
医薬の生物活性を改善するための方法
本発明は、一般式(I)または(II)、式中R1およびR2は水素、アルキルラジカルまたはアリールラジカルである、を有する部分構造を含むプロドラッグに関する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、薬剤の生物活性を改善するための方法に関する。
【背景技術】
【0002】
経口投与後の薬剤の治療効果に関する要件は、消化管からのそれらの吸収である。そのような効果のうち最も重要なメカニズムは、受動拡散である。受動拡散による吸収の程度は、とりわけ親油性に依存する。
【0003】
薬による多くの疾患の処置に伴う別の問題は、血液脳関門を通過する必要性である。血液脳関門は、脳内の物質の吸収に関する効果的なバリアを構成している。それは選択的な取り込みを保証し、物質が通過することを防ぐ。また、血液脳関門は、物理的なだけでなく、酵素的なバリアとしても機能する。様々なプロセスが、脳への物質の通過に関与している。他の適応と比較して、中枢神経系(CNS)でその効果を現す薬剤はわずかに販売されているにすぎない。これらのうち、多くは拡散によって中枢神経系に到達する。このような方法で、てんかん、慢性痛やうつ病などの疾患が処置される。他の重篤な機能障害、例えば脳腫瘍または筋萎縮性側索硬化症などは、現在この方法で処置するのは非常に困難である。
【0004】
受動拡散によって生体膜を突破することができるためには、物質は、親油性で、500Daより小さい分子量を有しているべきであり、また非荷電状態で存在すべきである。特にアミノ酸または糖などの小さい高極性分子を吸収するため、種々のトランスポーターシステム、例えばヌクレオシドトランスポーター、有機アニオンまたはカチオンの流入および流出トランスポーター、グルコーストランスポーター、ペプチドトランスポーターならびにアミノ酸トランスポーターなどが、バリア機能を有する生体膜(消化管、血液脳関門)で発現している。
【0005】
このような理由から、様々なプロドラッグシステムが、薬物動態学的特性を改善するために採用されている。プロドラッグは、薬理学的に不活性または実質的に不活性であり、かつそれが生体内で代謝されるまで、活性代謝物に変換されない薬剤である。
【0006】
N−ヒドロキシアミジン(アミドキシム)およびN−ヒドロキシグアニジンは、アミジン[Clement、B. Methoden zur Behandlung und Prophylaxe der Pneumocystis carinii Pneumonie (PCP) und anderen Erkrankungen sowie Verbindungen und Formulierungen zum Gebrauch bei besagten Methoden.(DE 4321444)][ニューモシスチス・カリニ肺炎(PCP)および他の疾患の処置および予防のための方法ならびにその方法に使用するための化合物および製剤]およびグアニジンの経口バイオアベイラビリティを高めるための既知のプロドラッグの原理を示す。アミノ基およびイミノ基の窒素原子は、アミジンおよびグアニジンの塩において、メソメリー平衡(mesomeric equilibrium)で存在し、そのコンセプトは、両方の窒素原子に採用することができる。
【0007】
活性代謝物への変換は、根底にあるプロドラッグの概念に応じて、種々の酵素システムを介して行われる。あらゆる生物(live)において実質的に生じる酵素システムはシトクロームP450(CYP450)であり、とりわけ、以下の反応:
芳香族および脂肪族化合物のN−酸化、S−酸化、N−脱アルキル、O−脱アルキル、S−脱アルキル、脱アミノ化、脱ハロゲン化およびヒドロキシル化
を触媒する。
【0008】
CYP450酵素システムの多様性が意味するところは、種々の基質と薬剤が変換時にシステムと競合するということである。これは、相互作用、相反的効果および望ましくない相互の影響をもたらす。このような理由からプロドラッグの開発の際には、CYP450−非依存性生物活性化が望まれる。
したがって、本発明の目的は、シトクロームP450(CYP450)酵素に依存しない生物活性化の経路を採用したプロドラッグシステムを提供することである。この目的は、特許請求の範囲に記載された事項によって達成される。従属クレームは、発明の有利な態様を提供する。
【発明の概要】
【0009】
本発明によれば、本発明の目的は、一側面において、一般式(I)または(II)
【化1】
式中、R1およびR2は、水素、アルキルラジカルまたはアリールラジカルである、
を有する部分構造を含むプロドラッグによって達成される。
【0010】
本発明の好ましい態様において、本明細書中で用いられている用語「部分構造」は、それぞれの式で示される構造的要素が物質、好ましくはプロドラッグの式の一部であることを示す。例えば、化合物O−カルボキシメチルベンズアミドキシム(1)は、薬剤ベンズアミジン(pharmaceutical benzamidine)に対応するプロドラッグを構成し、ここで部分構造は式(II)で表される部分構造であり、R1およびR2はそれぞれ水素原子を示す。この部分構造は、ベンゼン環上の置換基であり、それと一緒になって薬剤ベンズアミジンを構成する。
【0011】
本発明の好ましい態様において、本明細書中で用いられている用語「プロドラッグ」は、不活性または薬理学的に実質的に不活性であり、それが生体内で代謝されるまで、薬理学的に活性である薬剤に変換されないような物質を示す。プロドラッグは、実際の活性な薬剤よりも改善された経口バイオアベイラビリティを示すことができるが、示さなくてもよい。代替的に、プロドラッグは、薬剤と比較して改善された溶解性、生物活性化、血液脳関門の横断、物理化学的安定性、低い毒性および/または耐用可能なもしくはそれ以上の快適な風味を示すため、用いることができる。例えば、エリスロマイシンAの2’−エチルスクシナートは、必ずしもエリスロマイシンAの不十分な吸収または溶解性のためではなく、その苦みのために、プロドラッグとして子どもたちに投与されている(Bhadra et al. (2005)、J. Med. Chem.)。
【0012】
本発明のさらなる好ましい態様において、オリジナルのプロドラッグは、ワンステップ反応でプロドラッグから薬剤へと代謝されるのではなく、むしろ複数の反応工程により代謝され、一つの反応工程から得られた各代謝物は、オリジナルのプロドラッグより有利な、一種または二種以上の同一および/または異なる特性を示すことができる。この目的のために、すべての代謝物がプロドラッグ以上の有利な特性を示さなくてもよい。例えば、プロドラッグの第一の代謝産物は、プロドラッグと比較して改善された薬理活性を示すことができ、第一の代謝産物由来の第二の代謝産物は同様にプロドラッグと比較して改善された薬理活性を示すことができ、および第二の代謝産物由来の第三の代謝産物はプロドラッグと比較して改善された血液脳関門の横断および物理的−化学的安定性を示すことができる。
【0013】
本発明の好ましい態様において、本明細書中で用いられている用語「物理的−化学的安定性」は、例えば加水分解などの化学分解がなく、例えば水、バッファーまたは生理食塩水に溶解させるなどの関連する水溶液の形態で貯蔵されるおよび/または用いられる物質、例えばプロドラッグまたは薬剤の能力を示す。本発明のさらなる好ましい態様において、本明細書中で用いられているこの用語は、物質が安定した合成形で合成できることを示す。本発明のさらなる好ましい態様において、本明細書中で用いられているこの用語は、物質の合成中に、後続する合成産物または合成中間産物をより安定な形態で製造可能にするために、単離された関連する合成前駆体が、類似または同一の合成戦略に従って製造した他の物質の類似産物前駆体または中間産物よりも安定であるか、または最初から製造できることを示す。
【0014】
一態様において、本発明の目的はプロドラッグであって、プロドラッグが含む部分構造が、ヒドロキシルアミン、N−オキシド、ニトロン、ジアゼニウムジオラート(NONOat)または類似のN−O−含有一酸化窒素ドナー、ヒドロキサム酸、ヒドロキシ尿素、オキシム、アミドキシム(N−ヒドロキシアミジン)、N−ヒドロキシアミジノヒドラゾンあるいはN−ヒドロキシグアニジンの一部であることを特徴とする、前記プロドラッグによって達成される。
【0015】
薬剤ベンズアミジンのプロドラッグカルボキシメチルベンズアミドキシム(1)の場合において、例えば部分構造は式(II)の部分構造であり、R1およびR2はそれぞれ水素原子であり、およびプロドラッグが含む部分構造はアミドキシム(N−ヒドロキシアミジン)の一部である。
【0016】
一態様において、本発明の目的は、プロドラッグであって、プロドラッグが、一酸化窒素欠乏に関連する疾患の処置のための薬剤である薬剤へと代謝されることを特徴とする、前記プロドラッグによって達成される。
【0017】
一態様において、本発明の目的は、プロドラッグであって、プロドラッグまたは対応する薬剤が、プロテアーゼ阻害剤、DNA−インターカレート化合物およびRNA−インターカレート化合物、ウイルス酵素の阻害剤、ならびにN−メチル−D−アスパラギン酸受容体アンタゴニストからなる群から選択されることを特徴とする、前記プロドラッグによって達成される。
【0018】
本発明の好ましい態様において、用語「高次部分構造」(higher-level partial structure)は、この高次部分構造が一方では式(I)または(II)で表される部分構造を含み、他方では問題の物質の全体構造の一部であると理解するべきである。例えば、薬剤ベンズアミジン(2)のプロドラッグカルボキシメチルベンズアミドキシム(1)の場合は、部分構造の式(IIa)においてR1およびR2が水素原子である、ここで(Ia)で示される高次部分構造、ならびに部分構造の式(II)においてR1およびR2が同様に水素原子である、ここで(Ib)で示される部分構造を含む。
【0019】
【化2】
【0020】
一態様において、本発明の目的は、部分構造が、一般式IIaまたはIIbを有することを特徴とする、プロドラッグによって達成される。
【化3】
【0021】
例えば、薬剤ベンズアミジン(2)のカルボキシメチルベンズアミドキシム(1)プロドラッグの場合は、部分構造の式(IIa)においてR1およびR2が水素原子である、ここで(Ia)で示される高次部分構造、ならびに部分構造の式(II)においてR1およびR2が同様に水素原子である、ここで(Ib)で示される部分構造を含み、および薬剤は、プロドラッグにおける式(IIa)で表される部分構造に代えて構造(IIa−1)を有する。
【0022】
一態様において、本発明の目的はプロドラッグであって、プロドラッグが薬剤のプロドラッグであり、一般式IIaで表される部分構造が、代謝後に式
【化4】
を有する構造を含み、ならびに
一般式IIbで表される部分構造が、代謝後に式
【化5】
を有する構造を含むことを特徴とする、前記プロドラッグによって達成される。
【0023】
本発明のさらなる側面において、本発明の目的は、薬剤のプロドラッグであるプロドラッグの全体構造の一部として、
一般式(I)または(II)
【化6】
式中、R1およびR2は、水素、アルキルラジカルまたはアリールラジカルである、を形成する部分構造の使用によって達成される。
【0024】
一態様において、本発明の目的は、部分構造が、一般式(II)の構造を有し、ここで該部分構造は、薬剤のアミジン基またはグアニジン基に代わる高次の部分構造IIaまたはIIb
【化7】
の一部であり、溶解性、経口バイオアベイラビリティ、血液脳関門の横断、風味および/または物理的−化学的安定性を改善する、プロドラッグの使用によって達成される。
【0025】
一態様において、本発明の目的は、プロドラッグが
高次構造IIaの代わりにIIa−1またはIIa−2
【化8】
の部分構造の1つを含むか、または
高次構造IIbの代わりにIIb−1またはIIb−2
【化9】
の部分構造の1つを含むこと以外はプロドラッグと同一の構造を有する薬剤のプロドラッグである、プロドラッグの使用によって達成される。
一態様において、本発明の目的は、ペプチジルグリシンαアミド化モノオキシゲナーゼ(PAM)によって薬剤を活性化するための、プロドラッグの使用によって達成される。
【0026】
本発明の好ましい態様において、本明細書中で用いられている表現「ペプチジルグリシンαアミド化モノオキシゲナーゼ(PAM)によって薬剤を活性化する」、「PAM活性化経路によってプロドラッグを活性化する」、生物活性化などは、プロドラッグがPAMによって基質として認識され代謝されることを示す。本発明の好ましい態様において、本明細書中で用いられている表現「薬剤のプロドラッグの製造を含む、薬剤をPAM活性化経路に導入すること」は、対応するプロドラッグ形態がPAM活性化経路に導入される薬剤から製造され、このプロドラッグ形態がPAMによって認識され代謝されることを示す。好ましい態様において、PAMに対するプロドラッグの親和性は、当業者がKM値を用いて決定でき得るとおり、薬剤と比較して、1〜1000倍、2〜100倍、3〜50倍、4〜40倍、5〜20倍または6〜15倍高い。
【0027】
