c−Srcの阻害によって単独でまたは一部分媒介される腫瘍の予防または処置に有用であると考えられる、すなわち、本発明の活性物質は、このような処置を必要としている温血動物にc−Src酵素阻害作用を生じるのに用いることができる。具体的には、本発明の活性物質は、充実性腫瘍疾患の予防または処置に有用であると考えられる。
【0152】
したがって、本発明のこの側面により、充実性腫瘍疾患の封じ込めおよび/または処置において抗浸潤薬として用いるための薬剤の製造における、本明細書中の前に定義の本発明の活性物質の使用を提供する。
【0153】
本発明のこの側面のもう一つの特徴により、充実性腫瘍疾患の封じ込めおよび/または処置によって抗浸潤作用を生じる処置を必要としているヒトなどの温血動物に、充実性腫瘍疾患の封じ込めおよび/または処置によって抗浸潤作用を生じる方法であって、この動物に、有効量の本明細書中の前に定義の本発明の活性物質を投与することを含む方法を提供する。
【0154】
本発明のもう一つの側面により、ヒトなどの温血動物の充実性腫瘍疾患の予防または処置に用いるための薬剤の製造における、本明細書中の前に定義の本発明の活性物質の使用を提供する。
【0155】
本発明のこの側面のもう一つの特徴により、充実性腫瘍疾患の処置を必要としているヒトなどの温血動物の充実性腫瘍疾患の予防または処置の方法であって、この動物に、有効量の本明細書中の前に定義の本発明の活性物質を投与することを含む方法を提供する。
【0156】
本発明のもう一つの側面により、転移する腫瘍細胞の浸潤性および移動能力をもたらすシグナル伝達段階に関与するc−Srcキナーゼなどの非受容体チロシンキナーゼの阻害に感受性である腫瘍の予防または処置に用いるための薬剤の製造における、本明細書中の前に定義の本発明の活性物質の使用を提供する。
【0157】
本発明のこの側面のもう一つの特徴により、転移する腫瘍細胞の浸潤性および移動能力をもたらすシグナル伝達段階に関与するc−Srcキナーゼなどの非受容体チロシンキナーゼの阻害に感受性である腫瘍の予防または処置方法であって、この動物に、有効量の本明細書中の前に定義の本発明の活性物質を投与することを含む方法を提供する。
【0158】
本発明のもう一つの側面により、c−Srcキナーゼ阻害作用を与える場合に用いるための薬剤の製造における、本明細書中の前に定義の本発明の活性物質の使用を提供する。
本発明のこの側面のもう一つの特徴により、c−Srcキナーゼ阻害作用を与える方法であって、この動物に、有効量の本明細書中の前に定義の本発明の活性物質を投与することを含む方法を提供する。
【0159】
本明細書中の前に定義の抗癌処置は、単独の療法として適用されてよいし、または本発明のキナゾリン誘導体に加えて、慣用的な外科手術または放射線療法または化学療法を伴ってよい。このような化学療法には、次の分類の内の一つまたはそれを超える抗腫瘍薬が含まれてよい。
【0160】
(i)他の抗浸潤薬(例えば、N−(2−クロロ−6−メチルフェニル)−2−{6−[4−(2−ヒドロキシエチル)ピペラジン−1−イル]−2−メチルピリミジン−4−イルアミノ}チアゾール−5−カルボキサミド(ダサチニブ(dasatinib),BMS−3
54825;J. Med. Chem., 2004, 47, 6658-6661)のような他のc−Srcキナーゼファミリー阻害剤;マリマスタト(marimastat)のようなメタロプロテイナーゼ阻害剤;およびウロキナーゼプラスミノーゲンアクチベーター受容体機能の阻害剤);
(ii)医学腫瘍学で用いられるような増殖抑制薬/抗腫瘍薬およびそれらの組合せであって、アルキル化剤(例えば、シスプラチン、カルボプラチン、シクロホスファミド、ナイトロジェンマスタード、メルファラン、クロラムブシル、ブスルファンおよびニトロソ尿素);代謝拮抗薬(例えば、5−フルオロウラシルおよびテガフルのようなフルオロピリミジン類、ラルチトレキセド(raltitrexed)、メトトレキサート、シトシンアラビノ
シドおよびヒドロキシ尿素などの葉酸拮抗薬;抗腫瘍抗生物質(例えば、アドリアマイシン、ブレオマイシン、ドキソルビシン、ダウノマイシン、エピルビシン、イダルビシン(idarubicin)、マイトマイシンC、ダクチノマイシンおよびミトラマイシンのようなアントラサイクリン系);有糸分裂阻止薬(例えば、ビンクリスチン、ビンブラスチン、ビンデシンおよびビノレルビン(vinorelbine)のようなビンカアルカロイド類、およびタキ
ソールおよびタキソテールのようなタキソイド類(taxoids));およびトポイソメラー
ゼ阻害剤(例えば、エトポシドおよびテニポシドのようなエピポドフィロトキシン類、アムサクリン、トポテカン(topotecan)およびカンプトテシン)のようなもの;
(iii)細胞分裂抑制薬であって、抗エストロゲン(例えば、タモキシフェン、フルヴ
ェストラント(fulvestrant)、トレミフェン(toremifene)、ラロキシフェン(raloxifene)、ドロロキシフェン(droloxifene)およびヨードキシフェン(iodoxyfene))、抗アンドロゲン(例えば、ビカルタミド(bicalutamide)、フルタミド、ニルタミド(nilutamide)および酢酸シプロテロン)、LHRHアンタゴニストまたはLHRHアゴニスト(例えば、ゴセレリン、ロイプロレリン(leuprorelin)およびブセレリン(buserelin))、プロゲストゲン(例えば、酢酸メゲストロール)、アロマターゼ阻害剤(例えば、アナストロゾール(anastrozole)、レトロゾール(letrozole)、ボラゾール(vorazole)およびエクセメスタン(exemestane))、およびフィナステリドのような5α−レダクターゼの阻害剤のようなもの;
(iv)増殖因子機能の阻害剤:例えば、このような阻害剤には、増殖因子抗体および増殖因子受容体抗体が含まれる(例えば、抗erbB2抗体トラスツズマブ[HerceptinT
M]および抗erbB1抗体セツキシマブ(cetuximab)[C225]);このような阻
害剤には、更に、例えば、チロシンキナーゼ阻害剤、例えば、上皮増殖因子ファミリーの阻害剤(例えば、N−(3−クロロ−4−フルオロフェニル)−7−メトキシ−6−(3−モルホリノプロポキシ)キナゾリン−4−アミン(ゲフィチニブ(gefitinib),ZD
1839)、N−(3−エチニルフェニル)−6,7−ビス(2−メトキシエトキシ)キナゾリン−4−アミン(エルロチニブ(erlotinib),OSI−774)および6−アク
リルアミド−N−(3−クロロ−4−フルオロフェニル)−7−(3−モルホリノプロポ
キシ)キナゾリン−4−アミン(CI 1033)などのEGFRファミリーチロシンキナーゼ阻害剤;およびラパチニブ(lapatinib)などのerbB2チロシンキナーゼ阻害
剤);肝細胞増殖因子ファミリーの阻害剤;イマチニブ(imatinib)などの血小板由来増殖因子ファミリーの阻害剤;セリン/トレオニンキナーゼの阻害剤(例えば、ファルネシルトランスフェラーゼ阻害剤などのRas/Rafシグナリング阻害剤、例えば、ソラフェニブ(sorafenib)(BAY43−9006));およびMEKおよび/またはAkt
キナーゼによる細胞シグナリングの阻害剤が含まれる;
(v)抗血管新生薬であって、血管内皮増殖因子の作用を阻害するものなど[例えば、抗血管内皮細胞増殖因子抗体ベヴァシズマブ(bevacizumab)[AvastinTM];および4−(4−ブロモ−2−フルオロアニリノ)−6−メトキシ−7−(1−メチルピペリジン−4−イルメトキシ)キナゾリン(ZD6474;WO01/32651号中の実施例2)、4−(4−フルオロ−2−メチルインドール−5−イルオキシ)−6−メトキシ−7−(3−ピロリジン−1−イルプロポキシ)キナゾリン(AZD2171;WO00/47212号中の実施例240)、バタラニブ(vatalanib)(PTK787;WO98/
35985号)およびSU11248(スニチニブ(sunitinib);WO01/6081
4)号などのVEGF受容体チロシンキナーゼ阻害剤;および他の機構によって作用する化合物(例えば、リノマイド(linomide)、インテグリンαvβ3機能の阻害剤およびアンギオスタチン(angiostatin))];
(vi)血管損傷性薬であって、Combretastatin A4;および国際特許出願WO99/
02166号、WO00/40529号、WO 00/41669号、WO01/922
24号、WO02/04434号およびWO02/08213号に開示された化合物のようなもの;
(vii)アンチセンス療法、例えば、ISIS2503、抗rasアンチセンスのよう
な、上に挙げられた標的に向けられているもの;
(viii)遺伝子治療アプローチであって、例えば、異常p53または異常BRCA1またはBRCA2のような異常遺伝子を置き換えるアプローチ;シトシンデアミナーゼ、チミジンキナーゼまたは細菌ニトロレダクターゼ酵素を用いたものなどのGDEPT(遺伝子に支配される酵素プロドラッグ療法(gene-directed enzyme pro-drug therapy))ア
プローチ;および多剤耐性遺伝子治療のような、化学療法または放射線療法への患者耐性を増加させるアプローチを含めたもの;および
(ix)免疫療法アプローチであって、例えば、インターロイキン2、インターロイキン4または顆粒球−マクロファージコロニー刺激因子などのサイトカインでのトランスフェクションのような、患者腫瘍細胞の免疫原性を増加させる ex-vivo および in-vivo アプローチ;T細胞アネルギーを減少させるアプローチ;サイトカインでトランスフェクションされた樹状細胞のようなトランスフェクションされた免疫細胞を用いたアプローチ;サイトカインでトランスフェクションされた腫瘍細胞系を用いたアプローチ;および抗イディオタイプ抗体を用いたアプローチを含めたもの。
【0161】
このような共同処置は、個々の処置成分の同時の、逐次的または別々の投与によって達成することができる。このような組合せ製品は、本明細書中の前に記載の投薬量範囲内の本発明の化合物と、その承認された投薬量範囲内の他の薬学的活性剤を用いる。
【0162】
本発明のこの側面により、癌の共同処置のための医薬製品であって、本明細書中の前に定義の本発明の活性物質および本明細書中の前に定義の追加の抗癌薬を含む医薬製品を提供する。
【0163】
式Iの化合物の次の具体的な結晶形の製造方法、すなわち、
(i)AZD0530ジフマレートを製造する;
(ii)AZD0530セスキフマレート四水和物を製造する;
(iii)AZD0530三水和物を製造する;および
(iv)無水AZD0530を製造する方法は、本明細書中に開示されている。
【0164】
本発明のもう一つの側面により、実質的にAZD0530ジフマレートの形の式Iの化合物を製造する方法であって、
(a)4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンとフマル酸とを、AZD0530ジフマレートを形成するのに十分な時間接触させ;そして
(b)AZD0530ジフマレートを単離すること
を含む方法を提供する。
【0165】
直ぐ上の作業工程(a)において出発物質として用いられる4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンは、例えば、先行技術に記載のように製造される場合、またはAD0530三水和物のような本明細書中に記載の形の一つとして製造される場合、式Iの化合物のいずれの形であってもよい。
【0166】
好都合には、AZD0530ジフマレートへの変換は、一つまたはそれを超える適する溶媒中の4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン出発物質の溶液を製造し、そしてフマル酸を加えることによって行われる。好都合には、モル過剰のフマル酸を用いて、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン出発物質のAZD0530ジフマレートへの実質的に完全な変換を確実にすることができる(すなわち、フマル酸対キナゾリン化合物のモル比は、少なくとも2:1である)。フマル酸濃度の上限は、臨界的ではない。好都合には、僅かにモル過剰のフマル酸を用いる。例えば、フマル酸対キナゾリン化合物のモル比は、好適には、約2:1〜10:1、具体的には、約2:1〜3:1、より具体的には、約2.2:1である。
【0167】
具体的な態様において、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン出発物質の溶液は、水を補助溶媒として含有してよい、一つまたはそれを超える有機溶媒の混合物中で製造する。適する有機溶媒は、極性プロトン性溶媒、例えば、(1−4C)アルコール、具体的には、メタノール、エタノール、イソプロパノールおよびn−ブタノール;極性非プロトン性溶媒であって、脂肪族エステル、例えば、(1−4C)アルキル(2−3C)アルカノエートエステル、具体的には、酢酸エチル;脂肪族(3−6C)ケトン、具体的には、アセトンおよびメチルエチルケトン;脂肪族アミド、具体的には、N,N−ジメチルホルムアミド;およびニトリル、具体的には、アセトニトリルなどのような水混和性極性有機溶媒である。好都合には、水不混和性補助溶媒を、水混和性溶媒に加えてよい。適するこのような補助溶媒には、例えば、トルエンなどの芳香族溶媒が含まれる。特に好都合な有機溶媒には、例えば、イソプロパノールまたは酢酸エチル、またはそれらの混合物が含まれる。
【0168】
キナゾリン化合物について、用いられる有機溶媒の具体的な量は、選択される有機溶媒に、およびキナゾリン化合物をフマル酸と接触させる条件に依存するであろう。イソプロパノールまたは酢酸エチルなどの溶媒の場合、2〜20ml/gのような0.1〜30ml/gの範囲、具体的には、約10ml/gが、キナゾリン化合物に適しているが、ここにおいて、「ml/g」は、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−
4−イルオキシキナゾリン1g当たりの有機溶媒の容量を意味する。単一の有機溶媒を用いてよいし、または二つまたはそれを超える有機溶媒、例えば、酢酸エチルおよびイソプロパノールの(好適には、約1:1の容量比の)混合物を用いてよい。水は、補助溶媒として加えることができる。好都合には、有機溶媒(イソプロパノールなど)対水の適する容量比は、50:1〜2:1の範囲内、具体的には、10:1〜5:1の範囲内である。
【0169】
フマル酸について、用いられる有機溶媒の具体的な量は、水が補助溶媒として用いられようといまいと、選択される有機溶媒に、およびフマル酸をキナゾリン化合物と接触させる条件に依存するであろう。イソプロパノールまたは酢酸エチルなどの溶媒の場合、2〜30ml/gのような0.1〜60ml/gの範囲、具体的には、約15ml/gが、フマル酸に適している。単一の有機溶媒を用いてよいし、または二つまたはそれを超える有機溶媒、例えば、酢酸エチルおよびイソプロパノールの(好適には、約1:1の容量比の)混合物を用いてよい。水は、補助溶媒として加えることができる。好都合には、有機溶媒(イソプロパノールなど)対水の適する容量比は、50:1〜3:1の範囲内、具体的には、15:1〜5:1の範囲内、より具体的には、約10:1である。
【0170】
場合により、AZD0530ジフマレートの一つまたはそれを超える種結晶を加えて、AZD0530ジフマレートへの変換の開始および/または変換速度を増強させることができる。
【0171】
AZD0530ジフマレートへの変換に必要な時間は、温度、有機溶媒の存在、および播種結晶を用いるか否かのような、用いられる具体的な反応条件に依存する。概して、例えば、5分〜48時間の反応時間が適している。
【0172】
或いは、有機溶媒の量は、その方法の間中、スラリーが保持されているように、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン出発物質を完全に溶解させるのに不十分であってよい。好都合には、式Iの化合物をその方法中にスラリーで保持することにより、AZD0530ジフマレートは、例えば、混合物を冷却するまたは溶媒を蒸発させることによって結晶化を誘導することを必要とすることなく、形成させることができる。したがって、スラリー方法は、実質的に一定温度で操作することができる。理論によって拘束されたくはないが、その方法は、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン出発物質の限局的溶解および引き続きのAZD0530ジフマレートの結晶化の機構によって進行すると考えられる。したがって、本明細書中に記載のスラリー変換方法は、少量ずつの溶解と、出発物質のAZD0530ジフマレートへの少量ずつの変換であると考えられる。
【0173】
本発明のこの側面により、更に、実質的にAZD0530ジフマレートの形の式Iの化合物を製造する方法であって、
(a)化合物4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンを、有機溶媒および水を含む溶媒系中に溶解させ;
(b)有機溶媒および水を含む溶媒系中のフマル酸の溶液を加え;
(c)その溶媒系の温度を下げて、核生成を誘導し;
(d)その混合物を、核生成が開始した温度未満の温度で維持し;そして
(e)結晶性AZD0530ジフマレートを単離する
工程を含む方法を提供する。
【0174】
AZD0530ジフマレートを製造するこの結晶化方法は、ジフマレート塩を高純度で
製造することを可能にする。
溶媒系中の適する有機溶媒には、直ぐ上の作業工程(a)において出発物質を溶解させる温度で水溶性である有機溶媒が含まれる。適する有機溶媒には、例えば、弱極性有機溶媒であって、テトラヒドロフランなどの(4−7C)環状エーテルまたは脂肪族ジ−(1−6C)アルキルエーテルなど;より極性のプロトン性溶媒、例えば、エタノールおよびイソプロパノールなどの(2−6C)アルコール;極性非プロトン性溶媒であって、酢酸エチルなどの(1−4C)アルキル(2−3C)アルカノエートエステル;アセトンなどの脂肪族(3−6C)ケトン;N,N−ジメチルホルムアミドまたはN−メチルピロリジン−2−オンなどの脂肪族アミド;およびアセトニトリルなどのニトリルのようなものが含まれる。具体的な有機溶媒は、例えば、酢酸エチルである。単一の有機溶媒、または上の一つまたはそれを超える溶媒の混合物を用いてよい。
【0175】
好都合には、モル過剰のフマル酸を用いて、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン出発物質のAZD0530ジフマレートへの実質的に完全な変換を確実にすることができる(すなわち、フマル酸対キナゾリン化合物のモル比は、少なくとも2:1である)。フマル酸濃度の上限は、臨界的ではない。好都合には、僅かにモル過剰のフマル酸を用いる。例えば、フマル酸対キナゾリン化合物のモル比は、好適には、約2:1〜10:1、具体的には、約2:1〜3:1、より具体的には、約2.2:1である。
【0176】
キナゾリン化合物について、用いられる有機溶媒の具体的な量は、選択される有機溶媒に、およびキナゾリン化合物をフマル酸と接触させる条件に依存するであろう。イソプロパノールまたは酢酸エチルなどの溶媒の場合、2〜20ml/gのような0.1〜30ml/gの範囲、具体的には、約10ml/gが適している。単一の有機溶媒を用いてよいし、または二つまたはそれを超える有機溶媒、例えば、酢酸エチルおよびイソプロパノールの(好適には、約1:1の容量比の)混合物を用いてよい。水は、好都合には、補助溶媒として加える。好都合には、有機溶媒(イソプロパノールなど)対水の適する容量比は、50:1〜2:1の範囲内、具体的には、10:1〜5:1の範囲内である。
【0177】
フマル酸について、用いられる有機溶媒の具体的な量は、水が補助溶媒として用いられようといまいと、選択される有機溶媒に、およびフマル酸をキナゾリン化合物と接触させる条件に依存するであろう。イソプロパノールまたは酢酸エチルなどの溶媒の場合、2〜30ml/gのような0.1〜60ml/gの範囲、具体的には、約15ml/gが適している。単一の有機溶媒を用いてよいし、または二つまたはそれを超える有機溶媒、例えば、酢酸エチルおよびイソプロパノールの(好適には、約1:1の容量比の)混合物を用いてよい。水は、好都合には、補助溶媒として加える。好都合には、有機溶媒(イソプロパノールなど)対水の適する容量比は、50:1〜3:1の範囲内、具体的には、15:1〜5:1の範囲内、より具体的には、約10:1である。
【0178】
化合物4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンは、その方法の工程(a)において、溶媒系中で、実質的に全ての化合物が溶解するまで化合物を加熱することにより、溶解させることができる。同様に、フマル酸は、その方法の工程(b)において、溶媒系中で、実質的に全ての化合物が溶解するまで化合物を加熱することにより、溶解させることができる。好都合には、各々の化合物を、溶媒系のほぼ還流温度に、溶解を完了するのに十分な時間加熱する。より好都合には、各々の化合物を、約30〜100℃の範囲内、好ましくは、35〜80℃の範囲内の温度に加熱して、溶解を完了する。必要ならば、どちらかまたは双方の加温された溶液を濾過して、不溶性物質を除去することができる。キナゾリン化合物の溶液の温度を約50〜100℃の
範囲内、好都合には、約60〜90℃の範囲内に維持すると同時に、温フマル酸溶液を加える。次に、その混合物を、例えば、約50〜80℃の範囲内の温度に僅かに冷却させて、AZD0530ジフマレートの核生成を促進することができる。核生成は、自発的にかまたは、一つまたはそれを超える種結晶を加えることで起こりうるということは理解されるであろう。好都合には、その混合物を、約75℃の温度で維持し、種結晶を加えてAZD0530ジフマレートの核生成を促進し、そしてその混合物を、約75℃の温度で数時間維持して、結晶化方法を継続させる。次に、その混合物を、一定制御速度で周囲温度に冷却させることができる。適する冷却速度は、例えば、約20℃/時である。そのようにして得られた結晶性AZD0530ジフマレートは、いずれかの慣用法によって、例えば、濾過または遠心分離によって単離することができる。
【0179】
上記の結晶化方法において、一つまたはそれを超える種結晶を用いて核生成を開始させる場合、それら種結晶は、好ましくは、いずれか適する方法を用いて、例えば、以下の実施例中に記載の方法を用いて製造することができるAZD0530ジフマレートの結晶である。
【0180】
上記の手順を、常套の技術および情報を用いて変更することができるということは、当業者に理解されるであろう。例えば、逆添加手順を用いることができ、それによって、AZD0530の溶液をフマル酸の溶液に加える。更に、例えば、AZD0530ジフマレートが、いずれか他のAZD0530形を実質的に不含で得られるという条件ならば、反応する化合物4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンおよびフマル酸の量;溶媒およびいずれかの補助溶媒の性質および容量;溶媒混合物が用いられる場合の成分溶媒の比率;用いられる水の容量;および溶解相および結晶化相の温度はいずれも、変更することができる。例えば、ジフマレートの核生成は、例えば、若干の溶媒の蒸発によって誘導することができる。或いは、核生成は、ジフマレート化合物に適するアンチソルベントの添加によって誘導しうるが、それによって、溶液の過飽和を生じ、そこから、AZD0530ジフマレートが結晶化する。
【0181】
更に、実質的にAZD0530セスキフマレート四水和物の形の式Iの化合物を製造する方法であって、
(a)AZD0530ジフマレートを、有機溶媒および水を含む溶媒系または水中に溶解させ;
(b)溶媒系の部分蒸発を引き起こして、核生成を誘導し;
(c)その混合物を、周囲温度未満の温度に冷却し;そして
(d)結晶性AZD0530セスキフマレート四水和物を単離する
工程を含む方法を提供する。
【0182】
AZD0530セスキフマレート四水和物を製造するこの結晶化方法は、セスキフマレート四水和物塩を高純度で製造することを可能にする。
溶媒系中の適する有機溶媒には、直ぐ上の作業工程(a)において出発物質を溶解させる温度で水溶性である有機溶媒が含まれる。適する有機溶媒には、例えば、アセトニトリルなどのニトリル;および極性プロトン性溶媒、例えば、メタノール、エタノールおよびイソプロパノールなどの(2−6C)アルコールが含まれる。水との混合物で用いるための具体的な有機溶媒は、例えば、アセトニトリルである。単一の有機溶媒、または上の一つまたはそれを超える溶媒の混合物を用いてよい。好都合には、水不混和性補助溶媒を、水混和性溶媒に加えてよい。適するこのような補助溶媒には、例えば、トルエンなどの芳香族溶媒が含まれる。より好都合には、水を溶媒として用いる。
【0183】
溶媒の蒸発は、周囲温度で、例えば、その溶液を開放容器中で放置することによって行
うことができる。或いは、蒸発工程は、より高い温度で、例えば、約40〜80℃の範囲内の温度で、好都合には、約60℃で行うことができる。好都合には、空気または窒素などのガス流を、その溶液の表面中にまたはそれを越えて通過させて、溶媒蒸発を加速することができる。核生成が開始したら、結晶化混合物を、好都合には、周囲温度未満の温度に冷却して、結晶化を継続させる。好都合には、その混合物を、約10℃未満の温度に、より好都合には、約5℃の温度に冷却する。
【0184】
上記の手順を、常套の技術および情報を用いて変更することができるということは、当業者に理解されるであろう。例えば、AZD0530セスキフマレート四水和物が、いずれか他のAZD0530形を実質的に不含で得られるという条件ならば、AZD0530ジフマレートの量;用いられる水の容量;用いられるいずれかの補助溶媒の性質および容量;および溶解相、蒸発相および冷却相の温度はいずれも、変更することができる。例えば、核生成は、適するアンチソルベントの添加によって誘導しうるが、それによって、溶液の過飽和を生じ、そこから、AZD0530セスキフマレート四水和物が結晶化する。
【0185】
本発明のもう一つの側面により、実質的にAZD0530三水和物の形の式Iの化合物を製造する方法であって、
(a)化合物4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンを、水および有機溶媒を含む溶媒系中に溶解させ;
(b)その溶媒系の温度を下げて、核生成を誘導し;
(c)その混合物を、核生成が開始した温度未満の温度で維持し;そして
(d)結晶性AZD0530三水和物を単離する
工程を含む方法を提供する。
【0186】
溶媒系中の適する有機溶媒には、その方法の工程(a)において出発物質を溶解させる温度で水溶性である有機溶媒が含まれる。適する有機溶媒には、例えば、弱極性有機溶媒であって、テトラヒドロフランなどの(4−7C)環状エーテルまたは脂肪族ジ−(1−6C)アルキルエーテルなど;より極性のプロトン性溶媒、例えば、エタノールおよびイソプロパノールなどの(2−6C)アルコール;極性非プロトン性溶媒であって、酢酸エチルなどの(1−4C)アルキル(2−3C)アルカノエートエステル;アセトンなどの脂肪族(3−6C)ケトン;N,N−ジメチルホルムアミドまたはN−メチルピロリジン−2−オンなどの脂肪族アミド;およびアセトニトリルなどのニトリルのようなものが含まれる。具体的な有機溶媒は、例えば、酢酸エチルである。単一の有機溶媒、または上の一つまたはそれを超える溶媒の混合物を用いてよい。
【0187】
概して、モル過剰の水を溶媒系中に用いる(すなわち、水:4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンのモル比は、少なくとも3:1である)。水濃度の上限は、臨界的ではないが、しかしながら、概して、大モル過剰の水を用いる。例えば、水対4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンのモル比は、好適には、約3:1〜1000:1またはそれを超える、具体的には、約3:1〜約400:1である。
【0188】
場合により、補助溶媒を溶媒系中に用いてよい。適する補助溶媒には、例えば、トルエンなどの芳香族炭化水素;およびハロゲノ−(1−6C)アルカンなどの脂肪族ハロゲン化炭化水素、例えば、1,2−ジクロロエタンが含まれる。
【0189】
化合物4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メ
チルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンは、その方法の工程(a)において、溶媒系中で、実質的に全ての化合物が溶解するまで化合物を加熱することにより、溶解させることができる。好都合には、その方法の工程(a)における溶媒系中の化合物を、溶媒系のほぼ還流温度に、化合物を完全に溶解するのに十分な時間加熱する。次に、化合物4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンの溶液を、熱源から除去し、そして25〜60℃の範囲内の温度に冷却させて、AZD0530三水和物の核生成を促進することができるし、またはそれを更に、例えば、周囲温度に冷却することができる。好都合には、その溶液を、熱源から除去し、そして約50℃に冷却させて、AZD0530三水和物の核生成を促進することができる。その混合物は、約55℃に再加熱した後、一定制御速度で約50℃に冷却させることができる。適する冷却速度は、例えば、約10℃/時である。核生成は、自発的にかまたは、一つまたはそれを超える種結晶を加えることで起こりうるということは理解されるであろう。次に、その溶液を、約50℃の温度で保持して、生成物の結晶化を生じさせる。引き続き、その溶液を、一定制御速度で約20℃に冷却して、生成物の結晶化を終わらせることができる。適する冷却速度は、例えば、約10℃/時である。そのようにして得られた結晶性AZD0530三水和物は、いずれかの慣用法によって、例えば、濾過または遠心分離によって単離することができる。
【0190】
上記の結晶化/再結晶方法において、一つまたはそれを超える種結晶を用いて核生成を開始させる場合、それら種結晶は、好ましくは、AZD0530三水和物の結晶である。その一つまたは複数の種結晶は、AZD0530三水和物の製造についていずれか適する方法を用いて、例えば、非晶質AZD0530の試料を水中でスラリーにすることによって製造することができる。
【0191】
上記の手順を、常套の技術および情報を用いて変更することができるということは、当業者に理解されるであろう。例えば、AZD0530三水和物が、いずれか他のAZD0530形を実質的に不含で得られるという条件ならば、処理される化合物4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンの量;溶媒およびいずれかの補助溶媒の性質および容量;溶媒混合物が用いられる場合の成分溶媒の比率;用いられる水の容量および水対溶媒の比率;および溶解相および冷却相の温度はいずれも、変更することができる。例えば、その方法の工程(b)における、適する溶媒、例えば、エタノールなどの(2−6C)アルコール中の式Iの化合物の溶液の核生成は、例えば、若干のエタノール溶媒の蒸発によって誘導することができるし、或いは、核生成は、式Iの化合物に適するアンチソルベントの添加によって誘導しうるが、それによって、溶液の過飽和を生じ、そこから、AZD0530三水和物が結晶化する。
【0192】
