
北アメリカ薬用人参分留物の形成方法、それらを含む生成物および免疫賦活剤としての使用
【課題】北アメリカ薬用人参(Panax quinquefolium)から分留物を調製する化学的形成方法およびこれらの分留物を含有する薬学的組成物を提供する。
【解決手段】アメリカ薬用人参につき、アルコール、水等の溶媒により一定温度における数時間の抽出と、分離を繰り返すことによって得られた沈殿物を単離して薬用人参分留物PQ2、PQ223を生成する。
【解決手段】アメリカ薬用人参につき、アルコール、水等の溶媒により一定温度における数時間の抽出と、分離を繰り返すことによって得られた沈殿物を単離して薬用人参分留物PQ2、PQ223を生成する。
【発明の詳細な説明】
【技術分野】
【0001】
(発明の分野)
この発明は、北アメリカ薬用人参(Panax quinquefolium)からの分留物の化学的形成方法およびこれらの分留物を含有する組成物に関する。本発明の生成物は、抗体の生成を賦活するのに、あるいは、通常の風邪、インフルエンザ、慢性疲労症候群、AIDS、癌などの低免疫により特徴付けられる健康状態を対象とする治療法として、用いられ得る。また、本発明の生成物は、免疫系の深刻な抑制を引き起こす化学療法や放射線療法を受ける癌患者に対するサプリメントとして用いられ得る。
【背景技術】
【0002】
(発明の背景)
数百年の間、薬草化合物などのある種の無毒性薬品の使用は、さまざまな生理学的状態に、特に東洋で受け入れられてきた。Panax ginseng C.A.Meyerは最もよく知られた伝統的な中国薬である。薬用人参抽出物の重要な薬理学的活性は、単独で、あるいは他の薬品との組み合わせで、腎臓損傷の緩和、発癌の抑制およびストレスの予防を含む。また、固体の免疫学的反応性に対する薬用人参の影響について数多くの報告がなされている。報告されたある免疫賦活特性は、細胞レベルでのある基本的な免疫機能の賦活と同様に、感染に対する宿主抵抗の増強、抗炎症効果、腫瘍成長の抑制を含む。アメリカ薬用人参(Panax quinquefolium)は、多くの有益な健康上の効果を有する健康サプリメントとしての評判を得た薬用人参のもう一つの種である。いくつかの科学者グループが、薬用人参中に存在するポリサッカライドの単離および構造の特定を試みた。あるポリサッカライドは免疫系の賦活に活性があることが示された。
【0003】
薬用人参ポリサッカライドの単離、特徴付けおよび生物学的評価についての一連の研究が、共立薬科大学(日本)のTomodaのグループによりなされた。ある一連の研究では、薬用人参ポリサッカライドが酸性度に基づいて分留された。朝鮮薬用人参(Panax ginseng)の根から免疫学的に活性な2つの酸性のポリサッカライドが単離された[1,2]。スライスされた根がお湯で抽出された。抽出物は、硫酸ナトリウムの存在下においてセチルトリメチルアンモニウムブロマイド(CTAB)で処理された。沈殿物は、分離され、透析され、Sephadex G−25カラム、DEAE−Sephacel(Pharmacia)カラムに適用され、2つの純粋なポリサッカライドが得られ、それらはginsenan PAおよびginsenan PBと名付けられた。Toyopearl HW−55F上のゲルクロマトグラフィーは、ginsenan PAおよびginsenan PBの分子量として、それぞれ1.6×105、および、5.5×104の値を与えた。定量分析は、ginsenan PAが21.3%のアラビノース、53.4%のガラクトース、2.0%のラムノース、16.0%のガラクツロン酸および2.7%のグルクロン酸を含むことを示した。これらの成分糖のモル比は11:22:1:6:1であった。ginsenan PBは、11.0%のアラビノース、32.3%のガラクトース、8.1%のラムノース、39.9%のガラクツロン酸および5.0%のグルクロン酸を含んでいた。これらの成分糖のモル比は3:7:2:8:1であった。両ポリサッカライドとも、投与量に依存して、炭素除去試験における著しい細胞内皮系の有力な活性、際立った抗補体活性およびアルカリホスファターゼ誘導活性を示した。
【0004】
別の研究[3]において、CTABで処理された上記の抽出物の上澄みから2つの追加のポリサッカライドが単離された。即ち、上澄みがエタノール中に移された。沈殿物は分離され、DEAE−Sephadex A−25およびSephadex G−25の各カラムに適用され、S−IAおよびS−IIAと名付けられた2つの純粋なポリサッカライドを与えた。Toyopearl HW−55F上のゲルクロマトグラフィーは、S−IAおよびS−IIAの分子量として、それぞれ5.6×104、および、1.0×105の値を与えた。ginsenan S−IAは、42.3%のアラビノース、50.8%のガラクトース、6.9%のガラクツロン酸を8:8:1のモル比で含む。ginsenan S−IIAは、42.0%のL−アラビノース、32.6%のガラクトース、6.2%のグルコース、19,2%のガラクツロン酸を含む。モル比は15:10:2:5である。
【0005】
Panax ginsengの葉および根からのいくつかのポリサッカライド分留物は、日本の北里大学のオリエンタル・メディシン・リサーチセンターにおけるYamadaのグループにより分離された。化学特性および生物学的活性が調査および比較された[4]。
【0006】
中国産の薬用人参の根と葉は、それらのジンセノサイド(ginsenosides)を取り除くためにエタノールで処理した後、水で抽出され、残渣が0.5MのNaOHで抽出されて、水溶性およびアルカリ可溶性のポリサッカライド分留物(水溶性の分留物はGR−2およびGL−2、アルカリ可溶性の分留物はGRA−2およびGLA−2とそれぞれ名付けられる)を与えた。成分ポリサッカライドの酸性度に基づいて、セチルトリメチルアンモニウムとの処理により、全ての分留物が、強酸性(GR−3、GL−3、GRA−3、およびGLA−3と名付けられた)、弱酸性(GR−4、GL−4、GRA−4、およびGLA−4と名付けられた)、中性(GR−5、GL−5、GRA−5、およびGLA−5と名付けられた)のポリサッカライド分留物にさらに分留された。根は葉よりもポリサッカライドをより多量に含んでいた。根からの強酸性のポリサッカライド分留物は50%以上にもおよぶ高率のウロン酸含有量を有していた。似た成分糖は全ての分留物から検出された。それらは、ラムノース、アラビノース、ガラクトース、グルコース、ガラクツロン酸およびグルクロン酸であった。ガラクツロン酸は、主なウロン酸成分であった。
【0007】
最も高い抗補体活性を有する分留物であるGL−3は、DEAE−Sephadex、Sepharose CL−6B、DEAE Toyopearlのカラム処理、およびエタノール沈殿によりさらに分留されて、GL−PIからGL−PIVよ名付けられた分留物を与えた[5]。全ての分留物は32〜44%のウロン酸を含んでいた。分留物PIは50000という最も高い分子量を有していた。PIおよびPIIは主としてRha、Gal、およびGalAからなり、PIIIはさらにFucを含んでおり、一方、PIVはGal、Glc、およびGalAからなっていた。詳細な構造決定が行われた。
【0008】
抗潰瘍ペクチンポリサッカライド(GL−BIII)が、DEAE Sepharose CL−6BおよびSepharose CL−6Bのクロマトグラフィーによって、弱酸性のポリサッカライド分留物GL−4から単離された。主としてRha、Ara、Man、Gal、Glc、GalA、およびGlcAから、3:4:2:10:1:7:4のモル比で構成されていた。詳細な構造決定が行われた[6]。
【0009】
他のマクロファージFcレセプタ発現増強ポリサッカライド(GL−4IIb2)が、DEAE−Sepharose CL−6Bのアニオン交換クロマトグラフィーによって、GL−4から分離された。化学分析は、試料が65%の炭水化物と33.7%のウロン酸を含むことを示した。成分分析と構造決定が行われた[7]。
【0010】
抗補体活性を有する他のPanax ginseng抽出物、G−115が同じグループによって研究された[8]。G−115は、抗補体活性およびマイトジェン活性に対する活性物質を特徴付けるために分留された。最も効力のある抗補体活性は、未分留のポリサッカライド分留物G−115Gにおいて観測され、一方、水溶性で透析できる分留物であるG−115Eは最も効力のあるマイトジェン活性を示した。G−115Gは、さらにセチルトリメチルアンモニウムブロマイドとの沈殿形成、DEAE−Sepharoseでのアニオン交換クロマトグラフィー、およびSepharose CL−4Bでのゲル濾過によって精製され、目立って効力のある抗補体のポリサッカライドであるG−115I1−IIa−2−3が得られた。このポリサッカライドは均質であった。その分子量は3.68×105であると見積もられた。それは、主としてアラビノース、ガラクトースおよびグルコースと、これに加えて少量のガラクツロン酸、グルクロン酸、およびラムノースからなる。
【0011】
薬用人参ポリサッカライドが、日本の東北大学薬学部のHikinoのグループにより、韓国、中国、および日本の薬用人参から単離された。薬用人参ポリサッカライドの低血糖活性がテストされた。組成と構造のある特色が明らかにされた[9−14]。
【0012】
3つのポリサッカライド、quinquefolan A〜Cがアメリカの薬用人参から単離された[14]。それらの分子量は、Sephacryl S−500上のゲルクロマトグラフィーから2.0×106より大きいと見積もられた。中性糖成分は、quinquefolan Aに対してマンノースおよびグルコース(モル比は1.0:2.3)、quinquefolan Bに対してマンノースおよびグルコース(モル比は1.0:5.5)、quinquefolan Cに対してキシロースであった。quinquefolan A〜C中の酸性糖成分は、それぞれ10.8、11.7、および7.1%であることがわかった。これらのポリサッカライド中のペプチド部分の含有率は、quinquefolan A〜Cに対してそれぞれ2.7、2.9、および2.3%であった。これらのポリサッカライドの全てが、通常のマウスおよびアロキサンで減じられた低血糖のマウスにおいて低血糖効果を示した。
【0013】
分子量が150,000の酸性のポリサッカライドは、ginsanと呼ばれるが、韓国、ソウルの韓国癌センター病院の免疫学研究所(Laboratory of Immunology,Korean Cancer Center Hospital)の研究グループにより、Panaxginsengから単離された[15]。このポリサッカライドは3.7%のタンパク質、47.1%のヘキソース(グルコースおよびガラクトース)、43.1%のウロン酸(ガラクツロン酸)からなっていた。薬用人参は、T細胞とB細胞の増殖を誘発し、ナチュラルキラー細胞およびT細胞の両方から、内生的に生成されたマルチプルサイトカインを通してリンフォカイン活性化障害性細胞を発生させた[16]。
【0014】
中国のノースウエスト・ノーマル・ユニバーシティ(Northeast Normal University)のMiaoらは、アメリカの薬用人参からポリサッカライドを単離した。精製および構造解析が行われた[17]。
【0015】
アメリカの薬用人参からのポリサッカライドの生物学的活性は、ノーマン・ブスーン・ユニバーシティ・オブ・メディカル・サイエンス(Norman Bethune University of Medical Science)の研究グループにより研究された[18,19]。彼らは、アメリカの薬用人参からのポリサッカライド(PPQ)がリンパ球転換を増強することを発見した。マウスの脾臓リンパ球からのサイトカインのインビトロ(in vitro)での生成に対するPanax quinquefolium(PPQ−1)からのポリサッカライドの影響が研究された。データは、PPQ−1が免疫機能を調整することを示唆する。
【先行技術文献】
【非特許文献】
【0016】
【非特許文献1】Tomodaら、Biol.Pharm.Bull.,16,22−5(1993). 非特許文献1は、朝鮮薬用人参(Panax ginseng)の根から免疫学的に活性な2つの酸性のポリサッカライドが単離されることについて開示している。
【発明の概要】
【発明が解決しようとする課題】
【0017】
(発明のまとめ)
本発明者らは、あるアメリカの薬用人参の抽出物が免疫調整特性を有することを発見した。CVT−E002、およびそれから精製された分留物PQ2およびPQ223が特にマウスの脾臓細胞がB細胞を増殖するように刺激を与え、それは次に多量の抗体を生成させる。また、分留物は血清免疫グロブリン(例えばトータルIgG)レベルを増加させ、マクロファージにIL−1、IL−6、およびTNF−αを生成するように刺激を与える。これらの分留物は、一般的伝染病および他の免疫不全関連症の防止あるいは治療に使用される可能性がある。
【課題を解決するための手段】
【0018】
従って、本発明は、アメリカの薬用人参の試料から薬用人参分留物PQ2、PQ223、CVT−E002を調製する方法に注目する。
具体的に述べると、薬用人参分留物PQ2を調製する方法は、以下を有する:
−アメリカの薬用人参をアルコールを含む第1の溶媒に混合し、得られた溶液を約80〜100℃の温度で約2〜4時間の間加熱し、第1の薬用人参溶液を生成すること;
−その後で、上記第1の薬用人参溶液を分離して、アルコール/薬用人参溶液と第1の薬用人参残渣を生成すること;
−その後で、上記第1の薬用人参残渣を水に混合し、得られた溶液を約80〜100℃の温度で約2〜4時間の間加熱し、薬用人参残渣溶液を生成すること;
−その後で、上記薬用人参残渣溶液を分離して、第2の薬用人参残渣と、第1の薬用人参抽出物を含む第1の抽出物水溶液を生成すること;
−少なくとも上記第1の薬用人参抽出物の一部を含む第2の抽出物水溶液を提供すること、ここで、上記第2の抽出物水溶液中の上記第1の薬用人参抽出物の水に対する比率は約1:18〜1:22である;
−その後で、アルコールを含む第2の溶媒に上記第2の抽出物水溶液を混合して、第1の沈殿と第1の上澄みを生成すること、ここで、上記第2の溶媒の水に対する比率は約1:1〜3:5である;
−その後で、アルコールを含む第3の溶媒に以前の工程において生成された上記第1の上澄みを混合して、第2の沈殿と第2の上澄みを生成すること、ここで、上記第3の溶媒の第1の上澄みに対する比率は約3:2〜3:1である;および
−上記第2の沈殿物を単離して薬用人参分留物PQ2を生成すること。
【0019】
薬用人参分留物PQ223を調製する方法は、以下を有する:
−上述のように、薬用人参分留物PQ2を提供すること;
−上記薬用人参分留物PQ2を分留して第1溶出分留物および第2溶出分留物を生成すること、ここで、第1溶出分留物は溶出体積が35および50mlの間に観測される炭水化物ピークに合致し、第2溶出分留物は溶出体積が50および85mlの間に観測される炭水化物ピークに合致し、これらは下記の材料を用いたゲル濾過クロマトグラフィーにより決定される:
(1)アリルデキストランおよびN,N’−メチレンビスアクリルアミドからなる架橋した球形の共重合体のマトリックスを含み、16×600mmのベッド寸法および120mlのベッド体積と、球状のタンパク質に対して5000〜250,000、デキストランに対して1000〜80,000の分留範囲(MW)を有するクロマトグラフィーカラム、および
(2)0.1N HClおよびpHが7.0の0.3M NaClを含むトリス−HClの溶出バッファ;および
−上記第1溶出分留物および上記第2溶出分留物を単離し、かつ混合して、薬用人参分留物PQ223を生成すること。
【0020】
薬用人参分留物CVT−E002を調製する方法は、以下を有する:
−薬用人参1グラムあたり約7〜9mlの第1の溶媒の比率で、アルコールを含む第1の溶媒にアメリカの薬用人参を混合し、得られた溶液を約80〜100℃の温度で約2〜4時間の間加熱し、第1の薬用人参溶液を生成すること;
−その後で、上記第1の薬用人参溶液を分離して、アルコール/薬用人参溶液と第1の薬用人参残渣を生成すること;
−その後で、薬用人参残渣1グラムあたり約7〜9mlの水の比率で、上記第1の薬用人参残渣を水に混合し、薬用人参残渣溶液を約80〜100℃の温度で約2〜4時間の間加熱し、薬用人参残渣溶液を生成すること;
−その後で、上記薬用人参残渣溶液を分離して、第2の薬用人参残渣と、薬用人参抽出物を含む抽出物水溶液を生成すること;および
−上記抽出物水溶液を乾燥させて薬用人参分留物CVT−E002を生成すること。
【0021】
本発明は、また、上述の方法で調製された薬用人参分留物PQ2、PQ223、およびCVT−E002を含む。
【0022】
本発明は、特定の炭水化物内容物を有する薬用人参分留物をさらに含む。
第1の薬用人参分留物は、約2〜6モル%のラムノース、約41〜49モル%のガラクツロン酸、約12〜18モル%のグルコース、約16〜22モル%のガラクトース、および約12〜19モル%のアラビノースを含む炭水化物内容物を有する。
第2の薬用人参分留物は、約3〜8モル%のラムノース、約36〜44モル%のガラクツロン酸、約2〜7モル%のグルコース、約25〜33モル%のガラクトース、および約17〜25モル%のアラビノースを含む炭水化物内容物を有する。
第3の薬用人参分留物は、約0.5〜5モル%のラムノース、約11〜22モル%のガラクツロン酸、約40〜60モル%のグルコース、約10〜19モル%のガラクトース、および約11〜19モル%のアラビノースを含む炭水化物内容物を有する。