一態様において、本発明の目的は、部分構造が、ヒドロキシルアミン、N−オキシド、ニトロン、ジアゼニウムジオラート(NONOat)または類似のN−O−含有一酸化窒素ドナー、ヒドロキサム酸、ヒドロキシ尿素、オキシム、アミドキシム(Nヒドロキシアミジン)、N−ヒドロキシアミジノヒドラゾンあるいはN−ヒドロキシグアニジンの一部であることを特徴とする、プロドラッグの使用によって達成される。
【0028】
本発明のさらなる側面において、本発明の目的は、薬剤のプロドラッグの製造を含む、フリーアミジンまたはグアニジン官能基を含む薬剤をPAM活性化経路へと導入するための方法によって達成される。
本発明のさらなる側面において、本発明の目的は、患者へのプロドラッグの投与を含む、患者を処置するための方法によって達成される。
本発明のさらなる側面において、本発明の目的は、薬剤を製造するためのプロドラッグの使用によって達成される。
【0029】
本発明の好ましい態様において、インフルエンザなどのウイルス感染に対抗するための、HIV感染に対抗するための、内臓や皮膚リーシュマニア症の予防と処置のための、ニューモシスチス・カリニ肺炎(PCP)の予防のための、トリパノソーマ症(アフリカ睡眠病)を処置するための、マラリアを処置するため、バベシア症を処置するための、血液凝固を阻害するための、例えば静脈血栓塞栓症の一次予防のための、心房細動患者の脳卒中の予防のための、血圧を下げるための、悪性腫瘍の成長を抑制するための、神経保護のための、インフルエンザなどのウイルス感染に対抗するための、例えば心不全、肺水腫、中毒、腎不全または肝硬変などに対する、体から水を(利尿的)排除するための、アレルギーを処置するための、喘息を処置するための、炎症性疾患、例えばリウマチまたは膵炎を処置するための、あるいは虚血(不十分な血液供給)の予防のための、薬剤は薬剤であり、またはプロドラッグはプロドラッグである。
【0030】
本発明のさらなる側面において、本発明の目的は請求項7〜11および14のいずれか一項に記載のプロドラッグ、または請求項13に記載の方法により達成され、該使用または該方法は一酸化窒素欠乏に関連する疾患の処置のための使用または方法である。
一態様において、本発明の目的は、薬剤またはプロドラッグが、プロテアーゼ阻害剤、DNA−インターカレート化合物およびRNA−インターカレート化合物、ウイルス酵素の阻害剤、ならびにN−メチル−D−アスパラギン酸受容体アンタゴニストからなる群から選択されることを特徴とする、プロドラッグの使用によって達成される。
【0031】
一態様において、本発明はプロドラッグの使用によって達成され、ここで該使用は、内臓及び/または皮膚のリーシュマニア症、トリパノソーマ症、トリパノソーマ症の第2相またはPneumocystis cariniiによって引き起こされる肺炎の予防および/または処置のため、悪性腫瘍の成長を阻害するため、血液凝固を阻害するため、血圧を下げるため、神経保護のため、またはインフルエンザおよびHIV感染を含むウイルス感染に対抗するための使用である。
【0032】
本発明のさらなる側面において、本発明は一般式(I)または(II)、
【化10】
式中R1およびR2は水素、アルキルラジカルまたはアリールラジカルである、を有する部分構造を含む薬剤によって達成される。
【0033】
一態様において、本発明の目的は、部分構造が、ヒドロキシルアミン、N−オキシド、ニトロン、ジアゼニウムジオラート(NONOat)または類似のN−O−含有一酸化窒素ドナー、ヒドロキサム酸、ヒドロキシ尿素、オキシム、アミドキシム(Nヒドロキシアミジン)、N−ヒドロキシアミジノヒドラゾンあるいはN−ヒドロキシグアニジンの一部であることを特徴とする、一般式(I)または(II)を有する部分構造を含む薬剤によって達成される。
一態様において、本発明の目的は、一酸化窒素欠乏に関連する疾患の処置のために設計された薬剤であることを特徴とする、前記クレームのいずれか一項に記載の薬剤によって達成される。
【0034】
一態様において、本発明の目的は、薬剤が、プロテアーゼ阻害剤、DNA−インターカレート化合物およびRNA−インターカレート化合物、ウイルス酵素の阻害剤、ならびにN−メチル−D−アスパラギン酸受容体アンタゴニストからなる群から選択されることを特徴とする、薬剤によって達成される。
【0035】
本発明のさらなる側面において、本発明の目的は、薬剤の溶解性、バイオアベイラビリティ、血液脳関門の横断、生物活性および/または物理的−化学的安定性を改善するために、
一般式(I)または(II)
【化11】
式中R1およびR2は水素、アルキルラジカルまたはアリールラジカルである、を形成する部分構造を含む薬剤を製造するためのO−カルボキシアルキル化N−O−含有官能基の使用によって達成される。
【0036】
一態様において、本発明の目的は、ペプチジルグリシンαアミド化モノオキシゲナーゼ(PAM)によって薬剤を活性化するためのO−カルボキシアルキル化N−O−含有官能基を含む薬剤の使用によって達成される。
一態様において、本発明の目的は、部分構造が、ヒドロキシルアミン、N−オキシド、ニトロン、ジアゼニウムジオラート(NONOat)または類似のN−O−含有一酸化窒素ドナー、ヒドロキサム酸、ヒドロキシ尿素、オキシム、アミドキシム(Nヒドロキシアミジン)、N−ヒドロキシアミジノヒドラゾンあるいはN−ヒドロキシグアニジンの一部であることを特徴とする、薬剤の使用によって達成される。
【0037】
一態様において、本発明の目的は、薬剤が一酸化窒素欠乏に関連する疾患の処置のために設計されたことを特徴とする、薬剤の使用によって達成される。
【0038】
一態様において、本発明の目的は、薬剤がプロテアーゼ阻害剤、DNA−インターカレート化合物およびRNA−インターカレート化合物、ウイルス酵素の阻害剤、ならびにN−メチル−D−アスパラギン酸受容体アンタゴニストからなる群から選択されることを特徴とする、薬剤の使用によって達成される。
【0039】
一態様において、本発明の目的は、薬剤が、内臓及び/または皮膚はリーシュマニア症、トリパノソーマ症、トリパノソーマ症の第2相またはPneumocystis cariniiによって引き起こされる肺炎の予防および/または処置のため、悪性腫瘍の成長を阻害するため、血液凝固を阻害するため、血圧を下げるため、神経保護のため、またはインフルエンザおよびHIV感染を含むウイルス感染に対抗するために設計されていることを特徴とする、薬剤の使用によって達成される。
【0040】
本発明のさらなる側面において、本発明による化合物および/またはその塩を含む、医薬化合物、医薬組成物および医薬製品を提供する。医薬組成物は、好ましくは担体および/またはアジュバントを含み、理想的にはそれらは薬理学的に適合性がある。当業者はそのような担体およびアジュバントについて一般的に精通している。本発明による化合物は、医薬品における使用のためにも提供される。
【図面の簡単な説明】
【0041】
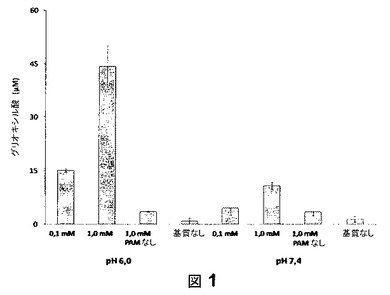
【図1】図1は、グリオキサル酸形成の比色定量の結果を示す。
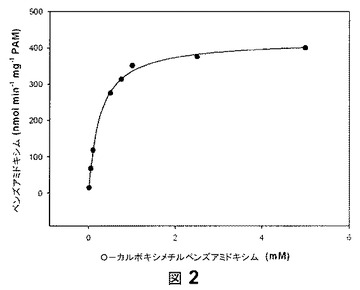
【図2】図2は、O−カルボキシメチルベンズアミドキシムを用いた比色アッセイにおける、KM値およびVmax値の決定の代表的な例示である。
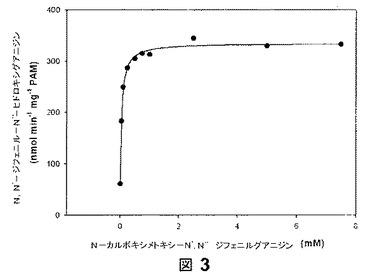
【図3】図3は、N−カルボキシメトキシ−N’,N”−ジフェニルグアニジンを用いた比色アッセイにおける、KM値およびVmax値の決定の代表的な例示である。
【発明を実施するための形態】
【0042】
薬剤が少なくとも一種または二種以上の活性アミジン、N−ヒドロキシアミジン(アミドキシム)、グアニジンまたはN−ヒドロキシグアニジン官能基を提案された形態で含めば、それで十分である。したがって、薬剤は、例えば複数のアミドキシム官能基(例えばペントキシム(pentoxime)エステルとして2つ)、またはN−ヒドロキシグアニジン官能基、ここでこれらの少なくとも一種の基が前述の方法で修飾されている、を含むことができる。同様に、薬剤の混合物もまた用いることができ、それらのうちの少なくとも一種は本発明にしたがって修飾されている。
【0043】
本発明による化合物は、ボーラス投与のように一度に、毎日、毎週または毎月投与することができる。投与の方法も同様に容易に決定することができる。一般的に、可能な投与形態は、経口、直腸、例えば静脈内、筋肉内、皮下、経皮投与、肺内投与などの非経口およびエアロゾルとしての投与、膀胱内注入、腹腔内や心臓内注射、粘膜や膣内の適用を介する吸収、例えば坐剤などが挙げられる。
【0044】
この目的のために、液体製剤に用いるこれらの態様に対して、有効成分、または有効成分混合物を、適切な非毒性溶媒、例えば水、一価アルコール、特にエタノール、多価アルコール、特にグリセリンおよび/またはプロパンジオール、ポリグリコール、特にポリエチレングリコールおよび/またはミグリオール、グリセリンホルマール(glycerinformal)、ジメチルイソソルビド、天然または合成油などに受容させる。
【0045】
従来のベース製品、例えばベントナイト、ビーガム、グアー粉(guar meal)および/またはセルロース誘導体、特にメチルセルロースおよび/またはカルボキシメチルセルロース、ビニルアルコールおよび/またはビニルピロリドン製ポリマー、アルギン酸塩、ペクチン、ポリアクリレート、固体および/または液体ポリエチレングリコール、パラフィン、脂肪アルコール、ワセリンおよび/またはワックス、脂肪酸および/または脂肪酸エステルが、半固形または固形剤を製造するために用いられる。
【0046】
また、既知の増量剤、例えばコロイド状ケイ酸、タルク、乳糖、でんぷん粉、砂糖、ゼラチン、金属酸化物および/または金属塩が固形製剤中に存在してもよい。さらなる添加剤、例えば安定剤、乳化剤、分散剤や防腐剤などは、当然選択されるものである。
【0047】
驚くべきことに、窒素(N)を介して医薬分子に結合し、シトクロームP450(CYP450)酵素非依存性の生物活性化の経路を利用する、一般式(I)または(II)
【化12】
ここで、(I)および(II)は、例えばヒドロキシルアミン、N−オキシド、ニトロン、ジアゼニウムジオラート(NONOat)または類似のN−O−含有一酸化窒素ドナー、ヒドロキサム酸、オキシム、アミドキシム(Nヒドロキシアミジン)、N−ヒドロキシアミジノヒドラゾンあるいはN−ヒドロキシグアニジンの一部であり、およびR1(Rは、pro−R構成されなければならない)およびR2は、水素、アルキルラジカルまたはアリールラジカルである、で表される、O−カルボキシアルキル化N−O−含有官能基が見出された。
【0048】
CYP450酵素は一般的に酸化的O−脱アルキル化を触媒することが知られており、これは本明細書で提案しているプロドラッグ原理の場合においてもまた実際の薬剤を放出するために必要であることから、これは予想外の結果をもたらしている。
【0049】
提案するカルボキシアルキルラジカルを有するN−O−含有官能基のエーテル化は、CYP450酵素とは異なる酵素を生物活性化のために利用することができるという特別な利点を提供する:ペプチジルグリシンαアミド化モノオキシゲナーゼ(PAM)。例えば、これは副作用および他の同時投与された薬剤との前述の相互作用を防ぐ。
【0050】
高等生物(脊椎動物)において、ペプチジルグリシンα−アミド化モノオキシゲナーゼ(PAM)は、モノオキシゲナーゼドメイン(PHM、ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼ、EC 1.14.17.3)およびリアーゼドメイン(PHL、ペプチジル−α−ヒドロキシグリシンα−アミド化リアーゼ、EC4.3.2.5)を含有する二官能性酵素を構成する。全体的に、PAMは、スプライシングと発現による強度に組織特異的および発達依存的(development dependent)な調節を受けやすい。翻訳後修飾の意味の範囲内で、PAMは、多様な生理的に発生するペプチドホルモン、神経伝達物質および成長因子(例えば、サブスタンスP、ニューロペプチドY、オキシトシン、バソプレシン、カルシトニン)を活性化することができる。このプロセスにおいて、ペプチドは、モノオキシゲナーゼ反応における酸化的N−脱アルキル化による末端グリシンの分離によって、C末端がアミド化されている。
【0051】
本発明により提案しているとおりのカルボキシアルキルラジカルを有するN−O−含有官能基の特別な利点は、生理的条件下(pH6〜8)で負に荷電するカルボン酸の挿入に起因する改善された溶解性である。
さらなる利点は、本発明により提案するN−O−含有官能基のエーテル化−(アルコキシカルボニル)アルキルエーテルまたは(アリールオキシ)アルキルエーテルを用いる−が、親油性を大きく増加させて受動拡散を可能にし、これによりバイオアベイラビリティおよび/または血液脳関門の横断が向上することである。
【0052】
プロドラッグ基として、比較的小さいラジカル−最も簡単な場合において、カルボキシメチルラジカル−を使用する可能性は、医薬分子を適度の大きさにしか大きくせず、同様に有利である。
Wand et al. [Metabolism 1985、34、11、1044]は、異なるヒト組織におけるPAMの活性を分析し、CNSの組織において(特に下垂体において)最も高い活性を検出した。対照的に、典型的な異物代謝の臓器、肝臓と腎臓において、活性は認められなかった。計画されたプロドラッグ概念をまた利用することができる活性が、血漿、心臓および肺で同様に検出された。