AZD0530三水和物を製造する結晶化方法は、三水和物を高純度で製造することを可能にする。更に、そのようにして得られたAZD0530三水和物の再結晶は、上記の方法を用いて行うことができる。再結晶は、その物質を更に精製する可能性を与える。
【0193】
本発明のもう一つの側面により、実質的にAZD0530三水和物の形の式Iの化合物を製造する方法であって、
(a)4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンと水とを、AZD0530三水和物を形成するのに十分な時間接触させ;そして
(b)AZD0530三水和物を単離すること
を含む方法を提供する。
【0194】
直ぐ上の作業工程(a)において出発物質として用いられる4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンは、式Iのいずれかの形の化合物、例えば、先行技術に記載の非晶形、または無水AD0530のような本明細書中に記載の結晶形のいずれか一つであってよい。
【0195】
好都合には、AZD0530三水和物への変換は、水中の4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン出発物質のスラリーを、場合により、一つまたはそれを超える適する有機溶媒の存在下で製造することによって行われる。概して、モル過剰の水を用いて、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン出発物質のAZD0530三水和物への実質的に完全な変換を確実にする(すなわち、水対キナゾリン化合物のモル比は、少なくとも3:1である)。水濃度の上限は、臨界的ではないが、しかしながら、概して、大モル過剰の水を用いる。例えば、水対キナゾリン化合物のモル比は、好適には、約3:1〜1000:1またはそれを超える、具体的には、約3:1〜約400:1である。
【0196】
具体的な態様において、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン出発物質のスラリーは、水および有機溶媒、そして場合により、一つまたはそれを超える補助溶媒の混合物中で製造する。本発明者は、有機溶媒の使用が、出発物質をAZD0530三水和物へ変換するのに必要な時間を有意に減少させるということを発見した。適する有機溶媒は、水混和性極性有機溶媒であって、極性プロトン性溶媒、例えば、(1−4C)アルコール、具体的には、エタノールおよびイソプロパノール;極性非プロトン性溶媒であって、脂肪族エステル、例えば、(1−4C)アルキル(2−3C)アルカノエートエステル、具体的には、酢酸エチル;アセトンなどの脂肪族(3−6C)ケトン;N,N−ジメチルホルムアミドなどの脂肪族アミドなどのようなものである。具体的な溶媒には、例えば、イソプロパノールまたは酢酸エチル、またはそれらの混合物が含まれる。
【0197】
用いられる有機溶媒の量は、その方法の間中、スラリーが保持されているように、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン出発物質を完全に溶解させるのに不十分であってよい。好都合には、式Iの化合物をその方法中にスラリーで保持することにより、AZD0530三水和物は、例えば、混合物を冷却するまたは溶媒を蒸発させることによって結晶化を誘導することを必要とすることなく、形成させることができる。したがって、スラリー方法は、実質的に一定温度で操作することができる。
【0198】
理論によって拘束されたくはないが、その方法は、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン出発物質の限局的溶解および引き続きのAZD0530三水和物の結晶化の機構によって進行すると考えられる。したがって、本明細書中に記載のスラリー変換方法は、少量ずつの溶解と、出発物質のAZD0530三水和物への少量ずつの変換であると考えられる。
【0199】
用いられる有機溶媒の具体的な量は、選択される有機溶媒に、およびスラリーを水と接触させる条件に依存するであろう。イソプロパノールまたは酢酸エチルなどの溶媒の場合、2〜10ml/gのような0.1〜20ml/gの範囲、具体的には、約5ml/gが
適しているが、ここにおいて、「ml/g」は、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン1g当たりの有機溶媒の容量を意味する。
【0200】
単一の有機溶媒を用いてよいし、または二つまたはそれを超える有機溶媒、例えば、酢酸エチルおよびイソプロパノールの(好適には、約1:1の容量比の)混合物を、水と一緒に用いてよい。
【0201】
場合により、補助溶媒を用いてよい。適する補助溶媒には、例えば、弱極性有機溶媒であって、芳香族炭化水素(例えば、トルエン)、ハロゲノ−(1−6C)アルカン(例えば、1,2−ジクロロエタン)および脂肪族ジ−(1−6C)アルキルエーテルまたは(4−7C)環状エーテル(例えば、テトラヒドロフラン)などが含まれる。具体的な補助溶媒は、トルエンである。補助溶媒(トルエンなど)対有機溶媒(イソプロパノールなど)の適する容量比は、50:1〜0.05:1の範囲内、好都合には、10:1〜0.5:1の範囲内、具体的には、約3:1〜1:1である。
【0202】
場合により、AZD0530三水和物の一つまたはそれを超える種結晶を、スラリーに加えて、AZD0530三水和物への変換速度を増強させることができる。
その方法は、ほぼ周囲温度で、例えば、約15〜30℃、具体的には、約20〜25℃で行われる。
【0203】
AZD0530三水和物への変換に必要な時間は、温度、有機溶媒の存在、および播種結晶を用いるか否かのような、用いられる具体的な反応条件に依存する。概して、例えば、5分〜48時間の反応時間が適している。
【0204】
本発明者は、更に、AZD0530三水和物は、結晶性無水AZD0530へと容易に変換することができるということを発見した。したがって、AZD0530三水和物の結晶化および引き続きの無水AZD0530への変換は、無水AZD0530を高純度で製造する手段を提供する。実質的に無水AZD0530の形の式Iの化合物のこのような製造方法は、本発明のもう一つの側面を提供する。
【0205】
更に、実質的に結晶性無水AZD0530の形の式Iの化合物を製造する方法であって、AZD0530三水和物を脱水する工程を含む方法を提供する。
以下、変換方法1と称されるこの変換の一つの態様は、実質的に乾燥した不活性ガス流を、AZD0530三水和物の試料の上におよび/またはそれを介して、水を駆逐し且つ無水AZD0530への変換を行うのに十分な時間および十分な温度で通過させる工程を含む。
【0206】
好都合には、変換方法1は、周囲温度(15〜25℃の範囲内、具体的には、約20℃の温度)で行われる。適する不活性ガスは、例えば、窒素ガスであるが、それは、必要ならば、実質的に乾燥するまで乾燥させるべきである。概して、変換方法1は、AZD0530三水和物を無水AZD0530へ変換するのに、5分〜50時間、好適には、1〜30時間の乾燥時間を必要とする。好都合には、AZD0530三水和物は、フィルター上に置くことができ、そして乾燥ガスを、フィルターを介して通過させることができる。好適には、変換方法1における乾燥工程は、所望の無水物形への実質的に完全な変換を確実にするのに十分な時間継続すべきである。実質的に完全な変換とは、式Iの化合物の少なくとも80%が、無水AZD0530の形であり、20%未満のいずれか他のAZD0530形が存在するということを意味する。具体的には、式Iの化合物の少なくとも90%、特に、少なくとも95%は、無水AZD0530の形である。必要な無水AZD0530への変換度は、常套技術、例えば、本明細書中に記載のXRPDを用いて評価すること
ができる。
【0207】
場合により、窒素などの不活性ガス流は、その物質の上におよび/またはそれを介して通過させる前に加温する。加温されたガスに適する温度は、例えば、25〜100℃、具体的には、40〜60℃の温度である。
【0208】
以下、変換方法2と称されるこの変換のもう一つの態様は、実質的にAZD0530三水和物の形の式Iの化合物を、水を駆逐し且つ無水AZD0530への変換を行うのに十分な時間および十分な温度で加熱する工程を含む。
【0209】
変換方法2は、好適には、AZD0530三水和物を50〜150℃、具体的には、80〜140℃、より具体的には、120〜130℃の温度で加熱することによって行われる。必要な加熱時間は、特に、試料のサイズおよび用いられる加熱方法に依存する。概して、AZD0530三水和物を無水AZD0530へと変換するには、5分〜100時間、好適には、1〜30時間の加熱時間で十分である。AZD0530三水和物は、慣用的な技法を用いて、例えば、適するオーブンまたは真空オーブン中で、または流動床乾燥器などの慣用的な乾燥システム中で加熱することができる。好適には、変換方法2における加熱工程は、所望の無水物形への本明細書中の前に定義の実質的に完全な変換を確実にするのに十分な時間および十分な温度で継続すべきである。
【0210】
以下、変換方法3と称されるこの変換のもう一つの態様は、
(a)実質的にAZD0530三水和物の形の式Iの化合物を、溶媒または溶媒混合物で洗浄して、実質的に水を除去し;そして
(b)そのようにして形成された無水AZD0530を単離することを含む。
【0211】
変換方法3において適する溶媒には、例えば、洗浄温度において式Iの化合物が可溶性に乏しい水混和性有機溶媒が含まれる。適する溶媒の例には、弱極性有機溶媒であって、テトラヒドロフランなどの(4−7C)環状エーテルまたは脂肪族ジ−(1−6C)アルキルエーテルなど;より極性のプロトン性溶媒、例えば、エタノールおよびイソプロパノールなどの(2−6C)アルコール;極性非プロトン性溶媒であって、酢酸エチルなどの(1−4C)アルキル(2−3C)アルカノエートエステル;およびアセトニトリルなどのニトリルのようなものが含まれる。このような溶媒の混合物を用いてもよい。具体的な溶媒は、イソプロパノールおよび/または酢酸エチルである。
【0212】
「洗浄」工程は、無水AZD0530への変換を行うのに適する時間を必要とするということは理解されるはずである。固体と洗浄溶媒との間の適する接触時間は、約5分〜1時間またはそれを超える範囲内である。より好都合には、接触時間は、約30分〜約2時間の範囲内、例えば、約1時間である。好都合には、固体および洗浄溶媒のスラリーを製造する。好都合には、そのスラリーを撹拌して、洗浄溶媒と固体の結晶との間の接触を改善する。洗浄溶媒は、例えば、約30〜50℃の温度に加温してよいが、しかしながら、概して、無水AZD0530への変換を行うには、ほぼ周囲温度での洗浄で十分である。
【0213】
場合により、変換方法3において一つまたは複数の溶媒洗浄工程後に単離された物質を乾燥させて、水の完全な除去および所望の結晶性無水AZD0530への変換を確実にする。変換方法1または変換方法2の方法を用いることができる。
【0214】
本発明を、次の実施例、データおよび図面によって以下に詳しく説明するが、ここにおいて、
(i)X線回折データは、Siemens D5000装置を用いて得た。試料は、メノウ乳棒・乳鉢を用いて結晶凝集体を穏やかに粉砕することによって調製した。その試料を、標準
的なホルダー(フラットリップを有する)中に充填し、顕微鏡スライドガラスでリップまで平らに圧縮した。その試料を、30回毎分(rpm)で回転させて、計数統計値を改善した。X線は、40kVおよび40mAで操作される銅ロングファインフォーカス管球によって発生させた。X線の波長は、1.5406Åであった。その計器を、θ−θ配置において2°2θ〜40°2θの走査範囲にわたって0.02°2θ増加分につき4秒暴露で操作した。実験は、Bragg-Brentano 配置で行ったが、それによって、X線ビームは、
V20で自動可変発散スリットを介して通過し、そして反射した放射線は、2mm散乱防止スリットおよび0.2mm検出器スリットによって方向づけられた。反射は、それらの重心値として引用される(DIFFRAC/ATなどの計算機パッケージによって計算される)。当XRPD業者は、約30ミクロンを超えるサイズの粒子および単一でない(non-unitary)アスペクト比を有する試料の分析が、ピークの相対強さに影響することがあ
りうるということを理解するであろう。当業者は、更に、反射位置は、試料が位置する回折計中の正確な高さおよび回折計のゼロ検定によって影響されるということを理解するであろう。試料表面の平坦さも、僅かに作用することがありうる。したがって、示された回折図形データを、絶対値とみなすべきではない。
【0215】
更に、異なった装置および/または条件は、僅かに異なったデータを生じさせることがありうる、例えば、ピークの位置および相対強さに変動がありうるということは理解されるであろう。概して、XRPDスペクトル中のピーク位置(回折角)の測定誤差は、約±0.3°2θであろう。このような程度の測定誤差は、XRPDスペクトルが実質的に同じであるか否かを評価する場合に考慮されるべきである。当業者は、正確な個々のピーク位置よりもむしろ、それらピークの相対位置が、試料が実質的に同じ結晶形に由来したか否かについてのより信頼できる指標であるということを理解するであろう。具体的には、XRPDを用いて測定されたピークの強さは、結晶性粒子をXRPDマウント中に充填する作用のために、粒子のサイズおよび形状の結果として異なることがありうる。このような充填作用は、当該技術分野において周知であり、しばしば、「選択配向」作用と称される。検体の選択配向は、いろいろな反射の強さに影響を与えるので、全くの無作為の試料により予想されうる強さと比較して、あるものはより強く、他のものはあまり強くない。選択配向作用は、サイズ減少が、より微細な針状結晶またはプレートレットを生じる場合、針様またはプレート様の結晶について特に明らかである。結果として、結晶形は、主に、X線ディフラクトグラム中の相対ピーク位置を最も確実に特徴とする。これら作用並びに標準的なX線回折分析方法は、例えば、Bunn, C. W. (1948), Chemical Crystallography, Clarendon Press, London;または Klug, H. P. & Alexander, L. E. (1974), X-Ray
Diffraction Procedures, John Wiley and Sons, New York に見出されうる。したがっ
て、本明細書中に引用された図面を、絶対値とみなすべきではない。したがって、本明細書中に開示されたXRPDスペクトルの一つと実質的に同じであるXRPDスペクトルを与えるいずれかの結晶が、本発明の範囲内であるということは理解されるはずである。
【0216】
(ii)融点およびTGAは、Mettler DSC820eおよび Mettler TG851装置
を用いて、それぞれ、TSO891ROロボットシステムを用いて決定した。
融点決定について、パンタイプは、穴のあいた蓋付きのアルミニウム(40μlサイズ)であった。試料重量は、約1〜5mgであった。手順は、窒素ガス流下(100ml/分)で行い、検討された温度範囲は、10℃/分の一定の温度上昇速度において25℃〜325℃であった。当業者は、融点の正確な値が、化合物の純度、試料重量、加熱時間および粒子サイズによって影響されるということを理解するであろう。したがって、別の融点読みが、他のタイプの装置によって、または記載されたものとは異なった条件を用いることによって与えられるかもしれないということは理解されるであろう。したがって、本明細書中に引用された図面を、絶対値とみなすべきではない。
【0217】
TGA決定については、各々の試料(約1〜12mg)を、開放型酸化アルミニウムパ
ン(70μlサイズ)に入れ、そして手順は、ヘリウムガス流下(50ml/分)で行い、検討された温度範囲は、10℃/分の一定の温度上昇速度において25℃〜325℃であった。僅かに異なったデータが、異なった装置および/または条件を用いた場合に生じるかもしれないということは理解されるであろう。したがって、本明細書中に引用された図面を、絶対値とみなすべきではない。
【0218】
(iii)DRIFT分光法データは、Nicolet 20SXC分光計において、粉末臭化カ
リウム中の試料の2%w/w分散を用いて、4000〜400cm−1の度数範囲にわたって得た。僅かに異なったデータが、異なった装置および/または試料調製条件を用いた場合に生じるかもしれないということは理解されるであろう。したがって、本明細書中に引用された図面を、絶対値とみなすべきではない。
【0219】
次の実施例に関して、概して、
(i)操作は、周囲温度で、すなわち、17〜25℃の範囲内で、そして特に断らない限り、窒素またはアルゴンなどの不活性ガスの雰囲気下で行った;
(ii)概して、反応経過は、薄層クロマトグラフィー(TLC)および/または分析高速液体クロマトグラフィー(HPLC)で追跡したが;与えられている反応時間は、必ずしも達成可能な最小値ではない;
(iii)必要な時に、有機溶液を無水硫酸マグネシウム上で乾燥させ、処理手順は、濾
過による残留固体の除去後に行い、蒸発は、真空中のロータリーエバポレーションによって行った;
(iv)収率は、示されている場合、必ずしも達成可能な最大値ではないが、必要な時に、更に多量の反応生成物が必要とされた場合、反応を繰り返した;
(v)概して、式Iの最終生成物の構造は、核磁気共鳴(NMR)および/または質量スペクトル法で確認した;エレクトロスプレー質量スペクトルデータは、正・負双方のイオンデータを獲得する Waters ZMDまたは Waters ZQ LC/質量分析計を用いて得たが、概して、親構造に関するイオンのみを報告している;プロトンNMR化学シフト値は、300MHzの場の強さで操作する Bruker Spectrospin DPX300分光計を用いてδスケールで測定した;次の略語を用いた:s,一重線;d,二重線;t,三重線;q,四重線;m,多重線;br,幅広;
(vi)中間体は、必ずしも完全に精製しなかったが、それらの構造および純度は、TLC、分析HPLC、赤外(IR)および/またはNMR分析によって評価した;
(vii)特に断らない限り、カラムクロマトグラフィー(フラッシュ法による)および
中圧液体クロマトグラフィー(MPLC)は、Merck Kieselgel シリカ(Art. 9385
)上で行った;
(viii)分取HPLCは、C18逆相シリカ上、例えば、Waters ‘Xterra’分取逆相
カラム(5ミクロンシリカ、19mm直径、100mm長さ)上で、溶離剤として減少極性混合物、例えば、水(1%酢酸または1%水性水酸化アンモニウム(d=0.88)を含有する)およびアセトニトリルの減少極性混合物を用いて行った;
(ix)次の分析HPLC法を用いた;概して、逆相シリカは、約1ml/分の流速で用い、そして検出は、230nmの波長でのUV吸光度によった。
【0220】
方法A:Phenomenex LUNAフェニルヘキシルカラム(Phenomenex, Macclesfield, UK;3ミクロンシリカ、2mm直径、50mm長さ)、溶媒Aは、0.05%トリフルオ
ロ酢酸を含有する水であり、溶媒Bは、0.05%トリフルオロ酢酸を含有するメタノールであり、そして95:5の溶媒Aおよび溶媒Bの混合物〜0:100の溶媒Aおよび溶媒Bの混合物の5分間にわたる溶媒勾配を用いた;
方法B:Phenomenex PRODIGY ODSカラム(5ミクロンシリカ、4.6mm
直径、150mm長さ)、溶媒Aは、900:100:0.5:0.5の水、アセトニトリル、トリフルオロ酢酸および酢酸の混合物であり、溶媒Bは、50:950:0.5:
0.5の水、アセトニトリル、トリフルオロ酢酸および酢酸の混合物であり、そして100%溶媒A〜60:40の溶媒Aおよび溶媒Bの混合物の8分間にわたる溶媒勾配、そして更に、60:40の溶媒Aおよび溶媒Bの混合物〜100%溶媒Bの10分間にわたる溶媒勾配を用いた。
【実施例】
【0221】
実施例1
5,7−ジフルオロ−3,4−ジヒドロキナゾリン−4−オン(経路1)
2,4,6−トリフルオロベンゾニトリル(10g)を、イソプロパノール中の4.9Mアンモニア撹拌溶液(220ml;イソプロパノール中にアンモニアを通気することによって調製される)に加え、得られた混合物を45℃に16時間加熱した。溶媒を蒸発させて、2:1の2−アミノ−4,6−ジフルオロベンゾニトリルおよび4−アミノ−2,6−ジフルオロベンゾニトリルの混合物を含む白色固体(11.9g)を残した。
【0222】
その混合物の一部分(5g)を、水(10ml)中に懸濁させ、濃硫酸水(80%;40ml)を加えた。得られた混合物を撹拌し、65℃に16時間加熱した。得られた溶液を、周囲温度に冷却し、水(60ml)で希釈し、10M水性水酸化ナトリウム(180ml)の添加によって塩基性にし、そして酢酸エチルで抽出した。有機溶液を、硫酸マグネシウム上で乾燥させ、蒸発させた。このようにして、2:1の2−アミノ−4,6−ジフルオロベンズアミドおよび4−アミノ−2,6−ジフルオロベンズアミドの混合物を含むクリーム色固体(4g)を得た。
【0223】
そのようにして得られた混合物を、オルトギ酸トリエチル(60ml)中に懸濁させた。濃塩酸水(0.1ml)を加え、得られた混合物を146℃に8時間加熱した。反応混合物を周囲温度に冷却させた。得られた濃厚懸濁液を濾過し、メチル tert−ブチルエー
テル(20ml)で洗浄した。そのようにして得られた物質を、真空中において35℃で3時間乾燥させた。このようにして、5,7−ジフルオロ−3,4−ジヒドロキナゾリン−4−オンを得た(1.61g;方法Aを用いて97%のHPLC純度、保持時間2.29分)。
【0224】
NMRスペクトル:(DMSOd6) 7.3-7.4 (m, 2H), 8.12 (s, 1H)。
実施例2
5,7−ジフルオロ−3,4−ジヒドロキナゾリン−4−オン(経路2)
実施例1に記載の2:1の2−アミノ−4,6−ジフルオロベンゾニトリルおよび4−アミノ−2,6−ジフルオロベンゾニトリルの混合物の一部分(0.5g)を、シリカ上のカラムクロマトグラフィーにより、塩化メチレンおよびメタノールの漸増極性混合物を溶離剤として用いて精製した。このようにして、2−アミノ−4,6−ジフルオロベンゾニトリル(0.15g)を得た。そのようにして得られた物質、濃硫酸水(80%;4ml)および水(1ml)の混合物を、100℃に15時間加熱した。得られた溶液を、周囲温度に冷却し、水で希釈し、10M水酸化ナトリウム水溶液の添加によって塩基性にし、酢酸エチル(10ml)で洗浄した。得られた水溶液を、希塩酸水溶液の添加によって中和し、酢酸エチル(20ml)で抽出した。有機層を、硫酸マグネシウム上で乾燥させ、蒸発させた。このようにして、2−アミノ−4,6−ジフルオロ安息香酸を無色固体として得た(0.11g;方法Bを用いて97%のHPLC純度、保持時間6.87分)。
【0225】
NMRスペクトル:(DMSOd6) 6.25 (m, 1H), 6.4 (m, 1H)。
そのようにして得られた物質、1,3,5−トリアゼン(0.044g)、メタノール(4ml)およびピペリジン(0.038ml)の混合物を、70℃に24時間加熱した。得られた混合物を、周囲温度に冷却し、蒸発させた。ジエチルエーテル(3ml)および酢酸エチル(1ml)を加え、得られた固体を単離し、ジエチルエーテル(1ml)で
洗浄した。このようにして、5,7−ジフルオロ−3,4−ジヒドロキナゾリン−4−オン(0.048g)を得た。
【0226】
実施例3
7−フルオロ−5−テトラヒドロピラン−4−イルオキシ−3,4−ジヒドロキナゾリン−4−オン
カリウム tert−ブトキシド(6.15g)を、THF(40ml)中の4−ヒドロキ
シテトラヒドロピラン(2.94g)の溶液に加え、その混合物を周囲温度で15分間撹拌した。得られた混合物を、加熱して還流しているTHF(60ml)中の5,7−ジフルオロ−3,4−ジヒドロキナゾリン−4−オン(5g)の撹拌溶液に加えた。追加部分のTHF(20ml)を加え、反応混合物を加熱して30分間還流した。第二部分のカリウム tert−ブトキシド(6.15g)を加え、反応混合物を加熱して40分間還流した
。第三部分のカリウム tert−ブトキシド(1.52g)を加え、反応混合物を加熱して
20分間還流した。反応混合物を周囲温度に冷却させた。水(50ml)を加え、大部分の有機溶媒を蒸発させた。残留物を、2M水性塩酸の滴加によってpH<2へと酸性にした。得られたスラリーを15分間撹拌した。混合物を濾過し、そして単離された固体を、水(20ml)で洗浄し、真空中において40℃で一晩乾燥させた。このようにして、7−フルオロ−5−テトラヒドロピラン−4−イルオキシ−3,4−ジヒドロキナゾリン−4−オンを得た(5.96g;方法Aを用いて96%のHPLC純度、保持時間3.34分)。
【0227】
NMRスペクトル:(DMSOd6) 1.6-1.75 (m, 2H), 1.9-2.0 (m, 2H), 3.5-3.6 (m, 2H),
3.85-3.95 (m, 2H), 4.8 (m, 1H), 6.9 (m, 1H), 7.05 (m, 1H), 8.0 (s, 1H)。
実施例4
7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシ−3,4−ジヒドロキナゾリン−4−オン
カリウム tert−ブトキシド(3.77g)を、THF(30ml)中の1−(2−ヒ
ドロキシエチル)−4−メチルピペラジン(国際出願WO01/94341号、実施例2、注記[9];1.78g)の溶液に加え、その混合物を10分間撹拌した。得られた溶液を、THF(50ml)中の7−フルオロ−5−テトラヒドロピラン−4−イルオキシ−3,4−ジヒドロキナゾリン−4−オン(2.96g)の撹拌スラリーに加え、得られた溶液を加熱して3時間還流した。第二部分のカリウム tert−ブトキシド(2.52g
)を加え、反応混合物を加熱して16時間還流した。反応混合物を周囲温度に冷却させた。水(25ml)を加え、大部分の有機溶媒を蒸発させた。残留物を、2M水性塩酸の滴加によって中和し、酢酸エチルで抽出した。有機相を硫酸マグネシウム上で乾燥させ、蒸発させた。残留物を、シリカ上のカラムクロマトグラフィーにより、塩化メチレンおよびメタノールの漸増極性混合物を溶離剤として用いて精製した。このようにして、7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシ−3,4−ジヒドロキナゾリン−4−オンを得た(3.1g;方法Bを用いて91%のHPLC純度、保持時間1.1分)。
【0228】
NMRスペクトル:(CDCl3) 1.9-2.0 (m, 2H), 2.0-2.15 (m, 2H), 2.35 (s, 3H), 2.4-2.8 (br m, 8H), 2.85 (t, 2H), 3.6-3.7 (m, 2H), 4.1-4.15 (m, 2H), 4.2 (t, 2H), 4.65 (m, 1H), 6.55 (s, 1H), 6.85 (s, 1H), 7.25 (s, 1H), 7.9 (s, 1H)。
【0229】
実施例5
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−5,7−ジフルオロキナゾリン(経路1)
塩化ホスホリル(3.32ml)を、5,7−ジフルオロ−3,4−ジヒドロキナゾリン−4−オン(5g)、ジイソプロピルエチルアミン(7.16ml)およびアセトニト
リル(120ml)の氷浴中で冷却された撹拌混合物に加えた。得られた反応混合物を、80℃に2時間加熱した。第二部分の塩化ホスホリル(1.52ml)を加え、反応混合物を加熱して更に2.75時間還流して、4−クロロ−5,7−ジフルオロキナゾリンの溶液を与え、それを、単離することなく用いた。アセトニトリル(15ml)中の6−クロロ−2,3−メチレンジオキシアニリン(国際出願WO01/94341号、実施例17、注記[30];4.95g)の溶液を加え、反応混合物を80℃に4時間加熱した。得られた反応混合物を、周囲温度で16時間撹拌した。アセトニトリル(5ml)中の6−クロロ−2,3−メチレンジオキシアニリン(1.18g)の第二部分溶液を加え、反応混合物を80℃に1時間再加熱した。反応混合物を周囲温度に冷却させ、1時間撹拌した。得られたスラリーを濾過し、そして単離された固体をアセトニトリル(20ml)で洗浄し、乾燥させた。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−5,7−ジフルオロキナゾリンを一塩酸塩として得た(7.88g、方法Aを用いて99.3%のHPLC純度、保持時間4.46分)。
【0230】
NMRスペクトル:(DMSOd6) 5.5-6.0 (br s, 1H), 6.15 (s, 2H), 7.0 (d, 1H), 7.1 (d, 1H), 7.6 (d, 1H), 7.8 (m, 1H), 8.7 (s, 1H).1.9-2.0 (m, 2H)。
実施例6
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−5,7−ジフルオロキナゾリン(経路2)
トリメチルアルミニウム(トルエン中の2M溶液、4.69ml)を、トルエン(10ml)中の6−クロロ−2,3−メチレンジオキシアニリン(1.07g)の撹拌溶液に加え、得られた溶液を周囲温度で15分間撹拌した。トルエン(10ml)中の2,4,6−トリフルオロベンゾニトリル(0.98g)の溶液を滴加し、得られた混合物を周囲温度で10分間撹拌した後、90℃に3時間加熱した。反応混合物を、周囲温度に冷却し、16時間撹拌した。反応混合物を、水(20ml)で洗浄した。有機溶液を、10%クエン酸水溶液で抽出した。水溶液を、2M水性水酸化ナトリウムで塩基性にし、塩化メチレン(50ml)で抽出した。有機溶液を、硫酸マグネシウム上で乾燥させ、蒸発させた。このようにして、N1−(6−クロロ−2,3−メチレンジオキシフェニル)−2,4,6−トリフルオロベンズアミジン(0.92g)を得た。
【0231】
ホルムアミジン酢酸塩(0.185g)を、トルエン(5ml)中のN1−(6−クロロ−2,3−メチレンジオキシフェニル)−2,4,6−トリフルオロベンズアミジン(0.204g)の撹拌溶液に加え、反応混合物を加熱して16時間還流した。第二部分のホルムアミジン酢酸塩(0.185g)を加え、反応混合物を加熱して更に16時間還流した。トリエチルアミン(0.25ml)を加え、反応混合物を加熱して更に3日間還流した。得られた反応混合物を、周囲温度に冷却し、塩化メチレン(25ml)と飽和重炭酸ナトリウム水溶液(25ml)とに分配した。有機溶液を、10%水性クエン酸(25ml)で洗浄し、硫酸マグネシウム上で乾燥させ、蒸発させた。得られた油状物を、シリカゲル上のカラムクロマトグラフィーにより、イソヘキサンおよび酢酸エチルの漸増極性混合物を溶離剤として用いて精製した。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−5,7−ジフルオロキナゾリン(0.068g)を得た。
【0232】
実施例7
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−5,7−ジフルオロキナゾリン
塩化ホスホリル(4.96ml)を、5,7−ジフルオロ−3,4−ジヒドロキナゾリン−4−オン(6.5g)、クロロベンゼン(64.9ml)、6−クロロ−2,3−メチレンジオキシアニリン(7.08g)およびジイソプロピルエチルアミン(7.47ml)の、窒素ガス雰囲気下において95℃に加熱された撹拌混合物に、40分間にわたって加えた。得られた反応混合物を、95℃で5時間加熱した。反応混合物を18℃に冷却