【0023】
本発明は、また、本発明の薬用人参分留物を有し、薬学的に許容なキャリアに混合した薬学的組成物を含む。
本発明は、本発明による薬用人参分留物の、低免疫で特徴付けられる健康状態を治療するのに適した薬学的組成物の調製において、単独でのあるいは他の薬剤との組み合わせての使用をさらに含む。
【0024】
本発明は、また、IL−1、IL−6、および/またはTNF−αの細胞中の生成を刺激するための本発明に係る薬用人参分留物の使用を含む。
本発明は、インビトロ(in vitro)またはインビボ(in vivo)での免疫グロブリンの生成を刺激するための本発明に係る薬用人参分留物の使用をさらに含む。
また、Bリンパ球の増殖とそれらからの抗体の生成を活性化するための本発明に係る薬用人参分留物の使用が含まれる。
【0025】
本発明は、また、健康状態を治療するのに効果的な量の本発明に係る薬用人参分留物を患者に投与することを含む、それを必要としている患者の低免疫で特徴付けられる健康状態を治療する方法を含む。
【0026】
(本発明の詳細な記述)
薬用人参分留物PQ2を調製する方法は、下記を含む:
(a)アメリカの薬用人参をアルコールを含む第1の溶媒に混合し、得られた溶液を約80〜100℃の温度で約2〜4時間の間加熱し、第1の薬用人参溶液を生成すること;
(b)その後で、上記第1の薬用人参溶液を分離して、アルコール/薬用人参溶液と第1の薬用人参残渣を生成すること;
(c)その後で、上記第1の薬用人参残渣を水に混合し、得られた溶液を約80〜100℃の温度で約2〜4時間の間加熱し、薬用人参残渣溶液を生成すること;
(d)その後で、上記薬用人参残渣溶液を分離して、第2の薬用人参残渣と、第1の薬用人参抽出物を含む第1の抽出物水溶液を生成すること;
(e)少なくとも上記第1の薬用人参抽出物の一部を含む第2の抽出物水溶液を提供すること、ここで、上記第2の抽出物水溶液中の上記第1の薬用人参抽出物の水に対する比率は約1:18〜1:22である;
(f)その後で、アルコールを含む第2の溶媒に上記第2の抽出物水溶液を混合して、第1の沈殿と第1の上澄みを生成すること、ここで、上記第2の溶媒の水に対する比率は約1:1〜3:5である;
(g)その後で、アルコールを含む第3の溶媒にステップ(f)において生成された上記第1の上澄みを混合して、第2の沈殿と第2の上澄みを生成すること、ここで、上記第3の溶媒の第1の上澄みに対する比率は約3:2〜3:1である;および
(h)上記第2の沈殿物を単離して薬用人参分留物PQ2 を生成すること。
【0027】
第1の溶媒、第2の溶媒、および第3の溶媒のそれぞれにおけるアルコールは、薬用人参に不活性で、所望の生成物から容易に分離可能であるアルコールを含む。当業者は、これらの必要条件を満たすアルコールをたやすく選ぶことができるだろう。好ましくは、それぞれのアルコールが、他と関係なく、飽和あるいは不飽和のC1〜C6アルコールを含む。より好ましくは、それぞれのアルコールが、他と関係なく、エタノールあるいはメタノールを含む。
【0028】
ステップ(a)と(c)のそれぞれにおいて、得られる溶液を約3時間の間加熱することが好ましい。ステップ(a)において、第1の溶媒および薬用人参が、薬用人参1グラムあたり約7〜9mlの第1の溶媒の比率で混合されることが好ましく、最も好ましくは薬用人参1グラムあたり約8mlの第1の溶媒の比率で混合される。ステップ(c)において、水および第1の薬用人参残渣が、薬用人参残渣1グラムあたり約7〜9mlの水の比率で混合されることが好ましく、最も好ましくは薬用人参残渣1グラムあたり約8mlの水の比率で混合される。
【0029】
上述の方法においてステップ(d)に続いてすぐ、第1の薬用人参抽出物が随意に濃縮されてもよい。これは、当業者によく知られた手順によるどんな方法によって行われてもよい。例えば、第1の薬用人参抽出物を含む第1の水溶液は遠心分離機で(例えば2500〜10000rpmの速さで約5〜15分)分離されてもよい。また、第1の水溶液は濾過されてもよい。第1の薬用人参抽出物の濃縮に替えて、あるいはこれに追加して、第1の薬用人参抽出物は後で使用するようにフリーズドライされてもよい。
【0030】
ステップ(e)において、第2の抽出物水溶液は、少なくとも第1の薬用人参抽出物の一部を含んでいる。第2の抽出物水溶液中の第1の薬用人参抽出物の水に対する比率は約1:18〜1:22であり、より好ましくは1:20であることが重要である。これは、多くの方法のいずれでも達成できる。上述のように、第1の薬用人参抽出物が、ステップ(d)に続いて濃縮される、および/または、フリーズドライされるならば、所望の比率を達成するように第1の薬用人参抽出物に水が加えられるべきである。あるいは、第1の抽出物水溶液の一部あるいは全部が第2の抽出物水溶液に用いられてもよい。必要なら、所望の比率を達成するために、追加の水を加えてもよい。所望の量の第1の薬用人参抽出物を含有する第1の抽出物水溶液の少なくとも一部を使用することは、ステップ(d)と(e)の間に追加の濃縮またはフリーズドライステップを実施する必要を逃れるだろう。
【0031】
ステップ(f)において、第2の溶媒の水に対する比率は約3:4であることが好ましい。
ステップ(g)において、第3の溶媒の第1の上澄みに対する比率は約2:1であることが好ましい。
【0032】
薬用人参分留物PQ223を調製する方法は、下記を含む:
(a)上述の方法によって生成された、薬用人参分留物PQ2を提供すること;
(b)上記薬用人参分留物PQ2を分留して第1溶出分留物および第2溶出分留物を生成すること、ここで、第1溶出分留物は溶出体積が35および50mlの間に観測される炭水化物ピークに合致し、第2溶出分留物は溶出体積が50および85mlの間に観測される炭水化物ピークに合致し、これらは下記の材料を用いたゲル濾過クロマトグラフィーにより決定される:
(1)アリルデキストランおよびN,N’−メチレンビスアクリルアミドからなる架橋した球形の共重合体のマトリックスを含み、16×600mmのベッド寸法および120mlのベッド体積と、球状のタンパク質に対して5000〜250,000、デキストランに対して1000〜80,000の分留範囲(MW)を有するクロマトグラフィーカラム、および
(2)0.1N HClおよびpHが7.0の0.3M NaClを含むトリス−HClの溶出バッファ;および
(c)上記第1溶出分留物および上記第2溶出分留物を単離し、かつ混合して、薬用人参分留物PQ223を生成すること。
【0033】
第1の溶出分留物はPQ2Aとしても知られており、第2の溶出分留物はPQ2Bとしても知られている。これらの溶出分留物は別々に単離され得る。また、追加の溶出分留物PQ2C(溶出体積が95および110mlの間に観測される炭水化物ピークに合致する)、およびPQ2D(溶出体積が120および250mlの間に観測される炭水化物ピークに合致する)を生成するために、上述と同じ方法が用いられてもよい。これらの分留物は別々に単離され得る。また、PQ223の他に、PQ2AからPQ2Dの2以上の分留物からなる組成物も形成されることができる。
【0034】
薬用人参分留物PQ2は、ゲル濾過クロマトグラフィーを用いて分留されることが好ましい。しかしながら、当業者に知られている他のいずれの種類の分留も適している。
ゲル濾過クロマトグラフィーを行う手順は、フローレート、試料体積、および手順が実施される温度に関して、製作者の推薦に従って、当業者によく知られている。これらの要因の製作者の指定範囲内の変動は、クロマトグラフィー試験の結果に対する明らかな影響はない。
【0035】
炭水化物内容物の決定は、当業者に知られたいずれの手順によって行われてもよい。分留物の炭水化物組成を決定するのに、マイクロフィルタープレートアッセイ(microfilter plate assay)が実施されることが好ましい。そのようなアッセイは当業者によく知られている。例えば、Duboisら、Anal.chem.28:350−56(1956)を参照されたい。この結果、参照により組み込まれる。マイクロフィルタープレートアッセイに用いるのにもたらされた試料の吸収は、好ましくは492nmで実施される。
【0036】
薬用人参分留物CVT−E002を調製する方法は、下記を含む:
(a)薬用人参1グラムあたり約7〜9mlの第1の溶媒の比率で、アルコールを含む第1の溶媒にアメリカの薬用人参を混合し、得られた溶液を約80〜100℃の温度で約2〜4時間の間加熱し、第1の薬用人参溶液を生成すること;
(b)その後で、上記第1の薬用人参溶液を分離して、アルコール/薬用人参溶液と第1の薬用人参残渣を生成すること;
(c)その後で、薬用人参残渣1グラムあたり約7〜9mlの水の比率で、上記第1の薬用人参残渣を水に混合し、薬用人参残渣溶液を約80〜100℃の温度で約2〜4時間の間加熱し、薬用人参残渣溶液を生成すること;
(d)その後で、上記薬用人参残渣溶液を分離して、第2の薬用人参残渣と、薬用人参抽出物を含む抽出物水溶液を生成すること;および
(e)上記抽出物水溶液を乾燥あるいは濃縮させて薬用人参分留物CVT−E002を生成すること。
【0037】
第1の溶媒におけるアルコールは、薬用人参に不活性で、所望の生成物から容易に分離可能であるアルコールを含む。当業者は、これらの必要条件を満たすアルコールをたやすく選ぶことができるだろう。好ましくは、このアルコールが飽和あるいは不飽和のC1〜C6アルコールを含む。より好ましくは、このアルコールがエタノールあるいはメタノールを含む。
【0038】
ステップ(a)において、第1の溶媒および試料が、試料1グラムあたり約8mlの第1の溶媒の比率で混合されることが好ましい。ステップ(c)において、水および第1の薬用人参残渣が、薬用人参残渣1グラムあたり約8mlの水の比率で混合されることが好ましい。
ステップ(a)と(c)において、第1の薬用人参溶液を約3時間の間加熱することが好ましい。
【0039】
本発明は、また、いくつかの薬用人参分留物を含む。
第1の薬用人参分留物は、約2〜6モル%のラムノース、約41〜49モル%のガラクツロン酸、約12〜18モル%のグルコース、約16〜22モル%のガラクトース、および約12〜19モル%のアラビノースを含む炭水化物内容物を有する。好ましくは、炭水化物内容物が、約3〜5モル%のラムノース、約43〜47モル%のガラクツロン酸、約14〜16モル%のグルコース、約18〜20モル%のガラクトース、および約14〜17モル%のアラビノースを含む。最も好ましくは、炭水化物内容物が、約4モル%のラムノース、約45モル%のガラクツロン酸、約15モル%のグルコース、約19モル%のガラクトース、および約15モル%のアラビノースを含む。
【0040】
本発明に係る第2の薬用人参分留物は、約3〜8モル%のラムノース、約36〜44モル%のガラクツロン酸、約2〜7モル%のグルコース、約25〜33モル%のガラクトース、および約17〜25モル%のアラビノースを含む炭水化物内容物を有する。好ましくは、炭水化物内容物が、約4〜7モル%のラムノース、約37〜42モル%のガラクツロン酸、約3〜6モル%のグルコース、約27〜32モル%のガラクトース、および約19〜24モル%のアラビノースを含む。最も好ましくは、炭水化物内容物が、約5モル%のラムノース、約39モル%のガラクツロン酸、約4モル%のグルコース、約29モル%のガラクトース、および約21モル%のアラビノースを含む。
【0041】
本発明に係る第3の薬用人参分留物は、約0.5〜5モル%のラムノース、約11〜22モル%のガラクツロン酸、約40〜60モル%のグルコース、約10〜19モル%のガラクトース、および約11〜19モル%のアラビノースを含む炭水化物内容物を有する。好ましくは、炭水化物内容物が、約1〜3モル%のラムノース、約13〜20モル%のガラクツロン酸、約42〜57モル%のグルコース、約12〜17モル%のガラクトース、および約13〜17モル%のアラビノースを含む。
【0042】
本発明は、また、本発明に係る薬用人参分留物のいずれかを有し、薬学的に許容なキャリアに混合した薬学的組成物を含む。当業者は、このことについて有効であるだろういずれの薬学的に許容なキャリアにも熟知しており、従って、本発明に係る薬学的組成物を形成する手順は詳細に議論しない。薬学的組成物は、適当に、タブレット、カプセル、液体、菱形(lozenge)、ローション、あるいは座薬の形態をとる。
【0043】
本発明は、通常の風邪、インフルエンザ、慢性疲労症候群、AIDS、癌などの低免疫で特徴付けられる健康状態を治療するのに適した薬学的組成物の調製における本発明に係る薬用人参分留物の使用を含む。薬用人参分留物は、単独で、あるいは他の薬剤を組み合わせて用いられ得る。癌患者は免疫系に深刻なサプレッションを有することが知られているので、本発明の薬用人参分留物は、化学療法の試薬とともに投与するのに、あるいは、放射線療法でのサプリメントとして、特に適している。
【図面の簡単な説明】
【0044】
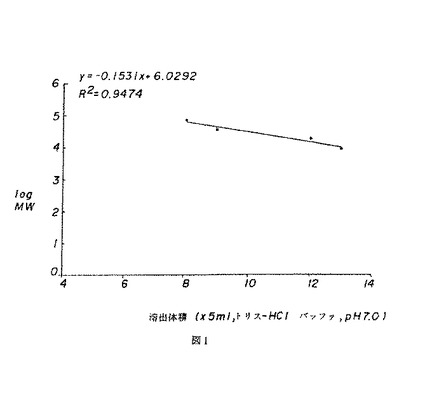
【図1】図1は、クロマトグラフィーカラムで溶出された標準デキストラン試料の分子量のlog値に対して溶出体積をプロットした図を示す。
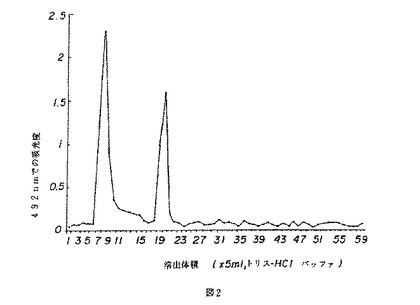
【図2】図2は、薬用人参分留物CVT−E002のクロマトグラムを示す。
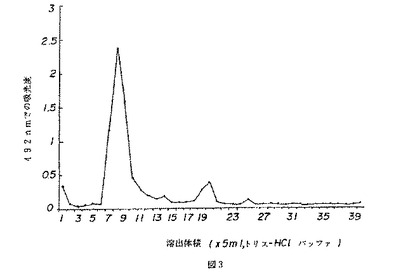
【図3】図3は、薬用人参分留物PQ1のクロマトグラムを示す。
【図4】図4は、薬用人参分留物PQ2のクロマトグラムを示す。
【図5】図5は、薬用人参分留物PQ3のクロマトグラムを示す。
【図6】図6は、薬用人参分留物PQ223のクロマトグラムを示す。
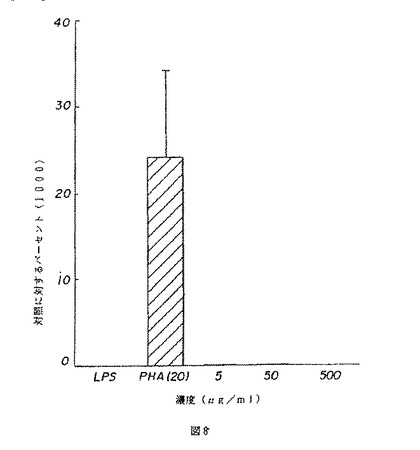
【図7】図7は、マウスの脾臓細胞のB細胞の増殖に対する薬用人参分留物PQ223の効果を示す。
【図8】図8は、マウスの脾臓細胞のT細胞の増殖に対する薬用人参分留物PQ223の効果を示す。
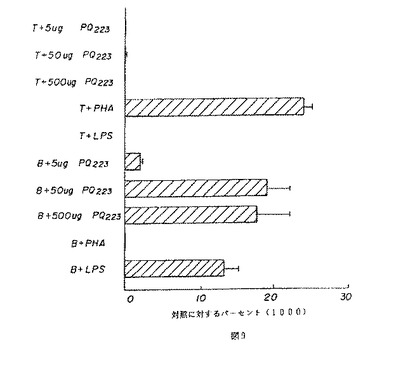
【図9】図9は、マウスの脾臓細胞のTおよびB細胞の増殖に対する薬用人参分留物PQ223の効果の特異性を示す。
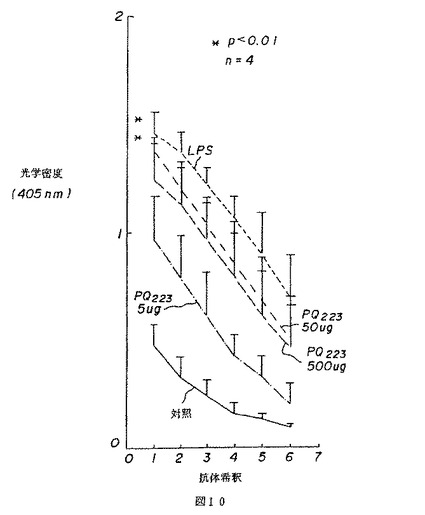
【図10】図10は、マウスの脾臓によるインビトロ(in vitro)抗体生成に対する薬用人参分留物PQ223の効果を示す。
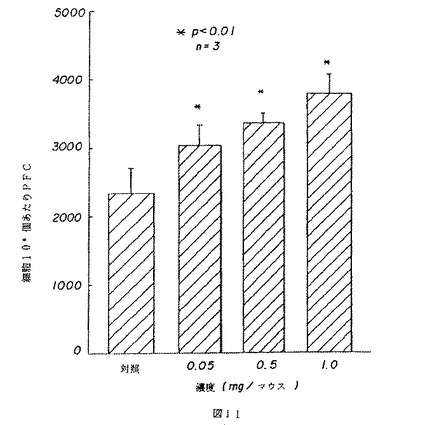
【図11】図11は、薬用人参分留物PQ223が注入されたマウスのプラーク形成細胞(PFC)の増強を示す。
【0045】
本発明は、また、健康状態を治療するのに効果的な量の本発明に係る薬用人参分留物を患者に投与することを含む、それを必要としている患者の低免疫で特徴付けられる健康状態の治療方法を含む。好ましくは、健康状態が、通常の風邪、インフルエンザ、慢性疲労症候群、AIDS、癌からなるグループから選択される。本発明に係る薬用人参分留物の投与量は、年齢、性別、および患者の一般的な健康状態はもちろん、治療される個々の健康状態にも依存する。しかしながら、適切な投与量は、1日に1〜10回の投与範囲において、1日あたり、1〜5000mg/kg体重の範囲に見つかるだろう。薬用人参分留物は、経口で、注射もしくは点滴で、局部的に、鼻から吸って、点眼で、膣に、あるいは直腸に、投与できる。
本発明は、下記の実施例により、さらに説明される。
【実施例1】
【0046】
(実施例1:分留物CVT−E002の調製およびこの分留物の精製のための第1の方法)
アメリカの薬用人参の根が化学的に抽出され、引き続いて精製され、分留物CVT−E001およびCVT−E002を与えた。CVT−E002の総量がさらに精製され、分留物G1、G2、およびG3を与えた。この手順の詳細な記述は以下の通りである。
【0047】
乾燥してすり砕いた薬用人参の根500グラムが、4リットルの85%エタノールあるいは3リットルの90%メタノールにより、80〜85℃の湯浴で3時間の間攪拌しながら抽出され、濾過されてアルコール溶液と残渣を与えた。アルコール溶液は濃縮され、スプレードライされて、全サポニンの生成物(CVT−E001)を与えた。残渣は4リットルの水により、95〜100℃の湯浴で3時間の間攪拌しながら抽出された。抽出物はモスリン袋を通して濾過され、遠心分離機で分離されて、CVT−E002を含む水溶液を与え、また、残りの残渣は廃棄された。
【0048】
水溶液部分はさらなる精製に用いられ、同体積の95%エタノールが水溶液中に加えられ、それは沈殿を引き起こした。沈殿物は遠近分離機で分離され、減圧下で凍結乾燥され、分留物G1を与えた。上澄みは蒸発濃縮により体積を減らされた。同体積の95%エタノールが加えられ、次の沈殿G2を与え、残りの上澄みはエタノールとともに除去され、減圧下で凍結乾燥され、粉末G3を生成した。
【0049】
G2のさらなる精製が以下のように実施された。2グラムのG2が80mlの水に溶解され、1200の分子量で切り離すSigma D−7884透析チューブ中で3倍の水に対して透析された。透析は、24時間毎に2回透析物を集めながら、4℃で72時間行われた。得られた透析物は15mlまで濃縮され、その後、同体積のメタノールで沈殿された。沈殿物は水に溶かされ、減圧下で凍結乾燥されて、粉末G22を生じた。
【0050】