【0053】
特に、CNSにおけるこの酵素の高い活性は、O−カルボキシアルキル化プロドラッグを、血液脳関門を通して輸送し、その後それらを変換するために利用することができる。しかしながら、経口適用および消化管からの吸収後の心血管系における生物活性化も可能である。
本発明によるプロドラッグシステムは、アミジンまたはグアニジン官能基を有する種々の薬剤に適用することができる。以下の薬剤が特に好ましい:
【0054】
ペンタミジン、ダビガトラン、BSF 411693(アボット)、イダゾキサン塩酸塩、イルベサルタン、リノグリリド、ロフェキシジン塩酸塩、テトラヒドロゾリン塩酸塩、トラゾリン、キシロメタゾリン塩酸塩、ペンタアミジンイセチオナート、タリバビリン、チアミン (ビタミンB1)、ボセンタン、ジブロモプロプアミジネイセチオナート、ヒドロキシスチルブアミジンイソチオナート、シブラフィバン、オルボフィバン、キセミロフィバン、アルガトロバン、キシメラガトラン、メラガトラン、2−ピペリジン酸、オルボフィバンアセタート、エピナスチン(レレスタット)、
【0055】
RO 43-8857、AB1(クロラムブシル類似体)、AMG-126737、AY-0068、B-623、BABIM、BIBT-986(べーリンガーインゲルハイム)、CI-1031(会社名:バイオサイエンス)、CJ-1332(会社名:キュラサイト)、CJ-463(会社名:キュラサイト)、CJ-672(会社名:キュラサイト)、CT50728(Portolla Pharmaceuticals)、CVS-3983、DX-9065a、ラミフィバン(ロシュ)、LB-30870(会社名:エルジー・ライフ・サイエンシーズ・リミテッド)、LY-178550(会社名:Lilly)、PHA-927Fおよび類似体、RO-44-3888(ロシュ)、セピモスタット、FUT-187(鳥居)、ビラミジン(Ribapharm)、WX-FX4(Wilex)、YM-60828(山之内製薬株式会社)、ZK-807191(Berlex バイオサイエンス)、NAPAP(SR 25477)、BIIL 315(べーリンガーインゲルハイム)、BIIL 260(べーリンガーインゲルハイム)、BIIL 284/260(べーリンガーインゲルハイム)、
【0056】
タノギトラン、モキシルバント、スチルバミジン、パナミジン(panamidine)、フラダフィバン、ジミナゼン、ロキシフィバン、フラミジン、PD0313052、PHA 927F、PHA 798、フィデキサバン、オタミキサバン、トロンブストップ(thromstop)(Thrombstop)、ザナミビル、アミロリド塩酸塩、アナグレリド塩酸塩、プログアニル、シメチジン、クロニジン塩酸塩、グアノキサン、ペラミビル、ロミフィジン、チラパザミン、チザニジン、トロニジン硝酸塩、メトホルミン、ジミナゼン、デブリソキン、スルファメタジン、エプチフィバチド、ファモチジン、バイエル薬品、ストレプトマイシン、ナファモスタット、FUT-175、イノガトラン、グアネチジン(Thilodigon)、
【0057】
3DP-10017、APC-366、CVS-1123、ジフェニルホスファート誘導体、E-64、FOY-305、MBGB、MIBG、RWJ-422521、シンタリン、WX-293、WX-340、BMS-189090、JTV-803(日本たばこ産業)、ナプサガトラン、イスメリン、Tan 1057A、Hydikal、Phenformix(Retardo)、ネトロプシン(シナノマイシン)、BIIB 722(サビポリド)、グアナドレル、デオキシスペルグアリン、BMS 262084、Siamformet(Orabet)、PPACK(Pebac)、メルゲトパ(プラマーカルボキシペプチダーゼ阻害剤)、ペラミビル、ファモチジン、ザルチジン。
【0058】
添付には、化学式、CAS登録番号および薬剤の効能を付したテーブルを含む。
【0059】
以下、本発明による4種のプロドラッグを例として示す:
【化13】
【0060】
【化14】
【0061】
【化15】
【0062】
【化16】
【0063】
非ペプチド性O−カルボキシアルキル化N−O−含有官能基が、PAMの基質として許容されるという驚くべき発見は、アミドキシム−ベースおよびN−ヒドロキシグアニジン−ベースのモデル化合物に基づく例示的な態様において実証される。
【0064】
O−カルボキシメチルベンズアミドキシム(1)をアミドキシムのモデル化合物として、そのPAM基質特性を試験した。O−カルボキシメチルベンズアミドキシムは、薬剤ベンズアミジンの有力なプロドラッグである。O−カルボキシメチルベンズアミドキシム(1)のベンズアミドキシム(2)へのPAM触媒による生物活性化は、同時にグリオキサル酸を放出しながら起こる。
【化17】
【0065】
図1に、グリオキサル酸形成の比色定量の結果を示す。決定したグリオキシル酸濃度は、平均値±2回ずつ測定された2つのインキュベーションからの標準偏差である。1のPAMの触媒作用の切断産物としてのグリオキサル酸の形成は、濃度依存的であることが確認された。PAMの最適pH(pH6.0)でのインキュベーションは、pH7.4でのインキュベーションと比べて極めて高い変換をもたらした。比色アッセイにおいて、グリオキシル酸の5点キャリブレーションを、1の試験と同時に行った。キャリブレーションは、決定された濃度の範囲で線形的であった(r2=1.000)。
【0066】
これらの結果から、O−カルボキシメチルベンズアミドキシム(1)は、PAMによる基質として許容されたため、反応を、KM値およびVmax値を決定することによって、より詳細に特徴づけた。
この目的のため、HPLC分析を開発した。ベンズアミドキシムに対するキャリブレーション線は、決定された濃度の範囲で線形的であり(r2=1.000)、回収率は130.6%であった(r2=0.999)。2つの独立した実験(n=2)は、KM値307±80μMおよびVmax値393±40nmolmin−1mg−1PAMとなった。図2はかかる決定の代表的な例示である。
【0067】
CYP450基質の研究のために、前述のHPLC分析を、さらに、ベンズアミドキシム(2)のN−還元(N−reduction)の生成物として考えられる代謝産物ベンズアミジンの検出が可能になるように改変した。pH6.0およびpH7.4で、ベンズアミドキシム(ベンズアミジンの有力なプロドラッグ)とベンズアミジンのいずれもCYP450酵素源において検出されなかった。
ベンズアミドキシムモデル化合物1に基づいて、O−カルボキシメチル官能基はPAMから除去されるが、モノオキシゲナーゼ反応の意味の範囲内において、シトクロームP450からは除去されない。
【0068】
N−カルボキシメトキシ−N’,N”−ジフェニルグアニジン(3)をヒドロキシグアニジンモデル化合物としてそのPAM基質特性について試験した。
【化18】
【0069】
N−カルボキシメトキシ−N’,N”−ジフェニルグアニジン(3)のN,N’−ジフェニル−N”−ヒドロキシグアニジンへのPAM触媒による生物活性化は、同時にグリオキサル酸を放出しながら起こる。
【0070】
3を用いた比色アッセイの結果は、アミドキシムモデル化合物1の結果に匹敵するものであった。KM値およびVmax値を決定するために、プロドラッグ3とヒドロキシグアニジン4をRPカラム上で15分以内に分離することができるHPLCを開発した。N,N’−ジフェニル−N”−ヒドロキシグアニジン(4)に対するキャリブレーション線は、決定された濃度の範囲で線形的であり(r2=0.999)、回収率は111.7%であった(r2=0.999)。2つの独立した実験(n=2)は、KM値37±5μMおよびVmax値373±53μmolmin−1mg−1PAMとなった。図3はかかる決定の代表的な例示である。
【0071】
変換速度が同等の場合、決定したKM値から、PAMに対する親和性が、アミドキシムプロドラッグ1と比較して約8倍以上であることが導かれ得る。
CYP450基質の研究のために、PAM基質のために開発したHPLC分析を、さらに考えられる代謝産物N,N’−ジフェニルグアニジンの検出が、ヒドロキシグアニジン4のN−還元の生成物として可能になるように改変した。pH6.0およびpH7.4で、4とN,N’−ジフェニルグアニジンのいずれも、180分のインキュベーションタイム後の用いたCYP450酵素源のいずれにおいても検出されなかった。
【0072】
O−カルボキシメチルベンズアミドキシム(1)と同様に、ヒドロキシグアニジンモデル化合物3に基づいて、O−カルボキシメチル官能基は、PAMからのみ除去されるが、モノオキシゲナーゼ反応の意味の範囲内において、シトクロームP450からは除去されない。
【0073】
材料および方法
改変されたO−カルボキシメチルベンズアミドキシム一水和物のナトリウム塩(1)Koch [Ber. Dtsch. Chem. Ges. 1889、22、3161]による手順:
【化19】
【0074】
5mlエタノール中の681mgベンズアミドキシム(5.0mmol)、1.04gブロモ酢酸(7.5mmol)および500mg水酸化ナトリウムペレット(12.5mmol)の溶液を還流下で5時間沸騰させる。その後、沈殿物が形成され始めるまで真空下で溶媒を除去する。沈殿物を完全に沈殿させ、濾過し、乾燥する。生成物をエタノール(96%)/水(95:5)から再結晶させる。
【0075】
【化20】
【0076】
N−カルボキシメトキシ−N’,N”−ジフェニルグアニジン(3)
【化21】
【0077】
546mgアミノオキシ酢酸セミクロリド(5mmol)および697μlトリエチルアミン(5mmol)を乾燥DMF(dryDMF)10ml中で30分間撹拌する。沈殿物をろ過し、濾液に970mgN,N’−ジフェニルカルボジイミド(5mmol)を加える。バッチを4時間室温で撹拌し、酢酸エチルで溶媒抽出し、生成物をエタノールから再結晶する。
【0078】
【化22】
【0079】
HPLCシステム
Waters 1525 ポンプ、Waters 2487吸収検出器、Waters 717 Plus オートサンプラーおよびBreeze記録および評価ソフトウェア(バージョン3.30)、Gynkotek STH 585カラムオーブンを備えたWaters Breeze HPLCシステム
【0080】
HPLCカラム:
C-18プレカラム(4 x 3mm)(Phenomenex)を備えたSynergiMax-RP80A(250 x 4.6mm、4μm);
LiChroCART、LiChrospher100、LiChrospher60プレカラムを備えたRP-8(125 x 4mm、5μm)、RP-selectB(4 x 4mm、5μm)(Merck);
LiChroCART、LiChrospher60プレカラムを備えたLiChrospher RP-selectB(250 x 4.6mm、5μm)、RP-selectB(4 x 4mm、5μm)(Merck)
【0081】
追加のデバイスおよび材料:
Cary 50紫外可視分光光度計(Varian);96−ウェルプレート(Greiner);GFL-1083振とう水浴(Gesellschaft fuer Labortechnik、Burgwedel);マイクロリットル遠心分離機(Hettich GmbH);LiQ Plast pH 電極(Hamilton)付InoLab pH Level 1 pH測定デバイス(Wissenschaftlich-Technische Werkstaetten GmbH、Weilheim);VF2ボルテクサー(vortexer)(Janke und Kunkel GmbH & Co. KG、Staufen);1.5 ml反応器(Sarstedt AG & Co.、Nuembrecht)
【0082】
酵素源:使用した組換えペプチジルグリシンα−アミド化モノオキシゲナーゼ(PAM、ラット、EC 1.14.17.3)は、Unigene Laboratories,Inc.(ニュージャージー州、米国)(比活性=5.8 106U/mgタンパク質)から提供された;牛肝臓カタラーゼ(EC 1.11.1.6)、比活性=12600U/mg固体(アルドリッチ)。
【0083】
使用したシトクロームP450酵素源は、Clement von Gruenewaldワーキンググループにおいて、以下の説明に従い入手した:
ブタ肝ミクロソームおよび上清9000g:
豚の肝臓は、地元の肉屋(Bordesholm)から調達し、臓器は、屠殺後、氷冷した20mM リン酸バッファー(1mM NA2 EDTA、pH7.4)中で、直接輸送した。さらなる処理のために、まず肝葉を50mM リン酸バッファー(1mM NA2 EDTA、pH7.4)で灌流し、洗浄した。組織片に切断し、市販の肉挽き器に通した。懸濁液を等量のリン酸バッファーで希釈し、フローホモジナイザーを用いてホモジナイズした。さらに、差動超遠心分離することによってミクロソームおよび上清9000グラムを得た。貯蔵のために、得られた標本は、小分けして−80℃で凍結した。
【0084】
ヒト肝ミクロソームおよび上清9000g:
ヒトミクロソームを得るために、クリスチャン−アルブレクト大学(Christian-Albrecht University)大学病院の外科の、部分的肝切除を受けなければならなかった癌患者からヒト肝組織を得た。
【0085】