し、30分間撹拌した。撹拌を止め、反応混合物を30分間放置した。その混合物を濾過し、そして単離された固体を、クロロベンゼン(2x23ml)で洗浄し、真空中において45℃で乾燥させた。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−5,7−ジフルオロキナゾリンを一塩酸塩として得た(8.9g、方法Aを用いて96.5%のHPLC純度、保持時間4.46分)。
【0233】
m.p.234〜237℃;
NMRスペクトル:(DMSOd6) 5.5-6.0 (br s, 1H), 6.15 (s, 2H), 7.0 (d, 1H), 7.1 (d, 1H), 7.6 (d, 1H), 7.8 (m, 1H), 8.7 (s, 1H)。
【0234】
実施例8
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−フルオロ−5−テトラヒドロピラン−4−イルオキシキナゾリン(経路1)
7−フルオロ−5−テトラヒドロピラン−4−イルオキシ−3,4−ジヒドロキナゾリン−4−オンの第一部分(0.25g)を、塩化ホスホリル(1.76ml)、ジイソプロピルエチルアミン(3.95ml)およびアセトニトリル(10ml)の80℃に加熱された撹拌混合物に加えた。得られた混合物を、80℃に3時間加熱した。第二部分の7−フルオロ−5−テトラヒドロピラン−4−イルオキシ−3,4−ジヒドロキナゾリン−4−オン(0.25g)を加え、その混合物を、加熱して更に90分間還流した。このようにして、4−クロロ−7−フルオロ−5−テトラヒドロピラン−4−イルオキシキナゾリンの溶液を得、それを、単離することなく用いた。アセトニトリル(3ml)中の6−クロロ−2,3−メチレンジオキシアニリン(0.32g)の溶液を加え、反応混合物を80℃に2.5時間加熱した。必要な変換が不完全であったので、反応混合物を蒸発させ、そしてトルエン(15ml)を反応溶媒として加えた。第二部分の6−クロロ−2,3−メチレンジオキシアニリン(0.32g)を加え、反応混合物を加熱して3時間還流した。反応混合物を周囲温度に冷却し、そして塩化メチレンと塩化ナトリウム水溶液とに分配した。有機相を水で洗浄し、硫酸マグネシウム上で乾燥させ、蒸発させた。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−フルオロ−5−テトラヒドロピラン−4−イルオキシキナゾリンを泡状物(0.73g)として得た。
【0235】
NMRスペクトル:(DMSOd6) 1.9-2.05 (m, 2H), 2.1-2.2 (m, 2H), 3.5-3.6 (m, 2H),
3.8-3.95 (m, 2H), 5.1 (m, 1H), 6.1 (s, 2H), 7.0 (d, 1H), 7.1 (d, 1H), 7.3 (d, 1H), 7.4 (m, 1H), 8.6 (s, 1H), 9.3 (s, 1H)。
【0236】
実施例9
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−フルオロ−5−テトラヒドロピラン−4−イルオキシキナゾリン(経路2)
カリウム tert−ブトキシド(5.42g)およびTHF(30ml)の混合物を、T
HF(30ml)中の4−ヒドロキシテトラヒドロピラン(1.53ml)の溶液に加え、得られた混合物を20分間撹拌した。THF(30ml)中の4−(6−クロロ−2,3−メチレンジオキシアニリノ)−5,7−ジフルオロキナゾリン塩酸塩(6g)のスラリーを加え、得られた混合物を加熱して1.75時間還流した。第二部分のカリウム tert−ブトキシド(1.81g)を加え、その混合物を加熱して更に2時間還流した。第二
部分の4−ヒドロキシテトラヒドロピラン(0.15ml)および第三部分のカリウム tert−ブトキシド(0.45g)を加え、その混合物を加熱して0.5時間還流した。第
四部分のカリウム tert−ブトキシド(0.9g)を加え、その混合物を加熱して更に2
0分間還流した。得られた反応混合物を50℃に冷却させ、そしてブライン(60ml)および水(30ml)を順次加えた。層を分離し、そして水溶液を、順次、THF(30ml)でおよび酢酸イソプロピル(30ml)で抽出した。有機抽出物を一緒にし、ブライン(30ml)で洗浄した。有機溶液を蒸発させた。残留する固体を、メチル tert−
ブチルエーテル(24ml)およびイソヘキサン(12ml)の混合物下で1時間撹拌した。その固体を単離し、1:1のメチル tert−ブチルエーテルおよびイソヘキサンの混
合物で洗浄し、真空中において40℃で一晩乾燥させた。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−フルオロ−5−テトラヒドロピラン−4−イルオキシキナゾリンを得た(5.02g、方法Aを用いて93%のHPLC純度、保持時間4.61分)。そのようにして得られた物質の一部分(3g)を、熱酢酸エチル(54ml)中に溶解させた。その熱溶液を濾過した。濾液を周囲温度に冷却させ、3時間撹拌した。得られた固体を、濾過によって単離し、真空中において周囲温度で乾燥させた。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−フルオロ−5−テトラヒドロピラン−4−イルオキシキナゾリンを得た(1.61g、方法Aを用いて99.2%のHPLC純度、保持時間4.51分)。
【0237】
NMRスペクトル:(DMSOd6) 1.9-2.0 (m, 2H), 2.1-2.2 (m, 2H), 3.5-3.6 (m, 2H), 3.8-3.95 (m, 2H), 5.1 (m, 1H), 6.1 (s, 2H), 6.95 (d, 1H), 7.1 (d, 1H), 7.2 (d, 1H), 7.3 (d, 1H), 8.4 (s, 1H), 9.3 (s, 1H)。
【0238】
実施例10
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−フルオロ−5−テトラヒドロピラン−4−イルオキシキナゾリン(経路3)
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−5,7−ジフルオロキナゾリン塩酸塩(80g)を、ナトリウムtert−ペントキシド(90.2g)およびN−メ
チルピロリジン−2−オン(500ml)の窒素ガス雰囲気下の撹拌混合物に少量ずつ加えた。4−ヒドロキシテトラヒドロピラン(23.5ml)およびN−メチルピロリジン−2−オン(35ml)を加え、得られた混合物を60℃に3時間加熱した。水(764ml)を、加熱された反応混合物に3時間で加え、その混合物を撹拌し、60℃に更に3時間加熱した。その温反応混合物を濾過し、単離された固体を水(2x230ml)で洗浄し、真空中において恒量まで乾燥させた。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−フルオロ−5−テトラヒドロピラン−4−イルオキシキナゾリンを得た(68.6g、方法Aを用いて95%のHPLC純度、保持時間4.6分)。
【0239】
m.p.209〜212℃;
NMRスペクトル:(DMSOd6) 1.9-2.0 (m, 2H), 2.1-2.2 (m, 2H), 3.5-3.6 (m, 2H), 3.8-3.95 (m, 2H), 5.05 (m, 1H), 6.1 (s, 2H), 6.95 (d, 1H), 7.05 (d, 1H), 7.1 (d,
1H), 7.3 (d, 1H), 8.4 (s, 1H), 9.3 (s, 1H)。
【0240】
実施例11
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン(経路1)
トルエン(3ml)中の第一部分の7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシ−3,4−ジヒドロキナゾリン−4−オン(0.19g)を、塩化ホスホリル(0.059ml)、ジイソプロピルエチルアミン(0.13ml)およびトルエン(3ml)の80℃に加熱された撹拌混合物に加え、得られた混合物を80℃に6時間加熱した。その混合物を、周囲温度に冷却させ、一晩撹拌した。その混合物を80℃に再加熱し、そしてトルエン(2ml)中の6−クロロ−2,3−メチレンジオキシアニリン(0.088g)の溶液を加えた。得られた混合物を撹拌し、80℃に1.5時間加熱した。混合物を周囲温度に冷却し、そして析出した油状ガムから溶媒を傾瀉した。その油状ガムを、DMF(3ml)中に懸濁させ、第二部分の7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−
4−イルオキシ−3,4−ジヒドロキナゾリン−4−オン(0.088g)を加え、反応混合物を100℃に9時間加熱した。混合物を周囲温度に冷却させ、そして酢酸エチルと2M塩酸水溶液(10ml)とに分配した。その水溶液を、10M水酸化ナトリウム水溶液(10ml)の添加によって塩基性にし、塩化メチレンで抽出した。有機溶液を硫酸マグネシウム上で乾燥させ、蒸発させた。得られた油状物を、シリカ上のカラムクロマトグラフィーにより、塩化メチレンおよびメタノールの漸増極性混合物を溶離剤として用いて精製した。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン(0.008g)を得た。
【0241】
NMRスペクトル:(DMSOd6) 1.85-1.95 (m, 2H), 2.1-2.2 (m, 2H), 2.2 (s, 3H), 2.2-2.4 (m, 4H), 2.4-2.6 (m, 4H), 2.87 (m, 2H), 3.5-3.6 (m, 2H), 3.8-3.9 (m, 2H), 4.2 (m, 2H), 5.1 (m, 1H), 6.1 (s, 2H), 6.85 (s, 1H), 6.9 (s, 1H), 6.95 (d, 1H), 7.05 (d, 1H), 8.35 (s, 1H), 9.2 (s, 1H)。
【0242】
実施例12
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン(経路2)
窒素ガス雰囲気下において、塩化ホスホリル(0.07ml)を、7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシ−3,4−ジヒドロキナゾリン−4−オン(0.2g)、ジイソプロピルエチルアミン(0.22ml)およびブチロニトリル(2ml)の96℃に加熱された撹拌混合物に加え、得られた混合物を96℃に4時間加熱した。第二部分の塩化ホスホリル(0.12ml)を加え、得られた混合物を96℃に1.7時間加熱した。6−クロロ−2,3−メチレンジオキシアニリン(0.098g)を加え、得られた混合物を96℃に2時間加熱した。混合物を周囲温度に冷却させた。水(2ml)を加え、有機層を分離した。水性層をブチロニトリル(1ml)で洗浄した。水性層を、濃水酸化ナトリウム水溶液(47%w/w)の添加によってpH9へと塩基性にし、n−ブタノール(2x2ml)で抽出した。得られた有機層を一緒にし、蒸発させた。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン(0.094g)を得た。
【0243】
実施例13
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン(経路3)
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−フルオロ−5−テトラヒドロピラン−4−イルオキシキナゾリン(0.5g)を、水酸化カリウム(0.168g)、1−(2−ヒドロキシエチル)−4−メチルピペラジン(0.69g)およびジ−(2−メトキシエチル)エーテル(10ml)の120℃に加温された撹拌混合物に加え、得られた反応混合物を120℃に12時間加熱した。反応混合物を周囲温度に冷却し、1M水性塩酸(9ml)の添加によってpH1〜3へと酸性にし、酢酸イソプロピル(20 ml)で洗浄した。その水溶液を撹拌し、そして2M水性水酸化ナトリウム(5ml)の添加によってpH13〜14へと塩基性にした。10分後、水(22ml)を加え、混合物を2時間撹拌して、固体の沈殿を終了させた。その混合物を10℃に冷却し、濾過した。得られた固体を、水(20ml)で洗浄し、真空中において40℃で乾燥させた。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンを得た(0.47g、方法Bを用いて92.5%のHPLC純度、保持時
間7.3分)。
【0244】
NMRスペクトル:(CDCl3) 1.65 (br s, 3H), 1.9-2.05 (m, 2H), 2.2-2.3 (m, 2H), 2.31 (s, 3H), 2.4-2.8 (m, 8H), 2.9 (m, 2H), 3.6-3.7 (m, 2H), 3.95-4.05 (m, 2H), 4.2-4.25 (m, 2H), 4.8 (m,1H), 6.05 (s, 2H), 6.55 (s, 1H), 6.75 (d, 1H), 6.85 (s,
1H), 7.0 (d, 1H), 8.55 (s, 1H), 9.25 (s, 1H)。
【0245】
実施例14
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン(経路4)
窒素ガス雰囲気下において、1−(2−ヒドロキシエチル)−4−メチルピペラジン(13.93g)を、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−フルオロ−5−テトラヒドロピラン−4−イルオキシキナゾリン(12.9g)、ナトリウム
tert−ペントキシド(9.87g)および1,2−ジエトキシエタン(37.5ml)
の撹拌混合物に加えた。水(1.34g)および1,2−ジエトキシエタン(25ml)を加え、得られた混合物を撹拌し、86℃に18時間加熱した。反応混合物を50℃に冷却し、そして真空蒸留下において約60ミリバール圧力で、約50mlの反応溶媒を留去した。濃塩酸水(36%,10ml)および水(84ml)の混合物を、反応混合物の温度を最大60℃で保持する速度で添加することにより、反応混合物をpH7.0〜7.6に中和した。反応混合物をの温度を60℃で保持しながら、反応混合物を酢酸エチル(225ml)で抽出した。有機溶液を水(50ml)で洗浄した。水(25ml)を加え、そして温度を60℃で保持しながら混合物を10分間撹拌後、30分間放置し、そして水性層を分離した。溶媒を大気圧下において約90℃で蒸留することにより、有機層を濃縮して約100mlの容量とした。残留する混合物を、45℃に1時間で冷却し、その温度で2時間保持して、生成物を結晶化させた。その混合物を55℃に短時間加温した後、18℃に4時間で冷却し、その温度で1時間保持した。結晶性沈殿を濾過によって単離し、そして順次、水(17ml)で、および tert−ブチルメチルエーテル(17ml)で洗
浄した。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンを三水和物として得た(11g;方法Bを用いて88%のHPLC純度、保持時間7.3分)。
【0246】
NMRスペクトル:(CDCl3) 1.65 (br s, 3H), 1.9-2.05 (m, 2H), 2.2-2.3 (m, 2H), 2.31 (s, 3H), 2.4-2.8 (m, 8H), 2.9 (m, 2H), 3.6-3.7 (m, 2H), 3.95-4.05 (m, 2H), 4.2-4.25 (m, 2H), 4.8 (m,1H), 6.05 (s, 2H), 6.55 (s, 1H), 6.75 (d, 1H), 6.85 (s,
1H), 7.0 (d, 1H), 8.55 (s, 1H), 9.25 (s, 1H)。
【0247】
そのようにして得られた物質の一部分(10g)を、フィルター上に置き、乾燥窒素ガス流中において周囲温度で乾燥させた。得られた物質を、乾燥窒素雰囲気を維持しながら、乾燥イソプロパノール(140ml)中に60℃で溶解させた。その溶液を、周囲温度に冷却させ、乾燥窒素雰囲気下で2日間放置した。得られた結晶性固体を、乾燥窒素雰囲気下の濾過によって単離した。そのようにして得られた物質(8g)は、無水物結晶形の4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンであった。
【0248】
m.p.142〜144℃。
実施例15
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピ
ペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンジフマレート塩
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン三水和物(27.1g)、イソプロパノール(200ml)および水(10ml)の混合物を、75℃に加熱した。フマル酸(12.8g)、イソプロパノール(200ml)および水(40ml)の混合物を、80℃に加熱した。
【0249】
その加温されたキナゾリン化合物溶液の一部分(80ml)を、フマル酸溶液に、温度を75℃で維持しながら加えた。得られた混合物を75℃で75分間撹拌した。残りのキナゾリン化合物溶液を、温度を75℃で維持しながら1時間で加えた。イソプロパノール(50ml)を加え、得られた混合物を75℃で7時間撹拌した。
【0250】
その混合物を、50℃に少なくとも25分間にわたって徐々に冷却し、その温度で6時間撹拌した。混合物を、20℃に少なくとも20分間にわたって徐々に冷却し、その温度で18.5時間撹拌した。結晶性固体を、濾過によって単離し、10:1のイソプロパノールおよび水の混合物で2回洗浄し(それぞれ、50mlおよび100ml)、真空中において45℃で恒量まで乾燥させた。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンジフマレート塩(37.0g)を得た。
【0251】
m.p.233〜237℃;
NMRスペクトル:(DMSOd6) 1.76-1.88 (m, 2H), 2.1-2.17 (m, 2H), 2.33 (s, 3H), 2.6 (br s, 8H), 2.78 (t, 2H), 3.51-3.6 (m, 2H), 3.83-3.9 (m, 2H), 4.24 (t, 2H), 4.98-5.07 (m, 1H), 6.07 (s, 2H), 6.6 (s, 4H), 6.83 (d, 1H), 6.84 (d, 1H), 6.91 (d, 1H), 7.05 (d, 1H), 8.33 (s, 1H), 9.18 (s, 1H)。
【0252】
実施例16
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンジフマレート塩
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン三水和物(27.1g)、イソプロパノール(210ml)および水(30ml)の混合物を、40℃に加熱し、混合物を濾過した。フィルターを、イソプロパノール(20ml)で洗浄し、そしてその洗液を、温濾液に加えた。得られた溶液を75℃に加温した。
【0253】
フマル酸(12.8g)、イソプロパノール(200ml)および水(20ml)の混合物を、70℃に加熱し、得られた混合物を濾過した。フマル酸溶液の一部分(110ml)を、加温された4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン溶液に、温度を75℃で維持しながら加えた。4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンジフマレート塩の種結晶(0.02g)を加え、得られた混合物を75℃で1時間撹拌した。残りのフマル酸溶液を、温度を75℃で維持しながら1時間で加え、得られた混合物を75℃で14時間撹拌した。
【0254】
その混合物を、20℃に少なくとも2時間にわたって徐々に冷却し、その温度で1時間
撹拌した。結晶性固体を濾過によって単離し、10:1のイソプロパノールおよび水の混合物で2回洗浄し(それぞれ、50mlおよび100ml)、真空中において45℃で恒量まで乾燥させた。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンジフマレート塩(35.8g)を得た。
【0255】
m.p.234〜237℃;
NMRスペクトル:(DMSOd6) 1.76-1.88 (m, 2H), 2.1-2.17 (m, 2H), 2.33 (s, 3H), 2.6 (br s, 8H), 2.78 (t, 2H), 3.51-3.6 (m, 2H), 3.83-3.9 (m, 2H), 4.24 (t, 2H), 4.98-5.07 (m, 1H), 6.07 (s, 2H), 6.6 (s, 4H), 6.83 (d, 1H), 6.84 (d, 1H), 6.91 (d, 1H), 7.05 (d, 1H), 8.33 (s, 1H), 9.18 (s, 1H)。
【0256】
実施例17
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンセスキフマレート塩
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンジフマレート(0.15g)および水(20ml)の混合物を、ヒートガンを用いて加温して、溶液を得た。その試料を、空気流下において周囲温度で徐々に24時間蒸発させて、約3mlの容量としたが、ここで、沈殿が形成し始めた。その混合物を、冷蔵庫中に4℃で2日間置いた。得られた沈殿を、濾過によって単離し、水で洗浄した。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンをセスキフマレート四水和物塩(0.084g)として得、それを、XRPD、DSC、TGA、FTIRおよび溶液NMR技法を用いて特性決定した。
【図面の簡単な説明】
【0257】
【図1】図1は、AZD0530ジフマレートについての、水平軸上にプロットされた2θ値および垂直軸上にプロットされた相対線強さ(計数)でのX線粉末回折図形を示す。
【図2】図2は、AZD0530セスキフマレート四水和物についての、水平軸上にプロットされた2θ値および垂直軸上にプロットされた相対線強さ(計数)でのX線粉末回折図形を示す。
【図3】図3は、AZD0530三水和物についての、水平軸上にプロットされた2θ値および垂直軸上にプロットされた相対線強さ(計数)でのX線粉末回折図形を示す。
【図4】図4は、無水AZD0530についての、水平軸上にプロットされた2θ値および垂直軸上にプロットされた相対線強さ(計数)でのX線粉末回折図形を示す。
【図5】図5は、AZD0530ジフマレートについての、水平軸上にプロットされた4000〜400cm−1の度数範囲および垂直軸上にプロットされた吸光度でのDRIFTスペクトルを示す。
【図6】図6は、AZD0530セスキフマレート四水和物についての、水平軸上にプロットされた4000〜400cm−1の度数範囲および垂直軸上にプロットされた吸光度でのDRIFTスペクトルを示す。
【技術分野】
【0001】
本発明は、抗腫瘍性を有するある種のキナゾリン誘導体またはそれらの薬学的に許容しうる塩の製造において有用な改善された化学的方法および中間体に関する。本発明は、更に、それら中間体の製造方法、およびそれら中間体を利用したこのようなキナゾリン誘導体の製造方法に関する。本発明は、更に、抗腫瘍性を各々有する、ある種のキナゾリン誘導体の特定の結晶形およびそれらの特定の結晶性の薬学的に許容しうる塩に関する。
【背景技術】
【0002】
具体的には、本発明は、化合物4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンの製造に有用な化学的方法および中間体に関するが、この化合物は、国際特許出願WO01/94341号の実施例14の表中に化合物番号73として開示されている。
【0003】
その化合物は、本明細書中に、式I
【0004】
【化1】
【0005】
によって、および化合物が知られているコード番号AZD0530として記載されている。
AZD0530は、非受容体チロシンキナーゼ酵素のSrcファミリーの阻害剤であり、したがって、腫瘍細胞の運動性の選択的阻害剤および哺乳動物癌細胞の播種および浸潤性の選択的阻害剤であって、転移性腫瘍成長の阻害をもたらす。具体的には、化合物AZD0530は、c−Src非受容体チロシンキナーゼの阻害剤であるので、ヒトまたは動物体の充実性腫瘍疾患の封じ込めおよび/または処置に用いるための抗浸潤薬として価値があるはずである。
【0006】
国際特許出願WO01/94341号に開示されている式Iの化合物を製造する経路は、化合物4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−ヒドロキシ−5−テトラヒドロピラン−4−イルオキシキナゾリンとアルキル化剤との、7位に2−(4−メチルピペラジン−1−イル)エトキシ側鎖を形成する反応を包含する。その反応生成物は、WO01/94341号に、二塩酸塩の形でおよび遊離塩基の形で開示されている。
【0007】
この既存の経路は、比較的少量の式Iの化合物の合成には十分であるが、その経路は、収束的よりもむしろ直線的な合成を包含し、多数のクロマトグラフィー精製工程の使用およびかなり多数の中間体の単離を必要とする。それだけでは、全体の合成収率は高くない。したがって、一層多量の式Iの化合物を製造するのに用いるのに適する一層有効なその
化合物の合成が要求されている。好ましくは、新しい合成は、高価で且つ時間のかかるクロマトグラフィー精製法を包含すべきではない。
【発明の開示】
【0008】
本発明により、本発明者は、ここで、式Iの化合物であるAZD0530の製造に適する方法を発明した。それら新しい方法は、最終生成物を高品質で且つ十分な収率で一層大規模に製造することを可能にするという点で好都合である。それら方法は、単離されるべき中間体の数の実質的な減少を可能にし、そして概して、従来の経路より収束的である。このような変化は、時間および費用について有意の利点を与える。好都合には、クロマトグラフィー精製法を必要としない。
【0009】
本発明により、更に、AZD0530の製造に用いることができる不可欠な中間体の製造方法を提供する。
本発明のもう一つの側面により、更に、抗腫瘍性を各々有する、AZD0530の特定の結晶形およびそれらの特定の結晶性の薬学的に許容しうる塩を提供する。
【0010】
本発明のもう一つの側面により、更に、式I
【0011】
【化2】
【0012】
を有する化合物であるAZD0530の製造方法であって、式II
【0013】
【化3】
【0014】
(式中、Lは、置換可能な基であり、そしてNH官能基は、必要ならば保護されている)を有するキナゾリンと、1−(2−ヒドロキシエチル)−4−メチルピペラジンとの、好都合には、適する塩基の存在下における反応を含み;その後、存在するいずれの保護基も、慣用的な手段によって除去し;そしてその後、必要ならば、遊離塩基の形で得られた式Iの化合物は、薬学的に許容しうる塩へと変換することができるし、塩の形で得られた式Iの化合物は、遊離塩基へと変換することができる方法を提供する。
【0015】
その反応は、好都合には、適する塩基、例えば、有機アミン塩基、例えば、ピリジン、
2,6−ルチジン、コリジン、4−ジメチルアミノピリジン、トリエチルアミン、N−メチルモルホリン、N−メチルモルホリンまたはジアザビシクロ[5.4.0]ウンデカ−7−エンなど;または例えば、アルカリ金属またはアルカリ土類金属の炭酸塩または水酸化物、例えば、炭酸ナトリウム、炭酸カリウム、炭酸カルシウム、水酸化ナトリウムまたは水酸化カリウム;または例えば、アルカリ金属アミド、例えば、ナトリウムヘキサメチルジシラザン;または例えば、アルカリ金属水素化物、例えば、水素化ナトリウム;または例えば、アルカリ金属またはアルカリ土類金属の(1−12C)アルコキシド、例えば、ナトリウムまたはカリウム tert−ブトキシド、ナトリウムまたはカリウム tert−ペントキシド、またはナトリウムまたはカリウム3,7−ジメチルオクトキシドの存在下で行うことができる。好都合には、適する塩基は、例えば、アルカリ金属水酸化物、例えば、水酸化ナトリウムまたは水酸化カリウム;または例えば、アルカリ金属(1−6C)アルコキシド、例えば、ナトリウムまたはカリウム tert−ブトキシド、またはナトリウムま
たはカリウム tert−ペントキシドである。より好都合には、適する塩基は、例えば、ア
ルカリ金属(1−6C)アルコキシド、例えば、ナトリウムまたはカリウム tert−ブト
キシド、またはナトリウムまたはカリウム tert−ペントキシドである。
【0016】
適する置換可能な基Lは、例えば、ハロゲノ基、(1−6C)アルコキシ基、アリールオキシ基またはスルホニルオキシ基、例えば、フルオロ基、クロロ基、ブロモ基、メトキシ基、エトキシ基、フェノキシ基、ペンタフルオロフェノキシ基、メタンスルホニルオキシ基またはトルエン−4−スルホニルオキシ基である。好都合には、置換可能な基Lは、ハロゲノ基である。より好都合には、置換可能な基Lは、フルオロ基である。
【0017】
その反応は、好都合には、適する不活性溶媒または希釈剤、または適する不活性溶媒または希釈剤の混合物の存在下で、例えば、ジブチルエーテル、メチル tert−ブチルエー
テル、ジ−(2−メトキシエチル)エーテル、1,2−ジメトキシエタン、1,2−ジエトキシエタン、テトラヒドロフランまたは1,4−ジオキサンなどの置換されていてよいジ−(1−6C)アルキルエーテルまたは環状アルキルエーテル;またはN,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリジン−2−オンまたはジメチルスルホキシドなどの双極性非プロトン性溶媒中で行われる。好都合には、50℃を超える沸点を有する適する不活性溶媒または希釈剤、例えば、ジ−(2−メトキシエチル)エーテルまたは1,2−ジエトキシエタンなどの置換されていてよいジ−(1−6C)アルキルエーテルを用いる。
【0018】
反応は、例えば、0〜250℃の範囲内、好都合には、50〜150℃の範囲内、より好都合には、75〜130℃の範囲内の温度で行われる。
好都合には、NH官能基を保護する必要はない。しかしながら、保護基を用いることが望まれる場合、このような基は、概して、問題の基の保護について適宜、参考文献に記載されているまたは当該化学者に知られているいずれかの基より選択することができるし、慣用法によって導入することができる。保護基は、問題の保護基の除去について適宜、参考文献に記載されているまたは当該化学者に知られているいずれかの好都合な方法によって除去することができるし、このような方法は、分子中のどこか他の基の妨害を最小限にして保護基の除去を行うように選択される。
【0019】
保護基の具体的な例を、便宜上、下に与えるが、ここにおいて、「低級」は、例えば、低級アルキルの場合のように、それが付けられている基が、好ましくは、1〜4個の炭素原子を有するということを意味する。これら例が、完全なものではないということは理解されるであろう。保護基の除去方法の具体例が下に与えられている場合、これらは、同様に完全なものではない。具体的に述べられていない保護基および脱保護方法の使用は、当然ながら、本発明の範囲内である。