G22のさらなる精製が以下のように実施された。1.5グラムのG22が60mlの水に溶解され、1000の分子量で切り離すフィッシャー・スペクトラル/ポー(Fisher Spectral/Por)モレキュラー・ポーラス・メンブレン・チューブ中で600mlの水に対して4℃で透析された。24時間後に集められた第1の透析物は、蒸発乾燥を用いて15mlに濃縮され、その後に減圧下で凍結乾燥された。得られた乾燥粉末はG221で示された。同様の方法で、48時間後に透析物の第2バッチが集められ、G222で示された。保持物質は濃縮され、減圧下で凍結乾燥されて、G223と呼ばれる粉末を与えた。
【実施例2】
【0051】
(実施例2:分留物CVT−E002の調製およびこの分留物の精製のための第2の方法)
本発明に係る薬用人参分留物CVT−E002を調製する別の方法は下記のようである。
乾燥してすり砕いたアメリカの薬用人参の根1000グラムが、8リットルの85%エタノールにより、95〜100℃の湯浴で3時間の間攪拌しながら抽出され、濾過されてアルコール溶液と残渣を与えた。残渣は、3時間の間攪拌しながら湯浴にて水と混合(1:8)された。室温まで冷却した後、混合物が濾過された。濾過液は5000rpmで10分間遠心分離機にかけられた。上澄みは濃縮され、フリーズドライされて、抽出物CVT−E002を与えた。この方法により生成されたCVT−E002の量は、実施例1において生成されたのとほぼ同じ量、即ち、元の生の薬用人参の重量の10%であった。
【0052】
CVT−E002部分は、以下のようにさらに分留された。100グラムのCVT−E002粉末を2000mlの水に溶いた溶液中に、1600mlの95%エタノールが加えられた。沈殿物はPQ1として単離された。上澄みは500mlにまで濃縮された。この濃縮された溶液中に、別の部分の95%エタノール(1000ml)が再び加えられ、第2の沈殿分留物を与えた。沈殿物は単離され、フリーズドライされて、分留物PQ2を与えた。上澄みは濃縮され、フリーズドライされてPQ3を与えた。
【実施例3】
【0053】
(実施例3:マイトジェン活性試験)
種々のレベルに精製したアメリカの薬用人参の分留物が、そのリンパ球のマイトジェン特性に基づいたインビトロ(in vitro)のスクリーニングにより選択された。分裂促進性(mitogenicity)は、免疫系においてインビボ(in vivo)で生じる通常のイベントに対して、むしろ人工的イベントであると考えられるが、マイトジェンは可能なエフェクタ機能のよい指示を提供する。
【0054】
マイトジェン活性のテスト方法は、以下の通りである:Balb/CあるいはC57B1/6Jのマウスがテストに用いられた。Balb/Cマウスは、ヘルス・サイエンス・ラボラトリー・アニマル・サービス・ファシリティ(ユニバーシティ・オブ・アルバータ、エドモントン、カナダ)(Health Sciences Laboratory Animal Services Facility(University of Alberta,Edmonton,Canada))から得られた。C57B1/6Jマウスは、ジャクソン・ラボラトリー(バー・ハーバー、ME)(Jackson Laboratory(Bar Harbar,ME))から得られた。生後7〜10週間のマウスが各実験に歳と性別を適合された。マウスは頸部のずれにより命を絶たれた。脾臓が無菌の技術を用いて取り出され、2枚のスライドガラスのすり加工された界面の間で押しつぶされた。ハンクス平衡塩溶液(Hanks Balanced Salt Solution)(HBSS)中での遠心分離により洗浄した後、細胞は、RPMI1640に中間のpH7.4で(Gibco,Grand Island,N.Y.)懸濁され、それは10%の牛の胎児の血清(Flow Labs)、50mMのメルカプトエタノール(ICN Pharmaceuticals,Plainview,N.Y.)、およびペニシリン−ストレプトマイシン(Gibco)を含んでいた。培養液は、96のウェルを持つ底部が平坦なLinbroプレートに、最終濃度の1mlあたり1.25×106個の細胞で3重にセットされた。実験グループは、前もって濾過で滅菌され、HBSSに溶解された試験分留物とともに、セットされた。対照培養液は、マイトジェンなしのグループと、フィトヘマグルチニン(PHA)を20μg/mlあるいはリポポリサッカライド(LPS)を25μg/ml有するグループからなっていた。PHAは特にT型の脾臓細胞を刺激し、一方でLPSはB型の脾臓細胞を刺激することが知られている。培養液は、37℃の温度、加湿された5%のCO2雰囲気下で72時間の間培養された。72時間の培養の終了時から4時間前に、1μCiのトリチウム化したチミジン(New England Nuclear,Boston,Massachusetts)が各ウェルに加えられた。細胞は自動試料採取機(Skatron,Virginia)により採取され、その後に組み込まれた放射能がシンチレーション計測により分析された。結果は対照に対する%として計算された(平均±標準偏差、3重で)。
【0055】
水中でのCVT−E002の濃度や沈殿に用いられるエタノールの体積など、実施例1および2に示される異なる抽出条件は、CVT−E002のさらなる分留に影響を与える。これら2つの方法から得られた生成物は、マイトジェン活性で比較された。結果は下記の表に示される。各テストは3つのバッチにより3重で実施された。
【0056】
【表1】

【0057】
【表2】
【0058】
表1および2が示すように、実施例1および2の間の抽出手順の変化は、G1分留物からPQ2分留物への生物学的活性の移動に相関する。
【実施例4】
【0059】
(実施例4:PQ2の分留および分留物PQ223の調製の方法)
実施例2に係るCVT−E002、PQ1、PQ2、およびPQ3は、分子量分布に従い、アリルデキストランおよびN,N’−メチレンビスアクリルアミドからなる架橋した球形の共重合体のマトリックスを含み、16×600mmのベッド寸法および120mlのベッド体積と、球状のタンパク質に対して5000〜250,000、デキストランに対して1000〜80,000の分留範囲(MW)を有するHiPrep 16/60 Sephacryl S−200ハイレゾリューションカラム(Pharmacia,Biotech,Cat.No.17−1166−01)上のゲル濾過クロマトグラフィーによって、さらに分留される。デキストラン試料(MW=71.4k、37.5k、19.5k、および9.5k)はSigmaから購入され、標準試料として用いられた。各試料5mgが1mlの水に溶解され、カラムに導入され、かつ、環境温度4℃で、0.1N HClおよびpHが7.0の0.3M NaClを含むトリス−HClバッファにより、フローレート0.3ml/分で溶出された。多くの溶出液がいくつもの個々の5ml部分として集められた。
【0060】
個々の溶出部分のそれぞれの炭水化物の内容物は、全て、炭水化物内容物全てに対してマイクロタイター(microtiter)プレートアッセイによりテストされた。これは、DuboisらのAnal.Chem.28:350−56(1956)に記載された方法を一部変更した方法である。溶出液中の炭水化物全ては、所定の吸収スペクトルで検出可能とするために誘導体に合成される。D−グルコースが標準試料として用いられ、また、用いられる他の材料は濃硫酸(98%)とフェノール(5%)であった。ポリスチレンマイクロタイタープレートはSARSTDT(Quebec,Canada)から購入された。マイクロタイタープレートの各ウェルに、1〜10μgのD−グルコースを含有する40μlの標準試料溶液が加えられ、40μlの5%フェノールが加えられて混合された。その後、200μlの濃硫酸が丁寧に加えられた。試薬と個々の溶出部分をマルチチャンネルピペットで混合した後、プレートが80℃の下、1時間の間培養された。室温まで冷却した後、マルチスキャン(Multiskan)マイクロタイタプレートリーダにより492nmにおいて試料の吸収が測定された。データは保存され、マイクロソフトエクセルプログラムを用いてゲル濾過クロマトグラフィーの結果がプロットされた。
【0061】
分子量がよく知られた一連の標準デキストラン(Sigma)を同一のカラムにかけることにより、標準曲線が形成された(図1)。CVT−E002、および、PQ1、PQ2、およびPQ3の組21に対するクロマトグラムが図2〜5に示される。各図において、X軸上の点は溶出体積を実際の体積の1/5に換算して示す。このため、正しい溶出体積の値を得るには、図の結果に5がかけられなければならない。
【0062】
CVT−E002のゲル濾過クロマトグラム(図2)は、主たる3つのピークを示した。第1のピーク(溶出分留7〜10)の分子量は、標準曲線(図1)によると70,000あるいはそれ以上と見積もられた。第3のピーク(溶出分留18〜22)の分子量は約1000と見積もられた。小さな幅広の第2のピーク(溶出分留10〜17)も観測された。これら3つのピークの比は57.2:8.5:34.4であった。
【0063】
PQ1のクロマトグラム(図3)は、CVT−E002の高分子量分留物に合致する、1つの主たるピーク(溶出分留7〜10)と、弱いピーク(溶出分留18〜21)を示した。この2つのピークの比は86.8:13.2であった。
【0064】
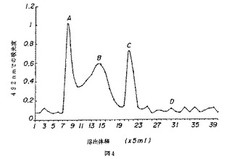
PQ2のクロマトグラム(図4)は、主たる3つのピークを示した。A(溶出分留7〜10)およびC(溶出分留19〜22)と名付けられたピークは、CVT−E002の2つの主たるピークに合致する。B(溶出分留10〜17)と名付けられた、もう一つの幅広のピークが、ピークAとCの間に観測された。このピークに対する分子量は約2,000〜60,000と見積もられた。3つのピークA〜Cの比は34.1:45.7:20.3であった。D(溶出分留24〜50であり、その部分は図4にて示されている)と名付けられた小さなピークの領域も観測された。
【0065】
PQ3のクロマトグラム(図5)は、1つの主たるピーク(溶出体積18〜21)のみを示し、これはCVT−E002の低い分子量分留物に合致する。
【0066】
PQ2の分留物A、B、C、およびDは、集められ、フリーズドライされる。乾燥された粉末は、それぞれPQ2A、PQ2B、PQ2C、およびPQ2Dと新たに命名される。また、分留物AおよびBは混合され、混合された分留物はPQ223(そのクロマトグラムは図6に示される)と呼ばれた。PQ2A、PQ2B、PQ2C、およびPQ2Dのマイトジェン活性がテストされ、分留物PQ223、PQ2およびCVT−E002の活性と比較された。結果は以下の表に示される。
【0067】
【表3】
【0068】
結果が示すように、有効性の序列はPQ223>PQ2>CVT−E002である。
【実施例5】
【0069】
(実施例5:CVT−E002、PQ1、PQ2、PQ3、およびPQ223の炭水化物総量およびタンパク質組成の化学的決定)
各分留物CVT−E002、PQ1、PQ2、PQ3、およびPQ223の3バッチが得られ、各分留物に対する炭水化物およびタンパク質の総量の組成が測定された。炭水化物の総量の含有量は、従来より知られたフェノール−サルファリック(sulfuric)法によって決定された。タンパク質の含有量は、従来より知られたLowry法によって検査され、ウシ血清アルブミンが標準として用いられた。結果は以下の表に示される。
【0070】
【表4】
【実施例6】
【0071】
(実施例6:CVT−E002、PQ1、PQ2、PQ3、およびPQ223のモノサッカライド組成の化学的決定)
炭水化物の総量の含有量は主にポリサッカライドとオリゴサッカライドの量を表しているので、所定の分留物に対するモノサッカライドのモル比もまた決定された。
【0072】
CVT−E002、PQ1、PQ2、PQ3、およびPQ223の分留物の組は、その構造単位であるモノサッカライドを遊離させるため、酸触媒加水分解された。遊離したモノサッカライドは、MPP(3−メチル−1−フェニル−2−ピラゾリン−5−オン)と誘導体を合成された後、その定性的および定量的解析がHPLCにより行われた。詳細な方法が以下の通りである:2NのHCl中の試料溶液が95〜100℃に6時間加熱された。混合物はNaOH溶液で中和された。内部標準が加えられた。MeOHおよびNaOH水溶液(0.3N)がこの混合物に加えられた。その後、これが50〜60℃で8時間(あるいは室温で1週間)攪拌された。混合物は水で(1:10)に希釈され、HPLCで解析された。MPPで誘導体が合成された試料は、C−18カラムで、0.1Mもリン酸バッファ(pH7)中の18%アセトニトリル溶液からなる溶離液を用い、フローレート1ml/分、245nmでのUV検出にて、解析された。下記の表は、CVT−E002、PQ1、PQ2、およびPQ223のモノサッカライドのモル比を示す。グルクロン酸のトレース量は、あるCVT−E002、PQ1、およびPQ2の試料中に見つかった。
【0073】
【表5】
【0074】
結果が示すように、グルコースの含有量の序列は次の通りである:PQ1>CVT−E002>PQ2>PQ3;また、ガラクツロン酸の含有量は次の通りである:PQ2>PQ223>CVT−E002>PQ1。
【実施例7】
【0075】
(実施例7:薬用人参分留物の免疫調節)
1.マクロファージ活性
CVT−E002、PQ2、G2、およびPQ223マウスのマクロファージに対する効果は、細胞性反応を優先的にマウントするC57B1/6マウスと、抗体性反応を優先的にマウントするBalb/cからの腹膜滲出マクロファージを用いて、インビトロ(in vitro)に研究された。マクロファージが100μg/mlのテスト試料で刺激された後、IL−1、IL−6、およびTNF−αの生成が測定された。その方法は下記の通りである。
【0076】
A.腹膜滲出細胞および細胞上澄みからのマクロファージの調製
1)3%チオグリコレート1mlがマウスの腹膜腔に注入され、3日後に腹膜腔から腹膜滲出マクロファージが採取された。
2)細胞懸濁液がHanksバッファとともに、4℃にて1100rpmで5分間遠心分離機にかけて2回洗浄された。
3)マクロファージがRPMI−10% FBS媒体中に懸濁された。
4)細胞が計測され、RPMI−10% FBS媒体で106/mlの最終濃度に希釈された。
5)マクロファージが、37℃、10×106/10ml RPMI−10%FBSにて2時間培養された。上澄みが廃棄され、沈殿したマクロファージが10mlのPBSで2回洗浄された。
6)マクロファージが採取され、RPMI−10% FBSに再び懸濁された。
7)マクロファージ(5×105)が、10mg/mlのLPSあるいは100mg/mlのテスト試料とともに、24あるいは48時間培養された。その後で、バイオアッセイ(IL−1、IL−6、およびTNF−α)のために、上澄みが集められ、濾過滅菌された。実験は3重に行われた。
【0077】
B.上澄み中のIL−1の決定
NOB1細胞系は、IL−1に応答して活性(IL−2)を刺激するCTLL成長を生成するEL4細胞系のTK変異種である。NOB1細胞は3HTdRを組み込まないので、IL−1への応答はCTLL系による取り込みの点から測定できる。手順は下記に示す通りである。
1)96のウェルを持つ底部が平坦なマイクロタイタープレートの各ウェルにおいて、104個のCTLL細胞が、IMDM中の5%FBS溶液200μl中で、4×104個のNOB1細胞と混合された。IL−1標準の4、8、16、または32倍、あるいは連続的な希釈液である一続きに希釈されたIL−1テスト試料が加えられた。各ウェルの最終的な体積は200μlであった。各試料は3重に調製された。
2)プレートが、37℃、加湿された5%CO2の培養器において、24時間の間培養された。
3)[3H]−チミジンが培養の最後の5時間の間加えられた。
4)細胞が採取され、液体シンチレーション計測により[3H]−チミジン組み込みが測定された。
5)IL−1濃度が算出された。結果は下記の表に示される。
【0078】
【表6】
【0079】
結果は、CVT−E002、PQ2、G2、およびPQ223は、C57B1/6マウスからのマクロファージを明らかに刺激してIL−1を生成したことを示す。全てのテスト試料はIL−1生成を刺激する同様な能力を示した。
【0080】
C.IL−6の決定
マウスのB細胞ハイブリドーマ細胞系であるB9の増殖はIL−6に依存する。B9細胞は、濃度を減らしているテスト試料を含む一連のマイクロウェル中で培養された。手順は下記に示す通りであった。
1)貯蔵培養フラスコから移した一部試料中のB9細胞の数が、それらの成長の対数期において計測された。
2)細胞は、180xg、4℃の卓上遠心分離機にて5分間遠心分離され、ペレットが、10mlの完全なRPMI−10媒体中に再懸濁された。この手順が2回繰り返され、細胞は、完全なRPMI−10媒体中に、2×103個の細胞/mlで再懸濁された。
3)洗浄された細胞(2×103個の細胞/ml)の100μlが、96のウェルを持つマイクロタイタープレートの各ウェルに加えられた。
4)テスト試料の2倍連続希釈液100μlが、各ウェルに加えられ、プレートの2または3列がIL−6標準のために残しておいた。
5)プレートが、加湿された、37℃、5%CO2の培養器で72時間培養された。
6)[3H]−チミジンが加えられ、プレートが37℃で4時間培養された。
7)細胞が採取され、液体シンチレーションカウンタを用いて、[3H]−チミジンの組み込みが決められた。
8)IL−6濃度が算出された。結果は下記の表に示される。
【0081】
【表7】
【0082】