肝臓の組織片は、ショ糖含有リン酸バッファー(10mM K2HPO4、10mM KH2PO4、250mM ショ糖、1mM NA2 EDTA、pH7.4、4℃)で急速冷凍した。臓器片の十分な量が(>3)得られたらすぐに、対応する小片を、個体間変動に起因する差異を相殺するために、解凍し、プールした。組織片を、4℃でより小さい片に切断し、バッファー(EDTAを含まない)で数回洗浄し、ホモジナイザーを用いて懸濁液へと処理した。さらに、分画超遠心分離することによってミクロソームおよび上清9000グラムを得た。貯蔵のために、得られた標本は、小分けして−80℃で凍結した。
【0086】
PAMアッセイ:インキュベーション条件
典型的な300μl(総量)のインキュベーションバッチに、25000 U/ml ペプチジルグリシンα−アミド化モノオキシゲナーゼ(PAM、会社名:Unigene Laboratories)、250U/ml カタラーゼ、1μM 銅(II)(酢酸/水和物として採用)、2mM アスコルビン酸ナトリウム、5mM ヨウ化カリウムおよび0.1mMまたは1mMの濃度のそれぞれの基質を、種々のpH値を有するバッファー中に入れた。使用したバッファー系は、pH6.0でのインキュベーションのために30mM MESならびにpH7.4のインキュベーションのために50mM HEPESであった。pH値は、それぞれの場合において希水酸化ナトリウムで調整した。インキュベーションを37℃で60分間振とう水浴中で行い、100μlを取り出し、反応を50μl 10%TFA(aq)/アセトニトリル(2:3)で停止させた。残りのバッチは、37℃で別に180分間インキュベートし、100μl 10%TFA(aq)/アセトニトリル(2:3)で停止させた。
【0087】
停止したサンプルは5分間振とうし(ボルテクサー)、−80℃で凍結させた。サンプルを分析するために、それらを解凍し、5分間振とうし、沈殿したタンパク質を、10000rpmで遠心分離した。上清を比色グリオキシル酸定量および/またはHPLCの測定に用いた。KMおよびVmaxの決定のために、100μlのバッチを、pH6.0で前述の条件下、しかしインキュベーション時間が30分という点で異なる、で処理した。
【0088】
グリオキシル酸の比色定量
タンパク質が除かれたインキュベーションバッチ200μlを20μlのフェニルヒドラジン溶液(2mlaqua bidest中20mg)と混合し、37℃で5分間振とう水浴中で振とうした。その後、混合物を15分間で0℃まで冷却し、100μl 氷冷6NのHClを加え、0℃でさらに5分間放置した。その後、20μl のヘキサシアノ鉄酸(III)カリウム溶液(2mlaqua bidest中100mg)を加えた。バッチを、室温で15分間放置し、200μlをプレートリーダー(Cary 50紫外可視分光光度計、520nm)を用いる測定のために取り出した。
キャリブレーション:
5点キャリブレーションについて、アッセイバッファー(pH6.0):10%TFA(aq)/アセトニトリル(2:3)の2:1混合物中の2、5、10、50および100μM濃度のグリオキサル酸を上記のとおり測定した。このキャリブレーションは、実施したテスト化合物の各アッセイに対して同時に行った。
【0089】
O−カルボキシメチルベンズアミドキシム(1)およびベンズアミドキシム(2)の分離のためのHPLC分析
【表1】
【0090】
キャリブレーションと回収
キャリブレーションのために、ベンズアミドキシムを0.1〜500μMの8つの濃度で溶解し、アッセイバッファー(30mM MES、1μM 酢酸銅(II)、2mM アスコルビン酸ナトリウム、5mM ヨウ化カリウム、pH6.0)中に溶解し、上述のHPLC法を用いて測定した。
回収を決定するために、アッセイバッファー(エンド容量=100μl)中に同濃度のものを製造した。さらに、O−カルボキシメチルベンズアミドキシム(0.5mM)および250U/mlカタラーゼを加え、50μl10%TFA(aq)/アセトニトリル(2:3)を続いて加えた。サンプルを、ボルテクサーを用いて振とうし、−80℃で凍結させた。サンプルを測定するために、それらを解凍し、ボルテクサーを用いて5分間振とうし、10000rpmで5分間遠心分離した。
【0091】
N−カルボキシメトキシ−N’,N”−ジフェニルグアニジン(3)およびN−ヒドロキシ−N’,N”−ジフェニルグアニジン(4)の分離のためのHPLC分析
【表2】
【0092】
キャリブレーションと回収
キャリブレーションのために、N−ヒドロキシ−N’,N”−ジフェニルグアニジン(4)を0.1〜500μMの8つの濃度で溶解し、アッセイバッファー(30mM MES、1μM 酢酸銅(II)、2mM アスコルビン酸ナトリウム、5mM ヨウ化カリウム、pH6.0)中に溶解し、上述のHPLC法を用いて測定した。
回収を決定するために、アッセイバッファー(エンド容量=100μl)中に同濃度のものを製造した。さらに、N−カルボキシメトキシ−N’,N”−ジフェニルグアニジン(3)(0.5mM)および250U/mlカタラーゼを加え、50μl10%TFA(aq)/アセトニトリル(2:3)を続いて加えた。サンプルを、ボルテクサーを用いて振とうし、−80℃で凍結させた。サンプルを測定するために、それらを解凍し、ボルテクサーを用いて5分間振とうし、10000rpmで5分間遠心分離した。
【0093】
CYP450アッセイ:インキュベーション条件
典型的な500μl(総量)のインキュベーションバスに、0.3mgタンパク質(豚またはヒト肝臓酵素源)、0.1mM(または1mM)のテスト化合物を、100mM リン酸バッファー(pH6.0またはpH7.4)および1mM NADH(またはNADPH)中に入れた。バッファー中の酵素およびテスト化合物の5分間プレインキュベーション後、インキュベーションを開始し、NADH(またはNADPH)を追加し、生成物を振とう水浴槽で37℃60分間または180分間振とうした。反応を同量のアセトニトリルを加えることによって停止し、ボルテクサーを用いて振とうし、−80℃で凍結させた。
サンプルを分析するために、それらを解凍し、ボルテクサーを用いて5分間振とうし、5分間10000rpmで遠心分離することによって、タンパク質を分離した。上清をHPLC分析に用いた。
【0094】
O−カルボキシメチルベンズアミドキシム(1)、ベンズアミドキシム(2)およびベンズアミジンの分離のためのHPLC分析
【表3】
【0095】
N−カルボキシメトキシ−N’,N”−ジフェニルグアニジン(3)、N−ヒドロキシ−N’,N”−ジフェニルグアニジン(4)およびN’,N”−ジフェニルグアニジンの分離のためのHPLC分析
【表4】
【0096】
以下に、本発明によるプロドラッグシステムを好ましく適用できる医薬品の表を示す。
【表5】
【表6】
【表7】
【表8】
【表9】
【表10】
【表11】
【表12】
【表13】
【表14】
【技術分野】
【0001】
本発明は、薬剤の生物活性を改善するための方法に関する。
【背景技術】
【0002】
経口投与後の薬剤の治療効果に関する要件は、消化管からのそれらの吸収である。そのような効果のうち最も重要なメカニズムは、受動拡散である。受動拡散による吸収の程度は、とりわけ親油性に依存する。
【0003】
薬による多くの疾患の処置に伴う別の問題は、血液脳関門を通過する必要性である。血液脳関門は、脳内の物質の吸収に関する効果的なバリアを構成している。それは選択的な取り込みを保証し、物質が通過することを防ぐ。また、血液脳関門は、物理的なだけでなく、酵素的なバリアとしても機能する。様々なプロセスが、脳への物質の通過に関与している。他の適応と比較して、中枢神経系(CNS)でその効果を現す薬剤はわずかに販売されているにすぎない。これらのうち、多くは拡散によって中枢神経系に到達する。このような方法で、てんかん、慢性痛やうつ病などの疾患が処置される。他の重篤な機能障害、例えば脳腫瘍または筋萎縮性側索硬化症などは、現在この方法で処置するのは非常に困難である。
【0004】
受動拡散によって生体膜を突破することができるためには、物質は、親油性で、500Daより小さい分子量を有しているべきであり、また非荷電状態で存在すべきである。特にアミノ酸または糖などの小さい高極性分子を吸収するため、種々のトランスポーターシステム、例えばヌクレオシドトランスポーター、有機アニオンまたはカチオンの流入および流出トランスポーター、グルコーストランスポーター、ペプチドトランスポーターならびにアミノ酸トランスポーターなどが、バリア機能を有する生体膜(消化管、血液脳関門)で発現している。
【0005】
このような理由から、様々なプロドラッグシステムが、薬物動態学的特性を改善するために採用されている。プロドラッグは、薬理学的に不活性または実質的に不活性であり、かつそれが生体内で代謝されるまで、活性代謝物に変換されない薬剤である。
【0006】
N−ヒドロキシアミジン(アミドキシム)およびN−ヒドロキシグアニジンは、アミジン[Clement、B. Methoden zur Behandlung und Prophylaxe der Pneumocystis carinii Pneumonie (PCP) und anderen Erkrankungen sowie Verbindungen und Formulierungen zum Gebrauch bei besagten Methoden.(DE 4321444)][ニューモシスチス・カリニ肺炎(PCP)および他の疾患の処置および予防のための方法ならびにその方法に使用するための化合物および製剤]およびグアニジンの経口バイオアベイラビリティを高めるための既知のプロドラッグの原理を示す。アミノ基およびイミノ基の窒素原子は、アミジンおよびグアニジンの塩において、メソメリー平衡(mesomeric equilibrium)で存在し、そのコンセプトは、両方の窒素原子に採用することができる。
【0007】
活性代謝物への変換は、根底にあるプロドラッグの概念に応じて、種々の酵素システムを介して行われる。あらゆる生物(live)において実質的に生じる酵素システムはシトクロームP450(CYP450)であり、とりわけ、以下の反応:
芳香族および脂肪族化合物のN−酸化、S−酸化、N−脱アルキル、O−脱アルキル、S−脱アルキル、脱アミノ化、脱ハロゲン化およびヒドロキシル化
を触媒する。
【0008】
CYP450酵素システムの多様性が意味するところは、種々の基質と薬剤が変換時にシステムと競合するということである。これは、相互作用、相反的効果および望ましくない相互の影響をもたらす。このような理由からプロドラッグの開発の際には、CYP450−非依存性生物活性化が望まれる。
したがって、本発明の目的は、シトクロームP450(CYP450)酵素に依存しない生物活性化の経路を採用したプロドラッグシステムを提供することである。この目的は、特許請求の範囲に記載された事項によって達成される。従属クレームは、発明の有利な態様を提供する。
【発明の概要】
【0009】
本発明によれば、本発明の目的は、一側面において、一般式(I)または(II)
【化1】
式中、R1およびR2は、水素、アルキルラジカルまたはアリールラジカルである、
を有する部分構造を含むプロドラッグによって達成される。
【0010】
本発明の好ましい態様において、本明細書中で用いられている用語「部分構造」は、それぞれの式で示される構造的要素が物質、好ましくはプロドラッグの式の一部であることを示す。例えば、化合物O−カルボキシメチルベンズアミドキシム(1)は、薬剤ベンズアミジン(pharmaceutical benzamidine)に対応するプロドラッグを構成し、ここで部分構造は式(II)で表される部分構造であり、R1およびR2はそれぞれ水素原子を示す。この部分構造は、ベンゼン環上の置換基であり、それと一緒になって薬剤ベンズアミジンを構成する。
【0011】
本発明の好ましい態様において、本明細書中で用いられている用語「プロドラッグ」は、不活性または薬理学的に実質的に不活性であり、それが生体内で代謝されるまで、薬理学的に活性である薬剤に変換されないような物質を示す。プロドラッグは、実際の活性な薬剤よりも改善された経口バイオアベイラビリティを示すことができるが、示さなくてもよい。代替的に、プロドラッグは、薬剤と比較して改善された溶解性、生物活性化、血液脳関門の横断、物理化学的安定性、低い毒性および/または耐用可能なもしくはそれ以上の快適な風味を示すため、用いることができる。例えば、エリスロマイシンAの2’−エチルスクシナートは、必ずしもエリスロマイシンAの不十分な吸収または溶解性のためではなく、その苦みのために、プロドラッグとして子どもたちに投与されている(Bhadra et al. (2005)、J. Med. Chem.)。
【0012】
本発明のさらなる好ましい態様において、オリジナルのプロドラッグは、ワンステップ反応でプロドラッグから薬剤へと代謝されるのではなく、むしろ複数の反応工程により代謝され、一つの反応工程から得られた各代謝物は、オリジナルのプロドラッグより有利な、一種または二種以上の同一および/または異なる特性を示すことができる。この目的のために、すべての代謝物がプロドラッグ以上の有利な特性を示さなくてもよい。例えば、プロドラッグの第一の代謝産物は、プロドラッグと比較して改善された薬理活性を示すことができ、第一の代謝産物由来の第二の代謝産物は同様にプロドラッグと比較して改善された薬理活性を示すことができ、および第二の代謝産物由来の第三の代謝産物はプロドラッグと比較して改善された血液脳関門の横断および物理的−化学的安定性を示すことができる。