【0020】
NH官能基のための保護基の例には、ホルミル基、アリール−低級アルキル基(例えば、ベンジル、および4−メトキシベンジル、2−ニトロベンジルおよび2,4−ジメトキシベンジルなどの置換ベンジル、およびトリフェニルメチル);ジ−4−アニシルメチル基およびフリルメチル基;低級アルコキシカルボニル基(例えば、tert−ブトキシカルボニル);低級アルケニルオキシカルボニル基(例えば、アリルオキシカルボニル);アリール−低級アルコキシカルボニル基(例えば、ベンジルオキシカルボニル、4−メトキシベンジルオキシカルボニル、2−ニトロベンジルオキシカルボニルおよび4−ニトロベンジルオキシカルボニル);トリアルキルシリル基(例えば、トリメチルシリルおよび tert−ブチルジメチルシリル);アルキリデン基(例えば、メチリデン)およびベンジリデ
ン基および置換ベンジリデン基が含まれる。
【0021】
NH官能基のための保護基の除去に適当な方法には、例えば、2−ニトロベンジルオキシカルボニルなどの基の酸、塩基、金属または酵素に触媒された加水分解;ベンジルなどの基の水素化;および2−ニトロベンジルオキシカルボニルなどの基の光分解が含まれる。
【0022】
読者は、反応条件および試薬の一般的な指針については、Advanced Organic Chemistry, 4th Edition, by J. March, published by John Wiley & Sons 1992 を、そして保護基の一般的な指針については、Protective Groups in Organic Synthesis, 2nd Edition, by T. Green et al., also published by John Wiley & Son を参照する。
【0023】
式Iの化合物は、この方法から遊離塩基の形で得ることができるし、または或いは、ハロゲン化水素酸塩などの酸付加塩の形で得ることができる。塩から遊離塩基を得ることが望まれる場合、その塩は、適する塩基、例えば、有機アミン塩基、例えば、ピリジン、2,6−ルチジン、コリジン、4−ジメチルアミノピリジン、トリエチルアミン、N−メチルモルホリンまたはジアザビシクロ[5.4.0]ウンデカ−7−エンなど;または例えば、アルカリ金属またはアルカリ土類金属の炭酸塩または水酸化物、例えば、炭酸ナトリウム、炭酸カリウム、炭酸カルシウム、水酸化ナトリウムまたは水酸化カリウムで処理することができる。式Iの化合物を薬学的に許容しうる塩の形で得ることが望まれる場合、その遊離塩基形を、慣用法を用いて適する酸と反応させて、例えば、塩酸、臭化水素酸、硫酸、トリフルオロ酢酸、クエン酸またはマレイン酸などの無機酸または有機酸との酸付加塩を形成することができる。
【0024】
Lが、本明細書中に前に定義の置換可能な基である式IIのキナゾリン出発物質は、国際特許出願WO01/94341号に開示されたものなどの慣用法によって得ることができる。具体的には、Lがフルオロ基である式IIのキナゾリン出発物質は、国際特許出願WO01/94341号に、例えば、実施例4の表中の化合物番号5の製造について開示されたものなどの慣用法によって得ることができる。
【0025】
本発明のもう一つの特徴により、式III
【0026】
【化4】
【0027】
を有するキナゾリンの製造方法であって、
(a)式IV
【0028】
【化5】
【0029】
を有するキナゾリノンと活性化剤との、好都合には、適する塩基の存在下における、式V
【0030】
【化6】
【0031】
(式中、L1は置換可能な基である)
を有するキナゾリンを形成する反応;
(b)式Vのキナゾリンと、6−クロロ−2,3−メチレンジオキシアニリンとの、好都合には、適する塩基の存在下における、式VI
【0032】
【化7】
【0033】
を有するキナゾリノンを形成する置換反応であって、その後、遊離塩基の形で得られた式
VIの化合物は、塩へと変換することができるし、塩の形で得られた式VIの化合物は、遊離塩基へと変換することができる置換反応;および
(c)式VIのキナゾリンと、4−ヒドロキシテトラヒドロピランとの、好都合には、適する塩基の存在下における、式IIIのキナゾリンを形成する反応
を含み;その後、遊離塩基の形で得られた式IIIの化合物は、塩へと変換することができ
るし、塩の形で得られた式IIIの化合物は、遊離塩基へと変換することができる方法を提
供する。
【0034】
作業工程(a)について、脱離基L1を形成するであろう適する活性化剤は、例えば、塩化ホスホリルまたは臭化ホスホリルなどのハロゲン化ホスホリル;または塩化チオニルなどのハロゲン化剤;または四塩化炭素およびトリフェニルホスフィンの混合物によって形成されるハロゲン化剤;または四塩化炭素およびトリフェニルホスフィンの混合物によって形成されるハロゲン化剤である。或いは、そのようにして得られるいずれかの4−ハロキナゾリンは、必要ならば、ペンタフルオロフェノールとの反応により、炭酸カリウムなどの適する塩基の存在下およびN,N−ジメチルホルムアミドなどの適する溶媒の存在下において4−ペンタフルオロフェノキシキナゾリンへと変換することができる。作業工程(a)の際に用いることができる適する塩基は、例えば、有機アミン塩基、例えば、ピリジン、2,6−ルチジン、コリジン、4−ジメチルアミノピリジン、トリエチルアミン、ジイソプロピルエチルアミン、N−メチルモルホリンまたはジアザビシクロ[5.4.0]ウンデカ−7−エンなどである。作業工程(a)に適する溶媒または希釈剤は、例えば、トルエン、キシレン、クメン、クロロベンゼン、アニソールまたはフェネトールなどの芳香族溶媒である。更に適する溶媒または希釈剤は、アセトニトリル、プロピオニトリル、ブチロニトリル、酢酸エチル、テトラヒドロフランまたは1,4−ジオキサンなどの極性非プロトン性溶媒;またはN,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリジン−2−オンまたはジメチルスルホキシドなどの双極性非プロトン性溶媒である。更に適する溶媒または希釈剤は、水;または第一級、第二級または第三級の(1−6C)アルキルアルコールなどの極性プロトン性溶媒、例えば、メタノール、エタノール、ブタノールまたはペンタノールである。このような適する溶媒または希釈剤の混合物を用いることができる。その反応は、例えば、10〜250℃の範囲内、好都合には、40〜160℃の範囲内の温度で行うことができる。
【0035】
好都合には、作業工程(a)について、適する活性化剤は、例えば、塩化ホスホリルなどのハロゲン化ホスホリルであり、そして反応は、トリエチルアミンまたはジイソプロピルエチルアミンなどの有機アミン塩基の存在下において、トルエン、クロロベンゼン、アニソールまたはアセトニトリルなどの溶媒または希釈剤を用いて、70〜160℃の範囲内、より好都合には、70〜120℃の範囲内の温度で行われる。
【0036】
作業工程(b)の置換反応は、適する酸の存在下または適する塩基の存在下で行うことができる。適する酸は、例えば、無機酸、例えば、塩化水素または臭化水素などである。適する塩基は、例えば、有機アミン塩基、例えば、ピリジン、2,6−ルチジン、コリジン、4−ジメチルアミノピリジン、トリエチルアミン、ジイソプロピルエチルアミン、N−メチルモルホリンまたはジアザビシクロ[5.4.0]ウンデカ−7−エンなど;または例えば、アルカリ金属またはアルカリ土類金属の炭酸塩または水酸化物、例えば、炭酸ナトリウム、炭酸カリウム、炭酸カルシウム、水酸化ナトリウムまたは水酸化カリウム;または例えば、アルカリ金属水素化物、例えば、水素化ナトリウムである。
【0037】
その置換反応は、好都合には、適する溶媒または希釈剤、例えば、イソプロパノール、sec−ブタノールまたは tert−ブタノールなどの第一級、第二級または第三級の(1−6C)アルキルアルコール;塩化メチレン、クロロホルムまたは四塩化炭素などのハロゲン化溶媒;トルエン、キシレン、クメン、クロロベンゼン、アニソールまたはフェネトール
などの芳香族溶媒;アセトニトリル、プロピオニトリル、ブチロニトリル、酢酸エチル、テトラヒドロフランまたは1,4−ジオキサンなどの極性非プロトン性溶媒;またはN,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリジン−2−オンまたはジメチルスルホキシドなどの双極性非プロトン性溶媒の存在下で行われる。このような適する溶媒または希釈剤の混合物を用いることができる。その反応は、好都合には、例えば、10〜250℃の範囲内、好適には、40〜160℃の範囲内、より好都合には、70〜120℃の範囲内の温度で行われる。
【0038】
典型的に、作業工程(b)の置換反応は、イソプロパノールなどのプロトン性溶媒の存在下において、例えば、25〜150℃の範囲内の温度、好都合には、反応溶媒の還流温度でまたはその付近の温度で行うことができる。場合により、その置換反応は、酸の存在下、例えば、ジエチルエーテル中の塩化水素ガス;または式IVの化合物を、塩化チオニルまたは塩化ホスホリルなどのハロゲン化剤である活性化剤と反応させた場合に形成される塩化水素の存在下において行うことができる。
【0039】
作業工程(c)について、反応は、好都合には、適する塩基、例えば、有機アミン塩基、例えば、ピリジン、2,6−ルチジン、コリジン、4−ジメチルアミノピリジン、トリエチルアミン、N−メチルモルホリンまたはジアザビシクロ[5.4.0]ウンデカ−7−エンなど;または例えば、アルカリ金属またはアルカリ土類金属の炭酸塩または水酸化物、例えば、炭酸ナトリウム、炭酸カリウム、炭酸カルシウム、水酸化ナトリウムまたは水酸化カリウム;または例えば、アルカリ金属アミド、例えば、ナトリウムヘキサメチルジシラザン;または例えば、アルカリ金属水素化物、例えば、水素化ナトリウム;または例えば、アルカリ金属またはアルカリ土類金属の(1−12C)アルコキシド、例えば、ナトリウムまたはカリウム tert−ブトキシド、ナトリウムまたはカリウム tert−ペントキシド、またはナトリウムまたはカリウム3,7−ジメチルオクトキシドの存在下で行うことができる。好都合には、適する塩基は、例えば、アルカリ金属水酸化物、例えば、水酸化ナトリウムまたは水酸化カリウム;または例えば、アルカリ金属(1−6C)アルコキシド、例えば、ナトリウムまたはカリウム tert−ブトキシド、ナトリウムまたはカリ
ウム tert−ペントキシドである。より好都合には、適する塩基は、例えば、アルカリ金
属(1−6C)アルコキシド、例えば、ナトリウムまたはカリウム tert−ブトキシド、
またはナトリウムまたはカリウム tert−ペントキシドである。
【0040】
作業工程(c)について、反応は、好都合には、適する不活性溶媒または希釈剤、または適する不活性溶媒または希釈剤の混合物の存在下で、例えば、ジブチルエーテル、メチル tert−ブチルエーテル、ジ−(2−メトキシエチル)エーテル、1,2−ジメトキシ
エタン、1,2−ジエトキシエタン、テトラヒドロフランまたは1,4−ジオキサンなどの置換されていてよいジ−(1−6C)アルキルエーテルまたは環状アルキルエーテル;またはN,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリジン−2−オンまたはジメチルスルホキシドなどの双極性非プロトン性溶媒中で行われる。好都合には、50℃を超える沸点を有する適する不活性溶媒または希釈剤、例えば、テトラヒドロフランなどの環状アルキルエーテル;またはN−メチルピロリジン−2−オンなどの双極性非プロトン性溶媒を用いる。
【0041】
作業工程(c)について、反応は、例えば、0〜250℃の範囲内、好都合には、25〜125℃の範囲内、より好都合には、40〜80℃の範囲内の温度で行われる。
より好都合には、式Vの中間体は、そのままで単離しないが、有機溶媒中の溶液またはスラリーとして用いる。それによって、式VIの化合物は、式IVの化合物からワンポット法で製造することができる。この方式での式IVの化合物の式VIの化合物への変換は、以下の実施例5に詳しく説明されている。より一層好都合には、式Vの中間体は、6−クロロ−2,3−メチレンジオキシアニリンの存在下で形成され、それてワンポット法において直
接的に反応する。この方式での式IVの化合物の式VIの化合物への変換は、以下の実施例7に詳しく説明されている。
【0042】
式VIのキナゾリンは、本発明のもう一つの側面を形成する新規な化合物である。
本発明のもう一つの側面により、AZD0530の製造方法であって、式VIのキナゾリンを製造する直ぐ上の工程(a)および工程(b)と、本明細書中の前に定義のようなAZD0530へのその変換を含む方法を提供する。
【0043】
更に、式VIのキナゾリンの別の製造方法であって、
(a)式VII
【0044】
【化8】
【0045】
を有する2,4,6−トリフルオロベンゾニトリルと、6−クロロ−2,3−メチレンジオキシアニリンとの、好都合には、適する有機金属触媒の存在下における、式VIII
【0046】
【化9】
【0047】
を有するアミジンを形成する反応、および
(b)式VIIIのアミジンと、ホルムアミジンまたはその塩との、式VIのキナゾリノンを形成する反応
を含み;その後、遊離塩基の形で得られた式VIの化合物は、塩へと変換することができるし、塩の形で得られた式VIの化合物は、遊離塩基へと変換することができる方法を提供する。
【0048】
直ぐ上の作業工程(a)について、適する有機金属試薬は、例えば、トリメチルアルミニウムなどの有機アルミニウム化合物;ジフェニルホスフィノフェロセンなどの有機鉄化合物;またはテトラキス(トリフェニルホスフィン)パラジウム(0)などの有機パラジウム化合物である。その反応は、好都合には、本明細書中の前に定義の適する不活性溶媒または希釈剤の存在下で行われる。好都合には、トルエンまたはキシレン、クメンまたはクロロベンゼンなどの芳香族溶媒を、反応溶媒として用いる。その反応は、好都合には、例えば、10〜250℃の範囲内、好適には、75〜125℃の範囲内の温度で行われる。
【0049】
直ぐ上の作業工程(b)について、反応は、好都合には、本明細書中の前に定義の適する不活性溶媒または希釈剤の存在下で、例えば、トルエンまたはキシレン、クメン、クロロベンゼン、アニソールまたはフェネトールなどの芳香族溶媒中において、例えば、10〜250℃の範囲内、好適には、75〜125℃の範囲内の温度で行われる。
【0050】
この方式での式VIIの化合物の式VIの化合物への変換は、以下の実施例6に詳しく説明
されている。
更に、式III
【0051】
【化10】
【0052】
を有するキナゾリンの別の製造方法であって、
(a)式IV
【0053】
【化11】
【0054】
を有するキナゾリノンと、4−ヒドロキシテトラヒドロピランとの、好都合には、本明細書中の前に定義の適する塩基の存在下における、式IX
【0055】
【化12】
【0056】
を有するキナゾリンを形成する反応であって、その後、遊離塩基の形で得られた式IXの化合物は、塩へと変換することができるし、塩の形で得られた式IXの化合物は、遊離塩基へと変換することができる反応;
(b)式IXのキナゾリンと、本明細書中の前に定義の活性化剤との、好都合には、適する塩基の存在下における、式X
【0057】
【化13】
【0058】
(式中、L1は、本明細書中の前に定義の置換可能な基である)
を有するキナゾリノンを形成する反応;および
(c)式Xのキナゾリンと、6−クロロ−2,3−メチレンジオキシアニリンとの、好都合には、適する塩基の存在下における置換反応
を含み;その後、遊離塩基の形で得られた式IIIの化合物は、塩へと変換することができ
るし、塩の形で得られた式IIIの化合物は、遊離塩基へと変換することができる方法を提
供する。
【0059】
直ぐ上の作業工程(a)について、反応は、好都合には、上の作業工程(c)について定義の(式VIのキナゾリンから式IIIのキナゾリンの製造に関する)適する塩基の存在下
、上の作業工程(c)について定義の(式VIのキナゾリンから式IIIのキナゾリンの製造
に関する)適する不活性溶媒または希釈剤、または適する不活性溶媒または希釈剤の混合物の存在下における、例えば、0〜250℃の範囲内、好都合には、25〜125の範囲内、より好都合には、40〜80℃の範囲内の温度で行うことができる。
【0060】
直ぐ上の作業工程(b)について、脱離基L1を形成するであろう適する活性化剤は、例えば、塩化ホスホリルまたは臭化ホスホリルなどのハロゲン化ホスホリル;または塩化チオニルなどのハロゲン化剤;または四塩化炭素およびトリフェニルホスフィンの混合物によって形成されるハロゲン化剤;または四塩化炭素およびトリフェニルホスフィンの混合物によって形成されるハロゲン化剤である。直ぐ上の作業工程(b)の際に用いることができる適する塩基は、例えば、有機アミン塩基、例えば、ピリジン、2,6−ルチジン、コリジン、4−ジメチルアミノピリジン、トリエチルアミン、ジイソプロピルエチルアミン、N−メチルモルホリンまたはジアザビシクロ[5.4.0]ウンデカ−7−エンなどである。直ぐ上の作業工程(b)に適する溶媒または希釈剤は、例えば、トルエン、キシレン、クメン、クロロベンゼン、アニソールまたはフェネトールなどの芳香族溶媒である。更に適する溶媒または希釈剤は、アセトニトリル、プロピオニトリル、ブチロニトリル、酢酸エチル、テトラヒドロフランまたは1,4−ジオキサンなどの極性非プロトン性溶媒;またはN,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリジン−2−オンまたはジメチルスルホキシドなどの双極性非プロトン性溶媒である。このような適する溶媒または希釈剤の混合物を用いることができる。その反応は、例えば、10〜250℃の範囲内、好都合には、40〜120℃の範囲内の温度で行うことができる。
【0061】
好都合には、直ぐ上の作業工程(b)について、適する活性化剤は、例えば、塩化ホスホリルなどのハロゲン化ホスホリルであり、そして反応は、トリエチルアミンまたはジイソプロピルエチルアミンなどの有機アミン塩基の存在下において、トルエン、クロロベンゼンまたはアセトニトリルなどの溶媒または希釈剤を用いて、70〜100℃の範囲内の温度で行われる。
【0062】
直ぐ上の作業工程(c)の置換反応は、適する酸の存在下または適する塩基の存在下で
行うことができる。適する酸は、例えば、無機酸、例えば、塩化水素または臭化水素などである。適する塩基は、例えば、有機アミン塩基、例えば、ピリジン、2,6−ルチジン、コリジン、4−ジメチルアミノピリジン、トリエチルアミン、ジイソプロピルエチルアミン、N−メチルモルホリンまたはジアザビシクロ[5.4.0]ウンデカ−7−エンなど;または例えば、アルカリ金属またはアルカリ土類金属の炭酸塩または水酸化物、例えば、炭酸ナトリウム、炭酸カリウム、炭酸カルシウム、水酸化ナトリウムまたは水酸化カリウム;または例えば、アルカリ金属水素化物、例えば、水素化ナトリウムである。
【0063】
直ぐ上の作業工程(c)の置換反応は、好都合には、適する溶媒または希釈剤、例えば、イソプロパノール、sec−ブタノールまたは tert−ブタノールなどの第一級、第二級または第三級の(1−6C)アルキルアルコール;塩化メチレン、クロロホルムまたは四塩化炭素などのハロゲン化溶媒;トルエン、キシレン、クメン、クロロベンゼン、アニソールまたはフェネトールなどの芳香族溶媒;アセトニトリル、プロピオニトリル、ブチロニトリル、酢酸エチル、テトラヒドロフランまたは1,4−ジオキサンなどの極性非プロトン性溶媒;またはN,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリジン−2−オンまたはジメチルスルホキシドなどの双極性非プロトン性溶媒の存在下で行われる。このような適する溶媒または希釈剤の混合物を用いることができる。その反応は、好都合には、例えば、10〜250℃の範囲内、好適には、40〜120℃の範囲内の温度で行われる。
【0064】
好都合には、式Xの中間体は、そのままで単離しないが、有機溶媒中の溶液またはスラリーとして用いる。それによって、式IIIの化合物は、式IXの化合物からワンポット法で
製造することができる。この方式での式IXの化合物の式IIIの化合物への変換は、以下の
実施例8に詳しく説明されている。
【0065】
本発明のもう一つの側面により、AZD0530の製造方法であって、式IXのキナゾリンを製造する直ぐ上の工程(a)と、本明細書中の前に定義のようなAZD0530へのその変換を含む方法を提供する。
【0066】
式IVのキナゾリノンのような必要な出発物質は、有機化学の標準法によって得ることができる。式IVのキナゾリノンの製造は、以下の代表的な実施例中に記載されている(実施例1および実施例2)。或いは、このような必要な出発物質は、当該化学者の常套技術の範囲内である、示されているのに類似した手順によって入手可能である。
【0067】
本発明のもう一つの側面により、式I
【0068】
【化14】
【0069】
を有する化合物であるAZD0530の製造方法であって、
(a)式XI
【0070】
【化15】
【0071】
を有するキナゾリンと、本明細書中の前に定義の活性化剤との、好都合には、本明細書中の前に定義の適する塩基の存在下における、式XII
【0072】
【化16】
【0073】
(式中、L1は、本明細書中の前に定義の置換可能な基である)
を有するキナゾリノンを形成する反応;および
(b)式XIIのキナゾリンと、6−クロロ−2,3−メチレンジオキシアニリンとの、
好都合には、本明細書中の前に定義の適する塩基の存在下における置換反応
を含み;その後、遊離塩基の形で得られた式Iの化合物は、薬学的に許容しうる塩へと変換することができるし、塩の形で得られた式Iの化合物は、遊離塩基へと変換することができる方法を提供する。
【0074】
好都合には、直ぐ上の作業工程(a)について、適する活性化剤は、例えば、塩化ホスホリルなどのハロゲン化ホスホリルであり、そして反応は、トリエチルアミンまたはジイソプロピルエチルアミンなどの有機アミン塩基の存在下において、ブチロニトリルまたはトルエンなどの溶媒または希釈剤を用いて、70〜120℃の範囲内の温度で行われる。
【0075】
好都合には、直ぐ上の作業工程(b)について、置換反応は、好都合には、ブチロニトリルまたはトルエンなどの適する不活性溶媒または希釈剤の存在下において、70〜120℃の範囲内の温度で行われる。
【0076】
好都合には、式XIIの中間体は、そのままで単離しないが、有機溶媒中の溶液またはス
ラリーとして用いる。それによって、式Iの化合物は、式XIの化合物からワンポット法で製造することができる。この方式での式XIの化合物の式Iの化合物への変換は、以下の実施例11および実施例12に詳しく説明されている。
【0077】
本発明のもう一つの側面により、式XIのキナゾリンの製造方法であって、
(a)式IV
【0078】
【化17】
【0079】
を有するキナゾリノンと、4−ヒドロキシテトラヒドロピランとの、好都合には、本明細書中の前に定義の適する塩基の存在下における、式IX
【0080】
【化18】
【0081】
を有するキナゾリンを形成する反応であって、その後、遊離塩基の形で得られた式IXの化合物は、塩へと変換することができるし、塩の形で得られた式IXの化合物は、遊離塩基へと変換することができる反応;および
(b)NH官能基が、必要ならば保護されている式IXのキナゾリンと、1−(2−ヒドロキシエチル)−4−メチルピペラジンとの、好都合には、本明細書中の前に定義の適する塩基の存在下における反応を含み;その後、存在するいずれの保護基も、慣用的な手段によって除去し;そしてその後、必要ならば、遊離塩基の形で得られた式XIの化合物は、塩へと変換することができるし;塩の形で得られた式XIの化合物は、遊離塩基へと変換することができる方法を提供する。
【0082】
直ぐ上の作業工程(a)について、反応は、好都合には、上の作業工程(c)について定義の(式VIのキナゾリンから式IIIのキナゾリンの製造に関する)適する塩基の存在下
、上の作業工程(c)について定義の(式VIのキナゾリンから式IIIのキナゾリンの製造
に関する)適する不活性溶媒または希釈剤、または適する不活性溶媒または希釈剤の混合物の存在下における、例えば、0〜250℃の範囲内、好都合には、25〜125の範囲内、より好都合には、40〜80℃の範囲内の温度で行うことができる。
【0083】
直ぐ上の作業工程(b)について、適する塩基は、例えば、アルカリ金属(1−6C)アルコキシド、例えば、ナトリウムまたはカリウム tert−ブトキシド、ナトリウムまた
はカリウム tert−ペントキシド、またはナトリウムまたはカリウム3,7−ジメチルオ
クトキシドである。その反応は、好都合には、適する不活性溶媒または希釈剤、または適する不活性溶媒または希釈剤の混合物の存在下で、例えば、ジブチルエーテル、メチル tert−ブチルエーテル、ジ−(2−メトキシエチル)エーテル、1,2−ジメトキシエタ
ン、1,2−ジエトキシエタン、テトラヒドロフランまたは1,4−ジオキサンなどの置換されていてよいジ−(1−6C)アルキルエーテルまたは環状アルキルエーテル;またはN,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリジン−2−オンまたはジメチルスルホキシドなどの双極性非プロトン性溶媒中で行われる。好都合には、50℃を超える沸点を有する適する不活性溶媒または希釈剤、例えば、テトラヒドロフランまたは1,4−ジオキサンなどの環状アルキルエーテル;またはジ−(2
−メトキシエチル)エーテルまたは1,2−ジエトキシエタンなどの置換されていてよいジ−(1−6C)アルキルエーテルを用いる。好都合には、その反応は、例えば、50〜150℃の範囲内の温度で、より好都合には、約70℃で行われる。
【0084】
式XIのキナゾリノンは、本発明のもう一つの側面を形成する新規な化合物である。
AZD0530結晶形
本明細書中の前に述べられたように、AZD0530として現在知られている化合物4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンは、非受容体チロシンキナーゼ酵素のSrcファミリーの阻害剤であり、したがって、腫瘍細胞の運動性の選択的阻害剤および哺乳動物癌細胞の播種および浸潤性の選択的阻害剤であって、転移性腫瘍成長の阻害をもたらす。具体的には、化合物AZD0530は、c−Src非受容体チロシンキナーゼの阻害剤であるので、ヒトまたは動物体の充実性腫瘍疾患の封じ込めおよび/または処置に用いるための抗浸潤薬として価値があるはずである。
【0085】
その化合物は、国際特許出願WO01/94341号の実施例14の表中に化合物番号73として開示された。その化合物は、二塩酸塩の形または遊離塩基の形で得られたということが述べられた。AZD0530の二塩酸塩形およびAZD0530の遊離塩基形の結晶化度は、述べられなかった。
【0086】
国際特許出願WO01/94341号には、そこに開示されたキナゾリン誘導体が、溶媒和した形で、更には、非溶媒和の形で存在しうるということは、特に述べられなかった。具体的には、特定の水和した形のAZD0530は開示されなかった。
【0087】
AZD0530の遊離塩基形についての引き続きの分析は、X線粉末回折分析、示差走査熱量測定または熱重量分析を用いて行った。AZD0530の遊離塩基形は、結晶形および非晶形の混合物であることが確かめられた。熱量測定は、約30〜85℃の広範な吸熱を示した。約65℃に開始し且つピークを約79℃に有する単一の広範な融解吸熱が存在した。重量分析は、約25〜120℃の温度範囲において原試料重量の約10%の損失重量を示した。
【0088】
薬学的に許容しうる塩に関して、国際特許出願WO01/94341号には、その中の式Iの化合物の適する薬学的に許容しうる塩は、例えば、その式Iの化合物の酸付加塩、例えば、塩酸、臭化水素酸、硫酸、トリフルオロ酢酸、クエン酸またはマレイン酸などの無機酸または有機酸との酸付加塩;または例えば、十分に酸性であるその式Iの化合物の塩、例えば、カルシウム塩またはマグネシウム塩などのアルカリ金属またはアルカリ土類金属の塩;またはアンモニウム塩;またはメチルアミン、ジメチルアミン、トリメチルアミン、ピペリジン、モルホリンまたはトリス−(2−ヒドロキシエチル)アミンなどの有機塩基との塩であるということが述べられた。
【0089】
国際特許出願WO01/94341号には、その中の式Iを有するいずれか特定の化合物またはそのいずれか特定の薬学的に許容しうる塩が、結晶性物理的形態のような驚くほど有益な物理的形態を有するということは述べられていない。
【0090】
多数の薬学的に活性な化合物は、製造および/または製剤化の方法中の単離および取扱いに適する物理的形態を有することがない。物理的形態のこのような欠陥を克服する一つの方法は、その適する薬学的に許容しうる塩が存在するか否かを決定することである。物理的形態のこのような欠陥を克服するもう一つの方法は、適する薬学的に許容しうる多形が存在するか否かを決定することである。物理的形態のこのような欠陥を克服するもう一つの方法は、適する形を有する溶媒和化合物または水和物を形成することである。好都合
には、このような形は、適度な融点を有するさらさらした結晶性固体を構成する。
【0091】
本発明者は、ここで、本明細書中の式Iの化合物であるAZD0530のある種の形であって、ある種のその薬学的に許容しうる塩を含めた形が、好都合な性質を有する結晶質であるということを発見した。このような結晶質は、非晶質を実質的に含まない。
【0092】
特定の結晶形の化合物は、いずれか他の結晶形または非晶形のものとは異なる物理的性質を有することがありうるが、このような性質は、特に、その化合物を商業規模で製造するまたは用いる場合に、その化合物の化学的および薬学的加工に顕著に影響を与えることがありうる。例えば、結晶形の化合物は各々、結晶のサイズおよび形状、融点、密度、吸湿性および安定性などの物理的性質の相違を示すことがありうる。このような相違は、化合物の機械的取扱適性(固体材料の流動性など)および化合物の圧縮特性を変更することがありうる。異なった結晶形の化合物は、異なった熱力学的安定性を有することがありうる。概して、より安定な形、例えば、より安定な多形形は、商業規模での製剤化および加工に一層適する物理的形態である。
【0093】
例えば、安定性に乏しい形、例えば、安定性に乏しい多形の加工において、問題が生じることがありうる。錠剤成形方法において用いられるような圧縮力は、若干の安定性に乏しい形をより安定な形へと変換して、製剤化された製品中においてより安定な形の結晶生長を引き起こすことがありうる。これは、いずれかこのような結晶化方法が、錠剤の保全性を破壊して、減少した錠剤強度を有する脆い錠剤を生じることがありうるので、望ましくないことがありうる。更に、二つのこのような形の可変混合物を与えようとした場合、一つまたは双方の活性化合物の溶解速度およびバイオアベイラビリティーは、例えば、各々の形が異なった粒度有することがありうるように変動することがありうる。粒度が、薬学的に活性な化合物の溶解速度およびバイオアベイラビリティーに影響することがありうるということは周知である。したがって、製品の品質は、不都合に影響を受けることがありうるし、投薬への生物学的作用について再現不可能の問題が起こりうる。
【0094】
更に、カプセル剤または錠剤の形での医薬化合物は、化合物の組成が調節されている且つ安定であるという適当な調節根拠を示す必要があるような準安定な形またはそのような形の混合物ではなく、安定な形、例えば、安定な塩または最も安定な多形を用いて製造されることが好ましい。熱力学的に安定性に乏しい形、例えば、安定性に乏しい多形を、錠剤中に単独でまたは熱力学的により安定な形との混合物で与えた場合、その熱力学的により安定な形の量は、貯蔵時に増加する傾向がありうるので、錠剤の組成、例えば、錠剤の多形組成を調節することはきわめて難しいであろう。