CVT−E002、PQ2、およびPQ223は、C57B1/6およびBalb/cからのマクロファージの上澄み中でのIL−6の生成を増加させた。能力は、PQ223>PQ2>CVT−E002である。
【0083】
D.TNF−αの測定
下記のプロトコルは、細胞培養物から得られた上澄み中のTNF活性を定量するため、TNFに感度があり、アクチノマイシンD処理されたマウスのL929繊維芽細胞を採用した。1)IMDM−5%FSC 50μl中の4×104個のL929細胞が各ウェルに加えられた。
2)テスト試料50μlが各列の2番目のウェル(カラム2)に加えられた。ウェル2の内容物を静かに混合させることで2倍連続希釈液が形成され、ウェル2からウェル3に50μl移された。ウェル3の内容物は静かに混合され、ウェル3から50μlがウェル4に移された。この手順がウェル12まで続けられた。最後に、カラム12の全てのウェルの内容物が静かに混合され、各ウェルから50μlが廃棄された。この点で、全てのウェルは50μlを含んだ。
3)アクチノマイシンD溶液50μlが各ウェルに加えられた(2μl ml)。
4)プレートが、37℃、5%CO2の空気中で24時間培養された。
5)ナチュラルレッド5μlが各ウェルに加えられ、プレートが2.5時間培養された。
6)各ウェルの上澄みが直ちに、注意深くすすがれ、移されて、プレートが200μlのPBSで2回洗浄された。
7)PBSが吸い出され、0.05MのNaH2PO4中50%エタノール溶液100μlが各ウェルに加えられ、室温で5分間攪拌された。
8)マイクロタイタープレートリーダにより、570nmにける吸光度において、各ウェルが直ちに読まれた。
9)TNF−α濃度が算出された。結果は下記の表に示される。
【0084】
【表8】
【0085】
TNF−αの生成は、Balb/cあるいはC57B1/6細胞系からのマクロファージの上澄みにおいてCVT−E002およびPQ223により誘発された。Balb/cマウスからのマクロファージの上澄みにおけるTNF−αは、明らかにG2により刺激を受けた。PQ2はマクロファージによるTNF−α生成に刺激を与えないようであった。
【0086】
2.血清免疫グロブリン生成
血清免疫グロブリン生成(トータルIgG)は、水を与えられたマウスに比べて、CVT−E002を与えられたマウスにおいては21%、PQ2 を与えられたマウスにおいては26%、PQ223を与えられたマウスにおいては31%、増加した。能力は、PQ223>PQ2>CVT−E002である。結果は表9に示される。
【0087】
【表10】
【0088】
3.PHAあるいはLPSなどのよく知られたマイトジェンは、それらが刺激する細胞の種類について特異性を示す。脾臓細胞のどの特定の種類が薬用人参分留物により刺激を受けるかを知ることは重要である。従って、われわれは、アフィニティカラムによってT細胞を分離し、T細胞を減らすことによって、B細胞を濃縮した。
【0089】
A.T細胞濃縮
赤い血液細胞を除去するため、脾臓細胞の単一細胞懸濁液がリンパ球−M(Cedarlane Labs)上に積層された。その後、マウスの免疫グロブリンで被覆されたビーズへの吸着によりB細胞を取り除くアフィニティクリマトグラフィーカラム(Biotex Co. Ltd.,Edmonton,Alberta)にリンパ球を通過させた。溶出液は、濃縮された集団のT細胞からなっていた。他の細胞種に対するT細胞の割合の決定は、抗thy1.2モノクローナル抗体と補体処理を用いた生存能力テストによってなされた。
【0090】
B.B細胞濃縮
脾臓細胞集団からの赤い血液細胞の除去の後で、リンパ球は、モノクローナル抗thy1.2抗体とともに37℃で1時間培養された。低毒のウサギの補体が加えられ、4℃で30分培養された。この手順はT細胞の細胞懸濁を減らし、B細胞集団を濃縮する結果となる。
【0091】
C.PQ223との培養
BまたはT細胞はPQ223の適用量を変えながら培養され、応答がトリチウム化されたチミジンの組み込みによって測定された。図7および8は、それぞれ、相対的に精製されたBおよびT細胞に対するPQ223の増殖効果を示す。対照グループは、B細胞に特異的なマイトジェンであるLPS、およびT細胞に特異的なマイトジェンであるPHAで処理された細胞を含んだ。PQ223はB細胞に特異的であることが発見された(図9参照)。
【0092】
4.インビトロ(in vitro)での抗体生成に対するPQ223の効果
脾臓細胞は、適用量を変えたPQ223の存在下において72時間培養された。対照培養物は、25μgのLPSが加えられたグループと、マイトジェンが加えられなかったもう一つのグループから構成された。上澄みは、エンザイム・リンクト・イムノソーベント・アッセイ(Enzyme Linked Immunosorbent Assay(ELISA))を用いて、可溶性の免疫グロブリンの存在がテストされた。可溶性の免疫グロブリンを含む上澄みは、0.1Mのトリスバッファ(pH9)中に連続的に希釈された。マイクロタイタープレートが、この上澄みで被覆され、4℃で終夜培養された。プレートは、PBS Tweenで3回洗浄され、その後、抗体−酵素複合Goat F(ab’)2抗マウスIgセイヨウワサビペルオキシダーゼ(Tago Inc.,Burlingame California)100μlが1:1000の希釈率で加えられた。プレートは室温で1時間培養された。プレートは再びPBSで3回洗浄され、その後、ABTSペルオキシダーゼ基質(Kirkegaard and Perry Lab Inc.)が各ウェルに加えられた。1時間後、405nmの波長での吸光度がフローマルチスキャン(Flow Multiscan)ELISAリーダによって測定された。
【0093】
図10は、50μg/mlのPQ223が、LPS25μgの場合と同程度に、免疫グロブリンの生成物を刺激したことを示す。高濃度のPQ223、500μg/mlは、より高い水準の抗体生成を誘い出さなかった。この適用量応答曲線は、異種の脾臓細胞集団および精製されたB細胞の増殖応答と一致する。これらの結果は、4つの実験に基づいた。
【0094】
5.インビボ(in vivo)抗体プラーク形成細胞
3つの濃度のPQ223がそれぞれ3匹のマウスからなる3グループに静脈注射された。対照グループは、平衡塩溶液が注射された。PQ223での静脈処理の7日後に、マウスはHBSS中の10%SRBC懸濁液0.2mlにより免疫性を与えられた。免疫性を与えられてから5日後に、マウスは犠牲となり、脾臓からの単一細胞懸濁液が調製され、1mlあたり4×106個の細胞に調節された。この細胞懸濁液の0.1mlの一部試料がテンジクネズミ補体の10%SRBC溶液0.3mlとRPMI媒体0.5mlと混合された。この混合物の70μlがカニンガム(Cunningham)チャンバ−に移された。チャンバーはパラプラスト(paraplast)ワックスで封止され、プラーク形成細胞を計測する前に、37℃で1時間培養された。全てのPFCレベルは100万個の脾臓細胞あたりとして表現される。図11は、三種の適用量が全て、化合物が注射されたマウス内で、明らかに抗体生成を増強したことを示す。
【産業上の利用可能性】
【0095】
まとめると、本研究の結果は、マクロファージが、IL−1、IL−6、およびTNF−αなどのサイトカインを生成するのを本発明の薬用人参分留物が活性化することを示す。また、それらは、リンパ球、特にBリンパ球の増殖および抗体生成も活性化する。体液が媒介する免疫系の全体が刺激され、感染に対する予防効果を示した。報告されたTNF−α生成の活動は、抗ウイルスおよび抗腫瘍の恩恵を含む。TNF−αは、また、種々の寄生感染の治療のためにもなると報告されている。
【0096】
以下の本発明の背景において言及された引用文献は、ここに参照により本発明に組み込まれる。
1.Tomodaら、Biol.Pharm.Bull.,16,22−5(1993).
2.Tomodaら、Biol.Pharm.Bull.,17,1287−91(1994).
3.Tomodaら、Biol.Pharm.Bull.,16,1087−90(1993).
4.Gaoら、Planta Medica,55,9−12(1989).
5.Gaoら、Carbohydr.Res.,181,175−87(1988).
6.Kiyoharaら、Carbohydr.Res.,263,89−101(1994).
7.Shinら、Carbohydr.Res.,300,239−49(1997).
8.Yamadaら、Phytotherapy Res.,9,264−9(1995).
9.Konnoら、Planta Medica,50,434−6(1984).
10.Hikinoら、未発表.
11.Oshimaら、J.Ethnopharmacology,14,255−9(1985).
12.Konnnoら、J.Crude Drug Res.,25,53−5(1987).
13.Konnnoら、J.Ethnopharmacology,14,69−74(1985).
14.Oshimaら、J.Natural Products,50,188−990(1987).
15.Leeら、Anticancer Res.,17,323−32(1997).
16.Kimら、Planta Medica,64,110−5(1998).
17.Miaoら、Shengwu Huaxue ZaZhi,9,610−4(1993).
18.Maら、Baiqiuen Yike Daxue Xuebao,23,236−8(1997).
19.Zhuら、Zhongguo Yaolixue Tongbao,13,76−8(1997).
【技術分野】
【0001】
(発明の分野)
この発明は、北アメリカ薬用人参(Panax quinquefolium)からの分留物の化学的形成方法およびこれらの分留物を含有する組成物に関する。本発明の生成物は、抗体の生成を賦活するのに、あるいは、通常の風邪、インフルエンザ、慢性疲労症候群、AIDS、癌などの低免疫により特徴付けられる健康状態を対象とする治療法として、用いられ得る。また、本発明の生成物は、免疫系の深刻な抑制を引き起こす化学療法や放射線療法を受ける癌患者に対するサプリメントとして用いられ得る。
【背景技術】
【0002】
(発明の背景)
数百年の間、薬草化合物などのある種の無毒性薬品の使用は、さまざまな生理学的状態に、特に東洋で受け入れられてきた。Panax ginseng C.A.Meyerは最もよく知られた伝統的な中国薬である。薬用人参抽出物の重要な薬理学的活性は、単独で、あるいは他の薬品との組み合わせで、腎臓損傷の緩和、発癌の抑制およびストレスの予防を含む。また、固体の免疫学的反応性に対する薬用人参の影響について数多くの報告がなされている。報告されたある免疫賦活特性は、細胞レベルでのある基本的な免疫機能の賦活と同様に、感染に対する宿主抵抗の増強、抗炎症効果、腫瘍成長の抑制を含む。アメリカ薬用人参(Panax quinquefolium)は、多くの有益な健康上の効果を有する健康サプリメントとしての評判を得た薬用人参のもう一つの種である。いくつかの科学者グループが、薬用人参中に存在するポリサッカライドの単離および構造の特定を試みた。あるポリサッカライドは免疫系の賦活に活性があることが示された。
【0003】
薬用人参ポリサッカライドの単離、特徴付けおよび生物学的評価についての一連の研究が、共立薬科大学(日本)のTomodaのグループによりなされた。ある一連の研究では、薬用人参ポリサッカライドが酸性度に基づいて分留された。朝鮮薬用人参(Panax ginseng)の根から免疫学的に活性な2つの酸性のポリサッカライドが単離された[1,2]。スライスされた根がお湯で抽出された。抽出物は、硫酸ナトリウムの存在下においてセチルトリメチルアンモニウムブロマイド(CTAB)で処理された。沈殿物は、分離され、透析され、Sephadex G−25カラム、DEAE−Sephacel(Pharmacia)カラムに適用され、2つの純粋なポリサッカライドが得られ、それらはginsenan PAおよびginsenan PBと名付けられた。Toyopearl HW−55F上のゲルクロマトグラフィーは、ginsenan PAおよびginsenan PBの分子量として、それぞれ1.6×105、および、5.5×104の値を与えた。定量分析は、ginsenan PAが21.3%のアラビノース、53.4%のガラクトース、2.0%のラムノース、16.0%のガラクツロン酸および2.7%のグルクロン酸を含むことを示した。これらの成分糖のモル比は11:22:1:6:1であった。ginsenan PBは、11.0%のアラビノース、32.3%のガラクトース、8.1%のラムノース、39.9%のガラクツロン酸および5.0%のグルクロン酸を含んでいた。これらの成分糖のモル比は3:7:2:8:1であった。両ポリサッカライドとも、投与量に依存して、炭素除去試験における著しい細胞内皮系の有力な活性、際立った抗補体活性およびアルカリホスファターゼ誘導活性を示した。
【0004】
別の研究[3]において、CTABで処理された上記の抽出物の上澄みから2つの追加のポリサッカライドが単離された。即ち、上澄みがエタノール中に移された。沈殿物は分離され、DEAE−Sephadex A−25およびSephadex G−25の各カラムに適用され、S−IAおよびS−IIAと名付けられた2つの純粋なポリサッカライドを与えた。Toyopearl HW−55F上のゲルクロマトグラフィーは、S−IAおよびS−IIAの分子量として、それぞれ5.6×104、および、1.0×105の値を与えた。ginsenan S−IAは、42.3%のアラビノース、50.8%のガラクトース、6.9%のガラクツロン酸を8:8:1のモル比で含む。ginsenan S−IIAは、42.0%のL−アラビノース、32.6%のガラクトース、6.2%のグルコース、19,2%のガラクツロン酸を含む。モル比は15:10:2:5である。
【0005】
Panax ginsengの葉および根からのいくつかのポリサッカライド分留物は、日本の北里大学のオリエンタル・メディシン・リサーチセンターにおけるYamadaのグループにより分離された。化学特性および生物学的活性が調査および比較された[4]。
【0006】
中国産の薬用人参の根と葉は、それらのジンセノサイド(ginsenosides)を取り除くためにエタノールで処理した後、水で抽出され、残渣が0.5MのNaOHで抽出されて、水溶性およびアルカリ可溶性のポリサッカライド分留物(水溶性の分留物はGR−2およびGL−2、アルカリ可溶性の分留物はGRA−2およびGLA−2とそれぞれ名付けられる)を与えた。成分ポリサッカライドの酸性度に基づいて、セチルトリメチルアンモニウムとの処理により、全ての分留物が、強酸性(GR−3、GL−3、GRA−3、およびGLA−3と名付けられた)、弱酸性(GR−4、GL−4、GRA−4、およびGLA−4と名付けられた)、中性(GR−5、GL−5、GRA−5、およびGLA−5と名付けられた)のポリサッカライド分留物にさらに分留された。根は葉よりもポリサッカライドをより多量に含んでいた。根からの強酸性のポリサッカライド分留物は50%以上にもおよぶ高率のウロン酸含有量を有していた。似た成分糖は全ての分留物から検出された。それらは、ラムノース、アラビノース、ガラクトース、グルコース、ガラクツロン酸およびグルクロン酸であった。ガラクツロン酸は、主なウロン酸成分であった。
【0007】
最も高い抗補体活性を有する分留物であるGL−3は、DEAE−Sephadex、Sepharose CL−6B、DEAE Toyopearlのカラム処理、およびエタノール沈殿によりさらに分留されて、GL−PIからGL−PIVよ名付けられた分留物を与えた[5]。全ての分留物は32〜44%のウロン酸を含んでいた。分留物PIは50000という最も高い分子量を有していた。PIおよびPIIは主としてRha、Gal、およびGalAからなり、PIIIはさらにFucを含んでおり、一方、PIVはGal、Glc、およびGalAからなっていた。詳細な構造決定が行われた。
【0008】
抗潰瘍ペクチンポリサッカライド(GL−BIII)が、DEAE Sepharose CL−6BおよびSepharose CL−6Bのクロマトグラフィーによって、弱酸性のポリサッカライド分留物GL−4から単離された。主としてRha、Ara、Man、Gal、Glc、GalA、およびGlcAから、3:4:2:10:1:7:4のモル比で構成されていた。詳細な構造決定が行われた[6]。
【0009】
他のマクロファージFcレセプタ発現増強ポリサッカライド(GL−4IIb2)が、DEAE−Sepharose CL−6Bのアニオン交換クロマトグラフィーによって、GL−4から分離された。化学分析は、試料が65%の炭水化物と33.7%のウロン酸を含むことを示した。成分分析と構造決定が行われた[7]。
【0010】
抗補体活性を有する他のPanax ginseng抽出物、G−115が同じグループによって研究された[8]。G−115は、抗補体活性およびマイトジェン活性に対する活性物質を特徴付けるために分留された。最も効力のある抗補体活性は、未分留のポリサッカライド分留物G−115Gにおいて観測され、一方、水溶性で透析できる分留物であるG−115Eは最も効力のあるマイトジェン活性を示した。G−115Gは、さらにセチルトリメチルアンモニウムブロマイドとの沈殿形成、DEAE−Sepharoseでのアニオン交換クロマトグラフィー、およびSepharose CL−4Bでのゲル濾過によって精製され、目立って効力のある抗補体のポリサッカライドであるG−115I1−IIa−2−3が得られた。このポリサッカライドは均質であった。その分子量は3.68×105であると見積もられた。それは、主としてアラビノース、ガラクトースおよびグルコースと、これに加えて少量のガラクツロン酸、グルクロン酸、およびラムノースからなる。