【0013】
本発明の好ましい態様において、本明細書中で用いられている用語「物理的−化学的安定性」は、例えば加水分解などの化学分解がなく、例えば水、バッファーまたは生理食塩水に溶解させるなどの関連する水溶液の形態で貯蔵されるおよび/または用いられる物質、例えばプロドラッグまたは薬剤の能力を示す。本発明のさらなる好ましい態様において、本明細書中で用いられているこの用語は、物質が安定した合成形で合成できることを示す。本発明のさらなる好ましい態様において、本明細書中で用いられているこの用語は、物質の合成中に、後続する合成産物または合成中間産物をより安定な形態で製造可能にするために、単離された関連する合成前駆体が、類似または同一の合成戦略に従って製造した他の物質の類似産物前駆体または中間産物よりも安定であるか、または最初から製造できることを示す。
【0014】
一態様において、本発明の目的はプロドラッグであって、プロドラッグが含む部分構造が、ヒドロキシルアミン、N−オキシド、ニトロン、ジアゼニウムジオラート(NONOat)または類似のN−O−含有一酸化窒素ドナー、ヒドロキサム酸、ヒドロキシ尿素、オキシム、アミドキシム(N−ヒドロキシアミジン)、N−ヒドロキシアミジノヒドラゾンあるいはN−ヒドロキシグアニジンの一部であることを特徴とする、前記プロドラッグによって達成される。
【0015】
薬剤ベンズアミジンのプロドラッグカルボキシメチルベンズアミドキシム(1)の場合において、例えば部分構造は式(II)の部分構造であり、R1およびR2はそれぞれ水素原子であり、およびプロドラッグが含む部分構造はアミドキシム(N−ヒドロキシアミジン)の一部である。
【0016】
一態様において、本発明の目的は、プロドラッグであって、プロドラッグが、一酸化窒素欠乏に関連する疾患の処置のための薬剤である薬剤へと代謝されることを特徴とする、前記プロドラッグによって達成される。
【0017】
一態様において、本発明の目的は、プロドラッグであって、プロドラッグまたは対応する薬剤が、プロテアーゼ阻害剤、DNA−インターカレート化合物およびRNA−インターカレート化合物、ウイルス酵素の阻害剤、ならびにN−メチル−D−アスパラギン酸受容体アンタゴニストからなる群から選択されることを特徴とする、前記プロドラッグによって達成される。
【0018】
本発明の好ましい態様において、用語「高次部分構造」(higher-level partial structure)は、この高次部分構造が一方では式(I)または(II)で表される部分構造を含み、他方では問題の物質の全体構造の一部であると理解するべきである。例えば、薬剤ベンズアミジン(2)のプロドラッグカルボキシメチルベンズアミドキシム(1)の場合は、部分構造の式(IIa)においてR1およびR2が水素原子である、ここで(Ia)で示される高次部分構造、ならびに部分構造の式(II)においてR1およびR2が同様に水素原子である、ここで(Ib)で示される部分構造を含む。
【0019】
【化2】
【0020】
一態様において、本発明の目的は、部分構造が、一般式IIaまたはIIbを有することを特徴とする、プロドラッグによって達成される。
【化3】
【0021】
例えば、薬剤ベンズアミジン(2)のカルボキシメチルベンズアミドキシム(1)プロドラッグの場合は、部分構造の式(IIa)においてR1およびR2が水素原子である、ここで(Ia)で示される高次部分構造、ならびに部分構造の式(II)においてR1およびR2が同様に水素原子である、ここで(Ib)で示される部分構造を含み、および薬剤は、プロドラッグにおける式(IIa)で表される部分構造に代えて構造(IIa−1)を有する。
【0022】
一態様において、本発明の目的はプロドラッグであって、プロドラッグが薬剤のプロドラッグであり、一般式IIaで表される部分構造が、代謝後に式
【化4】
を有する構造を含み、ならびに
一般式IIbで表される部分構造が、代謝後に式
【化5】
を有する構造を含むことを特徴とする、前記プロドラッグによって達成される。
【0023】
本発明のさらなる側面において、本発明の目的は、薬剤のプロドラッグであるプロドラッグの全体構造の一部として、
一般式(I)または(II)
【化6】
式中、R1およびR2は、水素、アルキルラジカルまたはアリールラジカルである、を形成する部分構造の使用によって達成される。
【0024】
一態様において、本発明の目的は、部分構造が、一般式(II)の構造を有し、ここで該部分構造は、薬剤のアミジン基またはグアニジン基に代わる高次の部分構造IIaまたはIIb
【化7】
の一部であり、溶解性、経口バイオアベイラビリティ、血液脳関門の横断、風味および/または物理的−化学的安定性を改善する、プロドラッグの使用によって達成される。
【0025】
一態様において、本発明の目的は、プロドラッグが
高次構造IIaの代わりにIIa−1またはIIa−2
【化8】
の部分構造の1つを含むか、または
高次構造IIbの代わりにIIb−1またはIIb−2
【化9】
の部分構造の1つを含むこと以外はプロドラッグと同一の構造を有する薬剤のプロドラッグである、プロドラッグの使用によって達成される。
一態様において、本発明の目的は、ペプチジルグリシンαアミド化モノオキシゲナーゼ(PAM)によって薬剤を活性化するための、プロドラッグの使用によって達成される。
【0026】
本発明の好ましい態様において、本明細書中で用いられている表現「ペプチジルグリシンαアミド化モノオキシゲナーゼ(PAM)によって薬剤を活性化する」、「PAM活性化経路によってプロドラッグを活性化する」、生物活性化などは、プロドラッグがPAMによって基質として認識され代謝されることを示す。本発明の好ましい態様において、本明細書中で用いられている表現「薬剤のプロドラッグの製造を含む、薬剤をPAM活性化経路に導入すること」は、対応するプロドラッグ形態がPAM活性化経路に導入される薬剤から製造され、このプロドラッグ形態がPAMによって認識され代謝されることを示す。好ましい態様において、PAMに対するプロドラッグの親和性は、当業者がKM値を用いて決定でき得るとおり、薬剤と比較して、1〜1000倍、2〜100倍、3〜50倍、4〜40倍、5〜20倍または6〜15倍高い。
【0027】
一態様において、本発明の目的は、部分構造が、ヒドロキシルアミン、N−オキシド、ニトロン、ジアゼニウムジオラート(NONOat)または類似のN−O−含有一酸化窒素ドナー、ヒドロキサム酸、ヒドロキシ尿素、オキシム、アミドキシム(Nヒドロキシアミジン)、N−ヒドロキシアミジノヒドラゾンあるいはN−ヒドロキシグアニジンの一部であることを特徴とする、プロドラッグの使用によって達成される。
【0028】
本発明のさらなる側面において、本発明の目的は、薬剤のプロドラッグの製造を含む、フリーアミジンまたはグアニジン官能基を含む薬剤をPAM活性化経路へと導入するための方法によって達成される。
本発明のさらなる側面において、本発明の目的は、患者へのプロドラッグの投与を含む、患者を処置するための方法によって達成される。
本発明のさらなる側面において、本発明の目的は、薬剤を製造するためのプロドラッグの使用によって達成される。
【0029】
本発明の好ましい態様において、インフルエンザなどのウイルス感染に対抗するための、HIV感染に対抗するための、内臓や皮膚リーシュマニア症の予防と処置のための、ニューモシスチス・カリニ肺炎(PCP)の予防のための、トリパノソーマ症(アフリカ睡眠病)を処置するための、マラリアを処置するため、バベシア症を処置するための、血液凝固を阻害するための、例えば静脈血栓塞栓症の一次予防のための、心房細動患者の脳卒中の予防のための、血圧を下げるための、悪性腫瘍の成長を抑制するための、神経保護のための、インフルエンザなどのウイルス感染に対抗するための、例えば心不全、肺水腫、中毒、腎不全または肝硬変などに対する、体から水を(利尿的)排除するための、アレルギーを処置するための、喘息を処置するための、炎症性疾患、例えばリウマチまたは膵炎を処置するための、あるいは虚血(不十分な血液供給)の予防のための、薬剤は薬剤であり、またはプロドラッグはプロドラッグである。
【0030】
本発明のさらなる側面において、本発明の目的は請求項7〜11および14のいずれか一項に記載のプロドラッグ、または請求項13に記載の方法により達成され、該使用または該方法は一酸化窒素欠乏に関連する疾患の処置のための使用または方法である。
一態様において、本発明の目的は、薬剤またはプロドラッグが、プロテアーゼ阻害剤、DNA−インターカレート化合物およびRNA−インターカレート化合物、ウイルス酵素の阻害剤、ならびにN−メチル−D−アスパラギン酸受容体アンタゴニストからなる群から選択されることを特徴とする、プロドラッグの使用によって達成される。
【0031】
一態様において、本発明はプロドラッグの使用によって達成され、ここで該使用は、内臓及び/または皮膚のリーシュマニア症、トリパノソーマ症、トリパノソーマ症の第2相またはPneumocystis cariniiによって引き起こされる肺炎の予防および/または処置のため、悪性腫瘍の成長を阻害するため、血液凝固を阻害するため、血圧を下げるため、神経保護のため、またはインフルエンザおよびHIV感染を含むウイルス感染に対抗するための使用である。
【0032】
本発明のさらなる側面において、本発明は一般式(I)または(II)、
【化10】
式中R1およびR2は水素、アルキルラジカルまたはアリールラジカルである、を有する部分構造を含む薬剤によって達成される。
【0033】
一態様において、本発明の目的は、部分構造が、ヒドロキシルアミン、N−オキシド、ニトロン、ジアゼニウムジオラート(NONOat)または類似のN−O−含有一酸化窒素ドナー、ヒドロキサム酸、ヒドロキシ尿素、オキシム、アミドキシム(Nヒドロキシアミジン)、N−ヒドロキシアミジノヒドラゾンあるいはN−ヒドロキシグアニジンの一部であることを特徴とする、一般式(I)または(II)を有する部分構造を含む薬剤によって達成される。
一態様において、本発明の目的は、一酸化窒素欠乏に関連する疾患の処置のために設計された薬剤であることを特徴とする、前記クレームのいずれか一項に記載の薬剤によって達成される。
【0034】
一態様において、本発明の目的は、薬剤が、プロテアーゼ阻害剤、DNA−インターカレート化合物およびRNA−インターカレート化合物、ウイルス酵素の阻害剤、ならびにN−メチル−D−アスパラギン酸受容体アンタゴニストからなる群から選択されることを特徴とする、薬剤によって達成される。
【0035】
本発明のさらなる側面において、本発明の目的は、薬剤の溶解性、バイオアベイラビリティ、血液脳関門の横断、生物活性および/または物理的−化学的安定性を改善するために、
一般式(I)または(II)
【化11】
式中R1およびR2は水素、アルキルラジカルまたはアリールラジカルである、を形成する部分構造を含む薬剤を製造するためのO−カルボキシアルキル化N−O−含有官能基の使用によって達成される。
【0036】
一態様において、本発明の目的は、ペプチジルグリシンαアミド化モノオキシゲナーゼ(PAM)によって薬剤を活性化するためのO−カルボキシアルキル化N−O−含有官能基を含む薬剤の使用によって達成される。
一態様において、本発明の目的は、部分構造が、ヒドロキシルアミン、N−オキシド、ニトロン、ジアゼニウムジオラート(NONOat)または類似のN−O−含有一酸化窒素ドナー、ヒドロキサム酸、ヒドロキシ尿素、オキシム、アミドキシム(Nヒドロキシアミジン)、N−ヒドロキシアミジノヒドラゾンあるいはN−ヒドロキシグアニジンの一部であることを特徴とする、薬剤の使用によって達成される。
【0037】
一態様において、本発明の目的は、薬剤が一酸化窒素欠乏に関連する疾患の処置のために設計されたことを特徴とする、薬剤の使用によって達成される。
【0038】
一態様において、本発明の目的は、薬剤がプロテアーゼ阻害剤、DNA−インターカレート化合物およびRNA−インターカレート化合物、ウイルス酵素の阻害剤、ならびにN−メチル−D−アスパラギン酸受容体アンタゴニストからなる群から選択されることを特徴とする、薬剤の使用によって達成される。
【0039】
一態様において、本発明の目的は、薬剤が、内臓及び/または皮膚はリーシュマニア症、トリパノソーマ症、トリパノソーマ症の第2相またはPneumocystis cariniiによって引き起こされる肺炎の予防および/または処置のため、悪性腫瘍の成長を阻害するため、血液凝固を阻害するため、血圧を下げるため、神経保護のため、またはインフルエンザおよびHIV感染を含むウイルス感染に対抗するために設計されていることを特徴とする、薬剤の使用によって達成される。
【0040】
本発明のさらなる側面において、本発明による化合物および/またはその塩を含む、医薬化合物、医薬組成物および医薬製品を提供する。医薬組成物は、好ましくは担体および/またはアジュバントを含み、理想的にはそれらは薬理学的に適合性がある。当業者はそのような担体およびアジュバントについて一般的に精通している。本発明による化合物は、医薬品における使用のためにも提供される。
【図面の簡単な説明】
【0041】
【図1】図1は、グリオキサル酸形成の比色定量の結果を示す。