【0095】
したがって、これら因子は、固相、すなわち、化合物の錠剤またはカプセル剤に、およびそれらの懸濁製剤に影響力を有することがありうる。
AZD0530の化合物の性質の研究は、結晶性塩および/または結晶性の溶媒和化合物または水和物が形成されうるか否か、および多形現象が起こるか否かを発見するために行った。例えば、次の薬学的に許容しうる酸を、AZD0530のメタノール性溶液に個々に加えて、いずれの結晶性塩が形成されるか否かを確かめた(塩酸、クエン酸、マレイン酸、コハク酸、リンゴ酸、アジピン酸、マロン酸、4−トルエンスルホン酸、メタンスルホン酸、サリチル酸、酒石酸、アスコルビン酸、フマル酸、グリコール酸およびリン酸)。
【0096】
本発明者は、ここで、驚くべきことに、化合物の医薬加工に価値があるように十分に安定であり且つ結晶性であるAZD0530の薬学的に許容しうる塩および/または溶媒和した形が、比較的少数しか存在しないということを発見した。具体的には、AZD0530結晶性塩形成は、最初は、リンゴ酸、メタンスルホン酸、フマル酸およびリン酸でのみ
示された。引き続きの研究は、リン酸との塩が非晶質であるということを示した。本発明者は、ここで、フマル酸で形成された塩が、好ましい性質を有するということを発見した。
【0097】
AZD0530の一つまたはそれを超える特定の結晶形の試料を、X線粉末回折(以下、XRPD)分析、示差走査熱量測定(以下、DSC)、熱重量分析(以下、TGA)、拡散反射赤外フーリエ変換(DRIFT)分光法、近赤外(NIR)分光法、溶液および/または固相核磁気共鳴分光法、および/またはカール・フィッシャー分析による水分定量法の組合せを用いて分析した。
【0098】
ジフマレート塩結晶形
本発明者は、AZD0530およびフマル酸が、以下、AZD0530ジフマレートと称されるジフマル酸塩の形で結晶性塩を形成するということを発見した。AZD0530ジフマレート塩は、それが、容易に単離されるし且つ十分に安定でもあって、商業規模において高レベルの純度および高収率で容易に製造することができる結晶性物理的形態を有するという点で異例である。
【0099】
本発明のこの側面により、実質的に均一な結晶形の、実質的にジフマレート塩の形の式Iの化合物である4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン(AZD0530ジフマレート)を提供する。
【0100】
本発明が、実質的に均一な結晶形の式Iの化合物に関すると述べられている場合、(XRPD手段によって決定することができる)結晶化度は、好都合には、約60%を超える、より好都合には、約80%を超える、好ましくは、約90%を超える、より好ましくは、約95%を超える。
【0101】
本発明のこの側面が、AZD0530ジフマレートに関すると述べられている場合、各々のAZD0530分子対各々のフマル酸分子のモル比は、1:1.7〜1:2.5の範囲内、好都合には、1:1.8〜1:2.3の範囲内、より好都合には、1:1.9〜1:2.1の範囲内にあり、好ましくは、約1当量のAZD0530対約2当量のフマル酸を有する。
【0102】
AZD0530ジフマレートは、式Iを有する安定な形の化合物である。具体的には、AZD0530ジフマレートは、実質的に非吸湿性であり、したがって、非晶形のAZD0530とは異なり、水蒸気に暴露されたとしても、貯蔵中に容易に形態変化することはない。このような形態変化はいずれも、熱力学的に安定性に乏しい形の熱力学的により安定な形への変換が、溶解速度の減少を引き起こすことがありうることから、問題でありうる。式Iを有する二つのこのような形の化合物の可変混合物を与えようとした場合、一つまたは双方の活性化合物の溶解速度およびバイオアベイラビリティーは、それら二つの形の異なった特性の結果として変動することがありうる。
【0103】
AZD0530ジフマレートは、結晶のサイズおよび形状、融点、密度および吸湿性などの、式Iを有する他の既知の形の化合物と比較した場合に異なる他の物理的性質を示す。このような相違は、製造の際に、改善された固体材料流動性および/または改善された濾過のような、好都合な化合物取扱適性を提供することができる。このような利点は、商業規模での式Iの化合物の改善された製剤化および加工を提供することができる。具体的には、AZD0530ジフマレートの晶癖は、好都合な濾過特性を有する材料を提供する。
【0104】
更に、AZD0530ジフマレートは、商業規模において高レベルの純度および高収率で容易に製造することができる。
AZD0530ジフマレートは、実質的には以下の図1に示されるような、下の表1に示される2θスケール上のピーク(最初の4個のピークと、より強い他のピークの内の6個を挙げている)を包含するX線回折図形を有する。
【0105】
【表1】
【0106】
具体的には、表1中の約7.1°、9.1°および10.6°にある一つまたはそれを超えるピークは、AZD0530ジフマレートについて顕著であると考えられる。
以下に述べられるように、XRPDスペクトル中のピーク位置の測定誤差は、約±0.3°2θであろう。このような程度の測定誤差は、XRPDスペクトルが同じ物理的形態に由来したか否かを評価する場合に考慮されるべきである。当業者は、個々のピーク位置よりもむしろ、それらピークの相対位置が、AZD0530ジフマレートの試料が実質的に同じであるか否かについてのより信頼できる指標であるということを理解するであろう。
【0107】
以下に述べられるように、XRPDディフラクトグラム中のピークの強さは、用いられる測定条件に依存して、若干の変動性を示すこともありうる。したがって、表1においておよび以下に引用されるように、相対強さは、数字で記述されていない。その代わり、強さについて次の定義を用いる。
【0108】
【表2】
【0109】
AZD0530ジフマレートのDSCサーモグラム分析は、その塩が、約231〜240℃の範囲内の融点を有する;言い換えると、融解の開始は約231℃であり、融点ピークは約237℃であるということを示した。具体的には、融点は、約233〜239℃の範囲内である。より具体的には、融点は、約234〜238℃の範囲内である。より具体的には、融点は、約237℃である。典型的に、DSC分析は、AZD0530ジフマレートが、融解開始を約235℃におよび融点ピークを約237℃に有する高融点固体であるということを示している。
【0110】
AZD0530ジフマレートのDRIFT分光法トレースは、以下の図5に示されるが、それは、約3359cm−1(N−H)、3100〜2700cm−1、1719cm−1(C=O)、1662cm−1、1616cm−1、1586cm−1、1523cm−1、1501cm−1、1360〜1200cm−1、1200〜1000cm−1および979cm−1にあるピークを包含する。具体的には、約3359cm−1および1719cm−1にある一つまたは双方のピークは、AZD0530ジフマレートについて顕著であると考えられる。
【0111】
非晶形のAZD0530ジフマレートは、その材料の試料をグラインダー中に入れ且つ約10分間またはそれを超えて粉砕した場合に得ることができる。粉砕された材料の非晶性は、XRPDスペクトル中の明瞭なピークの不存在によって示された。
【0112】
セスキフマレート塩結晶形
本発明者は、更に、AZD0530ジフマレートを水中でスラリーにした場合、またはAZD0530フマル酸塩製造の際に2当量未満のフマル酸を用いた場合、より少ないフマル酸含量を有するAZD0530フマル酸塩が形成されるということを発見した。本発明者は、このような塩が、結晶水を収容するのに十分な空間を有する結晶格子を形成するということに注目した。それによって、かなり均一な結晶性塩を、2〜6当量の水を含有するセスキフマル酸塩の形で得ることができる。
【0113】
具体的には、実質的に均一な結晶性塩を、以下、AZD0530セスキフマレート四水和物と称されるセスキフマル酸塩四水和物の形で得ることができる。AZD0530セスキフマレート四水和物は、単離可能であり且つ適度な安定性を有する結晶性物理的形態を有する。
【0114】
この実質的に均一な結晶形の(XRPD手段によって決定することができる)結晶化度は、好都合には、約60%を超える、より好都合には、約80%を超える、好ましくは、約90%を超える。最も好ましくは、結晶化度は、約95%を超える。
【0115】
AZD0530セスキフマレート四水和物の場合、各々のAZD0530分子対各々のフマル酸分子のモル比は、1:1.3〜1:1.7の範囲内、好都合には、1:1.4〜1:1.6の範囲内にあり、より好都合には、約1当量のAZD0530対約1.5当量のフマル酸を有する。
【0116】
AZD0530セスキフマレート四水和物の場合、各々のAZD0530分子対各々の水分子のモル比は、1:3.5〜1:4.5の範囲内、好都合には、1:3.7〜1:4.3の範囲内にあり、より好都合には、約1当量のAZD0530対約4当量の水を有する。
【0117】
AZD0530セスキフマレート四水和物は、実質的には図2に示されるような、下の表2に示される2θスケール上のピーク(最も強いXRPDピークの内の10個を挙げている)を包含するX線回折図形を有する。
【0118】
【表3】
【0119】
具体的には、表2中の約2.8°、10.3°および22.6°にある一つまたはそれを超えるピークは、AZD0530セスキフマレート四水和物に特有であると考えられる。
【0120】
AZD0530セスキフマレート四水和物のDSCサーモグラム分析は、その塩が、約25〜100℃の最初の熱イベントを有するということを示したが、それは、水和水の損失のためであると考えられる。発熱は、AZD0530ジフマレートの結晶化に対応して、約150℃を超えて生じる。引き続きの加熱で、更なる熱イベントは、AZD0530ジフマレートの融点に対応して、約230〜240℃で生じる。
【0121】
AZD0530セスキフマレート四水和物のTGAは、約4当量の水の損失に対応して、約30〜130℃で約8%〜10%の損失重量を示した。
AZD0530セスキフマレート四水和物のDRIFT分光法トレースは、以下の図6に示されるが、それは、約3345cm−1(N−H)、3100〜2700cm−1、1698cm−1(C=O)、1660〜1450cm−1および1400〜1000cm−1にあるピークを包含する。具体的には、約3345cm−1および1698cm−1にある一つまたは双方のピークは、AZD0530セスキフマレート四水和物について顕著であると考えられる。
【0122】
三水和物遊離塩基結晶形
本発明者は、更に、AZD0530を、以下、AZD0530三水和物と称される結晶性三水和物の形で、湿潤している有機溶媒から結晶化させることができるということを発見した。AZD0530三水和物は、それが、容易に単離されるし且つ十分に安定でもあって、商業規模において高レベルの純度および高収率で容易に製造することができる結晶性物理的形態を有するという点で異例である。
【0123】
本発明のこの側面により、実質的に均一な結晶形の、実質的に三水和物の形の式Iの化合物である4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン(AZD0530三水和物)を提供する。
【0124】
本発明が、実質的に均一な三水和物結晶形の式Iの化合物に関すると述べられている場
合、(XRPD手段によって決定することができる)結晶化度は、好都合には、約90%を超える、より好都合には、約95%を超える。
【0125】
本発明のこの側面が、AZD0530三水和物に関すると述べられている場合、各々のAZD0530分子対各々の水分子のモル比は、1:2〜1:4の範囲内、好都合には、1:2.5〜1:3.5の範囲内、より好都合には、1:2.75〜1:3.25の範囲内にあり、好ましくは、約1当量のAZD0530対約3当量の水を有する。
【0126】
AZD0530三水和物は、式Iを有する安定な形の化合物である。具体的には、AZD0530三水和物は、水の存在下で安定である。例えば、AZD0530を、水性懸濁液として製造する場合、得られた懸濁液は安定であるが、式Iを有する他の形の化合物を用いて製造された水性懸濁液は、水和した形のAZD0530へと部分的にまたは完全に変換する傾向がありうる。
【0127】
AZD0530三水和物は、商業規模において高レベルの純度および高収率で容易に製造することができる。更に、AZD0530三水和物は、無水物結晶形のAZD0530へと、およびAZD0530のある種の薬学的に許容しうる塩結晶形へと変換することができる。AZD0530三水和物の製造、その精製および他の結晶形への変換は、収率および純度の点で有益である。
【0128】
AZD0530三水和物は、実質的には以下の図3に示されるような、下の表3に示される2シータ(θ)スケール上のピーク(最も強いXRPDピークの内の10個を挙げている)を包含するX線回折図形を有する。
【0129】
【表4】
【0130】
具体的には、表3中の約16.0°にあるピークは、無水物結晶形と比較して(下を参照されたい)、AZD0530三水和物について顕著であると考えられる。
以下に述べられるように、XRPDスペクトル中のピーク位置の測定誤差は、約±0.3°2θであろう。このような程度の測定誤差は、XRPDスペクトルが同じ物理的形態に由来したか否かを評価する場合に考慮されるべきである。当業者は、個々のピーク位置よりもむしろ、それらピークの相対位置が、AZD0530三水和物の試料が実質的に同じであるか否かについてのより信頼できる指標であるということを理解するであろう。
【0131】
AZD0530三水和物のDSCサーモグラム分析は、約50〜94℃の広範な吸熱を示したが、それは、水の損失のためであると考えられる。その吸熱は、約75℃にピーク
を有する約65℃での開始を示した。
【0132】
AZD0530三水和物のTGAは、約3当量の水の損失に対応して、約30〜110℃で約9%の損失重量を示した。
無水遊離塩基結晶形
本発明者は、更に、AZD0530が、二つの無水物の形で、すなわち、一定の融点を有していない非晶質の非結晶形と、十分に規定された狭い融点を有する高結晶形とで得ることができるということを発見した。本発明者は、AZD0530三水和物を、以下、無水AZD0530と称される実質的に均一な無水物結晶形へと容易に変換することができるということを発見した。したがって、AZD0530三水和物の結晶化およびその後の無水AZD0530への変換は、無水AZD0530を高純度で製造する手段を提供する。無水AZD0530は、それが、容易に単離されるし且つ実質的無水条件下において十分に安定でもあって、商業規模において高レベルの純度および高収率で容易に製造することができる結晶性物理的形態を有するという点で異例である。
【0133】
この実質的に均一な結晶形の(XRPD手段によって決定することができる)結晶化度は、好都合には、約60%を超える、より好都合には、約80%を超える、好ましくは、約90%を超える、そしてより好ましくは、約95%を超える。
【0134】
無水AZD0530は、式Iを有する安定な形の化合物である。具体的には、無水AZD0530は、水の不存在下においてきわめて安定である。しかしながら、無水AZD0530は、実質的無水貯蔵条件が維持されない場合、貯蔵中にAZD0530三水和物へと変換する傾向がある。
【0135】
無水AZD0530は、商業規模において高レベルの純度および高収率で容易に製造することができる。更に、無水AZD0530は、AZD0530のある種の薬学的に許容しうる塩結晶形へと変換することができる。
【0136】
無水AZD0530は、実質的には図4に示されるような、下の表4に示される2θスケール上のピーク(最も強いXRPDピークの内の10個を挙げている)を包含するX線回折図形を有する。
【0137】
【表5】
【0138】
表4中の約15.1°、17.0°および18.0°にある一つまたはそれを超える強いピーク、具体的には、約20.4°および23.3°にある一つまたは双方のきわめて
強いピークは、三水和物形と比較して(上を参照されたい)、無水AZD0530について顕著であると考えられる。
【0139】
以下に述べられるように、XRPDスペクトル中のピーク位置の測定誤差は、約±0.3°2θであろう。このような程度の測定誤差は、XRPDスペクトルが同じ物理的形態に由来したか否かを評価する場合に考慮されるべきである。当業者は、個々のピーク位置よりもむしろ、それらピークの相対位置が、無水AZD0530の試料が実質的に同じであるか否かについてのより信頼できる指標であるということを理解するであろう。
【0140】
無水AZD0530のDSCサーモグラム分析は、約133〜152℃の熱イベントを示した。融解の開始は約142℃であり、約144℃に融点ピークを有した。
式Iの化合物の次の具体的な結晶形は、本明細書中に開示されている。
【0141】
(i)AZD0530ジフマレート;
(ii)AZD0530セスキフマレート四水和物;
(iii)AZD0530三水和物;および
(iv)無水AZD0530。
【0142】
これら物質は各々、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン(AZD0530)などの化合物について国際特許出願WO01/94341号に開示されたものと同じ薬理学的性質を有する。具体的には、これら物質は各々、c−Srcなどの非受容体チロシンキナーゼの阻害剤であり、それは、腫瘍細胞の運動性の選択的阻害および哺乳動物癌細胞の播種および浸潤性の選択的阻害を与えて、転移性腫瘍成長の阻害をもたらす。具体的には、これら物質は各々、充実性腫瘍疾患の封じ込めおよび/または処置に用いるための抗浸潤薬として価値があるはずである。式Iの化合物のこれら結晶形は、以下、集合的に、「本発明の活性物質」と記載される。
【0143】
ヒトを含めた哺乳動物の処置に本発明の活性物質を用いるために、それを、通常は、標準的な医薬慣例にしたがって医薬組成物として製剤化する。
本発明のもう一つの側面により、医薬組成物であって、本発明の活性物質を薬学的に許容しうる希釈剤または担体と一緒に含む医薬組成物を提供する。
【0144】
例えば、本発明の組成物は、経口投与に(例えば、錠剤、ロゼンジ、硬または軟カプセル剤、水性または油状懸濁剤、乳剤、分散性散剤または顆粒剤、シロップ剤またはエリキシル剤として)、局所投与に(例えば、クリーム剤、軟膏剤、ゲル剤、または水性または油状液剤または懸濁剤として)、吹入に(例えば、水性懸濁剤として)または非経口投与に(例えば、静脈内、皮下、腹腔内または筋肉内投与用の滅菌水性または油状液剤として、または直腸投与用の坐剤として)適合した形であってよい。
【0145】
好ましい投与方法は、経口投与である。本発明の活性物質は、好都合には、錠剤の形で経口投与される。
本発明のそれら組成物は、当該技術分野において周知の慣用的な医薬賦形剤を用いて慣用法によって得ることができる。したがって、経口使用を予定した組成物は、例えば、一つまたはそれを超える着色剤、甘味剤、着香剤および/または保存剤を含有してよい。例えば、その組成物は、一つまたはそれを超える増量剤、結合剤、崩壊剤および/または滑沢剤を含有してよい。適する増量剤には、例えば、ラクトース、糖、デンプン、化工デンプン、マンニトール、ソルビトール、無機塩類、セルロース誘導体(例えば、微結晶性セルロース、セルロース)、硫酸カルシウム、キシリトールおよびラクチトール(lactitol)が含まれる。適する結合剤には、例えば、ポリビニルピロリドン、ラクトース、デンプ
ン、化工デンプン、糖類、アラビアゴム、トラガカントゴム、グアーゴム、ペクチン、ワックスバインダー、微結晶性セルロース、メチルセルロース、カルボキシメチルセルロース、ヒドロキシプロピルメチルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース、コポリビドン、ゼラチンおよびアルギン酸ナトリウムが含まれる。適する崩壊剤には、例えば、クロスカルメロースナトリウム、クロスポビドン、ポリビニルピロリドン、ナトリウムデンプングリコラート、トウモロコシデンプン、微結晶性セルロース、ヒドロキシプロピルメチルセルロースおよびヒドロキシプロピルセルロースが含まれる。適する滑沢剤には、例えば、ステアリン酸マグネシウム、ステアリン酸、パルミチン酸、ステアリン酸カルシウム、タルク、カルナウバロウ(carnuba wax)、水素添加植
物油、鉱油、ポリエチレングリコールおよびステアリルフマル酸ナトリウムが含まれる。加えることができる追加の慣用的な賦形剤には、保存剤、安定化剤、酸化防止剤、シリカフローコンディショナー、付着防止剤または滑剤が含まれる。用いることができる他の適する増量剤、結合剤、崩壊剤、滑沢剤および追加の賦形剤は、次の参考文献に記載されている。Handbook of Pharmaceutical Excipients, 3rd Edition;The Theory and Practice of Industrial Pharmacy, 3rd Edition 1986;Pharmaceutical Dosage Forms 1998;Modern Pharmaceutics, 3rd Edition 1995;および Remington’s Pharmaceutical Sciences, 20th Edition 2000。
【0146】
一つまたはそれを超える賦形剤と混合されて単一剤形を生じる本発明の活性物質の量は、必然的に、宿主処置および具体的な投与経路に依存して異なるであろう。例えば、ヒトへの経口投与を予定した製剤は、好都合には、全組成物の約5〜約98重量%であってよい適切且つ好都合な量の賦形剤と配合される、例えば、0.5mg〜0.5gの活性剤(好都合には、1〜250mg、より好都合には、10〜200mgまたは25〜100mg)を含有するであろう。好ましくは、その製剤は、例えば、50mg〜500mgの活性物質を含むであろう。より好ましくは、その製剤は、例えば、100mg〜250mgの活性物質、特に、125mg〜225mgの活性物質を含むであろう。
【0147】
本発明の活性物質を治療目的または予防目的に用いる場合、それは、概して、必要ならば分割用量で与えられる、例えば、0.1mg/kg〜20mg/kg体重の範囲内の1日経口用量が与えられるように投与されるであろう。好ましくは、例えば、1mg/kg〜10mg/kg体重の範囲内の1日経口用量が与えられる。より好ましくは、例えば、2mg/kg〜8mg/kg体重の範囲内の1日用量が与えられる。
【0148】
本発明の活性物質は、許容しうる毒性プロフィールを示す。
本発明の活性物質は、国際特許出願WO01/94341号に、その中の式Iの化合物について開示されたものと同じ薬理学的性質を有する。具体的には、本発明の活性物質は、c−Srcなどの非受容体チロシンキナーゼの阻害剤であり、それは、腫瘍細胞の運動性の選択的阻害および哺乳動物癌細胞の播種および浸潤性の選択的阻害を与えて、転移性腫瘍成長の阻害をもたらす。具体的には、本発明の活性物質は、充実性腫瘍疾患の封じ込めおよび/または処置に用いるための抗浸潤薬として価値があるはずである。例えば、本発明の活性物質は、肺癌(小細胞性肺癌および非小細胞性肺癌を含めた)、乳癌、前立腺癌、卵巣癌、結腸直腸癌、胃癌、脳癌(グリオーマおよび下垂体腺腫を含めた)、頭頸部癌、膀胱癌、膵臓癌、食道癌、胃癌、腎癌、皮膚癌(悪性黒色腫を含めた)、婦人科学的癌(子宮頸部、子宮内膜、膣、外陰部および子宮を含めた)および甲状腺癌などの多数の一般的なヒト癌の処置に、およびCMLおよびALLなどの一定範囲の白血病およびリンパ系悪性疾患の処置に、および癌腫および肉腫などの充実性腫瘍の処置に有用である。
【0149】
本発明の活性物質の薬理学的性質は、例えば、国際特許出願WO01/94341号に開示された試験手順、または十分に当業者の領域の範囲内である同等の試験手順の一つまたはそれを超えるものを用いて評価することができる。その特許出願によるこのような試
験手順は、本明細書中に援用される。
【0150】
本発明のもう一つの側面により、ヒトまたは動物体の療法による処置方法に用いるための、本明細書中の前に定義の本発明の活性物質を提供する。
上述のように、c−Src非受容体チロシンキナーゼの主な役割は、限局性腫瘍が、血流中への播種、他の組織の浸潤および転移性腫瘍成長の開始の時期を経て進行するのに必然的に要求される細胞運動性を調節することである。本発明者は、本発明の活性物質が、転移する腫瘍細胞の浸潤性および移動能力をもたらすシグナル伝達段階に関与するc−Srcキナーゼなどの一つまたはそれを超える非受容体チロシン特異的プロテインキナーゼの阻害によって得られると考えられる強力な抗腫瘍活性を有するということを発見した。
【0151】
したがって、本発明の活性物質は、抗腫瘍薬として、具体的には、転移性腫瘍成長の阻害をもたらす哺乳動物癌細胞の運動性、播種および浸潤性の選択的阻害剤として価値がある。具体的には、本発明の活性物質は、充実性腫瘍疾患の封じ込めおよび/または処置において抗浸潤薬として価値がある。具体的には、本発明の活性物質は、転移する腫瘍細胞の浸潤性および移動能力をもたらすシグナル伝達段階に関与するc−Srcキナーゼなどの一つまたはそれを超える多数の非受容体チロシンキナーゼの阻害に感受性である腫瘍の予防または処置に有用であると考えられる。更に、本発明の活性物質は、酵素c−Srcの阻害によって単独でまたは一部分媒介される腫瘍の予防または処置に有用であると考えられる、すなわち、本発明の活性物質は、このような処置を必要としている温血動物にc−Src酵素阻害作用を生じるのに用いることができる。具体的には、本発明の活性物質は、充実性腫瘍疾患の予防または処置に有用であると考えられる。
【0152】
したがって、本発明のこの側面により、充実性腫瘍疾患の封じ込めおよび/または処置において抗浸潤薬として用いるための薬剤の製造における、本明細書中の前に定義の本発明の活性物質の使用を提供する。
【0153】
本発明のこの側面のもう一つの特徴により、充実性腫瘍疾患の封じ込めおよび/または処置によって抗浸潤作用を生じる処置を必要としているヒトなどの温血動物に、充実性腫瘍疾患の封じ込めおよび/または処置によって抗浸潤作用を生じる方法であって、この動物に、有効量の本明細書中の前に定義の本発明の活性物質を投与することを含む方法を提供する。
【0154】
本発明のもう一つの側面により、ヒトなどの温血動物の充実性腫瘍疾患の予防または処置に用いるための薬剤の製造における、本明細書中の前に定義の本発明の活性物質の使用を提供する。
【0155】
本発明のこの側面のもう一つの特徴により、充実性腫瘍疾患の処置を必要としているヒトなどの温血動物の充実性腫瘍疾患の予防または処置の方法であって、この動物に、有効量の本明細書中の前に定義の本発明の活性物質を投与することを含む方法を提供する。
【0156】
本発明のもう一つの側面により、転移する腫瘍細胞の浸潤性および移動能力をもたらすシグナル伝達段階に関与するc−Srcキナーゼなどの非受容体チロシンキナーゼの阻害に感受性である腫瘍の予防または処置に用いるための薬剤の製造における、本明細書中の前に定義の本発明の活性物質の使用を提供する。
【0157】
本発明のこの側面のもう一つの特徴により、転移する腫瘍細胞の浸潤性および移動能力をもたらすシグナル伝達段階に関与するc−Srcキナーゼなどの非受容体チロシンキナーゼの阻害に感受性である腫瘍の予防または処置方法であって、この動物に、有効量の本明細書中の前に定義の本発明の活性物質を投与することを含む方法を提供する。
【0158】
本発明のもう一つの側面により、c−Srcキナーゼ阻害作用を与える場合に用いるための薬剤の製造における、本明細書中の前に定義の本発明の活性物質の使用を提供する。
本発明のこの側面のもう一つの特徴により、c−Srcキナーゼ阻害作用を与える方法であって、この動物に、有効量の本明細書中の前に定義の本発明の活性物質を投与することを含む方法を提供する。
【0159】
本明細書中の前に定義の抗癌処置は、単独の療法として適用されてよいし、または本発明のキナゾリン誘導体に加えて、慣用的な外科手術または放射線療法または化学療法を伴ってよい。このような化学療法には、次の分類の内の一つまたはそれを超える抗腫瘍薬が含まれてよい。
【0160】
(i)他の抗浸潤薬(例えば、N−(2−クロロ−6−メチルフェニル)−2−{6−[4−(2−ヒドロキシエチル)ピペラジン−1−イル]−2−メチルピリミジン−4−イルアミノ}チアゾール−5−カルボキサミド(ダサチニブ(dasatinib),BMS−3
54825;J. Med. Chem., 2004, 47, 6658-6661)のような他のc−Srcキナーゼファミリー阻害剤;マリマスタト(marimastat)のようなメタロプロテイナーゼ阻害剤;およびウロキナーゼプラスミノーゲンアクチベーター受容体機能の阻害剤);
(ii)医学腫瘍学で用いられるような増殖抑制薬/抗腫瘍薬およびそれらの組合せであって、アルキル化剤(例えば、シスプラチン、カルボプラチン、シクロホスファミド、ナイトロジェンマスタード、メルファラン、クロラムブシル、ブスルファンおよびニトロソ尿素);代謝拮抗薬(例えば、5−フルオロウラシルおよびテガフルのようなフルオロピリミジン類、ラルチトレキセド(raltitrexed)、メトトレキサート、シトシンアラビノ
シドおよびヒドロキシ尿素などの葉酸拮抗薬;抗腫瘍抗生物質(例えば、アドリアマイシン、ブレオマイシン、ドキソルビシン、ダウノマイシン、エピルビシン、イダルビシン(idarubicin)、マイトマイシンC、ダクチノマイシンおよびミトラマイシンのようなアントラサイクリン系);有糸分裂阻止薬(例えば、ビンクリスチン、ビンブラスチン、ビンデシンおよびビノレルビン(vinorelbine)のようなビンカアルカロイド類、およびタキ
ソールおよびタキソテールのようなタキソイド類(taxoids));およびトポイソメラー
ゼ阻害剤(例えば、エトポシドおよびテニポシドのようなエピポドフィロトキシン類、アムサクリン、トポテカン(topotecan)およびカンプトテシン)のようなもの;
(iii)細胞分裂抑制薬であって、抗エストロゲン(例えば、タモキシフェン、フルヴ
ェストラント(fulvestrant)、トレミフェン(toremifene)、ラロキシフェン(raloxifene)、ドロロキシフェン(droloxifene)およびヨードキシフェン(iodoxyfene))、抗アンドロゲン(例えば、ビカルタミド(bicalutamide)、フルタミド、ニルタミド(nilutamide)および酢酸シプロテロン)、LHRHアンタゴニストまたはLHRHアゴニスト(例えば、ゴセレリン、ロイプロレリン(leuprorelin)およびブセレリン(buserelin))、プロゲストゲン(例えば、酢酸メゲストロール)、アロマターゼ阻害剤(例えば、アナストロゾール(anastrozole)、レトロゾール(letrozole)、ボラゾール(vorazole)およびエクセメスタン(exemestane))、およびフィナステリドのような5α−レダクターゼの阻害剤のようなもの;
(iv)増殖因子機能の阻害剤:例えば、このような阻害剤には、増殖因子抗体および増殖因子受容体抗体が含まれる(例えば、抗erbB2抗体トラスツズマブ[HerceptinT
M]および抗erbB1抗体セツキシマブ(cetuximab)[C225]);このような阻
害剤には、更に、例えば、チロシンキナーゼ阻害剤、例えば、上皮増殖因子ファミリーの阻害剤(例えば、N−(3−クロロ−4−フルオロフェニル)−7−メトキシ−6−(3−モルホリノプロポキシ)キナゾリン−4−アミン(ゲフィチニブ(gefitinib),ZD