【0011】
薬用人参ポリサッカライドが、日本の東北大学薬学部のHikinoのグループにより、韓国、中国、および日本の薬用人参から単離された。薬用人参ポリサッカライドの低血糖活性がテストされた。組成と構造のある特色が明らかにされた[9−14]。
【0012】
3つのポリサッカライド、quinquefolan A〜Cがアメリカの薬用人参から単離された[14]。それらの分子量は、Sephacryl S−500上のゲルクロマトグラフィーから2.0×106より大きいと見積もられた。中性糖成分は、quinquefolan Aに対してマンノースおよびグルコース(モル比は1.0:2.3)、quinquefolan Bに対してマンノースおよびグルコース(モル比は1.0:5.5)、quinquefolan Cに対してキシロースであった。quinquefolan A〜C中の酸性糖成分は、それぞれ10.8、11.7、および7.1%であることがわかった。これらのポリサッカライド中のペプチド部分の含有率は、quinquefolan A〜Cに対してそれぞれ2.7、2.9、および2.3%であった。これらのポリサッカライドの全てが、通常のマウスおよびアロキサンで減じられた低血糖のマウスにおいて低血糖効果を示した。
【0013】
分子量が150,000の酸性のポリサッカライドは、ginsanと呼ばれるが、韓国、ソウルの韓国癌センター病院の免疫学研究所(Laboratory of Immunology,Korean Cancer Center Hospital)の研究グループにより、Panaxginsengから単離された[15]。このポリサッカライドは3.7%のタンパク質、47.1%のヘキソース(グルコースおよびガラクトース)、43.1%のウロン酸(ガラクツロン酸)からなっていた。薬用人参は、T細胞とB細胞の増殖を誘発し、ナチュラルキラー細胞およびT細胞の両方から、内生的に生成されたマルチプルサイトカインを通してリンフォカイン活性化障害性細胞を発生させた[16]。
【0014】
中国のノースウエスト・ノーマル・ユニバーシティ(Northeast Normal University)のMiaoらは、アメリカの薬用人参からポリサッカライドを単離した。精製および構造解析が行われた[17]。
【0015】
アメリカの薬用人参からのポリサッカライドの生物学的活性は、ノーマン・ブスーン・ユニバーシティ・オブ・メディカル・サイエンス(Norman Bethune University of Medical Science)の研究グループにより研究された[18,19]。彼らは、アメリカの薬用人参からのポリサッカライド(PPQ)がリンパ球転換を増強することを発見した。マウスの脾臓リンパ球からのサイトカインのインビトロ(in vitro)での生成に対するPanax quinquefolium(PPQ−1)からのポリサッカライドの影響が研究された。データは、PPQ−1が免疫機能を調整することを示唆する。
【先行技術文献】
【非特許文献】
【0016】
【非特許文献1】Tomodaら、Biol.Pharm.Bull.,16,22−5(1993). 非特許文献1は、朝鮮薬用人参(Panax ginseng)の根から免疫学的に活性な2つの酸性のポリサッカライドが単離されることについて開示している。
【発明の概要】
【発明が解決しようとする課題】
【0017】
(発明のまとめ)
本発明者らは、あるアメリカの薬用人参の抽出物が免疫調整特性を有することを発見した。CVT−E002、およびそれから精製された分留物PQ2およびPQ223が特にマウスの脾臓細胞がB細胞を増殖するように刺激を与え、それは次に多量の抗体を生成させる。また、分留物は血清免疫グロブリン(例えばトータルIgG)レベルを増加させ、マクロファージにIL−1、IL−6、およびTNF−αを生成するように刺激を与える。これらの分留物は、一般的伝染病および他の免疫不全関連症の防止あるいは治療に使用される可能性がある。
【課題を解決するための手段】
【0018】
従って、本発明は、アメリカの薬用人参の試料から薬用人参分留物PQ2、PQ223、CVT−E002を調製する方法に注目する。
具体的に述べると、薬用人参分留物PQ2を調製する方法は、以下を有する:
−アメリカの薬用人参をアルコールを含む第1の溶媒に混合し、得られた溶液を約80〜100℃の温度で約2〜4時間の間加熱し、第1の薬用人参溶液を生成すること;
−その後で、上記第1の薬用人参溶液を分離して、アルコール/薬用人参溶液と第1の薬用人参残渣を生成すること;
−その後で、上記第1の薬用人参残渣を水に混合し、得られた溶液を約80〜100℃の温度で約2〜4時間の間加熱し、薬用人参残渣溶液を生成すること;
−その後で、上記薬用人参残渣溶液を分離して、第2の薬用人参残渣と、第1の薬用人参抽出物を含む第1の抽出物水溶液を生成すること;
−少なくとも上記第1の薬用人参抽出物の一部を含む第2の抽出物水溶液を提供すること、ここで、上記第2の抽出物水溶液中の上記第1の薬用人参抽出物の水に対する比率は約1:18〜1:22である;
−その後で、アルコールを含む第2の溶媒に上記第2の抽出物水溶液を混合して、第1の沈殿と第1の上澄みを生成すること、ここで、上記第2の溶媒の水に対する比率は約1:1〜3:5である;
−その後で、アルコールを含む第3の溶媒に以前の工程において生成された上記第1の上澄みを混合して、第2の沈殿と第2の上澄みを生成すること、ここで、上記第3の溶媒の第1の上澄みに対する比率は約3:2〜3:1である;および
−上記第2の沈殿物を単離して薬用人参分留物PQ2を生成すること。
【0019】
薬用人参分留物PQ223を調製する方法は、以下を有する:
−上述のように、薬用人参分留物PQ2を提供すること;
−上記薬用人参分留物PQ2を分留して第1溶出分留物および第2溶出分留物を生成すること、ここで、第1溶出分留物は溶出体積が35および50mlの間に観測される炭水化物ピークに合致し、第2溶出分留物は溶出体積が50および85mlの間に観測される炭水化物ピークに合致し、これらは下記の材料を用いたゲル濾過クロマトグラフィーにより決定される:
(1)アリルデキストランおよびN,N’−メチレンビスアクリルアミドからなる架橋した球形の共重合体のマトリックスを含み、16×600mmのベッド寸法および120mlのベッド体積と、球状のタンパク質に対して5000〜250,000、デキストランに対して1000〜80,000の分留範囲(MW)を有するクロマトグラフィーカラム、および
(2)0.1N HClおよびpHが7.0の0.3M NaClを含むトリス−HClの溶出バッファ;および
−上記第1溶出分留物および上記第2溶出分留物を単離し、かつ混合して、薬用人参分留物PQ223を生成すること。
【0020】
薬用人参分留物CVT−E002を調製する方法は、以下を有する:
−薬用人参1グラムあたり約7〜9mlの第1の溶媒の比率で、アルコールを含む第1の溶媒にアメリカの薬用人参を混合し、得られた溶液を約80〜100℃の温度で約2〜4時間の間加熱し、第1の薬用人参溶液を生成すること;
−その後で、上記第1の薬用人参溶液を分離して、アルコール/薬用人参溶液と第1の薬用人参残渣を生成すること;
−その後で、薬用人参残渣1グラムあたり約7〜9mlの水の比率で、上記第1の薬用人参残渣を水に混合し、薬用人参残渣溶液を約80〜100℃の温度で約2〜4時間の間加熱し、薬用人参残渣溶液を生成すること;
−その後で、上記薬用人参残渣溶液を分離して、第2の薬用人参残渣と、薬用人参抽出物を含む抽出物水溶液を生成すること;および
−上記抽出物水溶液を乾燥させて薬用人参分留物CVT−E002を生成すること。
【0021】
本発明は、また、上述の方法で調製された薬用人参分留物PQ2、PQ223、およびCVT−E002を含む。
【0022】
本発明は、特定の炭水化物内容物を有する薬用人参分留物をさらに含む。
第1の薬用人参分留物は、約2〜6モル%のラムノース、約41〜49モル%のガラクツロン酸、約12〜18モル%のグルコース、約16〜22モル%のガラクトース、および約12〜19モル%のアラビノースを含む炭水化物内容物を有する。
第2の薬用人参分留物は、約3〜8モル%のラムノース、約36〜44モル%のガラクツロン酸、約2〜7モル%のグルコース、約25〜33モル%のガラクトース、および約17〜25モル%のアラビノースを含む炭水化物内容物を有する。
第3の薬用人参分留物は、約0.5〜5モル%のラムノース、約11〜22モル%のガラクツロン酸、約40〜60モル%のグルコース、約10〜19モル%のガラクトース、および約11〜19モル%のアラビノースを含む炭水化物内容物を有する。
【0023】
本発明は、また、本発明の薬用人参分留物を有し、薬学的に許容なキャリアに混合した薬学的組成物を含む。
本発明は、本発明による薬用人参分留物の、低免疫で特徴付けられる健康状態を治療するのに適した薬学的組成物の調製において、単独でのあるいは他の薬剤との組み合わせての使用をさらに含む。
【0024】
本発明は、また、IL−1、IL−6、および/またはTNF−αの細胞中の生成を刺激するための本発明に係る薬用人参分留物の使用を含む。
本発明は、インビトロ(in vitro)またはインビボ(in vivo)での免疫グロブリンの生成を刺激するための本発明に係る薬用人参分留物の使用をさらに含む。
また、Bリンパ球の増殖とそれらからの抗体の生成を活性化するための本発明に係る薬用人参分留物の使用が含まれる。
【0025】
本発明は、また、健康状態を治療するのに効果的な量の本発明に係る薬用人参分留物を患者に投与することを含む、それを必要としている患者の低免疫で特徴付けられる健康状態を治療する方法を含む。
【0026】
(本発明の詳細な記述)
薬用人参分留物PQ2を調製する方法は、下記を含む:
(a)アメリカの薬用人参をアルコールを含む第1の溶媒に混合し、得られた溶液を約80〜100℃の温度で約2〜4時間の間加熱し、第1の薬用人参溶液を生成すること;
(b)その後で、上記第1の薬用人参溶液を分離して、アルコール/薬用人参溶液と第1の薬用人参残渣を生成すること;
(c)その後で、上記第1の薬用人参残渣を水に混合し、得られた溶液を約80〜100℃の温度で約2〜4時間の間加熱し、薬用人参残渣溶液を生成すること;
(d)その後で、上記薬用人参残渣溶液を分離して、第2の薬用人参残渣と、第1の薬用人参抽出物を含む第1の抽出物水溶液を生成すること;
(e)少なくとも上記第1の薬用人参抽出物の一部を含む第2の抽出物水溶液を提供すること、ここで、上記第2の抽出物水溶液中の上記第1の薬用人参抽出物の水に対する比率は約1:18〜1:22である;
(f)その後で、アルコールを含む第2の溶媒に上記第2の抽出物水溶液を混合して、第1の沈殿と第1の上澄みを生成すること、ここで、上記第2の溶媒の水に対する比率は約1:1〜3:5である;
(g)その後で、アルコールを含む第3の溶媒にステップ(f)において生成された上記第1の上澄みを混合して、第2の沈殿と第2の上澄みを生成すること、ここで、上記第3の溶媒の第1の上澄みに対する比率は約3:2〜3:1である;および
(h)上記第2の沈殿物を単離して薬用人参分留物PQ2 を生成すること。
【0027】
第1の溶媒、第2の溶媒、および第3の溶媒のそれぞれにおけるアルコールは、薬用人参に不活性で、所望の生成物から容易に分離可能であるアルコールを含む。当業者は、これらの必要条件を満たすアルコールをたやすく選ぶことができるだろう。好ましくは、それぞれのアルコールが、他と関係なく、飽和あるいは不飽和のC1〜C6アルコールを含む。より好ましくは、それぞれのアルコールが、他と関係なく、エタノールあるいはメタノールを含む。
【0028】
ステップ(a)と(c)のそれぞれにおいて、得られる溶液を約3時間の間加熱することが好ましい。ステップ(a)において、第1の溶媒および薬用人参が、薬用人参1グラムあたり約7〜9mlの第1の溶媒の比率で混合されることが好ましく、最も好ましくは薬用人参1グラムあたり約8mlの第1の溶媒の比率で混合される。ステップ(c)において、水および第1の薬用人参残渣が、薬用人参残渣1グラムあたり約7〜9mlの水の比率で混合されることが好ましく、最も好ましくは薬用人参残渣1グラムあたり約8mlの水の比率で混合される。
【0029】
上述の方法においてステップ(d)に続いてすぐ、第1の薬用人参抽出物が随意に濃縮されてもよい。これは、当業者によく知られた手順によるどんな方法によって行われてもよい。例えば、第1の薬用人参抽出物を含む第1の水溶液は遠心分離機で(例えば2500〜10000rpmの速さで約5〜15分)分離されてもよい。また、第1の水溶液は濾過されてもよい。第1の薬用人参抽出物の濃縮に替えて、あるいはこれに追加して、第1の薬用人参抽出物は後で使用するようにフリーズドライされてもよい。
【0030】
ステップ(e)において、第2の抽出物水溶液は、少なくとも第1の薬用人参抽出物の一部を含んでいる。第2の抽出物水溶液中の第1の薬用人参抽出物の水に対する比率は約1:18〜1:22であり、より好ましくは1:20であることが重要である。これは、多くの方法のいずれでも達成できる。上述のように、第1の薬用人参抽出物が、ステップ(d)に続いて濃縮される、および/または、フリーズドライされるならば、所望の比率を達成するように第1の薬用人参抽出物に水が加えられるべきである。あるいは、第1の抽出物水溶液の一部あるいは全部が第2の抽出物水溶液に用いられてもよい。必要なら、所望の比率を達成するために、追加の水を加えてもよい。所望の量の第1の薬用人参抽出物を含有する第1の抽出物水溶液の少なくとも一部を使用することは、ステップ(d)と(e)の間に追加の濃縮またはフリーズドライステップを実施する必要を逃れるだろう。
【0031】
ステップ(f)において、第2の溶媒の水に対する比率は約3:4であることが好ましい。
ステップ(g)において、第3の溶媒の第1の上澄みに対する比率は約2:1であることが好ましい。
【0032】
薬用人参分留物PQ223を調製する方法は、下記を含む:
(a)上述の方法によって生成された、薬用人参分留物PQ2を提供すること;
(b)上記薬用人参分留物PQ2を分留して第1溶出分留物および第2溶出分留物を生成すること、ここで、第1溶出分留物は溶出体積が35および50mlの間に観測される炭水化物ピークに合致し、第2溶出分留物は溶出体積が50および85mlの間に観測される炭水化物ピークに合致し、これらは下記の材料を用いたゲル濾過クロマトグラフィーにより決定される:
(1)アリルデキストランおよびN,N’−メチレンビスアクリルアミドからなる架橋した球形の共重合体のマトリックスを含み、16×600mmのベッド寸法および120mlのベッド体積と、球状のタンパク質に対して5000〜250,000、デキストランに対して1000〜80,000の分留範囲(MW)を有するクロマトグラフィーカラム、および
(2)0.1N HClおよびpHが7.0の0.3M NaClを含むトリス−HClの溶出バッファ;および
(c)上記第1溶出分留物および上記第2溶出分留物を単離し、かつ混合して、薬用人参分留物PQ223を生成すること。
【0033】
第1の溶出分留物はPQ2Aとしても知られており、第2の溶出分留物はPQ2Bとしても知られている。これらの溶出分留物は別々に単離され得る。また、追加の溶出分留物PQ2C(溶出体積が95および110mlの間に観測される炭水化物ピークに合致する)、およびPQ2D(溶出体積が120および250mlの間に観測される炭水化物ピークに合致する)を生成するために、上述と同じ方法が用いられてもよい。これらの分留物は別々に単離され得る。また、PQ223の他に、PQ2AからPQ2Dの2以上の分留物からなる組成物も形成されることができる。
【0034】
薬用人参分留物PQ2は、ゲル濾過クロマトグラフィーを用いて分留されることが好ましい。しかしながら、当業者に知られている他のいずれの種類の分留も適している。
ゲル濾過クロマトグラフィーを行う手順は、フローレート、試料体積、および手順が実施される温度に関して、製作者の推薦に従って、当業者によく知られている。これらの要因の製作者の指定範囲内の変動は、クロマトグラフィー試験の結果に対する明らかな影響はない。
【0035】
炭水化物内容物の決定は、当業者に知られたいずれの手順によって行われてもよい。分留物の炭水化物組成を決定するのに、マイクロフィルタープレートアッセイ(microfilter plate assay)が実施されることが好ましい。そのようなアッセイは当業者によく知られている。例えば、Duboisら、Anal.chem.28:350−56(1956)を参照されたい。この結果、参照により組み込まれる。マイクロフィルタープレートアッセイに用いるのにもたらされた試料の吸収は、好ましくは492nmで実施される。
【0036】
薬用人参分留物CVT−E002を調製する方法は、下記を含む:
(a)薬用人参1グラムあたり約7〜9mlの第1の溶媒の比率で、アルコールを含む第1の溶媒にアメリカの薬用人参を混合し、得られた溶液を約80〜100℃の温度で約2〜4時間の間加熱し、第1の薬用人参溶液を生成すること;
(b)その後で、上記第1の薬用人参溶液を分離して、アルコール/薬用人参溶液と第1の薬用人参残渣を生成すること;
(c)その後で、薬用人参残渣1グラムあたり約7〜9mlの水の比率で、上記第1の薬用人参残渣を水に混合し、薬用人参残渣溶液を約80〜100℃の温度で約2〜4時間の間加熱し、薬用人参残渣溶液を生成すること;