【図2】図2は、O−カルボキシメチルベンズアミドキシムを用いた比色アッセイにおける、KM値およびVmax値の決定の代表的な例示である。
【図3】図3は、N−カルボキシメトキシ−N’,N”−ジフェニルグアニジンを用いた比色アッセイにおける、KM値およびVmax値の決定の代表的な例示である。
【発明を実施するための形態】
【0042】
薬剤が少なくとも一種または二種以上の活性アミジン、N−ヒドロキシアミジン(アミドキシム)、グアニジンまたはN−ヒドロキシグアニジン官能基を提案された形態で含めば、それで十分である。したがって、薬剤は、例えば複数のアミドキシム官能基(例えばペントキシム(pentoxime)エステルとして2つ)、またはN−ヒドロキシグアニジン官能基、ここでこれらの少なくとも一種の基が前述の方法で修飾されている、を含むことができる。同様に、薬剤の混合物もまた用いることができ、それらのうちの少なくとも一種は本発明にしたがって修飾されている。
【0043】
本発明による化合物は、ボーラス投与のように一度に、毎日、毎週または毎月投与することができる。投与の方法も同様に容易に決定することができる。一般的に、可能な投与形態は、経口、直腸、例えば静脈内、筋肉内、皮下、経皮投与、肺内投与などの非経口およびエアロゾルとしての投与、膀胱内注入、腹腔内や心臓内注射、粘膜や膣内の適用を介する吸収、例えば坐剤などが挙げられる。
【0044】
この目的のために、液体製剤に用いるこれらの態様に対して、有効成分、または有効成分混合物を、適切な非毒性溶媒、例えば水、一価アルコール、特にエタノール、多価アルコール、特にグリセリンおよび/またはプロパンジオール、ポリグリコール、特にポリエチレングリコールおよび/またはミグリオール、グリセリンホルマール(glycerinformal)、ジメチルイソソルビド、天然または合成油などに受容させる。
【0045】
従来のベース製品、例えばベントナイト、ビーガム、グアー粉(guar meal)および/またはセルロース誘導体、特にメチルセルロースおよび/またはカルボキシメチルセルロース、ビニルアルコールおよび/またはビニルピロリドン製ポリマー、アルギン酸塩、ペクチン、ポリアクリレート、固体および/または液体ポリエチレングリコール、パラフィン、脂肪アルコール、ワセリンおよび/またはワックス、脂肪酸および/または脂肪酸エステルが、半固形または固形剤を製造するために用いられる。
【0046】
また、既知の増量剤、例えばコロイド状ケイ酸、タルク、乳糖、でんぷん粉、砂糖、ゼラチン、金属酸化物および/または金属塩が固形製剤中に存在してもよい。さらなる添加剤、例えば安定剤、乳化剤、分散剤や防腐剤などは、当然選択されるものである。
【0047】
驚くべきことに、窒素(N)を介して医薬分子に結合し、シトクロームP450(CYP450)酵素非依存性の生物活性化の経路を利用する、一般式(I)または(II)
【化12】
ここで、(I)および(II)は、例えばヒドロキシルアミン、N−オキシド、ニトロン、ジアゼニウムジオラート(NONOat)または類似のN−O−含有一酸化窒素ドナー、ヒドロキサム酸、オキシム、アミドキシム(Nヒドロキシアミジン)、N−ヒドロキシアミジノヒドラゾンあるいはN−ヒドロキシグアニジンの一部であり、およびR1(Rは、pro−R構成されなければならない)およびR2は、水素、アルキルラジカルまたはアリールラジカルである、で表される、O−カルボキシアルキル化N−O−含有官能基が見出された。
【0048】
CYP450酵素は一般的に酸化的O−脱アルキル化を触媒することが知られており、これは本明細書で提案しているプロドラッグ原理の場合においてもまた実際の薬剤を放出するために必要であることから、これは予想外の結果をもたらしている。
【0049】
提案するカルボキシアルキルラジカルを有するN−O−含有官能基のエーテル化は、CYP450酵素とは異なる酵素を生物活性化のために利用することができるという特別な利点を提供する:ペプチジルグリシンαアミド化モノオキシゲナーゼ(PAM)。例えば、これは副作用および他の同時投与された薬剤との前述の相互作用を防ぐ。
【0050】
高等生物(脊椎動物)において、ペプチジルグリシンα−アミド化モノオキシゲナーゼ(PAM)は、モノオキシゲナーゼドメイン(PHM、ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼ、EC 1.14.17.3)およびリアーゼドメイン(PHL、ペプチジル−α−ヒドロキシグリシンα−アミド化リアーゼ、EC4.3.2.5)を含有する二官能性酵素を構成する。全体的に、PAMは、スプライシングと発現による強度に組織特異的および発達依存的(development dependent)な調節を受けやすい。翻訳後修飾の意味の範囲内で、PAMは、多様な生理的に発生するペプチドホルモン、神経伝達物質および成長因子(例えば、サブスタンスP、ニューロペプチドY、オキシトシン、バソプレシン、カルシトニン)を活性化することができる。このプロセスにおいて、ペプチドは、モノオキシゲナーゼ反応における酸化的N−脱アルキル化による末端グリシンの分離によって、C末端がアミド化されている。
【0051】
本発明により提案しているとおりのカルボキシアルキルラジカルを有するN−O−含有官能基の特別な利点は、生理的条件下(pH6〜8)で負に荷電するカルボン酸の挿入に起因する改善された溶解性である。
さらなる利点は、本発明により提案するN−O−含有官能基のエーテル化−(アルコキシカルボニル)アルキルエーテルまたは(アリールオキシ)アルキルエーテルを用いる−が、親油性を大きく増加させて受動拡散を可能にし、これによりバイオアベイラビリティおよび/または血液脳関門の横断が向上することである。
【0052】
プロドラッグ基として、比較的小さいラジカル−最も簡単な場合において、カルボキシメチルラジカル−を使用する可能性は、医薬分子を適度の大きさにしか大きくせず、同様に有利である。
Wand et al. [Metabolism 1985、34、11、1044]は、異なるヒト組織におけるPAMの活性を分析し、CNSの組織において(特に下垂体において)最も高い活性を検出した。対照的に、典型的な異物代謝の臓器、肝臓と腎臓において、活性は認められなかった。計画されたプロドラッグ概念をまた利用することができる活性が、血漿、心臓および肺で同様に検出された。
【0053】
特に、CNSにおけるこの酵素の高い活性は、O−カルボキシアルキル化プロドラッグを、血液脳関門を通して輸送し、その後それらを変換するために利用することができる。しかしながら、経口適用および消化管からの吸収後の心血管系における生物活性化も可能である。
本発明によるプロドラッグシステムは、アミジンまたはグアニジン官能基を有する種々の薬剤に適用することができる。以下の薬剤が特に好ましい:
【0054】
ペンタミジン、ダビガトラン、BSF 411693(アボット)、イダゾキサン塩酸塩、イルベサルタン、リノグリリド、ロフェキシジン塩酸塩、テトラヒドロゾリン塩酸塩、トラゾリン、キシロメタゾリン塩酸塩、ペンタアミジンイセチオナート、タリバビリン、チアミン (ビタミンB1)、ボセンタン、ジブロモプロプアミジネイセチオナート、ヒドロキシスチルブアミジンイソチオナート、シブラフィバン、オルボフィバン、キセミロフィバン、アルガトロバン、キシメラガトラン、メラガトラン、2−ピペリジン酸、オルボフィバンアセタート、エピナスチン(レレスタット)、
【0055】
RO 43-8857、AB1(クロラムブシル類似体)、AMG-126737、AY-0068、B-623、BABIM、BIBT-986(べーリンガーインゲルハイム)、CI-1031(会社名:バイオサイエンス)、CJ-1332(会社名:キュラサイト)、CJ-463(会社名:キュラサイト)、CJ-672(会社名:キュラサイト)、CT50728(Portolla Pharmaceuticals)、CVS-3983、DX-9065a、ラミフィバン(ロシュ)、LB-30870(会社名:エルジー・ライフ・サイエンシーズ・リミテッド)、LY-178550(会社名:Lilly)、PHA-927Fおよび類似体、RO-44-3888(ロシュ)、セピモスタット、FUT-187(鳥居)、ビラミジン(Ribapharm)、WX-FX4(Wilex)、YM-60828(山之内製薬株式会社)、ZK-807191(Berlex バイオサイエンス)、NAPAP(SR 25477)、BIIL 315(べーリンガーインゲルハイム)、BIIL 260(べーリンガーインゲルハイム)、BIIL 284/260(べーリンガーインゲルハイム)、
【0056】
タノギトラン、モキシルバント、スチルバミジン、パナミジン(panamidine)、フラダフィバン、ジミナゼン、ロキシフィバン、フラミジン、PD0313052、PHA 927F、PHA 798、フィデキサバン、オタミキサバン、トロンブストップ(thromstop)(Thrombstop)、ザナミビル、アミロリド塩酸塩、アナグレリド塩酸塩、プログアニル、シメチジン、クロニジン塩酸塩、グアノキサン、ペラミビル、ロミフィジン、チラパザミン、チザニジン、トロニジン硝酸塩、メトホルミン、ジミナゼン、デブリソキン、スルファメタジン、エプチフィバチド、ファモチジン、バイエル薬品、ストレプトマイシン、ナファモスタット、FUT-175、イノガトラン、グアネチジン(Thilodigon)、
【0057】
3DP-10017、APC-366、CVS-1123、ジフェニルホスファート誘導体、E-64、FOY-305、MBGB、MIBG、RWJ-422521、シンタリン、WX-293、WX-340、BMS-189090、JTV-803(日本たばこ産業)、ナプサガトラン、イスメリン、Tan 1057A、Hydikal、Phenformix(Retardo)、ネトロプシン(シナノマイシン)、BIIB 722(サビポリド)、グアナドレル、デオキシスペルグアリン、BMS 262084、Siamformet(Orabet)、PPACK(Pebac)、メルゲトパ(プラマーカルボキシペプチダーゼ阻害剤)、ペラミビル、ファモチジン、ザルチジン。
【0058】
添付には、化学式、CAS登録番号および薬剤の効能を付したテーブルを含む。
【0059】
以下、本発明による4種のプロドラッグを例として示す:
【化13】
【0060】
【化14】
【0061】
【化15】
【0062】
【化16】
【0063】
非ペプチド性O−カルボキシアルキル化N−O−含有官能基が、PAMの基質として許容されるという驚くべき発見は、アミドキシム−ベースおよびN−ヒドロキシグアニジン−ベースのモデル化合物に基づく例示的な態様において実証される。
【0064】
O−カルボキシメチルベンズアミドキシム(1)をアミドキシムのモデル化合物として、そのPAM基質特性を試験した。O−カルボキシメチルベンズアミドキシムは、薬剤ベンズアミジンの有力なプロドラッグである。O−カルボキシメチルベンズアミドキシム(1)のベンズアミドキシム(2)へのPAM触媒による生物活性化は、同時にグリオキサル酸を放出しながら起こる。
【化17】
【0065】
図1に、グリオキサル酸形成の比色定量の結果を示す。決定したグリオキシル酸濃度は、平均値±2回ずつ測定された2つのインキュベーションからの標準偏差である。1のPAMの触媒作用の切断産物としてのグリオキサル酸の形成は、濃度依存的であることが確認された。PAMの最適pH(pH6.0)でのインキュベーションは、pH7.4でのインキュベーションと比べて極めて高い変換をもたらした。比色アッセイにおいて、グリオキシル酸の5点キャリブレーションを、1の試験と同時に行った。キャリブレーションは、決定された濃度の範囲で線形的であった(r2=1.000)。
【0066】
これらの結果から、O−カルボキシメチルベンズアミドキシム(1)は、PAMによる基質として許容されたため、反応を、KM値およびVmax値を決定することによって、より詳細に特徴づけた。
この目的のため、HPLC分析を開発した。ベンズアミドキシムに対するキャリブレーション線は、決定された濃度の範囲で線形的であり(r2=1.000)、回収率は130.6%であった(r2=0.999)。2つの独立した実験(n=2)は、KM値307±80μMおよびVmax値393±40nmolmin−1mg−1PAMとなった。図2はかかる決定の代表的な例示である。
【0067】
CYP450基質の研究のために、前述のHPLC分析を、さらに、ベンズアミドキシム(2)のN−還元(N−reduction)の生成物として考えられる代謝産物ベンズアミジンの検出が可能になるように改変した。pH6.0およびpH7.4で、ベンズアミドキシム(ベンズアミジンの有力なプロドラッグ)とベンズアミジンのいずれもCYP450酵素源において検出されなかった。
ベンズアミドキシムモデル化合物1に基づいて、O−カルボキシメチル官能基はPAMから除去されるが、モノオキシゲナーゼ反応の意味の範囲内において、シトクロームP450からは除去されない。
【0068】
N−カルボキシメトキシ−N’,N”−ジフェニルグアニジン(3)をヒドロキシグアニジンモデル化合物としてそのPAM基質特性について試験した。
【化18】
【0069】
N−カルボキシメトキシ−N’,N”−ジフェニルグアニジン(3)のN,N’−ジフェニル−N”−ヒドロキシグアニジンへのPAM触媒による生物活性化は、同時にグリオキサル酸を放出しながら起こる。