1839)、N−(3−エチニルフェニル)−6,7−ビス(2−メトキシエトキシ)キナゾリン−4−アミン(エルロチニブ(erlotinib),OSI−774)および6−アク
リルアミド−N−(3−クロロ−4−フルオロフェニル)−7−(3−モルホリノプロポ
キシ)キナゾリン−4−アミン(CI 1033)などのEGFRファミリーチロシンキナーゼ阻害剤;およびラパチニブ(lapatinib)などのerbB2チロシンキナーゼ阻害
剤);肝細胞増殖因子ファミリーの阻害剤;イマチニブ(imatinib)などの血小板由来増殖因子ファミリーの阻害剤;セリン/トレオニンキナーゼの阻害剤(例えば、ファルネシルトランスフェラーゼ阻害剤などのRas/Rafシグナリング阻害剤、例えば、ソラフェニブ(sorafenib)(BAY43−9006));およびMEKおよび/またはAkt
キナーゼによる細胞シグナリングの阻害剤が含まれる;
(v)抗血管新生薬であって、血管内皮増殖因子の作用を阻害するものなど[例えば、抗血管内皮細胞増殖因子抗体ベヴァシズマブ(bevacizumab)[AvastinTM];および4−(4−ブロモ−2−フルオロアニリノ)−6−メトキシ−7−(1−メチルピペリジン−4−イルメトキシ)キナゾリン(ZD6474;WO01/32651号中の実施例2)、4−(4−フルオロ−2−メチルインドール−5−イルオキシ)−6−メトキシ−7−(3−ピロリジン−1−イルプロポキシ)キナゾリン(AZD2171;WO00/47212号中の実施例240)、バタラニブ(vatalanib)(PTK787;WO98/
35985号)およびSU11248(スニチニブ(sunitinib);WO01/6081
4)号などのVEGF受容体チロシンキナーゼ阻害剤;および他の機構によって作用する化合物(例えば、リノマイド(linomide)、インテグリンαvβ3機能の阻害剤およびアンギオスタチン(angiostatin))];
(vi)血管損傷性薬であって、Combretastatin A4;および国際特許出願WO99/
02166号、WO00/40529号、WO 00/41669号、WO01/922
24号、WO02/04434号およびWO02/08213号に開示された化合物のようなもの;
(vii)アンチセンス療法、例えば、ISIS2503、抗rasアンチセンスのよう
な、上に挙げられた標的に向けられているもの;
(viii)遺伝子治療アプローチであって、例えば、異常p53または異常BRCA1またはBRCA2のような異常遺伝子を置き換えるアプローチ;シトシンデアミナーゼ、チミジンキナーゼまたは細菌ニトロレダクターゼ酵素を用いたものなどのGDEPT(遺伝子に支配される酵素プロドラッグ療法(gene-directed enzyme pro-drug therapy))ア
プローチ;および多剤耐性遺伝子治療のような、化学療法または放射線療法への患者耐性を増加させるアプローチを含めたもの;および
(ix)免疫療法アプローチであって、例えば、インターロイキン2、インターロイキン4または顆粒球−マクロファージコロニー刺激因子などのサイトカインでのトランスフェクションのような、患者腫瘍細胞の免疫原性を増加させる ex-vivo および in-vivo アプローチ;T細胞アネルギーを減少させるアプローチ;サイトカインでトランスフェクションされた樹状細胞のようなトランスフェクションされた免疫細胞を用いたアプローチ;サイトカインでトランスフェクションされた腫瘍細胞系を用いたアプローチ;および抗イディオタイプ抗体を用いたアプローチを含めたもの。
【0161】
このような共同処置は、個々の処置成分の同時の、逐次的または別々の投与によって達成することができる。このような組合せ製品は、本明細書中の前に記載の投薬量範囲内の本発明の化合物と、その承認された投薬量範囲内の他の薬学的活性剤を用いる。
【0162】
本発明のこの側面により、癌の共同処置のための医薬製品であって、本明細書中の前に定義の本発明の活性物質および本明細書中の前に定義の追加の抗癌薬を含む医薬製品を提供する。
【0163】
式Iの化合物の次の具体的な結晶形の製造方法、すなわち、
(i)AZD0530ジフマレートを製造する;
(ii)AZD0530セスキフマレート四水和物を製造する;
(iii)AZD0530三水和物を製造する;および
(iv)無水AZD0530を製造する方法は、本明細書中に開示されている。
【0164】
本発明のもう一つの側面により、実質的にAZD0530ジフマレートの形の式Iの化合物を製造する方法であって、
(a)4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンとフマル酸とを、AZD0530ジフマレートを形成するのに十分な時間接触させ;そして
(b)AZD0530ジフマレートを単離すること
を含む方法を提供する。
【0165】
直ぐ上の作業工程(a)において出発物質として用いられる4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンは、例えば、先行技術に記載のように製造される場合、またはAD0530三水和物のような本明細書中に記載の形の一つとして製造される場合、式Iの化合物のいずれの形であってもよい。
【0166】
好都合には、AZD0530ジフマレートへの変換は、一つまたはそれを超える適する溶媒中の4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン出発物質の溶液を製造し、そしてフマル酸を加えることによって行われる。好都合には、モル過剰のフマル酸を用いて、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン出発物質のAZD0530ジフマレートへの実質的に完全な変換を確実にすることができる(すなわち、フマル酸対キナゾリン化合物のモル比は、少なくとも2:1である)。フマル酸濃度の上限は、臨界的ではない。好都合には、僅かにモル過剰のフマル酸を用いる。例えば、フマル酸対キナゾリン化合物のモル比は、好適には、約2:1〜10:1、具体的には、約2:1〜3:1、より具体的には、約2.2:1である。
【0167】
具体的な態様において、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン出発物質の溶液は、水を補助溶媒として含有してよい、一つまたはそれを超える有機溶媒の混合物中で製造する。適する有機溶媒は、極性プロトン性溶媒、例えば、(1−4C)アルコール、具体的には、メタノール、エタノール、イソプロパノールおよびn−ブタノール;極性非プロトン性溶媒であって、脂肪族エステル、例えば、(1−4C)アルキル(2−3C)アルカノエートエステル、具体的には、酢酸エチル;脂肪族(3−6C)ケトン、具体的には、アセトンおよびメチルエチルケトン;脂肪族アミド、具体的には、N,N−ジメチルホルムアミド;およびニトリル、具体的には、アセトニトリルなどのような水混和性極性有機溶媒である。好都合には、水不混和性補助溶媒を、水混和性溶媒に加えてよい。適するこのような補助溶媒には、例えば、トルエンなどの芳香族溶媒が含まれる。特に好都合な有機溶媒には、例えば、イソプロパノールまたは酢酸エチル、またはそれらの混合物が含まれる。
【0168】
キナゾリン化合物について、用いられる有機溶媒の具体的な量は、選択される有機溶媒に、およびキナゾリン化合物をフマル酸と接触させる条件に依存するであろう。イソプロパノールまたは酢酸エチルなどの溶媒の場合、2〜20ml/gのような0.1〜30ml/gの範囲、具体的には、約10ml/gが、キナゾリン化合物に適しているが、ここにおいて、「ml/g」は、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−
4−イルオキシキナゾリン1g当たりの有機溶媒の容量を意味する。単一の有機溶媒を用いてよいし、または二つまたはそれを超える有機溶媒、例えば、酢酸エチルおよびイソプロパノールの(好適には、約1:1の容量比の)混合物を用いてよい。水は、補助溶媒として加えることができる。好都合には、有機溶媒(イソプロパノールなど)対水の適する容量比は、50:1〜2:1の範囲内、具体的には、10:1〜5:1の範囲内である。
【0169】
フマル酸について、用いられる有機溶媒の具体的な量は、水が補助溶媒として用いられようといまいと、選択される有機溶媒に、およびフマル酸をキナゾリン化合物と接触させる条件に依存するであろう。イソプロパノールまたは酢酸エチルなどの溶媒の場合、2〜30ml/gのような0.1〜60ml/gの範囲、具体的には、約15ml/gが、フマル酸に適している。単一の有機溶媒を用いてよいし、または二つまたはそれを超える有機溶媒、例えば、酢酸エチルおよびイソプロパノールの(好適には、約1:1の容量比の)混合物を用いてよい。水は、補助溶媒として加えることができる。好都合には、有機溶媒(イソプロパノールなど)対水の適する容量比は、50:1〜3:1の範囲内、具体的には、15:1〜5:1の範囲内、より具体的には、約10:1である。
【0170】
場合により、AZD0530ジフマレートの一つまたはそれを超える種結晶を加えて、AZD0530ジフマレートへの変換の開始および/または変換速度を増強させることができる。
【0171】
AZD0530ジフマレートへの変換に必要な時間は、温度、有機溶媒の存在、および播種結晶を用いるか否かのような、用いられる具体的な反応条件に依存する。概して、例えば、5分〜48時間の反応時間が適している。
【0172】
或いは、有機溶媒の量は、その方法の間中、スラリーが保持されているように、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン出発物質を完全に溶解させるのに不十分であってよい。好都合には、式Iの化合物をその方法中にスラリーで保持することにより、AZD0530ジフマレートは、例えば、混合物を冷却するまたは溶媒を蒸発させることによって結晶化を誘導することを必要とすることなく、形成させることができる。したがって、スラリー方法は、実質的に一定温度で操作することができる。理論によって拘束されたくはないが、その方法は、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン出発物質の限局的溶解および引き続きのAZD0530ジフマレートの結晶化の機構によって進行すると考えられる。したがって、本明細書中に記載のスラリー変換方法は、少量ずつの溶解と、出発物質のAZD0530ジフマレートへの少量ずつの変換であると考えられる。
【0173】
本発明のこの側面により、更に、実質的にAZD0530ジフマレートの形の式Iの化合物を製造する方法であって、
(a)化合物4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンを、有機溶媒および水を含む溶媒系中に溶解させ;
(b)有機溶媒および水を含む溶媒系中のフマル酸の溶液を加え;
(c)その溶媒系の温度を下げて、核生成を誘導し;
(d)その混合物を、核生成が開始した温度未満の温度で維持し;そして
(e)結晶性AZD0530ジフマレートを単離する
工程を含む方法を提供する。
【0174】
AZD0530ジフマレートを製造するこの結晶化方法は、ジフマレート塩を高純度で
製造することを可能にする。
溶媒系中の適する有機溶媒には、直ぐ上の作業工程(a)において出発物質を溶解させる温度で水溶性である有機溶媒が含まれる。適する有機溶媒には、例えば、弱極性有機溶媒であって、テトラヒドロフランなどの(4−7C)環状エーテルまたは脂肪族ジ−(1−6C)アルキルエーテルなど;より極性のプロトン性溶媒、例えば、エタノールおよびイソプロパノールなどの(2−6C)アルコール;極性非プロトン性溶媒であって、酢酸エチルなどの(1−4C)アルキル(2−3C)アルカノエートエステル;アセトンなどの脂肪族(3−6C)ケトン;N,N−ジメチルホルムアミドまたはN−メチルピロリジン−2−オンなどの脂肪族アミド;およびアセトニトリルなどのニトリルのようなものが含まれる。具体的な有機溶媒は、例えば、酢酸エチルである。単一の有機溶媒、または上の一つまたはそれを超える溶媒の混合物を用いてよい。
【0175】
好都合には、モル過剰のフマル酸を用いて、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン出発物質のAZD0530ジフマレートへの実質的に完全な変換を確実にすることができる(すなわち、フマル酸対キナゾリン化合物のモル比は、少なくとも2:1である)。フマル酸濃度の上限は、臨界的ではない。好都合には、僅かにモル過剰のフマル酸を用いる。例えば、フマル酸対キナゾリン化合物のモル比は、好適には、約2:1〜10:1、具体的には、約2:1〜3:1、より具体的には、約2.2:1である。
【0176】
キナゾリン化合物について、用いられる有機溶媒の具体的な量は、選択される有機溶媒に、およびキナゾリン化合物をフマル酸と接触させる条件に依存するであろう。イソプロパノールまたは酢酸エチルなどの溶媒の場合、2〜20ml/gのような0.1〜30ml/gの範囲、具体的には、約10ml/gが適している。単一の有機溶媒を用いてよいし、または二つまたはそれを超える有機溶媒、例えば、酢酸エチルおよびイソプロパノールの(好適には、約1:1の容量比の)混合物を用いてよい。水は、好都合には、補助溶媒として加える。好都合には、有機溶媒(イソプロパノールなど)対水の適する容量比は、50:1〜2:1の範囲内、具体的には、10:1〜5:1の範囲内である。
【0177】
フマル酸について、用いられる有機溶媒の具体的な量は、水が補助溶媒として用いられようといまいと、選択される有機溶媒に、およびフマル酸をキナゾリン化合物と接触させる条件に依存するであろう。イソプロパノールまたは酢酸エチルなどの溶媒の場合、2〜30ml/gのような0.1〜60ml/gの範囲、具体的には、約15ml/gが適している。単一の有機溶媒を用いてよいし、または二つまたはそれを超える有機溶媒、例えば、酢酸エチルおよびイソプロパノールの(好適には、約1:1の容量比の)混合物を用いてよい。水は、好都合には、補助溶媒として加える。好都合には、有機溶媒(イソプロパノールなど)対水の適する容量比は、50:1〜3:1の範囲内、具体的には、15:1〜5:1の範囲内、より具体的には、約10:1である。
【0178】
化合物4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンは、その方法の工程(a)において、溶媒系中で、実質的に全ての化合物が溶解するまで化合物を加熱することにより、溶解させることができる。同様に、フマル酸は、その方法の工程(b)において、溶媒系中で、実質的に全ての化合物が溶解するまで化合物を加熱することにより、溶解させることができる。好都合には、各々の化合物を、溶媒系のほぼ還流温度に、溶解を完了するのに十分な時間加熱する。より好都合には、各々の化合物を、約30〜100℃の範囲内、好ましくは、35〜80℃の範囲内の温度に加熱して、溶解を完了する。必要ならば、どちらかまたは双方の加温された溶液を濾過して、不溶性物質を除去することができる。キナゾリン化合物の溶液の温度を約50〜100℃の
範囲内、好都合には、約60〜90℃の範囲内に維持すると同時に、温フマル酸溶液を加える。次に、その混合物を、例えば、約50〜80℃の範囲内の温度に僅かに冷却させて、AZD0530ジフマレートの核生成を促進することができる。核生成は、自発的にかまたは、一つまたはそれを超える種結晶を加えることで起こりうるということは理解されるであろう。好都合には、その混合物を、約75℃の温度で維持し、種結晶を加えてAZD0530ジフマレートの核生成を促進し、そしてその混合物を、約75℃の温度で数時間維持して、結晶化方法を継続させる。次に、その混合物を、一定制御速度で周囲温度に冷却させることができる。適する冷却速度は、例えば、約20℃/時である。そのようにして得られた結晶性AZD0530ジフマレートは、いずれかの慣用法によって、例えば、濾過または遠心分離によって単離することができる。
【0179】
上記の結晶化方法において、一つまたはそれを超える種結晶を用いて核生成を開始させる場合、それら種結晶は、好ましくは、いずれか適する方法を用いて、例えば、以下の実施例中に記載の方法を用いて製造することができるAZD0530ジフマレートの結晶である。
【0180】
上記の手順を、常套の技術および情報を用いて変更することができるということは、当業者に理解されるであろう。例えば、逆添加手順を用いることができ、それによって、AZD0530の溶液をフマル酸の溶液に加える。更に、例えば、AZD0530ジフマレートが、いずれか他のAZD0530形を実質的に不含で得られるという条件ならば、反応する化合物4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンおよびフマル酸の量;溶媒およびいずれかの補助溶媒の性質および容量;溶媒混合物が用いられる場合の成分溶媒の比率;用いられる水の容量;および溶解相および結晶化相の温度はいずれも、変更することができる。例えば、ジフマレートの核生成は、例えば、若干の溶媒の蒸発によって誘導することができる。或いは、核生成は、ジフマレート化合物に適するアンチソルベントの添加によって誘導しうるが、それによって、溶液の過飽和を生じ、そこから、AZD0530ジフマレートが結晶化する。
【0181】
更に、実質的にAZD0530セスキフマレート四水和物の形の式Iの化合物を製造する方法であって、
(a)AZD0530ジフマレートを、有機溶媒および水を含む溶媒系または水中に溶解させ;
(b)溶媒系の部分蒸発を引き起こして、核生成を誘導し;
(c)その混合物を、周囲温度未満の温度に冷却し;そして
(d)結晶性AZD0530セスキフマレート四水和物を単離する
工程を含む方法を提供する。
【0182】
AZD0530セスキフマレート四水和物を製造するこの結晶化方法は、セスキフマレート四水和物塩を高純度で製造することを可能にする。
溶媒系中の適する有機溶媒には、直ぐ上の作業工程(a)において出発物質を溶解させる温度で水溶性である有機溶媒が含まれる。適する有機溶媒には、例えば、アセトニトリルなどのニトリル;および極性プロトン性溶媒、例えば、メタノール、エタノールおよびイソプロパノールなどの(2−6C)アルコールが含まれる。水との混合物で用いるための具体的な有機溶媒は、例えば、アセトニトリルである。単一の有機溶媒、または上の一つまたはそれを超える溶媒の混合物を用いてよい。好都合には、水不混和性補助溶媒を、水混和性溶媒に加えてよい。適するこのような補助溶媒には、例えば、トルエンなどの芳香族溶媒が含まれる。より好都合には、水を溶媒として用いる。
【0183】
溶媒の蒸発は、周囲温度で、例えば、その溶液を開放容器中で放置することによって行
うことができる。或いは、蒸発工程は、より高い温度で、例えば、約40〜80℃の範囲内の温度で、好都合には、約60℃で行うことができる。好都合には、空気または窒素などのガス流を、その溶液の表面中にまたはそれを越えて通過させて、溶媒蒸発を加速することができる。核生成が開始したら、結晶化混合物を、好都合には、周囲温度未満の温度に冷却して、結晶化を継続させる。好都合には、その混合物を、約10℃未満の温度に、より好都合には、約5℃の温度に冷却する。
【0184】
上記の手順を、常套の技術および情報を用いて変更することができるということは、当業者に理解されるであろう。例えば、AZD0530セスキフマレート四水和物が、いずれか他のAZD0530形を実質的に不含で得られるという条件ならば、AZD0530ジフマレートの量;用いられる水の容量;用いられるいずれかの補助溶媒の性質および容量;および溶解相、蒸発相および冷却相の温度はいずれも、変更することができる。例えば、核生成は、適するアンチソルベントの添加によって誘導しうるが、それによって、溶液の過飽和を生じ、そこから、AZD0530セスキフマレート四水和物が結晶化する。
【0185】
本発明のもう一つの側面により、実質的にAZD0530三水和物の形の式Iの化合物を製造する方法であって、
(a)化合物4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンを、水および有機溶媒を含む溶媒系中に溶解させ;
(b)その溶媒系の温度を下げて、核生成を誘導し;
(c)その混合物を、核生成が開始した温度未満の温度で維持し;そして
(d)結晶性AZD0530三水和物を単離する
工程を含む方法を提供する。
【0186】
溶媒系中の適する有機溶媒には、その方法の工程(a)において出発物質を溶解させる温度で水溶性である有機溶媒が含まれる。適する有機溶媒には、例えば、弱極性有機溶媒であって、テトラヒドロフランなどの(4−7C)環状エーテルまたは脂肪族ジ−(1−6C)アルキルエーテルなど;より極性のプロトン性溶媒、例えば、エタノールおよびイソプロパノールなどの(2−6C)アルコール;極性非プロトン性溶媒であって、酢酸エチルなどの(1−4C)アルキル(2−3C)アルカノエートエステル;アセトンなどの脂肪族(3−6C)ケトン;N,N−ジメチルホルムアミドまたはN−メチルピロリジン−2−オンなどの脂肪族アミド;およびアセトニトリルなどのニトリルのようなものが含まれる。具体的な有機溶媒は、例えば、酢酸エチルである。単一の有機溶媒、または上の一つまたはそれを超える溶媒の混合物を用いてよい。
【0187】
概して、モル過剰の水を溶媒系中に用いる(すなわち、水:4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンのモル比は、少なくとも3:1である)。水濃度の上限は、臨界的ではないが、しかしながら、概して、大モル過剰の水を用いる。例えば、水対4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンのモル比は、好適には、約3:1〜1000:1またはそれを超える、具体的には、約3:1〜約400:1である。
【0188】
場合により、補助溶媒を溶媒系中に用いてよい。適する補助溶媒には、例えば、トルエンなどの芳香族炭化水素;およびハロゲノ−(1−6C)アルカンなどの脂肪族ハロゲン化炭化水素、例えば、1,2−ジクロロエタンが含まれる。
【0189】
化合物4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メ
チルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンは、その方法の工程(a)において、溶媒系中で、実質的に全ての化合物が溶解するまで化合物を加熱することにより、溶解させることができる。好都合には、その方法の工程(a)における溶媒系中の化合物を、溶媒系のほぼ還流温度に、化合物を完全に溶解するのに十分な時間加熱する。次に、化合物4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンの溶液を、熱源から除去し、そして25〜60℃の範囲内の温度に冷却させて、AZD0530三水和物の核生成を促進することができるし、またはそれを更に、例えば、周囲温度に冷却することができる。好都合には、その溶液を、熱源から除去し、そして約50℃に冷却させて、AZD0530三水和物の核生成を促進することができる。その混合物は、約55℃に再加熱した後、一定制御速度で約50℃に冷却させることができる。適する冷却速度は、例えば、約10℃/時である。核生成は、自発的にかまたは、一つまたはそれを超える種結晶を加えることで起こりうるということは理解されるであろう。次に、その溶液を、約50℃の温度で保持して、生成物の結晶化を生じさせる。引き続き、その溶液を、一定制御速度で約20℃に冷却して、生成物の結晶化を終わらせることができる。適する冷却速度は、例えば、約10℃/時である。そのようにして得られた結晶性AZD0530三水和物は、いずれかの慣用法によって、例えば、濾過または遠心分離によって単離することができる。
【0190】
上記の結晶化/再結晶方法において、一つまたはそれを超える種結晶を用いて核生成を開始させる場合、それら種結晶は、好ましくは、AZD0530三水和物の結晶である。その一つまたは複数の種結晶は、AZD0530三水和物の製造についていずれか適する方法を用いて、例えば、非晶質AZD0530の試料を水中でスラリーにすることによって製造することができる。
【0191】
上記の手順を、常套の技術および情報を用いて変更することができるということは、当業者に理解されるであろう。例えば、AZD0530三水和物が、いずれか他のAZD0530形を実質的に不含で得られるという条件ならば、処理される化合物4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンの量;溶媒およびいずれかの補助溶媒の性質および容量;溶媒混合物が用いられる場合の成分溶媒の比率;用いられる水の容量および水対溶媒の比率;および溶解相および冷却相の温度はいずれも、変更することができる。例えば、その方法の工程(b)における、適する溶媒、例えば、エタノールなどの(2−6C)アルコール中の式Iの化合物の溶液の核生成は、例えば、若干のエタノール溶媒の蒸発によって誘導することができるし、或いは、核生成は、式Iの化合物に適するアンチソルベントの添加によって誘導しうるが、それによって、溶液の過飽和を生じ、そこから、AZD0530三水和物が結晶化する。
【0192】
AZD0530三水和物を製造する結晶化方法は、三水和物を高純度で製造することを可能にする。更に、そのようにして得られたAZD0530三水和物の再結晶は、上記の方法を用いて行うことができる。再結晶は、その物質を更に精製する可能性を与える。
【0193】
本発明のもう一つの側面により、実質的にAZD0530三水和物の形の式Iの化合物を製造する方法であって、
(a)4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンと水とを、AZD0530三水和物を形成するのに十分な時間接触させ;そして
(b)AZD0530三水和物を単離すること
を含む方法を提供する。
【0194】
直ぐ上の作業工程(a)において出発物質として用いられる4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンは、式Iのいずれかの形の化合物、例えば、先行技術に記載の非晶形、または無水AD0530のような本明細書中に記載の結晶形のいずれか一つであってよい。
【0195】
好都合には、AZD0530三水和物への変換は、水中の4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン出発物質のスラリーを、場合により、一つまたはそれを超える適する有機溶媒の存在下で製造することによって行われる。概して、モル過剰の水を用いて、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン出発物質のAZD0530三水和物への実質的に完全な変換を確実にする(すなわち、水対キナゾリン化合物のモル比は、少なくとも3:1である)。水濃度の上限は、臨界的ではないが、しかしながら、概して、大モル過剰の水を用いる。例えば、水対キナゾリン化合物のモル比は、好適には、約3:1〜1000:1またはそれを超える、具体的には、約3:1〜約400:1である。
【0196】
具体的な態様において、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン出発物質のスラリーは、水および有機溶媒、そして場合により、一つまたはそれを超える補助溶媒の混合物中で製造する。本発明者は、有機溶媒の使用が、出発物質をAZD0530三水和物へ変換するのに必要な時間を有意に減少させるということを発見した。適する有機溶媒は、水混和性極性有機溶媒であって、極性プロトン性溶媒、例えば、(1−4C)アルコール、具体的には、エタノールおよびイソプロパノール;極性非プロトン性溶媒であって、脂肪族エステル、例えば、(1−4C)アルキル(2−3C)アルカノエートエステル、具体的には、酢酸エチル;アセトンなどの脂肪族(3−6C)ケトン;N,N−ジメチルホルムアミドなどの脂肪族アミドなどのようなものである。具体的な溶媒には、例えば、イソプロパノールまたは酢酸エチル、またはそれらの混合物が含まれる。
【0197】
用いられる有機溶媒の量は、その方法の間中、スラリーが保持されているように、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン出発物質を完全に溶解させるのに不十分であってよい。好都合には、式Iの化合物をその方法中にスラリーで保持することにより、AZD0530三水和物は、例えば、混合物を冷却するまたは溶媒を蒸発させることによって結晶化を誘導することを必要とすることなく、形成させることができる。したがって、スラリー方法は、実質的に一定温度で操作することができる。
【0198】
理論によって拘束されたくはないが、その方法は、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン出発物質の限局的溶解および引き続きのAZD0530三水和物の結晶化の機構によって進行すると考えられる。したがって、本明細書中に記載のスラリー変換方法は、少量ずつの溶解と、出発物質のAZD0530三水和物への少量ずつの変換であると考えられる。
【0199】
用いられる有機溶媒の具体的な量は、選択される有機溶媒に、およびスラリーを水と接触させる条件に依存するであろう。イソプロパノールまたは酢酸エチルなどの溶媒の場合、2〜10ml/gのような0.1〜20ml/gの範囲、具体的には、約5ml/gが
適しているが、ここにおいて、「ml/g」は、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン1g当たりの有機溶媒の容量を意味する。
【0200】
単一の有機溶媒を用いてよいし、または二つまたはそれを超える有機溶媒、例えば、酢酸エチルおよびイソプロパノールの(好適には、約1:1の容量比の)混合物を、水と一緒に用いてよい。
【0201】
場合により、補助溶媒を用いてよい。適する補助溶媒には、例えば、弱極性有機溶媒であって、芳香族炭化水素(例えば、トルエン)、ハロゲノ−(1−6C)アルカン(例えば、1,2−ジクロロエタン)および脂肪族ジ−(1−6C)アルキルエーテルまたは(4−7C)環状エーテル(例えば、テトラヒドロフラン)などが含まれる。具体的な補助溶媒は、トルエンである。補助溶媒(トルエンなど)対有機溶媒(イソプロパノールなど)の適する容量比は、50:1〜0.05:1の範囲内、好都合には、10:1〜0.5:1の範囲内、具体的には、約3:1〜1:1である。
【0202】
場合により、AZD0530三水和物の一つまたはそれを超える種結晶を、スラリーに加えて、AZD0530三水和物への変換速度を増強させることができる。
その方法は、ほぼ周囲温度で、例えば、約15〜30℃、具体的には、約20〜25℃で行われる。
【0203】
AZD0530三水和物への変換に必要な時間は、温度、有機溶媒の存在、および播種結晶を用いるか否かのような、用いられる具体的な反応条件に依存する。概して、例えば、5分〜48時間の反応時間が適している。
【0204】
本発明者は、更に、AZD0530三水和物は、結晶性無水AZD0530へと容易に変換することができるということを発見した。したがって、AZD0530三水和物の結晶化および引き続きの無水AZD0530への変換は、無水AZD0530を高純度で製造する手段を提供する。実質的に無水AZD0530の形の式Iの化合物のこのような製造方法は、本発明のもう一つの側面を提供する。