(d)その後で、上記薬用人参残渣溶液を分離して、第2の薬用人参残渣と、薬用人参抽出物を含む抽出物水溶液を生成すること;および
(e)上記抽出物水溶液を乾燥あるいは濃縮させて薬用人参分留物CVT−E002を生成すること。
【0037】
第1の溶媒におけるアルコールは、薬用人参に不活性で、所望の生成物から容易に分離可能であるアルコールを含む。当業者は、これらの必要条件を満たすアルコールをたやすく選ぶことができるだろう。好ましくは、このアルコールが飽和あるいは不飽和のC1〜C6アルコールを含む。より好ましくは、このアルコールがエタノールあるいはメタノールを含む。
【0038】
ステップ(a)において、第1の溶媒および試料が、試料1グラムあたり約8mlの第1の溶媒の比率で混合されることが好ましい。ステップ(c)において、水および第1の薬用人参残渣が、薬用人参残渣1グラムあたり約8mlの水の比率で混合されることが好ましい。
ステップ(a)と(c)において、第1の薬用人参溶液を約3時間の間加熱することが好ましい。
【0039】
本発明は、また、いくつかの薬用人参分留物を含む。
第1の薬用人参分留物は、約2〜6モル%のラムノース、約41〜49モル%のガラクツロン酸、約12〜18モル%のグルコース、約16〜22モル%のガラクトース、および約12〜19モル%のアラビノースを含む炭水化物内容物を有する。好ましくは、炭水化物内容物が、約3〜5モル%のラムノース、約43〜47モル%のガラクツロン酸、約14〜16モル%のグルコース、約18〜20モル%のガラクトース、および約14〜17モル%のアラビノースを含む。最も好ましくは、炭水化物内容物が、約4モル%のラムノース、約45モル%のガラクツロン酸、約15モル%のグルコース、約19モル%のガラクトース、および約15モル%のアラビノースを含む。
【0040】
本発明に係る第2の薬用人参分留物は、約3〜8モル%のラムノース、約36〜44モル%のガラクツロン酸、約2〜7モル%のグルコース、約25〜33モル%のガラクトース、および約17〜25モル%のアラビノースを含む炭水化物内容物を有する。好ましくは、炭水化物内容物が、約4〜7モル%のラムノース、約37〜42モル%のガラクツロン酸、約3〜6モル%のグルコース、約27〜32モル%のガラクトース、および約19〜24モル%のアラビノースを含む。最も好ましくは、炭水化物内容物が、約5モル%のラムノース、約39モル%のガラクツロン酸、約4モル%のグルコース、約29モル%のガラクトース、および約21モル%のアラビノースを含む。
【0041】
本発明に係る第3の薬用人参分留物は、約0.5〜5モル%のラムノース、約11〜22モル%のガラクツロン酸、約40〜60モル%のグルコース、約10〜19モル%のガラクトース、および約11〜19モル%のアラビノースを含む炭水化物内容物を有する。好ましくは、炭水化物内容物が、約1〜3モル%のラムノース、約13〜20モル%のガラクツロン酸、約42〜57モル%のグルコース、約12〜17モル%のガラクトース、および約13〜17モル%のアラビノースを含む。
【0042】
本発明は、また、本発明に係る薬用人参分留物のいずれかを有し、薬学的に許容なキャリアに混合した薬学的組成物を含む。当業者は、このことについて有効であるだろういずれの薬学的に許容なキャリアにも熟知しており、従って、本発明に係る薬学的組成物を形成する手順は詳細に議論しない。薬学的組成物は、適当に、タブレット、カプセル、液体、菱形(lozenge)、ローション、あるいは座薬の形態をとる。
【0043】
本発明は、通常の風邪、インフルエンザ、慢性疲労症候群、AIDS、癌などの低免疫で特徴付けられる健康状態を治療するのに適した薬学的組成物の調製における本発明に係る薬用人参分留物の使用を含む。薬用人参分留物は、単独で、あるいは他の薬剤を組み合わせて用いられ得る。癌患者は免疫系に深刻なサプレッションを有することが知られているので、本発明の薬用人参分留物は、化学療法の試薬とともに投与するのに、あるいは、放射線療法でのサプリメントとして、特に適している。
【図面の簡単な説明】
【0044】
【図1】図1は、クロマトグラフィーカラムで溶出された標準デキストラン試料の分子量のlog値に対して溶出体積をプロットした図を示す。
【図2】図2は、薬用人参分留物CVT−E002のクロマトグラムを示す。
【図3】図3は、薬用人参分留物PQ1のクロマトグラムを示す。
【図4】図4は、薬用人参分留物PQ2のクロマトグラムを示す。
【図5】図5は、薬用人参分留物PQ3のクロマトグラムを示す。
【図6】図6は、薬用人参分留物PQ223のクロマトグラムを示す。
【図7】図7は、マウスの脾臓細胞のB細胞の増殖に対する薬用人参分留物PQ223の効果を示す。
【図8】図8は、マウスの脾臓細胞のT細胞の増殖に対する薬用人参分留物PQ223の効果を示す。
【図9】図9は、マウスの脾臓細胞のTおよびB細胞の増殖に対する薬用人参分留物PQ223の効果の特異性を示す。
【図10】図10は、マウスの脾臓によるインビトロ(in vitro)抗体生成に対する薬用人参分留物PQ223の効果を示す。
【図11】図11は、薬用人参分留物PQ223が注入されたマウスのプラーク形成細胞(PFC)の増強を示す。
【0045】
本発明は、また、健康状態を治療するのに効果的な量の本発明に係る薬用人参分留物を患者に投与することを含む、それを必要としている患者の低免疫で特徴付けられる健康状態の治療方法を含む。好ましくは、健康状態が、通常の風邪、インフルエンザ、慢性疲労症候群、AIDS、癌からなるグループから選択される。本発明に係る薬用人参分留物の投与量は、年齢、性別、および患者の一般的な健康状態はもちろん、治療される個々の健康状態にも依存する。しかしながら、適切な投与量は、1日に1〜10回の投与範囲において、1日あたり、1〜5000mg/kg体重の範囲に見つかるだろう。薬用人参分留物は、経口で、注射もしくは点滴で、局部的に、鼻から吸って、点眼で、膣に、あるいは直腸に、投与できる。
本発明は、下記の実施例により、さらに説明される。
【実施例1】
【0046】
(実施例1:分留物CVT−E002の調製およびこの分留物の精製のための第1の方法)
アメリカの薬用人参の根が化学的に抽出され、引き続いて精製され、分留物CVT−E001およびCVT−E002を与えた。CVT−E002の総量がさらに精製され、分留物G1、G2、およびG3を与えた。この手順の詳細な記述は以下の通りである。
【0047】
乾燥してすり砕いた薬用人参の根500グラムが、4リットルの85%エタノールあるいは3リットルの90%メタノールにより、80〜85℃の湯浴で3時間の間攪拌しながら抽出され、濾過されてアルコール溶液と残渣を与えた。アルコール溶液は濃縮され、スプレードライされて、全サポニンの生成物(CVT−E001)を与えた。残渣は4リットルの水により、95〜100℃の湯浴で3時間の間攪拌しながら抽出された。抽出物はモスリン袋を通して濾過され、遠心分離機で分離されて、CVT−E002を含む水溶液を与え、また、残りの残渣は廃棄された。
【0048】
水溶液部分はさらなる精製に用いられ、同体積の95%エタノールが水溶液中に加えられ、それは沈殿を引き起こした。沈殿物は遠近分離機で分離され、減圧下で凍結乾燥され、分留物G1を与えた。上澄みは蒸発濃縮により体積を減らされた。同体積の95%エタノールが加えられ、次の沈殿G2を与え、残りの上澄みはエタノールとともに除去され、減圧下で凍結乾燥され、粉末G3を生成した。
【0049】
G2のさらなる精製が以下のように実施された。2グラムのG2が80mlの水に溶解され、1200の分子量で切り離すSigma D−7884透析チューブ中で3倍の水に対して透析された。透析は、24時間毎に2回透析物を集めながら、4℃で72時間行われた。得られた透析物は15mlまで濃縮され、その後、同体積のメタノールで沈殿された。沈殿物は水に溶かされ、減圧下で凍結乾燥されて、粉末G22を生じた。
【0050】
G22のさらなる精製が以下のように実施された。1.5グラムのG22が60mlの水に溶解され、1000の分子量で切り離すフィッシャー・スペクトラル/ポー(Fisher Spectral/Por)モレキュラー・ポーラス・メンブレン・チューブ中で600mlの水に対して4℃で透析された。24時間後に集められた第1の透析物は、蒸発乾燥を用いて15mlに濃縮され、その後に減圧下で凍結乾燥された。得られた乾燥粉末はG221で示された。同様の方法で、48時間後に透析物の第2バッチが集められ、G222で示された。保持物質は濃縮され、減圧下で凍結乾燥されて、G223と呼ばれる粉末を与えた。
【実施例2】
【0051】
(実施例2:分留物CVT−E002の調製およびこの分留物の精製のための第2の方法)
本発明に係る薬用人参分留物CVT−E002を調製する別の方法は下記のようである。
乾燥してすり砕いたアメリカの薬用人参の根1000グラムが、8リットルの85%エタノールにより、95〜100℃の湯浴で3時間の間攪拌しながら抽出され、濾過されてアルコール溶液と残渣を与えた。残渣は、3時間の間攪拌しながら湯浴にて水と混合(1:8)された。室温まで冷却した後、混合物が濾過された。濾過液は5000rpmで10分間遠心分離機にかけられた。上澄みは濃縮され、フリーズドライされて、抽出物CVT−E002を与えた。この方法により生成されたCVT−E002の量は、実施例1において生成されたのとほぼ同じ量、即ち、元の生の薬用人参の重量の10%であった。
【0052】
CVT−E002部分は、以下のようにさらに分留された。100グラムのCVT−E002粉末を2000mlの水に溶いた溶液中に、1600mlの95%エタノールが加えられた。沈殿物はPQ1として単離された。上澄みは500mlにまで濃縮された。この濃縮された溶液中に、別の部分の95%エタノール(1000ml)が再び加えられ、第2の沈殿分留物を与えた。沈殿物は単離され、フリーズドライされて、分留物PQ2を与えた。上澄みは濃縮され、フリーズドライされてPQ3を与えた。
【実施例3】
【0053】
(実施例3:マイトジェン活性試験)
種々のレベルに精製したアメリカの薬用人参の分留物が、そのリンパ球のマイトジェン特性に基づいたインビトロ(in vitro)のスクリーニングにより選択された。分裂促進性(mitogenicity)は、免疫系においてインビボ(in vivo)で生じる通常のイベントに対して、むしろ人工的イベントであると考えられるが、マイトジェンは可能なエフェクタ機能のよい指示を提供する。
【0054】
マイトジェン活性のテスト方法は、以下の通りである:Balb/CあるいはC57B1/6Jのマウスがテストに用いられた。Balb/Cマウスは、ヘルス・サイエンス・ラボラトリー・アニマル・サービス・ファシリティ(ユニバーシティ・オブ・アルバータ、エドモントン、カナダ)(Health Sciences Laboratory Animal Services Facility(University of Alberta,Edmonton,Canada))から得られた。C57B1/6Jマウスは、ジャクソン・ラボラトリー(バー・ハーバー、ME)(Jackson Laboratory(Bar Harbar,ME))から得られた。生後7〜10週間のマウスが各実験に歳と性別を適合された。マウスは頸部のずれにより命を絶たれた。脾臓が無菌の技術を用いて取り出され、2枚のスライドガラスのすり加工された界面の間で押しつぶされた。ハンクス平衡塩溶液(Hanks Balanced Salt Solution)(HBSS)中での遠心分離により洗浄した後、細胞は、RPMI1640に中間のpH7.4で(Gibco,Grand Island,N.Y.)懸濁され、それは10%の牛の胎児の血清(Flow Labs)、50mMのメルカプトエタノール(ICN Pharmaceuticals,Plainview,N.Y.)、およびペニシリン−ストレプトマイシン(Gibco)を含んでいた。培養液は、96のウェルを持つ底部が平坦なLinbroプレートに、最終濃度の1mlあたり1.25×106個の細胞で3重にセットされた。実験グループは、前もって濾過で滅菌され、HBSSに溶解された試験分留物とともに、セットされた。対照培養液は、マイトジェンなしのグループと、フィトヘマグルチニン(PHA)を20μg/mlあるいはリポポリサッカライド(LPS)を25μg/ml有するグループからなっていた。PHAは特にT型の脾臓細胞を刺激し、一方でLPSはB型の脾臓細胞を刺激することが知られている。培養液は、37℃の温度、加湿された5%のCO2雰囲気下で72時間の間培養された。72時間の培養の終了時から4時間前に、1μCiのトリチウム化したチミジン(New England Nuclear,Boston,Massachusetts)が各ウェルに加えられた。細胞は自動試料採取機(Skatron,Virginia)により採取され、その後に組み込まれた放射能がシンチレーション計測により分析された。結果は対照に対する%として計算された(平均±標準偏差、3重で)。
【0055】
水中でのCVT−E002の濃度や沈殿に用いられるエタノールの体積など、実施例1および2に示される異なる抽出条件は、CVT−E002のさらなる分留に影響を与える。これら2つの方法から得られた生成物は、マイトジェン活性で比較された。結果は下記の表に示される。各テストは3つのバッチにより3重で実施された。
【0056】
【表1】
【0057】
【表2】
【0058】
表1および2が示すように、実施例1および2の間の抽出手順の変化は、G1分留物からPQ2分留物への生物学的活性の移動に相関する。
【実施例4】
【0059】
(実施例4:PQ2の分留および分留物PQ223の調製の方法)
実施例2に係るCVT−E002、PQ1、PQ2、およびPQ3は、分子量分布に従い、アリルデキストランおよびN,N’−メチレンビスアクリルアミドからなる架橋した球形の共重合体のマトリックスを含み、16×600mmのベッド寸法および120mlのベッド体積と、球状のタンパク質に対して5000〜250,000、デキストランに対して1000〜80,000の分留範囲(MW)を有するHiPrep 16/60 Sephacryl S−200ハイレゾリューションカラム(Pharmacia,Biotech,Cat.No.17−1166−01)上のゲル濾過クロマトグラフィーによって、さらに分留される。デキストラン試料(MW=71.4k、37.5k、19.5k、および9.5k)はSigmaから購入され、標準試料として用いられた。各試料5mgが1mlの水に溶解され、カラムに導入され、かつ、環境温度4℃で、0.1N HClおよびpHが7.0の0.3M NaClを含むトリス−HClバッファにより、フローレート0.3ml/分で溶出された。多くの溶出液がいくつもの個々の5ml部分として集められた。
【0060】
個々の溶出部分のそれぞれの炭水化物の内容物は、全て、炭水化物内容物全てに対してマイクロタイター(microtiter)プレートアッセイによりテストされた。これは、DuboisらのAnal.Chem.28:350−56(1956)に記載された方法を一部変更した方法である。溶出液中の炭水化物全ては、所定の吸収スペクトルで検出可能とするために誘導体に合成される。D−グルコースが標準試料として用いられ、また、用いられる他の材料は濃硫酸(98%)とフェノール(5%)であった。ポリスチレンマイクロタイタープレートはSARSTDT(Quebec,Canada)から購入された。マイクロタイタープレートの各ウェルに、1〜10μgのD−グルコースを含有する40μlの標準試料溶液が加えられ、40μlの5%フェノールが加えられて混合された。その後、200μlの濃硫酸が丁寧に加えられた。試薬と個々の溶出部分をマルチチャンネルピペットで混合した後、プレートが80℃の下、1時間の間培養された。室温まで冷却した後、マルチスキャン(Multiskan)マイクロタイタプレートリーダにより492nmにおいて試料の吸収が測定された。データは保存され、マイクロソフトエクセルプログラムを用いてゲル濾過クロマトグラフィーの結果がプロットされた。
【0061】
分子量がよく知られた一連の標準デキストラン(Sigma)を同一のカラムにかけることにより、標準曲線が形成された(図1)。CVT−E002、および、PQ1、PQ2、およびPQ3の組21に対するクロマトグラムが図2〜5に示される。各図において、X軸上の点は溶出体積を実際の体積の1/5に換算して示す。このため、正しい溶出体積の値を得るには、図の結果に5がかけられなければならない。
【0062】
CVT−E002のゲル濾過クロマトグラム(図2)は、主たる3つのピークを示した。第1のピーク(溶出分留7〜10)の分子量は、標準曲線(図1)によると70,000あるいはそれ以上と見積もられた。第3のピーク(溶出分留18〜22)の分子量は約1000と見積もられた。小さな幅広の第2のピーク(溶出分留10〜17)も観測された。これら3つのピークの比は57.2:8.5:34.4であった。
【0063】
PQ1のクロマトグラム(図3)は、CVT−E002の高分子量分留物に合致する、1つの主たるピーク(溶出分留7〜10)と、弱いピーク(溶出分留18〜21)を示した。この2つのピークの比は86.8:13.2であった。
【0064】