【0070】
3を用いた比色アッセイの結果は、アミドキシムモデル化合物1の結果に匹敵するものであった。KM値およびVmax値を決定するために、プロドラッグ3とヒドロキシグアニジン4をRPカラム上で15分以内に分離することができるHPLCを開発した。N,N’−ジフェニル−N”−ヒドロキシグアニジン(4)に対するキャリブレーション線は、決定された濃度の範囲で線形的であり(r2=0.999)、回収率は111.7%であった(r2=0.999)。2つの独立した実験(n=2)は、KM値37±5μMおよびVmax値373±53μmolmin−1mg−1PAMとなった。図3はかかる決定の代表的な例示である。
【0071】
変換速度が同等の場合、決定したKM値から、PAMに対する親和性が、アミドキシムプロドラッグ1と比較して約8倍以上であることが導かれ得る。
CYP450基質の研究のために、PAM基質のために開発したHPLC分析を、さらに考えられる代謝産物N,N’−ジフェニルグアニジンの検出が、ヒドロキシグアニジン4のN−還元の生成物として可能になるように改変した。pH6.0およびpH7.4で、4とN,N’−ジフェニルグアニジンのいずれも、180分のインキュベーションタイム後の用いたCYP450酵素源のいずれにおいても検出されなかった。
【0072】
O−カルボキシメチルベンズアミドキシム(1)と同様に、ヒドロキシグアニジンモデル化合物3に基づいて、O−カルボキシメチル官能基は、PAMからのみ除去されるが、モノオキシゲナーゼ反応の意味の範囲内において、シトクロームP450からは除去されない。
【0073】
材料および方法
改変されたO−カルボキシメチルベンズアミドキシム一水和物のナトリウム塩(1)Koch [Ber. Dtsch. Chem. Ges. 1889、22、3161]による手順:
【化19】
【0074】
5mlエタノール中の681mgベンズアミドキシム(5.0mmol)、1.04gブロモ酢酸(7.5mmol)および500mg水酸化ナトリウムペレット(12.5mmol)の溶液を還流下で5時間沸騰させる。その後、沈殿物が形成され始めるまで真空下で溶媒を除去する。沈殿物を完全に沈殿させ、濾過し、乾燥する。生成物をエタノール(96%)/水(95:5)から再結晶させる。
【0075】
【化20】
【0076】
N−カルボキシメトキシ−N’,N”−ジフェニルグアニジン(3)
【化21】
【0077】
546mgアミノオキシ酢酸セミクロリド(5mmol)および697μlトリエチルアミン(5mmol)を乾燥DMF(dryDMF)10ml中で30分間撹拌する。沈殿物をろ過し、濾液に970mgN,N’−ジフェニルカルボジイミド(5mmol)を加える。バッチを4時間室温で撹拌し、酢酸エチルで溶媒抽出し、生成物をエタノールから再結晶する。
【0078】
【化22】
【0079】
HPLCシステム
Waters 1525 ポンプ、Waters 2487吸収検出器、Waters 717 Plus オートサンプラーおよびBreeze記録および評価ソフトウェア(バージョン3.30)、Gynkotek STH 585カラムオーブンを備えたWaters Breeze HPLCシステム
【0080】
HPLCカラム:
C-18プレカラム(4 x 3mm)(Phenomenex)を備えたSynergiMax-RP80A(250 x 4.6mm、4μm);
LiChroCART、LiChrospher100、LiChrospher60プレカラムを備えたRP-8(125 x 4mm、5μm)、RP-selectB(4 x 4mm、5μm)(Merck);
LiChroCART、LiChrospher60プレカラムを備えたLiChrospher RP-selectB(250 x 4.6mm、5μm)、RP-selectB(4 x 4mm、5μm)(Merck)
【0081】
追加のデバイスおよび材料:
Cary 50紫外可視分光光度計(Varian);96−ウェルプレート(Greiner);GFL-1083振とう水浴(Gesellschaft fuer Labortechnik、Burgwedel);マイクロリットル遠心分離機(Hettich GmbH);LiQ Plast pH 電極(Hamilton)付InoLab pH Level 1 pH測定デバイス(Wissenschaftlich-Technische Werkstaetten GmbH、Weilheim);VF2ボルテクサー(vortexer)(Janke und Kunkel GmbH & Co. KG、Staufen);1.5 ml反応器(Sarstedt AG & Co.、Nuembrecht)
【0082】
酵素源:使用した組換えペプチジルグリシンα−アミド化モノオキシゲナーゼ(PAM、ラット、EC 1.14.17.3)は、Unigene Laboratories,Inc.(ニュージャージー州、米国)(比活性=5.8 106U/mgタンパク質)から提供された;牛肝臓カタラーゼ(EC 1.11.1.6)、比活性=12600U/mg固体(アルドリッチ)。
【0083】
使用したシトクロームP450酵素源は、Clement von Gruenewaldワーキンググループにおいて、以下の説明に従い入手した:
ブタ肝ミクロソームおよび上清9000g:
豚の肝臓は、地元の肉屋(Bordesholm)から調達し、臓器は、屠殺後、氷冷した20mM リン酸バッファー(1mM NA2 EDTA、pH7.4)中で、直接輸送した。さらなる処理のために、まず肝葉を50mM リン酸バッファー(1mM NA2 EDTA、pH7.4)で灌流し、洗浄した。組織片に切断し、市販の肉挽き器に通した。懸濁液を等量のリン酸バッファーで希釈し、フローホモジナイザーを用いてホモジナイズした。さらに、差動超遠心分離することによってミクロソームおよび上清9000グラムを得た。貯蔵のために、得られた標本は、小分けして−80℃で凍結した。
【0084】
ヒト肝ミクロソームおよび上清9000g:
ヒトミクロソームを得るために、クリスチャン−アルブレクト大学(Christian-Albrecht University)大学病院の外科の、部分的肝切除を受けなければならなかった癌患者からヒト肝組織を得た。
【0085】
肝臓の組織片は、ショ糖含有リン酸バッファー(10mM K2HPO4、10mM KH2PO4、250mM ショ糖、1mM NA2 EDTA、pH7.4、4℃)で急速冷凍した。臓器片の十分な量が(>3)得られたらすぐに、対応する小片を、個体間変動に起因する差異を相殺するために、解凍し、プールした。組織片を、4℃でより小さい片に切断し、バッファー(EDTAを含まない)で数回洗浄し、ホモジナイザーを用いて懸濁液へと処理した。さらに、分画超遠心分離することによってミクロソームおよび上清9000グラムを得た。貯蔵のために、得られた標本は、小分けして−80℃で凍結した。
【0086】
PAMアッセイ:インキュベーション条件
典型的な300μl(総量)のインキュベーションバッチに、25000 U/ml ペプチジルグリシンα−アミド化モノオキシゲナーゼ(PAM、会社名:Unigene Laboratories)、250U/ml カタラーゼ、1μM 銅(II)(酢酸/水和物として採用)、2mM アスコルビン酸ナトリウム、5mM ヨウ化カリウムおよび0.1mMまたは1mMの濃度のそれぞれの基質を、種々のpH値を有するバッファー中に入れた。使用したバッファー系は、pH6.0でのインキュベーションのために30mM MESならびにpH7.4のインキュベーションのために50mM HEPESであった。pH値は、それぞれの場合において希水酸化ナトリウムで調整した。インキュベーションを37℃で60分間振とう水浴中で行い、100μlを取り出し、反応を50μl 10%TFA(aq)/アセトニトリル(2:3)で停止させた。残りのバッチは、37℃で別に180分間インキュベートし、100μl 10%TFA(aq)/アセトニトリル(2:3)で停止させた。
【0087】
停止したサンプルは5分間振とうし(ボルテクサー)、−80℃で凍結させた。サンプルを分析するために、それらを解凍し、5分間振とうし、沈殿したタンパク質を、10000rpmで遠心分離した。上清を比色グリオキシル酸定量および/またはHPLCの測定に用いた。KMおよびVmaxの決定のために、100μlのバッチを、pH6.0で前述の条件下、しかしインキュベーション時間が30分という点で異なる、で処理した。
【0088】
グリオキシル酸の比色定量
タンパク質が除かれたインキュベーションバッチ200μlを20μlのフェニルヒドラジン溶液(2mlaqua bidest中20mg)と混合し、37℃で5分間振とう水浴中で振とうした。その後、混合物を15分間で0℃まで冷却し、100μl 氷冷6NのHClを加え、0℃でさらに5分間放置した。その後、20μl のヘキサシアノ鉄酸(III)カリウム溶液(2mlaqua bidest中100mg)を加えた。バッチを、室温で15分間放置し、200μlをプレートリーダー(Cary 50紫外可視分光光度計、520nm)を用いる測定のために取り出した。
キャリブレーション:
5点キャリブレーションについて、アッセイバッファー(pH6.0):10%TFA(aq)/アセトニトリル(2:3)の2:1混合物中の2、5、10、50および100μM濃度のグリオキサル酸を上記のとおり測定した。このキャリブレーションは、実施したテスト化合物の各アッセイに対して同時に行った。
【0089】
O−カルボキシメチルベンズアミドキシム(1)およびベンズアミドキシム(2)の分離のためのHPLC分析
【表1】
【0090】
キャリブレーションと回収
キャリブレーションのために、ベンズアミドキシムを0.1〜500μMの8つの濃度で溶解し、アッセイバッファー(30mM MES、1μM 酢酸銅(II)、2mM アスコルビン酸ナトリウム、5mM ヨウ化カリウム、pH6.0)中に溶解し、上述のHPLC法を用いて測定した。
回収を決定するために、アッセイバッファー(エンド容量=100μl)中に同濃度のものを製造した。さらに、O−カルボキシメチルベンズアミドキシム(0.5mM)および250U/mlカタラーゼを加え、50μl10%TFA(aq)/アセトニトリル(2:3)を続いて加えた。サンプルを、ボルテクサーを用いて振とうし、−80℃で凍結させた。サンプルを測定するために、それらを解凍し、ボルテクサーを用いて5分間振とうし、10000rpmで5分間遠心分離した。
【0091】
N−カルボキシメトキシ−N’,N”−ジフェニルグアニジン(3)およびN−ヒドロキシ−N’,N”−ジフェニルグアニジン(4)の分離のためのHPLC分析
【表2】
【0092】
キャリブレーションと回収
キャリブレーションのために、N−ヒドロキシ−N’,N”−ジフェニルグアニジン(4)を0.1〜500μMの8つの濃度で溶解し、アッセイバッファー(30mM MES、1μM 酢酸銅(II)、2mM アスコルビン酸ナトリウム、5mM ヨウ化カリウム、pH6.0)中に溶解し、上述のHPLC法を用いて測定した。
回収を決定するために、アッセイバッファー(エンド容量=100μl)中に同濃度のものを製造した。さらに、N−カルボキシメトキシ−N’,N”−ジフェニルグアニジン(3)(0.5mM)および250U/mlカタラーゼを加え、50μl10%TFA(aq)/アセトニトリル(2:3)を続いて加えた。サンプルを、ボルテクサーを用いて振とうし、−80℃で凍結させた。サンプルを測定するために、それらを解凍し、ボルテクサーを用いて5分間振とうし、10000rpmで5分間遠心分離した。
【0093】
CYP450アッセイ:インキュベーション条件
典型的な500μl(総量)のインキュベーションバスに、0.3mgタンパク質(豚またはヒト肝臓酵素源)、0.1mM(または1mM)のテスト化合物を、100mM リン酸バッファー(pH6.0またはpH7.4)および1mM NADH(またはNADPH)中に入れた。バッファー中の酵素およびテスト化合物の5分間プレインキュベーション後、インキュベーションを開始し、NADH(またはNADPH)を追加し、生成物を振とう水浴槽で37℃60分間または180分間振とうした。反応を同量のアセトニトリルを加えることによって停止し、ボルテクサーを用いて振とうし、−80℃で凍結させた。
サンプルを分析するために、それらを解凍し、ボルテクサーを用いて5分間振とうし、5分間10000rpmで遠心分離することによって、タンパク質を分離した。上清をHPLC分析に用いた。
【0094】
O−カルボキシメチルベンズアミドキシム(1)、ベンズアミドキシム(2)およびベンズアミジンの分離のためのHPLC分析
【表3】
【0095】
N−カルボキシメトキシ−N’,N”−ジフェニルグアニジン(3)、N−ヒドロキシ−N’,N”−ジフェニルグアニジン(4)およびN’,N”−ジフェニルグアニジンの分離のためのHPLC分析
【表4】
【0096】
以下に、本発明によるプロドラッグシステムを好ましく適用できる医薬品の表を示す。
【表5】
【表6】
【表7】
【表8】
【表9】
【表10】
【表11】
【表12】
【表13】
【表14】
【特許請求の範囲】
【請求項1】
一般式(I)または(II)
【化1】
式中、R1およびR2は水素、アルキルラジカルまたアリールラジカルである、
を有する部分構造を含む、プロドラッグ。
【請求項2】