【0205】
更に、実質的に結晶性無水AZD0530の形の式Iの化合物を製造する方法であって、AZD0530三水和物を脱水する工程を含む方法を提供する。
以下、変換方法1と称されるこの変換の一つの態様は、実質的に乾燥した不活性ガス流を、AZD0530三水和物の試料の上におよび/またはそれを介して、水を駆逐し且つ無水AZD0530への変換を行うのに十分な時間および十分な温度で通過させる工程を含む。
【0206】
好都合には、変換方法1は、周囲温度(15〜25℃の範囲内、具体的には、約20℃の温度)で行われる。適する不活性ガスは、例えば、窒素ガスであるが、それは、必要ならば、実質的に乾燥するまで乾燥させるべきである。概して、変換方法1は、AZD0530三水和物を無水AZD0530へ変換するのに、5分〜50時間、好適には、1〜30時間の乾燥時間を必要とする。好都合には、AZD0530三水和物は、フィルター上に置くことができ、そして乾燥ガスを、フィルターを介して通過させることができる。好適には、変換方法1における乾燥工程は、所望の無水物形への実質的に完全な変換を確実にするのに十分な時間継続すべきである。実質的に完全な変換とは、式Iの化合物の少なくとも80%が、無水AZD0530の形であり、20%未満のいずれか他のAZD0530形が存在するということを意味する。具体的には、式Iの化合物の少なくとも90%、特に、少なくとも95%は、無水AZD0530の形である。必要な無水AZD0530への変換度は、常套技術、例えば、本明細書中に記載のXRPDを用いて評価すること
ができる。
【0207】
場合により、窒素などの不活性ガス流は、その物質の上におよび/またはそれを介して通過させる前に加温する。加温されたガスに適する温度は、例えば、25〜100℃、具体的には、40〜60℃の温度である。
【0208】
以下、変換方法2と称されるこの変換のもう一つの態様は、実質的にAZD0530三水和物の形の式Iの化合物を、水を駆逐し且つ無水AZD0530への変換を行うのに十分な時間および十分な温度で加熱する工程を含む。
【0209】
変換方法2は、好適には、AZD0530三水和物を50〜150℃、具体的には、80〜140℃、より具体的には、120〜130℃の温度で加熱することによって行われる。必要な加熱時間は、特に、試料のサイズおよび用いられる加熱方法に依存する。概して、AZD0530三水和物を無水AZD0530へと変換するには、5分〜100時間、好適には、1〜30時間の加熱時間で十分である。AZD0530三水和物は、慣用的な技法を用いて、例えば、適するオーブンまたは真空オーブン中で、または流動床乾燥器などの慣用的な乾燥システム中で加熱することができる。好適には、変換方法2における加熱工程は、所望の無水物形への本明細書中の前に定義の実質的に完全な変換を確実にするのに十分な時間および十分な温度で継続すべきである。
【0210】
以下、変換方法3と称されるこの変換のもう一つの態様は、
(a)実質的にAZD0530三水和物の形の式Iの化合物を、溶媒または溶媒混合物で洗浄して、実質的に水を除去し;そして
(b)そのようにして形成された無水AZD0530を単離することを含む。
【0211】
変換方法3において適する溶媒には、例えば、洗浄温度において式Iの化合物が可溶性に乏しい水混和性有機溶媒が含まれる。適する溶媒の例には、弱極性有機溶媒であって、テトラヒドロフランなどの(4−7C)環状エーテルまたは脂肪族ジ−(1−6C)アルキルエーテルなど;より極性のプロトン性溶媒、例えば、エタノールおよびイソプロパノールなどの(2−6C)アルコール;極性非プロトン性溶媒であって、酢酸エチルなどの(1−4C)アルキル(2−3C)アルカノエートエステル;およびアセトニトリルなどのニトリルのようなものが含まれる。このような溶媒の混合物を用いてもよい。具体的な溶媒は、イソプロパノールおよび/または酢酸エチルである。
【0212】
「洗浄」工程は、無水AZD0530への変換を行うのに適する時間を必要とするということは理解されるはずである。固体と洗浄溶媒との間の適する接触時間は、約5分〜1時間またはそれを超える範囲内である。より好都合には、接触時間は、約30分〜約2時間の範囲内、例えば、約1時間である。好都合には、固体および洗浄溶媒のスラリーを製造する。好都合には、そのスラリーを撹拌して、洗浄溶媒と固体の結晶との間の接触を改善する。洗浄溶媒は、例えば、約30〜50℃の温度に加温してよいが、しかしながら、概して、無水AZD0530への変換を行うには、ほぼ周囲温度での洗浄で十分である。
【0213】
場合により、変換方法3において一つまたは複数の溶媒洗浄工程後に単離された物質を乾燥させて、水の完全な除去および所望の結晶性無水AZD0530への変換を確実にする。変換方法1または変換方法2の方法を用いることができる。
【0214】
本発明を、次の実施例、データおよび図面によって以下に詳しく説明するが、ここにおいて、
(i)X線回折データは、Siemens D5000装置を用いて得た。試料は、メノウ乳棒・乳鉢を用いて結晶凝集体を穏やかに粉砕することによって調製した。その試料を、標準
的なホルダー(フラットリップを有する)中に充填し、顕微鏡スライドガラスでリップまで平らに圧縮した。その試料を、30回毎分(rpm)で回転させて、計数統計値を改善した。X線は、40kVおよび40mAで操作される銅ロングファインフォーカス管球によって発生させた。X線の波長は、1.5406Åであった。その計器を、θ−θ配置において2°2θ〜40°2θの走査範囲にわたって0.02°2θ増加分につき4秒暴露で操作した。実験は、Bragg-Brentano 配置で行ったが、それによって、X線ビームは、
V20で自動可変発散スリットを介して通過し、そして反射した放射線は、2mm散乱防止スリットおよび0.2mm検出器スリットによって方向づけられた。反射は、それらの重心値として引用される(DIFFRAC/ATなどの計算機パッケージによって計算される)。当XRPD業者は、約30ミクロンを超えるサイズの粒子および単一でない(non-unitary)アスペクト比を有する試料の分析が、ピークの相対強さに影響することがあ
りうるということを理解するであろう。当業者は、更に、反射位置は、試料が位置する回折計中の正確な高さおよび回折計のゼロ検定によって影響されるということを理解するであろう。試料表面の平坦さも、僅かに作用することがありうる。したがって、示された回折図形データを、絶対値とみなすべきではない。
【0215】
更に、異なった装置および/または条件は、僅かに異なったデータを生じさせることがありうる、例えば、ピークの位置および相対強さに変動がありうるということは理解されるであろう。概して、XRPDスペクトル中のピーク位置(回折角)の測定誤差は、約±0.3°2θであろう。このような程度の測定誤差は、XRPDスペクトルが実質的に同じであるか否かを評価する場合に考慮されるべきである。当業者は、正確な個々のピーク位置よりもむしろ、それらピークの相対位置が、試料が実質的に同じ結晶形に由来したか否かについてのより信頼できる指標であるということを理解するであろう。具体的には、XRPDを用いて測定されたピークの強さは、結晶性粒子をXRPDマウント中に充填する作用のために、粒子のサイズおよび形状の結果として異なることがありうる。このような充填作用は、当該技術分野において周知であり、しばしば、「選択配向」作用と称される。検体の選択配向は、いろいろな反射の強さに影響を与えるので、全くの無作為の試料により予想されうる強さと比較して、あるものはより強く、他のものはあまり強くない。選択配向作用は、サイズ減少が、より微細な針状結晶またはプレートレットを生じる場合、針様またはプレート様の結晶について特に明らかである。結果として、結晶形は、主に、X線ディフラクトグラム中の相対ピーク位置を最も確実に特徴とする。これら作用並びに標準的なX線回折分析方法は、例えば、Bunn, C. W. (1948), Chemical Crystallography, Clarendon Press, London;または Klug, H. P. & Alexander, L. E. (1974), X-Ray
Diffraction Procedures, John Wiley and Sons, New York に見出されうる。したがっ
て、本明細書中に引用された図面を、絶対値とみなすべきではない。したがって、本明細書中に開示されたXRPDスペクトルの一つと実質的に同じであるXRPDスペクトルを与えるいずれかの結晶が、本発明の範囲内であるということは理解されるはずである。
【0216】
(ii)融点およびTGAは、Mettler DSC820eおよび Mettler TG851装置
を用いて、それぞれ、TSO891ROロボットシステムを用いて決定した。
融点決定について、パンタイプは、穴のあいた蓋付きのアルミニウム(40μlサイズ)であった。試料重量は、約1〜5mgであった。手順は、窒素ガス流下(100ml/分)で行い、検討された温度範囲は、10℃/分の一定の温度上昇速度において25℃〜325℃であった。当業者は、融点の正確な値が、化合物の純度、試料重量、加熱時間および粒子サイズによって影響されるということを理解するであろう。したがって、別の融点読みが、他のタイプの装置によって、または記載されたものとは異なった条件を用いることによって与えられるかもしれないということは理解されるであろう。したがって、本明細書中に引用された図面を、絶対値とみなすべきではない。
【0217】
TGA決定については、各々の試料(約1〜12mg)を、開放型酸化アルミニウムパ
ン(70μlサイズ)に入れ、そして手順は、ヘリウムガス流下(50ml/分)で行い、検討された温度範囲は、10℃/分の一定の温度上昇速度において25℃〜325℃であった。僅かに異なったデータが、異なった装置および/または条件を用いた場合に生じるかもしれないということは理解されるであろう。したがって、本明細書中に引用された図面を、絶対値とみなすべきではない。
【0218】
(iii)DRIFT分光法データは、Nicolet 20SXC分光計において、粉末臭化カ
リウム中の試料の2%w/w分散を用いて、4000〜400cm−1の度数範囲にわたって得た。僅かに異なったデータが、異なった装置および/または試料調製条件を用いた場合に生じるかもしれないということは理解されるであろう。したがって、本明細書中に引用された図面を、絶対値とみなすべきではない。
【0219】
次の実施例に関して、概して、
(i)操作は、周囲温度で、すなわち、17〜25℃の範囲内で、そして特に断らない限り、窒素またはアルゴンなどの不活性ガスの雰囲気下で行った;
(ii)概して、反応経過は、薄層クロマトグラフィー(TLC)および/または分析高速液体クロマトグラフィー(HPLC)で追跡したが;与えられている反応時間は、必ずしも達成可能な最小値ではない;
(iii)必要な時に、有機溶液を無水硫酸マグネシウム上で乾燥させ、処理手順は、濾
過による残留固体の除去後に行い、蒸発は、真空中のロータリーエバポレーションによって行った;
(iv)収率は、示されている場合、必ずしも達成可能な最大値ではないが、必要な時に、更に多量の反応生成物が必要とされた場合、反応を繰り返した;
(v)概して、式Iの最終生成物の構造は、核磁気共鳴(NMR)および/または質量スペクトル法で確認した;エレクトロスプレー質量スペクトルデータは、正・負双方のイオンデータを獲得する Waters ZMDまたは Waters ZQ LC/質量分析計を用いて得たが、概して、親構造に関するイオンのみを報告している;プロトンNMR化学シフト値は、300MHzの場の強さで操作する Bruker Spectrospin DPX300分光計を用いてδスケールで測定した;次の略語を用いた:s,一重線;d,二重線;t,三重線;q,四重線;m,多重線;br,幅広;
(vi)中間体は、必ずしも完全に精製しなかったが、それらの構造および純度は、TLC、分析HPLC、赤外(IR)および/またはNMR分析によって評価した;
(vii)特に断らない限り、カラムクロマトグラフィー(フラッシュ法による)および
中圧液体クロマトグラフィー(MPLC)は、Merck Kieselgel シリカ(Art. 9385
)上で行った;
(viii)分取HPLCは、C18逆相シリカ上、例えば、Waters ‘Xterra’分取逆相
カラム(5ミクロンシリカ、19mm直径、100mm長さ)上で、溶離剤として減少極性混合物、例えば、水(1%酢酸または1%水性水酸化アンモニウム(d=0.88)を含有する)およびアセトニトリルの減少極性混合物を用いて行った;
(ix)次の分析HPLC法を用いた;概して、逆相シリカは、約1ml/分の流速で用い、そして検出は、230nmの波長でのUV吸光度によった。
【0220】
方法A:Phenomenex LUNAフェニルヘキシルカラム(Phenomenex, Macclesfield, UK;3ミクロンシリカ、2mm直径、50mm長さ)、溶媒Aは、0.05%トリフルオ
ロ酢酸を含有する水であり、溶媒Bは、0.05%トリフルオロ酢酸を含有するメタノールであり、そして95:5の溶媒Aおよび溶媒Bの混合物〜0:100の溶媒Aおよび溶媒Bの混合物の5分間にわたる溶媒勾配を用いた;
方法B:Phenomenex PRODIGY ODSカラム(5ミクロンシリカ、4.6mm
直径、150mm長さ)、溶媒Aは、900:100:0.5:0.5の水、アセトニトリル、トリフルオロ酢酸および酢酸の混合物であり、溶媒Bは、50:950:0.5:
0.5の水、アセトニトリル、トリフルオロ酢酸および酢酸の混合物であり、そして100%溶媒A〜60:40の溶媒Aおよび溶媒Bの混合物の8分間にわたる溶媒勾配、そして更に、60:40の溶媒Aおよび溶媒Bの混合物〜100%溶媒Bの10分間にわたる溶媒勾配を用いた。
【実施例】
【0221】
実施例1
5,7−ジフルオロ−3,4−ジヒドロキナゾリン−4−オン(経路1)
2,4,6−トリフルオロベンゾニトリル(10g)を、イソプロパノール中の4.9Mアンモニア撹拌溶液(220ml;イソプロパノール中にアンモニアを通気することによって調製される)に加え、得られた混合物を45℃に16時間加熱した。溶媒を蒸発させて、2:1の2−アミノ−4,6−ジフルオロベンゾニトリルおよび4−アミノ−2,6−ジフルオロベンゾニトリルの混合物を含む白色固体(11.9g)を残した。
【0222】
その混合物の一部分(5g)を、水(10ml)中に懸濁させ、濃硫酸水(80%;40ml)を加えた。得られた混合物を撹拌し、65℃に16時間加熱した。得られた溶液を、周囲温度に冷却し、水(60ml)で希釈し、10M水性水酸化ナトリウム(180ml)の添加によって塩基性にし、そして酢酸エチルで抽出した。有機溶液を、硫酸マグネシウム上で乾燥させ、蒸発させた。このようにして、2:1の2−アミノ−4,6−ジフルオロベンズアミドおよび4−アミノ−2,6−ジフルオロベンズアミドの混合物を含むクリーム色固体(4g)を得た。
【0223】
そのようにして得られた混合物を、オルトギ酸トリエチル(60ml)中に懸濁させた。濃塩酸水(0.1ml)を加え、得られた混合物を146℃に8時間加熱した。反応混合物を周囲温度に冷却させた。得られた濃厚懸濁液を濾過し、メチル tert−ブチルエー
テル(20ml)で洗浄した。そのようにして得られた物質を、真空中において35℃で3時間乾燥させた。このようにして、5,7−ジフルオロ−3,4−ジヒドロキナゾリン−4−オンを得た(1.61g;方法Aを用いて97%のHPLC純度、保持時間2.29分)。
【0224】
NMRスペクトル:(DMSOd6) 7.3-7.4 (m, 2H), 8.12 (s, 1H)。
実施例2
5,7−ジフルオロ−3,4−ジヒドロキナゾリン−4−オン(経路2)
実施例1に記載の2:1の2−アミノ−4,6−ジフルオロベンゾニトリルおよび4−アミノ−2,6−ジフルオロベンゾニトリルの混合物の一部分(0.5g)を、シリカ上のカラムクロマトグラフィーにより、塩化メチレンおよびメタノールの漸増極性混合物を溶離剤として用いて精製した。このようにして、2−アミノ−4,6−ジフルオロベンゾニトリル(0.15g)を得た。そのようにして得られた物質、濃硫酸水(80%;4ml)および水(1ml)の混合物を、100℃に15時間加熱した。得られた溶液を、周囲温度に冷却し、水で希釈し、10M水酸化ナトリウム水溶液の添加によって塩基性にし、酢酸エチル(10ml)で洗浄した。得られた水溶液を、希塩酸水溶液の添加によって中和し、酢酸エチル(20ml)で抽出した。有機層を、硫酸マグネシウム上で乾燥させ、蒸発させた。このようにして、2−アミノ−4,6−ジフルオロ安息香酸を無色固体として得た(0.11g;方法Bを用いて97%のHPLC純度、保持時間6.87分)。
【0225】
NMRスペクトル:(DMSOd6) 6.25 (m, 1H), 6.4 (m, 1H)。
そのようにして得られた物質、1,3,5−トリアゼン(0.044g)、メタノール(4ml)およびピペリジン(0.038ml)の混合物を、70℃に24時間加熱した。得られた混合物を、周囲温度に冷却し、蒸発させた。ジエチルエーテル(3ml)および酢酸エチル(1ml)を加え、得られた固体を単離し、ジエチルエーテル(1ml)で
洗浄した。このようにして、5,7−ジフルオロ−3,4−ジヒドロキナゾリン−4−オン(0.048g)を得た。
【0226】
実施例3
7−フルオロ−5−テトラヒドロピラン−4−イルオキシ−3,4−ジヒドロキナゾリン−4−オン
カリウム tert−ブトキシド(6.15g)を、THF(40ml)中の4−ヒドロキ
シテトラヒドロピラン(2.94g)の溶液に加え、その混合物を周囲温度で15分間撹拌した。得られた混合物を、加熱して還流しているTHF(60ml)中の5,7−ジフルオロ−3,4−ジヒドロキナゾリン−4−オン(5g)の撹拌溶液に加えた。追加部分のTHF(20ml)を加え、反応混合物を加熱して30分間還流した。第二部分のカリウム tert−ブトキシド(6.15g)を加え、反応混合物を加熱して40分間還流した
。第三部分のカリウム tert−ブトキシド(1.52g)を加え、反応混合物を加熱して
20分間還流した。反応混合物を周囲温度に冷却させた。水(50ml)を加え、大部分の有機溶媒を蒸発させた。残留物を、2M水性塩酸の滴加によってpH<2へと酸性にした。得られたスラリーを15分間撹拌した。混合物を濾過し、そして単離された固体を、水(20ml)で洗浄し、真空中において40℃で一晩乾燥させた。このようにして、7−フルオロ−5−テトラヒドロピラン−4−イルオキシ−3,4−ジヒドロキナゾリン−4−オンを得た(5.96g;方法Aを用いて96%のHPLC純度、保持時間3.34分)。
【0227】
NMRスペクトル:(DMSOd6) 1.6-1.75 (m, 2H), 1.9-2.0 (m, 2H), 3.5-3.6 (m, 2H),
3.85-3.95 (m, 2H), 4.8 (m, 1H), 6.9 (m, 1H), 7.05 (m, 1H), 8.0 (s, 1H)。
実施例4
7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシ−3,4−ジヒドロキナゾリン−4−オン
カリウム tert−ブトキシド(3.77g)を、THF(30ml)中の1−(2−ヒ
ドロキシエチル)−4−メチルピペラジン(国際出願WO01/94341号、実施例2、注記[9];1.78g)の溶液に加え、その混合物を10分間撹拌した。得られた溶液を、THF(50ml)中の7−フルオロ−5−テトラヒドロピラン−4−イルオキシ−3,4−ジヒドロキナゾリン−4−オン(2.96g)の撹拌スラリーに加え、得られた溶液を加熱して3時間還流した。第二部分のカリウム tert−ブトキシド(2.52g
)を加え、反応混合物を加熱して16時間還流した。反応混合物を周囲温度に冷却させた。水(25ml)を加え、大部分の有機溶媒を蒸発させた。残留物を、2M水性塩酸の滴加によって中和し、酢酸エチルで抽出した。有機相を硫酸マグネシウム上で乾燥させ、蒸発させた。残留物を、シリカ上のカラムクロマトグラフィーにより、塩化メチレンおよびメタノールの漸増極性混合物を溶離剤として用いて精製した。このようにして、7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシ−3,4−ジヒドロキナゾリン−4−オンを得た(3.1g;方法Bを用いて91%のHPLC純度、保持時間1.1分)。
【0228】
NMRスペクトル:(CDCl3) 1.9-2.0 (m, 2H), 2.0-2.15 (m, 2H), 2.35 (s, 3H), 2.4-2.8 (br m, 8H), 2.85 (t, 2H), 3.6-3.7 (m, 2H), 4.1-4.15 (m, 2H), 4.2 (t, 2H), 4.65 (m, 1H), 6.55 (s, 1H), 6.85 (s, 1H), 7.25 (s, 1H), 7.9 (s, 1H)。
【0229】
実施例5
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−5,7−ジフルオロキナゾリン(経路1)
塩化ホスホリル(3.32ml)を、5,7−ジフルオロ−3,4−ジヒドロキナゾリン−4−オン(5g)、ジイソプロピルエチルアミン(7.16ml)およびアセトニト
リル(120ml)の氷浴中で冷却された撹拌混合物に加えた。得られた反応混合物を、80℃に2時間加熱した。第二部分の塩化ホスホリル(1.52ml)を加え、反応混合物を加熱して更に2.75時間還流して、4−クロロ−5,7−ジフルオロキナゾリンの溶液を与え、それを、単離することなく用いた。アセトニトリル(15ml)中の6−クロロ−2,3−メチレンジオキシアニリン(国際出願WO01/94341号、実施例17、注記[30];4.95g)の溶液を加え、反応混合物を80℃に4時間加熱した。得られた反応混合物を、周囲温度で16時間撹拌した。アセトニトリル(5ml)中の6−クロロ−2,3−メチレンジオキシアニリン(1.18g)の第二部分溶液を加え、反応混合物を80℃に1時間再加熱した。反応混合物を周囲温度に冷却させ、1時間撹拌した。得られたスラリーを濾過し、そして単離された固体をアセトニトリル(20ml)で洗浄し、乾燥させた。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−5,7−ジフルオロキナゾリンを一塩酸塩として得た(7.88g、方法Aを用いて99.3%のHPLC純度、保持時間4.46分)。
【0230】
NMRスペクトル:(DMSOd6) 5.5-6.0 (br s, 1H), 6.15 (s, 2H), 7.0 (d, 1H), 7.1 (d, 1H), 7.6 (d, 1H), 7.8 (m, 1H), 8.7 (s, 1H).1.9-2.0 (m, 2H)。
実施例6
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−5,7−ジフルオロキナゾリン(経路2)
トリメチルアルミニウム(トルエン中の2M溶液、4.69ml)を、トルエン(10ml)中の6−クロロ−2,3−メチレンジオキシアニリン(1.07g)の撹拌溶液に加え、得られた溶液を周囲温度で15分間撹拌した。トルエン(10ml)中の2,4,6−トリフルオロベンゾニトリル(0.98g)の溶液を滴加し、得られた混合物を周囲温度で10分間撹拌した後、90℃に3時間加熱した。反応混合物を、周囲温度に冷却し、16時間撹拌した。反応混合物を、水(20ml)で洗浄した。有機溶液を、10%クエン酸水溶液で抽出した。水溶液を、2M水性水酸化ナトリウムで塩基性にし、塩化メチレン(50ml)で抽出した。有機溶液を、硫酸マグネシウム上で乾燥させ、蒸発させた。このようにして、N1−(6−クロロ−2,3−メチレンジオキシフェニル)−2,4,6−トリフルオロベンズアミジン(0.92g)を得た。
【0231】
ホルムアミジン酢酸塩(0.185g)を、トルエン(5ml)中のN1−(6−クロロ−2,3−メチレンジオキシフェニル)−2,4,6−トリフルオロベンズアミジン(0.204g)の撹拌溶液に加え、反応混合物を加熱して16時間還流した。第二部分のホルムアミジン酢酸塩(0.185g)を加え、反応混合物を加熱して更に16時間還流した。トリエチルアミン(0.25ml)を加え、反応混合物を加熱して更に3日間還流した。得られた反応混合物を、周囲温度に冷却し、塩化メチレン(25ml)と飽和重炭酸ナトリウム水溶液(25ml)とに分配した。有機溶液を、10%水性クエン酸(25ml)で洗浄し、硫酸マグネシウム上で乾燥させ、蒸発させた。得られた油状物を、シリカゲル上のカラムクロマトグラフィーにより、イソヘキサンおよび酢酸エチルの漸増極性混合物を溶離剤として用いて精製した。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−5,7−ジフルオロキナゾリン(0.068g)を得た。
【0232】
実施例7
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−5,7−ジフルオロキナゾリン
塩化ホスホリル(4.96ml)を、5,7−ジフルオロ−3,4−ジヒドロキナゾリン−4−オン(6.5g)、クロロベンゼン(64.9ml)、6−クロロ−2,3−メチレンジオキシアニリン(7.08g)およびジイソプロピルエチルアミン(7.47ml)の、窒素ガス雰囲気下において95℃に加熱された撹拌混合物に、40分間にわたって加えた。得られた反応混合物を、95℃で5時間加熱した。反応混合物を18℃に冷却
し、30分間撹拌した。撹拌を止め、反応混合物を30分間放置した。その混合物を濾過し、そして単離された固体を、クロロベンゼン(2x23ml)で洗浄し、真空中において45℃で乾燥させた。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−5,7−ジフルオロキナゾリンを一塩酸塩として得た(8.9g、方法Aを用いて96.5%のHPLC純度、保持時間4.46分)。
【0233】
m.p.234〜237℃;
NMRスペクトル:(DMSOd6) 5.5-6.0 (br s, 1H), 6.15 (s, 2H), 7.0 (d, 1H), 7.1 (d, 1H), 7.6 (d, 1H), 7.8 (m, 1H), 8.7 (s, 1H)。
【0234】
実施例8
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−フルオロ−5−テトラヒドロピラン−4−イルオキシキナゾリン(経路1)
7−フルオロ−5−テトラヒドロピラン−4−イルオキシ−3,4−ジヒドロキナゾリン−4−オンの第一部分(0.25g)を、塩化ホスホリル(1.76ml)、ジイソプロピルエチルアミン(3.95ml)およびアセトニトリル(10ml)の80℃に加熱された撹拌混合物に加えた。得られた混合物を、80℃に3時間加熱した。第二部分の7−フルオロ−5−テトラヒドロピラン−4−イルオキシ−3,4−ジヒドロキナゾリン−4−オン(0.25g)を加え、その混合物を、加熱して更に90分間還流した。このようにして、4−クロロ−7−フルオロ−5−テトラヒドロピラン−4−イルオキシキナゾリンの溶液を得、それを、単離することなく用いた。アセトニトリル(3ml)中の6−クロロ−2,3−メチレンジオキシアニリン(0.32g)の溶液を加え、反応混合物を80℃に2.5時間加熱した。必要な変換が不完全であったので、反応混合物を蒸発させ、そしてトルエン(15ml)を反応溶媒として加えた。第二部分の6−クロロ−2,3−メチレンジオキシアニリン(0.32g)を加え、反応混合物を加熱して3時間還流した。反応混合物を周囲温度に冷却し、そして塩化メチレンと塩化ナトリウム水溶液とに分配した。有機相を水で洗浄し、硫酸マグネシウム上で乾燥させ、蒸発させた。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−フルオロ−5−テトラヒドロピラン−4−イルオキシキナゾリンを泡状物(0.73g)として得た。
【0235】
NMRスペクトル:(DMSOd6) 1.9-2.05 (m, 2H), 2.1-2.2 (m, 2H), 3.5-3.6 (m, 2H),
3.8-3.95 (m, 2H), 5.1 (m, 1H), 6.1 (s, 2H), 7.0 (d, 1H), 7.1 (d, 1H), 7.3 (d, 1H), 7.4 (m, 1H), 8.6 (s, 1H), 9.3 (s, 1H)。
【0236】
実施例9
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−フルオロ−5−テトラヒドロピラン−4−イルオキシキナゾリン(経路2)
カリウム tert−ブトキシド(5.42g)およびTHF(30ml)の混合物を、T
HF(30ml)中の4−ヒドロキシテトラヒドロピラン(1.53ml)の溶液に加え、得られた混合物を20分間撹拌した。THF(30ml)中の4−(6−クロロ−2,3−メチレンジオキシアニリノ)−5,7−ジフルオロキナゾリン塩酸塩(6g)のスラリーを加え、得られた混合物を加熱して1.75時間還流した。第二部分のカリウム tert−ブトキシド(1.81g)を加え、その混合物を加熱して更に2時間還流した。第二
部分の4−ヒドロキシテトラヒドロピラン(0.15ml)および第三部分のカリウム tert−ブトキシド(0.45g)を加え、その混合物を加熱して0.5時間還流した。第
四部分のカリウム tert−ブトキシド(0.9g)を加え、その混合物を加熱して更に2
0分間還流した。得られた反応混合物を50℃に冷却させ、そしてブライン(60ml)および水(30ml)を順次加えた。層を分離し、そして水溶液を、順次、THF(30ml)でおよび酢酸イソプロピル(30ml)で抽出した。有機抽出物を一緒にし、ブライン(30ml)で洗浄した。有機溶液を蒸発させた。残留する固体を、メチル tert−
ブチルエーテル(24ml)およびイソヘキサン(12ml)の混合物下で1時間撹拌した。その固体を単離し、1:1のメチル tert−ブチルエーテルおよびイソヘキサンの混
合物で洗浄し、真空中において40℃で一晩乾燥させた。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−フルオロ−5−テトラヒドロピラン−4−イルオキシキナゾリンを得た(5.02g、方法Aを用いて93%のHPLC純度、保持時間4.61分)。そのようにして得られた物質の一部分(3g)を、熱酢酸エチル(54ml)中に溶解させた。その熱溶液を濾過した。濾液を周囲温度に冷却させ、3時間撹拌した。得られた固体を、濾過によって単離し、真空中において周囲温度で乾燥させた。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−フルオロ−5−テトラヒドロピラン−4−イルオキシキナゾリンを得た(1.61g、方法Aを用いて99.2%のHPLC純度、保持時間4.51分)。
【0237】
NMRスペクトル:(DMSOd6) 1.9-2.0 (m, 2H), 2.1-2.2 (m, 2H), 3.5-3.6 (m, 2H), 3.8-3.95 (m, 2H), 5.1 (m, 1H), 6.1 (s, 2H), 6.95 (d, 1H), 7.1 (d, 1H), 7.2 (d, 1H), 7.3 (d, 1H), 8.4 (s, 1H), 9.3 (s, 1H)。
【0238】
実施例10
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−フルオロ−5−テトラヒドロピラン−4−イルオキシキナゾリン(経路3)