PQ2のクロマトグラム(図4)は、主たる3つのピークを示した。A(溶出分留7〜10)およびC(溶出分留19〜22)と名付けられたピークは、CVT−E002の2つの主たるピークに合致する。B(溶出分留10〜17)と名付けられた、もう一つの幅広のピークが、ピークAとCの間に観測された。このピークに対する分子量は約2,000〜60,000と見積もられた。3つのピークA〜Cの比は34.1:45.7:20.3であった。D(溶出分留24〜50であり、その部分は図4にて示されている)と名付けられた小さなピークの領域も観測された。
【0065】
PQ3のクロマトグラム(図5)は、1つの主たるピーク(溶出体積18〜21)のみを示し、これはCVT−E002の低い分子量分留物に合致する。
【0066】
PQ2の分留物A、B、C、およびDは、集められ、フリーズドライされる。乾燥された粉末は、それぞれPQ2A、PQ2B、PQ2C、およびPQ2Dと新たに命名される。また、分留物AおよびBは混合され、混合された分留物はPQ223(そのクロマトグラムは図6に示される)と呼ばれた。PQ2A、PQ2B、PQ2C、およびPQ2Dのマイトジェン活性がテストされ、分留物PQ223、PQ2およびCVT−E002の活性と比較された。結果は以下の表に示される。
【0067】
【表3】
【0068】
結果が示すように、有効性の序列はPQ223>PQ2>CVT−E002である。
【実施例5】
【0069】
(実施例5:CVT−E002、PQ1、PQ2、PQ3、およびPQ223の炭水化物総量およびタンパク質組成の化学的決定)
各分留物CVT−E002、PQ1、PQ2、PQ3、およびPQ223の3バッチが得られ、各分留物に対する炭水化物およびタンパク質の総量の組成が測定された。炭水化物の総量の含有量は、従来より知られたフェノール−サルファリック(sulfuric)法によって決定された。タンパク質の含有量は、従来より知られたLowry法によって検査され、ウシ血清アルブミンが標準として用いられた。結果は以下の表に示される。
【0070】
【表4】
【実施例6】
【0071】
(実施例6:CVT−E002、PQ1、PQ2、PQ3、およびPQ223のモノサッカライド組成の化学的決定)
炭水化物の総量の含有量は主にポリサッカライドとオリゴサッカライドの量を表しているので、所定の分留物に対するモノサッカライドのモル比もまた決定された。
【0072】
CVT−E002、PQ1、PQ2、PQ3、およびPQ223の分留物の組は、その構造単位であるモノサッカライドを遊離させるため、酸触媒加水分解された。遊離したモノサッカライドは、MPP(3−メチル−1−フェニル−2−ピラゾリン−5−オン)と誘導体を合成された後、その定性的および定量的解析がHPLCにより行われた。詳細な方法が以下の通りである:2NのHCl中の試料溶液が95〜100℃に6時間加熱された。混合物はNaOH溶液で中和された。内部標準が加えられた。MeOHおよびNaOH水溶液(0.3N)がこの混合物に加えられた。その後、これが50〜60℃で8時間(あるいは室温で1週間)攪拌された。混合物は水で(1:10)に希釈され、HPLCで解析された。MPPで誘導体が合成された試料は、C−18カラムで、0.1Mもリン酸バッファ(pH7)中の18%アセトニトリル溶液からなる溶離液を用い、フローレート1ml/分、245nmでのUV検出にて、解析された。下記の表は、CVT−E002、PQ1、PQ2、およびPQ223のモノサッカライドのモル比を示す。グルクロン酸のトレース量は、あるCVT−E002、PQ1、およびPQ2の試料中に見つかった。
【0073】
【表5】
【0074】
結果が示すように、グルコースの含有量の序列は次の通りである:PQ1>CVT−E002>PQ2>PQ3;また、ガラクツロン酸の含有量は次の通りである:PQ2>PQ223>CVT−E002>PQ1。
【実施例7】
【0075】
(実施例7:薬用人参分留物の免疫調節)
1.マクロファージ活性
CVT−E002、PQ2、G2、およびPQ223マウスのマクロファージに対する効果は、細胞性反応を優先的にマウントするC57B1/6マウスと、抗体性反応を優先的にマウントするBalb/cからの腹膜滲出マクロファージを用いて、インビトロ(in vitro)に研究された。マクロファージが100μg/mlのテスト試料で刺激された後、IL−1、IL−6、およびTNF−αの生成が測定された。その方法は下記の通りである。
【0076】
A.腹膜滲出細胞および細胞上澄みからのマクロファージの調製
1)3%チオグリコレート1mlがマウスの腹膜腔に注入され、3日後に腹膜腔から腹膜滲出マクロファージが採取された。
2)細胞懸濁液がHanksバッファとともに、4℃にて1100rpmで5分間遠心分離機にかけて2回洗浄された。
3)マクロファージがRPMI−10% FBS媒体中に懸濁された。
4)細胞が計測され、RPMI−10% FBS媒体で106/mlの最終濃度に希釈された。
5)マクロファージが、37℃、10×106/10ml RPMI−10%FBSにて2時間培養された。上澄みが廃棄され、沈殿したマクロファージが10mlのPBSで2回洗浄された。
6)マクロファージが採取され、RPMI−10% FBSに再び懸濁された。
7)マクロファージ(5×105)が、10mg/mlのLPSあるいは100mg/mlのテスト試料とともに、24あるいは48時間培養された。その後で、バイオアッセイ(IL−1、IL−6、およびTNF−α)のために、上澄みが集められ、濾過滅菌された。実験は3重に行われた。
【0077】
B.上澄み中のIL−1の決定
NOB1細胞系は、IL−1に応答して活性(IL−2)を刺激するCTLL成長を生成するEL4細胞系のTK変異種である。NOB1細胞は3HTdRを組み込まないので、IL−1への応答はCTLL系による取り込みの点から測定できる。手順は下記に示す通りである。
1)96のウェルを持つ底部が平坦なマイクロタイタープレートの各ウェルにおいて、104個のCTLL細胞が、IMDM中の5%FBS溶液200μl中で、4×104個のNOB1細胞と混合された。IL−1標準の4、8、16、または32倍、あるいは連続的な希釈液である一続きに希釈されたIL−1テスト試料が加えられた。各ウェルの最終的な体積は200μlであった。各試料は3重に調製された。
2)プレートが、37℃、加湿された5%CO2の培養器において、24時間の間培養された。
3)[3H]−チミジンが培養の最後の5時間の間加えられた。
4)細胞が採取され、液体シンチレーション計測により[3H]−チミジン組み込みが測定された。
5)IL−1濃度が算出された。結果は下記の表に示される。
【0078】
【表6】
【0079】
結果は、CVT−E002、PQ2、G2、およびPQ223は、C57B1/6マウスからのマクロファージを明らかに刺激してIL−1を生成したことを示す。全てのテスト試料はIL−1生成を刺激する同様な能力を示した。
【0080】
C.IL−6の決定
マウスのB細胞ハイブリドーマ細胞系であるB9の増殖はIL−6に依存する。B9細胞は、濃度を減らしているテスト試料を含む一連のマイクロウェル中で培養された。手順は下記に示す通りであった。
1)貯蔵培養フラスコから移した一部試料中のB9細胞の数が、それらの成長の対数期において計測された。
2)細胞は、180xg、4℃の卓上遠心分離機にて5分間遠心分離され、ペレットが、10mlの完全なRPMI−10媒体中に再懸濁された。この手順が2回繰り返され、細胞は、完全なRPMI−10媒体中に、2×103個の細胞/mlで再懸濁された。
3)洗浄された細胞(2×103個の細胞/ml)の100μlが、96のウェルを持つマイクロタイタープレートの各ウェルに加えられた。
4)テスト試料の2倍連続希釈液100μlが、各ウェルに加えられ、プレートの2または3列がIL−6標準のために残しておいた。
5)プレートが、加湿された、37℃、5%CO2の培養器で72時間培養された。
6)[3H]−チミジンが加えられ、プレートが37℃で4時間培養された。
7)細胞が採取され、液体シンチレーションカウンタを用いて、[3H]−チミジンの組み込みが決められた。
8)IL−6濃度が算出された。結果は下記の表に示される。
【0081】
【表7】
【0082】
CVT−E002、PQ2、およびPQ223は、C57B1/6およびBalb/cからのマクロファージの上澄み中でのIL−6の生成を増加させた。能力は、PQ223>PQ2>CVT−E002である。
【0083】
D.TNF−αの測定
下記のプロトコルは、細胞培養物から得られた上澄み中のTNF活性を定量するため、TNFに感度があり、アクチノマイシンD処理されたマウスのL929繊維芽細胞を採用した。1)IMDM−5%FSC 50μl中の4×104個のL929細胞が各ウェルに加えられた。
2)テスト試料50μlが各列の2番目のウェル(カラム2)に加えられた。ウェル2の内容物を静かに混合させることで2倍連続希釈液が形成され、ウェル2からウェル3に50μl移された。ウェル3の内容物は静かに混合され、ウェル3から50μlがウェル4に移された。この手順がウェル12まで続けられた。最後に、カラム12の全てのウェルの内容物が静かに混合され、各ウェルから50μlが廃棄された。この点で、全てのウェルは50μlを含んだ。
3)アクチノマイシンD溶液50μlが各ウェルに加えられた(2μl ml)。
4)プレートが、37℃、5%CO2の空気中で24時間培養された。
5)ナチュラルレッド5μlが各ウェルに加えられ、プレートが2.5時間培養された。
6)各ウェルの上澄みが直ちに、注意深くすすがれ、移されて、プレートが200μlのPBSで2回洗浄された。
7)PBSが吸い出され、0.05MのNaH2PO4中50%エタノール溶液100μlが各ウェルに加えられ、室温で5分間攪拌された。
8)マイクロタイタープレートリーダにより、570nmにける吸光度において、各ウェルが直ちに読まれた。
9)TNF−α濃度が算出された。結果は下記の表に示される。
【0084】
【表8】
【0085】
TNF−αの生成は、Balb/cあるいはC57B1/6細胞系からのマクロファージの上澄みにおいてCVT−E002およびPQ223により誘発された。Balb/cマウスからのマクロファージの上澄みにおけるTNF−αは、明らかにG2により刺激を受けた。PQ2はマクロファージによるTNF−α生成に刺激を与えないようであった。
【0086】
2.血清免疫グロブリン生成
血清免疫グロブリン生成(トータルIgG)は、水を与えられたマウスに比べて、CVT−E002を与えられたマウスにおいては21%、PQ2 を与えられたマウスにおいては26%、PQ223を与えられたマウスにおいては31%、増加した。能力は、PQ223>PQ2>CVT−E002である。結果は表9に示される。
【0087】
【表10】
【0088】
3.PHAあるいはLPSなどのよく知られたマイトジェンは、それらが刺激する細胞の種類について特異性を示す。脾臓細胞のどの特定の種類が薬用人参分留物により刺激を受けるかを知ることは重要である。従って、われわれは、アフィニティカラムによってT細胞を分離し、T細胞を減らすことによって、B細胞を濃縮した。
【0089】
A.T細胞濃縮
赤い血液細胞を除去するため、脾臓細胞の単一細胞懸濁液がリンパ球−M(Cedarlane Labs)上に積層された。その後、マウスの免疫グロブリンで被覆されたビーズへの吸着によりB細胞を取り除くアフィニティクリマトグラフィーカラム(Biotex Co. Ltd.,Edmonton,Alberta)にリンパ球を通過させた。溶出液は、濃縮された集団のT細胞からなっていた。他の細胞種に対するT細胞の割合の決定は、抗thy1.2モノクローナル抗体と補体処理を用いた生存能力テストによってなされた。
【0090】
B.B細胞濃縮
脾臓細胞集団からの赤い血液細胞の除去の後で、リンパ球は、モノクローナル抗thy1.2抗体とともに37℃で1時間培養された。低毒のウサギの補体が加えられ、4℃で30分培養された。この手順はT細胞の細胞懸濁を減らし、B細胞集団を濃縮する結果となる。
【0091】
C.PQ223との培養
BまたはT細胞はPQ223の適用量を変えながら培養され、応答がトリチウム化されたチミジンの組み込みによって測定された。図7および8は、それぞれ、相対的に精製されたBおよびT細胞に対するPQ223の増殖効果を示す。対照グループは、B細胞に特異的なマイトジェンであるLPS、およびT細胞に特異的なマイトジェンであるPHAで処理された細胞を含んだ。PQ223はB細胞に特異的であることが発見された(図9参照)。
【0092】
4.インビトロ(in vitro)での抗体生成に対するPQ223の効果
脾臓細胞は、適用量を変えたPQ223の存在下において72時間培養された。対照培養物は、25μgのLPSが加えられたグループと、マイトジェンが加えられなかったもう一つのグループから構成された。上澄みは、エンザイム・リンクト・イムノソーベント・アッセイ(Enzyme Linked Immunosorbent Assay(ELISA))を用いて、可溶性の免疫グロブリンの存在がテストされた。可溶性の免疫グロブリンを含む上澄みは、0.1Mのトリスバッファ(pH9)中に連続的に希釈された。マイクロタイタープレートが、この上澄みで被覆され、4℃で終夜培養された。プレートは、PBS Tweenで3回洗浄され、その後、抗体−酵素複合Goat F(ab’)2抗マウスIgセイヨウワサビペルオキシダーゼ(Tago Inc.,Burlingame California)100μlが1:1000の希釈率で加えられた。プレートは室温で1時間培養された。プレートは再びPBSで3回洗浄され、その後、ABTSペルオキシダーゼ基質(Kirkegaard and Perry Lab Inc.)が各ウェルに加えられた。1時間後、405nmの波長での吸光度がフローマルチスキャン(Flow Multiscan)ELISAリーダによって測定された。
【0093】
図10は、50μg/mlのPQ223が、LPS25μgの場合と同程度に、免疫グロブリンの生成物を刺激したことを示す。高濃度のPQ223、500μg/mlは、より高い水準の抗体生成を誘い出さなかった。この適用量応答曲線は、異種の脾臓細胞集団および精製されたB細胞の増殖応答と一致する。これらの結果は、4つの実験に基づいた。
【0094】
5.インビボ(in vivo)抗体プラーク形成細胞
3つの濃度のPQ223がそれぞれ3匹のマウスからなる3グループに静脈注射された。対照グループは、平衡塩溶液が注射された。PQ223での静脈処理の7日後に、マウスはHBSS中の10%SRBC懸濁液0.2mlにより免疫性を与えられた。免疫性を与えられてから5日後に、マウスは犠牲となり、脾臓からの単一細胞懸濁液が調製され、1mlあたり4×106個の細胞に調節された。この細胞懸濁液の0.1mlの一部試料がテンジクネズミ補体の10%SRBC溶液0.3mlとRPMI媒体0.5mlと混合された。この混合物の70μlがカニンガム(Cunningham)チャンバ−に移された。チャンバーはパラプラスト(paraplast)ワックスで封止され、プラーク形成細胞を計測する前に、37℃で1時間培養された。全てのPFCレベルは100万個の脾臓細胞あたりとして表現される。図11は、三種の適用量が全て、化合物が注射されたマウス内で、明らかに抗体生成を増強したことを示す。
【産業上の利用可能性】
【0095】
まとめると、本研究の結果は、マクロファージが、IL−1、IL−6、およびTNF−αなどのサイトカインを生成するのを本発明の薬用人参分留物が活性化することを示す。また、それらは、リンパ球、特にBリンパ球の増殖および抗体生成も活性化する。体液が媒介する免疫系の全体が刺激され、感染に対する予防効果を示した。報告されたTNF−α生成の活動は、抗ウイルスおよび抗腫瘍の恩恵を含む。TNF−αは、また、種々の寄生感染の治療のためにもなると報告されている。
【0096】
以下の本発明の背景において言及された引用文献は、ここに参照により本発明に組み込まれる。
1.Tomodaら、Biol.Pharm.Bull.,16,22−5(1993).
2.Tomodaら、Biol.Pharm.Bull.,17,1287−91(1994).
3.Tomodaら、Biol.Pharm.Bull.,16,1087−90(1993).
4.Gaoら、Planta Medica,55,9−12(1989).
5.Gaoら、Carbohydr.Res.,181,175−87(1988).
6.Kiyoharaら、Carbohydr.Res.,263,89−101(1994).
7.Shinら、Carbohydr.Res.,300,239−49(1997).
8.Yamadaら、Phytotherapy Res.,9,264−9(1995).
9.Konnoら、Planta Medica,50,434−6(1984).
10.Hikinoら、未発表.
11.Oshimaら、J.Ethnopharmacology,14,255−9(1985).
12.Konnnoら、J.Crude Drug Res.,25,53−5(1987).
13.Konnnoら、J.Ethnopharmacology,14,69−74(1985).
14.Oshimaら、J.Natural Products,50,188−990(1987).
15.Leeら、Anticancer Res.,17,323−32(1997).
16.Kimら、Planta Medica,64,110−5(1998).
17.Miaoら、Shengwu Huaxue ZaZhi,9,610−4(1993).
18.Maら、Baiqiuen Yike Daxue Xuebao,23,236−8(1997).
19.Zhuら、Zhongguo Yaolixue Tongbao,13,76−8(1997).