プロドラッグが含む部分構造が、ヒドロキシルアミン、N−オキシド、ニトロン、ジアゼニウムジオラート(NONOat)または類似のN−O−含有一酸化窒素ドナー、ヒドロキサム酸、ヒドロキシ尿素、オキシム、アミドキシム(N−ヒドロキシアミジン)、N−ヒドロキシアミジノヒドラゾンあるいはN−ヒドロキシグアニジンの一部であることを特徴とする、請求項1に記載のプロドラッグ。
【請求項3】
プロドラッグが、一酸化窒素欠乏に関連する疾患の処置のための薬剤へと代謝されることを特徴とする、請求項1または2に記載のプロドラッグ。
【請求項4】
プロドラッグまたは対応する薬剤が、プロテアーゼ阻害剤、DNA−インターカレート化合物およびRNA−インターカレート化合物、ウイルス酵素の阻害剤、ならびにN−メチル−D−アスパラギン酸受容体アンタゴニストからなる群から選択されることを特徴とする、請求項1〜3のいずれか一項に記載のプロドラッグ。
【請求項5】
部分構造が、一般式IIaまたはIIb
【化2】
を有することを特徴とする、請求項1〜4のいずれか一項に記載のプロドラッグ。
【請求項6】
プロドラッグが薬剤のプロドラッグであり、
一般式IIaで表される部分構造が、代謝後に式
【化3】
を有する構造を含み、ならびに
一般式IIbで表される部分構造が、代謝後に式
【化4】
を有する構造を含むことを特徴とする、請求項5に記載のプロドラッグ。
【請求項7】
薬剤のプロドラッグであるプロドラッグの全体構造の一部としての、
一般式(I)または(II)
【化5】
式中、R1およびR2は、水素、アルキルラジカルまたはアリールラジカルである、を形成する部分構造の使用。
【請求項8】
部分構造が、一般式(II)の構造を有し、ここで、該部分構造は、薬剤のアミジン基またはグアニジン基に代わる高次の部分構造IIaまたはIIb
【化6】
の一部であり、溶解性、経口バイオアベイラビリティ、血液脳関門の横断、風味および/または物理化学的安定性を改善する、請求項7に記載の使用。
【請求項9】
プロドラッグが、高次構造IIaの代わりにIIa−1またはIIa−2
【化7】
の部分構造の1つを含むか、または
高次構造IIbの代わりにIIb−1またはIIb−2
【化8】
の部分構造の1つを含む以外はプロドラッグと同一の構造を有する薬剤のプロドラッグである、請求項7または8に記載の使用。
【請求項10】
ペプチジルグリシンαアミド化モノオキシゲナーゼ(PAM)によって薬剤を活性化するための、請求項8または9に記載の使用。
【請求項11】
部分構造が、ヒドロキシルアミン、N−オキシド、ニトロン、ジアゼニウムジオラート(NONOat)または類似のN−O−含有一酸化窒素ドナー、ヒドロキサム酸、ヒドロキシ尿素、オキシム、アミドキシム(N−ヒドロキシアミジン)、N−ヒドロキシアミジノヒドラゾンあるいはN−ヒドロキシグアニジンの一部であることを特徴とする、請求項7〜10のいずれか一項に記載の使用。
【請求項12】
請求項1〜6のいずれか一項に記載の薬剤のプロドラッグの製造を含む、フリーのアミジンまたはグアニジン官能基を含む薬剤をPAM活性化経路へと導入するための方法。
【請求項13】
請求項1〜6のいずれか一項に記載のプロドラッグを患者に投与することを含む、患者を処置するための方法。
【請求項14】
薬剤を製造するための請求項1〜6のいずれか一項に記載のプロドラッグの使用。
【請求項15】
請求項7〜11および13のいずれか一項に記載の使用または請求項14に記載の方法であって、該使用または方法が、一酸化窒素欠乏に関連する疾患の処置のためのものである、前記使用または方法。
【請求項16】
薬剤またはプロドラッグが、プロテアーゼ阻害剤、DNA−インターカレート化合物およびRNA−インターカレート化合物、ウイルス酵素の阻害剤、ならびにN−メチル−D−アスパラギン酸受容体アンタゴニストからなる群から選択されることを特徴とする、請求項7〜11および15に記載の使用。
【請求項17】
使用が、内臓および/または皮膚のリーシュマニア症、トリパノソーマ症、トリパノソーマ症の第2相またはPneumocystis cariniiによって引き起こされる肺炎の予防および/または処置のため、悪性腫瘍の成長を阻害するため、血液凝固を阻害するため、血圧を下げるため、神経保護のため、またはインフルエンザおよびHIV感染を含むウイルス感染に対抗するための使用である、請求項7〜11および15〜17のいずれか一項に記載の使用。
【請求項18】
式
【化9】
で表される、N−アルコキシグアニジンの製造方法であって、
式R6−N=C=N−R7で表されるカルボジイミドと式H2N−O−R8で表されるアミノオキシ化合物またはその塩との反応を含み、ここでアミノオキシ化合物の塩を用いる場合には、反応を塩基の存在下で行なわれ、
式中R1、R2、R3およびR4、R6およびR7は、各々独立して、H、任意に置換されたアルキル、任意に置換されたアリール、任意に置換されたヘテロアリール、任意に置換されたアリールオキシカルボニル、任意に置換されたアミノアシル、任意に置換されたアルコキシカルボニル、任意に置換されたアルコキシカルボニルアルコキシ、任意に置換されたヘテロアルキル、任意に置換されたアルキルシクロアルキル、任意に置換されたヘテロアルキルシクロアルキル、任意に置換されたアラルキルおよび任意に置換されたシクロアルキルからなる群から選択され、
R5は、アルコキシカルボニル、(アルコキシカルボニル)アルコキシおよびカルボキシアルコキシからなる群から選択される、前記製造方法。
【請求項19】
反応が、塩基の存在下で行われ、塩基が好ましくはジイソプロピルアミンおよびトリエチルアミンからなる群から選択される、請求項18に記載の方法。
【請求項1】
一般式(I)または(II)
【化1】
式中、R1およびR2は水素、アルキルラジカルまたアリールラジカルである、
を有する部分構造を含む、プロドラッグ。
【請求項2】
プロドラッグが含む部分構造が、ヒドロキシルアミン、N−オキシド、ニトロン、ジアゼニウムジオラート(NONOat)または類似のN−O−含有一酸化窒素ドナー、ヒドロキサム酸、ヒドロキシ尿素、オキシム、アミドキシム(N−ヒドロキシアミジン)、N−ヒドロキシアミジノヒドラゾンあるいはN−ヒドロキシグアニジンの一部であることを特徴とする、請求項1に記載のプロドラッグ。
【請求項3】
プロドラッグが、一酸化窒素欠乏に関連する疾患の処置のための薬剤へと代謝されることを特徴とする、請求項1または2に記載のプロドラッグ。
【請求項4】
プロドラッグまたは対応する薬剤が、プロテアーゼ阻害剤、DNA−インターカレート化合物およびRNA−インターカレート化合物、ウイルス酵素の阻害剤、ならびにN−メチル−D−アスパラギン酸受容体アンタゴニストからなる群から選択されることを特徴とする、請求項1〜3のいずれか一項に記載のプロドラッグ。
【請求項5】
部分構造が、一般式IIaまたはIIb
【化2】
を有することを特徴とする、請求項1〜4のいずれか一項に記載のプロドラッグ。
【請求項6】
プロドラッグが薬剤のプロドラッグであり、
一般式IIaで表される部分構造が、代謝後に式
【化3】
を有する構造を含み、ならびに
一般式IIbで表される部分構造が、代謝後に式
【化4】
を有する構造を含むことを特徴とする、請求項5に記載のプロドラッグ。
【請求項7】
薬剤のプロドラッグであるプロドラッグの全体構造の一部としての、
一般式(I)または(II)
【化5】
式中、R1およびR2は、水素、アルキルラジカルまたはアリールラジカルである、を形成する部分構造の使用。
【請求項8】
部分構造が、一般式(II)の構造を有し、ここで、該部分構造は、薬剤のアミジン基またはグアニジン基に代わる高次の部分構造IIaまたはIIb
【化6】
の一部であり、溶解性、経口バイオアベイラビリティ、血液脳関門の横断、風味および/または物理化学的安定性を改善する、請求項7に記載の使用。
【請求項9】
プロドラッグが、高次構造IIaの代わりにIIa−1またはIIa−2
【化7】
の部分構造の1つを含むか、または
高次構造IIbの代わりにIIb−1またはIIb−2
【化8】
の部分構造の1つを含む以外はプロドラッグと同一の構造を有する薬剤のプロドラッグである、請求項7または8に記載の使用。
【請求項10】
ペプチジルグリシンαアミド化モノオキシゲナーゼ(PAM)によって薬剤を活性化するための、請求項8または9に記載の使用。
【請求項11】
部分構造が、ヒドロキシルアミン、N−オキシド、ニトロン、ジアゼニウムジオラート(NONOat)または類似のN−O−含有一酸化窒素ドナー、ヒドロキサム酸、ヒドロキシ尿素、オキシム、アミドキシム(N−ヒドロキシアミジン)、N−ヒドロキシアミジノヒドラゾンあるいはN−ヒドロキシグアニジンの一部であることを特徴とする、請求項7〜10のいずれか一項に記載の使用。
【請求項12】
請求項1〜6のいずれか一項に記載の薬剤のプロドラッグの製造を含む、フリーのアミジンまたはグアニジン官能基を含む薬剤をPAM活性化経路へと導入するための方法。
【請求項13】
請求項1〜6のいずれか一項に記載のプロドラッグを患者に投与することを含む、患者を処置するための方法。
【請求項14】
薬剤を製造するための請求項1〜6のいずれか一項に記載のプロドラッグの使用。
【請求項15】
請求項7〜11および13のいずれか一項に記載の使用または請求項14に記載の方法であって、該使用または方法が、一酸化窒素欠乏に関連する疾患の処置のためのものである、前記使用または方法。
【請求項16】
薬剤またはプロドラッグが、プロテアーゼ阻害剤、DNA−インターカレート化合物およびRNA−インターカレート化合物、ウイルス酵素の阻害剤、ならびにN−メチル−D−アスパラギン酸受容体アンタゴニストからなる群から選択されることを特徴とする、請求項7〜11および15に記載の使用。
【請求項17】
使用が、内臓および/または皮膚のリーシュマニア症、トリパノソーマ症、トリパノソーマ症の第2相またはPneumocystis cariniiによって引き起こされる肺炎の予防および/または処置のため、悪性腫瘍の成長を阻害するため、血液凝固を阻害するため、血圧を下げるため、神経保護のため、またはインフルエンザおよびHIV感染を含むウイルス感染に対抗するための使用である、請求項7〜11および15〜17のいずれか一項に記載の使用。
【請求項18】
式
【化9】
で表される、N−アルコキシグアニジンの製造方法であって、
式R6−N=C=N−R7で表されるカルボジイミドと式H2N−O−R8で表されるアミノオキシ化合物またはその塩との反応を含み、ここでアミノオキシ化合物の塩を用いる場合には、反応を塩基の存在下で行なわれ、
式中R1、R2、R3およびR4、R6およびR7は、各々独立して、H、任意に置換されたアルキル、任意に置換されたアリール、任意に置換されたヘテロアリール、任意に置換されたアリールオキシカルボニル、任意に置換されたアミノアシル、任意に置換されたアルコキシカルボニル、任意に置換されたアルコキシカルボニルアルコキシ、任意に置換されたヘテロアルキル、任意に置換されたアルキルシクロアルキル、任意に置換されたヘテロアルキルシクロアルキル、任意に置換されたアラルキルおよび任意に置換されたシクロアルキルからなる群から選択され、
R5は、アルコキシカルボニル、(アルコキシカルボニル)アルコキシおよびカルボキシアルコキシからなる群から選択される、前記製造方法。
【請求項19】
反応が、塩基の存在下で行われ、塩基が好ましくはジイソプロピルアミンおよびトリエチルアミンからなる群から選択される、請求項18に記載の方法。
【図1】

【図2】
【図3】
【図2】
【図3】
【公表番号】特表2012−514608(P2012−514608A)
【公表日】平成24年6月28日(2012.6.28)
【国際特許分類】
【出願番号】特願2011−544787(P2011−544787)
【出願日】平成22年1月8日(2010.1.8)
【国際出願番号】PCT/DE2010/000009
【国際公開番号】WO2010/078867
【国際公開日】平成22年7月15日(2010.7.15)
【出願人】(510109165)ドリッテ・パテントポートフォリオ・ベタイリグンスゲゼルシャフト・エムベーハー・ウント・コンパニー・カーゲー (8)
【氏名又は名称原語表記】Dritte Patentportfolio Beteiligungsgesellschaft mbH & Co. KG
【住所又は居所原語表記】Berliner Str. 1, 12529 Schoenefeld/Waltersdorf, Germany
【Fターム(参考)】
【公表日】平成24年6月28日(2012.6.28)
【国際特許分類】
【出願日】平成22年1月8日(2010.1.8)
【国際出願番号】PCT/DE2010/000009
【国際公開番号】WO2010/078867
【国際公開日】平成22年7月15日(2010.7.15)
【出願人】(510109165)ドリッテ・パテントポートフォリオ・ベタイリグンスゲゼルシャフト・エムベーハー・ウント・コンパニー・カーゲー (8)
【氏名又は名称原語表記】Dritte Patentportfolio Beteiligungsgesellschaft mbH & Co. KG
【住所又は居所原語表記】Berliner Str. 1, 12529 Schoenefeld/Waltersdorf, Germany
【Fターム(参考)】
[ Back to top ]