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−5,7−ジフルオロキナゾリン塩酸塩(80g)を、ナトリウムtert−ペントキシド(90.2g)およびN−メ
チルピロリジン−2−オン(500ml)の窒素ガス雰囲気下の撹拌混合物に少量ずつ加えた。4−ヒドロキシテトラヒドロピラン(23.5ml)およびN−メチルピロリジン−2−オン(35ml)を加え、得られた混合物を60℃に3時間加熱した。水(764ml)を、加熱された反応混合物に3時間で加え、その混合物を撹拌し、60℃に更に3時間加熱した。その温反応混合物を濾過し、単離された固体を水(2x230ml)で洗浄し、真空中において恒量まで乾燥させた。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−フルオロ−5−テトラヒドロピラン−4−イルオキシキナゾリンを得た(68.6g、方法Aを用いて95%のHPLC純度、保持時間4.6分)。
【0239】
m.p.209〜212℃;
NMRスペクトル:(DMSOd6) 1.9-2.0 (m, 2H), 2.1-2.2 (m, 2H), 3.5-3.6 (m, 2H), 3.8-3.95 (m, 2H), 5.05 (m, 1H), 6.1 (s, 2H), 6.95 (d, 1H), 7.05 (d, 1H), 7.1 (d,
1H), 7.3 (d, 1H), 8.4 (s, 1H), 9.3 (s, 1H)。
【0240】
実施例11
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン(経路1)
トルエン(3ml)中の第一部分の7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシ−3,4−ジヒドロキナゾリン−4−オン(0.19g)を、塩化ホスホリル(0.059ml)、ジイソプロピルエチルアミン(0.13ml)およびトルエン(3ml)の80℃に加熱された撹拌混合物に加え、得られた混合物を80℃に6時間加熱した。その混合物を、周囲温度に冷却させ、一晩撹拌した。その混合物を80℃に再加熱し、そしてトルエン(2ml)中の6−クロロ−2,3−メチレンジオキシアニリン(0.088g)の溶液を加えた。得られた混合物を撹拌し、80℃に1.5時間加熱した。混合物を周囲温度に冷却し、そして析出した油状ガムから溶媒を傾瀉した。その油状ガムを、DMF(3ml)中に懸濁させ、第二部分の7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−
4−イルオキシ−3,4−ジヒドロキナゾリン−4−オン(0.088g)を加え、反応混合物を100℃に9時間加熱した。混合物を周囲温度に冷却させ、そして酢酸エチルと2M塩酸水溶液(10ml)とに分配した。その水溶液を、10M水酸化ナトリウム水溶液(10ml)の添加によって塩基性にし、塩化メチレンで抽出した。有機溶液を硫酸マグネシウム上で乾燥させ、蒸発させた。得られた油状物を、シリカ上のカラムクロマトグラフィーにより、塩化メチレンおよびメタノールの漸増極性混合物を溶離剤として用いて精製した。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン(0.008g)を得た。
【0241】
NMRスペクトル:(DMSOd6) 1.85-1.95 (m, 2H), 2.1-2.2 (m, 2H), 2.2 (s, 3H), 2.2-2.4 (m, 4H), 2.4-2.6 (m, 4H), 2.87 (m, 2H), 3.5-3.6 (m, 2H), 3.8-3.9 (m, 2H), 4.2 (m, 2H), 5.1 (m, 1H), 6.1 (s, 2H), 6.85 (s, 1H), 6.9 (s, 1H), 6.95 (d, 1H), 7.05 (d, 1H), 8.35 (s, 1H), 9.2 (s, 1H)。
【0242】
実施例12
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン(経路2)
窒素ガス雰囲気下において、塩化ホスホリル(0.07ml)を、7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシ−3,4−ジヒドロキナゾリン−4−オン(0.2g)、ジイソプロピルエチルアミン(0.22ml)およびブチロニトリル(2ml)の96℃に加熱された撹拌混合物に加え、得られた混合物を96℃に4時間加熱した。第二部分の塩化ホスホリル(0.12ml)を加え、得られた混合物を96℃に1.7時間加熱した。6−クロロ−2,3−メチレンジオキシアニリン(0.098g)を加え、得られた混合物を96℃に2時間加熱した。混合物を周囲温度に冷却させた。水(2ml)を加え、有機層を分離した。水性層をブチロニトリル(1ml)で洗浄した。水性層を、濃水酸化ナトリウム水溶液(47%w/w)の添加によってpH9へと塩基性にし、n−ブタノール(2x2ml)で抽出した。得られた有機層を一緒にし、蒸発させた。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン(0.094g)を得た。
【0243】
実施例13
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン(経路3)
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−フルオロ−5−テトラヒドロピラン−4−イルオキシキナゾリン(0.5g)を、水酸化カリウム(0.168g)、1−(2−ヒドロキシエチル)−4−メチルピペラジン(0.69g)およびジ−(2−メトキシエチル)エーテル(10ml)の120℃に加温された撹拌混合物に加え、得られた反応混合物を120℃に12時間加熱した。反応混合物を周囲温度に冷却し、1M水性塩酸(9ml)の添加によってpH1〜3へと酸性にし、酢酸イソプロピル(20 ml)で洗浄した。その水溶液を撹拌し、そして2M水性水酸化ナトリウム(5ml)の添加によってpH13〜14へと塩基性にした。10分後、水(22ml)を加え、混合物を2時間撹拌して、固体の沈殿を終了させた。その混合物を10℃に冷却し、濾過した。得られた固体を、水(20ml)で洗浄し、真空中において40℃で乾燥させた。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンを得た(0.47g、方法Bを用いて92.5%のHPLC純度、保持時
間7.3分)。
【0244】
NMRスペクトル:(CDCl3) 1.65 (br s, 3H), 1.9-2.05 (m, 2H), 2.2-2.3 (m, 2H), 2.31 (s, 3H), 2.4-2.8 (m, 8H), 2.9 (m, 2H), 3.6-3.7 (m, 2H), 3.95-4.05 (m, 2H), 4.2-4.25 (m, 2H), 4.8 (m,1H), 6.05 (s, 2H), 6.55 (s, 1H), 6.75 (d, 1H), 6.85 (s,
1H), 7.0 (d, 1H), 8.55 (s, 1H), 9.25 (s, 1H)。
【0245】
実施例14
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン(経路4)
窒素ガス雰囲気下において、1−(2−ヒドロキシエチル)−4−メチルピペラジン(13.93g)を、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−フルオロ−5−テトラヒドロピラン−4−イルオキシキナゾリン(12.9g)、ナトリウム
tert−ペントキシド(9.87g)および1,2−ジエトキシエタン(37.5ml)
の撹拌混合物に加えた。水(1.34g)および1,2−ジエトキシエタン(25ml)を加え、得られた混合物を撹拌し、86℃に18時間加熱した。反応混合物を50℃に冷却し、そして真空蒸留下において約60ミリバール圧力で、約50mlの反応溶媒を留去した。濃塩酸水(36%,10ml)および水(84ml)の混合物を、反応混合物の温度を最大60℃で保持する速度で添加することにより、反応混合物をpH7.0〜7.6に中和した。反応混合物をの温度を60℃で保持しながら、反応混合物を酢酸エチル(225ml)で抽出した。有機溶液を水(50ml)で洗浄した。水(25ml)を加え、そして温度を60℃で保持しながら混合物を10分間撹拌後、30分間放置し、そして水性層を分離した。溶媒を大気圧下において約90℃で蒸留することにより、有機層を濃縮して約100mlの容量とした。残留する混合物を、45℃に1時間で冷却し、その温度で2時間保持して、生成物を結晶化させた。その混合物を55℃に短時間加温した後、18℃に4時間で冷却し、その温度で1時間保持した。結晶性沈殿を濾過によって単離し、そして順次、水(17ml)で、および tert−ブチルメチルエーテル(17ml)で洗
浄した。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンを三水和物として得た(11g;方法Bを用いて88%のHPLC純度、保持時間7.3分)。
【0246】
NMRスペクトル:(CDCl3) 1.65 (br s, 3H), 1.9-2.05 (m, 2H), 2.2-2.3 (m, 2H), 2.31 (s, 3H), 2.4-2.8 (m, 8H), 2.9 (m, 2H), 3.6-3.7 (m, 2H), 3.95-4.05 (m, 2H), 4.2-4.25 (m, 2H), 4.8 (m,1H), 6.05 (s, 2H), 6.55 (s, 1H), 6.75 (d, 1H), 6.85 (s,
1H), 7.0 (d, 1H), 8.55 (s, 1H), 9.25 (s, 1H)。
【0247】
そのようにして得られた物質の一部分(10g)を、フィルター上に置き、乾燥窒素ガス流中において周囲温度で乾燥させた。得られた物質を、乾燥窒素雰囲気を維持しながら、乾燥イソプロパノール(140ml)中に60℃で溶解させた。その溶液を、周囲温度に冷却させ、乾燥窒素雰囲気下で2日間放置した。得られた結晶性固体を、乾燥窒素雰囲気下の濾過によって単離した。そのようにして得られた物質(8g)は、無水物結晶形の4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンであった。
【0248】
m.p.142〜144℃。
実施例15
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピ
ペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンジフマレート塩
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン三水和物(27.1g)、イソプロパノール(200ml)および水(10ml)の混合物を、75℃に加熱した。フマル酸(12.8g)、イソプロパノール(200ml)および水(40ml)の混合物を、80℃に加熱した。
【0249】
その加温されたキナゾリン化合物溶液の一部分(80ml)を、フマル酸溶液に、温度を75℃で維持しながら加えた。得られた混合物を75℃で75分間撹拌した。残りのキナゾリン化合物溶液を、温度を75℃で維持しながら1時間で加えた。イソプロパノール(50ml)を加え、得られた混合物を75℃で7時間撹拌した。
【0250】
その混合物を、50℃に少なくとも25分間にわたって徐々に冷却し、その温度で6時間撹拌した。混合物を、20℃に少なくとも20分間にわたって徐々に冷却し、その温度で18.5時間撹拌した。結晶性固体を、濾過によって単離し、10:1のイソプロパノールおよび水の混合物で2回洗浄し(それぞれ、50mlおよび100ml)、真空中において45℃で恒量まで乾燥させた。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンジフマレート塩(37.0g)を得た。
【0251】
m.p.233〜237℃;
NMRスペクトル:(DMSOd6) 1.76-1.88 (m, 2H), 2.1-2.17 (m, 2H), 2.33 (s, 3H), 2.6 (br s, 8H), 2.78 (t, 2H), 3.51-3.6 (m, 2H), 3.83-3.9 (m, 2H), 4.24 (t, 2H), 4.98-5.07 (m, 1H), 6.07 (s, 2H), 6.6 (s, 4H), 6.83 (d, 1H), 6.84 (d, 1H), 6.91 (d, 1H), 7.05 (d, 1H), 8.33 (s, 1H), 9.18 (s, 1H)。
【0252】
実施例16
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンジフマレート塩
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン三水和物(27.1g)、イソプロパノール(210ml)および水(30ml)の混合物を、40℃に加熱し、混合物を濾過した。フィルターを、イソプロパノール(20ml)で洗浄し、そしてその洗液を、温濾液に加えた。得られた溶液を75℃に加温した。
【0253】
フマル酸(12.8g)、イソプロパノール(200ml)および水(20ml)の混合物を、70℃に加熱し、得られた混合物を濾過した。フマル酸溶液の一部分(110ml)を、加温された4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリン溶液に、温度を75℃で維持しながら加えた。4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンジフマレート塩の種結晶(0.02g)を加え、得られた混合物を75℃で1時間撹拌した。残りのフマル酸溶液を、温度を75℃で維持しながら1時間で加え、得られた混合物を75℃で14時間撹拌した。
【0254】
その混合物を、20℃に少なくとも2時間にわたって徐々に冷却し、その温度で1時間
撹拌した。結晶性固体を濾過によって単離し、10:1のイソプロパノールおよび水の混合物で2回洗浄し(それぞれ、50mlおよび100ml)、真空中において45℃で恒量まで乾燥させた。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンジフマレート塩(35.8g)を得た。
【0255】
m.p.234〜237℃;
NMRスペクトル:(DMSOd6) 1.76-1.88 (m, 2H), 2.1-2.17 (m, 2H), 2.33 (s, 3H), 2.6 (br s, 8H), 2.78 (t, 2H), 3.51-3.6 (m, 2H), 3.83-3.9 (m, 2H), 4.24 (t, 2H), 4.98-5.07 (m, 1H), 6.07 (s, 2H), 6.6 (s, 4H), 6.83 (d, 1H), 6.84 (d, 1H), 6.91 (d, 1H), 7.05 (d, 1H), 8.33 (s, 1H), 9.18 (s, 1H)。
【0256】
実施例17
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンセスキフマレート塩
4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンジフマレート(0.15g)および水(20ml)の混合物を、ヒートガンを用いて加温して、溶液を得た。その試料を、空気流下において周囲温度で徐々に24時間蒸発させて、約3mlの容量としたが、ここで、沈殿が形成し始めた。その混合物を、冷蔵庫中に4℃で2日間置いた。得られた沈殿を、濾過によって単離し、水で洗浄した。このようにして、4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンをセスキフマレート四水和物塩(0.084g)として得、それを、XRPD、DSC、TGA、FTIRおよび溶液NMR技法を用いて特性決定した。
【図面の簡単な説明】
【0257】
【図1】図1は、AZD0530ジフマレートについての、水平軸上にプロットされた2θ値および垂直軸上にプロットされた相対線強さ(計数)でのX線粉末回折図形を示す。
【図2】図2は、AZD0530セスキフマレート四水和物についての、水平軸上にプロットされた2θ値および垂直軸上にプロットされた相対線強さ(計数)でのX線粉末回折図形を示す。
【図3】図3は、AZD0530三水和物についての、水平軸上にプロットされた2θ値および垂直軸上にプロットされた相対線強さ(計数)でのX線粉末回折図形を示す。
【図4】図4は、無水AZD0530についての、水平軸上にプロットされた2θ値および垂直軸上にプロットされた相対線強さ(計数)でのX線粉末回折図形を示す。
【図5】図5は、AZD0530ジフマレートについての、水平軸上にプロットされた4000〜400cm−1の度数範囲および垂直軸上にプロットされた吸光度でのDRIFTスペクトルを示す。
【図6】図6は、AZD0530セスキフマレート四水和物についての、水平軸上にプロットされた4000〜400cm−1の度数範囲および垂直軸上にプロットされた吸光度でのDRIFTスペクトルを示す。
【特許請求の範囲】
【請求項1】
実質的に三水和物の形の式Iを有する4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンの実質的に均一な結晶形(AZD0530三水和物)。
【化1】
【請求項2】
一つまたはそれを超えるピークを2θスケール上の約13.8°および16.0°に有するX線回折図形を特徴とする、請求項1に記載のAZD0530三水和物。
【請求項3】
一つまたはそれを超えるピークを2θスケール上の約7.4°、13.8°、14.8°、16.0°および17.8°に有するX線回折図形を特徴とする、請求項1に記載のAZD0530三水和物。
【請求項4】
下に挙げるような2θスケール上の一つまたはそれを超えるピークを包含するX線回折図形を特徴とする、請求項1に記載のAZD0530三水和物。
【表1】
【請求項5】
約50〜94℃の広範な吸熱を有する示差走査熱量測定サーモグラムを特徴とする、請求項1に記載のAZD0530三水和物。
【請求項6】
約65℃に開始して、ピークを約75℃に有する吸熱を示す示差走査熱量測定サーモグラムを特徴とする、請求項1に記載のAZD0530三水和物。
【請求項7】
医薬組成物であって、請求項1に記載の式Iの化合物の結晶形および薬学的に許容しうる希釈剤または担体を含む医薬組成物。
【請求項8】
式I
【化2】
を有する化合物であるAZD0530の製造方法であって、式II
【化3】
(式中、Lは、置換可能な基であり、そしてNH官能基は、必要ならば保護されている)を有するキナゾリンと、1−(2−ヒドロキシエチル)−4−メチルピペラジンとの反応を含み;その後、存在するいずれの保護基も、慣用的な手段によって除去し;そしてその後、必要ならば、遊離塩基の形で得られた式Iの化合物は、薬学的に許容しうる塩へと変換することができるし、塩の形で得られた式Iの化合物は、遊離塩基へと変換することができる方法。
【請求項9】
反応を、適する塩基の存在下で行う、請求項8に記載の方法。
【請求項10】
置換可能な基Lが、ハロゲノ基である、請求項1に記載の方法。
【請求項11】
式III
【化4】
を有するキナゾリンの製造方法であって、
(a)式IV
【化5】
を有するキナゾリノンと活性化剤との、式V
【化6】
(式中、L1は置換可能な基である)
を有するキナゾリンを形成する反応;
(b)式Vのキナゾリンと、6−クロロ−2,3−メチレンジオキシアニリンとの、式VI
【化7】
を有するキナゾリノンを形成する置換反応であって、その後、遊離塩基の形で得られた式VIの化合物は、塩へと変換することができるし、塩の形で得られた式VIの化合物は、遊離塩基へと変換することができる置換反応;および
(c)式VIのキナゾリンと、4−ヒドロキシテトラヒドロピランとの、式IIIのキナゾ
リンを形成する反応
を含み;その後、遊離塩基の形で得られた式IIIの化合物は、塩へと変換することができ
るし、塩の形で得られた式IIIの化合物は、遊離塩基へと変換することができる方法。
【請求項12】
活性化剤が、ハロゲン化ホスホリルである、請求項11の作業工程(a)に記載の方法。
【請求項13】
反応を、適する塩基の存在下で行う、請求項11の作業工程(c)に記載の方法。
【請求項14】
式Vの中間体を、そのままで単離しないことによって、式VIの化合物を、式IVの化合物からワンポット法で製造することができる、請求項4に記載の方法。
【請求項15】
式IVのキナゾリノンを、6−クロロ−2,3−メチレンジオキシアニリンの存在下で式Vの中間体へと変換し、それで、式Vの中間体がワンポット法で直接的に反応する、請求項11に記載の方法。
【請求項16】
請求項11に記載の式VIの中間体。
【請求項17】
AZD0530の製造方法であって、式VIのキナゾリンを製造する請求項11に記載の工程(a)および工程(b)と、AZD0530へのその変換を含む方法。
【請求項18】
式III
【化8】
を有するキナゾリンの製造方法であって、
(a)式IV
【化9】
を有するキナゾリノンと、4−ヒドロキシテトラヒドロピランとの、式IX
【化10】
を有するキナゾリンを形成する反応であって、その後、遊離塩基の形で得られた式IXの化合物は、塩へと変換することができるし、塩の形で得られた式IXの化合物は、遊離塩基へと変換することができる反応;
(b)式IXのキナゾリンと活性化剤との、式X
【化11】
(式中、L1は置換可能な基である)
を有するキナゾリノンを形成する反応;および
(c)式Xのキナゾリンと、6−クロロ−2,3−メチレンジオキシアニリンとの置換反応
を含み;その後、遊離塩基の形で得られた式IIIの化合物は、塩へと変換することができ
るし、塩の形で得られた式IIIの化合物は、遊離塩基へと変換することができる方法。
【請求項19】
AZD0530の製造方法であって、式IXのキナゾリンを製造する請求項18に記載の工程(a)と、AZD0530へのその変換を含む方法。
【請求項20】
AZD0530の製造方法であって、
(a)式XI
【化12】
を有するキナゾリンと活性化剤との、式XII
【化13】
(式中、L1は置換可能な基である)
を有するキナゾリノンを形成する反応;および
(b)式XIIのキナゾリンと、6−クロロ−2,3−メチレンジオキシアニリンとの置
換反応
を含み;その後、遊離塩基の形で得られた式Iの化合物は、薬学的に許容しうる塩へと変換することができるし、塩の形で得られた式Iの化合物は、遊離塩基へと変換することができる方法。
【請求項21】
式XI
【化14】
を有するキナゾリンの製造方法であって、
(a)式IV
【化15】
を有するキナゾリノンと、4−ヒドロキシテトラヒドロピランとの、式IX
【化16】
を有するキナゾリンを形成する反応であって、その後、遊離塩基の形で得られた式IXの化合物は、塩へと変換することができるし、塩の形で得られた式IXの化合物は、遊離塩基へと変換することができる反応;および
(b)NH官能基が、必要ならば保護されている式IXのキナゾリンと、1−(2−ヒドロキシエチル)−4−メチルピペラジンとの反応を含み;その後、存在するいずれの保護基も、慣用的な手段によって除去し;そしてその後、必要ならば、遊離塩基の形で得られた式XIの化合物は、塩へと変換することができるし、塩の形で得られた式XIの化合物は、遊離塩基へと変換することができる方法。
【請求項22】
請求項21に記載の式XIの中間体。
【請求項1】
実質的に三水和物の形の式Iを有する4−(6−クロロ−2,3−メチレンジオキシアニリノ)−7−[2−(4−メチルピペラジン−1−イル)エトキシ]−5−テトラヒドロピラン−4−イルオキシキナゾリンの実質的に均一な結晶形(AZD0530三水和物)。
【化1】
【請求項2】
一つまたはそれを超えるピークを2θスケール上の約13.8°および16.0°に有するX線回折図形を特徴とする、請求項1に記載のAZD0530三水和物。
【請求項3】
一つまたはそれを超えるピークを2θスケール上の約7.4°、13.8°、14.8°、16.0°および17.8°に有するX線回折図形を特徴とする、請求項1に記載のAZD0530三水和物。
【請求項4】
下に挙げるような2θスケール上の一つまたはそれを超えるピークを包含するX線回折図形を特徴とする、請求項1に記載のAZD0530三水和物。
【表1】
【請求項5】
約50〜94℃の広範な吸熱を有する示差走査熱量測定サーモグラムを特徴とする、請求項1に記載のAZD0530三水和物。
【請求項6】
約65℃に開始して、ピークを約75℃に有する吸熱を示す示差走査熱量測定サーモグラムを特徴とする、請求項1に記載のAZD0530三水和物。
【請求項7】
医薬組成物であって、請求項1に記載の式Iの化合物の結晶形および薬学的に許容しうる希釈剤または担体を含む医薬組成物。
【請求項8】
式I
【化2】
を有する化合物であるAZD0530の製造方法であって、式II
【化3】
(式中、Lは、置換可能な基であり、そしてNH官能基は、必要ならば保護されている)を有するキナゾリンと、1−(2−ヒドロキシエチル)−4−メチルピペラジンとの反応を含み;その後、存在するいずれの保護基も、慣用的な手段によって除去し;そしてその後、必要ならば、遊離塩基の形で得られた式Iの化合物は、薬学的に許容しうる塩へと変換することができるし、塩の形で得られた式Iの化合物は、遊離塩基へと変換することができる方法。
【請求項9】
反応を、適する塩基の存在下で行う、請求項8に記載の方法。
【請求項10】
置換可能な基Lが、ハロゲノ基である、請求項1に記載の方法。
【請求項11】
式III
【化4】
を有するキナゾリンの製造方法であって、
(a)式IV
【化5】
を有するキナゾリノンと活性化剤との、式V
【化6】
(式中、L1は置換可能な基である)
を有するキナゾリンを形成する反応;
(b)式Vのキナゾリンと、6−クロロ−2,3−メチレンジオキシアニリンとの、式VI
【化7】
を有するキナゾリノンを形成する置換反応であって、その後、遊離塩基の形で得られた式VIの化合物は、塩へと変換することができるし、塩の形で得られた式VIの化合物は、遊離塩基へと変換することができる置換反応;および
(c)式VIのキナゾリンと、4−ヒドロキシテトラヒドロピランとの、式IIIのキナゾ
リンを形成する反応
を含み;その後、遊離塩基の形で得られた式IIIの化合物は、塩へと変換することができ
るし、塩の形で得られた式IIIの化合物は、遊離塩基へと変換することができる方法。
【請求項12】
活性化剤が、ハロゲン化ホスホリルである、請求項11の作業工程(a)に記載の方法。
【請求項13】
反応を、適する塩基の存在下で行う、請求項11の作業工程(c)に記載の方法。
【請求項14】
式Vの中間体を、そのままで単離しないことによって、式VIの化合物を、式IVの化合物からワンポット法で製造することができる、請求項4に記載の方法。
【請求項15】
式IVのキナゾリノンを、6−クロロ−2,3−メチレンジオキシアニリンの存在下で式Vの中間体へと変換し、それで、式Vの中間体がワンポット法で直接的に反応する、請求項11に記載の方法。
【請求項16】
請求項11に記載の式VIの中間体。
【請求項17】
AZD0530の製造方法であって、式VIのキナゾリンを製造する請求項11に記載の工程(a)および工程(b)と、AZD0530へのその変換を含む方法。
【請求項18】
式III
【化8】
を有するキナゾリンの製造方法であって、
(a)式IV
【化9】
を有するキナゾリノンと、4−ヒドロキシテトラヒドロピランとの、式IX
【化10】
を有するキナゾリンを形成する反応であって、その後、遊離塩基の形で得られた式IXの化合物は、塩へと変換することができるし、塩の形で得られた式IXの化合物は、遊離塩基へと変換することができる反応;
(b)式IXのキナゾリンと活性化剤との、式X
【化11】
(式中、L1は置換可能な基である)
を有するキナゾリノンを形成する反応;および
(c)式Xのキナゾリンと、6−クロロ−2,3−メチレンジオキシアニリンとの置換反応
を含み;その後、遊離塩基の形で得られた式IIIの化合物は、塩へと変換することができ
るし、塩の形で得られた式IIIの化合物は、遊離塩基へと変換することができる方法。
【請求項19】
AZD0530の製造方法であって、式IXのキナゾリンを製造する請求項18に記載の工程(a)と、AZD0530へのその変換を含む方法。
【請求項20】
AZD0530の製造方法であって、
(a)式XI
【化12】
を有するキナゾリンと活性化剤との、式XII
【化13】
(式中、L1は置換可能な基である)
を有するキナゾリノンを形成する反応;および
(b)式XIIのキナゾリンと、6−クロロ−2,3−メチレンジオキシアニリンとの置
換反応
を含み;その後、遊離塩基の形で得られた式Iの化合物は、薬学的に許容しうる塩へと変換することができるし、塩の形で得られた式Iの化合物は、遊離塩基へと変換することができる方法。
【請求項21】
式XI
【化14】
を有するキナゾリンの製造方法であって、
(a)式IV
【化15】
を有するキナゾリノンと、4−ヒドロキシテトラヒドロピランとの、式IX
【化16】
を有するキナゾリンを形成する反応であって、その後、遊離塩基の形で得られた式IXの化合物は、塩へと変換することができるし、塩の形で得られた式IXの化合物は、遊離塩基へと変換することができる反応;および
(b)NH官能基が、必要ならば保護されている式IXのキナゾリンと、1−(2−ヒドロキシエチル)−4−メチルピペラジンとの反応を含み;その後、存在するいずれの保護基も、慣用的な手段によって除去し;そしてその後、必要ならば、遊離塩基の形で得られた式XIの化合物は、塩へと変換することができるし、塩の形で得られた式XIの化合物は、遊離塩基へと変換することができる方法。
【請求項22】
請求項21に記載の式XIの中間体。
【図1】

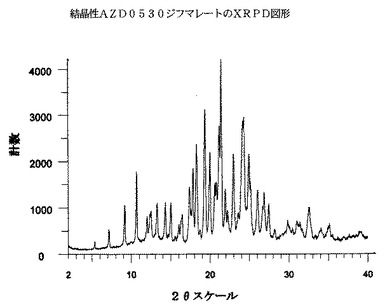
【図2】
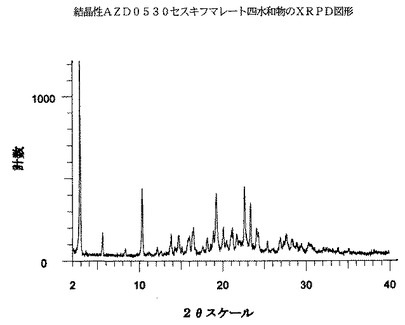
【図3】
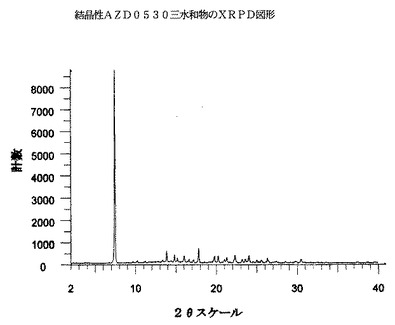
【図4】
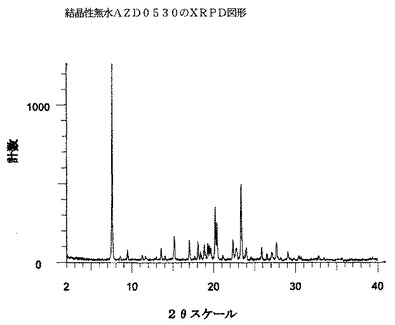
【図5】
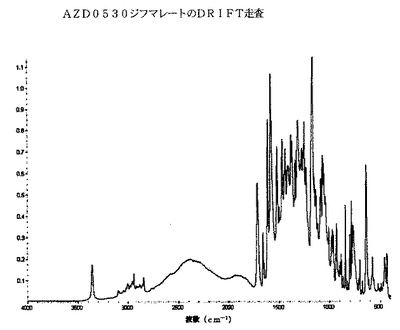
【図6】
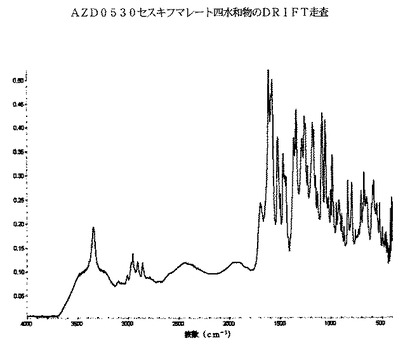
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2009−143920(P2009−143920A)
【公開日】平成21年7月2日(2009.7.2)
【国際特許分類】
【出願番号】特願2008−318186(P2008−318186)
【出願日】平成20年12月15日(2008.12.15)
【分割の表示】特願2007−546172(P2007−546172)の分割
【原出願日】平成17年12月14日(2005.12.14)
【出願人】(300022641)アストラゼネカ アクチボラグ (581)
【Fターム(参考)】
【公開日】平成21年7月2日(2009.7.2)
【国際特許分類】
【出願日】平成20年12月15日(2008.12.15)
【分割の表示】特願2007−546172(P2007−546172)の分割
【原出願日】平成17年12月14日(2005.12.14)
【出願人】(300022641)アストラゼネカ アクチボラグ (581)
【Fターム(参考)】
[ Back to top ]