【特許請求の範囲】
【請求項1】
薬用人参分留物PQ2を調製する方法であって、当該方法は、
(a)アメリカの薬用人参をアルコールを含む第1の溶媒に混合し、得られた溶液を約80〜100℃の温度で約2〜4時間の間加熱し、第1の薬用人参溶液を生成すること;
(b)その後で、上記第1の薬用人参溶液を分離して、アルコール/薬用人参溶液と第1の薬用人参残渣を生成すること;
(c)その後で、上記第1の薬用人参残渣を水に混合し、得られた溶液を約80〜100℃の温度で約2〜4時間の間加熱し、薬用人参残渣溶液を生成すること;
(d)その後で、上記薬用人参残渣溶液を分離して、第2の薬用人参残渣と、第1の薬用人参抽出物を含む第1の抽出物水溶液を生成すること;
(e)少なくとも上記第1の薬用人参抽出物の一部を含む第2の抽出物水溶液を提供すること、ここで、上記第2の抽出物水溶液中の上記第1の薬用人参抽出物の水に対する比率は約1:18〜1:22である;
(f)その後で、アルコールを含む第2の溶媒に上記第2の抽出物水溶液を混合して、第1の沈殿と第1の上澄みを生成すること、ここで、上記第2の溶媒の水に対する比率は約1:1〜3:5である;
(g)その後で、アルコールを含む第3の溶媒にステップ(f)において生成された上記第1の上澄みを混合して、第2の沈殿と第2の上澄みを生成すること、ここで、上記第3の溶媒の第1の上澄みに対する比率は約3:2〜3:1である;および
(h)上記第2の沈殿物を単離して薬用人参分留物PQ2を生成すること
を具備する薬用人参分留物PQ2を調製する方法。
【請求項2】
上記第1の溶媒、第2の溶媒、および第3の溶媒のそれぞれにおける上記アルコールが、他と関係なく、飽和あるいは不飽和のC1〜C6アルコールを含む
請求項1記載の方法。
【請求項3】
上記第1の溶媒、第2の溶媒、および第3の溶媒のそれぞれにおける上記アルコールが、他と関係なく、エタノールあるいはメタノールを含む
請求項1記載の方法。
【請求項4】
ステップ(e)において、上記第2の抽出物水溶液が、少なくとも上記第1の抽出物水溶液の一部を含む
請求項1記載の方法。
【請求項5】
ステップ(a)において、上記得られた溶液を約3時間の間加熱する
請求項1記載の方法。
【請求項6】
ステップ(c)において、上記得られた溶液を約3時間の間加熱する
請求項1記載の方法。
【請求項7】
ステップ(e)において、上記第1の薬用人参抽出物の水に対する比率が約1:20である
請求項1記載の方法。
【請求項8】
ステップ(f)において、上記第2の溶媒の水に対する比率が約3:4である
請求項1記載の方法。
【請求項9】
ステップ(g)において、上記第3の溶媒の第1の上澄みに対する比率が約2:1である
請求項1記載の方法。
【請求項10】
ステップ(a)において、上記第1の溶媒および上記薬用人参が、薬用人参1グラムあたり約7〜9mlの第1の溶媒の比率で混合される
請求項1記載の方法。
【請求項11】
ステップ(a)において、上記第1の溶媒および上記薬用人参が、薬用人参1グラムあたり約8mlの第1の溶媒の比率で混合される
請求項1記載の方法。
【請求項12】
ステップ(c)において、上記水および上記第1の薬用人参残渣が、薬用人参残渣1グラムあたり約7〜9mlの水の比率で混合される
請求項1記載の方法。
【請求項13】
ステップ(c)において、上記水および上記第1の薬用人参残渣が、薬用人参残渣1グラムあたり約8mlの水の比率で混合される
請求項1記載の方法。
【請求項14】
請求項1〜13のいずれかに記載の方法によって生成された、薬用人参分留物PQ2。
【請求項15】
薬用人参分留物PQ223を調製する方法であって、当該方法は、
(a)請求項1〜13のいずれかに記載の方法によって生成された、薬用人参分留物PQ2を提供すること;
(b)上記薬用人参分留物PQ2を分留して第1溶出分留物および第2溶出分留物を生成すること、ここで、第1溶出分留物は溶出体積が35および50mlの間に観測される炭水化物ピークに合致し、第2溶出分留物は溶出体積が50および85mlの間に観測される炭水化物ピークに合致し、これらは下記の材料を用いたゲル濾過クロマトグラフィーにより決定される:
(1)アリルデキストランおよびN,N'−メチレンビスアクリルアミドからなる架橋した球形の共重合体のマトリックスを含み、16×600mmのベッド寸法および120mlのベッド体積と、球状のタンパク質に対して5000〜250,000、デキストランに対して1000〜80,000の分留範囲(MW)を有するクロマトグラフィーカラム、および
(2)0.1N HClおよびpHが7.0の0.3M NaClを含むトリス−HClの溶出バッファ;および
(c)上記第1溶出分留物および上記第2溶出分留物を単離し、かつ混合して、薬用人参分留物PQ223を生成すること
を具備する薬用人参分留物PQ223を調製する方法。
【請求項16】
薬用人参分留物PQ2がゲル濾過クロマトグラフィーを用いて分留される
請求項15記載の方法。
【請求項17】
請求項15または16に記載の方法によって生成された、薬用人参分留物PQ223。
【請求項18】
約2〜6モル%のラムノース、約41〜49モル%のガラクツロン酸、約12〜18モル%のグルコース、約16〜22モル%のガラクトース、および約12〜19モル%のアラビノースを含む炭水化物内容物を有する
薬用人参分留物。
【請求項19】
上記炭水化物内容物が、約3〜5モル%のラムノース、約43〜47モル%のガラクツロン酸、約14〜16モル%のグルコース、約18〜20モル%のガラクトース、および約14〜17モル%のアラビノースを含む
請求項18記載の薬用人参分留物。
【請求項20】
上記炭水化物内容物が、約4モル%のラムノース、約45モル%のガラクツロン酸、約15モル%のグルコース、約19モル%のガラクトース、および約15モル%のアラビノースを含む
請求項18記載の薬用人参分留物。
【請求項21】
約3〜8モル%のラムノース、約36〜44モル%のガラクツロン酸、約2〜7モル%のグルコース、約25〜33モル%のガラクトース、および約17〜25モル%のアラビノースを含む炭水化物内容物を有する
薬用人参分留物。
【請求項22】
上記炭水化物内容物が、約4〜7モル%のラムノース、約37〜42モル%のガラクツロン酸、約3〜6モル%のグルコース、約27〜32モル%のガラクトース、および約19〜24モル%のアラビノースを含む
請求項21記載の薬用人参分留物。
【請求項23】
上記炭水化物内容物が、約5モル%のラムノース、約39モル%のガラクツロン酸、約4モル%のグルコース、約29モル%のガラクトース、および約21モル%のアラビノースを含む
請求項21記載の薬用人参分留物。
【請求項24】
約0.5〜5モル%のラムノース、約11〜22モル%のガラクツロン酸、約40〜60モル%のグルコース、約10〜19モル%のガラクトース、および約11〜19モル%のアラビノースを含む炭水化物内容物を有する
薬用人参分留物。
【請求項25】
上記炭水化物内容物が、約1〜3モル%のラムノース、約13〜20モル%のガラクツロン酸、約42〜57モル%のグルコース、約12〜17モル%のガラクトース、および約13〜17モル%のアラビノースを含む
請求項24記載の薬用人参分留物。
【請求項26】
請求項14、17、または18〜25のいずれかに記載の薬用人参分留物を有し、薬学的に許容なキャリアに混合した
薬学的組成物。
【請求項27】
請求項14、17、または18〜25のいずれかに記載の薬用人参分留物の、低免疫で特徴付けられる健康状態を治療するのに適した薬学的組成物の調製において、単独でのあるいは他の薬剤との組み合わせての使用方法。
【請求項28】
上記健康状態が、通常の風邪、インフルエンザ、慢性疲労症候群、AIDS、癌からなるグループから選択される
請求項27記載の使用方法。
【請求項29】
IL−1、IL−6、および/またはTNF−αの細胞中の生成を刺激するための請求項14、17、または18〜25のいずれかに記載の薬用人参分留物の使用方法。
【請求項30】
インビトロ(in vitro)またはインビボ(in vivo)での免疫グロブリンの生成を刺激するための請求項14、17、または18〜25のいずれかに記載の薬用人参分留物の使用方法。
【請求項31】
Bリンパ球の増殖とそれらからの抗体の生成を活性化するための請求項14、17、または18〜25のいずれかに記載の薬用人参分留物の使用方法。
【請求項32】
健康状態を治療するのに効果的な量の請求項14、17、または18〜25のいずれかに記載の薬用人参分留物を患者に投与することを含む、それを必要としている患者の低免疫で特徴付けられる健康状態の治療方法。
【請求項33】
上記健康状態が、通常の風邪、インフルエンザ、慢性疲労症候群、AIDS、癌からなるグループから選択される 請求項32記載の方法。
【請求項1】
薬用人参分留物PQ2を調製する方法であって、当該方法は、
(a)アメリカの薬用人参をアルコールを含む第1の溶媒に混合し、得られた溶液を約80〜100℃の温度で約2〜4時間の間加熱し、第1の薬用人参溶液を生成すること;
(b)その後で、上記第1の薬用人参溶液を分離して、アルコール/薬用人参溶液と第1の薬用人参残渣を生成すること;
(c)その後で、上記第1の薬用人参残渣を水に混合し、得られた溶液を約80〜100℃の温度で約2〜4時間の間加熱し、薬用人参残渣溶液を生成すること;
(d)その後で、上記薬用人参残渣溶液を分離して、第2の薬用人参残渣と、第1の薬用人参抽出物を含む第1の抽出物水溶液を生成すること;
(e)少なくとも上記第1の薬用人参抽出物の一部を含む第2の抽出物水溶液を提供すること、ここで、上記第2の抽出物水溶液中の上記第1の薬用人参抽出物の水に対する比率は約1:18〜1:22である;
(f)その後で、アルコールを含む第2の溶媒に上記第2の抽出物水溶液を混合して、第1の沈殿と第1の上澄みを生成すること、ここで、上記第2の溶媒の水に対する比率は約1:1〜3:5である;
(g)その後で、アルコールを含む第3の溶媒にステップ(f)において生成された上記第1の上澄みを混合して、第2の沈殿と第2の上澄みを生成すること、ここで、上記第3の溶媒の第1の上澄みに対する比率は約3:2〜3:1である;および
(h)上記第2の沈殿物を単離して薬用人参分留物PQ2を生成すること
を具備する薬用人参分留物PQ2を調製する方法。
【請求項2】
上記第1の溶媒、第2の溶媒、および第3の溶媒のそれぞれにおける上記アルコールが、他と関係なく、飽和あるいは不飽和のC1〜C6アルコールを含む
請求項1記載の方法。
【請求項3】
上記第1の溶媒、第2の溶媒、および第3の溶媒のそれぞれにおける上記アルコールが、他と関係なく、エタノールあるいはメタノールを含む
請求項1記載の方法。
【請求項4】
ステップ(e)において、上記第2の抽出物水溶液が、少なくとも上記第1の抽出物水溶液の一部を含む
請求項1記載の方法。
【請求項5】
ステップ(a)において、上記得られた溶液を約3時間の間加熱する
請求項1記載の方法。
【請求項6】
ステップ(c)において、上記得られた溶液を約3時間の間加熱する
請求項1記載の方法。
【請求項7】
ステップ(e)において、上記第1の薬用人参抽出物の水に対する比率が約1:20である
請求項1記載の方法。
【請求項8】
ステップ(f)において、上記第2の溶媒の水に対する比率が約3:4である
請求項1記載の方法。
【請求項9】
ステップ(g)において、上記第3の溶媒の第1の上澄みに対する比率が約2:1である
請求項1記載の方法。
【請求項10】
ステップ(a)において、上記第1の溶媒および上記薬用人参が、薬用人参1グラムあたり約7〜9mlの第1の溶媒の比率で混合される
請求項1記載の方法。
【請求項11】
ステップ(a)において、上記第1の溶媒および上記薬用人参が、薬用人参1グラムあたり約8mlの第1の溶媒の比率で混合される
請求項1記載の方法。
【請求項12】
ステップ(c)において、上記水および上記第1の薬用人参残渣が、薬用人参残渣1グラムあたり約7〜9mlの水の比率で混合される
請求項1記載の方法。
【請求項13】
ステップ(c)において、上記水および上記第1の薬用人参残渣が、薬用人参残渣1グラムあたり約8mlの水の比率で混合される
請求項1記載の方法。
【請求項14】
請求項1〜13のいずれかに記載の方法によって生成された、薬用人参分留物PQ2。
【請求項15】
薬用人参分留物PQ223を調製する方法であって、当該方法は、
(a)請求項1〜13のいずれかに記載の方法によって生成された、薬用人参分留物PQ2を提供すること;
(b)上記薬用人参分留物PQ2を分留して第1溶出分留物および第2溶出分留物を生成すること、ここで、第1溶出分留物は溶出体積が35および50mlの間に観測される炭水化物ピークに合致し、第2溶出分留物は溶出体積が50および85mlの間に観測される炭水化物ピークに合致し、これらは下記の材料を用いたゲル濾過クロマトグラフィーにより決定される:
(1)アリルデキストランおよびN,N'−メチレンビスアクリルアミドからなる架橋した球形の共重合体のマトリックスを含み、16×600mmのベッド寸法および120mlのベッド体積と、球状のタンパク質に対して5000〜250,000、デキストランに対して1000〜80,000の分留範囲(MW)を有するクロマトグラフィーカラム、および
(2)0.1N HClおよびpHが7.0の0.3M NaClを含むトリス−HClの溶出バッファ;および
(c)上記第1溶出分留物および上記第2溶出分留物を単離し、かつ混合して、薬用人参分留物PQ223を生成すること
を具備する薬用人参分留物PQ223を調製する方法。
【請求項16】
薬用人参分留物PQ2がゲル濾過クロマトグラフィーを用いて分留される
請求項15記載の方法。
【請求項17】
請求項15または16に記載の方法によって生成された、薬用人参分留物PQ223。
【請求項18】
約2〜6モル%のラムノース、約41〜49モル%のガラクツロン酸、約12〜18モル%のグルコース、約16〜22モル%のガラクトース、および約12〜19モル%のアラビノースを含む炭水化物内容物を有する
薬用人参分留物。
【請求項19】
上記炭水化物内容物が、約3〜5モル%のラムノース、約43〜47モル%のガラクツロン酸、約14〜16モル%のグルコース、約18〜20モル%のガラクトース、および約14〜17モル%のアラビノースを含む
請求項18記載の薬用人参分留物。
【請求項20】
上記炭水化物内容物が、約4モル%のラムノース、約45モル%のガラクツロン酸、約15モル%のグルコース、約19モル%のガラクトース、および約15モル%のアラビノースを含む
請求項18記載の薬用人参分留物。
【請求項21】
約3〜8モル%のラムノース、約36〜44モル%のガラクツロン酸、約2〜7モル%のグルコース、約25〜33モル%のガラクトース、および約17〜25モル%のアラビノースを含む炭水化物内容物を有する
薬用人参分留物。
【請求項22】
上記炭水化物内容物が、約4〜7モル%のラムノース、約37〜42モル%のガラクツロン酸、約3〜6モル%のグルコース、約27〜32モル%のガラクトース、および約19〜24モル%のアラビノースを含む
請求項21記載の薬用人参分留物。
【請求項23】
上記炭水化物内容物が、約5モル%のラムノース、約39モル%のガラクツロン酸、約4モル%のグルコース、約29モル%のガラクトース、および約21モル%のアラビノースを含む
請求項21記載の薬用人参分留物。
【請求項24】
約0.5〜5モル%のラムノース、約11〜22モル%のガラクツロン酸、約40〜60モル%のグルコース、約10〜19モル%のガラクトース、および約11〜19モル%のアラビノースを含む炭水化物内容物を有する
薬用人参分留物。
【請求項25】
上記炭水化物内容物が、約1〜3モル%のラムノース、約13〜20モル%のガラクツロン酸、約42〜57モル%のグルコース、約12〜17モル%のガラクトース、および約13〜17モル%のアラビノースを含む
請求項24記載の薬用人参分留物。
【請求項26】
請求項14、17、または18〜25のいずれかに記載の薬用人参分留物を有し、薬学的に許容なキャリアに混合した
薬学的組成物。
【請求項27】
請求項14、17、または18〜25のいずれかに記載の薬用人参分留物の、低免疫で特徴付けられる健康状態を治療するのに適した薬学的組成物の調製において、単独でのあるいは他の薬剤との組み合わせての使用方法。
【請求項28】
上記健康状態が、通常の風邪、インフルエンザ、慢性疲労症候群、AIDS、癌からなるグループから選択される
請求項27記載の使用方法。
【請求項29】
IL−1、IL−6、および/またはTNF−αの細胞中の生成を刺激するための請求項14、17、または18〜25のいずれかに記載の薬用人参分留物の使用方法。
【請求項30】
インビトロ(in vitro)またはインビボ(in vivo)での免疫グロブリンの生成を刺激するための請求項14、17、または18〜25のいずれかに記載の薬用人参分留物の使用方法。
【請求項31】
Bリンパ球の増殖とそれらからの抗体の生成を活性化するための請求項14、17、または18〜25のいずれかに記載の薬用人参分留物の使用方法。
【請求項32】
健康状態を治療するのに効果的な量の請求項14、17、または18〜25のいずれかに記載の薬用人参分留物を患者に投与することを含む、それを必要としている患者の低免疫で特徴付けられる健康状態の治療方法。
【請求項33】
上記健康状態が、通常の風邪、インフルエンザ、慢性疲労症候群、AIDS、癌からなるグループから選択される 請求項32記載の方法。
【図1】

【図2】
【図3】
【図4】
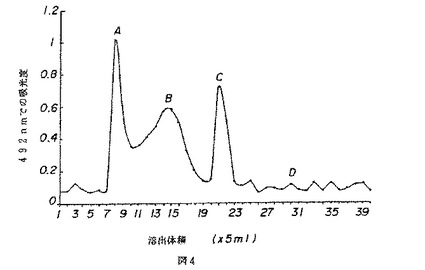
【図5】
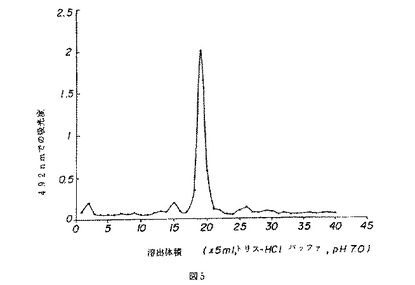
【図6】
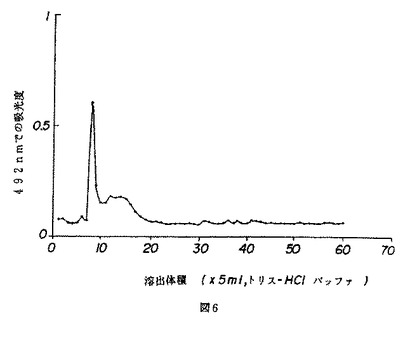
【図7】
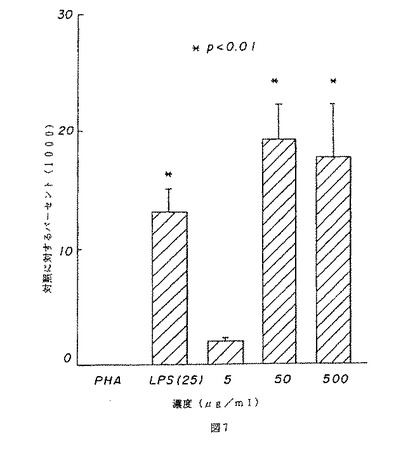
【図8】
【図9】
【図10】
【図11】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【公開番号】特開2009−256387(P2009−256387A)
【公開日】平成21年11月5日(2009.11.5)
【国際特許分類】
【出願番号】特願2009−186027(P2009−186027)
【出願日】平成21年8月10日(2009.8.10)
【分割の表示】特願2000−538705(P2000−538705)の分割
【原出願日】平成10年12月11日(1998.12.11)
【出願人】(509031408)エフエックス ライフ サイエンシズ アクチェンゲゼルシャフト (2)
【Fターム(参考)】
【公開日】平成21年11月5日(2009.11.5)
【国際特許分類】
【出願日】平成21年8月10日(2009.8.10)
【分割の表示】特願2000−538705(P2000−538705)の分割
【原出願日】平成10年12月11日(1998.12.11)
【出願人】(509031408)エフエックス ライフ サイエンシズ アクチェンゲゼルシャフト (2)
【Fターム(参考)】
[ Back to top ]